/



































































































































































































































































































































































































































































































































Текст
ЭНЦИКЛОПЕДИЯ
ПОЛИМЕРОВ
РЕДАКЦИОННАЯ КОЛЛЕГИЯ
В. А. КАБАНОВ (главный редактор), М. С. АКУТИН, Н. Ф. БАКЕЕВ,
Е. В. ВОНСКИЙ (ответственный секретарь), В. Ф. ЕВСТРАТОВ, Н. С. ЕНИ-
КОЛОПЯН, В. В. КОРШАК, М. М. КОТОН, Б. А. КРЕНЦЕЛЬ, А. Б. ПАК-
ШВЕР, В. С. СМИРНОВ, Г. Л. СЛОНИМСКИЙ (зам. главного редактора),
С. Я. ФРЕНКЕЛЬ, С. В. ЯКУБОВИЧ.
2
Л—Полинозные волокна
ИЗДАТЕЛЬСТВО «СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
54.01@3)
Э.68
РЕДАКТОРЫ ОТДЕЛОВ ЭНЦИКЛОПЕДИИ ПОЛИМЕРОВ
Полимеризационные процессы. Члены редколлегии: член-корр. АН СССР Н. С.
ЕНИКОЛОПЯН, член-корр. АН СССР В. А. КАБАНОВ. Редакторы-консультанты: канд. химич.
наук А. А. АРЕСТ-ЯКУБОВИЧ, доктор химич. наук Б. Л. ЕРУСАЛИМСКИЙ, доктор химич.
наук В. П. ЗУБОВ, доктор химич. наук Н. А. ПЛАТЭ.
Поликонденсационные процессы. Член редколлегии член-корр. АН СССР
В. В. КОРШАК. Редакторы-консультанты: канд. химич. наук П. М. ВАЛЕЦКИЙ, канд. химич.
наук В. А. ВАСНЕВ, доктор химич. наук Л. Б. СОКОЛОВ.
Физика и физик о-х имия полимеров. Члены редколлегии: доктор химич. наук
Н. Ф. БАКЕЕВ, доктор химич. наук Г. Л. СЛОНИМСКИЙ, доктор физ.-мат. наук С. Я. ФРЕН-
ФРЕНКЕЛЬ. Редакторы-консультанты: канд. физ.-мат. наук Ал. Ал. БЕРЛИН, канд. химич. наук
A. Б. ЗЕЗИН, доктор физ.-мат. наук А. Я. МАЛКИН, канд. химич. наук Э. Ф. ОЛЕЙНИК, доктор
физ.-мат. наук С. Б. РАТНЕР, канд. химич. наук Л. А. ШИЦ.
Индивидуальные полимеры и их классы, мономеры. Члены редколлегии:
член-корр. АН СССР М. М. КОТОН, доктор химич. наук Б. А. КРЕНЦЕЛЬ. Редакторы-консуль-
Редакторы-консультанты: доктор химич. наук А. А. ЖДАНОВ, доктор химич. наук К. С. МИНСКЕР, канд. технич.
наук М. В. ПРОКОФЬЕВА, канд. химич. наук Е. Ф. РАЗВОДОВСКИЙ, канд. химич. наук
B. П. ШИБАЕВ.
Пластмассы. Члены редколлегии: доктор технич. наук М. С. АКУТИН, канд. химич. наук
В. С. СМИРНОВ. Редакторы-консультанты: канд. химич. наук А. Б. ДАВЫДОВ, канд. химич.
наук В. В. КОВРИГА, доктор химич. наук П. В. КОЗЛОВ, канд. технич. наук А. А. ПЕШЕХО-
НОВ, доктор химич. наук Е. Б. ТРОСТЯНСКАЯ.
Каучуки и резины. Член редколлегии член-корр. АН СССР В. Ф. ЕВСТРАТОВ. Ре-
Редактор-консультант доктор технич. наук А. И. ЛУКОМСКАЯ.
Лакокрасочные материалы. Член редколлегии доктор химич. наук С. В. ЯКУ БО-
ВИЧ. Редактор-консультант канд. технич. наук М. М. ГОЛЬДБЕРГ.
Химические волокна. Член редколлегии доктор технич. наук А. Б. ПАКШВЕР.
Переработка полимерных материалов. Член редколлегии доктор технич. наук
М. С. АКУТИН. Редакторы-консультанты: канд. технич. наук В. К. ЗАВГОРОДНИЙ, канд.
химич. наук М. Л. КЕРБЕР, доктор технич. наук Р. В. ТОРНЕР.
РЕДАКЦИЯ
Заведующий редакцией В. М. САХАРОВ; научные редакторы: канд. химич. наук
Е. В. ВОНСКИЙ, Э. С. ДРАГУНОВ, Н. А. ДУБРОВСКАЯ, Р. Я. ПЕСЧАНСКАЯ,
Л. С. СОЛОДКИН; младшие редакторы: А. А. МЕРКУЛОВА, В. А. СОЛОМЕН-
НИКОВА.
Редактор-библиограф Е. И. ШАРОВА
Художественный редактор И. П. САХАРОВА
Технический редактор Г. В. СМИРНОВА
Указатель составил А. Б. ДМИТРИЕВ
r 20505-023 ,-, rr~
007@1)-74 П°Дписное (& ИЗДАТЕЛЬСТВО «COBETCI^JyfГМОЦЕДИЯ». 1974 •
л
ЛАВСАН — см. Полиэфирные волокна.
ЛАКИ И ЭМАЛИ (varnishes and enamels, Lacke und
Emaillen, vernis et emaux). Лаки (Л.) — р-ры при-
природных или синтетич. пленкообразующих веществ (плен-
кообразователей) в органич. растворителях. Э м а-
л и (Э.), или эмалевые краски, — суспензии
пигментов (иногда в смеси с наполнителями) в Л. После
нанесения тонким слоем на подложку и высыхания Л.
образуют прозрачные, Э.— непрозрачные лакокрасоч-
лакокрасочные покрытия. В зависимости от вида пленкообразую-
пленкообразующего различают масляные, алкидные, кремнийоргани-
ческие, полиэфирные, полиуретановые, полиакриловые,
хлоркаучуковые и др. Л. и Э.
Состав. Л. и Э. содержат нелетучие и летучие компо-
компоненты. К нелетучим относятся пленкообразующие, пиг-
пигменты, наполнители, сиккативы, отвердители, пласти-
пластификаторы, стабилизаторы, поверхностно-активные ве-
вещества и др. Летучие компоненты Л. и Э.— раство-
растворители и разбавители.
Пленкообразующие вещества — ос-
основные компоненты любого лакокрасочного материала,
которые после высыхания слоя Л. или Э. создают на
окрашиваемой поверхности прочное лакокрасочное по-
покрытие и обусловливают его адгезию к подложке. В
Э. пленкообразующие, кроме того, смачивают и проч-
прочно удерживают частицы пигментов и наполнителей.
Большинство пленкообразующих — олигомеры, пере-
переходящие в высокомолекулярные продукты в процессе
пленкообразования (превращаемые, или термореактив-
термореактивные, пленкообразующие). В нек-рых случаях они м. б.
высокомолекулярными продуктами, не претерпевающи-
претерпевающими при пленкообразовании химич. изменений (непре-
вращаемые, или термопластичные, пленкообразующие).
К непревращаемым пленкообразующим относятся эфи-
ры целлюлозы (см. Эфироцеллюлозные лаки и эмали),
битумы (см. Битумные лаки и эмали), перхлорвинило-
вые смолы (см. Перхлорвиниловые лаки и эмали) и др.;
к превращаемым — высыхающие масла (см. Масла ра-
растительные), алкидные смолы (см. Алкидные лаки и
эмали), ненасыщенные полиэфиры (см. Полиэфирные
лаки и эмали), полиуретаны (см. Полиуретановые лаки
и эмали) и др. См. также Пленкообразующие вещества.
Пигменты придают Э. цвет и укрывистость,
т. е. способность перекрывать цвет подложки (делать
ее невидимой). При изготовлении Э. используют гл.
обр. неорганич. пигменты (TiO2, ZnO, литопон, Fe2O3
и др.). Применяют также органич. пигменты (голубой
и зеленый фталоцианиновые и др.). В Л. иногда входят
растворимые органич. красители, к-рые позволяют
сохранить прозрачность окрашенной пленки. Тип
пигмента, а также количественное соотношение между
пленкообразующим и пигментом оказывают сущест-
существенное влияние на важнейшие свойства лакокрасочных
покрытий — свето-, атмосферо- и влагостойкость, анти-
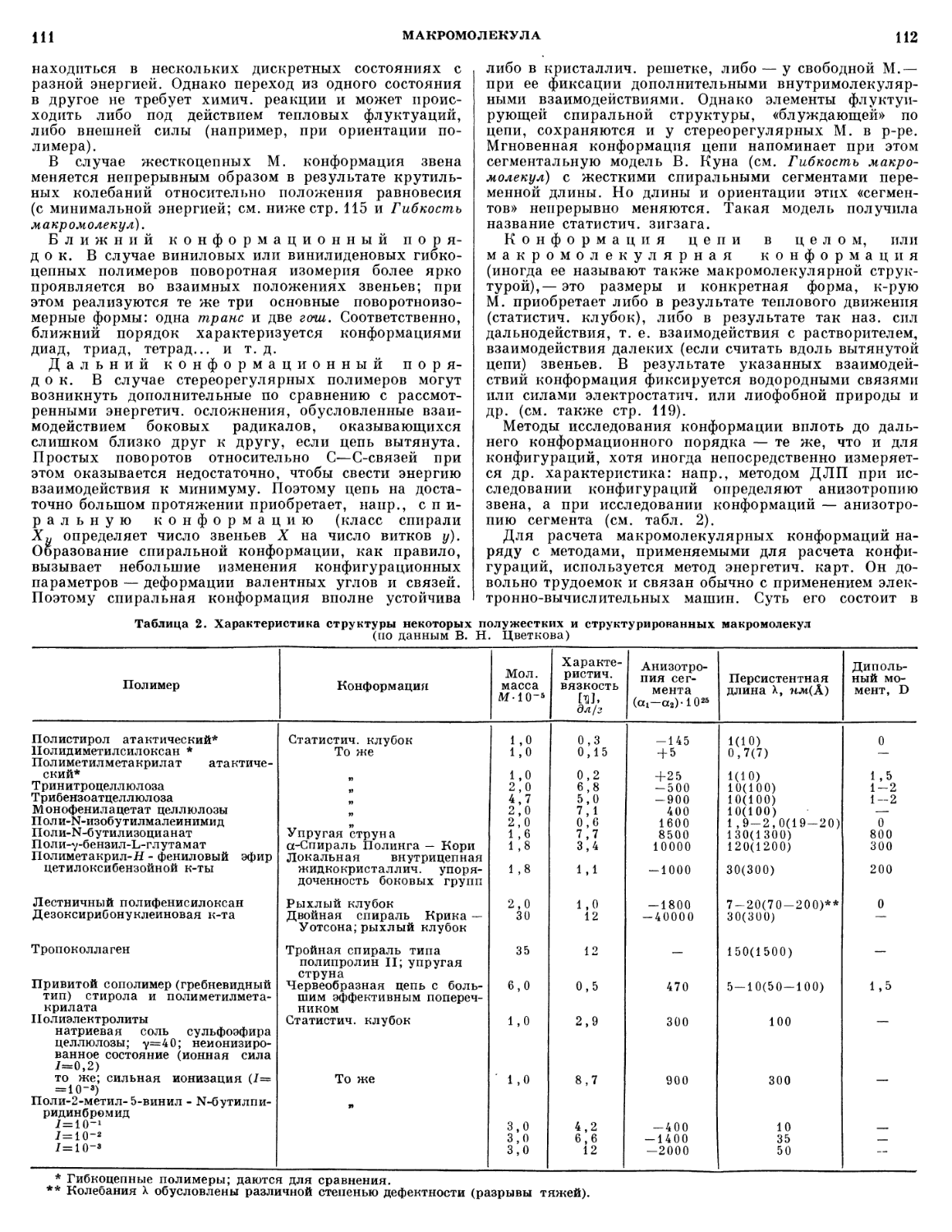
антикоррозионные свойства и др. Важной характеристикой
является критич. объемная концентрация пигмента,
вблизи к-рой происходит скачкообразное изменение
нек-рых свойств лакокрасочного покрытия. См. также
Пигменты лакокрасочных материалов, Красители.
Наполнители вводят в состав Э. гл. обр. для
улучшения их малярно-технич. свойств, повышения
прочности, влаго-, свето-, термостойкости и др. эксплуа-
эксплуатационных свойств покрытий, а также для экономии
пигментов. Наполнителями служат неорганич. тонко-
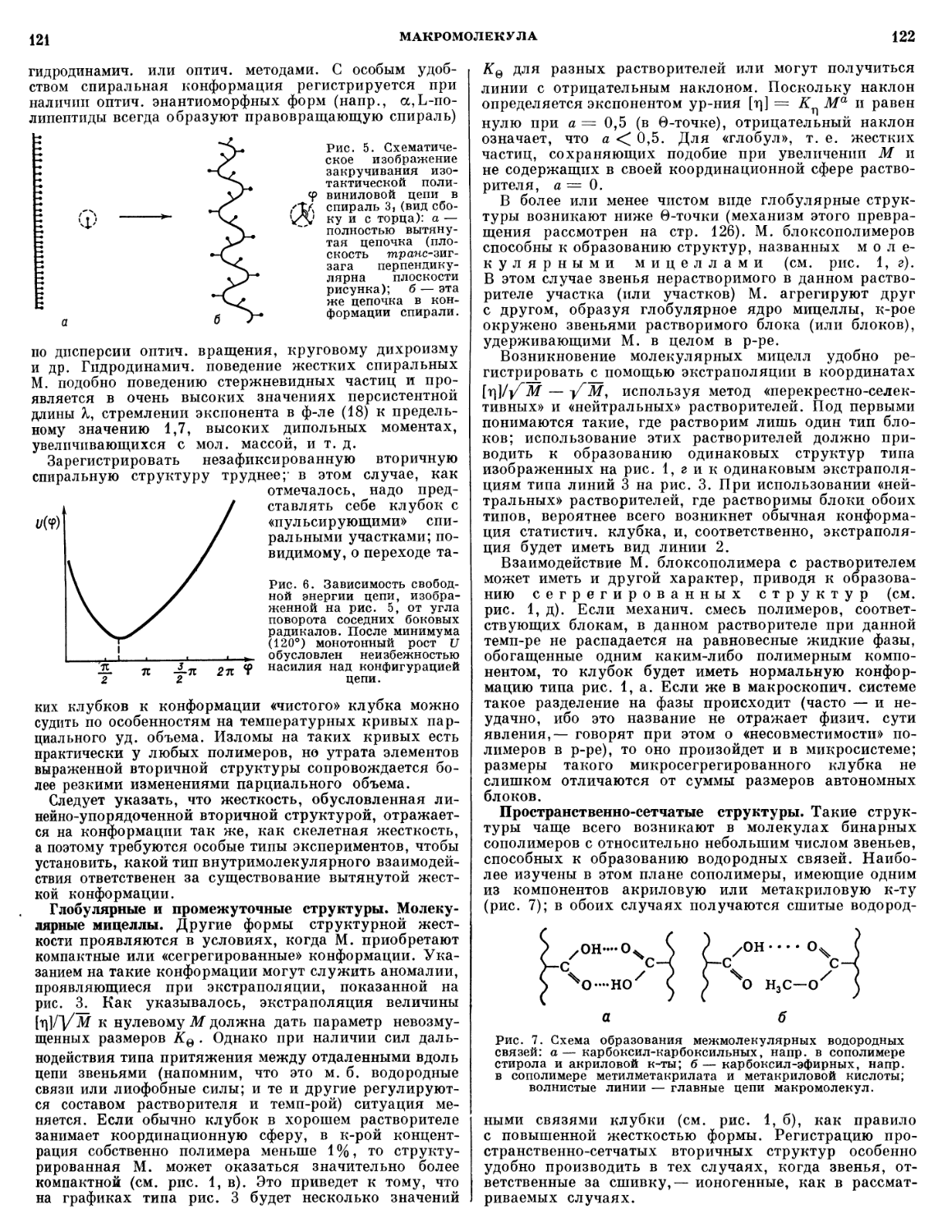
тонкодисперсные природные (мел, слюда, тальк, каолин) и
синтетич. (окись алюминия, карбонат бария и др.) про-
продукты. См. также Наполнители лакокрасочных мате-
материалов.
Пластификаторы улучшают технологиче-
технологические свойства лакокрасочного материала и расширяют
интервал вы со ко эластического состояния лакокрасочно-
лакокрасочного покрытия. Кол-во пластификатора может в зависи-
зависимости от природы пленкообразующего, содержания
пигмента и наполнителя изменяться в пределах 5—40%
(от массы пленкообразующего). Пластификаторами
Л. и Э. служат сложные эфиры (фталаты, себацинаты,
фосфаты), невысыхающее касторовое масло, невысыхаю-
невысыхающие алкидные смолы, а также хлорпарафин (при полу-
получении химстойких покрытий). См. также Пластифика-
Пластификаторы.
Сиккативы ускоряют высыхание Л. и Э. на
основе растительных масел. Сиккативы представляют
собой линолеаты, резинаты, нафтенаты, октоаты Со,
Mn, Pb, реже Zn, Ca и др. В состав лакокрасочного
материала часто вводят одновременно соли двух или
трех металлов, что дает синергич. эффект. Количество
сиккатива в Л. и Э. строго лимитируется, т. к. с повы-
повышением его концентрации ускоряется старение покры-
покрытий. См. также Сиккативы, Олифы.
Стабилизаторы замедляют окисление, дест-
деструкцию и др. превращения пленкообразующих, приво-
приводящие к ухудшению свойств как самих Л. и Э., так и
покрытий. Замещенные фенолы, гс-аминофенол и др.
стабилизаторы препятствуют образованию пленки окис-
окисленного продукта на поверхности Л. и Э. при их хра-
хранении; эпоксидированные масла связывают НС1, отще-
отщепляющийся при нагревании и действии УФ-света на Л.
и Э. на основе хлорсодержащих пленкообразующих и
ускоряющий их деструкцию (см., напр., Винилхлорида
полимеры), и т. д. Стабилизатор не должен замедлять
пленкообразование и изменять цвет пленки. См. также
А нтиоксиданты, Светостабилизаторы.
Растворителями служат органич. жидко-
жидкости, к-рые не вызывают химич. превращений пленкооб-
пленкообразующего и испаряются в процессе пленкообразова-
пленкообразования. В зависимости от темп-ры кипения различают
низко- (ниже 100 °С), средне- A00—150 °С) и высококи-
пящие (до 250 °С) растворители. Многие лакокрасочные
материалы содержат смеси нескольких растворителей
(иногда с добавкой нерастворителя, или разбавителя).
Составом смесей растворителей и разбавителей, зави-
зависящим от природы пленкообразующего, атмосферных
условий и способов нанесения, определяются важней-
важнейшие технологич. свойства Л. и Э. — вязкость, дли-
длительность сушки и др. См. также Растворители лако-
лакокрасочных материалов.
Прочие компоненты. В состав Л. и Э. вво-
вводят матирующие агенты (напр., воски, алюминиевые
соли жирных к-т или к-т канифоли); поверхностно-ак-
поверхностно-активные вещества (напр., алифатич. амины с длинной
углеводородной цепью), к-рые облегчают смачивание
пигментов при получении Э. (см. Краски) и препятст-
препятствуют осаждению пигментов из Э. при их хранении.
11
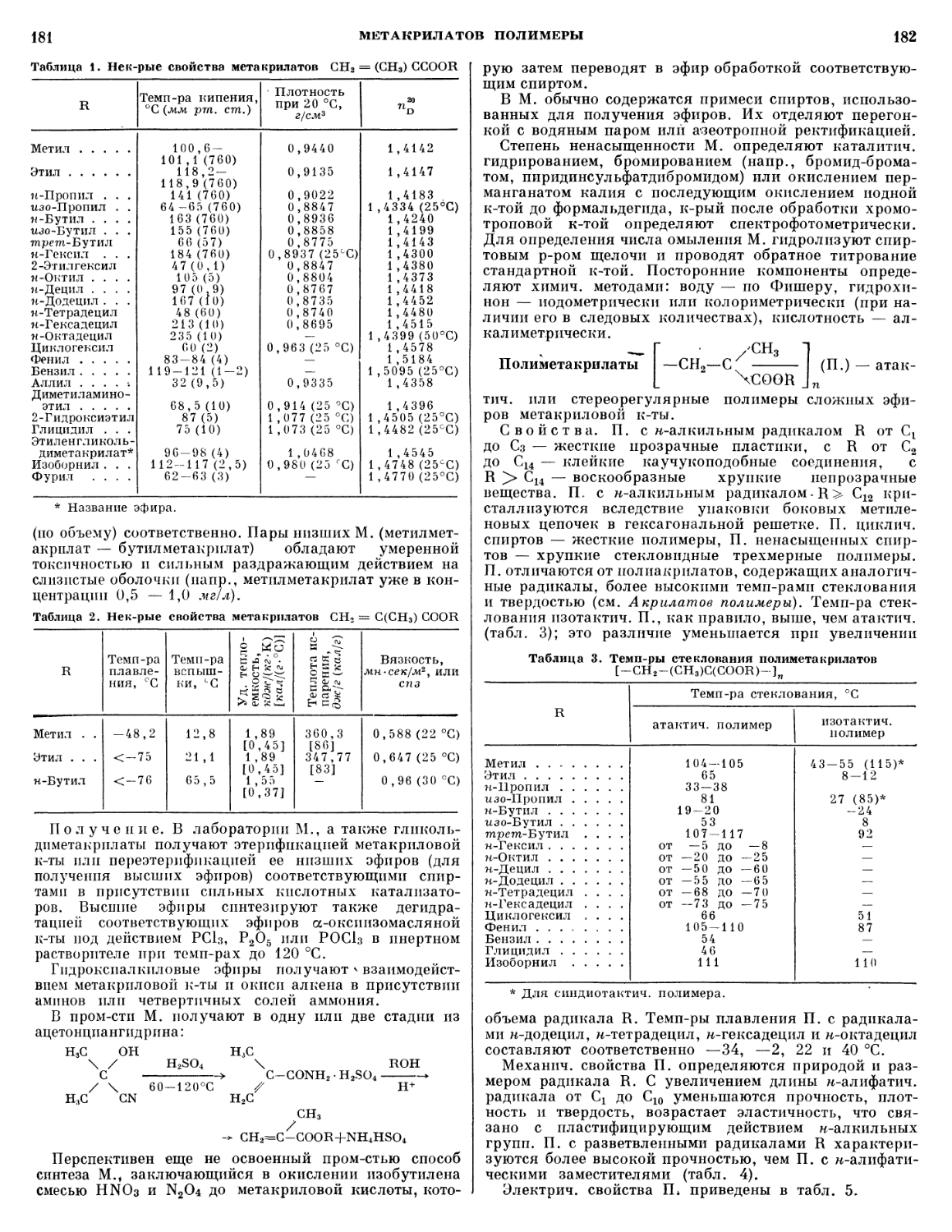
ЛАКОКРАСОЧНЫЕ МАТЕРИАЛЫ
12
При введении специальных добавок, напр, бентонита
или тонкодисперсной двуокиси кремпля «аэросил»,
образуются обратимые коагуляционные (тиксотропные)
структуры, благодаря которым лакокрасочный матери-
материал утрачивает текучесть и удерживается на вертикаль-
вертикальных поверхностях. Такие материалы называются тиксо-
тропными.
Пленкообразование. Высыхание слоя Л. и Э. на под-
подложке возможно в результате: 1) чисто физического про-
процесса улетучивания растворителя (достоинство таких
материалов — быстрое высыхание при комнатной тем-
температуре); 2) химического превращения пленкообразу-
пленкообразующего, к-рое может сопровождаться испарением рас-
растворителя.
Пленкообразование в результате
улетучивания растворителя происходит в три
стадии: 1) кратковременное испарение растворителя
из верхних слоев покрытия, сопровождающееся повы-
повышением концентрации и, следовательно, вязкости лако-
лакокрасочного материала; 2) более продолжительная диф-
диффузия растворителя из нижнего слоя покрытия к по-
поверхности; 3) длительное A0—15 сут) испарение остат-
остатков растворителя из практически высохшей пленки.
Испарение обусловливает понижение теми-ры лакокра-
лакокрасочного материала и, следовательно., возможность кон-
конденсации влаги на поверхности еще не высохшей плен-
пленки. Это нежелательное явление м. б. устранено введе-
введением в состав Л. и Э. высококипящих соединений
(бутил- или амилацетата, циклогексанона, бутилового
спирта и др.)- Формирование пленки происходит
вследствие сближения макромолекул пленкообразую-
пленкообразующего и их физич. взаимодействия, определяющего
когезионную прочность пленки (см. Ногезия). Возни-
Возникающие при формировании пленки внутренние напря-
напряжения могут вызывать ее растрескивание или даже
отслаивание от подложки. Пластификация пленки
позволяет устранить это нежелательное явление.
Пленкообразование вследствие
химических превращений м. б. обуслов-
обусловлено двумя процессами: полимеризацией и иоликонден-
сацией. Так, пленкообразование в Л. и Э. на основе
высыхающих масел или высыхающих алкидных смол
происходит в результате окислительной полимеризации
пленкообразующего. Полиэфирные лаки и эмали высы-
высыхают в результате сополимеризации нолиалкиленгли-
кольмалеинатов (фумаратов) со стиролом или с др. мо-
мономерами (в этих Л. и Э. мономеры служат одновре-
одновременно и растворителями).
Высыхание феноло-альдегидных лаков и эмалей обу-
обусловлено поликонденсацией пленкообразующего после
испарения растворителя, высыхание полиуретановых
лаков и эмалей — поликонденсацией диизоцианатов и
соединений с подвижными атомами водорода, напр, гли-
колей или олигоэфиров. Л. и Э., содержащие отверди-
тели или инициаторы сополимеризации, обладают, как
правило, низкой жизнеспособностью. Поэтому указан-
указанные компоненты вводят в Л. и Э. обычно непосредствен-
непосредственно перед их нанесением.
Применение. Л. и Э. — важнейшие промышленные про-
продукты, применяемые практически во всех отраслях
народного хозяйства, а также в быту. Л. и Э. исполь-
используют для получения защитных и декоративных покры-
покрытий но металлу, дереву, пластмассам и др. Важные
области применения Л.— приготовление эмалей, грун-
грунтовок, шпатлевок, а также пропитка электроизоляци-
электроизоляционных обмоток. Наиболее широко как в СССР, так и
за рубежом применяют алкидные Л. и Э., что объяс-
объясняется доступностью и сравнительной дешевизной ис-
исходного сырья, хорошими технологическими свойства-
свойствами этих материалов, а также возможностью их моди-
модификации.
Основные тенденции развития производства лакокра-
лакокрасочных материалов: 1) замена природных пленкообра-
пленкообразующих синтетическими; 2) сокращение потребления
органич. растворителей в результате применения водо-
водорастворимых пленкообразующих (см., напр., Водораз-
бавляемые грунтовки и эмали, Полиакриловые лаки и
эмали); 3) расширение применения полиуретановых,
эпоксидных, кремнийорганических пленкообразующих,
на основе к-рых получают покрытия, обладающие цен-
ценными специальными свойствами. О методах испытаний
Л. и Э. см. Испытания лакокрасочных материалов и
покрытий, о методах нанесения — Лакокрасочные по-
покрытия.
Лит.: Дринберг А. Я., Технология пленкообразую-
пленкообразующих веществ, 2 изд., Л., 1955; Лакокрасочные материалы,
Справочник, под ред. И. Н. Сапгира, М., 1961; Гольд-
б е р г М. М., Материалы для лакокрасочных покрытий, М.,
1972; Справочник по лакокрасочным покрытиям в машино-
машиностроении, под ред. М. М. Гольдберга, 2 изд., М., 1974; Ш а м-
петьв Г., Р а б а т э Г., Химия лаков, красок и пигмен-
пигментов, [пер. с франц.], т. 2, М., 1962; Лакокрасочные покрытия,
под ред. X. В. Четфилда, пер. с англ., М., 1968.
В. В. Чеботаревский.
ЛАКОКРАСОЧНЫЕ МАТЕРИАЛЫ (paint materi-
materials, Anstrichstoffe, peintures et vernis) — жидкие или
пастообразные составы, к-рые при нанесении тон-
тонким слоем на твердую подложку высыхают с образо-
образованием пленки (лакокрасочного покрытия), удержива-
удерживаемой на поверхности силами адгезии. К Л. м. отно-
относятся лаки (см. Лаки и эмали), краски, грунтовки,
шпатлевки.
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ (paint coatings,
Anstrichiiberzuge, revetements de peintures et vernis).
Содержание:
Строение покрытий 13
Подготовка поверхности 13
Методы нанесения лакокрасочного материала ... 17
Сушка покрытий 22
Отделочные работы 24
Лакокрасочные покрытия — покрытия, к-рые обра-
образуются в результате пленкообразования (высыхания)
лакокрасочных материалов, нанесенных на поверхность
изделий, зданий, сооружений. Основное назначение
Л. п.— защита металлов и неметаллич. материалов
(дерева, бетона, пластмасс и др.) от разрушения (кор-
(коррозии) под действием внешней среды и придание
поверхности изделий требуемого внешнего вида. См.
также Защитные лакокрасочные покрытия, Декора-
Декоративные лакокрасочные покрытия, Лакокрасочные по-
покрытия по дереву, Лакокрасочные покрытия по пласт-
пластмассам.
По стойкости в различных условиях эксплуатации
различают атмосферостойкие Л. и. (см. Атмосферо-
стойкость), бензо- и маслостойкие лакокрасочные по-
покрытия, водостойкие лакокрасочные покрытия, термо-
термостойкие лакокрасочные покрытия, химстойкие лако-
лакокрасочные покрытия, электроизоляционные лакокрасоч-
лакокрасочные покрытия. В особую группу выделяют покрытия
специального назначения, напр, противодействующие
обрастанию морскими организмами (см. Необрастаю-
щие лакокрасочные покрытия), антимикробные поли-
полимерные покрытия, флуоресцентные и фосфоресцентные
(см. Светящиеся лакокрасочные покрытия), сигнализи-
сигнализирующие о достижении температурного предела процес-
процесса (см. Термоиндикаторные лакокрасочные покрытия),
регулирующие темп-ру (см. Терморегулирующие лако-
лакокрасочные покрытия), а также стойкие к механическим
воздействиям и др.
Л. п. могут быть прозрачными и непрозрачными (ук-
рывистыми); прозрачные получают при нанесении ла-
лаков (см. Лаки и эмали), укрывистые — при нанесении
грунтовок, шпатлевок, красок (в том числе и эмалей).
В большинстве случаев наносят укрывистые Л. п.;
прозрачные применяют преимущественно для деревян-
деревянных изделий и заготовок из листового металла, дета-
деталей электро- и радиотехнич. назначения.
13
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ
14
Строение покрытий
Толщина Л. п. составляет обычно 60—100 мкм, иног-
иногда — ок. 300 мкм. Лакокрасочные материалы наносят,
как правило, несколькими слоями толщиной 10—
25 мкм и сушат после нанесения каждого слоя. Необ-
Необходимость нанесения нескольких слоев обусловлена
во многих случаях невозможностью получить Л. п.
с хорошими защитными свойствами, т. к. при нанесе-
нанесении одного утолщенного слоя затрудняются улетучива-
улетучивание растворителя и др. процессы пленкообразования
и может получиться Л. п. с подтеками и наплывами.
Слоем толщиной ~350 мкм м. б. нанесены густые шпат-
шпатлевки, тиксотропные лаки и эмали, а также материалы,
содержащие реакционноспособные растворители, напр.
полиэфирные лаки и эмали.
Во многих случаях отдельные функции Л. п. выпол-
выполняют различные его слои, для к-рых выбирают соответ-
соответствующие материалы. Нижние слои (грунтовоч-
(грунтовочные) обеспечивают прочное сцепление с подложкой
и антикоррозионную стойкость Л. п. и при необходи-
необходимости заполняют также поры подложки. Для их полу-
получения применяют грунтовки, к-рые наносят одним или
двумя слоями.
Верхние слои Л. п. (покровные) сообщают по-
поверхности нужные декоративные свойства, укрывис-
тость и стойкость к действию внешней среды. Число
покровных слоев обычно бывает не менее двух, а иногда
достигает пяти—восьми (напр., в химстойких Л. п.).
Для нанесения покровных слоев используют гл. обр.
краски. Верхний слой краски иногда покрывают слоем
лака, придающим Л. п. блеск или матовость.
Между грунтовочными и покровными слоями иногда
наносят промежуточные слои различного назначения,
напр, для выравнивания поверхности и заделки свар-
сварных и заклепочных швов (шпатлевочные),
для предотвращения набухания грунтовочного или др.
ранее нанесенного слоя в растворителе, содержащемся
в покровной краске. В зависимости от типа лакокрасоч-
лакокрасочного материала операции нанесения отдельных слоев
наз. соответственно грунтованием, шпатлеванием, ок-
окрашиванием, лакированием.
Отдельные слои Л. п. должны быть окрашены в раз-
разные цвета, т. к. в противном случае трудно отличить
их друг от друга при получении Л. п., его разрушении
во время эксплуатации, а также при ремонте. В то же
время цвета слоев не должны быть резко контрастными
во избежание увеличения числа слоев Л. п., необходи-
необходимых для достижения укрывистости.
Подготовка поверхности
Для обеспечения прочной адгезии Л. п. к подложке
необходима очистка окрашиваемой поверхности, а так-
также придание ей шероховатости глубиной 1 — 10 мкм.
Очистку осуществляют механич. и химич. способами.
Первые в большинстве случаев менее производительны,
однако, в отличие от химических, не связаны с после-
последующей промывкой водой и горячей сушкой поверх-
поверхности.
Технология подготовки поверхности под окраску
включает, помимо очистки, получение фосфатных, ок-
оксидных или металлич. подслоев, что повышает адге-
адгезию лакокрасочного материала к поверхности и за-
защитные свойства Л. п.
Механическая очистка. При очистке механи-
механизированным инструментом, к-рая в 5
и более раз производительнее ручной (стальными шпа-
шпателями, скребками, зубилами, молотками, металлич.
щетками), применяют легкие и компактные машинки
типа пневматич. молотков и дрелей и электрич. дрелей.
Первые исключают опасность пожаров и возможность
поражения током, более пригодны для использования
во влажной среде, при высокой температуре, но менее
экономичны, чем электрические. Рабочей частью ма-
машинок могут служить бойки молотков, шарошки, абра-
абразивные круги, щетки (металлич., из капрона и др.).
Инструмент надевают непосредственно на вал электрич.
или пневматич. двигателя или (в тех случаях, когда
двигатель нельзя поместить у рабочего места) на конец
гибкого вала, соединенного с двигателем. Недостатки
машинок — невозможность обработки углублений, за-
зазоров, а также быстрый износ рабочего инструмента.
Повышенной износостойкостью обладают жесткие ме-
металлич. щетки нового типа, т. наз. иглофрезы. Прово-
Проволоки иглофрезы, собранные в пакеты, с одного конца
опрессованы и сварены вместе, а с другого могут откло-
отклоняться при работе на очень небольшой угол, образуя
своеобразные полужесткие микрорезцы. При чередова-
чередовании направления вращения иглофреза самозатачивает-
самозатачивается. Срок службы иглофрез 200—300 ч (обычных метал-
металлич. щеток — 10—12 ч). Они пригодны для удаления
толстых слоев окалины.
Мелкие детали можно очищать во вращающихся ба-
барабанах (т. наз. галтовка), к-рые загружают на
80—85% их объема. Для ускорения очистки в барабан
загружают также звездочки из белого чугуна, рубле-
рубленую проволоку или др. Производительность галтовки
повышается в 2—3 раза в случае применения вибра-
вибрационных барабанов.
При гидроабразивной (гидропеско-
(гидропескоструйной) очистке поверхность обрабатывают
струей водной суспензии абразива (кварцевого песка,
молотого гранита). Во избежание коррозии очищаемой
поверхности в суспензию добавляют пассиватор (NaNO2,
К2Сг207) или после очистки промывают деталь р-ром
пассиватора. Производительность гидропескоструйной
очистки в 2—3 раза выше, чем очистки механизиро-
механизированным инструментом. Недостатки способа — сильное
загрязнение смежных необрабатываемых поверхнос-
поверхностей и необходимость последующей промывки и сушки
изделия.
Окрашиваемые изделия можно очищать также м е-
таллич. дробью @,5—0,8 мм) или песком
@,3—2,5 мм), к-рые подаются струей сжатого воздуха.
При этом поверхность приобретает микрошерохова-
микрошероховатость, обеспечивающую лучшую адгезию Л. п., чем при
использовании др. способов очистки. Дробеструйную
очистку применяют для изделий с толщиной стенок не
менее 3 мм. Изделия из черных металлов очищают чу-
чугунной и стальной дробью или песком, изделия из
цветных металлов — алюминиевой и латунной дробью
или алюминиевым песком. Иногда при очистке изделий
из цветных металлов взамен дроби или песка применяют
гранулят из скорлупы грецких орехов или фруктовых
косточек с частицами размером 0,5 —1,0 мм. Обрабо-
Обработанные поверхности рекомендуется грунтовать во избе-
избежание их коррозии на воздухе.
Очистку металлич. дробью или песком осуществляют
в камерах, снабженных вытяжными устройствами,
фильтрами и уловителями пыли, песка, дроби. Усовер-
Усовершенствованные аппараты снабжены устройствами для
отсасывания отработанного абразива, его воздушной
сепарации и системой подачи в аппарат для повторного
использования. Мелкие изделия очищают в дробе-
или пескоструйных барабанах.
Дробеметная очистка осуществляется
под действием крупной дроби или стальной рубленой
проволоки, выбрасываемой на поверхность изделий
лопатками ротора дробеметного аппарата. Частота
вращения ротора 2000—2500 об/мин, скорость, с к-рой
выбрасывается дробь, —50—70 м/сек. С помощью дробе-
метных установок очищают изделия с толщиной стенок
не менее 5 мм. Очищенные изделия после обдувки
воздухом по возможности быстро грунтуют. Крупные
и средние изделия чаще всего очищают в камерах не-
непрерывного действия, оборудованных вытяжной вен-
15
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ
16
тиляцией, транспортерами для загрузки и возврата
отработанной дроби, приспособлениями для транспор-
транспортировки изделий. Мелкие изделия очищают в дробемет-
ных барабанах. Из механич. способов очистки дробе-
метный — наиболее производительный, экономичный и,
кроме того, он не вызывает сильного пылеобразования.
Недостатки способа — быстрый износ узлов аппарата и,
прежде всего, лопаток ротора, а также шум при работе.
Химическая очистка. Способы химич. очистки поверх-
поверхности включают операции обезжиривания и травления.
При щелочном обезжиривании в ста-
стационарных ваннах применяют водные р-ры NaOH,
а также Na2CO3, INa3PO4 или смесей этих солей. Смеси
солей, в отличие от щелочи, не разрушают металлы
с амфотерными свойствами (напр., алюминий и его
сплавы) и дольше сохраняют щелочность р-ра на нуж-
нужном уровне. В р-р добавляют эмульгаторы — мыло,
жидкое стекло или неионогенные поверхностно-актив-
поверхностно-активные вещества типа ОП-7 или ОП-10. Оптимальные ре-
результаты получают при концентрации р-ра 50—150 г/л,
темп-ре 70—90 °С, продолжительности обработки
5—20 мин. Р-р омыляет растительные и животные жи-
жиры и эмульгирует нефтяные масла. Остатки щелочи
удаляют промывкой горячей и холодной водой. Во
избежание отложения на поверхности изделий нераст-
нерастворимых кальциевых и магниевых мыл в воду добав-
добавляют Триполи-, тетрапиро- или гексаметафосфат натрия
или применяют деминерализованную воду.
В условиях массового производства обезжиривание
осуществляют в т. наз. струйных агрегатах. Детали,
движущиеся на подвесном конвейере или на транспор-
транспортерной ленте, проходят сквозь камеры обезжиривания
и промывки, где их обрабатывают струями щелочного
р-ра или воды из контура труб, снабженных гидрофор-
гидрофорсунками или патрубками. Ударное действие струй
ускоряет обезжиривание и промывку и способствует
удалению прилипшей грязи. В нижней части камеры
установлена ванна, где стекающая жидкость подогре-
подогревается и с помощью насоса вновь нагнетается в обли-
обливающий контур, проходя по пути очистной фильтр.
Для очистки крупных объектов эффективен способ об-
обработки поверхности смесью горячего моющего р-ра
с перегретым паром, выходящей из распылительной
головки. Установки для такой очистки м. б. перенос-
переносными и стационарными.
Щелочное обезжиривание рационально применять
перед операциями, к-рые проводят в водных р-рах (трав-
(травление, фосфатирование и др.). В противном случае
обезжиренные этим способом поверхности необходимо
перед нанесением Л. п. подвергать горячей сушке
(исключение — нанесение водоразбавляемых лакокра-
лакокрасочных материалов).
Обезжиривание растворителями не
требует последующей горячей сушки, т. к. растворители
сравнительно быстро улетучиваются при комнатной
темп-ре. При использовании горючих и взрывоопасных
растворителей (бензина, уайт-спирита и др.) детали
протирают смоченными растворителем полотенцами.
Безопасные в пожарном отношении негорючие раство-
растворители (напр., три- или тетрахлорэтилен) токсичны, по-
поэтому при работе с ними применяют герметизированную
аппаратуру. Детали погружают в ванны, обрабатывают
струей жидкого растворителя или в паровой фазе.
Применяют также агрегаты, в к-рых детали последова-
последовательно обезжиривают всеми тремя способами. Наиболее
эффективно обезжиривание в паровой фазе. При этом
детали на конвейере проходят сквозь камеру, запол-
заполненную парами растворителя, к-рый испаряется из
ванны, расположенной в нижней части камеры. При
обезжиривании деталей, изготовленных из металлов
(напр., из алюминия и его сплавов), к-рые могут реа-
реагировать с трихлорэтиленом с большим тепловыделени-
тепловыделением, применяют тетрахлорэтилен.
Детали можно обезжиривать, погружая их в ванну
с эмульсией, к-рую готовят перемешиванием 60—
80% растворителя (уайт-спирита, бензина) с 40—20%
воды в присутствии 0,5% неионогенного эмульгатора.
Преимущества обезжиривания эмульсиями перед ис-
использованием горючих растворителей — меньшая пожа-
роопасность и возможность одновременного удаления
неорганич. загрязнений.
Для обезжиривания мелких деталей щелочными
р-рами, органич. растворителями или эмульсиями
применяют вращающиеся барабаны.
Травление поверхности (обычно предваритель-
предварительно обезжиренной) производят для удаления с нее слоя
окалины, ржавчины и др. продуктов коррозии. Выбор
состава для травления зависит от природы металла
и удаляемых продуктов. С углеродистых сталей окали-
окалину удаляют в 20%-ном р-ре H2SO4 при 70—80 °С или
18—20%-ном р-ре НС1 при 30—40 °С. Продолжитель-
Продолжительность процесса в обоих случаях 10—30 мин. Во избе-
избежание образования на поверхности металла травиль-
травильных раковин в р-р вводят 1—3% ингибитора кислотной
коррозии. По окончании процесса поверхность нейтра-
нейтрализуют 3—5%-ным р-ром Na2CO3 и промывают
водой. Травление проводят обычно в ваннах, стенки
и днище к-рых защищены кислотостойким материалом
(свинцом, винипластом и др.). При использовании
струйных агрегатов скорость травления увеличивается
в 3—5 раз. Имеются составы для одновременного обез-
обезжиривания и травления стальных деталей.
Для травления крупногабаритных изделий и конст-
конструкций применяют травильные пасты, для пассивиро-
пассивирования — пассивирующие пасты (дисперсии минераль-
минеральных наполнителей соответственно в р-ре к-ты или пасси-
ватора), к-рые наносят щетками, после чего поверх-
поверхность промывают водой. Для стальных поверхностей
разработаны т. наз. преобразователи ржавчины, нане-
нанесение к-рых исключает необходимость ее удаления (см.
Защитные лакокрасочные покрытия).
Медь и ее сплавы травят в 10%-ной HNO3, а затем
пассивируют в р-ре, содержащем 100 г/л К2Сг207 и
10 г/л H2SO4. Для травления алюминия и его сплавов
применяют водный р-р NaOH с концентрацией 100—
150 г/л, после чего поверхность осветляют 2—3 мин,
в 15—20%-ном р-ре HNO3.
Термическая (пламенная) очистка. Поверхность изде-
изделия обрабатывают пламенем кислородно-ацетиленовой
горелки. Образующаяся при этом окисная пленка рас-
растрескивается, что обусловлено различием коэфф. линей-
линейного расширения металла и его окислов, и отслаивается.
Остатки окислов удаляют проволочной щеткой. По-
Поверхность, остывшую *до 50—70 °С, грунтуют. Метод
применяют для изделий с толщиной стенки не менее
3 мм, покрытых толстым слоем окалины, ржавчины или
старым Л. п.
Получение фосфатных, оксидных и металлических
подслоев. Фосфатирование — процесс обра-
образования на поверхности металла микропористой плен-
пленки из нерастворимых в воде трехзамещенных ортофос-
фатов, напр. Zn3(PO4J-Fe3(PO4J. Это происходит при
взаимодействии металла с водорастворимыми одно-
замещенными ортофосфатами марганца — железа
Mn(H2PO4J-Fe(H2PO4J (препарат «мажеф»), цинка
или железа. Пленка имеет кристаллическое строение
и обладает низкой электрической проводимостью. Наи-
Наиболее часто фосфатируют черные металлы. При этом
преимущественно используют р-р Zn(H2PO4J A0—25
г/л), к к-рому добавляют ускоритель — NaNO3 или
Zn(NO3JB1—30 г/л), NaNO2 @,2—1,0 г/л), а также со-
соли никеля, меди и др. Изделие погружают на 5—15 мин
в ванну с р-ром при 65—98 °С. Имеются составы для
фосфатирования алюминия и его сплавов, одновремен-
одновременного фосфатирования стальных, оцинкованных, кад-
мированных и др. поверхностей.
17
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ
18
В массовом производстве детали, размещенные на
конвейере, фосфатируют в струйных камерах, совме-
совмещенных в одном агрегате с камерами струйного обез-
обезжиривания и промывки. Продолжительность процесса
составляет 1,5—3 мин при темп-ре р-ра 45—55 °С.
Фосфатную пленку для повышения ее защитных свойств
промывают водой и пассивируют 0,04%-ным р-ром хро-
хромовой к-ты, после чего промывают деминерализован-
деминерализованной водой и сушат.
Повышенной прочностью при ударе и изгибе обла-
обладают тонкие фосфатные пленки мелкокристаллич.
структуры: их масса составляет 1,5 г (вместо 5 г для
обычных пленок) на 1 м2 поверхности.
Качество кристаллич. фосфатных пленок сильно за-
зависит от режима нанесения. Значительно проще в этом
отношении щелочное фосфатирование (фосфатное пасси-
пассивирование), при к-ром под действием р-ра NaH2PO4
или NH4H2PO4 на поверхности образуется тонкий слой
аморфной Fe3(PO4J @,2—0,5 г/м2). Защитные свойства
таких слоев хуже, чем кристаллических; их достоинст-
достоинство — способность выдерживать значительные деформа-
деформации при ударе и изгибе, что особенно важно в произ-
производстве окрашенных металлич. листов и ленты.
Оксидирование имеет наибольшее значение
для изделий из алюминия и его сплавов. Чаще всего
применяют электрохимич. способ (анодное оксидиро-
оксидирование), т. к. он позволяет получать пленку наилучшего
качества. Процесс обычно проводят при постоянном
токе. Электролитом служит 20%-ный р-р H2SO4, ано-
анодом — оксидируемые детали, катодом — свинцовые
пластины. Темп-pa р-ра 20 ^ 5 °С, анодная плотность
тока 0,7—2,0 а/м2, продолжительность процесса 20—
60 мин, толщина образующейся пленки 5—25 мкм.
После промывки и пассивирования 10%-ным кипящим
р-ром К2Сг207 в течение 20 мин или уплотнения стенок
пор в подкисленной горячей воде изделия подвергают
горячей сушке.
При химическом оксидировании детали из алюминия
и его сплавов погружают, напр., в р-ры состава (в г/л):
Н3РО4 — 40—50; СгО3 — 8—10; NaF — 4—5. Темп-pa
р-ра 15—20 °С; продолжительность обработки 10—15
мин; толщина пленки 2—3 мкм. Пленку дополнитель-
дополнительно пассивируют 2—5 сек 2%-ным р-ром СгО3.
Магниевые сплавы чаще всего оксидируют химич.
способом. Напр., детали из литейных сплавов оксиди-
оксидируют в р-ре, содержащем (в г/л): К2Сг207 — 40—55;
HNO3 (плотн. 1,4 г/см3)—110; NH4C1 — 1,0. Темп-ра
р-ра 70—80 °С, продолжительность процесса 30—120
сек; толщина пленки 0,25—3 мкм. Пленку пассиви-
пассивируют р-ром К2Сг207.
Металлические подслои получают эле-
электролитическим способом на катоде (цинкование или
кадмирование). Значительное улучшение защитных
свойств и адгезии достигается при нанесении слоя
цинка или алюминия методом напыления. Сочетание
такого подслоя с Л. п. наз. комбинированным покры-
покрытием.
Методы нанесения лакокрасочного материала
Метод пневматического распыления
наиболее широко распространен в машиностроении.
Оборудование — пистолетообразные краскораспылите-
краскораспылители, снабженные распыляющими форсунками. Лако-
Лакокрасочный материал из стаканчика @,5—0,8 л), уста-
установленного на корпусе распылителя, или из бака ем-
емкостью 15—100 л нагнетается в распылитель под дав-
давлением очищенного от влаги и масла сжатого воздуха
[50 кн/м2 @,5 кгс/см2)]. В массовом производстве приме-
применяют системы централизованной подачи лакокрасоч-
лакокрасочного материала из краскозаготовительного отделения
к рабочим местам. Обычно применяют краскораспыли-
краскораспылители высокого давления [250—450 кн/м2 B,5—
4,5 кгс/см2)]\ррш^-^низкого давления [30—100 кн/м2
@,3—1,0 кгс/см2)]. Диаметр сопел распылителей нахо-
находится в пределах 1,4—2,5 мм. Оптимальное расстояние
от распылителя до окрашиваемой поверхности — 300—
500 мм при плоской струе и 250—300 мм при круглой.
В автоматизированных установках применяют рас-
распылители с автоматич. пусковым устройством дистан-
дистанционного управления. Разработаны автоматич. распы-
распылители для окраски внутренней поверхности труб,
а также небольшие распылители (аэрографы) с соплом
диаметром 0,4—1,2 мм для художественной окраски.
Двухупаковочные лакокрасочные материалы наносят
с помощью двухсопловых распылителей, в к-рых ком-
компоненты смешиваются внутри форсунки или на выходе
из нее.
Метод пневматич. распыления обеспечивает в 4 — 5 раз
большую производительность, чем при нанесении ки-
кистью, дает возможность получать Л. п. высокого каче-
качества на поверхностях разнообразной формы (за исклю-
исключением внутренних полостей сложного профиля), на-
наносить лакокрасочный материал на конвейерных ли-
линиях и автоматич. установках.
При пневматич. распылении лакокрасочные мате-
материалы разбавляют в 2,5—3 раза большим количеством
растворителя, чем при окраске кистью (рабочая вяз-
вязкость при 20 °С — 15—35 сек по вискозиметру ВЗ-4).
Это обусловливает больший расход растворителей, ог-
ограничивает толщину наносимого слоя и, соответст-
соответственно, вызывает необходимость увеличения числа слоев
для получения Л. п. требуемой толщины.
Применение метода пневматич. распыления с подо-
подогревом материала до 55—70 °С (в стаканчике или кра-
сконагревателе циркуляционной системы) позволяет
существенно уменьшить его вязкость и благодаря этому
сократить количество добавляемого растворителя или
исключить его совсем. В аппаратах нек-рых конструк-
конструкций предусмотрен подогрев не только материала, но
и воздуха, расходуемого на распыление. При использо-
использовании этого метода однократное нанесение дает возмож-
возможность получить слой толщиной 35—60 мкм.
Существенный недостаток пневматич. распыления —
большие потери материала B5—50%) на рассеивание
в воздухе и связанные с этим пожароопасность и плохие
санитарно-гигиенич. условия производства. Все это
вызывает необходимость осуществлять окрашивание
в камерах с местным отсосом загрязненного воздуха,
что сильно удорожает производство.
В машиностроении применяют распыление
под высоким давлением, создаваемым на-
насосом (безвоздушное распыление). При распылении
с подогревом материал, нагретый до 60—100 °С, нагне-
нагнетается к керамич. соплу распылителя диаметром 0,3—
0,5 мм под давлением 4—10 Мн/м2 D0—100 кгс/см2).
Способ имеет ряд преимуществ перед пневматич. рас-
распылением: потери материала на рассеивание в воздухе
уменьшаются до 15—30%, значительно сокращается
расход растворителей, при однократном нанесении
получаются слои большей толщины. Применение мето-
метода распыления под высоким давлением с подогревом
вызывает затруднения при окраске деталей сложной
формы.
При распылении без подогрева материал нагнетается
насосом к распылителю под давлением 10—25 Мн/м2
A00—250 кгс/см2). Установки компактнее, дешевле
и проще в обслуживании, чем установки с подогревом,
и могут применяться для более широкого ассортимента
лакокрасочных материалов. Недостатки метода — на
5—15% большие потери материала, худший внешний
вид и меньшая толщина слоев Л. п., чем при распыле-
распылении с подогревом.
Принцип метода распыления в электри-
электрическом поле высокого напряжения (—100 кв)
заключается в следующем. Частицы материала, рас-
распыленного тем или иным способом, направляются к от-
19
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ
20
рицательному коронирующему электроду, приобрета-
приобретают заряд и под действием сил электрического ноля оса-
осаждаются на противоположно заряженном электроде,
роль к-рого выполняет окрашиваемая деталь. Коро-
нирующим электродом в нек-рых установках служит
медная сетка. В этом случае материал подают в элект-
рич. ноле сжатым воздухом из автоматич. краскораспы-
краскораспылителя; частицы материала заряжаются, адсорбируя
ионы газов воздуха (ионная зарядка). В др. установ-
установках распыляющее устройство служит одновременно
коропирующим электродом. Такие устройства бывают
вращающиеся и неподвижные; первые имеют форму
чаши, воронки, диска, грибка. Материал, нагнетаемый
насосом, приобретает на них заряд и под его дейст-
действием, а при использовании вращающихся устройств —
также и центробежной силы распыляется с острой
кромки на мелкие частицы (контактная зарядка). В
неподвижных щелевых электрораспылителях, удобных
при окраске плоских изделий, материал распыляется
с коронирующей кромки стальной пластины, закреп-
закрепленной в щели корпуса распылителя.
В электрич. поле наносят многослойные Л. п. как
на металлы, так и на немета л лич. материалы, напр,
на дерево, влажность к-рого должна быть при этом не
менее 8%. При окраске стеклопластиков, во избежание
отталкивания частиц лакокрасочного материала из-за
скопления электрич. зарядов, окрашиваемую поверх-
поверхность смачивают р-ром поверхностно-активного веще-
вещества или для снижения сопротивления стеклопластика
устанавливают за изделием дополнительный отрица-
отрицательный электрод, создающий нейтрализующий по-
потенциал.
Окрашивание в электрич. поле можно осуществлять
автоматически на стационарных установках или с по-
помощью ручных электрораспылителей. В автоматических
установках ток поступает от высоковольтно-выпрями-
тельного устройства. Камеры, внутри к-рых на конвей-
конвейере двигаются детали, снабжены приспособлениями для
отсоса паров растворителей и устройствами для дози-
дозированной подачи лакокрасочных материалов, предуп-
предупреждения образования искры, системой автоблокиров-
автоблокировки, включающей или, в случае необходимости, снимаю-
снимающей высокое напряжение. Электрораспылители раз-
размещают в камерах на стойках, к-рые могут подниматься
и опускаться. Такие установки можно использовать
и для окрашивания непрерывно движущейся металлич.
ленты или полосы, из к-рой изготовляют изделия спо-
способом глубокой вытяжки.
Ручные электрораспылители, для к-рых источником
высокого напряжения служат электростатические ге-
генераторы, используют для окраски единичных или
разнотипных изделий. Такие распылители безопасны
в работе (сила тока короткого замыкания не превыша-
превышает 300 мка).
Недостаток метода электрораспыления — невозмож-
невозможность получения Л. п. равномерной толщины на по-
поверхностях сложного профиля с глубокими впадинами.
Это вызывает необходимость последующей ручной
окраски отдельных участков пневматич. распылителя-
распылителями. Для устранения этого недостатка сконструированы
комбинированные распылители, в к-рых материал
можно распылять как в электрич. поле, так и сжатым
воздухом или гидравлич. давлением, создаваемым на-
насосом.
Для распыления в электрич. поле пригодны материа-
материалы с уд. объемным электрич. сопротивлением в преде-
пределах 50—500 ком-м E-Ю6—5- 107ом-см) и диэлектрич.
проницаемостью 6—11. Материалы с такими парамет-
параметрами м. б. получены введением в их состав соответст-
соответствующих растворителей или поверхностно-активных
веществ. Материалы, не удовлетворяющие этим требо-
требованиям, обычно подают в электрич. поле пневматич.
или ультразвуковым распылителем. В электрич. поле
чаше всего наносят алкидные, а также мочевино- и ме-
ламино-алкидные материалы.
При электрораспылении потери материала на рассеи-
рассеивание в воздухе значительно меньше, чем при пневма-
пневматич. распылении. Так, при ионной зарядке они не
превышают 10%, при контактной — 5% от расхода
лакокрасочного материала.
Аэрозольное распыление производит-
производится под давлением сжиженных газов (иропеллентов),
заключенных вместе с лакокрасочным материалом
в баллончик из жести, алюминия или стекла. Емкость
баллончика 0,5—1 л, давление 80—250 кн/м2 @,8—
2,5 кгс/см2). В качестве пропеллента применяют инерт-
инертные низкокииящие нетоксичные фторхлорпроизводные
углеводородов — фреоны, напр, смесь фреона-11 и
фреона-12 (т. кип. соответственно 23,7 и 29,3 °С). Про-
пеллент должен быть разбавителем лакокрасочного ма-
материала и не вызывать коагуляции пленкообразующего.
Этим методом наносят алкидные, нитроцеллюлозные
и др. лакокрасочные материалы преимущественно хо-
холодной сушки при подкраске, восстановлении внешне-
внешнего вида мебели, легковых автомобилей и др.
Методом окунания в ванну с лакокрасочным
материалом Л. п. наносят преимущественно на кон-
конвейерной поточной линии. Избыток материала стекает
с извлекаемых из ванны деталей на наклонный лоток,
откуда снова попадает в ванну. Мелкие изделия наве-
навешивают в несколько ярусов на подвески или окраши-
окрашивают в сетчатых корзинах. Метод сравнительно прост,
дешев, позволяет быстро окрасить большое количество
продукции как снаружи, так и изнутри. Его используют
в основном в тех случаях, когда к качеству Л. п. не
предъявляют высоких требований. Недостаток метода —
необходимость применения больших объемов лако-
лакокрасочных материалов; поэтому он безопасен в пожар-
пожарном отношении только при работе с водоразбавляемы-
ми материалами.
Лакокрасочный материал можно наносить на детали,
движущиеся на конвейере, обливанием их из
контура труб с соплами диаметром 6—9 мм. Избыток
материала стекает в ванну, откуда насосом через фильтр
вновь нагнетается к соплам. В усовершенствованных
установках контур труб заменен одной трубой, уста-
установленной под деталями и снабженной 5—8 соплами,
из к-рых материал фонтанирует на высоту верхней
кромки деталей. Труба совершает возвратно-поступа-
возвратно-поступательное движение в направлении, перпендикулярном
направлению движения конвейера, и одновременно
качательное (в пределах 50°) относительно своей оси.
Преимущество обливания перед окунанием — возмож-
возможность окрашивания крупногабаритных деталей и при-
примерно десятикратное уменьшение количества краски
в системе.
Методы окунания и обливания пригодны лишь для
деталей, окрашиваемых со всех сторон в один цвет и
имеющих обтекаемую форму и гладкую поверхность.
Их используют для нанесения материалов горячей
сушки. Чтобы получить Л. и. равномерной толщины
без подтеков и наплывов, применяют ряд мер: подве-
подвешивают детали на конвейер в положении, обеспечи-
обеспечивающем максимальное стекание избытка лакокрасоч-
лакокрасочного материала; удаляют этот избыток быстрым враще-
вращением деталей после окраски; устанавливают в ванне
т. наз. «волнообразователь», к-рый удаляет (смывает)
избыток материала с нижних частей детали; создают
в установке электрич. поле постоянного тока высокого
напряжения, причем одним из электродов служат де-
детали, а другим — устанавливаемая под ними медная
сетка. Однако наиболее эффективный способ — про-
пропускание окрашенных деталей через туннель с контро-
контролируемым количеством паров растворителя, поступаю-
поступающих из сушильной камеры. Благодаря этому замед-
замедляется испарение растворителя из нанесенного слоя,
21
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ
22
материал загустевает медленнее и, следовательно, боль-
большее его количество успевает стечь с детали.
При использовании метода обливания с выдержкой
в парах растворителя потери лакокрасочного материа-
материала на 25—30% меньше, чем при пневматнч. распылении.
При работе с водоразбавляемыми материалами метод
безопасен в пожарном отношении.
Разновидность обливания — нанесение покрытий
с помощью лаконаливных машин. Потери материала при
этом на 10—20% меньше, чем при нневматич. распыле-
распылении (об этом методе см. Лакокрасочные покрытия по
дереву).
На очень мелкие изделия (болты, пряжки, крючки
для одежды и т. п.) Л. и. наносят в барабанах, вра-
вращающихся с частотой 20—80 об/мин, или в центрифугах
при частоте вращения ок. 200 об/мин. Барабаны м. б.
снабжены нагревателями для последующей сушки Л. п.
При нанесении Л. и. э л е к т р о о с а ж д е н и е м
изделия, движущиеся на конвейере, погружают в ван-
ванну с водоразбавляемым лакокрасочным материалом
(см. Водоразбавляемые грунтовка и эмали). Окраши-
Окрашиваемое изделие служит анодом; корпус ванны, а иног-
иногда, кроме того, и специальные пластины — катодом.
Частицы материала в результате электрофореза дви-
движутся к аноду и, разряжаясь, осаждаются на нем,
т. к. вследствие низкого значения рН в прианодной
зоне переходят в водонерастворимую кислую форму.
При этом, помимо электрофореза, протекают одновре-
одновременно электролиз и электроосмос воды. В начале
процесса материал осаждается преимущественно на
острых кромках и выступах окрашиваемой поверхности.
В дальнейшем силовые линии электрич. поля перерас-
перераспределяются вследствие изолирующего действия нане-
нанесенного слоя Л. п. Это обеспечивает образование на
всей поверхности, даже сложной формы, плотной плен-
пленки равномерной толщины B0—25 мкм), содержащей
не более 5% влаги. Изделия из черных металлов иног-
иногда предварительно фосфатируют для улучшения адге-
адгезии пленки и предотвращения сильного растворения
металлич. подложки на аноде.
Метод позволяет хорошо защитить от коррозии ост-
острые углы и кромки изделий, сварные швы, внутренние
полости, сократить потери лакокрасочных материалов,
улучшить санитарно-гигиенич. условия труда. Уста-
Установки безопасны в пожарном отношении. Недостаток
метода — возможность применения его только для на-
нанесения первого слоя Л. и. (напр., грунтовки), т. к.
этот изолирующий слой препятствует электроосажде-
электроосаждению след. слоя.
Лакокрасочный материал можно наносить также
ручными валиками, напр, в строительстве при
окраске стен и фасадов зданий. Систему валиков исполь-
используют для механизированного нанесения лакокрасоч-
лакокрасочного материала на металлич. листы или непрерывно
движущуюся металлич. ленту. Нанесение лакокрасоч-
лакокрасочного материала возможно как на одну, так и одновре-
одновременно на обе стороны изделия. Обычно для этого спосо-
способа применяют густые лакокрасочные материалы, т. к.
жидкие плохо удерживаются на валках.
Наиболее старые и малопроизводительные метода
нанесения Л. п. (кистью или шпателем)
применяют обычно при работе с медленно высыхающи-
высыхающими материалами.
Новые методы получения покрытий из адсорби-
адсорбированных на подложке мономеров
под действием электронного излучения или тлеющего
разряда позволяют получать топкие покрытия без при-
применения растворителей, обладающие хорошими ди-
электрич. свойствами, высокой адгезией к подложке
и химстойкостью. Покрытия можно получать на метал-
металлич. и неметаллич. подложках; на последние иногда
предварительно наоносят тонкий слой алюминия
[~ 100 нм (~ 1000А)] методом вакуумного напыления.
Механизм образования покрытий сложен. Предпола-
Предполагают, что при неупругих столкновениях электронов
с мономерами образуются возбужденные молекулы,
ионы, радикалы, к-рые адсорбируются на твердой под-
подложке и взаимодействуют друг с другом с образованием
полимерных пленок. Действие ионов и электронов на
полимер приводит, в свою очередь, к образованию
макроионов и макрорадикалов и протеканию в пленке
процессов сшивания и деструкции.
С помощью источника электронного излучения (элект-
(электронной пушки, ускорителя) на изделиях различной
конфигурации получают очень тонкие покрытия (до
1 мкм) из различных мономеров, в частности оргаиоси-
локсанов. Покрытия образуются в заполненной парами
мономера вакуум-камере [разрежение 133—13,3 мн/м2
A0~3—10~4 мм рт. ст.), энергия потока электронов
1—2 кэв, плотность тока не более 2 ма/см2].
Под действием тлеющего разряда [разрежение в ва-
вакуум-камере 1,333—666,6 н/м2 A0~2 — 5 мм рт. ст.),
напряжение переменного тока 500—700 в] полимери-
зуются стирол, акриловые соединения и др. с образо-
образованием покрытий толщиной 0,01—10 мкм. Преимуще-
Преимущества этого способа перед электронным облучением —
более простое оборудование, большая скорость роста
пленки (~1 мкм/мин). Кроме того, способ не требует
радиационной защиты.
Сушка покрытий
Во время сушки из нанесенного слоя лакокрасоч-
лакокрасочного материала улетучивается растворитель и могут
также протекать химич. процессы пленкообразования
(см. Лаки и эмали).
Холодную сушку A5 — 25 °С) применяют
преимущественно для быстровысыхающих термопла-
термопластичных материалов (нитроцеллюлозных, перхлорвинн-
ловых и др.). Ее используют также для медленно вы-
высыхающих материалов при их нанесении на крупные
конструкции и сооружения. Продолжительность холод-
холодной сушки быстровысыхающих материалов составляет
1—3 ч, медленно высыхающих — ~16—30 ч.
Без горячей сушки во многих случаях невозможно
получить Л. п. из материалов на основе термореактив-
термореактивных пленкообразующих (алкидных, меламино-алкид-
ных, феноло-формальдегидных и др.)- Наиболее рас-
распространенные способы такой сушки — конвективный,
терморадиационный, индукционный. Пары растворите-
растворителей, к-рые отсасываются из сушильных камер, м. б.
подвергнуты окислению на палладии, платине или др.
катализаторах. Тепловыделение при окислении обус-
обусловливает повышение темп-ры потока окисленных па-
паров, основная часть к-рых м. б. возвращена в камеру
для использования в качестве теплоносителя. Такой
рациональный способ использования растворителей
позволяет одновременно уменьшить загрязнение атмо-
атмосферы.
При конвективной сушке окрашенное
изделие нагревается циркулирующим горячим возду-
воздухом в камерах с теплоизоляционным ограждением
и с нагревательными приборами, к-рые м. б. помещены
внутри или вне камеры. Загрязненный парами раство-
растворителя рециркулирующий воздух подогревается и час-
частично заменяется свежим.
При конвективном способе сушки сначала высыхает
поверхностный слой Л. п., что затрудняет улетучивание
растворителя из нижних слоев и может приводить к об-
образованию покрытия с пузырями -и кратерами. Кроме
того, передача тепла к Л. п. происходит сравнительно
медленно, и сушка длится 0,5 — 0,3 ч.
Т е р м о р а д и а ц и о н и а я сушка осущест-
осуществляется под действием ИК-излучения. Процесс начи-
начинается от поверхности металла, который поглощает
ИК-лучи, прошедшие через покрытие, и поэтому про-
прогревается в начале сушки сильнее, чем Л. п. Такое
23
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ ПО ДЕРЕВУ
24
направление теплового потока обеспечивает беспрепят-
беспрепятственное улетучивание растворителя. Кроме того, ИК-
лучи ускоряют химич. процессы пленкообразования.
Источниками ИК-лучей служат ламповые и т. наз.
«темные» излучатели. Применение ламп накаливания
в сочетании с алюминиевыми рефлекторами позволяет
сократить длительность сушки в 3—6 раз по сравнению
с длительностью конвективной сушки. Ламповые су-
сушильные установки просты по конструкции, однако
они не обеспечивают равномерного обогрева изделий,
а расход электроэнергии при их эксплуатации на 15—
20% больше, чем при эксплуатации установок с тем-
темными излучателями. Кроме того, лампы хрупки, не-
недолговечны, через их стекло проникает И К-излучение
с максимальной длиной волны 0,5 мкм, недостаточной
для эффективной сушки. Вместо обычных осветитель-
осветительных используют также зеркальные лампы параболич.
формы, лампы с двумя спиралями накаливания разной
мощности и др. Для сушки отдельных подкрашенных
участков поверхности применяют передвижные щиты
с несколькими ламповыми излучателями.
Темные излучатели изготовляют в виде трубчатых
электронагревателей, к-рые снабжают рефлекторами из
полированного алюминия (трубчатый излучатель) или
заключают их в чугунную нагревательную плиту (па-
нельно-плиточный излучатель). Применяют также газо-
газовые генераторы излучения в виде нагревательных пане-
панелей с внутренним или выносным расположением горе-
горелок. Изделия сложного профиля сушат в терморадиа-
ционно-конвективных газовых установках (с рецирку-
рециркуляцией отходящих газов для дополнительного конвек-
конвективного нагрева). Такие установки в 2—3 раза дешевле
электрич. в эксплуатации. Очень эффективны установ-
установки с газовыми беспламенными горелками, керамич.
пористой излучающей поверхностью и алюминиевыми
рефлекторами.
Темные излучатели испускают основной тепловой по-
поток на волнах длиной 3,5—5 мкм, к-рые в одинаковой
степени отражаются и поглощаются лакокрасочными
материалами любого цвета и хорошо проникают в Л. п.
Оптимальное расстояние от генераторов излучения до
изделий 100—400 мм. Терморадиационная сушка не-
нерациональна для массивных отливок с толщиной стен-
стенки более 300 мм.
При индукционной сушке окрашенное
изделие помещают в переменное электромагнитное
поле, к-рое создается в индукторах токами повышенной
или промышленной частоты. Процесс длится несколько
минут. Метод рационально применять для сушки Л. п.
на металлич. лентах, листах обшивки, изделиях с об-
обмоткой, пропитанной лакокрасочными материалами.
Сушка с помощью УФ-излучения
и электронного излучения имеет огра-
ограниченное применение для материалов на основе р-ров
ненасыщенных полиэфиров и др. олигомеров в реак-
ционноспособных растворителях (мономерах). С по-
помощью УФ-излучения можно отверждать только про-
проницаемые для него материалы, напр, лаки, а также
шпатлевки, содержащие соответствующие наполнители
(микрослюду, бланфикс и др.). В состав материалов
должен быть введен фотосенсибилизатор. Источниками
излучения служат след. лампы: суперактиничные,
люминесцентные, синего света, ртутные высокого и
низкого давления. Продолжительность отверждения
1 —12 мин вместо 16—24 ч, необходимых в случае ис-
использования химич. инициатора и ускорителя сополи-
меризации.
Отверждение под действием образующегося в уско-
ускорителе мощного пучка электронных лучей с энергией
100—500 кэв можно применять как для наполненных,
так и ненаполненных лакокрасочных материалов в от-
отсутствие инициаторов. Покрытия отверждаются с боль-
большой скоростью. Так, при окрашивании непрерывно
движущейся металлич. полосы скорость конвейера
может достигать 200 м/мин (при сушке др. способами
она не превышает ~100 м/мин). Способ более дорог,
энерго- и трудоемок, чем отверждение под действием
УФ-излучения, и требует радиационной защиты.
Отделочные работы
После сушки Л. п. обычно шлифуют и полируют.
Шлифование промежуточных слоев способст-
способствует улучшению адгезии между ними благодаря появ-
появлению микроцарапин на их поверхности, а также удале-
удалению механич. включений. Кроме того, шлифование
придает Л. п. матовость. Для шлифования применяют
тканевые и бумажные шкурки с различными абрази-
абразивами. Процесс м. б. «сухим» и «мокрым»; в последнем
случае шкурку для ускорения шлифования смачивают
уайт-спиритом или водой.
Полирование применяют для получения Л. п.
с зеркальным блеском. Это достигается последователь-
последовательной обработкой покрытия нанесенными на мягкую
ткань полировочными пастами (смесью мягкого абра-
абразива, напр, трепела, со связующим, напр, минераль-
минеральным маслом), дисперсией мягкого абразива в водной
эмульсии уайт-спирита и сухой овечьей шкуркой.
Шлифование и полирование производят вручную или
с помощью аппаратов, рабочие инструменты к-рых
(жесткие или мягкие круги, абразивная лента и др.)
совершают вращательное, возвратно-поступательное
или поступательное движение. Л. п. на плоских по-
поверхностях полируют с помощью полировальных авто-
автоматов. Для получения высококачественных Л. п. по
дереву применяют шеллачные политуры, к-рые наносят
вручную ватным тампоном.
Помимо шлифования и полирования, выполняют
и др. отделочные работы, напр, наносят на Л. п. опоз-
опознавательные знаки, рисунки и т. д.
Л. п. применяют во всех отраслях народного хозяй-
хозяйства, а также в быту. По масштабам использования они
значительно превосходят металлические, стеклоэмале-
вые и др. покрытия, несмотря на то, что уступают этим
видам покрытий по твердости, абразиво- и термостой-
термостойкости. Широкое распространение Л. п. обусловлено
их высокой атмосферостойкостью, возможностью при-
придавать поверхности изделия разнообразный внешний
вид, простотой нанесения и ремонта, относительной де-
дешевизной. В 1972 во всем мире для получения Л. п.
было израсходовано ок. 14,5 млн. т лакокрасочных
материалов.
Лит.: Любимов Б. В., Специальные защитные покры-
покрытия в машиностроении, 2 изд., М.— Л., 1965; А р о н о в Н. В.,
Оборудование и механизация цехов металлических: защитных
покрытий, М., 1969; Справочник по лакокрасочным покрытиям
в машиностроении, под ред. М. М. Гольдберга, 2 изд., М.,
1974; Мачевская Р. А., Мочалова О. С, Подго-
Подготовка поверхности под окраску, М., 1971; Альбом оборудо-
оборудования окрасочных цехов, М., 1970; Аппаратура и приборы
для нанесения и испытания лакокрасочных покрытий, М., 1973.
М. М. Голъдберг.
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ ПО ДЕРЕВУ
(paint coatings on wood, Anstrichuberziige auf Holz,
revetements vernis des bois). Лакокрасочные покрытия
(Л. п.) по дереву выполняют две основные функции:
защищают дерево от гниения и придают изделиям
красивый внешний вид. В ряде случаев эти Л. п. пони-
понижают также горючесть дерева, повышают его стойкость
к агрессивным средам, улучшают эксплуатационные
свойства изделий и др.
Л. п. по дереву бывают прозрачными (на основе ла-
лаков) и непрозрачными, или укрывистыми (на основе
эмалей). Для отделки дерева применяют материалы
воздушной или низкотемпературной сушки, т. к. низ-
низкая теплостойкость дерева и склонность фанерованных
изделий к короблению исключают возможность их на-
25
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ ПО ДЕРЕВУ
26
грева выше 60 °С. Выбор лакокрасочного материала
(таблица) определяется требованиями к эксплуатаци-
эксплуатационным свойствам изделия и к его внешнему виду, фор-
формой изделия, способом нанесения материала на под-
подложку.
Технология отделки дерева лакокрасочными материа-
материалами включает: 1) подготовку поверхности древесины
(столярную обработку, отбелку, крашение, заполнение
пор древесины, нанесение грунтовок, шпатлевок);
2) нанесение лака или эмали; 3) сушку покрытия;
4) обработку (облагораживание) покрытия (шлифование,
полирование и др.)-
Подготовка поверхности древесины. Деревянная под-
подложка отличается от металлической рядом специфич.
особенностей: гигроскопичностью, пористостью, раз-
различной структурой вдоль и поперек волокон. Приме-
Применение влажной древесины обусловливает низкую адге-
адгезию Л. п. и его растрескивание вследствие деформации
подложки при высыхании в процессе эксплуатации.
Поэтому древесину сначала подсушивают (в сушилках
или в естественных условиях) до влажности 6—10%
(по массе). Не менее важно и качество столярной подго-
подготовки древесины, обеспечивающей создание гладкой
поверхности с чистотой не ниже 10 класса.
При необходимости изменения естественного цвета
древесины ее обрабатывают отбеливающими веществами
(окислителями) или красителями. Наибольшее приме-
применение в качестве окислителя нашла перекись водорода,
к-рая разлагается в щелочной среде с выделением ки-
кислорода. При отбелке древесину сначала обрабатывают
48%-ным р-ром NaOH, выдерживают на воздухе в тече-
течение 30—40 мин, а затем смачивают 30%-ным водным
р-ром Н2О2. После этого поверхность древесины нейт-
нейтрализуют 2—4%-ным водным р-ром уксусной или щаве-
щавелевой к-ты и сушат на воздухе в течение одних суток.
Отбелке обычно подвергают древесину наиболее свет-
светлых пород — березу, бук, ясень. Крашение в большин-
большинстве случаев проводят с целью имитации дешевых пород
под дорогостоящие, а также для выравнивания естест-
естественного цветового тона древесины. Чаще всего приме-
применяют 0,5—3,0%-ные водные р-ры коричневых и красных
кислотных азокрасителей широкой гаммы оттенков,
к-рые наносят распылением. Поверхность древесины,
применяемой для изготовления изделий, эксплуати-
эксплуатируемых в условиях влажного тропич. климата, проти-
протирают специальными пропиточными материалами, обла-
обладающими фунгицидными свойствами и предотвращаю-
предотвращающими образование плесени. К таким материалам отно-
относится т. наз. «окситерпеновый растворитель» — отход
от очистки скипидара, содержащий 55—60% терпено-
вых спиртов.
Проникновение лакокрасочного материала в поры
древесины («проседание») приводит к тому, что покры-
покрытие, нанесенное ровным слоем, становится по мере его
высыхания неровным. Для устранения этого явления
после описанных выше подготовительных операций по-
поры древесины заполняют специальными материалами.
Для прозрачной отделки применяют т. наз. поро-
заполнители, к-рые втирают тампоном (вруч-
(вручную или на станке). Избыток порозаполнителя удаляют,
тщательно протирая поверхность ветошью поперек
волокон. Порозаполнители (напр., марок Кф-1 и Кф-2)
для дуба, ореха, ясеня представляют собой конц. сус-
суспензию (пасту), получаемую диспергированием тонко-
тонкодисперсного стеклянного порошка или кремнезема
в р-ре смеси льняного масла с глицериновым эфиром
канифоли в нетоксичном высококипящем растворителе
(скипидаре, уайт-спирите, этилцеллозольве). Для крас-
красного дерева применяют порозаполнители (напр., марки
Кф-3), подкрашенные красными органич. или неорга-
нич. пигментами. Заполнение пор при отделке дерева
спиртовыми шеллачными лаками в мелких кустарных
мастерских или в домашних условиях производят
р-ром воска в скипидаре, к-рый наносят подогретым
до 50—60 °С вручную тампоном.
На поверхность древесины мелкопористых пород
(береза, бук), не требующей применения порозаполни-
телей, наносят прозрачные грунтовки, не содержащие
порошкообразных наполнителей, напр. 45—53%-ную
водную дисперсию поливинилацетата. Операцию за-
заполнения пор удается исключить при использовании
парафинсодержащэго ненасыщенного полиэфирного ла-
Лакокрасочные материалы для покрытий по дереву
Тип лака
Полиэфирный парафинсо-
держащий
Полиэфирный беспарафино-
беспарафиновый
Нитроцеллюлозный(для рас-
распыления с подогревом)
Нитроцеллюлозный (для рас-
распыления без подогрева)
Нитро-алкидно-карбамид-
ный кислотного отверж-
отверждения
Нитро-алкидно-эпоксидный
Алкидно-карбамидный кис-
кислотного отверждения
Алкидно-стирольный
Алкидный (пентафталевый)
Перхлорвиниловый
Пол иу рет анов ый
Характеристика покрытия
Глянцевое с зеркальным блеском, свето-
светопрочное, стойкое к спирту, ацетону, при
темп-pax от —40 до 100 °С
То же
Глянцевое с неоднородным блеском*,
стойкое при темп-pax до —12 °С, не
стойкое к спирту и ацетону
Глянцевое с открытыми порами
Глянцевое с неоднородным блеском, стой-
стойкое к спирту и при темп-pax до —20 °С
Полуматовое, атмосферостойкое при эк-
эксплуатации под навесом
Полуглянцевое, стойкое к спирту и при
темп-pax до —15 °С
Глянцевое, стойкое в условиях тропич.
климата
На основе лака — глянцевое, стойкое при
темп-pax до 40 °С, на основе эмали —
полуглянцевое, атмосферостойкое
Полуглянцевое, с пониженной" горючестью
Глянцевое, износостойкое
Окрашиваемые объекты
лаком
Щитовые элементы мебели
Футляры телевизоров в
собранном виде
Щитовые элементы мебели
Решетчатая мебель и др.
изделия в собранном ви-
виде, детские игрушки
Щитовые элементы мебели
Лыжи, стулья
Деревянная тара для при-
приборов
Сидения в вагонах город-
городского и железнодорож-
железнодорожного транспорта
Паркетные полы
эмалью
Мебель кухонная, меди-
медицинская, детская, щитовая,
мебель из древесностру-
древесностружечных плит, пианино, ро-
рояли
Мебель кухонная и дет-
детская в собранном виде
Мебель кухонная, меди-
медицинская, детская (щитовая
и в собранном виде)
Мебель садово-парковая,
устанавливаемая под кры-
крышей
Лыжи, мебель кухонная
Мебель садово-парковая
Деревянные детали в ваго-
вагонах городского и железно-
железнодорожного транспорта
* Покрытия на основе эмалей м. б. также полуглянцевыми, матовыми, с «молотковым» узором, с рисунком под крокодиловую
кожу (см. Декоративные лакокрасочные покрытия).
27
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ ПО МЕТАЛЛУ
28
ка (см. Полиэфирные лаки и эмали), т. к. этот лак нано-
наносят толстым слоем (порядка 350—500 мкм) и, кроме
того, он обладает малой усадкой в процессе сушки.
При укрывистой отделке поверхность древесины вы-
выравнивают теми же грунтовками и шпатлевками воз-
воздушной или низкотемпературной сушки, что и при от-
отделке металлич. поверхностей. Специально для дерева
разработаны шпатлевки ПЭШ, к-рые представляют
собой густую пасту, получаемую диспергированием
смеси талька, литопона и мела в полиэфирном лаке.
Перед нанесением на поверхность к ПЭШ добавляют
инициатор полимеризации — гидроперекись кумола
и ускоритель — нафтенат кобальта. Такую композицию
(жизнеспособность не менее 16 ч) наносят шпателем или
распылением и отверждают при 60 °С в течение 2—2,5 ч.
ПЭШ благодаря их низкой усадке можно применять
для выравнивания поверхности даже древесностружеч-
древесностружечных плит. Покрытие на основе ПЭШ толщиной 2 мм
не растрескивается при —40 °С в течение 25 ч.
Нанесение лака или эмали. Способы нанесения ла-
лаков и эмалей на подготовленную поверхность древесины
аналогичны тем, к-рые применяют при окраске металла
(см. Лакокрасочные покрытия). Специфическим для
дерева является метод налива, используемый только
при отделке плоских деталей, напр. щитовых элемен-
элементов мебели, футляров, радиоприемников и телевизоров.
Для нанесения Л. п. этим методом применяют л а к о-
наливные машины, снабженные одной или
двумя головками, конструкция к-рых аналогична кон-
конструкции фильер, испольауемых в поливочных машинах
для изготовления пленок методом полива (см. Пленки
полимерные). Головки размещают над движущейся
плоскостью, на к-рую помещают отделываемую деталь.
Лаконаливные машины с двумя головками применяют
для нанесения лаков с ограниченной жизнеспособ-
жизнеспособностью, напр. парафинсодержащих полиэфирных.
В этом случае в одну из головок загружают лак с уско-
ускорителем, в другую — лак с инициатором. Оба нолу-
фабрикатных лака наливают последовательно на дви-
движущийся щит; при смешении компонентов начинается
сополимеризация полиэфира с входящим в состав лака
мономером, обусловливающая отверждение покрытия.
При использовании метода налива потери лакокрасоч-
лакокрасочных материалов в 6—10 раз меньше, чем при их нане-
нанесении методом пневматич. распыления, и, кроме того,
в этом случае можно применять материалы с повышен-
повышенной рабочей вязкостью (т. е. с большим содержанием
пленкообразующих веществ). Лакокрасочный материал
можно наносить на высокопроизводительных поточных
линиях (скорость движения щитов 40—120 м/мин).
При нанесении методом распыления в электрич. поле
высокого напряжения поверхность древесины предва-
предварительно обрабатывают материалами, придающими
ей электрич. проводимость: 7 — 10%-ным р-ром продук-
продукта алкамон ОС-2 в уайт-спирите или р-рами других
поверхностно-активных веществ.
Сушка покрытия. Продолжительность воздушной
сушки (в естественных условиях) может в отдельных
случаях, особенно при нанесении многослойных Л. п.,
достигать ~72 ч. Поэтому такие покрытия сушат обыч-
обычно в искусственных условиях. Для того чтобы избежать
пересыхания, растрескивания или коробления древе-
древесины, Л. п. по дереву сушат в среде влажного воздуха.
Так, при темп-ре сушки 30—60 СС и влажности древе-
древесины 6—8% влажность воздуха в сушильной камере
должна составлять 30—40%.
Покрытия на основе нитроцеллюлозных лаков сушат
конвекционным способом при 40—60 °С;
при этом продолжительность высыхания в 2—3 раза
меньше, чем при комнатной темп-ре. Еще большее
ускорение процесса достигается при нанесении лака на
щиты, предварительно нагретые до 45 °С. Такой способ
сушки позволяет, кроме того, получать покрытия без
воздушных пузырьков, т. к. воздух удаляется из дре-
древесины при ее предварительном прогреве. На нагретый
щит можно наносить материалы с повышенной вязко-
вязкостью, уменьшая таким образом число слоев лака или
эмали.
Терморадиационный способ (под дей-
действием И К-излучения) наиболее пригоден для сушки
полиэфирных лаков. Сушка нитроцеллюлозных лаков
эффективна при толщине пленки до 100 мкм, т. к. при
большей толщине резко уменьшается проникновение
ИК-лучей в нижние слои покрытия.
Применительно к полиэфирным лакам развиваются
новые методы ускоренной сушки: с по-
помощью УФ-лучей и быстрых электронов. Продолжи-
Продолжительность отверждения в первом случае составляет
1 —12 мин, во втором — 0,1 — 2 сек. С помощью УФ-
лучей можно сушить однокомпонентные полиэфирные
лаки, обладающие ограниченной жизнеспособностью;
в случае сушки под действием быстрых электронов
из состава лака м. б. исключены т. наз. всплывающие
добавки, к-рые вводят для подавления ингибирующего
действия кргслорода воздуха на процесс отверждения
(см. также Полиэфирные лаки и эмали). О способах суш-
сушки см. также Лакокрасочные покрытия.
Обработка покрытий. Покрытия на основе парафинсо-
парафинсодержащих полиэфирных лаков шлифуют и полируют.
Эти операции выполняют с помощью тех же материалов,
что и при обработке покрытий по металлу (см. Лакокра-
Лакокрасочные покрытия), но на др. оборудовании; для Л. п.
по дереву чаще всего применяют ленточно-шлифоваль-
ные и ленточно-полировальные станки. При отделке
изделий сложной конфигурации, покрытых нитролака-
нитролаками, применяют специфич. операцию — «разравнива-
«разравнивание»: на отшлифованное покрытие вручную наносят
т. наз. распределительные жидкости — смеси актив-
активных растворителей с разбавителями (напр., 35% этил-
или бутилацетата и 65% этилового или бутилового
спирта). В случае получения матовых покрытий после
разравнивания на поверхность изделия наносят слой
матирующего лака, например нитролака, содержаще-
содержащего в качестве матирующих добавок стеараты цинка
или алюминия или ультратонкую двуокись кремния
(аэросил).
Лит.: Буглай Б. М., Технология отделки древесины,
М., 1962; Мищенко Г. Л., Н е й м а н А. Ф., Технология
прозрачной отделки щитовых элементов мебели, М., 1964;
Нагорская И. А., Отделка древесины лакокрасочными
материалами, М., 1965; Беляева К. П., Ш т а н ь-
к о Н. Г., Т о д о р о в а Т. В., Лакокрасочные материалы
для отделки изделий из дерева, М., 1971. К. П. Беляева.
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ ПО МЕТАЛ-
МЕТАЛЛУ — см. Защитные лакокрасочные покрытия, Лако-
Лакокрасочные покрытия.
ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ ПО ПЛАСТМАС-
ПЛАСТМАССАМ (paint coatings on plastics, Anstrichiiberztige
auf Kunststoffe, revetements vernis des matieres plasti-
ques). Лакокрасочные покрытия (Л. п.) наносят на изде-
изделия из пластмасс для их защиты от разрушения под
действием влаги, солнечной радиации, химич. реаген-
реагентов, а также для декоративной отделки и маркировки
изделий. В нек-рых случаях Л. п. наносят на пластмас-
пластмассы перед их металлизацией с целью повышения адге-
адгезии металлич. покрытия.
Подготовка поверхности пластмассы, необходимая
для обеспечения хорошей адгезии Л. п., во многих слу-
случаях сводится к обезжириванию органич. соединеиия-
ми с целью удаления разделительных смазок (восков,
стеарата цинка, кремнийорганич. жидкостей), мине-
минеральных масел и др. Используемые для этого соедине-
соединения не должны растворять пластмассу. Так, полисти-
полистирол, полиметилметакрилат, поликарбонат обезжири-
обезжиривают бензином или уайт-спиритом. Иногда для улуч-
улучшения адгезии Л. п. обезжиривающие вещества сме-
смешивают с небольшим количеством растворителя пласт-
29
ЛАКТАМОВ ПОЛИМЕРИЗАЦИЯ
30
массы, вызывающего набухание («растравливание») ее
поверхностного слоя.
Нек-рые пластмассы, напр, стеклопластики, подвер-
подвергают после обезжиривания механич. обработке абразив-
абразивными шкурками или пастами (напр., смесью пемзы,
мыла, растительного масла и воды), дробеструйной
очистке гранулятом из скорлупы фруктовых косточек,
гидроабразнвной очистке струей 15%-ной водной сус-
суспензии песка, галтовке в присутствии песка во вра-
вращающихся барабанах и др. Механическая обработка
стеклопластиков не должна вызывать повреждений
наполнителя.
Поверхность полполефинов, не содержащих функцио-
функциональных групп и имеющих гладкую малопористую по-
поверхность, перед нанесением Л. п. подвергают химия,
модификации, в результате к-рой макромолекулы при-
присоединяют полярные группы. Наиболее распростра-
распространенный способ модификации — обработка жидкими
или газообразными окислителями. Изделия предвари-
предварительно обезжиривают, выдерживают для набухания
поверхностного слоя пластмассы п смеси ацетона с то-
толуолом A : 1) и окисляют хромовой смесью D мин при
85 °С или 2 ч при 18—25 °С), хромовой к-той (полиэти-
(полиэтилен __ ю мин при 70 СС, полипропилен — 5 мин при
100 °С), смесью HG1 и HNO3 C : 1), конц. HNO3, гипо-
хлоритом натрия C0—90 °С), Н2О2 и др.
Кроме этого способа, используют также: 1) кратко-
кратковременную (не более 1 сек) обработку пламенем газовой
горелки с темп-рой 700—900 °С; 2) выдержку в элект-
рич. поле высокого напряжения при нормальном давле-
давлении или в поле низкого напряжения под вакуумом;
в этом случае поверхность пластмассы окисляется об-
образующимся озоном; 3) хлорирование при каталити-
каталитическом действии видимого света или сульфохлори-
рование смесью хлора и двуокиси серы при УФ-об-
лучении.
Специфич. способ модификации поверхности поли-
политетрафторэтилена — обработка р-ром металлич. нат-
натрия в жидком аммиаке или р-ром натрия и нафталина
A : 1) в тетрагидрофуране. При этом толщина модифи-
модифицированного слоя, содержащего ненасыщенные связи,
достигает нескольких мкм.
При выборе лакокрасочных материалов для окраски
изделий учитывают природу пластмассы, а также назна-
назначение и условия эксплуатации Л. п. (таблица). На^не-
ровную поверхность изделий (напр., из стеклопла-
стеклопластиков, пенопластов) предварительно наносят слой
шпатлевки. В состав грунтовок вводят обычно смесь
органич. жидкостей, содержащую небольшое количест-
количество растворителя пластмассы; напр., в состав грунтовки
для полистирола входит смесь бутанола с ксилолом.
В нек-рых случаях наносят также грунтовочные слои,
к-рые препятствуют миграции в Л. п. содержащихся
в пластмассе пластификаторов и красителей.
Улучшению адгезии Л. п. способствует нанесение
подогретого лакокрасочного материала или погруже-
погружение изделия перед его окраской на 10—15 сек в подогре-
подогретый растворитель. Изделия из нек-рых полимеров,
напр, из полистирола или полнакрилатов, перед окрас-
окраской подвергают термообработке, чтобы снять внутрен-
внутренние напряжения, возникающие в изделии при его из-
изготовлении. Томп-ра термообработки должна быть ни-
ниже темп-ры стеклования полимера (напр., для полисти-
полистирола _ не выше 60 СС). Л. п. на формованные изде-
изделия наносят спустя пек-рое время после их изготовле-
изготовления, т. к. в противном случае возможно отслоение
Л. п. из-за усадки изделия.
Л. п. наносят на пластмассы теми же методами, что
и на металлы (см. Лакокрасочные покрытия). Темп-ра
сушки Л. п. определяется как составом лакокрасоч-
лакокрасочного материала (см. Пленкообразующие вещества),
так и теплостойкостью пластмассы, формой изделия,
толщиной его стенки и др. Основная масса раствори-
Лакокрасочные материалы для покрытий
по пластмассам
Назначение покрытия
(предельная темп-ра
эксплуатации)
Лакокрасочные материалы,
входящие в состав покрытия
Стеклопластик
Защита от влаги и декора-
декоративная отделка (90 °С)
То же B00 °С)
То же C00 °С)
Гетинакс,
Защита от влаги, масла и
бензина
Защита от влаги, электро-
электроизоляция
Феноло-формальдегидная или
эпоксидная шпатлевка, поли-
полиакриловый лак или алкидная
грунтовка, полиакриловая
или перхлорвиниловая эмаль
Эпоксидная шпатлевка, эпок-
эпоксидная или фторопластовая
эмаль
Кремнийорганическая эмаль
текстолит
Эпоксидная или эпоксидно-уре-
тановая эмаль*
Эпоксидная эмаль или эпоксид-
эпоксидный или эпоксидно-уретано-
вый лак*, **
Органическое стекло
Отражение или поглощение
света
Декоративная отделка
Полиакриловая или эпоксидная
эмаль
Нитроцеллюлозная или алкид-
алкидная эмаль
П о л и а м и д-68
Защита от влаги и декора- I Перхлорвиниловая эмаль
тивная отделка |
Фтороплас т-4***
Декоративная отделка и I Эпоксидно-полиамидная или
маркировка | фторопластовая эмаль
Полиэтиле н*** и полиэтилентерефталат
То же I Эпоксидно-полиамидная или по-
I лиакриловая эмаль
Полистирол
Подслой под металлич. по-
покрытие, наносимое в ва-
вакууме
Покровный слой по метал-
металлич. покрытию
Меламино-алкидный лак
Полиуретановый лак
* Наносят также на стеклопластик, эксплуатируемый в
аналогичных условиях. **Покрытие предназначено для эксплу-
эксплуатации внутри помещения. ***Наносят на модифицированную
поверхность пластмассы.
теля должна улетучиваться из нанесенного слоя лако-
лакокрасочного материала при комнатной темп-ре. Макси-
Максимальная темп-ра сушки Л. п. на поливинилхлориде —
60 °С, на полистироле — 70 °С, на полиэтилене и поли-
полипропилене — 85 "С.
Лит.: Берлин А. А., Басин В. Е., Основы адгезии
полимеров, М., 1969; РотреклБ., Г у д е ч е к К., К о-
марекЯ., СтанекИ., Поверхностная обработка пласт-
пластмасс, пер. с чеш., Л., 1972. См. также лит. при ст. Лако-
Лакокрасочные покрытия. М. М. Гольдберг.
ЛАКТАМОВ ПОЛИМЕРИЗАЦИЯ (polymerization
of lactams, Polymerisation, von Laktamen, polymeri-
polymerisation des lactames).
Лактамы (Л.) полимеризуются под действием ката-
катализаторов катионного типа (вода, соли аминов, неор-
ганич. к-ты) и анионного типа (щелочные и щелочно-
щелочноземельные металлы, их гидриды, металлоорганич. сое-
соединения, сильные основания).
Термодинамика полимеризации. Л., как и др. сое-
соединения, полимеризуются при условии AG < 0. Здесь
AG — изменение термодинамич. потенциала системы,
причем AG = АН — TAS, где АН — изменение эн-
энтальпии, AS — изменение энтропии, Т — темп-ра
реакции, К. Уменьшение AG при полимеризации Л.
с небольшим числом звеньев в цикле определяется
в основном уменьшением энтальпии системы, при поли-
полимеризации больших слабо напряженных циклов —
возрастанием энтропии. Значение (— АН) реакции
для незамещенных Л. возрастает с увеличением раз-
размера цикла, достигая максимума для 9-членного Л.,
31
ЛАКТАМОВ ПОЛИМЕРИЗАЦИЯ
32
а затем падает практически до нуля (таблица). Наличие
заместителя у наиболее удаленного от гетеросвязи
углеродного атома (напр., для у~СН3-капролактама)
приводит к значительному уменьшению (—Д#) по срав-
Изменение энтальпии
A ккал/молъ
Лактам
сс-Пирролидон
а-Пиперидон
е-Капролактам . .
а-СНз-Капролактам . . .
3-СНз-Капролактам
Y-СНз-Капролактам . . .
6-СНз-Капролактам
е-СНд-Капролактам .
а-С2Н5-Капролактам
а-С3Н7-Капролактам
3-С2Н5-Капролактам
7-С2Н5-Капролактам
7-С3Н7-Капролактам
е-С2Н5-Капрол актам
е-С3Н7-Капролактам
N-СНз-Капролактам
^-Энантолактам , .
3-С2Н5-Энантолактам
е-С2Н5-Энантолактам
?-С2Н5-Энантолактам
!-С3Н:-Энантолактам
N-СНз-Энантолактам
•п-Каприллактам ... . .
Пеларгонлактам
Капринлактам
Ундеканлактам
Лауринлактам
при полимеризации лактамов
= 4,1868
Число
атомов в
цикле
5
6
7
7
7
7
7
7
7
7
7
7
7
7
7
7
8
8
8
8
8
8
9
10
11
12
13
кдж/молъ)
V АН
измерено*
0, 51
3,3-4,0
—
—
2,Э-2,5
3,8
3,9
3,5
—
—
3,5
3,4
—
5,2
5,2
5,1
6,8
4,9
—
7 ,8 — 9,6
—
0-1,4
, ккал/молъ
рассчитано
от —1,3 до 1,1
1,1-2,2
3,0-3,8
2,5—3,8
2,6—3,0
2,6-3,0
2,6-3,0
2,5-3,8
—
—
2,5
2,4
—
—
2,3
5 } 3—5 ,7
—
—
—
3,9
5,6
2,8
-0,5
0
* По данным калориметрии.
нению с незамещенным Л. Введение заместителя в по-
положение, соседнее с гетеросвязью, практически не из-
изменяет энтальпии реакции. Значительное влияние на
АН оказывает заместитель у атома азота: (—АН) для
]Ч-СН3-капролактама и N-GHg-энантолактама меньше,
чем для незамещенных Л. Значение ASrJgg возрастает
с увеличением лактамного цикла от —7,3 кал/'(моль-°С)
для Я-пирролидона до 4,0 кал/(моль -°С) для |-энан-
толактама.
Следует отметить, что на основании данных таблицы
нельзя делать окончательных выводов о реакционной
способности Л. в полимеризации. Необходимо учиты-
учитывать изменение энтальпии и энтропии, обусловленное
существованием между молекулами Л. водородных
связей. Разрушение этих связей может приводить
к «активации» плохо полимеризующихся мономеров.
Катионная полимеризация. Под действием катион-
ных катализаторов Л. полимеризуются в массе с замет-
заметной скоростью F0—70% превращения за 2—3 ч) при
220—260 °С и молярном содержании катализатора
в системе ок. 1%. Схема образования активного центра
при использовании в качестве катализатора воды
(т. наз. гидролитич. полимеризация) приведена ниже:
NH
+Н2О
NH3-(CH2)n-COO~
СО
Гидролитич. полимеризация носит ярко выраженный
автокаталитич. характер. Рост цепи происходит в ре-
результате присоединения мономера к активному центру,
а не за счет конденсации концевых групп аминокислоты
(этот путь роста начинает играть значительную роль
только на конечных стадиях реакции). Максимальная
скорость процесса уменьшается при увеличении числа
членов в цикле Л. от 7 до 13. Однако время достиже-
достижения максимальной скорости для 7-членных Л. больше,
нем для 8- и 9-членных.
Скорость полимеризации Л. под действием а м и-
н о с о л е й или безводных неорганич.
кислот монотонно убывает во времени, индукцион-
индукционный период отсутствует. Присоединение мономера
к активному центру происходит в результате конден-
конденсации групп — ГШз-Х~ с ,Л. При этом образуется
амидиновая соль, к-рая затем гидролизуется выделив-
выделившейся при конденсации водой:
NH NH
| \ -Н2О / I _^
RNH3X~+ (GH2)n — ^l(GH2)n ~_±.
СО G=NH—R
х-
NH-X-
+н2о
— ^>R-NH-CO-(CH2)n-NH2X- и т. д.
-NH-R
Начальные скорости полимеризации Л. с числом
членов в цикле от 7 до 13 в присутствии аминосолей
уменьшаются с увеличением числа членов.
Наличие заместителя у углеродного атома приводит
к резкому снижению скорости катионной полимери-
полимеризации. Наибольшее влияние оказывают заместители
в положении, соседнем с гетеросвязью. N-СНз-Капро-
лактам не полимеризуется совсем, N-СНз-энантолактам
полимеризуется гораздо медленнее незамещенного мо-
мономера.
Кинетика и механизм катионной полимеризации Л.
наиболее подробно изучены для капролактама и энан-
толактама.
Анионная полимеризация. Л. полимеризуются по
анионному механизму в массе с большой скоростью
F0—70% превращения за 20—30 мин) при 150—220 °С
и молярном содержании катализатора ок. 1%. Ини-
Инициатор процесса — соль лактама, к-рая образуется,
напр., при взаимодействии щелочного металла с Л.;
инициирование заключается в присоединении Л. к этой
соли с образованием N-ацилпроизводного:
/
NH
+Ме
СО
NMe+
+Н2
- NH
|
о
ю _
N-CO-(CH2)n-NH
СО
Образовавшийся в результате инициирования анион
реагирует с Л. с образованием соединения с концевыми
амино- и имидной группами и аниона Л. Последний
присоединяется к имидной группе, к-рая остается
в процессе реакции на конце цепи:
H2N-CO[NH-(CH2)n-CO]m -N-CO
N-CO
\/
(СН2)„
H2N-CO[NH-(CH2)n-CO]mN-(CH2)n-CO-N-CO
(СН)
NH-CO
\/
(СН2)„
N-CO
H2N-CO[NH-(CH2)nCO]m+1N-CO
(СН2)
'п
Для ускорения полимеризации процесс проводят
в присутствии т. наз. активаторов (сокатализаторов),
33
ЛАКТОНОВ ПОЛИМЕРИЗАЦИЯ
в качестве к-рых используют N-ацилпроизводные Л.
или соединения, способные ацилировать Л. в условиях
полимеризации (эфиры, ангидриды и галогенангидриды
карбоновых к-т). При этом механизм роста цепи такой
же, изменяются только концевые группы полимера. Ин-
Ингибиторы анионной полимеризации — соединения, спо-
способные реагировать с имидной группой или с сильно ос-
основными анионами, напр, вода и др. соединения с ак-
активным водородом. Активность 7-, 8- и 9-членных Л. в
анионной полимеризации отличается незначительно; 5-,
6- и 13-членные Л. полимеризуются значительно хуже.
Лит.: КоршакВ.В., ФрунзеТ.М., Синтетические
гетероцепные полиамиды, М., 1962; Бонецкая А. К.,
в сб.: Современные проблемы физической химии, т. 6, М.,
1972; Волохина А. В., Хим. волокна, № 4, 3 A966);
Вихтерле, Себенда, Краличек, Хим. и технол.
полимеров, № 7, 39 A961), Collection of Czechosl. chem. com-
munic, 36, 3391, 3275 A971); там же, 37, 263, ИЗО A972).
О. Б. Саламатина.
ЛАКТОНОВ ПОЛИМЕРИЗАЦИЯ (polymerization
of lactones, Polymerisation von Laktonen, polymerisa-
polymerisation des lactones). Лактоны (Л.), как и сложные эфиры,
способны реагировать с электрофильными и с нуклео-
фильными реагентами (см. Лаптопов полимеры). Поэ-
Поэтому они могут полимеризоваться на большинстве
катионных и анионных катализаторов. Л. п. осуществ-
осуществляется также под действием ионизирующих излучений
и инициаторов радикального типа. Наиболее полно
изучена катионная и анионная Л. п.
Катионная полимеризация. Л. п. под действием
катионных катализаторов легко протекает при ком-
комнатной темп-ре. Механизм процесса для 4-членных Л.
заключается в нуклеофильной атаке атома кислорода
цикла на активный растущий центр, представляющий
собой ацильный катион:
о сн2-сн2 о о
II I ! II II A)
~С+ + О С=О -> ~* С -О(СН2J-С+
Одновременно протекает обратимая дезактивация
активных растущих центров вследствие связывания
их молекулами мономера и полимера:
о сн2-сн2
о
+
СН2-СН2
I I
о=с—о
-о
о=с
С
\
о
II + /
-с-о-=с
B)
(СН2J~
\
(СН2J
Для этого механизма характерен разрыв по ацилкис-
лородной связи в лактонном цикле в процессе роста
цепи. Однако при полимеризации Р,Р-диметил-Р-про-
пиолактона разрыв кольца происходит по простой эфир-
эфирной связи С — О с образованием стабильного карбка-
тиона:
Рост цепи для 5-, б- и 7-членных Л. протекает при
атаке карбонильным атомом кислорода мономера соот-
сн3
с ]
сн3
с с
" * 1
—0 <
:-о
сн,
эс—сн9—с —ос—сн2—с+ х
II 2 I ч II 2 I
о сн, о сн3
=~О
сн
Н3С^СН,
сн2
—сн,—с . —о с=о
C)
ветственно у-, б- или е-углеродного атома активного
растущего иона:
R X + О=С
RO —C-
х"
х"
[
RO—C-
-(сн2)-
О X
г
о=с-
(сн2)
—о
-(сн2)-
RO-C(CH2)nO=C-
о х
00
Реакции ограничения роста цепи связаны с передачей
цепи на низкомолекулярные примеси нуклеофильного
характера (см. Катионная полимеризация, Передача
цепи). Для механизма C) характерна также передача
цепи с отщеплением протона с образованием концевой
двойной связи:
СНз СНз
/~ос-сн2-с+х- -> ~ос-сн=с+нх E)
II i II I
О СНз О СНз
Аналогичная реакция может иметь место при повы-
повышенных темп-pax и при др. механизмах роста цепи.
Кроме того, во всех случаях одной из реакций огра-
ограничения роста цепи м. б. обрыв цепи на противоионе:
где Me — металл, X — галоген.
Анионная полимеризация. Имеются два принци-
принципиально различных пути анионной Л. п., механизм
к-рых четко доказан. Механизм определяется гл. обр.
типом катализатора и строением Л.
В присутствии третичных аминов, фосфинов, бетаи-
бетаинов, галоидных и карбоксилатных солей щелочных
металлов расщепление лактонного цикла идет по про-
простой эфирной связи С — О (алкильное расщепление).
Активный растущий центр в этом случае представляет
собой карбоксилатный анион. По этому механизму
полимеризуются в основном Р-пропиолактон и его
производные:
СН2—СН2
II
Me X + О С=О
Инициирование
ХСН2СН2СОО"Ме+
Рост цепи
сн2—сн2
~М+
х [(сн2Jсоо]— (сн2Jсоо~ м?
»- Х[(СН2JСОО]..+ — (СН2JСОО~Ме
В случае инициирования Л. п. третичными аминами,
фосфинами или бетаинами растущая макромолекула
представляет собой цвиттер-ион, напр.:
Инициирование
-с=о
—*- R3N (CH2)n СОО
Рост цепи
(СН2)
+ г 1 Г(СН2)
R3N (СН2) СОО (СН9) СОО + О
с=о
R3N
(CH2)nCOO~
(8)
35
ЛАКТОНОВ ПОЛИМЕРЫ
36
Для механизма, характеризующегося алкильным
расщеплением Л. в акте роста цепи, весьма характерна
передача цепи на мономер:
г-(СН2)-,
- + г
~СОО Me + 0-
~соон +
СН2=СНСОО Me
(9)
В результате протекания акта передачи цепи на моно-
мономер в ходе полимеризации полимер содержит концевые
карбоксильные и винильные группы в соотношении
1 : 1.
При полимеризации а-замещенных E-пропиолактона
передача цепи на мономер не идет, поэтому образуются
значительно более высокомолекулярные полимеры, чем
при полимеризации Р-пропиолактона.
В присутствии алкоголятов щелочных и щелочнозе-
щелочноземельных металлов и металлалкилов расщепление лак-
тонного кольца идет по одинарной связи в группе
О — С = О (ацильное расщепление). Растущий актив-
активный центр представляет собой алкоксильный анион:
сн-сн2
О С OR
О Me
Инициирование
сн9—сн2
Ме-
I
—С-OR
—О
Рост цепи
— +
ROC (СНДО Me*
О
RO [С (СН2)О] -С— (СН2JО Me + СН2-СН2 ^
о J о о с-о
— RO [С (СН2JО] .+ГС-(СН2JО-Ме+
о о
Полимеризация протекает без обрыва и передачи цепей,
т. е. по существу с участием живущих полимеров.
По этому механизму полимеризуются 6- и е-Л. и E-Л.,
содержащие в а-положении электроноакцепторные за-
заместители, благоприятствующие координации алко-
ксигруппы с карбонильным атомом углерода.
Л. п. под действием гидроокисей щелочных металлов
протекает с ацильным расщеплением кольца на стадии
инициирования и с алкильным расщеплением на ста-
стадии роста цепи:
Инициирование
О
кон + сн,—сн2 -
о с=о
нос-сн2сн2ок
—*- кос-сн2сн2он
Рост цепи
носн2сн2соо~ к*
*- НОСН2СН2СООСН2СН2СОО~ К+
Радиационная аполимеризация. Исследована радиа-
радиационная полимеризация лишь E-пропиолактона. Про-
Процесс протекает как в твердой фазе, так и в р-ре при
действии облучения мощностью дозы 0,07—70 ма/кг
A03—10е р/ч). Выход полимера не превышает 15—20%.
Мол. масса полимера, образующегося при твердофаз-
твердофазной полимеризации, возрастает с увеличением раз-
размеров кристаллов мономера. При —78 °С образуется
кристаллич. полимер даже при облучении мелких кри-
кристаллов, а при —196 °С — аморфный полимер. Пред-
Предполагается, что акт инициирования происходит на сты-
стыке кристаллов и в местах дефектов кристаллической
решетки мономера. Выход и мол. масса полимера воз-
возрастают с увеличением мощности дозы излучения. По-
После прекращения облучения образца наблюдается
пост-полимеризация. При действии у-облучения пред-
предполагается, что процесс протекает по анионному ме-
механизму.
Полимеризация м. б. инициирована также и потоком
электронов.
Под действием у-лучей E-пропиолактон сополимери-
зуется с акрилонитрилом, метилметакрилатом, сти-
стиролом, дикетеном, E-бутиролактоном, а,а-бггс-(хлор-
метил)-Р-пропиолактоном.
Сополимеризация. Л. способны вступать в сополи-
меризацию с большим кругом мономеров (см. Лаптопов
полимеры).
Реакционная способность Л. при катионной сополи-
меризации изменяется симбатно с их основностью
(таблица).
Значения основности лактонов*
Лактон
б-Валеролактон
е-Капролактон
Y-Бутиролактон
3, З-Диметил-3-пропиолактон . . .
З-Метил-р-пропиолактон
З-Пропиолактон
а, а-бис-(Хлорметил)-|3-пропиолак-
тон
Q-D,
1 (ж
10100A01)
8000 (80)
7200 G2)
4100 D1)
3700 C7)
3200 C2)
5,10
5,31
6,12
9,53
9,59
10,06
12,05
* Основность оценивалась по сдвигу полосы О—D валентных
колебании молекулы дейтерометанола в присутствии исследуе-
исследуемого Л. Значение рК рассчитывалось по ур-нию Горди.
Р-Пропиолактон значительно менее реакционноспо-
собен, чем тетрагидрофуран и 3,3-бмс-(хлорметил)-
оксациклобутан, что связано с его низкой основностью.
Отметим, однако, что в случае сополимеризации E-про-
пиолактона с 3,3-6 ггс-(хлорметил)оксациклобутаном
образующийся сополимер имеет блочное строение,
что не соответствует полученным значениям констант
сополимеризации.
Л. сополимеризуются под действием катионных
катализаторов с виниловыми мономерами, однако
и в этом случае наблюдается нестатистический ха-
характер распределения сомономеров в цепи сополимера,
причем большое влияние на состав сополимера
оказывает природа растворителя и концентрация ка-
катализатора.
Активность Л. в катионной сополимеризации воз-
возрастает с увеличением числа алкильных заместите-
заместителей у Р-углеродного атома и понижается при увели-
увеличении числа алкильных заместителей у а-углеродного
атома.
Л. легко сополимеризуются с виниловыми мономе-
мономерами и под действием анионных катализаторов. Блоксо-
полимеры Л. с виниловыми и диеновыми мономерами
м. б. получены при инициировании полимеризации
живущими полимерами.
Лит.: С а з а н о в Ю. Н., Усп. хим., 37, в. 6, 1084 A968);
Джавадян Э. А., Розенберг Б. А., Е н и к о л о-
пян Н. С, Высокомол. соед., 15А, 1317 A973); Хомя-
Хомяков А. К., ЛюдвигЕ. Б, ДАН СССР, 201, № 4, 877
A971)- Ефремова А. И. [и др.1, Изв. АН СССР, сер. хим.,
№ 4, 807 A969); Yamashita V. [а. о.], Makromol. Chem.,
Hi, 139 A968); Ф р у н з е Т. М., Курашев В. В., Усп.
хим., 37, в. 9, 1600 A968). Б. А. Розенберг.
ЛАКТОНОВ ПОЛИМЕРЫ (polylactones, Polylak-
tone, polylactones).
Лактоны (Л.) — внутренние циклич. эфиры окси-
кислот. В зависимости от типа оксикислот, образующих
Л., различают р-, у-, б- и е-Л. Положение заместителей
у углеродных атомов лактонного кольца принято
37
ЛАКТОНОВ ПОЛИМЕРЫ
38
определять относительно карбонильного углерода.
В качестве мономеров используют, как правило, Л.
алифатич. ряда.
Свойства. Л. — бесцветные жидкости с резким
специфическим запахом (см. табл.); низшие Л. (напр.,
Р-пропиолактон) — лакриматоры.
Физические свойства наиболее распространенных лактонов
A мм рт. ст. = 133 , 322 н/м*)
Лактон
(З-Пропиолактон
СН2-СН2
1 1
Y-Бутиролактон
СН2- (СН2J
1 1
О С = О
6-Валеролактон
СН2- (СН2K
I I
ПС П
е-Капролактон
СН2- (СН2L
1 |
1 1
Темп-ра
плавле-
плавления, °С
-33,4
__
12,5
Темп-ра
кипения,
°С (мм
рт. ст.)
155G60)
51 A0)
203-204
G60)
89 A2)
218—220
G60)
88D)
98-99B)
Плотность
при 20 °С,
г/см3
1,1460
1,1441
@ °С)
1,0794
1 ,0693
Показа-,
тель пре-
преломления
1,4131
B0 °С)
_
1,4503
B0 °С)
1,4611
B2 °С)
Л. обладают многими свойствами сложных эфиров,
но, как правило, реакционноспособнее последних.
Реакции Л. сопровождаются раскрытием цикла. При
нагревании в воде, а также в р-рах к-т или щелочей
Л. гидролизуются в соответствующие оксикислоты;
со спиртами образуют эфиры оксикислот, с аминами —
амиды, с гидразинами — гидразиды оксикислот, т. е.
во всех этих реакциях Л. служат ацилирующими аген-
агентами. С ароматич. углеводородами в присутствии к-т
Льюиса Л. взаимодействуют как алкилирующие агенты:
СН2-(СН2)П
АгН -Н | п -> Аг(СН2)пСООН
Р-Л. могут реагировать с разрывом как ацил-кисло-
родной, так и алкил-кислородной связи с образованием
соответственно производных оксикислот или замещен-
замещенных к-т (см. Лактонов полимеризация).
При умеренном нагревании Р-Л. образуются олефины:
R2C-CR'2
I | -> R2C=CR2+CO2
о-с=о
Высшие Л. при нагревании изомеризуются в непре-
непредельные к-ты,
Л. сополимеризуются с виниловыми мономерами,
циклич. простыми эфирами, циклич. ацеталями, цик-
лич. аминами и амидами, N-карбоксиангидридами
аминокислот, дикетеном, изоцианатами, циклич. фос-
фосфитами, а также друг с другом, причем в этом случае
в качестве одного из сомономеров м. б. использован
Л., к-рый не способен к гомополимеризации (напр.,
7-бутиролактон).
Получение. В пром-сти и в лаборатории р-
пропиолактон и его моно- и дизамещенные в Р-положе-
нии получают обычно при взаимодействии кетена с со-
соответствующими карбонильными соединениями (альде-
(альдегидами или кетонами) в присутствии к-т Льюиса:
R2C-CH2
R2C=O+CH2=G=O -> | |
o-c=o
Газообразный кетен пропускают через р-р, содер-
содержащий карбонильное соединение и катализатор, при
0—20 °С. В качестве растворителя используют простые
эфиры, кетоны, галогеналкилы, однако реакцию пред-
предпочтительнее проводить в р-ре самого Л. Из реакцион-
реакционной смеси Л. выделяют перегонкой в вакууме.
а-Задоещенные Р-Л. получают из р-галогензамещен-
ных к-т (предпочтительнее использовать их соли, напр,
свинцовые):
R'
СН2-С
C1CH2-C(R\R")COOH—> I |\
-на I R"
о—с=о
Высшие Л. обычно синтезируют, окисляя соответст-
соответствующие циклич. кетоны надкислотами:
О
СН2-(СН2) +RC
\ / \
с=о оон
СН2-(СН2)„ + RGOOH
I I n
о с=о
Кроме того, все Л. могут быть получены циклизацией
галогено- и оксикислот и нагреванием серебряных
солей дикарбоновых к-т с галогенами.
Следы влаги из Л. удаляют путем кипячения с не-
небольшими количествами A—3%) изоцианатов (р-про-
пиолактон можно кипятить также с уксусным ангид-
ангидридом и бутанолом). Далее Л. перегоняют в вакууме.
Полилактоны (П.) — сложные полиэфиры общей
ф-лы:
[-OCHR"(CRR')nCO-]m
где R, R', R"= H, алкил или др. радикал.
Свойства. В зависимости от молекулярной массы
A-Ю3 — 5-105) П. —вязкие жидкости или твердые
бесцветные вещества. Наиболее полно изучены поли-
Р-Л., являющиеся полимерами высокой степени кри-
кристалличности.
Поли-р-пропиолактон — кристаллич. продукт белого
цвета; плотность при 20 °С 1,2—1,4 г/см3 (в зависимо-
зависимости от мол. массы). Он может существовать в двух
кристаллич. модификациях: а- и Р-формы с темп-рами
кристаллизации 65 и 20 °С соответственно. Строение
а-формы (период идентичности 7,02 А) 21-спираль,
Р-формы (период идентичности 4,28 А) — плоский зиг-
зигзаг. При медленном нагревании до 65 °С Р-форма
может переходить в а-форму. Р-Форма образуется при
получении пленки из р-ра путем медленного испарения
растворителя (хлороформа); при быстром испарении
преобладает а-форма.
Поли-Р-пропиолактон растворим в хлороформе и
муравьиной к-те, нерастворим в бензоле, ацетоне,
метаноле, гексане и воде. Поли-(р, Р-диметил)- и поли-
(а,а-диметил)-р-пропиолактоны растворяются в фе-
феноле, крезоле, трихлоруксусной кислоте лишь при на-
нагревании, а поли-(а,а-дихлорметил)-Р-пропиолактон
растворим лишь в дифенилоксиде и диметилфталате
при темп-pax кипения растворителей. С увеличением
мол. массы растворимость П. сильно уменьшается.
Полимеры высших Л. растворимы в ароматич. и хлори-
хлорированных алифатич. углеводородах.
Химич. свойства П. определяются наличием в основ-
основной цепи полимера сложноэфирной группы (см. Поли-
Полиэфиры сложные). П. характеризуются весьма высокой
термич. стабильностью. Так, поли-Р-пропиолактон
в инертной атмосфере разлагается при 230—250 °С,
поли-(а,а-дихлорметил)-р-пропиолактон — при 270—
280 °С. В присутствии кислорода скорость деструкции
значительно возрастает.
Получение. П. получают полимеризацией Л. в
присутствии большинства известных катализаторов
катионного и анионного типа. Температура катионной
полимеризации Л. (обычно под действием к-т Льюиса,
карбониевых и оксониевых солей, протонных к-т)
близка к комнатной. Полимеризация Л. под действием
анионных катализаторов (щелочных и щелочноземель-
щелочноземельных металлов, их окисей, гидроокисей, солей, а также
аминов, бетаинов и др.) протекает при 50—60 °С. Поли-
Р-пропиолактон м. б. получен также радиационной
полимеризацией Р-Л. при низких темп-pax (от —80
39
ЛАТЕКС НАТУРАЛЬНЫЙ
40
до —196 °С) или под действием инициаторов радикаль-
радикального типа. Подробно см. Лаптопов полимеризация.
Применение и переработка. В пром-сти
П. пока не применяют. Однако их можно перерабаты-
перерабатывать в волокна и пенопласты, использовать в качестве
пластификаторов, а также модификаторов, повы-
повышающих устойчивость полимеров к гидролизу, све-
то- и маслостойкость.
Изделия из П. можно изготавливать всеми извест-
известными способами переработки термопластов.
Впервые полимеризацию Л. осуществили Фихтер
и Байсвенгер в 1903.
Лит.: EtienneY., FischerN., в кн.: Heterocyclic
compounds, pt 2, N. Y.— [а. о.], 1964; Chemistry of carbon
compounds, ed. E. H. Rodd, v. 1, pt B, Amst.— [a. o.], 1952;
Сазанов Ю. Н., Усп. химии, 37, в. 6, 1084 A968).
Б. А. Розенберг.
ЛАТЕКС НАТУРАЛЬНЫЙ (natural latex, Natur-
latex, latex naturel) — млечный сок тропич. каучуко-
каучуконосных растений, представляющий собой водную дис-
дисперсию натурального каучука. Промышленный источ-
источник получения Л. н.— бразильская гевея.
Дисперсная фаза Л. н. состоит из частиц (глобул)
шарообразной или грушевидной формы размером
от 0,25 до 5 мкм; средний размер — ~ 2 мкм. Глобулы
Л. н. состоят из плотной эластичной каучуковой обо-
оболочки (гель-каучук), внутри к-рой находится жидкая
низкомолекулярная фракция (золь-каучук). Наруж-
Наружная поверхность каучуковой оболочки окружена защит-
защитным слоем, состоящим из белковых веществ (в свеже-
свежесобранном Л. н. их содержится ~ 4%), смол, мыл и
гидратно-связанной воды. По мере старения Л. н. бел-
белки постепенно гидролизуются в аминокислоты; по со-
содержанию последних можно судить о возрасте латекса.
Адсорбированная на поверхности глобул белковая
оболочка сообщает им отрицательный заряд, к-рый
вместе с гидратно-связанным слоем воды обеспечивает
агрегативную устойчивость Л. н. как коллоидной сис-
системы. Электрокинетич. потенциал глобул конц. Л. н.
составляет 100 мв. Нейтрализация заряда частиц или
разрушение защитной оболочки под воздействием
микроорганизмов, химич. или физич. факторов при-
приводит к коагуляции Л. н.
Л. н. извлекают путем надреза (т. наз. под-
подсочки) наружного слоя коры дерева по диагонали —
€лева направо. Выделяющийся латекс стекает к ниж-
нижнему концу надреза, а оттуда — в подставленный со-
сосуд. Для предотвращения самопроизвольной коагу-
коагуляции в Л. н. сразу после его получения вводят кон-
консервирующие агенты: аммиак @,5—0,7%), пентахлор-
фенолят натрия, едкий натр, буру и др.
Содержание сухого вещества в Л. н. составляет
37—41%; для последующей переработки Л. н. подвер-
подвергают концентрированию методами сливкоот-
деления (отстаивания), упаривания, центрифугирова-
центрифугирования. Наиболее широко применяют последний метод.
При сливкоотделении Л. н. сначала тщательно пере-
перемешивают с альгинатом натрия или аммония, поливи-
поливиниловым спиртом, карбоксиметилцеллюлозой или др.
агентом сливкоотделения (~ 0,3% от массы водной
фазы латекса, т. наз. серума), а затем заливают в спе-
специальные емкости. Под влиянием агентов сливкоотде-
сливкоотделения образуются крупные агрегаты частиц Л. н.,
всплывающие вследствие различной плотности каучука
и серума, составляющей 0,91 и 1,02 г/см3 соответствен-
соответственно. После отстаивания в течение 4—5 сут сливают
отделившийся серум, а затем концентрат Л. н.
Упаривание осуществляют нагреванием Л. н.
(~70 °С) при одновременном перемешивании. Для уско-
ускорения удаления воды процесс иногда проводят под
вакуумом или в токе горячего воздуха. Аппараты для
упаривания снабжают приспособлениями, предотвра-
предотвращающими возникновение пены и обеспечивающими
удаление пленки коагулюма, образующейся на поверх-
поверхности Л. н. В латекс перед упариванием вводят консер-
консервирующие агенты (нелетучую щелочь, обычно КОН),
защитный коллоид (казеинат калия), калийное мыло.
Концентрирование проводят также в центрифугах
с частотой вращения ~ 8000 об/мин. При этом частицы
каучука, обладающие меньшей плотностью, постепенно
переходят в слой, прилегающий к оси вращения. Кон-
Концентрат Л. н. и серум сливают через отверстия, рас-
расположенные в днище центрифуги на разном расстоянии
от оси.
• Свойства концентрированного Л. н. меняются в
довольно широких пределах в зависимости от способа
концентрирования (таблица).
Свойства натурального латекса
Показатели
Содержание, %
сухого вещества . . .
каучука
щелочи (в пересчете
на аммиак)
Вязкость по Брукфилду,
мн-сек/м2, или спз
Поверхностное натяже-
натяжение, мн/м, или
дин/см
Показатель рН
До кон-
центри-
центрирования
37-41
35-38
0,8-1,0
4-6
33-36
11-13
После концентрирования
сливко-
отделе-
нием
62-65
58—63
0,6-0,8
30-60
33-40
10 — 11
упари-
упариванием
72-75
65 — 69
9500
33 — 35
10-11
центрифу-
гирова-
гированием
61 — 62
60-61
0,5-0,7
30-50
35-42
10,2-10,8
Основную массу Л. н. применяют для полу-
получения натурального каучука, ок. 7% — для получения
латексных изделий (перчаток, метеорологич. оболочек,
различных медицинских изделий, резиновых нитей,
губчатых резин), а также латексных клеев. О свойствах
вулканизатов Л. н. см. Каучук натуральный.
Иногда изделия изготовляют из т. наз. в у л ь т е к-
с а — Л. н., в к-ром каучук свулканизован на стадии
латекса путем термич. обработки @,5—2 ч при 70 °С)
в присутствии серы, ультраускорителя вулканизации
(напр., дибутилдитиокарбамата цинка) и ZnO. При
использовании вультекса на заводах-изготовителях
упрощается технология производства изделий, т. к.
исключаются стадии изготовления латексных смесей
и вулканизации.
Нек-рые зарубежные торговые марки Л. н.: концен-
концентрированного сливкоотделением — MAP-DM-1135X,
упариванием — ревертекс стандарт и
ревертекс Т, центрифугированием — д а н л о п
G-60, гектолекс, квалитекс; наиболее рас-
распространенная марка вулканизованного Л. н.— р е-
вультекс.
Лит.: Нобль Р. Д ж., Латекс в технике, пер. с англ.,
Л., 1962; В 1 а с к 1 е у D. С h., High polymer latices, v. 1—2,
L._ N. Y., 1966; MadgeE.W., Latex foam rubber, L., 1962;
Aupetit A., Boucher M., Rev. gener. caout. et plast.,
45, № 9, 991 A968).
Д. П. Трофимович, В. В. Черная, М. И. Шепелев.
ЛАТЕКСНЫЕ ИЗДЕЛИЯ (latex articles, Latexar-
tikel, articles en latex). Широкий ассортимент Л. и.
(метеорологич. радиозондовые оболочки, перчатки раз-
различных типов, резиновые нити, изделия из губчатых
резин и др.) обусловлен простотой технологии их про-
производства и возможностью легкой механизации и авто-
автоматизации всех стадий процесса. Применение латексов
позволяет получать резиновые изделия, к-рые не могут
быть изготовлены из твердых каучуков, напр, тонко-
тонкостенные бесшовные. Кроме того, замена р-ров каучука
(резиновых клеев) латексом при изготовлении нек-рых
изделий исключает необходимость применения токсич-
токсичных и пожароопасных органич. растворителей.
Технология производства латексных изделий
Технологич. схема производства большинства Л. и.
включает след. основные стадии: 1) приготовление
41
ЛАТЕКСНЫЕ ИЗДЕЛИЯ
42
латексной смеси, 2) получение полуфабриката (геля),
3) уплотнение геля, 4) сушку изделия, 5) его вулкани-
вулканизацию.
Приготовление латексной смеси. Основная задача
при составлении латексных смесей — правильный
выбор типа латекса. Так, Л. и. с высокой
прочностью при растяжении получают при использо-
использовании натурального или синтетич. карбоксилатного
латекса. При изготовлении Л. и., стойких к действию
масел и растворителей, применяют бутадиен-нитриль-
ные или хлоропреновые латексы. Низкая газопрони-
газопроницаемость и высокая озоностойкость м. б. достигнуты
при использовании хлоропренового латекса или ис-
искусственного латекса бутил каучука.
Способы переработки латекса существенно зависят
от его коллоидно-химич. свойств, определяемых при-
природой и содержанием эмульгатора, степенью насыщен-
насыщенности поверхности глобул эмульгатором, размером
глобул, вязкостью, концентрацией, стойкостью к дейст-
действию высоких и низких темп-р и др. Латексы, стабилизи-
стабилизированные анионоактивными эмульгаторами, позволяют
получать Л. и. методами желатинирования, коагулянт-
ного макания и ионного отложения (см. ниже). Эти
методы неприменимы для латексов с неионогенными
эмульгаторами, обладающих высокой агрегативной
стабильностью. Высокое содержание эмульгатора, обес-
обеспечивающее полное насыщение поверхности глобул
латекса, позволяет вводить в смесь значительное коли-
количество наполнителей, но обусловливает низкие меха-
нич. свойства и невысокую водостойкость Л. и. Кроме
того, высокая (более 60%) степень насыщенности
поверхности глобул эмульгатором может отрицательно
сказываться на получении Л. и. указанными выше
методами. О свойствах латексов см. Латекс натураль-
натуральный, Латексы синтетические.
В состав латексных смесей, кроме обычных ингре-
ингредиентов резиновых смесей (вулканизующих агентов,
ускорителей вулканизации, активаторов вулканизации,
антиоксид анто в, наполнителей, пластификаторов
и др.), вводят поверхностно-активные вещества, так
наз. астабилизирующие добавки, способствующие сни-
снижению устойчивости коллоидной системы, загустители,
пеногасители, антисептики и др.
Для вулканизации смесей используют те же системы,
что и для изделий из твердых каучуков. Карбоксилат-
ные латексы вулканизуют с использованием солей Zn,
Са и Mg, являющихся одновременно астабилизирую-
щими агентами при ионном отложении.
Ускорителями вулканизации слу-
служат гл. обр. ультраускорители (дитиокарбаматы, тиу-
рамы), т. к. при изготовлении смесей из латексов
исключается опасность под вулканизации. Широко
используют также тиазолы и гуанидины. Ускорители
вулканизации, помимо своего основного назначения,
могут оказывать существенное влияние на коллоидно-
химич. и технологич. свойства смесей. Напр., в при-
присутствии цинковой соли меркаптобензтиазола повы-
повышается вязкость смесей из натурального латекса.
Введение активатора вулканизации
ZnO в латексы, содержащие аммиак, приводит к их
медленной астабилизации. В этом случае устойчивость
смеси повышают, удаляя предварительно аммиак из ла-
латексов или вводя в смесь дополнительный стабилизатор
коллоидной системы.
В латексные смеси вводят те же пластифика-
пластификаторы, а н т и о к с и д а н т ы и антиозо-
н а н т ы, что и в смеси на основе твердых каучуков.
Порошкообразные наполнители (каолин, мел, лито-
литопон, высокодисперсная SiO2, сажа) обычно не способст-
способствуют эффективному повышению механич. свойств
изделия. Введение этих ингредиентов позволяет сни-
снизить стоимость Л. и., повысить их жесткость, изменить
окраску. Для улучшения механич. свойств Л. и. при~
меняют резорцино-формальдегидные, феноло-формаль-
дегидные, мочевино-формальдегидные смолы, латексы
поливинилхлорида и полистирола, щелочной сульфат-
сульфатный лигнин. Действие смол наиболее эффективно в том
случае, когда их синтез происходит непосредственно
в латексе (в процессе гелеобразования или вулкани-
вулканизации) или при введении смолы в стадии резола в про-
процессе получения латекса. В последнем случае даль-
дальнейшая поликонденсация протекает одновременно с по-
полимеризацией мономера.
Важное значение в рецептуре латексных смесей
имеют поверхностно-активные веще-
вещества: 1) анионные (мыла олеиновой к-ты, синтетич.
жирных к-т и к-т канифоли, натриевая соль продук-
продукта конденсации Р-нафталинсульфокислоты с формаль-
формальдегидом — диспергатор НФ, казеин, карбоксиметил-
карбоксиметилцеллюлоза и др.), 2) неионогенные (продукты кон-
конденсации моноалкилфенолов с окисью этилена —
продукт ОП-7 или олеиновой к-ты с окисью этилена —
эмульфор А и др.) и 3) катионные (амины, соли чет-
четвертичного аммония и др.). Эти добавки служат сма-
смачивающими и диспергирующими агентами, стабилиза-
стабилизаторами латексных смесей, латексной пены, суспензий
и эмульсий ингредиентов п т. д.
В случае необходимости повышения вязкости латек-
латексов применяют так наз. загустители, к-рые
образуют вязкие водные р-ры или способствуют созда-
созданию тиксотропных структур. Загустителями служат
синтетич. полимеры (соли полиакриловой к-ты и ще-
щелочных металлов, полиакриламид, поливиниловый
спирт и др.), природные высокомолекулярные вещества
и их производные (карбоксиметилцеллюлоза, крахмал,
казеин и др.).
Для предотвращения образования пены при пе-
перемешивании латекса и латексных смесей применя-
применяют пеногасители — эмульсии кремнийорганиче-
ских жидкостей, высшие спирты С8—С12, минеральные
масла и др. При наличии в смесях ингредиентов, к-рые
могут разрушаться под влиянием бактерий (казеин,
костный клей, крахмал и др.), применяют антисеп-
антисептики — соединения фенил ртути, бутил олова, фтор-
силикат натрия, хлорированные фенолы.
Все ингредиенты вводят в латекс в виде водных
р-ров, суспензий или эмульсий. При этом размер
частиц суспензий или эмульсий должен быть близок
к размеру глобул латекса, а применяемый для их ста-
стабилизации диспергатор должен мало отличаться по
своей поверхностной активности от поверхностно-ак-
поверхностно-активного вещества в самом латексе; рН эмульсий пли
дисперсий, вводимых в латекс, должен быть равен
рН латекса.
Дисперсии твердых ингредиентов получают их из-
измельчением в водном р-ре диспергатора, гл. обр.
в шаровых мельницах, а также в вибро- и коллоидных
мельницах, мельницах «аттритор». В шаровых мельни-
мельницах получают дисперсии со средним размером частиц
0,4—10,0 мкм (продолжительность обработки 24—72 ч
при частоте вращения корпуса мельницы 20—50
об/мин). Поскольку разные вещества обладают раз-
различной способностью к диспергированию, дисперсии
каждого ингредиента чаще готовят отдельно, однако
в нек-рых случаях эти процессы можно совместить.
Типичные составы дисперсий нек-рых ингредиентов
(в мае. ч.): 1) сера — 50, диспергатор НФ — 5, бенто-
бентонит — 1, вода — 44; 2) сажа ДГ-100 — 50, дисперга-
диспергатор НФ — 5, вода — 95.
Водные эмульсии жидких ингредиентов готовят
в быстроходных мешалках (частота вращения 300—
1000 об/мин), коллоидных мельницах, дисковых эмуль-
сификаторах. Продолжительность процесса составляет
обычно 0,5—2 ч. Стабилизаторы эмульсий предвари-
предварительно растворяют в воде или синтезируют при эмуль-
эмульгировании. В последнем случае одно из исходных со-
43
ЛАТЕКСНЫЕ ИЗДЕЛИЯ
44
единений для синтеза (напр., жирную к-ту) предвари-
предварительно смешивают с эмульгируемым ингредиентом,
а второе (напр., щелочь) — с водной фазой. Типичный
состав эмульсии жидкого ингредиента (в мае. ч.): ди-
бутилсебацинат — 50, олеат триэтаноламина — 4,
вода — 46.
При изготовлении латексных смесей целесообразно
использовать свежеприготовленные дисперсии и эмуль-
эмульсии ингредиентов. В случае хранения их следует
непрерывно перемешивать (частота вращения мешалок
30—40 об/мин), не допуская вспенивания. Важное зна-
значение при изготовлении латексной смеси имеет порядок
введения ингредиентов. Обычно в латекс сначала вводят
р-ры стабилизаторов коллоидной системы, а затем дис-
дисперсии вулканизующих агентов, ускорителей вулка-
вулканизации, антиоксидантов, наполнителей, эмульсии
пластификаторов и, наконец, дисперсию ZnO.
Для смешения латекса с дисперсиями, эмульсиями,
р-рами ингредиентов используют емкости, снабженные
рубашками для охлаждения или нагрева и мешалками
с переменной частотой вращения. Внутренняя поверх-
поверхность емкости должна быть эмалированной или иметь
антикоррозионное покрытие. Р-ры, дисперсии и эмуль-
эмульсии ингредиентов вводят в латекс при непрерывном
перемешивании. Частота вращения мешалки в пределах
30—40 об/мин позволяет исключить пенообразование,
затрудняющее равномерное смешение. Продолжитель-
Продолжительность изготовления латексной смеси составляет 30—
60 мин.
В ряде случаев после введения всех необходимых
ингредиентов смесь «вызревает» 6—24 ч при 20—60 °С
и медленном перемешивании. При этом изменяются
коллоидно-химич. характеристики смеси (падает рН,
возрастает вязкость и др.), повышается ее однородность
и улучшаются технологич. свойства. В ходе вызрева-
вызревания (особенно при повышенной темп-ре) полимер
в латексе частично вулканизуется.
Получение полуфабрикатов. Большинство способов
получения Л. и.— ионное отложение, коагулянтное
макание, термосенсибилизация, электроотложение,
желатинирование, многократное макание — основано
на выделении (коагуляции) полимера в результате
астабилизации коллоидной системы (напр., под дейст-
действием электролитов) или удаления влаги из латексной
смеси (высушивания). Л. и., к-рые получают погру-
погружением формы в латексную смесь, наз. макаными.
При ионном отложении форму, моделирую-
моделирующую изделие, погружают сначала в т. наз. фиксатор,
представляющий собой водный р-р электролита [СаС12,
Ga(NO3J], загущенный каолином или «белой сажей»,
а затем сразу же в латексную смесь. После образова-
образования слоя геля необходимой толщины форму с гелем
извлекают из смеси. Способ отличается высокой про-
производительностью (при одно- или двукратном погру-
погружении формы в латексную смесь можно получать Л. и.
толщиной до 2 мм) и широко используется в пром-сти.
Разновидность ионного отложения — т. наз. коа-
коагулянтное макание; в этом случае фикса-
фиксатором служит незагущенный р-р электролита в лету-
летучем растворителе (спирте, ацетоне). Форму после
подсушивания на ней фиксатора (обычно 1—3 мин
при комнатной темп-ре) погружают в латексную смесь
и наращивают гель определенной толщины.
В способе, основанном на т. наз. термосенси-
термосенсибилизации, используют латексные смеси, к-рые
содержат агенты [поливинилметиловый эфир (лутанол
М-40, игевин М-50), меркаптобензимидазолят натрия
и др.], вызывающие астабилизацию латекса при погру-
погружении в него формы, нагретой до 60 —100 °С. Содер-
Содержание термосенсибилизирующего агента в смеси со-
составляет обычно 1—3 мае. ч. (здесь и далее — в расчете
на 100 мае. ч. каучука в латексе). Для обеспечения
стабильности латексных смесей при комнатной темп-ре
в них вводят казеин или др. стабилизаторы. Скорость
отложения геля при термосенсибилизации выше, чем
при ионном отложении. Напр., в случае получения
Л. и. с толщиной стенки 0,8 мм из вулканизованного
натурального латекса «ревультекс» (см. Латекс нату-
натуральный) продолжительность образования геля при
ионном отложении составляет 9 мин, при термосенси-
термосенсибилизации — 40 сек. Способ термосенсибилизации при-
применяют гл. обр. для изготовления различных изделий
медицинского назначения.
При желатинировании смесь заливают
в форму, конфигурация к-рой определяет форму гото-
готового изделия. Смесь содержит желатинирующие агенты
(чаще всего 2—10 мае. ч. ZnO в смеси с 1—5 мае. ч.
NH4C1 и 1—4 мае. ч. NH4OH или 1—3 мае. ч. Na2SiF6),
под действием к-рых гель образуется при комнатной
темп-ре за 1—5 мин. Этим способом можно изготовлять
Л. и., в том числе пустотелые, любых размеров и с раз-
различной толщиной стенки, а также изделия сложной
конфигурации [напр., рабочие органы («пальцы») чае-
чаесборочных машин, технич. толстостенные перчатки
с рифами на ладонной части, нек-рые виды резиновой
обуви]. Способ лежит в основе получения губчатых
резин из латекса.
При электроотложении глобулы латек-
латекса, имеющие отрицательный заряд, оседают в электрич.
поле на аноде, образуя слой геля. Достоинство спо-
способа — возможность быстрого получения прочного
геля сравнительно большой толщины при небольшом
расходе энергии. Так, при плотности электрич. тока,
равной 400 а/м2, можно за 1 мин получить слой тол-
толщиной 1,4 мм. Недостатки способа — необходимость
предотвращать газовыделение на аноде, обусловлен-
обусловленное электролизом солей серума, т. к. в противном
случае м. б. получены пористые пленки, а также
трудность получения разнотолщинных пленок. Способ
электроотложения не нашел широкого применения.
Наиболее целесообразная область его использования —
нанесение покрытий на металлич. детали.
При многократном макании форму,
моделирующую изделие, многократно C—5 раз в за-
зависимости от толщины стенки изделия) погружают
в латексную смесь. Слой смеси, остающийся на форме
после каждого ее погружения, сушат 5 — 10 мин при
комнатной темп-ре. Способ малопроизводителен и на-
находит применение только при получении Л. и. с тол-
толщиной стенки до 0,2 мм.
Уплотнение геля. При получении многих Л. и. гель
подвергают уплотнению, при к-ром происходит медлен-
медленное самопроизвольное сближение глобул (см. Синере-
зис). Процесс обычно проводят, выдерживая форму
с гелем в воде в течение 1—4 ч при 25—30 °С. Его ско-
скорость возрастает при повышении темп-ры, уменьшении
содержания в латексе поверхностно-активных веществ,
улучшении аутогезионных свойств полимера. В ре-
результате уплотнения геля повышаются его механич.
свойства (модуль и прочность при растяжении, отно-
относительное удлинение), что необходимо для проведения
дальнейших операций при изготовлении Л. и., и, кроме
того, ускоряется сушка изделий.
Сушка. Обычно Л. и. сушат 2—15 ч в сушильных
камерах в токе горячего воздуха при постепенном повы-
повышении темп-ры от 40 до 70—80 °С. Один из путей сок-
сокращения продолжительности сушки — применение
комбинированного нагрева токами промышленной ча-
частоты и ИК-лучами. Необходимость резкого увеличе-
увеличения продолжительности сушки при увеличении толщи-
толщины слоя геля препятствует получению Л. и. с толщи-
толщиной стенки более 2—3 мм. Быстрая сушка приводит
к удалению влаги только с поверхности геля, что
может вызывать его растрескивание и преждевременную
вулканизацию поверхностной пленки, препятствующую
дальнейшей диффузии влаги из геля к его поверхности.
45
ЛАТЕКСНЫЕ КЛЕИ
46
Вулканизацию проводят, как правило, в камерах
в среде горячего воздуха при 100—140 °С. Л. и. вул-
вулканизуют на формах или в свободном состоянии; в пос-
последнем случае их помещают в камеры на противнях,
заполненных тальком.
Виды латексных изделий и их свойства
Метеорологические радиозонд о-
вые и шаропилотные оболочки изго-
изготовляют из хлоропренового латекса, в к-рый вводят
25 мае. ч. дибутилсебацината. Гель латексной смеси,
полученный ионным отложением на ребристых формах,
подвергают уплотнению в воде, снимают с формы,
раздувают в 2—4 раза сжатым воздухом и сушат в раз-
раздутом состоянии 1,5—2 ч при постепенном повышении
темп-ры от 30 до 45 °С. Затем воздух из внутренней
части оболочки эвакуируют, опудривают оболочку
тальком и вулканизуют при 120 °С в сложенном со-
состоянии. Операции, связанные с получением геля,
автоматизированы. Раздувку, сушку и вакуумирова-
ние оболочки осуществляют также автоматически
на пульсирующем конвейере; при этом контролируют
напряжение, возникающее при раздувке, т. к. его
постоянство обеспечивает получение оболочек со ста-
стабильными свойствами (табл. 1).
Таблица
Марка
оболочки
100
150
200
1. Основные показатели
оболочек, выпускаемых
Началь-
Начальный диа-
диаметр, см
100
150
200
Масса, г
400
800-950
1500-1600
метеорологичес ких
в СССР
Фактор
качества*
0,75
0,70
0,65
Средняя
высота
подъема,
км
18
28
30
* Отношение удлинения при разрыве, полученного при
испытании готового изделия, к среднему удлинению при
разрыве образцов, вырезанных из оболочек той же партии.
Резиновые перчатки изготовляют из на-
натурального центрифугированного (хирургич. и ди-
электрич. перчатки), хлоропренового (анатомич. и тех-
нич. перчатки) и бутадиен-нитрильного (бензо- и масло-
стойкие перчатки) латексов. Состав латексной смеси
для получения хирургич. перчаток (в мае. ч.): каучук
натурального латекса («квалитекс») — 100; сера —
0,25; диэтилдитиокарбамат цинка — 1; тетраметилтиу-
рамдисульфид — 2; ZnO — 2; фенил-Р-нафти л амин —
0,5; КОН - 0,5.
Технология изготовления перчаток различного типа
(табл. 2) включает след. основные операции: получе-
получение геля на формах ионным отложением, сушку геля,
вулканизацию на форме, съем с формы, промывку
и сушку изделия. При изготовлении диэлектрич. пер-
перчаток гель перед сушкой отмывают от электролитов
для повышения электрич. прочности изделия. В тех
случаях, когда перчатки получают на трикотажной
основе, ионное отложение проводят на надетой на фор-
форму трикотажной перчатке, предварительно обработан-
обработанной фиксатором.
Легкую резиновую обувь изготовляют
из натурального и нек-рых синтетич. (напр., хлоро-
хлоропренового) латексов. Гель получают ионным от-
отложением на формах; для увеличения толщины подош-
подошвы после наращивания слоя геля определенной толщи-
толщины форму поднимают таким образом, чтобы в латексную
смесь была погружена только ее нижняя часть. Если
ионное отложение проводить на гравированных формах,
образуется изделие с рельефным рисунком. Гель после
подсушивания до содержания в нем 8—12% влаги
снимают с гравированной формы, выворачивают, наде-
надевают на другую (гладкую) форму, нагретую до 40—
50 °С, и вулканизуют.
Таблица 2. Основные показатели резиновых перчаток,
выпускаемых в СССР
Тип перчаток
Хирургиче-
Хирургические ....
Масло- и бен-
зостойкие .
Диэлектри-
Диэлектрические . . .
Толщина
стенки,
мм
0,2
0,7
0,7
Проч-
Прочность
при рас-
растяжении,
Мн/м2
(кгс/см2)
30C00)
10 A00)
15 A50)
Относи-
Относительное
удлине-
удлинение, %
840
700
700
Набуха-
Набухание в
смеси
бензина
с бензо-
бензолом
C:1), %
30
Сила тока
утечки
после пре-
пребывания
под на-
напряжением
3,5 кв в
течение
1 мин, ма
3,5
Для изготовления резиновых нитей при-
применяют натуральный латекс, концентрированный цент-
центрифугированием или сливкоотделением (см. Латекс
натуральный)у а также искусственный латекс изо-
пренового каучука (см. Латексы синтетические). Со-
Состав латексной смеси (в мае. ч.): каучук натурального
латекса — 100; сера — 1; диэтилдитиокарбамат цин-
цинка — 1,5; ZnO — 3; белый пигмент — 7,5; фенил-Р-наф-
тиламин — 2; светостабилизатор — 2; КОН — 0,6. Ни-
Нити изготовляют на агрегатах непрерывного действия.
Латексную смесь после ее вызревания в течение 20—
24 ч и вакуумирования (эту операцию проводят для
удаления из латексной смеси воздушных пузырей)
выдавливают через фильеры со скоростью 9—12 м/мин
в раствор коагулянта (напр., 25%-ную СН3СООН),
промывают (ванны длиной 7—10 м, темп-pa воды
80 °С), сушат и вулканизуют в вулканизационных ка-
камерах A—4 ч при 125 °С), опудривают, наматывают на
барабаны и довулканизовывают 4—6 ч при 80 °С. Дав-
Давление латексной смеси, поступающей в фильеры, авто-
автоматически поддерживается постоянным; этим обеспе-
обеспечивается получение нитей заданной толщины, к-рая
может изменяться в пределах 0,2—1 мм.
Лит.: Синтез латексов и их применение, Л., 1961; Проблемы
синтеза и переработки латексов, М., 1968; Радиозондовые обо-
оболочки, Л., 1968. См. также лит. при ст. Латекс натуральный,
Латексы синтетические.
Д. П. Трофимович, В. В. Черная, М. И. Шепелев.
ЛАТЕКСНЫЕ КЛЕИ (latex adhesives, Latexkleb-
stoffe, colles au latex) — клеи на основе натурального
или синтетич. латексов. Кроме обычных ингредиентов
латексных смесей — стабилизаторов коллоидной систе-
системы, вулканизующих агентов, ускорителей и активато-
активаторов вулканизации, антиоксидантов и др. (см. Латексные
изделия), в состав Л. к. вводят добавки, способст-
способствующие повышению их клеящей способности: расти-
растительные и минеральные масла, дибутилфталат, кани-
канифоль, животные клеи, казеин, альбумин крови, сили-
силикат натрия, крахмал и др. Для наиболее полного взаи-
взаимодействия полимера с этими добавками в латексную
смесь добавляют вещества (напр., этиловый спирт),
вызывающие частичную астабилизацию (разрушение
защитной оболочки) глобул полимера. Иногда клея-
клеящую способность Л. к. повышают предварительным
окислением полимера, напр, перекисью водорода. Для
повышения вязкости Л. к. применяют загустители
(напр., мездровый клей, казеинат аммония, полиакри-
латы), для повышения водостойкости — меламино-форм-
альдегидную смолу.
Л. к. изготовляют таким же образом, как и смеси
для латексных изделий.
Л. к. применяют гл. обр. вместо резиновых клеев
(р-ров каучуков или резиновых смесей в органич. раст-
растворителях). Основные преимущества Л. к. перед рези-
резиновыми клеями: 1) низкая вязкость при высоком (до
60% по массе) содержании сухого вещества (вследствие
высокой вязкости резиновых клеев их концентрация
47
ЛАТЕКСНЫБ КРАСКИ
48
м. б. не более 12%); 2) отсутствие в составе Л. к. ток-
токсичных и огнеопасных растворителей; 3) отсутствие
у большинства Л. к. неприятного запаха. В отличие от
"клеев животного происхождения (см. Клеи природные),
Л. к. не подвергаются плесневению. Недостаток Л. к.—
большая продолжительность сушки, чем для резиновых
клеев.
Прочность клеевых соединений, получаемых с при-
применением Л. к. и резиновых клеев, практически одина-
одинакова. Присутствие в Л. к. гидрофильных поверхностно-
активных веществ обусловливает склонность клеевых
соединений к набуханию в воде, приводящему к сниже-
снижению их прочности; после высыхания прочностные свой-
свойства таких соединений восстанавливаются.
Л. к. могут быть нанесены на склеиваемые поверх-
поверхности пульверизатором, кистью, а также способами
облива, макания или шпредингования. Поверхность
нек-рых материалов (напр., кожу, резину, металлы)
перед склеиванием предварительно очищают и обраба-
обрабатывают различными способами с целью повышения
адгезии. В случае необходимости (напр., при эксплуа-
эксплуатации изделия в условиях повышенных темп-р) Л. к.
могут быть свулканизованы.
Высокие адгезионные и когезионные свойства Л. к.
позволяют применять их в самых различных отраслях
пром-сти.
В производстве обуви с помощью Л. к. склеивают
кожаный верх с подошвой из каучука или резины,
вклеивают внутренние детали, собирают заготовки.
На предварительно очищенную и отшерохованную по-
поверхность кожи или резины наносят один слой Л. к.,
а через 20—30 мин — другой. Когда слои Л. к. стано-
становятся совершенно прозрачными, склеиваемые детали
соединяют друг с другом, прикатывая или обжимая
места склейки. Состав нек-рых обувных Л. к. приведен
ниже (в мае. ч.): 1) каучук в натуральном латексе —
100; сера — 1,75; ZnO — 5; каптакс — 1,25; неозон
Д — 0,5; натриевая соль к-т канифоли (смачива-
(смачиватель) — 0,5; 2) каучук в хлоропреновом латек-
латексе — 100; ZnO — 5; неозон Д — 0,5; натриевая соль
к-т канифоли — 2; 3) каучук в натуральном ла-
латексе — 100; костный клей — 200; формалин — 60;
а-нафтол — 10.
Л. к. применяют для наклеивания корешков перепле-
переплетов, брошюрования книг, заклеивания коробок, паке-
пакетов, наклеивания этикеток. Для ускорения «схватыва-
«схватывания» в Л. к. иногда вводят органич. растворители. Ко-
Корешки переплетов, предварительно отшерохованные
для лучшего поглощения клея, пропускают под про-
промазывающим валком. Состав некоторых Л. к., при-
применяемых в переплетном деле, приведен ниже (в мае. ч.):
1) каучук в натуральном латексе — 100; олеат натрия—
15,5; сера — 2; ZnO — 2; дибутилдитиокарбамат цин-
цинка — 1; неозон Д — 1; 2) каучук в бутадиен-стирольном
латексе —100; олеат натрия — 0,5; сера — 0,75; ZnO —
3; диэтилдитиокарбамат цинка — 1; казеинат аммо-
аммония — 1; меламино-формальдегидная смола — 1; 3) по-
ливинилацетат в дисперсии — 100; олеат натрия — 0,5;
дибутилфталат — 12; казеинат аммония — 0,5.
В деревообрабатывающей пром-сти Л. к. применяют
для склеивания деревянных поверхностей (шлифован-
(шлифованных, струганых, отпиленных), напр, при изготовлении
фанеры. Оптимальная влажность дерева, необходимая
для получения прочных клеевых соединений, 8—12%.
Слой Л. к. наносят на обе склеиваемые поверхности,
к-рые затем соединяют и прессуют от 15 мин до 2 ч
под давлением 0,1 —1,2 Мн/м2 A —12 кгс/см2). При из-
изготовлении фанеры применяют горячее прессование
[70—100 °С, избыточное давление 0,35 Мн/м2
C,5 кгс/см2)].
В строительной технике с помощью Л. к. приклеи-
приклеивают линолеум и плитки из полимерных материалов,
а также паркет к различным основаниям пола.
В производстве текстильных материалов и ковров
Л. к. применяют для соединения хлопчатобумажных,
шерстяных и льняных нитей без образования узлов.
Состав такого клея (в мае. ч.)\ каучук в натуральном
латексе — 100; этиловый спирт — 5; ZnO — 10.
В производстве абразивных материалов Л. к., содер-
содержащие меламино-формальдегидную смолу, наносят па
тканевую или бумажную основу, а затем распыляют
абразив.
Л. к. широко применяют для склеивания пенополи-
стирола, пенорезины, поливинилхлорнда, а также для
приклеивания керамич. материалов, стекла, металлов
к бумаге, полистиролу, тканям, коже и др. Ниже при-
приведен состав клея для приклеивания алюминиевой
фольги к бумаге (в мае. ч.): каучук в хлоропреновом
латексе — 100; ZnO — 15; неозон Д — 2; казеинат аммо-
аммония — 20; силикат натрия — 0,25.
При изготовлении клейких лент на бумагу наносят
клеи на основе акрилатного латекса, содержащие до-
добавки бутилацетата и монохлорбензола.
Лит. см. при ст. Латекс натуральный, Латексы синте-
синтетические. Д. П. Трофимович, В. В. Черная, М. И. Шепелев.
ЛАТЕКСНЫЕ КРАСКИ — см. Эмульсионные краски.
ЛАТЕКСЫ СИНТЕТИЧЕСКИЕ (synthetic latexes,
synthetische Latizes, latex synthetiques).
Содержание:
Свойства и методы испытаний 48
Получение 52
Агломерация глобул 54
Концентрирование 54
Модификация 5 5
Применение 5 5
Латексы синтетические — коллоидные системы, пред-
представляющие собой водные дисперсии синтетич. полиме-
полимеров. Макромолекулы полимера находятся в Л. с. в виде
глобулярных агрегатов (см. Глобулы). Коллоидная си-
система Л. с. стабилизирована поверхностно-активными
веществами (эмульгаторами). Большинство Л. с.—
водные дисперсии эластомеров, образующиеся непо-
непосредственно в результате эмульсионной полимериза-
полимеризации. Некоторые Л. с. изготовляют диспергированием
в воде «готовых» полимеров (напр., бутилкаучука,
синтетического изопренового каучука).Такие дисперсии
обычно называют искусственными латек-
латекса м и. К Л. с. относят также водные дисперсии тер-
термопластов (напр., поливинилацетата, поливинилхлори-
да), образующиеся при эмульсионной или суспензион-
суспензионной полимеризации.
Л. с. делят в зависимости от химич. состава полимера
на бутадиен-стирольные, бутадиен-нитрильные, хлоро-
преновые и др. (табл. 1).
Свойства и методы испытаний. Размер глобул
(см. табл. 1) оказывает существенное влияние на вяз-
вязкость латекса при высоком содержании в нем сухого
вещества (см. рисунок), а также на его стабильность.
Последняя тем выше, чем меньше размер глобул. Для
определения размера глобул применяют методы нефе-
нефелометрии и электронной микроскопии; при неполном
насыщении поверхности глобул эмульгатором пользуют-
пользуются методом адсорбционного титрования латекса р-ром
эмульгатора.
Концентрация Л. с. изменяется в пределах
18—75% (см. табл. 1); ее определяют по сухому остатку
навески латекса, высушенной при 70—105 СС до посто-
постоянного значения массы, или по количеству полимера,
выделенного из навески латекса спиртом и высушен-
высушенного до постоянного значения массы.
Вязкость Л. с. зависит не только от размера
глобул, но также и от способа получения Л. с, их
концентрации, типа и количества эмульгатора. Ла-
Латексы с концентрацией выше 30% ведут себя как ненью-
неньютоновские жидкости; их вязкость обычно определяют на
реовискометре Гепплера или вискозиметре Брукфилда.
49
ЛАТЕКСЫ СИНТЕТИЧЕСКИЕ
Таблица 1. Свойства некоторых синтетических латексов
50
Тип полимера в латексе
Тип эмульгатора
Средний диаметр
глобул, им (А)
РН
Сухой ос-
остаток, %
Марка (страна)
Б у т а д и е н-с тирольные латексы
Сополимеры бутадиена со стиролом
в соотношении (
70
70
50
50
мае. ч.)
30
30
50
50
Сополимеры стирола с бутадиеном
в соотношении (мае. ч.)
70
65
80
30
35
20
Натриевая соль син-
тетич. жирных к-т
—
Мыло к-т канифоли
То же
С т и р о л-б у т а
Аммониевая соль
жирной к-ты
Некаль
Анионоактивный*
80 (800)
300 C000)
150 A500)
160 A600)
диеновые
100 A000)
110 A100)
80 (800)
10
10
10
11
латекс
9,
9,
8,
,0
,5
,5
,5
ы
5
0
5
30
65
49
62
49
45
50
Сополимеры бутадиена с акрило-
нитрилом в соотношении (мае. ч.)
80
72
60
20
28
40
55 : 45
Сополимер со средним содержанием
акрилонитрила
Сополимер с высоким содержанием
акрилонитрила
Полихлоропрен
То же
Сополимер бутадиена с акрилонит-
рилом и с метакриловой к-той в
соотношении (мае. ч.) 60 : 40 : 3
То же
Сополимер бутадиена со стиролом
и с метакриловой к-той в соотно-
соотношении (мае. ч.) 70 : 30 : A,5-4)
То же
Сополимер бутадиена с метакрило-
метакриловой к-той в соотношении (мае.
ч.) 100 : 2
Сополимер бутадиена со стиролом
и 2-винилпиридином в соотноше-
соотношении (мае. ч.) 70:15:15
Сополимер бутадиена с 2-метил-5-
винилпиридином в соотношении
(мае. ч.) 90:10
Сополимер изобутилена с неболь-
небольшим количеством изопрена
То же
Б у т а д и е н-н итрильные латексы
Мыло жирных к-т
Калиевая соль син-
тетич. жирных к-т
Анионоактивный*
То же
160 A600)
60-80 F00—800)
60-80 F00 — 800)
250 B500)
90 (900)
120 A200)
10,3
9,0
8,5-9,5
9,0
9,0
8,5
Хлоропреновые латексы
Анионоактивный*
То же
80 (800)
120 A200)
130 A300)
10,0-
11,0
10
12,5
11,5 —
12,4
13
Карбоксилатные латексы
Некаль + лейканол — 4—5
Анионоактивный*
Некаль
Некаль
40 D00)
100 A000)
9,5
3,5-9,5
7,0
8,5-9,5
8,0-9,5
Винилпиридиновые латексы
Мыло жирной к-ты
и к-т канифоли
Мыло синтетич.
жирных к-т
80 (800)
80 (800)
10,5-
11,5
10,0-11,2
63
45
40-42
42,5
40
40
45 — 50
40—45
50
38,5
50
40
40
37
20
49
18
40
26
Искусственные латексы бутилкаучука
Олеат калия
300 C000)
350C500)
5,5
10,5
61-65
45
Искусственные латексы изопренового каучука
1,4-г^ис-Полиизопрен
То же
Полистирол
Анионоактивный*
Калиевая соль к-т
канифоли
700 G000)
500 E000)—
700 G000)
10,5
10,5
Полистирольный латекс
Анионоактивный* I — 18,0
65
60
50
СКС-ЗОШХП (СССР)
Полисар 72 5 (Канада)
2002 (США)
Южитекс 2107 (Франция)
Плиолит 170 (США)
СКС-6 5ГП (СССР)
Полисар 776 (Канада)
Полисар 761 (Канада)
Пербунан 2818 (ФРГ)
СКН-40П (СССР)
Кемигам 23 5 (США)
AL-911 (Великобритания)
Хайкар 1571 (США)
Наирит ЛНТ-1 (СССР)
Наирит Л-7 (СССР)
Неопрен 750 (США, Япо-
Япония)
Неопрен 736 (США, Япо-
Япония)
Пербунан С-МК (ФРГ)
СКН-40-1-ГП (СССР)
Хайкар латекс 1577 (США)
Бреон 15 74 (Великобрита-
(Великобритания)
СКС-3 0-1 (СССР)
Дау латекс 540 (США)
СКД-1 (СССР)
Джен-Тэк, Пиратекс
(США), Полисар 781
(Канада), Нипол SL85 4FS
(Япония)
ДМВП-ЮХ (СССР)
Инджейбутиллатекс 100
(США)
Латекс бутилкаучука
(СССР)
Карифлекс IR-7 0 0 (США)
Латекс ^ис-полиизопрена
(СССР)
Дау 586 (США)
Поливинилхлорид
То же
Поливинилхлоридные латексы
Анионоактивный*
Алкилсульфонат
натрия
8,0
6,9
52
45
Плиовиклатекс 30 0 (США)
Латекс ПВХ (СССР)
* Точный состав эмульгатора неизвестен.
51
ЛАТЕКСЫ СИНТЕТИЧЕСКИЕ
52
Вязкость менее конц. латексов м. б. определена с по-
помощью вискозиметра Гепплера. Снижают вязкость Л. с.
агломерацией глобул или добавлением небольших коли-
количеств электролита; повышение вязкости достигается
диализом с последующим концентрированием, введе-
введением загустителей и др. способами.
4000
2000
1000
600
400
200
' 100
: 60
40
20
I
40 50 60
Ионцентрация, X
70
Зависимость вязкости бутадиен-стирольного латекса от его
концентрации и размера глобул (в полулогарифмич. коор-
координатах): 1 — диаметр глобул 60 нм F00 А); 2 — 200 нм
B000 А); 3 — 250 нм B500 А).
Плотность Л. с, зависящую от их концентра-
концентрации, плотности полимера и водной фазы (серума),
определяют обычно с помощью ареометров.
Важнейший критерий агрегативной ста-
стабильности Л. с — степень насыщения поверх-
поверхности глобул эмульгатором. Ее определяют титрова-
титрованием Л. с. р-ром эмульгатора с одновременным конт-
контролем электрич. проводимости и поверхностного натя-
натяжения латекса. Значения последнего (обычно 30—
60 мн/м, или дин/см) уменьшаются с увеличением сте-
степени насыщения глобул.
Заряд глобул обусловлен диссоциацией эмульгато-
нон
ров, напр, по схеме RCOONa^i^ RCOO~ + Na+, где
R — алкил С12—С17. (Глобулы Л. с, стабилизиро-
стабилизированные неионогенными эмульгаторами, не имеют заря-
заряда.) Углеводородный радикал адсорбирован на поверх-
поверхности глобул; часть молекулы эмульгатора, несущая
электрич. заряд, вместе с диффузионным двойным
электрич. слоем ионов обусловливает электрокинетич.
потенциал глобул латекса (т. наз. С-потенциал).
Помимо заряда глобул (электростатич. фактор), на
агрегативную стабильность Л. с. оказывает влияние
гидратация адсорбционных оболочек глобул и струк-
турно-механич. барьер, обусловленный высокой проч-
прочностью коллоидных адсорбционных слоев эмульгатора
(неэлектростатич. факторы). Толщина слоя гидратиро-
ванного эмульгатора составляет 2—15 нм B0—150 А)
в зависимости от степени насыщения поверхности гло-
глобул эмульгатором, строения последнего, а также от ти-
типа полимера в латексе.
Большинство Л. с. стабильно при рН>7, т. к. только
в такой среде эмульгатор проявляет свои поверхностно-
активные свойства. Л. с, стабилизированные эмульга-
эмульгаторами на основе сильных к-т (напр., сульфоновых),
стабильны и при рН < 7.
Агрегативная стабильность Л. с. существенно зави-
зависит и от свойств полимера. Так, при наличии в полимере
достаточного количества полярных функциональных
групп можно получить стабильные Л. с. в отсутствие
эмульгаторов. Примером таких Л. с. может служить
латекс сополимера уксуснокислой соли 2-диметиламино-
этилметакрилата, винилацетата и бутилметакрилата.
Устойчивость этого латекса обусловлена присутствием
полярных групп на границе глобула — вода. Агрега-
Агрегативная стабильность Л. с. связана также со сродством
эмульгатора к полимеру. Так, стабильность бутадиен-
стирольного латекса, содержащего калиевое мыло к-т
канифоли, выше, чем бутадиен-нитрильного с тем же
эмульгатором.
Л. с. могут утрачивать агрегативную стабильность
при хранении в результате химич. реакций полимера.
Напр., коагуляция хлоропренового латекса м. б. обус-
обусловлена структурированием полимера вследствие от-
отщепления хлора, а также действия кислорода. Введе-
Введение активных антиоксидантов и уменьшение концент-
концентрации латекса способствуют повышению его стабиль-
стабильности.
В технологич. практике стабильность Л. с. оценива-
оценивают по их стойкости к механич. и химич. воздействиям.
Для определения механич. стабильности, к-рая харак-
характеризует способность Л. с. не коагулировать под воз-
воздействием механич. нагрузок (при перекачивании на-
насосами, транспортировке и др.), широко применяют
прибор Марона. В этом приборе латекс подвергают
деформации сдвига, создаваемой металлич. ротором
(диском), вращающимся с частотой 1000 об/мин и при-
прижимаемым к дну стакана пружинами с усилием, рав-
равным 1,11 Мн/м2 A1,3 кгс/см2). Мерой стабильности слу-
служит масса коагулюма, образовавшегося при испытании
порции латекса в течение 5 мин.
Химич. стабильность Л. с. устанавливают по т. наз.
порогу коагуляции — критич. концентрации электро-
электролита, вызывающей коагуляцию латекса.
Морозостойкость Л. с. определяют по
количеству коагулюма, образовавшегося в условиях
замораживания пробы латекса при темп-pax от —10
до —30 °С, или по числу циклов замораживание —
оттаивание, к-рые латекс выдерживает, не изменяя
своих свойств (не коагулируя, сохраняя размер гло-
глобул и др.). Морозостойкость Л. с. ухудшается с повы-
повышением мол. массы эмульгатора, снижением рН и
темп-ры замораживания. Этот показатель зависит так-
также от типа полимера. Напр., морозостойкость бутадиен-
стирольных латексов тем хуже, чем выше содержание
в сополимере винильного мономера. Для улучшения
морозостойкости Л. с. в них вводят небольшие количе-
количества низших спиртов, казеината аммония или насыщают
глобулы эмульгатором. Л. с, выпускаемые в пром-сти,
как правило, недостаточно морозостойки, и поэтому
их следует хранить при темп-pax выше О °С.
Свойства вулканизатов Л. с. оцени-
оценивают обычно по комплексу их механич. показателей
(модуль и прочность при растяжении, относительное
удлинение), определяемых при одномерной деформации
образцов в виде пленки (ГОСТ 12580—67). Механич.
показатели нек-рых изделий (напр., метеорологич.
радиозондовых оболочек) определяют при двумерной
деформации, к-рую создают раздувкой сжатым возду-
воздухом специально изготовленного или вырезанного из
изделия образца (пленки) вулканизата, зажатого по
периметру.
Получение. Латексы образуются как промежуточ-
промежуточные продукты при синтезе каучуков эмульсион-
эмульсионной полимеризацией (см., напр., Бута-
диен-стиролъные каучуки). При получении товарных
Л. с. процесс проводят в несколько иных условиях:
применяют эмульсии с высоким содержанием мономе-
мономера, доводят полимеризацию до большей глубины,
используют меньшее исходное количество эмульгато-
эмульгаторов и др. В зависимости от объема производства Л. с.
процесс м. б. периодическим или непрерывным. Первый
осуществляют в одном полимеризаторе, второй — в ба-
батарее из 10—11 полимеризаторов.
При получении Л. с. чаще всего применяют анионо-
активные эмульгаторы (натриевые или аммониевые
соли природных или синтетич. высших жирных к-т,
калиевые соли к-т канифоли, алкилсульфонат нат-
53
ЛАТЕКСЫ СИНТЕТИЧЕСКИЕ
54
рия, дибутилнафталинсульфонат натрия — некаль);
в нек-рых случаях их вводят в смеси с неионогенными
эмульгаторами, напр, с продуктом ОП-10. Количество
эмульгаторов в полимеризационной смеси составляет
обычно 4—5 мае. ч. на 100 мае. ч. мономеров.
Для получения Л. с. с крупными глобулами в начале
полимеризации вводят 2—2,5 мае. ч. эмульгатора, а ос-
остальное его количество дозируют по ходу процесса
вместе с инициатором и активатором или добавляют
в готовый латекс.
В большинстве случаев регуляторы мол. массы не
вводят, т. к. образование разветвленных полимеров не
препятствует дальнейшей переработке Л. с.
Бутадиен-стирольные латексы синтезируют при 5—
10 °С или при 50 °С, бутадиен-нитрильные и карбокси-
латные — при 30 °С, винилпиридиновые — при 5 °С.
Конверсия мономеров составляет в большинстве слу-
случаев 90—100%, продолжительность полимеризации
20—40 ч. В нек-рых случаях (напр., при получении
Л. с. для пропитки шинного корда) конверсия мономе-
мономеров не превышает 55—60%.
Важное требование, предъявляемое к Л. с, — низкое
содержание остаточных мономеров (в пределах 0,02—
0,05%). Для этого после полимеризации их отгоняют из
латексов с водяным паром на колоннах, работающих
по противоточной схеме. Понижение концентрации
мономера в Л. с. достигается также повышением темпе-
температуры в конце полимеризации, применением актив-
активных инициирующих систем и др. способами. В гото-
готовые Л. с. иногда вводят 1—2% (от массы полимера)
неокрашивающих антиоксидантов в виде водной
дисперсии.
Искусственные латексы можно изго-
изготовлять двумя способами. Наибольшее распростране-
распространение получил способ, основанный на растворении поли-
полимера при 15—50 °С в подходящем растворителе (обычно
в углеводородах линейного или циклич. строения
с темп-рой кипения ниже 100 °С — изопентане, гексане,
циклогексане, этилхлориде, четыреххлористом угле-
углероде). Полученный р-р с концентрацией 10—15%
и вязкостью не более 600 мн-сек/м2, или спз, эмульги-
эмульгируют в воде с растворенным в ней эмульгатором. Для
этого применяют ультразвуковые генераторы, высоко-
высокоскоростные смесители, центробежные насосы. Соотно-
Соотношение углеводородной и водной фаз составляет от
1,8 : 1 до 3:1, продолжительность процесса 4—12 ч.
Из образовавшейся эмульсии на колоннах при темп-рах
ниже 100 °С отгоняют растворитель под вакуумом или
с водяным паром. Для подавления ценообразования
применяют пеногасители. В растворитель часто вводят
небольшие количества A0—15%) полярных веществ
(низшие спирты, кетоны), снижающих вязкость р-ра
полимера и поверхностное натяжение на границе р-р
полимера — водная фаза и облегчающих получение
эмульсии. В нек-рых случаях полярные вещества обра-
образуют азеотропные смеси типа углеводород — спирт
или углеводород — спирт — вода, кипящие при более
низкой темп-ре, чем чистые углеводороды, что облег-
облегчает отгонку растворителя.
После отгонки растворителя водную дисперсию поли-
полимера (концентрация 14—20%, содержание остаточяого
растворителя 0,05—0,2%) стабилизируют в зависимо-
зависимости от ее назначения ионогенными эмульгаторами или
их смесями с неионогенными D—10 мае. ч. на 100 мае. ч.
полимера в дисперсии) и, при необходимости, подвер-
подвергают концентрированию (см. ниже). В нек-рых случаях
после отгонки растворителя можно получить дисперсию
с концентрацией полимера до 45%. Это достигается
увеличением соотношения между углеводородной и вод-
водной фазами и повышением концентрации полимера
в р-ре. Пример рецепта для получения такой дисперсии
(в мае. ч.): бутилкаучук — 100; циклогексан — 300;
этиловый спирт — 67; олеат калия — 4,5; вода — 150.
В случае проведения полимеризации в р-ре образую-
образующийся р-р полимера м. б. непосредственно использован
для получения дисперсии. В пром-сти искусственные
латексы получают периодич. способом; общая продол-
продолжительность процесса, включая концентрирование,
составляет 40—72 ч.
По др. способу получения искусственных латексов
полимер смешивают в течение 2 ч на вальцах или в ре-
зиносмесителе с водным р-ром диспергирующего агента
(натриевой соли высших жирных к-т или к-т канифоли,
казеина и др.) или с органич. к-тами С10— С20 с после-
последующим введением в смесь водного р-ра щелочи. Во
время смешения полимера с диспергирующим агентом
воду добавляют до тех пор, пока не образуется паста, в
к-рой вода является непрерывной фазой. При содержа-
содержании в смеси более 20—30% воды образовавшаяся пер-
первоначально эмульсия воды в полимере превращается
в дисперсию^тЙлимера в воде; последнюю разбавляют
водой до требуемой концентрации. Способ имеет ряд
существенных недостатков, из-за к-рых не получил
широкого распространения в пром-сти: 1) применение
энергоемкого оборудования; 2) введение больших коли-
количеств диспергирующих агентов (до 10% от массы поли-
полимера), что ограничивает возможности последующей пе-
переработки дисперсий; 3) возможность изготовления
только грубых дисперсий с размером частиц ~ 1000 им
A0 000А), имеющих низкую стабильность при хране-
хранении; 4) деструкция полимера при его обработке на сме-
смесительном оборудовании, что приводит к ухудшению
свойств изделий.
Агломерация глобул. Эта операция предшествует
концентрированию Л. с, получаемых эмульсионной
полимеризацией и содержащих, как правило, глобулы
сравнительно небольшого размера.
При агломерации замораживанием латекс непрерыв-
непрерывно подают на вращающийся охлаждающий барабан
(темп-pa от —12 до —14 °С). В результате замерзания
латекса создается высокая местная концентрация по-
полимера в промежутках между кристаллами льда, что
способствует слиянию глобул, поверхность к-рых не пол-
полностью насыщена эмульгатором. После оттаивания ла-
латекса получают продукт со средним размером глобул,
в 1,5—2 раза превышающим первоначальный. В про-
процессе агломерации повышается также степень насыще-
насыщения поверхности глобул эмульгатором и снижается по-
поверхностное натяжение латекса. Для Л. с, содержа-
содержащих глобулы, полностью насыщенные эмульгаторами,
способ агломерации замораживанием непригоден.
Укрупнение глобул происходит также при добавле-
добавлении в латекс водорастворимых полимеров (поливинил-
метилового эфира, поливинилового спирта, натриевых
солей полиакрилатов и др.) или при продавливании
его через узкое отверстие под давлением ок. 30 Мн/м2
(ок. 300 кгс/см2).
Концентрирование. При концентрировании с л и в-
коотделением в латекс добавляют 0,3—0,5%
гидрофильных коллоидов: альгинат натрия, щелочные
соли полиакрилатов, метилцеллюлозу, карбоксиметил-
цэллюлозу, поливиниловый спирт и др. Под влиянием
этих агентов система расслаивается. Концентрация
полимера в верхнем слое (при плотности полимера <1)
может достигать 40—45%. Эффективность сливкоотде-
ления, как правило, увеличивается при повышении
темп-ры, рН, снижении вязкости латекса. Продолжи-
Продолжительность процесса 24—96 ч.
Для концентрирования методом центрифуги-
центрифугирования применяют аппараты типа молочного
сепаратора или центрифуги типа Шарплес (частота
вращения ротора соответственно 8000 и 18 000—
20 000 об/мин). Этот метод наиболее эффективен для
Л. с. с достаточно крупными глобулами [более 300 им
C000 А)], напр, для искусственных латексов с концент-
концентрацией 20—40%. После центрифугирования концент-
55
ЛАТЕКСЫ СИНТЕТИЧЕСКИЕ
56
рация Л. с. может достигать 60%. Повторное центри-
центрифугирование предварительно разб. концентрата позво-
позволяет получить продукт с содержанием сухого остатка
65—67%. Концентрат отличается небольшим содер-
содержанием примесей и высоким качеством. За рубежом
этим методом концентрируют дисперсии 1,4-^мс-поли-
изопрена и бутилкаучука.
Концентрирование латекса упариванием осу-
осуществляют в выпарных аппаратах, снабженных мешал-
мешалками или др. приспособлениями для обновления по-
поверхности латекса. Эффективность процесса повышают,
проводя его в вакууме или пропуская над поверхностью
латекса горячий воздух. Пенообразование при упари-
упаривании предотвращают с помощью пеногасителей. Л. с,
сконцентрированные этим методом, отличаются наи-
наибольшей вязкостью, содержат больше всего примесей
и меньше всего остаточных мономеров, т. к. последние
удаляются вместе с парами воды. Содержание сухого
остатка в таких Л. с. составляет 55—65%.
Модификация. Для придания изделиям нек-рых
специфич. свойств латексы подвергают модификации.
Один из способов — получение привитых сополи-
сополимеров радикальной сополимеризацией винильного мо-
мономера (напр., винилацетата, стирола) с полимером
латекса. Вулканизованные пленки из таких сополиме-
сополимеров характеризуются повышенным модулем при растя-
растяжении и твердостью. Модификация Л. с. галогенсодер-
жащими веществами, напр, трихлорбромметаном, при-
придает изделиям из Л. с. огнестойкость. Для повыше-
повышения мягкости, пластичности и клейкости пленок из
Л. с. полимер окисляют, напр, путем нагревания
латекса острым паром в течение 3—4 ч в присутствии
перекиси водорода. Латексы, содержащие окисленный
полимер, применяют для получения клеев.
Применение. Области применения Л. с. разнообраз-
разнообразны и непрерывно расширяются. Значительная часть
Л. с. расходуется в производстве губчатых резин и др.
латексных изделий. На основе Л. с. изготовляют клеи
(см. Латексние клеи), краски. Важная область приме-
применения Л. с.— производство бумаги. При
этом латекс вводят в бумажную массу, пропитывают
им бумажное полотно или наносят на поверхность
бумаги (этот способ используют наиболее широко).
Применение Л. с. позволяет повысить прочность, гиб-
гибкость, влаго- и маслостойкость бумаги, а также
улучшить ее внешний вид и уменьшить растекание на
ней чернил.
Использование композиций на основе Л. с. для о б-
работки текстильных материалов
способствует улучшению их эксплуатационных свойств
(прочности, эластичности, износостойкости, водо- и га-
газонепроницаемости, стойкости к действию агрессивных
сред) и повышению адгезии к др. материалам. Обработ-
Обработка текстильных нитей или пряжи уменьшает их истира-
истирание при ткачестве и позволяет в нек-рых случаях ис-
использовать некрученые нити.
При пропитке адгезивами на основе латекса шинного
корда повышается прочность его связи с резиной (см.
Кордные нити и ткани). Пропитка шнуров и канатов
повышает их водостойкость, износостойкость и предот-
предотвращает разлохмачивание. Для пропитки наиболее при-
пригодны латексы на основе полимеров с функциональными
группами (напр., карбоксилатные, винилпиридиновые),
способные к химич. взаимодействию с волокном. Для
этой цели м. б. также использованы Л. с, стабилизи-
стабилизированные катионоактивными эмульгаторами. С помо-
помощью Л. с. осуществляют аппретирование.
Композиции на основе Л. с. применяют при изго-
изготовлении прошивных ковров, вор-
ворсовых тканей, искусственного меха
и др. с целью закрепления ворса и лучшего сохранения
формы изделий из этих материалов, а также при изго-
изготовлении дублированного (кашированного) текстиль-
текстильного полотна. При этом используют композиции на
основе Л. с, обладающие высокой вязкостью, механич.
стабильностью и не склонные к ценообразованию. Адге-
Адгезию Л. с. к ткани повышают введением в композицию
поливинилового спирта, водостойкость — введением
меламино-формальдегидных смол. См. также Нетканые
изделия.
Таблица 2. Области применения синтетических и искусственных латексов
Тип латекса
Маканые
перчатки
-
4-
+
it
—
—
-
—
+
+
4-
4- 4-1
латексные
изделия
радиозондо-
вые оболочки
-
-
4-1
—
—
-
—
-
-
медицинские
изделия
-
-
-
—
—
-
—
4-
_
в качестве
усиливающих
добавок
-
-
-
4-1
—
7
4-1 ¦
—
-
+
Губчатые резинь
4-
+
4-
_
—-
—
-
—
4-
+
Краски
-
4-
-
_
4-1
-
4-
__
+
•А-
Полимерцемент
14-
-
4-
_
4-1
-
—
-
+
-
Клеи
+
4-
+
4-
4-
4-1
+
+
4-
_
14- 4-
Отделка кожи
-
+
4-
_
—
-
4-
-
+
Искусственная
кожа
4-
4-
+
4-
-4-
4-1
4-1
—
-
14- 4-
Бумага
+
4-
+
4-1
-}-
4-4-
14-
4-
-
_
+
Нетканые изде-
изделия
+
1
~г
4-
4-
-{-
14-
4-1
4-
-
_
14- 4-
Аппретирование
текстиля
4-1
+
-
-(_
-j-
±
14-
4-
4-
~~~
+
Пропитка корда
14-
-
-
_
—
-
—
-
_
-
Битумы
-
+
+
_
—
I
-
—
-
4-
-
Картоны
4-1
-1-
+
4-
4-
4-1
4-1
—
-
_
4-
я
Антикоррозионн
покрытия
-
-h
4-
4-
4-
4-
-
4_
-
4-
+
Гру
Бутадиен-стирольный
Стирол-бутадиеновый
Бутадиен-нитрильный
Винилпиридиновый .
Хлоропреновый ....
Фторсодержащий . . .
Поливинилхлоридный
Полиакрилатный . . .
Поливинилацетатный
Полистирольный ....
Поливинилиденхлорид-
ный
Дисперсия полиэтилена
Дисперсия полиизобути-
лена
Дисперсия 1 ,4-г^ис-по-
лиизопрена
Дисперсия хлорсульфи-
рованного полиэти-
полиэтилена
Дисперсия бутилкаучука
Карбоксилатные
бутадиеновый .
бутадиен-стирольный
бутадиен-нитриль-
бутадиен-нитрильный
- - - 4-
Примечание. «4-» применяют; «—» не применяют.
57
ЛЕСТНИЧНЫЕ ПОЛИМЕРЫ
58
Л. с. применяют также в качестве связую-
связующего при изготовлении: 1) резино-асбестовых изде-
изделий (асбестовый картон, фрикционные изделия), в к-рых
используют асбестовое волокно, предварительно обра-
обработанное латексными композициями; 2) т. н. «прорези-
«прорезиненного волоса» — материала из растительных воло-
волокон, животного волоса или их смесей, скрепленных
композициями на основе Л. с. (из этого материала из-
изготовляют сиденья для автомобилей и мебели); 3) пресс-
материалов из пробковой муки, древесных опилок,
соломы, льняных очесов и др.
Л. с. используют для отделки натураль-
натуральной кожи с поврежденной лицевой поверхностью,
а также в качестве кожевенных лаков. При изготовле-
изготовлении искусственной кожи Л. с. служат грунтами (их
наносят на тканевую или нетканевую основу кожи
с целью повышения прочности ее связи с материалом
покрытия), лаками, а также компонентами водных
дисперсий измельченного кожевенного волокна, из
к-рых формуют листы (см. Кожа искусственная).
Л. с. применяют в строительной техни-
к е: 1) при изготовлении полимер цемента, использу-
используемого, в частности, при устройстве покрытий для
полов; 2) для панесения водонепроницаемого покрытия
на свежий бетон, что способствует ускорению его твер-
твердения; 3) в качестве компонентов битумных компози-
композиций, используемых в материалах для дорожных покры-
покрытий и для герметизации стыков строительных кон-
конструкций.
Л. с. вводят в состав композиций, используемых для
защиты почвы от ветровой эрозии.
При небольшом уд. расходе таких композиций через
2—3 ч после их распыления на поверхности почвы
образуется защитная пленка, не препятствующая про-
прорастанию семян. На основе Л. с. получают антикорро-
антикоррозионные полимерные покрытия для металлов, пасты для
герметизации швов консервных банок и др.
Потребление Л. с. благодаря их широкому ассорти-
ассортименту и разнообразию свойств превышает потребле-
потребление натурального латекса (см. Латекс натуральный).
Так, в первом полугодии 1972 в США было перерабо-
переработано ок. 113 тыс. т Л. с. и ок. 54 тыс. т натурального
латекса. Применимость Л. с. для изготовления раз-
различных материалов и изделий показана в табл. 2.
Лит.: Encyclopedia of polymer science and technology,
v. 8, N. Y.— [a. o.], 1968, p. 164; BlackleyD.Ch., High
polymer latices, v. 1—2, L.— N. Y., 1966; Современные дости-
достижения в области физико-химии латексов, структуры вулкани-
затов и технологии переработки латексов, М., 1971; Ней-
Нейман Р. Э., Коагуляция синтетических латексов, Воронеж,
1967; МюльстепВ., ПегеВ., Дисперсии пласто- и эла-
эластомеров, пер. с нем., М., 1967; Новые синтетические латексы
и теоретические основы процессов латексной технологии
(ЦНИИТЭНефтехим), М., 1973; Проблемы синтеза, исследования
свойств и переработки латексов, под ред. В. В. Черной и
М. И. Шепелева (ЦНИИТЭНефтехим), М., 1971; Справочник ре-
резинщика. Материалы резинового производства, М., 1971, с. 198.
Д. П. Трофимович, В. В. Черная,
М. И. Шепелев, Л. В. Космодемьянский.
ЛЕСТНИЧНЫЕ ПОЛИМЕРЫ, полимеры со
сдвоенной цепью, полимеры с ре-
регулярной линейной сеткой (ladder po-
polymers, Leiterpolymere, polymeres en echelle).
Содержание:
Терминология и классификация 57
Синтез и структура 58
Свойства 63
Переработка и применение 66
Терминология и классификация. Л. п.— высоко-
высокомолекулярные соединения, макромолекулы к-рых по-
построены из конденсированных циклов, соединенных меж-
между собой не менее чем через два общих атома (табл. 1).
Л. п. получили свое название из-за сходства схемы про-
проекции плоскости макромолекулы с лестницей.
В данной статье рассматриваются также близкие Л. п.
по структуре и свойствам спирополимеры —
Таблица 1. Основные типы лестничных и спирополимеров
Модельная структура
_LL
Лестничный полимер со
сдвоенной цепью (циклы
связаны через два или бо-
более общих атомов)
гтттт—птп
Частично лестничный (по-
(полулестничный, блоклест-
ничный) полимер
Спирополимер (циклы свя-
связаны через один общий
атом)
Типичный полимер
Лестничный полимер, полу-
полученный циклизацией полибу-
полибутадиена
Лестничный полипиримидин
Поли-(нафтоилен-бггс-бенз-
имидазол), лестничный поли-
(бензимидазобензофенантролин)
Полисп ирокеталь
полициклич. соединения, циклы в к-рых соединены
через один общий атом.
В зависимости от состава основной цепи макромолеку-
макромолекулы Л. п. могут быть 1) органическими; 2) элементоор-
ганическими (в циклах наряду с органогенами содер-
содержатся неорганогенные элементы, напр. Si, Ti, Al, Sn) и
3) неорганическими, построенными целиком из неорга-
неорганогенов (табл. 2). Неорганич. Л. п. в данной статье
не рассматриваются. О них, а также о неорганич. спи-
рополимерах см. Координационные полимеры.
Структура реальных Л. п. в большей или меньшей
степени отличается от «идеальной» структуры, изобра-
изображаемой ф-лой основного звена. Часто получают поли-
полимеры с незавершенной лестничной структурой (т. наз.
полулестничные, или блоклестничные, см. табл. 2),
представляющие собой своеобразные блоксополимеры,
в макромолекулах к-рых конденсированные полицик-
полициклич. фрагменты чередуются с линейными или развет-
разветвленными. Участок цепи, содержащий конденсирован-
конденсированные циклы, наз. лестничным сегментом;
длина такого сегмента определяется числом циклов
в нем. Иногда рассматривают среднюю длину конден-
конденсированного полициклич. фрагмента, т. к. при одной
и той же степени циклизации, т. е. соотношении лест-
лестничных и линейных участков, распределение и длина
конденсированных полициклич. фрагментов м. б. раз-
различными.
Синтез и структура. Л. п. могут быть получены как
циклизацией линейных полимеров, так и непосредствен-
непосредственно из мономеров поликонденсацией или полимериза-
полимеризацией (в том числе реакциями Дильса — Альдера, Вюр-
ца — Фиттига, Фриделя — Крафтса и др.).
При циклизации линейных полимеров наряду
с основным (внутримолекулярным) процессом проте-
протекает межмолекулярная реакция, приводящая к обра-
образованию трехмерных структур. Циклизации благо-
благоприятствует высокое разбавление (способствующее,
59
ЛЕСТНИЧНЫЕ ПОЛИМЕРЫ
60
^^v^v^v^^/^ v^v^^^vVy^ ^^^^v^v^^^^ ьлияние стерич. эффекта на структуру и
Т X X X X —•>• -i v i Jc x ~~~" х i X х х конечное значение мол. массы образующих-
I Т YT Y ^ Y Y Y ^> Y y" Y ся Л. п. особенно четко проявляется при
д
однако, возрастанию скорости обрыва цепи), соответ-
соответствующее стерич. расположение функциональных групп,
возникновение ненапряженных циклов, наличие «жест-
«жестких» групп (ароматич. циклы, непредельные связи)
в исходной линейной макромолекуле, образование
исходными соединениями промежуточных комплексов
с геометрич. формой, соответствующей возникающему
циклу, и др. Схема циклизации линейных (А) и блок-
лестничных (Г) полимеров приведена выше.
Переход А —> В в процессе внутримолекулярной по-
полимеризации боковых реакционноспособных групп
X—Y, к-рыми могут быть группы >С=С<, >С = О,
—С = N, >G=N — (полимеры II, IV, VII, XI; здесь
и далее римские цифры обозначают шифр полимера
в табл. 2), протекает статистически с образованием
вначале моноциклич. структур Б. Теория предсказы-
предсказывает, что максимальная степень завершенности таких
реакций, протекающих по закону случая, составляет
85%. Непрореагировавшие группы X—Y могут уча-
участвовать и в межмолекулярных реакциях. Длина лест-
лестничного сегмента, зависящая от соотношения скоростей
инициирования, роста и обрыва цепи, определяется
условиями реакции, природой инициатора и исходного
полимера. Так, скорость циклизации полидиенов в иден-
идентичных условиях уменьшается в ряду: поли-B,3-диме-
тилбутадиен-1,3) > полиизопрен > полибутадиен. Наи-
Наибольшую среднюю длину сегмента, возрастающую
с увеличением продолжительности циклизации, имеет
полибутадиен. При циклизации поли-B,3-диметилбу-
тадиена-1,3) образуются только моноциклич. струк-
структуры, что обусловлено стерич. препятствиями, связан-
связанными с наличием двух метильных групп в элементар-
элементарном звене полимера (см. также Циклизация каучуков).
Аналогичный переход от А к В через Б может осу-
осуществляться также циклизацией боковых реакционно-
способных групп [напр., — С(СН3) = О], связанных
с насыщенной линейной углеродной цепью (напр., III).
Синтез Л. п. из мономеров [напр., поли-
циклоконденсацией тетрафункциональных мономеров
(XIII — XXX) или по реакции Дильса — Альдера
(I, V, VI, XII)] не подчиняется указанным статистич.
закономерностям; в этом случае возрастает вероятность
получения полностью лестничной структуры, а стадии
образования полулестничной и лестничной структур
(переход Г -> Д) часто трудно разделить.
Успешное осуществление полициклоконденсации
определяется высокой тенденцией к образованию ста-
стабильных пяти- и шестичленных циклов. При этом доля
межмолекулярных процессов, особенно при выборе
соответствующих мономеров и условий синтеза, зна-
значительно уменьшается. Вероятность образования Л. п.
еще более возрастает при циклизации, приводящей
к образованию ароматич. полисопряженных структур.
При диеновом синтезе полимера I промежуточно обра-
образующиеся соединения норборненового типа являются
слабыми диенофилами, что обусловливает необходи-
необходимость проведения процесса при повышенной темп-ре.
В этих условиях реакция становится заметно обрати-
обратимой, и олигомеры деполимеризуются так же быстро,
как и образуются. Эта трудность в образовании сдвоен-
сдвоенных цепей успешно преодолена при синтезе Л. п. V,
VI и XII из мономеров, содержащих в молекуле одно-
одновременно диеновую и диенофильную группировки.
синтезе кремнийорганич. полимеров XXXI.
Если полициклич. соединения, полученные
из tt-G3H7SiCl3 и tt-C4H9SiCl3, струк-
структурируются с образованием трехмерных
полимеров, то полициклич. соединения
из C6H5SiCl3 и З-метил-1-бутенилтрихлорси-
лана образуют Л. п. высокой мол. массы.
Полициклы, содержащие большие алкильные замести-
заместители, образуют продукты невысокой мол. массы (R =
= R' = изо-СьНп) или совершенно не полимеризу-
ются под действием электрофильных и нуклеофильных
агентов (R = R' = изо-С6Н13, н-С7Н15, изо-С4Н9).
Аналогичное влияние оказывают обрамляющие крем-
кремний смешанные заместители (R ф R').
Таблица 2. Методы синтеза и структура некоторых
лестничных полимеров
Шифр
поли-
полимера
Формула полимера
Метод синтеза и ис-
исходные соединения
I
II
III
IV
V
VI
К ар б оцик л ичес к ие п
ц
* R R
ЛрСо
1
n
олимеры
Реакция Дильса—
Альдера (из цикло-
пентадиена)
Полициклизация
линейных полидиенов
(полибутадиена, по-
полиизопрена) или по-
полимеризация бутади-
бутадиена- 1,3 и изопрена;
R = Н, СНз
Полициклизация
полиметилвинилке-
тона
Дегидрирование
лестничного полибу-
полибутадиена (II, R = Н);
полимеризация
R(C==CJR, где R=OH,
СвН6, OGONH-a-наф-
тил
Реакция Дильса -
Альдера (из 2-винил-
бутадиена и тг-бензо-
хинона)
Реакция Дильса —
Альдера (из тетраме-
тиленциклогексана и
п-бензохинона)
VII
Гетероциклические полимеры
Полимеризация ак-
рилонитрила и мет-
акрилонитрила или
термообработка ли-
линейных полиакрило-
нитрила и полимет-
акрилонитрила (R=
= Н, СНз)
61
ЛЕСТНИЧНЫЕ ПОЛИМЕРЫ
Продолжение табл. 2
62
Шифр
поли-
полимера
Формула полимера
VIII
IX
XI
XII
XIII
XIV
XV
XVI
Метод синтеза и ис-
исходные соединения
Окислительное де-
дегидрирование поли-
полимера VII с R=H
(Х=СН); полимериза-
полимеризация GNGN или поли-
поликонденсация
(GONH2J, X=N
сн.
сн
Полимеризация 4 , 4-диметил-1,6-гептадиен-З , 5-
диона, полиоксимирование полученного полимера и
последующая дегидратация
rtrr
к R R R
хгкх
Полимеризация N-
винилполиизоциана-
та или поливинил-
изоцианата
Полимеризация ак-
акролеина (R = Н) или
метакролеина
(R=CH3); полицикли-
полициклизация линейных по-
полиакролеинов
О О
с-о—(сн2L—о—с
с-о-(сн2)-о-с
о о
Реакция Дильса-Альдера (из 2-винилбутадиена-1, 3
и циклич. димера тетраметиленфумарата)
Пол икон денсация
2,3, 7, 8-тетрахлор-
1,4,6, 9-тетраазаан-
трацена с 1,3-диами-
но-4, 6-диокси- (или
дитио)бензолом
(Х=О, S)
Гомополиконденса-
ция 2,З-диокси-6,7-
диаминохиноксалина
Поликонденсация
1, З-диамино-4, 6-ди-
оксибензола с 2,5-
диацетокси-п-бензо-
хиноном (Х=Н) или
2,5-диокси-3,6-ди-
хлор-п-бензохиноном
(Х=С1)
Поликонденсация
2,5-диокси-п-бензо-
хинона с 1, 2, 4, 5-
тетрааминобензолом
Продолжение табл. 2
Шифр
поли-
полимера
XVII
XVIII
XIX
XX
XXI
XXII
XXIII
XXIV
XXV
Формула полимера
ПХССС
н
- п
7
л
О
Y
п
J
о о J,
X N N
^n-^^v^t
н о о
9 н н
н А о
1
-п
' П
Метод синтеза и ис-
исходные соединения
Поликонденсация
2, 5-диокси-п-бензо-
хинона с 2, 3, 6, 7-
тетрааминодибензо-
диоксаном
Поликонденсация
1,2,6,7-тетракето-
пирена с 1,2,4,5-
тетрааминобензолом
Поликонденсация
1,2,5, 6-тетракето-
антрацена с 1 ,2,4,5-
тетрааминобензолом
Поликонденсация
диангидрида пиро-
меллитовой к-ты с
1,2,4, 5-тетраамино-
бензолом
Поликонденсация
1,4,5,8-нафталин-
тетракарбоновой к-ты
с 1,2,4, 5-тетраами-
нобензолом
Пол иконденсац ия
диангидрида 1,4,5,
8 -нафталинтетракар -
боновой к-ты с 1, 4,
5,8-тетрааминонаф-
талином
Поликонденсация
хлор- и броманило-
вых к-т (X=Y=O>
или обработка серой
полихиноноксидов
(Х=О, Y=S) и поли-
хинонаминов (X =
=NH, Y=S)
Полициклизация
полиамина, получен-
полученного поликонденса-
поликонденсацией диэтилсукцино-
сукцината с jvt-фени-
лендиамином
Полициклизация
ароматизированного
полиамина, получен-
полученного поликонденса-
поликонденсацией диэтилсукцино-
сукцината с jvt-фени-
лендиамином
63
ЛЕСТНИЧНЫЕ ПОЛИМЕРЫ
64
Продолжение табл. 2
Шифр
поли-
полимера
XXVI
XXVII
XXVIII
XXIX
XXX
XXXI
Формула полимера
HN
\/
II
-R—N
Метод синтеза и ис-
исходные соединения
N R Nl N—
II II ^^ J!
Г NH HN
I I NH
II II 1 II
N R Nj N—
или
H
N
_N==/' \=N_R_N
-N=^~)==N—R—N-
N
H
н
N
"ft
-В
NN^
Н
=N—
=N—
п
Взаимодействие 1,2,4, 5-тетрацианбензола с м-и
n-фенилендиамином или 4,4'
эфиром (R соответственно
п-С6Н4-О-С6Н4)
Г 0 1
-СН. У\ C—0-
X x
-сн2х \/4-o-
Г ^ l \
n
ОСН СН 0 1
К/ v 2\у 2 1
[Д /\ /°\ ^ J
о от
^ 1 И
^v^s^ у x
г г\ О "
~\ y—\ г
KKXDO:
n
Р' R В'Г Р 1
| 0 | 0 |
1 1 1
0 0 0
^SL /Si ^Si
1 0 # 0 '
R R R
/° 1
I1
0
1
/Si,
0 A.f
0
Q
1%
-диаминодифениловым
ж- и п-СвН4 или
Нагревание диэтил-
3, З-бис-(оксиметил)-
циклобутан-1,1-
дикарбоксилата
Поликонденсация
1,4-циклогександи-
она с пентаэритритом
Пол иконденсация
2,2-бис-(аминоме-
тил)-1,4-диамино-
циклогексана с пиро-
меллитовым дианги-
дридом
Поликонденсация
1,4-бис- (аминоме-
тил)-1,4-диамино-
циклогексана с диан-
гидридом 1, 4 , 5, 8-
нафталинтетракарбо-
новой к-ты
Гидролитич. поли-
поликонденсация RSiX3
и полимеризация (R=
=Rf) или сополиме-
ризация (R =т= R') крем-
нийорганич. поли-
циклич. соединений
(R и R = С6Н5,
п-СН3С6Н4, 3-метил-
1 -бутенил)
Среднее число циклов в лестничном сегменте м. б.
определено различными методами: анализом групп
пли связей, типичных для линейного и лестничного
участков; на основании молекулярных характери-
характеристик полимера с использованием теоретич. уравне-
уравнений зависимости размеров невозмущенной цепи от
числа циклов в лестничном сегменте; по кинетическим
данным или результатам химических превращений.
Т. к. большинство гетероциклических Л. п. нераство-
нерастворимо и неплавко, исследование процессов синтеза и
структуры таких полимеров основано главным обра-
образом на изучении синтеза и структуры модельных со-
соединений.
Свойства. Макромолекулы Л. п. имеют ограниченную
гибкость, занимая промежуточное положение между
абсолютно жесткими субмикроскопич. частицами
(напр., вирусами или глобулярными белками) и гиб-
гибкими цепными макромолекулами (напр., полистиро-
полистиролом). Жесткость цепи Л. п. вызвана не заторможенным
вращением вокруг валентных связей (или поворотной
изомерией), а микродеформациями макромолекулярной
«решетки» в процессе ее тепловых колебаний. Количе-
Количественной мерой равновесной жесткости (и соответствен-
соответственно гибкости, см. Гибкость макромолекул, Сегмент ма-
макромолекулы) могут служить длина статистич. сегмен-
сегмента А или персистентная длина цепи а = А/2 (табл. 3).
На примере блоклестничного поли-1Ч-бутилмалеини-
мида показано, что для увеличения жесткости макро-
макромолекул до величины, характерной для Л. п., доля
зациклизованных звеньев должна быть значительно
больше 0,5.
Таблица 3. Структурные характеристики макромолекул нек-рых
полимеров
Полимер
Полистирол . . .
Поли-К-бутилма-
леинимид . . .
XXXI
OS
• о
1-100
1,9-
34
0, 36—
33,5
о
ее"
о
%
20
40
180
о
в
-140
160
— 1800
о
•
1
Г!
-18
10
—25
О А А-5
о • 1 U
8-Ю-3—
4,4-Ю
0,42 —
4,8-10-»
Примечание. А — длина статистич. сегмента (ац — а^)—
анизотропия мономерного звена; (ах — а2) — разность поляризу-
емостей сегмента; Q — степень ориентационного порядка моле-
молекулы.
Таблица 4. Изменение темп-р плавления нек-рых лестничных
олигомеров с увеличением молекулярной массы
Темп-pa плавления, °С
Полициклопентадиен
Полиацен
1
2
3
4
60
190
270
373 (начало разложе-
разложения)
218
357
290 (разл.)
Разлагается выше 50 0 °С
до плавления
Л. п. растворимы хуже, чем их линейные аналоги.
Растворимость Л. п. увеличивается при уменьшении
жесткости цепи и ослаблении межмолекулярного вза-
взаимодействия, чему способствуют: увеличение числа ато-
атомов в цикле; введение в цикл или между лестничными
сегментами «шарнирных» атомов или групп, напри-
например О, S, СН2; переход от ароматических к алицик-
лическим системам; образование ассоциатов из мак-
макромолекул Л. п. и молекул растворителя. Если поли-
полимер V нерастворим в обычных органических раство-
растворителях, то полимер XXXI растворим даже в неполяр-
неполярных, а полимеры XII и XXVIII — в полярных раство-
растворителях. Растворы блоклестничного полимера II харак-
характеризуются низкой вязкостью при высокой молеку-
молекулярной массе.
Циклизация приводит обычно к значительному повы-
повышению темп-ры плавления, причем наибольшее повы-
65
лигнин
66
Таблица 5. Изменение свойств нек-рых полимеров после
циклизации
Полимер
Натуральный каучук ....
после циклизации . . .
3,4-Полиизопрен
после циклизации ....
Циклополиизопрен** . . .
после дополнительной
циклизации
Полибутадиен
после циклизации ....
Циклополибутадиен** . . .
после дополнительной
циклизации
Полидифенилбутадиин-1,3
после циклизации . . .
Темп-pa раз-
разложения, °С
_
95-130*
25*
140*
370
388
—
—
405
413
210*
Не размягча-
размягчается
Плотность при
25° С, г/см3
0,92
0,98-1 ,016 B0 °С)
0,91
0,96-0,97
1 ,082
—
0,90
1,07
0,966
—
1 ,26 B0 °С)
1 ,30 B0 °С)
* Темп-pa размягчения. ** Полимер получен полимериза-
полимеризацией мономера в присутствии системы СвН5М^Вг — ТЛС14.
шение наблюдается в том случае, когда циклы стабили-
стабилизированы за счет сопряжения (табл. 4), а также темп-ры
разложения и плотности (табл. 5). Темп-pa плавления
большинства Л. п. превышает их темп-ру разложения.
Высокие темп-ры стеклования (плавления) Л. п. опре-
определяют их более высокую по сравнению с линейными
полимерами теплостойкость.
Нек-рые Л. п. отличаются весьма высокими механич.
показателями (табл. 6), соизмеримыми или превышаю-
Таблица 6. Механические свойства нек-рых
полимеров
Полимер
3,4-Полиизопрен . .
Циклизованный
3,4-полиизолрен . .
XXI
XXVI (R =
7i-CeH4OCeH4) . . .
Прочность при
растяжении,
Мн/м2 (кгс/см2)
3,5 C5)
16-30A60-
300)
115 A150)
72G20)*
Относительное
удлинение, %
435
1-2
2,9
лестничных
Модуль упру-
упругости М-10~3,
Мн/м2 (кгс/см2)
3,5 C5)
1,75-2,45
A7,5-24,5)
7,7G7)
5,4 E4)
* Прочность при сжатии и срезе соответственно 6200 Мн/м2
F2000 кгс/см2) и 200 B0 000).
щими таковые для линейных полимеров (например,
для полистирола, полиимидов). Прочность при рас-
растяжении полимеров XXXI возрастает с уменьшени-
уменьшением размера и концентрации алкильного заместителя
(при переходе от R = R' = СбН5 к R = CeH5, Rr =
изо-С5Нп, ггзо-С4Н9), что объясняется изменением меж-
межмолекулярного взаимодействия. При повышенных тем-
температурах обнаружены значительные частично обрати-
обратимые деформации.
Разложение Л. п. связано с разрывом не менее двух
простых связей в одном цикле, в то время как для разло-
разложения линейного полимера достаточно разрыва одной
связи. Теоретич. рассмотрение возможных путей дест-
деструкции по закону случая карбоциклич. Л. п. и построе-
построение с использованием статистич. модели Монте-Карло
кривых изменения мол. массы полимера во времени
показали след.: 1) полимеры с идеальной лестничной
структурой должны быть более термостойки, чем поли-
полимеры, содержащие конденсированные полициклич.
фрагменты, разделенные линейными участками; 2) иде-
идеальный Л. п., построенный из шестичленных циклич.
звеньев, должен быть более термостоек, чем Л. п.,
построенный из четырехчленных циклов; 3) блоклест-
ничные полимеры, независимо от того, состоят ли их
лестничные сегменты из шести- или четырехчленных
циклов, должны быть более термостойки, чем аналогич-
аналогичные линейные полимеры. Частичная эксперименталь-
экспериментальная проверка этих теоретич. выводов проведена на
примере гидролиза полимера XII. Полифенилсилок-
саны XXXI более устойчивы к гидролизу, чем обычные
полисилоксаны, и почти одинаковы с ними по стойкости
к тепловому старению при обработке водяным паром
и горячим воздухом.
Высокая термостойкость — наиболее ценное свойст-
свойство Л. п. Многие из них, особенно ароматические, спо-
способны сохранять структуру до темп-ры не ниже 300 °С
на воздухе и до темп-ры не ниже 500 °С в инертной
атмосфере. Высокотемпературная карбонизация и гра-
фитизация Л. п. часто протекают с относительно не-
небольшими потерями массы. При этом образуются про-
продукты графитоподобной структуры, обладающие еще
более высокой термостойкостью.
Окисление диеновых циклополимеров типа II при
темп-pax выше 100 °С, в отличие от окисления линей-
линейных полимеров, к-рое носит автокаталитич. характер,
протекает сначала с высокой скоростью, а затем замед-
замедляется. Энергия активации окисления циклополиизо-
прена примерно в 8 раз меньше, чем натурального
каучука. Существенно, что окисление и распад вторич-
вторичных продуктов не инициируют цепных процессов с боль-
большой длиной кинетич. цепи. Скорость окисления умень-
уменьшается и при термоокислительной деструкции полимера
IV (R = С6Н5). Характер окисления зависит от длины
лестничного сегмента и природы концевых групп
в макромолекулах Л. п.
Переработка и применение. Л. п. могут быть исполь-
использованы прежде всего в виде волокон и пленок, устой-
устойчивых к действию света, тепла, радиации и химич. реа-
реагентов. Технич. использование большинства Л. п.
осложняется трудностями, возникающими при перера-
переработке ввиду их нерастворимости и неплавкости. В слу-
случае полимеров аценовой (IV) или гетероциклич. (напр.,
VIII, XXI) природы эти трудности разрешаются двух-
стадийным проведением процесса получения Л. п.
Сначала синтезируют растворимый промежуточный
продукт (форполимер) линейной или блоклестничной
структуры; из него обычными методами изготавливают
волокна, а затем термич. или ката литич. переработкой
переводят в нерастворимый и неплавкий Л. п. (см.,
напр., Полиимиды, Полиимидоэфиры). Л. п. находят
применение в тех случаях, когда к изделиям предъяв-
предъявляют требования особенно высокой теплостойкости
и термостойкости, гл. обр. в военной и космич. технике.
Лит.: В е р л и н А. А., Чау сер М. Г., в сб.: Успехи
химии полимеров, М., 1966, с. 256; Encyclopedia of polymer
science and technology, v. 8, N. Y.— [a. o.], 1968, p. 97;
OverbergerC. G., M о о г е J. A., Adv. Polymer Sci.,
7, № 1, 113 A970); Фрейзер А. Г., Высокотермостойкие
полимеры, пер. с англ., М., 1971, с. 246; Коршак В. В.,
Дорошенко Ю. Е., РенардТ. Л., в сб.: Итоги науки.
Химия и технология высокомолекулярных соединений, т. 3,
М., 1971, с. 5. М. Г.Чаусер.
ЛИГНИН (lignin, Lignin, lignine) — природный по-
полимер, инкрустирующее вещество одревесневших расти-
растительных тканей, скрепляющее целлюлозные волокна.
Л. вместе с гемицеллюлозой определяет прочность ство-
стволов и стеблей растений. Содержание Л. в древесине
составляет ок. 30%. Л. может быть выделен из древеси-
древесины двумя способами: растворением ее углеводных ком-
компонентов (напр., при гидролизе полисахаридов древе-
древесины) или растворением самого Л.
Способы выделения и растворимость. Природный Л.,
не выделенный из растительной ткани, нерастворим
в органич. растворителях. В водных р-рах щелочей Л.
растворяется медленно и только при нагревании.
Природный Л. приобретает способность частично рас-
растворяться в органич. растворителях после интенсив-
интенсивного размола древесной муки на шаровой вибрацион-
67
лигнин
68
ной мельнице в индифферентной жидкости, напр,
в толуоле. После длительного размола из еловой дре-
древесины можно извлечь диоксаном при комнатной
темп-ре до 50% содержащегося в ней Л. (т. наз. лигнин
Бьеркмана). Этот Л., по-видимому, является наименее
химически измененным видом изолированного Л. В
спирте он растворяется в очень незначительных коли-
количествах.
Для выделения Л. из растительных тканей с помощью
спиртов (алкоголиз) или фенола необходимо присут-
присутствие минеральных к-т и более или менее длительное
кипячение. При этом Л. претерпевает значительные
изменения, вступая со спиртами во взаимодействие.
Аналогично получают фенол-Л. и диоксан-Л. Природ-
Природный Л. растворяется в т. наз. гидротропных р-рах
(напр., в водных р-рах Na-соли бензол- или толуол-
сульфокислоты) при нагревании и под давлением. Л.
из таких р-ров может быть выделен разбавлением их
водой (гидротропный Л.). В виде кальциевых солей
лигносульфоновых к-т Л. получается в качестве отхода
при производстве целлюлозы по сульфитному способу.
Этот способ заключается в варке древесины при темп-ре
ок. 140 °С с бисульфитом кальция, магния, натрия или
аммония в присутствии свободного SO2. При сульфат-
сульфатной варке древесины Л. растворяется в щелочной ва-
варочной жидкости и может быть осажден из нее подкис-
лением р-ра (щелочной, или сульфатный, Л.).
Структура и состав. Сложность структуры Л. и его
химич. неустойчивость являются причинами того, что
строение Л. далеко еще не выяснено. Строение и свой-
свойства Л. резко отличают его от основных компонентов
древесины — целлюлозы и гемицеллголоз. Л.— нере-
нерегулярно построенный полимер с разветвленными мак-
макромолекулами. Структурная единица лигнина Бьерк-
Бьеркмана (из еловой древесины) — С9Н88О2?4 (OCH3Hi96-
Его мол. масса равна примерно И 000 ' (ул'ьтрацентри-
фугалъный метод).
Л. не является индивидуальным химич. соединением
со строго определенными свойствами,составом и строе-
строением. Л. различного происхождения заметно отличают-
отличаются друг от друга. Для Л. хвойных пород характерны
структурные элементы, представляющие собой произ-
производные гваяцилпропана. Л. лиственных пород наряду
с этими элементами содержит большое количество про-
производных сирингилпропана. В состав Л. однолетних
растений, кроме того, входят производные гс-оксифе-
нилпропана. Предполагают, что Л. хвойных пород
образован в основном из остатков кониферилового спир-
спирта (I), Л. лиственных пород — из кониферилового и
синапового (II) спиртов, а Л. однолетних растений,
кроме того, и из га-оксикоричного спирта (III), к-рый
в небольших количествах входит в состав также Л.
хвойных:
РСН3
НО
НО-/ V-R
R=CH=CH-CH2OH
Л. содержит разнообразные функциональные группы.
Число метоксильных групп в Л. различных типов
неодинаково, больше всего их в Л. из древесины лист-
лиственных пород. Содержание гидроксильных групп в Л.
разного происхождения тоже неодинаково и зависит
также от способа выделения Л. из растительных тканей.
Результаты определения тех или иных функциональ-
функциональных групп в Л. часто в значительной степени зависят
от используемого метода, что обусловлено сложностью
и неустойчивостью структуры Л., а также наличием
в нем функциональных групп, занимающих разное по-
положение в молекулах. Лигнин Бьеркмана содержит на
одну ОСН3-группу ок. 1,15 группы ОН, из них пример-
примерно 0,3— фенольные. Часть этих групп — первичные.
Значительные количества спиртовых групп — вто-
вторичные, из них часть находится в боковой цепи у атома
С, находящегося в а-положении к ароматич. ядру (бен-
зилспиртовые ОН-группы). Эти гидроксильные груп-
группы особенно реакционноспособны и их наличием обус-
обусловлены многие характерные для Л. реакции. К таким
реакциям относится сульфирование Л. бисульфитом
(сульфитная варка), когда сульфогруппа, по-видимому,
гл. обр. становится в боковую цепь к С-атому, находя-
находящемуся в а-положении к ядру, вместо соответствующих
ОН-групп. Эти группы легко, уже при 20 °С, реаги-
реагируют с метиловым спиртом в присутствии 0,5% НС1,
образуя эфир.
Количественное определение ОН-групп различной
природы в Л. связано с большими трудностями и до
сих пор для этой цели нет вполне надежных методик.
Содержание фенольных ОН-групп определяют спектро-
фотометрич. методом, основанным на смещении УФ-
спектров в щелочной среде, методом потенциометрич.
титрования в неводных растворителях, окислительным
деметоксилированием с помощью NaIO4 и др. Для Л.
Бьеркмана все эти методы дают сравнительно близкие
значения.
Л. содержит значительное число ОН-групп, связан-
связанных в форме простых эфиров. Эфирные мостики соеди-
соединяют в молекулах Л. отдельные мономеры друг с дру-
другом (наряду с эфирными мостиками в Л. имеются также
С — С-связи между мономерами). Большая часть фе-
фенольных ОН-групп Л. образует эфиры. В Л. хвойных
пород почти полностью этерифицированы (метиловым
спиртом) фенольные ОН-группы в ^-положении к бо-
боковой цепи и ок. 0,7 всех фенольных ОН-групп, находя-
находящихся в /г-положении к боковой цепи. Предполагают,
что значительная часть этих фенольных гидроксиль-
гидроксильных групп связана в виде алкиларильных эфи-
эфиров типа р-кониферилового эфира а -гваяцилглицери-
на (IV).
ОСНо
но
-f V-C
« ? У
CH(OH)-CH-CH2OH
о
•сн=сн-сн2-он
\=/
ОСН3
IV
В Л., кроме того, существуют и другие типы эфирных
связей: фенилкумарановые, связи алкиларильного ти-
типа, образованные за счет гидроксильной группы остат-
остатка гваяцил глицерина, находящейся в а-положении
к ядру, а также а-алкилалкильные связи (типа бензи-
ловых эфиров). Эти эфиры легко разлагаются, напр,
при сульфировании Л. в условиях сульфитной варки
и легко подвергаются переэтерификации. Л. содержит
карбонильные (кетонные и альдегидные) группы. Он
обладает характерными для СО-групп полосами погло-
поглощения в ИК-спектрах, указывающими на наличие очень
небольшого количества конъюгированных альдегид-
альдегидных групп типа кониферилового альдегида (одна на
35 структурных элементов), конъюгированных с двой-
двойной связью ароматич. кольца кетонных групп, а также
неконъюгированных кетонных групп, занимающих
в боковых цепях Р-положение по отношению к арома-
ароматич. кольцу. Число СО-групп, определенных в лигнине
Бьеркмана по гидроксиламинному способу, составляет
~0,2 на 1 ОСН3-группу. Двойных связей в лигнине
Бьеркмана мало. Их наличие объясняется присутствием
небольших количеств кониферилового альдегида, вхо-
входящего в структуру Л.
69
ЛИТЬЕ КОМПАУНДОВ
70
Попытки представить строение «природного» Л.
в виде химич. формулы (Фрейденберг, Адлер) отра-
отражают, однако, лишь возможные принципы строения
Л. и далеко еще не обоснованы экспериментальными
данными. Не ясно, состоит ли Л. из вполне идентичных
макромолекул или, что вероятнее, составляющие его
макромолекулы более или менее значительно отлича-
отличаются друг от друга порядком расположения отдельных
звеньев и связей между ними.
Химические свойства. Благодаря наличию большого
числа активных функциональных групп различного
типа Л. способен к многочисленным химич. превраще-
превращениям. Являясь фенолом, Л. легко нитруется. Даже
очень разб. C—8%-ная) HNO3 нитрует Л. При этом на
каждые 3 элементарных звена нитролигнина прихо-
приходится ок. 2 групп NO2. Одновременно Л. окисляется,
приобретая карбоксильные группы, число к-рых опре-
определяется условиями нитрования. Л. легко хлорирует-
хлорируется. Хлор вступает как в ароматич. ядро, так и в боко-
боковые цепи. Атом хлора в ароматич. ядрах (с закрытыми
фенольными группами) Л. хвойных пород занимает
положение 6 относительно боковой цепи. Положение
хлора в боковых цепях точно не установлено. В присут-
присутствии влаги одновременно с хлорированием происходит
окисление Л. Получаемый хлорлигнин содержит кар-
карбоксильные группы.
При сплавлении Л. со щелочью образуются пирока-
пирокатехин и протокатеховая к-та. Если метилированный Л.
сплавить со щелочью и снова метилировать, а затем
окислить КМпО4, получается ряд к-т, среди к-рых пре-
преобладают вератровая, изогемипиновая и дегидродиве-
ратровая. Каталитич. гидрирование Л. приводит к по-
получению мономерных продуктов гидрогенолиза Л.
В зависимости от условий реакции и типа катализатора
получаются смеси различных соединений ароматич.
или гидроароматич. рядов. Так, при гидрировании во-
водородом Л. осины в присутствии медно-хромового ка-
катализатора при 250—260 °С получается смесь произ-
производных пропилциклогексана: 4-«-пропилциклогекса-
нол-1, 4-н-пропилциклогександиол -1,2,3 -D-оксицикло-
гексил)пропанол-1. В присутствии никеля Ренея при
225—250 °С получаются также производные пропил-
пропилциклогексана, производные этил- и пропилбензола,
а также гидрированные полимеры. При более низкой
темп-ре в присутствии скелетного никеля образуются
фенолы.
Л. легко окисляется. При окислении Л. нитробензо-
нитробензолом в щелочной среде (при 160 °С) образуются ароматич.
альдегиды. Из Л. хвойных пород получается ванилин
(до 25% от массы Л.). Из Л. лиственных пород полу-
получается смесь сиреневого альдегида и ванилина, соот-
соотношения выходов к-рых несколько различаются в за-
зависимости от породы древесины. В небольшом количе-
количестве получается также гс-оксибензальдегид. Из изоли-
изолированных Л. выходы ванилина значительно ниже. Вани-
Ванилин может быть получен и окислением Л. кислородом
в щелочной среде в присутствии нек-рых окислов ме-
металлов (GuO, Ag2O, HgO). Из сульфитных щелоков ва-
ванилин с выходом до 4—6% получается также при ще-
щелочном гидролизе (нагревание с водными р-рами щело-
щелочей). При действии на Л. других окислителей (пере-
(перекиси водорода, перманганата, хромовой к-ты и др.)
получаются ароматические к-ты, в том числе неболь-
небольшое количество бензолполикарбоновых к-т и щаве-
щавелевая к-та.
Применение. Л. пока не нашел широкого примене-
применения. Отходы гидролизной пром-сти (гидролизный Л.)
и бумажной пром-сти (лигносульфоновые к-ты) яв-
являются сильно измененными, трудно используемыми
формами Л. Более интересен Л. сульфатных щелоков,
однако этот продукт нельзя считать отходом, т. к. он
участвует в цикле регенерации щелочи в сульфат-цел-
сульфат-целлюлозном производстве. Попытки найти рациональные
способы применения громадных отходов Л. пока еще
не привели к существенным успехам.
Гидролизный Л. может быть использован как напол-
наполнитель в производстве прессованных досок и плит. Л.,
особенно полученный осажцением к-той из черных суль-
сульфатных щелоков, может применяться в качестве актив-
активного усилителя каучуков взамен газовой сажи в резино-
резиновой пром-сти. Гидролизный Л. для этой цели следует
предварительно активировать, напр, нагреванием со
щелочью в автоклаве. Являясь трехмерным полимером
и обладая фенольными функциями, Л. может быть ис-
использован в производстве пластмасс как наполнитель
при получении прессизделий, а также при синтезе тер-
термореактивных смол взамен части фенола. Смолы баке-
бакелитового типа удовлетворительного качества м. б. полу-
получены при замене лигнином 30% фенола.
Из Л., в том числе гидролизного, путем его модифи-
модифицирования (напр., окислением) м. б. получены замени-
заменители природных и синтетич. дубителей (регуляторы
реологич. свойств, комплексообразователи и т. д.).
Гидролизный Л.— хорошее сырье для получения ак-
активных углей. Л. применяют в производстве пористого
кирпича. Лигносульфоновые к-ты, остающиеся в бар-
барде после сбраживания содержащихся в сульфитных
щелочах Сахаров на спирт, находят применение в
нек-рых отраслях пром-сти (как дешевые крепители
и связующие в литейном производстве, при формовании
керамич. и абразивных изделий, добавки к цементу,
пластифицирующие добавки в бетон, для брикетирова-
брикетирования угольной пыли, рудной мелочи, для улучшения
реологич. свойств глинистых р-ров при бурении, в до-
дорожном строительстве). Лигносульфоновые к-ты обла-
обладают слабым дубящим свойством, и поэтому сульфит-
спиртовая барда используется при дублении кожи.
Из Л. могут быть получены ценные продукты: вани-
ванилин, ванилиновая к-та, протокатеховый альдегид, про-
протокатеховая к-та, пирокатехин. Выходы этих ве-
веществ из гидролизного Л., однако, малы. Из природ-
природного Л., содержащегося в древесных опилках, м. б.
получены те же продукты с более высокими выходами.
Лит.: Никитины. И., Химия древесины и целлюлозы,
М. — Л., 1962; Химия древесины, под ред. Л. Э. Уайза,
Э. С. Джана, пер. с англ., т. 1, М.— Л., 1959, гл. XI; Груш-
н и к о в О. П., ЕлкинВ.Р., Резников В. М., Дости-
Достижения и проблемы химии лигнина, М., 1973; Lignin, ed.
К. V. Sarkanen, N. Y., 1971; Брауне Ф. Э., Бра-
Брауне Д. А., Химия лигнина, пер. с англ., М., 1964; Г р у ш-
ников О. П., Шорыгина Н. Н., Усп. хим., 39, в. 8,
1459 A970); 40, в. 8, 1394 A971); 41, в. 11, 2024 A972); Чу-
д а к о в М. И., Промышленное использование лигнина, 2 изд.»
М., 1972. Н. Н. Шорыгина.
ЛИНЕЙНЫЕ ПОЛИМЕРЫ (linear polymers, lineare
Polymere, polyraeres lineaires) — полимеры, в макро-
макромолекулах к-рых атомы или атомные группы распола-
располагаются в виде открытой цепи или вытянутой в линию
последовательности циклов. К числу Л. п. относятся,
напр., натуральный каучук и целлюлоза.
ЛИНОЛЕУМ — см. Покрытия для полов.
ЛИТЕРАТУРА О ПОЛИМЕРАХ (literature of poly-
polymers, Literatur von Polymere, literature des polymeres).
Список важнейших книг и периодич. изданий, посвя-
посвященных химии, физике и технологии полимеров и по-
полимерных материалов, приведен в конце III тома.
ЛИТЬЕ КОМПАУНДОВ, заливка компа-
компаундами (casting of compounds, FormgieBen von
Mischungen, coulee des melanges) — 1) метод герметиза-
герметизации и изоляции изделий электронной, радио- и элек-
электропромышленности компаундами полимерными, 2) спо-
способ формования изделий из компаундов.
В зависимости от вязкости компаунда литье осущест-
осуществляется без давления или под небольшим давлением [до
0,5 Мн/м2 E кгс/см2)]. В простейшем случае герметиза-
герметизации и изоляции компаунд из любой емкости заливают
до краев кожуха прибора (или до краев открытой фор-
формы, в к-рую помещают прибор, если он не имеет кожу-
71
ЛИТЬЕ ПОД ДАВЛЕНИЕМ
72
ха). При формовании изделий компаунд заливают в пу-
пустую открытую форму. Для повышения производитель-
производительности Л. к. и улучшения качества получаемых изделий
применяют специальное технологич. оборудование для
смешения компонентов компаунда, его дозирования
и подачи в форму. Последнюю изготовляют из политет-
политетрафторэтилена, кремнийорганич. каучуков и компаун-
компаундов на их основе, луженой жести, алюминия, стали
и др. материалов. Формы из полимерных материалов
сравнительно просты в изготовлении, однако менее
долговечны.
В нек-рых случаях вместо литья в форму или кожух
засыпают таблетированный порошкообразный компа-
компаунд, к-рый при нагревании формы расплавляется и за-
заполняет ее. Такой способ переработки более просто, чем
Л. к., поддается автоматизации. Л. к. под давлением
требует применения закрытых форм, имеющих литни-
литниковую систему для подачи материала в оформляющую
полость формы. Такие формы по конструкции отлича-
отличаются от обычных литьевых форм более тонкими стен-
стенками, т. к. работают под меньшим давлением. Компаунд
подают в формы снизу для вытеснения из них воздуха,
к-рый при заливке сверху может задерживаться под
выступами формы. Для предотвращения прилипания
компаунда на поверхность формы наносят разделитель-
разделительный слой, состоящий из кремнийорганич. каучука,
кремнийорганич. жидкости, воска, полиэтилена или
др. материалов.
Заполненные компаундом формы или кассеты с изде-
изделиями помещают в термошкафы или (если время от-
отверждения не превышает 2—3 ч) в конвейерные печи.
Длительность отверждения большинства компаундов —
существенный недостаток Л. к., т. к. при массовом про-
производстве требуется большое количество форм и боль-
большая производственная площадь для работы с ними.
Поэтому Л. к. в нек-рых случаях заменяют более про-
производительными методами — литьевым прессованием,
обволакиванием изделий тиксотропными компаундами,
напылением порошкообразных компаундов в элект-
электрическом поле или из псевдоожиженного слоя (см.
Напыление).
Лит.: X а р п е р Ч., Заливка электронного оборудования
синтетическими смолами, пер. с англ., М.— Л., 1964; Чер-
Черняк К. И., Эпоксидные компаунды и их применение, 3 изд.,
Л., 1967. А. М. Поймапов.
ЛИТЬЕ ПОД ДАВЛЕНИЕМ (injection moulding,
Spritzgup, moulage par injection) — метод формования
изделий из пластмасс и резиновых смесей в литьевых
машинах, заключающийся в размягчении материала до
вязкотекучего состояния и последующем перемещении
его в литьевую форму, где материал затвердевает при
изменении темп-ры, приобретая конфигурацию внут-
внутренней полости формы. Этим методом получают изде-
изделия массой от нескольких г до нескольких кг с толщиной
стенок 6—10 мм (в редких случаях 15—20 мм). См. так-
также Литье под давлением термопластов, Литье под
давлением реактопластов, Литье под давлением рези-
резиновых смесей.
При литье под давлением (рис. 1) материал в грану-
гранулированном или порошкообразном виде поступает
в пластикационный (инжекционный) цилиндр литьевой
машины, где прогревается и перемешивается вращаю-
вращающимся шнеком. По мере пластикации шнек отходит на-
назад (на рисунке показано положение при впрыске).
В поршневых машинах пластикация осуществляется
только в результате прогрева. При переработке термо-
термопластов цилиндр нагревают до 200—350 °С, при пере-
переработке реактопластов и резиновых смесей — до 80—
120 °С. Пластицированный материал при поступатель-
поступательном движении шнека или поршня нагнетается в литье-
литьевую форму, где термопласты в зависимости от их при-
природы и требований, предъявляемых к изделию, охлаж-
охлаждаются до 20—40 °С (полистирол, полиэтилен) или
до 80—120 °С (полиформальдегид, поликарбонат), а .
реактопласты и резины нагреваются до 160—200 °С.
В форме материал выдерживается под давлением для
уплотнения, что значительно снижает последующую
усадку при охлаждении изделия вне формы.
Объем изделий, получаемых Л. п. д., ограничивается
объемом материала, к-рый может быть выдавлен червя-
червяком или поршнем при наибольшем ходе. В разновид-
разновидности метода, наз. интрузией, удается на той же
машине изготовить изделия значительно большего объе-
объема (в 2 — 3 раза). При обычном режиме литья под давле-
давлением материал пластицируется вращающимся червя-
червяком, а нагнетается в литьевую форму невраща?ющимся.
При интрузии инжекционный цилиндр снабжают соп-
соплом с широким каналом, позволяющим расплаву пере-
перетекать в форму при вращении червяка до начала его
поступательного движения. Обычно 70—80% формы
заполняют под давлением вращающегося червяка.
Литьевые машины, работающие в режиме интрузии,
оснащены подпрессовочными устройствами для компен-
компенсации значительной усадки материала при его охлаж-
охлаждении. При интрузии скорость впрыска материала
в форму ниже, чем при обычном режиме литья, однако
.. У
Рис. 1. Схема шнековой литьевой машины с диаграммами
распределения давления (I) и темп-ры (II — для термопла-
термопластов, III — для реактопластов.) по длине машины, а также
по времени (после впрыска материала в форму): 1 — литье-
литьевая форма; 2 — литниковая втулка; 3 — сопло; 4 — головка
пластикационного цилиндра; 5 — шнек; б — пластикацион-
пластикационный цилиндр; 7 — бункер; 8 — привод; 9 — гидравлич.
цилиндр; ю — передаточный механизм; и — электрич.
нагреватели.
общая длительность цикла изготовления изделия не
увеличивается благодаря частичному совмещению во
времени отдельных операций.
Основные параметры процесса. Эффективное давле-
давление, действующее непосредственно на материал в форме,
из-за потерь в узлах машины ниже того, к-рое создается
шнеком (рис. 1, кривая I). В самой форме оно снижается
от входа к ее задней стенке. Значение давления в форме
и характер его распределения по длине зависят от фи-
зико-механич. свойств полимера, режима Л. п. д.,
а также от конфигурации и размеров изготавливаемого
изделия.
Кривая давление — время, построенная для одной
точки на входе в форму, представлена на рис. 2. Следует
отметить, что характер изменения давления в форме
зависит не только от продолжительности отдельных
стадий цикла, но и от расположения рассматриваемой
точки относительно входного отверстия. Развитие дав-
давления в форме наглядно описывается в трехмерных
координатах (рис. 3). По мере заполнения формы дав-
давление в каждой точке (от ах до ап) возрастает. В точке
73
ЛИТЬЕ ПОД ДАВЛЕНИЕМ
74
ап форма заполнена. Уплотнение материала (приток
новых порций из пластикационного цилиндра) вызы-
вызывает увеличение давления на каждом участке (Ь^,
b2c2'--bncn). В результате затвердевания материала
Рис. 2. Кривая давление—вре-
давление—время для одной точки литьевой
формы: участок 1—2—запол-
1—2—заполнение формы; 2—3 — нараста-
нарастание давления; з—4 — уплотне-
уплотнение материала; 4—5 — выте-
вытекание материала из формы; 5 —
затвердевание литника; 6—7—
падение остаточного давления
в изделии.
и его усадки происходит нек-рое снижение давления
в форме (участки cxdx, c2d2...cndn). Дальнейшее охлаж-
охлаждение материала происходит без притока новых порций,
в результате чего уменьшение давления происходит
быстрее (участки d^, d2e2).
Регулирование давления в цикле Л. п. д. позволяет
направленно изменять свойства изделий. Такую регу-
регулировку особенно легко
осуществить в шнековых
литьевых машинах, ос-
оснащенных механизмами,
обеспечивающими пере-
переключение давления в ги-
дравлич. системе маши-
машины по заданной про-
программе.
По одному из режимов
Л. п. д. давление в гид-
равлич. системе после за-
заполнения формы допол-
дополнительно поднимается и
затем поддерживается
постоянным. По др. ва-
варианту давление в гид-
гидравлической системе и
сопле пластикационного
цилиндра после запол-
заполнения формы в течение
нескольких сек сохраня-
сохраняется на прежнем уровне, а затем сбрасывается. Если
на входе в форму снизить давление до значения, рав-
равного давлению в конце формы в данный момент, то
можно добиться равномерного распределения давления
вдоль всей формы. Режим литья под давлением со
сбросом давления позволяет эффективнее использовать
усилие запирания литьевой машины (т. е. увеличить
площадь изготавливаемого изделия), а при заданной пло-
площади изделия и небольшом усилии запирания исклю-
исключить опасность раскрытия формы и образование на
изделии облоя.
Как уже отмечалось, при Л. п. д. давление на мате-
материал в форме в течение цикла формования падает
(см. рис. 1), что может привести к усадке изделия в фор-
форме при охлаждении, особенно значительной при изго-
изготовлении толстостенных изделий. Для того чтобы
предотвратить этот эффект, применяют специальные
формы, давление в которых создается с двух сторон:
шнеком и механизмом закрывания формы через шток
и пуансон (см. также Литьевые машины). Указанный
метод наз. инжекционным прессованием.
Перерабатываемый материал нагревается в пласти-
кационном цилиндре внешними нагревателями, уста-
установленными на цилиндре, а также за счет тепла трения,
выделяемого при пластикации в шнеке (см. рис. 1, кри-
кривые II и III). В передней части пластикационного
цилиндра материал разогревается дополнительно в уз-
узком пространстве между горячими стенками, а при
Рис. 3. Диаграмма давление —
время — длина литьевой формы.
впрыске в форму — за счет сдвиговой деформации
в узких каналах сопла и литниковой втулки. Темпе-
Температура термопластов при течении по форме почти не
меняется, а реактопластов и резин несколько уве-
увеличивается, т. к. течение осуществляется медленнее
и материал продолжает нагреваться от горячих сте-
стенок формы.
Затвердевание изделия начинается с поверхности.
При этом термопласты охлаждаются ниже темп-ры
стеклования, после чего изделие можно извлекать из
формы. Отверждение реактопластов сопровождается
повышением темп-ры. Скорость затвердевания материа-
материала в форме зависит от температуры формования, темп-ры
стенок формы, а также от толщины изделия и теплофи-
зич. свойств материала.
Различный характер изменения темп-ры и давления
на отдельных участках формы обусловливает неодина-
неодинаковое протекание физико-химич. процессов в перераба-
перерабатываемом материале и, так. обр., предопределяет
анизотропию свойств полученного изделия (см. об этом
Литье под давлением термопластов).
Кроме давления и темп-ры, на свойства готового
изделия оказывают влияние продолжительность от-
отдельных стадий цикла, а также конфигурация и разме-
размеры формы, свойства материала (вязкость, температуро-
температуропроводность, термостабильность, релаксационные свой-
свойства и др.), особенности его пластикации (характер
изменения темп-ры по объему порции материала, способ
передачи давления в форму и др.).
Математическое описание процесса. Многообразие
физич. и химич. процессов, протекающих при Л. п. д.,
определяет сложность математич. описания данного
метода переработки. Существующие способы описания
базируются, в основном, на использовании упрощенных
ур-ний гидродинамики и теплопередачи с учетом реоло-
гич. свойств материала.
С нек-рым допущением течение полимерного материа-
материала по каналам литниковой втулки и по литьевой форме
может рассматриваться как стационарное изотермиче-
изотермическое, описываемое ур-ниями установившегося ламинар-
ламинарного осесимметричного движения между двумя парал-
параллельными пластинами (для литьевой формы) или по
цилиндрич. каналу (для литника). Протекающие при
этом деформационные процессы характерны для несжи-
несжимаемых (неньютоновских) жидкостей и подчиняются
степенному закону изменения вязкостных свойств.
Теплообмен при течении материала по литьевой форме
рассматривают как одномерный тепловой поток от на-
нагретого материала с темп-рой Тм к охлаждаемой стенке
формы с постоянной темп-рой Тф (для термопластов)
или от нагретой стенки к менее нагретому материалу
(для реактопластов и резиновых смесей).
В общем виде ур-ния движения A), энергии B), рео-
логич. ур-ние вязкой жидкости C) и ур-ние неразрыв-
неразрывности D) для случая одномерного движения записы-
записываются:
дх
ду
дТ
dVx
дх
2- =0
B)
C)
D)
где р — давление, прикладываемое к материалу в на-
направлении х\ тху — напряжение сдвига в направлении
dVx
движения х; q— — градиент скорости в направлении
у; п — индекс течения, учитывающий отклонение
реологич. свойств расплава от ньютоновской жидкости;
75
ЛИТЬЕ ПОД ДАВЛЕНИЕМ РЕАКТОПЛАСТОВ
76
Т — темп-pa материала; а — коэфф. температуропро-
температуропроводности; S — диссипативная функция; \i — вязкость
материала; р — плотность материала; Ср — теплоем-
теплоемкость материала.
Совместное решение этих ур-ний при различных
граничных условиях, отражающих различную поста-
постановку задачи для Л. п. д. аморфных и кристаллич.
термопластов, реактопластов или резиновых смесей,
позволяет приближенно определить скорость движения
материала по литьевой форме, время ее заполнения,
перепад давления по длине формы в период ее запол-
заполнения и др. Различными интерпретациями этих ур-ний
пользуются для решения конкретных задач численны-
численными методами. Однако применение их для более общих
расчетов затрудняется сложностью зависимостей вяз-
вязкостных и теплофизич. свойств расплава от непрерывно
изменяющихся в цикле литья темп-р и напряжений
сдвига.
Для описания формования материала на стадии уп-
уплотнения применяют ур-ние состояния, связывающее
уд. объем (V) материала с температурой (Т) и давле-
давлением (р):
(V-<»){p+M)=KRT E)
где со, М, К — константы ур-ния состояния полиме-
полимера (определены для ряда материалов), R — универ-
универсальная газовая постоянная.
Это ур-ние м. б. использовано для определения изме-
изменения темп-ры и давления материала при постоянной
плотности или для определения усадки (изменение
объема материала) в процессе охлаждения при задан-
заданных значениях давления и темп-ры.
Стадия уплотнения материала характеризуется не-
небольшими градиентами скорости течения расплава
@,1 — 1 сек -1). Изменение плотности (р) материала во
времени (t) с учетом ур-ния E) в общем виде м. б. за-
записано:
ар
dt
1
киту
p+« J
I" RT dp _
дТ
dt
] F)
Ур-ние неразрывности для этого периода формования
имеет вид:
-f+ ^(PFx)=0 G)
Совместное решение ур-ний A) и C) (при значениях
параметров, характерных для процесса уплотнения)
с ур-ниями B) (при допущении одномерности теплово-
теплового потока), F) и G) при соответствующих граничных
условиях позволяет описать поведение материала на
этой стадии. Решение этих ур-ний в общем виде пред-
представляет значительные трудности. Для решения част-
частных задач принимают допущения и наиболее упрощен-
упрощенный характер изменения параметров и свойств материа-
материала в цикле литья.
Упрощенное моделирование теплообмена материала
в литьевой форме, протекающего при его охлаждении
после снятия давления (при допущении постоянства
теплофизич. коэфф. и отсутствия внутренних источни-
источников тепловыделения), проводится с помощью дифферен-
дифференциальных ур-ний теплопроводности с краевыми усло-
условиями первого рода (для неограниченной пластины):
(8)
где 0 — средняя относительная темп-ра;
я - 8
П~~ B711J2'
Решением ур-ния (8) можно определить изменение
темп-ры материала во времени T(t) или найти время т
охлаждения материала от темп-ры Гм, при к-рой он
поступает в форму, до нек-рой заданной темп-ры. При
этом Fo = j?- (a — коэфф. температуропроводности,
h — половина толщины детали).
Приведенные выражения A—8) позволяют проводить
приближенные инженерные расчеты, необходимые при
определении технологич. режимов Л. п. д. (тепловые,
скоростные, силовые), размеров оснастки и параметров
оборудования (усилия впрыска, запирания, площади
литья и др.). Существуют различного рода эмпирич.
соотношения, к-рые упрощают подобные расчеты, одна-
однако требуют знания в каждом конкретном случае харак-
характеристик перерабатываемого материала.
Лит.: Завгородний В. К., Калинчев Э. Л.,
МарамЕ.И., Литьевые машины для термопластов и реакто-
реактопластов, М., 1968; Т о р н е р Р. В., Основные процессы пере-
переработки полимеров, М., 1972; Мак-Келви Д. М., Пере-
Переработка полимеров, пер. с англ., М., 1965; Басов Н. И.,
Скуратов В. К., К и м В. А., Оборудование для про-
производства объемных изделий из термопластов, М., 1972; Л а п-
ш и н В. В., Основы переработки термопластов литьем под да-
давлением, М., 1974. Э. Л. Калинчев, Е. И. Марам.
ЛИТЬЕ ПОД ДАВЛЕНИЕМ РЕАКТОПЛАСТОВ
(injection moulding of thermosets, SpritzguP von Durop-
lasten, moulage par injection des thermodurcissables).
Основные принципы процесса приводятся в ст. Литье
под давлением. Здесь же описана специфика переработ-
переработки этим методом реактопластов, к-рая заключается
в необходимости строгой регулировки темп-ры. Если
темп-pa пластикации превышает оптимальную, происхо-
происходит отверждение материала еще до заполнения литьевой
формы. При темп-ре ниже оптимальной реактопласт
плавится долго и затвердевает, не превратившись в рас-
расплав с необходимой вязкостью (для нагнетания в фор-
форму высоковязкого расплава усилие впрыска, создавае-
создаваемое машиной, недостаточно).
Особенностью Л. п. д. р. является также то, что ма-
материал подается в обогреваемую форму, а не в охлаж-
охлаждаемую, как при литье термопластов. Условия Л^
п. д. р. приведены в таблице.
Литье на червячных литьевых машинах — наиболее
эффективный и экономичный метод переработки реакто-
реактопластов, позволяющий осуществить высококачествен-
высококачественную пластикацию материала и быстро отвердить его
в форме, предотвратив коробление готового изделия.
Метод позволяет получать изделия массой до 2—3 кг
с большой поверхностью и разнотолщинностью стенок.
По физико-механическим характеристикам такие из-
изделия не уступают изделиям, отформованным др. ме-
методами.
В поршневых литьевых машинах пластикация проис-
происходит только за счет тепла, подводимого к материалу
от стенок цилиндра. В этом случае не удается добиться
равномерного прогрева материала по всей массе и гомо-
гомогенизации расплава. В связи с указанными недостат-
недостатками разработан метод Л. п. д. р. на поршневых маши-
машинах (т. н. струйное формование), при к-ром
пластицированный реактопласт нагнетается в форму
через обогреваемое сопло. Проходящий по такому соп-
соплу материал быстро нагревается и с большой скоростью
заполняет форму. После окончания впрыска давление
в цилиндре снижается, а сопло охлаждается водой.
Этот метод позволяет получить на короткое время
расплав реактопласта пониженной вязкости, что дает
возможность отливать мелкие изделия с тонкой арма-
арматурой.
Л. п. д. р. начало осуществляться в 1960-е гг. поч-
почти одновременно в Великобритании, США, ФРГ и
Японии.
Лит.: Dietz R. G., Kunststoff — Berater, 15, № 8, 745
A970); 3 а в г о р о д н и й В. К., К а л и н ч е в Э. Л., Ма-
Марам Е. И., Литьевые машины для термопластов и реакто-
реактопластов, М., 1968; Woebcken W., в кн.: Kunststoff —
77
ЛИТЬЕ ПОД ДАВЛЕНИЕМ РЕЗИНОВЫХ СМЕСЕЙ
Условия переработки реактопластов литьем под давлением на червячных машинах
78
Связующее
Фенол о-формальдегидное
То же
То же
Наполнитель
Древесная мука
Асбест
Каучук
Темп-pa цилиндра, °С
на входе
60-70
60 — 70
60 — 70
на выходе
85-100
95
95
Темп-ра
сопла, °С
85 — 100
105
105
Темп-ра
расплава
на выходе
из сопла,
°С
110-120
110-115
120
Давление впрыска,
Мн/м2 (кгс/см2)
60-120 F00-1200)
120-150 A200 — 1500)
60-120 F00-1200)
Темп-ра
формы,
°С
160 — 180
170-185
160 — 180
Мочевино-формальдегидное
Меламино-формалъдегидное
То же
Полиэфирное
То же
Древесная мука
Древесная мука
Асбест
Стекловолокно
Асбест
55-65
55-65
50-60
60 — 80
85 — 100
90-100
80-90
60-80
100-110
100-110
90-100
60-80
115-120
115-120
100 — 115
70-90
70-130 G00-1300)
70 — 120 G00 — 1200)
100-160 A000-1600)
90-120 (900-1200)
150-165
150 — 165
160 — 170
170-190
Примечание. Давление пластикации во всех случаях не превышает 30 Мн/м2 C00 кгс/см*), частота вращения червяка
ок. 50 об/мин, длительность пребывания материала в инжекционном цилиндре 4—20 сек, продолжительность впрыска 2—7 сек.
Handbuch, Bd 10, Munch., 1968, S. 689; S с h a a f W., H a h-
n e m a n n A., Verarbeitung von Plasten, Lpz., 1968; 3 а в г o-
родний В. К., Механизация и автоматизация переработ-
переработки пластических масс, 3 изд., М., 1970; Вальковский
Д. Г., в сб.: Новое в переработке полимеров. Сб. переводов и
обзоров из иностр. периодич. литературы, М., 1969, с.45.
ЛИТЬЕ ПОД ДАВЛЕНИЕМ РЕЗИНОВЫХ СМЕ-
СМЕСЕЙ (injection moulding of rubber mixtures, SpritzguB
von Gummimischungen, moulage par injection des melan-
melanges de caoutchouc). Основные принципы процесса при-
приводятся в ст. Литье под давлением. В данной статье
описана специфика переработки этим методом резино-
резиновых смесей. Литье под давлением применяют для пере-
переработки резиновых смесей в фасонные изделия, гл.
обр. толстостенные (иногда с металлич. арматурой).
При этом вулканизация осуществляется в подогревае-
подогреваемой литьевой форме. При литье резиновых смесей мож-
можно применять литьевые машины, разработанные для пе-
переработки реактопластов. Однако лучшие результаты
дает использование специальных машин, характери-
характеризующихся след. параметрами: 1) длина червяка пласти-
катора не более 12—15 D (D — диаметр); 2) степень
сжатия материала в инжекционном цилиндре не более
1,25—1,50; 3) давление впрыска 60—170 Мн/м2 F00—
1700 кгс/см2); 4) уд. усилие смыкания литьевой формы
18—25 Мн/м2 A80—250 кгс/см2); 5) темп-ра формы
до 220 °С. Желательно также, чтобы литьевая ма-
машина имела зону декомпрессии для удаления летучих
продуктов в процессе переработки.
При течении резиновой смеси через сопло литьевого
устройства и литниковую систему формы давление те-
теряется и в гнездах литьевой формы оно составляет,
как правило, 50% от давления впрыска. Темп-ра мате-
материала в момент окончания его подачи в форму на
10—30 °С ниже темп-ры стенок формы. Материал со-
соприкасается с горячими стенками формы, нагревается
в ее замкнутой полости и его давление внутри гнезд
формы возрастает, достигая 100 Мн/м2 A000 кгс/см2).
Обычно зазор по плоскости разъема формы составляет
0,01—0,02 мм и при таком давлении резиновой смеси
невозможно избежать образования облоя. Прецизион-
Прецизионные формы, обеспечивающие получение безоблойных
изделий, дороже обычных в ~5 раз.
При конструировании форм следует учитывать зна-
значительную усадку материала, к-рая обычно на 20—25%
выше усадки резиновых смесей при прессовании. Диа-
Диаметр литниковых каналов в форме должен быть мини-
минимальным (обычно 3—4 мм) для уменьшения потерь
резиновой смеси, но достаточным для того, чтобы
гнезда успели заполниться за время впрыска.
Резиновая смесь для литья должна обладать след.
свойствами: 1) небольшой склонностью к подвулкани-
зации при повышенных темп-pax (время начала под-
вулканизации по Муни не менее 10 мин при 120 °С);
2) способностью к быстрой вулканизации по окончании
впрыска материала в литьевую форму; 3) способностью
к резкому разогреванию вследствие внутреннего тре-
трения в материале и трения о стенки сопла; 4) легкостью
истечения через сопло диаметром 3—4 мм при опреде-
определенной вязкости.
Для литья резиновых изделий широкого ассорти-
ассортимента лучше всего подходят резиновые смеси с вязко-
вязкостью по Муни 40—60 (при 100 °С). Смеси с вязкостью
выше 80—100 перерабатывать литьем нецелесообразно,
т. к. в этом случае необходимо значительно увеличивать
диаметр литниковых каналов, что приводит к повыше-
повышению отходов производства. Смеси с вязкостью ниже
20—30 плохо прогреваются в инжекционном цилиндре
и сопле литьевой машины.
Вязкость смесей можно уменьшить, понижая коли-
количество наполнителя в смеси (если это допустимо с точки
зрения эксплуатационных требований, предъявляемых
к полученному изделию) или вводя в смесь пластифи-
пластификатор. Даже небольшое количество последнего E—
10 мае. ч.) заметно улучшает литьевые свойства резино-
резиновых смесей. Хорошим пластификатором для резиновых
смесей на основе большинства неполярных каучуков
является низкомолекулярный полиэтилен. Кроме того,
для смесей на основе бутадиен-нитрильных каучуков
применяют стабилизированный поливинилхлорид, а для
смесей на основе бутадиен-стирольных каучуков —
полистирол. Вместе с тем введение в смесь плас-
пластификатора, как правило, ухудшает ее адгезион-
адгезионные свойства. Поэтому при изготовлении резинометал-
лич. изделий лучше снижать вязкость смеси, повышая
темп-ру в инжекционном цилиндре литьевой машины
или скорость вращения червяка.
Подбор вулканизующих систем для резиновых сме-
смесей, перерабатываемых литьем, должен быть особенно
тщательным, т. к. темп-ра в инжекционном цилиндре
машины достигает 120 °С, что вызывает опасность
подвулканизации. Рекомендуются ускорители вулкани-
вулканизации, обладающие замедленным действием в началь-
начальной стадии процесса и обеспечивающие высокую ско-
скорость вулканизации в литьевой форме. Хорошие ре-
результаты дают системы, содержащие сульфенамиды,
напр, комбинация сера — сантокюр — тиурам. Такая
система обеспечивает завершение вулканизации в литье-
литьевой форме за 50—70 сек. В то же время резиновые смеси,
содержащие только тиурам, склонны к подвулканизации
при высоких темп-pax. Кроме того, в резиновые смеси
можно ввести замедлители подвулканизации: органич.
к-ты, их ангидриды (напр., фталевый), соли, имиды и др.
Наилучшей литьевой способностью обладают рези-
резиновые смеси на основе изопреновых каучуков. Далее,
в порядке ухудшения литьевых свойств, следуют смеси
на основе мягких типов хлоропреновых и бутадиен-
нитрильных каучуков, бутадиен-стирольных, бутадиено-
бутадиеновых, жестких типов хлоропреновых и бутадиен-ни-
бутадиен-нитрильных каучуков и фторкаучуков.
79
ЛИТЬЕ ПОД ДАВЛЕНИЕМ ТЕРМОПЛАСТОВ
80
К основным преимуществам литья под давлением
перед прессованием резиновых смесей относятся: 1) воз-
возможность более равномерной вулканизации мас-
массивных изделий при высоких температурах (до 200 °С);
2) отсутствие стадии приготовления заготовки опреде-
определенной конфигурации и заданного объема (или массы);
3) снижение отходов резины до 5—15%; 4) большая
однородность свойств изделий по толщине, более высо-
высокая прочность при изгибе и растяжении, что объясняет-
объясняется равномерным прогревом материала при переработке;
5) возможность автоматизации процесса. Литье под да-
давлением не применяют для изготовления резино-ткане-
вых изделий, а также тонких изделий с большими
поверхностями (напр., листов, лент).
Лит.: Достижения науки и технологии в области резины.
Сб. статей, под ред. Ю. С. Зуева, М., 1969; Шварц А. И.,
Анализ процесса литьевого формования и выбор оптимальных
параметров при работе на литьевых машинах червячно-плун-
жерного типа, М., 1971; Ш в а р ц А. И., К о н г а р о в Ю. С,
Состояние и перспективы развития производства резиновых
изделий литьевым методом, М., 1968; Технико-экономические
основы проектирования современного процесса формования-
вулканизации резинотехнических изделий, М., 1969; 3 а-
харьев Г. А., Жданов И. М., Основные виды современ-
современного оборудования для литья резин под давлением и некоторые
рекомендации по его проектированию и подбору, М., 1968;
В о о t h D. A., Rev. gener. caout. et plast., 44, № 3, 331 A967).
И. М. Жданов.
ЛИТЬЕ ПОД ДАВЛЕНИЕМ ТЕРМОПЛАСТОВ (in-
(injection moulding of thermoplastics, SpritzguB von The-
rmoplasten, moulage par injection des thermoplastiqu-
es). Основные принципы процесса приводятся в ст. Литье
под давлением. В данной статье описана в основном спе-
специфика переработки этим методом термопластичных
материалов.
Физико-химические процессы, протекающие при ли-
литье термопластов. Пластикация полимера в материаль-
материальном цилиндре литьевой машины сопровождается пере-
переходом материала в вязкотекучее состояние. Гомогени-
Гомогенизация расплава завершается при течении полимера
с высокой скоростью через сопло, когда вследствие зна-
значительных сдвиговых напряжений темп-pa расплава
дополнительно повышается. Одновременно в сопле
происходит ориентация макромолекул и надмолеку-
надмолекулярных образований, к-рая продолжается при течении
расплава полимера в литьевой форме. При заполнении
формы макромолекулы ориентируются в направлении
движения потока материала, причем степень ориен-
ориентации растет с увеличением сдвиговых напряжений,
т. е. с увеличением давления литья, скорости заполне-
заполнения формы и с уменьшением сечения полости формы.
Ориентация сопровождается упрочнением материала
в направлении ориентации, что, при соответствующей
конструкции формы, позволяет получать изделия с по-
повышенной прочностью тех частей, к-рые несут наиболь-
наибольшую нагрузку в процессе эксплуатации.
Вследствие расширения потока расплава термопласта
в форме перпендикулярно направлению течения в нем
возникают соответствующие ориентационные напряже-
напряжения. Оба указанных процесса ориентации происходят
одновременно и, складываясь, могут привести к двух-
двухосной ориентации материала в изделии. При этом сте-
степень ориентации уменьшается по мере удаления от вход-
входного отверстия формы, что обусловливает анизотропию
свойств изделия в направлении течения. Различие
в степени ориентации по длине и в поперечном сечении
изделий приводит к возникновению внутренних оста-
остаточных напряжений, к-рые могут привести к деформа-
деформации изделий, их растрескиванию и др.
Др. причина возникновения внутренних остаточных
напряжений в изделиях из термопластов, полученных
литьем под давлением,— различия в скоростях и сте-
степени охлаждения материала в поверхностных и внут-
внутренних слоях. При соприкосновении с холодными
стенками формы полимер быстро затвердевает. Темп-ра
во внутренних слоях материала из-за низкой тепло-
и температуропроводности полимеров остается более
высокой, поэтому внутри изделия процессы релаксации
и структурообразования успевают пройти полнее.
Так, если поверхностные слои литых изделий из кри-
кристаллизующихся термопластов обычно аморфны или
имеют мелкокристаллич. структуру, то центральные
слои, особенно в толстостенных изделиях, характери-
характеризуются более высокой степенью кристалличности и
большими по размеру кристаллич. образованиями. Это
приводит к возникновению в материале термич. на-
напряжений и обусловливает его структурную неоднород-
неоднородность, что также отрицательно сказывается на проч-
прочностных и эксплуатационных свойствах изделий.
Ориентационные напряжения в готовом изделии без
изменения его конфигурации и размеров уменьшить
не удается, поэтому при разработке конструкции формы
и выборе режимов литья необходимо принимать меры
для уменьшения степени ориентации материала в форме.
Термич. напряжения можно снизить либо уменьше-
уменьшением перепада темп-р между материалом и формой,
либо прогревом готовых изделий (см. Термическая об-
обработка). В случае кристаллизующихся полимеров
одновременно происходит дальнейшая кристаллиза-
кристаллизация и повышается однородность структуры.
В ходе процесса под действием высоких темп-р и зна-
значительных механич. напряжений в полимере могут
происходить деструктивные процессы, сопровождаю-
сопровождающиеся выделением летучих продуктов, изменением цве-
цвета материала и др. Поэтому при литье под давлением
ряда термически нестабильных полимеров необходимо
использовать эффективные стабилизаторы.
При охлаждении изделия в форме и после его извле-
извлечения из нее наблюдается усадка — уменьшение линей-
линейных размеров изделия. Величина усадки зависит от
сжимаемости расплава полимера, его коэфф. объемного
термич. расширения, скорости релаксации и степени
кристалличности. Усадка в форме частично компенси-
компенсируется ее подпиткой расплавом до момента затвер-
затвердевания литника; поэтому материал усаживается
гл. обр. после извлечения изделия. Ориентация макро-
макромолекул и надмолекулярных образований при литье,
а также неравномерность охлаждения изделия в форме
обусловливают анизотропию усадки вдоль и поперек
направления течения расплава, на участках изделия
с различной толщиной и т. д. Анизотропия усадки осо-
особенно заметна у кристаллизующихся полимеров. Ма-
Малая скорость релаксации и кристаллизации приводит
к тому, что усадочные явления развиваются в течение
длительного времени, что препятствует получению изде-
изделий со стабильными размерами.
Введение в полимерные материалы струкпгурообра-
зователей приводит к формированию надмолекулярных
структур с повышенной стабильностью и однородно-
однородностью, что в конечном итоге может обеспечить изотро-
изотропию свойств литьевых изделий. Аналогичное влияние
оказывает термич. обработка готовых изделий, но она
значительно менее эффективна.
Режимы переработки. Режимы переработки распрост-
распространенных термопластов приведены в табл. и на рис.
Полистирол можно перерабатывать в широ-
широком диапазоне темп-р (от 150 до 250 СС). В этом диапазоне
он обладает хорошей текучестью, благодаря чему хо-
хорошо перерабатывается не только на червячных, но и
на поршневых машинах. Полистирол не нуждается
в предварительной сушке, однако предварительный по-
подогрев материала в бункере до 50—70 °С способствует
повышению производительности машины и улучшению
качества изделий.
Полиэтилен хорошо перерабатывается литьем
под давлением. При охлаждении полиэтилена в форме
происходит его кристаллизация. Степень кристаллич-
кристалличности влияет на твердость, прочность изделий и ха-
характер их деформации.
81
ЛИТЬЕ ИОД ДАВЛЕНИЕМ ТЕРМОПЛАСТОВ
82
328
280
240
Поликарбонат
Полиформальдегид
Полиэтилен высокой плотности
280
240
200
1601
Винипласт
Полистирол
240
200
160
240
200
160
Полианрилаты
%
.. j—-и-—-н"—Hi
Температурные области литья под давлением нек-рых тер-
термопластов. Заштрихованная зона — возможная область
переработки; жирная линия — оптимальный температурный
режим.
Поликарбонат в области темп-р переработки
отличается высокой вязкостью и термич. стабильностью.
При изготовлении изделий из поликарбоната (особен-
(особенно с низкой мол. массой) с повышением темп-ры литья
снижаются прочность изделий при растяжении и отно-
относительное удлинение. Поэтому при производстве слож-
сложных изделий со строгими размерными допусками литье
необходимо осуществлять при пониженной темп-ре
расплава и повышенной темп-ре формы (не менее 100 °С).
Перед загрузкой в бункер машины поликарбонат под-
подсушивают при 70—75 °С в течение 4—6 ч (если мате-
Нек-рые параметры переработки и усадка термопластов
при литье под давлением
Материал
Полистирол . . .
Полистирол уда-
ударопрочный . . .
Полиэтилен
низкой плот-
плотности
высокой плот-
плотности
Поликарбонат
Полиформальде-
Полиформальдегид
Полипропилен .
Полиакрилаты .
Поливинилхло-
рид
пластифициро-
пластифицированный ....
жесткий ....
Полиамиды . . .
Давление впрыска,
Мн/м2 (кгс/см2)
40-60 D00-600)
60-100 F00-1000)
50-100 E00-1000)
90 — 110 (900-1100)
80-120 (800-1200)
80 — 120 (800—1200)
80-140 (800—1400)
80-150 (800-1500)
50-90 E00—900)
80-150 (800-1500)
80 — 100 (800—1000)
Темп-ра
формы, °С
20-40
50-55
40-70
20-60
—
65—75*
40-70
45-65
20-60
40-50**
60 — 120
Усадка,
0/
/о
0,2 — 0,5
0,2-0,5
1-3
1 — 3
0,5-0,8
1 ,5 — 3,5
1,0-2,5
^1,5
<1
0,4 — 0,7
-2
* Для нек-рых
70-80 °С.
марок 90 —120 СС. ** Для нек-рых марок
риал был плотно упакован) или при 120—130 °С в те-
течение 12—20 ч (если материал был влажным). Влаж-
Влажность поликарбоната не должна превышать 0,05%.
Полиформальдегид очень чувствителен
к действию высокой темп-ры. Время его пребывания
при высокой темп-ре должно быть ограничено, иначе
изменяется окраска материала и выделяются пары фор-
формальдегида. Расплав полимера имеет низкую теку-
текучесть; скорость впрыска при заполнении формы должна
быть максимально возможной.Полиформальдегид отли-
отличается хорошими механяч. свойствами при повышен-
повышенных темп-pax, поэтому отлитые из него изделия можно
извлекать из формы в горячем состоянии.
Полипропилен хорошо перерабатывается
литьем под давлением в тонкостенные изделия; при из-
изготовлении же толстостенных изделий возможна их
значительная деформация после извлечения из формы.
Вязкость расплава полипропилена в большей степени
зависит от градиента скорости сдвига, чем от темп-ры.
Поэтому при литье толстостенных изделий и изделий
сложной конфигурации лучших результатов можно
достичь, повышая давление впрыска, а не темп-ру
расплава. Полипропилен склонен к образованию уса-
усадочных раковин в толще материала, поэтому выдержку
его в форме следует осуществлять под возможно бо-
более высоким давлением, а также применять формы,
конструкция которых препятствует образованию
раковин.
Полиакрилаты иполиметакрилаты
трудно перерабатывать литьем под давлением, т. к.
темп-ры их размягчения и разложения близки, а вяз-
вязкость расплавов высока. Из-за чувствительности этих
полимеров к изменению темп-ры переработки необхо-
необходим строгий контроль режима пластикации. При
охлаждении полиакрилатов и полиметакрилатов вяз-
вязкость их расплавов быстро растет, поэтому давление
при заполнении материалом формы должно быть вы-
высоким. Эти полимеры перед переработкой желательно
подсушивать при 65—90 °С в течение 2—3 ч. Для умень-
уменьшения внутренних напряжений изделия из полиакри-
полиакрилатов и полиметакрилатов подвергают термообработке
при 75—85 °С в течение 2 ч в термошкафах.
Литье под давлением жесткого полив и-
нилхлорида (винипласта) затруднено из-за низ-
низкой теплопроводности этого материала и небольшой
разницы между темп-рами плавления и начала разло-
разложения. При разложении этого материала выделяется
HG1, к-рый вызывает коррозию частей литьевой маши-
машины. Кроме того, экзотермич. реакция разложения ме-
мешает поддержанию необходимого темп-рного режима.
При литье жесткого поливянилхлорида скорость впры-
впрыска должна быть максимально возможной.
Полиамиды перед литьем подсушивают в тер-
термошкафу при 70—80 °С в течение 4—5 ч. Время пре-
пребывания полимера в материальном цилиндре должно
быть ограничено, т. к. при нагревании выше 80 °С
он окисляется. Расплавы полиамидов имеют очень
низкую вязкость, что позволяет отливать из них изде-
изделия сложной конфигурации. Для предотвращения
самопроизвольного вытекания расплава из сопла
литьевой машины оно снабжается запорным кла-
клапаном, открывающимся при определенном давлении
впрыска.
Литье термопластов (целлулоида) применяется
в пром-сти с последней четверти 19 века, однако ши-
широкое распространение этот метод получил только
в 1940-е гг., когда был создан необходимый парк
поршневых литьевых машин. С 1950-х гг. поршневые
литьевые машины заменяются червячными, литье на
к-рых стало важнейшим методом переработки термопла-
термопластов.
Лит.: Завгородний В. К., Калинчев Э. Л.,
М а р а м Е. И., Литьевые машины для термопластов и реакто-
пластов, М., 1968; Бернхардт Э. [сост.], Переработка
83
ЛИТЬЕ ЦЕНТРОБЕЖНОЕ
84
термопластичных материалов, пер. с англ., М., 1965; 3 а в г о-
р о д н и й В. К., Механизация и автоматизация переработки
пластических масс, 3 изд., М., 1970; Новое в переработке по-
полимеров. Сб. переводов и обзоров из иностранной периодической
лит-ры, М., 1969; Завгородний В. К., КалинчевЭ. Л.,
Махаринский Е. Г., Оборудование предприятий по пере-
переработке пластмасс, Л., 1972. В. К. Завгородний.
ЛИТЬЕ ЦЕНТРОБЕЖНОЕ — см. Центробежное фор-
формование.
ЛИТЬЕВОЕ ПРЕССОВАНИЕ, трансферное
прессование (transfer moulding, Spritzpressen,
moulage par transfert) — разновидность прессования
реактопластов, при к-рой пластицированный материал
впрыскивается в предварительно замкнутую прессфор-
Рис. 1. Схема процессов формования изделий из реакто-
реактопластов (а— компрессионное прессование; б—литьевое
прессование): 1 — верхний плунжер; 2 — оформляющие
гнезда прессформы; з — перерабатываемый материал; 4 —
поршень; 5 — трансферный цилиндр; б — загрузочное от-
отверстие; 7 — изделие; 8 — литниковые каналы; 9 —
прессформа.
му перемещающимся в осевом направлении поршнем
(в отличие от компрессионного прессования, при к-ром
материал загружается в полость открытой прессфор-
прессформы — рис. 1).
Для Л. п. применяют специальные трансферные гид-
гидравлич. прессы с двумя (верхним и нижним) рабочими
плунжерами или универсальные прессы с одним верх-
верхним плунжером. В первом случае (см. рис. 1,6) пред-
предварительно нагретый или ненагретый материал 3
загружают через отверстие 6 в камеру трансферного
цилиндра 5 при опущенном поршне 4, и прессформу
замыкают верхним плунжером 1. Материал 3 пластици-
руется за счет нагревания от горячих стенок трансфер-
трансферного цилиндра 5 и под действием поршня 4, соединен-
соединенного с нижним рабочим плунжером (на рис. не показан),
нагнетается в оформляющие гнезда 2 прессформы через
литниковые каналы 8. После отверждения изделия
прессформа раскрывается и
отформованные изделия с лит-
литниками выталкиваются порш-
поршнем 4 из прессформы, пор-
поршень опускается в исходное
положение, а камера транс-
^5 ферного цилиндра вновь за-
загружается материалом.
Рис. 2. Схема установки для ли-
литьевого прессования на универ-
универсальном прессе: 1 — поршень;
2 — камера; з—4 — полу матрицы;
5 — обойма; б — выталкиватель;
7 — оформляющее гнездо пресс-
формы; 8 — литниковый канал.
При Л. п. с помощью универсальных рамных или ко-
колонных прессов (рис. 2) на столе (нижней плите) прес-
пресса устанавливают обогреваемую обойму 5, в к-рую встав-
вставляют съемные полуматрицы 3 и 4. Прессматериал за-
загружают в камеру 2 и после нагрева до необходимой
темп-ры нагнетают поршнем 1 в гнезда прессформы.
После отверждения изделие выталкивают вместе с
матрицей выталкивателем 6.
Камеру трансферного цилиндра удобнее загружать
материалом не вручную, а с помощью червячного пла-
стикатора, к-рый соединен непосредственно с трансфер-
ным цилиндром или может перемещаться в зону разъе-
разъема прессформы для загрузки материала (рис. 3). При
этом трансферный цилиндр загружается однородно
пластицированным материалом, что обеспечивает по-
повышение производительности оборудования и качества
отпрессованных изделий. Подъем и опускание поршня 3
осуществляется установленным в гидравлич. цилиндре 2
плунжером 1. Под давлением материала, пластицируе-
а
Рис. 3. Схема трансферного пресса с двумя вариантами
загрузки материала (а — пластикатор соединен непосредст-
непосредственно с трансферным цилиндром; б — пластикатор может
перемещаться в зону разъема прессформы): 1 — плунжер;
2 — гидравлич. цилиндр; з — поршень; 4 — трансферный
цилиндр; 5 — оформляющие гнезда прессформы; 6 — лит-
литниковые каналы; 7 — прессформа; 8 — цилиндр пластика-
тора; 9 — червяк; ю — шибер; 11 — плунжер гидравлич.
цилиндра привода шибера.
мого вращающимся в обогреваемом цилиндре 8 червяч-
червячком Р, последний перемещается вправо. После накопле-
накопления необходимой для впрыска дозы материала червяк
перемещается влево под действием плунжера 1 гидрав-
гидравлич. цилиндра 2 и нагнетает пластицированный мате-
материал в полость трансферного цилиндра 4 (см. рис. 3,а).
Выходной торец перемещающегося червячного пла-
стикатора (см. рис. 3, б) закрыт шибером 10, к-рый
открывается только на время подачи материала из
пластицирующего цилиндра в трансферный. Пласти-
Пластикация новой порции материала осуществляется в от-
отведенном в сторону пластикаторе.
Поскольку при Л. п. материал впрыскивается в замк-
замкнутую форму, на отформованном изделии отсутствует
грат (заусеницы) по полости разъема формы. Прибыль
и литники удаляют из формы и материального ци-
цилиндра после каждого цикла.
Л. п. осуществляют при высоких уд. давлениях
150—200 Мн/м2 A500—2000 кгс/сль2), что в 5 — 10 раз
выше, чем при компрессионном прессовании. При этом
давление в прессформе составляет 50—65 Мн/м2 E00—
650 кгс/см2).
Благодаря высоким давлениям и скоростям впрыска
внутренние слои материала прогреваются лучше, чем
при компрессионном прессовании. Материал дополни-
дополнительно прогревается при соприкосновении с горячей
металлич. поверхностью также в литниковых каналах
и за счет внутреннего трения.
Предварительная пластикация материала и высокая
скорость впрыска позволяют значительно повысить
темп-ру процесса и сократить длительность отвержде-
отверждения материала в прессформе приблизительно в 2 раза
по сравнению с компрессионным прессованием. При
Л. п. удается изготовить изделия сложной конфигура-
конфигурации с недостаточно жесткой арматурой в прессформах
с тонкими перегородками и деталями.
При Л. п. необходимо строго регулировать темп-ру
пластикации. Если темп-pa превышает оптимальную,
отверждение материала происходит еще до заполнения
85
ЛИТЬЕВЫЕ МАШИНЫ
86
прессформы. При темп-ре ниже оптимальной реакто-
пласт плавится долго и затвердевает, не превратившись
в расплав с необходимой вязкостью. Это затрудняет или
делает невозможным впрыск материала в прессформу
даже при повышенных давлениях.
Наиболее пригодны для Л. п. реактопласты на основе
новолачных феноло-формальдегидных смол, расплавы
к-рых имеют малую вязкость и сравнительно долго
не отверждаются при темп-ре пластикации.
При использовании обогреваемых тиглей или транс-
ферных цилиндров, в к-рых пластикация происходит
только за счет тепла внешних нагревателей, очень
важен выбор оптимальной глубины заполнения тигля.
На практике установлено, что при загрузке в тигель
ненагретого материала глубина заполнения не должна
превышать 0,5 D, т. к. в этом случае достигается опти-
оптимальное с точки зрения теплообмена соотношение по-
поверхности теплообмена и объема нагреваемого -мате-
-материала. При предварительном нагреве таблетиро-
ванного материала глубина м. б. увеличена до ID,
однако в этом случае давление впрыска должно быть
повышено до ~200 Мн/м2 B000 кгс/см2). В том случае,
если трансферный цилиндр загружается пластициро-
ванным материалом из червячного пластикатора, глу-
глубина заполнения цилиндра м. б. больше. Однако при
чрезмерном увеличении глубины заполнения часть
материала отверждается раньше, чем вся доза пройдет
через литниковые каналы.
Из описанных выше особенностей Л. п. следует, что
этот метод переработки пластмасс занимает промежу-
промежуточное место между прессованием и литьем под давле-
давлением. Основное отличие Л. п. от литья под давлением
заключается в том, что в этом методе переработки
для изготовления отливки используется весь объем
пластицированного материала. Благодаря этому Л. п.
могут быть переработаны реактопласты, расплавы к-рых
могут находиться в вязкотекучем состоянии лишь срав-
сравнительно короткое время. С помощью Л. п. можно изго-
изготовлять изделия со сложной арматурой, к-рую невоз-
невозможно установить в литьевой форме.
Лит.: Завгородний В. К., Механизация и автома-
автоматизация переработки пластических масс, 3 изд., М., 1970;
Брацыхин Е. А., Мин длин С. С, С т р е л ь-
ц о в К. Н., Переработка пластических масс в изделия, М.— Л.,
1966; Гиберов 3. Г., Механическое оборудование заводов
пластических масс, М., 1967; Завгородний В. К., К а-
линчев Э. Л., Махаринский Е. Г., Оборудование
предприятий по переработке пластмасс, Л., 1972.
В. К. Завгородний.
ЛИТЬЕВЫЕ МАШИНЫ для полимерных
материалов (injection moulding machines,
Sprit zguflmaschinen, presses d'injection) — машины, ос-
основной рабочей частью к-рых является инжекционный
цилиндр, в к-ром перерабатываемый материал размяг-
размягчается (пластицируется) и под действием червяка или
поршня нагнетается в литьевую форму. Л. м. применя-
применяют для литья под давлением пластмасс и резиновых
смесей. Принцип работы Л. м. описан в ст. Литье под
давлением. Там же представлена схема червячной Л. м.
Машина поршневого типа показана на рис. 1.
В зависимости от расположения инжекционного ме-
механизма (см. ниже) Л. м. делят на горизонталь-
горизонтальные, вертикальные, угловые и комбинированные.
В отдельную группу принято выделять роторные Л. м.,
машины для литья двух- и многоцветных изделий и
нек-рые др. специфич. конструкции.
Машины для литья термопластов. Для переработки
термопластов выпускаются машины с объемом одной
отливки от 0,5 до 30 000 см3. Наибольшее распростра-
распространение нашли горизонтальные Л. м. с объемом отливки
30—125 см3 -(таблица). Л. м., предназначенные для вы-
выпуска изделии самого различного объема (от 15—20 до
1000—2000 см3), обычно являются универсальными и
пригодны для переработки различных термопластов в
разнообразные изделия.
12
Рис. 1. Литьевая машина поршневого типа: 1 —гидравлич.
цилиндр; 2 — плунжер; 3 — подвижная плита; 4 —литье-
—литьевая форма; 5 — неподвижная плита; 6 — сопло; 7 — тор-
торпеда; 8 — инжекционный цилиндр; 9 — бункер; ю —
поршень; 11 — плунжер; 12 — гидравлич. цилиндр; 13 —
электрообогрев.
Для производства изделий с вставными деталями и
арматурой более удобны вертикальные и угловые Л. м.
с вертикально расположенным механизмом смыкания
формы. Угловые машины позволяют осуществлять ин-
жекцию полимера как в плоскость разъема формы, так
и в перпендикулярном направлении. Это значительно
расширяет возможности выпуска изделий с арматурой
и сложной конфигурации.
Основными узлами Л. м., от конструкции к-рых за-
зависит выбор перерабатываемого материала и объема
формуемого изделия, являются инжекционный ме-
механизм и механизм замыкания (размыкания) формы.
В поршневых Л. м. пластикацию осуществляют в ин-
жекционном цилиндре гл. обр. за счет тепла внешних
нагревателей (см. рис. 1). Более совершенна конструк-
конструкция червячного инжекционного механизма, в к-ром
полимер нагревается также в результате деформации
сдвига, возникающей при вращении червяка.
Для предотвращения обратного течения пластициро-
пластицированного полимера по винтовым каналам червяка во
время впрыска на головке червяка устанавливают об-
обратный клапан или головку.
Применение червячной системы пластикации позво-
позволяет снизить мощность электрообогрева Л. м. и исклю-
исключает местные перегревы и глубокую деструкцию поли-
полимера. Червячные Л. м., в отличие от поршневых, обычно
не имеют торпед (см. рис. 1), т. к. равномерный прогрев
материала достигается и без них. Замена поршневого
инжекционного механизма червячным значительно по-
повышает возможную мощность машины и улучшает ка-
качество изделий. Кроме того, более тщательная пласти-
пластикация полимера по всему объему дает возможность
уменьшить усилие впрыска и, соответственно, снизить
усилие замыкания формы.
В мощных Л. м. требуется прогреть значительные
количества полимера, чего практически нельзя добиться
в поршневом инжекционном цилиндре. Поэтому часто
поршневые Л. м. (иногда и червячные) снабжают до-
дополнительным устройством (предпластикато-
р о м), обеспечивающим предварительный нагрев ма-
материала до темп-ры на 30—50 °С ниже темп-ры литья.
Предпластикатор представляет собой горизонтальную
или наклонную цилиндрич. камеру с обогреваемыми
стенками. Из этой камеры полимер в пластицирован-
ном состоянии поршнем или червяком подается в ин-
инжекционный цилиндр.
Конструкция сопла Л. м. определяется свойством
расплава перерабатываемого материала. Для боль-
большинства термопластов, напр, полиолефинов или поли-
полистирола, применяют сопло со скользящим клапаном
(рис. 2,а). При упоре сферич. поверхности сопла 1
в литниковую втулку 5 формы сжимается пружина 7
и канал сопла соединяется с инжекционным цилинд-
цилиндром. После отвода сопла от формы скользящий клапан
2 под давлением расплава перемещается влево и пере-
перекрывает доступ материалу из инжекционного цилиндра
в выходной канал сопла.
87
ЛИТЬЕВЫЕ МАШИНЫ
7 6
Нек-рые полимеры, напр, полиамиды, обладают на-
настолько низкой вязкостью расплава, что могут самопро-
самопроизвольно вытекать из сопла со скользящим клапаном.
Для литья таких полимеров применяют сопло с иголь-
игольчатым клапаном (рис. 2,6), к-рый открывается только
в том случае, когда давление расплава полимера в по-
полости сопла достигает заданной величины. При этом
пружина 7 сжимается, и запорная игла 2 отходит от
канала.
Открытое сопло с конич. головкой (рис. 2, в) предназ-
предназначено для формования изделий из полимеров с высокой
вязкостью расплава, напр. поливинил хлорида. Сопло
такой конструкции не имеет застойных зон и исклю-
исключает термодеструкцию полимера.
Для замыкания (т. е. смыкания и запирания)
иразмыкания формы применяют в основном
гидромеханич, рычажные или гидравлич. ступенчатые
механизмы. В первом случае (рис. 3) перемещение пли-
плиты 5 с одной полуформой 6 осуществляется под дейст-
действием гидравлич. цилиндра 8 и поршня, шток 7 к-рого
соединен с осью рычагов 2, 3. Для регулировки длины
рычагов в зависимости от толщины устанавливаемой
формы предназначено винтовое устройство 4.
В том случае, если форма смыкается и запирается
только под действием гидравлич. цилиндра 8 и переда-
передаточных рычагов 2, 3, очень трудно точно установить
длину регулируемого рычага, к-рая обеспечила бы за-
заданное усилие запирания формы. Поэтому коленно-
рычажные механизмы во многих случаях применяются
только для смыкания и размыкания формы.
Для запирания формы под заданным усилием обыч-
обычно применяют гидравлич. цилиндр 1. Коленно-рычаж-
Коленно-рычажные механизмы с гидравлич. или электромеханич. при-
приводом устанавливают на машинах небольшой мощности.
На мощных Л. м. применяют ступенчатые механизмы
замыкания формы с использованием промежуточной
плиты и раздельных гидравлич. цилиндров для смы-
смыкания, запирания и размыкания форм. Под действием
гидравлич. цилиндров дл*я смыкания и размыкания
форм передаются относительно небольшие усилия,
в то время как запирание формы требует значительных
усилий, превышающих давление впрыска. Вместе с тем
ступенчатый механизм обеспечивает ускоренное замы-
замыкание и размыкание формы за счет различных конструк-
конструктивных решений.
Роторные машины, на к-рых первоначаль-
первоначально изготовляли толстостенные изделия, применяют
7 2
5 6
Рис. 3. Гидроме-
Гидромеханический меха-
механизм замыкания
формы литьевой
машины: 1 — гид-
гидравлич. цилиндр;
2, 3 — рычаги;
4 — винтовое ус-
устройство; 5 — по-
подвижная плита;
6 — полуформа;
7 — шток; 8 —
гидравлический
цилиндр.
Рис. 2. Конструкции сопел литьевых ма-
машин: а) сопло со скользящим клапаном
A — наконечник сопла; 2 — скользящий
клапан; з — втулка; 4 — инжекционный
цилиндр; 5 — литьевая форма; 6 — ка-
канал; 7 — пружина); б) сопло с игольча-
игольчатым клапаном A — наконечник сопла;
2 — запорная игла; 3 — выточки; 4 —
втулка; 5 — литьевая форма; 6 — инже-
инжекционный цилиндр; 7 — пружина; 8 —
канал; 9 — отверстие); в) открытое соп-
сопло с конич. головкой A — инжекцион-
инжекционный цилиндр; 2 — наконечник сопла; 3 —
литьевая форма; 4 — конич. головка чер-
червяка).
теперь также для литья тонкостенных изделий массово-
массового производства, т. к. в этом случае наиболее полно
используется высокая пластицирующая способность
червячных инжекционных механизмов. Принцип ра-
работы роторной Л. м. (рис. 4) заключается в следующем.
Загруженный в бункер 1 материал непрерывно пласти-
цируется и нагнетается вращающимся червяком 2
в инжекционный цилиндр 4. Под давлением нагнетае-
нагнетаемого материала поршень 5 перемещается вправо. На
роторе 12 установлены шесть, восемь или десять мат-
матриц (полуформ) 13.
При размыкании (отведении полуформ друг от дру-
друга) и смыкании (подведении полуформ друг к другу)
формодержатели 8 перемещаются плунжерами 7 гид-
гидравлич. цилиндров 11. Когда предварительно сомкну-
10
Рис. 4. Литьевая машина роторного типа: 1 — бункер;
2 — червяк; з — гидравлич. цилиндр; 4 — инжекционный
цилиндр; 5 — поршень; б — кран; 7 — плунжер; 8 — фор-
модержатель; 9 — плунжер; Ю — гидравлич. цилиндр;
11 — гидравлич. цилиндр; 12 — ротор; 13 — матрица; I—
VI — позиции литьевой формы.
тая форма устанавливается в позицию I, она окончате-
окончательно смыкается и запирается гидравлич. цилиндром 10
через плунжер 9 и формодержатель 8. Кран 6 откры-
открывает сопло инжекционного цилиндра, и под действием
гидравлич. цилиндра 3 поршень 5 впрыскивает расплав
в форму. После инжекции плунжер 9 отходит влево
и форма перемещается в позицию II, удерживаясь в за-
закрытом состоянии только усилием, создаваемым гидрав-
гидравлич. цилиндром 11. Изделия охлаждаются, когда фор-
форма находится в позициях И — V. На участке между
позициями V и VI форма открывается и готовые изде-
изделия выталкиваются.
Более мощные роторные Л. м. оснащены горизон-
горизонтальным ротором. Установленные на роторе формы
иногда обслуживаются не одним, а двумя инжекцион-
ными механизмами, что позволяет изготовлять двух-
двухцветные изделия, а также изделия различной конфигу-
конфигурации и объема.
Для литья двухцветных изделий
из термопластов, помимо роторных машин, применяют
червячные и поршневые машины двух типов. На Л. м.
первого типа (рис. 5,а) возможно частичное или значи-
значительное смешение материалов различного цвета, по-
поскольку эти материалы одновременно или последова-
последовательно впрыскиваются в одну форму.
89
ЛИТЬЕВЫЕ МАШИНЫ
91)
Четкое разделение материалов разной окраски обе-
обеспечивается на Л. м. второго типа (рис. 5, б). Вначале
из инжекционного цилиндра 1 материал одного цвета
впрыскивается в первую форму, при этом отливается
4 Рис. 5. Литьевые машины для формо-
формования двухцветных изделий: а) машины
упрощенного типа A, 4 — материалы
различных цветов; 2 — инжекционные цилиндры; 3 —
литьевая форма); б) машина усовершенствованного типа
A,2 — инжекционные цилиндры; 3 — полуформы; 4 —
поворотная плита; б — подвижная плита; 6 — поворотное
гидромеханич. устройство; 7 — плунжер перемещения по-
подвижной плиты).
часть изделия. После разъема первой формы эта часть
изделия вместе с полуформой 3 переносится в другую
(на рис. нижнюю) форму, где из инжекционного цилинд-
цилиндра 2 отливается недостающая часть изделия др. цвета.
Плита 4 с установленными на ней полуформами 3
поворачивается гидромеханич. устройством #, к-рое
закреплено на подвижной плите 5. На Л. м. этого типа
можно не только изготовлять двухцветные изделия, но
и наносить ажурные рисунки и надписи на изделия из
термопластов.
Для лабораторных и исследова-
исследовательских работ очень удобны малогабарит-
малогабаритные, обычно верстачные, Л. м. упрощенной конструк-
конструкции с ручным или механизированным управлением.
Такие Л. м. можно легко разбирать и очищать от пере-
перерабатываемого материала. Для отливки изделий на
лабораторной Л. м. требуется очень мало материала,
что важно при исследовании литьевых свойств новых
материалов.
Машины для литья реактопластов и резиновых сме-
смесей. Для литья реактопластов применяют
червячные машины — горизонтальные, угловые и ро-
роторные. Конструкции этих машин должны обеспечи-
обеспечивать точный контроль темп-ры инжекционного цилинд-
цилиндра, сопла и литьевой формы. Степень сжатия материала
не должна превышать 0,8—1,0, т. к. при большем сжа-
сжатии резко увеличивается тепловыделение, к-рое может
вызвать преждевременное отверждение термореактив-
термореактивного материала.
Вращение червяка осуществляется через гидропере-
гидропередачу, обеспечивающую бесступенчатое регулирование
частоты вращения. Червяк имеет канал для охлажде-
охлаждения водой. Л. м. снабжена термостатом, обеспечиваю-
обеспечивающим двухзонный обогрев инжекционного цилиндра
горячей водой. Темп-pa обогрева обеих зон контроли-
контролируется автоматически. Головка и сопло инжекционного
цилиндра имеют электрич. обогрев. Полуформы также
оснащены автономными системами электрич. обогрева
и автоматич. контроля темп-ры (темп-pa формы обычно
составляет 160—180 °С). Л. м. для переработки реакто-
реактопластов обеспечивают формование изделий объемом от
1—2 см3 до 2000 см3.
Базовые модели Л. м. для переработки реактопластов
и резиновых смесей одинаковы. На машинах для
литья резиновых смесей вместо бункера
устанавливают бобину с намотанным жгутом из пред-
предварительно провальцованной или стрейнированной ре-
резиновой смеси. Поскольку пластицированные резино-
резиновые смеси обладают высокой вязкостью, Л. м. для их
переработки отличаются повышенными инжекционным
давлением [до 170—200 Мн/м2 A700—2000 кгс/см2)] и
мощностью двигателя для вращения червяка. Кроме
того, для повышения усилий сдвига применяют червя-
червяки с переменной глубиной винтового канала и пере-
переменным шагом нарезки. Отношение длины червяка
к его диаметру у машин для переработки резиновых
смесей обычно составляет не более (9—10) : 1. Посколь-
Поскольку резиновые смеси впрыскиваются в форму под повы-
повышенным давлением, Л. м. для их переработки должны
быть оснащены более мощными механизмами замыкания
форм. Кроме того, при литье резиновых смесей не тре-
требуется такая высокая точность контроля и регулирова-
регулирования темп-ры инжекционного цилиндра по зонам, как
при литье реактопластов.
Перспективы производства литьевых машин. Л. м.
относятся к наиболее распространенному типу оборудо-
оборудования для производства изделий из термопластов. Са-
Самые перспективные из них — универсальные Л. м.,
позволяющие формовать изделия методами инжекции,
интрузии, инжекционного прессования и литья предва-
предварительно сжатым расплавом. Производство малогабарит-
малогабаритных изделий из термопластов в многогнездных формах
на универсальных Л.м. пе всегда экономически выгод-
выгодно. Для этих целей целесообразно использовать мел-
мелкие специализированные машины, обеспечивающие от-
отливку изделий в одно- или малогнездных формах.
Основные технические характеристики литьевых машин
Характеристика
Объем отливки, см3
16
32
63
40
20-100
140
A400)
32X19
14—25
560 E6)
6
10
,2хО,8х
XI,6
2,5
125
45
20-90
160
A600)
40X32
17-31
1000
A00)
6
18
4,5Х1Х
Х2
4,9
250
60
25 — 100
160
A600)
50X40
24-37
1800
A80)
7
24
4,5Х1Х
Х2
7
500
1000
2000
4000
Диаметр червяка, лип
Частота вращения червяка,
об/мин
Инжекционное давление, Мн/м2
(кгс/см2)
Расстояние между колоннами,
см
Высота (толщина) форм, см . . .
Усилие запирания формы, пн (тс)
Мощность, кет
обогрева
электродвигателей . . . .
Габаритные размеры, м
Масса машины, т
25
20-100
120
A200)
20
до 16
180A8)
3
2X0,7Х
XI,4
1,2
30
20-100
140
A400)
25
до 22
320 C2)
3
4
2,5ХО,7Х
Xl,5
-2
80
25-90
180
A800)
63x50
28—50
3200
C20)
13,2
40
,6Х1,4Х
Х2
10,8
80
20-95
180
A800)
80X63
34-62
6300
F30)
13,2
40
,3x3,ЗХ
Х2,8
20
105
20-54
180
A800)
100X80
44-77
12500
A250)
38,4
57 и 30
, 1X3 ,2Х
Х2,9
37
125
20; 30; 44
180
A800)
120X100
44-80
12500
A250)
60
57 и 30
8,6ХЗ,ЗХ
Х2,9
38
91
ЛИТЬЕВЫЕ ФОРМЫ
92
Расширяется применение роторных Л. м., обеспечива-
обеспечивающих высокую производительность при отливке как тол-
толстостенных, так и тонкостенных изделий (в том числе
многоцветных из различных материалов).
Для управления Л. м. перспективно применение элек-
электронно-механических манипуляторов, связанных с вы-
вычислительными машинами. Электронные устройства
обеспечивают высокую точность воспроизведения техно-
технологических параметров, обладают малым временем сра-
срабатывания и почти неограниченным сроком службы.
Успешны попытки программировать работу Л. м. с по-
помощью перфокарт или магнитной записи. Большой ин-
интерес представляют самонастраивающиеся машины,
к-рые обеспечивают автоматическое регулирование про-
процесса в зависимости от одного или нескольких задан-
заданных параметров (давление расплава, его температура,
потребляемая электродвигателем червяка мощность
и т. п.).
Первый патент на Л. м. для переработки термопла-
термопластов был выдан в 1872 в Германии. Серийное производ-
производство Л. м. впервые налажено там же в 1922.
Лит.: Рябинин Д. Д.,
Л у к а ч Ю. Е., Червячные ма-
машины для переработки пластиче-
пластических масс и резиновых смесей,
М., 1965; Завгородний
В. К., Новое оборудование для
переработки пластмасс, М., 1967;'
ЗавгороднийВ. К., Ка-
линчев Э. Л., МарамЕ.И.,
Литьевые машины для термопла-
термопластов и реактопластов, М., 1968;
Г у р в и ч С. Г., И л ь я ш е н-
ко Г. А.,Свириденко G.X.,
Машины для переработки термо-
термопластических материалов, М.,
1965; Завгородний В. К.,
Механизация и автоматизация пе-
переработки пластических масс, 3
изд., М., 1970; Завгородний
В. К., Калинчев Э. Л.,
МахаринскийЕ. Г., Обо-
Оборудование предприятий по пере-
переработке пластмасс, Л., 1972.
В. К. Завгородний.
ЛИТЬЕВЫЕ ФОРМЫ (in-
(injection moulds, SpritzguMor-
men, moules pour injection) —
инструменты, которые уста-
устанавливаются на литьевую ма-
машину и в процессе литья обес-
обеспечивают получение изделий
заданной формы и размеров.
Принципиальная схема фор-
формы. Л. ф. (рис. 1) состоит из
двух полуформ: на неподвиж-
неподвижной расположена матрица 5,
на подвижной — пуансон 3.
Поверхности 20 и 21 пуансо-
пуансона и матрицы, к-рые непос-
непосредственно соприкасаются с /\ /
расплавом, наз. оформляю- 4""/|
щими. После замыкания фор- А\
мы между оформляющими по- ^
верхностями образуется офор-
оформляющая полость 6, или гнез-
гнездо, где собственно и формует-
формуется изделие.
При замыкании Л. ф. пру-
пружины возвращают выталки-
выталкиватели 4 и сбрасыватель 14 в
левое крайнее положение, а
колонки 18 входят во втул-
втулки 19. После этого сопло 22
цилиндра литьевой машины
сферической
поступает через литниковые каналы в оформляю-
оформляющие полости. Охлаждающая жидкость, циркули-
циркулирующая в каналах 15, вызывает затвердевание рас-
расплава, принявшего форму изделия. При размыкании
Л. ф. изделия и затвердевший в литниковых каналах
материал (литники) остаются на пуансоне. При даль-
дальнейшем перемещении подвижной полуформы шток 2
упирается в упор 1 машины и останавливает выталки-
выталкиватели и сбрасыватель, что вызывает сталкивание из-
изделий 24 и литников 25 в приемную тару. Отделение
изделий от литников производится в данном случае
вне формы.
Если изделие невозможно извлечь из формы из-за
наличия в нем выступов или резьбы на наружной
поверхности, матрицы делают разъемными. Один из
вариантов Л. ф. с разъемной матрицей представлен
на рис. 2. Полуматрицы 3 и 6, установленные на опор-
опорной плите 2, при размыкании Л. ф. перемещаются по
поводкам 8 до упоров 1. При этом полуматрицы стаски-
стаскивают изделие 5 со стержня 7, к-рое падает в приемную
тару через пространство, образовавшееся между пло-
3 4
17
19
14
18
А
12
Рис. 1. Принципиальная схема
многогнездной литьевой формы
в разомкнутом (а) и замкнутом
(б) виде: А — неподвижная по-
полуформа, В — подвижная полу-
полуформа, 1 — упор литьевой ма-
машины, 2 — шток, 3 — пуансон,
4 — выталкиватель, 5 — матри-
матрица, б — оформляющая полость,
7 — поднутренное отверстие,
8 — разводящий литниковый канал, 9 — центральная литниковая втулка, Ю — централь-
центральный литниковый канал, 11 — сферич. часть литниковой втулки, 12 — впускной литниковый
канал, 13 — пружина для возврата выталкивателей, 14 — сбрасыватель, 15 — каналы охлаж-
охлаждения, 16 — плоскость замыкания матрицы, 17 — плоскость замыкания пуансона, 18 — на-
15
прижимается к
части 11 литниковой втул- пРавля1°Щая колонка, 19 — направляющая втулка, 20 — оформляющая поверхность пуансона,
^ярттттяи ттпттттлтопа 21 ~ 0Ф°РмляюЩая поверхность матрицы, 22 — сопло литьевой машины, 23 — инжекционный
ки
и расплав полимера
цилиндр, 24 — изделие, 25 — литники.
93
ЛИТЬЕВЫЕ ФОРМЫ
94
S 6
Рис. 2. Литьевая форма с разъемной матрицей в разомк-
разомкнутом (а) и замкнутом (б) виде (стрелками указано направ-
направление перемещения полуматриц при размыкании формы;
литниковые каналы на схеме не показаны): 1 — упор, 2 —
опорная плита, 3 — левая полу-матрица, 4 — плоскость
замыкания полуматриц, 5 — изделие, 6 — правая полумат-
полуматрица, 7 — оформляющий стержень, 8 — наклонный пово-
Д0К) д — центрирующие и запирающие конич. поверхности,
20 — передний фланец, 11 — плоскость разъема, Z— вели-
величина перемещения каждой полуматрицы.
скостями 4. При замыкании Л. ф. поводки входят
в наклонные отверстия полуматриц и сближают их
на величину 21, а конич. поверхности 9 окончательно
центрируют и замыкают полуматрицы.
В Л. ф. отрыв литников, перемещение полуматриц,
свертывание резьбовых изделий и др. могут произво-
производиться автоматически.
Основные расчеты формы. Расчету оптимальной
гнездности Л. ф. предшествует предварительный выбор
типа литьевой машины в зависимости от геометрии и
материала изделия и потребности в изделиях. Для вы-
выбранной машины по 3 различным ур-ниям определяется
значение числа гнезд формы п. При этом исходят из
объема номинального впрыска машины (ур-ние 1),
необходимости предотвращения разхмыкания формы
в процессе литья (ур-ние 2) и соответствия длитель-
длительности охлаждения отливки тому времени, к-рое необ-
необходимо для пластикации объема материала, идущего
на изготовление одного изделия C):
v V
и_ Н Л. С D)
где 7Н — объем номинального впрыска машины за
1 цикл, м3 (по данным паспорта машины); 7ИЗД —
объем изделия, м3; 7Л. с,— выбранный приблизитель-
приблизительный объем литниковой системы, м3; к — коэфф., учиты-
учитывающий сжимаемость материала и его утечку по порш-
поршню или червяку; принимается равным 1,25—1,30.
^ ()
^изд рср °изд
где Р3ам —~ усилие замыкания литьевой машины,
Мн (кгс); ?изд — площадь проекции изделия на пло-
плоскость замыкания плит машины, м2 (см2); ?л. с. — вы-
выбранная приблизительно площадь литниковой системы,
м2 (см2); рср — среднее давление в Л. ф., Мн/м2
(кгс/см2); для литья вязких материалов на поршневых
машинах pcv принимается равным 60—70 Мн/м2
F00—700 кгс/см2), для низковязких материалов на
червячных машинах — 30—40 Мн/м2 C00—400 кгс/см2),
в многовпусковых Л. ф. с незатвердеваемьши литниками
Рср может достигать 20--25 Мн/м2 B00—250 кгс/см2).
71 —
0,8хт
C)
где тт — время, необходимое для охлаждения изде-
изделия до темп-ры, при к-рой его можно извлечь из Л. ф.,
ч; тпл — время, необходимое для пластикации объема
материала, идущего на изготовление одного изделия,
ч. Значения тт и тпл определяются из соотношений
где Гизд — объем изделия, м3; б — половина толщи-
толщины изделия, м; а — коэфф. температуропроводности
материала, м2/ч; tT — темп-pa в центре сечения изде-
изделия, при к-рой его можно извлечь из Л. ф., °С; t§—
темп-pa Л. ф., °С; tM— темп-pa впрыскиваемого ма-
материала, °С; Q — пластикационная производитель-
производительность литьевой машины для полистирола, кг/ч; кх—
коэффициент, учитывающий вязкость материала; для
полистирола принимается равным 0,8, для более вяз-
вязких материалов — 0,7, для менее вязких — 0,9; у —
плотность материала, кг/м3.
Если значения п, определенные по ур-ниям A) —C),
отличаются не более чем на 1—2, машина выбрана
правильно. При этом гнездность Л. ф. должна соответ-
соответствовать минимальному из 3 вычисленных значений.
Во избежание смятия соприкасающихся поверхно-
поверхностей пуансона и матрицы при замыкании Л. ф. удель-
удельные нормальные напряжения Рсм, действующие на
эти поверхности, не должны превышать 80—100 Мн/м2
(800—1000 кгс/см2). Поэтому после определения гнезд-
гнездности проводится проверочный расчет Л. ф. по этому
показателю: _ Р3ам
см ^"^отл
где F — площадь замыкания Л. ф., м2 (см2); ?отл —
площадь отливки, м2 (см2).
Основные функциональные группы деталей формы.
Литниковые системы предназначены для
подвода и распределения расплава по гнездам. Су-
Существуют 2 принципиально различных типа таких
систем: в одних материал, находящийся в литниковых
каналах, затвердевает при охлаждении изделия в
оформляющей полости (см., напр., рис. 1), в других
остается в расплавленном состоянии. Недостатком
системы затвердеваемых литников является значи-
значительное повышение в процессе литья вязкости распла-
ва в каналах, в результате чего невозможно получить
достаточно уплотненную отливку. Избежать этого
можно повышением темп-ры впрыскиваемого материала
и давления литья, что, однако, отрицательно сказы-
сказывается на свойствах материала. Кроме того, отливки,
полученные в системе с затвердеваемыми литниками,
требуют механич. обработки для отделения литников
от изделий, а иногда зачистки или даже полировки.
Этих недостатков в значительной мере лишена си-
система незатвердеваемых литников (рис. 3). В этой си-
системе все литниковые каналы, кроме впускных, разме-
размещены в специальном распределителе 7, где параллель-
параллельно разводящему каналу с двух сторон находятся на-
нагревательные элементы, к-рые с помощью терморегу-
терморегуляторов поддерживают постоянную темп-ру расплава
в каналах 6. Обогреваемый расплав подается в оформ-
оформляющую полость через сопло 3. Для вязких расплавов
применяют открытые, а для маловязких — самозапи-
самозапирающиеся сопла. Для улучшения условий теплопере-
теплопередачи от обогревателей к материалу сопла формы из-
изготавливают из бериллиевой бронзы. Вокруг сопел
предусматривают зазор 4 толщиной 2—3 мм, куда за-
затекает расплав, к-рый служит теплоизолирующим
слоем, предотвращающим нагревание дна матрицы 1
и матрицы 14 (последние в процессе литья должны
95
ЛИТЬЕВЫЕ ФОРМЫ
96
1,2 1 ?
LULL
Рис. 3. Многогнездная
литьевая форма с неза-
твердеваемыми литника-
литниками: 1 — дно матрицы,
2 — точечный впускной
канал, 3—сопло формы,
4 — зазор для изолиру-
изолирующего слоя, 5— запор-
запорный клапан, 6— разво-
разводящий литниковый ка-
распределитель, 8 — пружина клапана, 9 — со-
центральный литниковый ка-
канал, 7
пло литьевой машины, 10
нал, 11— сферич. заглушка, 12—каналы охлаждения, 13—
отверстия для нагревательных элементов, 14 — матрица.
охлаждаться). Особенно эффективно применение си-
системы с незатвердеваемыми литниками в многогнезд-
ных формах, с помощью к-рых можно изготовить более
100 изделий за 1 цикл.
Длина пути течения расплава по каналам в системе
с затвердеваемыми литниками должна быть минималь-
минимальной, а сами каналы не должны иметь поворотов, ост-
острых углов и тупиков. Оптимальная форма сечения
таких каналов — окружность, для системы с неза-
незатвердеваемыми литниками — прямоугольник. Площадь
сечения впускных каналов и их расположение должны
быть такими, чтобы Л. ф. заполнилась одновременно
по всему фронту течения. Но чрезмерное увели-
увеличение площади сечений обусловливает повышенное
давление в Л. ф., рост напряжений в изделии в зоде
впуска и увеличение времени выдержки изделия в Л. ф.
Если Л. ф. имеет только один впускной канал, как,
напр., на рис. 1, на ней можно отлить изделие сравни-
сравнительно небольшой площади (до ~1200 см2). Для литья
более крупных изделий используют многовпусковые
Л. ф., один из вариантов к-рых представлен на рис. 4.
Места впуска расплава в оформляющую полость вы-
выбирают с таким расчетом, чтобы обеспечить параллель-
параллельность течения отдельных потоков.
При литье изделий с отверстиями или с неравномер-
неравномерной толщиной стенок, а также литье во многовпусковых
Л. ф., расплав полимера в форме разбивается на от-
отдельные потоки, в местах «встречи» к-рых происходит
образование стыков — наиболее слабых участков в
изделии. Этого можно избежать при использовании
системы с незатвердеваемыми литниками.
Удовлетворительная методика расчета размеров лит-
литниковой системы отсутствует. Практические данные
приведены в литературе.
Газоотводящие каналы в Л. ф. необ-
необходимы для отвода воздуха и газов из оформляющей
полости по мере заполнения ее расплавом. Они выпол-
выполняются в виде щелей прямоугольного сечения и рас-
располагаются на плоскости разъема Л. ф., напротив
впускных литниковых каналов, т. к. в этих местах
наблюдается максимальное скопление газов. Чтобы
предотвратить выдавливание расплава через газоот-
газоотводящие каналы, толщина щели не должна превышать
0,02-0,06 мм.
Оформляющие поверхности имеют не-
негативное изображение поверхности формуемого изде-
изделия. Класс чистоты этих поверхностей должен соответ-
соответствовать V9 — V12, что позволяет обеспечить хоро-
хороший внешний вид изделия и снизить трение в момент
его извлечения из Л. ф. Оформляющие детали Л. ф.
изготавливают из углеродистых или цементируемых
сталей, к-рые подвергают термообработке с целью
повышения твердости по Роквеллу до 48—52 для угле-
углеродистых сталей и до 52—56 для цементируемых.
Такая твердость обусловливает способность оформля-
оформляющих деталей хорошо полироваться и незначительно
прогибаться при уплотнении отливки. Прогиб не дол-
должен превышать 0,02—0,03 мм, т. к. в противном
случае расплав вытечет через образовавшийся зазор
и получится изделие с заусенцем и неравномерной
толщиной стенок. Полированные оформляющие по-
поверхности для повышения их твердости, износостой-
износостойкости и химич. стойкости, уменьшения адгезии к от-
А-А
Рис. 4. Оформляющие детали многовпусковой двухгнезд-
ной литьевой формы с затвердеваемыми литниками: i —
разводящий канал, 2 — центральный литниковый ка-
канал, 3 — подводящие литниковые каналы. 4 — впускные
литниковые каналы, 5 — пуансон, б — каналы охлаждения,
7 — матрица, 8 — оформляющая полость.
литым изделиям хромируют или никелируют с после-
последующей термообработкой (толщина покрытия 6—9 мкм).
После этого поверхности вторично полируют до чи-
чистоты, соответствующей V10 — V12 классу.
Размеры оформляющих поверхностей расчитываются
с учетом усадки изделия при охлаждении и износо-
износостойкости формы по ф-ле:
где 1ф — размер оформляющей поверхности, мм;
1Ср — номинальный размер изделия при симметричном
расположении поля допуска, мм; Аизн — заданный
износ оформляющей поверхности за время эксплуата-
эксплуатации, мм, обычно принимается равным 0,02—0,1 мм;
Аизг — допуск на изготовление элемента оформляю-
оформляющей поверхности с размером /ф, обычно принимается
по 2—3 классу точности; х — предполагаемая усадка
элемента изделия, %.
Строгое соответствие во взаимном расположении
оформляющих поверхностей пуансона и матрицы до-
достигается с помощью центрирующих эле-
элемент о в — направляющих колонок и втулок (см.,
напр., рис. 1). При расположенил впускных каналов
на боковой грани изделия или при литье изделий,
имеющих разную толщину стенок, возникают значи-
значительные боковые усилия, к-рые вызывают изгиб ко-
колонок и смещение осей пуансона и матрицы, что в
свою очередь приводит к изменению заданной толщины
стенок. Этих недостатков лишена конструкция центри-
97
ЛИТЬЕВЫЕ ФОРМЫ
98
рующих элементов (рис. 5), представляющая собой
массивные конич. выступы 1 и уступы 4, к-рые предот-
предотвращают смещение оформляющих деталей 2 и 3. Такая
конструкция более долговечна, т. к. поверхности 1 и 4
соприкасаются лишь в момент замыкания формы и
разъединяются без трения скольжения.
Для отвода тепла от расплава полимера в оформля-
оформляющих деталях или в прилегающих к ним деталях раз-
размещают каналы охлаждения, в к-рых
циркулирует охлаждающая жидкость. Оптимальная
конструкция этих каналов должна обусловить одно-
одновременное окончание охлаждения изделия по всему
контуру до одинаковой темп-ры, что обеспечивает
минимальное коробление отлитых изделий. От интен-
интенсивности охлаждения изделия в Л. ф. зависит степень
усадки материала, уровень остаточных напряжений,
чистота поверхности изделия и производительность
процесса. Интенсивность охлаждения изделия зависит
от площади поверхности охлаждающих каналов,
темп-ры и скорости течения жидкости, ее теплофизич.
характеристик, материала изделия и формы.
Поскольку каналы охлаждения располагаются обыч-
обычно внутри охлаждаемых деталей и выполняются свер-
сверлением, они имеют цилиндрич. форму и прямолинейны.
Такие каналы не могут обеспечить оптимальных усло-
условий теплопередачи (при равной с прямоугольными
каналами площади сечения они' имеют меньшую по-
поверхность охлаждения). Кроме того, прямолинейные
каналы нельзя расположить достаточно близко ко
всем точкам криволинейного контура изделия. Более
прогрессивная система охлаждения с каналами прямо-
прямоугольного сечения представлена на рис. 6. Т. к. такие
каналы изготовляют точением или фрезерованием, их
располагают на открытых поверхностях матрицы 2
или прилегающей к ней детали S, а также на спе-
специальных вставных деталях 5 и 13. Последние изго-
изготавливают из специальных сплавов, обладающих вы-
высокой тепло- и температуропроводностью, напр, из
бериллиевой бронзы.
Темп-pa охлаждающей жидкости при литье полиоле-
финов составляет для изделий с толщиной стенки до
3 мм 15—20 °С, для изделий с толщиной стенки
8—10 мм 4—8 °С. При литье поликарбоната, полифор-
полиформальдегида, полиэтилентерефталата и др. полимеров
с высокой вязкостью расплава темп-pa жидкости со-
составляет для изделий с толщиной стенки 8—12 мм
Рис. 5. Литьевая форма с массивной центрирующей систе-
системой: 1 — конич. выступы на матрице, 2 — матрица, 3 —
пуансон, 4 — конич. уступы на пуансоне, 5 — конич. ко-
колонки, предохраняющие от продольного перемещения, 6 —
конич. отверстия для колонок.
70—80 °С, а для изделий с толщиной стенки 2—3 мм
120 °С. В последнем случае вместо воды применяют
минеральные масла, глицерин или кремнийорганич.
жидкости.
Выталкивающая система Л. ф. пред-
предназначена для извлечения изделий из матриц, сталки-
сталкивания их с пуансонов и сбрасывания через люк машины
Рис. 6. Литьевая форма с вставными деталями охлаждения
(стрелками показано направление течения охлаждающей
жидкости): 1 — опорная плита, 2 — матрица, з — пуансон,
4 — выталкиватель, 5 — вставная деталь со спиральными
каналами охлаждения на торцевой и наружной поверхности,
6 — резиновые уплотнения, 7 — вставная деталь из берил-
бериллиевой бронзы, 8 — передний фланец со спиральными кана-
каналами прямоугольного сечения, 9 — центральный литник,
10 — плоскость разъема, 11 — сферич. часть литниковой
втулки, 12 — вставная деталь матрицы с охлаждающими
каналами прямоугольного сечения, 13 — вставная деталь
в матрицу, оформляющая центральное отверстие в изделии
и являющаяся одновременно центральной литниковой втул-
втулкой, 14 — дисковый подводящий литник, 15 — кольцевой
впускной литник.
в тару. Детали выталкивающей системы располагают
с таким расчетом, чтобы они минимально изгибали и
не повреждали изделие, а также не оставляли следов
на его лицевой стороне. Рабочее движение деталей этой
системы производится за счет перемещения подвижной
части литьевой машины, пружин, рычагов, винтовых
и зубчатых соединений, пневматич., гидравлич. и
электрич. устройств. Выталкивание производится ци-
цилиндрическими, фасонными, щелевыми и др. выталки-
выталкивателями, сталкивающей плитой, закрытыми полу-
полуматрицами, давлением сжатого воздуха и др. В особых
случаях, напр, при необходимости получить изделие
с арматурой, для возврата деталей выталкивающей
системы применяют специальные устройства.
Наряду с автоматизированными Л. ф., где системы
и узлы перемещаются автоматически, в тех случаях,
когда автоматизация экономически не оправдана или
вызывает значительные усложнения при изготовлении,
применяются Л. ф. для полуавтоматич. и ручного
режимов работы. Для этого в Л. ф. предусматриваются
комплекты съемных кассет, знаков, резьбооформлякь
щих деталей и др. Они удаляются из Л. ф. вместе с
изделиями вручную и после охлаждения отделяются
от изделия вне формы. Несмотря на применение руч-
ручных приемов, процесс с такими Л. ф. достаточно про-
производителен, т. к. время, необходимое для охлаждения
изделия вне формы и удаления съемных элементов, ис-
исключается из общей продолжительности цикла. При
необходимости операции извлечения съемных знаков
из изделий м. б. механизированы.
Лит.: Завгородний В. К., Калинчев Э. Л.,
М а р а м Е. И., Литьевые машины для термопластов и реакто-
пластов, М., 1968; В и д г о ф Н. Б., Точечное литье термопла-
термопластов, Л., 1961; Брагинский В. А., Усадка и точность
деталей из пластмасс, ч. 1—2, Л., 1963. Я. Б. Видгоф.
м
МАКАНЫЕ ИЗДЕЛИЯ—см. Латексные изделия.
МАКРОИОНЫ (macroiones, Makroionen, macroions) —
макромолекулы, содержащие ионные функциональные
группы. При этом к М. не относят обычно ионизован-
ионизованные макромолекулы полиэлектролитов, т. наз. по-
поли и о н ы.
М. являются, напр., растущие цепи в процессах
ионной полимеризации, к-рые несут на одном или
обоих концах катионные или анионные активные
центры (в частности, карбкатионы или карбанионы).
Для генерирования М. в полимеризационную систему
вводят различные инициирующие агенты полярного
характера, подвергают систему действию ионизирую-
ионизирующего излучения или электрич. тока (см. Анионная
полимеризация, Катионная -полимеризация, Катали-
Катализаторы полимеризации). Растущие М., как правило,
активно реагируют с различными примесями (водой,
кислородом и другими), поэтому при работе с ними
необходимо тщательно очищать реагенты и аппа-
аппаратуру.
В среде органич. растворителей активные центры
ионной полимеризации, как правило, существуют не
в виде свободных ионов, а связаны с соответствующими
ионами противоположного знака (противоионами), об-
образуя с ними ионные пары или поляризованные ко-
валентные молекулы. Положение равновесия между
неионизованными формами, ионными парами и свобод-
свободными ионами в значительной степени определяется
свойствами растворителя. Напр., константа диссо-
диссоциации активных центров полистириллития на свобод-
свободные ионы в бензоле с 10% тетрагидрофурана состав-
составляет ок. 10~17 моль/л, в тетрагидрофуране — 10~7 моль/л,
а в сильно полярном гексаметилфосфортриамиде (ди-
электрич. проницаемость 30) степень диссоциации
приближается к 1.
Для большинства полимерных карбкатионов и карб-
анионов в той или иной степени характерны различ-
различные спонтанные реакции, приводящие к их полной
дезактивации (рекомбинация с противоионом, отщеп-
отщепление протона или гидрид-иона и др.) или превраще-
превращению в менее активные формы (изомеризация). Ско-
Скорость этих реакций и, следовательно, время жизни
соответствующих М. меняются в очень широких пре-
пределах в зависимости от природы ионной группы,
противоиона, растворителя и др. факторов. Обычно
время жизни карбанионов выше, чем аналогичных карб-
карбкатионов. В нек-рых случаях время жизни растущих
М. (в отсутствие воды, воздуха и др. активных приме-
примесей) может быть теоретически неограниченным и дости-
достигать суток и месяцев; такие М. обладают рядом снеци-
фич. свойств (см. Живущие полимеры).
Помимо М. с концевыми ионными группами, сущест-
существуют и такие, в к-рых ионогенные группы входят
в состав внутренних мономерных звеньев. Такие М.
получают, напр., металлированием полимеров; их
применяют как промежуточные продукты для синтеза
привитых сополимеров, введения в полимер различных
функциональных групп и др.
Лит.: Бреслер С. Е., Ерусалимский Б. Л.,
Физика и химия макромолекул, М.— Л., 1965; Шварц М.,
Анионная полимеризация, пер. с англ., М., 1971.
А. А. Ар ест-Якубович.
МАКРОМОЛЕКУЛА (macromolecule, Makromolekul,
macromolecule).
Содержание:
Основные понятия, общие характеристики 100
Мономерное и повторяющееся звенья 100
Линейные, разветвленные и сшитые макромолекулы 101
Способы изображения стереохимич. структуры макро-
макромолекулы. Характер соединения звеньев и стереоре-
гулярность 101
Первичная структура и конфигурация 102
Молекулярная масса, степень полимеризации,
полидисперсность 103
Размеры и форма. Конформация 103
Классификация макромолекул по химическим признакам
и химические свойства 104
Конфигурация 106
Конформация НО
Статистическая физика изолированных линейных и лест-
лестничных макромолекул. Жесткость ИЗ
Взаимодействие макромолекул с растворителем. Объемные
эффекты 115
Методы определения размеров и формы макромолекул.
Гидродинамические характеристики 116
Вторичные макромолекулярные структуры 119
Основные определения. Уровни макромолекулярной
организации 119
Линейно-упорядоченные структуры 120
Глобулярные и промежуточные структуры. Молеку-
Молекулярные мицеллы 121
Пространственно-сетчатые структуры 122
Явления микросегрегации 124
Термодинамика и статистическая механика структурных
переходов в макромолекулах 124
Термодинамика малых систем и переход клубок —
глобула 124
Переходы типа спираль — клубок 127
Влияние внешней силы на переходы в макромолеку-
макромолекулах 129
Преобразование химической энергии в механическую . . 131
Макромолекулы и информация. Молекулярная киберне-
кибернетика 132
Основные понятия, общие характеристики
В буквальном переводе «макромолекула» означает
«гигантская молекула». Однако в современной физич.
химии полимеров не всякая совокупность достаточно
большого числа атомов именуется макромолекулой.
М.— это молекула полимера; в таком
определении уже содержится требование об особом
способе объединения простейших элементов структуры
в М. Этот способ состоит в повторении одной и той же
структурной единицы или чередовании в достаточной
мере различающихся структурных единиц. Простей-
Простейшей наглядной моделью линейной М. является ожерелье
из одинаковых (гомополимер) или различных (сополи-
(сополимер) бусин. Эти бусины изображают простейшие эле-
элементы структуры, именуемые мономерными
или повторяющимися звеньями.
Мономерное и повторяющееся звенья. Хотя обычно
между этими понятиями не проводится различия,
в действительности они не эквивалентны, и второе
является более точным и общим. В самом деле, моно-
мономерное и повторяющееся звенья совпадают только
для М., полученных полимеризацией ненасыщенного'
или циклич. мономера, напр.:
nCH2=CHR -> [-СН2—CHR-]M A)
72СН2 CHR -> [—СН2- CHR — O-L B),
\ / я V /
о
101
МАКРОМОЛЕКУЛА
102
Если же при получении М. в ходе реакции выде-
выделяется низкомолекулярное соединение, повторяющееся
звено цепи будет отличаться от исходных компонен-
компонентов, напр, в случае М., синтезированных поликон-
поликонденсацией дикарбоновых к-т с диаминами:
nHOOC-R'-COOH + nH2N-R"-NH2 -»
-» [-R'-CO-NH-R"-b + (n—1)H,O C)
В сополимере с правильным чередованием звеньев
двух типов, полученном по реакции
nCH2=CHR' + nCH2=CHR" -» [-CH2-CHR'-CH2-CHR'']n D)
элементарные ф-лы мономеров и мо но мерных звеньев
хотя и совпадают, повторяющимся звеном цепи яв-
является совокупность СН2 — CHR' — СН2 — CHR".
В редких случаях повторяющимся звеном является
атом, напр, в полимерной сере Sn.
Линейные, разветвленные и сшитые макромолекулы.
При определенных условиях даже простейшая реак-
реакция A) может приводить к образованию не линейных,
а разветвленных М. При этом ветви могут иметь длину
того же порядка, что и основная цепь (длинноцепные
ветвления), или состоять лишь из нескольких повто-
повторяющихся звеньев (короткоцепные ветвления). Раз-
Разветвленные М. являются промежуточной формой меж-
между линейными и сшитыми М. (см. также Высокомоле-
Высокомолекулярные соединения). Примером линейных М. могут
служить М. каучука натурального, регулярного поли-
полиэтилена, полиамидов и полиэфиров сложных, получен-
полученных поликонденсацией бифункциональных мономеров,
целлюлозы, нек-рых белков, нуклеиновых кислот и др.
Примерами синтетических разветвленных М. явля-
являются полиэтилен, полученный при высоком давлении,
привитые сополимеры, полимеры, синтезированные
поликонденсацией с участием три- или тетрафунк-
циональных мономеров, природные М.— амилопектин
(разветвленный компонент крахмала), гликоген и др.
Сшитые М. получаются при образовании поперечных
связей между М. в процессах полимеризации или
поликонденсации, под действием химич. агентов (вул-
(вулканизация, отверждение) или ионизирующих излуче-
излучений на заранее синтезированные линейные или раз-
разветвленные полимеры. По мере развития процессов
сшивания в подобные агрегаты вовлекается все боль-
большее число цепей, и на определенном этапе исчезает
грань между М. и макроскопич. телом.
Способы изображения стереохимич. структуры ма-
макромолекулы. Характер соединения звеньев и стерео-
регулярность. Простые структурные ф-лы типа изо-
изображенных в ур-ниях A—4) часто недостаточны для
описания М., что связано со след. обстоятельствами:
1. Эти ф-лы не учитывают отличие концевых групп
от всех остальных звеньев. Концевые звенья могут
содержать двойные связи или радикалы, являющиеся,
напр., «остатком» инициатора полимеризации. Если
число п мономерных или повторяющихся звеньев до-
достаточно велико (превосходит 100), указанным отли-
отличием часто можно пренебрегать. Однако при малых п
(в случае т. наз. олигомеров) влияние концевых групп
на все свойства М. может быть очень велико.
2. Ф-лы A—4) подразумевают чередование звеньев,
но ничего не говорят о способе чередования.
Вместо ф-лы типа A) целесообразно бывает изобра-
изобразить только наиболее «типичный» участок цепи:
^CH2-CHR-CH2-CHR^ E)
Это часть М. винилового полимера, причем все повто-
повторяющиеся звенья в этой цепи соединены друг с другом
одинаково, в данном случае по типу «голова к хвосту».
В зависимости от условий полимеризации могут возни-
возникать различные типы нарушений такого расположения.
Одним из них является инверсия присоединения («го-
(«голова к голове» или «хвост к хвосту»), напр.:
^GH2-CHR-CHR-CH2-CHR- F\
Другой тип нарушений возникает, если лишь один
углеродный атом винилового мономера входит в основ-
основную цепь, а второй оказывается сбоку:
~CH2-CHR-CR-CH2-CHR~< G)
I
СНз
Характер расположения звеньев — очень важная
структурная характеристика М. Как правило, он
предопределяет способность М. к кристаллизации в
макроскопич. объеме полимера. Однако отсутствие
нарушений в расположении звеньев — лишь необ-
необходимое, но не достаточное условие для кристаллиза-
кристаллизации. Даже простейшие мономеры — виниловые соеди-
соединения СН2 = CHR можно собрать в М. двумя спо-
способами, различающимися ориентацией R по отношению
к плоскости, в к-рой лежат С—С-связи цепи главных
валентностей (т. наз. хребет цепи). Иными
словами, атом углерода в М., при к-ром расположен
радикал R, является асимметрическим, и звено встраи-
встраивается в цепь в d- или Z-положениях, т. е. в правой
или левой конфигурации.
Так. обр., для более углубленного представления
о структуре М. вместо «плоской» ф-лы участка М.
типа E) или F), позволяющей судить только о регуляр-
регулярности присоединения повторяющихся звеньев, полезно
использовать более сложную пространственную ф-лу,
по меньшей мере учитывающую ориентацию радикалов
R относительно хребта цепи, когда он образует вытя-
вытянутый плоский зигзаг:
R R R R
i i i
R R R
Здесь (jt — 6) — валентный угол 109°; для простоты
скелетные атомы С изображены точками, а атомы во-
водорода не показаны; зигзаг в ф-ле (8) соответствует
максимально возможному растяжению цепи без де-
деформации связей или валентных углов.
М. наз. стереорегулярной, если харак-
характеризуется периодич. порядком в ориентациях ради-
радикала R. Если все они ориентированы по одну сторону
плоскости зигзага (только d или только I), М. наз.
изотактической, а если имеет место правильное чере-
чередование d- и /-ориентации — синдиотактической. Сте-
реорегулярность — необходимое условие для того,
чтобы М. могли кристаллизоваться. Если, однако, ван-
дерваальсов радиус радикала R сопоставим с радиусом
атома водорода, то они оказываются взаимозаменяе-
взаимозаменяемыми в кристаллич. решетке, и отсутствие стереоре-
гулярности перестает быть препятствием для кристал-
кристаллизации. Примером является поливиниловый спирт.
Первичная структура и конфигурация. Как уже от-
отмечалось, ф-ла (8) дает исчерпывающее представление
о порядке и способе чередования повторяющихся
звеньев. Порядок и способ чередования звеньев наз.
первичной структурой М. Часто первич-
первичная структура бывает такова, что лишь отдельные
участки цепи стереорегулярны. Характеристикой та-
такой неполной стереорегулярности служит степень
стереорегулярности, определяемая как от-
отношение числа звеньев, входящих в стереорегулярные
участки цепи, к общему числу звеньев. Наряду со
степенью стереорегулярности широко используется
понятие тактичность, к-рое включает в себя
степень стереорегулярности, ее характер (изо-, син-
дио- или др. типы, к-рые будут рассмотрены ниже
в разделе «Конфигурация») и характер чередования
стереорегулярных участков.
Ф-ла (8) еще не является полным изображением про-
пространственного строения цепной М. Для исчерпываю-
103
МАКРОМОЛЕКУЛА
104
щего изображения следует показать атомы водорода
и раскрыть формулу «R». Пространственное распреде-
распределение атомов в М., определяемое длинами соответ-
соответствующих связей и значениями валентных углов, наз.
конфигурацией М. Конфигурация не может
быть изменена без хотя бы одной перестановки связей
или углов, т. е. химич. реакции. См. об этом также
ниже — раздел Конфигурация.
Молекулярная масса, стедень полимеризации, поли-
полидисперсность. Мол. масса является однозначной харак-
характеристикой обычных молекул и многих М. биополиме-
биополимеров. Мол. масса М. определяется числом п повторяю-
повторяющихся звеньев, к-рое наз. степенью (или к о-
эффициентом) полимеризации. Осо-
Особенность синтетич. М. заключается в том, что их
нельзя характеризовать одним определенным значе-
значением п или мол. массы М. Практически любой реаль-
реальный синтетический полимер представляет собой набор
полимергомологов, т. е. М. разной степени полиме-
полимеризации (см. Молекулярная масса, Молекулярно-массо-
вое распределение). Это свойство называется п о-
л и дисперсностью, или полимолеку-
лярност ью.
Хотя М и п связаны элементарным соотношением
М = топ, где т0 — «мол. масса» повторяющегося зве-
звена, они не являются вполне эквивалентными характе-
характеристиками М. Действительно, при очень больших mn,
оценивая макромолекулу только по значению М,
можно не «почувствовать» различия между полимером
и олигомером. С др. стороны, если т0 велико вслед-
вследствие большой длины повторяющегося звена (напр.,
в полиундеканамиде), М может оказаться более реа-
листич. характеристикой, позволяющей предсказать
физич. свойства полимера.
Пока мол. масса относительно мала все физич. и
химич. свойства молекулы быстро изменяются с уве-
увеличением М. Однако по достижении нек-рого предела
дальнейшее возрастание М перестает существенным
образом отражаться на этих свойствах. Этот предел
быстрее всего достигается для агрегатных состояний
(жидкие олигомеры становятся воскоподобными уже
при п порядка 20—30), несколько медленнее — для
химич. свойств (исчезает влияние концевых групп)
и еще медленнее — для механич. и релаксационных
свойств (см. также Олигомеры).
Размеры и форма. Конформация. Для исчерпываю-
исчерпывающего химич. описания М. вместо простой структурной
ф-лы типа A) необходимо изобразить ее конфигурацию
в вытянутой форме и указать функцию распределения
по степеням полимеризации.
Однако вытянутая форма реальной М. представляет
собой лишь крайность, и притом довольно маловероят-
маловероятную. Если изолированную М. поместить в вакуум,
благодаря силам когезии между звеньями цепь «свора-
«сворачивается на себя», образуя глобулу — другую
предельную форму М. Подобным способом (т. е. дис-
диспергируя разб. р-р полимера в вакууме) можно полу-
получить глобулярные М. и наблюдать их с помощью элек-
электронного микроскопа. Глобулы могут также возникать
в процессе полимеризации или при осаждении М. из
предельно разб. р-ра.
Возможность образования глобул обусловлена тем,
что мономерные звенья, объединенные в М., сохраняют
вращательные степени свободы. Однако обычно под
влиянием внутрицепного, или микробраунова (тепло-
(теплового), движения М. приобретает свою наиболее вероят-
вероятную форму статист и ч. клубка с непрерывно
изменяющимися размерами. Не следует смешивать
понятия «клубок» и «глобула». Плотность глобулы
может приближаться к «сухой» плотности соответст-
соответствующего полимера; напротив, объемная концентрация
звеньев в координационной сфере клубка не превы-
превышает 3% (а обычно составляет ок. 0,1%).
Вращательные степени свободы реализуются посред-
посредством поворотов звеньев вокруг простых связей хребта
цепи. В действительности свобода этих поворотов огра-
ограничена (см. стр. 114); чем сильнее эти ограничения,
тем меньше гибкость макромолекулы и тем больше
средние размеры клубка.
Способность к изменению размеров и формы в весьма
широких пределах — важнейшее физич. свойство М.,
во многом предопределяющее все особенности меха-
механич. и термомеханич. поведения полимеров. Эта спо-
способность обусловлена тем, что М. можно рассматри-
рассматривать как миниатюрное физич. тело, состоящее из ан-
ансамбля простых элементов (повторяющихся звеньев),
сохраняющих определенную «индивидуальность», т. е.
способность к взаимодействию с растворителем или
др. звеньями этой же М., далеко отстоящими от них,
если считать вдоль вытянутой цепи. Такой ансамбль
связанных элементов при достаточно больших п под-
подчиняется статистич. законам термодинамики малых
систем; как будет показано ниже (см. стр. 114), термо-
динамич. поведение М. в р-ре в нек-рых определяю-
определяющих чертах существенно разнится от поведения р-ра
в целом.
В каждый момент форма М., как и конфигурация,
м. б. описана пространственным распределением атомов
и атомных групп, постоянными значениями валентных
углов и переменными (из-за микробраунова движения)
ориентациями валентных связей. Такое распределе-
распределение, к-рое может непрерывным или дискретным обра-
образом меняться без химич. реакции, наз. к о н ф о р-
м а ц и е й М. Наряду с беспорядочной конформаци-
ей статистического клубка могут существовать упо-
упорядоченные, спиральные или складчатые конформа-
ции. Обычно такие конформации фиксированы не-
нехимическими «вторичными» (напр., водородными) свя-
связями; переход из одной фиксированной конформа-
конформации в другую или в конформацию статистического
клубка происходит прерывным (дискретным) обра-
образом в узком интервале темп-р, активности раство-
растворителя и т. п.
Обязательным условием кристаллизации полимера
является упорядоченность конформации по крайней
мере части М. В отличие от конфигурации, конформа-
конформация представляет собою переменную характеристику,
и для приобретения упорядоченной конформации,
помимо определенных равновесных (термодинамиче-
(термодинамических) условий, требуется время структурной релак-
релаксации (зависящее от мол. массы и гибкости М.). Поэто-
Поэтому кинетика кристаллизации очень сильно зависит
от целого комплекса условий, таких как степень пере-
переохлаждения, скорость охлаждения или изменения
состава растворителя, наличие внешних механич. или
электрич. полей и т. п.
Подводя итоги, можно сказать, что мол. масса (или
степень полимеризации), конфигурация и гибкость М.
предопределяют все остальные физич. характеристики
изолированных М., такие как размеры, форма, способ-
способность к спонтанному или вынужденному изменению
формы и др. Часто перечисленные характеристики
объединяются под названием «макромолеку-
л я р н ы е характеристики».
Классификация макромолекул по химическим
признакам и химические свойства
Классификация М. по происхождению или структуре
хребта цепи (карбоцепные, гетероцепные и др.) совпа-
совпадает с классификацией соответствующих полимеров
(см. Высокомолекулярные соединения).
То, что обычно именуется «химической
структурой» М., м. б., как мы видели, исчер-
исчерпывающим образом охарактеризовано ее конфигура-
конфигурацией. Для описания сополимеров дополнением к кон-
конфигурации и молекулярно-массовому распределению
105
МАКРОМОЛЕКУЛА
106
должны быть две функции, описывающие неоднород-
неоднородность М. сополимера по составу: 1) собственно компо-
композиционная неоднородность, определяемая как функ-
функция распределения М. одинаковой степени полиме-
полимеризации по содержанию в них мономерных звень-
звеньев разного сорта; 2) конфигурационная неоднород-
неоднородность, представляющая собой функцию распределе-
распределения сополимерных М. одинаковых степеней поли-
полимеризации и мономерных составов по конфигураци-
конфигурациям, т. е. характеру чередования звеньев разных
сортов.
Если М. содержат ионогенные группы (полиэлектро-
(полиэлектролиты) или, будучи сополимерными, группы резко
различающейся полярности (д и ф и л ь н ы е, или
амфифильные, М.), они при вариации рас-
растворителя могут приобретать различные фиксирован-
фиксированные конформации. Гомополимерные М. с достаточно
сложной структурой звеньев также могут проявлять
черты дифильности (внутренняя дифильность), приво-
приводящие к тем же последствиям.
При образовании М. полиэлектролитов или дифиль-
ных сополимеров объединение в одну цепь существен-
существенным образом модифицирует химич. свойства свобод-
свободных мономерных молекул или входящих в них групп.
Напр., полиэлектролиты, содержащие карбоксильные
группы (поликислоты), проявляют повышенную гид-
ролитич. активность по сравнению с набором такого
же числа мономерных к-т. Благодаря образованию
жестких конформации, наиболее сложные по составу
М. белков способны к проявлению каталитич. актив-
активности, превосходящей активность простых молекул
в 10е—109 раз. Используя способность сополимерных
синтетич. М. с ионогенными звеньями к образованию
фиксированных конформации, в нек-рых случаях
(напр., на М. неполностью замещенных четвертичных
оснований) удается смоделировать образование при-
примитивных центров каталитич. активности с коэфф.
ускорения реакций порядка 103—106 (см. Катализа-
Катализаторы полимерные).
Химич. р-ции М. могут идти с изменением и без
изменения степени полимеризации. К реакциям пер-
первого типа принадлежат деструкция и сшивание (напр.,
вулканизация, отверждение), к реакциям второго
типа — полимер аналогичные превращения, напр, об-
образование эфиров целлюлозы, омыление поливинил-
ацетата до поливинилового спирта. Особняком стоят
реакции изомеризации, при к-рых без изменения п
происходит изменение конфигурации М. (см., напр.,
Изомеризация каучуков).
Важным химич. свойством М. является чрезвычай-
чрезвычайная чувствительность производных макромолекуляр-
ных характеристик (т. е. конформации и связанных
с нею экспериментально определяемых величин; см.
стр. 117) к незначительным изменениям состава. В бел-
белках или нуклеиновых кислотах иногда замена одного
звена из нескольких сот приводит к катастрофич.
последствиям для целого организма. На уровне М.
белка эта замена обычно связана с не очень значитель-
значительным изменением конформации, но сильным изменением
растворимости и химич. свойств, обусловленных спе-
цифич. конфигурацией поверхностного рельефа бел-
белковой М.
В случае синтетич. полимеров замена звена или не-
нескольких звеньев приводит к последствиям того же
характера, хотя обычно и менее ярко выраженным.
Важно, однако, помнить, что при подобной незначи-
незначительной химич. модификации (даже если она затраги-
затрагивает лишь одно звено) следует с большой осторож-
осторожностью пользоваться ф-лами для определения мол.
массы по гидродинамич. характеристикам. Иногда
применение этих ф-л, эмпирич. коэфф. к-рых соответ-
соответствуют «неповрежденным» М., может приводить к
грубым ошибкам.
Конфигурация
Как уже отмечалось, конфигурация исчерпывающим
образом характеризует «химич. структуру» М., т. е.
является основной стереохимич. характеристикой М.
Конфигурация м. б. рассчитана или воспроизведена,
если известна первичная структура, т. е. общее число,
порядок и способ чередования звеньев в М. Однако
существование подобной однозначной связи еще не
означает эквивалентности этих двух стереохимич.
характеристик. Первичная структура изображается
плоскими структурными ф-лами типа E—7) или аксо-
нометрич. проекцией «объемной» ф-лы, в к-рой детали
пространственной ориентации ограничиваются распо-
расположением радикалов относительно нек-рой плоскости,
в данном случае — плоскости полностью вытянутой,
но не деформированной цепи (ф-ла 8).
Переход от такой ф-лы к изображению конфигурации
в истинном значении слова далеко не примитивен.
Важно, что конфигурация — это интегральная харак-
характеристика вытянутой цепи, к-рая складывается из
структурных элементов — локальных конфигураций.
Эти локальные конфигурации образуют своего рода
последовательность, или иерархию, конфигурационных
уровней. Число этих уровней может варьировать в
зависимости от характера первичной структуры: чем
она сложнее, тем больше локальных конфигураций,
суперпозиция к-рых образует конфигурацию М. в
целом.
Все же, не нарушая общности, можно выделить че-
четыре основных уровня, определяющих характер иерар-
иерархии конфигураций: 1) конфигурация звена, 2) ближ-
ближний конфигурационный порядок, т. е. конфигурация
присоединения соседних звеньев, 3) дальний конфигу-
конфигурационный порядок, характеризующий структуру боль-
больших участков М., напр, длину и распределение вет-
ветвлений, блоки и их чередование в блоксополимерах,
4) конфигурация вытянутой цепи в целом. Существо-
Существование подобной иерархии обусловлено тем, что кон-
конфигурация не является специфич. характеристикой
именно М.
Конфигурация звена. Если звено имеет
достаточно сложную структуру, можно принимать во
внимание и конфигурацию скелетных атомов цепи
в пределах звена (это важно для «длинных» звеньев,
напр, в полиарилатах) или конфигурацию боковых
радикалов. Необходимость учета последней достаточно
очевидна, если в качестве примера написать плоские
структурные ф-лы двух изомеров поликарбэтоксифе-
нилметакрил амида:
С1
I
I
N—Н
~СН9— СН~
(9)
о—с2н5
о=с-о-с2н5
п = изомер
о= изомер
Надо учитывать, что даже на рассматриваемом про-
простейшем конфигурационном уровне по стерич. или
энергетич. причинам возможна деформация углов и
связей, регистрируемая физич. методами. Простейший
полимер винилиденового ряда, у к-рого наблюдается
такая деформация,— полиизобутилен. Нек-рая де-
деформация связей и углов должна иметь место и в поли-
о-карбэтоксифенилметакриламиде в результате обра-
образования внутримолекулярной водородной связи.
107
МАКРОМОЛЕКУЛА
108
Таблица 1. Схемы некоторых макромолекулярных
конфигураций
Тип макромолекулы
Линейные макромо-
макромолекулы
с повторяющимся
звеном
-СН2-СНХ-
То же
с повторяющимся
звеном**
-СНХ-СНУ
То же
с повторяющимся
звеном
I I I I
-с-с=с-с—
I I
То же
Двухтяжевые макро-
макромолекулы
То же
Разветвленные ма-
макромолекулы
То же
Тип конфигурации
или характер стерео-
изомерии
Атактический
Изотактический
Синдиотактический
Дизотактический
Дисиндиотактиче-
ский
транс-Изомер
Лестничные
Спиронолимеры
С короткоцепными
ветвлениями
С длинноцепными
ветвлениями
статистические
гребневидные
звездообразные
Схема
ТП I 1 И!
1 И 1 1 1 1 И И I И 111
I I I I I I 11
1 I I I I I 1 1
ISISIilSINSISI!!!!
В диеновых полимерах важной характери-
характеристикой звеньев является их цис- или транс-
конфигурация (табл. 1), т. е. взаимное рас-
расположение 1-го и 4-го С-атомов по отношению
к двойной связи. В данном специальном слу-
случае уже этот простейший уровень конфигура-
конфигурации М. предопределяет основные физич. свой-
свойства полимера (напр., ^ггс-полиизопрен—каучук,
га/?бшс-полиизопрен — гуттаперча).
Ближний конфигурационный
порядок — конфигурация присоединения
соседних звеньев. В виниловых гомополимерах
или полиолефинах ближний порядок характе-
характеризует регулярность цепи и различные типы
тактичности. Как уже отмечалось, количествен-
количественной мерой тактичности является степень сте-
реорегулярности. Помимо интегрального выра-
выражения (т. е. отношения числа звеньев в сте-
реорегулярных участках к общему числу звень-
звеньев), тактичность может описываться числом
пар ближайших соседей (диад), троек (триад),
четверок (тетрад) и т. д. Распределение таких
диад, триад, тетрад и т. д., т. е. упорядочен-
упорядоченных последовательностей ближайших соседей
(отсюда и термин — ближний порядок), может
характеризоваться нек-рой аналитич. функцией,
параметры к-рой определяются эксперимен-
экспериментально. Аналогичным образом в сополимерах
функция конфигурационной неоднородности
(см. стр. 105) описывает распределение последо-
последовательностей одинакового состава.
Дальний конфигурационный
порядок характеризует структуру доста-
достаточно протяженных (включающих десятки и
сотни скелетных атомов цепи) участков М. Те
же функции распределения, к-рые описывают
ближний порядок, могут «перерасти» в функции
для описания дальнего порядка, если они рас-
распадаются на дискретные термы, характеризу-
характеризующие очень большие последовательности звень-
звеньев с одинаковой стереорегулярностью (стерео-
блоки), одинаковым составом (блоки в блок-
сополимерах) и т. п.
Конфигурация цепи в целом
определяется взаимным расположением круп-
крупных последовательностей звеньев, к-рые исчер-
исчерпывающим образом м. б. охарактеризованы
дальним конфигурационным порядком. Напр.,
в случае разветвленных М. конфигурация оп-
определяется характером чередования ветвей,
распределением их длин, их стереорегулярно-
стереорегулярностью, составом (в случае привитых сополиме-
сополимеров) и т. п.
Поскольку конфигурация цепи в целом яв-
является суперпозицией локальных конфигура-
конфигураций, к-рые сами по себе м. б. достаточно слож-
сложны, число возможных конфигураций М. весьма
велико.
В табл. 1 приведен возможный вариант клас-
классификации гомополимеров по их конфигура-
конфигурационным характеристикам. Наряду с линейны-
линейными и разветвленными М. в таблицу включены
М. с более сложными конфигурациями, напр,
лестничные и сетчатые. Для М. лестничных
полимеров характерен новый тип нарушения ре-
регулярности, относящийся к конфигурации М.
в целом (несмотря на определенную локализа-
локализацию этого нарушения): разрывы тяжей. Такое
нарушение может существенным образом по-
повлиять и на конформацию (табл. 2). Такие же
типы нарушения регулярности должны прини-
приниматься во внимание у плоских и пространст-
пространственных сетчатых М.
109
МАКРОМОЛЕКУЛА
110
Продолжение табл. 1
Типы макромо-
макромолекулы
Сетчатые макромо-
макромолекулы
То же
Тип конфигурации
или характер стерео-
изомерии
Плоские сетчатые
B-мерный аналог
лестничных)
Пространственно-
сетчатые
статистические
упорядоченные
Схема
Конформация
Конформация представляет собой физич. ха-
характеристику М., производную от конфигура-
конфигурации. Формально «переход» от конфигурации к
конформации производится посредством учета
микробраунова движения звеньев и боковых
групп. Конформация — это, в общем случае,
переменное распределение в пространстве ато-
атомов и атомных групп, образующих М. Кон-
Конформация характеризуется постоянными (в пер-
первом приближении) валентными углами и пере-
Вулканизационные сетки менными (благодаря возможности более или
Трехмерный аналог лест- менее заторможенного вращения около одиноч-
аналог крис- ных связей хребта цепи или боковых групп)
ориентациями валентных связей. При полно-
полностью заторможенном внутреннем вращении (как
ничных или
таллич. решетки
* для виниловых полимеров предполагается, что плоскость транс- rTTVuap N яятсттттттпттммчпттттяттятп^
зигзага (хребта цепи) перпендикулярна плоскости чертежа**. Пунктиром в слУчае гч-алкилполиизоцианатов;
изображены боковые радикалы в р-положении.
Количественной характеристикой конфигурации ста-
тистич. сетчатых М. является плотность сшивки, т. е.
средняя степень полимеризации участка цепи между
узлами сетки (см. Вулканизационная сетка, Трехмер-
Трехмерные полимеры). Регулярные сетки также обладают
практически бесконечной молекулярной массой, одна-
однако их удобнее характеризовать по аналогии с кри-
кристаллами.
Конфигурацию М. определяют методами рентгено-
структурного анализа и электронографии, ИК-спектро-
скопии, ЯМР, двойного лучепреломления в потоке
(ДЛП) и др. Нек-рые из этих методов «настроены»
на одну какую-нибудь конфигурационную характери-
характеристику (или, что эквивалентно, на один определенный
конфигурационный уровень), т. е. особенно чувстви-
чувствительны к ней. Так, ЯМР во многих случаях позво-
позволяет количественно охарактеризовать ближний кон-
конфигурационный порядок в гомо- или сополимерах;
ДЛП — анизотропию и, в конечном счете, конфигура-
конфигурацию звена.
Расчет геометрич. параметров конфигурации произ-
производится обычно на основании комбинированных дан-
данных нескольких методов с применением валентно-
оптич. схемы. Суть этой схемы сводится к следующему.
От ф-лы типа (8) переходят к более детализированному
изображению соответствующего участка М. (звена,
диады и т. п.) с учетом вандерваальсовых радиусов,
длин связей, величин валентных углов и т. п. Весьма
удобно использовать для этого макроскопич. атомные
модели типа Стюарта — Бриглеба или чертежи, пред-
представляющие собой изображение соответствующих
«атомных конструкций» (см., напр., рис. 4, б). Для
подобной модели можно рассчитать нек-рые опреде-
определяемые на опыте оптич. характеристики, напр, внут-
внутреннюю анизотропию звена. Несовпадение рассчи-
рассчитанных и измеренных значений указывает на неточ-
неточность построения модели; иногда для приведения в
соответствие экспериментальных и вычисленных харак-
характеристик оказывается необходимым учесть деформа-
деформацию одной или нескольких связей или углов. Так ме-
методами последовательных приближений удается до-
достигнуть максимального соответствия между структурой
модели и находимыми на опыте характеристиками.
Использование валентно-оптич. схемы существенно
упрощается, если М. обладают оптич. активностью.
Соответственно, измерения оптич. активности (диспер-
(дисперсия, ИК-дихроизм, эффект Коттона) являются важными
методами экспериментального изучения конфигура-
конфигураций нек-рых классов М.
В заключение этого раздела заметим, что пока из-
известен единственный случай (N-алкилполиизоцианаты —
см. табл. 2), когда конфигурация линейной М. предо-
предопределяет не только конформацию, но и ее жесткость.
ция совпадает с конфигурацией. Обычно же М.
способна принимать бесконечно большое число
«мгновенных» конформации, изменяющихся непрерыв-
непрерывным или прерывным образом только за счет внут-
внутреннего теплового движения (без химич. реакций).
Конформационные характеристики, будучи произ-
производными от конфигурационных, вместе с тем напоми-
напоминают их в том смысле, что здесь тоже имеет место
иерархия и соответственно суперпозиция уровней.
Кроме того, между конфигурацией и конформацией
существует обратная связь: приобретение опред. кон-
конформации в кристаллич. решетке или даже отдельной
М. зачастую связано с нек-рой деформацией валентных
углов и связей, легче всего регистрируемой спектро-
спектроскопически. Ниже приводится схема иерархии конфор-
конформации.
Конформация звена. Как и в случае
конфигурации звена, здесь возможны подуровни —
конформация боковых групп или конформация ске-
скелетных атомов. Та же ф-ла (9), к-рая была приведена
как пример сложности конфигурации боковой группы,
может служить примером и сложной конформации:
боковая группа может вращаться как целое или час-
частями. Для понимания конформации звена важно отме-
отметить, что число степеней свободы (а значит и возмож-
возможных конформации) боковой группы у орmo-изомера
меньше, чем у иа?>а-изомера, из-за водородной связи,
замыкающей псевдоцикл, смежный с бензольным коль-
кольцом. Как было отмечено ранее, образование этой водо-
водородной связи также влияет и на конфигурацию соответ-
соответствующего участка звена. Т. обр., упоминавшаяся
обратная связь проявляется и на уровне конформации
звена.
Конформация скелетных атомов не может рассмат-
рассматриваться в отрыве от связанных с ними радикалов;
она характеризуется по полной аналогии с конформа-
циями аналогичных звеньев простых молекул. Извест-
Известно, что такие простые молекулы, как «-бутан, дихлор-
дихлорэтан и др., представляют собой смесь поворотных изо-
изомеров. Непосредственной причиной возникновения по-
поворотной изомерии является то, что может существо-
существовать не одно, а несколько состояний с минимумами
свободной энергии, т. е. несколько стабильных кон-
формаций. В случае дихлорэтана минимуму соответ-
соответствует гарбшс-конформация, когда атомы хлора (если
рассматривать молекулу с торца) повернуты друг от-
относительно друга на 120°. Аналогичным образом мо-
можно характеризовать поворотноизомерные конфор-
конформации звена. В виниловых полимерах обычно при-
приходится иметь дело с тремя поворотными изомера-
изомерами: одним транс- и двумя «свернутыми», или гош-изо-
мерами.
М. в целом, из-за существования различных кон-
конформации звеньев, представляет собой сложную
«смесь» поворотных изомеров: каждое звено может
ill
МАКРОМОЛЕКУЛА
112
находиться в нескольких дискретных состояниях с
разной энергией. Однако переход из одного состояния
в другое не требует химич. реакции и может проис-
происходить либо под действием тепловых флуктуации,
либо внешней силы (например, при ориентации по-
полимера).
В случае жесткоцепных М. конформация звена
меняется непрерывным образом в результате крутиль-
крутильных колебаний относительно положения равновесия
(с минимальной энергией; см. ниже стр. 115 и Гибкость
макромолекул).
Ближний конформационный поря-
порядок. В случае виниловых или винилиденовых гибко-
цепных полимеров поворотная изомерия более ярко
проявляется во взаимных положениях звеньев; при
этом реализуются те же три основные поворотноизо-
мерные формы: одна транс и две гош. Соответственно,
ближний порядок характеризуется конформациями
диад, триад, тетрад... и т.д.
Дальний конформационный поря-
порядок. В случае стереорегулярных полимеров могут
возникнуть дополнительные по сравнению с рассмот-
рассмотренными энергетич. осложнения, обусловленные взаи-
взаимодействием боковых радикалов, оказывающихся
слишком близко друг к другу, если цепь вытянута.
Простых поворотов относительно С—С-связей при
этом оказывается недостаточно, чтобы свести энергию
взаимодействия к минимуму. Поэтому цепь на доста-
достаточно большом протяжении приобретает, напр., спи-
спиральную конформацию (класс спирали
Ху определяет число звеньев X на число витков у).
Образование спиральной конформации, как правило,
вызывает небольшие изменения конфигурационных
параметров — деформации валентных углов и связей.
Поэтому спиральная конформация вполне устойчива
либо в кристаллич. решетке, либо — у свободной М.—
при ее фиксации дополнительными внутримолекуляр-
внутримолекулярными взаимодействиями. Однако элементы флуктуи-
флуктуирующей спиральной структуры, «блуждающей» по
цепи, сохраняются и у стереорегулярных М. в р-ре.
Мгновенная конформация цепи напоминает при этом
сегментальную модель В. Куна (см. Гибкость макро-
макромолекул) с жесткими спиральными сегментами пере-
переменной длины. Но длины и ориентации этих «сегмен-
«сегментов» непрерывно меняются. Такая модель получила
название статистич. зигзага.
Конформация цепи в целом, или
макромолекулярная конформация
(иногда ее называют также макромолекулярной струк-
структурой),—это размеры и конкретная форма, к-рую
М. приобретает либо в результате теплового движения
(статистич. клубок), либо в результате так наз. сил
дальнодействия, т. е. взаимодействия с растворителем,
взаимодействия далеких (если считать вдоль вытянутой
цепи) звеньев. В результате указанных взаимодей-
взаимодействий конформация фиксируется водородными связями
или силами электростатич. или лиофобной природы и
др. (см. также стр. 119).
Методы исследования конформации вплоть до даль-
дальнего конформационного порядка — те же, что и для
конфигураций, хотя иногда непосредственно измеряет-
измеряется др. характеристика: напр., методом ДЛП при ис-
исследовании конфигураций определяют анизотропию
звена, а при исследовании конформации — анизотро-
анизотропию сегмента (см. табл. 2).
Для расчета макромолекулярыых конформации на-
наряду с методами, применяемыми для расчета конфи-
конфигураций, используется метод энергетич. карт. Он до-
довольно трудоемок и связан обычно с применением элек-
электронно-вычислительных машин. Суть его состоит в
Таблица 2. Характеристика структуры некоторых полужестких и структурированных
Полимер
Полистирол атактический*
Нолидиметилсилоксан *
Полиметилметакрилат атактиче-
атактический*
Тринитроцеллюлоза
Трибензоатцеллюлоза
Монофенил ацетат целлюлозы
Поли-К-изобутилмалеинимид
Поли-К-бутилизоцианат
Поли-7-бензил-Ъ-глутамат
Полиметакрил-Я - фениловый эфир
цетилоксибензойной к-ты
Лестничный полифенисилоксан
Дезоксирибонуклеиновая к-та
Тропоколлаген
Привитой сополимер (гребневидный
тип) стирола и поли метил мет а-
крилата
Полиэлектролиты
натриевая соль сульфоэфира
целлюлозы; y=40; неионизиро-
ванное состояние (ионная сила
1=0,2)
то же; сильная ионизация (/=
= 10)
Поли-2-метил- 5-винил - N-бутилпи-
ридинбромид
1= 10~х
/=ю-2
/=10
(по данным В. Н.
Конформация
Статистич. клубок
То же
п
и
п
п
п
Упругая струна
а-Спираль Полинга — Кори
Локальная внутрицепная
жидкокристаллич. упоря-
упорядоченность боковых групп
Рыхлый клубок
Двойная спираль Крика —
Уотсона; рыхлый клубок
Тройная спираль типа
полипролин II; упругая
Червеобразная цепь с боль-
большим эффективным попереч-
поперечником
Статистич. клубок
То же
Цветкова)
Мол.
масса
М-10-5
1,0
1,0
1,0
2,0
4,7
2,0
2,0
1,6
1,8
1,8
2,0
30
35
6,0
1,0
' 1,0
3,0
3,0
3,0
Характе-
ристич.
вязкость
Ш,
дл/з
0,3
0,15
0,2
6,8
5,0
7,1
0,6
7,7
3,4
1,1
1,0
12
12
0,5
2,9
8,7
4,2
6,6
12
Анизотро-
Анизотропия сег-
сегмента
-145
Н-5
+25
-500
-900
400
1600
8500
10000
-1000
-1800
-40000
470
300
900
-400
-1400
—2000
макромолекул
Персистентная
длина X, нм(А)
1A0)
0,7G)
1A0)
10A00)
10A00)
10A00)
1,9-2,0A9-20)
130A300)
120A200)
30C00)
7-20G0-200)**
30C00)
150A500)
5—10E0 — 100)
100
300
10
35
50
Диполь-
ный мо-
момент, D
0
—
1,5
1-2
1-2
—
0
800
300
200
0
—
_
1,5
—
—
—
* Гибкоцепные полимеры; даются для сравнения.
** Колебания X обусловлены различной степенью дефектности (разрывы тяжей).
113
МАКРОМОЛЕКУЛА
114
следующем. Макромолекулярная конформация пред-
представляет собой суперпозицию низших конформацион-
ных уровней, для к-рых равновесные (т. е. соответ-
соответствующие минимуму свободной энергии) конформации
м. б. сравнительно просто рассчитаны. Строится трех-
трехмерный график зависимости свободной энергии дис-
дискретных конформации от двух переменных — обычно
углов и расстояний между соответствующими группа-
группами. Такой график, естественно, содержит «хребты»
(энергетически «запрещенные» области) и «ложбины»
(«разрешенные» области). Плоское сечение графика
вполне аналогично географич. карте, описывающей
сложный рельеф, и представляет собой энергетич.
карту. Областям минимума энергии соответствуют раз-
разрешенные конформации. Метод энергетич. карт приме-
применяется и для расчета дальнего конфигурационного
порядка.
Как и в случае валентно-оптич. схемы, расчеты конт-
контролируются экспериментальными данными: необходи-
необходимые параметры поставляются такими физич. методами,
как дифракция рентгеновых лучей в больших и малых уг-
углах, рассеяние света, гидродинамич. методы, ДЛП и др.
Статистическая физика изолированных линейных
и лестничных макромолекул. Жесткость
М. не могут существовать в равновесии в газовой
фазе. Однако, как уже отмечалось, реалистич. моделью
газового состояния М. является разб. р-р. Чтобы пре-
пренебречь взаимодействием звеньев, надо рассматривать
не просто разб. р-р, а р-р в 6-растворителе (см. Флори
д-температура), где все внутрицепные взаимодейст-
взаимодействия компенсируются взаимодействиями звеньев с рас-
растворителем. В этих условиях М. приобретает невоз-
невозмущенные (квазиидеальные) размеры и конформацию.
Такая М. представляет собой статистич. клубок, раз-
размеры к-рого наглядно представляются с помощью
координационной сферы (рис. 1), т. е.
усредненного по времени и пространственным коорди-
координатам объема, к-рый занимает М., претерпевающая
внутреннее (микробрауново) и внешнее (макробрауно-
во) тепловое движение. Макробрауново движение скла-
складывается из поступательной и вращательной диффузии
М. как целого.
Координационная сфера — это занятая звеньями
М. область, к-рую удалось бы зафиксировать в вооб-
воображаемом опыте с фотографированием М. в течение
длительного времени при фиксированном положении
фото устройства относительно центра тяжести М.
В в-растворителе концентрация полимера в координа-
координационной сфере не превышает 3%, а в хороших раство-
растворителях м. б. на порядок меньше. В этом, как уже
отмечалось, состоит главное отличие статистич. клубка
от глобулы.
В линейных цепных М. микробрауново движение
реализуется в результате поворотов звеньев относи-
относительно друг друга. Если в воображаемом опыте рас-
растянуть М. за концы, в результате микробраунова дви-
движения она вновь приобретет конформацию клубка.
Поскольку эта конформация м. б. приобретена боль-
большим числом путей — тем большим, чем больше степень
полимеризации — наиболее вероятные размеры клубка
соответствуют максимуму энтропии. Соответственно,
сила, возвращающая растянутую цепь в конформацию
клубка, имеет энтропийную природу; в макроскопич.
полимерных телах она проявляется при реализации
высокоэластического состояния.
Статистич. рассмотрение реальных геометрич. свойств
М. сопряжено с большими математич. трудностями.
Простейшая модель М.— свободносочлененная цепь,
в к-рой ориентация каждого звена произвольна и
не зависит от ориентации предыдущего (см. Гибкость
макромолекул). На самом деле М.— кооперативная
система, и ориентация каждого звена зависит по край-
крайней мере от ориентации ближайших соседей. В про-
простейшем варианте это ограничение вводится фиксацией
валентных углов. В следующем приближении прини-
принимается во внимание заторможенность внутреннего вра-
Рис. 1. Типы макромолекулярных структур (пунктиром
всюду обозначены границы координационной сферы): а —
обычный статистич. клубок; б — клубок, частично сшитый
водородными связями (точечные линии); в — глобула; г —
молекулярная мицелла, образовавшаяся из блоксополимера
типа ABA; д — сегрегированная структура, образованная
таким же блоксополимером; е — структура разбавленного
р-ра М. (координационные сферы не перекрываются; кон-
концентрация собственно полимера внутри координационных
сфер мала, но все же больше средней концентрации р-ра).
щения, определяющая гибкость М. Полный учет коопе-
ративности следовало бы производить, учитывая корре-
корреляции внутренних вращений отдельных звеньев.
Безотносительно от выбора модели, главной геомет-
геометрич. характеристикой клубка является среднеквадра-
среднеквадратичное расстояние между его концами или связанная
с ним величина — среднеквадратичный радиус инер-
инерции (более удобная, когда «концов» много, напр,
в разветвленных М.). При отсутствии корреляций
между внутренними вращениями звеньев средний
квадрат расстояния между концами М. выражается
ф-лой
1+1
1
1— COSU 1 —TJ
A0)
где ft — угол, дополнительный к валентному (см. ф-лу
8), I — длина мономерного звена, N — степень поли-
полимеризации, t] — средний косинус угла внутреннего
вращения, в конечном счете и определяющий жест-
жесткость М.:
2ч
2ч 2тс
cos? = J exp [—U(y)/kT] cos ydyj j exp [—U(<p)/kT\d<?
i
(ii)
где U (ф) — потенциальная энергия внутреннего вра-
вращения. При U (ф) > кТ (высокая жесткость) ф~ла
115
МАКРОМОЛЕКУЛА
116
<10) переходит в ф-лу СЕ. Бреслера и Я. И. Френ-
Френкеля, к-рая соответствует модели М. в виде плоской
ленты, способной ограниченно закручиваться и совер-
совершать крутильные колебания:
4г A2)
Ситуация, описываемая ф-лой A2), приблизительно
соответствует большинству производных целлюлозы,
а наиболее близко — лестничным М. Таким образом,
лестничные М. должны обладать конформацией ста-
тистич. клубка, но с малой степенью скрученности;
наличие дефектов в одном из тяжей «лестницы» будет
приводить к увеличению степени скрученности и соот-
соответственно уменьшению </г2>.
Невозмущенные клубки гибкоцепных М. обычно на-
называют гауссовыми. Дело в том, что достаточно длин-
длинная реальная цепь м. б. заменена эквивалентной ей
по размерам и гидродинамич. свойствам свободно-
сочлененной, т. к. корреляции между ориентациями
достаточно удаленных друг от друга звеньев нет. Ины-
Иными словами, цепь из N звеньев длины I «подменяется»
цепью из Z сверхзвеньев — статистич. элемен-
элементов (сегментов) длиной А, ориентации к-рых
в пространстве независимы. При этом задача определе-
определения <&2> сводится к так наз. диффузионной задаче,
решаемой в теории брауновского движения. При Z > 1,
<^h2^> = 2Л2, а вероятность того, что размеры такой
цени заключены в интервале от h до h + dh выражается
гауссовой функцией:
W (h)dh= (-|- izZA^j'2 bn[exv(—3h2/2ZA2)]h2dh A3)
Взаимодействие макромолекул с растворителем.
Объемные эффекты
Более строгая теория, наряду с учетом корреляции
внутренних вращений, требует также учета объемных
эффектов, предусматривающих невозможность попада-
попадания в одну точку двух или более удаленных друг от
друга вдоль цепи звеньев. Объемные эффекты обуслов-
обусловливают набухание клубка, характеризуемого пара-
параметром набухания а:
«=:<
(hi >V
A4)
где индекс «в» соответствует невозмущенным разме-
размерам. Смысл параметра а м. б. понят из рассмотрения
координационной сферы рис. 1, если трактовать ее
как миниатюрную осмотич. систему, где мембрана за-
заменена связями между звеньями. При положительных
отклонениях от идеальности (взаимодействие звеньев
с растворителем сильней, чем друг с другом) осмотич.
силы раздувают координационную сферу, а при отри-
отрицательных (тенденция звеньев к агрегации) клубок и
соответствующая координационная сфера сжимаются
(о переходе клубок — глобула см. ниже). В первом
приближении теории эти обстоятельства выражаются
ф-лой:
fVi A5)
где в — в-температур'а в данном растворителе, ty1— эн-
энтропийный коэффициент, характеризующий тенден-
тенденцию к преимущественному взаимодействию звеньев
друг с другом или с растворителем, Ст — коэфф. раз-
размерности,
27
М
3/з
A6)
где М — мол. масса, Nд — число Авогадро, и1 — мо-
молярный объем растворителя, и — парциальный уд.
объем полимера.
Главным и не всегда оправдываемым на опыте допу-
допущением, определяющим характер приближения в ф-ле
A5), является предположение о неизменности <й^>
в любом в-растворителе. Кроме того, ф-ла A5) не при-
принимает во внимание существование у многих полимеров
второй, высокотемпературной 0-точки, к-рую, в отли-
отличие от 01-точки Флори, иногда называют 62-т о ч к о й
Роулинсона. Последняя часто расположена
вблизи крнтич. темп-ры жидкость — пар растворителя.
Хотя по нек-рым данным значения <&2в> ряда полиме-
полимеров в обеих 6-точках почти совпадают, нет оснований
полагать, что это — обязательное правило. (Поэтому
на рис. 2, б у 02-точки поставлен вопросительный знак.)
Иными словами, вопреки ф-ле A5), размеры реальных
М. изменяются с темп-рой немонотонно (см. рис. 2).
Рис. 2. Зависимость растворимости и размеров макромоле-
макромолекулы от темп-ры: а — типичная фазовая диаграмма с верх-
верхней (КО и нижней (К2) критич. темп-рами смешения; &t —
тета-точка Флори, в2 — тета-точка Роулинсона; ф2 — объ-
объемная доля полимера в р-ре; б — зависимость среднеквадра-
среднеквадратичного расстояния между концами клубка <h2>a/2 и пара-
параметра набухания а от темп-ры Т. < h2e>72 — невозмущен-
невозмущенные размеры; вопросительным знакам ниже горизонтальной
пунктирной прямой соответствуют мало исследованные
области; вопросительный знак при стрелке, указывающей
на размеры в в2, связан с неясностью вопроса о том, совпа-
совпадают ли размеры в в2 и Qlt
В еще более грубом приближении, используемом
при построении гидродинамич. теорий, принимают
а~Мг A7)
что является приближенным решением ур-ния A5);
8 — малый параметр порядка 0,1.
Таким образом, размеры реального клубка опреде-
определяются достаточно сложной комбинацией близкодей-
ствий (сводимых к жесткости цепи) и дальнодействий.
Последние в 8-точке можно не учитывать. Статистика
набухших клубков уже отличается от гауссовой. К от-
отклонениям от гауссовой статистики приводит повышен-
повышенная жесткость или возникновение элементов вторичной
структуры (напр., спиральной — см. ниже), когда
существенно усиливается кооперативность системы,
т.е. корреляция ориентации соседних звеньев. В слу-
случае полиэлектролитов комбинация эффектов близко-
действия и дальнодействия одной природы (электро-
статич. отталкивание) также приводит к резким от-
отклонениям от распределения размеров A3).
Методы определения размеров и формы макромолекул.
Гидродинамические характеристики
Со среднеквадратичными размерами М. связаны та-
такие важнейшие гидродинамич. характеристики поли-
полимеров, как вязкость характеристическая, коэфф. диф-
диффузии (см. Диффузионное движение макромолекул),
коэфф. седиментации. Эти параметры, служащие для
определения размеров и жесткости М., связаны также
с мол. массой соотношениями Марка — Куна — Ху-
винка, имеющими след. вид соответственно для харак-
117
МАКРОМОЛЕКУЛА
118
теристич. вязкости и коэфф. поступательного трения
(через к-рый выражаются коэфф. седиментации и
диффузии):
Ы-К^М* A8)
= KfMb
причем, как правило,
Ъ =
О+1
A9)
B0)
Ф-лы A8) и A9) получаются из соотношений, являю-
являющихся аналогами ур-ний Эйнштейна и Стокса для вяз-
вязкости и коэфф. поступательного трения:
f=P{ h\
B1)
B2)
где так наз. параметр Флори Ф зависит от
гибкости цепей и характера статистич. распределения
их размеров; для гауссовых цепей Ф = 2,84-1021
([т]] в дл/г) и убывает с увеличением жесткости или на-
набухания; другой параметр Флори — Р слабо зависит
от этих факторов, и его принимают обычно равным 5,1.
Из формул B1) и B2) непосредственно следует ра-
равенство B0).
Через [г|] выражаются и такие-характеристики гиб-
гибкости М., как длина статистич. элемента А или пер-
систентная длина А, (см. Гибкость макромолекул).
Часто возникают большие трудности с измерением [г\]
в ©-растворителе. В этом случае пользуются экстра-
экстраполяцией Штокмайера — Фиксмана, к-рая приобре-
приобретает особое значение как метод детектирования или
диагностирования вторичных структур М. Как следует
из ф-лы A5), при М->0 а —> 1, что вполне понятно,
ибо с укорочением цепи эффекты дальнодействия исче-
исчезают. Полагая, что в 8-точке ф-ла A8) преобразуется
в *"лу , I#i/
fo] = KB м'2 B3)
можно считать параметр Кв характеристикой невозму-
невозмущенных размеров. Достаточно очевидным образом К&
найдется из графич. построения [ц]/М^2 = F(Mx/z)
(рис. 3).
Тангенс угла наклона прямых на графиках рис. 3
пропорционален второму вириальному коэффициенту
f-njl Рис. 3. Экстраполяция по
1 IJ - Штокмайеру — Фиксману
для определения К@ в
формуле Гп]0 = к@ МХ12-
Прямая 1 соответствует 0-
точке, 2 — хорошему раст-
растворителю (при отсутствии
вторичных структур К® не
должно зависеть от раст-
растворителя); «?, 4,5 — раз-
различные варианты «анома-
«аномалий»: з — указывает на су-
существование «вулканиза-
ционной» или «конденса-
MV2
ционной» структуры (типа рис. 1, б или г); 4 — на появле-
появление «второго ©-размера», микросегрегацию или наличие
локальной вторичной структуры (как в полиметакриловой
к-те); 5 — на глобулярную структуру.
осмотич. ур-ния, к-рый характеризует степень и ха-
характер неидеальности р-ра. В свою очередь, второй
вириальный коэфф. связан однозначным соотношением
с параметром набухания а. Так. обр., наряду с разме-
размерами М. указанное построение позволяет определить
важные термодинамич. характеристики р-ров.
Расстояние между концами цепи не является един-
единственной характеристикой размеров и формы клубков.
Реальная геометрич. форма клубка примерно соответ-
соответствует трехосному эллипсоиду, или «бобу»; надо только
помнить, что форма эта все время флуктуирует из-за
микробраунова движения. При больших степенях
полимеризации поперечными размерами цепи обычно
можно пренебречь (во всяком случае, когда речь идет
о гидродинамич. свойствах). Однако с уменьшением М
эффективный поперечник цепи уже оказывается сопо-
сопоставимым с контурной длиной и среднеквадратичными
размерами, приводя к «аномалиям» гидродинамич. по-
поведения олигомеров. [На самом деле это не аномалии,
а результат пренебрежения запретом на использование
соотношений типа A8) или A9) при малых молекулярных
массах.]
Вопрос о влиянии эффективного поперечника (рис. 4)
на гидродинамич. свойства М. был решен еще В. и
Г. Кунами в опытах, где молекулярные клубки имити-
Рис. 4. Модели макромо-
макромолекул, используемые для
определения их эффектив-
эффективного гидродинамич. попе-
поперечника d: a — проволоч-
проволочная модель клубка Кунов;
б — модель изотактич.
триады в виниловой цепи (стрелка показывает, что вытя-
вытянутая цепочка должна свернуться в спираль — см. ниже
рис. 5; d—диаметр цилиндра, к-рый получился бы при
вращении цепи вокруг продольной оси); в — структура при-
привитого сополимера стирола (ветви) и метилметакрилата
(по В. Н. Цветкову).
ровались проволочными макроскопич. моделями с пря-
прямолинейными и дуговыми сегментами, а при расчетах
использовался принцип гидродинамич. аналогии. Для
[г|] и / были получены ф-лы, сходные с ф-лами наиболее
строгой теории Кирквуда — Райзмана, в к-рой при
расчетах использовалась модель «жемчужное ожерелье»
(расчет выполнялся для поливиниловой цепи; скелет-
скелетные атомы углерода изображались в виде «бисерин»).
Для модели с дуговыми сегментами ф-ла для [г[] имеет
вид:
AL*N ,
0,43
М
—= B4)
-1,6+2,3 lg(A/d)+ VL/A
где L — контурная длина, d — гидродинамич. толщи-
толщина, т. е. эффективный гидродинамич. поперечник. Ра-
Разумеется, в этой ф-ле эффекты набухания не прини-
принимаются во внимание (т. е. она соответствует как бы
в-точке). Поскольку L пропорционально М, при боль-
больших М ф-ла B4) сводится к [г|] ~ М1/2, но зато при
малых М легко определяется d. Аналогичным образом
d находится по данным измерений коэфф. диффузии
или седиментации. Переписывая ф-лу B4) в виде
М1Ы = -«1
(A/d)+as УМ
B5)
убедимся, что для нахождения d нужно произвести
графич. экстраполяцию к М = 0 на графике — =
= Fx (M^2). Значение А яри этом определяется из
независимых опытов. Построения такого рода весьма
полезны при исследовании олигомеров, тогда как ис-
использование ф-лы A8) в этих случаях зачастую лишено
смысла и приводит к ошибкам.
Позднее были предложены более строгие модели
макромолекулярной структуры и развиты методы не-
непосредственного определения гидродинамич. попереч-
поперечника (упоминавшаяся уже модель «жемчужное оже-
ожерелье» и различные ее модификации, а также пер-
систентная модель), но методы определения d, основы-
119
МАКРОМОЛЕКУЛА
120
вающиеся на использовании простой экстраполяции,
предписываемой ф-лой B5), остаются наиболее надеж-
надежными и точными.
Помимо гидродинамич. методов, размеры клубков
определяются по данным рассеяния света или малоугло-
малоуглового рассеяния рентгеновских лучей. Последний метод
позволяет также непосредственно определить персис-
тентную длину и иногда d.
Важной характеристикой клубков является также
анизотропия формы, определяемая методом ДЛП.
Этот же метод весьма чувствителен при обнаружении
агрегатов М. и вторичных молекулярных структур.
Вторичные макромолекулярные структуры
Основные определения. Уровни макромолекулярной
организации. Макромолекулярная конформация пред-
представляет собой результат суперпозиции ближнего и
дальнего конформационного порядка; макромолекуляр-
макромолекулярная конформация может реализоваться различными
способами. В тех случаях, когда возникают конфор-
мации, отличные от статистического клубка и харак-
характеризуемые жесткостью формы, или структурной
жесткостью, принято говорить о вторичных (синони-
(синонимы — внутрицепные, внутримолекулярные) структу-
структурах М.
Как уже указывалось, ответственны за образование
таких структур обычно водородные, электростатические
или лиофобные связи.
Во многих случаях в образовании вторичной струк-
структуры участвуют взаимодействия различной природы.
По аналогии с конформациями, вторичные макромоле-
макромолекулярные структуры также могут образовывать опре-
определенную иерархию уровней организации, к-рые спо-
способны к взаимным превращениям лишь при одновре-
одновременном распаде и перераспределении многих связей
нековалентной природы.
Представление о последовательных уровнях макро-
макромолекулярной организации было впервые выдвинуто
Линдерштрем-Лангом применительно к белкам и затем
обобщено Дж. Берналом на любые типы М. Под пер-
первичной структурой белка понимают общее
число пептидных связей в Мс и характер чередования
боковых радикалов аминокислотных остатков. Как
известно, а,Ь-полипептиды способны принимать упо-
упорядоченные конформации типа а-спирали, кросс-[3-
формы и др. Последовательность упорядоченных и
неупорядоченных участков белковой цепи (однозначно
предопределенную первичной структурой — см. ниже)
называют вторичной структурой. По-
Поскольку развернутая белковая М. в водной среде не-
нестабильна из-за обилия гидрофобных боковых радика-
радикалов, она сворачивается в относительно компактное об-
образование — квази-глобулу (отсюда термин «глобуляр-
«глобулярные белки») с устойчивой формой; эта внешняя форма
структурированной молекулы была названа третич-
третичной структурой.
На основе анализа взаимодействий, придающих
устойчивость каждому из названных уровней молеку-
молекулярной организации, предложено характеризовать эти
уровни относительной стабильностью связей. Так, пер-
первичная структура определяется ковалентными связями
полипептидной цепи, вторичная — водородными свя-
связями между витками а-спирали или складками кросс-
C-формы, третичная — «гидрофобными» связями между
неполярными радикалами, а также более слабыми
водородными связями между далекими вдоль цепи
звеньями и солевыми мостиками и т. п. Иными слова-
словами, восхождение от низших к высшим уровням моле-
молекулярной организации характеризуется постепенным
ослаблением связей; устойчивость же уровней с «ослаб-
«ослабленными» связями обусловлена эффектами коопера-
тивности (см. стр. 128). Бернал дополнил схему пред-
представлением о четвертичной структуре—
понятием, характерным для М. биополимеров. Во мно-
многих случаях (аллостерич. ферменты, гемоглобин) не-
несколько М., обладающих третичной структурой, спо-
способны образовывать стехиометрич. комплексы, к-рые
по гидродинамич. свойствам и по биологич. активности
действуют как автономные единицы (при этом говорят,
что М. состоят из субмолекул). В других случаях не-
несколько цепей, не обладающих третичной структурой,
объединяются в двойные или тройные спирали (ну-
(нуклеиновые к-ты, коллаген), к-рые тоже ведут себя
подобно самостоятельным одиночным М. Мультиспи-
ральные М. способны к дальнейшей агрегации друг
с другом или с глобулярными молекулами; поэтому
Бернал считал возможным говорить о структу-
структурах высших порядков, постепенно перехо-
переходящих в элементы надмолекулярной структуры (также
обладающие ограниченной автономностью).
Такая схема с нумерацией уровней, наряду с удоб-
удобствами, обладает и рядом недостатков. Так, мульти-
спиральные М. коллагена или нуклеиновых к-т, если
следовать букве классификации, «перепрыгивают» через
третичную структуру; М. глобулярных белков, отно-
относящиеся к категории пространственно-сетчатых, не
укладываются в «силовую» схему (к какой структуре
относить ковалентные связи между далекими вдоль
цепи соседями, образующие макроскладки?) и т. п.
Кроме того, при таком подходе теряется принципиаль-
принципиальное различие между конфигурационными и конформа-
ционными характеристиками.
В случае М. синтетич. полимеров удобнее говорить
о вторичных молекулярных структурах, не придавая
слову «вторичный» тот смысл, к-рый оно имеет в струк-
структурных схемах для биополимеров. Однако и здесь
характер этих структур существенно зависит от типа
ответственных за них внутрицепных связей: так, во-
водородные связи способствуют образованию более про-
протяженных структур, а лиофобные (аналог «гидрофоб-
«гидрофобных» в случае белков) — более компактных.
С определенной степенью условности можно подраз-
подразделить вторичные структуры линейных М. на линейно-
упорядоченные, компактные («глобулярные») и про-
пространственно-сетчатые (вулканизационные); имеются и
здесь промежуточные формы.
Линейно-упорядоченные структуры. Истинно стерж-
невидными М. (а, на первый взгляд, именно такие М.
должны были бы представлять линейно-упорядоченные
М.) м. б. лишь молекулы N-алкилполиизоцианатов и
нек-рых координационных полимеров. Большинство
известных линейно-упорядоченных жестких конфор-
конформации связано со спиральной упорядо-
упорядоченностью. При этом реализуется истинная, на-
напоминающая кристаллическую, периодичность струк-
структуры в направлении оси спирали (один из синонимов
линейно-упорядоченной структуры — линейно-
кристаллическая структура).
Как мы видели, закручивание цепи в спираль воз-
возможно только у достаточно стереорегулярных М.
(изо- и синдиотактических или с двойной тактичностью);
обусловлено оно энергетич. невыгодностью сближения
боковых радикалов, к-рое имело бы место, скажем,
при линейной гарбшс-конформации изотактич. вини-
виниловой цепи (рис. 5 и 6). Однако спирально-закручен-
спирально-закрученная цепь может сохранять конформацию статистич.
клубка (или зигзага) в р-ре, если только нет допол-
дополнительных причин, способствующих фиксации спи-
спирально-упорядоченной конформации.
Таких причин м. б. две: образование межвитковых
водородных связей, как в спиралях а,Ь-полипепти-
дов, или межвитковое сверхсопряжение (по существу,
также за счет водородной связи), как в изотактич.
высших полиолефинах, содержащих в боковой группе
двойную связь. Такие фиксированные спирали обла-
обладают жесткостью формы, к-рая м. б. зарегистрирована
121
МАКРОМОЛЕКУЛА
122
гидродинамич. или оптич. методами. С особым удоб-
удобством спиральная конформация регистрируется при
наличии оптич. энантиоморфных форм (напр., a,L-no-
липептиды всегда образуют правовращающую спираль)
Рис. 5. Схематиче-
Схематическое изображение
закручивания изо-
тактической поли-
ср виниловой цепи в
tA спираль 3, (вид сбо-
v ку и с торца): а —
' полностью вытяну-
вытянутая цепочка (пло-
(плоскость транс-зиг-
транс-зигзага перпендику-
перпендикулярна плоскости
рисунка); б — эта
же цепочка в кон-
формации спирали.
vi-
viпо дисперсии оптич. вращения, круговому дихроизму
и др. Гидродинамич. поведение жестких спиральных
М. подобно поведению стержневидных частиц и про-
проявляется в очень высоких значениях персистентной
длины X, стремлении экспонента в ф-ле A8) к предель-
предельному значению 1,7, высоких дипольных моментах,
увеличивающихся с мол. массой, и т. д.
Зарегистрировать незафиксированную вторичную
спиральную структуру труднее;- в этом случае, как
отмечалось, надо пред-
представлять себе клубок с
«пульсирующими» спи-
спиральными участками; по-
видимому, о переходе та-
Рис. 6. Зависимость свобод-
свободной энергии цепи, изобра-
изображенной на рис. 5, от угла
поворота соседних боковых
радикалов. После минимума
A20°) монотонный рост U
. i , . , обусловлен неизбежностью
я -тг Лж 9-тг Ф насилия над конфигурацией
2 z цепи.
ких клубков к конформации «чистого» клубка можно
судить по особенностям на температурных кривых пар-
парциального уд. объема. Изломы на таких кривых есть
практически у любых полимеров, но утрата элементов
выраженной вторичной структуры сопровождается бо-
более резкими изменениями парциального объема.
Следует указать, что жесткость, обусловленная ли-
линейно-упорядоченной вторичной структурой, отражает-
отражается на конформации так же, как скелетная жесткость,
а поэтому требуются особые типы экспериментов, чтобы
установить, какой тип внутримолекулярного взаимодей-
взаимодействия ответственен за существование вытянутой жест-
жесткой конформации.
Глобулярные и промежуточные структуры. Молеку-
Молекулярные мицеллы. Другие формы структурной жест-
жесткости проявляются в условиях, когда М. приобретают
компактные или «сегрегированные» конформации. Ука-
Указанием на такие конформации могут служить аномалии,
проявляющиеся при экстраполяции, показанной на
рис. 3. Как указывалось, экстраполяция величины
[т]]/~1/М к нулевому М должна дать параметр невозму-
невозмущенных размеров К® . Однако при наличии сил даль-
дальнодействия типа притяжения между отдаленными вдоль
цепи звеньями (напомним, что это м. б. водородные
связи или лиофобные силы; и те и другие регулируют-
регулируются составом растворителя и темп-рой) ситуация ме-
меняется. Если обычно клубок в хорошем растворителе
занимает координационную сферу, в к-рой концент-
концентрация собственно полимера меньше 1 %, то структу-
структурированная М. может оказаться значительно более
компактной (см. рис. 1, в). Это приведет к тому, что
на графиках типа рис. 3 будет несколько значений
К& для разных растворителей или могут получиться
линии с отрицательным наклоном. Поскольку наклон
определяется экспонентом ур-ния [т| ] = .ЙГ Ма и равен
нулю при а = 0,5 (в 8-точке), отрицательный наклон
означает, что а < 0,5. Для «глобул», т. е. жестких
частиц, сохраняющих подобие при увеличении М и
не содержащих в своей координационной сфере раство-
растворителя, а — 0.
В более или менее чистом виде глобулярные струк-
структуры возникают ниже 8-точки (механизм этого превра-
превращения рассмотрен на стр. 126). М. блоксополимеров
способны к образованию структур, названных моле-
молекулярными мицеллами (см. рис. 1, г).
В этом случае звенья нерастворимого в данном раство-
растворителе участка (или участков) М. агрегируют друг
с другом, образуя глобулярное ядро мицеллы, к-рое
окружено звеньями растворимого блока (или блоков),
удерживающими М. в целом в р-ре.
Возникновение молекулярных мицелл удобно ре-
регистрировать с помощью экстраполяции в координатах
b\VV~M — ~)/~М, используя метод «перекрестно-селек-
«перекрестно-селективных» и «нейтральных» растворителей. Под первыми
понимаются такие, где растворим лишь один тип бло-
блоков; использование этих растворителей должно при-
приводить к образованию одинаковых структур типа
изображенных на рис. 1, г и к одинаковым экстраполя-
циям типа линий 3 на рис. 3. При использовании «ней-
«нейтральных» растворителей, где растворимы блоки обоих
типов, вероятнее всего возникнет обычная конформа-
конформация статистич. клубка, и, соответственно, экстраполя-
экстраполяция будет иметь вид линии 2.
Взаимодействие М. блоксополимера с растворителем
может иметь и другой характер, приводя к образова-
образованию сегрегированных структур (см.
рис. 1, д). Если механич. смесь полимеров, соответ-
соответствующих блокам, в данном растворителе при данной
темп-ре не распадается на равновесные жидкие фазы,
обогащенные одним каким-либо полимерным компо-
компонентом, то клубок будет иметь нормальную конфор-
мацию типа рис. 1, а. Если же в макроскопич. системе
такое разделение на фазы происходит (часто — и не-
неудачно, ибо это название не отражает физич. сути
явления,— говорят при этом о «несовместимости» по-
полимеров в р-ре), то оно произойдет и в микросистеме;
размеры такого микросегрегированного клубка не
слишком отличаются от суммы размеров автономных
блоков.
Пространственно-сетчатые структуры. Такие струк-
структуры чаще всего возникают в молекулах бинарных
сополимеров с относительно небольшим числом звеньев,
способных к образованию водородных связей. Наибо-
Наиболее изучены в этом плане сополимеры, имеющие одним
из компонентов акриловую или метакриловую к-ту
(рис. 7); в обоих случаях получаются сшитые водород-
о н3с—о
Рис. 7. Схема образования межмолекулярных водородных
связей: о— карбоксил-карбоксильных, напр, в сополимере
стирола и акриловой к-ты; б — карбоксил-эфирных, напр,
в сополимере метилметакрилата и метакриловой кислоты;
волнистые линии — главные цепи макромолекул.
ными связями клубки (см. рис. 1, б), как правило
с повышенной жесткостью формы. Регистрацию про-
пространственно-сетчатых вторичных структур особенно
удобно производить в тех случаях, когда звенья, от-
ответственные за сшивку,— ионогенные, как в рассмат-
рассматриваемых случаях.
123
МАКРОМОЛЕКУЛА
124
Производя вискозиметрия, титрование, т. е. снимая
зависимость [х\] от состава растворителя, постепенно
увеличивая полярность последнего (рис. 8), можно
Рис. 8. Результаты вискози-
метрического титрования со-
лолимера метилметакрилата
и метакриловой к-ты с ма-
малым содержанием последней
(молярная концентрация ок.
5%). ДХЭ — дихлорэтан,
ДМФА — диметилформамид.
1—2 — переход от конфор-
мации подшитого клубка к
обычному клубку; з—4 —
превращение обычного клуб-
ЮОХЛМФА ка в ЙЙ
сказать, что СНз-группы «нерастворимы» в воде, но
зато растворимы в метаноле.
Явления микросегрегации. Эффекты микросегрега-
микросегрегации могут очень своеобразно проявляться в гребневид-
гребневидных М. как привитых сополимеров, так и гомополиме-
ров. Напр., привитой сополимер стирола и метилме-
наблюдать сначала превращение вулканизованного
клубка в обычный, а затем в полиэлектролитный.
Точка 1 на рис. 8 соответствует сильно «прошитому»
водородными связями клубку. Отрезок 2—3 указывает
на практическое постоянство объема клубков при ва-
вариации растворителя (напомним, что [г\] пропорцио-
пропорционально объему клубка), устанавливающееся после
скачкообразного изменения размеров (переход 1—2).
Скачок отображает распад большинства внутрицеп-
ных водородных связей. Переход 3—4 соответст-
соответствует новому резкому набуханию клубка в резуль-
результате ионизации карбоксилов (полиэлектролитное на-
набухание).
О превращении М. в полиэлектролит можно судить
также по изменению концентрационных кривых при-
приведенной вязкости (рис. 9): именно для М. полиэлек-
Рис. 9. Концентрационные
кривые приведенной вязко-
вязкости для того же сополимера
метилметакрилата и метак-
метакриловой к-ты, что и на рис.
8; цифры соответствуют ком-
композициям растворителей на
рис. 8.
тролитов характерно увеличение, а не уменьшение
приведенной вязкости r\YJS>/c при разбавлении. При до-
достаточном содержании внутримолекулярных водород-
водородных связей клубок может приобрести компактную
конформацию. Вискозиметрич. титрование в комбина-
комбинации с построением в координатах [г\]/М^— М^ может
дать более детальную информацию о структуре и
структурных превращениях М. с внутримолекулярны-
внутримолекулярными сшивками.
Вторичными структурами, скорее всего простран-
пространственно-сетчатого типа, обладают и нек-рые полиэлек-
тролиты-гомополимеры, напр, полиметакриловая к-та.
На графиках зависимости [т]]/М^ от М* при изме-
измерениях [г\] в подкисленной воде (рН < 4) и метаноле
получаются две параллельные линии с нулевым на-
наклоном (т. е. оба растворителя якобы в-системы),
но резко различающимися предельными значениями
Kq (в метаноле на порядок больше, чем в воде). Оче-
Очевидно, в подкисленной воде недиссоциированные кар-
карбоксилы образуют внутримолекулярную сетку водо-
водородных связей. Однако полиакриловая к-та не обна-
обнаруживает таких особенностей. Следовательно, нужно
принять во внимание и гидрофобные СНз-группы
полиметакриловой к-ты, отсутствующие в полиакри-
полиакриловой. Видимо, гидрофобное взаимодействие этих
групп является непосредственной причиной сворачи-
сворачивания если не всей М., то достаточно протяженных ее
участков, а водородные связи играют роль фиксаторов
этого сворачивания. Так. обр., здесь эффекты селек-
селективности разыгрываются на уровне мономера: можно
ветвями характеризуется резко повышенной скелетной
жесткостью хребта по сравнению с хребтом свободного
полиметилметакрилата. Это обусловлено сегрегацией
полистирольных ветвей; поведение молекулы в целом
в явлениях диффузии, вязкости и т. д. описывается
в терминах червеобразной модели с большой персис-
тентной длиной и с гидродинамич. толщиной, сопо-
сопоставимой по масштабу с линейными размерами клубка
(см. рис. 4, в). Эта толщина м. б. определена из по-
построения B5), но более прямым указанием на измене-
изменение скелетной жесткости хребта полиметилметакрилата
является резкое возрастание сегментальной анизотро-
анизотропии (см. табл. 2), определяемой в опытах по динамич.
двойному лучепреломлению в потоке.
Высшие полиалкилакрилаты и полиметакрилаты
тоже могут рассматриваться как гребневидные поли-
полимеры. Известно, что такие М. кристаллизуются боко-
боковыми группами, а не основной цепью. Обнаружить эту
тенденцию в разб. р-ре трудно. Однако если гребне-
гребневидный гомополимер содержит в качестве боковых
групп радикалы, способные в свободном виде образо-
образовывать жидкие кристаллы, то уже на молекулярном
уровне удается наблюдать особый тип вторичной струк-
структуры, характеризующийся внутримолекулярным жид-
кокристаллич. порядком. Природа этого эффекта близ-
близка к микросегрегации в привитых сополимерах, о чем
можно судить по изменению внутренней анизотропии
таких М. и персистентной длины по сравнению с поли-
метилметакрилатом (см. табл. 2).
В заключение этого раздела еще раз укажем на об-
обнаруженный В. Н. Цветковым эффект, родственный
сверхсопряжению, но в пределах первичной структу-
ы. Речь идет о полиизоцианатах с общей структурной
|-ЛОЙ
II I
О R
Как видно из табл. 2, если R — алкильный радикал
(в данном случае «-бутил), первичная структура цепи
характеризуется даже большей жесткостью, чем вто-
вторичная структура спирального полипептида поли-
"р-бензил-Ь-глутамата. Видимо, это связано с дефекта-
дефектами в структуре а-спирали (и тем более двойных спи-
спиралей дезоксирибонуклеиновой к-ты) и отсутствием
дефектов в первичной структуре. Если, однако, заме-
заменить алкильный радикал на арильный, М. полийзо-
цианата по жесткости не будет отличаться от М. боль-
большинства карбоцепных полимеров. По-видимому, аро-
матич. радикал нарушает цепь квазисопряжения.
Как указывалось, большой жесткостью первичной
структуры должны обладать лестничные полимеры;
из табл. 2, однако, видно, что она довольно умеренна,
хотя анизотропия (по абсолютному значению) высока.
Вероятно, это связано с локальными разрывами тя-
тяжей или образованием неправильных циклов.
Термодинамика и статистическая механика
структурных переходов в макромолекулах
Термодинамика малых систем и переход клубок —
глобула. М. и растворитель, содержащийся в ее коор-
координационной сфере, можно рассматривать как стати-
стич. ансамбль, подчиняющийся термодинамике малых
систем. Одной из особенностей этой термодинамики
является инверсия нек-рых интенсивных и экстенсив-
экстенсивных параметров. В частности, поскольку молярная
125
МАКРОМОЛЕКУЛА
126
E
J
f/
li
li
li
II
Jl
/
/ V
i
i
\
N
к
\
\
f'
\
\
\
\
V \
\ \ ,_
концентрация собственно полимера в координацион-
координационной сфере определяется ф-лой A5), она оказывается
переменной величиной, зависящей от темп-ры.
Рассмотрим фазовую диаграмму обычной макроско-
пич. системы (рис. 10). Т. к. такие диаграммы для р-ров
т полимеров всегда крайне
0| асимметричны, для удоб-
К|| ¦>—^ ства по оси абсцисс от-
отложена не концентра-
концентрация, а логарифм объем-
объемной концентрации поли-
полимера ф. Сплошная коло-
Рис. 10. Фазовая диаграмма
системы полимер — раство-
растворитель. Т — темп-pa, ф —
объемная доля полимера в
системе, @j—0-точка Фло-
W% ^T," ls*?2 ls<?2 Ри> Kl — верхняя критич.
темп-pa смешения.
колообразная кривая (бинодаль) разделяет область
полной взаимной смесимости (или растворимости)
полимера и растворителя. (Если сравнить рис. 10 и
рис. 2, я, станет ясно, что рассматривается лишь диа-
диаграмма состояний вблизи верхней критич. темп-ры
смешения.)
Пользуются фазовой диаграммой следующим обра-
образом. Допустим, нас интересует содтав фаз при нек-рой
темп-ре эксперимента Тэ. Пусть полная объемная
концентрация полимера в системе будет <рс (ей соответ-
соответствует точка С на рисунке). По точкам пересечения
изотермы Тэ с бинодалью (Ef и F') определяются кон-
концентрации полимера в разбавленной (<р,') и концентри-
концентрированной (ср^) фазах.
При другом содержании полимера в системе (точка D)
состав фаз будет тем же, но относительные объемы
концентрированной и разб. фаз другими. Двигаясь
вправо по оси концентраций, т. е. увеличивая кон-
концентрацию полимера в системе, мы, сохраняя состав
фаз, будем непрерывно увеличивать относительное
содержание конц. фазы.
Все точки «внутри» бинодали («под куполом») соот-
соответствуют распаду системы на две фазы, а вне ее —
гомогенной (однофазной) системе. Пунктирная кривая
(спинодаль) отграничивает области абсолютно неустой-
неустойчивых составов («внутри» спинодали) от метастабиль-
ных, где могут сосуществовать устойчивые микрофазы
или происходят т. наз. гетерофазные флук-
флуктуации, вызывающие явления критич. опа-
опале с ц е н ц ии, Устойчивость микрофаз имеет не
статич., а динамич. характер: это значит, что при кон-
концентрации полимера ух < <рх <<р" в равновесии с
р-ром находятся микрокапельки с концентрацией
<р2 < Ъ\^Чг- Эти капельки возникают и исчезают
(флуктуируют), но среднее их число или, точнее,
средняя их объемная доля, остается неизменной.
Для простых жидких смесей левая и правая ветви
бинодали и спинодали равноценны; в случае же поли-
полимеров приходится считаться с релаксационными явле-
явлениями. Фазовая диаграмма типа рис. 10 описывает
равновесную в термодинамич. смысле ситуацию. Однако
скорость установления равновесия «справа» и «слева»,
вследствие высокой вязкости р-ров полимеров, может
различаться на несколько порядков. Интервал состоя-
состояний между точками F" и F' может соответствовать
застеклованному р-ру. В этом случае термодинамически
нестабильная (или метастабильная) система может
оказаться абсолютно кинетически стабильной. То же,
разумеется, будет относиться и ко всем состояниям
правее точки F'.
Рассматриваемая фазовая диаграмма соответствует
разделению на аморфные фазы. Если полимер кри-
кристаллизующийся, возникают осложнения, к-рых мы
здесь касаться не будем. Осложнения возникают и
при полидисперсности, но их удается использовать
в методич. плане (см. Фракционирование).
В области малых концентраций р-р имеет дискретную
структуру типа изображенной на рис. 1, е: отдельные
М. занимают неперекрывающиеся координационные
сферы. Поэтому, следуя одному из основных постула-
постулатов термодинамики малых систем, для полного описа-
описания состояния разб. р-ра необходимо охарактеризовать
это состояние двумя точками — А и С (рис. 11), пер-
первая из к-рых соответст-
соответствует «большой» систе- т ас
ме — р-ру в целом, а вто-
вторая — «малой» (одной
координационной сфере
с М.).
При понижении темп-
ры, если пренебречь не-
незначительным термич.
сжатием, изменение со-
состояния раствора в це-
целом должно описываться
вертикальной прямой
АВ'В"\ в точке ?'долж- ?ис* И* к теории перехода клу-
, -a ^UJim бок — глобула; все пояснения &
но начаться, а в точке тексте.
В" — закончиться разде-
разделение на макроскопические фазы. Однако состояние
малой системы будет меняться совсем иным образомг
в соответствии с уравнением состояния Флори, вы-
выражающим зависимость параметра набухания а от Г
(см. стр. 115). Это изменение показано на рис. 11 кривой
CDE'\ после точки D начинается область отрицатель-
отрицательных объемных эффектов (а < 1), заканчивающаяся
при пересечении бинодали (?'). Вопрос этот пока до
конца не исследован, но все авторы сходятся на том,
что ниже в-точки характер кривой меняется; в мате-
матич. терминах это означает наличие особенности
в точке D (излом, разрыв первой и второй производ-
производных и др.).
Внутримолекулярное взаимодействие звеньев или
статистич. элементов цепи описывается такой же
функцией, как и межмолекулярное. Поэтому на участке
Е'Е" внутри координационной сферы должны разы-
разыграться примерно такие же события, как для большой
системы на участке В'В". При темп-ре Т2, когда р-р
в целом еще вполне стабилен, путь малой системы
пересечет спинодаль, и М. полностью утратит устой-
устойчивость. Но т. к. разделение на фазы в обычном смысле
при этом не происходит, то единственное, что может
сделать М.,— это выпасть «на себя», по полной анало-
аналогии с поведением нерастворимых блоков при образо-
образовании молекулярной мицеллы. Это и есть переход
клубок — глобула, резкость к-рого должна увеличи-
увеличиваться с увеличением гибкости.
Линия перехода на рис. И (E"FG) оборвана в точке
G; далее дан пунктир с вопросительным знаком. Этим
условно обозначены следующие обстоятельства:
1. Априори невозможно сказать, при какой сте-
степени компактности глобулы переход остановится.
Можно лишь предполагать, что плотность глобул очень
гибкоцепных полимеров типа алкилполисилоксанов
практически совпадает с «сухой» макроскопич. плот-
плотностью, а «выпавшие на себя» М. жесткоцепных поли-
полимеров типа поли-р-винилнафталина (их можно назы-
называть глобулами лишь с оговорками) содержат лишь
ок. 10% собственно полимера.
2. Пока трудно сказать что-либо определенное об
обратном переходе, но есть основания полагать, что
он не является полным обращением прямого перехода,
т. е. будет иметь место гистерезис. Это значит, что
для возвращения из точки G на какую-нибудь точку
линии CD требуется либо существенное повышение-
127
МАКРОМОЛЕКУЛА
128
темп-ры, либо длительное время (время структурной
релаксации), тем большее, чем выше мол. масса и
чем больше гибкость М.
Еще один вариант анализа вопроса о термодинамич.
природе перехода клубок — глобула связан с модели-
моделированием этого перехода на электронно-вычислитель-
электронно-вычислительной машине. При этом в программу вводятся сведения
о нек-ром ансамбле цепочек с определенным характе-
характером взаимодействия между звеньями, характеризуе-
характеризуемым функцией, к-рая далее обозначена га. Переход
от отталкивания к притяжению между звеньями (про-
(происходящий в точке D на рис. И) приводит к уменьше-
уменьшению средних размеров цепочки, соответствующему
уменьшению параметра набухания а с темп-рой и
увеличению т. Можно, далее, построить распределе-
распределение размеров М. в зависимости от значения га (рис. 12).
q(m)
Рис. 12. Схема статистико-механического анализа природы
конформационного перехода ниже в-температуры; т —
функция взаимодействия звеньев, q{m) — функция распре-
распределения макромолекул по числу внутрицепных контактов;
малым т соответствует конформация клубка, большим —
глобулярная конформация: 1 — очень гибкие макромоле-
макромолекулы (резкий переход по принципу «все или ничего»), 2 —
макромолекулы средней жесткости (слабо выраженный пере-
переход); 3 — жесткие макромолекулы (отсутствие перехода).
Если кривая распределения размеров распадается на
два независимых максимума, разделенных «пустым»
диапазоном га, можно говорить о двух различных фа-
фазах, в к-рых может существовать М. Аналогичный
подход возможен при анализе др. причин возникнове-
возникновения компактных вторичных структур.
Переходы типа спираль — клубок. Более разрабо-
разработана, хотя и не может быть сведена к простому пере-
пересечению кривой фазового равновесия, теория перехо-
переходов линейно-упорядоченных систем, напр, спираль —
клубок. Независимо от используемых здесь различных
математич. формализмов, суть всех теорий сводится
к прямому учету взаимодействия соседних элементов
в духе теории ферромагнетизма Изинга (для перехода
от порядка к беспорядку элементарные магнитики
должны разориентироваться, однако положение про-
произвольно выбранного магнитика зависит от состояния
его соседей, и он не может повернуться без поворота
его ближайших соседей). В случае а-спирали стаби-
стабилизация витков обеспечивается образованием во-
водородных связей между каждым первым и четвер-
четвертым звеньями. При этом каждая скелетная пептид-
пептидная группа — CONH — является одновременно доно-
донором и акцептором, т. е. фиксируется двумя водород-
водородными связями. Произвольно выбранный спиральный
участок цепи может быть переведен в неупорядоченное
состояние лишь при одновременном распаде всех во-
водородных связей, ограничивающих подвижность каж-
каждого витка.
Для количественного статистико-механич. описания
этого одновременного распада водородных связей и
соответственно исчезновения упорядоченных витков
вводится представление о параметре коопе-
рат явности а. Наглядно, хотя и не очень стро-
строго, физич. сущность о м. б. понята из след. соображе-
соображений. По существу, в кооперативной системе типа
а-спирали наиболее жестко реализуется принцип кор-
корреляции состояний соседних звеньев. Можно говорить
поэтому о некоторой корреляционной области, вклю-
включающей конечное количество звеньев, кинетически не-
независимой по отношению к соседним таким же об-
областям.
По аналогии с сегментом (при определении к-рого
корреляции состояния соседних звеньев сводили только
к их ориентациям в пространстве) можно считать
такую корреляционную область основной кинетич.
единицей, определяющей процесс структурного пре-
превращения. Другое определение: корреляционная об-
область представляет собой ту минимальную последо-
последовательность звеньев (связей, витков и др.), к-рая при
структурном превращении как целое переходит из
упорядоченного (спираль) в неупорядоченное состоя-
состояние. Значение о определяет размеры этой последова-
последовательности и поэтому имеет смысл нек-рой меры ее
степени полимеризации. С др. стороны, а должно
непосредственно зависеть от энергии взаимодействия
соседних звеньев. Соответственно, если F — свободная
энергия «инициирования связанного участка», опреде-
определяемая понижением энтропии при жестком ограниче-
ограничении конформации последовательности двух звеньев
(т. е. мера взаимодействия ближайших соседей), то о
определяется как константа равновесия для реакции
образования одного разрыва в последовательности
водородных (или ицых) связей:
о= ехр (—F/kT) B6)
Можно, далее, показать, что в температурной области
перехода средние числа последовательностей моно-
мономерных единиц в находящихся между собой в равно-
равновесии жестких и неупорядоченных участках (т. е.
степень полимеризации корреляционной области) при-
примерно равны:
^ж^св=~^а~1/2 B7)
Представим себе, что мы наблюдаем за переходом
спираль — клубок и можем определить константу
равновесия этого перехода (к-рый растянут по темп-ре).
Эта константа равна отношению числа упорядоченных
и неупорядоченных корреляционных областей и вы-
выражается обычной ф-лой
кр = ехр (-Н/кТ) B8)
Энергия активации Я, находимая из аррениусовского
построения, «не понимает», к молю каких элементов
она относится; согласно данному определению, это
должен быть «моль» корреляционных областей. Если,
с другой стороны, известна энергия, приходящаяся
на разрыв одной «независимой» водородной связи, от-
откуда можно определить энергию освобождения одного
звена АН, то v приближенно выразится как
^Я/АЯ B9)
Более строго о найдется при измерении интервала
плавления (см. рис. 13) AT:
|ДН|
Эта ф-ла верна при а« 1; отношение г— в C0)
соответствует Я в B8).
Как явствует из рис. 13, для экспериментального
определения о достаточно измерить температурную
зависимость в области перехода любого структурно-
чувствительного параметра (X на рисунке), зависящего
от соотношения упорядоченных и неупорядоченных
участков М. В случае а-спирали это м. б. ротационно-
оптич. характеристики, спектральные или конформа-
ционные, напр. [х\]. Значение а можно определить и
вариацией растворителя. Дело в том, что, по полной
129
МАКРОМОЛЕКУЛА
Рис. 13. К определению
параметра кооперативно-
сти по ширине интервала
плавления. Обычно кривая
перехода имеет S-образную
форму; за Гпл принима-
принимается точка перегиба; X —
любой конформационно-
чувствительный параметр;
Xt соответствует упорядо-
упорядоченному, Х2 — неупорядо-
неупорядоченному состоянию макро-
макромолекулы .
аналогии с классич.
выражением для пони-
понижения темп-ры замер-
замерзания раствора по
сравнению с чистым растворителем, понижение темпе-
температуры перехода вследствие специфического взаимо-
взаимодействия растворителя с М. дается ф-лой:
ТПЛ=Т° 11 +
ДЯ
-Ка'
C1)
где индекс «О» соответствует переходу в отсутствие
специфич. взаимодействия, а' — активность раствори-
растворителя, взаимодействующего с М. (это м. б. смешанный
растворитель; тогда а' относится лишь к его «эффек-
«эффективному» компоненту, константа* связывания к-рого
с М. равна К). Обычно АН < 0.
Очень схематично изложенные здесь принципы от-
отнюдь не ограничиваются линейно-упорядоченными фор-
формами вторичных молекулярных структур и примени-
применимы к любым переходам, напр, сшитый клубок (про-
(пространственно-сетчатая структура) — подвижный клу-
клубок (переход 1—2 на рис. 8) или подвижный клубок —
ионизованный клубок (переход 3—4 на рис. 8, к-рому
соответствует переход от кривой 3 к кривой 4 на рис. 9;
здесь кооперативный характер носит ионизация звеньев
метакриловой к-ты по достижении «критич.» концент-
концентрации диметилформамида). Кооперативными участками
{корреляционными областями) здесь м. б. уже ассо-
ассоциации далеких вдоль цепи звеньев, образующих
нек-рые локальные элементы структуры «вулканизо-
«вулканизованной» М. (сравни рис. 1,6; 4 таких участка).
Рассматриваемые переходы происходят в тем менее
широком интервале темп-р, чем больше кооператив-
ность системы. Сам по себе факт существования тем-
температурного интервала перехода еще не дает основа-
основания подвергать сомнению фазовую природу конфор-
мационных превращений. Теоретически кристаллиза-
кристаллизация в одномерных системах невозможна. Однако даже
в случае «линейно-кристаллических» систем следует
помнить, что у спирали или кросс-Р-формы есть протя-
протяженность в трех измерениях. Истинным критерием
природы перехода является его «конформационная пре-
прерывность» (в смысле рис. 12), к-рая никак не противо-
противоречит модифицированному правилу фаз Гиббса.
Влияние внешней силы на переходы в макромоле-
макромолекулах. Интересной особенностью конформационных
переходов, наиболее ярко проявляющейся в случае
линейно-упорядоченных систем, является выраженная
зависимость темп-ры перехода Гпл (безотносительно к
тому, в какой степени оправдан термин «плавление»,
следующий из индекса) от внешней нагрузки. По су-
существу, здесь реализуется принцип Ле Шателье —
Вант-Гоффа. В строгой форме его можно записать в
виде ур-ния равновесия фаз типа Клапейрона — Клау-
зиуса, в к-ром, по сравнению с обычными трехмерными
системами, объем заменен на длину, а давление —
на растяжение:
C2)
Где f __ растягивающая сила, AL и ЛЯ соответственно
изменение длины и энтальпии при переходе. Детали-
Детализированный расчет приводит к зависимости между
130
Рис. 14. К теории пере-
перехода спираль — клубок
под нагрузкой; / — растя-
растягивающая сила, Т —
темп-pa, L — длина.
длиной L, Тил и / типа
изображенной на рис.
14. На участке О А до-
достигается темп-pa пла-
плавления («кристаллиза-
(«кристаллизации»), соответствующая
заданной силе и длине.
I В точке В кристалли-
-*- зация завершается. Со-
Соответственно, увеличе-
увеличение / в растягиваемой упорядоченной системе сверх
критического значения f/T дестабилизирует систему,
а ниже — стабилизирует, повышает Гпл. Участок
«дестабилизации» (BCD) соответствует переходу в
другую упорядоченную форму через «расплав», т. е.
неупорядоченную форму. В этой области f/T рас-
растяжение понижает Гпл. В точке D возникает новая
устойчивая форма, и теперь растяжение снова повы-
повышает Гпл. Суть этого наглядно иллюстрируется при-
примером с переходом а-спираль — клубок — р-форма
(второе «линейно-упорядоченное» состояние полипеп-
полипептидной цепи).
Здесь (рис. 15) непосредственно показано приложение
принципа Ле Шателье. Устойчивость системы (к-рую
можно характеризовать конформационной энтропи-
энтропией) определяется тенденцией М. к сокращению или
удлинению в результате перехода из одной формы
(«фазы») в другую. . _9,, ..
Если при переходе спираль—клубок <А2 > /2 клуока
меньше длины спирали, то растяжение, препятствую-
препятствующее сокращению длины, повышает Гпл (участок ОА
рис. 14). Вытянутая р-форма существенно длиннее
а-формы. Но между ними лежит область неупорядо-
неупорядоченных состояний (рис. 15, в), где< h2 > ХЫ клубка
больше длины спирали. Поэтому растяжение, способ-
способствующее увеличению длины, понижает TnR. (Можно
также говорить, что растяжение позволяет преодо-
преодолеть систему кооперативных водородных связей между
витками.) Так будет, пока система не перейдет во вто-
вторую устойчивую форму с большей длиной. Дальней-
Дальнейшее растяжение должно опять стабилизировать упо-
упорядоченную фазу.
Очень важно, что подобное рассмотрение в равной
мере приложимо как к отдельным М., так и к макро-
макроскопически ориентированным системам (в последнем
f
Рис. 15. Схема, поясняющая влияние нагрузки на конфор-
мационные переходы. Слева указано направление растя-
растяжения. На каждом участке (аб, бв, вг) растяжение стабили-
стабилизирует конформацию с большей длиной: а — сильно сверну-
свернутый клубок, б — а-спираль, в — растянутый клубок, г —
антипараллельная Р-форма (белые кружки соответствуют
межцепным водородным связям). Снизу показаны зависи-
зависимости Гпл от / на участках аб, бв и вг.
131
МАКРОМОЛЕКУЛА
132
случае понятия «фаза», «кристаллизация», «плавление»
могут употребляться безоговорочно). На опыте су-
существование фазовых равновесий, обусловленных кон-
формационными переходами под действием внешней
силы, подтверждено как для макроскопич. систем, так
и для отдельных М. В последнем случае растягиваю-
растягивающая сила м. б. генерирована гидродинамич. полем,
электрически (в полиэлектролитах или М. с очень
большими дипольными моментами) и др. В случае М.
с «вулканизационной» или «конденсационной» вторич-
вторичной структурой гидродинамич. поле должно понижать
стабильность этой структуры, чему также имеются
экспериментальные подтверждения.
Преобразование химической энергии в механическую
Если изменение химич. характеристик среды, в
к-рой находится дифильная или амфифильная М.
(т. е. изменение рН, ионной силы, окислительно-вос-
окислительно-восстановительного потенциала и др.), приводит к изме-
изменению ее конформации и соответственно размеров, то
вызванное внешней силой изменение размеров приво-
приводит к соответствующему изменению химич. характе-
характеристик среды. Этот термодинамич. принцип, устанав-
устанавливающий существование обратных связей между
размерами и конформацией М., внешними и внутрен-
внутренними напряжениями, соответственно воздействующи-
воздействующими на М. или развивающимися в ней, и химич. харак-
характеристиками среды, был впервые сформулирован В.
и Г. Кунами и назван ими тейнохимическим
принципом.
Проще всего тейнохимич. принцип реализуется на
полиэлектролитах. В условиях сильной ионизации
происходит полиэлектролитное набухание М., при
к-ром ее линейные размеры могут увеличиться в не-
несколько раз. Источником этого набухания является
кулоново дальнодействие: электростатич. отталкива-
отталкивание одноименно заряженных и далеко (если считать
вдоль цепи) расположенных звеньев. Примером такого
набухания является резкое увеличение размеров М.
сополимера метилметакрилата и метакриловой к-ты,
изображенное на рис. 8 и 9 (переход 3—4). В водной
среде, где ионизация несравненно сильнее, чем в ди-
метилформамиде, такие переходы выражены гораздо
резче.
Если объединить М. полиэлектролита в слабо сши-
сшитую пленку или волокно, они будут претерпевать
макроскопич. изменения размеров, повторяющие изме-
изменения размеров отдельных М. при ионизации и деиони-
зации. На этом основано действие химич. машины, так
наз. «рН -мускула». Если к растянутому волокну
подвесить груз, а затем деионизовать образующие
волокно М., то волокно сократится (М. свернутся в
обычные клубки) и поднимет груз. Разумеется, сущест-
существует нек-рый предел, за к-рым волокно уже не способ-
способно поднять груз. Это означает, что под действием внеш-
внешней нагрузки, даже в условиях, благоприятствующих
сокращению, сокращение не произойдет.
Соответственно, если нагрузить сокращенное волокно
таким «сверхкритическим» грузом, оно растянется и
при этом как бы выжмет из себя нейтрализующие его
противоионы, к-рые перейдут в омывающий волокно р-р.
Простые опыты такого рода не только позволили
построить первую химич. машину, но и помогли по-
понять механизм возникновения обратных связей на
уровне изолированных М. По-видимому, возникнове-
возникновение таких (отрицательных) обратных связей сыграло
решающую роль в эволюции жизни на земле. В тех-
нич. плане реализация тейнохимич. принципа сулит
в будущем создание саморегулирующихся смешанных
механо-каталитич. систем, способных производить
энергию и «попутно» регулировать химич. процессы.
Заметим, что на прямое превращение химич. энергии
в механическую способны только полимеры.
Тейнохимич. принцип м. б. распространен на любую-
форму химич. активности М., включая и каталитиче-
каталитическую. Действительно, в образовании третичной струк-
структуры макромолекул биополимеров участвуют силы
различной природы, весьма чувствительные к измене-
изменениям внешней среды. В частности, электрохимич.
характеристики этой среды могут стабилизировать
или дестабилизировать третичную структуру белков
по полной аналогии с тем, как они могут вызывать
разворачивание или сворачивание молекул полиэлек-
полиэлектролитов.
Ввиду непрерывности третичной структуры даже
локальные конформационные изменения на одном участ-
участке белковой М., передаваясь и распространяясь по М.,
могут вызвать локальные же конформационные пре-
превращения, затрагивающие центр ферментативной ак-
активности. Т. обр., механическое воздействие может
«включать» или «выключать» каталитич. центр М.
Но этот центр, в свою очередь, будучи включенным,
может менять электрохимич. свойства среды (за счет
продуктов реакции; напомним, что все макромолекулы
биополимеров — полиэлектролиты). Можно представить
себе ситуацию, когда продукты реакции таким образом
меняют рН, что активная макромолекула деформирует-
деформируется и утрачивает тем самым активность; здесь даже
не требуется внешнее поле. После удаления продуктов
реакции М. возвращается в исходное состояние, центр'
снова включается, и начинается новый цикл.
Макромолекулы и информация. Молекулярная
кибернетика
Способность к реализации обратной связи — не един-
единственное «кибернетич.» свойство М. Не только высоко-
высокоорганизованные М. нуклеиновых к-т являются носите-
носителями и передатчиками сложнейшей (наследственной)
информации; по существу, любая М. уже является
информационной системой. Действительно, если обра-
обратиться к табл. 1, видно, что сам факт существования
различных конфигураций цепи, при одном и том же
числе повторяющихся звеньев, делает цепи различи-
различимыми. В принципе, атактич. линейная М. со степенью
полимеризации п может существовать в 2п~1 стереоизо-
мерных формах, или конфигурациях. По аналогии
с кодом Морзе, комбинации конфигураций в диадах,
триадах, тетрадах и т. п. вида
гоиссГи ииссиесие ири у Щ си ее .....
играют роль знаков. [Для простоты рассматривается
виниловая цепь с двумя возможными конфигурациями
соседей — изо- (И) и синдио- (С)]. Совершенно анало-
аналогичным образом «текст» м. б. записан на цепи бинар-
бинарного сополимера, состоящего из звеньев А и В:
ААВАВВВАААВВШАВААААВАВААВАВВ .
ААВАВВВАААВВАВрШАААВАВААВШ...^
Не трудно понять, что конфигурация цепи, ее первич-
первичная структура есть не что иное, как информация о
всех ее возможных конформациях, включая вторичные
структуры; о способности цепей к образованию тех
или иных надмолекулярных структур; об условиях,
в каких образуются эти структуры, а значит и о до-
достижимых физических и, в частности, механич. свойст-
свойствах полимера и др. Чем детализированнее первичная
структура, т. е. чем сложнее сополимер, тем более
однозначную информацию его М. несут. При этом нет
необходимости стремиться к увеличению числа «зна-
«знаков»: даже наследственные признаки человека коди-
133
МАКРОРАДИКАЛЫ
134
руются примерно 20 знаками (соответствующими при-
примерно 18 амино- и 2 иминокислотам, из к-рых состоят
все белки; на деталях, связанных с вырождением кода,
мы здесь не останавливаемся), составленными из ком-
комбинаций четырех типов звеньев в цепях дезоксирибо-
нуклеиновой и рибонуклеиновой к-т.
Молекулярная кибернетика вполне утвердила себя
как реально существующая ветвь науки о полимерах,
возникшая на стыке химии, физики, молекулярной
биологии и «обычной» кибернетики. Основные задачи
молекулярной кибернетики — изучение способов «за-
«записи» свойств полимера при синтезе М., физико-химич.
процессов передачи этой записи (реализации инфор-
информации), разработка методов «прочтения» записи, ис-
использование «текстов» для разработки рациональной
технологии и т. п. Правильное прочтение информации
о структуре, записанной на М., служит разработке
оптимальной технологии; неправильное — как всякая
дезинформация — приводит к «антитехнологии».
Лит.: Волькенштейн М. В., Конфигурационная
статистика полимерных цепей, М.— Л., 1959; его же, Моле-
Молекулы и жизнь, М., 1965; БреслерС. Е., Ерусалим-
ский Б. Л., Физика и химия макромолекул, М.— Л., 1965;
Цветков В. Н., Эскин В. Е., Френкель С. Я.,
Структура макромолекул в растворах, М., 1964; Мора-
вец Г., Макромолекулы в растворе, пер. с англ., М., 1967;
Бирштейн Т. М., Птицын О. Б., Конформации макро-
макромолекул, М., 1964; Hill Т., Thermodynamics of small systems,
pt. 1—2, N. Y.— Amst., 1963—64; Джейл Ф. X., Поли-
Полимерные монокристаллы, пер. с англ., Л., 1968; Флори П.,
Статистическая механика цепных молекул, пер. с англ., М.,
1971; К а р г и н В. А., Слонимский Г. Л., Краткие очер-
очерки по физико-химии полимеров, 2 изд., М., 1967; Френ-
Френкель С. Я., ЕльяшевичГ. К., ПановЮ. Н., в сб.:
Успехи химии и физики полимеров, М., 1970, с. 87.
С. Я. Френкель.
МАКРОПОРИСТЫЕ ИОНООБМЕННЫЕ СМОЛЫ-
см. Пористые ионообменные смолы.
МАКРОРАДИКАЛЫ (macroradicals, Makroradikale,
macroradicaux) — макромолекулы, обладающие неспа-
ренным электроном на внешней (валентной) орбитали.
Различают свободные (нейтральные) М. (R) и заряжен-
ные — анион-радикал (R~) и катион-радикал (R+).
Поскольку М.— парамагнитные частицы, то наиболее
прямым методом их обнаружения, изучения природы,
структуры и количественного анализа является элек-
электронный парамагнитный резонанс (ЭПР). Для иден-
идентификации М. особое значение имеет анализ сверхтон-
сверхтонкой структуры спектров ЭПР, возникающей в резуль-
результате взаимодействия неспаренного электрона с окру-
окружающими ядрами, имеющими собственные магнитные
моменты (ХН, 2D, 13C, 14N и др.) и расположенными
в а-, E- и ^-положениях по отношению к неспаренному
электрону. М. изучают также оптич. методами по
спектрам поглощения в УФ-, видимой и ИК-областях.
По сравнению с макромолекулами, поглощение М.
сдвинуто в длинноволновую область спектра. Иногда
М. исследуют с помощью акцепторов радикалов, ра-
радиоактивных добавок, путем инициирования привитой
сополимеризации.
М. образуются при деструкции полимеров в резуль-
результате разрыва химических связей в главной или боковых
цепях макромолекул, при радикальной полимериза-
полимеризации, при взаимодействии макромолекул со свободными
радикалами примесей, в реакциях гидрирования и т. д.
Природа и структура М., образующихся в полиме-
полимерах при действии ионизирующих излучений, опреде-
определяются химич. строением макромолекул, изотопным
составом полимера, темп-рой, дозой излучения и др.
При низкотемпературном радиолизе в большинстве
полимеров возникают преимущественно алкильные М.
со свободной валентностью, локализованной на атоме
углерода в середине полимерной цепи, напр. М. строе-
строения ~ СН2СНСН.2 ~ в полиэтилене. В этих условиях
образуются также аллильные М., ион-радикалы и
радикальные пары.
При 77 К радиационно-химич. выходы (G) М. лежат
в пределах от 0,1 до 6 (G — число М. на 100 эв погло-
поглощенной энергии). Так, для полиэтилена G = 5,8, для
поли-е-капролактама —2,4, для ДНК —1—2, для поли-
политетрафторэтилена —0,2. Значение G зависит от изотоп-
изотопного состава полимера: для дейтерополиэтилена и
дейтерополипропилена G в ~2 раза меньше, чем для
соответствующих «обычных» полимеров. Для синте-
тич. полимеров при низких темп-pax G больше, чем
при высоких. Для нек-рых природных полимеров на-
наблюдается обратная зависимость. Для белков G при
300 К в ~ 2 раза больше, чем при 77 К.
При нагревании М. вступают в реакции присоеди-
присоединения, замещения, изомеризации, рекомбинации и
диспропорционирования. Термич. устойчивость М. за-
зависит от их структуры, фазового состояния полимера,
строения макромолекул и др. В кристаллич. фазе по-
полимера М. более устойчивы, чем в аморфной. Кинетика
гибели М. при нагревании в области низких темп-р
имеет т. наз. «ступенчатый» характер, а при высоких
темп-pax описывается ур-нием второго порядка. При
нагревании наблюдается превращение активных М.
в более стабильные. Эффективная энергия активации
рекомбинации и превращений М. лежит в пределах
21—251 кдж/молъ E—60 ккал/молъ). Рекомбинацию
и превращение М. связывают с перемещением М. по
механизму миграции свободной валентности или по
механизму диффузии.
При действии видимого и УФ-света М. претерпевают
различные превращения. Заряженные М. погибают
или превращаются в нейтральные М. Аллильные М.
(^макс = 255 нм) в полиэтилене и полипропилене
превращаются в алкильные (^Макс =215 нм). При
действии света М. вступают в реакции замещения,
изомеризации, рекомбинации и диссоциации. Кине-
Кинетика фотохимич. превращений М. описывается ур-ния-
ми первого и второго порядка. Эффективная энергия
активации таких превращений аллильных и перекис-
ных М. в полипропилене и политетрафторэтилене соста-
составляет 0—12,6 кдж/молъ @—3 ккал/молъ).
М. играют определяющую роль в реакциях полиме-
полимеризации, при модифицировании полимеров и их раз-
разрушении в процессе эксплуатации. В нек-рых полиме-
полимерах М. легко присоединяют кислород: эффективная
энергия активации и предэкспонента реакции окисле-
окисления составляют соответственно 12,6—33,5 кдж/молъ
C—8 ккал/молъ) и 10~15 —10~17 см3/сек. Эффективная
энергия активации реакции перекисных М.
R'OO + HR -> R'OOH + R-
составляет 41,9—62,8 кдж/молъ A0—15 ккал/молъ).
В результате реакций перекисных М. на воздухе в
полимерах образуются кислородсодержащие группы.
Так, в полиэтилене при гибели 1 алкильного М. обра-
образуются 5 гидроксильных и 12 карбоксильных групп.
Образование и превращения М. под действием света
лежат в основе процессов старения полимеров. УФ-свет
вызывает различные реакции перекисных М. даже
при низких темп-pax. Так, в политетрафторэтилене М.
— CF2CF(O2)CF2 ~ термически устойчивы при темп-рах
до 470 К, а при УФ-облучении диссоциируют с раз-
разрывом цепи и образованием М. ~ CF2GF2 при 77 К.
М., образующиеся в полимерах при действии иони-
ионизирующих излучений, в значительной степени ответ-
ответственны за радиационно-химич. превращения полиме-
полимеров — сшивание, деструкцию, образование химич. нена-
ненасыщенных групп, окисление и др. Реакции М. с моно-
мономерами используются для модифицирования полимеров.
Лит.: ЭПР свободных радикалов в радиационной химии,
Б П Ю [ ] У 38 4, 593
Ней-
НейЛит.: ЭПР свободых радиао радиациоой х,
М., 1972; Б у т я г и н П. Ю. [и др.], Усп. хим., 38, в. 4, 593
A969); Бучаченко А. Л., Л е б е д е в Я. С, Н
ман М. Б., в сб.: Успехи химии и физики полимеров, М.
409 Б X С Т
, . ф р, ,
1970, с. 409; Багдасарьян X. С, Теория радикальной
полимеризации, 2 изд., М., 1966; Campbell D., J. Poly-
Polymer. Sci, D4, 91 A970). В. К. Милинчук.
135
МАКСВЕЛЛА МОДЕЛЬ
136
МАКСВЕЛЛА МОДЕЛЬ (Maxwell model, Maxwell-
sches Modell, modele de Maxwell) — простейшая меха-
нич. модель упруговязкой жидкости (см. Реология),
при течении к-рой накапливаются упругие (обратимые)
деформации; состоит (рисунок) из после-
последовательно соединенных пружины с жест-
жесткостью к и демпфера, при движении порш-
поршня в к-ром возникает сопротивление гх
(г — коэфф. вязкого сопротивления; х —
скорость смещения поршня относительно
окружающей жидкости). Связь между
^ 2 действующей на М. м. силой F и сме-
щением поршня в демпфере х выража-
Модель Максвелла: 1 — пружина; 2— цилиндр
с вязкой жидкостью; 3 — поршень.
F F
ется уравнением ~^+~ = х- Если смещение поршня
принять как характеристику деформации -у» а
вместо силы использовать напряжение т, то это урав-
уравнение принимает форму
-^-+ —=Y
где G — модуль упругости; т\ —вязкость среды. Это
ур-ние описывает поведение среды <<в точке». Переход
от ур-ния модели к математич. описанию поведения
реальных тел осуществляется методами механики
сплошной среды на основании ур-ния равновесия эле-
элемента среды, записанного для напряжений, а напряже-
напряжения связываются с деформациями с помощью ур-ния
М. м. При задании постоянной деформации у = const
напряжение в М.м. изменяется во времени (релаксирует)
по закону т/т0 = е—*/в , где т0 — значение т в момент
времени t = 0; 0 = ц/G — константа материала, наз.
временем релаксации. При произвольном изменении
скорости деформации у (t) во времени зависимость
т (t) для М. м. описывается ф-лой
j
о
М. м. позволяет предсказать существование частот-
частотной зависимости динамич. модуля, но не описывает
запаздывания в развитии ползучести, ибо при при-
приложении напряжения т = т0 = const M. м. ведет се-
себя как вязкая жидкость, в к-рой сохраняется по-
постоянная обратимая компонента деформации у = xo/G.
Особенности поведения М. м. зависят от соотношения
параметра 0, имеющего смысл внутреннего масштаба
времени материала, и длительности эксперименталь-
экспериментальной шкалы времени t*. Если t* > 0, М. м. ведет себя
как жидкость с вязкостью rj, если I* « 6 — оказы-
оказывается аналогичной твердому упругому телу.
М. м., как и Кельвина модель, используется для по-
построения обобщенной теории линейной вязкоупруго-
сти, описывающей поведение тел, механич. свойства
к-рых характеризуются не одним, а набором (спектром)
времен релаксации. Эта теория позволяет приблизиться
к описанию свойств реальных полимерных тел, однако
она не учитывает зависимость самих времен релакса-
релаксации от условий деформирования, обусловливающую
разнообразные нелинейные эффекты (см. Реология).
Тем не менее простейшая М. м. полезна для качествен-
качественного анализа релаксационных свойств вязкоупругих
жидкостей, т. к. она отражает нек-рые принципиаль-
принципиальные особенности их поведения. А. я. Малкип.
МАЛЕИНОВОГО АНГИДРИДА ПОЛИМЕРЫ И
СОПОЛИМЕРЫ (polymers and copolymers of maleic
anhydride, Polymere und Kopolymere von Malein-
saureanhydrid, polymeres et copolymeres d' anhydride
maleique).
Малеиновый ангидрид, ангидрид этилен-1,2-гшс-ди-
сн-со
карбоновой к-ты (М.а.)
I—СО
О — бесцветные кри-
кристаллы; плотность (тв.) 1,48 г/см3; т. пл. 52,8 °С; т. кип.
199,9 °С; теплота плавления 11,5 Мдж/кмолъ B,75
ккал/молъ); теплота испарения 43,96 Мдж/кмолъ A0,5
ккал/молъ); теплота нейтрализации 126,9 Мдж/кмолъ
C0,32 ккал/молъ). М. а. смешивается с диоксаном;
хорошо растворяется в ацетоне G0%), этилацетате
E3%), хлороформе C4%), бензоле C3%), ограничен-
ограниченно - в СС14 @,6%).
М. а.— чрезвычайно реакционноспособное соедине-
соединение. При смешении с водой и спиртами образуются
соответственно малеиновая к-та и диалкилмалеинаты;
при взаимодействии в мягких условиях с сопряженными
диенами — циклич. аддукты типа ангидридов цикло-
гександикарбоновых к-т. При действии на М. а. ам-
аммиака и аминов образуются соответственно аспараги-
новая к-та HOOCGH2CH(NH2)GOOH и ее N-алкилза-
мещенные. При высоких темп-pax М. а. взаимодей-
взаимодействует с алкилбензолами (напр., с толуолом) и несо-
несопряженными ненасыщенными соединениями, содержа-
содержащими метильные или метиленовые группы при кратной
связи, напр.:
сн2=сн-сн2-сн—со
сн2=сн-сн3
сн-со
\
сн-со
СН2-СО
При поликонденсации М. а. с гликолями образуются
ненасыщенные олигоэфиры (см. Полиалкиленгликолъ-
малеинаты и полиалкиленгликолъфумараты); сам М. а.
и эвтектич. (низкоплавкие) смеси его с ангидридами
др. карбоновых к-т служат отвердителями эпоксидных
смол. Полимеризация М. а. осложняется стерич. за-
затруднениями при образовании активированного ком-
комплекса в акте роста цепи, что характерно для всех
1,2-дизамещенных этиленов.
М. а. сополимеризуется со многими мономерами в
присутствии перекисей и др. свободнорадикальных
инициаторов, обнаруживая сильную тенденцию к че-
чередованию звеньев (см. таблицу).
Константы сополимеризации мал ей нового ангидрида (rt)
с другими мономерами (г2)
Сомономер
Метилакрилат .
Метилметакрилат
Стирол
Винилацетат ... • . . . .
0,02
0,03
0
0,01
2 , 8 ± 0 , 0 5
3,5
0,01
0,072 + 0,04
В ряде случаев при смешении М. а. с сомономерами,
напр. со стиролом A : 1), наблюдается образование
комплексов с переносом заряда. В таких системах обыч-
обычно образуются сополимеры с регулярным чередова-
чередованием звеньев в цепи.
М. а. получают парофазным каталитич. окислением
бензола или фурфурола над V2O5 при 450—500 °С.
М. а.— основной продукт перего"нки малеиновой, фу-
маровой (над Р2О5) и яблочной к-т; он образуется
также как побочный продукт при каталитич. окислении
олефиновых и ароматич. углеводородов и их произ-
производных.
Полималеиновый ангидрид [_сн—СН—]„ (П. а.) —
I I
с с
0 0 0
твердое аморфное вещество белого цвета. Мол.
масса П. а., измеренная методом светорассеяния
(для соответствующей полималеиновой к-ты), не пре-
137
МАСЛА РАСТИТЕЛЬНЫЕ
138
вышает обычно 30 000. Темп-pa размягч. 320 °С (с раз-
разложением). П. а. хорошо растворим в воде (с образо-
образованием полималеиновой к-ты), хуже — в уксусном
ангидриде, диоксане, низших спиртах, кетонах, эфи-
рах и нитропарафинах; нерастворим в ароматич. угле-
углеводородах, большинстве хлорсодержащих раствори-
растворителей и в высших парафинах.
Продукт гидролиза П. а.— п о л и м а л е и н о в а я
к-та [—СН—СН—]п легко растворима в воде, ог-
I I
соон соон
раниченно набухает в бутаноле. Мононатриевая
соль полималеиновой к-ты также растворима в воде.
Полималеиновая к-та — полиэлектролит с большой
плотностью зарядов, что обусловливает ее специфич.
кислотные и вязкостные свойства, солевой эффект,
а также влияние размера катиона титрующего р-ра
на диссоциацию к-ты. Так, при потенциометрич. тит-
титровании полималеиновой к-ты стандартным р-ром NaOH
титруется только половина карбоксильных групп,
вероятно вследствие влияния электростатич. поля,
препятствующего диссоциации более слабых (вторич-
(вторичных) кислотных групп.
П. а. можно получить полимеризацией малеинового
ангидрида в р-ре (напр., в уксусном ангидриде, диок-
диоксане) и в массе под действием у-об л учения (при 25—
75 СС), а также в присутствии, радикальных инициато-
инициаторов, напр, перекиси бензоила. М. а. полимеризуется
также в массе под давлением 2—4 Гн/м2 B0 000 —
40 000 тс/см2-) и 100—170 °С.
М. а. можно использовать в качестве сшивающего
агента при получении, напр., поливинилспиртових
волокон. П. а. используется для получения носителей
ферментов (примером может служить система, в к-рой
трипсин «вшит» в сетку из П. а.).
Лит.: LangJ.L., P a v е 1 i с h W. А., С 1 а г е у H.G.,
J. Polymer. Sci., 55, 531 A961); их же, там же, pt A, 1,
№ 4. 1123 A963); Хэм Д., Сополимеризация, пер. с англ.,
М., 1971; С а г d о n A., Goethals Е. J., J. Macromol.
Sci., A5, №6, 1021 A971); Pledger H., [а. о.], там
же, А5, № 8, 1339 A971). Д. А.Топчиев.
МАЛЕИНОВОЙ И ФУМАРОВОЙ КИСЛОТ СОПО-
СОПОЛИМЕРЫ (copolymers of maleic and fumaric acids,
Kopolymere von Malein- und Fumarsaure, copolymeres
des acides maleique et fumarique).
Малеиновая (I) и фумаровая (II) кислоты, цис-
и тр<шс-бутен-2-диовые-2,3-кислоты.
HC-GOOH НС-СООН
II II
НС-СООН НООС-СН
I, г^ис-изомер II, транс-изомер
Малеиновая (М. к.) и фумаровая (Ф. к.) к-ты — бес-
бесцветные моноклинные призмы; т. пл. соответственно
131 и 287 °С (в запаянной ампуле); т. кип. 160 и 290 °С
соответственно. М. к. легко растворима (в г на 100 г
растворителя) в воде G8,8; 25 °С) и спирте F9,9;
— 30 °С); трудно — в бензоле. Ф. к. плохо растворима
в воде @,69; 17 °С) и почти во всех органич. растворите-
растворителях; лучше — в спирте E,75; ~30 °С).
М. к. легко переходит в Ф. к. на свету или при нагре-
нагревании выше 200 °С. Этот процесс протекает и при син-
синтезе ненасыщенных полиэфирных смол на основе ма-
малеинового ангидрида, в результате чего существенно
улучшаются свойства продуктов (большая активность
Ф. к. при отверждении, лучшие физико-механич. свой-
свойства отвержденных продуктов; см. П олиалкиленгликоль-
малеинаты и полиалкиленгликолъфумараты). ф. к. пре-
превращается в М. к. при УФ-облучении. При нагревании
малеиновой и фумаровой к-т с Р2О5 образуется мале-
иновый ангидрид (ангидрид фумаровой к-ты неизве-
неизвестен). Двойная связь в обеих к-тах легко восстанавли-
восстанавливается, напр, амальгамой натрия, с образованием ян-
янтарной к-ты.
Малеиновая и фумаровая к-ты не полимеризуются
из-за стерич. затруднений, связанных с наличием
в молекулах этих к-т объемистых СООН-групп в 1,2-
положениях. Полималеиновая к-та м. б. получена толь-
только гидролизом полималеинового ангидрида (см. Ма-
Малеинового ангидрида полимеры и сополимеры).
В пром-сти получают ангидрид М. к. Ф. к. можно
получить кипячением 30—40%-ного водного р-ра
М. к. с HG1. Обе к-ты получаются при нагревании
яблочной к-ты.
Сополимеры. Малеиновая и фумаровая к-ты, а также
их соли (калиевые, магниевые и др.) сополимеризуются
в присутствии радикальных инициаторов, напр, пере-
перекиси бензоила, с рядом мономеров. Получены сополи-
сополимеры М. к. с винилацетатом, акриламидом, стиролом,
акрилонитрилом, метакриловой к-той, а также сополи-
сополимеры Ф. к. с 2-метил-5-винилпиридином, акриловой
к-той, стиролом, акрилонитрилом (см. таблицу). Содер-
Содержание к-т в сополимерах не превышает 50%. Тенден-
Тенденция к чередованию звеньев особенно заметна при сопо-
лимеризации этих к-т со стиролом и винилацетатом.
Константы сополимеризации малеиновой и фумаровой кислот
(г О с другими мономерами (г2)
Сомономер
Стирол ....
Винилацетат
Акрилонитрил
0,
Малеиновая к-та
ri
0
030±0,015
0
г2
0,58 + 0,
0,045±0,
17 ,0±0
05
015
,5
Фумаровая
0
0
г2
0,40±
15, 5±
к-та
0,03
:1,0
Сополимеры малеиновой и фумаровой к-т—как пра-
правило, светлые аморфные вещества невысокой мол. мас-
массы. Характеристич. вязкость их р-ров составляет обыч-
обычно 0,1—0,5 дл/г. Сополимеры малеиновой и фумаровой
к-т различаются по ряду свойств (напр., растворимо-
растворимости, вязкости). Сополимеры М. к. титруются как двух-
двухосновная к-та, сополимеры Ф. к.— как моноосновная
к-та, что обусловлено различной пространственной кон-
конфигурацией карбоксильных групп в макромолекулах.
Лит.: Лебедев В. С. [и др.], Высокомол. соед., 6, № 7,
1161 A964); 6, № 7, 1174 A964); 6, № 8, 1353 A964); Эль
С а й е д А. А., там же, АН, № 2, 282 A969); U 1 b г i с h t J.
[а. о.], Faserforschung und Textiltechnik, H. 7, 16, 331 A965);
T о п ч и е в Д. А. [и др.1, Высокомол. соед., 13Б, № И, 821
A971). Д. А. Топчиев.
МАРКА — ХУВИНКА УРАВНЕНИЕ — см. Вяз-
Вязкость характеристическая.
МАСЛА РАСТИТЕЛЬНЫЕ, жиры расти-
растительные (vegetable oils, Pflanzenole, huiles vege-
tales) — продукты, добываемые из семян и плодов
растений и состоящие главным образом из смесей пол-
полных сложных эфиров (триглицеридов) общей ф-лы
CH2(OCOR')—CH(OCOR")—CH2(OCORW), где R', R",
R"' — одинаковые или различные а л килы.
М. р. имеют важное пищевое значение. Вместе с тем
ок. V3 их мирового производства (в 1972 оно составляло
25 млн. т) используется для технич. целей. Это обус-
обусловлено гл. обр. способностью многих из них полиме-
ризоваться («высыхать») в тонком слое с образованием
прочных защитных покрытий (см. Пленкообразующие
вещества, Масляные лаки и эмали, Масляные краски,
Олифы), а также смазывающими, пластифицирующими
и поверхностно-активными свойствами многих произ-
производных жирных к-т. В данной статье М. р. рассматри-
рассматриваются в аспекте, связанном с их применением в качест-
качестве пленкообразующих для лакокрасочных материалов.
Наряду с М. р. нек-рое применение для этой цели нахо-
находят обработанные жиры рыб и др. морских животных.
Триглицериды М. р. содержат преимущественно не-
разветвленные одноосновные насыщенные (Сб—С24) и
ненасыщенные (С14—С18) жирные к-ты с четным чис-
числом атомов углерода (табл. 1,2). Из насыщенных к-т в
составе М. р. наиболее часто встречаются пальмитино-
пальмитиновая и стеариновая, из ненасыщенных — олеиновая, ли-
139
МАСЛА РАСТИТЕЛЬНЫЕ
140
нолевая и линоленовая. В триглицеридах М. р. преоб-
преобладают к-ты С18, в триглицеридах жиров рыб и мор-
морских животных — к-ты С14—С24; в последних присут-
присутствуют большие количества насыщенных к-т С14—С18
и высоконенасыщенные жирные к-ты С20—С24, содер-
содержащие в молекуле 4—10 двойных связей. Помимо три-
триглицеридов, в М. р. содержатся небольшие количества
свободных жирных к-т (~1%), фосфатидов (до 2—3%),
стеринов @,5—1,5%), антиоксидантов @,05—0,2%),
незначительные количества витаминов, восков, крася-
красящих и слизистых веществ и др. Нек-рые М. р. содержат
дубящие вещества, алкалоиды, эфирные масла и др.
Классификация. Общепринятой является классифи-
классификация М. р. по их ненасыщенности {йодному числу) и,
соответственно, способности к «высыханию» (см. табл.
1, 2): 1) высыхающие (йодное число >130); 2) полувы-
полувысыхающие (90—130) и 3) невысыхающие « 90). В ы-
сыхающие М. р. содержат значительные кол-ва
триглицеридов ненасыщенных жирных к-т с двумя и
тремя двойными связями (линолевой, линоленовой,
элеостеариновой). М. р. этой группы, в свою очередь,
подразделяют на масла, содержащие к-ты с изолиро-
изолированными двойными связями (льняное, перилловое,
конопляное, лаллеманциевое и др.) и с сопряженными
двойными связями (тунговое, ойтисиковое, дегидрати-
дегидратированное касторовое). В полувысыхающих
М. р. преобладают триглицериды к-т с одной или двумя
изолированными двойными связями (олеиновой, лино-
линолевой). К этим маслам относятся маковое, подсолнеч-
подсолнечное, ореховое, соевое, хлопковое, кукурузное, талло-
вое и др. Невысыхающие М. р. содержат боль-
большое количество триглицеридов насыщенных или моно-
мононенасыщенных к-т. Напр., насыщенные к-ты входят
в состав кокосового и пальмоядрового, мононенасы-
мононенасыщенные — в состав оливкового, арахисового и нек-рых
др. масел. Невысыхающее касторовое масло, содержа-
содержащее гл. обр. глицериды рицинолевой A2-оксиолеино-
вой) к-ты, обычно выделяют в особую группу; после
дегидратации оно приобретает свойства высыхающего
М. р.
Физические свойства. При комнатной темп-ре М. р.
и жиры — жидкие, твердые или мазеобразные вещест-
вещества; их консистенция определяется гл. обр. жирнокис-
лотным составом. Продукты, в к-рых преобладают три-
триглицериды ненасыщенных жирных к-т, при обычной
темп-ре являются жидкостями. М. р. и жиры кипят
только в высоком вакууме [остаточное давление
< 133 мн/м2 (< 10~3 мм рт. ст.)]. Нагревание М. р.
до 300—350 °С при атмосферном давлении приводит
к их разложению.
Таблица 1. Состав и свойства нек-рых растительных масел и жиров морских животных
Название продукта
Йодное
число
Число
омыле-
омыления
Плот-
Плотность
при
15СС,
г/см3
Показа-
Показатель пре-
ломле-
ломления п2Х
Кислотный состав, % по массе
Насыщенные к-ты
Миристи-
новая
С14
Пальми-
Пальмитиновая
С1в
Стеари-
Стеариновая
С18
Арахи-
новая
С2о
Ненасыщенные к-ты
Олеиновая
Линолевая
Лино-
лено-
леновая
Дегидратированное
касторовое
Конопляное
Лаллеманциевое . .
Льняное
Ойтисиковое . . . .
Перилловое
Тунговое
Подсолнечное . . . .
Соевое
Талловое (жирнокис-
лотная фракция)
Хлопковое
Касторовое
Кокосовое
Жир сельдей . . .
Жир сардин . . .
Китовый жир . .
Тюлений жир . .
112-135
140—143
162
170-204
140-152
180-208
154-176
127-136
120-141
165-170
103-111
81-90
8-10
148-185
170 — 190
94-145
165-190
188-194
1 90-
181-
-194
-185
191-195
186-
187-
195
197
188-197
186-
188-
194
195
175-185
194-196
176-187
246-268
189-193
189-193
175-220
170-195
0,948-
0,958
0,929
0,934
0,933
0,965
0,931
0,940
Высыхающие масла
1,4820
1,4517
1,4800
1,5140
1,4815
1,5200
3,0
5
6,5
8-Ю
17,0
3,7
1,2
Полувысыхающие масла
0,924 1,4680
D0 °С)
0,928 1,4745
0,920
1,4634
0,3-0,5
D0 СС)
Невысыхающие масла
8-10
2,4-6,
6-8
20-22
8
4,
—
4-7
2,0
,3
0,
0,
—
4-1
1-0
,0
,6
0,960-
0,967
0,925
0,923-
0,933
0,920-
0,934
0,910-
0,928
0,910-
0,929
1,4771
1,4497
D0 °С)
1,4787
1,4763-
1,4852
1 ,4630-
1,4710
D0 СС)
16,5-20,0
Жиры
4,3-7,5
3,0
0,8-5,0
32
30
2
3-9
14,0
5-20
6,0
14-20
10-15
20-30
,0-35,6
41—48
,5-35,2
3-9
0-10,3
до 86
65,0
93, 5а
25-50
12-18
-
до 60
51,5-57,0
36-39
41 ,7-44,0
2-3
1,0
3,0
16,0
21-45
77, 0б
65-70
~80В
—
2-3
-
3,0
-24,0 •
-22,0 ¦
9,2
4,7
15,6
12,1
1,9
2,0
0,6е
0,1s
30,0
10,0
а Содержание линолевой и эруковой (С22) к-т. б Ликановая к-та. в Элеостеариновая к-та. г Фракция, выделяемая вакуум-рек-
вакуум-ректификацией таллового «масла», к-рое получают из сульфатного мыла — побочного продукта производства целлюлозы из древе-
древесины хвойных пород сульфатным методом. Д Содержит также 80—85% рицинолевой к-ты. е Содержит также 10,7 — 22,2% к-т
Св—С10 (капроновая, каприловая, каприновая) и 45 — 51% лауриновой к-ты. ж Жир сельдей и сардин содержит соответственно 15
и 12% пальмитолеиновой к-ты, 19 и 22% к-т C2oH2B0-ccH2, 12 и 19% к-т С22Н2B2-сс)О2 (ее = 4 —10); тюлений и китовый ж,ир —
соответственно ненасыщенных к-тС14-3,3 и 2,5%, С1в-19,2 и 13,9%, С18-31,8 и 37,2%, С20-12,9 и 12%, С22-13,4 и 7,1%;
3 Содержание к-т С20—С22-
141
МАСЛА РАСТИТЕЛЬНЫЕ
142
Таблица 2. Свойства важнейших жирных кислот, входящих в
состав триглицеридов растительных масел и жиров животных
Название кислоты и
ее брутто-формула
Число
двойных
связей
Темп-ра
плавле-
плавления, °С
Кислотное
число
Йодное
число
Насыщенные кислоты
Лауриновая С1::Н24О2
Миристиновая
СХ4Н28О2
Пальмитиновая
С1вН3г0а
Стеариновая С18Н3в02
Нена сыщенные
Лальмитолеиновая
43,6
53,8
62,0
70,5
280,1
245,8
218,9
197,3
кислоты
Олеиновая С18Н34Ог
Линолевая С18Н32О2
Линоленовая СмН3оС
Элеостеариновая
хвзог
Клупанодоновая
С22Н34О,
Уруковая С22Н42О
14,0
9,5
48*
34
220,5
198,6
200,1
201 ,5
201,5
170,4
165,7
99,8
89,9
181
273
273,5
384,4
75,0
Замещенные ненасыщенные кислоты
85.
Рипинолевая С18Н34
Ликановая С18Я28О
4-5
188,0
191,9
260,4
* Данные для а-изомера; для 3-изомера 71 °С.
Все М. р. и жиры практически не растворимы в воде
и образуют с ней эмульсии. Они легко поглощают
многие газы (СО, СО2, N2, O2, Н2) и летучие продукты,
выделяемые различными пахучими веществами. Раст-
Растворители М. р. и жиров — эфир, бензол, бензин (за
исключением касторового масла), сероуглерод, хлоро-
хлороформ, четыреххлористый углерод и др. хлорированные
углеводороды. В низших спиртах растворяется только
касторовое масло. Жидкие М. р. и жиры обычно легче
растворяются в органич. растворителях, чем твердые.
Химические свойства. Гидролиз М. р. и жиров
происходит при их нагревании A05—200 °С) с водой
под давлением, а также при действии к-т, щелочей
и ферментов. При этом образуются свободные жирные
к-ты и глицерин. При омылении М. р. и жиров
образуются соответствующие соли жирных к-т (мыла)
и глицерин. На этой реакции основан промышленный
•способ получения мыл и сиккативов. Алкоголиз
М. р. одноатомными спиртами (метиловым, этиловым)
широко используют при исследовании жиров. Алкого-
Алкоголиз многоатомными спиртами, приводящий к образова-
образованию неполных эфиров, лежит в основе получения
-алкидных смол, маслорастворимых эмульгаторов и др.
Ацидолиз М. р. многоосновными к-тами приме-
применяют в произ-ве алкидных смол. Подобно алкоголиз у,
реакция обратима. Она идет при 240—260 °С в присут-
присутствии кислых катализаторов.
Гидрирование используют гл. обр. в произ-
производстве маргаринов. Более глубокое восстановление при
250—300 °С и давлениях 15—20 Мн/м2 A50—200 кгс/см2)
приводит к образованию высших насыщенных и нена-
ненасыщенных первичных спиртов.
По двойным связям М. р. и жиров могут присоеди-
присоединяться галогены, галогеноводороды, серная к-та, ро-
дан, кислород, сера и др. Реакции присоединения гало-
галогенов и родана применяют в аналитич. целях для опре-
определения содержания в М. р. двойных связей.
Двойные связи могут перемещаться (изомеризация)
и вступать в полимеризацию. Эти процессы имеют важ-
важное значение при хим. обработке (см. ниже) М. р. и жи-
жиров и их высыхании.
При высыхании М. р. поглощают кислород
A0—30% от массы масла), в результате чего образуют-
образуются гидроперекисные соединения и одновременно выделя-
выделяются летучие продукты окисления. Распад гидропере-
гидроперекисей ведет к возникновению свободных радикалов,
инициирующих окислительную полимеризацию. Окис-
Окисление протекает с индукционным периодом, к-рый свя-
связывают гл. обр. с присутствием в М. р. природных ан-
тиоксидантов. В процессе высыхания повышаются
вязкость, плотность, показатель преломления М. р.;
слой масла превращается в мягкую пленку, к-рая по-
постепенно затвердевает и теряет плавкость и раствори-
растворимость вследствие образования «сшитого» полимера.
Способность М. р. и жиров к высыханию возрастает
с увеличением числа двойных связей в их молекулах.
Однако очень большое число двойных связей (напр.,
в триглицеридах рыбьих жиров) приводит к быстрому
старению покрытий. М. р., содержащие жирные к-ты
с сопряженными двойными связями, высыхают быстрее
и образуют более твердые пленки, чем М. р. с изолиро-
изолированными связями. Высыхание ускоряется под дейст-
действием УФ-лучей, сиккативов, катализирующих разло-
разложение гидроперекисей, а также при нагревании.
Получение. М. р. и жиры получают из измельчен-
измельченных семян или из животных тканей экстракцией горя-
горячей водой, паром, органич. растворителями, прессова-
прессованием (из семян) или комбинацией этих методов. В ла-
лакокрасочной пром-сти чаще применяют М. р., получен-
полученные горячим прессованием (выше 80 °С).
Присутствие в маслах природных примесей ухудшает
качество лакокрасочных материалов (напр., антиок-
сиданты замедляют высыхание, фосфатиды — алкого-
алкоголиз). Для очистки (рафинации) М. р. и жиров, исполь-
используемых в производстве этих материалов, применяется
обычно комбинация трех методов: 1) обработка паром
или горячей водой (т. наз. гидратация), в результате
к-рой фосфатиды, белковые и слизистые вещества, по-
поглощая воду, набухают, теряют способность раство-
растворяться в масле и выпадают в виде хлопьев, удаляемых
фильтрацией; 2) обработка водными р-рами щелочей
(щелочная рафинация); образующиеся при этом мыла
обладают большой адсорбционной способностью и, осе-
оседая, увлекают фосфатиды, красящие вещества и др.
примеси; 3) адсорбционная отбелка природными и ис-
искусственными отбельными порошками (преимуществен-
(преимущественно активированными глинами), адсорбирующими нежи-
нежировые компоненты и слизистые вещества и одновремен-
одновременно обесцвечивающими М. р. Очищенные таким образом
М. р. наз. лаковыми маслами. Улучшение
пленкообразующих свойств М. р. и жиров м. б. до-
достигнуто путем отделения плохо высыхающих глице-
ридов насыщенных и мононенасыщенных к-т. Основные
методы отделения — кристаллизация (вымораживание),
экстракция растворителями, высоковакуумная дистил-
дистилляция.
Химическая обработка. Для улучшения качества
покрытий, получаемых на основе маслосодержащих
пленкообразующих, М. р. подвергают полимеризации,
оксидации, изомеризации, дегидратации, обработке
серой (фактизации), переэтерификации, сополимери-
зации с ненасыщенными мономерами.
Термическая полимеризация — наи-
наиболее часто применяемый способ обработки высы-
высыхающих и полувысыхающих М. р., в результате к-рого
уменьшается их ненасыщенность и возрастает вязкость
и плотность. Процесс проводят в отсутствие кислорода
при 280—300 °С (тунговое масло нагревают до 200—
230 °С). Скорость полимеризации повышается с уве-
увеличением ненасыщенности М. р. и содержания в них
к-т с сопряженными двойными связями. Катализаторы
реакции — сера, двуокись серы, трехфтористый бор,
соли никеля, кобальта, железа, металлич. никель,
антрахинон. Жирные к-ты взаимодействуют при поли-
полимеризации по реакции Дильса — Альдера (см. Диеновый
синтез).
Полимеризованные М. р. применяют в качестве полу-
полуфабрикатов для изготовления олиф, лаков, полиграфи-
полиграфических красок, линолеума; слабополимеризованные
143
МАСЛА РАСТИТЕЛЬНЫЕ
144
масла — в производстве алкидных смол. При исполь-
использовании полимеризованных М. р. улучшаются блеск,
твердость, водо- и атмосферостойкость покрытий. Из
этих М. р. выделяют т. наз. «димерные кислоты», пред-
представляющие собой смесь мономерных (С18), димерных
(С36) и тримерных (С54) к-т, к-рые используют в произ-ве
полиамидов и нек-рых полиэфиров.
Оксидация (окислительная полимеризация)
производится путем продувания воздуха через слой
масла при 50—200 °С в присутствии катализаторов
(соединений свинца, кобальта, марганца) или без них.
При этом вязкость и плотность М. р.возрастают, йодное
число уменьшается, растворимость в углеводородах
снижается, а в полярных растворителях — возрастает.
Оксидированные масла применяют в качестве полуфаб-
полуфабрикатов для изготовления масляных лаков и олиф.
Изомеризации способствует нагревание ма-
масел (или жирных к-т) в присутствии катализаторов —
едких щелочей, осажденного на активированном угле
никеля, сернистого ангидрида, окислов металлов,
активированных глин, сульфидов, хинонов и др. При
этом содержание к-т с сопряженными двойными свя-
связями достигает 30—50% (от общего содержания к-т),
что значительно повышает скорость полимеризации и
обусловливает лучшее высыхание масла. Изомеризо-
ванные М. р. рекомендованы для изготовления алкид-
алкидных смол.
Дегидратацию проводят для получения вы-
высыхающего масла из невысыхающего касторового,
содержащего оксикислоты. Масло нагревают выше
250 °С в присутствии катализаторов (гл. обр. к-т, напр,
фталевой, фосфорной, серной, или кислых солей серной
к-ты). В результате невысыхающие триглицериды ри-
цинолевой к-ты превращаются в высыхающие тригли-
триглицериды линолевых к-т, содержащие ок. 30% глицеридов
с сопряженными двойными связями.
Переэтерификация (обменная реакция
между эфирами) особенно широко используется для
улучшения пленкообразующих свойств полувысыхаю-
полувысыхающих масел; для этого их нагревают с высыхающими
маслами. Реакция часто протекает также при высоко-
высокотемпературной обработке М. р., напр, при термич.
полимеризации, изготовлении олиф, масляных лаков.
Сополимеризация с ненасыщен-
ненасыщенными мономерами. Для получения сополи-
мерных масел используют гл. обр. ненасыщенные сое-
соединения трех классов: 1) а, (З-ненасыщенные к-ты, напр,
акриловую, метакриловую, кротоновую, фумаровую,
малеиновую, итаконовую, их ангидриды и эфиры;
2) виниловые мономеры, преимущественно стирол и
винилтолуол; 3) 1,3-диены, преимущественно цикло-
и дициклопентадиен. Наибольшее значение имеют
малеинизированные масла, к-рые по-
получают нагреванием М. р. с 3—8% малеинового ангид-
ангидрида при 100—250 °С. Катализаторы — сильные к-ты
и перекиси. К маслам, содержащим жирные к-ты с со-
сопряженными двойными связями, малеиновый ангидрид
присоединяется по реакции Дильса — Альдера:
В тех случаях, когда триглицериды содержат к-ты
с • изолированными двойными связями, малеиновый
ангидрид присоединяется с образованием замещенных
янтарных к-т:
Малеинизированные масла имеют кислую реакцию и
обычно не используются без дальнейшей химич. обра-
обработки. Этерификацией их многоатомными спиртами по-
получают масляные лаки, нейтрализацией аминами —
водорастворимые масла, обработкой окисями (или гид-
гидроокисями) металлов — сиккативы.
М. р. с сопряженными двойными связями в сополи-
меризации с виниловыми мономерами более реакционно-
способны, чем М. р. с изолированными связями. Пер-
Первые образуют с мономером, напр, стиролом, два вида
продуктов — истинные сополимеры с высокой мол.
массой и аддукты реакции Дильса — Альдера. Присое-
Присоединение стирола к М. р. ускоряется перекисными ката-
катализаторами. Ценные свойства «стиролизован-
н ы х» масел — быстрое высыхание, светлая окрас-
окраска. Они образуют покрытия с хорошими водостойкостью
и электрич. свойствами. На основе стиролизованных
масел получают полиграфич. краски, строительные
эмульсионные краски, а также алкидные смолы.
Нагревание высыхающих или полувысыхающих
М. р. с 1,3-диенами при атмосферном или повышенном
давлении в присутствии полярных катализаторов
(напр., BF3 или его производных) приводит к получению
продуктов, к-рые близки к тунговому маслу по ско-
скорости высыхания и образуют покрытия примерно такой
же твердости. В присутствии BF3 высыхающие масла
сополимеризуются также с терпенами.
Обработкой ненасыщенных М. р. надуксусной или
надмуравьиной к-той при темп-ре ниже 80 °С в присут-
присутствии кислого катализатора осуществляют э п о к с и-
дирование масел:
О
-СН=СН ЬСНзСООН -> -СН СН- + СНзСООН
Реакцию широко используют в промышленных масшта-
масштабах для изготовления эпоксидированного соевого мас-
масла — важного стабилизатора и пластификатора хлорсо-
держащих полимеров, гл. обр. поливинилхлорида. При
взаимодействии эпоксидированных М. р. с дикарбоно-
выми к-тами образуются алкидные смолы.
В результате взаимодействия полиизоцианатов со»
смесями неполных сложных эфиров, получаемых при
алкоголизе высыхающих М. р. пентаэритритом или др.
многоатомным спиртом, образуются уретановые
масла. Эти полимеры высыхают на воздухе в резуль-
результате окислительной полимеризации с образованием по-
покрытий, обладающих исключительно высокой водо-,
щелоче- и абразивостойкостью.
Конденсацией ненасыщенных жирных к-т или
их смесей с насыщенными к-тами в присутствии борной
к-ты (обычно при 280 °С) получают синтетиче-
синтетические масла неэфирного типа (у-пироны).
Продукты конденсации ненасыщенных к-т близки по
свойствам к полимеризованным М. р., обладают хоро-
хорошей способностью к высыханию. Благодаря отсутст-
отсутствию сложноэфирных групп эти синтетич. масла стойки
к гидролизу и омылению.
Применение. Среди высыхающих М. р. наибольшее
значение в лакокрасочной пром-сти имеют льняное,,
тунговое и дегидратированное касторовое. Эти М. р.
применяют для изготовления олиф, масляных лаков,
алкидных смол, модифицирования эпоксидных смол.
Материалы на основе таких пленкообразующих высы-
высыхают на воздухе при комнатной темп-ре. К числу важ-
важнейших полувысыхающих М. р., к-рые используются
для изготовления олиф (преимущественно в смеси с вы-
высыхающими маслами), а также в производстве алкидных
и эпоксидных смол, относятся соевое, подсолнечное
и хлопковое масла и жирные кислоты таллового ма-
масла. Лакокрасочные материалы, изготовленные иа
этих М. р., высыхают при комнатной темп-ре медлен-
145
МАСЛЯНЫЕ КРАСКИ
146
нее и образуют пленки значительно менее твердые, чем
из высыхающих М. р. Невысыхающие М. р. непригодны
для изготовления олиф и масляных лаков, но в больших
количествах применяются в производстве алкидных
смол, используемых в смеси с мочевино- и мелами-
но-формальдегидными смолами для изготовления про-
промышленных эмалей горячей сушки, а также в качест-
качестве пластификаторов. Наибольшее промышленное зна-
значение в этой группе имеют кокосовое и касторовое
масла. Рыбьи жиры после соответствующей переработ-
переработки также применяются для изготовления олиф и мас-
масляных лаков.
Несмотря на непрерывное расширение производства
синтетич. пленкообразующих, маслосодержащие лако-
лакокрасочные материалы, особенно олифы и алкидные
смолы, используются очень широко. Среди сырья
органич. происхождения М. р. занимают по масштабам
потребления первое место в зарубежной и отечествен-
отечественной лакокрасочной пром-сти.
Наряду с маслами технич. назначения лакокрасочная
пром-сть использует большие количества пищевых ма-
масел (в США — преимущественно соевое, в СССР — под-
подсолнечное, хлопковое, соевое). Необходимость эконо-
экономии пищевых масел выдвигает задачу увеличения
производства высыхающих масел (в первую очередь
льняного и тунгового), касторового масла и более
широкого использования жирных к-т таллового масла
и рыбьих жиров.
В качестве заменителей жирных к-т природных масел
неоднократно предлагались синтетич. жирные к-ты.
Насыщенные к-ты фракций С6— С9 и Сп—С16, получае-
получаемые окислением парафинов, использовали во время
второй мировой войны в Германии (позднее и в СССР)
для синтеза алкидных смол, в мыловарении и для при-
приготовления нек-рых др. продуктов. Разработаны также
способы получения синтетич. жирных к-т разветвлен-
разветвленного строения, к-рые в промышленном масштабе приме-
применяются в производстве алкидных смол и сиккативов.
Большой интерес представляют исследования, прово-
проводимые в ряде стран, в том числе и в СССР, в области
разработки методов синтеза высших ненасыщенных
жирных к-т (типа линолевой, линоленовой, элеостеари-
новой и др.).
Лит.: Лакокрасочные покрытия, под ред. X. В. Четфилда,
пер. с англ., М., 1968; Основные направления развития масло-
жировой промышленности на 1971 —1975 гг., под общ. ред.
А. Г. Сергеева, Л., 1970; Analysis and characterization of oils,
fats and fat products, ed. by H. A. Boeokenoogen, vol. 1 — 2,
L._ [a. o.], 1964—68; H i 1 d i t с h T. P., W i 1 1 i a m s P.N.,
The chemical constitution of natural fats, 4 ed., L., 1964;
Knuth С h. J., Schiller A. M., в кн.: Encyclopedia
of polymer science and technology, v. 1, N. Y.— [a. o.], 1964,
p. 98; W e x 1 e г Н., там же, v. 5, N. Y.— [a. o.], 1966,
p. 216; Kirk — О t h m e r, Encyclopedia of chemical tech-
technology, 2 ed., N. Y.— [a. o.], 1965, v. 7, p. 398, v. 8, p. 776.
A. M. Лубман.
МАСЛОНАПОЛНЕННЫЕ КАУЧУКИ — см. На-
Наполненные каучуки.
МАСЛОСТОЙКОСТЬ полимерных мате-
материалов — см. Б ензо стойкость.
МАСЛЯНЫЕ КРАСКИ (oil paints, Olfarben, peintu-
res a l'huile) — суспензии пигментов и наполнителей
в олифах.
Состав. М. к. изготовляют на основе натуральных,
комбинированных, полунатуральных («оксоль») и ал-
алкидных олиф (об эмалевых М. к., к-рые получают на
основе масляных лаков, см. Масляные лаки и эмали).
Для придания М. к. цвета и укрывистости в их состав
вводят неорганич. пигменты (см. Пигменты лакокрасоч-
лакокрасочных материалов). Наполнителями, к-рые применяют
для экономии пигментов, а также для улучшения адге-
адгезии, механич. свойств и атмосферостойкости покрытий
из М. к., служат тальк, каолин, слюда, тяжелый шпат
и др. (см. Наполнители лакокрасочных материалов).
Вспомогательные компоненты в составе М. к.— ка-
катализаторы высыхания (сиккативы) и поверхностно-ак-
поверхностно-активные вещества; последние облегчают диспергирова-
диспергирование пигментов и наполнителей в пленкообразующем.
В качестве сиккативов применяют растворимые в оли-
олифах соли Со, Mn, Pb. Оптимальное содержание металла
в сиккативе (в % от массы масла в олифе): Со — 0,13;
Мп — 0,12; РЬ — 0,45. При более высоких концентра-
концентрациях ускоряется старение пленок М. к. Поверхностно-
активные вещества (лецитин, мыла меди, бария, цинка,
продукт алкамон ОС-2) вводят в М. к. в количестве
0,5—1,0% от массы пленкообразующего.
Получение. Пром-сть выпускает М. к. двух видов
(см. таблицу): густотертые (пастообразные) и готовые
к употреблению (жидкие). При изготовлении густотер-
Состав и свойства нек-рых масляных красок
Пигмент
Содержа-
Содержание плен-
кообразу-
кообразующего, %
Укрыви-
стость,
г/м2
Дисперс-
Дисперсность пиг-
пигмента по
прибору
«клин»
Густотертые краски
Белила цинковые.
Сурик железный .
Охра
Литопон
Белила свинцовые
Сажа
20-22
18
28
13-15
11 — 16
33
120
3 5
180
160-170
210-310
25
40
40
45
40
Краски, готовые к употреблению
Двуокись титана (рутил) .
Белила цинковые .....
Сурик железный
Охра (светлая)
27-35
25-32
23-30
28-38
90
175
35
180
40
40
80
80
тых М. к. (содержание пленкообразующего 11 — 33%
в зависимости от маслоемкости пигмента) олифу, пиг-
пигменты и наполнители тщательно перемешивают (иногда
в присутствии поверхностно-активных веществ) в сме-
смесителе до образования однородной пасты. Затем пиг-
пигменты и наполнители диспергируют в пасте до задан-
заданной степени дисперсности (обычно 40—80 по прибору
«клин»; см. Испытания лакокрасочных материалов и
покрытий) на 3-, 5- или 8-валковых краскотерках.
М. к., готовые к употреблению (содержание пленко-
пленкообразующего 23—38%), изготовляют разбавлением гу-
густотертых М. к. или смешением всех компонентов
в шаровых мельницах. Густотертую М. к. перемешивают
в смесителе с дополнительным количеством пленкооб-
пленкообразующего. В случае необходимости в этот же смеси-
смеситель для корректировки цвета (колеровки) М. к. до-
добавляют при перемешивании густотертую пасту требуе-
требуемого цвета.
Продолжительность изготовления готовых к употреб-
употреблению М. к. при использовании шаровых мельниц ко-
колеблется в пределах 2—3 сут в зависимости от мощно-
мощности мельницы, а также природы пигмента и наполни-
наполнителя. Готовые к употреблению М. к. на основе алкид-
алкидных олиф изготовляют только этим способом. Подробно
о технологии и оборудовании для изготовления красок
см. Краски.
Свойства. Пленкообразование при высыхании М. к.
обусловлено окислительной полимеризацией входящих
в их состав высыхающих или полувысыхающих расти-
растительных масел (см. Масла растительные, Олифы). Ско-
Скорость высыхания М. к. и свойства образующихся при
этом пленок зависят от типа масла и пигмента, темп-ры,
освещенности и др. факторов. М. к. на основе льняной
олифы высыхают в светлом помещении через 24 ч,
в темном — через 48 ч. При повышении темп-ры сушки
и яркости освещения возрастают твердость, эластич-
эластичность, влаго- и химстойкость пленок (в частности,
стойкость к воздействию слабых к-т). Пленки М. к.,
высушенных при 250—300 °С, стойки в слабых р-рах
щелочей. При сушке выше 100—150 °С пленки М. к.
белого или светлых тонов желтеют или темнеют.
147
МАСЛЯНЫЕ ЛАКИ И ЭМАЛИ
148
Нек-рые пигменты (ZnO, PbO, Pb3O4) ускоряют высы-
высыхание М. к., действуя как сиккативы; сажа и др. пиг-
пигменты с высокой адсорбционной способностью могут
адсорбировать сиккативы на своей поверхности и тем
самым замедлять высыхание М. к. Этот процесс замед-
замедляется также в среде влажного воздуха и паров раство-
растворителей. При высыхании красок выделяются летучие
побочные продукты окисления масел (СО, муравьиная
к-та, акролеин). Эти соединения, особенно акролеин,
обладают неприятным запахом, раздражают слизистые
оболочки глаз.
Нагревание и солнечная радиация ускоряют старе-
старение пленок М. к., сопровождающееся повышением их
твердости, уменьшением эластичности, и приводят
к растрескиванию пленок. Атмосферостойкость пленок
М. к. на основе натуральной олифы составляет 3—5 лет.
В условиях длительного воздействия влаги пленки на-
набухают и размягчаются; при этом уменьшается их
адгезия к подложке (исключение — пленки М. к. на
основе натуральной олифы, содержащих свинцовые
пигменты, т. к. в этом случае образуются водостойкие
свинцовые мыла). Пленки М. к., за исключением пле-
пленок на основе глифталевой олифы, полуматовые, их
нельзя шлифовать и полировать. Они легко подвергают-
подвергаются поражению нек-рыми бактериями и грибками.
Применение. М. к. применяют для получения защит-
защитных и декоративных покрытий по металлу, древесине,
пластикам и др. материалам. М. к. широко используют
в строительстве (для окраски крыш, стен, деревянных
и металлич. конструкций) и др. областях в тех случаях,
когда к декоративным свойствам лакокрасочных покры-
покрытий не предъявляются высокие требования.
Лит. см. при ст. Краски, Масла растительные, Олифы.
В. В. Чеботаревский.
МАСЛЯНЫЕ ЛАКИ И ЭМАЛИ (oil varnisches and
enamels, Ollacke und Emaillen, vernis et emaux a l'hui-
le). Масляные лаки — р-ры продуктов совме-
совмещения растительных масел и природных смол в орга-
нич. растворителях. В зависимости от содержания
масла лаки делят на тощие @,5—1,5 мае. ч. масла на
1 мае. ч. смолы), средней жирности A,6—2,5) и жирные
{2,6—5,0). Масляные эмали, или масля-
масляные эмалевые краски,— суспензии пиг-
пигментов и наполнителей в масляных лаках.
Состав. Масляные лаки изготовляют на основе ра-
рафинированных и полимеризованных до заданной вяз-
вязкости высыхающих масел — льняного, периллового,
ойтисикового, тунгового, конопляного; иногда приме-
применяют также полувысыхающие масла — подсолнечное
или хлопковое (см. Масла растительные). В состав
пленкообразующего входят канифоль или ее препара-
препараты, напр, глицериновые или пентаэритритовые эфиры,
канифольно-малеиновые аддукты, резинаты кальция
и цинка. Применяют также ископаемые смолы — ян-
янтарь, копалы и др. (см. Смолы природные). На основе
продуктов совмещения масел с битумами получают
черные битумно-масляные лаки (см. Битумные лаки).
Растворителями служат уайт-спирит, скипидар, соль-
вент-нафта, бензин, ксилол и др. Для ускорения высы-
высыхания М. л. и э. применяют сиккативы — нафтенаты,
резинаты, линолеаты Со, Mn, Pb. В состав эмалей, по-
помимо указанных компонентов, вводят пигменты и на-
наполнители. См. Пигменты лакокрасочных материалов,
Наполнители лакокрасочных материалов. В нек-рых
случаях М. л. и э. могут содержать также антиокси-
данты, фунгициды, поверхностно-активные вещества
и др. вспомогательные добавки.
Получение. Лаки на основе канифоли
и ее производных изготовляют обычно в стационарных
котлах путем сплавления смолы с полимеризованным
маслом при 250—280 °С. Процесс проводят в течение
нескольких часов до получения лаковой основы с опре-
определенной вязкостью. Затем в основу добавляют при
150—160 °С растворитель и сиккатив. Полученный лак
«ставят на тип», т. е. корректируют по содержанию
пленкообразующего (сухому остатку) и вязкости, до-
добавляя лаковую основу или растворитель, и выдержи-
выдерживают в течение 5—30 су т. Готовый лак очищают от
механич. примесей центрифугированием или фильтро-
фильтрованием на фильтрпрессе.
Лаки на основе копалов изготовляют
в переносных котлах A00—200 л) на открытых горнах.
Смолы перед их совмещением с маслами нагревают до
320—360 °С; образующиеся при этом летучие продук-
продукты деструкции, так наз. копаловое масло B0—30%
от массы смолы) отгоняют. Затем в расплав смолы до-
добавляют предварительно полимеризованное нагретое
масло. Процесс совмещения компонентов продолжают
до получения лаковой основы требуемой вязкости.
В основу лака добавляют при 150—160 °С скипидар
и раствор сиккатива. После «вызревания», продолжи-
продолжительность которого достигает 1 года, лак «ставят на
тип», а затем очищают от примесей на центрифугах или
фильтрпрессах.
При изготовлении эмалей пигменты, наполните-
наполнители и масляный лак предварительно смешивают в сме-
смесителях. Для диспергирования (перетира) пигментов
применяют краскотерки. Корректировку цвета (коле-
(колеровку) эмали производят в планетарных мешалках,
добавляя пигментные пасты. Иногда масляные эмали
изготовляют в шаровых мельницах; в этих же аппара-
аппаратах производят и колеровку эмали. Готовую эмаль
очищают от механич. примесей на одновалковых кра-
краскотерках с фильтрующим брусом или на металлич.
ситах (последний способ прост, но малопроизводите-
малопроизводителен). Очистку на центрифугах применяют реже, т. к.
в этом случае возможно расслоение эмали вследствие
различной плотности входящих в ее состав пигментов
и наполнителей и, следовательно, изменение цвета
эмали. Подробно о технологии и оборудовании для из-
изготовления эмалей см. Краски.
Свойства и применение. Пленкообразование при вы-
высыхании М. л. и э. происходит в результате химич.
превращений пленкообразующего, сопровождающихся
в начальной стадии улетучиванием растворителя. Вы-
Высыхание пленок при комнатной темп-ре обусловлено
гл. обр. окислительной полимеризацией жирных к-т
растительных масел, а при горячей сушке A50—
200 °С) — их термич. полимеризацией. Продолжитель-
Продолжительность высыхания «от пыли» возрастает с увеличением
жирности лаковой основы; при наличии в лаке быстро-
высыхающих масел (напр., тунгового) процесс уско-
ускоряется. Практически полное высыхание тощих лаков
при комнатной темп-ре наступает через 10—12 ч,
жирных — через 24 ч. Об определении времени высыха-
высыхания см. Испытания лакокрасочных материалов и по-
покрытий.
Свойства пленок масляных лаков зависят от состава
и содержания масляной части лака, условий сушки
покрытия и нек-рых др. факторов. Так, с увеличением
содержания масла повышается атмосферостойкость
пленок, но ухудшается их способность к шлифованию.
Пленки жирных лаков, содержащих смесь льняного и
тунгового масел (в соотношении 1 : 1 или 1 : 0,5), обла-
обладают высокой атмосфере- и влагостойкостью и являются
хорошим связующим для атмосферостойких масляных
эмалей. Пленки масляных лаков горячей сушки харак-
характеризуются высокой твердостью, хорошей адгезией,
влагостойкостью, удовлетворительными электроизоля-
электроизоляционными свойствами.
В условиях эксплуатации пленок М. л. и э. происхо-
происходят структурирование и деструкция пленкообразую-
пленкообразующего; последний процесс сопровождается выделением
летучих продуктов — окиси углерода, уксусной и му-
муравьиной к-т, альдегидов и кетонов. В закрытых по-
помещениях эти процессы протекают медленно и пленка
149
МАТРИЧНАЯ ПОЛИРЕАКЦИЯ
150
сохраняет эластичность в течение длительного времени
(до нескольких лет). При эксплуатации на солнечном
свету деструкция резко ускоряется; пленки теряют
эластичность и со временем растрескиваются. Пленки
эмалей белого цвета (особенно на основе лаков, содер-
содержащих льняное масло и марганцовые сиккативы) при
этом желтеют. Высокой атмосферостойкостыо отли-
отличаются пленки масляных эмалей, пигментированных
алюминиевой пудрой, окисью хрома, сажей.
Масляными лаками пропитывают обмотки электрич.
машин, лакируют металлы, древесные пластики. Их
наносят поверх пленок масляных эмалей для улучше-
улучшения декоративных свойств покрытий. Масляные лаки
служат, кроме того, пленкообразующим для водо- и
коррозионностойких грунтовок, а также шпатлевок.
М. л. и э. наносят на защищаемые поверхности с по-
помощью валика, кисти, краскораспылителя, а также оку-
окунанием и др. методами (см. Лакокрасочные покрытия).
Производство М. л. и э. постепенно сокращается
в связи с их заменой алкидными лаками и эмалями,
содержащими меньше масла (в среднем на 15—20%)
и образующими покрытия с более высокими эксплуата-
эксплуатационными свойствами.
Лит. см. при ст. Лаки и эмали, Масла растительные, Смолы
природные. В. В. Чеботаревский.
МАСС-СПЕКТРОСКОПИЯ полимеров, масс-
спектрометрия (mass-spectroscopy, Massen-
Spektroskopie, spectroscopie de masse) — метод изуче-
изучения химич. строения, состава и свойств полимеров путем
определения массы (чаще отношения массы к заря-
заряду т/е) и количества ионов, получаемых при иониза-
ионизации летучих продуктов разложения анализируемого ве-
вещества.
Приборы, используемые в М.-с, — масс-спект-
масс-спектрометры — состоят из трех основных частей: ион-
ионного источника, разделительного устройства (масс-
анализатора) и приемного устройства. В ионном источ-
источнике происходит ионизация исследуемого вещества
и формирование пучка ионов. В современных масс-
спектрометрах применяются различные способы иони-
ионизации: электронный удар, фотоионизация, лазерная
ионизация и т. д. В масс-анализаторе пучок ионов в за-
зависимости от величины т/е разделяется в пространстве
(ионы с различными т/е движутся одновременно по раз-
разным траекториям) или во времени (ионы с различными
т/е движутся по одной траектории, но попадают в при-
приемное устройство в разное время). Разделение ионов
происходит под действием электрического и магнитного
полей. В приемном устройстве ионы каждого вида
собираются вместе на коллекторе, при этом формиру-
формируется сигнал, пропорциональный ионному току, т. е.
количеству ионов, попадающих в единицу времени
в приемное устройство.
Каждое вещество при ионизации дает спектр ио-
ионов различной массы и заряда, обычно наз. масс-
спектром. Масс-спектр — индивидуальная харак-
характеристика вещества, позволяющая проводить его иден-
идентификацию. Т. обр., М.-с. позволяет проводить качест-
качественный и количественный анализ вещества.
Получить масс-спектры полимеров не удается, т. к.
полимеры нельзя перевести в газовую фазу без разло-
разложения. Поэтому масс-спектрометрич. исследованию
подвергают продукты разложения полимеров (чаще все-
всего продукты пиролиза). Состав продуктов пиролиза в
определенных условиях достаточно специфичен. Это по-
позволило применить М.-с. для идентификации
полимеров и даже для анализа состава
полимерных композиций. Так, напр.,
масс-спектрометрич. метод с успехом использовался для
изучения состава сополимеров этилена и пропилена.
Помимо анализа состава, М.-с. широко применяется
при исследовании механизма и кинетики
химических превращений в полимерах
(скорость образования летучих продуктов определяют
по высоте соответствующих пиков в масс-спектре; см.,
напр., рис.). Здесь большое значение приобретают такие
достоинства М.-с, как высокая чувствительность (в
нек-рых типах приборов для анализа достаточно та-
il
15
31
41
60 69
100
Масс-спектр летучих продуктов деструкции полиметилмет-
акрилата под действием света лампы ДРШ-100 при 90 °С;
спектр снят через 1 мин после облучения.
кое количество вещества, к-рое создает давление па-
паров в ионном источнике ~10~12 тор), быстрота анали-
анализа (сотни анализов в 1 сек), возможность наблюдения за
отдельным веществом в смеси. Эти достоинства обусло-
обусловили возможность исследования самых начальных ста-
стадий разрушения полимеров в процессах термической,
фотохимической, механической деструкции. Здесь с М.-с.
не может конкурировать ни один др. физич. метод.
В качестве примера можно привести результаты изуче-
изучения начальных стадий фотохим. деструкции полиме-
тилметакрилата. Как видно из рис., даже при непро-
непродолжительном облучении ничтожные количества про-
продуктов деструкции могут быть достаточно четко зафик-
зафиксированы масс-спектрометром.
При исследовании механич. деструкции полимеров
было показано, что выделение продуктов распада на-
начинается сразу же после приложения нагрузки, за-
задолго до разрыва образца (см. Долговечность).
М.-с. находит также применение при исследовании
деструкции полимеров под действием различных излу-
излучений; одновременное изучение состава и кинетики
образования летучих продуктов в этом случае позволя-
позволяет получить данные, характеризующие взаимодействие
излучения с полимерами. Получаемая методом М.-с.
информация о закономерностях различных видов де-
деструкции необходима для понимания природы этих
процессов и определения таких важных свойств поли-
полимеров, как термостойкость, фотостойкость и прочность.
Лит.: Бейнон Дж., Масс-спектрометрия и ее при-
применение в органической химии, пер. с англ., М., 1964; Б у д-
зикевичГ., ДжерассиК., Уильяме Д., Интер-
Интерпретация масс-спектров органических соединений, пер. с англ.,
М., 1966; Материалы I Всесоюзной конференции по масс-спект-
рометрии, Л., 1972; Коробейничев О. П., Усп. хи-
химии, 38, в. 12, 2113 A969); Регель В. Р., С л у ц-
к е р Я. И., Томашевский Э. Е., Усп. физич. наук,
106, в. 2, 193 A972); С о г n u A., MassoR., Compilation
of mass spectral data, L., 1967; Мадорский С, Термиче-
Термическое разложение органических полимеров, пер. с англ., М.,
1967; Труды Международного симпозиума по хромато-масс-
спектрометрии, 21 — 28 мая 1968 г., М., 1969.
В. Р. Регель, А. В. Амелин.
МАТРИЧНАЯ ПОЛИРЕАКЦИЯ (matrix polyreac-
tion, Matrizenpolyreaktion, polyreaction en matrice) —
химич. превращение мономеров или олигомеров в мак-
макромолекулы, направляемое в соответствии со струк-
турно-химич. информацией, к-рая заложена в последо-
последовательности звеньев др. макромолекул (матриц), при-
присутствующих в реакционной системе. М. п.— путь син-
синтеза биополимеров в живых организмах. Дезоксирибо-
нуклеиновые к-ты (ДНК), рибонуклеиновые к-ты (РНК)
и белки образуются в клетках или в модельных систе-
151
МАТРИЧНАЯ ПОЛИРЕАКЦИЯ
152
мах in vitro путем матричной поликонденсации, ката-
катализируемой соответствующими ферментами. Роль мат-
матриц выполняют макромолекулы ДНК или РНК.
Общие принципы матричных полиреакций. Матрич-
Матричная поликонденсация в живых клетках — очень важ-
важный, но лишь частный случай М. п., т. к. не существует
принципиальных ограничений для осуществления М. п.
иного типа вне клеток и для расширения круга участ-
участвующих в них мономеров или олигомеров. Поскольку
в упомянутых частных случаях М. п. доведены приро-
природой до пока еще далеко не превзойденного совершенства,
они приняты за принципиальные эталоны, а сведения
об их механизме использованы для формулирования
общих принципов матричного синтеза.
Передача информации при М. п. происходит благо-
благодаря тому, что матрица осуществляет структурно-
химич. контроль над совокупностью элементарных ак-
актов роста дочерней цепи, причем контакт между матри-
матрицей и растущей цепью м. б. прямым (как при реплика-
репликации ДНК или синтезе информационной РНК — см.
Нуклеиновые кислоты) или через посредников (как
в синтезе белка на информационной РНК с участием
транспортной РНК).
При М. п. в дочерней макромолекуле могут контро-
контролироваться: 1) состав и последовательность расположе-
расположения химич. звеньев (благодаря тому, что матрицы
«определяют» партнеров, участвующих в каждом эле-
элементарном акте роста); 2) степень полимеризации (мол.
масса), к-рая определяется степенью полимеризации
(мол. массой) матриц или их неблокированных участ-
участков; 3) изомерный состав и последовательность располо-
расположения возможных изомеров звеньев. Эти виды контроля
обусловливают структурные матричные
эффекты. Их можно обнаружить по окончании
синтеза путем сравнительного изучения строения и
свойств дочерних макромолекул и матриц.
В ходе синтеза могут проявляться и динамиче-
динамические матричные эффекты, к-рые выража-
выражаются в ускорении или замедлении М. п. по сравнению
с аналогичной полиреакцией в отсутствие матрицы.
Для структурно-химич. контроля роста цепи матри-
матрица должна обладать способностью адсорбировать исход-
исходные мономеры, а также удерживать на себе образовав-
образовавшиеся фрагменты дочерней цепи. Эта способность
привносится комплементарностью мономеров и дочерней
цепи по отношению к матрице.
Комплементарность в химич. смысле подразумевает
полностью или частично насыщаемое взаимное сродство.
Две частицы А и В комплементарны, если при их столк-
столкновении может образоваться комплекс А ... В, сродство
к-рого к А и к В заметно меньше, чем взаимное сродство
исходных компонентов. В ряде случаев насыщение
достигается только при условии, если структуры А
и В удовлетворяют определенным геометрич. тре-
требованиям. Фрагменты макромолекулы ~АААА~ и
~ВВВВ~, построенные из химически комплементар-
комплементарных звеньев А и В, вообще говоря, всегда химически
комплементарны. Однако для связывания друг с дру-
другом достаточно большой доли таких звеньев, принадле-
принадлежащих этим фрагментам, требования предъявляются
и к структуре фрагментов как целому. Жесткость требо-
требований тем выше, чем сильнее выражена направленность
связей между А и В и чем острее минимум на соответст-
соответствующей потенциальной кривой их взаимодействия.
Если эти требования выполнены, возникает возмож-
возможность прочной ассоциации фрагментов; каждая связь
А... В в ассоциате дополнительно стабилизируется
(кооперативное взаимодействие).
Ассоциация (и, следовательно, контакт) макромоле-
макромолекулы-матрицы с комплементарным фрагментом дочер-
дочерней цепи и сорбция незанятым звеном матрицы компле-
комплементарного мономера в непосредственной близости от
реакционноспособного конца растущего фрагмента,
предшествующая его включению в цепь,— необходимые
условия матричного синтеза. При их соблюдении м. б.
реализован весь комплекс как динамических, так и
структурных матричных эффектов.
Схема полиреакции на двухбуквенной матрице при-
приведена на рис. 1. Матрица и растущая цепь связаны
друг с другом и образуют участок двухтяжной струк-
структуры. Для включения в цепь мономера необходимо,
Рис. 1. Схема роста цепи на
двухбуквенной матрице (звез-
(звездочкой помечен активный ко-
конец цепи).
Дочерняя цепь
Матрица
чтобы он прежде прикрепился к свободному звену
матрицы. При этом, напр., к звену А может прикре-
прикрепиться только комплементарный ему мономер В, к-рый
затем образует химич. связь с концом дочернего фраг-
фрагмента (акт роста), и т. д. Таким образом контролирует-
контролируется последовательность звеньев в дочерней цепи. В идеа-
идеале процесс заканчивается после полной достройки
двухтяжной структуры. Тем самым длина дочерней
цепи коррелируется с длиной матрицы. Ориентация и
невалентные взаимодействия сорбированного мономера
с окружающими группами м. б. причиной, обусловли-
обусловливающей стереорегулирование в реакции роста. Наконец,
само «концентрирование» комплементарных мономеров
у концов растущих цепей и благоприятное (или небла-
неблагоприятное) ориентирование реакционных центров ве-
ведут к ускорению (или замедлению) М. п. по сравнению
с аналогичной полиреакцией в отсутствие матрицы.
Для отнесения какой-либо полиреакции к категории
матричных достаточно установить факт проявления
хотя бы одного из структурных или динамич. матричных
эффектов. Однако для создания полной принципиаль-
принципиальной модели матричного биосинтеза необходимо реали-
реализовать полиреакцию, в к-рой бы сочетались все матрич-
матричные эффекты. В остальных случаях М. п. можно рас-
рассматривать как частные модели.
Матричные полиреакции в небиологических системах.
Первый пример полиреакции, протекающей без участия
биополимеров, в к-рой установлен динамич. матрич-
матричный эффект,— поликонденсация 4-винилпиридина на
макроанионах поликислот (полиакриловой, полимета-
криловой, полистиролсульфокислоты, полиэтиленсуль-
фокислоты и др.) в относительно разб. водных р-рах.
Эта реакция происходит в области рН, в к-рой часть
мономера существует в виде незаряженных молекул,
а другая часть протонирована и присутствует в р-ре
в виде соли 4-винилпиридиния. В результате полиреак-
полиреакции образуется поли-1,4-пиридинийдиилэтилен (про-
тивоион на схеме не показан):
СН2=СН—/ V + СН2=СН— f NNH ——
\—/ \—/
СН2=СН—f NN~ СН2—СН—f NNH
V ¦/ \ /
сн2=сн—(;n—ch2—ch2—/)n
¦ СН2=СН-
153
МАТРИЧНАЯ ПОЛИРЕАКЦИЯ
154
Рост цепи состоит в попарном соединении фрагментов,
один из к-рых имеет на конце двойную связь, активи-
активированную по отношению к нуклеофильной атаке,
а другой — пиридиновой цикл:
2—СН
+ СИ,
СН2-СН2—( )N-CH2—CH
Далее карбанион II присоединяет протон из реакцион-
реакционной среды и превращается в очередное звено полимера I.
Для осуществления элементарного акта роста необ-
необходимо, чтобы положительно заряженный конец реаги-
реагирующего фрагмента цепи образовал ионную пару с анио-
анионом соли Х~. Тогда активированный комплекс III ста-
стабилизируется,
~СН2—СН2 СН2—СН2~
а конечный продукт присоединения представляет со-
собой квадруполь, т. е. частицу со скомпенсированными
зарядами. Если в реакционной системе присутствуют
макроанионы поликислот (т. е. противоионы связаны
друг с другом в молекулярные цепи), стабилизация
активированного комплекса отрицательным зарядом
реализуется благодаря кооперативной адсорбции прак-
практически всех реагирующих поликатионных фрагментов
на полианионах. Одновременно достигается благоприят-
благоприятная для сборки цепи ориентация реакционных центров.
Т. обр., полианионы выполняют функцию матриц. Если
полианионные матрицы заменить низкомолекулярны-
низкомолекулярными анионами, необходимые для роста цепи ионные па-
пары образуются лишь при случайных контактах про-
противоположно заряженных частиц. Исключается и фак-
фактор взаимной ориентации реакционных центров. Поэто-
Поэтому скорость матричной поликонденсации значительно
выше, чем скорость той же реакции в отсутствие мат-
матриц (рис. 2).
Динамич. матричные эффекты установлены также при
радикальной полимеризации метакриловои и акриловой
к-т на полиэтиленоксиде, поливинилпирролидоне, по-
поливиниловом спирте и в ряде др. случаев.
Структурные матричные эффекты удается моделиро-
моделировать, используя относительно простые объекты. Приме-
Примером служит радикальная поли-
полимеризация метилметакрилата в
диметилформамиде в присутствии
растворенного изотактич. поли-
метилметакрилата. Изотактич. по-
последовательности звеньев материн-
материнских цепей стимулируют образо-
образование на себе синдиотактич. по-
лиметилметакрилатных блоков,
связывающихся с ними в прочные
кооперативные стереокомплексы.
Контакт матрицы с растущей до-
дочерней цепью и сорбция на ней
Рис. 2. Зависимость начальной скоро-
скорости превращения (vn) 4-винилпиридина
в присутствии полиакриловой (/)и ук-
уксусной B) к-т от молярного отноше-
. р з , с с ния кислота/мономер при 20 °С; сум-
/ марная концентрация 4-винилпиридина
Кислота /мономер и 4-винилпиридиния 0,1 М.
о 5
молекул мономера обусловливают стереохимич. кон-
контроль элементарных актов роста. Влияние матриц на
изомерный состав дочерних цепей обнаружено и при
анионной полимеризации метилметакрилата, иницииро-
инициированной литийалкилом, в присутствии синдиотактич.
по лимети лметакри л ата.
Примером М. п. на двухбуквенных матрицах, при
к-рой, по-видимому, достигается контроль над последо-
последовательностью чередования химич. звеньев в дочерних
цепях, может служить радиационная сополимеризация
акриловой к-ты с винилиденхлоридом на поверхности
ориентированных волокон из полиамида-6,6. Надо пола-
полагать, что в поверхностном слое волокна, помещенного
в газообразную смесь мономеров, происходит упорядо-
упорядоченная адсорбция молекул к-ты на амидных группах
макромолекулы. При полимеризации промежутки меж-
между прочно сорбированными молекулами первого мономе-
мономера, приходящиеся на метиленовые группы, заполня-
заполняются звеньями относительно слабо сорбирующегося
винилиденхлорида, молекулы к-рого мигрируют по
поверхности и наталкиваются на активные центры це-
цепей, растущих вдоль оси волокна. После того как
поверхность волокна оказывается экранированной, роль
матриц начинают играть макромолекулы образовавше-
образовавшегося сополимера.
Матричный контроль длины дочерних цепей моде-
моделирован путем радикальной полимеризации акрилат-
ных и метакрилатных остатков, предварительно «со-
«собранных» в заготовки путем этерификации крезоло-
формальдегидных олигомеров соответствующими не-
непредельными к-тами:
СН
1з ^пз <-н3 СН3
При полимеризации в разб. р-рах образуются лест-
лестничные олигомеры:
YCH2-CH
СН2-СН
CH2—CHY
Их гидролиз позволяет выделить в свободном состоянии
олигомеры полиакриловой и полиметакриловой к-т,
степень полимеризации к-рых равна степени полимери-
полимеризации крезоло-формальдегидных матриц.
Исследования в области небиохимич. М. п. пока еще
находятся в начальной стадии. Поэтому преждевремен-
преждевременно обсуждать их будущие конкретные приложения.
Существенно, однако, что М. п. приводят к образованию
весьма жестких полимер-полимерных комплексов, про-
проявляющих свойства упорядоченных кооперативных
систем. Важнейшее из этих свойств — резкое обрати-
обратимое изменение характеристик (механич. свойств, рас-
растворимости и способности к набуханию, сорбционной
способности, проницаемости и т. п.) в весьма узких
интервалах изменения внешних условий (темп-ры, со-
состава среды, кислотности и др.). Надо полагать, что
уникальные свойства поликомплексов, образующих-
образующихся в результате М. п., выдвинут их в новый класс
практически важных полимерных материалов.
155
МЕДИЦИНСКИЕ НИТИ
156
Лит.: Каргин В. А. [и др.], ДАН СССР, 161, № 5,
1131 A965); Кабанов В. А. [и др.], Высокомол. соед.,
13А, № 2, 348 A971); Осада Е. [и др.], ДАН СССР, 191,
№2, 399 A970); Buter R., Tan Y. Y., Shall a G.,
J. Polymer Sci., pt A—1, 10, № 4, 1031 A972); International
symposium on macromol. chem., Helsinki, 1972, preprint, v. 1,
p. 35; G о 1 о v a L. K., A m e r i k V u. В., К г е n t-
s e 1 В. А., там же, v. 2, p. 555; К a m m e г е г Н. Lu. a.],
Makromol. Chem., 91, 1 A966); 101, 284 A967); 116, 62,
72A968); ЦетлинБ. Л., Г о л у б е в В. Н., ДАН СССР,
201, № 4, 881 A971). В. А. Кабанов.
МЕДИЦИНСКИЕ НИТИ (medical thread, Medizi-
nische Faden, medical fil). ИзМ.н. изготовляют перевя-
перевязочные и шовные материалы, протезы трубчатых орга-
органов, ленты и сетки для фиксации и реконструкции раз-
различных тканей и органов и др. изделия медицинского
назначения. Для изготовления М. н. применяют при-
природные волокна (целлюлозные — хлопковые, льня-
льняные; белковые — натуральный шелк), искусственные
(в основном вискозные) и синтетические (полиэфир-
(полиэфирные, полиамидные, поливинилспиртовые и др.)- Обычно
используют М. н. из филаментных или штапельных во-
волокон, а в нек-рых случаях жилку из синтетич. поли-
полимеров, кетгут и натуральный волос.
К М. н., по сравнению с нитями для обычных тек-
текстильных изделий, предъявляют ряд специфич. требо-
требований. Так, М. н. не должны оказывать на организм
местного раздражающего и общего токсического (в т. ч.
канцерогенного) действия. Кроме того, М. н. должны
отвечать ряду частных требований, определяемых спе-
спецификой назначения и особенностями конструкций из-
изготовляемых из них изделий.
Нити для изготовления перевязочных материалов
(средств). Эти нити и материалы на их основе наряду
с удовлетворительными механич. свойствами должны
обладать хорошими физико-гигиенич. характеристи-
характеристиками: достаточно хорошо впитывать кровь и раневой
экссудат (жидкость, проникающая сквозь стенки не-
неповрежденных кровеносных сосудов в окружающие
ткани при любом воспалении), быстро высыхать, не
раздражать рану и не прилипать к ее поверхности.
При выработке марли наиболее часто используют
хлопковую пряжу. Однако такая марля не отвечает
в достаточной мере всем предъявляемым к ней требова-
требованиям (высокая впитывающая способность, прочность
и др.). Лучшими свойствами обладают марли на основе
вискозных нитей, получаемых из штапельного волок-
волокна, к-рые комбинируют по основе и утку с нитями из
хлопка, капрона или лавсана. Хлопчато-вискозная
марля обладает довольно высокой способностью к по-
поглощению экссудата и повышенным коэфф. трения, что
обеспечивает хорошее сцепление отдельных слоев бин-
бинта в повязке и предотвращает ее сползание. Вискозно-
капроновая и вискозно-лавсановая марли лучше, чем
хлопковая, способствуют оттоку крови и раневого экс-
экссудата и менее болезненно отделяются от грануляции
(молодой соединительной ткани, заполняющей рану).
Однако, обладая более гладкой поверхностью, эти
марли хуже обеспечивают сцепление слоев в повязке.
Кроме того, применяют перевязочные материалы, из-
изготовленные из частично окисленных хлопковых или
вискозных нитей, способных растворяться в воде или
слабом р-ре соды. Этим облегчается отделение таких
материалов от раневой поверхности.
Для менее болезненного отделения перевязочных
средств от поверхности раны или обожженной ткани
предложено использовать марлю, изготовленную из
рассасывающегося материала — оксицеллюлозы или
солей альгиновой к-ты. Первую получают окислени-
окислением первичных гидроксильных групп целлюлозы (взя-
(взятой обычно в виде хлопчатобумажной пряжи или мар-
марли) окислами азота. Срок рассасывания оксицеллюло-
оксицеллюлозы зависит от содержания в ней СООН-групп: для
материала с 14—16%-ным содержанием этих групп он
составляет 15 дней. В США вата и марля из оксицел-
оксицеллюлозы выпускаются под названием седжицелл.
Перевязочные материалы яа основе альгиновой ки-
кислоты рекомендуется применять при лечении ожогов.
Срок рассасывания этих материалов под действием
выделяемого пораженной поверхностью экссудата ре-
регулируется содержанием ионов кальция, играющих
роль солевых сшивок между СООН-группами альгино-
альгиновой к-ты. Ионы кальция вводятся в альгинатное волок-
волокно на стадии прядения водных р-ров альгината натрия.
В Великобритании альгинатная марля выпускается
под названием калджитекс. Вата и марля на основе
оксицеллюлозы и альгината кальция обладают гемо-
статич. (кровоостанавливающим) действием.
Для изготовления перевязочных материалов (марли и
сеток) применяют также нити из антимикробных воло-
волокон. Антимикробная активность этих материалов сохра-
сохраняется при длительной эксплуатации. Наложение по-
повязок из антимикробных материалов на раны обеспе-
обеспечивает значительное уменьшение патогенной раневой
флоры (болезнетворных микроорганизмов, населяющих
рану) и тем способствует их заживлению. Для изго-
изготовления материалов такого типа применяют анти-
антимикробные поливинилспиртовые нити (отечественная
марка — летилан), к-рые содержат хпмиотерапевтич.
препараты нитрофуранового ряда. Для повышения
прочности изделий и улучшения условий дренирова-
дренирования экссудата эти нити комбинируют с нитями из
лавсана. Известны перевязочные материалы из анти-
антимикробных целлюлозных и гидратцеллюлозных нитей.
В качестве перевязочных средств используют так-
также трубчатые эластичные бинты сетчатой структуры
на основе хлопковых нитей в комбинации с резино-
резиновыми. В Италии их выпускают под названием ретэласт.
Нити для изготовления шовных материалов. М. н.,
применяемые для изготовления хирургич. шовного ма-
материала, должны отвечать указанным выше общим тре-
требованиям и наряду с этим обладать эластичностью, до-
достаточно большой прочностью, а также хорошо завязы-
завязываться при наложении швов, не скользить и не «рас-
«расслабляться» в узлах.
Для сшивания ран широко используют прочные и
эластичные нити из натурального шелка. Однако,
являясь белковыми веществами, такие нити способ-
способны фиксировать на себе микроорганизмы и служить ис-
источником нагноения раны.
В виде шовного материала используют также жилку
из кишок мелкого рогатого скота — кетгут. Последний
применяют в основном для наложения рассасываю-
рассасывающихся швов. Кетгут рассасывается в организме в тече-
течение 2—4 недель, причем скорость рассасывания регу-
регулируется условиями дубления и толщиной нити. Основ-
Основной недостаток кетгута — способность вызывать у
нек-рых больных аллергич. реакцию. В качестве
заменителя кетгута разработаны искусственные волок-
волокна из коллагена (белка соединительной ткани живот-
животных). Недостатков кетгута лишены рассасывающиеся
хирургич. нити из полигликолида.
Нити из лавсана, применяемые в качестве шовного
материала, имеют существенные преимущества перед
нитями из натурального шелка. Лавсановая нить проч-
прочна, гладка, эластична, легко стерилизуется, негигро-
негигроскопична, устойчива к гниению. Для придания этим
нитям антимикробных свойств их комбинируют с ни-
нитями из летилана. Известны хирургич. шовные мате-
материалы из капроновых, полипропиленовых фторлоно-
вых, льняных и др. нитей.
Нити для изготовления протезов трубчатых органов.
Протезы трубчатых органов (кровеносных сосудов,
пищевода и др.) изготовляют гл. обр. на основе волок-
волокнистых материалов. Сосудистые протезы делают в
виде плетеных, вязаных или тканых трубок, к-рые
обязательно подвергаются гофрировке, предохраня-
предохраняющей их от сжатия и крутых изгибов. Т. к. по физио-
логич. условиям работы этих протезов в организме
157
МЕДНОАММИАЧНЫЕ ВОЛОКНА
15S
на них действуют большие механич. нагрузки и они
подвергаются многократным деформациям, жела-
желательно, чтобы нити, из к-рых их изготовляют, имели
достаточную прочность (не менее 40—50 гс/текс при
разрывном удлинении 15—20%) и высокий модуль
эластичности — 9—10 Гн/м2 (900—1000 кгс/мм2). Эти
нити должны иметь небольшую толщину A,6—5 текс),
чтобы обеспечивать создание тонкой стенки искусст-
искусственного сосуда, и гладкую поверхность, чтобы не
способствовать тромбообразованию внутри протеза,
а также гемолизу (разрушению форменных элементов
крови). Они должны удовлетворять и всем общим
требованиям, предъявляемым к хирургич. нитям.
Для изготовления сосудистых протезов используют
нити из лавсана, полипропилена, фторлона. Лавса-
Лавсановые нити лучше, чем другие, обеспечивают сохра-
сохранение формы протезов, приданной при гофрировке.
Фторлоновые искусственные сосуды наиболее био-
биологически инертны, но плохо поддаются гофрировке.
Поэтому в ряде случаев используют фторлон — лав-
лавсановые протезы. Для регулирования кровопрони-
цаемости и «вживления» протезов их иногда импре-
гнируют различными составами, в т. ч. содержащими
гепарин и коллаген, к-рый также предложено вводить
в виде нитей непосредственно в структуру протезов
(т. наз. полубиологич. протезы). Известны комби-
комбинированные летилан — лавсановые протезы, обла-
обладающие антимикробными свойствами. Часто изготов-
изготовляют кровеносные сосуды с раздвоенными трубками —
т. наз. бифуркационные протезы.
Из нитей лавсана конструируют трубки для времен-
временного протезирования пищевода при неоперабельных
опухолях, а также различного вида катетеры, в т. ч.
рентгеноконтрастные, для зондирования сердца, дли-
длительного внутривенного введения жидкостей и др.
Перспективно применение нитей из полых волокон
в аппаратах «искусственная почка» (для диализа
крови) и «искусственное легкое» (для избирательного
извлечения СО2).
Нити для прочих медицинских материалов. М. н. при-
применяют для изготовления эластичных бинтов и чулок,
которые используют при заболеваниях вен ниж-
нижних конечностей. К этим изделиям предъявляют ряд
специфич. требований (хорошая растяжимость, возду-
воздухопроницаемость, равномерное распределение давле-
давления на всех участках поверхности тела и др.). Для их
изготовления используют гл. обр. нити из капрона
(обычные и эластик). При выработке чулок часто вместе
с капроновыми применяют резиновые или высокоэла-
высокоэластичные полиуретановые нити (спандекс).
Широко распространено лечебное белье из нитей
и пряжя хлорин, применяемое при заболеваниях пери-
ферич. нервной системы (радикулит, полиартрит и др.).
Это белье обладает физиотерапевтич. действием, к-рое,
как полагают, обусловлено электрич. свойствами во-
волокон хлорин.
Лит.: Вольф Л. А., М е о с А. И., Волокна специаль-
специального назначения, М., 1971; Biomedical polymers, ed. by A. Rem-
baum, M. Shen, N. Y., 1971; Encyclopedia of polymer science and
technology, v. 15, N. Y.— [a. o.], 1971, p. 270; Микиртиче-
в a 3. В. [и др.], Текст, пром-сть, № 12, 11 A961); № 5, 29
A962); Лебедев Л. В., Плоткин Л. Л., Ортопедия,
травматология и протезирование, № 2, 49 A961). Л. А. Вольф.
МЕДНОАММИАЧНЫЕ ВОЛОКНА (cupri-ammonic
fibers; Kupferfasern; fibres cupri-ammoniaques)— искус-
искусственные волокна, получаемые из целлюлозы. М. в.
производят в виде непрерывных (текстильных) нитей
и штапельного волокна.
Получение. Приготовление прядиль-
прядильного р-ра заключается в растворении химически
очищенной хлопковой или облагороженной древесной
целлюлозы в медноаммиачном р-ре. Последний полу-
получают из гидроокиси или основных солей меди и конц.
р-ра аммиака (содержание NH3 не менее 2Ь%), взятого
в избытке. В результате реакции между компонентами
р-ра образуется основание — куприаммингидрат
[Cu(NH3)w(OHJ, где тп < 4],к-рое в воде и р-рах аммиа-
аммиака диссоциировано на 65—70%. При взаимодействии
этого основания с целлюлозой образуется сложное
комплексное соединение, к-рое м. б. описано суммар-
суммарной ф-лой
СвН7О2(ОНK-(ОнГ
где у — число ОН-групп в 100 элементарных звеньях
макромолекулы целлюлозы, связанных с ОН'-ионами.
Обычно значение у производственных прядильных р-ров
колеблется от 200 до 220, а содержание целлюлозы
в р-ре — от 7,5 до 11%.
Применяемая для производства М. в. целлюлоза дол-
должна иметь степень полимеризации не ниже 800—1000
и содержать не более 3,0—3,5% низкомолекулярных
фракций, т. к. в противном случае получаются волокна
пониженной прочности. Перед растворением целлюлозу
для повышения ее реакционной способности измельчают
и увлажняют (добавляют до 100% воды от массы сухой
целлюлозы).
Прядильные р-ры приготавливают двумя способами:
однофазным и двухфазным. По первому из них вод-
водный кислый р-р медного купороса нейтрализуют р-ром
соды или аммиака. При этом осаждается гидроокись
меди, к-рую промывают, отжимают и смешивают
с конц. р-ром аммиака. В полученной смеси растворяют
влажную целлюлозу. На 0,4 кг металлич. меди и 0,7—
1,0 кг аммиака берут 1 кг целлюлозы; растворение пос-
последней происходит при 15—20 °С и интенсивном пере-
перемешивании. Вязкость полученного прядильного р-ра
составляет 800 пз\ без доступа воздуха он вполне стаби-
стабилен. После тщательного перемешивания прядильный р-р
фильтруют через частую никелевую сетку, а затем из
него под вакуумом, обычно при 20—25 °С, удаляют
пузырьки воздуха, вместе с к-рыми улетучивается
ок. 30—40% аммиака. При этом р-р становится более
стабильным и облегчается формование из него волокна
по водному способу (см. ниже). В результате перемеши-
перемешивания, фильтрации и дегазации прядильного р-ра сте-
степень полимеризации целлюлозы снижается до 400—450.
По двухфазному способу прядильный р-р готовят с ис-
использованием основной соли меди — 5Cu(OHJ'2CuSO4.
Для этого водный кислый раствор медного купоро-
купороса частично нейтрализуют содой или аммиаком, добав-
добавляя лишь 5/7 от необходимого количества основания.
Высадившуюся основную соль меди смешивают с
конц. р-ром аммиака и загружают в эту смесь влаж-
влажную целлюлозу. При этом целлюлоза набухает и лишь
частично переходит в р-р. Затем в растворитель добав-
добавляют NaOH для перевода основной соли меди в гидро-
гидроокись меди. После этого происходит полное растворе-
растворение целлюлозы. Последующие операции такие же,
как при однофазном способе получения.
Прядильный р-р, полученный по второму способу,
отличается более высокой (примерно на 20%) вязкостью
и меньшей стойкостью к обработке водой при формо-
формовании.
М. в. формуют по мокрому двухванному способу.
При водном способе формования при-
применяют герметически закрытые конич. прядильные
воронки, в верхней части к-рых расположены фильеры.
Вокруг фильеры расположены отверстия для подачи
в воронку осадительной ванны (умягченной воды).
Струйки прядильного р-ра, выходящие из фильеры,
опускаются вниз, увлекаемые током воды. При этом
толщина струек постепенно уменьшается и на выходе
из воронки становится в 100—200 раз меньше перво-
первоначальной. Одновременно вода частично разлагает мед-
но-целлюлозный комплекс. При этом аммиак и часть
меди переходят в осадительную ванпу; у снижается до-
значений ниже 100. Для прядильного р-ра, получен-
159
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ
160
яого по однофазному способу, темп-pa осадительной
ванны составляет 70—75 °С, по двухфазному — 45—
50 °С. При формовании текстильных нитей применяют
воронки высотой 150 мм и диаметром 60—70 мм и фи-
фильеры с числом отверстий от 10 до 100, при формовании
штапельного волокна — воронки высотой 500—600 мм
и диаметром 150—165 мм и фильеры с числом отверстий
от 1500 до 3000. Диаметр отверстий фильеры в обоих
случаях составляет 1,0 — 1,2 мм. Скорость формования
текстильных нитей 30—60, штапельного волокна —
35—50 м/мин. Выходящее из воронки волокно поступа-
поступает во вторую ванну A,5—2,0%-й р-р H2SO4).
Щелочной способ формования М. в.
осуществляют на тех же прядильных машинах и в тех
же условиях, чго и формование вискозного волокна.
Прядильный р-р из фильеры поступает в первую ванну
(р-р NaOH концентрацией 40 г/л), в к-ром медноаммиач-
ное комплексное соединение целлюлозы превращается
в нерастворимую меднонатронную целлюлозу с соотно-
соотношением (по массе) С6Н10О5: Си : Na^l,0 : 0,5 : 0,5.
Затем волокно поступает во вторую ванну (р-р H2SO4),
где формование заканчивается. Для получения тек-
текстильных нитей и штапельного волокна применяют
фильеры с таким же числом отверстий, как при фор-
формовании по водному способу. Однако в этом случае
диаметр отверстий фильеры не превышает 0,08 мм.
Вытяжка волокон в первой ванне составляет всего
20-30%.
Операции по отделке и сушке М. в.,
полученных различными способами формования, при-
примерно одинаковы. Текстильные нити или штапельные
жгуты обрабатывают слабым р-ром H2SO4 для удаления
меди, промывают, обрабатывают мылом или авиважным
составом и сушат при 65—75 °С. Если необходимо по-
получить волокна повышенной мягкости, эти операции
повторяют. Штапельные жгуты режут на отрезки 30—
40 мм (при переработке по хлопко-прядильной системе)
или 60—100 мм (при переработке по шерстяным систе-
системам) и дополнительно разрыхляют.
При формовании М. в. по обоим способам выделяются
менее вредные продукты и в меньших количествах, чем
при получении вискозного волокна. Однако большие
расходы меди, аммиака и воды (особенно при формо-
формовании М. в. водным способом) обусловливают необхо-
необходимость проведения дополнительных операций для
полной регенерации меди, аммиака и очистки воды.
Свойства и применение. Толщина М. в., формуемых
по водному способу, вследствие большой фильерной
вытяжки очень мала @,15—0,3 текс) и легко может
быть снижена до значения менее 0,1 текс. Такие М. в.—
юамые тонкие из всех известных химич. волокон. Кроме
того, они обладают равномерной структурой, большой
мягкостью, низкой плотностью,высокой скоростью на-
накрашивания, эластичностью, но невысокой прочно-
прочностью A5 — 18 гс/текс).
Свойства М. в., формуемых по щелочному способу,
аналогичны свойствам вискозных волокон. Наличие
ориентированного поверхностного слоя в структуре
таких волокон затрудняет диффузию красителей в глубь
волокон и уменьшает их эластичность и мягкость. Од-
Однако прочность М. в. щелочного формования обычно не-
несколько выше, чем у волокон, получаемых по водному
способу.
Медноаммиачные штапельные волокна применяют,
в основном,, для производства ковров и сукна, а тонкие
E—10 текс) комплексные нити — для выработки три-
трикотажных изделий, чулок и легких тканей. Для технич.
целей М. в. почти не применяют из-за низкой прочности,
хотя известны способы получения нитей с прочностью
-55 гс/текс.
Стоимость М. в. несколько выше, чем вискозных во-
волокон, гл. обр. из-за высокой стоимости химически очи-
очищенной хлопковой или высокооблагороженной древес-
древесной целлюлозы. Поэтому выпуск М. в. постепенно сни-
снижается и в 1973 составил лишь ок. 1% от мирового
производства химич. волокон. В сравнительно крупном
масштабе М. в. производят в СССР (штапельное во-
волокно), ФРГ и Японии (текстильные нити).
М. в.— одни из наиболее старых видов искусственных
волокон. В производственном масштабе эти волокна
впервые были выпущены в Германии в 1897.
Лит.: Пакшвер А. Б., Технология медноаммиачного
волокна, М.— Л., 1947. А. Б. Пакшвер.
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ, С И
(International System of Units, Internationale Einhei-
tensystem, Systeme International d'Unites) — универ-
универсальная система единиц физич. величин для всех отрас-
отраслей науки и техники; принята в I960 XI Генеральной
конференцией по мерам и весам и одобрена рядом Меж-
Международных организаций. К началу 1974 38 стран
(в том числе Австрия, Болгария, Венгрия, ГДР, Ита-
Италия, Канада, СССР, Франция, ФРГ, Чехословакия,
Швеция) ввели Международную систему единиц в ка-
качестве обязательной или факультативной. Внедрение
СИ устранит многообразие системных и внесистемных
единиц, вызывающее значительные затруднения в об-
обработке результатов научных исследований, при про-
проектировании, строительстве и эксплуатации производ-
производственных объектов и оборудования, а также в процес-
процессе преподавания научных дисциплин. Важнейшие до-
достоинства СИ по сравнению с применяемыми система-
системами — универсальность, унификация единиц, удобные
для практики размеры основных, дополнительных и
большинства производных единиц, когерентность (от-
(отсутствие коэффициентов пропорциональности в физич.
ур-ниях, определяющих размеры производных единиц),
простота записи расчетных ф-л. Кроме того, внедре-
внедрение СИ в науку и торговлю приведет к улучшению вза-
взаимопонимания при развитии научно-технич. и торго-
торговых связей между странами.
СИ положена в основу окончательной редакции госу-
государственного стандарта «Единицы физических вели-
величин», одобренной Государственным комитетом стандар-
стандартов Совета Министров СССР для обязательного приме-
применения во вновь разрабатываемых или пересматривае-
пересматриваемых стандартах всех видов (в том числе и на средства
измерений), в нормативно-технич. документации, при
преподавании во всех учебных заведениях (в средних
школах, в высших и средних специальных учебных
заведениях и др.) и в научно-технич., общественно-
политич. и экономич. литературе.
СИ содержит семь основных величин и соответственно
семь основных единиц (т.е. единиц, размеры к-рых уста-
устанавливаются по определениям), две дополнительные и
большое число производных величин и соответствующие
им дополнительные и производные единицы. К основ-
основным величинам СИ относятся: длина с единицей метр
(м), масса с единицей килограмм (кг), время
с единицей се к у н д а (с), сила электрич. тока с еди-
единицей ампер (А), термодинамич. темп-pa Кельвина
с единицей к е л ь в и н (К), сила света с единицей
к а н д е л а (кд) и количество вещества с единицей
моль (моль). Следует отметить, что масса и количе-
количество вещества — понятия не тождественные. Опреде-
Определение моля, принятое XIV Генеральной конференцией
по мерам и весам: «Моль — количество вещества систе-
системы, содержащей столько же структурных элементов,
сколько содержится атомов в нуклиде 12С массой
0,012 кг (точно). При применении моля структурные
элементы должны быть специфицированы и могут быть
атомами, молекулами, ионами, электронами и др. час-
частицами или специфицированными группами частиц».
К дополнительным величинам (т. е. к величинам, не
являющимся ни основными, ни производными) относят-
относятся: плоский угол с единицей радиан (рад) и телес-
телесный угол с единицей стерадиан (ср).
161
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ
162
Размер производных единиц принимается на основа-
основании физич. законов, устанавливающих связь между
физич. величинами; они образуются как когерентные
единицы. Так, единица силы — ньютон (Н) уста-
устанавливается из второго закона Ньютона F = та,
как сила F = 1 Н, сообщающая телу массой т = 1 кг
ускорение а = 1 м/с2 в направлении действия силы.
Единица теплопроводности устанавливается из ур-ния
для стационарного режима теплопроводности Я = d t
как теплопроводность вещества Я = 1Вт/(м-К), в к-ром
при стационарном режиме с поверхностной плотностью
теплового потока q = 1 Вт/м2 устанавливается темпе-
температурный градиент grad t = 1 К/м.
В М. с. е. размер электрич. и магнитных единиц уста-
устанавливается из ур-ний электромагнитного поля, запи-
записанных в рационализованной форме. Рационализация
ур-ний электромагнитного поля имеет целью исключе-
исключение безразмерных коэффициентов 4я и У4я из всех соот-
соотношений, в к-рых наличие этих коэффициентов не оп-
оправдано, и введение их в соотношения для явлений,
характеризуемых осевой или сферич. симметрией. Для
рационализации ур-ний электромагнитного поля, запи-
записанных в нерационализованной форме, приписывают:
1) множитель 4л к след. величинам — D (электрич.
смещение), tyD (поток электрич. смещения), Н (напря-
(напряженность магнитного поля), F (магнитодвижущая сила),
гт (магнитное сопротивление), 8а и е0 (абсолютная ди-
электрич. проницаемость и диэлектрич. постоянная),
2) множитель уАп — к Кт (магнитная восприимчивость),
gm (магнитная проводимость), ца и jli0 (абсолютная маг-
магнитная проницаемость и магнитная постоянная).
Решением XIV Генеральной конференцией по мерам
и весам приняты собственные наименования паскаль
(Па) для единицы давления и механич. напряжения
и сименс (См) — для единицы электрич. проводимости.
Определение паскаля: «Паскаль — давление, вызы-
вызываемое силой 1 Н, равномерно распределенной по нор-
нормальной к ней поверхности». Определение сименса:
«Сименс — электрич. проводимость проводника сопро-
сопротивлением 1 Ом».
Для сокращения количества значащих цифр в числе
при выражении значений величин, полученных в ре-
результате расчетов или измерений, следует применять
кратные и дольные единицы от единиц СИ, образуемые
умножением исходных единиц на число 10, возведенное
в соответствующие положительные или отрицатель-
отрицательные степени. Наименования десятичных кратных и
дольных единиц образуются присоединением при-
приставок к наименованиям исходных единиц. Так,
150 000 000 000 Ом-м = 150 Том-м (тераом-метров);
0,000 000 006 Вт=6 нВт (нановатт). Не допускается при-
применение подряд двух и более приставок к простому
наименованию единицы; так, вместо микромикрофара-
микромикрофарады, т. е. миллионной доли от миллионной доли фарады,
следует применять пикофараду (пФ), равную 10~12 Ф,
т. е. биллионной доле фарады. Допускается ограничен-
ограниченное применение приставок деци A0), санти A0~2),
дека A01), гекто A02) только в наименованиях дольных
и кратных единиц, получивших широкое распростране-
распространение (напр., дециметр, сантиметр, декалитр). В сложном
наименовании единицы приставку присоединяют к наи-
наименованию первой единицы; не рекомендуется приме-
применение приставок в знаменателе обозначения единицы
сложного наименования. Рекомендуется использовать
приставки таким образом, чтобы числовые значения
величин лежали в пределах 0,1 — 1000. Приставками
для образования кратных и дольных единиц служат:
тера (Т) — 1012, гига (Г) — 109, мега (М) — 106, кило
(к) — 103, милли (м) — 10~3, микро (мк) — 10~6, нано
(н) — 10~9, пико (п) — 10~12, фемто (ф) — 10~15, атто
(а) — 10~18. Вместо микрона (мк) следует применять
микрометр (мкм).
Наравне с единицами СИ допускается применение
ряда единиц, не могущих быть изъятыми в ближайшее
время, напр, минута (мин), час (ч), сутки (сут) — для
времени; центнер (ц) и тонна (т) — для массы; градус
Цельсия (°С) — для темп-ры; угловые градусы (...°),
минута (../) и секунда (...") — для плоского угла; гек-
гектар (га) — для площади; литр (л) — для объема и вме-
вместимости.
Допускается к временному применению с постепен-
постепенным изъятием из употребления ряд единиц — калория
(кал), килограмм-сила (кгс), рад, рентген (Р), кюри
(Ки), бар и основанные на них единицы ангстрем (А),
карат (кар), лошадиная сила (л. с.) и др. Обозначения
единиц, названных в честь ученых (напр., ампер, бел,
ватт, вольт), следует писать с большой (заглавной) буквы
в соответствии с решениями Генеральных конференций
по мерам и весам.
При прямом шрифте текста следует для обозначений
единиц применять прямой шрифт, а при курсивном
шрифте текста — курсивный шрифт (в «Энциклопедии
полимеров», подготовка к-рой началась до принятия
этого решения, почти все обозначения единиц наби-
набираются курсивом).
Особое внимание следует обратить на то, что кило-
килограмм (кг), грамм (г), тонна (т) являются единицами
массы, а не веса или силы тяжести, а килограмм на ку-
кубический метр (кг/м3), грамм на кубический сантиметр
(г/см3) — единицами плотности (в том числе средней
и насыпной), а не единицами удельного веса (насып-
(насыпного или объемного веса). Масса тела — скалярная
величина — в технике определяется как результат взве-
взвешивания тела на весах. Значение массы не зависит
от значения ускорения свободного падения в пункте
измерения или определения. Сила тяжести и вес тела —
векторные величины и в общем случае не являются си-
синонимами. Сила тяжести тела определяется в соответ-
соответствии со вторым законом Ньютона F = mg, где т —
масса тела в кг (в СИ), g — ускорение свободного паде-
падения в м/с2 (в СИ), F — сила тяжести в Н (в СИ). Зна-
Значение силы тяжести зависит от значения ускорения
свободного падения в пункте измерения или определе-
определения. Вес тела — сила, действующая на опору или на
нить подвеса. Вес тела м. б. определен по ф-ле Р =
= т(ё±^)^ гДе a — ускорение, сообщаемое телу.
Т. обр., вес тела зависит и от ускорения свободного паде-
падения g и от ускорения тела а. Если ускорение, сообщае-
сообщаемое телу, равно нулю (а = 0), вес тела равен силе тя-
тяжести. Силу тяжести и вес тела определяют с помощью
динамометрич. приборов в условиях относительного
покоя тела и динамометра.
Единицами веса и силы тяжести, как и любой силы,
являются в СИ ньютон (Н), кратные и дольные от нью-
ньютона; в системе СГС — дина; внесистемные единицы —
килограмм-сила (кгс), грамм-сила (гс) и тонна-сила (тс).
В основных технич. характеристиках полимерных тел
приводятся их масса (а не вес!) и плотность (а не удель-
удельный вес!).
Безразмерную величину, определяемую отношением
плотности рассматриваемого вещества к плотности
условного стандартного вещества (воды для твердых и
жидких тел и воздуха для газов в определенных физич.
условиях), следует называть относительной плотностью
(а не удельным весом или относительным удельным
весом). Аналогично неправильно применять термины
«атомный вес», «молекулярный вес», «эквивалент-
«эквивалентный вес», «закон сохранения веса веществ», «молекуляр-
но-весовое распределение», «весовая концентрация»,
«весовое содержание в % » вместо терминов «относитель-
«относительная атомная масса», «относительная молекулярная
масса», «эквивалентная масса», «закон сохранения
массы вещества», «молекулярно-массовое распределен
МЕЖФАЗНАЯ ПОЛИКОНДЕНСАЦИЯ
164
Соотношения
Величина
Сила; вес
Уд. сила; уд. вес
Давление; меха-
нич. напряжение
Кинематич.
вязкость
Динамич.
вязкость
Характеристи-
Характеристическая вязкость
Ударная вяз-
вязкость
Работа
Количество теп-
теплоты
Мощность
Газопроницае-
Газопроницаемость (объем-
(объемная)
Газопроницае-
Газопроницаемость (массовая)
Проницаемость
пористых сред
Линейная плот-
плотность нити
Прочность нити
на разрыв
Уд. теплота
Теплопровод-
Теплопроводность
. Уд. теплоем-
теплоемкость
Коэфф. тепло-
теплообмена (тепло-
(теплоотдачи) и теп-
теплопередачи
Температуро-
Температуропроводность
Электрич. мо-
момент диполя
Экспозиционная
доза фотонного
излучения
Мощность эк-
экспозиционной
дозы, фотонного
излучения
Доза излучения
Эквивалентная
доза излучения
Мощность дозы
излучения
Мощность экви-
эквивалентной дозы
излучения
Активность ну-
нуклида в радио-
радиоактивном источ-
источнике
между нек-рыми единицами, подлежащими
изъятию, и единицами СИ
Единица, подле-
подлежащая изъятию
1 кгс
1 тс
1 ГС
1 кгс/м3
1 гс/см3 1
1 тс/м3 |
1 кгс/см2 = 1 ат
1 кгс/мм2
1 мм вод. ст. =
= 1 кгс/м2
1 мм рт. ст. 1
1 торр /
1 бар
1 мб ар
1 гбар
1 кбар
1 Ст (стоке) =
= 1 СМ2/С
1 сСт (санти-
1 П (пуаз)
1 сП (сантипуаз)
1 дл/г (дециметр
на грамм)
1 кгс -см/см2
1 кге-м •
1 л-атм
1 кал
1 ккал
1 Мкал
• 1 Гкал
1 эВ
1 л. с.
1 ккал/ч
1 кал/с
1 см2/(с • кгс/см2)
1 г/(с- см- кгс/см2)
1 Д (дарси)
1 денье
1 гс/текс 1
1 ркм (раз- 1
рывной ки- |
лометр) J
1 ккал/кг =
= 1 кал/г
1 кал/(с-см-°С)
1 ккал/(ч-м-°С)
1 ккал/(кг-°С)=
= 1 кал/(г-°С)
1 ккал/(ч-м2-°С)
1 кал/(с-см2-°С)
1 М2/Ч
1 Д (дебай)
1 Р (рентген)
1 Р/с
1 Р/мин
1 Р/ч
1 рад
1 бэр
1 рад/с
1 рад/ч
1 бэр/с
1 Ки (кюри)
1 резерфорд
Значение в единицах СИ,
кратных и дольных от них
9,806 65Н «ЮН
9,806 65 кН « 10 кН
9,806 65 мН « 10 мН
9,806 65 Н/м3 « 10 Н/м3
9,806 6 5 кН/м3 « 10 кН/м3
98,066 5 кПа ж 100 кПа =
= 0,1 МПа
9,806 65 МПа « 10 МПа
9,806 65 Па « 10 Па
133,322 Па « 133 Па
100 кПа = 0,1 МПа
100 Па = 0,1 кПа
10 МПа
100 МПа
10~4 м*/с
1 мм2/с
0,1 Па-с
1 мПа-с
0 ,1 м3/кг =100 л/кг
980 , 665 Дж/м2 « 1 кДж/м2
9,806 65 Дж « 10 Дж
101 ,328 Дж ж 101 Дж
4, 1868 Дж
4,1868 кДж
4,1868 МДж
4,1868 ГДж
1 ,602 19-Ю-19 Дж
735,499 Вт
1 ,163 Вт
4,186 8 Вт
1,019 72-10-9м2/(с-Па)
1,019 72-10-6кг/(с-м-Па)
1,019 72-10~12м2 ж 1 мкм2
0,111-10-6кг/мж
ж 0 , 111 мг/м
ж 10 к11а-м3/кг
4,186 8 кДж/кг
418,68 Вт/(м-К) =
= 418,68 Вт/(м-°С)
1,163 Вт/(м-К) = 1,163
Вт/(м-°С)
4, 1868 кДж/(кг-К) =
= 4,1868 кДж/(кг-°С)
1 ,163 Вт/(м2-К) =
= 1 ,163 Вт/(м2-°С)
41,868 кВт/(м2-К) =
= 41,868 кВт/(м*.°С)
27 7,78-10"вм2/с
3,335 64-10-30Кл.м
2,58-10~4Кл/кг
2, 58-Ю-4 А/кг
4,30-1 О А/кг
7,17-10-» А/кг
0,01 Дж/кг
0,01 Дж/кг
0,01 Вт/кг
2, 778-Ю-6 Вт/кг
0,01 Вт/кг
3,7-Юю с
106с-х
ние», «массовая концентрация» (или «концентрация по
массе»), «массовое содержание в%»(или «массовая до-
доля в %»).
В Международном стандарте ИСО 31/VII приведены
след. определения: 1) «Относительная атомная масса
элемента есть отношение средней массы атома природ-
природной смеси изотопов элемента к г/12 массы атома нукли-
нуклида 12С». 2) «Относительная молекулярная масса веще-
вещества есть отношение средней массы молекулы природ-
природной смеси изотопов вещества к V12 массы атома нуклида
12С». При этом указывается, что ранее относительную
атомную массу называли атомным весом, а относитель-
относительную мол. массу — мол. весом.
При переводе системных или внесистемных единиц,
отличных от единиц СИ, в единицы СИ (см. таблицу)
должна быть сохранена их точность: число значащих
цифр должно быть таким, чтобы при переводе точность
не была уменьшена или увеличена. С этой целью задан-
заданное значение величины умножают на переводной коэф-
коэффициент без его округления, а затем полученный ре-
результат округляют до требуемого числа значащих цифр.
Л. Р. Стоцкий,
МЕЖФАЗНАЯ ПОЛИКОНДЕНСАЦИЯ, поли-
поликонденсация на границе раздела
фаз (interfacial polycondensation, Interphasen-Poly-
kondensation, polycondensation interfaciale) — поли-
поликонденсация, протекающая на границе раздела двух
несмешивающихся жидкостей или жидкости и газа.
М. п.— гетерогенный необратимый процесс, скорость
к-рого лимитируется скоростью диффузии реагентов.
Наиболее подробно изучена поликонденсация на гра-
границе раздела двух несмешивающихся жидкостей.
Поликонденсация на границе раздела двух несмеши-
несмешивающихся жидкостей. Для проведения М. п. этого типа
исходные, реагенты растворяют раздельно в двух не-
несмешивающихся жидкостях (фазах). Напр., при полу-
получении полиамидов или полиэфиров в качестве одной
из жидкостей, как правило, применяют воду, в к-рой
растворяют соответственно диамин или бисфенол. Дру-
Другой жидкостью служит органич. растворитель, химиче-
химически инертный к исходным реагентам (напр., бензол),
в к-ром растворяют дихлорангидрид дикарбоновой
к-ты. При контакте указанных р-ров на границе разде-
раздела фаз мгновенно образуется полимер. Для более
полного контакта реагирующих соединений фазы обыч-
обычно перемешивают.
При синтезе полиамидов или полиуретанов на гра-
границе раздела фаз образуется тонкая полимерная плен-
пленка, удаление к-рой немедленно приводит к образова-
образованию новой пленки. Так. обр., полимер может непре-
непрерывно удаляться из зоны реакции до полного исчезно-
исчезновения мономеров. В других случаях образующаяся
на границе раздела фаз пленка полимера, как правило,
не обладает достаточной прочностью и поэтому ее нель-
нельзя удалять из зоны реакции.
Для проведения М. п. наиболее удобны мономе-
мономеры с высокой реакционной способ-
способностью (дихлорангидриды дикарбоновых к-т, ди-
диамины и бисфенолы), т. к. время контакта реагентов
в этом случае значительно меньше, чем при проведении
поликонденсации др. методами. Кроме того, высокая
реакционная способность мономеров позволяет осущест-
осуществлять М. п. при низких темп-pax, когда побочные реак-
реакции практически не идут. При М. п. допустимо наличие
в мономерах инертных примесей, к-рые действуют как
разбавители и не вступают во взаимодействие с мономе-
мономерами. Такими примесями являются, напр., в диами-
диамине — вода и минеральные соли нейтрального характера,
в дихлорангидриде дикарбоновой к-ты — органич. раст-
растворители. Присутствие в реакционной среде монофунк-
монофункциональных реакционноспособных соединений влияет
1 на М. п. таким же образом, как и на поликонденсацию,
проводимую др. методами (см. Поликонденсация).
165
МЕЖФАЗНАЯ ПОЛИКОНДЕНСАЦИЯ
166
Последовательность введения мо-
мономеров в реакционную среду оказывает сущест-
существенное влияние на мол. массу и выход образующегося
полимера. Напр., при получении полиамидов значи-
значительная мол. масса достигается только при добавлении
р-ра дихлорангидрида дикарбоновой к-ты к р-ру ди-
диамина. Выход полимера и его мол. масса увеличиваются
также, если интенсивно перемешивать р-р мономера,
к к-рому приливают второй компонент.
Мол. масса и выход полимера зависят также от кон-
концентрации мономеров. Для достижения
максимальных значений мол. массы и выхода полимера
необходимо, чтобы молярные концентрации реагентов
в зоне протекания реакции были одинаковы (соблю-
(соблюдение принципа эквимолярности). Последние опреде-
определяются концентрацией мономеров в несмешивающихся
фазах и их диффузией в слой, прилежащий к поверх-
поверхности раздела.
Различное соотношение объемов фаз (при постоянной
концентрации мономеров в этих фазах) практически
не оказывает влияния на мол. массу образующихся
полимеров. В этом случае при любом избытке одного
из реагентов (объема фазы) в зоне реакции достигается
одно и то же соотношение исходных реагентов, опреде-
определяющееся только скоростью диффузии этих реагентов
в зону реакции. Скорость диффузии зависит от концент-
концентрации реагентов в р-ре и не зависит от объема фазы.
М. п. может осуществляться в водной и органич. фа-
фазах вблизи границы их раздела или на самой этой гра-
границе, т. е. везде, где возможно взаимодействие исходных
реагентов. Установлено, что рост макромолекул поли-
полиамидов начинается вблизи границы раздела фаз со сто-
стороны органич. фазы, а полиэфиров — со стороны вод-
водной фазы.
Влияние характера границы раз-
раздела фаз наМ.п. окончательно не выяснено. Уста-
Установлено лишь, что, как правило, чем больше поверхност-
поверхностное натяжение на границе раздела, тем больше мол.
масса образующегося полимера. Не исключено, что гра-
граница раздела может оказывать ориентирующее влияние
на исходные реагенты и способствовать регулированию
роста цепей макромолекул.
Константы скоростей М. п. близки к констан-
константам скоростей радикальных и ионных реакций, напр,
для наиболее быстрых реакций между дихлорангидри-
дами дикарбоновых к-т и диаминами они составляют
102—Ю6 л/(моль-сек).
М. п. протекает в диффузионной области, при этом
скорость процесса в основном определяется скоростью
обновления поверхности раздела фаз. Обычно М. п,
проводят при комнатной температуре. Повыше-
Повышение темп-ры реакции, как правило, приводит к умень-
уменьшению выхода и мол. массы образующегося полимера.
Однако при получении ароматич. полиамидов отмеча-
отмечается нек-рое увеличение мол. массы полимера при по-
повышении темп-ры.
Тип органич. растворителя определяет
распределение реагентов между фазами, скорость диф-
диффузии реагентов, скорость основной и побочных реак-
реакций, поверхностное натяжение на границе раздела
фаз, характер взаимодействия полимеров с раствори-
растворителем и др, В большинстве случаев для получения
полимеров с наибольшей мол. массой и наибольшим
выходом растворитель подбирается опытным путем.
Существует точка зрения, согласно к-рой наилучшим
растворителем является соединение, к-рое вызывает
нек-рое набухание образующегося полимера. В зави-
зависимости от характера взаимодействия растворителя
с образующимся полимером последний может нахо-
находиться в реакционной системе в виде ненабухшего или
частично набухшего осадка или в р-ре.
В качестве «второго» растворителя обычно исполь-
используют воду, однако известны и системы, состоящие из
двух несмешивающихся органич. • жидкостей [напр.,
тетрахлорэтилен — этиленгликоль, ксилол (или то-
толуол) — этиленгликоль]. Такие системы не обладают
какими-либо преимуществами по сравнению с водными
системами; их взаимная смешиваемость, как правило,
выше, чем у водных, и, кроме того, в этих системах
образуются полимеры с меньшим выходом и сравни-
сравнительно невысокой мол. массой.
Использование при М. п. эмульгаторов спо-
способствует увеличению поверхности раздела фаз, что
приводит к повышению выхода полимера и, в нек-рых
случаях, к увеличению мол. массы. Наиболее распро-
распространенный эмульгатор при М. п.— натриевая соль
сульфолауриновой к-ты.
Одно из условий успешного проведения М. п.— на-
наличие в реакционной среде акцепторов веществ (обычно
хлористый водород), выделяющихся в ходе реакции. Наи-
Наилучшие акцепторы хлористого водорода — сильные
основания (напр., NaOH, КОН); при этом молярное со-
соотношение акцептор : дихлорангидрид составляет 2:1.
Возможно применение и слабых оснований, напр.
Na2CO3, но в этом случае наибольшие мол. масса и
выход полимера достигаются при молярном соотноше-
соотношении карбонат натрия : исходный реагент, равном 4:1.
Бикарбонат натрия—слишком слабое основание для
использования его в качестве акцептора НС1. Напр.,
если получать полиамид в присутствии бикарбоната
натрия, диамин сам служит акцептором, что приводит
к образованию полимеров с более низким выходом
и меньшей мол. массой. Использование в качестве ак-
акцептора органич. оснований (напр., пиридина и др.
третичных аминов) приводит к образованию полимера
со значительно более низкими мол. массой и выходом,
чем при применении неорганич. оснований.
Синтез полимеров методом М. п. обычно сопровож-
сопровождается побочными реакциями, из к-рых
наиболее важны образование циклов и гидролиз исход-
исходных реагентов (см. Обменные реакции). Гидролизу мо-
могут подвергаться дихлорангидриды дикарбоновых к-т,
бмс-хлорформиаты, диизоцианаты и др. Наиболее под-
подробно изучен гидролиз дихлорангидридов. Энергия ак-
активации этой реакции в системе вода—органич. раство-
растворитель для алифатич. и ароматич. дихлорангидридов
составляет ок. 42—54 кдж/молъ A0—13 ккал/молъ).
Скорость гидролиза дихлорангидридов зависит от при-
природы органич. фазы, темп-ры реакции, количества осно-
основания в водной фазе, скорости диффузии дихлорангид-
дихлорангидридов из органич. фазы в водную. С увеличением числа
метиленовых групп в алифатич. дихлорангидридах
склонность последних к гидролизу в гетерофазной си-
системе уменьшается. Ароматич. дихлорангидриды более
стойки к гидролизу, чем алифатические.
М. п. на границе раздела двух несмешивающихся
жидкостей — удобный и быстрый препаративный метод
получения полимеров (в основном полиэфиров и поли-
полиамидов). Применение М. п. в пром-сти ограничено не-
необходимостью использовать дорогостоящие мономеры
с высокой реакционной способностью (напр., дихлор-
дихлорангидриды дикарбоновых к-т), большими объемами фаз
и затратами на регенерацию органич. растворителя.
Этот метод поликонденсации целесообразно использо-
использовать для получения продуктов, синтез к-рых другими
методами затруднен, напр, полимеров из термически
нестойких мономеров, высокоплавких полимеров, высо-
высокодисперсных полимерных порошков.
Поликонденсация на границе раздела жидкость —
газ. Для проведения этого варианта М. п. один из моно-
мономеров (диамин, бисфенол, дитиол) растворяют в воде,
а другой (недокись углерода, дихлорангидриды дикар-
дикарбоновых к-т) в смеси с газом-носителем пропускают
через этот р-р. В качестве газа-носителя применяют
азот, воздух, пары органич. растворителей и др. Вспе-
Вспенивание р-ра приводит к увеличению длительности кон-
167
МЕЛАМИНО-ФОРМАЛЬДЕГИДНЫЕ ЛАКИ И ЭМАЛИ
168
такта компонентов. Реагент газовой фазы полностью
вступает в реакцию за время контакта. Высокомолеку-
Высокомолекулярный продукт удается получить, если скорость ре-
реакции значительно превышает 102 л/(моль - сек).
Значение мол. массы и выход образующегося поли-
полимера подчиняются тем же закономерностям, что и в слу-
случае М. п. на границе раздела двух несмешивающихся
жидкостей. Исключение составляет влияние темп-ры:
наибольшие значения мол. массы и выхода полимера
наблюдаются при повышенной темп-ре. В случае при-
применения легкокипящих, нестойких к гидролизу реа-
реагентов (оксалилхлорида, фосгена и др.) выход полиме-
полимера не превышает 60% от теоретического. Если исполь-
используются высококипящие, стойкие к гидролизу дихлор-
ангидриды ароматич. к-т, выход высокомолекулярного
полимера близок к теоретическому.
Методом М. п. на границе раздела жидкость — газ
получают полиамиды, полимочевины, политиоэфиры
и др. классы полимеров. Применяя несколько однотип-
однотипных реагентов в жидкой или газовой фазах, легко полу-
получить сополимеры.
Непрерывный процесс с использованием пенного ре-
режима чрезвычайно производителен, легко управляем,
допускает значительные изменения условий синтеза при
стабильном качестве продукта, легко автоматизируется
и весьма перспективен для промышленного использо-
использования.
М. п. была открыта в 1898, когда немецкий ученый
Айнхорн описал способ получения полимеров при пере-
перемешивания 20%-ного толуольного р-ра фосгена с водно-
щелочным р-ром бифенола (гидрохинона, резорцина,
пирокатехина). Однако работа Айнхорна была забыта
и только с 1956—58 вновь привлекла внимание ис-
исследователей.
Лит.: Коршак В. В., Виноградова С. В., Не-
Неравновесная поликонденсация, М., 1972; Соколов Л. Б.,
Поликонденсационный метод синтеза полимеров, М., 1966;
Морган П. У., Поликонденсационные процессы синтеза
полимеров, пер. с англ., Л., 1970; Фрунзе Т. М., К у-
р а ш е в В. В., К о з л о в Л. В., Усп. химии, 30, в. 5, 593
A961). В. В. Курашев, В. 3. Никонов.
МЕЛАМИНО-ФОРМАЛЬДЕГИДНЫЕ ЛАКИ И
ЭМАЛИ, меламино-алкидные лаки и
эмали (melamine-formaldehyde varnishes and ena-
enamels, Melamin-Formaldehydlacke und Emaillen, vernis
of emaux melamine-formaldehyde) — лакокрасочные ма-
материалы на основе смесей частично бутанолизированной
меламино-формалъдегидной смолы (ее содержание в сме-
смеси пленкообразующих составляет обычно до 35%)
с тощей алкидной смолой. Отверждение этих материа-
материалов осуществляется в результате взаимодействия функ-
функциональных групп меламино-формальдегидной и алкид-
алкидной смол; последняя служит также пластификатором
лакокрасочных пленок. Обычно М.-ф. л. и э. отвержда-
ют при 100—140 °С в течение 20—60 мин после предва-
предварительной выдержки на воздухе в течение 10—20 мин.
В нек-рых случаях, напр, при исправлении дефектов
покрытий, применяют М.-ф. л. и э., высыхающие при
85 °С в течение 30 мин. Такие материалы содержат кис-
кислотный катализатор отверждения — дибутил- или бу-
бутил фосфорную к-ту (в виде 50%-ного р-ра в ксилоле).
Для улучшения «розлива» в состав М.-ф. л. и э. иног-
иногда вводят кремнийорганич. жидкость. Покрытия из
М.-ф. л. и э. превосходят покрытия из мочевино-фор-
мальдегидных лаков и эмалей по антикоррозионным
и декоративным свойствам, а также по стойкости к на-
нагреванию. См. также Алкидные лаки и эмали.
М. М. Гольдберг.
МЕЛАМИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ (mel-
(melamine-formaldehyde resins, Melamin-Formaldehyd-
Harze, resines melamine-formaldehyde) — олигомерчые
продукты поликонденсации меламина с формальдеги-
формальдегидом, способные превращаться в пространственные (сши-
(сшитые) полимеры.
С NHCH2OH
Получение. Представления о механизме образования
М.-ф. с. во многом сходны с теми, к-рые развиты для
мочевино-формалъдегидных смол. Особенность состоит
в том, что все шесть атомов водорода в молекуле мела-
меламина при взаимодействии с формальдегидом могут
замещаться метилольными группами. Характер обра-
образующихся начальных продуктов реакции в значитель-
значительной мере зависит от соотношения исходных компонен-
компонентов и темп-ры реакции. Присоединение первых трех
молекул формальдегида (моно- и диметилолмеламины
выделить не удалось) с ^
образованием тримети- HQC НК
лолмеламина (см. ф-лу)- HOCH2HN С
практически необрати- ^ N
мый экзотермический ^с^
процесс, протекающий с i
большой скоростью, не NHCH ОН
зависящей от рН среды. 2
Для мольного соотношения меламин/формальдегид,
равного г/3, константа равновесия &равн — 0,04
(80 °С). Последующее присоединение формальдегида
к триметилолмеламину, приводящее к образованию
пента- и гексаметилолмеламинов,— обратимый эндо-
термич. процесс. Для соотношения меламин/формаль-
меламин/формальдегид, равного 1/бУ кравн составляет 3,78 при 60 °С
и 1,46 при 80 °С. Т. обр., повышение темп-ры спо-
способствует образованию пента- и гексаметилолмелами-
гексаметилолмеламинов. Избыток формальдегида также приводит к сдви-
сдвигу равновесия реакции в сторону образования указан-
указанных соединений. Поэтому их получают обычно ири
мольном соотношении меламин/формальдегид соответ-
соответственно Vs и Vi2- Темп-pa существенно влияет не только
на скорость присоединения формальдегида к мел амину,
но и на скорость растворения последнего в используе-
используемом для реакции водном р-ре формальдегида. Выше
60 °С меламин растворяется быстро, и реакция практи-
практически протекает в гомогенной среде.
С очень высокой скоростью смолы образуются в кис-
кислой среде, причем процесс сопровождается гелеобразо-
ванием. В нейтральных и основных средах получается
гл. обр. смесь метилолмеламинов, осаждающихся пос-
после охлаждения реакционной смеси в виде рыхлых масс.
Поэтому синтез М.-ф. с. проводят сначала в слабооснбв-
ных средах (рН 8) для достижения высокой степени пре-
превращения меламина и высокого выхода метилолмелами-
нов, а затем вводят слабокислый катализатор и, во
избежание гелеобразования, быстро завершают процесс.
В молекулах М.-ф. с. содержатся как метиленовые,
так и диметилен-эфирные связи. Число простых эфир-
эфирных связей в процессе отверждения при темп-pax выше
130 °С начинает возрастать, однако при 150—180 °С
эти связи превращаются в метиленовые по схеме:
-сн2осн2- -* - сн2- + сн2о
В отличие от процесса синтеза мочевино-формальде-
гидных смол, содержание метанола в водных р-рах
формальдегида, используемых для гюликонденсации
с меламином, не оказывает существенного влияния на
свойства М.-ф. с. По-видимому, метилирование М.-ф. с.
практически не сопровождается увеличением их гидро-
фильности, т. к. плотность трехмерной сетки этих смол
значительно выше, чем у мочевино-формальдегидных.
Мольное соотношение меламин/формальдегид зависит
от назначения М.-ф. с. и для смол, используемых
в произ-ве пластмасс и лаков, составляет соответствен-
соответственно 1/2—х/з п Ve—Vi2- Т. обр., при произ-ве пластмасс ис-
исходят в основном из триметилолмеламинов, в то время
как при произ-ве лаков — из пента- и гексаметилолмел-
гексаметилолмеламинов, т. к. лаковыми смолами служат продукты
частичной этерификации метилолмеламинов спиртами
(при этом часть метилольных групп связывается спир-
спиртом и, следовательно, не участвует в образовании оли-
гомера). Немодифицированные М.-ф. с. для лаков не
169
МЕЛАМИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
170
производят ввиду их высокой хрупкости и несовме-
несовместимости с обычными лаковыми компонентами; о полу-
получении лаковых М.-ф. с. см. раздел «Модификация».
При получении М.-ф. с. для произ-ва пластмасс син-
синтез метил ол мел аминов обычно осуществляют при 80—
90 °С и рН 8,0—8,5. Завершению образования мети-
метило л ьных производных соответствует значение водного
числа 2—10 (количество воды в мл, пошедшее на титро-
титрование реакционного р-ра до появления помутнения,
обусловленного выпадением метилолмеламинов из разб.
водных р-ров). После этого р-р охлаждают до 50—60 °С
и вводят кислотный катализатор, напр, моноуреид
фталевой к-ты, служащий одновременно и катализато-
катализатором отверждения; р-р смолы перемешивают и охлаж-
охлаждают до комнатной темп-ры. М.-ф. с. получают в виде
водных р-ров концентрации 50—55% (по массе). Содер-
Содержание свободного формальдегида в М.-ф. с. 1,0—1,5%,
вязкость р-ра по вискозиметру ВЗ-4 90—180 сек. Вод-
Водные р-ры М.-ф. с. нестабильны, их можно хранить в те-
течение лишь 4—6 мес. Для увеличения продолжитель-
продолжительности хранения М.-ф. с. обезвоживают в вакууме. Су-
Сухие порошки хранят в плотно закрытой таре.
Свойства. М.-ф. с.— аморфные продукты белого
цвета, хорошо растворимые в воде и не растворимые
в органич. растворителях. Отверждение М.-ф. с. проис-
происходит не только в кислой, но и в нейтральной и слабо-
основной средах. Скорость и глубина отверждения
возрастают в присутствии кислотных катализаторов
и при нагревании. Достаточно высокая водостойкость
конечных продуктов, зависящая от степени отвержде-
отверждения, достигается уже после нагревания М.-ф. с. при
50—60 °С в отсутствие отвердителя или после смеше-
смешения с катализатором, напр, солями аммония, при ком-
комнатной темп-ре. Существенный недостаток М.-ф. с.—
выделение формальдегида в процессе переработки и
эксплуатации, а также относительно высокая стоимость
из-за относительной дороговизны мел амина.
Продукты отверждения М.-ф. с— бесцветные, про-
прозрачные, светостойкие, легко окрашивающиеся поли-
полимеры, характеризующиеся хорошей дугостойкостью,
теплостойкостью и относительно высокой водостой-
водостойкостью (изделия выдерживают кипячение в воде).
Свойства отвержденных меламино-формальдегидных
смол (без наполнителя) приведены ниже:
Плотность, г/см3
Модуль упругости, Мн/м2 [кгс/см2]
Теплостойкость по Мартенсу, °С .
Твердость по Роквеллу, шкала М .
Прочность, Мн/м2 (кгс/см2)
при растяжении
при сжатии
при изгибе
Относительное удлинение, %
Ударная вязкость, кдж/м2, или кгс • см/см2
Диэлектрич. проницаемость при 1 Мгц . .
Тангенс угла диэлектрич. потерь при
1 Мгц
Дугостойкость, сек
Водопоглощение за 24 -ч, %
1 ,45—1,56
E0-55)-10а
[E0-55)-103]
160—240
130-140
40-56
D00 — 560)
150—200
A500-2000)
55-98
E50-980)
0,5
2,5—6,0
4,8
0,09
100-145
0,3-0,5
О свойствах отвержденных смол см. также Аминопла-
сты, Карбамидные клеи.
Модификация. Для повышения диэлектрич. свойств,
придания способности растворяться в органич. раство-
растворителях, улучшения совместимости с компонентами,
входящими в состав лаков и эмалей, улучшения гигие-
нич. свойств и снижения стоимости М.-ф. с. обычно под-
подвергают модификации.
Получение модифицированных М.-ф. с. может быть
осуществлено в одну или две стадии. В первом случае
часть меламина заменяют какой-либо модифицирующей
добавкой, напр, мочевиной. Соотношение компонентов
можно варьировать в широких пределах, однако наи-
наиболее часто мольное соотношение меламин /мочевина/
формальдегид составляет 1/1/5. При сополиконденсации
меламина и бензгуанамина B,4-диамино-6-фенил-1,3,5-
триазина) с формальдегидом на 1 моль меламина берут
обычно 0,2—0,3 моль бензгуанамина. Технология син-
синтеза таких смол не отличается от принятой для М.-ф. с.
При получении модифицированных М.-ф. с. в две
стадии на первой синтезируют метилолмеламины или
олигомеры, на второй в их водные р-ры вводят модифи-
модификатор, напр, ди- или триэтаноламин, гс-толуолсульф-
амид, фурфурол; для частичной этерификации по ме-
метил ольным группам используют гл. обр. одноатомные
спирты (метиловый или бутиловый).
Процесс получения, напр., этерифицированных бути-
бутиловым спиртом М.-ф. с. осуществляют след. образом.
К предварительно нейтрализованному щелочью форма-
формалину добавляют меламин и смесь нагревают при 80 °С
в течение времени, необходимого для того, чтобы про-
прореагировало 60% загруженного формальдегида. За-
Затем, не прекращая перемешивания, вводят спирт
с растворенным в нем фталевым ангидридом (катали-
(катализатор). Реакцию при 85—90 °С продолжают до рас-
расслаивания смеси на водный и олигомерный слои. Пос-
Последний отделяют, промывают теплой водой, затем воду
и частично спирт отгоняют при остаточном давлении
40—80 мм рт. ст. A мм рт. ст.= 133,322 н/м2) до
получения р-ра нужной вязкости. В р-р вводят пласти-
пластификатор (напр., касторовое масло) или р-р полиэфир-
полиэфирной смолы (напр., глифталевой смолы в толуоле). Мо-
Молярное соотношение меламин/формальдегид/бутанол
обычно составляет 1/8/8.
Применение. Водные р-ры М.-ф.с. A2%-ные) исполь-
используют для пропитки бумаги и картона с целью придания
им высокой прочности во влажном состоянии. В каче-
качестве отвердителей при этом используют разб. сильные
к-ты или соли, напр, квасцы. Бумага, содержащая
4—8% смолы (в расчете на сухую бумажную массу),
обладает в мокром состоянии почти такой же проч-
прочностью, как и в сухом, а сопротивление излому воз-
возрастает даже в несколько раз. Прочность в сухом со-
состоянии увеличивается на 10—15%.
Метиловые эфиры метил олмеламинов используют для
противоусадочнои отделки тканей и придания им несми-
наемости. Усадка шерсти резко снижается при содер-
содержании смолы на ткани 3—15%. Отверждение осущест-
осуществляют после нанесения олигомера на ткань (катализа-
(катализатором служит диаммонийфосфат). Для придания ткани
водонепроницаемости используют метилолмеламины,
частично этерифицированные высшими спиртами, напр,
стеариновым или лауриловым. Модифицированные
спиртами М.-ф. с. широко используют в смеси, напр,
с алкидными смолами при приготовлении лаков и эмалей
(см. Алкидные лаки и эмали, Меламино-формалъдегид-
ные лаки и эмали).
М.-ф. с, модифицированные диэтаноламином, три-
этаноламином или гс-толуолсульфамидом, используют
при изготовлении дугостойких материалов с улучшен-
улучшенными электроизоляционными свойствами. Модифици-
Модифицирующие добавки служат также пластификаторами,
улучшающими литьевые свойства смолы. Аналогичные
свойства придает смоле фурфурол, способствующий,
кроме того, повышению теплостойкости материала
(см. Аминопласты). Для получения декоративного
слоистого пластика, клеев, древесно-стружечных и дре-
весно-волокнистых плит в качестве связующего ши-
широко используют меламино-мочевино-формальдегидные
смолы (см. Карбамидные клеи). Чем выше содержа-
содержание меламина, тем более в готовом полимере прояв-
проявляются свойства, присущие М.-ф. с, но тем выше его
стоимость.
М.-ф. с, модифицированная бензогуанамином (см.
Гуанамино-формалъдегидные смоли), обладает улучшен-
улучшенными санитарно-гигиенич. свойствами: при эксплуа-
171
МЕМБРАНЫ ИОНИТОВЫЕ
172
тации значительно меньше выделяется формальдегида
@>1—0,5 мг/л против 5 мг/л для аминонласта), изделия
не загрязняются несмываемыми пятнами от пищевых
продуктов (чая, кофе), характеризуются повышенной
поверхностной твердостью, блеском, легко моются.
Поэтому такие смолы рекомендованы для изготовления
посуды. Кроме того, эти смолы отличает хорошая теку-
текучесть, возможность широко варьировать скорости от-
отверждения, а отвержденные продукты — устойчи-
устойчивость к деструкции (термоокислительной, гидролити-
гидролитической).
Лит.: Николаев А. Ф., Синтетические полимеры
и пластические массы на их основе, 2 изд., М.— Л., 1966; Л а-
зарев А. И., Сорокин М. Ф., Синтетические смолы
для лаков, М.— Л., 1953; X у в и н к Р., Ставерман А.
[сост.], Химия и технология полимеров, пер. с нем., т. 2, М.—
Л., 1966; Голдинг Б., Химия и технология полимерных
материалов, пер. с англ., М., 1963; Bachmann A.,
В е г t z Т., Aminoplaste, 2 Aufl., Lpz., 1970; Технология пла-
пластических масс, под ред. В. В. Коршака, М., 1972.
Г. М. Цейтлин.
МЕМБРАНЫ ИОНИТОВЫЕ (ionite membranes,
Ionenaustauschermembranen, membranes echangeuses
d'ions) — пленки или пластины из ионитов или из ком-
композиций, включающих иониты.
Классификация. М. и. обычно классифицируют по
их структуре на две основные группы: гомогенные и
гетерогенные.
Гомогенные М. и. состоят только из. ионооб-
ионообменного компонента. При сильном увеличении (в 500—
1000 раз) в структуре гомогенных М. и. можно заме-
заметить неоднородности, однако, как правило, размеры
этих неоднородностей сравнимы с длинами волн ви-
видимой части света. Поэтому такие М. и. обычно про-
прозрачны.
Гетерогенные М. и. содержат два и более
компонентов. В таких мембранах частицы ионита раз-
размером 1—50 мкм распределены в инертном (т. е. не
обладающем ионообменными свойствами) термопластич-
термопластичном полимере, придающем М. и. эластичность и меха-
нич. прочность. Гетерогенные М. и. могут быть разде-
разделены физич. способами (напр., экстракцией) на два
компонента.
Близки по свойствам и строению гомогенным М. и.
интернолимерные и т. наз. привитые мембраны. В и н-
тер полимерные М. и., получаемых совмеще-
совмещением в р-ре двух полимеров, хнмич. связи между
макромолекулами обычно не образуются. П р и в и-
т ы е М. и. получают прививкой на инертную поли-
полимерную основу мономера с ионогенными группами или
группами, способными после соответствующих химич.
превращений придавать пленке ноногенные свойства.
М. и. подразделяют также по типу ио но генных групп
или способам их введения. По аналогии с ионитами
М. и. классифицируют: 1) по знаку заряда ионогенных
групп — на катноннтовые (катионообменные), аниони-
товые (анионообменные), амфотерные и биполярные;
последние представляют собой двухслойные пленки, на
противоположных сторонах к-рых находятся ионоген-
ные группы противоположной полярности; 2) по сте-
степени диссоциации ионогенных групп — на слабо-, средне-
и сильноосновные (кислотные). Кроме того, М. и.
группируют по специфич. свойствам, напр, по способ-
способности к комплексообразованию, электронообменным
свойствам ц др. Известны т. наз. «жидкие» М. и., пред-
представляющие собой пористые пластины, пропитанные
р-рами органич, веществ, содержащих в молекуле ионо-
генные группы, в несмешивающихся с водой жидкостях
(бензоле, ксилоле, нитробензоле и др.).
Получение. Гомогенные М. и. получают поли-
полимеризацией или поликонденсацией мономеров, содер-
содержащих ионогенные группы или группы, легко превра-
превращаемые в ионогенные (эфирные, амидные, хлорангид-
ридные и др.), Полимеризацию проводят в тонком слое
на антиадгездонном материале или между двумя стек-
стеклами. Лучшие М. и. этого типа с высокой механич. проч-
прочностью и малым электрич. сопротивлением получены
сополимеризацией стиролсульфокислоты и ее произ-
производных (в основном эфиров) с дивинильными соедине-
соединениями; инициаторы процесса — перекисные соедине-
соединения, УФ-свет или быстрые электроны. В качестве исход-
исходного сырья используют также акриловые, винилфосфо-
новую, фенилвинилфосфеновую к-ты, винилпиридины
и др. Сшивающими агентами служат дивинил бензол,
диметакрилаты гликолей, дивинилпиридиньь
Анионитовые гомогенные М. и. получают поликонден-
поликонденсацией меламина и гуанидина с формальдегидом. Для
получения катионитовых гомогенных М. и. вместо
формальдегида используют сульфофенолы или их
соли, а также производные бисфенолов. Известны М. и.
на основе продуктов поликонденсации фенола, формаль-
формальдегида и стирола, сульфированных по двойной связи.
Для повышения прочности гомогенных М. и. их арми-
армируют стекловолокном или синтетич. хемо- и термостой-
термостойкими волокнами (саран, виньон и др.) и тканями на их
основе.
М. и., получаемые активацией инертных пленок
(в данном случае инертными считают пленки, к-рые
не обладают ионообменными свойствами), можно услов-
условно разделить на две группы. К М. и. первой группы
относят пленки на основе сшитых сополимеров стирола,
активированные сульфированием, фосфорилированием,
хлорметилированием с последующим аминированием.
В синтезе М. и. этого типа используют также бутаднен-
стирольные латексы, к-рые наносят на ткани и после
этого сульфируют. При введении в пленку ионогенных
групп в ней возникают внутренние напряжения, что
обусловливает ее низкую механич. прочность. Для
устранения этого недостатка при получении пленок
используют пластифицированные полимеры. На по-
последующих стадиях в процессе химич. обработки
и промывки пластификатор удаляют. Для той же цели
иногда на стадии сополимеризации стирола и сшиваю-
сшивающего агента к стиролу добавляют форполимер стирола,
что позволяет снизить тепловой эффект сополимериза-
сополимеризации в массе.
Активация инертных термопластичных пленок (по-
лиолефиновых, фторопластовых, полившшлспиртовых)
возможна методом прививки к ним различных соедине-
соединений, в основном винилароматических, с ноногенными
группами или без них; в последнем случае после при-
прививки осуществляют химич. превращения с целью об-
образования ионогенных групп. Прививку инициируют
с помощью перекисных и гидроперекисных соединений,
облучением частицами больших энергий, УФ-светом
или механич. воздействием (о способах прививки см.
Привитые сополимеры). Ионогенные группы, вводимые
в боковые цепи полимеров, не лишают исходные пленки
эластичности.
М. и., полученные методом прививки, отличаются
хорошей механич. прочностью и эластичностью, высо-
высокой селективностью (способностью к избирательному
пропусканию ионов противоположного заряда), термич.
и химич. стабильностью, хорошими электрохимич. по-
показателями. Такие М. и. гомогенны, однако для них
характерна неоднородность прививки по объему из-за
различной доступности для прививаемого соединения
поверхностных и глубинных слоев пленки. Плотность
молекулярной упаковки в исходной пленке и степень
ее кристалличности сильно влияют на распределение
ионогенных групп по толщине и площади пленки. Осо-
Особенно это относится к М. и., полученным в присутствии
сшивающих агентов, когда наряду с прививкой идет
процесс образования трехмерного полимера в прост-
пространственных дефектах пленки.
Для получения т. наз. интерполимерных
М. и. используют два полимера, один из к-рых явля-
является полиэлектролитом или способен приобретать свой-
173
МЕМБРАНЫ ИОНИТОВЫЕ
174
ства полиэлектролита в результате последующей обра-
обработки, а другой — инертным пленкообразующим поли-
полимером. Оба компонента растворяют в общем раство-
растворителе и затем формуют пленки из этого р-ра мето-
методом полива. После удаления растворителя макромо-
макромолекулы и ассоциаты макромолекул полиэлектролита
и инертного полимера образуют прочные межмоле-
межмолекулярные контакты, что затрудняет, но не исключает
полностью возможность вымывания полиэлектролита
из М. и.
. Получены, напр., сильнокислотные интерполимер-
интерполимерные М. п. на основе стиролсульфокислоты и поливи-
поливинилового спирта. Однако такие мембраны не нашли ши-
широкого применения вследствие высокой набухаемости.
Интерполимерные М. и. с лучшими свойствами полу-
получены при использовании в качестве инертного пленкооб-
пленкообразующего компонента сополимера акрилонитрила и ви-
нилиденхлорида, а в качестве полиэлектролита — поли-
стиролсульфокислоты, продукта сополимеризации ма-
леинового ангидрида с винилметиловым эфиром, по-
полиакриловой к-ты, поливинилимидазола, превращен-
превращенного в соль четвертичного аммониевого основания с по-
помощью йодистого метила. Для получения р-ров поли-
полимеров используют высокополярные растворители: ди-
метилформамид, диметилсульфоксид, смесь циклогекса-
нона с метанолом, гексаметилтрифосфамид, бутиро-
лактон и др. Уд. поверхностное электрич. сопротивле-
сопротивление интерполимерных М. и. 0,28—1,5 мом-м2 B,8 —15
ом-см2), толщина 25—150 мкм.
Разработаны способы получения М. и. химич. активи-
активированием инертных интерполимерных пленок. Инерт-
Инертные полимеры совмещают различными способами:
растворением в общем растворителе, сплавлением или
вальцеванием в атмосфере азота, набуханием полимер-
полимерной пленки в мономере, напр, в стироле с добавкой ди-
дивинил бензола, и последующей полимеризацией мономе-
мономера внутри набухшей пленки. Химпч. активацию пленок
проводят сульфированием, хлорметилированием и ами-
нированием, или реакцией сшивки-сульфирования
с формальдегидом. По последней реакции получают
М. и. типа «анкалит», активируя инертную интерполи-
интерполимерную пленку с малым содержанием пленкообразую-
пленкообразующего компонента. Уд. объемное электрическое сопро-
сопротивление этих мембран в дистиллированной воде
0,3—0,7 ом-м C0—70 ом-см).
Гетерогенные М. и. обычно получают смеше-
смешением измельченного ионита с полимером-связующим
(полиэтиленом, фторопластом, синтетич. каучуками,
полиакрилонитрилом, полиметилметакрилатом и др.).
Смесь гомогенизируют на вальцах или в смесителях
в среде органич. растворителя. Пленку формуют при
повышенной темп-ре на прессах или каландрах. Воз-
Возможно формование пленки поливом на барабан или на
непрерывную ленту (о методах получения пленок см.
Пленки полимерные) с последующим уплотнением на
прессах. Метод полива технологически более сложен,
однако дает более однородные М. и.
Для улучшения механич. характеристик М. и. в про-
процессе прессования проводят одностороннее или двухсто-
двухстороннее их армирование технич. тканями. Так, гетеро-
гетерогенные катионитовые мембраны отечественной марки
МК-40 получают смешением на вальцах сульфополисти-
рольного катионита КУ-2 и полиэтилена с последующим
двухсторонним армированием листа капроновой тка-
тканью на прессе.
Сильноосновные аниониты подвергаются деструкции
при темп-pax выше 100 °С. Поэтому в случае получения
из таких ионитов мембран сначала вальцуют пленку
из смеси термопластичного связующего и сополимера
стирола с дивинилбензолом, а затем ее хлорметилируют
и аминируют третичными аминами. Таким образом
получают гетерогенные М. и. с малым электрич. сопро-
сопротивлением, причем в этом случае исключается стадия
термич. обработки анионита при прессовании и валь-
вальцевании.
Б и полярные 'М. и. получают поливом компо-
композиции термопластичного связующего с анионитом на
предварительно сформованную катионитовую М. и.
Такие М. и. получают также, прививая на одну сторону
полистиролыюй пленки мономер с катионогенными^
группами, а на другую — с анионогенными группами.
Подбором соответствующих мономеров для сополиме-
сополимеризации можно получить амфотерные М. и., содержащие
кислотные и основные группы (см. Амфотерные ионооб-,
менные смолы). Формование пленки осуществляют обыч-
обычными для гомогенных М. и. способами.
М. и. с комплексообразующими свойствами получают,
на основе мономеров с фосфонатными группами, напр.,
вннилфосфоновой кислоты (см. Комплексообразующие,
ионообменные смолы).
Свойства и методы испытаний. Большинство процес^
сов на М. и. подчиняется основным закономерностям
ионного обмена. Основное различие в работе слоя иони-
ионита и мембраны состоит в селективной нонопроницае-
мости последней. М. и. обладают способностью к пре-
преимущественному пропусканию противоионов, тогда как
через слой ионита проходят и сопутствующие ионы,
(коионы). Биполярные М. и. обладают только протоно-(
селективностью, т. е. способностью к преимуществен-,
ному переносу протонов, и показывают выпрямляющий
эффект на частотах до 100 гц.
Селективность М. и. (т. е. избирательность,
к переносу катиона или аниона через М. и. при наложе-
наложении электрич. поля) м. б. непосредственно измерена
при пропускании электрич. тока через мембрану, разде-
разделяющую два р-ра. Селективность определяется выходом
но току, т. е. отношением количества электричества,
переносимого противбионами, ко всему количеству
электричества, прошедшего через М. и. за счет переноса
противоионов и коионов, имеющих одноименный заряд
с фиксированными ионами. Селективность может выра-,
жаться в %. В реальных М. и. обычно только 85—95%,
проходящего тока переносится мигрирующими противо-
ионами, а 5—15% — за счет диффузии коионов в обрат-,
ном направлении из-за наличия в мембране нек-рого
количества поглощенного свободного электролита (см.
Ионный обмен).
Селективность М. и. существенно зависит от наличия
дефектов и полостей в их структуре. С увеличением,
степени сшивки и уплотнением структуры матрицы
М. и. показывают большую селективность. В порах
свободные ионы не испытывают существенного воздейст-
воздействия со стороны фиксированных ионов, что обусловли-
обусловливает особенно сильное ухудшение селективности по
отношению к малым и слабо гидратированным ионам.
Применение инертного полимера с более плотной
упаковкой молекул (с малым содержанием аморфной
фазы) в качестве связующего для гетерогенных мемб-
мембран также приводит к увеличению селективности. Этот*
же эффект достигается в ряде случаев при механич.
уплотнении материала прессованием.
Весьма сильно снижается селективность с увеличе-
увеличением концентрации внешнего р-ра, симбатно с к-рой'
растет концентрация поглощенного электролита в М. и.
Отрицательно на селективность влияет и HeK-pbmt
перенос р-ра, находящегося внутри мембраны, при пе-
перемещении свободного электролита противоионами в уз-
узких порах мембраны в направлении миграции противо-
противоионов.
Селективность М. и. зависит и от плотности электрич.
тока. При больших плотностях тока происходит умень-
уменьшение концентрации электролита в диффузном слое
(см. Ионный обмен) у одной из поверхностей мембраны,
что приводит к концентрационной поляризации мембра-
мембраны. Вследствие этого, напр., на катионитовой мембране
в диализаторе повышается рН на поверхности, теряю-
175
МЕМБРАНЫ ИОНИТОВЫЕ
176
щей ионы Н+, и уменьшается на поверхности, их при-
принимающей, что сильно снижает выход по току. При
этом селективность может падать до 0,1 для катионито-
вых и до 0,6 для анионитовых М. и. Подобные эффекты
были изучены с помощью хромопотенциометрич. ис-
исследований, к-рые позволяют определить время пере-
перехода ионов через мембрану и, сравнивая их с под-
подвижностью ионов в р-ре, оценить поляризационные
явления.
Основная причина переноса ионов через М. и.— раз-
различная вероятность перехода ионов в прямом и обрат-
обратном направлениях. Движущей силой процесса может
являться разность потенциалов между двумя р-рами
электролитов, разделенных М. и., или сила, приложен-
приложенная извне: гидростатич. давлением в случае гипер-
гиперфильтрации (т. е. фильтрации от ионов при пропуска-
пропускании под давлением р-ра электролита через М. и.),
внешним электрич. полем в случае электродиализа
(см. ниже).
Наряду с селективностью важнейшей технологич.
характеристикой М. и. является электрич. со-
сопротивление. Для гетерогенных мембран с уве-
увеличением степени наполнения этот показатель умень-
уменьшается; при этом селективность несколько увеличивает-
увеличивается. При уменьшении степени сшивки ионита электрич.
сопротивление мембраны резко падает при нек-ром
снижении селективности.
М. и. являются проводниками второго рода, для
к-рых электрич. проводимость носит чисто ионный ха-
характер и, как правило, отсутствует электронная про-
проводимость. Электрич. сопротивление М. и. определяется
так же, как для р-ров обычных электролитов:
r=RS/l; r'^RS
где г — уд. объемное сопротивление, ом-м (ом-см);
г' — уд. поверхностное сопротивление, ом-м2 (ом-см2);
R — сопротивление образца, ом; S — сечение образца,
м2(см2); I — длина проводящего слоя, м (см). Элект-
Электрич. сопротивление М. и. весьма существенно зависит
от регулярности и пространственного расположения
сегментов макромолекул с фиксированными ионами.
Известны способы уменьшения электрич. сопротивления
катионитовых мембран (частично и анионитовых) с по-
помощью предварительной оптимальной ориентации фик-
фиксированных ионов. С этой целью через М. и. в электро-
электродиализной ячейке пропускают электрич. ток плотно-
плотностью 5 ка/м2 @,5 а/см2) в течение 5—10 мин в зависи-
зависимости от материала мембраны. При этом М. и. разогре-
разогревается до темп-ры начала пластич. деформации мате-
материала, и происходит ориентация фиксированных ионов
в направлении электрич. тока, после чего плотность
тока снижают до 3 ка/м2 @,3 а/см2) и охлаждают мемб-
мембрану до нормальной темп-ры. В результате электрич.
уд. объемное сопротивление М. и. снижается с 1,4 до
0,5 мом-м2 (с 14 до 5 ом-см2). Аналогичный эффект
дает освещение окрашенных М. и. видимым светом.
Наряду с ионной проводимостью М. и., обусловлен-
обусловленной переносом противоионов, большое значение имеет
и миграционная ионная проводимость по Гротгусу,
когда заряд проходит по цепи молекул воды, связан-
связанных водородными связями, с миграцией протона.
Для электрич. проводимости реальных мембран м. б.
применима модель Шпиглера, в к-рой фиксируются
три пути тока: через ионит, через свободный электро-
электролит и «смешанный» путь — через ионит и свободный
электролит. Для М. и., показывающих высокую селек-
селективность, второй тип проводимости не характерен. Точ-
Точные измерения электрич. проводимости М. и. обычно
проводят на переменном токе A—2 пгц) при термоста-
тировании с точностью ±@,02—-0,1)°С.
Определение электрич. проводимости в обычной
электрохимич. ячейке является основным технологич.
методом контроля качества М. и. Мембрану помещают
в ячейку между двумя р-рами электролита, в к-рые
опущены контрольные электроды. Электрич. сопротив-
сопротивление М. и. находят по разности сопротивлений ячейки
с мембраной и без нее. Погрешность таких измерений
±D—6)%. Несколько меньшей погрешности удается
достичь измерениями с разбавлением электролита и
экстраполяцией полученных значений электрич. сопро-
сопротивления М. и. к нулевой концентрации электролита.
Однако с понижением концентрации р-ра от 0,01 н. до
0,001 н. погрешность измерения возрастает в 10 раз.
Этот метод дает суммарные значения электрич. проводи-
проводимости (собственно мембраны и р-ра полиэлектролита,
находящегося в мембране). К тому же постоянная ячей-
ячейки различна при наличии и в отсутствие мембраны и
сильно зависит от ее толщины. Рекомендуемое расстоя-
расстояние между электродами должно быть в 2—3 раза боль-
больше, чем толщина мембраны; отклонение от этого значе-
значения сильно увеличивает погрешность измерения.
Более точный метод определения электрич. проводи-
проводимости М. и.— контактный, по к-рому измеряют сопро-
сопротивление набухшей мембраны с помощью контактных
электродов из платинированной платины. Этот метод
позволяет измерять электрич. проводимость М. и. как
в электролитах, так и в дистиллированной воде. По-
Погрешность метода ±A—2)%. Недостатком метода явля-
является зависимость контактного потенциала на границе
мембрана — электрод от свойств мембраны и электрода.
Этот недостаток частично устраняется при использова-
использовании четырехэлектродных измерительных ячеек. В этом
случае измерения проводят с помощью двух подвижных
электродов, подобно тому, как это делается при измере-
измерении электрич. сопротивления в поляризующихся систе-
системах на постоянном токе.
Степень набухания М. и. оценивается как
соотношение объемов мембраны в набухшем и сухом
состоянии или как соотношение линейных размеров
М. и.
Показатель гидродинам и ч. прони-
проницаемости М. и. для растворителя оценивается на
конкретных р-рах в специальной ячейке по количеству
растворителя, проходящего через единицу площади
мембраны в единицу времени при приложении выбран-
выбранного избыточного давления по одну сторону мембраны.
Содержание ко ионов (т. е. ионов, несу-
несущих одноименный заряд с фиксированными ионами
мембраны) в М. и. определяют по количеству ионов,
вымываемых водой из мембраны, находившейся в кон-
контакте с 0,1—0,5 н. р-ром электролита. В качестве коио-
нов используют аналитически легко определяемые ионы
С1, Вг, Ва, Са. В табл. 1 и 2 приведены физико-химич.
показатели ряда отечественных и зарубежных мембран.
Применение. М. и. позволяют проводить химич. про-
процессы с минимальным расходом реактивов, использова-
использованием наиболее удобного и дешевого вида энергии —
электрич. энергии.
Электродиализ с использованием М. и. в качестве
сепараторов, не пропускающих или ограниченно про-
пропускающих ионы, широко применяют для опреснения
соленых вод и для удаления электролитов из коллоид-
коллоидных р-ров и суспензий под действием электрич. тока.
Солесодержание опресненных вод колеблется от 0,3
до 0,5 г/м3.
Известно использование электродиализа с М. и. для
регенерации травильных р-ров и в электрохимич. син-
синтезе при получении щелочей, к-т, замене ионов в солях.
Ряд стран успешно эксплуатирует подобные установки
для получения поваренной соли из морской воды. На-
Наряду с ионообменными процессами можно проводить
и окислительно-восстановительные, напр, электрохи-
электрохимич. синтез фторидов урана из уранилнитрата. В слу-
случае р-ров, содержащих ионы тяжелых металлов или
их комплексы большого объема, перенос металлов
через М. и. незначителен, и электродиализ используют
177
МЕТАКРИЛАМИДА ПОЛИМЕРЫ
Таблица 1. Свойства некоторых зарубежных ионитовых мембран*
178
Марка
Толщина,
мм
Уд. поверхностное элек-
трич. сопротивление,
МОМ'М2(ОМ-СМ2)
Селектив-
Селективность в
0,5—0,6н.
NaCl, %
Статич.
обменная
емкость,
мг • же/г
Степень
набуха-
набухания (по
объему),
%
Прочность при растя-
растяжении, Мн/м2 (кгс/см2)'
Амфион, тип 100
Амфион, тип 310
Нептон СР-61
Нептон АР-111
Ионак МС3142, МС 3235,
МС34ОХ
Ионак МА 3148, МА 3236,
МА3475Х
Асимплекс СК-1, ДК-1 . . . .
Асимплекс СА-1, ДА-1
Селемион СМ-10, CMV-10, CSG
Селемион АМТ-10, AST
Пермаплекс С-20
Пермаплекс А-20
Налфилм 1-4
15
15
15
6
0,15-0,3
0,17
0,
0,
0,2-
0,1-
-0,3
22
9')
0,25
0,25
США
0,7G)
0,9 (9)
0,5-0,8 E-8)
0,7 G)
1,1 A1)
1,0-1,5 A0-15)
1,8-3,5 A8-35)
0,2—0,4 B-4)
0,2—0,4 B-4)
0,6-1,4 F-14)
0,15-0,8 A,5-8)
90-93
90
85—90
65 — 70
65^70
95
90-94
92
95
90-95
90-95
1,1-1
0,6
2,5
1,8
1-1
0,75-1
2,8
1,6-2
,4
2
,0
15-20
12—14
30-35
30-35
15
15
36-38
26-31
Великобритания
0,6
0,6
0,09-0,11
0,8(8)
0,8 (8)
0,23-2,0 B,3-20)
90
90
90-95
2,8
2,1
1,6-1,2
30-40
30-40
6-20
0,310 C,16)**
0,380 C,86)
0,690 G,03)**
0,82 (8,4)
0,82 (8,4)
1,27 A3,0)***
1,30 A3,3)***
0,41-0,93 D,2-9,5)
0,55-0,86 E,6 — 8,8)
0,59—0,65 F-6,7)****
0,59-0,65 F-6,7)****
* В обозначении марок катионообменных М. и. присутствует буква «С», анионообменных М. и. — буква «А». ** М. и. на
основе фторопластов. *** Гетерогенная М. и. на основе поливинилхлорида. **** М. и., усиленные стекловолокном.
для регулирования кислотности среды и изменения ее
ионного состава.
Широко используют М. и. в пищевой пром-сти для
очистки электродиализом соков, сывороток, молока,
сиропов и т. п. Весьма интенсивно применяют М. и.
в медицине, напр, в аппаратах типа «искусственная
почка».
Электрохимич. регенерация ионообменных смол
представляет одну из разновидностей электродиализа,
когда вместо р-ра электролита обессоливанию подвер-
подвергают водную суспензию катионита или анионита. Элект-
Электрохимич. регенерация позволяет получить без затрат
химических реагентов концентрированные растворы
сорбированных ионитами веществ, что весьма важно
для процессов, где необходимо применять специаль-
специальные методы обезвреживания сбросных вод. Метод элек-
электрохимич. регенерации м. б. использован для реге-
регенерации смесей катионит — анионит и для регенера-
регенерации полиамфолитов.
Свойство М. и. при фильтрации водных р-ров под
гидростатич. давлением пропускать воду и задерживать
соли широко используется для получения чистой воды
(способ гиперфильтрации). Создание М. и. с высокими
механич. характеристиками и селективностью и успехи
технологии изготовления трубчатых мембран обеспе-
обеспечили широкое развитие гиперфильтрации. Этот способ
экономичнее др. способов обессоливания с помощью
М. и. и, кроме того, обеспечивает наименьшее содержа-
содержание солей в воде; в отличие от электродиализа его
экономическая эффективность возрастает с повышением
Таблица
Марка
МК-40
МК-100
РМК-100
МА-40
РМА
2. Свойства
Толщина,
мм
0,7-0,8
0,2
0,3
0,7-0,8
0,1
i некоторых отечественных ионитовых
мембран
Уд. объем-
объемное элек-
трич. со-
противле-
противление в 1 н.
NaCl, OM'M
(ОМ'СМ)
2,5 B50)
1,5 A50)
0,2 B0)
2,5B50)
1,04A04)
Селек-
Селективность
в 0,01 —
0,2 н.
NaCl, %
93
90-98
96
85-90
96
Степень
набуха-
набухания (по
линей-
линейным раз-
размерам),
%
20
10-15
20
15
34
Прочность
при рас-
растяжении,
Мн/м2
(кгс/см2)
12A20)
12-15
A20-150)
12 A20)
12 A20)
18 A80)
солесодержания в воде. Гиперфильтрация используется
также для обессоливания и очистки сточных вод.
Топливные элементы с использованием М. и. позво-
позволяют создать весьма малогабаритные, экономичные и
надежные источники энергопитания специальных си-
систем. В этом случае используется явление переноса
через М. и. различных ионов с последующей электро-
электрохимич. реакцией внутри или на поверхности М. и.
с превращением химич. энергии в электрическую.
В аналитич. практике для потенциометрич. титрова-
титрования большое применение имеют электроды на основе
М. и. (мембранные электроды). Их используют для
индикации конечной точки непосредственных потенцио-
потенциометрич. определений ионов серебра, бария, висмута,
алюминия, никеля, фосфатов, галогенов, сульфатов
и др. в присутствии посторонних ионов в широком
диапазоне концентраций. С помощью мембранных элект-
электродов наиболее просто определяют активность ионов
в растворах. Линейность кислотной функции некоторых
типов мембранных электродов в присутствии ряда
поливалентных металлов определяет преимущество
мембранного электрода в ряде случаев перед стеклянным.
Лит.: ЛаскоринБ. Н., Смирнова Н. М., Г а н т-
м а н М. Н., Ионообменные мембраны и их применение, М.,
1961; Савенко О. Д., Ш о ста к Ф. Т., Ласко-
ЛаскоринБ. Н., Усп. хим., 37, в. 11, 2094 A968); Ш п и г-
л е р К. С, в кн.: Ионообменная технология, пер. с англ.,
М., 1959; Block M. and Kitchener J. A., J. Elect-
rochem. Soc, ИЗ, № 9, 947 A966); Деминерализация методом
электродиализа, пер. с англ., М., 1963. Ю. А. Лейкин.
МЕМБРАНЫ РАЗДЕЛИТЕЛЬНЫЕ — см. Раздели-
Разделительные мембраны.
МЕРСЕРИЗАЦИЯ — см. Вискоза.
МЕТАКРИЛАМИДА ПОЛИМЕРЫ (polymethacryl-
amides, Polymethakrylamide, polymethacrylamides).
Метакриламид (и з о б у т е н а м и д, амид м е-
такриловой к-ты) СН2 = G(GH3)CONH2 (M.).
Свойства. М.— бесцветные кристаллы без запаха;
т. пл. 111 °С; теплота полимеризации 56,57 кдж/молъ
A3,5 ккал/молъ). М. хорошо растворим в воде, метило-
метиловом и этиловом спиртах, ацетоне, этилацетате; пло-
плохо — в гептане, бензоле, толуоле.
Химич. свойства М. определяются наличием амидной
группы и двойной связи, сопряженной с карбонилом.
М. гидролизуется до метакриловой к-ты; при дегидрата-
дегидратации образуется метакрилонитрил, при алкоголизе —
эфиры метакриловой к-ты. По двойной связи М. при-
179
МЕТАКРИЛАТОВ ПОЛИМЕРЫ
180
соединяются гидроксилсодержащие соединения, водо-
водород, галогены, аммиак, амины, меркаптаны, бисуль-
бисульфит-ион.
М. устойчив при 10—25 °С (полимеризуется при плав-
плавлении); поэтому его хранят при комнатной темп-ре
в отсутствие ингибиторов. М. сополимеризуется с акри-
лонитрилом, акриловой и метакриловой к-тами и их
эфирами, а также с др. виниловыми мономерами.
Константы сополимеризации: М. (rL — 0,49 ± 0,02)
с метилметакрилатом (г2 = 1,65 + 0,05); М. (гх = 0,3)
с метакриловой к-той (г2 = 2,0).
Получение. М. в лаборатории получают: 1) ам-
монолизом метнлметакрилата водным р-ром аммиа-
аммиака; из водного р-ра продукт экстрагируют органич.
растворителями; 2) омылением метакрилонитрила.
В нром-сти М. получается при синтезе метилмета-
крплата из ацетонцпангидрина:
H2SO4
(CH3JC(OH)CN >
125-140 С
-> CH2=C(CH3)CN -> CH2=C(CH3)-CONH2-H2SO4
Раствор амид-сульфата охлаждают, массу растворяют
в воде, нейтрализуют углекислым кальцием, отфильт-
отфильтровывают CaSO4 п, выпаривая досуха, получают М.
Выход 90—96%. М. очищают перекристаллизацией из
бензола, этилацетата или возгонкой в вакууме.
Чистоту М. определяют титрованием бромит-бромат-
ным методом или количественным гидрированием над
Pd [на СаСО'з]; содержание азота определяют методом
Къельдаля.
Полиметакриламид [ —СН2—С(СНз) — ]ЭТ(П.)—лиией-
I
CONH2
ный полимер, построенный но типу «голова к хвосту».
Свойства. П.— аморфное вещество белого цвета,
без запаха; температура стеклования ~ 200 °С; мол.
масса ~104. Зависимость между характеристич. вяз-
вязкостью (в дл/г) и мол. массой П. выражается ур-нием:
[т|] = 6,8-10~4-Mn °'66 (вода, 25 °С). П. растворим в во-
воде, формамиде; не растворим в спирте, ацетоне, гексане.
П. вступает в типичные реакции алифатнч. амидов.
При щелочном гидролизе амидные группы частично
превращаются в карбоксильные (образуются полиами-
докислоты). При нагревании П. выделяется аммиак
и происходит внутримолекулярная или межмолекуляр-
межмолекулярная имиднзация, приводящая к образованию соответ-
соответственно шестичленных циклов в цепи (т. е. линейно-
линейного кристаллического П.) или к сшиванию. Реакции
ускоряются в присутствии сильных к-т. Благодаря об-
образованию циклов в цепи термостабильность полимера
возрастает. Под действием радиоактивного излучения
П. деструктируется. Группы — CONH2 м. б. частично
превращены в аминогруппы при взаимодействии П.
с NaOBr (реакция Гофмана).
N-Ал кил- и N-арилзамещенные П. общей ф-лы
[ — СНо—С(СНз)—]п размягчаются при высоких
I
CONHR
темп-рах.Напр., темп-ры размягчения N-метил-, N-w-бу-
тил-, -М-тре/п-бутил- и N-циклогексилзамещенных П. со-
составляют соответственно -205 °С, -155 СС, 220—250 °С
и 235 — 250 °С. С увеличением длины алкилыю-
го заместителя температура размягчения понижается.
N-A л кил замещенные П., у к-рых в алкнльной группе
содержится не менее 8 атомов углерода, воскообразны.
Первые члены гомологич. ряда П. (до R = С3Н7) рас-
растворимы в воде; последующие члены растворяются в уг-
углеводородах. Поли-]Ч-арилметакриламиды хрупки; их
темп-ры размягчения повышаются с увеличением объема
и полярности заместителя. Сравнение молекулярно-
массового распределения орто- и дш/?а-изомеров П.
(R — С6Н4СООС2Н5) выявляет резкие различия в пове-
поведении радикальных концов растущих цепей в зависи-
зависимости от положения заместителя в бензольном кольце.
В случае о/??по-изомера обрыв цепи происходит по ме-
механизму рекомбинации, и его среднечисловая мол. мас-
масса на порядок выше, чем у wapa-изомера.
Получение. П. получают радикальной полиме-
полимеризацией М. в водном р-ре (концентрация М.~10%)
в присутствии персульфатов щелочных металлов или
динитрила азоизомасляной к-ты. Полученный водный
р-р П. обычно используют непосредственно; если необ-
необходимо, П. из р-ра выделяют выпариванием при низкой
темп-ре (в вакууме) или осаждением метиловым спиртом
либо ацетоном. При полимеризации М. в массе или в бо-
более концентрированных р-рах, а также при более высо-
высоких темп-pax вследствие протекания имидизации (гл,
обр., по-видимому^ межмолекулярной) образуются П.,
не растворимые в воде. Одновременно происходит час-
частичный гидролиз амидных групп до карбоксильных.
Под влиянием металлнч. натрия или алкоголята нат-
натрия происходит миграционная полимеризация М. с об-
образованием поли-а-метил-р-аланина (типа найлона-3):
CH2=C(CH3)-CONH2 -> [-GH2-CH(CH3)-CO-NH-]n
Применен и е. ]Ч,]Ч'-Алкш1ендиметакриламиды
применяют в качестве сшивающих агентов для получе-
получения макросетчатых ионообменных смол. М. и N-метил-
метакрилампд иногда сополимеризуют с метилметакри-
метилметакрилатом для получения органич. стекла с повышенной
теплостойкостью. П. можно применять в тех же обла--
стях техники, что и полнакрнламид (см. Акриламида
полимеры), т. е. в качестве коагулянта (флокулянта)
в цветной металлургии, химич. и горнодобывающей
пром-сти, для пропитки бумаги, аппретирования тка-
тканей. Широкого применения П. не нашел.
Лит.: Smets G., Makromol. Chem., 34, 190 A959);
ЧетыркинаГ.М., Соколова Т. А., КотонМ.М.,
в сб.: Карбоцепные высокомолекулярные соединения, М.,
1963, с. 213; Schildknecht С. Е., Vinyl and related
polymers, N. Y.— L., 1952; Савицкая М. Н., Холо-
Холодова Ю. Д., Полиакриламид, Киев, 1969; Марек О.,
Томка М., Акриловые полимеры, пер. с чеш., М.— Л.,
1966; Encyclopedia of polymer science and technology, v. 1,
N. Y.— [a. o.], 1964, p. 187. Г. А. Соколова.
МЕТАКРИЛАТОВ ПОЛИМЕРЫ (polymethacrylates,
Polymethakrylate, polymethacrylates).
Метакрилаты (M.) CH2 = (CH3)CCOOR (где R —
алифатич., карбоциклич., гетероциклич. радикал или
его функциональное производное).
Свойства. Низшие гомологи М.— бесцветные ле-
летучие (R до С4) и нелетучие (R — С5 — С12) легкогорю-
легкогорючие жидкости с резким запахом; пары раздражают сли-
слизистые оболочки глаз. Высшие гомологи — твердые
вещества. Все М. хорошо растворимы в органич. раст-
растворителях и практически нерастворимы в воде (за ис-
исключением метнлметакрилата, окси- и ампнопропзвод-
ных М.). Физич. свойства М. приведены в табл. 1 и 2.
М. легко подвергаются омылению (в кислой и щелоч-
щелочной средах) и переэтернфикации под действием кислот-
кислотных катализаторов. В присутствии сильных оснований
(алкоголяты, феноляты и др.) М. присоединяют но двой-
двойной связи галогены, галогеноводороды, спирты, мер-
меркаптаны, фенолы, алифатич. нитросоединения, аммиак,
амины, амиды к-т и др. Метилметакрилат — диенофиль-
ный компонент в диеновом синтезе.
Низшие гомологи М. легко полимеризуются при хра-
хранении, особенно под действием солнечного света; выс-
высшие гомологи М. самопроизвольно не полимеризуются.
Все М. легко сополимеризуются с большинством винило-
виниловых мономеров. Полимеризацию М. ингибируют медь
и ее соли (особенно резинаты), элементарная сера, фено-
фенолы, гидрохинон и пирогаллол, дифениламин и др. в ко-
количестве 0,005 — 0,5% (по массе). Хранят ингибирован-
ные М. в контакте с сухим воздухом при темн-рах не
выше 5 °С. Нижний предел взрывоопасных концентра-
концентраций паров метил- и этилметакрнлатов с воздухом при
25 °С и атмосферном давлении составляет 2,12 и 1,80%
181
МЕТАКРИЛАТОВ ПОЛИМЕРЫ
182
Таблица 1. Нек-рые свойства метакрилатов СН2 = (СН3) GGOOR
R
Метил
Этил ....
н-Пропил . . .
изо-Пропил . .
«-Бутил ....
изо-Бутил . . .
трет-Бутил
н-Гексил . . .
2-Этилгексил
н-Октил ....
и-Децил ....
н-Додецил . . .
н-Тетрадецил
н-Гексадецил
н-Октадецил
Циклогексил
Фенил
Бензил
Аллил . . . . .
Диметиламино-
этил
2-Гидроксиэтил
Глицидил . . .
Этиленгликоль-
диметакрилат*
Изоборнил . . .
Фу рил ....
Темп-ра кипения,
°С (мм рт. ст.)
100,6 —
101,1 G60)
118,2 —
118,9 G60)
141 G60)
64-65 G60)
163 G60)
155 G60)
66 E7)
184 G60)
47@,1)
105 E)
97 @.9)
167 A0)
48 F0)
213 A0)
235 A0)
60 B)
83-84 D)
119-121 A-2)
32 (9,5)
68,5 A0)
87E)
75 A0)
96-98 D)
112-117 B,5)
62-63 C)
Плотность
при 20 °С,
г/см3
0,9440
0,9135
0,9022
0,8847
0,8936
0,8858
0,8775
0,8937 B5СС)
0,8847
0,8804
0,8767
0,8735
0,8740
0,8695
—
0,96 3 B 5 °С)
—
—
0,9335
0,914 B5 °С)
1,077 B5 °С)
1,073B5 °С)
1 ,0468
0,980 B5 СС)
1,4142
1,4147
1 ,4183
1,4334 B56С)
1,4240
1 ,4199
1,4143
1,4300
1 ,4380
1,4373
1,4418
1,4452
1,4480
1 ,4515
1,4399 E0°С)
1,4578
1,5184
1,5095 B5°С)
1,4358
1,4396
1,4505 B5°С)
1,4482 B5°С)
1,4545
1,4748 B5СС)
1,4770 B5°С)
* Название эфира.
(по объему) соответственно. Пары низших М. (метилмет-
акрилат — бутилметакрилат) обладают умеренной
токсичностью и сильным раздражающим действием на
слизистые оболочки (напр., метилметакрилат уже в кон-
концентрации 0,5 — 1,0 мг/л).
Таблица 2. Нек-рые свойства метакрилатов СН2 = С(СН3) COOR
R
Метил . .
Этил . . .
н-Бутил
Темп-ра
плавле-
плавления, СС
-48,2
<-75
<-76
Темп-ра
вспыш-
вспышки, СС
12,8
21 ,1
65,5
2 S3
С А <Ъ °.
1,89
[0,45]
1,89
[0,45]
1,55
[0,37]
CJ ^
СЗ К" Q
С й w
1=5 ? ru
С ft-—
<и сз ^
360,3
[86]
347,77
Г О О
Вязкость,
мн-сек/м2, или
спз
0,588 B2 °С)
0,647 B5 °С)
0,96 C0 °С)
П о л у ч е и и е. В лаборатории М., а также гликоль-
дпметакрплаты получают этерпфпкацией метакриловой
к-ты или переэтерпфпкацией ее низших эфиров (для
получения высших эфиров) соответствующими спир-
спиртами в присутствии сильных кислотных катализато-
катализаторов. Высшие эфиры синтезируют также дегидра-
дегидратацией соответствующих эфиров а-оксиизомасляной
к-ты под действием РС1з, Р2О5 илп РОСЬ в инертном
растворителе при темп-pax до 120 °С.
Гидроксиалкиловые эфиры получают * взаимодейст-
взаимодействием метакриловой к-ты и окиси алкена в присутствии
аминов или четвертичных солей аммония.
В пром-сти М. получают в одну или две стадии из
ацетонциангидрина:
н3с он H^c
\ / H2SO4 \ ROH
С > C-CONH2 • H2SO4 >
/ \ 60-120°С / Н+
Н3С CN Н2С
СНз
-> CH2=C-COOR+NH4HSO4
Перспективен еще не освоенный пром-стью способ
синтеза М., заключающийся в окислении изобутилена
смесью HNO3 и N2O4 до метакриловой кислоты, кото-
которую затем переводят в эфир обработкой соответствую-
соответствующим спиртом.
В М. обычно содержатся примеси спиртов, использо-
использованных для получения эфиров. Их отделяют перегон-
перегонкой с водяным паром или азеотропной ректификацией.
Степень ненасыщенности М. определяют каталитич.
гидрированием, бромированием (напр., бромид-брома -
том, пиридинсульфатдибромидом) или окислением пер-
манганатом калия с последующим окислением йодной
к-той до формальдегида, к-рый после обработки хромо-
троповой к-той определяют спектрофотометрически.
Для определения числа омыления М. гидролиз уют спир-
спиртовым р-ром щелочи и проводят обратное титрование
стандартной к-той. Посторонние компоненты опреде-
определяют химич. методами: воду — по Фишеру, гидрохи-
гидрохинон — иодометрически или колориметрически (при на-
наличии его в следовых количествах), кислотность — ал-
алкал имет.рически.
Полиметакрилаты
Г /СНз 1
-СН2-С( (П.)-,
\соок
полимеры сложных эфи-
тич. или стереорегулярные
ров метакриловой к-ты.
Свойства. П. с м-алкильным радикалом R от Сг
до Сз — жесткие прозрачные пластики, с R от С2
до С14 — клейкие каучукоподобные соединения, с
R > С14 — воскообразные хрупкие непрозрачные
вещества. П, с w-алкильным радикалом-R> C12 кри-
кристаллизуются вследствие упаковки боковых метиле-
новых цепочек в гексагональной решетке. П. циклич.
спиртов — жесткие полимеры, П. ненасыщенных спир-
спиртов — хрупкие стекловидные трехмерные полимеры.
П. отличаются от полиакрилатов, содержащих аналогич-
аналогичные радикалы, более высокими темп-рами стеклования
и твердостью (см. Акрилатов полимеры). Темп-ра стек-
стеклования изотактич. П., как правило, выше, чем атактич.
(табл. 3); это различие уменьшается при увеличении
Таблица 3. Темп-ры стеклования полиметакрилатов
[-CH2-(CHS)C(COOR)-1
R
Метил
Этил
н-Пропил
изо-Пропил
к-Бутил
изо-Бутил
mpem-Бутил ....
н-Гексил
н-Октил
н-Децил
н-Додецил
н-Тетрадецил ....
?^Гексадецил ....
Циклогексил ....
Фенил
Бензил
Глицидил .
Изоборнил
Темп-ра стеклования, °С
атактич. полимер
104 — 105
65
33 — 38
81
19-20
53
107-117
от —5 до —8
от —20 до —25
от —50 до —60
от — 5 5 до — 6 5
от -68 до —70
от —73 до —75
66
105-110
54
46
111
изотактич.
полимер
43-55 A15)*
8-12
27 (85)*
-24
8
92
51
87
110
* Для синдиотактич. полимера.
объема радикала R. Темп-ры плавления П. с радикала-
радикалами и-додецил, w-тетрадецил, w-гексадецил и w-октадецил
составляют соответственно —34, —2, 22 и 40 °С.
Механич. свойства П. определяются природой и раз-
размером радикала R. С увеличением длины w-алифатич.
радикала от Сх до С10 уменьшаются прочность, плот-
плотность и твердость, возрастает эластичность, что свя-
связано с пластифицрфующим действием «-алкильных
групп. П. с разветвленными радикалами R характери-
характеризуются более высокой прочностью, чем П. с «-алифати-
«-алифатическими заместителями (табл. 4).
Электрич. свойства П* приведены в табл. 5.
183
МЕТАКРИЛАТОВ ПОЛИМЕРЫ
184
Таблица 4. Нек-рые свойства полиметакрилатов
[-CH2-(CH3)G(COOR)-]n
Метил . .
Этил . . .
н-Пропил
н-Бутил .
иао-Бутил
i
В* Q
затель
ения п
|i
489
484
484
483
,477
при
И
So
Кем
1 ,187
1,119
1 ,085
1 ,055
1 ,02
250 B5)
НО A1)
70 G)
*
90 (9)
10.
11
1,6
1
63,5 F35)
35 C50)
28 B80)
7 G0)
24 B40)
Ч
о И
о к
4
7
5
230
2
* Образцы слишком эластичны для испытаний.
Таблица 5. Электрические свойства полимета крилатов
[-CH2-(CH3)C(COOR)-]n
R
Метил
Этил
(i-Хлорэтил . . .
н-Пропил ....
изо-Пропил . . .
н-Бутил .....
я-Октил
н-Гексадецил . .
Тангенс угла диэлек-
трич. потерь при 1 кгц
дипольно-
сегменталь-
ные потери
3-10-2
8-Ю-2
7 -10-*
8- Ю-2
8,3- Ю-2
дипольно-
групповые
потери
7-Ю-2
6-Ю-2
3-Ю-2
2,5-10-2
Эффектив-
Эффективный ди-
польный
момент мо-
мономерного
звена мак-
макромолекулы
1,42
1,43
1,52
1,52
1,43
1 ,40
Корре-
ляци-
ляционный
пара-
параметр*
0,64
0,59
0,64
0,65
0,62
0,60
* Характеризует заторможенность вращения звеньев макро-
макромолекулы.
Таблица 6. Константы в ур-нии Марка—Хувинка [т\]=КМа
для полимета крилатов
[-GH2-(CH3)C(COOR)-L
R
Метил
»
»
Этил
н-Бутил
изо-Бутил
н-Гексил
»
н-Додецил
н-Октадецил
Циклогексил
Бензил
Растворитель
Бензол
Ацетон
Хлороформ
Метилэтилкетон
Метилэтилкетон
Ацетон
Метил этил кетон
То же
Бутилацетат
Тетрагидрофуран
Метилэтилкетон
Этилацетат
Бензол
Темп-pa,
°С
30
30
30
23
23
25
23
32,6
23
30
30
21
30
К-105
5,2
5,83
4,39
2,83
1 ,56
0,199
2,12
43
8,64
2,5
7,0
46
1 ,03
а
0,76
0,72
0,80
0,79
0,81
0,94
0,78
0,50
0,64
0,75
0,66
0,50
0,82
П. растворимы в собственных мономерах, в хлориро-
хлорированных и ароматич. углеводородах, сложных эфирах;
устойчивы к действию воды, разб. р-ров к-т и щелочей.
Водопоглощение П., за исключением окси- и аминопро-
изводных, невелико и не зависит от темп-ры в пределах
0—60 °С. П., содержащие w-алкильные радикалы с R
от Сх до С6, растворяются в ацетоне и этилаце-
тате; при дальнейшем увеличении числа атомов
углерода в радикале улучшается растворимость
в менее полярных растворителях и, соответст-
соответственно, снижается бензо- и маслостойкость П.
Зависимость между характеристич. вязко-
вязкостью [ц] и мол. массой выражается ур-нием
Марка — Хувинка, константы к-рого для ря-
ряда П. приведены в табл. 6.
Увеличение числа атомов углерода в боковой
эфирной группе вплоть до С8 в ряду П. «-али-
фатич. спиртов практически не влияет на жест-
кость основной цепи макромолекулы; у П. с чи- Лолиметилметакри-
слом атомов углерода в эфирной группе более атактич. .....
10 наблюдается возрастание жесткости, обу- изотактич
словленное взаимодействием боковых алкиль- акрилатТШШеТ~
ных радикалов, что способствует появлению вы- атактич
сокого ориентационного порядка в расположе- изотактич
нии боковых цепей. Наиболее совершенным акрил?КСИЛМеТ".
ориентационным порядком характеризуются П., Поли-н-октилм'ет-'
боковые заместители к-рых способны к образо- акрилат
ванию жидкокристаллич. структур. Так, поли- Метак"рГила?ДеЦИЛ" .
фенилметакримеловый эфир цетилоксибензойной Полифенилметакри-
к-ты обладает исключительно высокой отрица- ловый эфир цетил-
тельной сегментной анизотропией, сравнимой
с анизотропией кристаллоподобных молекул
(табл. 7).
При повышенных темп-рах (80—100 °С) П. подвер-
подвергаются кислотному или щелочному гидролизу до поли-
метакриловой к-ты (см. Метакриловой кислоты поли-
полимеры). П. низших спиртов легко переэтерифицируются.
П. устойчивы к свету и воздействию кислорода. Термо-
Термодеструкция B00—250 °С) П. алифатич. спиртов сопро-
сопровождается почти количественным выходом мономера,
за исключением П. третичных спиртов; напр., при раз-
разложении поли-трега-бутилметакрилата образуются по-
лиметакриловый ангидрид и изобутилен с выходом
100%. Тенденция к такому разложению наблюдается
и у нек-рых П. вторичных спиртов.
Под действием ионизирующих излучений П., содер-
содержащие «-алифатич. радикалы с числом атомов углерода
менее шести, деструктируются с образованием моно-
мономера; П., содержащие более длинные алкильные груп-
группы, образуют гели, причем тем легче, чем больше длина
радикала. П. с разветвленными и ароматич. кольцами
в боковой эфирной группе гелей не образуют. Кажущие-
Кажущиеся энергии активации разрыва связей при у-облучении
в вакууме П., содержащих метил ьный, этильный,
w-бутильный, трет-бутильпый, фенильный или бен-
бензил ьный радикал, составляют соответственно 56, 75,
226, 43, 228 и 711 эв.
Таблица 7. Данные, характеризующие анизотропию и гибкость
макромолекул полимета крилатов
Полиэфир
оксибензойной к-ты
ft
о; и
М0Н(
енье
ой
%«
7,
6,
8,
7,
19
25
X
I
0
7
6
9
ста
гмен
Л О !§
S р^
«ь2
15,1
16,6
21,7
20,0
43±2
65
-2500
Примечание: о, иа2 — поляризуемость сегмента макромолеку-
макромолекулы вдоль и перпендикулярно основной цени соответственно; а|| и ai — то
П. устойчивы в воде, щелочах, водных р-рах (-r2V/2
неорганич. солей и большинстве разб. к-т, за же для мономерного звена; { hj -невозмущенный размер полимерного
исключением HF. Конц. минеральные к-ты, клубка; (/i св ) — размер полимерного клубка при условии свободного
напр, серная, азотная и хромовая, разрушают П. вращения звеньев в макромолекуле.
185
МЕТАКРИЛОВОЙ КИСЛОТЫ ПОЛИМЕРЫ
186
Получение. Атактич. П. получают радикальной
полимеризацией в массе, эмульсии и суспензии, реже—
в р-ре. Полимеризация в массе — основной производ-
производственный способ получения листовых материалов, осо-
особенно из метил мета к р плата (см. Метилметакрилата
полимеры, Органическое стекло). Для инициирования
полимеризации широко используют перекиси, азосо-
единения, а также УФ- и у-облучение. Анионной по-
полимеризацией в присутствии в основном металлоор-
ганич. катализаторов в неполярных растворителях,
щелочных металлов в жидком аммиаке, комплексов
ароматич. углеводородов с щелочными металлами
или др. получают изотактич. П.; в присутствии метал л о-
органических катализаторов в полярных средах или ка-
каталитической системы А1(С2Н5)з — TiCU в толуольных
р-рах при темп-pax ниже О °С — синдиотактические
полимеры.
Сополимеризацию М. с др. мономерами чаще всего
проводят в суспензии; особенно широкое распростра-
распространение получили сополимеры метил- и бутилметакрила-
та с метакриловой к-той, винилхлоридомт стиролом,
акрилонитрилом, а -метилстиролом и др. В табл. 8
приведены нек-рые кинетич. параметры радикальной
полимеризации и сополимеризации М.
Таблица 8. Нек-рые кинетические параметры*
радикальной полимеризации и сополимеризации метакрилатов
[-СН2- (CHS)C(COOR)-~L
R
Метил
Этил
н-П решил
м-Бутил
¦изо-Бутил
шретп-Бутил
Циклогексил
Фенил
0
0,
0
о,
о,
10
104
07
116
20
0,13-0,
0,12-0,
0,16-0,
0
0
,22
,21
V.
F0)
F0)
C0)
C0)
F0)
18 F0)
15 E0)
21 G0)
F0)
F0)
Теплота поли-
полимеризации,
кдж/молъ
[пкал/молъ]
58,2[13,9]G6,8)
55,7 [13,3]G4,5)
57,8[13,8]B6,9)
60,4[14,4] A40)
59,6 [14,2]G4,5)
57,5 [13,7]G4,5)
60,0 [14,3]B6,9)
57,5 [13,7]G4,5)
60,0 [14,3]G4,5)
54,5 [13,0]B6,9)
53,2 [12,7]B6,9)
51,1 [12,2]G6,8)
51,6[12,31G6,8)
Параметры
Алфрея
и Прайса
Q
0,74
0,56
1,47
0,72
0,77
1,18
0,73
1 ,17
е
0,40
0,17
0,41
-0,23
-0,04
— 0,35
0,41
0,51
Бензил I 0,4F0) I 56,1 [13,4] G6,8I 0,8б| 0,42
* В круглых скобках указана темп-pa (в °С), при к-рой оп-
определены указанные параметры; kp и k0 — константы скорости
соответственно роста и обрыва цепи.
Для нек-рых М. вычислены абсолютные значения кон-
константы скорости роста цепи кр[л/(молъ-сек)], к-рые для
фотоинициированной полимеризации в массе метил-,
w-пропил-, н-бутил- и mp^m-бутилметакрилатов равны
224 D5 °С), 467 C0 °С), 369 C0 °С) и 350 B5 °С) соответ-
соответственно.
Применение. Наибольшее применение нашли по-
полимеры метил-, этил- и бутилметакрилатов и сополиме-
сополимеры их с метакриловой кислотой, а также друг с другом
в производстве безосколочных стекол, используемых в
авиационной, автомобильной пром-сти и др. областях
техники и быта. Полимеры и сополимеры М. широко
применяют в медицине для изготовления протезов
(хирургия, стоматология) и контактных линз для глаз
(на основе гидрофильных П.). Полимеры и-бутил-
и ш?о-бутилметакрилатов и их сополимеры используют
для приготовления клеев (см. Полиакриловые клеи)
и лаков (см. Полиакриловые лаки и эмали), а также как
связующее в произ-ве слоистых пластиков. Эмульсии
полибутилметакрилата используют для аппретирова-
аппретирования в текстильном и кожевенном производствах. М.
высших жирных спиртов используют как сомономеры
для «внутренней» пластификации жестких пластиков,
напр, поливинилиденхлорида. Циклогексилметакрилат,
усадка к-рого при полимеризации в 2 раза меньше, чем
метилметакрилата, применяют в приборостроении для
изготовления линз. На основе полибензилметакрила-
та изготовляют модели сложных конструкций и де-
деталей машин для изучения распределения и матема-
математической оценки внутренних напряжений методом
фотоупругости.
Промышленное производство пластмасс на основе М.
начато в Германии и США в конце 20-х годов нашего
века.
Лит.: Schildknecht G. E., Vinyl and related poly-
polymers, N.Y.—L., 1952; R i d d 1 e E. H., Monomeric acrylic esters,
N. Y., 1954; Марек О., Томка М., Акриловые полимеры,
пер. с чеш., М.— Л., 1966; Николаев А. Ф., Синтетиче-
Синтетические полимеры и пластические массы на их основе, М.— Л.,
1964; Цветков В. Н., Высокомол. соед., НА, № 1, 132
A969); Т а г е р А. А., Физико-химия полимеров, 2 изд.,
М., 1968; Шибаев В. П. [и др.], Высокомол. соед., 12А,
№ 1, 140 A970); П л а т э Н. А., Ш и б а е в В. П., Высоко-
Высокомол. соед., 13А, № 2, 410 A971); Shetter J. A., J. Polymer
Sci. pt. В, I, № 5, 209 A963); Успехи химии и физики поли-
полимеров, М., 1973. В. П. Шибаев.
МЕТАКРИЛОВОЙ КИСЛОТЫ ПОЛИМЕРЫ (poly-
methacrylic acid, Polymethacrylsaure, acide polymeth-
acrylique).
Метакриловая кислота (пропенкарбоновая-2, а-ме-
тилакриловая к-та) СН2= С(СН3) — СООН (М. к.) —
бесцветная жидкость с острым, раздражающим за-
запахом; т. пл. 16 °С; т. кип. 160,5 °С G60 мм рт. ст.),
106,6 °С A00 мм рт. ст.), 60 °С A2 мм рт. ст.); плот-
плотность при 20 °С 1,0153 г/см3; л^ 1,4314; теплота поли-
полимеризации 66,3 кдж/моль A5,8 ккал/моль). М. к. сме-
смешивается в любых соотношениях с горячей водой;
растворима в эфире, спирте и др. органич. растворите-
растворителях. Из водных р-ров ее выделяют высаливанием NaCl
или СаС12; константа диссоциации 4,79 -10~5 B5 °С).
М. к. сильно корродирует металлы; при действии амаль-
амальгамы натрия восстанавливается до изомасляной к-ты;
с HI дает Р-иодизомасляную к-ту; присоединяет бром,
образуя а,Р~дибромизомасляную к-ту; со спиртами об-
образует соответствующие эфиры.
М. к. синтезируют в присутствии ингибиторов поли-
полимеризации (хинонов). В лаборатории ее получают на-
нагреванием а -бромизомасляной к-ты с избытком D : 1)
25% -ного водного р-ра щелочи (выход 75% от теоретич.)
или окислением изопропенилметилкетона щелочным
р-ром гипохлорита натрия (выход 41% от теоретич.).
Чистую, практически безводную М. к. можно получить
гидролизом метил- или бутилметакрилата. В пром-сти
М. к. и ее эфиры получают из ацетона A), а также нагре-
нагреванием со щелочью продукта конденсации хлороформа
с ацетоном B):
-Н2О
(CH3JC=O+HCN -> (CH3JC(OH)CN >
ROH
CH2=C(CH3)-CN тг-r— GH2=G(GH3)-GOOR
H2SO4
KOH KOH
GH3GOGH3+HGG13 > (GH3JG(CG13)OH »
_> GH2=G(GH3)GOOH
A)
B)
Наиболее удобный метод оценки чистоты М. к.— алка-
лиметрич. титрование; наличие двойных связей опре-
определяют бромид-броматным способом в калиевой или
натриевой соли М. к. Для предотвращения полимери-
полимеризации при хранении в М. к. добавляют эффективные
радикальные ингибиторы, к-рые перед использованием
187
МЕТАКРИЛОНИТРИЛА ПОЛИМЕРЫ
188
к-ты удаляют обработкой адсорбентами или перегонкой
в вакууме.
СН3
I
Полиметакриловая кислота [—СН2—С—]п (П.к.)—
I
соон
бесцветный стеклообразный хрупкий и неплавкий по-
полимер.
Свойства. Наибольшая мол. масса (~ 1 • 107) дости-
достигается при полимеризации М. к. в водных р-рах при рН
2—3 (концентрация М. к. 5—20%). Зависимость между
характеристической вязкостью [г|] р-ров П. к. и мол.
массой (М) выражается ур-ниями: [г\] = 6,6-10~4
C0 °С, 0,002 н. водный р-р НС1); [х\] = 24,2- lO-
B6 °С, 0,1 н. р-р LiCl в метаноле). При определении мол.
массы по светорассеянию для неионизованной П. к.
дп/дс = 0,138 ± 0,002[А, = ЪАТнм E470 А), 30 °С,
в 0,002 н. водном р-ре НС1]; значение дп/дс возрастает
по мере ионизации поликислоты.
П. к. характеризуется высокой гигроскопичностью.
Она не плавится и не переходит в высокоэластич. со-
состояние. При нагревании ее в атмосфере азота B50—
260 °С) образуется нерастворимый полиметакриловый
ангидрид; выше 400 °С происходит разложение (без
выделения мономера). П. к. растворяется в воде, водно-
аммиачном р-ре, спирте, диметилформамиде, диоксане,
плохо растворяется в ацетоне, не растворяется в соб-
собственном мономере, хлороформе, уксусной к-те, этило-
этиловом эфире, циклогексане. С увеличением степени
изотактичности растворимость полимера снижается.
Макромолекулы неионизованной П. к. в водном р-ре
свернуты в клубки, стабилизованные внутри- и меж-
межмолекулярными водородными связями, вандерваальсо-
выми взаимодействиями, особенно гидрофобными
взаимодействиями неполярных метильных групп. При
ионизации П. к. ощутимо возрастает вязкость ее р-ра,
т. к. происходит заметное разворачивание клубков
вследствие электростатич. отталкивания ионизованных
карбоксильных групп. Кооперативный конформацион-
ный переход из плотного в более развернутый клубок
обнаруживается в узком интервале рН при степени
ионизации П. к. 0,1—0,15. Водный р-р П. к.—типич-
к.—типичный полиэлектролит (см. Макромолекула, Полиэлек-
Полиэлектролиты). Константа диссоциации (р/Га) П. к. заметно
зависит от природы растворителя, концентрации р-ра,
природы нейтрализующего (титрующего) агента, ми-
микротактичности и слабо зависит от мол. массы. Для
водного р-ра П. к. (концентрация 0,04 моль/л, 25 °С,
титрование 0,1 н. р-ром NaOH) эффективное значение
])Ка ок. 7,0. Макромолекулы П. к. в р-рах обнару-
обнаруживают способность к специфич. (избирательному) свя-
связыванию катионов (напр., Li+, Na+, Cu2+), к-рая за-
зависит от микротактичности цепей. П. к. взаимодейст-
взаимодействует с основаниями, образует полиэфиры.
Получение. П. к. получают радикальной полиме-
полимеризацией М. к. в массе, водных р-рах или в органич.
растворителях в присутствии радикальных инициато-
инициаторов. Чаще всего полимеризацию проводят в кислых
водных р-рах. Общая скорость полимеризации и, мол.
масса М образующегося полимера и константа ско-
скорости роста цепи /ср резко уменьшаются с увеличением
рН от 2 до 6, т. е. по мере ионизации мономера. Ми-
Минимальная скорость соответствует рН 6—7. При
рН > 7, когда П. к. ионизована, с увеличением рН на-
наблюдается возрастание у, М и /ср. Это обусловлено
тем, что с увеличением ионной силы р-ра, т. е. с рос-
ростом содержания в р-ре катионов (напр., Na+ в случае
р-ра NaOH), нивелируется электростатическое от-
отталкивание отрицательно заряженных мономера (мет-
акрилат-аниона) и макрорадикала в акте роста цепи.
При рН 9—10 в присутствии катионов аммония или
изобутиламмония образуется синдиотактич. П. к. (со-
(содержание синдиотактич. диад в макромолекулах дости-
достигает 95%).
Скорость полимеризации М. к. в органич. раствори-
растворителях и ее активность при сополимеризации во многом
зависят от способности М. к. к ассоциации (напр., от
способности образовывать циклич. димер или ассоцпаты
с молекулами растворителя).
П. к. можно получить также гидролизом ее полиэфи-
полиэфиров (легко гидролизуется поли-гаре/м-бутилметакрилат).
Изотактич. П. к. получают гидролизом изотактич.
полиизопропилметакрилата или поли-гарет-бутилмет-
акрилата.
М. к. легко сополнмеризуется с большим числом
мономеров; метакрилат-анион — с акриламидом, ди-
этиламиноэтилметакрилатом, винилпиридином, N-ви-
нилпирролидоном и др. Сополимеры М. к. можно также
получать полимер аналогичными превращениями.
Применение. Соли П. к. применяют как эмульга-
эмульгаторы, а также добавки, повышающие вязкость авиваж-
ных ванн, используемых для подшлихтовки синтетиче-
синтетического волокна. Сополимеры бутадиена с М. к., а также
тройные сополимеры стирол — М. к.— метакрилат
используются для приготовления клеев. Сополимеры
бутилметакрилата с М. к. в смеси с частично гидролизо-
ванным поливинилацетатом оказались эффективными
компонентами клеев, применяемых в произ-ве фанеры
и др. слоистых материалов. Промышленным продуктом
является сополимер бутадиена пли изопрена с неболь-
небольшим количеством М. к. (см. Карбоксилатные каучуки).
М. к. испольлуют в качестве сомономера в производстве
органических стекол» Сополимеры М. к. используются
также как ионообменные смолы.
Впервые П. к. была получена Фиттигом в 1877—80.
Лит.: Schildknecht G. E., Vinyl and related poly-
polymers, N. Y.— L., 1952; В а ц у л и к П., Химия мономеров,
пер. с чеш., т. 1, М., 1960; PinnerS. Н., A practical course
in polymer chemistry, N. Y., 1961; Мономеры, пер. с англ.,
М., 1953; Кабанов В. А., Топчиев Д. А., Высокомол.
соед., А13, № 6, 1324 A971). Д. А. Топчиев.
МЕТАКРИЛОНИТРИЛА ПОЛИМЕРЫ (polymeth-
acrylonitrile, Polymethakrylnitril, polymethacrylonit-
rile).
Метакрилонитрил (нитрил метакр иловой
к-ты) СН2— С(СНз) — CeeN (M.)— прозрачная, бесцвет-
бесцветная жидкость; плотность 0,7991 г/см3 A8 °С); п^ 1,3977;
ц20 0,392 мн-сек/м2, или спз; т. пл. —35,8 °С; т. кип.
90,3 °С; теплота испарения 31,8 кдж/моль G,6 ккал/моль);
теплота полимеризации ~64 кдж/моль A5,3 ккал/молъ);
темп-pa вспышки 12,8 °С; поверхностное натяжение
0,024 н/м B4,4 дин/см2) при 20 °С. М. образует азеотроп-
ную смесь с водой (84% М.), кипящую при 76,5 °С.
М. хорошо растворим во всех органич. растворителях.
В 100 г воды при 20 °С растворяется 2,59 г М., в 100 г
мономера — 1,62 г воды. Нитрильные группы М. омы-
ляются до амидных (в мягких условиях) и до карбок-
карбоксильных (в жестких условиях), гидрируются до амлн-
ных; при взаимодействии М, с формалином образуется
триазин. По двойной связи М. присоединяются спирты,
меркаптаны, фенолы, амины и др, под действием ще-
щелочных катализаторов (реакции цианэтилирования),
а также водород и др. М. легко сополимеризуется
по радикальному механизму с другими мономерами
(таблица),
М. хранят и транспортируют в алюминиевых боч-
бочках; ингибиторы — металлиламин или а-нитрозо-р-на-
фтол. В отсутствие ингибиторов М. полимеризуется
под действием света, кислорода и различных при-
примесей.
М. значительно токсичнее акрилонитрила, но, в от-
отличие от последнего, не обладает раздражающим дейст-
действием на слизистую оболочку глаз и на кожу.
Высокоэффективный способ получения М. в
пром-сти — окислительный аммонолиз изобутилена
189
МЕТАКРОЛЕИНА ПОЛИМЕРЫ
190
Константы сополимеризации метакрилонитрила
с различными сомономерами (г2)
Сомономер
Акрилонитрил
Метилметакрилат ....
а-Метилстирол
Бутадиен
2,68
0,70
0,35
0,04
0,32
0,74
0,20
0,36
Темп-ра
сополиме-
сополимеризации,
60
80
80
5
в газовой фазе при 300—600 °С; инертный разбави-
разбавитель — водяной пар или азот; катализатор — соедине-
соединения сурьмы, олова или молибдена. В лабораторных
условиях лучший способ синтеза М.— дегидратация
метакриламида путем перегонки его с фосфорным
ангидридом.
Полиметакрилонитрил [—СН2—С(СНз) — ]п (П.) —
CeeN
прозрачный полимер белого или светло-желтого цвета;
темп-pa размягчения ок. 115 °С. Мономерные звенья
в П. присоединены гл. обр. по типу «голова к хвосту».
В основной цепи макромолекулы содержатся в сравни-
сравнительно небольшом количестве кетениминные звенья
—СН2—C(CH3)=C=N—. П. растворим во многих ор-
ганич. растворителях и в собственном мономере. Рас-
Растворение в конц. серной к-те сопровождается частич-
частичным омылением П. Нерастворимые П. получаются в ре-
результате самопроизвольной полимеризации мономера
или при полимеризации в присутствии комплексных
металлоорганич. катализаторов.
Под действием к-т и щелочей нитрильная группа П.
способна омыляться до амидной, а затем до карбоксиль-
карбоксильной группы. При нагревании A60 °С и выше) образуется
лестничный полимер с системой сопряженных двойных
связей нафтиридинового типа.
Стереорегулярные кристаллич. полимеры и сополи-
сополимеры М. плавятся при 220—240 °С; они не растворяются
в обычных органич. растворителях, напр, в сероугле-
сероуглероде, спиртах, эфирах; растворяются в органич. и не-
органич. к-тах. Эти продукты характеризуются срав-
сравнительно высокими физико-механич. показателями.
П. и сополимеры М. получают как радикальной
(в р-ре или эмульсии), так и анионной полимеризацией.
В качестве катализаторов предложены также соедине-
соединения типа Li[Zn(C2H5JC4H9], способствующие образова-
образованию стереорегулярных полимеров.
Из-за низкой теплостойкости, изменения окраски
при нагревании и плохой текучести П. не нашел прак-
тич. применения. В очень ограниченном масштабе ис-
используются сополимеры М. с акриловой или метакри-
ловой к-тами и их эфирами для произ-ва, небьющихся
стекол. Тройные сополимеры М., стирола и бутадиена
(содержание М. от 10 до 90%), полученные в эмульсии,
в смеси с поливинил хлоридом используют в производ-
производстве пленок и электроизоляционных лаков и эмалей.
Прочность при растяжении такой пленки 0,06 Мн/м2
@,57 кгс/см2), модуль упругости 2,5 Мн/м2 B5 кгс/см2),
относительное удлинение 5,3%.
Лит.: Марек О., Томка М., Акриловые полимеры,
пер. с чеш., М.— Л., 1966; Химические реакции полимеров,
под ред. Е. Фвттеса, пер. с англ., т. 1—2, М., 1967; Рого-
Роговин 3. А. [и др.], Хим. пром-сть, № 5, 267 A958).
Н. М. Бедер.
МЕТАКРОЛЕИНА ПОЛИМЕРЫ (polymethacro-
lein, Polymethakrolein, polymethacroleine).
Метакролеин (изобутеналь, 2-метилпропеналь)
12 3 4
СН2=С(СН3)—СН=О (М.) — бесцветная жид-
жидкость с резким запахом, лакриматор; т. пл. ниже
—78 °С; т. кип. 68,6—68,8 °С; плотность 0,8721 г/см3
B0 °С); п^ 1,4312. М. плохо растворим в воде F,4 г
ОН
в 100 г воды), хорошо — в органических раствори-
растворителях. С водой образует азеотропную смесь. М. самопро-
самопроизвольно полимеризуется при хранении; фенолы ин-
гибируют полимеризацию. М. сополимеризуется с ме-
такрилонитрилом, стиролом, акролеином, винилаце-
татом и др.
В лаборатории М. синтезируют конденсацией пропио-
наля с формальдегидом в присутствии солянокислого
диэтиламина с добавлением соды (рН » 7) при нагре-
нагревании до 40—45 °С в течение 30 мин (реакция Манниха).
В пром-сти М. получают окислением изобутилена кис-
кислородом или воздухом при 500 °С и выше в присутствии
окисно-медных катализаторов. Очистка, анализ и физио-
логич. действие аналогичны акролеину (см. Акролеина
полимеры).
Полиметакролеин (П.). При радикальной полимери-
полимеризации М. с использованием воды и окислительно-вос-
окислительно-восстановительной системы мономерные звенья входят в
цепь в положении l,2;i в макромолекулах образую-
образующегося полимера содержатся в основном полуацеталь-
ные структуры 1-а G0—80%), а также свободные
альдегидные группы I:
Такую полимеризацию СН
осуществляют в эмульсии
или на границе раздела фаз
М.— вода в присутствии
соответственно системы
персульфат калия — азот-
азотнокислое серебро или пер-
персульфат калия — метабисульфит натрия.
При радикальной полимеризации М. в отсутствие
окислительно-восстановительной системы и воды обра-
образуется полимер, в макромолекулах к-рого содержатся
только свободные альдегидные группы. Циклизация
последних происходит под действием паров воды и
СО2, содержащихся в атмосфере.
При радикальной полимеризации на границе раздела
фаз М.— вода получены порошкообразные полимеры
с мол. массой 200 000 и более (темп-pa размягчения
210—230 °С для П. с мол. массой 200 000). П. раство-
растворимы в пиридине, диметилформамиде, тетрагидрофура-
не, диметилсульфоксиде и др. органич. растворителях
и нерастворимы в воде. Их можно использовать как
селективные комплексообразователи на ионы тяже-
тяжелых металлов.
Полимеризация М. под действием анионных катали-
катализаторов (нафтила натрия, тритила калия, натрийнафта-
лина или литийбутила) протекает одновременно по по-
положению 1,2 (структура I) и 3,4 (структура II); звеньев,
соответствующих присоединению молекул М. в поло-
положении 1,4, не обнаружено. Кроме того, наблюдается
образование лестничных (III) и лактонных (IV) струк-
структур:
СН,
Структура П. определяет его темп-ру плавления
A20—380 °С), к-рая повышается с увеличением количе-
количества звеньев III. Степень превращения М. в полимер
может достигать 100%. Анионную полимеризацию
проводят, как правило, в р-ре, и полимер получают
в виде порошка, растворимого в органических раство-
растворителях.
При катионной полимеризации М. в присутствии
газообразного BF3 П. получается в виде прозрачных
блоков, не растворимых в органич. растворителях
и воде; т. пл. 380—390 °С.
191
МЕТАЛЛИЗАЦИЯ ПЛАСТМАСС
192
Благодаря растворимости и наличию структур I и
I-а П. можно легко модифицировать. При восстановле-
восстановлении П. LiAlH4 получен высокомолекулярный поли-
металлиловый спирт с содержанием гидро-
ксильных групп ок. 80% (от теоретич.). Из такого спирта
можно изготовлять эластичные пленки. Взаимодействие
с LiAlH4 частично оксимированного (по свободным аль-
альдегидным группам) П. приводит к полимеру, содержа-
содержащему аминные и гидроксильные группы (а):
COONa
CH2NH2 CH2OH
а
Регулярные гидроксикарбоксилатные полимеры (б),
образующиеся при действии на П. спиртового р-ра ще-
щелочи, можно использовать как связующие, эмульга-
эмульгаторы, структурообразователи для почвы. П., в макро-
макромолекулах к-рых содержатся структуры IV — аппре-
аппреты для придания несминаемости тканям.
При деструктивном окислении П., полученного в вод-
водной среде, разб. водным р-ром Н2О2 образуется полимер,
к-рый можно использовать в качестве маскирующего
комплексообразователя солей тяжелых металлов.
Лит.: К о т о н М. М. [и др.], Высокомол. соед., 7, № 12,
2039 A965); А н д р е е в а И.' В. [и д р.], ЖПХ, 41, № 10,
2269 A968); их же, J. Polymer Sci., pt A—I, 10, № 5,
1467 A972); и х ж е, Высокомол. соед., 9А, №11, 2496 A967);
Андреева И. В., Медведев Ю. В., там же, 5А, № 4,
852 A973); А н д р е е в а И. В. [и др.], ЖПХ, 42, № 8,
1857 A969). И. В. Андреева.
МЕТАЛЛИЗАЦИЯ ПЛАСТМАСС (metallizing of
plastics, Metallisierung von Plasten, metallisation des
plastiques) — нанесение металлич. покрытий на изде-
изделия и полуфабрикаты из пластмасс. Первоначально
основным назначением М. п. было придание изделиям
декоративных свойств, однако в дальнейшем этот про-
процесс стали использовать для придания поверхности
полимерных материалов тепло- и электропроводности,
большей тепло-, атмосферо- и износостойкости, спо-
способности отражать электромагнитные волны. Кроме
того, в результате металлизации повышаются прочност-
прочностные характеристики пластмасс (таблица) и появляется
возможность соединять изделия из них с помощью
пайки.
Металлич. покрытия наносят на полистирол, поли-
этилентерефталат, полипропилен, полиамиды, поли-
полиэфиры, полиимиды, полисульфон, полиметилметакри-
лат и др. Для этого используют алюминий, медь, ни-
никель, хром, серебро, золото, цинк и др. металлы, а
также тугоплавкие металлы и сплавы.
Ниже описаны основные способы М. п. Из них
в пром-сти широко применяются термич. испарение
металлов в вакууме (вакуумная металлизация) и элект-
ролитич. осаждение металлов в сочетании с химич.
осаждением, а в полиграфии — горячее тиснение ме-
металлизированной полимерной пленкой. Первый способ
наиболее экономичен. Он применяется гл. обр. для при-
придания декоративного вида изделиям.
Термическое испарение металлов
в вакууме включает след. операции: 1) нанесение
лакового подслоя, 2) собственно металлизация, 3) на-
нанесение защитного лакового покрытия. В нек-рых
случаях лаковый подслой и защитное покрытие не на-
наносят. Лаковый подслой выравнивает изъяны поверх-
поверхности, повышает ее адгезию к металлу и уменьшает
газовыделение с поверхности в вакууме. Игрушки,
галантерейные изделия и др. товары широкого потреб-
потребления покрывают лаками холодной сушки, а изделия
технич. назначения — лаками холодной и горячей
A—3 ч при 80—180 °С) сушки. Последние обеспечи-
обеспечивают металлич. покрытиям лучшую адгезию, высокую
прочность, коррозионную стойкость и стойкость к исти-
истиранию, однако такой метод сушки связан с необходи-
необходимостью сооружения сушильных камер.
Металлизацию осуществляют в вакуумной камере [ос-
[остаточное давление 1,33— 133мн/м2 A -10~5—1 • 10~3
мм рт. ст.)], куда помещают металл и обрабатываемое
изделие. Для термич. испарения наиболее часто ис-
используют алюминий высокой чистоты (99,6—99,99%),
обеспечивающий высокое качество покрытия. Испа-
Испарителем обычно служит спираль, скрученная из воль-
вольфрамовой проволоки. Темп-pa испарения алюминия
поддерживается ок. 1400—1800 °С. Для испарения др.
металлов используют также ленточные испарители из
молибденовой, вольфрамовой или танталовой фольги.
Для М. п. этим методом используется самое различ-
различное оборудование, т. к. он применяется при обработке
изделий различной конфигурации, напр, пуговиц,
ручек, призм, оправ для очков, а также изделий в виде
непрерывной пленки или нитей. Разработаны вакуум-
вакуумные камеры диаметром 0,3—2,0 м с вертикальным или
горизонтальным расположением испарительной каме-
камеры, со спиральным или точечным испарительным эле-
элементом, с различными видами вращения изделия вокруг
испарительного элемента для равномерного нанесения
металла на изделие. Перечисленные установки позволя-
позволяют наносить металл на плоские, круглые или фасонные
изделия равномерным слоем. Толщина металлич. слоя
ок. 0,1 мкм достигается за ~15 сек. Для получения ме-
металлизированного рисунка изделие загораживают от
испарительного элемента специальными масками, от-
отверстия в к-рых соответствуют по форме необходимому
рисунку.
Металлич. покрытие обычно нуждается в защите от
коррозии, истирания и царапания. Поэтому его покры-
покрывают лаками (теми же, что и для подслоя). Окрашенные
лаки позволяют получить покрытие различных цветов.
Напр., для имитации золота изделие металлизируют
алюминием и окрашивают желтым лаком. Металлизи-
Металлизированные пленки для этих целей обычно дублируют
др. полимерными пленками.
Достоинства термич. испарения металлов в вакууме:
1) возможность нанесения широкого круга металлов
и сплавов, 2) простота металлизации цилиндрич. и фа-
фасонных изделий, 3) возможность металлизации без
разогрева пластмассы. Недостаток метода — трудность
закрепления на пластмассе покрытий толщиной более
1—2 мкм. Для изделий из термостойких пластмасс этот
недостаток устраняется нагреванием изделий во время
металлизации до 150—200 °С. Напр., при металлизации
фторопласта при нагревании удается получить прочное
покрытие толщиной ок. 20 мкм.
Электролитическое осаждение ме-
металлов лучше всего разработано для металлизации
АБС-пластиков, полисульфона, полипропилена, фторо-
фторопластов и др. медью, никелем и хромом. Нанесение по-
покрытий этим методом возможно только на электропро-
электропроводящую поверхность. Поэтому предварительно на ма-
материал необходимо нанести электропроводящий слой
каким-либо др. методом. Обычно это осуществляется
химическим осаждением металлов
(чаще всего меди или никеля), обеспечивающим высокую
адгезию электропроводящего слоя к пластмассе. Кроме
того, этот метод высокопроизводителен и не требует
сложного оборудования.
Обычно в пром-сти электролитич. и химич. осаждение
металлов объединены в единую технологич. схему.
Подготовка поверхности включает обезжиривание от-
отработанными травильными р-рами, ацетоном, спиртом,
бензином или синтетич. моющим средством, травление
и, иногда, нанесение адгезионного лакового подслоя.
Затем изделие промывают и подвергают сенсибилизации
для получения на полимерной поверхности свежевос-
становленного слоя каталитически активного благород-
благородного металла. Для этого изделие погружают на 1—2 мин
193
МЕТАЛЛОНАПОЛНЕННЫЕ ПЛАСТИКИ
Некоторые свойства пластмасс до и после металлизации*
194
Показатель
Феноло-форм-
альдегидный
прессматери-
ал с древес-
древесной мукой
Феноло-
формаль-
дегидный
гетинакс
Поливинил-
хлорид
Поливинили-
денхлорид
Полистирол
Полиметил-
метакрилат
АБС-
пластик
Прочность**, Мн/м2
при растяжении
при изгибе
Ударная вязкость, кдж/м2, или
кгс • см/см2
Теплостойкость, °С
Водопоглощение за 24 ч, % . .
55 F5)
85 A00)
15 B0)
135 B65)
0,44 @,12)
90 A00)
ПО A25)
40 D5)
160 B43)
3,5 @,12)
50 F0)
77 A07)
0,42 @,11)
50 F4)
105 A20)
50 F0)
82 A21)
0,08 @,02)
35 D5)
90 A05)
15 B0)
77 A13)
0,01 @,00)
70 (85)
70 (85)
20 B2)
68 A21)
0,35 @,03)
40 E0)
60 (85)
20 C5)
88 A01)
1,00 @,07)
* Без скобок даны показатели для пластмасс до металлизации,
медь 7,6 мкм и кадмий 12,7 мкм. * * 1 Мн/м2х 10 кгс/см2.
в скобках — после электролитич. металлизации в два слоя:
в р-р SnC]2, а затем в р-р соли благородного металла,
напр, серебра. Если металлизируют только часть изде-
изделия, то поверхности, к-рые не нужно металлизировать,
покрывают лаком, препятствующим закреплению ка-
талитич., слоя.
После сенсибилизации изделие промывают и подвер-
подвергают активации катализатором, для чего погружают
в р-р азотнокислого серебра. Сенсибилизацию и акти-
активацию можно совмещать, помещая материал в р-р, со-
содержащий двухвалентное олово и соль благородного
металла. Обработанные изделия тщательно промывают,
т. к. даже следы активатора мбгут вызвать преждевре-
преждевременное восстановление металла из солей в мёталлиза-
ционных р-рах. Подготовленные поверхности подвер-
подвергают химич. меднению из щелочных р-ров двухвалент-
двухвалентных комплексов меди. В качестве восстановителя ис-
используют формальдегид, способный восстанавливать
медь при комнатной темп-ре. В этом случае процесс
носит автокаталитич. характер и не прекращается даже
после того, как весь каталитич. слой закрыт медью.
Описанный метод позволяет получать высококачест-
высококачественные и красивые изделия из пластмасс, к-рые дешев-
дешевле и легче соответствующих изделий из металлов. Не-
Недостаток метода заключается в ограниченности круга
применяемых металлов и в разрушительном действии
электролитич. ванны на полимер. Последнее обстоя-
обстоятельство делает невозможным применение электроли-
электролитич. осаждения для металлизации нек-рых изделий,
напр, оптич. назначения.
Химич. металлизацию можно осуществить др. путем.
Для этого изделия подвергают обработке с целью об-
образования на поверхности полимера функциональных
групп (—SO3H,—ОН,—GOOH), способных обменивать
ионы металлов или их комплексы на ионы водорода.
Для улучшения металлизации поверхность дополни-
дополнительно активируют в р-рах солей благородных метал-
металлов. Затем изделие помещают в металлизационный р-р
на 3—5 мин, после чего восстанавливают ионы сорби-
сорбированного металла. Этот метод позволяет добиться
высокой адгезии металла к пластмассе, однако приго-
пригоден для обработки ограниченного круга материалов.
Катодное распыление металлов
в вакууме [остаточное давление воздуха 1,33—
13,3 н/м2 A-10~2—1-Ю мм рт.ст.)]. В этом процессе
катод, изготовленный из напыляемого металла, медлен-
медленно разрушается под воздействием ударяющихся о него
положительных ионов разреженного воздуха. Частицы
вещества катода осаждаются на изделии. К достоинст-
достоинствам метода следует отнести высокую прочность сцепле-
сцепления металла с пластмассой, к недостаткам — разогрев
изделий во время металлизации, ограниченный круг
металлов, пригодных для изготовления катода, и необ-
необходимость соответствия конфигурации катода форме
изделия.
При М. п. распылением расплавлен-
расплавленного металла струей воздуха или газа частицы
металла с большой силой ударяются о поверхность
изделия и прилипают к ней. Для расплавления металла
и управления металло-воздушной струей применяют
электродуговые и газопламенные аппараты, а также
аппараты, питаемые токами высокой частоты и работаю-
работающие с использованием низкотемпературной плазмы.
Этот метод позволяет покрывать металлами большие
поверхности, однако не обеспечивает необходимой рав-
нотолщинности и гладкости металлич. слоя и достаточ-
достаточной адгезии металла к пластмассе. Кроме того, в связи
с опасностью расплавления и деструкции полимера
необходимо пользоваться металлами и сплавами с низ-
низкими темп-рами плавления.
Горячее тиснение маркировки или рисун-
рисунков на изделиях из пластмасс с использованием ме-
металлизированной полимерной пленки осуществляют
в специальных прессах. Метод производителен и прост,
однако позволяет металлизировать только плоские по-
поверхности (см. Печать на полимерах).
При нанесении на поверхность из-
изделий лака или клея, смешанного
с металлич. пудрой, образуется электро- и
теплопроводное покрытие.
Металлизированные пластмассы находят широкое
применение. В машиностроении и электротехнике их
используют для изготовления электрообогреваемых,
антистатич. и антимагнитных изделий, а также электро-
люминесцирующих панелей со светящимися надписями
и различных указателей. Оптич. пром-сть выпускает
поглощающие и интерференционные фильтры, зеркала
и призмы. В радиотехнике и электронике металлизиро-
металлизированные изделия применяются для изготовления печат-
печатных схем, конденсаторов, электродов, микромодулей,
а также ручек управления и деталей внешней отделки
радиоприемников, телевизоров, магнитофонов и различ-
различной аппаратуры. В авиа-, автомобиле- и вагоностроении
применяют металлизированные детали внешнего и
внутреннего оформления. Значительная часть товаров
широкого потребления из пластмасс выпускается с по-
полированным или матовым металлич. декоративным по-
покрытием, а также с покрытием «мороз», «муар» или ме-
металлизированным рисунком.
Лит.: РотреклБ., Дитрих .3., Тамхина И.
Нанесение металлических покрытий на пластмассы, пер. с чеш.
«П., 1968; Катц Н. В., Металлизация тканей, 2 изд., М.
1972; Шалкаускас М. И., Вашкялис П. - А. Ю.
Химическая металлизация пластмасс, Л., 1972; Хол
л э н д Л., Нанесение тонких пленок в вакууме, пер. с англ.
М.» 1963; Narcus H., Metallizing of plastics, N. Y.—L.
1960. M. M. Гудимов, Э. Ф. Маркина
МЕТАЛЛОНАПОЛНЕННЫЕ ПЛАСТИКИ (metal
filled plastics, metallgefiillte Plaste, plastiques charges
du metal) — пластмассы с металлич. порошкообразным
или волокнистым наполнителем.
Состав. В качестве связующего для М. п. используют
большинство термопластов, феноло-формальдегидные,
эпоксидные и полиэфирные смолы, кремнийорганич.
полимеры, а также каучуки.
195
МЕТАЛЛОНАПОЛНЕННЫЕ ПЛАСТИКИ
196
Металлич. порошки (частицы их могут иметь сфери-
сферическую или чешуйчатую форму) изготовляют из железа,
меди, алюминия, серебра, никеля, олова, висмута,
кадмия, палладия и др. Железо и медь могут катали-
катализировать термоокислительную деструкцию связующего
в М. п., поэтому поверхность частиц порошков из этих
металлов иногда лакируют. Быстро окисляющиеся
с поверхности порошки алюминия, железа и меди не
применяют в производстве электропроводящих поли-
полимерных материалов.
Металлич. волокна изготавливают обычно из меди,
алюминия, стали, бериллия, бора, тантала, титана,
магния, вольфрама и молибдена. Диаметр волокон
колеблется от 0,01 до 0,2 мм, а длина от 6 до 25 мм.
М. п., содержащие в качестве наполнителя «усы» (мо-
(монокристаллы металлов в виде волокон длиной ок. 1 мкм),
обладают очень высокими физико-механич. характе-
характеристиками, однако изготовление «усов» сложно и по-
поэтому такие М. п. не нашли пока промышленного при-
применения.
Кроме связующего и металлич. наполнителя, М. п.
могут содержать др. наполнители минерального и орга-
нич. происхождения, стабилизаторы, красители, пла-
пластификаторы и поверхностно-активные в-ва, тип и
количество к-рых зависят главным образом от природы
связующего.
Производство. Простейший способ изготовления М. п.
заключается в смешении ме'галлич. наполнителя с по-
порошком, гранулами, расплавом твердого полимера или
жидким связующим в смесителях различного типа. Из
полученной смеси прессуют изделия. Этот способ наи-
наименее энерго- и трудоемок, однако он не обеспечивает
равномерного распределения наполнителя в материале.
М. п. можно получить также по след. технологии:
предварительно приготовленную суспензию металла
(чаще всего водную) смешивают с р-ром полимера или
латексом, а затем коагулируют. Этот способ прост
в аппаратурном оформлении, но полученный таким
образом материал содержит значительное количество
поверхностно-активных веществ (стабилизаторы сус-
суспензии). Менее распространены способы, основанные
на вибропомоле металла с одновременным его диспер-
диспергированием в полимере или мономере. Диспергирован-
Диспергированный в мономере металл часто является катализатором
полимеризации. М. п. с размером частиц наполнителя
менее 0,25 мкм можно получить также термическим
или электролитическим разложением металлооргани-
ческих соединений (например, карбонилов или форми-
атов), предварительно диспергированных в мономе-
мономерах, олигомерах, р-рах, расплавах или суспензиях
полимеров.
При изготовлении М. п. с волокнистым наполнителем
последний погружают в емкость с жидким связующим,
затем извлекают, высушивают и из полученного мате-
материала прессуют изделия. Этот метод позволяет полу-
получить М. п. более высокого качества, чем при смешении
волокнистого наполнителя с порошком или гранулами
полимера.
Изделия, обладающие анизотропией электро- и тепло-
теплопроводности, формуют в электромагнитном поле, в ре-
результате чего происходит ориентация наполнителя и
создание токопроводящих цепочек.
Свойства. Прочностные характеристики М. п. в зна-
значительной степени определяются адгезией металла к
связующему. Адгезия металлов к синтетическим полиме-
полимерам уменьшается в ряду: никель, сталь, железо, олово,
свинец. Адгезия металлов к полимерам м. б. вызвана как
физическим, так и химич. взаимодействием между ними
(см. Адгезия). Напр., в случае наполнения полимеров
металлами платиновой группы или золотом решающую
роль играют вандерваальсовы силы. Полимеры с нена-
ненасыщенными связями способны образовывать с металлами
комплексные соединения, а карбоксилсодержащие поли-
полимеры взаимодействуют с металлич. наполнителем в отсут-
отсутствие окислителей за счет свободных электронов. Кар-
Карбоксильные, фенольные и гидроксильные группы свя-
связующего образуют с окислами металлов, практически
всегда присутствующими на поверхности наполнителя,
связи ионного или ион-дипольного характера. Между
металлич. наполнителем и связующим возможно также
образование водородных связей.
Толстые окисные пленки с большим количеством де-
дефектов (напр., на поверхности медного наполнителя)
обладают низкими физико-механич. показателями; по
этим дефектам происходит разрушение пары металл —
полимер. С другой стороны, тонкие прочные окисные
пленки на поверхности алюминиевого или железного
наполнителя обеспечивают прочную связь металла с
полимером.
Кроме того, прочность связи компонентов в М. п.
зависит от способа их изготовления. Наибольшая проч-
прочность отмечена в том случае, когда при получении
М. п. частицы металла формируются в полимере, оли-
гомере или мономере, т. к. в момент образования они
обладают высокой реакционной способностью.
М. п. обладают более высокими, чем исходные поли-
полимеры, прочностными характеристиками, термостой-
термостойкостью и теплопроводностью (таблица).
Свойства металлонаполненных пластиков на основе
эпоксидной смолы типа ЭД-5 и металлического волокна
(содержание волокна 20% по объему, длина волокна 6,5 мм,
диаметр 0 ,1 мм)
Показатели
Прочность, Мн/м2
(кгс/см2)
при растяжении . . .
при сжатии
Модуль упругости при
сжатии, Мн/м2
(кгс/см2)
Теплопроводность,
вт/(м-К),
[кпал/(м - ч • °С)] ....
Исход-
Исходная
смола
6,1
F1)
91
(910)
740
G400)
20,3
[17,8]
Волокно
алюми-
алюминий*
22
B20)
115
A150)
3920
C9200)
116,3
[100]
медь
30
C00)
122
A220)
3360
C3600)
117,6
[102]
сталь
36
C60)
132
A320)
4890
D8900)
129,6
[114]
* Длина 9,5 мм.
Однако введение более 40% порошкообразного метал-
металлич. наполнителя обычно приводит к нек-рому сниже-
снижению прочности вследствие возрастания внутренних
напряжений в высоконаполненном пластике. Напол-
Наполнение полимера волокнистым наполнителем приводит
к большему возрастанию прочностных характеристик
и теплопроводности М. п., чем при наполнении порош-
порошком. Напр., введение в эпоксидную смолу 10% алюми-
алюминиевых волокон (длина 9,5 мм, диаметр 0,18 мм) при-
приводит к повышению прочности при растяжении на
110% и теплопроводности на 425%. Этот же эффект по
теплопроводности достигается при введении 32% алю-
алюминиевого порошка с размером частиц 0,02 мм; при
этом прочность пластика при растяжении не меняется.
С увеличением длины и диаметра волокна возрастает
прочность М. п. при сжатии и его теплопроводность,
к-рая может быть в десять раз выше, чем у чистого по-
полимера.
С увеличением содержания металлич. наполнителя
(кроме железа и меди) возрастает стойкость М. п.
к термоокислительной деструкции. Напр., введение в
полистирол 8% коллоидных частиц свинца повышает
темп-ру начала деструкции полистирола с 280 до
320 °С, а 45% свинца — до 350 СС.
М. п. обладают электрич. проводимостью, зависящей
от типа металла, степени наполнения, смачиваемости
наполнителя связующим и условий переработки мате-
197
МЕТАЛЛОПЛАСТ
198
риала. Максимальная проводимость достигается в том
случае, когда металлич. наполнитель не окисляется
и возможные его химич. реакции со связующим проис-
происходят только на поверхности контакта металл — поли-
полимер. При одной и той же степени наполнения электро-
электропроводность М. п. возрастает с увеличением дисперс-
дисперсности наполнителя. Напр., при наполнении полимера
серебряным порошком с размером частиц 2—6 мкм
можно достичь уд. электропроводности 103—10е ом~1-м~1
A03—104 ом'1-см'1). Сферич. частицы металличе-
металлического порошка частично отслаиваются от связую-
связующего, что обусловливает различную электропровод-
электропроводность в различных слоях материала. Лучших резуль-
результатов удается достичь при использовании порошков
с частицами чешуйчатой формы, к-рые распола-
располагаются в материале более равномерно и не отслаи-
отслаиваются.
-Зависимость электропроводности М. п. от степени
наполнения нелинейна. На примере эпоксидной смолы
с железным, свинцовыми и никелевым наполнителями
установлено, что при содержании наполнителя до
20% (по массе) частицы наполнителя располага-
располагаются в виде отдельных изолированных агрегатов.
В этом случае уд. электропроводность составляет 10~10—
10~7 ом^-м'1 A0~12—10~9 ом'1-см'1) и реализуется за
счет ионной проводимости через контакт полимер —
металл. С увеличением количества наполнителя все
большее число частиц металла образует токопроводя-
щие мостики, что ведет к резкому возрастанию удель-
удельной электропроводности материала до 10~2 ом~1-м~1
A0~4 ом-см'1). Максимальная электропроводность
достигается при степени наполнения М. п. ок. 40%.
При этом частицы наполнителя образуют токопрово-
дящую коагуляционную структуру. В этом случае
перенос зарядов осуществляется преимущественно че-
через контакт металл — металл (см. также Электропро-
Электропроводящие полимерные материалы).
Нек-рые металлич. наполнители придают полимерам
специфич. свойства, напр, порошки железа и его спла-
сплавов — ферромагнитные свойства, чешуйки алюминия,
никеля, серебра и др.— низкую газо- и паропроницае-
мость, порошки алюминия и медных сплавов — деко-
декоративность. М. п. на основе тонко дисперсных порошков
платины, палладия, родия, иридия и железа обладают
способностью катализировать реакции гидрирования
и часто превосходят по каталитич. активности метал-
металлич. порошки. Материалы, наполненные свинцом, кад-
кадмием и вольфрамом, пригодны в качестве защиты от
излучений высокой энергии.
Применение. М. п. дешевы, доступны и заменяют
цветные и драгоценные металлы при изготовлении под-
подшипников, втулок, вкладышей и др. изделий с высокой
теплопроводностью и низким температурным коэфф.
линейного и объемного расширения. Они применяются
в производстве магнитных лент, экранов для защиты
от высокочастотных электромагнитных помех, нагре-
нагревателей для точного термостатирования различных
приборов, устройств для отвода статич. электричества,
токопроводящих элементов на панелях из диэлектри-
диэлектриков, сопротивлений, конденсаторов и соединительных
проводов в печатных радио- и электрич. схемах.
Лит.: Электропроводящие полимерные материалы, М., 1968;
Натансон Э. М., У льбергЗ. Р., Коллоидные метал-
металлы и металлополимеры, Киев, 1971; Натансон Э. М.,
Б рык М. Т., Металлополимеры, Успехи химии, 41, 1465
A972). Л. В. Аристовская.
МЕТАЛЛОПЛАСТ (Metalloplast) — листовой кон-
конструкционный материал, состоящий из металлич. поло-
полосы (листа) с одно- или двухсторонним полимерным по-
покрытием. Толщина металлич. полосы обычно состав-
составляет 0,3—1,2 мм, полимерного покрытия — 0,05—
1,0 мм. Покрытие м. б. одноцветным или многоцветным,
гладким или рельефным, имитировать ценные породы
дерева, мрамор и др. М. не расслаивается и не коро-
коробится при механич. обработке, штамповке, гибке и
сварке, изделия из него не нуждаются в антикоррозион-
антикоррозионной защите и декоративной отделке.
Для изготовления М. пригодно большинство листовых
конструкционных металлич. материалов — из стали,
алюминия и его сплавов, титана и др. Полимерный
слой может состоять из полиолефинов, фторопластов,
полиамидов, пластифицированного поливинилхлорида
и др.
М. обычно производят на непрерывных линиях. Про-
Процесс включает след. технологич. операции: 1) обработ-
обработка металлич. полосы для повышения ее адгезии к по-
полимерам и предотвращения коррозии, напр, обезжи-
обезжиривание, травление, фосфатирование; 2) промывка
и сушка; 3) нанесение полимерного покрытия путем
наклеивания на металлич. полосу заранее изготовлен-
изготовленной полимерной пленки, намазывания пасты полимер-
полимерной или напыления полимера в порошкообразном состоя-
состоянии (методы лакирования и эмалирования металлич.
полос в данной статье не рассматриваются; соответст-
соответствующие материалы к М. не относятся); 4) оплавление
полимера (при напылении), нанесение рисунка и тис-
тиснение рельефа; 5) охлаждение М., разрезание его на
куски и упаковка. Существующие методы производства
М. отличаются в основном способом нанесения полиме-
полимера на металл.
Наиболее целесообразно объединить производство
металлич. полосы и нанесение на нее полимерного
покрытия в единый технологич. процесс, однако это
удается не всегда, т. к. скорость поступления металлич.
полосы с прокатного стана определяется ее толщиной
и часто отличается от скорости проведения др. опе-
операций.
Наклеивание полимерной пленки. В наибольшей сте-
степени пром-стью освоен процесс наклеивания на непре-
непрерывно движущуюся стальную полосу пленочного
пластиката. В качестве клея применяют р-р сополи-
сополимера винилхлорида с винилацетатом B ч.) в циклогек-
саноне E ч.). Во время эксплуатации М. пластификатор
из пластиката переходит в клеевой шов, что снижает
прочностные характеристики последнего. Поэтому поли-
винилхлоридный клей заменяют на эпоксидный или
феноло-формальдегидный, к-рые обеспечивают стабиль-
стабильность свойств М. во времени.
Технология наклеивания в установке непрерывного
действия заключается в следующем. На движущуюся
металлич. полосу намазывающим роликом наносят слой
клея, подогревают его в первой печи до 40—50 °С (для
поливинилхлоридного и феноло-формальдегидного кле-
клеев) или до 100 °С (для эпоксидных клеев), прика-
прикатывают валками пленку и направляют полученное
изделие во вторую печь для окончательного отверж-
отверждения клея. По выходе из нее разогретый М. подвер-
подвергают тиснению, после чего охлаждают. Скорость
движения стальной полосы в установке не превыша-
превышает 0,5 м/сек.
Полученный М. сочетает прочность стального листа
с химич. стойкостью пластиката. При комнатной темп-ре
он устойчив в р-рах серной и уксусной к-т, а также
щелочей. Пленка на поверхности М. не набухает в бен-
бензине, керосине и минеральных маслах. С повышением
темп-ры химич. стойкость полимерной пленки снижа-
снижается. Электроизоляционные свойства М. зависят от
толщины полимерного покрытия. Так, при толщине
0,12 мм электрич. прочность составляет 4500 в, при
0,25 мм — 7000 в и при 0,30 мм — 9000 в. М. этого типа
может длительно работать в интервале темп-р от —40
до 65 °С. Он легко штампуется и режется, а также сва-
сваривается электродуговой и рельефной односторон-
односторонней сваркой без удаления покрытия. Во время эксплуа-
эксплуатации изделий из таких М. пластификатор выпоте-
выпотевает на поверхность полимерной пленки и испаряется,
а пленка становится более жесткой и хрупкой.
199
МЕТИЛАКРИЛАТА ПОЛИМЕРЫ
200
Процесс плакирования непрерывно движущейся по-
полосы металла полиэтиленовой пленкой принципиально
не отличается от описанного выше плакирования пле-
пленочным пластикатом. Т. к. надежные клеи для полиэти-
полиэтилена отсутствуют, необходимой адгезии пленки к ме-
металлу добиваются частичным окислением ее поверх-
поверхности горячим воздухом, кислородом, к-тами, УФ-об-
лучением или др. методами. Установлено, что при вве-
введении в полиэтилен высокодисперсных минеральных
наполнителей и органических перекисей эффектив-
эффективность окисления резко возрастает. Описан, например,
метод прикатки полиэтиленовой пленки, содержа-
содержащей 8—10 об. ч. талька, к нагретой до 200—250 °С
стальной полосе.
Нанесение паст (пластизолей). Практич. значение
имеет нанесение пластизоля на металлич. полосу по
клеевому подслою. Применение пластизоля позволяет
исключить операцию изготовления пластикатовой
пленки, что снижает стоимость М. Пластизоль приклеи-
приклеивают теми же клеями, что и пластикат. Наносят его на
стальную полосу, покрытую слоем клея, при помощи
скребкового ножа или намазывающего ролика. Толщина
полимерного покрытия регулируется высотой подъема
ножа (ролика) над полосой металла. Затем М. попа-
попадает в печь, где поливини л хлорид набухает в пласти-
пластификаторе, после чего проходит тиснильные валки, ох-
охлаждается и направляется на разрезывание и упаковку.
Свойства полученного этим методом М. практически
не отличаются от свойств М., полученного наклеива-
наклеиванием пластиката на стальную полосу.
Напыление порошков. Порошки полимеров напы-
напыляют на металлич. полосу в псевдоожиженном слое
и в электрич, поле высокого напряжения с последую-
последующим оплавлением порошка.
В первом случае полоса металла проходит индукци-
индукционную печь, где нагревается на 30—80 °С выше темп-ры
плавления полимера, после чего попадает в камеру
напыления, где порошок полимера поддерживается
в псевдоожиженном состоянии потоком воздуха, посту-
поступающего через пористое дно камеры. Соприкасаясь
с горячей поверхностью металла, полимер оплавляется
и прилипает к ней, а затем уплотняется тиснильными
валками. Скорость движения полосы в агрегате не
превышает 0,2 м/сек. Этот способ пригоден только для
тех полимеров, темп-pa начала деструкции к-рых ле-
лежит намного выше темп-ры перехода их в вязкотеку-
чее состояние, напр, полиэтилена, иоливинилхлорида,
алифатич. полиамидов, поливинилбутираля. Процесс
прост в аппаратурном оформлении, но не обеспечивает
равномерной толщины полимерного покрытия.
При напылении полимеров в электрич. поле порошок
давлением воздуха или вибратором подается в камеру,
где около поверхности движущейся металлич. полосы
с помощью коронирующей электродной сетки, нахо-
находящейся под напряжением ок. 100 ив, и диэлектрич.
боковых экранов создается равномерное электрич. поле.
При напряженности поля 8—9 кв/см электрич. взаимо-
взаимодействие между заряженными частицами полимера и
полосой металла обеспечивает равномерное распреде-
распределение порошка по поверхности вплоть до скорости
движения полосы 2—3 м/сек. Производительность про-
процесса зависит в нек-рых случаях не только от скорости
напыления, но и от скорости охлаждения полимера.
Напр., достаточная адгезия фторопласта к стальной
полосе достигается при скорости охлаждения покры-
покрытия 1 — 2 °С/мин, к-рую удается реализовать только
при скорости движения полосы металла не выше
1 м/мин.
Наведенное в металле электрич. поле и поляризация
макромолекул приводят к значительному росту адге-
адгезии полимера к металлу и возникновению в полимерной
пленке пограничного слоя толщиной до 10—15 мкм,
твердость к-рого в 2—3 раза выше твердости «свобод-
«свободной» пленки. Кроме того, пограничный слой обладает
высокой эластичностью. Напр., разрушение фторопла-
фторопластового покрытия толщиной 70—90 мкм происходит
вместе с разрушением стальной полосы. Отслаивания
полимерной пленки или ее разрушения не наблюдается
при кипячении М. в течение нескольких ч в дистиллиро-
дистиллированной воде, 3%-ном р-ре поваренной соли или уксус-
уксусной к-ты.
Применение. В строительстве М. применяют для
отделки стен зданий, перил балконов, крыш, водо-
водосточных желобов, внутренней обшивки стен, изготов-
изготовления дверных и оконных рам и др. Из М. изготовляют
корпуса автомобилей, внутреннюю отделку вагонов
и автофургонов. Кроме того, М. применяют для про-
производства корпусов холодильников, стиральных ма-
машин, радиоприемников, телевизоров, тары для хране-
хранения агрессивных материалов и др.
Впервые производство М. было начато в Герма-
Германии в начале 40-х годов 20 в. В 1969 выпуск рулон-
рулонной стали с полимерным покрытием во всех капита-
капиталистических странах^ составил 300 млн. м2; предпо-
предполагается, что в 1974 половина всех изделий из ли-
листовых металлов будет выпускаться с полимерным
покрытием.
Лит.: Полякова К. К., Зельцер Ю. Г., Поли-
Полимерные цокрытия полосового проката, М., 1971; Файн-
штейн В. М., Оборудование для нанесения полимерных по-
покрытий на листовой прокат за рубежом, М., 1973.
А. А. Черников.
МЕТИЛАКРИЛАТА ПОЛИМЕРЫ [poly(methyl ас-
rylate), Polymethylakrylat, polyacrylate de methyle].
Метилакрилат CH2 = CH—COOCH3 (М.).
Свойства. М. — бесцветная прозрачная легко вос-
воспламеняющаяся жидкость; т. кип. 80,2 °С; плот-
плотность 0,9564 г/см3 B0 °С); л? 1, 4040; уд. теплоем-
теплоемкость 2,01 кдж/(кг-К) [0,48 кал/(г-°С)]; теплота испа-
испарения 385,18 пдж/кг (92 кал/г); темп-pa вспышки 10 °С;
пределы взрываемости при 25 °С в смеси с воздухом
2,8—25%. М. хорошо растворим во многих органич.
растворителях (спиртах, кетонах, эфирах, ароматич.
углеводородах и др.); растворимость М. в воде 5,2%,
растворимость воды в М. 2,44% (по массе). М. легко
образует азеотропные смеси с водой и др. гидроксил-
содержащими соединениями. М. токсичен, обладает
наркотическим и общеядовитым действием; пары его
действуют на слизистые оболочки; максимально допус-
допустимая концентрация М. в воздухе 0,001% (по объему).
М. легко присоединяет по двойной связи амины,
галогены и др., вступает в реакцию переэтерификации
с высшими спиртами. М. легко полимеризуется при
комнатной темп-ре, поэтому при хранении к нему
добавляют ингибитор (как правило, гидрохинон или
монометиловый эфир гидрохинона в количестве 0,0015—
0,1%). Без ингибитора М. можно хранить при темп-рах
не выше 10 °С в течение 3—4 недель. Ингибитор из М.
удаляют отмывкой разб. водным р-ром щелочи с после-
последующей перегонкой в атмосфере инертного газа.
М. легко сополимеризуется со всеми типами винило-
виниловых мономеров (таблица).
Константы сополимеризации метилакрилата (rt)
с некоторыми сомономерами (г2)
Сомономер
Акрилонитрил . . .
Винилацетат ....
Винилиденхлорид . .
Винилхлорид ....
Метилметакрилат . .
Метилметакрилат . .
Стирол
Стирол
1,22±0,20
9±2,5
0,84±0,06
5
>1
1,5±0,01
0,4±0,2
0,15±0,05
0,70±0,20
0, 1±0,1
0,99±0,10
0
<1
0,3±0,005
2,2±0,2
0,7±0,1
Темп-ра,
СС
20
60
60
60
20
65
20
60
201
З-МЕТИЛБУТЕНА-1 ПОЛИМЕРЫ
202
Получение. В пром-сти М. получают в основном
из этиленциангидрина A), ацетилена B, 3) и Р-пропио-
лактона D):
160°С ...
HOCH2GH2CN+CH3OH > СН2=СНСООСН3 (I)
H2SO4
4GH=CH+4GH3OH+2H++Ni(GOL->
-> 4GH2=GHGOOGH3+Ni(II)+2H
Тетрагидрофуран
CH===CH-fCO+H2O CH2=GH-COOH
Ni(II)
CH2=CHCOOCH3 (метод Реппе)
GH2—CH2-CO -> [-GHg-G
H2O
сн3он
-> CH2=GHGOOH > СН2=СНСООСН3 D)
н+ v '
Степень чистоты выпускаемого продукта 95—99,9%.
Полиметилакрилат [— СН2— СН — ]п (П.).
СООСНд
Свойства. П.— бесцветный прозрачный полимер.
Степень полимеризации П. в зависимости от способа
получения, количества инициатора и типа раствори-
растворителя колеблется от нескольких сотен до сотен тысяч.
Ниже приведены нек-рые свойства П.:
Плотность при 25°С, г/см3 . . .
25
Показатель преломления п^
Темп-pa стеклования, °С
Темп-pa хрупкости, °С
Уд. теплоемкость,
кдж/(кг • К) [ккал/(кг • °С)]
1,07
1,479
8
4
1,97—2,01
[0,47-0,48]
Прочность при растяжении, Мн/мг (кгс/см2) 7,0 G0,7)
Относительное удлинение, % 75 0
Влагопоглощение за 24 ч, % 18,8
Диэлектрич. проницаемость 5—6
Уд. поверхностное электрич. сопротивле-
сопротивление, Том[ом] 4—5
[D-5). 10"]
Электрич. прочность, кв/мм 28—34
П. растворим во многих органич. растворителях
(ароматич. углеводородах, кетонах, сложных эфирах,
хлоруглеводородах); нерастворим в воде. При нагре-
нагревании выше 200 °С он подвергается деструкции; при
этом выделяется только ~ 1% мономера; П. стано-
становится хрупким (вследствие сшивания).
Получение. В пром-сти получают только атактич.
П. Используют методы радикальной полимеризации
в массе и р-ре, реже — в эмульсии и суспензии.
В качестве инициатора обычно применяют азосоеди-
нения, перекиси, гидроперекиси; возможно также тер-
мич., фото- или радиационное инициирование. Наиболее
приемлемы азосоединения вследствие их высокой эффек-
эффективности; кроме того, они, в отличие от перекисей,
не образуют сшивок. При полимеризации М. в массе
получают р-р П. в метилакрилате. Для полимеризации
М. в массе с добавкой фотосенсибилизатора (диацетила)
при 30 °С константы скорости роста (кр) и обрыва (к0)
равны соответственно 720 и 2,15* 10е л/(моль* сек)',
kp'ko~x = 0,49; энергия активации роста цепи
29,7 кдж/молъ G,1 ккал/молъ).
При полимеризации в массе образуются полимеры
высокой мол. массы, т. к. константа передачи ради-
радикала П. на мономер низка @,7-10~5 — 4,0-10~5
при 60 °С). Для полимеризации М. в р-ре в присутствии
динитрила азоизомасляной к-ты k^-kQ~^ составляет
0,982 (при 44,1 °С) и 1,93 (цри 60 °С); теплота полимери-
полимеризации 78,7 кдж/молъ A8,8 ккал/молъ). П. низкой мол.
массы синтезируют в р-ре, применяя агенты передачи
цепи (ССЦ и др.). Полимеризацией М. в эмульсии
получают П. высокой мол. массы. Реакцию проводят
в присутствии 1—5% неионных или анионных поверх-
поверхностно-активных веществ; инициатором служат пер-
персульфаты или перекись водорода, растворенные в воде.
Изотактич. П. получен в лаборатории анионной поли-
полимеризацией М. при темп-pax от —60 до —80 °С в при-
присутствии и-бутиллития и др. анионных катализаторов.
При комнатной темп-ре и выше атактич. и изотактич.
полимеры аморфны.
Применение. М. применяют для «внутренней»
пластификации жестких пластиков (с этой целью его
сополимеризуют со стиролом, винилхлоридом, вини-
лиденхлоридом, акрилонитрилом, винилацетатом и др.).
П. используют в производстве клеев, лаков, искусст-
искусственной кожи, пленок и листов (при изготовлении без-
безосколочного стекла «триплекс»).
Впервые получили метилакрилат и сообщили о его
полимеризации Каспари и Толленс в 1873.
Лит.: Schildknecht G. E., Vinyl and related poly-
polymers, N. Y., 1952; Riddle E. H., Monomeric acrylic esters,
N. Y., 1954; JoshiR.M., Makromol. Chem., 66, 114 A963);
Mahadevan V., Santhappa M., там же, 16, № 2,
119 A955); M a t h e s о n M. S. [a. o.], J. Am. Ghem. Soc,
73, Jsfi 11, 5395 A951); Chapiro A., Radiation chemistry
of polymeric systems, N. Y., 1962; FurukawaJ., Polymer,
3, № 3, 487 A962); Matsuzaki K. [a. o.], J. Polymer
Sci., pt A—1, 5, № 8, 2167 A967). Б. С. Петрухин,
З-МЕТИЛБУТЕНА-1 ПОЛИМЕРЫ (polymethylbutene,
Polymethylbuten, polymethylbutene).
З-Метилбутен-i CH2 = СН—СН(СН3J (М.) — бес-
бесцветная легко воспламеняющаяся жидкость; темп-ра
кипения 20,06 °С; темп-pa плавления — 168, 50 °С;
плотность 0,630 г/см3 B0 °С); п^5 1,3672; упругость
пара 46,93 кн/м2, или 352,04 мм рт. ст. @ °С);
101,11 кн/м2, или 758,36 мм рт. ст. B0 °С); 438,7 кн/м2,
или 4,387 кгс/см2 G0 °С); ЛЯ°29816 — 28994,8 дж/молъ,
118,79 дж/(молъ-К), или
81в
в
333,94 дж/(молъ-К), или
или —6920 кал/молъ; Ср
28,35 кал/(моль-°С);
79,70 кал/(моль-°С).
М. обладает химич. свойствами, характерными для
алкенов (напр., вступает в реакции присоединения по
двойной связи, в реакции замещения в аллильном поло-
положении, образует комплексы с переходными металлами,
полимеризуется и сополимеризуется с др. а-олефинами
по катионному и координационно-ионному механизмам).
В пром-сти М. может быть выделен фракциониро-
фракционированием пентан-амиленовой фракции (т. кип. до 78 °С)
пиролиза керосина F70—680 °С; 100 кн/м2, или
1 кгс/см2). Во фракции A2—13%), перегоняющейся
в интервале 20—30 °С, содержание М. до 40%.
Смесь изомеров метилбутена с содержанием М. 5%
получают также дегидрированием изопентана при 560—
580 °С и давлении 100 кн/м2, или 1 кгс/см2. Продукты
отделяют от изопентана экстрактивной ректификацией
диметилформамидом.
В лаборатории М. получают дегидратацией соответст-
соответствующего спирта, а также пиролизом B20 °С, безводная
РЬО) изоамилацетата.
Поли-З-метилбутен-1 [— СН2 — СН — ]п (П.) — кри-
СН(СН3J
сталлизующийся (период идентичности 6,84 А, по рент-
геноструктурным данным) термопластичный полимер
белого цвета; т. пл. 240—310 °С, плотность 0,9 г/см9
B0 °С). По основным свойствам П. близок кристаллич.
полиолефинам, напр. полипропилену и полиэтилену,
однако темп-pa эксплуатации его существенно выше
(~ 200 °С). Термич. деструкция П. начинается при
300 °С; при этом образуются углеводороды различного
строения, причем первичным продуктом является,
по-видимому, диизопропилциклопропан. Термоокисли-
Термоокислительная деструкция П. наблюдается уже при темп-рах
ок. 100 °С. Энергия активации 60,8 f 5,1 кдж/молъ
A4,5 ± 1»2 ккал/молъ). Продукты деструкции — гл.
обр. ацетон, а также углеводороды и формальдегид.
Аморфный П. окисляется быстрее кристаллического.
Стабилизаторами служат замещенные фенолы, органич.
фосфиты и др. Кристаллич. полимер, полученный при
203
МЕТИЛВИНИЛПИРИДИНОВЫЕ КАУЧУКИ
204
полимеризации М. под действием А1СЬ в интервале
темп-р от —130 °С до —60 °С (ал кил хлорид в качестве
растворителя), имеет изомерную структуру:
сн3
[-G-GH2-CH2-]n
СНз
П. получают стереоспецифической полимеризацией
М. на катализаторах Циглера — Натта (С2Н5)зА1 —
a-TiClg [соотношение A,5—2) : 1] при 30—80 °С в сре-
среде насыщенного углеводорода; энергия активации
~ 41 дж/молъ (~ 10 ккал/молъ). Схема получения
типична для процессов стереоспецифич. полимериза-
полимеризации а-олефинов (см. Пропилена полимеры, Этилена
полимеры). Полимеры, получаемые при изомеризацион-
ной полимеризации М. в присутствии А1С1з, пока
практич. применения не нашли; интересен сам факт
получения из одного мономера полимеров различного
строения и с различными физич. свойствами.
В лаборатории П. получают полимеризацией сухого
М. в циклогексане в присутствии каталитич. системы
АШз (или тетрадециллитийалюминий) — TiCh. К полу-
полученному П. добавляют спирт, отфильтровывают, про-
промывают спиртом и сушат. Активность катализатора
можно повысить введением олефина С2 или Сз в коли-
количестве менее 1% от М.
Пока П. в промышленном масштабе не производится.
Из П. можно формовать пленки и волокна, ориентирую-
ориентирующиеся при растяжении B50—300 °С). Др. возможные
области использования аналогичны применению поли-
полипропилена, поли-4-метилпентена-1, но темп-ры экс-
эксплуатации П. существенно выше (~ 200 °С). Однако
изделия из П. характеризуются высокой хрупкостью.
Перспективны работы в области синтеза П. с меньшей
степенью кристалличности, сополимеризации М. с др.
а-олефинами, изомеризационной полимеризации М.,
а также в области разработки ударопрочных компози-
композиций на основе П., характеризующихся высокими
физико-механич. свойствами.
Лит.: Natta G., Angew. Ghem., 68, № 12, 393 A956);
Сёренсон У., Кемпбел Т., Препаративные методы
химии полимеров, пер. с англ., М., 1963, с. 254; Б е р д н и-
коваМ. П., КиссинЮ. В., Чирков Н. М., Высоко-
мол. соед., 5, № 1, 63 A963); Quynn R. G., Spra-
q u е В. S., J. Polymer Sci., pt A2, 8, № 11, 1971 A970).
МЕТИЛВИШШПИРИДИНОВЫЕ КАУЧУКИ — см.
Винилпиридиновые каучуки.
МЕТИЛМЕТАКРИЛАТА ПОЛИМЕРЫ [poly(methyl
methacrylate), Polymethylmethakrylat, polymethacry-
late de methyle].
Метилметакрилат (метиловый эфир метакриловой
к-ты) СН2==С(СН3)—СООСНз (М.).
Свойства. М.— бесцветная прозрачная жидкость.
Ниже приведены нек-рые свойства М.:
Плотность при 20°С, г/см3 0,9430
Показатель преломления п-р 1,4146
Вязкость при 20°С, мн• сек/м2, или спз . . 0,6322
Темп-pa плавления, °С —48,2
Темп-pa кипения, °С 100,6
Уд. теплоемкость при 20°С,
кдж/(кг.К)[кал/(г.°С)] 1,88±0,008
[0,45±0,002]
Скрытая теплота испарения,
кдж/молъ(ккал/молъ) 38,2 (9,1)
Теплота полимеризации,
кдж/молъ (кпал/молъ) 54,6 A3,0)
Темп-pa воспламенения, °С 10
Пределы взрывоопасных концентраций в
смесях с воздухом при 25°С, % (по объе-
объему) 2,12-12,5
М. неограниченно растворим в обычных органич.
растворителях, кроме этиленгликоля и глицерина.
Растворимость М. в 100 г воды 1,72 ± 0,06 г A0 °С),
1,59 4-0,06 г B0 °С); растворимость воды в 100 г М.
0,99 ± 0,001 г A0 °С), 0,15 ± 0,02 г B0 °С). М> обра-
зует азеотропные смеси с водой и метиловым спиртом,
кипящие соответственно при 83 °С A4% воды) и при
64,2 °С (84,5% метанола). М. обладает общеядовитым
и наркотич. действием; пары его раздражают слизистые
оболочки глаз; предельно допустимая концентрация
паров в воздухе 0,05 мг/л.
Химич. свойства М. обусловлены наличием сложно-
эфирной группы и этиленовой связи, сопряженной
с карбоксильной группой. М. гидролизуется как
в кислой, так и в щелочной среде с образованием
соответственно метакриловой к-ты и ее соли. При
нагревании М. с органич. к-тами (предпочтительно
с муравьиной) в присутствии нелетучих минеральных
к-т образуется метакриловая к-та и метиловый эфир
органич. к-ты. При нагревании М. с каким-либо безвод-
безводным спиртом, взятым в избытке, в присутствии силь-
сильных к-т (напр., серной либо ароматич. сульфокислоты)
или алкоголятов происходит переэтерификация. Этот
процесс используют в технике для получения ряда др.
эфиров метакриловой к-ты.
По двойной связи М. количественно присоединяются
водород (катализатор — палладий на поливиниловом
спирте), хлор, бромистый водород, хлорноватистая
к-та, соли двухвалентной ртути (не количественно).
В присутствии сильных оснований (напр., алкоголя-
алкоголятов или гидроокиси триметилбензиламмония) к М.
присоединяются многие соединения с подвижным ато-
атомом водорода, напр, меркаптаны, тиофенолы, первич-
первичные и вторичные алифатич. нитросоединения (в аци-
форме), цианистый водород (в присутствии цианистого
калия), производные р-кетокислот. М. может быть
использован в качестве диенофила в диеновом синтезе
циклич. соединений. М. легко полимеризуется и сопо-
лимеризуется (табл. 1) под действием свободных ра-
радикалов и анионных катализаторов.
Таблица 1. Константы сополимеризации
метилметакрилата (гг) с различными мономерами (г2)
Мономер
Акриловая к-та
Акрилонитрил
Бутадиен
Винилацетат
Винилиденхлорид
Винилхлорид
Метилакрилат
Стирол
1,25
1,35
0,25
20,0
2,53
10,0
0,3
0,46
0,225
0,18
0,75
0,015
0,24
0,1
1 ,5
0,52
Темп-ра,
°С
70
60
90
60
60
70
65
60
Получение. М. может быть получен этерифика-
цией метакриловой к-ты, дегидрогалогенированием
метиловых эфиров а- или Р-галогенизомасляной к-ты,
дегидратацией эфиров а- или Р-оксиизомасляной к-ты,
при действии метанола и фосфорного ангидрида на без-
безводную а-оксиизомасляную к-ту в среде сухого хлоро-
хлороформа. В большинстве европейских стран (в том числе
в СССР) М. получают в пром-сти из а-оксиизобутиро-
нитрила (ацетонциангидрина) — продукта взаимодейст-
взаимодействия ацетона и синильной к-ты:
О
60-90°С / 120-130°С
(GH3JG(OH)-GN + H2SO4 > (GH3JG С
I
OSO2OH
NH2
• СН2=С(СН3)-С
\
Н2О, СНзОН;
NH2-H2SO4 ~80°C
-* GH2=G(GH3)-GOOGH3+NH4HSO4
Образующийся мономер отгоняют с водяным паром,
промывают и ректифицируют. Выход ок. 90%. Основ-
Основные примеси: вода, метанол, метакриловая к-та, метило-
метиловый эфир а-оксиизомасляной к-ты. Процесс м. б.
осуществлен как периодически, так и непрерывно.
205
МЕТИЛМЕТАКРИЛАТА ПОЛИМЕРЫ
206
В др. варианте синтеза М. из ацетонциангидрина
(США) при дегидратации вместо метакриламида полу-
получают метакрилонитрил, при взаимодействии к-рого
с метанолом в присутствии серной к-ты образуется М.
(подобно синтезу акрилатов из акрилонитрила); выход
83% по ацетону. Для промышленного синтеза М. можно
использовать также метакрилонитрил, получаемый
окислительным аммонолизом изобутилена.
В США разработан промышленный способ получения
М. в две стадии. Изобутилен или смесь его с бутанами
окисляют четырехокисью азота в жидкой фазе при
0_5 °С до а-оксиизомасляной к-ты (выход 80—86%
по изобутилену):
N2O4
СН2=С(СН3J > [ONCH2-C(CH3J] -> НООС-С(СН3J.
I
ONO2
он
При окислении образуется также нек-рое количество
ацетона и уксусной к-ты. После обработки реакционной
массы насыщенным р-ром мочевины или сульфаминовой
к-ты (для удаления следов азотистых соединений)
а-оксиизомасляную к-ту подвергают одновременно
дегидратации (H2SO4) и этерификации (метиловый
спирт в дихлорэтане) при темп-ре не выше 50 °С.
В Японии и ряде других стран разрабатывается
процесс получения М. путем окисления изобутилена
в газовой фазе кислородом воздуха на твердом ката-
катализаторе.
Выпускаемый продукт должен содержать основного
вещества 99,8—99,9%. Допустимое содержание при-
примесей: воды 0,05% , метакриловой к-ты 0,005% и осталь-
остальных (ацетона, метанола метил акрил ата, метил-а-окси-
изобутирата) не более 0,15%. Степень чистоты М.
определяют газовой хроматографией, химич. метода-
методами — содержание воды (по методу Фишера), к-ты
(нейтрализацией), перекисных соединений (иодомет-
рич. методом) и наличие двойной связи (каталитич.
гидрированием или бромированием бромид-броматной
смесью).
Для предотвращения полимеризации в процессе
синтеза и при хранении М. ингибируют, чаще всего
гидрохиноном @,01—0,05% по массе) или его моно-
монометиловым эфиром. Содержание в М. ингибитора опре-
определяют методом колориметрии или полярографии. Мо-
Мономер перед полимеризацией обычно освобождают
от ингибитора промывкой сначала 2—5%-ным щелоч-
щелочным р-ром (в случае гидрохинона), а затем водой
с последующим высушиванием или ректификацией.
Для воспроизводимости результатов по полимериза-
полимеризации необходима тщательная очистка мономера; не со-
содержащий примесей М. в отсутствие воздуха и действия
света достаточно устойчив.
Полиметилметакрилат ([—СН2 — С (СНз) —]П(П.) —
СООСНд
линейный термопластичный полимер.
Свойства. В зависимости от условий полимериза-
полимеризации П. может быть атактическим, синдиотактиче-
ским, изотактическим, а также стереоблоксополимером
изо- и синдиоструктуры. Получаемый в пром-сти П.—
аморфный атактич. полимер, в макромолекулах к-рого
ок. 80% мономерных звеньев входит в синдиотактич.
последовательности [образование синдиотактич. струк-
структуры характеризуется меньшей энергией активации, чем
изотактической; разница в значениях энтальпии акти-
активации составляет ок. 4,19 кдж/молъ (ок. 1 ккал/молъ)].
Мол. масса П. может достигать нескольких млн.
(для полимера, полученного в массе под действием
УФ-света); плотность 1,19 г/сж3, показатель прелом-
преломления п р 1,492. Зависимость между мол. массой
М и характеристич. вязкостью [г\] выражается ур-нием
1ц] = К-Ма (табл. 2).
Таблица 2. Значения констант в уравнении [г{\=^К-М
Метод определения
мол. массы
Светорассеяние
Светорассеяние
Светорассеяние
Осмометрия
Осмометрия
Осмометрия
Седиментация
Растворитель
и темп-ра
Метилэтилкетон B0°С)
Хлороформ B0°С)
Толуол B0°С)
Бензол B5°С)
Хлороформ B0°С)
Ацетон C 0°С)
Бензол B0°С)
К
0,68-10-4
0,48-10-4
0,71 -Ю-4
0,75-10-4
0,63-10-4
0,77 -Ю-4
0,84-10-4
а
ос
0,72
0,80
0,73
0,76
0,80
0,70
0,73
П. растворяется в собственном мономере и др. слож-
сложных эфирах, ароматич. и галогензамещенных углево-
углеводородах, кетонах, муравьиной и ледяной уксусной
к-тах, образуя очень вязкие р-ры (вязкость 10%-ного
р-ра блочного П. в органич. растворителе 105—
106 мн-сек/м2, или спз). П. не растворим в воде, спир-
спиртах, алифатич. углеводородах и простых эфирах;
устойчив к действию разб. щелочей и к-т. Для полного
омыления водным р-ром щелочи П. необходимо нагреть
до темп-ры не ниже 200 °С (заметный гидролиз начи-
начинается только при 160 °С). Конц. серной к-той при
25 °С за 6 ч П. гидролизуется на 52%, при 75 °С менее
чем за 1 ч — полностью. Он подвергается ацидолизу
водным р-ром уксусной к-ты в присутствии /г-толуол-
сульфокислоты с образованием полиметакриловой
к-ты и метилацетата. П. физиологически безвреден
и стоек к биологич. средам.
П., получаемый радикальной полимеризацией в массе
(т. наз. органическое стекло), — бесцветный прозрачный
полимер, обладающий высокой проницаемостью для
лучей видимого и УФ-света, высокой атмосферостой-
костью, хорошими физико-механич. и электроизоля-
электроизоляционными свойствами.
При нагревании выше 120 °С П. размягчается, пере-
переходит в высокоэластич. состояние и легко формуется;
выше 200 °С начинается заметная деполимеризация П.,
к-рая с достаточно высокой скоростью протекает при
темп-pax выше 300 °С. Практически количественно
П. может быть деполимеризован при 300—400 °С в ва-
вакууме F6,7—266,6 н/м2, или 0,5—2 мм рт. ст.).
В пром-сти деполимеризацией • отходов П. получают
мономер.
Стереорегулярные П.— кристаллизующиеся поли-
полимеры с более высокой плотностью (табл. 3) и повы-
повышенной стойкостью к действию растворителей, чем
атактические П.
Таблица 3. Нек-рые свойства стереорегулярных
полиметилметакрилатов, полученных в присутствии
литийорганических катализаторов
Полимер
Синдиотактический . . .
Изотактический
Стереоблоксополимер . .
Темп-ра
стеклова-
стеклования, °С
115
45
60—95
Темп-ра
плавле-
плавления, °С
200
160
170
Плотность
при 30°С (для
аморфизованно-
го полимера),
г/см3
1,19
1 ,22
1,20-1,22
Изотактич. П. кристаллизуется легче синдиотакти-
ческого. Кристалличность дополнительно повышают
термообработкой или набуханием полимера в ксилоле,
диэтиловом эфире, метаноле или гептаноне-4. Стерео-
блоксополимеры характеризуются низкой степенью
кристалличности; при термообработке или набухании
они полностью аморфизуются. Химич. поведение раз-
различных стереорегулярных модификаций П. также раз-
различно. Напр., скорость щелочного гидролиза снижает-
снижается в ряду: изотактический > стереоблочный > син-
синдиотактический.
Получение. В пром-сти П. получают преиму-
преимущественно свободнорадикальной полимеризацией М.
207
МЕТИЛМЕТАКРИЛАТА ПОЛИМЕРЫ
208
при умеренных темп-pax. Инициаторами служат орга-
нич. и неорганич. перекиси (перекиси бензоила, лау-
рила, персульфат калия), азо-бис-изобутиронитрил
и нек-рые окислительно-восстановительные системы
(напр., перекись бензоила с третичными аминами или
с диметиланилином, персульфат аммония с гидросуль-
гидросульфитом натрия). Полимеризацию М. можно осуществ-
осуществлять в массе (блоке), суспензии, эмульсии или р-ре.
В пром-сти наибольшее распространение получили
блочный и суспензионный методы. При блочной поли-
полимеризации конфигурация полимеризационной формы
предопределяет конфигурацию получаемого П. (под-
(подробно об этом методе см. Органическое стекло).
Суспензионную («бисерную») полимеризацию М. осу-
осуществляют в водной среде в реакторах, снабженных
лопастными или турбинными мешалками. Получаемый
полимер имеет вид прозрачных шариков, размеры
к-рых (от 1-10~3 до нескольких мм) зависят от интен-
интенсивности перемешивания, природы и количества ста-
стабилизатора суспензии. В качестве стабилизаторов
суспензии используют желатину, водорастворимый
крахмал, соли полиакриловой и полиметакриловой
к-т, полиметакриламид, поливиниловый спирт, а также
неорганич. порошкообразные диспергаторы (напр.,
каолин, осажденный карбонат магния, гидроокись
алюминия). Инициаторами служат растворимые в мо-
мономере перекиси (гл. обр. пе.рекись бензоила). Средняя
мол. масса (80 000—120 000) суспензионного П. ниже,
чем у блочного. Для снижения мол. массы П. исполь-
используют растворимые в мономере алифатич. меркаптаны
(регуляторы мол. массы). Суспензионным способом
чаще всего получают сополимеры М. с небольшим коли-
количеством (менее 10%) низших акриловых эфиров, сти-
стирола или винил ацетата, а также П., пластифициро-
пластифицированный дибутилфталатом. Полученные продукты раз-
различаются по темп-рам размягчения и вязкостям рас-
расплава.
Константы скорости роста (/ср) и обрыва (к0 • 10~7)
цепи при радикальной полимеризации М. в массе состав-
составляют соответственно [л/(моль-сек)]: 0 °С— 41,6 и 0,26;
23,6 °С — 310 и 6,6; 30 °С — 143 и 0,61; 35,9 °С — 410
и 6,8; 50,5 °С — 580 и 6,9. Обрыв цепи происходит
как диспропорционированием, так и рекомбинацией.
Соотношения &диспр/^рекомб составляют 1,5 @ °С),
2,13 B5 °С), 5,75 F0 °С). Добавление в реакционную
систему комплексообразователей типа ZnCl2, А1С1з
и др. приводит к повышению /ср и резкому снижению
к0. Этот прием м. б. использован для управления про-
процессами радикальной полимеризации и сополимериза-
ции М.
При глубине превращения ок. 20% наступает сущест-
существенное возрастание скорости полимеризации вследствие
гелъ-эффекта. В процессе полимеризации в массе при
достижении гель-точки значительно возрастает вяз-
вязкость, резко повышается темп-pa и при плохом отводе
тепла может произойти взрыв. Поэтому при полимери-
полимеризации в массе требуется строго соблюдать темп-рный
режим процесса. При суспензионной полимеризации
обеспечивается хороший отвод тепла.
Полимеризацией и сополимеризацией М. в эмульсии
и р-ре получают композиции, используемые для при-
приготовления лаков (см. Полиакриловые лаки и эмали)
и в качестве пропитывающих составов. Поскольку
эмульсионный П. обладает пленкообразующими свойст-
свойствами только при содержании 40—50% дибутилфталата,
акриловые латексы либо содержат пластификатор,
либо чаще всего представляют собой дисперсии сопо-
сополимеров М. с этил- или бутилакрилатом и небольшим
количеством метакриловой к-ты. Эмульсионную поли-
полимеризацию проводят обычно в присутствии водораство-
водорастворимых перекисей или окислительно-восстановительных
инициаторов (напр., персульфата аммония и гидро-
гидросульфита натрия). В зависимости от требований, предъ-
предъявляемых к получаемой дисперсии, в качестве эмуль-
эмульгатора используют сульфаты жирных спиртов, алкил-
или арилсульфонаты, соли жирных к-т, продукты
конденсации алкилфенолов с окисью этилена и др.;
регуляторами мол. массы служат меркаптаны. Для
полимеризации М. в р-ре используют этилацетат, то-
толуол, ксилол и ряд др. растворителей. Значение мол.
массы П. зависит от типа растворителя, концентрацийМ.
и инициатора, а также от темп-ры реакции. Константы
передачи цепи на нек-рые растворители в процессе
полимеризации М. при 80 °С приведены ниже:
Бензол 0,075-Ю Хлороформ. ... 1,4-10—*
Циклогексанон 0,10-10~4 Четыреххлористый
Толуол 0,52-10-4 углерод .... 2,39-10~4
Этилбензол . . . 1,35-Ю-4 Тетрабромметан 3300-Ю
Изопропилбензол 1,90 • 10~4 Хлорбензол ... 0 ,2 • 10~4
Любым из рассмотренных выше методов получают
сополимеры М. с акрилатами, метакрилатами или др.
виниловыми мономерами (стиролом, а-метилстиролом,
акрилонитрилом, винилацетатом). Так, сополимериза-
сополимеризацией М. и акрилонитрила в массе получают сополимер,
отличающийся высокой ударной вязкостью и эластич-
эластичностью (см. Акрилонитрила сополимеры).
Сополимеризацией М. с небольшими количествами
полифункциональных соединений (напр., с гликольди-
метакрилатом, ангидридом метакриловой к-ты, триал-
лилциануратом и др.) получают сшитый полимер,
к-рый не растворяется в обычных растворителях
и не размягчается при темп-pax до 200 °С.
Синдиотактич. кристаллизующийся П. получают
полимеризацией М.: а) в присутствии инициаторов сво-
боднорадикального типа (УФ-свет, бензоин) в поляр-
полярных растворителях или в массе при низких темп-рах
(напр., при —40 °С); б) в присутствии анионных
катализаторов (алкоголятов или металлорганич. сое-
соединений щелочных металлов, соединений Гриньяра)
в сильнополярных средах (напр., в тетрагидрофуране,
гликольдиметиловом эфире, аммиаке) при темп-рах
ок. —60 °С. Изотактич. П. получают анионной поли-
полимеризацией М. в неполярных растворителях (в толуоле,
гексане, гептане или др.), напр, в присутствии 9-флуо-
рениллития или бутиллития как катализатора в толуо-
толуоле в темп-рном интервале от —60 до —70 °С либо
в присутствии реактива Гриньяра в толуоле с добавкой
бромистого магния при 0 °С. В смесях полярного и
неполярного растворителей образуются стереоблоксо-
полимеры.
Применение и переработка. Пром-стью П.
поставляется гл. обр. в виде листового органического
стекла. В качестве конструкционного материала П.
применяют в лазерной технике.
Суспензионный П., получаемый в виде порошка,
предварительно гранулируется на экструзионных ма-
машинах. Гранулированный Д. перерабатывают прессо-
прессованием, литьем под давлением или экструзией. Суспен-
Суспензионные полимеры используют в автомобильной
пром-сти (задние фонари, подфарники, шкалы, свето-
световые отражатели и др.), в приборостроении (линзы,
призмы, шкалы), для изготовления изделий широкого
потребления (посуда, пуговицы и др.) и канцелярских
принадлежностей. Экструдированные из суспензионных
полимеров и сополимеров листы используются для
изготовления светотехнич. изделий (напр., рассеива-
телей света для светильников), вывесок и т. п.
Суспензионный П. с размером частиц 0,05—0,15 мм
или высушенный эмульсионный П. применяют для
изготовления самоотверждающихся пластмасс E5—
60% П., 35—40% мономера, содержащего инициатор,
с добавкой красителя). Эти пластмассы используются
в произ-ве зубных протезов, для изготовления штампов,
литейных моделей, абразивного инструмента.
Акриловые дисперсии и полимеры, полученные
в р-ре, используются как лаки для кузовов автомо-
209
4-МЕТИЛПЕНТЕНА-1 ПОЛИМЕРЫ
210
билей, для отделки тканей, волокон, бумаги, кож и
т. д. В качестве клея для склеивания органического
стекла используют мономерно-полимерную смесь или
20—30%-ные р-ры П. (см. Полиакриловые клеи).
За рубежом блочный П. производится под названия-
названиями: плексиглас (США, ФРГ, Франция), пер с пек с
(Великобритания), к л а р е к с (Япония), леофлекс
(Швейцария) и др.; суспензионный — люсайт (США),
диакон (Великобритания), плексигум (ФРГ),
вед рил (Италия); сополимер М. с акрилонитрилом —
плексидур (ФРГ), имплекс (США).
Стереорегулярные полимеры М. не нашли пока
широкого практич. применения, хотя в Англии и США
выпускается изотактич. П. под названием к р и -
с т а к р и л, к-рый отличается от обычного П. высо-
высокой ударной вязкостью C0—40 кдж/м2у или тс» см/см2)
и устойчивостью к действию растворителей.
Лит.: Rauch-Puntigam H., Volker Th.,
Acryl- und Methacrylverbindungen, В.— [u. a.], 1967 (Chemie,
Physik und Technologie der Kunststoffe in Einzeldarstellungen,
Bd9); МарекО., ТомкаМ., Акриловые полимеры, пер.
с чеш., М., 1966; Encyclopedia of polymer science and techno-
technology, v. 1, N. Y.— [a. o.], 1964; H и к о л а е в А. Ф., Синте-
Синтетические полимеры и пластические массы на их основе, 2 изд.,
М.— Л., 1966; Хувинк Р., Ставерман А. [сост.],
Химия и технология полимеров, пер. с нем., т. 2, М.— Л.,
1965; Modern plastics, 46, № 5, 20—21 A969); № 9, 25 A969);
Дебский В., Полиметилметакрилат, пер. с польск., М.,
1972. Е. М. Лукина.
4-МЕТИЛПЕНТЕНА-1 ПОЛИМЕРЫ (polymethyl-
pentene, Polymethylpenten, polymethylpentene).
4-Метилпентен-1 CH2=CH—CH2—CH(CH3J (М.) —
бесцветная легковоспламеняющаяся жидкость; т.
кип. 53,6—53,9 °С, плотность 0,66370 г/см* B0 °С),
и^ 1,38267, темп-pa плавления —156,63 °С, давление
насыщенных паров (в кн/м2, или в мм рт. ст.): 29,47,
или 221,03, B0 °С), 43,88, или 329,15, C0 °С), 89,34,
или 670,13, E0 °С), АН°298Дв - 48855,5 дж/молъ
(-11660 кал/моль), С°рш 16126,'б7 дж/{молъ-К) [30,23
кал/(молъ-°С)], S°298le368,26 дж/(моль-К) [87,89
кал/(молъ-°С)], вязкость при 20 °С 0,288 мн-сек/м2,
или спз соответственно.
М. обладает химич. свойствами, характерными для
алкенов: вступает в реакции присоединения по двой-
двойной связи, замещения в аллильном положении, ком-
плексообразования с переходными металлами, окисле-
окисления, полимеризации и сополимеризации с др. а-олефи-
нами и т. д.
М. получают димеризацией пропилена при темп-рах
150—200 °СТ давлении 3—10 Мн/м2 C0—100 кгс/см2)
и большом времени контакта. В качестве катализатора
используют дисперсии щелочных металлов (Na, К,
Rb, Cs) в углеводородных средах [белое масло, содер-
содержащее 0,2% (по массе) олеиновой к-ты]. Для умень-
уменьшения индукционного периода к катализатору часто
добавляют в качестве промоторов алкилфенолы, эфиры,
алкилгалогениды.
В результате димеризации пропилена непрерывным
способом при 140 °С, давлении 10 Мн/м2 A00 кгс/см2)
и объемной скорости 0,7 ч на катализаторе Na/K2CO3
образуется смесь различных соединений, содержащая
85% 4-метилпентена-1. М. выделяют ректификацией
(на колонне со 100 теоретич. тарелками получают
дистиллат, содержащий 99,9% М.). Допустимое содер-
содержание примесей в М. (не выше): вода 0,001%, ацети-
ацетиленовые + аллены 0,02%, О2 0,0005 % (по массе).
Поли-4-метилпентен-1 [— СН2—СН — ]п (И.).
I
СН2СН(СН3J
Свойства. П.— прозрачный кристаллизующийся
термопластичный полимер (конформация цепи в кри-
кристаллическом состоянии 72-спираль; период идентич-
идентичности 1,385 нм, или 13,85 А, по рентгеноструктурным
данным). П. устойчив к действию F мес, 20 °С) воды,
насыщенных р-ров солей, конц. соляной и серной к-т,
разб. азотной, хромовой и уксусной к-т, разб. р-ров
щелочей, а также масел, спиртов, фенолов, диалкил-
фталатов и др. Не устойчив к воздействию бензола,
хлорбензола и многих др. ароматич. углеводородов,
алкилацетатов, четыреххлористого углерода и др. ве-
веществ. П. более прозрачен и термостоек, чем полиэти-
полиэтилен и полипропилен. При длительном нагревании
выше 280 °С П. подвергается деструкции, сопровождаю-
сопровождающейся резким снижением мол. массы (в 2,5—3 раза
за 8 ч). П. легко окисляется, поэтому его" не рекомен-
рекомендуют для эксплуатации на воздухе; в полученный П.
обязательно вводят стабилизаторы, напр, алкилзаме-
щенные фенолы или диоксидифенилсульфиды, органич.
фосфиты, азометины.
П. характеризуется хорошими физико-механич. и
электроизоляционными свойствами. Ниже приведены
нек-рые его свойства:
Плотность при 20°С, г/см3 0,83
Насыпная масса, г/л 200—340
Прозрачность, % 90
Темп-pa плавления кристаллов, СС . . . . 230—240
Прочность при растяжении со скоростью
50%/лшн, Мн/м2 (кгс/см2)
—20°С выше 50 E00)
20сС 28 B80)
100°С 7 G0)
Относительное удлинение,% 5—15
Модуль упругости при 0,2%-ном удлине-
удлинении B0°С), Мн/м2 (кгс/см2) 1000—1400
A0 000 — 14 000)
Твердость по Роквеллу 67—74
Теплостойкость по Вика, °С 160—180
Темп-рный коэфф. линейного расшире-
расширения, °С-1 11,7-10-*
Теп лопрово дност ь
вт/(м-К) 0,167
кал/(см ¦ сек • °С) 4,0-10-*
Уд. теплоемкость
кдж/(кг -К) 2,18
кал/(г-0С) 0,52
Водопоглощение за 24 ч, % 0,01
Усадка в форме, % 1—3
Диэлектрич. проницаемость при 25°С и
102—10е гц 2,12
Уд. объемное электрич. сопротивление,
Том'М (ом'См) более 102
(более 101в)
Газо- и паропроницаемость у П. несколько выше,
чем у полипропилена и полиэтилена.
Получение. В пром-сти П. получают полиме-
полимеризацией М. в присутствии катализаторов Циглера —
Натта. Реакцию осуществляют в w-гептане или др.
насыщенных углеводородах при темп-ре до 80 °С в те-
течение 2—5 ч. Схема получения типична для этих про-
процессов (см. Пропилена полимеры, Этилена полимеры).
П. получают в виде порошка белого цвета. В пром-сти
мол. массу получаемого П. регулируют введением в ре-
реакционную систему агентов передачи цепи (напр., Н2).
В лаборатории П. получают полимеризацией сухого
М. в циклогексановом р-ре в инертной атмосфере. Ка-
Катализатором служит АШз (или тетрадециллитий-алю-
миний) — TiCl4. К полученному П. добавляют спирт,
отфильтровывают, промывают спиртом и сушат.
При сополимеризации М. в сложных смесях метил -
пентенов, метил буте нов или с изобутиленом под дейст-
действием кислотных катализаторов (А1СЬ) получают жид-
жидкие или вязкотекучие продукты неопределенного строе-
строения, используемые в качестве специальных электро-
электроизоляторов или добавок к смазочным маслам (темп-ра
полимеризации от —20 до —60 °С). Сополимеры М.
с w-гексеном отличаются ударопрочностью и эластич-
эластичностью. При сополимеризации М. с 10—25% гексена-1
или с др. линейными а-олефинами образуются плавя-
плавящиеся при высокой темп-ре продукты, причем темп-ра
плавления зависит от содержания сомономера, напр,
при его содержании в П. 10—12% темп-ра плавления
сополимера 200 °С. Сополимеры хорошо растворимы
в циклогексане. хлороформе, хлорбензоле и т. п.,
211
МЕТИЛСТЙРбЛОВ ПОЛИМЕРЫ
212
в к-рых растворимость гомополимера П. незначительна.
При введении в сополимеризацию дивинил бензол а
образуется тройной сополимер, растворимый при обыч-
обычной темп-ре в перечисленных выше растворителях.
При нагревании B25 °С) он легко структурируется
с образованием нерастворимого полимера.
Переработка и применение. П. легко перера-
перерабатывается всеми известными для термопластов метода-
методами (экструзия, литье под давлением и др.). Оптималь-
Оптимальная область темп-р переработки 260—320 °С: прессова-
прессование н экструзию осуществляют при 260—270 °С (дав-
(давление в головке 2—3 Мн/м2, или 20—30 кгс/см2),
литье — при 260—300 °С. Особенность переработки
П.— высокая темп-pa переработки, но и резкая за-
зависимость вязкости расплава от темп-ры.
П. применяется для изготовления медицинского
мерного оборудования, способного переносить неодно-
неоднократную стерилизацию до 160 °С; печатных схем
в электро- и радиотехнике; осветительной арматуры
в автомобильной пром-сти; изделий светотехники (пла-
(плафоны, светильники); различных емкостей. Перспек-
Перспективны работы в области совершенствования производст-
производства М. и П., сополимеризации М. с др. а-олефинами
и разработки композиций, характеризующихся стой-
стойкостью к химич. и энергетич. воздействиям.
П. производится в Великобритании с 1968 под тор-
торговым названием TRX в количестве 2—3 тыс. т в год.
В ближайшие годы производство П. должно возрасти.
Лит.: PenfoldR.C, Brit. Plast., 38, № 4, 213 A965);
Hambling J. К., N о r t h с о t t R. P., Rubb. Plast.
Age, 49, № 3, 224, 225, 227, 170, 171, 173 A968); Пластмассы,
№ 10, 70 A968); V i s s e г P. J., Metaal en Kunststof, 6, № 31,
1438 A968); Burfield D. R., McKenzie I. D.,
Та it P. J. Т., Polymer, 13, № 7, 302, 307, 315, 321 A972).
К. С Muncnep.
МЕТИЛСТИРОЛОВ ПОЛИМЕРЫ — см. Стирола
производных полимеры.
МЕТИЛЦЕЛЛЮЛОЗА (methyl cellulose, Methyl-
zellulose, methylcellulose) — простой метиловый эфир
целлюлозы [СбН7О2(ОНK-а;(ОСНз)а;]п.
Физич. свойства. М.— твердое вещество белого цвета
без запаха и вкуса. Предельное содержание метоксиль-
ных групп в макромолекуле М. 45,6%, что соответст-
соответствует у — 300 (у — число замещенных гидроксильных
групп в 100 элементарных звеньях макромолекулы
целлюлозы).
М. со степенью замещения у = 140—
200 (содержание метоксильных групп 24—33%) имеет
наибольшее технич. значение; плотность 1,29—1,31
г/см3 B0 °С); насыпная масса 0,3—0,5 г/см3; плавится
с разложением при 290—305 °С. Водопоглощение М.
при 25 °С и относительной влажности воздуха 50, 75
и 100% составляет соответственно 3—5, И и 40—50%.
М. растворима в холодной воде, бензиловом спирте,
этиленхлоргидрине, пиридине, метилсалицилате, му-
муравьиной, молочной и ледяной уксусной к-тах, анилине,
Вязкость 2%-ного р-ра в смесях низших спиртов с
мети л целлюлозы (I) и ее водой, при нагревании — в
гликолях, глицерине, поли-
гликолях и их эфирах, эта-
этанол амине; нерастворима в
горячей воде. М. совмещает-
совмещается с др. водорастворимыми
эфирами целлюлозы, с при-
природными водорастворимыми
полимерами и поливинило-
поливиниловым спиртом. При набухании
в воде объем М. увеличива-
увеличивается в 40 раз. Макромолеку-
Макромолекулы М. даже в разб. вод-
водных р-рах склонны к силь-
сильной агрегации. Водные р-ры
М. стабильны при рН 2—12;
* При 20°с. их охлаждение способству-
характеристическая
вязкость в воде (II) при
различной степени полим
ризации продукта
Степень
полиме-
полимеризации
70
НО
140
220
340
460
580
650
750
I*
мн • сек/м2,
или спз
10
40
100
400
1500
4000
8000
15000
19000
II*,
дл/г
1,40
2,05
2,65
3,90
5,70
7,50
9,30
11 ,00
12,00
ет повышению раство-
растворимости М. Конц. р-ры
М. псевдопластичны и
почти не обладают тиксо-
тропными свойствами.
Примерные соотношения
между вязкостью водных
р-ров М. и степенью по-
полимеризации приведены
в таблице.
Зависимость между ха-
рактеристич. вязкостью
[ц] в дл/г водных р-ров
М. при 20 °С и ее мол.
массой М выражается
соотношением: [ц] =
2,8-10-3М°>63, которое
справедливо для мол.
масс 20 000—300 000.
Водные р-ры М.,в от-
отличие от р-ров др.водо-
Зависимость вязкости вод-
водных р-ров метилцеллюлозы
различной концентрации от
темп-ры.
5000
4000
3000
2000
0 10 20 30 40 50 60 70
Температура. °С
растворимых полимеров, при нагревании в интервале
35—56 °С желатинизируются; при понижении темп-ры
ниже темп-ры желатинизации гель разрушается. Чем
выше концентрация р-ра и степень полимеризации поли-
полимера, тем ниже темп-pa желатинизации (см. рис.). До-
Добавка мочевины к водным р-рам М. повышает теми-ру
желатинизации, добавка же определенных количеств
солей, а также танина и фенолов приводит к вы-
выделению М. из р-ра и понижению темп-ры желатини-
желатинизации.
В водных р-рах М. обладает хорошими поверхностно-
активными свойствами. Ниже приведены свойства вод-
водных р-ров М., содержащей 30% метоксильных групп:
Плотность при 20 °С, г/см3
1%-ный р-р
5%-ный р-р
10%-ный р-р
Показатель преломления
1 ,0012
1,0017
1,0245
1 ,336
B%-ный р-р) nD
Парциальный уд. объем*, см3/г
A сж»/г=10-3 м*/кг) 0,70-0,74
Уд. теплоемкость A0—25%-ный р-р, 20—
90°С), кдж/(кг-К)[кал/(г-°С)] 3,9±0,2
[0,93±0,05]
Поверхностное натяжение B5°С, 0,001 —
1%-ный р-р), мн/м, или дин/см
Межфазное натяжение (парафиновое масло,
25°С, 0,001 —1%-ный р-р), мн/м, или
дин/см ,
рН
Темп-pa замерзания B%-ный р-р), °С . .
19-23
47-58
5-7
0
* Парциальный уд. объем зависит от содержания мето-
метоксильных групп: i;=0,560+0,00 5 42-[OCH3]; [ОСН3] в %.
Свойства непластифицированных пленок из водорас-
водорастворимой М.:
Плотность при 20°С, г/см3 1,39
Показатель преломления п^ 1,49
Прочность при растяжении B4°С, относи-
относительная влажность воздуха 5 0%), Мн/м2
(кгс/см2) 60-80
F00-800)
Относительное удлинение B4°С, относитель-
относительная влажность воздуха 50%), % 10 — 15
Число двойных изгибов пленки толщиной 2 мм
до разрушения
с нагрузкой 10 нA тс) 12 000
без нагрузки 32 000
Проницаемость для водяных паров C7,8°С,
относительная влажность воздуха 90—
100%) за 24 чу кг • м/м2(г • мм/см2) . . . . 1,04 • 10~3( 0,104)
213
МЕТИЛЦЕЛЛЮЛОЗА
214
Проницаемость для кислорода B4°С) за
24 ч, м*-м/м2 (см3-мм/см*) 0,4 • 10~в @,04)
Пропускание УФ-лучей пленкой толщи-
толщиной 2 мм при различных длинах волн, %
400 мкм • . 54,6
290 мкм 49,0
210 мкм 25,7
Введение в макромолекулу М. этоксильных, окси-
этильных и оксипропильных групп повышает ее термо-
термопластичность, темп-ру же латинизации водных р-ров,
совместимость с органич. растворителями и солями.
М.— легковоспламеняющееся и взрывоопасное ве-
вещество; темп-pa воспламенения 360 °С, нижний предел
взрываемости 30 г/ж3.
М. фракционируют из водных р-ров дробным осаж-
осаждением насыщенными р-рами солей (напр., Na2SO4)
и ацетоном, а также дробным растворением в системах
вода — спирт, вода — ацетон.
М. со степенью замещения у — 10—110
(содержание метоксильных групп 2—19%) растворима
в 2—8%-ных р-рах едкого натра, нерастворима в воде
и органич. растворителях. Прочность при растяжении
пленок из такой М. 72—114 Мн/м2 G20—1140 кгс/см2)\
относительное удлинение 18—20%. Степень полиме-
полимеризации для указанной М. может быть определена
по ф-ле Штаудингера г)уД = КтР, где Р — степень
полимеризации; г)уд — уд. вязкость р-ра; Кт — по-
постоянная, равная 7-Ю для щелочных р-ров и
11-10 для ацетоновых р-ров полностью пронитро-
ванной М.
М. со степенью замещения у = 240—
280 (содержание метоксильных групп 37—43%) раство-
растворима в полярных органич. растворителях и смесях
спиртов с ароматич. углеводородами, нерастворима
в р-рах щелочей; частично набухает в воде.
Метилоксипропилцеллюлоза — твер-
твердое вещество белого цвета, по физич. и химич.
свойствам близкое к М. Промышленный продукт
выпускается с у= 140—170 по метоксильным и у =
= 10—25 по оксипропильным группам; т. пл. 240—
260 °С; темп-pa желатинизации водных р-ров 65—90 °С.
С повышением содержания оксипропильных групп
снижается темп-pa плавления, возрастают темп-ра
желатинизации водных р-ров, совместимость с органич.
растворителями и солями.
Метилоксиэтилцеллюлоза — твердое
вещество белого цвета, аналогичное по физич. и химич.
свойствам М. Промышленный продукт выпускается
с у = 150 по метоксильным и у — 12 по оксиэтильным
группам.
Метилэтилцеллюлоза — твердое вещест-
вещество белого цвета с у = 40 по метоксильным и у = 90
по этоксильным группам; темп-pa желатинизации вод-
водных р-ров 60 °С.
Химич. свойства. Метоксильные группы в М. устой-
устойчивы к действию щелочей и большинства к-т; отщеп-
отщепление происходит при обработкеМ. иодистоводородной
к-той (см. раздел «Анализ») или металлич. натрием,
растворенным в жидком аммиаке. В р-рах минераль-
минеральных к-т М. подвергается гидролитич. деструкции
по глюкозидным связям с сохранением метоксильных
групп. В щелочных р-рах М. стабильна в отсутствие
кислорода; на воздухе происходит ее деструкция,
к-рая ускоряется в присутствии соединений кобальта
и марганца. Продукты с у = 200 устойчивы к действию
микроорганизмов, пленки из М.— к УФ-излучению,
действию любых масел и большинства органич. раство-
растворителей. •'
При взаимодействии М. с алкилирующими реаген-
реагентами получены различные выпускаемые в пром-сти
продукты — смешанные простые эфиры целлюлозы; их
физич. свойства приведены выше.
При обработке М. муравьиной к-той и уксусным ан-
ангидридом синтезированы соответствующие смешанные
эфиры целлюлозы, содержащие сложные и простые
эфирные группы. Используя методы прививки rfa поли-
полимерные цепи, можно получить привитые сополимеры М.
Получение. В пром-сти М. получают взаимодействием
щелочной целлюлозы и хлористого метила:
NaOH
Целл. (ОНK + осСН3С1 > Целл. (ОСН3)х(ОНKа; -f ocNaCL
Одновременно протекают побочные реакции — омы-
омыление хлористого метила до метанола и метилирование
последнего с образованием диметилового эфира.
Технологич. схема произ-ва водорастворимой М.
включает следующие стадии: 1) получение щелочной
целлюлозы обработкой древесной или хлопковой цел-
целлюлозы 35—40%-ным водным р-ром едкого натра при
~ 20 °С в течение 1 — 1,5 ч; 2) измельчение полученного
продукта до насыпной массы 0,13—0,15 г/см3 и про-
проведение окислительной деструкции для регулиро-
регулирования его степени полимеризации; 3) алкилирование
щелочной целлюлозы хлористым метилом в автоклаве
при 60—90 °С и избыточном давлении 1,2—2 Мн/м2
A2—20 кгс/см2) в течение 6—8 ч; 4) промывка полу-
полученной М. водой при 70—90 °С для удаления побочных
продуктов реакции, сушка и дробление (или гранули-
гранулирование).
Щелочную целлюлозу готовят классич. периодич.
или непрерывным вискозным методом либо обработкой
целлюлозы в смесителе р-ром едкого натра (см. Вискоза).
Алкилирование можно проводить при одновременной
загрузке щелочной целлюлозы и СН3С1 в избытке или
при постоянной его циркуляции.
М. выпускают в виде мелкорезаной волокнистой
массы, порошка или гранул.
В лабораторных условиях М. может быть получена
взаимодействием щелочной целлюлозы с диметилсуль-
фатом или йодистым метилом либо взаимодействием
целлюлозы с диазометаном или метиловыми эфирами
органич. сульфокислот.
Анализ. Для количественного определения содержа-
содержания метоксильных групп в М. применяют объемный
микрометод, основанный на отщеплении метоксильных
групп конц. HI и выделении образующегося СН31.
Получаемый после ряда последовательных реакций иод
оттитровывают тиосульфатом натрия. Метоксильные
группы в М. могут быть определены также спектраль-
спектральным методом. Качественно М. может быть определена
с помощью хромотроповой к-ты или антрона (9, 10-
дигидро-9-оксоантрацена). При нагревании хромотро-
хромотроповой к-ты с М. в присутствии перекиси бензоила появ-
появляется фиолетовое окрашивание. Антрон в присутствии
серной к-ты с М. дает зеленое окрашивание. Р-р иода
в КТ окрашивает М. от фиолетово-коричневого до фио-
фиолетового цвета, пропадающего в присутствии сильной
щелочи. Для качественного определения можно исполь-
использовать способность танина осаждать М. из водных
р-ров в виде хлопьев.
Переработка и применение. Хотя по сравнению с др.
простыми эфирами целлюлозы М. более устойчива
при нагревании на воздухе, ее перерабатывают и при-
применяют в условиях, в к-рых исключены сильные термо-
термоокислительные воздействия. М. используют в основ-
основном в виде водных р-ров, при приготовлении к-рых
вначале замачивают М. в небольшом @,2—0,5 ч. от все-
всего необходимого объема) количестве горячей (80—90 °С)
воды, что обеспечивает равномерное смачивание всех
частиц. Полученную суспензию выдерживают непро-
непродолжительное время, затем добавляют оставшееся ко-
количество холодной @—10 °С) воды. Водным р-рам М.
свойственно сильное пенообразование, вследствие чего
в ряде случаев необходимо применять пеногасители
типа алканолов (С7—С10) и эфиров ортофосфорной к-ты.
Пленки из М. получают методом полива на барабан
или экструзией (см. Пленки полимерные). Пластифика-
215
МЕХАНИЧЕСКАЯ ДЕСТРУКЦИЯ
216
торами служат глицерин, гликоли и их эфиры или
полигликоли. Пленки м. б. переведены в нераствори-
нерастворимое состояние обработкой эпоксидными соединениями,
диальдегидами, многоосновными к-тами, мочевино- или
меламино-формальдегидными смолами.
Водорастворимая М. находит самое разнообразное
применение в различных областях техники. Ее исполь-
используют для изготовления различных клеев (для кожи,
обоев, пенопластов, бумаги и др.)- В керамич. произ-
производстве, строительных р-рах, бетонах М. применяют
как связующий, пластифицирующий и регулирующий
устойчивость масс к усадке агент; при производстве
карандашей — как добавку, связывающую и пласти-
пластифицирующую карандашную массу. М. широко исполь-
используют в качестве эмульгатора и стабилизатора для ла-
тексных красок и акварельных паст, при суспензион-
суспензионной и эмульсионной полимеризации, как стабилизатор
кремов, водно-жировых эмульсий, шампуней, в ка-
качестве загустителя и стабилизатора р-ров и суспензий
пестицидов, инсектофунгицидов и др., а также как
водорастворимую упаковочную пленку для гранули-
гранулированных удобрений, в произ-ве бумаги — для мело-
вания и придания жиро- и маслонепроницаемости. М.
входит в состав композиции для снятия автомобильных
лаков. М. используют для подшлихтовки волокон, как
загуститель полиграфия-, красок, как связующее и
пленкообразователь в табачной пром-сти. Разнообразно
применение М. в фармацевтич. и пищевой пром-сти
(капсулирование таблеток и пилюль, безжировая осно-
основа мазей, глазных капель, компонент в слабительных,
загуститель различных соков, стабилизатор мороже-
мороженого и т. д.).
М. выпускают в СССР; США (м е т о ц е л); Велико-
Великобритании (м е т о ф а с, ц е л а к о л); Японии (м е-
т о л о з а); ФРГ (т и л о з а). Области применения
смешанных эфиров целлюлозы такие же, что и М. Под
теми же названиями, что и М., производят метилокси-
пропилцеллюлозу в США, Японии, Англии; метилокси-
этилцеллюлозу — в ФРГ; метилэтилцеллюлозу под тор-
торговыми названиями целлофас и эдифас —
в Великобритании (гл. обр. для получения водных
эмульсий в косметике).
Выпуск М. (вместе с метилоксипропилцеллюлозой)
в США составляет более 10 тыс. т в год.
Впервые М. синтезирована Суида в 1905; первое
промышленное производство освоено в Германии в 1926.
Лит.: Роговин 3. А., Химия целлюлозы, М., 1972;
Cellulose and cellulose derivatives, ed. by E. Ott [a. o.], pt 2,
N. Y.— L., 1954, p. 930—37; Encyclopedia of polymer science
and technology, v. 3, N. Y.—[a. o.], 1965, p. 492—511; Melt-
zer J. L., Water-Soluble polymers, Technology and Applica-
Application, New Jersey, 1972, p. 129; Никитин Н. И., Химия
древесины и целлюлозы, М.— Л., 1962, с. 324—27.
М. В. Прокофьева.
МЕХАНИЧЕСКАЯ ДЕСТРУКЦИЯ полимеров
(mechanical degradation, mechanische Abbau, deg-
degradation mecanique) — снижение мол. массы поли-
полимеров при механич. воздействиях, вызванное разры-
разрывами упруго деформированных макромолекул. М. д.—
один из видов механохимич. превращений полимеров
(см. Механохимия); ее необходимо учитывать при ана-
анализе закономерностей деформирования и разрушения
полимеров (см. Течение химическое, Утомление, Дол-
Долговечность). М. д.— нежелательный процесс при экс-
эксплуатации изделий; предотвращение ее последствий —
одна из задач стабилизации полимерных материалов.
М. д. биополимеров изучается в биологии и медицине.
Деструкция макромолекул линейных и трехмерных
полимеров, находящихся в вязкотекучем, высокоэла-
высокоэластическом и стеклообразном состояниях, а также
макромолекул в разб. и конц. р-рах полимеров проис-
происходит при действии сравнительно малых напряжений.
Склонность к разрыву химич. связей в основной цепи
макромолекул не связана с их пониженной прочностью
или с существованием «слабых» звеньев. Основная
причина — неравномерность распределения напряже-
напряжений по отдельным связям и существование «перенапря-
«перенапряженных» участков цепей, где истинные нагрузки близки
к предельной прочности химич. связей на разрыв.
Перенапряжения возникают вследствие различий в на-
направлении и величине сил внутреннего трения, дейст-
действующих на отдельные сегменты, на участках «проход-
«проходных» цепей между элементами надмолекулярной струк-
структуры полимера, вблизи узлов физической или химич.
сетки и др. Перенапряженные связи, отличающиеся
значительным смещением частот скелетных колебаний,
обнаружены методом колебательной спектроскопии.
В образцах кристаллич. полимеров, не содержащих
дефектов структуры (т. наз. «усы»), напряжения рас-
распределены равномерно по всем связям, и деструкция,
вызывающая разрушение материала, происходит при
внешних нагрузках, приближающихся к теоретич.
прочности химич. связей.
Упругие напряжения в макромолекулярных систе-
системах складываются из энергетической и энтропийной
составляющих. Для М. д. основное значение имеет
энергетич. составляющая напряжения, обусловленная
искажением валентных углов, межатомных расстояний
и др. параметров внутри- и межмолекулярных связей.
Энтропийная составляющая напряжения связана с от-
отклонениями конформации цепей от равновесной. Воз-
Возможность косвенного влияния энтропийной составляю-
составляющей на скорость М. д. в достаточной мере не исследо-
исследована. Экспериментально установлено, что небольшие
энтропийные напряжения не ускоряют М. д. вулкани-
затов каучуков.
При растяжении энергия связи уменьшается (рис.
1,а). Теоретич. предельная прочность связей С — С
и С — О на разрыв Fo = (~) (U — энергия
or ' max
взаимодействия между атомами, г — расстояние между
ними, см. рис. 1 — б и в) по ориентировочным оценкам
равна D—6) • 10~4 дин на 1 связь, что соответству-
соответствует разрывным напряжениям порядка 105Мн/м2
A04 кгс/мм2). Разрыв свя-
U(F)
0.5
0,5
1,0
F
U(r)
зей под действием пре-
предельных напряжений
осуществляется только в
том случае, когда нагруз-
нагрузка, равная Fo, прило-
приложена за время, близкое
к периоду колебаний ато-
атомов цепи. Такой чисто
«механический» разрыв
реализуется в очень же-
жестких условиях нагру-
жения.
При длительном дей-
действии напряжений дест-
деструкция осуществляется
по термофлуктуацион-
ному механизму при на-
напряжениях, меньших
предельного (см. Долго-
Долговечность). Для элемен-
элементарного акта чисто тер-
мич. деструкции период
Рис. 1. Схемы, иллюстриру-
иллюстрирующие: а) изменение энергии
химич. связи при ее растя-
растяжении, б) зависимость энер-
энергии взаимодействия ив) си-
силы, действующей между ато-
атомами, от расстояния г меж-
между ними. Uo— энергия свя-
связи, U(F) — энергия связи,
деформированной силой F,
Fo — предельная теоретич.
прочность связи.
217
МЕХАНИЧЕСКАЯ ДЕСТРУКЦИЯ
218
полураспада зависит от энергии связи по закону:
тО5^тоехр
где т0 — период колебаний, по порядку величины
близкий к 10~13 сек; R — универсальная газовая
постоянная; Т — абсолютная темп-pa, К; Uo — энер-
энергия активации разрыва, близкая по величине к энергии
связи. Под действием внешней силы энергия связи
уменьшается, а, следовательно, уменьшается и энер-
энергия активации распада. По ориентировочным оценкам,
при действии силы F~1/2F0 период полураспада
связей С — С при комнатной темп-ре уменьшается
до 102—104 сек. Так. обр., нагрузки, меньшие Fo,
резко увеличивают скорость термич. разрыва связей.
Точно так же упругие напряжения повышают веро-
вероятность разрыва макромолекул под действием ионизи-
ионизирующего излучения, УФ-света и др. физических и хи-
мич. факторов. Как правило, энергетич. выход деструк-
деструкции в этих случаях значительно больше суммы выхо-
выходов чисто механического и чисто радиационного про-
процессов (см. Течение химическое).
Скорость М. д. характеризуют увеличением концент-
концентрации макромолекул п в единицу времени. Т. к. ве-
вероятность деструкции зависит от степени полимериза-
полимеризации, то
4г = 2 *(*)»(«
где суммирование производится по всем макромоле-
макромолекулам различной длины, п(Р) — концентрация макро-
макромолекул со степенью полимеризации Р, к(Р) — кон-
константа скорости деструкции, зависящая от степени
полимеризации. Из-за трудностей, возникающих при
анализе данных, чаще всего определяют усредненную
константу скорости деструкции /сср.
Прирост числа макромолекул в системе совпадает
с количеством связей, разорванных за данный проме-
промежуток времени t:
х- N ( 1
где N — число Авогадро, m — мол. масса звена, Ро
и Р| — среднечисловая степень полимеризации до на-
начала процесса и в момент времени t. Скорость разрыва
связей оценивают такжа по скорости образования пер-
первичных продуктов деструкции — свободных радика-
радикалов; их концентрацию измеряют по расходу акцептора
или методом электронно-
электронного парамагнитного резо-
резонанса.
Всегда при снижении
степени полимеризации
константа скорости дест-
деструкции падает практиче-
практически до 0 (рис. 2), что яв-
является следствием суще-
12
0.4
молекуляряо-массового распределения при деструк-
деструкции отражают особенности распределения напряжений
по связям в данной макромолекулярной системе. До-
Достаточно полно эти зависимости еще не исследованы.
2
Р-КГ3
Рис. 2. Зависимость констан-
константы скорости механич. дест-
деструкции полистирола от степе-
степени полимеризации (облучение
р-ров ультразвуком).
ствования предела деструкции Роо — минимальной
степени полимеризации, при достижении к-рой М. д.
резко замедляется или прекращается совсем. Значение
Роо зависит от интенсивности подвода энергии и ус-
условий деструкции; по различным оценкам, Рос ме-
меняется в пределах 102—103 звеньев. Наименьшее зна-
значение Роо ^ 70—80 получено при измельчении по-
порошка полиметилметакрилата в интенсивной вибра-
вибрационной мельнице в течение нескольких часов.
Вид зависимости константы скорости М. д. от степени
полимеризации при Р > Рос, а также изменения
ю4 ш5 ю6
Молекулярная масса
Рис. 3. Изменения молекулярно-массового распределения
при механической деструкции (пластикации) натурального
каучука. Цифры у кривых соответствуют продолжительности
обработки в мин.
Для ряда систем с ростом степени полимеризации
константа скорости деструкции увеличивается по за-
закону, близкому к квадратичному; разрывы макромоле-
макромолекул происходят вблизи середины цепей, и М. д. со-
сопровождается сужением молекулярно-массового рас-
распределения (рис. 3). Эти закономерности наблюдали
для разб. растворов полимеров, где макромолекулы
изолированы одна от другой, и при пластикации
каучуков.
При механич. обработке твердых полимеров коп-
станта скорости деструкции м. б. прямо пропорцио-
пропорциональна степени полимеризации. Тогда ур-ние скорости
деструкции после интегрирования принимает вид
соответствующий т. наз. деструкции по закону слу-
случая. Начальные участки кинетич. кривых деструкции
полимеров при измельчении часто удовлетворительно
описываются этим простым ур-нием. Иногда кинетика
деструкции подчиняется эмпирич. ур-нию Н. К. Ба-
рамбойма
Pt-Poo = (P0-Poo) exp (—k6t)
пользуясь к-рым оценивают значение Роо.
Разрыв связей при М. д. большинства полимеров
происходит по свободнорадикальному механизму. Сво-
Свободные радикалы обнаружены в продуктах М. д. основ-
основных классов синтетических и природных полимеров
(исключение — полидиметилсилоксан). Места разрыва
макромолекулярных цепей установлены для отдельных
полимеров в результате анализа структуры первичных
радикалов по их спектрам электронного парамагнит-
парамагнитного резонанса. Например, в полиэтиленоксиде пре-
преимущественно разрываются связи —С—С —, а не
—С—О—, а в поликапролактаме — связи —С—С—,
ближайшие к амидной группе.
Всегда вслед за возникновением первичных радика-
радикалов происходят их дальнейшие превращения (см. Меха-
нохимия). Первичные радикалы в момент своего обра-
образования обладают избыточной энергией, к-рая выделя-
выделяется в результате сокращения участков упруго напря-
напряженной цепи после разрыва. Избыточной энергии
достаточно для того, чтобы вызвать распад таких «горя-
«горячих» радикалов с образованием низкомолекулярных
соединений, гл. обр. мономера. По этой причине меха-
механич. разрушение полимеров обычно сопровождается
219
МЕХАНИЧЕСКАЯ ОБРАБОТКА
220
выделением летучих продуктов, регистрируемых хро-
матографическим и масс-спектроскопич. методами.
Для М. д. нек-рых полимеров важны также радикаль-
радикальные реакции передачи атома водорода~ R' + ~R"H~->
—> ~R'H + ~ R"~ и распада вторичных радикалов
~ R" ~ —> ~ R + Р (Р — остаток макромолекулы).
При упругом деформировании полимера вероятность
распада радикалов ~ R" ~ увеличивается, и появля-
появляется возможность цепного развития деструкции в ре-
результате чередования этих двух реакций. Т. обр.,
снижение мол. массы полимера при М. д. складывается
из первичных разрывов и вторичной деструкции (или
соединения) отрезков макромолекулярных цепей. Вто-
Вторичные реакции можно предотвратить, вводя в поли-
полимер ингибиторы свободных радикалов.
Лит. см. при ст. Механохимия. П. Ю. Бутягин.
МЕХАНИЧЕСКАЯ ОБРАБОТКА пластмасс
(machining of plastics, mechanische Behandlung von
Kunststoffen, traitement mecanique des plastiques).
Содержание:
Введение 219
Обработка резцом простейшей геометрии 220
Качество обработанной поверхности 22 0
Удельная сила и удельная работа резания 222
Критический передний угол резца 222
Критерии обрабатываемости 222
Температура резания ¦ 22 4
Остаточные напряжения в зоне обработки 225
Износ инструмента 225
Охлаждение инструмента и изделия 225
Обработка многолезвийными инструментами . . . .226
Вырубка 22 7
Галтовка 228
Разрезание разогретой проволокой 228
Режимы резания и геометрия инструмента 22 8
Точность обработки 228
Заключение 229
Введение. Основные способы М. о. различных изде-
изделий сведены в табл. 1.
Таблица 1. Типовые способы механической обработки
пластмасс
Вид изделия Способы обработки
Листовой материал
Трубы, прытки, слож-
сложные профили, получен-
полученные методом экструзии
Крупногабаритные из-
изделия, полученные ме-
методами намотки, кон-
контактного прессования,
пневмо- и вакуумформо-
вания
Средние и мелкие изде-
изделия, не требующие повы-
повышенной точности
Изделия, требующие
повышенной точности
или получаемые в основ-
основном механич. обработкой
Раскрой с помощью различных
ножниц, ленточных и циркульных
пил, отрезных фрез, абразивных
кругов, алмазных дисков, разогре-
разогретой проволоки (для термопластич-
термопластичных пенопластов). Вырубка
Отрезание с помощью циркуль-
циркульных пил, абразивных кругов, ал-
алмазных дисков
Удаление технологич. припусков,
зачистка терцов с помощью токар-
токарных резцов, абразивных кругов.
Сверление, развертывание, фрезеро-
фрезерование
Удаление грата сверлением, фре-
фрезерованием, обработкой во вращаю-
вращающихся барабанах, абразивными кру-
кругами и лентами. Шлифование и по-
полирование
Точение, сверление, развертыва-
развертывание, строгание, фрезерование, про-
протягивание, зубо-и резьбонарезание,
шлифование, полирование
М. о. следует рассматривать как вынужденную опе-
операцию. Она часто вызывает появление остаточных
напряжений в зоне резания и приводит к нарушению
поверхностного слоя полимерного материала. Это
в ряде случаев снижает прочность, ускоряет старение
материалов и приводит к изменению формы изделий
с течением времени.
Обработка резцом простейшей геометрии. С процес-
процессом резания пластмасс наиболее удобно познакомиться
на примере обработки заготовки резцом простейшей
геометрии (рис. 1). f?
Рис. 1. Схема сил, возникающих при резании пластмасс
(отдельно вынесена схема сил, действующих на резец):
А — передняя грань резца; Б — задняя грань резца; С —
обрабатываемая поверхность; D — обработанная поверх-
поверхность; К — зона упруго-эластич. восстановления материала;
г — радиус закругления режущей кромки, возникающий
при затуплении; у — передний угол резца; а — задний угол
резца; а — толщина срезаемого слоя; h3 — износ по задней
грани; Pi — усилие со стороны материала на переднюю
грань; Fi — сила трения материала о переднюю грань;
Р2 и F2 — вертикальная и горизонтальная составляющие
сил, действующих по задней грани; jx — коэфф. трения;
Дм— сила, действующая со стороны передней грани резца
на материал; Р —суммарная сила резания.
В этом случае отделение материала в виде стружки
вызывается в основном силон i?M, действующей со сто-
стороны передней грани резца на материал. Силы, дейст-
действующие на материал со стороны задней грани, влияют
на процесс резания только в тех случаях, когда обус-
обусловленная ими деформация вызывает изменения свойств
обрабатываемого материала. Величина я направление
/?м зависят от физико-механич., структурных и реоло-
гич. свойств полимерных материалов, величины пе-
переднего угла резца, темп-ры и режимов резания.
Пластмассы обладают специфич. свойствами, к-рые
существенным образом влияют на процессы их М. о.:
небольшой уд. теплоемкостью, низкой теплопровод-
теплопроводностью, большим температурным коэфф. линейного
расширения, резко выраженной способностью к упру-
упруго-эластич. восстановлению формы в подрезцовой зоне.
Кроме того, обработка пластмасс сопровождается боль-
большим износом режущего инструмента.
Мощность резания N в н-м/сек (или кгс-м/сек.
определяется из соотношения:
N = PZV
где Р г— горизонтальная составляющая суммарной си-
силы резания, н (или кгс)\ V — скорость резания, м/сек.
Усилие Ру воспринимает резцедержатель.
Качество обработанной поверхности. Обработанная
поверхность имеет макро- и микронеровности, геомет-
геометрия и количество к-рых зависят от типа образующейся
стружки (рис. 2).
Сливная стружка, характеризующаяся отсутствием
плоскостей скалывания, возникает при малых скоро-
скоростях резания в случае обработки материалов, имеющих
большую высокоэластич. деформацию и значительное уд-
удлинение при разрыве (полиэтилен, фторопласт и др.).
Толщина стружки примерно равна толщине срезаемого
слоя. Колебания силы резания незначительны. Качест-
Качество обработанной поверхности высокое.
221
МЕХАНИЧЕСКАЯ ОБРАБОТКА
222
Стружка скалывания образуется в результате воз-
возникновения в материале плоскостей сдвига (напр., для
полистирола). Стружка гладкая вследствие малых
интервалов между этими плоскостями. Как правило,
Рис. 2. Типы стружек, об-
образующиеся при резании
пластмасс: а — сливная;
б — скалывания (штрихов-
(штриховкой указаны плоскости
скалывания); в—элемент-
в—элементная; г — надлома; д — неоднородная элементная; а —
толщина срезаемого слоя; dc — толщина стружки.
толщина стружки больше толщины срезаемого слоя.
Качество обработанной поверхности высокое. Сливная
стружка и стружка скалывания являются непрерыв-
непрерывными.
Элементная стружка образуется при обработке по-
полиметил метак рил ата или слоистых пластиков в ре-
результате возникновения поверхностей скалывания на
относительно большом расстоянии друг от друга.
Стружка иногда принимает форму свернутых жгутов
различного диаметра. При обработке наблюдаются
колебания силы резания. Качество обработанной по-
поверхности плохое.
Стружка надлома образуется в результате больших
напряжений растяжения и сжатия в комбинации
с напряжениями сдвига при резании инструментом
с отрицательным передним углом. Качество обрабо-
обработанной поверхности весьма плохое.
Неоднородная элементная стружка образуется в ре-
результате хрупкого надлома при обработке жестких
материалов (напр., полиметилметакрилата, отвержден-
ных феноло-формальдегидных, полиэфирных и эпок-
эпоксидных смол) инструментом с большим передним уг-
углом при значительной толщине срезаемого слоя. В ма-
материале перед резцом возникает опережающая трещина,
в направлении к-рой и происходит отделение стружки.
Качество обработанной поверхности очень плохое.
При обработке одного и того же материала можно
получить стружку любого типа, изменяя геометрию
резца и режимы резания. Напр., при изменении перед-
переднего угла у изменяется величина и направление силы Рг
(см. рис. 1), вследствие чего видоизменяется напря-
напряженное состояние материала и тип образующейся
стружки. При возрастании скорости резания высоко-
эластич. деформации могут уступить место пластич.
сдвигу или хрупкому разрушению. Увеличение толщи-
толщины срезаемого слоя повышает усилие, необходимое для
образования непрерывной стружки, и разрушение
материала может начаться из-за возникновения опере-
опережающей трещины и хрупкого надлома. Этому также
способствует повышение скорости резания и увеличение
7- Ниже для различных материалов приведена толщина
срезаемого слоя а (в мм), при к-рой образуется стружка
непрерывного типа (V = 400 м/мин, у = 0):
Полиэтилен 0,30 Полипропилен . . . . 0,18
Винипласт 0,12 Гетинакс 0,01
Полиметилметакрилат 0,12 Литая полиэфирная
Фторопласт-4 0,10 смола 0,01
Поликарбонат 0,25
При резании наполненных реактопластов затуплен-
затупленным резцом на образующейся поверхности возникают
дефекты, тип и размер к-рых в значительной мере
зависят от характера взаимодействия связующего
с наполнителем. В случае больших внутренних напря-
напряжений (напр., в стеклопластиках) происходит хрупкое
разрушение материала с образованием глубоких тре-
трещин, сколов, отслаиванием значительных участков
материала и разлохмачиванием волокнистого напол-
наполнителя. Если связующее способно глубоко пропиты-
пропитывать наполнитель (гетинакс, текстолит), то дефекты
поверхности носят менее выраженный характер, без
элементов хрупкого разрушения.
Удельная сила и удельная работа резания. Под уд.
силой резания понимается усилие Рг (см. рис. 1),
приходящееся на единицу площади срезаемого слоя
материала. Численно уд. сила резания равна работе
отделения единицы объема материала, т. е. уд. работе
резания. Значение уд. силы резания уменьшается при
увеличении толщины срезаемого слоя. Особенно резкое
уменьшение наблюдается при изменении типа образую-
образующейся стружки. Ниже приведены значения уд. силы
резания при обработке различных материалов в усло-
условиях, обеспечивающих получение стружки непрерыв-
непрерывного типа:
Мн/м2 (пгс/мм2)
Полиэтилен 80—110 (8—11)
Винипласт 200 B0)
Полиметилметакрилат 200 B0)
Фторопласт-4 80 (8)
Поликарбонат 230 B3)
Полипропилен 190 A9)
Гетинакс 400 D0)
Текстолит на основе феноло-формаль-
дегидной смолы 350 C5)
Текстолит на основе меламино-форм-
альдегидной смолы 290 B9)
Литая полиэфирная смола 40 D)
Критический передний угол резца. Вблизи режущей
кромки обрабатываемый материал подвергается растя-
растягивающим или сжимающим напряжениям под дейст-
действием силы R'M (см. рис. 1). Направление Л'м в значи-
значительной степени определяется знаком и величиной
переднего угла у. Упруго-эластич. деформация мате-
материала во внешних слоях обработанной поверхности
снижает точность изделий и увеличивает силу трения
по задней грани резца. В результате может произойти,
напр., защемление пилы в пропиле или уменьшение
диаметра отверстия после сверления. Увеличение силы
трения материала о заднюю грань резца приводит
к повышению темп-ры резания, более быстрому износу
инструмента, а также требует увеличения силы Рг
и мощности резания.
Подбором у можно свести к нулю R'M. При этом
изменение формы обработанного изделия будет мини-
минимальным. Передний угол, при к-ром /?'м = 0, наз.
критич. передним углом. В большинстве случаев
устранение R'M позволяет добиться наивысшей точ-
точности изделий при минимальном износе инструмента.
Критич. передний угол зависит от силы Р1У с к-рой
материал воздействует на переднюю грань резца (см.
рис. 1), и силы трения материала F1 об эту грань, ко-
которые в свою очередь определяются условиями обработ-
обработки. Критич. передний угол находится эксперимен-
экспериментально для каждого материала при различных режи-
режимах резания (рис. 3).
Критерии обрабатываемости. Эти критерии характе-
характеризуют способность материала к механич. обработке.
В зависимости от требований, предъявляемых к про-
процессу, м. б. выбраны различные критерии, напр.:
7 = -^г5 7= -?4 ]=Н
где W — объемная производительность, N — мощ-
мощность резания, h3 — износ по задней грани при про-
223
МЕХАНИЧЕСКАЯ ОБРАБОТКА
224
50
40
30
20
10
Полиэтилен
414
0,01
14
12
10
50
40
30
20
0,05 0,10
Винипласт
,10
Полипропилен
70
Полистирол
Полиацетала
400
25
20
15
0,05 0,10 0,15
Полиметилметанрилат
-0,8
0,2
0.0!
-0,8
0,05 0,10
Политетрафтор э тилен
'0.76
0,05 .0,10 0,15
Ударопрочный сополимер
стирола АБС
30
20
10
0
400'
i i i
30
0,0! 0.05 0,10
¦218
765
0,01
0.1
Лолиамид-6
0.4
1,0
150
400
0,10
0,20
0 0,02 0,04 0,06 0.08 0,10
200
40
30
20
Гетинакс
0,05 0,10
Поликарбонат
15
400
30
20
0,06 0,08
Текстолит
70
' 150
40(
0,05 0,10
0,04 0,06 0,08 0,10
Рис. 3. Зависимость критического переднего угла резца -\ от толщины срезаемого слоя а при различных скоростях
резания пластмасс. По оси абсцисс — толщина срезаемого слоя вм; по оси ординат — критич. передний угол резца
в град; цифры на кривых — скорость резания в м/мин.
хождении резцом расстояния L, Н — средняя квадра-
квадратичная высота микронеровностей. Первые два крите-
критерия применяют при черновой обработке, когда оценка
материала производится с позиции обеспечения высо-
высокой производительности процесса при удовлетвори-
удовлетворительной стойкости инструмента. Третий критерий
используют при чистовой обработке, когда основное
внимание уделяется качеству обработанной поверх-
поверхности и стойкости инструмента. Критерием обрабаты-
обрабатываемости может служить также максимальная глубина
резания, при к-рой образуется стружка непрерывного
типа. В соответствии с этим критерием пластмассы де-
делят на след. группы: 1) отличная обрабатываемость —
полиэтилен, фторопласт-4, сополимеры АБС средней
и высокой ударной прочности, полиамид-6 и полиамид-
6,10, поликарбонат; 2) обрабатываемость между отлич-
отличной и хорошей — винипласт, полиакрилаты, сополи-
сополимеры АБС низкой ударной прочности, полиацетали,
полипропилен и полиамид-6,6; 3) хорошая обрабаты-
обрабатываемость — сополимеры акрилонитрила со стиролом;
4) удовлетворительная обрабатываемость — гетинакс
и текстолит; 5) плохая обрабатываемость — гетинакс
на основе меламино-формальдегидной смолы, стекло-
стеклотекстолит на основе феноло-формальдегидной смолы,
литые полиэфирные и эпоксидные смолы.
В качестве критерия обрабатываемости стеклопласти-
стеклопластиков используют также отношение скорости резания рас-
рассматриваемого материала к скорости резания эталон-
эталонного материала при равном износе инструмента. Обра-
Обрабатываемость стеклопластиков улучшается в след. ряду
(в скобках приведены рекомендуемые скорости резания
при обработке твердосплавным инструментом): а) стек-
стеклопластики с кремнеземным стекловолокном, получен-
полученные прессованием при высоком давлении B0—
40 м/мин); б) стеклопластики с алюмоборосиликатным
стекловолокном, полученные прессованием при высо-
высоком давлении G0—120 м/мин); в) стеклопластики
с алюмоборосиликатным стекловолокном, полученные
прессованием при низком давлении дли намоткой A50—
200 м/мин).
Температура резания. Вследствие низкой теплопро-
теплопроводности полимерных материалов выделяющаяся при
резании теплота нагревает стружку и заготовку только
в очень тонком слое и тепло отводится в основном
(на 95—99,5%) через инструмент. При этом резец
может значительно разогреваться. Тепловыделение
растет с увеличением мощности резания. Очень боль-
большое значение при этом имеет степень затупления
инструмента. При уменьшении толщины срезаемого
слоя возрастает уд. работа резания и повышается теп-
теплосодержание стружки.
При неблагоприятных условиях резания темп-pa ма-
материала может превзойти допустимые значения:
темп-ру размягчения для термопластов или темп-ру
термодеструкции для реактопластов. При этом возни-
возникают резкие колебания Pz, обусловливающие непо-
непостоянство качества обработанной поверхности. По мере
повышения темп-ры термопластов появляются задиры
поверхности, и материал налипает на переднюю грань
резца с образованием нароста. Периодич. возникно-
возникновение и исчезновение нароста отрицательно сказыва-
сказывается на процессе образования стружки и качестве
обработанной поверхности.
В зоне резания наблюдается значительный темпера-
температурный градиент, обусловленный низкой тепло- и тем-
температуропроводностью пластмасс. При обработке ге-
тинакса цилиндрич. фрезой (Sz = 0,32 мм/зуб, глу-
глубина резания — 20 мм, см. рис. 6) со скоростями
150 м/мин и 670 м/мин темп-pa на площадке контакта
материала с инструментом составляет соответственно
140 °С и 215 °С, но уже на глубине 0,15 мм в обоих
случаях она снижается до 30 °С. При износе инстру-
225
МЕХАНИЧЕСКАЯ ОБРАБОТКА
226
мента темп-pa материала резко возрастает. Напр.,
при обработке стеклотекстолита указанной выше фре-
фрезой со скоростью 670 м/мин темп-pa на контактной
площадке составляет 430 °С при износе по задней гра-
грани 0,02 мм и 590 °С при износе 0,30 мм.
Остаточные напряжения в зоне обработки. Как пра-
правило, механич. и тепловое воздействие на материал
в процессе резания увеличивает исходные остаточные
напряжения, нередко приводя к возникновению тре-
трещин в процессе обработки или через нек-рое время
после его завершения. Удары режущих кромок инстру-
инструмента и вибрация при М. о. разрушают обрабатываемый
материал, особенно при резании поперек слоев напол-
наполнителя.
При нагреве материала с кристаллич. структурой
возможна ее перестройка с уменьшением уд. объема.
В этом случае после охлаждения полимера в его наруж-
наружных слоях возникают опасные растягивающие напря-
напряжения. Эти напряжения, складываясь с монтажными
напряжениями (напр., при затягивании болтов), часто
приводят к появлению мельчайших поверхностных тре-
трещин на изделиях из термопластов. Такое же явление
наблюдается и при действии растворителей на обра-
обработанную поверхность.
Для уменьшения влияния М. о. на качество и струк-
структуру поверхностных слоев материала используют раз-
различные технологич. приемы: поджатие материала при
сверлении параллельно слоям наполнителя; попутное
безударное фрезерование; многооперационное сверле-
сверление отверстий, при к-ром последний проход произво-
производится специальной калибрующей разверткой с удли-
удлиненной заборной частью, снимающей минимальный
припуск при чистовых режимах резания.
Изделия из термопластов, к к-рым предъявляются
повышенные требования, после механич. обработки
подвергают умеренному нагреванию, в результате чего
снимается большая часть остаточных напряжений.
Износ инструмента. За критерий износа инструмента
принимают предельную величину износа по задней
грани (h3 — см. рис. 1), при к-рой возможно полу-
получение поверхности заданной чистоты. В зависимости
от обрабатываемого материала и вида обработки эта
величина составляет 0,1—0,3 мм.
Охлаждение инструмента и изделия. Жидкостное
охлаждение при обработке пластмасс может применять-
применяться только в тех случаях, когда оно не вызывает неже-
нежелательного изменения физико-механич. свойств мате-
материала. Напр., попадание воды на изделия из полиме-
тилметакрилата приводит к образованию сетки мелких
наружных трещин. Воздействие воды на гидрофильные
связующие или наполнители вызывает интенсивное
проникновение влаги внутрь материала, приводящее
к его разбуханию, возникновению остаточных напря-
напряжений, снижению механич. и ди-
электрич. свойств. Попадание во-
воды в уже имеющиеся в материале
микротрещины приводит к их
дальнейшему разрастанию.
Поэтому жидкостное охлажде-
охлаждение E%-ным р-ром эмульсола
Рис. 4. Схема процесса свер-
сверления: 2<р — угол при верши-
вершине; со = arctg ~t угол на-
наклона винтовой канавки; D —
диаметр сверла; I— шаг витка;
а — толщина срезаемого слоя;
So— подача на 1 оборот; dc—
толщина стружки (без учета
усадки).
в воде) применяют для ограниченного числа термо-
термопластов и не используют при обработке реактопластов.
Вместо этого охлаждают водой инструмент через от-
отверстия в его теле. Вращающийся инструмент охлаж-
охлаждают струей сжатого воздуха.
Обработка многолезвийными инструментами. Свер-
Сверление материалов (рис. 4) сопровождается снятием
стружки небольшой толщины (dc), вследствие чего уд.
работа резания в этом процессе велика и может достиг-
достигнуть нек-рого критич. значения, при к-ром сильно
возрастает темп-pa инструмента и начинается размяг-
размягчение термопластов или термодеструкция реактопла-
стов. С др. стороны, увеличение dc снижает качество
обрабатываемой поверхности.
Для спиральных сверл с двумя режущими кромка-
кромками толщина стружки определяется из ур-ния:
где dc — толщина стружки, мм; U—осевая подача свер-
сверла за 1 оборот, мм; п—частота вращения сверла, об/мин;
Ф — половина угла при вершине сверла. Т. о., ка-
качество обработки определяется осевой подачей и
частотой вращения или окружной скоростью режущих
кромок сверла (рис. 5). Качество обработки в значи-
2000 4000 6000 8000
Частота вращения сверла, об/мин
О 50 100 150
Окружная спорость, м/мин
Рис. 5. Влияние режимов сверления на качество обработки
полиметилметакрилата.
тельной степени зависит также от свойств обрабаты-
обрабатываемого материала. Сужение диапазона оптимальных
режимов по мере возрастания частоты вращения сверла
связано, с одной стороны, с упрочением материала при
повышении скорости его деформирования, а с другой —
с размягчением или термодеструкцией материала при
темп-ре обработки.
Для уменьшения дефектов краев отверстий приме-
применяют сверла с двойной заточкой. Уменьшение длины
поперечной кромки в 2—2,5 раза позволяет снизить
осевое давление на материал. Кроме того, для качест-
качественной обработки отверстий необходимо обеспечить
беспрепятственное удаление из них стружки. Это дости-
достигается применением сверл с полированными канавками,
имеющими больший угол наклона. Однако увеличение
этого угла вызывает одновременное увеличение пе-
переднего угла инструмента, что может привести к сни-
снижению качества обработки материала. Кроме спи-
спиральных сверл, для обработки пластмасс применяют
также перовые сверла.
При сверлении слоистых пластиков необходимо при-
принимать во внимание расклинивающее действие вершины
сверла. Если сверло входит в материал перпендику-
перпендикулярно слоям наполнителя, угол заточки сверла при
вершине B ф) должен быть 60°, если параллельно—
110—120°.
Для получения точных отверстий обработку следует
вести в несколько проходов.
Влияние толщины стружки на качество обработки
поверхности при фрезеровании аналогично
таковому при сверлении (рис. 6).
При обработке слоистых пластиков направление
резания должно совпадать с направлением подачи
227
МЕХАНИЧЕСКАЯ ОБРАБОТКА
228
материала (попутное фрезерование). При этом происхо-
происходит прижим наполнителя к поверхности обработки,
что предохраняет от возникновения трещин и расслаи-
расслаивания материала. Применение цилиндрич. фрез со спи-
спиральными зубьями обес-
обеспечивает безударное ре-
резание материала и об-
образование непрерывной
стружки. Одновременно
уменьшается запылен-
запыленность рабочего места.
Рис.6. Схема процесса фре-
фрезерования: В —толщина ма-
материала; R— радиус окруж-
окружности резания; t — шаг
зубьев; 0 — угол встречи;
Sz — подача на 1 зуб.
Абразивные отрезные' круги применяют для р а с-
к р о я реактопластов, содержащих износостойкие
наполнители. Круги обладают значительно большей
износоустойчивостью, чем отрезные фрезы и циркуляр-
циркулярные пилы. Особенно эффективно применение карборун-
карборундовых кругов средней твердости на вулканитовой связке
с размером зерен абразива 0,25—0,50 мм. Скорость
обработки — 1500—2400 м/мин.
Для шлифования пластмасс наиболее при-
пригодны высокопористые карборундовые круги средней
твердости на керамич. или бакелитовой связке с разме-
размером зерен абразива 0,8—0,5 мм для черновой обработки
и 0,25—0,16 мм для чистовой. Часто применяют также
шлифовальную шкурку с теми же размерами зерен
абразива. Шкурку закрепляют на вращающемся круге
или используют в виде бесконечной ленты. Для пред-
предотвращения быстрого засаливания шкурки обработку
производят со скоростью не менее 1500—2400 м/мин.
При шлифовании нельзя допускать длительного
контакта материала с инструментом во избежание раз-
размягчения термопластов или термодеструкции реакто-
реактопластов. Усилие прижима не должно превышать 50—
100 кн/м2 @,5—1,0 кгс/см2). При обработке термопластов
применяют обильное жидкостное охлаждение. Хорошие
результаты достигаются при использовании абразив-
абразивных отрезных кругов, облицованных по боковым сто-
сторонам рифленой металлической пленкой. Эта пленка
уменьшает трение круга о стенки пропила и хорошо
рассеивает тепло в окружающую среду. Такие круги
особенно эффективны при раскрое термопластов.
Для получения поверхностей особо высокого качест-
качества или придания им стойкости к окружающей среде
применяют полирование. Кроме того, поли-
полирование придает изделиям из пластмасс товарный вид.
Полирование производят мягкими кругами толщи-
толщиной до 120 мм. Круги изготовляют путем набора и
уплотнения пакета, составленного из муслиновых
дисков различного диаметра таким образом, что за каж-
каждым диском диаметром 350—450 мм располагаются два
диска диаметром 150—200 мм. Применяют также фет-
фетровые круги. Часть круги покрывают полировальной
пастой, содержащей в качестве абразивного элемента
окись хрома. Другая часть полировального круга
остается сухой и служит для протирки изделий. В про-
процессе полирования изделия нельзя сильно прижимать
к кругу во избежание размягчения термопластов или
термодеструкции реактопластов.
Вырубка. Этот метод М. о. осуществляют с целью
получения отверстий или зачистки кромок в листовых
изделиях. Для вырубки материалов толщиной более
0,5 мм, а также хрупких материалов любой толщины
применяют штампы с острой матрицей. Зазор между
пуансоном и матрицей не должен превышать 0,02—
0,04 мм. Реактопласты толщиной свыше 1,5—2,0 мм
(гетинакс, текстолит, стеклотекстолит) перед штампов-
штамповкой нагревают до 80—200 °С, в зависимости от вида
материала и толщины заготовки. Для нагревания целе-
целесообразно применять инфракрасные излучатели (см.
Штампование).
Галтовка. Процесс предназначен для удаления грата
(излишки материала, остающиеся на кромках изделия
после прессования, литья под давлением, раздува
и т. д.) с изделий небольших размеров или шлифования
и полирования таких изделий. Галтовку осуществляют
в горизонтальных или наклонных барабанах, к-рые
заполняют изделиями и вспомогательными телами и
приводят во вращение. В зависимости от толщины
грата, его распределения и конфигурации изделий в ка-
качестве вспомогательных тел применяют стальные ша-
шарики, шпильки, дробь или шары из плавленой окиси
алюминия. Грат снимается с изделий в результате уда-
ударов и трения и удаляется из барабана через сетчатые
стенки или дно. Более тонкая обработка поверхности
достигается при галтовке в герметичных барабанах в
присутствии воды и поверхностно-активных веществ.
Шлифование или полирование изделий осуществляют
при помощи кусочков пемзы, восковых шаров или
деревянных блоков, пропитанных полировальной па-
пастой. Изделия предварительно тщательно очищают,
промывают и сушат. Продолжительность галтовки —
0,5—1,5 ч.
Разрезание разогретой проволокой. Эту операцию
применяют для разделения блоков из термопластичных
пенопластов. Инструментом является нихромовая про-
проволока или электронож, изготовленный из тонкой
полосы сплава с высоким электрич. сопротивлением.
Проволоку или нож нагревают от источника напряже-
напряжения 5—36 в до темп-ры, зависящей от разрезаемого
материала (напр., для полистирола до 670 — 700 °С),
и протягивают через материал со скоростью 60—
100 мм/мин.
Режимы резания и геометрия инструмента. Выбор
скорости резания материалов различной толщины при
работе резцом заданной геометрии производят по гра-
графикам, аналогичным изображенным на рис. 3. В по-
подавляющем большинстве случаев при увеличении а
следует уменьшить скорость резания. Однако для
нек-рых материалов (напр., полиметилметакрилат)
наблюдается обратная зависимость. Такое явление
наблюдается в тех случаях, когда при увеличении тол-
толщины срезаемого слоя изменяется природа процесса
образования стружки. После выбора скорости резания
по графику необходимо убедиться, что при данной
скорости не происходит размягчения или термодест-
термодеструкции материала. В противном случае следует произ-
произвести корректировку скорости или применить охлаж-
охлаждение.
Значение у также находят по графикам, представлен-
представленным на рис. 3. Передние углы резцов при черновой и
чистовой обработке должны быть различными.
Значение а должно быть таким, чтобы, по возмож-
возможности, максимально уменьшить силы трения по задней
грани резца. Однако чрезмерное увеличение а при-
приводит к снижению жесткости инструмента.
В СССР проводятся работы по стандартизации режу-
режущего инструмента и установлению оптимальных режи-
режимов М. о. Эти данные отражаются в общесоюзных
стандартах и ведомственных нормалях.
Точность обработки. Между допуском на обработку
б и размером обрабатываемой поверхности D сущест-
существует зависимость:
где Кип — коэффициенты, характеризующие приро-
природу обрабатываемого материала и способ обработки
(табл. 2). Если заготовка не обладает достаточной
жесткостью, достижимая точность м. б. подсчитана
из ур-ния оэ=7тго, где т — коэффициент, учитываю-
учитывающий жесткость заготовки.
229
МЕХАНИЧЕСКИЕ СВОЙСТВА
230
Таблица 2. Значения коэффициентов в формуле для определения
допуска на обработку при точении
Материал
Полистирол
Полиметилметакрилат
Фторопласт-4
Гетинакс
Текстолит
Фенопласт с порошковым древесным
наполнителем*
Стеклотекстолит на основе феноло-
формальдегидной смолы и алюмобо-
росиликатного стекловолокна . . . .
0,014
0,017
0,012
0,021
0,011
0,015
0,010
0,43
0,39
0,49
0,25
0,48
0,25
0,40
* При др. видах М. о. коэфф. Кип для фенопласта с по-
порошковым древесным наполнителем значительно отличаются от
указанных в таблице. Напр., при сверлении они соответст-
соответственно равны 0,038 и 0,33, при фрезеровании 0,030 и 0,48.
Точность обработки изделий резко снижается при
работе затупленным инструментом или при непра-
неправильно выбранных условиях резания. При этом наблю-
наблюдается большое искажение формы поверхности в ре-
результате нестабильного упруго-эластич. восстановле-
восстановления материала в подрезцовой зоне.
Для повышения точности обработки особо твердых
материалов и материалов с относительно большим уд.
сопротивлением резанию (напр:, стеклопластиков, по-
полиамидов, поливинилхлорида) применяют алмазные
инструменты. Прочие инструменты должны быть зато-
заточены наостро с обязательной последующей доводкой их
пастой карбида бора или алмазным кругом. При обра-
обработке необходимо применять охлаждение.
В табл. 3 приведены достижимые классы точности
при М. о. пластмасс.
Таблица 3. Достижимые классы точности при механической
обработке термопластов и реактопластов
Класс
точности
2
2а
Способ обработки
Наружное и внут-
внутреннее шлифование.
Двукратное развер-
развертывание
Наружное и внут-
внутреннее шлифование.
Однократное раз-
развертывание. Чисто-
Чистовое точение
Класс
точности
3
За
4
5
Способ обработки
Сверление. Чистовое
точение
Сверление. Чистовое
фрезерование
Черновое точение.
Чистовое фрезерова-
фрезерование
Черновое фрезерова-
фрезерование
Заключение. При М. о. широко применяются метал-
металлообрабатывающие или деревообрабатывающие станки.
И те и др. сконструированы без учета специфич. осо-
особенностей пластмасс и условий их обработки и поэтому
не обеспечивают высокопроизводительного изготовле-
изготовления изделий высокой точности. В связи с этим наблю-
наблюдается стремление к созданию специализированных
станков для обработки изделий из пластмасс. В этих
станках предполагается использовать бесступенчатый
привод в кинематич. цепях главного движения и по-
подачи. Принимая во внимание относительно небольшую
жесткость пластмассовой заготовки, необходимо повы-
повысить требования к точности ее центрирования и рав-
равномерности зажимных усилий фиксирующих элемен-
элементов станка и приспособлений.
Одновременно с совершенствованием оборудования
и инструмента необходимо специально изучать усло-
условия М. о. пластмасс и выработать минимально необ-
необходимый комплекс показателей, который обеспе-
обеспечивал бы научно обоснованный выбор геометрии
инструмента и режимов резания полимерных мате-
материалов.
Лит.: Руднев А. В., Королев А. А., Обработка
резанием стеклопластиков, М., 1969; С е м к о М. Ф. [и др.],
Механическая обработка пластмасс. Фрезерование, М., 1965;
Бобры нин Б. Н., Технология штамповки неметалличе-
неметаллических материалов, М., 1962; Kobayshi Akira, Machi-
Machining of plastics, N. Y., 1967; Encyclopedia of polymer science
and technology, v. 8, N. Y.— [a. o.], 1968, p. 338—74; Справоч-
Справочник машиностроителя, т. 5, кн. 2, М., 1964, гл. 10.
Ю.Н .Казанский.
МЕХАНИЧЕСКИЕ СВОЙСТВА полимеров
(mechanical properties, mechanische Eigenschaften,
proprietes mecaniques).
Содержание:
Классификация и общая характеристика механических
свойств 231
Деформационные свойства 231
Прочностные свойства 233
Фрикционные свойства 233
Физическое состояние и механические свойства . . . .2 34
Аморфное состояние 234
Кристаллическое (аморфно-кристаллическое)
состояние 236
Ориентированное состояние 236
Зависимость механических свойств от химического строе-
строения, состава и надмолекулярной структуры 2 37
Зависимость механических свойств от нек-рых внешних
факторов 239
Механические свойства полимеров — комплекс
свойств, определяющих механич. поведение полимеров
при действии на них внешних сил.
Для М. с. полимеров характерны:
1. Способность развивать под действием внешних
механич. сил большие обратимые (высокоэластические)
деформации, достигающие десятков, сотен и даже ты-
тысяч процентов. Эта способность характерна только
для полимерных материалов.
2. Релаксационный характер реакции тела на меха-
механич. воздействие, т. е. зависимость деформаций и на-
напряжений от длительности (частоты) воздействия. Эта
зависимость обусловлена отставанием деформации от
напряжения (см., напр., Гистерезисные явления) и мо-
может проявляться в чрезвычайно широком временном
диапазоне (от долей секунды до многих лет — см. Ре-
Релаксационные явления).
3. Зависимость М. с. полимера от условий его полу-
получения, способа переработки и предварительной обра-
обработки. Это связано с существованием в полимерных
телах разнообразных форм надмолекулярной структу-
структуры, времена перестройки к-рых м. б. настолько велики,
что полимер при одних и тех же условиях может устой-
устойчиво существовать в состояниях с различной морфоло-
морфологией.
4. Способность под действием анизотропного меха-
механич. воздействия приобретать резкую анизотропию
М. с. и сохранять ее после прекращения воздействия
(см. Анизотропия свойств).
5. Способность претерпевать под действием механич.
сил химич. превращения (см. Механохимия).
Общий характер механич. поведения конкретного
полимерного тела определяется тем, в каком физич.
состоянии оно находится. Линейные и разветвленные
полимеры могут находиться в трех основных аморфных
состояниях — стеклообразном, высокоэластическом и
вязкотекучем, трехмерные (пространственные, сшитые)
полимеры — только в первых двух из этих состояний.
Многие полимеры могут также находиться в кристал-
кристаллическом состоянии, существенной особенностью к-рого
является то, что практически всегда в полимерном теле
наряду со строго упорядоченными кристаллич. обла-
областями сохраняются области с аморфной структурой
(поэтому такое состояние наз. также аморфно-кристал-
аморфно-кристаллическим, частично кристаллическим или полукри-
полукристаллическим). Строго кристаллич. состояние реали-
реализуется только в полимерных монокристаллах.
При рассмотрении М. с. полимеров в особую группу
выделяют ориентированное состояние, в к-ром могут
находиться как аморфные, так и кристаллич. полимеры
и для к-рого характерна анизотропия М. с.
Область применения полимера во многом определя-
определяется тем, в каком состоянии находится он в температур-
231
МЕХАНИЧЕСКИЕ СВОЙСТВА
232
ном интервале эксплуатации (обычно от —40 до 40 °С).
Полимеры, находящиеся в этом интервале в высоко-
эластич. состоянии, наз. эластомерами. Из эластомеров
широкое технич. применение находят резины. Полимер-
Полимерные материалы, находящиеся в условиях эксплуатации
в стеклообразном или кристаллич. состоянии, наз. пла-
пластическими массами. Последние используют в виде
объемных изделий и пленок. Одноосноориентированные
полимеры широко применяют в качестве волокон (см.
Волокна химические).
Классификация и общая характеристика
механических свойств
Под действием механич. сил все тела деформируются,
а при достаточно сильных или длительных воздействи-
воздействиях разрушаются. В соответствии с этим различают де-
деформационные и прочностные свойства. В отдельную
группу М. с. выделяют фрикционные свойства, прояв-
проявляющиеся при движении твердого полимерного тела по
поверхности др. тела.
Для изучения М. с. и определения механич. характе-
характеристик материалов проводятся по определенным мето-
методикам механич. испытания. Испытания различаются
типом деформации (одноосное и двухосное растяжение
и сжатие, всестороннее сжатие, изгиб, сдвиг, кручение,
вдавливание и др.) и режимом нагружения (постоянная
нагрузка, нагрузка, обеспечивающая линейный рост
деформации или ее постоянство, циклич. нагрузка,
удар и др.). Выбор метода испытаний определяется как
их целями, так и типом исследуемого материала. О ме-
методах испытаний различных полимерных материалов
см. Испытания лакокрасочных материалов и покры-
покрытий, Испытания пластических масс, Испытания ре-
резин, Испытания химических волокон.
Для качественного и количественного описания М. с.
полимеров пользуются теми же понятиями и характе-
характеристиками, что и для описания М. с. неполимерных
материалов. Вместе с тем особенности поведения поли-
полимеров требуют введения новых понятий, а иногда и
нек-рого изменения смысла принятых.
Деформационные свойства. Упругость и в ы-
сокоэластичность (эластичность) — свойст-
свойства тела восстанавливать свою форму и размеры после
прекращения действия внешних сил. В узком смысле
под «упругими» часто имеют в виду только мгновенно-
упругие (точнее, происходящие со скоростью звука)
деформации, к-рым отвечают модули упругости поряд-
порядка 103—105 Мн/м2 A04—106 кгс/см2). Для запаздываю-
запаздывающих механически обратимых деформаций, к-рым соот-
соответствуют существенно меньшие модули упругости
A—10 Мн/м2— для наполненных резин, 0,1 —10
Мн/м2 — для типичных «мягких» резин, 10~4—
0,1 Мн/м2 — для пластифицированных резин и гелей),
обычно употребляют термин «высокоэластические»,
относя его и к малым деформациям этих тел.
При равновесном деформировании упругих тел вся
работа внешних сил обратимо запасается в материале,
и в соотношения между напряжениями и деформациями
не входит временной фактор. В линейной области ме-
механич. поведения упругого тела компоненты тензора
деформаций 8rs выражаются как линейные комбинации
компонент тензора напряжений oik: ?rs = 2brsik oik
и наоборот: oih = 2a/ftr.tSers. В этих соотношениях коэфф.
пропорциональности a^rs наз. модулями упругости,
коэфф. brsik— податливостями. Для анизотропного тела
независимыми м. б. только 21 коэффициент, для изо-
изотропного сжимаемого тела число характеризующих
его параметров понижается до двух, для изотропного
несжимаемого тела — до одного. При одноосном растя-
растяжении линейного упругого тела связь между относи-
относительным удлинением 8 и растягивающим напряжением а
задается законом Гука: а = Ее, где Е — модуль Юнга.
Аналогично определяется модуль упругости при сдви-
сдвиге и модуль всестороннего сжатия. Для нелинейного
упругого тела связь между компонентами тензоров на-
напряжений и деформаций задается нелинейными соот-
соотношениями произвольного строения, причем в общем
случае может утрачиваться даже однозначность этих
соотношений.
Мгновенно-упругие деформации полимеров обуслов-
обусловлены небольшими взаимными смещениями атомов,
приводящими к изменению расстояний между валентно
не связанными атомами и валентных углов. Высоко-
эластич. деформации, будучи формально аналогичными
упругим, отличаются от них специфически «полимер-
«полимерным» механизмом: они связаны с перемещением от-
отдельных участков макромолекул, приводящим к изме-
изменениям их конформаций. Поэтому высокоэластич. де-
деформации приводят к изменениям энтропии системы,
обычно составляющим определяющую часть изменений
свободной энергии тела при деформировании, тогда как
мгновенно-упругие деформации сопровождаются изме-
изменением только внутренней энергии.
Развитие больших высокоэластич. деформаций про-
происходит в нелинейной области механич. поведения тела.
Однако и в этом случае для характеристики сопротив-
сопротивления тела деформированию используют термин модуль
упругости (или высокоэластичности), понимая под
этим отношение напряжения к деформации (см. Мо-
Модуль). В области высокоэластич. деформаций модуль
упругости на 3—4 десятичных порядка меньше модуля
всестороннего сжатия. Поэтому изменением объема
тела при высокоэластич. деформации обычно прене-
пренебрегают.
Понятия о мгновенно-упругих и высокоэластич. де-
деформациях представляют собой идеализацию, посколь-
поскольку деформирование реальных полимерных тел всегда
сопровождается диссипативными эффектами — часть
работы внешних сил необратимо рассеивается в виде
тепла. Поэтому реальные полимеры являются вязко-
упругими или упруговязкими (см. Кельвина модель,
Максвелла модель, Болъцмана — Волыперры уравнения).
Эффекты, связанные с вязкоупругими релаксационны-
релаксационными явлениями, наиболее резко выражены в переход-
переходных областях между стеклообразным и высокоэла-
высокоэластическим и высокоэластическим и вязкотекучим со-
состояниями.
Жесткость и мягкость — качественные
характеристики деформируемости твердых тел. Жест-
Жесткими обычно наз. полимерные материалы, имеющие
модуль Юнга выше 103 Мн/м2 A02 кгс/мм2), а мягки-
мягкими — менее 102 Мн/м2 A0 кгс/мм2).
Вынужденная высокоэластич-
высокоэластично с т ь (квазипластичность) — свойство твердых по-
полимерных тел испытывать большие деформации, имею-
имеющие тот же механизм, что и высокоэластич. деформации,
но после снятия напряжения восстанавливающиеся
только при повышении темп-ры образца (отжиге) или
его набухании. Для развития вынужденной высокоэла-
высокоэластич. деформации необходимо, чтобы напряжение пре-
превысило нек-рое значение ав —предел вынуж-
вынужденной высокоэластичности. Значение
ав увеличивается с ростом частоты или скорости воз-
воздействия (см. Высокоэластичностъ вынужденная).
Пластичность — свойство твердых тел раз-
развивать необратимые (истинно остаточные) деформации.
Необратимые деформации жидких тел (вязкое течение)
развиваются при любом напряжении. Для твердых
тел их осуществление требует достижения нек-рого
наименьшего напряжения, называемого пределом
текучее ти. Практически за предел текучести
принимают значение напряжения, при к-ром на кривой
зависимости напряжения от деформации наблюдается
точка максимума или выход на постоянное напряже-
напряжение. Часто пределом текучести наз. предел вынужден-
вынужденной высокоэластичности.
233
МЕХАНИЧЕСКИЕ СВОЙСТВА
234
Ползучесть (крип) — свойство твердых тел
медленно накапливать деформации при воздействии
постоянных напряжений. Ползучесть проявляется да-
даже у весьма жестких полимеров и обусловлена разви-
развитием как пластической, так и квазипластической де-
деформаций (см. Ползучесть).
Вязкость — свойство жидких тел сопротив-
сопротивляться необратимому изменению формы. Вязкое со-
сопротивление приводит к необратимому выделению
тепла при деформации. Скорость сдвиговой деформа-
деформации идеальной вязкой жидкости пропорциональна
приложенному напряжению сдвига: у = т/тд, где ц —
коэффициент вязкости, или просто вяз-
вязкость. Величину, обратную вязкости, называют т е-
кучестью.
Внутреннее трение — свойство твердого
полимерного тела, характеризующее рассеяние в нем
энергии при упругих и высокоэластич. деформациях.
Это свойство обусловливает релаксационный характер
развития этих деформаций. Если упругое твердое тело
имеет внутреннее трение, оно наз. вязкоупру-
г и м. При линейном вязкоупругом поведении соблю-
соблюдается пропорциональность деформации напряжению
в каждый момент времени. Полимерные жидкости,
проявляющие наряду с текучестью упругость формы,
наз. упруговязкими (см. Реология).
Прочностные свойства. Прочность — свойство
твердого тела сохранять целостность при действии
нагрузок. Обычно прочностные свойства материалов
характеризуют пределом прочности (или
просто прочностью) — величиной напряжения, при
котором происходит разрушение тела в условиях на-
гружения, ведущегося в определенном режиме рос-
роста деформации и обычно продолжающегося не бо-
более нескольких минут. При более длительных воз-
воздействиях разрушение происходит и при напряжени-
напряжениях, значительно меньших предела прочности (см. Проч-
Прочность).
Долговечность — прочностное свойство, ха-
характеризующее продолжительность времени от момента
нагружения до разрушения полимерного тела при
сохранении постоянного напряжения. Долговечность
резко уменьшается при увеличении напряжения и
темп-ры (см. Долговечность).
Хрупкость — свойство твердого тела разру-
разрушаться при малых упругих деформациях. Проявле-
Проявление хрупкости или пластичности определяется не толь-
только свойствами самого тела, но и временем воздействия:
тела, разрушающиеся хрупко при больших кратко-
кратковременных воздействиях, могут пластически деформи-
деформироваться до разрушения при более слабых длитель-
длительных воздействиях.
Ударная вязкость — свойство тел сопро-
сопротивляться кратковременным ударным воздействиям;
измеряется отношением работы, затрачиваемой на
разрушение образца, к площади поверхности, образо-
образовавшейся в результате разрушения. Ударная вязкость
является свойством, в известной мере противополож-
противоположным хрупкости. При переходе от хрупкого разрушения
к пластическому ударная вязкость возрастает.
Твердость — свойство твердого тела противо-
противодействовать внедрению в него другого тела. При вдав-
вдавливании предмета в материал возникают местные пла-
пластические и квазииластич. деформации, сопровождаю-
сопровождающиеся при дальнейшем увеличении давления локальным
разрушением. Твердость обычно повышается с рос-
ростом жесткости.
Усталостная прочность (выносли-
(выносливость) — свойство тела выдерживать, не разрушаясь,
многократные нагрузки (см. Утомление).
Фрикционные свойства. Для количественного описа-
описания этих свойств используют коэффициент
трения — отношение тангенциальной силы к нор-
нормальному усилию и износостойкость, харак-
характеризующую скорость разрушения материала при тре-
трении (см. Трение, Истирание).
Физическое состояние и механические свойства
Аморфное состояние. Различие между отдельными
физич. состояниями аморфных полимеров состоит
в разной реакции полимеров, находящихся в этих со-
состояниях, на механич. воздействие — упругой в стек-
стеклообразном состоянии, гл. обр. высокоэластической
в высокоэластическом и развитием необратимых дефор-
деформаций в вязкотекучем. Из-за релаксационного харак-
характера высокоэластич. деформации и вязкого течения ха-
характер реакции на механич. воздействие существенно
зависит от длительности воздействия. В определенном
диапазоне темп-р тело может реагировать на кратко-
кратковременное воздействие упруго, а при длительных
(порядка времени релаксации высокоэластич. дефор-
деформации или большего) проявлять высокоэластичность.
При более высоких темп-pax вследствие уменьшения
с ростом темп-ры времени релаксации тело может
проявлять высокоэластичность при кратковременных
воздействиях, а при длительных вести себя как вязкая
жидкость. Т. обр., разделение на стеклообразное, высо-
высокоэластическое и вязкотекучее состояния связано
с временным режимом воздействия.
Чтобы придать определенность разделению на со-
состояния, при нахождении темп-р переходов выбирают
некоторую скорость нагревания (напр., 1 сС/сек) и по
резкому изменению величины деформации определяют
температуры переходов (см. Термомеханическое иссле-
исследование). Поскольку упругая и высокоэластич. дефор-
деформации имеют характерные, сильно различающиеся
между собой значения модулей, деление на состояния
проводят также по значению модуля, измеряемого
в динамич. режиме (см. Динамические свойства) или
в режиме релаксации напряжений. Стеклообразному
состоянию отвечают значения модуля 10:3—104 Мн/м'1
A04—105 кгс/см2), высокоэластическому — порядка
\0~хМ н/ м2 A0 кгс/см2)', переход в вязкотекучее состояние
(темп-pa текучести) фиксируется по падению модуля до
значений менее К)-1»5 Мн/м2 A0~0'5 кгс/см2). При таком
способе разделения в особое физич. состояние (вязко-
упругое) выделяют иногда переходную область между
стеклообразным и высокоэластич. состояниями, к-рой
отвечают промежуточные значения модуля. Эта область
может охватывать десятки градусов.
В стеклообразном состоянии ниже
темп-ры хрупкости Гхр (см. Хрупкости температура)
полимер ведет себя как хрупкое твердое тело, разру-
разрушаясь при небольших, до нескольких процентов, отно-
относительных деформациях (рис. 1, кривая 1). Выше Тхру
при напряжениях, больших
/ ав — предела текучести (вы-
(вынужденной высокоэластич-
ности), развивается вынуж-
Рис. 1. Кривые растяжения амор-
аморфного полимера при различных
темп-рах: 1—ниже темп-ры хруп-
хрупкости, 2 — между темп-рой хруп-
хрупкости и темп-рой стеклования,
3 — выше темп-ры стеклования.
денная высокоэластич. деформация, к-рая может до-
достигать десятков и сотен процентов; при этом про-
происходит переход от хрупкого разрушения к квазипла-
квазипластическому, сопровождающийся обычно резким ростом
ударной вязкости (кроме тех случаев, когда падение
прочности происходит быстрее роста предельной де-
деформации). Растяжение полимера при темп-pax выше
Гхр (рис. 1, кривая 2) у многих полимеров протекает
неоднородно по образцу, образуется локальное суже-
сужение — шейка, в к-рой материал сильно ориентирован.
Удлинение
235
МЕХАНИЧЕСКИЕ СВОЙСТВА
236
По мере растяжения шейка распространяется на весь
образец (см. также Высокоэластичностъ вынужденная).
С ростом темп-ры модуль Юнга, прочность, твердость
падают, однако их изменение не превышает, как прави-
правило, одного порядка. С ростом темп-ры уменьшаются
также значения предела текучести, достигая нуля
при темп-ре стеклования Тс (см. Стеклования темпе-
температура). Восстановление формы образца достигается
нагреванием до темп-ры, несколько превышающей Тс.
В высокоэластическом состоянии
высокоэластич. деформация может развиться при любом
напряжении. Переход в это состояние при Тс сопро-
сопровождается быстрым изменением нек-рых равновесных
физич. свойств, в частности коэфф. теплового расшире-
расширения. Переход в стеклообразное состояние м. б. осущест-
осуществлен также изменением временного фактора воздейст-
воздействия на материал, напр, частоты деформирования.
В этом случае говорят о механич. стекловании. Каж-
Каждой частоте отвечает определенная темп-pa Тм, при
к-рой развитие деформаций сопровождается наибольши-
наибольшими механич. потерями. Положение максимума механич.
потерь определяет значение темп-ры стеклования, а его
зависимость от частоты — кинетический (релаксаци-
(релаксационный) характер стеклования.
Вблизи Тм рост деформации с темп-рой происходит
наиболее резко (рис. 2). Это связано с тем, что в этой
области время релаксации падает при линейном росте
темп-ры (вернее при линейном уменьшении обратной
темп-ры) по закону, близ-
близкому к экспоненциально-
экспоненциальному. Описать единым обра-
образом деформацию полимера
Рис. 2. Зависимость динамич.
модуля типичного аморфного
полимера от темп-ры (частота
порядка нескольких герц);
ТЧ — темп-pa текучести, Тм
и Тс — темп-ры соответствен-
соответственно механич. и структурного
стеклования.
Температура
в переходной области в определенном интервале времен
и частот воздействия позволяет суперпозиции принцип
температурно-временной (температурно-частотный),
устанавливающий количественно эквивалентность влия-
влияния роста темп-ры и уменьшения времени воздействия
(увеличения частоты, см. также Александрова — Ла-
зуркииа частотно-температурный метод). С ростом
темп-ры происходит уменьшение внутреннего трения,
приводящее к уменьшению времени релаксации, и при
достаточно высоких темп-pax развитие высокоэластич.
деформации происходит за доли секунды. Эту область
называют иногда плато высокоэластичности. Растяже-
Растяжение полимера в высокоэластич. состоянии носит суще-
существенно нелинейный характер и при больших деформа-
деформациях сопровождается ориентацией макромолекул, к-рая
может приводить к обратимой кристаллизации. При
больших деформациях проявляется существенное раз-
различие в поведении линейных и пространственных (сши-
(сшитых) полимеров. Если деформация сшитых полимеров
обратима, то у линейных полимеров развитие высоко-
высокоэластич. деформации сопровождается также развитием
необратимых деформаций.
В вязкотекучем состоянии доминирую-
доминирующим является вязкое течение, осуществляемое в ре-
результате необратимого перемещения целых макромоле-
макромолекул или даже агрегатов макромолекул. Особенностью
течения полимерных тел является то, что одновремен-
одновременно с ним развивается обратимая высокоэластич. дефор-
деформация. Это приводит к ряду специфич. эффектов, в част-
частности к разбуханию струи, вытекающей из трубы
(высокоэластическое восстановление), Вайссенберга эф-
эффекту и др. Для полимеров в вязкотекучем состоянии
характерно также явление тиксотропии — обратимое
разрушение структуры в процессе течения, приводя-
приводящее к падению вязкости.
К свойствам полимеров в вязкотекучем состоянии
близки свойства конц. р-ров полимеров (см. Вязкотеку-
чее состояние). М. с. разб. р-ров полимеров близки
к свойствам вязких простых жидкостей, причем с ростом
концентрации полимера, а также мол. массы вязкость
р-ров растет. Даже в очень разб. р-рах полимеров
наблюдается градиентная зависимость вязкости (см.
Вязкости аномалия).
Кристаллическое (аморфно-кристаллическое) состоя-
состояние. М. с. полимеров в аморфно-кристаллич. состоянии
во многом определяются тем, что в этом состоянии
полимеры представляют собой своеобразные микрокон-
микроконструкции, состоящие из связанных между собой
элементов (кристаллических и аморфных областей) с раз-
различными механич. характеристиками. Различные обла-
области полимера деформируются по-разному, а в пределах
одной области разные макромолекулы напряжены и
деформированы также различно. Физич. методы позво-
позволяют установить особенности реакции отдельных струк-
структурных элементов на механич. воздействие. В част-
частности, исследование смещения рефлексов на широко-
широкоугловых рентгенограммах кристаллич. полимеров при
их растяжении позволило рассчитать величины дефор-
деформации и модули Юнга кристаллич. участков. Рассчи-
Рассчитанные модули для всех полимеров превышали модули
Юнга, определенные по механич. испытаниям, причем
для полиэтилена при растяжении примерно на 10% на
долю кристаллич. участков пришлась деформация
всего в 0,1%, а модуль Юнга кристаллич. решетки до-
достиг значения 25 000 Мн/м2 B500 кгс/мм2), превысив
значение механич. модуля Юнга на 2 порядка.
При небольших напряжениях и деформациях бла-
благодаря существенному вкладу в общую деформацию
деформации аморфных областей М. с. аморфно-кристал-
аморфно-кристаллич. полимеров имеют сходство с М. с. аморфных
полимеров. При повышении темп-ры происходит умень-
уменьшение модуля Юнга, причем при переходе через темп-ру
стеклования аморфных участков иногда наблюдается
падение модуля, однако не на 4—5, как в случае аморф-
аморфных полимеров, а всего на 1 — 2 порядка. Ниже опреде-
определенной темп-ры аморфно-кристаллич. полимеры, как
и аморфные, разрушаются обычно хрупко (исключение
составляют полипропилен и нек-рые полиимиды, напр,
полипиромеллитимид, сохраняющие способность к боль-
большим деформациям до темп-ры —200 °С).
При больших напряжениях аморфно-кристаллич.
полимеры проявляют вынужденную высокоэластнч-
ность. При этом деформируются как аморфные, так и
кристаллич. области, разрушаются одни кристаллич.
образования и возникают другие. У многих полимеров
растяжение в кристаллич. состоянии идет с образова-
образованием шейки, в к-рой происходит ориентация макромоле-
макромолекул, сопровождающаяся обычно переходом от сферолит-
ной кристаллич. структуры к фибриллярной; при этом
происходит резкое изменение М. с. полимера.
Повышение темп-ры вызывает изменение механич. ха-
характеристик — уменьшение прочности, предела теку-
текучести, твердости и увеличение ударной вязкости. При
темп-ре плавления кристаллич. полимер переходит
в вязкотекучее состояние. Этот переход является фазо-
фазовым, но темп-pa плавления зависит от условий кристал-
кристаллизации. М. с. аморфно-кристаллич. полимеров зависят
от степени кристалличности. Так, с ростом степени
кристалличности растет модуль Юнга.
Ориентированное состояние. В одноосном и двухос-
двухосном ориентированных состояниях могут находиться
и кристаллические, и аморфные полимеры. М. с. ориен-
ориентированных полимеров существенно зависят от степени
ориентации. С повышением степени одноосной ориен-
ориентации возрастает прочность (более чем на порядок),
237
МЕХАНИЧЕСКИЕ СВОЙСТВА
238
а деформируемость, как правило, падает. Повышение
прочности носит четко выраженный анизотропный ха-
характер и происходит только в направлении ориентации;
в перпендикулярном направлении прочность, как пра-
правило, падает, причем иногда настолько сильно, что
может произойти расслоение полимера (волокна). См.
также Ориентированное состояние.
Зависимость механических свойств от химического
строения, состава и надмолекулярной структуры
Реализация при определенной темп-ре физич. состоя-
состояния полимера с присущим ему комплексом М. с, изме-
изменение состояния с изменением темп-ры и особенности
механич. поведения в определенном состоянии опреде-
определяются, в первую очередь, химич. строением и свойст-
свойствами составляющих полимер макромолекул: характе-
характером расположения мономерных групп (линейным, с раз-
разветвлениями или в виде пространственной сетки), гиб-
гибкостью полимерной цепи, степенью регулярности ее
строения, конкретным строением мономерного звена,
от к-рого зависит межмолекулярное взаимодействие,
а также мол. массой и молекулярно-массовым распреде-
распределением.
Выше уже отмечалось различие в реализующихся
физич. состояниях линейных и пространственных поли-
полимеров и их различное поведение в высокоэластич. со-
состоянии. Появление поперечных связей между макро-
макромолекулами приводит к образованию полимерного кар-
каркаса и вызывает понижение темп-ры стеклования,
в случае достаточно частых поперечных связей настоль-
настолько резкое, что полимер может перейти из вязкотеку-
чего состояния не только в высокоэластическое, но
и непосредственно в стеклообразное {отверждение
нек-рых термореактивных смол). В стеклообразном
состоянии влияние полимерного каркаса проявляется
особенно сильно при больших напряжениях (приводит
к подавлению вынужденной высокоэластичности). Реа-
Реализация у линейных полимеров вязкотекучего состоя-
состояния определяет их термопластичвость — способность
к многократному размягчению.
Следует отметить, что полимерный каркас может
образоваться не только за счет химич. связей, но также
благодаря присутствию в полимерной цепи участков,
между к-рыми существует сильное межмолекулярное
взаимодействие. При этом достигается термолабиль-
термолабильность поперечных связей между макромолекулами.
Примером таких материалов являются термоэласто-
пласты — полимеры, обладающие свойствами эласто-
эластомеров и термопластичностью, напр, блоксополимеры
эластомеров со стиролом, б локсопо лимеры, имеющие
кристаллизующиеся и некристаллизующиеся участки,
и нек-рые полиуретаны, у к-рых роль термолабильных
узлов играют сегрегированные диизоцианатные блоки.
К образованию полимерного каркаса может приводить
также введение в полимерную цепь кислотных групп
с последующей нейтрализацией их ионами металлов
(см. Мономеры).
Термодинамич. гибкость макромолекул является необ-
необходимым условием проявления высокоэластичности.
Однако для реализации этого состояния необходима
также достаточная кинетическая (механическая) гиб-
гибкость, зависящая от межмолекулярного взаимодейст-
взаимодействия и растущая с ростом темп-ры. Все эластомеры яв-
являются гибкоцепными полимерами. Жесткоцепные по-
полимеры и гибкоцепные при достаточно сильном межмо-
межмолекулярном взаимодействии являются пластиками.
Сочетание значительной жесткости со способностью
к большим деформациям реализуется при наличии меж-
между жесткими участками гибких сочленений (нек-рые
полиимиды и гетерополиарилены).
Стереорегулярность строения (см. Стереорегулярные
полимеры) в ряде случаев необходимое условие реали-
реализации кристаллич. состояния. Регулярность строения
макромолекулы может оказать решающее влияние
на М. с. полимера. Так, атактический полипропилен
является эластомером, а изотактический полимер кри-
кристаллизуется и обладает волокнообразующими свой-
свойствами.
Мол. масса влияет на темп-рный интервал реализации
различных физич. состояний аморфного полимера
(рис. 3). При малых мол. массах поведение полимера
сходно с поведением низ-
низкомолекулярных веществ.
У
ур щ
Увеличение мол. массы ве-
ведет к расширению интер-
интервала высокоэластичности
Рис. 3. Зависимость темп-р те-
текучести (Тт), стеклования
(Тс) и хрупкости (Тхр) от моле-
молекулярной массы; I — область
реализации вязкотекучего сос-
состояния, II — вьгсокоэластиче-
ского, III и IV — стеклообраз-
стеклообразного (в области IV полимер
разрушается хрупко).
Т,
Мал масса
вследствие более сильного влияния мол. массы на
темп-ру текучести, чем на темп-ру стеклования. В вя;*-
котекучем состоянии существует критпч. мол. масса,
при достижении к-рой особенно сильно проявляются
упруговязкие свойства (подробнее см. Вязко текучее
состояние). Реализация кристаллич. состояния с вы-
высокой степенью упорядоченности требует достаточно
узкого молекулярно-массового распределения с малой
долей низкомолекулярных фракций. Хорошими во-
волокнообразующими свойствами обладают полимеры с
мол. массой в интервале 1,5-10*—8-Ю4 (см. Волокно-
образующие полимеры).
М. с. полимерных материалов существенно зависят
от вводимых в них добавок: наполнителей, пластифика-
пластификаторов, стабилизаторов, сшивающих агентов, структуро-
образователей и др. (см. Ингредиенты полимерных ма-
материалов). Даже при небольших количествах добавок
изменение М. с. может быть очень сильным, как, напр.,
в случае введения сшивающих агентов, вызывающих
образование поперечных связей между макромолеку-
макромолекулами (см. Вулканизация, Отверждение).
Наполнение полимеров различными веществами орга-
нич. и неорганич. происхождения в количествах, до-
достигающих более 50% (по массе), а иногда и 90%, при-
приводит к получению композиционно неоднородных мате-
материалов с самыми разнообразными М. с.
Наполнение пластмасс волокнистыми материалами
является наиболее эффективным способом получения
жестких (высокомодульных) и высокопрочных материа-
материалов. Пластик на основе волокна бора, например, поч-
почти не уступает по прочности стали, имея в 4 раза
меньшую плотность. Особенностью армированных ма-
материалов является то, что прочность и модуль при
сдвиге для них м. б. более чем на порядок меньше
прочности и модуля при растяжении (см. Армирован-
Армированные пластики).
При заданных химич. строении и составе М. с. пла-
пластиков сильно зависят от реализующейся надмолеку-
надмолекулярной структуры, что обусловливает зависимость М. с.
от условий получения, переработки и последующей
обработки полимерного материала. Наиболее подробно
изучено влияние особенностей сферолитной структуры
на М. с. кристаллич. полимеров. Влияние надмолеку-
надмолекулярной структуры на М. с. аморфных полимеров пока
практически не изучено, хотя сам факт такого влияния
несомненен.
В таблице приведены для сравнения механич. ха-
характеристики нек-рых полимерных материалов. Дан-
Данные взяты из разных источников. Таблица носит ке
справочный, а иллюстративный характер.
239
МЕХАНИЧЕСКИЕ СВОЙСТВА
Механические характеристики нек-рых полимерных материалов при комнатной температуре
240
Полимер
Резина из натурального каучука
ненаполненная
наполненная
Резины из синтетич. каучуков ....
Полиэтилен
низкой плотности
высокой плотности
Феноло-формальдегидная смола отвер-
жденная
Полистирол
Полиметилметакрилат •
Поликапролактам
ненаполненный пластик (капрон)
наполненный стеклопластик (ка-
пролон GH)
волокно (капроновый корд) ....
Полиэтилентерефталат
пленка
. высокопрочное волокно (лавсан)
Полиимид ПМ (пленка)
Стеклопластик СВАМ
Армированный пластик на основе во-
волокна бора
Прочность при растя-
растяжении, Мн/м2 {тс/мм2)
30 C)
34 C,4)
20 — 50 B-5)
12—16 A,2-1,6)
22—33 B,2-3,3)
40-50 D-5)
30-70 C-7)
70 G)
55-70 E,5-7)
350-450 C5 — 45)
740-880 G4-88)
170 A7)
800 — 1000 (80 — 100)
160-180 A6-18)
450-470 D5-47)
2200 B20)
Модуль Юнга,
Мн/м* (кгс/мм2)
1,3* @,13)
7* @,7)
1-10* @,1-1)
150-250 A5-25)
550—800 E5—80)
2000-2500 B00-250)
2800 — 3500 B80—350)
2700 B70)
800-1000 (80-100)
2500 — 3000 B50 — 300)
3500 C50)
3500 C50)
5000 — 16000 E00 — 1600)
3500 C50)
35000 C500)
250 000 B5000)
Относи-
Относительное
удлинение
при растя-
растяжении, %
850
650
500-1000
100 — 600
200-900
2-3
V
100-150
15—20
100
10 — 20
50-100
-
-
Физич. состояние
Высокоэластичесьое
То же
То же
Кристаллическое
То же
Стеклообразное
Стеклообразное
То же
Кристаллическое
То же
То же
Кристаллическое
или стеклообразное
-
-
-
* При 300%-ном растяжении.
Зависимость механических свойств от некоторых
внешних факторов
Для полимеров характерно старение — изменение
структуры со временем, сопровождающееся изменением
механич. характеристик. Старение может вызываться
как химич. процессами (в основном деструкцией), так
и структурными перестройками, напр, медленной кри-
кристаллизацией. Одним из проявлений старения является
растрескивание полимерных материалов, т. е.
появление на поверхности изделий и в объеме наруше-
нарушений сплошности материала, происходящее при незна-
незначительных внешних напряжениях или даже в нена-
груженном материале. Растрескивание вызывается
внутренними напряжениями, обусловленными струк-
структурной неоднородностью полимерных материалов и
возникающими в процессе получения изделия и его
эксплуатации. В массивных изделиях из пластиков,
особенно армированных, важную роль в возникновении
внутренних напряжений играют градиенты темпера-
температур, появляющиеся в изделии при изменении температу-
температуры среды.
Скорость старения существенно зависит от среды,
в к-рой оно происходит, и от темп-ры (с ростом послед-
последней скорость резко возрастает). Изменение механич.
характеристик даже в обычных атмосферных условиях
м. б. очень сильным (см. Атмосферостойкостъ). Старе-
Старение часто идет неравномерно, наблюдается явление
насыщения — после резкого изменения М. с. в первые
несколько месяцев в последующие месяцы или даже
годы М. с. почти не меняются. В др. случаях процесс
носит ускоряющийся характер — нек-рое время не
наблюдается изменения М. с, а затем происходит их
быстрое изменение. Для борьбы с ухудшением меха-
механических характеристик в полимер вводят стаби-
стабилизаторы.
Влияние агрессивных сред на М. с. обусловлено тем,
что многие вещества, в частности к-ты и щелочи, вы-
вызывают протекание в полимерах химич. процессов. Вли-
Влияние среды м. б. значительным даже тогда, когда она
является химически нейтральной. Проникновение воды
в микропустоты, имеющиеся в полимере, вызывает
изменение деформационных и прочностных свойств,
причем действие воды м. б. как пластифицирующим,
так и расклинивающий (эффект Ребиндера). Действие
среды на поверхность обычно сказывается гл. обр. на
прочностных характеристиках.
Действие различных ионизирующих излу-
излучений при больших дозах приводит к уменьшению
прочности, в несколько меньшей степени влияя на де-
деформационные свойства. При этом действие излучения
часто носит критич. характер — до определенной дозы
прочность не изменяется (у ряда полимеров даже воз-
возрастает благодаря эффекту радиационного сшивания),
начиная же с некоторой дозы происходит резкое паде-
падение прочности (см. Радиационная стойкость).
Диапазон «рабочих» темп-р наиболее распространен-
распространенных полимерных материалов на основе карбоцепных
полимеров обычно не превышает 100—150 СС. При более
высоких темп-pax происходит резкое изменение М. с.
(уменьшение жесткости, прочности, твердости), свя-
связанное с приближением к темп-ре текучести аморфных
или темп-ре плавления кристаллич. полимеров (см.
Теплостойкость). Вплоть до темп-р 300—400 СС спо-
способны сохранять прочность и жесткость нек-рые гете-
роцепные полимеры, напр. кремнийорганические, но-
лифениленоксиды, полиимиды, полибензимидазолы. Из-
Изменение М. с. перечисленных полимеров обычно бывает
связано не с изменением агрегатного состояния, а с тер-
термической деструкцией (см. Термостойкость).
При низких темп-pax для полимеров характерна
хрупкость, что в случае ряда материалов, напр, резин,
241
МЕХАНОХИМИЯ
242
приводит к невозможности их эксплуатации (см. Мо-
Морозостойкость).
Высокое давление приводит к существенному изме-
изменению М. с. полимеров. Вызвано это тем, что модуль
Юнга и модуль всестороннего сжатия полимеров пре-
превышают прочность и предел текучести не на 3—4 по-
порядка, как у металлов, а всего на 1—2 порядка. Поэ-
Поэтому при напряжениях, значительно меньших разру-
разрушающего, может происходить изменение объема, сопро-
сопровождающееся существенным изменением взаимодейст-
взаимодействия структурных элементов полимера. У пластиков
с ростом давления происходит повышение темп-ры
стеклования, модуля Юнга, прочности при растяжении
и сжатии и предела текучести. У полистирола, напр.,
при комнатной темп-ре и давлении 200—300 Мн/м2
B000—3000 кгс/см2) наблюдается переход от хрупкого
разрушения к пластическому, причем предел текуче-
текучести в области давлений выше критического практически
линейно растет с повышением давления. Под влиянием
высокого давления в полимерах может совершаться
перестройка надмолекулярной структуры, как обра-
обратимая, так и необратимая.
Лит.: К о б е к о П. П., Аморфные вещества, М.— Л.,
1952; К а р г и н В. А., Слонимский Г. Л., Краткие
очерки по физико-химии полимеров, 2 изд., М., 1967; А л ф-
р е й Т., Механические свойства высокополимеров, пер. с англ.,
М., 1952; Ф е р р и Д ж., Вязкоупругие свойства полимеров,
пер. с англ., М., 1963; Тобольский А., Свойства и
структура полимеров, пер. с англ., М., 1964; У о р д Й., Ме-
Механические свойства твердых полимеров, пер. с англ., М.,
1974; Бартенев Г. М., Зуев Ю. С, Прочность и раз-
разрушение высокоэластических материалов, М.—Л., 1964;
Гуль В. Е., Кулезнев В. Н., Структура и механиче-
механические свойства полимеров, М., 1972; Гуль В. Е., Структура
и прочность полимеров, 2 изд., М., 1971; Бартенев Г. М.,
Лаврентьев В. В., Трение и износ полимеров, Л., 1972;
Лодж А., Эластические жидкости, пер. с англ., М., 1969;
Рабинович А. Л., Введение в механику армированных
полимеров, М.— Л., 1970; Малмейстер А. К., Т а-
мужВ.П., Тетере Г. А., Сопротивление жестких поли-
полимерных материалов, Рига, 1972; АйнбиндерС.Б. [и др.],
Свойства полимеров при высоких давлениях, М., 1973; Конст-
Конструкционные полимеры, под ред. П. М. Огибалова, М., 1972;
Регель В. Р., Слуцкер А. И., Т о м а ш е в-
с к и й Э. Е., УФН, 106, № 2, 193 A972); Kargin V. A.,
Slonimski G. L., Mechanical properties, кн.: Encyc-
Encyclopedia of polymer science and technology, v. 3, N. Y.—[a. o.],
1968, p. 445; F e г г у J. D., Viscoelastic properties of polymers,
2 ed., N. Y.—L., 1970; Bueche F., Physical properties of
polymers, N. Y., 1962. A.M.Елъяшевич.
МЕХАНОХИМИЯ полимеров (mechanochemis-
try, Mechanochemie, mecanochimie) — раздел хи-
химии полимеров, в к-ром рассматриваются инициирова-
инициирование и ускорение химич. превращений высокомолеку-
высокомолекулярных веществ, а также синтез макромолекул в ре-
результате поглощения системой упругой энергии. Про-
Процессы превращения химич. энергии в механическую
изучает хемомеханика полимеров.
Механохимич. реакции протекают при переработке
высоковязких и высокоэластичных полимерных мате-
материалов на вальцах, в экструдерах, различных смесите-
смесителях, при измельчении полимеров в мельницах, при
эксплуатации изделий в условиях статических и дина-
динамических механич. нагрузок, при различных видах меха-
механич. обработки, трении, под действием высоких давле-
давлений в сочетании с деформацией сдвига, при прохожде-
прохождении ударных волн, облучении полимеров и их р-ров
ультразвуком, интенсивном перемешивании р-ров, их
замораживании и др.
Энергетические аспекты М. еще четко не сформули-
сформулированы, экспериментального материала накоплено не-
недостаточно, хотя именно для М. подобные исследо-
исследования имеют первостепенное значение.
Ускорение химич. реакций в упруго напряженном
материале вызвано увеличением внутренней энергии
системы, т. е. смещением уровней энергии внутри-
и межмолекулярных взаимодействий. Так, по калори-
метрич. данным внутренняя энергия при растяжении
пленок полимеров может увеличиться на несколько
кдж/молъ, а при воздействии ударных волн — на де-
десятки кдж/молъ. Как правило, во всех реальных мате-
материалах избыточная энергия распределена неравномер-
неравномерно по их объему и всегда существуют локальные обла-
области (напр., аморфные прослойки в кристаллических
полимерах или др. типы дефектов структуры), где
плотность энергии может в десятки и сотни раз пре-
превышать средний уровень и соизмерима с энергией
химических связей.
Существует несколько механизмов влияния упругих
механич. напряжений на протекание химич. реакций:
1) уменьшение энергии химич. связей в упруго напря-
напряженном материале вызывает уменьшение энергии акти-
активации химич. реакции (см. рис., схема а); 2) сброс на-
Энергетические диаграммы термической (сплошная кривая)
и механохимической (пунктирная линия) реакций (Еп —
энергия активации термич. реакции; по оси абсцисс — коор-
координата реакции): а — уменьшение энергии связи на величи-
величину AEVnp под действием упругих напряжений; б— колеба-
колебательное возбуждение (уровень Д2?Кол) связей при рассеи-
рассеивании упругой энергии в тепло.
пряжений в условиях, когда система не успевает со-
совершить работу против внешних сил, сопровождается
превращением избыточной упругой энергии в энер-
энергию колебательного возбуждения химич. связей; в этом
случае реакция идет с более высокого энергетич. уров-
уровня (см. рис., схема б); 3) при рассеивании упругой энер-
энергии в тепло в малых локальных объемах вещества
(напр., на неровностях поверхностей трения, в кави-
тационных полостях, образующихся при ультразвуко-
ультразвуковом облучении) развиваются высокие давление и
темп-pa, благоприятные для химич. превращений;
4) деформирование, разрушение, адгезионное расслаи-
расслаивание и трение полимеров сопровождаются статич.
электризацией, электронной эмиссией, люминесцен-
люминесценцией; электроны эмиссии и газовый разряд между
заряженными поверхностями генерируют активные
частицы (радикалы, ионы и др.), способные возбуж-
возбуждать химич. реакцию.
Энергетич. выход механохимич. реакций, т. е. число
молекул, прореагировавших в результате затраты
определенного количества механической энергии, в
самых благоприятных условиях достигает 10—10
молекул на 100 эв (для нецепных реакций), прибли-
приближаясь к энергетическим выходам при радиационных
воздействиях.
Кинетика механохимических превращений, т. е. ха-
характер зависимости скорости от темп-ры, интенсивно-
интенсивности механич. воздействий, физико-механич. свойств
материала и др. факторов, определяется в значитель-
значительной мере соотношением скоростей двух последователь-
последовательных стадий механохимич. процесса: 1) приложения на-
нагрузки и распределения упругих напряжений в системе
и 2) собственно химич. реакции, инициированной на-
напряжением.
В постоянных механич. полях, когда полимер дли-
длительное время находится в напряженном состоянии,
первую стадию можно не учитывать. В этом случае
кинетич. закономерности процесса отражают влияние
темп-ры и действующего напряжения на скорость соб-
собственно химич. реакции. Тогда выражение для констан-
константы скорости реакции имеет вид:
Для мономолекулярных реакций к — константа ско-
243
МЕХАНОХИМИЯ
244
рости, сек'1; к0 — предэкспоненциальный множитель,
сек'1; R — универсальная газовая постоянная; Т —
абсолютная темп-pa, К; Е(о) — энергия актива-
активации механохимической реакции, зависящая от напря-
напряжения а.
Экспериментально установлено, что энергия актива-
активации реакций в напряженном полимере (напр., при рас-
растяжении в режиме а = const) уменьшена на величину,
пропорциональную среднему действующему напря-
напряжению:
где постоянная Ео близка по величине к энергии акти-
активации термич. реакции в отсутствие напряжений,
у — эмпирич. коэффициент, величина к-рого зависит
от химич. природы и надмолекулярной структуры ма-
материала. Предэкспоненциальный множитель к0 по
порядку величины близок к 1013 сек'1, т. е. в постоян-
постоянных механич. полях значения kQ и Ео в первом прибли-
приближении совпадают с соответствующими величинами
в ур-нии Аррениуса для обычных чисто термических
реакций.
В переменных механич. полях, напр, при циклич.
испытаниях материалов, измельчении полимеров в ви-
вибрационной мельнице, самой медленной стадией, опре-
определяющей скорость всего процесса, может оказаться
перераспределение упругих напряжений в системе.
Тогда константа скорости механохимич. процесса км
будет пропорциональна скорости перераспределения
А в сек'1 (величина постоянной А определяется релак-
релаксационными свойствами материала и частотой воздей-
воздействия) и вероятности накопления на данной связи
нек-рой критич. энергии деформации U*, достаточной
для инициирования реакции:
где А ?/упр = сбТр / — средний уровень запасенной уп-
упругой энергии; а — коэфф. пропорциональности, близ-
близкий по величине к. п. д. процесса; тр — время релак-
релаксации; / — интенсивность подвода механич. энергии.
(и* \
-rfj 1 в общем случае неизвестен.
„ А^Упр/
В нек-рых системах распределение вероятностей опи-
описывается экспоненциальным законом и тогда:
ехр
( U*
1 "p
В этом ур-нии в явном виде не содержится зависимо-
зависимости скорости процесса от темп-ры, но А и тр зависят от
темп-ры и резко изменяются в области структурных
переходов, прежде всего вблизи темп-ры стеклования.
Т. к. с ростом темп-ры время релаксации уменьшается,
в переменных механич. полях скорость механохимич.
превращений часто характеризуется отрицательным
температурным коэффициентом.
Внешне последнее ур-ние похоже на закон Аррениу-
Аррениуса, но величина и физич. природа постоянных здесь
другие: в показателе экспоненты фигурирует упругая
энергия, а предэкспоненциальный множитель А по
порядку величины близок к скорости релаксации на-
напряжений (для полимерных стекол—10~4 сек'1), что
во много раз меньше постоянной к0 в ур-нии Аррениуса
(~1013 сек'1).
Эффективность механохимич. превращений в пере-
переменных механич. полях увеличивается с ростом ин-
интенсивности подвода энергии. Поэтому при технологич.
оформлении механохимич. процессов более выгодно
оборудование с максимальной энергонапряженностью;
при этом частота воздействий должна попадать в гра-
границы спектра частот механич. потерь материала
(см. Релаксационные явления).
Рассмотренные случаи далеко не исчерпывают опи-
описания кинетики сложных многостадийных механохи-
механохимич. реакций в полимерах. Лимитирующей стадией мо-
может оказаться транспорт реагирующих частиц друг
к другу, процесс образования новой поверхности и др.
Константа скорости может изменяться в ходе процесса
из-за изменений надмолекулярной структуры, дисперс-
дисперсности, молекулярно-массового распределения и др.
причин.
Элементарные стадии механохимических превраще-
превращений. Как правило, механохимич. реакции в полимерах
и их р-рах, в мономерах и системах полимер — мо-но-
мер протекают по свободнорадикальному механизму.
В результате механич. воздействий возникают поли-
полимерные или низкомолекулярные радикалы, к-рые да-
далее вступают в реакции замещения, присоединения,
распада и гибели (рекомбинация или диспропорциони-
рование). Детальные данные о др. механизмах отсут-
отсутствуют.
При разрыве макромолекулы образуются два пер-
первичных радикала со свободной валентностью на конце
цепи. В полимерах винилового ряда это радикалы
~СН2 и CXY — (X и Y—атомы водорода или др. боко-
боковые заместители), в каучуках — аллильные радикалы
~CX=CY — СН2 и т. д. (см. Механическая деструкция).
Первичные радикалы ~СН2 отличаются высокой
химич. активностью, близкой к активности атомов Н
и радикалов СНз в реакциях передачи атомов водоро-
водорода: ~ СН2 + ~ СНХ > ~ СНз + - СХ ~. Энергия
активации этой реакции тем больше, чем больше проч-
прочность связи С — Н в звене ~ СНХ~. В полиэтилене
скорость передачи атома водорода сравнительно мала,
но и в этом случае период полупревращения при 130 К
составляет 102 —103 се.к. Поэтому первичные радика-
радикалы ~ СН2 можно зарегистрировать только в случае
проведения механохимич. реакции при темп-pax ниже
| 100 К или при механич. обработке замороженного
разб. р-ра полимера в химически инертном раство-
растворителе. Для аллильных радикалов в каучуках пере-
передача атома Н не столь характерна.
•
Реакционная способность радикалов —CXY подроб-
подробно исследована в процессах роста цепи. В первом
приближении константа скорости и энергия актива-
активации элементарных реакций присоединения различных
мономеров и распада радикалов (деполимеризация) со-
сохраняют свои значения и при механохимич. иницииро-
инициировании этих реакций в полимерах и системах полимер —
мономер при давлениях, близких к атмосферному.
В полимерах, содержащих боковые винильные груп-
группы, возможно присоединение радикалов ~CXY (а так-
также др. первичных радикалов) к двойным связям с обра-
образованием разветвлений или сшивок. При X = Н и
Y = СО(ОСНз), ОСО(СНз), СН3 и др. радикалы ~ CXY
активны при передаче атома водорода.
Вторичные радикалы со свободной валентностью
•
в середине цепи (~СХ~), как правило, могут участ-
участвовать в реакциях распада, передачи атома Н и гибели.
Их поведение при распаде в общих чертах сходно с по-
поведением радикалов ~~ CXY при деполимеризации.
Распад — реакция, обратная присоединению, и поэ-
поэтому чем меньше теплота полимеризации, тем меньше
энергия активации распада и тем больше вероятность
деструкции по месту локализации свободной валент-
валентности. В полиметилметакрилате [теплота полимери-
полимеризации метилметакрилата ок. 55 кдж/молъ A3 ккал/моль)]
энергия активации распада радикалов ~СН~ близка
к 67 кдж/молъ A6 ккал/молъ), и выше 0 °С эти радикалы
неустойчивы. Полиметилметакрилат и др. полимеры,
характеризующиеся низкой теплотой полимеризации
мономеров, относятся к сильно деструктирующимся
245
МЕХАНОХИМИЯ
246
не только при механич. воздействиях, но и в др. про-
процессах, протекающих через стадию образования ради-
радикалов ~СХ~ (напр., при радиолизе).
В полиэтилене и др. полимерах, для к-рых теплота
полимеризации мономеров достигает 88—92 кдж/молъ
B0—22 ккал/молъ), вероятность распада радикалов
~СХ~меныые вероятности др. возможных превраще-
превращений радикалов. В полиэтилене преобладает реакция
отрыва атома водорода от «слабой» связи С — Н:
Дальнейшая рекомбинация радикалов приводит к по-
появлению разветвлений или сшивок.
Первичные и вторичные радикалы со свободной ва-
валентностью, локализованной на атомах углерода, легко
присоединяют молекулу кислорода и переходят в пере-
кисную форму:
Часто эта реакция лимитируется скоростью диффузии
кислорода в полимере. Как правило, перекисные ради-
радикалы активнее алкильных при передаче атома водорода:
ROO -f HR -> ROOH + R*
Чередование реакций присоединения кислорода и пе-
передачи атома Н приводит к развитию цепного окисле-
окисления (см. Термоокислительная деструкция). Длина цепи
в большинстве случаев не превышает нескольких зве-
звеньев.
Гибель алкильных и перекисных радикалов во мно-
многих полимерах происходит при темп-pax ниже темп-ры
стеклования, когда подвижность цепей ограничена
и скорость диффузии ничтожно мала (коэфф. диффузии
j)^ 1Q-20 см1/сек). Возможность передвижения ради-
радикалов друг к другу в объеме полимера в этих случаях
объясняют различными химич. механизмами перемеще-
перемещения свободной валентности (чередование реакций пере-
передачи атома Н, деполимеризации, распада с выделением
низкомолекулярных радикалов, цепного окисления
и др.).
Так. обр., первичные радикалы всегда инициируют
дальнейшие химич. превращения, направление к-рых
определяется свойствами полимера, среды и добавлен-
добавленных мономерных соединений. В линейных и сетчатых
полимерах появляются новые концевые группы (напр.,
метильные или винильные), меняется концентрация
разветвлений и сшивок; в смесях полимеров при реком-
рекомбинации радикалов образуются блоксополимеры; в си-
системах полимер — мономер возникают новые функцио-
функциональные группы или инициируется полимеризация
и т. п. На этих реакциях основаны механохимич. про-
процессы модифицирования полимеров (см. Модификация
химическая), регенерации сетчатых полимеров (см.
Регенерация резины) и др. Вторичные реакции можно
предотвратить или замедлить, вводя в систему ингиби-
ингибиторы радикалов (см. Стабилизация).
Особенности механохимических превращений в раз-
различных механических полях. Роль механич. напряже-
напряжений заключается не только в инициировании активных
частиц, но и в ускорении (или торможении) отдельных
элементарных стадий химич. реакций. Пластич. де-
деформации увеличивают скорость бимолекулярных реак-
реакций, к-рые лимитируются скоростью перемещения реа-
реагирующих частиц в объеме материала; статич. сжатие,
наоборот, тормозит эти процессы; сдвиг и растяжение
ускоряют распад радикалов со свободной валентностью
в середине цепи и т. д. Изменения внутримолекулярной
подвижности и надмолекулярной структуры при де-
деформировании полимеров также влияют на протека-
протекание химич. реакций. Поэтому направление, скорость
и энергетич. выход механохимич. превращений раз-
различны в разнообразных механич. полях, отличающихся
величиной и продолжительностью действия напряже-
напряжений (т. е. уровнем избыточной упругой энергии в ма-
материале), характером напряженного состояния (сдвиг,
растяжение, сжатие) и интенсивностью процессов рас-
рассеивания упругой энергии (переход в тепло, пластич.
течение, перестройка надмолекулярной структуры
и др.).
При механич. обработке высоковязких и высокоэла-
высокоэластичных полимерных материалов на вальцах и в раз-
различных пластикаторах, при измельчении полимеров
механохимич. реакции инициируются гл. обр. радика-
радикалами, образующимися в результате разрыва макромо-
макромолекул. В мельницах при интенсивности подвода энер-
энергии порядка 10 дж/(см^-сек) скорость инициирования
достигает 10~5 моль/(л • сек), а энергетич. выход К)-1
радикалов на 100 эв, то есть к. п. д. «механического»
разрыва связей порядка Ю %. При пластикации
каучуков энергетич. выход меньше, т. к. большая часть
энергии расходуется на пластическое деформирова-
деформирование материала.
Рабочее давление в пластикаторах и мельницах,
как правило, меньше 10 Мн/м2 A02 кгс/см2), и констан-
константы скорости элементарных реакций радикалов не сильно
отличаются от значений, характерных для ненапря-
ненапряженного материала. При пластикации и измельчении
различных макромолекулярных систем осуществлены
синтезы многих блоксополимеров (напр., каучука или
целлюлозы с мономерами винилового ряда), привитых
сополимеров (системы полимер — наполнитель), меха-
механохимич. полимеризация и др.
Механохимич. превращения при облучении ультра-
ультразвуком р-ров полимеров или жидких мономеров свя-
связаны с возникновением кавитации. В момент смыка-
смыкания («захлопывания») кавитационной полости запол-
заполняющие ее газ и пар адиабатически сжимаются; в ре-
результате темп-pa внутри полости повышается до тысяч
°С, а давление — до 103 Мн/м2 A04 кгс/см2). В этих
условиях молекулы газа и пара диссоциируют; актив-
активные частицы затем переходят в жидкую фазу (в присут-
присутствии воды образуются радикалы Н и ОН, а также
электроны), где инициируют химич. реакции, напр,
полимеризацию. Химико-акустич. энергия, т. е. та
часть акустич. энергии, к-рая затрачивается на обра-
образование первичных активных частиц, составляет всего
0,08—0,1% (для водных р-ров), а энергетич. выход не
превышает 10~3—10~2 радикалов на 100 эв.
Захлопывание кавитациоыных полостей возбуждает
ударные волны с амплитудой давления до 103 Мн/м2
A04 кгс/см2). Предполагают, что ударные волны, рас-
распространяющиеся в р-ре полимера, разрывают макро-
макромолекулы, то есть в этом случае механохимич. превра-
превращения инициируются макрорадикалами. Химическое
применение ультразвука ограничено низким к. п. д.
процесса и рационально лишь для достижения ре-
результатов, которые нельзя реализовать другими ме-
методами (например, в медицине, где ультразвуковую
полимеризацию используют для сращивания костных
тканей).
При действии высокого давления [до 104 Мн/м2
A05 кгс/см2)] на твердые мономеры внутренняя энергия
вещества увеличивается на десятки кдж/молъ. Возни-
Возникающие в этих условиях химически активные частицы
(по-видимому, низкомолекулярные радикалы) могут
вызвать полимеризацию мономеров, однако рост це-
цепей возможен только при одновременном развитии
пластич. течения. При сочетании высокого давления
с деформацией сдвига выход полимера в первом прибли-
приближении пропорционален величине сдвиговой деформа-
деформации, обеспечивающей транспорт молекул мономера
к растущим цепям.
Столь же высокие давления [103—105 Мн/м2
A04—106 кгс/см2)] создаются в веществе при прохожде-
прохождении ударных волн. Кратковременное A0~7—10~5 сек)
247
МИКРОКАПСУЛИРОВАНИЕ
248
сжатие сопровождается пластич. течением вещества
во фронте ударной волны. Остаточный нагрев, как пра-
правило, не превышает 101—102 °С. При давлении
~104 Мн/м2 A05 кгс/см2) и продолжительности воз-
воздействия — 10~6 сек интенсивность подвода энергии до-
доходит до 108 дж/(см3-сек). При ударном сжатии по-
лимеризуются многие мономеры. В оптимальных ус-
условиях [например, для метакриламида давление около
1-Ю4 Мн/м2 A-Ю5 кгс/см2)] выход полимера достигает
70% ; при более высоких давлениях преобладает дест-
деструкция образующихся макромолекул. Ненасыщенные
высокомолекулярные соединения — каучуки — при
прохождении ударной волны мгновенно сшиваются,
причем плотность сетки можно варьировать в широких
пределах. Сшивание вызвано, по-видимому, разрывами
цепей и дальнейшим присоединением радикалов к двой-
двойным связям каучука. Энергетич. выход этой реакции
составляет ок. 10 сшивок на 100 эв.
* *
*
Исследования в области механохимии полимеров раз-
развиваются в след. направлениях: 1) изучение механиз-
механизмов и энергетики химич. превращений в различных
механич. полях; 2) применение механохимич. реакций
для синтеза и модифицирования полимерных материа-
материалов и 3) поиски путей торможения нежелательных ме-
механохимич. процессов, вызывающих падение проч-
прочности, повышенный износ и преждевременный выход из
строя различных деталей и конструкций из полимер-
полимерных материалов при их эксплуатации в статических
и динамических механич. полях (см. Утомление,
Долговечность).
Лит.: БарамбоймН. К., Механохимия высокомоле-
высокомолекулярных соединений, 2 изд., М., 1971; Симионес-
ку К., Опреа К., Механохимия высокомолекулярных
соединений, пер. с рум., М., 1970; Ц е р е з а Р., Блок-и при-
привитые сополимеры, пер. с англ., М., 1964; Берлин А. А.,
Усп. химии, 29, 1189, A960); Бутягин П. Ю., Усп. хи-
химии, 40, 1935 A971); Бутягин П. Ю., Дубин-
с к а я А. М., Р а д ц и г В. А., Усп. химии, 38, 593 A969);
Регель В. Р., Слуцкер А. И., Томашев-
ский Э. Е., Усп. физ. наук, 106, 193 A072); Журн. ВХО,
28, вып. 1, 73, 80, 90 A973); С a s a I e A., Porter P.,
Johson J., Criogenics, 11, 534, A971). П.Ю.Бутягин.
МИКРОКАПСУЛИРОВАНИЕ (microencapsulation,
Mikroverkapselung, microencapsulation).
Содержание:
Введение 247
Теоретические основы процесса. Технология 248
Пленкообразование из растворов 248
Пленкообразование из расплавов 253
Пленкообразование в результате поликонден-
поликонденсации и полимеризации 254
Применение микрокапсулированных продуктов. . .25 5
Введение
Микрокапсулирование — технологич. процесс заклю-
заключения мелких частиц вещества в тонкую оболочку
пленкообразующего материала. В результате М. ди-
диспергированное до нужной степени твердое или жидкое
вещество превращают в порошок, состоящий из микро-
микрокапсул размером от нескольких мкм до нескольких мм.
Содержание капсулируемого вещества (KB) в микро-
микрокапсуле обычно составляет 70—85% от массы микро-
микрокапсул, но может достигать и 95—99%. Форма индиви-
индивидуальной микрокапсулы определяется формой частицы
KB (микрокапсулированные жидкости состоят, как
правило, из шаровидных частиц). KB в микрокапсуле
может находиться в твердом, жидком или газообраз-
газообразном состоянии и представлять собой индивидуальное
вещество, твердые смеси различных веществ, суспен-
суспензии, эмульсии, р-ры.
Оболочка микрокапсул может иметь толщину от до-
долей мкм до десятков мкм, быть однослойной или много-
многослойной и, в зависимости от свойств пленкообразующе-
пленкообразующего, эластичной или жесткой. В качестве пленкообра-
пленкообразующего используют высокомолекулярные соединения
животного и растительного происхождения, напр, бел-
белки (желатин, альбумин, казеин), декстраны, производ-
производные целлюлозы (метил-, этил-, ацетил-, нитро-, карбо-
ксиметилцеллюлозу), природные смолы (камеди, шел-
шеллак), синтетич. полимеры и олигомеры — полиолефины,
поливиниловый спирт, поливинилацетат, поливинил-
хлорид, полиакриламид, эпоксидные и полиэфирные
смолы, полиамиды, полиорганосилоксаны, а также па-
парафины и стеарины. Пленкообразующим могут служить
также металлы, углерод, силикаты, карбиды и др., одна-
однако процессы образования микрокапсул с оболочкой из
неорганич. материалов в данной статье не рассматри-
рассматриваются.
KB из оболочек может высвобождаться при их меха-
механич. разрушении (раздавливании, истирании, ультра-
ультразвуковом воздействии, плавлении, разрыве изнутри
парами или газообразными веществами, выделяющи-
выделяющимися при изменении внешних условий), растворении
оболочек или в результате диффузии KB через стенку
микрокапсулы при ее набухании в окружающей жид-
жидкости. При этом диффузия KB во внешнюю среду опи-
описывается ур-нием первого порядка, а скорость процесса
обратно пропорциональна толщине стенки. Самопроиз-
Самопроизвольное выделение из микрокапсул жидких легколету-
легколетучих KB может быть довольно значительным. Предот-
Предотвратить этот нежелательный процесс можно, подбирая
специальные пленкообразующие, создавая многослой-
многослойные стенки микрокапсул и др.
Теоретические основы процесса. Технология
Существует большое число технологич. приемов
получения микрокапсулированных продуктов. Все они
основаны на процессах пленкообразования в гетеро-
гетерогенных системах — на границе раздела жидкость —
жидкость, жидкость — твердое тело, газ (пар) — жид-
жидкость, газ (пар) — твердое тело. По механизму пленко-
пленкообразования все способы М. можно разделить на 3 ос-
основные группы: 1) пленкообразование из р-ров пленко-
пленкообразующих за счет регулирования их растворимости
в данной среде, 2) пленкообразование из расплавов
пленкообразующих, 3) пленкообразование в резуль-
результате полимеризации или поликонденсации низкомоле-
низкомолекулярных веществ на поверхности КВ.
Пленкообразование из растворов. В процессах, ос-
основанных на этом механизме пленкообразования, ис-
используют р-ры пленкообразующего в органич. раство-
растворителе или в воде. Для осуществления М. добиваются
выделения из многокомпонентных систем, представляю-
представляющих собой дисперсию KB в выбранном р-ре, фазы, обо-
обогащенной пленкообразующим. Этого можно достичь,
изменяя темп-ру р-ра, его рН или испаряя часть раст-
растворителя. В результате мелкие капли фазы, обогащен-
обогащенной пленкообразующим,
отлагаются на поверхно-
поверхности частиц KB, образуя
сплошную оболочку.
Для выбора парамет-
параметров процесса использу-
используют предварительно по-
построенные диаграммы фа-
фазового состояния систем,
содержащих два или бо-
более компонентов. Свой-
Свойства двухкомпонентных
А А,
A00%)
В, В
A00%) систем полимер (В) —
Рис. 1. Диаграмма состояния растворитель (А) харак-
двухкомпонентной системы (А — теризуют диаграммой ра-
растворитель, В — полимер). створимости, в к-рой со-
составы фаз являются
функцией темп-ры (рис. 1). В области расслоения, ле-
лежащей ниже кривой Ах — Гкр — Вх, системы любого
состава разделяются на два р-ра. Так, напр., система,
характеризуемая нек-рой точкой с, разделяется на фа-
249
МИКРОКАПСУЛИРОВАНИЕ
250
зу состава Л 2, обогащенную растворителем, и фазу со-
состава В2, обогащенную пленкообразующим. Вне обла-
области расслоения смеси Л — В являются гомогенными
р-рами. Для осуществления М. используют гомоген-
гомогенные р-ры пленкообразующего (напр., состава Лз), в
к-рых вызывают разделение фаз, понижая темп-ру
(напр., от t\ до t2)- Иногда добиваются выделения
пленкообразующей фазы в изотермич. условиях, ис-
используя исходные гомогенные р-ры состава е с даль-
дальнейшим испарением растворителя, в результате чего
состав смеси изменяется, напр., по линии ее при по-
постоянной темп-ре г2- На практике образования двух фаз
достигают в результате одновременного действия двух
факторов: удаления растворителя и понижения темп-ры
системы.
Для выделения полимерной фазы из р-ров пленкооб-
пленкообразующего чаще добавляют третий компонент, являю-
являющийся осадителем. На рис. 2 приведена одна из типич-
типичных диаграмм состояния трехкомпонентной системы
растворитель — полимер — оса-
дитель, характеризующаяся изо-
изотермич. условиями расслоения.
Область расслоения лежит ниже
линии ткп. Исходным р-рам
пленкообразующего соответству-
Рис. 2. Диаграмма состоя-
состояния трехкомпонентной си-
системы при ограниченной
смешиваемости двух ком-
компонентов (А — раствори-
растворитель, В — полимер, С —
осадитель). Условия изо-
изотермические.
А A00%)
С A00%)
В A00%)
ют точки (напр., dl или d2), лежащие вне области рас-
расслоения. В результате добавления к р-ру состава dx
осадителя С добиваются изменения состава смеси до
состава d% с выделением фазы состава п, обогащен-
обогащенной пленкообразующим, и фазы состава т, содержа-
содержащей незначительное количество пленкообразующего.
К такому же результату приводит уменьшение со-
содержания растворителя в системе (напр., от состава
d2 до состава dz) в результате его испарения.
В таблице приведены типичные сочетания компонен-
компонентов, используемые при М. методом пленкообразования
из р-ров.
Компоненты трехкомпонентных систем, используемые при
микрокапсулировании методом пленкообразования из растворов
Полимер
Поливинилацетат
То же
»
»
Поливинилстеарат
То же
»
Поливинилхлорид
Нитроцеллюлоза
Ацетаты целлюлозы
Ацетобутират цел-
ТТЮТТЛ'ЧЫ
Бензил цел л юлоза
Этилцеллюлоза
То же
»
Сополимер стирола с
малеиновой к-той
Растворитель
Ацетон
Трихлорэтилен
Метанол
То же
Бензол
Хлороформ
Керосин
Циклогексан
Ацетон
То же
Метилэтилкетон
Трихлорэтилен
Ксилол
Этиловый спирт
Бензол
Этиловый или
метиловый спирт
Осадитель
Вода
Гексан
Бутиловый спирт
Изоамиловый спирт
Ацетон
Метилэтилкетон
Минеральное масло
Гликоль
Вода
То же
Изопропиловый эфир
н-Пропиловый спирт
Гексан, гептан
Вода
Кукурузное или ми-
минеральное масло
Этилацетат, метил-
метилэтилкетон, бутил-
этилкетон, изопро-
изопропиловый эфир
Для осуществления М. подготовленное KB вводят
в исходный р-р пленкообразующего, размешивают до
мелкодисперсного состояния и, пользуясь одним из
описанных выше приемов, вызывают возникновение
фазы, обогащенной пленкообразующим. Выделяющаяся
в виде мелких капелек фаза пленкообразующего при
дальнейшем перемешивании системы обволакивает
частицы KB с образованием непрерывной оболочки. Для
облегчения диспергирования KB в жидкой среде иногда
используют поверхностно-активные вещества (ПАВ).
При правильном подборе ПАВ их присутствие в системе,
содержащей гидрофобное пленкообразующее и гидро-
гидрофильное KB, препятствует т. наз. обращению фаз,
т. е. обволакиванию капсулирующей фазы частицами
КВ. Следует, однако, учитывать, что присутствие ПАВ
в оболочках микрокапсул снижает их прочность, уско-
ускоряет диффузию KB и растворение оболочек в жидких
средах.
Необходимыми условиями образования оболочки на
поверхности частиц KB являются: нерастворимость KB
в используемой для диспергирования среде и осадителе,
большее поверхностное натяжение на KB по сравнению
с поверхностным натяжением фазы, обогащенной плен-
пленкообразующим, и малое поверхностное натяжение на
границе раздела этих фаз. Важное условие успешного
осуществления М.— выделение капсулирующей фазы
в жидком виде, т. к. выпадение этой фазы в виде хло-
хлопьев приводит к получению микрокапсул с оболочками
низкого качества и с низким содержанием КВ.
Формирование пленки на поверхности диспергиро-
диспергированных частиц протекает в течение длительного вре-
времени, иногда до нескольких часов. После его заверше-
завершения добиваются затвердевания оболочек, понижая
темп-ру системы или разбавляя ее осадителем. При
необходимости повышения устойчивости оболочек мик-
микрокапсул (напр., к воздействию жидких сред) молеку-
молекулы пленкообразующего сшивают дифункциональным
реагентом. М. завершают отделением микрокапсул от
среды с помощью центрифугирования, фильтрования
или декантации с последующим промыванием и высу-
высушиванием микрокапсул.
Один из вариантов метода М. пленкообразованием
из р-ров основан на явлении коацервации —
возникновении в р-ре высокомолекулярного соедине-
соединения капель, обогащенных растворенным веществом
(коацервата). Различают простую и сложную коацер-
вацию. Простая коацервация — результат взаимодей-
взаимодействия растворенного высокомолекулярного вещества
с низкомолекулярным веществом, напр, желатины
с сульфатом натрия. Сложная коацервация наблюда-
наблюдается при взаимодействии двух полимеров, молекулы
к-рых несут противоположные заряды, напр, при сме-
смешении водных р-ров желатины и гуммиарабика.
Коацервация возможна при содержании полимера
в р-ре в количестве десятых и даже сотых долей
процента, причем концентрация вещества в коацерват-
ных каплях может достигать нескольких десятков
процентов.
Для осуществления М. с помощью сложной коацер-
коацервации кроме желатины и гуммиарабика применяют
альбумин, казеин, агар-агар, альгинаты, крахмал,
пектиновые вещества, карбоксиметилцеллюлозу, сопо-
сополимеры акриловой к-ты, малеинового ангидрида, по-
полиамиды и поликислоты. Из солей, вызывающих про-
простую коацервацию, наибольшей эффективностью обла-
обладает Na2SO4, наименьшей — LiCl. Достижение нуж-
нужного значения рН р-ра, требуемого для коацервации и
зависящего от изоэлектрич. точки одного из полимерных
компонентов, обеспечивается добавлением щелочных
или кислых р-ров или простым разбавлением р-ра.
Кроме наличия в системе по крайней мере одного ионо-
генного полимерного вещества необходимым условием
осуществления М. этим методом является возможность
затвердевания оболочек после их формирования на по-
поверхности обволакиваемых частиц. Поэтому обычно
используют пленкообразующие, к-рые обладают спо-
способностью к гелеобразованию при понижении темп-ры.
251
МИКРОКАПСУЛИРОВАНИВ
252
Наиболее удобен метод коацервации для М. гидро-
гидрофобных веществ. Типичным примером этого метода
служит М. масла в желатиновую оболочку (рис. 3).
Диспергирование масла в р-ре желатина, коацервацию
и вызревание оболочек проводят при 50—60 °С, т. е.
выше темп-ры плавления желатинового геля. При
этом происходит обволакивание частиц масла обильным
слоем коацервата, содержащего большое количество
Щ=ный р=р гуммиарабика
B00масч)
Щ = ный р = р желатина
(изоэл точка при рН~8
B00масч.)
Эмульгирование при 60 °С
Доведение рН до 5,0,
добавление 500масч воды
Доведение рН до 4,4,
вызревание
t
Добавление 37%=нэго формалина
13 8масч)
Охлаждение до 10 °С
в течение 30 мин
*
Доведение рН до 9,
перемешивание A-3 ч)
Отделение капсул от среды,
промывание
¦
Высушивание
Масло
(80 мзсч)
Рис. 3. Схема ми-
крокапсулирова-
ния масел мето-
методом коацервации.
водной фазы. Понижение темп-ры до 10—20 °С вызы-
вызывает гелеобразование оболочек, в результате чего ста-
становится возможным отделение микрокапсул без их
разрушения. Последующая обработка продукта альде-
альдегидами в щелочной среде приводит к сшиванию макро-
макромолекул с резким снижением набухаемости и прони-
проницаемости оболочек в водных средах. Окончательно обез-
обезвоживают оболочки, промывая продукт полярными
растворителями и высушивая.
При необходимости М. продуктов, хорошо раство-
растворимых как в органич. растворителях, так и в воде,
описанный процесс дополняют стадией т. н. экст-
экстракционного замещения. Для этого мик-
микрокапсулы, содержащие какой-либо липофильный орга-
органич. растворитель и имеющие проницаемую для низко-
низкомолекулярных веществ оболочку, обрабатывают жид-
жидким продуктом, предназначенным для окончательного
М. Этот продукт постепенно замещает первоначально
капсулированный растворитель.
Для М. водных р-ров и воды предложен метод, осно-
основанный на использовании т. наз. вторичных эмульсий.
В этом случае KB (водный р-р или воду) эмульгируют
в р-ре пленкообразующего в легкокипящем липофиль-
ном растворителе и полученную эмульсию вновь эмуль-
эмульгируют в большом избытке воды. Р-р пленкообразую-
пленкообразующего образует жидкую оболочку вокруг первоначально
сформированных капель KB, к-рая затвердевает при
испарении растворителя.
М. описанными выше способами проводят в обычных
емкостях или реакторах, снабженных эффективными
мешалками якорно-пропеллерного типа, рубашками
для нагревания и охлаждения смесей, обратными холо-
холодильниками и мерниками. При осуществлении М. из
р-ров полимеров в органических растворителях на
стадии образования полимерной фазы обычно наблю-
наблюдается нежелательное явление — внутренние полости
реактора покрываются электроизолирующей плен-
пленкой полимера. Поэтому в таких случаях все дета-
детали основного аппарата заземляют для предотвраще-
предотвращения возникновения разрядов статического электриче-
электричества (до 15 000 в).
Для отделения микрокапсулированных продуктов
используют гл. обр. центрифуги непрерывного дейст-
действия, в к-рых в последующем осуществляют промывку
готовых микрокапсул.
Один из вариантов метода М. пленкообразованием
из р-ра основан на высушивании при рас-
распыл е н и и. По этому способу KB диспергируют
в жидкой среде, содержащей в растворенном или дис-
диспергированном виде пленкообразующее. Для осущест-
осуществления стадии М. смесь распыляют в нагретой камере,
в результате чего происходит испарение среды с обра-
образованием полимерной пленки на поверхности КВ. При
этом необходимо, чтобы среда была легколетучей,
a KB низко летучим.
Способ М. напылением в псевдоо жи-
жиже ином слое заключается в том, что образующие
псевдоожиженный слой твердые частицы KB орошаются
сверху р-ром пленкообразующего в легколетучем раст-
растворителе. Процесс проводят в цилиндрич. аппарате
(рис. 4). Псевдоожижение создается потоком воздуха
или инертного газа, скорость подачи к-рого опреде-
определяется размером и плотностью частиц КВ. Толщина
оболочек микрокапсул зависит от скорости подачи и
концентрации р-ра пленкообразующего. Необходимый
температурный режим в аппарате создается газом-но-
газом-носителем, предварительно нагреваемым (или охлаждае-
охлаждаемым) до требуемой темп-ры. Одна из модификаций
способа (см. рис. 46) позволяет микрокапсулировать
очень мелкие частицы, склонные к слипанию даже при
диспергировании в псевдоожиженном слое.
Для этого используют аппарат с центральным усечен-
усеченным конусом, сквозь к-рый с большой скоростью про-
пропускают газ-носитель. Частицы KB, поступающие в зо-
зону I, разделяются на фракции газом-носителем: часть
KB, содержащая гл. обр. неслипшиеся частицы, увле-
увлекается в верхнюю часть зоны I и орошается в псевдо-
псевдоожиженном слое р-ром пленкообразующего. Агломе-
Раствор Раствор
пленкообразующего пленкообразующего
Иапсулируе-
мое вещество
Готовый
продукт
Напсулируе-
мое вещество
— Зона I
Зона II
Готовый
продукт
Газ—носитель
Газ —носитель
Рис. 4. Схемы аппаратов для осуществления микрокапсу-
лирования в псевдоожиженном слое: а — обычный вариант
аппарата, б — аппарат для микрокапсулирования веществ,
склонных к слипанию.
раты частиц KB поступают в нижнюю часть аппарата
и увлекаются потоком газа в усеченный конус (зона II).
Диспергуясь потоком газа-носителя, проходящего зо-
зону II с большой скоростью, они поступают в зону ороше-
орошения. Другая модификация способа основана на замо-
253
МИКРОКАПСУЛИРОВАНИЕ
254
раживании KB перед введением его в аппарат или в са-
самом аппарате потоком охлажденного газа. Таким обра-
образом осуществлено, напр., М. монофенилового эфира
этиленгликоля в полистирол.
При М. в псевдоожиженном слое производят предва-
предварительное измельчение (или гранулирование) и фрак-
фракционирование КВ. Размер частиц может колебаться
в широких пределах, но обычно он составляет от 0,1
до 1 мм. При этом скорость потока газа-носителя варьи-
варьирует от 10 до 200 м/мин. Отношение (по массе) KB: плен-
пленкообразующее для микрокапсул, полученных методом
напыления в псевдоожиженном слое, может колебаться
от 100 : 1 до 1 : 1. Продолжительность процесса состав-
составляет от 5 до 20 ч. Метод применяют преимущественно
для М. фармацевтич. препаратов с использованием
в качестве пленкообразующего зеина, воска, производ-
производных целлюлозы, сополимера винилиденхлорида с ви-
вини л хлорид ом.
Пленкообразование из расплавов. По одному из ва-
вариантов этого метода KB диспергируют в горячем масле,
содержащем диспергированный расплавленный воск
(все три компонента системы не должны смешиваться).
Частицы KB обволакиваются расплавом, к-рый затвер-
затвердевает при охлаждении. В результате образуются мик-
микрокапсулы, способные высвобождать KB при нагрева-
нагревании, раздавливании или действии неполярного раство-
растворителя. По этому способу можно получить капсулы
с оболочкой из любого термопластичного материала,
плавящегося при достаточно низкой темп-ре, напр,
полиэтилена, полистирола.
Ряд способов, относящихся ко второй группе про-
процессов М., основан на принципе «з а п л а в л е н и я
падающей капли». Общее для этих способов —
предварительное формирование тонкой вязкой пленки
на поверхности с отверстиями (соплами) малого диамет-
диаметра, сквозь к-рые продавливается КВ. После прохож-
прохождения сопла формируется капля в оболочке, затверде-
затвердевающей в дальнейшем в результате охлаждения или при
обработке сшивающими агентами. Для М. этим спосо-
способом используют центрифугу особой конструкции (рис.5),
состоящую из полого пер-
перфорированного ротора, по-
помещенного в центр резервуа-
резервуара с охлаждающим агентом.
KB, не смешивающееся с
пленкообразующим, подают
в виде жидкости, порошка
или р-ра на диск, размещен-
размещенный внутри ротора. Нап-
Направления вращения ротора
и диска противоположны.
Плоскость диска устанав-
Рис. 5. Схема центрифужного
аппарата для микрокапсулиро-
вания: 1 — ротор, 2 — вал ро-
ротора, 3 — вал внутреннего ди-
диска, 4 — охлаждающая среда,
5 — микрокапсулы.
ливают на одном уровне с рядом отверстий в стенках
ротора. Расплав пленкообразующего подают изнутри
на стенки ротора. Частицы KB попадают на вязкую
мембрану пленкообразующего, закрывающую отвер-
отверстия, продавливают ее и после обволакивания пленкой
вылетают из отверстий стенки ротора в сосуд с охлаж-
охлаждающей средой. Размер частиц при заданном размере
отверстий в роторе зависит от поверхностного натяже-
натяжения пленкообразующего и скорости вращения цилинд-
цилиндра. Этим методом были микрокапсулированы насы-
насыщенные р-ры солей, вода и глицерин в различные тер-
термопласты.
Для М. по способу «заплавления падающей капли»
используют также аппараты, основной элемент к-рых
представляет собой две трубки различных диаметров,
П'леннообразующее
I
расположенные одна в другой. По наружной трубке
подают расплав пленкообразующего, по внутренней —
КВ. Выходное отверстие трубок открывается с опреде-
определенной частотой (ок. 10 раз в сек). Образующиеся ми-
микрокапсулы попадают в охлаждающую среду. Таким
способом микрокапсулируют вазелиновое масло, со-
содержащее различные витамины, в желатину.
На принципе пленкообразования из расплавов осно-
основан также способ электростатической
коагуляции. На частицы KB и капли расплав-
расплавленного пленкообразующего наводят противоположные
по знаку электрич. заряды (этого достигают при раз-
раздельном пропускании их в виде аэрозолей сквозь слои
газа, ионизированные тлеющими разрядами). Затем
потоки разноименно заряженных частиц объединяют,
в результате чего происходит коалесценция с образова-
образованием микрокапсул. Основными требованиями, предъ-
предъявляемыми к материалам при проведении этого про-
процесса, являются: высокое поверхностное натяжение
жидкого KB, низкое поверхностное натяжение и хоро-
хорошая смачивающая способность пленкообразующего по
отношению к KB, а также высокая электрич. проводи-
проводимость обоих материалов. В ряде случаев этим способом
получают капсулы, содержащие до 90% KB, однако
чаще содержание KB не превышает 50%.
Пленкообразование в результате поликонденсации
и полимеризации. Для проведения поликонден-
поликонденсации на границе раздела фаз один
из мономеров растворяют в органич. р-рителе, другой —
в воде, содержащей незначительное количество катали-
катализатора. В одну из фаз вводят КВ. При этом органич.
фаза не должна растворять образующийся полимер
и смешиваться с водной фазой (см. также Межфазная
поликонденсация). При введении одной фазы в другую
через диспергирующее сопло на поверхности частиц,
содержащих KB, немедленно начинается поликонден-
поликонденсация с отделением всплывающих или погружающихся
микрокапсул.
Конденсацией дифункциональных хлорангидридов
с диаминами или гликолями получают капсулы с обо-
оболочками соответственно из полиамидов или полиэфиров.
М. можно осуществить также в результате проведе-
проведения на границе раздела двух фаз процессов т. наз. сту-
ступенчатой полимеризации. Так, напр., микрокапсулы
с оболочками из полиуретанов и полимочевин обра-
образуются при взаимодействии диизоцианатов соответст-
соответственно с гликолями или диаминами.
В качестве пленкообразующих компонентов м. б. ис-
использованы также полученные предварительно форпо-
лимеры, отверждаемые сшивающим реагентом, раство-
растворенным в одной из фаз. В среде диспергируют KB с пер-
первым реагентом. Второй реагент (или реагенты) отдельно
растворяют в дополнительном количестве среды и полу-
полученный р-р вводят в первоначально образованную
эмульсию. В этом случае происходит отложение нераст-
нерастворимого ПМ по мере его образования сразу на всех
диспергированных частицах. Таким образом капсу-
лируют, напр., краситель в среде минерального или
растительного масла или органич. растворителя, а
также магнитный порошок в среде метакрилатного
пластизоля в оболочки из полиэтилентерефталата.
М. веществ, устойчивых к воздействию повышенных
темп-р A50—200 °С), осуществляют, используя реак-
реакцию полирекомбинации гс-ксилелена. Процесс проводят
след. обр.: пары гс-ксилилена из камеры, где происхо-
происходит термич. распад димера (при темп-pax до 600 °С),
направляют в камеру, в к-рой находится в диспергиро-
диспергированном виде KB (при темп-ре ниже 200 °С). На поверх-
поверхности частиц KB происходит полирекомбинация с обра-
образованием полимера, к-рый сохраняет свои электроизо-
электроизоляционные свойства при очень низких темп-pax и,
кроме того, обладает высокой термо- и влагостойкостью
(см. также Поликсилилен).
255
МИКРОКАПСУЛИРОВАНИЕ
256
М. может осуществляться также в результате поли-
полимеризации мономеров на поверхности частиц КВ.
Последнее вводят в реакционную среду в твердом или
жидком виде. Напр., для М. волокон целлюлозы в по-
полиэтиленовую оболочку целлюлозу диспергируют в то-
толуоле. В дисперсию вводят катализатор типа Циг-
лера — Натта, а затем газообразный этилен. Процесс
осуществляют при 40—60 °С. Рост макромолекул проис-
происходит с поверхности волокон наружу. Толщина пленки
регулируется количеством вводимого этилена и может
составлять от 0,5 до 60 мкм, количество полиэтилена
в продукте может колебаться от 5 до 90% (по массе).
По окончании процесса катализатор дезактивируют
добавлением воды, спирта или обработкой воздухом,
продукт отфильтровывают или отделяют центрифуги-
центрифугированием, а затем высушивают или суспендируют в во-
воде для дальнейшего формования листов. Аналогично
капсулируют стеклянные или асбестовые волокна, по-
порошкообразные металлы, пигменты и соли. В США ма-
материалы, изготавливаемые этим методом, как и сам ме-
метод, получили общее коммерческое название «н а л-
к о н». Сходным образом осуществляют М. путем поли-
полимеризации на границе раздела двух жидких фаз.
При необходимости получения микрокапсул разме-
размером от долей мкм до нескольких мкм осуществляют
полимеризацию на границе с газообразной фазой.
Пользуясь методами получения аэрозолей, KB диспер-
диспергируют в среде инертного газа и совмещают со средой,
содержащей пары мономера, способного к каталитич.
полимеризации. В качестве мономеров используют гл.
обр. диолефины, триолефины и виниловые эфиры, отли-
отличающиеся большой упругостью паров и высокой реак-
реакционной способностью. Катализаторами полимериза-
полимеризации служат обычно газообразные соединения, напр.
NO2, BF3. Полимеризация должна завершаться в те-
течение 1—2 мин, что определяется непродолжитель-
непродолжительностью пребывания взвешенных частиц в зоне реакции.
Применение микрокапсулированных продуктов
Заключение различных продуктов в микрокапсулы
позволяет: 1) длительно хранить реакционноспособные,
неустойчивые и быстропортящиеся вещества, 2) сме-
смешивать реагирующие друг с другом или несмешиваю-
щиеся соединения, 3) снижать токсичность продуктов,
4) осуществлять постепенное введение продукта в тре-
требуемый момент времени и за необходимый период,
5) маскировать цвет, вкус, запах, 6) придавать жидким
веществам вид сыпучих продуктов и т. д. Наиболее
широкое применение М. находит в производстве клеев,
бумаг, фотоматериалов, герметиков, лаков, др. химич.
товаров и фармацевтич. продуктов.
М. компонентов клеев позволяет создавать компо-
композиции, не обладающие липкостью до момента использо-
использования. Такие композиции проявляют свои клеящие
свойства при высвобождении компонентов в результате
раздавливания капсул или их нагревания. Микрокап-
сулированные клеи выпускают в виде карандашей или
нанесенными на пленки, листы, конверты и т. п. Для
снижения липкости до момента использования капсули-
капсулируют один или несколько компонентов, являющихся
растворителями, пластификаторами (для композиций
на основе каучуков) или отвердителями. Типичными
двухкомпонентными капсулируемыми клеевыми систе-
системами являются: силикон—оловоорганич. соединение,
полисульфид — двуокись свинца, эпоксидная смола —
амин (или эфират BF3) и др. Из растворителей и пласти-
пластификаторов капсулируют толуол, дихлордифенил, ди-
алкилфталаты. В капсулированном виде получают
резиновые клеи (на основе хлоропренового или бутадиен-
нитрильного каучука), а также полиалкилакрилатные
и нитро- или этилцеллюлозные композиции.
Широкое применение находит самокопирую-
самокопирующая бумага, позволяющая изготавливать копии
без использования углеродистой копировальной бума-
бумаги. Этот эффект достигается нанесением на оборотную
сторону листа бумаги тонкого слоя микрокапсул, со-
содержащих в хрупких оболочках бесцветный краситель,
окрашивающийся при раздавливании капсул и контакте
со слабокислым или щелочным агентом. Такой агент
в виде тонкого порошка адсорбента наносится на лице-
лицевую сторону листа.
С использованием микрокапсул получены фото-
фотобумага, чувствительная к УФ-лучам, и магни-
магнитофонная пленка. М. фототропных материа-
материалов позволило исключить из фотографич. процесса
стадию проявления и довести разрешающую способ-
способность фотослоев до 1 : 40 000. При создании однослой-
однослойных материалов для цветной фотографии капсулируют
отдельные компоненты, необходимые для получения
цветного изображения, а также воду, вводимую в фо-
фотослои для безбачкового проявления.
Микрокапсулированные герметики типа поли-
полисульфида и вещества, обладающие антикоррозионными
свойствами, такие, как хромат цинка, широко приме-
применяют в капсулированном виде для нанесения на корро-
корродирующие детали и крепежные элементы в самолето-
самолетостроении. На алюминиевые заклепки наносят микрокап-
микрокапсулы размером 105—420 мкм, содержащие в желати-
ново-каррагениновых оболочках полисульфид или
хромат цинка. Монослой из капсул закрепляют на
поверхности заклепок алкидными смолами, а затем
дополнительно покрывают его защитным слоем поли-«-
бутилметакрилата. Содержимое капсул высвобожда-
высвобождается при их раздавливании в месте крепления в момент
создания заклепочного соединения. Производство таких
заклепок за рубежом достигло десятков млн. штук.
Весьма перспективно применение микрокап-
микрокапсулированных эластомеров, к-рые вво-
вводят в жесткие пластики на стадии синтеза материала
или его переработки с целью улучшения комплекса
механич. свойств (в первую очередь ударной проч-
прочности).
Аналогично капсулированным клеевым композициям
изготавливают лаки, содержащие в капсулированном
виде один из компонентов (растворитель или катализа-
катализатор). Микрокапсулируют различные красители
(напр., флуоресцентные) с целью повышения их устой-
устойчивости к воздействию внешней среды и для облегчения
их расфасовки и дозировки. Оболочки микрокапсул
таких красителей создают из полимеров, растворимых
в используемых для окрашивания жидких средах.
Наиболее хорошо разработана технология микрокап-
сулирования неполярных органич. растворите-
растворителей, углеводородных топлив и ма-
с е л. Микрокапсулированный бензин в виде брикетов
может храниться в открытых местах, не требует тары
для перевозки, свободно плавает на воде без растекания
и допускает сбрасывание с парашютом в труднодоступ-
труднодоступные районы. Его горение протекает без взрыва и пре-
прекращается при использовании обычных средств пожа-
пожаротушения. Кроме бензина, получают микрокапсули-
микрокапсулированные бутан, лигроин, керосин, дизельное топливо,
смазочные материалы, алифатич., ароматич. и алици-
клич. углеводороды, к-рые превращают таким образом
в сыпучие нелетучие продукты. Осуществляют также
М. катализаторов и инициаторов
полимеризации (триэтилентетрамин, ди^ти-
лентриамин, эфират трехфтористого бора, диэтилани-
лип, перекись бепзоила), синтетич. олигомеров
и полимеров (полиэфиров, полиамидов, эпоксид-
эпоксидных смол) и др. М. применяют для временного изо-
изолирования гидридов легких металлов (Be, Li, Mg)
от высокореакционноспособных компонентов твердых
топлив.
Осуществление М.щелочи позволило создать т. н.
ленточные сухие батареи — компактные устройства
257
МИПОРА
258
для получения электрич. тока. Основой таких батарей
является полипропиленовая лента, покрытая с одной
стороны перекисью серебра (отрицательный полюс),
а с противоположной — цинком (положительный по-
полюс). Помимо этого, на поверхность ленты наносят
микрокапсулы с р-ром КОН. Ленту, хранящуюся до
момента использования в рулонах, активируют раз-
раздавливанием капсул между двумя роликами, что при-
приводит к высвобождению р-ра щелочи, в результате че-
чего начинается электрохимич. реакция. Такие элемен-
элементы предназначены для индивидуального использова-
использования космонавтами при проведении работ в космосе.
В фармацевтич. пром-сти М. нашло
наиболее широкое применение. С помощью М. стаби-
стабилизируют неустойчивые препараты (витамины, антибио-
антибиотики, вакцины, сыворотки, ферменты), маскируют вкус
горьких и тошнотворных лекарств (касторовое масло,
рыбий жир, экстракт алоэ, кофеин, хлорамфеникол,
бензидрин), регулируют скорость высвобождения пре-
препаратов или обеспечивают их высвобождение в нужном
участке желудочно-кишечного тракта, создают новые
типы продуктов диагностич. назначения (капсулиро-
ванные нестабильные реагенты для анализа крови
и мочи, терморегистрируюшие пленки, а также акти-
активированный уголь, ионообменные смолы и т. п.).
Большинство фармацевтич. препаратов подвергают
М. с целью удлинения времени тераневтич. действия
с одновременным снижением максимального уровня
концентрации препарата в организме. Таким образом
сокращают число приемов препарата и ликвидируют
раздражающее действие на ткани, вызываемое прили-
прилипанием таблеток к стенкам желудка. Гастролабильные
препараты заключают в оболочки, устойчивые в кислых
средах и разрушающиеся в слабо щелочных или нейт-
нейтральных средах кишечника. Напр., оболочки из ацето-
фталата целлюлозы защищают лекарственные препа-
препараты при рН 1,2 и быстро разрушаются при рН 7,5.
Важная область применения М. в фармацевтике —
совмещение в общей дозировке лекарственных веществ,
не совместимых при смешении в свободном виде. С этой
целью, напр., готовят смеси микрокапсулированных
аспирина и малеата хлорфениламина. Для диагностич.
целей изготавливают пасты и пленки, содержащие ми-
микрокапсулы с т. н. жидкими кристаллами нек-рых
эфиров жирных к-т и холестерина, изменяющие окрас-
окраску в момент плавления. Правильный подбор смесей раз-
различных эфиров позволяет изготавливать составы, к-рые
изменяют цвет в пределах всего видимого спектра
в узких интервалах темп-ры. За рубежом выпускаются
пленки, изменяющие цвет от голубого до красного при
темп-pax от 38 до 37 °С, от 24 до 21 °С и от 51 до 45 °С
с точностью измерения температуры 0,25, 1 и 3 °С
соответственно. С помощью этих пленок или мазей
можно изучать распределение темп-ры на поверхности
тела пациентов с целью установления места локализа-
локализации плаценты, воспалительных процессов, опухолевых
образований и в нек-рых др. случаях.
Осуществлено М. активированного угля, ионообмен-
ионообменных смол, уреазы и нек-рых др. ферментов, что откры-
открывает широкие перспективы для лечения заболеваний
печени и почек путем искусственного очищения крови
от токсич. веществ и избыточного количества продуктов
обмена. М. активированного угля и уреазы позволило
создать опытные образцы нового компактного аппарата
типа «искусственная почка» с большой рабочей поверх-
поверхностью и малым объемом, заполняемым кровью. М.
ферментов защищает их от дезактивации макромоле-
макромолекулами сыворотки крови, повышая эффективность
ферментативного лечения почечной недостаточности.
М. используют с целью предотвращения разложения
отбеливателей белья при стирке, для изготовления кре-
кремов для обуви, мазей от загара, косметич. товаров
и др.
Для космических аппаратов в США разрабатывают
защитное покрытие, закрывающее пробоины от попа-
попадания микрометеоритов, состоящее из кремнийорга-
нического олигомера и капсулированного катализа-
катализатора его отверждения. Разрабатывают смеси, предназ-
предназначенные для создания пеноматериалов в условиях
космоса, состоящие из двух мономерных компонен-
компонентов или из олигомера и воды в микрокапсулирован-
ном виде.
Выпускаются микрокапсулированные продукты для
химич. чистки одежды и антисептич. средства, нанесен-
нанесенные на бумажные подложки (листы и салфетки). Мик-
рокапсулируют бактерии и вирусы для изучения воз-
воздействия на них космич. лучей, родентициды (напр.,
хлора лозу), различные удобрения, ароматизирующие
добавки к сигаретам, гигиенич. салфеткам, пищевым
смесям, а также пищевые продукты.
Лит.: LuzziL. A., J. Pharma Sci., 59, № 10, 1367
A970); К on do Т., Koishi M., Toso To Toryo, № 191,
77A970); FlinnJ.E., NackH, Battelle Technol. Rev.,
16, № 2 A967); HerbigJ.A., в кн.: Kirk— Othmer,
Encyclopedia of chemical technology, v. 13, 2 ed., N. Y.—
[a. o.], 1967, p. 436; Gardner G. L., Ghem. Eng. Pro-
Progress, 62, №4, 87 A966); Mattson H. W., Internat. Sci.
Technol., № 40, 66 A965); HuberH. F., Stroble
H. G., Tappi, 49, № 5, 41—A A966).
А. Б. Давыдов, В. Д. Солодовник.
МИКРОПОРИСТЫЕ РЕЗИНЫ - см. Кожа искусст-
искусственная.
МИПОРА (mipora) — жесткий пенопласт на основе
мочевино-формальдегидной смолы. Технологич. про-
процесс производства М. состоит из след. стадий: 1) полу-
получение р-ра модифицированной мочевино-формальдегид-
мочевино-формальдегидной смолы, 2) приготовление водного р-ра пенообразо-
пенообразователя, 3) получение пенопласта, 4) отверждение пено-
пенопласта.
Для получения водного р-ра мочевино-формальдегид-
мочевино-формальдегидной смолы мочевину и формальдегид (в виде 30%-ного
формалина) берут в молярном соотношении 1 : A,7 —
1,8). Для модификации смолы в реакционную смесь до-
добавляют многоатомные спирты (обычно глицерин) в ко-
количестве от 20 до 50% от массы мочевины. Формалин
нейтрализуют 10%-ным водным р-ром едкого натра.
Содержимое реактора доводят до кипения, затем в реак-
реакционную смесь вводят 10%-ный р-р муравьиной к-ты
до достижения рН 4,5—5,5. Конденсацию продолжают
до образования р-ра смолы с вязкостью 25—30 мн-сек/м2,
или спз, к-рый нейтрализуют водным р-ром щелочи.
Р-р охлаждают и разбавляют водой до концентрации
ок. 30% по смоле.
В качестве пенообразователя при производстве М.
применяют водный р-р натриевой соли нефтяных суль-
фокислот, к к-рому добавляют резорцин — стабилиза-
стабилизатор пены. В р-р пенообразователя добавляют также
ортофосфорную к-ту, к-рая является катализатором
отверждения. Для предотвращения преждевременного
образования пены первоначально готовят конц. р-р
пенообразователя, к-рый перед употреблением разбав-
разбавляют водой.
Вспенивание мочевино-формальдегидной смолы про-
проводят в цилиндрич. аппарате с вертикальной многоло-
многолопастной мешалкой. Первоначально в течение 2—3 мин
взбивают пену из одного пенообразователя. Пена воз-
возникает вследствие диспергирования воздуха в р-ре
типичного поверхностно-активного вещества — соли
нефтяных сульфокислот. Затем к полученной пене до-
добавляют р-р мочевино-формальдегидной смолы, взби-
взбивают пенопласт в течение 15—20 сек, после чего сли-
сливают его в металлич. формы, где подвергают частичной
сушке с отверждением. Частично отвержденную М.
извлекают и дополнительно отверждают в сушильных
камерах в течение 3 сут при 30—50 °С.
По др. технологии неотвержденный пенопласт, со-
содержащий дополнительный катализатор отверждения
(напр., 5%-ный р-р щавелевой к-ты), получают на месте
259
МИЦЕЛЛА
260
применения и заливают в заполняемый объем. Отверж-
Отверждение пенопласта происходит без нагревания.
Нек-рые физич. свойства М. приведены ниже:
Объемная масса, г/см3 не более 0,02
Коэфф. теплопроводности, вт/(м-К)
[ккал/(м-ч-°С)] 0,03 [0,026]
Показатель паропроницаемости (т = 24 ч),
г/ж* (г/дм*) 460D,6)
Прочность при сжатии, Мн/мг (кгс/см2). . 0,025—0,05
@,25-0,50)
Влажность, % 12
М. обугливается, но не загорается в открытом пла-
пламени при 500 °С; после введения в нее солей ортофос-
форной к-ты или др. антипиренов не воспламеняется
даже в среде кислорода. М. обладает нек-рой эластич-
эластичностью (при сжатии на 20% не разрушается), хорошо
поглощает звук, особенно в диапазоне от средних до
высоких частот. Однако этот материал недостаточно
устойчив к действию агрессивных химич. реагентов
и легко впитывает влагу. Поэтому при хранении и экс-
эксплуатации М. следует защищать пленками из целло-
целлофана, полиэтилена и т. п. М. выпускается в виде бло-
блоков, плит, крошки или заливочной вспененной компо-
композиции. Ее применяют в качестве тепло- и звукоизоля-
звукоизоляционного материала в строительстве, для изоляции
холодильных установок, хранилищ и сосудов для пе-
перевозки жидкого кислорода, для заполнения пустоте-
пустотелых конструкций в транспортном машиностроении.
Жидкий заливочный мочевино-формальдегидный пено-
пенопласт применяют для улучшения структуры почвы,
борьбы с ветровой эрозией почвы и др.
Существует тенденция замены блочной М. заливоч-
заливочным' мочевино-формальдегидным пенопластом. Объем
производства пенопластов на основе мочевино-формаль-
дегидных смол незначителен. Напр., в США он состав-
составляет менее 1% от общего объема производства полимер-
полимерных пеноматериалов.
Промышленное производство пенопласта на основе
мочевино-формальдегидной смолы было впервые орга-
организовано в США. За рубежом блочные пенопласты на
основе мочевино-формальдегидной смолы производят
под названиями к о л ф о а м (США), ф а ф ф (Анг-
(Англия), п и а т е р м (ГДР), ипорка (ФРГ) и др.,
заливочные — изошаум (ФРГ), гризошаум
4 (Швейцария).
• МИЦЕЛЛА (micelle, Mizelle, micelle) — отдельная
частица дисперсной фазы золя, т. е. микрогетероген-
микрогетерогенной (коллоидной) системы с жидкой дисперсионной сре-
средой. М. состоит из кристаллич. или аморфного ядра
и поверхностного слоя, включающего сольватно свя-
связанные молекулы окружающей жидкости. В зависимо-
зависимости от интенсивности межмолекулярного взаимодейст-
взаимодействия веществ дисперсной фазы и дисперсионной среды
различают лиофобные и лиофильные золи. Строение
их мицелл различно.
Лиофобные золи термодинамически не-
неустойчивы. Дисперсная фаза в них слабо взаимодейст-
взаимодействует со средой. Вследствие избытка свободной межфаз-
межфазной энергии такие системы постоянно сохраняют тен-
тенденцию к распаду путем самопроизвольного укрупне-
укрупнения коллоидных частиц. Однако агрегативно устойчи-
устойчивые лиофобные золи способны сохраняться годами
и десятилетиями без видимых изменений дисперсности.
Объясняется это тем, что поверхностный слой М. лио-
фобных золей образован адсорбированными молеку-
молекулами или ионами стабилизатора — третьего компонен-
компонента системы, растворенного в дисперсионной среде.
Слой стабилизатора препятствует сближению и сли-
слипанию (коагуляции) М. под влиянием близкодействую-
близкодействующих сил притяжения. Препятствием к сближению
частиц м. б. различные факторы: расклинивающее дав-
давление жидкой дисперсионной среды, сольватно свя-
связанной молекулами или ионами стабилизатора; электро-
статич. отталкивание одноименно заряженных ионов,
окружающих ядро М.; повышенная структурная вяз-
вязкость защитного поверхностного слоя. Во многих кол-
коллоидных системах действуют одновременно все эти
факторы.
В случае «классических» лиофобных гидрозолей,
стабилизованных электролитами, М. имеет следующее
строение. Ядро ультрамикроскопич. размеров окружено
двумя слоями противоположно заряженных ионов,
т. наз. двойным электрич. слоем. Число положитель-
положительных и отрицательных зарядов в нем одинаково и поэто-
поэтому в целом М. электронейтральна. Непосредственно
у поверхности ядра расположены потенциалопределяю-
щие ионы. Они с частью ионов другого знака (противои-
нов) образуют уплотненный адсорбционный слой. Ос-
Остальные противоионы находятся в диффузном слое.
Сильно гидратированный диффузный слой создает во-
вокруг М. ионное «облако», плотность которого постепен-
постепенно падает по мере удаления от поверхности ядра.
Именно диффузный слой обеспечивает защиту М. от
коагуляции.
М. лиофобных золей, стабилизованных неэлектроли-
неэлектролитами, напр, неионогенными поверхностно-активными
веществами (ПАВ), защищены от коагуляции сильно
сольватированными лиофильными группами молекул
стабилизатора. Среди многокомпонентных полимерных
систем примером типично лиофобных коллоидов могут
служить синтетич. латексы — гидрозоли высокомоле-
высокомолекулярных соединений, полученные эмульсионной поли-
полимеризацией. Лиофобными коллоидами являются также
высокодисперсные системы, возникающие при выделе-
выделении новой фазы из пересыщенных р-ров полимеров или
диспергировании полимерных материалов в присутствии
ПАВ-стабилизаторов.
Лиофильные золи — термодинамически ус-
устойчивые системы. Их агрегативная устойчивость не
связана с наличием стабилизатора. Поверхностный
слой М. в таких системах образован лиофильными
группами молекул вещества самой дисперсной фазы.
Коллоидные частицы лиофильных золей интенсивно
взаимодействуют с окружающей жидкостью и межфаз-
межфазная свободная энергия чрезвычайно мала. Лиофильные
золи образуются в результате самопроизвольного дис-
диспергирования твердых тел или жидкостей под влиянием
теплового движения и не разрушаются со временем
при сохранении условий их возникновения. Таковы,
напр., системы типа критич. эмульсий, возникающих
вблизи критич. темп-ры смешения двух жидкостей,
водные дисперсии бентонитовых глин, коллоидные дис-
дисперсии мыл и смнтетич. моющих веществ, а также
нек-рых органич. пигментов и красителей.
Типично лиофильные золи образуются при смешении
с водой блоксополимеров окиси этилена и окиси про-
пропилена, карбоцепных полимеров с привитыми полиэти-
леноксидными боковыми цепями, полиэтиленгликоле-
вых производных жирных к-т, спиртов, аминов и т. п.
Все эти соединения составляют обширный класс неионо-
генных ПАВ. Аналогично обычным жировым мылам
они образуют гидрозоли, М. к-рых представляют собой
ассоциаты дифильных, т. е. по-разному взаимодейст-
взаимодействующих со средой различными своими частями, моле-
молекул. В каждой такой молекуле длинный углеводород-
углеводородный (гидрофобный) радикал связан с полярной (гидро-
(гидрофильной) группой. Молекула может иметь сложное
строение и состоять из нескольких гидрофильных и гид-
гидрофобных участков. Десятки или сотни молекул объе-
объединяются так, чтобы гидрофобные радикалы сформи-
сформировали ядро, а гидрофильные группы — поверхност-
поверхностный слой М. Если дисперсионной средой является не-
неполярная органич. жидкость, ориентация молекул
в М. м. б. обратной: в ядре сосредоточиваются поляр-
полярные группы, а гидрофобные радикалы обращены во
внешнюю фазу. М.-ассоциаты обратимо распадаются
261
МОДЕЛИ РЕЛАКСАЦИИ МЕХАНИЧЕСКОЙ
262
на отдельные молекулы или димеры при разбавлении
системы ниже критической концентра-
концентрации мицеллообразования. С ростом кон-
концентрации структура и форма М. могут существенно
меняться.
Термин «лиофильные коллоиды» можно встретить
как устаревшее название растворов высокомолекуляр-
высокомолекулярных соединений, представляющих собой гомогенные
(однофазные) системы. Это связанно с тем, что перво-
первоначально полимеры рассматривались как особый класс
коллоидных систем и им приписывали мицеллярное
строение. Причиной заблуждения послужила общность
многих свойств (оптич., осмотич., реологич. и др.)
золей и р-ров полимеров, а также отсутствие в то вре-
время достоверных данных о молекулярном строении по-
последних. Представления о М. как фазовых частицах
было перенесено из коллоидной химии в учение о струк-
структуре полимерных тел.
" Гипотезы о мицеллярном строении полимеров выдви-
выдвигались и после того, как было доказано существование
цепочечных макромолекул. Причем на различных эта-
этапах развития химии и физики полимеров понятие «М.»
трактовалось по-разному. Так, в конце 20-х гг. нашего
столетия К. Майер и Г. Марк определяли М. как пучок
макромолекул, устойчивый вследствие межмолекуляр-
межмолекулярных взаимодействий и сохраняющийся при растворе-
растворении полимера. Г. Штаудингер уже в те годы критически
относился к мицеллярной теории, считая основной
структурной и кинетич. единицей полимерных систем
палочкообразную макромолекулу. В дальнейшем но-
новые данные о строении и свойствах полимеров и их
р-ров вызывали неоднократный пересмотр взглядов,
в основе к-рых лежало представление о М. как основ-
основной и универсальной структурной единице полимерных
тел. Одной иа наиболее распространенных явилась
предложенная Фрей-Висслингом модель бахром-
бахромчатой М. Такая М. представлялась пучком длинных
гибких макромолекул, к-рые на отдельных своих отрез-
отрезках настолько хорошо взаимно ориентированы, что
образуют кристаллиты. Проходя кристаллиты на-
насквозь, макромолекулы перепутываются друг с дру-
другом, образуя аморфные области, а затем вновь попада-
попадают в кристаллич. область и т. д.
Представления о строении полимерных тел прошли
сложную эволюцию от мицеллярных теорий к совре-
современным концепциям структурной физики полимеров
(см. Структура, Надмолекулярная структура, Кри-
Кристаллическое состояние, Аморфное состояние, Коллоид-
Коллоидные полимерные системы). Несостоятельность мицел-
мицеллярных теорий строения линейных гомополимеров
с однородными по строению цепями макромолекул
(напр., целлюлозы, натурального каучука) заключает-
заключается в отсутствии физич. причин существования устой-
устойчивых фазовых частиц коллоидных размеров. Разви-
Развитие представлений о макромолекулах, не отличающих-
отличающихся от малых молекул природой сил межмолекулярного
взаимодействия, исключило возможность научного обо-
обоснования мицеллярных представлений о строении по-
полимеров и их р-ров. Здесь следует еще раз подчерк-
подчеркнуть, что имеются в виду макромолекулы, лишенные
дифильности в упомянутом выше смысле. Гибкие мак-
макромолекулы, содержащие разнородные по полярности
участки, в определенных условиях могут давать микро-
микрогетерогенные системы типа лиофильных золей. При
этом лиофобные группы макромолекул объединяются
в ядре коллоидной частицы (напр., белковой глобулы),
а лиофильные образуют ее поверхностный слой.
Представления о мицеллярном строении полимеров
уступили место новой концепции пачечного строения
полимеров, развитой В. А. Каргиным, А. И. Китай-
Китайгородским и Г. Л. Слонимским (см. Пачки).
Лит.: П а с ы н с к и й А. Г., Коллоидная химия, М., 1968;
ВоюцкийС.С, Курс коллоидной химии, М., 1964; К а р-
гин В. А., Слонимский Г. Л., Краткие очерки по фи-
зико-химии полимеров, 2 изд., М., 1967; Штаудин-
Штаудингер Г., Высокомолекулярные органические соединения, пер.
с нем., Л., 1935, Г. Л. Слонимский, Л.А.Шиц.
МОДЕЛИ РЕЛАКСАЦИИ МЕХАНИЧЕСКОЙ (mo-
(models of a mechanical relaxation, Modelle von mechani-
schen Nachwirkung, modeles de relaxation mecanique) —
механические, электрические и др. физич. системы,
характеризующиеся совпадением математич. описания
типичных для них процессов с описанием релаксацион-
релаксационных явлений в полимерных телах (оригиналах). Это
выражается в одинаковости соответствующих ур-ний
и однозначности соотношений между переменными
в ур-ниях модели и оригинала. Модели механич. ре-
релаксации (М.) используются для наглядного представ-
представления и изучения сложных релаксационных механич.
явлений в полимерных телах, для установления феноме-
феноменологических и структурных, качественных и количе-
количественных закономерностей.
Для описания линейных релаксационных процессов
применяются М., состоящие из линейных составляющих
элементов. В случае, напр., механич. М. такими эле-
элементами служат пружины и жидкостные демпферы. Для
нелинейных процессов возможны различные приемы
построения М., напр, введением в них нелинейных
упругих и неупругих элементов, что эквивалентно заме-
замене постоянных коэффициентов в ур-ниях, описывающих
механич. поведение элементов моделей, на коэффици-
коэффициенты, зависящие от деформации, механич. напряжения,
скоростей их изменения и т. п. Данная статья посвя-
посвящена наиболее разработанным линейным М.
Следует различать формальные и физич. М. При
формальном подходе, напр, в случае механич. М., про-
произвольно комбинируют пружины с различными коэфф.
упругости (жесткостями) и жидкостные демпферы
с различными коэфф. сопротивления (вязкостями), под-
подбирая систему той или иной сложности, сколь угодно
близкую по релаксационным свойствам к реальным
полимерным телам. При построении физич. модели
выбор числа и расположения пружин и демпферов,
а также их характеристик производят с учетом реаль-
реальной структуры оригинала. Физич. моделирование воз-
возникло значительно позднее формального, поскольку
строение и свойства макромолекул стали известны
лишь в 30-х годах текущего столетия. Опираясь на эти
данные, удалось развить представления о природе
механич. релаксационных процессов и деформации
полимеров (Я. И. Френкель, П. П. Кобеко, Е. В. Кув-
шинский, Г. И. Гуревич).
Механические модели. Это наиболее распространен-
распространенный вид М. Массой составляющих их элементов (пру-
(пружин и жидкостных демпферов) обычно пренебрегают,
т. к. для релаксационных процессов в полимерных те-
телах характерны достаточно медленные изменения де-
деформаций и напряжений, при к-рых силы инерции пре-
пренебрежимо малы по сравнению с вязкими силами (если
же требуется учет сил инерции, напр, при акустич.
исследованиях, то в М. вводят дополнительные массив-
массивные элементы).
Простейшими механич. М. являются Максвелла
модель и Кельвина модель, описывающие свойства двух
основных типов релаксирующих тел — соответственно
упруговязкого (текучего тела, обладающего упруго-
упругостью) и вязкоупругого (упругого тела с внутренним
трением). Однако эти модели дают только качествен-
качественное и далеко не полное описание релаксационных яв-
явлений в полимерных телах. Формально соединяя в еди-
единую систему большое число моделей Максвелла и Кель-
Кельвина с различными характеристиками входящих в них
пружин и демпферов, получают М., способные описать
механич. релаксационные процессы в полимерных те-
телах с любой степенью точности. При этом любое число
параллельно соединенных различных моделей Кельви-
Кельвина полностью эквивалентно одной модели Кельвина
263
МОДЕЛИ РЕЛАКСАЦИИ МЕХАНИЧЕСКОЙ
264
с жесткостью и вязкостью, соответственно равными
суммам жесткостей и вязкостей соединенных моделей.
Аналогично, последовательное соединение любого числа
моделей Максвелла эквивалентно введению одной моде-
модели Максвелла, у к-рой обратные величины жесткости
и вязкости соответственно равны суммам обратных ве-
величин жесткостей и вязкостей соединенных моделей.
Однако такое произвольное комбинирование моделей,
несмотря на достигаемую этим точность количествен-
количественного описания, ни в коей мере не соответствует физич.
картине изучаемых процессов.
Первая физически обоснованная приближенная М.
полимерного тела, учитывающая его упругие, вязкие
и высокоэластич. свойства, была предложена в 1939
(А. П. Александров и Ю. С. Лазуркин). Эта модель,
если изобразить ее в механич. форме (авторы дали лишь
ее математич. описание), имеет вид последовательно
соединенных пружины, модели Кельвина и демпфера,
что соответствует представлению об аддитивности
упругой, высокоэластичной и необратимой (вязкой)
слагающих деформации полимерного тела. Эта М.,
хорошо передавая основные качественные закономер-
закономерности механич. релаксации полимерного тела, оказала
серьезное влияние на развитие представлений о его
механич. свойствах.
В 1947 была предложена (В. А. Каргин и Г. Л. Сло-
Слонимский) первая физич. М., учитывавшая как адди-
аддитивность трех упомянутых слагающих деформации,
так и др. особенности строения полимерного тела (вза-
(взаимосвязанность, длину и гибкость макромолекул).
В этой модели (рис.) каждый сегмент макромолекулы
; _Ai ^ кг~ ' " к.
характеризуется обычной упругостью, высокоэластич-
ностью и вязким сопротивлением окружающей его
полимерной среды. Упругость в модели отражена пру-
пружиной (жесткостью к), высокоэластичность — моделью
Кельвина (жесткость кг и коэфф. вязкого сопротивле-
сопротивления rt); вязкое сопротивление окружающей среды
движению сегмента (коэфф. вязкого сопротивления г)
учтено введением взаимодействующего с ней шара;
силы инерции не учитываются. Эти три элемента моде-
модели сегмента соединены последовательно в соответствии
с аддитивностью слагающих деформации. Линейное
строение макромолекулы отражено последовательным
соединением всех моделей сегментов в единую цепь,
а число сегментов п определяется отношением масс
макромолекулы и сегмента.
Т. о., для полимера однородного химич. строения,
т. е. с одинаковыми сегментами, модель вводит пять
физич. параметров — к, кг, г, гх и п; значения первых
четырех м. б., в принципе, вычислены из молекулярных
констант (подобно тому, как рассчитывается модуль
упругости кристалла или вязкость жидкости), а пя-
пятый задается гибкостью и длиной макромолекул.
Следует заметить, что при переходе от сосредоточен-
сосредоточенных постоянных к распределенным эта М. превращается
в систему, состоящую из струны и вязкой жидкости,
причем струна обладает упругостью и высокоэластич-
ностью. Соответствующей упрощенной моделью явля-
является упругая струна в вязкой жидкости.
Приведенная на рис. М. упруговязкого полимерного
тела превращается в М. вязкоупругого тела, если левый
конец цепи прикрепить при помощи пружины к обо-
оболочке М., уничтожив этим возможность необратимых
перемещений.
Эти М. позволили теоретически проанализировать
особенности механич. релаксационных свойств поли-
мерных тел и связать их со степенью полимеризации
и гибкостью макромолекул, охарактеризовать темпера-
температурные зависимости релаксационных процессов (в част-
частности, раскрыть смысл трех физич. состояний аморф-
аморфных полимеров). Одним из важнейших результатов
было физич. обоснование возникновения релаксацион-
релаксационного спектра в макромолекулярных системах, ранее
вводимого только формально.
Позднее различные формы этой М., по-видимому не-
независимо, предлагались зарубежными учеными (напр.,
наиболее соответствующая упрощенной форме модели
Каргина — Слонимского известная модель «ожерелья»),
успешно изучавшими с их помощью свойства р-ров
полимеров.
Другие виды моделей. Наряду с механич. М. воз-
возможно использование линейных и нелинейных элект-
электрических, магнитных, акустических, тепловых и др.
Из них чаще встречаются линейные электриче-
электрические М., применение к-рых основано на полной иден-
идентичности описания замкнутых электрич. цепей, состоя-
состоящих из конденсаторов и омич. сопротивлений (а также
катушек индуктивности, если необходимо ввести аналог
массы), и описания механич. М. Так, при сопоставле-
сопоставлении заряда Q и механич. перемещения х, силы тока
/ = dQidt (t — время) и скорости перемещения
и = dx/dt, обратной величины емкости конденсатора
1/С и жесткости пружины к, электрич. омического со-
сопротивления R и коэфф. вязкого сопротивления демп-
демпфера г, коэфф. самоиндукции L и массы т оказывается,
что известные выражения для электрич. величин по
форме эквивалентны соотношениям, связывающим ме-
механич. параметры. А именно, ур-ния для разности по-
потенциалов, приложенной к обкладкам конденсатора
Uс = QIC, и падения напряжения на омич. сопротивле-
сопротивлении Ur = BI = RdQldt по форме соответственно экви-
эквивалентны выражениям для растягивающей упругую
пружину силы Fk = кхи преодолевающей вязкое сопро-
сопротивление демпфера силы Fr — rv = rdxldt, а электродви-
электродвижущая сила индукции Е^ = —Ldlldt = —Ld2Q/dt2 и
сторонние электродвижущие силы ЕСТ эквивалентны
соответственно силе инерции fm = — mdv/dt =
= —md2x/dt2 и внешним силам /вн, действующим на ме-
механич. систему. При этом для электрических и механич.
величин справедливы одинаковые по форме основные
соотношения: ЕСТ + EL = Uc + UR и /вн + fm =
= Fk + Fr.
Задав в модельной электрич. цепи начальные рас-
распределения электрич. величин, соответствующие реаль-
реальной механич. системе, можно экспериментально иссле-
исследовать электрич. релаксационный процесс, совпадаю-
совпадающий по своим качественным и количественным харак-
характеристикам с механич. релаксационным процессом
в изучаемом полимерном теле.
При построении идентичных электрических и меха-
механич. М. необходимо учитывать, что в случае последова-
последовательного соединения в цепь механич. элементов адди-
аддитивными свойствами обладают перемещения, а силы,
действующие на любой из этих элементов, равны одна
другой, в то время как при параллельном соединении
элементов складываются силы, а перемещения одина-
одинаковы. Поэтому первому случаю соответствует аддитив-
аддитивность зарядов, т. е. параллельное соединение электрич.
элементов М., а второму — аддитивность электрич.
напряжений, т. е. последовательное соединение эле-
элементов. Как и в случае механич. М., возможен переход
от сосредоточенных постоянных к распределенным.
Так, напр., в случае упрощенной механич. модели Кар-
Каргина — Слонимского такой эквивалентной электрич.
М. с распределенными постоянными оказывается теле-
телефонная линия, т. е. два провода (омические сопротив-
сопротивления), обладающие электрич. емкостью.
Т. о., возможность построения механических, элект-
электрических и других М. открывает, при достаточно хоро-
265
МОДИФИКАЦИЯ СТРУКТУРНАЯ
266
шем соответствии модели оригиналу, пути экспери-
экспериментального моделирования релаксационных процес-
процессов в полимерных телах. Однако существует еще дру-
другая, не менее важная особенность М. Она заключается
в том, что ур-ния, описывающие свойства М. и идентич-
идентичные по форме ур-ниям релаксирующего полимерного
тела, м. б. проанализированы чисто математич. мето-
методами. Это позволяет проводить исследование релакса-
релаксационных процессов путем анализа свойств одних толь-
только ур-ний. Возникло понятие о математиче-
математических М., позволяющих вообще не привлекать на-
наглядные механические, электрические и др. физич.
образы.
Математич. линейные и нелинейные М. лежат в ос-
основе современной теории релаксации, теории конст-
конструирования и расчета полимерных изделий, в т. ч. де-
деталей машин, а также теории переработки полимеров
в материалы и изделия. Они позволяют отразить мно-
многие тонкие структурные особенности полимерных тел,
моделирование к-рых с помощью физич. систем пред-
представляет сложную, почти невыполнимую задачу. Это
стало особенно очевидным после обнаружения в поли-
полимерах надмолекулярных структур, учет к-рых потребо-
потребовал существенного усложнения обычных моделей.
Большие возможности в области разработки матема-
математич. моделей открыло применение преобразования Ла-
Лапласа (Б. Гросс, А. Тобольский, Дж. Ферри), а также
дробных интегральных и дифференциальных операто-
операторов (Г. Слонимский).
Разработка разных форм М. сыграла и продолжает
играть важную роль в создании и развитии современ-
современной физики твердых и жидких полимерных тел.
Лит.: А л ф р е й Т., Механические свойства высокопо-
лимеров, пер. с англ., М., 1952, гл. 2; Каргин В. А.,
Слонимский Г. Л., Краткие очерки по физико-химии
полимеров, 2 изд., М., 1967, с. 134; ДАН СССР, 62, № 2, 239
A948); Александров А. П., Лазуркин Ю. С,
ШТФ, 9, в. 14, 1249 A939); Слонимский Г. Л., ДАН
СССР, 140, № 2, 343 A961); J. Polymer Sci., pt С, №16, 1667
A967); Высокомол. соед., 13А, № 2, 450 A971).
Г. Л. Слонимский.
МОДИФИКАЦИЯ СТРУКТУРНАЯ полиме-
полимеров, модификация физическая (struc-
(structural modification, strukturelle Modifizierung, modi-
modification structurale) — направленное изменение физиче-
физических (прежде всего механических) свойств полимеров,
осуществляемое преобразованием их надмолекулярной
структуры под влиянием физич. воздействий. В отли-
отличие от модификации химической, при М. с. химич.
строение макромолекул сохраняется.
Введение в полимер значительных количеств пласти-
пластификатора, наполнителя, др. полимера, широко исполь-
используемое в пром-сти (см. Пластификация, Наполнение,
Совместимость), не является М. с, поскольку при этом
изменение свойств системы обусловлено не только из-
изменением ее физич. структуры, но и состава.
М. с. может быть осуществлена различными путями:
внешними механич. воздействиями на твердое поли-
полимерное тело (см., напр., Ориентированное состояние)]
изменением температурно-временнбго режима струк-
структурообразования твердого полимерного тела из расп-
расплава; изменением природы растворителя и режима его
удаления при образовании из р-ров полимеров покры-
покрытий, пленок и волокон; введением в полимер малых
количеств (не более нескольких %) др. веществ, влияю-
влияющих на кинетику образования и морфологию надмоле-
надмолекулярной структуры модифицируемого полимера (см.
Структур эобразователи).
Успешное развитие М. с. обусловлено обнаружением
многих сосуществующих форм надмолекулярных обра-
образований, многообразия соответствующих им физич.
структурных превращений. Это потребовало пересмот-
пересмотра упрощенных представлений о полимерных телах
как о системах перепутанных макромолекул. Влияние
механич. воздействий, ранее понимавшееся лишь как
влияние на форму и ориентацию гибких макромолекул,
оказалось гораздо более сложным. Напр., при одноос-
одноосном растяжении полимерного тела происходят различ-
различные преобразования элементов его надмолекулярной
структуры (разрушение, образование, деформация),
особенно разнообразные у кристаллич. полимеров.
Соответственно оказалось, что и тепловые воздействия
влияют не только на подвижность макромолекул,
но и на скорость образования и роста первичных цент-
центров разных форм элементов надмолекулярной структу-
структуры, а также на скорость их взаимных превращений.
Модифицирование изменением условий получения
твердого тела. Структурно-механич. исследования по-
позволили получить представление о влиянии на физич.
структуру полимерного тела его механической и тепло-
тепловой предыстории. Это открыло эффективные приемы
М. с, основанные на выборе рационального темпера-
температурно-временнбго механич. режима формования поли-
полимерного тела из расплава (темп-pa расплава, длитель-
длительность его выдержки при повышенных темп-pax, режим
охлаждения, давление и др.), а также на преобразова-
преобразовании уже сложившейся надмолекулярной структуры
материала или изделия путем механич. воздействий на
нее в соответственно выбранных температурных усло-
условиях (напр., многократными деформациями, прокат-
прокаткой).
В случае получения изделий из р-ров полимеров
структурно-механич. подход позволил раскрыть влия-
влияние природы растворителя и условий его испарения
или условий осаждения полимера на процессы струк-
структурообразования, что привело к возможности управ-
управления ими и формирования изделий (напр., волокон
и пленок) с желательной надмолекулярной структурой.
Модифицирование введением малых количеств ве-
веществ иного строения. Для воздействия на процессы
образования и агрегирования различных элементов
надмолекулярной структуры предложено введение в по-
полимер малых количеств поверхностно-ак-
поверхностно-активных веществ, к-рые влияют на процессы
структурообразования в полимерах так же активно,
как и в хорошо известных случаях структурообразова-
структурообразования низкомолекулярных веществ. Добавками поверх-
поверхностно-активных веществ можно изменять размеры и
форму. элементов надмолекулярной структуры.
Как известно, центрами роста различных надмоле-
надмолекулярных образований являются неоднородности
флюктуационной природы, существующие в расплавах
и р-рах полимеров (см. Пачки, Кристаллизация). Сле-
Следовательно, регулирование структуры м. б. осущест-
осуществлено изменением числа таких центров (в случае кри-
кристаллизации — это зародыши кристаллизации).
Введение в полимер химически не взаимодействую-
взаимодействующих с ним высокодисперсных инородных частиц орга-
нич. или неорганич. природы, не растворяющихся в его
расплавах или р-рах, может вызывать появление в по-
полимере собственных зародышей структурообразования,
располагающихся на границе раздела между частицей
и полимером.
Такие вещества, получившие название искус-
искусственных зародышеобразователей,
приводят к ускорению структурообразования и воз-
возникновению более однородной надмолекулярной струк-
структуры. В случае кристаллич. полимеров такое воздейст-
воздействие на физич. структуру и свойства проявляется, напр.,
в уменьшении размеров сферолитов и соответствующем
изменении механич. свойств (рис. 1 и 2). Кроме того,
наличие в кристаллич. полимере частиц искусствен-
искусственного зародышеобразователя имеет другим своим важ-
важным следствием стабилизацию физич. структуры поли-
полимерного тела, восстанавливающейся после плавления
и повторной кристаллизации.
Существенно, что для М. с. при помощи искусствен-
искусственных зародышеобразователей не требуется соответ-
267
МОДИФИКАЦИЯ СТРУКТУРНАЯ
268
вие их собственной структуры и структуры полимера.
Они м. б. твердыми (кристаллическими и аморфными),
жидкими и даже газообразными, низкомолекулярными
или высокомолекулярными веществами. Одно и то же
Рис. 1. Влияние частиц искусственного зародышеобразова-
теля на надмолекулярную структуру полипропилена: а —
образец, закристаллизованный без зародышеобразователя;
б— образец, содержащий 1% (по массе) индиго.
вещество может служить искусственным зародышеобра-
зователем для одного полимера и не являться им для
другого, что указывает на существенную роль химич.
природы полимера и вводимой частицы, несмотря на
отсутствие химич. взаимодействия между ними при
М. с. Хотя этот прием М. с. уже имеет практич. значе-
значение, механизм зародышеобразования на вводимых
в полимер инородных
200-
200 300 400 500 z,%
частицах пока еще из-
изучен мало.
Введение в полимер
крупных частиц (воло-
Рис. 2. Диаграмма растя-
растяжения образцов полипро-
полипропилена, закристаллизован-
закристаллизованных в присутствии заро-
зародышеобразователя и без
него. Обозначения см. рис.
1 {? — напряжение, = —
деформация) Aкгс/сж2^0,1
Мн/м2).
конец, отдельных кристаллов) зародышеобразователей
приводит к М. с. граничащих с этими частицами слоев
полимера (рис. 3), измененные свойства к-рых влияют на
свойства полимерного тела в целом, препятствуя, напр.,
развитию трещин. Эти эффекты обусловлены не самими
частицами зародышеобразователей, а именно М. с.
окружающих их слоев полимера. Весьма интересна
М. с. поверхностного слоя полимерного тела (рис. 4),
осуществляемая контактом
его поверхности с плен-
пленкой искусственного заро-
зародышеобразователя в про-
процессе формования (напр.,
при прессовании такая
пленка м. б. помещена на
поверхности прессформы).
Поверхностная М. с. поз-
позволяет получать твердые
полимерные тела с повы-
повышенной износостойкостью,
_ п _ а также с пониженной га-
Рис. 3. Картина кристаллиза- олпппттиттярмпртт m
ции изотактического иолисти- зопроницаемостью.
рола на крупных частицах ин- Упомянутые приемы
диго. М. с. полимерных тел при
помощи зародышеобразо-
зародышеобразователей, позволяющие получать как однородные, так
и неоднородные по типам и размерам надмолекуляр-
надмолекулярные образования, еще более эффективны при их соче-
сочетании. При этом обеспечивается улучшение комплекса
свойств полимерного тела в результате М. с. его
объема в целом, улучшение механических и др.
свойств в наиболее опасных частях изделия вследст-
вследствие их армирования модифицированными внутрен-
внутренними слоями, а также создание желательной струк-
структуры поверхностного слоя изделия в наиболее ответ-
ответственных его наружных частях.
М. с. при помощи зародышеобразователей оказалась
особенно эффективной при переработке кристаллизую-
кристаллизующихся полимеров. Значительно меньше изучена такая
М. с. в случае стеклообразных и высокоэластич. поли-
полимеров, а также расплавов, хотя и для них обнаружен
эффект модифицирования.
Следует отметить эффективность сочетания М. с.
с приемами химич. стабилизации полимеров, осущест-
осуществляемого, напр., в случае кристалллч. полимеров ис-
искусственным зародышеобразованием частицами стаби-
стабилизаторов.
Для кристаллизующихся полимеров возможно так-
также совмещение М. с. с синтезом макромолекул пу-
путем введения зародышеобразователя в мономер (вы-
(выбирается зародышеобразователь, не влияющий замет-
заметно на химич. строение образующихся макромолекул)
или покрытия зародышеобразователем внутренней по-
поверхности реакционного сосуда. М. с. как кристалличе-
кристаллических, так и аморфных полимеров в процессе синтеза
может осуществляться также путем варьирования ре-
режима реакции и изменением химич. природы или фи-
зич. структуры среды, в к-рых происходит образова-
образование макромолекул. Естественно, что во всех случаях
М. с, осуществляемой в ходе синтеза или переработки,
необходимо выбирать такие модифицирующие вещества,
к-рые не вызывают коррозию оборудования.
Введение в полимер жидких или высокоэластич. ве-
веществ др. строения является эффективным приемом
М. с. не только для кристаллич. полимеров, но и для
аморфных. Хотя механизм их действия в этих случаях
не выяснен, но установлено их существенное влияние
на механич. свойства полимера. Отнесение такого вида
модифицирования к М. с. обусловлено тем, что малое
количество добавки вызывает значительное измене-
изменение механич. свойств, что м. б. понято только как
результат изменения либо самих элементов надмо-
надмолекулярной структуры, либо взаимодействия между
ними. Пример такой М. с— структурная пластифика-
пластификация, позволяющая введением малого количества пла-
пластификатора (десятые и сотые доли %) резко изменить
механич. свойства жест-
коцепных и нек-рых др.
Рис. 4. Надмолекулярная
структура, видимая в поля-
поляризованном свете в попереч-
поперечном срезе с пленки полипро-
полипропилена (толщина 140 мкм),
поверхностно модифицирован-
модифицированной с обеих сторон.
аморфных полимеров, а также приемы повышения
ударной прочности аморфных твердых полимеров вве-
введением в них небольшого количества эластомера, обра-
образующего включения микроскопического размера.
М. с. применяется для улучшения качества волокон
(напр., при ориентациоином упрочнении, реализуемом
вытяжкой в отсутствие или в присутствии зародыше-
зародышеобразователей), для борьбы с самопроизвольным рас-
растрескиванием твердых полимеров, для повышения
ударной прочности пластмасс, для регулирования
структуры полимеров в процессах изготовления из-
изделий (напр., при экструзии, прессовании, образовании
полимерных покрытий), для стабилизации их физич.
структуры.
Лит.: Карги н В. А., Слонимский Г. Л., Крат-
Краткие очерки по физико-химии полимеров, 2 изд., М\, 1967; С о -
голова Т. И., Механика полимеров, jsft 1, 5 A965), № 5,
269
МОДИФИКАЦИЯ ХИМИЧЕСКАЯ
270
643 A966), № 3, 395 A972); ее же, в кн.: Успехи химий
и физики полимеров, М., 1970, с. 232; Андрианова Г.П.,
БакеевН. Ф., Козлов П. В., Высокомол. соед., 13 А,
266 A971). Т. И. Соголова.
МОДИФИКАЦИЯ ХИМИЧЕСКАЯ полиме-
полимеров (chemical modification, chemische Modifizierung,
modification chimique) — направленное изменение
свойств полимеров при введении в состав макромоле-
макромолекул малого количества фрагментов иной природы. Все
методы химич. модификации (М.) полимеров м. б. клас-
классифицированы след. образом:
1. Модификация, основанная на химич. превращени-
превращениях уже синтезированных макромолекул: а) реакции по-
полимера с низкомолекулярным соединением (модифика-
(модификатором), не способным к полимеризации или поликонден-
поликонденсации в выбранных условиях. Сюда относятся процес-
процессы, не сопровождающиеся изменением длины цепи
(полимер аналогичные превращения, внутримолекуляр-
внутримолекулярные превращения, реакции концевых групп), сшивание
макромолекул низкомолекулярными соединениями;
к этой же группе процессов можно отнести образование
пространственно-сетчатых структур под действием УФ-
облучения или радиации, хотя оно может протекать
и в отсутствие химич. агентов; б) реакции полимера
с мономером, когда в ходе процесса генерируются рас-
растущие цепи, взаимодействующие с полимером с обра-
образованием разветвленных или пространственно-сетчатых
структур; в) взаимодействие полимера с высокомоле-
высокомолекулярным модификатором.
2. Модификация на стадии синтеза полимера.
Модификация, основанная на химических превраще-
превращениях уже синтезированных макромолекул. Для ряда
полимеров наличие концевых групп с подвижными
атомами водорода обусловливает сравнительно легкое
протекание деполимеризации и, следовательно, отно-
относительно низкую термостабильность таких полимеров.
Это делает необходимым блокирование концевых групп
органическими радикалами, достаточно стабильными
в условиях переработки и применения полимера.
Так, ацетилирование или метилирование концевых
ОН-групп полиформальдегида повышает его термоста-
термостабильность при глубоком вакууме и темп-ре 200 °С
в 8 — 10 раз. Блокирование концевых силанольных
групп в полидиметилсилоксановых каучуках, гидро-
ксильных и карбоксильных групп в простых и сложных
ароматич. полиэфирах также повышает темп-ру разло-
разложения соответствующих материалов.
Значительно большие возможности для М. полиме-
полимеров открываются при использовании реакций присое-
присоединения по кратным связям макромолекул (при сравни-
сравнительно малых степенях превращения), полимеранало-
гичных превращений, протекающих с заменой функцио-
функциональных групп или раскрытием напряженных циклов.
Так, при вальцевании натурального каучука с 0,5—
2 моль тиобензойной или тиотрихлоруксусной к-ты
(в расчете на 1 моль звена) и инициатором получается
материал более регулярной структуры, содержащий
звенья различной природы и пространственного распо-
расположения:
X'+RCOSH -» ХН + RCOS*
СН3 СН3 SGOR
RGOS- l
СН3 SGOR
+RCOSH I I
> ~<СН2-СН СН-СНа^ + RGOS'
где R — фенил или СС13, X* — радикал, образующийся
при распаде инициатора. Одновременно с реакцией
присоединения протекает цис-транс-изомеризаищя кау-
каучука. На основе таких модифицированных каучуков
получаются резины, обладающие повышенной морозо-
морозостойкостью, масло- и бензостойкостью.
Перспективы практич. использования имеет метод
М., основанный на обработке синтетич. каучуков
(стереорегулярного бутадиенового и изопренового) низ-
комолекулярнымР1 меркаптанами в присутствии ди-
нитрила азоизомасляной к-ты, напр.:
SR
RSH
С
CHS
При помощи этой реакции можно получить эластомеры
с повышенной озоностойкостью и улучшенными физи-
ко-механич. свойствами.
В результате взаимодействия натурального или
синтетич. каучука с 3—5% ангидрида малеиновой или
другой а,Р-ненасыщенной к-ты образуется продукт,
к-рый по ряду свойств отличается от исходного кау-
каучука. Он нерастворим в бензоле, образует прочные
вулканизаты с окислами свинца, цинка и ряда др. ме-
металлов и обладает меньшей газопроницаемостью. Про-
Процесс можно проводить в р-ре в присутствии источника
свободных радикалов (напр., перекиси бензоила) или без
инициатора при вальцевании. В последнем случае
реакция инициируется полимерными радикалами, обра-
образующимися при механической деструкции каучука на
вальцах.
В результате превращений, протекающих с частич-
частичным замещением, нарушается, как и в случае рассмот-
рассмотренных выше реакций присоединения, химич. однород-
однородность и регулярность расположения звеньев, а, следо-
следовательно, характер и тип надмолекулярных структур.
Так, введение 10—15% связанного хлора в полиэтилен
и полипропилен приводит к получению трудно кристал-
кристаллизуемых эластичных материалов, для к-рых характер-
характерно наличие дефектных сферолитов и ленточных образо-
образований, а при большом содержании хлора — фибрилляр-
фибриллярных структур.
Хлорсульфирование полиэтилена смесью сернистого
ангидрида и хлора позволяет получить каучукоподоб-
ный материал, т. н. хайпалон, при введении 1,3—1,75%
связанной серы и 26—29% хлора, т. е. одной хлорсуль-
фоновой группы и одного атома хлора на соответственно
90 и 7 атомов углерода (см. Полиэтилен хлорсульфиро-
ванный). Хлорсульфирование м. б. применено и для
модификации свойств полистирола, полибутадиена и
стереорегулярных олефинов.
С использованием реакций замещения модифицируют
не только насыщенные полимеры, но и эластомеры, со-
содержащие в небольших количествах непредельные зве-
звенья. Так, введение в бутилкаучук 1,1 —1,3% хлора или
2—3% брома облегчает его вулканизацию (благодаря
присутствию весьма реакционноспособного аллильного
галогена, входящего в звенья изопрена) и позволяет
получать более прочные резины.
Реакции замещения м. б. применены и для М. насы-
насыщенных полимеров, содержащих атомы галогена или
др. группы, замещаемые при нуклеофильной атаке.
Так, для повышения термостабильности поливинил-
хлорида или хлорированных полиолефинов представ-
представляет интерес введение в их боковые цепи оловоорганич.
групп. Это достигается обработкой полимера трифенил-
или трибутилоловолитием:
+R SnLi
-'СНа-СН-СНа-СН-
I I
Gl R
> —СН2-СН-СН2-СН~'
I I
SnR' R
где R — Н,С1. Введение в поливинилхлорид одноц
станнильной группы на 50 звеньев уменьшает кон-
константу скорости дегидрохлорирования при 175 °С
в два раза.
Интересным примером использования нуклеофиль-
ного замещения для М. полимеров является ониевая ин-
интерполимеризация хлорсодержащих полимеров с по-
полимерами, содержащими нуклеофильные группы. Эта
271
МОДИФИКАЦИЯ ХИМИЧЕСКАЯ
272
реакция лежит в основе промышленного способа полу-
получения ударопрочного поливинилхлорида. Оказалось,
что в процессе смешения с поливинилхлоридом бу-
тадиен-2-метил-5-винилпиридинового каучука, содер-
содержащего 15—25% винилпиридиновых звеньев, или
бутадиен-нитрильного каучука с 15—18% акрилонит-
рильных звеньев протекают следующие реакции:
~СН2-СНС1— СН2— СНС1-
~(СН2— СН=СН—СН2) — СН-—СН2~
CN
^СН2-СН—(СН2—СН=СН-СН2)/? ~
-(сн2-сн=сн-сн2)/7-сн2-
~сн2—сн—сн2—chci<~
I
СН2— СН—СН2— СНС1~
-сн-сн -Ген -сн-гн-
В результате при введении 8—10% каучука дости-
достигается более чем десятикратное повышение ударной
вязкости без заметного изменения темп-ры стеклова-
стеклования. Образующийся интерполимер представляет собой
равномерно распределенную фазу системы, играющую
роль эластичного армирующего наполнителя.
Реакции функциональных групп макромолекул яв-
являются, пожалуй, наиболее широко применяемым мето-
методом М. полимеров. Чаще всего таким путем удается
направленно влиять на гигроскопичность материала,
его адгезию, способность к накрашиванию и структу-
структурированию, ионо- и электронообменные свойства, био-
биоактивность и прочность.
При обработке целлюлозы, поливинилового спирта
или др. полимеров, содержащих в макромолекуле
группы с «подвижным» водородом (—ОН, —NH2,
— NHR, —CONH2 и др.)? веществами, способными
в мягких условиях замещать атом водорода на длинные
алифатические или алкилсилильные радикалы, можно
придать материалу гидрофобные свойства и стойкость
к действию микроорганизмов. При этом химич. обра-
обработка не вызывает деструкции и изменения морфоло-
гич. структуры полимера. Так, при поверхностной об-
обработке волокон, пленок и др. изделий из целлюлоз-
целлюлозных материалов 1—3%-ной водной коллоидной дис-
дисперсией окта- или гексадецилхлорметилата пиридина
(«велан») протекает реакция:
+ ' 110-120°С +
ROCH2NR3C1- + (OH)Cell > ROCH2OCell -f NHR3C1"
Повышение влагостойкости и устойчивости при экс-
эксплуатации в атмосферных условиях достигается, напр.,
при мягкой обработке целлюлозных и белковых мате-
материалов кремнийорганич. мономерами (алкилтриэтокси-
силан, алкилтриацетоксисилан, стеароксисилан и др.)
или полимерами (полиэтилгидросилан, полиэтилгидро-
ксисилан). Подробно об этом см. Гидрофобизаторы.
Наличие в полимере гидроксильных или аминогрупп
делает возможным получение на его основе волокон,
химически связанных с красителем. Такой эффект до-
достигается при обработке волокнистого или пленочного
материала красителями, содержащими цианурилхло-
ридные, альдегидные или др. реакционноспособные
группы. См. также Крашение волокон.
Большую актуальность приобретает проблема М.
полимеров с целью придания им биологич. активности.
Для введения низкомолекулярных биоактивных сое-
соединений в макромолекулы также используются реак-
реакции функциональных групп. В случае нерастворимой
в воде полимерной основы (целлюлоза и ее производ-
производные) получаются ткани, волокна и пленки с антимик-
антимикробным, гемостатическим и др. действием, находя-
находящие все возрастающее применение в медицине (см.
Антимикробные волокна, Медицинские нити). При М.
водорастворимых полимеров (декстран, поливиниловый
спирт) введением в них фрагментов низкомолекуляр-
низкомолекулярных лекарственных веществ удается пролонгировать
их действие (см. Полимеры в медицине).
Наличие в макромолекулах гидроксильных, кар-
карбоксильных, карбонильных, амидных, эпоксидных
групп позволяет осуществить их сшива-
сшивание даже при малых дозировках реаген-
реагентов-модификаторов. Так, большое зна-
значение в лакокрасочной пром-сти приобре-
приобретают лаковые композиции, состоящие из
карбоксилсодержащих сополимеров (напр.,
сополимеры бутилметакрилата с 5—10%
метакриловой к-ты) и метилольных произ-
производных мочевины или меламина. При на-
нагревании происходит взаимодействие СООН-
групп с СН2ОН-группами указанных сое-
соединений, в результате чего образуется не-
неплавкий и нерастворимый полимер, по-
видимому, пространственно-сетчатого строения.
О сшивании (вулканизации) макромолекул каучука,
содержащих карбоксильные группы, см. Карбокси-
латные каучуки.
М., направленная на создание пространственно-сет-
пространственно-сетчатой структуры, имеет большое значение также в тех-
технологии волокнистых материалов. Так, для повышения
устойчивости к сминанию целлюлозных тканей их
пропитывают водными р-рами или дисперсиями бисме-
тилолциклоэтиленмочевины, тетраметилолацетиленди-
мочевиной (продукт взаимодействия глиоксаля и моче-
мочевины), ]Ч,]>Г-диметилол-]Ч,]>Г-этилентриазоном и др.
при рН<7 и затем 3—5 мин выдерживают при 160 °С.
В этом случае происходит сшивание макромолекул,
напр.:
СН2-СН2
2Се11(ОН) + НОСН2—N
со
СН2-СН2
N J1С
J
со
сн2-сн2
-н2О
сн2—сн2
—*~ Се11-О-СН2—N N—CH2-N N —CH2O—Cell
СО СО
Аналогичный эффект достигается и при использова-
использовании сол-еобразных продуктов взаимодействия хлорме-
тиловых эфиров гликолей или глицерина с пиридином
(сшивание осуществляется при рН>7—8 и 110—
120 °С):
nch2oroch2n( } —*-
сГ сГ
Cell— OCH2OROCH2O — Cell +
Cl
Наряду с описанными выше методами превращения
макромолекул в результате взаимодействия с модифи-
модификатором, не проявляющим полиреакционности, большое
значение для М. имеют методы привитой сополимериза-
ции. Т. к. привитые сополимеры содержат более или
менее протяженные фрагменты прививаемого вещества-
модификатора, они могут сочетать свойства соответст-
соответствующих полимеров.
М. методами привитой сополимеризации может осу-
осуществляться по различным технологич. схемам: 1) в
процессе получения полимерного вещества, 2) при
его переработке в полимерные материалы и изделия
и 3) путем обработки готовых изделий. Второй и третий
пути наиболее перспективны с технико-экономич. точ-
273
МОДИФИКАЦИЯ ХИМИЧЕСКАЯ
274
ки зрения, т. к. исключают предварительную стадию
получения привитого сополимера. Осуществление при-
привитой сополимеризации в условиях сохранения струк-
структурной упорядоченности и морфологич. особенностей
полимерного вещества позволяет широко варьировать
его свойства. Так, напр., прививка атактич. полисти-
полистирола на изотактический дает материал, сочетающий
высокую кристалличность и темп-ру плавления, ха-
характерную для изотактич. полистирола, с четко прояв-
проявляющимися высокоэластич. свойствами атактич. поли-
полимера. При этом меняется тип надмолекулярных струк-
структур (фибриллярные образования вместо сферолитов
и единичных кристаллов).
В отличие от этого М. в разбавленных р-рах, когда
привитая сополимеризация может затрагивать каждую
отдельную макромолекулу, приводит к получению
некристаллизующегося полимера с надмолекулярной
организацией, характерной для аморфных высокомоле-
высокомолекулярных соединений.
Интересен процесс, в к-ром сам тетра- или поли-
полифункциональный мономер или олигомер образует
пространственно-сетчатые агрегаты, равномерно рас-
распределенные в массе полимера и химически связанные
с его макромолекулами, напр.:
X СН2=СН
-СН2-С~ + R -
А СН2=СН
~сн2—с~
~сн2-сн
I
1
~сн2—с~
X
(А — элементарное звено модифицируемого полимера
с активным центром, образовавшимся в результате
действия на полимер инициатора радикальной полиме-
полимеризации или радиации; пунктиром обведен пространст-
пространственно-сетчатый агрегат).
Микрогетерогенная структура полученной таким
образом системы характеризуется весьма интересным
комплексом физико-химических и механич. свойств,
в основном обусловленных наличием связанных с поли-
полимерными цепями сетчатых образований заданной химич.
структуры, играющих роль активного наполнителя-
модификатора. Так, взаимодействие каучуков с олиго-
эфиракрилатами позволяет получать высокопрочные
резины без применения наполнителей. Такие резины
превосходят стандартные по стойкости к тепловому ста-
старению, динамич. выносливости, диэлектрическим и ря-
ряду др. свойств и характеризуются меньшими гистере-
зисными потерями. Кроме того, введение в каучуки
10—50% (от массы эластомера) жидкого термореактив-
термореактивного олигоэфиракрилата в 5—7 раз снижает вязкость
смеси, что резко облегчает переработку и позволяет
создать более рациональные методы формования ре-
резиновых изделий.
Описанный метод М. осуществляется на стадии пере-
переработки. Он оказался весьма перспективным для улуч-
улучшения технологических и эксплуатационных свойств
различных промышленных полимеров — поливинил-
хлорида, полиакрилатов, эфиров целлюлозы, поливи-
нилбутираля.
Образование микрогетерогенной структуры происхо-
происходит и при инициированной привитой сополимеризации
дифункциональных мономеров с каучуками. На этом
основан промышленный метод получения ударопроч-
ударопрочного полистирола: 5—8% бутадиенового или изопре-
нового каучука растворяют в стироле и далее ведут
полимеризацию при повышении темп-ры до 150 °С.
В результате образуется материал с высокой прочно-
прочностью при динамич. изгибе (в 8—10 раз большей, чем
у немодифицированного полимера).
Используя методы радиационной привитой сополи-
сополимеризации, введение в полимер групп, распадающихся
с образованием радикалов, применяя мягкие окисли-
окислители или смеси перекисных инициаторов с восстанови-
восстановителями удается осуществить М. готовых полимерных
материалов и изделий. Так, промышленное значение
получил способ прививки полиакрилонитрила к вискоз-
вискозному штапельному волокну путем его пропитки водо-
водорастворимой инициирующей системой (Н2О2+ Fe+2)
и последующим взаимодействием с мономером. Такое
волокно сочетает свойства гидратцеллюлозных волокон
(высокая гидрофильность, накрашиваемость, устойчи-
устойчивость к истиранию и др.) со свойствами, типичными
для полиакрилонитрильных волокон (шерстеподобный
гриф, устойчивость к действию микроорганизмов,
высокая светостойкость и др.).
Для М. полиолефиновых волокон и пленок оказался
перспективным метод радиационной привитой сопо-
сополимеризации мономеров (акрилонитрил, винилхлорид,
акриловая к-та и др.) в р-ре или из газовой фазы. Ми-
Миграционная привитая сополимеризация окиси этилена
нашла применение для повышения гидрофильности и
накрашиваемости полиамидов, напр.:
2N NHCO(CH2LCO~'
о
/ \
+СН2-СН2
о
/ \
+СН2-СН2
-* ^NH(GH2NNCO(CH2LCO- > и т. д.->
СН2СН2ОН
-» ^NH(CH2)eNCO(CH2LCO~>
СН2СН2(ОСН2СН2)ПОН
Модификация на стадии синтеза полимера. Введе-
Введение в макромолекулы небольшого числа звеньев, со-
содержащих эпоксидные, гидроксильные, амино- или кар-
карбоксильные группы, повышает адгезию полимеров
к полярным поверхностям, температуру стеклова-
стеклования, твердость и способность к накрашиванию. Наряду
с этим такие полимеры обладают способностью к раз-
различным реакциям с низкомолекулярными вещества-
веществами, что расширяет возможности их М. Так, наличие
в полимере звеньев с пероксидными группами придает
ему способность к привитой сополимеризации и сшива-
сшиванию. В результате радикальной эмульсионной сополи-
сополимеризации бутадиена и стирола с 5—10% мономера,
содержащего га/?ега-бутилпероксидную группу, обра-
образуются каучукоподобные сополимеры, способные к вул-
вулканизации за счет перекисных групп и аллильных водо-
родов.
Принципиально иной эффект м. б. достигнут при
синтезе полимеров, содержащих малые количества зве-
звеньев с группами, способными разрушать гидропереки-
гидроперекиси, инициирующие окислительную деструкцию поли-
полимеров. В этом случае резко повышается фото-, термо-
и радиационная стабильность полимера. Так, в ре-
результате радикальной сополимеризации метилметакри-
лата или стирола с 1—3% тиоалкилакрилата общей
ф-лы CH2=C(X)COORSR' (X = Н, СН3; R — алкил с 1 —
4 атомами С; R' — алкил с 2—12 атомами С) образу-
образуются сополимеры, выдерживающие длительное нагрева-
нагревание при 280 °С, тогда как немодифицированные контро-
контрольные полимеры в этих условиях почти полностью
деструктируются.
Большой интерес представляют сополимеры а-оле-
финов с непредельными моно- или дикарбоновыми
к-тами, в к-рых часть групп нейтрализована ионами
275
МОДИФИКАЦИЯ ХИМИЧЕСКИХ ВОЛОКОН
276
металлов I или II групп периодич. системы. Такие со-
сополимеры обладают высокой прочностью, адгезией,
стойкостью к растворителям и рядом др. ценных свойств,
по-видимому обусловленных сильным межмолекуляр-
межмолекулярным взаимодействием (см. также Мономеры).
Лит.: Б е р л и н А. А., Усп. хим., 29, в. 10, 1189 A960);
Б е р л и н А. А. [и др.], Хим. пром-сть, № 2, 20 A962); Б е р-
л и н А. А. [и др.], Кауч. и рез., № 6, 9 A969); П л а т э Н.А.,
в кн.: Кинетика и механизм образования и превращения макро-
макромолекул, М., 1968, с. 250; П у р и н с о н Ю. А. [и др.], Высоко-
мол. соед., 10Б, № 4, 257 A968); Лившиц Р. М., Рого-
в и н 3. А., в кн.: Прогресс полимерной химии, 1968, под ред.
В. В. Коршака, М., 1969, с. 158; Химические реакции поли-
полимеров, под ред. Е. Феттеса, пер. с англ., т. 1—2, М., 1967;
П у ч и н В. А. [и др.], Высокомол. соед., 10Б, № 7, 530 A968);
П у чин В. А. [и др.], там же, НА, № 4, 789 A969); В е-
г 1 i n A. A., Plast. u. Kaut., 20, № 10, 728 A973).
МОДИФИКАЦИЯ ХИМИЧЕСКИХ ВОЛОКОН
(modification of chemical fibres, Modifizierung von Che-
miefasern, modification des fibres chimiques) — направ-
направленное изменение строения или состава волокон, в ре-
результате чего они приобретают новые свойства. М.х. в.—
основное направление усовершенствования выраба-
вырабатываемых в производственных условиях искусствен-
искусственных и синтетич. волокон. Путем модификации можно
значительно улучшить потребительские свойства отдель-
отдельных типов химич. волокон и тем самым расширить обла-
области их применения, повысить конкурентоспособность
с др. типами природных и химич. волокон.
В зависимости от того, какие процессы протекают
при М. х. в., все методы, ведущие к направленному
изменению свойств волокон, м. б. разделены на физи-
физические и химические (это соответствует общей класси-
классификации методов модификации полимеров — см. Мо-
Модификация структурная, Модификация химическая).
Химическая модификация волокон. Эта группа мето-
методов получила применение при производстве различных
типов синтетич. волокон, а в последние годы — ив про-
производстве искусственных волокон. Ниже описаны наи-
наиболее распространенные методы химической моди-
модификации.
Синтез привитых сополимеров. Этот
метод М. х. в. получает все более широкое применение,
особенно для вискозных волокон. Прививка может осу-
осуществляться по различным технологич. схемам: 1) к
исходному волокнообразующему полимеру с последую-
последующим формованием волокна из образовавшегося приви-
привитого сополимера, 2) к сформованному волокну и 3) к
готовым текстильным изделиям. Свойства модифициро-
модифицированных волокон могут широко варьироваться в зависи-
зависимости от типа прививаемого мономера и условий прове-
проведения процесса.
Первая из упомянутых технологич. схем проверена
в опытно-промышленных масштабах. Прививка к ис-
исходному полимеру м. б. использована, напр., для мо-
модификации эфироцеллюлозных волокон, в частности
ацетатного волокна.
Как правило, прививка осуществляется на готовые
волокна или, что наиболее целесообразно, непосредст-
непосредственно на прядильной машине в процессе формования
волокна. Последний вариант реализован, напр., путем
прививки мономеров к свежесформованному вискозному
волокну, содержащему нек-рое количество тиокарбоно-
вых группировок.
Осуществление прививки значительных количеств
др. полимеров к готовым изделиям (тканям, трикотажу)
в обычных случаях нецелесообразно, т. к. при этом
толщина нити увеличивается, соответственно повышает-
повышается плотность изделия и увеличивается внутреннее на-
напряжение в материале. Последнее резко ухудшает
один из важнейших показателей, определяющих экс-
эксплуатационные свойства изделия,— т. наз. устойчи-
устойчивость к раздиру. Этот вариант оформления процесса
м. б. рекомендован только при прививке небольшого
количества синтетич. полимера (не более 3—5% от
массы исходного материала), локализуемого на поверх-
поверхности материала и обеспечивающего придание ему
требуемых свойств, напр, при получении тканей, обла-
обладающих водо- и маслоотталкивающими свойствами,
путем прививки фторсодержащих мономеров.
В зависимости от требуемых результатов и условий
процесса к исходному полимеру м. б. привито различ-
различное количество синтетич. полимера, причем масса при-
привитого полимера м. б. даже больше массы исходного.
Однако модификацией обычно называют процесс, при
к-ром это количество не превышает 30—50%.
В зависимости от характера привитого полимера,
в частности от содержания в нем полярных или непо-
неполярных групп, абсолютная прочность волокна может
снижаться или несколько повышаться. Уд. прочность
волокон, рассчитанная на единицу сечения, обычно
снижается в результате увеличения диаметра (толщины)
нити после прививки.
Для синтеза привитых сополимеров могут быть ис-
использованы, в зависимости от химич. состава исходного
полимера и наличия в нем различных реакционноспо-
собных групп, разнообразные методы (см. Привитые
сополимеры). Так, для синтеза привитых сополимеров
целлюлозы, содержащей реакционноспособные ОН-
группы, наряду с полимеризацией м. б. применена
поликонденсация. Однако в промышленном масшта-
масштабе используется только метод радикальной полиме-
полимеризации.
Для образования макрорадикала в волокнообразую-
щем полимере принципиально м. б. использованы все
известные методы инициирования полимеризации, од-
однако для практич. использования наиболее перспек-
перспективны окислительно-восстановительное инициирование
и обработка ионизирующим излучением (см. Иницииро-
Инициирование полимеризации, Радиационная полимеризация).
Первый метод м. б. осуществлен только на полимерах
с реакционноспособными группами, к-рые могут яв-
являться одним из компонентов окислительно-восстано-
окислительно-восстановительной системы, в частности для целлюлозы, поли-
поливинилового спирта и полиамидов.
С точки зрения технико-экономич. показателей
М. х. в. методом привитой радикальной сополимериза-
ции особенно целесообразна при выполнении след.
условий:
1. Реакция проводится в водной среде — в водных
р-рах или водных эмульсиях мономеров. Использование
органич. растворителей допустимо только в отдельных
случаях, когда образование стабильных водных эмуль-
эмульсий мономеров невозможно (напр., при прививке
нек-рых типов фторсодержащих мономеров). Перспек-
Перспективен метод непрерывной прививки к нитям или гото-
готовым изделиям с использованием паров мономеров.
При удачном аппаратурном оформлении процесса этот
вариант может получить практич. применение.
2. Образование гомополимера в результате побочной
реакции устранено или сведено к минимуму. В про-
противном случае значительно повышается непроизводи-
непроизводительный расход прививаемого мономера и появляется
необходимость в дополнительной сложной операции —
экстракции гомополимера из привитого сополимера ор-
органич. растворителями. Выполнение этого требования
возможно путем подбора соответствующих условий
процесса и, в частности, состава окислительно-восста-
окислительно-восстановительной системы, определяющей соотношение ско-
скоростей инициирования (образование макрорадикала),
роста и обрыва растущей цепи.
3. Прививка производится в условиях, исключающих
значительную деструкцию и, соответственно, снижение
показателей механических и физико-химич. свойств
исходных волокон.
4. По возможности процесс осуществляется в аппа-
аппаратуре, имеющейся на заводах химич. волокон или
на текстильных (отделочных) фабриках.
'277
МОДУЛЬ
278
В зависимости от требований к свойствам получаемого
химически модифицированного волокна и условий про-
процесса прививаемые цепи синтетич. полимера м. б.
локализованы на поверхности или распределены по
всему объему волокна. Для ряда модифицированных
волокон, обладающих масло- и водоотталкивающими,
кислотостойкими, бактерицидными и гнилостойкими
свойствами (см. Антимикробные волокна), достаточно
осуществить прививку на поверхности волокна, в то
время как при получении, напр., ионообменных воло-
волокон необходимо, чтобы процесс проходил во всем объеме
полимера.
Используя метод синтеза привитых сополимеров,
удалось получить химич. волокна с самыми различными
свойствами. Нек-рые из этих свойств м. б. приданы
и др. способами, в частности путем пропитки или вве-
введения добавок, однако только химически модифициро-
модифицированные текстильные материалы устойчиво сохраняют
приобретенный эффект при различных обработках,
в частности при стирке, истирании, многократных
деформациях и др.
На основе привитых сополимеров созданы, в част-
частности, огнезащитные химические волокна (с различ-
различными фосфорсодержащими соединениями — см. Огне-
Огнестойкость), водоотталкивающие (с фторорганич. поли-
полимерами, полидиеиами, полистиролом или др.), кислото-
кислотостойкие (с различными гидрофобными полимерами,
затрудняющими диффузию водных р-ров внутрь во-
волокна), маслоотталкивающие (с фторсодержащими по-
полимерами), биологически активные, антимикробные и
ионообменные.
Синтез линейных сополимеров, со-
содержащих в макромолекуле небольшое количество
E—15% от массы сополимера) второго мономера. При
этом можно, напр., повысить эластичность соответст-
соответствующего гомополимера и, следовательно, получаемого
из него волокна. В результате введения в макромоле-
макромолекулу второго мономера, содержащего полярные функ-
функциональные группы, в большинстве случаев улучшается
гигроскопичность и накрашиваемость волокна. Этот
вариант широко используется для модификации свойств
волокон из карбоцепных синтетич. полимеров и м. б.
применен для модификации нек-рых синтетич. гетеро-
цепных волокон, в частности полиамидных (изменение
темп-ры плавления, накрашиваемости, растворимости).
Образование «сшивок» (химич. свя-
связей) между макромолекулами путем
обработки волокон различными би- или полифункцио-
полифункциональными соединениями. Достигаемый результат —
значительное повышение термостойкости, устойчивости
к многократным деформациям, снижение набухаемости
и растворимости. Это пока единственный метод, обес-
обеспечивающий получение несминаемых тканей как из
природных, так и из искусственных целлюлозных во-
волокон.
Превращение реакционноспособ-
ных функциональных групп в мо-
молекуле полимера (метод полимер аналогичных
превращений). Этот метод нашел известное практич.
применение для повышения прочности в мокром состоя-
состоянии и эластичности упрочненного вискозного штапель-
штапельного волокна путем его поверхностного ацетилирова-
ния, для увеличения гидрофильности и улучшения на-
накрашиваемости полиакрилонитрильного волокна в ре-
результате частичного омыления нитрильных групп.
Физическая модификация волокон. Среди этой груп-
группы методов М. х. в. наибольшее значение имеют след.:
а) ориентация путем вытягивания волокна, осущест-
осуществляемая, как правило, с полной или частичной релакса-
релаксацией волокна (см. также Формование химических во-
волокон)', в результате улучшаются механич. свойства
волокна— повышаются прочность в сухом и мокром
состояниях, начальный модуль, устойчивость к много-
многократным деформациям; б) введение малых добавок в р-р
или расплав волокнообразующего полимера — стаби-
стабилизаторов, в частности антиоксид актов и светостабили-
заторов (при этом значительно повышается срок службы
изделий), бактерицидных веществ (см. Антимикроб-
Антимикробные волокна) и др.; в) формование волокон из смесей
полимеров (из р-ров или расплавов), позволяющее полу-
получать волокна, сохраняющие ценные свойства каждого
из компонентов, входящих в состав смеси.
Ряд методов физич. М. х. в. осуществляется обработ-
обработкой уже готового волокна. Сюда, напр., относятся все
пути получения высокообъемных нитей, в том числе
бикомпонентных (см. Викомпонентные волокна).
Лит.: Роговин 3. А., Химия целлюлозы, М., 1972;
его же, Основы химии и технологии производства хими-
химических волокон, 4 изд., М., 1974; его же, Химические пре-
превращения и модификация целлюлозы, М., 1967.
3. А. Роговин.
МОДУЛЬ (модуль упругости) в тех-
технологии и физике полимеров (modulus,
Modul, module) — мера жесткости материала, харак-
характеризующая сопротивление развитию упругих (обра-
(обратимых) деформаций и равная отношению напряжения
к обратимой деформации, отвечающей этому напряже-
напряжению. В зависимости от вида напряженного состояния
различают модуль растяжения (сжа-
(сжатия), модуль сдвига и объемный мо-
модуль (модуль всестороннего с ж а-
т и я).
Модули идеально упругих тел. Понятие о М. как
характеристике упругости возникло для идеально
упругих тел, у к-рых напряжение а пропорционально
относительной обратимой деформации е, и тогда М.
определяется как отношение о7е. Для изотропного
идеально упругого тела между модулями одноосного
растяжения (модулем Юнга) Е, сдвига G
и всестороннего сжатия К существуют след. соотно-
соотношения:
где \х — коэфф. Пуассона, представляющий собой
отношение относительного поперечного сжатия к отно-
относительному продольному удлинению при одноосном
растяжении. Для несжимаемого тела |л = 0,5 и поэ-
поэтому G = Е!Ъ и К -> оо .
Для анизотропного тела число М. зависит от поряд-
порядка симметрии. В наиболее общем случае (кристаллы
триклмнной сингонии) число независимых М. состав-
составляет 21, по мере повышения порядка симметрии оно
снижается до 3 (для кубич. кристаллов) и для пол-
полностью изотропного тела — до 2.
Эти общие представления о М. широко используют
при описании механич. свойств любых тел, способных
к накоплению обратимых деформаций. Применительно
к полимерам понятие о М. было обобщено для таких
систем и режимов деформирования, когда а непропор-
непропорционально е, изменяется во времени при е = const
и т. д. Для полимерных материалов вследствие доми-
доминирующей роли релаксационных явлений в проявле-
проявлении комплекса их механич. свойств М. зависит от ре-
режима деформирования (продолжительности, скорости,
частоты и т. п.), темп-ры, а также от особенностей
строения полимерного материала и его термомеханич.
предыстории. В анизотропных полимерах, в частности
в ориентированных и армированных пластмассах, М.
зависит от направления деформирования при его изме-
измерении.
Т. к. в реальных полимерных материалах обычно
отсутствует пропорциональность о и е, определение
М. как отношения о/г носит эффективный (эквивалент-
(эквивалентный) характер. Наиболее близки к идеально упругим
телам аморфные полимеры в стеклообразном состоянии
и до нек-рой степени в высокоэластич. состоянии
в средней части плато высокоэластичности (когда низки
279
МОДУЛЬ
280
диссипативные потери) в области малых деформаций,
а также монокристаллы кристаллич. полимеров.
Модули в равновесных условиях. Зависимость сг(е)
в равновесных условиях определяют при статич. режи-
режимах нагружения (когда один из механич. параметров,
т. е. напряжение или деформация, поддерживаются
постоянными) после полного завершения процессов
механич. релаксации. Вычисленное из этой зависи-
зависимости отношение о7е наз. равновесным мо-
модулем. Для стеклообразных полимеров, когда
релаксация практически исключена, имеют место
«квази-равновесные» условия и вместо равновесного
модуля следует пользоваться понятием «мгновен-
«мгновенного модуля упругости», значение к-рого
для очень многих полимеров при растяжении близко
к 3-Ю9 н/м2 C-Ю10 дин/см2). Для материалов, нахо-
находящихся в высокоэластич. состоянии, отношение сг/е
наз. равновесным модулем высоко-
эластичности. Его величина при малых
деформациях составляет обычно 0,1 — 1 Мл/м2 A—
10 кгс/см2), меняясь в зависимости от химич. природы
полимера (полярности, наличия боковых заместителей
и т. д.), густоты сетки, содержания пластификаторов,
наполнителей и т. п. При этом \х ^ 0,5 и равновесные
М. при растяжении и сдвиге связаны простым соотно-
соотношением: Е = 3G.
При больших деформациях равновесный М. высоко-
эластичности меняется в зависимости от 8, что в наи-
наиболее явном виде показывает эффективный характер
определения М.
Различают «начальный модуль», соответствующий
начальному, прямолинейному, участку кривой зави-
зависимости ст(е) (строго говоря, наклону касательной,
проведенной к этой кривой в точке г — 0), и «секущий
модуль», соответствующий любой точке на кривой
и определяемый как отношение о7е в этой точке. В тех-
нологич. практике часто используют величину секу-
секущего модуля при е 100 или 300%. Начальный и се-
секущий (для деформации 8 = га — рис. 1) модули рав-
равны соответственно тангенсам углов а и р.
Характер зависимости М. от 8 определяется видом
ур-ния состояния материала, т. е. видом функции
ст(е). В широко распространенной и описывающей
в первом приближении действительную картину дефор-
деформирования высокоэластич. материалов (резин) теории
высокоэластичности, основанной на предположении
об энтропийной природе больших деформаций (Г.
Джеймс, Г. Марк, Э. Гут, В. Кун и др.), соотно-
Рис. 1. Зависимость напря-
напряжения а от деформации 8 в
равновесном режиме нагру-
нагружения высокоэластич. мате-
материала: тангенсы углов аир
отвечают значениям началь-
начального и секущего (эффектив-
1;еформаЕ
дулей.
шения между а и 8 для различных видов напряженного
состояния оказываются нелинейными. Напр., для
растяжения напряжение, отнесенное к сечению образца
в недеформированяом состоянии, / выражается в зави-
зависимости от степени растяжения а ф-лой
Входящая в нее константа GB наз. модулем высоко-
высокоэластичности, хотя при больших деформациях она
не равна а/е и поэтому ее не следует путать с вели-
величинами М., определенными как а/е и не являющимися
константами. Лишь при сдвиговых деформациях, со-
согласно этой теории, о" = GBe, т. е. а — ей поэтому
GB имеет смысл М. упругости (высокоэластичности)
при сдвиге, к-рый в этой теории не зависит от дефор-
деформации. Согласно физич. смыслу названной теории,
GB выражается через основной структурный параметр
материала N — число участков макромолекулярных
цепей, находящихся между узлами вулканизационной
сетки: GB = yN/RT (R — универсальная газовая по-
постоянная; Т — абс. темп-pa; у — числовой множитель
порядка 1, точное значение к-рого зависит от вида
принятой физич. модели материала). Расчеты N по из-
измеренной величине GB показывают, что для реальных
материалов N следует рассматривать как нек-рую
эквивалентную величину, характеризующую густоту
сетки, образованной как химическими, так и физич.
связями.
Энтропийная теория высокоэластичности и предска-
предсказываемые ею формы зависимости о(е) в равновесных
условиях для различных видов напряженного состояния
описывают деформационные свойства резин лишь в пер-
первом приближении, поскольку не учитывают изменение
внутренней энергии макромолекулярных цепей при
деформировании и возможность их совместного (коопе-
(кооперативного) деформирования.
При описании зависимости сг(е) для резин в равновес-
равновесных условиях более сложными теоретич. ф-лами понятие
о М. высокоэластичности теряет прямой смысл, по-
поскольку в эти ф-лы входят не одна, а несколько тео-
теоретич. или эмпирич. констант, не являющихся М.,
который по-прежнему должен определяться в равновес-
равновесных условиях как а/е.
Модули в неравновесных условиях. Понятие о М.
может быть распространено на любые неравновесные
режимы деформирования, когда отношение о/в ста-
становится зависящим от временного фактора (продолжи-
(продолжительности или частоты деформирования).
Наиболее распространены измерения неравновесных
характеристик полимерных материалов при малых
деформациях, когда а ~ е (но отношение а/е зависит
от временного фактора) и применимы соотношения ли-
линейной теории вязкоупругости (см. Болъцмана —
Вольтерры уравнения, Реология). Определение М. в не-
неравновесных режимах деформирования позволяет со-
сопоставить значения М. с релаксационным спектром по-
полимера, что дает возможность перейти к общей ха-
характеристике механич. свойств материала.
Известно большое число разнообразных способов
измерений свойств полимерных материалов, описывае-
описываемых М. в неравновесных условиях. Важнейшими
из них являются метод создания гармонич. колебаний
в установившемся режиме (что связано с разработан-
разработанностью соответствующего математич. аппарата и про-
простотой применения определяемых в таком эксперименте
величин к расчету зависимостей а или е от времени t
при произвольном режиме нагружения), измерение
релаксации напряжения o(t) при сохранении посто-
постоянной деформации и метод измерения деформации в ре-
режиме ползучести г(г) при задании постоянного напря-
напряжения.
При задании гармонич. колебаний с частотой со
вначале осуществляется переходной (неустановивший-
(неустановившийся) режим деформирования и затем достигается устано-
установившийся режим колебаний, в к-ром измеряемые
характеристики материала зависят только от со, но
не от продолжительности деформирования. Если мате-
материал подвергается нагр ужению по закону o(t) =
=а0 е ibit [где o(t) — напряжение, изменяющееся во вре-
времени t по гармонич. закону; а0 — амплитудное значение
напряжения; со — круговая частота (со — 2:rcv, v —
число циклов в единицу времени); i ="|/ — 1], то в
установившемся режиме при достаточно малых зна-
значениях а0 изменение деформации во времени описыва-
описывается ур-нием:
281
МОДУЛЬ
282
где § _ разность фаз (или угол механич. потерь);
е0 — амплитудное значение деформации. Величина б
в общем случае зависит от частоты и темп-ры, но не от
а0 или е0. Под комплексным модулем
упругости G* понимают отношение G* =
= o(tj/E(t) = G' +i G", где G' и G" — действительная
и мнимая компоненты комплексного модуля упруго-
упругости, часто наз. упругим модулем (или моду-
модулем запаса) и модулем потерь соответствен-
соответственно. Обратное отношение 1* =1/G* = е/а наз. комп-
комплексной податливостью, эта величина
также используется для характеристики механич.
свойств материала. Компоненты /* = /' — И" связаны
с компонентами G* соотношениями:
Абсолютное значение комплексного модуля
| G* | =
= сго/ео
I 8
^® 6
иногда наз. динамическим модулем.
Значение G" характеризует энергию А — -j- е20 coG",
рассеиваемую при динамич. деформировании в единицу
времени; Gr — энергию, упруго запасаемую при дина-
динамич. деформировании и отдаваемую материалом при
снятии нагрузки. Вместо G" иногда пользуются про-
пропорциональной ему величиной модуля внутреннего
трения К, равной 2nG".
Типичные зависимости Gf от частоты при сдвиговых
деформациях для основных классов полимерных мате-
материалов приведены на рис. 2. Он иллюстрирует сущест-
Рис. 2. Зависимости уп-
упругого модуля G' («мо-
(«модуля запаса») от частоты
деформирования о) для
важнейших классов по-
полимерных материалов:
1 — низкомолекулярный
полимер, не обладаю-
обладающий высокоэластич. свой-
свойствами; 2 — аморфный
линейный полимер, к-рый
^ при низких частотах ве-
lgoi-В дет себя как упруговяз-
кая жидкость, при сред-
средних как высокоэластич. материал и при высоких — как
стекло; 3 — сшитый полимер (резина), для к-рого плато
высокоэластичности продолжается до сколь угодно низ-
низких частот; 4 — полимер с высокой степенью кристаллич-
кристалличности. Константы В выбраны так, чтобы эксперименталь-
экспериментальные данные совмещались в одном диапазоне значений
lgco. A дин/см* =0,1 н/м2).
вование основных областей релаксационных состоя-
состояний — вязкотекучего с G' < 104 н/м2 A05 дин1см2)\
высокоэластического, к-рому отвечают примерно посто-
постоянные значения G' — плато с G ъ 105 н/м2 A0° дин/см2)
и стеклообразного с постоянными значениями G' поряд-
порядка 109 н/м2 A010 дин/см2), а также переходных обла-
областей, соединяющих области вязкотекучего и стеклооб-
стеклообразного состояний с плато высокоэластичности. Макси-
Максимальное значение G' (теоретически при м—>эо) наз.
мгновенным модулем упругости. Эта
величина практически отвечает квази-равновесному
М. полимера в стеклообразном и кристаллич. состоя-
состояниях. Минимальное значение G' для сшитых полимеров
отвечает уровню плато высокоэластичности, а для
несшитых (текучих) равно 0.
Темп-рная зависимость G' аналогична его частотной
зависимости. Это лежит в основе принципа температур-
но-временной суперпозиции, причем области низких
частот отвечает высокая темп-ра.
Зависимости G" от частоты и темп-ры в целом подобны
аналогичным зависимостям для G' тех же полимеров,
но темп-рные зависимости G" или отношения G"/Gr =
= tg6 (наз. тангенсом угла механич.
потерь) обнаруживают ряд максимумов, соот-
соответствующих тем или иным характерным временам
релаксации или их группам, что обусловлено специ-
фич. релаксационными переходами в полимере (под-
(подробно см. Динамические свойства).
При измерении неравновесных свойств полимеров
в режиме релаксации задают постоянную деформацию
е = const и по изменяющемуся во времени t напряже-
напряжению вычисляют т. наз. релаксационный М.:
где o(t) — напряжение в момент времени t. Зависи-
Зависимость Gr(t) (рис. 3) подобна ?'(со), причем в первом
Рис. 3. Зависимость ре-
релаксационного модуля
Gr от длительности ре-
релаксации. Константа В
выбрана так же, как на
рис. 2. Обозначение кри-
кривых также см. на рис. 2
A дин/см2 = 0,1 н/м2).
Ж 8
\gt-B
приближении Gr ^ G' при t == со; известЕЫ и более
точные ф-лы, связывающие 6> с G'. Зависимость
Gr(T), определяемая при t = const, подобна G'(T) при
со = const.
Измерение развития деформации a(t) при а = const
(см. Ползучесть) также дает информацию о свойствах
материала, качественно подобную получаемой при
измерениях С (со) или Gr(t), а количественная связь
между характеристиками материала, измеряемыми
во всех этих режимах нагружения, устанавливается
точными или приближенными соотношениями линей-
линейной теории вязкоупругости (см. также Реология по-
полимеров).
Все описанные выше зависимости неравновесных
значений М. от частоты, времени и темп-ры относились
к области малых деформаций, когда М. не зависит
от значения деформации. Важное значение приобре-
приобретают также определения М. в неравновесных нелиней-
нелинейных режимах деформирования — при колебаниях или
в статических условиях при больших деформациях.
Хотя определение М. в этом случае не всегда одно-
однозначно, но общей закономерностью является то, что
при увеличении деформации происходит разрушение
внутренней структуры полимерного материала, вслед-
вследствие чего обычно жесткость материала падает и
уменьшаются эффективные (определевные тем или
иным способом) значения М., характеризующие эту
жесткость.
Методы экспериментального определения модулей..
Методы измерений М. представляют собой частные
случаи механич. испытаний полимерных материалов
(подробно см. Испытания пластических масс, Испыта-
Испытания резин, Испытания химических волокон), отличаю-
отличающиеся необходимостью задания или вычисления таких
количественных характеристик режима деформирова-
деформирования, как напряжения и деформации. Соответственно
задачам измерений опыты могут проводиться в рав-
равновесных режимах, когда главной экспериментальной
задачей является достаточно длительная выдержка
образца в заданных условиях, чтобы завершились про-
процессы релаксации, или в неравновесных режимах,
когда существенно достижение установившегося режи-
режима и выполнение измерений в очень широком времен-
временном диапазоне — при частотах от 10~3 до 109 гц или
продолжительностях нагружения от малых долей сек
до многих су т. Для расширения частотного (временно-
(временного) режима нагружения важной является возможность
взаимного пересчета различных М., измеренных в ли-
283
МОЛЕКУЛЯРНАЯ МАССА
284
нейной области, и использование суперпозиции прин-
принципа. Наиболее общими методами измерений М. при
динамич. испытаниях являются способ возбуждения
вынужденных гармонич. нерезонансных или резонанс-
резонансных колебаний, создаваемых механическими или элект-
электромагнитными способами, измерение частоты и интен-
интенсивности снижения амплитуды свободно затухающих
колебаний, а также (в области высоких частот) опре-
определение скорости распространения и интенсивности
поглощения импульсов продольных или поперечных
волновых колебаний. Для стеклообразных, высокоэла-
высокоэластических и кристаллич. тел наиболее часто измеряются
М. при растяжении, для жидких вязкоупругих тел —
при сдвиговых деформациях.
Измерения М. выполняют: 1) для оценки темп-рных
и частотных границ различных областей физических
(релаксационных) состояний полимеров и температур-
но-временных областей работоспособности материала,
в частности для прогнозирования долговременного
поведения материала при эксплуатации; 2) для изу-
изучения механич. свойств и релаксационных переходов
полимеров, что позволяет судить о химическом и фи-
зич. строении материала («механическая спектроско-
спектроскопия»); 3) для наблюдения за физико-химич. процесса-
процессами, происходящими в материале при его технологич.
обработке (при вулканизации каучуков, отверждении
термореактивных смол, кристаллизации и др.), с цэлыо
контроля производства, качества готовой продукции
и т. п., а также стабильности ее эксплуатационных
характеристик. А. я. Малкин.
МОЛЕКУЛЯРНАЯ МАССА полимера (mole-
(molecular mass, Molekularmasse, masse moleculaire). Прак-
Практически каждый синтетич. полимер состоит из макро-
макромолекул различной длины. Поэтому М. м. полимер-
полимерного образца является средней статистич. величиной
и определяется видом молекулярно-массоеого распреде-
распределения (ММР) и способом усреднения. В зависимости
от способа усреднения различают среднечисловую
(среднечисленную), среднемассовую и z-среднюю М. м.
Среднечисловая молекулярная
масса выражается ф-лой
_ N
Мп= 2 ^Mi A)
г=1
где v; — числовая доля фракции макромолекул
i-ro вида с М. м., равной Mt\ N — число фракций. При
больших М. м. с достаточной степенью надежности
суммирование можно заменить интегрированием, по-
поскольку от реального, прерывного ММР в данном
случае с хорошим приближением можно перейти
к непрерывному. Тогда числовая доля макромолекул,
М. м. к-рых лежит в интервале от М до М + dM,
оказывается равной pn(M)dM, где рп(М) непрерывная
дифференциальная числовая функция ММР. Тогда
Мп= \ ?(M)MdM
B)
Этот способ усреднения дает среднее арифметич. зна-
значение М. м. Значение Мп экспериментально опреде-
определяют методами осмометрии, эбулиоскопии, криоскопии,
а также по данным количественного определения кон-
концевых групп макромолекул спектроскопич. и химич.
методами.
Средне массовая молекулярная
масса представляется ур-нием
N
2
г=1
C)
где u?i — массовая доля фракции макромолекул ?-го
вида с М. м., равной Mt. Для непрерывных ММР
«=J
J pn(M)MdM
о
D)
где pw (M) —. непрерывная дифференциальная массо-
массовая функция распределения, равная рп(М) ^-. Сред-
м п
немассовую М. м. получают при измерении методом
светорассеяния, а также из данных измерения седи-
ментационного равновесия (см. Седиментация).
2-Средняя молекулярная масса
равна:
pw(M)M4M J pn(M)M3dM
E)
?w(M)MdM j pn(M)M*dM
О
z-Среднюю M. м. получают при измерении методом
седиментационного равновесия.
Рассмотренные выше усреднения можно свести к об-
общему виду т. наз. g-средних М. м.:
o(M)Mq dM
p(M)MqdM
*q oo
F)
pw(M)Mq 2 dM
dM
При q, равном 1, 2 и 3, получают соответственно Мп,
Mw и Мг. Любые g-средние М. м. могут быть рас-
рассчитаны, если известны вид ММР или определяющие
его характеристики.
К др. типам усреднения приводят методы исследова-
исследования гидродинамич. свойств полимеров. Соответствую-
Соответствующие средние М. м. наз. среднегидродина-
м и ч е с к и м и. Их определяют по данным измерения
вязкости (Мц, т. наз. средневязкостная М. м.),_ кон-
константы седиментации (Ms) или коэфф. диффузии (Md), и
они отвечают таким типам усреднений:
I/O
G)
[] 1/A-5)
[ ?w(M)Ml~b dM I (8)
О
J
, = J ?w(M)M~b dM
(9)
Для вычисления Mt\ Ms, Md используют эмпирич.
соотношения
Ы=къ м« (Ю)
(И)
D=KDM~b A2)
где fnl — характеристич. вязкость, ж3/кг (дл/г); S —
константа седиментации, сек/кг (ед. Сведберга); D —
коэфф. диффузии, м2/сек (ед. Фика); Кт\, Ks, Kd,
а и Ъ — эмпирич. константы, зависящие от конформа-
ции и конфигурации макромолекулы в р-ре и характера
взаимодействия полимера с растворителем; эти констан-
константы определяют, подставляя значения [г|], 5, D и М. м.
(измеренное независимым методом) для узких фракций
полимера в соответствующие ур-ния. Ур-ния A0) —
A2) с определенными значениями констант применимы
285
МОЛЕКУЛЯРНО-МАССОВОЕ РАСПРЕДЕЛЕНИЕ
286
только в том диапазоне М. м., для к-рого получены эти
значения. Об ур-нии A0), к-рое наз. ур-нием Марка ¦=¦
Хувинка, см. также Вязкость характеристическая.
Т. наз. двойную с р е д н е м а с с о в у ю М. м.
можно определить также методом Сведберга, основан-
основанным на одновременном измерении коэфф. диффузии
и константы седиментации, связанных с М. м. соот-
соотношением
(где v — уд. парциальный объем полимера в р-ре, м3/кг;
d — плотность, кг/м3). Этой М. м. соответствует та-
такой тип усреднения:
, 1'"
(М)М ъ dM
Рп(М)М bdM
A4)
¦ Одновременное определение параметров поступа-
поступательного и вращательного движения макромолекул
в р-ре также позволяет оценить среднюю М. м. полиме-
полимера. Для этого случая Ф. Флори и Л. Манделькерн
предложили след. ур-ния:
nlV«
Г дтфУзр-1 "I3
L ^NA J
A5)
A6)
где т]0 — вязкость растворителя, м3/кг(дл/г); N a —
число Авогадро, кмоль ~ь, Ф'/зР = 2,5-10~6 (Ф
и Р — параметры, зависящие от формы макромолекул
в р-ре при вращательной и поступательной диффу-
диффузии соответственно), когда макромолекулу в р-ре мож-
можно моделировать гауссовым непроницаемым клубком;
So и Do — соответственно константа седиментации
\сек/кг (ед. Сведберга)] и коэфф. диффузии [м2/сек (ед.
Фика)] при концентрации полимера, равной 0.
Средние М. м., полученные различными методами, раз-
различаются между собой в тем большей степени, чем шире
ММР полимера [см., напр., ур-ния A) и C), иллюстри-
иллюстрирующие зависимость М. м. от характера суммирования].
По относительному значению они располагаются
в ряд Mn<^Mw<^Mz. Место, к-рое в этом ряду
занимают средние гидродинамич. М. м., зависит от зна-
значения параметров в ур-ниях A0) — A2). Так, при а =1
Мь — Mw; _при а < 1 Мп < М-ц < Mw\ при 1 < а < 1,7
Значение М. м. и вид ММР зависят от способа и
условий получения полимера.
Средняя М. м., а особенно вид ММР в свою очередь
во многом определяют механич, свойства полимеров.
Темп-pa текучести линейных полимеров (Тт) быстро
возрастает с повышением М. м. Для аморфных линей-
линейных полимеров существует след. зависимость:
т — тЛ
где Тс — темп-pa стеклования; А, В и С — константы
для данного полимергомологич. ряда.
С ростом М. м. резко возрастает вязкость расплавов
и конц. р-ров линейных полимеров и одновременно
расширяется темп-рный интервал высокоэластич. со-
состояния. Зависимость вязкости расплавов и конц.
р-ров полимеров от М. м. во мн. случаях выражается
ур-нием i)=K(c)M«' A8)
где коэф. К(с) и а' зависят от природы полимера (или
системы полимер — растворитель) и соотношения кон-
концентрации с и степени полимеризации. При нек-ром
критич. соотношении этих величин К становится про-
пропорционально с5, а а' —> 3,4. Впрочем, для жесткоцеп-
ных полимеров это эмиирич. правило иногда не вы-
выполняется. Ф-ла A8) позволяет оценить среднегидро-
динамич. мол. массу, к-рая при предельных значениях
К и а' меньше A/z, но больше Mw . Хэрактеристич.
вязкость р-ров полимеров с хаотично разветвленными
макромолекулами зависит от М. м. в меньшей степени,
чем для линейных полимеров. О зависимости свойств
полимерных материалов от значения М. м. и вида
ММР см. также Молекулярно-массовое распределение.
Лит. см. при ст. Молекулярно-массовое распределение.
С. А. Павлова, И. И. Твердохлебова.
МОЛЕКУЛЯРНО-МАССОВОЕ РАСПРЕДЕЛЕНИЕ
(molecular-weight distribution, Molekulargewichtsver-
teilung, distribution des poids moleculaires)^
Содержание:
Введение *****„ 286
Экспериментальные методы определения молекулярно-
массового распределения 290
Свойства полимеров и молекулярно-массовое распределе-
распределение 293
Молекулярно-массовое распределение полимеров, полу-
получаемых по различным механизмам 2 93
Радикальная полимеризация 293
Ионная и координационно-ионная полимеризация 295
Поликонденсация 297
Обратимые процессы полимеризации и поликонден-
поликонденсации (равновесные молекулярно-массовые рас-
распределения) • 297
Молекулярно-массовое распределение полимеров в про-
процессах деструкции 299
Введение
Молекулярно-массовое распределение — соотноше-
соотношение количеств макромолекул различной мол. массы
(М. м.) в данном образце полимера, т. е. состав поли-
полимера по М. м. Существование М.-м. р. характерно
только для полимеров, особенно синтетических. Боль-
Большинство биополимеров — индивидуальные вещества,
все макромолекулы к-рых имеют одинаковую М. м.
Наличие М.-м. р. у синтетич. и модифицированных
природных полимеров — следствие случайного (ста-
(статистического) характера реакций их образования,
деструкции и модификации. Понятие М.-м. р. теряет
смысл для трехмерных (сшитых) полимеров, в к-рых
участки полимерного тела макроскопич. размеров
представляют собой как бы целые «молекулы».
Интерес к теоретич. и экспериментальным исследо-
исследованиям М.-м. р. определяется тем, что наряду с химич.
строением молекул оно оказывает существенное влия-
влияние на механические, в частности реологические, и др.
макроскопич. свойства полимеров. Кроме того, изуче-
изучение М.-м. р. позволяет получить дополнительную
информацию о механизмах образования и превращения
макромолекул.
Любой образец полимера представляет собой смесь
макромолекул различной длины (длина макромолекулы
определяется степенью полимеризации, т. е. числом
мономерных звеньев, из к-рых она состоит), а следо-
следовательно, и различной М. м. и может быть охарактери-
охарактеризован кривой распределения по длинам или no M. м.
При теоретич. анализе М.-м. р. удобно пользоваться
кривыми в координатах плотность распределения
lv—dM~ ^М —число макромолекул с М. м., равной
М; No — общее число макромолекул в образце) —
dN
мол. масса. Функция -^—-щ- от М носит название
числового дифференциального М.-м. р. Для количест-
количественного сравнения М.-м. р. различных образцов поли-
полимеров вводят понятие среднечисловой (сред-
нечисленной) молекулярной массы Мп.
Она равна отношению общей массы полимера т0 к об-
общему числу макромолекул {No). Графически среднечис-
ловую М. м. можво представить как абсциссу центра
тяжести площади, ограниченной кривой числового
287
МОЛЕКУЛЯРНО-МАССОВОЕ РАСПРЕДЕЛЕНА
288
распределения и осью абсцисс. Однако среднечисловая
М. м. не является исчерпывающей характеристикой
М.-м. р., так как полимеры с одинаковой Мп могут
иметь совершенно различные распределения (рис. 1),
1 dNM
v мтт М Мтах
Молекулярная масса
Экспериментально обычно
Рис. 1. Кривые числово-
числового молекулярно-массового
распределения образцов
полимеров с широким (i) и
узким B) распределением,
но одинаковой среднечи-
словой мол. массой Мп
(А — центр тяжести пло-
площадей, ограниченных кри-
кривыми 1 и 2 и осью абс-
абсцисс).
измеряют массу макро-
макромолекул с данной М. м. и строят кривые массового рас-
распределения но М. м. (т. наз. массового дифферен-
дифференциального молекулярно-массового
распределения). Переход от кривых число-
числового к кривым массового распределения осуществляется
1 dNM
умножением каждой плотности-^- dM на отноше-
отношение МШп. При этом форма кривой распределения
меняется (рис. 2). Это связано с тем, что макромолекулы
различной М. м. вносят различный вклад в числовое
и массовое М.-м. р. Низкомолекулярная фракция поли-
полимера играет относительно большую роль в числовом
М.-м. р., высокомолекулярная — в массовом. Поэтому
числовое распределение более точно описывает М.-м. р.
в области малых М. м., а массовое — в области больших
при одной и той же относительной ошибке в функции рас-
распределения. Для кривой массового распределения суще-
существует свое значение средней М. м.— с р е_д н е м а с-
совая молекулярная масса Mw, Графи-
Графически она представляет собой абсциссу центра тяжести
площади, ограниченной кривой массового распределе-
распределения и осью абсцисс. Для любых полимеров Mw>Mn,
~ »- мп только в предельном случае узкого
причем
А
1
Мп Мш
Молекулярная масса
Рис. 2. Кривые числового A) и массового B) молекулярно-
массовых распределений одного образца полимера (гадо»
т0 — масса молекул с М. м., равной М, и общая масса
полимерного образца соответственно; Мп , Mw — соответст-
соответственно среднечисловая и среднемассовая мол. массы; А, В —
центры тяжести площадей, ограниченных соответственно
кривыми 1 и 2 и осью абсцисс).
М.-м. р., когда все макромолекулы имеют одинаковую
М. м. (монодисперсное распределение). Чем шире рас-
распределение, тем больше различие в форме кривых
числового и массового распределений и тем больше
различаются Mw и Мп. Т. о., отношение M-jMn
может служить характеристикой ширины М.-м. р.
На рис. 1 и 2 показаны кривые распределения
с одним максимумом (унимодальные). Встречаются и
кривые распределения, имеющие большее число макси-
максимумов (би- и мультимодальные). Полимеры с унимо-
унимодальным М.-м. р. и Mw IMn = 2 обычно получаются
в результате процессов, подчиняющихся простым ста-
тистич. законам; мультимодальные М.-м. р.— вследст-
вследствие неоднородных условий получения или превраще-
превращения полимера, напр, при изменении темп-ры и кон-
концентрации реагентов по объему образца или в ходе
процесса, вследствие гетерофазности процесса, наличия
нескольких видов активных центров или при протека-
протекании реакции по нескольким механизмам одновременно.
Наложение нескольких унимодальных М.-м. р. с раз-
различными средними М. м. может привести как к мульти-
модальному, так и к унимодальному суммарному
М.-м. р. в зависимости от положения максимумов и ши-
ширины каждого индивидуального М.-м. р. Поэтому уни-
унимодальность М.-м. р. не свидетельствует однозначно
о простом механизме образования полимера. Широкое
унимодальное М.-м.р. может быть также следствием
зависимости эффективных констант скоростей реак-
реакций от длины макромолекул.
При экспериментальном определении М.-м. р. иногда,
напр, при применении метода турбидиметрич. титро-
титрования, измеряют суммарное количество Атм поли-
полимера, имеющего М. м. меньше (или больше) нек-рой
определенной М. м., равной М, причем последняя
величина постепенно изменяется от Мт\п до Мтах.
Получаемое распределение наз. интегральным
молекулярно-массовы м (рис. 3).
т0 dM
О Mmin Молекулярная масса Мтах
Рис. 3. Кривые дифференциального A) и интегрального
B) массового молекулярно-массового распределения образца
полимера.
Для теоретич. анализа М.-м. р. и связи его со свойст-
свойствами полимеров и механизмами их образования и пре-
превращения удобно пользоваться аналитическими (а не
графическими) выражениями функции М.-м. р.
Для описания М.-м. р. используют дискретные или
непрерывные функции от М. м. (или степени полиме-
полимеризации). В первом случае предполагают, что М. м.
полимера принимает значения, кратные массе моно-
мономерного звена, во втором — что М. м. может принимать
любые значения. Функция, характеризующая отноше-
отношение числа молекул данной М. м. к общему числу мак-
макромолекул, наз. дискретной дифференциальной число-
числовой функцией М.-м. р. Рп(М). Функция, характери-
характеризующая отношение массы молекул данной М. м.
ко всей массе полимера, наз. дискретной дифферен-
дифференциальной массовой функцией М.-м. p. Pw (M). Соот-
Соответствующие функции, характеризующие число или
массу всех макромолекул, имеющих М. м. меньше дан-
данной, наз. дискретными интегральными числовой Qn{M)
или массовой Qw (M) функциями М.-м. р. Каждая
из этих функций однозначно описывает М.-м. р. и мо-
может быть выражена через другие:
м
PW(M)= -lL-Pn(M); Qn(M)= У, Рп{М)
М
м
min
Pn(M) = Qn(M)-Qn(M-M0); PW(M)=QW(M)-QW(M-MO)
где Мо — масса мономерного звена.
289
МОЛЕКУЛЯРНО-МАССОВОЕ РАСПРЕДЕЛЕНИЕ
290
Непрерывная дифференциальная числовая функция
М.-м. p. pn (M) определяется как отношение числовой
доли dn макромолекул, имеющих М. м. в интервале
от М до М + dM, к размеру этого интервала dM,
т. е. рп(М) = dnidM. Непрерывные дифференциальная
массовая pw (M), интегральные числовая qn{M) и мас-
массовая qw (M) функции М.-м. р. определяются аналогич-
аналогично соответствующим дискретным функциям:
м
м
min
М
9w(M)dM;
М
dM
min
Любая из перечисленных выше функций полностью
описывает М.-м. р. полимера. Однако переход от ин-
интегральных к дифференциальным функциям М.-м. р.
требует графического или численного дифференциро-
дифференцирования, что ведет к появлению значительных ошибок.
Поэтому экспериментальная дифференциальная кривая
точнее описывает реальное М.-м. р., чем интегральпая.
При решении кинетич. задач часто используют функ-
функции распределения по степеням полимеризации /
[pn(/)> Pw (/)» PnU) и т- Д-]- В этом случае все непрерыв-
непрерывные и дискретные функции при />1 совпадают одна
с другой. Непрерывные функции, однако, можно
использовать только при условии,что для большинства
макромолекул выполняется условие / > 1.
Аналитич.' выражения для среднечисловой и средне-
массовой М. м. имеют вид
рп(М)
мрп(М)
используют средне-
(z - с р е д н ю ю) мо-
м
Кроме Mw и Мп на практике
седиментационную
лекулярную массу:
М
mz=
М
тах
2
Ширину М.-м. р., следовательно, можно характери-
характеризовать также отношением Mz/Мгг. Др. характеристи-
характеристиками ширины М.-м. р. могут служить отношение сред-
невязкостной (см. Молекулярная масса) к среднечисло-
среднечисловой М. м. или отношение средневязкостных М. м. для
различных растворителей. Средневязкостная М. м.
{Мч\) находится из значения характеристич. вязкости
по ур-нию Марка — Хувинка [ц] = КМах (см. Вяз-
Вязкость характеристическая). Отношения средневязкост-
ной к среднечисловой М. м. или средневязкостных
М. м., определенных в различных растворителях, тем
чувствительнее к ширине М.-м. р., чем больше а или
разница между значениями а для разных раствори-
растворителей.
Перечисленные характеристики хотя и не являются
полными, часто оказываются достаточными для уни-
унимодальных М.-м. р. при анализе реакций образования,
превращения и свойств полимеров. Мультимодальные
М.-м. р. следует характеризовать, по крайней мере,
положениями максимумов и шириной каждого макси-
максимума.
Для аналитич. выражения экспериментальных уни-
унимодальных М.-м. р. наиболее часто используют модель-
модельные функции, приведенные в таблице. Функции 1, 2
(при к < 0), 5, 7 и 8 (последняя при Ъ < 1) имеют
максимальные значения при наименьшей М. м. и моно-
монотонно уменьшаются с ростом М. м. (напр., кривая 1
на рис. 4). Остальные функции имеют более или менее
широкий максимум. Функции 2 (при к > 0), 3 и 4
используют обычно для описания узких (MwlMn
Рис. 4. Молекулярно-массовое
распределение полимеров, об-
образующихся при радикальной
полимеризации: 1 — в отсут-
отсутствие рекомбинации (распре-
(распределение Флори, см. табл.);
2—при обрыве цепи в резуль-
результате рекомбинации (распреде-
(распределение Шульца, см. табл.).
а 5—8 — широких (Mw/Mn^> 2) М.-м. р. Функции 7
и 8 не имеют смысла в области малых М. м., и теоре-
тич. значение среднечисловых М. м. для них равно
нулю.
Подбор модельной функции для описания эксперимен-
экспериментальной кривой М.-м. р. проводят в след. порядке:
1) вычисляют из экспериментальной кривой М.-м. р.
или измеряют независимым методом средние М. м.
(Мп, Mw и Mz)\ 2) подбирают типы функций, соотно-
соотношения средних М. м. для к-рых лучше всего совпадают
с найденными из эксперимента; 3) вычисляют парамет-
параметры этих функций (см. табл.) из экспериментальных
средних М. м.; 4) выбирают ту функцию, в коорди-
координатах к-рой (см. табл.) наилучшим образом спрям-
спрямляется экспериментальная кривая М.-м. р.
Практически любое экспериментальное унимодаль-
унимодальное М.-м. р. можно «подогнать» хотя бы под одну
из модельных функций. Однако такое описание имеет
ограниченный смысл, т. к. одна и та же эксперименталь-
экспериментальная функция м. б. описана несколькими модельными
функциями, лишь нек-рые из к-рых имеют четкий ки-
кинетич. смысл (см. ниже). С др. стороны, функции
М. м.-р., имеющие кинетич. смысл, часто более сложны
и либо не м. б. сведены к приведенным в табл., либо
являются их линейными комбинациями.
Экспериментальные методы определения
молекулярно-массового распределения
Как уже отмечалось, неполной характеристикой
М.-м. р. могут служить различные средние М. м. и их
соотношения (о методах определения средних М. м.
см. Молекулярная масса, Осмометрия, Эбулиоскопия).
Ниже кратко рассмотрены методы, позволяющие оп-
определить всю функцию М.-м. р. К этим методам от-
относятся скоростная седиментация, фракционирова-
фракционирование, адсорбционная тонкослойная и гель-проникающая
хроматография, деструкционный метод (описан на
стр. 299).
Метод скоростной седиментации основан на зависи-
зависимости скорости седиментации макромолекул в центро-
центробежном поле от их М. м. Непосредственно в опыте
получают кривую распределения концентрации поли-
полимера по коэфф. седиментации, однозначно связанным
с М. м. Фракционирование полимеров, т. е. разделение
на части с различными средними М. м. и сравнительно
узкими М.-м. р., возможно благодаря зависимости
растворимости макромолекул при данных условиях
(теми-pa, состав растворителя и др.) от их длины.
Фракционирование обычно осуществляют, изменяя со-
состав смеси растворитель — осадитель или темп-ру. Для
разделения полимера на фракции используют также
зависимость коэфф. распределения полимера между
двумя несмешивающимися растворителями от М. м.;
различие в зависимости коэфф. диффузии и термодиф-
термодиффузии от М. м. (метод термодиффузии) и др. Выделяе-
Выделяемые фракции имеют довольно широкое М.-м. р. Для
лучшего разделения фракции рефракционируют. Тем
Таблица 1. Функции молекулярно-массового распределения и их характеристики (приведены в наиболее употребительной форме)
Название
функции
Аналитическое выражение
функции
Мг
м0
MZ:MW: Mn
Характерные соотноше-
соотношения средних молекуляр-
молекулярных масс
Координаты спрямления графика функции
Распределение
Флори
?п О') = 4e~~'lj
3:2:1
Mn=Mw-Mn=Mz~Mw
lg рп, М или lg = , М
1 + М/Мп
Распределение
Шульца
vft+1
eke~V
k+ I
М
h+1
Mw--Mn=Mz-
-Mh
мп
мп J
Распределение
Пуассона
3V + 1 . V + 1 .
V2 + V
Mw = Mn + Af о
MwMn - M2n
О
н
I
s
1
3
н
ЕЯ
Распределение
Гаусса, нор-
нормальное
1 +¦
n+ Mo
: Рп, (М - iVr^J или
- Л1Г
MwMn -
: 1
Распределение
Бизли
lg р7г, lg 1 H
3 A -p) . 2A--?) .
- 3? 1 - 2,3
M | или
: 1
Mm-2Mr
1 +"
MMJMr
Распределение
Крэмера—Лан-
Крэмера—Лансинга
exp -
e 0,25,32
e?2: e0-5?1: 1
Af
v
Распределение
Веслау
2:1:0
А/ или lg(l - qw)t M
Распределение
Танга
¦к.
: 1 : 0
7/b
Г1+ —
Afz -A/^^ AlMJ»Mn
-gu,), JgAf
Обозначения: Г(х) = J e
x V 0
/
—полная гамма-функция,
= A! при целых k; ^(a, x)= J e~
0
—неполная гамма-функция; ф (х) =
= r n J e d^ — интеграл вероятности; y, ft, v, ;x, [3, ?> — параметры, характеризующие средние М. м. и ширину распределения; Мо — масса мономерного звена в
полимере.
293
МОЛЕКУЛЯРНО-МАССОВОЕ РАСПРЕДЕЛЕНИЕ
294
не менее отношение MjMn для конечных фракций
все же обычно довольно велико A,2—1,3). Поэтому при
построении кривых М.-м. р. исходного полимера при-
прибегают к специальной обработке данных с учетом
ширины каждой фракции. Хроматографич. методами
удается разделить полимер на 30—40 узких фракций
(Mw/Mn = 1,01 — 1,02). При определении М.-м. р.
с помощью гель-проникающей хроматографии р-р поли-
полимера пропускают через колонку с набухшим в раство-
растворителе сшитым полимером, к-рый представляет собой
молекулярное сито. Применение полимеров с различ-
различной частотой сетки или микропористых стекол позво-
позволяет фракционировать этим методом полимеры разной
средней М. м.
Свойства полимеров и молекулярно-массовое
распределение
Количественных теорий, связывающих М.-м. р.
с макроскопич. свойствами полимеров, Пока не су-
существует. Для полимерных систем с сильными межмо-
межмолекулярными взаимодействиями это объясняется зна-
значительным влиянием на свойства надмолекулярных
структур. Количественный учет этого влияния пред-
представляет значительные трудности. Для систем со сла-
слабыми межмолекулярными взаимодействиями (распла-
(расплавы, каучуки) обнаружены нек-рые соотношения, свя-
связывающие их свойства со средними М. м. и М.-м. р.
Наиболее подробно изучена связь между М. м.,
М.-м. р. и вязкостью расплава полимера (см. Молеку-
Молекулярная масса). Вязкость расплавов т] при нулевой ско-
скорости сдвига обычно зависит только от среднемассовой
М. м. (т) = ЯМи.3-4, где К — константа) и при одной и
той же Ми, не зависит от особенностей М.-м. р. Распла-
Расплавы полимеров — неньютоновские жидкости, и их вяз-
вязкость зависит от скорости сдвига, причем форма зави-
зависимости определяется М.-м. р. полимера. Вязкость
полимеров с узким (MwIMn — 1-^-1,2) М.-м. р. в ши-
широком диапазоне скоростей сдвига мало изменяется,
а при нек-рой критич. скорости сдвига наблюдается
срыв течения (см. Вязкость). Вязкость полимеров с
более широким распределением уменьшается при уве-
увеличении скорости сдвига, причем характер изменения
зависит от М.-м. р.
М.-м. р. и М. м. влияют на механич. свойства поли-
полимеров непосредственно или косвенно, определяя крис-
таллич. структуру, плотность, степень ориентации
и пр. Исследования зависимостей прочности при рас-
растяжении, удлинения при разрыве, прочности при из-
изгибе полистирола, полиэтилена, полипропилена, поли-
винилхлорида, полиамида, производных целлюлозы
и др. показали, что прочность растет при увеличении
Mw и Мп до нек-рых критич. значений М. м., а затем
сохраняется постоянной. Критич. значения Mw и
Мп для каждого полимера определяются эксперимен-
экспериментально. Если как MWJ так и Мп выше критических,
то прочностные характеристики полимера не зависят
от М.-м. р.
Деформационные характеристики аморфных поли-
полимеров (в частности, модули при растяжении и изгибе)
слабо зависят от М. м. и М.-м. р. за исключением очень
низких значений М. м.
Молекулярно-массовое распределение полимеров,
получаемых по различным механизмам
Радикальная полимеризация. Для радикальной по-
полимеризации характерно медленное инициирование
и быстрый рост одной цепи по сравнению со временем
всей реакции (время полупревращения). Кроме того,
химич. структура растущего радикала в обычных слу-
случаях одинакова для всех полимерных молекул и не за-
зависит от природы инициатора. Поэтому при анализе
М.-м. р. предполагают, что: 1) реакция характери-
характеризуется только одним набором констант скорости (один
сорт растущих радикалов); 2) константы скорости
роста, передачи и обрыва цепи не зависят от длины
(степени полимеризации) макрорадикала; 3) для мак-
макрорадикалов любой длины выполняется условие ста-
стационарности dP*t (j)ldt = 0. При соблюдении этих
условий функция распределения макрорадикалов по
их длинам имеет вид
) = Y(l-Y)j или
ПРИ
A)
(где Р%, р% — функции распределения макроради-
макрорадикалов), т. е. получается экспоненциальное распределе-
распределение Флори. Вероятность гибели растущего радикала
определяется соотношением
Y=
где wr, wn и Шр — соответственно скорости гибели
радикалов, передачи и роста цепи. Для вывода ур-ний
A) существенно, что от массы макрорадикала не зави-
зависит именно величина у, а не константы скорости.
М.-м. р. «мертвого» полимера, образующегося в дан-
данный момент (мгновенное М.-м. р.), зависит от доли
радикалов, погибающих в результате рекомбинации:
?nU) - (l-p)ye-tj+py2je-V B)
где р и A — р) — соответственно доли молекул, обра-
образовавшихся в результате рекомбинации и др. реакций
ограничения роста цепи. В отсутствие рекомбинации
М.-м. р. полимера описывается функцией Флори (см.
табл. и рис. 4, кривую 1). Если все молекулы обра-
образуются только в результате соединения макрорадика-
макрорадикалов, М.-м. р. представляет распределение Шульца при
к = 1 (см. табл. и рис. 4, кривую 2) и Мг : Mw : Мп =
= 4:3:2. В общем случае, зная отношение средних
М. м., напр. Mw/Mn, можно определить р по ур-нию
мп
Для небольших степеней превращения « 10%)
при гомогенной радикальной полимеризации обычно
можно не учитывать изменения у из-за уменьшения кон-
концентрации мономера, инициатора, передатчика цепи
или изменения физич. параметров среды. Тогда М.-м.«р.
всего полимера (суммарное М.-м. р.) описывается
ур-нием B). При больших степенях превращения
мгновенное М.-м. р. также описывается ур-нием B),
а массовая функция М.-м. р. всего полимера представ-
представляет сумму мгновенных М.-м. р. типа, представленного
ур-нием B), и ее можно найти, проинтегрировав соот-
соответствующую массовую мгновенную функцию [pw (J)]
по времени или степени превращения. Если у непре-
непрерывно изменяется во времени, то, независимо от того,
увеличивается или уменьшается у, М. м. р. расширя-
расширяется и отношение Мю/Мп увеличивается. При этом
обычно сохраняется унимодальный характер М.-м. р.
Только в нек-рых случаях, напр» при скачкообразном
изменении у из-за смены темп-рного режима полимери-
полимеризации, могут возникать мультимодальные М.-м. р.
Если при радикальной полимеризации происходит
передача цепи на полимер, приводящая к появлению
разветвленных макромолекул, то М.-м. р. полимера
значительно изменяется. Вероятность того, что про-
произойдет передача цепи на данную макромолекулу, тем
больше, чем больше масса этой молекулы. Это приво-
приводит к увеличению доли длинных молекул. В ходе про-
процесса увеличивается количество полимера и, следова-
следовательно, вероятность передачи цепи на полимер, и М.-
м. р. расширяется. Оно в этом случае описывается
функцией распределения Виз ли (см. табл.), где р -*-
295
МОЛЕКУЛЯРНО-МАССОВОЕ РАСПРЕДЕЛЕНИЕ
296
среднее число разветвлении на одну ветвь макромоле-
макромолекулы. При Р —> 0 распределение Б из ли переходит
в распределение Флори. Параметр Р можно найти,
зная ширину М.-м. р.:
м
2A—Р
Наибольшее значение передача цепи на полимер имеет
при промышленном синтезе полиэтилена низкой плот-
плотности, имеющего широкое М.-м. p. (MwlMn > 20).
При гетерофазной радикальной полимеризации часто
образуются полимеры с мультимодальным М.-м. р. Это
связано с наличием дискретного набора различных
у для каждой фазы и межфазной рекомбинацией.
Ионная и координационно-ионная полимеризация.
Приведенные выше при разборе радикальной полиме-
полимеризации условия выполняются также и в нек-рых слу-
случаях ионной полимеризации и координационно-ионной
полимеризации. При этом справедливы все выводы отно-
относительно М.-м. р. получающегося полимера, сформу-
сформулированные выше. Однако для ионной и координацион-
координационно-ионной полимеризации характерно нарушение усло-
условий стационарности и наличие нескольких видов актив-
активных центров, каждый из к-рых имеет свой набор кон-
констант скорости элементарных стадий и может или
не может превращаться в др. вид. Наиболее простой
для анализа случай — ионная полимеризация с обра-
образованием живущих полимеров. При этом может сохра-
сохраняться стационарная скорость расходования мономера,
т. е. постоянная суммарная концентрация активных
центров, но не выполняться условие стационарности
для растущих полимерных молекул данной длины
( 2L = 0; —2 ф 0 ) . При такой полимеризации обыч-
\ dt dt J
но образуются полимеры с узким М.-м. р., описы-
описываемым функцией Пуассона (смотри таблицу для слу-
случая v = fcp mf, где v — средняя степень полимеризации;
/ср—константа скорости роста цепи; т — концентрация
мономера; t — время). Полимеры с таким распределе-
распределением получаются при выполнении след. условий:
1) &р не зависит от длины макромолекулы; 2) иниции-
инициирование проходит быстро (кш ^ /ср, где кИ— константа
скорости инициирования); 3) гибель и передача цепи
не происходят; 4) каждая макромолекула имеет один
активный конец; 5) все активные центры идентичны
(одно значение fep); 6) реакционная система гомогенна;
7) деполимеризация не протекает (полимеризация не-
необратима). Отношение MwlMn для такого распределе-
распределения выражается как 1 + — и практически равно 1
для полимеров с высокой средней М. м. Для больших
v функция распределения Пуассона совпадает с рас-
распределением Гаусса (см. табл.), причем дисперсия
равна средней степени полимеризации. Нарушение
условий 2, 3, 5, 6 и 7 приводит к расширению М.-м. р.
Увеличение константы скорости роста цепи с длиной
(условие 1) также приводит к расширению распределе-
распределения, уменьшение — к сужению. Полимеризация на мак-
макромолекулах, имеющих два активных конца, с соблю-
соблюдением условий 1 — 3, 5—7 приводит к еще более узкому
М.-м. р.
При медленном инициировании низкомолекулярная
ветвь кривой М.-м. р. становится более пологой (рис. 5),
т. к. молекулы полимера, начавшие расти через нек-рое
время после начала реакции, имеют меньшую длину.
Чем меньше константа скорости инициирования (по
сравнению с константой скорости роста цепи), тем доль-
дольше протекает инициирование и, следовательно, более
пологой становится низкомолекулярная ветвь кривой
М.-м. р.; распределение при этом расширяется. В пре-
предельном случае, при инициировании с постоянной
скоростью в течение всего процесса, площадь, ограни-
ограниченная кривой М.-м. р. и осью абсцисс, принимает вид
практически правильного прямоугольника. Отношения
MwlMn для такого М.-м. р. равно 4/з» в остальных
случаях в зависимости от значения ка/кр и v оно мо-
может составлять от 1 до 4/3.
Если в системе протекают реакции ограничения роста
цепи (передача или гибель цепи), в полимере наряду
Рис. 5. Влияние скорости р
инициирования на молеку-
лярно-массовое распреде-
распределение при полимеризации
с образованием «живущих»
полимеров: 1 — мгновен-
мгновенное инициирование, 2,
3— медленное иницииро-
инициирование, 4— инициирование
с постоянной скоростью.
с фракцией «живущего» полимера высокой М. м. (рас-
(распределение Пуассона) присутствует и фракция «мерт-
«мертвого» полимера с низкой М. м. и широким распределе-
распределением (рис. 6, кривая 1). В ходе процесса доля первой
фракции постепенно уменьшается и М. м. «живущего»
полимера повышается. Если кол-во передатчика или ин-
ингибитора превосходит кол-во активных центров, в кон-
конце реакции эта фракция полностью пропадает, М.-м. р.
конечного полимера ока-
оказывается достаточно ши-
широким и описывается
ур-ниями A), если веро-
Рис. 6. Влияние гибели и
передачи цепи на молеку-
лярно-массовое распределе-
распределение: 1 — постоянная вероят-
вероятность гибели цепи; 2 — не-
небольшое количество передат-
передатчика цепи или ингибитора.
ятность гибели у не меняется в ходе процесса, или
более сложными ур-ниями в общем случае. При
небольшом количестве эффективного ингибитора (мень-
(меньшем, чем количество активных центров) конечный по-
полимер имеет бимодальное М.-м. р. (см. рис. 6, кри-
кривую 2) с узкой высокомолекулярной частью и широкой
низкомолекулярной. При этом отношение Mw/Mn
может изменяться в широких пределах в зависимости
от того, какая доля активных центров погибает. Когда
эта доля мала, Mw/Mn близко к 1. Если в начале реак-
реакции у макромолекулы оба конца активны, то в при-
присутствии небольшого количества эффективного инги-
ингибитора одна часть молекул активна лишь с одного
конца, другая полностью «погибает», а третья сохра-
сохраняет активность обоих концов. В результате последние
молекулы растут в два раза быстрее первых и кривая
М.-м. р. конечного полимера имеет два узких макси-
максимума и низкомолекулярную ветвь.
Обычно при ионной и координационно-ионной поли-
полимеризации реакция протекает по крайней мере на двух
(ионы и ионные пары) или более типах активных цент-
центров, каждый из к-рых характеризуется своим набором
констант скорости роста, передачи и обрыва цени.
По мере роста цепи может происходить превращение
активного центра одного вида в другой. Причем, если
за время роста цепи происходит многократное превра-
превращение активного конца этой цепи, то справедливо все
сказанное выше о М.-м. р. при ионной полимеризации;
система в этом случае м. б. охарактеризована одним
типом активных центров, имеющих нек-рые эффектив-
эффективные константы скорости элементарных актов. В част-
частности, при выполнении условий 1—3, 6 и 7 полимер
имеет узкое пуассоновское М.-м. р. Если за время роста
цепи превращение активного центра не успевает про-
произойти, полимер получает бимодальное М.-м. р. При
297
МОЛЕКУЛЯРНО-МАССОВОЕ РАСПРЕДЕЛЕНИЕ
298
соизмеримости скоростей превращения активных цент-
центров и присоединения к ним мономера возникает нек-рое
уни- или бимодальное М.-м. р. с промежуточным зна-
значением Mw/Mn, причем по Mw/Mn можно найти от-
отношение скоростей этих процессов.
Гетерогенные комплексные катализаторы характе-
характеризуются большим набором активных центров различ-
различного типа. При полимеризации на таких катализаторах
обычно получаются полимеры с довольно широким
(Mw/Mn > 10) унимодальным М.-м. р. Так, для про-
промышленных образцов полиэтилена и полипропилена,
полученных на катализаторах Циглера — Натта,
MwJMn =¦ 10 -f- 30. Возможным объяснением особен-
особенностей М.-м. р. таких полимеров является зависимость
констант скорости ограничения роста цепи от длины
макромолекулы. Если выполняется условие стацио-
стационарности для активных центров различной длины
и вероятность ограничения цепи у зависит от длины
макромолекулы, то М.-м. р. полимера имеет вид
з
PnG)=Y(/> °
При увеличении у с ростом / М.-м. р. сужается, при
уменыпепии расширяется по сравнению с простым
экспоненциальным. При отрыве макромолекулы от по-
поверхности катализатора увеличивается энтропия, т. к.
растущая макромолекула, связанная с поверхностью,
может принимать лишь те конформации, к-рые позво-
позволяют ей располагаться в объеме, не занятом самим
катализатором. Такой выигрыш тем больше, чем длин-
длиннее макромолекула. Следовательно, с увеличением /
скорость обрыва цепи и у должны увеличиваться, а
М.-м. р. сужаться. С др. стороны, для того чтобы
произошел обрыв цепи, молекула полимера должна
продиффундировать от поверхности катализатора. Ско-
Скорость диффузии, а следовательно, и константа скорости
обрыва уменьшаются с длиной макромолекулы, т. е.
М.-м. р. должно расширяться. В зависимости от усло-
условий реакции (темп-ры, растворителя и пр.) может
преобладать тот или иной процесс. Кроме того, величи-
величина у может меняться с глубиной реакции, что приво-
приводит к соответствующему усложнению вида М.-м. р.
Поликонденсация. При анализе М.-м. р. полимеров,
образующихся при гомогенной поликонденсации, обыч-
обычно принимают, что реакционная способность концевых
групп всех молекул не зависит от их длины. При этом
должен образовываться полимер с экспоненциальным
М.-м. р. [ур-ния A)], причем величина A — у) в этом
случае будет иметь смысл степени завершенности реак-
реакции, т. е. отношения числа прореагировавших конце-
концевых групп к числу концевых групп в исходной смеси.
Экспериментальные данные, полученные для многих
поликонденсационных систем при степенях завершен-
завершенности реакций ~ 0,99 (у — 0,01), в основном подтверж-
подтверждают предсказания теории. Однако в нек-рых случаях
(например, для полиарилатов) на более глубоких ста-
стадиях процесса образуются полимеры с узким М.-м. р.
(Mw/Mn = 1,5-1,7).
При межфазной поликонденсации получаются поли-
полимеры либо с экспоненциальным, либо с более сложным
М.-м. р., что, по-видимому, связано с гетерогенностью
процесса и значительным влиянием диффузии.
Обратимые процессы полимеризации и поликонден-
поликонденсации (равновесные молекулярно-массовые распределе-
распределения). При ионной полимеризации и поликонденсации
могут протекать обратимые процессы, приводящие
к равновесному М.-м. р. полимеров, к-рое соответст-
соответствует минимуму химич. потенциала полимера в данных
условиях. К таким обратимым процессам относятся,
напр., полимеризация с образованием «живущих»
полимеров и их деполимеризация. В реакции с уча-
участием «живущих» полимеров равновесное М.-м. р. уста-
устанавливается, конечно, только для «живой» части
полимера. При образовании гетероценных полимеров
по катионному механизму к равновесному М.-м. р.
приводит передача цепи на полимер с разрывом (см.
Катионная полимеризация, Передача цепи). В резуль-
результате этой реакции, во-первых, происходит обратимое
перераспределение молекул по длинам (при бимолеку-
бимолекулярной реакции) и, во-вторых, образование циклич.
макромолекул (при мономолекулярной реакции). При
достаточно большой скорости этой реакции в системе
независимо от др. процессов должно устанавливаться
равновесное М.-м. р. как линейных, так и циклич.
макромолекул. Перераспределение молекул по длинам
коснется всех молекул, а не только тех, к-рые в дан-
данный момент имеют активную концевую группу, т. к.
последняя может переходить от одной макромолекулы
к другой.
При иоликонденсации аналогом такой реакции яв-
является межцеиной обмен (см. Обменные реакции).
Кроме того, при обратимой поликонденсации может
происходить разрыв образующихся макромолекул под
действием выделяющихся в реакции низкомолеку-
низкомолекулярных продуктов. Все эти реакции приводят к равно-
равновесному М.-м. р. Наименее эффективна, с этой точки
зрения, деполимеризация, т. к. в каждом акте реак-
реакции происходит изменение степени полимеризации
данной молекулы всего на единицу. При всех осталь-
остальных реакциях в результате каждого акта длина ма-
макромолекулы изменяется сильно, и достаточно всего
нескольких актов реакции в расчете на одну макромо-
макромолекулу, чтобы установилось равновесное М.-м. р.
Если принять, что константа скорости всех реакций,
приводящих к равновесию, не зависит от длины моле-
молекул, то равновесным М.-м. р. линейных макромолекул
будет экспоненциальное распределение [ур-ние A)].
Промышленные образцы полимеров и сополимеров
формальдегида имеют распределение, близкое к экс-
экспоненциальному (Mw/Mn ж 2), что, по-видимому,
объясняется протеканием реакции передачи цепи с
разрывом.
Принцип независимости реакционной способности
макромолекул от их длины был высказан Флори и обос-
обоснован им с точки зрения решетчатой теории конц.
р-ров полимеров. С термодинамич. точки зрения этот
принцип означает энергетич. и энтропийную равно-
равноценность мономерных единиц в макромолекулах раз-
различной длины. Это условие не выполняется для циклич.
макромолекул, в к-рых не м. б. реализован тот же
набор конформации, что и в линейных макромолеку-
макромолекулах. Энтропия циклич. молекул меньше, чем линейных,
и эта разница увеличивается с ростом мол. массы.
М.-м. р. циклич. макромолекул, находящихся в рав-
равновесии с линейными молекулами со средней степенью
полимеризации и экспоненциальным М.-м. р., имеет
вид
где А — нормировочный множитель. Такой вид распре-
распределения справедлив лишь для больших ненапряжен-
ненапряженных циклов. Для малых циклов следует учитывать
также изменение энтальпии при образовании цикла
данной М. м. Общее число циклов, как и средняя
М. м., при поликонденсации определяется условиями
равновесия.
Принцип Флори не выполняется и для линейных
макромолекул в разб. р-ре, где макромолекулы нахо-
находятся в виде клубков различной плотности. Чем длин-
длиннее макромолекула, тем более рыхлый клубок она
образует, тем, следовательно, в ней меньше контактов
мономерных звеньев друг с другом и больше контактов
с растворителем. В «хорошем» растворителе контакты
звеньев с ним более выгодны, равновесное М.-м. р.
299
моноволокно
300
шире распределения Флори и в первом приближении
имеет вид
рпG)=Л ехр (- -А- 52 + In 53-Y/
где Л — нормировочный множитель; у — параметр,
характеризующий среднюю М. м.; 5 — степень набуха-
набухания клубка в данном растворителе, зависящая от /.
Т. о., вид равновесного М. м.-р. в разб. р-ре зависит
от природы растворителя, темп-ры и т. д. В 6-точке
(идеальный растворитель — см. Флори ^-температу-
^-температура) Ь = 1 и равновесное М.-м. р. представляет собой
распределение Флори.
Молекулярно-массовое распределение полимеров
в процессах деструкции
Переработка и эксплуатация полимеров сопровож-
сопровождаются их деструкцией, приводящей, в частности,
к изменению М.-м. р. и, следовательно, свойств про-
продуктов. В свою очередь, М.-м. р. исходного полимера
оказывает существенное влияние на кинетику деструк-
деструкции и изменение средних М. м. Характер изменения
М.-м. р. и влияние исходного М.-м. р. на кинетич.
характеристики процесса зависят от конкретного меха-
механизма деструкции.
При анализе М.-м. р. обычно принимают, что кон-
константы скорости деструкции не зависят от длины цепи.
Если деполимеризация, начавшись с одного конца
цепи, быстро распространяется до ее др. конца, то
в ходе процесса не меняются ни М.-м. р., ни средние
М. м., и скорость реакции не зависит от вида М.-м. р.,
Mw, Mn и т. д. При медленной ступенчатой деполи-
деполимеризации с конца цепи все молекулы практически
с одинаковой скоростью укорачиваются и общее число
их уменьшается. Это приводит к тому, что М.-м. р.
в каждый момент времени является нек-рой частью
от исходного М.-м. р. (рис. 7). В зависимости от шири-
ширины начального М.-м. р. средние М. м. могут уменьшать-
уменьшаться (узкое начальное М.-м. p., Mwl'Мп < 2), не изме-
изменяться (наиболее вероятное М.-м. p., Mw/Mn = 2) или
даже увеличиваться (широкое М.-м. р., -Л/и>/Л/п>2),
если начальное М.-м. р. имеет значительную низко-
низкомолекулярную часть наряду с высокомолекулярной.
Скорость деполимеризации в каждый момент време-
времени пропорциональна числу оставшихся макромоле-
макромолекул, т. е. числу молекул, имевших в начале процес-
процесса степени полимеризации больше Агд t (где &д —
эффективная константа скорости деполимеризации,
t — время от начала процесса). Т. о., изучая кинети-
кинетику процесса, можно определить интегральную число-
числовую функцию М.-м. р. исходного полимера (деструк-
ционный метод).
При полимеризации или деполимеризации по «закону
случая» М.-м. р. в большинстве случаев стремится
к экспоненциальному виду [ур-ния A)]. Проводя частич-
частичную деструкцию образца полимера с широким М.-м. р.
и большой средней М. м., можно достигнуть сужения
М.-м. р. и улучшения механич. свойств полимера.
Такой метод предлагался для модификации полипро-
полипропилена .
Кинетика процесса и характер изменения средних
М. м. в начале реакции существенным образом зависят
от вида М.-м. р. исходного полимера и не зависят от
от него на глубоких стадиях. В частности, чем шире
М.-м. р., тем быстрее уменьшается среднемассовая
М. м. В тех случаях, когда длина кинетич. цепи депо-
деполимеризации определяется длиной макромолекулы,
оказывается возможным находить функцию М.-м. р.
исходного полимера по кинетич. данным или по изме-
изменению средних М. м.
При различных воздействиях на полимер происхо-
происходит как деструкция, так и сшивание макромолекул.
Если сшивание происходит чаще, чем разрыв, то М. м.
полимера постепенно возрастает, М.-м. р. расширяется
и в нек-рый момент, когда число сшивок станет равным
общему числу макромолекул, образуется трехмерная
структура. Изменение среднемассовой М. м. и характе-
ристич. вязкости полимера в начале процесса зависит
от ширины начального М.-м. р. Так, при достаточно
широком М.-м. p. (Mw/Mn > 2) сначала наблюдается
нек-рое падение характеристич. вязкости, а затем про-
Рис. 7. Изменение моле-р°
кулярно-массового распре- гп
деления полимера при сту-
ступенчатой деполимеризации.
Справа от оси ординат
Рп — кривая распределе-
распределения в момент времени t,
отрезок kj^t соответствует
укорочению длинных ма- ^<7Г J
кромолекул; кривая слева
от этой оси соответствует начальному распределению корот-
коротких молекул, «выгоревших» к моменту t.
исходит ее возрастание и образуется трехмерная струк-
структура. В случае полимера с узким М.-м. р. характери-
характеристич. вязкость монотонно возрастает.
Процессы деструкции реальных полимерных материа-
материалов осложнены существованием в них надмолекулярных
структур, наличием неоднородностей в молекулярной
структуре цепи (разветвления, соединения «голова
к голове», стереоизомерия и т. д.), а также протеканием
процессов диффузии и теплопередачи.
Лит.: Френкель С. Я., Введение в статистическую
теорию полимеризации, М., 1965; Flory P. J., Principles
of polymer chemistry, Ithaca, 1953; Шварц М., Анионная
полимеризация, пер. с англ., М., 1971; Б рее л ер С. Е.,
Ерусалимский Б. Л., Физика и химия макромолекул,
М.— Л., 1965; Кинетика радикальной полимеризации винило-
виниловых соединений, пер. с англ., М., 1961; Берлин А. А.,
Ениколопян Н. С, Высокомол. соед., 10А, № 7, 1475
A968); Рафиков СР., Павлова С. А., Твердо-
хлебоваИ. И., Методы определения молекулярных весов
и полидисперсности высокомолекулярных соединений, М., 1963;
Martini. R., Johnsonl.F., Cooper A. R. J., Mac-
romal. Sci.— Revs macromol. chem., C8(l), 57 A972).
Ал. Ал. Берлин.
МОНОВОЛОКНО, мононить (monofilament, Мо-
nofil, monofil) — одиночное элементарное химич. во-
волокно большой длины.
Получение. М. можно формовать из большинства
волокнообразующих полимеров. Однако чаще всего ис-
используют полиамиды, полиэтилентерефталат, поли-
олефины и сополимеры винилиденхлорида с винилхло-
ридом (см. Винилиденхлорида сополимеры). М. формуют
через фильеру с одним или несколькими отверстиями,
чаще всего из расплавов полимеров, т. к. при формо-
формовании из р-ров получают М. со значительной порис-
пористостью и, следовательно, невысокой прочностью. О ме-
методах формования и применяемом оборудовании см.
Формование химических волокон, Прядильные машины.
М. условно подразделяют на волокна малого
« 0,1 мм) и большого О 0,1 мм) диаметра. М. малого
диаметра незначительно отличаются по свойствам от
текстильных элементарных волокон. Получают оба
эти типа волокон по одинаковой технологической
схеме — формованием в воздушную охлаждающую
среду.
М. большого диаметра получают по след. схеме. По-
Полимер в виде крошки или гранул загружают в пла-
вильно-формовочное устройство, откуда он выдавли-
выдавливается через фильеру в виде расплавленных нитей.
Последние, пройдя короткий A—30 см) путь по возду-
воздуху, попадают в жидкую (обычно водную) охлаждаю-
охлаждающую ванну, где быстро затвердевают. Использование
такой ванны необходимо для того, чтобы быстро отвес-
отвести тепло от формуемых нитей, толщина к-рых довольно
значительна и может в 10—100 раз превышать толщину
текстильных элементарных волокон. Форму и диаметр
поперечного сечения М. регулируют, изменяя темп-ру
301
МОНОМЕРЫ КРЕМНИЙОРГАНИЧЕСКИЕ
302
ванны, расстояние от фильеры до жидкости, диаметр
и глубину погружения т. н. «заправочного» ролика в
жидкость.
Повышение темп-ры охлаждающей ванны до 40—
60 °С обычно способствует образованию М. с сечением,
наиболее близко приближающимся к форме круга,
и предотвращает извивание нитей из-за неравномер-
неравномерного охлаждения. В зависимости от темп-ры ванны мо-
могут изменяться структура и механич. свойства М. Уве-
Увеличение расстояния от фильеры до охлаждающей жид-
жидкости приводит к уменьшению толщины М., а увеличе-
увеличение диаметра заправочного ролика способствует обра-
образованию более круглого поперечного сечения М.
Сформованное М. с целью упрочнения подвергают
ориентационному вытягиванию (о конструкции вы-
вытяжных механизмов см. Прядильные машины). Напр.,
полиамидные М. вытягивают в 3,5—5,0 раз, полиэфир-
полиэфирные — в 4,5—6,0, полиолефиновые — в 5—7. Вытяги-
Вытягивание проводят в нагретой воде, паре, горячем воздухе.
Полиамидные и полученные из сополимеров винилиден-
хлорида с винилхл'оридом М. сравнительно небольшого
диаметра иногда вытягивают при нормальной темп-ре.
Вытяжку М. сравнительно небольшого диаметра
обычно осуществляют между цилиндрами, вращающи-
вращающимися с различной скоростью. М. большого диаметра
чаще всего вытягивают на горизонтальном вытяжном
агрегате, к-рый иногда располагают отдельно от пря-
прядильной машины; для облегчения ориентации макро-
макромолекул в нек-рых случаях вытяжку осуществляют
в две стадии.
Для выравнивания М. по диаметру иногда исполь-
используют метод волочения, аналогичный тому, к-рый при-
применяют при калибровке металлич. проволоки. Напр.,
при получении М. из полиэтилентерефталата вначале
проводят волочение в горячей воде, а затем — допол-
дополнительное ориентационное вытягивание. При этом по-
получают М., отличающееся равномерным диаметром но
всей длине и повышенной прочностью — 500 мн/текс
E0 гс/текс). Приспособление для калибровки М. рас-
располагают перед зоной ориентационного вытягивания.
Вытянутое М. подвергают термообработке в обогре-
обогреваемой камере. В результате может возрастать устой-
устойчивость М. к двойным изгибам, но одновременно увели-
увеличиваться жесткость в результате дополнительной кри-
кристаллизации полимера. Готовое М. принимают на намо-
намоточное устройство. В зависимости от назначения М.
выпускают на катушках или в мотках.
Свойства. В зависимости от вида исходного полиме-
полимера, параметров формования и вида последующей об-
обработки можно получить М. с различными свойствами
(таблица).
Свойства основных видов моноволокон
Виды моноволокон
Полиамидные ....
Полиэтилентерефта-
латные(полиэфир-
латные(полиэфирные)
Полиолефиновые . .
Из сополимеров ви-
нилиденхлорида с
винилхлоридом . .
Основные показатели
диаметр,
ММ
0,03-1 ,0
0,15-1 ,5
0,1-1 ,0
0,5-1
прочность,
мн/текс
(гс/текс)
300-600 C0-60)
300-500 C0-50)
350-700 C5-70)
150-300 A5-30)
относи-
относительное уд-
удлинение,
%
20-40
10-35
15-30
15-45
Полиамидные М. обладают высокой усталостной проч-
прочностью, устойчивостью к истиранию, хорошей проч-
прочностью в петле и узле, однако недостаточной атмосферо-
стойкостью. Высокий модуль упругости, хорошая
износо- и атмосферостойкость характеризуют полиэфир-
полиэфирные М.; основные недостатки этих волокон — невысо-
невысокая прочность в петле и узле, недостаточно хорошее
восстановление после изгиба. Высокой прочностью и
устойчивостью к двойным изгибам, небольшой плот-
плотностью, гидрофобностью обладают полиолефиновые
волокна, к-рые, однако, имеют низкую устойчивость
к истиранию и плохую атмосферостойкость. К досто-
достоинствам М. из сополимеров винилиденхлорида с винил-
хлоридом относят высокую устойчивость к истиранию,
хорошие электроизоляционные свойства, гидрофоб-
ность, к недостаткам — низкую разрывную и усталост-
усталостную прочность.
Применение. М. всех видов используют при изготов-
изготовлении щеток взамен натуральной щетины (напр., для
щеток мельничных и уборочных машин, малярных кис-
кистей, щеток для чистки одежды и обуви). М. применяют
также для производства фильтровальных сеток и ви-
вибросит в нефтяной, угольной и химич. пром-сти. Поли-
Полиэфирные М. используют для изготовления сеток бума-
бумагоделательных машин (взамен металлических) и транс-
транспортерных лент, полиамидные—в качестве рыболовной
лески (диаметром от 0,1 до 1 мм), в произ-ве рыболов-
рыболовных сетей, канатов; полиолефиновые — для изготовле-
изготовления легких канатов, сетей, обивочных материалов.
Лит.: Кларе Г., ФрицшеЭ., ГребеФ., Синте-
Синтетические полиамидные волокна, пер. с нем., М., 1966; Hen-
sen F., Utermann W., Kunststoff u. Gummi., 3, № 12,
455 A964); Геллер В. Э. [и др.], Механика полимеров,
№ 6, 146 A965). В. Э. Геллер.
МОНОКРИСТАЛЛЫ п о л и м е р о в — см. Кри-
Кристаллическое состояние.
МОНОМЕР (monomer, Monomer, monomere) — низ-
низкомолекулярное вещество, молекулы к-рого способны
вступать в реакцию друг с другом или с молекулами
др. веществ с образованием полимера. О различных
типах М. и их свойствах см. Поликонденсация, Поли-
Полимеризация, Реакционная способность мономеров. Функ-
Функциональность мономеров.
МОНОМЕРЫ КРЕМНИЙОРГАНИЧЕСКИЕ (orga-
nosilicon monomers, Organosilikonmonomere, monome-
res organosiliciques).
Содержание:
Методы получения 302
Прямой синтез органохлорсиланов 302
Пиролитические способы получения органохлорси-
органохлорсиланов 303
Термокаталитическое силилирование 304
Гидросилилирование непредельных соединений . . . 3 04
Диспропорционирование заместителей у кремния . . 305
Металлорганический синтез .305
Получение мономеров на основе органохлорсиланов 3 06
Свойства 307
Мономеры кремнийорганические — соединения, име-
имеющие в молекуле один или несколько атомов кремния
и способные превращаться в кремнийорганические поли-
полимеры или олигомеры.
Наибольшее значение среди всех М. к. имеют орга-
нохлорсиланы RnSiCl4_n и органохлоркремнийгидри-
ды RnSiCl3_nH. При получении кремнийорганич. моно-
мономеров и полимеров часто используют также чисто неор-
ганич. соединения — SiCl4 и SiHCl3. Все остальные
М. к. получают теми или иными превращениями ука-
указанных кремнийсодержащих соединений.
Методы получения
Прямой синтез органохлорсиланов. В пром-сти
органохлорсиланы получают гл. обр. по т. наз. «пря-
«прямому методу», заключающемуся во взаимодействии
алкил- или арилхлоридов при 200—600 °С с элементар-
элементарным кремнием. Последний входит в состав т. наз. кон-
контактных масс (сплавы или смеси порошков кремния и
катализатора — меди в количестве от 3 до 30% по
массе). Сокатализаторами служат такие элементы как
Ag, Zn, Gd, Mg, Ga, Al и др. Для активирования кон-
контактных масс можно также до начала процесса или
вместе с органохлоридами пропускать через реактор
водород, хлористый водород, хлор. Прямой синтез
303
МОНОМЕРЫ КРЕМНИЙОРГАНИЧЕСКИЕ
304
органохлорсиланов осуществляют по непрерывной схе-
схеме в аппаратах с механич. перемешиванием контактной
массы (реакторы типа «вращающийся барабан» или
с мешалкой), а также в высокопроизводительных аппа-
аппаратах в кипящем слое. Состав получаемого конденсата
и скорость реакции определяются составом и способом
приготовления контактных масс, способом их активи-
активирования, наличием примесей в исходном органохлори-
де, аппаратурным оформлением процесса, а также вре-
временем контакта, темп-рой и давлением.
Важнейшие классы промышленных органохлорсила-
органохлорсиланов — метилхлорсиланы, этилхлорсиланы и фенилхлор-
силаны. Получают их след. образом. При взаимодейст-
взаимодействии метилхлорида с контактной массой образуется
сложная смесь продуктов:
200-250сС
СН3С1 + Si > (CH3JSiCl2 + CH3SiCl3 + (CH3KSiCl +
+ CH3SiHCl2 + (GH3JSiHCl
В состав продуктов реакции могут входить также
и др. кремнийсодержащие вещества, углеводороды, во-
водород. Направленный синтез позволяет получать кон-
конденсат с содержанием диметилдихлорсилана > 80%.
Добавление к метилхлориду водорода приводит к уве-
увеличению выхода метилхлоркремнийгидридов. Модифи-
Модификация контактной массы позволяет обогатить продукты
реакции триметилхлорсиланом. Во всех случаях в за-
заметных количествах образуются SiHGl3, SiCl4, а также
метилхлорполисиланы и метилхлорполисилметилены.
Взаимодействие хлорбензола с контактной массой
происходит при более высокой темп-ре:
450-500°С
С6НБС1 + Si > (C6H5JSiCl2 + C6H5SiCl3 + (G6H5KSiGl +
+ G6He + СбН5С6Н5 + др. продукты
В качестве сокатализатора весьма эффективно се-
серебро, позволяющее проводить процесс при более низ-
низких темп-pax; используют также контактные массы
с цинком и кадмием. Содержание дифенилдихлорсила-
на может достигать 80%. При добавлении НС1 основ-
основным продуктом реакции является фенилтрихлорсилан;
в этом случае в конденсате содержится значительное
количество SiHCl3 и SiCl4.
Прямой синтез используют также для производства
этилхлорсиланов из этилхлорида. Варьируя различные
факторы, получают конденсат с содержанием 20—65%
диэтилдихлорсилана, 15—60% этилдихлорсилана, 10 —
50% этилтрихлорсилана и незначительного количества
диэтилхлорсилана. Прямой синтез м. б. использован
и для получения ряда иных органохлорсиланов, в част-
частности с аллильными, высшими алкильными и арильны-
ми заместителями у кремния. Из дихлорпроизводных
алифатич. углеводородов образуются М. к. с двумя
хлорсилильными группами, а также дихлорсилацикло-
алканы. Использование в прямом синтезе органоброми-
дов позволяет получать органобромсиланы.
По реакции НС1 с кремнием, ферросилицием или
кремнийсодержащими контактными массами в пром-сти
получают SiHCl3. Взаимодействие кремния или ферро-
ферросилиция с хлором приводит к получению SiCl4. Трихлор-
силан и четыреххлористый кремний являются исход-
исходными продуктами для приготовления различных М. к.;
в нек-рых случаях их используют в качестве компонен-
компонентов смеси хлорсиланов при получении полимеров.
Пиролитические способы получения органохлорсила-
органохлорсиланов. Большое практич. значение имеют пиролитич.
способы получения органохлорсиланов. Процесс про-
проводят в проточной системе в газовой фазе при 500—
750 °С. Реактором служит полая труба из стали, меди,
керамики или кварца. Метод весьма удобен для тех-
нологич. оформления. Выходы М. к. достигают 80%.
Наибольшее значение имеет термич. конденсация,
заключающаяся во взаимодействии арил- или алкенил-
хлоридов с трихлорсиланом или органохлоркремний-
гидридами. Основные направления взаимодействия от-
отражаются схемой:
п + HG1 (К)
RnSiCl4_n (B)
Реакцию, ведущую к целевому М. к., наз. конденса-
конденсацией (К), ведущую к образованию углеводорода — вос-
восстановлением (В). Соотношение К/В определяется при-
природой исходных органохлоридов и кремнийгидридов,
условиями процесса и материалом реактора. Значение
К/В уменьшается по мере замены атомов хлора в три-
хлорсилане органич. радикалами. Термич. конденса-
конденсация нашла промышленное использование для получения
винилтрихлорсилана, метилвинилдихлорсилана и фе-
нилтрихлорсилана:
СН2=СНС1 + HSiCl3 -» CH2=CHSiCl3 + HG1
GH2=CHG1 + HSiCH3Cl2->CH2=CHSiCH3Cl2 -f HG1
G6H5G1 -j- HSiCl3 -» GeH6SiGl3 + HG1
Термич. конденсация — удобный метод синтеза орга-
органохлорсиланов с двумя различными органич. замести-
заместителями у атома кремния, напр.:'
C6H5Cl-f HSiCH3Cl2 _> C6H5SiCH3Cl2+HCl
Пиролиз позволяет из органохлорсиланов синтези-
синтезировать др. классы М. к., напр, из фенилхлорсиланов —
соединение с фениленовым мостиком между силильными
группами:
C3H5SiCl3 -> Cl3SiC6H4SiCl2C6H5 + C6H5Gl2SiG6H4SiGl2G6H5 +
+ CeH5Cl2SiC6H4SiC]2CeH4SiCl2C6H6 + др. продукты
Термокаталитическое силилирование. Для получения
арилтрихлорсиланов и арилорганохлорсиланов опреде-
определенное значение имеет термокаталитич. силилирование
ароматич. соединений в жидкой фазе:
RH-j-HSiR^Cl3_n -» RR^SiCl3n+H2
где R — арил; R' — метил, этил, фенил. Реакция про-
проходит под давлением при темп-ре выше 200 °С в присут-
присутствии ВС13, BF3, A1C13, соединений, образующихся при
взаимодействии борной к-ты с хлорсиланами, и др.
При более высоких темп-рах D00 °С) процесс может
осуществляться и без катализаторов. Термокаталитич.
силилирование используют в пром-сти для получения
метилфенилдихлорсилана.
Гидросилидирование непредельных соединений. Для
синтеза карбофункциональных алкилхлорсилапов, а так-
также алкилхлорсиланов. с высшими алкилами наиболь-
наибольшее распространение получила реакция гидросилилиро-
вания, заключающаяся в присоединении простейших
М. к., имеющих связь Si — Н, по кратным связям не-
непредельных органич. соединений, напр.:
Gl3SiH + НС=СН ~» Cl3SiCH=CH2
(C2H6OKSiH + CH2=GH-GH2NH2 -» (G2H5OKSiGH2GH2GH2NH2
Gl2(GH3)SiH + GH2=GH-GN -» Gl2(GH3)SiCH2CH2GN
Gl2(GH3)SiH + GH2=GHGF3 -> Cl2(CH3)SiCH2CH2CF3
Представители класса а, to-дикарбофункциональных
мономеров типа O(CH2)nSi(CH3J0Si(CH3J(CH2)n(p, где
Ф = ОН, SH, COOH, NH2 и др., легко образуются
при использовании в качестве органохлоркремнийгид-
рида диметилхлорсилана, напр.:
-HG1
7iGH2=GHGH2OH + nHSi(CH3JGl >
Кат. Н2О
-> nCH2=CHCH2OSi(CH3JH > [-Si(GH3J(GH2KO -]n
^n/2HO(GH2KSi(GH3JOSi(CH3J(GH2KOH
Гидросилилирование можно проводить как по периодич.
схеме в обычных аппаратах с мешалками, так и непре-
непрерывно в проточной системе. Наиболее широко исполь-
используются платиновые катализаторы, в частности р-ры
платинохлористоводородной к-ты в спиртах; примени-
305
МОНОМЕРЫ КРЕМНИЙОРГАНИЧЕСКИЕ
306
ют также различные соединения переходных металлов,
а в нек-рых случаях — амины, амиды, фосфины и др.
Гидросилилирование можно инициировать перекисями,
УФ- и 7-об л учением, а также проводить при повышен-
повышенных темп-pax без инициаторов и катализаторов. В зави-
зависимости от природы исходных компонентов реакцию ве-
ведут при темп-pax от 0 до 150 °С. Во многих случаях
гидросилилирование сопровождается экзотермич. эф-
эффектом. Выходы М. к. могут достигать 90—95%.
Диспропорционирование заместителей у кремния.
Для получения органохлорсиланов с различными заме-
заместителями у атома кремния большое значение имеет
реакция диспропорционирования. Использование в этой
реакции органохлорсиланов, получаемых прямым син-
синтезом, делает доступными ценные М. к. с метильными
и фенильными заместителями у одного атома кремния:
CeH5SiCl3 + (CH3KSiCl -^ C6H5(CH3)SiCl2 +
+ CeH5(CH3JSiCl + (CH3JSiCl2 + CH3SiCl3
CHsSiCl3 + (GeH5KSiCl -^ CeH5(CH3)SiCl2 + (CeH5JCH3SiCl +
+ (CeH5JSiCl2 + CeH5SiCl3
Варьируя соотношение исходных реагентов и усло-
условия реакции, можно добиться преимущественного об-
образования одного из органохлорсиланов. Реакцию обыч-
обычно проводят под давлением при 250—400 °С. Катализа-
Катализаторами диспропорционирования служат к-ты Льюиса.
В органосиланах с алкокси- и ацилоксигруппами орга-
нич. заместители легко диспропорционируют под дейст-
действием щелочей и алкоголятов.
Металлоорганический синтез. Большое распростра-
распространение в лабораторной практике получил металлоорганич.
синтез М. к. на основе SiCl4, SiHGl3 и органохлорсила-
органохлорсиланов. По синтетич. возможностям этот метод наиболее
универсален и позволяет в одну или несколько стадий
получить М. к. практически любого типа. Наибольшее
применение нашел синтез М. к. с использованием реак-
реактива Гриньяра:
mRMgHal + R^SiCl4_n -> RmR^S
mMgHalGl
Магнийорганич. синтез широко используется для
приготовления органохлорсиланов с различными орга-
нич. заместителями у атома кремния, напр.:
CH3SiCl3 + CeH5MgBr -> (C6H5)CH3SiCl2+(C6H5JCH3SiCl + MgClBr
CH2=CHSiCl3 + G6H5]VIgBr -> (CeH5)(CH2=CH)SiCl2 + MgCIBr
Этот метод позволяет синтезировать силациклоалканы:
СН2
Mg / \
Cl(CH3JSiCH2CH2CH2Hal > (GH3JSi GH2
\ /
GH2
GH2
Mg / \
2Cl(CH3JSiCH2Cl > (GH3JSi Si(GH3J
GH2
M. к. можно получать через магнийорганич. соеди-
соединения как в одну, так и в две стадии (с приготовлением
на первой стадии реагента Гриньяра). В качестве раст-
растворителей обычно используют органич. простые эфиры.
В одностадийных процессах с эфирами кремниевой к-ты
роль растворителя выполняет кремнийорганич. компо-
компонент реакции. В лабораторной практике М. к. часто
приготовляют с помощью органич. соединений щелоч-
щелочных металлов (гл. обр. лития). Значительно меньшее
распространение имеют синтезы с помощью цинк- и
алюминийорганич. соединений.
Металлоорганич. синтез М. к. имеет ограниченное
использование в пром-сти вследствие взрыво- и пожаро-
опасности растворителей, необходимости отделения
реакционной массы от образующихся солей металлов,
трудности регулирования процесса. Как правило, ме-
метод используют в тех случаях, когда др. способы не
м. б. применены, напр, для получения моно- или диси-
лациклобутанов.
Получение мономеров на основе органохлорсиланов.
Реакциями замещения по связям Si — С1 органохлорси-
ланы м. б. превращены в М. к. с различными функцио-
функциональными группами у атома кремния. Наибольшее зна-
значение среди этих М. к. имеют органоалкокси-, орга-
ноацилоксисиланы и силанолы. Последние являются
промежуточными соединениями при получении поли-
органосилоксанов гидролитич. поликонденсацией (см.
Кремнийорганические полимеры).
Нек-рые силанолы м. б. получены гидролизом в мяг-
мягких условиях М. к. с различными кремнефункцио-
нальными группами:
-сн3он
(G6H5JSi(OGH3)o+2H20 > (GeH5JSi(OHJ
[(GH3KSi]2NH f 2H2O •
RSi-H+H,0
I
-NH3
-H8
2(GH3KSi0H
Кат. (Ni, Pt, Pd)
RSiOH
Силанолы с алифатич. заместителями, как правило,
в результате межмолекулярного взаимодействия легко
отщепляют воду, превращаясь в соответствующие сило-
ксаны. Еще более нестабильны силандиолы, содержащие
две гидроксигруппы у атома кремния. Менее склонны
к отщеплению воды силанолы с разветвленными алифа-
алифатич. и ароматич. заместителями у кремния.
Органоалкокси- и органоацилоксисиланы получают
взаимодействием органохлорсиланов соответственно со
спиртами и к-тами или их производными (алкоголяты,
соли, ангидриды и др.)» напр.:
—2HG1
(CH3JSiCl2-f 2С2Н5ОН > (GH3JSi(OG2H5J
-3NaCl
C6H6SiCl3-f 3CH3COONa > С6Н58}(ОСОСН3K
О
О О || О
! || || -СН3СС1 | ||
RSi-Cl+CH3C-O-CCH3 > RSi-OC-CH3
I I
Взаимодействием органохлорсиланов с аминами син-
синтезируют органоаминосиланы. Аммонолизом органо-
органохлорсиланов получают органосилазаны:
-2HG1
2(CH3KSiCl+NH3 > (GH3KSiNHSi(GH3K
При аммонолизе диорганодихлорсиланов наряду с ли-
линейными силазанами образуются органоциклосилазаны
(—R2SiNH—)n с п>Ъ.
В качестве М. к. при получении линейных полиорга-
носилоксанов, в частности эластомеров (см. Кремний-
органические каучуки), во многих случаях используют
циклосилоксаны. Последние образуются при гидроли-
гидролитич. конденсации диорганодихлорсиланов:
(CH3JSiCl2
+н2о
> [(GH3JSi(OHJ;
Г СН3 1
-Н2О I
> -Si-O-
L сн3
-f
-HG1
+ линейные полиметилсилоксаны
Образованию циклосилоксанов способствует проведе-
проведение реакции в кислой среде. Циклосилоксаны с раз-
различными диорганосилоксизвеньями м. б. синтезирова-
синтезированы гетерофункциональной конденсацией, напр.:
н5с6 сн3
\/
H5G. GeH5 Н,С СН3 H5Ge OSiO GH3
! Г "I I -2HG1 \/ \/
HOSiOSiOH -f- GISiOSi > Si Si
II II / \ / \
H3C CH3 H3G GH3 H3G OSiO GH3
H3G CH3
Низкомолекулярные циклосилоксаны м. б. получены
термокаталитич. деполимеризацией линейных и выс-
высших циклич. полиорганосилоксанов:
f-(GH3JSi-O-]n
Щелочь
— [-(CHa)aSi-O-l
307
МОНОМЕРЫ КРЕМНИЙОРГАНИЧЕСКИЕ
308
Перспективным является получение циклосилокса-
нов взаимодействием диметилдихлорсилана с метано-
метанолом:
ZnCl2
(CH3JSiCl2 + 2СН3ОН ^ 2СН3С1 + [-(GH3JSi-O-] + Н2О
Образующийся метилхлорид может быть направлен в
прямой синтез для получения метилхлорсиланов, что
делает весь цикл производства экономически весьма
выгодным.
Реакциями замещения в органич. радикале органо-
хлорсиланы м. б. превращены в различные карбофунк-
циональные М. к. Среди этих реакций наибольшее зна-
значение имеет хлорирование алкил- и арилхлорсиланов.
Алкилхлорсиланы хлорируют в присутствии ини-
инициаторов (перекиси, динитрил азоизомаслянойк-ты)
или под действием облучения. Варьируя соотноше-
ние реагентов и условия процесса (в частности, вы-
выводя целевой продукт из зоны реакции), можно на-
направленно синтезировать моно-, ди-, три- или поли-
хлорпроизводные алкилхлорсиланов.
Арилхлорсиланы хлорируют молекулярным хло-
хлором с использованием в качестве катализаторов
к-т Льюиса. Этим путем в пром-сти получают моно-,
ди-, три- и тетрахлорфенилтрихлорсиланы и соот-
соответствующие производные метилфенилдихлорси-
лана. Наиболее мягкими катализаторами, позво-
позволяющими значительно уменьшить степень побоч-
нои реакции
связи Si — С
'фенила
являются SbCl3 и 12. Хлоралкилхлорсиланы м. б.
расщепления
h "
использованы в реакциях дегидрохлорирования
для получения алкенилхлорсиланов, для крем-
неалкилирования ароматич. соединений и в ряде
др. реакций.
Проводя замещение по связи С — CI органич. ра-
радикала хлорорганоалкокси- или хлорорганоаци-
локсисиланов, можно получить М. к. с различными
карбофункциональными группами, напр.:
-НС1
(C2H6OJSi(CH3)CH2Cl + NH2GeH5 >
> (C2H5OJCH2SiCH2NHCeH5
О
||
(CH3COOJSi(CH3)CH2Cl + NaOG-СНз
О
II
» (CH3COOJCH3SiCH2OC -CH3
-NaCl
(G2H5OKSiGH2Cl + Р(ОС2Н5)з
-С2НбС1
(
II
(C2H5OKSiCH2P(OC2H5J
Свойства
При получении полиорганосилоксанов в боль-
большинстве случаев органич. заместители в молекулах
М. к. не претерпевают изменений, и природа их
в значительной степени определяет многие свой-
свойства полимеров. Важнейшими свойствами заме-
заместителей являются их химич. стабильность и проч-
прочность связи с кремнием. Прочность связи Si — С
(характеризуемая ее энергией) для различных
заместителей варьирует в широких пределах [в
кдж/молъ (в скобках — ккал/молъ)]:
Si-CeH5 -310 G4), Si-GH3-314 G5),
Si-CH=CH2 —297 G1), Si—G2H5 -260 F2),
Si-G3H7 -239 E7), Si-G4H9-21 8 E2 )
Фенилхлорсиланы и метилхлорсиланы имеют вы-
высокую термич. стабильность: заметный пиролиз
их происходит при темп-pax выше 500 °С. Нали-
Наличие функциональных групп в органич. замести-
заместителе понижает термич. устойчивость молекулы,
что особенно заметно для алкильных групп с
функциональностью у р-углеродного атома. Так, (Р-
хлорэтил)хлорсиланы разлагаются при 350—400 °С,
а (р-фторэтил)хлорсиланы уже при 150—250 °С. Как
правило, связь Si — С достаточно стабильна к оки-
окислению; в первую очередь окисляются связи С — Н.
Связь Si — С вследствие разницы в электроотрица-
электроотрицательности кремния и углерода A,8 и 2,5 в шкале По-
линга) достаточно полярна и склонна к расщеплению
под действием электрофильных и нуклеофильных аген-
агентов. Алкильные заместители легко отщепляются под
действием таких реагентов, как иод или галогенводо-
роды в присутствии каталитич. количеств к-т Льюиса.
Конц. H2SO4 может отщеплять метальные (наиболее
Свойства кремнийорганических мономеров
Мономер
Четыреххлористый кремний
SiCl4
Трихлорсилан
1,
HSiCl,
Метилтрихлорсилан
GH3SiGl3
Диметилдихлорсилан
(CH3JSiCl2 ....
Триметилхлорсилан
(GH3KSiGl
Метилдихлорсилан
GH3SiHGl2
Диметилхлорсилан
(GH3JSiHGl
Этилтрихлорсилан
253
Диэтилдихлорсилан
(G2H5JSiGl2
Этилдихлорсилан
G2H5SiHGl2
Диэтилхлорсилан
(G2H6JSiHGl
Винилтрихлорсилан
СН2 = CHSiCl3
Метилвинилдихлорсилан
(СН2 = GH) CH3SiCl2 . . .
Фенилтрихлорсилан
CeH5SiCl3
Дифенилдихлорсилан
(CeH5JSiCl2
Трифенилхлорсилан
(GeH5KSiGl
Фенилдихлорсилан
GeH5SiHGl2
Метилфенилдихлорсилан
(CeH5)CH3SiCI2
Диметилфенилхлорсилан
(С6Нб) (CH3JSiCl
Метилдифенилхлорсилан
(CeH5JCH3SiCl
Хлорметилтрихлорсилан
ClCH2SiCl3
Хлормэтил метилдихлорси-
метилдихлорсилан ClCH2(CH3)SiCl3 . . .
Хлорметилдиметилхлорси-
лан С1СН2 (СН3J SiGl . .
Хлорфенилтрихлорсилан
ClCeH4SiCl3
Метил(хлорфенил) дихлор-
силан (С1С6Н4) GH3SiCl2 .
Дихлорфенилтрихлорсилан
Gl2GeH3SiGl3
Дихлорфенилметилдихлор-
силан (Gl2GeH3)GH3SiGl2 .
тФТ°т
хлорсилан
F3GCH2GH2SiGl3
Метил-ууу-тРиФт°РпРопил~
дихлорсилан
(F3GGH2GH2) GH3SiCl2 . . .
{З-Цианэтилтрихлорсилан
GNGH2GH2SiGl3
Метил C-цианэтил) дихлор-
дихлорсилан
(GNGH2GH2) CH3SiCl2 . . .
Y-Аминопропилтриэток-
сисилан
H2NGH2GH2GH2Si (ОС2НбK-
Темп-ра
плавле-
плавления, °С
-70
-128
-90
-86
-40
-92,5
-111
-105,6
-96,5
-107
— 143
94-95
34
Темп-ра
кипения,
°С
57,6
31,8
66,1
72
57,7
41
36
100,5
129
75,4
99,7
92,5
92,5-93,0
201 ,5
304
378
182
205,5
193,5
295
118
121 ,5
115
230-245
230-240
267-275
262-270
113,5
121 ,5
109*
215
122*
d20
4
1,4810
1 ,3417
1,2750
1,0637
0,8580
1 ,1047
0,8874
1,2373
1 ,0504
1,0926
0,8895
1 ,2426
1,0868
1 ,3240
1,2216
-
1,2115
1 ,1866
1 ,0320
1,1277
1,4646
1,2858
1 ,0865
1
1
1
1
1
1
1
1
4280-
4570
2920-
3170
5450-
5520
4060 —
4200
1,3990
1 ,2610
-
1,1900
0
94 70
* При 30 мм рт. ст.
309
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
310
легко), этильные, циклоалкильные и другие замести-
заместители, напр.:
-сн4 н2о
O(CH2)nSi(CHa)s + H2SO4 > <D(CHa)n(CH8),Si(SO,H) >
—-> [O(GH2)n(GH3JSi]20
где Ф = NH2, COOR и др. Эта реакция дает возмож-
возможность легко получать различные бискарбофункцио-
нальные дисилоксаны. Наличие электроноакцепторных
заместителей в алкильном радикале способствует ста-
стабилизации связи Si — С к действию электрофильных
реагентов, однако облегчает отщепление карбофунк-
циональной алкильной группы нуклеофильными реа-
реагентами. Так, хлорметильная группа легко отщепляется
сильными щелочами, трихлорметильная — даже хо-
холодной водой.
Гетеролитич. отщепление арильных заместителей
происходит значительно легче, чем алкильных. Нали-
Наличие у кремния наряду с арильным радикалом электро-
электроотрицательных заместителей стабилизирует связь
Si — Сарил. Так, если фенильная группа в фенилтри-
алкилсиланах отщепляется под действием HG1 в мяг-
мягких условиях, то дифенилдихлорсилан стабилен к та-
такому воздействию даже в кипящей уксусной к-те. К-ты
Льюиса, особенно А]С13, катализируют отщепление
арильной группы в арилсиланах при действии галоге-
галогенов, галогенводородов, галогенацилов, BF3 и др. реа-
реагентов. Хлористый алюминий при нагревании с экви-
молярным количеством фенилтрихлорсилана легко раз-
разрушает последний с образованием SiCl4. Электроноак-
цепторные заместители в арильной группе, как и для
алкильных групп, стабилизируют связь Si — С к дейст-
действию электрофильных реагентов и облегчают ее расщеп-
расщепление под действием нуклеофильных.
Органохлорсиланы за небольшим исключением пред-
представляют собой жидкости, легко гидролизующиеся
во влажной атмосфере с выделением НС1. Органоалкок-
си- и органоацилоксисиланы менее чувствительны к вла-
влаге воздуха. Органоциклосилоксаны и циклокарбосила-
ны водой не гидролизуются. Нек-рые физико-химич.
константы основных М. к., выпускаемых пром-стью,
приведены в таблице.
Лит.: Андрианов К. А., Методы элементоорганиче-
ской химии. Кремний, М., 1968; П е т р о в А. Д. [и др.], Синтез
кремнийорганических мономеров, М., 1961; Борисове. Н.,
ВоронковМ.Г., Лукевиц Э. Я., Кремнеэлементоорга-
нические соединения. Производные неорганогенов, Л., 1966;
Voorhoeve R. J. H., Organohalosilanes. Precursors to si-
licones, Amst.— [a. o.l, 1967; E a b о r n C, Organosilicon com-
pounds,L., 1960; Bazant V. [a. o.], Organosilicon compounds,
v. 1, v. 2 A); v. 2 B), Prague, 1965. E. А. Чернышев.
МОРОЗОСТОЙКОСТЬ полимерных мате-
материалов (freeze resistance, Frostbestandigkeit, resi-
resistance au froid)—способность этих материалов сохранять
при низких темп-pax свои эксплуатационные свойства.
Критерии М. различны в соответствии с различием
требований к материалу, связанных как с условиями
его работы, так и с его исходными свойствами. Для
стеклообразных полимеров М.— это отсутствие хруп-
хрупкости. Для высокоэластич. полимеров (наряду с этим
критерием) основным является требование ограничен-
ограниченного снижения деформируемости (податливости или
разрывного удлинения) или ограниченного повышения
твердости (модуля упругости), сохранения восстанав-
восстанавливаемости или коэфф. термич. расширения, отсутст-
отсутствия значительной кристаллизации (для кристаллизую-
кристаллизующихся каучуков). Т. обр., для эластомеров М. означает
сохранение высокой эластичности. Поэтому для них
температурной границей М. является стеклования
температура, тогда как для стеклообразных полиме-
полимеров — хрупкости температура.
Для практич. целей важна не только температурная
граница М., но и степень сохранения тех или иных
свойств при данной (низкой) темп-ре по сравнению
с этими же свойствами при комнатной темп-ре. Этот
критерий обычно выражают коэфф. М. К = -^— (где
ХТ и Х2о — значения данного показателя при низкой
температуре Т и комнатной температуре). Величина
коэффициента М. (заключенная в пределах от 0 до 1)
не одинакова при оценке М. по изменению различ-
различных показателей.
Т. обр., М. данного материала м. б. охарактеризована
либо в целом — температурной границей появления
хрупкости или стеклования, либо частично — величи-
величиной коэффициента М. для данного показателя при дан-
данной температуре.
На практике М. выражают также способностью мате-
материала выдерживать без растрескивания разовое охлаж-
охлаждение до заданной темп-ры в течение определенного
времени или многократные циклы охлаждения и нагре-
нагревания. Эти испытания проводят, когда материал нахо-
находится в ненапряженном состоянии либо деформируется
будучи охлажденным (так испытывают пленки, изгибае-
изгибаемые вокруг стержня с определенным диаметром). Иног-
Иногда представляет интерес сохранение электрич. сопро-
сопротивления, напр, для иоли-га-ксилилена, в к-ром при
темп-ре жидкого гелия можно ожидать появления
сверхпроводимости.
М. полимерных материалов зависит от продолжи-
продолжительности (скорости, частоты) нагружения, поскольку
от нее зависят темп-ры стеклования и хрупкости,
а также другие (испытуемые) свойства материала.
Темп-pa Тк, отвечающая данному коэфф. М., связана
с продолжительностью t или частотой со нагружения
соотношением ^-; = А + Blgt = С — i?lgco (где А,
В, С — константы), в чем проявляется суперпозиции
принцип температурно-временной.
Методы определения М. резин и пластмасс, т. е. тех
классов полимерных материалов, для к-рых этот пока-
показатель наиболее важен, унифицированы (см. Испытания
резин, Испытания пластических масс). с. Б. Ратнер.
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ ЛАКИ И ЭМА-
ЭМАЛИ, мочевино-алкидные лаки и
эмали, алкидно-карбамидные лаки
и эмали (urea-formaldehyde varnishes and enamels,
Harnstoff-Foraialdehydlacke und Emaillen, vernis et
emaux uree-formaldehyde) — лакокрасочные материалы
на основе смесей частично бутанолизированной мочеви-
но-формалъдегидной смолы (ее содержание в смеси плен-
пленкообразующих составляет обычно до 35%) с гидроксил-
содержащим полиэфиром, чаще всего с тощей алкидной
смолой. Отверждение таких материалов осуществляется
в результате взаимодействия функциональных групп
мочевино-формальдегидной и алкидной смолы; послед-
последняя служит также пластификатором лакокрасочных
пленок. М.-ф. л. и э. могут отверждаться при 100—
140 °С или при обычных темп-рах.
Материалы холодной сушки изготовляют одно- или
двухкомпонентными (двухупаковочными). Последние
состоят из лаковой основы и кислотного катализатора
отверждения, к-рый вводят в лакокрасочный материал
непосредственно перед его нанесением. Однокомпонент-
ные материалы содержат в своем составе нитроцеллю-
нитроцеллюлозу, к-рая ускоряет пленкообразование, пластифика-
пластификаторы (фосфаты, фталаты или касторовое масло) и отвер-
дитель — ортофосфорную к-ту. Жизнеспособность та-
таких материалов достигает 12 мес. Продолжительность
их отверждения при комнатной темп-ре 1,0—1,5 ч,
при 40—60 °С — 20—30 мин. Покрытия из этих мате-
материалов, применяемые гл. обр. для отделки деревянной
мебели, превосходят нитроцеллюлозные по твердости,
водостойкости и стойкости при высоких и низких
темп-pax. См. также Алкидные лаки и эмали.
М. М. Голъдберг.
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ (urea-
formaldehyde resins, Harnstoff-Formaldehyd-Harze, re-
311
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
312
sines uree-formaldehyde) — олигомерные про-
продукты поликонденсации мочевины с формаль-
формальдегидом, способные превращаться в простран-
пространственные (сшитые) полимеры.
Получение. Механизм образования М.-ф. с.
детально не выяснен. Однако установлено, что
при поликонденсации мочевины с формальдеги-
формальдегидом в водном р-ре в зависимости от рН среды,
соотношения исходных компонентов, продол-
продолжительности реакции и темп-ры м. б. получе-
получены различные продукты. Так, в щелочной
среде (рН 11 — 13) даже в разб. р-рах обра-
образуется монометилолмочевина H2NCONHCH2OH;
в нейтральных и слабощелоч-
слабощелочных средах (рН 7—8) в зависимости от
соотношения исходных компонентов — моно- и
диметилолмочевины OG(NHGH2OHJ. Последняя
образуется также при взаимодействии мономе-
тилолмочевины с формальдегидом. Положение
равновесия этих реакций не зависит от рН
среды, а определяется концентрацией реаги-
реагирующих веществ и темп-рой.
При взаимодействии мочевины с формальде-
формальдегидом в сильнокислой среде (рН<3)
образующиеся метилолмочевины сразу же под-
подвергаются дегидратации, давая метиленмочеви-
ны, например NH2 — C=N, которые в условиях
I I
О—СН2
реакции быстро превращаются в полиметилен-
мочевины общей ф-лы (G2H4N2O)n — неплавкие
и нерастворимые аморфные продукты, не имею-
имеющие практич. значения. Поэтому для получения
М.-ф. с. процесс следует проводить в условиях,
способствующих образованию метилольных про-
производных мочевины.
Моно- и диметилолмочевины — белые кри-
сталлич. продукты, растворимые в воде и мета-
метаноле; первый плавится при 111 °С (из этано-
этанола), второй — при 121 — 126 °С (из 80%-ного
этанола); диметилолмочевина при нагревании
растворяется также в этаноле. При нагревании
безводные моно- и диметилолмочевины превра-
превращаются в полиметиленмочевииы; первая
ностью переходит в нерастворимый продукт при
100 °С, вторая — выше 140 °С. В водных кислых
р-рах (рН 4,5—6,0) метилолмочевина способна
к дальнейшим превращениям с образованием,
вероятно, метилен-бггс-амида (I), метилолметилен-
бис-амида (II) или простого эфира (III) и азоме-
тилена (IV), к-рый сразу же тримеризуется:
-н2о
NH2CONHGH2OH + NH2GONH2 * NH2GONHGH2NHGONH2
GH2OH
-H2O /
> NH2GONHGH2N
NH-CHjf-NH
CO CO
I I
NHCH.OH NE
NH2
CO
I y CO CO
I / NH2 NH—CH2-
~N— 6h2-NH
NH-CH2-N-
NH-CH2-N-CH2OH
CO
CO
NH,
NH9
CO
-NH
NH2
CO
I
CO
NH2
NH2
CO
I
nh—ch2—n—сн2— n~
H-CH
—CH2-N-CH2-N—CH2-N ~
I
CO
NH2
CO
I
NH,
NH2CO—N
CH
CH2
N—CONH2
N
CONH2
CH
~HNOC—N
NH-
co
N
? CH2
I
N-CO
/ I
CH2 NH
-CH2
CH2O
NH
CO
N
CH?
I 2
9—N
сн2
^CH, NH
~HNOC—N N—CO
CH2 CH2
Y
CONHCH2-
? CH2
I 2 I
C9—N N--CONHCH2~
NH CH2
CH2
NH CH2
I
I
CO
N
CH2
N— CONHCH2«v
ПОЛ- HOCH2HNOC
N
CH2
CH2
N
CONHCHo-
CONHCH2OCH2NHCONH- CONHCH2~
N N
сн2 сн2 сн2 сн2
N-CONHCH2OCH2NHCO—N N-CONHCH2NHCO—N N—CONH-
CH2
N
2NH2GONHGH2OH -
\
G
GONH2
>NH2CONHCH2OCH2NHCONH2
-H2O III
GONH2
N
-H2O / \
3NH2GONHGH2OH > 3NH2GON=CH2—> GH2 GH2
IV I i
H2NOGN N-CONHa
GH2
В аналогичных условиях скорость гомополиконденса-
ции диметилолмочевины очень низка. Она также вза-
взаимодействует с мочевиной и монометилолмочевиной.
Предполагается, что основная реакция, приводящая
к М.-ф. с,— бимолекулярная, и скорость ее пропорцио-
пропорциональна концентрации водородных ионов. Далее приве-
приведены вероятные схемы образования М.-ф. с.
По-видимому, наиболее вероятна последняя схема,
предусматривающая наличие в структуре полимера
кислородных мостиков, присутствием к-рых можно
объяснить выделение формальдегида при хранении и
эксплуатации изделий из М.-ф. с.
Направление реакции мочевины с формальдегидом за-
зависит также от темп-ры. Повышение ее выше 40 °С
(оптимальная темп-pa для получения метилолмочевины)
способствует образованию нежелательных продуктов —
метиленмочевин. Вероятность получения последних
существует и в том случае, когда процесс начинают
в условиях, благоприятных для синтеза метилолмоче-
вин; это связано с изменением рН среды в ходе реакции.
Так, мочевина, способная образовывать с к-тами нестой-
нестойкие соли, связывает муравьиную к-ту, всегда содер-
содержащуюся в формалине. В результате этого при добавле-
добавлении мочевины в р-р формалина рН реакционной смеси
повышается. Однако по мере расходования мочевины
к-та высвобождается и рН понижается. Кроме того,
в условиях реакции муравьиная к-та образуется из
формальдегида (реакция Канниццаро — Тищенко):
2СН2О + NaOH -* CH3OH -f HGOONa
313
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
314
По этой причине сначала получают метилольные произ-
производные мочевины, поддерживая нейтральную или слабо-
слабощелочную среду (рН 7—8), а затем, не выделяя метилол-
мочевины из р-ра, в слабокислой среде (рН 3—6,5) осу-
осуществляют их поликонденсацию. Слабокислая среда
предотвращает чрезмерное нарастание вязкости смолы
и преждевременное гелеобразование (особенно на ста-
стадии сушки). Для регулирования рН среды используют
буферные вещества (ацетат натрия и его смесь с лимон-
лимонной к-той, карбонат аммония и др.), а также уротро-
уротропин. При нагревании мочевины и формальдегида в при-
присутствии последнего р-р с течением времени приобре-
приобретает ту кислотность, к-рую он имел до прибавления
уротропина. По-видимому, уротропин образует с му-
муравьиной к-той буферные соли, разрушающиеся при
нагревании. Проведение реакции в водных средах пре-
препятствует отщеплению воды от метилолмочевин, что
предотвращает их переход в метиленмочевины.
Технология производства М.-ф. с.
в значительной мере определяется их назначением.
Для синтеза М.-ф. с. обычно используют 30%-ные вод-
водные р-ры формальдегида, содержащие ок. 1% мета-
метанола. С повышением концентрации формадьдегида уве-
увеличивается содержание метанола; напр., 40%-ные р-ры
содержат его до 10%. Метанол способствует образова-
образованию метилированных смол невысокой мол. массы, лег-
легко разрушающихся водой после отверждения. Кроме
того, с увеличением содержания метанола в формалине
возрастает абсорбция паров воды отвержденными смо-
смолами (с 2% при использовании безметанольного вод-
водного р-ра формальдегида до 6—10% при содержании
метанола 8%). Молярное соотношение мочевина/
формальдегид колеблется в пределах 1/1,3—1/1,8.
Темп-рный режим существенно зависит от назначения
смолы. Проводя поликонденсацию при темп-ре ок.
40 °С, получают связующее для пресс-порошков (см.
Аминопласты). Смолы, используемые в произ-ве сло-
слоистых пластиков, и смолы, модифицированные спир-
спиртами (см. ниже), синтезируют при 70—120 °С.
Содержание свободного формальдегида в М.-ф. с. ко-
колеблется в пределах 0,9—4,0%. Смолу, содержащую
минимальное количество свободного формальдегида,
получают методом поликонденсации с постепенным
вводом мочевины (тремя порциями) с таким расчетом,
чтобы исходное молярное соотношение мочевина/фор-
мочевина/формальдегид, равное 1/2,2, после загрузки третьей порции
мочевины составляло 1/1,55. Процесс проводят при рН
6—6,2 и темп-ре 90—92 °С.
Смолы с мол. массой, в 2—2,5 раза превышающей
обычное значение, получают в присутствии полимерных
карбоксилсодержащих катионитов, в ионогенные груп-
группы к-рых введены ионы переходных металлов с неза-
незаполненной d-орбиталью. Такие М.-ф. с. содержат
всего 0,5—1% формальдегида, а после отверждения при
кипячении в воде теряют лишь 12—13 мг/дм2 связанного
формальдегида (обычно 40—50 мг/дм2), при кипячении
в 1%-ном р-ре уксусной к-ты — до 3—4 мг/дм2 (обыч-
(обычно 15—20 мг[дм2).
Полученный олигомер обрабатывают аммиачной во-
водой, что увеличивает продолжительность хранения го-
готового продукта, к-рая составляет обычно 2—12 мес.
Основная масса М.-ф. с. выпускается и находит приме-
применение в виде водных р-ров.
Порошкообразные («сухие») смолы более стабильны
при хранении (до года и более), чем водные р-ры. Кро-
Кроме того, их удобнее транспортировать. Однако произ-
производство таких смол весьма сложное и дорогостоящее.
Их получают путем центробежного, механич. или пнев-
матич. распыления жидкой смолы и сушки ее в потоке
горячего воздуха, перегретого пара и др. Объем про-
производства таких смол невелик.
Свойства. М.-ф. с.— твердые продукты белого цвета,
легко растворимые в воде и не растворимые в органич.
растворителях. Отверждение М.-ф. с. ускоряется в при-
присутствии кислотных катализаторов и с повышением
темп-ры. В качестве катализаторов используют как ор-
органические (щавелевая, фталевая), так и минеральные
(фосфорная, соляная) к-ты и нек-рые соли (А1С13,
ZnCl2). Продукты отверждения — бесцветные, свето-
светостойкие, легко окрашивающиеся полимеры (см. также
Аминопласты, Карбамидные клеи). Смолы, отверж-
денные при низких темп-pax даже в присутствии боль-
больших количеств катализатора, имеют пониженную во-
водостойкость. При повышении темп-ры отверждения
водостойкость возрастает. Однако продукты, получен-
полученные в оптимальном режиме A20—140 °С, катализатор),
все же частично разлагаются под действием горячей
воды или водных р-ров солей. Это обусловлено недоста-
недостаточной разветвленностью цепей и малым кол-вом попе-
поперечных связей, о чем свидетельствует низкое коксовое
число продуктов отверждения A4—21,5%) и их быст-
быстрая деструкция при нагревании без доступа воздуха.
Существенный недостаток М.-ф. с.— выделение фор-
формальдегида в процессе переработки и при эксплуатации
отвержденных смол. Это объясняется наличием в мате-
материале формальдегида, не прореагировавшего при поли-
поликонденсации, а также образованием его вследствие на-
наличия в полимере метилольных групп и метиленэфир-
ных связей, превращающихся в метиленовые. Формаль-
Формальдегид оказывает токсич. действие на организм человека
и вызывает растрескивание изделий.
Отвержденные М.-ф. с. (в отличие от феноло-формаль-
дегидных) прозрачны даже тогда, когда в них содер-
содержится 10—15% воды. Вода находится в полимере в дис-
диспергированном состоянии и химически не связана с ним.
Она постепенно испаряется даже при комнатной темп-ре,
причем происходит усадка и растрескивание материала.
Поэтому для удержания нек-рого количества воды
в М.-ф. с. иногда вводят гидрофильные добавки (поли-
(поливиниловый спирт, крахмал, белковые вещества) и твер-
твердые наполнители (напр., древесную муку), препятст-
препятствующие усадке полимера.
Модификация. Для повышения водостойкости, при-
придания способности растворяться в органич. растворите-
растворителях, увеличения гидрофобности и адгезии или для
улучшения совместимости с др. полимерами и компо-
компонентами, входящими в состав лаков, эмалей, клеев,
М.-ф. с. обычно модифицируют. Модификация м. б.
осуществлена на стадии синтеза олигомеров путем ча-
частичной этерификации по метилольным группам одно-
одноатомными (бутиловым, фуриловым) или многоатомными
(чаще всего этилен-, диэтилен- и триэтиленгликолями)
спиртами, а также путем замены части мочевины в ре-
реакционной смеси на фенол или меламин. При этери-
этерификации сначала образуются, вероятно, неполные
эфиры диметилолмочевины HOCH2NHCONHCH2OR,
к-рые вступают в поликонденсацию, превращаясь в
смолу, растворимую в органич. растворителях:
NHGH2OR
п СО
NHGH2OH
ROGH2NH
!
GO
NHGH2OR
NHGH2OH
NH-CII
[I "I I
GO CO
-N-CH2- -N-CH2
J n-2
-N-CH2OH
Отверждение такого олигомера сопровождается от-
отщеплением спирта.
Модификация М.-ф. с. бутиловым спиртом
м. б. осуществлена несколькими методами. Для метода,
основанного на нагревании предварительно синтезиро-
синтезированных эфиров диметилолмочевины, характерны отно-
относительно невысокие выходы олигомеров (до 60%).
Лучшие выходы достигаются при поликонденсации
мочевины с формальдегидом (молярное соотношение г/2)
в присутствии 10-кратного количества бутилового спир-
спирта (от массы мочевины). Реакция протекает при темп-ре
кипения смеси, причем из сферы реакции непрерывно
315
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
316
выводятся пары воды и спирта (бутиловый спирт обра-
образует азеотроп с водой), к-рые, конденсируясь, направ-
направляются в отстойник; из последнего бутиловый спирт
непрерывно возвращается в зону реакции. По мере уда-
удаления воды темп-pa реакционной смеси непрерывно по-
повышается, и при 117,5 °С (темп-pa кипения бутилового
спирта) начинает отгоняться спирт. Процесс заканчи-
заканчивается при получении 60—70%-ного р-ра олигомера
в бутиловом спирте. Иногда для предотвращения обра-
образования неплавких и нерастворимых продуктов в про-
процессе поликонденсации добавляют небольшое количест-
количество регулятора рН среды — гексаметилентетрамина.
М.-ф.с,модифицированные бутиловым спиртом, мож-
можно получать также и по иной технологии. Сначала при
кипячении формалина со спиртом синтезируют бутил-
формаль. Затем добавляют водный р-р мочевины и
продолжают кипячение. Спустя 1 ч отгоняют в вакуу-
вакууме азеотропную смесь воды и бутилового спирта, а
затем спирт до достижения требуемой вязкости р-ра.
Модификация бутиловым спиртом придает М.-ф.с. спо-
способность хорошо совмещаться с алкидными, полиэфир-
полиэфирными и эпоксидными смолами, с нитроцеллюлозой и др.
полимерами, а также с растительными маслами. Лаки
на основе таких смол образуют светостойкие, с высо-
высокой поверхностной твердостью покрытия.
При модификации М.-ф. с. фуриловым спир-
спиртом [молярное соотношение мочевина/формальдегид
составляет A —1,75)/3,0] сначала осуществляют конден-
конденсацию мочевины с формальдегидом в щелочной среде
(рН 8,0—9,0) при кипении реакционной смеси (96—
98 СС). При этом контактные газы (после каталитич.
окисления метана или метанола), содержащие формаль-
формальдегид, поглощают водным р-ром мочевины. Завершают
процесс в том же темп-рном интервале в кислой среде
(рН 4,9—5,3). После достижения вязкости по вискози-
вискозиметру ВЗ-4 13,3—14,0 сек (коэфф. рефракции 1,419 —
1,422) рН р-ра доводят 2 н. р-ром щелочи до 7,0—8,0
и затем упаривают в вакууме до вязкости 18—20 сек
(по ВЗ-4). К этому т. наз. конденсационному р-ру до-
добавляют фуриловый спирт в количестве 15—50% от
общей массы р-ра и доводят рН до 7,5—8,5. Получен-
Полученные р-ры обладают низкой вязкостью. Применение
газофазного метода позволяет исключить стадию полу-
получения формалина, использовать тепло, выделяющееся
при растворении формальдегида, на проведение поли-
поликонденсации и исключить стадию выпаривания лишней
воды, снизив тем самым энергозатраты.
Модифицированные фуриловым спиртом М.-ф. с.
(т. наз. карбамидно-фурановые смолы) легко совме-
совмещаются с наполнителем (песком), обеспечивая проч-
прочность и термостойкость отвержденных композиций
(в частности, стержней для литья).
Модификация М.-ф. с. гликолями обеспечи-
обеспечивает повышение адгезии. Произ-во таких олигомеров
мало отличается от технологии получения немодифи-
цированных М.-ф.с. и осуществляется на том же обору-
оборудовании (см. рисунок).
Смесь исходных компонентов (рН 6,5—7) из смеси-
смесителя закачивается насосом в напорную емкость, из
к-рой самотеком поступает в реактор; туда же подается
гликоль. Темп-pa греющей поверхности реактора со-
составляет 120—140 °С, а темп-pa реакционной смеси
105—115 °С. Реактор снабжен холодильником^ к-рый
при пуске аппарата работает как обратный, а в тече-
течение всего процесса — как прямой, что обеспечивает
постоянную отгонку воды из получаемого р-ра смолы.
Готовый олигомер непрерывно поступает в аппарат,
куда также подается 25%-ная аммиачная вода A ч. на
3 ч. смолы). Содержание сухого вещества в готовом
олигомере составляет не менее 70%, вязкость по виско-
вискозиметру ВЗ-1 30—50 (мн-сек)/м2, или спз.
Модификация М.-ф. с. поливинилацетат-
ной эмульсией осуществляется путем добав-
добавления эмульсии в готовую смолу или в реакционную
смесь при поликонденсации. В последнем случае взаи-
взаимодействие мочевины с формальдегидом осуществляют
вначале в щелочной среде, вводят эмульсию и завер-
завершают процесс в кислой среде (рН снижается при введе-
введении эмульсии). Молярное соотношение мочевина/фор-
мочевина/формальдегид составляет A —1,5)/2,0; с увеличением
молярной доли формальдегида увеличивается продол-
продолжительность отверждения и снижается содержание сво-
Глиноль
Мочевина
i
Схема непрерывного процесса
производства мочевино-формаль-
дегидной смолы, модифициро-
модифицированной гликолем: 1 — смеси-
смеситель; 2 — сборник конденсаци-
конденсационного р-ра; з, 7 — сетчатые фильтры; 4 — реактор; 5 —
холодильник кожухотрубный; 6 — весовой мерник гликоля;
8 — аппарат для стабилизации.
бодного формальдегида в готовой смоле. Модификация
клеевых М.-ф. с. поливинилацетатной эмульсией спо-
способствует снижению хрупкости клеевого шва и повыше-
повышению его прочности.
Применение. М.-ф. с. применяют в качестве связую-
связующих в производстве аминопластов, слоистых пластиков
и для изготовления клеев (см. Карбамидные клеи).
М.-ф. с, модифицированные бутиловым спиртом, ис-
используют в смеси с др. пленкообразующими для приго-
приготовления электроизоляционных, декоративных, анти-
антикоррозионных лакокрасочных материалов (см. Моче-
вино-формальдегидные лаки и эмали).
М.-ф. с, модифицированные фуриловым спиртом, ис-
используют для изготовления литейных стержней для
алюминиевого и чугунного литья. Они экономически
выгодны в высокопроизводительном литейном произ-
производстве.
М.-ф. с, модифицированные поливинилацетатной
эмульсией, используют в производстве карбамидных
клеев и в качестве связующих в произ-ве слоистых
пластиков, к к-рым предъявляются высокие требования
по водостойкости.
Водоэмульсионные М.-ф. с, модифицированные глице-
глицерином, способствующим снижению хрупкости полиме-
полимера, используют для получения тепло- и звукоизоляци-
звукоизоляционного пенопласта — мипоры.
В СССР производятся М.-ф. с. марок МФ, М, УКС,
б а р т р е в. За рубежом М.-ф. с. выпускают под на-
названиями: аутосет, формасет, циакор
резин, урак резин, америте (США);
фиталак, каларок (Великобритания); и п о р-
ка,бек уро л, фестопас, элмопласт (ФРГ);
г е д а м и н (Франция); аминоцел (Италия);
с и д о н у р (США, Канада, Великобритания, ФРГ),
д е л а м и н (Австрия) и др.
Лит.: Николаев А. Ф., Синтетические полимеры и
пластические массы на их основе, М., 1964; X у в и н к Р.,
Ставерман А. [сост.], Химия и технология полимеров,
пер. с нем., т. 2, ч. 2, М., 1966; Голдинг В., Химия и
технология полимерных материалов, пер. с англ., М., 1963;
Bachmann A., Bertz Т., Aminoplaste, 2 Aufl., Lpz.,
1970; Технология пластических масс, М., 1972. Г. М. Цейтлин.
МЯГЧИТЕЛИ — см. Пластификаторы.
н
НАБУХАНИЕ полимеров (swelling, Quellung,
gonflement) — увеличение объема (массы) полимеров
в результате поглощения низкомолекулярной жид-
жидкости или ее паров. Неограниченным Н. иногда наз.
процесс растворения полимера, однако правильнее
применять термин «набухание» только в тех случаях,
когда жидкость поглощается ограниченно, или к стадии,
предшествующей растворению, поскольку, как правило,
представление о Н. связывается с сохранением общей
формы образца.
Количественные характеристики Н.— степень Н.
и скорость Н. Степень Н. (а) определяют отно-
отношением массы (объема) поглощенной полимером жид-
жидкости к массе (объему) исходного полимера (пг{)):
т0 .
где т — масса (объем) набухшего образца. В случа-
случаях, когда т > т0, степень Н. часто выражают отно-
отношением га/га0.
Скорость Н. dxldx (т — продолжительность Н.)
обычно имеет максимальное значение в начале процесса.
При ограниченном Н. по мере приближения системы
к состоянию равновесия dx/dT—> 0, а а—> атах (атах —
равновесная, или максимальная, стейень Н.). Кине-
Кинетика Н. может быть описана различными ур-ниями.
Для простейшего случая (кривая 1 на рис. 1) широко
используют дифференциальное ур-ние
dx/dz = Цатах—ссх )
или его интегральную форму
к = -1 In amax
т amax—ат
где к — константа скорости Н.; ат — степень Н. ко
времени т. В нек-рых случаях (см. ниже) после дости-
достижения максимальной степени Н. полимер «отдает»
часть поглощенной жидкости в результате перестройки
своей структуры и протекающих при этом релаксаци-
релаксационных процессов (см. рис.
1, кривую 2).
Изотропные полимер-
У2 ные материалы, у к-рых
нет ориентации макромо-
, лекул в каком-либо на-
правлении, сохраняют при
Н. исходную форму. При
Рис. 1. Кинетические кривые
набухания полимеров: 1 —
типичная кривая набухания;
2—кривая набухания, ослож-
осложненного релаксационными
процессами.
Продолжительность набухания
Н. анизотропных полимерных материалов (пленок и
особенно волокон) размеры образца в направлении,
перпендикулярном оси ориентации, изменяются значи-
значительно больше, чем вдоль этой оси. В нек-рых случаях
наблюдается даже усадка материала вдоль оси. Коли-
Количественно анизотропия Н. полимеров характеризуется
отношением продольного удлинения (усадки) к измене-
изменению поперечных размеров. Это отношение может слу-
служить косвенным показателем степени ориентации по-
Рис. 2. Схематичес-
Схематическая кривая продоль-
продольной усадки при на-
набухании искусственного волокна,
подвергнутого ориентационной вы-
вытяжке.
лимерного материала. При Н. искусственных волокон,
сохраняющих после формования внутренние напря-
напряжения, размеры вдоль оси волокна изменяются во
времени по сложной кривой (рис. 2). При этом сте-
степень Н. волокна на
всех стадиях процесса Продолжительность набухания
больше 1 (здесь и да-
далее за степень Н. при-
принято отношение т/га0).
При Н. в условиях
сохранения постоянно-
постоянного объема полимера
внутри образца разви-
развиваются очень высокие
давления. При погло-
поглощении полимером пер-
первых порций жидкости
(до 3—5% по массе)
давление набухания составляет несколько
сот Мн/м2 (несколько тысяч кгс/см2), а при поглощении
двух-четырехкратного количества оно уменьшается до
0,15—0,2 Мн/м2 A,5—2 кгс/см2). Установлению равно-
равновесия отвечает нулевое давление Н.
При взаимодействии полярных полимеров с поляр-
полярными низкомолекулярными жидкостями поглощение
первых порций жидкости сопровождается выделением
довольно большого количества тепла; дальнейшее по-
поглощение сопровождается незначительным тепловым
эффектом. Н. гибкоцепных неполярных полимеров в не-
неполярных жидкостях не связано с существенными
тепловыми эффектами, т. к. происходит в основном за
счет изменения энтропии системы.
Набухание аморфных полимеров. Теоретически сте-
степень Н. аморфного полимера, свободного от остаточных
внутренних напряжений и с однородным молекулярно-
массовым распределением по всему объему, определя-
определяется положением точки на диаграмме состояния систе-
системы полимер — низкомолекулярная жидкость (рис. 3).
Если через х0 обозна-
обозначена доля
(по массе)
при темп-ре
полимера
в системе
пень набухания по мае- о
Ж
Рис. 3. Диаграмма состоя-
состояния системы полимер (JT)—
низкомолекулярная жид-
жидкость (Ж): Тк — критич.
темп-ра совместимости
компонентов; Го— темп-ра
опыта; хо — равновесная
концентрация полимера
в набухшей системе при
темп-ре опыта. Область
распада системы на две
фазы заштрихована.
се (в %) составит — 100. Выше критич. темп-ры сме-
х0
шения понятие о равновесной степени Н. не имеет
смысла, т. к. полимер и низкомолекулярная жидкость
совместимы в любых соотношениях.
Состояние равновесия при Н. аморфного полимера
термодинамически отвечает равенству химич. потенциа-
потенциалов низкомолекулярной жидкости в чистом виде и в на-
Состав
319
НАДМОЛЕКУЛЯРНАЯ СТРУКТУРА
320
бухшем полимере. Для чистой низкомолекулярной жид-
жидкости относительное давление паров над набухшим по-
полимером в положении равновесия равно единице. Сни-
Снижение этого давления в результате введения в жид-
жидкость каких-либо растворимых веществ должно приво-
приводить, в принципе, к уменьшению степени Н. полимера.
Это явление осложняется, однако, если вводимый
в жидкость компонент активно взаимодействует с поли-
полимером (напр., является растворителем для полимера).
Схематически изменение степени Н. при введении ново-
нового компонента можно передать диаграммой состояния
трехкомпонентной системы (рис. 4). Пусть чистая низ-
низкомолекулярная жидкость
Жх вызывает Н. полимера Л
до значения, определяемого
положением точки а. При
Рис. 4. Диаграмма состояния
системы полимер (Я) — низко-
низкомолекулярная жидкость (Жх) —
растворитель (Ж2): а — началь-
начальный состав набухшего поли-
полимера: б — суммарный состав
системы после введения раство-
растворителя ; в — конечный состав
набухшего полимера.
введении третьего компонента Ж2, к-рый является раст-
растворителем для полимера, в количестве, отвечающем точ-
точке б, концентрация полимера в р-ре и соответственно
степень Н. его определится положением точки в на соот-
соответствующей кривой равновесия.
Степень Н. аморфных полимеров обычно возрастает
с темп-рой, что объясняется увеличением подвижности
компонентов системы. Но для нек-рых систем, напри-
например целлюлоза (или ее низкозамещенный эфир) —
вода, степень Н. понижается с повышением температу-
температуры. Это является следствием разрушения при по-
повышенных температурах водородных связей между во-
водой и полимером.
Степень Н. аморфных полимеров в существенной
степени зависит от предыстории образца. В пленках и
волокнах, полученных испарением растворителя из
однофазных р-ров полимера, могут сохраниться внут-
внутренние напряжения, возникшие в момент перехода
в стеклообразное состояние. При этом промежуточные
значения степени Н. могут оказаться более высокими,
чем равновесная величина ее (см., напр., рис. 1, кри-
кривую 2). Более значительным оказывается влияние на Н.
предыстории образца полимера в тех случаях, когда
при его получении система застудневала. В студне,
полученном из р-ра полимера в результате охлаждения
или добавления вещества, не являющегося растворите-
растворителем, при высушивании сохраняются большие внутрен-
внутренние напряжения, вызванные неравновесным характе-
характером удаления растворителя, а также распадом системы
на две фазы. Набухающий образец стремится восста-
восстановить тот объем и форму, к-рые были присущи студню
перед высушиванием. Так, степень Н. желатины тем
выше, чем ниже была исходная концентрация р-ра, из
к-рого получался студень. Если же получить образец
желатины из р-ра путем испарения воды при темп-ре
выше точки застудневания (расположенной в интерва-
интервале 33—38 °С), то степень последующего Н. в воде рез-
резко уменьшается.
Гистерезисные эффекты при Н. полимеров, получен-
полученных высушиванием студней, отчетливо проявляются
также при растворении желатины. Если пластинку же-
желатины (или столярного клея) погрузить в горячую во-
воду, то на ее поверхности образуется слой гомогенного
конц. р-ра, к-рый препятствует проникновению воды
в толщу пластинки, и требуется очень продолжитель-
продолжительное время для перевода всей массы образца в р-р. С дру-
другой стороны, Н. пластинки в холодной воде протекает
очень быстро благодаря релаксационному восстанов-
восстановлению объема, к-рый студень занимал до сушки. Про-
Проникновение воды внутрь пластинки способствует по-
последующему быстрому растворению желатины при на-
нагревании.
Степень Н. трехмерных полимеров зависит от часто-
частоты сетки. Так, для слабовулканизованного каучука
степень Н. в углеводородах может составлять 500—
1000%, для эбонита (высокая частота сетки) — 150—
250%. При соблюдении определенных условий по сте-
степени Н. сшитого полимера можно оценить частоту
сетки. Подробнее о термодинамике Н. сшитых полиме-
полимеров см. Студни.
Набухание кристаллизующихся полимеров. Пол-
Полностью закристаллизованный полимер не должен,
в принципе, набухать в жидкостях. Однако вследствие
того, что реально ни один из полимеров полностью
не кристаллизуется, и благодаря наличию дефектов
в кристаллитах кристаллич. полимеры также набу-
набухают. Если по условиям получения полимера кристал-
кристаллизация не была завершена до момента стеклования его
аморфной составляющей, то Н. в жидкости может при-
привести к смещению точки стеклования ниже темп-ры
опыта и к дополнительной кристаллизации аморфной
части полимера. При этом кривая в координатах сте-
степень Н.— время может иметь максимум (т. е. происхо-
происходит уменьшение степени Н. во времени).
Практическое значение явления набухания. Н. яв-
является необходимой стадией во мн. процессах модифи-
модификации и переработки полимеров, напр, при их пласти-
пластификации. При модификации полимеров в результате Н.
облегчается доступ реагентов внутрь частиц полимера.
Напр., в произ-ве вискозного волокна и целлофана для
ускорения образования ксантогената целлюлозу обра-
обрабатывают водными р-рами щелочей, вызывающими
сильное Н. волокон. В связи с этим большое внимание
уделяется условиям получения исходного материала,
поскольку на степень Н. оказывает существенное влия-
влияние предыстория полимера. Технологич. процессы
крашения волокон, варки древесины, дубления кожи,
переработки продуктов питания и мн. другие также
связаны с явлениями Н.
Во мн. случаях Н. полимерных материалов нежела-
нежелательно. Так, Н. природных и искусственных волокон,
кожи приводит к изменению размеров и формы изделий
из них после смачивания. Интенсивное Н. резины в мас-
маслах ограничивает использование изделий из натураль-
натурального и нек-рых видов синтетич. каучука в качестве
амортизационных деталей в приборах и машинах.
Упаковочные материалы из целлофана при Н. в воде
не только меняют размеры, но и теряют до 50—60%
первоначальной механич. прочности. Лакокрасочные
покрытия в результате Н. легко отслаиваются от под-
подложки. Для предупреждения этих отрицательных явле-
явлений изделия из полимеров защищают покрытиями,
стойкими в агрессивной среде, либо подвергают поли-
полимер структурной или химич. модификации, в частности
сшиванию. Структурная модификация, приводящая
к резкому уменьшению Н. в воде, происходит, напр.,
при ориентационной вытяжке поливинилспиртовых
волокон.
Лит.: КаргинВ.А., Слонимский Г. Л., Краткие
очерки по физико-химии полимеров, 2 изд., М., 1967; Т а-
г е р А. А., Физико-химия полимеров, 2 изд., М., 1968; Коз-
Козлов П. В., Физико-химия эфиро-целлюлозных пленок, М.,
1948, с. 286; JoplingD. W., J. Appl. Ghem., 6, pt. 2, 79
A956). С. П. Папков.
НАДМОЛЕКУЛЯРНАЯ СТРУКТУРА полиме-
полимеров (supermolecular structure, tibermolekulare struk-
tur, structure supermoleculaire) — физическая струк-
структура полимерных тел, обусловленная различными
видами упорядочения во взаимном расположении ма-
макромолекул.
В 1957 В. А. Каргин, А. И. Китайгородский и
Г. Л. Слонимский на основании анализа данных элект-
321
НАДМОЛЕКУЛЯРНАЯ СТРУКТУРА
322
роншшикроскопических и рентгеноструктурных иссле-
исследований, изучения механич. релаксационных явлений
и др. физич. процессов в полимерах сделали вывод о не-
невозможности полностью хаотич. расположения макро-
макромолекул ни в одном из физич. состояний полимеров,
как аморфных (включая расплавы, р-ры и студни), так
и кристаллических (см. Структура). Так, напр., было
установлено, что цепные макромолекулы в одних слу-
случаях свернуты «сами на себя» в клубки (молекулярные
глобулы), собирающиеся в более крупные агрегаты
(гроздья), а в других — образуют системы гибких или
жестких цепных макромолекул (пачек), в к-рых каж-
каждая цепь в основном граничит по всей своей длине с од-
одними и теми же соседними цепями. Это обусловливает
продолговатую форму и необычно большую длину роя
более или менее вытянутых макромолекул, т. е. пачки,
по сравнению с роем малых молекул.
Обнаружение Н. с. позволило объяснить казавшиеся
противоречивыми данные, указывающие, с одной сто-
стороны, на гибкость макромолекул и хаотичность их теп-
теплового движения, а с другой — на сложную микроне-
микронеоднородность строения полимерных тел.
Представление о Н. с. в течение нескольких лет по-
получило многочисленные экспериментальные подтверж-
подтверждения и стало эффективной руководящей идеей в совре-
современной структурной физике полимеров.
Многоступенчатость форм надмолекулярной
структуры и их превращения
В аморфных полимерных телах (полимерных стеклах,
эластомерах, расплавах и р-рах полимеров) пачки и
глобулы подвергаются дальнейшей агрегации, приво-
приводящей к образованию более сложных и крупных эле-
элементов Н. с. (фибрилл, дендритных образований и др.),
разнообразные формы к-рой (см. рис.) пока изучены
мало. Естественно, что возможность образования раз-
разных форм Н. с. одного и того же полимерного вещества
обусловливает существование одинаковых по химич.
строению аморфных полимерных тел, обладающих
различными механическими и др. физич. свой-
свойствами.
Пачки и глобулы в определенных, зависящих от
химич. строения полимера условиях, способны перехо-
переходить одна в другую. Так, напр., в случае р-ра полимера
изменение природы растворителя может привести к раз-
развертыванию скрученных гибких макромолекул линей-
линейного строения и агрегации их в пачки, а также к обрат-
обратному процессу дезагрегации пачки и образования гло-
глобул. При достаточной гибкости пачек может происхо-
происходить их свертывание, т. е. образование более крупных
глобулярных форм Н. с.
Механическими и тепловыми воздействиями можно
вызывать превращения одних форм Н. с. в другие. При-
Примерами являются возникновение ориентированного
состояния аморфных полимеров, в частности разверты-
развертывание некоторых глобулярных форм Н. с. при их одно-
одноосном растяжении, образование из р-ра полимера физич.
пространственной сетки из макромолекул, пачек или
др. элементов Н. с. (см. Студни). Такие физич. струк-
структурные превращения в аморфных полимерах во мно-
многом напоминают фазовые превращения.
Несравненно большее разнообразие форм Н. с. и со-
соответствующих структурных превращений реализуется
в кристаллич. полимерах.
200 400
Относительное удлинение, %
Некоторые формы надмолекулярной структуры полимеров: глобулярная (а), фибриллярная (б) и дендритная (в) формы в
аморфном полимере (сополимер диэтилового эфира винилфосфиновой кислоты с акриловой кислотой); фибриллярный сферолит
полиамида (г); пластинчатый сферолит изотактического полистирола (д); отдельный сферолит (е) и сферолитная лента (ж) изо-
изотактического полистирола (в поляризованном свете); сферолиты и кристаллы изотактического полибутилена (з); монокристалл
полиэтилена (и); глобулярный монокристалл вируса некроза табака (к); различные формы надмолекулярной структуры
изотактического кристаллического полипропилена (л, м, н) и соответствующие им диаграммы растяжения (о).
323
НАДМОЛЕКУЛЯРНАЯ СТРУКТУРА
324
Н. с, возникающая при кристаллизации аморфных
полимеров глобулярного строения, изучена мало. Из-
Известно, что в пределах самой глобулы степени упоря-
упорядоченности звеньев макромолекул могут быть настолько
различны, что можно говорить об аморфных и закри-
закристаллизованных глобулах. При этом, если глобулы оди-
одинаковы, как, напр., у ряда природных полимеров, то
они сами могут располагаться взаимно упорядоченно
в узлах кристаллич. решетки. При этом образуются
макроскопич. размера ограненные кристаллы, перио-
периоды идентичности к-рых очень велики соответственно раз-
размерам глобул.
Н. с, возникающая при кристаллизации аморфных
полимеров пачечного строения, изучена значительно
лучше. Известны две формы кристаллизации пачек,
а именно с образованием кристаллич. микрофибрилл
с выпрямленными макромолекулами и с образованием
т. наз. лент в результате многократного перегибания
и складывания макромолекул самих на себя. Кристал-
Кристаллич. образования обоих типов агрегируются в более
крупные, имеющие весьма разнообразное строение и
способные к дальнейшей агрегации (см. Кристалли-
Кристаллизация). Наиболее часто образуются сферолиты, но мо-
могут возникать и др. формы, в частности монокристал-
монокристаллы, сложные нерегулярные формы, состоящие из
надмолекулярных образований, не доросших до пра-
правильных сферолитов или монокристаллов. Сферолиты
также способны агрегироваться, образуя сферолитные
ленты и пластины.
В реальном кристаллич. полимере могут сосущество-
сосуществовать все возможные в заданных условиях формы над-
надмолекулярных образований, а также аморфные области
с характерными для них формами надмолекулярной
структуры.
Способность различных элементов Н. с. к структур-
структурным превращениям (самопроизвольным или вынуж-
вынужденным внешними воздействиями) обусловливает неста-
нестабильность механических и др. физич. свойств полиме-
полимеров. Это проявляется, напр., в образовании шейки и
в возникновении ряда сложных релаксационных про-
процессов в механически напряженном теле, в постепен-
постепенном росте хрупкости кристаллического полимерного
тела вследствие укрупнения и др. изменений элемен-
элементов его Н. с.
Ограничение применимости к полимерам ряда физи-
физических понятий. Многообразие форм и размеров эле-
элементов Н. с. требует весьма критич. отношения к ряду
традиционных физич. понятий при их применении к по-
полимерам. Здесь прежде всего следует рассмотреть по-
понятия «поверхность раздела», «термодинамич. фаза»,
«степень кристалличности».
Представление о поверхности раздела требует, чтобы
размеры образующих эту поверхность частиц были
столь малы, чтобы ими можно было пренебречь по срав-
сравнению с размерами самой поверхности. Это выполняет-
выполняется для частиц, состоящих из многих малых молекул,
напр, для частиц лиофобных коллоидов. Однако в слу-
случае пачки макромолекул, содержащей в поперечном
сечении лишь десятки или немногие сотни макромоле-
макромолекул и к тому же имеющей флюктуационную природу,
представление о поверхности раздела становится край-
крайне неопределенным.
В случае кристаллизации пачек, остающихся в окру-
окружении какого-то числа макромолекул, характеризую-
характеризующихся упорядоченностью, свойственной аморфному
состоянию полимерного тела, представление о поверх-
поверхности раздела физически значительно более определен-
определенно, что позволяет рассматривать фазовые состояния
полимеров и их фазовые превращения (напр., кристал-
кристаллизацию, плавление, рекристаллизацию), но требует
осторожного использования привычных понятий, осо-
особенно термодинамических. Это же следует и из сложного
строения кристаллич. фазы, в к-рой для наиболее мел-
мелких структурных элементов, образуемых сегментами
макромолекул, понятие «фаза» применяется просто,
поскольку эти элементы имеют те же размеры, что и
обычные малые молекулы, но по мере роста размеров
структурных образований, составляющих кристаллич.
полимер, положение усложняется вследствие потери
гомогенности, замедления процессов установления рав-
равновесий и возрастания значения поверхностных явле-
явлений. Именно этот переход от явлений молекулярных
и равновесных к коллоидным и неравновесным и далее
к макроскопическим заставляет переосмысливать мно-
многие понятия, казалось бы твердо установленные для
молекулярных систем.
Понятие «степень кристалличности», основанное на
схематичном представлении о сосуществовании в одном
теле двух фаз (аморфной и кристаллической), становит-
становится физически неопределенным при рассмотрении Н. с.
реального полимерного тела. Такое тело всегда весьма
дефектно вследствие наличия пограничных областей
между различными его структурными элементами, на-
нарушений кристаллич. порядка в самих кристаллич.
элементах структуры, напр. в области складок, сохра-
сохранения части незакристаллизовавшегося вещества и
т. п. Однако многие из относительно малоупорядо-
ченных (аморфных) областей являются неотъемлемой
частью безусловно кристаллич. элементов Н. с. и не
могут считаться аморфной фазой. Наряду с этим в том
же кристаллич. полимерном теле возможно присутствие
аморфных элементов Н. с. (напр., аморфных глобул),
допускающих отделение от кристаллич. элементов и
поэтому представляющих собой аморфную фазу. При
любом способе оценки степени кристалличности раз-
различия всех этих форм малоупорядоченных (аморфных)
и хорошо упорядоченных (кристаллических) областей
не учитываются. Это существенно снижает ценность
такой оценки и приводит к необходимости характери-
характеризовать систему, состоящую из многих сосуществующих
форм упорядочения макромолекул, не одним, а многи-
многими параметрами.
Образование и преобразование надмолекулярной
структуры при синтезе, переработке и эксплуатации.
Формы Н. с, с одной стороны, определяют комплекс
физич. свойств полимерного тела, а с другой — сами
зависят от химич. строения макромолекул. Для обра-
образования всего богатства форм Н. с. в кристаллическом
состоянии тела необходима возможность самого про-
процесса кристаллизации. Для этого требуется достаточно
регулярное строение макромолекул и высокая их гиб-
гибкость, обеспечивающая заметную скорость процессов
упорядочения. Нарушение этих условий приводит к об-
образованию аморфных тел с присущими им формами
Н. с. Очень жесткие макромолекулы продолговатой
формы вследствие действия межмолекулярных сил
образуют стеклообразные полимерные тела, обнару-
обнаруживающие во многих случаях весьма высокую упоря-
упорядоченность макромолекул, сравнимую с порядком
в кристаллич. телах. Для таких стеклообразных тел
характерны фибриллярные формы Н. с. и соответст-
соответствующие им комплексы свойств. Очень жесткие макро-
макромолекулы скрученной формы, изменить к-рую они без
химич. воздействий не могут, образуют стеклообразные
тела с глобулярной Н. с, отличающиеся от фибрилляр-
фибриллярных тел того же химич. строения повышенной хруп-
хрупкостью.
Макромолекулы полимеров в высокоэластич. состоя-
состоянии обязательно обладают достаточной гибкостью и за-
заметной подвижностью. Поэтому элементы Н. с. эласто-
эластомеров менее стабильны и слабее выражены, чем в стек-
стеклообразных, а тем более в кристаллич. полимерных те-
телах. Это затрудняет их наблюдение и исследование.
Еще менее стабильны и более подвижны элементы Н. с.
расплавов и р-ров полимеров. Их существование до-
доказано, но детально они пока не изучены.
325
НАПОЛНЕНИЕ
326
Н. с, возникающая уже в условиях синтеза макро-
макромолекул, может претерпевать самопроизвольные или
вынужденные глубокие изменения при переработке
полимера в материалы и изделия, а также в условиях
длительного хранения и эксплуатации. Глубина и ско-
скорость таких изменений существенно зависят не только
от типа и интенсивности внешних воздействий, но и от
физического состояния полимера.
В случае высокой гибкости макромолекул Н. с, соз-
созданная при образовании твердого полимерного тела
(в процессе синтеза, затвердевания расплава, при
удалении растворителя из р-ра), м. б. в дальнейшем
изменена на всех ее уровнях. Это достигается механич.
воздействиями, нагреванием, плавлением (или растворе-
растворением) с последующим образованием твердого тела в оп-
определенном режиме охлаждения (или удаления раст-
растворителя). В зависимости от характера межмолекуляр-
межмолекулярного взаимодействия и от интенсивности воздействия
изменяются и различные уровни надмолекулярной ор-
организации полимерного тела. В одних случаях преобра-
преобразование Н. с. происходит при разрушении крупных
ее элементов, но с сохранением наиболее мелких и ме-
менее сложных. В других происходит полная перестрой-
перестройка всех уровней Н. с. полимерного тела.
В случае очень жестких макромолекул, не способных
без химич. превращений изменять свою форму, весьма
существенное влияние на Н. с. оказывают условия син-
синтеза. В зависимости от этих условий могут образовать-
образоваться, напр., преимущественно скрученные или, наоборот,
выпрямленные макромолекулы. Заданные при синтезе
формы жестких макромолекул остаются в дальнейшем
неизменными, и это определяет возможные формы Н. с.
Естественно, что из скрученных жестких макромолекул
возникают глобулярные формы Н. с, а из вытянутых —
фибриллярные.
Поскольку при переработке происходит изменение
Н. с. и свойств полимера, то получаемые из одного и
того же полимера материалы и изделия могут значи-
значительно различаться по физич. характеристикам, если
применяются разные приемы переработки. Кроме того,
может различаться и стабильность физич. структуры
и свойств таких материалов или изделий. Поэтому изу-
изучение закономерностей физич. структурообразования
в полимерных телах и возможностей управления им
является одной из важнейших проблем структурной и
технической физики полимеров (см. Модификация
структурная).
Методы исследований надмолекулярной структуры.
Важнейшими прямыми методами изучения Н. с. поли-
полимерных тел являются электронная и световая микро-
микроскопия, электронная, рентгеновская и световая дифрак-
тометрия. Существенные сравнительные данные полу-
получают из механич. и др. физических исследований поли-
полимерных тел с различной Н. с. Наиболее плодотворным
оказалось комплексное использование всех этих мето-
методов, позволяющее устанавливать связи характеристик
Н. с. (типа, размеров и числа элементов Н. с.) с физич.
свойствами полимерных тел. Быстрое развитие ис-
исследований Н. с. полимеров уже принесло много |
принципиально важных научных и прикладных резуль-
результатов.
Лит.: К а р г и н В. А., Китайгородский А. И.,
Слонимский Г. Л., Колл. журн., 19, № 2, 131 A957);
К а р г и н В. А., Слонимский Г. Л., Краткие очерки
по физико-химии полимеров, 2 изд., М., 1967, с. 24, 147;
Д ж е й л Ф. X., Полимерные монокристаллы, пер. с англ.,
[Л.], 1968. Г. Л. Слонимский.
НАИРИТ — см. Хлоропреновые каучуки.
НАЙЛОН — см. Полиамидные волокна.
НАПОЛНЕНИЕ полимеров (filling, Fiillung,
chargement) — сочетание полимеров с твердыми, жид-
жидкими или газообразными веществами, к-рые относи-
относительно равномерно распределяются в объеме образую-
образующейся композиции и имеют четко выраженную границу
раздела с непрерывной полимерной фазой (матрицей).
Н.— один из основных способов создания пластмасс,
резин, лакокрасочных материалов, клеев синтетиче-
синтетических и др. полимерных материалов с заданными техно-
логич. и эксплуатационными свойствами. Особенно
важное значение Н. имеет при получении резин на осно-
основе большинства синтетич. каучуков, характеризующих-
характеризующихся низким межмолекулярным взаимодействием, а так-
также композиций из термореактивных смол (феноло-,
мочевино-, меламино-формальдегидных, полиэфирных,
эпоксидных и др.), отверждение к-рых сопровождается
значительной усадкой и приводит к образованию трех-
трехмерных полимеров с большой частотой сетки. Наряду
с этими материалами широко используют также на-
наполненные термопласты конструкционного назначе-
назначения — полиамиды, полиэтилен, полипропилен, поли-
поликарбонаты, политетрафторэтилен и др.
В подавляющем большинстве случаев для получения
наполненных полимерных материалов применяют твер-
твердые наполнители: тонкодисперсные с частицами зерни-
зернистой (сажа, двуокись кремния, древесная мука, мел,
каолин и др.) или пластинчатой (тальк, слюда, графит
и др.) формы, а также разнообразные волокнистые ма-
материалы. Последние применяют в виде элементарных
волокон, нитей, прядей, жгутов, тканей, холстов, ма-
матов, бумаги, шпона, прутков, сеток. В особую группу
среди твердых наполнителей выделяют т. наз. э л а-
стификаторы, к-рыми служат полимеры с низ-
низким модулем упругости (гл. обр. эластомеры), исполь-
используемые в сочетании с такими жесткими полимерами, как
полистирол и большинство реактопластов. Подробно
о твердых наполнителях см. Наполнители пластмасс,
Наполнители резин, Наполнители лакокрасочных ма-
материалов.
Тип и природа наполнителя находят отражение
в классификации полимерных материалов. Так, разли-
различают, напр., асбопластики, графитопласты, древесные
пластики, стеклопластики, газонаполненные пластмас-
пластмассы и др. Полимерные материалы с волокнистыми напол-
наполнителями наз. обычно армированными (см. Армиро-
Армированные пластики).
Способы наполнения определяются фи-
физич. состоянием полимера и типом наполнителя. Основ-
Основной способ Н. при использовании твердых тонкодисперс-
тонкодисперсных наполнителей или рубленого волокна — смешение
наполнителя: 1) с высокомолекулярным полимером,
находящимся в высокоэластич. (каучуки) или вязкоте-
кучем (термопласты) состоянии; 2) с расплавом или
р-ром термореактивного связующего с последующим
отверждением (или сушкой с отверждением) наполнен-
наполненной системы; 3) с мономером, форполимером (или с их
р-рами) с последующей полимеризацией или поликон-
поликонденсацией; этот способ особенно широко используют
при получении клеевых и лакокрасочных композиций.
Основное оборудование для смешения наполнителя со
связующим — смесители различной конструкции, а
также вальцы.
Непрерывные волокна, нити, жгуты, ткани и др. во-
волокнистые заготовки пропитывают расплавами или
р-рами полимера, форполимера или термореактивной
смолы (см. Пропитка наполнителей). Если наполни-
наполнитель пропитывают форполимером или олигомером или
смешивают с ними, то последующая полимеризация или
поликонденсация может сопровождаться образованием
химич. связей между связующим и реакционноспособ-
ными группами на поверхности наполнителя и, следо-
следовательно, повышением адгезии между полимером и на-
наполнителем. Этот эффект м. б. достигнут и модифика-
модификацией поверхности наполнителя, напр, заменой одних
функциональных групп другими, имеющими большее
сродство к полимеру, хемосорбцией поверхностью на-
наполнителей аппретов (см., напр., Стеклянные волокна)
или поверхностно-активных веществ.
327
НАПОЛНЕНИЕ
328
Эластификаторы обычно распределяют в матрице же-
жесткого полимера механич. перетиранием. Более благо-
благоприятна полимеризация мономера будущей матрицы на
поверхности частиц эластомера, используемого в виде
латекса, или сплавление эластификатора с олигомером
и отверждение последнего. В этих случаях на границе
раздела частиц эластификатора со связующим обра-
образуются блок- или привитые сополимеры, включающие
блоки обоих полимеров, что обеспечивает совместную
работу эластификатора и связующего при внешних воз-
воздействиях.
Газонаполненные материалы получают вспениванием
составов с помощью специальных агентов (порообразо-
вателей) или механич. вспениванием (напр., при полу-
получении пенорезины из латекса). Пенистая структура
полимерного материала фиксируется охлаждением ком-
композиции ниже темп-ры стеклования полимера, отверж-
отверждением или вулканизацией — см. Пенопласты, Губча-
Губчатые резины.
Пример материала с жидким наполнителем — зали-
заливочные составы с малой плотностью, получаемые меха-
механич. эмульгированием воды в полиэфирной смоле. От-
Отверждение таких составов превращает дисперсионную
среду в жесткую матрицу без разрушения первоначаль-
первоначальной структуры эмульсии.
Свойства наполненного полимер-
полимерного материала определяются свойствами по-
полимерной матрицы и наполнителя, характером распре-
распределения последнего, природой взаимодействия на гра-
границе раздела полимер — наполнитель. Материалы
с жидкими и газообразными наполнителями, как прави-
правило, изотропны; с твердыми наполнителями — изотроп-
изотропны или анизотропны в зависимости от вида наполни-
наполнителя и характера его распределения. Свойства напол-
наполненного полимерного материала существенно зависят
также от дисперсности и формы частиц наполнителя,
степени и условий Н., фазового или физич. состояния
полимера, природы его звеньев, частоты пространст-
пространственной сетки. Деление наполнителей на активные
(упрочняющие, усиливающие) и неактивные (инертные)
в известной мере условно, поскольку, улучшая какую-
либо характеристику системы, наполнитель может
ухудшать др. ее свойства. Напр., большинство саж по-
повышает одновременно прочность и модуль (жесткость)
резин, однако увеличение жесткости во многих случаях
нежелательно. Кроме того, активность наполнителя
проявляется только при его определенном содержании
в системе.
При Н. полимеров твердыми наполнителями в ре-
результате стерич. ограничений, обусловленных присут-
присутствием твердой поверхности, а также взаимодействия
полимера с этой поверхностью существенно уменьшает-
уменьшается молекулярная подвижность макромолекул в гранич-
граничном слое. Это, в свою очередь, приводит к изменению
структуры и свойств граничного слоя, что проявляется
в повышении темп-ры стеклования и текучести полиме-
полимера (рис. 1), изменении релаксационных свойств систе-
системы и др. Ограничение молекулярной подвижности при
Н. обусловливает возрастание средних времен релак-
релаксации; они тем больше, чем выше степень Н. и дисперс-
дисперсность наполнителя, меньше гибкость макромолекул
(в случае линейных полимеров) или больше плотность
трехмерной сетки. При этом возрастающая жесткость
макромолекул в адсорбционных слоях может привести
к кажущемуся уменьшению средних времен релакса-
релаксации, поскольку большие времена релаксации не смогут
реализоваться. Выше темп-ры стеклования полимера
возникают дополнительные релаксационные процессы,
вызванные отрывом макромолекул от частиц наполни-
наполнителя и перегруппировкой частиц.
Уменьшение числа возможных конформаций макромо-
макромолекулы в граничном слое приводит к изменению плот-
плотности упаковки аморфных полимеров и к изменению
условий кристаллизации. С увеличением степени Н.
эластомеров плотность и жесткость матрицы возраста-
возрастают, температурный коэфф. расширения, коэфф. диффу-
диффузии и проницаемости обычно уменьшаются. При Н.
жесткоцепных пласти-
пластиков плотность полиме-
полимера в граничном слое
уменьшается, модуль
упругости, температур-
температурный коэфф. расшире-
расширения, сорбция, коэфф.
диффузии и проницае-
Рис. 1. Термомеханические
кривые полистирола: 1 —
ненаполненный полимер;
2 — 5% стекловолокна;
3 — 20% стекловолокна.
150 200
Температура, °С
мости матрицы возрастают. Сочетание наполнителя с
кристаллич. полимерами сопровождается изменением
типа и размеров кристаллитов. При очень малых
концентрациях наполнитель м. б. искусственным за-
зародышем структурообразования (см. Структурообра-
зователи). С возрастанием Н. степень кристаллично-
кристалличности полимера уменьшается. Темп-pa плавления поли-
полимера при Н. существенно не изменяется. Из-за раз-
разницы температурных коэфф. расширения жесткоцеп-
ного полимера и наполнителя в материале могут
возникать значительные напряжения, приводящие
к разрушению связующего в граничном слое.
Отверждение (вулканизация), протекающее в мат-
матрице на поверхности частиц наполнителя, может су-
существенно отличаться от того же процесса в объеме
полимера. Это различие обусловлено тем, что на по-
поверхности наполнителя изменяются соотношения ско-
скоростей элементарных реакций, происходит избиратель-
избирательная адсорбция компонентов полимерной фазы, участ-
участвующих в отверждении, а с другой стороны, в эту ре-
реакцию вступают функциональные группы на поверх-
поверхности наполнителя и др. Изменение свойств полимера
в граничном слое (увеличение структурной и физич.
микрогетерогенности и даже химич. неоднородности)
м. б. столь существенным, что вклад наполнителя
в свойства композиции окажется нивелированным.
Поэтому наполненный полимер целесообразно рассмат-
рассматривать как трехкомпонентную систему, состоящую из
наполнителя, граничного слоя с измененными свойст-
свойствами и полимера, свойства к-рого аналогичны свойст-
свойствам ненаполненного. Однако определить объемную
долю граничного слоя в наполненном полимере прак-
практически невозможно, поскольку понятие «толщина гра-
граничного слоя» условно, и ее эффективное значение для
того или иного свойства системы может изменяться
в широких пределах.
Существенное изменение свойств наполненных поли-
полимеров, особенно реологических и физико-механических,
обусловлено не только влиянием наполнителя на струк-
структуру и свойства граничного слоя полимера, но и взаимо-
взаимодействием частиц наполнителя между собой. Повыше-
Повышение степени Н. и степени асимметрии частиц наполните-
наполнителя приводит к их агрегированию вплоть до образования
непрерывной сетки, созданной в результате непосредст-
непосредственного контакта частиц наполнителя или слияния
адсорбционных слоев полимера, окружающих контакти-
контактирующие частицы. В результате резко возрастает вяз-
вязкость материала вплоть до потери текучести.
Обоснованная физич. теория, позволяющая математи-
математически описать изменение свойств материала с измене-
изменением степени Н., пока не разработана; имеются лишь
ограниченно применимые соотношения, к-рые с учетом
особенностей конкретной пары полимер — наполни-
наполнитель м. б. использованы для прогнозирования направ-
направленного изменения свойств системы с изменением типа
329
НАПОЛНЕНИЕ
330
и соотношения компонентов. Так, допустив, что между
связующим и наполнителем отсутствует специфич. взаи-
взаимодействие, частицы наполнителя не деформируются
под нагрузкой, имеют сферич. форму, не взаимодейст-
взаимодействуют друг с другом и полностью смачиваются связую-
связующим, вязкость ц наполненных композиций можно опи-
описать ур-нием:
гДе тюв — вязкость связующего, Н'сек/м2, или пз\
С — объемная доля наполнителя.
При тех же допущениях приведенное ур-ние м. б.
использовано для расчета зависимости модуля высоко-
эластичности системы от модуля высокоэластичности
ненаполненного полимера и объемной доли наполни-
наполнителя. В реальных композициях модуль высокоэластич-
высокоэластичности всегда выше, поскольку часть связующего нахо-
находится в адсорбционном слое и, кроме того, под внешним
воздействием деформируются элементы структур, обра-
образованные частицами наполнителя.
Модуль упругости Е композиции с порошкообраз-
порошкообразным наполнителем лежит между верхним и нижним
пределами, соответственно Е = A—C)EQB + СЕН и
Е = ECB.EH/[{i - С)ЕН + СЕСВ], где Есв и Ен —
модули упругости связующего и наполнителя. Верх-
Верхние предельные значения модуля соответствуют случаю
одинаковой деформации обоих компонентов при нали-
наличии адгезии между ними по всей поверхности контакта,
нижние отвечают случаю одинаковых напряжений
в компонентах при отсутствии адгезионного взаимо-
взаимодействия между ними.
Температурные коэфф. расширения полимеров, на-
наполненных частицами, форма к-рых близка к сфери-
сферической, изменяются в зависимости от концентрации
наполнителя согласно ур-нию:
где Р, рсв, рн — соответственно коэфф. объемного рас-
расширения композиции, связующего и наполнителя,
°С~1#, в — поправочный коэфф.
Для композиций, в к-рых связующее находится
в стеклообразном состоянии и модуль упругости напол-
наполнителя выше, чем модуль упругости связующего, со-
соблюдается соотношение 1 < в < 3. В наполненных
композициях со связующим, находящимся в высоко-
эластич. состоянии, значение в приближается к еди-
единице.
Для прогнозирования диэлектрич. проницаемости
композиций, наполненных не взаимодействующими
друг с другом сферич. частицами одинакового размера,
можно использовать ур-ние Максвелла:
ея есв
где есв и jbh — соответственно диэлектрич. проницае-
проницаемость связующего и наполнителя.
На основании формального подобия законов перено-
переноса вещества, энергии или заряда в результате гради-
градиента концентраций, темп-р или электрич. потенциала
приведенное соотношение можно использовать для
расчета коэфф. теплопроводности и газопроницаемости
в зависимости от значения этих констант для полимера
и наполнителя и объемной доли последнего. Для коэфф.
газопроницаемости ур-ние справедливо лишь в том слу-
случае, когда сорбция газа подчиняется закону Генри,
т. е. прямо пропорциональна давлению газа в окру-
окружающей среде.
Диэлектрич. проницаемость и теплопроводность ре-
реальных композиций с твердыми наполнителями, как
правило, ниже рассчитанной из-за наличия воздушных
включений, обусловленных неполным смачиванием
поверхности наполнителя связующим. Если теплопро-
теплопроводность наполнителя намного выше, чем у связую-
связующего, то при агрегировании частиц наполнителя обра-
образуются тепловые мостики и теплопроводность компози-
композиции резко возрастает. Неполное смачивание наполни-
наполнителя связующим и механич. дефекты в граничном слое
повышают газопроницаемость композиции.
Упрочнение материала в результате Н. наблюдается
в тех случаях, когда наполнитель прочнее полимера,
а условия Н. обеспечивают совместную работу обоих
компонентов под действием механич. напряжений. По-
Последнее условие выполняется при достижении прочного
контакта между полимерной матрицей и частицами на-
наполнителя по всей поверхности раздела.
С термодинамич. точки зрения для упрочнения необ-
необходимо, чтобы энергия адгезии полимер — наполнитель
была больше энергии когезии полимера. Этот вывод сде-
сделан на том основании, что при условии адгезионного на-
нарушения контакта разделение фаз сопровождается
исчезновением поверхности раздела между поли-
полимером и наполнителем с образованием равных по
площади поверхностей обеих фаз. Свободная энергия
адгезии между двумя фазами выражается ур-нием:
Ya=Yi + Y2—Yi,2
где Yi и Y2 — свободная поверхностная энергия соот-
соответственно полимера и наполнителя, у12 — свободная
межфазная энергия. Если принять (по аналогии с жид-
жидкостью), что свободная энергия когезии полимера
7к = 2yi, to 7a —Yk = Y2 — Yi~ Yi,2» т. е. условие
упрочнения сводится к (у 12 + у1 — 72) < 0- Поскольку
Рис. 2. Влияние ти-
типа и содержания на-
наполнителя на энер-
энергию упругости вул-
вулканизованного кау-
каучука: 1 — окись маг-
магния; 2 — газовая ка-
канальная сажа; 3 —
каолин; 4 — барит
A кгс-м/см2 =
0,1 Мдж/м*).
25
Содержание, X по объему
поверхностная энергия большинства наполнителей вы-
выше, чем у полимеров, то Н. должно, как правило, при-
приводить к упрочнению полимера. Согласно П. А. Ребин-
ДеРУ> упрочнение является следствием перевода поли-
полимера в адсорбционный слой с большой поверхностью
раздела. Необходимая для этого работа преодоления
сил поверхностного натяжения полимера вносит допол-
дополнительный вклад в суммарную работу разрушения
материала. На рис. 2 показано, что упрочняющее дейст-
действие наполнителя зависит от его природы и степени Н.
Существуют нек-рые специфич. особенности в меха-
механизме упрочнения эластомеров и жестко-
цепных линейных и сетчатых полимеров. В частности,
существенное упрочнение эластомеров достигается при
использовании высокодисперсных наполнителей, пре-
преимущественно сажи, прочные первичные агрегаты к-рой
создают в среде эластомера цепочечные структуры (см.
также Наполнители резин). Действие этих структур
объясняется гл. обр. тем, что их элементы являются
той матрицей, на к-рой ориентирована макромолекула.
Чем больше развита цепочечная структура, тем в боль-
большей степени проявляется ее ориентирующее и упроч-
упрочняющее действие. Образующиеся в ходе смешения хао-
тич. связи каучук — наполнитель при деформации
под напряжением разрываются и вновь восстанавли-
восстанавливаются в новых положениях, закрепляя на поверхности
наполнителя макромолекулы каучука, частично ориен-
ориентированные в направлении действия напряжений. В ре^
зультате происходит выравнивание местных перена-
перенапряжений. Чем выше прочность связи каучук — на-
331
НАПОЛНЕННЫЕ КАУЧУКИ
332
полнитель, тем значительнее эффект упрочнения.
Однако эта зависимость наблюдается лишь до опреде-
определенных значений адгезии между каучуком и поверх-
поверхностью наполнителя, не превышающих тот предел, при
к-ром разрушение материала происходит преимущест-
преимущественно на границе раздела. В этом случае одновременно
образуются многочисленные очаги разрушения, что
сопровождается повышенным рассеянием энергии при
деформации. Разрушение происходит скачкообразно
от одной поверхности раздела каучук — наполнитель
к другой по зигзагообразной линии. Т. о., повышение
прочности резин при Н. обусловлено прежде всего
выравниванием напряжений в ходе деформации и уд-
удлинением пути разрушения, а следовательно, и уве-
увеличением работы разрушения. Дополнительный вклад
в упрочнение вносит повышенный механич. гистерезис
наполненных резин, обусловленный энергией, к-рая
выделяется при разрыве физич. связей между напол-
наполнителем и каучуком, уменьшением подвижности мак-
макромолекул у поверхности наполнителя, а также раз-
разрушением агломератов частиц и цепочечных структур
последнего. Вследствие более высокого механич. гисте-
гистерезиса степень релаксации напряжения наполненных
резин в области больших деформаций перед вершиной
разрастающейся трещины всегда больше, чем в нена-
полненных.
Влияние дискретного наполнителя на проч-
прочность жесткоцепных полимеров м. б.
объяснено с точки зрения статистич. теории распреде-
распределения внутренних дефектов в твердом теле. Упрочняю-
Упрочняющее действие наполнителя связано с изменением усло-
условий перенапряжения на краях трещин, с релаксацией
напряжений и перераспределением их на большее чис-
число центров прорастания микротрещин. Это должно
увеличивать среднее напряжение, ведущее к разруше-
разрушению тела. Микротрещина, развиваясь в наполненном
полимере, может «упереться» в наполнитель и, следо-
следовательно, ее дальнейшее развитие будет требовать по-
повышения напряжения. Чем больше концентрация на-
наполнителя в полимере, тем больше создается препятст-
препятствий для развития трещин; благодаря этому тормозится
процесс разрушения. Кроме того, в тонких слоях по-
полимера, согласно статистич. теории прочности, число
дефектов, приводящих к разрушению, должно быть
меньше; в определенных пределах увеличение прочно-
прочности пропорционально уменьшению толщины слоя по-
полимера.
В случае систем, содержащих большое количество
дисперсных наполнителей, их частицы образуют, как
и в наполненных резинах, непрерывную коагуляцион-
ную структуру, пронизывающую весь объем. Т. обр.,
наполненная система состоит из первичной структуры,
к-рую образуют частицы наполнителя, и вторичной,
создаваемой макромолекулами, ориентированными на
поверхности этих частиц и образующими поверхност-
поверхностный слой с измененными свойствами. Это приводит
к повышению прочности и одновременному увеличению
жесткости композиции в тем большей степени, чем вы-
выше дисперсность и асимметрия частиц наполнителя.
Предельно возможное Н. определяется из условий
сохранения формуемости материала и минимальной
толщины граничного слоя. Оно может быть повышено
при увеличении размеров частиц наполнителя или
изменения распределения частиц по размерам. На этом
основано, в частности, получение таких высокона-
полненных материалов как графитопласты, аман (см.
Антифрикционные полимерные материалы), полимер-
бетон.
В случае наполнения (армирования) волокнами, при-
применяемыми в виде ровницы, матов, тканей и др., в ме-
механизме упрочнения все большую роль начинают иг-
играть макроструктура наполнителя и его прочностные
свойства. Роль полимерного связующего в таких
материалах сводится к обеспечению равномерности
нагружения и одновременной работы всех воло-
волокон в материале. Связующее склеивает волокна
и защищает их от воздействия внешней среды.
Первостепенное значение приобретает прочность адге-
адгезионного взаимодействия между полимером и напол-
наполнителем. В процессе деформации волокно удлиняется
и одновременно уменьшается в диаметре. При этом оно
должно или оторваться по всей поверхности от окру-
окружающей его пленки связующего, или растянуть эту
пленку. В плоскости, перпендикулярной приложен-
приложенной силе, создается растягивающее напряжение, пре-
препятствующее удлинению волокна. Значение напряже-
напряжения определяется адгезией полимера к поверхности
наполнителя и свойствами граничной пленки полимера.
Следовательно, для разрушения материала необхо-
необходимо преодолеть не только суммарную прочность воло-
волокон, но и силы, препятствующие уменьшению их диа-
диаметра. Эти силы тем больше, чем выше упругие свойст-
свойства полимера.
Хотя высокая адгезия между жесткоцепным полиме-
полимером и наполнителем — одно из условий получения
упрочненного материала, повышение адгезионного взаи-
взаимодействия допустимо лишь в определенных пределах,
поскольку оно сопровождается увеличением жесткости
системы. Это приводит к возрастанию и «заморажива-
«замораживанию» в материале остаточных напряжений (термич.
и усадочных), понижающих его физико-механические
свойства.
Лит.: Липатов Ю. С, Физико-химия наполненных
полимеров, Киев, 1967; Липатов Ю. С, Сергее-
Сергеева Л. М., Адсорбция полимеров, Киев, 1972; Современные
композиционные материалы, под ред. Л. Браутмана и Р. Крока,
пер. с англ., М., 1970; Усиление эластомеров, под ред. Г. Крау-
са, пер. с англ., М., 1968; Попов В. А., Производство и
переработка пластмасс, синтетич. смол и стеклянных волокон,
№ 10, ЦНИИТЭХИМ, М., 1968; Андреевская Г. Д.,
Высокопрочные ориентированные стеклопластики, М., 1966;
Л у к о м с к а я А. И., Колл. ж. 23, в. 4, 428 A961); Мул-
линз Л., Кауч. и рез., № 10, 20 A970); Ц е т л и н Б. Л.
Ы др.], ДАН, 114, № 1, 146 A957). Ю.С. Липатов.
НАПОЛНЕННЫЕ КАУЧУКИ (filled rubbers, ge-
iillte Kautschuke, caoutchoucs charges). В данной
статье рассматриваются смеси каучуков с нефтяными
минеральными маслами (маслонаполненные каучуки),
сажей (саженаполненные каучуки), с теми и др. одновре-
одновременно (сажемаслонаполненные каучуки), а также менее
распространенные смеси каучуков с синтетич. пласти-
пластиками или лигнином.
Н. к. изготовляют гл. обр. на основе наиболее мас-
массовых синтетич. каучуков общего назначения — бута-
диен-стирольных, синтезируемых эмульсионной поли-
полимеризацией при низких темп-pax, и стереорегулярных
бутадиеновых. Кроме того, производят наполненные
изопреновые, этилен-пропиленовые, бутадиен-стироль-
ные (получаемые полимеризацией в. р-ре) каучуки, бу-
бутадиеновые каучуки эмульсионной полимеризации,
бутилкаучук и др. При получении Н. к. указанные вы-
выше ингредиенты смешивают с каучуками, как правило,
в технологич. процессе производства последних.
Маслонаполненные каучуки
Введение масел в каучуки приводит к повышению
их пластичности (понижению вязкости по Муни), что
обусловливает улучшение технологич. свойств каучу-
каучуков при переработке (см. Пласто-эластические свойст-
свойства). Благодаря этому для изготовления маслонаполнен-
ных каучуков м. б. использованы полимеры с высокой
мол. массой, что позволяет, несмотря на введение масла,
получать резины с достаточно хорошими технич. свойст-
свойствами. Применение маслонаполненных каучуков дает
также значительный экономич. эффект, поскольку
часть полимера заменяется в них более дешевым мас-
маслом. Ассортимент этих Н. к. (табл. 1—4) отличается
большим разнообразием (далее в таблицах и в тексте
333
НАПОЛНЕННЫЕ КАУЧУКИ
334
Таблица 1. Ассортимент наиболее распространенных маслонаполненных бутадиен-
стирольных каучуков эмульсионной полимеризации, выпускаемых в СССР
и др. социалистических странах
Марка каучука*
СКС-ЗОАРКМ-15 или
СКМС-ЗОАРКМ-15
СКС-ЗОАРКМ-27 или
СКМС-ЗОАРКМ-27
Буна S 040
KERS-3014
Каром 1712
Страна
СССР
»
ГДР
ПНР
СРР
Тип масла
Высокоароматическое
То же
Смесь высокоароматиче-
высокоароматического и парафинового
Высокоароматическое
То же
Количест-
Количество масла,
мае. ч.
20
37,5
40**
22**
37,5**
Жесткость
каучука,
н (гс)
4,5-7,5
D50-750)
5,0-7,5
E00-750)
7,5
G50)
5,0-8,0
E00-800)
Вязкость
каучука
по Муни
при 100 °С
32-58
35-58
50
45-65
* О принципах обозначения марок отечественных каучуков см. Бутадиен-стиролъные
паучуки. ** В % по массе (в расчете на смесь каучука и масла).
определяющие безопасность и
удобство применения масел при
получении Н. к. Лучшей совме-
совместимостью с бутадиен-стироль-
ными каучуками обладают аро-
ароматич. масла; с бутадиеновыми
и этилен-пропиленовыми — па-
парафиновые и нафтеновые масла.
На совместимость с каучуком
влияет также структура угле-
углеводородов (напр., число колец
в молекуле, длина боковых це-
цепей), входящих в состав масла.
Так, с бутадиен-стирольным кау-
каучуком лучше всего совмещают-
совмещаются т. наз. «легкие» (коэфф. реф-
рефn20D = 1,49 — 1,53)
B0 154 15
Таблица 2. Ассортимент наиболее распространенных
маслонаполненных бутадиен-стирольных каучуков
эмульсионной полимеризации, выпускаемых
в капиталистических странах
каучу-
II
1708
1712
1713
1714
1778
Страна
США, Италия
США, Канада, Австрия,
Италия, Нидерланды,
США, Италия, Нидер-
Нидерланды, ФРГ, Индия
США, Италия, Нидер-
Нидерланды, ФРГ
США, Канада, ФРГ,
Япония, Австралия
Тип масла
Нафтеновое
Высокоарома-
Высокоароматическое
Нафтеновое
Высокоарома-
Высокоароматическое
Нафтеновое
ичест-
асла,
ч.
37,5
37,5
50
50
37,5
юсть
[ука по
и при
С
Is Si
60
55
52
52
55
* О принципах подразделения каучуков на типы см. Бу-
Бутадиен-стиролъные каучуки.
Таблица 3. Основной ассортимент зарубежных
маслонаполненных стереорегулярных бутадиеновых каучуков
Марка каучука
Америпол СВ-441
Америпол СВ-442
Полисар Тактин-125 0
Полисар Тактин-1251
Полисар Тактин-12 52
Карифлекс BR-1251
Нипол BR-1441
Нипол BR-14 42
Страна
США
»
Канада
»
»
Франция
Япония
Тип масла
Высокоарома-
Высокоароматическое
Нафтеновое
Высокоарома-
Высокоароматическое
Нафтеновое
Высокоарома-
тическое
Нафтеновое
Высокоарома-
Высокоароматическое
Нафтеновое
й S ?
37,5
50
50
50
37,5
50
37,5
50
к р s
35
30
35
30
35
25-35
35
30
количество ингредиентов указано в расчете на 100
мае. ч. каучука).
В производстве Н. к. применяют минеральные масла
трех типов: 1) парафиновые, в состав к-рых входят гл.
обр. парафиновые углеводороды; 2) нафтеновые, содер-
содержащие в основном циклопарафины; 3) ароматические,
состоящие преимущественно из конденсированных аро-
матич. соединений. К высокоароматич. обычно относят
масла, содержащие не менее 80% ароматич. углеводоро-
углеводородов. Выбор типа масла определяется его совместимо-
совместимостью с полимером, влиянием на технологич. свойства
смесей и технич. показатели резин, а также стоимостью
и доступностью. К важным характеристикам масел
относятся также темп-ры их вспышки и застывания,
ракции D , ,)
«средние» (n20D = 1,54 — 1,59)
углеводороды, содержащие 2—4 кольца в
ароматич.
молекуле.
Получение. При изготовлении маслонаполненных
эмульсионных каучуков масло вводят в латекс (после
добавления регулятора мол. массы и отгонки непрореа-
гировавших мономеров) в виде водной эмульсии. По-
Таблица 4. Основной ассортимент зарубежных
маслонаполненных бутадиен-стирольных (получаемых
полимеризацией в растворе), изопреновых и этилен-
пропиленовых каучуков
Марка
каучука
Страна
Тип масла
Количест-
Количество масла,
мае. ч.
Бутадиен-стирольные каучуки
Солпрен 37 5
Солпрен 376
Солпрен 377
Стереон 7 50
AR-1710*
AR-1730*
IR-500
AR-2710**
AR-2740**
США
Япония
Нафтеновое
Нафтеновое
Высокоароматическое
37,
50
37,
37,
37,
Изопреновые каучуки
Нидерланды
Япония
Нафтеновое
Высокоароматическое
Этилен-пропиленовые каучуки
ДютpaлTER/54 5
Келтан 520x50
ОР-375
ОР-5 00
Италия
Нидерланды
ФРГ
Нафтеновое
Нафтеновое
37,5
25
37,5
37,5
100
50
37,5
50
* Сополимеры с различным содержанием стирола. ** Смеси
маслонаполненных изопреновых и бутадиеновых каучуков в
различных соотношениях.
следнюю приготовляют в специальных аппаратах с ме-
мешалками в присутствии эмульгаторов (напр., стеарата
триэтаноламина). Каучуки, синтезируемые в р-ре (напр.,
стереорегулярные бутадиеновые), можно смешивать
с маслами непосредственно после полимеризации.
Свойства. При введении в бутадиен-стирольный кау-
каучук ароматич. масел получают резиновые смеси с луч-
лучшей клейкостью и вулканизаты с более высокой проч-
прочностью при растяжении, но с меньшей эластичностью по
отскоку и морозостойкостью, чем при использовании
парафиновых и нафтеновых масел. При переходе от ма-
масел, содержащих «легкие» ароматич. углеводороды,
к маслам с «тяжелыми» (тг20?)>1,59) ароматич. углево-
углеводородами повышаются прочность при растяжении, со-
сопротивление раздиру, относительное удлинение, изно-
износостойкость вулканизатов, ухудшаются их эластичность
по отскоку и морозостойкость. Т. обр., вулканизаты
с оптимальными свойствами м. б. получены при напол-
335
НАПОЛНЕННЫЕ КАУЧУКИ
336
нении каучуков маслами, содержащими смеси различ-
различных углеводородов в соотношении, к-рое должно опре-
определяться областью применения изделий. С увеличением
содержания масла в Н. к. механич. свойства вулкани-
затов несколько ухудшаются (табл. 5).
Таблица 5. Влияние количества высокоароматического масла
на свойства вулканизатов бутадиен-стирольного каучука
Показатели
Модуль при растяжении 300%,
Мн/м* (кгс/см2)
Прочность при растяжении,
Мн/м2 (кгс/см2)
Относительное удлинение, %
Сопротивление раздиру, кн/м,
или кгс/см
при 20 °С
при 100 °С
Эластичность по отскоку, %
при 20 °С
при 100 °С
Темп-pa хрупкости, °С
Маслонаполнен-
ный каучук
20 мае.
ч. мас-
масла*
6,8 F8)
27 B70)
640
46
38
31
49
-49
37,5 мае.
ч. мас-
масла**
6,0 F0)
24 B40)
660
39
20
28
48
-45
Непапол-
ненный
кау-
каучук***
7,3 G3)
31 C10)
680
54
40
38
50
-53
* Состав смеси (мае. ч.): каучук—100; ZnO— 5; стеарино-
стеариновая к-та —2; сажа ДГ-100— 50; альтакс—1,5; дифенилгуани-
дин —0,3; сера —2; вулканизация 80 мин при 143 °С. **Состав
смеси (мае. ч.): каучук—100; ZnO—5; сажа ДГ-100 —40; аль-
альтакс—2,75; сера —2; вулканизация 60 мин при 143 СС. ***Сос-
тав смеси (мае. ч.): каучук—100; ZnO —5; стеариновая к-та —
1,5; сажа ДГ-10 0 —40; альтакс —3; сера —2; вулканизация
80 мин при 143 °С.
Применение. Маслонаполненные каучуки широко
применяют для изготовления протектора и каркаса
шин, при восстановительном ремонте протектора,
в производстве транспортерных лент, плоских привод-
приводных и клиновых ремней, резиновой обуви, губчатых
резин и др. Производство маслонаполненных каучу-
каучуков в различных странах составляет существенную
долю в общем объеме производства наиболее массовых
каучуков общего назначения. Так, в 1970 в США вы-
выпуск бутадиен-стирольных каучуков, наполненных вы-
высокоароматическими или нафтеновыми маслами, соста-
составил (в расчете на полимер) ок. 560 тыс. т; т. е. ок. 42%
от общего объема производства этих каучуков.
Саже- и сажемаслонаполненные каучуки
Благодаря равномерному распределению сажи такие
Н. к. обладают хорошими технологич. свойствами (рези-
(резиновые смеси на их основе легко шприцуются и каланд-
руются). Вулканизаты этих Н. к. имеют высокие тех-
нич. показатели. Продолжительность смешения саже- и
сажемаслонаполненных каучуков с ингредиентами в
среднем на 25—30% меньше, чем при изготовлении ре-
резиновых смесей из ненаполненных каучуков. Это не
только обусловливает повышение производительности
смесительного оборудования, но и уменьшает опас-
опасность под вулканизации резиновых смесей из Н. к. вслед-
вследствие их меньшего разогрева при смешении. При
работе с саже- и сажемаслонаполненными каучуками
облегчается изготовление резиновых смесей непре-
непрерывным методом, в среднем на 30% сокращается об-
общий расход электроэнергии, повышается культура
производства.
Применение саже- и сажемаслонаполненных стереоре-
гулярных бутадиеновых каучуков — наиболее эффек-
эффективный способ улучшения их технологич. свойств,
позволяющий повышать содержание этих каучуков в
резиновых смесях и улучшать т. обр. эксплуатацион-
эксплуатационные свойства изделий.
Для изготовления саженаполненных каучуков при-
применяют гл. обр. тонкодисперсные печные сажи из
жидкого сырья [напр., N 220(ISAF), N 330 (HAF)], что
объясняется лучшей диспергируемостью этих саж в ла-
тексах или р-рах каучуков, чем в твердых каучуках,
а также возможностью получения из этих Н. к. износо-
износостойких резин. Саженаполненные каучуки большинст-
большинства типов содержат небольшие количества E—12,5
мае. ч.) масла, к-рое вводят для облегчения после-
последующей переработки Н. к. Количество масла в са-
сажемаслонаполненных каучуках изменяется в преде-
пределах 35—62,5 мае. ч. в зависимости от содержания в них
сажи (табл. 6, 7).
Таблица 6. Основной ассортимент зарубежных саже-
и сажемаслонаполненных бутадиен-стирольных каучуков
эмульсионной полимеризации
Тип
Количе-
Количество
сажи,
мае. ч.
Количе-
Количество
высоко-
арома-
тич.
масла,
мае. ч.
Вязкость
каучука
по Муни
при
100 °С
1605
1606
1608
1609
1613
1805
1808
1809
1813
1814
1815
1820
1823
1824
1831
Саженаполненные каучуки
США, Италия,
Нидерланды
США, Канада,
Италия, Нидер-
Нидерланды
США, Италия
То же
США
N 330
(HAF)
То же
N 220
(ISAF)
N 110
(SAF)
То же
50
52
52
40
40
_
10
12,5
5
10
46 — 56
50-60
Сажемаслонаполненные каучуки
США, ФРГ
США, Канада,
Италия, Нидер-
Нидерланды, Индия То же 75 50 45 — 57
США, Канада
США
США, Канада,
Нидерланды То же 7 5 5 0 58
США, Нидерлан-
Нидерланды
США
США, Италия
США
США
N 330
(HAF)
То же
»
N 220
(ISAF)
То же
N 330
(HAF)
N 550
(FEF)
N 330
(HAF)
N220
(ISAF)
N 242
(ISAF-
-HS)
75
75
75
60
75
75
68,75
82,5
82,5
82,5
37,5***
50
37,5
37,5
50
50***
37,5***
62,5
62,5
62,5
* О принципах подразделения каучуков на типы см. Бута-
диен-стиролъные каучуки. ** О принципах обозначения типа
сажи см. Наполнители резин. *** Нафтеновое масло.
Получение. Технология получения саженаполненных
каучуков включает стадии приготовления суспензии
сажи в воде или в углеводороде и смешения этой сус-
суспензии с латексом или с р-ром каучука. В случае полу-
получения сажемаслонаполненных каучуков масло чаще
всего вводят в виде эмульсии.
При получении Н. к. на основе эмульсионных
бутадиен-стирольных каучуков ди-
дисперсию сажи готовят елед. образом. Сухую сажу сме-
смешивают в высокоскоростном смесителе или в аппарате
с мешалкой с водным р-ром диспергирующего агента.
При этом образуется 10—22%-ная суспензия сажи.
Диспергирующими агентами служат натриевая или ка-
калиевая соль продукта конденсации Р-нафталинсульфокис-
лоты с формальдегидом (лейканол, даксад), лигносуль-
фоновая к-та и ее соли, мыла к-т канифоли и др.; коли-
количество этих агентов (обычно 0,2—4,0% от массы сажи)
337
НАПОЛНЕННЫЕ КАУЧУКИ
338
Таблица 7. Основной ассортимент зарубежных
сажемаслонаполненных бутадиеновых каучуков
Марка каучука
Тип сажи*
Коли-
Количество
сажи,
мае. ч.
Количест-
Количество высоко-
ароматич.
мазла,
мае. ч.
Каучук и, полученные полимеризацией
в растворе
Цис-4-1350**
Цис-4-1351**
Америпол СВ-микроблек
СВ-460***
Америпол СВ-микроблек
СВ-464***
N 220 (ISAF)
То же ....
N 347 (HAF-HS)
N 242 (ISAF-HS)
80
90
75
90
35
50
37,5
58
Каучуки эмульсионной полимеризации
Синпол E-BR 8454А
Синпол E-BR 8455А
Синпол 845 8
N 330 (HAF) . . .
N 220 (ISAF) . .
N 3 47 (HAF-HS)
75
69
82,5
37,5
37,5
62,5
* О принципах обозначения типа сажи см. Наполнители ре-
резин. * * Получены смешением р-ра каучука с суспензией сажи в
углеводороде. *** Получены смешением р-ра каучука с водной
суспензией сажи.
определяется уд. поверхностью, рН и зольностью са-
сажи. Суспензию сажи перемешивают с латексом до обра-
образования однородной смеси, к-рую затем коагулируют
(напр., с помощью H2SO4). Наиболее равномерное рас-
распределение сажи в каучуке достигается в том случае,
когда коагулирующий агент вводят в саже-латексную
смесь при быстром перемешивании (т. наз. «мгновен-
«мгновенная» коагуляция).
При изготовлении суспензии в отсутствие дисперга-
тора сажу (обычно гранулированную) подают в измель-
измельчитель, где ее одновременно смешивают с водой. Обра-
Образовавшуюся 5—6%-ную суспензию сажи смешивают
с латексом.
При получении сажемаслонаполненных каучуков
латекс можно одновременно смешивать с суспензией
сажи и эмульсией масла или производить смешение
в две стадии. В последнем случае сначала получают
смесь латекса и эмульсии масла. После интенсивного
перемешивания смесь всех компонентов поступает в ем-
емкость с коагулирующим агентом, где происходит «мгно-
«мгновенная» коагуляция.
В США разработан эффективный способ диспергиро-
диспергирования сажи в аппарате непрерывного действия типа
«Джет-о-Майзер» (этот способ применяют также мно-
многие фирмы Италии и Канады). В аппарат подают сажу
и перегретый водяной пар, к-рый конденсируется на са-
саже. Смоченная паром сажа поступает в размольную
камеру. Мелкие частицы сажи A—2 мкм) образуют
с водой пастообразную массу, к-рая немедленно вводит-
вводится в латекс, а частицы большего размера поступают на
повторное измельчение.
Нек-рые зарубежные фирмы изготовляют саже- и са-
жемаслонаполненные эмульсионные каучуки путем
введения сухой сажи в частично астабилизированный
(флокулированный) и загущенный латекс или в мокрый
коагулюм каучука в резиносмесителях или в др. аппа-
аппаратах, где создаются большие напряжения сдвига,
позволяющие одновременно интенсивно перемешивать
каучук с сажей и удалять влагу из Н. к. Этот метод
дешевле рассмотренных выше, дает возможность изго-
изготовлять различные Н. к., но имеет и ряд недостатков:
1) сажа хуже распределяется в каучуке, чем в случае
предварительного изготовления ее суспензии; 2) Н. к.
не могут быть получены непрерывным способом; 3) ис-
исключается стадия промывки Н. к., что ухудшает их ка-
качество.
При изготовлении Н. к. на основе каучуков,
синтезируемых полимеризацией
в растворе, сажа м. б. предварительно дисперги-
диспергирована в углеводороде (обычно в том же растворителе,
к-рый используют для полимеризации) или в воде. Пер-
Первый способ применяют при получении нек-рых напол-
наполненных стереорегулярных бутадиеновых каучуков (см.
табл. 7), второй — при получении наполненных изопре-
новых, этилен-пропиленовых, бутадиеновых каучуков,.
бутилкаучука.
Аппаратурное оформление процесса получения Н. к.
при диспергировании сажи в воде сложнее, чем в слу-
случае ее диспергирования в углеводороде. Однако этот
способ более экономичен и позволяет изготовлять Н. к.,
содержащие одновременно бутадиен-стирольный и сте-
реорегулярный бутадиеновый каучуки. Такие комби-
комбинированные Н. к. можно получать, напр., перемеши-
перемешиванием р-ра бутадиенового каучука с латексом бута-
диен-стирольного каучука, введением в эту смесь вод-
водной суспензии сажи и эмульсии масла, гомогенизацией
всей системы в скоростных смесителях, типа коллоид-
коллоидных мельниц, коагуляцией латекса к-тами (напр.,
H2SO4) и выделением смеси Н. к. путем отгонки раст-
растворителя или осаждения в горячей воде (95—97 °С).
Комбинированные саженаполненные Н. к. весьма пер-
перспективны для производства шинных протекторов.
Напр., в протекторах из резин на основе таких Н. к., со-
содержащих свыше 30% бутадиенового каучука, прак-
практически устраняются растрескивание канавок, сколы
и др. дефекты. В промышленном масштабе Н. к., по-
получаемые смешением латексов и р-ров каучуков, не
производят. В Японии выпускают сажемаслонапол-
ненную смесь каучуков марки СН-45, содержащую 50
мае. ч. бутадиенового каучука, 50 мае. ч. бутади-
ен-стирольного каучука, 100 мае. ч. сажи типа N 330<
(HAF) и 30 мае. ч. высокоароматич. масла. Смесь по-
получают введением сажи и масла в твердые каучуки
в резиносмесителе.
Свойства. Выше уже отмечалось, что Н. к. обладают
лучшими технологич. свойствами, чем исходные кау-
каучуки. Кроме того, из саже- и сажемаслонаполненных
каучуков получают вулканизаты, обладающие высокой
износостойкостью и выносливостью при многократных
деформациях. На механич. свойства вулканизатов
Н. к. оказывает влияние не только тип сажи, но также
и способ ее введения в каучук (табл. 8).
Применение. Основная область применения саже-
и сажемаслонаполненных каучуков — производства
шинных протекторов. Эти Н. к. используют также для
изготовления различных резино-технич. изделий, рези-
резиновой обуви и др. Способы получения саже- и сажемас-
сажемаслонаполненных каучуков постоянно совершенствуются;
объем их потребления непрерывно растет. Так, если
в 1957 производство саже- и сажемаслонаполненных
бутадиен-стирольных каучуков в США составляло
(в расчете на полимер) 56 тыс. т, то к 1970 оно достиг-
достигло — 260 тыс. т (соответственно 8,4 и 25,0% от выпуска
бутадиен-стирольных каучуков низкотемпературной
полимеризации).
Каучуки, наполненные синтетическими пластиками
В качестве наполнителей наибольшее распростране-
распространение получили продукты эмульсионной сополимериза-
ции стирола с бутадиеном, акрилонитрилом или
нек-рыми др. мономерами, содержащие более 50%
стирола. Применяют также феноло-, анилино-, резор-
цино-, мочевино- и меламино-формальдегидные смолыг
поливинилхлорид и др.
Каучуки, наполненные сополиме-
сополимерами стирола, получают смешением их латексов
с латексами каучуков и последующей совместной коа-
коагуляцией. Введение сополимеров стирола в натураль-
натуральный или синтетич. каучуки облегчает каландрование
339 наполнители лакокрасочных материалов 340
Таблица 8. Влияние типа сажи и способа ее введения на свойства вулканизатов бутадиен-стирольного каучукаа
Показатели
Сажа типа N 3 30 (HAFN
Саженаполненный каучук
типа 1606в
II
Ненаполнен-
ный каучук
типа 15 00г
Сажа типа N 220 (ISAFN
Саженаполнен-
Саженаполненный каучук
типа 1608д
Ненаполненный
каучук типа
1500е
Модуль при растяжении 300%, Мн/м2,
(кгс/см2)
Прочность при растяжении, Мн/м2 (кгс/см9-)
при 20°С
при 100°С
Сопротивление раздиру, кн/м, или кгс/см
при 20°С
при 100°С
Эластичность по отскоку, %
при 20°С
при 100°С
Относительная износостойкость, усл. ед. . . .
Выносливость при многократном растяжении
на 100%, тыс. цикловж
11,5A15)
26,5 B65)
11,5 A15)
57
30
40
53
122
60,5
14,5 A45)
25,5 B55)
9,5 (95)
54
22
25
40
109
27,5
14 A40)
24,5 B45)
10 A00)
57
25
42
56
100
60
12 A20)
27,4 B74)
12 A20)
60
45
33
52
116
11,5 A15)
23 B30)
10,5 A05)
60
40
34
53
100
а Обозначение типов Н. к. см. в ст. Бутадиен-стиролъные каучуки. Обозначение типов сажи см. в ст. Наполнители резин.
в Состав смесей (мае. ч.): каучук+сажа—147,5; стеариновая к-та—1,5, альтакс—2,0; ZnO—5,0, сера—2,0; I—сажа дисперги-
диспергирована перед введением в латекс с помощью перегретого пара в аппарате непрерывного действия, II—сухая сажа введена в ре-
зиносмесителе во флокулированный латекс. г Состав смеси (мае. ч.): каучук-91,0; сажа—47,5; масло ПН-6—9,0; стеариновая
к-та—1,5; альтакс—2,0; ZnO—5,0; сера—2,0. д Состав смеси (мае. ч.): каучук+сажа—146,0; стеариновая к-та—1,5; альтакс—2,0;
ZnO—5,0; сера—2,0; е Состав смеси (мае. ч.): каучук—89,0; сажа—46; масло ПН-6—11,0; стеариновая к-та—1,5; альтакс—2,0;
ZnO—5,0; сера—2,0. ж Испытания на образцах с проколом; отношение гост/М30о=0,25 A0СТ—остаточное удлинение, М300—модуль
при растяжении 300%).
и шприцевание резиновых смесей. Вулканизаты таких
Н. к. характеризуются более высокой твердостью, изно-
износостойкостью, стойкостью к действию воды, к-т, щело-
щелочей, алифатич. растворителей, чем вулканизаты соот-
соответствующих ненаполненных каучуков. Резины из
бутадиен-стирольного каучука, наполненного сополи-
сополимерами стирола, обладают высокой прочностью при
статич. и динамич. нагрузках. Улучшение свойств ре-
резин объясняют участием сополимеров стирола, способ-
способных структурироваться под действием обычных вулка-
вулканизующих агентов, в образовании вулканизационной
сетки. Усиливающее действие этих сополимеров про-
проявляется при введении их в каучук в количестве не
менее 10 мае. ч. Из каучуков, содержащих 20—30 мае. ч.
сополимеров стирола, получают эластичные вулкани-
вулканизаты, до 50 мае. ч.— кожеподобные материалы, свыше
50 мае. ч.— пластики.
Бутадиен-стирольные каучуки, наполненные сопо-
сополимерами стирола, выпускают в промышленном мас-
масштабе в ряде стран. Основная область их применения —
изготовление резины для подошвы.
Каучуки, наполненные терморе-
термореактивными смолами, м. б. получены след.
способами: 1) введением в латекс каучука заранее
синтезированных смол (форполимеров); 2) синтезом
смол в среде латекса. Смеси каучуков со смолами
выделяют из латекса желатинированием или коагуля-
коагуляцией. Такие Н. к. легко обрабатываются на вальцах,
хорошо смешиваются с обычными ингредиентами. Ре-
Резиновые смеси на их основе характеризуются хорошей
клейкостью, но повышенной склонностью к подвул-
канизации. В результате введения смол повышаются
твердость, прочность при растяжении, износо- и тепло-
теплостойкость вулканизатов, а также их стойкость к дейст-
действию масел и растворителей. Усиливающее действие
смол объясняется их способностью образовывать химич.
связи с макромолекулами каучука. На основе Н. к.,
содержащих до 30 мае. ч. смол (оптимальное количест-
количество 10—15 мае. ч.), получают эластичные вулканизаты,
до 60 мае. ч.— кожеподобные материалы, свыше
60 мае. ч.— порошкообразные пластики.
Каучуки, наполненные смолами на стадии латекса,
в промышленном масштабе не выпускают. Несомненно,
однако, что такой способ получения Н. к. более эффек-
эффективен, чем совмещение твердого каучука со смолой на
смесительном оборудовании, т. к. в первом случае
образуются более однородные композиции с лучшим
комплексом свойств.
Каучуки, наполненные лигнином
При получении этих Н. к. водный р-р щелочного суль-
сульфатного лигнина смешивают с латексом каучука при
нагревании и затем коагулируют смесь. Кроме каучу-
каучуков, наполненных одним лигнином, м. б. также полу-
получены лигниномаслонаполненные каучуки. Достоинства
лигнинонаполненных каучуков — небольшая плот-
плотность и возможность изготовления на их основе цвет-
цветных изделий. В резиновые смеси на основе этих Н. к.
можно вводить значительные количества др. наполни-
наполнителей, в том числе сажу, и получать т. обр. высокока-
высококачественные и дешевые резиновые изделия. Наполнен-
Наполненный бутадиен-стирольный каучук типа 1500, содержа-
содержащий 70 мае. ч. лигнина (марка каучука LG-70), выпу-
выпускают в промышленном масштабе в Японии.
Лит.: Справочник резинщика. Материалы резинового про-
производства, М., 1971; Синтетический каучук, под ред. Г. С. Уит-
би, пер. с англ., Л., 1957; Усиление эластомеров, под ред. Дж.
Крауса, пер. с англ., М., 1968; Производство, свойства и при-
применение саже- и сажемаслонаполненных каучуков эмульсион-
эмульсионной и растворной полимеризации, ЦНИИТЭНефтехим, М., 1973;
Janssen H. J. J., Weinstock К. V., Rubb. Chem.
Technol., 34, № 5, 1485 A961); Canad. Chem. Process., 55, № 3,
39 A971); Pickett W., Rubb. Age, 103, № 6, 69 A971);
Бурджесс, Хиршфилд, Стоке, Химия и технол.
полимеров, № 8, 138 A966); Rubb. J., 154, № 2, 30 A972);
Godfrey N. Н., Polymer Age, 3, № 7, 270 A972).
В. И. Гусева, Ф. С. Кантор, Н. И. Троицкая, Л. Е. Полуэктова.
НАПОЛНИТЕЛИ ЛАКОКРАСОЧНЫХ МАТЕРИА-
МАТЕРИАЛОВ (extenders of paint materials, Fullstoffe fiir Anstri-
chstoffe, charges des peintures et vernis) — высокодис-
высокодисперсные неорганич. продукты, нерастворимые в пленко-
пленкообразующих веществах. Назначение Н. л. м.— улучше-
улучшение малярно-технич. свойств лакокрасочных материалов
и эксплуатационных характеристик лакокрасочных по-
покрытий, а также экономия пигментов лакокрасочных
материалов. Наиболее часто используют белые Н. л. м.
(белизна не менее 90% по отношению к MgO, служаще-
служащему эталоном белого цвета). Серые и цветные Н. л. м,
находят лишь ограниченное применение.
По происхождению Н. л. м. делят на природные
(минеральные) и синтетические (осажденные из p-poB)s
341
НАПОЛНИТЕЛИ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
Свойства наиболее распространенных наполнителей лакокрасочных материалов
342
Название и химич. ф-ла
Форма
частиц
Размер
частиц,
мкм
Маслоем-
кость, г/кг
(г на 100 г)
Показатель
преломле-
преломления nD
Плотность,
г/см3
рН водной
вытяжки
Твердость
по минера-
логич.
шкале (по
Моосу)
П р и р о д н о-д исперсные наполнители
Бентонит (монтмориллонит)
Al2O3-4Si(V2H2O ....
Диатомит (инфузорная земля, кизельгур)
SiO2 (аморфная)
Доломит CaMg (СО3J
Каолин Al2O3-2SiO2-2H2O , .
Мел** СаСОз , . . ,
Пластинчатая
Неправильная
зернистая
Зернистая
Пластинчатая
Зернистая
0,2-2,0
1-6
5-20
0,5-10,0
2-50
300—700
C0-70)
900-1400*
(90-140)
150-190
A5-19)
280-600
B8-60)
90-160
(9-16)
1,58
1,45-1,49
1 ,62
1 ,56
1 ,48 — 1 ,68
1,90-2,40
2,3
2,85
2,58
2,6-2,9
Природные механически диспергируемые наполнители
Ангидрит (легкий шпат) CaSO4
Асбест хризотиловый, антофилитовый
3MgO-2SiO2-2H2O
Барит (тяжелый шпат) BaSO4
Вермикулит
Mg (Fe2+, Fe3+K [Si4 А14О10]ЧОНJ.пН2О
Витерит Ва-СОз
Волластонит (метасиликат кальция)
CaSiO3
Гипс CaSO4-2H2O
Пирофиллит Al2O3-4SiO2-H2O
Слюда (мусковит) K2O-3Al2O3-6SiO2'2H2O
Тальк 3MgO-4SiCvH2O
Зернистая
Волокнистая
Зернистая
Чешуйчатая
(пачки)
Зернистая
Волокнистая
игольчатая
Моноклинная
крупнокрис-
таллич.
Волокнистая
Пластинчатая
слоистая
Чешуйчатая
(листованная)
2-30
10-70
2-3
10-35
5-10
15 — 100
5-40
200-250
B0-25)
155A,55)
60-140
F-14)
300—400
C0-40)
250 — 300
B5 — 30)
300 C0)
400-900
D0-90)
150-700
A5-70)
1 ,60-1 ,65
1,53-1,57
1,64
1,53-1,67
1,63
1,53
1,57
1,57-1,59
1,57
Аэросил*** SiO2 . .
Белая сажа SiO2-H2O
Бланфикс BaSO4 . .
Гидроокись алюминия (типографские бе-
белила) А1(ОН)з
Карбонат бария ВаСО3 , . . .
Окись алюминия А12О3
Синтетические наполнители
1,45
1,45-1,55
Зернистая
Зернистая
0,005 —
0,040
0,02-
0,10
0,5-2,0
0,01-
0,20
0,2-5,0
1700 — 3000
A70-300)
1000-2000
A00 — 200)
10 0 — 65 0
A0-65)
550-1500
E5-150)
170-220
A7-22)
1,64
1 ,55 — 1 ,97
1,63
1 ,76
2,85-3,0
2,4-3,2
4,45-4,53
2,4-2,7
4,2-4,3
2,8-2,9
2,3-2,35
2,7
2,7-3,1
2,77 — 3,20
1 ,95 — 2,3
2,1-2,2
4,4
2,42
4,3
3,9
4-6
7-9,9
9,9 — 10
4,5
9,3
6,5 — 7
6,5-7
9,6
7-8
6, 3
7,5-7,7
8,1-9,6
4-6
8-10
7,0
3,5-4,0
1
3
3 — 3,5
2-3
3-3,5
1-1,5
3-3,5
4,5-5
1 ,5
1
2-3
* Маслоемкость кизельгура 1000—2200 г/кг A00—220 г на 100 г). ** Аналогичными свойствами обладает механически диспер-
диспергируемый кальцит. *** Продукт пиролитич. разложения галогенидов кремния (подробно см. Наполнители резин).
Первые, в свою очередь, разделяют на природно-дис-
персные и механически диспергируемые (таблица). По
степени дисперсности Н. л. м. делят на обыкновенные
(размер частиц 5—100 мкм) и микронизированные
(до 90% частиц с размером менее Б мкм при отсутствии
частиц более 20 мкм), получаемые измельчением обыкно-
обыкновенных Н. л. м. в струйных мельницах.
Свойства Н. л. м. характеризуют теми же пока-
показателями, что и свойства пигментов: формой и размером
частиц, маслоемкостью, твердостью, рН водной вытяж-
вытяжки, уд. поверхностью, диспергируемостью в пленкооб-
пленкообразующих, адсорбционной способностью, нераствори-
нерастворимостью в воде и в органич. растворителях. Основное
отличие Н. л. м. от пигментов — меньший показатель
преломления (nD = 1,45—1,65), близкий к показателю
преломления растительных масел и синтетич. смол.
Этим обусловлена малая'укрывистость (см. Пигменты
лакокрасочных материалов) наполнителей в лакокрасоч-
лакокрасочных материалах на основе указанных пленкообразую-
пленкообразующих. В средах с меньшим показателем преломления,
чем у Н. л. м., последние становятся укрывистыми
(напр., мел в клеевых красках). Т. обр., разделение
компонентов лакокрасочных материалов на наполните-
наполнители и пигменты в известной мере условно. Обычно
Н. л. м. вводят в количестве 25—50% от массы белых
(ZnS, TiO2, ZnO) или цветных пигментов. Укрывистость
пигментов при этом практически не ухудшается.
Наполнители не только заменяют в лакокрасочных
материалах часть пигментов, но и выполняют собствен-
собственные специфич. функции. Так,Н. л. м., образующие коа-
гуляционные структуры (бентонит, каолин, аэросил),
обусловливают повышение вязкости («загущение»),
а Н. л. м. с низкой маслоемкостью (барит, кварц, слю-
слюда) — уменьшение вязкости («разжижение») лакокра-
лакокрасочных материалов. Добавки бентонита, аэросила, ми-
кронизированных Н. л. м. (микротальк, микрослюда,
микродоломит), а также нек-рых специальных продук-
продуктов (стеараты или нафтенаты Al, Zn, Ca, Mg) изменяют
тиксотропные свойства лакокрасочных материалов (см.
Тиксотропия). Небольшие добавки Н. л. м. A,0—1,5%
от массы пигментов), образующих коагуляционные
структуры, уменьшают скорость осаждения пигментов
из лакокрасочных систем при их хранении. Эффектив-
Эффективность стабилизирующего действия Н. л. м. повышает-
повышается в случае их модификации органич. поверхностно-
активными веществами. Н. л. м. основного характера
343
НАПОЛНИТЕЛИ ПЛАСТМАСС
344
модифицируют анионными (напр., жирными к-тами),
Н. л. м. кислого характера — катионными (напр., ами-
аминами с длинной углеводородной цепью) поверхностно-
активными веществами. В результате модификации
снижается также маслоемкость Н. л. м. и улучшается
их совместимость с пленкообразующими.
При совместном применении пигментов и Н. л. м.
с частицами различной формы и размера достигается
более равномерное распределение частиц одного мате-
материала между частицами другого и увеличивается плот-
плотность их упаковки, а, следовательно, и т. наз. объемная
концентрация пигмента в пленке. Благодаря этому
уменьшается расход дорогих пленкообразующих и
улучшается качество пленок, в частности повышается
их атмосферостойкость и прочность. Последнее особен-
особенно важно для шпатлевок и грунтовок, т. к. покрытия
из этих материалов обычно шлифуют. Для увеличения
объемной концентрации пигмента наиболее широко при-
применяют тяжелый и легкий шпат, доломит, мел, тальк,
слюду, кварц. Оптимальное содержание каждого Н. л. м.
устанавливают по данным маслоемкости его компо-
композиций с пигментами в данном пленкообразующем.
Высокоукрывистые пигменты с большой красящей
способностью (особенно органические) окрашивают под-
подложку в свой цвет при толщине пленки 5—10 мкм,
недостаточной для эффективной защиты изделия от
атмосферных и др. вредных воздействий. Наполнение
лакокрасочных материалов тяжелым шпатом, блан-
бланфиксом, слюдой, тальком, асбестом приводит к увели-
увеличению толщины покрытия и повышению его атмосфере-,
влаго-, кислото- и термостойкости. В случае приме-
применения в качестве пигмента TiO2 рутильной модифика-
модификации атмосферостойкость покрытий резко возрастает
при введении 25% слюды или 35—50% талька (от
массы TiO2).
Частицы Н. л. м. пластинчатой или волокнистой
формы (слюда, вермикулит, волластонит, тальк, асбест
и др.) армируют пленку, позволяют, уменьшить уса-
усадочные явления и, следовательно, снизить опасность
растрескивания покрытий. Введение волокнистых
Н. л. м. предотвращает также стекание лакокрасочных
материалов при их нанесении на окрашиваемую по-
поверхность, придает пленкам эластичность и способ-
способность поглощать вибрацию.
Высокомаслоемкие Н. л. м. (диатомит, аэросил, ки-
кизельгур, каолин, микронизированные тальк и мел)
матируют пленки (уменьшают их глянец) и устраня-
устраняют неприятный неравномерный блеск волнистых по-
покрытий.
При использовании т. наз. наполнителей-
пигментов (окись и гидроокись алюминия, суль-
сульфат кальция и др.) получают прозрачные лакокрасоч-
лакокрасочные материалы (шпатлевки, полиграфические краски,
порозаполнители для дерева). Нек-рые Н. л. м. при-
придают лакокрасочным пленкам специальные свойства.
Так, графит и высокодисперсные металлич. порошки
повышают, а слюда и кварц понижают электрич. про-
проводимость пленок, углекислый цинк (витерит) и угле-
углекислый барий снижают их горючесть, бланфикс и
окислы свинца сообщают пленкам непроницаемость
к рентгеновским лучам.
При выборе Н. л. м. следует учитывать их реакцион-
реакционную способность. Так, напр., сульфат кальция может
ускорять коррозию в грунтовках по металлу, карбо-
карбонаты кальция и глиноземы — вызывать загустевание
масляных и алкидных связующих с высокими кислот-
кислотными числами, а также разрушать пигменты, не стой-
стойкие к действию щелочей. В водоразбавляемых латекс-
ных красках высокодисперсные Н. л. м. могут адсор-
адсорбировать поверхностно-активные вещества и нарушать
т. обр. агрегативную стабильность системы. Присут-
Присутствие в Н. л. м. водорастворимых солей может вызы-
вызывать коагуляцию водных дисперсных систем.
Лит.: П э й н Г. Ф., Технология органических покрытий,
пер. с англ., т. 2, Л., 1963, с. 114; Hunk a r D., Deutsche
Farben-Zeitschrift, 20, Н. 2, 39 A966); 21, № 3, 133 A967);
Meyer G., там же, 21, Н. 1, 7 A967); Остроумо-
Остроумова Т. С, К и з н е р Н. А., Ш т е р н М. А., Лакокрасочные
материалы и их применение, № 3, 1 A969); Белень-
Беленький Е. Ф., в кн.: Природные минеральные наполнители,
[ч.] 2, вып. 95, М., 1963, с. 104. П. И. Ермилов.
НАПОЛНИТЕЛИ ПЛАСТМАСС (fillers of plastics,
Ftillstoffe fur Kunststoffe, charges des plastiques) —
твердые, жидкие, газообразные неорганические или
органические вещества, к-рые вводят в термо- и реак-
топласты. Наполнители используют для улучшения
эксплуатационных характеристик пластмасс, придания
им различных специфич. свойств и снижения стоимо-
стоимости. Наполненные пластмассы применяют гл. обр. как
конструкционные материалы, механич. прочность
к-рых определяется прочностными и деформационными
характеристиками полимерной матрицы и наполнитечя
(см. также Наполнение). От наполнителя в значитель-
значительной степени зависят также технологич. свойства пласт-
пластмасс и возможные способы переработки их в изделия.
Содержание наполнителя в пластмассах может изме-
изменяться в широких пределах. Во многих случаях оно
составляет 45—50% (в расчете на массу полимера). Су-
Существуют также высоконаполненные пластмассы,
в к-рых содержание наполнителя может в 3 и более
раз превышать содержание полимера.
Наиболее распространенные Н. п.— твердые (табл.
1—3), представляющие собой высокодисперсные поро-
порошки, волокна, зерна (гранулы) различной формы,
листы и др. Многие Н. п. (напр., графит, стекло, ме-
металлы) применяют как в виде порошков или зерен, так
и в виде волокон, в том числе монокристаллических
(«усы»). В зависимости от характера взаимодействия
с полимером Н. п. условно делят на инертные (не из-
изменяющие свойств полимера) и активные (упрочняю-
(упрочняющие). Активные волокнистые Н. п. наз. также армирую-
армирующими (см. Армированные пластики).
Помимо общих требований к наполнителям для поли-
полимерных материалов (способность совмещаться с поли-
полимером или диспергироваться в нем с образованием од-
однородных композиций, хорошая смачиваемость распла-
расплавом или р-ром полимера, стабильность свойств при
хранении, а также при переработке и эксплуатации
материала), к Н. п. предъявляют нек-рые специальные
требования, определяемые типом перерабатываемого
полимера.
Различный характер процессов, протекающих при
получении изделий из термо- и реактопластов (см. Пе-
Переработка пластических масс), обусловливает нек-рые
отличия в требованиях к наполнителям для этих пла-
пластиков.
Наполнители для реактопластов, к-рые перерабаты-
перерабатывают обычно в виде расплавов или р-ров с относительно
невысокой вязкостью, м. б. более грубодисперсными
и менее однородными по размеру частиц, чем наполни-
наполнители для термопластов. Наполнители для реактопла-
реактопластов (или содержащиеся в этих Н. п. примеси) не долж-
должны оказывать каталитич. действия на отверждение
полимера. Желательно также, чтобы эти Н. п. содер-
содержали функциональные группы, способные участво-
участвовать в образовании химич. связи полимер — наполни-
наполнитель (см. Наполнение). Частицы наполнителей для
термопластов должны иметь шероховатую поверхность,
т. к. это обеспечивает прочное механич. сцепление на-
наполнителя с поверхностью полимера. Наполнители
для пластифицированных термопластов должны обла-
обладать минимальной пористостью, т. к. в противном слу-
случае они могут поглощать содержащийся в пластмассе
пластификатор.
Порошкообразные наполнители. Полимерные мате-
материалы, содержащие эти Н. п., как правило, изотропны.
Для получения высокопрочных пластмасс целесообраз-
целесообразно применять Н. п. с наибольшей уд. поверхностью,
345
НАПОЛНИТЕЛИ ПЛАСТМАСС
346
т. е. с наименьшим размером частиц. Однако при вы-
выборе оптимальных размеров частиц Н. п., особенно
используемых для наполнения низковязких термореак-
термореактивных олигомеров, необходимо учитывать два фак-
фактора: 1) склонность частиц к агломерации, к-рая воз-
возрастает с ростом уд. поверхности Н. п.; 2) седимента-
седиментацию частиц, к-рая ускоряется с уменьшением уд. по-
поверхности, повышением плотности Н. п. и снижением
вязкости связующего. Введение тонкодисперсных Н. п.
связано с большими технологич. трудностями, т. к. со-
сопровождается загустеванием композиций и снижением
их текучести (см., напр., Компаунды полимерные). Как
правило, размер частиц Н. п. не должен превышать
40 жкж, чаще всего он составляет 1 —15 мкм. Для улуч-
улучшения технологич. свойств высоконаполненных компо-
композиций в отдельных случаях применяют Н. п. с разме-
размером частиц до 200—300 мкм.
Органические порошкообразные
наполнители. Наиболее распространенный на-
наполнитель — древесная мука, содержащая в качестве
основных компонентов целлюлозу и лигнин. Этот деше-
дешевый наполнитель широко применяют для получения
фенольных и мочевино-формальдегидных (карбамид-
ных) прессматериалов общего назначения. Недостатки
древесной муки (особенно из древесины лиственных
пород) — низкие тепло-, влаго- и химстойкость. За
рубежом применяют также муку из скорлупы орехов;
этот наполнитель позволяет получать пластмассы с
повышенными прочностными и электроизоляционны-
электроизоляционными свойствами.
Для повышения тепло- и химстойкости, а также жест-
жесткости пластмасс используют газовую канальную сажу
(см. Наполнители резин), измельченный кокс и
графит (см. Графитопласты). Химстойкость нек-рых
пластмасс м. б. повышена введением в их состав тонко-
тонкодисперсных поливинилхлорида, полиформальдегида,
полиэтилена. Для снижения коэфф. трения пластмасс
применяют политетрафторэтилен.
Неорганические порошкообраз-
порошкообразные наполнители. Наибольшее значение сре-
среди этих Н. п. имеют мел, каолин, тальк, слюда. В каче-
качестве Н. п. применяют мел различной дисперсности: мо-
молотый (размер частиц 5—20 мкм), дезинтегрированный
E—8 мкм), отмученный B—5 мкм), химически осаж-
осажденный (—0,4 мкм). Мел — один из важнейших на-
наполнителей для полиэтилена и поливинилхлорида.
Каолин (размер частиц —2 мкм) используют для напол-
наполнения этих же термопластов, а также при получении
премиксов. Тальк (размер частиц 3—5 мкм) и слюду
применяют для наполнения как термо-, так и реакто-
пластов, особенно при получении электроизоляцион-
электроизоляционных материалов.
Таблица 1. Влияние нек-рых наполнителей на свойства пластмасс
Наполнитель
ffl и
Свойства, придаваемые полимерному материалу
I
II
ран
II
п
л
а> о
со в
Древесные материалы
Древесная мука
Измельченная древесная
кора
Целлюлоза
p
p
T, P
+
+
:
+
+
+
Природные, искусственные, синтетич. волокна, порошкообразные
синтетич. полимеры
Джут
Сизаль
Хлопковое волокно
а-Целлюлоза
Вискозное волокно
Полиакрилонитрильное во-
волокно
Полиамидное волокно . . .
Политетрафторэтилен по-
порошкообразный
ft
ft -ftftft
T, P
T, P
T, P
+
i
i
x
+
+
+
+
+
+
+
i
+
+
+
i
I
+
i
t
i
Графит
Кокс порошкообразный .
Сажа газовая
Углеродное волокно . .
Алюминий пластинчатый
Алюминий порошкообраз-
порошкообразный
Окись алюминия порошко-
порошкообразная
Бронза порошкообразная . .
Асбест
Каолин
Каолин кальцинирован-
кальцинированный
Метасиликат кальция . .
Силикат кальция
Слюда
Стеклянное волокно . . .
Тальк
Двуокись кремния ....
Карбонат кальция ....
T, P
p
T, P
p
Углеродные материалы
+
+
Металлы, их окислы и сплавы
4-
+
+
Т, Р
р
т
р
4-
+
4-
+
4-
4-
4-
4-
4-
4-
4-
4-
+
4-
С
Т, Р
Т, Р
Т, Р
Т, Р
р
Т, Р
Т, Р
Т, Р
Т, Р
Т, Р
иликаты, двуо
+
4-
4-
+
4-
+
+
+
4-
+
4-
4-
¦+¦
+
+
4-
4-
4-
+
4-
+
4-
к и с ь
+
4-
+
+
+
+
4-
к р е м i
+
1 И Я, К
+
+
+
арб
4-
4-
+
+
4-
4-
4-
4-
4-
о н а т ы
+
+
4-
+
+
4-
4-
4-
4-
+
4-
4-
+
4-
+
4-
-г
4-
+
+
4-
+
4-
4-
*Р — реактопласт, Т — термопласт.
347
НАПОЛНИТЕЛИ ПЛАСТМАСС
348
Таблица 2. Свойства нек-рых волокон, применяемых в качестве наполнителей
Наполнитель
Плотность
при 25 °С,
г/см3
Диаметр
волок-
волокна3, мкм
Прочность при рас-
растяжении, Мн/м2
(кгс/см2)
Модуль упруго-
упругости, Ми/м2
(кгс/смг)
Бумага .
Сизаль .
Хлопок
Шерсть
Природные волокна
Вискозное волокно (рей-
он)
Полиакрилонитрильное
волокно (орлон) . . . .
Полиамидное волокно
(найлон)
Полиэфирное волокно
(дакрон)
Фторволокно
Волокна иг
Алюминий
Бор . .
Вольфрам^
Молибден0
Титан6
Окись^алюминия . . . .
Сталь6
Карбид бериллия
Углерод . . .
Графит . . .
То же («усы»)
Асбест .
Кварц .
Стекло .
0,7-1,2
1,3
1,6
1,3
к у с с т в е
1,5
1,2
1,14
1,4
2,2
металл
2,7
2,3
19,3
10,2 •
4,72
3,97
7,9
2,44
1-4
19
17
28
—
340 C400)
350—770
C500-7700)
20 B00)
иные и синтетич. вол
10—40
10-25
10—40
10-25
20
560E600)
350 C500)
840 (8400)
700 G000)
330 C300)
о в, их окислов, с п л a i
4-20
—
20
5-20
0,02
1-25
—
630 F300)
3,5-103C5-103)
4,3-103D3-103)
14 -103 A40 • 103)
2103B0 10з)
700 G000)
4,2-103D2-103)
1 ,1 • 103 A1 • 103)
Углеродные волокна
2,5
1,6-2,2
2,0-2,2
Вол
1-100
2- 30
-
3,5-103 C5-Ю3)
140 A400)
21 -Ю3 B10-Ю3)
окна из силикатов
—
окна
2,8-103B8.10*)
2,8-Ю3 B8-Ю3)
2,8-103B8-103)
4 , 2-103 D2-103)
2,8-Ю3 B80-103)
зов, карбидов
7-Ю3 G0-Ю3)
45.103D50.Ю3)
41-103 D10-103)
36-103C60-103)
12-Ю3 A20-Ю3)
53-103E30-103)
20-103B00-103)
3,2-103 C20-103)
20-103B00-103)
35-103C50-103)
-
135
100
135
100
200
230
250
250
270
660
2120
3430
2630
1680
2100
1600
2100
3700в
3700в
3700в
2,5
2,2
2,49-2,54
1-10
8-10
5-15
1 ,4-Ю3 A4-103)
7-103G0-103)
C,5-5)-103
[C5 —50)-103]
18-103A80-103)
7-103G0-103)
G,4-8,9)-Ю3
[G4-89)-103]
1510
1940
1310—
1630
Поверхность порошкооб-
порошкообразных Н. п. часто обраба-
обрабатывают р-рами или эмуль-
эмульсиями поверхностно-актив-
поверхностно-активных веществ, напр, стеаратов
металлов. Такая обработка
улучшает смачиваемость на-
наполнителя полимером, снижа-
снижает склонность частиц напол-
наполнителя к агломерации и по-
поглощению пластификаторов,
улучшает водостойкость и ди-
электрич. свойства полимер-
полимерных материалов. О свойствах
неорганич. порошкообразных
наполнителей см. также На-
Наполнители лакокрасочных ма-
материалов.
Содержание порошкообраз-
порошкообразных наполнителей составляет
обычно 25—50 мае. ч., в высо-
конаполненных пластмассах
оно может достигать 200—
300 мае. ч. на ^00 мае. ч. по-
полимера. Для введения таких
Н. п. применяют смесители
различных типов (валковые,
роторные, лопастные), вальцы
и др. Наполненные термопла-
термопласты обычно гранулируют;
прессматериалы на основе
реактопластов вальцуют для
более тщательной пропитки
наполнителя связующим, а
затем измельчают.
Волокна. В качестве Н. п.
могут применяться как не-
непрерывные, так и рубленые
(штапельные) волокна дли-
длиной от нескольких десятков
мкм до нескольких десятков
Максимальная длина волокон бумаги и молибдена 15 мм, стекла 5 мм, остальных волокон мм (см. табл. 2). В зависимо-
50 мм. ° Проволока. В бескислородной атмосфере.
Дешевые природные двуокись кремния (песок, кварц,
диатомиты) и силикаты (асбест, бентонит, вермикулит,
нефелин, пемзу и др.), к-рые имеют сравнительно невы-
невысокую плотность и хорошо совмещаются различными
полимерами, применяют для наполнения полиолефи-
нов, поливинилхлорида, полиамидов, полиуретанов,
эпоксидных и фенольных смол и др.
Все более широкое применение находят синтетич.
неорганич. Н. п.— SiO2, получаемая различными спосо-
способами (см. Наполнители резин), силикаты Al, Ca, Mg, Sr
и др. Синтетич. неорганич. Н. п. отличаются от природ-
природных большей чистотой, а также повышенной дисперсно-
дисперсностью и большей однородностью по размерам частиц.
Фториды и сульфаты нек-рых металлов, напр. Ва, Са,
повышают тепло- и химстойкость полимеров, дисуль-
дисульфид молибдена понижает коэфф. трения.
Порошки металлов и их сплавов (Fe, Си, Al, Pb,
бронза) придают пластмассам нек-рые специальные
свойства. При определенной концентрации такого на-
наполнителя, необходимой для непосредственного кон-
контакта между его частицами, резко повышаются тепло-
теплопроводность и электрич. проводимость полимерного
материала и, кроме того, материал становится стойким
к действию электромагнитного и проникающего излу-
излучений. Пластмассы, наполненные металлич. порошком
или стружкой (опилками), можно применять для изго-
изготовления различного инструмента и оснастки, заделки
дефектов в металлич. литье и т. д. (см. Металлонапол-
ненные полимеры).
сти от соотношения показате-
показателей механических свойств
полимера и наполнителя, размеров волокон, а также
от характера взаимодействия на поверхности разде-
раздела полимерная матрица — волокно последние могут
проявлять свойства как обычных дисперсных, так и
армирующих наполнителей, упрочняющее действие
к-рых весьма значительно вследствие реализации опре-
определенной доли прочности наполнителя. Для эффектив-
эффективного армирования термопластов длина волокна долж-
должна быть не менее 200 мкм; при наполнении реактопла-
реактопластов применяют волокна различной длины. Волокни-
Волокнистые наполнители пластмасс позволяют значительно
повысить физико-механич. свойства, тепло-, износо-,
химстойкость и др. показатели пластмасс. При исполь-
использовании волокон в виде непрерывных нитей получа-
получают изделия с исключительно высокими прочност-
прочностными показателями (см. Армированные пластики, Сте-
Стеклопластики).
Из органических волокон наиболее
широко применяют хлопок — в виде текстильных отхо-
отходов (коротковолокнистый линтер, очесы), измельчен-
измельченного волокна, нитей, обрезков ткани и др. Хлопок —
важнейший наполнитель карбамидных пресс-материа-
пресс-материалов (см. Аминопласты). Он легко окрашивается, обла-
обладает удовлетворительными физико-химич. и хорошими
диэлектрич. свойствами; его недостатки — значитель-
значительное водопоглощение и низкая химстойкость. Находят
применение и др. природные волокна — джут, сизаль,
рами, лен. Использование этих волокон в смеси с по-
порошкообразными наполнителями повышает ударную
349
НАПОЛНИТЕЛИ ПЛАСТМАСС
350
вязкость материала; их применение вместо стеклово-
стекловолокна позволяет уменьшить плотность, улучшить ди-
намич. характеристики пластмассы, однако химстой-
кость материала при этом снижается. Существенное до-
достоинство хлопковых волокон — дешевизна и доступ-
доступность.
В качестве Н. п. все более широко применяют синте-
тич. волокна, напр, полиамидные, полиэфирные, поли-
полиакрил онитри л ьные. Пластмассы, содержащие эти во-
волокна, характеризуются исключительно высокой кор-
коррозионной и химич. стойкостью, малым коэфф. трения
и высокой износостойкостью. Благодаря хорошей
адгезии синтетич. волокон к наполняемым поли-
полимерам такие пластмассы стойки к действию воды.
Недостаток этих Н. п.— сравнительно невысокая
теплостойкость, а также ограниченный выбор свя-
связующих, т. к. многие из них могут изменять структуру
и механич. свойства волокна. Повышение теплостой-
теплостойкости и механич. характеристик пластмасс достигается
применением полиимидных и полиимидазольных воло-
волокон, а также углеродных нитей; последние способны
выдерживать темп-ры выше 2000 °С (см. также Органо-
волокниты, Термостойкие волокна).
Важнейшие неорганические волоки а—
стеклянное и асбестовое. Ассортимент стеклянных во-
волокон очень широк. Их вводят в реакто- и термопласты,
иногда в со.четании с порошкообразными или с др.
волокнистыми наполнителями (см. Стеклопластики,
Стекловолокниты). При введении стеклянного волокна
повышаются физико-химич. показатели, понижается
коэфф. трения, улучшаются диэлектрич. свойства,
тепло-, износо- и химстойкость материала. Недостатки
стекловолокна как наполнителя — низкая адгезия
к нек-рым связующим, заметное снижение прочности
во влажных средах, а при наполнении термопластов —
анизотропия свойств получаемого изделия вследствие
ориентационных эффектов при переработке наполнен-
наполненного материала.
В качестве Н. п. применяют асбест двух видов —
змеевиковый (хризотил) и рогообманковый (крокидо-
лит). Первый имеет длинноволокнистую структуру и
характеризуется повышенной прочностью; волокна
второго значительно короче и отличаются повышенной
кислотостойкостью. Толщина асбестового волокна ок.
10 мкм, прочность достигает 3 Гн/м2 C00 кгс/мм2),
теплостойкость превышает 1000 °С. Волокно применяют
как наполнитель для термо- и реактопластов, а также
в производстве различных мастик, замазок и др. При
наполнении асбестом, который может быть использо-
использован как в виде распушенных измельченных волокон,
так и нитей или тканей, повышаются тепло-, огне-,
атмосфере- и химстойкость, а также ударная вязкость
пластмасс (см. Асбоволокнит, Асбопластики, Асбо-
текстолит).
Для наполнения пластмасс применяют волокна из
кварца, базальта, керамики (нитрид бора), а также ме-
таллич. проволоку (сталь, Fe, W, Ti) и волокна В, Be,
Mo, W. Особый интерес представляет применение мо-
нокристаллич. волокон (нитевидных кристаллов, или
«усов» — whiskers), к-рые получены из различных
металлов, их окислов, карбидов, нитридов и др., а так-
также т. наз. вискеризованных волокон, т. е. волокон из
различных материалов, гл. обр. углеродных, на по-
поверхности к-рых создан слой из нитевидных кристаллов.
Диаметр «усов» может достигать нескольких мкм, дли-
длина — нескольких мм] их относительное удлинение при
разрыве составляет 1—2%. Монокристаллич. волокна
отличаются исключительно высокими модулем упру-
упругости и прочностью при растяжении (см. табл. 3). При
их использовании в сочетании с высокопрочными
термореактивными связующими (содержание напол-
наполнителя может составлять 80% и выше) получают мате-
материалы, в к-рых удается реализовать до 50—75% проч-
Таблица 3. Прочность при растяжении объемных образцов
и монокристаллических волокон («усов»; из нек-рых
материалов [в Мн/м2 (кгс/см2)]
Материал
Алюминий
Железо
Медь
Карбид бора
Кремний
Объемные образцы
560 E600)
28 B80)
1,4 A4)
158 A580)
35 C50)
Монокристаллич.
волокна
43-103D30.103)
13-10» A30-10»)
2,8-103 B8-10»)
6,8-103 F8-1 О3)
3 , 8 • 1 О3 C8-Ю3)
ности наполнителя. Одновременно заметно возрастают
теплостойкость и др. свойства материала. Применение
«усов» и вискеризованных волокон ограничивается их
высокой стоимостью.
Содержание волокнистых наполнителей в термопла-
термопластах составляет обычно 15—40%, в реактопластах —
30—80% от массы полимерного материала. Способы
приготовления наполненных композиций м. б. самыми
различными. Так, в производстве волокнита наполни-
наполнитель пропитывают связующим с последующим удале-
удалением растворителя. При получении, напр., наполнен-
наполненных полиамидов непрерывное волокно покрывают на
экструдере оболочкой полимера, а затем материал дро-
дробят на гранулы. В нек-рых случаях рубленое стекло-
стекловолокно целесообразно вводить в мономер до полиме-
полимеризации или на промежуточной стадии синтеза полиме-
полимера (напр., при осаждении поликарбоната из его р-ра
в метиленхлориде). Твердые (порошкообразные) поли-
полимеры или их расплавы смешивают с наполнителями
в смесителях различных типов. В специальных методах
формования, напр, при намотке из нитей или лент, на-
нанесение связующего на наполнитель совмещается с про-
процессом собственного формования. См. также Армирован-
ные пластики, Стеклопластики, Органоволокнитыу
Стекловолокниты.
Наполнители в виде зерен. К этим новым Н. п. отно-
относятся: полые сферы (микробаллоны), получаемые из
стекла, углерода, полимеров и др.; стеклянные чешуй-
чешуйки и гранулы различной формы, гранулированные по-
полимеры и др. Размеры частиц таких Н. п. могут изме-
изменяться в широких пределах: диаметр полых сфер —
от 2 до 500 мкм, размер гранул может достигать не-
нескольких мм. Наполнители этого типа придают поли-
полимерным материалам коррозионную стойкость и благода-
благодаря наличию граней изменяют их оптич. характеристики
и регулируют коэфф. трения (устраняют про-
проскальзывание). При использовании полых сфер умень-
уменьшается плотность пластмасс, улучшаются их теплоизо-
теплоизоляционные свойства (см. также Пластики с полыми на-
наполнителями) .
Зернистые Н. п. смешивают с порошкообразным по-
полимером или вводят в его расплав; при смешении и по-
последующем гранулировании полимерного материала
применяют минимальные нагрузки во избежание раз-
разрушения наполнителя (особенно микробаллонов). По-
Потребление Н. п. в виде зерен непрерывно возрастает.
Листовые наполнители. Такие Н. п. служат основой
для получения слоистых пластиков. К листовым Н. п»
относятся бумага, ткани, шпон, холсты, сетки, пленки,
маты и др. материалы, имеющие, как правило, пористую
или волокнистую структуру. Материалы, полученные
с такими Н. п., обладают значительной анизотропией
свойств. Листовые наполнители применяют почти
исключительно для наполнения термореактивных поли-
полимеров. При получении листовых полимерных материа-
материалов эти Н. п. имеют ряд технологич. преимуществ перед
др. наполнителями. Обычно Н. п. пропитывают или по-
покрывают с одной стороны (лакируют) р-ром или рас-
расплавом связующего (см. Пропитка наполнителей).
В нек-рых случаях порошкообразный полимер наносят
поверх каждого слоя наполнителя.
351
НАПОЛНИТЕЛИ РЕЗИН
352
Жидкие наполнители. В качестве Н. п. применяют,
например, воду и минеральные масла. Воду использу-
используют при получении жестких материалов на основе по-
полиэфирных смол; минеральные масла — для сохране-
сохранения слоя смазки на поверхностях трения. Пластмассы
с жидкими наполнителями получают отверждением
стабильных эмульсий, в которых наполнитель явля-
является дисперсной фазой, а полимер — дисперсионной
средой.
Газообразные наполнители. К этим Н. п. относятся
газы (СО2, N2, NH3) и низкокипящие углеводороды
(пентан, изооктан и др.), применяемые для вспенива-
вспенивания полимерных материалов. Углеводороды вводят
обычно на стадии полимеризации или приготовления
полимерной композиции, газы (или вещества, разла-
разлагающиеся при нагревании с выделением газообразных
продуктов) — непосредственно при формировании изде-
изделий. Газонаполненные материалы характеризуют обыч-
обычно кажущейся плотностью, к-рая может составлять
0,01—0,02 г/см3 и менее. См. также Порообразователи,
Пенопласты.
Лит.: Николаев А. Ф., Синтетические полимеры и
пластические массы на их основе, 2 изд., М.— Л., 1966; Пет-
Петров Г. С, Левин А. Н., Термореактивные смолы и пла-
пластические массы. М., 1959; П л о т к и н Л. Г., Ша-
Шалун Г. Б., Декоративные бумажно-слоистые пластики, [М.],
1968; Современные композиционные материалы, под ред. Л. Бра-
утмана и Р. Крока, пер. с англ., М., 1970; Modern plastics
encyclopedia, N. Y., 1963, 69, 70; Materials and compounding
ingredients for rubber and plastics, N. Y., [1965]; Encyclopedia
of polymer science and technology, v. 6, N. Y.— [a. o.], 1967,
p. 740. M. Л. Кербер.
НАПОЛНИТЕЛИ РЕЗИН (fillers of rubbers, Ful-
Istoffe fur Kautschuk, charges des caoutchoucs) — вы-
высокодисперсные неорганич. или органич. вещества,
к-рые вводят в твердый каучук или в латекс. Н. р.,
занимающие существенную долю общего объема рези-
резиновой смеси (чаще всего 10—25%), могут выполнять
следующие основные функции: 1) изменять механичес-
механические показатели резин и придавать им нек-рые спе-
специальные свойства (напр., электрич. проводимость,
химич. стойкость); 2) облегчать обработку резиновой
смеси; 3) снижать стоимость изделий, т. к. наполните-
наполнители, как правило, значительно дешевле, чем каучук.
Н. р., улучшающие механич. свойства резин, наз.
активными, или усиливающими, в отличие от инерт-
инертных наполнителей (разбавителей), к-рые не изменяют
или в нек-рых случаях даже несколько ухудшают свой-
свойства резин.
Активными Н. р. служат сажа, синтетич. дву-
двуокись кремния (часто наз. «белой сажей») и силикаты
металлов, нек-рые органич. продукты (синтетич. поли-
полимеры, лигнин). Важнейший и наиболее распространен-
распространенный Н. р.— сажа. Активные Н. р. повышают модуль
резин, их прочность при растяжении, сопротивление
раздиру и износостойкость. Напр., при введении сажи
в смесь на основе бутадиен-стирольного каучука проч-
прочность вулканизата при растяжении увеличивается
в 10 и более раз. Активные Н. р. сильно влияют также
и на пласто-эластические свойства резиновых смесей.
К инертным Н. р. относятся гл. обр. различ-
различные неорганич. продукты природного происхождения:
мел, каолин и др. Эти же наполнители применяют в про-
производстве пластмасс и лакокрасочных материалов.
Подробно о них см. Наполнители пластмасс, Наполни-
Наполнители лакокрасочных материалов.
Сажа
Сажа — высокодисперсный углеродный материал,
образующийся при неполном сгорании или термич.
разложении углеводородов, к-рые содержатся в природ-
природном или промышленном газах и в жидких продуктах
(маслах) нефтяного или каменноугольного происхож-
происхождения. Пром-сть производит сажу с различными свойст-
свойствами, что позволяет выбирать для каждого резинового
изделия наиболее пригодный тип.
Классификация. По способу производства сажи
делят на три группы: канальные, печные и термичес-
термические. Каждая группа включает несколько типов (марок)
сажи.
Канальные (диффузионные) сажи
получают при неполном сгорании природного газа или
его смеси с маслом (напр., антраценовым) в т. наз.
горелочных камерах, снабженных щелевыми горелками.
Внутри камер расположены охладительные поверхно-
поверхности (каналы), на к-рых сажа осаждается из диффузи-
диффузионного пламени.
Печные сажи получают при неполном сжи-
сжигании масла, природного газа или их смеси в факеле,
создаваемом специальным устройством в реакторах
(печах). Сажа в виде аэрозоля выносится из реактора
продуктами горения и охлаждается водой.
Термические сажи получают в специаль-
специальных генераторах при термич. разложении природного
газа или ацетилена без доступа воздуха.
Согласно классификации, принятой в СССР (табл.
1), в обозначении марки сажи первая буква указы-
указывает на способ производства сажи (Д — диффузион-
Таблица 1. Обозначения приблизительно эквивалентных
типов (марок) сажи, вырабатываемых в СССР и за рубежом
Обозна-
Обозначения
отечест-
отечественных
марок
Обозначения зару-
зарубежных марок
Распрост-
Распространенные
хг " л\ KJJY1С И.
ттГПЭО Ы"и"РЛР
Д \J d d ri xl olC
ASTM
Канальные из газа
ДГ-100
МРС, ЕРС| S 301
Канальные из
смешанного сырья
ДМГ-80
П еч н
ПМ-130
пм-юов
ПМ-100
ПМ-100Н
ПМ-75В
ПМ-7 5
ПМ-75Н
-
_
ые из жидкого
сырья
SAF
ISAF-HS
ISAF
ISAF-LS
HAF-HS
HAF
HAF-LS
N 110
N 242
N 220
N 219
N 347
N 330
N 326
Обозначе-
Обозначения оте-
чествен-
чественных ма-
марок
ПМ-50
ПМ-ЗОВ
ПМ-30
ПМ-15
П е ч
Обозначения зару-
зарубежных марок
Распрост-
Распространенные
FEF
—
Рекомен-
дован-
дованные
ASTM
N 550
z
—
кые из газа
и смешанного
О Ы Т1 f» Я
пгм-зз
Терм
ТГ-10
ТА*
GPF
SRF
и ч е с к и е
газа
FT, MT
Acet
N 660
N 770
и з
N 880 —
N 990
* ТА (термич. ацетиленовая) — обозначение сажи с различ-
различной уд. поверхностью, получаемой из ацетилена.
нал, П — печная, Т — термическая), следующие одна
или две буквы — на вид сырья (Г — газ, М — масло,
МГ — смесь масла с газом). Числа после букв соот-
соответствуют значению уд. геометрич. поверхности (см.
ниже). Последние буквы указывают на степень струк-
структурности (см. ниже) сажи (В — высокая, Н — низкая);
отсутствие этих букв означает, что сажа имеет нор-
нормальную структурность.
По классификации США, к-рой пользуются во мно-
многих странах, первые две буквы в обозначении типа
сажи (см. табл. 1) указывают на свойства, к-рые са-
сажа придает резинам [напр., НА (high abrasion) —
высокая износостойкость] или резиновым смесям
[напр., ЕР (easy processing) — легкая обрабатывае-
обрабатываемость], а третья буква — на способ ее производства
[С (channel) — канальная, F (furnace) — печная, Т
(thermal) — термическая]. Буквы HS и LS в обозна-
обозначении нек-рых саж указывают на их структурность:
в первом случае — высокую (high structure), во вто-
втором — низкую (low structure).
В 70-х гг. начали применять также классификацию
ASTM, согласно к-рой сажу подразделяют на типы
в зависимости от ее влияния на скорость вулканизации:
N (normal) — нормальная, S (slowly) — замедленная.
Приводимая в обозначении первая цифра представ-
353
НАПОЛНИТЕЛИ РЕЗИН
354
ляет собой порядковый номер в таблице, в к-рой сажи
расположены по возрастанию диаметра их частиц
[напр., саже типа SAF (диаметр частиц 11 — 19 нм)
присвоен номер 1, саже типа HAF B6—30 нм) — но-
номер 3]; две другие цифры косвенно характеризуют
структурность сажи; напр., низкоструктурной саже
HAF-LS присвоен индекс 26, высокоструктурной
HAF-HS — индекс 47.
Строение. По степени кристалличности сажа зани-
занимает промежуточное положение между кристаллич.
графитом и аморфным углеродом; ее часто называют
турбостратической (неупорядоченно-слоевой) формой
углерода. Частицы сажи, имеющие приблизительно
сферич. форму и связанные между собой в первичные
агрегаты, состоят из параллельно-слоевых пакетов
(кристаллитов), образованных обломками графитовых
плоскостей (слоев), беспорядочно расположенных во-
вокруг общей для всех слоев нормали. Слои кристалли-
кристаллитов сажи образованы правильными спаянными шести-
шестиугольниками (гексагонами), в вершинах к-рых на рас-
расстоянии 0,142 нм A,42 А) друг от друга находятся ато-
атомы углерода. Кристаллиты упакованы в частице сажи
беспорядочно; степень упорядоченности в их располо-
расположении увеличивается в направлении от центра частицы
к ее поверхности.
Расстояние между слоями в кристаллите сажи зна-
значительно больше, чем в кристалле графита [соответст-
[соответственно 0,345—0,370 и 0,335 нм C,45—3,70 и 3,35 А)].
Кристаллиты большинства типов сажи состоят из 3—
5 слоев, ацетиленовой сажи — из 6—8 слоев. Их разме-
размеры вдоль плоскости слоя 1,5—3,0 нм A5—30 А), пер-
перпендикулярно плоскости слоя 1—2 нм A0—20 А).
Среднее число кристаллитов в частице колеблется
от 1600 для наиболее дисперсных до 5,4-106 для наи-
наименее дисперсных (термических) саж.
Наряду с кристаллитами в частице сажи имеются
единичные слои и не входящий в структуру слоя или
кристаллита так наз. неорганизованный углерод (гл.
обр. в виде углеводородных цепей, связанных с крае-
краевыми атомами кристаллитов и единичных слоев).
Неполная валентная насыщенность краевых углерод-
углеродных атомов обусловливает связь кристаллитов не-
непосредственно друг с другом или через боковые угле-
углеводородные цепи. Краевые атомы поверхностного слоя
частицы сажи являются активными центрами, в част-
частности центрами окислительных процессов, приводящих
к образованию на поверхности частиц различных
химич. групп. Нагревание сажи при темп-ре ок. 3000 °С
(графитирование) ослабляет связи между кристалли-
кристаллитами, увеличивая их подвижность. При этом мелкие
кристаллиты срастаются в более крупные, повышается
степень упорядоченности в их расположении и унич-
уничтожаются поверхностные химич. группы и активные
центры. Графитированная сажа уже не является актив-
активным наполнителем и по свойствам приближается к гра-
графитовому порошку.
Элементарный состав. Кроме углерода, сажа содер-
содержит (табл. 2) водород, серу, к-рые переходят в сажу
из сырья и распределены по всему объему частицы,
кислород, к-рый связывается с сажей при ее окислении,
концентрируясь преимущественно в поверхностном
слое частицы, а также минеральные вещества, к-рые
могут попадать в сажу при охлаждении саже-газовой
смеси промышленной водой.
Свойства. Усиливающее действие сажи на каучук
определяется в основном след. ее свойствами: 1) дис-
дисперсностью, к-рая характеризуется величиной частиц
сажи, ее уд. поверхностью и так наз. уд. числом ча-
частиц; 2) шероховатостью; 3) структурностью; 4) уд.
активностью (энергией поверхности).
Величина частиц. Поскольку промышлен-
промышленная сажа состоит из частиц разных размеров, для
характеристики их диаметра пользуются тремя сред-
Таблица 2. Элементарный состав сажи наиболее распространен-
распространен(% )
Тип сажи
ДГ-100 . . .
ДМГ-80
ПМ-75 .
ПМ-5 0 .
ПГМ-33
ИМ-ЗОВ
ПМ-15 .
ТГ-10 .
ных
С
94,8
94,7
97,3
98,6
98,9
98,4
99,0
99,3
типов (^
Н
0,9
0,9
0,5
0,4
0,4
0,7
0,5
0,5
& по массе)
S
<0 1
0,'б
0,8
0,4
о|4
0,3
О
4,3
3,8
1,2
0,4
0,6
0,3
0,1
0,1
Минераль-
Минеральный оста-
остаток
<0,1
<0 1
0,2
0,2
0,1
0,2
0,1
0,1
ними значениями: среднеарифметическим Da (табл. 3),
среднеповерхностным Dn и среднечастичным D4:
где щ — число частиц с диаметром Z),-. Значения D,-
обычно определяют методом электронной, микроскопии
и измеряют в нм, мкм или А A нм = 10 А). Удель-
Удельная поверхность определяется суммой по-
поверхностей частиц, приходящихся на единицу массы
сажи. Различают геометрическую (Sr) и адсорбцион-
адсорбционную (*Sa) уд. поверхность:
^г = гр- Sg, = Ам No coM
где р — плотность сажи, кг/мъ (г/см3); Ам — количест-
количество вещества, адсорбированного единицей массы сажи
в виде плотного мономолекулярного слоя, моль/кг
(моль/г); No — число Авогадро, моль'1; сом — пло-
площадь, занимаемая одной молекулой вещества, адсор-
адсорбированного на поверхности сажи, м2 (см*)', Sr и Sa
имеют размерность мг1кг (смУг).
Удельное число частиц N, определяе-
определяемое числом частиц в единице массы сажи в кг'1 (или
г), подсчитывают по ф-ле:
С увеличением уд. поверхности сажи, а также ее
уд. активности (см. ниже) повышаются прочность при
растяжении, сопротивление раздиру и износостойкость
резин, уменьшается их эластичность и возрастает теп-
теплообразование при многократных деформациях.
Высокодисперсные сажи имеют малую насыпную
массу, что затрудняет их транспортировку и введение
в резиновую смесь. Поэтому такие сажи после их
получения подвергают гранулированию или уплот-
уплотнению.
Шероховатость. Выходя из реакционной
зоны, сажевые частицы (преимущественно их активные
центры) могут подвергаться действию кислорода воз-
воздуха, в результате чего поверхность частиц сажи ста-
становится шероховатой. Уд. адсорбционная поверхность
такой сажи больше, чем уд. геометрическая. Отношение
Sa/Sr наз. коэфф. шероховатости (см. табл. 3). Шеро-
Шероховатость присуща гл. обр. канальной саже, при
получении к-рой поверхность частиц может подвер-
подвергаться действию кислорода при достаточно высокой
темп-ре (>100 °С).
«Шероховатые» сажи труднее, чем «нешероховатые»,
распределяются в резиновых смесях. Кроме того, та-
такие сажи могут замедлять вулканизацию вследствие
адсорбции компонентов вулканизующих систем в углуб-
углублениях на поверхности частиц.
Структурность — свойство сажи, обуслов-
обусловленное наличием в ней первичных агрегатов различных
размеров и формы, образующихся в реакционной зоне
в результате столкновения и дальнейшего совместного
роста частиц. Возникновение между частицами сажи
химич. связей углерод — углерод обусловливает выео-
355
НАПОЛНИТЕЛИ РЕЗИН
356
кую прочность первичных агрегатов. Особенно благо-
благоприятны для образования агрегатов условия печного
процесса при использовании жидкого сырья.
Структурность сажи характеризуют так наз. «масля-
«масляным числом» (см. табл. 3) — объемом (в мл) или массой
(в г) дибутилфталата или льняного масла, абсорбиро-
абсорбированным 1 г (или 100 г) сажи при ее легком растирании
с этими веществами до образования «лепешки».
Сажи с высокой структурностью быстрее и лучше,
чем низкоструктурные, распределяются в резиновой
смеси при ее изготовлении, улучшают обрабатываемость
смеси при ее каландровании и шприцевании, снижают
усадку, повышают модуль резин, но уменьшают их
эластичность и сопротивление разрастанию трещин.
Крупные первичные агрегаты затрудняют гранулиро-
гранулирование сажи.
Первичные агрегаты, соприкасаясь, образуют менее
прочные вторичные; прочность последних повышается
с увеличением шероховатости частиц сажи. При доста-
достаточно высоком содержании сажи в резиновой смеси
вторичные агрегаты могут образовывать сетчатую са-
сажевую структуру, обусловливающую электрическую
проводимость резин. Наибольшая электрическая про-
проводимость характерна для резин, содержащих ацети-
ацетиленовую сажу.
Удельная активность сажи (средняя
активность, или энергия, 1 ж2 поверхности) зависит
от концентрации и типа активных центров и химич.
групп на поверхности ее частиц. Ребра и углы кри-
кристаллитов, инородные атомы и группы (в том числе
возникающие в результате окисления), внедренные
в углеродный скелет, и др. искажения
решетки кристаллита образуют актив-
активные центры поверхности. Мерой энер-
энергии этих центров может служить диф-
Типичные кривые зависимости
дифференциальных теплот ад-
адсорбции низкомолекулярного
углеводорода от степени запол-
заполнения поверхности саши: 1 —
активная саша с неоднородным
распределением энергии поверх-
поверхности; 2 — неактивная сажа
(графитированная) с практиче-
практически однородным распределени-
распределением энергии поверхности.
Степень заполнения поверхности
сами адсорбатом
ференциальная теплота адсорбции, к-рая выделяется
при постепенном заполнении поверхности сажи низко-
низкомолекулярным углеводородом (адсорбатом). Взаимодей-
Взаимодействие первых порций адсорбата с наиболее активными
центрами сопровождается выделением наибольших теп-
теплот адсорбции. По мере заполнения поверхности сажи
адсорбатом значения теплот адсорбции уменьшаются.
На участках поверхности сажи, занятых «нормальны-
«нормальными» слоями, теплоты адсорбции мало отличаются от теп-
теплот конденсации адсорбата в данных условиях. По зна-
значениям дифференциальных теплот адсорбции и по фор-
форме кривой их зависимости от степени заполнения по-
поверхности сажи молекулами адсорбата (рисунок)
можно судить об уровне энергии активных центров и их
распределении по поверхности частиц сажи. Как пра-
правило, удельная активность сажи возрастает с повы-
повышением ее дисперсности. Так, средние значения теплот
адсорбции бутена-1 при заполнении 5% поверхнос-
поверхности сажи типа ПМ-100, ПМ-75 и ПМ-50 составляют
соответственно 42; 38,5 и 33,9 кдж/молъ A0; 9,2 п 8,1
ккал/молъ).
На поверхности сажи имеются свободные (неспарен-
ные) электроны, к-рые только частично связаны с ак-
активными центрами. Обнаруженные на поверхности
сажи гидроксильные (фенольные) и карбонильные
(хинонные) группы образуются на ранних стадиях
окисления; более глубокое окисление ведет к обра-
образованию лактонных и карбоксильных групп, после-
последующая декарбоксилация обусловливает шерохова-
шероховатость сажи. Наличие в саже карбоксильных групп
понижает рН ее водной суспензии (см. табл. 3), что
влечет за собой замедление вулканизации резиновых
смесей.
Применение. Сажу применяют в производстве боль-
большинства резиновых изделий. Выбор типа сажи и сте-
степени наполнения определяется ее влиянием на свойства
резиновой смеси и условиями эксплуатации готового
изделия.
Ассортимент углеродных саж претерпел существен-
существенные изменения после второй мировой войны. В пред-
предвоенные и военные годы единственной активной сажей
для резины была канальная, изготовляемая из при-
природного газа; в 1942 объем производства этой сажи
в США составлял 90% от общего выпуска сажи. После
войны наметились след. основные тенденции.
1. Природный газ вытесняется жидким сырьем,
гл. обр. нефтяного происхождения, что обеспечивает
организацию производства сажи в странах, не имеющих
собственных источников газа. Дешевизна и легкая
транспортабельность жидкого сырья обусловливает
перевод производства сажи на это сырье и в богатых
газом странах.
2. Широко внедряется печной способ производства
сажи, позволяющий на одном типовом технологич.
оборудовании, при изменении параметров процесса,
производить сажу с различными свойствами. Благо-
Благодаря применению печных саж из жидкого сырья стало
возможным освоение новых синтетич. каучуков, в
частности стереорегулярных бутадиеновых (см. Бута-
Бутадиеновые каучуки).
В 1971 производство сажи в капиталистич. странах
составляло (в тыс. т): США — 1372, Япония — 288,
Великобритания — 283, ФРГ — 281, Франция — 176,
Италия — 123, Нидерланды — НО.
сажи
Тип
ДГ-100
ДМГ-80
ПМ-75
ПМ-5 0
пгм-зз
пм-зов
ПМ-15
ТГ-10
||\
о сг о
с* ?
30
34
35
48
70
81
130
210
Уд. геометрич.
поверхность Sp
м2/кг (м21г)
100
80
75
50
33
30
15
10
103 A00)
103 (80)
103G5)
103E0)
103C3)
103C0)
103A5)
103A0)
Таблица 3. Свойства сажи наиболее распространенных '
Уд. адсороцион-
ная поверх-
поверхность, Sa, м/2к&
130
90
73
40
33
27
12
И
•103A30)
103(90)
103G3)
103 D0)
103 C3)
103B7)
103A2)
103A1)
6
«¦I
IK-
1,30
1,12
0,97
0,80
1,00
0,90
0,82
1,10
*
ГНОСТ1
si
1,80
1,76
1 ,86
1,87
1,88
1,82
1,82
1,90
6 а
*§
if
Macj
ло, .
мл/г
0,8
0,9
1 ,0
1 ,05
0,7
1,1
0,95
0,25
]
Насыпная
масса
рану-
>ва-
до г
лире
ния
0,
0,
0,
0,
0,
0,
о,
о,
060
057
061
067
144
067
095
285
г/см3
1
поел
0
0
0
0
0
о
?§
32
31
36
30
45
гипов
Объемное число, м3/пг
(ежз/г)
до гранулиро-
0
0
0
0
0
0
0
0
вания
,0165 A6
,0175 A7
,0165 A6
,015 A5)
,007 G)
,015 A5)
,0105 A0
,0035 C,
,5)
,5)
,5)
,5)
после грану-
гранулирования
0
0
0
0
0
0031 C
0032 C
0028 B
0033 C
0022 B
—
,1)
,2)
,8)
,3)
,2)
,
о
'с
i
4,
4,
8,
8,
8,
8,
8,
7,
зии
спен
0
5
0
0
5
5
5
5
* В этаноле.
357
НАПЫЛЕНИЕ
358
Двуокись кремния и силикаты металлов
Высокодисперсные двуокись кремния и силикаты
металлов (гл. обр. А1 и Са) образуют группу синтетич.
светлых активных минеральных наполнителей. Про-
Процессы производства этих Н. р. делят на пирогенети-
ческие (напр., получение высокодисперсной SiO2 раз-
разложением галогенидов кремния в среде перегретого
водяного пара при темп-ре выше 1000 °С) и так наз.
«мокрые» (напр., осаждение силиката кальция при
взаимодействии водных р-ров СаС12 и Na^SiOs).
меров (или композиций на их основе) на поверхность
детали или формы, используемый для получения
покрытий различного назначения, а также в произ-
производстве нек-рых изделий. Для напыления могут быть
использованы практически все полимеры; композиции
на их основе могут содержать наполнители, пласти-
пластификаторы, стабилизаторы, отвердители, порообразова-
тели и др. ингредиенты.
В данной статье описаны промышленные способы
Н. твердых порошкообразных композиций, основой
Таблица 4. Химический состав и свойства высокодисперсных двуокиси кремния и силикатов металлов нек-рых типов
Наполнитель
Фирменное
название
Обозначение типа*
Содержание,
%
SiO2
СаО
я к
И
Уд. адсорбционная поверх-
поверхность Sa, ж2/«а [ж2/г]
<я
В
чис-
К
5*"
11
о
О
«а
о а
« м
Двуокись кремния
Аэросил . .
Кабосил . .
Хайсил . .
Ультрасил .
Дуросил . .
Силтег . . .
Калсил . .
Силен . . .
PP-SiO2-ER
PP-SiO2-ER
WP-SiOo—SR
WP-SiCV-SR
WP-SiO2-HR
WP—Al/SiO2—HR
WP-Ca/SiO2-MR
WP-Ca/SiO3—MR
99,3-
99,8
99,0
87,5
87,0
84,0
0,75
1,0
10,0
11,0
12,0
5-40
8-15
22
15—28
20
A75-380)-103[175-380]
B00-300)-10»[200-300]
150-103 [1 50]
A20-234)-103[120-234]
Силикаты металлов
72,
74
65,
70
65
0-
,0
0-
,0
,0
7.
8,0
и,
19
20
0-
•k-k-k
0-
,0
,0
и,
13
15,
17
12,
14
0-
,0
0-
,0
0 —
, о
3 0—35
10—40
30
E5 — 1 30)-1 О3 [55 —130]
87-Ю3 [87]
2,36
2,10
2,00
1,90-
1 ,95
2,10
1 ,95 —
2,00
1 ,95 —
2,05
2,10
1,5-2,0
1,45
2,4
1,5
1 ,0
4,0
3,9
6,9
5,5-
7,0
7,0
10,5-
11,0
10,0
* РР и WP — наполнители, получаемые соответственно пирогенетич. и «мокрым» способами; ER — наполнитель чрезвычайно
высокоусиливающий, SR — сверхвысокоусиливающий; HR — высокоусиливающий, MR — среднеусиливающий. ** Определено по
абсорбции льняного масла. *** Содержание А12О3.
Свойства SiO2 и силикатов металлов определяются
в основном теми же показателями (табл. 4), что и для
сажи. По усиливающему действию эти Н. р. практи-
практически не уступают активным углеродным сажам; осо-
особенно эффективно их влияние на сопротивление резин
раздиру и старению.
Высокодисперсные SiO2 и силикаты металлов при-
применяют гл. обр. в производстве белой и цветной рези-
резиновой обуви (для изготовления подошв и каблуков
с высокой износостойкостью) и др. изделий народного
потребления, а также для изготовления кабельных
и электроизоляционных резин. Высокодисперсную
SiO2 в смеси с углеродной сажей иногда применяют
в производстве шин и нек-рых других изделий.
Органические наполнители
К этой группе Н. р. относятся синтетич. смолы
(феноло-, анилино-, меламино- и мочевино-формальде-
гидные), структурированные продукты полимеризации
стирола или его производных (так наз. виниловые
наполнители), а также лигнин, выделенный из черного
щелока (продукта сульфатной обработки древесины),—
порошок с плотн. 1,3 г/см3, содержащий ок. 1,5% серы.
При использовании органич. Н. р. эффект усиления
достигается при их введении в латекс; нек-рые синте-
синтетич. смолы получают непосредственно в латексе. См.
также Наполненные каучуки.
Лит.: Зуев В. П., Михайлов В. В., Производство
сажи, 3 изд., М., 1970; Производство и свойства углеродных
саж, под ред. В. Ф. Суровикина, вып. 1, Омск, 1972; Печ-
ковская К. А., Сажа как усилитель каучука, М., 1968;
Усиление эластомеров, под ред. Дш. Крауса, пер. с англ., М.,
1968; Справочник резинщика, Материалы резинового про-
производства, М., 1971. К.А.Печковская.
НАПЫЛЕНИЕ (spraying, Aufspritzen, pulverisation) —
способ нанесения порошкообразных или жидких поли-
к-рых могут служить полиэтилен, сополимеры этилена
с винилацетатом, поливинилбутираль, эфиры целлю-
целлюлозы, фторопласты, поликарбонаты, полиакрилаты,
эпоксидные смолы, полиамиды, пентапласт и др. (см.
Порошковые краски). Н. осуществляется под действием
сил электрич. поля, а также пневматических или ме-
механических сил.
Сущность способа напыления в электри-
электрическом поле заключается в том, что распылен-
распыленным частицам порошка [обычно с уд. объемным элект-
электрич. сопротивлением 108—1012 ом-м A010—1014 ом-см)]
и заземленному изделию сообщают заряды противопо-
противоположного знака (порошок заряжают, как правило, отри-
отрицательно). Для распыления применяют заряжающую
распылительную головку или ручной пистолет. Прак-
тич. применение нашли два способа зарядки частиц—
контактный и ионный. В первом случае частица при-
приобретает заряд в результате контакта с металлич.
электродом, соединенным с источником высокого на-
напряжения. При ионной зарядке с источником высокого
напряжения соединяются тонкие металлич. элект-
электроды, к-рые коронируют и создают в воздухе поток
ионов. Последние, оседая на частицах, сообщают им
заряд. Существуют также устройства, в к-рых соче-
сочетаются контактная и ионная зарядка. Слой полимер-
полимерного материала образуется в результате осаждения
частиц порошка на поверхности противоположно заря-
заряженного изделия и их последующего сплавления (для
этого изделие помещают в печь).
Процесс осуществляют обычно в камере с конвейе-
конвейером, на к-ром размещают детали. Источником высокого
напряжения служат роторные электростатич. генера-
генераторы или трансформаторы с выпрямителями (частота
10—20 кгц), обеспечивающие напряжение 50—120 кв
и силу тока 100—250 мка. В случае контактной зарядки
359
НАПЫЛЕНИЕ
360
(рис. 1, а) порошок подается воздухом по винтовым
каналам головки к острой заряжающей кромке, соеди-
соединенной с источником высокого напряжения (при про-
прохождении порошка по винтовым каналам развивает-
развивается центробежная сила, обеспечивающая контакт частиц
с головкой). Около острой кромки напряженность
электрич. поля наибольшая, и поэтому частица порош-
порошка приобретает максимальный заряд. При ионной
зарядке (рис. 1,. б) порошок подается воздухом к го-
головке с коронирующими иглой и кольцом.
Рис. 1. Схемы головок для контактной (а) и ионной (б) за-
зарядки порошковой краски: 1 — корпус; 2 — металлич. за-
заряжающая головка; 3 — винтовые каналы; 4 — металлич.
коронирующая игла; 5 — кольцо.
Способом Н. в электрич. поле можно наносить по-
порошковые материалы на тонкостенные изделия, напр,
из фольги, что трудно осуществить при использовании
др. способов, и получать покрытия толщиной от 10—
20 мкм до ~ 1,0 мм. Недостаток способа — трудность
получения покрытия в углубленных местах изделия
из-за уменьшения в этих местах напряженности элект-
электрич. поля. Применением для Н. камер непрерывного
действия, внутри к-рых установлены вентиляторы,
поддерживающие непрерывную циркуляцию порошка,
удается получить равномерное покрытие на изделиях
сложной конфигурации и ускорить процесс Н.
Способ Н. в электрич. поле получил в пром-сти
наибольшее распространение. Это обусловлено воз-
возможностью его автоматизации, а также минимальными
потерями порошкового материала.
При использовании способа напыления в
псевдоожиженном слое порошка
деталь, предварительно нагретую на 50—150 °С выше
темп-ры плавления полимера (в зависимости от ее
теплоемкости и массы), помещают на несколько секунд
в емкость с порошком, находящимся во взвешенном
состоянии. При соприкосновении с поверхностью де-
детали порошок нагревается, прилипает к ней и сплав-
сплавляется. Окончательное сплавление нанесенного слоя
происходит в печи.
Псевдоожижение порошка м. б. достигнуто с по-
помощью воздуха (или др. газа) или вибрации, а также
при их одновременном действии. Выбор метода опре-
определяется способностью порошка к псевдоожижению.
При воздушном псевдоожижении («вихревой» метод)
используют емкость с расположенной в ее нижней час-
части пористой перегородкой, на которую помещают по-
порошок, а снизу подают под давлением воздух, соз-
создающий псевдоожиженный (взвешенный) слой порош-
порошка, объем которого превышает объем исходного слоя
в 1,4—1,7 раза.
Пористые перегородки изготовляют из войлока тол-
толщиной 25—35 мм, 3—4 слоев стеклоткани, а также
из др. материалов с размером пор 40—150 мкм при
пористости ~ 50%. Давление воздуха под перегород-
перегородкой составляет 0,2—0,4 Мн/м2 B—4 кгс/см2), его ско-
скорость в емкости — 3—5 м/сек (скорость тем больше,
чем крупнее частицы порошка). Метод пригоден для
порошков, к-рые легко переходят в псевдоожиженное
состояние, напр, для поливинилбутиральных. Если
псевдоожижение затруднено (напр., в случае полиэти-
полиэтиленовых порошков), то используют вибрационный или
вибро-вихревой метод. В первом случае псевдоожи-
псевдоожижение происходит в результате вибрации (частота
50—100 гц, амплитуда 1—3 мм) всей емкости или
только ее дна. Вибро-вихревой метод, представляющий
собой сочетание вибрационного и вихревого, позволяет
увеличить объем порошка в 2—2,7 раза.
Способ Н. в псевдоожиженном слое широко распрост-
распространен в пром-сти. Это объясняется простотой оборудова-
оборудования и технологии, а также вполне удовлетворительным
качеством покрытий. Недостатки способа — трудность
получения слоев равномерной толщины на деталях
сложной конфигурации, сепарация частиц псевдоожи-
женного порошка по размерам, охлаждение нагретой
детали струей воздуха. В связи с тем, что максималь-
максимальная толщина псевдоожиженного слоя составляет
~~ 15 см, способ применяют при Н. порошков на срав-
сравнительно малогабаритные изделия.
В усовершенствованных комбинированных способах
Н., напр, электровихревом, псевдоожижение
порошка совмещается с его ионной зарядкой (рис. 2):
псевдоожиженный порошок
заряжается от коронирую-
щей сетки и осаждается на
заземленной детали. При
этом получается значитель-
значительно более равномерный слой
Рис. 2. Схема ванны для «вих-
«вихревого» напыления с ионной за-
зарядкой порошковой краски: I —
деталь; 2 — пористая перегород-
перегородка; з — труба для подачи газа;
4 — коронирующая проволочная
сетка.
и ускоряется процесс. Для Н. могут быть также ис-
использованы ванны с игольчатым электродом, в к-рых
как псевдоожижение порошка, так и его перенос на
заземленное изделие осуществляются под действием
сил электрич. поля.
При газопламенном напылении струя
воздуха со взвешенными в ней частицами порошка
выбрасывается из сопла распылительного пистолета
и проходит сквозь пламя газовой горелки автогенного
типа, смонтированной вместе с пистолетом (рис. 3).
К пистолету по одному шлангу струей воздуха подается
из питательного бачка порошок, по второму шлангу —
горючий газ (напр., ацетилен), по третьему — воздух.
Под действием тепла горелки порошок нагревается
до темп-ры размягчения полимера, а на поверхности
детали сплавляется, образуя сплошной слой (обычно
Рис. 3. Схема пистолета
для газопламенного на-
напыления: 1 — шланг для
подачи порошковой крас-
краски струей воздуха из пи-
питательного бачка; 2 —
шланг для подачи горючего газа; 3 — шланг для подачи
воздуха; 4 — инжектор; 5 — корпус пистолета; 6 — факел
горелки; 7 — изделие.
0,1—3 мм). Продолжительность контакта порошка
с горючим газом (темп-pa ок. 1500 °С) — доли секунды,
и поэтому значительная деструкция полимера, как
правило, не происходит.
Струя, выходящая из горелки, должна быть направ-
направлена перпендикулярно покрываемой поверхности, т. к.
при этом достигается большая равномерность нанесен-
нанесенного слоя. Для лучшей адгезии покрытия поверхность
изделия перед Н. прогревают горелкой. Недостатки
способа — малая производительность C—5 м2/ч);
неравномерная толщина слоя; большие потери краски,
к-рая сгорает или не попадает на изделие; коробление
тонких материалов, напр, листов, при их прогреве;
невозможность К. порошка на изделия большой тол-
361
НЕОБРАСТАЮЩИЕ ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ
362
щины из-за трудности их прогрева. Способ газопла-
газопламенного Н. применяют в ремонтном деле, напр, при
заделке дефектов покрытий или раковин в металле,
облицовке сварных швов, а также при нанесении
покрытий на крупные металлич. конструкции.
Разновидность газопламенного Н.— плазмен-
плазменное напыление. Способ состоит в нагревании
порошка плазмой (ионизированным инертным газом —
аргоном, гелием, азотом) с темп-рой 15 000—30 000 °С,
образующейся в пламени вольтовой дуги. Порошок
инжектируется в пламя также с помощью инертного
газа. Несмотря на высокую темп-ру, полимер не дест-
руктируется, т. к. находится в инертной среде, а про-
продолжительность его контакта с плазмой составляет
доли секунды. Этим способом можно наносить порошки
на основе любых полимеров, в том числе и с высокой
темп-рой плавления.
При струйном напылении порошковый
материал наносят на предварительно нагретое изде-
изделие из специальных пневматических распылителей —
ручных или механических. Способ отличается просто-
простотой, сравнительно большой производительностью и
позволяет получать покрытия хорошего качества. Од-
Однако его широкое использование ограничивается не-
необходимостью предварительного нагрева изделия.
При получении покрытий порошок наносят на по-
поверхность, к-рую предварительно очищают и обезжи-
обезжиривают теми же методами, что и при получении лако- \
красочных покрытий; в частности, металлы часто под- •
вергают пескоструйной обработке и обезжириванию
органич. растворителями. После образования покры-
покрытия изделие охлаждают в воде, в маслах или на воздухе.
Скорость процесса и охлаждающая среда влияют на ад-
адгезию и механич. свойства пленок. Так, медленное
охлаждение покрытий на основе аморфных полимеров,
особенно в средах, пластифицирующих полимер, умень-
уменьшает внутренние напряжения в пленке и повышает
ее адгезию к подложке. Улучшение свойств покрытий
на основе кристаллич. полимеров достигается их бы-
быстрым охлаждением (закалкой), приводящим к умень-
уменьшению структурной упорядоченности в пленке. Покры-
Покрытия, получаемые напылением, контролируют теми же
методами и приборами, что и лакокрасочные (см. Ис-
Испытания лакокрасочных материалов и покрытий).
Н. имеет ряд преимуществ перед нанесением лако-
лакокрасочных покрытий: можно использовать более широ-
широкий ассортимент полимеров и, следовательно, полу-
получать покрытия с самыми разнообразными свойствами;
нет необходимости в применении растворителей, что
важно не только с экономич., но и с санитарно-гигие-
нич. точки зрения (выброс горячих газов из нагрева-
нагревательных устройств в атмосферу уменьшается в несколь-
несколько раз); при использовании усовершенствованных
способов Н. процесс сопровождается минимальными
потерями порошка и м. б. легко автоматизирован.
Технико-экономич. эффективность Н. обусловлена по-
повышением сроков службы изделий, защищенных покры-
покрытием, а также экономией цветных металлов и нержавею-
нержавеющих сталей (вместо изделий из этих материалов можно
применять изделия из углеродистых сталей, защищен-
защищенных покрытием). Трудоемкость Н. ниже трудоемкости
нанесения гальванич. и лакокрасочных покрытий
соответственно в 5—10 и 2—3 раза. Однако производи-
производительность механизированных способов нанесения лако-
лакокрасочных покрытий выше, а их декоративные свойства
лучше, чем у покрытий, получаемых напылением.
Описанными выше способами можно также изготов-
изготовлять полые тонкостенные изделия (например, емко-
емкости), сплавляя заготовки, полученные напылением по-
порошкового материала на поверхность формы. Особенно
перспективно применение Н. для производства изде-
изделий из полимеров с высокой темп-рой плавления. При
Н. в электрич. поле высокого напряжения порошок
равномерно распределяется по форме, образуя плотные
заготовки, к-рые прочно фиксируются на стенках
формы. Необходимая толщина стенки изделия дости-
достигается напылением нескольких слоев порошка, повыше-
повышением темп-ры формы или изменением напряженности
электрич. поля. Частично сплавленную заготовку
подвергают дополнительной термообработке.
Формы изготовляют из легко вытравляемого металла
(напр., алюминия), растворимой соли (напр., NaNO2)
или из стекла, к-рое растрескивается при быстром
охлаждении формы с нанесенным слоем после его
сплавления. Эффективность процесса в случае Н. по-
порошка на стеклянную форму повышается при исполь-
использовании заземленного экрана, расположенного на внут-
внутренней поверхности формы.
Показатели механич. свойств и монолитности стенки
изделий (напр., из фторопластов), полученных Н.
в электрич. поле и последующим сплавлением загото-
заготовок, на 20—40% выше, чем соответствующие показа-
показатели таких же изделий, изготовленных литьем под дав-
давлением или экструзией.
При использовании способа напыления концентра-
концентрация распыленных порошков в производственных по-
помещениях не должна превышать допустимые нормы.
Помимо вредного влияния на здоровье работающих,
превышение норм запыленности может приводить
к пожарам, т. к. смеси распыленных порошков с воз-
воздухом (аэродисперсии) взрывоопасны. Поэтому при
Н. необходимы механизация и автоматизация техноло-
гич. процесса, дистанционный контроль и управление,
полная герметизация оборудования и очистка выбра-
выбрасываемого воздуха. При движении аэродисперсий
по трубопроводам следует соблюдать правила защиты
от возникающего в этих условиях статич. электричест-
электричества, в частности при Н. в электрич. поле применять
источники питания с небольшой силой тока (до 150—
200 мка).
Н. нашло практич. применение в пром-сти в 50-е гг.
В 1972 в промышленно развитых странах Западной
Европы этим способом получали ок. 14% защитных
покрытий. По прогнозам зарубежных специалистов до-
доля покрытий, получаемых напылением, к 1980 достиг-
достигнет 30—50%, а в том случае, если этот способ найдет
применение при наружной отделке автомобилей, то
и 60—70%.
Лит.: Генель С. В. [и др.], Применение полимерных
материалов в качестве покрытий, М., 1968; Яковлев А. Д.,
Зцор В. Ф., Каплан В. И., Порошковые полимерные
материалы и покрытия на их основе, Л., 1971; Поляко-
Полякова К. К., Пайма В. И., Технология и оборудование для
нанесения порошковых полимерных покрытий, М., 1972.
Е. В. Моисеев, А. А. Черников.
НАТРИЙ-БУТАДИЕНОВЫЙ КАУЧУК — см. Бута-
Бутадиеновые каучуки.
НАТУРАЛЬНЫЙ КАУЧУК - см. Каучук нату-
натуральный.
НЕОБРАСТАЮЩИЕ ЛАКОКРАСОЧНЫЕ ПОКРЫ-
ПОКРЫТИЯ (antifouling paints coatings, Antifoulinganstrich-
iiberziige, revetements de peintures et vernis antisallisan-
tes) — покрытия, предотвращающие обрастание подвод-
подводной части судов и гидротехнич. сооружений морскими
организмами (т. наз. обрастателями). Н.л.п. представля-
представляют собой многослойную систему, состоящую из грунта,
3—4 слоев антикоррозионной и 1—2 слоев «необрастаю-
щей» краски. В состав последней, кроме пленкообра-
пленкообразующих веществ, растворителей, нейтральных пигмен-
пигментов и пластификаторов, вводят яды — преимущественно
окислы меди, ртути, цинка и др. металлов, а также ор-
органич. соединения олова [напр., бис-(трибутилолово)-
окись], мышьяка (напр., хлорфеноксарсин), ртути, свин-
свинца (напр., трифенилацетат свинца), производные триа-
зина, хиноксалина и др.
Эффективность защитного действия Н. л. п. зависит
от скорости выщелачивания яда из покрытия, а т^акже
363
НЕОПРЕН
от его способности проникать через оболочку личинок
обрастателей. Скорость выщелачивания ядов из Н. л. п.
зависит от вида пленкообразующего вещества, а так-
также от объемной концентрации яда в покрытии. При
контакте с морской водой из Н. л. п. может постепен-
постепенно выщелачиваться только яд или одновременно яд
и пленкообразующее.
Использование для изготовления «необрастающих»
красок синтетич. полимеров вместо ранее применяв-
применявшихся масляно-смоляных пленкообразующих позволило
получить быстросохнущие Н. л. п. с высокой концен-
концентрацией яда. Эти краски можно наносить при темп-
рах ниже О °С.
Наиболее часто «необрастающие» краски готовят
на основе перхлорвиниловой смолы, сополимера ви-
нилхлорида с винилацетатом, хлоркаучука, полиизо-
бутилена, политрибутилоловометакрилата. Скорость
выщелачивания яда из красок можно регулировать
добавлением канифоли, к-рая постепенно растворяет-
растворяется в морской воде, имеющей слабощелочную реак-
реакцию (рН 8,1). Растворителями и разбавителями для
таких красок служат ксилол, сольвент-нафта, ацетон,
бутилацетат и др.
В качестве антикоррозионного подслоя в Н. л. п.
применяют покрытия на основе диванилацетиленовых
лаков и эмалей, эпоксидных эмалей (см. Эпоксидные
лаки и эмали), а также покрытие, состоящее из одного
слоя фосфатирующего грунта и 3—4 слоев эмали на ос-
основе сополимера винилхлорида (см. также Защитные
лакокрасочные покрытия). При использовании тиксо-
тропных лакокрасочных материалов число слоев м. б.
уменьшено до 1 — 2.
На объекты, эксплуатируемые в теплых морях,
«необрастающие» краски наносят в два слоя, в север-
северных — в один слой. Для эффективной защиты в теп-
теплых морях толщина пленки, образованной «необра-
стающей» краской, должна быть не менее 90—100 мкм,
а толщина всего подводного покрытия — 250—350 мкм.
При этом эффективная защита обеспечивается в тече-
течение 2 лет. При нанесении поверх этого покрытия
дополнительного слоя «необрастающей» краски на ос-
основе пластифицированных препаратов канифоли (напр.,
КФ-751) срок службы Н. л. п. может быть увеличен
до 3 лет и более.
«Необрастающие» краски токг7:*тпы ч^я человека.
Поэтому их наносят на скрашиваемые поверхности,
как правило, кистью или валиком. При соблюдении
необходимых мер предосторожности они м. б. также
нанесены методом безвоздушного распыления (см.
Лакокрасочные покрытия).
Лит.: Морское обрастание и борьба, с ним, пер. с англ.,
М., 1957; Гуревич Е. С, Природа, № 11, 66 A966);
Проблемы биологических повреждений и обрастания материа-
материалов, изделий и сооружений, М., 1972; Коррозия и защита мор-
морских судов, Л., 1973. Е. С. Гуревич.
НЕОПРЕН — см. Хлоропреновые каучуки.
НЕОРГАНИЧЕСКИЕ ПОЛИМЕРЫ (inorganic poly-
polymers, anorganische Polymere, polymeres inorganiques)—
полимеры, макромолекулы к-рых имеют неорганич.
главные цепи и не содержат органич. боковых радика-
радикалов (обрамляющих групп).
Подобно органич. и элементоорганич. полимерам (см.
Высокомолекулярные соединения, Макромолекула) Н. п.
классифицируют по след. признакам: по происхожде-
происхождению — синтетич. и природные; по конфигурации мак-
макромолекул — линейные, разветвленные, лестничные,
регулярные («паркетные») и нерегулярные плоско-
плоскосетчатые, регулярные (ковалентные кристаллы) и не-
нерегулярные пространственно-сетчатые; по химич.
структуре главной цепи — гомоцепные (гомоатомные)
и гетероцепные (гетероатомные).
Природные Н. п., относящиеся к группе сетчатых,
чрезвычайно распространены и в виде минералов вхо-
входят в состав земной коры. Часть их перерабатывается
и используется в виде неорганич. стекол или керамич.
материалов. К этой категории относится подавляю-
подавляющее большинство известных Н. п.; их характерным
физич. отличием от органич. или элементоорганич.
полимеров является неспособность существовать в кау-
чукоподобном (высокоэластическом) состоянии. В рав-
равной мере, лишь незначительная часть линейных Н. п.
способна к образованию характерных для кристалли-
кристаллизующихся органич. полимеров морфоз (надмолекуляр-
(надмолекулярных структур) — сферолитов, ламелярных кристаллов,
дендритов и др.
Н. п. отличаются по химич. и физич. свойствам
от органических или элементоорганич. полимеров
гл. обр. вследствие различной электронной структуры
главной цепи и отсутствия органич. обрамляющих
групп. Электронная структура однозначно определяет
вообще возможность образования более или менее
устойчивой цепи. Обрамляющие группы модифицируют
электронную структуру, защищают главную цепь
от атаки нуклеофильными или электрофильными аген-
агентами и, наконец, определяют характер межцепных
взаимодействий. Последние, в свою очередь, предопре-
предопределяют возможность использования достаточно высо-
высокомолекулярных Н. п. в виде каучуков или волокон.
Электронная структура главных цепей. Из данных
табл. 1 видно, что область существования Н. п. огра-
ограничена элементами III—VI групп периодич. системы.
Внутри группы с увеличением номера ряда по мере
возрастания степени «металлизации» (т. е. делокали-
зации электронов или их коллективизации) способ-
способность элементов к образованию гомоатомных цепей
или к вхождению в гетероатомные цепи резко убывает.
Элементы VII группы, известные в химии органич.
полимеров как агенты обрыва цепи, играют ту же
роль и в Н. п. и могут быть лишь концевыми атомами.
Элементы VIII группы встречаются лишь в координа-
координационных Н. п.
Для понимания основных химич. свойств Н. п.
уместно сравнить их с полимерами углерода. Как
известно, в таблице Д. И. Менделеева углерод зани-
занимает особое положение, состоящее в способности
к образованию только чисто ковалентных связей
(за счет неспаренных электронов). Элементы слева
от IV группы образуют донорно-акцепторные связи
М <— L (за счет вакантных орбиталей атома М), а справа
от IV группы — дативные связи М —> L (за счет непо-
деленных пар атома М). При образовании таких гетеро-
атомных связей возникают соответствующие смещения
электронной плотности (между донором и акцептором
электронов), связанные иногда с возникновением за-
заметной полярности. Такие «частично ионные» связи
обычно прочнее чисто ковалентных. Кроме того, если
атом М принадлежит к элементам второго периода
(В, N, О), а атом L — к элементам третьего или
последующих периодов, т. е. имеет валентные d-орби-
тали, то кратность и реальная прочность связи типа
М — L м. б. выше, чем в цепях органич. полимеров.
Поэтому среди линейных Н. п. такого типа должны
встречаться весьма термостойкие вещества.
Термодинамич. устойчивость связей в вакууме или
инертной атмосфере, выраженная в их энергии, при-
приведена в табл. 2. Энергия связи может служить кос-
косвенным критерием способности соответствующих пар
атомов к образованию цепи. Однако таблица не отра-
отражает реакционной способности соответствующих свя-
связей, к-рая является не менее существенным критерием
стабильности цепи, чем термодинамич. стабильность.
Сильная поляризация связей типа М — L, как правило,
резко повышает их способность к различным реакциям
замещения (при атаке нуклеофильными или электро-
электрофильными реагентами) и окисления—восстановления
в присутствии влаги или кислорода воздуха. Разу-
Разумеется, с повышением темп-ры эта кинетич. нестабиль-
365
НЕОРГАНИЧЕСКИЕ ПОЛИМЕРЫ
366
ность тоже возрастает, из чего следует, что табл. 2
не дает даже реального критерия термостойкости.
Что касается аналогов углерода по IV группе, то
необходимо заметить следующее. В углероде простая
с-связь С — С заметно прочнее, чем тс-компоненты
кратных связей С = С или С = С. Уже одно это об-
обстоятельство объясняет способность производных эти-
этилена или ацетилена к образованию свободных радика-
радикалов и последующей полимеризации. По мере продвиже-
продвижения вниз по IV группе баланс энергий а- и хс-связей
меняется неблагоприятным образом из-за упоминавшейся
постепенной «металлизации», к-рая уже у Ge достаточ-
достаточно сильно выражена. В целом (это верно для любой
комбинации атомов), как только энергия тс-компонент
становится больше, чем а-компонент, на образование
цепей по любому известному в химии органич. поли-
полимеров механизму накладывается «запрет». Этому экви-
эквивалентно утверждение о невозможности образования
длинных цепей с простыми связями; длинные же цепи
с кратными связями уже сами по себе неустойчивы,
в первую очередь по кинетич. причинам. Соответст-
Соответственно, аналоги углерода, а также его сосед слева —
бор способны лишь к образованию олигомерных ана-
аналогов полиэтилена (см. ниже).
Роль обрамляющих групп. В элементоорганич. поли-
полимерах обрамляющие группы (обычно алкильные или
арильные радикалы) не только выполняют функции
стабилизации электронной структуры главных цепей
и защиты их от атаки электрофильными или нуклео-
фильными реагентами, но и снижают межцепное взаимо-
взаимодействие до уровня (характеризуемого соответствую-
соответствующими энергиями вандерваальсовых связей), присущего
органич. полимерам. Поэтому длинноцепочечные эле-
элементоорганич. полимеры, в первую очередь полиорга-
носилокеаны (см. Кремнийорганические полимеры), про-
проявляют типичный для органич. полимеров комплекс
физич. и физико-химич. свойств: они растворимы в ор-
органич. растворителях и могут существовать в трех
основных релаксационных («физических») состояниях
аморфных полимеров — стеклообразном, высокоэла-
высокоэластическом и вязкотекучем (см. Аморфное состояние).
Наличие обрамляющих групп в низкомолекулярных
исходных соединениях существенно облегчает также
синтез полимеров из них вследствие предотвращения
ряда побочных реакций.
В Н. п. добиться подобного эффекта, в общем случае,
невозможно. Сохраняя способность к стабилизации
электронной структуры главной цепи, неорганич.
обрамляющие группы не могут «защитить» цепи от
внешней атаки. Кроме того, вследствие полярной
природы этих групп межцепное взаимодействие обычно
усиливается, что обусловливает нерастворимость или
ограниченную растворимость и ограниченную сегмен-
сегментальную подвижность цепей.
В этом плане отсутствие обрамляющих групп в гомо-
цепных полимерах VI группы даже благоприятно:
указанные полимеры можно получить в кристаллич.
и всех трех аморфных состояниях, а их кристаллич.
морфозы — те же, что у кристаллизующихся органич.
полимеров.
Получение. Методы получения линейных Н. п. долж-
должны в определенной степени имитировать геохимич. про-
процессы образования минералов, но с исключением реак-
реакций, приводящих к образованию разветвленных и сет-
сетчатых структур. Др. сложность, возникающая при
синтезе Н. п., связана с повышенной тенденцией неор-
неорганич. соединений к образованию простых или слож-
сложных олигомерных циклов, стабилизирующих структуру
цепи; и, наконец, еще одна сложность определяется
неспособностью атомов (за исключением С) образовы-
образовывать ненасыщенные соединения, к-рые могли бы поли-
меризоваться. Поэтому огромного класса потенциаль-
потенциальных аналогов органич. полимеров просто не сущест-
существует. По более или менее привычному радикальному
механизму с термич. раскрытием циклов полимери-
зуются лишь сера и ее аналоги, а также нек-рые фос-
фазены. Следовательно, если вообще есть смысл в по-
поисках аналогий между неорганич. и органич. полиме-
полимерами, эти аналогии следует искать в основном для ли-
линейных полимеров, полученных поликонденсацией.
Использование элементоорганич. полимеров как
полупродуктов для получения Н. п. (т. е. использо-
использование метода блокирующих групп, широко распростра-
распространенного в органич. синтезе) вряд ли возможно, т. к.
в большинстве случаев удаление обрамляющих групп
будет приводить к дестабилизации главных цепей и,
в частности, к деполимеризации или циклоолигомери-
зации.
Гомоцепные неорганические полимеры. Поли-
Полимеры углерода и его аналогов. Со-
Согласно дефиниции, полимеры углерода без обрамляю-
обрамляющих групп м. б. отнесены к Н. п. Хорошо известными
природными полимерами углерода являются алмаз
(упорядоченный пространственно-сетчатый полимер) и
графит (упорядоченный плоско-сетчатый полимер).
Из линейных полимеров углерода известны кумулены
=С=С=С=С= и карбин—С = С—СееС—СееС—С = С—.
Кумулены, как правило, представляют собой олиго-
мерные соединения. Карбин способен существовать
в виде длинных цепей с умеренной конформационной
жесткостью.
Как факты, так и теоретич. оценки показывают, что
из-за «металлизации» при переходе от С к РЬ ката-
катастрофически ослабляются тс-компоненты связей типа
MIV — MIV, что делает невозможным образование
кратных связей уже у кремния. Однако аналоги угле-
углерода способны к образованию ковалентных кристаллов
алмазоподобного типа. Впрочем, по мере металлизации
устойчивость этих кристаллов и величина образующих
их истинно полимерных доменов убывают; уже олово
способно при понижении темп-ры переходить в заве-
заведомо неполимерную модификацию.
Длинноцепочечных аналогов углеводородов также
не обнаружено. Число звеньев (атомов Si) в линейных
полисиланах [— SiH2— ]n не превышает 20. Известны
олигомеры с германием в главной цепи: полигерман
[ —GeH2—]п, полиоксигерман [ — Ge(OH)(H) — ]n и по-
лихлоргерман [— Ge(Cl)(H) — ]n. Эти полимеры весь-
весьма неустойчивы; приближенные оценки позволяют за-
заключить, что п в них также не превышает 20 (см.
также Германийсодержащие полимеры). Олово еще
способно к образованию гомоатомных цепей (очень
коротких!), а свинец уже нет.
Полимеры бора. Хотя существуют о л иго-
мерные циклич. аналоги бороводородов и углеводоро-
углеводородов, линейные длинноцепочечные бороводороды не из-
известны. Высший олигобороводород — декаборан имеет
«корзинчатую» структуру, включающую различные
внутренние и внешние циклы, обусловленные образо-
образованием ковалентных многоцентровых связей (бор —
водород — бор и бор — бор), утилизирующих вакант-
вакантный уровень атома В в исходной молекуле «мономера»
ВН3. Образование таких многоцентровых связей вооб-
вообще типично для систем с дефицитом электронов, т. е.
там, где число пар валентных электронов меньше числа
возможных двухцентровых взаимодействий. См. также
Борсодержащие полимеры, Поликарбораны.
Полимеры элементов VI группы
существуют только без обрамляющих атомов или ради-
радикалов. Образуются они при нагревании расплава серы,
селена или теллура выше нек-рой критич. темп-ры
в результате разрыва 8-членных колец и радикальной
полимеризации. Это кристаллизующиеся гибкоцепные
полимеры, образующие обычные для этого класса
полимеров кристаллич. морфозы. Полиселен и полител-
полителлур устойчивы в полимерной кристаллич. модифика-
367
НЕОРГАНИЧЕСКИЕ ПОЛИМЕРЫ
368
ции при комнатных темп-pax; благодаря соответствую-
соответствующим электрич. свойствам их применяют гл. обр.
в полупроводниковой технике. В аморфной модифи-
модификации полиселен и полителлур способны существовать
во всех трех релаксационных состояниях. Все три
полимера элементов VI группы могут содержать в цепи
нек-рые гетедооатомы, напр, элементы V группы. Вве-
Введение в расплавы атомов галогенов приводит, как уже
указывалось, к обрыву цепи и снижению степени поли-
полимеризации. Судя по реологическим и морфологическим
(величина рентгеновского большого периода) оценкам,
степень полимеризации полиселена может достигать
нескольких тысяч. Полителлур менее изучен.
Существует представление о том, что полисера ста-
стабильна в аморфной модификации. Однако то, что при-
принимают за аморфную серу, в действительности явля-
является р-ром полимерной серы в низкомолекулярной.
Избавиться при низких темп-pax от «пластификатора»
S8 не представляется возможным, ибо полимерная
сера устойчива как полимер в относительно узком
темп-рном интервале (примерно 130—250 °С). Поэтому
при комнатных темп-pax непрерывно генерируется
молекулярная сера.
Особого внимания заслуживает наличие ограничен-
ограниченного сверху и снизу темп-рного интервала устойчивости
полисеры; при низких темп-pax равновесие сдвинуто
в сторону образования восьмичленной циклич. формы
S8, а при высоких — в сторону двухвалентного аналога
кислорода S2. Эта особенность полисеры и в меньшей
степени полиселена (константа деполимеризации к-рого
при комнатной темп-ре пренебрежимо мала) — убеди-
убедительное доказательство того, что полимеризация пред-
представляет собой типичный фазовый переход, специфика
к-рого определяется характером возникающей струк-
структуры (конфигурации) макромолекулы. Естественно
поэтому, что наиболее выраженный фазовый характер
полимеризация имеет, если макромолекула линейна
и состоит из ковалентно связанных атомов без обрам-
обрамляющих групп.
В чистом виде Sn — типичный кристалло-аморфный
полимер, обычно низкой степени кристалличности
из-за неустойчивости при пониженных темп-pax. В этом
плане уместно указать на единственный известный
в физике полимеров парадокс: темп-pa плавления
кристаллич. полисеры ниже нижнего темп-рного пре-
предела ее устойчивости как полимера.
Гетероцепные неорганические полимеры. В силу при-
причин, изложенных выше, гетероцепные Н. п. распрост-
распространены значительно шире, чем гомоцепные. Сопостав-
Сопоставление табл. 1 и 2 позволяет уяснить термодинамич.
причины, способствующие этому. Что касается кине-
тич. критериев, то, утрируя, можно сформулировать
след. правило: повышенной кинетич. устойчивостью
должны обладать те полимеры типа М — L, у к-рых
один из атомов является кислородом. Действительно,
в этих случаях окисление как бы «уже состоялось»,
и дальнейшая атака главной цепи кислородом мало-
маловероятна. С др. стороны, кислородсодержащие Н. п.
во многих случаях можно трактовать как_ полимерные
ангидриды (это видно из реакций^их образования),
поэтому они м. б. подвержены гидролитич. атаке.
Защитить главную цепь от гидролиза может либо
система обрамляющих групп, либо, что случается чаще,
стабилизация полимера путем перехода в трехмерную
(упорядоченную или неупорядоченную) форму. Этот пе-
переход не является необратимым: именно при переработ-
переработке природных трехмерных Н. п. удается в нек-рых
случаях перевести их в линейную или умеренно раз-
разветвленную форму.
Для более детального анализа термодинамич. и ки-
кинетич. стабильности гетероцепных Н. п. пришлось бы
в отдельности анализировать электронную структуру
цепей каждого конкретного (химического) класса, что
В рамках данной ста- Таблица 1. Комбинации элементов,
тьи нр ттпрттгтявттяртгя образующие неорганические
тьи не представляется полимеры типа [—М— L-"L,
возможным. п
Ниже рассмотрены
нек-рые наиболее ха-
характерные представи-
представители линейных и про-
пространственных Н. п.,
обладающие каучуко-
подобными, стекло- и
волокнообразующими
свойствами и относите-
льно легко синтезируе- Эле~е'нты
мые или (если речь идет
о природных Н. п.) пе-
перерабатываемые. Такое
ограничение обуслов-
обусловлено двумя причинами: Элементы
1) большинство комби- ——- л
наций, представленных
в табл. 1, в действите-
действительности приводит лишь
к олигомерным или ци-
клич. структурам; 2)
из числа длинноцепо-
чечных линейных по-
полимеров выбраны те, физические и морфологич. свой-
свойства к-рых сопоставимы с привычными свойствами ор-
ганич. или элементоорганич. полимеров.
Линейные полимеры. В данном подраз-
подразделе рассмотрены линейные полимеры, являющиеся
каучуками или стеклами.
Наиболее известный неорганич. каучук —
полидихлорфосфазен (полифосфонитрилхлорид)
[— N — Р(С12) — ]п (см. Полифосфазены). В приведенной
ф-ле показаны двойные связи между азотом и фосфо-
фосфором. Однако все известные экспериментальные данные
м
Элементы
группы III
В
А1
Элементы
группы IV
Si
Ge
Sn
Pb
Элементы
группы V
Р
As
Sb
s
Se
Те
L
M
X
X
X
X
X
X
X
X
X
X
X
a
о
о
Й
X
X
X
X
X
X
X
X
X
X
X
X
<
X
X
X
X
X
X
X
X
X
0)
со 3
p
в
Таблица 2. Энергия простои а-связи в гомо-и
структурах
Гомоатомные
Связь
С-С
S-S
р-р
Se-Se
Те—Те
Si-Si
Sb-Sb
Ge-Ge
As-As
N—N
O-O
Энергия связи,
кдж /моль
(ккал/молъ)
336(80,0)
264 F3,0)
222 E3,0)
210 E0,0)
205 D9,0)
189 D5,0)
176 D2,0)
163 C9,2)
163 C9,0)
155 C7,0)
142C4,0)
гетероатомных
Гетероатомные
Связь
В-О
B-N
С-В
Si-O
Р-О
С-О
C-N
As—О
А1-С
C-S
Si—S
C-Si
C-As
N-O
Энергия связи,
пдж/молъ
(ккал/молъ)
499 A19,3)
436 A04,3)
420 A00,0)
373* (89,3)
343 (81,7)
331 G9,0)
277 F6,0)
270 F4,5)
258 F1,6)
258 F1,5)
256 F0,9)
241 E7,6)
229 E4,6)
155 C7,0)
* По др. данным 44 3 пдж'молъ A06,0 ккал/молъ).
(напр., рентгеноструктурного анализа и инфракрас-
инфракрасной спектроскопии) указывают на полную выравнен-
ность всех связей фосфор—азот. Более того, полимер
с сопряженными двойными связями, согласно всем
существующим критериям, не мог бы обладать скелет-
скелетной гибкостью, характерной для макромолекул каучу-
ков. Но именно здесь в образование главной цепи
включаются d-орбитали, благодаря чему возникают
«размазанные» вдоль всей макромолекулы связи по-
повышенной кратности, хотя и не двойные.
Объяснить высокоэластич. свойства полифосфонит-
рилхлорида позволяет также представление об изо-
электронном строении (конфигурации) цепи этого по-
полимера и типичного элементоорганич. каучука, напр.
369
НЕОРГАНИЧЕСКИЕ ПОЛИМЕРЫ
370
полидиметилсилоксанового. Появление за счет 3d-
орбитали у «партнеров» Si и Р соответственно в цепях
Si — О и Р — N лишней я-связи (dn -связи) приводит
к существенно осевой симметрии распределения элек-
электронной плотности и тем самым к малой скелетной (кон-
формационной) жесткости цепей. В обоих случаях
эти цепи, имея нелинейную кон-фигурацию, м. б. пред-
представлены как цепи квазикумулированных связей
II II
II II
что и подчеркивает их изоэлектронный характер и де-
делает понятной способность этих столь несхожих между
собой полимеров проявлять аналогичные каучукопо-
добные свойства. В то же время написание привыч-
привычных (со «счетом валентностей») структурных ф-л
—О—Si—O-Si— и — P=N-P=N—
II II
не отражает участия неподеленных электронных пар
атомов О и N в образовании связей и приводит к про-
противоречию с фактами (цепь сопряженных связей
не м. б. гибкой).
Полифосфонитрилхлорид набухает (а при умерен-
умеренных п растворяется) в бензоле; деполимеризация на-
начинается при нагреве выше 300 °С; при деполимери-
деполимеризации устанавливается равновесие между полимером
и циклич. три- или тетрамером.
Степень полимеризации полифосфонитрилхлорида
может достигать 15 000, однако, в соответствии со ска-
сказанным выше, его гидролитич. стабильность мала.
Последняя м. б. повышена лишь при замене атомов
хлора на нек-рые органич. радикалы, но при этом
полимер, по дефиниции, уже переходит в категорию
элементоорганических.
Каучукоподобными свойствами обладает также поли-
полимерная окись серы (сульфан)
[-Н
Сульфан, обладая достаточной термостойкостью, гид-
гидролитически нестабилен и сравнительно легко превра-
превращается в серную к-ту.
На примере поведения серы в реакциях образования
органич. и неорганич. полимеров можно лишний раз
убедиться в неполноте «аналогий», основывающихся
на периодич. таблице элементов. Так, в органич.
полимерах связи С — ОиС — S во многом аналогич-
аналогичны. При замене углерода на кремний эта аналогия
исчезает, а попутно становится ясной ограниченность
аналогии самих элементов О и S. Действительно,
никакого «тиоаналога» силикатов не существует, хотя
при взаимодействии с кремнием сера и не проявляет
свои «дополнительные» (по сравнению с кислородом)
валентности. Известен циклолинейный спирополимер
силикондисульфид, образующийся при взаимодействии
сульфида алюминия и окиси кремния:
Поскольку этот полимер нерастворим и неплавок,
трудно что-либо сказать о степени его полимеризации.
По физич. свойствам он ближе к минералам и керамич.
материалам, чем к привычным полимерам.
К линейным полимерам относятся силикатные стекла
(включающие, по-видимому, умеренно разветвленные
макромолекулы и относительно подвижные нерегуляр-
нерегулярные сетки), соли полифосфорной и полиборной к-т
(соответственно полифосфаты и полибораты), а также
Н. п., содержащие третьи атомы в главной цепи (боро-
силикаты, металлосиликаты и др.; см. табл. 1). Эти
атомы обязательно должны быть способны к генери-
генерированию хотя бы олигомерных продуктов; в этом случае
они могут благоприятно влиять на электронную струк-
структуру главной цепи, стабилизируя ее и способствуя
достижению более высоких степеней полимеризации.
В противном случае (если эти атомы выпадают за пре-
пределы III—VI групп) они могут располагаться лишь
на концах цепей.
Все эти полимеры являются продуктами поликонден-
поликонденсации соответствующих к-т, т. е. полиангидридами
того же типа, что и сульфан. В то же время располо-
расположение В, Si и Р в таблице Д. И. Менделеева обуслов-
обусловливает повышенную гидролитич. стабильность этих
полиангидридов.
Нек-рые аналогии между неорганич. силикатными
стеклами и органич. стеклами давно известны. В част-
частности, реологич. свойства расплавов силикатных сте-
стекол, их способность к образованию высокопрочных
и высокомодульных волокон являются типичными
проявлениями линейной полимерной структуры.
Из-за недостаточной регулярности цепей полибора-
полиборатов и силикатов в них не удавалось наблюдать кристал-
лич. полимерных морфоз типа сферолитов или ламе-
лярных кристаллов. В то же время известны т. наз.
кристаллизационные катастрофы в стеклах, при к-рых
внезапная кристаллизация порождает столь сильные
внутренние напряжения, что происходит своего рода
взрыв, при к-ром стекло рассыпается в пыль. Напротив,
в полифосфатах легко наблюдаются все характерные
элементы надмолекулярной организации кристалли-
кристаллизующихся гибкоцепных полимеров.
Полифосфорная к-та — продукт поликонденсации
мета- или ортофосфорной к-ты общего строения
-О-Р^О-
Натриевая и калиевая соли полифосфорной к-ты пред-
представляют собой типичные полиэлектролиты, применяе-
применяемые в качестве ионитов. В р-ре макромолекулы этих
солей вследствие полиэлектролитного набухания имеют
почти развернутую конформацию.
Трехмерные полимеры. Как уже отме-
отмечалось, большинство Н. п. относится к категории мине-
минералов или керамич. материалов. Наиболее распростра-
распространенным представителем таких полимеров является
трехмерный вариант полимерной окиси кремния —
кварц. Образование пространственных структур Н. п.
в природе произошло в результате «геологич. естест-
естественного отбора» — как форма стабилизации цепей
без обрамляющих групп илд с «незащищающими»
обрамляющими группами. В зависимости от геологич.
условий трехмерные структуры природных Н. п. ре-
регулярны или нерегулярны. В мягких условиях более
вероятен рост трехмерных ковалентных кристаллов,
не отличимых, с одной стороны, от гигантских сверх-
сверхмолекул, а с другой — по физич. свойствам — от обыч-
обычных молекулярных кристаллов. Отличием является
лишь то, что в случае регулярных трехмерных Н. п.
плавление и деполимеризация оказываются единым
процессом.
Н. п., образовавшиеся при геологич. катастрофах,
напр, при извержениях вулканов, чаще представляют
собой аморфные сетки. Однако из-за отсутствия непо-
неполярных обрамляющих групп эти сетки практически
неподвижны (очень сильное межмолекулярное взаимо-
взаимодействие) и лишены каких-либо физич. черт сходства
с вулканизационными сетками в органич. полимерах.
Однако в процессе переработки можно «заставить»
такие вещества вести себя подобно органическим
волокно- или стеклообразующим полимерам: это озна-
означает, что при переработке, обязательно включающей
плавление (а значит и деполимеризацию), баланс
между линейными (или слабо разветвленными) и выра-
женно-сетчатыми структурами смещается в сторону
371
НЕРАВНОВЕСНАЯ ПОЛИКОНДЕНСАЦИЯ
372
первых; в предельном варианте трехмерные Н. п. пре-
превращаются в линейные и в этом новом качестве исполь-
используются как волокна или стекла. Процесс получения
керамич. материалов связан, по-видимому, лишь с из-
изменением характера пространственной сетки, но не с
ее ликвидацией.
Мы не можем здесь входить в химич. детали соот-
соответствующих технологич. процессов; ясно лишь, что
во многих случаях для перевода Н. п. из пространст-
пространственной в линейную модификацию необходимо снаб-
снабдить ее стабилизирующими обрамляющими группами;
это достигается, напр., введением всякого рода «до-
«добавок» при получении стекла.
Лит.: Андрианов К. А., Полимеры с неорганически-
неорганическими главными цепями молекул, М., 1962; Коршак В. В.,
Прогресс полимерной химии, М., 1965; Неорганические поли-
полимеры, под ред. Ф. Стоуна и В. Грэхема, пер. с англ., М., 1965;
Ван Везер Д ж., Усп. хим., 28,1108A969); Барте-
н е в Г. М., Строение и механические свойства неорганических
стекол, М., 1966; Шусторович Е. М., Электронное
строение полимерных молекул с кратными связями в основной
цепи, М., 1967; Encyclopedia of polymer science and technology,
v. 7, N. Y.— [a. o.], 1967, p. 664. С. Я. Френкель.
НЕРАВНОВЕСНАЯ ПОЛИКОНДЕНСАЦИЯ — см.
Поликонденсация.
НЕТКАНЫЕ ИЗДЕЛИЯ, нетканые мате-
материалы, нетканые полотна (nonwoven
fabrics, Faservliesstoffe, tissus non tisses) — текстиль-
текстильные изделия из волокон или нитей, соединенных меж-
между собой без применения методов ткачества.
Крупное промышленное производство современных
Н. и. появилось в 40-е гг. нашего века, а сейчас они
являются одним из основных видов текстильной про-
продукции во многих странах. В 1972 во всем мире было
выпущено более 3 млрд. м2 Н. и., для их изготовления
израсходовано более 200 тыс. т текстильных волокон.
С 1962 по 1972 объем мирового производства Н. и.
возрос в 5 раз.
Нетканые материалы, получаемые физико-
химическими способами
Большинство Н. и. получают физико-химич. спосо-
способами, основанными на использовании связующих
веществ для склеивания волокон (или нитей) друг
с другом. Полученные таким образом изделия наз.
клееными неткаными материалами.
Наиболее распространены клееные Н. и., основой
к-рых является т. наз. волокнистый холст (слой тек-
текстильных волокон, масса 1 м2 к-рого составляет от 10
до 1000 г и более). При формировании холста приме-
применяют различные методы. Наиболее распространен
механич. метод (рис. 1), к-рый основан на образовании
Рис. 1. Схема устрой-
устройства для получения
холста механическим
методом: 1 —съемный
барабан чесальной
машины, 2 — прочес,
з — раскладчик про-
прочеса, 4 — сформиро-
сформированный холст.
холста из нескольких слоев прочеса (поступающего
€0 съемного барабана чесальной машины, используемой
в классич. текстильном производстве), укладываемых
друг на друга. Часто холст формируют также аэроди-
намич. методом (рис. 2). В этом случае волокна сни-
снимаются с барабана чесальной машины потоком воздуха
и переносятся к поверхности сетчатого барабана (кон-
(конденсора) или на горизонтальную сетку, где из волокон
формируется холст необходимой массы. Устройства
такого типа дают возможность получать холст со ско-
скоростью 5—10 м/мин и более. Максимальная скорость
(до 100 м/мин и более) достигается при формировании
холста гидравлическим методом, когда холст отли-
отливают из водной дисперсии волокон на сетке бумагоде-
бумагоделательной машины. При этом используют технологию,
которая сходна с технологией получения бумаги.
Иногда при изготовлении Н. и.
функции волокнистого холста вы-
выполняет материал, полученный рас-
расщеплением полимерной пленки,
Рис. 2. Схема уст-
устройства для по-
получения холста
аэродинамическим
методом: 1 — во-
волокно, 2 — съем-
съемный барабан че-
чесальной машины, 3 — диффузор, 4 — конденсор, 5 — вывод-
выводной транспортер, 6 — сформированный холст.
которая имеет структуру, сходную со структурой во-
волокнистого холста из слабо связанных волокон (о
таком материале смотри Бумага из синтетических
волокон).
Основой Н. и. может служить не холст, а система
нитей, напр, несколько слоев пряжи (нитей), нало-
наложенных друг на друга таким образом, чтобы каждая
нить пересекалась большим числом других нитей с об-
образованием «сетки».
Волокна или нити, образующие основу Н. и., склеи-
склеивают друг с другом связующим веществом, содержание
к-рого в готовом Н. и. составляет обычно от 20 до 60%.
Иногда связующие не используют; в этом случае осно-
основу Н. и. подвергают специальной обработке (тепловой,
химич. реагентами и др.), способствующей появлению
«липкости» волокон (нитей), к-рые после этого соеди-
соединяются друг с другом в местах пересечения. В зависи-
зависимости от особенностей склеивания волокон (нитей)
различают несколько основных способов получения
клееных Н. и.
Широко распространен в производстве клееных Н. и.
способ пропитки, хотя он и не относится
к числу наиболее перспективных. Способ заключается
в пропитке холста жидким связующим (латексом,
в том числе вспененным, или р-ром полимера) с после-
последующей сушкой (для удаления влаги или растворителя)
и термообработкой, способствующей упрочнению Н. и.
Чаще всего при пропитке холст окунают в ванну
со связующим или последнее распыляют над поверх-
поверхностью холста. При изготовлении Н. и. небольшой
массы пропитку ведут по технологии, сходной с тех-
технологией нанесения рисунка на поверхность ткани
при ее отделке методом печати. Этот способ дает воз-
возможность осуществлять процесс с высокой скоростью
E0 м/мин и более) и, кроме того, в этом случае мож-
можно совместить процессы пропитки и отделки Н. и.
Пропитанный материал высушивают и подвергают
термообработке в термокамерах, нагреваемых горя-
горячим воздухом или с помощью инфракрасных излу-
излучателей.
В качестве основы связующих, применяемых при
изготовлении Н. и. способом пропитки, чаще всего
используют синтетич. каучуки, полиакрилаты, поли-
винилхлорид, поливинилацетат, полиуретаны и др.
В СССР наиболее распространенным связующим при
изготовлении таких Н. и. является латекс на основе
карбоксилатного бутадиен-нитрильного каучука (напр.,
марки СКН-40-1ГП). Холст обычно формируют из хлоп-
хлопка или из смеси вискозных и полиамидных волокон
(чаще всего в соотношении 70 : 30). В значительных
373
НЕТКАНЫЕ ИЗДЕЛИЯ
374
Вол он на
количествах для изготовления этого вида Ы. и. при-
применяют отходы классич. текстильного производства,
в том числе непрядомьте. Получаемые Н. и. использу-
используются в швейной промышленности (в качестве борто-
вочных и прокладочных материалов), для фильтров и
изделий медицинского назначения, в автомобильной
промышленности (тепло- и звукоизоляционные материа-
материалы) и др.
Способ пропитки не обеспечивает равномерного рас-
распределения связующего по всей толщине Н. и., поэто-
поэтому по механич. свойствам такие материалы часто усту-
уступают Н. и., полученным др. способами. К недостаткам
способа относится также то, что он связан с «мокрыми»
процессами, поэтому такие Н. и. часто по внешне-
внешнему виду больше похожи на бумагу, чем на ткань.
Кроме того, при использовании способа пропитки
возникает необходимость принимать специальные меры
по очистке промышленных вод.
Способ горячего прессования более
перспективен, т. к. в этом случае не применяются
жидкие связующие и имеется больше возможностей
получить Н. и. лучшей структуры и, соответственно,
лучшего качества. Способ основан на склеивании
волокон термопластичными связующими (полиамиды,
полиэтилен, поливинилхлорид и др.) под давлением
до 2 Мн/м2 B0 кгс/см2) при повышенных темп-рах,
обычно на специальных ка-
каландрах. Горячему прессова-
прессованию предшествует термообра-
термообработка слоя волокон, содержа-
содержащего связующее. Последнее
вводят в холст на стадии его
формирования (в виде легко-
легкоплавких волокон, сетки, нитей
и др.) или в уже сформирован-
сформированный холст (в виде порошка).
Н. и. лучшего качества полу-
получают при использовании хол-
холста, содержащего бикомпоненгп-
ные волокна (при условии, что
один компонент волокон яв-
является легкоплавким и спо-
способен выполнять функции свя-
связующего). Иногда при изготов-
изготовлении Н. и. связующее синтезируют непосредственно
в сформированном холсте из введенных туда мономе-
мономеров, а затем осуществляют горячее прессование, после
к-рого получают Н. и. точечной структуры (связующее
распределено преимущественно в местах пересечения
волокон — рис. 3).
Бумагоделательным (мокрым) спо-
способом Н. и. получают с помощью бумагоделатель-
бумагоделательных машин, позволяющих работать с высокой произво-
производительностью A00 mi мин и более). В состав Н. и. при
этом, кроме текстильных волокон и связующего, часто
входит древесная целлюлоза. Связующее вводят в мас-
массу, поступающую на бумагоделательную машину, или
в уже отлитое полотно. При использовании обычных
машин полотно материала формируют из сравнительно
коротких волокон B—6 мм). Машины с наклонной сет-
сеткой дают возможность использовать текстильные во-
волокна обычной длины C0—35 мм).
В качестве связующего в бумагоделательном спо-
способе применяют латексы, легкоплавкие волокна, фи-
бриды (см. Бумага из синтетических волокон) и биком-
понентные волокна. Получаемые Н. и. дешевы, а
поэтому широко используются в производстве изделий
однократного применения (постельного белья в го-
гостиницах, полотенец, скатертей, перевязочных мате-
материалов).
Фильерный спосо б—один из наиболее пер-
перспективных при получении Н. и. Волокна (нити),
образующиеся на выходе из фильер прядильной машины
•язующее
Рис. 3. Элемент нетка-
нетканого материала точечной
структуры.
(рис. 4), аналогичной применяемым на заводах химич.
волокна, проходят через каналы, где вытягиваются
в воздушном потоке, а затем укладываются на движу-
движущийся транспортер с образованием полотна Н. и. Чаще
всего сформированный материал закрепляют с помощью
связующего, однако известны схемы, где оно не при-
применяется. В последнем случае используется липкость
свежесформованных волокон.
Перспективны Н. и., получае- I Ь^_
мые фильерным способом из
смеси полимеров, т. е. на ос-
основе биКОМПОНентнЫХ ВОЛОКОН, Воздух
один из компонентов к-рых
выполняет роль связующего.
Фильерным способом изго-
изготовляют Н. и. из полиамидов,
полипропилена, полиэтилена и
Рис. 4. Схема получения неткано-
нетканого материала фильерным способом:
1 — расплав полимера, 2 — филье-
фильера, 3 — устройства для вытяжки
волокон, 4 — волокна, образующие
холст, 5 — транспортер для уклад-
укладки сформированного холста.
др. волокнообразующих полимеров. Перспективны по-
получаемые этим способом вискозные Н. и., к-рые могут
заменить нек-рые виды бумаги, получаемой по клас-
классич. бумажной технологии, а это позволит исключить
сброс в водоемы больших количеств промышленных
вод и, следовательно, обойтись без строительства очист-
очистных сооружений.
Структурообразующий способ свя-
связан с получением Н. и. без использования волокон.
При этом Н. и. формируют путем образования конден-
конденсационных структур из р-ров или аэрозолей полимеров
(в виде осадка пористой, иногда волокнистой структу-
структуры, к-рый может содержать наполнители, затем из него
вымываемые), отверждением пены, полимеризацией
мономеров в среде, содержащей вымываемые наполни-
наполнители, и др. После нанесения рисунка тиснением такие
Н. и. по внешнему виду напоминают традиционные
текстильные материалы (ткань, вой-
войлок и др.). Они «дышат» подобно
ткани. Их можно использовать вмес-
вместо ткани или высококачественной
бумаги в технике (для фильтров и
др.) и для бытовых целей. ||11ЙУ№$Ш*—2
Существуют и др. способы полу- ~^^^^
чения клееных Н. и. При введении,
напр., в систему нитей связующего
вещества (путем пропитки, при ис-
использовании бикомпонентных нитей —l^—
и др.) получают Н. и. для декора- П7А?У1И^$§§1—2
тивных целей и для использования
в технике (вместо тканых сеток).
Иногда в клееный нетканый материал
Рис. 5. Элемент структуры нетканого ма-
материала со вспомогательным элементом:
1 — холст, 2 — вспомогательный элемент,
з — связующее.
на основе холста из волокон связующее вводят с помощью
вспомогательных элементов (нитей, полотна ткани,
пленки и др.). При изготовлении, напр., Н. и., заменяю-
заменяющих тканый ватин, жидкое связующее (расплав, ла-
латекс и др.) сначала наносят на поверхность вспомога-
вспомогательного элемента — нитей (пряжи); при прессовании
холста, пронизанного такими нитями, связующее пере-
переходит с поверхности нитей в холст и склеивает волокна.
Особенности структуры такого Н. и. показаны на
375
НЕТКАНЫЕ ИЗДЕЛИЯ
376
рис. 5. Таким способом получают, напр., ковровые изде-
изделия, приклеивая систему нитей к поверхности ткани.
Нетканые изделия, получаемые механическими
способами
Наиболее известны след. виды Н. и., получаемых
механич. способами: вязально-прошивные, иглопро-
иглопробивные и валяльно-войлочные.
К числу вязально-прошивных Н. и.
относятся холстопрошивные, нитепрошивные и полот-
нопрошивные материалы. При изготовлении холсто-
прошивных Н. и. (технология маливатт — ГДР,
а р а х н е — Чехословакия и др.) применяют вязаль-
вязально-прошивные машины, действие к-рых основано
на тех же принципах, что и основовязальной трико-
трикотажной машины. В движущемся через вязально-про-
шивную машину холсте волокна закрепляются в ре-
результате прошивания его нитями, к-рые укладываются
и соединяются так, как при основовязании. Стежки
длиной 2—4 мм образуются со скоростью более 1000
в 1 мин при рабочей ширине прошиваемого материала
180 см и более. Получаемые Н. и. используются в ка-
качестве теплоизоляционных (взамен тканого ватина
и др.) или упаковочных материалов, как основа в про-
производстве кожи искусственной и др. Их изготавливают
со скоростью 3—8 м/мин и более.
Нитепрошивные Н. и. (материалы м а-
л и м о — ГДР) получают прошиванием одной или
нескольких систем нитей вспомогательной системой
швейных нитей. В одних случаях вспомогательные
нити соединяют основные и уточные нити (путем их
провязывания), а в других материал изготавливают
только из двух систем нитей (уточных и швейных).
В готовом Н. и. на долю швейных нитей приходится
50—60%. Н. и. шириной до 240 см выпускаются со ско-
скоростью 5—10 м/мин и более. Этот материал успешно
используется для декоративных целей, в производстве
пляжной и верхней одежды, полотенец и др. Особый
интерес представляют нитепрошивные Н. и. с ворсо-
ворсовыми провисающими петлями (полупетлями), к-рые
успешно конкурируют с махровыми тканями (материа-
(материалы типа фротте).
Полотнопрошивные Н. и. получают прошиванием
текстильного полотна ворсовой пряжей (материал
Это способствует улучшению
структуры и свойств полотна,
в качестве к-рого используют
ткань, материал типа малимо
и др. Схема переплетения вор-
ворсовых нитей (без полотна) по-
показана на рис. 6. Прошивани-
Прошиванием хлопчатобумажного полот-
полотна с массой 1 м2 160—180 г
получают при использовании
хлопчатобумажной ворсовой
пряжи махровые Н. и. с мас-
массой 1 м2 350—400 г при дли-
длине ворса 2—5 мм. С примене-
применением шерстяной пряжи изготавливают Н. и. для из-
изготовления пальто и юбок.
К числу полотнопрошивных иногда относят Н. и., по-
получаемые петлистым прошиванием основы (ткани или
Н. и., изготовленного фильерным способом) с после-
последующим закреплением петель с изнаночной стороны
с помощью связующего вещества (проклеиванием).
В данном случае используются методы как химической,
так и механической технологии. Таким комбинирован-
комбинированным методом получают преимущественно ковры (т. н.
тафтинг-ковры), выпуск к-рых увеличивается во всем
мире высокими темпами. При изготовлении тафтинг-
ковров прошивание основы ковровой пряжей осу-
осуществляется с помощью игл, протаскивающих пряжу
через ткань (рис. 7). При обратном движении иглы
малиполь — ГДР).
Рис. 6. Схема перепле-
переплетения нитей в полотно-
прошивном материале.
пряжа захватывается держателем, что ведет к образова-
образованию петли. В момент, когда игла находится над осно-
основой, последняя продвигается вперед, и далее цикл
повторяется. Имеется возможность регулировать вы-
высоту и частоту петель. При раз-
разрезании петель получают ковер с
разрезным ворсом. Для закрепле-
закрепления петель на изнанку ковра на-
носят связующее — каучуки или
полиуретаны. Известны машины
для изготовления тафтинг-ковров
шириной 550 см со скоростью
5 м2/мин и более.
4 I II I 5
Рис. 7. Схема образования петель при
изготовлении тафтинг-ковра: 1—тка-
1—тканая основа ковра, 2—нить (ковровая
пряжа), з — игла, протаскивающая
нить через тканую основу, 4 — держатель, удерживающий
петли при движении иглы вверх, 5 — петли, образующие
лицевой слой.
С помощью вязально-прошивных машин изготавли-
изготавливают Н. и. без применения нитей (материалы в о л ь-
т е к с — ГДР, арабева — Чехословакия и др.).
Такие Н. и. могут состоять, напр., из ткани (каркаса)
и холста из длинных волокон. При образовании Н. и.
петли из волокон, закрепляющие волокна на ткани,
образуются по тому же принципу, как петли из нитей
в нитепрошивных Н. и. После протаскивания волокон
из холста сквозь тканый каркас на изнаночной сторо-
стороне Н. и. образуются прочные петли, а на лицевой
стороне — пушистый и высокий ворс. Иногда волокно
дополнительно закрепляется петлями из пряжи. Этот
вид Н. и. успешно используется в качестве утепляю-
утепляющей ворсовой прокладки в спортивной одежде и деми-
демисезонных пальто, заменяет мех при изготовлении го-
головных уборов и теплой обуви и др.
К числу наиболее перспективных относятся игло-
иглопробивные Н. и., изготавливаемые путем перепутыва-
ния волокон в холсте при многократном прокалывании
Рис. 8. Схема иглопрокалывания холста: 1 — слой воло-
волокон, 2 — питающий транспортер, з — подвижная доска,
4 — иглы, 5 — решетка, б — выводящий транспортер, 7 —
готовый материал.
его специальными иглами с зазубринами (рис. 8)
на иглопробивной машине. Иглы укрепляют в доске,
совершающей возвратно-поступательное движение в
вертикальном направлении. При движении доски
вниз (до упора) иглы прокалывают холст, а в момент,
когда иглы находятся над поверхностью холста, мате-
материал продвигается вперед. В СССР для изготовления
таких Н. и. применяют иглопробивные машины с ра-
рабочей шириной 180 см, производительность к-рых
может достигать 5 м/мин и более при числе проколов
более 200 на 1 см2 поверхности холста. Масса 1 м2
Н. и. может достигать 1500 г. Известны иглопробив-
иглопробивные машины с рабочей шириной до 15 м.
Большинство иглопробивных Н. и. используется
в качестве ковров, к-рые успешно конкурируют не
только с ткаными, но и с тафтинг-коврами, т. к.
для своего изготовления не требуют пряжи. Иглопро-
Иглопробивные Н. и. применяют также в качестве одеял, сукон
бумагоделательных машин, фильтров и др. Особенно
377
НИТРАТЫ ЦЕЛЛЮЛОЗЫ
378
перспективны иглопробивные Н. и., при изготовлении
к-рых применяют усадочные волокна (способные за-
закручиваться в спираль после специальной обработки
материала), в том числе бикомпонентные.
К числу Н. и. относят и валяльно-войлоч-
ные текстильные материалы, при изго-
изготовлении к-рых используется способность волокон
шерсти к свойлачиванию (при механической или теп-
ловлажностной обработке). В состав этих Н. и. иногда
вводят каркас из ткани. Эта технология имеет много-
многовековую историю; таким образом изготовляют, напр.,
валенки.
Большинство Н/*и. получают в виде полотна. Однако
известны Н. и., к-рые имеют форму швейных изделий.
Напр., при напылении на форму смеси коротких воло-
волокон и связующего получают нек-рые виды спецодежды,
колпаки для шляп и др.
Лит.: Тихомиров В. Б., Химическая технология
производства нетканых материалов, М., 1971; Krcma R.,
Manual of nonwovens, Manchester, 1971; Тихомиров В. Б.,
Физико-химические основы получения нетканых материалов,
М., 1969; Технология производства нетканых материалов, М.,
1967; Сухарев М. И., Свойства нетканых текстильных
материалов и методы их исследования, М., 1969; BureshF.,
Nonwoven fabrics, N. Y., 1962; Перепелкина М. Д.,
Щербакова М. Н., Золотницкая К. Н., Механиче-
Механическая технология производства нетканых материалов, М., 1973.
В. Б. Тихомиров.
НИТРАТЫ ЦЕЛЛЮЛОЗЫ, нитроцеллю-
нитроцеллюлоза (cellulose nitrates, Zellulosenitrate, nitrates
de cellulose) — сложные азотнокислые эфиры целлю-
целлюлозы общей ф-лы [CeH7O2(OHK-x(ONO2)x]n.
Свойства. Н. ц.— твердое волокнистое вещество бе-
белого цвета, без запаха, по внешнему виду очень напо-
напоминающее целлюлозу. Степень этерификации, или сте-
степень замещения (С. 3.), Н. ц. может изменяться от О
до 3; ее рассчитывают на основании содержания азо-
азота [N] (в %) по ф-ле:
. С.З. = 3,6[N]/C1,1 — [N])
Степень замещения — одна из важнейших характери-
характеристик, в значительной степени определяющая физико-
механич., химич. и технологич. свойства этого полиме-
полимера. Предельное содержание азота, соответствующее эте-
этерификации трех ОН-групп, составляет 14,14%, при
этерификации двух ОН-групп — 11,1% и одной ОН-
группы — 6,8%. Различают след. основные виды тех-
нич. Н. ц.: коллоксилин A0,7—12,2% N),
пироксилин №2 A2,2—12,5% N) и п и р о-
к си лин №1 A3,0—13,5% N).
Плотность Н. ц^ незначительно зависит от содержа-
содержания азота. Так, плотность при 20 °С коллоксилина,
содержащего 11,8 и 12,2% N, составляет соответствен-
соответственно 1,58 и 1,65 г/смъ. Средняя степень полимеризации
и, следовательно, мол. масса Н. ц. колеблются в ши-
широких пределах. Напр., степень полимеризации Н. ц.,
используемого в производстве целлулоида, составляет
400—600 (мол. м. 100 000—150 000), в производстве
лака — 150—300 (мол. масса 38 500—80 000), бездымно-
бездымного пороха — 1000—2000 (мол. масса 250 000—500 000).
Н. ц.— неоднородные по мол. массе продукты, что
обусловлено гл. обр. неоднородностью по мол. массе
исходной целлюлозы, а также ее гидролитич. распадом
при синтезе Н. ц. Проведение синтеза при 0 °С и не-
небольшом содержании E%)
Таблица 1. Значения констант
в ур-нии Марка—Хувинка
[ц]=КМ* (для М=68 000—
240 000)
Растворитель
Ацетон
Циклогексанон
Метилацетат . .
к-Бути л ацетат
Нитробензол . ,
К-10*
2,53
2,24
1,83
0,92
0,61
а
0,795
0,810
0,835
0,905
0,945
воды в нитрующей смеси
позволяет сохранить мо-
лекулярно-массовое рас-
распределение исходной цел-
целлюлозы. Этот метод ши-
широко используют для изу-
изучения молекулярно-мас-
сового распределения цел-
целлюлозы и ее производных.
Мол. массу (М) Н. ц.,
содержащего 12,2% N,
рассчитывают по характеристич. вязкости [г|] (в дл/г)
его р-ра при 25 °С, используя ур-ние Марка — Ху-
Хувинка (табл. 1).
Зависимость между [т]] и степенью полимеризации Р
нитратов целлюлозы описывается ур-нием [ц] = КРа
(табл. 2).
Таблица 2. Значения констант в
Полимер
Нитроцеллюлоза
A3,8% N)
Тринитрат целлю-
целлюлозы
Тринитрат целлю-
целлюлозы
Растворитель
Ацетон
Ацетон
н-Бутилацетат
ур-нии 1ц]=КР0
Темп-
ра, °С
20
25
25
К-102
1,36
1,42
1,41
а
0,948
0,933
0,969
Темп-ры стеклования и плавления лежат выше
темп-ры разложения. С введением пластификаторов
темп-pa стеклования понижается. При содержании
в Н. ц. 10 и 40% пластификатора (дибутилфталата)
темп-pa стеклования составляет соответственно 120
и 55 °С. Теплота плавления Н. ц. (С. 3. = 2,44) со-
составляет 5,65 Мдж/кмолъ A350 кал/молъ), энтропия
плавления 6,322 кдж/(кмоль-К) [1,51 кал/(моль-°С)].
Коэфф. рефракции для Н. ц., содержащих 11,8—
12,2% N, равен 1,51.
Ниже приведены нек-рые свойства непластифициро-
ванной пленки из Н. ц., содержащих 11,8—12,2% N:
Прочность при растяжении,
Мн/м2 80 — 120
кгс/мм2 8 —12
Относительное удлинение, % 12—30
Число двойных перегибов до разрушения 40—150
Ударная вязкость, кдж/мг, или кгс-см/смг 200—400
Диэлектрич. проницаемость при 25 °С
частота 60 гц 7,5
>> 1 кгц 7,0
» 1 Мгц ........... 6,0
Тангенс угла диэлектрич. потерь
60 гц 0,03
1 кгц 0,06
Водопоглощение* при 21 °С за 24 -ч, % . . 2—3
Влагопроницаемость при 21 °С,
кг/(м-сек-н/м2) 0,04—0,56
г-см/(см2-ч-мм рт.ст.) @ , 2—2 , 8) • 10~в
Водопроницаемость при 21 °С,
см2/(сек>н/м2) 0,23-10~12
см3 • см/(см2 • сек • кгс/см2) 2,3-10~8
* Водопоглощение Н. ц. возрастает с уменьшением сте-
степени этерификации.
Нестабилизированные Н. ц. характеризуются низ-
низкой атмосферостойкостью и очень низкой термостой-
термостойкостью (особенно Н. ц., не содержащие воды). Так, при
эксплуатации в обычных атмосферных условиях Н. ц.
полностью разрушаются менее чем через 3 мес. При
нагревании Н. ц. начинают разлагаться уже при 40 —
60 °С, причем скорость разложения быстро возрастает
с повышением темп-ры, а также в присутствии следов
к-т, щелочей и примесей, образующихся при синтезе
Н. ц. (напр., продуктов окисления и гидролиза целлю-
целлюлозы). Разложение сопровождается выделением окис-
окислов азота, формальдегида, глиоксаля, нитрилов, му-
муравьиной и синильной к-т. Термич. разложение Н. ц.—
самоускоряющийся процесс, особенно в присутствии
кислорода; при быстром нагреве распад Н. ц. может
закончиться вспышкой и взрывом. Темп-pa воспламе-
воспламенения зависит от скорости подвода тепла: 190 °С —
при достаточно медленном нагревании, 160—170 °С —
при быстром. Энергия активации термич. распада кол-
коллоксилина на воздухе 119—142 Мдж/кмолъ B8,4—
33,9 ккал/моль), в атмосфере азота 142—162 Мдж/кмолъ
C3,6—38,8 ккал/молъ); тепловой эффект распада пиро-
ксилинов на воздухе и в вакууме соответственно 3.15
и 2,15 Мдж/кг G50 и 515 кал/г). Введение в Н. ц. стаби-
379
НИТРИЛОВ ПОЛИМЕРЫ
380
лизаторов (напр., дифениламина, производных мочеви-
мочевины) повышает их атмосферостойкость и термостойкость.
Растворимость Н. ц. в различных растворителях за-
зависит от степени замещения. Низкозамещенные Н. ц.
@,5—2% N) растворимы при комнатной темп-ре только
в 6%-ном р-ре NaOH, при содержании 9—11% N —
в этиловом спирте, в смеси спирта и толуола. Н. ц.,
содержащие 11 — 12,7% N (С. 3. = 2—2,5), растворимы
в кетонах, сложных эфирах (напр., этил ацетате), ук-
уксусной к-те, фурфуроле, метаноле, диоксане, этилен-
гликоле, циклогексаноне, нитробензоле. Универсаль-
Универсальным растворителем Н. ц., содержащих 10,5—14,4% N
(С. 3. = 1,8—3), служит ацетон. Н. ц. любой степени
замещения нерастворимы в воде и в неполярных рас-
растворителях (напр., в бензоле, СОЦ).
Н. ц. не устойчивы к действию к-т и щелочей. Разб.
минеральные к-ты вызывают медленную денитрацию
Н. ц. Щелочи, к тому же, омыляют и разрушают Н. ц.,
особенно быстро — спиртовые р-ры щелочей. Поэтому
при денитрации щелочью получают обычно деструкти-
рованную целлюлозу. Для снижения деструкции до
минимума Н. ц. омыляют водными р-рами гидросуль-
гидросульфида натрия (NaSH) или аммония (NtUSH) при 35 °С.
Получение. Для получения Н. ц. применяют коротко-
волокнистую хлопковую целлюлозу (линт). Техноло-
Технология получения Н. ц. включает след. стадии: 1) приго-
приготовление нитрующей смеси; 2) подготовка (рыхление
и сушка) целлюлозы; 3) нитрация целлюлозы; 4) ста-
стабилизация и обезввживание. Разрыхленную и высушен-
высушенную целлюлозу при получении, напр., коллоксилина
нитруют смесью, состоящей из 20—25% азотной к-ты,
55—60% серной к-ты и 18—20% воды, в специальных
аппаратах (нитраторах) или в центрифугах при 30—
45 °С и модуле ванны 40—50 (отношение массы нитрую-
нитрующей смеси к массе целлюлозы). В зависимости от
темп-ры продолжительность процесса составляет 20—
60 мин. При получении пироксилинов состав нитрующей
смеси B0-30% HNO3, 60-70% H2SO4 и 5-10% Н2О),
а также условия нитрации другие. Чем выше содержа-
содержание воды, тем ниже степень полимеризации. Макси-
Максимальная B,8) степень замещения Н. ц. достигается при
содержании воды в нитрующей смеси 3,5—5%. После
завершения нитрации образовавшийся продукт отжи-
отжимают от кислотной смеси и многократно промывают хо-
холодной и горячей водой, слабым р-ром соды и снова
водой. Затем И. ц. отжимают и выпускают в виде рых-
рыхлой волокнистой массы желтоватого цвета. Хранят
Н. ц. с содержанием воды 20—35%. Если необходимо,
Н. ц. обезвоживают, вытесняя воду спиртом. Требуе-
Требуемая степень полимеризации, а соответственно и вяз-
вязкость получаемых р-ров Н. ц. достигаются их кипя-
кипячением в воде при 125—140 °С под давлением 2—3 Мн/м2
B0—30 кгс/см2).
Н. ц. фракционируют растворением или (что более
целесообразно) дробным осаждением водой или гепта-
гептаном из ацетоновых р-ров.
Применение. Области применения Н. ц. зависят от
содержания в них азота. В СССР коллоксилин приме-
применяют для производства пластмасс (этролов), целлулоида,
для приготовления лаков и эмалей (см. Эфироцеллю-
лозные лаки и эмали), клеев. Коллоксилин, содержа-
содержащий 11,5—12,2% N, используют в производстве без-
бездымного пороха, динамитов и др. взрывчатых веществ,
для желатинизации жидких нитроэфиров (напр., нит-
нитроглицерина). Пироксилины применяют для получения
бездымного пороха.
За рубежом Н. ц. применяют для производства цел-
целлулоида, лаков и в очень небольших количествах
кино- и фотопленки. Основные фирменные названия
Н. ц.: америт, дуко, геркулес CN, гер-
к у л о и д (США); эксонайт (Великобрита-
(Великобритания); камфолоид (Япония), гелларидин
(ФРГ), т о к о л и т (Австрия).
Основной недостаток Н. ц.— горючесть, поэтому они
вытесняются др. полимерами (напр., ацетатами целлю-
целлюлозы и различными синтетич. полимерами).
Продукт взаимодействия целлюлозы (древесины)
с конц. HNO3 впервые получен в 1832 А. Бракконо.
Лит.: Ушакове. Н., Эфиры целлюлозы и пластические
массы на их основе, М.— Л., 1941; Р о г о в и н 3. А., Химия
целлюлозы, М., 1972; Закощиков А. П., Нитроцеллю-
Нитроцеллюлоза, М., 1950; С е д л и с В. О., Эфиры целлюлозы и пласти-
пластические массы на их основе, Л., 1958; Козлов П. В., Физи-
ко-химия эфироцеллюлозных пленок, М., 1948; Ники-
Никитин Н. И., Химия древесины и целлюлозы, М.—Л., 1962;
Справочник по пластическим массам, под ред. М. И. Гарбара
[и др.], т. 2, М., 1969, с. 196. В. Н. Кряжев.
НИТРИЛОВ ПОЛИМЕРЫ (polynrtriles, Polynitrile,
polynitriles).
Нитрилы (Н.)— соединения, содержащие в молекуле
одну (мононитрилы), две (динитрилы) или более нитри-
льных групп — CN, связанных с алифатич. или арома-
тич. радикалом. Свойства Н. приведены в таблице.
Свойства нитрилов
Нитрил
Ацетонитрил CH3C=N . . .
Пропионитрил C2H5C=N . .
Бутиронитрил h-C3H7C=N
Бензонитрил CeH5C=N . . .
Трифторацетонитрил
CF3C=N
Темп-ра
плавле-
плавления, °С
—44,9
-91,85
—111,9
— 13,8
Темп-
81,6
97,1
117,6
191 ,1
-63,9
Показа-
Показатель пре-
ломле-
ломления п D20
1 ,3442*
1,3683
1,3860
1 ,5306
Плот-
Плотность
при
15°С,
г/см3
0,7828*
0,7867
0,7954
1,0095
N.
С
N
* При 20 °С.
Н. довольно токсичны. Напр., предельно допусти-
допустимое содержание паров ацетонитрила в воздухе
0,002 мг/л.
Несмотря на высокую реакционную способность нит-
рильной группы, при действии каталитич. кол-в ве-
веществ кислого или основного характера при обычном и
при повышенных давлениях образуются не полимеры,
а димеры или циклич. тримеры (см. ур-ние реакции).
Основные методы полу- г> хт р
чения Н.: дегидратация
амидов или аммонийных со- 3 RCN *¦
лей карбоновых к-т; алки-
алкилирование щелочных солей
синильной к-ты галогенал-
килами или алкилсульфа-
тами; окислительное алкилирование (одновременное
алкилирование и дегидрирование) углеводородов или
нек-рых их производных.
Полинитрилы (П.) — полимеры общей формулы
[—С = N—]п, где R — алифатич. или ароматич. ради-
I
R
кал. В макромолекулах П. может содержаться также
довольно значительное число связей —С = С —, а в П.,
полученных полимеризацией RCH2C = N, — триази-
новые или пиридиновые циклы.
П.— аморфные вещества, цвет к-рых в зависимости
от мол. массы и степени сопряжения двойных связей
изменяется от красно-коричневого до черного. Мол.
масса растворимых П. не превышает нескольких тысяч.
Характеристич. вязкость р-ров П. в конц. H2SO4 до-
достигает 0,2 дл/г. П. растворимы в сильных конц.
минеральных к-тах (H2SO4, HC1, Н3РО4), несколько
хуже — в нек-рых органич. к-тах (в конц. муравьиной,
уксусной, трифторуксусной) и нерастворимы в боль-
большинстве органич. растворителей. Вследствие высокой
жесткости полисопряженных цепей и сильного межмо-
межмолекулярного взаимодействия большинство П. неплавки
и хрупки за исключением П. на основе высших алифа-
алифатич. Н. (напр., пропионитрила, капронитрила); по-
381
НУКЛЕИНОВЫЕ КИСЛОТЫ
382
следние размягчаются при темп-pax не ниже 180 °С.
Для П. характерны парамагнетизм и полупроводнико-
полупроводниковые свойства. Электрич. проводимость П. при ком-
комнатной темп-ре составляет 10~п—10~5 ом~1-см~1ч энер-
энергия активации проводимости — от 0,1 до 0,5 эв.
П. характеризуются высокой химич. и термич. ста-
стабильностью. Напр., полибензонитрил гидролизуется по
связям —С = N— только при темп-pax выше 200 °С и
в присутствии катализаторов [H2SO4C0%) + ZnCl2
или А1С1з]. Продукт гидролиза — бензойная к-та.
Устойчивость к термоокислительной деструкции оп-
определяется природой боковых заместителей (R) и сте-
степенью сопряжения. Темп-pa разложения политрифтор-
ацетонитрила выше 500 °С.
П. получают полимеризацией комплексов Н. с к-тами
Льюиса (напр., с ZnCl2, A1C13, GaCl3, S11CI4) при темп-pax
выше 200 °С. Сокатализаторами служат те же вещества,
что и при обычной катионной полимеризации винило-
виниловых мономеров. В качестве одного из промежуточных
продуктов полимеризации образуются циклич. тримеры.
Полимеры, подобные П., получаются также при по-
поликонденсации амидов или амонийных солей карбоно-
вых к-т с отщеплением воды в присутствии больших
количеств к-т Льюиса, карбидов металлов и др. во-
доотнимающих агентов.
П. впервые синтезированы В. А. Каргиным, В. А. Ка-
Кабановым и В. П. Зубовым в 1964. Промышленного при-
применения пока не нашли.
Лит.: OikawaE., MoniK., SaitoG., Bull. Chem.
Soc. Japan, 39, № 6, 1172 A966); К о м а р о в а О. П. [и др.],
Высокомол. соед., 9А, № 2, 336 A967). В. П.Зубов.
НИТРИЛСИЛОКСАНОВЫЕ КАУЧУКИ — см. Крем-
нийорганические каучуки.
НИТРОЛАКИ — см. Эфироцеллюлозные лаки и эмали.
НИТРОН — см. Полиакрилонитрильные волокна.
НИТРОЦЕЛЛЮЛОЗА — см. Нитраты целлюлозы.
НИТРОЦЕЛЛЮЛОЗНЫЕ ЛАКИ И ЭМАЛИ — см.
Эфироцеллю лозные лаки и эмали.
НОВОЛАЧНЫЕ СМОЛЫ, новолаки (novolaks,
Novolake, novolaques) — термопластичные олигомерные
продукты поликонденсации фенолов с альдегидами (гл.
обр. с формальдегидом). Получают их в кислой среде
в присутствии катализаторов — соляной, щавелевой
к-т, реже H2SO4, при избытке фенола. Н. с.— линей-
линейные олигомеры, в молекулах к-рых фенольные ядра
соединены друг с другом метиленовыми мостиками. В
отличие от резольных смол, в Н. с. практически отсутст-
отсутствуют метилольные группы, вследствие чего для их
отверждения при переработке необходимо вводить от-
вердитель (обычно гексаметилентетрамин).
Н. с. выпускают в виде стеклообразных кусков, че-
чешуек или гранул от светло-желтого до темно-коричне-
темно-коричневого цвета; средняя мол. масса 500—900; темп-pa кап-
лепадения по Уббелоде 90—130 °С; они легко раство-
растворяются в спиртах, сложных эфирах, кетонах и водных
р-рах едких щелочей, обычно содержат 1 — 7% несвя-
несвязанного фенола. Н. с. применяют в производстве пресс-
материалов (см. Фенопласты), абразивных изделий, ли-
литейных форм, лаков и пенопластов. Подробнее см. Фе-
ноло-формалъдегидные смолы. Ю. В. Горохолинский.
НУКЛЕИНОВЫЕ КИСЛОТЫ (nucleic acids, Nuk-
leinsauren, acides nucleiques).
Содержание:
Способы выделения 382
Химическое строение 382
Макромолекулярная структура дезоксирибонукле-
иновой к-ты 385
Матричный синтез 386
Физические свойства 388
Функции дезоксирибонуклеиновой к-ты. Ее хими-
химическая модификация 391
Функция рибонуклеиновой к-ты. Ее роль в синтезе
белка 394
Анализ 395
Синтез 396
Нуклеиновые кислоты — один из двух важнейших
классов биополимеров; входят в состав каждой клетки,
каждого вируса. Название Н. к. происходит от слова
nucleus (ядро).
Н. к. делятся на 2 химически различных типа: де-
зоксирибонуклеиновую кислоту (ДНК) и рибонуклеи-
рибонуклеиновую кислоту (РНК). У высших организмов ДНК со-
сосредоточена гл. обр. в клеточном ядре. У бактерий нет,
строго говоря, отдельного дифференцированного ядра
и ДНК собрана в специальной органелле — хромо-
хромосоме. Роль ДНК в природе — хранение и передача по-
потомству генетич. информации, т. е. программирование
структуры всех синтезируемых клеткой белков. Однако
непосредственно в синтезе белков ДНК не участвует.
Эту работу выполняет сосредоточенная в основном
в цитоплазме РНК, к-рая особым образом копируется
с ДНК (см. ниже). Так. обр., ДНК есть хранилище ге-
генетич. информации в клетке, а РНК — инструмент,
с помощью к-рого информация реализуется. Оба типа
Н. к. обязательно входят в состав всех живых организ-
организмов. Особняком стоят вирусы, к-рые могут содержать
одну РНК. Содержание ДНК в клетках чрезвычайно
постоянно и инвариантно: после деления все клетки
данного вида содержат ее в одинаковом количестве.
Перед новым делением ДНК в точности удваивается.
Бурно растущие, быстро размножающиеся и синте-
синтезирующие много белка клетки (бактерии, дрожжи,
эмбриональные ткани, опухоли, железы) содержат
относительно много РНК (у дрожжей до 10% от су-
сухой массы). В клетках, прекративших рост, ее гораз-
гораздо меньше.
Способы выделения
Источником получения ДНК обычно является ви-
лочковая железа (тимус) телят, т. к. в ее клетках ядра
составляют больше половины объема. Для получения
РНК удобнее всего использовать дрожжи. Н. к. всегда
тесно связаны с белками в нуклеопротеидных структу-
структурах. Для их освобождения проводят денатурацию бел-
белков добавлением к взвеси клеток (предварительно
вскрытых путем разрушения их оболочки ) р-ра фенола
высокой концентрации, а также нек-рых детергентов
(напр., додецилсульфата) или хлороформа. Обычно фе-
нольная депротеинизация ведется так, что фенола да-
дается большой избыток и после отстаивания или центри-
центрифугирования образуются два слоя: внизу — фенол,
насыщенный водой, сверху — вода, насыщенная фено-
фенолом. Денатурированные белки сосредоточиваются
в нижнем слое и у границы раздела, Н. к.— в верхнем
водном слое, откуда их осаждают спиртом. При фе-
нольной экстракции возможно частичное фракциониро-
фракционирование Н. к. на ДНК (растворяется преимущественно
при рН9) и РНК (растворима и при рН5). Однако пол-
полностью отделить ДНК и РНК друг от друга удается
только дополнительным воздействием специфическими
гидролитич. ферментами: ДНК-азой и РНК-азой. Эти
ферменты добываются из поджелудочной железы рога-
рогатого скота, подвергаются тщательной очистке и кри-
кристаллизации. Действуя ДНК-азой, разрушают ДНК
и получают чистую РНК. С помощью РНК-азы очи-
очищают ДНК от РНК. После ферментативной обработки
полученные продукты снова подвергают фенольной
депротеинизации с последующим осаждением Н. к.
спиртом.
Химическое строение
Цепи главных валентностей Н. к. образованы попе-
попеременно молекулами Сахаров (пентоз) и молекулами
фосфорной к-ты, соединяющими 3'- и б'-атомы сосед-
соседних пентоз фосфодиэфирными связями. Разница между
РНК и ДНК (рис. 1) состоит в том, что в последней
отсутствуют 2'-гидроксилы в пентозном цикле (дезок-
сирибоза вместо рибозы). Это отличие делает ДНК хи-
383
НУКЛЕИНОВЫЕ КИСЛОТЫ
384
мически более инертной, сообщает ей устойчивость
к щелочному гидролизу; концевой пентозный цикл ДНК
оказывается устойчивым к окислению НЮ4 (требую-
(требующему двух гидроксилов в ^-положении). В цепи глав-
главных валентностей к Г-атому пентозного цикла присо-
РНК
лу концевого звена (этот гидроксил должен быть сво-
свободен от фосфатной группы). Следовательно, рост
цепей Н. к. происходит всегда в направлении от 5'-
атома С к З'-атому. Такова специфичность биокатали-
биокатализаторов-ферментов.
Гуанин
Урацид
3—конец
Рис. 1. Первичные структуры нуклеиновых кислот.
3-конец
единены боковые группы — 2 различных пуриновых
основания (аденин и гуанин) и 2 пиримидиновых (ци-
(цитозин и тимин — в ДНК, цитозин и урацил — в РНК;
в этом еще одно различие между 2 типами Н. к.). Н. к.
способны диссоциировать на протоны (в каждой фос-
фосфатной группе) и полианионы. Они довольно сильные
к-ты, показатель диссоциации этих дважды этерифици-
рованных фосфатных групп рК = 1,5.
Т. обр., Н. к.— это сополимеры 4 типов мономер-
мономерных звеньев, называемых нуклеотидами. На
рис. 1 изображены все 4 разных нуклеотида, следую-
следующие друг за другом. На самом деле в цепи Н. к. различ-
различные нуклеотидные звенья могут чередоваться любым,
самым замысловатым образом. Существенно другое —
порядок чередования 4 типов звеньев в этом сополимере
детерминирован с абсолютной точностью и не допускает
никаких флуктуации. Если в половой клетке стойко
изменяется даже одно звено в цепи ДНК из десяти
миллиардов звеньев, то это уже наследственная м у-
т а ц и я, т. е. передаваемое по наследству изменение,
к-рое чаще всего гибельно для организма.
Нуклеотид состоит из ковалентно связанных моле-
молекул пентозы, фосфорной к-ты и азотистого основания.
Если от такого звена отщепить фосфорную к-ту, полу-
получим нуклеозид (их названия: аденозин, гуанозин,
тимидин, цитидин и уридин). Цепь Н. к. несимметрична
вследствие того, что фосфорная к-та, соединяющая по-
попарно все нуклеозиды, присоединена к пентозе в двух
структурно различных положениях: к З'-атому С и
к 5'-атому С. Поэтому полинуклеотидная цепь векто-
риальна, ей можно условно приписать направление.
Все известные сейчас синтетич. ферменты, ведущие
реакции наращивания полинуклеотидных цепей, при-
присоединяют очередное звено всегда только к З'-гидрокси-
Наряду с указанными главными пуриновыми и пири-
мидиновыми основаниями в состав ДНК входят в не-
небольшом количестве т. наз. минорные осно-
основания: 5-метилцитозин, б-И-метиладенин, 1-метил-
гуанин, №-диметилгуанин. В разных типах РНК (тран-
(транспортной и рибосомной) суммарное количество минор-
минорных оснований достигает 10% и они весьма разнообраз-
разнообразны. По большей части это продукты метилирования
главных оснований (напр., 1-метилгуанин, №-диметил-
гуанин) или их гидрирования E,6-дигидроурацил); про-
продукт дезаминирования аденина — гипоксантин и его
метилированная форма — 1-метилгипоксантин. Нако-
Наконец, в небольших количествах имеются азотистые осно-
основания, сильно отличающиеся от главных по своему
строению, напр, псевдоура-
цил (I) и пиримидины, со-
держащие тиогруппу (II).
Роль минорных основа-
оснований окончательно не выяс-
выяснена. Возможно, что свое-
своеобразие в расположении
минорных оснований важно
для того, чтобы деструктив-
деструктивные клеточные ферменты могли отличать свои Н. к.
от чужеродных, напр, бактериальных или вирусных.
Анализ Н. к. начинают с того, что расщепляют по-
полимерную цепь в основном до нуклеозидов (с помощью
70% -ной HCIO4 в течение 1 ч при нагревании до 100 °С)
и образовавшуюся смесь четырех типов нуклеозидов
разделяют хроматографией (на бумаге или на ионооб-
меннике) или электрофорезом.
В ДНК (но не в РНК) содержание различных зве-
звеньев подчиняется правилам Чаргафа:
АТГЦ
О
X
385
НУКЛЕИНОВЫЕ КИСЛОТЫ
386
где А, Т, Г, Ц — количества соответственно аденина,
тимина, гуанина и цитозина. Эти правила были прове-
проверены на громадном числе объектов и оказывались точ-
точными за очень редкими исключениями (напр., для ДНК
бактериофага ФХ174). Из правил Чаргафа, в совокуп-
совокупности с рентгеноструктурными данными, можно сделать
очень важные заключения о макромолекулярной струк-
туре ДНК.
Соотношение между А и Г зависит от конкретного
объекта. Т. наз. фактор специфичности
ДНК (Г/А) меняется у разных видов микроорганизмов
в пределах 0,45—2,8 (Белозерский и Спирин). У выс-
высших растений и животных это соотношение составляет
0,55-0,93.
Макромолекулярная структура дезоксирибонуклеиновой
к-ты
В выяснении строения макромолекулы ДНК решаю-
решающую роль сыграл рентгеноструктурный анализ воло-
волокон, вытянутых из литиевой соли ДНК (Уилкинс).
Рентгенограммы этих волокон бедны (всего ок. 100 реф-
рефлексов), т. к. кристаллическая структура несовер-
несовершенна. Однако основные топологические принци-
принципы упаковки получаются довольно непосредственно из
анализа рентгеноструктурной картины: 1) полимер-
полимерная цепь ДНК — двухзаходная спираль, 2) боковые
группы, т. е. азотистые основания, заполняют всю
внутреннюю часть спирали, укладываясь в ней подоб-
подобно столбику монет.
Выше уже рассмотрена т. наз. первичная структура
Н. к., т. е. ковалентные связи между атомами и звенья-
звеньями полимерной цепи (см. рис. 1). Но наряду с первич-
первичной у ДНК существует вторичная структура, т. е. ре-
регулярная пространственная конформация полимерной
цепи, определяемая молекулярными силами между ко-
валентно насыщенными атомами и группами. Существо-
Существование двухспиральной вторичной структуры ДНК
(рис. 2) было доказано Уотсоном и Криком в 1953. При-
Причины образования такой своеобразной структуры за-
заключаются в следующем.
О Н
00
) С в углевод-
углеводно-фосфатной
цепи
) С и N в основа-
основаниях
Ч3,4А
Рис. 2. Схема вторичной структуры дезоксирибонуклеино-
дезоксирибонуклеиновой кислоты (модель Уотсона и Крика).
Боковые группы ДНК, а именно пиримидиновые и
пуриновые основания, стремятся создать параллель-
параллельные слои внутри спирали, т.к. таким путем лучше всего
насыщаются их вандерваальсовы взаимодействия. Кро-
Кроме того, из соображений равенства поперечных сечений
спирали явствует, что рядом с пиримидиновым основа-
основанием всегда должно быть пуриновое и наоборот. Между
этими основаниями могут возникать водородные свя-
связи. Детальное рассмотрение всех возможностей приво-
приводит к однозначному решению, изображенному на рис. 3.
Аденин способен образовать 2 водородные связи только
с тимином, а гуанин — 3 водородные связи с цитозином.
Поскольку реализуется структура с максимально воз-
возможным числом водородных связей, то против каждого
аденина, принадлежащего одной из нитей двухзаход-
К цеп
К цепи
К цепи
Рис. 3. Комплементарность ос-
оснований во вторичной струк-
структуре дезоксирибонуклеиновой
к-ты.
ной спирали, лежит тимин во второй нити и против
каждого гуанина находится цитозин. Подобная кар-
картина спаривания оснований А — Т и Г — Ц носит на-
название принципа комплементарности
Уотсона — Крика.
Т. о., обе спирали в двухниточной структуре ДНК
зависимы друг от друга. Структура одной из них пол-
полностью детерминирует структуру второй.
Матричный синтез
Ни один из известных химич. приемов получения ре-
регулярных сополимеров не позволяет синтезировать
длинную полимерную цепь с полным повторением всего
чередования различных мономеров при соблюдении ма-
тематич. точности и безошибочности. Клетка же при
делении передает обеим дочерним клеткам весь свой
комплект генетич. информации, т. е. полные цепи ДНК,
идентичные той цепи, к-рая содержится в материнской
клетке. Единственное возможное объяснение этому
явлению можно дать на основе идеи о матричном синте-
синтезе, т. е. о принудительном наборе цепи ДНК из 4 ти-
типов мономеров под влиянием шаблона, или матрицы,
к-рая навязывает порядок чередования звеньев в со-
сополимере.
Идея о матричном синтезе высказывалась давно как
некая умозрительная абстракция. Принцип компле-
комплементарности придает ей вполне ясный физич. смысл-
При синтезе новых цепей ДНК двойная спираль нару-
387
НУКЛЕИНОВЫЕ КИСЛОТЫ
388
шается на нек-ром участке. Взамен разорванных водо-
водородных связей возникают новые благодаря сорбции
мономеров на освободившихся местах. Мономеры бу-
будут сорбироваться из р-ра с соблюдением принципа
комплементарности. Затем происходит поликонденса-
поликонденсация (мономерами являются нуклеозидтрифосфаты и
при каждом акте реакции отщепляется пирофосфат)
и вместо одной двойной спирали ДНК образуются
2 идентичные двойные спирали. Каждая дочерняя спи-
спираль имеет в своем составе одну материнскую непо-
неповрежденную нить, послужившую матрицей, и одну вновь
синтезированную. Но обе дочерние спирали струк-
структурно неотличимы друг от друга. Этот процесс наз.
полуконсервативной редуплика-
редупликацией (или удвоением). Термин «полуконсерватив-
«полуконсервативная» указывает на то, что макромолекула материнской
ДНК разделилась пополам между потомством.
Синтез ДНК — ферментативный процесс. Это озна-
означает, что существует специальный фермент (белок
ДНК-полимераза), к-рый образует комплекс с концом
растущей цепи и с молекулой мономера и ускоряет
поликонденсацию. Реакция не идет без фермента и без
ДНК-матрицы. Фермент не селективен по отношению
к 4 типам мономеров. Он может образовать комплекс
с любым из них. Отбор правильного мономера происхо-
происходит автоматически благодаря принципу комплементар-
комплементарности.
В самой структуре двойной спирали заложен прин-
принцип матричного синтеза и редупликации ДНК. Физич.
основой правильного функционирования матрицы при
синтезе ДНК являются молекулярные силы — взаимо-
взаимодействие А с Т и Г с Ц посредством водородных связей.
Принцип полуконсервативной редупликации ДНК под-
подвергся весьма детальной и полной проверке и сейчас
является одним из самых значительных завоеваний
биологич. науки.
Второй не менее важный процесс — синтез РНК по
ДНК — наз. транскрипцией. Этот процесс осу-
осуществляется с помощью фермента РНК-полимеразы.
Образование водородных связей между нитью ДНК
и нитью РНК происходит так же, как между двумя
комплементарными нитями ДНК. Тот факт, что одно
из пиримидиновых оснований, тимин, заменено на
урацил, никак не сказывается на способе спаривания,
т. к. метильная группа, отличающая тимин от урацила,
находится в положении 5 и не участвует в образовании
водородных связей.
Транскрипция в принципе протекает почти так же,
как редупликация. Главное отличие ее в том, что транс-
транскрибируется в каждой области молекулы ДНК лишь
одна из двух нитей, вторая остается инертной. При этом
транскрипция разных областей ДНК может происхо-
происходить с разных нитей. Но в каждом месте матрицей слу-
служит лишь одна из двух комплементарных нитей ДНК.
ДНК содержит особые области, т. наз. промоторы,
к к-рым прикрепляется фермент РНК-полимераза —
специфич. катализатор поликонденсации рибонуклео-
зидтрифосфатов. Этот фермент получен в чистом виде
и изучен в модельных внеклеточных условиях. От об-
областей, специфически узнаваемых ферментом, начи-
начинается транскрипция, причем фермент перемещается
вдоль вновь синтезируемой цепи РНК в направлении
от 5'- к З'-атому рибозы.
Выше уже отмечалось, что каждая цепь ДНК имеет
направление, т. е. представляет собой вектор (или
асимметрична в пространстве). Более того, обе цепи
ДНК в двойной спирали антипараллельны, т. е. на-
направлены в противоположные стороны. Транскрип-
Транскрипция происходит в одних местах с одной, в других — со
второй цепи ДНК. При этом молекулы фермента дви-
движутся то в одном, то в обратном направлениях, как это
указывает положение соответствующего промотора.
В силу однониточности РНК, она не может образовать
Рис. 4. Вторичная структура
рибонуклеиновой кислоты.
длинные спирали Уотсона — Крика. В цепи РНК обра-
образуются, благодаря наличию случайных комплементар-
комплементарных отрезков цепи, внутренние короткие спиральные
участки, перемежающиеся с бесструктурными обла-
областями типа статистических клубков (рис. 4). Отметим,
что у небольших бак-
бактериофагов типа ФХ174
ДНК также однониточ-
на. При этом ее втори-
вторичная структура такая
же, как у нитей РНК.
Кроме случайных ко-
коротких автокомплемен-
автокомплементарных участков, у ни-
нитей РНК имеются и бо-
более длинные законо-
закономерные области комплементарности, к-рые заставляют
соответствующие участки цепей РНК образовывать
спиральные складки. Функциональное значение этих
складок еще не ясно. Возможно, что в этих участках
происходит «узнавание» цепей РНК специфич. белками.
Матричный синтез в живой природе не ограничивает-
ограничивается случаями редупликации ДНК и транскрипции РНК.
Синтез белка происходит также путем набора амино-
аминокислотной последовательности на матрице. В этом слу-
случае матрицей служит РНК, и физич. механизм, осущест-
осуществляющий работу матрицы, сводится все к тому же
принципу комплементарности. О матричном синтезе
в небиологич. системах см. Матричная полиреакция.
Физические свойства
Макромолекулы ДНК ведут себя как деформируемые
жесткие стержни. Полужесткие цепи изгибаются при
тепловом движении плавно, как стальная проволока.
Однако при большой длине они многократно меняют
свое направление и образуют в конечном счете клубки.
Размеры этих клубков таковы, как если бы полимер
представлял собой свободно-сочлененную цепь, состоя-
состоящую не из мономерных, а из гораздо более длинных
звеньев — т. наз. статистич. сегментов (см. Гибкость
макромолекулы). У ДНК статистич. сегмент имеет дли-
длину порядка 900—1000 А, т. е. содержит ок. 300 моно-
мономерных звеньев. Следовательно, ДНК — жесткая пру-
пружина, к-рая на больших протяжениях все же обнаружи-
обнаруживает известную гибкость.
Цепи ДНК очень велики. Установлено, что каждая
хромосома содержит одну двухнитевую цепь ДНК.
Следовательно, у бактерий длина цепи составляет
10 000—20 000 статистич. звеньев, а у высших орга-
организмов в десятки раз больше. Однако при извлечении
ДНК из клеток она чаще всего подвергается фрагмен-
фрагментации до кусков порядка 100—300 статистич. звеньев.
Все эти цепи ДНК достаточно велики, чтобы вести
себя как гауссовы клубки.
У РНК статистич. сегмент гораздо короче (ок. 100 А),
что вполне согласуется с рассмотренной выше вторич-
вторичной структурой РНК. Для ДНК была установлена
связь между гидродинамич. свойствами, характери-
стич. вязкостью, константой седиментации и мол. мас-
массой. В большинстве препаратов ДНК, получаемых из
клеток бактерий или высших организмов, мол. масса
достигает 107—3-Ю7. По-видимому, в процессе осво-
освобождения ДНК от белка длинные цепи рвутся случай-
случайным образом на подобные фрагменты. Разрывы вызы-
вызываются гидродинамич. сдвиговыми силами или гради-
градиентом скорости.
С помощью гидродинамич. методов (включая обра-
обработку ультразвуком) удается разбить макромолекулы
ДНК на фрагменты все более низкой мол. массы. По-
Построены диаграммы констант седиментации и характе-
ристич. вязкостей, пригодные для измерения мол.
масс ДНК, не превосходящих 107. У ДНК вирусов,
в частности бактериофагов, мол. массы гораздо более
389
НУКЛЕИНОВЫЕ КИСЛОТЫ
390
высокие. Соблюдая необходимые предосторожности,
удается получить фаговую ДНК неповрежденной и мо-
монодисперсной, в отличие от ДНК бактерий и высших
организмов, к-рая фрагментируется и образует стати-
стич. набор цепей различной длины. Для вирусных
ДНК вполне обычны мол. массы от 3-Ю7 до 13-Ю7.
Для определения таких мол. масс по константам седи-
седиментации S пользуются ф-лой Бурги и Херши, к-рая
была выведена на основании измерений, произведенных
для ДНК ряда бактериофагов:
S = 2,4-Ю-3 Мо,54з
Калибровка мол. массы ДНК фагов производилась
с помощью аналитич. определения количества ДНК
в расчете на один вирион. Принято считать, что вся
ДНК бактериальной хромосомы состоит из одной длин-
длинной непрерываемой двойной цепи (ее масса в случае
Escherichia coli составляет 2,9-109).
Выделить ДНК очень высокой мол. массы без раз-
разрывов молекулы весьма затруднительно. Из бактерий
удалось получить фрагменты ДНК мол. массы 109,
что составляет значительную часть от хромосомы.
РНК отличается гораздо более низкой мол. массой,
поскольку транскрипция с ДНК идет по относительно
небольшим областям. Большую часть клеточной РНК
(по массе) составляет РНК рибосом — специальных
телец, в к-рых синтезируется белок. В рибосомы вхо-
входят три типа РНК. В бактериях рибосомная
РНК (рРНК) имеет константы седиментации 5, 16
и 23 единицы Сведберга, что соответствует мол. массам
35 000, 550 000, 1 100 000. В высших организмах соот-
соответственно рибосомы содержат РНК с мол. массами
40 000, 700 000, 1 700 000.
Мол. масса РНК (среднемассовая) вычисляется из
константы седиментации по эмпирич. ф-ле (при ионной
силе 0,15):
0,56
5 = 0,98 10~2Mw
Кроме рибосомной РНК, в клетке всегда имеются
транспортные РНК (тРНК) — семейство близ-
близких по структуре полимеров сравнительно низкой мол.
массы B5 000—30 000, что соответствует степеням
полимеризации 75 — 90). тРНК, число к-рых в клетке
порядка 60, составляют приблизительно Vio от массы
всей клеточной РНК.
Матрицами для синтеза белков служат молекулы
матричной РНК (мРНК), к-рая транскрибиру-
транскрибируется с многочисленных областей хромосомы. Мол.
массы мРНК достигают 3-Ю6— 5Ю6. Содержание
мРНК в бактериальной клетке составляет всего ок. 3%
от всей РНК. мРНК в бактериальной клетке быстро
расщепляется под действием ферментов, но очень быст-
быстро синтезируется. Наблюдаемая в среднем концентра-
концентрация мРНК является результатом установившегося ста-
стационарного процесса распада и синтеза. Остальные
типы РНК, как и ДНК, являются вполне стабильными
и заметно не разрушаются в течение многих клеточных
поколений.
Пуриновые и пиримидиновые основания имеют поло-
полосы поглощения в близком ультрафиолете. В результа-
результате суммирования парциальных поглощений отдельных
нуклеотидов Н. к. обнаруживают полосу поглощения
с максимумом вблизи 2600 А.
Важным свойством Н. к. является гипохром-
ный эффект — уменьшение уд. экстинкции в по-
полосе поглощения, когда ДНК находится в регулярной
спиральной конфигурации, по сравнению с экстинк-
цией в состоянии «аморфного» клубка. По значению
гипохромного эффекта (он может достигать 40—45%)
можно приближенно оценивать степень регулярности
или степень спиральности ДНК. Следует только учи-
учитывать, что гипохромный эффект обусловлен паралле-
параллельным расположением плоскостей молекул гетероцик-
лич. оснований. Поэтому он не полностью исчезает
при нарушении регулярной структуры ДНК, т. к.
корреляция между ближайшими звеньями цепи части-
частично сохраняется.
Другой метод оценки степени спиральности основан
на измерении оптич. активности. Н. к. образуют спира-
спирали только правой конфигурации и вращают плоскость
поляризации света вправо. Оптич. активность ДНК
лишь в малой степени объясняется оптич. активностью
асимметрич. атомов углерода, входящих в сахара.
Главный вклад дает асимметрич. структура спираль-
спиральных цепей как целое. Поэтому при переходе спираль —
клубок происходит резкое падение оптич. активности
ДНК (при исследовании в видимой области спектра,
т. е. вдали от полосы поглощения ДНК).
Образование двойной спирали напоминает кристал-
кристаллизацию, т. к. при этом возникает система с ближним,
и дальним порядком. Но кристаллизация эта своеоб-
своеобразна, т. к. в ней участвуют две объединившиеся поли-
полимерные цепи. При нагревании ДНК в р-ре происходит
плавление упорядоченной спиральной структуры. Плав-
Плавление ДНК (переход спираль — клубок) — это коопе-
кооперативный переход, напоминающий фазовые переходы
в трехмерных слстемах (см. Макромолекула). Однако
переход происходит хотя и резко, но во вполне измери-
измеримом темп-рном интервале (в отличие от плавления обы-
обычных кристаллов). У ДНК бактериофагов, к-рая впол-
вполне гомогенна по своему составу, ширина интервала
плавления ДГ (вычисленная путем проведения касате-
касательной в центре кривой перехода) составляет ок. 3 °С.
Цепи ДНК бактерий гетерогенны по составу (т. е.
по показателю Г/А). Такую гетерогенность полимерных
цепей можно назвать блочной, понимая под блоками
участки с различным средним составом. В этом случае
ДГ достигает 5 °С. У высших организмов степень
блочной гетерогенности значительно больше, чем у бак-
бактерий, вследствие большей дисперсии по составу. Соот-
Соответственно и ДГ повышается до 10 °С. Под точкой плав-
плавления ДНК, обозначаемой Гт, понимают середину ин-
интервала перехода. Эта величина есть функция фактора
специфичности ДНК, т. е. отношения Г/А. Между
парами Г — Ц образуются 3 водородные связи, а меж-
между А — Т и А — У — всего 2. Поэтому Тт линейно
растет с ростом содержания пар Г — Ц.
Следить за плавлением ДНК можно по исчезновению
гипохромного эффекта или вращения плоскости поля-
поляризации, а также по падению характеристич. вязкости
р-ра. Цепи ДНК сильно повышают вязкость р-ра вслед-
вследствие своей вытянутой формы. Переход спираль —
клубок вызывает резкое уменьшение вязкости. ДНК
в состоянии клубков наз. денатурирован-
денатурированной. Как правило, она теряет при денатурации свою
функциональную биологич. активность.
В баланс молекулярных сил, поддерживающих двой-
двойную спираль, входят водородные связи между азоти-
азотистыми основаниями в каждой паре, электростатич. силы
взаимодействия ионизируемых групп и вандерваальсо-
вы (дисперсионные) силы между параллельно располо-
расположенными пуриновыми и пиримидиновыми циклами.
Влияя на те или иные силы, ослабляя или усиливая их,
можно сдвигать точку плавления цепей ДНК. В част-
частном случае можно заставить ДНК плавиться при ком-
комнатной темп-ре.
ДНК превращается в клубок при смещении рН как
в кислую, так и в щелочную области. В первом случае
это объясняется ионизацией NH2-rpynn гуанина, цито-
зина и аденина и возникающими дополнительными си-
силами кулоновского отталкивания. Во втором случае
при рН12 происходит ионизация енольных групп, обра-
образующихся из кетогрупп гуанина и тимина, вследствие
чего разрываются водородные связи.
Влияние растворителя на водородные связи между
основаниями также весьма ясное. Так, замена воды на
391
НУКЛЕИНОВЫЕ КИСЛОТЫ
392
этиленгликоль вызывает укрепление водородных свя-
связей и повышает Тт на несколько десятков °С (она
становится выше 100 °С). Замена воды на формамид
вызывает ослабление водородных связей. В 80%-ном
формамиде ДНК хорошо растворима, но переходит
в клубок уже при комнатной темп-ре. То же наблюда-
наблюдается и при добавлении мочевины к воде. Денатуриро-
Денатурированная ДНК способна к обратному переходу клубок —
спираль, т. е. к ренатурации при медленном изменении
состояния (медленном охлаждении р-ра, называемом
«отжигом», или медленном удалении мочевины, фор-
мамида или к-ты). При этом ренатурация вирусной
ДНК происходит быстро (время порядка минуты) и
полно, т. к. в среднем одно столкновение макромоле-
макромолекул из двух дает нужную комбинацию комплементар-
комплементарных цепей.
Бактериальная ДНК ренатурируется гораздо мед-
медленнее (время порядка часа) и значительно менее полно,
т. к. в смеси из 300 сортов фрагментов лишь одно столк-
столкновение из 3002, т. е. из 105, эффективно. В ДНК выс-
высших организмов имеется некоторая часть (достига-
(достигающая 30—40%), способная ренатурироваться довольно
быстро (за время того же порядка, что и бактериа-
бактериальная ДНК). Эта часть ДНК содержит повторяющи-
повторяющиеся или многократно дублированные последовательно-
последовательности нуклеотидов. Наличие «повторов» в цепи ДНК
высших организмов, а возможно и бактерий, имеет,
по-видимому, глубокое функциональное значение и
связано с явлениями регулирования процесса транс-
транскрипции.
Функции дезоксирибонуклеиновой к-ты.
Ее химическая модификация
Как уже отмечалось выше, в ДНК в форме линейной
последовательности нуклеотидов зашифрована струк-
структура всех клеточных белков. Т. к. белки составлены
из 20 типов аминокислот, а нуклеиновые к-ты из 4 ти-
типов нуклеотидов, должен существовать специальный
генетич. код, связывающий обе последовательности
звеньев. Этот код триплетный: тройка соседних нуклео-
нуклеотидов образует к о д о н, соответствующий одному ами-
аминокислотному остатку белковой цепи. Число возмож-
возможных кодонов составляет 43 = 64, т. е. оно избыточно
для кодирования 20 аминокислот. Поэтому генетич.
код является «вырожденным»: в большинстве случаев
несколько кодонов соответствуют одной аминокислоте.
Генетич. код во всей живой природе универсален, но
картина его вырожденности носит на себе отпечаток
эволюционной истории.
Код для нек-рых простейших аминокислот вырож-
вырожден многократно (например, для лейцина — шестикрат-
шестикратно) . Самый распространенный тип вырожденности кодо-
кодонов — различие в третьем нуклеотиде при фиксиро-
фиксированных первых двух. Это дает чаще всего встреча-
встречающееся 4-кратное вырождение. Одна из самых слож-
сложных аминокислот, триптофан, представлена всего од-
одним кодоном.
Генетич. код представлен в таблице. Всякое химич.
изменение ДНК, затрагивающее хотя бы одно из азо-
азотистых оснований, может привести к мутации, т. к.
изменяется соответствующий кодон, а следовательно,
в одном из белков происходит замена аминокислотного
звена, изображаемого измененным кодоном. Получает-
Получается белок с «ошибкой» в одном звене цепи и это измене-
изменение наследуется, т. к. при редупликации ДНК изме-
измененный кодон повторяется в потомстве. Мутации м. б.
спонтанными (самопроизвольными) и индуцирован-
индуцированными.
Из уровня спонтанных мутаций у бактерий в расчете
на одно поколение рассчитано, что вероятность одной
репликационной ошибки при синтезе ДНК составляет
порядка 10~9. Эту величину можно рассматривать как
отношение скоростей реакций правильной репликации
Генетический код
Первый
нуклео-
тид
У
Ц
А
г
Второй нуклеотид
У
\ Фен
1 Фен
\ Лей
J Лей
) Лей
1 Лей
( Лей
J Лей
\ Изо
1 Изо
( Изо
J Мет*
) Вал
1 Вал
[ Вал
J Вал
Ц
Сер
Сер
Сер
Сер
Про
Про
Про
Про
Тре
Тре
Тре
Тре
Ала
Ала
Ала
Ала
А
Тир
Тир
«Охра»
«Янтарь»
Гис
Гис
Глн
Глн
Асн
Асн
Лиз
Лиз
Асп
Асп
Глу
Глу
Г
Цис
Цис
«Опал»
Три
Apr
Apr
Apr
Apr
Сер
Сер
Apr
Apr
Гли
Гли
Гли
Гли
Трет ий
нуклео-
нуклеотид
У
Ц
А
Г
У
ц
А
Г
У
ц
А
Г
У
ц
А
Г
* У 5'-конца цепи кодон служит инициатором роста бел-
белковой цепи и кодирует N-формилметионин у бактерий или ме-
тионин у высших организмов.
Примечание. Ала—аланин, Apr—аргинин, Асн—аспара-
гин, Асп — аспарагиновая к-та, Вал—валин, Гис — гистидин,
Гли—глицин, Глн—глутамин, Глу—глутаминовая к-та, Изо—
изолейцин, Лей—лейцин, Лиз—лизин, Мет—метионин, Про—
пролин, Сер—серии, Тир—тирозин, Тре—треонин, Три—трипто-
Три—триптофан, Фен—фенилаланин, Цис—цистеин.
ДНК (с соблюдением принципа комплементарности)
и ошибочной репликации.
Реальный «уровень шумов», создающий спонтанные
мутации, согласуется с тем энергетич. барьером, к-рый
защищает правильную репликацию матрицы от оши-
ошибочной. Спонтанные мутации — главный фактор измен-
изменчивости живых организмов.
Яркие примеры мутированных белков человека —
разнообразные гемоглобины, встречающиеся при забо-
заболеваниях крови. Изменения одного аминокислотного
звена гемоглобина из 287 оказывается достаточно,
чтобы вызвать тяжелейшие недуги вследствие наруше-
нарушения функции этого белка. Химич. анализ белковой
цепи позволяет изучить материальную природу каж-
каждой конкретной мутации, т. е. понять, какая замена
аминокислоты и в каком месте белковой молекулы
вызвала нарушение функции. Зная генетич. код, мож-
можно легко выяснить, какое изменение претерпел соответ-
соответствующий кодон. Чаще всего модифицируется лишь
одно нуклеотидное звено ДНК.
Большая часть мутаций нарушает функцию белков
и приносит вред организму. Постепенно такие мутации
исчезают в результате естественного отбора. Лишь
в редких случаях мутации могут несколько усовершен-
усовершенствовать функциональную активность. Такие полезные
для организма мутации закрепляются естественным
отбором.
Три кодона в таблице, помеченные символами «ян-
«янтарь», «охра» и «опал», являются «бессмысленными»,
т. е. им не соответствует какая-либо аминокислота.
Когда значащий кодон превращается в «бессмыслен-
«бессмысленный», синтез белковой цепи обрывается в соответст-
соответствующей точке. По всем данным, «бессмысленные» ко-
доны выполняют свою необходимую функцию — они
являются сигналами к окончанию синтеза белковой
цепи.
Индуцированные мутации вызываются мутагенными
веществами или излучениями. Особенно ясные резуль-
результаты получаются при изучении мутагенеза на самых
простых объектах — бактериофагах и вирусах. Проще
всего оказалось действие азотистой к-ты на Н. к.,
которое сводится к обычному дезаминированию осно-
оснований:
RNH2+HNO2->ROH+H2O+N2
393
НУКЛЕИНОВЫЕ КИСЛОТЫ
394
В итоге аденин превращается в гипоксантин:
^*NV .NHo xr О
HNO, \ ^С>~*С\
.N
NH
к-рыи спаривается водородными связями с цитозином,
а не с тимином. Цитозин после дезаминирования пре-
превращается в урацил
NH2
С
О
СН—С
)
НС
HNO-?
СН—С
НС
С
N
NH
)
N—С
к-рый спаривается с аденином вместо гуанина. Поэтому
под действием HNO2 возникают мутации типа замен пар
А — Т на Г — Ц или наоборотДНа нек-рых очень яс-
ясных примерах, напр, мутагенном действии HNO2 на
вирус табачной мозаики, эту схему удалось хорошо
подтвердить путем анализа химич. изменений, произо-
произошедших в мутированных белках.
Модифицирующее действие гидроксиламина сосредо-
сосредоточено на цитозине:
N
С
NH2 NH2
СН NH2OH N СН2
СН " I '
NHOH
С
NH2OH N СН2
СН—NHOH
И
i
с
\
СН2
CH-NHOH
С СН—NHOH
О N
NOH
с
hi/ Чсн2
С CH-NHOH
О N
I
В результате появления группы NH в положении
3 кольца пиримидин начинает спариваться водород-
J) Н—О
СН
4N-C
С—С
X
С—С
N-H
C
C-N
4N-H 0х
Н
Гуанин
Тимин (енольная форма)
'с—с
/
Н Н
AN H-N
NH ]
с
.с—с
СН
с—^
Аденин (имино-форма)
Цитозин
Рис. 5. «Неправильное» спаривание оснований, служащее
причиной спонтанных мутаций.
ной связью с аденином вместо гуанина. Следовательно,
этот мутаген стремится создавать замены только в сто-
сторону Г — Ц -* А — Т.
В самой структуре пуринов и пиримидинов содержат-
содержатся возможности для неправильных спариваний вследст-
вследствие таутомерных превращений, кето-енольных и амино-
иминных переходов. На рис. 5 изображены «неправиль-
«неправильные» пары, способные образоваться вследствие тауто-
таутомерии. Правда, статистич. вес таутомерных форм очень
низок, но и мутации образуются очень редко, если на
ДНК не воздействуют мутагенными агентами. Хоро-
Хорошим подтверждением роли таутомерии оснований в му-
мутагенезе служит след. факт. Если бактерии подпиты-
подпитывать 5-бромурацилом, то этот пиримидин частично
включается в ДНК наместо тимина. 5-Бромурацил ока-
оказывает при этом сильное мутагенное действие на клетки:
вследствие электроотрицательности брома происходит
сильное смещение равновесия в пользу таутомерной
(енольной) формы, и это основание начинает гораздо
чаще образовывать «ошибочную» пару с гуанином, чем
это делал тимин. Суммарное число мутаций у бактерий
под действием этого агента может достигать несколь-
нескольких процентов на поколение.
Функции рибонуклеиновой к-ты.
Ее роль в синтезе белка
Как уже отмечалось, функцией РНК является реа-
реализация матричного синтеза белка. Выше мы рассмот-
рассмотрели принципы кодирования белковой цепи. Разберем
теперь механизм реализации этого принципа. Ин-
Информация о. структуре белка содержится в матричной
РНК, к-рая является копией одной из цепей ДНК (с за-
заменой дезоксирибозы на рибозу и тимина на урацил,
что не отражается на спаривании оснований). Матрич-
Матричную РНК можно себе представить разбитой на трипле-
триплеты (кодоны). Нужная последовательность аминокислот
на матрице набирается с помощью транспортной рибо-
рибонуклеиновой к-ты.
Каждая аминокислота присоединяется в клетке
к своей тРНК эфирной связью к концевому З'-гидрок-
силу рибозы. Образовавшиеся соединения носят на-
название аминоацил-тРНК. Молекулы тРНК имеют
вторичную и третичную структуру. Они свертываются
так, чтобы образовать возможно большее число водо-
водородных связей по принципу Уотсона — Крика. Выра-
Выращены монокристаллы отдельных тРНК, и одна из них
изучена рентгеноструктурным анализом. тРНК обра-
образует две неполностью спирализованные цилиндриче-
цилиндрические палочки, сложенные концами под прямым углом.
В «аморфной части» центрального участка цепи тРНК
находится триплет оснований, комплементарный кодо-
ну (а н т и к о д о н), к-рый и служит для набора ами-
аминокислот на матрице.
На рис. 6 изображена работа «машины», синтези-
синтезирующей белок. В рибосоме, состоящей из двух слип-
слипшихся субъединиц разного размера, имеются два участ-
участка для сорбции тРНК. В одном из них располагается
тРНК, связанная со строящейся, еще незавершенной
белковой (пептидной) цепью. Последняя прикреплена
к З'-углероду концевой рибозы сложноэфирной свя-
связью. Это соединение наз. пептидил-тРНК. Сама тРНК
закреплена своим антикодоном на соответствующем
кодоне матричной РНК. Во втором участке сорбируется
аминоацил-тРНК с антикодоном, комплементарным
к соседнему кодону матрицы. Далее происходит удли-
удлинение пептидной цепи на одно звено. Свободная ами-
аминогруппа аминоацил-тРНК осуществляет нуклеофиль-
ную атаку эфирной связи между пептидной цепью и
тРНК в пептидил-тРНК. При этом образуется новая
пептидная связь с вновь добавленной аминоацил-тРНК,
к-рая превращается в пептидил-тРНК, удлиненную на
одно звено.
395
НУКЛЕИНОВЫЕ КИСЛОТЫ
396
Т. о., пептидная цепь передается от одной молекулы
тРНК, т. наз. донорной, ко второй, акцепторной. Осво-
Освободившаяся тРНК переходит в р-р. Далее удлиненная
на одно звено пептидил-тРНК перемещается в тот спе-
Рис. 6. Циклически работающая «машина», ведущая синтез полипептидной цепи на рибосоме
цифич. участок рибосомы, где она может вновь высту-
выступить в роли донора пептидной цепи. Кроме того, мРНК
продвигается относительно рибосомы точно на один
триплет, и тогда машина готова к приему след. амино-
ацил-тРНК. Каждый шаг этого процесса, включая и
перемещение (транслокацию), доказан много-
многочисленными экспериментами.
Т. о., разные типы РНК являются орудиями, осу-
осуществляющими матричный набор аминокислотной по-
последовательности в белковой цепи. Точность набора
зависит от связи кодон — антикодон, т. е. в конечном
счете определяется принципом комплементарности ос-
оснований.
Идентификация антикодона в тРНК проведена весьма
убедительными экспериментами. Получены мутанты,
содержащие мутационную ошибку в самом антикодоне
определенной тРНК. Затем путем полного химич. ана-
анализа цепи тРНК показано, какой именно нуклеотид
оказался замененным. Здесь и находился антикодон.
Связь кодон — антикодон осуществляется тремя нук-
леотидами, а не длинной цепочкой. Поэтому возмож-
возможность ошибок, или «уровень шумов», в процессе транс-
трансляции (так называют синтез белка) выше, чем
при редупликации ДНК или транскрипции РНК. Ве-
Вероятность ошибки при наборе белковой цепи достигает
10~4 (вместо 10~9 при матричном синтезе дезоксирибо-
нуклеиновой кислоты).
Анализ
Задача определения точного порядка чередования
нуклеотидов вдоль цепи исключительно сложна. Мож-
Можно идти по пути постепенного отщепления звеньев с то-
того и другого концов цепи, для чего служат ферменты
экзонуклеазы. Существуют ферменты различной спе-
специфичности, ведущие ступенчатый гидролиз,— одни
с 5'-, другие с З'-конца полимерной цепи. Однако
возможность постепенного отщепления концевых зве-
звеньев ограничена, и чем да-
далее оно продолжается, тем
больше возможность путани-
путаницы. Поэтому чаще полимер-
полимерную цепь расщепляют на
относительно большие блоки,
к-рые разделяют затем хро-
матографически и исследуют
последовательность в преде-
пределах каждого блока. Чтобы
правильно расставить блоки
в цепи, приходится прибегать
к различным приемам. Один
из них заключается в том,
что разбивку на блоки осуще-
осуществляют двумя независимыми
методами. Затем после пол-
полного анализа каждого из
блоков пользуются сопостав-
сопоставлениями первой и второй се-
серии фрагментов. Другой при-
прием основан на том, что путем
очень краткого импульса ра-
радиоактивного предшественни-
предшественника ДНК или РНК (обычно
одного из меченых нуклео-
зидтрифосфатов) осуществля-
осуществляют преимущественную метку
полимерной цепи с одного
конца.
Провести в жизнь эту про-
программу удалось пока только в
случае сравнительно корот-
коротких цепей (ок. 400 звеньев).
Для тРНК известно уже ок.
30 структурных формул. Для
успеха решающее значение имело нахождение фер-
ферментов, т. н. эндонуклеаз (они выделяются
из микроорганизмов), специфично расщепляющих внут-
внутренние эфирные связи цепи РНК. Примером удачного
фермента служит гуанил-РНК'аза, способная расщеп-
расщеплять РНК только по остаткам гуаниловой кислоты.
При относительно низких темп-pax и за короткое вре-
время подобные эндонуклеазы позволяют осуществить раз-
разбиение цепи рибонуклеиновой кислоты на большие и
притом воспроизводимые блоки.
Из наиболее трудных задач, решаемых в настоящее
время, можно упомянуть анализ вирусной РНК с дли-
длиной цепи порядка 4500 звеньев и рибосомальной РНК
кишечной палочки. Наибольшим достижением явилась
расшифровка последовательности нуклеотидов в пре-
пределах целого гена C87 звеньев) в вирусной РНК. Для
анализа ДНК эта программа еще практически не осу-
осуществлялась. Та часть цепи, к-рая кодирует белки, нам
известна, т. к. можно определить структурные ф-лы
белков (это сделать гораздо легче), а генетич. код изу-
изучен. Но в полинуклеотидной цепи имеются дополни-
дополнительные участки, служащие целям регуляции — запу-
запускающие редупликацию и транскрипцию, помогающие
управлять этими процессами, а также и другими не
менее важными генетич. процессами (напр., рекомбина-
рекомбинацией — образованием смешанного потомства). Изуче-
Изучение структурных формул Н. к. обещает пролить свет
на многие неизвестные стороны биологич. явлений.
Синтез
Как уже упоминалось, в природе Н. к. синтезируются
ферментативно при участии матриц. Исходными моно-
397
НУКЛЕИНОВЫЕ КИСЛОТЫ
398
мерами для дезоксирибонуклеиновой и рибонуклеиновой
кислот служат нуклеозидтрифосфаты:
О
_ II
О О
II II
п О—Р-О-Р—О—Р—О—СН
о
-р—о—р—о—р—о—сн2
I- 1-1-
п Н4Р2О7
Эти процессы удается реконструировать и вос-
воспроизвести in vitro на выделенных ферментах и
индивидуальных матрицах (вирусных Н. к.).
Безошибочность синтеза подтверждается тем, что
синтезированная in vitro ДНК или РНК бакте-
бактериофага обладает нормальной инфекциозностью (Корн-
берг, Шпигельман).
При изучении редупликации ДНК столкнулись вна-
вначале с серьезным противоречием. Опыты с изотопной
(тритиевой) меткой показали (Кэрнс), что хромосома
редуплицируется постепенно, начиная с определенной
точки и путем продвижения точки редупликации вдоль
матрицы. Это трудно согласовать с тем, что ДНК-поли-
мераза наращивает цепь ДНК только в направлении от
5'- к З'-атому дезоксирибозы, а обе цепи в одной макро-
макромолекуле ДНК антипара л лельны. Следовательно, фер-
фермент должен был бы двигаться в разные стороны вдоль
обеих цепей.
Оказалось, что дело обстоит иначе. В месте реплика-
репликации двойная спираль ДНК слегка расплетается и обе
цепи реплицируются небольшими фрагментами по
500—1000 мономерных единиц в длину (Оказаки, Су-
гимото). Эти блоки образуются действительно путем
встречного движения ферментов вдоль цепей. Но затем
образовавшиеся блоки соединяются встык ковалент-
ными (фосфоэфирными) связями. В итоге происходит
как бы направленное продвижение точки репликации
вдоль хромосомы. Стыкование блоков осуществляется
с помощью специального фермента — полинуклеотид-
лигазы. Этот фермент получен в чистом виде и хорошо
изучен (Гурвиц, Вейс, Ричардсон).
В области синтеза Н. к. без копирования заданной
природной матрицы также имеются значительные успе-
успехи. Т. к. методы классич. органич. химии применитель-
применительно к Н. к. оказываются чрезмерно сложными, то с их
помощью удается синтезировать лишь относительно не-
небольшие олигомеры (до 10—12 нуклеотидов). Решение
проблемы синтеза полинуклеотидных цепей достигнуто
путем комбинирования химических и
биохимических методов. Получение ря-
ряда ферментов в чистом виде позволило
использовать их для синтеза полинук-
полинуклеотидных цепей заданной структуры.
Первые успехи в этой области отно-
относились еще к синтезу полирибонуклео-
тидов (гомополимеров) с помощью
фермента полинуклеотидфосфорилазы
(Очоа и Гринберг-Манаго). Мономера-
Мономерами служили нуклеозиддифосфаты, по-
поликонденсация шла с выделением ор-
тофосфата.
Удалось получить сразу ряд гомо-
гомополимеров по ли А, полиГ, полиУ, по-
лиЦ достаточно высокой мол. массы
(до 106). Эти полимеры оказались чрезвычайно полез-
полезными в качестве простых моделей Н. к. Они образуют
двойные комплексы по-
О лиА — полиУ и полиГ —
полиЦ, к-рые имеют струк-
структуру двойных спиралей
Уотсона — Крика, обна-
обнаруживают гипохромный
эффект, оптич. активность
и др. свойства природных
Н. к. Кроме того, эти по-
полимеры послужили самы-
самыми простыми матрицами
для синтеза полипептидов
и позволили расшифро-
расшифровать сразу же четыре ко-
дона — УУУ, ААА, ГГГ,
ЦЦЦ, что дало огромный
толчок работам по расши-
расшифровке генетич. кода и
стимулировало дальней-
дальнейшие усилия по осуществле-
осуществлению полного синтеза по-
полинуклеотидных цепей заданной структуры.
Последние успехи связаны с синтезом небольших
областей ДНК, являющихся генами, с к-рых могут
транскрибироваться тРНК (Корана). Структурные фор-
формулы ряда тРНК сейчас уже известны. Следовательно,
можно «спроектировать» ту область ДНК, ск-рой транс-
транскрибируется данная тРНК (рис. 7). Это и есть ген для
синтеза тРНК. На схеме обе комплементарные цепи, каж-
каждая длиной в 77 звеньев, разбиты на блоки длиной от
8 до 12 нуклеотидов. Эти блоки были синтезированы по
заданной программе методами классич. органич. син-
синтеза. Далее соседние блоки соединялись друг с другом
и стыковка их производилась ферментативным путем
с помощью полинуклеотидлигазы.
Ряд обстоятельств очень важен для успеха всей про-
программы. При синтезе блоков обеих комплементарных
цепей ДНК необходимо предусмотреть, чтобы в пре-
пределах каждого блока одна из цепей после соединения
со своей комплементарной несколько выступала, т. е.
имела бы однониточный кончик для присоединения во-
20 19 18 17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1
У W Г У Ц Ц А Ц \\ А
Г А У У \\ Ц Г Г А Ц
-DJ-
—Ц—7—А— А—-Г—Г—Ц—Ц Г-Г-А-Г-Ц-А-Г-Г-Г-Г-Г-Г
I I I I I I I I I I I I I I I I
Ц—Ц—Г—Г—А—Ц—Т—Ц—Г—Т Ц-Ц-А-Ц-Ц-А
"CJ-
47 46 45 44 43 42 41 40 39 38 37 36 35 34 33 32 31 30 29 28 27 26 25 24 23 22 21 20 19 18 17
Me
г
Ц У Ц \\ \\ У У И Г Ц И
(9Ь
Г Г Г А Г А
Г У Ц У Ц Ц Г Г Т
¦F)-
Г А У У
— Г— А - А—Т— LI (;—Т—А—Ц—Ц-Ц — Т—Ц—Т—Ц-А—Г—А—Г—Г— Ц-Ц-А—А- Г-
I I У i Т I i i i И i Г i i I i i nil I I I
—Г—U—Т—Ц—Ц—Ц-Т-Т-А-Г-Ц-А-Т—Г—Г-Г А—Г—А—Г—Т—Ц—Т—Ц—Ц—Г—Г—Т—Т—Ц—Г—А—Т—Т
4 (8J ' ' G) " E) '
C'J Рибо
E'] Дезокси
C ) Дезокси
(Зг) Рибо
E ) Дезокси
(З') Дезокси
77 76 7Ь 74 73 72 71 70 69 68 67 66 65 64 63 62 61 60 59 58 57 56 55 54 53 52 51 50 49 48 47 46
Me Н2 Н2 Ме2
Г Г Г Ц Г У Г У Г Г Ц Г Ц Г У А Г У Ц Г Г У А Г Ц Г Ц Г Ц У \\ Ц- (Зг) Рибо
Ц-Ц-Ц-Г-Ц-А-Ц-А-1НК-Ц Г—Ц—А—Т—Ц—А—Г—Ц—Ц—А" "т—Ц—Г—Ц—Г—Ц—Г—А—Г— Г— E'J Дезокси
I I I I I I I I I I I I I I I I I I I I I I I I I I I
г-г-г-ц-г-т-г т-г—г—ц—г—ц—г—т—а—г т-ц-г-г-т-а-г-ц-г-ц-
-A3)-
C') Дезокси
Рис. 7. Схема синтеза гена транспортной
РНК аланина по Коране. Вся двойная цепь
ДНК разбита на 15 олигонуклеотидов, к-рые
были синтезированы химически. На схеме
видны «липкие концы», соединяющие от-
отдельные блоки, к-рые затем стыкуются с
помощью полинуклеотидлигазы. В нижней
строке показан др. вариант разбиения на
блоки.
77 76 75 74 73 72 71 70 69 68 67 66 65 64 63 62 61 60 59 58 57 56 55 54 53 52 51 50 49 48 47 46
Me H2 Н2 Ме2
Г Г Г Ц Г У Г У Г Г Ц Г Ц Г У А Г У Ц Г Г У А Г \\ Г Ц Г Ц У Ц Ц- [3'J Рибо
A4) . . A2') .. A0') * ,
Ц—Ц—Ц—Г—Ц—А—Ц—А-Ц-Ц-Г—Ц Г—Ц—А—Т—Ц—А—Г—Ц—Ц—А—Т—Ц Г— Ц— Г—Ц—Г—А—Г—Г— [5 ] Дезокси
I I I I I I I I I i I I I I I I I I I I I I I I I I I
Г—Г—Г—Ц—Г—Т—Г Т-Г—Г—Ц—Г—Ц—Г—*—А—Г Т—Ц—Г—Г—Т—А—Г—Ц—Г—Ц— [У] Дезош
399
НУКЛЕИНОВЫЕ КИСЛОТЫ
400
дородными связями соседнего блока, у к-рого также
предусмотрен комплементарный однониточный конец
(метод «липких концов»). Соединение соседних блоков
ДНК водородными связями должно предшествовать
ковалентному связыванию под действием лигазы.
Эксперимент показал, что «липкие концы» должны
содержать не менее 3—4 нуклеотидных звеньев. Разби-
Разбивая цепочку ДНК на блоки, предусмотрели невозмож-
невозможность каких бы то ни было ошибочных присоединений,
т. е. тщательным образом исключали любые «двусмыс-
«двусмысленности» при сочетании липких концов.
Далее ступенчато, звено за звеном, шел химич. син-
синтез отдельных олигомеров строго детерминированной
структуры. Все активные функциональные группы в мо-
нонуклеотидах защищали реагентами, к-рые по окон-
окончании синтеза отщепляли в мягких условиях. Только
реагирующие группы, а именно ОН-группа в фосфате
одного из нуклеотидов (мономерами служили 5'-нук-
леотиды, т. е. нуклеозид-5'-фосфаты) и ОН-группа
в положении 3' у дезоксирибозы следующего нуклео-
тида, оставались свободными. Далее следовало дейст-
действие конденсирующего агента, способного активировать
эти гидроксилы, т. е. отнять от них молекулу воды,
соединив нуклеотиды фосфоэфирной связью. Синтезы
велись в безводном пиридине в качестве растворителя,
конденсирующим агентом служил . мезитиленсульфо-
хлорид:
СН3
СН,
О
О
H3C-(~Vjj-OH
М о
HC1
СН3
Применяли маскирующие группы^ 5'-гидроксил дез-
дезоксирибозы, не участвующий в реакции, защищали
монометокситритилом I, З'-гидроксил — ацетилом
Н3С—С—О—, N6-aMHHorpynny цитозина — анизоилом
II
О
II, N6-aMHHorpynny аденина — бензоилом 111, №-ами-
ногруппу гуанина — изобутирилом (СН3JСН—С—.
II
О
После каждого сочетания продукт очищали в вод-
водном растворе с помощью хроматографии на ДЕАЕ-
целлюлозе (анионите). Чтобы хро-
хроматография протекала исключи-
исключительно по ионообменному меха-
С—
Н3СО—«
низму, к раствору добавляли мочевину. Защитные груп-
группы снимали кислотным или щелочным гидролизом.
Синтезированные химически блоки фосфорилирова-
ли с 5'-конца специальным ферментом (фосфокиназой),
затем соединяли попарно водородными связями и сты-
стыковали с помощью полинуклеотидлигазы. Самый боль-
большой из синтезированных генов представлял собой двой-
двойную цепь ДНК в 85 нуклеотидов.
Вполне реален синтез гена, кодирующего структуру
простого белка, например инсулина. На очереди стоят
задачи синтеза генов для ряда белков, а также вве-
введения этих искусственных генов в клетки в таких ус-
условиях, когда они могут «выражаться», т. е. подверга-
подвергаться транскрипции, и запустить синтез соответствую-
соответствующих белков. Решение этих задач должно составить со-
содержание новой области, получившей название «ген-
«генной инженерии».
Лит.: Микельсон А., Химия нуклеозидов и нуклео-
нуклеотидов, пер. с англ., М., 1966; Органическая химия нуклеиновых
кислот, под ред. Н. К. Кочеткова и Э. И. Будовского, М., 1970;
Нуклеиновые кислоты, под ред. И. Б. Збарского, М., 1966;
Бреслер С. Е., Молекулярная биология, Л., 1973; Инг-
р э м В., Биосинтез макромолекул, пер. с англ., М., 1966;
Дэвидсон Д., Биохимия нуклеиновых кислот, пер. с англ.,
М., 1968; Методы исследования нуклеиновых кислот, пер.
с англ., под ред. А. Н. Белозерского, М., 1970; Methods in
enzymology, v. 12 — Nucleic acids, pt A — B, N. Y.— L.,
1967—68. С. E. Бреслер.
о
ОБМЕННЫЕ РЕАКЦИИ гетероцепных по-
полимеров (exchange reactions, Austauschreaktionen,
reactions d'echange). В настоящей статье рассмотрены
реакции, в к-рые вступают образующиеся в ходе син-
синтеза гетероцепные полимеры (взаимодействие макромо-
макромолекул между собой, с мономерами, низкомолекулярным
продуктом реакции и др. веществами, присутствующи-
присутствующими в реакционном объеме), а также подобные реакции,
происходящие с полимерами при их хранении, пере-
переработке и эксплуатации.
О. р. играют особенно важную роль при поликонден-
поликонденсации, т. к. большинство гетероцепных полимеров син-
синтезируют именно этим методом. Учет этих реакций
позволяет предсказать молекулярно-массовое распреде-
распределение полимеров, а при сополиконденсации — порядок
чередования звеньев в макромолекуле.
О. р. происходят также в процессах ионной полиме-
полимеризации с раскрытием цикла (см. об этом, например,
Передача цепи, Циклических мономеров полимеризация).
Взаимодействие макромолекул с низкомолекуляр-
низкомолекулярными веществами. Реакции этого типа часто наз. о б-
менными деструктивными реакция-
м и, т. к. они ведут к снижению мол. массы образую-
образующегося полимера. Эти реакции, как правило, катали-
катализируются к-тами и щелочами. С ростом темп-ры их
скорость увеличивается. Степень деструкции полимера
пропорциональна концентрации деструктирующего низ-
низкомолекулярного вещества.
Гетероцепные полимеры способны вступать в О. р.
с водой, органич. к-тами, аммиаком, спиртами,
нек-рыми органич. растворителями (напр., N-замещен-
ными амидами). Этим объясняется пониженная хим-
стойкость таких полимеров. Кроме того, нек-рые ге-
гетероцепные полимеры деструктируют под действием
остатков мономеров; в подобных случаях требуется
особенно тщательное удаление мономера из готового
продукта.
Гидролиз протекает в тех случаях, когда вода
является низкомолекулярным продуктом реакции или
попадает в реакционный объем с растворителем, моно-
мономерами или др. путем, а также при контакте с водой
готового полимера, напр.:
~NH(CH2)nNH|oC(CH2)mCO~ -> ~NH(CH2)nNH2 +
н4-он
НООС(СН2)тСО-
В связи с этим полнота удаления воды в процессе по-
поликонденсации определяет предельное значение мол.
массы образующегося полимера. Напр., степень поли-
полимеризации Р полимера при полиэтерификации опреде-
определяется количеством оставшейся в системе воды пв
согласно ур-нию Шульца:
-/4 •
где К— константа поликонденсационного равновесия.
Аналогичная зависимость установлена для полиами-
дирования.
Скорость гидролиза полимеров зависит от природы
гидролизуемой связи, строения полимера и его физич.
состояния. Напр., в ряду полиэфиров наиболее легко
гидролизуются алифатич. эфиры угольной и щавелевой
к-т. При переходе к полиэфирам высших дикарбоновых
и, особенно, ароматич. к-т стойкость к гидролизу воз-
возрастает. Кристаллич. полимеры гидролизуются значи-
значительно медленнее, чем аморфные.
Ацидолизом наз. деструктивные реакции гете-
гетероцепных полимеров под действием карбоновых к-т.
Ацидолизу при поликонденсации подвержены полиме-
полимеры, для синтеза к-рых используют к-ты, напр, сложные
полиэфиры:
~O(CH2)n O-j-OC(CH2)mCO~ -> ~O(CH2)nOCOR-h
rgo-Loh
+ HOOC(CH2)mCO~
На скорость ацидолиза влияет тип к-ты. Напр., поли-
арилаты подвергаются деструкции адипиновой к-той
в несколько раз быстрее, чем изофталевой.
Сложные полиэфиры могут подвергаться а л к о г о-
л и з у по схеме:
~O(CH2)nO-j-OC(CH2)mCO > ~O(CH2)nOH + ROOC(CH2)mCO~
H-j-OR
При этом деструктирующим агентом может оказаться
как мономер (при синтезе полиэфира из гликоля и ди-
карбоновой к-ты), так и низкомолекулярный продукт
реакции (спирт при синтезе полиэфира из гликоля и
эфира дикарбоновой к-ты). Наиболее быстро алкоголиз
происходит у поликарбонатов. При переходе от али-
алифатич. сложных полиэфиров к ароматическим скорость
алкоголиза уменьшается.
Аналогично алкоголизу протекает ф е н о л и з по-
лиарилатов под действием бисфенолов.
Аминолиз протекает, напр., при синтезе поли-
полиамидов, полиамидоэфиров, анилино-формальдегидных
смол и при одностадийном получении полиимидов в ре-
результате действия соответствующих мономеров (диами-
(диаминов, аминокислот, анилина), напр.:
~NH(CH2)nNHJ-OC(CH2)mCO~ -> ~NH(CH2)nNH2 +
h1nh-r-nh2
+NH2RNHOC(GH2)mGO ~
Подобно аминолизу протекает и аммонолиз полиме-
полимеров под действием аммиака, выделяющегося в процессе
поликонденсации (напр., при синтезе полиамидов из
амидов аминокислот).
Полимеры с дисульфидными связями в основной цепи
макромолекулы претерпевают деструкцию под дейст-
действием низкомолекулярных дисульфидов или меркапта-
меркаптанов (с у л ь ф и д о л и з):
R-S-S-R~
I
Na+
>2~R-S-S—R'
~R-S-!-S-R~
R-S-S-R'
403
ОБМЕННЫЕ РЕАКЦИИ
404
Координационный полимер на основе 4,4'-бмс-(аце-
тоацетил)дифенилоксида и ацетилацетоната бериллия
подвержен деструкции ацетилацетоном.
Синтез полимеров из дихлорангидридов дикарбоно-
вых к-т может сопровождаться каталитич. переацилиро-
ванием образующегося полимера дихлорангидридом.
Такая О. р. протекает, напр., при поликонденсации
дихлорангидридов дикарбоновых к-т с б^с-(о-аминофе-
нолами) в среде диметилацетамида, причем растворитель
служит катализатором О. р.
Феноло-формальдегидные смолы могут вступать
в О. р. с фенолом. Эту реакцию иногда называют фено-
лизом, однако ее механизм отличается от механизма
фенолиза полиарилатов бисфенолами.
О. р. между гетероцепным полимером и низкомолеку-
низкомолекулярным веществом впервые наблюдали в 1833 Гей-
Люссак и Пелуз на примере гидролиза полилактида
(полиэфира молочной к-ты).
Взаимодействие макромолекул между собой (м е ж-
цепной обмен). Такое взаимодействие может
осуществляться двумя путями:
1) Концевая группа одной из макромолекул реагиру-
реагирует с внутренним звеном др. макромолекулы по типу
деструктивной О. р., напр.:
~R-NHCO—R'~+ ~NHCO-R"—GOOH — ~R-NHCO-R"~ -f
+ ~NHCO-R'-COOH
-R—NHGO—R'~+ ~NHCO-R"— NH2 — ~R-NHCO—R"—Ь
+ ~NHCO-R'-NH2
Доля О. р. этого типа будет падать с ростом мол.
массы полимера.
2) Взаимодействие происходит между двумя внутрен-
внутренними звеньями (функциональными группами) двух
различных макромолекул, напр.:
~R-NH CO-R"~ ~R -NHCO-R"~
I + I -» +
~R'—GO NH-R'"~ ~ R'—NHGO—R"'~
О. р. этого типа идут гл. обр. в присутствии катали-
катализаторов. Так, взаимодействие сложного полиэфира с по-
полиамидом (катализатор РЬО) протекает следующим об-
образом. Нуклеофильная атака амидной группы полиами-
полиамида на углеродный атом поляризованной катализатором
карбонильной группы полиэфира приводит к возникно-
возникновению четырехзвенного промежуточного комплекса, пе-
перераспределение связей к-рого дает новую пару амидной
и сложноэфирной групп.
При взаимодействии функциональных групп одной и
той же макромолекулы образуются макроциклы, напр,
при нагревании в присутствии катализатора р-ра коор-
координационного полимера на основе б^с-р-дикетона
и ацетилацетоната бериллия или расплава полиэтилен-
терефталата.
Условия, при к-рых взаимодействие макромолекул
между собой будет протекать с достаточно высокой ско-
скоростью, зависят от характера функциональных и кон-
концевых групп, участвующих в О. р. Скорость этих реак-
реакций определяется гл. обр. полярностью связей, а также
наличием катализаторов или растворителей, поляри-
поляризующих эти связи. Так, полимеры фосфорных к-т,
фосфатов натрия, ангидридов и эфиров фосфорных к-т
подвержены О.р. в расплаве, причем скорости образова-
образования и разрыва связей в группе Р — О — Р выше, чем
в группе С—О — Р, что соответствует соотношению их
полярностей.
О. р. в полимерах с достаточно полярными функцио-
функциональными группами (производные полифосфорных к-т,
координационные соединения бериллия и др.) проте-
протекают с достаточно высокой скоростью при темп-рах
до 100 °С. Напр., перераспределение звеньев координа-
координационного полимера из 4,4'-б^с-(ацетоацетил)дифенил-
оксида и ацетилацетоната бериллия начинается при
комнатной темп-ре и достигает высокой скорости при
70—100 °С. Перераспределение звеньев а,со-диметокси-
полиметилфосфонатов при 35 °С протекает с малой ско-
скоростью, а при 85 °С — с высокой.
Обменное взаимодействие макромолекул диметил-
полисульфатов общей ф-лы СН3О — [SO2—О — ]п—СН3
начинается при комнатной темп-ре и достигает высокой
скорости при 70 °С.
О. р. между макромолекулами различных полимеров
первоначально приводят к образованию блоксополи-
меров, а затем — статистич. сополимеров (рис.).
Межцепной обмен по-
полиамидов осуществляют, юо-[
как правило, в распла-
ве при 230—290 °С, по-
полиэфиров — в расплаве
выше 200 °С, серусодер-
жащих полимеров (напр.,
полиэтилендисульфида и
политригликольдисуль-
12
Время, ч
Основные стадии межцепно-
межцепного обмена полигексаметилен-
адипинамида и полигекса-
метиленизофталамида: 1 — содержание в смеси гомополи-
меров, 2 — содержание блоксополимера, з — содержание
статистического сополимера.
фида) — в водной дисперсии в присутствии ди-
дисульфида натрия при 90 °С. О. р. длятся обычно не-
несколько часов.
При межцепном обмене полиорганосилоксанов (на-
(нагревание в присутствии сильных неорганич. к-т или
щелочей) обычно устанавливается динамич. равновесие
между линейными и циклич. цепями. Блоксополимер
с участками политетраметил-м-силфениленсилоксана и
полидиметилсилокеана при 175 °С в присутствии окиси
калия за 30 ч превращается в статистич. сополимер.
Аналогичные О.р. характерны также для полисилазанов.
Поскольку непосредственные кинетич. исследования
реакций межцепного взаимодействия осуществить труд-
трудно, обычно исследуют модельные соединения, хотя не
существует полной аналогии между реакциями с уча-
участием низкомолекулярных и высокомолекулярных со-
соединений.
Межцепной обмен — удобный метод синтеза сополи-
сополимеров, особенно блоксополимеров. Применение этого
метода приобретает особое значение в тех случаях,
когда сополимеры трудно получить непосредственно из
мономеров. Иногда таким путем удается получить со-
сополимеры с разнообразными концевыми функциональ-
функциональными группами.
Др. направление применения межцепного обмена —
утилизация отходов производства изделий из полиме-
полимеров. О. р. в нек-рых полимерах (напр., в полифенилен-
сульфиде при 370 °С) играют существенную роль при
переработке их в изделия.
Межцепной обмен впервые наблюдал Флори в 1942 на
примере взаимодействия между собой макромолекул
полидекаметиленадипината с различной молекулярной
массой.
Лит.: Химические реакции полимеров, под ред. Е. Феттеса
пер. с англ., т. 1, М., 1967, с. 452—498; Коршак В. В.
Виноградова С. В., Равновесная поликонденсация, М.
1968, с. 54—97; К о р ш а к В. В. [и др.], Высокомол. соед.
АН, 16 A969); Дубровина Л. В. [и др.], там же, 12
1308 A970); Якубович А. Я., Вознесенская Н.И.
Б р а з Г. И., ДАН СССР, 194, 116 A970); Van Wazer J.R.
Grant D., Dungan С. Н., J. Am. Chem. Soc, 87, 333
A965); G ran t D., Van WazerJ. R., DunganC.H.
J. Polymer Sci., A5, 63 A967); M e r k er R. L., S с о t t M. J.
H a b e г 1 a n d G. G., там же, А2, 31 A964); Carmi
с h a e 1 J. В., H e f f e 1 J., J. Phys. Chem., 69, 2213 A965)
Bruno J., Compt. rend., C268, 319 A969); S h о r t J. N.,
Hill H. W., Chem. Technol., 2, № 8, 481 A972); D i w о v
K., G e о г g i e v J., Faserforsch. u. Textiltechnik, 24, 120
A973); Г у р ы л е в а А. А. [и др.], в сб.: Некоторые пробле-
проблемы органической и физической химии, Казань, 1972, с. 94.
М. М. Тепляков-
405
ОБЪЕМНЫЕ НИТИ
406
ОБРЫВ ЦЕПИ при полимеризации
(chain termination, Kettenabbruch, terminaision de chai-
ne) — элементарный акт, приводящий к необратимой
химич. дезактивации реакционных центров, необходи-
необходимых для продолжения роста цепи. Исключение из сферы
реакции активных центров в результате каких-либо
физических процессов, например окклюдирование их
в массе образующегося полимера, статистически ана-
аналогично О. ц. Поэтому это явление часто также от-
относят к О. ц.
При радикальной полимеризации
химич. дезактивация радикалов в результате мономоле-
мономолекулярного процесса наблюдается редко и обычно об-
обусловлена изомеризацией активных частиц. Интерес-
Интересный пример мономолекулярного О. ц. — дезактива-
дезактивация радикалов при полимеризации монозамещенных
ацетиленовых мономеров, обусловленная увеличени-
увеличением сопряжения неспаренного электрона в ходе рос-
роста цепи.
Более характерен для радикальной полимеризации
квадратичный О. ц., к-рый происходит по двум реак-
реакциям: 1) путем соединения двух радикалов с образова-
образованием одной неактивной «мертвой» макромолекулы (ре-
(рекомбинация радикалов):
R;- + ГЦ —> Rf-f;
2) путем отрыва одним радикалом от второго подвиж-
подвижного атома (обычно водорода) или группы с образова-
образованием двух «мертвых» макромолекул, одна из к-рых со-
содержит на конце цепи двойную связь (диспропорциони-
рование радикалов):
Rj-CH2—СНХ + R$—СН2—СНХ-> R;—СН,—СНЯХ+
+ Ri-CH^CHX
где R(- и Rj — радикалы степени полимеризации i и
/ соответственно. При этом один из радикалов м. б.
низкомолекулярным (радикал осколка инициатора,
примеси и др.)- Диспропорционирование обычно на-
наблюдается при полимеризации 1,1-дизамещенных оле-
финов и обусловлено стерич. затруднениями. Рекомби-
Рекомбинация характерна для виниловых мономеров, за исклю-
исключением мономеров, содержащих подвижный атом (груп-
(группу). Энергия активации диспропорционирования вы-
выше, чем энергия активации рекомбинации, поэто-
поэтому повышение температуры благоприятствует первой
реакции.
Суммарная энергия активации квадратичного О. ц.
при радикальной полимеризации обычно не превышает
6,3 кдж/моль A,5 ккал/молъ), константа скорости на-
находится в пределах 106—108 л/'(моль-сек).
Квадратичный О. ц.— диффузионно контролируемая
реакция, состоящая, согласно теоретич. представлени-
представлениям, из трех стадий: поступательной диффузии двух
макрорадикалов с образованием объединенного клуб-
клубка, взаимного сближения активных концов за счет
диффузии отдельных сегментов и звеньев и непосредст-
непосредственно химич. взаимодействия реакционных центров
с образованием продуктов реакции. Для большинства
исследованных виниловых мономеров константа ско-
скорости квадратичного О. ц. обратно пропорциональна
вязкости исходной системы, а стадией, определяющей
скорость процесса, является сегментальная диффузия
концов макрорадикалов. Факторы, снижающие сегмен-
сегментальную подвижность цепи (напр., введение в полиме-
ризационную систему модификаторов — веществ, спо-
способных образовывать комплексы со звеньями полимер-
полимерной цепи, или использование второго сомономера, уве-
увеличивающего жесткость цепи), значительно влияют
на скорость квадратичного О. ц.
Увеличение вязкости реакционных систем в ходе
полимеризации, приводящее к уменьшению подвиж-
подвижности радикалов роста, особенно заметное в случае
образования трехмерных сетчатых структур, обуслов-
обусловливает уменьшение константы скорости обрыва це-
цепи, что в свою очередь приводит к увеличению кон-
центрации радикалов и в конечном счете к значите-
значительному повышению скорости полимеризации (см.
Гель-эффект).
Следует отметить, что О. ц.— обязательная стадия
радикальной полимеризации в силу специфики самого
процесса. Единственным исключением пока является
фотополимеризация метилметакрилата в присутствии
больших количеств Н3РО4, для к-рой на стадии пост-
постэффекта О. ц. отсутствует. Это явление обусловлено
структурирующим влиянием к-ты на образующиеся
макромолекулы.
В случае ионной полимеризации
О. ц.— явление весьма редкое. Химизм реакций О. ц.
при ионной полимеризации изучен недостаточно. При
катионной полимеризации О. ц. может происходить
при взаимодействии растущего макрокатиона с противо-
противоионом А~:
R^ А
ВА~ -*
Rj A
j A + В
где В — к-та Льюиса. При этом, естественно, предпола-
предполагается, что В в отсутствие сокатализаторов не способна
инициировать полимеризацию.
При анионной полимеризации О. ц. может осущест-
осуществляться путем отрыва гидрид-иона от растущей цепи
противоионом — щелочным металлом с образованием
неактивного гидрида щелочного металла:
~СН2-СНХМе+ -> ~СН=СНХ + МеН
Цепь может обрываться также при взаимодействии
функциональных групп мономера или полимера с ра-
растущей макромолекулой. Так, при полимеризации
галогенсодержащих мономеров возможен О. ц. при
взаимодействии атома галогена в концевом звене
растущей цепи с противоионом с образованием неактив-
неактивной соли:
СН2Ме+ —> ~СН=СН2 + МеХ
При анионной полимеризации энергия активации
О. ц. часто превышает энергию активации роста цепи,
поэтому при понижении темп-ры относительная роль
реакций О. ц., как правило, снижается.
В случае анионной полимеризации реакцией, факти-
фактически эквивалентной О. ц., является изомеризация ра-
растущего карбаниона с образованием иона с пониженной
реакционной способностью.
При ионной полимеризации, как и при радикальной,
возможен также О. ц. вследствие замуровывания рас-
растущего макроиона в полимерном клубке. Особенно ча-
часто подобный тип обрыва цепи наблюдается при гетеро-
гетерогенной полимеризации, приводящей к образованию
кристаллических полимеров.
Отмеченные выше случаи О. ц. следует рассматривать
как естественные, присущие самой природе того или
иного процесса. Следует, однако, отметить, что О. ц.
может осуществляться при взаимодействии растущего
активного центра с веществами, присутствующими
в полимеризационной системе в качестве примесей
или специальных добавок (см. Ингибирование полиме-
полимеризации).
Лит.: Гладышев Г., Гибов К., Полимеризация
при глубоких степенях превращения и методы ее исследования,
Алма-Ата, 1968; БамфордК. [и др.], Кинетика радикаль-
радикальной полимеризации виниловых соединений, пер. с англ., М.,
1961; Гарина Е. С. [и др.], ДАН СССР, 209, № 2, 380
A973); Катионная полимеризация, под ред. П. Плеша, пер.
с англ., М., 1966; Шварц М., Анионная полимеризация,
пер. с англ., М., 1971; Structure and mechanism in vinyl poly-
polymerization, ed. T. Tsuruta, K. F. O'Driscoll, N. Y., 1969.
Б. А. Розенберг, М. Б. Лачинов.
ОБЪЕМНЫЕ НИТИ — то же, что еысокообъемные
нити.
407
ОГНЕСТОЙКОСТЬ
408
ОГНЕСТОЙКОСТЬ полимеров (fire-resistance,
Feuerbestandigkeit, resistance au feu) — способность по-
полимеров противостоять действию огня. Характеризуя
полимеры по огнестойкости, часто говорят об их горю-
горючести (возгораемости). В СССР по О. (возгораемости)
материалы делят на горючие, трудносгораемые и него-
негорючие, за рубежом — на горючие, трудновоспламеняе-
мые, трудносгораемые, самозатухающие, негорючие.
Эта классификация условна, так как О. зависит не
только от вида материала, но и от условий поджига-
поджигания, характера пламени.
Методы испытания О. полимеров настоль-
настолько разнообразны, что сравнительная оценка полимеров
по этому показателю на основании данных разных
стран очень затруднительна. Кроме того, ошибка экс-
эксперимента, как правило, довольно высока. Нередко
одни и те же полимеры на основании результатов
испытания по одному из методов относят к негорю-
негорючим, по другому — к самозатухающим или даже
горючим.
В СССР для оценки О. полимеров имеется несколько
методов. По одному из них (калориметрическому)
определяют показатель возгораемости
QjQzi гДе (?i — количество тепла (в кдж или ккал),
выделившееся при горении образца полимера, (?2 —
количество тепла, затраченное на поджигание образца.
В соответствии со значением этого показателя полимеры
делят на негорючие, или огнестойкие {QjQ2 < 0Д)>
трудносгораемые @,1—0,5) и горючие ( >0,5).
Показатели возгораемости ряда полимерных мате-
материалов приведены ниже:
Политетрафторэтилен 0,1
Сополимер винилиденфторида с гекса-
фторпропиленом 0,13
Фторкаучук 0,16
Текстолит на основе феноло-формальде-
гидной смолы 1,39
По др. методу О. характеризуют кислородны-
кислородными индексами воспламеняемости (минималь-
(минимальным содержанием кислорода в азотно-кислородной
смеси, при к-ром полимер еще может загореться).
Ниже приведены кислородные индексы ряда полиме-
полимеров (в %):
Полиэтиленоксид ....15,0
Полиметилметакрилат . 17,3
Полиэтилен 17,4
Полистирол 17,4
Хлорированный поли-
полиэтилен 21,1
Поливиниловый спирт 22,5
Поливинилфторид . . . 22,6
Полифениленоксид . . . 28,0
Полиамид 29,0
Поливинилиденфторид . 43,7
Поливинилхлорид . . . 49,0
Поливинилиденхлорид 60,0
Политетрафторэтилен 95,0
Кислородные индексы огнестойких полимеров близ-
близки к 100% [или к 1, если выражены в мольных долях
( по2 У\
\nO2+mN2JJ*
Существует ряд приближенных методов оценки О.
полимеров. При этом о принадлежности полимера по
горючести (т. е. по способности под воздействием огня
и высоких темп-р гореть с выделением тепла) к тому или
иному типу судят по следующим показателям: 1) вре-
времени самостоятельного горения (тления) образца и
потере его массы и 2) скорости распространения пла-
пламени. Первые показатели определяют одним из обще-
общепринятых экспресс-методов, т. н, методом «огневая
труба» (образец полимера располагают в трубе вер-
вертикально). Полимеры считают горючими, если потеря
массы при испытании этим методом превышает 20%,
а продолжительность самостоятельного горения со-
составляет 60 сек.
Второй показатель определяется длиной сгоревшей
(за 2 мин) части образца, к-рый располагают горизон-
горизонтально и поджигают с одного конца. Из группы горю-
горючих материалов, испытываемых по этому методу, вы-
выделяют легковоспламеняющиеся материалы, у к-рых
горение распространяется по всей длине образца, т. е.
на 300 мм.
За рубежом О. полимеров, как и в СССР, оценивают
по скорости горения образца, а также по времени его
самостоятельного горения после нескольких последо-
последовательных поджиганий. При определении О. полиме-
полимеров нек-рыми методами используют образцы большого
размера; так, метод ASTM E84-61 используется для
определения О. строительных материалов и конструк-
конструкций. Для полной характеристики возгораемости поли-
полимерного материала определяют температуры воспла-
воспламенения, тления, самовозгорания, самонагревания, а
также способность к дымовыделению, образованию
раскаленного плава и токсичность продуктов разло-
разложения.
При горении полимеров протекает ряд
химич. и физич. процессов. Для удобства рассматри-
рассматривают три зоны: 1) газовый слой; в нем происходит гл.
обр. термоокислительная деструкция продуктов раз-
разрушения поверхностного слоя полимера и наблюдается
интенсивный массо- и теплообмен; 2) поверхностный
слой полимера, подверженный действию пламени;
3) внутренние слои полимера, прилегающие к поверх-
поверхностному слою; здесь протекает в основном термич.
деструкция полимера. От природы продуктов, образую-
образующихся при пиролизе в третьей зоне, и скорости диффу-
диффузии их 'к поверхности зависит дальнейшее протекание
процессов воспламенения и горения.
На основании результатов изучения процессов горе-
горения различных полимеров установлено: 1) самогаше-
самогашение материала может происходить вследствие испаре-
испарения с его поверхности большого количества негорючих
частиц или образования на поверхности защитных по-
полимерных пленок, не поддерживающих горения; 2) вве-
введение фосфора в состав полимера способствует увеличе-
увеличению доли эндотермич. процессов («охлаждению» мате-
материала) и образованию в ряде случаев прочного кокса
(чем быстрее коксуется полимер, тем выше его О.), введе-
введение галогенов приводит к понижению темп-ры пламени
в газовом слое у поверхности полимера и ингибированию
воспламенения; 3) О. галогенсодержащих полимеров
в зависимости от природы галогена уменьшается в ря-
ряду: Вг>С1 > F; 4) совместное присутствие в полимер-
полимерном материале атомов фосфора и галогена (особенно
брома), галогена и сурьмы оказывает синергич. дейст-
действие на повышение О. (при определенном соотношении
соответствующих пар); у близких по химич. природе
полимеров О. повышается с увеличением термостойко-
термостойкости; 6) О. определяется химич. структурой полимера;
напр., при введении ароматич. звеньев, замене
группировок Р—О— С на Р—С, при уменьшении дли-
длины алкильной цепи у атома фосфора О. полимера воз-
возрастает; 7) с повышением плотности упаковки макро-
макромолекул О. у близких по химич. природе полимеров
возрастает.
К огнестойким или негорючим относятся след. груп-
группы полимеров: неорганические и нек-рые элементоорга-
нические; органические, содержащие в макромолекуле
ароматич. или гетероциклич. группировки; полностью
фторированные, галогенфторированные или сполна га-
логенированные.
Огнестойкие полимеры самозатухают при вынесении
их из пламени (самозатухающие) или вообще не горят
(негорючие). При воздействии пламени на нек-рые
элементоорганич. или неорганич. полимеры горючие
газы образуются в незначительном количестве или не
образуются совсем. Такие полимеры характеризуются
также повышенной термостойкостью. Напр., полиме-
таллоорганосилокеаны (металл — А1 или Ti), в макро-
макромолекулах к-рых в боковых ответвлениях содержат-
содержатся диалкилфосфинатные группировки, выдерживают
темп-ру до 800 °С. Однако такие полимеры могут на-
накаливаться (непламенное горение) и в ряде случаев
409
ОЗОННОЕ СТАРЕНИЕ
410
в результате разбрызгивания расплава с поверхности
стать источником горения. Полимеры, в структуре
к-рых имеются конденсированные ароматич. или гете-
роциклич. кольца, быстро коксуются, что обеспечивает
им пониженную горючесть или полную негорючесть.
Особое место в этом ряду занимают графитизирующиеся
и карбонизованные полимеры (подробно см. Карбони-
Карбонизация). И, наконец, горение ряда полимеров сопровож-
сопровождается большим эндотермич. эффектом, связанным
с испарением с поверхности негорючих частиц, как,
напр., у нек-рых сполна фторированных или гало-
генированных полимеров. Однако выделяющиеся при
горении фторсодержащих полимеров газы часто оказы-
оказываются токсичными.
Для придания или повышения О. имеется несколько
способов: 1) нанесение огнезащитного покрытия на
поверхность изделия; 2) введение наполнителей, имею-
имеющих пониженную горючесть, в композицию на основе
полимера; 3) введение огнезащитных добавок антипи-
ренов — инертных (не вступающих в химич. реакцию
с полимером) и химически активных (химич. моди-
модификация).
Первые два способа малоэффективны. При горении
огнезащитные покрытия (напр., на основе жидкого
стекла) могут накаливаться и отслаиваться от основ-
основного материала. Наполнитель (асбест, каолин, цемент
и др.) в ряде случаев может выполнять роль своеобраз-
своеобразного фитиля и способствовать распространению пламе-
пламени. Более эффективен третий способ. Количество анти-
пирена зависит от типа материала. Так, введение 2%
красного фосфора в пенополиуретан приводит к зна-
значительному повышению его О. вплоть до негорючести.
По-видимому, содержащийся в антипирене фосфор
превращается в фосфорную к-ту, к-рая на поверхности
материала образует защитную пленку полифосфорной
к-ты. Для придания самозатухаемости пластмассам на
основе полиэфиров в материал необходимо ввести
уже 5—6% фосфора или (равноценные по действию)
30% хлора, либо 8—10% брома. При одновременном
присутствии в огнезащитной добавке атомов галогена
(X) и фосфора количество того и др. элемента, необхо-
необходимое для придания О. материалу, м. б. уменьшено,
особенно если соотношение Х/Р таково, что проявля-
проявляется синергич. эффект.
Для придания О. горючим полимерам широко исполь-
используют химически активные антипирены, при взаимодейст-
взаимодействии к-рых с полимером в его состав вводятся атомы хло-
хлора, брома, фосфора, азота, бора, нек-рых металлов
(напр., кальция, бария, магния). При горении таких
модифицированных полимеров образуются ингибито-
ингибиторы воспламенения, горения (галогеноводороды, азот-,
бор- и фосфорсодержащие соединения), а также защит-
защитные пленки (напр., окислов металлов) на поверхности
материала. Так, при использовании полиакрилатов
бария в полимерных композициях на поверхности ма-
материалов при горении образуется пленка окисла ба-
бария, способствующая самогашению и предотвращающая
дальнейшее разрушение материала.
Широко применяют такие химически активные анти-
антипирены, как тетрабромфталевая к-та и ее производные,
различные производные бромфенолов и др. При выборе
модифицирующего агента необходимо учитывать, что
с повышением концентрации галогена возрастает ско-
скорость окисления полимера. Последнее было отмечено
при использовании бромированных полиэфиров в ком-
композициях на основе полиэфирных пластмасс.
Лит.: Пожарная опасность веществ и материалов, Справоч-
Справочник, [ч. 1—21, М., 1966—70; Н ind ers inn R. R., Wag-
Wagner G., Fire retaraancy, в кн.: Encyclopedia of polymer science
and technology, v. 7, N. Y.—L. [a. o.], 1967; КодоловВ.И.,
СапоговаЛ.А., СпасскийС.С, Пласт, массы, № 10,
40 A969); Nametz R. С, Ind. and Eng. Chem., 59, № 5,
99 A967); Einhorn I. N., J. Macromol. Sci., D 1, № 2,
113 A971); V о g e 1 H., Flammfestmachen von Kunststoffen,
Heidelberg, 1966. В. И. Кодолов, Я. С. Никитина.
ОЗОННОЕ СТАРЕНИЕ, озонное растре-
растрескивание (ozone cracking, Ozonripbildung, vieill-
issement а Г ozone) — растрескивание растянутых ре-
резин под действием озона. О. с.— один из видов т. н.
коррозионного растрескивания, к-рое наблюдается
при действии химически или физически активных сред
на напряженные материалы (напр., аммиака на латунь,
детергентов на полиэтилен, к-т или щелочей на резины
из полисулъфидных каучуков, HF на резины из кремний-
органических каучуков). Растягивающие напряжения
возникают в резинах при статическом или динамиче-
динамическом одно -или двумерном растяжении или при дефор-
деформации сдвига.
Для того чтобы произошло О. с, достаточно присут-
присутствия даже следов озона, к-рый всегда содержится
в атмосфере [B—6)-10~6 %; здесь и далее указана объ-
объемная концентрация озона] и, кроме того, может обра-
образоваться в определенных условиях в закрытых поме-
помещениях. Основная причина присутствия озона в атмо-
атмосфере — воздействие коротковолновой части солнеч-
солнечной радиации на кислород воздуха.
Озон образуется также в результате фотохимич.
окисления содержащихся в воздухе органич. примесей
с участием двуокиси азота. Особенно интенсивно этот
процесс протекает в больших городах, где загрязнение
воздуха выхлопными газами двигателей обусловливает
высокую концентрацию озона [до E0—100)-Ю % ].
В закрытых помещениях озон может образоваться под
действием УФ-света, у-лучей, рентгеновских лучей,
при электрич. разрядах, а также при окислении орга-
органич. соединений.
Механизм. Сущность О. с. заключается в резком уско-
ускорении разрушения напряженных резин, обусловлен-
обусловленном присоединением озона по кратным связям макро-
макромолекул каучука:
о3 /°-°\
>с=с<—> >с—с<-»>с с<
\/
о
I II
При этом образуется озонид (I), изомеризация к-рого
в изоозонид (II) может сопровождаться деструкцией
напряженной макромолекулы.
Напряжение, возникающее в резине при малых де-
деформациях, способствуя деструкции макромолекулы
и препятствуя рекомбинации макрорадикалов, уско-
ускоряет образование и разрастание микротрещин, перво-
первоначально направленных вдоль оси растяжения. Разрыв
слабых перемычек между этими микротрещинами при-
приводит к возникновению видимых глазом поперечных
трещин. При больших деформациях (сотни процентов)
трещины в процессе их роста остаются продольными,
т. к. вследствие эффекта ориентации (см. Ориентиро-
Ориентированное состояние) перемычки между трещинами приоб-
приобретают большую прочность.
Кинетика. При статич. напряжении а (или деформа-
деформации е) в процессе О. с. можно выделить 2 основные ста-
стадии: 1) индукционный период ти, окончание к-рого
практически совпадает с моментом появления трещин;
2) период развития видимых трещин твт, к-рое проис-
происходит в основном на стадии стационарной скорости
их роста тст (рис. 1). С ростом напряжения его разру-
разрушающее действие увеличивается, однако развивающая-
развивающаяся одновременно ориентация макромолекул приводит
к упрочнению полимера, что затрудняет его дальнейшее
разрушение. Поскольку в первой стадии О. с, проис-
происходящего на поверхности резины, разрушающая роль
напряжения усиливается из-за возрастания доли све-
Жей, вновь образованной поверхности, то ти обычно
монотонно уменьшается с ростом 8 (см. рис. 1). В раз-
развитии трещин в глубине образца состояние его поверх-
411
ОЗОННОЕ СТАРЕНИЕ
412
ности не играет роли; на этой стадии О. с. в большей
степени проявляется ориентационное упрочнение, в свя-
связи с чем скорость роста трещин проходит через макси-
максимум в области т. н. критической деформации екр (рис. 2).
Время до разрыва
тр = ти+твт зависит от
а (или е) так же, как хи
(см. рис. 1), или прохо-
проходит через минимум в
области 8кр (при боль-
больших деформациях —
через максимум, обус-
обусловленный исчерпани-
исчерпанием эффекта ориента-
80
>мя, мин
Рис. 1. Кинетика озон-
озонного растрескивания ре-
резины из хлоропренового
каучука (объемная кон-
концентрация озона 0,01%):
1 — деформация 100%, 2 — 57%, 3 — 50%, 4 — 44%;
S — площадь растрескавшейся части поперечного сечения
образца; ти и тст соответственно индукционный период
и стадия стационарной скорости роста трещин (для 5 7%-ной
деформации).
ционного упрочнения — см. рис. 2). Первая зависи-
зависимость, характерная для озоностойких резин, наблюда-
наблюдается в том случае, когда тр определяется продолжи-
продолжительностью хи (ти/тр^1), вторая — если тр определя-
определяется продолжительностью периода твт (хи/хр<1).
Значение екр определяется двумя
факторами: степенью уменьшения тр
с ростом а и степенью увеличения
Тр с развитием эффекта ориентации.
Увеличение межмолекулярного взаи-
! Г \\^1 модействия, затрудняя ориентацию
I* / \ макромолекул при деформации и спо-
способствуя повышению долговечности
0 20 40 60 80 100
'200 300 400 500
| 300
§¦ 200
I
100
0 20 40 60 80 100 120J 150 200 300 400 500
Деформация, %
Рис. 2. Зависимость скорости роста трещин и времени
до разрыва от значения деформации при озонном растрески-
растрескивании резин на основе различных каучуков (в скобках ука-
указана объемная концентрация озона): 1 — натуральный
каучук @,003%); 2 — бутадиен-стирольный каучук @,002%);
3 — хлоропреновый каучук @,01 и 0,02% при малых и
больших деформациях соответственно).
резин, может привести к сдвигу екр в сторону ее
больших значений. Такая зависимость наблюдается,
в частности, в ряду ненаполненных вулканизатов
следующих полимеров: натуральный каучук < гутта-
гуттаперча < хлоропреновый каучук. Значение екр воз-
возрастает также и при введении активных наполните-
наполнителей (см. Наполнители резин) в каучуки со сравнитель-
сравнительно слабо выраженным межмолекулярным взаимодейст-
взаимодействием. Так, при увеличении количества газовой каналь-
канальной сажи в натуральном каучуке от 0 до 90 мае. ч.
8кр возрастает от 15 до 50%. В случае значительного
уменьшения межмолекулярных взаимодействий (напр.,
при введении дибутилфталата в хлоропреновый кау-
каучук) значение екр резко уменьшается. Изменением меж-
межмолекулярного взаимодействия объясняется также
влияние на значение екр темп-ры, набухания и др.
факторов.
В сравнении со скоростью О. с. при статич. деформа-
деформациях, при многократных деформациях с постоянной
частотой может наблюдаться как ускорение О. с.
(в резинах из бутадиен-нитрильных каучуков), так
и его замедление (в резинах из натурального каучука).
В нек-рых резинах с увеличением частоты деформации
проявляется релаксационное упрочнение, приводящее
к уменьшению О. с. В области малых частот (до 100
колебаний в минуту) наибольшая скорость О. с. боль-
большинства резин наблюдается при частоте 10 колебаний
в минуту. Резины, содержащие воскообразные вещест-
вещества, слой к-рых на поверхности резины при многократ-
многократных деформациях легко разрушается, значительно
сильнее подвержены в этих условиях О. с, чем при
статич. деформациях.
Уменьшение концентрации озона С резко замедляет
О. с, причем вплоть до его атмосферных концентраций
сохраняется зависимость т = кС~п, где кип — по-
постоянные, а т может быть как ти, так и тр. В случае
больших т (годы) применение этой зависимости ослож-
осложняется изменением условий экспозиции резин (релак-
(релаксация напряжения, миграция на поверхность резин
антиозонантов и др.), оказывающих влияние на зна-
значения к и п.
Концентрация озона не влияет на положение екр
и значение энергии активации О. с. Последняя очень
мала (десятки кдж/молъ, или несколько ккал/молъ) и,
следовательно, изменение скорости О. с. с темп-рой
обусловлено гл. обр. изменением подвижности макро-
макромолекул. Это подтверждается тем, что скорость разра-
разрастания трещин подчиняется ур-нию Вильямса —
Лэндела — Ферри (см. Вязкотекучее состояние), опи-
описывающему релаксационные процессы. Понижение
температуры приводит к резкому замедлению О. с;
в условиях испытаний при постоянном значении е
О. с. практически прекращается при температурах,
на 15—20 °С превышающих температуру стеклования
полимера.
Солнечное излучение сильно ускоряет О. с. вследст-
вследствие фотоокисления резины, сопровождающегося дест-
деструкцией макромолекул, увеличения подвижности мак-
макрорадикалов, а также в результате общего повышения
темп-ры резины. Влага, сорбируясь сравнительно
гидрофильными резинами (напр., из натурального или
хлоропренового каучука) и способствуя более равно-
равномерному распределению напряжений на их поверхности,
несколько замедляет О. с. этих резин.
Озоностойкость резин. Способность резин сопротив-
сопротивляться О. с. существенно зависит от типа каучука.
По стойкости к О. с. (в условиях статич. деформации
до 50%) резины на основе различных каучуков можно
условно разделить на четыре группы: особо стойкие,
стойкие, умеренно стойкие, нестойкие.
Особо стойкие резины не разрушаются
в течение длительного времени (годы) при атмосферных
концентрациях озона и устойчивы более 1 ч при его
концентрациях порядка 0,1 — 1%. Такими свойствами
обладают резины на основе насыщенных каучуков —
фторсодержащих, этилен-пропиленовых, полиизобути-
лена, хлорсульфированного полиэтилена и, в меньшей
степени, резины из кремнийорганич. каучука; послед-
последние разрушаются веществами кислого характера, легко
образующимися в присутствии озона.
Стойкие резины не разрушаются в течение
нескольких лет в атмосферных условиях и устойчивы
более 1 ч при концентрациях озона ок. 0,01%. К этой
группе относятся резины на основе каучуков, слабо
413
ОКИСЕЙ ОРГАНИЧЕСКИХ ПОЛИМЕРИЗАЦИЯ
414
взаимодействующих с озоном вследствие небольшого
содержания в них кратных связей (напр., резины из бу-
тилкаучука) или благодаря присутствию связей, мало
активных к озону (напр., резины из уретановых и по-
полисульфидных каучуков), а также резины из хлоро-
преновых каучуков, стабилизированных антиозонан-
тами.
Умеренно стойкие резины устойчивы
в атмосферных условиях от нескольких мес до 1—2 лет,
а при концентрациях озона ок. 0,001% — более 1 ч.
В эту группу входят резины из нестабилизированного
хлоропренового каучука и из др. ненасыщенных кау-
каучуков (натурального, синтетич. изопренового, бута-
бутадиен-стирол ьных, бутадиен-нитрильных), содержащих
антиозонанты. Большая стойкость хлоропренового кау-
каучука к О. с. объясняется особенностями его физич.
структуры (легкой кристаллизуемостью, сильными меж-
межмолекулярными полярными взаимодействиями), обус-
обусловливающими образование тупоугольных, округлых,
медленно растущих трещин.
Нестойкие резины устойчивы в атмосфер-
атмосферных условиях от нескольких дней до 1 мес, а при кон-
концентрациях озона — 0,0001% — более 1 ч. К нестой-
нестойким относят резины из нестабилизированных каучуков
предыдущей группы, за исключением резин из хлоро-
хлоропренового каучука.
Повышение стойкости резин этой группы к О. с.
достигается введением в них антиозонантов и восков,
нанесением на резины озоностойких покрытий из
хлоропренового каучука, хлорсульфированного поли-
полиэтилена и др., химической обработкой (например, гид-
гидрированием) поверхности резин для уменьшения со-
содержания в макромолекулах ненасыщенных связей,
а также изменением конструкции изделий с целью
снижения в условиях их эксплуатации растягива-
растягивающих напряжений.
О способах защиты резин от О. с. см. также Анти-
Антиозонанты.
Помимо типа каучука, на стойкость резин к О. с.
влияет состав резиновых смесей. Так, в условиях
испытаний при одинаковой деформации 8 значения ти
и Тр для резин, содержащих наполнители и пластифи-
пластификаторы, будут меньше, чем для ненаполненных. Ухуд-
Ухудшение озоностойкости обусловлено след. причинами:
ростом напряжения, связанным с введением наполни-
наполнителей, и снижением прочностных свойств резин вследст-
вследствие введения пластификаторов.
Стойкость резин к О. с. оценивают по изменению
след. характеристик растянутых образцов: 1) степени
растрескивания (для этого по фотографиям образцов
составляют условную 4-, 6- или 10-балльную шка-
шкалу); 2) времени до появления трещин ти; 3) времени
до разрыва тр. За кинетикой развития трещин удоб-
удобно следить по спаду усилия Р в растянутом озони-
озонируемом образце. При этом тр соответствует моменту,
когда Р = 0.
Испытание в среде озона — эффективный метод
исследования долговечности резин при малых дефор-
деформациях (десятки процентов), характерных для условий
эксплуатации большинства резиновых изделий. Ре-
Результаты испытаний при повышенных концентрациях
озона позволяют также прогнозировать долговечность
резин, нестойких к действию озона, поскольку в этом
случае долговечность определяется сопротивляемостью
резин озонному старению.
О методах определения стойкости резин к О. с. см.
также Испытания резин.
Лит.: Зуев Ю. С, Разрушение полимеров под дейст-
действием агрессивных сред, 2 изд., М., 1972. Ю. С. Зуев,
ОКИСЕЙ ОРГАНИЧЕСКИХ ПОЛИМЕРИЗАЦИЯ
(polymerization of organic oxides, Polymerisation von
organischen Oxiden, polymerisation des oxydes organi-
ques). Органич. окиси (О. о.) обладают высокой реак-
реакционной способностью и полимеризуются под влиянием
широкого круга катализаторов, в основном ионного
типа. Катионная и координационно-катионная поли-
полимеризация характерна для всех О. о., по анионному
механизму полимеризуются лишь а-окиси. Значитель-
Значительно менее склонны О. о. к полимеризации по радикаль-
радикальному механизму.
Полимеризация О. о. протекает с разрывом СО-связи
в цикле и приводит к линейным простым полиэфирам:
- [ — (СН2)ХО—]„
Особенности механизма полимеризации О. о. обуслов-
обусловлены спецификой их электронной структуры: напря-
напряженностью цикла, являющейся следствием искажения
валентных углов атомов, и донорными свойствами
эфирного кислорода.
Таблица 1. Некоторые св-ва простейших органических окисей
Показатели
Валентный угол GOC . .
Энергия СО-связи
кдж/молъ
ккал/молъ
Теплота полимеризации
жидк. —тв. (—Д#п )
кдж/молъ
ккал/молъ
Свободная энергия по-
полимеризации газ — газ
<-*Fn)
кдж/молъ
ккал/молъ
Окись
этилена
Оксацик-
лобутан
61,6°
238
56,9
109
26,1
93,02
22, 17
7,91
94,5°
245
58,6
80,8
19,3
90,0
21,5
3,13
Тетраги-
дрофу-
ран
111°
329
78,5
19,3
4,6
2,14
5,00
Тетра-
гидропи-
ран
111,6°
348
83,0
-9,21
2,2
-7,08
-1,69
5,42
* Константа основности, рассчитанная по ур-нию: pKg =
= — 0,102AvQD + 2-6,8, где AvQD — сдвиг полосы ОШколеба-
ний в дейтеро-метаноле.
Термодинамика процесса. Полимеризация О. о., осо-
особенно напряженных а- и р-окисей, идет со значитель-
значительным выделением тепла. Причина этого — различие в
энергиях СО-связи в полиэфире [ок. 352 кдж/молъ
(84 ккал/молъ)] и в мономере; мерой этого раз-
различия является .теплота полимеризации в случае
образования достаточно длинных цепей, когда вклад
концевых групп пренебрежимо мал (табл. 1). Свобод-
Свободная энергия полимеризации в идеальном р-ре опреде-
определяется ур-нием:
где [Д/]р — равновесная концентрация мономера при
данной темп-ре Т. Значение AF°n является потенциаль-
потенциальной мерой способности мономера к полимеризации.
В тех случаях, когда AFL° близка к нулю, полимери-
полимеризация заметно обратима, и в системе в равновесии
в заметном количестве присутствует непрореагировав-
ший мономер. Это наиболее ярко проявляется при
полимеризации тетрагидрофурана, где мономер явля-
является самым стабильным продуктом деполимеризации.
Измерение равновесных концентраций мономера при
различных темп-pax позволяет определить термодина-
мич. параметры полимеризации; предельная темп-ра
(AFn° = 0) составляет здесь 85 °С. При А/^> 0 поли-
полимеризация практически неосуществима либо проте-
протекает до очень малой конверсии. По этой причине не по-
415
ОКИСЕЙ ОРГАНИЧЕСКИХ ПОЛИМЕРИЗАЦИЯ
416
Таблица 2. Свободные энергии полимеризации и константы основности для ряда а-окисей
Б—СН—СН2—О с различными радикалами R
Показатели
Свободная энергия по-
полимеризации (—Л-Рд)
кдж/молъ
ккал/молъ ...
*
Р
С2Н
71 ,
17,
6,
б —
^Ч
10
84
СН
7=S
17,
7,
з—
1 9
96
02
СвНбСНг—
82
19
7
,90
,80
,70
Н-
22
7
,82
,17
,91
СН3ООН2-
93
22
7
,87
,42
,97
С.]
Q6
23
8
н.-
72
,10
,11
свнбосн2-
105 ,8
25,26
8,57
СН,
112
26
8
G1-
Q
,97
,94
* См. примечание к табл. 1.
лимеризуются шестичленные циклы (диоксан, тетра-
гидропиран) и замещенные тетрагидрофураны. Их уча-
участие в сополимеризации с более активными мономера-
мономерами, однако, возможно.
Предельные явления термодинамич. характера имеют
место и в тех системах, где деполимеризация ведет
к др. циклич. продуктам. При полимеризации а-окисей
(эпоксидов), где образование мономера термодинами-
термодинамически нереально, в равновесии с полимером находятся
димеры (диоксан и его производные), тримеры, тетра-
меры. Аналогичная картина наблюдается при полиме-
полимеризации Р-окисей, где обнаружен циклич. тетрамер.
Реализация потенциальной склонности мономера
к полимеризации требует соответствующего кинетич.
механизма, в к-ром существенную роль играет стадия
активации мономера. При полимеризации О. о. акти-
активация осуществляется, как правило, через атом кисло-
кислорода, обладающий донорными свойствами. Основность
мономеров является, т. о., не менее важным фактором
их полимеризуемости, чем напряженность. Оба эти
параметра в рядах мономеров изменяются под влия-
влиянием заместителей обычно антибатно (табл. 2).
Катионная полимеризация. В качестве катализаторов
используют протонные и апротонные к-ты, металлоор-
ганич. соединения, соли стабильных карбониевых,
оксониевых и т. п. ионов.
Протонные кислоты приводят в стадии
инициирования к вторичному оксониевому иону (I):
ir х~ но (с
I
Концентрация I возрастает с основностью исходного
мономера. Активность этой частицы, очевидно, опреде-
определяется напряженностью мономерного цикла, т. е. па-
падает от эпоксидов к тетрагидрофурану. Вторичный
оксониевый ион может далее моно- или бимолекулярно
превращаться в собственно активный центр полимери-
полимеризации. Бимолекулярный нуклеофильный механизм бо-
более вероятен, поскольку протонизация повышает
частичный положительный заряд на замещаемом угле-
углероде. Низкая эффективность протонных к-т обуслов-
обусловлена именно тем, что их анионы обычно также доста-
достаточно нуклеофильны и участвуют в конкурирующей
стадии обрыва цепи:
НХ + О (СН2)Х
-«а
4- X"
О(СН2L X
В ряде случаев, однако, использование к-т с более
стабильными анионами дает возможность нолучить
относительно высокомолекулярные продукты.
При инициировании триалкилоксоние-
выми солями обменная реакция с мономером,
напр, тетрагидрофураном, приводит к активному цик-
циклич. оксониевому иону (II), дающему начало цепи:
О | « ~ R2O
Обычно в качестве ини-
инициатора используют три-
этилоксоний с BF4,
А1С1", SbCle, PFe.
Инициирование ста-
стабильными карбо-
ниевыми иона-
м и, напр, солями три-
фенилметилкарбония,
может происходить дво-
двояко: 1) непосредствен-
непосредственным присоединением ио-
иона к молекуле мономера:
Ph3C+ +
П
O(C
Ph3C—
+/—I
O^
2) путем гидридного переноса с образованием трифе-
нилметана и активного центра (III):
Ph3C
-а
Ph3CH
°0
Образование трифенилметана установлено эксперимен-
экспериментально при полимеризации тетрагидрофурана. По ана-
аналогичному механизму действует тропилий- и диоксо-
лений-ионы.
Эти методы инициирования особенно удобны для
детального исследования механизма полимеризации,
т. к. позволяют количественно генерировать активные
центры.
Наиболее сложным является механизм формирова-
формирования активных центров полимеризации при иницииро-
инициировании кислотами Льюиса (BF3, SbCl5, PF6
и др.). Вслед за стадией комплексообразования, экви-
эквивалентной реакции инициирования с помощью протон-
протонных к-т, происходит раскрытие мономерного цикла.
Х„Ме
¦О
—О
IV
Скорость и детальный механизм этого акта существенно
зависят от наличия в системе сокатализаторов, а также
от природы мономера и противоиона.
В случае а-окисей, обладающих высокой напряжен-
напряженностью, комплекс IV крайне неустойчив и быстро
превращается в цвиттер-ион карбониевого (V) или
оксониевого (VI) типа, напр.:
»- F3BOCH2CH2
V
« *- F3BOCH2CH2O
VI
Аналогичный процесс у тетрагидрофурана затруднен
и идет лишь на сильных к-тах; это обусловливает
низкие скорости полимеризации. Добавки эпоксидов,
однако, существенно облегчают образование активных
центров и резко повышают таким образом скорость
полимеризации.
417
ОКИСЕЙ ОРГАНИЧЕСКИХ ПОЛИМЕРИЗАЦИЯ
418
Вода и спирты также промотируют полимеризацию,
но участвуют одновременно в передаче цепи. Актив-
Активными передатчиками являются эфиры. В катионных
системах обнаружено несколько видов процессов гибели
активных центров, приводящих в нек-рых случаях
к прекращению полимеризации задолго до исчерпания
мономера или достижения термодинамич. предела.
По этой же причине измеряемая концентрация актив-
активных центров редко соответствует исходной концентра-
концентрации инициатора.
Вопрос о механизме роста цепи тесно связан с вопро-
вопросом о природе активного центра. Если активный центр
имеет карбониевый характер, то реакция следует
механизму SnI, где лимитирующей стадией является
раскрытие цикла в (VII):
-сн2 +
Быстро
' СН« —(
VII
Медленно
-сн2
Если же активный центр оксониевый, то акт роста цепи
является истинно бимолекулярным (Sn2):
~сн2— о
о
-СН2О(СН2L
Очевидно, что в любой полимеризующейся системе
могут присутствовать оба типа центров, находящиеся
в равновесии, а их соотношение, зависящее от значе-
значений элементарных констант скорости, будет опреде-
определяться особенностями мономера. Предполагается, что
полимеризация эпоксидов протекает по механизму
SnI, в то время как тетрагидрофуран, оксациклобутан
и бициклич. окиси полимеризуются по оксониевому
механизму.
Для катионной полимеризации и сополимеризации
О. о. весьма характерно образование циклов в резуль-
результате передачи цепи на полиэфирный кислород
СНо
-о
~сн2
о
а также аналогичные процессы межцепного обмена.
Это приводит к ряду своеобразных статистич. и кине-
тич. последствий. Участие полимерного кислорода и др.
присутствующих в системе доноров (D) в равновесиях
типа
А-М + D^: AD + М
уменьшает концентрацию активных центров роста А
и понижает скорость процесса.
Преимущество катионной полимеризации О. о.—
относительно высокие скорости даже при низких
темп-рах. Однако ряд осложняющих процессов (обрыв
и передача цепи, образование циклических фрагментов)
и неясности в механизме затрудняют направленное
осуществление этой реакции.
Анионная полимеризация. Иод действием анионных
катализаторов полимеризуются лишь напряженные
циклы.
Ряд процессов полимеризации a-окисей, напр, поли-
полимеризация окиси этиленд, инициированная натрий-
нафталином или щелочью в присутствии этиленгли-
коля, следует кинетич. модели Флори для реакции
без обрыва и передачи цепи, т. е. с образованием
«живущего» полимера. Схема Флори предполагает одно-
одновременный рост постоянного числа кинетически идентич-
идентичных активных частиц, результатом чего является уз-
узкое молекулярно-массовое распределение (распределение
Пуассона).
В более общем виде механизм анионной полимери-
полимеризации эпоксидов не укладывается в рамки этой схемы.
Основной причиной этого является тот факт, что при
наиболее распространенном способе инициирования
полимеризации щелочами, алкоголятами или аминами
стадия инициирования часто оказывается существенно
более медленной, чем рост цепи. Независимо от того,
обусловлен этот эффект различием истинных констант
скорости или связан с изменением характера среды
и сольватации активных частиц, он искажает кинетику
процесса, а молекулярно-массовое распределение ста-
становится более широким.
Другое осложняющее обстоятельство — наличие
в полимеризующейся системе обменных реакций расту-
растущих макроанионов PjO~ с гидроксилсодержащими
соединениями (вода, спирты, гликоли), эквивалентных
передаче цепи, напр.:
PjO- + ROH ^ PjOH + RO-
Гидроксилсодержащие соединения часто специально
вводят в систему для регулирования мол. массы поли-
полимера, поскольку обменная реакция делает возможным
участие всех алкоксильных групп в росте цепи. Сте-
Степень полимеризации в этом случае определяется
по ур-нию
о _ |МЬ-[М]
*п — [р.о-] + [ROH]
где [М]о и [М] — соответственно исходная и текущая
концентрация мономеров. Соединения, содержащие
ОН-группы, образуются также при передаче цепи
на мономер:
Р.-О- + СНз-СН- СН2 -> РЮН + СН2=СН-СН2СГ
Наличие реакций обмена и передачи цепи на моно-
мономер приводит к существованию в системе различных
по активности центров, а также ограничению роста
макромолекул. Спирты, кроме того, участвуют в
сольватации активных центров, комплексообразова-
нии и т. п.
Катализаторы анионной полимеризации эпоксидов —
производные щелочных и щелочно-
щелочноземельных металлов: гидроокиси, алкого-
ляты, амиды и т. п. Менее эффективны в качестве ката-
катализаторов амины. Полимеризацию проводят в массе
или среде эфирных растворителей (диоксан, тетрагидро-
тетрагидрофуран), часто с добавками спиртов, повышающих
растворимость катализаторов, а также в полярных сре-
средах (диметилсульфоксид, гексаметилфосфамид и др.).
Рост цепи осуществляется путем последовательных
актов нуклеофильного присоединения мономера к ак-
активному центру, представляющему собой чаще всего
ионную пару:
ОМе + Н2С — СН2
+
Me
I
Н2СГ
+
• ~ОСН2СН2ОМе
Свободные алкоксоанионы образуются, видимо, лишь
в сильно сольватирующих средах (спиртах, диметил-
сульфоксиде, гексаметилфосфамиде) и в небольших
количествах. Характерная особенность оксоанионов
по сравнению с карбанионами — высокая степень
локализации отрицательного заряда, обусловливает
особую прочность ионных пар и их высокую склонность
к ассоциации. В тетрагидрофуране, напр., сольвати-
рующая способность к-рого невелика, активные центры
полимеризации окиси этилена практически полностью
ассоциированы.
По анионному механизму осуществляется, видимо,
полимеризация на нек-рых твердых катализаторах —
окислах Be, Mg и щелочноземельных
металлов, амидах, карбонатах, суль-
сульфатах Са, Sr, Ва и др., приводящих к исключи-
исключительно высокомолекулярным полимерам окиси этилена.
Здесь, однако, возможна предварительная донорно-
акцепторная координация мономера, поскольку окись
419
ОКИСЕЙ ОРГАНИЧЕСКИХ ПОЛИМЕРИЗАЦИЯ
420
пропилена также образует высокомолекулярные кри-
сталлич. полимеры (при анионной полимеризации в р-ре
образуются только аморфные олигомеры).
Высшие циклы в обычных условиях вполне устой-
устойчивы к действию анионов.
Полимеризация под действием металлоорганических
соединений. Многие металлоорганич е-
ские катализаторы обладают специфич.
активностью при полимеризации эпоксидов, особенно
замещенных, приводя в последнем случае к стереоре-
гулярным полимерам.
Имеется ряд систем, катализирующих полимериза-
полимеризацию любых окисей, напр, триалкилалюминий с со-
катализаторами.
Механизм полимеризации окисей в присутствии алю-
минийалкилов сложен и, видимо, различен для боль-
больших и малых циклов. Так, система AlEt3 — Н2О до со-
соотношения компонентов 1 : 1 обладает кислотными
свойствами, полимеризует оксациклобутан и тетрагид-
рофуран в присутствии добавок а-окисей, склонна к об-
образованию циклич. форм, т. е. проявляет черты, харак-
характерные для катионной полимеризации. Введение ами-
аминов полностью подавляет способность этой системы
полимеризовать 4- и 5-членные циклы, но увеличивает
ее эффективность в отношении а-окисей, повышая
мол. массу полимеров и их стереорегулярность. К ана-
аналогичному эффекту приводит увеличение содержания
воды. Эти данные предполагают существование двух
типов активных центров — катионных и анионно-
координационных, однако детальный механизм их
возникновения не ясен. Аналогичные закономерности
характерны для цинкорганич. катализаторов. Катали-
тич. система ZnEt2 — Н2О при низком содержании
воды обладает ярко выраженной кислотностью по отно-
отношению к простым эфирам, полимеризует оптически
активную окись пропилена со значительной рацемиза-
рацемизацией; ведет полимеризацию 3,3-дихлорметилоксацик-
лобутана и тетрагидрофурана. Катионный характер
этой системы резко подавляется при соотношении
H2O:ZnEt2, превышающем 1:1.
Функция сокатализатора в цинкорганич. системах
(Et2Zn, Bu2Zn, Ph2Zn) сводится в большинстве случаев
к созданию поляризованных связей Zn — О (вода,
спирты, Н2О2, кислород), Zn — S (молекулярная сера),
Zn — N (амины, гидразин, нитраты). При сокатализе
кетонами поляризация создается карбонильной груп-
группой (VIII), а в случае диметилсульфоксида — сильным
донорно-акцепторным взаимодействием (IX):
RZn — СН2 — С— R
VIII
О
ъ ъ
RZn — CH2R
OS(CH3J
IX
Полимеризация эпоксидов идет путем внедрения по по-
поляризованной связи с предварительной координацией
мономера. Кинетика полимеризации и молекулярно-
массовое распределение образующихся полимеров, как
правило, сложны.
Для металлоорганич. катализа полимеризации О. о.
характерно образование стереорегулярных полимеров
из замещенных эпоксидов. Механизм этих процессов
подробно изучен на примере окиси пропилена. Поли-
Полимеризация представляет собой в этом случае истинную
сополимеризацию оптич. D- и L-изомеров. При кати-
катионной или анионной полимеризации в р-ре активные
центры не способны к отбору мономера какой-либо
конфигурации, и полимеризация рацемич. смеси приво-
приводит к атактич. полимерам. Если же в качестве моно-
мономера использовать только один из оптич. изомеров,
то при анионной полимеризации образуется стереоре-
гулярный изотактич. полимер, поскольку присоеди-
присоединение идет только к незамещенному углероду, а кон-
конфигурация асимметрического атома (отмечен звездоч-
звездочкой) сохраняется:
н2с
~СН2 — сн — О 4- *| /О -
I НС
СН3 I
сн.
— с?1 — о — сн2 — сн — о
сн3 сн3
Рентгенографически подтверждена изотактич. струк-
структура всех известных до сих пор стереорегулярных
полиэпоксидов. Исключение — поли-трега-бутилэти-
леноксид, образующийся при анионном инициировании
и являющийся синдиотактич. полимером.
При полимеризации на металлоорганич. катализато-
катализаторах (Zn, Al, Fe) в системе одновременно присутствуют
активные центры обеих конфигураций — Ad и Al,
способные специфично «выбирать» из смеси оптич. изо-
изомер определенной конфигурации. Соотношение кон-
концентраций энантиоморфных центров м. б. изменено
введением различных асимметрич. соединений, взаи-
взаимодействующих с катализатором. Образующийся поли-
полимер будет представлять собой соответственно эквива-
эквивалентный (Ad = Al) или неэквивалентный (Ad ф Al)
набор D- или L-макромолекул, т. е. будет оптически
активным или неактивным.
Действительная схема стереоспецифич. полимери-
полимеризации а-окисей более сложна, и ее развитие во многом
связано с разработкой количественных методов ана-
анализа стереорегулярности полимеров этого типа.
Полимеризация под действием радикальных инициа-
инициаторов. Большинство окисей не полимеризуется по ра-
радикальному механизму. Это связано со склонностью
образующихся при инициировании радикалов к изо-
изомеризации в а-кеторадикал, к-рый «предпочитает»
отрыв водорода присоединению к циклу:
RCH —СН2
О
Инициирование
RC—СН2
V
RC (СН2)ХСН2О и т.д.
О
Радикальную полимеризацию, приводящую к корот-
коцепным разветвленным полимерам, наблюдали лишь
в случае окиси стирола и фенилглицидилового эфира,
где радикалы, видимо, более стабильны. Окись стирола
полимеризуется радикально и при радиационном ини-
инициировании в жидкой и твердой фазах. Известна также
чередующаяся сополимеризация окиси этилена с пер-
фторпропиленом под действием перекисей, а также
теломеризация а-окисей с олефинами.
Полимеризация под действием излучений. Радиа-
Радиационным путем полимеризуются очень немногие окиси:
3,3-дихлорометилоксациклобутан и его аналоги в твер-
твердой и жидкой фазе, окись пропилена (в присутствии
добавок окиси стирола), а также окиси циклогексена,
изобутилена и тетрафторэтилена. Предполагается рост
цепи по катионному механизму.
Появились сообщения о возможности фотохимич.
инициирования полимеризации окисей в присутствии
малеинового ангидрида или карбонила марганца.
Лит.: Фурукава Д., Саегуса Т., Полимериза-
Полимеризация альдегидов и окисей, пер. с англ., М., 1965; Ring-opening
polymerization, ed. by К. С. Frisch and S. L. Reegen, N. Y.—
L., 1969; Катионная полимеризация, под ред. П. Плеша, пер.
с англ., М., 1966; Дрейфус П., Дрейфус М., Хим.
и технол. полимеров, № 11, 20 A967); Addition and Condensation
421
ОКИСЕЙ ОРГАНИЧЕСКИХ ПОЛИМЕРЫ
422
polymerization processes. Adv. in Chem. Series, № 91, 1969;
TanakaY., J. Macromol. Sci., Al, 1059 A967); E н и-
колопянН. С, И р ж а к В. И., РозенбергБ. А.,
Усп. хим., 35, в.4, 714 A966); Saegusa Т., J. Macromol.
Sci., 6A, 997 A972). К. С. Казанский.
ОКИСЕЙ ОРГАНИЧЕСКИХ ПОЛИМЕРЫ (poly-
(polymers of organic oxides, Polymere von organischen Oxiden,
polymeres des oxydes organiques). Органич. окиси
(О.) — циклич. мономеры, содержащие в цикле один
атом кислорода. В данной статье рассматриваются
полимеры моноциклических (I) и бициклических (II)
окисей, а также диэпоксидов (III).
<СН,)те-О (CH,)m-CH-O-CH-(CH,)n
i н
СН2—СН-( СН2)т- СН—СН2
III
Моноцикли ч. окиси (синонимы для а-оки-
сей — эпоксиды, оксираны; для Р-окисей — оксетаны,
оксациклобутаны) полимеризуются с образованием
линейных простых полиэфиров. Характерной особен-
особенностью таких полимеров, определяющей весь комплекс
их физич. свойств, является высокая термодинамич.
и кинетич. гибкость макромолекул. Это обусловливает
низкие значения темп-р плавления и стеклования (таб-
(таблица), высокоэластичность и др'.
В ряду полиэфиров [ —(СН2)т О — ]п все показатели
гибкости макромолекул — темп-ры плавления и стек-
стеклования, размеры клубков, модули высокоэластично-
сти имеют минимальные значения при т = 2 — 3,
т. е. для полиэтиленоксида и полиоксациклобутана
(см. рисунок). Растворимость полимеров этого типа
проявляет ту же тенденцию при изменении т. Введение
в цепь заместителей, особенно больших по объему или
полярных, понижая гибкость молекул, способствует
тем самым увеличению темп-р плавления полимеров,
их прочности, снижению высокоэластичности и т. п.
В кристаллич. структуре незамещенных полиокисей,
имеющих конформацию плоского зигзага (т > 4),
наблюдается изменение с ростом т типа кристалли-
кристаллической решетки от моноклинной, характерной для
политетрагидрофурана (см. Тетрагидрофурана поли-
полимеры), к орторомбической (полиэтилен, т = оо). Для
полиоктаметиленоксида (т = 8) могут реализоваться
обе кристаллич. модификации. С ростом т в этом интер-
интервале монотонно возрастает темп-pa плавления поли-
полимеров.
6 8 10 12
Значение т
Изменение темп-ры плавления A) и размера клубков макро-
макромолекул B) для полиэфиров [—(СН2)тО —]п в зависимости
от значения т.
Способность к кристаллизации у полимеров моноза-
мещенных О. зависит от взаимной ориентации замести-
заместителей, регулируемой способом синтеза полимера. Напр.,
анионная полимеризация замещенной О. пропилена
в р-ре приводит к нерегулярным некристаллизующим-
ся полимерам, тогда как координационно-анионная —
к кристаллич. изотактич. полимерам (см. Окиси про-
пропилена полимеры). Известно большое число кристал-
кристаллич. полимеров замещенных О. этилена, наибольший
интерес среди к-рых представляют полимеры галоген-
гидринов и 2,3-эпоксибутана.
Из пятичленных окисей полимеризуется лишь тетра-
гидрофуран, причем его полимеризация обратима
(см. Окисей органических полимеризация). Замещенные
О. этого типа вообще не удается превратить в полимер,
в основном по термодинамич. причинам, но эти соеди-
соединения участвуют в сополимеризации с более активными
мономерами. Это'же в полной мере относится к высшим
окисям с числом групп СН2, равным 5 и более.
Свойства линейных полимеров органических
Полимер
Полиэтиленоксид
[-(СН,), О-]п
Полипропиленок-
сид
СНз
1
[-СН2СНО-]П
Полиоксациклобу-
тан
Г-(СН2KО-]П
Поли-3,3-дихлор-
метилоксацикло-
бутан
СН2С1
1
[~СН2-С-СЯ2О-]п
СН2С1
Политетрагидрофу -
ран
[-(СН,LО-]Г|
Темп-ра
плавле-
плавления, °С
66
70
35—37
170-180
40-41
се вГ
as
Темп
лова!
-67
-75
—
7
0
Модуль упру-
упругости крис-
кристаллич. фазы
Гн/м2 (кгс/см2)
10 A05)
25B5-Ю4)*
—
Ю2(Ю6)
5,5 E,5-105)
окисей
Характери-
Характеристика равно-
равновесной гиб-
гибкости макро-
макромолекул**
<h2>1/2/(h2>01/2
1 ,38
1,59
1,43
—
1 ,58
* Значение для полиизобутиленоксида. ** Обозначение вели-
величин см. Гибкость макромолекул.
О. образуют сополимеры друг с другом, а также
с мономерами др. типов — циклическими, виниловыми,
карбонилсодержащими. Введение кислородного мостика
в углеводородную очень повышает гибкость макромо-
макромолекул, растворимость полимера, повышает адгезию.
Возможно получение блоксополимеров различного
типа (см. Окисей органических сополимеры).
В связи с низкими темп-рами плавления большинство
полимеров моноциклических О. не находит прямого
практич. использования. Исключение составляет водо-
водорастворимый полиэтиленоксид (см. Окиси этилена
полимеры) и поли-3,3-дихлорметилоксациклобутан
(пентапласт — СССР, пентон — США, Великобритания).
Наиболее широкое применение полиэфиры на основе
О. находят в виде гидроксилсодержащих олигомеров
при синтезе полиуретанов и уретановых каучуков.
Перспективно также введение в макромолекулы поли-
полиэфиров боковых ненасыщенных групп, обусловливаю-
обусловливающих способность полимера к серной или радикальной
вулканизации. Образующиеся каучуки сохраняют цен-
ценные свойства исходных полиэфиров: морозостойкость,
устойчивость к действию масел и т. п. Возможность
синтеза полимеров и сополимеров О. с широкой вариа-
вариацией состава, мол. массы и разветвленности позволяет
тонко регулировать свойства конечных вулканизатов.
Применение сополимеров предпочтительно, поскольку
они исключают кристаллизацию каучуков.
423
ОКИСЕЙ ОРГАНИЧЕСКИХ СОПОЛИМЕРЫ
424
Мостиков ые бициклические эфи-
р ы, а также нек-рые диэпоксиды нолимери-
зуются с образованием полиэфиров, содержащих циклы
в основной цепи и обладающих в связи с этим повы-
повышенной термич. стабильностью.
В случае 1,3- или 1,4-ориентации в цепи цикл накла-
накладывает серьезные конформационные ограничения,
в результате чего повышается жесткость и темп-pa пла-
плавления полимеров, понижается их растворимость. Так,
темп-pa плавления полимера 1,4-эпоксициклогексана
(II, т = п = 2) составляет ок. 450 °С. Этот полимер
получают катионной полимеризацией в присутствии
к-т Льюиса и сокатализаторов. Сополимеризация 1,4-
эпоксициклогексана с тетрагидрофураном, окисью про-
пропилена и эпихлоргидрином приводит к более низко-
низкоплавким, растворимым и эластичным полимерам.
Полимеризация таких бициклич. О., как окиси цик-
лопентена, циклогексена (II, т = 0), приводит к обра-
образованию полимеров с циклами в положении 1,2, прак-
практически не отличающихся от линейных полимеров.
Цикл в этом случае практически не ограничивает гиб-
гибкости цепи, темп-pa плавления составляет 60—70 °С,
полимеры хорошо растворимы.
Полимеризация бициклич. мономеров, содержащих
функциональные группы в периферич. части цикла,
открывает возможность модифицировать затем обра-
образующийся полимер.
Диэпоксиды сопряженных диенов полимеризуются
с образованием сшитых полимеров. Так, моноокись
дивинила (III, m = 0) полимеризуется, в основном, с
образованием твердого сшитого полиэфира. Полиме-
Полимеризация диэпоксидов несопряженных диенов приводит,
напротив, к растворимым полимерам с циклами в основ-
основной цепи. Рост цепи происходит по меж- и внутримоле-
внутримолекулярному механизмам, причем возможность цикли-
циклизации определяется термодинамич. причинами, а струк-
структура образующихся циклов — конкуренцией путей
раскрытия второй эпоксигруппы атакой на а- или
Р-углерод:
СН2-СН2
СН2-СН2
I |а |3
сно- сн—сн2
~СН,—СН СН—СН,—СГ
/СНЧ
сн2 сн-о-
-СН2-СН
сн2
При полимеризации 1,2,5,6-диэпоксигексана (III,
т = 2) образуются, в основном, тетрагидрофурано-
вые циклы в цепи.
Полимеры диэпоксидов обладают повышенной жест-
жесткостью и низкой растворимостью. Их термич. разло-
разложение начинается вблизи 200 °С. Метод циклополимери-
зации диэпоксидов и др. подобных мономеров с окис-
ными циклами сулит определенные возможности регу-
регулирования структуры цепи.
Лит.: Polyethers, ed. N. G. Gaylord, v. 3, pt 1 — Polyal-
kylene oxides and other polymers, N. Y., 1962; Encyclopedia of
polymer science and technology, v. 6, N. Y.—[a. o.J, 1967, p. 103.
См. также лит. при ст. Окисей органических полимеризация.
К. С. Казанский.
ОКИСЕЙ ОРГАНИЧЕСКИХ СОПОЛИМЕРЫ (соро-
lymers of organic oxides, Mischpolymere von organischen
Oxiden, copolymeres des oxydes organiques). Будучи
активными мономерами, органич. окиси (О.) легко
сополимеризуются друг с другом, а также с мономерами
др. типов — циклическими, карбонилсодержащими
и виниловыми. С практической точки зрения наиболее
важным направлением в сополимеризации О. является
синтез гидроксилсодержащих олигомеров для полиуре-
полиуретанов. В зависимости от способа проведения процесса
м. б. получены би- или полифункциональные олигоме-
ры. Использование сополимеров с широкой вариацией
природы мономерных звеньев и состава позволяет
более тонко регулировать физико-механич. свойства
уретановых вулканизатов и, что не менее важно, их
склонность к кристаллизации.
Сополимеризацию а-О. с циклами большего размера
(оксетаны, тетрагидрофуран) проводят обычно катион-
ным путем, причем активность сомономеров определя-
определяется гл. обр. их основностью. По анионному механизму
получают статистич. и блоксополимеры различных
а-О. Последние используют как поверхностно-актив-
поверхностно-активные вещества', соотношением окиси этилена и второго
сомономера (окиси пропилена или бутилена) регули-
регулируют гидрофильность сополимера.
Другое перспективное направление — сополимериза-
сополимеризация простых а-О., чаще всего окиси пропилена (см.
Окиси пропилена полимеры), с мономерами этого же типа,
содержащими ненасыщенные группы, с последующей
серной или радикальной вулканизацией. Получаю-
Получающиеся каучуки, как и аналогичные им полисульфидные,
обладают прекрасными механич. характеристиками
и высокой термостойкостью (см. Эпоксидные каучуки).
О. могут сонолимеризоваться также с Р-пропиолак-
тоном, циклич. формалями и ацеталями, N-карбоксиан-
гидридами а-аминокислот (ангидриды Лейкса) и цик-
циклич. ангидридами дикарбоновых к-т. Статистич.
характер сополимеров установлен лишь в одиночных
случаях; часто в системе образуются гомополимеры.
С ангидридами к-т эпоксиды образуют чередующиеся
сополимеры состава 1:1. При сополимеризации эпок-
сидов с азиридинами происходит прививка гликолевых
групп по NH-гругшам гомополимера азиридина.
Сополимеризация О. с альдегидами систематически
не исследована, хотя определенно установлено поло-
положительное влияние ацетальных групп на термич. ста-
стабильность полиметиленоксида и др. полиальдегидов.
Введение эпоксидов в полиметиленоксидную цепь м. б.
достигнуто их сополимеризацией с формальдегидом или
триоксаном при катионном инициировании. Известен
также двухстадийный способ синтеза блоксополимера
окиси этилена с ацетальдегидом, обладающего повы-
повышенной термич. стабильностью. При сополимеризации
эпоксидов с окисью углерода образуется чередующийся
сополимер сложноэфирной структуры.
Другую группу образуют сополимеры О. с винило-
виниловыми мономерами. Эта область лишь начинает разви-
развиваться, однако ясно, что введение простых эфирных
связей в углеродные цепочки виниловых полимеров
позволит улучшить нек-рые их свойства: эластичность,
гидрофильность, адгезию. Относительно просто обстоит
дело с привитой и блоксополимеризацией, к-рые обычно
проводят анионным путем, причем основой чаще явля-
является «живой» виниловый полимер (полистирол, поливи-
нилнафталин, полиакрилонитрил). Возможна прививка
винилового мономера к полиэтиленоксиду при его ме-
ханодеструкции. Синтез блоксополимеров путем реком-
рекомбинации «живущих» анионов и катионов осуществлен
на примере полистирола и политетрагидрофурана.
Возможность синтеза статистич. сополимеров О.
с виниловыми мономерами определяется, в основном, их
способностью полимеризоваться по одинаковому меха-
механизму. Наиболее общим способом синтеза является
в связи с этим катионная полимеризация, охватываю-
охватывающая большинство непредельных и циклич. мономеров.
а- и р-О. сополимеризуются в различных комбинациях
со стиролом и его производными, изобутиленом, акри-
лонитрилом, простыми виниловыми эфирами в при-
присутствии к-т Льюиса с образованием, как правило,
низкомолекулярных полимеров. В ряду а-О. склон-
склонность к сополимеризации со стиролом растет с увели-
увеличением карбониевого характера активного центра.
Тетрагидрофуран и его производные с трудом вступают
в такую сополимеризацию в связи с существенно иным
425
ОКИСИ ПРОПИЛЕНА ПОЛИМЕРЫ
426
(оксониевым) механизмом роста цепи. Введение циклич.
формален, занимающих промежуточное положение,
может улучшать результаты сополимеризации «оксо-
ниевых» мономеров со стиролом, изобутиленом и др.
По радикальному механизму сополимеры образуют
только а-О., как более напряженные. Известны их
сополимеры с этиленом, винилиденхлоридом, перфтор-
пропиленом и хлортрифторэтиленом, но только в двух
последних случаях образуется статистич. сополимер.
Лит.: Patsiga R. A., J. Macromol. Sci., pt С. 1, 223
A967); Саундерс Дж. X., К. Фриш, Химия поли-
полиуретанов, пер. с англ., М., 1968. См. также лит. при ст. Окисей
органических полимеризация. К. С. Казанский.
ОКИСИ ПРОПИЛЕНА ПОЛИМЕРЫ, полиок-
си пропилены, полипропиленоксиды
[poly (propylene oxide), Polypropylenoxid, polyoxypropy-
lene].
Окись пропилена (пропиленоксид, 1,2-эпо-
ксипропан) СН3СН — СН2 (О. п.) -- бесцветная
жидкость, с эфирным запахом; т. кип. 34,6 °С; т. пл.
-112,13 °G; ci240 0,8311; я?° 1,3664; критич. темп-ра
215,3 °С; давление пара р и абсолютная темп-pa Т связа-
связаны в интервале от —30 до 33,4 °С ур-нием Igp = 10,615—
1722,7/Г (р в н/м2) или Igp = 8,49 —1722,7/Г (р
в мм pm. cm.)', теплота испарения 28,9 кдж/молъ
F670 кал/молъ); теплота сгорания Qv 1916 кдж/молъ
D57,68 ккал/молъ), стандартная теплота образования
—121,7 кдж/молъ (—28,84 ккал/молъ)[жщк.]; энтропия
281,1 дж/(молъ -К) [67,15 кал/'(моль -К)]; уд. тепло-
теплоемкость с°р 72,68 дж/(молъ-К) [17,36 кал/(молъ -°С)];
теплота полимеризации (жидк.— тв.) ок. 105 кдж/молъ
B5 ккал/молъ); вязкость при 20 °С 0,38 мн-сек/м2,
или спг\ дипольный момент 6,50• 10~30 к-м A,95 D).
О. п. хорошо смешивается с водой A : 1,5), спиртами,
эфиром и многими органич. растворителями; при —5 °С
образует с водой кристаллогидрат (т. пл. —3 °С).
Азеотропные смеси: Н2О, 99% О. п. (т. кип. 39,8 °С);
СН2С12, 23% О. п. (т. кип. 40,6 °С); С5Н12, 57% О. п.
(т. кип. 27,5 °С).
Чистая О. п. вполне устойчива к нагреванию: ее
термич. разложение до окиси углерода и этана наблю-
наблюдают вблизи 500 °С. В контакте с активными поверх-
поверхностями (А12О3, Gr2O3-W O3 и др.) разложение, а также
изомеризация в пропионовый альдегид, ацетон или
аллиловый спирт идут при более низких темп-рах.
В присутствии воды, щелочных или кислотных агентов
возможна частичная изомеризация О. п. при нор-
нормальных условиях.
О. п. токсична, но в меньшей степени, чем окись
этилена. При попадании на кожу вызывает ожоги.
Летучесть О. п., а также низкая темп-pa воспламенения
(—28,9 °С) требуют особых предосторожностей при хра-
хранении и работе.
О. п. получают дегидрохлорированием 1,2-пропи-
ленхлоргидрина под действием щелочи:
кон
СН3-СНС1-СН2ОН > СНз—GH—СН2
\/
или прямым окислением пропилена. Очистку осуществ-
осуществляют ректификацией.
Полипропиленоксиды (П.). Полимеризация О. п.
идет с раскрытием цикла по эфирной связи. В связи
с асимметрией молекулы мономера возможны два
направления этого процесса, приводящее к образова-
образованию мономерных звеньев различной структуры:
С
с
СНз—GH— CH2
., ~сн-сн2-о~
у i
II
Благодаря стерич. эффекту C-разрыв предпочтите-
предпочтителен. При анионной полимеризации вероятность рас-
раскрытия цикла с образованием звеньев типа II очень
близка к единице, тогда как полимеры, образующиеся
в катионных процессах, содержат статистич. набор
звеньев различной структуры.
Изотактический П. м. б. получен поли-
полимеризацией О. п. в присутствии металлоорганич. ка-
катализаторов (См. Окисей органических полимеризация).
Существует в оптически активной и рацемич. форме
(см. Оптически активные полимеры). Изотактич. П.
кристаллизуется в ячейке орторомбич. типа, вклю-
включающей 2 полимерных цепи, взаимное расположение
к-рых таково, что наличие противоположных по кон-
конфигурации (D или L) макромолекул не препятствует
кристаллизации. Плотность полностью кристаллич.
полимера 1,157 г/см3, степень кристалличности варьи-
варьирует в очень широких пределах и зависит, в основ-
основном, от способа синтеза. Большинство полимеров это-
этого типа структурно неоднородно и содержит фрак-
фракции, различающиеся по стереорегулярности; фракци-
фракционирование м. б. осуществлено путем охлаждения
р-ров П. в «-гексане, ацетоне, изооктане.
Мол. масса П. лежит в интервале от нескольких
десятков тысяч до нескольких миллионов, молекуляр-
но-массовое распределение является широким.
В связи с низкой темп-рой плавления (не выше
75 °С) стереорегулярный П. не находит непосредст-
непосредственного практич. применения.
Полипропиленгликоль — низкомолеку-
низкомолекулярный П., имеющий две концевые гидроксильные
группы, получают анионной или кислотно-каталитич.
полимеризацией в присутствии воды и гликолей, регу-
регулирующих состав концевых групп и мол. массу поли-
полимера.
В отсутствие гидроксилсодержащих добавок П.,
получаемый анионной полимеризацией, содержит зна-
значительную долю ненасыщенных концевых групп
(аллильных и пропенильных), что отрицательно ска-
сказывается при использовании этого полимера в синтезе
полиуретанов. Причина появления таких концевых
групп в полимере — передача цепи на мономер:
- сн2сно- + сн3сн-сн2 -> ~сн2снон
СНз ° СН3
сн2=сн-сн.о~
Полипропиленгликоль представляет собой бесцвет-
бесцветную вязкую жидкость и характеризуется след. пока-
показателями:
Мол. масса
Плотность, кг/м9 {г/'см3)
150-4000
990 — 1020
@,99-1 ,02)
1,443-1,451
Показатель преломления nj)
Уд. теплоемкость,
пджЦпг-К) . . 1,76-1,97
[кал/(г-°С)] . . [0,42-0,47]
Вязкость, мн-сек/м'г, или спз
при 25 °С 60-1200
при 100 °С . 3-80
Темп-pa стеклования*, °С .от —60 до —75
Диэлектрич. проницаемость 6—7
Дипольный момент, к-м (D)
(бензол, 20 °С)
при мол. массе 400 12 • 10~30 C , 6)
при мол. массе 4000 28, 4 • 1О30 (8,5)
* По данным различных авторов.
Коммерческий полипропиленгликоль имеет узкое
молекулярно-массовое распределение: (Mw/Mn ^
^ 1,14 — 1,25). Коэфф. для расчета мол. массы по дан-
данным характеристич. вязкости приведены в таблице.
Растворителями полипропиленгликоля являются ке-
тоны, хлорпроизводные, бензол, толуол. Раствори-
Растворимость в воде резко падает с увеличением мол. массы;
склонность к растворению в углеводородных средах
427
ОКИСИ ЭТИЛЕНА ПОЛИМЕРЫ
428
Коэффициенты в ур-нии [т]]=КМа, связывающем
характеристическую вязкость и молекулярную массу
полипропиленоксида
Растворитель
Бензол
Толуол
Гексан
Метанол
Тетрагидрофуран
Циклогексанол
Темп-
ра, °С
25
25
46
20
20
21
К, дл/г
1,12-10-*
1,29-10-*
1,97-10-*
4,06-10-*
5,50-10-*
1,0.10-3
а
0,77
0,75
0,67
0,64
0,62
0,50
Интервал
мол. масс
3-10*-о-10в
То же
до 3 • 103
То же
»
при этом, напротив, растет. Так, w-гексан при 40—
45 °С растворяет высокомолекулярные образцы П.
Полипропиленгликоль используют, в основном, как
полупродукт в синтезе полиуретанов.
Прочие гомо- и сополимеры окиси
пропилена. В синтезе сшитых полиуретанов
применяют полиоксипропиленполиолы, получаемые
присоединением О. п. к многоосновным спиртам (гли-
(глицерин, сорбит, ксилит, маннит и др.) в присутствии
щелочных катализаторов; их свойства зависят от сте-
степени разветвленности и длины цепи. Аналогичным
образом м. б. получены полиоксипропиленовые про-
производные спиртов, фенолов, аминов, меркаптанов
и др. соединений с подвижным водородом. Хорошо
известны также используемые как поверхностно-актив-
поверхностно-активные вещества блоксополимеры О. п. с окисью этилена
(«плюроник», «тетроник» и др.). Их свойства регули-
регулируются соотношением длины блоков. Статистич. сопо-
сополимеры О. п. с тетрагидрофураном, имеющие гид рок-
сильные концевые группы и используемые также в син-
синтезе полиуретанов, получают кислотно-каталитич. пу-
путем в присутствии спиртов и гликолей.
Наряду с синтезом полиуретанов успешным направ-
направлением использования свойств П. является создание
каучуков на основе сополимера О. п. с непредельными
органич. окисями — моноокисью дивинила, аллилгли-
цидиловым эфиром, глицидилметакрилатом, окисью
винилциклогексена. Их введение в цепь несколько
снижает способность полимера к кристаллизации и
делает возможной вулканизацию серой. Каучуки этого
типа по морозостойкости, устойчивости к действию
масел и озона, механич. свойствам [прочность при
растяжении до 20 Мн/м2 B00 кгс/см2), относительное
удлинение до 600%], эластичности при низких темп-рах
не уступают др. каучукам, в том числе натуральному.
Попытки прямого сшивания П. свободными радика-
радикалами, фотохимически и радиационно приводят обычно
к его деструкции.
П. весьма склонны к деструкции с разрывом цепи,
что обусловлено наличием третичного углеродного
атома. При пиролизе П. выше 270—280 °С образуются
ацетальдегид, ацетон, пропилен. Термоокислительная
деструкция в атмосфере кислорода идет с образованием
воды (до 60%), уксусного и муравьиного альдегидов
(до 40%), ацетона, метана, СО и водорода. Для ста-
стабилизации полимера используют обычные антиокси-
данты (фенолы, амины). Ниже нек-рой критич. кон-
концентрации @,2% по массе) ингибирующая эффектив-
эффективность антиоксидантов мала.
Лит.: Саундерс Д ж. X., Фриш К., Химия поли-
полиуретанов, пер. с англ., М., 1968; Грубер [Е.] [и др.],
Хим. и технол. полимеров, № 6, 119 A965). См. также лит. при
ст. Окисей органических полимеризация. К. С. Казанский.
ОКИСИ ЭТИЛЕНА ПОЛИМЕРЫ, полиокси-
этилены, полиэтиленоксиды [poly (ethy-
lene oxide), Polyathylenoxid, polyoxyethylene].
Окись этилена (этиленоксид, эпоксиэтан) Н2С — СН2
(О. э.) — бесцветная жидкость со специфич. запахом,
т. кип. 10,73 °С; т. замерз.-112,55 °С; 4°'? 0,8827;
л|/41,360; вязкость при 10 °С 0,283 мн-сек/м2,
или спз] критич. параметры: давление 4,98 мн/м2
E0,7 кгс/см2), темп-ра 195,8 °С; давление пара и абсо-
абсолютная темп-pa связаны ур-нием:
lgp=—2045,7/Г — 0,0215Г + 2,3328-105Г2 + 15,441
где р в н/м2, или Igp = — 2045,7/Г — 0,0215Г +
+ 2,3328-105 Г2 +13,3163, где р в мм рт. ст.; теплота
испарения 25,54 дж/молъ F,101 ккал/моль) [10,7 °С];
молярная теплоемкость с0 48,19 дж/(молъ • К)
[11,51 кал/(моль -°С)], удельная 1,84 кдж/(кг-К)
[0,44 кал/(г • °С)] [жидкость от —25 до 10 °С],
1,12 кдж/{кг-К) [0,268 кал/{г • °С)] [газ, 34 °С,
10 кн/м2]; теплота сгорания Qp 1310 кдж/молъ
C12,5 ккал/моль); стандартная теплота образования
(Д//р —51,04 кдж/молъ (—12,19 ккал/моль) для газо-
газообразного и —76,8 кдж/молъ (—18,29 ккал/моль) для
жидкого мономера; стандартная энтропия 243,4
дж/(молъ-К) [58,13 кал/(молъ-К)]; диэлектрич. проницае-
проницаемость 13,9 при — 1 °С; дипольный момент 6,27 • 10~30
к-м A,88 D).
О. э. хорошо растворима в большинстве органич.
растворителей (спиртах, эфирах, диоксане, кетонах,
бензоле, хлорпроизводных), а также в воде. Пары
О. э. устойчивы при нагревании до 370—380 °С; при
более высоких темп-pax идет разложение до окиси
углерода D0—50%), метана C5—40%), водорода и
этана. Изомеризация О. э. в ацетальдегид происходит
лишь при 500 °С, но в контакте с А12О3 — уже при
200—300 °С.
Смеси О. э. с воздухом взрывоопасны в широких
концентрационных пределах — от 3 до 100% О. э. при
нормальных условиях. В присутствии инертных раз-
разбавителей (азот, СО2, низшие углеводороды) пределы
существенно изменяются.
При работе с О. э. необходимо удаление источников
взрыва — нагретых поверхностей, открытого пламени,
статич. электричества, искр и т. п. Пары О. э. крайне
токсичны: в малых количествах обладают наркотич.
действием, в значительных — приводят к раздражению
слизистых оболочек, удушью и отеку легких. Корот-
Короткое пребывание в атмосфере, содержащей 0,025%
(по объему) О. э., не является безвредным и признаки
отравления появляются через несколько часов. Пре-
Предельно допустимая концентрация О. э. в воздухе —
0,001 г/м3 (мг/л), а порог восприятия запаха О. э.
человеком равен 1,26 г/м3 (мг/л). Жидкая О. э. в при-
присутствии влаги вызывает сильные ожоги.
В пром-сти О. э. получают через хлоргидрин
С1/Н2О КОН
сн2=сн3 > сн2он-сн2а > сн2—сн2
\
либо прямым каталитич. окислением этилена:
Ag/pt
СН2=СН2 + 72О2 > СН2— СНа
Второй способ более прогрессивен технологически и
дает чистую О. э. Окончательную очистку мономера
производят ректификацией.
Полиэтиленоксиды (П.)-О. э. полимеризуется под воз-
воздействием большого числа катализаторов. Термическая
и радикальная полимеризация О. э. практически
не наблюдаются. Полимеризация идет с раскрытием
эпоксидного цикла по С — О-связи
пСН2—СН2 -» [-СН2СН2О-]„
и сопровождается большим тепловыделением: теплота
полимеризации жидкого мономера составляет ок.
109 кдж/моль B6 ккал/моль). Высокая теплота реакции,
а также склонность к полимеризации в присутствии
429
ОКИСИ ЭТИЛЕНА ПОЛИМЕРЫ
430
|40
>20
разнообразных агентов являются причиной «спонтан-
«спонтанной» полимеризации, протекающей в редких случаях
как тепловой взрыв. В связи с этим хранение О. э.
требует особых предосторожностей.
П. имеет широкий интервал мол. масс — от 102 (ди-
этиленгликоль) до 107. В зависимости от мол. массы
это бесцветные жидкости, воскообразные или твердые
продукты. Изменение темп-ры плавления П. со сте-
степенью полимеризации (Р) дано на рис. 1. Темп-ра
стеклования П. лежит
вблизи — 60 °С.
П. кристаллизуется
в моноклинной ячейке,
включающей четыре
цепи, конформация —
спираль 7/2; плотность
о . полностью кристаллич.
s- I полимера 1330 кг/м3
A,33 г/см3). Степень
кристалличности поли-
полимеров достигает, по дан-
Рис. 1. Зависимость темп-ры плав- ?ым ЯМР, 92 95 /о.
ления полиэтиленоксида от степени Теплота плавления со-
полимеризации. ставляет по разным
данным 7,64—8,92 кдж/
моль A825—2130 кал/моль), энтропия 4,22 (при посто-
постоянном объеме) и 5,85 (при постоянном давлении)
кал/(моль -°С).
Плотность П. лежит в пределах 1,16—1,30 г/см3 и
зависит, в основном, от степени кристалличности [плот-
[плотность полностью аморфного полимера 1120 кг/м3
A,12 г/см3)] и мол. массы; темп-рный коэфф. уд. объе-
объема 6.10-6 м3/(кг-К) [6-Ю-3 см3/(г.°С)].
П. растворимы в воде и в большинстве органич.
растворителей, исключая парафиновые углеводороды,
причем растворимость в воде сохраняется до самых
высоких мол. масс. Коэфф. для расчета мол. масс
из характеристич. вязкости приведены в таблице.
Коэффициенты в уравнении [у\]=КМа, связывающем
характеристическую вязкость и молекулярную массу
полиэтиленоксида
Растворитель
Бензол
Вода
Вода
Метанол
СС14
Темп-
ра, °С
25
25
30
20
20
К, дл/г
3,97-10-*
2,4-10-*
1,25-10-*
3,3-Ю-4
6,9-Ю-4
а
0,686
0,73
0,78
0,72
0,61
Интервал
мол. масс
8 • 104—5 • 106
102 —105
10*-107
до 2-10*
до 2-104
Полимеры О. э. склонны к различным видам деструк-
деструкции: термической, окислительной, химической. Термо-
Термодеструкция П. при 320—370 °С приводит к формальде-
формальдегиду, этанолу, СО2 и воде; О. э. в продуктах не наблю-
наблюдалось. Потеря в массе полимера (мол. масса 10 000)
за 30 мин при 324 °С составляет ок. 7%, а при 363 °С—
98,6%. Основные продукты окислительной деструк-
деструкции — Н2О, формальдегид, СО2. Разрыв цепи, приво-
приводящий к увеличению содержания гидроксильных и
карбонильных групп и падению вязкости полимера,
интенсивно идет даже при комнатной темп-ре. Деструк-
Деструкция протекает и в р-рах; она сильно ускоряется ионами
Fe3+ и Сг3+, особенно на свету. Окислительная дест-
деструкция сильно замедляется добавками обычных анти-
оксидантов (фенолов, аминов, стабильных радикалов);
в водном р-ре эффективны добавки пропилгаллата,
изопропилового, этилового и аллилового спиртов
@,2-0,5%).
Полимеры О. э. деструктируют под действием надук-
сусной к-ты, перекиси водорода, хлора, брома, ради-
радикальных инициаторов, озона. Таким путем из высоко-
высокомолекулярных полимеров м. б. получены более низко-
низкомолекулярные. Полимеры с мол. м. 106—107 разруша-
разрушаются при высокоскоростном перемешивании р-ров;
процесс носит радикальный характер, а образующие-
образующиеся радикалы способны инициировать полимеризацию
других мономеров, например метилметакрилата, с
образованием блоксополимеров. При у~°блучении од-
одновременно с деструкцией идет образование сшито-
сшитого геля.
По физико-механич. свойствам и сферам применения
полимеры О. э. можно разделить на две группы: низ-
низкомолекулярные полимеры с гидроксильными конце-
концевыми группами (полиэтиленгликоли) и высокомолеку-
высокомолекулярные полимеры.
Полиэтиленгликоли (мол. масса до 40
тыс.) — жидкие или воскообразные продукты с низ-
низкой упругостью пара, вязкостью от 4 до 1000 ммУсек
(ест) A00 °С), плотностью 1120—1200 кг/м3 A,12—
1,20 г/см3), Пр 1,459—1,467, уд. теплоемкостью
2,05 — 2,18 кдж/(кг-Щ [0,49—0,52 кал/(г-°С)], по-
поверхностным натяжением 43—45 мн/м (дин/см), тепло-
теплотой плавления ок. 170 кдж/кг D0 кал/г). Эти полимеры
под маркой «карбовакс» (США) получают по след.
технологии. Инициирующий гликоль с катализатором
загружают в реактор, в к-рый затем при 100—150 °С
подают О. э. до получения полимера заданной мол.
массы, согласуя скорость подачи мономера с возмож-
возможностями отвода тепла.
Полиэтиленгликоли используют в качестве смачива-
смачивателей, умягчителей и антистатич. агентов в текстиль-
текстильной пром-сти, связующего — в фармацевтич. пром-сти
и косметике, как компонент моющих средств, а также
в производстве уретановых каучуков, где они обеспе-
обеспечивают высокую прочность, эластичность и низкую
темп-ру хрупкости вулканизатов.
Аналогичные функции выполняют продукты кон-
конденсации О. э. со спиртами, алкилфенолами, карбоно-
выми к-тами, аминами, меркаптанами. Большинство
этих продуктов, как и блоксополимеры О. э. и окиси
пропилена («плюроник», «тетроник»), являются неио-
ногенными поверхностно-активными веществами, в ко-
которых полиоксиэтиленовые блоки выполняют гидро-
гидрофильную функцию.
Высокомолекулярные полимеры
О. э. (мол. масса от 500 тыс. до 10 млн.) — водораство-
водорастворимые термопластичные полимеры, обладающие хоро-
хорошими механич. свойствами:
Прочность при растяжении*, Мн/м2 13 — 17
{кгс/см2) . '. A30 — 170)
Модуль при растяжении, Мн/м* (кгс/см2) 200 — 500
B000-5000)
Относительное удлинение, % 700 — 1200
Твердость по Шору (шкала А) 99
Темп-ра плавления, °С 66—68
* Прочность при растяжении ориентированных пле-
пленок может достигать 70 — 100 Мн/м2 G00—1100 кгс/см2).
Полимеры такого типа под маркой «п о л и о к с»
(США), «алкокс» (Япония) в виде порошка или мелких
гранул получают суспензионной полимеризацией при
20—50 °С в среде осадителей полимера в присутствии
амида, амид-алкоголята или аммиаката Са, а также Zn-
и Mg-органич. соединений. Полиокс поддается любым
способам переработки — литью, экструзии, каландро-
ванию, прессованию. Он образует нити и пленки, обла-
обладающие высокой прочностью и эластичностью. Меха-
Механические свойства полимера мало изменяются при на-
нахождении в воздухе с влажностью до 90%; а при более
высокой влажности прочность полимера резко ухуд-
ухудшается. Полиокс совмещается с многими природными
и синтетич. полимерами, а также с рядом пластифика-
пластификаторов. С другой стороны, он устойчив к действию масел
и смазок.
431
ОКИСЛИТЕЛЬНАЯ ПОЛИКОНДЕНСАЦИЯ
432
В связи с высокой кристалличностью полиокс резко
теряет прочностные свойства вблизи темп-ры плавле-
плавления, однако и выше этой темп-ры сопротивление сдвигу
достаточно велико. Вязкость расплава полиокса слабо
зависит от темп-ры и характеризуется резко аномаль-
аномальной зависимостью от скорости сдвига. Псевдопластич-
Псевдопластичность обнаруживают также водные р-ры полиокса даже
при очень низких концентрациях полимера. При высо-
высоких концентрациях полиокс образует с водой эластич-
эластичные гели.
Для водных р-ров полиокса характерно наличие
верхнего температурного предела растворимости. Этот
Рис. 2. Зависимость
темп-ры осаждения по-
полиокса из водных р-ров
от его концентрации и
мол. массы.
0,01 0.1 1 10
Нонцентрация полимера, zJdA
предел зависит от концентрации полимера и его мол.
массы (рис. 2). Предел растворимости сдвигается в об-
область более низких темп-р при добавках различных
солей (рис. 3), к-рые понижают также характеристич.
вязкость полимера. Нерастворимы в воде кристаллич.
комплексы полиокса с HgCl2, полиакриловой к-той,
мочевиной и тиомочевиной.
Рис. 3. Зависимость тем-
темп-ры осаждения полиок-
полиокса из водных р-ров от
концентрации и природы
добавленной к р-ру соли
(концентрация полимера
0,5% по массе): 1 — LiGl;
2 — NaCl; 3 — KF; 4 —
КОН; 5 — Li2SO4; 6 —
MgSO4, K2SO4; 7 —
Na2GO3, K2GO3; 8 —
Na3PO4.
0,2 0,4 0,6 0,8 1,0
Концентрация соли, моль /л
Полиокс растворим при комнатной темп-ре в ацето-
нитриле, этилен- и метилендихлориде, трихлорэтилене
и СС14, а при повышенной темп-ре — в кетонах, бен-
бензоле, метаноле. Полимер не растворим в парафинах,
гликолях, глицерине.
Область технич. применения полиокса непрерывно
расширяется. Он рекомендуется для применения в тек-
текстильной промышленности (шлихтование тканей, не-
нетканые материалы, антистатик), как загуститель с вы-
высокой вязкостью (латексы, латексные краски), упако-
упаковочный материал или защитные покрытия для водораст-
водорастворимых препаратов (удобрения, чернила, краски) или
пищевых продуктов, связующего в керамич. и др. от-
отраслях пром-сти. Низкая токсичность и устойчивость
к действию биологич. кислорода допускают разнооб-
разнообразные применения полиокса в медицине, фармацев-
фармацевтической и пищевой пром-сти, косметике. Известны
моющие и аэрозольные композиции на основе полиокса.
Наиболее высокомолекулярные образцы полиокса обла-
обладают хорошими коагулирующими и флокулирующими
свойствами и могут эффективно использоваться в этом
направлении. Их действие в меньшей степени чувстви-
чувствительно к рН среды, чем, напр., у полиакриламида.
Большой интерес представляет способность полиокса
значительно (до 70%) снижать гидродинамич. сопро-
сопротивление в водных и водно-органических р-рах при
концентрациях полимера 0,001—0,003%. Этот эффект
возрастает с увеличением мол. массы, но снижается
при повышении темп-ры. Эта способность полиокса
и ряда др. водорастворимых полимеров используется
для снижения сопротивления трубопроводов при пере-
перекачке жидкостей, р-ров и пульп.
Лит.: Окись этилена, М., 1967; Davidson R.L.,
S i t t i g M., Water-soluble resins, N. Y.— L., 1962; Nonionic
surfactants, Ed. by M. Schick, N. Y., 1967 (Surfactant sciences
series, v. 1); Hoyt I. V., Polymer Lett., 9, 851 A972).
К. С. Казанский.
ОКИСЛИТЕЛЬНАЯ ПОЛИКОНДЕНСАЦИЯ - см.
Д егидрополиконденсация.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ ИНИ-
ИНИЦИИРОВАНИЕ ПОЛИМЕРИЗАЦИИ - см. Иниции-
Инициирование полимеризации.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОЛИ-
ПОЛИМЕРЫ, редокситы (redox polymere, Re-
doxpolymere, polymeres redox).
Содержание:
Введение 4 32
Классификация 4 32
Основные количественные характеристики . . .4 33
Кинетика окислительно-восстановительных реак-
реакций 434
Получение и свойства редокс-полимеров . . .4 35
Получение и свойства редокс-ионитов 4 39
Регенерация 442
Применение 442
Введение. Окислительно-восстановительные поли-
полимеры — полимеры, макромолекулы к-рых содержат
группы, способные к окислительно-восстановительным
превращениям. О.-в. п. часто наз. также электрон о-
обменниками, однако этот термин неточен,
т. к. в ходе окислительно-восстановительной реакции
происходит не обмен, а перенос электронов. При пере-
переносе каждого электрона либо образуется (или исче-
исчезает) положительный заряд, либо исчезает (или обра-
образуется) положительно заряженный ион. Типичные
окислительно-восстановительные реакции приведены
ниже:
а. Система гидрохинон (восстановленная форма)/хинон
(окисленная форма):
б. Система сульфгидрил (восстановленная форма)/ди-
сульфид (окисленная форма):
Y
SH
Y Y
s—s
Помимо групп, способных к окислительно-восстано-
окислительно-восстановительным превращениям, в макромолекулы О.-в. п.
могут также входить ионообменные группы (—SO3H,
—СООН и др.).
Классификация. О.-в. п. делят обычно на два класса
в зависимости от наличия или отсутствия в них ионо-
ионообменных групп, а также от функций, к-рые выполня-
выполняет в полимерах углеводородная матрица и от типа связи
между матрицей и реакционноспособными группами:
1. Собственно О.-в. п., или редокс-поли-
м е р ы, углеводородная матрица к-рых содержит
только окислительно-восстановительные группы. Эти
полимеры м. б. получены поликонденсацией, полимери-
полимеризацией или методом полимераналогичных превращений.
В последнем случае окислительно-восстановительную
систему вводят в предварительно синтезированный
«инертный» полимер, к-рый может присоединять эту
систему ковалентно, в результате комплексообразова-
ния, адсорбции и др.
433
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОЛИМЕРЫ
434
2. О.-в. п., содержащие окислительно-восстанови-
окислительно-восстановительные и ионообменные группы,— ок и'слитель-
но-восстановительные иониты, или
редокс-иониты (иногда их наз. также
электроноионообмен н и к а м и). Эти
полимеры м. б. получены теми же способами, что и ре-
докс-полимеры (синтетич. редокс-иониты), а также
путем сорбции (редокс-иониты на основе сорбентов)
или осаждения (редокс-иониты на основе носителей)
окислительно-восстановительной системы в порах ио-
нита. Процессы окисления — восстановления и ион-
ионного обмена в редокс-ионитах могут протекать одно-
одновременно или независимо друг от друга.
О.-в. п. могут быть растворимыми (в воде, спирте,
неорганич. к-тах, органич. растворителях и др.) или
нерастворимыми. Линейные или слабосшитые О.-в. п.
обычно растворимы, полимеры с большим числом по-
поперечных связей — нерастворимы. Существуют О.-в.п.,
к-рые при восстановлении переходят в р-р, а при
окислении выпадают в осадок.
Основные количественные характеристики. Окис-
Окислительно-восстановительная ем-
емкость характеризует число активных (обратимых)
окислительно-восстановительных групп в полимере.
Обычно ее определяют окислением или восстановлением
О.-в. п. Для определения емкости восстановленных
О.-в. п. часто используют соли Fe3+ (сульфат, хло-
хлорид). Число этих ионов, восстановленных полимером
до ионов Fe2+, устанавливают титрованием р-ром пер-
манганата калия и выражают в мг-экв/г сухого поли-
полимера или в мг-экв/л р-ра или набухшего полимера.
По изменению емкости после нескольких последова-
последовательных циклов окисления — восстановления судят
о хим. -и термостойкости О.-в. п. В редокс-ионитах
определяют также ионообменную емкость (см. Ионооб-
Ионообменные смолы).
Нормальный потенциал Е°, характери-
характеризующий реакционную (окислительно-восстановитель-
(окислительно-восстановительную) способность О.-в. п., определяют по разности меж-
между значениями потенциала платинового электрода
в р-ре исследуемой окислительно-восстановительной
системы и потенциала нормального водородного элект-
электрода и выражают в в. Потенциал нерастворимых поли-
полимеров нельзя измерить непосредственно, поскольку
такие полимеры не могут получать электроны от элект-
электрода или отдавать их электроду. В этом случае подби-
подбирают растворимую окислительно-восстановительную
систему, удовлетворяющую условию Ее > Ей (ин-
(индексы «с» и «п» относятся соответственно к окислителю
и полимеру). Потенциал системы, к-рая находится
в равновесии с наполовину окисленным нерастворимым
полимером, принимают равным потенциалу полимера.
Потенциал обратимой системы в реакции окисления—
восстановления определяют из ур-ния:
rt 1°К1
Ь (!)
где R — газовая постоянная, равная 8,3202 дж/(моль-К)
[1,9872 кал/(моль-°С)]; Т — абсолютная темп-pa, К;
F — число Фарадея, равное 96 491 f 1,1 к/г-экв; п —
число электронов, участвующих в элементарном акте
реакции окисления — восстановления; [Ок] и [Вое] —
мол. концентрация соответственно окисленной и вос-
восстановленной форм системы. Если [Ок] = [Вое],
то Е = Е°.
Разность потенциалов О.-в. п. и окислителя опреде-
определяют из ур-ния:
rt
[Ок]с
Чем больше разность Ес — EIV тем полнее и с боль-
большей скоростью протекает процесс окисления — восста-
восстановления. В случае слабых окислителей эта разность
мала; реакция идет чрезвычайно медленно и не доходит
до конца. С увеличением рН на единицу значение Е°
полимера уменьшается на 0,059 #, т. е. полимер в кис-
кислом р-ре более сильный окислитель и более слабый
восстановитель, чем в нейтральном. В сильно щелоч-
щелочных средах значение Е° от рН не зависит. В условиях
термодинамич. равновесия между двумя окислительно-
восстановительными системами, напр, восстановленной
формой полимера и окислителем, Еп = Е°с.
Значение нормального потенциала О.-в. п. (табл. 1,
2) определяется природой окислительно-восстанови-
окислительно-восстановительной системы. Напр., в ряду замещенных хинонов
донорные заместители уменьшают, а акцепторные уве-
увеличивают потенциал. Это обусловлено тем, что первые
уменьшают, а вторые увеличивают сродство хинона
к электронам, с приобретением к-рых хинон превраща-
превращается в ион гидрохинона через промежуточную стадию
образования ион-радикала семихинода:
=о :^г о
При наличии в молекуле хинона двух или более
заместителей важен не только их характер, но и взаим-
взаимное расположение. Напр., значение Е° для гидрохи-
нона-1,2 выше, чем для гидрохинона-1,4. Бензольное
кольцо, конденсированное с циклом д-бензохинона
(нафтохинон), уменьшает энергию системы и ослабляет
сопряжение в хинонном цикле вследствие включения
одной двойной связи в ароматич. сопряжение. В еще
большей степени этот эффект проявляется в молекуле
антрахинона. Поэтому в ряду n-бензохинон, нафтохи-
нон-1,4, антрахинон-9,10 наименьший нормальный
потенциал имеет антрахинон. Дифенохинон, у к-рого
сопряженная хиноидная система включает оба кольца,
характеризуется высоким значением Е° и является
намного более сильным окислителем, чем гс-бензохй-
нон. Высокие значения нормального потенциала ха-
характерны также для о-хинонов. Аналогичное влияние
заместителей наблюдается и в др. органич. окислитель-
окислительно-восстановительных системах, напр, на основе краси-
красителей.
Кинетика окислительно-восстановительных реак-
реакций. В отличие от реакций в мономерных окислительно-
восстановительных системах, скорость реакций в
О.-в. п. лимитируется диффузией реагирующих веществ
в глубину фазы полимера. С наибольшей скоростью
окислительно-восстановительные процессы протекают
в р-рах. В нерастворимых О.-в. п. они замедляются
с увеличением степени сшивки матрицы. Напр., при
окислении трехвалентным железом слабо- и сильно-
сшитых сульфогидрохинонных О.-в. п. время установ-
установления равновесия составляет соответственно около
2 и 7 ч.
Высокие кинетич. свойства слабосшитого полимера
обусловлены сильным набуханием гелевидной струк-
структуры, приводящим к ускорению диффузионных про-
процессов. Скорость окислительно-восстановительных реак-
реакций можно повысить путем создания слабонабухаю-
щих полимеров макропористой структуры, обеспечи-
обеспечивающих довольно легкий доступ реагирующих групп
к функциональным группам О.-в. п.
Кинетич. свойства О.-в. п. на ионитовых носителях
зависят гл. обр. от размера и конфигурации пор,
содержащих окислительно-восстановительную систему,
а также от общей пористости и проницаемости гелевид-
ных областей полимерной матрицы. Скорость реакций
в редокс-ионитах макропористой структуры выше,
чем в таких же ианитах гелевидной структуры. Незави-
Независимо от структуры ионита кинетич. свойства О.-в. п.
на его основе ухудшаются с увеличением степени сшив-
сшивки матрицы. Эти свойства О.-в. п. зависят также от их
435
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОЛИМЕРЫ
436
термостойкости: чем выше термостойкость, тем больше
скорость окислительно-восстановительных реакций.
Процессы окисления — восстановления можно ката-
катализировать добавлением в р-р мономерных хинонов,
ионов хлора и железа. Обычно о скорости реакций
судят по изменению потенциала О.-в. п. во времени.
Получение и свойства редокс-полимеров. Поликон-
Поликонденсацией синтезируют О.-в. п., в к-рых ковалентно
связанные органич. окислительно-восстановительные
системы (хиноны, красители, ферроцен) находятся
в основной цепи макромолекулы. Этим методом полу-
получают, напр., гидрохинон-формальдегидные полимеры.
Полимеризацией или методом полимераналогичных
превращений синтезируют гл. обр. полимеры, содер-
содержащие окислительно-восстановительные системы в бо-
боковых цепях. При полимеризации мономеров, обла-
обладающих окислительно-восстановительными свойствами,
особенно винилгидрохинонов, может проявляться их
ингибирующее действие на этот процесс, приводящее,
как правило, к образованию химически нестойких,
растворимых низкомолекулярных продуктов (димеров
и тримеров). С целью получения высокомолекулярных
соединений гидроксильные группы винилгидрохинонов
блокируют бензоатными, ацетатными, этоксильными
и др. группами.
О.-в. п., содержащие систему бензохинон/гидрохи-
нон, обладают недостаточной химстойкостью; более
химстойкие О.-в. и. получают при использовании
хинонов, в к-рых реакционноспособные атомы водорода
замещены метильными группами. Для повышения спо-
способности О.-в. п. к набуханию, а следовательно, для
увеличения скорости реакции окисления — восстанов-
восстановления синтезированные продукты сульфируют. При
этом они приобретают дополнительно свойства сильно-
сильнокислотных катионитов (см. Катионообменные смолы).
Введением при синтезе О.-в. п. специальных добавок
(инертных растворителей, или разбавителей), хорошо
совмещающихся с исходными мо-
мономерами, можно получать О.-в. п.
макропористой структуры. В
качестве разбавителей применя-
применяют изоамиловый спирт, олеино-
олеиновую к-ту, метилэтилкетон, нат-
натриевую соль о-бензамидосульфо-
кислоты, ZnCl2, СаС12-6Н2О,
МпС12 и др. Степень макропори-
макропористости О.-в. п. можно регулиро-
регулировать подбором соотношений моно-
мономера и разбавителя. Макропорис-
Макропористые О.-в.п. характеризуются повы-
повышенной химстойкостью и высоки-
высокими кинетич. свойствами в процес-
процессах окисления — восстановления.
Ниже описаны способы полу-
получения и свойства наиболее харак-
характерных представителей редокс-
полимеров (см. табл. 1).
Система хинон/гид-
р о х и н о н. Синтез большого
числа полимеров, содержащих
эту окислительно-восстановитель-
окислительно-восстановительную систему, обусловлен лег-
легкостью ее введения в макромоле-
макромолекулу и способностью системы не-
необратимо восстанавливаться в со-
соответствующие гидрохинонные
формы. Кроме того, окислительно-
восстановительные свойства моно-
мономерных хинонов хорошо изучены,
а стандартные окислит, потенциа-
потенциалы системы можно варьировать в
широких пределах путем введения
соответствующих заместителей.
н
N-
Макромолекулы наиболее важных О.-в. п. это-
этого типа, получаемых поликонденсацией, содержат
звенья I, II (М, М', М"= — О —, —S —, — NH—,
—СН2—, — СН = СН— и другие):
Эти полимеры — порош- Он он
ки темно-коричневого или / I мг
черного цвета. При вое- / \_м_
становлении они перехо- \ /
дят в растворимую гидро- / |
хинонную форму, что по- ^н | ОН
зволяет отделять их от
образующихся при синтезе сшитых продуктов. Поли-
Полимеры устойчивы к термич. и термоокислительной дест-
деструкции до 300 °С.
ОН
•NH—M—NH-
II
~R—/^—R-CO—R—COO -
\ it/
б. м
IV
1 ЯнсТ с!
оо
Конденсацией n-бензохинона или его производных
с жирными, жирноароматич. и ароматич. диаминами
или с солями соответствующих бис-диазотированных
ароматич. диаминов получены полиариленаминохиноны
а. Аг =
б. Аг =
N
—С-
I—N
-N
Н
IX
-"Л
а. X=-NH—
б. Х=— S—
Аг =
м
М=—О—1 SO2 или отсутствует
437
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОЛИМЕРЫ
438
Таблица 1. Свойства нек-рых редокс-полимеров
Полимер*
Нормальный
потенциал
Е°, в
slss л-
Система хино н/г
Полигидрохиноноксид
Полигидрохинонамин
Полигидрохинонметилен
Поли-2,5-диоксиксилилен ....
Полидифениленгидрохинон ....
Полигидрохинондиоксин
Полигидрохинонтиоксин
Полигидрохинонтиазин
Полифениленаминогидрохинон . .
Полинафтоиленаминогидрохинон
Лолигидрохинонфенодитиазин . . .
Продукты поликонденсации фор-
«.п ттт» ТТОТ^ТЖ 7TQ
МсШЬДС! И Да
гидрохинон-фенол-формальде-
гидрохинон-фенол-формальдегид A : 1 : 2)
гидрохинон-резорцин-формаль-
гидрохинон-резорцин-формальдегид A:1:2)
пирокатехин-фенол-формальде-
пирокатехин-фенол-формальдегид A : 0,5 : 1,5)
п ирога л л о л -фенол -формал ь де -
гид A : 1 : 3)
юглон-фенол-формальдегид
A:1:3)
хризазин-фенол-формальдегид
A • 1 • 4)
Поли-4-винил-2', 5'-диоксидифе-
нилсульфона
ПоливинилB,5-диоксифенил)-
сульфона
Поли-4-винил-2', 5' -диоксидифе-
нил (производное полистиролаN
Поли-3,4-диоксистирол (производ-
(производное поли-п-аминостиролаN . . .
идрохинон
0,585-0,594
0,552
0,628-0,730
~0,730
—
0,525-0,544
0,520
0,473
0,615
0,570
0,539
0,695 — 0,710
-0,700
0,727
0,680-0,745
0,528
0,753
0,890
Система на основе красителе
Поли-2-винилфентиазина
Полииндиго
Политиоиндиго
Поли-6-винилизатин (производное
полистиролаN
Поли-6-винилиндирубин (производ-
(производное полистиролаN
ПолиF-винилиндол-3)-(тионаф-
тен-2)индиго (производное поли-
полистиролаN
Поли-4-винил -4',4"-бис-(диметил-
амино)трифенилкарбинола . . .
0,420-0,430
0,098
0,166
0,372
0,400
-0,210
8,3-10,6
11,7
14-16
14,75
0,95
9,4-10,0
9,9
9,9-10,0
13,17
11,60
9,8-12,4
6,5-7,0
7,19
9,0-9,5
5,5-7,7
4,5
2,5
7,24
4,08
8,77
7,0-9,5
6,82
5,85
4,2
4,5
4,2
1,25
Система сульфгидри л/д исульфид
Поли-п-тиол стирол а
То же (производное полистиролаN
Поли(п-тиометил)стирол(производ-
ное полистиролаN
Поливинилтиола 0,37—0,41
То же (производное поливинилового
спиртаN
Система на основе ферроцена
Производное поли-п-аминости-
,6
46
рола6
0,415
3,6-7,0
4,5-5,0
5,4
* Полимеризационные полимеры обозначены индексом «а»,
полученные методом полимераналогичных превращении — ин-
индексом «б».
(напр., Ilia и IIIб) — темные порошки, полностью
растворимые лишь в конц. H2SO4. Восстановленная
водорастворимая форма полимеров окисляется кисло-
кислородом воздуха и выпадает при этом в осадок. Введение
в макромолекулу сульфо- или карбоксильных групп
придает полимерам растворимость в диметилформамиде
и в водных р-рах щелочей.
Поликонденсацией д-бензохинон-2,5-диолов и бис-
ацилхлоридов (напр., оксалил-, сукцин-, себацоил-,
изофталоил- и терефталоилхлоридов) получают раство-
растворимые в органич. растворителях полиэфиры IV. Вос-
Восстановленные полимеры, напр, гидросульфитом натрия
или водородом на палладиевом катализаторе, раство-
растворяются значительно лучше, чем окисленные. Взаимо-
Взаимодействием 7,7'-дипиридила с хлоранилом или терефта-
лонитрилоксида с д-бензохиноном получают соответст-
соответственно полимеры V и VI.
К поликонденсационным О.-в. п., содержащим антра-
хиноновые циклы и обладающим высокой термич. ста-
стабильностью, относятся полипирроны VII и VIII,
полибензимидазолы IX, Ха, полибензтиазолы Хб и
полихиноксалины XI. Полимеры VII м. б. получены
из 1,2,5,6-тетраминоантрахинона и пиромеллитового
диангидрида, VIII — из 1,2,5,6-бис-(а,р-дикарбоксил-
пиразино)антрахинона и тетраминобензола или 3,3'-
диаминобензидина, IX — из 1,2,5,6-тетраминоантрахи-
1,2,5,6-тетраминоантрахинона и изо- или терефталевого альдегида (или хлор-
ангидрида), Ха и Хб — из дифенил-1,5-антрахинонди-
карбоксилата и соответственно 3,3'-диамино- или 3,3'-
димеркаптобензидина, XI — из 1,2,5,6-тетраминоантра-
1,2,5,6-тетраминоантрахинона и 2,3,7,8-тетрахлор-1,4,6,9-тетразаантрацена.
Хим- и термостойкие набухающие в воде продукты
получают сульфалкилированием полимеров или сопо-
сополимеров винилзамещенных хинонов с аннелированными
пиразольными циклами (полипиразолохиноны).
Синтез О.-в. п., содержащих систему хинон/гидро-
хинон, методом полимераналогичных превращений
обычно ведут по схеме:
~СН—СН2~
HNO3
Sn+HCl
NH2
~СН—
-он
В качестве «инертных» полимеров используют поли-
поливиниловый спирт, поливинилацетат, поли(а-гидрокси-
метил)акриламид, полистирол, сополимер стирола
с малеиновым ангидридом и хлорметилированный
сополимер стирола с дивинилбензолом, к-рые обраба-
обрабатывают n-бензохиноном и его производными, 1- или
2-аминоантрахиноном и 2-формил- или метоксиантра-
хиноном.
Системы на основе красителей. О.-в. п.,
содержащие эти системы, представляют особый инте-
интерес, т. к. переход полимеров из окисленной (нераство-
(нерастворимой) в восстановленную (растворимую) форму сопро-
сопровождается резким изменением окраски, что позволяет
визуально наблюдать за процессом окисления — вос-
восстановления. Путем различных химич. превращений
из линейного или сшитого полистирола (монолитной
и пористой структуры) получены поли-6-винилизатин,
поли-6-винилиндирубин, полиF-винилиндол-3)-(тио-
нафтен-2)индиго — блестящие тонкодисперсные по-
порошки красного или красно-фиолетового цвета. Син-
Синтезированы О.-в. п., содержащие в качестве обратимой
системы красители ди- и трифенилового ряда. К ним
относятся, напр., продукты взаимодействия поли-д-ами-
ностирола с фуксином, метилвиолетом или кристалл-
виолетом, сополимер стирола с малеиновым ангидри-
ангидридом, обработанный 4-амино-4',4"-бис-диметиламинотри-
фенилметаном.
Система сульфгидрил/дисульфид
Полимеры, макромолекулы к-рых содержат сульфгид-
439
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОЛИМЕРЫ
440
рильные (тиольные) группы, синтезированы поликон-
поликонденсацией тиофена, окситиофена или аминотиофена,
полимеризацией n-винилфенилтиоацетата и др. Хим-
стойкий полимер получен обработкой хлорметилиро-
ванного сополимера стирола с дивинилбензолом тио-
мочевиной и спиртовым р-ром щелочи. Большое число
таких полимеров синтезировано на основе поливини-
поливинилового спирта.
Системы на основе пиридина. При-
Примером таких О.-в. п. может служить полимер XIII,
к-рый получают двукратной последо-
последовательной обработкой гомополимера
2-винилпиридина или его сополимера с
дивинилбензолом сначала спиртовым
р-ром СН31, а затем спиртовым р-ром
щелочи до полного удаления ионов 1~.
О.-в. п., содержащие дигидропири-
диновые циклы в боковой цепи ма-
макромолекулы, получают кипячением
хлорметилированного сополимера стирола и дивинил-
дивинилбензола с безводным пиридином или никотинамидом
~СН—СН2~
XIV
с последующим восстановлением водным р-ром дитио-
нита и карбоната натрия до дигидропроизводных. Эти
О.-в. п. восстанавливают д-бензохинон, тионин, ме-
тиленовый голубой и малахитовый зеленый.
Система на основеферроцена. По-
Поликонденсацией ферроцена с различными алифатич. и
ароматич. альдегидами синтезированы растворимые
полимеры XIV с мол. массой до 5000. Полимеризацией
винил ферроцена, а также сополимеризацией его со сти-
стиролом, метилметакрилатом, хлоропреном или винил-
изобутиловым эфиром (напр., в присутствии катализа-
катализаторов Фриделя — Крафтса) получены О.-в. п. XV,
нерастворимые в обычных полярных растворителях,
с максимальной мол. массой 3500.
Получение и свойства редокс-ионитов. Перенос элект-
электронов редокс-ионитами, окислительно-восстановитель-
окислительно-восстановительная система к-рых находится в состоянии низшей
валентности, протекает по схеме:
SO3H окисление
Г SO3H1
Ка<
Же71
Ka-(SO3)n+1 Me
где Ка — матрица катионита, Me — металл.
В случае поглощения кислорода в реакции участвует,
по-видимому, гидроокись металла, к-рая образуется
при гидролизе катионита. При сорбции ионитом орга-
нич. окислительно-восстановительных групп процесс
идет по схеме:
окисление
RH > R+ + Н+ + е-
где RH — органич. восстановитель, поглощенный ка-
тионитом.
Синтетические редокс-иониты. Ти-
Типичный пример получения этих О.-в. п. (см. табл. 2) —
поликонденсация гидрохинона с фенолсульфокислотой
и формальдегидом (мол. соотношение 1:1:2). Наи-
Наиболее химстойкий и реакционноспособный редокс-
ионит получают из 4-пирогаллолкарбоновой к-ты, а так-
также из винилгидрохинонов. В последнем случае поли-
полимер выдерживают в дихлорэтане, обрабатывают хлор-
сульфоновой или конц. серной к-той, а затем подвер-
подвергают гидролизу и обработке р-ром КС1. Полученный
Таблица 2. Свойства нек-рых синтетических редокс-ионитов
Полимер*
Поли-ди(сульфофенилен)-
гидрохинон
Поликонденсационный по-
полимер 77'-ДипиРиДшт с
хлоранилом
Поликонденсационный по-
полимер гидрохинона с
фенолсульфокислотой и
формальдегидом A : 1 : 2)
Сополимер винилгидрохи-
нона со стиролом (суль-
(сульфированныйK
Полигидрохинонсульфон
(сульфированный)8 . .
Сополимер 2-винилантра-
хинона со стиролом
(сульфированный)а . . .
Полипиразолохинона
Поли(п-хлорметил)сти-
рол, обработанный тио-
нином (сульфирован-
(сульфированныйN
Поли-6-винилиндирубин
(сульфированныйN . . .
Поли-2 -винил-Ы-метил-
пиридон6
монолитный
пористый ... о ...
Нормальный
потенциал
Е°, в
0,751
—
^0, 700
0,50-0,52
(рН =0,8)
-0,175
-0,130
0,375
—
Окислитель-
но-восстано-
но-восстановительная
емкость,
мг-эпв/г
2,8
2,1
8,9
1,88-2,14
3,58
4,75
5,0
4,0-4,2
E0 СС)
4,0-4,1
5,13
3,15
Ионооб-
М6ННЗ.Я РМ—
ITXVslX 1 АСА./J. V^ITJ.
КОСТЬ,
мг-.тв/г
3,5
3,0
0,9
3,41 -3,56
2,85
2,02
2,65-3,36
—
5,07
5,05
* См. примечание к табл. 1.
Таблица 3. Свойства нек-рых медных редокс-ионитов
на основе ионитовых носителей
Марка носителя
Ионоген-
ные груп-
группы
Содержание
меди*
г/л
мг-экв/л
Ионо-
Ионообменная
емкость,
мг-экв/л
Редок с-и ониты наоснове
полимеризационных катионитов
Гелевидные
КУ-2-2 (слабости -
тый)
КУ-2-8
КУ-2-2 0 (сильно-
сшитый)
Макропористые* *
КУ-23 (8/60) .
КУ-23 C0/60).
КУ-23 A5/40) .
КБ-41 B0/50) .
КФ-11 C0/60).
-SO3H
-SO3H
-SO3H
-SO3H
-SO3H
-SO3H
-соон
-РО3Н2
86
141
229
90
164
203
210
294
2690
4420
7160
2810
5130
6370
6570
9200
Редок с-и ониты на основе
поли конденсационных ионитов
Катиониты
КУ-1
КУ-6
КУ-36 . . .
Анионит АН-31
-SO3H;
-ОН
-SOSH
-SO3H
=N; =NH
240
221
175
161
7500
6920
5480
5030
990
1290
1870
1900
1360
1740
2120
1200
650
460
610
1000
* Образцы получены в результате 10-кратного осаждения
меди. ** Первая цифра в скобках — содержание дивинилбензола,
вторая — содержание инертного растворителя, введенного при
синтезе макропористой модификации: например, катионит
КУ-23 (8/60) содержит 8% дивинилбензола и 60% растворителя.
таким способом полимер приобретает свойства катио-
катионита. Окислительно-восстановительными свойствами
обладают, напр., поликонденсационные катиониты,
к-рые синтезируют из формальдегида и фенолсульфо-
кислоты (КУ-1) или нафталинсульфокислоты (КУ-5),
а также аниониты, получаемые поликонденсацией
полиэтиленполиаминов, фенола и формальдегида
(АН-2Ф), триметилолмеламина и формальдегида (АН-1)*
мочевины, мел амина, гуанидина и формальдегида.
441
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ПОЛИМЕРЫ
442
Редокс-иониты на основе ионито-
вых сорбентов. Для введения окислительно-
восстановительной системы ионообменный полимер
обрабатывают р-рами органич. соединений с окислитель-
окислительно-восстановительными свойствами или р-рами солей
металлов переменной валентности. Большой ассорти-
ассортимент таких редокс-ионитов создан на основе промыш-
промышленных марок ионитов. Они получены, напр., сорбцией
ионов Sn2+, Gr3+, Ti3+, метилвиолета, фуксина на ка-
тионитах, гидрохинона, дигидрохлоранила на аниони-
тах или ионов S2O3~, SO32~ и др. на сильноосновных
анионитах. Известен также способ получения редокс-
ионита насыщением анионита, содержащего сильно-
и слабоосновные ионогенные группы (в соотноше-
соотношении 1:1) аммиачным р-ром гидросульфата натрия.
Существенный недостаток редокс-ионитов на основе
сорбентов (этот недостаток в большей или меньшей
степени присущ всем О.-в. п.) — постоянный, обуслов-
обусловленный гидролизом переход окислительно-восстанови-
окислительно-восстановительных ионов в р-р. Редокс-иониты с улучшенными
свойствами получают на основе сильнокислотных ка-
тионитов типа КУ-2, КУ-23 и сильноосновных аниони-
тов типа АВ-17 и АВ-170.
Изменяя условия обработки ионитов р-рами окисли-
окислительно-восстановительных соединений, можно регули-
регулировать реакционную способность редокс-ионита. Так,
при полном насыщении ионогенных групп окислитель-
окислительно-восстановительными ионами получают полимеры,
обладающие только окислительно-восстановительными
свойствами, при неполном насыщении — как окисли-
окислительно-восстановительными, так и ионообменными
свойствами. Сорбцией окислительно-восстановитель-
окислительно-восстановительных ионов на хим- и термостойких комплексообразую-
щих ионитах могут быть созданы О.-в. п., к-рые можно
использовать для одновременного проведения процессов
гетерогенного окисления — восстановления, комплек-
сообразования и ионного обмена.
Системы на основе ионитовых носите-
носителей. Получение этих редокс-ионитов (табл. 3) состо-
состоит из след. стадий:
1. Обработка катионита р-ром соли металла — восста-
восстановителя с целью перевода катионита в солевую форму:
Ка
SO3H
so3
CuSO4 -> Ka\ >Cu + H2SO4
4SO/
2. Вытеснение поглощенного иона реагентом, вос-
восстанавливающим ион в момент вытеснения:
.SO, Г SO3Nal
Ка; NCu + NaSO + 2NaOH > KaC
Na2S2O4 + 2NaOH -> KaC -Cu + 2NaHSO3
xSO3Na.J
редокс-ионит
3. Перевод носителя в Н-форму:
SO3H"|
а( • Си + Na2SO4
SOH J
SO3H J
Г SO.Na]
|Ка< .Cu + H2SO4-
L \SO3NaJ
При многократном повторении двух первых стадий
обработки получают редокс-иониты с большой восста-
восстановительной емкостью. О.-в. п. на ионитовых носите-
носителях м. б. получены на ионитах различных классов
и типов, независимо от химич. состава матрицы, при-
природы и заряда ионогенных групп. Их окислительно-
восстановительная и ионообменная емкость, а также
кинетич. и др. свойства определяются особенностями
структуры носителя. Так, термостойкие, высокоемкие
и обладающие хорошими кинетич. свойствами редокс-
иониты получают, напр., на основе макропористого
катионита КУ-23. При осаждении на этом катионите
до 200—250 г/л меди скорость ионного обмена почти
не изменяется.
При введении окислительно-восстановительной си-
стедоь напр. металлич. меди, ускоряется термогидро-
лиз (десульфирование) О.-в. п. на основе любых катио-
нитовых носителей. Однако по мере окисления металла
происходит миграция образующихся ионов Си2+ и
Си+ к сульфогруппам катионита:
Cu2+ + 2KaSO3H-->>(KaSO3JCu + 2Н2О
что сопровождается резким замедлением десульфиро-
вания.
Регенерация. Способ регенерации О.-в. п. зависит
от их назначения. Полимер, служащий восстановите-
восстановителем, регенерируют восстановителем, используемый
в качестве окислителя,— окислителем. Регенерация про-
протекает тем быстрее, чем больше разность между нор-
нормальными потенциалами О.-в. п. и регенерирующего
агента, напр, окисленного полимера и восстановителя.
О.-в. п., особенно используемые в пром-сти, должны
иметь потенциал, к-рый позволяет восстанавливать их
с максимальной скоростью при использовании деше-
дешевого восстановителя. Чаще всего для этой цели при-
применяют воднощелочной р-р Na2S2O4, р-ры TiCl3 в H2SO4,
KI в НС1 или в H2SO4, р-р Сг(СН3СООJ, тиогликоле-
вую к-ту. Такие доступные восстановители, как Na2SO3,
NaHSOg, H2SO3 и др. для регенерации О.-в. п. непри-
непригодны из-за относительно высоких значений их потен-
потенциала. Регенерация редокс-полимеров не вызывает
обычно особых затруднений.
При регенерации редокс-ионитов, особенно содержа-
содержащих в качестве окислительно-восстановительной систе-
системы ионы металлов или их окислов, должны быть пра-
правильно подобраны как восстановитель, так и реагент
для регенерации ионообменных групп. Оптимальные
условия процесса устанавливают обычно опытным
путем. Напр., при регенерации медного редокс-ионита
на основе катионита КУ-2 сначала восстанавливают
окислительно-восстановительные группы, а затем реге-
регенерируют ионообменные:
Г
L
SO3Na]
Ка\ -Cu(OHJ
XSO3NaJ
Г SO3Na]
2S2O4-H Ка\ -Си + NaHSO3
L XSO3NaJ
Ka( -Си + H2SO4 -> Ка
SO3H
SO3H
•Си + Na2SO4
Процесс проводят в хроматографич. колонке с целью
удаления р-ров продуктов, образующихся в ходе этих
реакций.
В нек-рых случаях, напр, при регенерации редокс-
ионитов на основе катионитов — носителей, содержа-
содержащих систему Fe2+ /Fe3+ , полимер обрабатывают смесью
Na2S2O4 со щелочью, что позволяет регенерировать
одновременно обе функциональные системы О.-в. п.
После регенерации редокс-ионит тщательно отмывают
в хроматографич. колонке от образующихся продуктов
обескислороженной водой. Подбор и соблюдение опти-
оптимальных условий регенерации особенно необходимы
при регенерации редокс-ионитов на основе сорбентов
во избежание вытеснения в р-р окислительно-восста-
окислительно-восстановительных ионов.
Применение. Наиболее широкое применение среди
О.-в. п. нашли редокс-иониты на основе носителей,
содержащие в качестве окислительно-восстановитель-
окислительно-восстановительной системы ионы металлов переменной валентности.
Так, хим- и термостойкие редокс-иониты на основе
макропористых катионитов, содержащие систему
Cu/Cu2+ и обладающие высокой способностью к погло-
поглощению кислорода, используют в атомной энергетике
и в теплоэнергетике при обескислороживании воды,
предназначенной для котлов высоких и сверхвысоких
параметров, а также при обескислороживании техно-
логич. воды в производстве полистирола, поливинил-
хлорида и др. Перспективно применение этих редокс-
ионитов для глубокой очистки азота от микропримесей
кислорода, углеводородов — от примесей сероводорода
443
ОКИСНОМЕТ АЛ Л ИЧ ЕСКИЕ КАТАЛИЗАТОРЫ
444
и др. соединений перед их подачей на каталитич. син-
синтез. Возможно также применение редокс-ионитов в про-
производстве винилацетата для очистки уксусной к-ты от
активного кислорода и др. химич. продуктов.
Синтетич. О.-в. п. применяют ограниченно из-за их
невысоких кинетич. характеристик и низких химстой-
кости и механич. прочности. О.-в. п. с низким Е°
(напр., поливинилантрахинон, поливинилиндирубин)
используют для получения перекиси водорода. В этом
случае восстановленный полимер окисляют воздухом
или кислородом; образующуюся Н2О2 извлекают водой
(выход 85% от теоретич.). Для удаления перекисей
из р-ров органич. соединений исходный р-р пропускают
через слой сильнокислотного катионита, содержащего
систему Fe3+/Fe2+, или через слой редокс-полимера,
содержащего краситель, напр, метиленовый голубой.
Редокс-полимеры используют для селективного вос-
восстановления различных катионов (Ag+, Au+, Hg2+,
Pb2+, Bi3+, Pt4+, V6+ и др.) и последующего раз-
разделения восстановленных металлов. Напр., при мно-
многократном пропускании р-ра, содержащего ионы Ag+,
через слой гидрохинон-формальдегидного полимера
образуется металлич. серебро, к-рое в результате
электрохимич. десорбции переходит в р-р и выделяется
на катоде в виде порошка чистого металла.
О.-в. п. можно применять для избирательного вос-
восстановления или окисления альдегидов соответственно
до спиртов или кислот; для удаления перекисных или
др. легко восстанавливающихся веществ из смазочных
масел, для дегалогенирования водных р-ров, напр,
дехлорирования питьевой воды. Фильтровальную бу-
бумагу, пропитанную бесцветным полимером с различ-
различными красящими окислительно-восстановительными
компонентами, используют в качестве индикаторной.
О.-в. п. применяют как модельные системы для изу-
изучения ферментов и различных биохимич. окислительно-
восстановительных процессов; в медицине — для за-
защиты от действия радиации, в качестве ингибиторов
роста вирусов и как противоязвенные препараты и
кровезаменители, обладающие анти окислительными
свойствами. Опытами на мышах установлено антилей-
кемическое действие полисульфофениленхинонов.
Гидрохинон-формальдегидные полимеры в окислен-
окисленной форме — катализаторы полимеризации винил ьных
мономеров, а в восстановленной — ингибиторы этого
процесса, проявляющие антиокислительные свойства.
Полисульфофениленхиноны обладают каталитич. актив-
активностью в процессах разложения гидразина и перекиси
водорода; полигидрохинонметилены. — ингибиторы
окисления нек-рых органич. соединений, напр, изопро-
пилового эфира. Полисульфофениленхиноны исполь-
используют также в качестве катализаторов полиэтерифика-
ции в синтезе олигоэфиракрилатов.
О.-в. п. применяют как высокотемпературные ста-
стабилизаторы термич. и термоокислительной деструкции
нек-рых промышленных полимеров. Так, хинонсодер-
жащие полимеры (полиариленхиноны, полихинондиок-
сины, полихинонтиазины, полихинонамины, полихи-
нонтиоксины, полиариленаминохиноны и др.) — эф-
эффективные стабилизаторы деструкции полиарилатов,
поликарбонатов, поливинилхлорида и др. полимеров
при темп-pax до 350—400 °С. Напр., примесь NaCl
(до 1,5—2%) в промышленных поликарбонатах значи-
значительно ускоряет деструкцию полимера при высоких
темп-pax, что объясняется, по-видимому, взаимодейст-
взаимодействием NaCl с концевыми фенольными гидроксилами
макромолекулы. Выделяющийся при этом НС1 ката-
катализирует гидролиз эфирных связей в поликарбонате.
В присутствии полихинонов в окисленной форме (~ 2%
от массы поликарбоната) НС1 образует с атомами
О, S, N О.-в. п. соответственно ионы оксония, суль-
фония и аммония. Особенно высокие ингибирующие
свойства имеют полихиноны лестничной структуры.
Электроды, приготовленные нанесением на пластин-
пластинку пленки гидрохинон-формальдегидного полимера,
предварительно окисленного на 50%, можно приме-
применять для измерения рН. Возможно использование
О.-в. п. в качестве диэлектриков в конденсаторах
и для гальванич. элементов.
Лит.: Кассиди Г. Дж., Кун К. А., Окислительно-
восстановительные полимеры (редокс-полимеры), пер. с англ.,
Л., 1967; Кожевников А. В., Электроноионообменники,
Л., 1972; Гельферих Ф., Иониты, пер. с нем., Л., 1962,
с. 468; Салдадзе К. М., Химически активные полимеры
и их применение, Л., 1969; Салдадзе К. М., Паш-
Пашков А. Б., Титов В. С, Ионообменные высокомолеку-
высокомолекулярные соединения, М., 1960; ЕргожинЕ.Е., Шо-
стак Ф. Т., Усп. хим., 34, в. 12, 2220 A965); Салдад-
Салдадзе К. М., Лукьянова Н. Л., в сб.: Успехи хромато-
хроматографии, М., 1972; Manecke G., Redoxharze, в кн.: Houben-
Weyl, Methoden der organischen Chemie, Bd 1, Tl. 1, Stuttgart,
1958, S. 601; StackelbergM., Elektrochemische Poten-
tiale organischer Stoffe, там же, Bd 3, Tl. 2, Stuttgart, 1955
S. 255. К. М.Салдадзе, А.А.Гуров, Г. К. Салдадзе.
ОКИСНОМЕТАЛЛИЧЕСКИЕ КАТАЛИЗАТОРЫ
полимеризации (metal oxide catalysts, Metal-
loxidkatalysatoren, catalyseurs des oxydes metalliques).
Полимеризация этилена и др. олефинов, а так-
также диенов до высокомолекулярного полимера в при-
присутствии катализаторов, представляющих собой пере-
переходные металлы или их окислы на носителях, была от-
открыта в начале 50-х годов XX в. Из катализаторов этого
класса наиболее изучены окиснохромовый, окисномо-
либденовый и никелевый.
Окиснохромовый катализатор (ина-
(иначе — катализатор фирмы «Филлипс петролеум») — са-
самый активный среди О. к. В его присутствии этилен
с высокой скоростью полимеризуется до высокомолеку-
высокомолекулярного полиэтилена. Полимеризация пропилена про-
протекает со скоростью примерно на 2 порядка ниже, чем
полимеризация этилена; основным продуктом (свыше
70%) является атактич. полимер. При полимеризации
на этом катализаторе бутадиена и изопрена получают-
получаются полимеры с 1,4-трawe-структурой. О производстве
полиэтилена на окиснохромовом катализаторе см. Эти-
Этилена полимеры.
Существует большое число методов приготовления
окиснохромового катализатора, однако во всех случаях
в состав его входят соединения шестивалентного хрома.
Наиболее активные катализаторы получаются при при-
применении в качестве носителей силикагеля или алюмо-
алюмосиликатов с низким (до 10%) содержанием А12Оз.
В качестве носителей предложены также окислы А1,
Th, Zr, Ti, Ge, но катализаторы на этих носителях мало-
малоактивны и не находят промышленного применения. Но-
Носитель для приготовления активных О. к. полимериза-
полимеризации должен обладать невысокой прочностью, чтобы
катализатор легко дробился в процессе полимеризации.
Дробление приводит к увеличению работающей поверх-
поверхности катализатора. Для того чтобы получить катали-
катализатор невысокой прочности, целесообразно использо-
использовать носители с большим объемом пор: силикагели и
алюмосиликаты с объемом пор не менее 1 см3/г и с уд.
поверхностью >300 м2/г. Оптимальный состав, тип но-
носителя и способ приготовления окиснохромового ката-
катализатора полимеризации этилена связаны с условиями
его применения. Однако во всех случаях можно выде-
выделить основные стадии приготовления катализатора:
1. Нанесение соединений хрома. Способ нанесения за-
зависит от типа используемого носителя: для относитель-
относительно прочных носителей применима пропитка водными
р-рами хромовой к-ты; для широкопористых носителей
с целью сохранения пористой структуры рекомендуется
пропитка р-рами СгО3 в органич. растворителях или на-
нагревание носителя с растертым хромовым ангидридом.
2. Сушка катализатора (если нанесение хрома прово-
проводилось из водных или органич. р-ров).
3. Активация катализатора, в процессе к-рой проис-
происходит удаление с поверхности катализатора кислорода
445
ОКСИСТИРОЛОВ ПОЛИМЕРЫ
446
и воды, являющихся каталитич. ядами. В ходе актива-
активации также образуются поверхностные соединения (ак-
(активный компонент катализатора), при последующем
взаимодействии к-рых с реакционной средой возникают
центры роста. Первый этап активации может включать
прокаливание катализатора в токе сухого воздуха при
400—700 °С с последующей продувкой азотом или на-
нагреванием в вакууме. Для повышения активности ка-
катализатора, особенно при его использовании в суспен-
суспензионной полимеризации (при < 90 °С), м. б. проведен
второй этап активации — восстановление катализа-
катализатора. Наиболее активные катализаторы получаются
при использовании в качестве восстановителя осушен-
осушенной окиси углерода. Восстановление проводится при
300—500 °С с последующим удалением СО откачкой или
продувкой.
Используемые в пром-сти катализаторы могут содер-
содержать 0,5—3% хрома. С уменьшением концентрации хро-
хрома активность, отнесенная к 1 г хрома, повышается.
Как и в общем случае полимеризации на гетерогенных
катализаторах, активность окиснохромового катали-
катализатора А пропорциональна поверхностной концентра-
концентрации центров роста 7V, работающей поверхности катали-
катализатора S и активности одного активного центра (или
константе скорости роста ир): A~kpNS. В случае при-
применения активных окиснохромовых катализаторов
скорость полимеризации этилена, как правило, изме-
изменяется в ходе процесса, проходя через максимум, что
связывается с изменением числа центров роста — об-
образованием их в начальный период из активного компо-
компонента катализатора и последующей дезактивацией при-
примесями. Максимальная скорость полимеризации про-
пропорциональна концентрации мономера. Энергия акти-
активации реакции роста —17 кдж/молъ (~4 ккал/молъ).
При изменении носителя &р возрастает в ряду
А12О3 < TiO « ZrO2 < SiO2
При полимеризации диенов на окиснохромовом ка-
катализаторе образуются живущие полимеры.
Окиснохромовый катализатор относится к одноком-
понентным катализаторам полимеризации олефинов,
в к-рых образование центров роста не требует обра-
обработки соединения металла переменной валентности ме-
таллоорганич. соединением. Существует точка зрения,
согласно к-рой активный компонент окиснохромового
катализатора — поверхностные соединения хрома в сте-
степени окисления 6 (предположительно поверхностные
хроматы). Образование центров роста связано с восста-
восстановлением до соединений, в к-рых степень окисления
ионов хрома не превышает 3. При алкилировании моно-
мономером координационно ненасыщенных ионов хрома
в низкой степени окисления происходит образование
активной связи металл — углерод.
Согласно другой точке зрения, основанной на сопо-
сопоставлении активности катализаторов с их спектрами
ЭПР, особую роль в полимеризации играют пятива-
пятивалентные ионы хрома. Предложен также след. состав
центров роста окисных катализаторов:
^>м-он L
°^>М—о—Me
где М — катион носителя, Me — металл переменной ва-
валентности, L — лиганд, к-рый может входить в состав
центра роста при восстановительной активации.
Окисномолибденовый катализа-
катализатор получается нанесением окиси молибдена на А12Оз
с последующим восстановлением водородом при
—500 °С. Рекомендуется использовать этот катализатор
в сочетании с металлоорганич. соединениями или гид-
гидридами щелочных и щелочноземельных металлов.
Окисномолибденовый катализатор применяется для
получения полиэтилена высокой плотности полиме-
полимеризацией этилена в интервале темп-р 70—300 °С при
давлениях 1 — 7 Мн/м2 A0—70 кгс/см2).
Никелевый катализатор представляет
собой никель, нанесенный на активированный уголь;
используется для полимеризации этилена при 70—
150 °С и давлении <5 Мн/м2 (<50 кгс/см2).
Лит.: Clark A., Catalysis Rev., 3, 145 A969); Ерма-
ковЮ. И., Окиснохромовые катализаторы глубокой полиме-
полимеризации, Новосибирск, 1969; Долгоплоск Б. А. Высоко-
мол. соед., 9А, № 7, 1602 A967); Ермаков Ю. И., За-
Захаров В. А., Усп. хим., 41, 377 A972). Ю. И. Ермаков.
ОКСИСТИРОЛОВ ПОЛИМЕРЫ (polyoxystyrene,
Polyoxystyrol, polyoxystyrene). .
Оксистиролы, винилфенолы (О.) СН=СН2
2- и 4—О.— бесцветные кристаллич. ве- ^-L
щества, легко полимеризующиеся и легко | л ои
окисляющиеся на воздухе с изменением \^
окраски; З-О. — бесцветная, легко поли-
меризующаяся, вязкая жидкость. Их свойства приве-
приведены в табл. 1.
Таблица 1. Некоторые свойства оксистиролов
A мм рт. ст. = 133,322 при2)
Показатели
Темп-pa кипения, °С(при
1 мм рт. ст.)
Темп-pa плавления, °С . .
Плотность, г/см3
Показатель преломления
-Б
2-Оксисти-
рол
100
29,0—29,5
1,029
(переохл.)
1,5808
З-Окси-
стирол
75,5-77*
—
1,035
1,5812
4-Окси-
стирол
—
71,5-72
—
—
* При 2 мм рт. ст.
2-О. получают декарбоксилированием 2-оксикорич-
ной к-ты при 260 °С A5 мм рт. ст.) или пиролизом
2,4-диметил-1,3-бенздиоксана. З-О. синтезируют ка-
каталитич. (Сг/А1) дегидрированием 3-этилфенола при
500—600 °С, дегидратацией 3-оксифенилметилкарбино-
ла или декарбоксилированием 3-бензоилкоричной ки-
кислоты с последующим омылением З-бензоилоксистирола.
4-О. получают дегидратацией 4-ацетоксифенилмети.л-
карбинола до 4-ацетоксистирола, к-рый подвергают
омылению.
Полиоксистиролы — полимеры общей формулы I.
П.— твердые .продукты белого цве-
цвета; мол. масса -10 000—70 000; быст-
быстро темнеют на воздухе в результате
окисления; растворимы в метиловом и
этиловом спиртах, ацетоне, метилэтил-
кетоне, тетрагидрофуране, диоксане,
—СН—СН2-
ОН
щелочи; нерастворимы в
диэтиленгликоле, в водной
воде, бензоле, СНС13, СС14.
П. вступают во все реакции, характерные для сое-
соединений, содержащих фенольный гидроксил, напр,
окрашиваются при действии солей железа, образуют
феноляты при реакции с водной щелочью, метилируют-
метилируются, ацетилируются. О. сополимеризуются с метилакри-
латом, стиролом, метил-
Таблица 2. Константы Алфрея
и Прайса для сополимеризации
оксистиролов с метилметакри-
латом
Оксистирол
метакрилатом (табл. 2).
П. получают ради-
радикальной полимеризацией
О. в присутствии азо-
бис-и зобутирони трила
или перекиси бензоила,
под действием УФ-облу-
чения и при нагревании
без инициатора. 4-О. по-
лимеризуется уже при
плавлении. Полимери-
Полимеризация сопровождается передачей цепи на мономер.
При полимеризации в одинаковых условиях из
3- и 4-О. образуются П., мол. масса к-рых в 2—
2-Оксистирол
3-Оксистирол
4-Оксистирол
1,15
1,02
1,14
— 1,23
-0,93
-1,16
447
ОКСИЭТИЛЦЕЛЛЮЛОЗА
448
3 раза превышает мол. массу П. из 2-0. Поли-
2-0. мол. массы 9000 образуется также по катионному
механизму под действием BF3-O(C2H5J в р-ре метилен-
хлорида. Поли-4-О. получается также при омылении
соответствующих полиэфиров, напр, поли-4-винилфе-
нилбензилового. П. очищают переосаждением водой
из метанола или бензолом из спирта. П. можно приме-
применять для получения окислительно-восстановительных
полимеров и в качестве пролонгаторов лекарственных
веществ.
Лит.: Corson В. [а. о.], J. Org. Chem., 23, № 4, 544
<1958); К a t о М. [а. о.], J. Polymer Sci., pt A-l, 4, 1773
A966); pt A-l, 7, 2175 A969); Bonsai I E. [a. o.], Trans.
Faraday Soc, 48, pt 8, 763 A952); MarvellC, R а о N. S.,
J. Polymer Sci., 4, № 6, 703 A949); F e r r u t i P., F e г г е A.,
J. Polymer Sci., pt A-l, 9, № 12, 3671 A971).
Е.Ф. Разводовский.
ОКСИЭТИЛЦЕЛЛЮЛОЗА (hydroxyethyl cellulose,
Hydroxyathylzellulose, hydroxyethylcellulose) — про-
простой эфир целлюлозы, образующийся при взаимодей-
взаимодействии ее с окисью этилена; общая формула этого эфи-
эфира {С6Н7О2(ОН)з_я [(ОСНаСН^ ОН]Х}П.
О. характеризуют молярным показателем замеще-
замещения — средним количеством молей окиси этилена, при-
присоединившейся к одному элементарному звену макро-
макромолекулы целлюлозы, степенью замещения гидроксиль-
ных групп в элементарном звене х и средним числом
элементарных звеньев у в привитой цбпи.
Свойства. Практич. значение имеет О. с молярным
показателем замещения 1,5—2,5 (х = 0,85—1,20;
у = 1,5—3), содержащая 28—40% связанной окиси
этилена (высокозамещенная О.), и О. с молярным пока-
показателем замещения 0,3—0,4 (х = 0,2—0,3; у = 1,25—
1,3), содержащая 7—9% связанной окиси этилена (низ-
козамещенная О.). О.— порошкообразное или волокни-
волокнистое аморфное вещество белого цвета, без запаха и вку-
вкуса; насыпная масса 0,20—0,37 г/см3.
Т. к. при алкилировании окисью этилена число гид-
роксильных групп в элементарном звене целлюлозы не
уменьшается, О. вступает во все реакции, характерные
для целлюлозы, отличаясь от нее более высокой реак-
реакционной способностью. Повышенная реакционная спо-
способность О. обусловлена более рыхлой структурой,
а также более легким набуханием во многих раствори-
растворителях, большими гидрофильностью и сорбционной
способностью, чем у целлюлозы.
При этерификации и алкилировании О. различными
реагентами получен ряд смешанных эфиров целлюло-
целлюлозы: бензилоксиэтил-, бутилоксиэтил-, карбоксиметил-,
оксиэтил-, метилоксиэтил-, цианэтилоксиэтилцеллюло-
за и др. Нек-рые из них, напр, этил оксиэтил- и метил-
оксиэтилцеллюлозу, производят в промышленном
масштабе.
Высокозамещенная О. имеет темп-ру раз-
мягч. 135—140 °С; она темнеет при 205—210 °С и пол-
полностью обугливается при 250 °С; плотность при 23 °С
и 50%-ной относительной влажности воздуха 1,34
г/см3. Содержание золы 4—6%. Равновесное вла-
гопоглощение при 23 °С и относительной влажности
воздуха 50 и 88% составляет соответственно 6 и 41 %.
Зависимость между характеристич. вязкостью ([ц]
в дл/г) р-ров высокозамещенной О. (молярный показа-
показатель замещения 1,57—1,67; х = 0,88) при 25 °С и сте-
степенью ее полимеризации (Р) выражается ур-ниями (для
р = .330—2650): [ц] = 1,3-10-2Р°>79 (кадоксен); [ц] =
= 1,Ы072/> °'87<вода).
Высокозамещенная О. растворима в воде, смеси эта-
этанол — вода C0 : 70 по массе), 90%-ной муравьиной
кислоте, кадоксене, диметилсульфоксиде, этиленхлор-
гидрине. Набухает и частично растворяется в холод-
холодных и горячих E0—60 °С) этиленгликоле, глицерине,
пропиленгликоле, смесях этанол — вода G0 : 30 и
60 : 40 по массе), диметилформамиде. Наиболее важное
для технич. применения свойство О.— ее раствори-
растворимость в воде. Поверхностное натяжение 0,001 — 1%-вых
р-ров О. при 25 °С составляет 60—70 мн/м, или дин/см;
межфазное натяжение (парафиновое масло — 0,001 —
1%-ный р-р О. в воде, 25 °С) равно 23—27 мн/м,
или дин/см.
Добавление небольших количеств (ок. 0,01%) по-
поверхностно-активных веществ повышает растворимость
О. в воде. При приготовлении водных р-ров почти не
наблюдается пенообразования. Водные р-ры О. ста-
стабильны при рН 2—12. При нагревании водных р-ров О.
гель не образуется (в противоположность метилцел-
люлозе).
В водных р-рах О. легче подвергается микробиоло-
микробиологическому воздействию, чем др. водорастворимые эфи-
ры целлюлозы. В качестве консервантов водных р-ров
используют формальдегид, хлорированные фенолы, про-
производные бензойной кислоты, сорбиновую кислоту,
иод и др.
В водных р-рах О. совмещается со многими водораст-
водорастворимыми природными и синтетич. полимерами, с ря-
рядом солей (NaCl, MgCl2, NH4Cl,ZnCl2 и др.), глицерином,
этиленгликолем, пропиленгликолем, этаноламином,
сульфированным касторовым маслом. Указанные орга-
нич. соединения употребляют в качестве пластифика-
пластификаторов высокозамещенной О.
Пленки из высокозамещенной О. устойчивы при на-
нагревании до 100 °С, а также к действию масел и многих
растворителей. В нерастворимое состояние их перево-
переводят обработкой мочевино- и меламино-формальдегид-
ными смолами, глиоксалем и многоосновными к-тами.
Тенденция пленок к слипанию наблюдается при отно-
относительной влажности воздуха более 50%. В табл.
приведены нек-рые свойства пленок.
Высокозамещенную О. фракционируют осаждением
из водных р-ров ацетоном.
Низкозамещенная О. растворима в 2—
10%-ных растворах едкого натра, кадоксене и 40%-ном
р-ре мочевины; плотность при 25 °С 1,49 г/см3. Зависи-
Зависимость между характеристич. вязкостью (в дл/г) р-ра
низкозамещенной О. (молярный показатель замеще-
замещения 0,3—0,4; х = 0,2—0,3) в кадоксене при 25 °С
и степенью ее полимеризации выражается ур-нием
(для /> до 6000): [т|] = 1,84-10~а Р°>76.
Свойства непластифицированных пленок из низкоза-
низкозамещенной О. приведены в таблице.
Свойства пленок из о к си эти л целлюлозы при 25 СС
и относительной влажности воздуха 50%
Свойства
Показатель преломления п^
Прочность при растяжении,
Мн/м2
кгс/см2
Относительное удлинение, % . .
Число двойных перегибов до
разрушения
Пленки из
высокозаме-
высокозамещенной окси-
этилцеллюло-
зы толщиной
2 мм
1 ,510
21-25
210-250
14-30
>10 000
Пленки из
низкозаме-
низкозамещенной окси-
этилцеллю-
лозы
1,534
80
800
6
1340
Получение. В пром-сти О. получают взаимодейст-
взаимодействием щелочной целлюлозы с окисью этилена по схеме:
NaOH
Целл. (ОН) + пСН2—СН2 > Целл. О(СН2 СН2О)Г1Н
\ /
ЧУ
Одновременно протекает побочная реакция окиси
этилена с водой, присутствующей в целлюлозе:
NaOH
Н2О + пСН2 —СН2 > НО(СН2СН2О)ПН
449
ОЛЕФИНОВ ПОЛИМЕРИЗАЦИЯ
450
Технологич. схема процесса получения водораство-
водорастворимой высокозамещенной О. включает следующие ста-
стадии: 1) подготовка щелочной целлюлозы — обработка
древесной или хлопковой целлюлозы 18—20%-ным
р-ром NaOH с последующим отжимом и измельчением;
2) обработка щелочной целлюлозы окисью этилена при
25—50 °С и давлении 93—96 кн/м2 G00—720 мм рт. ст.)
в течение 3—8 ч; 3) отмывка О. от щелочи и гликолей
органич. растворителем, напр, смесью метанола с аце-
ацетоном, нейтрализация оставшейся NaOH серной или
уксусной к-той, сушка и дробление продукта.
Применение. Водорастворимую высокозамещенную О.
применяют: 1) в качестве загустителя для латексных кра-
красок, т. к. она способствует хорошему совмещению компо-
компонентов и придает покрытиям устойчивость к растрескива-
растрескиванию и морозостойкость; 2) как эмульгатор при эмульси-
эмульсионной полимеризации винилацетата; 3) как высококаче-
высококачественную шлихту и носитель пигмента в текстильной
пром-сти; 4) в композиции с глиоксалем для придания
бумаге жиронепроницаемости и прочности во влажном
состоянии; 5) в качестве защитного коллоида в лито-
литографии и гальванопластике; 6) как связующее в произ-
производстве керамики, стеклянных изделий, литейной
пром-сти.
Щелочерастворимую низкозамещенную О. можно
использовать: 1) в качестве добавок к вискозным пря-
прядильным р-рам для улучшения накрашиваемости волок-
волокна; 2) как несмываемый аппрет и шлихту в текстиль-
текстильной пром-сти; 3) как флотореагент. Добавка продукта
к бумажной массе повышает прочность бумаги в мок-
мокром состоянии.
О. производят в промышленном масштабе в США.
Торговые названия водорастворимого продукта —
натрозол и целлозайс, щелочерастворимо-
го продукта — авкосет иэталоза.
О. впервые синтезирована Губертом в 1920.
Лит.: Роговин 3. А., Химия целлюлозы, М., 1972,
с. 394; Никитин Н. И., Химия древесины и целлюлозы,
М.—Л., 1962, с. 339—45; Quinchon J., Contribution k
l'etude des hydroxyethylcelluloses, P., 1960; В г о w n W. J.,
Tappi, 49, № 8, 367—379 A966); Prokof'evaM. V.[a. o.],
Cellulose Chem. Technol., 4, 357—371 A970). M.В.Прокофьева.
ОЛЕФИНОВ ПОЛИМЕРИЗАЦИЯ (polymerization
of olefines, Polymerisation von Olefinen, polymerisa-
polymerisation des olefines).Олефины (О.) полимеризуются с раз-
разрывом я-связи, образуя линейные карбоцепные поли-
полимеры:
nCH2=CHR »[-CH2-CHR-]n
где R — алкил или Н (см. Олефинов полимеры). Энер-
Энергия, выделяющаяся при полимеризации, определяется
разностью между энергией образования двух связей
С — С и энергией разрыва двойной связи С = С. Та-
Такого рода реакции идут с выделением энергии от 55
до 105 кдж/моль (от 13 до 25 ккал/моль) в зависимости
от характера О. Для большинства реакций присоедине-
присоединения к О. по кратной связи характерны очень низкие
значения энергии активации. Поэтому при сравнитель-
сравнительно низких темп-pax скорость их значительно превос-
превосходит скорость реакций замещения.
О. могут полимеризоваться по радикальному, ионно-
ионному и координационно-ионному механизмам.
Предельная темп-pa полимеризации О., т. е. темп-ра,
при к-рой свободная энергия процесса равна нулю, для
большинства олефинов выше 0 °С, и высокомолекуляр-
высокомолекулярные полиолефины могут образовываться при комнат-
комнатных темп-рах.
Радикальная полимеризация. По радикальному ме-
механизму с образованием высокомолекулярных продук-
продуктов полимеризуются этилен, тетрафторэтилен и его
производные (CF2 = CX2, CF2 = CFX, где X — гало-
галоген или водород). Зависимость энергии активации Е
присоединения свободных радикалов к двойной связи
О. от теплоты реакции (Q) описывается полуэмпирич.
правилом Поляни — Семенова: Е = А — aQ, где А
и а — константы, равные соответственно 10,0 и 0,25.
Т. к. Q колеблется от 54 до 105 кдж/моль (от 13 до
25 ккал/моль), то Е обычно не превышает 42 кдж/моль
A0 ккал/моль). Поэтому для инициирования радикаль-
радикальной полимеризации О. используют обычные способы
генерирования свободных радикалов (гомолитич. распад,
окислительно-восстановительные реакции, радиацию и
др. — см. Инициирование полимеризации).
Рост цепи осуществляется путем последовательного
присоединения молекул О. к радикалам R, возникаю-
возникающим в результате инициирования:
R + CH2=CHR >RCH2-CHR
RCH2-CHR + CH2=CHR >RCH2-CHR—CH2—CHR и т. д.
Обрыв цепи при радикальной О. п. может произойти
в результате рекомбинации двух растущих макрора-
макрорадикалов:
~СН2—CHR + CHR—СН2~ > ~СН2—CHR—CHR—СН2~
или их диспропорционирования
~CH2-CHR + CHR—CH2~ > ~СН2- CH2R 4- ~CH=CHR
Энергия активации этих реакций не превышает 4,2—
8,4 кдж/моль A—2 ккал/моль). Для радикальной О. п.
характерна передача цепи на образовавшуюся макро-
макромолекулу:
~CH2-CHR + CHR >~CH2-CH2R
Эта реакция является одной из причин образования раз-
разветвления в полиолефинах:
~CR ~CR-CH2-CHR
| + CH2=CHR —> I
~CH2 ~CH2
Передача цепи может осуществляться также на моно-
мономер:
~CH2-CHR 4- CH2=CHR->~CH=CHR 4- CH3-CHR
или, если R = СНз
~СН2-СНСН3 + СН2=СНСН3 —> ~СН2-СН2СН3 4- СН2=СНСН2
Передача цепи особенно характерна для радикальной
полимеризации а-О. (пропилена, бутилена и др.),
т. к. энергия связи С — Ну третичного атома углерода
Н
в макромолекулах типа ~ С ~ примерно на 21 кдж
R
E ккал) меньше, чем в группе —СН2—, а передача на
мономер увеличивается из-за большой лабильности
Н-атомов, находящихся в а-положении к двойной связи.
Наиболее важный процесс радикальной О. п.— по-
получение полиэтилена низкой плотности полимериза-
полимеризацией этилена при давлении не менее 120 Мн/м2 A200
кгс/см2) в трубчатых реакторах или автоклавах с ме-
мешалкой. См. Этилена полимеры.
Ионная полимеризация. Ккатионной поли-
полимеризации склонны изобутилен, а-бутилен, про-
пропилен, т. е. те О., к-рые имеют повышенную элект-
электронную плотность у двойной связи. Катионная поли-
полимеризация а-О. при темп-pax выше 0 °С приводит
к синтезу низкомолекулярных продуктов (олигоме-
ров). Полимеры с высокой мол. массой могут образо-
образовываться лишь при низких темп-pax (см., напр., Изобу-
тилена полимеры).
Элементарные акты катионной О. п. можно рассмот-
рассмотреть на примере изобутилена. Инициирование
сн3 СНз
I I
СН2=С 4- Н+Х—> СН3-С+Х-
СНз
СНз
451
ОЛЕФИНОВ ПОЛИМЕРИЗАЦИЯ
452
(где Х~— противоион) приводит к образованию карб-
катиона. Эта стадия может протекать через быстрое
равновесное образование промежуточного я-комплекса
протона с О., который затем изомеризуется в карб-
катион.
Энергия активации инициирования изменяется анти-
батно энтальпии инициирования. Изменение АН ини-
инициирования в ряду О. определяется гл. обр. различием
в сродстве О. к протону, к-рое зависит от степени заме-
замещения и природы заместителей вблизи двойной связи
(см. таблицу).
Сродство к протону нек-рых олефинов
(реакция в газовой фазе X + Н+ -> ХН+)
х
с2н4
с3нв
С3Нв
С2Н5СН=СН2
C2H5CH=CjH2
(СН3JС=СН2
(СНзJС=СН2
хн+
с2н5+
н-С3Н7+
изо-СзН7+
н-С4Н9+
изо-С4Н9+
изо-С4Н9+
mpem-C4H9+
—Л Щ98, кдж/молъ
(ккал/молъ)
641 A53)
691 A65)
758 A81)
682 A63)
792 A89)
696 A66)
821 A96)
Для быстрого инициирования под действием обычных
катионных катализаторов О. должен иметь сродство
к протону большее, чем пропилен.
Полимерная цепь растет путем последовательного
присоединения молекул а-О. к катиону:
СН3 СНз СНз СН3
I III
СН3-С+Х- + СН2=С ->СН3-С-СН2-С+Х- и т. д.
I III
СНз СНз СНз СНз
Для образования линейных полимеров с высокой мол.
массой необходимо, чтобы скорость роста цепи была
значительно выше, чем скорость всех др. реакций ра-
растущего конца цепи. Для катионной полимеризации
а-О., содержащих третичные атомы, характерна тен-
тенденция к изомеризации карбкатионов различного строе-
строения с образованием более устойчивых структур (см.
также Изомеризационная полимеризация). Обрыв цепи
при катионной полимеризации а-О. происходит в ре-
результате отщепления протона противоионом (Х~) от
макроиона
СН2
СН3
-сн2-с++х-
СНз
II
~СН2-С
СНз
СНз
I
1=С -\- Н+Х"~
I
СНз
или передачи цепи на мономер:
сн3 сн3
~СН2-С+ +
I
СНз
СН2=С ->
СН3
СНз
I
1=С + СНз-
с+
СНз СНз
Суммарная энергия активации катионной полимери-
полимеризации а-О. часто бывает отрицательной. В этих случа-
случаях скорость и степень полимеризации возрастают с по-
понижением темп-ры.
Катионная гомо- и сополимеризация а-О. использу-
используются для получения полиизобутилена, моторных топ-
лив, присадок к смазочным маслам и различных загу-
загустителей, бутилкаучука.
Анионная полимеризация для О. ме-
менее характерна, чем катионная и радикальная. Первич-
Первичные и вторичные карбанионы присоединяются к этиле-
этилену очень медленно. Однако третичный карбанион при-
присоединяет этилен с достаточно высокой скоростью.
В углеводородных растворителях в жестких условиях
[давление мономера 950 Мн/м2 (9500 кгс/см2), темп-ра
20—26 °С, время 17 ч] в присутствии алкилов лития из
этилена образуются лишь низкомолекулярные продук-
продукты с небольшим выходом. Замена растворителя на те-
трагидрофуран позволяет получить при среднем давле-
давлении полиэтилен с характеристич. вязкостью от 0,03
до 0,35 дл/г. Использование хелатирующих агентов,
напр, тетраметилэтилендиамина, в качестве сокатали-
заторов приводит к образованию полиэтилена в угле-
углеводородных растворителях с мол. массой до 40 000.
Координационно-ионная полимеризация. С помощью
катализаторов Циглера—Натта (как гомогенных, так
и гетерогенных) можно получать высокомолекулярные
полимеры на основе а-О. (полипропилен, полибутилен),
к-рые другими путями практически не образуются.
Растворителями, пригодными для проведения полиме-
полимеризации этилена и высших а-О., служат как алифати-
алифатические, так и ароматич. углеводороды, такие, как бен-
бензин, пропан, гексан, циклогексан, тетралин, декалин,
а также галогенпроизводные углеводородов.
Полимеризация О. на комплексных катализаторах
идет по след. механизму. При взаимодействии компо-
компонентов каталитич. системы происходит алкилирование
переходного металла (Me) и затем постепенное его вос-
восстановление. Инициирование заключается во внедре-
внедрении первой молекулы мономера по связи Me — С. Рост
цепи осуществляется путем многократного внедрения
молекулы мономера по связи переходный металл —
углерод:
yMe-R + nCH2=CHR ->^Me-(CH,-CHR)n - R
Для активного центра комплексных катализаторов
О. п. необходимым является наличие связи переходный
металл — углерод, свободной низколежащей орбитали
и отсутствие стирич. препятствий для координации О.
При координации О. с алкилированным переходным
металлом происходит одновременное уменьшение энер-
энергии связи Me — С, что обусловливает низкое значение
энергии активации реакции внедрения.
Число активных центров обычно очень невелико по
сравнению с общей концентрацией исходного переход-
переходного металла. Константы скорости элементарных реак-
реакций внедрения О. по связи переходный металл — угле-
углерод достаточно высоки и могут достигать значений
104—106л/(моль-сек). Эффективные константы скорости
роста цепи, рассчитанные на исходный переходный ме-
металл, имеют значения порядка 10—102 л/(моль-сек).
В большинстве систем на 1 атом переходного металла
образуется 1 цепочка полиолефина. Регенерация ак-
активных центров или эффективная передача цепи осуще-
осуществляется лишь в присутствии таких окислителей, как
О2, RCI, HC1, или большого избытка сокатализа-
тора — металлоорганич. соединения. В этих случаях
процесс протекает долгое время с постоянной ско-
скоростью.
Обрыв цепи осуществляется по реакциям внутри-
или межмолекулярного диспропорционирования, без
образования свободных радикалов:
\ /СН2—СН2—... — R \
)МеС -> ^Ме + CH2=CH-...-R + R'H
2^>Me-CH2-CH2-...-R —> ^>Ме + CH2-...-R + СН3СН2--...—R
Известны реакции спонтанного обрыва цепи и ограни-
ограничения цепи с участием мономера:
/Me-CH2-CH2-...-R —> ^>Ме-Н + CH2=CH-...-R
/Me-CH2-CH2-...-R -f C2H4 -> /Ме-С2Н5
CH2=CH-...-R
В последнем случае активный центр образует новую
макромолекулу. Существуют и др. типы передачи цепи,
453
ОЛЕФИНОВ ПОЛИМЕРЫ
454
напр, на АШз или среду, содержащую атомы (Н илиС1)
или группы (ОН, NH2), к-рые могут реагировать с ком-
комплексом Me — цепь.
Вследствие малой скорости передачи цепи на макро-
макромолекулу синтезированные с помощью комплексных
катализаторов полиолефины обладают малой развет-
вленностью и высокой кристалличностью.
Полимеризация а-О. на комплексных катализаторах
может приводить к образованию макромолекул с
высокоупорядоченной структурой полимерной цепи.
Напр., полипропилен, полученный в присутствии ка-
талитич. системы a-TiCl3 + A1R2X, имеет преимуще-
преимущественно изотактич. макромолекулы.
В присутствии комплексных катализаторов может
протекать изомеризационная полимеризация как а-,
так и C-олефинов. Напр., на тройной каталитич. си-
системе VC14 + Fe(acacK 4- A1R3 из пропилена в зависи-
зависимости от условий процесса можно синтезировать линей-
линейный полиэтилен
СН2=СН
СН,
Кат.
СНо—СН —Кат.
нсн2
—СНо—СНо СНо
-Кат. •
н т.д.
или сополимер этилена с пропиленом.
При полимеризации и сополимеризации р-О. на комп-
комплексных катализаторах могут образовываться полиме-
полимеры и сополимеры а-О. Так, сополимеризация транс-
бутена-2 и пентена-2 на каталитич. системе VC13 +
+ Ni(DMS) + АШз при 80 °С приводит к образованию
сополимеров бутена-1 и пентена-1.
Под действием комплексных катализаторов может
осуществляться сополимеризация этилена с пропиле-
пропиленом и др. высшими а-О. (см., напр., Этилен-пропиле-
новые каучуки). Константы сополимеризации опреде-
определяются стерич. препятствиями, возникающими при
внедрении О. по связи переходный металл — углерод.
Так, для одной и той же каталитич. системы скорость
полимеризации падает в ряду этилен, пропилен, а-бу-
тилен.
Для различных комплексных катализаторов значе-
значение констант сополимеризации для одной и той же пары
мономеров определяется природой переходного метал-
металла и мало зависит от характера металлорганических
соединений (см. также Циглера — Натта катализа-
катализатор ы).
О полимеризации О. на окислах металлов см. Окисно-
металлические катализаторы.
Лит.: БреслерС. Е., Ерусалимский Б. Д.,
Физика и химия макромолекул, М.— Л., 1965; Семе-
Семенов Н. Н., О некоторых проблемах химической кинетики и
реакционной способности, 2 изд., М., 1958; Багдасарь-
я н X. С, Теория радикальной полимеризации, 2 изд., М.,
1966; Катионная полимеризация, под ред. П. Плеша, пер. с
англ., М., 1966; Гейлорд Н., Марк Г., Линейные сте-
реорегулярные полимеры, пер. с англ., М., 1962; X э м Д.,
Сополимеризация, пер. с англ., М., 1971. Ф. С. Дьячковский.
ОЛЕФИНОВ ПОЛИМЕРЫ (polyolefines, Polyolefine,
polyolefines) — высокомолекулярные соединения, об-
образующиеся при гомо- или сополимеризации олефинов.
R
Гомополиолефины имеют общую ф-лу [—СН2—С—]п.
R'
В зависимости от природы заместителей R и R' они
м. б. разделены на 3 группы.
1. R = R' = Н, или R = Н, R' — алкил линейного
строения, или Rn R' — алкилы линейного строения,
напр. СНз, С2Н5. К этой группе относятся, в частности,
полиэтилен и полипропилен (см. Этилена полимеры,
Пропилена полимеры), полибутен (см. Бутенов полиме-
полимеры), а также полиизобутилен (см. Изобутилена поли-
полимер ы).
2. R = Н, R' — алкил разветвленного строения.
К этой группе принадлежат, напр., поли-4-метилпен-
тен-1 и поли-З-метилбутен-1 (см. А-Метилпентена-i
полимеры, З-Метилбутена-1 полимеры).
3. R = Н, R' — циклогексил или др. циклич. либо
полициклич. углеводородный радикал. Из О. п. этой
группы наиболее полно изучен поливинилциклогексан
(см. Винилциклоалканов полимеры).
Особую группу составляют полимеры циклоолефи-
нов, в частности циклопентена.
Среди сополимеров олефинов наибольшее значение
имеют этилен-пропиленовые эластомеры (см. Этилен-
пропиленовые каучуки), а также кристаллич. блоксопо-
лимеры, т. наз. и о л и а л л о м е р ы.
Кристаллич. О. п. обладают достаточно высокой ме~
ханич. прочностью, высокими диэлектрич. показателя-
показателями, устойчивы к действию агрессивных сред (за исклю-
исключением сильных окислителей, напр, азотной к-ты),
способны образовывать легко ориентируемые пленки
и в ряде случаев (напр, полипропилен) волокна, могут
перерабатываться любыми способами, обычно исполь-
используемыми в пром-сти пластмасс. Существенный недоста-
недостаток О. п.— плохая адгезия, обусловленная отсутствием
полярных групп, и сравнительно невысокая жесткость,
из-за к-рой часто ограничивается применение этих по-
полимеров как конструкционных материалов (для изго-
изготовления деталей машин). С другой стороны, отсутст-
отсутствием полярных групп объясняется повышенная химич.
стойкость О. п.
Введение разветвленных алифатич. или циклич. за-
заместителей заметно повышает темп-ру плавления и
теплостойкость полиолефинов. Так, если теплостой-
теплостойкость по Вика полиэтилена высокого давления 110 °С,
полиэтилена низкого давления 130 °С, полипропилена
150 °С, то у поли-4-метилпентена-1 и у поливинилцикло-
гексана она составляет соответственно 179 и 225 °С.
Поли-З-метилбутен-1 характеризуется особенно высо-
высокой степенью кристалличности; темп-pa плавления его
выше 300 °С. Однако полимеры такого типа деструкти-
руются, что создает затруднения при их переработке.
В данной статье подробно описаны только полимеры
циклсюлефинов и полиалломеры, т. к. всем остальным
промышленно важным О. п. в «Энциклопедии полиме-
полимеров» посвящены специальные статьи.
Полимеры циклоолефинов. В зависимости от условий
реакции циклоолефины могут полимеризоваться по
двойной связи или с раскрытием цикла. В первом
случае образуется полимер, содержащий циклы в цепи,
во втором — каучукоподобный полимер с открытой це-
цепью, напр.:
^ [-СН2-СН2-СН = СН-СН2-]а
Направление полимеризации существенно зависит от
природы переходного металла, входящего в состав ка-
каталитич. комплекса. Так, на примере циклобутена пока-
показано, что в присутствии V- или Cr-содержащих катали-
катализаторов реакция протекает по винильным связям,
в присутствии W- , Та-, Мо- или Ti-содержащих —
с раскрытием цикла.
По строению основной цепи полимеры с открытой
цепью аналогичны полидиенам, т. к. содержат двойную
связь в каждом элементарном звене, и, следовательно,
существенно отличаются по свойствам от обычных по-
полиолефинов. Наибольший практич. интерес пока пред-
представляет [f цс-полипентамер — эластомер,
отвечающий современным требованиям, предъявляе-
предъявляемым к каучукам массового назначения. Он медленно
кристаллизуется при растяжении; темп-pa стеклова-
455
ОЛЕФИНОВ ПОЛИМЕРЫ
456
ния ок. —НО °С, что на 40—50 °С ниже темп-ры стек-
стеклования стереорегулярного бутадиенового каучука.
if^-Полипентамер вулканизуется серой. По морозо-
морозостойкости он превосходит все известные углеводород-
углеводородные каучуки. Высокоэластич. свойства его наилучшим
образом проявляются при низких темп-pax (табл. 1).
Таблица 1. Свойства вулканизатов »<ис-полипентамера
Свойства
Прочность при рас-
растяжении,
Мн/м2 (кгс/см2) . .
Относительное удли-
удлинение, %
Модуль высокоэлас-
тичности, Мн/м2
(кгс/см2)
при 10 Мн/м2
A00 кгс/см2) . .
20 Мн/м2
B00 кгс/см2) . .
30 Мн/м2
C00 кгс/см2) . . .
Температура, °С
23
17A70)
500
2,5 B5)
5,0 E0)
9,0 (90)
-20
15 A50)
420
2,5 B5)
5,0 E0)
9,0 (90)
-50
22,5 B25)
490
3,0 C0)
6,5 F5)
11,5 A15)
-90
39 C90)
495
8,0 (80)
13,0 A30)
21,5 B15)
^ггс-Полипентамер получают стереоспецифич. поли-
полимеризацией циклопентена в присутствии МоС15 —
А1(С2Н5)з; время —4 ч. Выход полимера и содержа-
содержание трawe-звеньев в нем зависят также от темпера-
температуры полимеризации. Оптимальные результаты дости-
достигаются при молярных соотношениях МоС15: А1(С2Н5)з=
1,5—2,5 и циклопентен : МоС15 = 500 : 1.
тргшс-Полипентамер образуется при полимеризации
циклопентена в присутствии катализатора WCle —
— АШз. Для активации каталитич. систем в ряде слу-
случаев используют, напр., перекиси или гидроперекиси.
О свойствах О. п., получаемых полимеризацией цик-
лоолефинов по связи С = С, сведений пока нет.
Циклоолефины способны также сополимеризоваться
с олефинами, в частности с этиленом, обычно в присут-
присутствии ванадийсодержащих каталитич. систем:
О
Свойства сополимеров пока не изучены.
Полиалломеры. Кристаллич. структура этих блоксо-
полимеров обычно близка к соответствующим характе-
характеристикам гомополимера основного сомономера. Наиболее
полно исследованы (и практически используются) пока
полиалломеры на основе пропилена и этилена. Получе-
Получены и изучены также полиалломеры пропилена со стиро-
стиролом, винилциклогексаном, изобутиленом, 4-метилпен-
теном-1, винилхлоридом и др. Введение небольшого ко-
количества какого-либо сомономера в полипропилен су-
существенно снижает температуру хрупкости; при этом в
нек-рых случаях теплостойкость почти не изменяется,
улучшаются адгезионные свойства (в случае примене-
применения полярного сомономера) и уменьшается усадка.
Поэтому в большинстве случаев полиалломеры обла-
обладают лучшим комплексом свойств, чем соответствую-
соответствующие гомополимеры (табл. 2, 3).
На примерах пропилен-этиленовых полиалломеров
показано значительное влияние на их свойства мол.
массы (табл. 4).
Синтезированы также полиалломеры, содержащие
в макромолекулах одновременно полиолефиновые и по-
полиацетиленовые блоки. Благодаря наличию двойных
связей такие блоксополимеры способны сшиваться с об-
образованием более жестких материалов, чем полиэтилен
и полипропилен.
В основе синтеза полиалломеров лежит известный
принцип образования живущих полимеров. Полиалло-
Таблица 2. Свойства пропилен-этиленовых полиалломеров
(для сравнения приведены свойства полипропилена и
полиэтилена)
Свойства
Индекс расплава при
230 °С
Плотность, г/см3 . . . .
Темп-ра хрупкости, °С
Прочность при растя-
растяжении, Мн/м* (кгс/см2)
Модуль при изгибе,
Мн/м2 (кгс/см2) ....
Твердость по Роквеллу
Относительное удлине-
удлинение, %
Теплостойкость по Ви-
Вика, °С
Ударная вязкость (с
надрезом) при 230 °С,
кдж/м2, или кгс/см2. .
Поли-
пропи-
пропилен
2,4
0,91
33
C30)
998
(9980)
93
360
145
2,7
Поли-
Полиэтилен
2,4
0,97
-78
21,5
B15)
710
G100)
54
290
123
7,0
Полиалломеры с раз-
различным содержанием
этиленовых звеньев
0,6% j 2,0% | 0,3%
2,8
0,91
— 5
27
B70)
700
G000)
87
550
140
4,9
2,9
0,90
-22
25
B50)
650
F500)
68
600
127
10,3
2,5
0,90
-35
21,5
B15)
500
E000)
60
650
125
19,0
Таблица 3. Свойства полиалломеров пропилена с небольшими
количествами указанных сомономеров
Свойства
при
Индекс расплава
230 °С
Плотность (после вакал-
ки), г/см3
Темп-ра хрупкости, °С
Прочность при растя-
растяжении, Мн/м2 (кгс/см2)
Модуль при изгибе,
Мн/м2 (кгс/см2) . . . .
Относительное удлине-
удлинение, %
Теплостойкость по
ка, °С
Твердость по Роквеллу
Ударная вязкость (с
надрезом) при 230 °С,
кдж/м2, или кг-см/см2
Ви-
Бутен-1
2,4
0,91
-4
83 (830)
980
(9800)
225
142
90
3,27
Изопрен
1,4
0,91
-2
37C70)
1190
A1900)
75
147
99
3,27
Стирол
2,5
0,92
22
37 C70)
1400
A4000)
145
102
Винил-
хлорид
4,5
0,92
20
36 C60)
1400
A4000)
147
97
Таблица 4. Влияние молекулярной массы (индекса расплава)
пропилен-этиленовых полиалломеров, содержащих ок. 2%
этилена, на их свойства
Свойства
Темп-ра хрупкости, °С
Прочность при растяжении, Мн/м2
(кгс/см2)
Теплостойкость по Вика, °С
Ударная вязкость (с надрезом) при
230 °С, кдж/м2, или кгс-см/см2 ....
Индекс расплава
при 230 °С
2,9
-22
25 B50)
127
10
0,6
-32
23 B30)
128
58
меры получают последовательной полимеризацией вы-
выбранных мономеров в присутствии комплексных ме-
таллоорганич. каталитич. систем. В качестве катализа-
катализаторов используют гл. обр. комплексы алюминийалки-
лов с соединениями переходных металлов (в основном
с хлоридами титана или ванадия). Реакцию осущест-
осуществляют в р-ре, напр, «-гептане, посредством поперемен-
попеременной подачи разных сомономеров в реакционную зону
в течение определенного времени. В результате полу-
получают кристаллич. блоксополимеры, блоки в к-рых име-
имеют определенные размеры и определенным образом
распределены в макроцепи.
Большое влияние на структуру и свойства блоксопо-
лимеров оказывают условия введения в зону реакции
сомономеров. Так, каждый последующий сомономер
457
ОЛИГОМЕРЫ
458
необходимо вводить лишь после того, как полностью
прореагирует предыдущий. Иначе образуются блоки
статистич. сополимеров, нарушающих кристаллич.
структуру и ухудшающих свойства полиалломеров.
Развитие промышленного производства полиолефи-
нов. По масштабу промышленного производства и раз-
разнообразию областей применения первые два места сре-
среди О. п. принадлежат соответственно полиэтилену и
полипропилену. Это обусловлено как ценными технич.
свойствами указанных полимеров, так и наличием
дешевого и доступного нефтехимич. сырья — этилена
и пропилена. Мировое производство О. п. в 1972 пре-
превысило 8,0 млн. т, в том числе полиэтилена низкой
плотности ок. 5,5 млн. яг, полиэтилена высокой плот-
плотности свыше 1,5 млн. т, полипропилена свыше
1,0 млн. т. Существует тенденция к дальнейшему росту
производства этих полимеров.
Во многих странах налажено промышленное произ-
производство этилен-пропиленового каучука. В США и
Канаде в промышленном масштабе получают кристал-
кристаллич. блоксополимеры пропилена с этиленом. Из др.
полиолефинов, не содержащих заместителей в цепи,
наиболее перспективен полибутен-1. Для синтеза его
используют также дешевое сырье — бутиленовые фрак-
фракции продуктов нефтепереработки. Кроме того, открыт
и реализован процесс каталитич. димеризации этилена
в бутен-1 в мягких условиях с практически количест-
количественным выходом. Все это создало прочную сырьевую
базу для организации производства этого полимера.
В сравнительно небольших количествах его уже
получают (данные за 1972) в США (—10 тыс. т) и ФРГ
(ок. 5 тыс. т).
Поиски полиолефинов, отличающихся от полиэтиле-
полиэтилена и полипропилена повышенной теплостойкостью,
привели к развитию исследований по полимеризации
высших а-олефинов, содержащих разветвленные или
циклич. заместители. Уже производится в пром-сти
поли-4-метилпентен-1 (Великобритания, США). Разрабо-
Разработан метод получения поливинилциклогексана (СССР),
изучаются процессы полимеризации З-метилбутена-1,
5-метилгексена-1 и др.
Большое внимание уделяется разработке технологии
полимеризации циклоолефинов с целью получения
особого типа морозостойких каучуков.
Исследования в области синтеза чередующихся сополи-
сополимеров а-олефинов, напр. пропилена с бутадиеном, могут
привести к созданию новых типов каучуков, аналогич-
аналогичных по свойствам этилен-пропиленовым; однако, вслед-
вследствие наличия в макромолекуле двойных связей, их
можно будет вулканизовать обычным способом. Изу-
Изучается также возможность синтеза олигоолефинов, со-
содержащих реакционноспособные группы. При отверж-
отверждении (вулканизации) таких олигомеров можно будет
получать материалы более жесткие, чем полиэтилен
и полипропилен; к тому же при переработке олигомеров
не требуются высокие темп-ры, что исключает дест-
деструкцию, обычно протекающую при переработке выс-
высших полиолефинов.
Лит.: Кристаллические полиолефины, пер. с англ., т. 1—2,
М., 1970; Кренцель Б. А., Клейнер В. И., в сб. «Хи-
«Химия и технология высокомолекулярных соединений», М., 1974;
D е 1 Г A s t a G., Motroni G., Angew. macromol. Chem.,
16—17, 51 A971). Б. А. Кренцель.
ОЛИГОМЕРЫ (oligomers, Oligomere, oligomeres).
Содержание:
Введение 458
Свойства и структура 459
Фракционирование 460
Молекулярно-массовое распределение и распре-
распределение по функциональности 460
Получение 461
Полимеризационные методы 461
Поликонденсационные методы 464
Деструктивные методы 464
Ступенчатый синтез 465
Получение циклоолигомеров 4 66
Применение 466
Введение
Олигомеры — члены гомологич. рядов, занимающие
по размеру молекул область между мономерами и высо-
высокомолекулярными соединениями. Приставка «олиго»
была впервые использована Гельферихом в 1930;
он назвал олигосахаридами кристаллич. углеводы, со-
содержащие 2—6 остатков моноз.
Верхний предел мол. масс О. зависит от их химич.
природы и по порядку величины совпадает с мол.
массой сегмента (см. Гибкость макромолекул), при до-
достижении к-рой начинают проявляться высокоэластич.
деформации, вынужденная высокоэластичность и др.
свойства, характерные для высокомолекулярных ве-
веществ. Полярные О. и О. с жесткими цепями охваты-
охватывают более широкий интервал мол. масс .(примерно до
1,5-104), чем неполярные (примерно до 5-Ю3).
О. обладают рядом свойств, характерных как для
мономеров, так и для высокомолекулярных соединений.
Физико-химич. свойства соседних олигомерных гомо-
гомологов заметно различаются и зависят от природы кон-
концевых групп, причем по мере увеличения мол. массы
влияние концевых групп и разница в физич. константах
соседних членов ряда уменьшаются. О. характеризуют-
характеризуются значительными межмолекулярными взаимодейст-
взаимодействиями, повышенной вязкостью и высокими временами
релаксации, возрастающими с увеличением размера
молекул.
Из сказанного следует, что низшие члены олигомер-
олигомерных рядов м. б. легко получены или выделены в виде
химически индивидуальных соединений, в то время как
для высших олигомерных гомологов идентификация
связана со значительными трудностями. Это побудило
нек-рых ученых предложить для последних особый
термин — плейномеры. Однако деление об-
области низкомолекулярных полимеров на олигомеры и
плейномеры, по-видимому, искусственно и не имеет
достаточной физич. основы, т. к. современные методы
синтеза и разделения смесей гомологов позволяют по-
получать в виде химически индивидуальных веществ О.
со значительными степенями полимеризации (п =
= 25—30, среднечисленная мол. масса 2000—3000),
и не имеется принципиальных ограничений для полу-
получения индивидуальных соединений более высокой мол.
массы.
Введение понятия «плейномер» иногда обосновывают
появлением зависимости большого периода, опреде-
определяемого методом малоуглового рентгеноструктурного
анализа, от условий кристаллизации О., начиная
с нек-рой длины цепи, к-рую и принимают за нижний
предел области плейномеров. Действительно, для низ-
низших членов гомологич. рядов величина большого пе-
периода зависит только от размера молекулы, увеличи-
увеличиваясь с ростом последней, но не зависит от условий
кристаллизации. Начиная с нек-рой длины, определяе-
определяемой химич. природой О., цепи приобретают способ-
способность складываться, причем размер складок (а сле-
следовательно и большой период) определяется не только
длиной и гибкостью цепи, но и термодинамикой и усло-
условиями кристаллизации. Однако при определенных усло-
условиях (малая скорость кристаллизации; температура,
близкая к температуре плавления образца; высокие
давления; использование узких фракций О.) можно
закристаллизовать длинноцепочечные О. в выпрям-
выпрямленном состоянии [при этом образуются кристал-
кристаллы толщиной>100 им {> 1000 А)]. Тогда величина
большого периода, как и в случае низших гомо-
гомологов, достигает размеров, соответствующих длине
молекулы.
Следует различать реакционноспособные О., к-рые
могут превращаться в высокомолекулярные полимеры,
и О., не содержащие реакционноспособных групп или
центров. Реакционноспособные О. могут вступать в ре-
реакции полимеризации (олигоэфиракрилаты, олигодие-
459
ОЛИГОМЕРЫ
460
ны и их сополимеры, эпоксидные смолы, циклоолиго-
эфиры, циклоолигоамиды и др.), а также поликонденса-
поликонденсации и полиприсоединения (вещества, содержащие кар-
карбоксильные, амино-, изоцианатные и др. группы, фено-
ло-формальдегидные, карбамидные, алкидные смолы
и др.)- Реакционноспособные О., используемые для
получения высокомолекулярных полимеров, иногда
наз. форполимерами, или предполи-
мерами.
Свойства и структура
Как уже отмечалось, для низкомолекулярных О.-го-
О.-гомологов при переходе от одного представителя к после-
последующим наблюдается существенное изменение свойств.
Однако по мере возрастания степени полимеризации п
эти изменения становятся все менее выраженными. Для
неразветвленных О. наблюдается линейная зависимость
нек-рых свойств (парахора, рефракции, уд. объема,
характеристич. вязкости, величины, обратной темп-ре
стеклования, и др.) от мол. массы М, что м. б. выражено
ур-нием:
ср = а I М + Ъ
где ср — удельная характеристика О., являющаяся
аддитивной величиной, М — молекулярная масса,
а — константа, учитывающая «вклад» концевых групп,
Ъ — константа, учитывающая структуру повторяющего-
повторяющегося звена.
Темп-pa плавления О. зависит от степени полимери-
полимеризации. Принимая во внимание, что концевые группы
длинноцепочечных О. полностью исключены из кри-
сталлич. решетки, темп-ру плавления Тпл и среднечис-
ловую степень полимеризации п можно связать след.
соотношением:
1 1 2R
где Т° — т. пл. образца с бесконечно большой мол.
массой, К; R — газовая постоянная, дж/(молъ-К)
[кал/(моль-°С)]; АН — теплота плавления в расчете
на мономерное звено цепи, дж/молъ (кал/моль). Вели-
Величина 2/п представляет собой молярную долю некристал-
лизующихся звеньев. Влияние молекулярной массы
О. на его температуру плавления по мере возрас-
возрастания п заметно уменьшается. Зависимость Гпл от
п часто имеет экстремум в области низких значений,
при которых наблюдается изменение типа кристалли-
кристаллической решетки.
О. с малой степенью полимеризации образуют кри-
кристаллы с молекулярной решеткой, свойственной соот-
соответствующим индивидуальным соединениям. При боль-
больших значениях п О. образуют кристаллич. структуры,
характерные для соответствующих высокомолекуляр-
высокомолекулярных полимеров. Обычно узкие фракции О. относитель-
относительно большой мол. массы кристаллизуются, склады-
складываясь на себя. Добавка более низкомолекулярной
фракции приводит к формированию кристаллов мень-
меньшей толщины, часто равной длине добавленного О.
При кристаллизации полидисперсных О. молекулы
низкомолекулярных гомологов выталкиваются с гра-
граней растущих кристаллов, происходит отбор молекул
по их длинам и образуются кристаллы различной
толщины.
В отличие от р-ров высокомолекулярных соединений
для разб. р-ров различных О. (олигоэтиленгликоль,
олигостирол, олигопропиленгликоль, олигодиметил-
силоксан и др.) в хороших растворителях графики за-
зависимости lg[r]] — \gM в области малых значений мол.
массы показывают увеличение кривизны, т. е. уменьше-
уменьшение экспонента в ур-нии Марка — Хувинка [т]] = КМа,
где [ц] — характеристич. вязкость, М — мол. масса,
К и а — константы, зависящие от природы О. и раст-
растворителя. Многие О. характеризуются областью зна-
значений М, в к-рой [г\] = КОМ°>Ъ, несмотря на то, что
термодинамич. условия далеки от условий 0-раствора
(см. Растворы).
Константа Хаггинса к', связывающая приведенную
вязкость Т1уд/с с концентрацией р-ра с соотношением
Луд/с = I1)] + ^'[Л]2^ ПРИ уменьшении мол. массы до
определенного значения остается постоянной @,3—0,5),
после чего возрастает и достигает нескольких единиц
для О. с малой степенью полимеризации.
Ранее постулировалась независимость реакционной
способности функциональных групп от мол. массы. Од-
Однако в последние годы на ряде примеров было пока-
показано, что с увеличением длины молекулы активность О.
может возрастать или падать в зависимости от природы
О. и среды. Так, при изучении кинетики полимеризации
в массе диметакрилаталкиленгликолей
СН2 = С (СН3) СОО (СН2)ПОСОС (СН3) = СН2
где п = 4, 6, 10, обнаружено увеличение констант
роста цепи с возрастанием длины метиленовой цепочки
гликоля. С ростом мол. массы а,со-диметакриловых эфи-
ров олигоэтиленгликольформалей и олиготетрамети-
ленгликолей также наблюдается увеличение полиме-
ризационной способности. Так, при возрастании мол.
массы первых от 880 до 2200 и вторых от 700 до 2600 на-
начальные скорости полимеризации G0 °С) увеличиваются
в 2 и 3 раза соответственно.
Константа скорости реакции олигодиэтиленгликоль-
адипинатов с фенилизоцианатом в массе или в среде
модельного растворителя (дибутилиолидиэтиленгли-
кольадипинат) возрастает с увеличением мол. массы
от 300 до 2500 приблизительно в 2 раза. Однако при
проведении этой реакции в р-ре хлорбензола реакцион-
реакционная способность концевых гидроксильных групп па-
падает с увеличением мол. массы в этих же пределах и
асимптотически приближается к нек-рому постоянно-
постоянному значению. Изменение реакционной способности О.
связывается с различной степенью ассоциации молекул,
к-рая может как благоприятствовать протеканию реак-
реакции, так и препятствовать ей.
Фра кционирование
Фракционирование О. осуществляют, используя ме-
методы, применяемые для разделения на фракции высо-
высокомолекулярных соединений (см. Фракционирование),
а также хроматографич. методы (электрофорез, тонко-
тонкослойная и гель-проникающая хроматографии и др.—
см. Хроматография). Последние позволяют разделить
смеси О. не только на узкие фракции, но и получать
О.-гомологи в виде индивидуальных соединений. Элект-
Электрофорез на бумаге используют для разделения О. с кис-
кислотными или основными группами. Тонкослойную хро-
хроматографию и хроматографию на бумаге применяют
для разделения и идентификации О. различных клас-
классов (олигоэфиров, олигоамидов, олигофениленов, фе-
ноло-формальдегидных смол и др.). Благодаря своей
универсальности получила широкое распространение
гель-проникающая хроматография, позволяющая не
только разделить О.-гомологи, но одновременно оце-
оценить молекулярно-массовое распределение смесей.
Мо леку лярно-массовое распределение
и распределение по функциональности
Подобно высокомолекулярным соединениям, О. ха-
характеризуются различным молекулярно-массовым рас-
распределением (ММР) в зависимости от способа получе-
получения и условий проведения процесса. Во многих слу-
случаях ММР для О. более узкое, чем для соответствую-
соответствующих высокомолекулярных полимеров.
461
ОЛИГОМЕРЫ
462
При получении О. с функциональными группами
часто образуются продукты с различной функциональ-
функциональностью, т. е. отношение мол. масс, определенных фи-
зич. методом и по концевым группам, выражается не
целым числом и зависит от механизма роста цепи и
природы неконтролируемых реакций обрыва. Напр.,
олигобутадиен-а,со-диолы, полученные в присутствии
6\6-азо-бмс-F-цианпентанола), в диапазоне средних
мол. масс от 1000 до 4000 (в зависимости от соотноше-
соотношения мономер : инициатор) являются практически би-
бифункциональными продуктами и характеризуются уни-
унимодальным и сравнительно узким ММР (Mw/Мп =
= 1,22—1,32), не зависящим от соотношения мономер :
инициатор.
О. бутадиена (Мп=2000), полученные в присутствии
перекиси водорода, содержат наряду с бифункциональ-
бифункциональными моно- и полифункциональные О., причем функ-
функциональность их возрастает с увеличением мол. массы
фракции и для фракции Мп = 10 000 составляет —5 при
отношении MwlМп ок. 2.
Олигобутадиены и олигостиролы, синтезированные по
анионному механизму, характеризуются узким ММР;
в этом случае отношение Mw/Mn близко к 1.
Форма кривой ММР олигостирола, полученного оли-
гомеризацией мономера в присутствии динитрила азо-
бмс-(изомасляной) к-ты, близка к кривой ММР высоко-
высокомолекулярного полистирола, однако ММР олигомера
более узкое, чем у полимера, для к-рого Mw/Мп = 2.
Олигоэфирдиолы, полученные поликонденсацией ади-
пиновой к-ты с различными гликолями (этилен-, диэти-
лен- и триэтиленгликолями), характеризуются равно-
равновесным, отличным от наиболее вероятного, распре-
распределением. Для каждой среднечисловой мол. массы
имеется свое собственное ММР, и полидисперсность
возрастает с увеличением Мп. Отношение MjMn
олигоэтиленадипинатов меняется от 1,15 для Мп =
= 550 до 1,85 для Мп= 3470, а в случае олиготриэтилен-
адипината — от 1,18 для - Мп = 330 до 1,71 для Мп-=
= 3600. ММР олигоэфирдиолов на основе ди- и
триэтиленгликолей идентичны и_достигают наиболее
вероятного распределения (MwlMn = 2) при степени
полимеризации —15, а олигоэфирдиолов, полученных
из этиленгликоля, лишь при степени полимеризации
-25.
Фракционирование олигоэтиленадипината на колонке
с силикагелем позволило обнаружить в нем наличие
дифункциональных, моно- и безфункциональных (цик-
(циклических) соединений.
Получение
Процессы получения О. обычно наз. о л и г о м е-
ризацией. Большинство методов синтеза О. осно-
основано на реакциях ограничения роста макромолекул
в процессах полимеризации и поликонденсации. Кроме
того, О. получают деструкцией высокомолекулярных
полимеров в контролируемых условиях. Для синтеза
монодисперсных О. разработаны специфич. методы, осу-
осуществляемые ступенчато с выделением продуктов ре-
реакции на каждой стадии.
Полимеризационные методы. Полимеризация с обра-
образованием О. м. б. осуществлена по радикальному, ион-
ионному и анионно-координационному механизмам. Мол.
массу образующегося продукта регулируют введением
агентов передачи цепи или изменением концентрации
инициатора (катализатора). Олигомеризация, осущест-
осуществляемая в присутствии передатчиков цепи (телогенов),
определяющих природу концевых групп, наз. теломе-
ризацией.
При инициировании радикальной полимеризации
виниловых мономеров фрагменты инициатора входят
в макромолекулу в виде концевых группировок.
О. с концевыми функциональными группировками полу-
получают при использовании в качестве инициаторов сое-
соединений, распадающихся на радикалы, каждый из
к-рых содержит еще и реакционноспособную функцио-
функциональную группу, напр.:
ОН НО
он
он
CN CN CN
I I I
HOOC (CH2JC - N = N - С (CH2J COOH -* HOOC (CH2J C*+ N2
I I I
CH3 CH3 CH3
CN CN CN
I I I
HO (CH2KC - N = N - С (CH2KOH -* 2HO(CH2K C'+ N,
i I I
CH3 CH3 СНз
В случае олигомеризации мономеров, для к-рых об-
обрыв цепи осуществляется в основном за счет рекомби-
рекомбинации растущих радикалов (скорости реакций передачи
цепи на мономер и растворитель незначительны), обра-
образуются О. с функциональными группами на обоих
концах молекулы. Этим методом были получены а,со-
диокси-, а,со-дикарбоксипроизводные олигостиролов
(п = 12—190), олигоакрилонитрилов (п = 13—95),
олигобутадиенов (п = 30—150) и др. При олигомериза-
олигомеризации мономеров, для к-рых характерен обрыв цепи
диспропорционированием растущих радикалов, обра-
образуется смесь моно- и бифункциональных О. Природа
одной из функциональных групп определяется типом
используемого инициатора, вторая концевая группа
у бифункционального О. является двойной связью.
При катионной олигомеризации олефинов на катали-
катализаторах Фриделя — Крафтса и сильных к-тах обра-
образуются О. со степенью полимеризации от нескольких
единиц до нескольких десятков единиц. Ограничение
роста цепи макромолекулы в этом случае связано
с относительно высокой скоростью передачи цепи на
мономер по сравнению со скоростью роста. Вследствие
нестабильности и высокой реакционной способности
первичного карбкатиона значительное развитие полу-
получают вторичные реакции (изомеризация и перенос ка-
катиона), в результате чего образуются О. низкой мол.
массы и чрезвычайно разветвленной структуры. Широ-
Широко известным примером реакции такого типа является
получение синтетич. моторных топлив и смазочных ма-
масел олигомеризацией этилена и а-олефинов с примене-
применением в качестве катализаторов А1С13, BF3, а также сер-
серной и фосфорной к-т.
Анионно-координационная олигомеризация исполь-
используется для получения олигомерных олефинов (этилена,
пропилена, а-бутилена, изобутилена и др.). О. с мол.
массой до 3000—5000 синтезируют, осуществляя оли-
гомеризацию в присутствии агентов передачи цепи —
водорода или галогенсодержащих алифатических или
ациклич. углеводородов. С увеличением парциального
давления водорода мол. масса олигоолефинов умень-
уменьшается. При олигомеризации этилена на системе TiCl4—
А1(С2Н5K при давлении водорода 5—10 Мн/м2 E0—
100 кгс/см2) образуется высококристаллический низко-
низкоплавкий изотактический олигоэтилен с молекулярной
массой —1000.
Пропилен и высшие олефины более чувствительны
к добавкам водорода. Низкомолекулярные олигопро-
пилены от жидких масел до восков образуются при дав-
давлении водорода 0,25—0,30 Мн/м2 B,5—3 кгс/см2).
Природа катализатора оказывает существенное влия-
влияние на мол. массу олиго-а-олефинов. На системе
TiCl4 — С12А1(С2Н5) при прочих равных условиях
образуются более низкомолекулярные О., чем на
системе TiCl4 — А1(С2Н5),.
463
ОЛИГОМЕРЫ
464
При олигомеризации в р-ре галогенсодержащих раст-
растворителей мол. масса О. тем ниже, чем более активным
агентом передачи цепи является растворитель. С по-
повышением темп-ры процесса мол. масса О. уменьшается
и повышается степень его ненасыщенности. Олигомер-
ные олефины представляют собой вязкие жидкости,
воскоподобные или низкоплавкие кристаллич. вещест-
вещества, хорошо растворимые во многих органич. раствори-
растворителях.
Ряд особенностей анионной полимеризации, проте-
протекающей с образованием «живых» цепей (безобрывность
процесса, протекание реакции до полного исчерпания
мономера, легкость регулирования мол. массы, высо-
высокая реакционная способность растущих макроанионов),
позволяет широко использовать эту реакцию для целе-
целенаправленного получения О. с узким молекулярно-
массовым распределением (см. Живущие полимеры).
Реакционноспособные О. получают обработкой «жи-
«живых» цепей соответствующими реагентами, обеспечи-
обеспечивающими введение заданных концевых группировок,
напр.:
~СН—СНСН2ОН +NaX
R R
~СН— СО ОН + NaX
R
-СН-СОСН2СН2СООН
R
NaX
СН—СО
R
NH2 + R'ONa
~CHSi(CH3JCH=CH2+ NaCl
R
Анионной олигомеризацией получены различные
а,со-производные олигостирола, олигобутадиена, оли-
гоизопрена и др. О. и их гидрированные аналоги.
Широко распространенный способ получения гетеро-
цепных О.— олигомеризация гетероциклов (циклич.
окисей, тиоокисей, лактамов, иминов, сил океанов и
др.). Так, анионной олигомеризацией окисей этилена,
пропилена или их смесей получают олигогликоли с мол.
массой от 300 до 8000, известные под фирменными на-
названиями «карбовакс», «плюроник» и «тетроник». Олиго-
Олигомеризацией е-капролактона или его смеси с метил-е-кап-
ролактоном в присутствии этилен- или диэтиленглико-
ля под действием щелочей синтезируют олигоэфирдио-
лы, используемые в производстве термо- и водостойкого
полиуретанового волокна.
Реакционноспособные О. часто получают катионной
полимеризацией гетероциклич. мономеров в присутст-
присутствии регуляторов мол. массы — агентов передачи цепи
(вода, гликоли, органич. к-ты, их ангидриды, эфиры и
др.). Этим способом получают олигогликоли на основе
окисей этилена, пропилена, тетрагидрофурана и их сме-
смесей, олигоацетали на основе этиленгликольформаля,
диэтиленгликольформаля, пропил енгликольформаля
и др. Регулируемой полимеризацией гетероциклов
в присутствии непредельных соединений, способных
генерировать карбкатион и не претерпевающих в ус-
условиях реакции каких-либо изменений, связанных
с двойными связями, получают полимеризационноспо-
собные олигомеры, напр.:
/я о с„ + [сн2=с(сн3)со] о —¦
ch2=c(ch3)co[ocJ осос(сн3)=сн2
сн2-оЧсн
CH2— 0/
С - 9H2~CH2\
¦п Лн -си /
^П2 ^Гт2
Kat — BF3, SbCI5
Катионной олигомеризацией тетрагидрофурана, 1,3-
диоксолана и 1,3,6-триоксокана в присутствии метакри-
лового ангидрида получены а,со-диметакриловые про-
производные олиготетраметиленгликолей, олигоэтилен- и
олигодиэтиленгликольформалей с мол. массой от 500
до 10 000.
Олигомеризацией низкомолекулярных циклич. оли-
госилоксанов в присутствии дигалогеналкилсиланов
получают линейные а,со-дигалогенолигосилоксаны.
Путем взаимодействия последних с соответствующими
реагентами синтезируют олигосилоксаны с различны-
различными концевыми группами:
R R
\ /
О
Si
О
I
Si
R +
г 1
Cl2SiRj —*- Cl -(R2)Si0- CI—*-'
L J/7
-CH2=CH f—(R2) Si O-l Si (R2) CH=CH2
*HO f-(R2) SiO—1 Si (Rj) OH
П 1
Г П 1 .
-(R2)Si0~ Si(R'2)CH2O(CH2JOH
Г 1
CH2-C(X)COOCH2 -(R2)Si0- Si(R2)CH2OCOCX=CH2
Мол. масса О. возрастает с увеличением молярного
соотношения I : II.
Поликонденсационные методы. В процессе поликон-
поликонденсации регулирование мол. массы образующихся
продуктов можно осуществлять след. способами: 1) пре-
прекращением реакции при низких степенях превращения;
этот принцип получения различных О. широко исполь-
используется при производстве таких крупнотоннажных про-
продуктов, как феноло-формальдегидные, карбамидные,
эпоксидные, алкидные и др. смолы; 2) использованием
избытка одного из компонентов; по этому способу полу-
получают олигоэфирдиолы с мол. массой 1000—3000, при-
применяемые в производстве полиуретанов, а также не-
непредельные олигоэфиры с мол. массой от 600 до 5000,
нашедшие широкое применение в качестве компонен-
компонентов связующего для стеклопластиков; 3) введением
в реакционную смесь монофункционального соедине-
соединения, блокирующего функциональные группы одного
типа; в качестве примера можно привести синтез оли-
гоэфиракрилатов, осуществляемый конденсацией двух-
двухосновных к-т с гликолями в присутствии телогена —
акриловой к-ты.
Деструктивные методы. Одним из методов синтеза
О. является деструкция высокополимеров. Напр.,
озонолизом полиизобутилена (мол. м. 80 000), содер-
содержащего 2% (молярная концентрация) изопреновых
остатков в положении 1,4 , получен а,со-олигоизобути-
лендиол с мол. м. 2850, что отвечает средней мол. массе
465
ОЛИГОМЕРЫ
466
отрезков цепи, находящихся между изопреновыми
остатками. Олигосульфиды с мол. массой от 600 до 8000
получают обработкой высокомолекулярных полисуль-
полисульфидов неорганич. моносульфидами, бутилмеркаптаном,
меркаптоэтанолом и др. соединениями, вызывающими
расщепление дисульфидных связей.
Ступенчатый синтез. О., полученные по всем описан-
описанным выше методам, представляют собой смесь гомоло-
гомологов. Для получения химически индивидуальных О.
разработаны специальные методы, основанные на сту-
ступенчатом проведении процесса с выделением продуктов
реакции на каждой стадии. Молекулярная однород-
однородность таких О. доказана методами тонкослойной хро-
хроматографии, бумажного электрофореза или гель-хро-
гель-хроматографии. Ниже рассмотрены три различных ва-
варианта ступенчатого синтеза.
1. Ступенчатая конденсация дифункционального
соединения с др. дифункциональным реагентом, одна
из функциональных групп к-рого блокирована. Для
осуществления каждой последующей стадии блокирую-
блокирующую группу удаляют, восстанавливая функциональ-
функциональность. Так, синтез олигоэтилентерефталата с п = 1 —5
осуществлен взаимодействием диолов с эфирхлорангид-
ридом терефталевой к-ты или же монопроизводного
диола с хлорангидридом терефталевой к-ты.
Блокированием одной из функциональных групп
оксикислот, аминокислот и др. бифункциональных
соединений были получены соответствующие О. Напр.,
олигооксипивалаты (п = 1—8)
ZOCH2C (СН3JСОС1 + Н [ОСН2С (CH3JCO]WOR —
н+
-* Z [OCH2C (CH3JGO]n+1R —> Н [ОСН3С (CH3JCO]n+1OR
где Z — блокирующая группа, олигокапроамиды
(п = 2—10)
NH2(CH2MGOOH SO2C12
СвН5СН2ОСОС1 > GeH6GH2OCONH (CH2MGOOH >
—* СвНБСН2ОС(ЖН(СН2)БСОС1
NH2(CH2MCOOH
SO2G12
> GeH5CH2OCONH (CH2MCONH (GH2MCOOH >
> CeH5CH2OCO [NH (CH2MCO]2G1
H[NH(CH2MCO]nOH
H2/Ni
-* CeH5CH2OGO [NH (CH2NCO]n+2OH-
-» H [NH (СН2NСО]И+2ОН
олигоаланины (n = 2,3,5,10), олиго-у-метил-Ь-глута-
маты (п = 2 — 9), олигодиэтил-Ь-аспарагинаты (п =
= 2 — 7,11,14), а также смешанные олигопептиды
с заданным чередованием остатков аминокислот (о сту-
ступенчатом синтезе олигопептидов см. Полипептиды).
2. Исключительно плодотворным является ступен-
ступенчатый синтез олигомеров на полимерах-носителях,
разработанный сначала для нерастворимых сетчатых
полимеров, а затем модифицированный для линейных
растворимых полимеров. По этому способу можно
получать монодисперсные олигомеры с высокими сте-
степенями полимеризации. В качестве полимера-носителя
часто используют хлорметилированный полистирол.
Синтез олигоамидов проводится согласно схеме:
' СвН4СН2С1 + ВиОСО(ВА)ОН
> ~> СвН4СН2О(АВ)Н
CeH4CH2O(AB)OGOBu
BuOCO (BA) ОН
CF3COOH
> ~> GeH4GH2O(ABJOGOBu -
GF3COOH
BuOCO (AB)nOH
CF3COOH
> ~ CeH4GH2O(AB)n+2COOBu > НВг-Н (BA)n+2OH
НВг
где
Bu
a-, В = - NH (CH2),NH -,
- ОС (СН2LСО -
Этим методом были синтезированы олигогексамети-
ленадипамид (п = 2,3,4,5,10), олигоундеканамид (п =
= 2,3,4,5,10), олигокапроамид (п = 2 — 25) и ряд др.
олигоамидов и олигопептидов.
3. Т. наз. метод удвоения (дубликации) заключается
в постадийном взаимодействии бифункциональных со-
соединений, одно из к-рых взято в большом избытке
(до 200-кратного), что исключает развитие реакции
роста цепи. Метод применим лишь для необратимых
полиреакций, напр, для получения олигоуретанов
из диолов и диизоцианатов:
А + В (избыток) > ВАВ
ВАВ + А (избыток) > (АВJА
(АВJА + В (избыток) > (АВKВ
(АВJА + ВАВ (избыток) > (АВLВ
где А — диол, В — диизоцианат.
Этим методом были синтезированы диололигоуретаны
на основе гексаметилендиизоцианата и 1,4-бутандиола
Н [- О (CH2LOCONH (CH2)8NHCO -]nO (CH2LOH
где п = 1—7,9, на основе диэтиленгликоля и гекса-
гексаметилендиизоцианата
Н [- О (CH2JO(CH2JOCONH (CH2)eNHCO -]nO (CH2JO (CH2JOH
где п= 1—5,7,15, и на основе бутандиола-1,4 и о-ани-
зидиндиизоцианата
Hf-O(CH2LOCONH-/)Kyy-NHCO-]o(CH2) ОН
НоС
сн3
где п = 1—3,7,15.
Получение циклоолигомеров. Циклоолигомеры —
макроциклич. олигомерные соединения, содержащие
два или более повторяющихся мономерных звеньев.
Эти соединения часто образуются в результате проте-
протекания побочных процессов при получении высокомоле-
высокомолекулярных полимеров методами полимеризации или
поликонденсации. Циклоолигокапроамид с п — 2 —8
и циклоолигоундеканамид с п = 2,3 выделены методом
гель-хроматографии из полиамида-6 и полиамида-11
соответственно. Циклич. тример метакрилонитрила
и циклич. тетрамер оксациклобутана обнаружены
в продуктах анионной олигомеризации метакрило-
метакрилонитрила и катионной полимеризации оксациклобутана.
Циклич. три-, тетра- и пентамеры выделены из поли-
этилентерефталата, циклоолигомеры с п =2 — 9 —
из продуктов гидролитич. поликонденсации дихлор-
силанов (см. Кремнийорганические полимеры) и т. д.
Целенаправленный синтез циклоолигомеров осу-
осуществляют циклизацией соответствующих линейных
олигомеров в сильно разб. р-рах A—2%-ных). Напр.,
циклич. олигоамиды получают циклизацией тиоэфиров
A) или разложением азидов соответствующих к-т B):
CeH5SH
С6НБОСО [NH (СН2NСО]„ОН >
НВг, CH.COONa
»CeH5OCO [NH (CH2NCO]nSCeHB >
(CH3JNCOH
» Н [NH (CH2MCO]nSCeH5 >
> p[NH (CH2MCO]n -1
CeH5OCO [NH(CH2MCO]n NHNH2 *
> H [NH(CH2NCO]nN3 - ,-[NH (CH2MCO]n-.
A)
B)
Циклоолигомеры под различными воздействиями могут
превращаться в высокомолекулярные соединения.
Применение
Одна из наиболее важных областей применения
О.— производство полимерных изделий и материалов.
Использование реакционноспособных О. в этой обла-
области создает ряд преимуществ по сравнению с техноло-
467
ОЛИГОЭФИРАКРИЛАТЫ
468
гией производства полимерных изделий и материалов,
основанной на переработке высокомолекулярных по-
полимеров: 1) процессы получения высокомолекулярного
полимера и изготовления из него изделия можно осу-
осуществлять в одну операцию, 2) жидкие или легко-
легкоплавкие О., обладающие высокой текучестью в жидком
состоянии, превращаются в полимерные изделия лю-
любого размера и формы в сравнительно мягких условиях
без применения высоких давлений, темп-р и раствори-
растворителей; 3) процесс получения высокомолекулярных
соединений из О. сопровождается значительно мень-
меньшими тепловыми эффектами и усадками, чем при
полимеризации мономера (т. к. в последнем процессе
число элементарных актов присоединения значительно
больше), что позволяет форсировать скорость перехода
жидкость — твердый полимер и создавать высокопро-
высокопроизводительные процессы изготовления изделий без при-
применения сложного технологич. оборудования. Особый
интерес с этой точки зрения представляют полимери-
зационноспособные О., отверждающиеся без выделе-
выделения побочных продуктов (в случае поликонденсацион-
поликонденсационных процессов обычно необходимо отводить побочные
продукты реакции).
Отличительной особенностью высокомолекулярных
полимеров, полученных из О. с концевыми реакцион-
носпособными группами, является регулярность строе-
строения. При этом величина блоков, ответвлений и меж-
межузловых цепей соответственно линейных, разветвлен-
разветвленных или сетчатых полимеров определяется мол. массой
исходных продуктов. О. со случайным расположением
реакционноспособных групп или полифункциональные
О. образуют нерегулярные разветвленные и сетчатые
полимеры со статистич. распределением ответвлений
и сшивок.
Из большого числа различных реакционноспособных
О., нашедших широкое применение в различных отрас-
отраслях пром-сти, можно упомянуть эпоксидные, феноло-
формальдегидные, алкидные, карбамидные и др. смолы,
применяемые для производства слоистых материалов,
пенопластов, лаков, клеев, компаундов, для обработки
тканей, бумаги, в производстве древесноволокнистых
материалов, для модификации др. полимеров и т. д.;
олигоалкиленгликоли на основе этилен-, пропилен-
оксидов, тетрагидрофурана и их смесей (см. Окисей
органических полимеры), олигоэфирдиолы на основе
гликолей и двухосновных к-т, используемые для полу-
получения уретановых каучуков, пенополиуретанов и др.;
полиэфирные смолы (см. Полиалкиленгликолъмалеинаты
и полиалкиленгликолъфу Мараты)', олигоэфиракрилаты
и др. ненасыщенные олигоэфиры, применяемые в про-
производстве стеклопластиков, лаков и эмалей, литьевых
и пропиточных компаундов и др.; жидкие каучуки,
используемые в качестве компонентов электроизоля-
электроизоляционных лаков, герметизирующих и виброгасящих
материалов, для гуммирования тканей и др.
Продукты радикальной и ионной теломеризации
этилена и др. олефинов используются как исходное
сырье в производстве разнообразных органич. веществ;
продукты катионной олигомеризации олефинов при-
применяются в качестве синтетич. моторных топлив и сма-
смазочных масел. Полиэтиленовые воски, получаемые
олигомеризацией этилена в присутствии водорода,
галогеналканов и др. регуляторов мол. массы, исполь-
используются для гидрофобизации бумаги, картона, как ком-
компоненты полировальных паст, наполнители резин
и др. О. тетрафторэтилена и трифторхлорэтилена при-
применяются как высококипящие масла, термостойкие
теплоносители, жидкости для гидроприводов, пласти-
пластификаторы для высокомолекулярных полимеров и пр.
О. на основе окисей олефинов и их смесей нашли широ-
Jtoe применение в качестве неионогенных поверхност-
поверхностно-активных веществ, вспомогательных веществ в тек-
текстильной пром-сти, смазочных материалов и др. Оли-
гоэтиленимины используют для увеличения накраши-
ваемости и придания гидрофильности тканям, бумаге,
пластикам и др.
Олигомерные вещества играют немаловажную роль
в природе. Напр., окситоцин (гормон задней доли
гипофиза) представляет собой нонапептид: грамици-
грамицидин С — это циклодекапептид; многочисленные бак-
бактерии вырабатывают антибиотики, являющиеся цик-
лоолигопептидами; из плазматич. глобулина выделены
нонапептидбрадикинин и декапептид — каллидин Ш.
Лит.: Цан [Г.], Глайтсман [Д ж.], Хим. и тех-
нол. полимеров, № 5, 46 A964); Heidemann G., в кн.:
Encyclopedia of polymer science and technology, v. 9, N. Y.—
[a.o.], 1968, p. 485; Б е р л и н А. А., М а т в е е в а Н. Г.,
в кн.: Успехи химии и физики полимеров, М., 1970; Ц у т и-
д а Э., Синохара И., Хим. и технол. полимеров, № 10,
16 A965); Энтелис С. Г., Евреи нов В. В., К у за ев А. И.,
в сб.: Успехи химии и физики полимеров, М., 1973.
Я. Г. Матвеева, А. А. Берлин.
ОЛИГОЭФИРАКРИЛАТЫ, полиэфиракри-
л а т ы (oligoesteracrylates, Oligoesterakrylate, oligoester-
acrylates) — олигомерные сложные и простые эфиры
с концевыми или регулярно чередующимися акрило-
акриловыми (метакриловыми, хлоракриловыми и др.) груп-
группами.
Получение. О., при синтезе к-рых образуются с л о ж-
ноэфирные группы, получают в пром-сти об-
обратимой конденсационной теломеризацией. Метод осно-
основан на введении в конденсацию наряду с ди- или поли-
полифункциональными реагентами (спиртами и карбоно-
выми к-тами) регуляторов роста цепи — телогенов.
Последние содержат группу, принимающую участие
в конденсации, а также полимеризационноспособную
группу, не участвующую в конденсации; телогенами
служат к-ты акрилового ряда и их моноэфиры с диокси-
соединениями.
Образующаяся при полиэтерификации вода удаляет-
удаляется из сферы реакции азеотропной отгонкой с приме-
примененным растворителем, что обусловливает сдвиг рав-
равновесия в сторону образования О. Ниже приведена
схема конденсационной теломеризации для дифунк-
циональных реагентов:
Н*
nHOOGRCOOH + (n + 1) HOR'OH + 2СН2=С(Х)СООН «±
О 0 0 О
II II II II
«± CH2=C(X)COR'O [- CRCOR'O-]nCC(X)=CH2 + 2(n+l)H2O
где R и R' — алкильные или арильные двухвалентные
радикалы; X = Н, СН3, Hal, CN и др.
Средняя степень (п) полимеризации О. зависит
от концентрации исходных компонентов, но опреде-
определяется гл. обр. соотношением:
- 2_щ кг
[В] ' и2
где [L] и [В] — число молей соответственно двухоснов-
двухосновной и одноосновной к-т; кх — константа скорости роста
цепи при реакции дифункциональных мономеров;
к2 — константа скорости обрыва цепи при реакции
с монофункциональным соединением (телогеном).
Для получения О. с п = 1 при функциональности
гликоля (или полиола) F > 2 исходные компоненты
должны быть взяты в соотношении:
где [D] — число молей гликоля (или полиола). При
получении более высокомолекулярных теломеров и
F > 2 компоненты реакции берутся в соотношении:
Щ : [D]: [В] = п : (я + 1): [я (F - 2) + F)
Исходными соединениями для синтеза О. служат:
1) гликоли, полиолы или двухатомные фенолы (напр.,
моно- ди- и триэтиленгликоли, 1,3-пропиленгликоль,
1,4-бутиленгликоль, 1,6-гексаметиленгликоль, триме-
тилолпропан, глицерин, пентаэритрит, ксилит, дифе-
нилолпропан, фенолфталеин);
469
ОЛИГОЭФИРАКРИЛАТЫ
470
2) алифатич. дикарбоновые насыщенные к-ты с чис-
числом метиленовых групп в цепи 2—8 (в том числе содер-
содержащие сульфидные и сульфоновые группы), а также
ароматич. двух- или полиосновные к-ты либо их ангид-
ангидриды (напр., изофталевая, терефталевая, тримеллито-
вая к-ты, фталевый ангидрид);
3) а,р-ненасыщенные монокарбоновые _>?л
к-ты акрилового ряда (чаще всего мета-
криловая или акриловая к-та). Для синтезаО. с огра-
ограниченной горючестью и рядом др. специфич. свойств
используют галогенсодержащие компоненты.
Процесс осуществляют в среде инертных раствори-
растворителей (ароматич. углеводородов, хлорпроизводных
ароматич. или алифатич. углеводородов) при 80—
140 °С и интенсивном перемешивании в присутствии
в качестве катализаторов минеральных к-т или орга-
органич. сульфокислот (бензолсульфокислоты, га-толуол-
сульфокислоты, а-нафталинсульфокислоты или др.),
а также нек-рых типов катионообменных смол (напр.,
марок КУ-2, КУ-8). Весьма перспективны полисуль-
фофениленхиноны — катализаторы гетерогенного типа,
к-рые являются также ингибиторами полимеризации
образующегося О. и телогена. При использовании др.
катализаторов в реакционную смесь дополнительно
вводят ингибитор (гидрохинон, бензохинон, метилено-
вый синий и др.).
Технологич. процесс включает стадии: конденсаци-
конденсационной теломеризации, нейтрализации и промывки р-ра
полученного О., отгонки растворителя.
Выход в зависимости от природы исходных соеди-
соединений и вязкости О. обычно составляет 65—75%.
Принцип конденсационной теломеризации может
быть использован для получения О. переэтерифика-
цией, напр.:
О 0 0
II II II
2СН2 = С(Х)С-ОСН, + nCH3OCRCOCH3 + (n+l)H0R'0H «±
О 0 0 0
II II II II
CH2=C(X)C-OR'OCRCOR'OC(X)C
2(п+1)СН3ОН
Образующийся метанол отгоняют вместе с инертным
растворителем. Достоинства метода — возможность
использования в качестве телогена метилметакрилата,
проведение процесса при более низкой темп-ре и за бо-
более короткий срок, чем этерификацию, более свет-
светлая окраска получаемых О. Недостатки — возмож-
возможность миграции водорода гидроксильной группы к не-
ненасыщенной группе О. с образованием соединений,
не способных к дальнейшей полимеризации. Промыш-
Промышленное применение метода лимитировано дефицит-
дефицитностью метиловых эфиров дикарбоновых к-т.
О. могут быть получены также методами необрати-
необратимой конденсационной теломеризации:
а) Межфазной поликонденсацией', реакцию проводят
между хлорангидридами двух- и моноосновной к-т,
растворенными в гидрофобном органич. растворителе,
и водным, чаще всего щелочным, р-ром бисфенола или
гликоля:
О
II NaOH
2СН2 =С(Х)-С - Gl + nR(GOGlJ + (п +1) R'(OH), *
О 0 0 0
II II II II
•— CH2=C(X)COR'OCRCOR'OCC(X)=CH2 + 2(n+l)NaCl
Этот метод особенно целесообразен, когда применение
азеотропного метода затруднено, напр, вследствие
плохой растворимости или малой реакционной способ-
способности исходных веществ.
б) Низкотемпературной теломеризацией в р-ре орга-
органич. растворителей в присутствии акцепторов выделяю-
выделяющегося НС1 (органич. оснований). Этот метод исполь-
использован для получения олигокарбонатакрилатов — ана-
аналогов О., содержащих карбонатные группы. Реакцию
осуществляют между ди- или полиоксисоединениями,
их хлоругольными эфирами и моноэфирами диокси-
соединений и к-т акрилового ряда; темп-pa начала ре-
реакции от —8 до —5 °С, среда — низшие хлоралканы,
в присутствии пиридина:
2СН2 = G(X)GOOROH -f- п HOR'OH + (п + 1) ClOCOR^OGOGlC^H5N
G(X)COOROGOOR//O[GOOR'OGOOR//O]nGOOROGOG(X) = GH2+2(n-|-l)C5H5N-HC
(R,R',R" — алкильные или арильные радикалы;
X = Н, СН3; п = 0 или целое число >1).
Этим же методом при использовании хлорангидрида
алкилфосфиновой к-ты получают фосфорсодержащие
О., при отверждении к-рых образуются самозатухающие
полимеры.
О., при синтезе к-рых образуются простые
эфирные группы, получают регулируемой ка-
тионной теломеризацией циклич. мономеров (напр.,
тетрагидрофурана, диоксолана, 1,3,6-триоксакана). В
качестве телогенов используют метакриловый ангид-
ангидрид, акриловые к-ты, их хлорангидриды или диэфиры,
а также различные аллиловые производные; катали-
катализаторами служат протонные к-ты или к-ты Льюиса.
Варьируя соотношение между количествами моно-
мономера, телогена и катализатора, удается получить О.
заданной мол. массы E000—15 000) — а,ш-димет-
акриловые эфиры олиготетраметиленгликолей, а также
олигоэтилен- и диэтиленгликольформалей общей ф-лы
СН2= C(CH3)CO[OR]nOGOC(GH3)=CH2
где R — упомянутые выше двухвалентные радикалы.
Строение и свойства. О. могут быть линейными
(из бифункциональных исходных компонентов) или
разветвленными (из три- или полифункциональных
соединений). Промышленные О. могут характеризо-
характеризоваться небольшими кислотными числами @,5—5,0),
обусловленными природой катализатора, темп-рой и
присутствием влаги; значения этих чисел в нек-рых
случаях могут возрастать при хранении из-за гидролиза
образующихся нестойких сульфоэфиров гликолей. Вы-
Выделяющаяся к-та, по-видимому, может катализировать
гидролиз О., для предотвращения к-рого вводят ста-
стабилизирующие добавки, напр, эпоксидные смолы в ко-
количестве 0,5—1,0% (по массе), не ухудшающие физико-
механич. свойства полимеров.
О.— бесцветные прозрачные нелетучие жидкости
различной вязкости [E0—8000)-10~6 м2/сек, или
50—8000 ест] или твердые бесцветные кристаллич.
продукты; мол. масса 300—5000; плотность 1,08—
1,30 г/см3 B0 °С); w2D0 1,4600—1,5480. О., синтезиро-
синтезированные из низших алифатич. или ароматич. о-дикар-
боновых к-т и гликолей (или полиолов) — вязкие жид-
жидкости, не перегоняющиеся под высоким вакуумом. О.
на основе себациновой к-ты и различных гликолей —
легкоплавкие кристаллич. продукты (т. пл. 33—40 °С).
Темп-pa плавления О. на основе ароматич. м- или
/г-дикарбоновых к-т и бисфенолов составляет 150—
180 °С; у О. на основе тиодивалериановой к-ты, содер-
содержащей сульфидную группу, темп-pa плавления ниже,
а на основе сульфондивалериановой к-ты, содержащей
сульфогруппу — SO2—, выше, чем у О. на основе себа-
себациновой к-ты. _
Большинство О. с п < 4 хорошо растворяется
в спирте, ацетоне, эфире, ароматич. углеводородах,
хлорбензоле, тетрагидрофуране, дихлорэтане, хлоро-
хлороформе и др. органич. растворителях, а также в мономе-
мономерах винилового и аллилового типа и не растворяется
в воде. Нек-рые свойства О. приведены в табл. 1.
О., синтезированные азеотропной конденсационной
теломеризацией, не являются монодисперсными соеди-
соединениями, т. к. в ходе процесса из-за различия скоро-
скоростей взаимодействия компонентов наряду с целевым О.
образуются его ближайшие гомологи, различающиеся
471
ОЛИГОЭФИРАКРИЛАТЫ
472
Таблица 1. Физико-химические свойства наиболее распространенных олигоэфиракрилатов
Исходные компоненты
as
аи о !
UoCi
Условное
обозначе-
обозначение
Средняя мол.
масса
найде-
найдено
Плот-
Плотность
при
20 °С,
г/см3
Пока-
Показатель
пре-
ломле-
ломления
20
D
П,
Эфирное чис-
найде-
Бромное чис-
найде-
но
Вязкость** при
20 °С, мЧсек
Фталевый ангидрид, триэти-
ленгликоль, метакриловая
к-та
Фталевый ангидрид, диэти-
ленгликоль, метакриловая
к-та
Фталевый ангидрид, этилен-
гликоль, метакриловая к-та
Фталевый ангидрид, 1,4-бу-
тиленгликоль, метакрило-
метакриловая к-та
Фталевый ангидрид, пента-
эритрит, метакриловая к-та
Фталевый ангидрид, глице-
глицерин, метакриловая к-та . . .
Фталевый ангидрид, ксилит,
метакриловая к-та
Адипиновая к-та, пентаэрит-
рит, метакриловая к-та . . .
Себациновая к-та, 1,4-бути-
ленгликоль, метакриловая
к-та
Тиодивалериановая к-та, ди-
этиленгликоль, метакрило-
метакриловая к-та
Триэтиленгликоль, метакри-
метакриловая к-та
МГФ-9
МДФ-1
МДФ-2
МЭФ-1
МБФ-1
7-3
ТМГФ-11
МКФ-1
7-20
МБС-1
МДТВ-1
ТГМ-3*
540,0
492
728
360
430
740
591
623,0
1150
474,0
509
281
566,0
478
714
390
446,5
810
586
670,6
1170
482,5
546,7
286,3
1,165
1,165
1,2130
1,180
1,135
1,2090
1,1800
1 ,2600
1,2190
1,0215
(при
50 °С)
1,1250
1,0922
1 ,4891
1,4930
1,5150
1,500
1,4940
1,5092
1,4890
1,5070
1,4924
1,4953
1,4850
1,4621
392,9
466,6
466,5
570,0
499,2
515,0
550,0
482,0
540
458,8
406,4
296,0
395,5
469,6
470,0
575,0
502,7
553,0
573,0
501,2
574
465,6
410,5
391,0
60,0
67,6
43,2
81,6
70,3
107,0
82,0
82,7
90,0
58,0
112,6
56,5
66,8
44,8
81,9
71,6
118,0
122,8
95,7
109,0
66,4
112,0
A00—150). 10-в
D0 — 80). 1 0~в
(800-900). 10-*
B0—60). 10~в
A00—130).Ю-»
C000—5000).10"в
E000).10-«
A0-30).Ю-»
* Продукт представляет собой тетрафункциональный мономер — диметакрилат триэтиленгликоля. ** 1 м2/сек=10* ест. *** По
средней формуле. **** Твердое вещество, т. пл. 33—40 °С. ***** Вязкость 18 500 мн-сек/м2, или сп (при 50 °С).
на 1—4 звена. Фракционный состав О. зависит так-
также от природы гидроксилсодержащего компонента,
обусловливающей способность его к различным поли-
полиреакциям.
О. способны полимеризоваться и сополимеризоваться
(т. е. отверждаться) с образованием неплавких и не-
нерастворимых полимеров пространственно-сетчатой
структуры.
Физико-химич., термомеханич., диэлектрич. и др.
свойства сшитых полимеров на основе О. в основном
определяются степенью полимеризации, физич. струк-
структурой полимера, химич. природой макромолекул, при-
природой и величиной поперечных «сшивающих» групп,
а также концентрацией мостичных связей (степенью
сшивания). С увеличением длины и гибкости О. сни-
снижаются темп-pa стеклования, модуль упругости и
твердость полимера, возрастают прочность к динамич.
нагрузкам, адгезия, диэлектрич. потери, теплопровод-
теплопроводность и равновесное набухание. Зависимость прочно-
прочности при растяжении и статич. изгибе от длины и гиб-
гибкости О. носит экстремальный характер, обусловлен-
обусловленный степенью сшивания полимера. С увеличением
функциональности О. увеличиваются термостойкость,
твердость и теплостойкость полимера, несколько сни-
снижаются уд. ударная вязкость и прочностные свойства.
Введение в молекулу О. ароматич. ядер (особенно
содержащих заместитель в .мета-положении), замена
метакриловых групп на акриловые, алкиленоксидных
групп на метиленовые и особенно сульфидные при-
приводит к образованию теплостойких и прочных поли-
полимеров. Нек-рые свойства отвержденных О. приведены
в табл. 2.
Плотность приведенных в табл. 2 отвержденных
О. составляет 1,23—1,35 г/см3 B0 °С); уд. объемное
электрич. сопротивление 1,5-1014—1,0«1015, тан-
тангенс угла диэлектрич. потерь при 1 Мгц 0,014—0,035,
диэлектрич. проницаемость при 1 Мгц 3,0—4,5, водо-
поглощение за 30 сут 0,35—6,5%.
О. способны сополимеризоваться друг с другом,
а также с большим числом ди- и полифункциональных
мономеров и олигомеров (напр., с полиэфирными эпок-
эпоксидными и глифта левыми смолами, непредельными
кремнийорганич. соединениями). О. хорошо совмеща-
совмещаются с различными высокомолекулярными соединения-
соединениями (эфирами целлюлозы, поливинилхлоридом, поли-
акрилатами, синтетич. каучуками и др.), образуя
материалы, обладающие ценными свойствами.
Путем совмещения и сополимеризации О. с высоко-
высокомолекулярными соединениями осуществляют модифи-
модификацию технологич. и физико-механич. свойств послед-
последних. В этом случае О. служит «реакционным пласти-
пластификатором», облегчающим переработку термопластов
Таблица 2. Свойства отвержденных олигоэфиракрилатов *
Показатели
МЭФ-1
МДФ-1 МДФ-2 МБФ-1 МГФ-9 МБС-1 МКФ-1
ТГМ-3
Прочность, Мн/м2 (кгс/см2)
при растяжении
при статич. изгибе
Относительное удлинение, %
Ударная вязкость, кдж/м2 (кгс • см/см2) .
Твердость по Бринеллю, Мн/м* (кгс/мм2)
Модуль упругости, Мн/мг {кгс/мм2) . . .
60 F00)
86 (860)
1,5
27
200 B0)
62 F20)
11,5 A150)
12,0
50
135 A3,5)
3300 C30)
86 (860)
16 A600)
15,0
100
86 (8,6)
84,5 (845)
35
128 A2,8)
2900 B90)
47 D70)
68 F80)
90
19 A,9)
750 G5)
11A10)
20B00)
95
9 @,9)
74 G40)
19
269 B6,9)
40 D00)
2,8
50
181 A8,1)
* Состав О. приведен в табл. 1.
473
ОЛИФЫ
474
и способным в процессе формирования изделия превра-
превращаться в сетчатый полимер, химически или физически
связанный с исходным полимером. При этом прост-
пространственно-сетчатый привитой сополимер образуется
в том случае, когда в линейном модифицируемом поли-
полимере имеются реакционные группы или атомы, уча-
участвующие в передаче цепи при переработке. В про-
противном случае осуществляется захват (иммобилиза-
(иммобилизация) линейного полимера пространственной сеткой
отвержденного О. с образованием трехмерных кла-
тратных («змейка в клетке») полимеров, сочетающих
свойства, присущие линейному полимеру и сетчатому
полимеру О.
Отверждение и применение. Использование О., как
и др. реакционноспособных олигомеров, создает ряд
преимуществ и, в частности, возможность совмещения
в одной операции процессов полимеризации и изготов-
изготовления изделий. В технике отверждение достигается пу-
путем радикальной полимеризации или сополимеризации
О. Переход от жидкого олигоэфиракрилата к прост-
пространственно-сетчатому полимеру наблюдается при очень
малых степенях превращения @,25—1%) и сопровож-
сопровождается скачкообразным нарастанием вязкости и поте-
потерей текучести. Элементарные реакции протекают в диф-
диффузионной области. Начальная скорость полимериза-
полимеризации в изотермич. условиях зависит от физич. свойств
О., гл. обр. от вязкости и способности молекул О.
к ассоциации. С увеличением глубины полимеризации
скорость определяется плотностью пространственной
сетки и гибкостью ее межузловых цепей; плотность
и гибкость, в свою очередь, зависят гл. обр. от размера
молекул и природы исходного О.
После достижения максимальной скорости наблю-
наблюдается резкое автоторможение полимеризации, обус-
обусловленное уменьшением подвижности молекул, содер-
содержащих двойные связи. Предельная степень превраще-
превращения, при к-рой начинается автоторможение, различна
для различных типов О., причем наблюдается корре-
корреляция предельной степени превращения с физич.
свойствами олигомера. Так, с увеличением длины и
гибкости молекул О., обусловленной присутствием
групп (напр., О или S), обладающих низкими барье-
барьерами вращения, эффективная скорость полимеризации
и предельная степень превращения увеличиваются.
Увеличение функциональности О. приводит к значи-
значительному уменьшению эффективной скорости процесса
при высоких степенях превращения. В случае О. с ко-
короткими и жесткими цепями предельная степень пре-
превращения при умеренных темп-pax характеризуется
низкими значениями, возрастающими с повышением
темп-ры (напр., при отверждении в адиабатич. усло-
условиях). Требуемая технологичность исходных компози-
композиций, а также необходимые эксплуатационные свойства
полимерных материалов и изделий обычно достигаются
путем сочетания различных марок О., а также О.
с др. типами полимеризационноспособных олигомеров
или мономеров. Отверждение О. в условиях перера-
переработки их в изделия обычно проводят в массе при ат-
атмосферном давлении; при этом побочные продукты
реакции не образуются. Усадка О. уменьшается с уве-
увеличением длины молекулы О.; для различных типов
О. объемная усадка при полимеризации составляет
4-10%.
Инициаторами отверждения при 50—120 °С (горячее
отверждение) служат перекиси бензола и дикумила,
дициклогексилпероксидикарбонат и др. Для отверж-
отверждения при комнатных темп-pax (холодное отверждение)
применяют бинарные инициирующие системы (напр.,
перекись бензоила 4- диметиланилин; гидроперекись
кумола -f- нафтенаты или линолеаты кобальта). Для
отверждения О., содержащих сульфидные группы,
используют инициаторы неперекисного типа, напр,
динитрил азодиизомасляной к-ты. Отверждение О.
может быть также инициировано светом, излучением
высокой энергии (у-лучи, быстрые электроны) и ката-
катализаторами ионной полимеризации.
Весьма перспективен метод полимеризации в адиа-
адиабатич. условиях, когда отверждение проводят при
изменяющейся во времени темп-ре. Образующийся
полимер находится в высокоэластич. состоянии, что
создает благоприятные условия для роста и обрыва
цепи, вследствие чего достигаются и более высокие
степени превращения.
О. применяют как связующее в производстве стекло-
стеклопластиков, защитных, отделочных и электроизоля-
электроизоляционных лакокрасочных материалов, клеев для склеи-
склеивания металлов и неметаллич. материалов, литьевых
пластмасс, компаундов, полимербетонов для бесшовных
покрытий полов и стен, а также печатных форм и ана-
анаэробных герметиков. Водные эмульсии О. можно ис-
использовать для пропитки волокнистых материалов
или совмещать с латексами различных синтетических
полимеров.
Впервые О. разработаны и освоены в промышленном
масштабе в СССР.
Лит.: Берлин А. А., Кефели Т. Я., Коро-
Королев Г. В., Полиэфиракрилаты, М., 1967; Берлин А. А.,
Коровин Л. П., Сумин И. Г., Высокомол. соед., il Б,
Кя 6, 424 A969); Р а с к и н а Л. П., Туровская Л. Н.,
Берлин А. А., Пласт, массы, № 12, 7 A966); Берлин
А. А. [и др.], ЖПХ, 42, в. 4, 973 A969); и х ж е, там же,
42, в. 5, 990 A969); Б е р л и н А. А., М а т в е е в а Н. Г.,
в сб.: Успехи химии и физики полимеров, М., 1970; Бер-
Берлин А. А. [и др.], Высокомол. соед., 13 А, № 12, 2676 A970).
А. А. Берлин, Т. Я. Кефели.
ОЛИФЫ (boiled oils, Firnisse, huiles cuites) — жид-
жидкие пленкообразующие вещества, получаемые на основе
масел растительных или жирных алкидных смол. О.—
прозрачные жидкости от желтого до вишневого цвета.
Они хорошо смачивают металлич. и деревянные поверх-
поверхности, легко наносятся кистью или валиком и высы-
высыхают в тонком слое на воздухе, образуя мягкую эла-
эластичную пленку, нерастворимую в воде и в органич.
растворителях. Высыхание слоя О. обусловлено поли-
полимеризацией масла, к-рая инициируется органич. гидро-
гидроперекисями, образующимися при взаимодействии масла
с кислородом воздуха. Для ускорения высыхания в со-
состав О. вводят сиккативы — преимущественно лино-
линолеаты, резинаты или нафтенаты РЬ, Мп, Со. Содержа-
Содержание в О. активных металлов составляет (в расчете на
массу масла): РЬ — 0,1—0,5%, Мп — 0,04—0,12%,
Со — 0,01— 0,10%. Некоторые О. содержат органиче-
органические растворители (уайт-спирит, скипидар и др.), кото-
которые вводят для разбавления полимеризованных масел.
К О. предъявляют ряд общих требований: 1) полная
прозрачность после отстаивания в течение 24 ч при
20 °С; отстой — не более 1% (по объему); 2) кислотное
число в пределах 6—12 (при более высоких кислотных
числах возможна желатинизация красок на основе О.,
обусловленная ее взаимодействием со щелочными пиг-
пигментами); 3) отсутствие токсичных компонентов;
4) время высыхания «от пыли» и время полного высыха-
высыхания (при 18—22 °С и относительной влажности воздуха
60—70%) — соответственно не более 12 и 24 ч (об опре-
определении времени высыхания см. Испытания лакокра-
лакокрасочных материалов и покрытий). Кроме того, О. долж-
должны обеспечивать длительную (до нескольких лет)
службу лакокрасочных покрытий на воздухе и внутри
помещений в различных климатич. условиях. Цвет О.
не должен резко влиять на цвет краски.
О. применяют для изготовления масляных красок
(густотертых и готовых к употреблению для нанесения
кистью), а также для разбавления масляных красок до
рабочей вязкости при необходимости их нанесения не
кистью, а др. способами, напр, распылением (см. также
Краски). Кроме того, О. используют для поверхностной
пропитки древесины перед ее окрашиванием масля-
масляными красками.
475
ОЛИФЫ
476
О. на основе растительных масел делят в зависимости
от содержания в них масла на натуральные A00% мас-
масла), комбинированные (~70% масла и ~30% раство-
растворителей) и полунатуральные E0—55% масла и 45—50%
растворителей). Алкидные О. подразделяют в зависи-
зависимости от типа смолы на глифталевые, пентафталевые и
ксифталевые.
Натуральные олифы. Эти О. готовят из высыхающего
масла (чаще всего из льняного или конопляного),
в к-рое добавляют сиккатив. Масло предварительно
очищают от фосфатидов гидратацией или щелочной
рафинацией. В зависимости от способа изготовления на-
натуральные О. делят на окисленные (оксидированные)
и полимеризованные.
При получении окисленной О. масло сначала
быстро нагревают до 100 °С, а затем постепенно (в те-
течение нескольких ч) до 150—160 °С. Процесс проводят
при перемешивании и продувании воздуха. Сиккатив
B,5—4,5% от массы масла) вводят после удаления вла-
влаги из масла (прекращения пенообразования). При этом
нагрев и продувание воздуха прекращают во избежа-
избежание потемнения О.
Полимер и зованную О. получают нагре-
нагреванием масла при 140—150 °С с целью его обезвожива-
обезвоживания и последующей частичной полимеризацией при
270—280 °С без продувания воздуха (сиккативы вво-
вводят при 220 °С).
Как окисленную, так и полимеризованную О. можно
получать периодическим и непрерывным способами.
Степень полимеризации масла при получении натураль-
натуральных О. невысока. Охлажденную О. очищают центри-
центрифугированием или отстаиванием с последующей фильт-
фильтрацией.
Натуральная О. может быть изготовлена и без нагре-
нагревания (т. наз. «холодным смешением»). В этом случае
к маслу добавляют 7—8% сиккатива в виде р-ра в лету-
летучем растворителе (уйат-спирите, скипидаре). Однако
такая О. уступает по качеству продукту, полученному
«горячим смешением», т. к. масло остается неполимери-
зованным, что ухудшает блеск и прочность пленки.
При хранении в открытой таре О. покрывается кор-
коркой линоксина (продукта окислительной полимериза-
полимеризации масла), нерастворимого в масле и препятствующего
дальнейшему окислению О. Перед употреблением О.
корку тщательно удаляют. В присутствии влаги О.
мутнеет, поэтому ее нельзя хранить в открытой таре
вне помещения или наливать во влажную тару. Помут-
Помутнение О. возможно также при ее хранении ниже 0 °С
вследствие выпадения сиккатива или вымораживания
из масла глицеридов насыщенных жирных к-т. Вата
и тряпки, пропитанные О., при хранении могут само-
самовозгораться.
Нек-рые технич. требования к натуральной О. при-
приведены ниже:
Плотность при 20 °С, г/см3
окисленная льняная 0,939—0,945
окисленная конопляная 0,930—0,940
полимеризованная 0,935
Кислотное число, не более 7
Число омыления, не менее 185
Содержание неомыляемых, %, не более . . 1
Йодное число, не менее
окисленная льняная 160
окисленная конопляная 150
Содержание Р2О5 (в расчете на лецитин),
%, не более
окисленная 0,3
полимеризованная 0,2
Содержание золы, %, не более %
окисленная 0 ^4
полимеризованная 0,3
О. на основе конопляного масла темнее, чем льняная;
поэтому первую не применяют для изготовления белой
и светлых красок.
Несмотря на пригодность натуральных О. для всех
указанных выше областей применения, потребление
этих О. сокращается вследствие их замены более
экономичными О., содержащими меньше масла и обра-
образующими в ряде случаев лакокрасочные покрытия
более высокого качества.
Комбинированные олифы. Такие О. представляют
собой р-ры в уайт-спирите смесей окисленных масел
с маслами, обезвоженными при 120—130 °С (табл. 1).
Комбинированные О. изготовляют холодным смеше-
смешением масел, сиккатива и растворителя. Кислотное чис-
число О. не более 10; вязкость по ВЗ-4 B0 °С) 22—42 сек
(для О. марки К-3 — 12—25 сек). Комбинированные О.—
полуфабрикаты лакокрасочной пром-сти, применяемые
с целью экономии масел при изготовлении густотертых
масляных красок.
Таблица 1. Состав масляной части нек-рых комбинированных
олиф отечественного производства
Марка олифы
К-2
К-3
К-4
К-5
К-6
Тип
окисленное
Полувысыхающее
Высыхающее
Полувысыхающее
Высыхающее или по-
полувысыхающее
Полувысыхающее
масла
обезвоженное
Льняное или др. вы-
высыхающее
Высыхающее
Полу высыхающее
Тунговое
Тунговое
Полунатуральные олифы. Эти О. готовят на осно-
основе препарированных растительных масел с последую-
последующим введением в них сиккативов и растворителей —
уайт-спирита, скипидара и др. Наиболее распростра-
распространенные полунатуральные О.— оксоль и касторовая
(табл. 2).
Таблица 2. Состав масляной части и не^-рые свойства
полунатуральных и алкидных олиф
Марка олифы*
Состав масляной части
Вязкость
по ВЗ-4
при
20 е С, сек
| si
нн ТО
Полунатуральные олифы
Оксоль В
Оксоль СМ
Оксоль ПВ
Касторовая
Льняное
Смесь льняного или коноп-
конопляного (не менее 70%) и под-
подсолнечного (не более 30%)
Подсолнечное, соевое или
рыжиковое
Касторовое
18-22
19-23
19-25
7-10**
8
10
Глифталевая
Пентафталевая
ПФ-К
Пентафталевая
ПФ-ПВ
Ксифталевая
Алкидные олифы
Подсолнечное, рыжиковое
или их смесь с льняным
Касторовое
Соевое, подсолнечное или
рыжиковое
Полувысыхающее
20 -25
25-35
22-30
22—42
12
11
12
8
* Темп-pa вспышки касторовой О. не ниже 36 °С, остальных
О.-туне ниже 32 СС. ** Вязкость по Энглеру, СЕ.
Оксоль — р-р глубоко окисленного высыхающе-
высыхающего или полувысыхающего масла в уайт-спирите. При
получении этой О. масло нагревают A40—160 °С) до
образования продукта с вязкостью его 55%-ного р-ра
в уайт-спирите 19—21 сек (по ВЗ-4 при 20 °С). Процесс
проводят при продувании воздуха и перемешивании.
После охлаждения ниже 120 °С масло вливают при пе-
перемешивании в 2/3 требуемого количества уайт-спирита,
затем добавляют сиккатив и недостающее количество
растворителя и продолжают перемешивание до полу-
477
ОЛОВООРГАНИЧЕСКИЕ ПОЛИМЕРЫ
478
чения однородного прозрачного продукта. Касторо-
Касторовая О. —р-р дегидратированного и затем полимери-
зованного (при 270—280 °С) касторового масла в уайт-
спирите, содержащий сиккатив.
Краски, изготовленные на полунатуральной О.,
образуют после улетучивания растворителя более
тонкие и менее долговечные пленки, чем краски на
основе натуральной О. Полунатуральные О. не при-
пригодны для изготовления густотертых масляных кра-
красок, т. к. вызывают их быстрое загустевание. Эти О.
применяют только для разбавления красок, тертых
на натуральной О., к-рыми окрашивают различные
строительные деревянные или неответственные метал-
лич. конструкции, оштукатуренные фасады и внутрен-
внутренние помещения зданий, и т. д. Оксоль В используют
для наружных и внутренних работ, остальные О. этого
типа — только для внутренних. Оксоль не применяют
при получении красок для покрытия полов из-за низ-
низкой механической прочности образующихся покры-
покрытий.
Алкидные олифы. Эти О. представляют собой р-ры
низковязких жирных алкидных смол (~60—65% мас-
масла) в уайт-спирите (см. табл. 2).
Глифталевую О. E0%-ный р-р) изготовляют
нагреванием масла до 230—240 °С, его переэтерифика-
цией глицерином (до образования продуктов, раство-
растворимых в этиловом спирте) и последующей этерифика-
цией фталевым ангидридом при 240—250 °С. К получен-
полученной глифталевой смоле добавляют при перемешивании
сиккатив и уайт-спирит. Предварительная термич. об-
обработка масла при 275—285 °С или добавление к нему
10—15% тунгового масла способствуют уменьшению
потерь фталевого ангидрида и улучшению качества
покрытий на основе глифталевой О.
Пентафталевую О. E0%-ный р-р) изготов-
изготовляют таким же образом, как и глифталевую; для пере-
этерификации масла применяют пентаэритрит. Преи-
Преимущество этой О. перед глифталевой — большая ско-
скорость высыхания (<16 ч). Для приготовления к с и-
фталевой О. E5%- или 70%-ный р-ры) применяют
алкидные смолы, спиртовым компонентом к-рых служит
ксилит. Эта О., высыхающая «от пыли» за 6 ч и полно-
полностью за 24 ч, имеет более темную окраску, чем др. ал-
алкидные О.
На изготовление алкидных О. расходуется меньше
масла, чем при получении О. др. типов; покрытия на
их основе (особенно пентафталевые) наиболее атмосфе-
ростойки и долговечны. Благодаря этому краски, со-
содержащие алкидные О., пригодны как для внутренних,
так и для наружных работ.
Лит.: Дринберг А.Я., Технология пленкообразующих
веществ, 2 изд., Л., 1955; Варламов В. С, Производство
олиф и сиккативов, М., 1957; Киселев В. С, А б а ш-
кина А. Ф., Производство лаков, олиф и красок, 2 изд.,
М., 1961; Справочник по лакокрасочным покрытиям в машино-
машиностроении, под ред. М. М. Гольдберга, 2 изд., М., 1974.
М.М.Гольдберг.
ОЛОВООРГАНИЧЕСКИЕ ПОЛИМЕРЫ (organo-tin
polymers, Organozinnpolymere, polymeres organoeta-
ins) — высокомолекулярные соединения, содержащие
в основной или боковых цепях макромолекул атомы Sn.
Полимеры с атомом олова в основной цепи. П о л и-
R
I
диалкил(арил)станнилены [ — Sn — ]п, где
I
R'
R и R'— алкил и арил (одинаковые или разные), м. б.
получены следующими методами:
R
I
nR2Sn -»[— Sn—]n (n=2-10)
R
Амин |
п (R2SnH2) > nH2 + [-Sn
]n(n=6-9)
C6H5
nR2Sn [N (GeH5) CH = O]2
n (C6H5JSnH2 -»[- Sn- Sn-]n
I I
R CeH,
Полидиалкил(арил)станнилены—воскоподобные или
жидкие полимеры низкой молекулярной массы, плохо
растворимые в органических растворителях и нераст-
нерастворимые в воде; легко разлагаются при действии О2
и галогенов, а также при нагревании выше 200 °С.
В некоторых исследованиях указывается на цикличе-
циклическое строение этих полимеров.
R
I
Полиорганостанноксаны [—Sn—О — ]п
I
R
получают по схеме:
R
nR2SnX2 + п NaOH
I
[ — Sri — О — ]п
R
п NaX
Эти полимеры — бесцветные твердые неплавкие веще-
вещества, нерастворимые в воде и органич. растворителях.
Степень полимеризации их невелика {п = 2—7). По-
Полиорганостанноксаны отличаются высокой реакционной
способностью: активно взаимодействуют со спиртами
и к-тами, алкилируются магнийорганич. соединениями.
При сополиконденсации диалкил(арил)дихлоридов
или диалкил(арил)оксидов олова с бифункциональны-
бифункциональными производными кремния (напр., с диалкилдисилано-
лами) получают полиорганостанносилок-
с а н ы, например:
Н2О, NEL
(G2H5JSiCl2 + (G2H5JSnCl2 >
G2H5
С2Н5
» - [- Si - О - n - [- Sn - О -]m -
C2H5 C2H5
Линейные и разветвленные полиорганостанносилок-
саны — в основном жидкости с темп-рой стеклования
от —100 до —120 °С и мол. массой от 1000 до 5000. Они
хорошо растворяются в полярных и неполярных раст-
растворителях; при нагревании способны отверждаться.
В водной среде при 95—96 °С за 10 ч гидролизуется
10% связей —Sn—О—Sn—.
1 1
I
О. п., содержащие связь — Sn — С — в основной
I I
цепи, получают полиприсоединением оловоорганич. ди-
алкил- или диарилгидридов к диенам или производным
ацетилена. С диенами реакция протекает по схеме:
R2SnH2 + СН2 = CHR'CH = СН2 -+
R
-+ [- SnCH2GH2R'CH2CH2 -]n
R
Взаимодействие дигидридов олова с диацетиленами
протекает более активно, чем с диенами. При этом обра-
образуются О. п. с двойными связями в основной цепи
R
I
[—SnCH = CHR'CH = CH —]п. Напр., присоедине-
I
R
ние 7г-фенилен-бис-(диметилоловогидрида) к 1,5-гекса-
диину или 1,8-нонадиину приводит к получению хоро-
479
ОМЫЛЕНИЯ ЧИСЛО
480
шо растворимых в бензоле эластомеров с мол. м.
~ 100 000.
О. п., содержащие в основной цепи кроме атомов
Sn атомы Ge и РЬ (полиорганостанноме-
таллоксаны), можно получить по схеме:
(GeH5JSnH2 + (CeH5JMe (СвН4СН = СН2J -
I I
— [- Sn CH2CH2GeH4MCeH4GH2CH2-]n.
I I
CeH5 CeH5
Эти полимеры — твердые стеклообразные продукты
с мол. массой до 27 000.
Для введения в состав основной цепи О. п. атомов
серы используют взаимодействие дигидридов олова
с дивинилсульфоном, приводящее к получению полиме-
полимеров низкой мол. массы [ —(R2)SnCH2CH2SO2CH2CH2—]п
Полимеры с атомами олова в боковых цепях. К этому
классу относятся гомо- и сополимеры, содержащие
в составе макромолекул звенья типа
- СН2 - CR"- - СН2 - CR"—
SnR'
I
SnR'
I II
где R — G6H4, COO и др.; R' — алкил, фенил, цикло-
гексил, меркаптил, алкоксил; R" — Н, СН3 или
= СН-СН2.
О. п., содержащие звенья I, получают радикальной
гомо- и сополимеризацией оловосодержащих стиролов
СН2 = СН — С6Н4 — SnR'3, акрилатов СН2 = СН —
— COO — SnR'3 (или соответствующих метакрила-
тов), а также малеатов и оловоорганич. производных
нек-рых др. непредельных к-т. В качестве сомоно-
меров при этом используют акрилаты и метакрилаты,
стирол, винилхлорид и др.
О. п., содержащие звенья типа II, получают, в основ-
основном, в виде сополимеров винильных соединений строе-
строения СН2 = СН со стиролом, метилметакрилатом,
I ,
SnR3
винилхлоридом.
При полимеризации 2-триэтилстаннилбутадиена-
1,3 (инициатор азо-бис-изобутиронитрил) происходит
1,4-присоединение с образованием полимеров, со-
содержащих звенья — СН2НС = ССН2 —, а при поли-
Sn(C2H5K
меризации 1-триэтилстаннилбутадиена-1,3 — 3,4-присое-
динение с образованием звеньев — СНСН2—
CH=CHSn(C2H5K
Оловосодержащие диены весьма активны в радикаль-
радикальной полимеризации (более активны, чем стирол).
О. п. получают также обработкой р-ров полимеров,
содержащих реакционноспособные боковые группы
(напр., атомы галогена), низкомолекулярными оловоор-
оловоорганич. соединениями. Так, хлорированный полиэтилен
или поливинилхлорид обрабатывают в р-ре тетрагидро-
фурана соединениями типа LiSnR3 (R — фенил или
бутил), что приводит к замещению части атомов хлора
на оловоорганич. группы. Полученные таким способом
О. п. содержат от 2 до 17% (по массе) олова.
Применение оловоорганических полимеров. Одно из
основных направлений использования О. п.— приме-
применение их в качестве стабилизаторов различных полиме-
полимеров, в особенности галогенсодержащих, напр, поли-
винилхлорида. Преимущество О. п. перед низкомоле-
низкомолекулярными оловоорганич. стабилизаторами — нелету-
нелетучесть и лучшая совместимость с полимером. Для ста-
стабилизации используют как О. п., содержащие олово
в основной цепи (напр., полиорганостанноксаны), так
и полимеры с боковыми оловоорганич. группами [напр.,
политриалкилстаннилакрилаты или сополимеры ви-
нилхлорида с триалкил(арил)винильными производны-
производными олова].
О. п., получаемые сополимеризацией основного моно-
мономера (напр., винилхлорида) с небольшими относитель-
относительными количествами A—3%) винильного оловоорганич.
мономера, представляют собой гомогенные «самоста-
«самостабилизированные» системы, не требующие дополни-
дополнительного введения стабилизатора. Хймич. связывание
оловоорганич. групп с макромолекулами таких сополи-
сополимеров (взамен механич. добавок) позволяет резко сни-
снизить или полностью предотвратить выделение олово-
оловоорганич. соединений из полимера при его переработке
и эксплуатации. Последнее обстоятельство особенно
важно, если учесть высокую токсичность низкомоле-
низкомолекулярных оловоорганич. соединений.
Хорошие результаты дает использование О. п., раст-
растворимых в смазочных маслах, в качестве присадок. Эти
присадки не улетучиваются при высоких темп-pax и
существенно снижают износ трущихся поверхностей
деталей. О. п., получаемые радикальной гомо- и сопо-
сополимеризацией оловоорганич. акрилатов, обладают анти-
антимикробной и антигрибковой активностью и могут при-
применяться в качестве компонентов красок, образующих
необрастающие лакокрасочные покрытия.
Лит.: К о ч к и н Д. А., Азербаев И. Н., Олово и
свинецорганические мономеры и полимеры, Алма-Ата, 1968,
с. 209; Андрианов К. А., Полимеры с неорганическими
главными цепями молекул, М., 1962; Minoura Y. [а. о.],
J. Polymer Sci, A. 1, 4, № 11, 2757 A966). В. В. Мальцев.
ОМЫЛЕНИЯ ЧИСЛО (saponification number, Ver-
seifungszahl, indice de saponification) — число мг КОН,
прореагировавшего со свободными кислотами и слож-
сложными эфирами, содержащимися в 1 г исследуемого ве-
вещества. О. ч. равно сумме кислотного числа и эфир-
эфирного числа (последнее представляет собой число
мг КОН, необходимое для омыления 1 г сложного эфи-
эфира). Для сложных эфиров, не содержащих свободных
карбоксильных групп, О. ч. совпадает с эфирным
числом.
Для определения О. ч. в две конич. колбы емкостью
250 мл помещают точно отвешенные 0,5—2 г образца
и добавляют по 25 мл 0,5—2 н. р-ра КОН в этаноле.
Наряду с этанолом (обычно 95%-ным) для растворения
КОН используют метанол, пропанолы, бутанолы, ами-
амиловый спирт, диэтиленгликоль, а также смеси этанола
с бензолом или пиридином, диэтиленгликоля с фенето-
лом. Параллельно в третьей колбе ставят контрольный
опыт (без навески образца). К колбам присоединяют
вертикальные холодильники, закрытые трубками с нат-
натровой известью, и реакционную смесь кипятят в тече-
течение 1—24 ч. Избыток КОН оттитровывают 0,5—1 н.
р-ром НС1 в присутствии фенолфталеина. Титрование
темноокрашенных р-ров удобно проводить с универ-
универсальным индикатором или потенциометрич. методом.
Если полимер полностью не растворяется, то при тит-
титровании для полной нейтрализации КОН содержимое
колбы энергично встряхивают. О. ч. определяют по
ф-ле
0 q ^ 56,108 (NlVl-N2V2)
а
где Ni и N2 — нормальности р-ров соответственно КОН
и НС1; Vx и F2 — объемы р-ров соответственно КОН
и НС1, мл; а — навеска образца, г; 56,108— содержа-
содержание КОН в 1 мл 1 и. р-ра КОН, мг.
Из О. ч. может быть рассчитан эквивалент
омыления — число г вещества, взаимодействую-
взаимодействующего с 1 л 1 н. р-ра КОН:
56 108
Э. о. =¦
О. ч.
Разработан метод определения О. ч. с использованием
двойного индикатора, исключающий необходимость
проведения контрольного опыта.
481
ОПТИЧЕСКАЯ МИКРОСКОПИЯ
482
Лит.: Аналитическая химия полимеров, пер. с англ., т. 1,
3,М., 1963—66 (т. 1, с. 226, 508, 568; т. 3, с. 72); Л о с е в И. П.,
Федотова О. Я., Практикум по химии высокополимер-
высокополимерй М 1962 88 М ГЧ
д , ру
ных соединений, М., 1962, с. 88.
ОПТИЧЕСКАЯ МИКРОСКОПИЯ
р
М. Г.Чаусер.
полимеров
(optical microscopy, optische Mikroskopie, microscopie
optique).
Методы исследования. Каковы бы ни были модифи-
модификации методов О. м. или конкретные задачи, разрешае-
разрешаемые с ее помощью, в конечном счете главную роль
играет шкала линейных размеров, в пределах к-рой
микроскопич. объект доступен для наблюдения и к-рая
однозначно определяется разрешающей спо-
способностью микроскопов. Так, возможность ха-
характеристики оптич. свойств полимерного волокна
как целого все же ограничивается его толщиной: ниже
нек-рого предела, определяемого разрешающей способ-
способностью, дифракция от волокна делает его наблюдение
невозможным.
Разрешающая способность микроскопа, т. е. наи-
наименьшее различимое в нем расстояние между двумя
точками объекта (Zmjn), однозначно определяется раз-
разрешающей способностью объектива и выражается ф-лой
где X — длина световой волны; А — числовая апертура
объектива, равная rasina, где п — показатель преломле-
преломления среды между объективом и объектом, a — поло-
половина апертурного угла объектива. Для «сухих» и им-
иммерсионных объективов 1т{П колеблется в видимой об-
области света от 2,7-10~5 до 1,6-10 см.
Числовая апертура и собственное увеличение явля-
являются основными характеристиками объективов; с ними
связаны рабочее расстояние и величина поля зрения.
При достаточно большом увеличении объектива раз-
разрешаются очень мелкие детали, но они слишком плотно
располагаются в задней фокальной плоскости, где воз-
возникает первичное изображение. Поэтому первичное
изображение должно быть увеличено настолько, чтобы
разрешающая («воспринимающая») способность регист-
регистрирующего устройства (глаза, фотопленки и т. п.) мог-
могла быть использована полностью. Эту функцию п о-
лезного увеличения выполняют окуляры,
сочетание к-рых с объективом и образует, собственно,
микроскоп.
Следует, однако, помнить, что максимальное
увеличение микроскопа ограничено разрешаю-
разрешающей способностью объектива, а последующее повыше-
повышение увеличения окуляра уже не выявляет новых дета-
деталей; при этом отчетливость увеличенного изображения
понижается. Как правило, микроскопы бывают снаб-
снабжены объект-микрометром с ценой деления 0,01 мм,
по к-рому определяют увеличение. При визуальном
наблюдении для той же цели применяются окуляр-
микрометры.
Максимальное увеличение микроскопов, как прави-
правило, не превышает в видимом обычном свете 1200Х,
а в поляризованном — 700X. Большие увеличения м. б.
достигнуты при применении тонкодисперсных объектов
и иммерсионных объективов. Др. возможность (см.
выше ф-лу) заключается в уменьшении Х\ поэтому ульт-
ультрафиолетовые микроскопы (требующие, однако, спе-
специальной системы регистрации) имеют существенно по-
повышенное полезное увеличение. Третий — косвенный
способ заключается в повышении контрастности изобра-
изображения (фазовый контраст, темнопольная микроско-
микроскопия и др.).
Из всех модификаций О. м. наиболее широкое рас-
распространение получила поляризационная
микроскопия. Этим методом определяют обычно
локальные или усредненные значения оптич. анизо-
анизотропии объекта А/г. Проще всего это сделать сравнением
интерференционных окрасок объекта при скрещенных
и параллельных поляроидах,— т. наз. методом допол-
дополнительных цветов. Разность хода обыкновенного и не-
необыкновенного лучей Г определяется по таблицам,
а An = Yld, где d — толщина двулучепреломляющего
объекта. На практике обычно удобнее определять Г
посредством компенсации двойного лучепреломления
кварцевым клином или плоскопараллельной кальцито-
вой пластинкой, дающей переменную разность хода
при повороте на некоторый угол (компенсатор Берека).
Компенсаторы различных систем м. б. использованы
для исследования объектов, характеризующихся
Г<1600 нм. Для прецизионных измерений Г (с точ-
точностью до ^3 нм) при фиксированной длине волны ис-
используется метод Сенармона, основанный на анализе
ориентации электрич. векторов световой волны, про-
прошедшей через двулучепреломляющий объект (сферо-
лит, фибриллу, единичный кристалл и т. п.).
Абсолютные значения показателей преломления мож-
можно определять иммерсионным методом, используя стан-
стандартный набор жидкостей с известными показателями
преломления. Сравнение показателей преломления ани-
анизотропного объекта и «матрицы», в к-рую он погружен,
возможно с помощью т. наз. полоски Бекке, т. е. свет-
светлой полосы, окаймляющей объект (обычно кристалл)
там, где он соприкасается с матрицей (и вообще средой,
включая калибровочные жидкости). При подъеме ту-
тубуса микроскопа полоска Бекке, наблюдаемая в отсут-
отсутствие анализатора, всегда смещается от края кристалла
в сторону более преломляющей среды, а при опуска-
опускании — в сторону менее преломляющей.
Знак двойного лучепреломления кристаллич. агре-
агрегатов (напр., сферолитов) определяется с помощью
кварцевой или гипсовой пластинки. При введении та-
такой пластинки в ход лучей микроскопа разность ее хо-
ходов будет либо складываться, либо вычитаться из раз-
разности хода, создаваемой исследуемым агрегатом, в за-
зависимости от его ориентации. В результате меняется
интерференционная окраска, к-рую дают пластинки в
скрещенных поляроидах. Для определения знака двой-
двойного лучепреломления агре-
агрегатов сравнительно большо-
большого размера м. б. использо-
использован также кварцевый клин.
Для решения ряда спе-
специальных задач необходим
анализ т. наз. эллипсоида
поляризуемостей в кристал-
кристаллич. объекте; такой анализ
м* б- произведен при наблю-
дении интерференционных
картин в сходящемся свете
(наблюдение коноскопич.
фигур). Схема формирова-
формирования и наблюдения коноско-
Схема наблюдения интерференции
при помощи линзы Бертрана: On —
окуляр; Об — объектив; 3 — зеркало;
Б —линза Бертрана; А — анализатор;
П — поляризатор; Пр — препарат
(объект); К — конденсор; Фь Ф2 —
первичная и вторичная интерферен-
интерференционные картины; Ф3 — видимое мни-
мнимое изображение интерференционной
картины.
пических фигур приведена на рисунке. Первичная
интерференционная картина образуется непосредствен-
непосредственно за объективом и наблюдается при удаленном оку-
окуляре. Введение в ход лучей окуляра и линзы Бертра-
Бертрана, которой снабжены хорошие поляризационные ми-
микроскопы (например, МИН-8), образует схему «сла-
«слабого» микроскопа, лишь увеличивающего интерферен-
интерференционную картину (мнимое изображение Ф3). В случае
монокристалла интерференционная картина возникает
из-за различия, обусловленного двойным лучепрелом-
483
ОПТИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРЫ
484
лением, оптич. путей лучей и регистрируемая коно-
скопич. фигура дает информацию об ориентации и па-
параметрах эллипсоида поляризуемости.
Используя принципы коноскопич. анализа, удается
наблюдать и регистрировать с помощью поляризацион-
поляризационного микроскопа картины рассеяния (дифракции)
поляризованного света под малыми углами. Оптич.
схема для наблюдения рассеяния не отличается от изо-
изображенной на рис. Попутно переход от непосредствен-
непосредственного наблюдения прямого изображения к картине
рассеяния позволяет избежать упоминавшихся выше
трудностей, связанных с дифракцией от объекта (тон-
(тонкого волокна) как целого. Совмещение на одном при-
приборе двух приемов наблюдения открывает дополни-
дополнительные возможности изучения локальной дифракции
или решения обратной задачи: выбора участка, ди-
дифракция от к-рого почему-либо представляет специаль-
специальный интерес, «изоляции» этого участка с помощью ди-
диафрагмы и затем прямого его наблюдения.
При наличии в объекте двух или нескольких типов
структур можно, помещая в плоскости Фх (см. рис.)
различные диафрагмы, выделить рефлексы, соответст-
соответствующие той или иной структуре и, используя принци-
принципы темнопольной микроскопии, сформировать изобра-
изображение элементов только этой структуры. Это позво-
позволяет раздельно изучать наложенные друг на друга
структуры или тонкие детали их строения.
При введении в ход лучей двух- или многолучевого
интерферометра обычный оптический микроскоп преоб-
преобразуется в интерференционный.
Области применения оптической микроскопии. С по-
помощью поляризационной О. м. можно прежде всего
найти линейные и угловые размеры структурных эле-
элементов, поскольку величина An непосредственно свя-
связана с толщиной объекта d (см. вышеприведенную
ф-лу). Помимо этого, метод позволяет определять важ-
важные оптич. характеристики (показатели преломления,
знак двулучепреломления) как структурных элемен-
элементов, так и полимерных систем в целом. Установление
знака An в элементе надмолекулярной структуры весь-
весьма существенно, ибо позволяет определить ориентацию
молекулярных цепей в нем. В свою очередь (напр., при
появлении положительных, отрицательных и «ано-
«аномальных» сферолитов в полиэтилентерефталате), зна-
знание ориентации цепей позволяет сделать важные выво-
выводы о кинетике и морфологии кристаллизации в разных
режимах. Не менее важные выводы на основе измене-
изменений знака А/г, сопровождающих деформацию сфероли-
сферолитов в растягиваемых волокнах или пленках, м. б. сде-
сделаны о кинетике и морфологии ориентационных про-
процессов. По поводу значимости определения An в аморф-
аморфных полимерах см. Фотоу пру гость.
Изучение картин дифракции света дает возможность
решить проблемы, возникающие при работе «на гра-
границе» обычного оптич. разрешения. Круг таких про-
проблем довольно широк, поскольку все сферолиты, обна-
обнаруживаемые в технич. волокнах или пленках, имеют
величину порядка 0,5 —1,0 мкм. Метод позволяет так-
также описать изменение структуры образца в нек-рых
усредненных терминах (средний радиус сферолитов,
средняя их концентрация, средняя длина фибрилл
и т. п.). Переход к картине дифракции позволяет оха-
охарактеризовать препараты, содержащие большое число
анизотропных элементов надмолекулярной структу-
структуры. Таким путем удается, в частности, регистрировать
и идентифицировать упорядоченные надсферолитные
агрегаты, возникающие при нек-рых кристаллизацион-
кристаллизационных процессах и образованные из очень малых сфероли-
сферолитов. В комбинации с высокоинтенсивными источниками
света и скоростной киносъемкой описываемый метод
позволяет изучать кинетику, динамику и морфологию
быстро протекающих структурных превращений, напр,
при плавлении и кристаллизации, ориентации в усло-
условиях, близких к технологическим, ориентационной кри-
кристаллизации и т. п.
Интерференционная микроскопия наряду с такими
стандартными технич. приложениями, как определение
An и (или) степени ориентации химич. волокон и пле-
пленок, с успехом применяется для исследования диффу-
диффузии растворителей в полимер (и, соответственно, для
изучения кинетики набухания), сложных процессов,
связанных с суперпозицией диффузии низкомолекуляр-
низкомолекулярного вещества и стимулированной ею кристаллизации
полимера, для определения толщины монокристаллов
с точностью до нескольких десятых долей им и преци-
прецизионного измерения темп-рного коэфф. линейного рас-
расширения. В последнем случае интерференционный ми-
микроскоп играет роль дилатометра и его удобно исполь-
использовать для изучения релаксационных процессов при
размягчении и стекловании полимеров. С использова-
использованием специальных методов контрастирования на ин-
интерференционном микроскопе можно получать стерео-
скопич. изображения, не уступающие по четкости и
разрешению фотографиям, получаемым на сканирую-
сканирующем электронном микроскопе. Однако круг задач, ре-
решаемых этим методом, более ограничен.
Лит.: К а р г п н В. А., Современные проблемы науки о
полимерах, М., 1962; К а р г и н В. А., Слонимский Г. Л.,
Краткие очерки по физико-химии полимеров, 2 изд., М., 19Н7;
Джейл Ф., Полимерные монокристаллы, пер. с англ., Л.,
1968; Меланхолии Н. М., Методы исследования опти-
оптических свойств кристаллов, М., 1970; Ф е к л и ч е в В. Г.,
Микрокристалломорфологический анализ, М., 1966; Ф р а н-
с о н М., Фазово-контрастный и интерференционный микро-
микроскопы, пер. с франц., М., 1960. В. Г. Баранов, С. Я. Френкель,
ОПТИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРЫ (optically
active polymers, optisch aktive Polymere polymeres
optiquement actifs).
Содержание:
Введение 48 4
Синтез 485
Синтез из оптически недеятельных полимеров . . 486
Синтез из оптически активных мономеров ... 487 •
Синтез из оптически активных полимеров ... 488
Синтез из рацемических мономеров. Стерео-
селективные процессы 488
Синтез из симметричных мономеров. Стерео-
специфические процессы 490
Исследование методом спектрополяриметрии . . . 4(.J
Применение 41) 4
Введение
Оптически активные полимеры — высокомолекуляр-
высокомолекулярные соединения, обладающие способностью повора-
поворачивать плоскость поляризации света, проходящего
через их р-ры, расплавы или прозрачные стекла. По
происхождению О. а. п. могут быть природными (белки
и полипептиды, полисахариды, нуклеиновые кислоты)
и синтетическими. В настоящей статье рассмотрены
только синтетические О. а. п.
Оптич. активность полимеров обусловлена наличием
диссимметрических (хиральных) структур в макромоле-
макромолекулах: асимметрич. центров (чаще всего — асимметрич.
атомов углерода), атропоизомерных звеньев или участ-
участков макромолекул со спиральной конформацией. Пер-
Первые два типа структур локализованы в элементарном
звене О. а. п. (в основной цепи или в боковых ветвях)
и характеризуются конфигурацией звеньев полимера —
его первичной структурой. Последний тип связан со
вторичной структурой макромолекулы — ее конформа-
конформацией, к-рая зависит как от строения элементарного зве-
звена, так и от природы и силы межмолекулярных взаимо-
взаимодействий. При наличии в макромолекуле сразу не-
нескольких типов хиральных структур их вклады в оп-
оптич. активность полимера суммируются.
Количественной мерой оптич. активности полимера
являются значения его удельного и моляр-
молярного вращений:
100
485
ОПТИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРЫ
486
где I—толщина слоя р-ра полимера, дм] с — концент-
концентрация р-ра в г на 100 мл р-ра; а — угол поворота плос-
плоскости поляризации; М — мол. масса элементарного
звена макромолекулы; t и X — индексы, обозначающие
темп-ру, при к-рой проводится измерение, и длину вол-
волны поляризованного света.
Дисперсия оптического враще-
вращения — зависимость [а] от К в большинстве случаев
передается ур-нием Друде:
где i — индекс «о. а. полосы поглощения» в электрон-
электронном спектре полимера; %i и Ki — константы, характе-
характеризующие положение максимума г-той полосы погло-
поглощения и ее вращательную способность. Если в изме-
измеряемой области спектра оптич. активность проявляет
только один электронный переход, характеризующийся
вращательной константой Ко и дисперсионной констан-
константой Хо, для описания кривой дисперсии оптич. вращения
достаточно одночленного ур-ния Друде:
= к°
Константы Ко и Ко находят из линейного графика,
представляющего собой зависимость
[а]хХ2 от [а]х
Если макромолекула имеет спиральную конформа-
цию, что наиболее ярко выражено у полипептидов,
кривая дисперсии оптич. вращения подчиняется дву-
двучленному ур-нию Моффита и Янга:
1 -»2+2 Х,_х: ¦ (»-ф
где п — коэфф. преломления растворителя, а0 и Ъо—
константы, характеризующие вклады соответственно
элементарного звена и спиральной конформации цепи
в оптич. активность полимера.
В области поглощения о. а. хромофора кривая дис-
дисперсии оптич. вращения, как правило, меняет знак,
проходя последовательно через максимум, нуль (при
Хо) и минимум (эффект Коттона). Характер
кривой в этой области дает богатую информацию о кон-
формации отдельных участков макромолекулы. В по-
последнее время все чаще исследуются кривые циркуляр-
циркулярного дихроизма О. а. п., которые имеют максимумы в об-
области эффекта Коттона и с большой надежностью поз-
позволяют разделить вклады различных о. а. электронных
переходов в суммарную активность полимера.
Оптическая чистота полимера опреде-
определяется отношением избытка имеющихся в нем асиммет-
асимметрич. центров одной конфигурации (R) над числом цент-
центров противоположной конфигурации (S) к суммарному
числу асимметрич. центров (в %):
Степень стереоселективности или стереоспецифичности
процессов и оптич. выход асимметрич. синтеза численно
равны оптической чистоте образующихся в этих про-
процессах О. а. п.
Синтез
Методы получения О. а. п. могут быть подразделены
наследующие основные группы: 1) полимер аналогичные
превращения неактивного полимера, приводящие к вве-
введению в его боковые радикалы оптически активных
групп или к созданию в макромолекуле асимметрич.
центров путем асимметрич. синтеза; 2) полимеризация
и поликонденсация о. а. мономеров, к-рая, естественно,
должна проходить в условиях, исключающих рацеми-
рацемизацию; 3) полимераналогичные превращения О. а. п.;
4) стереоселективная полимеризация одного из двух
оптич. изомеров, содержащихся в рацемич. смеси моно-
мономера; 5) асимметрич. синтез — стереоспецифич. полиме^
ризация (или полиприсоединение) симметрич. мономе-
мономеров. В последнем случае под влиянием о. а. инициато-
инициаторов или сомономеров в процессе полимеризации в каж^
дом элементарном звене возникают асимметрич, атомы
(в основной цепи) преимущественно одной из двух воз-
возможных конфигураций.
Первые три метода наиболее часто используются для
практич. синтеза О. а. п.; стереоселективная полимери-
полимеризация и асимметрич. синтез исключительно интересны
с научной точки зрения.
Синтез из оптически недеятельных полимеров. Поли-
мераналогичными превращениями с о. а. низкомолеку-
низкомолекулярными соединениями многие неактивные полимеры
м. б. превращены в диссимметрические. Так, нейтрали-
нейтрализацией поливинилпиридина D-винной к-той, D-кам-
форсульфокислотой или L-миндальной к-той получены
о. а. полимерные соли. Значение уд. вращения солей,
синтезированных из атактич. и тактич. ноливинилпири-
динов, оказались различными. Аналогично получены
полимерные соли хинина с двумя образцами полиакри-
полиакриловой к-ты, приготовленными радикальной полимери-
полимеризацией акриловой к-ты и сополимеризацией ее с ангид-
ангидридом акриловой к-ты с последующим гидролизом
ангидридных групп. Уд. вращения и константы Друде
двух солей оказались различными. Такие различия
могут использоваться для изучения микротактичности
полимерных цепей.
Ацилированием поливинилового спирта хлорангидри-
дом ]Ч-Gг-толуолсульфонил)-Ь-валина получен соответ-
соответствующий о. а. поливиниловый эфир. Наоборот, при
ацилировании полиакрилхлоридом о. а. спиртов (напр.,
D-глюкозы, хинина) или о. а. первичных и вторичных
аминов (например, этилового эфира L-лейцина, L-эфе-
дрина) получают о. а. эфиры или амиды полиакрило-
полиакриловой кислоты.
Полимераналогичными превращениями асимметрич.
группировки могут вводиться и в нерастворимые трех-
трехмерные полимеры. Большое практич. значение приоб-
приобретают синтезируемые таким путем диссимметрические
ионообменные смолы. Так, использование хелатообра-
зующих ионообменников, получаемых обработкой о. а.
а-аминокислотами или их производными галогенмети-
лированных сополимеров стирола с дивинилбензолом,
позволило разработать новый перспективный метод
разделения оптич. изомеров. Сорбенты, содержащие
L-пролин или L-оксипролин в качестве асимметрич. хе-
латообразующих группировок, способны с высокой
степенью стереоселективности образовывать в присут-
присутствии ионов меди смешанные комплексы с D-изомерами
а-аминокислот. Благодаря этому при пропускании рас-
раствора рацемической аминокислоты через колонну с
таким сорбентом происходит отделение D-изомеров ами-
аминокислоты от L-изомеров (процесс лигандообмешюй
хроматографии).
Полимераналогичные превращения позволяют вво-
вводить в боковые ветви неактивных полимеров не только
группировки с асимметрич. атомами, но и хиральные
атропоизомерные группировки. Так,
ацилированием поливинилового спир-
спирта хлорангидридом (+)-2-метил-6-нит-
родифенил-2-карбоновой к-ты получен
с 68%-ной степенью превращения по-
полимер I, обладающий высокой опти-
= +490,
~СН2-СН
активностью:
20
ческой
20 380
[а] = + 2500; в диоксане. Его актив-
250
ные элементарные звенья не содержат асимметричных
атомов.
К этой же группе методов получения О. а. п. следует
отнести и прямой асимметрич. синтез на полимере. При
487
ОПТИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРЫ
этом асимметричными могут становиться углеродные
атомы как боковых ветвей, так и основной цепи поли-
полимера. Так, при восстановлении поли-/г-винилацетофенона
в присутствии хинина с оптическим выходом 9% полу-
получен правовращающий ([а] = +5,9; в тетрагидрофу-
ране) полимерный спирт (звездочкой здесь и далее
отмечен асимметрич. атом):
~СН2-СН~ ~СН2-СН~
*снон
си,
Примером создания асимметрич. атомов в основной
цепи может служить гидрирование над скелетным ни-
никелевым катализатором, модифицированным о. а. ве-
веществами (L-глутаминовая к-та, D-винная к-та), поли-
полимерных [3-кетоэфиров:
[- с - сн - (сн2)в-]п <± [- с = с - (сн2)в-]п —
о соос2н5
ОН СООС2Н5
- [- СН - СН - (СН2)в-]п
ОН СООС2Н5
Образующиеся полимерные р-оксиэфиры обладают за-
заметной правовращающей способностью. Асимметрич.
гидрирование, вероятно, связано с участием о. а. ком-
компонента в образовании переходного активного комп-
комплекса, присоединяющего водород.
Синтез из оптически активных мономеров. Полимери-
Полимеризация или поликонденсация о. а. мономеров обладает
по сравнению со всеми остальными методами синтеза
О. а. п. тем преимуществом, что гарантирует наличие
о. а. группировок в каждом элементарном звене. Это
справедливо, естественно, только в том случае, если
исходный мономер оптически чист, а в процессе поли-
полимеризации не затрагивается асимметрич. центр и не
происходит его частичной рацемизации. Эксперимен-
Экспериментальная проверка последнего условия связана с боль-
большими трудностями.
Первым синтезом О. а. п., описанным в литературе,
является проведенная Вальденом в 1896 спонтанная
полимеризация о. а. диамилового эфира итаконовой к-ты
СН2 = С—СН2—СООС5НИ. До сих пор полимеризация
СООС5НИ
ненасыщенных соединений с о. а. боковыми ветвями
является наиболее распространенным методом синтеза
О. а. п. В качестве активных мономеров использовались:
а-олефины; эфиры акриловой, метакриловой, малеи-
новой и др. ненасыщенных к-т (ментиловые, борнило-
вые, втор -бутиловые, а-метилбензиловые и др.); ами-
амиды указанных к-т с а-метилбензиламином и различными
а-аминокислотами в качестве аминного компонента;
производные стирола с разнообразными о. а. замести-
заместителями в ароматич. ядре; сложные виниловые эфиры
таких к-т, как миндальная, р-фенилмасляная; различ-
различные виниламины и др.
Оптич. активность образующихся полимеров с асим-
асимметрич. центрами в боковых ветвях обычно не зави-
зависит от степени полимеризации и глубины превращения.
В ряде случаев она значительно отличается от актив-
активности исходного мономера. Это различие неоднократно
использовалось для изучения кинетики полимеризации
поляриметрич. методом [напр., в случае полимеризации
D-ягаор-бутил-а-хлоракрилата или винил-(—)-р-фенил-
бутирата]. Дисперсия оптич. вращения полимеров,
как правило, хорошо подчиняется одночленному ур-нию
Друде, но вращательная константа А", как и [а]\ силь-
сильно зависит от механизма полимеризации, т. к. находит-
находится в непосредственной связи со степенью стереорегу-
лярности полимера.
Полимеризацией о. а. альдегидов или сополимери-
зацией а-олефинов с двуокисью серы получают гетеро-
цепные О. а. п. с асимметричными центрами в боковых
ветвях.
Широко используется полимеризация о. а. гетеро-
циклов, таких как окись пропилена, транс-2, 3-эпокси-
бутан, пропиленимин, дилактид, N-карбоксиангидри-
ды а-аминокислот, (З-метил-е-капролактам и др. В этих
случаях получают гетероцепные О. а. п. с асимметрич.
атомами углерода в основной цепи. Т. к. полимериза-
полимеризация протекает с раскрытием цикла, активность полиме-
полимера нередко отличается даже по знаку от активности
мономера. Получение О. а. п. полимеризацией о. а.
трехчленных гетероциклов (замещенных этиленокси-
дов, этилениминов и этиленсульфидов) говорит о том,
что разрыву подвергается связь гетероатома с первич-
первичным атомом углерода, тогда как асимметрич. вторич-
вторичный атом остается незатронутым. Именно так с помо-
помощью поляриметрии был установлен Sn2 механизм поли-
полимеризации окиси пропилена под воздействием КОН
или FeCl3.
Поликонденсацией м. б. получены как карбоцепные
О. а. п. (напр., поликонденсацией формальдегида
с N-толуолсульфонил-Ь-тирозином), так и различные
гетероцепные полимеры. К последним относятся поли-
полиамиды и полиэфиры D-винной, D-камфарной, 1,2-
циклогександикарбоновой, р-метиладипиновой к-т, про-
продукты конденсации природных углеводов, аминокислот
или оксикислот и др. Этим методом удается создавать
макромолекулы с очень сложным, но регулярным чере-
чередованием отдельных звеньев в цепи. Напр., взаимодей-
взаимодействием L-лизина с дихлорангидридом адипиновой к-ты
в присутствии ионов меди получают о. а. мономер след.
строения:
H2NCH (CH2LNH - СО (СН2L СО - NH (CH2LCHNH2
соон
соон
Методом межфазной поликонденсации его с дихлоран-
гидридами дикарбоновых к-т получают сложные регу-
регулярные о. а. полиамиды.
Получен ряд О. а. п. с атропоизомерными звеньями,
напр, конденсацией терефталоилхлорида с (+)-2,2'-
диаминобинафтилом-1,1' (полимер II).
-HN
• //—\
NH-CO-/ V-CO-
\=/
U
Этот полимер обладает очень высокой оптич. актив-
активностью подобно др. соединениям с собственно диссим-
метричным хромофором, т. е. соединениям, у кото-
которых «о. а. полоса поглощения» в электронном спектре
принадлежит непосредственно диссимметрической груп-
группировке.
Синтез из оптически активных полимеров. Если при
химич. превращениях О. а. п. не происходит рацеми-
рацемизация асимметрич. центров, то этим путем м. б. полу-
получены полимеры, отличающиеся по своей оптич. актив-
активности от исходных. Напр., гидрированием поли-(8)-4-
метилгексина-1 с [a]D = +2,0° получен поли-4-метил-
гексен-1 с [a]D= +178°.
Синтез из рацемических мономеров. Стереоселектив-
ные процессы. Полимеризация рацемич. мономера
в общем случае может протекать либо как сополимери-
489
ОПТИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРЫ
490
зация двух энантиомеров, либо как гомополимериза-
ция каждого из них в отдельности. В последнем случае
образующийся полимер представляет собой смесь мак-
макромолекул, состоящих либо из (+)-, либо из (—^изо-
(—^изомерных звеньев. Такой продукт м. б. разделен на
2 фракции с противоположными знаками вращения.
Процессы, в к-рых реагирующая асимметрия, частица
способна отбирать для взаимодействия только один
из двух стереоизомеров партнера, наз. стереоселектив-
ными. В случае стереоселективной по-
полимеризации такой активной асимметрич. ча-
частицей является растущий конец макромолекулы (пос-
(последнее звено, как и все остальные, содержит асиммет-
асимметрич. центр) или его комплекс с катализатором.
Первым прямым доказательством возможности про-
проведения стереоселективной полимеризации было разде-
разделение изотактич. поли-A\, S)-4-метилгексена-1 на фрак-
фракции с молекулярным вращением, равным —17,5 и
+62,5 (Натта, 1955). Разделение было проведено путем
хроматографии полимера на о. а. сорбенте — нераст-
нерастворимом изотактич. поли-(8)-3-метилпентене-1. Позд-
Позднее аналогичным образом были разделены на фракции
с противоположной активностью кристаллич. полиме-
полимеры рацемич. З-метилпентена-1, 3,7-диметилоктена-1,
1-метилпропилвинилового эфира и окиси пропилена.
О стереоселективности полимеризации свидетельствует
идентичность физических свойств и кристаллической
структуры полимеров, полученных из о. а. и рацеми-
рацемических мономеров.
При стереоселективной полимеризации образующий-
образующийся полимер и еще неизрасходованный мономер оптиче-
оптически неактивны, т. к. число растущих (+)- и (—)-цепей
одинаково. Если же сам катализатор, обеспечивающий
стереоселективную полимеризацию, содержит асим-
асимметрич. группировки, то число растущих (+)-цепей
или скорость их роста будут отличаться от числа и ско-
скорости роста (—)-цепей. В этом случае полимер обога-
обогащается одним изомером, а непрореагировавший моно-
мономер — другим. И продукт реакции, и оставшийся мо-
мономер приобретают оптич. активность. Процессы тако-
такого типа принято наз. стереоэлективной
полимеризацией.
При инициировании полимеризации рацемич. окиси
пропилена с помощью асимметрич. катализатора, полу-
полученного модификацией диэтилцинка о. а. ментолом,
борнеолом или а-аминокислотами, образуется о. а.
иолипропиленоксид, а непрореагировавший мономер
обогащается др. энантиомером. (+)-Борнеол способст-
способствует преимущественной полимеризации (+)-окиси про-
пропилена, а L-аминокислоты — (—)-изомера. Стерео-
элективная полимеризация окиси пропилена иниции-
инициируется и др. комплексными катализаторами: триэтил-
алюминий — N-карбоксиангидрид L-аланина, FeCl3—
(+)-борнилэтиловый эфир и др. Оптич. чистота поли-
полимера достигает 20%. В этих случаях асимметрич. ча-
частицей, осуществляющей отбор антиподов мономера,
является комплекс катализатора с растущим концом
цепи. Стереоэлективная полимеризация протекает по
механизму «каталитического контроля».
Др. пример такого процесса — стереоэлективная по-
полимеризация а-олефинов преимущественно S-конфи-
S-конфигурации, инициируемая комплексом (+)-6wc-[(S)-
2-метилбутил]цинка с хлоридом титана. Интересно, что
комплекс, осуществляющий каталитич. контроль, спо-
способен различать изомеры а-олефинов с асимметрич.
углеродом только в а-положении к двойной связи
(напр., у З-метилпентена-1 или 3,7-диметилоктена-1),
но не в (J-положении. Алюминий- и индийорганиче-
ские катализаторы не обеспечивают в этой реакции
столь высокой стереоселективности, как цинкорга-
нические.
Значительно реже конфигурационный контроль
в стереоэлективном процессе осуществляется саморас-
саморастущей цепью без участия катализатора, как, напр.,
при полимеризации N-карбоксиангидридов а-амино-
кислот.
Полимеры с высокой оптич. активностью получе-
получены при стереоэлективной полимеризации рацемич. со-
соединений: 1\-(г-бутил)пропиленимина под действием
D-винной к-ты, пропиленсульфида под действием систе-
системы диэтилцинк — (+)-борнеол или L-лейцин, а также
р-фенилпропилизоцианата в присутствии (—)-менти-
лата натрия.
Кроме о. а. катализатора, стереоэлективность поли-
полимеризации рацемич. мономера м. б. обусловлена при-
присутствием о. а. сомономера. Так, при сополимери-
зации (S)-4-метилгексена-1 с рацемич. 4-метилпенте-
ном-1 в присутствии каталитич. системы TiCl4 +
+ 2п(н-С^Н9J обнаружено, что в полимерную цепь
преимущественно встраивается (8)-изомер метилпен-
тена. Аналогичную стереоэлективность следует ожи-
ожидать при сополимеризации рацемич. окиси пропилена
с N-карбоксиангидридом L-аланина, инициируемой
А1(С2Н6K.
Стереоселективная и стереоэлективная полимериза-
полимеризация и сополимеризация являются примерами кинети-
кинетического «расщепления» рацематов, поскольку один
из оптических изомеров рацемического мономера при-
присоединяется к растущей полимерной цепи быстрее
другого.
Синтез из симметричных мономеров. Стереоспецифи-
ческие процессы. Процессы, в к-рых реагирующая асим-
асимметрич. частица способна диктовать определенную кон-
конфигурацию образующемуся в результате реакции ново-
новому асимметрич. центру, т. е. способна индуцировать
его асимметрию, наз. стереоспецифически-
м и. Третичные углеродные атомы каждого отдельно
взятого элементарного звена полимера винильного ти-
типа асимметричны, тогда как молекулы исходного моно-
мономера имеют плоскость симметрии. Конфигурация асим-
асимметрич. углерода последнего звена растущего конца
полимерной цепи может диктовать присоединяющемуся
звену либо противоположную конфигурацию (в этом
случае образуются синдиотактические полимеры), либо
идентичную конфигурацию (тогда образуются изо-
тактические цепи). Поэтому стереоспецифическую по-
полимеризацию справедливо считать асимметрическим
синтезом.
Особенность этой реакции заключается в том, что
в случае мономеров типа СН2 = СНХ, СН2 = CXY и
GHX = GHY она не может привести к образованию о. а.
макромолекул, даже если с помощью асимметризую-
щего агента добиться образования асимметрич. атомов
только одной конфигурации. Объясняется это тем, что
в изотактических и синдиотактич. гомополимерах и чере-
чередующихся сополимерах винилового типа все третичные
углеродные атомы основной цепи (за исключением кон-
концевых звеньев) имеют симметричное ближайшее окру-
окружение, т. е. псевдоасимметричны. Асимметрия только
концевых звеньев недостаточна для придания макро-
макромолекулам заметной оптич. активности.
Иначе обстоит дело при образовании политактич.
полимеров. При сополимеризации винилового мономе-
мономера с 1,2-дизамещенным олефином в каждом звене воз-
возникают три истинно асимметрич. атома. Используя
о. а. компонент, можно управлять их конфигурациями.
Так, при радикальном инициировании получен сополи-
сополимер Lra-метилбензилметакрилата и малеинового ан-
ангидрида, имеющий после удаления L-a-метилбензиль-
ных групп положительное вращение:
' СН2 - С (СН3) - [- СН
I \ I
COOR ОС-О-СО
СН - СН2-С (СН3)-]-СН-СН /
I I \ I
COOR OCOCO
где r = - н или сн (СН3) с6н5
491
ОПТИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРЫ
492
Механизм асимметрической индукции при асиммет-
асимметрической стереоспецифической сополимеризации тако-
такого типа показан схемой, иллюстрирующей акт при-
присоединения молекулы малеинового ангидрида к ра-
радикальному концу цепи его сополимера с L-a-метил-
бензилвиниловым эфиром: п
На этой стадии возникают ера- / \
зу два новых асимметрич. центра. Н Н ОС СО
Теоретически возможно образова- н—С--С*—С- С
ние четырех пространственных ill ^
изомеров (на схеме показаны два R О Н Н
из них), однако стереоспецифич- __ 1*
ность полимеризации и асиммет-
Н-С— СН3
Н—С—СН,
рич. индукция о. а. бокового за-
заместителя приводят к образова-
образованию только одного. Даже после
удаления боковых групп поли-
полимер проявляет оптич. активность.
О. а. компонент, ответственный за асимметрич. ин-
индукцию при полимеризации, может не являться ча-
частью молекулы мономера, а входить в состав катали-
катализатора полимеризации; при этом катализатор должен
контролировать стадию роста цепи. Если же катализатор
способен контролировать только стадию инициирова-
инициирования (как, напр., в случае использования о. а. ацильных
перекисей), то его влияние быстро падает с ростом це-
цепи. Статистич. рассмотрение
такого процесса показывает,
что оптич. активность полиме-
полимера стремится к нулю с ростом
мол. массы.
Пример успешного асиммет-
асимметрич. синтеза полимера с по-
помощью о. а. катализатора ка-
тионно-координационного ти-
типа (контролирующего стадию
роста) — полимеризация бен-
зофурана на катализаторе А1С13-Ь-C-фенилаланин.
Полимер III имеет диизотактич. структуру, все
атомы углеродной цепи асимметричны.
Бензофуран относится к циклич. мономерам, гомо-
полимеры к-рых имеют истинные асимметрич. атомы
в цепи. Проведена асимметрич. полимеризация мономе-
мономеров, аналогичных бензофурану — а- и р-нафтофуранов
(IV,V), однако получить О. а. п. индена (VI), 1-метил-
циклопентена (VII) или 4,5-дигидро-1-метилфурана
(VIII) пока не удалось.
СН3
VII!
Очевидно они образуют полимеры дисиндиотактич.
структуры. В синдиотактйч. цепях вклады асиммет-
асимметрич. R-HS-атомов цепи в оптич. активность полимера
взаимно компенсируются. .
К др. типу мономеров, образующих при гомополиме-
ризации цепи с истинными асимметрич. атомами, отно-
относятся сопряженные диены с заместителями в положе-
положении 1 или 1,4. Присоединение таких диенов к цепи
приводит к возникновению одного или двух асимхмет-
рич. атомов соответственно:
СН :
X
= СН — СН = СН -
Y
-> - [- СН - СН = СН - СН -]п- СН - СН = СН - СН -
X Y X Y
Так, о. а. изотактич. полимер гарамс-пентадиена-1,3,
имеющий 1,4-^ис-структуру, получен при использо-
использовании смешанного катализатора триэтилалюминий —
(—)-тетраментилат титана. Полимеризация метиловых
эфиров р-винилакриловой к-ты (IX), транс,транс-$-
стирилакриловой к-ты (X) и транс, транс-сотрбпио-
вой к-ты (XI) в неполярных растворителях под воздей-
воздействием о. а. A1)-2-метилбутиллития или комплекса
бутиллития с (—)-ментилатом натрия или (—)-ментил-
этиловым эфиром также приводит к О. а. п. 1,4-транс-
apwwpo-диизотактич. структуры:
=
XI
Г
L
Озонолизом полимеров получены о. а. фенил-и метил-
янтарная к-ты (оптич. чистота 6%), что подтверждает
осуществление асимметрич. синтеза. По катионному
механизму с использованием комплекса BF3 с (—)-мен-
то л ом осуществлена асимметрич. полимеризация 1-фе-
нил-4-метилбутадиена-1,3.
Миграционной полимеризацией бензальацетона и
a-бензальметилэтилкетона в присутствии (—)-менти-
лата лития получены о. а. поликетоны:
сн = с - с - сн3 —[- сн - сн - с - сн2-]п
*б И О
С6Н6 R О С6НГ< R О
Т. о., для осуществления асимметричной стереоспе-
цифич. полимеризации симметричных мономеров не-
необходимо выполнение след. условий: 1) в полимерной
цепи возникают истинно асимметрич. атомы; 2) ин-
индуцирующий асимметрич. центр имеет "сильно разли-
различающиеся по размеру заместители; 3) индуцирующий
асимметрич. центр располагается возможно ближе к
активному комплексу (растущий конец цепи— инициа-
инициатор — мономер) или, лучше, входит в его состав. Ката-
литич. системы гетерогенного или гомогенного ионно-
координационного типа обеспечивают образование
таких комплексов и, как следствие, высокую стереоре-
гулярность полимеризации; 4) темп-ра полимеризации
возможно более низкая, чтобы уменьшить конформа-
ционную подвижность реагирующих компонентов.
Последние три условия справедливы и для стереосе-
лективной полимеризации рацемич. мономеров.
Попытка осуществления «абсолютного» асимметрич.
синтеза полимера (без применения химич. асимметри-
зующих веществ) оказалась безуспешной: сополимери-
зация стирола с малеиновым ангидридом под воздейст-
воздействием поляризованного р-излучения привела к оптически
недеятельному полимеру.
Исследование методом спектрополяриметрии
Спектрополяриметрия — один из наиболее эффектив-
эффективных методов изучения конформации О. а. п. в растворе.
Наиболее успешно этот метод применялся при ис-
исследовании полипептидов, где удалось установить
прямую зависимость между степенью спирализации
макромолекулы и значением константы Ьо в ур-нии
Моффита и Янга (Ьоъ — 630° для 100%-ной правоспи-
ральной конформации). Явления перехода спираль —
статистич. клубок, диссоциации, ассоциации, денату-
денатурации и ренатурации полипептидов и белков исключи-
исключительно сильно отражаются на оптич. активности их
493
ОПТИЧЕСКИЕ СВОЙСТВА
494
р-ров. Кроме полипептидов, спиральные конформа-
ции предполагаются у полимеров о. а. молочной к-ты,
алкилвиниловых эфиров и пропиленимина, т. к. кри-
кривые дисперсии оптич. вращения их р-ров подчиняются
ур-нию Моффита и Янга.
Изучение р-ров поли-а-олефинов с о. а. боковыми
группами показало, что молярная оптич. активность
мзотактич. фракций этих полимеров по абсолютной
величине более чем на порядок превышает молярное
вращение соответствующих низкомолекулярных соеди-
соединений (таблица). Знак оптич. активности исследован-
исследованных поли-а-олефинов определяется абсолютной конфи-
конфигурацией асимметрич. углеродного атома мономера:
все полимеры (8)-а-олефинов характеризуются поло-
положительным вращением. Значительное увеличение оптич.
активности было отмечено при полимеризации альде-
альдегидов, содержащих те же о. а. заместители, что и при-
приведенные в таблице а-олефины.
Сравнение оптической активности модельных
низкомолекулярных парафинов и поли-а-олефинов
Ни;'.комоле-
кулярный
парафин
<?)-2,3-Диме-
тилпентан
(8)-2.4-Диме-
тилгоксан
<8)-2,5-Диме-
тилгептан
<8)-2,6-Диме-
тилоктан
25
-11,4
+21,3
+ 11,7
+ 14,4
Полимер
Поли-(8)-3-ме-
тилпентен-1
Поли-(8)-4-ме-
тилгексен-1
Поли-(8)-5-ме-
тилгептен-1
Поли-(8)-6-ме-
тилоктен-1
аморфная
или очень
слабокри-
сталлич.
фракция
+29,4
+ 116
+ 13,1
+ 12,8
высоко-
кри-
сталлич.
фракция
+ 161
+288
+68*
+20*
* Данные относятся к растворимым фракциям, имевшим
наибольшую степень изотактичности.
Столь высокие значения уд. вращения поли-а-оле-
поли-а-олефинов м.б. вызваны двумя причинами: наличием в р-рах
мзотактич. полимеров участков со спиральной конфор-
мацией цепи (такие структуры обнаружены в кристал-
лич. состоянии) либо существованием заторможенных
конформаций отдельных звеньев, обладающих высокой
оптич. активностью. Известно, что уд. вращение полиме-
полимеров и температурный коэфф. вращения быстро растут
до предельных значений с ростом стереорегулярности;
эти значения максимальны для полиолефинов с асим-
асимметрич. атомами в а-положении к цепи; кривые диспер-
дисперсии оптич. вращения в доступной для измерения обла-
области длин волн подчиняются простому ур-нию Друде.
Полуэмпирич. расчеты конформаций цепи показыва-
показывают, что цепи могут существовать преимущественно
в виде правой спирали, если ей соответствует больший
набор доступных конформаций асимметрич. боковых
групп. Однако имеющихся данных все еще недостаточ-
недостаточно, чтобы сделать однозначный выбор между двумя
возможными причинами аномально высокой оптич.
активности поли-а-олефинов.
Как и в случае поли-а-олефинов, оптич. активность
большинства полимеров винилового типа сильно зави-
зависит от их тактичности. Особенно велико влияние так-
тактичности на значение вращательной константы Ко
в ур-нии Друде.
Зависимость [а] от мол. массы полимера обычно ука-
указывает на частичную рацемизацию о. а. катализатора,
мономера или полимера в условиях полимеризации.
Этот факт также м. б. использован при выяснении ме-
механизма процесса. Существование значительной зави-
зависимости [а] от типа растворителя м. б. использовано
для изучения взаимодействия полимер — растворитель.
Измерение [а] продуктов сополимеризации активного
и неактивного мономеров позволяет определять кон-
константы их сополимеризации.
Применение
Наиболее важная область использования О. а. п.
в настоящее время — разделение с их помощью оптич.
изомеров методами жидкостной и газовой хроматогра-
хроматографии. В качестве сорбентов испытаны многочисленные
природные и синтетич. О. а. п., полученные модифика-
модификацией оптически недеятельных полимеров или полиме-
полимеризацией активных мономеров. Наиболее перспектив-
перспективными в этом плане оказались хелатообразующие О. а. п.,
пригодные для хроматографии рацемич. лигандов.
Асимметрич. циангидриновым синтезом (с оптич.
выходом 19%) в присутствии сшитого поли-(8)-изобу-
тилэтиленимина продемонстрирована принципиальная
возможность использования О. а. п. как катализаторов
в асимметрич. органич. синтезе. О. а. п. могут служить
матрицей и в асимметрич. синтезе полимеров. Так,
в присутствии того же поли-(8)-изобутилэтиленимина
полиприсоединением тетраметилендимеркаптана к эти-
ленгликольдиметакрилату получен полимер с [a]D = 1,5°:
СН2 = ССОО (СН2JООСС = СН2 + HS (GH,LSH -*
! I
СН3 СН3
-*[ - S (CH2LSCH2CCOO (CH2JOOGGCH2-j
i I
CH3 CH3
Полимеры, полученные из о. а. мономеров, обладают
более высокими механич. свойствами и повышенной
теплостойкостью по сравнению с рацемич. продукта-
продуктами, что представляет определенный практич. интерес.
О. а. п. пригодны для изготовления стекол и пленок,
обладающих способностью вращать плоскость поляри-
поляризации проходящего света (см. Поляроидпые пленки).
Лит.: Schulz R. С, К а i s е г Е., Adv. Polymer Sci.,
4, Н. 2, 236—315 A965): Goodman M., A b e A.,
Fan Y.—L., Macromoi. Revs, 1, 1—33 A967); П и н о П.,
Хим. и технол. полимеров, № 12, 54 A967); Пи но П.,
ЧиарделлиФ., Кие л лини Б., в кн.: Дисперсия
оптического вращения и круговой дихроизм в органической
химии, под ред. Г. Снатцке, пер. с англ., М., 1970, с. 333; К л а-
буновский Е. И., Л а то в В. К., в кн.: Химия и
технология высокомолекулярных соединений, М., 1971 (Итоги
науки. Сер. хим., т. 3). Б. А. Даванков, И. Я. Топчиева.
ОПТИЧЕСКИЕ СВОЙСТВА полимеров (opti-
(optical properties, optische Eigenschaften, proprietes opti-
ques) — свойства, характеризующие взаимодействие по-
полимера с электромагнитным излучением оптич. диапа-
диапазона.
Спектральный диапазон, изучаемый оптикой, охва-
охватывает УФ-область в диапазоне длин волн А от 3-.10~7
до 4-10~5 см, видимую (воспринимаемую глазом чело-
человека) область в диапазоне X от 4-10~5 до 8-10~5 см,
и ИК-область в диапазоне от 8-10~5 до ~10~2 см. Ча-
Частоты оптич. диапазона спектра электромагнитных
волн лежат в интервале от 1017 до 1012 гц. С коротко-
коротковолновой стороны этот диапазон граничит с рентгенов-
рентгеновским излучением (X > 5-10~7 см), а с длинноволновой
стороны — с микроволновым диапазоном (к < 10~2 см).
УФ-область спектра принято разделять на диапазон
вакуумного ультрафиолета (К = 5-10~7—1,7-10~5 см)
и нормального УФ-излучения (к= 1,7• 10~5—А-10~ъ см).
ИК-диапазон также разделяют на близкую область
(к < 25-10~4 см) и далекую область(Х > 25-Ю см).
При изучении О. с. полимеров обычно рассматри-
рассматривают преломление, отражение и поглощение света изо-
изотропными и анизотропными средами, дисперсию, рассея-
рассеяние и деполяризацию рассеяния света, естественную
оптич. вращающую способность, а также ряд явлений,
возникающих в полимерных средах иод воздействием
силового поля — электрического, магнитного, ультра-
ультразвукового, статического механического и др. Все эти
макроскопич. явления м. б. поняты только на основе
молекулярных представлений и поэтому рассматри-
рассматриваются в молекулярной оптике.
495
ОПТИЧЕСКИЕ СВОЙСТВА
496
Точное выражение молекулярных оптич. постоянных
возможно лишь в рамках квантово-механич. анализа.
Для этого необходимо иметь сведения об энергетич.
уровнях макромолекулы и вероятностях перехода меж-
между ними, т. е. ключ к раскрытию О. с. молекул дают
частоты и интенсивности линий в ее спектре. В основе
молекулярной оптики лежит спектроскопия.
Несмотря на ряд достижений квантово-механич. опи-
описания нек-рых О. с. вещества (напр., дисперсии света),
такое описание не удается осуществить из-за матема-
тич. трудностей. Однако квантово-механич. рассмот-
рассмотрение явлений молекулярной оптики показало прин-
принципиальную возможность применения именно в оптике
классич. моделей. Такие модели непригодны для точных
расчетов, однако соответствие между результатами
квантово-механич. и классич. рассмотрений оптич.
явлений дает возможность построить полуэмпирич. тео-
теорию молекулярно-оптич. явлений, дающую не только
качественное, но и количественное их объяснение.
О. с. среды определяются строением электронной обо-
оболочки молекул, из к-рых построена эта среда. При
частотах видимого диапазона порядка 1014 гц «оптиче-
«оптический электрон» (т. е. электрон, слабо связанный с ато-
атомом, но не свободный) способен следовать за колебания-
колебаниями поля. Его смещение I создает дипольный момент р
A)
—> —>
l
где е — заряд электрона.
Оптич. постоянные среды (напр., показатель прелом-
преломления) связаны с усредненными значениями молеку-
молекулярных постоянных, из к-рых наиболее важной явля-
является поляризуемость а
Г=«? B)
где Е — напряженность электрич. поля. Поляризуе-
Поляризуемость молекулы а характеризует способность ее
электронной оболочки деформироваться под действием
внешнего электрич. поля. Средняя поляризуемость а
тесно связана с др. важной оптич. постоянной среды —
молекулярной рефракцией R
п2—1 М лт 4 тс —
п*+2 р ~ А 3
C)
где п — показатель преломления, М — мол. масса,
р — плотность вещества, NА — число Авогадро. Пра-
Правая часть выражения C) показывает независимость
R от р, а следовательно от темп-ры и давления. Важно
отметить, что R относительно мало меняется при изме-
изменении агрегатного состояния вещества. Для молекул
органич. соединений в большинстве случаев R можно
представить в виде суммы рефракций (а значит и поля-
ризуемостей) различных элементов этой молекулы,
напр, атомов, ионов или валентных связей. Аддитив-
Аддитивность R выполняется не всегда, однако отклонения во
многих случаях незначительны. Это объясняет большую
роль рефракции при изучении структуры молекул.
Величина R зависит от длины волны света, однако адди-
аддитивность R, измеренных для одних и тех же длин
волн, сохраняется. Значения Я, экстраполированные
к бесконечной длине волны, сопоставимы с молекуляр-
молекулярной поляризацией в статич. поле.
Отклонение от аддитивности (разность наблюдаемой
R и вычисленной в предположении аддитивности ре-
рефракций связей #адд) наз- экзальтацией
рефракции:
ДЯ=Я-Яадд D)
Значение A R характеризует сильное взаимодействие
электронов, формально принадлежащих различным
связям в молекуле, и может служить мерой такого взаи-
взаимодействия, напр, мерой эффективности сопряжения.
Оптические постоянные макромолекул
Цепная макромолекула — сложный статистич. ан-
ансамбль мономерных звеньев; при его описании поль-
пользуются средними эффективными параметрами, величины
к-рых зависят от степени заторможенности внутреннего
вращения атомных групп и атомов вокруг единичных
связей цепи. Строгая оценка О. с. макромолекулы чрез-
чрезвычайно сложна.
Оптическая поляризуемость. Важнейшие О. с. поли-
полимеров зависят от тензорной величины — оптич. поля-*
ризуемости, и многие из этих свойств представляют
собой проявление анизотропии этого тензора. Тензор
поляризуемости молекул низко- и высокомолекуляр-
высокомолекулярных соединений является тензором второго ранга.
О. с. полимеров и близких им по строению низкомо-
низкомолекулярных соединений, как правило, различаются
не сильно. Лишь нек-рые О. с, связанные с размером
макромолекул, их цепным строением и огромным кон-
формационным набором, выражены для полимерных
систем сильнее, чем для соответствующих низкомоле-
низкомолекулярных соединений. К таким свойствам относятся
рассеяние света р-рами полимеров, двойное лучепре-
лучепреломление в потоке, фотоупругость и нек-рые др. динамо-
оптич. свойства. В то же время поглощение света, ком-
комбинационное рассеяние и др. свойства слабо отличаются
от свойств соответствующих низкомолекулярных моде-
моделей. При решении конкретных задач молекулярной
оптики низкомолекулярных соединений необходимо
статистич. усреднение тензора поляризуемости молекул
по их различным пространственным ориентациям. При
рассмотрении составляющих тензора поляризуемости
макромолекул, кроме вышеуказанного усреднения,
суммируют тензорные составляющие отдельных связей
или групп по конформациям макромолекулы и находят
инварианты тензора, определяющие рассматриваемое
О. с. В основе такого усреднения лежит поворотно-
изомерная модель макромолекулы, предложенная
М. В. Волькенштейном.
Оптическая анизотропия (О. а.) и анизотропия тен-
—»
зора поляризуемости (А. т. п.). Вектор г, соединяющий
концы макромолекулы, является направлением пре-
преимущественной ориентации составляющих ее статистич.
сегментов. Если поляризуемость сегмента вдоль его
оси ах не равна поляризуемости в поперечных направ-
направлениях а2, то это приводит к различию поляризуемо-
стей рх и р2 всей макромолекулы соответственно вдоль
и перпендикулярно г, причем О. а. макромолекулы
Pi-p»= -f- к - «i) (S)
близка к О. а. сегмента.
Согласно определению, статистич. сегмент состоит
из жестко сочлененных мономерных звеньев, находя-
находящихся в предельно вытянутой конформации. Поэтому
2=S(a{l —a
±
где а ц и а. — соответственно продольная и попереч-
поперечная поляризуемости мономерного звена, S — коэфф.,
характеризующий положение системы координат, свя-
связанной с макромолекулой, по отношению к лаборатор-
лабораторной системе координат.
Оптически анизотропные связи, особенно несиммет-
несимметрично расположенные в мономерном звене, вызывают
О. а. всей макромолекулы. Аддитивность средней по-
поляризуемости (рефракции) есть отражение свойств
инварианта тензора поляризуемости — его следа. Др.
инвариант тензора поляризуемости — его А. т. п.
(Y2 — см. ниже) — не обладает свойством аддитив-
аддитивности, т. к. зависит от валентных углов между связями
в макромолекуле.
А. т. п. макромолекулы зависит только от А. т. п.
отдельных связей, но не от их следов. Аддитивность
497
ОПТИЧЕСКИЕ СВОЙСТВА
векторов дипольных моментов и тензоров поляризуе-
поляризуемости (а также их производных по нормальным коорди-
координатам) лежит в основе т. н. валентно-оптич. схемы.
Инварианты — след и Y2 ~~ дают молекулярную
рефракцию и двойное лучепреломление, появляющееся
под действием внешних силовых полей. Первая вели-
величина определяется через среднюю поляризуемость
- _ с^+аа+яз ^ вторая равна
о
у2=0,5[(ах— а2J+(а1—азJ+(а2—азJ] G)
Обе величины эффективные.
При определении тензора поляризуемости макромо-
макромолекулы за основу принимают тензор поляризуемости
связи, к-рый в системе координат этой связи не зависит
от конформации цепи. Затем рассматривают группу
связей г. Внутри группы рассчитывается суммарный
тензор поляризуемости группы af-, не зависящей в си-
системе координат группы от конформации всей цепи.
Взаимная ориентация связей в группе должна быть
фиксирована и задается геометрией цепи (длинами свя-
связей и валентными углами), т. е. в группу должны вхо-
входить связи, исходящие из одного атома. Тензор поля-
поляризуемости макромолекулы получается суммированием
по всем конформациям цепи вкладов всех связей или
групп, входящих в цепь.
Анизотропия статистич. сегмента в реальной поли-
полимерной цепи — величина эффективная, зависящая от
условий внутреннего вращения. Цепи с заторможенным
внутренним вращением и фиксированными валентными
углами более анизотропны, чем свободно-сочлененные.
Рассмотренные оптич. постоянные изолированной
макромолекулы и служат основой для характеристики
О. с. полимеров.
Оптические постоянные упруго растянутой сетки.
Физич. картина растяжения аморфных нестеклообраз-
нестеклообразных полимеров как равновесного процесса предпола-
предполагает объединение макромолекул в сетку. При отсутст-
отсутствии сетки как системы контактов анизотропия, вноси-
вносимая растяжением, устранялась бы вследствие релакса-
релаксационных явлений. Цепная молекула становится элемен-
элементом сетки, поперечные связи к-рой сохраняются доста-
достаточно долго по сравнению со временем воздействия и
наблюдения. В нерастянутой сетке система цепей счи-
считается изотропной. Общее рассмотрение О. а. изотроп-
изотропно и однородно растянутой сетки макромолекул прово-
проводится также методом конформационного усреднения.
Растянутая сетка в среднем анизотропна; в общем слу-
случае средний тензор поляризуемости |сс| отдельных цепей
не является скалярным тензором (т. е. он не изотропен).
Макроскопич. поляризуемость представляет собой сум-
сумму средних вкладов различных цепей, входящих в ис-
исследуемый образец.
Отправной геометрич. характеристикой при расчете
компонентов тензора поляризуемости вдоль вектора
длины цепи г служит средний квадрат косинуса углов
между г и направлениями связей. Три главных значе-
значения тензора деформации или соответствующие им три
главных растяжения \г, Х2, Х3 полностью характери-
характеризуют деформацию. Реальная сетка в состоянии покоя
характеризуется функцией распределения векторов
длин цепей сетки W (г). До растяжения функция сим-
симметрична. После растяжения структура сетки харак-
характеризуется новой функцией распределения W'(r')y
причем векторы г преобразуются в векторы г", слагаю-
слагающие к-рых связаны соотношением
r'i =Vi (8)
Цепи, характеризуемые одним и тем же вектором длины
г, могут иметь большое число различных конформации,
имеет
= 0
L0
согласующихся с г. Можно считать, что по отношению
к такому набору цепей вектор г является в среднем
осью цилиндрич. симметрии. Отсюда следует, что усред-
усредненный тензор оптич. поляризуемости цепей должен
иметь диагональную форму в такой системе координат,
где одна из осей параллельна г (две другие перпендику-
перпендикулярны). Средний тензор поляризуемости в такой систе-
системе координат имеет вид
0 0 1
C;-аг)/2 0 (9)
0 Ca-ar)/2 J
где аг — среднее значение поляризуемости в направ-
направлении г. Тензор а можно также определить как усред-
усредненное по всем конформациям цепей значение поляри-
поляризуемости, когда цепи имеют фиксированное значение
вектора г и не подвергнуты каким-либо др. ограниче-
ограничениям вследствие переплетений с соседними цепями.
В теориях О. с. предполагается, что тензоры поля-
поляризуемости групп аддитивны и инвариантны относи-
относительно изменения конформации цепи.
Трудности применения модели свободно-сочленен-
свободно-сочлененной цепи к растягиваемой сетке частично преодолевают-
преодолеваются поворотно-изомерной теорией. При растяжении про-
происходит изменение исходного конформационного набора
макромолекул и затем уменьшение амплитуд крутиль-
крутильных колебаний около положения каждого звена, отве-
отвечающего минимуму потенциала внутреннего вращения.
Основные оптические свойства полимеров
Прозрачность (П.) — характеристика полимера, по-
показывающая, какая доля падающего на его поверхность
светового потока проходит без изменения направления
через слой определенной толщины.
П. учитывает как поглощение, так и рассеяние света
и отличается от пропускания — более общей характе-
характеристики (тонкий слой непрозрачного вещества может
пропускать рассеянные на неоднородностях лучи, из-
изменившие свое первоначальное направление). Макси-
Максимальная П. полимеров в видимом диапазоне спектра
составляет 92—94%. См. также Органическое стекло.
Стеклообразные аморфные полимеры обычно про-
прозрачны и бесцветны (при темп-рах ниже темп-ры стек-
стеклования). П. высокоэластич. полимеров встречается
реже. Кристаллизация вызывает увеличение мутностиг
у полимеров появляется молочно-белый оттенок (напр.,
в полнодефинах). Уменьшение П. связано с появлени-
появлением оптич. неоднородностей как на поверхности (сколы,
трещины, серебрение, царапины), так и в объеме поли-
полимерных материалов.
Р-ры полимеров при концентрациях менее 1% обычно
прозрачны. С увеличением концентрации мутность воз-
возрастает плавно или скачком в области фазовых перехо-
переходов. Эти переходы м. б. связаны с внутримолекулярны-
внутримолекулярными конформационными переходами или с разделением
р-ра на фазы при достижении критич. концентрации
смешения полимера с растворителем (при данных
темп-ре и мол. массе). Титрование по мутности — метод
определения мол. массы, полидисперсности. Регистра-
Регистрация отдельной фракции при фракционировании осно-
основана на изменении П. р-ра полимера соответствующей
мол. массы при добавлении осадите ля.
В р-рах полимеров при определенной темп-ре, близ-
близкой к темп-ре, при к-рой происходит высаживание
полимера, появляется критич. опалесценция —
резкое возрастание мутности вследствие рассеяния
света на флуктуациях плотности и концентрации в об-
области фазового перехода. Обратимое изменение П.
при термич. воздействиях наблюдается в нек-рых кол-
коллоидных р-рах полимеров; молекулярный механизм
явления — термокоагуляция дисперсной фазы полиме-
499
ОПТИЧЕСКИЕ СВОЙСТВА
500
С sin cp
П~~ 1Г~ ~ sin с?'
pa, имеющего ограниченную растворимость. Производ-
Производные поливинилового спирта (поливинилэтилаты) в сме-
смеси метиловый спирт — вода прозрачны при комнатной
темп-ре и снижают светопропускание до 2—3% при
30 СС. Возможно изменение светопропускания до 30 —
40% на 1 СС в интервале 2—3 °С.
П. гетерогенных полимерных систем определяется их
совместимостью: появление мутности указывает на недо-
недостаточную совместимость. Количественная характери-
характеристика П.— коэфф. светопропускания —
отношение потока излучения, вышедшего из слоя по-
полимера, к потоку, падающему на его поверхность.
П. органич. полимеров сравнима с П. лучших сортов
неорганич. стекол, а в УФ-диапазоне превышает ее.
Показатель преломления (П. п.). Известно, что свет
в веществе движется медленнее, чем в вакууме. Этот
факт учитывается введением показателя преломления
A0)
где С — скорость света в вакууме, С' — скорость света
в веществе, ср и ср' — углы падения и преломления
света соответственно.
Под действием электрич. поля Е световой волны
в молекулах среды индуцируются электрич. диполи, со-
совершающие вынужденные колебания с частотой, рав-
равной частоте воздействующей световой волны. Эти ди-
диполи являются источниками вторичных световых волн.
В результате сложения первичной волны и всех вто-
вторичных волн возникает результирующая волна, к-рая
внутри среды распространяется в соответствии с зако-
законами преломления, а вне ее — в соответствии с закона-
законами отражения. Молекулярная теория объясняет т. обр.
отсутствие света по направлениям, отличным от направ-
направлений преломленного и отраженного лучей. Это спра-
справедливо лишь при условиях равномерного распреде-
распределения молекул вещества по объему и малости расстоя-
расстояний между ними по сравнению с длиной световой вол-
волны. Только при этих условиях (вследствие интерферен-
интерференции когерентных вторичных волн) свет гасится по всем
направлениям, кроме предписанных законами геомет-
рич. оптики.
При прохождении через среду электромагнитная
волна затухает, т. е. выходит из среды, потеряв часть
своей энергии. Для описания этого явления пользуют-
пользуются понятием комплексного П. п.
n—nf—in" A1)
где п' и п" вещественны.
Величина п' определяет преломленную часть свето-
световой волны, а п" описывает поглощение (или «ослабле-
«ослабление») электромагнитной волны. Величина п" учитывает
процессы поглощения, отражения и аномальной дис-
дисперсии света. Ее иногда наз. коэффициентом
поглощения.
С П. п. связана молекулярная рефракция нерастя-
нерастянутого аморфного полимера (см. ур-ние 3). П. п. зави-
зависит от длины волны падающего излучения и плотности
полимера, меняющейся с темп-рой. В области олигоме-
ров п зависит от мол. массы М. По достижении
нек-рого М эта зависимость исчезает.
Существование поворотных изомеров не оказывает
влияния на п, т. к. их молекулярные рефракции одина-
одинаковы. Конфигурационная стереоизомерия, напротив,
сказывается на величине п.
Для р-ров полимеров п возрастает с ростом концент-
концентрации с линейно в достаточно большой области кон-
концентраций:
dn\
где пс — П. п. при данной концентрации с; п0 — П. п.
растворителя; dn/dc — инкремент показа-
показателя преломления (И. п. п.).
Первая производная п по выбранному направлению
наз. градиентом показателя прелом-
преломления (Г. п. п.) по данному направлению. Г. п. п.
регистрируется в оптич. методах исследования макро-
макромолекул. Непрерывность регистрации п возможна, т. к.
dn dn dc f.o.
dx — dc ' dx ^lo)
т. е. распределение п вдоль координаты х следует
за распределением концентрации.
Измерение И. п. п.— необходимый этап определения
мол. массы по рассеянию света и независимый способ
определения концентрации полимера в р-ре. И. п. п.
бинарных сополимеров определяется соотношением:
где Х2 — «молярная» доля второго компонента в мак-
макромолекуле сополимера. Аддитивность dn/dc позволяет
в макромолекуле сополимера определить средний со-
состав сополимеров с точностью до 2—3% . Однако ме-
метод не отличает истинный сополимер от примесей (го-
мополимеров).
Наиболее распространенные методы определения
п — рефрактометрия и интерферометрия. Для спе-
специальных условий п определяют по отношению
к стандартной атмосфере. У большинства полимеров
п близок к 1,5; для полиметилметакрилата при 20 °С
п^у = 1,4881 (D — желтая линия спектра натрия,
А = 589,3 нм). Максимальные п = 1,6—1,7 имеют фе-
ноло-фурфурольные и феноло-формальдегидные поли-
полимеры; минимальные п = 1,34—1,35 — фторопласты.
Учет п полимеров в первую очередь требуется в пле-
пленочной электронике (печатные схемы) и оптоэлектро-
нике; при автоматизации химич. процессов (контроль
процесса по непрерывному измерению п)\ при конструи-
конструировании двумерных (плоских) оптич. схем (в оптич.
ЭВМ и т. д.); в волоконной оптике (особенно в оптич.
преобразователях).
Дисперсия показателя преломления (Д.) — зависи-
зависимость п от частоты со падающего света. Эта зависимость
выражается соотношением
Nk
п = 1 + Const
A5)
k h ~ k
где cofe — собственная круговая частота электронов,
число к-рых в единице объема равно N^', Ьк —коэфф.
затухания. Выражение A5) показывает, что п изменя-
изменяется с частотой падающего света. По этой причине
призмы разлагают («диспергируют») свет в спектр. По-
Появление члена, описывающего затухание, объясняет
мнимую часть показателя преломления (см. ур-ние И).
Выражение A5) для показателя преломления справед-
справедливо для большого числа веществ.
Везде, за исключением области, где со очень близко
к одной из резонансных частот со^ исследуемой системы,
зависимость п от частоты падающего света «нормальная»,
т. е. наклон кривой п = f (со) положителен. Вблизи
резонансных частот этот наклон становится отрица-
отрицательным. Убывание п при увеличении со наз. ано-
аномальной дисперсией.
Д. необходимо учитывать при конструировании по-
полимерных деталей оптич. приборов; при изготовлении
фотопроводящих полимерных слоев; фото чувствитель-
чувствительных слоев из взвешенных в слое пластика частиц суль-
сульфида или селенида кадмия; при передаче световых изо-
изображений по оптич. волноводам. Измерение Д.— метод
исследования биополимеров.
Отражение света (О.). Если свет падает из среды
а и отражается от оптически менее плотной среды
Ь, то щ/па<^1. Определенному значению предельного
угла О. соответствует полное внутреннее
отражение (П. в. о.) — когда практически весь
световой поток возвращается в среду а.
501
ОПТИЧЕСКИЕ СВОЙСТВА
502
Однослойные полимерные пленки не могут быть не-
непосредственно использованы в качестве хороших отра-
отражателей (необходима металлизация задней поверхности
прозрачного покрытия). Однако многослойные поли-
полимерные пленки, толщина слоев к-рых сравнима с дли-
длиной световой волны, имеют очень высокую отражатель-
отражательную способность, сравнимую с этой характеристикой
для металлизированных полимерных пленок. При
этом многослойные пленки взаимодействуют со светом
как дифракционная решетка.
На матовой поверхности полимера свет диффузно от-
отражается от нерегулярно расположенных шероховато-
шероховатостей. При регулярном нанесении штрихов на металл
с последующим снятием с него реплики — затвердев-
затвердевшей (после нанесения расплава) полимерной пленки —
образуются периодич. дефекты поверхности, способные
давать дифракционные спектры О. При определенных
соотношениях ширины штриха и расстояния между
штрихами можно получить полимерные реплики, при-
пригодные для спектральных приборов.
Селективное О. применяется для дискретного откло-
отклонения светового луча в системах оптич. обработки ин-
информации, в растровой оптике. Зонные пластинки для
дифракционной оптики изготовляются из прозрачных
полимеров.
П. в. о. в полимерах используется при конструирова-
конструировании отражателей на автомашинах, дорожных заказах.
В оптич. волокнах световой поток после многократных
отражений выходит через торец волокна практически
без потери энергии. Полистирольные волокна перемен-
переменного сечения идут на изготовление фокусирующих (фо-
коны) и увеличивающих апертуру (афоконы) гибких
волоконных кабелей.
Поглощение света (П. с.) и цветность. П. с.— поте-
потеря энергии потоком света, проходящим через среду,
вследствие превращения этой энергии в различные фор-
формы внутренней энергии или в энергию вторичного из-
излучения. Направление потока вторичного излучения
и его спектральный состав (совокупность частот) отли-
отличаются от падающего излучения.
Интенсивности падающего света / (частота v0) и про-
прошедшего слой z — /2 связаны соотношением Ламбер-
Ламберта — Бугера
1г = Ie~kz A6)
Для р-ров полимеров во многих случаях справедлив
закон Бера, согласно к-рому поглощение kv света
р-ром пропорционально концентрации с полимера:
К = КчС A7)
Коэфф. пропорциональности i?v связан со строением
индивидуальной молекулы и с межмолекулярным взаи-
взаимодействием.
Метод изучения мутности — исследование зависи-
зависимости интегрального П. с. от частоты света v — позво-
позволяет в конц. р-рах и дисперсных системах определять
размеры и полидисперсность частиц.
Квантово-механич. теории рассматривают строение
электронных оболочек атомов и молекул в нормальных
и возбужденных состояниях для объяснения П. с.
и цвета. Основными представлениями являются поня-
понятие о гибридизации электронных орбит с образованием
атомом углерода тс-связей (сопряженных двойных свя-
связей) и а-связей, различающихся по пространственной
конфигурации. Органич. полимеры, не содержащие со-
сопряженных связей, прозрачны и неокрашены. В поли-
полимерах с сопряженными связями тс-электроны имеют
высокую подвижность в пределах цепи сопряжения
и сравнительно небольшую энергию возбуждения.
Это объясняет цвет полимеров с сопряженными связя-
связями (черный или коричневый). Часть энергии световой
волны, проходящей через такие полимеры, теряется,
что вызывает сильное П. с. Цвет м. б. обусловлен так-
также входящими в состав макромолекулы хромофорными
группами, содержащими тг-электроны и неподеленные
электронные пары гетероатомов. Стабильность цвета
зависит от химич. стабильности хромофорных групп,
условий переработки и светостойкости полимера.
Спектры — см. Спектроскопия, Колебательная спект-
спектроскопия.
Дихроизм — см. Дихроизм.
Люминесценция (Л.) — явление, при к-ром вещество,
возбужденное световым или др. электромагнитным из-
излучением, само испускает свет, причем время жизни
вещества в возбужденном состоянии составляет не
менее 10~10 сек.
В зависимости от длительности испускания света
(после прекращения действия возбуждающего излуче-
излучения) Л. делят на флуоресценцию (время
испускания < 10~3 сек) и фосфоресценцию
(>10~3 сек). При понижении темн-ры флуоресцирую-
флуоресцирующее вещество может стать фосфоресцирующим.
В зависимости от вида возбуждающего излучения Л.
подразделяют на фотолюминесценцию (возбуждение
видимым светом), катодолюминесценцию (катодными
лучами), рентгенолюминесценцию (рентгеновскими лу-
лучами), радиолюминесценцию (радиоволнами), радио-
термолюминесценцию (смотри Термолюминесценция),
электролюминесценцию (электрич. полем). Иногда све-
свечение появляется в результате химич. реакции (х е -
ми люминесценция). Наибольшее распростра-
распространение при исследовании полимеров получили хемилю-
минесценция, радиотермолюминесценция и катодолю-
минесценция. Последний метод позволяет наблюдать
свечение твердых полимеров непосредственно в скани-
сканирующем электронном микроскопе и определять по ха-
характеру свечения и распределению люминесцирующих
частиц надмолекулярную структуру полимеров. Разре-
Разрешающая способность такого метода —1000 А.
В соответствующих условиях Л. проявляют многие
полимеры. Л. не удается наблюдать лишь в тех случаях,
когда возбужденное состояние молекул способно к бы-
быстрому внутреннему перераспределению энергии воз-
возбуждения по собственным нормальным колебаниям.
Среди полимеров таким свойством обладают, по-види-
по-видимому, все карбоцепные и нек-рые гетероцепные поли-
полимеры, не содержащие в макромолекуле полиароматич.
или линейных сопряженных структур.
Исследование Л. может дать ценную информацию при
изучении свойств макромолекул, структуры надмоле-
надмолекулярных образований и релаксационных процессов
в полимерах.
Специфич. О. с. люминесцирующих веществ описы-
описываются набором различных характеристик: спектры
поглощения и люминесценции, поляризация высвечи-
высвечивания, поляризационные спектры, квантовый выход
Л., время жизни вещества в возбужденном состоянии,
затухание свечения, термич. высвечивание.
Поляризованная Л. — наиболее распро-
распространенный люминесцентный метод исследования поли-
полимеров. Он основан на частичной поляризации света,
излучаемого люминесцирующей системой. Для наблю-
наблюдения Л. в удобной для регистрации области спектра
в полимерную систему часто вводят люминесцирующие
метки (напр., производные антрацена), химически свя-
связанные или не связанные с макромолекулами. Изуче-
Изучение спектров люминесцентных меток, деполяризации
Л. и поведения ориентированного полимера с меткой
дает возможность определять конформационные харак-
характеристики и релаксационные свойства макромолекул
в р-рах и блоке.
Рассеяние света (Р. с.) — распространение света
в нек-рой среде по направлениям, отличным от направ-
направлений отраженной и преломленной волн. Различают
два вида Р. с— классическое (рэлеевское) и комбина-
комбинационное. В отличие от люминесценции, классич. Р. с.
503
ОРГАНИЧЕСКОЕ СТЕКЛО
504
происходит без изменения длины волны. В однородной
изотропной среде Р. с. не происходит.
Для полимеров характерно существование угловой
зависимости интенсивности Р. с. (асимметрия) от изоли-
изолированной молекулы во всем интервале X (от видимого
до рентгеновского диапазона). Интенсивность Р. с.—
суммарная величина рассеяния от отдельных оптич.
неоднородностей. Совокупность изотропных осцилля-
осцилляторов (даже при падении естественного света) дает пло-
плоско поляризованный рассеянный свет. Деполяризация
Р. с. под углом 90е к направлению исходного пучка
(другая особенность макромолекулы) — проявление
анизотропии тензора ее поляризуемости.
В оптически однородных средах малые по сравнению
с л оптич. неоднородности — термич. флуктуации плот-
плотности — источники местных изменений показателя пре-
преломления и причина молекулярного (рэлеевского)
Р. с. В области фазовых переходов оптич. неоднород-
неоднородности (долгоживущие гетерофазные флуктуации) —
причина критич. опалесценции (Р. с. высокой интен-
интенсивности). Интенсивность Р. с. особенно велика в кол-
коллоидных системах, когда размеры частиц сравнимы с X.
Р. с. полимерами под большими углами
(>30с) — метод определения размеров макромолекулы,
мол. массы, конформации, разветвленности, полидис-
полидисперсности, взаимодействия с растворителем (см. Свето-
Светорассеяние).
Малоугловое Р. с. полимерами появляется
при преимущественном Р. с. в области малых углов
(до 30е). Для него характерно образование диффузных
пятен (оптич. неоднородности не имеют четких границ)
или специфич. дифракционных картин: четырехлепест-
ковых симметричных (для сферолитов), колец и рефлек-
рефлексов (для жидких кристаллов блоксополимеров). Мало-
Малоугловое Р. с.— метод исследования кинетики образова-
образования, размеров (и формы) зародышей и надмолекулярных
образований при кристаллизации, их деформации и
разрушения при ориентации волокон и пленок; оно
используется также при изучении образования долго-
живущих ассоциатов и молекулярных комплексов
в р-рах, реальных сеток в гелях, при исследовании
фазовой сегрегации (микросинерезиса); при определе-
определении размеров монокристаллов полимеров в их взвесях,
их полидисперсности и агрегации.
Комбинационное Р. с. (К. р. с). Рассеяние,
обусловленное флуктуациями поляризуемости молекул,
проявляется как эффект модуляции рассеянного света
собственными колебаниями молекул.
На практике исследуемую среду облучают монохро-
матич. светом и на фоне диффузного рассеяния регист-
регистрируют дискретные максимумы, частоты к-рых отли-
отличаются от частоты падающего света на величины собст-
собственных частот нормальных колебаний молекул. Интен-
Интенсивное развитие К. р. с. полимеров обусловлено введе-
введением в практику исследований лазерных источников
света. Этот метод дает информацию о структуре рассеи-
рассеивающих молекул и о межмолекулярном взаимодействии.
Двойное лучепреломление — см. Двойное лучепре-
лучепреломление.
Эффект Керра — вынужденное постоянным и одно-
однородным электрич. полем двойное лучепреломление све-
света (направленного поперек поля). Действие электрич.
поля на молекулу обусловлено тем, что она обладает
постоянным дипольным моментом и способна к статич.
поляризации. Взаимодействие поля с дипольным мо-
моментом молекулы аналогично действию силы, к-рая
приводит к растяжению полимерной цепи и обуслов-
обусловливает разность средних поляризуемостей в направле-
направлениях, параллельном и перпендикулярном этой силе.
Для гибкоцепных полимеров (число мономерных зве-
звеньев в сегменте к-рых невелико) действие внешнего
поля приводит к эффекту, мало отличающемуся от на-
наблюдаемого в р-рах соответствующих низкомолекуляр-
низкомолекулярных соединений. Количественный анализ эффекта Кер-
Керра основан на усреднении величины Да по всем ори-
ентациям и внутренним конформациям рассматривае-
рассматриваемой цепи в поле.
В жесткоцепных макромолекулах ориентации по-
полярных групп коррелированы на большом участке
(сегмент велик), векторная сумма локальных группо-
групповых моментов значительна, т. е. молекула может иметь
большой дипольный момент и проявлять анизотропию
под действием электрич. поля.
Изучение эффекта Керра — метод определения вра-
вращательной подвижности, жесткости и упорядоченности
макромолекул.
Фотоупругость (Ф.). В отличие от низкомолекуляр-
низкомолекулярных соединений, аморфные полимеры проявляют Ф.—
вынужденное механич. полем двойное лучепреломление.
Ф. зависит от напряжения, молекулярной ориентации
и анизотропии статистич. сегмента полимера, от сте-
степени сшивки. На определении Ф. основан метод иссле-
исследования пространственных сеток в гетерогенных поли-
полимерных материалах, а также эластооптич. метод моде-
моделирования (изучение распределения напряжений на де-
деталях из прозрачных пластмасс, имитирующих крупные
детали и строительные конструкции), применяемой в тех-
технике и строительстве. См. Фотоупругость.
Оптическая активность — см. Оптически активные
полимеры.
Магнитооптические явления. Эффект Котто-
на — Мутона — ориентационное двойное лучепре-
лучепреломление в магнитном поле, аналогичное эффекту Кер-
Керра. Оно объясняется анизотропией тензора оптич. по-
поляризуемости и диамагнитной восприимчивости. Это
явление использовано для исследования полимеризации
стирола. По Эффекту Коттона — Мутона или дихроиз-
дихроизму в магнитном поле можно определять коэфф. враща-
вращательного трения макромолекулы.
Лит.: ВолькенштейнМ. В., Молекулярная оптика,
М. — Л.,1951; Цветков В. Н., ЭскинВ.Е., Френ-
Френкель С. Я., Структура макромолекул в растворах, М.— Л.,
1964; Волькенштейн М. В., Конфигурационная ста-
статистика полимерных цепей, М.— Л., 1959; Флор и П., Ста-
Статистическая механика цепных молекул, пер. с англ., М., 1971;
Encyclopedia of polymer science and technology, v. 9, N. Y.—
[a. o.], 1968; Новейшие методы исследования полимеров, под
ред. Б. Ки, пер. с англ., М., 1966.
ОРГАНИЧЕСКОЕ СТЕКЛО (organic glass, glasklarer
Kunststoff, verre organique) — технич. название про-
прозрачных твердых материалов на основе органич. поли-
полимеров. К этой группе относят полиакрилаты, полисти-
полистирол, полимеры аллиловых соединений, поликарбонаты,
сополимеры винилхлорида, сополимеры нек-рых эфиров
целлюлозы и др. В пром-сти под «органическим стеклом»
чаще всего понимают листовой материал, получаемый
полимеризацией в массе метилметакрилата. Производст-
Производство этого материала покрывает основные потребности в
О.с; выпуск остальных видов О. с. относительно невелик.
Получение. Полиметилметакрилатное О. с. получают
радикальной полимеризацией метилметакрилата в мас-
массе в присутствии перекиси бензоила, перекиси лаури-
лаурила, динитрила азоизомасляной к-ты и др. В зависи-
зависимости от назначения О. с. в состав полимеризационной
смеси могут входить пластификаторы, красители, за-
мутнители, стабилизаторы, а также др. акриловые мо-
мономеры. Наиболее распространенные пластификаторы—
эфиры фталевой к-ты. Для окрашивания О. с. приме-
применяют жирорастворимые и дисперсные красители, рас-
растворимые в мономере и совместимые с полимером. Воз-
Возможно применение нерастворимых в мономере пигмен-
пигментов. Замутнителями в производстве светорассеиваю-
щего О. с. служат полистирол и пигменты. Эфиры сали-
салициловой к-ты, производные бензотриазола, диоксибен-
зофенона и т. п. являются светофильтрующими вещест-
веществами, при использовании к-рых получают О. с, погло-
поглощающее ультрафиолетовое излучение Сополимеризация
505
ОРГАНИЧЕСКОЕ СТЕКЛО
506
метилметакрилата с др. акриловыми мономерами или
стиролом, а также введение термостабилизирующих
добавок позволяют получить О. с. с термостойкостью
до 200 °С.
Полимеризация метилметакрилата сопровождается
значительной усадкой реакционной массы (до 23%),
что могло бы привести к получению листов с дефектами.
Поэтому процесс обычно проводят в два этапа. Вначале
получают полимер невысокой мол. массы (т. наз. фор-
полимер), представляющий собой сиропообразную жид-
жидкость с вязкостью 50—200 мн-сек/м2, или спз. Затем
форполимер заливают в форму для получения листа;
дальнейшая полимеризация форполимера сопровож-
сопровождается значительно меньшей усадкой. Аналогичный
эффект достигается, если полимеризации подвергают
р-р полиметилметакрилата в мономере (т. наз. сироп-
раствор). Применение форполимера или сироп-раствора
предотвращает также утечку реакционной массы из
недостаточно уплотненных форм. Полимеризацию
мономера в один этап осуществляют только в тех слу-
случаях, когда необходимо получить полиметилметакри-
латное О. с. очень высокой оптич. прозрачности.
Все необходимые ингредиенты О. с. вводят в форпо-
форполимер или сироп-раствор. Полученную смесь тщательно
перемешивают, вакуумируют для удаления пузырьков
газа и фильтруют. Полимеризацию проводят в формах,
собранных из двух листов полированного силикатного
стекла, стали или алюминия, скрепленных зажимами,
с проложенными между ними эластичными прокладка-
прокладками. Толщина эластичных прокладок определяет буду-
будущую толщу листа О. с. Полированное силикатное стек-
стекло применяют, если необходимо получить О. с. с по-
поверхностью хорошего качества. В качестве материала
для эластичных прокладок используют различные ре-
резины, пластики и пр. Форму либо оклеивают по краям
плотной бумагой, либо ио периметру формы укладыва-
укладывают резиновые или поливинилхлоридные трубки, изолиро-
изолированные от мономера покрытием или слоем бумаги,
исключающим непосредственное соприкосновение мо-
мономера с трубкой. Устройство формы обеспечивает
возможность усадки в одном направлении—по толщине
формы. Форму, находящуюся в наклонном положении,
заполняют через воронку точно отмеренной порцией
полимеризационной смеси, герметически закрывают
или заклеивают и помещают в камеры с циркулирую-
циркулирующим теплым воздухом или в ванны с теплой водой
(в нек-рых случаях темп-pa воздуха или воды может
быть 18—20 °С).
Полимеризацию проводят в изотермич. условиях.
Нарушение изотермич. режима может привести к пере-
перегреву формы, вскипанию мономера и т. наз. «разгора-
нию», т. е. образованию пузырчатой массы. Поэтому
чем толще лист, тем медленнее и при более низкой
темп-ре проводят полимеризацию.
Если отвод тепла осуществлять неравномерно, то
глубина полимеризации в различных частях формы бу-
будет различной. В результате в листе возникнут напря-
напряжения, приводящие после нагревания к его дефор-
деформированию и, следовательно, ухудшению оптических
свойств. Темп-pa процесса зависит от типа форполиме-
форполимера, концентрации инициатора и толщины формы.
Обычно полимеризацию в формах проводят медленно,
часто в течение 24—48 ч, а в толстых слоях (более
50 мм) — неделями при 20—50 °С до конверсии моно-
мономера свыше 90%. Процесс завершают при темп-рах,
близких к темп-ре размягчения полиметилметакрилата,
т. к. при низких темп-pax диффузия непрореагировав-
шего мономера затруднена и поэтому даже за большой
период времени невозможно полное превращение мо-
мономера.
По окончании полимеризации формы охлаждают до
50 °С и отделяют силикатные стекла от органического.
Ориентацию осуществляют с помощью машин и прес-
прессов различной конструкции, равномерно растягивая
(обычно на 50—70%) или сжимая заготовки, разогретые
до темп-ры, на 10—12 °С превышающей темп-ру раз-
размягчения. Ориентированные листы охлаждают под на-
нагрузкой.
В отдельных случаях листы О. с. получают методом
фотополимеризации с использованием сенсибилиза-
сенсибилизаторов, напр, бензоина. Заполненные формы облучают
УФ-светом до образования геля, после чего осущест-
осуществляют процесс по обычной схеме.
Для производства стержней из полиметилметакрилата
ного О. с. полимеризационную смесь заливают в гори-
горизонтальные вращающиеся алюминиевые трубы, к-рые
затем в вертикальном положении помещают в водяную
ванну. Режимы полимеризации те же, что и при полу-
получении листового О. с. Изделия сложной конфигура-
конфигурации получают литьем под давлением или экструзией
гранулированного полиметилметакрилата, синтезиро-
синтезированного полимеризацией в массе или суспензии в
присутствии регуляторов мол. массы.
При получении литьевого О. с. из сополимера ме-
метилметакрилата с акрилонитрилом сополимеризацию
осуществляют в массе по такой же технологии, как и
в производстве полиметилметакрилатного стекла. Поли-
Полимеризацией в формах в присутствии перекисного ката-
катализатора получают изделия на основе диэтиленгли-
коль-бш:-(аллилкарбоната); образующийся полимер
практически не формуется и получить из него изделия
сложной конфигурации др. методом не удается. Листы
из полистирола, поликарбоната, сополимеров винил-
хлорида и эфиров целлюлозы получают экструзией,
а изделия сложной конфигурации — литьем под дав-
давлением гранулированных или порошкообразных поли-
полимеров, полученных обычными методами.
Свойства. О. с. обладает сравнительно невысокой
плотностью и малой хрупкостью, что является сущест-
существенным преимуществом перед силикатным стеклом. Од-
Однако темп-pa размягчения О. с. значительно ниже,
чем у силикатного стекла. Свойства нек-рых О. с. пред-
представлены в табл. 1 и 2.
Таблица 1. Свойства отечественных марок органического
стекла из неориентированного полиметилметакрилатаа
Показатели
Плотность, г/см3 ....
Прочность, Мп/м2
(кгс/см2)
при растяжении . . .
при статич. изгибе
Ударная вязкость,
кдж/м2, или тс-см/см2
Теплостойкость, °С
по методу ИФП . . .
по Вика
по Мартенсу
Относительное удлине-
удлинение , % • . .
Стрела прогиба, мм . .
Твердость по Бринеллю,
Мн/м2 (кгс/мм2) ....
Уд. теплоемкость,
кдж/(кг • К) [кал/(г • °С)]
Темп-рныйкоэфф. линей-
линейного расширения а • 106,
СОЛ6
1,206
71 G10)
99(990)
13,0
90-95
105
3,6
6,1
215
B1,5)
1,256
[0,413]
71
СТ-1В
1,189
78G80)
118
A180)
13,8
110-120
130
85
4,0
10,8
237
B3,7)
1,729
[0,413]
77
2-55г
1 ,200
92,5
(925)
121
A210)
14,8
133 — 138
144
120
2,8
8,1
299
B9,9)
1 ,691
[0,404]
69
Т-2-55Д
1 ,204
100
A000)
120
A200)
15,0
133 — 138
—
3,3
6,3
274
B7,4)
1,450
[0,375]
75,5
аФизико-механич-. показатели, характеризующиешластичность
ориентированного полиметилметакрилатного О. с. со степенью
вытяжки 50%, превосходят таковые для неориентированного О. с.
в 3 —5 раз. ^ Полиметилметакрилат, пластифицированный ди-
бутилфталатом. в Полиметилметакрилат с добавкой фенилсали-
цилата для поглощения УФ-излучения. г Сополимер на основе
метилметакрилата. д Термостабилизированный сополимер на
основе метилметакрилата.
507
ОРГАНИЧЕСКОЕ СТЕКЛО
508
Таблица 2. Свойства сополимеров винилхлорида
с метилметакрилатом (I) и поцидиэтиленгликоль-бмс-
ал лил карбонат а (II)
Показатель
Плотность, г [см3
Прочность при растяжении, Мн/м2
(кгс/см2) . .
Теплостойкость по Мартенсу, °С . . .
Температурный коэфф. линейного рас-
расширения а-106, СС~*
I
1,40
50 E00)
40
80
II
1,32
35-42
C50-420)
87
О свойствах О. с. на основе сополимеров метилмета-
крилата с акрилонитрилом см. Акрилонитрила
сополимеры, на основе полистирола — Стирола поли-
полимеры, на основе поликарбоната — Поликарбонаты, на
основе эфиров целлюлозы — Этролы.
Полиметилметакрилатное О. с. удовлетворительно
переносит пребывание на воздухе в условиях 97%-ной
влажности в течение 12 месяцев и старение в атмо-
атмосферных условиях от 5 до 10 лет и более. Сополимер
метилметакрилата с акрилонитрилом, поликарбонат,
эфиры целлюлозы и сополимеры винилхлорида с
эфирами метакриловой к-ты также обладают достаточ-
достаточной атмосферостойкостью. Полистирол менее атмосфе-
ростоек; при длительном воздействии .солнечного света
он желтеет и становится хрупким.
Среди оптич. свойств О. с. наиболее важны пока-
показатель преломления, оптич. прозрачность (светопроз-
рачность), оптич. искажения и фотоупругость.
Ниже приведены показатели преломления нек-рых
О. с, измеренные относительно воздуха в свете жел-
желтого дуплета Na [Х=589—589, 6 им E890—5896 А)]:
Полиметилметакрилат
непластифицированный 1,4895
пластифицированный 1,4920
Полиакрилат повышенной термостойкости . . . . 1,5466
Полиэтилметакрилат 1,4866
Сополимер метилметакрилата с акрилонитрилом 1,5 096
Поликарбонат 1,5896
Полидиэтиленгликоль-бис-(аллилкарбонат) . . . 1,4996
Ацетобутират целлюлозы 1,4754
Полистирол 1,5924
Феноло-формальдегидная отвержденная смола . . 1,7004
От одной поверхности листового полиметилметакри-
латного стекла отражается 3,5—4% падающего светово-
светового потока, а от двух — 8%. Т. обр., оптич. прозрач-
прозрачность полиметилметакрилатного О. с. не может пре-
превышать 92% при условии, что рассеяние и поглоще-
поглощение света равны нулю.
Оптич. прозрачность полистирола, поликарбоната и
полидиэтиленгликоль-бис-(аллилкарбоната) составляет
до 90%, сополимера метилметакрилата с акрилонитри-
акрилонитрилом— 85%, сополимера винилхлорида с метилмет-
метилметакрилатом (винипроза)—75%. Эфироцеллюлозные О. с.
пропускают свет различной длины волны неодинако-
неодинаково. Так, оптич. прозрачность для излучений с длиной
волны 300, 400, 500 и 600 нм составляет 12—20, 23—55,
70—80 и 85—90% (толщина образца 2,5 мм).
По оптич. прозрачности О. с. делят на прозрачные
в блоке и прозрачные только в пленках (тонких листах).
К первой группе относятся полимеры и сополимеры
метилметакрилата, полистирол, поликарбонат и др.
полимеры, обладающие незначительным поглощением
света; ко второй — О. с. на основе эфиров целлюлозы,
винипроз, литые эпоксидные и феноло-формальдегид-
ные стекла.
Рассеяние света с поверхности изделий из О. с. мож-
можно свести практически к минимуму при условии, что
качество обработки поверхности аналогично качеству
обработки полированного силикатного стекла.
Ненаполненные О. с. прозрачны для рентгеновско-
рентгеновского и у-излучения, а в тонких листах — для а- и Р-из-
л учения.
Под действием механич. нагрузок в первоначально
изотропном О. с. возникает оптич. анизотропия и свя-
связанное с ней двойное лучепреломление (т. наз. явление
фотоупругости). В результате на поверхности напря-
напряженного О. с. возникают радужные эллиптич. картины,
к-рые мешают наблюдению через стекло. Наибольшей
фотоупругостью обладают эпоксидные, феноло-формаль-
дегидные и термостойкие полиакрилатные стекла; наи-
наименьшей — полиметилметакрилатные, полистироль-
ные и поликарбонатные.
Оптич. искажения предметов, наблюдаемых сквозь
О. с, связаны гл. обр. с невозможностью изготовить
изделия из этих стекол с истинно плоскопараллельны-
плоскопараллельными поверхностями. В результате этого любое изделие
из О. с. является в той или иной мере призмой, обла-
обладающей абсолютным (угловым) оптич. искажением. При
падении пучка параллельных лучей света на изделие
из О. с. с одинаковой или мало меняющейся по всей
поверхности величиной абсолютного оптич. искажения
прошедший свет останется параллельным. В этом слу-
случае единственным результатом оптич. искажения будет
одностороннее смещение наблюдаемого предмета.
Приближение или удаление рассматриваемого пред-
предмета, его раздвоение или исчезновение, искривление
линий, а также нарушение пропорций и формы пред-
предмета возможны только в том случае, когда изделие из
О. с. обладает различной величиной абсолютного оп-
оптич. искажения в разных частях поверхности. На гра-
границе соединения двух листов О. с. с различной величи-
величиной абсолютного оптич. искажения возникает т. наз.
искривление, или излом, линии горизонта.
Переработка. Листовое О. с. перерабатывают ваку-
умформованием, пневмоформованием и штампованием.
Заготовку перед формованием нагревают до теми-ры,
к-рую выбирают в интервале между темп-рами размяг-
размягчения и деструкции полимера. В случае полиметилмет-
полиметилметакрилатного О. с. продолжительность разогрева заго-
заготовки t (в мин) м. б. ориентировочно определена по
ур-нию t = 10 + 3d, где d — толщина листа в мм.
Оптимальная темп-pa формования зависит от метода
формования, вида формовочного инструмента, окружаю-
окружающей темп-ры и времени выдержки. При формовании
изделий из ориентированного О. с. перед разогревом
лист закрепляют в массивной раме, в к-рой затем осу-
осуществляют формование. Используют также метод холод-
холодного формования.
Листовое О. с. можно подвергать всем видам механи-
механической обработки специально разработанным для этой
цели инструментом. Напр., сверление отверстий в ли-
листовом полиметилметакрилате следует осуществлять
сверлом с углом при вершине 120° при низких скоро-
скоростях резания и подачах инструмепта, охлаждая его
5%-ным водным р-ром эмульсола. При отсутствии спе-
специального инструмента механич. обработку О. с. осу-
осуществляют с применением инструментов и режимов,
обычно используемых при обработке легких сплавов
и дерева.
О. с. склеивают клеями, предназначенными для
склеивания соответствующих полимеров, и сварива-
сваривают встык или внахлест. При выборе правильных ре-
режимов сварки прочность шва при отрыве достигает
90% когезионной прочности материала. См. Сварка,
Склеивание.
Для крепления листового О. с. в рамах остекляемых
проемов разработаны различные способы. При жестком
болтовом креплении О. с. прижимают к раме или
каркасу при помощи болтов, винтов или заклепок,
пропущенных через отверстия в стекле или специаль-
специальные вырезы (фестоны). Этот способ относительно сло-
сложен, т. к. требует высокой точности совпадения отвер-
отверстий или фестонов в О. с. с отверстиями в раме. Заво-
Заворачивание болтов можно осуществлять только специ-
специальными тарированными гаечными ключами. При не-
509
ОРГАНОВОЛОКНИТ
510
соблюдении этих условий в стекле возникают напряже-
напряжения, к-рые нельзя снять последующим отжигом смон-
смонтированной конструкции, т. к. жесткое крепление
стекла препятствует его сокращению при остывании.
Кроме того, О. с, жестко закрепленное болтами через
отверстия, не имеет возможности перемещаться при
эксплуатационных нагрузках и температурных пере-
перепадах.
При жестком безболтовом креплении О. с. прижим
листа 1 (рис. 1) к каркасу или раме 4 осуществляют
металлич. накладками 2, к-рые крепятся болтами 3,
Рис. 1. Конструктивные варианты жесткого безболтового
крепления листового органического стекла: 1 — лист орга-
нич. стекла; 2 — металлич. накладка; 3 — болт; 4 — кар-
каркас или рама; 5 — эластичная прокладка.
Этот вид крепления более совершенен, т. к. нет необ-
необходимости рассверливать отверстия в листе стекла,
а возникающие в нем напряжения распределены более
равномерно, чем в предыдущем случае.
Мягкое безболтовое крепление листового О. с. не
приводит к возникновению напряжений в стекле в мо-
момент крепления и не препятствует деформации листа
при эксплуатации. Мягкое крепление осуществляют
след. образом: лента из полиамидной или полиэфир-
полиэфирной ткани 2 (рис. 2,а) приклеивается к краю листа 1
при помощи иолиуретанового или феноло-резорцино-
Рис. 2. Мягкое безболтовое крепление листового органиче-
органического стекла (а — с шомполом; б — без шомпола): 1 — лист
органич. стекла; 2 — лента из синтетич. ткани; 3 — шом-
шомпол; 4 — каркас или рама; 5 — болт; б — накладка.
вого клея. Образуемая лентой петля вводится в канав-
канавку продольного профиля рамы 4 и закрепляется в ней
шомполом 3. По др. варианту (рис. 2,6) синтетич.
ткань 2 наклеивают по всему периметру листа и закреп-
закрепляют ее между накладкой 6 и рамой 4 при помощи бол-
болта 5. Место соединения ленты с рамой промазывается
тиоколовыми или кремнийорганич. герметиками.
Применение. Применение полиметилметакрилатного
О. с. чрезвычайно разнообразно: в авиа-, автомобиле-
и судостроении — как конструкционный материал;
в пром-сти и гражданском строительстве — для остек-
остекления куполов, окон, веранд и декоративной отделки
интерьеров зданий; в сельском хозяйстве — для остек-
остекления парников, теплиц и т. п.; в светотехнич.
пром-сти — для изготовления защитных колпаков све-
светильников; в медицинской пром-сти — для изготовле-
изготовления деталей приборов и инструментов, а также проте-
протезов; в химич. машиностроении и пищевой пром-сти —
для изготовления труб; в оптике — в производстве
линз и призм; в искусстве — для создания гравюр,
скульптур и т. д.
О. с, поглощающее УФ-излучение, применяют в му-
музеях для защиты экспонатов от разрушающего действия
коротковолнового излучения.
Сополимер метилметакрилата с акрилонитрилом ис-
используют для изготовления корпусов приборов, безо-
безопасных смотровых куполов, деталей остекления само-
самолетов, вагонов, автобусов и пр., предназначенных для
работы под повышенными нагрузками.
Основные области применения прозрачного полисти-
полистирола — изготовление мелких деталей для электро-
и радиоприборов, линз для карманных фонарей, све-
светильников, увеличительных стекол и предметов до-
домашнего обихода.
Из полидиэтиленгликоль-бис-(аллилкарбоната) изго-
изготавливают линзы и стекла для очков. Поликарбонат
применяют прежде всего там, где требуется высокая
ударопрочность и теплостойкость, в частности для
изготовления смотровых стекол, сигнальных светиль-
светильников, защитных экранов, деталей и корпусов прибо-
приборов и др.
Винипроз служит преимущественно для производст-
производства листов, прутков, труб. Его используют также для
изготовления шкал, чертежных приборов, логариф-
мич. линеек, клише и матриц для типографских работ,
для защиты фотосхем, светокопировальных работ,
в картографии и для др. целей. Матированный про-
продукт применяют для снятия копий с планов и вычерчи-
вычерчивания на нем копий несмываемой тушью.
О. с. на основе эфиров целлюлозы применяют для
изготовления защитных очков, светозащитных окон-
оконных стекол и штор, а также для покрытия рекламных
щитов. Прозрачные формованные детали используют
в производстве магнитофонов, радиоприемников и те-
телевизоров.
Прозрачные модели из отвержденной эпоксидной или
феноло-формальдегидной смолы, обладающие высокой
фото упругостью, применяют для определения напря-
напряженного состояния деталей машин и строительных
конструкций.
Лит.: НогпМ. В., Acrylic resins, N. Y.— L., 1960;
Перов Б. В., Гудимов М. М., Ориентированное орга-
органическое стекло, М., 1961; Марек О., Томка М., Акри-
Акриловые полимеры, пер. с чеш., М.— Л.,» 1966; Справочник по
пластмассам, под ред. М. И. Гарбара [и др.], т. 1 — 2, М., 1967—
1969; D e b s k i W., Polimetakrylan metylu, Warsz., 1969;
Конструкционные свойства пластмасс, под ред. Э. Бэра, пер.
с англ., М., 1967; Конструкционные материалы, т. 2, М., 1964.
М. М. Гудимов, М. И. Фролова.
ОРГАНОВОЛОКНИТ (organovoloknit) — пластик,
в к-ром наполнителем служат материалы из синтети-
синтетических волокон (полиамидного, полиэфирного, поли-
акрилонитрильного, поливинилспиртового, полиимид-
ного и др.). К О. не относятся слоистые материалы
на основе бумаги из синтетич. волокон (о таких мате-
материалах см. Органогетинакс).
Термореактивный органоволокнит. Наполнителем
для О. этого типа служат ткани различного плетения,
нетканые материалы, маты, войлоки, жгуты, нити
и др. В качестве связующего чаще всего используют
полиэфирные и эпоксидные смолы, а для получения О*
511
ОРГАНОВОЛОКНИТ
512
с повышенной термостойкостью — феноло-формальде-
гидные смолы и полиимиды.
Модули упругости и температурные коэфф. линей-
линейного и объемного расширения наполнителя и связующе-
связующего в О. близки. Поэтому в таких материалах остаточ-
остаточные напряжения, возникающие при изготовлении изде-
изделий, в 4—6 раз ниже, чем, напр., в стеклопластиках на
основе того же связующего. Пористость отвержденных
О. (отношение объема газовых микровключений к объе-
объему материала) не превышает 1—3%. Кроме того, О.
обладает высокой ударной вязкостью и хорошей устой-
устойчивостью к распространению трещин. Указанные осо-
особенности обеспечивают высокую стабильность механич.
свойств О. при резкой смене темп-р, а также при дейст-
действии циклич. и ударных нагрузок.
По абсолютным величинам прочностных характери-
характеристик О. занимает промежуточное положение между
стекловолокнитом и волокнитом. Однако его плот-
плотность в среднем на 30% меньше плотности стекловолок-
нита, так что О. по уд. прочностным характеристикам
близок к стекловолокниту, а в нек-рых случаях пре-
превосходит его (табл. 1).
питывают в открытых ваннах с последующей сушкой
и намоткой на оправку. В отдельных случаях про-
пропитанный и высушенный наполнитель разрушается под
действием связующего, поэтому его нельзя долго хра-
хранить в виде полуфабриката.
Термореактивные О. находят применение в электро-
и радиопромышленности в качестве изоляционного и
конструкционного материала, в авиа- и автострое-
автостроении для изготовления перегородок, крыш, внутрен-
внутренних обшивок, сбрасываемых топливных баков, пуле-
защитной брони и пр. Высокая химич. стойкость и
монолитность О. позволяет применять его в производ-
производстве труб, емкостей для хранения реактивов и др.,
а также в качестве коррозионностойкого покрытия
корпусов судов, защиты фюзеляжа и крыльев самоле-
самолетов от эрозии песком и образования вмятин на обшив-
обшивке при взлете и посадке. О. используют в качестве
теплозащитных покрытий ракет и космич. кораблей.
Термопластичный органоволокнит. Наполнителем
для О. этого типа обычно служат ткани, маты, войлоки
(степень наполнения 50—70% по объему) или мелко-
мелкорубленое волокно (степень наполнения 10—25% но
Таблица 1. Свойства отвержденных органоволокнитов на основе феноло-формальдегидной смолы
Показатели
Волокнит с
наполнителем
из хлопковой
ткани
Органоволокнит с различными наполнителями
Ткани из син-
тетич. волокон
низкой проч-
прочности*
Ткани из син-
тетич.волокон
средней проч-
прочности**
Ткани из син-
тетич. волокон
высокой проч-
прочности***
Маты на осно-
основе синтетич.
волокон
Однонаправ-
ленно выло-
выложенное син-
синтетич. волокно
Плотность, г /см3
Теплостойкость по Мартенсу, °С .
Прочность, Мн/м2 (кгс/см2)
при растяжении
при сжатии
при изгибе****
Модуль упругости при растяже-
растяжении, Гн/м2 [кгс/см2}
Относительное удлинение, % . . .
Ударная вязкость, кдж/м2, или
кгс • см/см2
Водопоглощение за 24 -ч, % . . . .
1,3-1,4
125
85-100
(850-1000)
150A500)
160 A600)
9,5-10
[(95 —100)-103]
1.0
35-40
1,5
1,15-1,3
110-115
100-190
A000-1900)
75 G50)
100-180
A000-1800)
2,5-8
[B5-80). Ю3]
10-20
500-600
0,15 — 0,2
1,2-1,3
120
190-300
A900-3000)
НО A100)
160-250
A600-2500)
11-15
[(НО —150).1Оа
3-8
1 ,3 — 1,4
120
650-700
F500—7000)
180-200
A800-2000)
400-450
D000—4500)
35
[C5 0-103)]
2-5
1,3
120-140
70-75
G00-750)
140-150
A400 — 1500)
110—130
A100-1300)
25-35
0,7-1,2
1,4-1,5
120
1300 — 1700
A3000-17000)
250 — 300
B500—3000)
6000-9000
F0000-90000)
80
[800000]
2
* Полиамидное, полиэфирное и полиакрилонитрильное волокна, применяемые в производстве товаров широкого потребления.
**Поливинилспиртовое волокно. ***Волокно на основе ароматич. полиамидов. ****Максимальное напряжение.
О. имеет сравнительно низкую прочность при сжатии
и высокую ползучесть. Большинство О. обеспечивает
длительную работу конструкций при нагружении,
величина к-рого составляет не более 20—40% от раз-
разрушающего.
Большинство О. может длительно эксплуатироваться
при 100—150 °С, а материалы на основе полиимидных
и полиоксадиазольных волокон (см. Термостойкие
волокна) — при 200—300 °С (после прогрева в течение
100—200 ч при 300 °С прочностные характеристики
этих О. снижаются только на 50%). О. обладает высо-
высокой устойчивостью в агрессивных средах и во влаж-
влажном тропич. климате. Уд. объемное и поверхностное
электрич. сопротивление О. на 2—4 порядка, а элект-
рич. прочность на порядок выше, чем у волокнита на
основе хлопкового волокна и того же связующего.
Технологич. схема производства О. такая же, как
для стекловолокнита; она включает стадии подготовки
наполнителя (удаление замасливателя и термофиксация
волокна), пропитки его связующим в пропиточных ма-
машинах, открытых ваннах, смесителях и, наконец, суш-
сушки. Из пропитанного наполнителя прессуют листы и
изделия. Фигурные изделия из О. изготавливают про-
пропиткой сухого наполнителя связующим под давлением.
При изготовлении труб моноволокно или ленту про-
объему). В качестве связующего обычно используют
полиэтилен, полипропилен, поливинилхлорид, поли-
метилметакрилат, фторопласты.
Плотность О. этого типа на 40—60% ниже плотности
стеклопластика на основе того же связующего, в то
время как прочностные характеристики этих материа-
материалов (табл. 2) близки. Термопласты в результате напол-
наполнения их рубленым волокном значительно снижают
ползучесть. Напр., ползучесть под нагрузкой 8 Мн/м2
(80 кгс/см2) полиэтилена низкой плотности, наполнен-
наполненного на 20% по объему полиэфирным волокном, в 100
раз ниже, чем ненаполненного.
Химстойкость О. зависит в основном от типа связую-
связующего. Так, О. на основе полиэтилена и полипропилена
стоек к действию воды, минеральных масел, большин-
большинства органич. растворителей, р-ров к-т; менее стоек
в р-рах щелочей. Теплофизич. свойства О. также опре-
определяются в основном свойствами связующего.
Листовые О. с тканевым наполнителем, войлоком
или матами изготовляют прессованием пакета [давле-
[давление до 2 Мн/м2 B0 кгс/см2)], состоящего из пленок тер-
термопласта, переложенных наполнителем. Детали не-
несложной конфигурации готовят из описанных выше
листов пневмо- или вакуумформованием. При произ-
производстве изделий сложной конфигурации заготовку из
513
ОРГАНОГЕТИНАКС
Таблица 2. Свойства термопластичных органоволокнитов
514
Показатели
Полиамид 11-68 +
ткань из волокон-
ароматич.
полиамида
Полиоле-
фин + ткань
из поливинил-
спиртового
волокна
Полиамид П-68+
рубленое волок-
волокно ароматич.
полиамида
Фторопласт -f-
ткань из по-
лиэтиленте-
рефталатного
или полиимид-
ного волокна
Полиэтилен +
рубленое гю-
ливинилспир-
товое волокно
Полиэти-
лен+руб-
леное
стеклово-
стекловолокно
Плотность, г /см3
Прочность, Мн/м2 (кгс/см2)
при растяжении
при изгибе
Модуль упругости при растяже-
растяжении, Мн/м2 (кгс/см2)
Ударная вязкость, кдж/м2, или
кгс' см/см2
1,1-1,2
450-550
D500-5500)
450 D500)
36 000 C60 000)
120
1,10
135-150
A350-1500)
110 — 120
A100-1200)
6800 F8000)
95
1,1
130-150
A300-1500)
140 A400)
И 000 (НО 000)
26
1,76
90 (900)
60 F00)
3200 C2 000)
0,98
78 G80)
60F00)
3200C2 000)
40
1,3
61 F10)
77G70)
4000
D0 000)
17,4
пропитанного наполнителя собирают в матрице пресс-
формы, где и прессуют под давлением 0,25—2,0 Мн/м2
B,5—20 кгс/см2). Изделия из О., наполненного рубле-
рубленым волокном, получают экструзией, литьем под дав-
давлением и прессованием.
Термопластичные О. применяют в машиностроении
для изготовления бункеров, баков, крышек, труб,
фланцев, внутренних конструкций изделий и агрега-
агрегатов, работающих в контакте с агрессивными средами.
Изделия из О. на основе фторопластов работоспособны
до 1000 ч при темп-pax от —60 до 250 °С на воздухе или
до 500 ч при 200—250 °С в керосине или его парах.
Лит.: ГендриксГ., в кн.: Армированные полимер-
полимерные материалы, сб. переводов и обзоров из иностранной перио-
периодической литературы, М., 1968, с. 181 — 211; Пластики кон-
конструкционного назначения, под ред. Е. Б. Тростянской, М.,
1974. В. В. Павлов, Г. П. Машинская, Л. А. Тетерев.
ОРГАНОГЕТИНАКС (organogetinaks) — слоистый
пластик на основе бумаги из синтетических волокон.
Наполнителем для О. чаще всего служит бумага на
основе волокон из ароматич. полиамидов и поливинило-
поливинилового спирта, связующим — феноло-формальдегидные
и эпоксидные смолы, полиимиды, а иногда также поли-
полиэфирные смолы и кремнийорганич. полимеры.
Комплекс физико-механич. свойств О. (таблица) оп-
определяется близостью характеристик наполнителя и
связующего (напр., температурных коэфф. линейного
Основные характеристики гетинаксов
Показатели
Плотность, г/см3
Прочность, Мн/м2 (кгс/см*)
при растяжении
при изгибе
Модуль упругости при изгибе, Гн/м2
(кгс/см*)
Ударная вязкость, кдж/м2, или
кгс - см/см2
Деформационная теплостойкость, °С
Темп-рный коэфф. линейного расши-
расширения, °С~1
Водопоглощение за 24 ч, %
Уд. объемное электрич. сопротив-
сопротивление, ом-см
Электрич. прочность, Мв/м, или
кв/мм
Диэлектрич. проницаемость при
1 Мгц
Тангенс угла диэлектрич. потерь
при 1 Мгц о .,
Органогетинакс
бумага из воло-
волокон ароматич.
полиамида+по-
лиимидное свя-
связующее
бумага из по-
ливинилсгшр-
тового волок-
на-гфеноло-
формальдегид-
ное связую-
связующее
B
1,3
155 A550)
170 A700)
5,5 E5000)
18,8
280
1—8 , 6) • 10~5
0,3-1,1
К)*»—1017
25-30
2,8
0,015
и объемного расширения, модуля упругости). В ре-
результате этого при нагревании и под действием различ-
различных нагрузок в О. не возникают растягивающие уси-
усилия, к-рые приводили бы к его растрескиванию. Оста-
Остаточные напряжения в органогетинаксе также доста-
достаточно низки.
Основные преимущества О. перед гетинаксом обна-
обнаруживаются при работе в условиях повышенных темп-р
(при комнатной темп-ре механич. характеристики этих
материалов близки) и воздействии агрессивных сред.
Напр., при нагревании до 120 °С в течение 30 мин гети-
накс с феноло-формальдегидным связующим сохраняет
только 50% исходной прочности при изгибе, в то вре-
время как для О. на основе бумаги из ароматич. полиами-
полиамидов такая же потеря прочности за указанное время про-
происходит при прогреве до 500 °С. Усадка О. в этих усло-
условиях не превышает 1—2%. Под действием прямого пла-
пламени О. разрушается, но не горит. Он устойчив к дли-
длительному A0 лет) термостарению при 220 °С.
О. обладают высокой водостойкостью. Размеры из-
изделий из О. на основе бумаги из ароматич. полиамидов
при работе на воздухе с 95%-ной влажностью увеличи-
увеличиваются менее чем на 2%.
Диэлектрич. характеристики О. мало меняются в ус-
условиях повышенной влажности и повышенных темп-р
до 250 °С. Напр., электрич. прочность О. из бумаги на
основе ароматич. полиамида и поли-
имидного связующего практически не
меняется при нагревании до 200 °С
и уменьшается на 5%
1,2-1,25
70 G00)
135 A350)
16
, 1 -Ю
2,0
Гетинакс (бу-
ло-альдегид-
ное связую-
связующее)
на 5% в результате
мага целлю- прогрева при 250 °С. Диэлектрич. про-
лозная+фено- ницаемость О. практически не зави-
зависит от частоты и мало изменяется с
ростом темп-ры до 200 °С, а тангенс уг-
угла диэлектрич. потерь в диапазоне
температур от 20 до 220 °С и частот
от 100 до 105 гц на 2—3 порядка ниже,
чем у гетинакса.
Технологич. схема изготовления ли-
листового О. включает пропитку бумаги
на пропиточных машинах или в ван-
ваннах, сборку пакета из пропитанной
бумаги и его прессование. При этом
обычно соотношение (по массе) напол-
наполнитель : связующее составляет 1:1.
При использовании бумаги с анизо-
анизотропией свойств в продольном и попе-
поперечном направлениях (прочностные
характеристики бумаги могут изме-
изменяться в 1,5—2 раза в зависимости от
направления) пакет собирают таким
образом, чтобы обеспечить получение
О. с изотропными свойствами в плос-
плоскости листа.
1,25-1,4
70-80
G00-800)
80 — 100
(800-1000)
13
150
2-10
107—10"
16
6,0
1,05 — 0,1
515
ОРИЕНТИРОВАННОЕ СОСТОЯНИЕ
516
Температурный режим прессования определяется
природой связующего и обычно не отличается от ре-
режима прессования др. пластиков на основе этого же
связующего. Время выдержки под давлением опреде-
определяется из расчета 5 —10 мин на 1 мм толщины листа.
Особенность прессования О. заключается в примене-
применении большого числа ноднрессовок для обеспечения
лучших условий удаления летучих (наполнитель в
О. характеризуется низкой газопроницаемостью).
О. применяют для изготовления электро- и радио-
радиоаппаратуры, химич. аппаратуры (труб, змеевиков
и др.), а также различных прессованных и штампован-
штампованных изделий в машиностроении.
Лит.: ЛиГ., С т о ф ф и Д., Н е в и л л К., Новые
линейные полимеры, пер. с англ., М., 1972; Фрейзер А. Г.,
Высокотермостонкие полимеры, пер. с англ., М., 1971; Пластики
конструкционного назначения, под ред. Е. Б. Тростянской,
М., 1974. Г. П. Машинская.
ОРИЕНТИРОВАННОЕ СОСТОЯНИЕ поли м е-
р о в (oriented state, orientierter Zustand, etat oriente).
Содержание:
Введение ....515
Получение ориентированных полимеров . . . с .516
Ориентационная вытяжка . .516
«Направленная» полимеризация ...... 522
Строение ориентированных полимеров ..„.,. 523
Свойства ориентированных полимеров 52 5
Механические свойства ..........525
Термические свойства 527
Введение
Ориентированное состояние — сиецпфич. состояние
тел из линейных полимеров, характеризуемое тем, что
составляющие эти тела макромолекулы имеют преиму-
преимущественное расположение своих осей (как правило —
на отдельных участках молекулы) вдоль нек-рых на-
направлений — осей ориентации — во всем объеме тела.
(Микрообъемы любого твердого полимерного тела всег-
всегда находятся в О. с.— см. ниже).
Простейшим и наиболее часто встречающимся на
практике видом ориентации полимеров является од-
одноосная ориентация, к-рой в основном и посвящена
настоящая статья.
Имеются и др. виды О. с. Так, в кристаллических
полимерных пленках может образоваться т. наз. пло-
плоскостная текстура, когда совпадают направления двух
различных осей всех кристаллитов, напр, оси макро-
макромолекулы направлены но одному направлению в плос-
плоскости пленки и, кроме того, нормаль к какой-либо
кристаллографич. плоскости у всех кристаллитов рас-
расположена перпендикулярно к плоскости пленки. Сле-
Следует отметить, однако, что в промышленных образ-
образцах пленок возникает та или иная дисперсия ориен-
ориентации около направлений, соответствующих идеально
правильной плоскостной текстуре. Чаще всего оси ма-
макромолекул распределены более или менее равномерно
по всем направлениям в плоскости пленки, тогда
как нормаль к определенной кристаллографич. плос-
плоскости у всех кристаллитов направлена перпендику-
перпендикулярно пленке.
Помимо «искусственно» ориентированных полимеров,
существуют и биологические ориентированные поли-
полимерные объекты. Они распространены и в растительном
мире (напр., хлопок, лен, волокна в стеблях), и в жи-
животном мире (паутина, шелковые нити, волосы, сухожи-
сухожилия, мышечная ткань и др.). Можно считать, что почти
всюду, где природе требовались гибкие и прочные «де-
«детали», она формировала ткани из одноосноориентиро-
ванных полимеров. Их строение сейчас интенсивно ис-
исследуется, причем в нек-рых случаях отмечаются об-
общие черты с искусственно ориентированными полиме-
полимерами (периодич. гетерогенность надмолекулярного
строения, фибриллизация, сочетание кристаллических
и аморфных областей и др.— см. ниже). Принципы
«строительства» и роста ориентированных биологич.
тканей пока еще изучены недостаточно. Возможно,
что в дальнейшем такое изучение даст полезную био-
бионическую информацию, к-рую можно будет использо-
использовать для разработки новых способов получения ориен-
ориентированных синтетич. полимеров.
Получение ориентированных полимеров
Существуют два основных способа получения одно-
осноориентнрованных полимерных тел: а) перевод
неориентированного полимерного тела в ориентирован-
ориентированное состояние под воздействием внешнего растягиваю-
растягивающего усилия (ориентационная вытяжка, формование);
б) «направленная» полимеризация, т. е. осуществление
синтеза полимера в условиях, когда сразу получается
полимерное тело с ориентированной структурой.
Ориентационная вытяжка — наиболее распростра-
распространенный в настоящее время способ получения одноосно-
ориентированных полимеров. Заключается он в тем,
что зажатое с двух концов полимерное тело растяги-
растягивается, причем степень растяжения может варьировать
от нескольких десятков до сотен и тысяч процентов.
Внутри растягиваемого полимерного тела, как прави-
правило, идут процессы перестроения, охватывающие как
отдельные цепные молекулы, так и надмолекулярные
структуры. Эти перестроения м. б. разного типа —
с той или иной степенью распада и трансформации
исходных структурных образований (см. ниже).
Межатомные силы сцепления в полимерном теле
можно разделить на 2 группы: прочные (химические)
связи между атомами внутри полимерных молекул
и слабые (вандерваальсовы или водородные) — между
атомами разных макромолекул или частями одной мак-
макромолекулы. Т. обр., цепные молекулы в полимерном
теле в определенной мере сохраняют свою индивидуаль-
индивидуальность. Под действием растягивающих усилий межмоле-
кулярные связи нарушаются, и молекулы изменяют
свою конформацию — распрямляются. Этот процесс
и составляет молекулярную основу ориентационноп
вытяжки. Подобное ориентирование макромолекул
происходит только при условии их достаточной гиб-
гибкости, т. е. тогда, когда полимер находится в высоко-
высокоэластическом состоянии (о нек-рых исключениях см.
ниже), а последнее реализуется при достаточно высо-
высоких темп-pax, соответствующих скорости растягиваю-
растягивающего полимер воздействия (см. Релаксационные явле-
явления). При этом тепловое движение играет в процессе
ориентирования многостороннюю роль.
Прежде чем начать рассмотрение процессов ориен-
ориентирования твердых полимеров, следует кратко остано-
остановиться на ориентировании одиночных макромолекул.
Этот случай имеет
прежде всего большой
научный интерес, по-
поскольку на результа-
результатах таких «модельных»
экспериментов можно
основывать фундамен-
фундаментальные заключения об
ориентировании цеп-
цепных молекул. В р-ре
Рис. 1. Схема одиночной
линейной макромолекулы
в растворе («) и сеточная
модель аморфного полиме-
полимера (б) до и после растяже-
растяжения (/—/ — направление
растяжения).
полимера невысокой концентрации и достаточно «све-
«свежем» (т. е. таком, в к-ром процессы образования над-
надмолекулярных структур не зашли еще далеко) макро-
макромолекулы будут существовать как отдельные более
или менее свернутые клубки (рис. \а). При этом взаимо-
517
ОРИЕНТИРОВАННОЕ СОСТОЯНИЕ
518
действие разных участков молекулы между собой можно
практически не учитывать (р-р в состоянии в-точки).
Обеспечив достаточную вязкость такого р-ра (подбо-
(подбором темп-ры), его можно тем или иным способом растя-
растягивать, и тогда макромолекулы полимера будут ориен-
ориентироваться, т. е. молекулярные клубки начнут дефор-
деформироваться. Растворитель служит здесь средством пе-
передачи на отдельные полимерные молекулы растяги-
растягивающего усилия. Для сохранения ориентированного
состояния полимерных молекул в данном случае тре-
требуется либо поддерживать растягивающее усилие (под-
(поддерживая «течение» р-ра), либо охладить растянутый
р-р и «заморозить» деформированные полимерные клуб-
клубки. Отметим, что в подобных случаях высокая степень
ориентации не достигается, т. к. цепные молекулы
«растягиваются» сразу во многих точках, что не позво-
позволяет длинной молекуле распрямиться по всей длине.
В твердом полимерном теле макромолекулы хотя и
сохраняют в определенной степени свою «механиче-
«механическую» индивидуальность, на каждом участке практи-
практически вплотную сближены с др. макромолекулами или
же с др. участками этой же макромолекулы. Т. обр.,
в этом случае изменяются (по сравнению с ориентацией
одиночной макромолекулы) и условия действия внеш-
внешней растягивающей силы на молекулы, и возможности
ориентационной подвижности участков молекулы.
Влияние на ориентацию плотной упаковки макромо-
макромолекул, присущей твердым полимерам, следует рассмат-
рассматривать применительно к двум уровням — молекуляр-
молекулярному и надмолекулярному — строения полимеров (по
масштабу).
На молекулярном уровне это влияние учитывается
сеточной моделью строения полимеров. Цепные моле-
молекулы в твердом полимере, соприкасаясь, образуют кон-
контакты — узлы за счет межмолекулярных сил сцепле-
сцепления. В точках же «перехлеста» молекулярных цепей
образуются узлы с прочностью, приближающейся
к прочности хим. связей. В результате можно предста-
представить себе объем полимера в виде своеобразной трех-
трехмерной сетки с узлами разной степени устойчивости
(рис. 16). Подобное описание являлось доминирующим
ранее, когда прямое изучение строения полимеров еще
не приобрело значительного развития. Следует под-
подчеркнуть, что сеточная модель содержит в своей основе
реалистич. положения о взаимодействии макромолекул,
что и позволяет с успехом применять ее в довольно ши-
широкой области деформирования полимеров. В соответ-
соответствии с сеточной моделью строения полимеров ориен-
тационная вытяжка заключается в том, что передавае-
передаваемое через узлы сетки внешнее усилие распрямляет
и поворачивает в направлении оси действия силы уча-
участки молекул между узлами (см. рис. 16). Этот процесс
может идти как при «фиксированных» узлах, так и при
значительном изменении их концентрации и вида, что
определяется условиями ориентирования (темп-рой,
скоростью растягивания, напряжением) или свойствами
полимера.
Сеточная модель удовлетворительно описывает ориен-
ориентирование при не очень высоких степенях растяжения
аморфных пластиков (напр., полиметилметакрилата,
атактич. полистирола), эластомеров и значительно
хуже — ориентационную вытяжку кристаллизующихся
полимеров. Слабой стороной этой модели раньше было
отсутствие прямых данных об узлах сетки и о длине
и состоянии отрезков молекул между узлами. Теперь,
в связи с развитием многих прямых физич. методов
изучения полимеров (инфракрасная спектроскопия,
ядерный магнитный резонанс, ультразвук и др.), есть
возможность восполнить эти пробелы.
Однако есть и другая причина ограниченной при-
применимости сеточной модели при описании ориентиро-
ориентирования полимеров, а именно существование в твердых
полимерах надмолекулярных структур. Наличие таких
структур означает существование ориентированных
микрообъемов уже и в неориентированных полимерных
телах. Действительно, во всех случаях элементами
надмолекулярной структуры являются агрегаты поли-
полимерных молекул, характеризуемые тол или иной сте-
степенью внутренней упорядоченности во взаимном рас-
Рис. 2. Схемы молекул в составе надмолекулярных образо-
образований неориентированных полимеров: а — аморфный поли-
полимер, б, в — кристаллич. полимеры соответственно ламел-
лярного и сферолитного строения.
положении молекул. А наиболее естественной и рас-
распространенной для линейных «одномерных» молекул
формой упорядоченности является параллельное при-
прилегание их друг к другу на участках молекул (протя-
(протяженностью в десятки и сотни ангстрем).
Важным достижением физики полимеров в послед-
последние десятилетия является установление наличия над-
надмолекулярной структуры практически у всех твердых
полимеров: и аморфных, и кристаллических. У аморф-
аморфных полимеров — это образования с зачатками упоря-
упорядоченности, чаще всего — с одномерным (и то далеко
не совершенным) порядком (рис. 2а); у кристаллич.
полимеров — это области с достаточно совершенным
трехмерным порядком — кристаллиты, к-рые вдобавок
упорядоченно объединяются в более крупные морфо-
логич. образования: ламеллы и сферолиты (рис. 26,в).
Существуют и различные переходные случаи между
аморфными и кристаллич. полимерами.
Наличие надмолекулярной структуры весьма силь-
сильно влияет на протекание орпентационных процессов.
Прежде всего, это влияние проявляется в том, что
процессы ориентирования могут идти двумя путями:
1) поворотом уже готовых ориентированных участков
и «выстраиванием» их вдоль оси ориентирования;
здесь, т. обр., на первый план выступает «надмоле-
«надмолекулярное» ориентирование, поведение отдельных моле-
молекул в этом процессе менее существенно; 2) распадом
исходных элементов надмолекулярной структуры,
ориентированием полимерных молекул «поодиночке»
и затем уже формированием надмолекулярных образо-
образований, присущих ориентированному полимерному телу.
Кроме того, существование надмолекулярной струк-
структуры меняет представление о сеточном строении поли-
полимерного тела. Действительно, раньше при описании
строения полимеров как клубка «перепутанных»,
хаотически располагающихся ценных молекул на-
наиболее распространенными и важными считали уз-
узлы, образующиеся в результате «перехлеста» мо-
молекул (и такие узлы в системе перепутанных молекул
действительно были самыми естественными). В усло-
условиях параллельной укладки макромолекул внутри
элементов надмолекулярной структуры существова-
существование перехлестов молекул значительно менее вероятно.
Зато узлы-контакты за счет сил сцепления между моле-
молекулами оказываются и более распространенными, и бо-
более прочными, т. к. при параллельной укладке молекул
вероятность «прилегания» друг к другу отдельных мо-
молекул, естественно, возрастает.
Все отмеченные обстоятельства и требуют иного, чем
для случая простой сеточной модели, описания ориен-
ориентирования полимеров.
Наиболее детально изучены в структурном отноше-
отношении ориентационные процессы в кристаллизующихся
519
ОРИЕНТИРОВАННОЕ СОСТОЯНИЕ
520
полимерах. Это обусловлено большой информативно-
информативностью прямых структурных методов: электронной мик-
микроскопии и электронной дифракции в больших и малых
углах, рентгеновской дифракции в больших и малых
углах, светорассеяния и др. применительно к полиме-
полимерам, содержащим области высокой упорядоченности —
кристаллиты. С помощью этих методов можно просле-
проследить изменения в строении полимеров при их вытяжке,
начиная от исходного, неориентированного состояния
и кончая моментом завершения ориентационного про-
процесса.
Для кристаллизующихся полимеров ориентационная
вытяжка проводится, как правило, в интервале темп-р
между темп-рой стеклования аморфных областей и
темп-рой плавления кристаллов полимера. Ниже
темп-ры стеклования неориентированные кристаллич.
полимеры чаще всего оказываются хрупкими и при
нагружении разрушаются, не переходя в ориентиро-
ориентированное состояние. Выше точки плавления кристаллов
неориентированный полимер переходит в «полужидкое»
состояние и легко растягивается. Но при этом степень
ориентации молекул оказывается весьма низкой, т. к.
они проскальзывают друг по другу без эффективного
распрямления. Разумеется, все отмеченные процессы
следует понимать в релаксационном плане — с учетом
соотношения скоростей внешнего воздействия и тепло-
тепловой подвижности макромолекул.
В указанном интервале темп-р ориентирование кри-
кристаллич. полимеров в основных чертах протекает след.
образом.
На начальных стадиях растяжения (относительные
удлинения от нескольких процентов до нескольких де-
десятков процентов) под действием растягивающей силы
происходят повороты и перемещения кристаллитов,
к-рые остаются практически неизменными. Полимер
растягивается, и кристаллиты перемещаются вслед-
0,5 мм
2 мнм
-f ,
Рис. 3. Структурные перестроения при ориентировании
кристаллических полимеров (ось ориентации вертикальна;
электронная микроскопия; реплики с поверхности застывших
расплавов): а — ориентируемый ламеллярный полиэтилен;
видны остатки ламелл и участки новой фибриллярной
структуры; б — ориентируемый сферолитиый поликапро-
амид; между сферолитами образуются области с новой
фибриллярной структурой >
ствие высокоэластич. деформирования межкристаллит-
ных аморфных прослоек, состоящих из участков мак-
макромолекул, переходящих из одного кристаллита в дру-
другой. В этих условиях достигается определенная (иног-
(иногда довольно значительная) степень ориентации полимер-
полимерного тела. Если на этой стадии снять с полимера рас-
растягивающую нагрузку, то он быстро восстановит ис-
исходные размеры при той же темп-ре. Дальнейшее рас-
растяжение полимера вызывает распад исходных кристал-
кристаллов и надкристаллитной структуры, одновременно фор-
формируется новая, ориентированная структура (рис. 3).
Соотношение между реологич. составляющей ориен-
тационной вытяжки и стадией перестроения, глубина
распада исходных кристаллов и степень «обновления»
кристаллитной структуры в большей мере зависят
и от условий вытяжки (темн-ры, скорости растяжения
и др.), н от свойств полимера (вида межмолекулярных
связей, жесткости и длины молекул и др.). Так, один
и тот же полимер может при более высоких темп-рах
ориентироваться с незначительным распадом исходных
кристаллитов, а при сравнительно низких темп-рах —
почти с полным их перестроением (полиэтилен, поли-
винилиденфторид).
С особенностями и глубиной подобных структурных
переходов при ориентировании связано то, насколько
резко образуется «шейка» — локальное сужение растя-
растягиваемого полимера, в к-ром значение растяжения и
степень ориентации намного выше, чем в остальной ча-
части растягиваемого полимера. После образования шей-
шейки дальнейшее ориентирование образца идет путем
распространения шейки на всю длину полимера.
По завершении структурных перестроений растяну-
растянутый ориентированный образец кристаллич. полимера
уже не возвращается к исходному состоянию при той
же темп-ре: новая кристаллитная структура как бы
зафиксировала ориентированное состояние полимера.
Нагревание такого полимера вызовет его усадку, к-рая
м. б. как почти полной, так и незначительной (тогда
полимер расплавится в вытянутой но сравнению с исход-
исходной форме). Степень усадки, т. е. степень обратимости
ориентационной вытяжки, также зависит от свойств
полимера и условий вытяжки.
В аморфных полимерах надмолекулярные образова-
образования выражены меньше и изучены значительно слабее,
чем в кристаллических. Поэтому о прохождении здесь
ориентационного процесса столь же детально говорить
пока нельзя. Но, очевидно, все стадии, к-рые наблю-
наблюдаются при ориентировании кристаллич. полимеров
присущи и аморфным полимерам, только в значительно
менее явной форме. Аморфные полимеры также могут
растягиваться как с хорошо выраженной шейкой, так
и практически без нее (равномерно). Из-за «рыхлости»
надмолекулярной структуры у аморфных полимеров
элементы этой структуры не могут удержать полимер
в растянутом состоянии при снятии нагрузки (при
теми-ре вытяжки), и поэтому образец будет сокращать-
сокращаться. Чтобы сохранить аморфный полимер ориентиро-
ориентированным, надо его охладить — ослабить дезориенти-
дезориентирующую роль теплового движения. Отметим, что если
растягивать аморфный полимер при темп-ре много
выше темп-ры стеклования, то он будет «течь» — удли-
удлиняться без эффективного распрямления своих молекул.
Т. обр., как для кристаллических, так и для аморф-
аморфных полимеров должны выполняться следующие об-
общие положения для того, чтобы растяжение полиме-
полимера вызывало его ориентирование: в полимерном теле
должна обеспечиваться достаточная гибкость и под-
подвижность макромолекул; в то же время темн-ра вытяж-
вытяжки не должна быть слишком высокой, чтобы не распа-
распались контакты между молекулами (кристаллиты, узлы
и др.), к-рые и «ответственны» за нагружение отрезков
макромолекул, их распрямление и ориентирование.
Как известно, деление полимеров на аморфные и
кристаллические в значительной мере условно. Струк-
Структурное состояние полимера определяется как условиями
его перехода к данному состоянию (быстрое или мед-
медленное охлаждение, внешнее давление и др.), так и сте-
степенью стереорегулярности цепной молекулы (атак-
тичность, изотактичность, синдиотактичность). Поэ-
Поэтому при ориентационной вытяжке могут наблюдаться
своеобразные аморфно-кристаллические переходы.
Так, при быстром растяжении кристаллич. нолиэти-
лентерефталата в условиях невысоких темп-р можно
получить ориентированный аморфный полимер (хоро-
521
ОРИЕНТИРОВАННОЕ СОСТОЯНИЕ
522
шее доказательство распада кристаллитов при ориен-
тационной вытяжке). С другой стороны, растягивая
аморфный каучук, получаем ориентированные кри-
кристаллиты (к-рые тут же плавятся при разгрузке и со-
сокращении образца).
Все это показывает, что ориентирование полимеров
является процессом, носящим черты фазовых переходов.
С известной приближенностью ориентирование при
вытяжке можно назвать «направленной перекристал-
перекристаллизацией» полимера.
Для анализа и характеристики ориентационной вы-
вытяжки полимеров часто пользуются диаграммами рас-
растяжения. Типичный вид такой диаграммы представ-
представлен на рис. 4. Переход в области «о» отвечает началу
интенсивных структур-
структурных перестроений (в ча-
частности, образованию
шейки). Область «б» со-
соответствует завершению
построения ориентиро-
ориентированной структуры, к-рая
в данных условиях менее
податлива дальнейшему
Рис. 4. Типичный вид диаг-
диаграммы растяжения (зависи-
(зависимости напряжения о от рас-
растяжения е) кристаллич. по-
полимера, ориентируемого при
различных темп-рах(Т1<Т2<
<Т3<Т4).
перестроению и значительному растяжению (напряжение
на образце, а также на отдельных элементах его струк-
структуры быстро нарастает и он рвется). Из рис. 4 видно,
что с повышением темп-ры напряжение структурных
перестроений падает. Это является следствием того
важного обстоятельства, что распад исходной струк-
структуры полимера есть процесс не чисто механический, а
кинетический, в к-ром решающую роль играет тепло-
тепловое движение (см. Долговечность). Распад исходной
структуры (напр., кристаллитов) происходит вследст-
вследствие тепловых флуктуации, разрывающих напряженные
внешней силой межмолекулярные связи. Более высо-
высокая темп-pa вытяжки обусловливает более частые
флуктуации большой энергии, что и приводит к быст-
быстрому распаду кристаллитов при меньшем напряжении.
Т. обр., роль теплового движения в процессе ори-
ориентационной вытяжки полимеров очень многообразна.
Прежде всего, полимер должен быть нагрет — тепло-
тепловое движение молекул должно быть настолько интен-
интенсивным, чтобы обеспечивалась достаточная подвиж-
подвижность молекул. Уже здесь роль теплового движения
в ориентационной вытяжке двоякая. С одной стороны,
оно обеспечивает флуктуационные перескоки неболь-
небольших участков молекул таким образом, что при наличии
внешней силы участки молекул в аморфных областях
кристаллич. полимера или между узлами в аморфных
полимерах постепенно распрямляются и вытягиваются
приблизительно вдоль направления действия силы.
С другой стороны, это же тепловое движение «стремит-
«стремится» дезориентировать молекулы, вернуть их к более
скрученному состоянию. Поэтому уже на начальной
стадии ориентирования требуется подбор определен-
определенных оптимальных условий растяжения для успешного
проведения ориентационного процесса.
Далее, как уже отмечалось, вследствие тепловых
флуктуации разрушается напряженная исходная
структура. Но тут же, также вследствие теплового
движения, идет противоположный кинетич. процесс —
построение новой, ориентированной структуры.
Последующее растяжение полимера ведет к более
глубокому распаду исходной структуры и дальнейшей
ориентации. В то же время нарастает напряжение на
уже сформированных элементах ориентированной
структуры, к-рые больше «не могут» перестраиваться.
Повышению локальных перенапряжений способствует
то обстоятельство, что с ростом вытяжки постепенно
развивается надмолекулярная нерегулярность — дис-
дисперсность размеров «больших периодов» в фибриллах
ориентированного полимера. И тогда начинается кине-
кинетич. разрушение ориентированной структуры вследст-
вследствие термофлуктуационных разрывов химич. связей,
напряженных ориентирующей силой, т. е. за счет раз-
разрывов цепных молекул. В результате образец разру-
разрушается.
Итак, для эффективной ориентационной вытяжки
необходим учет всех названных тепловых факторов,
что обеспечивает оптимальный выбор теплового режима
процесса, скорости нагружения и растяжения исход-
исходного структурного состояния полимера.
В заключение раздела отметим интересный случай
обратимого растяжения полимера при очень низких
теми-pax (ниже —100 °С). В этих условиях аморфный
полимер (напр., атактич. полипропилен) очень хрупок
и при нагружении разрушается практически без за-
заметной деформации. Однако, напр., изотактическнй
хорошо закристаллизованный полипропилен, будучи
охлажденным столь же глубоко, способен растяги-
растягиваться на многие десятки процентов, причем это растя-
растяжение оказывается обратимым: при снятии нагрузки
и нагревании полимер полностью восстанавливает ис-
исходные размеры. Очевидно, хорошо выраженная кри-
кристаллич. структура позволяет проходить специфическим
скольжениям и сдвигам, когда одна часть кристаллита
«съезжает» по другой. Но в отличие от низкомолекуляр-
низкомолекулярных кристаллов здесь остается связь между частями
кристаллита (цепные молекулы «переходят» из одной
части в другую), что и обусловливает восстановление
формы кристаллита при нагревании.
При формовании полимера, когда происходит течение
струи его р-ра или расплава, также достигается та или
иная степень ориентации отвердевшего материала (см.,
напр., Литье под давлением термопластов). Этот спо-
способ более эффективен для жесткоцепных полимеров,
интерес к к-рым все более возрастает ввиду их высокой
термостойкости и прочности.
«Направленная» полимеризация. Этот способ получе-
получения ориентированных полимеров начал разрабатывать-
разрабатываться сравнительно недавно. Из известных методов про-
проведения «направленной» полимеризации можно отме-
отметить следующие: 1) полимеризация в твердой фазе, ког-
когда мономер существует в виде монокристалла; 2) по-
полимеризация жидкого полярного мономера в постоян-
постоянном электрич. поле; 3) полимеризация из газовой фазы,
когда мономер в виде газа окружает уже ориентирован-
ориентированный полимер (подложку) и полимеризуется на его по-
поверхности.
Полимеризация монокристаллов мономера позволяет
в принципе получить максимально ориентированное
полимерное тело, в к-ром цепные молекулы с совершен-
совершенно прямыми осяхми образуют монокристалл полимера.
Инициирование процесса может осуществляться раз-
разными способами: в результате облучения, катализа,
под действием ударных волн и др. Реакция развивается
вдоль определенных кристаллографич. направлений
в мономерном монокристалле. Образующееся полимер-
полимерное тело имеет игольчатую форму.
Степень ориентации полимера, образующегося при
жидкофазной полимеризации полярного мономера в по-
постоянном электрич. поле, не очень высока, т. к. тепло-
тепловое движение оказывает дезориентирующее влияние,
и при напряженности реальных полей порядка
100 кв/см достичь высокой направленности молекул
нельзя. Повышение же напряженности поля затрудни-
затруднительно из-за пробоев. Тем не менее, этот способ разра-
523
ОРИЕНТИРОВАННОЕ СОСТОЯНИЕ
524
батывается, т. к. даже частичная исходная ориентация
полимерного тела может представить большой интерес.
Полимеризация мономера из газовой фазы на ориен-
ориентированной полимерной подложке также создает инте-
интересные возможности. В этом случае на подложке по-
постепенно нарастает ориентированная полимерная «ру-
«рубашка». Полимеризация может идти как на подложке
из того же, так и из другого полимера. Степень ориен-
ориентации «наращиваемого» полимера здесь примерно такая
же, как и у полимера подложки. Скорость полимери-
полимеризации и мол. массу образующегося полимера можно
регулировать, изменяя условия процесса.
Способы получения ориентированных полимеров пу-
путем «направленной» полимеризации интересны тем, что
при их использовании удается избежать сложного этапа
«расплетания» ценных молекул, уже упакованных
в структуре неориентированного полимера.
Строение ориентированных полимеров
О строении ориентированных полимеров на мо-
молекулярном уровне (т. е. о конформации
и ориентации макромолекул) дают сведения следующие
прямые методы: поляризационная ИК-спектроскопия,
двойное лучепреломление, ЯМР, рентгеновская и
электронная дифракция в больших углах. Наиболее
информативным является первый метод, к-рый по
дихроизму различных полос поглощения в ИК-спектре
позволяет определять степень ориентации участков
макромолекул отдельно в аморфных и кристаллич.
областях полимера, ориентацию различных боковых
групп, распределение и ориентацию участков макромо-
макромолекул с различными последовательностями звеньев
(цис-, гош- и транс-формы). Подобной детальной инфор-
информации с помощью др. методов получить не удается, од-
однако применение каждого из них в каких-то случаях
оказывается весьма полезным. Напр., метод двойного
лучепреломления отличается простотой и доступно-
доступностью, метод рентгеновской дифракции очень чувствите-
чувствителен к ориентации кристаллитов.
Применение всех указанных методов позволяет на-
находить количественные характеристики степени ориен-
ориентации. Наиболее распространенной характеристикой
является величина cos26, где О — угол между осью
данного участка молекулы и осью ориентации образ-
образца. Используются также след. величины: средний угол
разориентации в у, (угол полуспада кривой распреде-
распределения молекул по ориентации) и фактор ориентации
F = ^со^н--1 # Пределы изменения этих величин (от
неориентированного до идеально ориентированного по-
полимера): ~з<. cos2B <1; оо >вз^ >0; O<F<1.
Отметим, что значения характеристик ориентации,
полученные разными методами для одного и того же
образца, оказываются различными ввиду различной
чувствительности методов к «спектру» структурной
упорядоченности в полимере.
В аморфных ориентированных полимерах ориентация
молекул всегда остается неполной (cos2H редко дости-
достигает 0,5), т. е. сохраняется достаточно широкое распре-
распределение сегментов молекул по направлениям вокруг
оси ориентации. Это связано в первую очередь со сте-
рич. затруднениями для молекулярных переупаковок
(теми же стерич. затруднениями, к-рые не дают воз-
возможности данным полимерам кристаллизоваться), а так-
также с дезориентирующим влияниехМ теплового движения.
В кристаллизующихся ориентированных полимерах
имеются две степени молекулярной ориентации: для
кристаллитов и для аморфных участков. В кристалли-
кристаллитах степень ориентации м. б. очень высокой (cos26
может достигать 0,95 и даже почти 1; 0з< составляет
2—3°), тогда как для аморфных областей значение
cos2B, как правило, составляет 0,6—0,7 (лишь изредка
-0,8).
Информацию о надмолекулярном строе-
н и и ориентированных полимеров дают методы элект-
электронной микроскопии и дифракции (в больших и малых
углах), рентгеновской дифракции (в больших и малых
углах), светорассеяния, онтич. микроскопии и др.
Электронная микроскопия показывает, что ориенти-
ориентированным полимерам присуще более или менее ярко
выраженное фибриллярное строение (рис. 5). Вдоль
0,5 мнм
0,5 ммм
Рис. 5. Фибриллярное строение ориентированных поли-
полимеров (ось ориентации вертикальная, электронная микро-
микроскопия): а — реплика с поверхности скола аморфного
полимера — полиметилметакрилата (вытяжка в 2,5 раза);
б — реплика с поверхности застывшего расплава кристал-
кристаллич. полимера — полиэтилена (вытяжка в 9 раз).
оси ориентации полимера идут примерно параллельно
друг другу удлиненные образования — фибриллы,
с поперечником в десятки — сотни ангстрем. Причины
такой фибриллизации до конца не выяснены. Имеется
пока лишь ряд гипотез: фибрилла в какой-то мере яв-
является растянувшимся или перестроившимся образо-
образованием, имевшимся в неориентированном теле; попе-
поперечные размеры фибрилл задаются размером области
коррелляции в расположении макромолекул; фибрил-
фибрилла — чисто статистич. агрегат макромолекул и др.
Этот вопрос относится к актуальным задачам физики
ориентированного состояния.
С помощью электронного микроскопа надежные дан-
данные о внутреннем надмолекулярном строении фибрилл
получены сравнительно недавно. Основную же инфор-
информацию об этом строении дают рентгенодифракционные
исследования в малых углах.
Для ориентированных аморфных полимеров в области
малых углов отсутствует дискретное рассеяние рентге-
рентгеновских лучей (малоугловые рентгеновские рефлексы)
(рис. 6а), что свидетельст-
свидетельствует о надмолекулярной
гомогенности фибрилл и
позволяет изобразить схе-
схему расположения макро-
макромолекул в них так, как
на рис. 1а.
Рис. 6. Дифракция рентгеновских лучей под малыми угла-
углами для ориентированных полимеров (ось ориентации вер-
вертикальна, первичный пучок направлен перпендикулярно
оси ориентации полимеров): а — аморфный полимер — по-
лиметилметакрилат (вытяжка в 2,5 раза, малоугловые
рефлексы отсутствуют); б — кристаллич. полимер — поли-
е-капроамид (вытяжка в 7 раз, видны четкие малоугловые
меридиональные рефлексы).
Малоугловая рентгенодифракционная картина для
ориентированного кристаллизующегося полимера в
большом количестве случаев содержит слоевые мери-
525
ОРИЕНТИРОВАННОЕ СОСТОЯНИЕ
526
К
дианальные рефлексы (см. рис. 66), что свидетельствует
о продольной гетерогенности фибрилл этих полимеров.
Здесь с периодом в десятки — сотни ангстрем череду-
чередуются области с разной плотностью: кристаллиты и
аморфные прослойки между ними (см. рис. 76). Кри-
Кристаллиты занимают большую часть периода — 60—
80%. Аморфные прослойки образованы участками
цепных молекул, пере-
переходящих из одного кри-
кристаллита в другой («про-
(«проходные» молекулы). Чис-
Число проходных молекул
составляет нек-рую до-
долю от числа молекул в
кристаллитах, т. к. часть
молекул «возвращается»
Рис. 7. Схемы внутреннего
строения фибрилл ориенти-
ориентированных полимеров: а —
аморфный полимер (надмо-
(надмолекулярная гетерогенность
отсутствует); б — кристал-
лич. полимер (продольная аморфно-кристаллич. гетероген-
гетерогенность); К — кристаллит; А — аморфная межкристаллитная
прослойка; d — «большой период» (/—/ — направление рас-
растяжения).
обратно в кристаллит, реализуя способность к склад-
складчатой конформации (см. Кристаллическое состояние).
Часть молекул, кроме того, «уходит» в соседние фиб-
фибриллы, осуществляя боковые связи.
В ряде случаев для ориентированных кристаллич.
полимеров, полученных при очень больших вытяжках,
при вытяжках в условиях сравнительно низких темп-р,
при «направленной» полимеризации в монокристаллах
мономера, не наблюдаются малоугловые рентгеновские
рефлексы. Это означает, что продольная аморфно-кри-
аморфно-кристаллич. гетерогенность в полимерах либо стала весьма
нерегулярной, либо вообще исчезла. Изучение подоб-
подобных объектов только начинается.
Свойства ориентированных полимеров
Любой распрямленный отрезок цепной молекулы
линейного полимера обладает сильной анизотропией
многих свойств, обусловленной ориентированным рас-
расположением атомов и атомных групп в макромолекуле,
являющейся фактически одномерным кристаллом. Бо-
Более или менее параллельная упаковка распрямленных
отрезков молекул при образовании одноосноориентиро-
ванного полимерного тела переносит анизотропию
свойств отдельной молекулы на все тело (см. Анизо-
Анизотропия).
Механические свойства. Высокая механич. прочность
в сочетании со способностью к большим обратимым де-
деформациям — наиболее широко используемые специ-
фич. свойства ориентированных полимеров. Эти свойст-
свойства реализуются гл. обр. в одноосноориентированных
кристаллизующихся полимерах, к-рые применяют
в виде волокон, пленок. Надмолекулярное строение
таких полимеров, а именно чередование кристаллич.
и аморфных участков вдоль оси ориентации, приводит
к появлению особой формы состояния полимерного ве-
вещества в аморфных межкристаллитных прослойках:
высокоэластнческого ориентированного состояния.
Как уже отмечалось, аморфный полимер в высоко-
эластич. ориентированном состоянии сам по себе не
удержится: после снятия растягивающей или удер-
удерживающей силы он сократится и перейдет в высоко-
оластич. неориентированное состояние, характеризуе-
характеризуемое более высокой энтропией. В ориентированном
кристаллич. полимере кристаллиты образуют своеоб-
своеобразный каркас, препятствующий дезориентации участ-
участков макромолекулы в аморфных прослойках (к тому же
эти участки молекул, как правило, невелики по дли-
длине — десятки ангстрем). Именно ориентированное вы-
сокоэластнч. состояние аморфных областей в значи-
значительной мере определяет прочностные и деформацион-
деформационные свойства ориентированных кристаллич. полимеров
в широком диапазоне темп-р.
Прочность отдельной полимерной молекулы, растя-
растягиваемой вдоль ее оси, как правило, весьма высока.
Так, для молекулы с С — С-связями разрывное напря-
напряжение составляет 2000—2500 кгс/мм2. Прочность же
полимерного тела, естественно, будет зависеть от рав-
равномерности распределения внешней силы но ценным мо-
молекулам в теле (т. е. от того, насколько «солидарно»
будут работать молекулы). Равномерность загрузки
молекул определяется параллельностью и равнодлин-
ностью отрезков молекул между местами их закрепле-
закрепления и прочностью этих «зажимов». В этом смысле вы-
высокоориентированные кристаллич. полимеры являются
наиболее совершенной системой: сами кристаллиты
оказываются достаточно прочными «зажимами» для
проходных молекул (к-рые и несут всю нагрузку),
а в аморфных прослойках при ориентационной вытяж-
вытяжке получается не очень широкая разнодлинность отрез-
отрезков молекул. В результате для ориентированных поли-
полимерных тел достигаются значения прочности при рас-
растяжении до 5—6 Гн/м- E00—600 кгс/мм2).
Яркая отличительная особенность ориентированных
полимеров — способность к большой обратимой дефор-
деформации. Это обусловлено способностью к высокоэласти-
высокоэластическому деформированию (растяжению или сжатию)
аморфных межкристаллитных прослоек при малом
деформировании кристаллитов — «микрозажимов»
(рис. 8). Ориентированные кристаллические полимеры
\ N
в
Рис. 8. Схема поведения надмолекулярной структуры фиб-
фибриллы в ориентированном кристаллич. полимере при его
растяжении вдоль оси ориентации: а — исходное состояние;
б — обратимое растяжение фибриллы за счет высокоэластич.
деформирования аморфных прослоек; в — начало разруше-
разрушения полимера: разрывы проходных молекул в отдельных
аморфных прослойках по объему полимера (О—О — ось
ориентации, /—/ — направление растяжения).
способны, как правило, обратимо растягиваться вдоль
оси ориентации на —5—20%. При этом аморфные про-
прослойки, составляющие в сумме меньшую часть длины
полимерного тела B0—30% ) и «принимающие на себя»
практически всю эту деформацию, растягиваются соот-
соответственно в несколько раз больше (т. е. на —20 —100%).
Опираясь на схему строения ориентированных кри-
кристаллич. полимеров и связь этого строения с их меха-
механич. свойствами, схемы строения ориентированных
полимеров с прочностью на уровне теоретической, но
с различными деформационными свойствами, можно изо-
изобразить так, как показано на рис. 9.
Возможность сочетания очень высокой прочности при
растяжении с большой эластичностью — отличитель-
отличительная особенность ориентированных кристаллич. поли-
полимеров. Ориентированные аморфные полимеры отли-
отличаются меньшей прочностью (степень ориентации моле-
молекул в них не доводится до высоких значений — см. вы-
выше) и, в общем, меньшей эластичностью, т. к. они
527
ОРЛОН
528
остаются ориентированными лишь при пониженных
(относительно их темп-ры стеклования) темп-рах.
Поэтому аморфные полимеры в ориентированном состо-
состоянии практически не используются, тогда как ориен-
ориентированные кристаллич. полимеры (прежде всего во-
волокна) находят самое широкое применение.
\4\i\ \
Рис. 0. Схемы строения ориентированных кристаллич. по-
полимеров с различным сочетанием прочностных и деформа-
деформационных свойств: а — «обычный» ориентированный поли-
полимер (число проходных молекул в аморфных прослойках
меньше 100%; отрезки проходных молекул в этих прослой-
прослойках разной длины); б — идеально ориентированный поли-
полимер — монокристалл с полностью распрямленными цепными
молекулами; обладает высокой (теоретической) проч-
прочностью, но не эластичен; в — ориентированный полимер
с оптимальным соотношением прочностных и высокоэластич.
свойств (число проходных молекул в аморфных прослойках
~100%; отрезки проходных молекул в этих прослойках
равной длины).
Отметим, наконец, своеобразную анизотропию меха-
нич. устойчивости ориентированных кристаллических
полимеров. Так, высокоориентированный полимер в на-
направлении оси ориентации, как правило, растягивается
обратимо, и вплоть до разрыва полимера (разрушение
начинается с разрыва отдельных, наиболее «слабых»
прослоек — см. рис. 8в) его строение практически не
меняется, так что после распада тела на части и со-
сокращения разгрузившихся частей их строение совпа-
совпадает со строением исходного «целого» ориентированного
образца. Т. обр., структура полимера устойчива при
нагружении вдоль оси ориентации.
Если же ориентированное тело (пленку, пластину)
растягивать в направлении, перпендикулярном оси
ориентации (при темп-pax выше темп-ры стеклования),
то оно сохраняет способность к небольшим E — 10% )
обратимым деформациям лишь на начальном этапе
нагружения, обусловленном растяжением межфибрил-
межфибриллярных аморфных прослоек. В дальнейшем начина-
начинается распад исходной структуры (кристаллитной и над-
кристаллитной) и происходит переориентация, к-рой
завершается формирование также ориентированной
структуры, но уже по новому направлению. Т. обр.,
структура ориентированного полимера оказывается
неустойчивой при растяжении его вдоль отличных от
оси ориентации направлений (переориентация разви-
развивается и при растяжении не только под прямым, но
и под косым углом к оси ориентации).
Термические свойства. Прежде всего следует отме-
отметить парадоксальное с точки зрения свойств обычных
твердых тел температурное поведение ориентированного
полимерного тела: вдоль оси ориентации во многих
случаях полимеры обладают отрицательным коэффи-
коэффициентом термич. линейного расширения, т. е. при на-
нагревании сокращаются в этом направлении. Это вызы-
вызывается «стягивающим» действием энтропийных сил, воз-
возрастающих пропорционально темп-ре в аморфных уча-
участках полимеров, и кристаллизационными процессами.
Ориентированным кристаллизующимся полимерам в до-
довольно широком интервале темп-р (десятки градусов)
присущи обратимые температурные изменения разме-
размеров. Аморфные ориентированные полимеры при нагре-
нагревании сокращаются, как правило, необратимо, воз-
возвращаясь постепенно к неориентированному состоянию.
Необратимые изменения при нагревании происходят
и для ориентированных кристаллизующихся полиме-
полимеров, но здесь они часто сопровождаются специфич.
процессами. Дело в том, что для таких полимеров повы-
повышение темп-ры может играть не только дезориентирую-
дезориентирующую, но и упорядочивающую роль. Действительно, при
наличии кристаллитов-зародышей (к тому же и ориен-
ориентированных) нагревание полимера ведет к увеличению
его степени кристалличности, т. е. к совершенствованию
решетки кристаллитов и, главное, к увеличению их
размеров. В ориентированных полимерах при этом мо-
может происходить увеличение доли полимерного веще-
вещества, входящего в состав ориентированных кристаллич.
областей, т. е. увеличение общей степени ориентации
полимерного тела. Разумеется, значительно чаще нагре-
нагревание приводит к ухудшению ориентации кристаллитов
и, следовательно, к уменьшению степени ориентации
полимерного тела.
Для проявления тех или иных особенностей термич.
свойств ориентированных полимеров весьма важно,
в каких условиях ведется нагревание: полимер «сво-
«свободен» или же концы его закреплены (в последнем
случае нагревание можно вести в условиях различных
растягивающих нагрузок). Именно от этих условий
зависит, в каких соотношениях будут реализоваться
разупорядочивающее и упорядочивающее действия
теплового движения в кристаллизующихся ориенти-
ориентированных полимерах.
При нагревании ориентированных кристаллизую-
кристаллизующихся полимеров, особенно в «свободном» состоянии,
могут происходить и фазовые переходы в кристаллич.
решетках, и изменение типа ориентированного состоя-
состояния. Так, состояние, когда вдоль оси ориентации на-
направлены оси цепных молекул в кристаллитах, может
перейти в состояние, когда вдоль оси ориентации на-
направлена др. ось решетки полимера.
Для случаев нагревания ориентированных полимеров
в фиксированном состоянии (в том числе в условиях
растягивающих нагрузок) интересным является часто
наблюдающееся повышение темп-ры плавления кри-
кристаллов полимера по сравнению с неориентированными
системами кристаллитов. Это является следствием
внутренних динамич. полей, возникающих в ориенти-
ориентированном полимерном теле за счет нарастания энтропий-
энтропийных сил при нагревании.
Лит.: Кобе к о П. П., Аморфные вещества, М.— Л.,
1952; Т а г е р А. А., Физико-химия полимеров, 2 изд., М.,
1968; Каргин В. А., Слонимский Г. Л., Краткие
очерки по физико-химии полимеров, 2 изд., М., 1967;
Джейл Ф. X., Полимерные монокристаллы, пер. с англ.,
Л., 1968; Манделькерн Л., Кристаллизация полиме-
полимеров, пер. с англ., М.—Л., 1966. А. И. Слуцкер.
ОРЛОН — см. Полиакрилопитрилъиые волокна.
ОРТОНОВОЛАКИ (orthonovolaks, Orthonovola-
cke, orthonovolaques) — новолачные смолы, в молеку-
молекулах к-рых фенольные ядра соединены друг с другом
метиленовыми мостиками преимущественно в орто-
положении. О. получают иоликонденсацией фенола
с формальдегидом в слабокислой среде при избытке
фенола. Катализаторами служат соли, окиси и гидро-
гидроокиси Zn, Mg и нек-рых др. металлов (см. Фенола-фор-
малъдегидные смолы). От др. новолачных смол О. от-
отличаются высокой скоростью отверждения; исполь-
используются гл. обр. для производства быстроотверждаю-
щихся пресспорошков (см. Фенопласты). О. впервые
описаны Бендером в 1947. ю. в. Горохолинский.
ОРТОТРОПНЫЕ ПОЛИМЕРНЫЕ МАТЕРИАЛЫ
(orthotropic polymer materials, orthotropische Kunst-
stoffe, materiaux orthotropiques polymeriques) — группа
анизотропных материалов, обладающих тремя ортого-
ортогональными плоскостями симметрии физич. свойств. По-
Полимерные материалы могут приобрести ортотропию
вследствие ориентации молекул в процессе переработки
и вытяжки. Однако наибольшее значение имеют О. и. м.,
529
ОРТОТРОПНЫЕ ПОЛИМЕРНЫЕ МАТЕРИАЛЫ
530
к-рые получают армированием полимерного связующего
волокнистыми материалами. О. и. м. обычно изготовля-
изготовляют армированием полимерной матрицы волокнами в двух
взаимно перпендикулярных направлениях (материалы
продольно-поперечной намотки), тканью (текстолиты),
специальными многослойными тканями объемного иле-
Рис. 1. Схематическое изображение ортотропного мате-
материала, армированного в трех направлениях.
тения, а также путем укладки армирующих волокон
в трех взаимно перпендикулярных направлениях (т.
н. «пространственно сшитые материалы»). Плоскости
симметрии материала определяются положением ар-
армирующих нитей или направлением укладки армирую-
армирующей ткани. На рис. 1 для примера схематически показан
материал, армированный в трех направлениях. Плос-
Плоскости симметрии упругих свойств материала совпа-
совпадают с координатными плоскостями. В частном случае
армирования волокнами в одном направлении полу-
получается монотронный материал, свойства к-рого одина-
одинаковы во всех направлениях, перпендикулярных арми-
армирующим волокнам.
Для получения армированных О. п. м. применяют
стекловолокно, асбестовые, углеродные и борные волок-
волокна. Применение последних позволяет достичь очень
высокой прочности при растяжении или сжатии
[2000 MhIm1 B00 кгс/мм2)] и большой жесткости (зна-
(значение модуля упругости при растяжении или сжатии
Е = 2-105 Мн/м2 B-Ю6 кгс/см2). В качестве полимер-
полимерного связующего обычно применяют термореактивные
(эпоксидные, полиэфирные, феноло-формальдегидные
и кремнийорганич. смолы), а также термопластичные
полимеры (полиамидные смолы). Наличие податливой
и менее прочной по сравнению с армирующими волок-
волокнами матрицы обусловливает ряд отрицательных в кон-
конструкционном отношении свойств О. п. м.— гл. обр.
низкую жесткость и прочность при сдвиге. Для расчета
свойств О. п. м. необходимо знать константы упруго-
упругости uijki, входящие в обобщенный закон Гука
=aijhlahl
7 = 1,2,3)
где t(j и Ом — тензоры деформаций и напряжений соот-
соответственно. Если оси координат ортогональны плоско-
плоскостям симметрии, то в матрице а,^/ имеется 9 различных
констант. При повороте осей координат число констант
увеличивается, но все они выражаются по закону пре-
преобразования тензоров через исходные 9 величин.
В развернутом виде закон Гука для О. п. м. с исполь-
использованием вместо aijki T- наз- технических констант за-
записывается так:
? а а ^ а
?22 — -gT- cfu + -g- a22 -—- a33,
?33 = — IF" aH ~ IT G22 + -ЁГ a33>
?12 — ~q~2 al2' ?13 — -q— a13? ?23 —
. ' E3 Ег ' E3
Здесь ?п, е22, ?33 — деформации, а аи, а22, а33 — на-
напряжения растяжения по осям, перпендикулярным
плоскостям симметрии; Е1ч Е2, Е3 — модули упругости
при растяжении или сжатии; м12, ..., v23 — коэффициен-
коэффициенты Пуассона; е12, е13, е23, а12, а13, а23 — соответственно
деформации и напряжения сдвига; G12, Gl3, G23 — моду-
модули упругости при сдвиге. Показателем относительной
жесткости при сдвиге является отношение G/E, к-рое
может достичь для стеклопластиков 1/1О-т-1/5о, а для бо-
ропластиков даже 1/100. Эту особенность необходимо
учитывать при расчетах свойств О. п. м. Жесткость при
растяжении О. п. м. под разными углами определяется
по закону преобразования компонент тензора ац^ и
существенно зависит от направления растяжения. По-
Полярная диаграмма податливости О. п. м. показана на
рис. 2.
Определение для О. п. м.
модулей ?\, Е2, Е3, С12, С13,
G23 и коэффициентов v12, v13,
v23 по соответствующим ха-
характеристикам арматуры и
связующего, а также исхо-
исходя из относительного со-
содержания арматуры и свя-
Рис. 2. Относительное изменение
податливости (характеризуемое
величиной Ei/Ei') в одной плос-
плоскости ортотропного полимерного материала. Податливости
в направлениях измерения и оси Хх характеризуются вели-
величинами соответственно 1/EV и i/Ei, где Ех и Et — соответ-
соответствующие модули).
зующего сделано только в простейших случаях. При
этом предполагалась регулярная (в частности, гекса-
гексагональная) укладка идеально прямых волокон арма-
арматуры, а также принималось, что полимерная матрица
и волокна деформируются совместно.
Для учета ползучести О. п. м. коэффициенты а-^уа
должны быть заменены нек-рыми временными опера-
операторами (см. Больцмана — Волътерры уравнения).
В первом приближении принимается, что только сдви-
сдвиговые деформации связаны с напряжениями времен-
временными зависимостями. Такое предположение часто
оправдано, поскольку пол-
ползучесть характерна в ос-
основном для связующего.
Прочность О. п. м. оп-
определяется прочностью ар-
армирующих волокон, проч-
прочностью полимерной матри-
матрицы и величиной адгезии
между волокнами и свя-
Растяшение °° Сжатие
/7
-•/7, нгс/мм2
200
зующим.
Для полного описания
прочности О. п. м. при
сложном напряженном со-
состоянии необходимо знать
Рис. 3. Полярная диаграмма
прочности при растяжении
(П+) и сжатии (.77") стеклопла-
стеклопластика, армированного в одном
A) и в двух взаимно перпен-
перпендикулярных B) направле-
направлениях.
45
45°
нек-рые основные параметры: прочность при растяжении
и сжатии в направлении армирующих волокон, проч-
прочность при сдвиге в направлении волокон и в диагональ-
диагональном направлении. Через полученные экспериментальные
точки в пространстве главных напряжений (оси коорди-
координат ортогональны плоскостям симметрии материала)
проводят поверхность прочности. Прочность О. п. м. су-
531
ОСАДИТЕЛЬНАЯ ВАННА
532
щественно зависит от направления нагружения (при
повороте осей координат поверхность прочности транс-
трансформируется).
На рис. 3 показаны типичные полярные диаграммы
прочности стеклопластика при растяжении и сжатии
в разных направлениях.
Лит.: Малмейстер А. К., Т а м у ж В. П., Те-
Тетере Г. А., Сопротивление жестких полимерных материа-
материалов, 2 изд., Рига, 1972; Т а р н о п о л ь с к и й Ю. М.,
Розе А. В., Особенности расчета деталей из армированных
пластиков, Рига, 1969; Сопротивление стеклопластиков, М.,
1968. В. П. Тамуж.
ОСАДИТЕЛЬНАЯ ВАННА — см. Формование хи-
химических волокон.
ОСМОМЕТРИЯ полимеров (osmometry, Osmo-
mctrie, osmometrie) — метод определения среднечисло-
вой молекулярной массы полимера, основанный на
измерении осмотич. давления.
Если р-р полимера отделить от растворителя мем-
мембраной, не проницаемой для растворенного вещества,
но проницаемой для растворителя, то последний диф-
диффундирует сквозь мембрану в р-р. Это явление наз.
осмосом. Оно вызвано тем, что химич. потенциал раст-
растворителя в р-ре меньше, чем у чистого растворителя.
Для того чтобы система пришла в равновесие, необхо-
необходимо выровнять значения химич. потенциала раство-
растворителя но обе стороны полупроницаемой мембраны. Это
можно осуществить, приложив избыточное давление
к р-ру, равное по величине осмотич. давлению. Осмо-
Осмотич. давление п связано с мол. массой М растворенного
полимера ур-нием Вант-Гоффа
где с — концентрация полимера в р-ре; 7? — универ-
универсальная газовая постоянная; Т — абсолютная темп-ра.
Если в р-ре содержатся макромолекулы с мол. масса-
массами М1ч 3/2, М3, ..-, концентрации к-рых равны сг, с2,
Сз,..., соответствующие им парциальные осмотич. дав-
давления -/ складываются и из ур-ния A) следует
Так как с = 2 с,- — общая концентрация макромо-
макромолекул, то 1
S (С| / М0
Jim — = RT — к '
с=0 С г- С>
Поскольку с/ пропорционально щМ^щ — число молей
г-го растворенного вещества), то ур-ние B) можно за-
записать след. образом:
lim — = RT
RT
C)
То е. осмотическое давление является мерой среднечис-
ловой молекулярной массы полимера Мп.
Для р-ра нейтрального полимера в термодинамически
хороших растворителях, где наблюдается взаимодейст-
взаимодействие между растворителем и полиме-
полимером, осмотич. давление при больших
концентрациях полимера растет зна-
значительно быстрее, чем при низких
(рис. 1). В этом случае ур-ние Вант-
Гоффа неприменимо и наиболее удоб-
Рис. 1. Зависимость осмотического давле-
давления от концентрации для р-ра нейтраль-
нейтрального полимера в термодинамически хоро-
^ с шем растворителе.
ным способом выражения отношения rjc (в области
разб. р-ров) является представление его в виде степен-
степенного ряда
\ = RT (Аг + А2с + А#* f ...) D)
\gA2
где А1ч А2, ...— первый, второй и т. д. вириальные
коэффициенты:
Лх = 1/Mn, A2 = yuNAl2Min
(V« — объем, из к-рого сегменты макромолекулы дан-
данного вида вытесняют сегменты др. макромолекул, т. наз.
эффективный исключенный объем; Nд — число Аво-
гадро).
Как видно из рис. 2, на к-ром схематично изображен
ход зависимости л/с от с, на оси ординат отсекается отре-
отрезок, равный 1-7") = —. Тангенс угла наклона соот-
\ /с~-о Л/п
ветствующей прямой—А2-
Практически наблюдаемая
линейная зависимость л/с
от с показывает, что А3
мал. Коэфф. А2 характе-
характеризует парные взаимодей-
взаимодействия полимер — раство-
растворитель (степень откло-
отклонения р-ра от идеального
поведения). Значение А2
зависит от ряда факто-
факторов, в том числе от струк- Рис< 2. Характер изменения ос-
туры макромолекул поли- мотического давления с кон-
мера, их размеров и фор- центрацией.
мы, темп-ры измерения.
Мол. масса полимера так-
также влияет на значение А2.
Как видно из рис. 3, lgA2
для р-ров полимеров в хо-
хороших растворителях при
повышении мол. массы
уменьшается, причем эм-
эмпирически установлено,
что А2 = 11Мьп (Ъ нахо-
находится в пределах 0,05 —
0,25).
Для измерения осмотич.
давления полимеров могут
применяться статистиче-
статистические и динамические ме-
методы и осмометры различных конструкций. На рис. 4
приведена простейшая схема осмометра. В сосуд 2 зали-
заливают чистый растворитель, в
сосуд 1 — р-р. Растворитель
переходит из одного сосуда
в другой до тех пор, пока
давление, пропорциональное
высоте столба раствора h в
трубке 4, не уравновесит по-
потока, обусловленного избыт-
избытком химич. потенциала в
растворителе. Высота стол-
Рис. 4. Схема осмометра: 1 — р-р;
2 — растворитель; 3 — мембрана;
4 — измерительный капилляр;
5 — капилляр сравнения.
ба р-ра h = л/d, где d — плотность р-ра. Прак-
Практически измеряют значения % при 3—4 концентрациях с
и строят прямую в координатах -/с от с, с помощью
к-рой находят величину (— 1 ==- (см.,напр., рис.2).
Для термодинамически хороших растворителей кри-
кривая зависимости л/с от с не параллельна оси абсцисс
и тангенс угла наклона кривой равен второму вириаль-
ному коэффициенту в ур-нии D). Это т. наз. статич.
метод измерения осмотич. давления. Он сравнительно
прост, но вследствие длительности установления рав-
равновесия может произойти деструкция растворенного
Рис. 3. Зависимость второго
вириального коэффициента в
ур-нии для осмотического дав-
давления от молекулярной мас-
массы полимера.
533
СТВЕРДИТЕЛИ
534
полимера, приводящая к повышению содержания низ-
низкомолекулярных частиц в р-ре. Кроме того, при дли-
длительном соприкосновении р-ра полимера с полупрони-
полупроницаемой мембраной последняя может адсорбировать
растворенное вещество и т. о. понизить концентрацию
полимера в р-ре. Поэтому в последние годы усилия
экспериментаторов были направлены на конструирова-
конструирование таких статич. осмометров, измерение в к-рых за-
занимает незначительное время.
Первоначально мембрана в осмометрах закреплялась
горизонтально. Для ускорения осмоса Р. Фуосс и
Д. Мид, а позднее X. Хельфриц предложили закреп-
закреплять две мембраны вертикально. Осмометр Хельфрица,
получивший наибольшее распространение,— целиком
металлический, высокогерметичный, малогабаритный.
Равновесие в нем достигается через 2—3 ч.
При измерении динамич. методом осмотич. давление
компенсируется точно измеряемым противодавлением,
приложенным извне. Основное преимущество этого
метода — быстрота измерений. Однако в динамич. осмо-
осмометрах необходимо тщательно укреплять мембрану,
чтобы свести к минимуму эффект ее прогиба (баллон-
эффект). Из таких осмометров наибольшее распрост-
распространение получили приборы, в к-рых небольшие изме-
изменения в осмотич. давлении фиксируются металлич.
фольгой; изгиб фольги приводит к изменению емкости
конденсатора, фиксируемому при помощи специаль-
специальной электрич. схемы.
Основная трудность при определении мол. массы
полимеров осмотич. методом (особенно для полимеров
с низкой мол. массой) заключается в выборе удовлетво-
удовлетворительной мембраны. Идеальная полупроницаемая
мембрана должна быть непроницаемой для молекул
растворенного вещества и обладать высокой проницае-
проницаемостью для растворителя. Изменение тзща мембраны
может привести к расхождениям при определении
мол. массы (и второго вириального коэффициента) для
одного и того же полимера. Лучшие мембраны — из
целлофана. Применяют также пленки из поливинило-
поливинилового спирта, поливинилбутираля, коллодиевые мемб-
мембраны, мембраны из стекла, бактериальные мембраны.
Перед началом измерения проверяют пористость мем-
мембран, т. е. степень ее проницаемости для данного раст-
растворителя. Меняя продолжительность и способ обра-
обработки, любой мембране (кроме стеклянной) можно при-
придать желаемую пористость.
Существующие мембраны и аппаратура позволяют
определять мол. массу полимеров в диапазоне от 10 000
до 2 000 000 с точностью -J- 10%
Лит.: Рафиков С. Р., Павлова С. А., Твер-
Твердо х л ^ б о в а И. И., Методы определения молекулярных
весов и полидисперсности высокомолекулярных соединений,
М., 1963; Тенфорд Ч., Физическая химия полимеров,
пер. с англ., М., 19E5, с. 249; Мелвин-ХьюзЭ. А.,
Физическая химия, пер. с англ., кн. 2, М., 1962, с. 697, 959;
Цветков В. Н., Э с к и н В. Е., Френкель С. Я.,
Структура макромолекул в растворах, М., 1964, с. 50, 334.
И. И. Твердохлебова.
ОТВЕРДИТЕЛИ (curing agents, Hartungsmittel,
durcisseurs) — вещества, обусловливающие отвержде-
отверждение реакционноспособных олигомеров. По характеру
действия эти вещества делят на след. группы:
1. Собственно отвердители, молекулы к-рых, реаги-
реагируя с функциональными группами олигомера, входят
в структуру образующегося полимера. Такие О. при-
применяют при отверждении олигомеров с небольшим чис-
числом функциональных групп или с функциональными
группами, не способными реагировать между собой
с образованием трехмерного полимера.
2. Инициаторы и катализаторы отверждения. Ини-
Инициаторы вызывают отверждение олигомеров, содержа-
содержащих ненасыщенные группы, по механизму радикальной
полимеризации. Катализаторы ускоряют взаимодейст-
взаимодействие олигомеров между собой или с отвердителем пер-
первой группы; эти реакции могут протекать по механиз-
механизму поликонденсации или ионной полимеризации (см.
также Катализаторы полимеризации).
Отверждение с использованием О. первой группы со-
сопровождается меньшими стерич. и диффузионными
препятствиями, чем непосредственное взаимодействие
молекул олигомеров в этом процессе. Поэтому с при-
применением таких О. получают обычно полимеры с боль-
большей частотой сетки.
В качестве о т в е р д и т е л е й используют поли-
полифункциональные вещества, выбор к-рых определяется
типом функциональных групп отверждаемого олиго-
олигомера. Высокой реакционной способностью отличаются
эпоксигруппы (—НС—СН2) и изоцианатные группы
О
(— N = C—О), реагирующие практически с любыми
функциональными группами. Поэтому О. олигомеров
с эпоксидными группами могут служить самые различ-
различные вещества. Наибольшее распространение среди них
получили алифатич. и ароматич. первичные и вторич-
вторичные амины, низкомолекулярные алифатич. полиамиды
и ангидриды к-т. Низкомолекулярные полифункцио-
полифункциональные соединения с эиоксигруппами могут, в свою
очередь, служить О. олигомеров, содержащих, напр.,
карбоксильные или гидроксильные группы.
Низкомолекулярные ди- и полиизоцианаты наиболее
широко используют в качестве О. для олигомеров
с гидроксильными концевыми группами. С другой сто-
стороны, О. олигомеров с концевыми изоцианатными груп-
группами (макроизоцианатов) могут служить низкомоле-
низкомолекулярные полифункциональные спирты или амины.
Благодаря возможности выбора разнообразных О.
удается в широких пределах регулировать технологич.
и эксплуатационные свойства отверждающихся мате-
материалов на основе эпоксидных смол и полиуретанов.
Среди др. технически важных олигомеров примене-
применения О. требуют новолачные феноло-формальдегидные
смолы, к-рые отверждаются гексаметилентетрамином,
параформом или эпоксидными олигомерами, и кремний-
органич. олигомеры, отверждающиеся алкоксисилана-
ми. В отверждении могут участвовать т. наз. «активные
растворители», напр, фурфурол и фуриловый спирт
для феноло-формальдегидных смол или ненасыщенные
мономеры (стирол, метилметакрилат, диметакрилаты
гликолей, эфиры аллилового спирта и др.) для поли-
алкиленгликольмалеинатов и олигоэфиракрилатов.
Количество О. определяется числом функциональных
групп в олигомере и отвердителе. Для систем, отвер-
отверждающихся по механизму ноликонденсации, оно обыч-
обычно близко к стехиометрическому.
Инициаторы отверждения — вещест-
вещества, распадающиеся в условиях отверждения с об-
образованием свободных радикалов (см. Иницииро-
Инициирование полимеризации). Эти О. вводят в количестве
0,1—5,0% (в расчете на массу полимера). Часто приме-
применяют не один инициатор, а т. наз. отверждающую систе-
систему — инициатор и ускоритель его распада, а иногда
и соускоритель, или промотор. Напр., для отверждения
полиэфирных смол часто используют систему, содер-
содержащую перекись метилэтилкетона (инициатор) и наф-
тенат кобальта (ускоритель); при этом темп-pa отверж-
отверждения снижается до —20 °С.
Выбор катализатора отверждения
определяется характером реакций функциональных
групп олигомеров. Полимеризация олигомеров ио
эпоксидным группам, а также реакции этих групп с гид-
гидроксильными, карбоксильными, ангидридными и др.
группами наиболее эффективно катализируются тре-
третичными аминами и к-тами Льюиса. Каталитич. актив-
активность третичных аминов резко повышается в присутст-
присутствии протонодонорных веществ (спиртов, фенолов, к-т)
и снижается в присутствии протоноакцепторных (ами-
(амидов к-т, сложных эфиров, альдегидов и кетонов). К-ты
535
ОТВЕРЖДЕНИЕ
536
Льюиса, гл. обр. BF3, чаще всего используют в виде
комплексов с аминами или эфирами (напр., комплекс
BF3 — моноэтиламин). В присутствии полярных ве-
веществ (воды, диметилформамида, спиртов и др.) ката-
литич. действие к-т Льюиса и их комплексов умень-
уменьшается. Для эпоксидных смол катализаторы обычно
используют как единственные О. или с целью ускорения
отверждения этих смол ангидридами к-т.
Для отверждения феноло-, мочевино- и меламино-
формальдегидных смол применяют к-ты (сульфокисло-
ты, серную, соляную, щавелевую и др.), реже — осно-
основания (аммиак, гидроокиси щелочных и щелочнозе-
щелочноземельных металлов). Основаниями и к-тами катализи-
катализируются также реакции отверждения олигомеров с изо-
цианатными группами. Катализаторами отверждения
кремнийорганич. олигомеров служат олово- и титанор-
ганич. соединения, амины и их комплексы, органич.
соли щелочных металлов, свинца, железа, кобальта.
Катализатор, не участвуя в образовании трехмерной
сетки, остается в отвержденном материале и может вли-
влиять на его свойства, особенно на влагопоглощение и ди-
электрич. характеристики. Кол-во катализатора не
связано с функциональностью олигомера и лежит обыч-
обычно в пределах 2—5 мае. ч. на 100 мае. ч. олигомера.
Все О. должны удовлетворять нек-рым общим техно-
логич. требованиям: растворяться в исходном олиго-
мере, обеспечивать оптимальное сочетание жизнеспо-
жизнеспособности отверждаемого материала, скорости и глу-
глубины отверждения, быть нетоксичными и др. С точки
зрения растворимости технологич. преимущества пе-
перед др. О. имеют жидкие при нормальных условиях или
легкоплавкие вещества. Понижение темп-ры плавле-
плавления м. б. достигнуто модификацией химич. струк-
структуры или применением эвтектич. смесей О. Для уве-
увеличения жизнеспособности отверждающегося материа-
материала при сохранении необходимой скорости отвержде-
отверждения используют т. наз. «скрытые» О.— микрокапсули-
рованные О., к-рые проявляют свою активность только
при разрушении оболочки микрокапсулы в условиях
отверждения (см. Микрокапсулирование), или комплек-
комплексы О., распадающиеся с выделением активного продук-
продукта при темп-pax, близких к темп-рам отверждения.
Лит. см. при ст. Отверждение.
Л. Н. Седов, П. Г. Бабаевский.
ОТВЕРЖДЕНИЕ (curing, Aushartung, durcissement) —
процесс, при к-ром жидкие (или используемые в виде
расплавов и р-ров) реакционноспособные олигомеры
необратимо превращаются в твердые, нерастворимые
и неплавкие трехмерные полимеры. [Термин «отверж-
«отверждение» используют обычно применительно к процес-
процессам образования трехмерных полимеров при перера-
переработке пластмасс, лаков, клеев, герметиков, компаун-
компаундов. Образование трехмерных полимеров в результате
соединения (сшивания) поперечными связями ранее
синтезированных макромолекул эластомеров наз. вул-
вулканизацией.}
О. происходит в результате взаимодействия реак-
ционноспособных групп олигомеров между собой или
со специально добавляемыми реагентами (отвердите-
лями) под действием тепла, УФ-света, излучений высо-
высокой энергии или катализатора. В отдельных случаях
отверждаться могут олигомеры, в молекулах к-рых
реакционноспособные группы возникают только в ус-
условиях О.
Механизм О. определяется природой реакцион-
носпособных групп в олигомере, типом отвердителя и
условиями процесса. О. может протекать по механизму
поликонденсации, сопровождающейся выделением низ-
низкомолекулярных продуктов (напр., О. феноло-, моче-
мочевино- и меламино-формальдегидных смол или кремний-
кремнийорганич. олигомеров) или без выделения таких продук-
продуктов (напр., О. эпоксидных смол первичными аминами).
По механизму гомо- или сополимеризации могут от-
отверждаться, напр., полиалкиленгликолъмалеинаты и
полиалкиленгликолъфумараты, а также олигоэфир-
акрилаты. В отдельных случаях в одном процессе могут
сочетаться полимеризационныи и поликонденсационный
механизмы О. (напр., при О. эпоксидных смол ангидри-
ангидридами к-т в присутствии катализаторов — третичных
аминов).
В общем процессе О. можно выделить две стадии:
1) начальную — до момента возникновения трехмер-
трехмерной сетки и, следовательно, потери отверждающимся
материалом текучести и растворимости — т. наз. точ-
точки гелеобразования (см. Жизнеспособность)', 2) ко-
конечную — после точки гелеобразования до предельных
стадий О.
Если О. протекает по механизму полимеризации, то
на начальной стадии наблюдается достаточно длитель-
длительный индукционный период. В этот период вязкость
отверждающегося материала существенно не изменяет-
изменяется. В период роста цепей материал превращается в
трехмерный полимер практически мгновенно. Продол-
Продолжительность индукционного периода можно регулиро-
регулировать в широких пределах подбором инициирующей си-
системы или используя ингибиторы. После точки геле-
гелеобразования процесс развивается обычно с самоуско-
самоускорением, обусловленным гель-эффектом, а затем пре-
прекращается в результате обрыва растущих цепей или
резкого уменьшения их подвижности.
О. по механизму поликонденсации протекает с по-
постепенным нарастанием вязкости до точки гелеобразо-
гелеобразования, соответствующей достижению определенной
степени конверсии функциональных групп олигомера.
В этом случае О. может быть прервано на любой задан-
заданной глубине. После точки гелеобразования скорость О.
обычно замедляется, и процесс заканчивается вследствие
исчерпания функциональных групп или резкого воз-
возрастания стерич. или диффузионных препятствий их
взаимодействию.
Существенный фактор, определяющий кинетику О.,—
темп-pa. Снижение теми-ры — важная практич. за-
задача, решение к-рой позволяет значительно упростить
технологию О. Однако при темп-pax, лежащих ниже
нек-рого предела, во многих случаях не удается до-
достигнуть необходимой степени О. и получить трехмер-
трехмерные полимеры с требуемым комплексом свойств, в част-
частности с высокой теплостойкостью. Важнейшее значение
для выбора режима О. имеет соотношение между
темп-рой процесса и темп-рой стеклования Тс образую-
образующегося полимера. Если при определенной степени О.
Тс окажется выше темп-ры процесса, то О. практически
прекратится. Для того чтобы произошло полное О.,
темп-pa этого процесса должна быть выше Тс предельно
отвержденного полимера (Гсоо). Поэтому олигомеры, обра-
образующие в результате О. трехмерные полимеры с высо-
высокой Тс (с высокой теплостойкостью), напр, феноло-
формальдегидные или меламино-формальдегидные, от-
верждают обычно при темп-pax до 180—200 СС. При
более высоких темп-pax начинается термоокислителъ-
ная деструкция полимера.
Темп-pa О. в значительной степени определяется
тепловыми эффектами реакций. Степень влияния экзо-
термич. эффекта на О. зависит от массы материала,
его теплоемкости и условий теплообмена. Самора-
Саморазогрев материала может способствовать практически
полному О. даже в том случае, если темп-pa О. будет
значительно ниже ГсОо. Этим объясняется, напр., срав-
сравнительно высокая степень холодного О. полиалкилен-
гликольмалеинатов и олигоэфиракрилатов, а также О.
эпоксидных смол третичными или алифатич. первичны-
первичными аминами. Однако экзотермич. характер процесса
обусловливает его невоспроизводимость, возникнове-
возникновение в материале неравномерных температурных полей,
что приводит к неравномерному О. в объеме материала
и к появлению в нем локальных напряжений.
537
ОТДЕЛКА ХИМИЧЕСКИХ ВОЛОКОН
538
Для контроля скорости О. широко при-
применяют метод торзионного маятника, в к-ром фикси-
фиксируется изменение жесткости нити, пропитанной иссле-
исследуемым материалом. Используют также визуальный и
различные вискозиметрич. методы (напр., определяют
скорость подъема пузырька воздуха или падения ме-
таллич. шарика в отверждаемом материале). Иногда
скорость О. устанавливают с помощью пластометров
ротационного типа (напр., системы Канавца), имити-
имитирующих технологич. процесс формования изделий.
Для контроля скорости О., сопровождающегося за-
заметным экзотермич. эффектом, применяют методы опре-
определения тепловыделения — дифференциальный терми-
термический анализ, дифференциальную сканирующую кало-
калориметрию.
Количественная оценка скорости О. возможна по
степени конверсии реакционноспособных групп олиго-
меров или но частоте сетки образующегося трехмерного
полимера. Конверсию реакционноспособных групп
оценивают методами колебательной спектроскопии,
дилатометрии или дифференциального термич. ана-
анализа, а также химич. методами, напр, проведением
соответствующих реакций в равновесно набухшем
отвержденном материале или контролем количества
выделяющихся низкомолекулярных продуктов.
Частоту сетки отвержденного полимера, находящего-
находящегося в высокоэластич. состоянии, рассчитывают по де-
деформационным свойствам полимера или по степени его
набухания в растворителе (см. Вулкаиизационная сет-
сетка). Степень О. часто характеризуют также содержа-
содержанием фракции, экстрагируемой кипящим растворите-
растворителем (золь-фракция). Иногда на глубоких стадиях О.
содержание этой фракции очень мало и соизмеримо
с ошибкой анализа. Присутствие в отверждающемся
материале различных ингредиентов, особенно напол-
наполнителей, затрудняет количественную оценку степени О.
В тех случаях, когда отвержденный полимер не на-
находится в высокоэластич. состоянии, степень О. можно
оценить лишь качественно по изменению какого-либо
свойства отверждающегося материала, чувствительно-
чувствительного к степени О. Наиболее распространено определе-
определение деформационных свойств при статич. или динамич.
нагружениях (напр., с помощью весов Каргина — см.
Термомеханическое исследование, торзпошюго маятника
и др.).
К числу важнейших характеристик процесса О. отно-
относятся также объемная усадка отверждаемого материа-
материала и количество выделяющихся летучих веществ —
низкомолекулярных продуктов реакций, легкоиспаряю-
щихся компонентов (напр., остатков растворителей),
продуктов частичной деструкции полимеров. Усадка,
обусловленная повышением плотности материала вслед-
вследствие возникновения большого числа новых химич.
связей, возрастает с уменьшением мол. массы исход-
исходных олигомеров и увеличением в них числа функцио-
функциональных групп. Существенное влияние на величину
усадки оказывает механизм О. Напр., при О. по меха-
механизму поликонденсации, не сопровождающейся выде-
выделением низкомолекулярных продуктов, она составляет
3—6%, при О., обусловленном полимеризацией олиго-
олигомеров с ненасыщенными связями, — 5 —12%, при поли-
поликонденсационном О. с выделением низкомолекуляр-
низкомолекулярных продуктов — 15—25%. Объемная усадка и давле-
давление внутри материала, создаваемое летучими про-
продуктами, обусловливают появление в отвержденных
полимерах остаточных напряжений и дефектов, а так-
также изменение их размеров.
Лит.: Химия и технология полимеров, пер. с нем., т. 2,
ч. 2, М. — Л., 1966; Химические реакции полимеров, под ред.
Е. Феттеса, пер. с англ., т. 1 — 2, М., 1967; Б е н и г Г. В.,
Ненасыщенные полиэфиры. Строение и свойства, пер. с англ.,
М., 1968; Берлин А. А., КефелиТ. Я., Королев
Г. В., Полиэфиракрилаты, М., 1967; Ли Г., Невилл К.,
Справочное руководство по эпоксидным смолам, пер. с
англ., М., 1973; Методы испытания, контроля и исследования
машиностроительных материалов, т. 3, Методы исследования
неметаллических материалов, М., 1973, с. 119; Encyclopedia of
polymer science and technology, v. 4, N. Y.— [a. o.], 1966, p. 528.
П. Г. Бабаевский, Л. Н. Седов.
ОТДЕЛКА ХИМИЧЕСКИХ ВОЛОКОН (aftertreat-
ment of chemical fibers, Nachbehandlung der Chemiefa-
sern, finissage des fibres chimiques) — ряд технологич.
операций, следующих после формования волокон. Без
отделки большинство химич. волокон нельзя использо-
использовать для производства тканей, трикотажных изделий
или пряжи (при переработке штапельного волокна).
О. х. в. состоит из след. операций: 1) промывки во-
водой; 2) очистки от примесей, не растворимых в воде;
3) отбелки; 4) авиважной обработки; 5) сушки; 6) вы-
вытяжки; 7) термообработки. В зависимости от вида во-
волокна и метода его получения ту или иную операцию
О. х. в. можно не проводить. Текстильные операции —
кручение, перемотка, текстурирование, сновка и тка-
ткачество, к-рые часто также выполняют на заводах хи-
химич. волокон, не относятся к О. х. в.
Текстильные и технич. нити подвергают отделке на
бобинах и в куличах (т. е. сохраняя форму паковки,
полученную на прядильной машине) или в мотках
(т. е. изменяя форму паковки). На машинах непре-
непрерывного формования обработке подвергают одиночную
нить. При производстве штапельного волокна отделы-
отделывают жгуты (пучки) волокна или нарезанное (штанели-
рованное) волокно. Различные операции О. х. в. (кро-
(кроме вытяжки и термообработки) чаще всего проводят
на отделочно-сушильных агрегатах.
Водой промывают в основном волокна,
сформованные из р-ров полимера по мокрому способу,
т. к. такие волокна содержат остатки растворителя,
соли, к-ты. Волокна, сформованные по др. способам,
водой не промывают. Исключение составляет волокно
капрон, к-рое содержит остатки мономера (капролак-
тама). О способах формования см. Формование химиче-
химических волокон.
Для лучшей отмывки водорастворимых примесей стре-
стремятся увеличить степень отжима волокон между сек-
секциями и число промывных секций, а также многократ-
многократно использовать промывную воду, направляя ее из
последней промывной секции через промежуточные
к первой. Многократное использование воды особенно
необходимо, если примеси, отмываемые водой, направ-
направляют на регенерацию. Обычно для промывки приме-
применяют умягченную воду, т. к. соли, содержащиеся в
жесткой воде, вызывают образование осадков, к-рые
мешают повторному использованию веществ, отмывае-
отмываемых из волокна, и придают волокну жесткость.
Для ускорения промывки воду часто подогревают до
50—80 °С. При этом тепло «отработанных» промывных
вод используют в теплообменниках. Расход промывной
воды зависит от вида волокон и примесей и составляет
0,2—0,8 м3 на 1 кг волокна.
Промывку волокон часто совмещают с их п л а с т и-
фикационной вытяжкой (на 80% и более),
к-рая заключается в пропускании свежесформованного
волокна через вытяжной механизм (см. Прядильные
машины), погруженный в промывную воду, подогре-
подогретую до 80 °С. Повышенная темн-ра облегчает ориента-
ориентацию макромолекул полимера, вследствие чего полу-
получаются волокна высокой прочности.
Очистке от нерастворимых в воде
примесей подвергают в основном гидратцеллю-
лозные волокна. Так, медноаммиачные волокна очи-
очищают от соединений Си, вискозные — от соединений Fe,
обрабатывая волокна водными р-рами минеральных
к-т. При этом образуются растворимые в воде соли
Си и Fe. После такой обработки волокна тщательно
промывают водой.
Свежесформованные вискозные волокна подвергают
десульфурации для удаления остатков @,2 —
0,3% от массы сухого полимера) элементарной серы,
539
ОТДЕЛКА ХИМИЧЕСКИХ ВОЛОКОН
540
к-рая придает волокнам грязно-желтый или серый отте-
оттенок. Десульфурация основана на превращении эле-
элементарной серы в водорастворимые соединения. Для
этого волокна обрабатывают р-рами NaOH (концент-
(концентрацией 10 — 15 г/л при 40—50 °С или 4—6 г/л при 60 СС),
Na2S C—5 г/л при 60 °С) или Na2SO3 B0—25 г/л при
70—75 °С). Продолжительность десульфурации при
использовании р-ров NaOH или Na2S — 10—12 мин,
для р-ра Na2SO3 — 20—25 мин.
При десульфурации р-рами NaOH сера удаляется
наиболее интенсивно, но прочность и относительное
удлинение волокон снижаются на 10%. Р-ры Na2S мень-
меньше влияют на прочность волокон, но ухудшают усло-
условия труда и часто придают волокнам сероватый отте-
оттенок (из-за образования FeS). Для десульфурации тек-
текстильных нитей наиболее пригоден р-р Na2SO3, не вли-
влияющий на их качество. После десульфурации вискоз-
вискозные волокна подвергают тщательной промывке водой.
Отбелка волокон необходима только при полу-
получении изделий белого цвета или окрашиваемых в свет-
светлые тона. Для отбелки вискозных и иолиакрилонит-
рильных волокон обычно применяют р-ры NaCIO или
Н2О2 в щелочной или NaClO2 в кислой среде, к-рые
разрушают окрашенные примеси. После отбелки вис-
вискозных волокон при 20—25 °С р-ром NaCIO (содержа-
(содержание активного хлора 1,0—1,2 г/л) в щелочной среде
@,2 г/л NaOH) степень белизны достигает 80%. Этот
процесс ведут при рН 8—9, т. к. при рН>9 отбеливаю-
отбеливающее действие NaCIO значительно ослабляется, а при
рН<8 возможно разрушение целлюлозы, приводящее
к снижению прочности волокон. Поэтому в отбеливаю-
отбеливающий р-р обычно добавляют буферные вещества (NaHCO3,
Na2HPO4). Отбелку вискозных волокон перекисями
A—2 г/л Н2О2) проводят в слабощелочной среде
B г/л NaOH) в присутствии буферных веществ C —
5 г/л Na2Si03) при рН8—8,5 и теми-ре 60 — 70 СС.
Степень белизны волокна достигает 70 — 75%.
Отбелку вискозных волокон р-ром NaClO2 @,5 —
1,0 г/л) в кислой среде (рНЗ,5) и темп-ре 85—95 СС
проводят редко из-за сильной коррозии аппаратуры,
хотя после такой отбелки волокно не теряет прочности,
а степень его белизны достигает 90%. Полиакрилонит-
рпльные волокна обычно отбеливают р-рами Н2О2
в нейтральной среде.
После отбелки р-рами окислителей волокна тщатель-
тщательно промывают водой.
Широко применяют оптич. методы отбелки, пригод-
пригодные для всех видов химич. волокон. При этом достигают
степени белизны 98—100%. Действие оптич. отбелива-
отбеливателей основано на их способности поглощать УФ-лучи
и вместо них излучать фиолетовые и синие лучи види-
видимой части спектра. В присутствии таких отбеливателей
степень белизны волокна (особенно при освещении лу-
лучами дневного света) может оказаться больше 100%,
т. к. видимых лучей они отражают больше, чем погло-
поглощают. В качестве оптич. отбеливателей используют не-
неокрашенные производные диаминостильбендисульфо-
кислоты, кумарона, диоксикумарона, нек-рых антрахи-
нон- и нафтиламинсульфокислот, бензимидазола и др.
Эти вещества можно вводить в прядильный р-р или
расплав, а также обрабатывать ими готовые волокна.
Авиважная обработка заключается в нане-
нанесении на поверхность волокна антистатиков, мягчите-
лей, регуляторов трения, поверхностно-активных и др.
веществ, облегчающих текстильную переработку нитей
или штапельного волокна (см. также Авиважная обра-
обработка).
Сушка — обязательная стадия для волокон, под-
подвергаемых при отделке «мокрым» обработкам. Волокна
сушат в свободном состоянии или под натяжением. Во
время сушки, помимо удаления воды с поверхности и из
внутренних пор волокна, происходит фиксация струк-
структуры волокна. При сушке гидрофобных волокон (напр.,
полиамидных, полиолефиновых) вода удаляется в одну
стадию, т. к. она не связана с полимером химич. свя-
связями (т. н. «свободная» вода). Для ускорения процесса
такие волокна сушат при высоких темп-pax (выше
100 °С), верхний предел к-рых ограничен только
темп-рой термодеструкции полимера.
Сушку гидрофильных волокон (напр., вискозных,
медноаммиачных, поливинилспнртовых) проводят в две
стадии. Вначале при 100—120 °С удаляют свободную
воду, затем при 70—80 °С—«связанную» воду, к-рая об-
образует с полимером межмолекулярные связи. Содержа-
Содержание «связанной» воды достигает в гидратцеллюлозных
волокнах 35% от массы сухого полимера. Применение
относительно низкой темп-ры на второй стадии сушки
связано с тем, что при более высокой темп-ре происхо-
происходит пересыхание (т. н. «ороговение») поверхности воло-
волокон и, следовательно, резкое снижение скорости сушки.
При сушке одновременно с удалением влаги из воло-
волокон происходит уплотнение их структуры и значитель-
значительная усадка по длине, напр, на 10—15% для волокон,
высушенных в свободном состоянии. В зависимости от
условий сушки (степени натяжения волокон, темп-ры
сушильных зон и др.) молекулярная структура во-
волокон уплотняется различно. Последнее обстоятельст-
обстоятельство сильно влияет на интенсивность последующего кра-
крашения волокон. Поэтому различное натяжение волокон
и нитей, колебание темп-ры и особенно чрезмерно высо-
высокая темп-pa сушки могут повлечь за собой резко выра-
выраженную неравномерность цвета при крашении.
Обычно сушку волокон проводят в многозонных тон-
тоннельных сушильных агрегатах (сушилках), работаю-
работающих по противоточной схеме. При переходе из зоны
в зону холодный воздух постепенно подогревается и до-
достигает наивысшей темп-ры в первой (по ходу волокна)
зоне, где встречается с наиболее влажным волокном,
менее чувствительным к действию высоких темл-р.
Вытяжку сухого волокна применяют только при
получении высокопрочных волокон, т. к. при такой
обработке макромолекулы и надмолекулярные струк-
структуры полимера ориентируются вдоль оси волокон, уве-
увеличивая их прочность. Синтетич. волокна вытягивают
в 2 —10 раз на холоду или при повышенных темп-рах.
Напр., полиамидные волокна вытягивают при 20 °С
в 3,5—4,5 раза или при 120—140 СС в 5,0—5,2 раза.
При этом их прочность достигает соответственно 600—
650 и 700 — 750 мн/текс F0 — 65 и 70 — 75 гс/текс).
Полиэфирные волокна, вытянутые при 100 —120 °С
в 5 раз, достигают прочности до 700 мн/текс G0 гс/текс).
Прочность полиакрилонитрильных, поливинилсиир-
товых и полиолефиновых волокон, вытянутых при
100 —140 СС, увеличивается до 500—700 мн'текс E0—
70 гс/текс). Вытяжку проводят в одну или несколько
стадий с помощью вытяжных машин, на к-рых эта опе-
операция часто совмещается с кручением нитей. От усло-
условий вытяжки (темп-ры, скорости, кратности и равно-
равномерности натяжения) зависит не только прочность,
но и равномерность цвета волокон при последующем
крашении, а также их усадка при нагревании.
Искусственные волокна на основе целлюлозы, за
исключением ацетатных, не способны после сушки до-
дополнительно вытягиваться в сухом виде.
Термообработка придает волокнам и изде-
изделиям из них т. наз. формоустойчивость (напр., сниже-
снижение усадки и удлинения при повышенных темп-рах).
На этой стадии волокна прогревают при более высоких
темп-рах, чем те, при к-рых эксплуатируются изготов-
изготовленные из них изделия (см. также Термообработка
химических волокон).
Лит.: Груздев В. А., Пакшвер А. Б., Отделка
вискозного волокна, М., 1956; Роговин 3. А., Основы
химии и технологии производства химических волокон, 3 изд.,
т> 1 — 2, М.—Л., 1964; Пакшвер А. Б., Физико-химические
основы технологии химических волокон, М., 1972.
А. Б. Накшвер.
п
ПАСТЫ ПОЛИМЕРНЫЕ (polymer pastes, polymere
Pasten, pates polymeriques) — индивидуальные поли-
полимеры или смеси на их основе, обладающие свойствами
пластично-вязкой среды (см. Бингама тело). «Пас-
«Пасты» — эмиирич. понятие, распространяемое на любые
пластично-вязкие (тестообразные) тела вне зависимости
от их состава, структуры и назначения.
Пасты м. б. гомогенными или микрогетерогенными
(двух- или многофазными) системами. Примеры гомо-
гомогенных паст — нек-рые блоксополимеры окиси этиле-
этилена и окиси пропилена (см. Окисей органических сополи-
сополимеры), полпглпколевыс эфиры алифатич. к-т, спиртов
или алкилфенолов. Микрогетерогенные П. п.— конц.
дисперсные системы с жидкой дисперсионной средой,
в к-рых одна из фаз (среда или диспергированное в ней
вещество) образована полимером. Многие полимерные
формовочные композиции (напр., неотвержденный фао-
лит, нек-рые эпоксидные компаунды) представляют со-
собой пасты, в к-рых связующее образует дисперсионную
среду, а наполнитель — дисперсную фазу. Пасты с по-
полимерной дисперсной фазой в свою очередь подразде-
подразделяются на водные и неводные. Примеры водных паст —
коагуляты латексов некаучукоподобных полимеров,
крахмальный клейстер и др. Неводные П. п.— конц.
дисперсии полимеров в органич. жидкостях (пласти-
лоли). В технологич. практике понятие «пасты» отно-
относят гл. обр. к пластизолям (П.).
Создание П. обусловлено необходимостью переработ-
переработки в изделия плохо растворимых и нестойких при на-
нагревании полимеров. П. обладают относительно высо-
высокой текучестью при больших напряжениях сдвига
и невысоких темп-pax, что позволяет изготовлять из
них изделия относительно сложной формы. При этом
для них характерна очень высокая вязкость или даже
полная нетекучесть при низких напряжениях сдвига,
благодаря чему изготовленные изделия не теряют фор-
формы до затвердевания П. Отформованные изделия из П.
подвергают желатинизации (гелеобразованию) при на-
нагревании, в результате чего П. затвердевает во всем
объеме без нарушения однородности системы (о меха-
механизме желатинизацни см. ниже). Нек-рые пластизоли
затвердевают в результате испарения дисперсионной
среды.
Для приготовления пластизолей используют спе-
специальные марки полимеров и органич. жидкости,
Составы нек-рых пластизолей
Диспергируемый полимер
Гомо- и сополимеры винил-
хлорида
Ацетат целлюлозы
Нитрат целлюлозы
Этилцеллюлоза, бензилцел-
люлоза
Полимеры эфиров метакри-
ловой к-ты
Феноло-формальдегидная
смола
Поливинилбутираль
Политетрафторэтилен
Дисперсионная среда
Алкилфталаты, хлорированные
углеводороды и др.
Диэтилфталат, этилфталат,
этилгликолят
Диэтилфталат, дибутилфталат,
касторовое масло
Диэтилфталат, дибутилсебаци-
нат
Эфиры метакриловой к-ты
Ароматич. углеводороды
Этнленгликоль
Тяжелые бензины, вазелиновое
масло
в к-рых эти полимеры не набухают при комнатной
теми-ре, но набухают при нагревании. Наиболее часто
используют промышленные пластификаторы. Посколь-
Поскольку практически не существует таких пластификаторов,
в к-рых полимеры совершенно не набухали бы при ком-
комнатных темн-рах, при хранении П. происходит частич-
частичная желатннизация, что приводит к росту вязкости
и частичной потере текучести нластизолем. В резуль-
результате по истечении нек-рого времени, называемого жиз-
жизнеспособностью П., материал уже нельзя перерабо-
переработать по стандартной (пластизольной) технологии. Жиз-
Жизнеспособность П. зависит также от скорости седимен-
седиментации частиц полимера в пластификаторе.
Известно значительное количество различных пласти-
пластизолей (таблица), однако широкое промышленное при-
применение нашли только пластизоли на основе поливи-
нилхлорида.
Поливини л х лори дные пластизоли
Состав. Для производства поливинилхлоридных
пластизолей применяют гомо- и сополимер ы
в и н и л х л о р и д а с мол. массой 150 000 —180 000.
В производстве особопрочных изделий используют П.
на основе более высокомолекулярных полимеров. Пас-
тообразующий поливинилхлорид получают суспензион-
суспензионной или эмульсионной полимеризацией винилхлорида.
При суспензионной полимеризации может образо-
образоваться полимер с частицами двух различных морфо-
логпч. типов: 1) индивидуальные глобулярные частицы
со средним диаметром 1—3 мкм; распределение частиц
но размеру м. б. мономодальным (диаметр ок. 1 мкм)
или полнмодалъным; 2) неоднородные пористые комки
неправильной формы. П. па основе суспензионного по-
полимера первого тина обладают малой вязкостью и
жизнеспособностью до 6 мее.
Латекс, образующийся при эмульсионной полиме-
полимеризации винилхлорида, сушат распылением. При этом
частицы полимера спекаются в агломераты диаметром
5—70 мкм, представляющие собой полые сферы (цено-
сферы) и их осколки или компактные сферич. комки
(нленосферы). В ценосферах частицы сплавлены проч-
прочно, а в иленосферах они распадаются при нагревании,
что облегчает желатинизацию пластизоля.
Эмульсионный поливинилхлорид, высушенный при
высокой темп-ре, меньше набухает в пластификаторах
:"?
при хранении П., однако значительные размеры частиц,
образующихся при этом, приводят к их быстрой седи-
седиментации. Поэтому П. на основе эмульсионного поли-
полимера обладают жизнеспособностью не более 6—8 не-
недель.
На поверхности высушенных распылением частиц
полимера остаются эмульгаторы и электролиты, вво-
вводимые в полимеризационную среду для придания ей бу-
буферных свойств, а также сода, к-рая добавляется перед
сушкой в латекс для термостабилпзации. Природа
и кол-во этих веществ влияют на свойства П. Часто
в латекс перед сушкой добавляют вещества, предотвра-
предотвращающие термодеструкцию П. и замедляющие набухание
полимера. Влажность полимера не должна превышать
0,3% т. к. вода снижает жизнеспособность П. и каче-
качество изделий.
В состав П. обычно входит 40—150% пластификатора
(от массы полимера). Для приготовления П. пригодны
543
ПАСТЫ ПОЛИМЕРНЫЕ
544
обычные первичные и вторичные пластификаторы, при-
применяемые в композициях на основе поливинилхлорида
(см. Пластификаторы, Пластикат). Растворяющая
способность вторичных пластификаторов (по отноше-
отношению к ноливинилхлориду) при комнатной темп-ре
меньше, чем первичных. Поэтому приготовленные на
них П. имели бы большую жизнеспособность. Однако
вторичные пластификаторы плохо совмещаются с по-
ливинилхлоридом, что не дает возможности ввести их
в композицию в необходимых количествах. Поэтому
на практике чаще пользуются смесями первичных и
вторичных пластификаторов. Смеси обычно готовят на
основе октил- и децилфталатов, различных фосфатов,
жидких хлорированных парафинов, метилацетилрезор-
цинолеата, тетрагидрофурфурилолеата и полимерных
пластификаторов. Следует отметить, что хлорирован-
хлорированные парафины придают изделиям высокую стойкость
к к-там и щелочам.
Для регулирования вязкости П. используют разба-
разбавители или загустители. Наиболее эффективно вяз-
вязкость снижают летучими разбавителями, в к-рых поли-
полимер не набухает даже при нагревании, а также поляр-
полярными органич. летучими жидкостями, способными ча-
частично сольватировать полимер. Для этой цели приме-
применяют парафиновые, терпеновые и ароматич. углеводо-
углеводороды, спирты, диизобутил- и метилизобутилкетон. П.
с большим содержанием летучих разбавителей наз.
о р г а н о з о л я м п. Разновидностью органозолей
являются р и г и з о л и — композиции с уменьшен-
уменьшенным (обычно менее 30%) содержанием пластификато-
пластификаторов и небольшим количеством органич. разбавителей.
Для приготовления ригизолей пригоден поливинилхло-
рид с глобулярной формой частиц и пластификаторы,
образующие с ним смеси низкой вязкости (напр., фта-
латы). Рпгизоли можно перерабатывать по обычной
пластизольной технологии и при этом получать жест-
жесткие изделия. Введение небольших количеств A — 3%)
поверхностно-активных веществ, напр. полиэтилен-
гликольмоноолеата, синтанола-ДТ-7, снижает вязкость
и повышает жизнеспособность П.
Для значительного повышения вязкости П. к ним до-
добавляют гелеобразователи: мыла (соли жирных к-т и
многовалентных металлов), гидрофобизованный бенто-
бентонит и различные наполнители с высокой маслоем-
костью. Лучшим гелеобразователем считается дистеа-
рат алюминия. П., содержащие гелеобразователь, наз.
и л а с т и г е л я м и. В отличие от собственно П.,
они сохраняют приданную им форму без желатини-
зации.
Для термостабилизации П. обычно применяют те же
стабилизаторы, что и для др. материалов на основе
поливинилхлорида (см. Винилхлорида полимеры). Пред-
Предпочтение отдается жидким стабилизаторам, к-рые,
в отличие от порошкообразных, не повышают вязкости
П. Стеараты бария, кадмия и кальция редко вводят в
П., т. к. они при комнатной темп-ре со временем при-
приводят к гелеобразованию. Нек-рые барий-, кадмий-и
цинксодержащие жидкие стабилизаторы не применяют
для стабилизации поливинилхлорида, латекс которого
перед сушкой был термостабилизирован содой, посколь-
поскольку они вступают с ней во взаимодействие с образова-
образованием газообразных продуктов, приводящих к воз-
возникновению в изделиях пузырей. Серусодержащие
органич. стабилизаторы не применяют, т. к. они вы-
вызывают образование на гладкой поверхности изделий
матовых пятен.
Наполнители вводят в П. для модификации свойств
и удешевления готовых изделий. Кроме того, наполни-
наполнители могут служить для изменения вязкости П. Напр.,
2— 5% коллоидной окиси кремния (аэросила) или не-
небольшие добавки бентонитов значительно увеличивают
вязкость П. Напротив, СаСО3 и BaSO4 с малой маслоем-
костью даже при содержании ок. 20% почти не влияют
на вязкость. Часто для снижения вязкости в качестве
наполнителя применяют суспензионный поливинил-
хлорид. Влажные наполнители снижают жизнеспособ-
жизнеспособность П.
В производстве П. применяют те же пигменты, анти-
пирены, антистатики и др. добавки, к-рые вводят в др.
композиции на основе поливинилхлорида (см. Винил-
хлорида полимеры).
В нек-рых случаях в П. вводят вещества, изменяю-
изменяющие технологич. свойства материала. Так, добавление
15% порошкообразного полиэтилена снижает просачи-
ваемость П. через трикотаж, а окись кальция или
магния поглощает влагу. Ок. 1% кремнийорганич.
жидкости снижает поверхностное натяжение П. и тем
самым способствует более быстрому удалению из него
пузырей воздуха. Для придания П. адгезии к металлу
и стеклу используют олигоэфиракрилаты, диаллило-
вые эфиры с инициаторами (напр., с гидроперекисями)
и эпоксидные смолы с отвердителями (напр., с мел-
амином).
Свойства пластизолей и механизм желатинизации.
Характер течения П. может изменяться в широких
пределах в зависимости от состава, условий получения
и скорости сдвига при переработке. Поэтому реологич.
свойства иластизолей нельзя строго характеризовать
вязкостью при одной скорости сдвига (эффективной
вязкостью). Тем не менее, для практич. целей П. ус-
условно классифицируют на низковязкие [1—3 и-сек/м'1
A0—30 пз)], средневязкие [10 —15 н-сек/м2 A00—
150 пз)] и высоковязкие [100 — 1000 н-сек/м2 A000 —
10 000 пз)]. При этом вязкость измеряется при низкой
скорости сдвига, напр, при 1 сек'1.
Механизм желатинизации состоит в следующем. При
повышении темп-ры пластификатор медленно прони-
проникает в частицы полимера, к-рые увеличиваются в раз-
размере. Агломераты распадаются на первичные частицы.
В зависимости от прочности агломератов распад может
начаться при комнатной или повышенной темп-ре.
По мере увеличения темп-ры до 80 —100 °С вязкость
пластизоля сильно растет, свободный пластификатор
исчезает, а набухшие зерна полимера соприкасаются.
На этой стадии, наз. преджелатинизацией, материал
выглядит совершенно однородным, однако изготовлен-
изготовленные из него изделия не обладают достаточными физико-
механич. характеристиками. Желатинизация завер-
завершается лишь тогда, когда пластификатор равномерно
распределится в поливинплхлориде, и П. превратится
в однородное тело. При этом происходит сплавле-
сплавление поверхности набухших первичных частиц поли-
полимера и образование пластифицированного поливинил-
поливинилхлорида.
Желатинизацию характеризуют температурой, при
к-рой завершается процесс. Изделия из пластизоля,
подвергнутого нагреванию при этой темп-ре, обладают
максимальными физико-механич. характеристиками.
В зависимости от конкретных требований П. могут быть
изготовлены с высокой или низкой жизнеспособностью.
П. с высокой жизнеспособностью B—6 мес) в технике
иногда называют товарными, или специальными, П.
Их можно транспортировать на большие расстояния
и затем перерабатывать распылением, окунанием, рота-
ротационным формованием и др. методами.
Пластизоли с жизнеспособностью 2—6 нед наз. тех-
техническими. Их перерабатывают на месте изготовления
преимущественно в кожу искусственную. Материалы
с оптимальными жизнеспособностью и способностью
к желатинизации производят из ноливинилхлорида
с мол. массой 150 000—180 000. Наличие в полимере
низкомолекулярных фракций или глобул диаметром
менее 1 мкм и повышение темп-ры хранения выше
25 °С снижают жизнеспособность П.
Производство. Существует два способа производства
пластизолей: одностадийный и многостадийный.
545
ПАЧКИ
546
Одностадийный процесс осуществляют в турбосмсси-
теле при разрежении 66,6—1,33 и/м2 E-10 —1-10~2
мм рт. ст.) и вращении мешалки с частотой
800—1800 об/мин. Этим способом изготовляют П. из
суспензионного поливинилхлорида с индивидуальными
глобулярными частицами. Смеситель охлаждается во-
водой, чтобы темп-pa П. не превышала 26—28 °С. Крупно-
Крупнодисперсные компоненты предварительно измельчают
в среде пластификатора на краскотерке (см. Краски).
Длительность смешения одной партии 15—30 мин.
Многостадийный процесс используют при получении
П. на основе эмульсионного поливинилхлорида и ком-
комков суспензионного поливинилхлорида. На первой ста-
стадии полимер с жидкими компонентами и предваритель-
предварительно перетертыми крупнодисперсными стабилизаторами
и пигментами перемешивают в тихоходном смесителе.
Гомогенизацию полученной высоковязкой массы осу-
осуществляют преимущественно на трехвалковых краско-
краскотерках с охлаждаемыми валками. Для созревания П.
выдерживают при комнатной темп-ре в емкости
любой конструкции в течение 2—24 ч. Для удаления
из П. воздуха созревшую массу вакуумируют при пе-
перемешивании в планетарных смесителях со съемной
чашей (см. С'мecumели).
Переработка и применение. Пластизоли перерабаты-
перерабатывают следующими методами: макание, заливка в фор-
формы, ротационное формование, экструзия, распыление
и шпредннгование.
Макание заключается в том, что модели или изде-
изделия погружают в ванну с П., затем извлекают и нагре-
нагревают до 170—180 СС. Модель или изделие может иметь
комнатную темп-ру или быть нагрето до 80 — 180 °С.
В последнем случае за одно окунание можно получить
изделие толщиной 0,5—3 мм. Ванну с П. рекомендуется
охлаждать, чтобы темп-pa в ней не поднималась выше
25 СС, а материал периодически осторожно переме-
перемешивать. Этим способом перерабатывают П. низкой или
средней вязкости, к-рые начинают течь при достаточно
больших напряжениях сдвига (оставаясь твердыми
при малых). Они также должны обладать достаточно
высокой жизнеспособностью, т. к. время пребывания
П. в ванне может быть продолжительным.
Маканием получают перчатки, пипетки, втулки,
прокладки и др. Этим методом наносят антикоррозион-
антикоррозионные легкоснимаемые покрытия на запасные части ма-
машин и инструменты. Изделия из металлов плакируют
П., содержащим адгезив. Покрытия из П. предотвра-
предотвращают разлетание осколков при взрыве стеклянных
флаконов с аэрозолями.
Существуют два способа переработки пластизолей
заливкой в формы: заливка в открытые формы
и заливка с выливанием («обратное макание»). Этим
методом перерабатывают П. низкой или средней вяз-
вязкости, имеющие псевдопластичный или близкий к нью-
ньютоновскому характер течения. Формы для заливки
штампуют из алюминия или получают гальванич. мето-
методом из слоев серебра, никеля и меди.
Заливку в открытые формы осуществляют на кон-
конвейере, лента которого проходит вначале заливоч-
заливочную машину, а затем печь и участок охлаждения.
Способ пригоден для производства монолитных изде-
изделий. Иногда используют закрытые формы, куда П. на-
нагнетается под давлением через узкое отверстие.
При заливке с выливанием П. помещают в предва-
предварительно нагретую до 80 —100 СС форму, где выдержи-
выдерживают нек-рое время, достаточное для того, чтобы при-
пристенный слой материала образовал пленку. После этого
избыток жидкого П. сливают, а форму с прилипшей
к ней пленкой помещают в печь для желатинизации.
Готовое изделие после частичного охлаждения легко
удаляется из формы. Метод применяют для изготовле-
изготовления емкостей, сапог и др. полых изделий.
Ротационным формованием изготов-
изготовляют емкости, манекены, куклы, поплавки и др. полые
изделия. Для этого дозированную порцию П. загружа-
загружают в металлич. форму, к-рую герметично закрывают
и приводят во вращение в трех взаимно перпендику-
перпендикулярных плоскостях, одновременно нагревая в печи.
После окончания желатинизации П. форму переносят
в охлаждающую камеру для охлаждения материала.
Затем форму останавливают, открывают и извлекают
готовое изделие. Подробнее см. Ротационное фор-
формование.
Экструзией П. получают гл. обр. изоляцию
для проводов и эластичные профили. При медленной
экструзии со скоростью сдвига 10—100 сек'1 перераба-
перерабатывают П. с вязкостью 15—18 н-сек/м2 A50—180 пз).
Экструзия со скоростью сдвига 1000 —10 000 сект1
позволяет использовать П. с вязкостью 20 — 25 н-сек/м2
B00 — 250 пз) и пластигели.
Для переработки П. этим методом применяют спе-
специальные экструдеры с удлиненным шнеком, снабжен-
снабженным мелкой нарезкой. Темп-pa цилиндра экструдера
должна быть ок. 150 °С, а темп-pa на выходе из мунд-
мундштука — ок. 180 °С. Самопроизвольное вытекание П.
из машины предотвращают сеткой, установленной перед
мундштуком.
Распылением перерабатывают П. с вязкостью,
уменьшающейся от 1000 до 11 п-сек/м1 (от 10 000 до
110 пз) с ростом скорости сдвига от 0,1 до 150 сек'1.
Процесс осуществляют с помощью пневмонасосов со
степенью сжатия, напр., 24 : 1 через пистолет безвоз-
безвоздушного распыления. Распыление применяется для
нанесения покрытий, защищающих днище кузова ав-
автомобиля от коррозии и истирания, а также для изо-
изоляции от шума. На этом же оборудовании можно шпри-
шприцевать П. через пистолет в виде жгута на сварные швы
для их герметизации. Жгут желатинизируют при
130 — 140 °С.
Осуществляют также распыление П. в постоянном
электрич. поле высокого напряжения. При таком рас-
распылении частицы П. попадают в зону коронирующего
отрицательного электрода, приобретают заряд и под
действием сил электрич. поля осаждаются на противо-
противоположно заряженном электроде, роль к-рого выполняет
покрываемое изделие. При этом способе распыления
потери материала на рассеивание в воздухе значитель-
значительно снижаются.
Шпредингование — основной способ изго-
изготовления искусственной кожи из П. Он заключается
в намазывании материала на движущуюся тканевую
ленту ножом или мажущим валиком.
П. из поливинилхлорида выпускаются в различных
странах под след. торговыми названиями: д и п л а-
з о л ь (СССР), велвик декорт (Великобрита-
(Великобритания), корогель (США), д и б е н о л (ФРГ), в и-
пласт (Италия) и др.
П. низкой жизнеспособности были впервые изготов-
изготовлены в Германии в 1938, высокой жизнеспособности —
в Великобритании в 1942.
Лит.: Технология пластических масс, под ред. В. В. Кор-
шака, М., 1972, с. 112; Encyclopedia of polymer science and
technology, v. 14, N. Y.— L.— [a. o.], 1971, p. 407.
В. Л. Балакирская.
ПАЧКИ (chain-packet, Kettenbundel, chaine-paqu-
ets)— название областей ближнего порядка в располо-
расположении макромолекул аморфных полимеров, предложен-
предложенное В. А. Каргиным, А. И. Китайгородским и Г. Л.
Слонимским в рамках их гипотезы о строении поли-
полимеров A957). Эта гипотеза, получившая широкое рас-
распространение, основана на предположении о том, что
в аморфных полимерах, как и в низкомолекулярпых
547
ПЕКТИНОВЫЕ ВЕЩЕСТВА
548
жидкостях, существует нек-рая упорядоченность в
расположении соседних молекул (рои молекул).
Упорядоченность цепных макромолекул должна про-
проявляться в близости ориентации соседствующих отрез-
отрезков макромолекул, длина к-рых определяется степенью
гибкости цепей.
В пользу существования П. свидетельствуют: плот-
плотность полимерных тел, к-рая значительно больше допу-
допустимой с точки зрения представлений о хаотич. располо-
расположении макромолекул; стремление систем макромолекул
к упорядочению; происходящая во многих полимерах
быстрая кристаллизация, невозможная в хаотич. си-
системе макромолекул (поскольку перегруппировки мак-
макромолекул, как это следует из скоростей механич.
релаксационных явлений, очень замедленны).
Особенностями П. по сравнению с низкомолекуляр-
низкомолекулярным роем должны быть большие размеры, обусловлен-
обусловленные значительной длиной макромолекул, и большая
длительность существования, определяемая подвиж-
подвижностью макромолекулы в целом, к-рая намного меньше,
чем у малой молекулы. Длительность жизни П. в раз-
разных физических состояниях полимеров должна быть
весьма различна: меньше всего в вязкотекучем состоя-
состоянии и в конц. р-рах полимеров и сколь угодно велика
в стеклообразных полимерах (конечно, в отсутствие
внешних воздействий, способных изменить структуру
полимера).
Являясь флюктуационным образованием, П. (в от-
отличие от мицеллы) не может рассматриваться как час-
частица, обладающая поверхностью раздела с окружаю-
окружающей средой, т. е. как частица другой фазы. Между
тем, наличие упорядоченности, большие размеры и
продолжительность жизни П. не позволяют рассмат-
рассматривать аморфные полимеры и их р-ры только как гомо-
гомогенные однофазные системы. В ряде процессов, более
кратковременных, чем срок жизни П., и затрагивающих
объемы вещества, сравнимые с объемом П., однофазные
аморфные полимеры необходимо рассматривать как
микрогетерогенные системы и даже учитывать наличие
«поверхностей раздела» между П. Особенно ярко это
проявляется в стеклообразных полимерах.
Кристаллизация полимеров должна, согласно па-
пачечной гипотезе, начинаться внутри П., внутренняя
упорядоченность к-рых обеспечивает большую скорость
фазового превращения. После кристаллизации П. ста-
становится частицей кристаллич. фазы и приобретает,
естественно, поверхность раздела с окружающей аморф-
аморфной полимерной средой (не успевшей или не способной
закристаллизоваться). Тем не менее такие кристаллы
иногда наз. кристаллическими П. Дальнейшая кристал-
кристаллизация происходит путем построения более сложных
форм многоступенчатой надмолекулярной структуры
полимеров.
При растворении аморфного полимера малые моле-
молекулы растворителя должны легче проникать в менее
упорядоченные области между П., что может приво-
приводить к распаду полимерного тела при переходе в р-р
на агрегаты П. и даже отдельные П. Вследствие гибко-
гибкости макромолекул П. могут принимать различные внеш-
внешние формы и изменять их со временем. В зависимости от
природы растворителя может также происходить огра-
ограниченное или неограниченное (вплоть до разделения на
макромолекулы) набухание П. Если скорость распада
тела на П. намного больше скорости их распада на
макромолекулы, то образуется коллоидная полимер-
полимерная система, в к-рой длительно существующие П. в пе-
период их жизни по всем свойствам должны быть анало-
аналогичны мицеллам. При других соотношениях скоростей
упомянутых процессов образуется молекулярный р-р
полимера, обладающий тем не менее, даже при сравни-
сравнительно низких концентрациях, пачечным строением
вследствие флюктуационных изменений упорядочен-
упорядоченности.
В ряде случаев (напр., когда достаточно длинные
участки линейных макромолекул различаются по хи-
мич. составу или когда в разветвленных макромолеку-
макромолекулах химич. состав ответвлений отличается от состава
основной цепи) межмолекулярное взаимодействие мо-
может приводить к упорядоченным агрегатам макромоле-
макромолекул с практически неограниченным сроком существо-
существования как в самом полимере, так и в его р-рах. Устой-
Устойчивость таких агрегатов связана с образованием у них
поверхностей раздела с окружающей средой, если
снаружи агрегата располагаются участки макромоле-
макромолекул, отличающиеся по химич. составу от участков,
расположенных внутри агрегата. Подобного рода
структурные элементы являются переходной формой
между П. и мицеллами.
Лит.: Каргин В. А., Китайгородский А. И.,
Слонимский Г. Л., Колл. журн., 19, № 2, 131 A057);
Каргин В. А., Слонимский Г. Л., Краткие очерки
по физико-химии полимеров, 2 изд., М., 1967. Г. Л. Слонимский.
ПЕКТИНОВЫЕ ВЕЩЕСТВА (pectines, Pektine,
matieres pectiques) — высокомолекулярные углеводы
растительного происхождения, главным структурным
компонентом к-рых является D-галактуроновая к-та:
н
ОН
соосн3
соон
н
н
соон
П. в. встречаются в тканях наземных растений и в
нек-рых водорослях; их содержание в одних расте-
растениях может достигать 30% от сухого вещества (напр.,
в белой части кожуры цитрусов), в других не превы-
превышать долей процента. К П. в. относятся: пектовая
кислота — поликислота, построенная из остатков
D-галактуроновой к-ты, связанных а-1,4-гликозидными
связями в длинные цепи (является основой всех П. в.);
пектаты — соли пектовой к-ты; пектины —
продукты этерификации метиловым спиртом пектовой
к-ты по карбоксильным группам (растворяются в воде
с образованием плотных гелей); пектинаты—
соли пектинов; протопектины — не раствори-
растворимые в воде вещества, представляющие собой сшитые
пектины.
В растениях П. в. присутствуют преимущественно
в виде протопектина, к-рый содержится гл. обр. в стен-
стенках растительной клетки, в межклеточном цементирую-
цементирующем материале, играя роль опорных элементов тканей.
В клеточном соке содержатся пектины и пектинаты.
При извлечении пектинов из растительного материала
протопектин разрушают горячей соляной к-той, и об-
образующийся пектин осаждают спиртом. Пектины раз-
различных растений характеризуются неодинаковой мол.
массой, степенью этерификации карбоксильных групп,
распределением метильных групп. В П. в. могут при-
присутствовать в небольших количествах D-галактоза,
L-арабиноза и др. моносахариды. Пектины довольно
стойки к действию к-т, под действием щелочей раз-
разрушаются; легко подвергаются окислительному рас-
расщеплению.
Ферменты, действующие на пектины, делятся на
п ектинэстеразы (старое название — пекта-
зы), гидролизующие сложноэфирные группировки, от-
отщепляя метильные группы, иполигалактуро-
н а з ы (старое название — пектиназы), расщепляю-
расщепляющие цепь до олигоуронидов и далее до D-галактуроно-
D-галактуроновой к-ты. Эти ферменты, различные представители
к-рых встречаются в микроорганизмах и растениях,
используют для расщепления П. в. при осветлении
фруктовых соков.
Количественно пектины определяют по объему С02,
образующегося при нагревании их с к-той; спектрофо-
549
ПЕНОПЛАСТЫ
550
тометрически — по реакции с карбазолом и др. Про-
Промышленные препараты характеризуют вязкостью пекти-
пектинов и их способностью образовывать гели.
В пром-сти пектины получают из кожуры цитрусов,
яблок и кормового арбуза. П. в. применяют в пищевой
пром-сти (гл. обр. при получении мармелада и дже-
джемов), а также в фармацевтич. пром-сти как гелеобра-
зователи.
Лит.: ХенглейнФ., в кн.: Биохимические методы
анализа растений, пер. с нем., М., 1960; Comprehensive bioche-
biochemistry, ed. M. Florkin, E. H. Stotz, v. 5, N. Y.— L., 1963;
Industrial gums, ed. R. L. Whistler, N. Y.— L., 1959; Пектино-
Пектиновые вещества и ггектолитические ферменты, М., 1971 (Итоги
науки, сер. Биохимия, под ред. В. Л. Кретовича); Химия угле-
углеводов, Библиографический указатель A965—1968), М., 1971,
с. 277. См. также лит. к ст. Полисахариды. Л.И.Линевич.
ПЕНОПЛАСТЫ (foamed plastics, Schaumkunststoffe,
mousses plastiques) — вид газонаполненных пластмасс.
Последние обычно подразделяют на П. и поропла-
сты. К П. относят материалы, в к-рых газ заполняет
несообщающиеся между собой ячейки; к поропластам —
материалы, в к-рых заполненные газом полости сооб-
сообщаются между собой. Такое деление газонаполненных
пластмасс условно, поскольку практически не удается
получить материал только с открытыми или только
с закрытыми ячейками, причем даже для одного типа
материала соотношение открытых и закрытых пор м. б.
очень различным. В настоящей статье рассмотрены как
П., так и поропласты.
К П. относят также пластики с полым наполните-
наполнителем, к-рые, в отличие от остальных П., получают без
вспенивания (см. ниже). Эти материалы имеют только
закрытую структуру ячеек.
П. условно делят на легкие (высоковспененные) с ка-
кажущейся плотностью до 500 кг/м3 и облегченные (час-
(частично вспененные, низковспененные, подвспененные)
с кажущейся плотностью 500—800 кг/м3. Различают
также эластичные, полужесткие и жесткие П.
Получение. Технология получения П. состоит из
операций приготовления композиции, введения газо-
газовой фазы в полимерную среду (чаще всего путем вспе-
вспенивания), придания вспененной массе необходимой
формы с последующей ее фиксацией. Операция фор-
формообразования может предшествовать процессу вспе-
вспенивания.
В процессе приготовления композиции совмещают
ингредиенты пластич. массы или резиновой смеси
(форполимеры, олигомеры, полимеры, отвердители, ка-
катализаторы, пластификаторы, красители, стабилиза-
стабилизаторы, наполнители, газообразователи и т. д.).
Для введения газовой фазы в полимерную среду при-
применяют след. способы: 1) смешивание композиции, на-
находящейся в вязкотекучем состоянии, с газом при нор-
нормальном давлении (механич. вспенивание); 2) насыще-
насыщение композиций, находящихся в вязкотекучем состоя-
состоянии (расплавы полимеров, олигомеры, пасты, сырые
резиновые смеси), газом при высоком давлении; 3) на-
насыщение композиций легкокипящими жидкостями,
к-рые при нагревании превращаются в пар; 4) введе-
введение в композицию веществ (газообразователей), разла-
разлагающихся при нагревании с выделением большого коли-
количества газообразных продуктов; 5) совмещение компо-
компонентов, взаимодействующих между собой с выделением
большого количества газообразных продуктов; 6) по-
получение заготовок прессованием из композиций, напол-
наполненных растворимыми в воде веществами, впоследствии
вымываемыми; 7) спекание неуплотненных порошкооб-
порошкообразных материалов или пористых заготовок, полученных
из порошков прессованием в холодных формах.
При механич. вспенивании в р-ры олигомеров или
полимеров вводят поверхностно-активные в-ва, способ-
способствующие равномерному распределению в объеме ма-
материала пузырьков газа и обеспечивающие устойчи-
устойчивость пены в течение времени, достаточного для от-
отверждения смолы. Вспенивание осуществляют в верти-
вертикальных цилиндрич. аппаратах, снабженных многоло-
многолопастными мешалками. В нижнюю часть аппарата при
работающей мешалке подается сжатый воздух. Взбитая
пена через отверстие в днище аппарата сливается в фор-
формы, в к-рых происходят сушка и отверждение (затвер-
(затвердевание) блоков П. Полученные рассмотренным спосо-
способом материалы имеют преимущественно открытую
структуру ячеек, т. к. растворитель, удаляясь в про-
процессе сушки и отверждения из стенок ячеек, разру-
разрушает их. Этим методом получают П. преимуществен-
преимущественно из карбамидных смол и поливинилхлорида (см.
Мипора, Пенополивинилхлорид).
Газом при высоком давлении насыщают резиновые
смеси, расплавы полимеров и полимерные насты (су-
(суспензии полимеров в пластификаторе или мономере).
Предварительно резиновой смеси экструдированием
или каландрованием придается необходимая форма,
полимерные пасты (напр., пластизоли) заливаются в
открытые формы. Подготовленные полуфабрикаты
помещают в автоклав, где производится насыщение
их газом (N2, CO2) под давлением. Величина давле-
давления зависит от вязкости полуфабриката. Полуфабри-
Полуфабрикаты резиновой смеси насыщают при давлении
до 30 Мн/м" C00 кгс/см2), полимерных паст — 3 Мн/м2
C0 кгс/см2). Под давлением происходит растворение
газа в композиции до образования насыщенного р-ра.
При последующем повышении теми-ры, необходимом
для вулканизации резиновой смеси или желатини-
зации полимерных паст, и сбросе давления раство-
растворимость газа в композиции резко снижается и он на-
начинает выделяться в виде газовых пузырьков, равно-
равномерно распределенных по объему композиции. При
этом объем композиции увеличивается. В зависимос-
зависимости от степени насыщения и режима вспенивания по-
получают П. преимущественно с открытой или закры-
закрытой структурой ячеек.
Насыщение расплавов полимеров газом и получение
из них вспененных изделий производят на червячных
литьевых машинах и в экструдерах. Расплав насыщают
газом в материальном цилиндре литьевой машины или
экструдера, в к-рый инертный газ подают через кана-
каналы в шнеках под давлением до 150 кгс/см2. При литье
под давлением порция насыщенного газом расплава
полимера впрыскивается в холодную литьевую фор-
форму, где вследствие резкого снижения давления проис-
происходит выделение газообразной фазы и вспенивание.
Экструдер, предназначенный для получения газона-
газонаполненных изделий, снабжается формообразующей
и калибрующей головками, геометрически подобными,
но имеющими различные размеры. Температурный ре-
режим строят т. обр., чтобы выходящий из формообразую-
формообразующей головки экструдат находился в высокоэластич.
состоянии. Вспенивание изготовленных профилей мо-
может проходить вне экструдера при нагреве отформо-
отформованного изделия или на выходе из головки экструдера.
В первом случае получается, как правило, П. низкой
плотности со смешанной структурой ячеек и пор, во
втором — с более высокой плотностью и закрытой
структурой ячеек. Для предотвращения вытягивания
пены при выходе из экструдера внутри нее протягива-
протягивается усиливающая сердцевина.
В производстве кабельных изделий такой сердцеви-
сердцевиной служит металлич. проводник. При этом рецептура
и режим получения вспененного на проводнике покры-
покрытия выбираются такими, чтобы обеспечить преимуще-
преимущественно закрытую структуру ячеек. При изготовле-
изготовлении более плотных пен одностадийным вспениванием
не требуется усиливающей сердцевины, но необхо-
необходима повышенная скорость экструзии и надежные при-
приемные устройства, не тормозящие выход материала из
головки.
Литьем под давлением и экструзией получают П.
с преимущественно закрытой структурой ячеек и на-
551
ПЕНОПЛАСТЫ
552
ружнон монолитной оболочкой (см. Пеиополивинилхло-
рид, Пенополиорганосилоксаны).
Насыщение термопластичных полимерных материа-
материалов, имеющих форму гранул, легкокипящими жидко-
жидкостями производится в процессе их получения, напр,
при суспензионной полимеризации. Мономер и легко-
кипящую жидкость (изопентан, метиленхлорид) под-
подбирают т. обр., чтобы жидкость растворяла мономер,
но не растворяла образующийся полимер. Выделение
легкокипящей жидкости в виде отдельной фазы проис-
происходит в момент превращения капелек мономера в по-
полимер. Поэтому в образующихся гранулах полимера
появляются вкрапления равномерно распределенных
капелек легкокипящей жидкости.
Насыщенные т. обр. гранулы засыпают в перфориро-
перфорированные стальные ограничительные формы и нагревают
насыщенным водяным паром до темн-ры, превышающей
темп-ру стеклования полимера. При этом происходит
вспенивание гранул под давлением пара, образовавше-
образовавшегося из низкокинящей жидкости. Кажущаяся плот-
плотность получаемых П. регулируется степенью заполне-
заполнения ограничительной формы или содержанием низко-
кпиящей жидкости в гранулах.
Для получения П. более равномерной структуры
гранулы подвергают предварительному иодвспенива-
нию в среде водяного пара до насыпной массы, равной
требуемой кажущейся плотности П. После подсушки
подвсиененыые гранулы засыпаются в перфорирован-
перфорированные формы, где производят их окончательное вспени-
вспенивание в среде водяного пара при темп-ре ок. 100 °С
и спекание. Получаемые П. имеют смешанную струк-
структуру ячеек (см. Пенополистирол).
С помощью газообразователей получают П. из термо-
термопластичных и термореактивных полимерных материа-
материалов. Изготовление П. из термопластов производят обыч-
обычным литьем, прессованием, вальцеванием, литьем под
давлением, экструзией. При изготовлении П. обычным
литьем применяют мономеры или полимер-мономерные
пасты, в к-рых растворяют газообразователь. Полу-
Полученный компаунд заливают в форму и производят поли-
полимеризацию его при темп-ре ниже темп-ры разложения
газообразователя. После этого блок полимера, являю-
являющийся по форме миниатюрой будущего изделия, из-
извлекают из формы и нагревают до темп-ры выше темп-ры
стеклования полимера и темп-ры разложения газооб-
газообразователя. Очень важно, чтобы разложение газооб-
газообразователя происходило в тот момент, когда полимер
находится в высокоэластичном состоянии; в против-
противном случае может произойти разрушение полимер-
полимерной фазы.
Под действием выделяющихся при разложении газо-
газообразователя газов блок высокоэластич. полимера рав-
равномерно увеличивается в размерах во всех направле-
направлениях. Вспененная заготовка охлаждается до комнатной
темп-ры для фиксации формы и предотвращения усадки
изделия в результате ухода газа через стенки ячеек.
Такой способ изготовления изделий из П. наз. мас-
масштабным формованием.
Миниатюры для масштабного формования можно
также получать прессованием из порошкообразных тер-
термопластов или полимер-мономерных паст. Компоненты
полимерного материала тщательно перемешивают с по-
порошкообразным газообразователем в шаровых мель-
мельницах или лопастных мешалках. Полученную компо-
композицию загружают в прессформу закрытого типа, в
к-рой при темп-ре, превышающей темп-ру плавления
полимера, формуется монолитный блок необходимой
конфигурации. Выделяющийся при разложении газо-
газообразователя газ равномерно распределяется и раст-
растворяется в расплаве полимерного материала. После
прессования заготовка охлаждается под давлением до
комнатной темп-ры и извлекается из формы. Т. к. газ
внутри такой заготовки находится под высоким давле-
давлением, масштабное формование изделия из нее должно
быть произведено не позже чем через 1 — 2 су т.
Плоские заготовки, полученные прессованивхМ, мож-
можно перерабатывать методом «самоформования» в газо-
газонаполненные изделия самой разнообразной конфигура-
конфигурации. Для этого заготовку закрепляют но контуру при-
прижимной рамкой над ограничительной формой соответ-
соответствующей конфигурации. При нагревании заготовки
водяным паром до темп-ры, превышающей темп-ру
стеклования полимера, происходит ее вспенивание.
Увеличению размеров заготовки в плоскости препятст-
препятствует прижимная рамка, поэтому заготовка искривля-
искривляется и, расширяясь, достигает стенок ограничительной
формы. Небольшое избыточное давление на заготов-
заготовку сверху препятствует ее вспучиванию вверх. Фик-
Фиксация формы изделия осуществляется охлаждением
до комнатной темп-ры (см. Пеиополистирол).
Заготовки в виде листов из полимер-мономерных
паст получают вальцеванием. Затем листы помещают
в герметичные формы и прогревают до завершения
процесса полимеризации. Полученные при этом моно-
монолитные заготовки вспенивают в ограничительных фор-
формах при темп-ре выше темп-ры стеклования полимера
(см. Пенополивинилхлорид).
Порошкообразные термопласты, совмещенные с га-
газообразователем, а также пластмассы, имеющие форму
гранул, поверхность к-рых онудривается газообразо-
газообразователем, м. б. переработаны в П. литьем под давлением
или экструзией. Разложение газообразователя и насы-
насыщение газом расплава полимера происходят в мате-
материальном цилиндре соответствующих машин. Насыщен-
Насыщенный газом расплав впрыскивается в литьевую форму
или выдавливается через головку экструдера так же,
как в рассмотренном выше методе насыщения распла-
расплавов сжатым газом.
П. из термореактивных пластмасс изготавливают
вспениванием полуфабриката в ограничительной форме.
Полуфабрикат получают, смешивая твердые и плавкие
компоненты (см. Reno фенопласты и Пенополиорганоси-
Пенополиорганосилоксаны) на вальцах или в шнековых смесителях; в
случае вязких компонентов (см. Пеноэпоксиды) исполь-
используют лопастные смесители.
Если полуфабрикат изготавливают из твердых тер-
термореактивных смол, то после смешения компонентов
на вальцах получают жесткую пленку толщиной
2—4 мм. Пленку дробят, а затем экструдируют через
головку, позволяющую получать полуфабрикат в виде
монолитного или полого прутка. Прутки разрезают
затем на гранулы различной длины (шприцованный
полуфабрикат). Для получения П. с заданной кажу-
кажущейся плотностью в ограничительную форму загру-
загружают полуфабрикат с насыпной массой, равной кажу-
кажущейся плотности будущего изделия. Ограничительная
форма нагревается сначала до темп-ры вспенивания
(темп-ры перехода полуфабриката в вязкотекучее
состояние и разложения газообразователя), а затем до
темп-ры отверждения смолы. Обычно нагревание про-
проводят с такой скоростью, чтобы нарастание вязкости
отверждающейся смолы несколько опережало разложе-
разложение газообразователя. Это способствует получению П.
преимущественно с закрытой структурой ячеек. В про-
процессе вспенивания внутри ограничительной формы раз-
развивается давление, достигающее 0,3—0,5 Мн/м2 C—
5 тс/см2). Поэтому ограничительные формы должны
быть достаточно жесткими. Композиции, составлен-
составленные из жидких термореактивных смол, совмещенных
с газообразователем, поверхностно-активным вещест-
веществом и др. компонентами, вспениваются и отверждают-
ся также в ограничительных формах (см. Пенофено-
пласты, Пеноорганосилоксаны, Пеноэпоксиды).
При смешении компонентов нек-рых заливочных
компаундов, находящихся в вязкотекучем состоянии
(см. Пенополиуретаны, 11 енофенопласты), или поли-
553
ПЕНОПЛАСТЫ
554
мерных наст (см. Пенополивинилхлорид) происходит
выделение газообразных продуктов реакции (напри-
(например, СО2, Н2), вспенивающих массу. Процесс изготов-
изготовления П. из таких композиций м. б. одно- и двухстадий-
ным. Одностадийный способ предусматривает одновре-
одновременное смешивание всех компонентов в смесителе
(якорные, пропеллерные, планетарные скоростные ме-
мешалки) и последующее вспенивание, в частности в ог-
ограничительной форме, к-рое начинается спустя 10—
15 сек после смешения и заканчивается за 1—2 мин.
Длительность отверждения пены определяется обт^е-
мом массы и ее химическим составом и колеблется
от нескольких ч до нескольких су т. Вспенивание и
отверждение могут происходить при обычной и повы-
повышенной темп-ре.
При двухстадийном способе сначала изготавливают
форполимер, а затем производят вспенивание в резуль-
результате взаимодействия компонентов композиции с
нек-рыми низкомолекулярными веществами (напр.,
водой) и отверждение.
Произ-во П. из таких компаундов осуществляется
непрерывным способом, заливкой в формы или напы-
напылением на изделие. Непрерывный способ предназначен
для произ-ва пен в виде листов, блоков или полотен.
Напылением создают тепло- или звукоизолирующие
покрытия на готовых изделиях (трубопроводах, холо-
холодильных установках, строительных панелях, контей-
контейнерах и т. п.).
Для получения П. прессованием заготовок из термо-
термопластов и реактопластов, наполненных растворимыми
в воде веществами, вначале тщательно смешивают по-
порошкообразный полимер с порошком растворимого ве-
вещества. Затем полученную композицию спрессовывают
в монолитный блок в иреесформах, нагреваемых до
темп-ры плавления полимера или отверждения смолы.
Блок охлаждают до комнатной темп-ры и помещают
в нагретую воду, к-рая вымывает находящиеся в поли-
полимере водорастворимые вещества. После этого блок по-
ропласта высушивают.
Методом спекания пористых заготовок получают
поропласты из полимеров, темп-pa плавления к-рых
лежит выше темп-ры их деструкции (напр., из фторо-
пласта-4). В этом случае порошкообразный полимер
уплотняют в холодных формах в таблетки определен-
определенной конфигурации и плотности. Затем таблетки спе-
спекают в нечах в пористые заготовки. Порошкообразные
частички соединяются одна с другой в процессе спе-
спекания только в местах контакта.
Для повышения жесткости и прочности П. арми-
армируют листовыми материалами (фанерой, металлом,
слоистыми пластиками), металлич. прутками, проволо-
проволокой, сеткой, сотами.
Свойства. П. обладают лучшими теплоизоляционны-
теплоизоляционными свойствами, чем традиционные теплоизолирующие
материалы. Природа полимера незначительно влияет
на величину коэфф. теплопроводности. В большей мере
этот показатель зависит от природы газа, заполняю-
заполняющего ячейки. Лучшими теплоизоляционными свойст-
свойствами обладают П., имеющие преимущественно закры-
закрытую структуру ячеек.
Природа полимера мало влияет также на диэлектрич.
проницаемость П., но значительно сказывается на тан-
тангенсе угла диэлектрич. потерь. Диэлектрпч. показатели
зависят также от природы др. компонентов, входящих
в композицию (газообразователей, поверхностно-ак-
поверхностно-активных веществ и т. п.).
П. с высоким содержанием закрытых ячеек имеют
низкие показатели влаго- и водопоглощения. Наиболее
быстро они поглощают воду в первые 7 —10 сут,
далее поглощение увеличивается очень медленно.
Для П. с открытой структурой ячеек характерно нара-
нарастание водопоглощения во времени. П. с высоким со-
содержанием закрытых ячеек хорошо сохраняют перво-
первоначальную плавучесть при пребывании в воде в тече-
течение нескольких лет.
П. с малой кажущейся плотностью имеют удовлет-
удовлетворительные механич. показатели, к-рые зависят не
только от свойств материала стенок ячеек, но и от
размеров и формы ячеек. Поэтому для оценки механич.
свойств П. не всегда легко выбрать правильный кри-
критерий. Прочность большинства жестких П. имеет
вполне определенную величину только при растяжении,
когда в процессе испытания происходит разрыв образ-
образца. Сжатие П. в большинстве случаев не приводит
к хрупкому разрушению всего образца при опреде-
определенной нагрузке. Поэтому для характеристики проч-
прочности П. в этом случае определяют нагрузку, вызы-
вызывающую заданную относительную деформацию ма-
материала.
П. обычно обладают анизотропией механич. свойств,
обусловленной в основном вытянутой формой ячеек
и ориентацией их стенок в направлении течения компо-
композиции при вспенивании. Степень анизотропности зави-
зависит от технологии получения П. Свободное вспенива-
вспенивание композиции приводит к образованию направлен-
направленных ячеистых структур. Вспенивание в замкнутых
объемах позволяет получать П. с более изотропными
свойствами. Для ряда П. различие в прочностных ха-
характеристиках в направлении вспенивания и в перпен-
перпендикулярном направлении составляет 25—30% .
Химич. стойкость П. и их сопротивляемость дейст-
действию открытого пламени в значительной мере опреде-
определяются природой полимера. Огнестойкость П. м. б.
улучшена введением в композицию специальных цла-
мягасящпх добавок.
Применение. В качестве легкого конструкционного
и теплоизоляционного материала П. широко исполь-
используются для изготовления двух- и многослойных конст-
конструкций. Такие конструкции применяются в качестве
несущих и навесных стеновых панелей и перегородок,
а также для защитной и декоративной облицовки. Наи-
Наиболее пригодны для этих целей пенополиуретаны, пено-
фенопласты и пенополистирол. Как теплоизоляционный
материал, П. широко используются при изготовлении
домашних холодильников, морозильных установок,
рефрижераторов, газоразделительных установок, раз-
различных емкостей.
В радиотехнике и электронике П. применяются для
электроизоляции узлов в приборах, каисулирования и
герметизации деталей, модулей, блоков, для изготовле-
изготовления антенных обтекателей, коаксиальных кабелей.
Низкое водопоглощение П. позволяет использовать
их при изготовлении плавучих средств (буи, бакены,
понтоны), спасательных средств (плотики, пояса, на-
нагрудники), легких лодок, катеров.
П. с полностью открытыми ячейками нашли примене-
применение для фильтрации газов и жидкостей. П. широко при-
применяют также в качестве упаковочных материалов.
Эластичные П. используются как амортизационный
материал для изготовления мягкой мебели (подушки
сидений, спинки, валики). Эластичные пенополиурета-
пенополиуретаны в виде листов используются для дублирования
с тканями, идущими на пошив теплой одежды, а также
в качестве подкладки под ковры. Амортизирующие,
вибродемифирующие и звукоизоляционные свойства
эластичных полиуретановых и поливинилхлоридных П.
позволяют применять эти материалы в качестве про-
прокладок в касках и шлемах, в защитных рукавицах
при работе с отбойными молотками, в вентиляционных
установках и различных глушителях.
Перспективно применение П. для структурирования
тяжелых и дренажа болотистых почв, удержания влаги
в почвах, укрепления песчаных грунтов, в качестве
искусственной почвы.
В 1964 мировое произ-во П. составляло 4 — 5% от об-
общего произ-ва пластмасс, в 1970 оно возросло до 6 — 7%
555
ПЕНОПОЛИВИНИЛХЛОРИД
556
A,8—2,1 млн. т) и существует тенденция к его дальней-
дальнейшему увеличению. См. также Губчатые резины.
Лит.: Берлин А. А., Основы производства газонапол-
газонаполненных пластмасс и эластомеров, М., 1954; Романеи ков
И. Г., Физико-механические свойства пенистых пластмасс,
М., 1970; Справочник по пластическим массам, т. 2, М., 1969;
В en n ing С. J., Plastics foams, v. 1 — 2, N. Y. —
[a. o.J, 1969; Кауфман Б. Н., К о с ы р е в а 3. С,
Шмидт Л. М., Яхонтова Н. Е., Строительные поро-
пласты, М., 1965; Пенопласты в машиностроении. Сб. ст., М.,
1962; Пенопластмассы, сб. статей, под ред. А. А. Моисеева,
В. В. Павлова и М. Я. Бородина, М., 1960; ЮдинЕ. Я.
[и др.], Звукопоглощающие и звукоизоляционные материалы,
М., 1966; Воробьев В. А., Андрианов Р. А., По-
Полимерные теплоизоляционные материалы, М., 1972; Алек-
Александров А. Я., Бородин М. Я., Павлов В. В.,
Конструкции с заполнителями из пено пластов, М., 1972; Химия
и технология вспененных пластмасс (труды ин-та), Владимир,
1970; Bender R. J., Handbook of foamed plastics, Liber-
tyville, 1965; HomannD., Kunststoffschaumstoffe, Munch.,
1966; Вспененные пластические массы, Каталог, Черкассы,
1973. В. М. Виноградов.
ПЕНОПОЛИВИНИЛХЛОРИД [poly(vinyl chloride)
foam, Polyvinylchloridschaumstoff, mousse de chlo-
rure de polyvinyle] — газонаполненный материал на
основе поливинилхлорида или сополимеров винил-
хлорида.
Состав. В производстве П. используется эмульсион-
эмульсионный поливинилхлорид (напр., отечественных марок
Е-62, Е-66П), иногда смесь его с перхлорвиниловой
смолой (хлорированный поливинилхлорид) или сопо-
сополимеры винилхлорида с метилметакрилатом и др. Ввиду
низкой темп-ры деструкции поливинилхлорида и от-
отсутствия текучести ниже этой темп-ры, что создает боль-
большие трудности при формовании П., обязательным ком-
компонентом смеси являются пластификаторы (напр.,
дибутилфталат, трикрезилфосфат, диоктилфосфат) и
мономеры (метилметакрилат и т. п.). Такие компози-
композиции наз. полимер-мономерными пастами (пластизоля-
ми). Соотношение пластификатора и мономера опре-
определяет степень жесткости П.
В качестве газообразователей применяют 2,2'-азо-
б^с-изобутиронитрил, тг,гг'-окси-б^с-бензосульфогидра-
зид, азодикарбонамид в сочетании с активаторами раз-
разложения (карбамид, стеараты или бензоаты цинка, кад-
кадмия, свинца и др.), бикарбона-
бикарбонаты натрия и аммония и др. В
нек-рых случаях вспенивающим
агентом являются легкокипящие
жидкости (напр., фреоны) или
газы, к-рыми предварительно
насыщают композицию.
Способы изготовления. При
рид. Экструзией можно изготавливать эластичный и по-
полужесткий П., известный в СССР под названием «пено-
эласт». Пена, выходящая из головки экструдера, в
этом случае не требует специальной поддержки или
усиления сердцевины. Легкость и конструкционную
прочность П. придает модификация поливинилхло-
поливинилхлорида каучуком.
Промышленное значение имеет экструзионныи метод
изготовления легких жестких П. с применением так
называемых временных пластификаторов (легколетучих
соединений, напр. ацетона), удаляемых из материала
в процессе его получения. П., производимый этим спо-
способом, имеет преимущественно закрытую структуру
ячеек.
Большое распространение получил способ изготовле-
изготовления из поливинилхлоридных композиций тонких вспе-
вспененных эластичных покрытий типа кожи искусствен-
искусственной. Они готовятся из пластизолей (см. Пасты поли-
полимерные) или каландрированных пленок с применением
газообразователей. По этому способу получают П.
со смешанной структурой ячеек.
Насыщение газами или низкоки-
п я щ и м и жидкостями. Пластизоль насыща-
насыщается азотом, двуокисью углерода, фреонами и после
этого выдавливается через сливные устройства на
транспортер, где вспенивается, желатинизуется и сплав-
сплавляется.
Этим способом в СССР получается эластичный и жест-
жесткий П., известный под названием «винипор».
Эффективный способ изготовления эластичного П.—
механическое вспенивание. Этот спо-
способ не получал промышленного развития из-за высокой
кажущейся плотности образующихся пен, пока не бы-
были подобраны поверхностно-активные вещества, позво-
позволяющие придать пене легкость и стабильность. В каче-
качестве таких веществ применяют соли щелочных метал-
металлов, кремнийорганич. соединения и др.
Вспенивание под действием га-
газов, выделяющихся при реакции
между компонентами смеси. Этим ме-
методом можно получать как эластичный, так и жесткий
Таблица 1. Свойства жесткого пенополивинилхлорида отечественных марок
Показатель
ПХВ-1
ПХВ-2
ПВ-1
Винипор (по-
(полужесткий)
структура
Преобладаю щая
ячеек
Кажущаяся плотность, кг/м3
получении П. используют боль- Прочность, Мн/м* (кгс/см*)
при растяжении
сжатии
Модуль упругости, Мн/м2
(кгс/см2)
при растяжении . ,
сжатии
проницаемость
шинство способов, применяемых
в производстве пенопластов.
Вспенивание газо-
образователями осу-
осуществляют при изготовлении
жесткого и эластичного П. пре- Ударная вязкость, кдж/м*
имущественно с закрытой струк- ди^л^ктрич!
турой ячеек. П. формуют прес- приЮ6гг|.
сованием или экструзией. Прес- Тангенс угла ^диэлектрич. по-
сованием получают заготовки эл^ктричПрочность, кв/мм '. '.
при 170 С и давлении около Коэфф. теплопроводности,
\Ъ Мн/м2 A50 кгс/см2). Моно-
Монолитная заготовка вспенивается
при повторном нагревании в
ограничительной форме при
100—110 °С, т. е. в зоне высо-
высокоэластичных деформаций поли-
полимера.
Широко применяют экстру-
экструзионныи способ изготовления
П., позволяющий получать как
высоковспененный, так и низко-
вспененный пенополивиыилхло-
фф родо
вт/(м ' К) [ккал/(м • ч -
Рабочая температура, °С . . . .
Линейная усадка при 60°С за
24 ч, %, не более
Водопоглощение за 24 ч, кг/м2,
не более
Щелочность (в пересчете на
Na2CO3), %, не более
Содержание ионов С1~, %, не
более
Горючесть
70-130
2B0)
0,4-0,7
D-7)
85(850)
80(800)
0,7-0,9
1,6
0,015
3,9
0,030-0,043
@,026 -
0,037)
Замкнутая
130-220
4,5D5)
0,8-1,5
(8-15)
180A800)
205B050)
1,7
1,8
0,016
4,1
0,052-0,056
@,045-
0,048)
50-110
0,2-0,4
B-4)
0,6-0,7
0,029-0,031
@,025-
0,027)
1,0
0,25
5,0
2,0
от —60 до +60
1,0
0,3
1,0
0,25
1,0
0,5
Не горит при вынесении из пламени
Открытая
100-120
0,64F,35)
1 ,5
1,0-1,2
0,0088
0,051
@,044)
от — 1 0 до 55
0,5
Трудновос-
пламеняемый
557
ПЕНОПОЛИВИНИЛХЛОРИД
558
П. Для получения эластичного П. в состав композиции
вводят боргидрид натрия, к-рый реагирует с водой с
выделением газообразного водорода. Кажущаяся плот-
плотность образующегося П. не менее 120 кг1мъ.
Жесткий П. с повышенной теплостойкостью получа-
получается из поливинилхлорида, диизоцианата, малеинового
ангидрида, мономера, катализаторов и наполнителей.
Компоненты смеси тщательно перемешивают и нагрева-
нагревают при 170 °С и давлении около 30 Мн/м2 C00 кгс/см2)
до перехода полимера в гелеобразное состояние. Од-
Одновременно начинается сополимеризация малеинового
ангидрида с мономером и прививка сополимера к по-
ливинилхлориду. Соиолимеризацию прекращают, пресс-
форму охлаждают под давлением и извлекают заготов-
заготовку из формы, когда полимер еще термопластичен.
Вспенивание осуществляют, прогревая заготовку го-
горячей водой или водяным паром. На этой стадии
происходит размягчение полимеров и вспенивание уг-
углекислым газом, образующимся при взаимодействии
изоцианата с водой и с кислотными группами сопо-
сополимера. Параллельно со вспениванием проходит ре-
реакция сшивания макромолекул поливинилхлорида. В
результате получается легкий и прочный П. с пре-
преимущественно закрытой структурой ячеек, известный
в СССР под маркой «пеноизовинил».
Вымыванием растворимых напол-
наполнителей (минеральных солей, крахмала и др.) из
отформованной прогретой заготовки получают пори-
пористый эластичный и жесткий пеыополивинилхлорид.
Однако этот способ не находит широкого применения.
Свойства и применение. По химич. стойкости, горю-
горючести, устойчивости к действию растворителей, окис-
окислителей и света П. аналогичен соответствующим не-
вспененным полимерам (см. Винилхлорида полимеры,
Винилхлорида сополимеры).
П. легко обрабатывается на деревообрабатывающих
станках и ручным столярным инструментом. Плиты П.
могут быть склеены между собой, а также с др. мате-
материалами (металлом, древесиной, пластмассой и т. д.)
различными клеями (преимущественно фенольно-кау-
чуковыми). Недостаток П.—корродирующее действие
на незащищенные металлич. поверхности. Основные
свойства П. представлены в табл. 1—4.
Жесткий П. с замкнутой структурой ячеек исполь-
используется как конструкционный, звуко- и теплоизоляцион-
теплоизоляционный материал. Этот П. достаточно прочен, водостоек
и не тонет в воде. В основном его применяют в строи-
строительстве, авиационной пром-сти, судостроении, для
изготовления плавучих изделий (спасательных средств,
буйков, плотов и т. п.).
Жесткий П. с преимущественно открытыми ячейками
обладает отличными звукопоглощающими св-вами в
широком диапазоне частот и применяется для глушения
шумов. Из него создают либо специальные конст-
конструкции, либо облицовывают им внутреннюю поверх-
поверхность помещения, где расположены источники шума.
Из такого П. готовят фильтры, а также разделители
и сепараторы для электротехнич. пром-сти.
Эластичный П. с замкнутой структурой ячеек при-
применяется для изготовления амортизаторов, упругих,
мягких и звукоизолирующих прокладок, плавучих и
спасательных средств. Из него готовят виброзащитные
устройства, эластичные ограждающие средства, дета-
детали для мебели, подошвы для обуви, спортивный инвен-
инвентарь. Эластичный П. с открытыми ячейками применя-
применяется как мягкий амортизирующей и звукопоглощающий
материал, гл. обр. при изготовлении декоративных
рельефных обивок мебели и автомашин, подкладок под
ковры, линолеум, ткань, одежду и т. д. Рулонный П.
типа искусственной кожи используется для пошива
одежды, обтяжки мебели, обивки сидений, изготовле-
изготовления обуви и т. д. Из эластичного П., получаемого пу-
путем экструзии, изготовляют вспененную изоляцию на
Таблица 2. Свойства жесткого пенополивинилхлорида зарубеж-
зарубежных марок
Показатель
Преобладающая структура
ячеек
Кажущаяся плотность, кг/м3
Прочность, Мн/м2 (кгс/см2)
при сжатии
» сдвиге
» растяжении . , . .
Теплостойкость, °С . . , . , .
Коэфф. теплопроводности,
вт/(м-К) [ккал/(м-ч-°С)] . .
Клежесель
(Франция)
3 а
40 — 75
0,4—0,95
D-9,5)
0,3-0,7
C-7)
0,8 — 1 ,5
(8-15)
Не менее
О а
У U
0,035-
0,047
@,03-
0,04)
Фи-ви
(Велико-
(Великобритания)
м к н у т
32
0,14
A,45)
—
0,6F,2)
60
-—•
Эарекс
(Швей-
(Швейцария)
а я
50-125
0,16-
0,33
A,6-
3,3)
—
60
0,037
@,032)
Таблица 3. Свойства эластичного пенополивинилхлорида
отечественных марок
Показатель
Преобладающая структура
ячеек
Кажущаяся плотность, кг/м6
Прочность при растяжении,
Мн/м2 (кгс/см2)
Относительное удлинение при
разрыве % ... ...
Эластичность по отскоку, %
Относительная остаточная де-
деформация при 5 0%-ном сжа-
сжатии за 72 ч, %
Водопоглощение за 24 ч,
кг/м2, не более
Рабочая температура, °С . . .
Усадка объемная за 24 ч, %,
не более
при 5 0°С
» 60°С
ПХВ-Э
Замкнутая
100-270
0,2-0,6
B-6)
80 — 105
0,05
от —10
до +40
20 — 1 5
Винипор
Открытая
80-180
0,08-
0,15
/Q g
1,5)
80 — 150
20-25
5-15
—
от —10
до +60
1
Пеноэласт
Замкнутая
80-300
0,5 — 1 ,5
E-15)
1 50 — 2 50
25-30
10-20
0,09
от —20
до +60
8-12
Таблица 4. Свойства эластичного пенополивинилхлорида
зарубежных марок
Показатель
Эласто-
фоум
(США)
Преобладающая структура
ячеек
Кажущаяся плотность, кг/м3
Прочность при растяжении,
Мн/м2 (кгс/см2)
Относительное удлинение
при разрыве, %
Модуль упругости при
5 0%-ном сжатии, Мн/м2
(кгс/см2) , . . . .
Эластичность по отскоку, °/{
Остаточная деформация, %
при 60°С
» 20°С . •
Дуфлекс
Велико-
брита-
британия)
Откры-
Открытая
90
0,17
A,69)
180
0,006
@,06)
55
10
Трови-
пор
(ФРГ)
Динапор
(США)
Смешан-
Смешанная
80-120
Не менее
0,12
A,20)
Не менее
100
Не более
20
Открытая
100 32-43
0,13
A,30)
100
0,006
@,06)
7-13
0,03 —
0,05
@,35 -
0,56)
559
ЛЕНОПОЛИОЛЕФИНЫ
560
токопроводящих жилах, профильные изделия и облег-
облегченные листы, пористые шланги для распылительной
поливки и т. д. Основной недостаток эластичного П.—
высокая кажущаяся плотность. Несмотря на это, в ряде
областей применения он выдерживает конкуренцию
с др. пенопластами благодаря своим специфич. свойст-
свойствам — способности к высокочастотной сварке с декора-
декоративными пленками, тканями, кожей, а также высокой
упругости.
П. занимает 3-е место среди др. пенопластов по объе-
объему мирового произ-ва (после пенополиуретанов и пе-
нополистирола).
Наиболее известными являются след. марки П.:
П X В - 1, П X В - 2, ПВ-1, П X В - Э, вини-
пор, пеноэласт (СССР); э л а с т о ф о у и,
д и н а п о р (США); д у ф л е к с, в и т а с е л,
фи-ви, аэролам (Великобритания); к л е ж е-
сель, армосель (Франция); тровипор
(ФРГ); э а р е к с (Швейцария); к а д о р и т (Ита-
(Италия); в и н и к о р к (Япония).
Лит. см. при статьях Пеиопласты и Винилхлорида поли-
полимеры. И. II. Ротеиберг.
ПЕНОПОЛИОЛЕФИНЫ (polyolefine foams, Polyole-
finschaumstoffe, polyolefines mousses).
Состав. В производстве П. обычно применяют поли-
полиэтилен низкой и высокой плотности, реже полипропи-
полипропилен, полиизобутилен или сополимеры этилена с винил-
ацетатом. Вспенивающими агентами служат азодикар-
бонамид (порофор 4X3-21), N, N'-динитрозопентамети-
лентетрамин (порофор 18), азодикарбоксилат бария,
минеральные газообразователи (углекислый аммоний,
углекислый натрий и др.), а также легкокипящие
жидкости (например, 1,2-дихлортетрафторэтан). Чаще
всего используется азодикарбонамид, так как для не-
него характерно наиболее высокое газовое число A94—
220 см3/г). Кроме того, этот газообразователь неток-
нетоксичен, скорость и температурный интервал его рас-
распада можно изменять, вводя такие вещества, как стеа-
раты.
Для достижения заданных физико-химич. свойств
и теплостойкости в состав композиции вводят напол-
наполнители — сажу, графит, окись титана, окись кремния.
Для придания П. негорючести в композицию вво-
вводят трехокись сурьмы, хлор- или бромсодержащие ве-
вещества.
Способы изготовления. Подавляющее количество П.
изготавливают вспениванием с помощью
газообраз ователей. При этом получают П.
с кажущейся плотностью от 30—60 кг/м3 (высоковспе-
ненные) до 400—500 кг/м3 (низковспеиенные). Заготов-
Заготовки для вспенивания формуют прессованием, экстру-
экструзией или литьем под давлением. О методах формова-
формования заготовок см. Пенопласты.
Вспениванием отпрессованных заготовок получают
П. с кажущейся плотностью 60 кг/м3 и выше, характе-
характеризующиеся равномерным распределением изолирован-
изолированных газовых ячеек в материале. Обычно в состав ком-
композиций вводят органич. перекиси (перекись дикумнла,
перекись трет.-бутилкумила и др.), к-рые, реагируя с
полимером в процессе прессования и вспенивания,
придают ему сетчатую структуру.
Экструзией композиции с одновременным или по-
последующим вспениванием изготавливают профильные
изделия. При одновременном вспенивании получают
П. с кажущейся плотностью 400 кг/мд и выше; они име-
имеют замкнутую структуру ячеек. Материал такой же
плотности получают литьем под давлением. Экструзи-
Экструзией с последующим вспениванием удается получить ма-
материал с кажущейся плотностью до 100 кг/м3.
Вспениванием с помощью легко-
кипящей жидкости создают пенополиэти-
лен с кажущейся плотностью 50 кг/м3 и более. Изделия
формуют экструзией со вспениванием по выходе из го-
головки экструдера. Метод разработан недавно и не на-
нашел еще должного применения.
Пенополиэтилен с кажущейся плотностью 500 кг/м3
производят также насыщением композиции азотом или
углекислым газом под давлением 3 Мн/м2 C0 кгс/см2)
при 160 СС. Полученный таким способом П. содержит
большое число ячеек открытой структуры.
Пористый полиэтилен с кажущейся плотностью
400 — 600 кг/м3 и малой газопроводностыо изгота-
изготавливают из композиций, наполненных водораствори-
водорастворимыми веществами (хлористым натрием, крахмалом
и другими).
Ионизирующее излучение вызывает сшивание моле-
молекул полиэтилена с выделением водорода, вспенивающего
полимер. При этом получается мелкопористый матери-
материал с кажущейся плотностью 500—600 кг/м3.
Свойства и применение. Высокие диэлектрич. свой-
свойства пенополиэтилена (табл. 1) позволяют применять
его для изготовления эластичной электрич. изоляции.
Низковспененный П. обладает удовлетворительными
механич. свойствами (табл. 2). Он характеризуется от-
Таблица 1. Диэлектрические свойства изоляции
из низковспененного и монолитного полиэтилена
Показатель
Кажущаяся плотность, кг/м3 ....
Диэлектрич. проницаемость при
10* гц
Тангенс угла диэлектрич. потерь
при 106 гц, не более
Электрич. прочность, кв/мм ....
Уд. объемное электрич. сопротив-
сопротивление, Том-м (ом-см)
Низковспе-
Низковспененный по-
полиэтилен
Монолит-
Монолитный поли-
полиэтилен
450-470
1,48-1 ,50
0,0008
9,0-9,5
4 50
D,5- 1016)
920
О ""> ') О
0,0004
20-30
510
E,1 -1016)
Таблица 2. Механические свойства труб из низковспененного
и монолитного полиэтилена
Показатель
Кажущаяся плотность, кг/м3 . . .
Прочность при растяжении, Мн/м2
(кгс/см2)
Относительное удлинение, % ...
Низковзпе-
ненный
полиэтилен
500-600
5-6
E0-60)
60-80
Монолитный
полиэтилен
920
10,5-12
A0 5 -120)
18 0 - 2 0 0
сутствием усадки, не коробится, его поверхности
м. б. придан красивый рисунок. Из этого П. изготов-
изготовляют достаточно прочные и легкие трубы и листы.
Механич. свойства листов из нпзковспененного поли-
полиэтилена с кажущейся плотностью 450—500 кг/м3 при-
приведены ниже:
Прочность при растяжении, Мн/м2 (кгс/см2), не менее
вдоль направления экструзии 4 D 0)
поперек » » ->,6 C 6)
Относительное удлинение при растяжении, 0/0, не менее
вдоль направления экструзии 1JH)
поперек » » 7 0
Основные свойства высоковспененного полиэтилена
приведены в табл. 3.
Пенонолиэтилен используют в качестве фильтрую-
фильтрующих элементов, обладающих высокой химич. стой-
стойкостью. Ниже приведены нек-рые показатели полиэти-
полиэтиленовых фильтров отечественной марки ФЭП-1 с разме-
размерами пор 30—40 мкм:
Кажущаяся плотность, кг/м3 300 — 3 20
Пористость, %, не менее 6 5
Газопроницаемость, м3-см/мм вод. сгп.-м2-ч. . . 50-60
Производительность (при фильтрации р-ра ме-
тилцеллюлозы с вязкостью 1500 — 2000 си при
давлении 4-5 кгс/см2), л/м2-ч 700-800
561
ПЕНОПОЛИОРГАНОСИЛОКСАНЫ
562
Таблица 3. Свойства различных марок высоковспененного
полиэтилена
Показатель
Кажущаяся плотность,
кг/м3
Прочность при растя-
растяжении, Мн/м2
(кгс/см'г)
Относительное удлине-
удлинение, %
Эластичность по отско-
отскоку, %
Коэфф. теплопроводно-
теплопроводности, в?п/(д1-К)
[ккал/(м-ч-°С)] . . .
Водопоглощение за
24 ч, %, не более . .
Этафоум
(США)
45-60
0,14-0,42
A ,4-4,2)
30-35
0,035-0,047
@,03-0 ,04)
1,0
Санфоум
(Япония)
60-70
0,5-0,72
E,0-7,2)
200-230
37
0,047
@,04)
1 ,0
ППЭ-2
(СССР)
55-65
0,52-0,83
E,2-8,3)
210-230
36-41
0,041 —
0,047
@,035-
0,040)
1 ,0
П. используют для изготовления тары для различных
деталей в автомобилестроении и мебельной пром-сти,
в качестве упаковочного материала, уплотнителей
и теплоизоляции в строительстве и сельском хозяйстве,
для изготовления плавучих средств (рыболовных снас-
снастей, поплавков, спасательных жилетов и кругов) и спор-
спортивного инвентаря (водных лыж, обивки спортивных
лодок, гимнастич. матов) и др.
Поропласты могут применяться в качестве пневмоглу-
шителей в машиностроении.
Впервые пенополиэтилен получен в 40-х годах в Ан-
Англии. Наиболее известными марками П. являются:
ППЭ-2, ФЭП-1 (СССР), этафоум (США),
пластазот (Великобритания), софтлон, х и
этилен С, с а н ф о у м (Япония).
Лит.: SPE journal, № 9, 18 A962); Japan plast. age, 7,
№ 6, 31, A969); KadowakiY., S a s a j i m a J., Chem.
Economy and Eng. Rev., 2, № 3, 54 A970); В e n n i n g C. J.,
Plast. Foams, v. 1, 262 A9 69). См. также литературу при
статьях Пенопласты, Этилена полимеры, Пропилена полимеры,
Изобутилена полимеры. А. И.. Ларионов.
ПЕНОПОЛИОРГАНОСИЛОКСАНЫ (silicone foams,
Silikon-Schaumstoffe, mousses de silicone) — газо-
газонаполненные материалы на основе кремнийорганиче-
ских полимеров.
Эластичные П. (пенорезины, или губчатые резины,
а также пеногерметики) изготавливаются на основе
сополимеров диметилсиландиола с метилвинил(или
феыил)силандиолом молекулярной массы 300 000—
800 000 (см. Кремнийорганические каучуки). Жесткие
П. получают из сополимеров метилфенилсиландиола
и фенил(или метил)силантриола (см. Кремпийорганиче-
ские полимеры).
Способы изготовления. Основной метод получения
П.— вспенивание композиций, содержащих газообразо-
ватели. (О принципиальных основах методов получе-
получения газонаполненных материалов см. Пенопласты.)
В качестве последних применяют вещества, разлагаю-
разлагающиеся при нагревании с выделением газообразных про-
продуктов (динитрил азо-бис-изомасляной к-ты, динитрозо-
пентаметилентетрамин, азодикарбонамид, аминогуа-
нидиыбикарбонат, карбонат аммония, бикарбонат нат-
натрия), или полиалкилгидросилоксаны (напр., поли-
полиэтил- или полиметилгидросилоксаны), при реакции
которых с амино- или гидроксилсодержащими соеди-
соединениями выделяется водород, вспенивающий компо-
композицию.
Вспенивание можно производить также, насыщая
полиорганосилоксановую композицию инертным газом
(N2) иод давлением. После частичного отверждения
или вулканизации внешнее давление снимается и раст-
растворенный газ «вспенивает» массу. Затем производится
окончательное отверждение. Однако на практике этот
метод находит ограниченное применение.
Для согласования скоростей отверждения и газовы-
газовыделения в состав композиций вводят катализаторы и ус-
ускорители: четвертичные аммонийные соли силанолов,
соли и гидроокиси щелочных металлов, оловянные или
свинцовые соли карбоновых к-т, амины или олигоамиды,
соединения платины и др. Активными катализаторами
отверждения являются нек-рые газообразователи: ди-
нитрозопентаметилентетрамин, аминогуанидинбикар-
бонат.
Пенопласты на основе полиорганосилоксанов обла-
обладают низкой прочностью. Для получения более проч-
прочных материалов, особенно для повышения их стойкости
к ударным нагрузкам и резкой смене темп-р, в состав
П. вводят волокнистые или чешуйчатые наполнители
(стеклянные или асбестовые волокна, молотый асбест,
кварц, окислы металлов, мелкодисперсный алюминий
и др.). Наполнители могут существенно влиять и на
скорость отверждения кремнийорганич. полимеров. Пе-
Пенопласты из смесей кремнийорганич. полимеров с эпок-
эпоксидными и феноло-формальдегидными смолами, поли-
полиуретанами также имеют более высокие прочностные
характеристики но сравнению с П., но в этом случае
снижается термоокислительная стойкость материалов,
увеличиваются диэлектрич. потери и теплопроводность.
Заготовки для произ-ва изделий из эластичных П.
формуют прессованием композиции, полученной смеше-
смешением компонентов на вальцах. Во время прессования
в закрытых прессформах при 110 — 120 СС происходит
формообразование массы, ее частичная вулканизация,
разложение газообразователя и насыщение массы га-
газом. Заготовку охлаждают под давлением, переносят
в ограничительную форму, предназначенную для мас-
масштабного формования, и вновь нагревают. Под давле-
давлением расширяющегося газа объем заготовки возрастает,
форма заполняется и структура пены фиксируется
вулканизацией.
Термостойкие и морозостойкие пеногерметикн (напр.,
отечественных марок ВПГ-1 и ВПГ-2) получают из ком-
композиций, содержащих полиметил- или полиметилфенил-
силоксановый каучук, упрочняющий наполнитель, во-
водород- и гидроксилсодержащие компоненты, оловоор-
ганич. катализатор холодной вулканизации. Перечис-
Перечисленные компоненты перемешивают, заливают или вво-
вводят с помощью шприца в герметизируемую емкость. Пос-
После короткого индукционного периода начинается вспе-
вспенивание и одновременная вулканизация массы. Вул-
Вулканизация пеногерметика протекает при комнатной
темп-ре в течение 10—15 ч.
При изготовлении жестких П. в виде плит или фор-
формованных изделий смесь полиметилфенилсилоксана
с динитрозопентаметилентетрамином, диазодикарбона-
мидом и тонкоизмельченным хризотиловым асбестом
(или алюминиевой пудрой) гомогенизируется при
80—100 СС на вальцах, после чего измельчается (до
насыпной массы —600 кг/м3). Порошкообразный полу-
полуфабрикат сохраняет способность к вспениванию в те-
течение 6 мес. При 80—100 СС порошок плавится, при
120 —130 °С происходит разложение газообразователя,
вспенивающаяся масса заполняет объем ограничитель-
ограничительной формы, одновременно отверждаясь. Нагрев продол-
продолжается до 170 °С, причем степень отверждения дости-
достигает 20—30% , а линейная усадка ок. 0,5% . После этого
изделие извлекают из формы и прогревают на воздухе
при 200 °С в течение 12 ч или при 250 СС в течение 6 ч
до полного отверждения. Линейная усадка при этом
составляет еще ^2%.
Вследствие значительной усадки этот вид П. мало-
малопригоден для непосредственного вспенивания в конст-
конструкциях, т. к. в материале возникают значительные
напряжения, к-рые могут привести к растрескиванию
или отрыву заполнителя от стенок. Вспенивание жест-
жестких П. в конструкциях можно осуществлять только
при изготовлении изделий однократного использова-
использования. Процесс в этом случае останавливают после пер-
первой стадии отверждения, когда усадка еще невелика.
563
ПЕНОПОЛИСТИРОЛ
564
Дальнейшее отверждение происходит непосредственно
во время эксплуатации конструкции при высоких
темп-pax, когда усадка компенсируется термич. удли-
удлинением, к-рое весьма велико (температурный коэфф.
линейного расширения жесткого П. —12• 10~5 °С~1).
Для вспенивания и отверждения в конструкциях,
многократно подвергающихся нагреву при эксплуата-
эксплуатации, можно применять полуфабрикат, приготовленный
указанным выше способом из смолы, полученной из
смеси три- и дифункциональных мономеров. П. этого
типа обладают упругоэластич. свойствами. Однако
после длительной эксплуатации на воздухе при 250—
300 С в результате термоокислительных процессов
происходит повышение их жесткости и усадка.
Жесткие П. холодного отверждения изготовляют так-
также с применением полиметилгидросилоксанов или
поливинилгидросилоксанов в качестве газообразова-
телей. и отвердителей. Кажущаяся плотность таких
материалов от 300 до 1200 кг/м3. В состав компози-
композиций вводят наполнители: окислы железа, кремния,
алюминия.
Свойства. П. характеризуются высокой тепло- и
термостойкостью. Они способны длительно (сотни ч)
сохранять хорошие диэлектрические и теплоизоляцион-
теплоизоляционные свойства при 250 °С и кратковременно (несколь-
(несколько ч) — при 350 °С. Введение наполнителей повышает
термостабильность П. (они приобретают способность
выдерживать длительный нагрев при 350 °С и кратко-
кратковременный — при 450 °С).
Жесткие П. без наполнителей вследствие большого
термич. линейного расширения выдерживают тепловой
удар с перепадом темп-р до 150 °С. В случае наполнен-
наполненных П. этот показатель повышается до 250 °С.
При темп-pax выше 350 °С на воздухе П. быстро
окисляются с выделением органич. соединений, к-рые
при 750—800 °С воспламеняются. П. без наполнителей
после этого превращаются в пористый остаток (SiO2). П.
с 10—20 мае. ч. мелкодисперсного алюминия после тер-
термообработки при 400—500 °С теряет органич. радика-
радикалы и превращается в материал керамич. типа, сохра-
сохраняющий механич. прочность до темп-ры 500 °С.
П. не вызывают коррозии черных и цветных металлов,
набухают в органич. неполярных и слабополярных
растворителях. Их можно склеивать с различными ма-
материалами. Жесткие П. склеивают с металлами клеями
холодного отверждения во избежание растрескивания
материала. Обычно на металл перед склейкой наносит-
наносится подслой феноло-каучукового клея (напр.,отечест-
(напр.,отечественной марки ВК-32-200).
Свойства одного из эластичных П.— губчатой резины
ВРП-1 приведены ниже:
Кажущаяся плотность, кг/м3 300 — 500
Темп-pa хрупкости, °С —76
Рабочий интервал темп-р, °С —60 до
+300
Водопоглощение, %
за 24 ч 2-3
за 48 ч 5-7
Относительное сжатие, % 50 — 60
Коэфф. высокоэластич. восстановления, % .... 85
Характеристики нек-рых пеногерметиков приведены
в табл. 1, жестких П.— в табл. 2.
Применение.]!, применяются в качестве теплоизоля-
теплоизоляционных, электроизоляционных материалов и заполни-
заполнителей в конструкциях, длительно работающих при
темп-рах 200—350 °С и кратковременно при 400—450 °С,
а также в абляционностоиких покрытиях.
Эластичные П. широко применяют для герметизации
разъемных соединений при темп-pax до 300 °С, в каче-
качестве амортизирующего и уплотняющего материала.
Кроме того, за рубежом они рекомендованы в качестве
материала для противопожарных перегородок и тепло-
теплозащитных экранов самолетов, космич. кораблей, тепло-
теплоизоляции топливных баков, огнезащитной упаковки,
для электро- и звукоизоляции. Пеногерметики приме-
применяют для заполнения электронных блоков, герметиза-
герметизации неразъемных соединений и др. целей. При этом по
сравнению с невспененными герметиками достигается
снижение массы изделий и расхода материалов.
Таблица 1. Основные свойства пеногерметиков отечественных
марок
Показатель
ВПГ-1
ВПГ-2
Кажущаяся плотность, кг/м3 ....
Водопоглощение за 10 су?п, % ...
Темп-pa хрупкости, °С
Диэлектрическая проницаемость
A06 гц) при
20°С
250°С A00 ч)
Тангенс угла диэлектрических потерь
A06 гц) при
20°С
250°С A00 ч)
Коэфф. теплопроводности, вт/(м-К)
[ккал/(м • ч • °С)]
500 — 700
1,2
-76
3,81
3,57
0,0059
0,0021
0,17
@,15)
400-600
3,2
-76
4,05
3,60
0,0041
0,0019
0,11
@,09)
Таблица 2. Некоторые свойства жестких пенопластов на основе
полиметил(фенил)силоксанов (кажущаяся плотность
250 — 300 кг/м3)
Показатель
Прочность при
сжатии, Мн/м2
(кгс/см2), при
20°С ....
250°С ....
300°С ....
Линейная усадка
за 100 ч, %, при
250°С ....
300°С ....
Диэлектрич. про-
проницаемость при
1О10 гц
Тангенс угла ди-
диэлектрич. по-
потерь при 1 О10 гц
Коэфф. теплопро-
теплопроводности при
20 0°С, em/(JW-K)
[ккал/(м -ч-°С)]
Температурный
коэфф. линей-
линейного расшире-
расширения при 20 —
200°С, °С . .
К-40
без напол-
наполнителя
1,4A4)
0,28B,8)
0,15A ,5)
0,4
1 ,9
1,31
0,002
0,058
@,050)
12- 10
с 12 мае. ч.
алюминия
1,94A9,4)
0,58E,8)
0,54E,4)
0,5
2,0
2,1
0,013
0,088
@,075)
G, 7 — 1 0I 0
К-9
с 10 мае. ч.
асбеста
2,2B2)
1 ,05A0,5)
0,73G,3)
0,2
0,9
1 ,35
0,0015
0,076
• @,065)
G,5-10,5I0-5
Жесткие и упругоэластичные П., как уже указыва-
указывалось, находят широкое применение в различных конст-
конструкциях, используются в качестве тепло- и электроизо-
электроизоляционных материалов в строительстве, машинострое-
машиностроении, авиационной пром-сти.
Наиболее широко известны след. марки эластичных
П.— ВРП-1 (СССР), с и ластик РТВС-
5 3 7 0, с и л а с т и к - 5 9, с и л а с т и к-6 9, Р Т В - 7,
Р Т В - 7 5 7 (США); пеногерметиков — ВПГ-1,
ВПГ-2 (СССР); жестких П.—Р -7001,Р-7002,
Р-700 3, кор-фоум (США), К-40, К-9 (СССР).
Лит.: Козловская Л. Н., Р у д е н к о Н. И.,
Кауч. и рез., № 4, 19 A962); С о л о м а т и н А. В., Ша-
Шалаева И. А., там же, № 7, 16 A967); Plast. World, 22, № И,
73 A964); Metal Progress, 88, № 1, 8 A965); Aircraft Eng., 37,
№ 11, 354 A965); Materials in Design Eng., 64, № 7, 60 A966);
J. Macromol. Sci., A3, № 4, 585 A969). См. также лит. при
статье Пенопласты. М. Я. Бородин.
ПЕНОПОЛИСТИРОЛ (foamed polystyrene, Schaum-
polystyrol, polysterene mousses) — газонаполненный ма-
материал на основе полистирола, его производных или
сополимеров стирола.
Состав. В производстве П. используют обычно поли-
полистирол, реже — полимонохлорстирол, полидихлорсти-
565
ПЕНОПОЛИСТИРОЛ
566
рол и сополимеры стирола с акрилонитрилом, бутадие-
бутадиеном и акрилонитрилом (АБС-сополимер). Для вспени-
вспенивания применяют газообразователи—-2,2'-азо-бис-изобу-
тиронитрил (порофор N), диазоаминобензол, углекислый
аммоний, бикарбонат натрия и др., или легкокипящие
жидкости (изопентан, метиленхлорид, петролейный
эфир и др.).
Способность к самозатуханию П. достигается введе-
введением в композицию антипиренов (напр., тетрабромпа-
раксилола, 1,2-дибромэтилбензола или соединений
сурьмы и фосфора). В композицию могут входить кра-
красители, пластификаторы, наполнители (напр., тонкодис-
тонкодисперсный порошок алюминия).
Способы изготовления. Подавляющее количество
П. (напр., отечественные марки ПСБ, ПСБ-С) произво-
производится вспениванием парами легкокипящей жидкости.
Метод высокопроизводителен и прост в аппаратурном
оформлении. Для получения П. этим методом произво-
производят гранульную полимеризацию стирола в присутствии
углеводорода, растворимого в стироле и нерастворимо-
нерастворимого в полистироле (о принципиальных основах этого
метода см. Пенопласты). Для предварительного вспени-
вспенивания гранулы нагревают горячей водой, паром или
горячим воздухом до 90—120 СС в аппаратах периоди-
периодического (вертикальные резервуары с мешалкой, ванны)
или непрерывного действия (шнековые, механические,
барабанные вспениватели). Объем гранул увеличи-
увеличивается в 10—30 раз в зависимости от количества жид-
жидкости в полистироле. Гранулы высушивают (если пред-
предварительное вспенивание производилось водой или во-
водяным паром) и выдерживают при комнатной темп-ре
в течение 6—24 ч с тем, чтобы частичное разрежение
в полостях гранул, вызванное изменением объема жид-
жидкости в них при охлаждении, было скомпенсировано
в результате проникновения внутрь гранул воздуха.
Спекание гранул с одновременным формованием изде-
изделий осуществляют на оборудовании периодического
(переносные и стационарные перфорированные формы,
автоклавы, оформляющие конструкции) и непрерыв-
непрерывного действия (конвейерные линии, карусельные маши-
машины, установки горизонтального и вертикального типа).
В качестве теплоносителя используют водяной пар,
токи высокой частоты, ИК-облучение. Гранулы нагре-
нагревают до 100—120 СС. При этом полистирол переходит
в высокоэластич. состояние, а в гранулах создается
давление паров низкокипящей жидкости и воздуха,
в результате чего гранулы увеличиваются в объеме
и заполняют ограничительную форму. Под внутрен-
внутренним давлением паров и воздуха стенки образующихся
ячеек деформируются и свариваются в местах контакта
друг с другом. Отформованное изделие быстро охлаж-
охлаждают в формах до 40—50 СС, фиксируя структуру пены
и форму изделия.
Вспениванием композиций на основе полистирола
с газообразователями изготовляют П. отечественных
марок ПС-1, ПС-2, ПС-4 и др. Компоненты (эмульси-
(эмульсионный полистирол, газообразователь, в нек-рых слу-
случаях пластификатор, краситель, наполнитель) тщатель-
тщательно перемешивают 10—14 ч в шаровых мельницах, ло-
лопастных мешалках или ротационных аппаратах. Из
смеси формуют заготовки в прессформах закрытого ти-
типа при 140—170 СС и давлении 15—20 Мн/м2 A50—
200 кгс/см2). Заготовки вспенивают в ограничительных
формах с одновременным масштабным формованием
или самоформованием изделий при 110—140 °С.
Если заготовка должна быть изготовлена заливкой
массы в форму, то композиции придают текучесть, до-
добавляют в нее нек-рое количество стирола или метил-
метакрилата. Компоненты (включая инициатор поли-
полимеризации и газообразователь) смешивают в смесителе
или на вальцах, получая т. наз. полимер-мономерную
пасту. Смесь заливают в формы или полость изделия.
Полимеризация мономера происходит ниже темп-ры
разложения газообразователя (обычно при 40—80 °С).
После этого заготовку вспенивают при 100—140 °С.
Если вспенивание сочетают с формованием изделий
литьем под давлением или экструзией, смесь эмульси-
эмульсионного полистирола и газообразователя подается в ма-
материальный цилиндр, где происходит размягчение поли-
полистирола, разложение газообразователя и насыщение га-
газом расплава. Насыщенная газом масса впрыскивает-
впрыскивается в холодную литьевую форму или выдавливается из
головки экструдера.
Существует также способ получения П., в к-ром по-
полимеризация стирола и вспенивание полученного по-
полистирола совмещены в одном непрерывном автомати-
автоматизированном процессе. В этом случае используется газо-
газообразователь (напр., 2,2'-азо-бис-изобутиронитрил или
циклогексиловый эфир азодикарбоновой к-ты), к-рый
вначале инициирует полимеризацию (или сонолимери-
зацию) стирола, а затем вспенивает полимер.
Насыщение полимерной композиции газом (СО2, N2)
с последующим сбросом давления не находит широкого
применения, т. к. связано с использованием сложного
и дорогого оборудования (специальных автоклавов).
Свойства и применение. П., полученный вспенива-
вспениванием парами легкокипящей жидкости, представляет
собой тонкоячеистые гранулы, спекшиеся одна с дру-
другой. Внутри каждой частицы имеются микропоры, а
между частицами — пустоты различных размеров. П.,
полученный в присутствии газообразователя, имеет
преимущественно закрытую структуру ячеек. Все
механич. свойства этих П. зависят от кажущейся плот-
плотности; с ее повышением увеличиваются прочностные
показатели, снижаются водопоглощение, гигроскопич-
гигроскопичность, воздухо- и паропроницаемость (см. табл.).
Свойства пенополистирола отечественных марок
Показатель
Кажущаяся плотность,
кг/ж3
Предельная темп-pa при-
применения, °С
Коэфф. теплопроводности
B 0 °С), вт/(м-К)
[ккал/(м • ч • °С)]
Прочность, Мн/мг (кгс/см2)
при сжатии
при статич. изгибе . .
при растяжении ....
Ударная вязкость, кдж/м2
[кгс-см/см2]
Водопоглощение за 24 ч,
кг/м*, не более
Диэлектрич. проница-
проницаемость при 1 О10 гц ...
Тангенс угла диэлектрич.
потерь при 1010 гг^ ...
ПС-1
7 0 — 20 0
60
0,033-
0,032
@,028-
0,045)
0,5-3
E-30)
1 ,2-6,5
A2-65)
0,7-4,2
G-42)
ДО 1 ,9
0,3
1,1 — 1,28
0,0012-
0,0024
ПС-4
40 — 65
60
0,029-
0,044
@,025-
0,038)
0,25-0,6
B,5-6,0)
—
0,8 — 1 ,2
(8-12)
ДО 1 ,0
0,3
1,08-1,15
0,0015-
0,0020
ПСБ-С
15 — 50
60
0,028 —
0,041
@,024-
0,035)
0,05-0,35
@,5 — 3,5)
0,15-0,5
A,5-5,0)
—
—
0,02 — 0,06
1,06-1,12
0,0012 —
0,0020
Химстойкость, устойчивость к термоокислительному
воздействию, световому облучению определяются свой-
свойствами исходного полимера (см. Стирола полимеры).
Основной недостаток П.— малая термостабильность и
повышенная горючесть (даже у самозатухающих сортов,
поскольку при воздействии огня они расплавляются).
Низкая кажущаяся плотность в сочетании с относи-
относительно высокими прочностными показателями, малая
теплопроводность, а также наличие развитой сырье-
сырьевой базы и относительно невысокая стоимость опреде-
определили широкое применение П. в качестве теплоизоля-
теплоизоляционного и конструкционного материала в строитель-
строительстве, судо-, вагоно- и авиастроении.
Значительные количества П. расходуются для упа-
упаковки прецизионных и оптич. приборов, товаров народ-
567
ПЕНОПОЛИУРЕТАНЫ
568
ного потребления (цветов, вин, яиц, битой птицы
и замороженных фруктов). Высокие электроизоляцион-
электроизоляционные свойства П. обусловили его применение в прибо-
приборостроении и радиотехнике. Нек-рые сорта П. исполь-
используют для структурирования почв. П., изготовляемый
прессовыми методами, успешно применяется для изго-
изготовления плавучих средств (плавающие буи, бакены),
спасательного снаряжения (плотики, пояса, нагруд-
нагрудники), непотопляемых судов, катеров, понтонов. П.
применяют также для изготовления специальных сор-
сортов бумаги, деталей мебели, при литье по выплавляе-
выплавляемым моделям, в качестве материала, имитирующего
древесину.
Впервые П. получили во Франции в 1928, а его
гроиз-во начато в 40-х гг. в Германии и США.
Наиболее известные след. торговые марки П.: П С - 1,
ПС-4 и ПС Б (СССР), стирофоуми руф-
м е й т (США), стиропор и стиромулъ
(ФРГ), сорбланит (ФРГ и ГДР), к а р и н е к с,
с т и росе л, монтопор, с а н т о ф о у м (Ве-
(Великобритания), рестичел (Италия), л о р к а-
с е л ь (Франция). Крупнейшими производителями П.
за рубежом в 1970 были США A20 тыс. т), ФРГ, Япо-
Япония G5 тыс. т), Италия, Великобритания (~ 25 тыс. т)
и Франция.
Лит.: Cube H. L., Р о h 1 К. Е., Die Technologie des
schaumbaren Polystyrols, Hdlb., 1965; Павлов В. А., Пе-
нополистирол, М., 1973. См. также лит. при статьях Пеиопла-
сты и Стирола полимеры. Ю. С. Мурашов,
Л. В. Аристовспая.
ПЕНОПОЛИУРЕТАНЫ (polyurethane foams, Poly-
urethanschaumstoffe, mousses de polyurethane).
Состав. Композиции для производства П. содержат
изоцианаты, гидроксилсодержащие олигомеры, воду,
катализаторы, эмульгаторы, а в нек-рых случаях
наполнители, красители и антипирены.
Для получения П. применяют 2,4-толуилендиизоциа-
нат, его смесь с 2,6-толуилендиизоцианатом в соотно-
соотношении 80 : 20, 4,4'-диизоцианатдифенилметан, 4,4',4"-
триизоцианаттрифенилметан и др.
В состав композиций для производства эластичных
П. входят простые олигоэфиры с мол. м. 750 — 6000,
синтезируемые из окисей алкиленов (этилена, пропи-
пропилена), тетрагидрофурана и гликолей. Реже используют
сложные олигоэфиры дикарбоновых к-т (адипиновой,
себациновой, янтарной) и гликолей (напр., диэтилен-
гликоля). Жесткие П. получают из простых олигоэфи-
олигоэфиров разветвленной структуры на основе окисей алки-
алкиленов и триолов (глицерина, триметилолпропана и др.)
или сложных олигоэфиров на основе дикарбоновых к-т
(адипиновой. фталевой и др.) и триолов или их смесей
с диэтиленгликолем. Плотность образующихся П. за-
зависит от соотношения изоцианатов и гидроксилсодер-
жащих олигомеров в исходной смеси. При избытке
изоцианатов П. содержат больше мочевинных групп,
чем при недостатке изоцианатов, когда образуется
больше уретановых групп. Поскольку полимочевины
обладают более низкой плотностью A,05—1,23 г/см3),
чем полиуретаны A,28 г/см3), в первом случае получа-
получаются П. с меньшей плотностью.
При взаимодействии изоцианатов с гидроксилсодер-
жащими олигомерами образуются уретановые звенья:
2OCN — R — NCO + НО - R'- ОН -^
-> OCN-R- NHCOO-R'—OOCHN-R-NCO
A)
При избытке изоцианата в реакционной среде на кон-
концах растущих макромолекул оказываются изоцианат-
ные группы, к-рые могут вступать в реакцию с водой:
п OCN- R-NHCOO —R'-OOCHN-R-NCO + г. Н2О ->
-* [-NHCONH-R-NHCOO-R'-OOCHN-R-Jn+ v. CO2 \ B)
При этом выделяется углекислый газ, вспенивающий
композицию, а макромолекулы присоединяются друг
к другу через мочевинные группы.
Взаимодействие изоцианатных групп с гидроксил-
содержащими олигомерами и водой — конкурирующие
реакции. Роль катализатора сводится к регулированию
скорости указанных выше реакций. При этом выделе-
выделение газа и рост полимерных молекул должны проис-
происходить с такими скоростями, чтобы газ оставался
в полимере и образовавшаяся пена была бы достаточно
прочной и не опадала. Наиболее часто в качестве ката-
катализаторов применяют соединения олова (олеат и окто-
ат, соли дибутилолова и др.), регулирующие реакцию
образования уретановых звеньев, и третичные амины
(триэтиламин, триэтаноламин, диметилбензиламин и
др.), катализирующие реакции образования трехмер-
трехмерной структуры и выделения углекислого газа. На прак-
практике используют каталитич. смесь, состоящую из соеди-
соединения олова и одного или нескольких аминов. Вспени-
Вспенивать полиуретановую композицию можно также легко-
кипящими жидкостями, обычно фреонами.
Химизм образования эластичных и жестких П. оди-
одинаков. Жесткие пены отличаются от эластичных тем,
что состоят из полимеров с большим числом поперечных
связей. В жестких П. средняя «молекулярная масса»
структурной единицы, приходящаяся на один узел
разветвления сетки, составляет 400 — 700, в эластичных
П. — 2500—20 000. Поэтому композиции для произ-
производства эластичных П. не содержат трифункциональ-
ных гидроксилсодержащих олигомеров (или содержат
их в небольшом количестве), а также содержат меньше
третичных аминов.
Обязательным компонентом композиции является
эмульгатор, к-рый способствует высокой степени дис-
диспергирования компонентов в массе и выполняет роль
стабилизатора пены в момент вспенивания. Для этого
используют сульфоспирты, сульфокислоты, кремний-
органич. жидкости и др. Нек-рые стабилизаторы (напр.,
парафиновые углеводороды, кремнийорганич. жидко-
жидкости) определяют характер (открытые или закрытые)
и размер образующихся пор.
В качестве антипиренов применяют трехокнсь сурь-
сурьмы, трихлорэтилфосфат, порошкообразный иоливинил-
хлорид и др. Для окрашивания П. пригодно большинст-
большинство органич. красителей. Наполняют П. тальком, ке-
керамзитом, суспензионным полистиролом, волокнами
различной природы.
Производство. П. изготовляют вспениванием компо-
композиции газами, выделяющимися в результате реакций
между компонентами исходной смеси (см. выше), или
с помощью легкокипящих жидкостей. Поскольку при
образовании П. по первому методу выделяется значи-
значительное количество тепла, внутренние слон крупнога-
крупногабаритных изделий могут обугливаться. Поэтому первый
метод применим только для изготовления изделий
небольшой толщины.
Во втором методе выделяющееся тепло затрачивается
на испарение легкокипящей жидкости, что позволяет
предотвратить местные перегревы и обугливание П.
Процесс производства П. может быть одностадийным
или двухстадийным. При одностадийном способе диизо-
цианат, гидроксилсодержащий олигомер, воду или
амин и др. компоненты композиции подают в смеситель
одновременно. Взаимодействие компонентов происхо-
происходит сразу же, причем подъем пены начинается прибли-
приблизительно через 10 сек и заканчивается через 1—2 мин
после смешения. Окончательное отверждение иены
продолжается от нескольких ч до нескольких су т. При
двухстадийном (форполимерном) способе сначала про-
проводят реакцию диизоцианата с олигоэфиром, а полу-
полученный форполимер затем превращают в П. при сме-
смешении с водой или амином. Изготовление пеноиоли-
уретановых изделий осуществляют по непрерывной или
периодич. схеме (заливкой в бумажные формы), а также
напылением. В любом случае предъявляются жесткие
требования к соблюдению соотношения исходных
569
ПЕНОФЕНОПЛАСТЫ
570
компонентов. В периодической схеме отклонения в
подаваемой порции по каждому компоненту должны
быть не более 1% (но массе), а в непрерывном смеси-
смесителе запаздывание или опережение подачи одного из
компонентов не должно быть более 1 — 2 мсек. За-
Заданная темп-pa процесса должна поддерживаться с точ-
точностью ± 1 °С.
Свойства. П. обладают хорошей атмосферостой-
костью, устойчивостью к действию света и окислителей.
На свету они темнеют, но другие свойства заметно
не ухудшаются. П. физиологически инертны и хорошо
совмещаются с бактерицидными добавками. П., полу-
полученные на основе простых олнгоэфиров, устойчивы
Таблица 1. Свойства жестких пенополиуретанов отечественных
марок
Показатель
Кажущаяся плот-
плотность, кг/м3
Прочность, Мп/м2
(кгс/см2) не менее
при сжатии ....
при изгибе ....
Ударная вязкость,
кдж/м2 или кгсх
Хсм/см2, не менее
Коэфф. теплопровод-
теплопроводности, вт/(м-К) . .
[ккал/(м • ч • °С)] . . .
Водопоглощение за
24 ч, %, не более
Темп-pa применения,
°С
Диэлектрич. проница-
проницаемость при 1О10 гц
Тангенс угла диэлек-
диэлектрич. потерь при
1010 гц
о
ь
10 0-200
1,0-1,9
A0-19)
0,8-1 ,5
(8-15)
0,4
0,031 —
0,03 5
@,027 —
0,0 3 0)
0,3
от —50
до 150
1 , 1-1 ,2
0,0015
О
к
150-250
2,0-4,2
B0-42)
1 , 5 — 3 ,5
A5-35)
0,5-0,8
0,033-
0,047
@,028-
0,040)
0,3
от —60
до 20 0
1,1-1,3
0,0 016—
0 , 0020
о
:ип
50
0,25
B,5)
0,2
B)
0,6
0,033-
0,038
@,028-
0,0 3 3)
0,3
от - 60
до 60
—
:(LН
Й
Й
30-50
0,15-0,5
A ,5-5)
0,2-0,9
B-9)
0,4-0,6
0,023 —
0 ,035
@,02 —
0,0 3)
0,3
от —60
до 100
—
Таблица 2. Свойства эластичных
пенополиуретанов
отечественных марок
Показатель
Кажущаяся плотность,
кг/ж3 . . .
Прочность при растяже-
растяжении, Мп/м2 (кгс/см2) . . .
Относительное удлинение,
%
Эластичность по отскоку,
%
Относительная остаточная
деформация при 5 0%-ном
сжатии в течение 72 ч
при 20 °С, %
Напряжение сжатия при
40%-ной деформации,
Мп/м2 (кгс/см2)
Темп-pa применения, °С . .
Потеря массы при горении
(метод «огневая труба»),
%
Коэфф. звукопоглощения
при
25 0 гц
1000 гц
40 00 гц ..... , .
ППУ-Э
25 — 60
0,12
A,2)
150
15
10
0,0025 —
0,0075
@,025 —
0,07 5)
от —15
до 100
0,3 5
0,80
0 ,75
ППУ-ЭТ
30 — 40
0,1
A,0)
100
15
15
0 , 003 —
0,01
@,03 —
0,1)
от -2 0
до 100
22
0,3 6
0,85
0,80
ППУ-ЭМ-1
30 — 50
0,11-0,13
A,1-1,3)
150 — 170
20 — 40
10
0,004 —
0,01
@,04-0, 1)
от -50
до 100
—
—
даже в кипящей воде. П. на основе сложных олигоэфи-
ров менее стойки к действию воды. Жесткие П. устой-
устойчивы в дизельных и смазочных маслах при темп-рах
до 100 СС, бензине, керосине, хлорированных углево-
углеводородах, спиртах, водных р-рах солей, 30%-ной уксус-
уксусной к-те. Они разрушаются ацетоном, этилацетатом,
крезолом, 30%-ной соляной к-той, 10%-ной серной
к-той и 15—20%-ными р-рами едкого натра.
Жесткие П. можно пилить, обтачивать, обрабаты-
обрабатывать на токарном станке. Эластичные П. можно резать.
П. моют мылом или синтетич. моющими средствами,
на них не действуют бактерии, их не ест моль. П., при-
применяемый в качестве изолирующего материала, можно
эксплуатировать при любых низких темп-pax, при этом
верхняя рабочая темп-pa достигает 200 СС. В условиях
двухстороннего нагрева изделия из П. можно кратко-
кратковременно эксплуатировать при 220 °С, а при односто-
одностороннем нагреве — при 450 — 500 °С. Основные свойства
П. приведены в табл. 1 и 2.
Применение. Эластичные П. применяют в произ-
производстве изделий широкого потребления: мягкой мебели,
одежды, подошв для обуви, ковров, губок, щеток и
т. п. Кроме того, их используют для упаковки бьющих-
бьющихся изделий, в качестве звуко- и теплоизоляционных
материалов.
Жесткие П. применяют как электроизоляционный,
теплоизоляционный и упаковочный материал. Они при-
пригодны также для заполнения полых конструкций
в авто-, самолето- и судостроении. Заполненная жест-
жестким П. радиоаппаратура надежно защищена от вибра-
вибрации, плесени, коррозии и проникновения влаги.
По объему производства в капиталистич. странах П.
почти в 2 раза превосходит др. пенопласты на основе
синтетич. полимеров. П. выпускается иод следующими
торговыми названиями: поролон (эластичный
П.— СССР); локфоам, вибрафоам, фоа-
м е к с (США); мольтопрен (ФРГ); а л л о-
ф о а м (Канада).
Промышленное производство П. было впервые осу-
осуществлено в 1952 в ФРГ.
Лит.: Булатов Г. А., Пенополиуретаны и их приме-
нение в летательных аппаратах, М., 1970.
См. также лит. при статье Пенопласты, Полиуретаны.
Ю. С. Липатов.
ПЕНОФЕНОПЛАСТЫ (phenolic foams, Phenol-
Schaumstoffe, mousses phenoliques) — газонаполненные
материалы на основе феноло-альдегидных смол.
Состав. В производстве П. наибольшее применение
находят феноло-дюрмалъдегидные смолы резольного и
новолачного типа, реже — феноло-анилино-формаль-
дегидная смола резольного типа.
Вспенивающим агентом обычно служит 2,2'-азо-бис-
изобутиронитрил (гюрофор N или ЧХЗ — 57), легко-
кипящие жидкости (четыреххлористый углерод, фрео-'
ны, м-пентан) или тонкодисперсные порошки алюми-
алюминия, магния в сочетании с к-тами. Отвердителем но-
волачных смол является гексаметилентетрамин. Ре-
зольные смолы отверждаются при нагревании или в
присутствии кислотных катализаторов. В случае запол-
заполнения пенофенопластами конструкций из металла, дере-
дерева или бетона рекомендуется использовать в качестве
катализатора отверждения продукты взаимодействия
арилсульфокислот или сульфоноволаков с карбонил-
содержащими соединениями, аминами, амидами, циан-
цианамидами или нитрилами (напр., продукты ВАГ), т. к.
они не вызывают коррозии вышеперечисленных мате-
материалов.
В композицию для получения пенопластов вводят
поверхностно-активные вещества (ОП-7, ОП-10 и др.),
антипирены, наполнители (стекловолокно, алюминие-
алюминиевая пудра, высокодисперсный асбест и др.). Сниже-
Снижения хрупкости П. добиваются введением в состав ком-
композиции бутадиен-нитрильного каучука, для вулка-
вулканизации к-рого используют серу.
571
ПЕНОФЕНОПЛАСТЫ
572
Способы изготовления. П. можно получать всеми
методами, применяемыми в производстве пенопластов.
(О принципиальных основах методов получения газо-
газонаполненных материалов см. Пенопласты). Наиболее
распространены вспенивание с помощью газообразо-
газообразователеи, легкокипящих жидкостей и газов, выделяю-
выделяющихся при взаимодействии исходных компонентов.
Технология получения и переработки П. в значитель-
значительной степени определяется агрегатным состоянием
исходной композиции.
При получении П. из р-ров и эмульсий (т. наз. за-
заливочных пенопластов) компоненты смешивают и за-
заливают в специальные формы или во внутреннюю
полость изделия. Вспенивание (указанными выше спо-
способами) идет в течение нескольких мин при комнатной
темп-ре и атмосферном давлении, что позволяет реали-
реализовать непрерывные, полностью автоматизированные
процессы формования.
Технология изготовления П. из порошкообразных
феноло-формальдегидных смол предусматривает сме-
смешивание всех порошкообразных компонентов (смолы,
отвердителя, газообразователя) в шаровой мельнице
в течение 2—12 ч. При получении жестких П. смесь
поступает непосредственно на вспенивание.
Для создания П. пониженной жесткости композицию
эластифицируют каучуком. Полуфабрикат получают
в виде пленок, порошка или прутков. Вспенивание
осуществляют в ограничительных формах с помощью
газообразователеи или легкокипящей жидкости, повы-
повышая темп-ру сначала до 80—90 °С, а затем до 90—
110 °С. Отверждение завершают при 150—200 СС. Пол-
Полный цикл вспенивания и отверждения П. составляет
6,5—8 ч.
Из пленочного полуфабриката вырабатывают П.
с кажущейся плотностью 300—400 кг/м3, из порошко-
порошкообразного — 400—500 кг/м3, из прутков — 100—
350 кг/м3.
Многоступенчатость такого процесса изготовления
П., необходимость использования малопроизводитель-
малопроизводительного громоздкого оборудования, большие трудности
в механизации и автоматизации процесса вспенивания
сильно удорожают производство. Но несмотря на пере-
перечисленные недостатки описанный способ находит при-
применение, т. к. с его помощью на обычном оборудова-
оборудовании можно изготавливать П. с различной прочностью,
жесткостью и теплостойкостью.
Рассмотренными методами получают П. в виде плит,
блоков или изделий сложной конфигурации. Плиты или
Таблица 1. Свойства пенофенопластов из твердых новолачных смол (ФФ), а также
сочетаний смолы с бутадиен-нитрильным каучуком (ФК-20, ФК-40), алюминиевой пудрой
(ФК-20-А-20) и стекловолокнистым наполнителем (ФС)
блоки можно раскраивать, применяя любые спосо-
способы резания, и вклеивать в конструкцию (изделие), за-
заполняя зазоры порошкообразным полуфабрикатом,
который при нагревании вспенивается и надежно со-
соединяет все заготовки между собой и со стенками кон-
конструкции.
Свойства и применение. Большинство П.— жесткие
газонаполненные материалы со смешанной структурой
ячеек; однако существуют и такие П., в к-рых коли-
количество замкнутых или открытых ячеек приближается
к 100%. По химстойкости П. не отличаются от моно-
монолитных фенопластов.
Таблица 2. Свойства
Показатель
Кажущаяся плотность,
кг 1 м?
Предельная темп-ра
применения, °С . . .
Коэфф. теплопроводнос-
теплопроводности при 20сС, вт/(м-К)
[ккал/(м*Ч'°С)] ....
Прочность, Мн/м2
(кгс/см2)
при сжатии
при статическом
изгибе
при растяжении
Влагопоглощение
за 24 ч в среде
98%-ной влажности,
%, не более
пенофенопластов заливочного
ФРП-1
30 — 80
150
0,032 —
0, 043
[0,027-
0,037]
0,05-
0,032
@,5-3,2)
0, 06—0,21
@,6-2,1)
0,04-0,22
@,4-2,2)
25
ФРП-
2М
1 00
200
0,046
[0,04]
0,25
B,5)
25
ФЛ-1
40 — 60
120
0,041
[0,035]
0,08—
0,2 5
@,8—
2,5)
0,05-
0,15
@,5 —
1,5)
30
типа
ФЛ-3
60—200
200
0,081
[0,07]
0,43-
4,5
D,3-
4,5)
0,3-
1,5
C-15)
30
Показатель
Кажущаяся плотность, кг/м3
Прочность, Мн/м2 (кгс/см2)
при сжатии
при изгибе
при растяжении . . .
Ударная вязкость,
кдж/м2 (кгс- см/см2) ....
Водопоглощение за 24 ч,
кг/м2, не более
Предельная темп-ра
применения, °С
Линейная усадка да 24 ч, %,
не более
Коэфф. теплопроводности,
вт/(м - К)[ккал/(м • ч • °С)]
ФФ
190-230
2 0 — 4,0
B0-40)
1 ,5A5)
1,2A2)
0,2
0,1-0,3
150
0,6
A50°С)
0,042-
0,061
@,036-
0,052)
ФК-20
190-230
1 ,5 — 3,0
A5-30)
2,5-3,0
B5 — 30)
2,0B0)
0,8
0,1-0,3
120-130
0,1
A30°С)
0,041 —
0,061
@,035 —
0,052)
ФК-20-А-20
140-200
0,85 — 2, 3
(8,5—23)
0,9-2,9
(9-29)
0,8—1 ,5
(8-15)
0,64-0,73
0,09
200-250
2,0
B00°С)
0,064—
0,074
@,055 —
0,063)
ФК-40
200-230
0 2 — 0 4
B-4)
0,98-1,05
(9,8-10,5)
0,78G,8)
2,16
0,2
80-100
1,0
(80°С)
0,061-
0,074
@,052 —
0,063)
ФС
70—100
0,2-0,4
B-4)
—
0,3-0,5
100
0,8
A00°С)
0,053-
0,058
@,045-
0,050)
Свойства П. зависят от их структуры и кажущейся
плотности и изменяются в зависимости от темп-ры
эксплуатации. Основные механич., теплофизич. и др.
свойства отечественных марок П., формуемых из твер-
твердых композиций, и П. заливочного типа приведены
в табл. 1 и 2.
Недостаток П.— сравнительно малая прочность при
динамич. нагрузках. Хрупкость П. пытаются преодо-
преодолеть, модифицируя исходные фе-
нольные смолы полиуретанами,
каучуками и латексами, эпо-
эпоксидными смолами, сополимера-
сополимерами акрилонитрила, поливинило-
поливиниловым спиртом и его производны-
производными.
Благодаря способности сох-
сохранять форму как в области
низких (до —200 °С), так и вы-
высоких темп-р (в отсутствие кон-
контакта с воздухом до 350 СС для
ненаполненных П. и до 500 °С —
для П., наполненных алюми-
алюминиевой пудрой), а также сравни-
сравнительно невысокой стоимости П.
находят самое широкое приме-
применение.
П. используют в качестве теп-
тепло-звукоизоляционного и кон-
конструкционного материала в
строительстве. Значительные ко-
количества П. расходуются для
изоляции теплотрасс, нефте- и
газопроводов, гелионагревателей
573
ПЕНОЭПОКСИДЫ
574
и промышленных^ холодильников. Др. обширные обла- нов, ди-D-аминофенил)сульфон и др.] применяют для
получения П., характеризующихся повышенной тепло-
сти применения П.— судо- и вагоностроение, авиацион-
авиационная пром-сть, произ-во автофургонов, контейнеров,
различных емкостей и рефрижераторов. Кроме того,
П. применяют для очистки воды от нефтепродуктов,
в качестве силового демпфирующего материала, а так-
также для изготовления непотопляемых спасательных
средств.
Перспективно использование П. для создания быст-
ровозводимых противопожарных перемычек, для за-
закрепления горных выработок и осыпей в шахтных ла-
лавах, в целях снижения утечки подаваемого в штреки
вентиляционного воздуха.
Особые сорта П., полученные путем карбонизации
обычных марок П. в результате ступенчатой термооб-
термообработки при 150—300 и 350—500 СС в течение несколь-
нескольких ч, обладают термостабильностью до 1000 СС и
находят применение в космич. технике.
П. впервые получены в Германии в 1926. Наиболее
распространенные марки П.: золь, фенекспон
(Франция), дин, фенодюр (ФРГ), бакелит
(Великобритания), ф е н о л и т (Италия), и н с у л-
фоум (США), ФФ, ФК, ФРП-1 (СССР).
Лит. см. при статьях Пенопласты, Фенопласты.
Л. В. Аристовская.
ПЕНОЭПОКСИДЫ (ероху foams, Epoxyschaumstoffe,
mousses ёроху) — газонаполненные материалы на ос-
основе эпоксидных смол. П. представляют собой жесткие
материалы, преимущественно с замкнутой структурой
ячеек.
Основой композиций при получении П. служат
эпоксидные смолы, образующиеся при реакции 2,2'-ди-
(га-оксифенил)пропана (диана) с эпихлоргидрином.
Наиболее широко применяют жидкие смолы, имеющие
при 25 °С вязкость в пределах 10—100 н -сек/м2 A00—
1000 пз). Кроме диановых, для получения П. нек-рое
значение приобрели эпоксидированные новолачные
смолы, из к-рых получают материалы, отличающиеся
повышенной термич. стабильностью.
Технологич. условия вспенивания и отверждения
эпоксидных смол, а также свойства П. в значительной
стойкостью. При этом вспенивание осуществляют с по-
помощью газообразователей — карбаматов полиаминов,
азосоединений, гидразидов, боргидридов и др. соедине-
соединений с темп-рой разложения ниже 150 СС. Отверждение и
параллельно протекающее вспенивание осуществляют
обычно при 100—150 °С в течение 3—5 ч. При исполь-
использовании нек-рых кислотных катализаторов (салици-
(салициловой, муравьиной к-т и др.) темп-ру отверждения
можно снизить примерно до 60 СС при длительности
процесса 2—3 ч. Катализаторы особенно необходимы
при вспенивании смесей фреонами, кипящими ниже
0 СС, например фреоном-12 (с температурой кипения
— 29 °С).
Для ускорения отверждения в рецептуры П. с от-
вердителями аминного типа вводят также диизоциана-
ты, напр, толуилендиизоцианат. Амин при этом берется
примерно в стехиометрич. количестве в расчете на реак-
реакцию с диизоцианатом и эпоксидной смолой. Отверж-
Отверждение обеспечивается благодаря быстро проходящей
реакции диизоцианата с полиамином с образованием
соответствующих полимочевин. В результате обра-
образуется устойчивая пена.
Вспенивание можно осуществлять с помощью газов,
выделяющихся при взаимодействии компонентов ком-
композиции. К числу таких компонентов относятся смеси
триалкилбороксинов с первичными полиаминами. Ком-
Композиция в этом случае состоит из диановой эпоксидной
смолы, первичного полиамина D,4'-диаминодифенил-
сульфона) и триалкилбороксина (триметоксиборокси-
на), причем на 1 моль бороксина берут ок. 3 моль ди-
диамина. В результате реакции триметоксибороксина
с амином выделяется метанол C моль) и образуется
сложный полиамин, отверждающий эпоксидную смолу.
Реакции сильно экзотермичны, и испаряющийся мета-
метанол обеспечивает вспенивание смеси. Для улучшения
вспенивания в композицию включают фреоны. Самыми
ценными качествами эпокси-бороксиновых пенопластов
являются исключительно высокая темп-pa размягче-
размягчения (до 300 °С) и высокая термостабильность (до 200 °С
в течение 48 ч).
Отвердитель и вспенивающий агент
Показатель
мере определяются применяемым отвердителем. Из от-
вердителей практич. зна-
значение приобрели ДИ- И по- Свойства и области применения основных типов пенопластов на основе диановой эпоксидной смолы
лиамины. При этом али- —_
фатич. полиамины приме-
применяют для получения П.
при комнатной темп-ре
или при умеренном нагре-
нагревании (до 100 °С). Время
гелеобразования при при-
применении нек-рых алифа-
тич. полиаминов измеря-
измеряется долями мин, и поэ-
поэтому такие смеси можно
наносить на защищаемые
поверхности напылением.
При использовании др.
рецептур с алифатич. по-
Кажущаяся плотность,
кг/ж3
Прочность, Мн/м2 (кгс/см2)
при сжатии
при статич. изгибе
лиаминами П. получают Ударная вязкость, кдж/м*
путем заливки смесей в (юс-см/см2) . . • . . .
формы или конструкции. П^ниТ?СТеМП"Ра.ЛРИМе
Вспенивание осуществля- Коэфф. теплопроводности
ется обычно в результате вт/(м-К) [ккал/м-Ч'°С].
испарения жидкости, вхо-
входящей в состав смеси и
кипящей при сравнитель- Электрич.^прочность, кв/мм
но низких темп-pax, напр,
фреона.
Ароматич. полиамины
[ж-фенилендиамин, 4,4'-
диаминодифенилметан, эв-
тектич. смеси этих ами-
при 5 0 гц
при постоянном токе . .
Диэлектрич. проницаемость
при 106 гц
Тангенс угла диэлектрич.
потерь при 106 гц ....
Алифатич.
полиамины;
фреоны
60-200
0,25-3
B,5-
30,0)
0,55-3,5
E,5-35,0)
0,4-1,2
90-100
0,029-
0,047
[0,025-
0,040]
3,0-3,5
4,0-4,5
1,07-1,35
0,002 —
0,0 06
Ароматич.
полиамины;
газообразова-
тели
80-300
0,4-5
D,0-
50,0)
1,2-6
A2,0-60,0)
0,5-1,5
100-200
0,031 —
0,058
[0,027-
0,050]
3,0-5,0
4,0 — 5,5
1,10-1,50
0,002 —
0,007
Эвтектич.
смеси арома-
ароматич. гюли-
аминов с ка-
тализатора-
тализаторами отвержде-
отверждения; фреоны
60-300
0,3-6
C,0-
60,0)
0,7-7
G,0-70,0)
0,4-1,5
110 — 140
0,029-
0,047
[0,025-
0,040]
3,0-5,0
4,0-5,5
1,10-1,50
0,002-
0,007
Полиами-
Полиамины с три-
метоксибо-
роксином;
фреоны
30-300
0,05-4
@,5-
40,0)
200-300
0,029-
0,058
[0,025 —
0,050]
Комплексы
BF3 со
спиртами,
эфирами и
аминами;
фреоны
20—50
0,03 — 0,15
@,3 — 1 ,5)
0,07 — 0,2
@,7-2,0)
100
0,023-
0,035
[0,020 —
0,030]
1,05-1,10
0,001 —
0,002
575
ПЕНТАПЛАСТ
576
Для получения сверхлегких П. B0—30 кг/м3) в ка-
качестве отвердителей применяют комплексы BF3 со спир-
спиртами, эфирами или аминами. При этом вспенивающими
агентами служат фреоны, чаще всего фреон-11. Вспе-
Вспенивание в присутствии поверхностно-активных веществ
происходит при комнатной темп-ре, причем отвержде-
отверждение длится в течение нескольких минут. Получаемые
П. имеют малую хрупкость, упруги, устойчивы к дей-
действию света, имеют белый цвет и в отличие, напр.,
от пенополиуретанов со временем не желтеют.
Свойства П. (таблица) и, следовательно, области их
применения изменяются в широком диапазоне в зави-
зависимости от рецептур и кажущейся плотности. По комп-
комплексу свойств П. сравнимы с жесткими пенополиуре-
пенополиуретанами. Они имеют высокую механич. прочность
в расчете на единицу массы. Их получают с любой за-
заданной кажущейся плотностью в пределах от 20 до
400 кг/м3.
П. малой кажущейся плотности (менее 100 кг/м3),
получаемые при вспенивании фреонами, по эффек-
эффективности тепловой изоляции превосходят пенополиу-
пенополиуретаны благодаря чрезвычайно низкой проницаемости
паров фреонов через отвержденные пленки эпоксидных
смол. П. с отвердителями аминного типа отличаются
высокой стойкостью к действию растворителей и неф-
нефтепродуктов, в том числе при повышенных темп-рах,
и поэтому находят применение для изготовления по-
поплавков и др. приспособлений, работающих в среде
топлива.
Благодаря хорошим диэлектрич. свойствам П. ши-
широко применяют в радиотехнич. пром-сти для изоля-
изоляции различных изделий.
Объем произ-ва П. пока невелик из-за их высокой
стоимости, к-рая обусловлена высокой ценой эпоксид-
эпоксидных смол или же несовершенством технологии полу-
получения.
Лит.: Валгин В. Д., Лебедев В. С, Василье-
Васильева Э. А., Пласт, массы, № 2, 34—36 A967); Л!> 3, 23—25
A967), Х° 4, 26—28 A967); Николаев А. Ф. [и др.], Пласт,
массы, № 10, 7 A971); Т у р е ц к и й Л. В. [и др.], Пласт,
массы, № 4, 23 A972).
См. также лит. при статье Пенопласты. В. Д. Валгин.
ПЕНТАПЛАСТ (pentaplast) — принятое в СССР
торговое название поли-[3,3-бмс-(хлорметил)оксетана].
3,3-бмс-(Хлорметил)оксетан, 3,3-дихлорметилоксаци-
клобутан, (X.) — бесцветная жид-
СН2 кость; т. пл. 18,9 °С, т. кип. 203 °С,
{СЯ2С\JС// о nD 1,4857 — 1,4860, плотность
ХСН2/ 1,2996—1,3000 г/см3. X. сравните-
сравнительно легко окисляется на воздухе,
поэтому в него вводят антиоксиданты (обычно арома-
тич. амины), к-рые могут окрашивать мономер. Взаи-
Взаимодействие X. с НС1, спиртами, фенолами и аминами
сопровождается раскрытием оксетанового цикла.
В пром-сти X. получают из пентаэритрита в две
стадии:
С(СН2ОНL
HCI
ДСН2С1K
CH2OCOR
сн9
сн2
Гидрохлорирование проводят в среде органич. к-ты
(обычно уксусной или масляной) при 80—140 °С в те-
течение 6—8 ч. Образующийся трихлоргидрин обраба-
обрабатывают 20%-ным водным р-ром NaOH при 90—95 °С
и получают X.
Пентапласт [ — ОСН2С(СН2С1JСН2—]п (П.) — термо-
термопластичный линейный полимер.
П.— рогоподобный бесцветный не имеющий запаха
продукт; степень кристалличности ~ 30% , мол. масса
в зависимости от технологии получения составляет
70—200 тыс. Соотношение между мол. массой М и ха-
рактеристич. вязкостью [г\] в р-ре циклогексанона при
40 °С выражается ур-нием: [ц] = 3,81-10~3- Af0*837.
В пром-сти мол. массу П. характеризуют приведенной
вязкостью 0,5%-ного р-ра П. в циклогексаноне. Эта
величина колеблется в пределах 1,2—2,0. По проч-
прочностным показателям П. близок полипропилену. Ниже
приведены нек-рые свойства П.:
Плотность при 20°С, г/см3
аморфной фазы 1,390 — 1,41
кристаллич. фазы 1,47
Темп-pa, СС
плавления кристаллич. фазы ок. 185
стеклования 5
хрупкости 2 0
Теплостойкость, °С
по Вика 160-168
по Мартенсу 45
под нагрузкой 1,85 Ми/м2 A8,Ькгс/см2) 99
Коэфф. теплопроводности, вт/(м-К) ... 0,13
кал/(см-сек-°С) 3,13-Ю
Темп-рный коэфф. линейного расшире-
расширения, ''С E-8)-Ю-3
Прочность, Ми/м2 (кгс/см2)
при растяжении при 2 0°С 4 0 — 55
D00 — 550)
при 100°С ок. 25 (ок.250)
при статич. изгибе при 20 "С 6 5 — 80
F50-800)
Ударная вязкость, кдж/м2, или
кгс • см/см2 8 0
Модуль упругости при изгибе,
Мн/м2 (кгс/см2) 900 — 1200
(9000 — 12000)
Относительное удлинение, % 10 — 15
Диэлектрич. проницаемость
при 1 пгц 3,3 — 3,1
при 1 Мгц 3,0-3,1
Тангенс угла диэлектрич. потерь
при 60 г^ 0,011
при 1 Мгц 0,009 — 0,012
Уд. объемное электрич. сопротивление,
Том-м[ом-см] A — 3) • 1 О2
[A-3). 10"]
Уд. поверхностное электрич. сопротивле-
сопротивление, Том [ом] @ ,8 —6,5)-КL
[@,8-6, 5). 106]
Электрич. прочность, Мв/м, или кв/мм . . 21—27
П. устойчив к действию большинства растворителей;
он растворяется лишь в циклогексаноне и хлорбензоле,
а также в кипящем диоксане и диметилформамиде при
темп-ре выше 110 °С, но при снижении темп-ры ниже
60 °С полностью выпадает из р-ра. Р-ры выше 5%-ной
концентрации желатинизируются.
По химстойкости П. превосходит поливини л хлорид
и несколько уступает фторопластам. Напр., он стоек
к действию конц. минеральных к-т при нагревании
до 100 °С, однако разрушается под действием сильных
окисляющих агентов, напр, азотной к-ты, олеума. Для
предотвращения деструкции П. при нагревании (осо-
(особенно в условиях переработки в изделия) его стабили-
стабилизируют ароматич. аминами, напр. К,К'-ди-C-нафтил-
га-фенилендиамином, фенолами, напр. бмс-E-метил-3-
тредп-бутил-2-оксифенилметаном) и эпоксидными сое-
соединениями. Для повышения химстойкости П. часто
наполняют инертными мелкодисперсными наполните-
наполнителями, напр, окисью хрома.
В пром-сти П. получают катионной или анионной
полимеризацией X. под действием соответственно к-т
Лыоиса или металлоорганич. соединений. Процесс
проводят в суспензии в органич. растворителях.
П. хорошо перерабатывается литьем под давлением,
экструзией с раздувом, вакуум- и пневмоформованием;
хорошо сваривается в токе горячего воздуха (контакт-
(контактным методом или с использованием прутка из П.)
и склеивается полярными клеями, напр, эпоксидными.
Так как П. может деструктироваться при темп-pax пере-
переработки, его следует перерабатывать на оборудовании,
на к-ром обеспечивается кратковременный нагрев
до необходимой темп-ры и отсутствуют «застойные»
577
ПЕРЕДАЧА ЦЕПИ
578
зоны. Вязкость расплава П.резко изменяется с темп-рой,
поэтому переработку осуществляют в узких темп-рных
пределах. В зависимости от мол. массы П., конфигура-
конфигурации и величины изделия, типа машины и д.р. литье под
давлением проводят при 190—240 °С (темп-pa формы
85-100 °С); экструзию — при 180 — 235 °С. Из-за срав-
сравнительно низкой вязкости расплава давление при пере-
переработке должно быть 80—100 Мн/м2 (800—1000 кгс/см2).
П. с приведенной вязкостью до 1,6 применяют гл.
обр. для нанесения антикоррозионных покрытий
на химич. аппаратуру и трубы; П. с вязкостью более
1,6— для изготовления различных машиностроитель-
машиностроительных деталей повышенной точности (в основном литьем
под давлением; при этом усадка составляет 0,3—0,5%).
Футеровку крупногабаритных аппаратов осуществ-
осуществляют наклеиванием тонкого листа П. при помощи клея
с последующей сваркой швов. Покрытия толщиной
до 0,5 мм получают путем вихревого или газопламен-
газопламенного напыления порошкообразного П.; покрытия мень-
меньшей толщины — распылением или нанесением кистью
суспензий П. в органич. растворителях с последующим
спеканием при 200—220 °С (см. также Антикоррози-
Антикоррозионные полимерные покрытия). Нек-рое применение П.
находит и в качестве кабельной химстойкой изоляции.
В США П. выпускают иод названием п е н т о н.
Впервые поли-3,3-бмс-(хлорметилоксетан) получил
Фартинг в 1954.
Лит.: Николаев А. Ф., Синтетические полимеры и
пластические массы на их основе, М.— Л., 1966; Справочник
по химии полимеров, Киев, 1971; Зарубежные промышленные
полимерные материалы и их компоненты. Толковый словарь-
справочник, М., 1963; Справочник по пластическим массам,
под ред. М. И. Гарбара [и др.], т. 2, М., 1969. Г. А. Балаев.
ПЕНТАФТАЛЕВЫЕ ЛАКИ И ЭМАЛИ — см.
Алкидные лаки и эмали.
ПЕНТАФТАЛЕВЫЕ СМОЛЫ — см. Алкидные смолы.
ПЕНТОЗАНЫ (pentosanes, Pentosane, pentosanes) —
полисахариды, построенные из остатков пентоз. В при-
природе распространены П. двух типов — а р а б а н ы,
построенные из остатков L-арабинозы, и к с и л а н ы,
построенные из остатков D-ксилозы. Нек-рые ксиланы,
кроме ксилозы, содержат незначительные количества
L-арабинозы, D-глюкозы, D-глюкуроновой к-ты и ее
4-О-метильного производного; ксиланы с высоким
содержанием арабинозы наз. арабоксилана-
м и. Арабаны в растениях сопутствуют пектиновым
веществам; ксиланы входят в состав гемицеллюлоз,
чрезвычайно широко распространены в наземных расте-
растениях, встречаются и в водорослях.
В арабанах (мол. масса 5000—6000) остатки ара-
арабинозы в фуранозной форме связаны преимущественно
а-1,3- и а-1,5-связями. Ксиланы объединяют группу
полисахаридов, значительно различающихся по строе-
строению; мол. масса порядка 10 000—30 000. Молекулы
нек-рых ксиланов представляют собой линейные или
слабо разветвленные цепи, состоящие из остатков кси-
ксилозы в ппранозной форме, соединенных |3-1,4-связями;
остатки др. моносахаридов образуют в ксиланах
обычно боковые цени.
П.— бесцветные аморфные вещества. Арабаны хорошо
растворимы в воде, ксиланы в воде не растворяются.
Восстанавливающими свойствами П. не обладают. Они
оптически активны, вращают плоскость поляризован-
поляризованного света влево: [a]D арабанов от —60 до —180°,
[а]о ксиланов от —90 до —110°. Получают П. из расти-
растительного сырья (соломы, кукурузных початков, подсол-
подсолнечной шелухи). Пентозансодержащее сырье исполь-
используется для получения фурфурола и производных фурана.
Лит.: Н и к и т и н Н. И., Химия древесины и целлю-
целлюлозы, М. — Л., 1962; Роговин 3. А., Шорыги-
н а Н. Н., Химия целлюлозы и ее спутников, М.— Л., 1953;
Химия древесины, 8. Химия гемицеллюлоз, Рига, 1971; Cellulose
and cellulose derivatives, ed. E. Ott [a. o.], pt. 1 — 2, 2 ed.,
N. Y. — L., 1954; Methods in carbo-hydrate chemistry, ed.
R. L. Whistler, v. 5 — General polysaccharides, N. Y.— L.,
1965. См. также лит. к ст. Полисахариды. Л. И. Липевич.
ПЕНТОН — см. Пентапласт.
ПЕРЕДАЧА ЦЕПИ при полимеризации
(chain transfer, Ketteniibertragung, transfert de chaine) —
элементарный акт процесса полимеризации, приводя-
приводящий к переносу активного центра от растущей полимер-
полимерной молекулы на другую частицу (т. наз. передатчик
цепи). Схематически П. ц. изображают след. образом:
Р*п + А Ь pn + л*
где Рп, Рп — соответственно активные (растущие)
и неактивные полимерные молекулы, имеющие степень
полимеризации п, А — молекула передатчика, Л* —
новая активная частица, к1Т — константа скорости
П. ц. В роли передатчика цепи могут выступать моле-
молекулы мономера, полимера, растворителя или инициа-
инициатора, компоненты каталитич. комплекса (в частности,
противоион), а также молекулы специально введенных
низкомолекулярных добавок или примесей.
Реакции П. ц. часто характеризуют относи-
относительной константой передачи цепи,
представляющей собой отношение кп/кр, где кр —
константа скорости роста цени.
Обычно под П. ц. понимают только такой процесс,
в результате к-рого получается частица Л* с той же
или большей активностью, чем исходный растущий
активный центр Р*п. Поэтому П. ц. приводит к про-
продолжению кинетич. цепи и прекращению (ограниче-
(ограничению) роста материальной цепи (макромолекулы). Если
же активность частицы Л* значительно меньше, чем
jP*, т. е. константа скорости реакции Л* + М _},Р\
значительно меньше константы скорости роста цепи
* А*
Pn -f М_Р ^п-и (^ —молекула мономера), мы
имеем дело с т. наз. вырожденной передачей цепи, или
ингибированием (см. Ингибирование полимеризации).
П. ц. практически не влияет на скорость полимери-
полимеризации, т. к. при кг^ /ср большая часть активных цент-
центров находится в виде растущих полимерных молекул
2.Р* > Л* , и скорость определяется их реакци-
реакционной способностью. При к^ < кр (ингибирование)
значительная часть активных центров находится в виде
малоактивных частиц Л*, скорость полимеризации
снижается и зависит от количества передатчика (инги-
(ингибитора), константы передачи (ингибирования) и кг.
С другой стороны, П. ц. на низкомолекулярные
вещества увеличивает число полимерных молекул,
уменьшает среднюю мол. массу, влияет на молекуляр-
но-массовое распределение продукта и характер конце-
концевых групп. П. ц. на полимер приводит к образованию
разветвленных продуктов, макроциклов, статистиче-
статистических, блок- или привитых сополимеров (см. ниже).
П. ц. сопровождает в той или иной степени боль-
большинство полимеризационных процессов. Лишь в
нек-рых случаях анионной полимеризации виниловых
мономеров и катионной полимеризации гетероциклов
удается полностью исключить акты передачи и гибели
цепи и получить т. наз. живущие полимеры.
Химизм П. ц. во многих случаях сходен как в ради-
радикальной, так и в ионной и координационно-ионной
полимеризации. Большинство реакций П. ц. представ-
представляет собой перенос водорода (при радикальной поли-
полимеризации), протона или гидридного иона (при ионной
полимеризации) от передатчика к активному центру
полимеризации или от активного центра к передатчику:
или — R* + нв-> ~ rh -f в*
н
-С - R*
НВ*
579
ПЕРЕДАЧА ЦЕПИ
580
где R* — активная концевая группа в полимере, НВ
или В — передатчик цепи; В* или НВ* — новая
активная частица. Имеется предположение о передаче
целой группы (СН3) при радикальной полимеризации.
Однако эти случаи весьма редки и доказательства этого
предположения недостаточно убедительны. При ионной
полимеризации возможна П. ц. с переносом целой
группы (реакции замещения групп у гетероатомов).
Скорость П. ц. и место отщепления водорода (в виде
атома, протона или гидридиого иона) определяются
подвижностью последнего или энергией связи водоро-
водорода с углеродом или др. атомом молекулы передатчика.
Напр., при радикальной полимеризации наибольшая
активность наблюдается для соединений, имеющих тре-
третичный атом водорода, меньше — у соединений с вторич-
вторичным и еще меньше — с первичным атомом водорода.
В частности, для производных бензола активность
изменяется в ряду: бензол <^ толуол <^ этилбензол <^
< изопрогшлбензол. В спиртах, кетонах, к-тах и
сложных эфирах преимущественно отщепляется водо-
водород в а-иоложении. Весьма эффективные передатчики
цепи — меркаптаны.
Скорость П. ц. увеличивается с ростом реакционной
способности растущего конца цени или энергии обра-
образующейся связи R—Н. Так, эффективность П. ц. на уг-
углеводороды при радикальной полимеризации растет при
переходе стирол —> метилметакрилат -> акрилонит-
рил —> винилацетат —> этилен. В случае ионной поли-
полимеризации относительная эффективность П. ц. в зна-
значительной степени зависит также от природы раствори-
растворителя и каталитич. системы.
Известна П. ц. с отрывом атома галогена. Наиболее
распространенный случай такой реакции — радикаль-
радикальная полимеризация виниловых мономеров в присутст-
присутствии СС14. В нек-рых условиях при этом образуются
олигомеры, содержащие на одном конце молекулы
атом хлора, на другом — группировку СС13. Эта реак-
реакция наз. теломеризацией. Галогеналкилы и др. гало-
генсодержащие соединения м. б. передатчиками цени
как в радикальной, так и в катионной полимеризации,
где они часто применяются в качестве растворителей.
В ионной полимеризации широко распространена
П. ц. на нротивоион, осуществляемая с переносом про-
протона при катионной или гидрид-иона при анионной
полимеризации, напр.:
R R
! I
~> СН2-С+, [BF,OH]~ -> ~'CH = C-bBF3OH2
н н
При этом обычно восстанавливается первичная ини-
инициирующая частица, быстро присоединяющая моле-
молекулу мономера и продолжающая расти дальше.
В нек-рых случаях реакции П. ц. специфичны для
полимеризационных систем определенного типа. Напр.,
при полимеризации на катализаторах Циглера —
Натта агентами П. ц. являются металлалкилы:
Кат - СН,-СН - + RjAl—>KaT-R + R,A1-CH2- CH-
I
СНз СНз
Особое место занимает П. ц. на мономер и полимер.
П. ц. на мономер определяет максимальное значение
мол. массы полимера, к-рое м. б. достигнуто при дан-
данных темп-ре и давлении. В частности, П. ц. на моно-
мономер не позволяет получить при радикальной полиме-
полимеризации (при 60 °С) стирола полимеры со степенью
полимеризации выше 1,5-104, металметакрилата —
КM, винилхлорида — 5-Ю4, винилацетата — 5-Ю3,
аллилацетата — 14. Энергия активации П. ц. обычно
выше, чем энергия активации роста цепи. Поэтому
увеличение темп-ры полимеризации приводит к сниже-
снижению мол. массы полимера вследствие П. ц.
Роль реакций П. ц. на полимер в процессе получе-
получения полимера увеличивается по ходу реакции, т. е.
по мере увеличения концентрации полимера. Такие
процессы можно разделить на два типа.
1. Перенос водорода (при радикальной полимериза-
полимеризации), протона или гидридного иона (при ионной) про-
происходит на конец растущей полимерной молекулы
от внутреннего звена др. макромолекулы, напр.:
2 *'.
—R +НоС—> —RH + НС
тт ~} (
Дальнейшее присоединение молекул мономера к ра-
радикалу — С — дает разветвленную макромолекулу.
Эта реакция не меняет числа макромолекул, однако
приводит к увеличению доли более длинных макромо-
макромолекул в полимере, т. е., не изменяя среднечисленной
мол. массы, расширяет молекулярно-массовое распреде-
распределение. Если концентрация полимера достаточно велика,
то возможно взаимодействие двух радикалов — СН~
с образованием поперечных сшивок. Относительная
скорость П. ц. на полимер в этом случае определя-
определяется подвижностью атома водорода в полимерной цени.
Напр., относительная константа П. ц. на полимер
при радикальной полимеризации винилацетата равна
1,02-10~3, метнлметакрнлата — 0,04-10~3 и сти-
стирола — 0,20-10-3 (при 50 СС).
В нек-рых случаях радикал^ СН ~, не успевая при-
присоединить молекулу мономера, распадается. Такая
реакция не может привести к образованию разветвлен-
разветвленного полимера, а ведет лишь к увеличению числа мак-
макромолекул и, следовательно, уменьшению мол. массы
полимера. Существование такой реакции было доказано
для гюлиизобутилена и для полиизопрена. Аналогичная
реакция, по-видимому, происходит при катиопной
полимеризации цпклич. ацеталей (триоксан, диоксолан
и др.) в результате гидрндного переноса от полимера:
н н + н н
I ••• II
-'О-С-О-С -+ Н2С~—О--->-О-С-О-С—fH,C-O-->
I i I
н н н
—>~О—~СН2 -{- О=СН^ 4- НзС-0 ~*
2. При образовании гетероцепных полимеров про-
протекает обменная реакция активной концевой группы
растущей макромолекулы с гетсроатомом основной
цепи. В результате происходит разрыв атакуемой цепи,
и один из осколков присоединяется к атакующей моле-
молекуле, напр.:
Б'-О-С+, A--hR"-O-R"' +± R'-O-R'" -f- R"-O-C+, A~
Эта реакция паз. передачей цеии с р а з-
р ы в о м. Аналогичная реакция идет при радикальной
полимеризации дисульфидов:
R"
R -S ¦
'RS- SR" + S - R"
Следствием такой реакции м. б. образование блоксо-
полимеров, если атакующая и атакуемая макромоле-
макромолекулы состоят из различных мономерных звеньев, пли
циклич. олигомеров и высокомолекулярных макроцик-
макроциклов, если активная концевая группа атакует внутрен-
внутреннее звено той же макромолекулы. Циклич. олигомеры
образуются, в частности, при катионной полимериза-
полимеризации формальдегида и триоксана, макроциклы — при
катионной полимеризации диоксолана.
П. ц. с разрывом (за исключением тех случаев, когда
образуются макроциклы) не приводит к изменению
среднечисленной степени полимеризации, но изменяет
молекулярно-массовое распределение полимера. Такие
реакции обратимы и стремятся привести линейные
и цпклич. макромолекулы к равновесному молекуляр-
но-массовому распределению. Кроме того, эти реакции
581
ПЕРЕРАБОТКА ПЛАСТИЧЕСКИХ МАСС
582
приводят к перераспределению концевых групп в по-
полимере и различных мономерных звеньев вдоль цепи
в сополимере, причем и в этом случае в конце концов
распределение становится равновесным, т. е. термо-
термодинамически наиболее выгодным. П. ц. с разрывом на
боковые группы макромолекулы может привести к раз-
разветвленным макромолекулам и привитым сополимерам.
По механизму, аналогичному П. ц. с разрывом, идет
П. ц. на низкомолекулярные кислородсодержащие ве-
вещества при катионной полимеризации, напр.:
^-0 - С+, А" + СН,ОСН2ОСН, —> —О — С - О - СЫз +
-j-A-,+C-O-CH,
Наиболее интенсивно П. ц. с разрывом протекает в си-
системах, содержащих ацетали и полиацетали. При поли-
полимеризации триоксана относительная константа пере-
передачи цепи на олигомеры диметоксиметиленгликоля
равна 1, что свидетельствует о равноценности ацеталь-
ных связей в мономере и олигомере. При радикальной
полимеризации аналогичным образом происходит пере-
передача на дисульфиды и перекиси, к-рые одновременно
м. б. и инициаторами полимеризации.
Идентификация реакций П. ц. осно-
основана на структурно-химич. методах (анализ концевых
групп, разветвленности, структуры сополимера и т. д.)
и кинетпч. .методах (исследование средних мол. масс
п молекулярно-массового распределения). Примером
использования первой группы методов является изме-
измерение количества галогена в полимере при П. ц. на га-
логенсодержащпе соединения или анализ двойных свя-
связей в полимере при отщеплении водорода с конца цепи.
При этом используют обычные химические или фи-
физико-химические методы анализа (ИК-сиектроскопия,
ЯМР и др.).
Разветвленность полимера обнаруживается при со-
сопоставлении характеристич. вязкости и среднечислен-
ной или среднемассовой мол. массы, измеренных др.
методами, или при измерении размеров полимерного
клубка в разб. р-ре полимера. Разветвленные полимеры
имеют значительно меньшие вязкости и размеры клубка
в р-ре, чем линейные полимеры той же мол. массы.
П. ц. на полимер можно идентифицировать и но обра-
образующемуся привитому сополимеру, если полимериза-
полимеризация мономера проводится в присутствии полимера,
построенного из других мономерных звеньев.
Один из наиболее распространенных способов ис-
исследования П. ц.— изучение закономерностей измене-
изменения мол. массы в зависимости от концентраций реаген-
реагентов. Кинетпч. обработка различна для квазистационар-
квазистационарной полимеризации (время роста одной цени значитель-
значительно меньше времени реакции) и нестационарного про-
процесса.
При квазистацттоиарном процессе среднечисленная
степень полимеризации (Рп) полимера, образующегося
в нек-рый момент времени, равна отношению скорости
роста цепи к сумме скоростей всех реакций ограниче-
ограничения роста цепи (передача и гибель):
где Up, vo, ulT — соответственно скорости процессов
роста, гибели и передачи цепи.
При радикальной полимеризации и бимолекулярном
обрыве справедливо выражение для средней степени
полимеризации:
&о1' , А\<
М
Рп ~ к\М* ' Лр Л/
где М, S, X — соответственно концентрации мономера,
растворителя и передатчика ценя, ки, kg, k0, ks, kM,
kh кх — соответственно константы скорости иници-
инициирования, роста, гибели и передачи цепи на раствори-
растворитель, мономер, инициатор и передатчик цепи, и — об-
общая скорость полимеризации. П. ц. па инициатор
обнаруживается по параболич. зависимости 1/Рп от и.
Относительные константы передачи на растворитель
(ks/kp) или передатчик (кх/кр) определяются из на-
наклона прямых зависимостей \/Рп от SlM или Х/М.
Относительную константу скорости передачи на моно-
мономер можно найти из графика зависимости 1/Рп от и/М'1.
Последняя величина варьируется измерением концент-
концентрации мономера или инициатора. Отрезок, отсекаемый
прямой на оси ординат, равен км/кр в отсутствие др.
актов передачи.
При ионной квазистационарпой полимеризации реак-
реакция гибели цепи мономолекулярна, и выражение для
средней степени полимеризации имеет вид:
-1 I' ' h h <ч Ь к Т h Y
о — п р s ,м I /" I х
рЦ= крМ ' к^ ~М r~k^rFpW~rT9Tl
где ^пр — константа скорости П. ц. на противоиоп,
/ — концентрация инициатора.
Константы П. ц. находятся при помощи :>того ур-ния
из зависимостей мол. массы от концентраций мономера,
растворителя, катализатора или передатчика. Вообще
говоря, при полимеризации до глубоких стадий мол.
масса полимера, образующегося в каждый момент
времени, меняется и для вычисления средней мол.
массы всего полимера, полученного за большой про-
промежуток времени, необходимо интегрировать соответ-
соответствующие ур-ния. Получающиеся выражения сложны,
поэтому предпочитают измерять мол. массы на началь-
начальных стадиях реакции, пока можно пренебречь измене-
изменением концентраций реагентов.
При нестационарной полимеризации для среднечис-
ленной степени полимеризации следует пользоваться
интегральным выражением:
\vpdt
Это ур-нне анализируется с учетом конкретных
механизмов полимеризации и позволяет в иек-рых
случаях определять константы П. ц.
Реакции П. ц. широко применяются в промышлен-
промышленных процессах радикальной и ионной полимеризации
для регулирования средней мол. массы полимера, для
введения в макромолекулы реакционноспособных кон-
концевых групп, получения привитых сополимеров (см.
Регуляторы молекулярной массы, Теломеризация).
Лит.: Бреслер С. Е., Е р у с а л и м с к и й Б. Л.,
Физика и химия макромолекул, М., 1965; Багдасарь-
я и X. С, Теория радикальной полимеризации, 2 изд., М.,
1966; Кинетика радикальной полимеризации виниловых соеди-
соединений, пер. с англ., М., 1961; II л е ш П., [ред.], Катпонная
полимеризация, пер. с англ., М., 1966; Е н и коло п я н Н. С,
И р ж а к В. И., Р о з е н б е р г Б. А., Усп. хим., 35, 714
A966); Шварц М., Анионная полимеризация, лор. с англ.,
М., 1971. Ал. Ал. Берлин.
ПЕРЕРАБОТКА ПЛАСТИЧЕСКИХ МАСС (plastics
processing, Verarbeitung von Kunststofien, traitement
des plastiqnes) — комплекс процессов, обеспечивающий
получение изделий или полуфабрикатов из пластмасс
с заданными свойствами на специальном оборудовании.
Переработке пластмасс предшествует проектирование
рациональной конструкции изделия и формующего
инструмента (формы, головки и др.), выбор оптималь-
оптимального метода переработки и условий его осуществления,
разработка рецептуры материала, наиболее пригодной
для данного метода и последующих условий .жеплуа-
тацип. Собственно П. и. м. включает приготовление
материала и подготовку его к формованию (гранулиро-
(гранулирование, таб л е жирование), формование изделий и их
последующую обработку с целью улучшения свойств
полимера (термич. обработка, радиационное сши-
сшивание и др.).
Переработка пластмасс возникла в середине XIX в.
одновременно с появлением первых искусственных;
583
ПЕРЕРАБОТКА ПЛАСТИЧЕСКИХ МАСС
584
материалов, прежде всего нитроцеллюлозы. Первыми
методами формования изделий из иластмасс были прес-
прессование и выдавливание (плунжерная экструзия),
применявшиеся ранее для получения изделий из
нек-рых видов материалов, обладающих высокой
пластичностью (глина, мягкие металлы). Вплоть до на-
начала XX в. методы переработки полимеров в изделия
копировали известные способы переработки тради-
традиционных материалов (литье, прессование, штамповка),
и лишь к середине XX в. появились новые приемы
формования пластмасс, использующие снецифич. осо-
особенности свойств полимеров,— вакуумформование,
каландрование и др. Сейчас число разнообразных мето-
методов и приемов П. и. м. исчисляется многими десятками.
Классификация методов переработки. При выборе
методов П. п. м. можно использовать след. их класси-
классификацию, основанную на физич. состоянии материала
в момент формования:
1. Формование из полимеров, находящихся в вязко-
текучем состоянии,— литье под давлением, экструзия,
прессование, спекание и др.
2. Формование из полимеров, находящихся в высоко-
высокоэластическом состоянии, обычно с использованием
листов или пленочных заготовок (вакуумформование,
пневмоформовапие, горячая штамповка и др.).
3. Формование из полимеров, находящихся в твер-
твердом (кристаллическом или стеклообразном) состоянии,
основанное на способности таких полимеров про-
проявлять высокоэластичностъ вынужденную (штам-
(штамповка при комнатной темп-ре, прокатка п др.).
4. Формование с использованием р-ров и дисперсий
полимеров (получение пленок методом полива — см.
Пленки полимерные, формование изделий окунанием
формы, ротационное формование пластизолей и др.).
Изделия и полуфабрикаты из пластмасс можно под-
подвергать также механической обработке, сварке (изделия
из термопластов), склеиванию. Специфич. методы пере-
переработки существуют для реактопластов — намотка,
контактное формование и др. (см. Стеклопластики).
К этой группе примыкают также методы формования
изделий непосредственно из мономеров или нолимер-
мономерных композиций (полимеризация в форме —
см., напр., Капролактама полимеры, пропитка в форме
под давлением и др.). В ряде случаев в едином потоке
сочетается несколько методов формования.
Особую группу составляют методы формования изде-
изделий из вспененных материалов, при изготовлении
к-рых находят применение как упомянутые выше
методы (литье, экструзия, прессование и др.), так
и нек-рые специальные приемы (см. Пенопласты).
Характеристика основных методов переработки. Мно-
Многие изделия из термопластов и реактопластов м. б.
изготовлены несколькими различными методами. Вы-
Выбор метода переработки для каждого конкретного изде-
изделия определяется большим числом факторов, важней-
важнейшими из к-рых являются конструктивные особенности
изделия, особенности свойств и технологич. возмож-
возможности выбранного полимера, условия эксплуатации
изделия и вытекающие из них требования к нему
(чистота и качество поверхности, точность размеров,
наличие арматуры, резьб, знаков и др.)> предполагае-
предполагаемая тиражность изделия, а также экономич. факторы —
стоимость оборудования и оснастки, их производитель-
производительность и срок эксплуатации, затраты труда и его ква-
квалификация и др. В ряде случаев определяющим фак-
фактором может оказаться тиражность — для выпуска
небольших партий изделий можно использовать мало-
малопроизводительные методы формования и применять
при этом более дешевую оснастку, тогда как крупносе-
крупносерийное производство оправдывает значительные рас-
расходы на изготовление оснастки, связанные с использо-
использованием наиболее производительных методов перера-
переработки.
Литьем под давлением можно с высокой
производительностью получать из термопластов изде-
изделия массой от долей г до десятков кг, из реактопластов—
от нескольких г до нескольких кг. Применение много-
гнездных форм, предварительный подогрев сырья
и прочие усовершенствования позволяют достигнуть
высокой эффективности использования оборудования.
Степень автоматизации процесса достаточно высока —
уже имеются полностью автоматизированные линии,
управляемые с помощью ЭВМ. Современные конструк-
конструкции литьевых машин позволяют получать изделия
двух и более цветов, пористые изделия с различной
плотностью по сечению изделия, многослойные изде-
изделия и др. Недостатки метода — высокая стоимость
формующего инструмента, сравнительно низкая про-
производительность при изготовлении армированных изде-
изделий и изделий сложной конфигурации.
Реактопласты льют под давлением реже, чем термо-
термопласты, однако этот метод их переработки весьма
прогрессивен. Благодаря интенсивному перемешива-
перемешиванию материала в процессе формования скорость и сте-
степень отверждения материала выше, чем при прессова-
прессовании. Этим методом можно изготавливать широкий
ассортимент изделий. Наиболее эффективен он при
производстве толстостенных деталей.
Экструзией можно получать непрерывные
изделия — пленки, профили самого разнообразного
типа, листы, трубы и шланги, а также объемные изде-
изделия. Масса погонного метра изделий, полученных
этим методом, может составлять от нескольких г до
100 кг и более; получены пленки шириной до 25 м и тру-
трубы диаметром до 1,2 м. Экструзией можно получать
многослойные изделия, вспененные изделия с поверх-
поверхностью, имитирующей различные декоративные мате-
материалы, и др. Производительность крупных экструде-
ров достигает 3—3,5 т/ч, степень автоматизации про-
производства также достаточно высока. Недостатки мето-
метода — сложность управления процессом и высокая стои-
стоимость оборудования.
Прессованном можно получать изделия слож-
сложной формы, разнообразных размеров pi толщины. Недо-
Недостатки метода — низкая производительность и труд-
трудность достижения высокого уровня автоматизации,
однако в последние годы в этом направлении достигну-
достигнуты большие успехи.
Вакуумформование — сравнительно но-
новый метод, появившийся в конце 40-х гг.; он позволяет
получать из листовых и пленочных материалов объем-
объемные изделия, как крупногабаритные, так и мелкие,
от ванн до мелкой тары для упаковки. Его преимущест-
преимущества — низкая стоимость формующего инструмента,
возможность автоматизации и организации непрерыв-
непрерывных процессов. Недостатки — большое количество
отходов, разнотолщинность получаемых изделий, отно-
относительно невысокая производительность.
Уд. вес основных методов переработки пластмасс
характеризуется данными таблицы.
Доля основных методов переработки в структуре пром-сти
переработки пластмасс нек-рых стран (и %, данные за 1907)
Метод
Литье под да»влением
Экструзия
Экструзия с раздувом
Прессование
Вакуумформование .
Каландрование ...
США
22,4
18,0
0,0
5,0
7,0
Япо-
Японии
18,0
47,7
2,1
9,3
1 ,8
16,3
Фран- ссср*
29, О I
7,1
9 , 3
8,0
24,1
31,4
34, Г»
2,7
3,9
* Данные за 1972.
Физические и химические процессы, происходящие
при переработке. Производительность методов и е-
реработки термопластов в основном
585
ПЕРЕРАБОТКА ПЛАСТИЧЕСКИХ МАСС
586
лимитируется скоростью происходящих при переработ-
переработке физич. и физико-химич. процессов (плавление, кри-
кристаллизация, нагрев и охлаждение, релаксация и др.).
Полнота и характер протекания этих процессов в зна-
значительной мере определяют также качество готового
изделия. Кроме того, на качество изделий оказывают
влияние деструктивные процессы, протекающие с по-
повышенной скоростью в полимере при его переработке
вследствие термических и механических воздей-
воздействий на материал со стороны рабочих органов
машин.
Придание формы изделию из термопластов м. б.
достигнуто в результате развития в полимере пласти-
пластической или высокоэластич. деформации. Из-за высокой
вязкости материала эти процессы деформирования про-
протекают с низкой скоростью. В зависимости от физич.
состояния, в к-ром полимер находится в момент фор-
формования, в готовом изделии достигается различная
степень неравновесности из-за неполной релаксации
внутренних напряжений. Это накладывает опреде-
определенные ограничения на температурный интервал экс-
эксплуатации изделий, полученных различными методами.
Увеличение доли высокоэластич. составляющей дефор-
деформации ведет к снижению верхнего температурного пре-
предела эксплуатации вплоть до темп-ры стеклования. Это
особенно заметно проявляется при обработке изделий
и полуфабрикатов из полимерных материалов, нахо-
находящихся в стеклообразном состоянии, при напряже-
напряжениях, превышающих предел вынужденной высокоэла-
стичности. Такой прием позволяет в значительной
степени увеличить прочностные показатели вследствие
ориентации надмолекулярных образований и уплот-
уплотнения рыхлой структуры полимера (напр., при про-
прокатке пленок и труб). Однако изделия, полученные
этим методом, должны эксплуатироваться при темн-рах
ниже темп-ры стеклования полимера, т. к. при более
высоких темп-pax они начинают необратимо деформи-
деформироваться из-за резкого ускорения релаксационных
процессов.
Особенность методов переработки реак-
то п ластов — сочетание физич. процессов собст-
собственно формования с химич. реакциями образования
трехмерных полимеров (отверждением), причем свойст-
свойства изделий определяются скоростью и полнотой отверж-
отверждения. Неполное исчерпывание при отверждении реак-
ционноспособных групп обусловливает нестабильность
свойств изделий из реактопластов во времени, а также
протекание деструктивных процессов в готовых изде-
изделиях. Низкая вязкость олигомеров в процессах формо-
формования ведет к снижению ориентационных эффектов,
увеличению скорости релаксации и меньшему влиянию
процессов деструкции при переработке на качество
и свойства готовых изделий из реактопластов.
В зависимости от метода переработки отверждение
совмещается с формованием изделия (в случае прессо-
прессования реактопластов), происходит после оформления
изделия в полости формы (литьевое прессование, литье
под давлением реактопластов) или при термич. обработ-
обработке сфорхмованной заготовки (при формовании крупно-
крупногабаритных изделий — см. Стеклопластики). Дости-
Достижение необходимой полноты отверждения нек-рых
типов олигомеров даже в присутствии катализаторов
и при повышенных темн-рах требует значительного
времени — до нескольких часов. Однако окончатель-
окончательное отверждение может проводиться вне формующей
оснастки, т. к. устойчивость формы приобретается
задолго до завершения этого процесса. По этой же
причине изделия можно извлекать из формы без охлаж-
охлаждения.
Процессы П. н. м., в первую очередь те из них,
в к-рых формование осуществляется в высокоэласти-
высокоэластическом и вязкотекучем состоянии, связаны с ориента-
ориентацией макромолекул и надмолекулярных образований.
В производстве пленок, при нанесении кабельной изо-:
ляции, литье тонкостенных изделий реализуются весь-
весьма высокие степени ориентации. Это позволяет значи-
значительно повысить прочностные характеристики в направ-
направлении ориентации, а при двухосной ориентации достиг-
достигнуть эффекта упрочнения без возникновения анизотро-
анизотропии свойств, характерной для одноосной ориентации
(см. Ориентированное состояние). Наряду с этим, раз-
различие в степени ориентации на разных участках, не-
неоднородных по сечению или длине изделий, ведет к
возникновению структурной неоднородности и разви-
развитию внутренних напряжений.
Наличие при переработке температурных перепадов
по сечению изделия ведет к еще большему возрастанию
структурной неоднородности и появлению дополни-
дополнительных напряжений, связанных с различием в ско-
скоростях охлаждения, кристаллизации, релаксации
в различных частях изделия, а также с различной
степенью отверждения (в случае реактопластов).
Указанные особенности обусловливают неоднород-
неоднородность свойств материала в изделии, что не всегда допу-
допустимо, и являются причиной значительного числа видов
брака в изделиях (нестабильность размеров, коробле-
коробление, растрескивание). Существование внутренних на-
напряжений, в первую очередь ориентационных, ограни-
ограничивает также температурный интервал эксплуатации.
Нек-рого повышения однородности надмолекулярной
структуры и снижения внутренних напряжений уда-
удается достигнуть в результате термич. обработки готово-
готового изделия, однако более эффективно использование
методов направленного регулирования структур в про-
процессах переработки.
В ряде случаев умелое использование ориентации
позволяет повысить прочностные показатели в направ-
направлении действия наибольших нагрузок. Это учитывается
при проектировании оформляющего инструмента и
оснастки. Так, при литье под давлением этого удается
достичь правильным расположением литников (см.
Литьевые формы), при производстве труб методом эк-
экструзии — поперечной ориентацией горячей заготовки
или использованием формующего инструмента с допол-
дополнительной поперечной ориентацией потока расплава
(за счет вращения, циклич. деформирования и др.).
Значительные тепловые и механич. воздействия,
к-рые испытывает полимер при переработке (особенно
при вальцевании, литье иод давлением, смешении,
экструзии), могут приводить к деструкции полимера,
сопровождающейся изменением мол. массы, молеку-
лярно-массового распределения, степени разветвлен-
ности макромолекул, их химич. строения, а в ряде
случаев, особенно при неоднократной переработке,
к заметным изменениям всего комплекса свойств поли-
полимера. Кроме того, накопление макрорадикалов в мате-
материале в ходе переработки может значительно ускорить
старение и привести к преждевременному выходу
изделий из строя. Интенсивность деструкции зависит
от химич. строения полимера, жесткости его цени и др.
факторов (см. Старение). Высокая скорость деструк-
деструкции делает невозможной переработку ряда полимеров
без предварительного введения стабилизаторов, а для
нек-рых полимеров исключает повторную переработку
отходов.
Оптимизация рецептуры материала и режимов пере-
переработки. В состав материала необходимо ввести все
ингредиенты, обеспечивающие направленное регули-
регулирование свойств полимера при переработке и в готовом
изделии. Особое внимание уделяется микродобавкам,
позволяющим снизить вязкость расплава, уменьшить
остаточные напряжения, повысить адгезию связуки
щего к поверхности наполнителя.
Оптимизация рецептуры или режимов переработки
в ряде случаев производится с применением методов
планирования экстремальных экспериментов, резуль-
587
ПЕРЛОН
588
тативность к-рых тем выше, чем четче определен кри-
критерий оптимальности.
Количественное описание и расчеты параметров
переработки охватывают три группы задач: 1) расчет
усилий, действующих в рабочих органах перерабаты-
перерабатывающего оборудования (давление, крутящие моменты);
2) расчет основных характеристик технологии, процес-
процесса — производительности, темн-ры, давления и др.;
3) расчет параметров процесса (темп-pa, давление,
время пребывания материала в аппарате и др.), обеспе-
обеспечивающих формирование необходимой структуры поли-
полимера. Решение первых двух групп задач основывается
на гидродинамич. подходах к описанию движения
расплава полимера; полученные результаты приме-
применяют для расчета экструдеров, литьевых машин, прес-
прессов, смесителей, каландров, вальцев и др. При реше-
решении третьей группы задач исходят из результатов
количественного анализа процессов формирования над-
надмолекулярной структуры, учитывающего ориентацию
и нестационарный теплообмен, а в случае реактопла-
стов и процессы отверждения.
Теоретич. основа решения упомянутых выше задач
базируется на достижениях механики сплошной среды,
реологии полимеров, структурной механики, тепло-
теплофизики и термодинамики полимеров, химич. кинетики
и механохимии полимеров. Сложность математпч. мо-
моделей реальных процессов переработки заставляет при
их исследовании и расчетах широко применять быстро-
быстродействующие ЭЦВМ.
Наряду с детерминированными физически обосно-
обоснованными моделями применяют статистич. модели в виде
степенных полиномов, коэффициенты к-рых определяют
методами многофакторного эксперимента с применением
дробных реплик.
Широко распространены методы моделирования
реальных процессов на мелкомасштабных установках,
на к-рых эмпирически устанавливаются оптимальные
параметры конструкции и технологич. режима с после-
последующим пересчетом этих параметров с использованием
методов теории подобия для крупнотоннажных уста-
установок.
Методы регулирования свойств изделий в процессах
переработки. При формовании возможно значительное
изменение структуры полимера. Как уже отмечалось,
важнейшими факторами, оказывающими влияние
на структуру, являются параметры процесса переработ-
переработки — темп-pa, давление, градиенты скоростей и напря-
напряжений сдвига, режимы охлаждения и др. Правильный
учет и подбор этих параметров позволяет достигнуть
в готовом изделии однородной структуры, минималь-
минимального уровня остаточных напряжений, высокой степени
завершенности процессов кристаллизации и др.
Дополнительные возможности регулирования свойств
полимеров дает химическое и структурное модифици-
модифицирование (см. Модификация химическая, Модификация
структурная), а также пластификация, наполнение,
смешение полимеров друг с другом (см. Совместимость),
вспенивание (см. Пенопласты) и др.
Тенденции развития промышленности переработки
пластмасс. Структура перерабатывающей иром-сти
за рубежом (см. таблицу) стабилизировалась и меняется
весьма незначительно. В СССР, в отличие от др. стран,
значительное место среди различных методов перера-
переработки занимает прессование, что связано с большой
долей реактопластов в общем объеме производства
пластмасс. Однако уд. вес термопластов в нашей стране
будет увеличиваться, в результате чего структура
перерабатывающей пром-сти также постепенно изме-
изменится.
Наибольшее распространение в будущем получат
литье под давлением, экструзия, вакуумформование.
При этом пром-сть переработки пластмасс будет раз-
развиваться как в результате расширения мощностей
в крупных потребляющих отраслях (строительство
специализированных заводов), так и создания крупных
заводов для выпуска крупнотоннажных изделий меж-
межотраслевого применения, специализирующихся по тех-
технологич. принципу.
В технич. отношении важнейшая тенденция — резкое
повышение уровня автоматизации и механизации про-
процессов подготовки сырья, переработки, обработки и
сборки готовых изделий, вплоть до появления автома-
автоматизированных линий и цехов. В этой связи особое зна-
значение приобретает разработка методов контроля про-
процессов переработки, возникают дополнительные тре-
требования к сырью — по однородности его свойств
и состава, формы и размера частиц н др.
Лит.: Торнер Р. В., Основные процессы переработки
полимеров, М., 1972; Б е р н х а р л т Э., Переработка тер-
термопластичных материалов, М., 1965; М а к - К е л в и Д. М.,
Переработка полимеров, пер. с англ., М., 1905; 3 а в г о-
р о д н и й В. К., Механизация и автоматизация перера-
переработки пластических масс, 3 и-зд., М., 1970; Основы кон-
конструирования изделий из пластмасс, под ред. Э. Бэра, пер.
с англ., М., 1970; Основы конструирования и расчета деталей
из пластмасс и технологической оснастки для их изготовления,
Л., 1972; Новое в переработке полимеров, под ред. 3. А. Рого-
Роговина и М. Л. Кербера, М., 1969. М. С. Акутии, М. Л. Кербср.
ПЕРЛОН — см. Полиамидные волокна.
ПЕРХЛОРВИНИЛОВЫЕ ЛАКИ II ЭМАЛИ [chlo-
[chlorinated po!y(vinyl chloride) varnishes and enamels, La-
cke nnd Emaillen aus nachchloriertem Polyvinylchlo-
rid, vernis et emaux de chlorure de polyvinyl surchlore]—
лакокрасочные материалы на основе р-ров перхлорвини-
ловых смол в органич. растворителях.
Состав. Для приготовления П. л. и э. применяют
перхлорвшшловые смолы, образующие р-ры средней
(ПСХ-С; средняя мол. масса ~ 57 000) и низкой
(ПСХ-Н; ~ 31 000) вязкости. Часто в целях повышения
концентрации смолы, улучшения декоративных свойств
(напр., блеска) и теплостойкости покрытий, а также
для повышения адгезии (особенно к металлу) в состав
:шалей вводят модификаторы C0—60% от массы плен-
пленкообразующего). Последними в большинстве случаев
служат высыхающие алкидные смолы средней жирно-
жирности, иногда — модифицированные канифолью, или
алкидно-акриловые смолы (см. Алкидные смолы).
В состав П. л. и э. входят также пластификаторы —
фталаты и фосфаты различных спиртов, хлорпарафин
и др. C0—50% от массы смолы), в нек-рых случаях —
термостабилизаторы (эиоксидированное соевое или
подсолнечное масло, низкомолекулярная эпоксидная
смола) и сиккативы (при наличии в рецептуре алкид-
ной смолы). Растворителями и разбавителями для
П. л. и э. служат кетоны, сложные эфиры, ароматич.
углеводороды. Для материалов на основе ПСХ-С в ка-
качестве растворителя чаще всего применяют смесь бутил-
ацетата A2%) с ацетоном B6%) и толуолом F2%),
для материалов на основе ПСХ-Н — смесь ксилола
C5%), сольвента E0%) и ацетона A5%). При получении
эмалей в состав лаков вводят минеральные (напр.,
двуокись титана, цинковые белила) и органические
(напр., фталоцианиновые) пигменты, а также различ-
различные наполнители — барит, тальк и др.
Получение и нанесение. Перхлорвшшловые лаки
готовят растворением смолы в органич. растворите-
растворителях. Эмали получают по обычной технологии путем
смешения предварительно изготовленной пигментной
пасты с лаком. Для приготовления насты пигменты
и наполнители диспергируют чаще всего в р-ре алкид-
ной смолы, иногда — в р-ре перхлорвиниловой смолы
или пластификатора. В последнем случае для интен-
интенсификации процесса рекомендуется добавлять поверх-
поверхностно-активные вещества.
Черные металлы перед нанесением П. л. и э. под-
подвергают обработке металлич. песком, дробеструйной
или гидропескоструйной очистке, цветные металлы —
гидропескоструйной очистке или травлению; после
589
ПЕРХЛОРВИНИЛОВЫЕ СМОЛЫ
590
очистки металл тщательно обезжиривают (подробно
о подготовке поверхности см. Лакокрасочные покрытия).
При окраске металлич. изделий эмали наносят обя-
обязательно по грунтам на основе глнфталевых, алкидно-
фенольных смол, сополимеров винилхлорида с винил-
ацетатом или винилиденхлоридом, а также но фосфати-
рующим грунтам на основе поливиыилбутираля. Иног-
Иногда применяют перхлорвиниловые грунты. Обычно
наносят один слой грунта толщиной 15—20 мкм (для
химстойких покрытий — несколько слоев). Одно из до-
достоинств П. л. и о.— возможность сушки при комнат-
комнатной теми-ре. Поэтому для предварительного грунтова-
грунтования поверхности наиболее целесообразно применять
быстросохнущие грунты холодной сушки на основе
сополимеров винилхлорида с винилиденхлоридом или
винил ацетатом.
П. л. и э. наносят на окрашиваемые поверхности
распылением — нневматич., безвоздушным или в элек-
трич. поле, в несколько слоев (эмали на основе ПСХ-Н
в 2 слоя, на основе ПСХ-С — в 3—4 слоя; для химстой-
химстойких покрытий число слоев может достигать 10). При
использовании эмалей, модифицированных алкидными
смолами, сверху наносят дополнительный слой пер-
перхлорвинил ового лака для повышения химетойкости
покрытия. Возможно нанесение последующего слоя
по невысохшему предыдущему. Толщина покрытий
на основе П. л. и э. в зависимости от назначения состав-
составляет 40—150 мкм.
Образование пленки П. л. и э. происходит в резуль-
результате испарения растворителей, а при наличии в рецеп-
рецептуре алкидной смолы — также и вследствие отверж-
отверждения последней под воздействием кислорода воздуха.
Покрытия на основе П. л. и э. сушат обычно при
комнатной темп-ре. При этом практич. высыхание до-
достигается через 1 ч после нанесения. Однако полное
высыхание в естественных условиях (удаление всего
растворителя) происходит в течение 5 су т. В ряде
случаев применяют горячую сушку; при этом вре-
время полного высыхания при 60 °С сокращается до
120 мин, при 120 СС — до 20 мин. Кроме того, при
горячей сушке повышается адгезия покрытия к под-
подложке.
Свойства покрытий. П. л. и э. образуют покрытия,
к-рые обладают очень высокой атмосферостойкостью,
прочностью и эластичностью (твердость по маятнико-
маятниковому прибору 0,3—0,5, прочность к удару 20—50 см,
эластичность по шкале гибкости 1—3 мм).
Покрытия характеризуются очень низкой паро-
нроницаемостыо, большой стойкостью к минеральным
к-там и щелочам, не горят, не растворяются в жирах,
маслах, спиртах и алифатич. углеводородах. Недостат-
Недостатки перхлорвиниловых покрытий — слабый блеск, спо-
способность размягчаться при повышенной F0 °С) темн-ре,
приводящая к их загрязнению, а также низкая адге-
адгезия к подложке. Поэтому при нанесении П. л. и э.
необходимо тщательно подготавливать поверхность
под окраску.
Применение. П. л. и э. применяют для защиты раз-
различных изделий и конструкций из дерева, металла
и бетона от воздействия влаги, атмосферы, жидких
и газообразных агрессивных сред и др. Их используют
для окраски сельскохозяйственных машин, железно-
железнодорожных вагонов и цистерн, металлорежущих стан-
станков, мостов, дорожных и строительных машин, при-
приборов, различных металлоконструкций, бетонных со-
сооружений, оборудования химических заводов. Наибо-
Наиболее широко эти материалы применяют для получения
хим- и атмосферостойких покрытий в различных кли-
климатических зонах, в том числе в условиях влаж-
влажных тропиков, а также для получения огнезащитных
покрытий.
Доля перхлорвиниловых материалов в общем объеме
производства лакокрасочных материалов в СССР в 1973
составила ок. 3%. Перхлорвиыиловые лакокрасочные
материалы применяют также в ГДР, Югославии, Ру-
Румынии и Польше. Во многих др. странах распростране-
распространены близкие к ним по свойствам лакокрасочнпе мате-
материалы на основе различных сополимеров внннлхлорида.
Лит.: Эрман В. Ю., Жури. ВХО, 12, № 4, 398 A967);
ГольдбергМ. М., Материалы для лакокрасочных по-
покрытий, М., 1972, с. 219.
См. также лит. при ст. Перхлорвипиловые смолы.
В. 10. Эрман.
ПЕРХЛОРВИНИЛОВЫЕ СМОЛЫ [chl orinated
poly(vinyl chloride), nachchloriertes Polyvinylchlorid,
chlorure de polyvinyl surchlore] — принятое в СССР
торговое название продуктов ограниченного хлориро-
хлорирования поливинилхлорида, содержащих E2,5—64,5%
связанного хлора. Название ото, однако, противоречит
смыслу приставки «пер», относящейся к сполна хлори-
хлорированным продуктам. Кроме того, термин «смола»
в современной отечественной научной литературе при-
применяется только по отношению к термореактивным
олигомерам (смолам синтетическим), но не к термо-
термопластичным продуктам, каковыми являются П. с.
Поливпнилхлорид (ПВХ) подвергают хлорированию
с целью улучшения растворимости и повышения теп-
теплостойкости (о свойствах ПГЗХ см. Винилхлорида поли-
полимеры). Кроме того, с повышением содержания хлора
возрастает устойчивость ПВХ к действию агрессивных
сред.
В пром-сти, помимо П. с, производится в небольшом
масштабе и др. продукт хлорирования ПВХ, к-рый
содержит 04—66% хлора, ограниченно растворяется
и обладает повышенной теплостойкостью. Этот продукт
также рассматривается в данной статье.
Получение. Перхлорв и н иловую смолу
получают хлорированием р-ров ПВХ (гомогенное хло-
хлорирование) обычно но периодич. схеме. В качестве
растворителей используют тетрахлорэтан, хлорбензол,
дихлорэтан, смесь хлороформа с СС14. Хлорирование
можно осуществлять под действием радикальных ини-
инициаторов (азо-бгге-изобутиронитрила, перекисей), УФ-
или у-излучения. Процесс включает следующие ста-
стадии: 1) очистка растворителя (от высших хлоридов
железа и воды); 2) получение р-ра ПВХ; 3) хло-
хлорирование р-ра ПВХ; 4) дегазация кислых газов;
5) фильтрация р-ров П. с; 6) выделение П. с. из
р-ров и ее сушка.
Т. к. хлорирование ингибируется кислородом и при-
примесями железа, аппаратуру и коммуникации защи-
защищают антикоррозионными покрытиями или исполь-
используют в качестве конструкционного материала никель
либо богатые никелем сплавы. Допустимое содержание
железа в р-ре не более 0,0001%, воды — 0,01%.
ПВХ растворяют в аппарате, снабженном мешалкой,
при 70—80 °С, получая р-ры в среднем 10%-ной кон-
концентрации (по массе). Затем при тщательном переме-
перемешивании р-ра в аппарат подают хлор (через барботаж-
ное устройство) в количестве 110—120% от теорети-
теоретически необходимого. Хотя требуемая глубина хлори-
хлорирования м. б. достигнута при темп-ре, близкой к темп-ре
растворения, процесс ведут при 100 —115 °С, что обес-
обеспечивает получение продукта с лучшей раствори-
растворимостью. В этом случае при использовании низкокнпя-
щих растворителей процесс необходимо проводить
при повышенном давлении [например, для дихлор-
дихлорэтана — 0,3 Мн/м2 C кгс/см2), для смеси СНС13 и
СС14 — > 1,0 Мн/м2 A0 кгс/см2)]. В зависимости
от условий (инициатор, давление, темп-pa) продолжи-
продолжительность хлорирования колеблется от 4—6 до 40—
50 ч. Хлорированию подвергается и сам растворитель:
монохлорбензол — на 1—2%, тетрахлорэтан — на 5 —
10%, дихлорэтан и хлороформ — на 20—30%. Процесс
заканчивается, когда достигается необходимое содер-
содержание хлора в полимере, что соответствует увеличению
массы исходного полимера на 20%.
591
ПЕРХЛОРВИНИЛОВЫЕ СМОЛЫ
592
После окончания хлорирования для удаления непро-
реагировавшего хлора и образовавшегося HG1 полу-
полученный р-р П. с. продувают азотом при 60—80 °С до
полного удаления НС1. Затем из дегазированного р-ра
отфильтровывают взвешенные частицы. Образовавший-
Образовавшийся полимер выделяют из р-ра с помощью осадителя
(метанола) или отгоняют растворитель в среде кипящей
воды в колонне непрерывного действия. Первый способ
принят для тетрахлорэтановых р-ров, второй — для
р-ров в более легкокипящих растворителях (ди-
(дихлорэтан, трихлорэтан и хлорбензол). При исполь-
использовании осадителя образуется порошок со сред-
средним диаметром частиц до 100 мкм, в случае отгон-
отгонки растворителя — пористая крошка с частицами до
200 мкм.
Ограниченно растворимый тепло-
теплостойкий хлорированный ПВХ получают в гетерогенных
условиях — хлорированием порошкообразного ПВХ
или различных его суспензий. Обычно процесс осу-
осуществляют в водной солянокислой среде (концентрация
НС1 5—20%). Для ускорения хлорирования к во-
воде иногда добавляют хлорорганические растворите-
растворители, вызывающие набухание полимера, например СС14,
СНС13.
Свойства. Хлорированный ПВХ — аморфный само-
самозатухающий полимер; мол. масса зависит от мол. массы
исходного ПВХ (обычно 40 000—80 000) и способа хло-
хлорирования. Промышленная П. с. неоднородна (см.
табл.).
Характеристики перхлорвиниловой смолы и ее фракций (фрак-
(фракции выделены методом фракционного осаждения и растворения)
Фракция
Исходный
полимер
2
3
4
5
Выход, %
58,06
26,66
5,49
3,37
6,15
Уд. вяз-
вязкость*
0,233
0,159
' 130
; 109
0,074
0
Мол.
масса**
73250
89540
04 38 0
43660
3 6260
29230
Содержание
хлора, %
64,52
64,42
64,39
64,48
64,55
64,45
* Для 0,2%-ного р-ра в хлорбензоле. ** Мол. масса рас-
рассчитана но ур-нию Марка — Хувинка: [ц]=КМа, где К= 1 • 10~4.
Механизм хлорирования окончательно не выяснен,
однако большинство исследователей считает, что хло-
хлорируются гл. обр. группы — СН2 — исходного ПВХ.
В условиях гомогенного хлорирования до невысокого
содержания хлора в полимере « 65%) образуются бо-
более или менее длинные последовательности!—СНС1—]к,
распределенные по цепи статистически. При высоком
содержании хлора G0—72%), а также при любом со-
содержании хлора в условиях гетерогенного хлорирова-
хлорирования, когда периферийные участки частичек поли-
полимера имеют более высокое содержание хлора, чем
центральные, в макромолекуле появляются трихлорэти-
леновые фрагменты, окруженные неизмененными моно-
мономерными звеньями. Наряду с этим в макромолекуле
присутствуют неизмененные участки ПВХ.
П. с. хорошо растворяются (образуя 10—30%-ные
р-ры) в хлористом метилене, хлороформе, дихлорэтане,
тетрахлорэтане, хлорбензоле, о-дихлорбензоле, толуо-
толуоле, ксилоле, ацетоне, циклогексаноне, толуидине,
пиридине, этил- и бутилацетате, диметилформамиде,
хлористом тиониле; набухают в четыреххлористом
углероде, бензоле, анилине, эфире, дибутилфталате,
трпкрезплфосфате; не растворяются в гексане, петро-
лейном эфире, метаноле, этаноле, изопропаноле, бута-
ноле и воде. Коагуляционные числа (количество оса-
осадителя в мл, к-рое требуется для высаживания поли-
полимера из р-ра) для П. с. с содержанием хлора 64,3%
(в скобках указан растворитель): 6,45 (тетрахлорэтан),
7,83 (ацетон), 10,31 (хлороформ), 15,68 (хлористый
метилен), 19,67 (дихлорэтан), 21,05 (хлорбензол), 48,32
(циклогексанон). Максимум растворимости в ацетоне
наблюдается при содержании хлора в П. с. 63—64%,
а в циклогексаноне — при 63—65%. П. с. становятся
нерастворимыми в хлорбензоле и ацетоне в резуль-
результате нагревания при 130 °С в течение 8 и 4 ч соот-
соответственно.
Вязкость р-ров П. с. изменяется в широких пре-
пределах; при прочих равных условиях она сильно зави-
зависит от тина растворителя. Напр., значения г| от„ 1%-ных
р-ров П. с, содержащей 64,3% хлора, при 20 СС состав-
составляют: 1,010 (ацетон), 1,115 (дихлорэтан), 1,220 (хло-
(хлористый метилен), 1,278 (хлороформ), 1,325 (хлорбен-
(хлорбензол), 1,529 (тетрахлорэтан), 1,889 (циклогексанон).
При хлорировании в гомогенных условиях вязкость
р-ра хлорированного ПВХ, как правило, незначитель-
незначительно (на 2—3%) увеличивается но сравнению с вязкостью
р-ра исходного ПВХ; при гетерогенном хлорировании
вязкость уменьшается на 10—15%. Увеличение вяз-
вязкости объясняется повышением жесткости цепей с воз-
возрастанием степени хлорирования, уменьшение — де-
деструкцией макромолекул, приводящей к снижению мол.
массы. Деструкция полимера особенно интенсивна,
если образующийся НС1 удаляется из сферы реакции.
При хлорировании в р-ре или в водной солянокислой
среде в присутствии агента набухания мол. масса
ПВХ, как правило, уменьшается незначительно. При
хлорировании в воде без агента набухания мол. масса
хлорированного ПВХ составляет 70% от мол. массы
исходного ПВХ; при хлорировании порошкообразного
ПВХ мол. масса снижается иногда более чем на 50%
(при высоком содержании хлора).
Физич. свойства хлорированного ПВХ зависят от
способа хлорирования, типа исходного ПВХ и содер-
содержания хлора. Ниже приведены нек-рые свойства П. с:
Плотность при 20°С, г/см3 1,47-1,5 0
Насыпная масса, г/см3 0,2 — 0,25
Вязкость (в дихлорэтане, 20°С)
абсолютная,жк-сек/м2, или спз 1,23
удельная 0,12
Темп-pa, СС
стеклования 8 5 — 95
разложения 130 — 145
Теплостойкость, °С
по Мартенсу 70
по Вика • . . . . 120
Темп-рный коэфф. линейного расшире-
расширения, СС~Х F-8)-Ю-5
Морозостойкость, °С ок. —45
Твердость, Ми/м2 {тс/см2) 7 0G00)
Прочность, Мн/м* (кгс/мм2)
при изгибе 110A1)
при растяжении • .... 65 — 75F,5 — 7,5)
Относительное удлинение, % 4-0
Модуль упругости, Мн/м2 {кгс/см2) . . . 3200C2000)
Ударная вязкость, кдж/м2, или
кгс-см/см2
без надреза 400
с надрезом 5
Влагопоглощение (через 7 сут при 20°С,
100 см2), мг Ю
Влагопроницаемость (пленка толщиной
70 — 80 мкм, 24 ч), мг/см2 0,5—0,7
Диэлектрич. проницаемость (при 50 гц) ~3,0
Тангенс угла диэлектрич. потерь 1-10~2
Уд. объемное электрич. сопротивление,
Том-м (ом-см) >102(>1016)
Уд. поверхностное электрич. сопротивле-
сопротивление, ом >10*
Электрич. прочность B0сС, 50 гц), кв/мм 20,0
Повышенное содержание хлора в суспензионном
хлорированном ПВХ способствует нек-рому увеличе-
увеличению прочности при растяжении, однако одновременно
возрастает хрупкость материала. Поэтому в пром-сти
используют полимер, хлорированный лишь до 64—
66%, часто в композиции с полиэтиленом, хлорирован-
хлорированным полиэтиленом и поливинилхлоридом. Ниже при-
приведены нек-рые свойства ПВХ, хлорированного в гете-
593
ПЕЧАТЬ НА ПОЛИМЕРАХ
594
рогенных условиях (в скобках указано содержание
хлора в %):
Плотность при 20°С, г/см3 1,400—1,605
E6,7 — 70,6)
Темп-pa, °С
стеклования 78—115,5
E6,7-72,6)
текучести 175—190
F2,1 — 66,6)
Теплостойкость по Вика, СС 106—144
F1,0 — 68,2)
Относительное удлинение A00 С, нагруз-
нагрузка 5 Мн/м2, или 50 кгс/см2, 1000 мин), % 2 , 25—0 , 65
F4,7-68,2)
По химич. свойствам хлорированный ПВХ аналоги-
аналогичен исходному полимеру. П. с. относительно устойчива
при комнатной теми-ре к действию хромовой смеси,
царской водки, фосфорных к-т, окислителей (гипохло-
рит, КМпО4 и др.), р-ров различных солей; к щелочам
П. с. менее устойчива, чем к к-там. С повышением
темп-ры до 80 °С устойчивость к к-там снижается.
Разложение нестабилизированной П. с, сопровождаю-
сопровождающееся отщеплением НС1, начинается при 90 °С.
С целью повышения относительного удлинения и
прочности волокон и изделий П. с. модифицируют;
обычно проводят радиационную привитую сополиме-
ризацию с метакриловой к-той, винилфторидом и акри-
лонитрилом.
Нагревание хлорированного ПВХ в инертной атмо-
атмосфере при темп-pax < 330 °С сопровождается выделе-
выделением HG1. Появляющаяся при этом окраска полимера
обусловлена возникновением системы сопряженных
связей. Длительное нагревание приводит к сшиванию
макромолекул. Зависимость Igk от 1/ Т (к — константа
скорости дегидрохлорирования) имеет перегиб в обла-
области 220—240 °С. Ниже этой темп-ры энергия активации
дегидрохлорирования составляет ок. 100 кдж/молъ
B4 ккал/молъ), выше — ок. 10 кдж/молъ E0 ккал/молъ).
Участок с низкой энергией активации обусловлен нали-
наличием в макромолекулах П. с. третичных атомов угле-
углерода, концевых двойных связей и др. Длительное хло-
хлорирование улучшает термостойкость вследствие раз-
разрыва цепей у третичного атома углерода, присоедине-
присоединения хлора по двойным связям. При темп-pax выше
330 °С образуются бензол, моно- и дихлорбензолы.
Чем выше содержание хлора, тем выше выход хлор-
бензолов. Конечным продуктом термораспада явля-
является пористый уголь.
Чем выше мол. масса исходного ПВХ и чем меньше
разветвлений и нарушений в цепи конечного хлориро-
хлорированного ПВХ, тем выше термостойкость последнего.
Деструкция П. с. ускоряется в присутствии окислов
металлов (Fe2O3, ZnO, Cr2O3), солей тяжелых ме-
металлов (Fe, Ni, Си, Zn) и в результате действия УФ-
излучения.
Для придания большей термостойкости изделиям
на основе хлорированного ПВХ в полимер в процессе
получения и переработки вводят стабилизаторы —
оловоорганич. соединения, соли (Pb, Ba, Gd, Ca) выс-
высших жирных к-т, эпоксидные соединения, амины, фос-
фины, гликоли и др.
Переработка и применение. Из П. с. формуют во-
волокна (см. Поливипилхлоридпые волокна), приготовляют
лаки (см. Пер хлор виниловые лаки и эмали) и клеи.
Суспензионные хлорированные ПВХ перерабаты-
перерабатывают экструзией, каландрованием, литьем иод давле-
давлением. Из них изготовляют трубы для транспортировки
горячих (вплоть до 100 °С) и агрессивных жидкостей,
ианр. трубы для центрального отопления и канализа-
канализации, контейнеры, ванны и различные конструкции
в химич. машиностроении.
Хлорированный ПВХ производится за рубежом
под след. назв.: и г е л и т PC (ГДР), ренофлекс,
т р о в и д у р, в и н и д у р, в и н и ф о л ь (ФРГ),
сольвнтерм (Франция), н и к а темп (Япония).
Лит.: AJroldi G., Га. o.j, Advan. Chem. St., 99, 119
A971); Kolinsky М., [а. о.], J. Polymer Sci., pt A-1,9,
№ 3, 791 A971); Berticat Ph., Rev. Gener. Cautch,
Plast., 48, 1361 A971); AllenV.R., Young R. D.,
J. Polymer Sci., pt 1-A, 8,3123A970); В а н л с б е р г Э.,
Пластмассы в промышленности и технике, пер. с ном., М., 1964.
А. Л. Энглин, Н. Н. Мельникова, Н. М. Викторова.
ПЕЧАТЬ НА ПОЛИМЕРАХ (printing on polymers,
Bedrucken an Polymeren, impression sur polymeres).
В настоящей статье рассмотрены способы нанесения
рисунков и текстов на изделия из полимерных мате-
материалов — упаковочные материалы, шкалы для раз-
различных приборов, альбомы наглядных пособий из про-
прозрачной полимерной пленки.
На изделия из вискозы, ;зфиров целлюлозы, поли-
полипропилена, полиамидов, поливинилхлорида, полиэти-
лентерефталата, поликарбоната и полистирола печать
м. б. нанесена без затруднений. Печать на полиэтилене
и политетрафторэтилене невозможна без специальной
обработки (активации) их поверхности. Так, полиэти-
полиэтилен обрабатывают иерманганатом или др. сильным окис-
окислителем. Однако после такой обработки полимер
не может быть использован для упаковки пищевых
продуктов из-за токсичности адсорбированных в-в.
Поэтому предпочитают обработку полиэтилена откры-
открытым пламенем или в электрич. поле. В последнем случае
пленку помещают между двумя электродами, подклю-
подключенными к генератору переменного тока высокого
напряжения. В результате разрядов между электро-
электродами происходит ионизация воздуха с образованием
атомарного кислорода и озона. При их воздействии
на поверхность полиэтиленовой пленки образуются пе-
рекисные и гидроперекисные группы, после чего плен-
пленка становится восприимчивой к полиграфич. краскам.
Для активации полиэтиленовой пленки в электрич.
поле обычно используют автоматич. машины, к-рые
обеспечивают обработку пленки шириной до 50 см
со скоростью до 100 м/мин. Активированная пленка
сохраняет свои свойства до б мес.
Методы печати. П. н. п. осуществляют преимущест-
преимущественно эластографским (флексографским), этмограф-
ским (трафаретным) или глубоким (ротагравюрным)
методом. Применяют также эластосферич. печать и ти-
тиснение красочной или металлизированной пленкой.
Типографский и офсетный методы печати для этой
цели используют редко, т. к. они не обеспечивают
хорошего качества печати из-за специфнч. свойств
применяемых в них красок.
Эластографская печать производится
с помощью эластичных рельефных (напр., резиновых)
печатных форм (рис. 1). Краска наносится на поверх-
Рис. 1. Принципиальная
схема одной из секций эла-
стографской машины: 1 —
резервуар для краски; 2 —
красочный валик; з —нанос-
—наносной валик; 4 — эластичная
рельефная печатная форма;
5 — формный цилиндр; б —
печатный цилиндр; 7 — по-
полимерная пленка.
ность печатной формы наносным валиком 3 и отпеча-
отпечатывается на пленке 7 под давлением печатного цилинд-
цилиндра. Эластографские машины состоят из двух или боль-
большего числа секций.
Машины для печати на полиэтиленовой пленке снаб-
снабжены дополнительным фрикционным устройством,
к-рое компенсирует деформацию пленки во время пе-
печати. Эти машины оборудуют также сварочным аппа-
аппаратом для изготовления полиэтиленовых мешков.
Этмография — способ печатания, основанный
на применении формы-трафарета из капроновой или
металлич. сетки (рис. 2). Пробельные элементы формы-
трафарета закрыты непроницаемой пленкой (напр.,
595
ПЕЧАТЬ НА ПОЛИМЕРАХ
596
полнвшшлспиртовой), а печатающие элементы откры-
открыты, и через них проходит краска. При печати краска
наносится на внутреннюю поверхность формы-трафа-
формы-трафарета и продавливается с помощью резинового ракеля
на поверхность полимерного материала. Отмография
пригодна для печати на полимерных баллонах, труб-
трубках и т. п. Главное достоинство способа — возможность
\
Рис. 2. Принципиальная схема этмографской печати: 1 —
печатная форма (шаблон-трафарет)-. 2 — резиновый ракель;
з — краска; 4 — рама печатной формы; 5 — полимерный
материал.
нанесения на полимер плотного толстого (до 100 мкм)
непрозрачного слоя краски. Поэтому этмография при-
применяется преимущественно для нанесения различного
рода изображений и надписей на просвечивающие
шкалы различных приборов.
Широкое распространение получили полуавтоматич.
машины для этмографской печати на пленках и листах.
Автоматпч. машины для этой цели не применяются,
т. к. материал с нанесенной на него печатью нельзя
укладывать в стоики без предварительной сушки.
Для этмографии на баллонах и трубках применяется
несложное устройство (рис. 3). При движении ци-
Рис. 3. Устройство для эт-
этмографской печати на по-
полимерных цилиндрич. изде-
изделиях: 1 — стол; 2 — тор-
тормозная стойка; 3—цилиндр;
4 — изделие; 5 — печатная
форма-трафарет; б —ракель;
7 — краска; 8 — цилиндр;
9 — соединительная пленка;
а — направление движения
цилиндров з и 8; б— направ-
направление движения формы-тра-
формы-трафарета 5.
-EL
J
линдров 3 и 8 в направлении, указанном стрелкой
«а», цилиндрич. изделие 4 будет вращаться. При этом
форма-трафарет 5 перемещается в направлении, ука-
указанном стрелкой «б».
Глубокая печать позволяет наносить
на изделия из полимерных материалов тексты, одно-
одноцветные или многоцветные рисунки. В последнем слу-
случае печатание осуществляют последовательно с не-
нескольких форм, каждая из к-рых служит для воспро-
воспроизведения рисунка в одном цвете. Печатающие эле-
элементы формы в этом методе (рис. 4) представляют
J
4 5
Рис. 4. Схема формы для глубокой печати: 1 — печатная
форма; 2 — краска; з — ракель; 4 — печатающие элементы
формы; 5 — пробельные элементы формы.
собой ячейки различной глубины (но не более 40 мкм),
отделенные друг от друга пробельными опорными
(растровыми) элементами. Избыток краски снимается
с формы ракелем. Так. обр. краской заполняются
только углубления формы, из к-рых при печати она
переходит на полимерную подложку иод давлением
печатного цилиндра. При этом толщина слоя краски,
нанесенного на изделие из полимерного материала,
пропорциональна глубине ячейки печатной формы.
Это позволяет при одноцветной печати получать ри-
рисунки с различными оттенками (напр., краски на ос-
основе ферроцианида железа в толстом слое имеют темно-
синий цвет, в тонком — небесно-голубой).
При печатании многоцветных рисунков благодаря
различной глубине ячеек в формах удается получить
качественные отпечатки с использованием только трех
цветов (и соответственно трех форм): голубого, пур-
пурпурного и желтого и отказаться от использования чет-
четвертой формы для серой краски, к-рую при др. мето-
методах печати используют для получения глубоких теней.
Глубокая печать предполагает применение маловяз-
маловязких [ок. 0,1 н-сек'м'2 A пз)\ красок на летучих раство-
растворителях.
Краска, нанесенная на полимерную подложку, не-
несколько растекается и растровые опорные липни про-
пропадают. В результате получается изображение с посте-
постепенными переходами от глубоких теней до очень свет-
светлых полутонов (подобно фотографич. снимкам).
Глубокую печать на одной или обеих сторонах поли-
полимерной пленки осуществляют обычными многокрасоч-
многокрасочными ролевыми машинами. С их помощью можно
печатать не только простую рекламу на упаковочных
материалах, но делать копии с цветных художествен-
художественных фотографий и репродукции картин.
Высокая разрешающая способность глубокой печати,
широкий цветовой охват и отличная градационная
передача позволяют воспроизводить практически лю-
любые цветные оригиналы, а современные ролевые рота-
ротационные машины глубокой печати, оборудованные
мощными системами сушки, обеспечивают точное сов-
совпадение изображений лицевой и обратной сторон,
надежное закрепление красочного слоя на оттисках.
При двухсторонней многокрасочной печати на полимер-
полимерных пленках обязателен белый или цветной раздели-
разделительный фон, иначе изображение одной стороны будет
искажать изображение другой. При печати изображе-
изображение с лицевой стороны просматривается через пленку,
а с обратной — непосредственно. Красочный слой
обычно покрывают защитным лаком.
Эластосферическая печать позволяет
наносить четкие изображения на изделия из пластмасс
сложной конфигурации. Печатающим элементом в этом
способе служит сфера («груша»), изготовленная из эла-
эластичного материала, чаще всего композиции на основе
желатины D3—45% по массе), глицерина D3—45%),
воды A0 — 14%) и сахара (до 5%). Раздуваемую возду-
воздухом сферу прижимают сначала к поверхности рельеф-
рельефного типографского клише, формы для глубокой печати
или офсетной плоской формы, покрытых краской.
Изображение1 переходит на сферу, к-рую зате*м с по-
помощью воздуха плотно прижимают к поверхности
изделия. Для эластосферич. печати применяют высо-
высоковязкие [ок. 20 н-сек/м2 B00 пз)] краски, содер-
содержащие синтетич. смолы (напр., алкидные), высококипя-
щие органич. растворители и высыхающие раститель-
растительные масла.
Тиснение красочной или метал-
металлизированной пленкой применяется для
нанесения несложных изображений на изделия из по-
полистирола и полиметилметакрилата. Красочная двух-
двухслойная пленка представляет собой полиэтилентере-
фталатную основу толщиной ок. 20 мкм, к-руго грун-
грунтуют воском, а сверху покрывают краской, состоящей
из пигмента и поливинилацетатной эмульсии или фе-
ноло-формальдегидного клея, модифицированного
поливинилбутиралем. Металлизированную четырехслой-
ную пленку, имитирующую золото, готовят след. обра-
образом: на полиэтилентерефталатную основу наносят
597
ПИГМЕНТЫ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
598
последовательно восковой и прозрачный желтый кра-
красочный слой. Сверху наносят алюминий в вакууме
(см. Металлизация пластмасс), а затем лакируют
р-ром иолибутилметакрилата в этилацетате или спир-
спиртовым р-ром нитрата целлюлозы. Металлизированная
пленка, имитирующая серебро, не содержит желтого
красочного слоя.
Для тиснения на изделиях из пластмасс применяют
универсальные механич. прессы малой мощности, обо-
оборудованные штампами с металлич. печатной формой-
пуансоном. Тиснение цифр, знаков или несложных
рисунков м. б. выполнено одновременно со штампова-
штампованием (напр., тиснение цифр на номерках для гардеро-
гардеробов из органич. стекла) или на уже готовом изделии.
Последний способ применяют чаще. На изделие накла-
накладывают красочную (металлизированную) пленку кра-
красочным слоем к пластмассе и помещают заготовку
в пресс. При опускании верхней подвижной плиты
пресса печатная форма-пуансон, нагретая до 100—
150 С, вырубает из пленки цифры или рисунок и вдав-
вдавливает их в поверхность изделия. При этом поверхность
изделия, соприкасающаяся с пленкой, слегка расплав-
расплавляется и приваривается к расплавленному красочному
слою. Восковой слой также расплавляется и позволяет
удалить полиэтнлентерефталатную основу.
Краски для печати на полимерах. Для П. н. п. при-
пригодны полиграфические краски, представляющие собой
дисперсии пигментов в лаках на основе низкокипящих
органпч. растворителей (напр., спирты, бензин, толуол,
ацетаты, хлорированные углеводороды и др.). В ка-
качестве плеикообразователей используют поливинил-
ацетат, перхлорвиниловую смолу (преимущественно
для печати на поливинилхлориде), нитрат целлюлозы
и др. Перспективным является применение полибутил-
метакрллата, к-рый с успехом может заменить шеллак
и нек-рые синтетич. пленкообразователи.
Краски этого тина прочно удерживаются на поверх-
поверхности изделий из пластмасс благодаря адгезии плен-
кообразователя к полимеру; проникновения органич.
растворителя в полимер практически не наблюдается.
Для печати на полиэтилене нужны специальные
краски. В случае оластографии и глубокой печати их
готовят на основе нитратов целлюлозы, иерхлорвини-
ловой смолы или алкидно-акриловых смол. Для этмо-
графии, кроме указанных выше, пригодны краски на ос-
основе поливинилацетатной эмульсии, нентафта левых
и эпоксидно-масляных смол. Для эластосферич. печати
лучше всего подходят офсетные краски. Однако наи-
наилучшие краски для печати на полиэтилене готовят
на основе шеллака.
Лит.: Анилиновая печать, М., 1963; 3 о т к и н С. Ф.,
К а л и и и ь Э. Я., Трафаретная печать, М., 1965; Неми-
ровский Е. Л.. Новые способы печати, М., 1956; Особые
виды печатных работ, Сб. переводов, под ред. А. Н. Черны-
Чернышева, М., 1960; СовременЕгая флексографская печать, обзор
отечественной и иностранной литературы, М.— Киев, 1969;
Левченко В. Т. [и др.], Флексографические краски, М.
1971; Попов В. В., Общий курс полиграфии, М., 1964
CermakW., Handbuch fur den Siebdruck, 3 Aufl., Lpz.
1961; К 1 e i 1 e i n O., Das Siebdruckverfahren, Lpz., 1955
BowlesR. F., Printing ink manual, 2 ed., Camb., 1969
Б. И. Бе резин
ПИГМЕНТЫ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
(pigments for paint materials, Pigmente fur Anstrich-
stoffe, pigments des peintures et vernis)— высокодиснерс-
ные окрашенные (в том числе белые и черные) порошки,
нерастворимые в воде и в пленкообразующих веществах
и имеющие высокий показатель преломления. При
диспергировании в пленкообразующих П. л. м. обра-
образуют стабильные слабофлокулированные дисперсные
системы — краски, грунтовки, шпатлевки, применяе-
применяемые для получения декоративных и защитных лако-
лакокрасочных покрытий. Вещества, используемые в качест-
качестве П. л. м., применяют также для окрашивания в мас-
массе и др. полимерных материалов (см. Красители, Кра-
Крашение волокон в массе).
Помимо своих основных функций, П. л. м. выпол-
выполняют также и ряд других. Адсорбируя на своей поверх-
поверхности пленкообразующие, частицы П. л. м. влияют
на характер надмолекулярной структуры образующей-
образующейся пленки, способствуя повышению механич. и защит-
защитных свойств покрытий. Поглощая, отражая и рассеи-
рассеивая световые лучи, в том числе и ультрафиолетовые,
П. л. м. защищают полимер в пленке от старения.
Нек-рые П. л. м. служат иассиваторами металла под-
подложки лакокрасочного покрытия, обусловливая его
антикоррозионные свойства (см. об этом Защитные
лакокрасочные покрытия).
П. л. м. могут придавать лакокрасочным покрытиям
также бактерицидные свойства (см. Антимикробные
лакокрасочные покрытия), способность предотвращать
обрастание подводной части судов и гидротехнич. соору-
сооружений морскими организмами (см. Необрастающие
лакокрасочные покрытия), способность светиться (см.
Светящиеся лакокрасочные покрытия). Они могут ре-
регулировать темп-ру (см. Терморегулирующие лакокра-
лакокрасочные покрытия), изменять окраску при нагревании
(см. Термоиндикаторные лакокрасочные покрытия),
повышать огнестойкость покрытий и др.
П. л. м. разделяют: 1) но химич. составу — на неор-
неорганические и органические; 2) по происхождению —
на природные (минеральные) и синтетические; 3) по
цвету — .на ахроматические (белые, серые, черные)
и хроматические (все цветные).
Неорганические П. л. м.— окислы, сред-
средние или основные соли или комплексные соединения
металлов, высокодисперсные порошки металлов (А1,
Си, Zn, Fe, Ni) п их сплавов (напр., бронзы, латуни).
К неорганич. П. л. м. относится также сажа. Орга-
Органические П. л. м.— нерастворимые формы син-
синтетич. органич. красителей. В данной статье рассмат-
рассматриваются только неорганич. пигменты (см. таблицу),
к-рые благодаря высокой устойчивости к воздействию
света, тепла, влаги, хнмич. реагентов, а также относи-
относительной дешевизне имеют в лакокрасочной пром-сти
наибольшее значение. Об органич. пигментах см. Кр-а-
сители.
Многочисленные способы производства
П. л. м. можно разделить на три принципиально раз-
различные группы: 1) осаждение кристаллов П. л. м.
из р-ров; 2) получение П. л. м. в газовой фазе с
последующей конденсацией возгонов; в этом случае
образуются высокодисперсные продукты, не требую-
требующие дополнительной обработки (напр., ZnO из метал-
металлич. цинка, TiO2 из TiCl4, сажа из природного газа);
3) получение П. л. м. термич. разложением, спека-
спеканием, окислением, восстановлением (напр., красные
железоокисные пигменты из железного купороса,
ультрамарин из каолина и серы, TiO2 из метатитановой
кислоты).
Для придания товарной (выпускной) формы и мяг-
мягкой текстуры П. л. м. подвергают размолу, нек-рые
П. л. м. — микронизации (измельчению в струйных
мельницах), а также модифицированию силикатами
натрия, кремнийорганич. соединениями, четвертичны-
четвертичными аммониевыми соединениями с длинной углеводород-
углеводородной цепью, неионогенными поверхностно-активными
веществами. В результате модифицирования П. л. м.
не агрегируются после измельчения, легко смачиваются
пленкообразующими и диспергируются в них.
К основным характеристикам П. л. м. относятся:
укрывистость, красящая способность (интенсивность),
цвет, кристаллич. структура, дисперсность, смачивае-
смачиваемость, маслоемкость, способность к взаимодействию
с пленкообразующими веществами, диснергируемость,
свето- и атмосферостойкость, химич. стойкость, физио-
логич. действие.
Укрывистость (кроющая способность) —
способность пигмента, диспергированного в связую-
599
ПИГМЕНТЫ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
Свойства важнейших неорганических пигментов для лакокрасочных материалов
600
Название
Основной ком-
компонент
Кристаллич.
решетка (фор-
(форма частиц)
Плотность,
г/см3
Показатель
преломления
Размер час-
частиц, мкм
Уд. повер-
поверхность,
мг/г
Укрыви-
стость,
г/ж2
Ма слоем-
кость 1-го
рода, г на
100 г
Белые пигменты
Двуокись титана
Двуокись титана
Белила цинковые
муфельные печные
Белила цинковые
ветерилльные*
Литопон
Белила свинцовые
Крон свинцовыг
лимонный
Крон свинцовый
желтый
Крон свинцовый
оранжевый
Крон цинковый
(малярный)
Крон молибдатньш
красный
Желтый железоокис-
ный
Красный железоокис-
ный
Охра**
Железный сурик**
Мумия**
Ультрамарин
Железная лазурь
Окись хрома
Изумрудная зелень
Свинцовые зелени
смешанные
TiO2 (рутил)
ТЮ2 (анатаз)
ZnO
ZnO
с примесью РЬ
ZnS+
(вюртцит)
+ BaSO4
2РЬСО3-РЬ (ОНJ
PbGrO4 + PbSO4
HPbCrO4 + PbSO4
PbGrO4 + PbO
3ZnCrO4-Zn(OHJ-
•K2GrO4.2H2O
7PbCrO4-PbMoO4-
• PbSO4
FeOOH (гетит)
Fe2O3 (гематит)
Алюмосиликат -J-
-)-r euuxi
Fe2O3 (гематит)
Fe2O3 + A12O3 +
+SiO2
Na6Al4Si6S4O24
KxFey[Fe(CNN]z-
¦?гН2О
Gr2O3
Сг2О3-пН2О
Смеси свинцовых
кронов, желез-
железной лазури и
BaSO4
Тетрагональ-
Тетрагональная (сдвоен-
(сдвоенные иглы)
Тетрагональ-
Тетрагональная (пирами-
(пирамиды)
Гексагональ-
Гексагональная (призмы)
Гексагональ-
Гексагональная (иглы)
ZnS — гекса-
гексагональная,
BaSO4 —ром-
—ромбическая
Гексагональ-
Гексагональная
Желтые
Ромбическая
(зерна)
Моноклинная
(иглы-волок-
(иглы-волокна)
Тетрагональ-
Тетрагональная
—
Тетрагональ-
Тетрагональная
Ромбическая
(иглы)
Гексагональ-
Гексагональная
—
_
С
Тетраэдриче-
ская
Кубическая
4,2
3,85
5,5
5,6-5,7
4,2-4,3
6,50-6,85
и краен
5,6-6,0
6,12
6,3
3,73
6,05
3,85-4,00
4,85-5,00
2,7-3,4
3,7-4,4
2,5
и н и е п и г
2,3-2,4
1,85
Зеленые пи
Гексагональ-
Гексагональная
—
4,65-5,00
4,8-5,0
3,4-5,0
2
2
2
2
1,84
2
ы е п
2
2
2
2,
2
2,
3,
2,
3,
мент
1,
1,
г м е н
2
1
,72
,55
02
37
-2,00
09
и г м е н
42
55
45
20
4
2
2
0
22
ы
52
6
т ы
»5
,5
0
0
0,
о,
о,
т ы
0
0
0
0
о,
,2-1,0
,2-0,8
15-2,00
15-10,00
1-3
50—1 ,25
,5-3,0
1,5
1-5
—
5 — 10
,3-0,8
,3-2,0
1-10
1-30
—
,5—2,0
ДО 4 0)
05 — 0,20
-
3—10
—
0
1
0
0
0
0
5-8
5-10
4-6
1-4
1-3
1 — 2
,5-2,0
1-2
0-1 ,5
2-4
2-1,0
8-12
4-10
5-5,0
1-1,2
5-1,0
—
50-70
5-7
—
—
15—25
20 — 30
100 — 120
120
120-140
120 — 150
60
36
45
120-150
10 — 20
10-15
3-8
60 — 90
10
15 — 30
—
10-20
8-12
50
10 — 15
17-20
25
14
10
12-15
9-12
10
15
_
—
80
40-60
20 -40
25-35
14-20
20-30
34
50-60
25
70
13—25
Сажа ДГ-150
Закись-окись железа
Алюминиевая пудра
Углерод
Fe3O4
А1
Серые, черные
Полиэдраль-
Полиэдральная (сферы)
Кубическая
Кубическая
(листки)
2,07-2,20
4,7-5,2
2,64
пигменты
2,0
2,0
—
0,015 —
0,030
0,2 — 0,3
10 — 30
140—180
—
—
3-G
4-5
—
180-200
3 0-35
—
Имеют светло-серый цвет. ** Природные П., взе остальные П. — синтетические.
щем, перекрывать цвет подложки, т. е. делать ее неви-
невидимой. Это свойство обусловлено диффузным отраже-
отражением (рассеянием) и поглощением света в пленке и за-
зависит от разности показателей преломления пигмента
(пат) и пленкообразующего (пи). Чем больше разность,
тем выше укрывистость. Поскольку обычно пп^ 1,5—1,6,
то укрывистыми м. б. вещества с ппГ > 1,6. В укры-
вистости черных П. л. м. основную роль играет погло-
поглощение света.
Укрывистость возрастает с уменьшением размера ча-
частиц П. л. м. до определенного предела @,20—0,40 мкм);
она зависит также от объемной концентрации П. л. м.
в пленке. О методе определения укрывистости см. Ис-
Испытания лакокрасочных материалов и покрытий.
Красящую способность, т. е. способ-
способность П. л. м. передавать свой цвет (окраску) др. те-
телам, оценивают визуально или с помощью специальных
приборов — колориметров, компараторов цвета. Опре-
Определение основано на сравнении оттенка смеси испытуе-
испытуемого П. л. м. и разбеливающего компонента с оттенком
эталонного образца. Высокая красящая способность
позволяет уменьшить расход П. л. м., заменив их
частично более дешевыми наполнителями лакокрасоч-
лакокрасочных материалов.
Цвет. В видимой части спектра @,380—0,760 мкм)
белые П. л. м. одинаково отражают 90—98% (MgO —
100%) лучей всех длин волн. Черные П. л. м. одинаково
поглощают все лучи; цветные отражают лишь к.-л.
часть спектра. Цвет П. л. м. характеризуют тремя
параметрами: цветовым тоном (длиной вол-
волны, соответствующей максимуму отражаемой пигмен-
пигментом части спектра), его насыщенностью (чис-
601
ПИГМЕНТЫ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
602
тотой тона) и яркостью. П. л. м. тем ярче, чем
больше падающего света он отражает. Цвет красок
оценивают визуально, сравнивая с эталонами, или оп-
определяют по трем координатам х, у, z в соответствии
с системой Международного светотехнического коми-
комитета. Для этого используют фотоэлектрич. приборы —
спектрофотометры, колориметры, компараторы цвета,
к-рые объективно характеризуют окраску.
Кристаллическая структура. Вслед-
Вследствие полиморфизма, т. е. способности кристаллизо-
кристаллизоваться в различных системах, вещества одинакового
химич. состава могут резко различаться по цвету,
ксюфф. поглощения, показателю преломления, плот-
плотности, твердости, термостойкости и др. свойствам.
Так, хроматы свинца ромбич. системы имеют лимонно-
желтую окраску, моноклинной — желтую, тетраго-
тетрагональной — красно-оранжевую; модификации TiO2
«рутил» и «анатаз» являются хорошими белыми П. л. м.,
а «брукит» свойствами П. л. м. не обладает.
Дефекты кристаллич. решетки П. л. м. обусловли-
обусловливают мозаичность свойств различных участков поверх-
поверхности, влияют на цвет, смачиваемость, электрич. про-
проводимость, реакционную и адсорбционную способность
и др. свойства П. л. м. Поэтому при получении П. л. м.
необходимо строго соблюдать условия кристаллизации
и последующей обработки. С целью фиксации опреде-
определенной кристаллич. решетки вводят зародыши кри-
кристаллизации.
Дисперсность. Размеры и форма первичных
частиц, степень агрегации и прочность агрегатов
П. л. м. в значительной мере определяют их укрыви-
стость, красящую способность, оттенок, способность
диспергироваться в пленкообразующем и образовывать
блестящие покрытия. Для каждого пигмента сущест-
существует оптимальный размер частиц. Напр., оптимальный
(по укрывистости) радиус частиц белых П. л. м. вычис-
вычисляют по ф-ле гпг = 0,447 XI л (паг — ип), где X —
длина волны падающего света, мкм.
Дисперсность П. л. м. характеризуется кривой диф-
дифференциального или интегрального распределения
частиц по размерам или уд. поверхностью порошка
б П ? 012 2/
?уд.
ц рр у
Для обычных П. л. м.
220
р р
= 0,1—2 м2/г, для
микронизированных — 2—20 м2/г, для высокодисперс-
высокодисперсных (сажа, железная лазурь, органические пигмен-
пигменты) — 50—200 мг/г. Знание 5уд, характеризующей
площадь взаимодействия П. л. м. с пленкообразую-
пленкообразующим, позволяет правильно составлять рецептуру кра-
красок. Об определении дисперсности пигментов см. Ис-
Испытания лакокрасочных материалов и покрытий.
Маслоем кость. Удельная поверхность, рас-
распределение частиц по размерам, их форма и смачивае-
смачиваемость П. л. м. определяют кол-во адсорбированного
на поверхности П. л. м.
пленк ообразующего
(масла), плотность упа-
упаковки частиц в пленке,
а, следовательно, и ко-
количество масла, к-рое
Зависимость свойств лако-
лакокрасочных покрытий от
объемной концентрации
пигмента: 1 — газо- и па-
ропроницаемость; 2 — уд.
электрич. проводимость;
4 — появление пузырей в
Объемная концентрация
3 — антикоррозионные свойства;
пленке; 5 — блеск покрытия; 6 — прочность пленки при
растяжении; 7 — относительное удлинение пленки; А —
критическая объемная концентрация пигмента.
необходимо для заполнения пустот между твердыми
частицами пигмента. Массу льняного масла (в г), не-
необходимую для превращения 100 г сухого порошка
П. л. м. в пасту (комок), наз. маслоемкостью
первого родаМ (маслоемкостью второго рода
наз. массу масла, необходимую для получения лако-
лакокрасочного материала рабочей консистенции). Макси-
Максимальной плотности упаковки П. л. м. в пленке соот-
соответствует т.
к о н ц с н т
рисунок).
Значения
наз. крит
рация п
М и КОКП
р.
= 4 Р^
КОКП
и ч с с
и г м е
можно
с
• ~*~
100
к
н
а
т
я
а
О
К
б'
О
вычислить
h
2
ъ е
К
по
м н а я
П (см.
ф-лам:
1,43 + 5УД рпг —
где ?уД — уд. поверхность пигмента, см2/г; рс и рПГ —
соответственно плотность среды и пигмента, г/см3;
h — толщина граничного слоя, разделяющего частицы
пигмента, см.
Взаимодействие с пленкообразую-
пленкообразующими. Молекулы пленкообразующих хемосорби-
руются на поверхности частиц П. л. м., создавая
структурированный слой толщиной 8—20 нм (80—
200 А). Избирательный характер хемосорбции (на раз-
различных активных центрах П. л. м. сорбируются моле-
молекулы олигомеров и полимеров с различными полярными
группами) обусловливает выбор как самого П. л. м.,
так и способа его обработки применительно к данному
пленкообразующему. При отсутствии или недостатке
в последнем полярных групп, способных взаимодейст-
взаимодействовать с активными центрами частиц П. л. м., а также
больших молекул, к-рые могут образовывать толстые
адсорбционные слои, затрудняется диспергирование
пигмента и происходит его флокуляция. Это приводит
к получению материалов с низкими малярно-технич.,
защитными и декоративными свойствами. Пленки
на основе таких материалов не имеют блеска, в них
возникают внутренние напряжения, ускоряющие ста-
старение и разрушение покрытий.
Выбор П. л. м. определяется также агрегатным
состоянием пленкообразующего (р-ры, латексы, рас-
расплавы). Так, в эмульсионных красках следует приме-
применять П. л. м., не содержащие растворимых солей,
способных разрушать эмульсию; в красках для нане-
нанесения покрытий методом электрофореза недопустимо
применение П. л. м., к-рые реагируют с водораствори-
водорастворимыми полимерами, содержащими аммониевые группы
(PbO, ZnO, свинцовый сурик, сернистые соединении,
железная лазурь и др.).
Диспергируемость в пленкообра-
пленкообразующем («перетираемость») зависит от размера
частиц, твердости П. л. м., прочности агрегатов, обра-
образовавшихся в результате спекания П. л. м. при их
сушке, и прочности коагуляционных структур (сажа,
железная лазурь, органич. пигменты). Диспергирова-
Диспергирование П. л. м. сопровождается лишь частичным разру-
разрушением агрегатов; размер первичных частиц, опреде-
определяемый технологией получения П. л. м., при этом
практически не уменьшается. Лакокрасочные материа-
материалы, обладающие удовлетворительной агрегативной
устойчивостью, образуются только в случае адсорбции
достаточно толстого слоя пленкообразующего на всей
поверхности частицы. При неполной смачиваемости
поверхности П. л. м. и избытке влаги (на гидрофиль-
гидрофильных П. л. м.) диспергирование сопровождается агре-
агрегацией и флокуляцией частиц пигмента. Наилучшей
дисиергируемостью обладают П. л. м., полученные
осаждением из р-ров в присутствии поверхностно-актив-
поверхностно-активных веществ, гидрофобизирующих поверхность П. л. м.
В этом случае при смешении с пленкообразующим
П. л. м. переходят из водной среды в «масляную», что
устраняет необходимость сушки и, следовательно,
спекание первичных частиц П. л. м. Улучшение
диспергируемости сухих П. л. м. может быть достиг-
603
ПИПЕРИЛЕНА ПОЛИМЕРЫ
604
нуто их модификацией поверхностно-активными ве-
веществами в процессе микронизации.
С в е т о-, а т м о с ф е р о- и х и м с т о й к о с т ь. Не-
органич. П. л. м., как правило, более светостойки, чем
органические. Нек-рые неорганич. П. л. м. изменяют
под действием света свою окраску: желтые свинцовые
кроны зеленеют, литопон желтеет. ТЮ2 (анатаз), ZnO
отличаются высокой фотохимич. активностью и сенси-
сенсибилизируют окислительно-восстановительные процес-
процессы. Это приводит не только к выцветанию органич.
П. л. м., применяемых в смеси с TiO2 или ZnO, но
и к деструкции макромолекул полимера и обусловли-
обусловливает т. наз. «меление» пленки. Фотохимич. активность
TiO2 и нек-рых других П. л. м. подавляют модифика-
модификацией их поверхности гидроокисями или фосфатами
Al, Zn и др.
В качестве П. л. м. используют продукты, не реаги-
реагирующие с др. компонентами красок, устойчивые к крат-
кратковременному нагреванию в условиях горячей сушки
лакокрасочных покрытий (до ~ 240 СС) и имеющие
реакцию водной вытяжки, близкую к нейтральной.
Соединения, к-рые образуются в результате хемосорб-
ции полярных групп на поверхности П. л. м., должны
быть нерастворимы в растворителях данной лакокра-
лакокрасочной кохмиозпцни и в пленкообразующих веществах.
Так, нерастворимые свинцовые соли жирных к-т
(мыла), образующиеся на свинцовых белилах, придают
пленкам высокую атмосферостойкость даже в условиях
морского троиич. климата, растворимые же цинковые
мыла вызывают загустевание красок и ухудшение
атмосферостойкости покрытий. При выборе П. л. м.
необходимо учитывать влияние на них рН среды.
Напр., железная лазурь разлагается при воздействии
самых слабых оснований; ультрамарин и ZnO не стойки
к действию к-т. П. л. м., содержащие РЬ, чернеют
под действием H2S, алюминиевая пудра и цинковая
пыль реагируют с водой. П. л. м. основного характера
(ZnO, PbO) реагируют с алкидными смолами, имею-
имеющими высокие кислотные числа, и т. д.
Физиологическое действие. При
производстве лакокрасочных материалов широкого
назначения недопустимо применение токсичных П. л. м.
В СССР запрещено применение ядовитых свинцовых
белил, медномышьяковистых зеленей, ртутных П. л. м.,
ограничено применение свинцовых кронов.
Лит.: Беленький Е. Ф., Р и с к и и И. В., Химия
и технология пигментов, 3 изд., Л., I960; II э й и Г. Ф., Тех-
Технология органических покрытий, пер. с англ., т. 2, Л., 1963;
Wagner H., Pigmente, Stuttgart, I960; Ермилов П. И.,
Диспергирование пигментов, М., 1971; Пигменты. Введение
$ физическую химию пигментов, пол ред. Д. Паттерсогга, Л.,
1971. П. И. Ермилов.
ШШЕРЫЛЕНА ПОЛИМЕРЫ (polypiperylene, Poly
piperylen, polypiperylene).
Пиперилен A-мстилбутадиен-1,3, нентадиен-1,3)
СН3 — GH == СН — СН = СН2; существует в цис- и
тра /^-формах:
GHj-GH
СН3-СН
СН2=СН-СН НС-СН=СН2
ijuc-изомер трешс-изомер
Свойства пиперилеиа приведены в табл. 1.
Таблица 1. Физические свойства пиперилеиа
Показатели
Тектт-ра, СС
плавления
кипения ....
Плотность ^4°
Показатель преломления
?7J) ...
D
у ис- Изомер
— 140 82
4 4,07
0,6910
1 ,4363
трап с -Изомер
— 82, 47
42,03
0,6760
1,4301
Пиперплен не растворяется в воде; хорошо раство-
растворим в ацетоне, четыреххлористом углероде, бензоле,
эфире, спирте. Вступает в реакции, характерные для
диенов: полимеризуется и сополимеризуется с бутадие-
бутадиеном, стиролом и др. сомономерами, гидрируется, окис-
окисляется, присоединяет SO2 с образованием сульфона:
сн3-сн=сн-сн=сн2+ so2
so
Реакцию используют для отделения пнперплена
от его изомера — изопрена, т. к. сульфон легко раз-
разлагается при нагревании на SO2 и траис-нииерилен.
Этот изомер, в отличие от г^гс-пиперилена, реагирует
с малеиновым ангидридом:
СН
СН
СН,
нс—со
НС-СО
сн2
нсх хсн—со
1 1 >с
нс сн—со
СН
I
СН3
Пиперилен м. б. получен дегидратацией 2,3-иен-
тандиола, каталитич. дегидрированием /«-понтонов.
Удобный лабораторный метод синтеза шшерилена —
«исчерпывающее» метилирование пиперидина с после-
последующим расщеплением продукта метилирования.
Ппперилен содержится в продуктах крекинга и пи-
пиролиза нефти и нефтепродуктов. Он образуется в ка-
качестве побочного продукта в процессе синтеза изопрена
дегидрированием изопентаяа и изоамиленов и при син-
синтезе бутадиена из спирта но способу С. В. Лебедева.
Получаемый в пром-стн пиперилен содержит 75—81%
транс- и 19 — 25% ^мс-нзомеров.
Полнпиперилены (П.). Гомо- и сополимеры пипери-
лена м. б. получены полимеризацией: 1) в массе (ка-
(катализаторы — щелочные металлы); 2) в р-ре иа литий-
органич. катализаторах или Циглера — Натта катали-
катализаторах', 3) в эмульсии.
Наибольший нрактич. интерес представляет и о л и-
м е р и з а ц н я п и п е р и л е н а в р - р а х с ис-
использованием л и т и и о р г а н и ч. к а т а-
л и заторов (;)тил-, бутил- или гексиллитпя).
Процесс проводят в инертных растворителях — алифа-
алифатических, циклических или ароматических углеводо-
углеводородах — при 50—90 С до конверсии мономера 80—
95%. П. выделяют нз р-ра обычными приемами (см.
Полимеризация в растворе). Мол. массу получаемого
каучукоподобного П. регулируют, изменяя соотноше-
соотношение между количеством мономера и катализатора.
Благодаря отсутствию реакций разветвления и сшива-
сшивания исключается образование нерастворимого полиме-
полимера. Важное преимущество этого метода перед методом
его синтеза на катализаторах Циглера — Натта (см.
ниже) — возможность полимеризации промышленных
смесей цис- и 7драш?-изо1Aеров пинерилена с образова-
образованием высокомолекулярных продуктов (табл. 2), обла-
обладающих достаточно хорошими механич. свойствами
(табл. 3). Отличительная, особенность П., получаемого
из смеси цис- и транс-ииперилена,— значительно более
широкое молекулярно-массовое распределение и, сле-
следовательно, лучшие технологнч. свойства, чем у ноли-
трамс-пиперплена (см. табл. 3).
Каучуки, синтезируемые иа литийорганич. катали-
катализаторах, практически не содержат примесей и имеют
светлую окраску. Этим обусловлена целесообразность
их применения, в первую очередь, в производстве
резиновой обуви, а также нек-рых резино-технич.
и кабельных изделий. Использование шшерилена, вы-
выделяемого из фракции С5 пиролиза нефтепродуктов,
должно обеспечить получение каучука, имеющего
более низкую себестоимость, чем себестоимость синте-
синтезируемого этим же способом бутадиенового каучука.
605
ПИРИДИНА ПОЛИМЕР
606
Таблица 2. Структура и физические свойства полипиперилена,
полученного на литийорганических катализаторах
Показатели
Содержание звеньев, %
1,^-цис и 1,2
1,Ь-транс и 1 ,2 . . .
3,4
Темп-pa стеклования, СС
Мол. масса* М • 10~3 . . .
Содержание
транс - 99,
цис— 0
38,2
61,3
0,5
4 00 — 600
изомеров в пинерилене
6%
4%
транс — 7 5 — 81%
цис-19-25%
4 0,5
59,1
0,4
— 5 0 , 5
200-500
* Определена по данным вискозиметрии; растворитель —
бензол.
Таблица .3. Свойства резиновых смесей и вулканизатов
на основе нолиниперилена*, полученного на литийорганических
катализаторах
Показатели
Содержание изомеров в пиперплене
транс -99 , 6%
цис —0 , 4%
траке-75 - 81%
цис— 19 -2 5%
Резиновые смеси
Вязкость по Муни при
100 С
50 — 71
G-9
В у л к а н и з a t ы*
32-48
1-4
Прочность при растяже-
растяжении, Мн'м2 (кгс/см2) . .
Относительное удлине-
удлинение, %
Остаточное удлинение, %
Эластичность по отскоку,
16-19
A60-190)
560-750
12-16
30-38
16,5-19
A65-190)
560-730
10- 18
32-39
* Состав смеси (мае. ч.): каучук—100; рубракс-5; стеари-
стеариновая к-та —2; сантокюр —1 ,2; ZnO-5; сажа ПМ-70 —50; се-
сера- 2.** По 1 0-балльной системе; лучший балл 1. *** Вулкани-
Вулканизация 4 0 мин при 143 СС.
При полимеризации и и и е р и л е н а
в р-ре на катализаторах Циглера —
Н а т т а высокомолекулярные полимеры получены
только из /и/;я/*с-изомера. В частности, из этого изоме-
изомера синтезированы синдиотактич. полимеры, содержащие
80—94% звеньев \,^±-цис, способные к кристаллизации
при растяжении. Ненаполненные вулканизаты на ос-
основе таких П. характеризуются высокой прочностью
при растяжении и эластичностью.
Некоторые свойства 1,4-^мс-П. и его наполненных вул-
вулканизатов E0 мае. ч. сажи ДГ-100) приведены ниже:
Исходный полимер
Содержание звеньев, %
1,4-цис 90,0
1,4-транс 2,5
1,2 7,5
Темп-pa стеклования, °С —57,5'
Характеристич. вязкость в 0?а:юле, дл/г 2,5'
Вязкость по Муни при 100°С 38,2
В у л и а н и з а т
Прочность при растяжении, Мн/м2 (кгс/см2)
до старения 28 B80)
после старения 48 ч при 100 СС 22 B20)
Относительное удлинение, "„
до старения 800
после старения 4 8 ч при 100 °С 7 70
Остаточное удлинение, °„ 3 0
Сопротивление раздиру, кн!м, или кгс/см .... 85
Твердость по ТМ-2 96
Эластичность по отскоку, % 33
Истираемость, с.ч3/(квт • ч) 77
Сопротивление разрастанию порезов, тыс. цик-
циклов 360
В случае стереоспецифич. сополимеризации 90%
бутадиена и 10% трапс-пиперилена образуются сопо-
сополимеры, отличающиеся от стереорегулярного бутадие-
бутадиенового каучука СКД несколько более высокой темпера-
температурой стеклования и замедленной скоростью кристал-
кристаллизации.
Опыты но эмульсионной г о м о- и с о н о-
л и м е р и з а ц и и п и п е р и л о н а не дали прак-
практически интересных результатов. Гомополимер и трой-
тройные сополимеры бутадиена, шшерилена и стирола
(а-метилстирола) но комплексу технич. свойств не-
несколько уступали гомо- и сополимерам бутадиена.
Удовлетворительные результаты получены при исполь-
использовании шшерилена вместо бутадиена в сополимерах
со стиролом, применяемых для изготовления кожеподоб-
кожеподобных резин. Наиболее высокими показателями характе-
характеризовались резины на основе сополимеров, содержащих
10—20% звеньев пииерилена, 70—85% стирола и
5 —10% акрилонитрила. Бутаднен-шшернленоные ла-
тексы нашли ограниченное применение в качестве свя-
связующего в производстве фрикционных асбестовых
и нек-рых других изделий.
Полимеризацией и и и е р и л е и а в
массе в присутствии щелочных металлов в СССР
в 50-х гг. был получен шшериленовый каучук марки
СКП. Однако вследствие технологич. недостатков этого
способа полимеризации и невысокого качества каучу-
ков промышленное производство СКП не было орга-
организовано.
Высокие темпы развития процессов пиролиза нефте-
нефтепродуктов с целью получения олефннов и диенов
(в частности, изопрена), в к-рых образуется большое
количество шшерилена, обусловливают расширение
исследовательских работ в области синтеза полимеров
пиперилепа во многих странах, в том числе и в СССР.
Исследования полимеризации шшернлена были на-
начаты практически одновременно с изучением полимери-
полимеризации бутадиена (Беркенгейм, 1895; Тиле, 1901; Лебе-
Лебедев, 1913).
Лит.: Лившиц И. А., Ильина С. И., Рейх В. П.,
Хим. пром-сть, № 6, 22 A957); М а к е е в а А. Р. [и др.],
Кауч. и рез., .N° 9, 25 A958); J. Polymer Sci., 31, № 2 A963);
БреслерЛ. С, Д о л г о и л о с к Б. А., К р о и а ч е-
ваЕ. Н., ДАН СССР, 155, № 5, 1101 A964); Л и в ш и ц И. А.,
А ф а л а с ь е в И. Д., Г е р га т е й н Е. Р., Кауч. и рез.,
№ 2, 4 A969); Лившиц И. А. [и др.], Кауч. и рез., Л?» 2 7
A973). Я. В. Бородина.
ПИПЕРИЛЕНОВЫЙ КАУЧУК — см. Пиперилена
полимеры.
ПИРИДИНА ПОЛИМЕР, н о л и и и р и д и и
(polypyridine, Polypyridine, polypyridine)
[ = СН— СН=СН— СН=СН— N = ]n (П.) — аморфный
неплавкий полимер черного цвета с металлич.
блеском, обладающий высокой термостойкостью. Ха-
рактеристпч. вязкость П. в серной к-те 0,02—0,2 дл/г
(в зависимости от условий полимеризации). П. раст-
растворим в конц. минеральных к-тах; после длительной
термообработки при темп-pax выше 400 °С становится
нерастворимым. Обладает полупроводниковыми свойст-
свойствами. При комнатной темп-ре электрич. проводимость
П., полученного при 390 °С, изменяется симбатно
с изменением молекулярной массы и составляет
10-ю —jQ-7 ом~1-см~1. П.— парамагнитное вещество.
Полимеризация ненапряженных азотсодержащих аро-
матич. гетероциклов (пиридина или хиполина) м. б.
осуществлена путем нагревания их комплексов с коор-
координационно-ненасыщенными соединениями, напр, [пи-
[дин]2^пС12, [хинолин]2^пС12. Комплексы нагре-
нагревают в присутствии протонсодержащих возбудителей
(НРО3, Н2О, хлоргидрата пиридина): в случае пири-
пиридина — при темп-ре не ниже 330 °С, в случае хиноли-
на — при темп-pax не ниже 250 °С. Полимеризация
происходит в результате раскрытия гетероцикла. Роль
комплексообразователя (ZnCl2) сводится к поляриза-
поляризации связи азот — углерод в гетероцикле, что облегчает
ее гетеролитич. расщепление. Полимеризация носит
автокаталнтич. характер; скорость процесса заметно
607
ПИРРОНЫ
608
возрастает при введении в исходную реакционную
смесь заранее полученного полимера.
Комплексы пиридина и хинолина сополимеризуются
друг с другом, а также с таким насыщенным гетеро-
циклич. соединением, как е-капролактам. В последнем
случае получаются сополимеры, в макромолекулах
к-рых участки сопряжения чередуются с гибкими
насыщенными участками; темп-ры плавления (размяг-
(размягчения) таких сополимеров 450—500 °С.
О свойствах пиридина см. Амины.
Полимеры пиридина A963) и хинолина A964) впер-
впервые получены В. А. Каргиным, В. А. Кабановым и сотр.
Лит.: Кабанов В. А., в сб. Кинетика и механизм
образования и превращения макромолекул, М., 1968, с. 35.
Д. А. Топчиев.
ПИРРОНЫ — см. Поли(ароилен-бис-бензимидазолы).
ПЛАВЛЕНИЯ ТЕМПЕРАТУРА полимеров
(melting point, Schmelztemperatur, point de fusion).
У полимеров различают две П. т.— равновесную (Ти°л)
и экспериментальную {Тпл), которую обычно называют
просто П. т.
Равновесная П. т. соответствует точке фазо-
фазового равновесия между монокристаллами полимера,
размеры к-рых достаточно велики, чтобы поверхностная
свободная энергия была пренебрежимо мала по срав-
сравнению с объемной, и расплавом при нормальном дав-
давлении. Она определяется термодинамич. соотношением
Тп°л = Д#ПЛ/Д5ПЛ, где А#пл и А?пл — соответственно
теплота и энтропия плавления (табл.).
Параметры плавления некоторых полимеров
Полимер
Полиамид-6,6
Полистирол . . .
Полиформальде-
Полиформальдегид
Полипропилен .
Полиэтилен . . .
Полиэтиленоксид
Полипропилен-
оксид . , . .
Каучук натура-
натуральный
Равно-
Равновесная
темп-ра
плавле-
плавления
Ул' К
548
523
460
456
416
340
3 55
303
Теплота
плавления,
А "ил-
дж/молъ
(кал/молъ)
46200
A1000)
8400
B000)
7140
A700)
10080
B400)
8064
A920)
8400
B000)
8400
B000)
4410
A050)
Энтропия
плавления,
дж/молъ- К
(пал/молъ-иС)
84
B0,0)
16
C,9)
15 5
C,7)
2 2,3
E,3)
19,3
D,6)
24,8
E,9)
23,5
E,6)
14 7
C,5)
Экспери-
менталь-
темп-ра
плавле-
плавления*
1 ПЛ' JV
538
515
454
449
411
339
346
284
* Экспериментальные П. т. могут различаться в зависимости
от условий кристаллизации, метода определения и др.
Существенное влияние на Гп°л оказывает жесткость
макромолекул. Низкие значения 7*п°л обычно харак-
характерны для полимеров с гибкими макромолекулами,
к-рые в расплавах сильно свернуты. Плавление таких
полимеров связано со значительным увеличением кон-
конформационной энтропии. Полимеры с жесткими цепями
имеют сравнительно высокие значения Гпл при сопо-
сопоставимых с др. полимерами теплотах плавления. Мак-
Макромолекулы этих полимеров в расплавах находятся
в развернутых конформациях, что приводит к мень-
меньшему возрастанию конформационной энтропии. Суще-
Существует ряд факторов, снижающих Гпл и обычно наз.
«примесными». Такими факторами являются любые
нарушения регулярности цепи — наличие разветвле-
разветвлений и сшивок, концевых групп, конфигурационные
неоднородности (цис-, транс- и т. д.).
Кристаллизация полимеров, в отличие от низкомо-
низкомолекулярных веществ, проходит обычно не полностью
и при этом образуются метастабильные кристаллиты.
При нагревании они плавятся в некотором интервале
темп-р (АГдд, иногда до десятков градусов). На прак-
практике верхнюю границу этого интервала и принимают
за экспериментальную П. т. Тпл обычно
ниже ТпЛ примерно на 5—20 °С. Наблюдаемые зна-
значения Тил и АТил зависят от химич. природы макро-
макромолекул, молекулярно-массового распределения, усло-
условий кристаллизации и способа определения (см. табл.).
В интервале плавления происходят процессы т. наз.
«частичного» плавления, связанные с постепенным
расплавлением наиболее дефектных граней кристал-
кристаллитов и постадийным плавлением кристаллитов раз-
разных размеров и различной степени дефектности.
На определенной стадии плавления, когда темп-ра
достаточно высока и степень кристалличности заметно
уменьшилась, образовавшийся расплав может начать
рекристаллизоваться. Частичное плавление полимера,
за к-рым следует рекристаллизация, часто наз. отжи-
отжигом. Возможностью рекристаллизации при нагреве
обусловливается зависимость Тпл от условий плавле-
плавления, прежде всего от скорости нагрева. В зависимости
от изменения последней величины может происходить
как возрастание, так и уменьшение Тпл. Эксперимен-
Экспериментальные значения Тпл хорошо воспроизводятся и наи-
наиболее приближаются к ТпЛ, когда кристаллизация
проводится вблизи П. т., а измерения осуществляются
при очень медленном нагреве.
Существует несколько экстраполяционных мето-
методов определения Т^л полимеров. Первый
метод основан на зависимости jM
П. т. кристаллитов от их раз-
размеров (рис. 1). Тпл ламеляр-
ного полимерного кристаллита
толщиной I (длина складки)
определяется соотношением
2ат
где ат — поверхностная энер-
энергия торцов кристаллитов; рнр —
плотность кристаллитов. Дли- Рис ^ 1/i
на складки I определяется ли-
либо по данным малоугловой рентгеновской дифракции,
либо электронномикроскопически. Измерения прово-
проводятся в таком режиме, чтобы избежать рекристал-
рекристаллизации.
Второй метод основан на зависимости Тпл от темп-ры
кристаллизации (Т^, рис. 2). Значение Тп°л находится
как точка пересечения прямых Тил = f (Гкр) и Тп°л —
= jTHp. Этим методом определены значения Тпл мно-
многих полимеров.
Рис. 2
Рис. 3
Vx.
Третий метод (рис. 3) основан на зависимости П. т.
от мол. массы (степени полимеризации), к-рая дается
ур-нием
1___ _1 2Д
Тпл Тпл ДНпл Хп
где R — универсальная газовая постоянная; А#пл —
теплота плавления в расчете на мономерное звено;
609
ПЛАСТИКАТ
610
Хп — среднечисловая степень полимеризации. Этот ме-
метод позволяет получить Т°ил полимера исходя из П. т.
его низкомолекулярных аналогов.
Наиболее распространенными методами определения
Тпл являются тепловые (дилатометрия, калоримет-
калориметрия, дифференциальный термический анализ), прямые
оптические и спектроскопические, механические и др.
Значения Тпл, полученные разными методами, могут
различаться, что скорее всего обусловлено различной
чувствительностью методов.
Лит.: Манделькерн Л., Кристаллизация полиме-
полимеров, М.— Л., 1966; Уббелоде А., Плавление и кристал-
кристаллическая структура, М., 1969; Шарплез А., Кристалли-
Кристаллизация полимеров, М., 1968; Van KrevelenD. W.,
Properties of polymers, Amst. —L. — N. Y., 1972.
Ю. К. Годовский.
ПЛАСТИ30ЛИ — см. Пасты полимерные.
ПЛАСТИКАТ (plasticat) — технич. название термо-
термопластичных смесей пластифицированного поливинил-
хлорида (ПВХ), подвергнутого пластикации.
Состав. Кроме ПВХ и пластификатора, в состав П.
входят стабилизаторы (термостабилизаторы и антиок-
сиданты), наполнители, смазки и пигменты.
Важнейшее требование ко всем компонентам смесей
для П.— отсутствие или минимальное содержание
ионогенных примесей, к-рые вызывают ухудшение
его диэлектрич. свойств (ионогенные вещества могут
также образовываться при разложении ПВХ и пласти-
пластификаторов в процессе переработки композиции или
эксплуатации материала). Размер частиц порошкооб-
порошкообразных ингредиентов (стабилизаторов, наполнителей,
пигментов и смазок) не должен превышать 2 —10 мкм.
Для этого их перед использованием дробят в коллоид-
коллоидных, шаровых, бисерных или кавитационных мельни-
мельницах, на валковых краскотерках и т. п.
В производстве П. используют поливинилхло-
р и д, полученный суспензионной полимеризацией или
полимеризацией в массе, со среднечисловой мол. массой
90—115 тыс. (константа Фикентчера 70—90, см. Ви-
нилхлорида полимеры). Для нек-рых назначений основу
П. составляют сополимеры винилхлорида, чаще всего
с винилацетатом.
В качестве пластификаторов (от 30 до
90 мае. ч.; здесь и далее колич. ингредиентов указано
в расчете на 100 мае. ч. ПВХ) применяют вещества,
хорошо или ограниченно совместимые с ПВХ. Хорошо
совместимые с ПВХ пластификаторы — диалкилфта-
латы и трикрезилфосфат. Ограниченно совместимые
пластификаторы — диоктилсебацинат, адипинаты и
азелаинаты, триоктилфосфат, полипропиленадипинат и
полипропиленсебацинат (см. также Пластификато-
Пластификаторы). Для получения П. с высокой морозостойкостью
(до —60 СС) применяют смеси ограниченно совместимых
низкомолекулярных пластификаторов с хорошо сов-
совместимыми пластификаторами. Весьма эффективный
способ повышения морозостойкости П.— введение
в композицию бутадиен-нитрильного каучука. При
этом повышается также масло- и бензостойкость П.
Если П. находится в контакте с полиэтиленом,
резиной и др. (напр., в кабеле с изоляцией проводов
из полиэтилена и оболочкой из П.), пластификатор
из П. может мигрировать в контактирующий материал.
При этом повышается жесткость П., снижаются моро-
морозостойкость и др. его свойства. В свою очередь, погло-
поглощение пластификатора приводит к ухудшению диэлект-
диэлектрич. свойств контактирующего материала. Меньшую
склонность к миграции из П. имеют смеси низкомоле-
низкомолекулярного и полимерного пластификаторов. Однако
морозостойкость П., содержащих такие пластификато-
пластификаторы, низка (от —30 до 5 СС).
Для получения П. с высоким электрич. сопротивле-
сопротивлением п морозостойкостью до —40 °С применяют фта-
латы — самые дешевые пластификаторы. В произ-
производстве П. для высокотемпературной изоляции при-
применяют пластификаторы, обладающие очень малой лету-
летучестью — додецилфталат, дидодецилфталат, дитриде-
цилфталат, производные пентаэритрита, сложные эфи-
ры пиро- и тримеллитовой к-т и полимерные пласти-
пластификаторы. Комбинируя
2 — 3 пластификатора,
добиваются сочетания
требуемых свойств.
Для снижения стоимо-
стоимости П. и придания ему
негорючести часть плас-
пластификатора (до 30%)
Зависимость уд. объемного
электрич. сопротивления р
пластиката при 25 °С от со-
содержания аэросила (в полу-
логарифмич. координатах):
1 — сухие образцы; 2 — об-
образцы после выдержки в ди-
дистиллированной воде в те-
течение 24 ч\ 3 — образцы, не
содержащие свинцовых со-
солей, после выдержки в ди-
дистиллированной воде в те-
течение 24 ч.
А
О 5 10 15 20 25 30
Содержание аэросила, мае. ч.
иногда заменяют хлорированными парафинами. По-
Последние имеют незначительную совместимость с ПВХ,
поэтому их применяют только в смеси с хорошо со-
совместимыми пластификаторами.
Термостабилизаторы вводят в компо-
композицию в количестве от 3 до 15 мае. ч. Основное их
назначение — связывание НС1, выделяющегося при
термоокислительной деструкции ПВХ в процессе пере-
переработки и эксплуатации при высокой темп-ре. Хлори-
Хлористый водород катализирует дальнейшее разложение
ПВХ, снижает электрич. сопротивление П. и корро-
корродирует металлич. части перерабатывающего оборудо-
оборудования. Эффективные термостабилизаторы — одно-,
двух- и трехосновные соли свинца (основной карбонат,
двухосновной фталат, двухосновной фосфит, трехоснов-
трехосновной сульфат и др.), к-рые используют для получения
непрозрачных П. В производстве прозрачного П. при-
применяют соли бария, кадмия и кальция. Часто исполь-
используют синергич. смесь двух стабилизаторов (напр., двух-
двухосновного фталата и двухосновного стеарата свинца).
Повышение атмосферо- и светостойкости П. дости-
достигается при использовании антиоксидантов
(фенолов, бензофенонов, бензтриазолов, эпоксидиро-
ванных растительных масел, дибензилсульфида, три-
нонилфенилфосфита и др.). Антиоксиданты предотвра-
предотвращают деструкцию ПВХ и пластификаторов. Их вводят
в количестве 0,02—0,5% от массы пластификатора.
Наполнители (каолин, аэросил, мел, дву-
двуокись титана, тальк, асбест и др.) вводят в П. в коли-
количестве не более 30 мае. ч. (см. рис.). Чрезмерное уве-
увеличение концентрации наполнителей в П. может при-
привести к ухудшению его морозостойкости, водостой-
водостойкости, перерабатываемости. Обычно морозостойкие П.
содержат не более 20 мае. ч. наполнителя.
Смазки облегчают переработку П. и улучшают
внешний вид изделий. В качестве смазок применяют
стеарин, стеариновую к-ту, стеараты кальция, кадмия,
бария и свинца в количестве 1—3 мае. ч.
Для получения окрашенного П. в состав компози-
композиции вводят органические или минераль-
минеральные пигменты в количестве от 0,1 до 3 мае. ч.
Как правило, для получения яркой окраски минераль-
минерального пигмента требуется в 3—4 раза больше, чем
органического. Минеральные пигменты хуже диспер-
диспергируются в композиции, однако они более атмосферо-
стойки, не мигрируют на поверхность и не снижают
электрич. сопротивление П. Среди органич. пигментов
для окраски П. наибольшее распространение находят
зеленый и голубой фталоцианиновые, оранжевый,
611
ПЛАСТИКАТ
612
бордо и синий антрахиноновые. Наибо-
Наиболее распространенные минеральные пиг-
пигменты — свинцовые и молибдатные кро-
кроны, кадмиевые, марганцовые, кобаль-
кобальтовые и др. Черный цвет придают П.
введением в композицию сажи, белый—
рутильной формы двуокиси титана.
Производство. Технологич. схема по-
получения П. включает след. основные
стадии: смешение, пластикацию и гра-
гранулирование.
При смешении компонентов про-
происходит поглощение пластификатора
полимером, разрушение агломератов по-
порошкообразных ингредиентов и равно-
равномерное распределение их между части-
частицами ПВХ. Дальнейшее дробление твер-
твердых ингредиентов осуществляют на ста-
стадии пластикации. Смешение осуществ-
осуществляют в больших смесителях периодич.
действия, напр, в Z-образном смеси-
смесителе, ленточном смесителе или смесителе
турбинного типа. В большинстве слу-
случаев сначала загружают ПВХ и порош-
порошкообразные стабилизаторы и смешивают
их в течение 3—5 мин, после чего по-
подают пластификатор. При 80—90 СС
ПВХ начинает интенсивно поглощать
пластификатор. Затем темп-ру повыша-
повышают до 110—130 °С. В самом конце сме-
смешения вводят наполнители и получен-
полученную смесь перемешивают еще 10—20
мин. Весь цикл смешения длится 30—
60 мин.
Порядок загрузки м. б. иным; напр.,
наполнители загружают сразу после
ПВХ, а стабилизаторы и пигменты, ес-
если их перед смешением необходимо раз-
размельчить, поступают в смеситель из
мельницы или краскотерки в виде су-
суспензии или пасты в пластификаторе
(мельницы тонкого помола или крас-
краскотерки производят размол твердых тел
в жидкости, в данном случае в пласти-
пластификаторе). Если порошкообразные ком-
компоненты имеют склонность к агломера-
агломерации и поступают в смеситель в виде су-
суспензии в пластификаторе, целесообраз-
целесообразно предварительно «смочить» ПВХ не-
небольшим колич. пластификатора. Это
позволяет исключить слишком быстрое
поглощение полимером пластификатора,
содержащегося в суспензии, и препят-
препятствует агломерации порошкообразных
компонентов, к-рые еще не успели рав-
равномерно распределиться в смеси. Рас-
Растворимые в пластификаторе антиокси-
данты целесообразно вводить в смеси-
смеситель в виде р-ра.
Выгрузку горячей смеси производят
в «холодную» емкость двухстадийного
смесителя или сразу на вальцы или в
экструдер, если используют смеситель
одностадийного типа.
Пластикацию смеси сразу же
после смешения осуществляют на валь-
вальцах, в экструдере или в смесителе ти-
типа «Бенбери». Последний аппарат обес-
обеспечивает очень высокое качество плас-
пластикации, но после него пластицирован-
ную массу необходимо провальцевать
для получения листового материала.
При пластикации набухшие в пласта-
пласта,2 .
СЮ
lag
R§3
С И н
fisg
IS
i
Is
|1!
Щ
If II
(ill
I I
lO "«-< lO
~* CM CM CM
05 00 о
w ^2 ^
II!!!
I 7 I
!M "
COO °
Ю -гнО О
•гч СО
7J '
оо
°^СМ
о°оо
00
I I
000
-CM
I I
11
ooo^ 00
CDO^^h CO M
ooo о c-o
^ ? О О -ун
^ю о
* 'со •
"гН •*"• со
г^О О С
ооо° ^
00 О О ° -С
оо м о ^«
I I I
I I
см см
I I
2&
н В
.2 с,-
* » * ее
:51
;?^^
.а *
• I I
ш
i=J U CM J3 a ;
8Игн ?> ;
н «со 5 2^
О О wO Я*
613
ПЛАСТИКАЦИЯ КАУЧУКОВ
614
фикаторе частицы ПВХ разрушаются до размера
1—2 мкм и менее, в результате чего поверхность кон-
контакта ПВХ со стабилизаторами и наполнителями воз-
возрастает в десятки раз. На вальцах и в головке экс-
трудера поддерживают темп-ру, при к-рой ПВХ на-
находится в вязкотекучем состоянии A55—180 °С).
Гранулирование. Сформованные после
пластикации листы, ленты или жгуты охлаждают во-
водой, осушают воздухом, а затем гранулируют. Готовые
гранулы размером 3—5 мм поступают на упаковку.
Иногда П. не гранулируют, а выпускают в виде лент
или полос различной толщины и ширины. Нек-рые
экструдеры снабжены гранулирующей головкой с вра-
вращающимся режущим устройством. При этом выходя-
выходящие через головку экструдера жгуты в горячем виде
режутся на гранулы, к-рые охлаждаются воздухом.
В этом случае вторая и третья технологич. стадии
совмещаются.
Окрашивают П. двумя способами: 1) пигмент
вводят в П. на стадии смешения компонентов; 2) кра-
крашение П. осуществляют с помощью окрашенного кон-
концентрата на стадии переработки пластиката в изде-
изделия. Концентрат представляет собой смесь пластифи-
пластифицированного ПВХ с большим количеством пигмента
B0—50 мае. ч.). Для получения качественной окраски
П. необходимо, чтобы концентрат обладал более низ-
низкой темп-рой текучести, чем П. Поэтому для его полу-
получения применяют ПВХ с константой Фикентчера
65-70.
Свойства. Основные свойства П. представлены в таб-
таблице. П. относительно устойчив к действию воды, к-т,
щелочей, частично растворим в нитробензоле, диокса-
не, галогенпроизводных углеводородов. Специальные
марки П. достаточно устойчивы к действию масел
и бензина. Такие П. после выдержки в течение 24 ч
в бензине при нормальной темп-ре или в масле при
100 СС сохраняют механич. свойства на 85—90%.
Полимерный компонент П. и пластификаторы хорошо
растворимы в дихлорэтане, циклогексаноне, тетрагид-
рофуране, диметилформамиде. П. огнестойки — они
затухают при вынесении из пламени.
Верхняя темп-pa эксплуатации изделий из П. обычно
не превышает 70 °С. Однако изоляция авто- и авиа-
авиапроводов, а также проводов обогревательных прибо-
приборов работает длительное время при темп-ре 105 °С.
Ухудшение основных свойств П. в течение всего срока
эксплуатации не должно превышать 20%. К П. быто-
бытового и некабельного назначения предъявляются менее
жесткие требования.
Переработка и применение. П. перерабатывают
в изделия на экструдерах, каландрах, прессах и литье-
литьевых машинах. Темп-pa переработки гранул П. должна
быть приблизительно на 5 СС выше, чем при пласти-
пластикации.
П. широко применяют в качестве изоляции, а также
для изготовления уплотняющих прокладок, мягких
резервуаров, тары, трубок, лент, пленок, колпачков,
втулок, мембран, профильных и погонажных изделий
и др. Электропроводящие П. применяют для экрани-
экранирования высоковольтных кабелей, изготовления токо-
проводящих жил, нагревательных элементов и др.
За рубежом материал, подобный П., выпускают
под след. торговыми названиями: в е л в и к (Вели-
(Великобритания), с и к р о н (Италия), с а м и к о н VM,
винихлон (Япония), кохинор, джеон
(США), люколен G (Франция), полвинит
(Польша) и др.
Лит.: Получение и свойства поливинилхлорида, под ред.
Е. Н. Зильбермана, М., 1970; Электрические свойства полиме-
полимеров, под общ. ред. Б. И. Сашина, Л., 1970; Справочник по
пластическим массам, под ред. М. И. Гарбара [и др.], т. 1 — 2,
М., 1967—69; Николаев А. Ф., Синтетические полимеры
и пластические массы на их основе, 2 изд., М. — Л., 4966; Т и-
ниус К., Пластификаторы, пер. с нем., М.— Л., 1964.
В. В. Гузеев.
ПЛАСТИКАЦИЯ КАУЧУКОВ (mastication of rub-
rubbers, Mastikation von Kautschuken, mastication des
caoutchoucs) — технологич. процесс резинового произ-
производства, в результате к-рого уменьшается высоко-
высокоэластическая и увеличивается пластическая составляю-
составляющая деформация каучука. В реальных условиях пла-
пластикации (П.) эти изменения пласто-эластических
свойств каучука обусловлены гл. обр. деструкцией
его макромолекул.
П. проводят для облегчения дальнейшей обработки
каучуков — смешения с ингредиентами, формования
и др.; наибольшее значение она имеет при переработке
натурального каучука. П. подвергают также нек-рые
синтетич. каучуки: стереорегулярные изопреновые,
получаемые на литиевых катализаторах, бутадиен-
нитрильные, хлоропреновые нек-рых типов л др.
Широко используемые в пром-сти стереорегулярные
бутадиеновые и изопреновые каучуки, получаемые
на комплексных (координационно-ионных) катализа-
катализаторах, не пластицируют. Не подвергают П. и бута-
диен-стирольные каучуки низкотемпературной поли-
полимеризации, т. к. их нластич. свойства (мол. массу)
регулируют в ходе синтеза.
Различают два способа П.— механическую и термо-
термоокислительную (без механич. воздействия на каучук).
Основное значение в пром-сти имеет механич. П.,
ускоренная введением в каучук нек-рых химич. аген-
агентов — ускорителей пластикации (см. ниже); такой
способ иногда наз. химической П. Степень П. оцени-
оценивают обычно показателями пластичности, жесткости,
вязкости по Муни (см. Пласто-эластические свойства).
Механическая пластикация. При этом способе П.
могут происходить как деструкция, так и активиро-
активирование химических связей в макромолекулах под вли-
влиянием механических напряжений (см. Механохимия).
Соотношение между скоростями обоих процессов за-
зависит от температуры, среды (воздух, кислород, азот),
интенсивности механических воздействий, типа поли-
полимера. С повышением температуры скорость П. сна-
сначала уменьшается, а затем возрастает. Температу-
Температура, соответствующая минимальной скорости П., зави-
зависит от типа полимера; например, для натурального кау-
каучука она составляет 70—80 СС (рисунок). Интенсивная
П. при темп-pax ниже 70 °С
обусловлена в основном §
механич. разрывом цепей. ^ 2о
Атмосферный кислород §
Зависимость интенсивности
пластикации натурального ка-
каучука от темп-ры (продолжи-
(продолжительность обработки на валь-
вальцах 30 мин).
; 40
60
50 70 90 110 130
Температура пластикации, °С
играет в этом случае роль стабилизатора образующихся
макрорадикалов и препятствует, т. обр., образованию
в полимере разветвленных и сшитых структур. Уско-
Ускорение П. выше 80 °С связано с тем, что в этих усло-
условиях основным процессом становится окисление поли-
полимера, активированное тепловой и механич. энергией.
Чем выше темп-pa, тем больше роль тепловой энергии
и меньше роль механической.
Как и в случае окисления каучуков, при высокотем-
высокотемпературной П. происходят одновременно деструкция
и сшивание. Соотношение между скоростями этих про-
процессов зависит от типа полимера. Напр., при П. нату-
натурального и синтетич. изопренового каучуков превали-
превалирует деструкция, при П. бутадиен-нитрильных каучу-
каучуков — сшивание. Существенную роль при высокотемпе-
высокотемпературной П. играют антиоксиданты, в присутствии
к-рых сшивание замедляется.
К наиболее распространенным ускорителям
пластикации относятся: пентахлортиофенол
615
ПЛАСТИКАЦИЯ КАУЧУКОВ
616
(ренацит V) и его цинковая соль (ренацит IV) — ф-лы
соответственно 1 и 2; о,о'-дибензамидодифенилдисуль-
о,о'-дибензамидодифенилдисульфид (пептон 22) и его цинковая соль (пептон 65) — ф-лы
соответственно 3 и 4; ди-B,4,5-трихлорфенил)дисуль-
фид (бистри) — ф-ла 5.
С1 С1
ci-/~~Y-sh
С1 С1
С1 С1
Cl Cl
2
H5C6OCHNK
Zn
/NHCOC6H5
S
H5C6OCHN\
Zn
4
Кроме того, применяют цинковые соли высших
жирных к-т (актипласт), а также нек-рые ускорители
вулканизации — меркаптобензтиазол (каптакс), дибенз-
тиазолилдисульфид (альтакс), гуанидины. Оптималь-
Оптимальное количество ускорителей П. составляет 0,1—0,3 мае.
ч. на 100 мае. ч. каучука. При П. натурального и син-
тетич. изопренового каучуков м. б. использованы все
названные выше соединения, при П. хлоропренового
каучука — гл. обр. дифенил- и ди-орто-толилгуанидин,
каптакс, альтакс.
Применение ускорителей приводит к повышению
скорости процесса как при низких, так и при высоких
темп-pax, а также к снижению темп-ры минимальной
скорости П. При низкотемпературной П. натурального
каучука эффективность действия ускорителей умень-
уменьшается в следующем ряду: тиофенолы > цинковые со-
соли тиофенолов > дисульфиды. Чем выше темп-pa П.,
тем меньше разница в активности этих соединений
(при 135 — 140 °С дисульфиды приближаются по эф-
эффективности к тиофенолам). В наибольшей степени
ускорители проявляют свою активность при темп-рах
ок. 80 °С и выше. На свойства резиновых смесей и вул-
канизатов они, как правило, не влияют.
Механизм действия ускорителей окончательно
не установлен. На примере окисления (пластикации)
натурального каучука при 70—100 °С в присутствии
меркаптанов показано, что последние в этом процессе
окисляются, превращаясь в дисульфиды. Эта реакция
протекает с большой скоростью и инициирует сравни-
сравнительно медленное окисление каучука. Кроме того, реа-
реагируя с макрорадикалами каучука, меркаптаны пре-
препятствуют сшиванию. Основные представления о ме-
механизме действия меркаптанов могут быть распрост-
распространены и на дисульфиды. Для образования тиофениль-
ных радикалов из дисульфидов присутствие кислорода
не обязательно, т. к. эти радикалы могут образо-
образоваться в результате распада дисульфидов под влия-
влиянием высокой темп-ры.
Условия механической пласти-
пластикации определяются как типом каучука, так и мас-
масштабами его переработки. Натуральный каучук пла-
пластицируют гл. обр. в резиносмесителях и в червячных
пластикаторах; в случае переработки небольших коли-
количеств каучука — на вальцах. Для пластикации син-
тетич. изопренового каучука чаще всего используют
резиносмесители и реже вальцы.
При П. на вальцах наибольшее повышение пластич-
пластичности наблюдается в течение первых 10—15 мин обра-
обработки. Поэтому натуральный каучук пластицируют
обычно в несколько приемов с «отдыхом» (~ 6—8 ч)
и охлаждением после каждого цикла. В соответствии
с этим различают каучук одно-, двух- и трехкратной
пластикации — П-1, П-2, П-3 (таблица).
Наиболее производительное оборудование — чер-
червячный пластикатор, в к-ром пластикаты П-1 и П-2
получают соответственно после одно- и двукратного
(с промежуточным «отдыхом» и охлаждением) пропуска
через машину при температуре в цилиндре 60 — 70 °С,
в головке 105—115 °С. Сокращение продолжительнос-
продолжительности обработки натурального и синте-
синтетического изопренового каучуков в
резиносмесителях в присутствии ус-
ускорителей позволяет в некоторых слу-
случаях совмещать П. с приготовлением
резиновой смеси.
Бутадиен-нитрильные каучуки, гл.
обр. жесткие, пластицируют на валь-
вальцах B0—40 мин) при минимальном за-
зазоре между валками. Так же, как и
для натурального каучука, целесо-
целесообразна двухстадийная П. Однако да-
даже в оптимальных условиях этого
процесса жесткость каучука снижает-
снижается не более чем на 4—5 н D00—500 гс). Скорость
механич. П. бутадиен-нитрильных каучуков возрастает
с увеличением содержания в них акрилонитрила. Иног-
Иногда пластицируют также и мягкие бутадиен-нитриль-
бутадиен-нитрильные каучуки; благодаря этому получают смеси с бо-
более гладкой поверхностью и с меньшей усадкой.
Режимы пластикации натурального каучука
Тип плас-
тицирован-
ного кау-
каучука
П-1 ....
П-2 , . , .
П-3 ....
Продолжительность обработки,
мин
на вальцах
E0-55 °С)
12
24 BX12)
36 CX12)
в скоростном резиносме-
сителе A40 — 180°С)
без ускори-
ускорителя
8
16 BX8)
с ускори-
ускорителем
4-5
6-8
Пределы
пластичности
0,21-0,30
0,31-0,40
0,41-0,50
Хлоропреновые каучуки (получаемые с применением
серы в качестве регулятора) пластицируют на вальцах
G—10 мин; 30—40 °С) или в резиносмесителях. В пос-
последнем случае П. можно совмещать с процессом сме-
смешения. Продолжительность обработки в отсутствие
и в присутствии ускорителей составляет соответственно
5 — 6 и 3—4 мин, темп-pa — не выше 100 °С.
Термоокислительная пластикация. В пром-сти такой
способ П. применяют только при переработке бутади-
ен-стирольных каучуков высокотемпературной поли-
полимеризации, вырабатываемых в небольших масштабах
(механич. П. этих каучуков малоэффективна). Измене-
Изменения пласто-эластич. свойств каучуков при термоокис-
термоокислительной П. обусловлены термоокислительной дест-
деструкцией макромолекул. В реальных условиях П.
одновременно, но с различными скоростями разви-
развиваются деструкция и сшивание; на первых стадиях П.
превалирует первый, на более позднюс — второй про-
процесс. Сшивание, к-рое наиболее отчетливо проявля-
проявляется при малых концентрациях кислорода и высоких
темп-pax, тормозится при введении в каучук антиокси-
дантов, солей железа, а также при снижении темп-ры
(в пределах, не вызывающих резкого замедления дест-
деструкции).
Оптимальные условия термоокислительной П., к-рую
проводят в котлах с циркуляцией воздуха, — 120—
140 °С и давление — 0,3 Мн/м2 (~ 3 кгс/см2). Однако
и в этих условиях в каучуке образуются разветвлен-
разветвленные и сшитые структуры. Поэтому смеси из термопла-
стицированного каучука имеют худшие технологич.
свойства, чем смеси из каучука, подвергнутого меха-
механич. П. при низких темп-pax (в частности, повышен-
повышенное эластич. восстановление), а резины — пониженные
механич. характеристики.
617
ПЛАСТИКИ С ПОЛЫМ НАПОЛНИТЕЛЕМ
618
Лит.: Догадки н Б. А., Химия эластомеров, М.,
1972; Кошелев Ф. Ф., Корн ев А. Е., К л и-
мов Н. С, Общая технология резины, 3 изд., М., 1968;
Справочник резинщика. Материалы резинового производства,
М., 1971, с. 359; Bateman L. [ed.], The chemistry and
physics of rubber-like substances, N. Y., 1963. д. Я. Девирц.
ПЛАСТИКАЦИЯ ПЛАСТМАСС (plastication
of plastics, Plastizierung von Kunststoffen, plastication
des plastiques) — процесс превращения материала
в расплав в ходе его переработки. П. п. связана с по-
повышением темп-ры материала в результате подвода
тепла к рабочим органам перерабатывающего оборудо-
оборудования от внешних нагревателей или вследствие выде-
выделения тепла при внешнем и внутреннем трении. Про-
Процесс осуществляют в таких режимах, чтобы в макси-
максимальной степени предотвратить деструкцию полимера
(сравни с пластикацией каучуков).
Пластикация м. б. самостоятельной стадией в пере-
переработке пластмасс (напр., в литьевых машинах сущест-
существует специальный узел пластикации, т. наз. пластика-
тор) или осуществляться одновременно с др. техноло-
гич. процессами (напр., с гомогенизацией материала
в зоне пластикации экструдеров). М. л. Кербер.
ПЛАСТИКИ С ПОЛЫМ НАПОЛНИТЕЛЕМ, син-
синтактические пены (sintactic foams, Kunst-
stoffe mit Hohlkorper-Fullstoff, plastiques aux charges
creux) — вид газонаполненных пластмасс, в к-рых
полые наполнители равномерно распределены в поли-
полимерном связующем.
Виды полых наполнителей. В качестве полых напол-
наполнителей используются частицы сферич. формы диа-
диаметром 20—70 мкм с толщиной стенки ~ 1,5—3% диа-
диаметра, насыпной массой 0,2—0,5 г/см3 (такие частицы
наз. микросферами, микробаллон а-
м и) или же сферы диаметром 10—40 мм (макро-
(макросферы). Введение полых наполнителей позволяет
получать более легкие пластики, чем с наполнителя-
наполнителями, имеющими монолитные частицы (каолин, квар-
кварцевая мука, тальк и др.), а также способствует обра-
образованию более текучих композиций и получению
пластиков с меньшими остаточными напряжениями в
материале.
Полые сферич. наполнители м. б. полимерными,
стеклянными, из керамики и металлов. Наиболее часто
используют наполнители из отвержденной феноло-фор-
мальдегидной смолы и стекла. Полые сферы из феноло-
формальдегидных смол получают на дисковых распы-
распылительных сушилках. Композиция, состоящая из смо-
смолы (в виде р-ра, эмульсии или тонкоизмельченного
порошка), в к-рую введены газообразователь, поверх-
поверхностно-активное вещество и др. добавки, с помощью
форсунок подвергается тонкодисперсному распылению
и током горячего воздуха переносится в сушилку.
Попадая в зону высоких темп-р, частички смолы пла-
плавятся и приобретают форму сферы. Одновременно с этим
происходит разложение газообразователя с выделением
продуктов, к-рые увеличивают размеры сферич. части-
частицы, и нарастание вязкости расплавленной смолы вплоть
до потери текучести в результате отверждения. Части-
Частицы наполнителя не должны иметь отверстий в обо-
оболочке. Это достигается подбором соответствующих
газообразователей и др. добавок, а также выбором
температурного режима.
Макросферы получают во вращающихся горизон-
горизонтальных аппаратах, снабженных перемешивающими
устройствами, путем нанесения связующего и порош-
порошкового наполнителя (напр., измельченного стеклово-
стекловолокна) на предварительно вспененные гранулы, напр,
полистирола. Отверждение оболочки макросфер (свя-
(связующего) и сплавление ее с вспененными гранулами
проводят при повышенных темп-р ах.
Сферич. частицы из стекла получают в вертикальных
трубчатых печах, тепловой режим в к-рых поддержи-
поддерживается сжиганием газа или пропан-бутановой смеси.
Тонкодисперсную порошкообразную композицию
на основе стекла и соединений, разлагающихся при
темп-ре плавления стекла с образованием газообразных
продуктов, распыляют в нижнюю часть камеры печи.
Композиция плавится во взвешенном состоянии; в про-
процессе плавления разлагается газообразователь и выде-
выделяется газ, к-рый раздувает оплавленные частицы
стекла. Образующиеся полые сферич. частицы пере-
перемещаются горячими газами в верхнюю (холодную)
зону печи и там охлаждаются. Для повышения хим-
стойкости, темп-ры размягчения, а также для отделе-
отделения частиц с низкой плавучестью сферы подвергают
кислотной обработке с последующей отмывкой водой.
Получение пластиков. В качестве связующих для
получения пластиков с полым наполнителем (П.) мож-
можно использовать практически любые полимерные свя-
связующие. Чаще всего применяют эпоксидные и поли-
полиэфирные смолы, реже феноло-формальдегидные и крем-
нийорганич. смолы, поливинилхлорид. К связующим
предъявляется ряд технологич. требований: опреде-
определенная вязкость, адгезия к сферам, способность отверж-
даться в больших блоках без значительного экзотер-
мич. эффекта. Связующее должно иметь такую жизне-
жизнеспособность при темп-ре переработки, к-рая позволяла
бы провести процессы совмещения компонентов и фор-
формование полученной композиции; при этом легкий
наполнитель не должен «всплывать» на поверхность
изделия. Для придания специфич. свойств в состав П.
вводят различные модифицирующие добавки (каучуки,
антипирены, разбавители, красители).
В зависимости от соотношения связующего и полого
наполнителя полуфабрикат П. может представлять
собой вязкую жидкость (литьевой тип) или пасту
(прессовочный тип). Получение полуфабрикатов обоих
типов можно осуществить двумя способами. По пер-
первому способу компоненты смешивают под вакуумом
в герметичной емкости для предотвращения проникно-
проникновения воздуха в композицию. Полученную композицию
заливают в формы (желательно под вакуумом) и от-
верждают в зависимости от типа связующего без под-
подвода тепла извне или при повышенных (80 —120 °С)
темп-pax. Способ характеризуется высокой производи-
производительностью и применяется для изготовления больших
отливок, а также для заливки крупногабаритных конст-
конструкций. В производстве полуфабрикатов по второму
способу сферы загружают в герметичную форму и соз-
создают в ней разрежение, в результате чего под действием
атмосферного давления промежутки между сферами
заполняются связующим. Полученную композицию
либо отверждают в той же форме, либо используют
для формования изделий др. способами.
Для получения П. из полуфабриката литьевого типа
можно использовать различные виды смесителей пе-
риодич. действия или заливочные установки полу-
полунепрерывного действия. Процесс смешения компонен-
компонентов необходимо проводить так, чтобы равномерно
распределить сферы в связующем и при этом не до-
допустить их механич. разрушения.
Изделия из вязкотекучих композиций получают,
заливая последние в негативные формы или напыляя
композиции на поверхность оснастки с последующим
отверждением.
Из пастообразных композиций изделия получают
в прессформах под давлением 0,5—1,5 Мн/м2 E—
15 кгс/см2) или в формах без применения давления.
Загрузка материала и уплотнение его в последнем
случае осуществляются шпателем. П., полученные та-
таким образом, подвергаются любым видам механич.
обработки (резка, сверление и др.), легко полируются.
Свойства и применение. П. имеют ряд преимуществ
перед др. газонаполненными пластиками: у них самая
большая уд. прочность, равномерная плотность по объе-
объему, отсутствие расклинивающих давлений при заливке,
619
ПЛАСТИФИКАТОРЫ
620
Таблица 1. Характеристики пластиков литьевого типа с феноло-формальдегидными сферном, так и при достаточно высоких
(пластики ЭДМ и СПБ) и стеклянными (пластики ЭДС, СПС) сферами (табд 3) давлениях. Особенно водостойки
Показатель
Кажущаяся плотность,
кг/мА
Прочность, Мн/м2 (кгс/см2)
при сжатии
при статич. изгибе . . .
при растяжении . . . .
Модуль упругости при сжа-
сжатии Е- Ю-3, Мн/м2
(кгс см2)
Ударная вязкость, кдж/м2,
или кгс • см/см-
Тангенс угла диэлектрич.
потерь при 5 • 1 О5 гц . . .
Диэлектрич. проницаемость
Электрич. прочность, кв/мм
Потеря массы после выдерж-
выдержки в течение 150 -ч, %
при 200 °С
при 150 СС
при 100 X
Линейная усадка за 150 ч, %
при 10 0 сС
при 150 СС .......
при 200 °С
Эпоксидные связующие
ЭДМ
ЭДС
600-750
29-55
B90-550)
\ 5 — 25
A50-250)
12-14
A20-140)
0,8 — 1 ,-6
(8-15)
1-3
0,014-0,020
1 ,8-2,0
10-8
3.7
1,8
0,2
0
0,5
0,27
600-750
55-100
E50 — 1000)
25-42
B50 — 420)
18-25
A80-250)
1,5-3
A5-30)
3-7
0,014-0,020
2,0-2,5
11-13,8
1 ,5
0,4
0
0
0,6
0,25
Полиэфирные связующие П. на основе эпоксидных связующих и
СПБ
СПС
600-750
18-25
A80-250)
10-12
A00-120)
5-8
E0-80)
0,3-0,5
C-5)
1-2
0,03-0,04
2,4
12-13
5,8
36
после
3,6}24 ч вы-
2,4 держки
600-750
стеклянных сфер (тип ЭДС). Ни один
из известных пенопластов не может кон-
конкурировать с ними в этом отношении.
Однако при длительной выдержке в во-
воде прочность этих пластиков несколько
малая усадка; они способны находиться длительное
время в контакте с жидкостями без заметного их погло-
поглощения. Недостаток П.— более высокая (в сравнении
Таблица 2. Характеристика пластиков прессовочного типа
Показатель
Кажущаяся плотность,
кг/м3 ...
Прочность, Мн/м2 (кгс/см2)
при статич. изгибе . .
при сжатии
при растяжении ....
Модуль упругости при
сжатии Е-10~3,
Мн/м2 (кгс/см2)
Ударная вязкость, кдж/мг,
или кгс ¦ см/см2
ЭДМ
260 — 450
3,5-10
C5-100)
6-23
F0-230)
—
0,15-0,43
A ,5-4,3)
0,5 — 1 ,0
ЭДС
280 —Г) 0 0
4 — 15
D0-150)
7-27
G0-270)
—
0,4-1,0
D-10)
1 ,5-2,5
СПБ
400 — 480
3-5
C0-50)
8-16
(80-160)
2-3
B0 — 30)
0,22—0,43
B,2-4,3)
0,3-0,4
с др. пенопластами) кажущаяся плотность. Кроме
того, их свойства в значительной мере определяются
качеством полых сферич. наполнителей и характером
их совмещения со связующим
(равномерность распределе-
распределения в полимере, отсутствие
механич. разрушения сфер
и др.).
Свойства П. приведены в
табл. 1 и 2. Пластики всех
типов, указанных в табл. 1,
устойчивы к действию масел,
Таблица 3. Изменение
прочности пластика
марки ЭДС*
0,1 A)
30 C 0 0)
40 D00)
50 E00)
Прочность при
сжатии [Мн/м2
(кгс/см2)] после
выдержки под
давлением в те-
течение
зо сут | 60 сут топлив, нефтепродуктов (при
62 F20)
64 F40)
56 E60)
51 E10)
обычных и повышенных
53E30) темп-pax), не поражаются
41 D10) микроорганизмами, не под-
32 C20) вергаются действию морского
тумана; они обладают хоро-
*Кажущаяся плотность шеи адгезией к металлам и
650кг/.из; исходная проч- стеклопластикам,
ность при сжатии 63 Мн/м* R п имртпт nwrnVArrn nn
F30 кгс/см2); размер образ- исе и* имеют высокую во-
цов 20x20x20 мм. достоикость как при атмо-
4 0—5 5 снижается (см. табл. 3) вследствие ослаб-
D00 — 550) ления адгезионной прочности на границе
j205 наполнитель — связующее.
10-13 Наиболее эффективным методом, по-
A00 — 13 0) зволяющим значительно снизить водопо-
глощение таких пластиков и т. обр. ста-
0,9-1,8 билизировать их прочностные характери-
характеристики в воде и влажной среде, является
предварительная модификация поверх-
поверхности наполнителя глдрофобно-адгезион-
ными веществами (аппретами), к-рые
усиливают взаимодействие на границе
полимер — наполнитель. Табл. 4 дает
представление о водостойкости пластиков
типа ЭДС и ЭДС-А (наполнитель — мик-
микросферы с аппретированной поверх-
поверхностью).
Водоиоглощение в значительной сте-
0'80 пени зависит от размеров образцов. На
практике используют блоки, имеющие
большие размеры, для к-рых водопогло-
щение значительно меньше величин, при-
приведенных в табл. 4. Сохранение прочности при сжа-
сжатии после месячного пребывания под давлением 60
Мн/м2 F00 кгс/см2) для пластиков типа ЭДС и ЭДС-А
составляет 80 и 95% соот-
соответственно. Таблица 4. Водопоглощение
Ич ТТ ичготяиттитеяют (в % гю массе) пластиков
и; п* изготавливают с полым наполнителем*
глубоководные поплавки,
буи и различные плавучие Давление,
(9-18)
1-2
0,03-0,04
2,3-3,0
10-13
22,5
3,9
0,8
,80
3,70
Мн/м*
(кгс/см2)
0,1 A)
20 B00)
40 D00)
50 E00)
60 F00)
ЭДС-А
0,4 — 0,6
0,8-1 ,2
1,2-1,4
1 ,5
Не более
2,5-3
средства др. типа, а так-
также системы для проведе-
проведения подводных спасатель-
спасательных работ и подъема за-
затонувших судов, замазки
для ремонта гидротехнич.
сооружений. Широко ис-
используются легкие проч-
прочные сэндвич-конструкции * продолжительность экспо-
с «начинкой» из таких пла- зиции в воде 3 0 сут (размер
стиков; кроме того, П. образцов 20x20x20 мм).
могут имитировать дерево
и мрамор; из них изготавливают светоотражающие си-
системы для маркировки дорог, абляционностойкие по-
покрытия, тепло- и звукоизоляцию и др.
Наиболее известны след. торговые названия П.,
выпускаемых в США: эккофоумз, эккосток,
стайкаст, эккофлоут.
Лит.: Ferrigno Т. Н., Rigid plastics foams, 2 ed.,
N. Y.— [a. o.], 1967.
См. также лит. при ст. Пенопласты. Т. В. Красникова.
ПЛАСТИФИКАТОРЫ (plasticizers, Weichmacher,
plastifiants) — вещества, вводимые в полимеры с целью
придания (или повышения) эластичности и (или) пла-
пластичности в условиях переработки и эксплуатации.
П. облегчают диспергирование в полимерах сыпучих
ингредиентов, снижают темп-ру переработки полимер-
полимерных материалов; нек-рые П. придают полимерным
материалам такие ценные свойства, как негорючесть,,
термо- и светостойкость.
Продукты, используемые в качестве П., должны обла-
обладать след. общими свойствами: 1) способностью совме-
совмещаться с полимером, т. е. образовывать с ним" устой-
устойчивые композиции при введении достаточно больших
количеств П.; 2) малой летучестью (низким парциаль-
621
ПЛАСТИФИКАТОРЫ
622
ным давлением), бесцветностью, отсутствием запаха;
3) способностью проявлять пластифицирующее дейст-
действие не только при нормальной, но и при пониженной
темп-ре; 4) химич. стойкостью, к-рая должна быть
не ниже, чем у пластифицируемого полимера. Кроме
того, П. не должны экстрагироваться из полимера масла-
маслами, растворителями, мылами, моющими средствами,
а также ухудшать его диэлектрич. свойства. Естест-
Естественно, что универсальных П., удовлетворяющих всем
перечисленным выше требованиям, не существует.
Свойства важнейших П. приведены в табл. 1.
Для применения в качестве П. предложено свыше
500 продуктов, однако промышленное значение имеют
не более 100. Наиболее широко П. используют при пе-
переработке пластмасс (ок. 70% от общего объема про-
производства П.— при переработке поливинилхлорида).
Важную роль П. играют и в резиновой промышленно-
промышленности (несмотря на то, что высокоэластич. свойства каучу-
ков проявляются в более широком температурном ин-
интервале, чем у пластиков, применение П. необходимо
как для переработки каучуков в изделия, так и для
придания последним нек-рых специфич. свойств). П.
вводят также в лакокрасочные материалы (см. Лаки
и эмали).
П. классифицируют обычно по их химич. природе
(см. табл. 1) и по степени совместимости с полимером
(табл. 2). По второму признаку П. делят на первичные
и вторичные (обладающие соответственно хорошей или
ограниченной совместимостью с полимером). Вторич-
Вторичные П. могут со временем выделяться («выпотевать»)
на поверхность полимерного материала в виде жидко-
жидкости или кристаллич. образований. Совместимость зави-
зависит от строения и полярности полимера и П. Этот
показатель м. б. определен визуально, по характеру
диаграмм фазового равновесия системы полимер —
пластификатор или др. методами. Деление П. на пер-
первичные и вторичные в известной мере условно, т. к.
совместимость П. с полимером может существенно
зависеть от темп-ры, давления, влажности воздуха,
интенсивности солнечной радиации и др. факторов.
Вторичные П. вводят в полимерные материалы, как
правило, вместе с первичными. Они могут придавать
материалам нек-рые специфич. свойства (напр., негорю-
негорючесть, термостойкость) или служить дешевыми замени-
заменителями первичных П. Подробно о механизме действия
П. см. Пластификация.
Пластификаторы для пластмасс. К числу важнейших
П. относятся эфиры ароматич. и алифатич. карбоновых
к-т, эфиры гликолей и монокарбоновых к-т, эфиры
фосфорной к-ты, полиэфиры, эпоксидированные со-
соединения.
Эфиры ароматических кислот. Ос-
Основная, группа промышленных П. — эфиры фталевой
к-ты и алифатич. спиртов (фталаты). Производство
этих П. в наиболее развитых капиталистич. странах
составляет 65—85% от общего выпуска П. Фталаты
отлично совмещаются со многими полимерами, отно-
относительно легко вводятся в композиции, обладают
хорошей тепло- и светостойкостью и дешевле других
П. эфирного типа. Основной универсальный П.—
ди-B-этилгексил)фталат. Композиции на основе поли-
поливинилхлорида, содержащие этот П., обладают высо-
высокими электроизоляционными свойствами, а также мо-
розо-, тепло- и светостойкостью. Ди-B-этилгексил)-
фталат применяют также для пластификации нитро-
и этилцеллюлозы, ацетобутирата целлюлозы и др.
" Фталаты изооктилового, изононилового, изодецило-
вого спиртов и смеси спиртов С7—С9 близки по свойст-
свойствам к ди-B-этилгексил)фталату. Фталаты синтетич.
нормальных спиртов С6—С10 и С8— С10 придают поли-
винилхлоридным композициям лучшую морозостой-
морозостойкость, чем ди-B-этилгексил)фталат. Низкая летучесть
фталатов изододецилового и тридецилового спиртов
позволяет использовать их для приготовления термо-
термостойких композиций. Особенно ценными свойствами
обладает бутилбензилфталат, к-рый широко применяют
в производстве масло- и бензостойких полимерных мате-
материалов. Ди-B-этилгексил)фталат, бутилбензилфталат,
дициклогексилфталат нетоксичны и допущены к при-
применению в изделиях пищевого и медицинского назна-
назначения.
Эфиры ароматич. поликарбоновых к-т (тримеллито-
вой и пиромеллитовой) обладают низкой летучестью,
придают полимерам высокую теплостойкость и стой-
стойкость к окислению. Это обусловливает их применение
при получении электроизоляционных материалов для
кабельной пром-сти, эксплуатируемых при повышенных
темп-рах.
Эфиры алифатических кислот. К
этой группе П. относятся адипинаты, себацинаты,
азелаинаты, а также стеараты и олеаты. Наибольшее
значение имеют ди-B-этилгексил)азелаинат и дибутил-
себацинат, используемые гл. обр. в композициях, к-рые
должны сохранять пластнч. свойства при низких
темп-pax. Применение себацинатов ограничивается их
высокой стоимостью, а адипинатов — повышенной
летучестью. Обычно эти П. используют в смеси с фта-
латами.
Эфиры стеариновой к-ты отличаются устойчивостью
к термич. воздействию и к облучению. Основная об-
область применения стеаратов и олеатов — пластифи-
пластификация производных целлюлозы. Пленки нитрата цел-
целлюлозы, пластифицированного бутилстеаратом, отли-
отличаются высокой прочностью, морозо- и водостойкостью;
тетрагидрофурфурилолеат служит П. для триацетата
целлюлозы.
Эфиры гликолей и монокарбоно-
монокарбоновых кислот. Среди этих П. наибольшее значение
имеют эфиры триэтиленгликоля и алифатич. монокар-
монокарбоновых к-т С6 — С9, а также эфиры бензойной к-ты,
применяемые для пластификации поливинилхлорида,
поливинилбутираля и др. полимеров. Триэтиленгли-
кольдикаприлат используют в производстве шахтных
конвейерных лент; триэтиленгликоль-ди-B-этилбути-
рат) и триэтиленгликоль-ди-B-этилгексоат) — для полу-
получения пленок поливинилбутираля, используемых в про-
производстве триплекса.
Эфиры фосфорной кислоты. Среди П.
этой группы наибольшее значение имеют трикрезил-,
крезилдифенил-, трибутил- и три-B-хлорэтил)фосфат.
Фосфаты хорошо совмещаются с иоливинилхлоридом,
поливинилацетатом и большинством производных эфи-
ров целлюлозы. Важнейшее свойство этих П., особенно
три-B-хлорэтил)фосфата — способность придавать
композициям негорючесть. Арилфосфаты характери-
характеризуются, кроме того, низкой летучестью, хорошими
антикоррозионными свойствами и стойкостью к экст-
экстракции маслами; недостаток композиций с этими П.—
низкая морозостойкость. При получении композиций,
к-рые должны обладать одновременно негорючестью
и морозостойкостью, используют алкилфосфаты, напр.
три-B-этилгексил)фосфат. В алкиларилфосфатах сочета-
сочетаются лучшие свойства алкил- и арилфосфатов.
Полиэфиры. В качестве П. в пром-сти приме-
применяют низкомолекулярные (мол. масса ~~ 2000) сложные
полиэфиры, получаемые поликонденсацией дикарбоно-
вых к-т (адипиновой, се^ациновой, азелаиновой, реже
фталевой) с полиолами (диэтиленгликолем, 1,2-пропан-
диолом, 1,3-бутандиолом, 2,2-диметилпропандиолом)
или переэтерификацией низших эфиров дикарбоновых
к-т полиолами.
Среди нелюдифицированных полиэфиров (т. е. содер-
содержащих свободные гидроксильные или карбоксильные
группы) лучшей совместимостью с нитратом целлю-
целлюлозы и поливинилхлоридом обладает эфир пропилен-
гликоля и себациновой к-ты. Модификация полиэфиров
623
ПЛАСТИФИКАТОРЫ
Таблица 1. Физические свойства пластификаторов
624
Наименование пластификатора
Плотность
при 25 °С,
г/еж3
Показа-
Показатель пре-
преломления
п
D
Вязкость
при 20сС
мн • сек/м2,
или спз
Парциальное
давление па-
пара при 150°С,
мм рт.ст.^
Темп-ра
вспышки,
СС
Темп-pa ки-
кипения,
'С/'мм рт.ст.а
Темп-ра
плавле-
плавления, СС
Эфиры ароматических карбоновых кислот и алифатических
Диметилфталат С10Н10О4 1,190 1,514б
Диэтилфталат С12Н14О4 1,120б 1,500б
Дибутилфталат Ci6H22O4
Бутилоктилфталат С2оНзо04
Бутилизодецилфталат С22Н34О4. . .
Дикаприлфталат С24Н38О4
Диалкилфталат-789 С24Н38О4 . . .
Ди-B-этилгексил)фталат С24Н38О4
Динонилфталат С26Н42О4
Диизодецилфталат С28Н46О4 . . . .
Дитридецилфталат Сз4Н58О4 . . . .
Дидодецилфталат С32Н54О4
Бутилбензилфталат Ci9H2o04 . . . .
Дициклогексилфталат С2оН2604
Триоктилтримеллитат СзоНатОв
Эфиры
190
120б
042-
049
991 —
001б
995
970
975б
986б
980
954
,053б
,950
,111-
,119
,148б
0
алифатических
1,492
1,484
1,486
1,480б
1,484
1,487,.
1,483б
1 ,483
1 ,482
1,534-
1,538
Диизобутиладипинат С14Н26О4
Ди-B-этилгексил)адипинат Gi
Диизооктиладипинат С22Н42О4
Октилдециладипинат
H42O4
Диизодециладипинат С2вН50О4 ....
Бензилоктиладипинат С21Н32О4 . . .
Ди-B -этилгексил)азелаинат С26Н48О4
Ди-B-этилбутил)азелаинат С21Н40О4
Диизооктилазелаинат С25Н48О4 ....
Диизобутилазелаинат С17Н32О4. . . .
Дибутилсебацинат С18Н34О4 .....
Диоктилсебацинат С26Н5о04
Бутилолеат С22Н42О2 .........
Тетрагидрофурфурилолеат С23Н42О3 .
Триэтиленгликоль-ди-B-этилбутират)
С18Н34Ов
Триэтиленгликоль-ди-B-этилгексоат)
987 1,485
карбоновых
950
9268L
922
16,3
12,6В
19-23
38
67
70-80
77-82
113-123
113-123
190в
297
55-65
б
0
0
0,
0,915
0,917
0,998
0,915б
0,934б
0,918б
0,932
0,934
0,912
0,865
0,922
1,429
1,447б
1,445б
1,447
1,4495
1,479
,4445
,445
,435
,442б
,450
,451б
,462
286
кислот
13-15
15-18
24 — 30
16-20
17-22
20
7-11
18-24
7,7В
17в
12,5
8,5
1,0-1,2
0,14
0,07
0,05
0,05 — 0,16
0,015
2,7•10~3
0,16
0,09
146
152 — 163
175
188
193
205
200
206
232
256
226
199
207
260
и алифатических
160
196
200
Эфиры гликолей
Триацетат глицерина С9Н14Ов
Этилфталилэтилгликолят С14Н16Ов . .
Бутилфталилбутилгликолят С,8Н24Ов.
Трибутилфосфат С12Н27О4Р
Три-B-этилгексил)фосфат G24H5i04P . . . .
Трифенилфосфат С18Н15О4Р . ,
0,995б
0,968б
1,160
1,180
1,097
1,4404б
1,444б
1,429
1,498
1 ,490
11,5
16,1
16В
65
<0,15
0,13
0,04
0,02
0,60
2,8- Ю
0,19
0,41
<0,22
0,47
0,10
Эфиры фосфорной кислоты
Крезилдифенилфосфат Gi9Hl4O4P
Трикрезилфосфат G2lH2lO4P . . «
Алкиларилфосфат С22Н39О4Р . ,..
0,975
0,926б
,201д
208
165
Полидиэтиленгликольадипинат, модифи-
модифицированный 2-этилгексиловым спиртом
Полидиэтиленгликольадипинат, модифи-
модифицированный каприловой кислотой . . . .
Полидиэтиленгликольсебацинат, модифи-
модифицированный 2-этилгексиловым спиртом
0,990
1,06б
1,10
3,4В
13,8
1,4226б
1.44346
1 ,552—
1 ,563
1 ,560
1 ,555
1,475
Полиэфиры
кД
39,5В
110-120
0,23
0,15
0,08
0,04
400 —500в
350-450
80 —120г
Алкилэпоксистеарат
Эпоксидированное соевое масло
2-Этилгексилэпокситаллат . . .
1,0235
Эпоксидированные соединения
0,899 1,454 35-40
0,995 1,471 800-1000
0,922б 1,451б 49
Хлорированные соединения
200-215
236
200
231
185
213-219
179
180
215
180
210
197
207
133
190
199
193
210
225
232
276
195
220
220
212
265
320
235
спиртов
282
298
340
220/5
215-240/4
231/5
413
255/1
235/25
370
218/5
260/1
спиртов
135 — 147/4
214/5
220/4
235/5
245/5
232-255/10
237/5
230/5
225—244/4
164-177/4
344
248/4
190-230/65
210/5
196/5
219/5
260
190/5
219/5
177/27
220/5
407
390
265/5
230/5
240/2
0
_4
-40
-50
-50
-60
-46
-37
-35
от 58
до 65
-46
-20
-70
-40
от —60
до —4
-43
-65
-76
-65
24
-12
-40
— 10
—65
-65
-78
-80
-90
43
-40
-36
-60
-13,5
Хлорпарафины С23Н42С1в
>> ci?4cil8
1,16
1 ,24
1 ,63
—
-
—
-
—
-
_
-
—
—
• 1 мм рт. ст. = 133,322 н/м*, б При 20 °С. вПри 25 °С. гПри 50 °С. дПри 60 °С.
625
ПЛАСТИФИКАТОРЫ
626
Таблица 2. Совместимость нек-рых пластификаторов с полимерами при комнатной темп-ре и нормальном давлении
(в мае. ч. на 100 мае. ч. полимера)
Пластификатор
Поли-
Поливинил-
хлорид
100
100
100
30
100
100
50
20
100
100
20
100
100
40
Поли-
вини-
лиден-
хлорид
75
25
10
20
75
15
10
20
75
75
75
75
50
Поли-
Поливинил-
ацетат
100
1
1
50
100
1
1
80
40
50
100
100
1
100
Поли-
Полистирол
100
50
100
20
100
100
25
20
15
50
30
30
20
Этил-
целлю-
целлюлоза
100
100
100
50
100
100
100
30
70
100
75
75
100
60
Нит-
Нитрат
целлю-
целлюлозы
100
100
100
50
100
35
35
75
100
100
100
100
100
80
Ацетат
целлю-
целлюлозы
25
1
1
1
10
1
1
35
15
20
100
50
1
90
Ацето-
бути-
рат
целлю-
целлюлозы
ю-о
50
50
20
100
25
50
50
30
80
100
50
1
50
Поли-
Полиамиды
25
25
20
10
25
25
25
10
25
25
1
20
_
50
Поли-
Полиэфиры
20
20
15
20
20
1
1
10
20
20
5
20
10
Эпок-
Эпоксидные
смолы
25
1
1
10
25
1
1
10
25
25
25
25
_
50
Алкид-
ные
смолы
70
25
25
25
50
50
25
50
70
70
70
70
_
25
Поли-
урета-
уретаны
Дибутилфталат
Ди-B-этилгексил)фталат . .
Ди-(тридецил)фталат . . . .
Дициклогексилфталат . . . .
Бутилбензилфталат
Ди-B-этилгексил)адипинат .
Диизодециладипинат
Трифенилфосфат
Трикрезилфосфат
2 -Этилгексил дифенилфосфат
Метилфталилэтилгликолят .
Бутилфталилбутилгликолят
Эпоксидированное соевое
масло
о,п-Диэтилтолуолсульфамид
25
25
25
10
25
15
15
10
2S
25
25
25
20
(этерификация концевых групп высшим алифатич.
спиртом или монокарбоновой к-той) позволяет улуч-
улучшить их совместимость с поливинилхлоридом; в случае
модификации спиртами совместимость возрастает с уве-
увеличением длины алкильного радикала концевой алко-
ксильной группы.
Полиэфирные П. обладают низкой летучестью,
не экстрагируются растворителями и др. средами. Это
обусловило их применение в качестве вторичных П.
в производстве масло- и бензостойких изделий (шланги
для бензина, трубопроводы для горючего, масло- и жи-
ростойкие упаковочные материалы, нек-рые виды элек-
электроизоляции), деталей из поливинил хлорида, находя-
находящихся в контакте с др. полимерами (прокладки для
холодильников, изоляция электрооборудования, обив-
обивка сидений в автомобилях и др.).
Эпоксидированные соединения. К
П. этой группы относятся эпоксидированные расти-
растительные масла (напр., соевое) и эфиры жирных к-т
таллового масла. Наиболее ценные свойства эпоксиди-
рованных П.— термо- и светостойкость, низкая лету-
летучесть, способность придавать композициям эластичность
при низких темп-pax. Эти П. служат также стабилиза-
стабилизаторами поливинил хлорида, т. к. реагируют с выделяю-
выделяющимся НС1, подавляя его каталитич. действие на раз-
разложение полимера, и образуют эффективные синергич.
композиции с такими стабилизаторами, как соли бария
и кадмия. Нек-рые эпоксидированные растительные
масла допущены для применения в изделиях пищево-
пищевого и медицинского назначения. Эпоксидированные со-
соединения обычно применяют в качестве вторичных
П. C—8 мае. ч. на 100 мае. ч. полимера) в производст-
производстве электроизоляционных материалов, покрытий, упа-
упаковочных пленок, линолеума, плащей, детских игру-
игрушек, транспортерных лент и др.
Пластификаторы для каучуков. Введение П. в кау-
чуки облегчает их переработку, повышает пластич-
пластичность резиновой смеси, способствует уменьшению разо-
разогрева при смешении и снижает опасность подвулкани-
зации. Благодаря введению П. снижается расход
электроэнергии на смешение и последующую обработку
резиновых смесей. Правильный выбор типа и количест-
количества П. позволяет существенно понизить твердость,
гистерезисные потери и теплообразование при много-
многократных деформациях резин. Иногда П. для каучуков
условно делят на собственно пластификаторы и «м я г-
ч и т е л и». При этом к первым относят только те про-
продукты, к-рые понижают темп-ру стеклования каучуков,
т. е. улучшают морозостойкость резин, ко вторым —
продукты, понижающие темп-ру текучести резиновых
смесей, но не оказывающие на морозостойкость резин
заметного влияния.
Общее содержание П. в резиновых смесях зависит
от типа и количества применяемых ингредиентов,
а также от химич. природы каучука и его исходной
пластичности. Большинство П. применяют в количестве
2—10 мае. ч. на 100 мае. ч. каучука. Для получения
изделий со специфич. свойствами содержание П. может
быть увеличено до 30 мае. ч. и более. Наряду с синте-
тич. продуктами (гл. обр. сложными эфирами) в ка-
качестве П. для каучуков широко применяют продукты
переработки нефти, каменного угля, лесохимических
материалов, а также растительные масла, жирные кис-
кислоты и др.
Продукты нефтепереработки— ос-
основные П., используемые в производстве шин и резино-
технич. изделий. Наиболее широко применяют парафи-
но-нафтеновые и ароматич. нефтяные масла, парафины,
нефтеполимерные смолы (инден-алкилароматические
и др.), асфальто-битумные продукты (рубракс), хло-
хлорированные парафиновые углеводороды и др. Эти П.
ограниченно совмещаются с каучуками (особенно с бу-
бутадиен-нитрил ьными) и характеризуются меньшей
пластифицирующей активностью, чем эфирные П. Од-
Однако они облегчают переработку смесей и придают
резиновым смесям и вулканизатам ряд ценных сиеци-
фич. свойств. Напр., рубракс облегчает диспергирова-
диспергирование сажи в резиновой смеси, улучшает монолитность,
каркасность (способность сохранять форму) и влаго-
влагостойкость изделий; хлорированные парафины повы-
повышают огнестойкость изделий; жидкие хлорпарафины
B4% хлора) улучшают также морозостойкость резин,
особенно на основе хлоропренового каучука.
Нефтяные масла часто вводят в синтетич. каучуки
в процессе их получения. Применение таких маслона-
полненных каучуков исключает в нек-рых случаях
необходимость введения П. при изготовлении резино-
резиновых смесей (см. Наполненные каучуки). Нефтяные П.
имеют небольшую стоимость, и поэтому их применение
экономически более выгодно, чем использование эфир-
эфирных П., особенно для неморозостойких изделий.
Сложные эфиры (фталаты, себацинаты, ади-
шшаты и др.) хорошо совмещаются с большинством
каучуков. Эти П. применяют гл. обр. в производстве
морозостойких резиновых изделий. Использование
сложных эфиров для др. целей нецелесообразно не толь-
только из-за их относительно высокой стоимости, но также
и вследствие снижения при введении этих П. механич.
свойств вулканизатов (прочности и модуля при растя-
растяжении, сопротивления раздиру).
Из числа продуктов переработки
лесохимич. материалов наиболее широка
применяют модифицированную (гидрированную) кани-
канифоль, сосновую смолу, из продуктов» и ер е-
€27
ПЛАСТИФИКАЦИЯ
628
работки каменного угля — кумароно-ин-
деновые смолы. Основное назначение этих П.— по-
повышение клейкости резиновых смесей.
Растительные масла (льняное, суреп-
сурепное) применяют для снижения усадки эбонитов при
вулканизации. К наиболее важным П. этой группы
относятся фактисы — продукты реакции расти-
растительных масел с серой, которые существенно облегча-
облегчают процессы экструзии и каландрования, способству-
способствуют сохранению формы полуфабрикатов и готовых из-
изделий.
Жирные кислоты (стеариновая, олеиновая)
служат диспергаторами наполнителей и одновременно
активаторами вулканизации.
Лит.: Т и н и у с К., Пластификаторы, пер. с нем., М.—
Л., 1964; Bruins P. F. [ed.], Plasticizer technology, v. 1,
N. Y.— L., 1965; D о о 1 i t t 1 e A. K., The technology of
solvents and plasticizers, N. Y.— L., 1954; P e n n W. S.,
PVC technology, L., 1962; Encyclopedia of polymer science and
technology, v. 10, N. Y.— [a. o.], 1969; Справочник резинщика.
Материалы резинового производства, М., 1971.
А. И. Нуценко, Л. М. Болотина, Т. В. Литвинова.
ПЛАСТИФИКАЦИЯ полимеров (plastici-
zation, Plastifizierung, plastification).
Содержание:
Способы пластификации 627
Влияние пластификаторов на темп-ры стеклова-
стеклования и текучести полимеров 628
Влияние пластификаторов на механич, свойства
полимеров 630
Влияние пластификаторов на диэлектрич. свой-
свойства полимеров 631
Структурная пластификация 632
Антипластификация 633
Пластификация — введение в полимеры веществ
(пластификаторов), повышающих эластичность и (или)
пластичность материала в условиях его эксплуатации
и (или) переработки.
В обычных случаях непременное условие П.— тер-
модинамич. совместимость пластификатора с полиме-
полимером, т. е. образование истинного р-ра пластифика-
пластификатора в полимере.
Совместимость зависит от природы полимера и плас-
пластификатора и м. б. охарактеризована диаграммой фа-
фазового состояния компонентов системы в координатах
состав — темп-pa, давлением набухания полимера в
пластификаторе или относительным понижением упру-
упругости пара над системой полимер — пластификатор,
характером изменения темп-ры стеклования полимера
при П. и рядом др. методов (см. Раствори, Совмести-
Совместимость). На практике широко применяют как хорошо
совместимые, так и ограниченно совместимые g поли-
полимером пластификаторы, часто в смесях друг с другом.
Если количество введенного пластификатора превы-
превышает концентрацию, соответствующую равновесному
пределу его совместимости с полимером, избыток
пластификатора может выделиться из системы при
переработке, хранении и эксплуатации материала.
В специальных случаях эффект пластифицирования
м. б. достигнут введением очень небольших количеств
веществ, не способных равномерно распределяться в
объеме полимера. В этом случае изменение механи-
механических и термомеханич. свойотв происходит, по-види-
по-видимому, благодаря воздействию пластификатора на гра-
границы раздела между относительно крупными элемен-
элементами надмолекулярной структуры (структурная пла-
пластификация).
Способы пластификации. Свойства системы поли-
полимер — пластификатор могут зависеть от способа П.
Эти способы можно классифицировать след. образом:
1) растворение полимера в р-ре пластификатора (про-
(производство пленок полимерных, изготовление кожи ис-
искусственной и лакокрасочных материалов)', 2) сорбция
пластификатора полимером или полимерным материа-
материалом из эмульсий или р-ров пластификатора (П. про-
производных целлюлозы, поливинилхлорида, полиамидов
и др.)'» 3) добавление пластификатора к мономерам
перед их полимеризацией или поликонденсацией (П.
феноло-формальдегидных и мочевино-формальдегидных
полимеров, полиэфиров и др.); 4) введение пластифи-
пластификатора в эмульсию полимера перед его переработкой
(П. поливинилхлорида и др.); 5) непосредственная
переработка полимера с пластификатором (производ-
(производство изделий из нитрата целлюлозы, пластизолей —
см. Пасты полимерные, и др.).
Влияние пластификаторов на температуры стекло-
стеклования и текучести полимеров. Введение пластифика-
пластификаторов существенно изменяет весь комплекс свойств
полимера. Большое значение с практической и теоре-
тич. ч^очки зрения имеет понижение темп-ры стеклова-
стеклования Тс и темп-ры текучести полимера Тт. Снижение
Тс при введении пластификаторов позволяет расши-
расширить температурную область высокоэластического со-
состояния полимеров, т. е. повысить их морозостойкость.
Понижение ТТ и вязкости полимерных расплавов
позволяет существенно облегчить переработку поли-
полимеров. Большое технологич. значение понижение Тс
и Тт имеет для переработки таких полимеров, у к-рых
эти характеристики лежат вблизи или даже выше
темп-ры их химич. разложения.
При П. полярных полимеров полярными пластифи-
пластификаторами, как показал С. Н. Журков, снижение Тс
пропорционально числу молекул пластификатора п,
сорбированных полярными группами полимерной цепи:
Д7С = к (с/М) = кп
где к — коэффициент, не зависящий от природы плас-
пластификатора, с — концентрация пластификатора, М —
его мол. масса. В этом случае механизм П. сводится
к экранированию молекулами пластификатора актив-
активных (полярных) групп полимерной цепи, к-рые осу-
осуществляют взаимодействие и определяют Тс полимера
до введения пластифицирующих компонентов. Это
ур-ние достаточно строго соблюдается лишь для по-
полярных низкомолекулярных веществ довольно про-
простого строения при сравнительно небольшой их кон-
концентрации в полимере.
В соответствии с правилом Каргина — Малинского
эффективность П. неполярных и слабополярных поли-
полимеров зависит от объемной доли пластификатора ф.
Введение равных объемов различных пластификаторов
понижает Тс полимера на одну и ту же величину. Это
правило выражается соотношением:
дгс = ц
где к — коэффициент, не зависящий от природы пла-
пластификатора. Правило выведено из предположения
о том, что основную роль при П. играет не ослабление
межмолекулярного взаимодействия, а чисто геометрич.
эффект уменьшения пространственных затруднений
при перемещении сегментов макромолекул.
Приведенные выше ур-ния справедливы лишь для
двух предельных случаев П.
На основании теории свободного объема предложено
ур-ние, связывающее TG системы с темп-рами стеклова-
стеклования чистого полимера и пластификатора (свободный
объем — разность между уд. объемами вещества при
данной темп-ре и абсолютном нуле). Как правило,
введение в полимеры пластификаторов существенно
повышает свободный объем системы.
Исходя из того, что все полимерные системы при
Тс характеризуются практически одинаковым значе-
значением относительного свободного объема (отношение
свободного объема к удельному при данной темп-ре),
а свободный объем р-ра является суммой свободных
объемов компонентов, выведено след. соотношение:
j __ ctn Ф2 Ггп + аР A — Ф2) Гер
с
аи ф2
A
629
ПЛАСТИФИКАЦИЯ
630
где Тсп и Гср — темп-ры стеклования соответственно
полимера и растворителя (пластификатора), ап и
ар— коэфф. объемного расширения соответственно
полимера и пластификатора; ф2 — объемная доля поли-
полимера в системе.
Согласно этому ур-нию, при прочих равных услови-
условиях снижение Тс тем больше, чем ниже темп-pa стек-
стеклования пластификатора.
Взаимодействие пластификатора с полимером учи-
учитывается в теории П., предложенной Г. Канигом и
также основанной на теории свободного объема. В этом
случае ур-ние для понижения Тс имеет вид:
Д^с == *сп — *с == "-1 у г^г (^пп ^пр)~г ^з (^*пп ^рр)
где Тс и Тсп — темп-ры стеклования пластифициро-
пластифицированной системы и полимера соответственно; Fp — сво-
свободный объем пластификатора; Лпп, Лпр, Арр — ве-
величины, характеризующие термодинамич. сродство
между молекулами полимера, полимера и пластифи-
пластификатора и молекулами пластификатора соответственно;
&!, /с2. к3 — коэффициенты, зависящие от состава р-ра
и свободных объемов компонентов. Из ур-ния, в част-
частности, следует, что эффективность пластификатора
(оцениваемая величиной Тсп — Тс) тем больше, чем
меньше свободный объем самого пластификатора Ур,
т. е. чем меньше размеры его молекул. Кроме того,
эффективность пластификатора тем больше, чем боль-
больЛ А А 4 б
фф
ше разности Л
— А
пр
и А
т. е. чем более
р рр
резко различается сродство между молекулами поли-
полимера и пластификатора. Это означает, что хорошим
пластификатором м. б. вещество, плохо совмещаю-
совмещающееся с полимером.
Большое влияние на Тс оказывают конфигурация и
конформация молекул пластификатора. При прочих
равных условиях значительно эффективнее пластифи-
пластификаторы с гибкими молекулами, способные принимать
различные конформации. В гомологич. рядах пласти-
пластификаторов, когда гибкость молекул постоянна, пласти-
пластифицирующее действие понижается с увеличением мол.
массы пластификатора.
Все приведенные выше закономерности справедливы
лишь в том случае, если полимер и пластификатор
совместимы друг с другом. При содержании пласти-
пластификатора, большем его предела совместимости с поли-
полимером, Тс не зависит от концентрации пластификато-
пластификатора. Так, напр., чем длиннее алкильный радикал в мо-
молекуле эфира фталевой к-ты и чем в меньшей степени
последний совместим с
ацетатом целлюлозы, тем
при меньших его кон-
концентрациях происходит
! Рис. 1. Схематич. изображе-
j ние изменений темп-р пере-
т ходов пластифицированного
гибкоцеиного полимера: 1—
пластифицированные полимеры
для кривой 3 выше); 4 —
Тс и Тт показаны только
Температура
чистый полимер; 2 и з
(концентрация пластификатора
р-р полимера в пластификаторе.
для кривой 1.
отклонение от прямолинейной зависимости в координа-
координатах Тс—концентрация пластификатора. При П. понижа-
понижаются как Тс, так и TTJ определенная термомеханич. ме-
методом. При этом для гибкоцеиных полимеров протя-
протяженность области высокоэластич. состояния уменьша-
уменьшается непрерывно по мере увеличения содержания
пластификатора (рис. 1).
Из рис. 1 следует, что при повышении содержания
пластификатора разность Тт — Тс может стать равной 0.
Такая система не обладает высокоэластич. свойствами
выше Тс и сразу переходит в вязкотекучее состояние.
Т. о., введение больших количеств пластификатора
может существенно снизить эксплуатационные харак-
характеристики материала, используемого в высокоэластич.
состоянии.
Для жесткоцепных полимеров высокоэластич. со-
состояние может не проявляться совсем или проявлять-
проявляться очень слабо. В этом случае пластификатор перево-
переводит полимер из стеклообразного состояния в вязко-
текучее. В нек-рых случаях разность Тт — Тс может
оставаться неизменной
или даже увеличиваться J
при введении относи- §-
тельно небольших коли- 1|
честв (до 20—30%) плас- §J
Рис. 2. Схематич. изобра-
изображение изменений темп-р
переходов пластифициро-
с
Температура
ванного жесткоцеиного полимера: 1 —чистый полимер; 2,
3 и 4 — пластифицированные полимеры (в порядке возрас-
возрастания концентрации пластификатора); 5 — р-р полимера в
пластификаторе. Тс и Гт показаны только для кривой 3.
тификатора (рис. 2). Для полярных полимеров слож-
сложного строения процесс стеклования может обуслов-
обусловливаться потерей подвижности одних групп атомов,
а потеря текучести — взаимодействием др. полярных
групп. Поэтому для П. таких систем можно исполь-
использовать смеси пластификаторов различных типов, одни
из к-рых влияют на положение области стеклования,
другие — на положение области текучести.
Темп-pa стеклования кристаллич. полимера при П.
снижается совершенно аналогично тому, как это на-
наблюдается для аморфных полимеров. Пластификатор
располагается в аморфных областях кристаллич. по-
полимера. Понижение темп-ры плавления ТПЛ в таких
системах обусловлено изменением термодинамич. усло-
условий растворения кристаллич. областей. Тпл кристал-
кристаллич. полимеров (а также Тт, близкая к Тпл) снижается,
но значительно меньше, чем Тс. Температурный ин-
интервал между Тпл и Тс расширяется по мере увеличе-
увеличения концентрации пластификатора.
Влияние пластификаторов на механические свойства
полимеров. В результате П. возрастает способность
материала к большим высокоэластическим и вынуж-
вынужденно высокоэластич. деформациям. Модуль упругости,
прочность и долговечность полимера при П. непрерыв-
непрерывно снижаются с увеличением концентрации пластифи-
пластификатора. Однако в ряде случаев прочность повышается
при введении небольших количеств пластификатора.
Это характерно для полимеров при темп-pax как выше,
так и ниже Тс. Для эластомеров нек-рое повышение
прочности наблюдается одновременно с повышением
удлинения при разрыве и предположительно связано
с облегчением ориентации макромолекул при растяже-
растяжении. О механизме повышения прочности полимеров
при темп-pax ниже Тс см. раздел «Антинластифика-
ция» (стр. 633).
Понижение прочности в присутствии пластификато-
пластификатора сказывается на положении темп-ры хрупкости Гхр,
определяемой точкой пере-
пересечения кривой зависимо-
зависимости предела вынужденной
высокоэластичности и хруп-
хрупкой прочности от темп-ры.
При пластификации Txv из-
меняется очень мало, и раз-
Рис. 3. Влияние пластификато-
пластификаторов на повышение темп-ры хруп-
хрупкости: а — непластифицирован-
ный полимер; б — пластифици-
пластифицированный полимер.
1
ф
•5
ее
1
а
«¦¦.
Ч V.
\\
т т1 т'
'хр 'хр 'с
Температура
ность Тс — ГХр уменьшается по мере увеличения со-
содержания пластификатора. В нек-рых случаях ТХХ) мо-
может даже несколько повышаться (рис. 3). Значитель-
631
ПЛАСТИФИКАЦИЯ
632
ное снижение Гхр м. б. достигнуто лишь введением
больших количеств пластификатора, т. е. ценой значи-
значительного уменьшения теплостойкости и прочности по-
полимера.
П. кристаллич. полимеров приводит, как уже отме-
отмечалось, к понижению их Тс без существенного изме-
изменения Тпл, т. е. интервал их нехрупкого поведения
расширяется по мере увеличения концентрации пла-
пластификатора (для ряда кристаллич. полимеров, напр,
полистирола и полипропилена, Тс близка к ^хр)-
Однако специфич. особенностью П. кристаллич. поли-
полимеров, находящихся при темп-pax выше Тс, является
уменьшение прочности и удлинения при разрыве по
сравнению с исходным полимером, находящимся в том
же состоянии, т. е. при темп-pax, равноудаленных
от темп-ры стеклования системы полимер—пластифика-
полимер—пластификатор. Это м. б. связано как с образованием более круп-
крупных надмолекулярных структур при кристаллизации
полимера в присутствии пластификатора, так и с умень-
уменьшением степени ориентации пластифицированных об-
образцов по сравнению с исходным при одинаковых
степенях растяжения.
В результате П. уменьшаются времена релаксации
полимера. Для пластифицированных полимеров, как
и для непластифицированных, функция ат, представ-
представляющая собой отношение времен релаксации при
данной темп-ре Т и темп-ре приведения Ts, следует
ур-нию Вильямса — Лэндела — Ферри (см. Супер-
Суперпозиции принцип температурно-временной):
х* "т - ^ С2 + (Г - Г,)
где Сг и С2 — эмпирич. константы; Тя лежит примерно
на 50 ± 4 °С выше Тс полимера. В приближении, соот-
соответствующем этому ур-нию, влияние пластификаторов
на температурную зависимость времен релаксации
связано с его влиянием на Ts, к-рое, в свою очередь,
приближенно соответствует влиянию на Тс полимера.
Для пластифицированного полимера, у к-рого Тс
понизилась на нек-рую величину, Ts понизится при-
приблизительно на эту же величину, и температурная
зависимость времен его релаксации будет выражена
менее резко, чем для чистого полимера, если обе систе-
системы сравниваются при одной и той же темп-ре, превы-
превышающей Тс каждой из них.
При П. изменяются потери на внутреннее трение
при динамич. испытаниях полимеров. Максимум тан-
тангенса угла механич. потерь при фиксированной частоте
испытаний смещается в сторону низких темп-р по мере
увеличения концентрации пластификатора. Потери на
внутреннее трение при динамич. испытаниях каучуков
при введении пластификаторов сначала уменьшаются
резко, а при достижении определенной концентрации
пластификатора — незначительно.
Сопротивление утомлению при динамич. испытаниях
зависит как от величины потерь энергии на внутрен-
внутреннее трение, так и от прочности вулканизатов.
Прочность вулканизатов, как уже отмечалось,
уменьшается при введении больших количеств пласти-
пластификатора. Наложение этих двух эффектов приводит
к тому, что сопротивление утомлению каучуков при
повышении концентрации пластификатора сначала
увеличивается, а затем, пройдя через максимум, умень-
уменьшается.
Влияние пластификаторов на диэлектрические свой-
свойства полимеров. Как правило, введение в полимер
пластификаторов ухудшает диэлектрич. характеристики
полимеров. При П. максимум тангенса угла диэлек-
диэлектрич. потерь смещается в сторону более низких темп-р.
Значения тангенса угла диэлектрич. потерь и диэлек-
диэлектрич. проницаемости тем выше, чем более полярна
молекула пластификатора. Введение пластификаторов,
особенно полярных, понижает уд. электрич. сопротив-
сопротивление полимера, что связано, с одной стороны, с по-
повышением подвижности уже имеющихся в полимере
носителей электрич. зарядов, а с другой — с увеличе-
увеличением числа носителей. Наконец, П. снижает электрич.
прочность полимеров.
Структурная пластификация. В нек-рых случаях
существенное изменение свойств полимера достигается
введением небольших количеств пластификатора. Так,
при введении всего
0,05% (по массе) касто- 16о
рового масла темп-ра
размягчения нитрата
целлюлозы снижается на
80 °С (рис. 4). Введение
небольших количеств
(до 1%) нек-рых пласти-
пластификаторов в полимеры,
находящиеся как в сте- ;
клообразном, так и в
высокоэластическом со-
Рис. 4. Зависимость темп-ры
размягчения Гр нитрата
целлюлозы от типа и кон-
концентрации с пластификато-
пластификатора: 1 —дибутилфталат; 2 —
касторовое масло; 2' — на-
начальный участок кривой 2.
160
120
80
п
о 120
к ^ ВО
\ 40
\
\
V
0.G5
\
X
0.1
t
\
2'
J
0,2
V -
0,3
15 \
с.Х
25 30
стоянии, приводит к существенному улучшению их ус-
усталостных свойств (см. таблицу).
Изменение свойств резин на основе хлоропренового каучука
в результате структурной пластификации
Компоненты системы
Сопротив-
Сопротивление раз-
разрыву, % к
исходному
хлоропреново-
Резина на основе
го каучука . .
То же с добавкой 0,1% крем-
нийорганич. жидкости . . . .
То же с добавкой 0,1% масла*
Резина с наполнителем C 0% са-
сажи)
То же с добавкой 0,1% кремний-
органич. жидкости
То же с добавкой 0,1% масла*
100
136
124
100
120
116
Относите-
Относительное уд-
удлинение,
% к исход-
исходному
100
107
100
106
Утом-
Утомление,
тыс.
циклов
25
360
360
37
83
720
* Продукт деполимеризации политрифторхлорэтилена.
В присутствии небольших количеств (до 1%) нек-рых
веществ на кривых зависимости индекса расплава
полиэтилена от содержа-
содержания добавки наблюдает-
наблюдается максимум. При вве-
введении в полипропилен
десятых долей процента
Рис. 5. Зависимость эффек- *г
тивной вязкости г] расплава
полипропилена при постоян-
постоянном напряжении сдвига
A-Ю6 дин/см2) от содержа-
содержания с полиэтилсилоксано-
вой жидкости № 5 A) и
№ 3 B).
Ч
t
I
Ч У
0.2
0,5
полиэтилсилоксановой жидкости вязкость расплава по-
полимера уменьшается в десять раз (рис. 5).
Согласно теории и многим экспериментальньш дан-
данным, введение пластификатора в количестве до 1% не
должно приводить к существенному изменению свойств
полимера (напр., Гс, вязкости расплавов). Поэтому
«аномальное» влияние малых добавок нек-рых пласти-
пластификаторов рассматривается с точки зрения современ-
633
ПЛАСТИЧЕСКИЕ МАССЫ
634
ных представлений о надмолекулярной структуре.
Предполагается, что механизм этого явления состоит
в распределении добавки между надмолекулярными
образованиями, ослаблении связи между ними и повы-
повышении их подвижности. П. малыми добавками наз.
структурной. Как правило, для эффективной структур-
структурной П. используются вещества, имеющие небольшое
термодинамич. сродство к полимеру. Очень часто
пластификаторы, совмещающиеся с полимером, дей-
действуют по механизму структурной П. лишь при малых
концентрациях. Следствие этого — появление экстре-
экстремумов на кривых зависимости ряда свойств полимера
от концентрации пластификатора.
Антипластификация. При введении в полимер, нахо-
находящийся в стеклообразном состоянии, относительно
небольших количеств нек-рых веществ его модуль
упругости (а иногда и прочность) может возрастать.
При этом удлинение при разрыве и ударная вязкость,
как правило, уменьшаются. Такое изменение механич.
свойств полимера противоположно изменению, наблю-
наблюдаемому при П., поэтому этот эффект наз. антипласти-
антипластификацией. Возрастание модуля упругости и прочности
при постоянных темп-ре и скорости растяжения про-
происходит лишь до определенной концентрации введен-
введенного вещества, дальнейшее увеличение содержания
добавки приводит, как и при П., к уменьшению значе-
значений этих характеристик. Один из отличительных при-
признаков антипластификаторов — способность уменьшать
или полностью подавлять молекулярную подвижность,
связанную со вторичным релаксационным переходом
ниже Тс. При антипластификации Тс понижается.
В нек-рых случаях это понижение заметно меньше,
чем при П.
Антипластификация свойственна жесткоцепным по-
полярным полимерам (поликарбонаты, гетероцепные по-
полиэфиры, триацетат целлюлозы и др.). Наиболее
эффективные антипластификаторы — совместимые с
полимером вещества, содержащие полярные атомы
(напр., хлор, азот, кислород, серу) и имеющие высо-
высокую Тс.
По-видимому, характер действия полярного вещест-
вещества на полярный полимер зависит от того, в каком
физич. состоянии находится система полимер—добавка.
Установлено, что увеличение концентрации ряда вво-
вводимых в поливинилхлорид добавок приводит к увели-
увеличению скорости звука и динамич. модуля, если система
полимер — добавка находится ниже Тс, и к уменьше-
уменьшению этих характеристик, если система находится
выше Тс.
Предполагается, что антипластификация — резуль-
результат влияния нескольких эффектов, к числу к-рых от-
относятся уменьшение свободного объема полимера,
усиление взаимодействия между полярными группами
полимера и антипластификатора и повышение жест-
жесткости полимера вследствие введения в полимер жестких
молекул антипластификатора.
Лит.: К а р г и н В. А., Слонимский Г- Л., Крат-
Краткие очерки по физико-химии полимеров, 2 изд., М., 1967; Т а-
г е р А. А., Физико-химия полимеров, 2 изд., М., 1968; Фер-
ри Д ж., Вязкоупругие свойства полимеров, М., 1963; Ти-
н и у с К., Пластификаторы, М.— Л., 1964; П е р е п е ч-
к о И. И., Акустические методы исследования полимеров,
М., 1973; Козлов П. В., Журн. ВХО, 9, № 6, 660 A964);
НатовМ. А., Джагарова Е. Хр., Высокомол. соед.,
8, № ю, 1841 A966); А н д р и а н о в а Г. П., БакеевН.Ф.,
Козлов П. В., Высокомол. соед., 8, № 2, 266 A971).
П. В. Козлов, А. В. Ефимов.
ПЛАСТИЧЕСКИЕ МАССЫ, пластмассы,
пластики (plastics, Plaste, matieres plastiques) —
материалы, основу к-рых составляют полимеры, нахо-
находящиеся в период формования изделий в вязкотеку-
чем или высокоэластич. состоянии, а при эксплуата-
эксплуатации — в стеклообразном или кристаллич. состоянии.
В зависимости от характера процессов, сопутствую-
сопутствующих формованию изделий, П. м. делят на термопласты
и реактопласты. К числу реактопластов от-
относят материалы, переработка к-рых в изделия соп-
сопровождается химич. реакциями образования трехмер-
трехмерного полимера — отверждением] при этом пластик
необратимо утрачивает способность переходить в вяз-
котекучее состояние. При формовании изделий из
термопластов не происходит отверждения, и
материал в изделии сохраняет способность переходить
в вязкотекучее состояние.
Состав пластмасс. П. м. обычно состоят из несколь-
нескольких взаимно совмещающихся и несовмещающихся ком-
компонентов. При этом, помимо полимера, в состав П. м.
могут входить наполнители (см. Наполнители пласт-
пластмасс), пластификаторы, стабилизаторы, красители
и др. Обо всех возможных компонентах П. м. см.
Ингредиенты полимерных материалов.
П. м. могут быть однофазными (гомогенными) или
многофазными (гетерогенными, композиционными) мате-
материалами. В гомогенных пластиках полимер является
основным компонентом, определяющим свойства мате-
материала. Остальные компоненты растворены в полимере.
В гетерогенных пластиках полимер выполняет функ-
функцию дисперсионной среды (связующего) по отношению
к диспергированным в нем компонентам, составляю-
составляющим самостоятельные фазы. Для распределения внеш-
внешнего воздействия на компоненты гетерогенного пла-
пластика необходимо обеспечить прочное сцепление на
границе контакта связующего с частицами наполни-
наполнителя, достигаемое адсорбцией, химич. реакцией свя-
связующего с поверхностью наполнителя. Чем большая
доля связующего находится в сфере влияния поверх-
поверхности наполнителя, тем резче изменяются свой-
свойства материала: понижается ползучесть, возрастает
темп-pa стеклования, изменяются степень кристал-
кристалличности и морфология кристаллов, скорость и сте-
степень отверждения, повышается вязкость расплава. См.
также Наполнение.
Типы наполненных пластмасс. Наполнитель в П. м.
может быть в газовой или конденсированной фазе.
В последнем случае его модуль упругости м. б. ниже
(низкомодульные наполнители) или выше (высокомо-
(высокомодульные наполнители) модуля упругости связующего.
К числу газонаполненных пластиков относятся пено-
пласты и поропласты. Такие П. м.— наиболее легкие
из всех пластиков; их кажущаяся плотность состав-
составляет обычно от 0,02 до 0,8 г/см3. В пенопластах газовые
пузырьки изолированы друг от друга пленкой связую-
связующего. Это придает таким материалам высокие элект-
электроизоляционные свойства (диэлектрич. проницаемость
1,1 — 1,3, тангенс угла диэлектрич. потерь 2,4-Ю —
3,0-10~3), плавучесть, высокие звуко- и теплоизоля-
теплоизоляционные характеристики. Так, коэфф. теплопровод-
теплопроводности для них составляет ок. 4,7-10~2 вт/(м-К) [ок.
4-10~2 ккал/(м-ч-°С)]. Поропласты пронизаны сквоз-
сквозными каналами и, в зависимости от их диаметра, изби-
избирательно проницаемы для частиц различных размеров.
См. также Пенопласты, Пористые ионообменные смолы.
Низкомодульные наполнители (их иногда называют
эластификаторами), в качестве к-рых обычно исполь-
используют эластомеры, не понижая теплостойкости и твер-
твердости полимера, придают материалу повышенную
устойчивость к знакопеременным и ударным нагруз-
нагрузкам, предотвращают прорастание микротрещин в свя-
связующем. Так, ударная вязкость полистирола после
эластификации возрастает с 15 до 80 кдж/м2, или
кгс-см/см2. Однако коэфф. термич. расширения эласти-
фицированных П. м. выше, а деформационная устой-
устойчивость несколько ниже, чем монолитных связующих.
Эластификатор диспергируют в связующем в виде
частиц размером 0,2—10 мкм. Это достигается полиме-
полимеризацией мономера на поверхности частиц синтетич.
латексов, отверждением олигомера, в к-ром дисперги-
диспергирован эластомер, механич. перетиранием смеси жест-
635
ПЛАСТИЧЕСКИЕ МАССЫ
636
коцепного полимера с эластомером. Наполнение должно
сопровождаться образованием блок- или привитого
сополимера на границе раздела частиц эластификатора
со связующим. Это обеспечивает кооперативную реак-
реакцию связующего и эластификатора на внешнее воздей-
воздействие в условиях эксплуатации материала.
Чем выше модуль упругости наполнителя и степень
наполнения материала, тем выше деформационная
устойчивость наполненного пластика. Однако введе-
введение высокомодульных наполнителей в подавляющем
большинстве случаев способствует возникновению
остаточных напряжений в связующем, а следователь-
следовательно, понижению прочности и монолитности полимер-
полимерной фазы.
Свойства пластиков с твердым наполнителем опреде-
определяются степенью наполнения, типом наполнителя и
связующего, прочностью сцепления на границе кон-
контакта, толщиной пограничного слоя, формой, размером
и взаимным расположением частиц наполнителя.
Пластики с частицами наполнителя малых размеров,
равномерно распределенными по материалу, характери-
характеризуются изотропией свойств, оптимум к-рых достигает-
достигается при степени наполнения, обеспечивающей адсорб-
адсорбцию всего объема связующего поверхностью частиц
наполнителя. При повышении темп-ры и давления
часть связующего десорбируется с поверхности на-
наполнителя, благодаря чему материал можно формовать
в изделия сложных форм с хрупкими армирующими
элементами. Мелкие частицы наполнителя, в зависи-
зависимости от их природы, до различных пределов повыша-
повышают модуль упругости изделия, его твердость, проч-
прочность при нагружении, придают ему фрикционные или
антифрикционные качества (см. Антифрикционные по-
полимерные материалы, Фрикционные полимерные ма-
материалы), теплоизоляционные, теплопроводящие или
электропроводящие свойства (см. Диэлектрические
свойства, Электропроводные полимерные материалы,
Металлонаполненные пластики).
Для получения пластиков низкой плотности приме-
применяют наполнители в виде полых частиц. Такие мате-
материалы (иногда называемые синтактическими пенами)
обладают хорошими звуко- и теплоизоляционными
свойствами (см. Пластики с полым наполнителем).
Применение в качестве наполнителей природных и
синтетических органич. волокон, стеклянных, квар-
кварцевых, углеродных, борных, асбестовых, хотя и огра-
ограничивает выбор методов формования и затрудняет из-
изготовление изделий сложной конфигурации, но резко
повышает прочность материала (см. Армированные
пластики). Упрочняющая роль волокон (диаметром
3 —12 мкм) в волокнитах и органоволокнитах, стекло-
волокнитах, асбоволокнитах, карбоволокнитах (см.
Углеродопласты) проявляется уже при длине волокна
в 2—4 мм. С увеличением длины волокон прочность
возрастает благодаря взаимному их переплетению и
понижению напряжений в связующем (в случае высо-
высокомодульного наполнителя), локализованных по кон-
концам волокон.
В тех случаях, когда это допускается формой изде-
изделия, волокна скрепляют между собой в нити и в ткани
различного плетения. П. м., наполненные тканью
(текстолиты), принадлежат к числу слоистых пласти-
пластиков, отличающихся анизотропией свойств, в частности
высокой прочностью вдоль слоев наполнителя и низкой
в перпендикулярном направлении. Этот недостаток
слоистых пластиков отчасти устраняется применением
т. наз. объемнотканых тканей, в к-рых полотна перепле-
переплетены между собой в перпендикулярном направлении.
Связующее заполняет неплотности переплетений, и,
отверждаясь, фиксирует форму, приданную заготовке
из наполнителя (см. Стеклопластики).
В изделиях несложных форм, и особенно в полых
телах вращения, волокна-наполнители укладывают по
направлению действия внешних сил. Прочность таких
пластиков в заданном направлении определяется в
основном прочностью волокон; связующее лишь фик-
фиксирует форму изделия и равномерно распределяет на-
нагрузку по волокнам. Модуль упругости и прочность
при растяжении изделия вдоль расположения волокон
зависят от степени наполнения. Эти показатели со-
соответственно достигают: для стекловолокнитов — 50
и 1,9 Гн/м2 E000 и 190 кгс/мм'2), для карбоволокни-
тов — 250 и 1,2 Гн/м2 B5 000 и 120 кгс/мм2), для бо-
роволокнитов — 275 и 1,4 Гн/м2 B7 500 и 140
кгс/мм2).
Для панельных конструкций удобно использовать
слоистые пластики с наполнителем из древесного шпо-
шпона или бумаги, в том числе из синтетич. волокна (см.
Древесно-слоистые пластики, Гетинакс, Органогети-
накс). Значительное понижение массы панелей при со-
сохранении жесткости достигается применением мате-
материалов трехслойной, или сэндвичевой, конструкции,
в к-рых сотопласты защищены тонкими листами об-
обшивки из слоистого пластика.
Основные виды термопластов и особенности их
свойств. Среди термопластов наиболее разнообразно
применение материалов из полиэтилена, поливинил-
хлорида и полистирола, преимущественно в виде гомо-
гомогенных или эластифицированных материалов, реже
газонаполненных и наполненных минеральными по-
порошками или короткими стеклянными, углеродными
либо синтетическими органич. волокнами.
Пластики на основе полиэтилена легко формуются
и свариваются в изделия сложных форм, они устой-
устойчивы к ударным и вибрационным нагрузкам (ударная
вязкость 100—120 кдж/м2, или кгс-см/см2), химически
стойки, отличаются высокими электроизоляционными
свойствами (диэлектрич. проницаемость 2,1—2,3) и
низкой плотностью. П. м. с особенно удачным соче-
сочетанием свойств получаются при наполнении полиэти-
полиэтилена коротким (до 3 мм) стекловолокном. При степени
наполнения 20% прочность при растяжении возрастает
в 2,5 раза, при изгибе — в 2 раза, ударная вязкость —
в 4 раза и теплостойкость — в 2,2 раза.
Жесткий пластик на основе поливинилхлорида —
винипласт, в том числе эластифицированный (ударо-
(ударопрочный), формуется значительно труднее полиэти-
полиэтиленовых пластиков, но прочность его к статич. нагруз-
нагрузкам много выше [напр., прочность при растяжении
60 Мн/м2 F00 кгс/см2) по сравнению с 15 Мн/м2
A50 кгс/см2)], ползучесть ниже и твердость выше.
Наиболее широкое применение находит пластифици-
пластифицированный поливинилхлорид — пластикат. Он легко
формуется и надежно сваривается, а требуемое сочета-
сочетание в нем прочности, деформационной устойчивости и
теплостойкости достигается изменением количества
пластификатора и твердого наполнителя. См. также
Поливинилхлоридные пластмассы.
Пластики на основе полистирола формуются много
легче, чем из винипласта, их диэлектрич. свойства
близки к свойствам полиэтиленовых П. м., они опти-
оптически прозрачны и по прочности к статич. нагрузкам
мало уступают винипласту, но более хрупки, менее
устойчивы к действию растворителей и горючи. Низкая
ударная вязкость A0—12 кдж/м1, или кг-см/см2) и
разрушение вследствие быстрого прорастания микро-
микротрещин устраняются при наполнении полистирольных
пластиков полимерами или сополимерами с темп-рой
стеклования ниже —40 °С. Эластифицированный (уда-
(ударопрочный) полистирол наиболее высокого качества
получают полимеризацией стирола на частицах латек-
латекса из сополимеров бутадиена со стиролом или с акрило-
нитрилом. Материал, названный АБС (см. Стирола
сополимеры), содержит около 15% гель-фракции, со-
состоящей из блок- и привитых сополимеров полистирола
и указанного сополимера бутадиена, эластифицирую-
637
ПЛАСТИЧЕСКИЕ МАССЫ
638
щего эластомера и полистирола, создающего жесткую
матрицу. Морозостойкость материала ограничивает
темп-pa стеклования эластомера, теплостойкость —
темп-pa стеклования полистирола.
Теплостойкость перечисленных термопластов (нена-
полненных) лежит в пределе 60—80 °С, коэфф. термич.
расширения высок и составляет —-10~4 °С~1, их свойства
резко изменяются при незначительном изменении
темп-ры, низка деформационная устойчивость под на-
нагрузкой. Этих недостатков ненаполненных термопла-
термопластов отчасти лишены иономеры, напр, сополимеры эти-
этилена, пропилена, стирола с мономерами, содержащими
ионогенные группы (обычно ненасыщенные карбоно-
вые к-ты или их соли). Ниже темп-ры текучести бла-
благодаря взаимодействию ионогенных групп между
макромолекулами создаются прочные узлы и образует-
образуется сетчатая структура, к-рая разрушается при размяг-
размягчении полимера. В иономерах удачно сочетаются свой-
свойства термопластов, благоприятные для формования
изделий с повышенными деформационной устойчиво-
устойчивостью и жесткостью, со свойствами, характерными
для сетчатых полимеров. Однако с повышением кон-
концентрации ионогенных групп в составе полимера
ухудшаются его диэлектрич. свойства и понижается
влагостойкость.
Пластики с более высокой теплостойкостью A00—
130 СС) и менее резким изменением свойств с повышени-
повышением темп-ры производят на основе полипропилена, по-
полиформальдегида, поликарбонатов, полиакрилатов,
ароматич. полиамидов. Особенно быстро расширяется
номенклатура изделий, изготавливаемых из поликар-
поликарбонатов, в том числе наполненных стекловолокном.
Для деталей, работающих в узлах трения, широко
применяются пластики из алифатич. полиамидов, на-
наполненных теплопроводящими материалами (см. По-
Полиамидные пластмассы, Г рафитопласты, Металлона-
полненные пластики).
Особенно высоки химич. стойкость, прочность к
ударным нагрузкам и диэлектрич. свойства пластиков
на основе политетрафторэтилена и сополимеров тетра-
фторэтилена. В материалах на основе полиуретанов
удачно сочетается износостойкость с морозостойкостью
и длительной прочностью в условиях знакопеременных
нагрузок. Полиметилметакрилат используют для из-
изготовления оптически прозрачных атмосферостойких
материалов, применяемых в качестве ударопрочных,
легких, легко штампуемых, механически обрабаты-
обрабатываемых и свариваемых органических стекол. Объем
производства термопластов с повышенной теплостой-
теплостойкостью и органич. стекол составляет ок. 10% общего
объема всех полимеров, предназначенных для изготов-
изготовления П. м.
Отсутствие реакций отверждения во время формова-
формования термопластов дает возможность предельно интен-
интенсифицировать процесс переработки, производить ва-
вакуум- и пневмоформование ранее изготовленных лис-
листов и профилей, раздув трубчатых заготовок в плен-
пленки и полые изделия, сборку сложных конструкций свар-
сваркой и повторное формование амортизированных изде-
изделий. Поскольку вязкость расплава высокомолекуляр-
высокомолекулярных полимеров велика, формование термопластов на
литьевых машинах или экструдерах требует уд. дав-
давлений 30—130 Мн/м2 C00 — 1300 кгс/см2).
Дальнейшее развитие производства термопластов
направлено на создание материалов из тех же поли-
полимеров, но с новыми сочетаниями свойств, применением
эластификаторов, порошковых и высокомодульных ко-
ротковолокнистых наполнителей.
Основные виды реактопластов и особенности их
свойств. После окончания формования изделий из
реактопластов полимерная фаза приобретает сетчатую
(трехмерную) структуру с высокой плотностью сетки
(см. Трехмерные полимеры). Благодаря этому отвер-
жденные реактопласты имеют более высокие, чем
термопласты, показатели по твердости, модулю упру-
упругости [до 4,5 Гн/м2 (до 450 кгс/мм2)], теплостойкости
(до 250—300 °С), усталостной прочности, более низкий
темп-рный коэфф. расширения. Однако неспособность
отвержденных реактопластов переходить в вязкотеку-
чее состояние вынуждает проводить синтез полимера
в несколько стадий.
Первую стадию оканчивают получением олигомеров
(смол) с мол. массой 500—1000. Благодаря низкой вяз-
вязкости р-ра или расплава олигомеры легко совмещать
с наполнителем и равномерно распределять по его
поверхности даже в том случае, когда степень напол-
наполнения достигает 80—85% (по массе). После введения
всех компонентов текучесть неотвержденного реакто-
пласта остается настолько высокой, что изделия из
него можно формовать заливкой (литьем), контактным
формованием, намоткой (см. Литье компаундов, Стекло-
Стеклопластики). Такие реактопласты наз. компаундами по-
полимерными и премиксами — в том случае, когда они
содержат наполнитель в виде мелких частиц, и пре-
прегами — если наполнителями являются непрерыв-
непрерывные волокна, ткань, бумага.
Технологич. оснастка для формования изделий из
компаундов, премиксов и препрегов проста и элерге-
тич. затраты невелики, но процессы связаны с длитель-
длительной выдержкой материала в индивидуальных формах
для отверждения связующего. При этом происходит
сильная усадка материала и в нем возникают значи-
значительные остаточные напряжения, а монолитность,
плотность и прочность далеко не достигают предель-
предельных значений (за исключением изделий, полученных
намоткой с натяжением), особенно в случае отвержде-
отверждения по реакции поликонденсации.
Чтобы избежать этих недостатков, в технологии
получения материала предусмотрена дополнительная
стадия (после смешения компонентов) — предотвержде-
ние олигомера, осуществляемое при вальцевании или
сушке. При этом сокращается длительность выдержки
материала в формах и повышается качество изделий,
однако заполнение форм из-за понижения текучести
связующего становится возможным только при давле-
давлениях 25—60 Мн/м2 B50—600 кгс/см2).
Олигомеры в реактопластах могут отверждаться
самопроизвольно (с тем большей скоростью, чем выше
темп-pa) или с помощью полифункционального низко-
низкомолекулярного вещества — отвердителя. Отверждение
может осуществляться по механизму поликонденсации
(напр., в случае феноло-альдегидных, эпексидных, кар-
бамидных, кремнийорганич. смол) и полимеризации
(полиалкиленгликольмалеинаты, олигоэфиракрилаты,
форполимеры диаллилфталата).
Реактопласты с любым наполнителем изготавливают,
применяя в качестве связующего феноло-формальдегид-
ные смолы, часто эластифицированные поливинилбу-
тиралем, бутадиен-нитрильным каучуком, полиами-
полиамидами, поливинилхлоридом (такие материалы наз. фе-
фенопластами), и эпоксидные смолы, иногда модифици-
модифицированные феноло- или анилино-формальдегидными
смолами или отверждающимися олигоэфирами.
Высокопрочные пластики [прочность при изгибе
1,0—1,9 Гн/м2 A00—190 кгс/мм2), ударная вязкость
100—150 кдж/м2, или кгс-см/см2] с термостойкостью
до 200 °С производят сочетанием стеклянных волокон
или тканей с отверждающимися олигоэфирами, феноло-
альдегидными или эпоксидными смолами. В произ-
производстве изделий, длительно работающих при 300 °С,
применяют стекло- или асбопластики с кремнийорга-
кремнийорганич. связующим; при 300—340 °С — полиимиды в со-
сочетании с кремнеземным, асбестовым или углеродным
волокном; при 350—500 °С в воздушной и при 2000—
2500 °С в инертной средах — фенопласты или пластики
на основе полиимидов, наполненные углеродным волок-
639
ПЛАСТМАССЫ
640
ном и подвергнутые карбонизации (графитизации) по-
после формования изделий.
Высокомодульные пластики [модуль упругости 250—
350 Гн/м2 B5 000—35 000 кгс/мм2)] производят сочета-
сочетанием эпоксидных смол с углеродными, борными или
монокристаллич. волокнами. Монолитные и легкие
материалы, устойчивые к вибрационным и ударным
нагрузкам, водостойкие и сохраняющие диэлектрич.
свойства и герметичность в условиях сложного нагру-
нагружения, изготавливают сочетанием эпоксидных, поли-
полиэфирных или меламино-формальдегидных смол с син-
тетич. волокнами, тканями, бумагой из синтетич. во-
волокон (см. Органоволокнит, Органогетинакс).
Наиболее высокие диэлектрич. свойства в сухой
среде (диэлектрич. проницаемость 3,5—4,0) характер-
характерны для материалов на основе кремнеземных или квар-
кварцевых волокон и полиэфирных или кремнийорганич.
связующих.
Древесно-слоистые пластики особенно широко ис-
используют в строительной технике и в судостроении.
Объем производства и структура потребления пласт-
пластмасс. В 1972 мировое производство полимеров для
П. м. достигло ~37 млн. т. Из них ок. 75% прихо-
приходилось на долю термопластов B5% полиэтилена, 20%
поливинилхлорида, 14% полистирола и его производ-
производных, 16% прочих пластиков). Существует тенденция
к дальнейшему увеличению доли термопластов (в ос-
основном полиэтилена) в общем выпуске П. м.
Хотя доля термореактивных смол в общем выпуске
полимеров для П. м. составляет всего ок. 25%, факти-
фактически объем производства реактопластов выше, чем
термопластов, из-за высокой степени наполнения
{60—80%) смолы.
Ниже представлены объемы производства П. м. в
нек-рых промышленно развитых странах (данные за
1972, в млн. т):
США 10,7
Япония ....... 5,6
ФРГ . « 5,5
Италия „ ,...„. 2,1
Франция 2,0
СССР г . , 1,9
Великобритания . . 1,6
Применение П. м. в различных областях техники ха-
характеризуют данные табл. 1.
Таблица 1. Структура потребления пластмасс в различных
странах (в % от общего потребления, данные за 1971)
Область применения
Электротехника и электрони-
электроника
Машиностроение
•Строительство
Легкая пром-сть и товары на-
народного потребления
Сельское хозяйство
СССР
10
25
35
24
6
США
12
23
28
31
6
ния
10
25
28
35
2
ФРГ
8
20
33
35
4
ГДр
16
18
28
32
Производство П. м. развивается значительно интен-
интенсивнее, чем таких традиционных конструкционных
материалов, как чугун и алюминий (табл. 2).
Таблица 2. Развитие мирового производства пластмасс,
черных металлов и алюминия (в млн. т)
Наименование
материала
Пластмассы
Черные металлы . .
Алюминий
1950
1,5
133,6
1,5
1960
7,5
258,6
4,5
1965
14,5
324,7
6,1
1970
30
560
11,3
1980
(прогноз)
100
900
32
См. также Переработка пластических масс, Испыта-
Испытания пластических масс, Экономика промышленности
пластических масс.
Лит.: Николаев А. Ф., Синтетические полимеры и
пластические массы на их основе, 2 изд., М.— Л., 1964; Техно-
Технология пластических масс, под ред. В. В. Коршака, М., 1972;
Современные композиционные материалы, под ред. Л. Браут-
мана и Р. Крока, пер. с англ., М., 1970; Конструкционные свой-
свойства пластмасс, под ред. Э. Бэра, пер. с англ., М., 1967'Спра-
1967'Справочник по пластическим массам, под ред. М. И. Гарбара [и др.],
т. 1—2, М., 1967—69; Лосев И. П., Т р о с т я н-
ская Е. Б., Химия синтетических полимеров, 3 изд., М.,
1971; Пластики конструкционного назначения, под ред. Е. Б.
Тростянской, М., [в печати]. Е. Б. Тростянская.
ПЛАСТМАССЫ— см. Пластические массы.
ПЛАСТОГРАФ БРАБЕНДЕРА — см. Брабендера
пластограф.
ПЛАСТОМЕТРЫ — см. Пласто-эластические свой-
свойства.
ПЛАСТО-ЭЛАСТИЧЕСКИЕ СВОЙСТВА (plasto-
elastic properties, plastoelastische Eingenschaften, pro-
prietes plasto-elastiques) — способность каучуков и
резиновых смесей к деформированию. В технологич.
практике П.-э. с. характеризуют соотношением между
необратимой (пластической) и обратимой (преимущест-
(преимущественно высокоэластической) составляющими деформа-
деформации. От П.-э. с. зависят такие технологич. свойства
каучуков и резиновых смесей, как скорость обработки,
способность сохранять приданную форму, гладкость
поверхности, склонность к под вулканизации. Кроме
того, определение П.-э. с. в стандартных условиях
испытаний служит для контроля соответствия каучу-
каучуков требованиям стандартов или технич. условий,
а также качества смешения каучуков с ингредиентами.
Пластичность каучуков и резиновых сме-
смесей обусловливает возможность придания полуфабри-
полуфабрикату резинового изделия той или иной формы, эла-
эластичность — восстановление после переработки
первоначальных размеров и формы полуфабриката;
она является также причиной нежелательного явле-
явления — усадки.
Пластическая деформация 8ПЛ развивается непрерыв-
непрерывно во времени и не исчезает после прекращения дей-
действия на материал механич. нагрузки. Высокоэласти-
Высокоэластическая (обратимая) деформация 8ВЭЛ тоже развивается
во времени и исчезает (также во времени) после снятия
нагрузки. Пренебрежимо малую долю в общей дефор-
деформации е занимает упругая деформация 8упр.
Т. обр., 8=8
Упр"
+
Соотношение меж-
ДУ 8вэл и 8пл зависит от режима нагружения: значе-
значений напряжений и деформаций, их вида и характера
изменения во времени, а также от длительности нагру-
нагружения и темп-ры.
В условиях нагружения при заданных (постоян-
(постоянных) темп-ре Т и напряжении а (рис. 1) мгновенно
Рис. 1. Развитие деформации е во времени t при постоянных
напряжении а и темп-ре Т: t0 — момент приложения меха-
механич. нагрузки; f — момент завершения развития высоко-
эластич. деформации; и — момент разгружения; еуПр —
упругая деформация; евэл — высокоэластич. деформация;
8пл — пластич. деформация; е°° — стационарная (равновес-
(равновесная) высокоэластич. деформация; сплошной линией пока-
показана общая деформация 8.
устанавливается 8упр, развивается во времени евэл,
достигая в момент времени f предельного (равновес-
оо
ного, или стационарного) значения 8ВЭЛ, и непрерывно
641
ПЛАСТО-ЭЛАСТИЧЕСКИЕ СВОЙСТВА
642
возрастает необратимая деформация епл. Период уста-
установления евэл, так наз. нестационарный период де-
деформации, зависит от релаксационных свойств мате-
материалов. После снятия нагрузки в момент t1 мгновенно
исчезает еупр, уменьшается во времени евэл (происхо-
(происходит эластич. восстановление) и полностью сохраняется
епл. Развивающееся после установления е°° течение
вэл
происходит в стационарный период (t > t ). Течение
каучуков и резиновых смесей в стационарный период
не является ньютоновским и в сравнительно неболь-
небольших диапазонах скоростей деформации ds/dt м. б.
приближенно описано эмпирич. степенным законом:
de/dt = ф-ап
где а — напряжение, п и 6 — константы. Для неваль-
цованного натурального каучука при 80 СС в интер-
интервале значений о 1 —100 кн/.ч2 A04—106 дин /см2),
если de/dt выражена в рад/сек, то п = 3,7, ф = 0,0036;
для бутадиен-стнролыюго каучука в этих же условиях
испытаний п = 4,05, Ф = 0,83. При расширении пре-
пределов скоростей деформаций характеристики степен-
степенного закона течения п и d» меняются. Для описания тем-
пературно-скоростной зависимости течения применим
принцип температурно-временной суперпозиции, или
метод приведенных неременных (см. Суперпозиции
принцип температурио-временной).
В условиях заданной постоянной скорости деформа-
деформации (или угловой скорости со) напряжение о в неста-
нестационарный период изменяется по кривой с максиму-
максимумом (рис. 2), т. к. в этом периоде протекают два про-
процесса, оказывающие на
а противоположное вли-
влияние: 1) развитие высо-
высокоэластической дефор-
деформации, повышающее на-
^i пряжение; 2) обратимое
Рис. 2. Изменение напряже-
напряжения сг во времени t при пос-
^ тоянных скорости деформа-
^ ции (о и темп-ре ((ot>@2).
(тиксотропное) разрушение структуры материала при
механич. воздействии, приводящее к снижению напря-
напряжения. В стационарный период о постоянно и тем вы-
выше, чем больше со.
Показатели пласто-эластических свойств определяют:
1) при сжатии материалов между плоскопараллель-
плоскопараллельными плитами (рис. 3, а); 2) при неограниченном сдвиге
запрессованного материала между коаксиально распо-
расположенными статором и ротором (рис. 3, б); 3) при про-
давливанни материала через отверстия — мундштуки,
сопла, капилляры и др. (рис. 3, в); 4) при вдавливании
(пенетрации) в испытуемый материал инденторов с
твердыми наконечниками различной формы (конусная
игла с затупленной вершиной, шарик); 5) при растя-
растяжении образцов материала, закрепленных в зажимах;
6) при простом сдвиге между плоскопараллельными
плитами.
Стандартизованные методы определения П.-э. с.
принадлежат гл. обр. к группе экспресс-методов, с по-
помощью к-рых эти свойства характеризуют условными
показателями (для каучуков и резиновых смесей —
нелинейных упруговязких систем, изменяющих струк-
структуру и свойства при деформировании, не найдены опре-
определяющие уравнения, или уравнения состояния, ис-
используя к-рые можно получить абсолютные характе-
характеристики и рассчитать механич. поведение этих мате-
материалов при любых условиях и режимах нагружения,
в том числе и при переработке на технологич. обору-
оборудовании).
Испытание на сжатие (ГОСТ 415—53)
производят на пластометре с илоскопараллельными
плитами при заданной сжимающей нагрузке Q. Регла-
Регламентированы: форма и размеры образца (цилиндр диа-
диаметром 16 мм и высотой /г0 =10 мм), нагрузка Q =
= 49 п E кгс), темп-ра 70 °С, время сжатия и восста-
восстановления 3 мин. Необратимая деформация харак-
-tj—r I I
--wlf *
3 2 1
0
1
Рис. 3. Принципиальная схема различных видов деформа-
деформации каучуков и резиновых смесей при определении пласто-
эластических свойств: а—сжатие; 1, 3— сжимающие
плиты; 2 — образец; Q — сжимающая нагрузка; /i0, hi,
h2 — соответственно высоты образца до сжатия, при сжа-
сжатии и после восстановления; б — неограниченный сдвиг;
1 — статор; 2 — материал; 3 — ротор; a, b — зазоры между
статором и ротором; b = Ro — R; Яо, R — соответственно
радиусы статора и ротора; h — высота ротора; М = Q • #t—
вращающий момент; Rt, Q — плечо и груз при тарировке
прибора; стрелкой показано направление вращения ротора;
в — продавливание; 1 — контейнер; 2 — поршень; з — ма-
материал; Q — выдавливающая нагрузка; So — площадь
отверстия.
теризуется условным показателем пластичности, пред-
представляющим собой отношение (/г0 — h2)^(hl -j- Л2),
где /г0, /?г и h2 — соответственно высоты образца (см.
рис. 3, а): начальная, при сжатии под нагрузкой Q
и после восстановления по снятии нагрузки. Обратимая
деформация, к рая характеризуется разностью высот
(h2—h^ в мм, наз. эластическим восстановлением.
"При испытании на сжатие на так наз. дефометре
(ГОСТ 10201—62) находят при постоянной темп-ре
80 °С нагрузку Q (гс), вызывающую заданную дефор-
деформацию стандартного цилиндрич. образца диаметром
10 мм и высотой /г0 = 10 мм за 30 сек. Стандартизо-
Стандартизована деформация (/г0 — h1)/h0= 0,6. Нагрузка Q ха-
характеризует общее сопротивление материала сжатию
и наз. жесткостью (иногда ее наз. также «дефо-твер-
достью»). Эластич. восстановление (/г2 — /гг) в мм
измеряют через 30 сек после снятия нагрузки (иногда
этот показатель наз. «дефо-эластичностью»).
Недостатки испытаний на сжатие: неоднородность
деформации (т. е. различная деформация по сечению
образца), обусловливающая зависимость резулг/гатов
испытаний от формы, размеров образца и значения
деформации; искажение формы и размеров образца
при его нагреве, влияющее на результаты; малая де-
деформация и невозхможность достиженрш стационарного
периода деформации из-за кратковременности испыта-
испытаний; возможность проведения испытаний только при
одних (стандартных) условиях; несоответствие режима
деформации при испытании режиму деформирования
материала на перерабатывающем оборудовании. По-
Показатели испытаний на сжатие могут быть в основном
использованы для контроля пластичности (или жест-
жесткости) каучуков и полуфабрикатов резинового произ-
производства, но недостаточны для оценки кривых те-
течения этих материалов.
Испытания на неограниченный
сдвиг, осуществляемые на ротационных сдвиговых
вискозиметрах, позволяют широко варьировать ско-
скорость деформации, теми-ру, давление на испытуемый
материал, время нагружения и создавать определен-
определенную (в том числе однородную) деформацию. Если при
испытании задана угловая скорость вращения ротора
со, пропорциональная скорости деформации d&fdt (в ди-
намич. режимах испытаний задают частоту колеба-
643
ПЛАСТО-ЭЛАСТИЧЕСКИЕ СВОЙСТВА
644
ыий), то измеряют момент сопротивления вращению
ротора в материале М, пропорциональный а. Если
задан М, вызывающий вращение ротора в испытуемом
материале, то в зависимости от режима испытаний
измеряют угловую скорость вращения или частоту
колебаний. Коэффициенты пропорциональности между
dddt и со или между а и М зависят от формы и раз-
размеров статора и ротора (о типах вискозиметров см.
Вискозиметрия).
При испытании согласно ГОСТ 10722—64 на сдвиго-
сдвиговом дисковом ротационном вискозиметре (см. рис. 3,6)
определяют (при различных постоянных темп-рах
Т и со) зависимость М от t, аналогичную показанной
на рис. 2 зависимости а от t. При стандартных разме-
размерах испытательной камеры и ротора [2Л = 38,1 мм,
h = 5,54 мм 2Л0 = 2 (R + Ъ) = 50,93 мм; Bа + К) =
= 10,6 мм] и со = 2 об/мин результаты испытаний
выражают в единицах «вязкости по Муни». За единицу
«вязкости по Муни» принят момент сопротивления
сдвигу М, равный 0,083 н-м @,846 кгс-см). Эластич.
восстановление измеряют углом обратного поворота
ротора в испытуемом материале после снятия вращаю-
вращающего момента (прекращения принудительного враще-
вращения ротора). «Перепад вязкости» / = (М3 — Mt)/Mf,
где М3 и Mt — соответственно значения вязкости
через 3 сек и t мин (обычно 3—5 мин) вращения рото-
ротора, характеризует тиксотропные свойства материала
(см. рис. 2).
Указанная продолжительность испытаний опреде-
определяется тем, что за 3 сек для большинства материалов
достигается максимальное значение момента сопро-
сопротивления сдвигу, а за 3—5 мин — минимальное ста-
стационарное значение М.
Испытания на сдвиговых вискозиметрах наиболее
перспективны, т. к. позволяют получать общие законо-
закономерности механич. поведения каучуков и резиновых
смесей. Их методич. преимущества: применение высо-
высоких давлений и закрытой испытательной камеры, что
исключает искажение деформации; возможность созда-
создания неограниченной деформации и непрерывного на-
наблюдения за результатами во времени; возможность
испытания как в нестационарном, так и в стационар-
стационарном периоде деформации.
Принципиальные преимущества испытания на сдвиг
при заданной скорости деформации: проведение испы-
испытаний в условиях, близких к условиям переработки
каучуков и резиновых смесей на оборудовании, и,
следовательно, возможность характеризовать показа-
показателями испытаний технологич. свойства этих мате-
материалов. Так, по «вязкости по Муни» судят об общем
сопротивлении каучуков и резиновых смесей дефор-
деформации (в частности о мощности, потребляемой обору-
оборудованием при переработке); по эластич. восстановле-
восстановлению — об усадке; по «перепаду вязкости» — о неодно-
неоднородности структуры материала и шероховатости по-
поверхности изделия. На основании результатов, полу-
полученных в широком диапазоне темп-р и скоростей де-
деформации, пользуясь методом температурно-временной
суперпозиции, находят кривые течения и вычисляют
характеристики степенного закона течения, исполь-
используемые при расчетах производительности оборудова-
оборудования. Возможность реализации на сдвиговых вискози-
вискозиметрах неограниченных во времени деформаций по-
позволяет также наиболее эффективно испытывать смеси
на подвулканизацию (см. ниже).
При испытании на продавливание
(см. рис. 3, в) задают нагрузку Q в н (кгс), измеряют
количество материала, вышедшего за определенное вре-
время через отверстие заданного сечения Su в м2 (см2),
и находят также усадку материала после продавли-
вания, характеризующую его эластич. восстановление.
Можно также задавать режим, при к-ром измеряют
нагрузку Q, обеспечивающую определенную скорость
выдавливания. При испытании на вдав-
вдавливание (пенетрацию) определяют глубину погру-
погружения наконечника в испытуемый материал под дей-
действием определенной нагрузки за определенное время
(или нагрузку, обеспечивающую погружение наконеч-
наконечника на заданную глубину за определенное время).
Испытание на растяжение чаще всего
проводят при заданной скорости деформации и изме-
измеряют зависимость между нагрузкой и деформацией.
При испытании на простой сдвиг
находят зависимость между нагрузкой и углом сдвига
для заданной малой деформации. Испытания каучуков
и резиновых смесей при продавливании, пенетрации,
растяжении и простом сдвиге не стандартизованы.
Их основные недостатки: ограниченность деформации
во времени (невозможность выхода на стационарный
период); неоднородность деформации и искажения,
к-рые вносит проскальзывание материала (при продав-
продавливании и пенетрации); зависимость результатов ис-
испытаний от размеров и формы образца.
Поскольку результаты определения П.-э. с. зависят
от условий испытаний и вида деформации, между по-
показателями, к-рые получают при использовании раз-
различных методов, не существует взаимосвязи общего
вида. Качественно, чем ниже пластичность па
ГОСТ 415—53, тем выше жесткость по ГОСТ 10201—62
и вязкость по Муни.
Наряду с лабораторными методами оценки П.-э. с.
существуют также способы их оценки с помощью т. наз.
«технологических проб» непосредственно на перераба-
перерабатывающем оборудовании: определение вальцуемости и
шприцуемости по качеству поверхности, усадке и др.
показателям. Кроме того, в лаборатории частично
имитируют работу оборудования на специальных ис-
испытательных приборах; напр., согласно стандарту
США ASTM 2230—63Т смеси продавливают через спе-
специальные профилирующие отверстия (метод Гарвея).
Качество резиновых смесей оценивают визуально по
десятибалльной системе, сравнивая их с эталоном. В
зависимости от пористости резиновой смеси, измене-
изменения ее размеров («разбухания» или усадки), вида по-
поверхности и др. наихудшие свойства оценивают бал-
баллом 1, наилучшие — баллом 10.
Склонность резиновых смесей к подвулканизации,
приводящей к необратимым изменениям их П.-э. с,
оценивают обычно при 100—125 °С по степени измене-
изменения пластичности, жесткости, вязкости по Муни или
эластич. восстановления .
в зависимости от дли-
тельности прогрева сме-
смеси. Согласно ГОСТ
10722—64, характерис-
характеристиками склонности сме-
Рис. 4. Зависимость «вяз-
«вязкости по Муни» М от вре-
времени прогрева образцов t
при испытании на подвул-
подвулканизацию; Мтш — мини-
минимальное значение «вязкости по Муни»; tb — время начала под-
подвулканизации при М = Мп
ДМ, где ДМ = 5 единиц
«вязкости по Муни»; ?,5 — время, при к-ром М = Mm\n + AM,
где AM = 35 единиц «вязкости по Муни»; tc — время, при
к-ром М = 2Mmjn.
сей к подвулканизации при испытании на сдвиговом дис-
дисковом ротационном вискозиметре (рис. 4) при выбранной
постоянной темп-ре служат время начала подвулканиза-
подвулканизации t5 в мин, т. е. время, за к-рое вязкость по Муни М
превысит минимальную Мт\п на 5 единиц, и разность
tc — t5, обратная скорости подвулканизации (tc — время
в мин, соответствующее значению М = 2Mmjn). Со-
Согласно рекомендации ИСО, вместо (tc — t5) принят
645
ПЛЕНКИ ПОЛИМЕРНЫЕ
646
показатель (t35 — tb), где t3b — время в м ин, за
к-рое значение М превысит Мт\п на 35 единиц.
Переход из каучукоподобного в резиноподобное со-
состояние (вулканизация) связан с существенным изме-
изменением соотношения между обратимой и необратимой
деформацией. Практич. исчезновение в вулканизатах
(резинах) обратимой деформации приводит к резкому
повышению сопротивления материала деформирова-
деформированию; при этом осуществление неограниченной дефор-
деформации (течения) на сдвиговых ротационных вискози-
вискозиметрах становится невозможным.
Испытания резиновых смесей при темп-pax вулкани-
вулканизации (выше 130 °С) на вулкаметрах, реометрах, вис-
кюрометрах, кюрометрах и др. аналогичных приборах
в условиях динамич. гармонич. деформации сдвига
с малой амплитудой сдвигового смещения, позволяют
исследовать всю кинетику вулканизации, включая
время достижения оптимума, плато вулканизации и
период реверсии. Испытания на реометрах могут также
служить для экспресс-контрольной оценки качества
смешения. Так, по характеру отдельных участков ки-
нетич. кривой устанавливают нарушения в содержании
вулканизующих агентов, пластификаторов, наполните-
наполнителей и др. ингредиентов резиновой смеси.
Лит.: Т р е л о а р Л., Физика упругости каучука, пер.
с англ., М., 1953; Ф е р р и Д ж., Вязкоупругие свойства
полимеров, пер. с англ., М., 1963; К а р г и н В. А., Сло-
Слонимский Г. Л., Краткие очерки по физико-химии поли-
полимеров, 2 изд., М., 1967; РезниковскийМ. М., Лу-
комская А. И., Механические испытания каучука и ре-
резины, 2 изд., М., 1968; ЛукомскаяА. И., в сб.: Пнев-
Пневматические шины, М., 1969, с. 340; Мак-Келви Д. М.,
Переработка полимеров, пер. с англ., М., 1965.
А. И. Лукомская.
ПЛЕЙНОМЕРЫ — см. Олигомеры.
ПЛЕНКИ ПОЛИМЕРНЫЕ (polymer films, Poly-
merfilme, films polymeres). К П. п. относят обычно
сплошные слои полимеров толщиной до 0,2—0,3 мм.
Более толстые слои полимерных материалов наз. ли-
листами или пластинами.
П. п. производят из природных, искусственных и
синтетич. полимеров. К первой группе относятся плен-
пленки, изготовляемые из белков (см. Белковые пластики),
натурального каучука, целлюлозы и нек-рых др. при-
природных полимеров. Наибольшее распространение в этой
группе П. п. получили гидратцеллюлозные пленки,
среди к-рых наиболее известен целлофан. Пленкооб-
Пленкообразующий полимер этих пленок — целлюлоза, регене-
регенерируемая в процессе формования из ее эфира — ксан-
тогената.
Вторую, более обширную группу составляют П. и.
из искусственных полимеров, т. е. продуктов химич.
модификации природных полимеров. К этой группе
относятся пленки, полученные на основе простых и
сложных эфиров целлюлозы (см. Эфироцеллюлозные
пленки), а также пленки из натурального каучука,
предварительно подвергнутого гидрохлорированию
(см. Гидр о хлор ид каучуковые пленки).
Самую обширную группу П. п. составляют пленки
на основе синтетич. полимеров. Среди П. п. этой груп-
группы наибольшее распространение получили полиамид-
полиамидные пленки, поливинилиденхлоридпые пленки, поливи-
нилхлоридные пленки, полиимидные пленки, полиоле-
финовые пленки, полис т и рольные пленки и полиэти-
лентерефталатные пленки.
Методы формования. Существуют след. промышлен-
промышленные методы изготовления П. п.: 1) экструзия расплава
полимера; 2) полив р-ра полимера или форполимера на
полированную металлич. или др. поверхность (в нек-рых
случаях р-р полимера подают в осадительную ванну);
3) полив дисперсии полимера на полированную поверх-
поверхность; 4) каландрование; 5) строгание заготовок;
6) прессование.
Экструзия расплава полимера при-
пригодна в тех случаях, когда перерабатываемые материа-
материалы при переходе в вязкотекучее состояние не подвер-
подвергаются термич. деструкции. Большинство синтетич.
полимеров перерабатывается в пленки именно этим
методом. Для его осуществления используют экстру-
деры с кольцевой или плоскощелевой головкой. В пер-
первом случае расплав полимера экструдируется в ви-
виде рукава, который растягивается сжатым воздухом,
что приводит к двухосной ориентации пленки. Рукав-
Рукавный способ — наиболее производительный и экономич-
экономичный процесс изготовления П. п.
Плоскощелевой способ позволяет формовать неориен-
неориентированные (изотропные) пленки, а с помощью спе-
специальных растягивающих устройств — одноосно ориен-
ориентированные или двухосно ориентированные П. п.
В некоторых случаях аморфные П. п. дополнитель-
дополнительно подвергаются разглаживанию или полированию на
гладильных валках. Этот способ предпочтителен в
тех случаях, когда требуется получить равнотол-
щинную пленку с высоким качеством поверхности.
П. п. из кристаллизующихся полимеров (напр., из
полиэтилентерефталата) после ориентации подвергают
кристаллизации, к-рая резко улучшает прочностные
свойства пленок и стабилизирует структуру, образо-
образованную при ориентации.
Производство П. п. поливом раствора
полимера на холодную или нагреваемую полиро-
полированную поверхность — один из старейших промыш-
промышленных способов, имеющий теперь ограниченное при-
применение. Этим способом выпускают в основном плен-
пленки на основе целлюлозы и ее производных, а так-
также нек-рые пленки из синтетич. полимеров (напр., из
полиимидов, поливинилового спирта, поликарбонатов
и нек-рых др.)- Способ включает след. операции:
1) приготовление р-ра полимера; 2) полив р-ра поли-
полимера на гладкую полированную поверхность (беско-
(бесконечная металлич. лента или барабан); 3) отделение
растворителя от полимера. Последнюю операцию мож-
можно осуществить различными методами: а) осаждением
полимера в осадительной ванне специального состава,
напр, в производстве гидратцеллюлозных пленок,
при к-ром осадительная ванна вызывает химич. раз-
разложение (омыление) растворенного ксантогената цел-
целлюлозы с выпадением нерастворимой целлюлозы; б) ис-
испарением (высушиванием). При изготовлении П. п. из
пластизолей (см. Пасты полимерные) вместо стадии
отделения растворителя от полимера осуществляют
гелеобразование, при к-ром пластизоль отвердевает,
превращаясь в гель. В случае органозолей на этой
стадии происходит гелеобразование и испарение лету-
летучего разбавителя.
П. п., полученную поливом р-ра полимера, подвер-
подвергают термич. обработке для снятия внутренних напря-
напряжений, а в нек-рых случаях для повышения физико-
механич. характеристик П. п. осуществляют одноос-
одноосную или двухосную ориентацию (см. Ориентированное
состояние).
При изготовлении П. п. на основе термостойких
гетероциклич. полимеров (полиимидов, полибензимида-
золов и др.) процесс включает две стадии: 1) изготовле-
изготовление пленки поливом р-ра фориолимера по описанной
выше технологии; 2) циклизация форполимера.
Производство П. п. поливом дисперсий
полимеров на твердую поверхность практически
не отличается от описанного выше. Для этой цели
обычно применяют коллоидные системы (напр., латек-
сы), в к-рых дисперсионной средой служит вода, а
дисперсной фазой — частицы полимера. Производство
П. и. из дисперсий полимеров обладает рядом сущест-
существенных преимуществ перед способом, при к-ром при-
применяются р-ры полимеров: 1) отпадает необходимость
в дорогостоящих, токсичных и огнеопасных органич.
растворителях; 2) возникает возможность непосред-
непосредственно использовать эмульсии и суспензии, получен-
647
ПЛЕНКИ ПОЛИМЕРНЫЕ
648
ные в результате эмульсионной и суспензионной поли-
полимеризации без промежуточной операции выделения
полимера.
К недостаткам метода произ-ва П. п. поливом дис-
дисперсий полимеров следует отнести трудность в преодо-
преодолении препятствий к слипанию полимерных частиц
в процессе высыхания дисперсии, что необходимо
для получения структурно однородной монолитной
пленки. При этом большое значение имеет природа
эмульгатора, к-рый применялся для стабилизации
дисперсии: если в процессе формирования пленки (при
к-ром происходит удаление воды) эмульгатор раство-
растворяется в полимерных частицах, можно ожидать образо-
образования монолитной пленки с хорошими физико-меха-
нич. характеристиками. Эти рассуждения не относятся
к природному латексу, в к-ром белковые высокомоле-
высокомолекулярные стабилизаторы вне зависимости от их взаи-
взаимодействия с частицами полимера обеспечивают обра-
образование монолитных пленок с высокими физико-меха-
нич. характеристиками.
Иногда для повышения качества полученных П. п.
их подвергают дополнительной термомеханич. обработ-
обработке или обработке органич. растворителями, что способ-
способствует слипанию частиц в пленке.
Полив дисперсий полимеров применяют, в частности,
для изготовления пленочных санитарно-гигиенических
изделий.
Каландрованием получают гл. обр. плен-
пленки из поливинилхлорида (см. Калаидрование).
Строганием заготовок производят срав-
сравнительно толстые пленки, гл. обр. из политетрафтор-
политетрафторэтилена и целлулоида. При изготовлении пленок из
политетрафторэтилена первоначально прессуют ци-
линдрич. заготовку, к-рую вращают вокруг своей оси,
срезая ножом непрерывное пленочное полотно. Полу-
Полученную пленку раскатывают на специальном прокат-
прокатном станке. Из целлулоида прессуют блок, из к-рого
строгают пленку.
В отдельных случаях П. п. можно получить др.
методами. Напр., пленку из политетрафторэтилена
прессуют без нагревания или вальцуют на холодных
вальцах с последующим спеканием ее при высокой
темн-ре в печи.
Применение. Шире всего П. п. применяются в ка-
качестве упаковочных материалов для пищевых продук-
продуктов, товаров широкого потребления, жидких и сыпу-
сыпучих химич. и нефтехимич. товаров, для бытовых целей.
Для изготовления упаковочных пленок используют
полиэтилен, полипропилен, целлюлозу и ее эфиры,
полимеры и сополимеры винилхлорнда, полистирол,
полиамиды, полиэфиры, гидрохлорид натурального
каучука и др. Нек-рыми специфич. свойствами обла-
обладают упаковочные многослойные материалы типа
пленка — пленка, пленка — бумага или пленка —
фольга, а также вспененные пленки. См. также Поли-
Полимеры в пищевой промышленности.
Широкое распространение получили электроизоля-
электроизоляционные пленки, используемые для изготовления обмо-
обмоточных и монтажных проводов и кабелей, в производ-
производстве конденсаторов и для пазовой изоляции электрич.
машин. Наиболее широко в электротехнике и электро-
электромашиностроении применяют полистирольные, полиэти-
лентерефта латные, поликарбонатные, политетрафтор-
этиленовые, полиимидные и полиолефиновые пленки.
В кабельной пром-сти получили распространение П. п.
в сочетании со специальными сортами бумаги, стекло-
стекловолокном или синтетич. волокнами. В электромашино-
электромашиностроении П. п. комбинируют со специальными сортами
бумаги и картона, применяют также электроизоляцион-
электроизоляционные материалы, представляющие собой П. п., на к-рые
наклеены очень тонкие чешуйки стекла и слюды или
асбестовые волоконца. См. также Полимеры в электро-
электротехнике.
Кинофотопленки — многослойные системы, состоя-
состоящие из П. п. (основы, подложки) с нанесенным на нее
светочувствительным слоем. К этой же группе пле-
пленочных материалов относятся магнитные ленты для
записи и воспроизведения звука и изображения; на-
нанесенный на них слой содержит тонкодисперсный фер-
ферромагнитный порошок. В качестве основы для кинофо-
кинофотопленок и магнитных лепт используют главным об-
образом ацетатцеллюлозные и полиэтилентерефталатные
пленки. Последние должны быть двухосно ориентиро-
ориентированными и закристаллизованными. Предпринимаются
также попытки применить поликарбонатные и поли-
полистирольные П. п.
Указанные выше типы П. п. могут также служить
основой при изготовлении материалов, предназначен-
предназначенных для записи информации без использования гало-
генидов серебра и ферромагнитных материалов. В их
чувствительных слоях происходят др. химич. реакции
(напр., разрушение под действием света солей диазо-
ния с последующим образованием азокрасителей при
диазотипии, к-рая широко применяется для копирова-
копирования чертежей) или физические явления (напр., фа-
фазовые переходы в сегнетоэлектриках, применяемых в
качестве элементов памяти электронных счетных
машин).
Из атмосферостойких прозрачных полимерных пле-
пленок (полиэтиленовых, полиамидных, поливинилхло-
ридных и полнэтилентерефталатных, в нек-рых слу-
случаях армированных стекловолокном или тканями на
основе синтетич. волокон) изготовляют парниковые
рамы, крыши теплиц, переносные атмосферозащит-
ные покрытия, предохраняющие растения в откры-
открытом грунте от заморозков или создающие внутри по-
покрытия микроклимат, благоприятный для вегетации
растений. См. также Полимеры в сельском и водном
хозяйстве.
Гидроизоляционные пленки используют в строи-
строительстве при сооружении искусственных водоемов и
каналов и для др. целей.
Поляроидные пленки — П. п., к-рые способны поля-
поляризовать проходящий свет. Изготовляют поляроидные
нитратцеллюлозные, ацетатцеллюлозные или иоливи-
нилспиртовые трехслойные пленки, в среднем слое
к-рых диспергированы и определенным образом
ориентированы игольчатые кристаллы геранатнта (кис-
(кислого сульфат-трииодида хинина). См. также Поляри-
Поляризующие пленки.
Поляроидные пленки широко применяются в ка-
качестве светофильтров для борьбы с ослеплением шофе-
шоферов светом фар встречных машин, для регулирования
степени освещенности при постоянной мощности источ-
источника света, для разнообразных способов сигнализа-
сигнализации, замены николей оптнч. приборов, изготовления и
демонстрации стереоскопнч. фильмов, создания худо-
художественных изображений в интерференционных цветах
и для др. целей.
Ионообменные пленки применяют для извлечения
веществ с помощью электролиза, опреснения соленой
воды, при очистке органич. соединений или их р-ров
(напр., сахарных или гидролизных сиропов от мине-
минеральных примесей), для концентрирования р-ров, раз-
разделения и идентификации различных соединений и др.
целей. См. также Мембраны ионитовые.
В большинстве случаев П. п. из синтетич. полимеров
по комплексу физико-механич. н химич. свойств пре-
превосходят пленки из природных и искусственных поли-
полимеров, поэтому их промышленное произ-во и потреб-
потребление непрерывно возрастают.
Первое место по объему мирового произ-ва занимают
полиолефиновые пленки, второе — поливинилхлорид-
ные. Так, в 1970 в США полиэтиленовые пленки
составляли ок. 62,3% объема пленочной продукции,
поливинилхлоридные — 25,1%, полипропиленовые
649
ПЛЕНКИ ПОЛИМЕРНЫЕ
Таблица 1. Механические свойства полимерных пленок
650
Полимер
Прочность при растяжении,
Мн/м* (кгс/см2)
Относи-
Относительное
удлинение давливании
при разры-
разрыве, %
Прочность при про-
по Мюл-
Мюллеру, Мн/м2 (кгс/см2)
Стойкость
к распро-
распространению
надрыва,
Стойкость к много-
многократному двойно-
двойному изгибу (циф-
(цифры — число циклов
до разрушения)
Полиэтилен
низкой плотности
средней плотности
высокой плотности
Поливинилхлорид
жесткий
мягкий
Ацетобутират целлюлозы
Ацетопропионат целлюлозы
Этилцеллюлоза
Целлофан
Диацетат целлюлозы
Иономер (сополимер этилена с мет-
акриловой к-той)
Сополимер этилена с винилацетатом .
Полипропилен
неориентированный
двухосно ориентированный . . .
Полистирол двухосно ориентированный
Триацетат целлюлозы
Сополимер винилхлорида с винилаце-
винилацетатом
жесткий
мягкий
Сополимер винилиденхлорида с ви-
нилхлоридом
Поливиниловый спирт
Поливинилфторид
Политетрафторэтилен
Политрифторхлорэтилен
Полиамид-6
Полиэтилентерефталат
Поликарбонат
Гидрохлорид полиизопрена
10,5 - 21 A05 - 210)
14 — 25,5 A40 - 255)
17 - 43 A70 - 430)
49 - 70 D90 - 700)
10 - 40 A00 - 400)
35 — 63 C50 — 630)
28 — 35 B80 - 350)
36 - 70 C60 - 700)
49 - 126 D90 - 1260)
56 - 115 E60 — 1150)
24,5-39 B45 — 390)
10,5 - 21 A05 — 210)
31,5- 70 C15 - 700)
84 - 231 (840 - 2310)
56 — 84 E60 - 840)
63-112 F30 — 1120)
38,5 — 56 C85 — 560)
17,5 — 35 A75 - 350)
56 — 140 E60 — 1400)
28 — 70 B80 - 700)
49 — 126 D90 - 1260)
10,5 — 28 A05 - 280)
35 — 56 C50 — 560)
63 - 126 F30 - 1260)
140 — 210 A400 — 2100)
58 - 61 E88 - 616)
24,5 - 35 B45 - 350)
100-700
50-650
10-650
25
150-500
50-100
60-80**
20-30
10-50
15-70
200-600
500-800
5 5 0-1000
50 — 200
3-40
10-4 0
3-100
100-500
35-110
180-600
115—2 50
100-350
5 0-150
250-550
7 0—120
85-105
200 — 800
1-2 A0 - 12)
3-
4
5
5
5,5
3
4 C0 — 40)
2 B0)
7 D0 - 70)
7 E0 - 70)
8,5 E0 - 85)
6,5 E5 — 65)
6 C0 - 60)
1 — 2 A0 - 12)
1,6
5
3,5 A6 — 35)
•7E0 — 70)
2B0)
2,5 - 3,5 B5 - 35)
1,9-7,0A9-70)
2,3 - 3, 1 B3 - 31)
5, 5 — 8,0 E 5 — 80)
2,5 - 3,5 B5 — 35)
растягивается Г)ез
разрушения
100-500
50-300
15-300
10-700
60-1400
5-10*
100
47-36
2 — 20
4-10
30
50-300
7-20
4-10*
10-30
30-1400
10-100
250-800
12-4 0
10-100
8-29
50-90
12-27
20-25
60-1600
очень высокая
800-1200*
80**
очень высокая
500-2000*
очень высокая
500 000
5000-47 000
хорошая
250 000
100 000
250 — 400
250 0 00
* Для пленки толщиной 25 мкм. ** Для пленки толщиной 125 мкм.
Таблица 2. Электрические свойства полимерных пленок
Полимер
Диэлектрич. проницаемость
при частоте
103 гц
10й гц
109 гц
Тангенс угла диэлектрич. потерь при
частоте
1С3 гц
106 гц
109 гц
Электрич.
прочность,
Мв/м, или
кв/мм
"«uSSc
Полиэтилен
низкой плотности . .
средней плотности . .
высокой плотности
Поливинилхлорид
жесткий
мягкий
Ацетобутират целлюлозы
Ацетопропионат целлюло-
целлюлозы
Этилцеллюлоза
Целлофан нелакированный
Диацетат целлюлозы ....
Иономер (сополимер этиле-
этилена с метакриловой к-той)
Сополимер этилена с винил-
ацетатом
Полипропилен
неориентированный
двухосно ориентиро-
ориентированный
Полистирол двухосно ориен-
ориентированный
Триацетат целлюлозы . . .
Сополимер винилхлорида с
винилацетатом
жесткий
мягкий
Сополимер винилиденхлори-
винилиденхлорида с винилхлоридом . . .
Поливинилхлорид
Политетрафторэтилен ....
Политрифторхлорэтилен . .
Полиамид—6 (сухой) ....
Полиэтилентерефталат . . .
Поликарбонат
Гидрохлорид полиизопрена
2,2
3,0-
4,0-
-3,3
-8,0
9
2,9
2,0
-3,3
2 2 — 2 7
3,0 -3,3
4,0-8,0
3,9-4,5
8,5
2,0-2,1
2,5-2,7
3,7
3,2
2,99
3
2,3
2,8-3,1
3,3-4,5
2 , 5
3,2
3,0
3,2
2,5
2,8-3,1
2,0-2, 1
2,4-2,7
3,3
2,8-3,1
3,3-4,5
3,0-4,0
7,4
2,0-2, 1
2,3-2,4
3,4
3,0
2,93
3
2,2
2 2
3
2,8
2,8
3,2
2,5
2,0-2,1
,4-2,7
3,2
2,*
2,7
,0-2, 1
2,3
2,89
0,0003
0,0003
0,0005
0,009-0,017
0,07-0, 16
0,013
0,002-0,020
0,015
0,013
0,0015
0,009-0,02
0,0003
0,0005
0,016
0,009-0,017
0,070-0, 160
0,052-0,063
1,6
0,0002
0,022-0,024
0,016
0,005
0,0015
0,0003
0,0003
0,0005
0,006-0,017
0,04-0, 14
0,030
0,015
0,010 — 0,060
0,038
0,0015
0,02-0,06
0,0003
0,0005
0,033
0,006-0,019
0,04-140
0,05-0,08
0,0002
0,009-0,017
0,025
0,016
0,010
0,006
0,0003
0,0003
0,0005
0,019
0,046
0,0015
0,0003
0,0005
0,019
0,016
0,0002
0,000 4
0,008
0,012
188
20
20
17-54
10-40
124
73,6
60*
80-100
128-200
40
120-180
100
148
17 — 54
10-40
120-280
140-180
17,2
40-148
52-60*
300**
60**
1014
1(I4
1Q14
1014
10»—10'^
1 О9—1 01а
1013
1013
10 '•>
108-Ю12
1017
1013
1014
10"—1013
1 О14
109-Ю12
1 О10— 1013
1011
10Ю
10Ю
Ю10
1016
Ю14
10"
* Для толщины 50 мкм и относительной влажности воздуха 50%. ** Для толщины 25 мкм. *** 1 ом-м= 102 ом*см.
651
ПЛЕНКООБРАЗУЮЩИЕ ВЕЩЕСТВА
652
Таблица 3. Стойкость полимерных пленок к различным воздействиям*
Полимер
Стойкость по отношению к
химическим реагентам
силь-
сильные
кисло-
кислоты
щелочи
жиры
органиче-
органические рас-
растворители
Водостойкость
водопоглоще-
ние, за 24 -ч,
%
при
высо-
высокой
влаж-
влажности
Стойкость
к солнеч-
солнечному све-
свету
Тепло-
стой-
стойкость,
Морозо-
Морозостойкость,
Горючесть
Полиэтилен
низкой плотности .
средней плотности
высокой плотности
Поливинилхлорид
жесткий
мягкий
Ацетобутират целлюлозы . .
Ацетопропионат целлюлозы
Целлофан лакированный . .
Этилцеллюлоза
Диацетат целлюлозы . . . .
Иономер (сополимер этиле-
этилена с метакриловой к-той)
Сополимер этилена с ви-
нилацетатом
Полипропилен
неориентированный
двухосно ориентиро
ванный
Полистирол двухосно ориен
тированный
Триацетат целлюлозы . . .
Сополимер винилхлорида
с винилацетатом
жесткий
мягкий
Сополимер винилиденхло-
рида с винилхлоридом . .
Поливиниловый спирт . .
Поливинилфторид ....
Политетрафторэтилен .
Политрифторхлорэтилен
Полиамид-6
Полиэтилентерефталат .
Поликарбонат
Гидрохлорид
на
полиизопре-
-н-
от —
ДО +
+4-
-t-
t
от -+-
ДО +
4~f
F0cC)
(80 °C)
от — до -f-
OT — ДО +
от ++
до
<0,01
<0,01
0
о
о
1-2
1 ,5-2,5 (тол
щи на 2 5 мкм
45-115
2,5-7,5
5-9
0,4
0,01
0,005
<0, 005
0,04-0,06
2,4-4,5
80
<0,5
0,005
0
9,5
0,8
0,3 5
++
++
т + +
до
от- до +
от— до +
от— до +
+
+
+
+
от— до-f
+
_
+
+
+
++•
+4-
4-+
4-4-
от-f до—
>т ± до +4-
-
8 0 — 90
104
120
66-93
66-93
50-80
60-90
13 0
120
60-90
60-70
плохая
130 —
1 50
140
80-95
15 0 —
200
плохая
66-93
66-93
(сухой)
150
(влаж-
(влажный)
хоро-
хорошая
105-
120
260
15 0-
2 0 0
9 0-
^ о о
»_ и v
150
130
80-95
-57
-5 7
-46
-46
-46
-57
-18
-57
80
очень
хорошая
-18
-50
от —56 до
-70
хорошая
-18
-73
-90
-2 00
-70
-60
— 100
-30
медленно
горит
то же
медленно
горит
до с а мо зату-
затухания
медленно
горит
то же
горюч
медленно
горит
медленно
горит
до самозату-
самозатухания
то же
медленно
горит
то же
медленно
горит
до самозату-
самозатухания
медленно
горит
до самозату-
самозатухания
самозатухает
медленно
горит
медленно
горит
до самозату-
самозатухания
негорюч
то же
самозатухает
медленно
горит
до самозату-
самозатухания
самозатухает
* Условные обозначения: +4- очень хорошая; + хорошая;
может быть нестойким.
± умеренная; — плохая; — — очень плохая. ** Лаковое покрытие
пленки — 2,4%, полиамидные пленки — 0,1%. На до-
долю остальных П. п. приходилось ок. 10% общего
объема пленочной продукции.
Лит.: Козлов П. В., Брагинский Г. И., Хи-
Химия и технология полимерных пленок, М., 1965; Пап-
ков С, П., Физико-химические основы переработки раство-
растворов полимеров, М., 1971; ВоюцкийС. С, ШтархБ.В.,
Физико-химия процессов образования пленок из дисперсий
полимеров, М., 1954; Т а к а х а с и Г., Пленки из полиме-
полимеров, пер. с япон., Л., 1971; Гуль В. Е., Полимерные пле-
пленочные материалы, М., 1972. В. Е. Гуль, П. В. Козлов.
ПЛЕНКООБРАЗУЮЩИЕ ВЕЩЕСТВА, плен
к о о б р а з о в а т е л и, пленкообразую-
пленкообразующие (film-forming compounds, film bi Id end e Stoffe,
matieres filmogenes) — основные компоненты любых
лакокрасочных материалов, придающие этим материа-
материалам способность к образованию пленки при нанесении
на твердую подложку. Основные тины П. в. представ-
представлены в таблице. Наибольшее значение имеют синтетич.
П. в., на основе к-рых получают покрытия с более
653
ПЛЕНКООБРАЗУЮЩИЕ ВЕЩЕСТВА
654
стабильными и разнообразными характеристиками, чем
на основе природных П. в. Кроме того, синтетич.
П. в. могут придавать покрытиям нек-рые специфич.
¦свойства, напр, термо- и химстойкость, отсутствующие
у покрытий из природных П. в. Поэтому во многих
странах, в том числе и в СССР, синтетич. П. в. почти
полностью заменили природные смолы. Растительные
масла также постепенно утрачивают роль самостоятель-
самостоятельных П. в.; их используют преимущественно для син-
синтеза алкидпых смол, эпоксиэфиров и др., а также как
пластифицирующие добавки к синтетпч. П. в. (см.
Масла растительные).
Наряду с органическими возрастающее значение
приобретают элементоорганич. П. в., в частности
кремнийорганические полимеры (см. Кремнийоргани-
ческие лаки и эмали). Из неорганических веществ не-
некоторое применение находит жидкое стекло (см. Сили-
Силикатные краски).
Большинство П. в.— реакционноспособные олиго-
меры разветвленного или линейного строения (ал-
кидные, феноло-формальдегидные, полиэфирные и эпок-
эпоксидные смолы, нек-рые сополимеры акрилатов и др.).
В качестве П. в. используют также сравнительно
низкомолекулярные полимеры, обычно не содержащие
реакционноспособных групп (нитроцеллюлозу, термо-
термопластичные полиакрилаты, перхлорвиниловые смолы
и др. хлорсодержащие полимеры).
Ок. 90% П. в. используют в виде р-ров в летучих
органич. растворителях (см. Растворители лакокрасоч-
лакокрасочных материалов). Для таких П. в. справедливо эмпи-
рич. правило — «подобное растворяется в подобном».
Напр., полярные гидроксилсодержащие пленкообразу-
пленкообразующие обычно хорошо растворимы в спирте, неполяр-
неполярные — в неполярных углеводородах. Нек-рые П. в.
применяют без растворителей (напр., высыхающие
растительные масла), в виде водных р-ров (напр.,
водорастворимые синтетич. смолы) или водных дис-
дисперсий (напр., иоливинилацетат, полиакрилаты).
Функции П. в. в порошковых красках выполняют
высокомолекулярные полимеры, напр, поливинилбути-
раль. Нанесенные на твердую поверхность мономеры
(стирол, акрилаты) также могут образовывать пленки
под действием облучения мощным пучком электронов
{см. Лакокрасочные покрытия).
В зависимости от характера процессов, протекаю-
протекающих при пленкообразовании, различают П. в. двух
типов: 1) непревращаемые (неотверждаемые, термопла-
термопластичные, обратимые); 2) превращаемые (отверждаемые,
термореактивные, необратимые). Первые не претер-
претерпевают при высыхании химич. превращений и образуют
пленку в результате физич. процессов: испарения орга-
органич. растворителя, воды и др. Превращаемые П. в.
содержат в макромолекуле функциональные группы
(гидроксильные, карбоксильные, эпоксидные, амино-
аминогруппы, двойные связи) и образуют пленку в резуль-
результате химич. процессов — поликонденсации или поли-
полимеризации.
П. в. должны обладать след. общими свойствами:
1) хорошо смачивать защищаемую поверхность и легко
и равномерно распределяться на ней; 2) не содержать
водорастворимых веществ, к-рые могут вызывать кор-
коррозию металла; 3) высыхать (отверждаться) при ком-
комнатной или повышенной (80—200 СС) темп-ре за срав-
сравнительно короткое время (от нескольких мин до
1 суш). В пигментированных лакокрасочных материа-
материалах (красках, грунтовках, шпатлевках) П. в. должны
смачивать поверхность частиц пигментов и наполни-
наполнителей, адсорбироваться на этих частицах и служить
дисперсионной средой (связующим). После нанесения
на поверхность П. в. должны прочно удерживать дис-
диспергированные частицы в формирующейся пленке
силами адгезии, действующими на границе раздела
пигмент — пленкообразующее.
Пленки неиигментированных П. в. прозрачны; они
м. б. бесцветными или иметь окраску от желтой до
коричневой (исключение — черные и непрозрачные
пленки битумов—см. Битумные лаки). Темная окраска
сильно ограничивает применение П. в. Большинство
П. в. образует блестящие пленки; блеск нек-рых пле-
пленок м. б. усилен полированием.
Для достижения необходимого комплекса свойств
лаков, пигментированных материалов и покрытий на
их основе широко используют совмещение в лакокра-
лакокрасочном материале двух и более П. в. При таком совме-
совмещении может происходить не только смешение, но и
химич. взаимодействие различных П. в. Вводя в макро-
макромолекулу П. в. полярные функциональные группы
(гидроксильные, метилольные, карбоксильные) или,
наоборот, блокируя эти группы алкильными или ариль-
ными радикалами, можно регулировать растворимость,
а также совместимость различных П. в.
Широкое использование в качестве П. в. реакцион-
реакционноспособных олигомеров обусловлено не только хоро-
хорошим качеством пленок, но также и растворимостью
олигомеров во многих растворителях. Кроме того,
олигомеры, вследствие их сравнительно небольшой
мол. массы, образуют маловязкие конц. р-ры, к-рые
после разбавления до рабочей вязкости могут содер-
содержать 30—50% П. в. (но массе). Благодаря этому при
нанесении одного слоя лакокрасочного материала воз-
возможно образование пленки толщиной 15—35 мкм.
При использовании р-ров олигомеров в реакционно-
способных растворителях (напр., полиалкиленгликоль-
малеинатов в стироле или в диметакриловом эфире
триэтиленгликоля) П. в. сополимеризуется с раствори-
растворителем (см. Полиэфирные лаки и эмали). При однократ-
однократном нанесении таких р-ров можно получить пленку
толщиной до 500 мкм, исключить потери растворите-
растворителей и улучшить условия труда.
Наряду с природными водорастворимыми П. в.—
казеином, декстрином и др. (см. Клеевые краски),
к-рые издавна используют при изготовлении строи-
строительных и художественных красок, все более широкое
применение находят водорастворимые синтетич. оли-
олигомеры (алкидные, феноло- и амино-формальдегидные
смолы и др.). На основе этих П. в. изготовляют лако-
лакокрасочные материалы, не содержащие токсичных и
горючих растворителей и пригодные для нанесения
высокопроизводительными методами, в частности элек-
электроосаждением (см. В одоразбавляемые грунтовки и
эмали, Лакокрасочные покрытия).
При изготовлении лакокрасочных материалов на
основе полимеров исходят из необходимости получения
р-ра рабочей вязкости с концентрацией П. в. не менее
10—15% (но массе). Этим определяется выбор опти-
оптимальной мол. массы полимера, т. к. применение поли-
полимеров со слишком низкой мол. массой может не обеспе-
обеспечить требуемой прочности пленки; использование же
менее конц. р-ров практический нецелесообразно из-за
большого расхода растворителя и образования при
однократном нанесении очень тонких пленок. Поли-
Полимеры обычно хуже растворяются, чем олигомеры, и
выбор растворителей для них затруднен. В отличие
от олигомеров, полимеры образуют непревращаемые
(обратимые) пленки, обладающие низкой стойкостью
к действию растворителей и повышенных темп-р. До-
Достоинство таких П. в.— способность высыхать при
комнатной или сравнительно невысокой F0—80 "С)
теми-ре в короткие сроки. Применение водных диспер-
дисперсий (латексов) высокомолекулярных полимеров (поли-
(поливинил ацетата, сополимера стирола с бутадиеном и
др.) позволяет не только исключить органич. раство-
растворители, но и получать системы с содержанием сухого
остатка 50—60% (см. Эмульсионные краски).
Важнейшие характеристики пленок — адгезию к
подложке, твердость, прочность при растяжении и из-
655
ПЛЕНКООБРАЗУЮЩИЕ ВЕЩЕСТВА
Наиболее распространенные пленкообразующие вещества
656
Пленкообразующее
Растворитель
Условия высыхания
темп-ра,
°С
время, ч
Способность
к образованию
необратимой
пленки
Адгезия
пленки к
металлу
Условия эксплуатации
пленок
Природные материалы и продукты
Растительные и животные клеи
Высыхающие растительные
масла
Природные смолы (копалы, ян-
янтарь, эфиры канифоли) в со-
сочетании с растительными
маслами
Битумы, гудроны, пеки . . . .
Битумы и гудроны в сочета-
сочетании с растительными масла-
маслами
Нитроцеллюлоза
Хлоркаучук .
Циклокаучук
Алкидные смолы . .
Полиэфирные смолы
Мочевино-формал ьдегидные
смолы в сочетании с алкид-
ными смолами и др. поли-
полиэфирами
Меламино-формал ьдегидные
смолы в сочетании с алкид-
ными смолами
Феноло-формальдегидные смо-
смолы (резольные)
Феноло-формальдегидные смо-
смолы в сочетании с раститель-
растительными маслами
Эпоксидные смолы
Эпоксиэфиры
Фу рано вые смолы .
Полиорганосилоксаны
Перхлорвиниловые смолы . . .
Сополимеры винилхлорида с
винилиденхлоридом
Сополимеры винилхлорида с
винилацетатом
Акриловые сополимеры термо-
термопластичные
Акриловые сополимеры термо-
термореактивные
Поливинилацетали
Поливинилацеталн в сочетании
с меламино-формальдегидны-
ми смолами
Вода
Скипидар, уайт-спирит
Сольвент, ксилол
То же
Ацетон, сложные эфиры
Ароматич. углеводоро-
углеводороды, сложные эфиры, ке-
тоны
Алифатич. и ароматич.
углеводороды
18-20
18-20
18—20
130-180
18-20
120-200
18-20
18 — 20
18-20
24
0,5 — 3
3
0,5-1
1
2-3
2-3
их переработки
Не способны
Способны
То же
Не способны
Способны
Не способна
Не способен
Способен
Синтетические продукты
Сольвент, ксилол, уайт-
спирит и их смеси
Стирол, акрилаты
Смесь бутанола с кси-
ксилолом
То же
Спирт
Ароматич. углеводоро-
углеводороды, ацетон, сложные
эфиры, бутанол
Кетоны, сложные эфиры,
этилцеллозольв
Ароматич. углеводо-
углеводороды
Смесь ацетона со спир-
спиртом A:1)
Ароматич. углеводороды
и их смеси с эфирами,
спиртами, кетонами
Сложные эфиры, кетоны
То же
Кетоны, сложные эфи-
эфиры, ароматич. углево-
углеводороды
Бутанол
Спирт
Смесь этилового спирта
с циклогексаноном или
этилцеллозольвом
18-20
70-2 00
18-20
6 0-70
18-20
100-130
90-130
18-20
18-20
150—180
150-190
18 — 20
150
18-20
150-200
18-20
60
18-20
60
18-20
60
18-20
80 — 100
18-20
120
24-48
1-6
24
0,5-1
2
1—1,5
0,5 — 1 ,5
24
24
1-2
1-3
1
4 — 5 (с по-
следую-
следующей вы-
выдержкой
7 — 8 сут)
1-2
1 ,5-3
1
1,5-3
1
1,5-3
1
1-2
4
3-5
1-4
Способны
То же
То же*
Способны
Способны**
Способны
Не способны
Не способны
То же
То же
Способны
Не способны
Способны
Хорошая
То же
Плохая
Удовлет-
воритель-
ворительная
Хорошая
Хорошая
Удовлет-
воритель-
ворительная
То же
Хорошая
Удовлет-
воритель-
ворительная
Плохая
Удовлет-
воритель-
ворительная
Хорошая
То же
То же
То же
То же
Внутри помещения
Внутри помещения и на
открытом воздухе
То же
Внутри помещения, б
воде, к-тах, щелочах
Внутри помещения и на
открытом воздухе
Внутри помещения, на
открытом воздухе, в
бензине
Внутри помещения, на
открытом воздухе, в
воде, к-тах, щелочах
При темп-pax до 200 СС,
в воде, к-тах, щело-
щелочах
Внутри помещения к
на открытом воздухе
Внутри помещения
Внутри помещения
На открытом воздухе
Внутри помещения, при:
темп-рах 16 0-170 'С,
в к-тах
На открытом воздухе
(в грунтовках)
Внутри помещения, при:
темп-рах 16 0—170 С,
в к-тах, щелочах
На открытом воздухе
(в грунтовках)и внут-
внутри помещения
При темп-рах 150—
160 СС, в к-тах, ще-
щелочах
При темп-рах от —50
до 200 — 250 °С, дей-
действии солнечной ра-
радиации, тропич. влаж-
влажности
Внутри помещения, на
открытом воздухе, не-
непродолжительно в
к-тах и щелочах
В к-тах и щелочах
Внутри помещения, на
открытом воздухе, в
воде
Внутри помещения, на
открытом воздухе
То же
В бензине, маслах
То же
657
ПНЕВМОФОРМОВАНИЕ
658
Продолжение табл.
Пленкообразующее
Полиуретаны
Полимеры дивинилацетилена
Поливинилацетат
Сополимеры стирола с бута-
бутадиеном
Фторсодержащие сополимеры
(растворимые)
Растворитель
Кетоны, сложные
эфиры
Ксилол
Вода***
Вода***
Кетоны, сложные
эфиры
Условия высыхания
темп-ра,
СС
18-20
60-120
18-20
18-20
18-20
150-200
время, ч
9-24
1 ,5 — 3
12
2
1
Способность
к образованию
необратимой
пленки
То же
То же
Не способен
То же
Не способны
Адгезия
пленки к
металлу
То же
То же
Удовлет-
воритель-
ворительная
Условия эксплуатации
пленок
Внутри помещения, на
открытом воздухе, при
интенсивном истира-
истирании
В воде
Внутри помещения, на
открытом воздухе
Внутри помещения
Внутри помещения, на
открытом воздухе, при
темп-pax до 200 °С,
в к-тах, щелочах, при
высокой влажности
* Материалы холодной сушки отверждаются в присутствии кислотного катализатора. ** Отверждаются в присутствии отвер-
дителя. *** Дисперсионная среда.
пюе, влагонепроницаемость, термостойкость и др.—
можно варьировать, применяя П. в. соответствующего
химич. состава, или модификацией П. в. Так, поляр-
полярные группы в макромолекулах П. в. обусловливают
хорошую адгезию пленок к металлич. поверхностям.
Поэтому пленки, получаемые из реакционноспособ-
ных П. в., обычно превосходят по адгезии пленки
на основе нереакционноспособных П. в. Адгезию по-
последних можно повысить, добавляя в лакокрасочный
материал П. в. с хорошей адгезией (напр., алкидные
смолы в р-р нитроцеллюлозы), а также частичным омы-
омылением П. в. (напр., сополимера винилхлорида с ви-
нилацетатом) или применением сополимеров, в к-рых
один из мономеров содержит карбоксильные или др.
полярные группы.
Повышение гибкости пленок достигается внутренним
пластифицированием П. в. в процессе их синтеза
(напр., применением для поликонденсации моно- или
бифункциональных соединений с длинной алкильной,
этиленоксидной или пропиленоксидной цепью; вы-
выбором при сополимеризации соответствующих моно-
мономеров и их соотношений) или добавлением пластифи-
пластификаторов — эфиров фталевой, себациновой, фосфорной
к-т, хлорпарафина, касторового масла, невысыхаю-
невысыхающих алкидных смол и др.
Присутствие в макромолекулах П. в. бензольных,
триазиновых или др. циклов способствует повышению
твердости и термостойкости пленок, но одновременно
приводит к снижению их гибкости. Такие же законо-
закономерности наблюдаются и при повышении функцио-
функциональности П. в., приводящей к увеличению частоты
сетки в отвержденыой пленке. С увеличением гидро-
фильности П. в. (напр., при повышении содержания
гидроксильных групп в их макромолекулах) и с умень-
уменьшением частоты сетки повышается влагопроницае-
мость и ухудшаются электроизоляционные свойства
пленок. Атомы Si, A1 и др., присутствующие в ма-
макромолекулах элементоорганич. П. в., обусловливают
термостойкость пленок; хлор- и фосфорсодержащие
П. в. образуют огнестойкие, фторсодержащне П. в.—
термо- и химстойкие покрытия.
Лит.: Дринберг А. Я., Технология пленкообразую-
пленкообразующих веществ, 2 изд., Л., 1955; Пэйн Г. Ф., Технология орга-
органических покрытий, т. 1, Л., 1959; Ш а м п е т ь е Г., Р а-
б а т э Г., Химия лаков, красок и пигментов, пер. с франц
т. 1, М., 1960; Верхоланцев В. В., Водные краски
на основе синтетических полимеров, Л., 1968; Соло-
Соломон Д. Г., Химия органических пленкообразователей пер
с англ., М., 1971; Г о л ь д б е р г М. М., Материалы для
лакокрасочных покрытий, М., 1972; Полимеризационные плен-
кообразователи, под ред. В. И. Елисеевой, М., 1971.
М. М. Гольдберг.
ПНЕВМОФОРМОВАНИЕ, формование сжа-
сжатым воздухом (pneumoforming, Pneuma-
tikverformung, i'ormage pneumatique) — способ формова-
формования листовых (иногда пленочных) термопластов. При
П. заготовку листового материала закрепляют по кон-
контуру формы и нагревают до темп-ры, при к-рой поли-
полимер находится в высокоэластическом состоянии. После
этого под действием подогретого и сжатого воздуха лист
оформляется в изделие (рис. 1). Для облегчения фор-
формования, особенно при переработке толстых листов,
П. можно комбинировать с
механич. формованием (напр.,
в прессах) или с вакуумформо-
ванием, т. е. создавать в поло-
полости формы разрежение.
Машины для изготовле-
изготовления изделий методом П. клас-
классифицируют: 1) по степени ав-
автоматизации — на машины с
ручным управлением, маши-
машины-автоматы и машины-полу-
машины-полуавтоматы (автоматы использу-
используют гл. обр. для массового
производства небольших изде-
изделий); 2) по числу рабочих по-
позиций — на однопозиционные
(все технологич. операции осу-
осуществляются последовательно
на одном и том же участке аг-
агрегата) и многопозицпонные
(производимые машиной техно-
технологич. операции осуществля-
осуществляются одновременно на раз-
Рис. 1. Схема негативного формова-
формования: а — нагрев; б — формование
(вытяжка); в — выталкивание; 1 —
матрица; 2 — зажимная рама; з —
нагреватель; 4 —пневмокамера; 5— u
заготовка; б—каналы для воздуха. в
воздух
Воздух
личных участках агрегата). Машины с числом пози-
позиций более трех подразделяют на роторные, или ка-
карусельные (позиции размещены на вращающемся столе),
и ленточные (позиции размещены на транспортере).
Оборудование для П. имеет след. общие элементы:
нагревательные и зажимные устройства, систему смы-
смыкания, пневмосистему, оформляющий инструмент и
станину. Для разогрева заготовок применяют тепло-
радиационные нагреватели и термокамеры, в к-рых
теплоносителем служит горячий воздух. С помощью
теплорадиационных нагревателей (стационарных или
подвижных) может осуществляться одно- или двух-
двухсторонний разогрев заготовки. Рабочими органами на-
нагревателей обычно являются элементы сопротивления
или кварцевые лампы. Широко применяют нагреватели
со сплошными, фигурными и сетчатыми экранами.
€59
ПНЕВМОФОРМОВАНИЕ
G60
В большинстве конструкций машин предусматривает-
предусматривается регулировка расстояния между нагревателем и
заготовкой; часто регулируется также темн-ра в от-
отдельных зонах нагревательных элементов.
Зажимное устройство (рама) служит для прочного
закрепления заготовки, а часто и для герметизации
формовочной камеры и пневмокамеры. Зажим листа
в раме производится с помощью запорных устройств
с ручным или пневматич. приводом. Конструкция этих
устройств должна обеспечивать равномерное распре-
распределение усилий по всей поверхности стыка.
Система смыкания необходима для надежного
уплотнения пневмокамеры, зажимного устройства с лис-
листом и формовочной камерой при оформлении изделия.
Для создания необходимого усилия используют прес-
прессовое устройство с механич., пневматич. или гидрав-
лич. приводом.
Пневмосистема состоит из компрессора, ресивера,
редукторов давления, клапанов и трубопроводов и
служит для подачи сжатого воздуха при формовании
заготовки. Сжатый воздух часто используют также для
привода органов машины.
К оформляющему инструменту относятся формы
(матрицы, пуансоны), специальные съемные устройства
для создания поднутрений, диафрагмы (тонкие пласти-
пластины из резины на основе натурального каучука). Диа-
Диафрагмы применяют обычно при изготовлении крупно-
крупногабаритных изделий для уменьшения их разнотолщин-
ности, а также для лучшего оформления поверхности
изделия, не входящей в непосредственный контакт
с матрицей или пуансоном. Избыточное давление при
формовании с применением диафрагмы создают обычно
с помощью воздушно-паровой смеси, к-рая является
одновременно и теплоносителем при разогреве листовой
заготовки. Основной недостаток диафрагм — недолго-
недолговечность. В ряде случаев (как правило, при формова-
формовании упаковки) в качестве оформляющего инструмента
используют сам объект упаковки — прототип. При оп-
определении размеров оформляющего инструмента учиты-
учитывают возможность усадки изделия при его охлаждении.
Выбор материала формы зависит от требуемой
чистоты поверхности изделия и срока эксплуатации
формы. Материалом для форм, рассчитанных на корот-
короткие сроки службы, служит дерево твердых пород.
Для улучшения качества рабочих поверхностей такие
формы часто покрывают эпоксидными смолами. Формы
со средними сроками службы отливают из фенольных
или эпоксидных смол, армированных стеклотканью или
металлом. Для длительных сроков службы применимы
металлнч. формы, гл. обр. из алюминиевых и магние-
магниевых сплавов. В этих случаях применяют также формы
из бетона или гипса, покрытые металлом, нанесенньш
гальванич. способом. Формы, изготовленные из мате-
материалов с высокой теплопроводностью, имеют каналы
для циркуляции хладагента, чаще всего воды.
При получении небольших партий изделий приме-
применяют, как правило, необогреваемые пуансоны, изготов-
изготовляемые из материала с низкой теплопроводностью
(дерево, пластмасса). Однако в результате соприкосно-
соприкосновения холодного пуансона с листом термопласта
темн-ра соответствующих участков заготовки все же
снижается. Следствием этого м. б. появление пятен
на изделии, а также несколько большая толщина его
дна по сравнению с толщиной стенок. При использо-
использовании металлич. обогреваемого пуансона материал
при вытяжке проскальзывает по пуансону и распреде-
распределяется по его поверхности более равномерно. Теми-ра
пуансона должна быть ниже темп-ры, при к-рой лист
термопласта начинает прилипать к нему, т. к. в про-
противном случае резко ухудшается взаимное проскальзы-
проскальзывание пуансона и формуемой заготовки.
Станины машин для П. состоят обычно из легкой,
малонагруженной металлич. конструкции с более
массивной прессовой частью системы смыкания, т. к.
усилие смыкания составляет в отдельных случаях мил-
миллионы н (сотни тс).
Пневмоформовочные агрегаты снабжают устройства-
устройствами для вырубки изделия, пробивки отверстий, отгиба
борта, заполнения отформованной тары сыпучим или
жидким продуктом и ее закупорки, предварительной
двухосной вытяжки заготовки. Зачистку мелких дета-
деталей можно производить на ленточных или дисковых
зачистных станках. Для крупных деталей, обработка
к-рых на стационарных станках неудобна, применяют
ручные шлифовальные машины с пневмо- или электро-
электроприводом. При обработке изделий со сложным профи-
профилем линии зачистки применяют ручные методы, чаще
всего опиливание.
Листовой материал (полуфабрикат) толщиной до
1,5—2 мм разрезают на заготовки на гильотинных
ножницах; хрупкий материал такой же толщины (напр.,
нолиметилметакрилат) разрезают на строгальном стан-
станке или специальным резаком вручную. Листы толщи-
толщиной 3—10 мм распиливают на дисковых или ленточных
пилах; последние применяют также для распиливания
более толстых листов и получения фигурных загото-
заготовок. При раскрое листа дают припуск A0—40% от
полезной площади заготовки), необходимый для креп-
крепления заготовки в зажимной раме пневмоформовочной
машины и обеспечения зазора между этой рамой и
наружным контуром оформляющего инструмента (за-
(зазор необходим для более равномерного распределения
материала заготовки по стенкам изделия).
В ряде случаев для получения изделий с улучшен-
улучшенными физико-механпч. свойствами заготовку подвер-
подвергают предварительной двухосной ориентации. Эта
операция может осуществляться как на специальных
машинах, так и с помощью приспособлений, входящих
в состав формующих агрегатов.
Основными методами и н е в м о ф о р-
м о в а н и я являются негативное, позитивное и сво-
свободное. Наибольшее практич. значение имеют различ-
различные комбинации этих методов (напр., негативно-по-
негативно-позитивное), т. к. при использовании каждого из них
в отдельности не удается получить высококачественное
изделие с большой глубиной вытяжки.
При негативном формовании, или формовании в мат-
матрицу (см. рис. 1), заготовку 5 укрепляют в зажимной
раме 2 и нагревают. Затем над заготовкой устанавли-
устанавливают пневмокамеру 4 и создают избыточное давление,
под действием к-рого лист термопласта принимает
форму будущего изделия. Для фиксирования получен-
полученной формы изделие охлаждают. Негативное П. позво-
позволяет получать изделия, наружная поверхность к-рых
воспроизводит форму, размер и рисунок внутренней
поверхности матрицы.
При позитивном формовании вместо матрицы в фор-
формовочную камеру устанавливают выпуклый оформляю-
оформляющий пуансон, форма, размер и рисунок к-рого воспро-
воспроизводятся на внутренней поверхности изделия. Фор-
Формование на пуансонах с выпукло-вогнутой поверх-
поверхностью наз. негативно-позитивным.
При свободном формовании заготовка, предваритель-
предварительно нагретая и укрепленная над так наз. проймой
(зажимной рамой, имеющей специальную прорезь),
формуется, не входя в контакт ни с оформляющим
инструментом, ни с пневмокамерой. При достижении
необходимой глубины вытяжки термопласта давление
воздуха уменьшают и поддерживают постоянным до
полного остывания изделия. В этом случае формооб-
формообразование происходит за счет равновесия между на-
напряжением, возникающим в формуемом термопласте, и
избыточным давлением воздуха, приложенным к заго-
заготовке. Свободное формование применяют, как прави-
правило, для получения изделий с высокими оптическими
свойствами.
661
ПНЕВМОФОРМОВАНИЕ
662
В СССР разработай и получил значительное распро-
распространение метод механопневмоформования, к-рый по-
позволяет исключить вспомогательные и отделочные
операции и получить полностью готовое изделие за
один рабочий цикл в одном агрегате. Такие агрегаты
создаются на базе стандартных гидравлич. прессов для
пластмасс [усилие 630, 1000, 1600 и 2500 кн F3, 100,
160, 250 тс)]. Агрегаты комплектуют электропе-
электропечью для разогрева заготовок и устройством, обеспе-
обеспечивающим автоматическую подачу листового материа-
материала из штабеля в печь и из печи к формующему
инструменту.
Совмещение основных и вспомогательных операций
при механопневмоформовании достигается примене-
применением универсальной нневмокамеры, вариант к-рой
показан на рис. 2. Камера состоит из двух частей:
в одной из них м. б. закреплен пуансон 1, а вторая
(неподвижная) приспособлена для установки в ней
матрицы 2. По периметру камеры размещен пресс-
кант 3, с помощью к-рого при смыкании верхней и
нижней частей камеры вырубается и, при необходимо-
необходимости, отгибается борт изделия. В универсальной ка-
Рис. 2. Схема универсальной
камеры для мэханопневмо-
формования: а — предвари-
предварительная механич. вытяжка;
б—вырубка борта изделия,
приварка бобышки и пневма-
тич. раздув; 1 — пуансон; 2 — матрица; 3 — пресс-кант; 4 —
заготовка; 5 — бобышка; б — канал для подачи воздуха при
пневматич. раздуве; 7 — каналы для выхода воздуха; 8 —
канал для подачи воздуха при выталкивании готового из-
изделия.
мере можно осуществить след. технологич. операции:
1) предварительную механич. вытяжку заготовки 4
пуансоном 1 (рис. 2,а); 2) приварку заранее отпрессо-
отпрессованной бобышки 5, изготовленной из того же мате-
материала, что и изделие (при использовании др. методов
П. эта операция невозможна из-за недостаточного
усилия), вырубку борта изделия и пневматич. раздув
термопласта воздухом, к-рый подается по каналу 6
(рис. 2,6); 3) выталкивание готового изделия из формы
с помощью воздуха, к-рый подается но каналу 8 в
нижнюю часть камеры. Для получения изделий с пер-
перфорацией камеру снабжают специальным подвижным
плунжером с пробойниками.
К основным технологическим па-
параметрам метода П. относятся: 1) темн-ра заго-
заготовки, 2) темп-pa оформляющего инструменту; 3) пе-
перепад давления; 4) скорость вытяжки (формования)
материала.
Темп-pa заготовки (таблица) должна соответство-
соответствовать области ее высокоэластич. состояния. Правиль-
Правильный выбор темп-ры внутри этой области с помощью
термомеханич. кривой данного материала (см. Термо-
механическое исследование) позволяет регулировать в
определенных пределах механич. свойства и разнотол-
щинность формуемого изделия. (Для регулирования
этих свойств изделия материал заготовки часто под-
подвергают механич. воздействиям, напр, предваритель-
предварительной вытяжке, а также термообработке).
Темп-ры заготовки и оформляющего инструмента при
переработке различных термопластов методом нневмоформования
Полимер
Полиметилметакрилат
Полистирол ударопрочный . . .
Поливинилхлорид
Полиэтилен высокой плотности .
Полиэтилен низкой плотности
Полипропилен
Полиформальдегид
Полиэтилентерефталат
Поликапроамид
Поликарбонат ^на основе бисфе-
нола А)
Темп-ра
заготовки,
°С
120-200
110 — 150
100-160
12 0 — 135
90-135
15 0 — 200
185-200
150-180
210-220
225-245
Темп-ра
оформляюще-
оформляющего инстру-
инструмента, °С
40-70
50 — 70
35-45
65-90
50-80
65-75
75 — 95
Темп-ра оформляющего инструмента должна быть
несколько ниже (— на 10—30 СС) темп-ры стеклования
перерабатываемого термопласта. Это необходимо для
того, чтобы зафиксировать форму изделия; вместе с
тем материал не должен быть переохлажден до оконча-
окончания процесса оформления.
Перепад давления определяется механич. свойствами
материала заготовки, ее толщиной и геометрией фор-
формуемого изделия. При недостаточном перепаде давле-
давления может произойти неполное оформление детали,
при слишком большом — возрастает энергоемкость
процесса и металлоемкость оборудования, увеличи-
увеличивается опасность механич. разрыва заготовки. В
пром-сти применяют избыточные давления в пределах
50—2500 кн/м2 @,5 — 25 кгс/см2).
Скорость вытяжки материала (обычно 100 —
200 мм/сек) зависит от свойств термопласта, геометрии
заготовки и перепада давления. Минимальная скорость
вытяжки должна быть такой, чтобы материал во время
формования не успел остыть и находился в высоко-
высокоэластич. состоянии. При выборе максимальной скорости
вытяжки необходимо учитывать предельное значение
скорости деформации материала, при к-рой может
наступить его разрыв. Скорость вытяжки определяет
значение остаточных напряжений в готовом изделии
и, следовательно, его склонность к растрескиванию и
короблению при эксплуатации.
Значительное влияние на технологич. параметры
П. оказывают глубина вытяжки и предыстория заго-
заготовки (способ получения, наличие или отсутствие
предварительной ориентации или термообработки).
Глубина вытяжки (отношение высоты изделия к его
среднему условному диаметру) косвенно определяет
значение деформации материала в процессе оформления
заготовки в изделие. Заготовки, в к-рых сохраняются
остаточные напряжения, требуют больших усилий для
закрепления в зажимном устройстве, склонны к обра-
образованию волнистой поверхности при нагреве и нерав-
неравномерной вытяжке.
Свойства изделия в значительной степени зависят от
режима его охлаждения. Для увеличения производи-
производительности формующего оборудования необходимо по-
повышать скорость охлаждения, однако при жестком ре-
режиме охлаждения в изделии сохраняются неравномер-
неравномерно распределенные остаточные напряжения, обуслов-
обусловливающие малую формоустойчивость изделия ири экс-
эксплуатации. Охлаждение с небольшими скоростями при-
приводит к понижению производительности машин, однако
в этом случае напряжения, возникающие в термопласте
в процессе его деформации, успевают частично отре-
лаксировать, их распределение становится равномер-
равномернее, и изделие получается более формоустойчивым.
Свойства изделий, получаемых методами пневмо- и
вакуумформования, идентичны. К преимуществам ва-
куумформования следует отнести меньшую стоимость
и металлоемкость оборудования. Однако при исполь-
использовании этого метода избыточное давление на заготов-
663
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
664
ку не может превышать 0,1 Мн/м2 A кгс/см2). При
большой толщине заготовки такое давление оказы-
оказывается недостаточным для полного оформления изде-
изделия. В этих случаях необходимо применять метод П.,
позволяющий варьировать избыточное давление в ши-
широких пределах.
Все изделия, полученные методом П., имеют разно-
толщинность. При использовании этого метода прак-
практически невозможно получить деталь с заданным «за-
«законом» изменения толщины стенки, трудно получить
и идеально равнотолщинную деталь. Кроме того,
в изделиях, полученных методом П., сохраняются вы-
высокие остаточные напряжения. Это обусловливает не-
недостаточную стабильность изделий при их эксплуата-
эксплуатации в условиях повышенных темп-р. Применение мето-
метода литья под давлением позволяет получать изделия
с заданным «законом» изменения толщины стенки и
с меньшими остаточными напряжениями. Однако
получение тонкостенных изделий литьем под давлени-
давлением весьма затруднено, в то время как метод П. дает
возможность получать изделия с самой различной тол-
толщиной стенки. Литье под давлением крупногабарит-
крупногабаритных изделий связано с конструкционно-технологич.
трудностями, применяемые машины громоздки (см.
Литьевые машины), требуют использования узлов
смыкания большой мощности; цикл изготовления изде-
изделий очень длительный. Прямые капитальные затраты
при организации производства крупногабаритных из-
изделий методом литья под давлением в 4—5 раз больше,
чем при использовании метода П. Благодаря малой
стоимости оснастки П. предпочтительнее литья иод
давлением при производстве малых партий изделий.
Методом П. получают обширный ассортимент изде-
изделий технич. и бытового назначения: детали для остек-
остекления негерметизированных кабин самолетов, изделия
санитарно-технич. назначения (ванны, раковины и
др.), контейнеры, различную упаковку, рельефные де-
декоративные панели, часовые стекла, корпуса и др.
детали холодильников и т. д.
Метод П. получил значительное распространение
как в СССР, так и за рубежом. Напр., за период
1960—70 объем производства изделий этим методом
в США возрос с 48 до 500 тыс. т.
Лит.: Бернхардт Э. [сост.]. Переработка термопла-
термопластичных материалов, пер. с англ., М., 1965; Стрель-
Стрельцов К. Н., Пневматическая переработка термопластов, Л.,
1963; его же, Переработка листовых термопластов методом
механопневмоформования. Производство и переработка пласт-
пластмасс, синтетических смол и стеклянных волокон, в. 7, 51 A968).
К. А. Салазкин, М. А. Шсрышев.
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА (sur-
(surface active substances, oberflachenaktive Stoffe, agents
tensio-actifs).
С о д е р ж а н и е:
Введение 66 3
Классификация 604
Молекулярное строение и получение ... ... 665
Ионогенные ПАВ . . . .* 66 5
Неионогенные ПАВ 6E6
Фторзамещенные ПАВ E 7 0
Высокомолекулярные ПАВ 670
Свойства 671
Применение 677
Введение
Поверхностно-активные вещества — вещества, ад-
адсорбирующиеся на поверхности раздела двух фаз
(тел) и образующие на ней слой повышенной концент-
концентрации (адсорбционный слой). При соответствующих
условиях поверхностно-активным м. б. любое вещество,
являющееся компонентом жидкого р-ра или газа (пара),
поскольку оно способно иод действием межмолекуляр-
межмолекулярных сил сгущаться на тех или иных межфазных по-
поверхностях. Однако в понятие «поверхностно-активные
вещества» (ПАВ) обычно вкладывают более узкий
смысл, относя его лишь к группе органич. соединений,
адсорбция к-рых из их р-ров даже очень малой кон-
концентрации (десятые и сотые доли %) приводит к рез-
резкому снижению поверхностного (межфазного) натяже-
натяжения на поверхности раздела р-ра с газом (паром), др.
жидкостью или твердым телом. [Термин «поверх-
«поверхностное натяжение» принято употреблять
по отношению к поверхности раздела конденсирован-
конденсированной фазы с газом (собственным паром), а термин
«м е ж ф а з н о е натяжение» — по отношению
к поверхности раздела двух конденсированных фаз].
Накопление и ориентация в адсорбционном слое
молекул или ионов ПАВ — следствие их д и ф и л ь-
н о с т и (двойственности свойств). Каждая молекула
типичных ПАВ имеет олеофильную, или липофиль-
ную, часть (один или несколько углеводородных
радикалов) и гидрофильную часть (одну или несколько
полярных групп). Олеофильная часть молекулы опре-
определяет ее тенденцию к переходу из полярной (напр.,
водной) среды в неполярную (напр., углеводородную),
а гидрофильные группы, наоборот, удерживают моле-
молекулу в полярной среде или, если молекула находится
в углеводородной фазе, определяет ее тенденцию к
переходу в полярную среду. Т. обр., поверхностная
активность ПАВ, растворенных в углеводородных жид-
жидкостях, обусловлена гидрофильными группами, а
растворенных в воде — олеофильными (гидрофобными)
радикалами.
Классификация
По типу гидрофильных групп ПАВ делят на ионные,
или ионогенные, и неионные, или неионогенные.
Ионогенные ПАВ диссоциируют в р-ре на
ионы, одни из к-рых обладают адсорбционной актив-
активностью, другие (противоионы) — адсорбционно не ак-
активны. Если адсорбционно активны анионы, ПАВ наз.
анионными, или анионоактивными, в противоположном
случае — катионными, или катионоактивными.
Нек-рые ПАВ содержат как кислотные, так и основ-
основные группы; такие ПАВ обладают амфотерными свой-
свойствами и в зависимости от рН среды проявляют себя
как анионоактивные или как катионоактивные. Их наз.
амфотерными, или а м ф о л и т н ы м и, ПАВ.
Неионогенные ПАВ не диссоциируют при
растворении на ионы; носителями гидрофилыюсти
в них обычно являются гидроксильные группы и поли-
гликолевые (полиоксиэтиленовые, или полиэтилен-
оксидные) цени различной длины.
Существуют также ПАВ, в к-рых наряду с неионо-
генными гидрофильными атомными группами присут-
присутствуют ионогенные.
В отдельный класс выделяют фторуглерод-
н ы е ПАВ (т. наз. ф т о р т е н з и д ы) — соеди-
соединения с полным или частичным замещением атомов
водорода в гидрофобных радикалах на атомы фтора.
Кр. того, как отдельную группу следует рассматривать
высокомолекулярные ПАВ — адсорбцион-
адсорбционно активные водорастворимые полимеры ионогенного
(полиэлектролиты) и неионогенного типов.
Все ПАВ можно разделить на две категории по типу
систем, образуемых ими при взаимодействии с раство-
растворителем (водой в наиболее практически важном слу-
случае). К одной категории относятся м и ц е л л о о б-
р а з у ю щ и е (полуколлоиды ы е, мы л о-
подобные) ПАВ, к другой — не образующие
мицелл. ПАВ первой категории в р-ре выше нек-рон
(определенной для каждого вещества) «критической»
концентрации образуют мицеллы, т. е. молекулярные
или ионные ассоциаты с числом молекул (ионов) от
нескольких десятков до нескольких сотен. Ниже
критической концентрации м и ц е л-
лообразования (К КМ) вещество находится
в истинно растворенном состоянии, а выше ККМ —
как в истинно растворенном, так и в мицеллярном.
G65
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
666
Мицеллы ПАВ находятся в обратимом термодинамич.
равновесии с молекулами; при разбавлении р-ра они
распадаются, а при увеличении концентрации вновь
возникают. Обычно такие р-ры обладают моющей
способностью. ПАВ второй категории не образуют
мицелл ни в р-рах, ни в адсорбционных слоях. При
любой концентрации они находятся в истинно рас-
растворенном состоянии.
Молекулярное строение и получение
Ионогенные ПАВ. Анионоактивные ве-
вещества составляют большую часть мирового про-
производства ПАВ. Промышленные ПАВ этого типа можно
разделить на след. основные группы: карбоновые к-ты
и их соли (мыла), алкилсульфаты (сульфоэфиры),
алкилсульфонаты и алкиларилсульфонаты, прочие
продукты.
В производстве мыл и многих ионо- и неионогенных
мылоподобных ПАВ используют карбоновые к-ты,
получаемые гидролизом из растительных и животных
жиров, и синтетич. жирные к-ты. Промышленное зна-
значение имеют также смоляные и жирные к-ты талло-
вого масла — побочного продукта целлюлозного про-
производства — и смоляные к-ты канифоли, среди к-рых
преобладает абиетиновая.
Наибольшее значение как ПАВ из солей монокарбо-
новых к-т имеют мыла (натриевые, калиевые и аммо-
аммонийные) жирных к-т RCOOH, где R — насыщенный
или ненасыщенный нормальный алифатич. радикал
с числом атомов углерода 12—18, и мыла (натриевые,
реже калиевые) смоляных к-т. Практич. значение
имеют также д и карбоновые к-ты, напр,
алкенилянтарные, получаемые в пром-сти конденса-
конденсацией непредельных углеводородов с мале-
иновым ангидридом, и их соли общей фор-
формулы
С?гН2п-СН-СО(Жа
I
CH2-COONa
где п — 8 — 16. ПАВ тина алкиларомати-
алкилароматических карбоновых кислот и их солей
имеют ограниченное применение.
Алкилсульфаты синтезируют обычно сульфоэтерифи-
кацией высших жирных спиртов или а-олефинов с по-
последующей нейтрализацией соответственно первичных
или вторичных алкилсерных кислот.
Алкиларилсульфонаты, гл. обр. моно- и диалкил-
бензолсульфонаты, а также моно- и диалкилнафталин-
сульфонаты составляют большую часть синтетич. анио-
ноактивных ПАВ. Ниже приведены типичные поверх-
поверхностно-активные сульфонаты на основе бензола и
нафталина:
SO^Na SO3Na
Н9С4
SO3Na
Помимо карбоксильных, сульфоэфирных и сульфо-
групп, в молекулярную структуру анионоактивных
ПАВ могут входить кислотные или соответствующие со-
солевые группы др. типов. Известны такие ПАВ, как орга-
нич. фосфаты, фосфонаты, тиофосфаты, арсенаты и др.
Катионоактивные ПАВ можно разде-
разделить на след. основные группы: амины различной сте-
степени замещения и четвертичные аммониевые основания,
др. азотсодержащие основания (гуанидины, гидразины,
гетероциклич. соединения и т. д.), четвертичные фос-
фониевые и третичные сульфониевые основания. Ни-
Ниже приведены ф-лы типичных катионоактивных ПАВ:
RNR'R"HX, RNR'R"R'"X~
4-
RN'
RPR'R"R"X~
RSR'R"X~
RSR'R"X
где R — алифатич. радикал С10 — C18; R', R" и Rw—-
короткие алифатич., алкилароматич. или ароматнч.
радикалы (напр., группы СН3—, С2Н5—, СбН5СН2 —,
СбН5—), в случае аминосоединений Rr и R" м. б. ато-
атомами водорода; X — анионы (хлорид, бромид, сульфат,
ацетат и др.; в щелочном р-ре — гидроксил-ионы).
Сырьем для катионоактивиых ПАВ, имеющих хозяй-
хозяйственное значение, служат амины, получаемые из
жирных к-т и спиртов, алкилгалогенидов, а также
алкилфенолов. Четвертичные аммониевые соли синте-
синтезируют из соответствующих длинноцепочечных галоид-
галоидных алкилов реакцией с третичными аминами, из ами-
аминов хлоралкилированием или др. путями из синтетич.
спиртов, фенолов и фенольных смесей. Примерами
таких четвертичноаммониевых ПАВ могут служить
приведенные ниже соединения:
C16H33N(C2H5) CH
О
О
+сГ,
с|6н33м!
+Вг".
\ /
cH2N(C2H5K
0СгН40С2Н4Ы(СгН5JСН2
+сГ
SO3Na
RCH
где R — алкильные радикалы с числом атомов угле-
углерода 8—18. Синтез этих веществ включает получение
алкилароматич. углеводородов, их сульфирование и
нейтрализацию сульфокислот.
Алкилсульфонаты обычно получают из насыщенных
углеводородов С12 — С18 нормального строения, к-рые
сульфохлорируют или сульфоокисляют с последующим
омылением или нейтрализацией продукта. Выпуска-
Выпускают первичные и вторичные сульфонаты:
R' ч
RSO3Na и
R
Большое значение как катионоактивные ПАВ и как
исходные продукты в синтезе неионогенных ПАВ (см.
| ниже) имеют не только моно-, но и диамины, полиами-
полиамины и их производные.
Амфотерные ПАВ м. б. получены из анио-
анионоактивных введением в них аминогрупп или из ка-
катионоактивных введением кислотных групп.
Такие соединения, например RNHCH2CH2COONa и
RN(CH2CH2COONaJ, получают взаимодействием пер-
первичного амина и метилакрилата с последующим омы-
омылением сложноэфирной группы щелочью.
Среди амфотерных ПАВ много веществ с различными
типами кислотных и основных групп и разным их
соотношением; есть и внутримолекулярные соли, напр,
алкилбетаины:
R-CHCGO-
'n (CHs)a
Пром-стью амфотерные ПАВ выпускаются в неболь-
небольшом количестве, и их потребление расширяется мед-
медленно.
Неионогенные ПАВ. Это наиболее перспективный и
быстро развивающийся класс ПАВ. Не менее 80—90%
таких ПАВ получают присоединением окиси этилена
к спиртам, алкилфенолам, карбоновым к-там, аминам
и др. соединениям с реакционноспособными атомами
водорода.
Пол и оке и эти леновые эфиры ал-
алкилфенолов — самая многочисленная и распро-
распространенная группа неионогенных ПАВ, включающая
более сотни торговых названий (из отечественных про-
667
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
668
дуктов наиболее известны препараты ОП-4, ОП-7 и
ОП-10). Типичное сырье — октил-, нонил- и додецил-
фенолы; кр. того, используют крезолы, крезиловую
к-ту, ^-нафтол и др. Если в реакцию взят индиви-
индивидуальный алкилфенол, готовый продукт представляет
собой смесь ПАВ общей ф-лы RC6H4O(CH2CH2O)mH,
где т — степень оксиэтилирования, зависящая от
молярного соотношения исходных компонентов.
По л иокс и этиленовые эфиры жир-
жирных к-т RCOO(CH2CH2O)mH синтезируют пря-
прямым оксиэтилированием к-т или этерификацией к-т
предварительно полученным полиэтиленгликолем.
Пол и оке и эти леновые эфиры спир-
спиртов RO(CH2CH2O)mH приобрели важное промыш-
промышленное значение, т. к. они легко поддаются биохимич.
разложению в природных условиях. Их получают
оксиэтилированием высших жирных спиртов, реакцией
алкилбромида с мононатриевой солью полиэтиленгли-
коля и др. путями.
Полиокс и этиленовые эфиры мер-
меркаптанов, как и спиртов, получают обычно
оксиэтилированием третичных алкилмеркаптанов, а
также первичных «-алкилмеркаптанов и нек-рых ал-
килбензолмеркаптанов. Наиболее распространен из
них оксиэтилированный тргт-додецилмеркаптан
C12H25S(CH2CH2O)WH. Эти продукты дешевы и устой-
устойчивы в нейтральных и щелочных р-рах, но их исполь-
использование ограничено из-за характерного неприятного
запаха.
Полиоксиэт и леновые производ-
производные алкиламинов составляют весьма разно-
разнообразную группу ПАВ, многие из к-рых выпускают
в пром-сти. Эти ПАВ, будучи по своей природе ка-
тионоактивными, с увеличением длины полиоксиэтиле-
новой цепи приобретают ярко выраженные свойства
неионогенных веществ. Наиболее важны в практич.
отношении продукты оксиэтилирования первичных
«-алкиламинов, mpem-алкиламинов и дегидроабиетил-
аминов. Так, в США под названием «этомины» выпус-
выпускают продукты на основе аминов, полученных из
смеси природных жирных к-т или индивидуальных
к-т алифатич. ряда:
/(С2Н4О) Н
rch2n/ х
где R — радикал С8 — С18; общее число этиленоксид-
ных звеньев (х + у) варьирует от 5 до 20. Все большее
значение приобретают «этодуомины», получаемые из
акрилонитрила и к-алкиламинов:
(СН2СН2О) Н
RNCH2CH2CH2N< x
I Х(СН2СН2О) Н
(GH2GH2O)ZH у
Выпускают также продукты на основе полиэтиленпо-
лиаминов, напр, диэтилентриамина, но они не имеют
широкого применения. В промышленном или полупро-
полупромышленном масштабе производят ПАВ с третичным
алифатич. радикалом RC(CH3JNH(CH2CH2O)WH,
содержащим 12—22 атома углерода, и т = 1 — 25;
полиоксиэтилендегидроабиетиламины (на основе к-т
канифоли и таллового масла); полиоксипропиленовые
производные аминов — «иропомины»:
сн3
RN
И
ЧСЕЬСНО^Н
СНз
Полиоксиэтиленалкиламиды
RCONH(CH2CH2O)mH и RGON
обычно получают оксиэтилированием амидов или
предварительно полученных моно- или дкэтилоламидов
жирных к-т (лауриновой, пальмитиновой, олеиновой,
стеариновой и др.).
Ряд неионогенных ПАВ получают на основе поли-
полиатомных спиртов, частично этерифицирован-
ных жирными к-тами. Используют спирты, содержа-
содержащие от 2 до 6 гидроксильных групп, пентаэритрит,
полиглицерины, углеводы. При оксиэтилировании к
свободным гидроксильным группам исходного продук-
продукта присоединяются полиоксиэтиленовые цепи разной
длины.
Большой интерес представляют оксиэтилированные
сложные моноэфиры ангидрогекситов и жирных к-т
(одно из торговых названий — «твины»), напр, поли-
оксиэтиленсорбитанмоностеарат:
Н (OCH2CH2V, ОСН-СН2Ч
Н (ОСН2СН2) ОСН-СН /
у I
нсо (сн2сн2о)гн
Н2СОСОС17Н35
где х -+- у + z = 20.
Др. путь получения ПАВ из полиатомных спир-
спиртов — сначала оксиэтилирование, а затем этерифи-
кация.
Практич. значение блоксополимеров
окиси этилена и окиси пропилена
как ПАВ постоянно возрастает. Их получают ступен-
ступенчатой полимеризацией, используя в качестве «затрав-
«затравки» соединения, содержащие реакционноспособные ато-
атомы водорода.
Монофункциональные исходные соединения для син-
синтеза таких ПАВ — одноатомные спирты, к-ты, меркап-
меркаптаны, вторичные амины, N-замещенные амиды и др.
Гидрофобной частью молекулы служит остаток исход-
исходного вещества, если оно имеет достаточно длинный
алифатич. радикал, и полипропиленоксидный блок.
Промышленный интерес из этой группы ПАВ предста-
представляют соединения общей ф-лы RO(C3H6O):x.(C2H4O)yH,
где R — остаток алифатич. спирта, х = 7 — И; по-
лиэтиленоксидный блок составляет 40—80% от общей
массы молекулы. Выпускаются также ПАВ («т е р-
г и т а л ы», США), в структуре к-рых гидрофобный
блок содержит мономерные звенья разных тиноб:
СпН2гг+1 ° KCsHeO)x (С2Н4Оу (C2H4O)z
где п = 1 — 9; отношение х:у варьирует от 85 : 15
до 95 : 5; полиэтиленоксидный блок составляет 45—
55% от массы всей молекулы.
Комплексом ценных технич. свойств обладают т.
наз. «п л ю р о н и к и» (США, Франция); аналогич-
аналогичные отечественные продукты наз. «п р о к с а н о л а-
м и». Их получают последовательным присоединением
к прониленгликолю сначала окиси пропилена, а затем
окиси этилена. Образуются сополимеры, состоящие
из трех блоков:
НО (С
(С3Н6О)у (С2Н4ОJН
где мол. масса полипропиленоксидной (гидрофобной)
части не менее 900 (у > 15), а оба полиэтиленоксидных
блока (гидрофильная часть) составляют от 20 до 90%
мол. массы соединения. Промышленные марки плю-
роников и их состав приведены на рис. 1.
Помимо плюроников на основе бифункциональ-
бифункционального исходного соединения выпускают др. ПАВ, в том
числе плюродаты (США). Они имеют такое же
строение, как и плюроники, но средний гидрофобный
блок содержит 10% этиленоксидных звеньев, а боко-
боковые гидрофильные — 10% оксипропиленовых звеньев.
Исходными веществами с тремя функциональными
группами в синтезе блоксополимерных неионогенных
669
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
670
Последняя цифра числового инденса
/234567
-L \2\—L 122—PI23 1 1 1 f 127-
0 10 20 30 40 50 60 70
Доля полиэтиленонсидных цепей в общей массе молекулы, X
Рис. 1. Торговые марки и состав блоксополимерных ПАВ—
«плюроников». Буквенный индекс указывает на физич.
состояние продукта: L — жидкий, Р — пастообразный,
F — хлопьевидный.
ПАВ могут быть глицерин и др. В США, напр., выпус-
выпускают такие ПАВ на кремнийорганич. основе:
сн3
C2H5Si [О (SiO)Y (С2Н4О)?у (СзН6О).
I х у
сн3
С4Н9]3
где каждая из 3 алкиленоксидных цепей содержит
по 50% этиленоксидных и пропиленоксидных звеньев
и заканчивается бутоксигруппой. Более устойчив к
гидролизу сополимер R'
R3SiO (SiO)^ SiR'2CH2CH2CH2OR"
R'
где R" — полиалкиленоксидная цепь, R и R' — али-
фатич. радикалы.
Из тетрафункциональных соединений для синтеза
блоксополимерных ПАВ чаще всего используют алифа-
тич. первичные диамины. Наиболее известны т е т р о-
н и к и (отечественные аналоги — проксами-
н ы), к-рые получают на основе этилендиамина:
н (осн) (ОСзНб)^ H2CH2N/c3HeO)x (с2н4о)?/ н
^ (ОСзНб^-7 2 2i ^(СзНбО)^ (С2Н4О)у Н
Мол. масса каждой полипропиленоксидной цепи
225—6250, полиэтиленоксидные цепи составляют 20—
90% от общей массы продукта.
Получают также блоксополимеры окисей алкилена
на основе пентаэритрита, диэтилентриамина, гекси-
тов (сорбита и маннита), сахарозы и др.
Неионогенные ПАВ различных типов используют
как исходные продукты для получения ряда ионоген-
ных ПАВ. На основе оксиэтилированных алифатич.
спиртов, алкилфенолов и др. рассмотренных выше ве-
веществ синтезируют поверхностно-активные сульфаты,
фосфаты, карбоксилаты и четвертичные аммониевые
соединения:
R @C2H4)mOSO3H, [R
О]3РО, R (OC2H4)wOCH2COOH,
\ос
,нл hJ c1"
К большинству оксиэтилированных продуктов можно
присоединить акрилонитрил с последующим переводом
полученного амина в четвертичное аммониевое основа-
основание обычными методами.
Фторзамещенные ПАВ составляют обширный класс
соединений, промышленное использование к-рых стало
возможным лишь в последние годы. Многие фторзаме-
фторзамещенные ПАВ разных типов получают на основе фтор-
ангидридов перфторкарбоновых и перфторсульфоно-
вых к-т. Нек-рые из них приведены ниже:
C7F15COOH, C7F15CH2OH, [C7F15CONHC3H6N (CH3JR]+ X~,
C7F15CONHC3H6N+ (CH3J С2Н4СОО~, C8F17SO2OH,
C8F17SO2N (R) CH2COOH, C8F17SO2N (R) (C2H4O)m H,
C8F17SO2N (R) C2H4OSO2OH, [C8F17SO2NHC3H6N (CH3J R]+ X~
Высокомолекулярные ПАВ — растворимые карбо-
или гетероцепные полимеры ионогенного или неионо-
генного типа с мол. массой от нескольких тысяч до
нескольких сотен тысяч. Среди них есть природные
соединения (белки, альгинаты, пектиновые вещества
и т. д.), продукты химич. обработки природных поли-
полимеров (напр., производные целлюлозы) и синтетич.
полимеры.
В структуре типичных высокомолекулярных ПАВ
должно быть четкое разграничение гидрофильных и
гидрофобных участков, как, напр., в рассмотренных
выше блоксополимерах окисей этилена и пропилена.
ПАВ являются сополимеры или гомополимеры, в к-рых
вдоль длинной гидрофобной основной цепи располо-
расположены через определенные интервалы гидрофильные
боковые цепи или группы. Типичные представители
анионоактивных ПАВ этой группы — полиакриловая
и полиметакриловая к-ты, их соли и нек-рые производ-
производные, а также карбоксилсодержащие полимеры на осно-
основе поливинилового спирта, полиакриламида, сополи-
сополимеров малеинового ангидрида с др. непредельными
соединениями. Поверхностной активностью обладают
сульфированные и сульфоэтерифицированные полиме-
полимеры (полистирол, поливиниловый спирт, оксиэтилиро-
ванный поликонденсат ?г-алкилфенола с формальдеги-
формальдегидом и др.).
Катионоактивные полимерные ПАВ получают хлор-
метилированием, а затем аминированием полистирола,
поливинилтолуола и др. виниловых полимеров. Осо-
Особенно высока поверхностная активность солей поли-
полимерных четвертичных аммониевых оснований, в том
числе солей поливинилпиридиния. Для получения
высокомолекулярных ионогенных ПАВ — раствори-
растворимых полиэлектролитов — пригодно большинство мето-
методов и исходных продуктов, к-рые применяют при син-
синтезе ионообменных смол.
Неионогенные высокомолекулярные ПАВ можно
получить оксиэтилированием практически из любого
полимера, содержащего гидроксильные или др. функ-
функциональные группы с реакционноспособными атомами
водорода. Так получены приведенные ниже соедине-
соединения, обладающие нек-рыми свойствами мылоподобных
веществ:
~СН-СН2
1(К20
CqHig
о=с—о(сн2сн2о) сн3
Нек-рые неионогенные поверхностно-активные по-
полимеры не содержат полиэтиленоксидных групп, как
в случае поливинилпирролидона.
671
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
672
Свойства
Поверхностная активность, т. е. способность пони-
понижать в результате адсорбции свободную поверхност-
поверхностную энергию — важнейшее общее свойство ПАВ.
В случае обратимой (термодинамически равновесной)
адсорбции количественным выражением поверхност-
поверхностной активности м. б. наибольшее значение производной
поверхностного натяжения по концентрации, к-рое,
в соответствии с адсорбционным ур-нием Гиббса, опре-
определяется наибольшим значением отношения поверхно-
поверхностного избытка (количества адсорбированного веще-
вещества) Г к равновесной концентрации с:
дс
= RT\
с
Gm — поверхностная активность в гиббсах A гиббс=
= эрг-см-моль, о — поверхностное натяжение,
эрг-см2', с — концентрация, молъ-см~3) Г — адсорб-
адсорбция, молъ-см~2; R — универсальная газовая постоян-
постоянная, эрг-моль'1-^С'1', Т — абсолютная темп-pa, К;
индекс m означает, что соответствующие величины
имеют максимальное значение; знак минус перед про-
производной указывает на падение значений а с увели-
увеличением с.
В случае возможности экспериментального определе-
определения а значения Gm находят графически или аналити-
аналитически по тангенсу угла наклона касательной, прове-
проведенной к изотерме а (с) в точке, соответствующей
*"дГ) w- В отсутствие перегиба на кривой эта точка
совпадает с началом изотермы (рис. 2), т. е. Gm =
= (— -t—)Cz=q. Если же изотерма в области малых
\ ас I
концентраций имеет S-образный вид, что характерно
Ночцетгрсция ПАВ
Нонцентрация ПАВ
Рис. 2. Типичные изотермы поверхностного натяжения
растворов ПАВ: 1 — не образующих мицелл; 2,3 — ми-
целлообр азующих.
для высокоактивных веществ (высших членов гомоло-
гич. рядов ПАВ), касательную для определения Gm
проводят в точке перегиба.
Определить поверхностную активность ПАВ, не об-
образующих мицелл, можно с помощью уравнения
Шишковского, устанавливающего связь между
понижением поверхностного натяжения и концентра-
концентрацией вещества в р-ре:
/ с
Да = а0 — а = b In +
где а0 и а — соответственно поверхностное натяжение
растворителя и р-ра; Ь/а — (—да,'дс)т. Постоянная Ь
почти не меняется в пределах одного гомологич. ряда
ПАВ, тогда как постоянная а при переходе от преды-
предыдущего (низшего) члена гомологич. ряда к последую-
последующему (высшему) изменяется в одинаковое число раз.
Эта закономерность обусловлена правилом
Т pay бе - Дюкло, по к-рому при постоянной
темп-ре поверхностная активность в гомологич. ряду
ПАВ возрастает в геометрич. прогрессии с возрастанием
длины гидрофобного радикала на каждую метиленовую
группу в нормальной углеводородной цепи. Напр.,
если (Gm)n — поверхностная активность ?г-го гомо-
гомолога, a (Gm)n+i — следующего за ним, то
(Gm)n/(Gm)n+i = Р = const-
Значение коэфф. Р определяется работой адсорбции
и сильно зависит от темп-ры и свойств растворителя.
Для большинства водных р-ров обычных ПАВ при
20 СС р^3,3 - 3,5.
Поверхностную активность удобно оценивать по
наибольшему понижению поверхностного натяжения
(Аа)т = а0— crmjn, деленному на соответствующую
концентрацию — ККМ (ст) в случае мицеллообразую-
щих ПАВ. Минимальное поверхностное натяжение
от\п водных р-ров различных мылоподобных веществ
на границе с воздухом при комнатной темп-ре обычно
составляет 28—30 эрг см~2, поэтому для них (Да) тъ
^const и, следовательно, приближенно поверхност-
поверхностная активность обратно пропорциональна ККМ:
Образование мицелл происходит в узком интервале
концентраций, к-рый становится уже и определенней
по мере удлинения гидрофобных радикалов. Выше
ККМ увеличение содержания ПАВ сопровождается
быстрым возрастанием равновесной концентрации ми-
мицелл без существенного изменения концентрации моле-
молекулярного р-ра вплоть до возникновения новых струк-
структурных изменений в системе.
Простейшие мицеллы типичных полуколлоидных
ПАВ, напр, солей жирных к-т, при концентрациях,
не слишком превышающих ККМ, имеют сфероидаль-
сфероидальную форму. В водных р-рах центральная часть (ядро)
таких мицелл образована углеводородными цепями,
а поверхность занята гидратированными полярными
группами. При повышении концентрации структура
отдельных мицелл и системы в целом меняется. В об-
области достаточно высоких концентраций возникают
пластинчатые многослойные мицеллы, к-рые можно
рассматривать как жидкокристаллич. образования
смектич. типа (смектическими наз. жидкие кристаллы,
в к-рых параллельно ориентированные палочковид-
палочковидные молекулы расположены слоями).
Структурные изменения в мицеллярных системах
отражаются на их реологич. характеристиках. В об-
области существования сфероидальных равномерно гид-
ратированных и слабо взаимодействующих между со-
собой мицелл системы практически не отличаются от
ньютоновских жидкостей. Появление же в системе
с ростом концентрации Пх\В анизометричных мицелл
сопровождается резким возрастанием структурной вяз-
вязкости (см. Вязкости аномалия), приводящей в нек-рых
случаях к гелеобразованию, т. е. полной потере теку-
текучести.
Мицеллообразование, как и адсорбция, является
следствием стремления системы принять состояние,
отвечающее наименьшему значению свободной энер-
энергии. Это значит, что в соответствии с принципами тер-
термодинамики, мицеллы образуются при условии
AZm = АЛт - ТА Sm < 0
где AZm, AHm и AS^ — изменения свободной энер-
энергии, теплосодержания (энтальпии) и энтропии системы
в области ККМ. Значения АНт для большинства
ПАВ невелики и редко выходят за пределы^Зккал/молъ
при комнатных темп-pax. Поэтому уменьшение свобод-
свободной энергии системы обусловлено, гл. обр., увеличе-
увеличением энтропии ASm.
Одним из главных факторов, определяющих как
поверхностную активность, так и ККМ водораствори-
673
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
674
мых ПАВ, является длина гидрофобного радикала.
Для ряда гомологов справедливо ур-ние:
lgcm = A-Bn A)
где п — число атомов углерода в парафиновой цепи
молекулы. При одинаковом п ККМ зависит от типа
гидрофильной группы, а в случае неионогенных
ПАВ — от числа отиленоксидных групп т в полиэти-
ленгликолевой цепи:
Ы ст = А'- В'т B)
Постоянные ур-ний 1 и 2 для гомологич. рядов
нек-рых ПАВ приведены в табл. 1.
Помимо молекулярной структуры ПАВ на ККМ
сильно влияют состав растворяющей среды, характер
и количество примесей (электролитов, полярных и
неполярных органич. веществ). В случае ионогенных
ПАВ установлена след. линейная зависимость
In ст = — К In ci + const
где ci — общая концентрация противоионов; экспе-
экспериментальная постоянная К « 0,4 — 0,6 для ПАВ
с одной ионогенной группой в молекуле; в случае
ПАВ с двумя ионогенными группами К « 1. ККМ
ионогенных ПАВ с ростом темп-ры возрастает, неионо-
неионогенных — понижается.
Для многих ПАВ существуют температурные преде-
пределы мицеллообразования. Так, ниже точкиКраф-
т а — темп-ры, соответствующей тройной точке, где
Рис. 3. Фазовая диаграмма раст-
растворов ПАВ вблизи точки Краф-
Крафта: I — истинный, II — мицел-
лярный р-р (коллоидная дис-
дисперсия), III — твердая макро-
макрофаза, находящаяся в равнове-
равновесии с истинным раствором; 1 —
кривая растворимости, 2 —кри-
—кривая ККМ; стрелкой указана
точка Крафта.
Ill 1
/
У
у
у
/
II
/
— ( -
|
-
Температура,°С
в равновесии находятся истинный (молекулярный) р-р
ПАВ, мицеллярная фаза (иногда ее наз. квазифазой)
и макрофаза ПАВ в твердом кристаллич. или гелеоб-
разном аморфном состоянии — существование мицелл
термодинамически невыгодно. При темп-pax ниже точки
Крафта в равновесии с насыщенным р-ром ПАВ на-
находится только твердая макрофаза, выше точки Краф-
Крафта — только мицеллы, к-рые обладают свойствами
жидкой фазы (рис. 3). В узкой области темп-р вблизи
тройной точки увеличение концентрации ПАВ может
Таблица 1. Постоянные уравнений A) иB) для некоторых
гомологических рядов поверхностно-активных веществ *
Общая формула
Сп Н2п+1 COONa . . .
Сп В.2П+1 OSO3Na . . .
Cn H2?i+i SO3Na . . . .
Сп H2n+i C6H4SO3 Na .
Сп H2n+l СН(СООКK .
CnH2n+lNH2.HCl . .
Сп Н2п+1 N(GHs)sBr .
Сп Н2п+1 СНСОСГ . .
N+(CH3K
Ci2H2o(OC2H4)mOH ' •
С16Н33(ОС2Н4)ШОН . .
C9H19C6H4(OC2H4)mOH
C12H2r,(OC2H4)TOOSO3Na
C14H37(OC2H4)mOSO3Na
А илиА' В или Б' Темп-ра,
1,96 *'
1 ,43
1,53
0,084
1 ,30
1 ,51
1,98
2,75
-4,4 0
-5,95
-4,54
2 12
-3,78
0,296**
0,290
0,290
0,253
0,219
0,286
0,311
0,469
0,009
0,028
0,014
0, 198
0,165
20-70
25-60
25-80
20-75
20-25
25-60
25-60
27-60
25
25
55
5 0
25
* В литературных данных м. б. расхождения. ** Прибли-
Приблизительно те же значения и для солей К.
несколько сместить эту точку в сторону более высоких
температур.
Большинство неионогенных ПАВ не имеет точки
Крафта, т. к. вплоть до темп-ры замерзания водной
среды они сохраняют жидкое или жидкообразное со-
состояние. Однако для них есть верхний температурный
предел мицеллообразования — точка помутне-
помутнения. Гидрофильность и, следовательно, раствори-
растворимость неионогенных ПАВ сильно уменьшается при
высоких темп-pax вследствие дегидратации. Поэтому
при постепенном нагревании водные р-ры неионоген-
неионогенных ПАВ внезапно мутнеют в узком температурном
интервале, а затем расслаиваются на две макрофазы.
С приближением к точке помутнения концентрацион-
концентрационная область существования мицелл сужается и степень
агрегации (число молекул в мицелле) увеличивается.
Выше точки помутнения мицеллы в системе отсутст-
отсутствуют. При малых концентрациях (до нескольких %)
точка помутнения не зависит от содержания вещества
в р-ре. Она понижается с уменьшением длины полиэти-
леноксидных цепей в молекулах неионогенных ПАВ,
а также при добавлении в р-р солей.
Температурную область мицеллообразования важно
учитывать, напр., при выборе ПАВ — эмульгаторов
для проведения эмульсионной полимеризации винило-
виниловых и диеновых мономеров. Процесс следует вести
при темп-pax, не выходящих за пределы этой области,
и при концентрациях ПАВ выше ККМ.
Точки Крафта и точки помутнения нек-рых мицелло-
образующих ПАВ приведены в табл. 2.
Таблица 2. Точки Крафта и точки помутнения для некоторых
мицеллообразующих поверхностно-активных веществ
Ионогенные ПАВ
CuH23COONa
C^H^OSOsNa
C12H25SO3Na
C12H25CH(CH3)C6H4SO3Na
C18H37(OCH2CH2LOSO3Na
C12H25NH2HC1
C3
&h
3 6
2 0
3 3
28
30
26
Неионогенные ПАВ
(VH,5(OCH2CH2)eOH
C16H{,(OCH2CH,NOH
CnH,,CO(OCH2CH2)eOCH3
С9Н19С6Н4(ОСН,СН,)юОН
C9H19C6H4(OCH2CH2J0OH
Точка
помутне-
помутнения. °G
4 8
32
31
fiO
82
ККМ
Примечание. Среди лит. данных м. б. значительные
расхождения
ККМ — важный технологич. показатель. Его можно
определять различными методами, т. к. в области
ККМ более или менее резко меняются многие физико-
химич. свойства системы
(рис. 4). ККМ находят по
характерным изменениям по-
поверхностного натяжения,
светорассеяния, электропро-
электропроводности, вязкости, диффу-
диффузии, солюбилизации (см. ни-
ниже), спектральных характе-
характеристик р-ра и т. д.
Солюбилизацня—
растворение в водных р-рах
Рис. 4. Типичные кривые изме-
изменения нек-рых свойств р-ров
ПАВ в области ККМ: 1 — мо-
моющее действие, 2 — плотность,
3 — поверхностное натяжение,
Концентрация
осмотич. давление, 5 — эквивалентная электропровод-
электропроводность, б — межфазное натяжение на границе водный р-р —
жидкий углеводород, 7 — мутность.
ПАВ нерастворимых или плохо растворимых в во-
воде органич. веществ. Это явление, наз. также
коллоидным растворением, характерно только для
675
ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
676
полуколлоидных ПАВ при концентрациях выше ККМ.
Оно обусловлено самопроизвольным и обратимым про-
проникновением солюбилизуемого вещества в углеводород-
углеводородное ядро мицеллы. (Солюбилизацию следует отличать
от гидротропии — повышенной растворимости
гидрофобных соединений в водных р-рах веществ, не
образующих мицелл.)
Для систем со сфероидальными мицеллами моляр-
молярная солюбилизация постоянна, т. е. в условиях термо-
динамич. равновесия cj{c — ст) & const, где концентра-
концентрации солюбилизированного углеводорода cs и ПАВ
выражены в молъ/л.
Солюбилизация имеет важное значение в различных
технологич. и биологич. процессах. Так, при эмуль-
эмульсионной полимеризации необходимо, чтобы часть мо-
мономера солюбилизировалась в мицеллах эмульгатора.
Чрезвычайно важна в практич. отношении способ-
способность ПАВ, гл. обр. мылоподобных и высокомолеку-
высокомолекулярных, стабилизовать высокодисперсные (микрогете-
(микрогетерогенные) системы. Мельчайшие частицы твердого тела,
капельки жидкости или пузырьки газа, образующие
дисперсную фазу и равномерно распределенные в
окружающей (дисперсионной) среде, имеют тенденцию
укрупняться вследствие слипания (коагуляции) или
слияния (коалесценции) при соударении в процессе
броуновского движения. Сделать систему агрегативно
устойчивой можно с помощью стабилизаторов — ве-
веществ, создающих вокруг частиц моно- или полимоле-
полимолекулярный защитный слой. Особенно эффективны ста-
стабилизаторы, к-рые образуют защитный слой, обладаю-
обладающий повышенной структурной вязкостью (что может
иметь место в случае высокомолекулярных и полукол-
полуколлоидных ПАВ). Такой слой предельно сольватирован
с внешней стороны, куда обращены лиофильные груп-
группы молекул стабилизатора. Вследствие сольватации
(гидратации — в случае водной дисперсионной среды)
слипания частиц по внешним поверхностям защитных
оболочек не происходит из-за ослабления межмолеку-
межмолекулярных сил сцепления. Вытеснение же защитного
слоя из зазора между сближающимися частицами за-
затруднено из-за повышенной поверхностной вязкости,
к-рая является следствием когезионного взаимодей-
взаимодействия лиофобных (гидрофобных в случае водной дис-
дисперсионной среды) частей молекул ПАВ в толще адсорб-
адсорбционной оболочки. Т. обр., наиболее эффективные ста-
стабилизаторы водных дисперсий (суспензий, эмульсий,
латексов, пен) — ПАВ, к-рые наряду с высокой гидро-
фильностью обладают достаточно длинными углеводо-
углеводородными радикалами, что обусловливает прочное коге-
зионное сцепление молекул в адсорбционном слое.
В гомологич. рядах алифатич. ПАВ, напр., способ-
способность стабилизовать водные дисперсии появляется
у соединений с числом углеродных атомов в углеводо-
углеводородной цепи более 9 — 11.
Понижая в результате адсорбции поверхностное на-
натяжение, ПАВ уменьшают работу образования новых
поверхностей, т. е. облегчают диспергирование раз-
различных материалов. К этому сводится механизм
адсорбционного понижения прочности твердых тел
(эффект Ребиндера). В случае снижения
межфазного натяжения до критич. значений от (де-
(десятые доли эрг-см~2 при комнатной темп-ре) и меньше
для диспергирования тела достаточно энергии тепло-
теплового движения, т. е. система самопроизвольно превра-
превращается в термодинамически устойчивую (лиофильную)
дисперсию с частицами коллоидных размеров F — 10~5 —
10 см). Условие самопроизвольного (спонтанного)
диспергирования можно выразить соотношением:
кТ
°12^°Ш = У —
где у — безразмерный множитель, к — постоянная
Больцмана, Т — абсолютная темп-pa и б — средний
размер частиц в см. Примером такой системы может
служить водный р-р (правильнее — коллоидная дис-
дисперсия) мыла или мылоподобного вещества при с > с
и темп-ре выше точки Крафта. При самопроизвольном
эмульгировании и пептизации осадков под действием
теплового движения в присутствии высокоактивных
ПАВ также образуются лиофильные системы.
В насыщенном адсорбционном слое на поверхности
раздела жидкость — газ и жидкость — жидкость моле-
молекулы ПАВ нормально ориентируются гидрофильными
группами в сторону полярной среды, а гидрофобными —
в противоположном направлении. При этом на свобод-
свободной поверхности водного р-ра каждая молекула жир-
жирной к-ты, амина или спирта занимает площадь 20,5—
21,5 А2, большинства омыл, алифатич. сульфатов и
сульфонатов — 20—60 А2, неионогенных ПАВ — 40—
120 А2. В случае адсорбции на поверхности твердого
тела нормальная ориентация не обязательна. Если
энергия связи гидрофильных групп с твердой поверх-
поверхностью окажется достаточно большой, что имеет место
при хемоадсорбции, то возможна обратная ориентация
молекул ПАВ — гидрофобными радикалами в сторону
полярной среды.
Нормальная ориентация приводит к гидрофилизации
поверхности и улучшению ее смачиваемости водой и
водными р-рами. Хорошими смачивателями являются
ПАВ с разветвленными и не слишком длинными
(С10 — С12) углеводородными радикалами. Обратная
ориентация молекул гидрофобизует поверхность, ухуд-
ухудшая ее смачиваемость водой. Если гидрофобные ради-
радикалы при этом имеют углеводородную природу, то
одновременно улучшается смачиваемость поверхности
органич. неполярными жидкостями, маслами, р-рами
и расплавами неполярных полимеров.
Рассмотренные выше общие свойства ПАВ определяют
т. наз. комплекс моющего действия мыл и мылоподоб-
мылоподобных веществ. Он включает смачивание загрязненной
поверхности, адсорбционное вытеснение, солюбили-
солюбилизацию и эмульгирование жировых или масляных
загрязнений, а также диспергирование и стабилиза-
стабилизацию частиц твердых загрязнений. Коллоидно-химич.
свойства р-ров неионогенных ПАВ зависят не столько
от длины (массы) гидрофобной части, хотя и это важ-
важно, сколько от соотношения длин (масс) гидрофобной
и гидрофильной частей молекулы. Такое соотношение,
выраженное в единицах условной шкалы (по Гриффи-
ну), наз. гидрофильно-липофильным
(т. е. олеофильным) балансом (ГЛБ). Числовые
значения ГЛБ определяют экспериментально по стан-
стандартной методике или расчетным путем. Определить
число ГЛБ можно и для ионогенных ПАВ, но большей
частью им характеризуют неноногенные вещества.
Продукты оксиэтилирования обычно имеют число
ГЛБ не выше 20, для олеата калия оно равно 20, а для
чистого лаурилсульфата — ок. 40. Ниже приведены
приблизительные интервалы чисел ГЛБ, отвечающие
тому или иному назначению ПАВ:
3—6 . . .эмульгатор для эмульсий типа «вода в масле».
7—9 : . .смачиватель
8—15 . . .эмульгатор для эмульсий типа «масло в воде»
13—15 . . .моющее средство
15—18 . . .солюбилизатор
Число ГЛБ иногда рассматривают как обобщенную
характеристику комплекса технологич. свойств ПАВ,
предопределяющую область их практич. использова-
использования. Однако следует подчеркнуть, что такой подход
не всегда справедлив, т. к. ГЛБ — условная и чисто
эмпирич. характеристика, не претендующая на уни-
универсальность.
Очень специфичны по свойствам фтортензиды, непо-
неполярная часть молекулы к-рых образована фторуглерод-
ными цепями. Вследствие слабого межмолекулярного
взаимодействия низкомолекулярные фторуглероды об-
677
ПОДАТЛИВОСТЬ
678
ладают чрезвычайно малой поверхностной энергией
(9—18 эрг-см'2). Поэтому фтортензиды, образуя насы-
насыщенный ориентированный адсорбционный слой, сни-
снижают поверхностное натяжение водных р-ров до
16—18 эрг-см~2, тогда как углеводородные ПАВ —
только до 28—30 эрг-см~2. Поверхностная активность
фторуглеродных ПАВ намного выше, чем у углеводо-
углеводородных аналогов. Напр., соли перфторалкановых к-т
С,—С9 в 20—30 раз более активны, чем соли аналогич-
аналогичных к-т жирного ряда.
Особенность фторуглеводородных ПАВ — соедине-
соединений с фторуглеродными и углеводородными радика-
радикалами — высокая поверхностная активность в непо-
неполярных органич. жидкостях с низкой поверхностной
энергией. Производные амидов перфторкарбоновых к-т,
к примеру, снижают поверхностное натяжение с 28—32
до 12—26 эрг-см~2. На межфазных поверхностях вод-
водный р-р — углеводородная жидкость фторзамещенные
ПАВ также проявляют исключительно высокую актив-
активность. Адсорбционный слой перфорированных ПАВ
на твердой поверхности, ориентированный фторугле-
фторуглеродными радикалами наружу, снижает крити-
критическое поверхностное натяжение
смачивания (определение см. в ст. Когезия)
до значений меньших, чем поверхностное натяже-
натяжение углеводородных жидкостей. Это значит, что та-
такая поверхность становится не только гидрофобной,
но и олеофобной, т. е. не смачиваемой маслами и др.
жидкими углеводородами. Фторуглеродные цепи,
вследствие высокой энергии межатомной (внутримоле-
(внутримолекулярной) связи, химически инертны и термостойки;
они не разлагаются при темп-pax выше 400 °С. Поэтому
термостойкость фторуглеродных ПАВ определяется
полярной группой. Фторуглеродные сульфонаты, напр.,
устойчивы почти до 350 °С, а карбоновые к-ты и их
соли — до 175—250 °С.
Применение
ПАВ находят широкое применение в пром-сти,
с.х-ве, медицине и быту. Мировое производство ПАВ
растет с каждым годом, причем в общем выпуске продук-
продукции постоянно возрастает доля неионогенных веществ.
Широко используют все виды ПАВ при получении
и применении синтетич. полимеров. Важнейшая об-
область потребления мицеллообразующих ПАВ — произ-
производство полимеров методом эмульсионной полимери-
полимеризации. От типа и концентрации выбранных ПАВ
(эмульгаторов) во многом зависят технологич. и физи-
ко-химич. свойства получаемых латексов (см. Эмуль-
Эмульсионная полимеризация, Латексы синтетические). ПАВ
(гл. обр. высокомолекулярные) применяют также для
облегчения концентрирования каучуковых латексов
методом сливкоотделения, для повышения агрегатив-
ной устойчивости натурального или синтетич. латек-
латекса. Иногда в латекс с целью его сенсибилизации, т. е.
увеличения чувствительности к действию коагулирую-
коагулирующих факторов, вводят ПАВ, ослабляющие защитное
действие стабилизаторов. ПАВ используют также при
суспензионной полимеризации. Обычно применяют вы-
высокомолекулярные ПАВ — водорастворимые полимеры
(поливиниловый спирт, производные целлюлозы, ра-
растительные клеи и т. п.). ПАВ как обязательные
компоненты содержатся в водных дисперсиях поли-
полимеров, получаемых механич. диспергированием или
путем образования новой полимерной фазы из пересы-
пересыщенного р-ра. Смешением лаков или жидких масляно-
смоляных композиций с водой в присутствии эмуль-
эмульгаторов получают эмульсии, применяемые при изго-
изготовлении пластмасс, кожзаменителей, нетканых мате-
материалов, импрегнированных тканей, водоразбавляемых
красок и т. д.
В производстве лакокрасочных материалов и пласт-
пластмасс большое значение имеют процессы подготовки
пигментов и наполнителей. Прежде всего здесь при-
приходится решать проблему тонкого измельчения твер-
твердых материалов. При этом ПАВ облегчают диспергиро-
диспергирование, понижая прочность (эффект Ребиндера), и,
адсорбируясь на поверхности частиц, препятствуют
их слипанию, т. е. выполняют функцию стабилизатора.
Кроме того, ПАВ используют для адсорбционного мо-
модифицирования пигментов и наполнителей, напр,
с целью увеличения адгезии полимеров. ПАВ добав-
добавляют в лаки, краски, пропиточные композиции и
жидкие связующие как смачиватели, вводят в р-ры,
расплавы и дисперсии полимеров для регулирования
их реологич. характеристик.
Разнообразные ПАВ применяют для поверхностной
обработки волокнистых (тканых и нетканых) и пленоч-
пленочных материалов (см., напр., Авиважная обработка),
как антистатики, модификаторы прядильных р-ров,
моющие средства. Среди ПАВ, применяемых как гидро-
фобизаторы, наиболее перспективны кремнийоргани-
ческие (см. Гидрофобизаторы кремнийорганические) и
фторуглеродные соединения. Последние при соответ-
соответствующей ориентации молекул в поверхностном слое
способны предотвратить смачивание материала не
только водой, но и углеводородными жидкостями.
В производстве губчатых резин и пенопластов ПАВ
применяют как стабилизаторы пен.
Высокомолекулярные водорастворимые ПАВ, по-
помимо использования в указанных выше технологич.
процессах, применяют как флокулянты в различных
видах водоочистки. С их помощью из сточных и тех-
технологич. вод, а также из питьевой воды удаляют за-
загрязнения, находящиеся во взвешенном состоянии.
Лит.: Ребиндер П. А., Жури. ВХО, 11, № 4, 362
A966); его же, там же, 4, № 5, 554 A959); Шварц А.,
Перри Дж., Берч Дж., Поверхностноактивные ве-
вещества и моющие средства, пер. с англ., М., 1960; Коллоидные
поверхностноактивные вещества, пер. с англ., под ред.
А. Б. Таубмана, 3. Н. Маркиной, М., 1966; Ш т ю п е л ь Г.,
Синтетические моющие и очищающие средства, пер. с нем., М.,
1960; Шенфельд Н., Неионогенные моющие средства,
пер. с нем., М., 1965; Н е в о л и н Ф. В., Химия и техноло-
технология синтетических моющих средств, 2 изд., М., 1971; Nonionic
surfactants, ed. by M. J. Schick, v. 1, N.Y., 1967; G e r r e n s H.,
в кн.: Polymer handbook, ed. J. Brandrup, E. H. Immergut,
N. Y.— La. o.], 1966, p. 11—399; H a g g e W., Chemie, physi-
physique et applications pratiques des agents de surface, Comptes
rendus du V-eme Congres International de la detergence, Barcelo-
Barcelona, v. 1, 1969, p. 1; В г у с е Н. G., в кн.: Fluorine chemistry,
ed. by J. H. Simons, v. 5, N. Y.— L., 1964; Успехи коллоидной
химии, М., 1973. Л.А.Шиц.
ПОДАТЛИВОСТЬ полимеров (compliance,
resilience, Nachgiebigkeit, resilience) — величина, ха-
характеризующая способность полимерных систем к раз-
развитию обратимых деформаций под действием прило-
приложенных напряжений. Количественно П. определяется
ур-нием
t
е @ = f I{t-z)G (x) dx
где / (t — т) — податливость; 8 (t) — деформация;
a (t) — напряжение; t — длительность его действия.
В случае a (t) — const П. определяется отношением
-?—, т. е. обратна значению модуля высокоэластичности.
При изменении напряжения по гармонич. закону
с частотой со деформация изменяется с той же часто-
частотой, но запаздывает по фазе относительно изменения
а на угол 6. Если амплитуды деформаций е0 и напря-
напряжений а0 малы, то s0 ~ a0 и комплексную П. /* = г/о
можно представить в виде суммы действительной /'
и мнимой /" компонент: /* = /' — И"; при этом
/' = e0cos6/a0 и /" = е0 sin д/о0. Характеристики
П. /' и V зависят от со. Величина /* обратна комплекс-
комплексному модулю G*, измеряемому при гармонич. измене-
изменении деформации, так что I*G* = 1. Поэтому компо-
компоненты комплексной П. выражаются исходя из компо-*
679
ПОДВУЛКАНИЗАЦИЯ
680
нент модуля G* = G'-\- iG"
по формулам /' =
G'
Функции / (t) или /' (со) и /" (со) (со — круговая
частота) определяются спектром распределения времен
запаздывания (ретардации) и через них связаны со всем
комплексом механич. свойств материала, описываемых
линейной теорией вязкоупругости (см. Реология).
Величина /", наз. податливостью потерь,
определяет интенсивность тепловыделений при перио-
дич. нагружении полимерной композиции. С помощью
этой величины характеризуются также критич. усло-
условия, при к-рых происходит переход к неконтролируе-
неконтролируемому разогреву и изделие теряет механич. устойчи-
устойчивость. Характер темп-рной зависимости /' и /", как и /
(см. Ползучесть), позволяет судить о границах релак-
релаксационных областей физич. состояния полимеров, а
также о структурных и фазовых переходах. Измерение
П.— один из основных способов оценки механич.
свойств полимерных систем. Широко применяемым
методом измерения П. является термомеханическое ис-
исследование полимеров, при к-ром определяется дефор-
деформируемость материала при определенной продолжи-
продолжительности нагружения в регламентированных усло-
условиях испытания. А. Я. Малкин.
ПОДВУЛКАНИЗАЦИЯ, преждевремен-
преждевременная вулканизация, скорчинг (scor-
(scorching, Anvulkanisation, prevulcanisation) — необрати-
необратимое изменение пласто-эластических свойств резиновой
смеси при ее изготовлении, технологич. обработке
(каландровании, шприцевании и др.) или хранении.
П. сопровождается падением пластичности, повыше-
повышением вязкости и эластич. восстановления резиновой
смеси. Вследствие этих изменений затрудняется, а
иногда даже становится невозможной дальнейшая
переработка смеси в изделие.
Склонность резиновых смесей к П.
характеризуют временем, в течение к-рого нагревае-
нагреваемая при данной темп-ре (обычно 100—125 °С) смесь
сохраняет требуемые пласто-эластич. свойства. Напр.,
при использовании вискозиметра Муни определяют
величину tb, т. е. время в минутах, за к-рое вязкость
образца, нагреваемого при определенной темп-ре, пре-
превысит минимальную на 5 единиц. Подробно о прибо-
приборах и методах оценки склонности смесей к П. см.
Пласто-эластические свойства.
Причины под вулканизации. Воздействие тепла на
резиновую смесь обусловливает возможность химич.
превращений каучука, характерных для вулканизации,
на стадиях технологич. процесса, предшествующих
этой заключительной операции. В случае применения
серусодержащих вулканизующих систем П. обуслов-
обусловлена гл. обр. взаимодействием каучука с серой; сте-
степень изменения пласто-эластич. свойств смеси опреде-
определяется количеством серы, присоединенной к каучуку.
Так, полная потеря пластичности при нагревании
A20 °С) ненаполненной смеси из бутадиен-стирольног о
каучука наблюдается при присоединении ~0,5% серы.
В присутствии высокодисперсных саж смесь теряет
пластичность при связывании ~0,3% серы, что объяс-
объясняется участием сажи в сшивании макромолекул.
Стабильность пласто-эластич. свойств смесей на
основе каучуков, вулканизуемых серой, определяется
гл. обр. типом применяемых ускорителей вулканиза-
вулканизации. Влияние последних на П. уменьшается в след.
ряду: ксантогенаты > дитиокарбаматы > тиурамы >
смесь меркантобензтиазола (каптакс) с дифенилгуани-
дифенилгуанидином > смесь дибензтиазолилдисульфида (альтакс) с
дифенилгуанидином > меркаптобензтиазол > дифенил-
гуанидин > дибензтиазолилдисульфид > сульфенамид-
ные производные меркантобензтиазола.
Большое влияние на П. оказывают также наполни-
наполнители. Склонность саженаполненных смесей к П. повы-
повышается с увеличением рН водной суспензии сажи. При
близких значениях рН склонность к П. возрастает
(табл. 1) с увеличением структурности (масляного
числа) и степени дисперсности (уд. поверхности) сажи.
Эти эффекты связаны с влиянием сажи на процесс
серной вулканизации и с возникновением химич. свя-
связей сажа — каучук в результате механохимич. воздей-
воздействий (подробно о свойствах сажи см. Наполнители
резин). Высокая дисперсность сажи, как и высокая
степень наполнения резиновой смеси, обусловливают
повышенное теплообразование при ее переработке, что
увеличивает опасность П.
Таблица 1. Влияние типа сажи на подвулканизацшо резиновых
смесей на основе натурального (НК) и бутадиен-стирольного
(БСК) каучуков
Термическая (МТ) . .
То же(FT) ....
Полуусиливающая
печная (SRF) . . .
Высокоструктурная
печная (FEF) . . .
Газовая канальная
(ЕРС)
рН
8,5
9,0
—
8,0
4,3
Уд. гео-
метрич.
ность,
м2/г
6
13
23
42
116
Масля-
ло, мл/г
0,35
0,40
0,76
1,30
1,12
t-o при
120°С,
мин
НК
21
12
10
9
14
BGK
29
21
23
18
31
П. резиновых смесей на основе нек-рых каучуков,
вулканизуемых окислами металлов (ZnO, MgO), также
связана с взаимодействием каучуков с этими вулкани-
вулканизующими агентами на стадиях, предшествующих вул-
вулканизации (см., напр., Карбоксилатные каучуки).
Способы защиты резиновых смесей от подвулканиза-
ции. Внедрение высокоскоростных и высокотемпера-
высокотемпературных процессов производства резиновых изделий
непосредственно связано с решением проблемы защиты
смесей от П. Эта проблема м. б. решена: а) примене-
применением ускорителей вулканизации замедленного действия
и сочетанием их с донором серы (дитиодиморфолином)
при полной или частичной замене элементарной серы;
б) использованием молекулярных сит (цеолитов) —
адсорбентов активных компонентов вулканизующей
системы (см. также Вулканизующие агенты); в) введе-
введением в резиновую смесь специальных добавок — за-
замедлителей П. (антискорчингов).
Замедлители П. широко применяют как
самостоятельно, так и в сочетании с ускорителями вул-
вулканизации замедленного действия (в тех случаях, когда
последние не устраняют полностью опасность П.).
Основное требование к замедлителям П.— способность
замедлять сшивание макромолекул при темп-pax пе-
переработки резиновых смесей, не оказывая при этом
влияния на время достижения оптимума вулканизации
и на физико-механич. свойства вулканизатов. К замед-
замедлителям П. относятся органич. к-ты, их ангидриды и
соли, производные тиофталимида, ??г/жс-алкилсульфе-
нилметана, а также нек-рые галогенсодержащие и
нитрозосоединения. Из них только немногие (табл. 2)
применяют в пром-сти.
Среди органич. к-т и их производных наиболее эф-
эффективен фталевый ангидрид (табл. 3). Предполагают,
что при темп-pax переработки смесей эти замедлители
П. ингибируют реакции элементарной серы с ускори-
ускорителями и активаторами вулканизации. С повышением
темп-ры и продолжительности нагревания резиновой
смеси тормозящее действие органич. к-т и фталевого
ангидрида на эти реакции уменьшается.
Бензойная, салициловая к-ты и фталевый ангидрид
трудно диспергируются в каучуке; поэтому их часто
681
ПОД ВУЛКАНИЗАЦИЯ
682
Таблица 2. Замедлители подвулканизации для смесей на основе
натурального и синтетических каучуков, вулканизуемых
серусодержащими вулканизующими системами
Название
N-Нитрозо д ифенил-
амин (НДФА, вул-
калент А)
Фталевый ангидрид
Бензойная к-та**
Салициловая к-та**
N-Циклогексилтио-
N-Циклогексилтиофталимид (санто-
гард PVI) ....
Трихлормеламин* * *
Химич. ф-ла
ОгО
N0
1 о
\
соон
¦6
соон
о
^0
H\ /н
N—C-N=C—N^
Темп-
pa
плав-
плавления,
°C
130
122
150
90
Рекомен-
Рекомендуемые
количест-
количества,
мае. ч.*
0,25-1,0
0,1 — 1 ,0
0,5 — 1 ,0
0,5-1,0
0, 1 — 1 ,0
0,1-1,0
* Здесь и далее — в расчете на 100 мае. ч. каучука. ** При-
Пригодна для применения в резинах пищевого назначения.
*** Выпускают в виде смеси с инертным наполнителем (BaSO4)
в соотношении 1:3 (продукт ТХМ-25).
Таблица 3. Эффективность действия органических кислот
и их производных как замедлителей подвулканизации в смесях
на основе натурального каучука *
Показатели
Пластичность
(ГОСТ 415-53)
И0ХОПН8.Я . .
после прогрева
при 100°С
1Омин ....
20жгш ....
ЗОжин ....
Оптимум вулканизации
при 143СС, мин ....
Прочность при растяже-
растяжении, Мп/мг(кгс/см2) . .
Относительное удлине-
Остаточное удлинение, %
Без за-
медли-
медлителя П.
Бензой-
Бензойная
к-та —
0,48
мае. ч.
Р е з и н о в i
0 , 60
0,55
0, 14
0,00
10-20
30C00)
810
15
0,73
0,63
0,59
0,14
10-20
Булка
30C00)
790
17
Салици-
Салициловая
к-та —
0,55
мае. ч.
\ я с м е (
0,73
0,69
0,68
0,67
20-30
н и з а т
28B80)
800
10
Фтале-
Фталевый
ангид-
ангидрид —
0 30
мае. ч.
г ъ
0,63
0,63
0,65
0,62
20
30C00)
820
12
* Ускоритель вулканизации — каптакс @,7 мае. ч.).
вводят в смеси в виде сплава с диспегаторами, напр,
со стеариновой к-той. Они малоэффективны в присут-
присутствии высокодисперсных печных саж, резко повышаю-
повышающих склонность резиновых смесей к П. В смесях с
такими сажами применяют ускорители вулканизации
замедленного действия в сочетании с N-нитрозодифенил-
амином, к-рый практически равноценен фталевому
ангидриду в присутствии ускорителей типа тиазолов,
гуанидинов или их комбинаций и значительно эффек-
эффективнее в резиновых смесях с ускорителями вулкани-
вулканизации класса сульфенамидов (табл. 4).
Таблица 4. Влияние фталевого ангидрида и N-нитрозодифенил-
амина на подвулканизацию смесей на основе натурального
каучука, содержащих различные ускорители вулканизации
Замедлитель подвулканизации
Фталевый ангидрид
N-Нитрозодифениламин
Без замедлителя
Количе-
Количество,
мае. ч.
0,5
0,5
tb при 120°С, мин
Сульфен-
амид
Ц @,5
мае. ч.)
26
32
26
Альта кс
@,8
мае. ч.)
16
18
15
При температурах переработки N-нитрозодифенил-
амин распадается на радикалы окиси азота (I) и
дифенилазота (II). Ра-
Радикал II реагирует с *—* .—.
N-нитрозодифенилами- / \-n—/ \ —*- no' + *n^
ном с образованием ста- ^"^ N0 ^=^
бильных азотокисных
радикалов, к-рые дез- "
активируют реакции в каучуке, взаимодействуя с
макрорадикалами; последние могут возникнуть при
распаде гидроперекисей, а также слабых связей, об-
образовавшихся в результате взаимодействия каучука
с ускорителями вулканизации. Кроме того, I оказы-
оказывает при 100—130 °С дезактивирующее действие на
серусодержащие радикалы, осуществляющие реакции
вулканизации, а II при более высоких темп-pax акти-
активирует эти реакции. В результате такого двоякого
действия фрагментов молекулы N-нитрозодифенилами-
на он в меньшей степени, чем фталевый ангидрид, за-
замедляет скорость вулканизации в главном ее перио-
периоде. N-Нитрозодифениламин не применяют в смесях,
вулканизуемых горячим воздухом (из-за возможно-
возможного порообразования), а также в белых и цветных
резинах.
N-Циклогексилтиофталимид—высокоэффективный не-
окрашивающий замедлитель П., мало влияющий на
скорость и степень вулканизации,— получил широкое
распространение в смесях с различными вулканизую-
вулканизующими системами (табл. 5). Предполагают, что при
Таблица 5. Сравнительная эффективность N-циклогексилтио-
фталимида и других замедлителей подвулканизации в резиновых
смесях различного состава *
Замедлитель П.
Фталевый ангидрид
Салициловая к-та
N-Нитрозодифениламин
N-Циклогексилтиофталимид ....
Кол-во,
мае. ч*.
1,0
1,0
1 , 0
0,4
tb при 121°С, мин
I
20
19
2 3
37
II
18
18
30**
*1 — смесь на основе маслонаполненного бутадиен-стироль-
ного каучука; ускоритель вулканизации — сульфенамид Ц; II—
смесь на основе композиции натурального и оутадиен-стироль-
ного каучука A:1); ускоритель вулканизации — смесь альтакса
с дифенилгуанидином. ** 0,5 мае. ч. замедлителя П.
темп-pax переработки резиновых смесей A20—130 °С)
это соединение взаимодействует с ускорителями вул-
вулканизации или продуктами их распада с образованием
неактивных промежуточных продуктов, к-рые в ус-
условиях вулканизации A50 СС и выше) распадаются на
фрагменты, активно участвующие в образовании вул-
канизационной сетки.
Трихлормеламин — один из наиболее эффективных
замедлителей П. среди галогенсодержащих соедине-
683
ПОКРЫТИЯ ДЛЯ ПОЛОВ
684
ний. В смесях с ускорителями вулканизации типа
тиазолов он значительно превосходит по защитному
действию фталевый ангидрид (рисунок). Эффектив-
Эффективность действия гало-
генсодержащих замед-
замедлителей П. опреде-
определяется наличием в
них подвижного ато-
атома хлора, выделяю-
выделяющегося при нагрева-
о!бнии и выступающего,
Влияние типа и количе-
количества замедлителя подвул-
канизации на время под-
вулканизации (а) и оп-
оптимальное время вулка-
\J низации (б) смеси на ос-
основе натурального кау-
каучука (ускоритель вулка-
вулканизации — каптакс): 1
0,1 0.2 0.3 0,4 0,5 0,6 без ~ Тамедлителя,' 2 —
Количество замедлителя подвулнанизации, Фталевый ангидрид, з —
мас.ч. трихлормеламин.
по-видимому, в роли ингибитора вулканизации при
темп-pax обработки резиновых смесей. Трихлормел-
Трихлормеламин целесообразно использовать в быстровулканизую-
щихся резиновых смесях, в смесях для тонкостенных
резиновых изделий, где важно хорошее распределение
ингредиентов, а также в смесях для белых и цветных
резин.
В принципе возможности предотвращения П. при
помощи замедлителей ограничены. Достижение боль-
большого эффекта замедления П. неизбежно связано с за-
замедлением вулканизации, что при тенденции к интен-
интенсификации этого процесса крайне нежелательно.
Устранение явления П. в условиях интенсифициро-
интенсифицированных технологич. процессов резинового производст-
производства — важная проблема, для решения к-рой необхо-
необходимо использовать весь комплекс эффективных средств,
снижающих тенденцию резиновых смесей к П. Основ-
Основное направление — создание широкого ассортимента
ускорителей вулканизации замедленного действия,
обеспечивающих возможность выбора соответствую-
соответствующей вулканизующей системы применительно к усло-
условиям технологич. обработки смесей и к требуемым
свойствам вулканизатов.
Лит.: Блох Г. А., Органические ускорители вулкани-
вулканизации каучуков, Л., 1972; Догадкин Б. А., Химия эла-
эластомеров, м., 1972, гл. 5; Гофманн В., Вулканизация и
вулканизующие агенты, пер. с нем., Л., 1968, с. 380; Догад-
Догадкин Б. А. [и др.], Высокомол. соед., 3, № 4, 497 A961);
№ 10, 1572 A961); 5, № 2, 164 A963); 11 А, № 7, 1590 A969);
14А, № 1, 278 A972); Гринберг А. Е., Золотарев-
ская Л. К., Замедлители подвулканизации, М., 1970;
Leib R. I., Sullivan А. В., Trivette С. D., Rubb.
Chem. and Technol., 43, № 5, 1188 A970); Davies J. R.,
D a v i e s K. M., L 1 о у d D. G., Kaut. und Gummi, Kunstst.,
24, № 5, 213 A971). Л. К. Золотарвеская.
ПОКРЫТИЯ ДЛЯ ПОЛОВ полимерные (flo-
(floorings, Bodenbelage, revetements de plancher). Материа-
Материалы, предназначенные для устройства верхнего покры-
покрытия пола, подразделяют на три группы: 1) рулонные;
2) плиточные; 3) наливные. Первая группа включает
линолеум, а также ворсовые ковровые материалы,
к-рые, как и линолеум, укладывают непосредствен-
непосредственно по железобетонному основанию на всю площадь пола.
Ворсовые материалы в данной статье не рассматривают-
рассматриваются; о технологии их получения см. Нетканые изделия.
Линолеум
Линолеум (Л.) изготовляют из оксидированных ра-
растительных масел, алкидных смол, поливинил хлорида,
синтетич. каучуков и др. полимеров с применением
тканевой основы или без нее. [За рубежом «линолеу-
«линолеумом» (от лат. linium — полотно, oleum — масло) наз.
только рулонный материал, представляющий собой
ткань (обычно джутовую), покрытую с одной стороны
слоем оксидированного льняного масла. В СССР такой
Л. выпускают в ограниченных масштабах].
Поливинилхлоридный линолеум. Л. этого вида полу-
получают из суспензионного или эмульсионного (латексно-
го) поливинилхлорида. В состав композиции (т. наз.
линолеумной массы) входят пластификаторы, напол-
наполнители, стабилизаторы, пигменты (о принципах со-
составления поливинилхлоридных композиций см. Пла-
Пластикат). В качестве основы Л. применяют ткани (джу-
(джутовую, льняную и др.) или нетканые волокнистые
материалы. Изготовляют также безосновный Л. Для
получения Л. применяют три способа — промазной,
вальцево-каландровый, экструзионный.
Промазной способ состоит в нанесении
высоконаполненной линолеумной массы на движущую-
движущуюся тканевую или др. основу с последующей термооб-
термообработкой в специальных (т. наз. терможелировочных)
камерах. Примерный состав линолеумной массы (в %):
эмульсионный (иастообразующий) поливинилхлорид —
42,0; диоктилфталат — 18,0; веретенное масло — 1,1;
тальк — 35,0; ZnO — 2,6; пигменты — 1,1; электро-
электрокорундовый порошок — 0,2.
Линолеумную массу получают перемешиванием в
лопастных смесителях пасты поливинилхлорида, кра-
красящей насты и наполнителей. Пасту поливинилхлорида
готовят смешением полимера с пластификаторами в
лопастных смесителях (для набухания полимера смесь
выдерживают при комнатной темп-ре в течение 2—12 ч);
красящую пасту — смешением пигмента с пластифика-
пластификатором в смесителе и последующей многократной обра-
обработкой в трехвалковой краскотерке (см. Краски).
Износостойкая пленка поливинилхлоридной пластмас-
пластмассы (толщина 1,5—2 мм) образуется при одно- или мно-
многократном нанесении линолеумной массы на ткане-
тканевую основу. В первом случае массу наносят с помощью
ракельного устройства на т. наз. грунтовально-жели-
ровочном агрегате, снабженном термокамерами. Про-
Проходя через термокамеры, масса желатинирует, а затем
на установленном в конце агрегата двухвалковом ка-
каландре из нее формуется (калибруется) пленка задан-
заданной толщины.
Агрегат для получения Л. методом многократного
нанесения массы имеет три последовательно располо-
расположенных блока, каждый из к-рых снабжен ракельным
устройством и терможелировочной камерой. В этом
случае формование материала на каландре не тре-
требуется, и вместо каландра в конце агрегата м. б. уста-
установлено устройство для нанесения печатного рисунка
или тиснения.
Достоинства промазного способа — простота, надеж-
надежность и возможность полной автоматизации техноло-
технологич. процесса, сравнительно низкая энерго- и метал-
металлоемкость оборудования; возможность получения Л.
на теплоизоляционной основе без применения дубли-
дублирующего агрегата. Недостатки способа — сравнитель-
сравнительно небольшая производительность F—10 м/мин); воз-
возможность использования только эмульсионного (пас-
тообразующего) поливинилхлорида.
Вальцево-каландровый способ,
к-рый используют для производства безосновного Л.,
заключается в обработке высоконаполненных поливи-
поливинилхлоридных композиций в специальных смесителях
и на вальцах и последующем их формовании на четы-
рехвалковых каландрах. Примерный состав линолеум-
линолеумной массы (в %): суспензионный поливинилхлорид —
40; диоктилфталат — 18; стабилизаторы (стеараты
кальция или бария, силикат свинца) — 1,0; кумароно-
инденовая смола — 2; белые пигменты (TiO2, ZnO,
литопон) — 10; цветные пигменты — 2; асбест — 27.
Компоненты линолеумной массы сначала смешивают
в лопастных смесителях, а затем обрабатывают в сме-
685
ПОКРЫТИЯ ДЛЯ ПОЛОВ
686
сителях-пластикаторах роторного типа при 150—160 °С.
После этого обработку (пластикацию) смеси продол-
продолжают при 130—135 °С на двух последовательно распо-
расположенных вальцах: на первых — 5 — 7 мин, на вто-
вторых — 10—12 мин\ зазор между валками — соответ-
соответственно 0,8—1,1 и 2,2—2,4 мм. Л. формуют на Z-об-
разном каландре при 130—135 °С. Таким образом
получают однослойный Л.
При изготовлении многослойного материала отдель-
отдельные пленки, полученные на каландре, дублируют на
барабанном прессе непрерывного действия в полотно
толщиной ок. 2 мм. Вальцево-каландровым способом
изготовляют также теплозвукоизоляционный Л. В этом
случае пленки дублируют на барабанных прессах
с основой из нетканых материалов. Для повышения
адгезии на пленку предварительно накладывают тон-
тонкий слой пасты поливинилхлорида.
Основные достоинства вальцево-каландрового спо-
способа: высокая производительность A0—30 м/мин)]
возможность получения на одном и том же оборудо-
оборудовании как однослойного безосновного Л. толщиной до
2 мм, так и пленок различной толщины @,05—0,5 мм).
Однако этот способ более энергоемок, чем др. способы
изготовления поливинилхлоридного Л. Кроме того,
для установки громоздкого и металлоемкого оборудо-
оборудования требуются мощные фундаменты и большие про-
производственные площади. Для получения многослой-
многослойного Л. или Л. на теплоизолирующей волокнистой
основе необходим дублирующий агрегат.
Экструзионный способ изготовления Л.
состоит в выдавливании линолеумной массы на спе-
специальных экструдерах с щелевыми головками. При-
Примерный состав массы для верхнего и нижнего слоев
приведен ниже (в %):
Верхний Нижний
слой слой
Суспензионный поливинилхлорид 4 0,0 32,0
Гидрофобный мел 24,0 35,0
Стабилизаторы (маточная смесь с
пластификатором) 15,7 —
Диоктилфталат 20,0 12,0
Хлорированный парафин — 7,0
Пигменты 0,3 6,0*
Измельченные отходы поливинил-
поливинилхлорида — 8,0
* Маточная смесь с поливинилхлоридом
Компоненты линолеумной массы смешивают (раз-
(раздельно для каждого слоя) в течение 20 мин в двухка-
двухкамерных вертикальных смесителях при темп-pax в верх-
верхней и нижней камерах соответственно 150 и 40 °С.
Готовые смеси подают в экструдеры для верхнего слоя
(диаметр шнека 30 мм, производительность 110 кг/ч)
и нижнего слоя A50 мм, 350 кг/ч). Иногда применяют
также двухшнековый экструдер. Выходящие из эк-
струдеров массы попадают в общую щелевую головку,
в к-рой при 160 °С оба слоя Л. свариваются в моно-
монолитное полотно. Иногда сваренное полотно дублируют
с прозрачной пленкой поливинилхлорида, на к-рую
нанесен печатный рисунок, напр, имитирующий пар-
паркет. Затем Л. проходит через горячие гладильные
валки, установку для снятия напряжений с камера-
камерами нагрева F0—120 °С) и охлаждения (до 30 °С) и
устройство, в к-ром одновременно производится об-
обрезка продольной кромки Л., поперечная резка
полотна и намотка его в рулоны. Оборудование для
получения поливинил хлоридного Л. экструзионным
способом отличается малой металлоемкостью. По-
Поскольку способ позволяет получать сразу двухслой-
двухслойный Л., то установка дублирующего агрегата необхо-
необходима только в случае изготовления Л. на теплоизоли-
теплоизолирующей войлочной основе. Производительность спо-
способа невысока A—3 м/мин).
Алкидный линолеум. Верхний слой этого Л. изго-
изготовляют из алкидных смол, к-рые получают из смесей
льняного или подсолнечного масла с кубовыми остат-
остатками синтетич. жирных к-т. Процесс изготовления
Л. включает след. стадии: 1) оксидация растительных
масел и кубовых остатков синтетич. жирных к-т;
2) получение алкидной смолы; 3) приготовление лино-
линолеумной массы; 4) наложение линолеумной массы на
тканевую основу, 5) приготовление т. н. «жидкого
сиккатива» для пропитки (грунтовки) оборотной сто-
стороны ткани с целью ее предохранения от гниения и
др. повреждений при эксплуатации Л.; 6) грунтовка
оборотной стороны ткани; 8) термообработка и «вызре-
«вызревание» Л.
Растительные масла и кубовые остатки синтетич.
жирных к-т оксидируют в специальных (т. н. линокси-
новых) аппаратах, снабженных мешалкой и рубашкой.
Масло, содержащее 0,2—0,3% сиккатива (РЬО), вы-
выдерживают в аппарате при 85—90 °С и непрерывной
подаче воздуха в течение 6—8 ч до получения продукта
с вязкостью не менее 250 сек (по ВЗ-4). По окончании
процесса масло охлаждают до 45—50 °С и сливают
в сборник. Кубовые остатки синтетич. жирных к-т
оксидируют в расплавленном виде в присутствии
0,3% свинцового глета A2—15 ч при ~90 °С).
Алкидную смолу готовят в цилиндрич. вертикаль-
вертикальных котлах с лопастными мешалками при 220 °С в те-
течение 6—7 ч. Смесь оксидированных растительных
масел и кубовых остатков синтетич. жирных к-т сна-
сначала переэтерифицируют смесью глицерина и ксили-
тана, а затем конденсируют с фталевым ангидридом.
Готовую смолу выливают тонким слоем на металлич.
пол, посыпанный древесной мукой. Затвердевшую
охлажденную смолу (т. н. «линолеумный цемент»)
режут на куски и используют для приготовления ли-
линолеумной массы, имеющей след. состав (в %): алкид-
ная смола — 27,0; пробковая мука — 7,5; древесная
мука — 23,5; асбест — 20,0; тальк — 1,0; измельчен-
измельченные обрезки Л.— 9,0; пигменты (охра, сурик, мумия) —
11,0; кумароно-инденовая смола — 1,0.
Линолеумная масса м. б. также приготовлена непре-
непрерывным способом. В этом случае алкидную смолу син-
синтезируют в нескольких последовательно расположен-
расположенных аппаратах. Смолу с заданной вязкостью, выходя-
выходящую из последнего аппарата, перемешивают в смеси-
смесителе с остальными ингредиентами.
Линолеумную массу наносят на джутовую или льня-
льняную ткань на каландрах. После каландра Л. поступает
сначала на медный охлаждающий барабан, а затем на
намоточный валик. При намотке для предохранения
поверхности Л. прокладывают с помощью промежуточ-
промежуточного валика слой бумаги с плотностью 35—40 г/ж2.
«Жидкий сиккатив» изготовляют из смеси льняного
масла с измельченной канифолью C5% от массы масла)
при постепенном нагревании до 230 °С (скорость подъ-
подъема темп-ры 20—30 °С/ч), затем в смесь добавляют
сиккатив (свинцовый глет), поднимают темп-ру до
250 °С и вводят второй сиккатив (пиролюзит). Коли-
Количество глета и пиролюзита составляет ~3 % от массы
реакционной смеси. После полного растворения пиро-
пиролюзита нагревание прекращают, смесь охлаждают до
100 °С и сливают в расходные баки. Общая продолжи-
продолжительность изготовления «жидкого сиккатива» — 12—
14 ч. Грунтовку оборотной стороны ткани «жидким
сиккативом» производят на грунто-промазочном агре-
агрегате.
После грунтовки Л. развешивают в сушильных ка-
камерах на железных ригелях ровными фалдами. Про-
Продолжительность сушки и «вызревания» Л.— 3—5 сут,
темп-ра — 45—62 °С. По окончании сушки на Л.
наносят с помощью печатных машин рисунок (при
получении «узорчатого» Л.) и сматывают полотнища
Л. в рулоны.
Резиновый линолеум (релин) представляет собой
двухслойный материал. Верхний (лицевой) слой Л.
687
ПОКРЫТИЯ ДЛЯ ПОЛОВ
688
получают гл. обр. из цветной резиновой смеси на основе
синтетич. каучука, не содержащего окрашивающих
антиоксидантов. В состав смеси вводят наполнители
(напр., белую сажу, каолин, мел), пластификаторы
(напр., вазелиновое масло, канифоль), серу, ускори-
ускорители вулканизации (напр., каптакс, тиурам), пигмен-
пигменты. Нижний слой Л. может быть изготовлен: 1) из ре-
резиновой смеси (напр., на основе бутадиен-стирольного
каучука CKMG-30APKM-15), в к-рую наряду с обычны-
обычными ингредиентами вводят измельченные отходы резины;
2) из вулканизующейся композиции измельченных от-
отходов резины с битумом.
Свойства линолеума
Показатели
Износостойкость
по уд. изменению массы
(ГОСТ 426-66), г/см*,
не более .... . .
по изменению толщины
(ГОСТ 11529 — 65), мкм,
не более
Упругость (ГОСТ 12729-67),
% , не менее
Водопоглощение за 24 ч, %,
не более
Поливинилхло-
ридный линолеум
на тка-
тканевой
основе*
0,06
45
45
5
одно-
однослойный
безоснов-
безосновный**
0,05
40
1,5
Алкид-
ный
лино-
линолеум
60
35
10***
Рези-
Резиновый
лино-
линолеум
0,05
40
75
1,0
*Промазной способ. ** Вальцево-каландровый способ.
••'•"** Для Л. толщиной 2,5 — 3 ли*.
Смеси изготовляют обычно в резиносмесителях (см.
Смесители), вулканизующие агенты вводят на листо-
вальных вальцах. Резиновое полотно для верхнего и
нижнего слоев Л. получают на трехвалковых каланд-
каландрах. Заключительная стадия процесса — дублирова-
дублирование верхнего и нижнего слоев и одновременная вулка-
вулканизация Л. (темп-pa ~160 СС) на барабанных вулкани-
вулканизаторах непрерывного действия (см. Вулканизацион-
ное оборудование).
Основные показатели Л. приведены в таблице.
Плиточные материалы
Эти материалы изготовляют из композиций на основе
поливинилхлорида, синтетич. каучуков, кумароно-
инденовых смол, коллоксилина и нек-рых др. связую-
связующих. Достоинство плиточных материалов — возмож-
возможность создания декоративных покрытий. Плитки вы-
вырубают из соответствующих рулонных материалов
(линолеума) или изготовляют но специальной техно-
технологии, включающей приготовление плиточной массы,
ее вальцевание или каландрование для получения листа
нужной толщины и вырезку плиток. Напр., двухслой-
двухслойные поливинилхлоридные плитки вырубают на спе-
специальных прессах из двухслойного поливинилхлорид-
ного линолеума, изготовляемого экструзионным или
вальцево-каландровым способом. Однослойные плитки
получают по упрощенной технологии: из приготовлен-
приготовленной в смесителе массы на двух парах вальцов формуют
лист толщиной 2—3 см, охлаждают его и вырезают
плитки нужной формы.
В состав плиточной массы входят смесь суспензион-
суспензионного и эмульсионного поливинилхлорида, наполни-
наполнитель (напр., тальк), красители, небольшое количест-
количество абразива (напр., электрокорунда), применяемого для
повышения износостойкости покрытия, и нек-рые др.
компоненты. В случае получения резиновых плиток вы-
вырезанную заготовку вулканизуют. Иногда плитки из-
изготовляют прессованием порошкообразных компози-
композиций при повышенных температурах, однако этот способ
малопроизводителен.
Наливные составы
Применение наливных составов позволяет полу-
получать бесшовные покрытия, более дешевые, чем др.
полимерные покрытия иолов, и превосходящие их но
простоте устройства и эксплуатационным свойствам.
Благодаря этому наливные материалы широко исполь-
используют в отечественной и зарубежной строительной прак-
практике. Различают две группы материалов: 1) мастичные
составы, к-рые наносят на железобетонное перекрытие
распылением или поливом; 2) бетонные составы на
основе полимер цемента, для нанесения к-рых приме-
применяют укладочные машины или виброириспособления.
Мастичные составы. Для их получения водную дис-
дисперсию полимера, напр, поливинилацетата, смешивают
с пластификатором (дибутилфталатом) и разбавляют
водой. Наполнитель (напр., песок) промывают, сушат,
измельчают в вибромельнице с керамич. шарами и
смешивают в вибросмесителе с пигментами и стабили-
стабилизаторами (напр., с формалином). После этого окрашен-
окрашенный наполнитель и разб. дисперсию перемешивают
в смесителе с Z-образными лопастями и фильтруют
через вибросито. Изготовляют мастичные составы
двух типов: 1) шпатлевочный, или выравнивающий
(павилит Ш), образующий нижний слой покры-
покрытия, и 2) отделочный (навилит О). Шпатлевочный
состав содержит в 4—5 раз больше наполнителя
D мае. ч. в расчете на 1 мае. ч. 50%-ной дисперсии),
чем отделочный. Покрытие из мастичного состава тол-
толщиной 3—4 мм высыхает при 18—25 °С в течение 24 ч.
Пленка отделочного состава имеет прочность при рас-
растяжении ок. 3 Мн/м2 (ок. 30 кгс/см2). Мастичные мате-
материалы применяют для устройства полов в жилых и
общественных зданиях, а также в таких производст-
производственных помещениях, где полы подвергаются слабым
механич. воздействиям (напр., при передвижении толь-
только ручных тележек на резиновых шинах). Составы
на основе дисперсии поливинилацетата пригодны для
получения масло- и бензостойких покрытий полов.
Бетонные составы на основе полимерцемента. Для
получения этих составов используют гл. обр. водную
дисперсию поливинилацетата или бутадиен-стирольный
латекс (см. Латексы синтетические). Наполнителями
служат мраморный или гранитный щебень (фракция
7 —10 мм) и каменный песок из этих же пород (фрак-
(фракция 0,25—1,5 мм).
В строительной практике применяют составы, к-рые
поставляются в двух упаковках. В одной из них со-
содержится стабилизированная дисперсия полимера, в
другой — сухая минеральная часть, к-рую получают
предварительным перемешиванием цемента с пигмен-
пигментом в вибромельнице и последующим смешением окра-
окрашенного цемента с наполнителями в обычном смеси-
смесителе. Бетонный состав приготовляют непосредственно
на строительстве. Напр., состав б е т о л и т полу-
получают, смешивая компоненты в след. соотношениях
(по массе): минеральная часть — 5,0; 50%-ная диспер-
дисперсия — 0,4; вода — 0,4. Покрытия, к-рые образуются
в результате затвердевания этого состава, характери-
характеризуются след. механич. свойствами: прочность при
сжатии 25—40 Мн/м2 B50—400 кгс/см2), прочность
при изгибе 10—13 Мн/м2 A00—130 кгс/см2). Прочность
покрытия возрастает во времени: через 3 мес после
нанесения состава она примерно на 10% превышает
прочность обычного бетона.
Бетонные составы на основе полимерцемента реко-
рекомендуется применять в производственных помещениях,
где покрытия полов эксплуатируются в условиях уме-
умеренных механич. воздействий. Составы на основе
дисперсии поливинилацетата более пригодны для
устройства полов с сухим режимом эксплуатации, со-
составы на основе бутадиен-стирольного латекса — для
устройства полов с влажным режимом эксплуатации.
689
ПОЛЗУЧЕСТЬ
690
0 применении полимерных материалов для покрытия
полов см. также Полимеры в строительстве.
Лит.: Линолеум, М., 1960; Данцин М. И., Теплозву-
коизоляционный линолеум, М., 1964; Быков А. С, Дан-
Данцин М. И., 3 о х и н Г. И., Строительные материалы и из-
изделия на основе синтетического сырья, 2 изд., М., 1970.
М. И. Дсищин.
ПОЛЗУЧЕСТЬ полимеров, крип (creep,
Kriechen, fluage) — процесс нарастания во времени
деформации материала в режиме постоянного истинного
напряжения о. В узком смысле речь идет только о раз-
развитии обратимых деформаций, хотя часто не разделяют
полную накопленную деформацию на обратимую и не-
необратимую (пластическую) составляющие. П. наз. так-
также процесс накопления деформации в режиме постоян-
постоянной нагрузки, т. е. постоянного условного, а не ис-
истинного напряжения.
П. можно количественно характеризовать отноше-
отношением деформации 8, развившейся до данного момента
времени t, к действующему постоянному напряжению
а. Это отношение наз. податливостью 1 (t), к-рая не
зависит от о при малых его значениях и в этом случае
является характеристикой данного материала, опреде-
определяемой только длительностью действия напряжения
и темп-рой. При больших о происходит переход в не-
нелинейную область механич. поведения и I(t) обычно
возрастает с увеличением напряжения.
Измерение П. (т. е. ее характеристики — податли-
податливости) при малых о широко используют как экспери-
экспериментальный метод определения физич. состояний по-
полимеров и темп-р переходов. В этом отношении опреде-
определение податливости представляет собой основной ва-
вариант общего метода термомеханич. исследования, при
к-ром измеряется I(t) при фиксированном значении
времени для различных темп-р; температурная зависи-
зависимость податливости, измеренная таким образом, пред-
представляет собой одну из основных физико-механич.
характеристик материала и определяет положение
областей его релаксационных состояний на темпера-
температурной шкале.
Типичные зависимости I(t) для различных полимер-
полимерных систем, полученные при малых о в широком
диапазоне изменения t, показаны на рис. 1. Для аморф-
аморфных полимеров большой мол. массы с широким моле-
кулярно-массовым распределением характерна зависи-
зависимость I(t) типа представленной кривой 2, но без плато
"-10
-10
-5
0
-5
0
Рис. 1. Зависимости I(t) для различных полимерных систем:
1— аморфные линейные полимеры с низкой мол. массой (пун-
(пунктир соответствует податливости I без учета составляющей
вязкого течения); 2 — аморфный линейный полимер с большой
мол. массой и узким молекулярно-массовым распределением;
3 — тот же полимер в области темп-р ниже темп-ры стекло-
стеклования (т. обр., 3 — продолжение 2 в области более низких
значений t)\ 4 — разб. сшитые гели; 5 — слабо сшитые
эластомеры; б — закристаллизованные полимеры. Констан-
Константа А на оси абсцисс выбрана так, чтобы экспериментальные
данные были представлены в одном масштабе значений t.
в средней части [в этом случае наблюдается постепен-
постепенное возрастание I(t)]. Для структурированных и сши-
сшитых полимеров характерно существование конечного
предельного, равновесного значения податливости /оо
(рис. 1, кривые 4 и 5). Значение /оо для пространст-
пространственно структурированных полимеров (см. кривую 5)
или значение /(?), соответствующее области плато (см.
кривую 2), для аморфных линейных полимеров отве-
отвечает высокоэластич. состоянию, а резкое возрастание
податливости для аморфных полимеров (в отсутствие
химич. процессов) — переходу в вязкотекучее состоя-
состояние. Для закристаллизованных полимеров (см. кривую
6') наблюдается очень медленное возрастание податли-
податливости в широком диапазоне значений времени. При
t ->¦ 0 для всех полимеров типичны близкие мгновен-
мгновенные значения податливости; они составляют ок.
103 Мн/м2 A010 дин/см2), что соответствует обычным зна-
значениям / твердых аморфных тел.
Вид функции I(t) определяется характером спектра
распределения времен запаздывания (ретардации) си-
системы и через эту фундаментальную характеристику ма-
материала связан со всеми остальными релаксационными
функциями, описывающими механич. свойства мате-
материала при малых о и произвольных режимах нагруже-
ния (см. Реология). Такой подход связан с использова-
использованием для описания зависимости e(t) широко распростра-
распространенной т. наз. теории наследственности. Согласно этой
теории, деформация в момент времени t зависит от
предшествующей истории изменения напряжений. Для
теории наследственности в линейной области справед-
справедлив принцип линейной суперпозиции Больцмана —
Вольтерры (см. Больцмана — Волъпгерры уравнения),
к-рый для обратимых деформаций при малых напря-
напряжениях о м. б. записан в виде
t
где Е — мгновенный модуль упругости. При высоких
напряжениях для описания П. применяют нелинейные
интегральные ур-ния. Из эмпирич. ур-ний, используе-
используемых для прикладных целей при описании эксперимен-
экспериментальных данных по за-
зависимости деформации е
от напряжения о и вре-
времени t (особенно приме-
применительно к конструк-
Рис. 2. Типичные деформа-
деформационные кривые конструк-
конструкционных полимеров. тк —
время достижения области
критич. ползучести.
ционным полимерным материалам, типичные дефор-
деформационные кривые к-рых показаны на рис. 2), рас-
распространение получили след. ф-лы, достаточно хорошо
описывающие реальные опытные данные в интервале
изменения t до 2—3 десятичных порядков:
? ~ ат in; ? ~ tn sh (<i/am);
? = кг ат A — e-tf) + к2 ат t; в ~ вх + в2 In t
(здесь кх, к2, т, п, am, ex, e2, q — эмпирич. постоянные).
Зависимость деформации от времени обычно м. б.
разделена на три характерных участка (см. рис. 2) —
начальной, установившейся и критич. П. Для практич.
целей не всегда требуется знать характер развития П.,
но важно установить время тк достижения области кри-
критич. П., когда начинается резкое возрастание скорости
накопления деформации. Нек-рыми авторами тк наз.
деформационной долговечностью
материала при заданном напряжении.
Форма ур-ний, описывающих зависимость тк и
долговечности от а и абсолютной темп-ры, одинакова,
хотя численные значения констант, входящих в эти
ур-ния, вообще говоря, не совпадают.
При П., поскольку она протекает при наложении де-
деформирующего напряжения, одновременно происходит
и разрушение материала (преобразование надмолеку-
надмолекулярной структуры, появление и рост микротрещин).
691
ПОЛИАЗАПОРФИНЫ
692
Пока нет ясности, какой из этих макро-процессов
(ползучесть или разрушение) первичен, а какой — вто-
вторичен. Если деформация протекает быстрее, чем раз-
разрушение, это приводит к макроползучести; в обратном
случае происходит разрушение тела. Равенство значе-
значений долговечности и деформационной долговечности
отвечает переходу от вынужденной высокоэластич. де-
деформации к хрупкому разрушению. Пластификация по-
полимера ускоряет, а ориентация замедляет П. Пониже-
Понижение П. равноценно повышению теплостойкости и пре-
предела вынужденной высокоэластичности.
П. обычно резко возрастает при наложении на по-
постоянную или медленно меняющуюся нагрузку ви-
вибраций с относительно небольшой амплитудой. В этом
случае развивается процесс т. наз. виброползу-
виброползучести, проявляющийся в значительном увеличе-
увеличении скорости П. по достижении нек-рых амплитуд и
частот налагаемых нагрузок. Причина этого явления
часто связана с саморазогревом материала из-за дис-
диссипации энергии колебаний, происходящим после
нек-рого индукционного периода, что приводит к росту
](t). Др. причиной ускорения развития деформаций
м. б. изменение релаксационных свойств материала
в режиме виброползучести из-за нелинейности режима
деформирования при наложении колебаний.
Лит.: Буга ков И. И., Ползучесть полимерных мате-
материалов, М., 1973. А. Я. Малкин.
ПОЛИАЗАПОРФИНЫ, полипорфиразины
(polyazaporphins, Polyazaporphine, polyazaporphines) —
полимеры, в макромоле-
макромолекулах к-рых содержатся
повторяющиеся азапор-
финовые макроциклы (I).
сн=сн
=С С—N
\У^ II
N=C
сн=с
сн=
NH
¦i »
CH=
I
I
/С—СН
/
HN
х
|
с-сн
А
сн
П. могут быть безметаллическими (не содержат атома
металла в азапорфиновом цикле) и металлсодержащими
(представляют собой комплексы с металлом); последние
П. могут содержать металл одного, двух или трех ти-
типов (напр., медь, же-
железо или никель, медь
и железо, медь и ни-
никель). Такие П. наз.
соответственно моно-,
би- или триметалличе-
скими.
В макромолекуле
азапорфиновые мак-
роциклы могут соеди-
соединяться между собой
через ароматич. или
гетероциклич. коль-
кольца [полифталоциани-
ны (II—IV), полина-
фталоцианины, поли-
фураноцианины (V),
политиофеноцианины
(VI), через кислород, связанный с металлом двух
соседних звеньев, а также непосредственно — через
пиррольные циклы соседних макроколец [полимеры
тетрацианэтилена (VIII, IX)].
Различают П. ленточного (поясного) строения (поли-
(полимеры III, IV, VII) и П. зашитого (плоскостного, или пар-
кетообразного) и стопочного строения (полимеры II, V,
—С- -С
—С--С
IX). П. характеризуются системой сопряженных связей,
к-рая м. б. непрерывной, напр. в полимерах II, IV—IX,
что создает благоприятные возможности для обобщест-
обобществления я-электро-
нов по всей макро-
макромолекуле, или пре-
прерываться атомами
О, S, а также груп-
группами СО, SO2 (напр.,
полимер III).
П. обладают ком-
комплексом специфич.
физико-химич. свой-
свойств, обусловленных
наличием 18 неспа-
ренных д-электро-
нов, к-рые делока-
лизованы в плоско- ^
сти азапорфинового макроцикла и придают ему арома-
тич. характер, а также высокую устойчивость. П.— не-
неплавкие, труднорастворимые, гигроскопичные вещест-
вещества, окрашенные в различные цвета (от сине-зеленого до
черного); устойчивы при нагревании на воздухе до
300—350 °С и в инертной
атмосфере — до 500—600 С0.
П. частично растворимы в
CN
V,X=O;
VI,X=S
диметилформамиде, пиридине, диметилсульфоксиде,
конц. H2SO4. Металлич. П., содержащие Са, Mg или
РЬ, в результате обработки конц. H2SO4 теряют
атомы металла, превращаясь в безметаллические про-
продукты; комплексы с Си, Ni или Со при этом практи-
практически не изменяются. При длительном воздействии
разбавленной H2SO4 П. гидролизуются.
/CN
,С=С-
NC
/С=с.
С—
—С--С
МеХ
с=с-
с=с-
Для металлич. и безметаллич. П. возможны взаим-
взаимные переходы. Схематически такие превращения по-
показаны на примере полимеров тетрацианэтилена
(VIII, IX).
П. обладают полупроводниковыми свойствами. В за-
зависимости от структуры, метода синтеза и условий
очистки уд. электрич. проводимость составляет
ТТОЛИАЗИНЫ
<>94
10-ю— \0-*сим/м (Ю-12 — 1(Г4 ом-1-см-1) при энергиях
активации проводимости 0,5—0,15 эв. П.— высокоселек-
высокоселективные катализаторы нек-рых реакций окислительно-
восстановительного тина (окисления алкиларомати-
ческих углеводородов, разложения перекиси водоро-
водорода и др.).
П. получают при взаимодействии тетрафункциональ-
ных к-т ароматич. или жирного ряда, а также их произ-
производных (напр., пиромеллитовая к-та, 3,3',4,4'-тетра-
карбоксидифениловый эфир, тетрацианэтилен, тетра-
цианбензол) с металлами или их солями в р-ре, расплаве
и парах мономера при 200—350 °С. При проведении
реакции в парах мономера П. получают в виде пленоч-
пленочных покрытий на изделиях любого размера и профиля,
что открывает пути практич. использования этих поли-
полимеров.
Сополимерные азапорфины получают аналогичным
образом, вводя в реакцию смеси соответствующих к-т
или их производных. Так, для получения ленточных
полимеров используют смеси ди- и тетранитрилов,
напр, фталонитрила и тетрацианэтилена (полимер VII),
фталонитрила и тетрацианбензола.
П. получены впервые в 1949—52 в США.
Лит.: Berlin A. A., Sherle A. I., Inorganic Macro-
raol. Revs, I, № 3, 235 A971). А. А. Берлин, А. И. Шерле.
ПОЛИАЗИНЫ (polyazines, Polyazine, polyazines) —
полимеры, содержащие звенья =N — N = G—R—С=,
I I
R' R"
где R = - CH2-, - C6H4 -, -CH2C6H4CH2 -,
-C6H4OC6H4—, — CeH4SC6H4— и др.; R' и R"=H,
одинаковые или разные органич. радикалы, напр.
—СН3, —С2Н5, —С6Н5.
П.— кристаллич. или аморфные полимеры линейной
или циклич. структуры. Мол. масса П. может достигать
20 000, однако у П. с системой сопряженных связей,
охватывающей всю макромолекулу, степень полимери-
полимеризации обычно не превышает 8—12. П. ограниченно раст-
растворимы в метиловом спирте, бензоле, хлороформе,
несколько лучше — в тетрахлорэтане, диметилформа-
миде, нитробензоле, крезолах, камфоре, а также
вконц. серной и муравьиной к-тах. П. могут быть окра-
окрашены в цвета от желтого до темно-коричневого. Глу-
Глубина окраски зависит от длины системы сопряженных
связей. П., в макромолекулах к-рых участки сопряже-
сопряжения разделены, напр., атомами О или S, группами
—СН2— или — СН2О —, плавятся в интервале 220—
350 СС.
Темп-pa плавления П. с системой сопряженных свя-
связей, охватывающей всю макромолекулу, обычно выше
темп-ры их разложения. Пиролиз полимеров этого типа
сопровождается значительными экзотермйч. эффекта-
эффектами. Разложение П., протекающее в узком A0—20 °С)
интервале темп-р, приводит к образованию термостой-
термостойких продуктов, практически не содержащих азота. На-
Наличие ароматич. циклов в цепи П., характеризующихся
системой сопряженных связей, охватывающей всю
макромолекулу, способствует повышению термостой-
термостойкости полимеров, но не изменяет общего характера дест-
деструкции. По-видимому, термич. распаду П. этого ти-
типа предшествует их перегруппировка в полиазосое-
динения:
C
Существенно по иному протекает термодеструкция
П., в макромолекулах к-рых короткие участки сопря-
сопряжения разделены гетероатомами или др. группами.
Термич. распад этих П. начинается при 350—400 °С
и не сопровождается значительными тепловыми эф-
эффектами; образующиеся твердые продукты пиролиза
содержат азот. В результате озонолиза П. образуются
сложные, трудно идентифицируемые азотсодержащие
продукты окисления.
Формирование кристаллич. областей в П., в к-рых
система сопряженных связей охватывает значительные
участки макромолекулы, часто приводит к ослаблению
таких типичных для полисопряженных систем свойств,
как поглощение в длинноволновой области спектров,
парамагнетизм, способность к образованию комплексов
с переносом заряда и др. Это происходит, если кристал-
кристаллизация сопровождается выходом из копланарности
отдельных блоков сопряжения в макромолекулах П.
Нарушение же регулярности чередования одинаковых
структурных элементов предотвращает кристаллиза-
кристаллизацию и способствует тем самым появлению у П. свойств,
типичных для полисопряженных систем.
Аморфные П. с системой сопряженных связей, охва-
охватывающей значительные участки цепи полимера, обла-
обладают полупроводниковыми свойствами и, в частности,
высокой фотоэлектрич. чувствительностью в видимой
и ультрафиолетовой областях спектра. Высокая фото-
фотоэлектрич. чувствительность часто сочетается у П. с вы-
высоким электрич. сопротивлением в темноте. П. с си-
системой полисопряжения образуют с акцепторами элект-
электронов (напр., с Вг2, 12, хлоранилом, тетрацианэтиле-
ном) комплексы с переносом заряда, к-рые являются па-
парамагнитными веществами, обладающими высокой
электрич. проводимостью A -10~6 — 7-10~4 ом^-см'1).
Некоторые П. характеризуются значительной фотосен-
сибилизирующей активностью, напр, в реакции фотоини-
циированного окисления аскорбиновой к-ты кислородом.
П. получают поликонденсацией по след. схемам:
nO=C-R-C=O+nH2N-NH2
I |
R'
R"
O[=C-R-C=N-N=1
I I
R' R"
n
Bn- 1)H2O
nH,N-N=C-R-C=O ->
I I
R' R"
->H8r=N-N=C-R-C=L O+(n - 1)H2O
I ! П
R' R"
Реакцию осуществляют в расплаве (с отгонкой воды)
или в среде органич. к-т (напр., уксусной или масляной)
при 95—110 °С.
При взаимодействии гидразина с а-дикарбонильны-
ми соединениями образуются линейные П., с у-дикарбо-
нильными соединениями — как линейные, так и цик-
циклические. Образованию циклич. П. способствует уве-
увеличение числа атомов углерода между карбонильными
группами дикарбонильного компонента. П. могут быть
получены также и по реакциям т. наз. двойного обмена:
тЮ=С-Аг-С=О + ?iCeH5-C=N-N=C-CeH5 ->
II II
R' R' R" R"
О
-l) CeH5=C-R"
nH2N-NH2 +
> Bn-l) CeH5NH2
О [ = C-Ar-C=N-N=l С-С6Н5
II п 1
R' R' R"
eH5N=:C—Ar—C=sNCeHs->
| I
R' R'
CeH5N[=C-Ar-C=N-N=] H2,
| n
|
R'
R'
nCeH5N=C-Ar-C=NC6H5 + 7iC6H5-C=N-N=C-CeH5
II I '
R' R' R" R"
-> Bn-l) C6H5C=NC6H5 +
I
R"
+ G6H5NC=G-Ar-C=N-N=] G - CeH5
i I n I
R' R' R"
695
ПОЛИАКРИЛАМИД
696
Эти реакции проводят в р-рах (напр., в бензоле) или
в расплавах (при 130—150 СС).
Впервые П. были получены Курциусом в 1889.
Лит.: Давыдов Б. Э. [и др.], Изв. АН СССР, сер.
хим., № 9, 1664 A963); Д а в ы д о в Б. Э. [и др.], ДАН
СССР, 160, х» 3, 650 A965); Давыдов Б. Э. [и др.],
Изв. АН СССР, сер. хим., № 9, 1664 A963); Топ-
ч и е в А. В. [и др.], ДАН СССР, 147, № 3, 645 A962); D'A I e -
1 i о G. F., Shoenig R. К., J. Macromol. Sci., СЗ, № 1,
105—234 A969); А2, № 5, 979—1043 A968). Б. Э.Давыдов.
ПОЛИАКРИЛАМИД — см. Акриламида полимеры.
ПОЛИАКРИЛАТЫ — см. Акрилатов полимеры.
ПОЛИАКРИЛОВАЯ КИСЛОТА — см. Акриловой
кислоты полимеры.
ПОЛИАКРИЛОВЫЕ КЛЕИ, акриловые клеи (acrylic
adhesives, Akrylklebstoffe, colles acryliques). В на-
настоящей статье рассмотрены клеи, основным компонен-
компонентом к-рых являются акриловая и метакриловая к-ты,
их производные или полимеры этих соединений (см.
Акриловой кислоты полимеры и Метакриловой кислоты
полимеры).
Клеи на основе акриловой и метакриловой кислот и
их производных. Клеи этого типа м. б. разделены на
три подгруппы: 1) клеевые композиции на основе р-ров
полимеров в инертных растворителях; их отверждение
происходит за счет испарения растворителя; 2) клее-
клеевые композиции на основе мономеров; их затвердева-
затвердевание происходит в результате полимеризации; 3) сме-
смешанные клеевые композиции на основе р-ров полиме-
полимеров в мономерах или смесях мономеров с инертными
растворителями; их отверждение происходит в резуль-
результате как испарения инертного растворителя и части
мономера, так и полимеризации остальной части мо-
мономера.
В качестве инертных растворителей для приготов-
приготовления клеевых композиций этого типа наиболее часто
применяют ацетон, толуол, хлороформ и метиленхло-
рид. Растворителем для акриламида и полиакриламида
обычно служит вода (см. Акриламида полимеры).
Полимеризацию акриловой и метакриловой к-т,
а также их производных, входящих в клеевую компо-
композицию, осуществляют под действием перекисей, гидро-
гидроперекисей, персульфата калия, нафтената кобальта,
стеарата кальция и др. веществ.
В клеевые композиции могут входить пластифика-
пластификаторы (эфиры фталевой и фосфорной к-т, канифоль) и на-
наполнители (цемент, кварцевая мука и др.). В качестве
модификаторов в П. к. вводят виниловые мономеры
(винилацетат, стирол, бутадиен и др.) или феноло-фор-
мальдегидные, мочевино-формальдегидные и эпоксид-
эпоксидные смолы.
Клеи этой подгруппы разработаны для решения узко-
узкоспециальных задач и отличаются друг от друга соста-
составом, свойствами, технологией применения и др. Ниже
рассмотрены наиболее распространенные отечествен-
отечественные П. к. этой подгруппы.
Клей ПК-5 предназначен для склеивания сталь-
стальных деталей с деревянными, оклеенными тканью. М. б.
использован для склеивания дуралюмина с дуралюми-
ном, древесиной, стеклопластиками, фанерой и сили-
силикатным стеклом. В состав клея ПК-5 входит ацетоно-
ацетоновый р-р полиметилакрилата, диметилакрилат-бг/с-три-
этиленгликольфталат, перекись бензоила и цемент.
Клей готовят последовательным смешением компонен-
компонентов в емкостях из эмалированного металла или стекла.
Его наносят на склеиваемые поверхности из расчета
250—300 г/м2, выдерживают на воздухе в течение 10 мин
при 20 °С и еще 10 мин при 60 °С (возможна выдержка
только при 20 °С в течение 40—50 мин), после чего склеи-
склеиваемые поверхности соединяют и выдерживают под дав-
давлением 0,5 Мн/м2 E кгс/см2) при 80 °С в течение 6 ч.
Клеевые соединения работоспособны при длитель-
длительном воздействии темп-р до 80 °С или переменном воз-
воздействии темп-р ± 60 СС, не разрушаются керосином
и минеральными маслами, однако не выдерживают дли-
длительного воздействия воды (см. также Метилакрилата
полимеры). В табл. 1 приведена прочность при сдвиге
клеевого соединения различных материалов клеем
ПК-5.
Таблица 1. Прочность при сдвиге [в Мн/м2 {кгс/см2)] клеевых
соединений различных материалов, полученных с применением
клея ПК-5
Склеиваемые материалы
Сталь ЭЯ1Т
Дуралюмин Д16АТ
Д16АТ -f дельта-древесина
Д16АТ -f стеклотекстолит
Д16АТ + фанера
Д16АТ -f силикатное стекло
Темп-pa, °С
— 60
6,5F5)
6,5F5)
20
15 A50)
14,5A45)
13,6A3 6)
13,3A33)
14,6A46)
7,9( 79)
60
7,2G2)
7,0G0)
Клей ВК-32-70 существует в виде двух
модификаций, одна из к-рых (ВК-32-70а) предназначена
для склеивания при подогреве листов или формован-
формованных изделий из органич. стекла силового назначения,
а другая (ВК-32-706) — для склеивания без нагрева-
нагревания изделий из органич. стекла несилового назначения.
Этот клей можно использовать для склеивания нек-рых
др. термопластов. Клей состоит из тетраметакрилат-
бг/с-глицерофталата, перекиси бензоила и сиккатива.
Клей наносят с помощью кисти на склеиваемые по-
поверхности из расчета 100—150 г/м'2, после чего выдержи-
выдерживают их на воздухе в течение 5—40 мин и соединяют
под давлением 0,01—0,15 Мн/м2 @,1 — 1,5 кгс/см2). От-
Отверждение происходит при комнатной темп-ре в течение
72 ч, при 65 °С — 4 ч. Клеевые соединения обладают
хорошей прозрачностью, водо-, масло- и топливостой-
костью, не подвержены действию грибков. Основным
преимуществом клея ВК-32-70 по сравнению с др. клея-
клеями, применяемыми для склеивания органич. стекла,
является то, что он не содержит растворителя и ком-
компонентов, разрушающих органич. стекло и др. термо-
термопласты. При склеивании этим клеем на поверхности
стекла длительное время не возникает «серебра».
Клей КС-609 предназначен для клеесварных
соединений металлов. В клеевую композицию входит
полибутилметакрилат, бутилметакрилат, кварцевая му-
мука и инициирующая окислительно-восстановительная
система из перекиси бензоила и диметиланилина. Клей
готовят растворением полимера в мономере, причем
полученный сироп можно хранить 7—10 су т. После
введения в него инициатора и наполнителя жизне-
жизнеспособность клея составляет ок. 4 ч. Клей наносят на
соединяемые поверхности слоем толщиной ок. 0,5 мм,
детали соединяют и сваривают по жидкому клею. При
сварке клей равномерно заполняет зазор и частично
выдавливается из-под нахлестки, образуя валик шири-
шириной ок. 3 мм. Через 5 ч после сварки клей обладает
уже достаточной прочностью, однако максимальная
прочность достигается через 7 су т.
Прочность при растяжении и сжатии клеесварных
соединений в 1,5 раза выше прочности сварного сое-
соединения тех же металлов. Важное значение имеет
эластичность клея в отвержденном состоянии; в сое-
соединениях, работающих при усталостных нагрузках,
клеевой слой не растрескивается и не отслаивается от
металла. Клеесварное соединение устойчиво в усло-
условиях различных процессов пассивирования металлов,
клеевая пленка надежно защищает сварной шов от по-
попадания на него электролитов. Клеесварное соедине-
соединение на основе клея КС-609 не склонно к старению: через
3 года после изготовления прочность и эластичность сое-
соединения сохраняются.
Водно-эмульсионный клей применя-
применяется в текстильном производстве при изготовлении
697
ПОЛИАКРИЛОВЫЕ ЛАКИ И ЭМАЛИ
698
нетканых изделий. В клеевую композицию входит
тройной сополимер алкнлакрилата, метилольного про-
производного акриламида и акриловой к-ты, вода и эмуль-
эмульгатор. Клей поставляется потребителю в готовом виде.
Нетканые материалы, изготовленные с применением
П. к., не мнутся и обладают хорошей светостойкостью.
Для склеивания бумаги разработан
П. к., представляющий собой 10%-ный водный р-р
сополимера, состоящего из 90% полиметакриламида и
10% аммонийной соли полиметакриловой к-ты. Клей
поставляется потребителю в готовом виде и может хра-
храниться при комнатной темп-ре продолжительное время.
Для склеивания кожи применяют П. к.,
представляющий собой 20% -ный водный р-р сополиме-
сополимера, состоящего из 70% полиметакриламида и 30% ам-
аммонийной соли полиметакриловой к-ты. По свойствам
клей не отличается от описанного выше.
Цианакрилатные клеи. Основу клеев этой подгруппы
составляют эфиры цианакриловой к-ты общей ф-лы
CN
I
СН2 = С — COOR, где R — алкил. Эфиры цианакри-
цианакриловой к-ты обладают высокой реакционной способно-
способностью и быстро отверждаются на воздухе под влиянием
следов влаги или в присутствии веществ основного ха-
характера. Благодаря высокой реакционной способности
цианакрилатов отпадает необходимость введения
в клеевые композиции специальных инициаторов или
катализаторов полимеризации. Для регулирования
вязкости клеев, эластичности клеевого соединения
и его устойчивости к воздействию повышенных темп-р
и жидких сред в клеевые композиции вводят пласти-
пластификаторы (дибутилфталат, простые поливинилбутило-
вые эфиры и ДР-Ь полимерные добавки (полиэтилциан-
акрилат, поливинилацетат и др.) и сшивающие агенты
(эфиры цианакриловой к-ты и ненасыщенных спиртов
и др.). „
Цианакрилатные клеи представляют собой низковяз-
низковязкие прозрачные жидкости, поставляемые в готовом
для употребления виде в полиэтиленовых бутылках
или ампулах. Срок хранения клея в герметизирован-
герметизированной таре при темп-pax от 0 до 4 °С составляет 6 мес.
Клеи этой подгруппы применяют для склеивания
металлов, стекла, пластмасс и тканей живого организ-
организма при операциях на органах дыхания и пищеварения,
в сердечно-сосудистой хирургии, хирургии внепече-
ночных желчных путей, печени и т. д.
Перед склеиванием материалы обезжиривают, нано-
наносят на них небольшие количества клея, совмещают
склеиваемые поверхности и выдерживают их под кон-
контактным давлением 1 — 5 мин без нагревания. За это
время происходит практически полное отверждение
клея, но максимальная прочность клеевого соедине-
соединения достигается спустя 24—48 ч.
При склеивании цианакрилатными клеями живых
тканей их предварительно осушают с помощью сухих
стерильных тампонов или тампонов, смоченных спир-
спиртом и эфиром. Клей наносят на склеиваемые поверх-
поверхности выдавливанием из ампул или распылением из
специального пульверизатора, затем поверхности сов-
совмещают и выдерживают под контактным давлением
несколько мин. При заживлении соединяемые ткани
срастаются, а клей постепенно рассасывается в орга-
организме в течение 1—6 мес.
Отвержденные цианакрилатные клеи в зависимости
от строения мономеров имеют различные физико-ме-
ханич. свойства и различную устойчивость к действию
воды (табл. 2).
Клеевые соединения на основе цианакрилатных кле-
клеев работоспособны при длительном воздействии темп-р
порядка 70—80 °С; при темп-pax выше 120 °С и при
минусовых темп-pax прочность их снижается почти
в 2 раза за 24 ч.
Таблица 2. Зависимость физико-механических свойств клеевых
соединений дуралюмина от природы цианакрилатов
Мономер
Этил-ос-цианакрилат
7^Пропил-а-цианакри-
лат
н-Бутил-а-цианакри-
лат
н-Амил-а-цианакрилат
Аллил-а-цианакрилат
р-Бутоксиэтил-а-циан-
акрилат
Модуль упру-
упругости,
Мп1м-
(кгс/см2)
1570A5700)
1000A0000)
750G500)
500E000)
750G500)
350C500)
Прочность при сдвиге,
Мн/м2 (кгс/см'г)
исходная
18,0A80)
13,0A30)
11 ,0A10)
8,0(80)
13 ,0A30)
9,0(90)
после 1 0
сут выдерж-
выдержки в воде
8,5(85)
7,0G0)
8,0(80)
10 A00)
Цианакрилатные клеи относительно дороги, что огра-
ограничивает области их применения радиоэлектроникой,
электротехникой, приборостроением и медициной.
В дальнейшем следует ожидать расширения областей
применения цианакрилатных клеев.
За рубежом цианакрилатные клеи выпускаются под
след. фирменными марками: истмен-910 (США),
цианобонд- 5000 (Япония), гистоакрил
(ФРГ).
Лит.: Кардашов Д. А., Синтетические клеи, 2 изд.,
М., 1968; X р у л е в В. М., Синтетические клеи и мастики,
М., 1970; Katz I., Adhesive materials, v. 1 — 2, N. Y.,
1964—66; Skeist I., Handbook of adhesives, N. Y., 1962;
HouwinkR., Salomon G. [eds], Adhesion and adhesi-
adhesives, 2 ed., v. 1, Amst.— [a. o.], 1965.
A. JEJ. Давыдов, А. Я. Акимова.
ПОЛИАКРИЛОВЫЕ ЛАКИ И ЭМАЛИ, акри-
акриловые лаки и эмали (acrylic varnisches
and enamels, Polyakryllacke und Emaillen, vernis et
emaux acrylyques). Эти лакокрасочные материалы под-
подразделяют на две группы в зависимости от типа плен-
пленкообразующего:
1. Материалы на основе непревращаемых (термопла-
(термопластичных) высокомолекулярных акрилатов (метакрила-
тов) — гомо- или сополимеров эфиров акриловой и
метакриловой к-т (см. Акрилатов полимеры, Метакри-
латов полимеры), образующих покрытия при комнат-
комнатной темп-ре в результате улетучивания растворителей.
Такие пленкообразующие применяют для получения
как лаков, так и эмалей.
2. Материалы на основе превращаемых (термореак-
(термореактивных) олигомеров, к-рые синтезируют сополимериза-
цией акрилатов и метакрилатов с акриловым мономе-
мономером, содержащим функциональные группы (карбок-
(карбоксильные, гидроксильные, эпоксидные и др.), а также
с третьим, обычно виниловым, сомономером, напр,
стиролом или винилтолуолом. Эти материалы образуют
покрытия, как правило, при повышенных темп-рах
в результате химич. взаимодействия функциональных
групп пленкообразующего друг с другом или с реак-
ционноспособными группами др. компонентов (напр.,
эпоксидных смол, изоцианатов), вводимых в состав
лакокрасочного материала. Превращаемые пленкооб-
пленкообразующие используют гл. обр. для получения эмалей.
Полиакриловые лаки (П. л.) — р-ры полиакриловых
пленкообразующих в смесях органич. растворителей:
сложных эфиров (обычно ацетатов), ароматич. углево-
углеводородов, кетонов, спиртов. В состав лаков входят
также пластификаторы (гл. обр. фталаты, адипинаты,
себацинаты).
Полиакриловые эмали (П. э.) — суспензии пигмен-
пигментов и наполнителей в П. л. Содержание этих компонен-
компонентов в П. э. может изменяться от 30 до 100% в расчете
на массу сухого пленкообразующего; с увеличением
содержания пигментов и наполнителей ухудшается
блеск покрытий. В состав П. э. могут также входить
ускорители высыхания — неорганич. или органич.
к-ты и др.
699
ПОЛИАКРИЛОВЫЕ ЛАКИ И ЭМАЛИ
700
П. л. получают в аппаратах с мешалками растворе-
растворением пленкообразующего или разбавлением его конц.
р-ров, к-рые получают при синтезе полиакриловых
пленкообразующих т. н. лаковым методом. После вве-
введения пластификаторов П. л. корректируют но вязко-
вязкости и содержанию сухого остатка, добавляя раство-
растворитель или пленкообразующее, и очищают от примесей
фильтрованием.
П. э. готовят диспергированием пигментов и напол-
наполнителей в конц. р-рах пленкообразующего в шаровых,
бисерных или песочных мельницах. К пигментным пас-
пастам добавляют в смесителе необходимое количество
пластификатора и р-ра пленкообразующего, корректи-
корректируют эмаль по вязкости и цвету (для этого добавляют
т. н. подцветочные пасты) и после тщательного пере-
перемешивания фильтруют. Для получения П. э. можно
также использовать суховальцованные пигментные
пасты (подробно о получении эмалей см. Краски).
Полиакриловые лакокрасочные материалы, как пра-
правило, стабильны в условиях хранения, транспортиров-
транспортировки и применения. Материалы на основе пленкообра-
пленкообразующих с карбоксильными группами рекомендуется
хранить в стеклянных или алюминиевых емкостях,
т. к. в другой таре, напр, из оцинкованной жести,
возможна желатинизация таких лаков и эмалей. П. л.
горючи, и поэтому при работе с ними, а также при хра-
хранении необходимо строго соблюдать правила противо-
противопожарной безопасности.
Полиакриловые лакокрасочные материалы приме-
применяют преимущественно для защиты метал л ич. поверх-
поверхностей. Основной метод нанесения — пневматич. рас-
распыление. Иногда используют также методы нанесения
в электрическом поле, валками, окунанием и др. П. э.
наносят на загрунтованную (иногда и зашпатлеванную)
поверхность; наиболее распространенные грунтовки —
полиакриловые, алкидно-меламиновые, эпоксидные. Оп-
Оптимальная толщина лаковых пленок 15—20 мкм, эма-
эмалевых, к-рые наносят обычно в 2—3 слоя, — 50 мкм
(о методах нанесения см. Лакокрасочные покрытия).
Материалы на основе непревращаемых пленкообра-
пленкообразующих. Для получения этих материалов применяют
гл. обр. сополимеры метилметакрилата с метил- или
бутилакрилатом, а также сополимеры метилметакри-
метилметакрилата с этил-, этилгексил- или стеарилметакрилатом
и др. (сомономеры с длинной цепью используют для
внутренней пластификации лакокрасочных пленок).
Содержание пленкообразующего в лаках с рабочей
вязкостью, необходимой для нанесения краскорас-
краскораспылителем A4—16 сек по ВЗ-4 при 20 °С), составляет
8-12%.
При необходимости улучшения адгезии и механич.
свойств пленок, а в нек-рых случаях и повышения со-
содержания пленкообразующего в лакокрасочном мате-
материале полиакрилаты совмещают с меламино-формаль-
дегидными, низковязкими эпоксидными, перхлорвини-
ловыми, маслосодержащими глифталевыми смолами (с
последними полиакрилаты ограниченно совместимы).
Продолжительность высыхания пленок П. л. на
подложке ~1 ч. Этим П. л. выгодно отличаются от
нек-рых алкидных и полиуретановых лаков (в част-
частности, используемых для получения атмосферостойких
покрытий), к-рые высыхают при комнатной темп-ре
в течение 12—24 ч.
Недостаток лаковых пленок, получаемых из непре-
непревращаемых полиакрилатов,— склонность к размягче-
размягчению при повышенных темп-pax. Термопластичность по-
покрытий м. б. существенно уменьшена при использова-
использовании сополимеров акрилатов с небольшими количест-
количествами E—7%) карбоксилсодержащих сомономеров,
напр, метакриловой к-ты. П. л. на основе таких плен-
пленкообразующих модифицируют конденсационными смо-
смолами, напр, меламино-формальдегидными. В результа-
результате взаимодействия карбоксильных групп полиакрило-
полиакрилового пленкообразующего с метилольными группами
конденсационной смолы в покрытии образуются попе-
поперечные связи; благодаря этому увеличивается твер-
твердость пленок, их стойкость к повышенным темп-рам
и бензину и улучшается также адгезия к защищаемой
поверхности. Эти покрытия сушат обычно нри 80—
90 СС.
Пленки П. л. бесцветны, обладают высокой свето-
и атмосферостойкостыо. Они сохраняют свои свойства
нри темп-pax от —50 до 150—170 °С. Благодаря отсут-
отсутствию в полиакрилатах гидроксильных групп пленки
отличаются высокой водостойкостью. Напр., набуха-
набухание пленки П. л. после выдержки в воде в течение
1 сут составляет 0,55—0,65% , пленок алкидных и по-
полиуретановых лаков — соответственно 4,4 и 3,5%.
Паропроницаемость пленок П. л. выше, чем у алкидных
и перхлорвиниловых: после испытаний в течение 4 сут
она составляет соответственно ~30, ~22 и — 4 мг/см'2.
Это ограничивает применение П. л. для защиты метал-
металлов, обладающих низкой коррозионной стойкостью,
напр, магниевых сплавов.
Наиболее широко П. л. применяют для защиты алю-
алюминия и его сплавов. Благодаря такой защите сохра-
сохраняется цвет металла. Для повышения адгезии лаковых
пленок и коррозионной стойкости металла рекомен-
рекомендуется предварительно анодировать алюминий в сер-
серной к-те с последующим уплотнением анодной пленки
в воде. Покрытия надежны в различных климатич.
условиях, включая тропические, в течение несколь-
нескольких лет. Пленки П. л. обладают хорошей адгезией
к покрытиям на основе многих пленкообразующих,
напр, эфиров целлюлозы, перхлорвиниловых смол.
Поэтому П. л. часто используют в качестве промежу-
промежуточного слоя в нек-рых многослойных покрытиях.
П. э. на основе термопластичных пленкообразующих
образуют покрытия, к-рые отличаются чистотой и яр-
яркостью тона, высокой атмосферо-, свето- и термостой-
термостойкостью. Они хорошо шлифуются и полируются, сохра-
сохраняют блеск в течение длительного времени. Недостаток
покрытий темных цветов, обусловленный их низким
коэфф. отражения,— склонность к размягчению под
действием солнечного излучения.
П. э. сушат обычно при комнатной темп-ре в течение
1 — 2 ч. При необходимости сокращения продолжитель-
продолжительности сушки (напр., в поточном производстве) темп-ру
повышают до 100 — 110 СС.
П. э. широко применяют для окраски самолетов. Све-
Светостойкие П. э., пигментированные двуокисью титана
рутильной модификации, имеющие коэфф. отражения
>80%, наносят на различные металлич. конструкции,
напр, кабины пассажирских самолетов, резервуары
для хранения топлива, купола обсерваторий, с целью
уменьшения их нагрева под действием солнечного из-
излучения. Яркие флуоресцентные П. э., хорошо разли-
различимые на дальних расстояниях и при плохом освеще-
освещении, наносят на крылья, оперение и фюзеляжи самоле-
самолетов, а также на ограничительные полосы взлетных до-
дорожек аэродромов. Их используют, кроме того, для
оформления витрин, выставочных стендов и др. Для
сохранения яркости покрытия на него наносят П. л.,
содержащий добавку светостабилизатора, например
2,2-диокси-4-метоксибензофенон. Лаковая пленка слу-
служит светофильтром, поглощающим УФ-лучи. Такие
П. л. можно наносить на некоторые пигментирован-
пигментированные лакокрасочные покрытия для уменьшения их
«меления».
Материалы на основе превращаемых пленкообразую-
пленкообразующих. Наибольшее распространение получили пленко-
пленкообразующие с метилольными группами, к-рые полу-
получают, напр., сополимеризацией акриламида со стиро-
стиролом и акрилатами и последующим взаимодействием
сополимера с формальдегидом. Такие соединения отно-
относительно дешевы и, кроме того, они хорошо смачивают
701
ПОЛИАКРИЛОНИТРИЛЬНЫЕ ВОЛОКНА
702
пигменты. Карбоксилсодержащие сополимеры синте-
синтезируют из акрилатов, акриловой (метакриловой) к-ты
и стирола (винилтолуола). При получении сополимеров
с эпоксидными группами в состав смесей мономеров
вводят глицидилметакрилат, сополимеров с гидроксиль-
ными группами — оксиэтил- или оксипропилметакри-
лат.
Благодаря сравнительно невысокой мол. массе тер-
термореактивных полиакриловых пленкообразующих ла-
лакокрасочные материалы на их основе, разбавленные
до рабочей вязкости, могут содержать ~50% сухого
остатка. Покрытия на их основе отверждаются при
125—150 °С в течение 30—40 мин.
Покрытия, получаемые из П. э. с метилольными и
эпоксидными группами, способны отверждаться без
добавления др. реакционноспособных соединений. Для
снижения темп-ры и ускорения отверждения метилол-
содержащих сополимеров акрил амида применяют к-ты,
напр, фосфорную или тг-толуолсульфоновую @,15—
2,0% от массы пленкообразующего). П. э. на основе
карбоксилсодержащих пленкообразующих отвержда-
отверждаются эпоксидными смолами, на основе гидроксилсо-
держащих — частично бутанолизированной меламино-
формальдегидной смолой. Снижение темп-ры отверж-
отверждения гидроксилсодержащих пленкообразующих до
комнатной (или резкое ускорение процесса при высо-
высоких темп-pax) достигается введением в них изоциана-
тов. Ускорителями отверждения в этом случае служат
октоаты или нафтенаты цинка, нек-рые амины. Такие
системы обладают низкой жизнеспособностью (до 10 ч),
и поэтому компоненты лакокрасочного материала сме-
смешивают непосредственно перед его нанесением.
При использовании указанных отвердителеи улучша-
улучшаются многие свойства покрытий. Так, при введении
в состав П. э. частично бутанолизированной меламино-
формальдегидной смолы улучшаются водо- и атмосферо-
стойкость пленок, повышается их твердость. Эпоксид-
Эпоксидные смолы повышают химстойкость пленок. П. э., мо-
модифицированные изоцианатами, образуют покрытия,
к-рые превосходят по светостойкости полиуретановые,
получаемые на основе ароматич. изоцианатов. Свойст-
Свойства покрытий на основе П. э. можно также варьировать
в широких пределах, изменяя состав сополимера.
Основное преимущество покрытий, к-рые образуются
при отверждении П. э. на основе превращаемых пленко-
пленкообразующих, перед покрытиями на основе непревра-
щаемых полиакрилатов — способность сохранять
твердость при повышенных темп-pax. Такие покрытия
превосходят меламино-алкидные по декоративным и
эксплуатационным свойствам (таблица). П. э. на основе
Нек-рые свойства покрытий, получаемых из эмалей на основе
сополимеров акриламида, модифицированных формальдегидом,
и на основе меламино-алкидных смол
Показатели
Твердость по карандашу
Стойкость
к механич. загрязнениям
к влаге A000 ч)
к моющим средствам A00 ч при
60 СС)
к образованию чернильных пятен
после контакта в течение 100 ч
Полиакри-
Полиакриловая
эмаль
ЗН
Отл.
Отл
Отл.
Отл.
Меламино-
алкидная
эмаль
НВ
Уловл.
Уловл (мел-
кие пузыри)
Неудовл.
Неудовл.
превращаемых пленкообразующих широко используют
для окраски кузовов автомобилей, а также тракторов
и др. сельскохозяйственных машин, строительных
конструкций из алюминиевых сплавов, шкал приборов,
рулонного металла, консервной тары и др.
Все большее значение приобретают П. э., получаемые
из водорастворимых превращаемых пленкообразую-
пленкообразующих, к-рые синтезируют, напр., сополимеризацией
70—87% бутил акрилата, 5—6% акриловой к-ты,
8—24% акриламида (третьим сомономером м. б. также
неакриловое соединение, содержащее карбоксильные,
гидроксильные, простые эфирные или амидогруппы).
Такие сополимеры становятся водорастворимыми пос-
после их обработки формальдегидом в спиртовой среде и
нейтрализации карбоксильных групп аммиаком или
триэтаноламином.
П. э. на основе водорастворимых пленкообразующих
безопасны в пожарном отношении, менее токсичны,
чем обычные полиакриловые лакокрасочные материа-
материалы. В отсутствие отвердителеи они высыхают при
темп-pax ок. 170 °С, при введении отвердителеи (напр.,
водорастворимых меламино-, мочевино- или феноло-
формальдегидных смол) — при более низких темп-рах.
Водорастворимые П. э. используют гл. обр. для получе-
получения покрытий методом электроосаждения. Образую-
Образующиеся при этом пленки обладают всеми характерными
свойствами полиакриловых покрытий и отличаются
лучшей адгезией к защищаемой поверхности, чем по-
покрытия из П. э., к-рые наносят др. методами, напр,
окунанием.
Лит.: Полимеризационные пленкообразователи, под ред.
В. И. Елисеевой, М., 1971; Гольдберг М. М., Материа-
Материалы для лакокрасочных покрытий, М., 1972; Соломон Д.Г.,
Химия органических пленкообразователей, пер. с англ. М.
1971; Шаров М. Я., Денкер И. И., Калини-
Калинина Е. П., Лакокрасочные материалы и их применение, № 5
25 A960); Дуброва Б. М., там же, № 2, 80 A962); Д е н-
к е р И. И., там же, № 4, 50 A964); Deutsche Farben-Zeit-
schrift, 22, Н. 12, 567 A968); J. Oil and Colour Chem. Assoc. 52
№ 6, 577 A969). И. И. Денкер.
ПОЛИАКРИЛОНИТРИЛ - см. Акрилонитрила
полимеры.
ПОЛИАКРИЛОНИТРИЛЬНЫЕ ВОЛОКНА (acrilic
fibers, Polyakrilnitrilfasern, fibres polyacryliques) —
синтетич. волокна, получаемые из полиакрилонитрила
или сополимеров, содержащих более 85% (по массе)
акрилонитрила. Волокна из сополимеров, содержащих
40—85% акрилонитрила, принято называть м о д а-
криловыми. П. в. в основном (на 99%) выпус-
выпускаются в виде штапельных волокон.
Получение. П. в. формуют только из р-ров. В качест-
качестве растворителей для приготовления прядильных р-ров
в пром-сти используют диметилформамид (ДМФ), ди-
метилацетамид (ДМА), водный р-р роданистого нат-
натрия, конц. азотную к-ту, водный р-р хлористого цинка,
диметилсульфоксид (ДМСО) и этиленкарбонат (ЭК).
Технологич. схема производства П. в. включает ста-
стадии получения полимера, формования волокна и реге-
регенерации растворителя. Существует большое число ва-
вариантов этой основной схемы, различающихся по виду
применяемых растворителей, способу получения во-
локнообразующего полимера, методам формования во-
волокна и регенерации растворителя.
В пром-сти применяют два способа синтеза полимера,
используемого для формования П. в.: полимеризация
в р-ре и суспензии (подробно об этом см. Акрилонитри-
Акрилонитрила полимеры, Акрилонитрила сополимеры). Прядиль-
Прядильный р-р, полученный по первому из этих способов, под-
подвергают демономеризации, а затем очищают в одну
или две стадии на рамных фильтр-прессах. Кроме то-
того, его подвергают вакуумной десорбции для полного
удаления газовых пузырьков и частичного удаления
растворенных газов. Эта операция проводится в аппа-
аппаратах пленочного типа, принцип действия к-рых осно-
основан на том, что прядильный р-р в них протекает в виде
тонких пленок и тем самым обеспечивается большая по-
поверхность удаления газов. В зависимости от вида раст-
растворителя подготовка прядильного р-ра к формованию
проводится при различных темп-pax: от 0 до 5 СС при
использовании HNO3, до 50—80 °С при использовании
ЭК, ДМФ, ДМА.
703
ПОЛИАКРИЛОНИТРИЛЬИЫЕ ВОЛОКНА
704
При получении полимера суспензионным способом
для приготовления прядильного р-ра необходима двух-
стадийная операция растворения полимера. В зависи-
зависимости от вида растворителя ее проводят на холоду
(HNO3) или при нагревании (ДМФ, ДМ А). На первой
стадии порошок полимера суспендируют в растворителе.
При этом в зависимости от скорости набухания полиме-
полимера и степени его измельчения применяют различные
аппараты: малого объема и с быстродействующим пе-
перемешивающим устройством или большого объема
и с более тихоходной мешалкой, рассчитанной на высо-
высоковязкую среду. На второй стадии происходит раство-
растворение полимера при нагревании в аппаратах непрерыв-
непрерывного действия, пленочных аппаратах с мешалками
или в шнеках (в последнем случае разогрев может про-
происходить в результате действия сил трения, возникаю-
возникающих при работе шнека в автогенном режиме).
При формовании П. в. по сухому методу нагретый до
90—120 °С прядильный р-р (в этом случае в качестве
растворителя применяют ДМФ или ДМ А) при помощи
дозирующих насосиков продавливается через отверстия
фильеры в обогреваемую воздушную шахту. Высажи-
Высаживание полимера из р-ра происходит в результате испа-
испарения растворителя. Сухое формование возможно толь-
только из конц. прядильных р-ров B0—35% полимера) и
при сравнительно высокой темп-ре в шахте B00—
280 °С). Из прядильной шахты волокно выходит пласти-
пластифицированным с содержанием растворителя 8—12%.
Основное достоинство сухого метода — высокая ско-
скорость формования B00—600 м/мин). Волокна отли-
отличаются малой напряженностью внутренней структуры
и поэтому мягки и эластичны. Недостатки сухого ме-
метода — ограниченное число отверстий в фильере (не-
(несколько сотен) и необходимость поддерживать в шахте
концентрацию паровоздушной смеси выше предельно
допустимой взрывной концентрации.
При мокром методе формования прядильный р-р
(в этом случае возможно применение любого раствори-
растворителя) продавливается дозирующими насосиками через
отверстия фильеры в жидкую осадительную ванну,
содержащую смесь растворителя (из к-рого приготов-
приготовлен прядильный р-р) с осадителем полимера. В зависи-
зависимости от вида осадителя, его количества и темп-ры
ванны получаются волокна с различными свойствами.
В пром-сти в качестве осадителей чаще всего приме-
применяют воду, обладающую наибольшей осадительной спо-
способностью, а также различные органич. жидкости, не
смешивающиеся с водой, но смешивающиеся с раство-
растворителем (керосин, жирные спирты, гексантриол и др.).
Из осадительной ванны выходит волокно, представ-
представляющее собой полимерный студень, инклюдированный
осадительной ванной. Содержание полимера в таком
волокне составляет 17 —15%. Оно обладает высокопо-
высокопористой структурой (внутренняя поверхность порядка
сотен м2/г).
Основные достоинства мокрого метода формования —
применение фильер с очень большим числом отверстий
(более 100 000) и более широкие возможности варьиро-
варьирования свойств получаемых волокон. Недостаток этого
метода — ограниченная скорость формования D0—
60 м/мин).
Для придания волокну необходимой прочности его
подвергают вытяжке. Эту операцию для П. в. можно
проводить на различных стадиях его изготовления:
после осаждения, во время и после промывки и, нако-
наконец, после сушки.
После осаждения полимер в волокне находится
в пластифицированном состоянии (пластифицирован
растворителем). Такое волокно растягивается легче
и при более низких темп-pax, однако в этом случае
эффективность вытягивания, т. е. прирост прочности
на единицу деформации, ниже, чем при вытягивании
волокна, не содержащего пластификатора. Вытяжка
пластифицированного П. в. составляет 600—1300%
в зависимости от условий осаждения. Темп-pa выби-
выбирается обычно в пределах 80—100 °С.
Вытяжка промытого волокна, лишенного раствори-
растворителя, но инклюдированного водой, происходит труд-
труднее, не превышает 600% и проводится при 100—130 СС.
Вытяжке только на этой стадии подвергают волокна,
сформованные из р-ров в HNO3, т. к. их нежелательно
нагревать до высоких темп-р без предварительного уда-
удаления растворителя.
Сухое волокно вытягивается при более высоких
темп-рах A50 —160 °С). Этот способ наиболее эффек-
эффективен, однако применяется в основном при получении
волокна технич. назначения, т. к. при 150—160 °С
П. в. приобретают слегка кремовый оттенок из-за хи-
мич. изменений макромолекул (группы — C = N пре-
превращаются в последовательности групп —С = N—
с сопряженными двойными связями). Часто применяют
комбинированные режимы придания ориентированной
структуры полимеру в волокне. В этом случае свеже-
сформованное волокно незначительно вытягивается для
придания ему нек-рой прочности после осаждения, за-
затем следует отмывка от растворителя и далее, перед
сушкой, волокно подвергается основной вытяжке.
В случае необходимости повышения прочности волок-
волокна его вытягивают еще раз в сухом состоянии.
Вытяжка волокна производится на различных маши-
машинах и в разных средах. Волокна, сформованные в вод-
водные осадительные ванны, вытягивают после осаждения
как в горячих водных р-рах растворителя, так и в па-
паровых шахтах. Болокна, сформованные в осадительные
ванны, содержащие высококипящие органич. осади-
тели, вытягиваются или на горячих поверхностях или
в нагретой смеси осадителя и растворителя. Вытяги-
Вытягивание волокна перед сушкой производится в кипящей
воде или паровых шахтах, сухого волокна — на го-
горячих поверхностях, с применением инфракрасных на-
нагревателей, различных теплоносителей (жидких и гра-
гранулированных твердых), в газовых шахтах и др. ме-
методами.
П. в., сформованные по любому способу, после осаж-
осаждения всегда содержат растворитель. При сухом фор-
формовании его содержание составляет 8—12%, при мок-
мокром — до 300%. Для удаления растворителя П. в.
промывают водой и водным р-ром растворителя или
смесью растворителя с органич. осадителем, к-рые за-
затем направляются на регенерацию. В результате про-
промывки не только удаляется растворитель, но и изменя-
изменяется структура волокна. Концентрация полимера в нем
повышается до 30—45% (но объему), т. е. волокно сжи-
сжимается; несколько снижается его пористость и повы-
повышается темп-pa стеклования благодаря увеличению
межмолекулярных связей в полимере. Время промывки
колеблется от 15 сек до 3 мин в зависимости от вида
применяемого растворителя, условий формования и
конструкции машины.
Цикл по растворителю на производствах П. в. замк-
замкнут. Поэтому необходимо, чтобы расход воды на про-
промывку был возможно более низким. В этом случае
промывные воды, идущие на регенерацию, будут иметь
наивысшую концентрацию растворителя. Промывка
производится обессоленной водой методом противото-
противотока, применяются машины с большим числом секций
и промежуточных отжимов.
В процессе производства П. в. в циркуляции нахо-
находится большое количество растворителя, к-рый подвер-
подвергается регенерации. При сухом методе формования
ДМФ или ДМА сначала конденсируется из паровоздуш-
паровоздушной смеси, а оставшийся в смеси ДМФ или ДМА абсор-
абсорбируется промывными водами, к-рые подвергаются ре-
регенерации. При мокром методе растворитель регенери-
регенерируется, как правило, из водного р-ра (смесь осадитель-
осадительной ванны и промывных вод); летучие растворители
705
ПОЛИАКРИЛОНИТРИЛЬНЫЕ ВОЛОКНА
706
(ДМФ, ДМСО и ДМА) — ректификацией, нелетучие
или мало летучие — упариванием. В последнем случае
все накапливающиеся примеси остаются в растворителе.
Для их удаления применяют различные методы: кри-
кристаллизацию, ионный обмен, экстракцию, химич. об-
обработку и др.
Сушка П. в. может производиться в аппаратах раз-
различного типа; наибольшее распространение получили
сушилки с перфорированными барабанами. При сушке
П. в. необходимо обеспечить возможность свободной
усадки волокна, к-рая происходит при любых темп-рах
и определяется механизмом капиллярной контракции
(усадки), т. к. мокрое волокно является высокопори-
высокопористым материалом. Пористость волокна, в свою очередь,
определяется условиями его формования, вытягивания
и промывки. Для сохранения заданной надмолекуляр-
надмолекулярной полимерной структуры волокна нежелателен нагрев
его в сушилке, особенно в последних зонах, выше
темп-ры стеклования (80—90 °С). Во время сушки кар-
каркасная структура полимера контрактирует и содержа
ние полимера увеличивается до 85—99% (по объему).
Содержание влаги в волокне перед сушкой составляет
150% от массы полимера; после сушки — 0,5—2%.
Для придания П. в. устойчивости к нагреванию (до
150 СС) и при кипячении в воде их подвергают термо-
термообработке (кратковременный прогрев сухого волокна
в условиях натяжения). Во время этой операции мож-
можно снизить или, наоборот, увеличить усадочность во-
волокна, подбирая условия натяжения, изменить его
блеск, сделать более однородным по механич. свойст-
свойствам. Аппаратурно операцию термообработки часто
оформляют в блоке с сушилкой, чтобы не охлаждать
волокно после сушки. Темп-рный режим при термооб-
термообработке м. б. различным (от 100 до 160 °С) в зависимо-
зависимости от требований, предъявляемым к готовым волокнам.
Для повышения способности перерабатываться на
текстильных машинах волокну придается извитость
(гофр). Для этого волокно быстро нагревают (для
нек-рого размягчения), сминают и затем охлаждают в
сжатом состоянии. Гофрировка производится на спе-
специальных машинах, имеющих нагревающее устройст-
:
операций, т. к. полимер при нагревании в щелочной
среде желтеет. Количество препаратов, наносимых на
волокно, колеблется от 0,2 до 0,3% от массы волокна.
П. в. могут производиться в окрашенном виде. При
крашении в массе к прядильному р-ру подмешивается
тонко размолотый пигмент (величина частиц менее
0,5 мкм). При формовании весь пигмент остается в во-
волокне, обеспечивая его равномерную окраску. Таким
образом окрашивают волокна в глубокие темные тона
(чаще всего в черный цвет). Окраска, полученная та-
таким методом, отличается высокой устойчивостью к раз-
различного рода обработкам, к свету и атмосферным воз-
воздействиям. Поверхностное крашение П. в. осущест-
осуществляют после стадии его промывки (до сушки), исполь-
используя высокую пористость влажных П. в. и их большую
внутреннюю поверхность. На волокно кратковремен-
кратковременным окунанием жгута наносят р-р красителя. Краси-
Краситель в течение нескольких секунд сорбируется порами
волокна. После сушки поры волокна закрываются,
прочно фиксируя краситель. При этом можно исполь-
использовать красители любого класса. Предпочтительнее
применять специальные основные красители, т. к. на
П. в. они дают наиболее яркие и устойчивые выкраски.
При поверхностном крашении волокна приобретают
менее равномерную окраску; этот метод более пригоден
для окраски в светлые тона. Оба метода крашения П. в.
экономичны, т. к. обеспечивают 100%-ное использова-
использование красителей.
См. также Крашение волокон, Крашение химических
волокон в массе.
Свойства. Ниже приведены нек-рые физич. свойства
П. в.:
Темп-pa размягчения, °С ......... 180 — 200
Плотность, г/см3 1,12 — 1,20
Влагосодержание, % (при относитель-
относительной влажности воздуха 65%). . . . , 0,7—2,0
Показатель преломления 1,52
Темп-pa стеклования, °С 80 — 90
По своим механич. свойствам (табл. 1) П. в. очень
близки к шерсти; в этом отношении они превосходят
все остальные химич. волокна.
Название волокна
(страна-изготови-
(страна-изготовитель)
во, питающее вальцы и закрытую
камеру, в к-рой волокно прессует-
прессуется при заданном давлении (подроб-
(подробно об этом см. Высокообъемные ни-
нити). Величину извитости и ее ус-
устойчивость регулируют темп-рой
нагрева волокна, давлением в ка-
камере и условиями охлаждения. Для
различных видов П. в. произво- Орлон 4 2 (США) .
дится различная гофрировка как кУРтель (Англия).
по степени извитости, так и по Кашмилон (Япония)
устойчивости извитка при нагру- Анилана (ПНР)
женин. Нитрон (СССР)
Таблица 1. Механические свойства полиакрилонитрильных волокон
Проч-
Прочность,
гс/текс
Отно-
ситель-
сительное уд-
лине-
линение, %
27-30
24-29
23-25
22-24
35 — 35
П. в. обладают довольно высокой
жесткостью и электризуемостью. Для уменьшения этих
недостатков на поверхность волокна наносят два вида
поверхностно-активных веществ: 1) неионогенные (про-
(производные полиоксиэтилена)—для смягчения и 2) ионо-
генные (полифункциональные амины и их соли) — для
антистатич. обработки. Препараты наносят на волок-
волокно из водных эмульсий погружением волокна в эмуль-
эмульсию и пропусканием волокна по поверхности валика,
смоченного эмульсией. Эти операции могут произво-
производиться как одновременно (из эмульсии, содержащей
оба вида поверхностно-активных веществ), так и раз-
раздельно. Препараты наносят на отмытое волокно. Воз-
Возможно нанесение их до сушки волокна, после сушки,
после стабилизации и после гофрировки. При нанесении
эмульсии до сушки волокно сорбирует большое количе-
количество препарата, часть к-рого остается после сушки внут-
внутри волокна. Антистатич. препараты, особенно основ-
основного характера, наносят, как правило, после тепловых
Прочность
в мокром
состоянии,
% от проч-
прочности су-
сухого
34-28 80
3 7-33 85
39-25
3 5-31
25-33
89
80
Прочность
в петле,
гс/текс
12-16
9-10
7 — 12
9-10
10-12
Начал ьный
модуль, Ми/м2
(кгс/мм2)
30C)
15-30
A ,5-3,0)
30-40C — 4)
10-20A-2)
30-50C-5)
Работа
разрыва,
н-м, или
гс-см
5-2,0
5-2,0
5—2,0
5-3,0
5-3,0
П. в. устойчивы к действию сильных к-т средней
концентрации даже при нагревании (табл. 2), а также
к щелочам средней концентрации (табл. 3).
Растворители, применяемые для стирки и чистки
одежды — бензин, ацетон, четыреххлористый углерод,
Таблица 2. Условия обработки полиакрилонитрильных волокон
в кислотах, при которых прочность волокон падает на 5%
Кислота
Серная .
Соляная
То же . .
Азотная
То же .
' -
55 К
онце
ради
0
10
18
37
18
2 0
40
20
20
рем я
ЗраОс
и, су
ffl о к
3
280
3
14
280
7 0
35
4
!
г-1 О.
6 5
20
20
7 5
2 0
20
50
75
Кислота
Азотная . . .
Ортофосфор-
ная
Фтористоводо-
Фтористоводородная . . .
Уксусная ле-
ледяная
нцен-
щия,
о ^
20
70
35
емя
забот-
, сут
ffl о к
3
3
3
3
9 0
В 5
65
65
707
ПОЛИАКРОЛЕИН
708
Таблица 3. Зависимость прочности полиакрилонитрильных
волокон от условий их обработки щелочами
Щелочь
Едкий натр
То же . . .
Сода
Аммиак
То же .
Концент-
Концентрация ще-
щелочи, %
5
10
50
5
5 0
Разб. и
нонц.
Конц.
10
Тем-
п-ра,
СС
20
20
50
79
75
100
20
20
Время
обработки,
сут
35
0,6
0,8
1 ,0
0,6
0,6
3
0,6
Потеря
прочности,
до 10
0
0-6,6
Полимер раз-
разлагается
То же
0
до 5
0
дихлорэтан и др. не влияют на прочность П. в.; фенол,
ж-крезол и формалин их разрушают.
Применение. П. в. применяют для изготовления
верхнего трикотажа, ковров, платяных и костюмных
тканей. Кроме того, их можно использовать для изго-
изготовления белья (в смеси с хлопком и вискозным волок-
волокном), гардин, брезентов, обивочных и фильтроваль-
фильтровальных тканей и др.
Штапельные П. в. выпускают в различных модифи-
модификациях: резаное волокно и жгут шерстяного или
хлопчатобумажного типа, высокоусадочное (способное
усаживаться в кипящей воде) и безусадочное, окраши-
окрашиваемое кислотными или основными красителями, с раз-
различной степенью извитости, невоспламеняющееся,
имеющее постоянную извитость. Толщина ГГ. в. может
колебаться от 0,26 до 6 текс.
В СССР П. в. выпускается под названием «н и т-
р о н». Наиболее распространенные зарубежные тор-
торговые названия П. в.: о р л о н, а к р и л а н (США),
кашмилон (Япония), к у р т е л ь (Англия), д р а-
л о н (ФРГ), вольпрюла (ГДР), а н и л а н а
(ПНР). Модакриловые волокна выпускаются под след.
названиями: из сополимера акрилонитрила с винил-
хлоридом в соотношении 40: 60 — канекалон
(Япония), д а й н е л (США); из сополимера акрило-
акрилонитрила с винилиденхлоридом— в е р е л (США).
Мировое производство П. в. неуклонно растет:
1962 — 167 тыс. т, 1967 — 541 тыс. т, 1971 —1175
тыс. т, 1972—1269 тыс. т.
Лит.: Пакшвер А. Б., Геллер Б. Э., Химия и
технология производства волокна нитрон, М., 1960; Кали-
Калинов с к и Е., Урбанчик Г. В., Химические волокна,
М., 1966; Монкрифф Р. У., Химические волокна, пер.
с англ., М., 1964; Карбоцепные синтетические волокна, под
ред. К. Е. Перепелкина, М., 1973. Э. А. Пакшвер.
ПОЛИАКРОЛЕИН — см. Акролеина полимеры.
ПОЛИАЛКИЛЕНАРИЛЕНЫ, полиалкилен-
а р и л ы (polyalkylene-arylenes, Polyalkylen-arylene,
polyalcylene-arylenes) — полимеры общей ф-лы I или II:
Г 1 Г /сн»>»-1
-Аг-(СН,)Я- -Ар\
L \x L х(сн2)-1
(СН2)„—J
II
Аг =
О
(/77 = 0,1,2,3),
[где x-°'s'(-c«.-)J
Получение. П. получают поликонденсацией но реак-
реакции Фриделя — Крафтса: \) ароматич. соединений, со-
содержащих не менее двух хлоралкильных групп, с аро-
ароматич. углеводородами; 2) монохлоралкилзамещенных
арилов; 3) дигалогеналканов с ароматич. соединениями:
Аг + Hal-(CH2)n-Hal -> [-(СН2)п-Ат~]х + HHal
(Hal=Cl, Br; n = 1—4) C)
Реакции проводят в присутствии к-т Льюиса (моляр-
(молярная концентрация 3—12% от содержания ароматич.
компонентов) при 70 —130 °С в р-ре (получаются гл.
обр. сильноразветвленные, потенциально термореак-
термореактивные продукты) или в расплаве (при эквивалентном
соотношении компонентов степень разветвления П.
наименьшая). Растворителями служат 1,2-дихлорэтан,
о-дихлорбензол, нитробензол.
Для предотвращения образования сшитого продукта
рекомендуется применять мягкие каталитич. агенты
(BF3 или SnCl4) либо проводить реакцию при низких
темп-pax. Так, полибензилы (I; Аг = С6Н5, п = 1)
со строением молекулярной цепи, близким к линейному,
получают в присутствии А1С13 при —100 С или в при-
присутствии VCl4 + А1(С2Н5K при 0 °С. Строго линейные
П. образуются лишь из ароматич. соединений, в цик-
циклах к-рых все активные к реакции электрофильного
замещения положения, кроме двух [в реакциях A)
и C)] или одного [в реакции B)], замещены или блоки-
блокированы соседними объемными заместителями. Актив-
Активность ароматич. колец к замещению снижается с уве-
увеличением электроотрицательности заместителей и мо-
стиковых групп и не зависит от длины цепи полимера.
Методы получения высокомолекулярных (мол. масса
ок. 500 000) строго линейных П. разработаны лишь
для поли-п-ксилилена и его производных.
Известны методы получения П. из олигофениленов
(см. Полифенилены) и га-ксилиленгликоля в присут-
присутствии минеральных к-т.
Свойства. П.— вещества белого цвета. Линейные
П.— кристаллич. полимеры с высокими темп-рами
плавления. Так, темп-pa плавления ряда П. на основе
хлорметилдурола 260—280 °С, полибензила 142 °С,
поли-2,5-диметилбензила выше 300 °С. Линейные П.
нерастворимы или плохо растворимы в органич. рас-
растворителях, хотя, за исключением поли-га-ксилиленов,
и не обладают высокой мол. массой (ок. 1000).
Разветвленные П., как правило,— вязкие жидкости
или сравнительно легкоплавкие твердые вещества,
мол. масса к-рых зависит от метода получения. П.
наиболее высокой мол. массы (до 8—9 тыс.) получают
по реакции C) при взаимодействии триалкилбензолов,
содержащих алкилы с 1 — 3 атомами углерода, с дигало-
геналканами с 2—4 атомами углерода в цепи. Мол.
масса разветвленных П. составляет 2—4 тыс. Они рас-
растворимы в обычных органич. растворителях (спиртах,
хлорированных углеводородах). Плотность П. (I,
Аг = тг-дифениленоксид, п = 1) 1,2—1,35 г/см3.
Сшитые П. не размягчаются при нагревании в отсут-
отсутствие воздуха до 400 СС и обладают высокой термич.
и химич. стойкостью. Так, после теплового B00 °С)
старения прочность на изгиб слоистых асбопластиков
на основе полибензила (в течение 4000 ч) и полимети-
лентри-д-фенилена A0 000 ч) возрастает и достигает
соответственно 170 Мн/м2 A700 кгс/см2) и 100 Мн1мг
A000 кгс/см2). Высокая прочность на изгиб у этих
пластиков увеличивается и после длительной D000 ч)
обработки конц. D00 г/л) едкими щелочами: у пер-
первого — до 190 Мн/м2 A900 кгс/см2), у второго -
до 200 Мн/м2 B000 кгс/см2).
Сшитый П., полученный поликонденсацией моно-
хлорметилдифениленоксида, остается твердым при на-
709
ПОЛИАЛКИЛЕНГЛИКОЛЬМАЛЕИНАТЫ И ПОЛИ А ЛКИЛЕНГЛИКОЛЬФУМ АРАТЫ
710
гревании на воздухе до 300 СС даже при воздействии
едких щелочей, минеральных к-т и солей; он обладает
высокой водо- и маслостойкостью, стоек к действию
органич. растворителей. Введение в макромолекулу
этого П. блоков дурофениленоксида улучшает меха-
нич. и электрич. свойства полимера при высоких
темп-рах.
Теми-ры разложения П. в инертной атмосфере пре-
превышают 400—450 °С. Темп-ры заметной термоокисли-
термоокислительной деструкции разветвленных и линейных П.
лежат в интервале 300—350 СС. Скорость термоокис-
термоокислительной деструкции у П. с п=2 (ф-лы I и II, см. выше)
уменьшается в ряду: нафтилен-2,6 > ?г-фенилен > ан-
трилен-9,10. Устойчивость к термоокислению у раз-
разветвленных полибензилов ниже, чем у разветвленных
поликсилиленов и др. полиотиленариленов, а у сши-
сшитых П. выше, чем у разветвленных.
Сшитые П., особенно содержащие полифениленовые
блоки, характеризуются высокой абляционной стойко-
стойкостью. Все П. являются самозатухающими полимерами.
Коэфф. теплопроводности полимера из монохлорметил-
дифениленоксида 0,43—0,46 дж/(м-сек-К) [0,37—0,4
ккал/(м*ч- °C)J. П. не сорбируют влагу (в этом отноше-
отношении они значительно превосходят отвержденные фено-
ло-формалъдегидные смолы), устойчивы к действию ра-
радиации. Вязкость жидких разветвленных П. (Аг =
= C6H6_mRm; R = алкил Сг— С5, т = 1 — 5; п =1—4)
после воздействия ^-радиации (интенсивность
2-Ю7 дж/кг, или 2-Ю9 рад) возрастает лишь на 150%
(минеральных масел — на 500%).
П.— хорошие диэлектрики. Диэлектрмч. проницае-
проницаемость и тангенс угла диэлектрич. потерь П. (I, Аг=ди-
фениленоксид, п = 1) составляют соответственно 3,38
и 0,024; для П. (I, Аг = три-м-фенилен, п=1) — соот-
соответственно 3,10 и 0,009. П. обладают хорошей адге-
адгезией к армирующим волокнам (синтетическим и стек-
стеклянным), к-рая у полибензилов сохраняется при на-
нагревании B50 °С) в течение 3000 ч.
Разветвленные П., содержащие в цепи достаточное
число галогеналкильных групп, можно отверждать
простым нагреванием до 120—140 °С в присутствии не-
неотделенного катализатора поликонденсации. Если не-
необходимо, вводят, кроме того, сшивающие агенты —
дигалогеналканы или дигалогеналкиларилы (напр., а,
а'-дихлорксилол). Скорость отверждения разветвлен-
разветвленного полиэтиленфенилена (мол. м. 5000) в присутствии
катализаторов Фриделя — Крафтса в зависимости от
природы дигалогеналканов возрастает в ряду: 1,4-ди-
бромбутан ^> 1,3-хлорбромпропан > 1,6-дибромгек-
сан > бромистый метилен> 1,2-дибромэтан. Чем выше
мол. масса полимера, тем меньше требуется сшиваю-
сшивающего агента.
П. могут отверждаться серой при 200—250 °С.
Процесс протекает по радикальному механизму по-
посредством соединения алкиленовых групп цепи мости-
мостиками —Sx—. Скорость отверждения снижается при
наличии в макромолекуле электроотрицательных за-
заместителей. Реакцию можно проводить в две стадии:
сначала нагревать П. с серой до получения раствори-
растворимого сульфидированного форполимера, а затем при
темп-ре выше 200 °С перевести его в сшитую форму.
Применение А1С13 снижает темп-ру сульфидирования
до 80—90 СС, при этом полисульфидные мостики об-
образуются между ароматич. циклами полимерных цепей.
Отвержденные серой П. термо- и химстойки, обла-
обладают хорошей адгезией к металлам. Стеклопластики
на основе таких П. по физико-механич. показателям
превосходят стеклопластики на основе феноло-формаль-
дегидных смол. Сульфидированные П. проявляют
стабилизирующий эффект подобно низкомолекуляр-
низкомолекулярным соединениям типа RSSR (R — алкил или арил).
Переработка и применение. П. можно перерабаты-
перерабатывать прессованием. При изготовлении армированных
пластиков на основе разветвленных полимеров, содер-
содержащих в макромолекулах хлоралкильные группы,
в присутствии катализатора Фриделя — Крафтса да-
давление прессования составляет 0,14 Л/и/ле2A,4 кгс/см2)
для стеклопластиков и 3,58 Мн/м2 C5,8 кгс/см2) для
пластиков, армированных алюмоборосиликатными во-
волокнами. Последующее отверждение изделия проводят
сначала в течение 2 ч при 180—200 °С, затем в течение
48 ч при 200—240 СС.
В пром-сти выпускают П. на основе хлорметилдифе-
ниленоксида (смола д о р и л и ее модификация, со-
содержащая в макромолекулах блоки дурофениленокси-
дурофениленоксида, — США), поли-/г-ксилилен и его монохлорзаме-
щенное производное. Дорил применяют для приготов-
приготовления электроизоляционных лаков, электроизоляцион-
электроизоляционных прессматериалов (наполнители — стекловата, ас-
асбестовая ткань, окись магния), пенопластов, исполь-
используемых как теплоизоляционный строительный мате-
материал. Этой смолой пропитывают ткани для придания
им негорючести. Стеклопластики на основе дорила
применяют для обшивки ракет, работающих на жид-
жидком топливе.
Жидкие П. используют как смазочные масла и тепло-
теплоносители. Сульфидированные форполимеры рекомен-
рекомендованы для изготовления лаков, отверждаемых
термически, а также как связующее в производстве
стеклопластиков.
Лит.: Ф и л л и п с Л., Хим. и технол. полимеров, № 11
13 A965); Гуров А., Зеленецкий А., там же, № И
115 A965); Юкельсон П. И., Глуховский В. С.
Усп. хим., 38, в. 12, 2155 A969); В i 1 о w N.. М i 1 1 е г L. J.
J. Macromol. Sci., A3, № 3, 501 A969); Юкельсон П. И.
Назарова А. Б., Гармонов В. И., Высокомол
соед., А13, 821 A971). А. Н. Зеленецкий
ПОЛИАЛКИЛЕНГЛИКОЛЬМАЛЕИНАТЫ И ПО-
ЛИАЛКИЛЕНГЛИКОЛЬФУМАРАТЫ, поли м а
леинаты и полифумараты [poly(alkylene
glycol maleates) and poly(alkylene glycol fumarates),
Polyalkylenglykolmaleinate und Polyalkylenglykolfuma-
rate, maleates et fumarates des polyalcylene-glycols] —
продукты поликонденсации малеиновой и фумаровой
к-т с алифатическими (реже арилалифатическими) гли-
колями. Общая ф-ла полималеинатов (ПМ) и полифу-
маратов (ПФ) имеет след. вид:
H-[-(OR'OCCH=CHC) -(OR'OCR"C) -1 -ОН
II II х . II IIу п
о о оо
где R' и R" — радикалы, входящие в состав молекул
гликолей и модифицирующих двухосновных к-т;
х = 1 — 5; у = 0 — 5; п = 1 — 20. Иногда ПМ и
ПФ наз. соответственно п о л и э ф и р м а л е и н а-
тами и полиэфирфу Маратам и. Эти тер-
термины неточны, т. к. суффикс «ат» уже свидетельствует
о принадлежности данного соединения к классу эфи-
ров, так что нет необходимости вводить в название сло-
слово «эфир».
Получение
Исходные вещества. Для синтеза ПМ и
ПФ, помимо малеиновой и фумаровой к-т, используют
малеиновый ангидрид (о свойствах этих соединений
см. Малеинового ангидрида полимеры и сополимеры,
Малеиновой и фумаровой кислот сополимеры). Аналоги
ПМ и ПФ — полиэфиры др. ненасыщенных двухоснов-
двухосновных к-т, напр, итаконовой, цитраконовой, мезаконовой
и хлормалеиновой, имеют значительно меньшее прак-
тич. значение.
Для регулирования свойств ПМ и ПФ (напр., для
улучшения их растворимости в органич. соединениях,
способных к согюлимеризации с ними, повышения ог-
огнестойкости или химич. устойчивости их сополиме-
сополимеров) в поликонденсацию вместе с основными реаген-
реагентами вводят обычно т. наз. модифицирующие к-ты
или ангидриды: фталевый ангидрид, изофталевую и
терефталевую к-ты, тетра- и гексагидрофталегый,
711
ПОЛИАЛКИЛЕНГЛИКОЛЬМАЛЕИНАТЫ И ПОЛИАЛКИЛЕНГЛИКОЛЬФУМАРАТЫ
712
тетрахлор- и тетрабромфталевый, эндометилентетрагид-
рофталевый и хлорэндиковый ангидриды, янтарную,
адипиновую, азелаиновую, себациновую и др. к-ты.
Из двухатомных спиртов, используемых для полу-
получения ПМ и ПФ, наиболее распространены этилен-,
1,2-пропилен-, диэтилен- или триэтиленгликоль, нео-
пентилгликоль, гидрированный и оксипропилирован-
ный дифенилолпропан. Иногда для блокирования кон-
концевых групп полиэфиров с целью повышения их сов-
совместимости с сомономерами, а также для увеличения
водостойкости сополимеров в реакцию вводят в неболь-
небольшом количестве одноатомные спирты и одноосновные
кислоты.
Синтез. ПМ и ПФ получают поликонденсацией,
чаще всего при 170—230 °С в расплаве или при более
низких темп-рах A00—150 °С) с добавлением 10—20%
(от массы смеси реагентов) растворителя (толуола,
ксилола или др.)> образующего азеотропную смесь
с выделяющейся в реакции водой. Если в синтезе ис-
используют ангидриды двухосновных к-т, процесс проте-
протекает в две стадии: 1) при 50—150 °С с большой ско-
скоростью идет экзотермич. [тепловой эффект 25 —
38 кдж/моль F—9 ккал/молъ)] реакция присоедине-
присоединения ангидридов к гликолям с образованием кислых
моно- и диэфиров; 2) поликонденсация (эндотермич.
реакция) этих эфиров, напр.:
НОСН2СН2ОН + СН=СН ?± НОСН2СН2ОССН=СНСОН (i)
II II II w
о
с
4 о чо
7iHOCH2CH2OCCH=CHCOH
II II
о о
: Н-[-ОСН2СН2ОССН=СНС-1п-ОН +
«- 1) Н2О
B)
Модифицирующие к-ты не участвуют в реакциях при-
присоединения на первой стадии. Они вступают только
в поликонденсацию с кислыми эфирами и гликолями,
не прореагировавшими на первой стадии. Поликонден-
Поликонденсация подчиняется закономерностям реакций второго
порядка. Эффективная константа скорости реакции
при 200 °С C0—120) АО-* г/(моль-сек) [2—7 г/(моль-мин)],
энергия активации 50—84 кдж/молъ A2—20 ккал/молъ).
Для ускорения процесса можно применять катализа-
катализаторы: соляную к-ту, тг-толуолсульфокислоту, кислот-
кислотные иониты, ацетат натрия и др.
Процесс получения этих полиэфиров осуществляют
по периодич. схеме в реакторах из нержавеющей кис-
кислотоупорной стали (рис. 1) при интенсивном переме-
перемешивании реагентов якорной или пропеллерной мешал-
мешалкой в токе инертного газа; это предотвращает окисле-
окисление продукта и способствует удалению паров выде-
выделяющейся в реакции воды. Последняя вместе с нек-рой
частью летучих гликолей проходит через обогреваемый
(выше 100 °С) конденсатор; гликоли конденсируются
и возвращаются в реактор. Водяные пары конденси-
конденсируются в холодильнике. По количеству воды в сбор-
сборнике конденсата судят о степени завершенности реак-
реакции. Для ускорения процесса на глубоких стадиях
реакцию можно проводить под вакуумом для отгонки
остатков непрореагировавших исходных в-в и воды.
После получения реакционной массы, имеющей кис-
кислотное число (критерий степени поликонденсации)
порядка 25—45, синтез завершают, продукт охлаж-
охлаждают и стабилизируют, вводя в реактор небольшое
количество ингибитора полимеризации, напр, гидро-
гидрохинона, гс-бензохинона или гс-трет-бутилпирокатехи-
гс-трет-бутилпирокатехина. В зависимости от степени завершенности реакции
и состава исходных веществ, в частности от того, в ка-
какой форме применяли кислотные реагенты (ангидрид
или к-та), выход продукта колеблется в пределах
85—95% (от массы введенных в реакцию веществ).
Продолжительность процесса 6 — 20 ч. Товарный про-
продукт — раствор полиэфира в мономере, так называе-
называемая полиэфирная смола.
ПМ и ПФ могут быть также получены взаимодей-
взаимодействием окисей олефинов с ангидридами двухосновных
к-т, фенолятов двухатомных фенолов с хлорангидри-
дами двухосновных к-т или нек-рыми др. методами.
Глиноль
Полиэфирная смола
Рис. 1. Технологическая схема производства полиалкилен-
гликольмалеинатов и полиалкиленгликольфуМаратов, а так-
также их р-ров в стироле (полиэфирных смол): 1 — мерник для
гликоля; 2 — реактор; з — аппарат (смеситель) для раство-
растворения полиэфира в мономере; 4 — конденсатор; ,5 — холо-
холодильник; б — сборник выделившейся в поликонденсации
воды.
Синтез ПМ и ПФ сопровождается побочными реак-
реакциями: превращением эфиров малеиновой к-ты в фу-
мараты (tywc-mpa/ic-изомеризация), присоединением гли-
гликолей по двойным связям с образованием производных
оксиалкоксиянтарных к-т, димеризацией и полимери-
полимеризацией ненасыщенных эфиров.
Отверждение. ПМ и ПФ отверждают, сопо-
лимеризуя их с различными мономерами, напр, со
стиролом, метилметакрилатом, диметакрилатами гли-
гликолей, винилтолуолом, хлорстиролом, диаллилфтала-
том, триаллилциануратом, или со способными к сопо-
лимеризации олигомерами, напр, с олигоэфиракрша-
тами. Мономеры и олигомеры служат, как правило,
растворителями полиэфиров и, кроме того, «сшиваю-
«сшивающими агентами», т. к. они сополимеризуются с ПМ и
ПФ с образованием трехмерных сополимеров (см.
Отверждение, Отвердители). В случае использования
в качестве растворителя стирола схему отверждения
можно представить в след. виде:
'OROOCCH==CHCOOROOCCH==CHCO~ СН9=СН
~OROOCCH=CHCOOROOCCH==CHCQ'
Инициатор
OROOCCH —CHCOOROOCCH —СНСО-'
OROOCCH—CHCOOROOCCH—СНСО-
713
ПОЛИАЛКИЛЕНГЛИКОЛЬМАЛЕИНАТЫ И ПОЛИАЛКИЛЕНГЛИКОЛЬФУМАРАТЫ
714
где R — остаток гликоля, хну — число звеньев сти-
стирола в сшивающих мостиках.
Отверждение полиэфирных смол протекает но ради-
радикальному механизму, обычно в присутствии перекисных
инициаторов (напр., перекисей бензоила, дикумила,
метилэтилкетона или циклогексанона, гидроперекиси
кумола) при 80—150 °С; в случае использования уско-
ускорителей — при 20—30 °С и даже более низких теми-рах
(отверждение на холоду). Эффективные ускорители,
применяемые в сочетании с перекисью бензоила,—
третичные амины, напр, диметил-, диэтил- или диэта-
ноланилин; с перекисями на основе кетонов и гидро-
гидроперекисями — нек-рые соли Со и V. В качестве уско-
ускорителей сополимеризации можно применять также
меркаптаны, jx-комплексы металлов, аскорбиновую и
диоксималеиновую к-ты и др.
Сополимеры стирола с ПМ и ПФ обычного состава
(немодифицированные или модифицированные фтале-
вой к-той), характеризующиеся оптимальными меха-
нич. свойствами, получают в присутствии инициирую-
инициирующих систем, содержащих нафтенат кобальта с гидро-
гидроперекисью кумола или перекисью метилэтилкетона.
Известны многокомпонентные инициирующие системы,
состоящие из двух инициаторов (напр., гидроперекиси
кумола и перекиси бензоила), одного инициатора и
двух ускорителей (напр., гидроперекиси кумола, наф-
тената кобальта и диметиланилина), двух инициаторов
и двух ускорителей (напр., перекиси бензоила, гидро-
гидроперекиси кумола, диметиланилина и нафтената ко-
кобальта). Выбор инициатора и ускорителя, а также их
дозировка зависят от состава полиэфира и мономера,
их соотношения и условий переработки. Процесс экзо-
термичен [теплота сополимеризации 210—500 кдж/кг
E0-120 ккал/кг)].
Полиэфиры можно отверждать также в присутствии
неперекисных инициаторов: соединений, склонных к
кето-енольной таутомерии (напр., фенилацетальдегида,
дезоксибензоина, гидратропового альдегида), динитрила
азоизомасляной к-ты, окиси стирола или ионных ка-
катализаторов, напр. TiCl4, SnCl2 или BF3. Возможно
также отверждение фотохимич. и радиационным ме-
методами.
Свойства
В зависимости от состава, химич. строения и мол.
массы (к-рая обычно лежит в пределах 500—3000)
ПМ и ПФ — вязкие жидкости или твердые вещества
различной окраски (бесцветные, светло-желтые, янтар-
янтарные, темно-красные, коричневые); плотность 1100—
1500 кг1м*, или 1,1 — 1,5 г/см3 B0 °С). Показатель пре-
преломления полиэфиров увеличивается при повышении
степени поликонденсации и замене алифатич. модифи-
модифицирующих к-т ароматическими. Этот показатель связан
линейной зависимостью с кислотным числом (КЧ)
полиэфиров. Так, для полидиэтиленгликольмалеинатов
и полидиэтиленгликольмалеинатфталатов A,1:0,67:0,33)
такая зависимость выражается соответственно ф-лами:
и? =1,5024-89-10-е КЧ;
л? = 1,5221 —108-10~6КЧ
Большинство промышленных ПМ и ПФ аморфно,
однако при использовании нек-рых видов сырья и
специальных методов синтеза получают полимеры,
содержащие кристаллич. фазу, напр, полиэтиленгли-
кольфумарат, полиэфиры на основе полиоксиэтилен-
гликолей, нек-рые полиэфиры, модифицированные се-
бациновой и терефталевой к-тами, в частности полу-
полученные двухстадийной поликонденсацией. Темп-pa раз-
размягчения твердых ПМ и ПФ достигает 100—130 °С.
Мол. массу ПМ и ПФ определяют обычно химич. ме-
методом (по концевым группам); возможно применение
эбулиоскопии и криоскопии. Зависимость характери-
стич. вязкости [г]] в дл/г полигексаметиленмалеината
и полигексаметиленфумарата в хлороформе от степени
их поликонденсации (Р) выражается соответственно
ур-ниями:
[г,] = 2,79-10-4
Вязкость расплава полиэфиров возрастает пропор-
пропорционально Рх (где х = 2 — 6), а при Р = const —
с увеличением соотношения между концевыми карбок-
сильными и гидроксильными группами. При введении
в макромолекулы ПМ или ПФ остатков ароматич. к-т
вязкость их расплавов значительно возрастает; моди-
модифицирование же полиэфиров нек-рыми алифатич.
к-тами, напр, янтарной или адипиновой, приводит
к уменьшению вязкости
расплава.
ПМ и ПФ растворяют-
растворяются в кетонах, эфирах,
хлорированных углево-
углеводородах и многих вини-
виниловых и аллиловых мо-
мономерах с образованием
р-ров различной естест-
естественной окраски, анало-
аналогичной окраске поли-
полиэфиров. Вязкость р-ров
этих полиэфиров в моно-
мономерах снижается с уве-
увеличением содержания
мономера и особенно рез-
резко при повышении
темп-ры (рис. 2). Нек-рые
Рис. 2. Зависимость вязко-
вязкости полиэфирных смол оте-
отечественных марок от
темн-ры: 1 — ПН-2; 2 —
ПН-3; 3 — ПН-1; 4 — ПН-4.
750
500
1 250
30 50 70 90 ПО
Температура, °С
полиэфирные смолы проявляют тиксотропные свойства;
эти свойства можно также придать введением специаль-
специальных добавок, напр, бентонита (глина), белой сажи,
поливинилхлорида и др.
Свойства отвержденных полиэфирных смол (сополи-
(сополимеров) зависят от химич. состава и строения сомоно-
меров, мол. массы, природы концевых групп полиэфи-
полиэфиров, условий сополимеризации и др. Типичные свойства
р-ров ПМ и ПФ в стироле, применяемых в качестве
связующих, а также отвержденных (ненаполненных)
полиэфирных смол приведены ниже:
Растворы
Плотность при 20еС, кг/м3 (г/см3) . . .
Вязкость при 20LC, мн-сек/м2, или спз
•го
Показатель преломления r?D
Отвержденные смол
Плотность при 20е С, кг/м3 (г/см3)
1 000-1 400
A,0-1,4)
75-7 000
1,49-1,55
Показатель преломления nD .
Уд. теплоемкость, кдж/(кг-К)
[ккал/(пг-сС)]
Теплопроводность, вт/(м • К)
/(^С)]
Темп-рный коэфф. линейного расширения,
СС
Теплостойкость, LC
по Вика
по Мартенсу
Прочность, Мн/м2(кгс/см2)
при растяжении
при сжатии
при изгибе
1 100 — 1 500
A,1-1,5)
1,52-1,57
1,05 — 2,30
[0,25-0,55]
0,15-0,22
[0,13-0,19]
G0 — 200)-10-°
55-250
40—100
20-100
B00 — 1 000)
80-200
(800-2 000)
30-130
C00-1 300)
715
ПОЛИАЛКИЛЕНГЛИКОЛЬМАЛЕИНАТЫ И ПОЛИАЛКИЛЕНГЛИКОЛЬФУМАРАТЫ
716
Относительное удлинение, % 0,2 — 7
Модуль упругости при растяжении,
Гн/м2 (тс/см2) 1,5 — 4,5
A5 000 — 45 000)
Твердость по Бринеллю, Мн/м2 (тс/мм2) 10 0 — 350
A0-35)
Ударная вязкость, кдж/м2, или тс• см/см2 2 — 15
Диэлектрич. проницаемость при 1 Мгц . . 2,8—5,0
Тангенс угла диэлектрич. потерь при \Мгц 0,01—0,04
Уд. электрич. сопротивление
поверхностное, Том (ом) 1—104
(Ю121(I0)
объемное, Гом-м(ом-см)
Электрич. прочность, Мв/м, или кв/мм
()
10 —104
A012-Ю18)
15 — 25
Одно из важнейших свойств сополимеров ПМ и ПФ
с различными мономерами, определяющих условия
применения этих продуктов,— теплостойкость. По-
Последняя возрастает при увеличении плотности сшивки
ПМ или ПФ вследствие повышения содержания двойных
связей в исходном полиэфире и увеличения степени
их превращения при сонолимеризации. Степень пре-
превращения возрастает в результате изомеризации ма-
леиновой к-ты в фумаровую при синтезе полиэфира,
а также в результате правильного выбора соотношения
реагентов и условий сополимеризации. Максимальная
теплостойкость большинства сополимеров ПМ и ПФ
со стиролом наблюдается при содержании стирола
30—40% (рис. 3). Как правило, при одинаковой плот-
плотности сшивки тепло-
теплостойкость сополимеров
полиэфиров, модифици-
модифицированных ароматич.
или алициклич. к-тами,
выше, чем у сопо-
сополимеров на основе ПМ
и ПФ, модифицирован-
модифицированных алифатич. к-тами.
Аналогичное влияние
на свойства отвержден-
ных смол оказывает
состав диолов. В част-
частности, весьма тепло-
теплостойкие сополимеры
160
140
120
100
80
60
40
15
25
35
45
55
Рис. 3. Влияние содер-
содержания стирола на теп-
лостойкость его сополи-
Содермание стирола, Л меров с различными по-
полиэфирами: 1 — ПОЛИДИ-
этиленгликольмалеинат; 2 — полиэтиленгликольмалеинат-
адининат C:2:1); з — полиэтиленгликольмалеинатфта-
лат B:1:1); 4 — полипентаэритритдихлоргидринмалеи-
натфталат B:1:1); ,5 — полидиэтиленгликольмалеинат-
фталат C:2: 1); б — полидиэтиленгликольмалеинатдифе-
нат C:2: 1).
получены из полиэфиров на основе гидрированного и
оксипропилированного дифенилолпропана (теплостой-
(теплостойкость по Мартенсу выше 100 °С).
С повышением мол. массы ПМ и ПФ темп-pa стекло-
стеклования и теплостойкость сополимеров возрастают. Теп-
Теплостойкие сополимеры, у к-рых в значительной мере
сохраняется прочность при нагревании до темп-р
выше 200 °С, м. б. также синтезированы сополимери-
зацией полиэфиров с полифункциональными мономе-
мономерами, напр, триаллилциануратом.
Обычно прочность, твердость и модуль упругости
при растяжении сополимеров достигают максимальных
значений при содержании модифицирующих к-т~ 0,15—
0,50 молъ на 1 молъ суммарного количества кислотных
реагентов. Жесткость сополимеров увеличивается с
повышением концентрации двойных связей в исходных
ПМ и ПФ, а при постоянстве последней — с повы-
повышением содержания в них ароматич. циклов.
Оптимальные механич. свойства сополимеров со сти-
стиролом немодифицированных ПМ и ПФ, а также моди-
модифицированных фталевой к-той, достигаются при мол.
массе исходных ПМ и ПФ 700 — 1200. С увеличением
концентрации концевых карбоксильных групп при
постоянстве мол. массы полиэфиров прочность и мо-
модуль упругости при растяжении сополимеров воз-
возрастают.
Оптимальное содержание мономера в полиэфирной
смоле зависит от степени ненасыщенности и состава
ПМ и ПФ. Как правило, максимум твердости и проч-
прочности сополимеров полиэфиров со стиролом соответ-
соответствует 30—40%-ной концентрации мономера в исход-
исходных растворах. Если степень ненасыщенности поли-
полиэфиров мала [молярная концентрация ненасыщенной
кислоты (или ангидрида) менее 50% от суммы кис-
кислотных реагентов], прочность отвержденных продук-
продуктов монотонно возрастает с увеличением содержания
стирола.
Для получения сополимеров, характеризующихся
повышенными ударной вязкостью A5—100 кдж/м2,
или же-см/см2), деформируемостью (относительное
удлинение 20—600%) и морозостойкостью (темп-ра
хрупкости от —20 до —70 °С), ПМ и ПФ модифицируют
алифатич. к-тами с длинной цепью, применяют гликоли,
содержащие простые эфирные группы, напр, нолиокси-
алкиленгликоли, или уменьшают степень ненасыщен-
ненасыщенности полиэфиров (т. е. содержание ненасыщенной
к-ты). Иногда с этой же целью полиэфиры синтезируют
специальными методами, напр, двухстадийной поли-
поликонденсацией. Термич. обработка продуктов, получен-
полученных отверждением «на холоду», способствует увеличе-
увеличению глубины сополимеризации и, следовательно, по-
повышению их прочности и модуля упругости при рас-
растяжении.
Большая часть отвержденных полиэфирных смол —
хорошие электроизоляционные материалы. С повыше-
повышением темп-ры, а также под воздействием влаги тангенс
угла диэлектрич. потерь и диэлектрич. проницаемость
отвержденных полиэфирных смол возрастают (рис. 4).
При получении прозрачных листовых материалов
из полиэфирных стеклопластиков необходимо, чтобы
оптич. свойства компонентов системы были близки
между собой, а между компонентами осуществлялся
тесный контакт. Т. к. отвержденные стиролсодержащие
смолы обычного состава имеют более высокий показа-
&0.04
) о 0,03
0.01
120
5 10 20 30 50 90
Время, сутки
Рис. 4. Зависимость тангенса угла диэлектрич. потерь при
частоте 7 Ггц стеклопластиков на основе полиэфирных смол
отечественных марок ПН-3 и ПН-10 от продолжительности
пребывания их в воде: 1 — полиэфирняя смола ПН-3; 2 —
полиэфирная смола ПН-10.
тель преломления, чем стекло (для малощелочного
стекла п^ = 1,548), в составе связующих для прозрач-
20
ных пластиков часть стирола (п^ = 1,547) заменяют
20
метилметакрилатом (ио = 1,415) или другим мономе-
мономером с более низким показателем преломления, чем у
стирола.
Полиэфиры, содержащие остатки алифатических и
алициклич. двухосновных к-т, более устойчивы к дей-
действию УФ-лучей, чем ПМ и ПФ, модифицированные
717
ПОЛИАЛКИЛЕНСУЛЬФИДЫ
718
ароматич. к-тами. Частичная замена стирола, входя-
входящего в состав полиэфирных смол, метилметакрилатом
приводит к уменьшению поглощения УФ-излучения.
Для повышения светостойкости изделий на основе поли-
полиэфирных смол в них вводят светостабилизаторы (про-
(производные 2-оксибензофенона и бензотриазола, салол
или др.) и отбеливающие агенты на основе бензотриазо-
лов, бенздиазолов и нек-рых др. соединений.
Химич. свойства ПМ и ПФ определяются наличием
в составе их макромолекул двойных связей, сложно-
эфирных и концевых групп — карбоксильных и гидро-
ксильных. Так, полиэфиры взаимодействуют, напр.,
с циклопентадиеном, гексахлорциклопентадиеном, ан-
антраценом, дитерпенами или др. с образованием аддук-
тов (реакция Дильса — Альдера). Оту реакцию при-
применяют для модификации полиэфиров в процессе их
синтеза. ПМ и ПФ присоединяют по двойным связям
водород, галогены и меркаптаны, что используют для
определения содержания двойных связей в полиэфи-
полиэфирах. Реакции концевых групп с монофункциональными
соединениями, напр, с к-тами и спиртами, служат для
регулирования мол. массы полиэфиров, придания им
повышенной совместимости с мономерами, а сополиме-
сополимерам — повышенной химич. стойкости. При взаимодей-
взаимодействии концевых групп с полифункциональными соеди-
соединениями, напр, с полиизоцианатами или диэпоксидами,
может происходить сшивание макромолекул полиэфи-
полиэфиров или образование блоксополимеров.
Сополимеры ПМ и ПФ со стиролом отличаются высо-
высокой водостойкостью и устойчивостью к действию мине-
минеральных и органич. к-т, бензина, масел и окислителей;
стойкость к действию щелочей, как правило, невысока.
Разработаны полиэфиры на основе производных ди-
фенилолпропана; прочность их сополимеров со стиролом
после отверждения существенно не изменяется при
длительном пребывании в щелочных средах. Обычно
термическая и термоокислительная деструкция от-
вержденных смол протекает с большой скоростью при
темп-pax выше 200 °С. Поэтому материалы на основе
полиэфирных смол можно использовать в условиях
длительной эксплуатации при темп-pax до 100 °С и
лишь в отдельных случаях — при 150—190 СС.
Применение
ПМ и ПФ выпускают в пром-сти в виде 50 — 70%-ных
р-ров в различных мономерах или олигомерах (ненасы-
(ненасыщенные полиэфирные смолы), т. е. в виде продуктов,
пригодных к непосредственному использованию. Боль-
Большую часть ПМ и ПФ используют в качестве связующих
для армированных пластиков, гл. обр. стеклопласти-
стеклопластиков. Ненасыщенные полиэфирные смолы (без армирую-
армирующих наполнителей) используют для заливки различных
деталей радио- и электротехнич. оборудования (см.
Компаунды полимерные), медицинских, биологических
и музейных препаратов, изготовления кабельных муфт,
листовых и стержневых заготовок для галантерейных
изделий. Полиэфирные смолы находят широкое при-
применение для приготовления полиэфирных лаков и эма-
эмалей, используемых для отделки мебели, корпусов
радиоприемников и телевизоров и др. Кроме того, эти
смолы применяют для пропитки древесины и пористых
металлич. отливок с целью их герметизации, а^также
для пропитки катушек трансформаторов и обмоток
электрич. машин.
Полиэфирные смолы применяют как основу при
изготовлении наливных полов промышленных зданий;
замазок для футеровки и ремонта химич. оборудования;
шпатлевочных масс для гидро- и пароизоляции бетона
и для заделки крупных дефектов металлич. литья;
клеев для склеивания стеклопластиков друг с дру-
другом, с асбоцементными и древесноволокнистыми пли-
плитами, сотопластами и др. материалами; клеев для склеи-
склеивания оптич. деталей и т. д. (см. Полиэфирные клеи).
Нек-рые ПМ и ПФ с гибкими молекулами вводят в
небольших количествах в полиэфирные и эпоксидные
смолы, образующие при отверждении жесткие продук-
продукты, с целью повышения их деформируемости, ударной
прочности и морозостойкости.
Впервые синтезированные в 1894 Форлендером ПМ
и ПФ стали производиться в промышленном масштабе
лишь в середине 40-х годов XX в. (США и Великобри-
Великобритания). С тех нор их производство во всем мире непре-
непрерывно расширяется. В 1972 мировое производство поли-
Нек-рые свойства полиэфирных смол отечественных и анало-
аналогичных им зарубежных марок, выпускаемых в промышленности
Полиэфирная смола
(страна-изготовитель)
Плотность при 20°С,
кг/м3 (г/см3)
Вязкозть
при 20сС,
мн-сек/м2,
или сиз
ПН-1 (СССР) ....
AR 403 (США) . .
Селлобонд АХ 2621
(Великобритания)
Смолы общего назначения
1 130-1 160 A,13-1,16)
1 140- 1 180 A,14 — 1,18)
ПН-3 (СССР) ....
Селлобонд АХ 266 0
(Великобритания)
1 150-1 160 A,15-1,16)
Теплостойкие смолы
1 120 — 1 160 A,12 — 1,16)
1 110-1 120 A,11-1,12)
ПН-62 (СССР) . . .
Родэстер 1511 L
(Великобритания)
Самозатухающие смолы
1 260-1 290 A,26-1,29)
1 310 A,31*)
5 0 0 — 80 0
2 000-3 000*
800 — 1 200
500-1 000
800-1 200
1 000-1 500
1 000-1 400*
* При 25 °С.
эфирных смол превысило 1 млн. га. Многие страны
выпускают широкий ассортимент полиэфирных смол,
в том числе смолы общего назначения, теплостойкие,
самозатухающие, химически стойкие, повышенной эла-
эластичности и др. (см. таблицу).
Лит.: Polyesters and their applications, N. Y.— L., 1956;
Б е н и г Г. В., Ненасыщенные полиэфиры. Строение и свой-
свойства, пер. с англ., М., 1968; Коршак В. В., Виногра-
Виноградова С. В., Гетероцепные полиэфиры, М., 1958; их же,
Равновесная поликонденсация, М., 1968; Ли II. 3., Михай-
Михайлова 3. В., Седов Л. Н., в кн.: Справочник по пласт-
пластмассам, т. 2, под ред. М. И. Гарбара [и др.], М., 1969, с. 7;
их же Вестн. технической и экономической информации
(НИИТЭХИМ), в. 11, 1961; DemmlerK., в кн.: Glasfa-
serverstarkte Kunststoffe, В.- [u. a.], 1967; Klosowska-
WolkowiczZ., KrolikowskiW., PenczekP.,
Zywice in laminaty poliestrowe, Warszawa, 1969; Седов Л. Н.
[и Др.], Синтез ненасыщенных полиэфиров, в сб. «Обзоры по
отдельным производствам химической промышленности»,
НИИТЭХИМ, вып. 42, 1973, с. 107. Л. Я. Седов.
ПОЛИАЛКИЛЕНСУЛЬФИДЫ [poly(alkylene sul-
fides), Polyalkylensulfide, sulfures des polyalcylenes].
Алкиленсульфиды, т и и р а н ы (А0 — соедине-
соединения, содержащие в молекуле трехчленный цикл с од-
одним атомом серы \q q/
/4
Наибольшее распространение в качестве мономеров
получили этиленсульфид и пропиленсульфид — бес-
бесцветные жидкости (табл. 1) с резким неприятным зана-
Таблица 1. Свойства этиленсульфида и пропиленсульфида
Свойства
Темп-pa кипения, °С . . . .
Показатель преломления
20
nD
Теплота сгорания, кдж/молъ
(ккал/моль)
Теплота образования,
кдж/молъ (ккал/молъ) . . .
Дипольный момент, D . . .
Этиленсульфид
СН.-СН,
V '
55--5G
1,4914
-2013,9
(—481 ,02)
51,83A2,38)
1 ,66
Пропиленсульфид
СН3-СН-СН2
75-76
1 ,4730
— 2653 4
(-633,75)
11 ,47B,74)
1,95
719
ПОЛИАЛКИЛЕНСУЛЬФИДЫ
720
хом, хорошо растворимые в органич. растворителях и
очень ограничено — в воде.
Электронная плотность на атоме S и энергия напря-
напряжения (82,81 кдж/молъ, или 19,78 ккал/молъ) тиирано-
вого цикла — наименьшие в ряду насыщенных трех-
трехчленных гетероциклов.
Упругость паров (в мм рт. ст.; 1 мм рт. ст.=
= 133,322 н/м2) этиленсульфида в интервале t =
= 18—88 °С подчиняется ур-нию:
log р = 7,03725 — 1194,37/(* + 232,42)
Теплота парообразования при 25 СС 30,31 ^ 0,209
кдж/молъ G,24 ^ 0,05 ккал/молъ).
Пропиленсульфид может существовать в двух опти-
оптически активных формах; величина уд.оптич. вращения
[a]D 37,5° (измерена в хлороформе).
Аналогично оксиранам все реакции тииранов про-
протекают с раскрытием цикла. В радикальных реакциях
А. не участвуют.
Реакции присоединения несимметричных А., напр,
пропил енсульфида, сопровождающиеся раскрытием
цикла, дают смесь двух возможных изомеров, причем
для анионного (нуклеофильного) присоединения харак-
характерно «нормальное» раскрытие цикла (у первичного
углеродного атома), а для катионного (электрофиль-
ного) — «нормальное» и «аномальное» (у вторичного
углеродного атома).
А. оказывают вредное воздействие на печень и ночки.
Летальная концентрация в воздухе 0,035%.
В лаборатории в полупромышленном масштабе А.
получают гл. обр. взаимодействием соответствующего
циклич. карбоната с тиоцианатом щелочного метал-
металла при 100—200 СС в атмосфере азота. Выход этилен-
сульфида 80—85% , прониленсульфида 50—60% .
Др. важный метод синтеза А.— разложение циклич.
монотиокарбонатов в присутствии малых количеств
щелочи. Так, при разложении этиленмонотиокарбоната
в присутствии 1% Na2CO3 при 200 °С образуется эти-
ленсульфид (выход 88%). А. могут быть получены так-
также взаимодействием соответствующих окисей и тиомоче-
вины или неорганич. тиоцианатов при 0—20 °С в вод-
водных или спиртовых р-рах. Выход А. 40—75%.
Полиалкиленсульфиды (П.) — полимеры общей ф-лы
[—СН2 — СН — S —]п, где R = Н или алкил.
R
Получение. П. получают поликонденсацией
или полимеризацией соответствующих мономеров с
раскрытием цикла.
Поликонденсацией получаются П. относительно низ-
низкой мол. массы (не более 10 000) по след. схемам:
п Вг—СН2—СН2—Вг + п K2S -> f-CH2-CH2-S-]n + 2?г КВг
п HO-CH2-CH2-SH
Н+
[-CH2-CH2-S-1 + п Н2О
Катионная полимеризация А. с раскрытием цикла и
образованием линейных П. протекает в присутствии,
напр., минеральных к-т, BF3, PF5, TiCl4, A1C13. Эти-
ленсульфид гюлимеризуется очень легко (даже при
комнатной темп-ре). П., полученные в присутствии
кислотных катализаторов, быстро деструктируются
под действием остатков, катализатора (поэтому мол.
масса этих П. не превышает 10 000). Меркаптаны инги-
бируют полимеризацию. Энергия активации катионной
полимеризации пропиленсульфида 75,4 ^ 6,3 кдж/молъ
A8 ±- 1,5 ккал/молъ); она в основном определяется
энергией разрыва цикла в актах роста цепи под влия-
влиянием активного центра. Скорость полимеризации про-
пропорциональна концентрациям мономера и катализатора
в первой степени.
Полимеризация А. в хлористом этиле в присутствии
эфирата BF3 протекает но механизму «живущих»
цепей, причем на глубоких стадиях превращения про-
происходит передача цепи с разрывом. Об этом свидетель-
свидетельствует бимодальный характер кривых молекулярно-
массового распределения, существенное расширение
его с увеличением степени превращения и образование
блоксополимеров при полимеризации этиленсульфида
в присутствии полипропиленеульфида.
Анионная полимеризация А. протекает с образова-
образованием «живущих» цепей. Кинетика и механизм детально
изучены на примере полимеризации пропиленсульфида
под действием Na-нафталинового комплекса в тетра-
гидрофуране, а также C4H9Li в различных раствори-
растворителях. В результате анионной полимеризации А.
образуются П. молекулярной массы до нескольких со-
сотен тысяч.
На анионных катализаторах типа Zn(C2H5J или
C4H9Li с оптически активными добавками, напр.
(—)-ментол, (+)-борнеол или L-лейцин, осуществлена
стереоэлективная полимеризация рацемич. пропилен-
пропиленсульфида. В присутствии комплекса C4H9Li«LiOR*
одновременно с полимеризацией происходит десульфи-
рование пропиленсульфида, разложение полимерных
цепей и образование полимеров с дисульфидными свя-
связями, обладающих высокой оптич. активностью (см.
Оптически активные полимеры).
При полимеризации А. на модифицированных ме-
таллоорганич. катализаторах, напр.
Zn (С2Н5J 4- Н2О, Zn (С2Н5J + S, Zn (С2Н5J + Н2О2,
А1 (С2Н5K 4- (n-C4H9SJ Fe,
получаются высокомолекулярные (~200 000) кристал-
лич. П. Активными катализаторами полимеризации А.
служат также ацетилацетонаты переходных металлов,
напр. Мп, Си, Со, Cr, Ш и их комплексы с алкилами
алюминия или тетраалкилалюмоксанами.
При сополимеризации пропиленсульфида с 1-аллил-
окси-2,3-эпитиопропаном в присутствии Zn(C2H5J +
+ Н2О или сополимеризации пропиленсульфида с
этиленсульфидом и ненасыщенным соединением (напр.,
аллилтиоглицидиловым эфиром, эписульфидом пента-
диена-1,4 или гексадиена-1,5) получен новый тип
эластомеров; процесс проводят в воде, катализато-
катализаторы — карбонаты Zn и Cd.
Раскрытие тииранового цикла с образованием линей-
линейных П. может происходить в результате донорно-ак-
цепторного взаимодействия между мономером и орга-
органич. акцепторами электронов (напр., малеиновым ан-
ангидридом, хинонами, полинитросоединениями, я-
к-тами). Начальный акт инициирования полимериза-
полимеризации — образование комплекса с переносом заряда
типа мл, степень переноса электрона в к-ром зависит
от силы акцептора и полярности среды.
Свойства и переработка. Полиэтилен-
сульфид — кристаллич. полимер белого цвета; мол.
масса до нескольких сотен тысяч; плотность 1,33—
1,34 г/см3 B0 °С); темп-pa плавления абсолютно кри-
кристаллич. полимера 215—220 °С, темп-pa стеклования
от —40 до —50 °С; теплота плавления 14,07 ^
± 2,1 кдж/молъ C,36 ± 0,5 ккал/молъ). Полиэтилен-
сульфид растворяется только при повышенных темп-рах
A40—175 °С) в этилентритиокарбонате, дитиолане-1,3,
дитиане-1,4, а-метилнафталине, нитробензоле, диметил-
сульфоксиде; химически стоек к действию к-т и щело-
щелочей; обладает хорошими механич. свойствами, осо-
особенно при повышенных темп-pax (см. табл. 2), вслед-
вследствие чего представляет интерес для пром-сти; во Фран-
Франции производится под названием политиоэтен.
Полиэтиленсульфид можно перерабатывать экструзи-
экструзией при 215—250 °С; лучшие стабилизаторы — поли-
полиамины с высокой температурой кипения или их про-
производные.
Полипропиленсульфид, образующийся на анионных
катализаторах,— аморфное каучукоподобное вещество,
растворимое в ароматич. углеводородах и циклич.
721
ПОЛИАМИДНЫЕ ВОЛОКНА
722
Таблица 2. Зависимость механич. * свойств полиэтиленсульфида
от температуры
Темп-
20
60
100
Модуль упругости,
М н/м° (кгс/см2)
1800A8-103)
1060A0,6-103)
790G,9-Ю3)
Прочность при рас-
растяжении,
Мн/м2 (крс/см2)
70-78 G00-780)
56—62 E60—620)
38—48 C80—480)
Относитель-
Относительное удлине-
удлинение, ?б
10-14
12
25-40
* Ползучесть под действием нагрузки 35,0 Мн/м2{ЗЪ0 кгс/см2)
в течение 500 ч составляет 1%.
эфирах и не растворимое в эфире, ацетоне, спирте, воде;
плотность 1,10—1,12 г/см3 B0 СС); темп-pa стеклова-
стеклования —48 °С. Зависимость характеристич. вязкости [y,]
от среднемассовой мол. массы (Мго ) выражается
ур-нием:
[т)] =0,33.10-4A/w°>86 (бензол, 20 СС).
Полимер обладает повышенной радиационной устой-
устойчивостью.
Полипропиленсульфид, образующийся на координа-
координационных металлоорганич. катализаторах,— частично
или полностью кристаллич. продукт. Изотактнч. фрак-
фракция П. нерастворима в метилэтилкетоне при 100 СС;
т. пл. 40—41 СС, плотность 1,152 г/см3 B0 °С).
Тройной сополимер на основе этиленсульфида B8%),
пропиленсульфида F9%) и аллилтиоглицидилового
эфира C%) (мольная концентрация) — эластомер бе-
белого цвета с мол. массой до нескольких сотен тысяч;
вулканизуется серой. Свойства вулканизата:
Прочность при растяжении,
Мн/м2(кгс/см2) 15,7A60)
Относительное удлинение, % 36 0
Твердость (но Шору) 81
Теми-pa стеклования, СС —40
Свойства вулканизата не изменяются после выдержи-
выдерживания его в течение 3 сут при 120 °С (в присутствии
2% N-фенил-р-нафтиламина как стабилизатора); вул-
канизат обладает повышенной масло-, бензо- и озоно-
стойкостыо.
Полиэтиленсульфид впервые получили К. Левиг и
С. Вейдман в 1839, полипропиленсульфид — Дж.
Крафтс в 1863.
Лит.: ЗандерМ., Усп. хим., 37, в. 3, 433 A968);
Davis F. О., Fettes E. М., в кн.: Polyethers, v. 13,
pt 3, N. Y., 1962, p. 1—37; L a 1 J., в кн.: The Chemictry of
cationic polymerization, ed. P. H. Plesch, Oxf.— [a. o.], 1963;
Ерусалимский Б. Л., Ионная полимеризация поляр-
полярных мономеров, Л., 1970; С и т н и к о в а Т. А. [и др.1,
ДАН СССР, 181, 401 A968); European Polymer J., 7, л» 12,
1661 A971); С о л о м а т и н а И. П., Алиев А. Д.,
Крен цель Б. А., Высокомол. соед., 13, № 4, 252 A971);
European Polymer J., 7, 1721 A971). Л. Л. Стойкая.
ПОЛИАЛКИЛЕНСУЛЬФОНЫ (polyalkylene sulpho-
nes, Polyalkylensulfone, polyalkylenesulfones) — линей-
линейные полимеры, содержащие в основной цепи макромо-
макромолекулы группы SO2. Лучше всего изучены П. строе-
строения [—R—SO2—]п, где R — алкиленовые или цикло-
алкиленовые остатки. Получают П. сополимеризацией
ненасыщенных углеводородов с SO2, напр.:
п RCH=CHR' + п SO2 -> [- СН (R)-CH (R')-SO2—]n
Сополимеризация часто характеризуется относитель-
относительно низкими верхними предельными темп-рами. Ее вы-
вызывают, используя обычные приемы инициирования
радикальных реакций.
Процесс м. б. проведен в массе, р-ре (напр., в толуо-
толуоле, циклогексаноне) или водной эмульсии.
Большинство олефинов образует П. состава R : SO2=
= 1:1; м. б. получены также полимеры состава
R : SO2 = 2:1. П., содержащие в боковой группи-
группировке SO2, получают сополимеризацией диаллила,
диаллилформаля, диаллилацеталя или 1,5-диенов с
SO2. В результате образуются полимерные продукты,
содержащие в цепи циклоалкилсульфоновые группи-
группировки, напр.:
п СН2=СН-СН2-СН3-СН=СН2 + 2п SOa
so2
-> Г-СН2-СН СН2
! I
СН2 CH-SO2-
L \ /
сн2
П. общей ф-лы [— (CH2)W — SO2 —]п, где т > 2,
получающиеся сополимеризацией SO2 с этиленом при
избытке последнего, и их симметрично замещенные
производные способны кристаллизоваться. Однако боль-
большинство полиолефинсульфонов — аморфные вещества.
Темп-ры стеклования их лежат в пределах 40—290 СС
(они тем ниже, чем больше длина бокового замести-
заместителя), напр, для поли(бутен-1)- и полн(октен-1)сульфо-
нов соответственно 83 и 44 °С. Наиболее высокой теп-
теплостойкостью обладают П., содержащие циклич. груп-
группировки в основной цепи [циклогексенсульфон B00 —
205 СС, с разл.), норборненсульфон B40 — 290 °С, с
разл.)]. Относительное удлинение П. 3 — 6%, прочность
при растяжении 28—50 Мн/м2 B80—490 тс/см2).
Растворимость П. изменяется в широких пределах
в зависимости от химич. структуры. Напр., полиизо-
бутиленсульфон, полиC-метилбутен-1)сульфон, ноли-
D,4-диметилпентен-1)сульфон не растворяются ни в
одном из известных органич. растворителей; поли-
(гексен-1)сульфон, поли(октен-1)сульфон и поли(до-
децен-1)сульфон растворимы, напр., в СС14, бензоле,
толуоле, СНС13, диоксане. П., содержащие карбоксиль-
карбоксильные и оксигруппы, растворяются в воде. П. относитель-
относительно устойчивы к действию к-т, однако легко разрушают-
разрушаются водными р-рами щелочей; при нагревании начинают
разлагаться в темп-рном интервале 190 — 275 СС, при-
причем полиолефинсульфоны деполимеризуются.
П. легко деструктируются при облучении у- и
рентгеновскими лучами; при этом сшивания полимеров
не наблюдается. Практич. применения П. пока не
нашли.
Лит.: I v i n К. J., Rose J. В., Adv. Macromol. Chem.,
1, 335 A968); G о е t h a 1 s E. J., J. Macromol. Sci., C2,
73 A968); Fettes E. M., DavisF. О., в кн.: High
Polymers, eds R. A. V. Raff, K. Doak, v. 13, N. Y., 1962, p. 225.
P. Д. Кацарава.
ПОЛИАЛКОКСИСТИРОЛЫ — см. Алкоксистиролов
полимеры.
ПОЛИАЛЛИЛОВЫН СПИРТ — см. Аллиловых со-
соединений полимеры.
ПОЛИАЛЛОМЕРЫ (polyallomers, Polyallomere, po-
lyallomeres) — кристаллич. блоксополимеры а-олефи-
нов. По сравнению с соответствующими гомонолиме-
рами П. характеризуются лучшим комплексом физико-
механич. свойств. Подробнее см. Олефинов полимеры.
ПОЛИАМИДНЫЕ ВОЛОКНА (polyamide fibres, Po
lyamidfasern, fibres de polyamide). В качестве волок-
нообразующих полиамидов при получении П. в. исполь-
используют поликапроамид (см. Капролактама полимеры),
полигексаметиленадипинамид, полиэиаптамид, поли-
ундеканамид, полидодеканамид, полигексаметиленсеба-
цинамид, полипирролидонамид (продукт полимеризации
лактама масляной к-ты), а также полимеры, получае-
получаемые анионной полимеризацией E-лактамов. В конце
60-х гг. XX в. началось производство волокон из поли-
полиамидов, получаемых поликонденсацией ароматич. мо-
мономеров — производных различных изомеров фени-
лендиамина с фталевыми к-тами (см. об этом Полиами-
Полиамиды). Выпуск таких волокон, обладающих высокой
термостойкостью и в ряде случаев исключительно вы-
высокими механич. показателями, пока ограничивается
трудностью их формования из р-ра (см. ниже). Наибо-
Наиболее широкое применение нашли волокна из полиамидов
на основе алифатич. мономеров, к-рые имеют достаточно
723
ПОЛИАМИДНЫЕ ВОЛОКНА
724
высокую термостойкость и вместе с тем могут формо-
формоваться из расплава.
Получение. Технологич. процесс получения П. в.
состоит из трех основных этапов: синтеза полимера,
формования волокна, текстильной обработки волокна.
Синтез полимера обычно осуществляется
на том же предприятии, где производится волокно.
При получении поликапроамида по окончании поли-
полимеризации е-капролактама в системе образуется нек-рое
равновесное для данной темп-ры количество низкомо-
низкомолекулярных соединений (ок. 10%), в основном моно-
мономера и его низших олигомеров. Присутствие их в поли-
полимере затрудняет текстильную переработку волокна и
отрицательно сказывается на потребительских свойствах
готовых изделий. В связи с этим гранулят полимера
перед сушкой подвергается демономеризации путем
водной обработки при повышенных темп-pax. С этой же
целью вакуумируют расплав полимера или обрабатыва-
обрабатывают его перегретым водяным паром или азотом, чтобы
волокно содержало не более 3% низкомолекул. соеди-
соединений.
Для получения волокна из поликапроамида созданы
высокоавтоматизированные непрерывные технологич.
процессы, в к-рых демономеризованный расплав поли-
полимера непрерывно транспортируется по обогреваемым
расплавопроводам к прядильной машине. Удаляемые
из демономеризатора пары низкомолекулярных соеди-
соединений конденсируются, после чего м. б. возвращены
в технологич. цикл. Такая схема процесса позволяет
резко сократить число технологич. операций, получить
более равномерное по физико-химич. свойствам волокно.
Помимо непрерывного процесса получения П. в. непо-
непосредственно из расплава полимера, реализована схема,
в к-рой осуществлены непрерывные операции водной
экстракции низкомолекул. соединений из гранулята
и последующей его сушки в токе инертного газа.
Формование П. в. может быть осуществлено
двумя принципиально различными способами: 1) из
расплава — в тех случаях, когда темп-pa плавления
полимера ниже темп-ры его термич. разложения, и
2) из р-ра — когда темп-pa плавления полимера выше
его темп-ры разложения (для большинства ароматич.
и нек-рых алициклич. полиамидов).
При формовании из расплава гранулят полиамида
плавят в атмосфере инертного газа в устройствах экс-
трузионного или змеевикового типа при 250—300 °С
с последующим продавливанием расплава с помощью
прядильных насосиков через отверстия фильеры. По вы-
выходу из фильеры струйки жидкого полимера попадают
в специальные шахты, куда для их охлаждения подает-
подается воздух.
При охлаждении струек расплава в прядильной шах-
шахте происходят процессы начальной ориентации макро-
макромолекул в волокне и структурообразования полимера.
Ориентация макромолекул вдоль оси волокна обуслов-
обусловливается напряжениями, возникающими вследствие
разности скоростей вытекания расплава из отверстия
фильеры и намотки нити на приемное устройство пря-
прядильной машины. Происходит т. наз. фильерная вы-
вытяжка, степень к-рой составляет 3000 — 6000%. По
выходу из шахты на сформованную нить наносится за-
заданное количество влаги и поверхностно-активных ве-
веществ для предотвращения электризации и распуше-
ния нити, а также для придания ей необходимой ком-
компактности. Пучки волокон поступают в приемное уст-
устройство, где наматываются на цилиндрич. бобину или
укладываются в контейнер. Приемка волокон происхо-
происходит со скоростью от 400 до 1800 м/мин в зависимости
от вида П. в. и конструкции прядильной машины.
Волокно из поликапроамида после формования в ряде
случаев подвергается дополнительной отмывке от низ-
низкомолекулярных соединений, к-рые образуются в поли-
полимере при его плавлении после демономеризации.
При формовании П. в. из р-ра (мокрое или сухо-
мокрое формование) приготавливается прядильный р-р
полиамида с концентрацией полимера от 5 до 20%.
Полиамиды, используемые для формования волокон
из р-ра, плохо растворимы; поэтому в качестве раство-
растворителя используют конц. серную к-ту, диметилацет-
амид, диметилсульфоксид или смесь двух соединений,
напр, диметилформамид с LiCl. Р-р из фильеры попада-
попадает в осадительную ванну, состоящую из того же раство-
растворителя, к-рый использовался для приготовления пря-
прядильного р-ра, но разбавленного водой. Процесс осу-
осуществляется обычно при комнатной темп-ре со скорос-
скоростью 40—100 м/мин. После формования волокно подвер-
подвергается промывке, сушке и препарированию (нанесение
определенного кол-ва влаги и поверхностно-активных
в-в). Т. к. способ формования из р-ра менее экономичен,
чем нз расплава, и связан с преодолением значительных
технич. трудностей его применяют сравнительно редко.
Текстильная обработка П. в. состоит
из операций ориентационного вытягивания, кручения
и перемотки. При ориентационном вытягивании, крат-
кратность к-рого составляет 300 — 500% , в П. в. происходят
глубокие процессы структурообразования, в результате
чего волокно приобретает необходимые текстильные
свойства — прочность при растяжении, эластичность,
усталостную прочность. Технологич. процесс произ-
производства П. в. заканчивается перемоткой их на товар-
товарную паковку, масса и форма к-рой определяются
областью применения П. в. Известны технологич. про-
процессы, в к-рых формование волокна из расплава сов-
совмещается на машине с его текстильной обработкой.
Крашение. Для крашения П. в. (пряжи, тка-
тканей, изделий) наибольшее практич. значение имеют
дисперсные красители и их водорастворимые производ-
производные, кислотные красители и пигменты. С технико-
экономич. точки зрения для нек-рых П. в. перспектив-
перспективным является крашение П. в. в массе. См. также Кра-
Крашение волокон, Крашение химических волокон в массе.
Свойства. Физич. и химич. свойства П. в. зависят
от химич. природы и мол. массы исходного полиамида,
структурных особенностей самого волокна. Обычно
для производства П. в. используют линейные полиами-
полиамиды с мол. массой от 15 000 до 30 000. Повышение мол.
массы полимера улучшает прочность при разрыве,
усталостную прочность, модуль упругости и др. физи-
ко-механич. показатели волокна.
Полиамиды, сформованные в волокно, легко кристал-
кристаллизуются, и многие свойства П. в. определяются со-
состоянием кристаллич. решетки полимера. Нарушение
кристаллич. решетки, напр, введением заместителей
в цепь макромолекулы или сополимеризацией различ-
различных мономеров, приводит к снижению механич. и тер-
термич. свойств волокна. Чувствительность П. в. к изме-
изменению структурных параметров и химич. состава по-
позволяет в широких пределах изменять свойства П. в.
П. в. характеризуются высокой прочностью при
растяжении и усталостной прочностью, отличным со-
сопротивлением к истиранию и ударным нагрузкам.
К недостаткам П. в. можно отнести низкую гигроско-
гигроскопичность, что является причиной их повышенной
электризуемости, сравнительно низкий модуль упру-
упругости (для волокон из алифатич. полиамидов) и низкую
устойчивость к термо- и фотоокислительному воздей-
воздействию. Для повышения устойчивости П. в. к окисли-
окислительным воздействиям в полиамиды вводят различные
антиоксиданты (ароматич. амины и фенолы, органич.
и неорганич. соли металлов переменной валентности
и др.). Помимо кислорода, деструктирующее влияние
на П. в. оказывают нек-рые др. окислители, напр, р-ры
гипохлорита натрия, перекиси водорода. Свойства
наиболее распространенных П. в. приведены в табл. 1.
Максимальная рабочая темп-pa для волокон из али-
алифатич. полиамидов составляет 80—150 °С (темп-pa на-
725
ПОЛИАМИДНЫЕ ВОЛОКНА
Таблица 1. Свойства полиамидных волокон
726
Показатель
Толщина, тепе
Плотность, г/см*
Равновесная влажность, %, при
относительной влажности воз-
воздуха
65%
95%
Прочность, гс/текс
Прочность при растяжении в
мокром состоянии, % от проч-
прочности сухого волокна ....
Прочность в узле, % от абсо-
абсолютной прочности
Прочность в петле, % от абсо-
абсолютной прочности
Устойчивость к многократным
изгибам при напряжении 5 0
Мн'м2 E кгс/мм2) на приборе
ДП-5-1-24, циклы
Устойчивость к истиранию при
нагрузке 0,Зк C0 гс), циклы
Относительное удлинение, %
в сухом состоянии ....
в мокром состоянии . . .
Темп-pa плавления, СС
Поликапро-
п it я- ХЖ TV / Т J* n IT V4 f~\ T_T Ч
амид ^капрон;
1 ,7-187,0
1,13-1,15
3,5-4,5
7 , 0 — 8,5
40-90
85-90
83-93
75-80
25000-30000
1500-2200
14-60
17-65
212-219
Полигексамети-
ленадипинамид
(анид)
1 ,7-187,0
1,15
3,5-4,5
5 ,8 — 6,1
40-80
88-94
80-85
70-75
12000-35000
1000-4000
14-30
14 — 34
240-253
Полиэнанто-
амид
(энант)
5,0-29,0
1 ,1
2 2 — 2 4
2*, 6 — 3', 8
40-80
95-99
—
—
27000-50000
1000-4500
13 — 65
—
205-225
Полипелар-
гонамид
(найлон-9)
5,0-6,7
1 ,06
1 ,2
1 , 3
35-38
98-99
—
—
4500
250-700
24—26
26-27,5
185-205
Полиунде-
канамид
(найлон-11)
5,0-6,7
1,0-1 ,03
0,7-0,9
1,1—1,2
30 — 36
10 0
—
—
13000 — 18000
700-1000
26-27
3 3-34
180-189
Полипирро-
лидонамид
(найлон-4)
—
5,0
45-50
—
—
—
—
—
30-40
—
235 — 237
Поли-3-
лактам
(найлон-3)
1,6
4,5-8,6
18-50
—
—
—
—
—
7-35
—
296-340
чала интенсивного термич. разложения 300—320 СС),
для волокон из ароматич. полиамидов — 350—600 °С.
П. в. неустойчивы к действию конц. к-т, особенно
минеральных, в к-рых легко растворяются. Раствори-
Растворителями для П. в. являются также фенолы, крезолы,
ксилол, трихлорэтан, хлороформ, бензиловый спирт,
нитробензол, диметилацетамид, диметилформамид (осо-
(особенно в сочетании с LiCl), а также нек-рые фторпро-
изводные спиртов и карбоновых к-т. Щелочи при ком-
комнатной теми-ре не оказывают воздействия на П. в.
При повышении темп-ры и концентрации щелочей их
деструктирующее действие на П. в. усиливается. Для
NaOH концентрация, вызывающая максимальную де-
деструкцию, составляет 10 — 12%. Высококонцентриро-
Высококонцентрированные р-ры щелочей на холоду не оказывают сущест-
существенного влияния на прочность П. в. Р-ры соды с кон-
концентрацией 10—30% при темп-pax до 70 °С и аммиач-
аммиачные р-ры любой концентрации при комнатной темп-ре
не снижают прочность П. в. На П. в. не оказывают
влияния такие органич. растворители, как алифатич.
спирты, ацетон, СС14, трихлорэтилен, углеводороды
и эфиры. Волокно энант (найлон-7) отличается повы-
повышенной эластичностью, что определяет его высокую
устойчивость к действию многократных изгибов, а
также более высокой устойчивостью к УФ-лучам.
Вследствие наличия длинной углеводородной цепоч-
цепочки в макромолекулах полиундеканамида и полидоде-
канамида волокна из этих полимеров обладают рядом
специфич. свойств, в частности пониженной гигроско-
гигроскопичностью, более высокой химстойкостью, понижен-
пониженной адгезией к резине. Эти волокна имеют приятный
гриф. Основной областью их применения являются
текстильная и трикотажная пром-сть. Часть этих во-
волокон используют также для технич. целей. Волокно
из нолипирролидонамида (найлон-4) отличается повы-
повышенным сродством к красителям и более высокой, чем
остальные П. в., гигроскопичностью.
В общем виде свойства П. в. в ряду найлон-3 — най-
найлон-12 изменяются след. образом: снижаются гидро-
фильность (приблизительно с 10 до \ %) и модуль упру-
упругости, повышаются химстойкость и эластичность. Вве-
Введение в алифатич. цепь полиамидов ароматич. или
алициклич. звеньев при условии изоморфного замеще-
замещения приводит к повышению жесткости цепи, темп-ры
плавления, модуля упругости, термостойкости волокон.
П. в. на основе поли-р-лактамов (типа найлон-3)
вследствие наличия в макромолекуле большого числа
амидных связей характеризуются высокой гигроско-
гигроскопичностью, меньшим, чем остальные П. в., удлине-
удлинением при разрыве, более высокой темп-рой плавления,
теплостойкостью, стойкостью к термоокислительной
и фотодеструкции. Эти волокна близки ио свойствам
натуральному шелку.
Волокна из нолигексаметиленсебацинамида имеют
темп-ру плавления 214 °С, в нормальных условиях
сорбируют 2,6% влаги. Они обладают большей, чем
волокна из полигексаметиленадипинамида, эластич-
эластичностью, приближаясь по этому показателю к шерсти.
Волокна из полностью ароматич. полиамидов (напр.,
из продукта поликонденсации производных гс-амино-
бензойной к-ты или д-фенилендиамина с терефталевой
к-той) благодаря высокой степени ориентации жестких
симметричных цепей и регулярной сетке межмолеку-
межмолекулярных водородных связей обладают исключительно
высокой прочностью и высокими значениями модуля
упругости (приближающимися к максимальным теоре-
теоретически возможным значениям — табл. 2).
Таблица 2. Свойства волокон из ароматических полиамидов
Показатель
Прочность, гс/текс ....
Модуль начальный, Гн/м2
(кгс/мм2)
Относительное удлинение, %
Поли-ж-фе-
ниленизофта-
ламид (номекс)
54
18,5
A850)
15
Волокно
«В»
180
60
F000)
5
Волокно
PRD-49
200
1360
A3600)
Применение. Наибольшее распространение П. в.
получили как сырье для производства товаров широ-
широкого потребления, а также шинного корда и резино-
технич. изделий. В первом случае П. в. применяются
в виде бесконечных нитей, число филаментов к-рых
колеблется от 1 до 39, а толщина от 2,2 до 16 текс,
и штапельного волокна с различной длиной резки
в зависимости от назначения (для смеси с хлопком или
шерстью). Часть П. в. текстильного назначения окра-
окрашивают в массе в широкую гамму цветов.
Широкое распространение получили текстурирован-
ные нити на основе П. в., высокая объемность к-рых
достигается за счет термомеханич. обработок волокна
(см. Высокообъемные нити), путем химич. модифика-
модификации полимера (применение разноусадочных соноли-
727
ПОЛИАМИДНЫЕ КЛЕИ
728
капрон (СССР),
(США), л и л и о н,
(ФРГ), д е д е р о н
меров), а также в результате получения бикомпонент-
пых волокон. Обычные П. в. имеют гладкую поверх-
поверхность и круглое поперечное сечение. Это обстоятельство
в ряде случаев снижает потребительские свойства
изделий из П. в. С целью ликвидации указанного не-
недостатка часть П. в. текстильного ассортимента вы-
выпускают с поперечным сечением в виде треугольника,
звездочки с различным числом лучей и т. д., что до-
достигается путем применения фильер со специальным
профилем отверстий. Выпускаются моноволокна тол-
толщиной от 0,05 до 2 мм.
Для производства шинного корда и резино-технич.
изделий П. в. выпускают в виде бесконечных нитей
с числом филаментов от 78 до 280 (см. также Кордные
нити и ткани). П. в. используют также для произ-
производства фильтровальных материалов, рыболовных се-
сетей, щетины, канатов и технич. тканей.
П. в. выпускают во многих странах под след. тор-
торговыми названиями: 1) волокно из поликапроамида —
найлон-6, капролан
г е л и о н (Италия), перлон
(ГДР), стилон (ПНР), р е-
лон (СРР), грилон, боданил (Швейцария),
а к у л о н (Голландия), а м и л а н (Япония) и др.;
2) волокно из полигексаметиленадипинамида — анид
(СССР), н а й л о н - 6,6 (США), родиа-найлон
(ФРГ), BNS-найлон (Великобритания), н и-
п л о н (Япония) и др.; 3) волокно из полиэнант-
амида — э н а н т (СССР), н а й л о н - 7 (США); 4)
волокно из полиундеканамида — ундекан (СССР),
найлон-11 (США), р и л ь с а н (Франция, Ита-
Италия), р е л с и н (Великобритания) и др.; 5) волокно
из полидодеканамида — найлон-12 (США) и др.;
6) волокно из полипирролидонамида — найлон-4
(США); 7) волокна из ароматич. полиамидов — н о-
м е к с, волокно «В», PRD-49 (США); 8) волокна из
полиамидов, получаемых на основе алициклич. моно-
мономеров, — к и а н а (США), к в и а н а (Япония);
9) волокна из поли-р-лактамов — найлон-3
(США); 10) волокна из полигексаметиленсебацинами-
да — найлон- 6,10, зайтел-31 (США). По-
Последние два типа волокон производят в незначитель-
незначительных масштабах.
П. в.— наиболее распространенный класс синтетич.
волокон. Мировое производство П. в. в 1972 составило
2424,5 тыс. т (ок. 38% от общего объема производства
синтетич. волокон).
Лит.: Фишман К. Е., Хрузин Н. А., Производ-
Производство волокна капрон, 2 изд., М., 1967; Кларе Г., Ф р и ц-
шеЭ., ГребеФ., Синтетические полиамидные волокна,
пер. с нем., М., 1966; Производство шинного корда, М.— Л.,
1964; Калиновский Е., Урбанчик Г. В., Химиче-
Химические волокна, М., 1966; Сигал М. Б., Производство поли-
полиамидных волокон, М., 1966; Монкрифф Р. У., Химиче-
Химические волокна, пер. с англ., М., 1961; Пакшвер А. Б.,
Физико-химич. основы технологии химич. волокон, М., 1972.
См. также лит. при ст. Полиамиды.
В. М. Харитонов, А. А. Сперанский.
ПОЛИАМИДНЫЕ КЛЕИ (polyamide adhesives, Po-
lyamidklebstofie. colles de polyamide).
Клеи на основе олигоамидов. Олигоамиды (за рубе-
рубежом их называют версамидами) представляют
собой продукты взаимодействия оквимолярных коли-
количеств жирных дикарбоновых к-т, содержащих от 25
до 40 метиленовых звеньев, с диаминами (чаще всего
с этилендиамином). Это твердые в-ва, растворимые
в спиртах, ацетоне, бензоле и др. органич. раствори-
растворителях.
Из олигоамидов готовят р-ры, в к-рые могут вхо-
входить также канифоль или ее эфиры (для увеличения
адгезии), парафин (для повышения устойчивости кле-
клеевого соединения к действию влаги), дибутилфталат
и др. пластификаторы. Кроме того, П. к. на основе
олигоамидов выпускают в виде клеевых пленок, к-рые
получают поливом на металлич. барабан р-ров или
расплавов.
Жидкие клеи поставляются потребителю в алюминие-
алюминиевых или жестяных бидонах. Они могут длительное
время храниться в сухом прохладном помещении при
темп-ре не ниже —5 СС и не выше 30 СС. Их приме-
применяют для склеивания бумаги, целлофана, нек-рых
видов полимерных пленок, для приклеивания этикеток
к стеклянной таре и т. п. Клей наносят на склеивае-
склеиваемые поверхности кистями, валками и др., подсушивают
и совмещают склеиваемые поверхности при контакт-
контактном давлении.
Клеевые пленки помещают между предварительно
нагретыми до 90—190 °С склеиваемыми поверхностями
изделий из металлов, керамики и др. и выдерживают
иод давлением 0,1—0,3 Мн/м2 A—3 кгс/см'2) до затвер-
затвердевания олигоамида.
Композиции на основе олигоамидов используют чаще
всего в качестве неконструкционных клеев, т. к. кле-
клеевые соединения, полученные при их использовании,
обладают невысокими прочностными характеристика-
характеристиками и термостойкостью.
Клеи на основе гомополиамидов и сополиамидов.
Гомополиамиды (полиамид-6; полиамид-6,6; полиамид-
6,10; полиамид-11 и др.) используют обычно в виде пле-
пленок и порошков для склеивания металлов, керамики,
древесины и др. Технология склеивания клеевыми
пленками такая же, как и П. к. предыдущей группы.
Порошки полиамидов наносят на склеиваемые поверх-
поверхности распылением струей горячего газа из специаль-
специальных форсунок. Адгезия гомоиолиамидов к стали со-
составляет 35—50 Мн/м2 C50—500 кгс/см2), к цветным
металлам несколько меньше, а к большинству др.
материалов — плохая.
Иногда готовят клеи-растворы из гомополиамидов
и спирторастворимых сополиамидов (напр., сополимера
соли АГ и е-капролактама в соотношении 60 : 40 по
массе). Растворителями в этом случае служат смеси
спирта с водой, резорцином и др. Клеи этого типа
применяют для соединения между собой изделий из
полиамидов. Прочность при неравномерном отрыве
склеенной полиамидной пленки составляет 0,5 Мн/м
E кгс/см).
Клеи на основе метилолполиамидов. Метилолиоли-
амиды получают при обработке полиамидов формаль-
формальдегидом:
[-HN-R-HN-CO-]n + п СН2О -> [-HN-R-N-CO-Jn
СН2ОН
Эти полимеры хорошо растворимы в спиртах, гликолях
или водно-спиртовых смесях. Метилолполиамиды от-
верждаются при высоких темп-pax или в присутствии
органич. к-т (щавелевой, малеиновой и др.).
Клеевые композиции состоят обычно из полимера,
смеси из 80% этанола и 20% воды и катализатора от-
отверждения A —10% по массе щавелевой, малеиновой и
др. к-т). Кроме того, в состав клеевой композиции
могут входить феноло-формальдегидные, эпоксидные,
кумароно-инденовые и др. смолы, канифоль, парафин,
шеллак и др. компоненты.
Клеи этой группы выпускают в виде прозрачных
р-ров или пленки. Поставляют р-ры потребителю
в герметически закрытых бутылях, где они могут
храниться в течение 6 мес при 10—30 СС. Отвердители
хранят в отдельной таре и вводят в клей непосредст-
непосредственно перед употреблением.
Клеевые пленки имеют неограниченный срок хране-
хранения в сухом неотапливаемом помещении. Не допускает-
допускается попадание на них воды, масел и органич. раствори-
растворителей.
Технология склеивания с помощью П. к. на основе
метилолполиамидов не отличается от общепринятой.
Клеевые соединения на основе этих клеев работоспо-
работоспособны длительное время в температурном интервале
от —60 до 60 °С, устойчивы к действию углеводородов,
729
ПОЛИАМИДНЫЕ ПЛАСТМАССЫ
730
масел, топлив, спиртов и конц. щелочей. Они обладают
высокой прочностью при различных нагрузках, в том
числе отдирающих и ударных. В таблице приведены
прочностные характеристики отечественного клея
МПФ-1.
Прочность склеивания дюралюмина
Показатели
Прочность при сдвиге, Мн/м2
(кгс'см2)
исходный образец
после 5 суш выдержки в
воде B0сС)
после 5 сут выдержки в
трансформаторном масле
С>0 °С)
Прочность при равномерном
отрыве, Мн/м2 (кгс/см2) . . .
Прочность при неравномерном
отрыве, кн/м, или кгс/см . .
клеем МПФ-1*
Температура испытаний
-60 °С
13,1A31)
—
48,2D82)
36
20 °С
17,5A75)
12,4A24)
16,8A68)
31,8C18)
65
60 °С
9,4(94)
5,2E2)
7,7G7)
13,7A37)
42
* Клей состоит из спиртового
феноло-формальдегидной смолы.
р-ра метилолполиамида и
Клеи этой группы благодаря хорошим физико-
механпч. и технологич. свойствам широко применяют
для склеивания цветных и черных металлов, силикат-
силикатного и органич. стекла, кожи, ткани, керамики, бе-
бетона, древесины, резины и нек-рых пластмасс.
Лит.: Кардашов Д. А., Синтетические клеи, 2 изд.,
М., 1968; Р о д и в и л о в а Л. А., в кн.: Новые синтетиче-
синтетические лаки и клеи, М., 1961; Handbook of adhesives, ed. by
I. Skeist, N. Y.— L., 1967; Adhesion and adhesives, ed. by
R. Houwink, G. Salomon, Amst.— [a. o.], 1965.
А. Б. Давыдов.
ПОЛИАМИДНЫЕ ПЛАСТМАССЫ (polyamide plastics,
Polyamidplaste, plastiques polyamides). В качестве пласт-
пластмасс можно применять полиамиды, не содержащие ни-
никаких добавок; о таких материалах см. Полиамиды.
В данной статье рассмотрены П. п., содержащие на-
наполнители, пластификаторы, стабилизаторы, антипи-
рены. Введение указанных добавок облегчает перера-
переработку полиамидов, придает им требуемые эксплуата-
эксплуатационные свойства и тем самым расширяет области их
применения.
Состав и свойства. Волокнистые напол-
наполнители (напр., стекловолокно, асбест, синтетич. и
углеродные волокна) в значительно большей степени
упрочняют материал, чем дисперсные, вследствие обра-
образования собственных структур, выполняющих роль
несущего каркаса. Наи-
Наибольшее практич. приме-
применение имеет стекловолок-
стекловолокно. Оптимальное количе-
количество его в полиамиде со-
составляет 30—33% (по мае-
Рис. 1. Зависимость износа
полиамида-6,10 от времени ис-
истирания: 1 — без наполните-
наполнителя; 2 — 20% графита; з —
10% графита; 4 — 30% стек-
стекловолокна.
J
50
се). Механич. прочность, теплостойкость полиамидов
при введении стекловолокна возрастает в 2—3 раза, в
то время как при введении дисперсных наполнителей —
лишь в 1,5 раза (табл. 1). Значительно уменьшается пол-
ползучесть и повышается износостойкость (рис. 1). Благода-
Благодаря близким показателям преломления полиамида и стек-
стекла, а также равномерному распределению наполнителя
стекловолокно в П. п. не видно невооруженным глазом.
Изделия из стеклонаполненного полиамида имеют ров-
ровную гладкую поверхность.
Для получения высокопрочных П. п. в качестве на-
наполнителя применяют стеклоткань в количестве 50 —
70% (по массе). Ниже приведены свойства армирован-
армированного стеклотканью полиамида-6 (капролона):
Плотность, г/см3
Прочность, Мн/м2 (кгс/см2)
при сжатии
при растяжении
при изгибе . . .
Ударная вязкость, кдж/м-, или кгс-см/см2
Твердость по Бринеллю, Мн/м2 (кгс/мм2) . .
Теплостойкость по Мартенсу, СС
Водопоглощение за 24 ч, %
1,01
280-300
B800-3000)
400-430
D000-4300)
450 — 500
D500 — 5000)
25 0 — 300
300 — 350
C 0 — 35)
210 — 220
0,6
В качестве дисперсных наполнителей
применяют графит, дисульфид молибдена, тальк, кварц,
нитрид бора и др. в количестве 1,5—60% (по массе).
Графит и дисульфид молибдена улучшают антифрикци-
антифрикционные свойства — снижают коэфф. трения и повышают
-40 -20
20 40 60 80 100 120 140 160 180 200
Температура. °С
Рис. 2. Зависимость ударной вязкости наполненного поли-
полиамида-6,10 от темп-ры: 1 — 20% талька; 2 — 20% стекло-
стекловолокна; 3 — 30% стекловолокна; 4 — 40% талька.
износостойкость полиамида (см. Графитопласты)',
тальк и кварц улучшают электроизоляционные свой-
свойства, уменьшают деформацию под нагрузкой (см.
табл. 1). Применение дешевых и доступных наполни-
наполнителей снижает стоимость П. п. Введение как волок-
волокнистых, так и дисперсных наполнителей повышает
стабильность свойств П. п. в условиях повышенных
темп-р (рис. 2), а также их водостойкость. Наполнен-
Наполненные П. п., особенно содержащие стекловолокно или
графит, обладают повышенной атмосферостойкостью
(табл. 2).
Пластификаторы вводят в полиамиды в ко-
количестве до 20% (по массе). В качестве пластификато-
пластификаторов применяют сульфонамиды, хотя испытаны также
третичные амины, эфиры разветвленных жирных к-т,
фосфорной к-ты и многоатомных фенолов, а также
нек-рые высокомолекулярные соединения, напр, со-
сополимеры винилового мономера с диеновым каучуком.
При введении пластификаторов значительно снижается
модуль упругости при растяжении, возрастает относи-
относительное удлинение, сопротивление удару (см. табл. 1).
Варьируя количество пластификатора, получают мате-
материалы, сочетающие гибкость с высоким сопротивле-
сопротивлением разрыву и удару при пониженных темп-рах.
В качестве антиоксидантов для полиами-
полиамидов применяют ароматич. диамины и синергич. смеси
соединений меди с йодистым калием в количестве 0,5%
(по массе). Окрашивают полиамиды фталоцианиновы-
ми красителями, двуокисью титана, окисью хрома,
сульфатом кадмия и др. Чаще всего красители
(до 0,5% по массе) вводят в уже готовый полимер,
к-рый затем пропускают через экструдер и гранули-
гранулируют. В полиамиды вводят также др. ингредиенты
@,5—1,5% по массе): антипирены (соединения фосфо-
фосфора, трехокись сурьмы и др.), антистатики (металлич.
731
ПОЛИАМИДНЫЕ ПЛЕНКИ
Таблица 1. Свойства пластмасс на основе алифатических полиамидов
732
Полиамидная пластмасса
Полиамид
Полигексаметиленсебацинамид (по-
лиамид-6,10)
То же
Поли-е-капроамид (полиамид-6)
То же
»
Полидодеканамид (полиамид-12)
Добавка (количество,
% по массе)
Тальк A0-40)
Стекловолокно C0)
Графит A0)
Дисульфид молибдена
A,5)
Стекловолокно C0)
Пластификатор A5)
о
Д
||
1,15-
1 ,38
1 ,35
1,11
1,11
1,36
1 ,08
s
г^ Ю ? ?
80-10
20-50
50-80
60-100
30-60
55-65
Прочность при
статич. изгибе,
Мн/м2 (кгс/см^)
80-120
(800 — 1200)
160-200
A600-2000)
9 0-100
(900 — 1000)
85-90
(850 — 900)
180-250
A800-2500)
~
Прочность при
растяжении,
Мн/м2 (кгс/см2)
50-60
E00-600)
100 — 120
A000-1200)
5 0-60
E00-600)
5 0-60
E00 — 600)
130-200
A300-2000)
46
D60)
Теплостой-
Теплостойкость при
изгибе (на-
(нагрузка 1,85
Мн/м0-, или
18,5 кгс 'см2),
50-80
190-200
50
50
190-200
40-45
порошки, производные четвертичных аммониевых осно-
оснований и др.)- Для облегчения переработки используют
парафин, воски, нек-рые олигомеры, к-рые снижают
трение материала о стенки перерабатывающего обору-
оборудования и обеспечивают получение изделий с ровной
гладкой поверхностью.
Переработка. Наиболее распространенный метод фор-
формования изделий из П. п.— литье под давлением. Этим
методом перерабатывают гранулированные П. п.,
имеющие индекс расплава 4—12 (мол. масса полиамида
20—30 тыс.). Для ненаполненных или содержащих
до 10% наполнителя П. п. давление при литье 80—
100 Мн/м2 (800—1000 кгс/см2). С увеличением содержа-
содержания наполнителя B0—40%) давление повышают до
100—140 Мн/м2 A000 — 1400 кгс/см2). Литье произво-
производят в нагретую до 80—120 °С форму. П. п. хорошо
перерабатываются в тонкостенные изделия толщиной
до 0,3 мм сложной конфигурации. Благодаря близким
коэфф. линейного расширения металлов и высокона-
полненных П. п. из них можно изготавливать детали,
содержащие большое кол-во металлич. арматуры; по-
последнюю помещают в литьевую форму перед отливкой.
Гранулированные П. п., имеющие высокую мол.
массу C0—60 тыс.) и индекс расплава 1—4, перераба-
перерабатывают экструзией, в том числе экструзией с разду-
раздувом. Из П. п., синтезируемых анионной полимериза-
полимеризацией, получают крупногабаритные изделия или блоки
непосредственно на стадии синтеза из мономера. Де-
Детали сложного профиля можно изготавливать механич.
обработкой блоков; из этих П. п. получают также
трубы и др. полые изделия центробежным или рота-
ротационным формованием и др. методами.
Таблица 2. Изменение физико-механических свойств полиамида-
6,10 без наполнителя (а) и содержащего 30% стекловолокна (б)
после атмосферного старения в различных климатических зонах
в течение 12 мес
Свойства
Прочность при
растяжении,
Мн/м2 ....
кгс/см2 ....
Прочность при
изгибе,
Мн/м2 ....
кгс/см2 ....
Ударная вязкость,
кдж/м2, или
кгс -см/см2 . . .
До
а
58,
585
79,
790
97,
испыта-
испытания
5
0
0
б
99,0
990
150,5
1505
32,0
Средние
широты
а
21 ,5
215
58,0
580
4,15
б
77
775
136
1360
20
,5
,0
,0
Влажные
субтро-
субтропики
(Батуми)
б
74,5
745
110,0
1100
14,9
Сухие
субтро-
субтропики
(Ферга-
(Фергана)
б
86,0
860
148,5
1485
20,7
Применение. П. п., наполненные дисперсными на-
наполнителями (до 10% по массе), применяют для изго-
изготовления деталей, работающих в узлах трения с за-
затрудненной смазкой или без смазки при темп-pax от
—60 до —100 °С (шестерни, вкладыши, сепараторы
подшипников, лопасти винтов). Из П. п. наполненных
дисперсными наполнителями в количестве 20—40%,
изготавливают детали с повышенной жесткостью и
устойчивостью к деформациям под действием нагру-
нагрузок в диапазоне темп-р от —60 до —120 С (упоры реле,
каркасы катушек, корпуса приборов и т. д.). Стекло-
наполненные П. п. применяют вместо цветных метал-
металлов и реактопластов [типа АГ-4, ДСВ (стеклонапол-
ненный материал на основе анилино-феноло-формаль-
дегидной смолы) и др.] для изготовления конструк-
конструкционных деталей, таких, как платы, подшипники, втул-
втулки, корпуса, каркасы, эксплуатируемых при темп-рах
от —60 до 150 °С.
Из блоков полиамидов, получаемых анионной поли-
полимеризацией, изготавливают вкладыши, втулки под-
подшипников скольжения (для узлов с обедненной смаз-
смазкой), шестерни для станков металлообрабатывающей и
автомобильной пром-сти. Из пластифицированных П. п.
изготавливают эластичные конструкционные детали,
работающие при пониженных темп-pax. П. п. на основе
ароматич. полиамидов (напр., фенилона) применяют
в электротехнике, машиностроении, приборостроении
и др. отраслях пром-сти для изготовления микровы-
микровыключателей, плат, пластин, элементов электрооборудо-
электрооборудования, подшипников и др. деталей, работающих при
темп-pax до 250 °С.
Лит.: Справочник по пластическим массам, под ред.
М. И. Гарбара [и др.], т. 1, 1967; Липатов Ю. С, Физико
химия наполненных полимеров, Киев, 1967; Plastic Today;
22, 9—11, 1964; Пласт, массы, № 1, Ю A971); там же, Л?> 4,20
A964). Л.А.Носова.
ПОЛИАМИДНЫЕ ПЛЕНКИ (polyamide films, Po
lyamidfilme, films de polyamide). П. и. изготовляют
гл. обр. из алифатич. полиамидов, сополимеров е-капро-
лактама с солями АГ и СГ и из метилолполиампдов
(о метилолполиамидах см. Полиамидные клеи), реже
из ароматич. полиамидов. Для производства П. п. при-
пригодны полимеры с мол. массой не менее 10 000. Иногда
в них вводят стабилизаторы (соли галогеноводородных
к-т) и пластификаторы (до 1% многоатомных спиртов,
фенолов и др.).
Производство. Алифатич. гомополиамиды не раство-
растворяются в обычных органич. растворителях, применяе-
применяемых для производства пленок методом полива. Поэтому
П. п. из этих полимеров изготовляют экструзией.
Из-за малой вязкости расплава полимера П. п.
вблизи головки экструдера обладают малой прочностью
и могут деформироваться. Для предотвращения этого
по выходе из головки П. п. быстро охлаждают на
733
ПОЛИАМИДНЫЕ ПЛЕНКИ
734
~60 СС ниже температуры плавления (это препятст-
препятствует также образованию в полимере мелкокристал-
мелкокристаллической структуры, из-за чего пленка теряет про-
прозрачность).
Экструзию П. п. осуществляют по обычной
технологич. схеме (см. Экструзия) или с помощью
шестеренчатого насоса, подающего низковязкий рас-
расплав полиамида в щелевую фильеру, расположенную
в нижней части полимеризатора, в к-ром синтезиро-
синтезировался полиамид. По выходе из головки экструдера или
из фильеры П. п. попадает на охлаждаемый металлич.
барабан или в водяную ванну. Для предотвращения
окисления горячего полиамида кислородом воздуха
охлаждающую ванну располагают как можно ближе
к фильере, а барабан помещают в кожух, в к-рый
подают азот.
Полученную пленку снова нагревают до темп-ры
размягчения и подвергают одноосной или двухосной
ориентации на специальном оборудовании. В резуль-
результате прочность П. п. при растяжении значительно по-
повышается. Для предотвращения усадки ориентирован-
ориентированную П. п. подвергают термофиксации в течение не-
нескольких мин при 170 — 180 °С, после чего этот пока-
показатель снижается до 0,5—3% .
Установки, применяемые для получения П. п. мето-
методом экструзии рукава с раздувом, характеризуются
след. параметрами: 1) отношение длины шнека к диа-
диаметру — не менее 1 : 12—1 : 14; 2) степень сжатия
полиамида в пластпкаторе — 1:4. Для изготовления
пищевых и медицинских П. п. гранулы полиамидов
перед экструзией отмывают от остатков мономеров
горячей водой.
Методом полива получают пленки из ароматич.
полиамидов, сополимеров е-капролактама с солями
АГ и СГ и из метилолполиамидов. Ароматич. полиамиды
Свойства полиамидных пленок*
сел, однако неустойчивы к действию к-т. Газопро-
Газопроницаемость П. п. меньше, чем у полиэтилентерефта-
латных и нек-рых др. пленок, однако она становится
значительной при высокой влажности окружающей
среды. Паропроницаемость П. п. существенно зависит
от температуры, например при 205 СС она в 25 раз вы-
выше, чем при нормальной температуре. П. п. на осно-
основе модифицированных полиамидов обладают исключи-
исключительной водостойкостью.
Распространенный способ улучшения физико-меха-
нич. показателей П. п., особенно прочности при раз-
дире,— армирование тканями из синтетич. волокон,
гл. обр. из полиамидов. Др. способ модификации —
изготовление многослойных пленок. Напр., трехслой-
трехслойная пленка, внешние слои к-рой состоят из пластифи-
пластифицированного сополимера е-капролактама с солью АГ,
а внутренний из полиамида-6, обладает высокой эла-
эластичностью и атмосферостойкостью. Двухслойные
пленки полиамид — полиэтилен и трехслойные пленки
поливинилиденхлорид — полиамид — полиэтилен или
полиэтилентерефталат — полиамид — полиэтилен моро-
морозостойки и выдерживают продолжительное кипячение
в воде.
Кроме того, используют П. п., дублированную алю-
алюминиевой фольгой. Этот материал прочен, термостоек
и гигиеничен.
П. п. сваривают термич. методом, ультразвуком и
токами высокой частоты. Их можно также склеивать
р-рами полиамидов в феноле или муравьиной к-те,
однако из-за токсичности растворителей этот метод
соединения П. п. не находит широкого распростране-
распространения. На П. п. можно наносить печать, они хорошо
подвергаются металлизации.
Применение. Пленки из полиамида-6,10 применяют
для упаковки и хранения растительного, сливочного и
топленого масла, а также др. жиров.
Пленки на основе полиамида-11 и по-
лиамида-12 используют в качестве
оболочек для колбас и сосисок, для
упаковки замороженного мяса, рыбы и
др. продуктов. Из П. п. (в том числе
дублированных полиэтиленом и трех-
трехслойных) готовят мешки для замора-
замораживания продуктов, к-рые можно не
удалять при варке или запекании про-
продуктов. Высокая термостойкость и
вместе с тем способность при повы-
повышенных темп-pax пропускать водяные
пары дает возможность применять их
для стерилизации медицинского инст-
инструмента при 100 —130 СС в течение 30—
. _ „ 60 мин. Кроме того, инструменты в та-
* Физико-механич. характеристики П. п. зависят от степени их ориентации и „лд. а^г* туп mw ^ nrwr
направления приложенных усилий. ** Условия испытания: время — 24 ч, толщи- кии упаковке можно подвергать хи-
Показатели
Плотность, г/см3 ....
Прочность при растяже-
растяжении, Мн/м2 (кгс/см2) . .
Относительное удлине-
удлинение, %
Термостойкость, СС . . .
Морозостойкость, °С . .
Водопоглощение за 24 ч,
Показатель паропрони-
цаемости**, г/м2(?/дм2)
Полиамид-6
1,13
63-140
F30-1400)
250-550
93-213
ДО -73
9,5
83-310
@,83-3,1)
Полиамид-6,6
1 ,14
63-98
F30-980)
200-450
250
-73
8,9
46-92
@,46-0,92)
Полиамид-11
1 ,04
60-80
F00-800)
250-400
180
-45
0,8-1 ,2
-
Полиамид-12
1,01
50-60
E00-600)
250-300
79,5-106
-73
0,25
1,0-1 ,7
@,01-0,017)
на пленки — 25 мкм.
растворяют в диметилацетамиде или диметилсульф-
оксиде, отливают на бесконечную ленту поливочной ма-
машины или на металлич. барабан и высушивают при
200 СС. Во избежание прилипания П. п. к барабану или
к бесконечной ленте и для получения пленок с высоким
качеством поверхности их покрывают слоем триаце-
триацетата целлюлозы.
В качестве растворителей для алифатич. сополиами-
дов обычно используют спирты, их смеси с водой или
метиленхлорид. Готовую пленку высушивают для уда-
удаления растворителя. Удаление и регенерация раство-
растворителя значительно удорожают процесс, к-рый на-
находит все меньшее применение.
Свойства и методы переработки. П. п. имеют высо-
высокие физико-механич. показатели (таблица). Они гиб-
гибки и прозрачны, обладают износостойкостью и хоро-
хорошей сопротивляемостью излому. П. п. устойчивы к
действию щелочей, органических растворителей и ма-
мич. стерилизации при помощи окиси
этилена. Полиамидные мешки совер-
совершенно непроницаемы для бактерий, поэтому стериль-
стерильный инструмент хранится в них длительное время.
Пленки на основе полиамида-6 применяют в технике
в качестве изоляции для изделий, работающих в среде
растворителей, для изготовления эластичных емкостей,
в качестве обмоточного материала для трубопроводов,
как чехлы и тенты для покрытия складских помеще-
помещений. П. п. лучше всего подходят в качестве раздели-
разделительного слоя при прессовании слоистых пластиков
на основе полиэфирных смол. Многослойными и арми-
армированными П. п. покрывают парники и теплицы. П. п.
на основе метилолполиамидов применяют в производ-
производстве искусственной кожи и обуви. Пленки на основе
ароматич. полиамидов используют в качестве электро-
электроизоляционных материалов.
Лит.: Такахаси Г., Пленки из полимеров, пер. с
японск., Л., 1971; Спюавочник по пластическим массам, под
ред. М. И. Гарбара [и др.], т. 1 — 2, М., 1967—69; Modern
plastics encyclopedia, oct. 1968. А. А. Пешехонов.
735
ПОЛИАМИДОЭФИРЫ
736
ПОЛИАМИДОЭФИРЫ (polyamidesters, Polyamid-
esteren, polyamide-esteres) — полимеры, содержащие
в основной цепи макромолекулы амидные — СО — NH —
и сложноэфирные —СО — О — группы. П. синтезируют
след. методами:
1. Поликонденсацией смеси низкомолекулярных ве-
веществ, способных образовывать амидные и сложно-
офирные связи (из дикарбоновых к-т и их производных,
гликолей, бисфенолов, диаминов, аминофенолов и др.)?
напр.:
п HOOCRCOOH + п H2NRf ОН -> [ - OCRCOHNR'O — ]п + 2пН2О
2. Полнконденсацией олигомеров, содержащих слож-
сложноэфирные или амидные связи, соответственно с диами-
диаминами или диоламн, напр.:
п ClOCR'COOROOCR'COCl + n H2NR"NH2 ->
-> [-OCRrGOOROOCR'COHNR"NH-]n + 2n HG1
3. Обменными реакциями из высокомолекулярных
соединений: аминолизом сложных полиэфиров, межцен-
межценным обменом сложных полиэфиров с полиамидами,
напр.:
*/2п H,NRNH2 + [ - OCR'COOR"O — ]n ->
_» [_OCR'COOR"OOGR'GOHNRNH-]^, + 72 n HOR"OH
4. Сополимеризацией N-замещенных циклич. иминов
с ангидридами дикарбоновых к-т, напр.:
V /
N-CA
п о:
,со
со
—*- f
-CH2—CH2—N—ОС
5. Взаимодействием б^с-оксазолонов с гликолями:
ос —с
I I
О N
С —
С —СО
N
О
п HOR ОН
— СОС—NH —ОС—R—CO—HN—С—COOR—О —
/\ <\ „
R' R" R R"
6. Сополимеризацией лактонов с лактамами, напр.:
HN—СО
(сн2M
о—со
(сн2M
—-*- f-HN - (СН2)-CO-I — f-O-(CH2M—СО-1
Поликонденсацию проводят в расплаве или р-ре при
240—280 °С, а также низкотемпературными методами
(от —30 до 80 °С) в р-ре и на границе раздела фаз.
Реакция в расплаве приводит к образованию стати-
стич. сополимеров, что обусловлено протеканием при
высоких темп-pax различных обменных реакций. Низ-
Низкотемпературными методами, как правило, получают
чередующиеся сополимеры или блоксополимеры.
П.— продукты белого цвета. В зависимости от хи-
мич. строения, мол. массы и фракционного состава они
м. б. воскообразными или твердыми, аморфными или
кристаллическими. С введением сложноэфирных свя-
связей в макромолекулы полиамида твердость, жесткость,
степень кристалличности и темп-pa плавления поли-
полимера иногда понижаются. Темп-pa плавления П. по-
повышается с увеличением количества амидных группи-
группировок, причем особенно резко уже при введении в
макромолекулу полиэфира сложного сравнительно не-
небольшого количества диамина.
П., полученные из соединений (дикарбоновых к-т,
диаминов, гликолей или др.), содержащих четное число
атомов углерода в молекуле, обладают более высокими
темп-рами плавления и прочностью, чем П. на основе
компонентов с нечетным числом атомов углерода.
Влияние «фактора четности» в П. менее ярко выраже-
выражено, чем в полиамидах. П., полученные из алифатич.
соединений, плавятся при сравнительно невысоких
темп-рах A00 —170 °С). Более высокими темп-рами
плавления характеризуются П., содержащие арома-
тич. звенья — п о л и а м и д о а р и л а т ы (напр.,
темп-pa плавления П. из ?г-аминофенола и хлорангид-
рида терефталевой к-ты выше 360 СС). Темп-ры плавле-
плавления блоксополимеров всегда выше, чем соответствую-
соответствующих статистич. сополимеров.
П. обладают лучшей растворимостью, чем полиами-
полиамиды: большинство из них растворимо в смеси хлориро-
хлорированных углеводородов с этиловым спиртом, циклогек-
саноне, бензиловом спирте и др., причем блоксополи-
блоксополимеры хуже растворимы, чем статистич. сополимеры.
В воде П. нерастворимы.
П. вступают во все реакции, характерные для поли-
полиамидов и сложных полиэфиров (ацидолиз, аминолиз,
гидролиз, алкоголиз, обменные реакции по сложно-
эфирным н амидным группам и др.).
Из р-ра и расплава П. формуют пленки (прочность
при растяжении пленки П. на основе сополимера
капролактама с капролактоном, полученной из р-ра
в бензиловом спирте, 60—80 Мн/м2, или 600—800 кг с/ см2)
и волокна (прочность при растяжении волокна из П.
на основе бутандиола-1,4, 4,4'-дикарбоэтоксибензани-
лида и 4,4'-дикарбобутоксибензанилида 270 Мн/м2,
или 2700 кгс/см2, относительное удлинение 30%). Ли-
Линейные П. можно превратить в трехмерные, отверждая
их альдегидами, ангидридами к-т или днэпоксидами.
Продукты, получаемые отверждением формальдегидом,
наз. в у л к а п р е н а м и.
Лит.: Коршак В. В., Виноградова СВ.,
Равновесная поликонденсация, М., 1968; Коршак В. В.
[и др.], Изв. АН СССР, сер. хим., № 3, 637 A968); Мор-
Морган П. У., Поликонденсационные процессы синтеза полиме-
полимеров, пер. с англ., Л., 1970; Коршак В. В., Виногра-
Виноградова С. В., Неравновесная поликонденсация, М., 1972.
Я. С. Выгодский.
ПОЛИАМИДЫ (polyamides, Polyamide, polyami-
des).
Содержание:
Получение
Свойства
Алифатические полиамиды . . ,
Ароматические полиамиды . . .
Применение
737
739
739
744
746
Полиамиды — гетероцепные полимеры, содержащие
в основной цепи макромолекулы амидные группы
—СО—NH—. Карбоцепные полимеры с боковыми амид-
ными группами—СО—NH2, напр, полиакриламид, обыч-
обычно к П. не относят. Полиамидами являются также бел-
белки и полипептиды, которые, однако, резко отличаются
от «обычных» П. по структуре, физическим и хими-
химическим свойствам, вследствие чего их и выделяют
в особые классы соединений. Амидные группы содер-
содержат также полимочевины и полиуретаны.
П. могут быть алифатич. или ароматич. в зависи-
зависимости от того, с алифатич. или ароматич. радикалами
связаны группы —СО — NH —. Основную группу П.
составляют г о м о п о л и а м и д ы, получаемые по-
поликонденсацией из диамина и дикарбоновой к-ты, из
со-аминокислоты и полимеризацией из лактама амино-
аминокислоты. Смешанные П. представляют собой
сополимеры, получаемые сополиконденсацией, т. е.
из двух или более диаминов с одной дикарбоновой к-той,
из одного диамина с двумя или более дикарбоновыми
к-тами, а также из аминокислоты или лактама со смесью
737
ПОЛИАМИДЫ
738
диамина и дикарбоновой к-ты. В зависимости от химич.
отроения П. могут быть линейными, разветвленными
или сшитыми.
Названия П. складываются из приставки «поли»,
двух корней, первый из к-рых обозначает диамино-
вый компонент, а второй — кислотный, и окончания
«амид». Напр., П., полученный из гексаметилендиами-
на и адипиновой к-ты, наз. полигексаметиленадипин-
амидом, из ж-фенилендиамина и терефталевой к-ты —
поли-ж-фенилентерефталамидом. В названии П., полу-
полученных из со-аминокислот или их лактамов, после при-
приставки «поли» следует корень, обозначающий амино-
аминокислоту. Напр., П., полученный из е-аминокапроновой
к-ты (или е-капролактама), наз. поли-е-капроамидом.
Для обозначения химич. состава алифатич. П. ши-
широко применяют след. обозначение: рядом со словом
«полиамид» (или «найлон», как часто принято в зару-
зарубежной литературе) ставят одну или несколько цифр,
обозначающих число атомов углерода в исходных
продуктах. Так, П., полученный из 8-аминокапроно-
вой к-ты (или е-капролактама), наз. полиамидом-6
(или найлоном-6), из гексаметилендиамина и адипино-
адипиновой к-ты — полиамидом-6,6 (или найлоном-6,6). Сопо-
Сополимеры обозначают комбинацией соответствующих чи-
чисел; в скобках указывается соотношение реагентов
в мае. ч. Напр., полиамид-6,6/6 F0 : 40) получается
из компонентов полиамида-6,6 F0 мае. ч.) и капролак-
тама D0 мае. ч.).
Получение
Как уже отмечалось, П. могут быть получены поли-
поликонденсацией или полимеризацией. Поликонденсация,
приводящая к образованию П., наз. п о л и а м и-
дированием.
Пол и конденсацией получают П. из со-
аминокарбоновых к-т или их эфиров A), а также из
дикарбоновых к-т (или их эфиров) и диаминов B) по
схеме:
n H2NRCOOH <± [ - HNRCO - ]м + п Н2О A)
п H2NRNH2 + n HOOCR'COOH «±
«± [-NHRNHCOR'CO-]n + 2nH2O B)
где R — алифатич. или ароматич. радикал.
Поликонденсацию можно проводить в расплаве ис-
исходных соединений или в р-ре высококипящего
растворителя, инертного по отношению к мономерам
и образующемуся полимеру, а в нек-рых случаях и
в твердой фазе (см. Поликонденсация в расплаве, Поли-
Поликонденсация в растворе, Твердофазная поликонденса-
поликонденсация). Если исходные соединения и образующийся по-
полимер устойчивы в расплавленном состоянии, пред-
предпочтителен метод поликонденсации в расплаве как
более удобный и экономичный, т. к. в этом случае не
требуется выделения полимера из р-ра и регенерации
растворителя. Способ получения П. в расплаве наибо-
наиболее изучен и его широко используют в пром-сти для
синтеза алифатич. П. Однако этот способ практически
не применяют для синтеза ароматич. П., т. к. послед-
последние в большинстве случаев разлагаются при темп-рах
ниже темп-р их плавления, что приводит к частичной
деструкции и сшиванию образующихся полимеров.
Реакции A) и B) обратимы; поэтому синтез доста-
достаточно высокомолекулярного П. возможен лишь при
удалении из сферы реакции выделяющейся воды или
др. низкомолекулярных веществ. Для удаления низко-
низкомолекулярных продуктов реакционную массу нагре-
нагревают в атмосфере инертного газа при постепенном по-
повышении темп-ры до определенного предела. При слиш-
слишком высокой темп-ре (особенно выше 300 °С) могут
протекать нежелательные побочные процессы: деструк-
деструкция и структурирование. Часто с целью получения П.
высокой мол. массы нагревание на последних стадиях
реакции проводят в вакууме. Обычно мол. массы обра-
образующихся алифатич. П. составляют 10 000—30 000.
Мол. масса П. в значительной степени зависит от
соотношения исходных реагентов. Избыток одного из
реагентов, а также присутствие в реакционной смеси
монофункциональных реагентов (напр., аминов или
монокарбоновых к-т) приводят к резкому уменьшению
мол. массы П. Для достижения при поликонденсации
эквимолярного соотношения между диамином и дп-
карбоновой к-той применяют их соли, напр, так наз.
соли АГ и СГ используют при получении соответствен-
соответственно полигексаметиленадипинамида и полигексаметилен-
себацинамида. При производстве П. монофункциональ-
монофункциональные соединения часто служат регуляторами мол. мас-
массы: количество добавляемого монофункционального
соединения определяет мол. массу получаемого П.
(см. Поликонденсация). На практике для регулирова-
регулирования мол. массы часто применяют уксусную к-ту или
берут в избытке один из компонентов.
Обычно при поликонденсации бифункциональных
мономеров (диамина и дикарбоновой к-ты) образуются
линейные макромолекулы. Если в реакции участвуют
мономеры, имеющие больше двух функциональных
групп в молекуле, образуются П. разветвленного или
трехмерного строения. К разветвлениям и сшиванию
могут вести также побочные реакции, инициируемые,
напр., кислородом воздуха. При получении П. из диа-
диаминов и дикарбоновых к-т используют такие соедине-
соединения, к-рые при темп-ре поликонденсации не склонны
циклизоваться. По этой причине нельзя применять,
напр., янтарную и глутаровую к-ты, этилендиамин и
триметилендиамин.
П. можно получать также поликонденсацией дигало-
генангидрида дикарбоновой к-ты (обычно дихлорангид-
рида) с диамином:
п H2NRNH2 + n GlCOR'COGl -> [ - HNRNHCOR'CO - ]„ +
+ 2nHCl C)
Эта реакция необратима; ее можно осуществлять
методом межфазной поликонденсации либо поликонден-
поликонденсацией в р-ре при относительно низких темп-pax (от
—5 до 20 °С). Во втором случае процесс проводят обыч-
обычно в присутствии органич. основания (пиридина, три-
этиламина, метилморфолина или др.), к-рое является
акцептором выделяющегося при поликонденсации хло-
хлористого водорода. Роль акцептора может выполнять
амидный растворитель, напр, диметилацетамид, N-ме-
тил-а-пирролидон, К,К,]Ч',К'-тетраметилмочевина. Аро-
Ароматич. П. получают гл. обр. методом низкотемпе-
низкотемпературной поликонденсации. При этом образуются П.
более высокой мол. массы, чем при межфазной поли-
поликонденсации.
П. можно получать также полиприсоеди-
полиприсоединением по след. схемам:
п O=G=NRN=C=O + nHO - Rr — ОН ->
-*¦ [— CONHRNHCOOR' О - ]п D)
п O=C=NRN=C=O + nNH2 — R' — NHs ->
->[ - CONHRNHCONHR'NH - ]n E)
' n H2NR "NH2 *-
" o=(r? V R'^?-?=o
ON NO
-R-
F)
f— COCR'Rr'NHCORCONHCR'R"CONHR'"NH—1
ртт с" ^Г ГН
» I )n—r-n( I
сн,—c
C—CH,
n H2NR'NH2
2
4No o"
G)
[—COCH2CH2CONHRNHCOCH2CH2CONHR;NH—1
Na
n CH2=G(R) - GONH2 > [ - GH2CH(R)GONH - ]n (8-)
739
ПОЛИАМИДЫ
740
/7CH2=CHCON
сн-сн2
n H2NRNH2-
(9)
CH,f-CH
сн
¦ \2 \
/NCOCH2CH2NHRNH—
nNC-R-CN+nHCHO
H2NRNH2->[-COCH2CONHRNH-;b
+n H2O
....
[ - HNCORCONHCH2 - ]n (И)
По реакциям D) и E) образуются своеобразные пред-
представители класса П.— полиуретаны и полимочевипы,
по реакциям F) и G) — смешанные П. со строго регу-
регулярным чередованием в элементарном звене макромо-
макромолекулы остатков нескольких диаминов и дикарбоно-
вой к-ты.
К пол и мер изационным методам
получения П. относятся:
1. Гидролитическая и каталитическая (анионная, ка-
тионная) полимеризация лактамов со-аминокислот (см.
Капролактама полимеры, Лактамов полимеризация)'.
x HN(CH2) CO
—HN(CH2)nCO—
A2)
Реакция протекает в присутствии гидролитич. аген-
агентов (напр., воды, к-т) и соединений, способных вызы-
вызывать гидролиз амидной связи в цикле, или ионных ка-
катализаторов.
2. Полимеризация N-карбоксиангидридов а-амино-
а-аминокислот (см. N-Карбоксиангидридов а-аминокислот поли-
полимеризация):
RCH-CO
п \ /O-»[-HNCH(R)CO-]n-fnCO2 A3)
HN - СО
3. Анионная полимеризация изоцианатов, приводя-
приводящая к образованию N-замещенных П. (см. Изоциана-
Изоцианатов полимеры):
nR-N=C=O-»[-NR-CO-]n A4)
В пром-сти П. синтезируют по реакциям A) — C)
и A2). Исходными соединениями для получения П.
могут служить различные алифатические, ароматиче-
ароматические, жирноароматические, алициклические дикарбо-
новые к-ты и диамины (и их производные), со-амино-
карбоновые к-ты и их лактамы. Таким образом, су-
существуют очень широкие возможности для получения
П. различного строения и состава. Однако в промыш-
промышленном масштабе производится лишь несколько П.,
что связано с недоступностью многих исходных соеди-
соединений. В пром-сти в качестве сырья для получения П.
широко используют е-капролактам, гексаметилендиа-
мин, адипиновую, себациновую и аминоундекановую
к-ты, а из ароматич. компонентов — терефталевую и
изофталевую к-ты, п- и ле-фенилендиамины.
Свойства
Алифатические полиамиды. Свойства алифатич. П.
могут изменяться в широких пределах в зависимости
от химич. структуры. Одни П.— твердые, рогообраз-
ные, в большинстве случаев кристаллические продукты
белого цвета, другие — аморфные, прозрачные, стекло-
стеклообразные вещества.
Макромолекулы П. в твердом состоянии обычно
имеют конформацшо плоского зигзага (рис. 1). Благо-
Благодаря наличию амидных групп макромолекулы П. свя-
связаны между собой водородными связями, к-рые обус-
обусловливают относительно высокие темп-ры плавления
кристаллич. П. Максимальная степень кристаллично-
кристалличности П. зависит от симметрии звеньев и от регулярно-
регулярности их расположения в макромолекуле; высокой сте-
степенью кристалличности D0—60%) обладают регуляр-
регулярен
ные алифатич. гомо-
полиамиды, напр.
полиамид-6,6, поли-
амид-6,10 и полиамид-
'6, а также нек-рые
П., содержащие в сво-
своем составе чередую-
чередующиеся алифатич. и
ароматич. звенья,
напр, политерефтал-
амиды, полученные
на основе алифатич.
диаминов, содержа-
содержащих четное число ато-
атомов углерода в моле-
молекуле. N-Замещенные
П. характеризуются
низкой степенью кри-
кристалличности и отно-
относительно невысокими
Рис. 1. Структура кри-
кристалла полигексамети-
ленадипинамида; макро-
макромолекулы в кристалле
соединены водородными
связями.
темп-рами плавления вследствие снижения симметрии
звена и менее сильного межцепного взаимодействия.
Многие N-замещениые алифатич. П. представляют со-
собой каучукоподобные полимеры. Высокая кристаллич-
кристалличность и высокие темп-ры плавления П., у к-рых отсут-
отсутствуют межцепные водородные связи, напр, у поли-
терефталамидов пиперазина и алкилзамещенных пипе-
разина, обусловлены жесткостью и высокой симметри-
симметрией макромолекул. Энтропия плавления таких полиме-
полимеров низка.
В гомологич. рядах П., полученные из дикарбоно-
дикарбоновых к-т и диаминов с четным числом атомов углерода
в молекуле, обладают более высокими темп-рами плав-
плавления, чем П. из дикарбоновых к-т и диаминов с не-
нечетным числом атомов углерода (рис. 2). Темп-ра
плавления повышается с
уменьшением числа метиле-
метиленовых групп в звеньях ма-
макромолекул П., что связано
с увеличением числа водо-
водородных связей между от-
отдельными макромолекулами
д ру
в единице объема. Темп-ры
плавления алифатич. П.
Рис. 2. Зависимость темп-ры
плавления полиамидов различ-
различного строения от числа метиле-
новых групп в элементарном
звене макромолекулы: 1 — по-
полиамиды из гексаметилендиами-
на и различных дикарбоновых
кислот; 2—полиамиды из а-ами-
а-аминокислот.
320
^300
g 280
I260
с 240
200
180
160
140
А
4 6 8 10 12 14 16
Число метиленовых групп
в звене полимера
можно найти из эмпирич. ф-лы: у = 1х + 110, где
у — темп-pa плавления (в °С), х — отношение числа
амидных групп к числу метиленовых (в %).
Приводим темп-ры плавления (в °С) нек-рых П.:
[-NH-CH2-CH2 —КН-СО(СН2)8СО-]„ 260
[- N-CH2-CH2- К-СО(СН2)8СО-]„ 100
I I
СН3 СНз
, 180
С Н о С Н о
—N
\
сн
а—сн
N—СО-
741
ПОЛИАМИДЫ
742
Кривая зависимости Тпл смешанных П. от состава
исходных соединений имеет млнимум (рис. 3). Поло-
Положение минимума отвечает составу, при к-ром средняя
длина регулярных последовательностей одинаковых
звеньев в макромолекулах сополимеров минимальна.
Сополимеры такого строе-
строения характеризуются наи-
наименьшей степенью кристал-
кристалличности и наиболее высо-
высокой растворимостью по
сравнению с соответствую-
соответствующими гомополимерами. Од-
Однако известны сополимеры
с изоморфными звеньями,
для к-рых кривая изменения
Рис. 3. Зависимость темп-ры
плавления смешанных полиами-
полиамидов от состава бинарных систем:
1 — соль гексаметилендиамина
с адипиновой (АГ) и азелаиновой
АГ, моль
(АзГ)к-тами; 2 — соль гексаметилендиамина с адипиновой (АГ)
и себациновой (СГ) к-тами; з — соль гексаметилендиамина с
адипиновой (АГ) и пробковой (ПГ) к-тами.
темп-ры плавления от состава исходных соединений
не имеет минимума, т. е. Тпл плавно изменяется меж-
между темп-рами плавления соответствующих гомополи-
меров. Изоморфизм в смешанных П. возникает в тех
случаях, если их звенья имеют весьма близкие разме-
размеры и форму. Тогда отсутствие химической однород-
однородности цепи не препятствует плотной упаковке макро-
макромолекул.
Алифатич. П. растворяются лишь в сильнополярных
растворителях, способных специфически сольватиро-
вать амидные группы, причем растворимость в значи-
значительной мере определяется строением полимера. Рас-
Растворимость П., как правило, уменьшается с уменьше-
уменьшением числа метиленовых групп в звеньях макромоле-
макромолекул. Алифатич. П. растворяются при комнатной темн-ре
в конц. к-тах, напр, в серной, азотной, муравьиной,
монохлоруксусной, трихлоруксусной, в фенолах (фе-
(фенол, крезол, ксиленол, тимол), хлорале, конц. р-ре
хлористого кальция и в спиртах. Универсальные
растворители для П.— трифторэтиловый и 2,2,3,3-тет-
рафторпропиловый спирты. Высококипящие спирты
(напр., бензиловый, фенилэтиловый, этиленгликоль) —
более слабые растворители.
Гомополиамиды не растворяются в воде, бензоле
и др. углеводородах, низших спиртах. N-Замещенные
П. характеризуются лучшей растворимостью и рас-
растворяются даже в менее полярных растворителях.
Напр., поли-К-этилундеканамид в этаноле растворяет-
растворяется при комнатной темп-ре, а в диоксане — при нагре-
нагревании. Смешанные П. обладают значительно лучшей
растворимостью, чем соответствующие гомополимеры.
Так, смешанный полиамид-6,6/10,6 D0 : 60) раство-
растворяется в смеси хлороформа с метанолом, хотя соот-
соответствующие гомополимеры нерастворимы в этой сме-
смеси. П., содержащие в своем составе алифатич. и аро-
матич. звенья, растворяются несколько хуже, чем
алифатич. П.; они растворяются, как правило, толь-
Показатели
ко в трифторуксусной и серной кислотах.
Высокая кристалличность обусловливает хорошие
физико - механические
Таблица 1. Свойства полиамидов, выпускаемых промышленностью свойства П. Прочность при
~~ ~~~~ ~ растяжении, модуль уп-
фенилен"- ругости при растяжении
Плотность, г/см3
Темп-pa, СС
плавления
размягчения
Водопоглощение при насыще-
насыщении, %
Прочность*, Мн/м2
при растяжении
при сжатии
при изгибе
Ударная вязкость, кдж/м2, или
кгс- см/см2
Относительное удлинение, %
Теплостойкость по Вика, СС . .
Твердость* по Бринеллю,
Мн/м2
Диэлектрич. проницаемость . .
Тангенс угла диэлектрич. по-
потерь
Поли-е-
капро-
амид
1,13
225
210
10,9
60
75-90
90
150—170
150-400
160—180
140-150
4,5
0,3
Полигек-
самети-
ленади-
пинамид
1,14
264
250
10,0
80
46
100
80-100
220-230
0,04
Полигек-
самети-
ленсеба-
цинамид
1,09-
1,11
213-220
3,5
45—60
70-90
70-90
100-120
100 — 150
195-205
100—150
0,03
Поли-
энанто-
амид
1,13
223
60
25
120-150
100—200
200
140—150
4,4
0,02
Поли-
ундекан-
амид
1,10
185
175
1,6
60-80
НО
90-120
46
3,2
0,02
1 ,33 —
1,36
430
270
80-120
до 320
340
* 1 Мн/м2 ^ 10 кгс/см2.
Таблица 2. Свойства нек-рых смешанных полиамидов*, выпускаемых промышленностью СССР
Показатели
Плотность при 20 СС, г/см3
Темп-pa плавления, СС
Прочность**, Мн/м2
при сжатии
при растяжении
при статич. изгибе
Модуль упругости при растяжении**, Мн/м2.
Ударная вязкость, кдж/м2, или кгс-см/см2. .
Относительное удлинение, %
Твердость по Бринеллю, Мн/м2 (кгс/мм2) . . .
Теплостойкость по Вика, СС
Усадка при литье, %
П-54
1,12
168-175
45—50
28-30
560
300-350
45—50
D,5-5,0)
115
1,0-1,2
П-548
1,12
150-160
35-40
18—19
340
350 — 400
38-32
C,8—3,2)
85-87
1,1-1,2
П-АК7
1,14
240-243
90-110
70-73
100-120
1500 —
1600
130-140
100
150—180
A5-18)
205
* О составе смешанных П. см. раздел «Применение и переработка». ** 1 Мн/м2 ~ 10 кгс/см2.
изофтал- и твердость П. возрастают
амид с увеличением степени
кристалличности, в то вре-
время как абсорбция влаги
и ударная вязкость не-
несколько уменьшаются.
Свойства нек-рых П. при-
приведены в табл. 1 и 2.
П. можно подвергать
холодной вытяжке; при
этом образуется «шейка».
В результате вытяжки
длина волокна или плен-
пленки возрастает на 400—
600%. При холодной вы-
вытяжке происходит ориен-
ориентация макромолекул П. в
направлении растяжения,
что способствует повыше-
повышению степени упорядочен-
упорядоченности макромолекул и,
следовательно, прочности
(прочность при растяже-
растяжении ориентированных во-
волокон или пленок П.
300—400 Мн/м2, или
3000—4000 кгс/см2, а не-
неориентированных 50—70
Мн/м2, или 500 — 700
кгс/см2).
Химич. свойства П. оп-
определяются в основном на-
наличием амидных групп в
макромолекулах. Поляр-
Полярный характер амидной свя-
связи обусловливает боль-
большую чувствительность П.
к различным полярным
П-АК80/20
1,13
83-86
60-70
85—90
110-130
150—200
743
ПОЛИАМИДЫ
744
агентам (к к-там, щелочам, аминам, воде и др.), под
воздействием к-рых могут протекать деструктивные ре-
реакции: гидролиз, ацидолиз, аминолиз и др. (см. Обмен-
Обменные реакции). При комнатной темп-ре П. устойчивы к
действию гидролизующих агентов. Так, вода совер-
совершенно не гидролизует П.; вконц. к-тах, напр, в сер-
серной и муравьиной, П. растворяются без заметной де-
деструкции. При повышении темп-ры скорость гидро-
гидролиза резко возрастает.
Водород амидной группы П. способен замещаться
на алкильные и др. радикалы. Это свойство использу-
используется для получения N-метилольных производных П.
(метилолполиамидов) при действии на П.
формальдегида. Такие П. термореактивны и харак-
характеризуются лучшей растворимостью и более высо-
высокой эластичностью по сравнению с исходными П.
Концевыми группами П., как правило, являются
аминные и карбоксильные. П., содержащие свободные
группы —СООН, способны образовывать соли метал-
металлов, а П., содержащие свободные группы — NH2,—
аммонийные соли. На этом свойстве основано примене-
применение П. для абсорбции к-т, щелочей, красителей, дуби-
дубителей и т. п. из р-ров. Эффективность абсорбции за-
зависит от рН среды. Так, с уменьшением рН увеличи-
увеличивается абсорбция к-т, достигающая максимума, напр.,
для соляной к-ты при рН 2,2—2,6. Это значение рН со-
соответствует моменту нейтрализации аминных групп в П.
Под действием ионизирующей радиации, при озони-
озонировании или нагревании в атмосфере воздуха в макро-
макромолекулах П. возникают свободные радикалы, к-рые
могут инициировать образование привитых П., напр.:
nCH2=GHR
^СН2 - СО - NH - СН2- -> ^СН2-СО - NH - СН - >
-> -СН2 - СО - NH - СН^
При интенсивном облучении П. рентгеновскими
лучами протекают одновременно процессы деструкции
и образования поперечных связей между макромолеку-
макромолекулами. Последний процесс иногда используют для
уменьшения растворимости и повышения темп-ры раз-
размягчения П.
При нагревании на воздухе происходит термоокис-
термоокислительная деструкция П., скорость к-рой резко воз-
возрастает под влиянием ультрафиолетового и солнечного
света. При нагревании в отсутствие кислорода П.
претерпевают термич. деструкцию, глубина к-рой воз-
возрастает с повышением темп-ры. Термостойкость зави-
зависит от строения П. Наименее термостойки поли-
метиленамиды. Напр., полиметиленазелаинамид
[—NHGH2NHCO(CH2OGO—]п при нагревании до
темп-ры плавления, в отличие от других П., структу-
структурируется. С увеличением числа метиленовых групп в
исходных молекулах диамина и дикарбоновой к-ты ста-
стабильность П. возрастает. Так, термостойкость П. на
основе себациновой к-ты выше, чем П. на основе ади-
пиновой к-ты. Среди П., получаемых из аминокислот,
наименее термостойки П. из а-аминокислот; с увеличе-
увеличением цепи аминокислоты термостойкость возрастает.
Для идентификации П. широко используют спектро-
скопич. методы исследования (см. Колебательная спек-
спектроскопия). Особенность инфракрасного спектра П.—
наличие полос поглощения, характерных для амид-
амидной связи: полоса поглощения при 3,03 мкм C050 см'1),
обусловленная валентными колебаниями группы N — Н;
полоса при 6,06 мкм A650 см'1), соответствующая в
основном валентным колебаниям группы С=О (обычно
наз. амидной связью I), и полоса при 6,45 мкм
A550 см'1), характерная для деформационных коле-
колебаний группы N—Н (обычно наз. амидной связью II).
Для идентификации П. подвергают гидролизу, а также
испытанию в пламени, исследуют растворимость, про-
проводят колориметрич. испытания и определяют конце-
концевые группы. Кроме того, для разделения и идентифи-
идентификации П. используют метод бумажной хроматографии.
Ароматические полиамиды. П. этой группы — в ос-
основном бесцветные высоконлавкие кристаллич. веще-
вещества, как правило плохо растворимые в органич. ра-
растворителях. Ароматич. П. обычно растворяются
только в основных растворителях, таких как диметил-
ацетамид или N-метилпирролидон (иногда при добав-
добавлении солей, напр. СаС12 или LiCl). Хорошие раство-
растворители для П.— конц. серная или хлоруксусная к-та.
/га/?а-3амещенные звенья П. обусловливают высокую
кристалличность благодаря жесткости и симметрии
цепей таких полимеров. Однако и мета-з&мещеяные
П. (вероятно, в этом случае нарушение симметрии
не очень велико) характеризуются достаточно высокой
степенью кристалличности, как, напр., поли-ж-фени-
ленизофталамид и сополимеры, построенные из чере-
чередующихся звеньев пара- и жегаа-замещенных циклов.
Благодаря высокой кристалличности ароматич. П.
также обладают резко выраженной Тил. Темп-ры плав-
плавления ароматич. П. значительно выше, чем алифати-
алифатических. С введением ароматич. звеньев в макромоле-
макромолекулы алифатич. П. возрастают темп-ры плавления
(из-за повышения жесткости макромолекул), снижа-
снижаются растворимость и способность к абсорбции влаги.
Темп-pa плавления особенно резко изменяется при
замещении алифатич. звеньев гаара-замещенными аро-
ароматич. компонентами. Напр., при замещении в П.
звеньев адипиновой к-ты звеньями терефталевой к-ты
темп-pa плавления повышается на 100 —170 °С, а при
замещении на арилалифатич. звенья — на 50—70 °С
(в этом случае в образующихся П. ароматич. циклы
не сопряжены с карбонильной группой амидной связи):
Темп-ра
плавления, °С
295
[_NH-(CH2L-NHCO-(CH2L-CO-]n
[-NH—(СН2) — NHCO—-(У-СО— | .... 455
I 4 \——У Jn
[-NH-(CH2)e-NHCO(CH2LCO-]n 265
—NH—(CH2)-NHCO
-Q-co-
371
t-NHCH2—<f V-CH2NHCO(CH2\CO-J . . 333
В табл. З приведены температуры плавления и раз-
размягчения некоторых ароматических П.
100 200 300 400 500 600 700 800
Температура,°С
Рис. 4. Относительная термостойкость ароматич. полиами-
полиамидов: 1 — из хлор ангидрида фталевой к-ты и м-фениленди-
амина; 2 — из хлорангидрида фталевой к-ты и о-фенилен-
диамина; з — из хлорангидрида фталевой к-ты и п-фенилен-
диамина; 4 — из хлорангидрида изофталевой к-ты и о-фе-
нилендиамина; 5 — из хлорангидрида терефталевой к-ты
и о-фенилендиамина; 6 — из хлорангидрида изофталевой
к-ты и ж-фенилендиамина; 7 — из хлорангидрида тере-
терефталевой к-ты и п-фенилендиамина; 8 — из хлорангидрида
изофталевой к-ты и п-фенилендиамина; 9 — из хлорангидри-
хлорангидрида терефталевой к-ты и лг-фенилендиамина.
745
ПОЛИАМИДЫ
746
Таблица 3. Температуры плавления и размягчения нек-рых
ароматических полиамидов
Полиамид
HN. .NH—СО СО—
HN NH—СО—f V-CO—
-HN—
Темп-ра
размягче-
размягчения, °С
260
260
270
290
300
520
Темп-ра
плавле-
плавления, °С
— 185
-200
-240
300
300
430
470
470
-600
Ароматич. П. характеризуются высокой термостой-
термостойкостью: темп-ры разложения многих П. составляют
—400 °С (рис. 4). Благодаря ряду ценных свойств
(см., напр., табл. 1) — высокой термостойкости, со-
сохранению физико-механич. и электрич. показателей
при высоких темп-pax, устойчивости к действию рас-
растворителей и химич. реагентов, низкой воспламеняе-
воспламеняемости, высокой твердости, высокой диэлектрич. про-
проницаемости и устойчивости к ионизирующему облуче-
облучению — П. можно использовать в качестве термо- и
теплостойких материалов.
Практически важную группу представляют П., со-
содержащие реакционноспособные заместители в орто-
положении ароматич. ядра. Эти полимеры относитель-
относительно легко растворимы (напр., в муравьиной и серной
к-тах, диметилформамиде), их можно перерабатывать
из р-ра в волокна и пленки. При нагревании таких
П. происходит полициклизация с образованием гете-
роциклов в макромолекулах. Полученные таким об-
образом полимеры — термостойкие материалы, характе-
характеризующиеся высокими темп-рами плавления и плохой
растворимостью. В зависимости от природы реакцион-
носпособных групп при полициклизации могут обра-
образовываться различные гетероциклы. Напр., из П.,
содержащих в бензольных кольцах в ор mo-положении
карбоксильные, гидроксильные или аминные груп-
группы, м. б. получены соответственно полиимиды, поли-
бензоксазолы и полибензимидазолы.
Применение
В пром-сти пока применяют гл. обр. алифатич. П.
П. используют в различных отраслях пром-сти и,
в первую очередь, в производстве синтетических во-
волокон (см. Полиамидные волокна), обладающих вы-
высокой прочностью, устойчивостью к истиранию, гни-
гниению, действию бактерий и моли. П. применяют так-
также для изготовления пленок (см. Полиамидные плен-
пленки), деталей машин — шестерен, подшипников, вту-
втулок и др.
К наиболее распространенным алифатич. П., произ-
производимым в промышленном масштабе, относятся: поли-
гексаметиленадипинамид, полигексаметиленсебацин-
амид, поли-е-капроамид (см. Капролактама полимеры),
поли-(О-энантоамид, поли-(?>-ундеканамид и полидоде-
канамид, сравнительно низкомолекулярные П. (мол. м.
2000—10 000), получаемые из полимеризованных нена-
ненасыщенных жирных к-т и различных полиаминосоеди-
нений. Низкомолекулярные П. используют как отвер-
дители для эпоксидных смол', в композиции с эпоксид-
эпоксидными смолами — для изготовления заливочных ком-
компаундов, в качестве клеев (см. Полиамидные клеи) и
связующих для стеклопластиков. В пром-сти выпус-
выпускаются также сополимер (отечественная марка П-54)
гексаметилендиаммонийадипината E0% соли АГ) с
е-капролактамом E0%), применяемый для изготовле-
изготовления прокладочных материалов и изделий, используе-
используемых в кабельной, химич. и авиационной пром-сти;
сополимер (марка П-548) гексаметилендиаммонийадипи-
гексаметилендиаммонийадипината и гексаметилендиаммонийсебацината C7% соли
АГ и 19% соли СГ) с е-капролактамом D4%), приме-
применяемый для изготовления пленок, прокладочного ма-
материала; сополимер (марка П-АК7) 93% соли АГ и 7%
е-капролактама, применяемый как водо-, масло- и
бензостойкий материал, устойчивый к истиранию, об-
обладающий абразивостойкостью и низким коэффици-
коэффициентом трения; сополимер (марка П-АК80/20) 80%
соли АГ и 20% е-капролактама, применяемый как
конструкционный материал для изготовления шесте-
шестерен для зубчатых передач, втулок, вкладышей под-
подшипников и т. д., а также изделий массового по-
потребления.
Для повышения эксплуатационных свойств П. в них
вводят различные антифрикционные наполнители (см.
Полиамидные пластмассы).
Из ароматич. П. промышленное применение нашел
поли-м-фениленизофталамид. Весьма перспективны, в
частности для изготовления высокопрочных высокомо-
высокомодульных волокон, ароматич. П. с симметричными
звеньями, напр, поли-тг-фениленамид, поли-/г-фенилен-
терефталамид.
Первый синтетич. П. получен Хорбердом в 1862 при
нагревании ж-аминобензойной к-ты в присутствии хло-
хлористого водорода. Развитие исследований п,о синтезу
и применению П. началось после работ У. Карозерса,
показавшего возможность получения из этого класса
высокомолекулярных соединений прочных волокон.
Лит.: КоршакВ. В., Фрунзе Т. М., Синтетиче-
Синтетические гетер оцепные полиамиды, М., 1962; Кларе Г., Фриц-
шеЗ., ГребеФ., Синтетические полиамидные волокна,
пер. с нем., М., 1966; Волокна из- синтетических полимеров,
под ред. Р. Хилла, пер. с англ., М., 1957; X о п ф ф Г.,
Мюллер А., Вагнер Ф., Полиамиды, пер. с нем., М.,
1958; Флойд Д. Б., Полиамиды, пер. с англ., М., 1960;
Соколов Л. Б., Поликонденсационный метод синтеза по-
полимеров, М., 1966; Морган П. У., Поликонденсационные
процессы синтеза полимеров, пер. с англ., Л., 1970; Справоч-
Справочник по пластическим массам, под ред. М. И. Гарбара [и др..!,
т. 1—2, М., 1967—69; Encyclopedia of polymer science and tech-
technology v. 10, N. Y. [a. 0.], 1969, p. 483. B.B. Курашев.
747
ПОЛИАМИНОТРИАЗОЛЫ
ПОЛИАМИНОТРИАЗОЛЫ — см. Политриазолы.
ПОЛИАМИНЫ (polyamines, Polyamine, polyami-
nes) — полимеры, содержащие в макромолекулах пер-
первичные, вторичные или третичные аминогруппы. П.
могут быть алифатическими, ароматическими или гете-
гетероциклическими.
Наиболее распространенный метод синтеза П.—
полимеризация виниловых мономеров, содержащих
аминогруппу; при этом образуются П. с атомом азота
в боковой цепи (см., напр., Аминостиролов полимеры,
Аллиловых соединений полимеры, Винилпиридинов по-
полимеры, Винилхинолина полимеры, Винилимидазолов
полимеры, Винилкарбазола полимеры). Некоторые
др. виниловые мономеры, способные полимеризовать-
ся и сополидшризоваться под действием радикаль-
радикальных инициаторов с образованием П., приведены в
таблице.
П. с аминогруппой в основной цепи получают поли-
полимеризацией циклич. иминов (см., напр., Этиленимина
полимеры), их N-замещенных производных или азот-
азотсодержащих бициклов. Высшие циклич. и бициклич.
имины полимеризуются по катионному механизму под
действием неорганич. к-т, напр. НС1, НВг, хлорной,
к-т Льюиса [BF3-O(C2H5J, A1C13, SnCl4, FeCl3], алки-
алкилирующих соединений (диметилсульфата, алкилгало-
генидов), а также аммониевых солей, в том числе чет-
четвертичных аммониевых солей самих мономеров. Пир-
ролидин, пиперидин, гексаметиленимин полимеризуют-
полимеризуются при 280 °С, гептаметиленимин, октаметиленимин,
додекаметиленимин и его N-метильное производное
(I) — при 225 °С с образованием разветвленных оли-
гомеров (степень полимеризации 10—50). Бициклич.
имины, напр. 1-азабицикло[3, 1, 0]гексан (II); 1-аза-
бицикло[4,2,0]октан, или конидин [III]; 1-азабици-
кло[2,2,2]октан, или хинуклидин [IV] и 1,4-диазаби-
цикло[2,2,2]октан, или триэтилендиамин [V], полиме-
полимеризуются с образованием высокомолекулярных линей-
линейных продуктов. Имины IV и V полимеризуются при
темп-pax не ниже 180 °С
с образованием П., рас-
растворимых только в к-тах,
напр, муравьиной, ук-
уксусной, соляной, II и
III — не ниже 30 °С с
образованием живущих
полимеров. Последние
растворимы в бензоле,
IV V метаноле и др. органич.
растворителях. Теплоты
полимеризации II, III, V равны: 98,2 кдж/молъ B3,4
ккал/молъ), 70,5 кдж/молъ A6,8 ккал/молъ) и 9,8 кдж/молъ
B,3 ккал/молъ) соответственно. Для П., полученного
из III, найдена зависимость между мол. массой и ха-
рактеристич. вязкостью: [г|] =• 2,4*10~4 М°>7. При по-
полимеризации пиридина и хинолина получены П. с со-
сопряженными двойными связями (см. Пиридина поли-
полимеры).
Высокомолекулярные П. с аминогруппами в основ-
основной цепи можно синтезировать также поликонденса-
поликонденсацией алифатич. вторичных диаминов и дивинильных
соединений, двойные связи в к-рых активированы элек-
троноакцепторными группировками, напр.:
R R
! I
HN-R'-NH + GH8=CHGONH - R"-NHOCCH=CH2 ->
R R
-»[-N-R'-N-CH2- GH2CONH - R" - NHOGGH2GH2 - ]n
Скорость реакции и мол. масса образующегося П.
зависят от полярности растворителя. При взаимодей-
взаимодействии пиперазина и дивинилсульфона образуются вы-
высокомолекулярные полиаминосульфоны. Вместо вто-
R=H; CH3
I I _j
II
/ \
ричных диаминов можно использовать и первичные;
в этом случае образуются разветвленные П. Низкомо-
Низкомолекулярные П. получены при реакции первичных
алифатических диаминов с дигалогенпроизводными,
например:
H2NRNH2 + С1СН2СН=СНСН2С1 -> [-HNRNHCH2GH=GHCH2 - ]п
Мол. масса П. достигает 2000—3000. Поскольку про-
процесс протекает по реакции Гофмана, П. содержат так-
также третичные и четвертичные аминогруппы. При низко-
низкотемпературной поликонденсации тетраметилендиамина
с диацетиленом получены окрашенные, содержащие
сопряженные двойные связи П. (мол. масса ~7000):
H2N(GH2LNH2 + СНеееС - С=СН ->
-+ [-HN(GH2LNHGH=GH-GH=CH - ]п
П. можно синтезировать также из анилина и диэпок-
сидов.
При реакции ароматич. диальдегидов, напр, тере-
фталевого альдегида, с первичными диаминами получе-
получены П., содержащие в основной цепи макромолекулы
связи —HC=N— (см. Полишиффовы основания). По-
Поликонденсацией получают высокотермостойкие П. с
азотсодержащими гетероциклами в цепи (см., напр.,
Полибензимидазолы, Полибензоксазолы, Полихинокса-
лины).
П. синтезируют также полимер аналогичными превра-
превращениями', напр., восстановлением полинитростирола
получают полиаминостирол, восстановлением поли-N-
диалкилакриламидов — поли-1Ч-диалкилаллиламины,
омылением поли-1Ч-винилфталимина или поли-]Ч-ацил-
этиленимина — поливиниламин или полиэтиленамин
соответственно. Термостойкие П. образуются при тер-
мич. обработке полиакрилонитрила (см. Акрилонитри-
ла полимеры).
Аналогично низкомолекулярным аминам, П., содер-
содержащие первичные и вторичные аминогруппы, при вза-
взаимодействии с ацилирующими или алкилирующими
агентами образуют полимерные N-ацильные и N-ал-
кильные производные. При исчерпывающем алкилиро-
вании галогеналкилами получаются полимерные чет-
четвертичные соли (см. Полиэлектролиты); последние
также образуются при действии алкилирующих соеди-
соединений на П., содержащие третичные аминогруппы.
Окисление полимерных третичных аминов приводит
к полимерным N-окисям (см. Винилпиридина полиме-
полимеры). Для всех линейных П. характерно образование
с неорганич. к-тами полимерных аммонийных солей,
растворимых в воде.
В отличие от мономерных аминов, П. способны обра-
образовывать стабильные комплексы с поликислотами,
напр, поливинилпиридины или полиэтиленимин с по-
полиакриловой или полиметакриловой к-тами.
П. применяют для синтеза слабоосновных анионо-
обменных смол. Для сшивки используют различные
дифункциональные соединения, напр, дигалогеналки-
лы. П. применяют в бумажной пром-сти для улучше-
улучшения обезвоживания бумажной массы, а также для
пропитки бумаги; как добавки к смазочным маслам,
обеспечивающие их пониженную активность при кор-
коррозии, и к топливу для увеличения вязкости; как
эмульгаторы при эмульсионной полимеризации. П.,
содержащие четвертичные группы, применяют в цвет-
цветной фотографии для закрепления красителя в желати-
желатиновой эмульсии. Предложено использовать П. как
связующее при произ-ве твердых ракетных топлив.
Перспективно применение П. в качестве физиологиче-
физиологически активных полимеров. Так, полимерные четвертич-
четвертичные соли — антагонисты гепарина, N-окиси П., напр.
поли-2-винилпиридин-1Ч-оксид,— эффективные противо-
силикозные препараты (см. Физиологически активные
полимеры). П.— исходные вещества для создания мате-
материалов на основе полимер-полимерных комплексов.
749
ПОЛИАМИНЫ
Физические свойства и способность к полимериза-
полимеризации нек-рых азотсодержащих виниловых мономеров
и свойства образующихся из них полиаминов
A мм рт. ст.=133,322 h/jh2)
Мономер
Физич. констан-
константы мономера
2-Винилпиррол
НС—СН
И И
не с—сн=сн2
К-Винил-3,5-ди-
метил пиразол
НС —С —СНЖ
II II •
Н3С —С N
Ч /
N
сн=сн2
З-Метил-5-изо-
пропенилпира-
золин
N —NH сНз
н3с-с с-с-сн2
сн
N-Винил-! ,2,
4-триазол
N-CH
ч
N
сн=сн2
Т. кип. 64-67°С
A8 мм рт. ст.);
пъ 1,5562
Т. кип. 51 —53°С
D мм рт. ст.);
d° 0,9618;
4
п^ 1,5151
Т. кип. 62,5 °С
Условия полимеризации
и свойства полиамина
Легко полимеризуется
уже при хранении с об-
образованием твердого по-
полимера.
Полимеризуется под
действием АБН* и пере-
перекиси бензоила. Сополиме-
Сополимеризуется со стиролом
(Q=0,18; е=— 1,27). По-
Полимер хорошо растворим
в спиртах, бензоле, аце-
ацетоне, хлорированных уг-
углеводородах, нераство-
нерастворим в воде. В отличие
от поливинил-2-метил-
имидазола, не образует
четвертичных солей.
Полимеризуется
действием АБН.
под
Т.кип.
202-203 °С;
Пр 1,4868
2-Винил-4,5-ди-
метилоксазол
(R^=H) и 2-изо-
пропенил-4,5-
диметилоксазол
(R=CH3)
Н3С-С—N R
2-Винил-4,4-ди-
метил-5-оксазо-
лон
о—сн—сн=сн.
ЧСН3
2-Винилтиазол
НС— N
И И
Для R=H т. кип
61 °СB0 мм рт
ст.), п^ 1,4875;
дляК=СН3т. кип
75 °С B5 мм рт.
ст.), п^у 1,489
Т. пл. 6 °С;
п^ 1,4575
Т. кип. 47 °С
A мм рт. ст.);
Пр 1,5435
Полимеризуется под
действием АБН и пере-
перекиси бензоила с образова-
образованием высокомолекуляр-
высокомолекулярных бесцветных гигро-
скопич. продуктов, рас-
растворимых в воде, диме-
тилформамиде, диметил-
сульфоксиде, уксусной
к-те; нерастворим в уг-
углеводородах, эфирах, ке-
тонах, спиртах.
Полимеризуются и со-
полимеризуются со сти-
стиролом под действием
АБН (для R=HQ=2,15,
е=—0,40); полимери-
полимеризуются под действием
Na-нафталина. Полиме-
Полимеры растворимы в бензо-
бензоле.
Полимеризуется в мас-
массе в присутствии АБН
при 65 °С. Полимер рас-
растворим в этилацетате и
ацетоне, нерастворим в
гексане. Сополимеризу-
Сополимеризуется со стиролом и п-ме-
токсистиролом.
Полимеризуется под
действием АБН в массе.
Сополимеризуется со сти-
стиролом (Q = 3 , 5; е =
4-0,08). Полимер рас-
растворим в бензоле.
750
Продолжение табл.
Мономер
Физич. констан-
константы мономера
Винилпиразин
НС СН
4-Винилпирими-
дин
сн=сн,
НС
N
2-]Ч-Диметил-
амин-4-винил-
пиримидин
сн=сн2
НС
N
II
C
2,4-Диамино-6-
винилтриазин
N N
Т. кип. 63—64 СС
B1 .мм рт. ст.);
п^ 1 ,5565
Т. кип. 56 — 58 °С
A0 мм рт. ст.);
d25 1,0598;
4
Пр 1 ,5405
Условия полимеризации
и свойства полиамина
Полимеризуется под
действием перекиси бен-
бензоила с образованием по-
полимера коричневого цве-
цвета, растворимого в воде.
Полимеризуется под
действием АБН с обра-
образованием полимера бело-
белого цвета, растворимого
в воде.
Т. кип. 95 — 98 °С
14 мм рт. ст.);
п^ 1,5608
Т. кип. 300 —
310 °С (с разло-
разложением)
N-Винилиндол
N
сн=сна
2-Винилбензти-
азол
)>-сн=сн2
2-Винилбензок-
сазол
Т. кип. 71 — 72 °С
A мм рт. ст.);
/° 1,0658;
т. пл. 31,5 — 32 °С,
Пр 1 ,6300
Т. кип. 73 — 75 °С
@,05 мм рт.
ст.); т. пл.
40,5 — 41,5 °С
Т. кип. 60—61 °С
A мм рт. ст.);
т. пл. — 16 °С;
d25 1,116;
4
n2Q 1,5581
Полимеризуется под
действием АБН с обра-
образованием полимера, рас-
растворимого в бензоле, не
растворимого в воде. Со-
Сополимеризуется со сти-
стиролом.
Полимеризуется под
действием персульфата
натрия в воде при 90 °С
с образованием полиме-
полимера, растворимого только
в минеральных к-тах.
Полимеризуется под
действием АБН. Сополи-
Сополимеризуется с метилмет-
акрилатом и метакри-
ловой к-той. Полимери-
Полимеризуется под действием
катионных катализато-
катализаторов [BF3-O(C2H5J,SnCl4];
полимер растворим в бен-
бензоле, толуоле; нераство-
нерастворим в метаноле.
Полимеризуется при
хранении и под дейст-
действием радикальных ини-
инициаторов; ингибитор
mpem-бутшширокате-
хин.
Полимеризуется под
действием перекиси бен-
бензоила, АБН. Сополимери-
Сополимеризуется при нагревании и
под действием перекиси
бензоила со стиролом и
метилметакрилатом.
751
ПОЛИАНГИДРИДЫ
Продолжение табл.
752
Мономер
8-Винил-2-мер-
каптобензтиазол
Физич. констан-
константы мономера
Т. кип. 135 —
136 СС
B мм рт. ст.);
d° 1,2629
4
I I >-s—сн=сн2
N-Винил-!,2,3-
бензтриазол
сн=сн2
fY \<
^**/ IN
N-Виниладенин
NH2
1 м
1 И J?
6-Метиламино-
9-винилпурин
NHCH3
N iTN\
L JkN/
1
CH=CH2
N-Диметиламино-
этилметакрилат
CHi
1
CH2=C—COOCH2CH2
1 , З-бис-Диме-
тиламиноизопро-
пилметакрилат
Т. пл. 199 —
200 °С
Т. пл. 178 —
179 °С
Т. кип. 65,5 °С
(8 мм рт. ст.);
rl Л Q49 1 •
и, U , v о i-> i ,
4
20
п j-j 1,4395
N(CH3)
Т. кип. 117 —
117,5 °СB0 лип
рт. ст.);
d° 0 9239"
4
n^L 1 4478
CH3 /CH2N(CH3J
сн2=с-соосн
CH2N(CH3J
2 -Пири дилм ет-
v акрилат
Т. кип. 90-99 °С;
т. пл. —15 °С;
п^ 1,5168
2
Условия полимеризации
и свойства полиамина
Полимеризуется и со-
сополимеризуется под дей-
действием АБН с метилмет-
акрилатом и стиролом.
Полимеризуется под
действием АБН, переки-
перекиси бензоила с образова-
образованием низкомолекуляр-
низкомолекулярных окрашенных продук-
продуктов, растворимых в уг-
углеводородах, диметил-
формамиде, уксусной
к-те, не растворимых
в воде, спиртах, эфирах,
кетонах.
Полимеризуется под
действием персульфата
калия или АБН в диме-
тилсульфамиде. Полимер
растворим в воде и ди-
мети л формами де. Сопо-
Сополимеризуется с акрил-
амидом, винилпирроли-
доном, малеиновым ан-
ангидридом.
Мономер полимеризу-
полимеризуется в диметилсульфок-
сиде под действием АБН
с образованием полиме-
полимера, растворимого в воде;
сополимеризуется с ак-
риламидом.
Легко полимеризуется
при нагревании или под
действием АБН и др.
Полимеризацию ингиби-
руют гидрохинон, фен-
тиазин. Полимер рас-
растворим в бензоле, этано-
этаноле. При действии алки-
лирующих веществ на
полимер получены поли-
полимерные четвертичные со-
соли.
Полимеризуется тер-
термически и под действи-
действием АБН. Полимер рас-
растворим в гептане, бензо-
бензоле, этаноле. При дейст-
действии алкилирующих ве-
веществ переходит в поли-
полимерные четвертичные со-
соли.
Полимеризуется под
действием АБН с обра-
образованием полимера, рас-
растворимого в пиридине;
сополимеризуется с ме-
тилметакрилатом и сти-
стиролом (Q=— 1,5, e=-j-0,8
и Q=—1,4, е= + 0,5 со-
соответственно).
Продолжение
Мономер
Метакрилат-N-
/3 -оксиэтиладенин
Физич. констан-
константы мономера
Т. пл. 201
203 °С
6с:>
Т 1 Р1'
СН2СН2ОС—С=СН2
Условия полимеризации
и свойства полиамина
Полимеризуется под
действием АБН. Поли-
Полимер растворим в диме-
тилсульфоксиде; сополи-
сополимеризуется с малеиновым
ангидридом и ацетони-
трилом.
* АБН — азо-бис-изобутиронитрил.
П. применяют для синтеза полупроводниковых и элек-
электропроводящих материалов. Сополимеры, содержащие
аминогруппу, обладают бактерицидными свойствами,
способны окрашиваться кислотными красителями; вве-
введение в макромолекулы каучуков аминогруппы уве-
увеличивает их адгезию к шинному корду и к металлам;
уменьшает поверхностный электростатич. заряд.
Лит.: Overberger С. G. [а. о.], J. Amer. Chem. Soc,
76, Кя 7, 1879 A954); 80, 898A958); 87, 296 A965); К а т а 1 М.
[а. о.], J. Organ. Chem., 27, № 4, 1363 A962); TakemotoK.
[а. о.], Makromol. Chem., 148, 131 A971); Разводов-
с к и й Е. Ф. [и др.], ДАН СССР, 198, № 4, 894 A971); Нои-
ben-Weyl., 14/2, 455, 477 A963). Е. Ф. Разводовский.
ПОЛИАНГИДРИДЫ (polyanhydrides, Polyanhyd-
ride, polyanhydrides) — полимеры, содержащие в ос-
основной цепи макромолекулы повторяющиеся ангид-
ангидридные группы — COOGO—.
Получение. П. получают поликонденсацией
или полимеризацией. Поликонденсация, приводящая
к образованию П., наз. полиангидридиза-
ц и е й. Полиангидридизацию проводят в расплаве,
р-ре или на границе раздела фаз (см. Межфазная
поликонденсация). Наиболее распространен метод поли-
ангидридизации алифатич. или ароматич. дикарбоно-
вых к-т с уксусным ангидридом. Обычно этот процесс
проводят в несколько стадий:
а) ацетилирование дикарбоновой к-ты (нагревание
при нормальном давлении):
HOOC-R-COOH + 2(СН3СОJО -> CH3COOCO-R-COOCOCH3 +
+2CH3COOH
б) образование низкомолекулярного а-П. [нагрева-
[нагревание в вакууме при 10—15 мм рт. ст. A мм рт. ст.—
= 133,322 н/м2)]:
лгСНзСООСО — R - СООСОСНз ->
-> СНзСОО— [—COR—COO—]x—СОСНз + (х— 1)(СН3СОJО
в) синтез высокомолекулярного со-П. (нагревание
в вакууме при 0,01 мм рт. ст.):
СНзСОО - [ - COR - СОО - З^СОСНз-*
-> СНзСОО - [ - COR - СОО - ]п - СОСН3+(СН3СОJО
где п > х.
Полиангидридизацию можно осуществлять также
по след. схемам:
2R'COC1
п НООС - R - СООН >[ - COR - СОО - ]п,
яНООС - R - СООН+я С1СО - R' - СОС1 + 2nRN ->
-»[-CO-R-COOCOR'COO-]n+2nRN.HCl,
nClCO-R-COCl + яН2О + 2nR'3N -> [-CO-R-COO -]nf
+ 2nR'3N-HCl
П. получают также полимеризацией циклич. ангид-
ангидридов:
/СО
п (ск2L^о-
f—CO(CH2) СОО—1
Свойства. П. — твердые вещества белого цве-
цвета, отличающиеся чаще всего высокой степенью кри-
кристалличности. Они не растворяются в воде и, как
753
ПОЛИАНГИДРИДЫ
754
правило, плохо растворяются в органич. растворите-
растворителях. Так, П. терефталевой к-ты не растворим в воде
и органич. растворителях; его можно растворить
(с разложением) лишь при нагревании в водном р-ре
щелочи и конц. серной к-те. Растворимость ароматич.
П. в органич. растворителях улучшается при введении
в основную цепь макромолекулы простых эфирных
связей (—0—) и объемистых полярных циклич. груп-
группировок, напр, фталидных.
В ряду П. на основе алифатич. дикарбоновых к-т
типа НООС(СН2)ПСООН темп-ры плавления возрастают
с увеличением в макромолекуле числа метиленовых
групп между ангидридными связями. Так, темп-ры
плавления П. с тг = 4 и тг = 16 составляют соответ-
соответственно 70 и 98 °С. П., полученные из алифатич. ди-
дикарбоновых к-т с четным числом атомов углерода в
молекуле, плавятся при более высоких темп-pax, чем
П., полученные из соседних дикарбоновых к-т с нечет-
нечетным числом атомов углерода. В отличие от алифатич.
П., ароматич. П.— достаточно теплостойкие полимеры
(см. табл.). Так, П. терефталевой и 2,6-нафталиндикар-
боновой к-т плавятся соответственно при 400 и 450 °С.
П., содержащие в основной цепи мета-замещеппые
бензольные ядра, плавятся при более низких темп-рах,
чем П. с гсара-замещенными бензольными ядрами.
Темп-pa плавления П. на основе дикарбоновых к-т
типа НООС—/~~\-R—/""Л-СООН существенно зави-
сит от химической природы мостиковои группы R; для
R = C=O темп-pa плавления составляет 338 °С;
для R = СН2 — 332 °С; для R= О — 290—296 °С; для
R = ОСН2 - 260 °С; для R = C (CH3J- 230-235 °С;
для R = ОСН2О — 220 °С. С увеличением числа мети-
метиленовых групп между бензольными ядрами темп-ра
плавления П. понижается, причем в полимергомоло-
гическом ряду при более высокой темп-ре плавятся
Свойства некоторых
П., содержащие четное число метиленовых групп
(СН2)пмежду бензольными ядрами, чем П., получен-
полученные из соседних дикарбоновых к-т с нечетным числом
групп (СН2)П. Для ароматич. П., содержащих между
бензольными ядрами группы — О(СН2)ДО —, как пра-
правило, наблюдается про-
противоположная зависи-
зависимость. При введении в
макромолекулы арома-
ароматич. П. остатков али-
алифатич. дикарбоновой
к-ты темп-pa плавле-
плавления значительно сни-
снижается.
Алифатич. П. отли-
отличаются от ароматиче-
20[|
§50
1-
§30
ских значительно бо-
Устойчивость различных
полиангидридов и поли-
этиленгликольтерефтала-
та к щелочному гидро-
гидролизу A н. р-р NaOH):
10
50 100
Время, ч
— П. себациновой к-ты; 2 и з — соответственно аморфный
и кристаллич. П. бие-D-карбоксифенил)-1,3-пропана; 4 —кри-
—кристаллический полиэтиленгликольтерефталат.
лее высокой ацилирующей способностью по отноше-
отношению к воде, спиртам и др. соединениям с подвижным
атомом водорода. Ввиду высокой гидролизуемости али-
алифатич. П. их получение и хранение можно осуществ-
осуществлять только в сухой атмосфере. Волокна и пленки,
изготовленные из алифатич. П., из-за гидролиза ан-
ангидридных связен очень быстро теряют прочность и
эластичность.
Ароматич. П. устойчивы к гидролизу, причем кри-
кристаллич. П. несколько более устойчивы к действию во-
воды, чем аморфные П. того же химич. строения (см. рис.).
полиангидридов
Полиангидрид
Формула
Характери-
стич.
вязкость,
дл/г
Темп-pa
плавле-
плавления, °С
Растворитель
Политетраметиленангидрид
Полиоктаметиленангидрид
Поли-[бис-(этилен)-п-фениленангидрид]
Поли-[бис-(этилен)-2,5-фуриленангидрид]
Поли- п-фениленангидрид
Поли-ж-фениленангидрид
Поли- 4,4' -дифенилфталидангидрид
Поли-[бггс-(п-оксифенилен)-
триметиленангидрид]
[-СО(СН2LСОО-]П
[-СО(СН2)8СОО-]П
|-СО(СН2) -^ Д-(СН2) СОО-1
О г h
[-со-/~~\-соо~|
(Мп=5000)
C1^=10 000)
0,23
0,14
[~c°-0rco°-i
[-со-/~Л-с~/~Л-соо-|
C
Поли-[бис-(п-амидофенилен)-
метиленангидрид]
[—га
1
0,11
ONH-CH2—NHCO—
COO —
0,14
70
85
90
70
400
260
220
260
325
Бензол, ацетон
Бензол, ацетон
л*-Крезол
м-Крезол
Нерастворим
Нерастворим
Тетрахлорэтан,
нитробензол,
трикрезол, ци-
клогексанон
м-К резол
ж-Крезол
755
ПОЛИАРИЛАТЫ
756
Из ароматических П., отличающихся значительной
химич. инертностью, получены пленки и волокна.
Так, изготовлены пленки из высоконлавких и устойчи-
устойчивых к гидролизу ароматич. П. на основе дикарбоно-
вых к-т типа НООССбН4 — R — С6Н4СООН, где R =
- (СН2)П—; —O(CH2)nO —; -S(CH2)nS- и др. Меха-
нич. свойства пленки П. (R = — О(СН2KО—), полу-
полученной из расплава при 110 °С, следующие: прочность
при растяжении 400 Мн/м2, или 40 кгс/мм2 (при удли-
удлинении 17,2%), модуль Юнга 5050 Мн/м2 E05 кгс/мм2).
П. применения в пром-сти пока не нашли.
Лит.: Cottier R. J., Matzner M., Chem. weekbl.,
63, № 11, 113A967); Коршак В. В., Виноградо-
Виноградова С. В., Равновесная поликонденсация, М., 1968; Кор-
Коршак В. В., Термостойкие полимеры, М., 1969.
В. А. Васнев.
ПОЛИАРИЛАТЫ (pclyarylates, Polyarylate, poly-
arylates) — сложные полиэфиры двухатомных фено-
фенолов общей ф-лы [ — OCRCOOR'O—]п, где R — остаток
дикарбоновой к-ты, R'— остаток двухатомного фенола.
Частный случай П.— поликарбонаты (полиэфиры уголь-
угольной к-ты и двухатомных фенолов).
Получение. П. можно получать поликонденсацией
по след. схемам:
п H3COCOR'OCOCH3 + n HOOCRGOOH -»
-> [ - OCRCOOR ГО - ]п + 2п СНзСООН
п ROOGRGOOR + п HOR'OH -> [-OCRCOOR'O-]n + 2n ROH
п C1OCRCOC1 + п HOR'OH -> [ - OCRCOOR'O - ]п + 2n HC1
Для синтеза полностью ароматич. П. целесообразно
использовать в качестве исходных кислотных агентов
дихлорангидриды. Такие П. могут быть получены тре-
тремя способами:
1. Высокотемпературной поликонденсацией (чаще все-
всего при 180—220 °С) в атмосфере инертного газа в сре-
среде высококииящего растворителя (например, динила,
дитолилметана, совола, а-хлорнафталина, трихлор-
бензола). Природа реакционной среды оказывает боль-
большое влияние на формирование надмолекулярной струк-
структуры полимера. Так, П. фенолфталеина и изофталевой
к-ты (Ф-1), полученный в среде дитолилметана, в к-рой
полимер не растворяется, имеет глобулярную надмоле-
надмолекулярную структуру; П., синтезированному в среде,
в к-рой он хорошо растворяется (а-хлорнафталин,
нитробензол), свойственна фибрилярная надмолекуляр-
надмолекулярная структура.
Изменением условий процесса можно регулировать
степень кристалличности П. Так, при взаимодействии
смеси 9,9-б^с-D-оксифенил)антрона-10 с дихлоранги-
дридом терефталевой к-ты в соволе при 320 °С в тече-
течение 60—90 мин и последующем быстром охлаждении
реакционной массы до 100 °С и ниже получают аморф-
аморфный П. При поликонденсации в соволе при 220 °С
в течение 7—10 ч образуется П. со степенью кристал-
кристалличности до 30%. Последний способ используется
в пром-сти для произ-ва П. марок Ф-1 и Ф-2 (из фенол-
фенолфталеина и дихлорангидридов изофталевой и терефта-
терефталевой к-т соответственно).
2. Акцепторно-каталитической, или низкотемпера-
низкотемпературной (от —20 до 50 °С), поликонденсацией. Процесс
проводят в среде органич. растворителя (напр., ацето-
ацетона, дихлорэтана) в присутствии стехиометрич. количе-
количества третичного амина (чаще всего триэтиламина).
Поликонденсация протекает по двум конкурирующим
механизмам:
~RfCOCl-fHO-R~ + R.N
f RC0C1-
Общий основной катализ
Преимущественное направление реакции зависит от
природы исходных веществ и третичного амина.
3. Межфазной поликонденсацией. В пром-сти по это-
этому методу получают смешанные П. из диана D,4'-ди-
оксидифенил-2,2-пропана) и дихлорангидридов терефта-
терефталевой и изофталевой к-т, взятых в различном моля-
молярном соотношении (марки Д-4 и Д-4С).
В соответствующих условиях м. б. получены П. весь-
весьма высокой мол. массы. Так, П. марки Ф-2, синтезиро-
синтезированные высокотемпературной поликонденсацией в со-
соволе и в 1,2,4-трихлорбензоле, имеют приведенные
вязкости (г|Пр, в тетрахлорэтане при 25 °С) — 1,5 дл!г
(мол. масса —100 000) и —2,5 дл/г соответственно.
Низкотемпературной поликонденсацией в смеси ацето-
ацетона с бензолом удается получить П. марки Ф-2 с т]пр-
^10 дл/г (тетрахлорэтан, 25 °С). Межфазной иоликон-
иоликонденсацией дихлорангидрида изофталевой к-ты с диа-
ном получены Ц. марки Д-1 с т)пр— 1,7 дл/г (трикре-
зол, 25 °С), что соответствует мол. массе —160 000.
Свойства П. во многом определяются их химич.
строением. П. алифатич. дикарбоновых к-т в большин-
большинстве случаев размягчаются при сравнительно невысо-
невысоких темп-pax (напр., темп-ры размягчения II. себаци-
новой к-ты с гидрохиноном, дианом или фенолфталеи-
фенолфталеином составляют соответственно 164, 26 и 100 °С). В ряду
П. на основе янтарной, адипиновой и себациновой к-т
темп-pa размягчения понижается с увеличением числа
атомов углерода в к-те; так, темп-ры размягчения П.
гидрохинона и приведенных выше к-т составляют соот-
соответственно 230, 190 и 164 °С. П. фумаровой к-ты раз-
размягчаются при более высоких темп-pax, чем большин-
большинство П. полиметиленовых дикарбоновых к-т (напр.,
темп-pa размягчения П. фумаровой к-ты и фенолфта-
фенолфталеина 220 °С).
Наибольший практич. интерес представляют П.
ароматич. дикарбоновых к-т. Этим П. свойственна
высокая тепло- и термостойкость, хорошие диэлектрич.
показатели в широком диапазоне теми-р, устойчивость
к действию многих химич. агентов, УФ- и ионизирую-
ионизирующего излучения.
П. гидрохинона, 4,4'-диоксидифенила и таких к-т,
как терефталевая, изофталевая, а также гомополимер
/г-оксибензойной к-ты — кристаллич. высокоплавкие
полимеры, не растворимые в обычных органич. раство-
растворителях. При наиболее высоких темп-pax размяг-
размягчаются П. на основе мономеров, содержащих функцио-
функциональные группы в гсара-положении ароматич. ядра.
Напр., полигидрохинонтерефталат не плавится до
500 °С, темп-pa размягчения полигидрохинонизофталата
320 °С, П. марок Д-1 и Д-2 (П. диана и соответственно
изофталевой и телефталевой к-т), Ф-1 и Ф-2 — соответ-
соответственно 275, 350, 270 и 320 °С. Темп-ры размягчения
П. на основе мономеров, содержащих функциональ-
функциональные группы в гсара-положении ароматич. ядра, воз-
возрастают при увеличении в молекулах мономеров числа
ароматич. ядер. Так, П. диана и 4,4'-дифени л дикарбо-
дикарбоновой к-ты размягчается при темп-ре выше 400 °С.
Свойства П. дикарбоновых к-т и бисфенолов типа
I во многом определяются *—* ,—.
химич. строением А. Наибо- но—f у А \ V-ОН
лее подробно исследованы \=/ \=/
П. бисфенолов с терефтале- х
вой и изофталевой к-тами. П. 4,4'-диоксидифенилме-
тана (А=СН2) — кристаллич. высокоплавкие поли-
полимеры, плохо растворимые в органич. растворителях.
При замене атомов водорода у
центрального углеродного атома
бисфенола более объемными заме-
R
-*- ~R COOR ~ + R,N • НС1
С! +HOR—J
NR3
Нунлеофильный катализ
стителями (А=-
I
-G-
I
R'
темп-ры
757
ПОЛИАРИЛАТЫ
758
размягчения П. понижаются, уменьшается склонность
их к кристаллизации (вплоть до полной аморфи-
зации), улучшается растворимость. П. диана [А=
С(СН3J] с терефталевой и изофталевой к-тами еще
сохраняют склонность к кристаллизации и прио-
приобретают растворимость в нек-рых растворителях
(напр., в тетрахлорэтане, ж-крезоле, в смеси фенол —
тетрахлорэтан). П. бисфенолов, содержащих у цен-
центрального углеродного атома фенильный заместитель
[А=— СН(СбН5) —, —С(СН3)(С6Н5)],— аморфные поли-
полимеры, хорошо растворимые в хлороформе, трикрезоле,
тетрахлорэтане, тетрагидрофуране. Темп-ры размяг-
размягчения П. терефталевой к-ты и бисфенолов, у к-рых
А=-СН(С6Н5)-, -С(СН3)(С6Н5)-, _С(С6Н5J-, со-
составляют 220, 270 и 290 °С соответственно.
Введение в молекулу бис-
фенола алкильных заместите-
ле1ч в Орто_ПОЛОжение к гид-
роксильной группе приводит
к «внутренней пластифика-
пластификации» П. на их основе. П. терефталевой к-ты и таких
бисфенолов размягчаются при более низких темп-рах
(темп-pa размягчения П. диметилдиана 180 °С), харак-
характеризуются более высоким сопротивлением ударной
нагрузке, лучшей растворимостью и более легко пере-
перерабатываются в монолитные изделия, чем П. соответ-
соответствующих бисфенолов, не содержащих заместителей
в ароматич. ядрах.
П. бисфенолов, центральный углеродный атом к-рых
входит в состав объемной боковой циклической (кар-
довой) группировки, т. е. А имеет строение:
сочетают высокую теплостойкость с хорошей раствори-
растворимостью в органич. растворителях. Напр., П. фенолфта-
фенолфталеина растворимы в хлороформе, дихлорэтане, мети-
ленхлориде, диоксане, циклогексаноне, тетрагидрофу-
тетрагидрофуране и др. Большинство таких П. аморфно.
К кардовым П. по свойствам близко примыкают эле-
ментоорганич. П., содержащие в цепи о- и м-карбора-
новые группы. Для синтеза таких П. использованы
1,2- и 1,7-бмс-D-карбоксифенил)карборан и 1,2-бис-
(оксифенил)карборан. Такие П. в аморфном состоя-
состоянии растворяются в большинстве органич. растворите-
растворителей и размягчаются до 300 °С; в кристаллич. состоя-
состоянии они не плавятся до темп-ры их разложения
(>400 СС). О таких полимерах см. также Поликарбо-
раны, Борорганические полимеры. П. на основе кардо-
вых бисфенолов, содержащих у центрального углерод-
углеродного атома симметрично построенный циклич. замести-
заместитель, проявляют тенденцию к кристаллизации. Осо-
Особенно ярко это выражено у П. терефталевой к-ты и
9,9-бггс-D-оксифенил)антрона-10 (фенолантрона); струк-
структуру таких П. можно изменять, варьируя условия
синтеза или соответствующим образом обрабатывая
готовый полимер.
Свойства П. зависят также от их физич. структуры.
Так, если аморфный П. терефталевой к-ты и фенолан-
фенолантрона размягчается при 330—360 °С и хорошо раство-
растворяется в хлороформе, метиленхлориде, дихлорэтане,
тетрахлорэтане, циклогексаноне, смеси фенол — тетра-
тетрахлорэтан, диоксане и в ряде др. органич. растворите-
растворителей, то кристаллич. П. не размягчается до 400 °С
(вплоть до разложения) и растворяется только в смеси
фенол — тетрахлорэтан. Аморфные П., имеющие фибрил-
фибриллярную надмолекулярную структуру, характеризуют-
характеризуются лучшим комплексом физико-механич. свойств, чем
соответствующие полимеры, обладающие глобулярной
надмолекулярной структурой. Так, для П. марки Ф-1
(мол. масса 28 000) глобулярной структуры характер-
характерны: темп-pa размяг. 270 °С, прочность при растяжении
неориентированной пленки 64 Мн/м2 F40 кгс/см2),
относительное удлинение 10—20% ; для П. фибрилляр-
фибриллярной структуры: темп-pa размяг. 290 °С, прочность
при растяжении 74 Мн/м2 G40 кгс/см2), относительное
удлинение 80%, ударная вязкость 30 кдж/м2, или
кгс • см/см2.
Свойства смешанных П. зависят не только от строе-
строения исходных веществ, но и от их соотношения. Так,
темп-pa размягчения смешанных П. диана, адипиновой
и терефталевой к-т с увеличением содержания терефта-
терефталевой к-ты повышается от —60 °С до —350 °С. В ряду
смешанных^ П. резорцина, диана и терефталевой к-ты
наибольшей эластичностью (относительное удлинение
пленки 60%) обладает П., содержащий 60% (молярная
концентрация) резорцина.
При применении для синтеза П. соединений, содер-
содержащих хромофорные группы (например, II, где R = ж-,
П-СОС1; о-, 72-ОН; диоксиан- .—. .
трахинонов; 7,8-диокси-5,6- ( V—N=N—/ \
фталилхинолина) можно полу- ^^\=/ \==1>\^
чать окрашенные П. различ- 1Т
ных заданных оттенков, характеризующиеся высо-
высокой стойкостью окраски к действию света. В ря-
ряду смешанных П. на основе бесцветных и содер-
содержащих хромофорные группы мономеров интенсив-
интенсивность поглощения полимеров в видимой области
спектра изменяется пропорционально изменению со-
содержания в них окрашенного компонента. Таким сме-
смешанным П. свойственна достаточно высокая интенсив-
интенсивность окраски даже при сравнительно небольшом со-
содержании окрашенного компонента @,05—0,1 моль).
П. обладают высокой светостойкостью, к-рая может
быть дополнительно увеличена посредством включения
в полимерную цепь небольших количеств серы или
фосфора [при использовании для синтеза, наряду" с
обычными мономерами, таких соединений, как фенол-
сульфофталеин, дихлорангидрид окиси б^с-(д-карбокси-
фенил)метилфосфина и др.], а также осуществления
перегруппировки Фриса, приводящей к возникновению
в полимерной цепи фрагментов строения III:
Для ряда П. определены константы а и К в ур-нии
Марка — Хувинка: [ц] = КМа . Напр., для П. марок
Ф-2, Ф-1, изофталевой к-ты и фенолфлуорена, синтези-
синтезированных высокотемпературной поликонденсацией, а
и К составляют соответственно: 0,696 и 2,421 • 10~4; 0,53
и 16,70-Ю; 0,495 и 23,05-Ю (тетрахлорэтан, 25 °С)
и 0,596 и 6,357-Ю-4; 0,400 и 48,4-10~4; 0,407 и
50,24-Ю'4 (тетрагидрофуран, 25 °С). Для П. марки
Д-1, синтезированных методами высокотемпературной
и межфазной поликонденсации, а и К равны соответ-
соответственно: 0,605 и 9,44-10~4; 0,745 и 2,04-10~4 (тетрахлор-
(тетрахлорэтан, 25 °С).
П. обладают пленко- и волокнообразующими свой-
свойствами. Прочность при растяжении неориентированных
пленок П. составляет 60—100 Мн/м2 F00—1000 кгс/см2);
ее можно увеличить до 150—200 Мн/м2 A500 —
2000 кгс/см2) путем ориентации пленок в двух взаимно
перпендикулярных направлениях. Ценное свойство
пленок П.— способность сохранять хорошие механич.
показатели при повышенных темп-pax и после дли-
длительного нагревания. Так, прочность пленки П. марки
Ф-2 при 180 °С составляет свыше 50% от ее прочности
759
ПОЛИАРИЛЕНАЛКИЛЕНЫ
760
при 20 °С; прочность при растяжении пленки П. тере-
фталевой к-ты и фенолфлуорена при комнатной темп-ре
равна 84 Мн/м2 (840 кгс/см2), при 300 °С — 44 Мн/м2
D40 кгс/см2). Прочность пленок смешанного П. диана
с терефталевой и изофталевой к-тами не изменяется
после нагревания в течение 1000 ч при 180 °С.
60 100 140 180
Температура, °С
Зависимость напряжения
сжатия от темп-ры (скорость
нагрева 4 °С/мин), для по-
лиарилатов дихлордиана и
терефталевой к-ты, получен-
полученных акцепторно-каталитич.
полиэтерификацией в ди-
дихлорэтане при 30 °С A) и вы-
высокотемпературной поли-
полиэтерификацией в а-хлорна-
фталине при 220 °С B).
.1 кгс/см2 ^ 0,1 Мн/м2.
потерь П. марок Ф-1 и
+250 °С
Тангенс угла диэлектрич р
Ф-2 в интервале темп-р от —60 до +250 °С при частоте
10 кгц составляет E—8)-10~3, уд. объемное электрич.
сопротивление при 200 °С 1013—1014 ом-см. Следова-
Следовательно, по диэлектрич. свойствам и теплостойкости
П. превосходят полиэтилентерефталат и поликарбо-
поликарбонат диана.
П. ароматич. дикарбоновых к-т свойственна высокая
термостойкость. Согласно данным термогравиметрич.
анализа, П. терефталевой к-ты и диана, фенолфталеина,
фенолантрона или фенолфлуорена начинают разла-
разлагаться в инертной атмосфере при ~350—360 °С (ско-
(скорость повышения темп-ры 5 °С/мин). П., содержащие
1,2- и 1,7-дифенилкарборановые фрагменты в цепи,
начинают разлагаться при 400—410 °С. Как и др.
свойства, термостойкость П. зависит от степени кри-
кристалличности (зависимость носит сугубо кинетич.
характер). Потери массы высококристаллич. П. те-
терефталевой к-ты и гидрохинона обнаруживаются лишь
при 440 °С (термогравиметрич. анализ в атмосфе-
атмосфере гелия). Высококристаллич. поли-(га-оксибензоат),
получаемый из /г-оксибензойной к-ты и известный под
названием «эконол», по термостойкости превосходит
полиимиды и пирроны. В изотермич. условиях потери
массы для этого П. (порошкообразного) составляют
(в %/ч): 0,06 C16 °С), 0,1 C43 °С), 0,5 D00 °С), 3,0
D55 °С). Темп-pa длительной работоспособности эко-
нола на воздухе 315 °С.
П. устойчивы к длительному воздействию разб.
водных р-ров минеральных и органич. к-т, нек-рых
окислителей, петролейного эфира, бензина, керосина и
не стойки к действию р-ров щелочей, конц. H2SO4?
аммиака, диметилацетамида, диметилформамида.
Высококристаллич. поли-(д-оксибензоат) устой-
устойчив также к действию углеводородов, хлоруглево-
дородов, спиртов, простых и сложных эфиров, ке-
тонов, фенолов; он слегка набухает после нагре-
нагревания при 320 °С в течение 24 ч в хлорированном
дифениле (соволе) и разрушается при нагревании
под действием конц. H2SO4 и конц. щелочи.
При нагревании с органич. к-тами, гидроксилсодер-
жащими соединениями, эфирами П. способны к обмен-
обменным деструктивным реакциям алкоголиза, ацидолиза,
эфиролиза.
При нагревании в присутствии катализаторов Фриде-
ля — Крафтса или под действием УФ-излучения П.
претерпевают перегруппировку Фриса. Линейные тер-
термопластичные П. способны переходить в трехмерное
состояние пги взаимодействии с формальдегидом или
новолаком. Термореактивные П., содержащие в макро-
макромолекуле реакционноспособные группы (напр., гидро-
ксильную, двойную связь), способны к различным
химич. превращениям по этим группам.
Акцепторно-каталктич. полиэтерификацией на основе
о,о'-дизамещенных двухатомных фенолов впервые по-
получены нового типа стереорегулярные поликонденса-
поликонденсационные П., представляющие собой полимерные кон-
формеры (поворотные изомеры). Соотношение таких
изомеров в молекулах П., зависящее от природы ре-
реакционной среды и третичного амина, влияет на физи-
ко-механич. свойства П. Кривые релаксации напряже-
напряжения, измеренной на блочных образцах этих П., имеют
не один, а два максимума и, соответственно, два участ-
участка быстрого уменьшения напряжения (см. рис.).
Переработка и применение. П. можно перерабаты-
перерабатывать литьем под давлением (марки Д-4, Д-4С, Ф-1),
литьевым и компрессионным прессованием (марки Ф-2
и Д-9 — на основе фенолфлуорена). Растворимые П.,
прежде всего кардового типа, можно перерабатывать
из р-ров в пленки и волокна. Кардовые П. хорошо сов-
совмещаются с нек-рыми гетероцепными полимерами.
Изделия из практически неплавкого поли-(га-оксибен-
зоата) формуют спеканием порошкообразного полимера
при 425—450 °С и давлении 35—140 Мн/м2 C50-
1400 кгс/см2). Его подвергают также горячей (адиабати-
(адиабатической) ковке. При этом заготовку нагревают до 150 °С
и проковывают в течение 6—10 сек с общим расходом
механич. энергии 1400—14 000 кгм; выделяющегося
тепла достаточно для поддержания необходимой
темп-ры. П. можно напылять газопламенным, а эконол
и плазменным (в атмосфере гелия) методами.
П. находят применение как конструкционные изде-
изделия, антифрикционные самосмазывающиеся пластмас-
пластмассы (при наполнении графитом, дисульфидом молибде-
молибдена, нитридом бора, фторопластами), в виде пленок,
фильтрующих материалов Петрянова и др.
Лит.: К о р ш а к В. В., В и н о г р а д о в а С. В., По-
лиарилаты, М., 1964; их же, Polyesters, N. Y.—L. La. о.],
1965; их же, Неравновесная поликонденсяция, М., 1972;
Коршак В. В., Термостойкие полимеры, М., 1969, с. 132;
Аскадский А. А., Физико-химия полиарилатов, М.,
1968; Виноградова С. В., Выгодский Я. С, Усп.
химии, 42, № 7, A973); Морган П. У., Поликонденсационные
процессы синтеза полимеров, пер. с англ., Л., 1970; Вино-
Виноградова СВ., в кн.: Технология пластических масс,
под ред. В. В. Коршака, М., 1972, с. 408; KorshakV. V.
[а. о.], J. Polymer Sci., 10В, 429 A972). С. В. Виноградова.
ПОЛИАРИЛЕНАЛКИЛЕНЫ — см. Полиалкилен-
арилены.
ПОЛИАРИЛЕНСУЛЬФОНЫ [poly(arylene sulpho-
nes), Polyarylensulfone, polyarylenesulfones] — полиме-
полимеры общей ф-лы:
[-ArSO2Ar'SO2-]n
I
где Аг и А г' — ари леновые группы (одинаковые или
различные), напр.:
XJ.OO О-б.-О-~О
s—f У- —/ \-co-< V-
\=/ , \=/ \_/ ,
К П. также относят полиэфирсульфоны общей ф-лы:
"Л Г
где Аг =
с(сн3J,
(x«o,s,ch2,
C(CF3J
.С(С6Н5J,С(СН3)(СВН5) и др.)
Получение. Полиариленсульфоны (I)
получают поликонденсацией по реакции Фриделя-
Крафтса ароматич. моносульфонилхлоридов A) или
761
ПОЛИАРИЛЕНСУЛЬФОНЫ
762
ароматич. дисульфонилхлоридов с ароматич. углево-
углеводородами B):
п HArSO2Cl -» [ - ArSO2 - ]п + п HG1 или п HArSO2Cl +
+ mHAr'SO1Gl-*[-ArSO,-]n-[-Ar'SO1-]m + (n+m)HQ A)
п ClSO2Ar'SO2Cl + п ArH2 -> [ — ArSO2Ar'SO2 — Jn + 2n HG1 B)
Реакцию проводят в расплаве при 230—320 °С, а
также в р-ре сероуглерода, нитробензола или хлори-
хлорированного дифенила при 45, 120—140 и 160 °С соответ-
соответственно. Наиболее эффективные катализаторы — FeCl3,
SbCl5, InCl3. В присутствии каталитических коли-
количеств (до 4%) этих веществ реакция протекает количе-
количественно.
Мономеры, содержащие электроноакцепторные мости-
ковые группы, напр. —SO2— или —СО — , а также мо-
мономеры, у к-рых обе сульфонилхлоридные группы на-
находятся в одном фениленовом кольце, не образуют
полимеров высокой мол. массы. При поликонденсации
моносульфонилхлоридов присоединение протекает ис-
исключительно в паря-положение (>99%, по данным
ЯМР). Поликонденсация дисульфонилхлоридов с аро-
ароматич. углеводородами, не содержащими электроноак-
цепторных групп, дает смесь (80 : 20) пара- и орто-
замещенных структур. Снижение способности арома-
ароматич. кольца после присоединения группы —SO2—
к дальнейшему замещению приводит к образованию
практически линейных П., однако с увеличением мол.
массы полимера возможно сульфонирование и основ-
основной цепи, причем вторично замещаются преимущест-
преимущественно орmo-замещенные фенильные кольца. Содержа-
Содержание разветвленных звеньев достигает 1,6%.
Побочные реакции: поликонденсация ароматич. цик-
циклов под действием к-т Льюиса (напр., FeCl3) и окисли-
окислителя с образованием полифениленовых блоков (см.
Полифенилены), ответственных за темную окраску П.;
десульфонирование, также приводящее к полифениле-
новым цепям.
Последняя реакция сводится к минимуму при раз-
разбавлении сульфонилхлорида растворителем, а также
в присутствии меди или нек-рых ее солей и катализи-
катализируется следами А1 или Fe.
Полиэфирсульфоны (II) получают поли-
поликонденсацией щелочных солей дифенолов с ароматич.
дигалогенидами, в к-рых атомы галогена активированы
электроноакцепторной группой —SO,—:
Ar(OMe) +X
—ОАгО
2МеХ
\=/
где Me = Na, К или Cs; X = F, C1 или Вг.
Процесс проводят в апротонных биполярных раство-
растворителях (наилучшие — диметилсульфоксид и тетраме-
тиленсульфон) при 130—140 °С в отсутствие катализа-
катализатора. Реакция протекает быстро и практически не со-
сопровождается побочными процессами. Реакционная
способность фенолятов возрастает с увеличением основ-
основности фенолов. Фторпроизводные дигалогениды актив-
активнее хлорпроизводных, а цезиевые и калиевые соли
дифенолов активнее натриевых. Наличие примесей
воды и др. нуклеофилов (напр., спиртов) приводит
к уменьшению мол. массы П. Применение меди, ее
окислов или солей как катализатора ускоряет реак-
реакцию; использование этих соединений необходимо при
поликонденсации сильнокислых фенолов. При наличии
следов щелочи происходят гидролиз дигалогенида и
расщепление полимерной цепи по кислородному мо-
мостику.
Этим методом можно получить также I (Аг = Аг' =
= и, га'-дифениленоксид), однако более низкой мол.
массы, чем при поликонденсации по Фриделю —*
Крафтсу.
Свойства. П.— твердые, в основном аморфные термо-
термопласты белого цвета. Темп-ры стеклования П. до 250 °С
(табл. 1). В зависимости от метода получения мол.
масса П. может изменяться от 3 до 230 тыс. При при-
приведенной вязкости 1%-ного р-ра П. в диметилформ-
амиде, большей 0,4 дл/г, темп-pa стеклования П. не
зависит от их мол. массы.
Таблице
1 1.
-с
/—\
Cl/
-о-/~Л-
К>
Температуры стеклования
Полиариленсульфоны
кнн
Ю-О-—
п
¦-O--CW-O
-«снл-О-о-О-80!
\—/ \—/
С1
-(снЛ-С^Оо
х=сн2
с(сн3J
C(CF3J
С(СН3)(С6Н5)
с(с6н5J
б
0
со
S
so2
полиариленсульфонов
¦О!.
'п
Температура
стеклования,°С
250
240
C75-410)*
210 C10)*
205
235
180
195
205 B55)*
200
230
205
250
180
205
175
245
* Кристаллизуется, в скобках приведена темп-pa плавления
кристаллич. фазы.
П. нерастворимы в воде и простых органич. раство-
растворителях; растворяются в полярных ароматич. и хло-
хлорированных углеводородах и апротонных биполярных
растворителях; устойчивы к действию щелочей, силь-
сильных минеральных к-т и р-ров минеральных солей,
растворимы в конц. H2SO4, к-рая, однако, сульфирует
и расщепляет цепь П.
П. стабильны при нагревании на воздухе до 400 °С
(II не изменяются при многократном плавлении при
300—400 °С). Быстрое разложение начинается при
460—500 °С как на воздухе, так и в аргоне, и в вакууме.
При этом прежде всего выделяется SO2 и образуется
полифениленовая цепь. При наличии в цепи групп
—СН3 возможно их отщепление или разрыв по С—С-
связи между алифатич. мостиком и фениленовым цик-
циклом. Дальнейший распад протекает по связям С—О—С
с образованием полифенолов сложного строения. П.,
содержащие в цепи мостики —О— и —SO2—,
763
ПОЛИ(АРОИЛЕН-бмс-БЕНЗИМИДАЗОЛЫ)
764
Таблица 2. Механические
и теплофизические свойства
полиариленсульфонов зарубежных марок
Свойства
Плотность, г/см3
Показатель преломления п^
Прочность, Мн/м2 (кгс/см2)
при растяжении
при изгибе
Модуль упругости,
Мн/м2(кгс/смг)
при растяжении
при изгибе
Относительное удлинение, %
Ударная вязкость по Изоду с
надрезом » B3 °С), кдж/м2,
или кгс-см/см
надрез V4 ч. бруска . . .
надрез i/8 ч. бруска . . .
Деформационная теплостой-
теплостойкость (нагрузка 1,85 Мн/м2,
или 18,5 кгс/см2), "С ....
Твердость по Роквеллу, шка-
шкала R
Коэфф. трения
по стали
по полимеру
Теплопроводность, вт/{м • К)
[кал/(см-сек-сС)'\
Усадка при формовании,мм/мм
Водопоглощение при 23 СС за
24 ч, %
Арилон
1 ,14
—
52,5E25)
77G70)
2-1 О13
B-1 О14)
1 9 • 1 О3
(Г, 9-1 О4)
—
24,4
24,4
149
117
—
0,007
Поли-
Полисульфон
1,24
1,633
71 ,5G15)
108A080)
2,5-Юз
B,5-104)
2 7-Ю3
B ,'7-1 О4)
50-100
3,0
4,0
174
120
0 4
0,67
4,5[0,24]
0, 007
0, 22
Астрел
360
1 ,36
91(910)
120A200)
2,6-Ю3
B, 6-104)
2 8-Ю3
B', 8-Ю4)
10
10-20*
275
—
—
0,008
1,8
* Без надреза.
Таблица 3. Диэлектрические свойства полиариленсульфонов
зарубежных марок
Диэлектрич. проницаемость
при 6 0 гц и 23 еС ....
при 60 гц и 150 СС . . . .
при 1 Мгц и 23 СС . • . .
Уд. объемное электрич. сопротив-
сопротивление, ом-см
23 еС
50 СС
225 ^С
Тангенс угла диэлектрич. потерь
при 60 гг/ и 23 СС
при 60 гц и 225 СС
при 1 Мгц и 23 °С
Диэлектрич. прочность, кв/м, или
в/мм
Поли-
Полисульфон
3,14
2,98
3,10
9,1-10й
2,2-Ю14
0,0003
0,0034
425
Бис-S-
полиэфир
Астрел
360
3,92
3,45
3,62
8,9-1016
3,1-1016
0,00092
0,0204
0,0257
3,94
3,2-1016
14000
более термостабильны, чем П., содержащие лишь
мостики — SO2-*-.
Физич. и электрич. свойства П. очень незначительно
изменяются в широком интервале темп-р. Так, свойст-
свойства II, применяемого в пром-сти под названием «поли-
«полисульфон» (Аг = п, га'-дифенилендиметилметаы, мол. м.
25—60 тыс.), не изменяются в интервале от —100 до
175 СС, а также при длительном нагревании при 140 °С
на свету. В табл. 2 приведены нек-рые свойства вы-
выпускаемых в США самозатухающих П. марок «п о-
лисульфо н», «астрел 360» и «а р и л о н»
(последний относится к группе полиэфирсульфонов;
точная ф-ла и метод получения его не раскрыты).
Полисульфон и арилон по механич. свойствам близки
поликарбонатам, однако значительно менее склонны
к ползучести (см. рисунок). П.— хорошие диэлектрики
(табл. 3).
Астрел 360 (видимо, I, Аг = м,м'-дифенилен,
Аг' = гс-дифениленоксид; получен по Фриделю —
Крафтсу) можно эксплуатировать кратковременно при
350 °С и длительно при 260 °С [при 280 °С модуль
упругости при сжатии 900 Мн/м2 (9000 кгс/см2) и при
изгибе 1700 Мн/м2 A7 000 кгс/см2)].
Полисульфон, армированный ^30% короткорезан-
ного стекловолокна, имеет модуль упругости при рас-
растяжении 7600 Мн/м2 G6 000 кгс/см2), относительное
удлинение 2%, плотность 1,4 г/см3.
Кривые ползучести для
образцов полисульфона,
полученных прессованием:
1 — 167 °С, макс, напря-
напряжение 7 Мн/м2 G0 кгс/см2);
2—111 °С, макс, напряже-
напряжение 21 Мн/м2 B10 кгс/см2);
3 — 40 °С, макс, напряже-
напряжение 28 Мн/м2 B80кгс/сж2);
1 кгс/см2 ^=0,1 Мн/м2.
100 1000
Время, ч
10000
Переработка и применение. Все П. можно перераба-
перерабатывать прессованием B00 °С для полисульфона, 360—
380 °С для астрела), литьем под давлением (темп-ра
расплава 345—400 °С, 230—330 °С и 360—440 °С,
темп-pa формы 100 СС, 80—100 СС и 230—260 СС соот-
соответственно для полисульфона, арилона и астрела 360)
и экструзией C15—370 °С и вплоть до 410 °С при экст-
экструзии тонких пленок из полисульфона). Волокна и
пленки формуют также из р-ров в хлороформе. Про-
Промышленные марки П. выпускают в виде прозрачных
бесцветных или окрашенных гранул.
Полисульфон применяют обычно без добавления
модификаторов и пластификаторов для изготовления
конструкционных деталей (для автомобилей, станков,
бытовых машин и т. п.), электротехнич. изделий, а
также изделий для упаковки пищевых продуктов, ме-
металлизированных матриц для типографских клише,
плит и труб, для приготовления клеев и лаков, связую-
связующих в производстве стеклопластиков. Из арилона из-
изготовляют корпуса электрохимич. батарей спутников,
трубопроводы для пищевой пром-сти, соединительную
арматуру конструкционных деталей, листы, трубы,
защитные шлемы.
Астрел 360 рекомендуют для изготовления электро-
электротехнич. изделий (катушки, выключатели, переключа-
переключатели и др.), а его модификацию астрел 380 — для полу-
получения пленок, волокон, клеев, лаков и покрытий.
Полисульфон выпускают в пром-сти США с 1965
(в 1967 5000 т, в 1970 ок. 6500 т).
Лит.: Johnson R. N. [а. о.], J. Polymer Sci., pt
А-1,5, №9, 2375A967); Ли Г., С т о ф ф и Д., Н е в и л л К.,
Новые линейные полимеры, пер. с англ., М., 1972; Rose J.B.,
Ghem. Ind., jSfi 15, 461 A968); J о п е s J. I., J. Macromol.
Sci., С 2, jsr» 2, 319 A968); Goethals E. J., Makromol.
Ghem., 109, 132 A967); Новые поликонденсационные полимеры,
под ред. 3. А. Роговина и П. М. Валецкого, М., 1969; В и-
gel Т. Е., WaltonR. К., Machine design, 37, № 17,
195 A965); Лапшин В. В., Пласт, массы, № 9, 74 A972);
Newton A., Rose J., Polymer, 13, 465 A972).
А. К. Зеленецкий.
ПОЛЩАРОИЛЕН-быс БЕНЗИМИДАЗОЛЫ) [ро-
ly(aroylene-fo>benzimidazoles), Polyaroylen-6w-benz-
imidazole, polyaroylene-6w-benzimidazoles] — полимеры
общей ф-лы I.
N
О
С
R
R и R' -
¦"XX
CH2,SO2,C = O или отсутствует)
765
ПОЛИ(АРОИЛЕН-бггс-БЕНЗИМИДАЗОЛЫ)
766
CH=N-/~~\— R'—.
(R'=H, C6H5)
Наиболее подробно изучены полибензимид-
азопирролоны, или и и р р о н ы (содержат в
макромолекулах о/?яго,ор/7го-радикалы R и FT), и и о-
ли(нафтоилен-бмс-бензимидазолы).
Получение. П. получают обычно полициклоконденса-
полициклоконденсацией диангидридов ароматич. тетракарбоновых к-т
с ароматич. тетрааминами в одну или несколько стадий.
В ходе реакции последовательно образуются полиами-
ноамидокислота (II), иолиаминоимид (III) и П. (IV):
cov
,co
NH2
+
>
УК
с0
NH2
^CO — NH-
4COOH
t
-2лН2О
О О
I! II
<сжс>
< Y У
NH2q О
\p/
N —
-2лН2О
Основная побочная реакция — взаимодействие сво-
свободного диангидрида с аминогруппами макроцепей II,
что приводит к образованию сшитых структур. Для
предотвращения сшивания используют мономеры с
двумя активными функциональными группами (напр.,
2,5-дикарбометокситерефталоилхлорид, 3,3'-динитро-
4,4'-диаминодифенилоксид, диацетильные производные
тетрааминов); две др. функциональные группы, необхо-
необходимые для циклизации в П., генерируются при химич.
обработке II или активируются при проведении второй
стадии реакции в более жестких условиях. В присут-
присутствии акцепторов HG1, напр, третичных аминов, вместо
свободных оснований тетрааминов можно использовать
их солянокислые соли.
При проведении полициклоконденсации в две
стадии на первой синтезируют II. Реакцию про-
проводят при темп-pax от —30 до 85 °С в сухих диметил-
формамиде, диметилсульфоксиде, диметилацетамиде
или N-метилпирролидоне в инертной атмосфере в те-
течение 2—10 ч при эквимолекулярном соотношении
реагентов или небольшом избытке диангидрида (общая
концентрация мономеров 4—15%). Мол. масса II
возрастает при введении в реакционный р-р неорганич.
солей, напр. LiCl.
Вторую стадию проводят в изделиях нагреванием
до 250—400 СС на воздухе или в вакууме A0~3—10 мм
рт. ст.; 1 мм рт. ст.= 133,322 н/м2). Скорость реак-
реакции снижается по мере увеличения жесткости цепей,
из-за чего при получении полибензимидазопирролонов
степень циклизации составляет только 60—80%. Сте-
Степень циклизации поли(нафтоилен-бггс-бензимидазолов)
достигает 100% благодаря тому, что шестичленный
гетероцикл образуется легче, чем, например, пяти-
членный; к тому же их молекулярная масса уве-
увеличивается вследствие поликонденсации по концевым
группам.
Одностадийный метод (без выделения II)
применяют гл. обр. для получения П. на основе шести-
членных диангидридов (напр., диангидрида нафталин-
1,4,5,8-тетракарбововой к-ты). Реакцию проводят
в полифосфорной к-те при 180—230 СС и эквимолеку-
эквимолекулярном соотношении исходных веществ (инертная атмо-
атмосфера, 8—12 ч). П. после выделения нагревают при
250— 350 °С в вакууме.
П. можно получать термич. обработкой солей типа
[(H2NJR(NHJ J.(ROOCJR'(COO-J]; горячим прес-
прессованием эквимолекулярных смесей мономеров при
450 °С и давлении 28 Мн/м2 B80 кгс/см2); проведением
реакции в расплаве мономеров C00 °С) или в р-ре ки-
кипящего пиридина либо диметилового эфира диэтилен-
гликоля B30—300 °С).
Свойства. П. на основе шестичленных диангидридов—
твердые неплавкие полимеры лестничной структуры.
Они глубоко окрашены, поскольку являются анало-
аналогами индантреновых красителей; растворимы в конц.
к-тах (серной, полифосфорной, метилсерной). В разб.
р-рах макромолекулы П. ведут себя как гибкие спи-
спирали с относительно свободным вращением вокруг
простых связей. П. из диангидрида нафталин-1,4,5,8-
тетракарбоновой к-ты и 3,3'-диаминобензидина имеют
мол. массу 100—170 тыс. ([ц] = 2,7—5,5 дл/г, H2SO4,
25 °С).
Полибензимидазопирролоны, как правило, неплавки
и нерастворимы, однако П., макромолекулы к-рых
содержат объемные боковые заместители, растворимы
в конц. к-тах и амидных растворителях.
П. стабильны к термоокислительной деструкции до
450—570 °С. Для полибензимидазопирролонов, неза-
независимо от структуры, максимальные скорости выделе-
выделения Н2О, СО и HCN наблюдаются при 390, 550 и
730 °С соответственно. При нагревании до 900 °С в
инертной атмосфере или в вакууме (в динамич. усло-
условиях) П. теряют в массе 10—30%.
В изотермич. условиях D00 °С) П. на основе нафта-
нафталин-1,4,5,8-тетракарбоновой к-ты не изменяются в те-
течение 7 ч. Волокна из таких П. можно длительно эк-
эксплуатировать при темп-pax до 370 °С; после нагрева-
нагревания при 800 °С в течение 1 мин их прочность при рас-
растяжении уменьшается на 50% . Прочность при растя-
растяжении стеклопластиков с такими П. в качестве свя-
связующего после 200 ч нагревания на воздухе при 230,
260, 290 и 315 °С составляет соответственно 97, 85, 55
и 15% от исходной; после нагревания 200 ч в инертной
атмосфере при 315 °С прочность уменьшается на 5 —7%.
Прочность при растяжении пленок из поли(нафтоилен-
б^с-бензимидазолов) при 20 °С составляет 110—
140 Мн/м2 A100—1400 кгс/см2), модуль упругости при
растяжении D—6)• 103 Мн/м2, или D—6)• 104 кгс/см2,
при 200 °С соответственно 80 Мн/м2 (800 кгс/см2) и
4-Ю3 Мн/м2 D-Ю4 кгс/см2); относительное удлинение 3
и 7% при 20 и 200 °С соответственно. Прочность и
гибкость волокон из П. невелики; так, прочность
при растяжении волокон 32—40 гс/текс C,5—
4,5 г/денъе), относительное удлинение 3—8%.
Прочность при сжатии пенопластов (плотность
0,48 г/см3) из полибензимидазопирролона на основе
767
ПОЛИАЦЕНАФТИЛЕН
768
диангидрида 3,3',4,4'-бензофенонтетракарбоновой к-ты
и 3,3'-диаминобензидина составляет 16 Мн/м2
A60 кгс/см2) при 20 °С и 10 Мн/м2 A00 кгс/см2) при
370 °С. Прочность при растяжении стеклопластиков
на основе указанного П. 3,5-102 Мн/м2 C,5-103 кгс/см2)
при 20 °С; модуль упругости при растяжении 2,8-
•104 Мн/м2 B,8-105 кгс/см2) при 20 СС; прочность при
изгибе 600 Мн/м2 F000 кгс/см2) при 20 °С, 500 Мн/м2
E000 кгс/см2) при 300 °С; прочность на сдвиг между
слоями 10 Мн/м2 A00 кгс/см2) при 20 °С. Для прессо-
прессовочных изделий прочность при изгибе 90 Мн/м2
(900 кгс/см2) при 20 °С; 80 Мн/м2 (800 кгс/см2) при
260 °С.
П.— абляционностойкие полимеры; они длительно
противостоят потоку нагретого D25 °С) воздуха со
скоростью 300 м/сек.
П. на основе диангидридов пиромеллитовой и 3,3%
4,4'-бензофенонтетракарбоновой к-т — хорошие ди-
диэлектрики; уд. объемное электрич. сопротивление
102 —106 Гом-м A013—1017 ом-см), электрич. прочность
при 25 °С 108 в/м A06 в/см), при 300 °С 107 в/м A05 в/см),
диэлектрич. проницаемость 2,3—3,2, фотопроводимость
в области длин волн 200—1300 нм при 25 и 300 °С
соответственно 10~13 и 10~8 —10~10 сим/м A0~15 и
100—10~12 ом-см*1). П. на основе нафталин- или
перилентетракарбоновых к-т и 3,3'-диаминобензидина
проявляют полупроводниковые свойства: энергия акти-
активации проводимости 0,3—0,5 эв, уд. проводимость
10-2—Ю-4 сим/м A0—10~6 ом^-см-1).
Облучение пленок полибензимидазопирролонов эле-
электронами с энергией 3 Мэв и суммарной дозой
21 000 Мрад (мощность дозы 5500 Мрад/ч) повышает
их прочность при растяжении на 13%, а облучение
суммарной дозой 58 000 Мрад снижает ее только на 10% .
Полиаминоамидокислоты имеют мол.
массу 7—20 тыс. ([ц] = 0,6 — 1,5 дл/г, в диметилформ-
амиде, 25 °С). Они растворимы в амидных раствори-
растворителях, а также в хлорфенолах, крезолах и др.
Лит.: Усп. хим., 40, № 3, 513 A971); J. Polymer Sci., A-l,
10, № 9, 2727 A972); Л и Г., Стоффи Д., Н е в и л л К.,
Новые линейные полимеры, пер. с англ., М., 1972.
Б. И. Западипский.
ПОЛИАЦЕНАФТИЛЕН— см. Аценафтилена поли-
полимеры.
ПОЛИАЦЕТАЛЬДЕГИД —см. Ацеталъдегида по-
полимеры.
ПОЛИАЦЕТИЛЕН— см. Ацетилена полимеры.
ПОЛИБЕНЗИМИДАЗОЛЫ (polybenzimidazoles, Po-
lybenzimidazole, polybenzimidazoles) — полимеры, со-
содержащие в основной цепи макромолекулы бензимид-
бензимидазольные циклы.
з Получение. П. получают гл. обр.
^ одностадийной полициклоконденсаци-
СН ей тетрааминов с ди-
NH^ карбоновыми к-тами
7 i или их производными,
а также производных карбокси-о-ари-
лендиаминов. Наиболее распространен-
распространенный способ — твердофазная полицикло-
полициклоконденсация ароматич. тетрааминов с
дифениловыми эфирами дикарбоновых
к-т или фениловых эфиров о-диамино-
замещенных карбоновых к-т, напр.:
Реакцию проводят в инертной атмосфере при 220—
280 °С A—2 ч) и затем в вакууме A0—10~6 мм рт. ст.;
1 мм рт. ст.= 133,322 н/м2) при постепенном повы-
повышении темп-ры до 300—400 °С (-6—10 ч).
Известны и др. методы синтеза П., из к-рых наиболее
интересны следующие:
1. Полициклоконденсация тетрааминов (в виде хлор-
гидратов) с дикарбоновыми к-тами или их производ-
производными (сложными эфирами, амидами, нитрилами) в
р-ре полифосфорной к-ты. Реакцию проводят при
длительном нагревании A00—200 °С) в инертной ат-
атмосфере. Полимер осаждают, выливая реакционный р-р
в воду.
2. Полициклоконденсация дииминоэфиров с тетра-
аминами или их хлоргидратами в диметилацетамиде
при 100—165 °С с последующим прогревом образую-
образующегося П. в вакууме при 250 °С.
3. Полициклоконденсация тетрааминов с тг-диацетил-
бензолом сначала в расплаве в инертной атмосфере
при 250 °С, а затем в вакууме при 300 °С для превра-
превращения образующегося полибензимидазолина в П. с
выделением СН4.
4. Полициклоконденсация амидов, напр. 3-амино-
4-анилинобензамида, способных к образованию рас-
растворимых и плавких до 350 °С П., в расплаве или в р-ре
при 150—300 °С.
5. Взаимодействие б^с-бензоиленбензимидазолов с
тетрааминами в р-ре полифосфорной к-ты при 140—
200 СС с последующим прогреванием П. в вакууме при
200—300 °С. Этим способом можно получить П., содер-
содержащие также боковые бензимидазольные заместители:
П., содержащие в цепи различные функциональные
группы, синтезируют поликонденсацией мономеров,
имеющих в молекуле бензимидазольный цикл. Поли-
имиды, содержащие бензимидазольные циклы, полу-
получают, напр., из соответствующих диаминов и диан-
диангидридов:
-N
y"V-c
NH
•NH2
+ О
NH
чсо
NH'
СО'
H, СН3или С6Н5
Трехмерные П. получают из полифункциональных
мономеров, напр, при взаимодействии тетрааминов с
фениловыми эфирами три- и тетрафункциональных
карбоновых к-т или путем сшивания линейных П.,
напр, при обработке р-ра П. формальдегидом с после-
последующим прогревом полимера в твердом состоянии
в вакууме.
Свойства. Большинство П.— бесцветные полимеры.
Однако П. с явно выраженной системой сопряжения в
цепи — продукты темного (вплоть до черного) цвета.
П. могут иметь кристаллическое или аморфное строе-
769
ПОЛИВЕ НЗИМИ Д АЗО Л Ы
770
ние. Так, П. на основе диаминобензидина и тсрефтале-
вой к-ты — кристаллич. полимер, а соответствующий
П. на основе изофталевой к-ты — аморфный. П. могут
кристаллизоваться под влиянием растворителей, спо-
способных, по-видимому, образовывать с П. кристалло-
сольваты. Для 1Юли-2,2/-(.ч-фенилен)-5,5/-дибензимид-
азола известны К и а в ур-нии Марка — Хувинка:
[г|] = КМа , равные 1,73-10~3 и 0,63 соответственно
(диметилацетамид, 30 °С).
Все линейные П. растворимы в конц. H2SO4. Многие
П. растворимы также в НСООН, днметилсульфоксиде,
диметилацетамиде, диметилформамиде и др., причем
добавление солей, напр. LiCl, в амидные растворители
повышает их растворяющую способность. Ароматич. П.
с мостиковыми связями между фенильными ядрами
[-(СН2)П-, -О-, -S-,-SO2-, -РО(СН3)-,сило-
ксановые] или с боковыми метильными, фенильными,
бензимидазольными заместителями в цепи, а также сме-
смешанные П. и П., содержащие др. функциональные
связи, напр, полиамидобензимидазолы, как правило,
хорошо растворимы в диметилсульфоксиде, диметил-
диметилацетамиде. Полностью ароматич. П. на основе тетраами-
нобензола или диаминобензидина и терефталевой или
нек-рых др. к-т практически нерастворимы в органич.
растворителях. Растворимость П. резко уменьшается
вследствие нарушения линейности П., к-рое может
произойти, напр., при нетщательном удалении воздуха
в процессе их синтеза.
П.— термостойкие полимеры (таблица). Замена во-
водорода в имидазольном цикле на фенил практически
не снижает термостойкости П., однако заметно умень-
уменьшает их теплостойкость.
Наилучшими прочностными показателями обладают
П. на основе алифатич. полиметиленовых дикарбоновых
к-т, напр, себациновой к-ты, затем следуют П., содер-
содержащие между бензольными ядрами мостиковые груп-
группы, например кислородные, и, наконец, аромати-
ароматические П.
П. характеризуются высокой гидролитич. устойчи-
устойчивостью, обусловленной стойкостью самого бензимид-
азольного цикла. Так, поли-2,2'-(ж-фенилен)-5,5'-ди-
бензимидазол не изменяется при кипячении в 70% -ной
H2SO4 или 25%-ном р-ре NaOH в течение 10 ч. На-
Начальная прочность на сдвиг клеевых соединений, вы-
выполненных внахлестку при помощи клея на основе
П., сравнима с прочностью эпоксифенольных клеев.
При повышенных темп-pax клеи из П. превосходят
по прочности эпоксифенольные, однако при длитель-
длительном воздействии высоких температур прочность кле-
клеевого шва уменьшается, особенно при склеивании ме-
металлов, содержащих железо, вследствие термоокисли-
термоокислительной деструкции П. Так, при нагревании в тече-
течение 100 ч при 300 °С прочность клеевого шва умень-
уменьшается в 2 раза. Клеи стабилизуют тиоарсенатом
мышьяка.
Стеклопластики со связующим на основе П. харак-
характеризуются высокой прочностью при статич. изгибе,
к-рая, однако, заметно уменьшается после длительной
выдержки при повышенных темп-pax, что обусловлено
как термоокислительной деструкцией, так и низкой
плотностью П- из-за образования летучих веществ при
отверждении. По термостабильности стеклопластики
на основе П. превосходят стеклопластики на основе
фенольного связующего, но уступают стеклопластикам
с полиимидным связующим. Электрич. свойства стекло-
стеклопластиков на основе П. почти не изменяются при высо-
высоких темп-pax (напр., при повышении темп-ры от 24 °С
до 315 °С ди;)лектрич. проницаемость изменяется от
4.ПО до 4,77 при 9,375 Мгц).
Эластичность пленок из П., содержащих полимети-
леновые участки в цепи, не изменяется при минусовых
темп-pax. Прочность при растяжении изотропной плен-
пленки из поли-2,2'-(октаметилен)-5,5'-дибензимидазола со-
составляет 120—130 Мн/м1 A200 — 1300 кгс/см2), относи-
относительное удлинение 18—20%. После ориентации проч-
прочность пленок возрастает до 500 Мн/м2 E000 кгс/см2);
пленки характеризуются хорошими диэлектрич. свой-
свойствами (тангенс угла диолектрич. потерь при 5-Ю5 гц
0,003; уд. объемное олектрнч. сопротивление 1016 ом-см;
электрич. прочность 200 кв/мм).
Прочность термостойких волокон из поли-2,2'-(ле-
фенилен)-5,5г-дибензимидазола составляет 52,3 гс/текс
E,81 г/денье), удлинение при разрыве 20%. После на-
нагревания на воздухе до 450 С прочность снижается
до 9 гс/текс (до 1 г/денье). Бог .:но обладает высо-
высокой огнестойкостью. Ткани из ;)т;,;о волокна впиты-
впитывают около 15% влаги, удобны в ::оске и стойки к ис-
истиранию.
Применение. П. используют для изготовления клеев,
лаков, пленок, волокон, связующих для стеклопласти-
стеклопластиков, абляционной теплозащиты, антифрикционных ма-
материалов и др. Для указанных целей используют гл.
обр. поли-2,2'-(ж-фенилен)-5,5'-дибензимидазол.
Стеклопластики на основе П. используются для из-
изготовления деталей ракет и самолетов; ткани из поли-
бензимидазольного волокна — для изготовления лет-
летных и др. специальных костюмов, наспинных ранцев,
привязных ремней для летчиков, надувных спасатель-
спасательных жилетов.
Свойства нек-рых полибензимидазолов
Полибензимидазол
NH
NH
'NH'
NH
-О),
Ц
II
1 , 10
14,0
3,34
1 ,00
470
(с разлош.)
500-600
300-400
-500
-500
сн3 сн3
I 3 I 3
Si—О—S
сн3 сн
1 NH/4/
i
с6н5
I I /"I
^-^NH 1
JkJw С
с6н5
^J "
0
1
2
0
,49
,26
,50
,86
420-450*
~390
~490
-300
>450
-5 00
490
-500
* Темп-pa плавления.
771
ПОЛИБЕНЗОКСАЗИНОНЫ
772
Для абляционной теплозащиты космич. кораблей
используют композиции на основе поли-2,2'-(л*-фени-
лен)-5,5'-дибензимидазола, а также сшитых П., полу-
получаемых введением в мономерную систему для синтеза
указанного П. сшивающих агентов (трифенилового
эфира тримезиновой к-ты или полифункционального
амина, образующегося при низкотемпературном окис-
окислении диаминобензидина). В последнем случае компо-
композиция обладает лучшей абляционной стойкостью. Для
получения термостойких антифрикционных материалов
(АСП-пластиков) применяют П. в смеси с наполните-
наполнителями, гл. обр. сульфидом молибдена. Связующее для
стеклопластиков и клей на основе поли-2,2'-(ж-фени-
лен)-5,5'-дибензимидазола выпускаются в США под
названием и м и д а й т.
Лит.: Коршак В. В., Термостойкие полимеры, М.,
1969; КардашИ. Е., Телешов Э. Н., Синтез, свой-
свойства и применение высокотермостойких гетероциклических по-
полимеров, М., 1971; Л и Г., Стоффи Д., Н е в и л л К.,
Новые линейные полимеры, пер. с англ., М., 1972; К о г-
shak V. V., Teplyakov M. M., J. Macroraol. Sci. (Revs.),
С5, № 2, 409 A971); Singleton R. W., Appl. Polymer
Symposia, № 9, 133 A969); Helminiak Т. Е., Ben-
nerC. L., GibbsW. E., Polymer Preprints, 11, № 1,
291 A970). M. M. Тепляков.
ПОЛИБЕНЗОКСАЗИНОНЫ (polybenzoxazinones,
Polybenzoxazinone, polybenzoxazinones) — линейные
полимеры, содержащие в основной цепи макромоле-
макромолекулы бензоксазиноновые циклы:
Получение. П. получают полициклоконденсацией аро-
матич. дикарбоновых к-т или их производных (хлоран-
гидридов, амидов, сложных эфиров, нитрилов) и аро-
матич. дикарбоксидиаминов или их хлоргидратов, со-
содержащих карбоксильные группы в ор/по-положении
к аминогруппам, в две стадии, напр.:
НООС
-2пНС1
Первую стадию — синтез п о л и а м и д о к и с-
л о т (I) — проводят в р-ре соединений амидного типа
(напр., в N-метилпирролидоне, N-метилкапролактаме,
гексаметилфосфортриамиде) с добавкой LiCl в течение
2—3 ч при 20—40 °С или в полифосфорной к-те; в этом
случае ступенчато повышается темп-pa от 60 до 140—
160 °С и реакционная смесь выдерживается при по-
последней 4—5 ч. Соотношение исходных веществ экви-
молярное. Значения логарифмич. приведенной вязко-
вязкости т|лог полиамидокислот, полученных обоими мето-
методами, составляют 1,50 и 2,60 (в конц. H2SO4) соот-
соответственно.
Низкотемпературной поликонденсацией хлорангид-
ридов дикарбоновых и 2,4-диаминобензойной к-т в
диметилацетамиде при 0 °С и концентрации исходных
веществ 1 молъ/л синтезированы полиамидокислоты
[выход 90% , приведенная вязкость г)пр 1,4 дл/г (в конц.
H2SO4)], из к-рых можно получить бесцветные, про-
прозрачные пленки с прочностью при растяжении 85—
110 Мн/м2 (850—1100 кгс/см2) и относительным удли-
удлинением 15 — 19%.
Вторую стадию — синтез П. (II) — проводят путем
нагревания полиамидокислот в различных условиях:
1) в вакууме при 180—360 °С в течение нескольких
часов, 2) в присутствии химич. агентов, напр, смеси
пиридина и уксусного ангидрида, при комнатной
темп-ре и кратковременном нагревании при ISO-
ISO °С, 3) в полифосфорной к-те при 220—250 °С в те-
течение 1—3 ч.
Свойства. П.— термостойкие неплавкие и нераство-
нерастворимые в воде и обычных органич. растворителях поли-
полимеры. По данным динамич. термогравиметрич. анализа,
потери в массе П. в инертной атмосфере и на воздухе
начинаются при 550 и 450 СС соответственно; потери
в массе П. на основе дикарбоксибензидина и хлоран-
гидрида изофталевой к-ты, нагретого до 900 °С в инерт-
инертной атмосфере и на воздухе, составляют 24 и 30% со-
соответственно. П. растворимы с разложением только в
конц. H2SO4 и дымящей HNO3; при комнатной темп-ре
устойчивы к действию 10%-ного р-ра NaOH и 3%-ной
H2SO4.
Полиамидокислоты — твердые вещества белого цве-
цвета, растворимые в N-метилиирролидоне и др. раство-
растворителях амидного типа с добавкой LiCl. Из р-ра в N-
метилнирролидоне полиамидокислоты I образуют про-
прозрачные прочные пленки. В отличие от полиамидокис-
полиамидокислот, получаемых при поликонденсации ангидридов
тетракарбоновых к-т и диаминов (см. Полиимиды),
I — гидролитически устойчивые соединения.
Лит.: Коршак В. В., Термостойкие полимеры, М.»
1969; Кудрявцев Г. И. [и др.], Высокомол. соед., 10Б,
295 A968); Л и Г., Стоффи Д., Н е в и л л К., Новые
линейные полимеры, пер. с англ., М., 1972. Я.С.Выгодский.
П0ЛИБЕН30КСА30ЛЫ (polybenzoxazoles, Poly-
benzoxazole, polybenzoxazoles) — полимеры, содержа-
содержащие в основной цепи макромолекулы бензоксазольные
циклы
7
Получение. П. получают по лицикло конденсацией
в одну или две стадии:
1) ароматич. бггс-(о-оксиаминов) и дикарбоновых к-т
или их производных:
ноч .он
X-R—X
NH,
ч°х°
яЧ
где R' — алифатич. или ароматич. радикал, X =
= С(О)ОН, C(O)Hal, C(O)OR, C(NH)OR'" и др.;
R"— алкил или арил, R"' — алкил.
2) ароматич. о-аминооксикарбоновых к-т или их
производных:
к-х
N
где X = С(О)ОН, С(О)ОСбН5, CN.
3) б^с-(о-ациламинофенолов) и дикарбоновых к-т или
их производных:
4NHC(O)R
О
+ X(O)C-R-C(O)X
кхн.
где R'— алифатич. или ароматич. радикал, R" = Н,
алкил или арил, X = ОН, OR'", OCOR"', NH2, Hal;
R'" — алкил или арил.
773
ПОЛИБЕНЗОКСАЗОЛЫ
774
При полициклоконденсации в расплаве в одну ста-
стадию мономеры нагревают в инертной атмосфере сначала
при ~200 °С, а затем при ~400 °С (иногда в вакууме).
Продолжительность реакции от нескольких ч до 2 су т.
тилпирролидоне). В результате (за 1—2 ч) образуются
поли-о-оксиамиды (I), к-рые на второй стадии подвер-
подвергают циклодегидратации при 200—400 °С с целью полу-
получения П. (II):
С(О)-
Мол. масса (логарифмич. приведенная вязкость г]лог
до 1) П., как правило, ниже, чем у П., синтезирован-
синтезированных др. способами. Получаемые П. обычно неплавки
и нерастворимы, вследствие чего не пригодны к пере-
переработке; лишь полимеры, содержащие алифатич. груп-
группы в основной цепи, можно перерабатывать из распла-
расплава. Этим способом синтезирован также ряд раствори-
растворимых в органич. растворителях П. строения:
R = C(CH3J, CH2,C(CH3)(C6H5) идр;
R = H,CH3>Cl; R —алифатич. или ароматич. радикал
обладающих низкой мол. массой (приведенная вязкость
Т]пР= 0,1—0,4 дл/г).
Полициклоконденсацию в полифосфорной к-те осу-
осуществляют также в одну стадию путем нагревания,
напр., дихлоргидрата ароматич. б^с-(о-оксиамина) с ди-
карбоновой к-той в течение 2—8 ч при 200 °С (в случае
ароматич. к-т) и при 100—120 °С (в случае алифатич.
к-т). По этому способу получаются П. высокой мол.
массы (г|лОг>2), однако также практически не пригод-
пригодные для переработки.
При синтезе П. в две стадии вначале проводят низко-
низкотемпературную @—20 °С) поликонденсацию, напр.
3,3'-диоксибензидина с дихлорангидридом изофтале-
вой к-ты в р-ре (напр., в Г^^-диметилацетамиде, N-ме-
Свойства. П.— как правило, кристаллич. полимеры.
Плотность ароматич. П. 1,35 — 1,40 г/см3 B0 °С). Боль-
Большинство ароматич. П. растворимо в конц. H2SO4 и не
растворимо в органич. растворителях. П., содержащие
алифатич. группы в цепи, растворимы также в ж-крезо-
ле и муравьиной к-те. П. обладают яркой люминесцен-
люминесценцией; р-ры их в конц. серной и полифосфорной к-тах
флуоресцируют.
Ароматич. П. устойчивы при нагревании на воздухе
(по данным термогравиметрич. анализа, нагревание со
скоростью —7 °С/мин) до —450 °С, в вакууме — до
— 500 °С. П. с алифатич. группами в цепи заметно раз-
разлагаются на воздухе уже при 300 °С. Термостойкость
ароматич. П. практически не зависит от положения
B,5 или 2,6) бензоксазольной группировки в макромо-
макромолекуле и от наличия в цепи фениленовых циклов. Аро-
Ароматич. П., содержащие в основной цепи атомы —О —,
—S— или группы —SO2 —, имеют более высокие на-
начальные скорости деструкции, а П., содержащие
SO2-rpynny,— несколько более низкие темп-ры начала
деструкции.
П. характеризуются довольно высокой гидролитич.
стойкостью; напр., пленка полимера II выдерживает
кипячение в 10%-ных водных р-рах H2SO4 и NaOH в те-
течение 4,5 ч. По гидролитич. стабильности П. заметно
уступают полибензимидазолам.
Полиоксиамиды (промежуточные продукты
поликонденсации) — линейные высокомолекулярные,
как правило, аморфные полимеры,растворимые в амид-
ных растворителях, диметилсульфоксиде, водных р-рах
щелочей, конц. H2SO4. Полиоксиамиды, содержащие
Свойства пленок из некоторых полиоксиамидов
НО ОН
—HN NH-C(O)—R'—С(О)-
1и полибензоксазолов I—/ ^r^ \— r'—I
R
XXX
XXX
xxx-
xwcc
R'
X7
-o-
Полиоксиамиды
логариф-
мич. при-
приведенная
вязкость
1,7
1,1
1.3
0,8
1,0
проч-
прочность* при
растяже-
растяжении, Мн/м2
165
160
145
НО
120
относи-
относительное
удлине-
удлинение, %
32
50
65
90
13
Полибензоксазолы
прочность*
растяжении,
20 °С
170
160
120
110
120
300 °С
90
47
40
3
50
при
Мн/м2
400 °С
40
17
-
--
16
относительное
удлинение, %
20 °С
7
8
25
Э0
12
300 °С
9
30
135
230
8
400 °С
150
100
-
-
100
темпера-
температура**
стекло-
стеклования/ С
425
370
285
240
-
* 1 Мн/м? с~ 10 кгс/см2. ** По данным термомеханич. исследования.
775
ПОЛИБЕНЗТИАЗОЛЫ
776
в цепи гетероатомы или группы, повышающие ее гиб-
гибкость (напр., О, S, SO2), могут растворяться также
в ж-крезоле, тетраметиленсульфоне и др. При создании
полибензоксазольной структуры прочность полиокси-
амидных пленок практически не меняется, уменьша-
уменьшаются относительное удлинение, модуль упругости
[напр., с 5900 Мн/м2 E9 000 кгс/см2) для I до 3100 Мн/м2
C1 000 кгс/см2) для II] и, как правило, плотность (см.
табл.). Прочность при растяжении и относительное уд-
удлинение не меняются после нагревания пленки полиме-
полимера II на воздухе при 400 °С в течение 3 ч, при 300 °С
в течение суток, при 400 СС в вакууме в течение более
200 ч.
Уд. объемное электрич. сопротивление пленки из
полимера II 1014 ом-см B50 °С), тангенс угла диэлект-
рич. потерь и диэлектрич. проницаемость соответствен-
соответственно 0,0025 и 3,5 B0 °С, 400—5000 гц), электрич. проч-
прочность — 24 кв/мм E00 °С).
Волокно из II характеризуется след. показателями:
Прочность, гс/текс
Относительное удлинение, %
Начальный модуль, Мн/м2 (кгс/мм2) . .
Сохранение прочности, % к исходному
значению
при 300 °С
при 400 °С
после нагревания при 3 00 сС в тече-
течение 48 -ч
30 — 50
1-18
15000-17000
A500-1700)
до 80
30
80
Волокно не претерпевает усадки после нагревания
при 300 °С в течение 48 ч и при 400 °С в течение 2,5 ч.
Лит.: КардашИ. Е., Телешов Э. Н., Синтез,
свойства и применение высокотермостойких гетероциклических
полимеров, М., 1971. В. С. Якубович.
ПОЛИБЕНЗТИАЗОЛЫ (polybenzothiazoles, Poly-
benzthiazole, polybenzthiazoles) — полимеры, содержа-
содержащие в основной цепи макромолекулы бензтиазольные
циклы:
Вторую стадию — полициклизацию преполимеров
с образованием П. (II) — проводят в твердой фазе при
150—300 °С в инертной атмосфере или в вакууме.
2. Одностадийной полициклоконденсацией хлоргид-
ратов о,о'-димеркаптодиаминов с дикарбоновыми к-тами
или их производными, а также аминомеркаптобензой-
ных к-т или их производных (сложных эфиров, амидов,
нитрилов, а также солей, напр, хлоргидратов, фосфа-
фосфатов, сульфатов, цинковых солей). Реакцию проводят
в полифосфорной к-те при длительном нагревании
A00—200 °С) в инертной атмосфере. Полимер осажда-
осаждают, выливая реакционный р-р в воду.
При использовании в качестве кислотных компонен-
компонентов дифениловых эфиров или иминоэфиров дикарбоно-
вых к-т реакцию можно осуществлять соответственно
в р-ре кипящего диэтиланилина B15 °С) с последую-
последующим нагреванием образовавшегося продукта при 400 °С
или в диметилацетамиде при 100 °С с последующим на-
нагреванием при 165 °С.
3. Полициклоконденсацией аминомеркаитобензойиых
к-т или их амидов, а также 2,2'-диметилтиазолиновых
производных в расплаве в инертной атмосфере при по-
постепенном повышении темп-ры до 300—400 °С или
при кипячении в органич. растворителе (ж-крезоле
или диметилацетамиде).
4. При постепенном нагревании (до 200 — 260 °С)
в инертной атмосфере метиламиноариленов, напр.
тг-то л ундина, с серой
сн,—
+ S
__h2S
С —
с-н
Процесс сопровождается побочными реакциями сши-
сшивания.
5. Взаимодействием терефталоилен-бггс-(бензимид-
азола) с о,о'-димеркаптодиаминами:
H2Nv
+ п
HS-
Получение. П. получают след. основными метода-
методами: 1. Двухстадийной полициклоконденсацией о,о'-ди-
меркаптодиаминов или их хлоргидратов с хлорангид-
ридами дикарбоновых к-т, напр.:
-2 л на
На первой стадии обычно осуществляют поликонден-
поликонденсацию в р-ре в амидных растворителях, напр. в диме-
диметилацетамиде, при темп-ре ниже 20 °С в сухой инерт-
инертной атмосфере. Амидный растворитель служит одно-
одновременно акцептором выделяющегося HCL Продукт
реакции (пренолимер) — полимеркаптоамид (I). Мож-
Можно осуществлять также межфазную поли конденсацию,
используя в качестве акцептора HG1 щелочь или Na2S.
В этом случае продуктом реакции является полиами-
нотиоэфир.
Реакцию проводят в р-ре полифосфорной к-ты при 140—
200 °С с последующим нагреванием образовавшихся
полимеров в вакууме при 200—300 °С. В полученных
П. содержатся боковые бензимидазольные циклы.
П., содержащие в цепи наряду с бензтиазольными
циклами др. функциональные группы, получают из
смеси соответствующих исходных компонентов, напр.
из о,о'-димеркаптодиамина и диамина, или из мономе-
мономеров, содержащих в молекулах бензтиазольные циклы,
напр, при получении полиимидобензтиазолов из 2,5-
диаминобензтиазола и пиромеллитового диангидрида:
О
17 О
777
ПОЛИВИНИЛАМИН
778
Свойства. П.— термостойкие полимеры от темно-
желтого до коричневого цвета (в зависимости от строе-
строения и методов получения). Полностью ароматич. П.
начинают разлагаться на воздухе при темн-рах выше
500 °С, П. на основе алифатич. к-т — при темн-рах
выше 300 °С (см. табл.). Ароматич. П., за исключением
П., содержащих в макромолекулах боковые бензимид-
азольные циклы, неплавки и не растворимы в органич.
растворителях (растворимы лишь в полифосфорной
и конц. серной к-тах).
П. с боковыми бепзимидазольными циклами раство-
растворимы в диметилсульфоксиде и гексаметилфосфорамиде.
П. на основе алифатических дикарбоновых кислот
растворяются обычно в муравьиной кислоте, крезоле
и растворителях амидного тина, например в диметил-
ацетамиде. П. обладают высокой гидролитической
устойчивостью. Например, вязкость поли-2,2'-(тг-фе-
нилен)-6,6'-дибензтиазола после кипячения при 120 °С
в течение 5 ч в 10 н. H2SO4 или 5 н. КОН снижается не-
незначительно (напр., приведенная вязкость от 1,02
до 0,61 и 0,97 дл/д соответственно).
Применение. П. на основе алифатических дикарбо-
дикарбоновых кислот можно рекомендовать для получения тер-
^ 1 мостойких рленок. П. общей ф-лы I
можно использовать в качестве абля-
ционностойкого материала. П. не об-
обладают хорошими адгезионными свой-
свойствами; свойства стеклопластиков на
их основе при темп-ре выше 360 °С
неудовлетворительны. Из растворов нолиамидобенз-
тиазолов, полученных на основе 3,3'-димеркаптобен-
зидина, ж-фенилендиамина и изофталевой кислоты,
можно формовать прочные эластичные пленки, стой-
стойкие к ультрафиолетовому излучению. Из нерчоторых по-
лиамидобензтиазолов получены термостойкие волок-
волокна с прочностью 20 гс/текс B,2 гуденье)', из полиими-
добензтиазолов, содержащих в цени регулярно рас-
расположенные бензтиазольные и имидные участки, изго-
изготовлены термостойкие и стойкие к ультрафиолето-
ультрафиолетовому излучению волокна с прочностью 42 гс/текс
D,7 г/денье).
Свойства некоторых полибензтиазолов
с—
I
Аминотиофенольный
компонент
HCI -H2N\^%
I 1
H2N^%y-CONH2
H2Nv/^ ^-yNH2
To же
To же
1 • 2 HCI
Кислотный компонент
HOOG-(GH2)8—COOH
HN NH
^C-(CH2)8-C^
Q,H3O OG2H5
C6H5OCO^^V ,^уСООС6Н5
KJ-o-kJ
hooc^^ycooh
Приве-
Приведенная
вязкость,
дл/г
2,03
0,31
1 ,33
0,31
0,59
0,50
0,43
0,47
1,51
Темп-pa
размяг-
размягчения,
LG
>450
— 250
-300
>400
527*
910*
Темп-pa нача-
начала разложе-
разложения на возду-
воздухе, СС (% ле-
летучих при на-
нагревании в ус-
условиях ТГА)
500
600
550
300
538 B,1);
593 D,8)
450
593 (8,3)
593 @)
* Темп-pa стеклования.
Лит.: Коршак В. В., Термостойкие полимеры, М.,
1969; Прогресс полимерной химии, 1968, под ред. В. В. Кор-
шака, М., 1969, с. 198; Фрейзер А., Высокотермостойкие
полимеры, пер. с англ., М., 1971; KorshakV. V., Tep-
1 у а к о v M. M., J. Macrorno]. Sci. (Revs.), C5, Xs 2, 409 A971);
Preston J. [a. o.l, Appl. Polymer Symposia, Л1> 9, 75, 145
A969); Коршак В. В., Максимов А. Д., Итоги науки,
сер. Химия, Химия и технология высокомол. соед., 3, М.,
1971, с. 60. М. М. Тепляков.
ПОЛИБИТИАЗОЛЫ — см. Лолитиазолы.
ПОЛИБУТАДИЕН — см. Бутадиена полимеры,
Бутадиеновые каучуки.
ПОЛИБУТЕН — см. Б у те но в полимеры.
ПОЛИВИНИЛАМИН (polyvinylaminc, Polyvinyl-
amin, polyvinylamine) — полимер общей ф-лы
[_СН2-СН-]П.
NH2
Свойства. П. — гигроскопичный порошок белого цве-
цвета, хорошо растворимый в воде, метиловом, этиловом
и бензиловом спиртах, а также в уксусной к-те. При
хранении на воздухе он поглощает нары воды (до 40%)
и углекислый газ F%). Мол. масса П. 5000—100 000.
П. обладает хорошей адгезией к стеклу, фарфору,
керамике и др. материалам. В результате нагревания
он начинает при 140 °С постепенно желтеть, при 170—
180 °С разлагаться с выделением аммиака (при этом
размягченный полимер затвердевает и становится не-
нерастворимым). Водные р-ры П., как и др. полиэлектро-
полиэлектролитов, показывают аномальную зависимость приведен-
ной вязкости от концентрации р-ра, что свидетельст-
свидетельствует о его значительной ионизации, обусловленной на-
наличием групп — NH2.
П.— слабое основание. Соли П. с HG1, НВг и НСЮ4,
получаемые сливанием водных или спиртовых р-ров П.
и соответствующих к-т,— кристаллич. вещества бе-
белого цвета, хорошо растворимые в воде; проявляют
свойства полиолектролитов. Соль П. с НС1О4 в сухом
состоянии при нагревании или ударе взрывается.
П. легко вступает во все реакции, свойственные пер-
первичным аминам: с алифатич. или ароматич. однооснов-
одноосновными к-тами образует поливинилалкил(арил)амиды,
с двухосновными к-тами — поливинилалкил(арил)ими-
ды (см. Винилимидов поли-
полимеры); с ароматич. и гете-
роциклнч. альдегидами —
полишиффовы основания.
Из П. и формальдегида в
муравьиной к-те (реакция
Эшвейлера— Кларка) син-
синтезирован и о л и - N - д и-
м е т и л в и п и л а м и нг
из П. и галогеналкилов—
п о л и - N - д и а л к и л-
в и н и л а м и н ы.
Поли- N - б е н з и-
л и д е н в и н и л а м и н,
п о л и - N - с а л и ц и л и-
деивиниламин и
поли-]М-фурфури-
лиденвиниламин
общей ф-лы:
[-СН2-СН-]П -
N=CH-R
порошкообразные полиме-
полимеры белого цвета, раство-
растворимые в бензиловом спир-
спирте, диметилформамиде и
уксусной к-те; темп-ра
стеклования 60—80 °С,
темп-pa текучести 180—
200 °С. Для них характер-
характерны химич. св-ва низкомо-
низкомолекулярных азометинов.
779
ПОЛИВИНИЛАНТРАЦЕНЫ
780
Поли^-диметилвиниламин
[—СН2—GH—]п — порошкообразный полимер белого
I
N(GH3J
цвета, хорошо растворимый в воде, низших спиртах,
пиридине и уксусной к-те. Он близок по свойствам по-
ливиниламину, но является более сильным основанием.
Получение. П. не может быть получен полимериза-
полимеризацией виниламина, т. к. последний в момент образования
изомеризуется в этиленимин. Поэтому его синтезируют
гл. обр. с помощью полимераналогичных превращений,
напр, гидролизом поли-]Ч-винилдигликольимида в вод-
водных р-рах минеральных или органич. к-т при нагрева-
нагревании до темп-ры кипения; сольволизом поли-Г^-винил-
фталимида гидразингидратом при темп-ре кипения
и поли-]Ч-винил-тре/?г-бутилкарбамата хлористоводо-
хлористоводородной кислотой в среде ледяной уксусной кислоты
при нагревании до 100 °С; обработкой полиакрилами-
да в водном растворе щелочью и бромом при слабом
нагревании.
Сополимеры виниламина с различными непредельны-
непредельными соединениями м. б. получены аналогичным образом
из соответствующих сополимеров N-винилфталимида
или Г^-винил-ягре/п-бутилкарбамата. Напр., синтези-
синтезированы сополимеры виниламина с виниловым спиртом
и дивинилбензолом.
Применение. Полимеры и сополимеры виниламина
могут представить интерес, напр., для получения рас-
растворимых полиэлектролитов, ионообменных смол, по-
поверхностно-активных веществ, пленок и волокон, обла-
обладающих высокой реакционной способностью по NH2-
группам, а также, возможно, и в синтезе лекарственных
веществ.
Лит.: Николаев А. Ф., Бондаренко В. М.,
в кн.: Химические свойства и модификация полимеров. Сб.
статей, М., 1964, с. 146; Николаев А. Ф., Бонда-
Бондаренко В. М., Высокомол. соед., 6, № 10, 1825 A964); 7,
№ 12, 2105 A965); 7, № 10, 1743 A965); SugiuraM.,
OchiM., TaniY., J. Chem. Soc. Japan Industr. Chem.
Sec, 72, № 8, 1926 A969); Л о м а к о Л. А., Т о л м а-
ч е в В. Н., Высокомол. соед., 13Б, 49 A971); Никола-
Николаев А. Ф., Терещенко Г. П., Бондаренко В. М.,
ЖПХ, 44, 1372 A971); G u i I b а и 1 t L. J., MuranoM.,
Н а г w о о d Н. J., J. Macromol. Sci.— Chem., A7, 1065
A973). А. Ф. Николаев.
ПОЛИВИНИЛАНТРАЦЕНЫ — см. Винилантра-
цена полимеры.
ПОЛИВИНИЛАЦЕТАЛИ — см. Ацетали поливини-
поливинилового спирта.
ПОЛИВИНИЛАЦЕТАЛЬНЫЕ ЛАКИ И ЭМАЛИ,
поливинилацеталевые лаки и эма-
л и (polyvinylacetal varnishes and enamels, Polyvi-
nylazetallacke und Emaillen, vernis et emaux des acetals
polyvinyliques) — лакокрасочные материалы на основе
ацеталей поливинилового спирта, гл. обр. поливинилфор-
поливинилформаля, поливинилформальэтилаля и поливинилбутира-
ля. В состав пленкообразующих для поливини л-
формалевых и поливинилформаль-
этилалевых лаков, кроме соответствующего
поливинилацеталя, входит резольная феноло-форм-
альдегидная смола. Растворителями при получении
поливинилформалевых лаков служат фенол, ди- или
трикрезол, разбавителями D0% в расчете на массу
летучей части) — сольвент, ксилол. Содержание плен-
пленкообразующих в лаке составляет 15—16%, соотно-
соотношение поливинилформаль: феноло-формальдегидная
смола — 2:1 (по массе). Такие лаки выпускают в
ряде стран: СССР — метальвин (ВЛ-941),
США — формекс, ФРГ — формадур, Фран-
Франция — формтеналь.
Лаки на основе поливинилформальэтилаля (фини-
флекса) м. б. получены без применения токсичных и де-
дефицитных фенолов. Напр., разработанный в СССР лак
ВЛ-931 содержит в качестве растворителя этилцел-
лозольв. Количество пленкообразующих в этих лаках
20—22% , соотношение растворитель : разбавитель
(хлорбензол, ксилол) —1:1.
Поливинилформалевый и поливинилформальэтила-
левый лаки применяют гл. обр. в кабельной пром-сти
для электроизоляции (т. наз. эмалирования) проводов.
Лаки имеют большую вязкость (до 800 сек по ВЗ-1).
Поэтому их наносят на проволоку с помощью калибров,
к-рые снимают избыток лака после прохождения про-
проволоки через лаковую ванну; иногда для этой цели
применяют фетровые обжимы. Отверждение покрытия
происходит в печи C00—500 °С) в результате взаимо-
взаимодействия гидроксильных групп поливинилацеталя с ме-
тилольными группами феноло-формальдегидной смо-
смолы (отверждению предшествует улетучивание раство-
растворителей).
Поливинилацеталевая изоляция отличается высокой
механич. и электрич. прочностью, эластичностью. Она
не разрушается при значительном растяжении прово-
провода; пробивное напряжение провода диаметром 1 мм
при толщине изоляции 0,06 мм составляет 3—5 кв.
Благодаря этим достоинствам провода с поливинил-
ацеталевой изоляцией получили массовое применение в
производстве электрич. двигателей, приборов и аппа-
аппаратов. Предельно допустимая темп-pa длительной
эксплуатации этих проводов 105 °С.
На основе поливинилформаля получают модифициро-
модифицированные лаки, к-рые, кроме феноло-формальдегидных
смол, содержат изоцианаты, меламино-формальдегид-
ные и эпоксидные смолы. Покрытия, образующиеся
при нанесении таких лаков, отличаются повышенной
стойкостью к действию фреонов (это позволяет исполь-
использовать их при изоляции проводов для холодильных
устройств), а также трансформаторного масла.
На основе поливинилбутираля приго-
приготовляют различные лаки и эмали. Растворителями при
их изготовлении служат смеси спиртов, альдегидов,
кетонов и сложных эфиров; иногда в такие смеси до-
добавляют в качестве разбавителей ароматич. углеводо-
углеводороды. Эмали, помимо пленкообразующих и раствори-
растворителей, содержат пигменты (напр., алюминиевую пудру,
красный железоокисный пигмент) и наполнители
(напр., тальк). Для получения эластичного покрытия
в состав лакокрасочной композиции добавляют пласти-
пластификаторы — сложные эфиры (фталаты, фосфаты), ка-
касторовое масло и др. На окрашиваемую поверхность
лаки и эмали наносят краскораспылителем, окунанием
и др. методами (см. Лакокрасочные покрытия).
Поливинилбутиральные лаки и эмали широко при-
применяют для изготовления бензо- и маслостойких и анти-
антикоррозионных покрытий по металлу. Напр., лак
ВЛ-51 на основе поливинилбутираля, крезоло-формаль-
дегидной смолы и моноглицерида льняного масла при-
применяют для антикоррозионной защиты металлич.
деталей. Эмаль ВЛ-515 на основе поливинилбутираля
и крезоло-формальдегидной смолы, содержащую крас-
красный железоокисный пигмент и тальк, используют для
защиты металлич. деталей от действия бензина, сма-
смазочных масел и горячей воды (90—95 °С). Для защиты
цветных металлов (например, магния, алюминия)
применяют эмаль ВЛ-725 на основе поливинилбути-
поливинилбутираля, меламино-формальдегидной и невысыхающей
алкидной смолы, пигментированную алюминиевой
пудрой.
Поливинилбутираль применяют также для изготовле-
изготовления фосфатирующих грунтовок, к-рые наносят на сталь,
цинк, алюминий, медь, олово, сплавы магния и др.
В состав грунтовок входят р-р поливинилбутираля,
хроматы (напр., тетраоксихромат цинка) и фосфорная
к-та. Фосфатирующие грунтовки поставляют обычно
в виде двух компонентов, к-рые смешивают перед упот-
употреблением. Первый компонент (основа) содержит сус-
суспензию пигментов в р-ре поливинилбутираля в органич.
растворителях, второй (кислый разбавитель) — спир-
781
ПОЛИВИНИЛБУТИРАЛЬ
782
товой р-р ортофосфорной кислоты. Соотношение между
основой и кислым разбавителем составляет от 4 : 1
(для черных металлов) до 10 : 1 (для цветных метал-
металлов). Жизнеспособность грунтов после добавления
кислого разбавителя — от 4 до 24 ч (при темп-pax от
-10 до 40 °С).
Лит.: П э й н Г. Ф., Технология органических покры-
покрытий, пер. с англ., т. 1 — 2, Л., 1959—63; Ушаков С. Н.,
Поливиниловый спирт и его производные, т. 1 — 2, М.— Л.,
1960; Николаев А. Ф., Синтетические полимеры и пла-
пластические массы на их основе, М.—Л., 1964; МайофисИ. М.,
Химия диэлектриков, М., 1970; Привезенцев В. А.,
Пешков И. В., Обмоточные и монтажные провода, М., 1971.
Е. С. Гуревич, И. М. Майофис.
ПОЛИВИНИЛАЦЕТАТ — см. Винилацетата поли-
полимеры.
ПОЛИВИНИЛАЦЕТАТНЫЕ КРАСКИ — см. Эмуль-
Эмульсионные краски.
ПОЛИВИНИЛВЕНЗОЙНАЯ КИСЛОТА — см. Ви-
нилбензойной кислоты полимеры.
ПОЛИВИНИЛБРОМИД — см. Винилбромида по-
полимеры.
ПОЛИВИНИЛБУТИРАЛЬ [poly(vinyl butyral), Po-
lyvinylbutyral, butyral polyvinylique] — карбоцепной
полимер, ацеталь поливинилового спирта и масляного
альдегида общей ф-лы:
[-сн-сн2-сн-сн2-]п
о—сн
G
о
GH2GH2GH3
Свойства. П.— аморфный полимер белого цвета.
Степень полимеризации П. составляет обычно 500 —
1600. В макромолекулах технич. П. содержатся винил-
бутиральные F5—78%), винилспиртовые C2—19%)
и винилацетатные (не более 3%) звенья. Нек-рые свой-
свойства технич. П. приведены ниже:
Плотность при 20° С, г/см3
2
Показатель преломления nr
Темп-pa стеклования, °С
Тй °С
p
Теплостойкость, °С
по Вика
по Мартенсу
Уд. теплоемкость, ?едж/(кг-К) [ккал/(кг-°С)]
Темп-рный коэфф. линейного расширения,
СС~1 •
Водопоглощение при 20 °С за 24 ч, % . .
Влагопроницаемость, кг/(м-сек-н/м2) . . .
г/(см>ч-мм рт. cm)
Прочность, Мн/м2 (кгс/см2)
при растяжении
при статич. изгибе .
Модуль упругости при изгибе, Мн/м*
(кгс/смг)
Ударная вязкость, кдж/м*, или кгс- см/см2
Относительное удлинение, %
Уд. электрич. сопротивление
поверхностное, Том(ом)
объемное, Том-м (ом-см)
Диэлектрич. проницаемость
при 1 пгц
при 1 Мгц
Тангенс угла диэлектрич. потерь
при 1 пгц
при 1 Мгц
Электрич. прочность, кв/мм
1,1
1 ,485
57
60-75
48-54
1,67@,4)
9,2-Ю
0,4—3,0
2 ,5-Ю-14
1,2-Ю-7
28,0-59,5
B80—595)
80-140
(800—1400)
2000-2200
B0 000-22 000)
60 — 130
15—25
>100(>10и)
>1(>10")
3,4
3,33
0,007
0,024
16
П. растворим в спиртах, кетонах, сложных эфирах,
хлорированных углеводородах, уксусной к-те, пири-
пиридине, в смесях растворителей (спирт — бензол, ди-
дихлорэтан — этанол, метиленхлорид — метанол и др.);
не растворим в бензине, керосине, жирах, этиловом
эфире. П. хорошо совмещается с пластификаторами
(напр., с дибутилфталатом, дибутилсебацинатом, три-
крезилфосфатом), природными смолами (шеллаком,
копалами), синтетич. смолами (феноло-, мочевино-, мел-
амино-формальдегидными и алкидными). П. средней
мол. массы совмещается с 16—18% пластификатора.
П. обладает очень хорошими оптическими свойства-
свойствами (прозрачностью, бесцветностью, светостойкостью),
высокой адгезией к углеродистой и нержавеющей ста-
стали, алюминию, цинку, кадмию, хрому, никелю, ме-
меди, оцинкованному железу, оксидированному алюми-
алюминию и магнию, стеклу, дереву, бумаге, тканям и
пластмассам.
Химич. свойства П. определяются наличием в его
макромолекулах прежде всего гидроксильных групп.
По этим группам П. может взаимодействовать с диэпо-
ксидами и диизоцианатами, с феноло- и мочевино-фор-
мальдегидными смолами, с многоосновными к-тами,
солями меди, хроматами и бихроматами металлов и др.
В результате образуются сшитые нерастворимые про-
продукты. При обработке минеральными к-тами в жестких
условиях (выше 100 °С) П. разлагается с выделением
масляного альдегида. П. отличается высокой атмосфе-
ростойкостью, стойкостью к действию солнечного света,
кислорода и озона, высокой устойчивостью при исти-
истирании. При нагревании выше 160 °С П. разлагается
с выделением воды и масляного альдегида. Кислород
воздуха способствует образованию гидроперекисей,
выделению масляного альдегида и структурированию
полимера. В П., подвергнутом термостарению, обна-
обнаружены группы >С—О —, >С=О и >С=С<. С по-
появлением системы сопряженных связей связано окра-
окрашивание полимера. Для замедления термич. распада
в П. вводят стабилизаторы, напр, азометины, фенолы,
производные салициловой к-ты, амины.
Получение. Синтез П. может быть осуществлен любым
из методов, используемых для получения поливинил-
ацеталей (см. Ацетали поливинилового спирта).
В пром-сти П. обычно получают конденсацией поли-
поливинилового спирта с масляным альдегидом в водной
среде в присутствии соляной к-ты как катализатора
и постепенном повышении темп-ры от 8 до 55 °С. Об-
Образующийся полимер выпадает из р-ра в виде мелко-
мелкодисперсного порошка, к-рый тщательно отмывают
водой от катализатора, нейтрализуют остаток к-ты
небольшим количеством щелочи и сушат.
П. получают также обработкой масляным альдеги-
альдегидом поливинилацетата, растворенного в органич. рас-
растворителе, в присутствии катализатора (минеральной
к-ты); образовавшийся полимер осаждают из р-ра водой.
Описано омыление и одновременное ацеталирование
масляным альдегидом дисперсий поливинилацетата
в воде в присутствии минеральных к-т.
Содержание ацетатных групп в П. определяют мето-
методом щелочного омыления в спиртовом р-ре, бутираль-
бутиральных групп — оксиметрич. методом (взаимодействием
с солянокислым гидроксиламином), а гидроксильных—
ацетилированием в смеси уксусного ангидрида и пири-
пиридина или методом фталирования.
Применение и переработка. Промышленность выпу-
выпускает П., различающиеся по содержанию бутиральных
групп и мол. массе. П. в виде пластифицированных пле-
пленок применяют для изготовления безосколочных стекол
триплекс, используемых в автомобиле- и самолетострое-
самолетостроении (см. Стекло многослойное). Пленки получают
экструзией полимера, предварительно пластифициро-
пластифицированного дибутилсебацинатом A6—18%) или триэтилен-
гликольди-2-этилбутиратом B8—30%). Полимер с пла-
пластификатором можно совмещать непосредственно в экс-
трудере.
Реже получают пленку методом полива из р-ра П.
с пластификатором в соответствующем растворителе
(низшие спирты и др.).
В виде спиртовых р-ров П. применяют для изготов-
изготовления клеев типа БФ; в состав этих клеев входит ре-
зольная феноло-формальдегидная смола (см. Феноло-
формалъдегидные клеи).
П. смешивают с нек-рыми синтетич. каучуками,
напр, с полиизобутиленом, хлоропреновым или поли-
изопреновым каучуком, для получения композиций,
783
ПОЛИВИНИЛДИФЕНИЛ
784
обладающих повышенными ударной вязкостью и мо-
модулем упругости; из них формуют изделия для машино-
машиностроения и электротехники. Методом экструзии из
композиций на основе П. изготавливают шланги, труб-
трубки, прутки.
Широко применяют П. для получения лаков и эмалей
(см. II оливинилацеталъные лаки и эмали). Методом га-
газопламенного напыления на металлич. изделиях полу-
получают антикоррозионные и декоративные покрытия из
П. Композиции П. с феноло-формальдегидной смолой
и наполнителем используют в автомобильной иром-сти
для образования нижних слоев лакокрасочных покры-
покрытий с целью выравнивания поверхности кузовов и ка-
кабин автомобилей. В виде р-ра в смеси спирта и этил-
ацетата П. применяют для получения противопригарных
покрытий на литейных формах. Водные дисперсии
П., содержащие пластификаторы, используют в каче-
качестве аппретуры для тканей, искусственного волокна и
хлопка.
За рубежом П. выпускают под след. фирменными
названиями: б у т в а р (Канада, США, Великобрита-
Великобритания), в и и и л и т, XYSG, XYHL, б у т а ц и т (США),
м о в и т а л ь В (ФРГ), и и о л о ф о р м В (ФРГ),
р е в и л ь В (Франция), ров и нал ь В (Франция),
S-1 о с В (Япония).
Впервые П. синтезирован В. О. Германом и В. Гене-
лем в 1927; первое промышленное производство начато
в Канаде и Германии ок. 1930.
Лит.: У ш а к о в С. Н., Поливиниловый спирт и его
производные, т. 1 — 2, М.— Л., 1960; Николаев А. Ф.,
Синтетические полимеры и пластические массы на их основе,
2 изд., М.— Л., 1966; Справочник по пластическим массам,
под ред. М. И. Гарбара [и др.], т. 1, М., 1967; Ethylene and its
industrial derivatives, ed. S. A. Miller, L., 1969, p. 1041.
M. Э. Розенберг.
ПОЛИВИНИЛДИФЕНИЛ — см. Винилдифенила
полимеры.
ПОЛИВИНИЛЕНГЛИКОЛЬ [polyfvinylene glycol),
Polyvinylenglykol, polyvinylene-glycol] — карбоцепной
полимер общей ф-лы
[-СН-СН-]
I i n
он он
П.— твердый аморфный полимер белого цвета. Мол.
масса зависит от степени полимеризации исходного по-
ливиниленкарбоната и может достигать 300 000. При
темп-pax выше 200 СС П. разлагается, не плавясь. П.
нерастворим в органич. растворителях и воде, раство-
растворим только в конц. водных р-рах щелочей; сильно по-
поглощает и удерживает воду. Прочность при растяжении
набухших в воде тонких полосок пленок П. после вы-
вытяжки их на 600—800% составляет 58 гс1 текс(Ь,Ьгс/ денъе).
При взаимодействии П. с уксусным ангидридом
в расплаве мочевины при 140 СС образуется иол и-
ацетоксиметилен — твердый аморфный по-
полимер, размягчающийся при темп-pax выше 200 °С;
из р-ров в ацетоне он образует хрупкие пленки. Слож-
Сложные эфиры П. и ароматич. к-т можно получить при про-
проведении реакции на границе раздела фаз (щелочной
водный р-р П.— углеводородный р-р хлорангидрида
ароматич. к-ты). Напр., с хлорангидридом д-толуило-
вой к-ты получено полимерное производное [степень
замещения ок. 70% (молярная концентрация), т. пл.
210—220 °С], образующее из р-ра в бензоле хрупкие
пленки. Синтезированы также нек-рые простые эфиры
и ацетали низкомолекулярного П. Так, при арилиро-
вании П. бензилхлоридом получен растворимый
в органич. растворителях полимер, содержащий боко-
боковые бензоксигрунпы [степень замещения 80% (моляр-
(молярная концентрация), т. размягч. 80 СС]. Полный мети-
метиловый эфир П. можно получить катионной полимери-
полимеризацией 1,2-диметоксиэтилена. Омылением сополимеров
виниленкарбоната с винилацетатом получены сополи-
сополимеры виниленгликоля с виниловым спиртом; их свой-
свойства подробно изучены.
П. синтезируют из поливиниленкарбоната (см. Ви-
ииленкарбоната полимеры) с помощью полимер анало-
аналогичных превращений — гидролизом разб. водными
р-рами щелочей при 5 —15 С или переэтерификацией
метанолом в присутствии метилата натрия при 50 — 60 °С
сн_сн-
| | \
0
|
0
CH3ONa+CH3OH
сн-сн
| |
ОН ОН
II
о
Оба процесса протекают в гетерогенных условиях.
Конечный продукт промывают водой до нейтральной
реакции.
Пленки и волокна П. получают омылением соответст-
соответственно поливиниленкарбонатных пленок и волокон.
Степень кристалличности ориентированных пленок П.
достигает 30%. Волокна из П. упрочняют, подвергая
их дополнительной ориентационной вытяжке в пере-
перегретом водяном паре. Пленки П. прозрачны; волокна
но внешнему виду похожи на вол.окна из поливинило-
поливинилового спирта, но характеризуются более низкими проч-
прочностью и удлинением.
П. предложено использовать в качестве высокоэф-
высокоэффективного избирательного сорбента для извлечения
бора из рассолов и природных вод, а также для произ-
производства волокон, нитрат П.— как компонент реактив-
реактивного топлива.
П. синтезирован впервые Ньюменом и Аддором
в 1953.
Лит.: Field N. D., SchaefgenJ. R., J. Polymer
Sci., 58, № 166, 533 A962); С к о р о х о д о в С. С, Ле-
Левин С. 3., Шапиро А. Л., Хим. волокна, До 4. 1 A963);
Encyclopedia of polymer science and technology, v. 14, N. Y.—
[a. o.], 1971, p. 498. С. С. Скороходов.
ПОЛИВИНИЛЕНКАРБОНАТ — см. Виниленкарбо-
Виниленкарбоната полимеры.
ПОЛИВИНИ ЛЕНЫ, полнены, п о л и м е т и-
н ы (polyvinylenes, Polyvinylene, polyvinylenes) —
олигомерные и полимерные соединения, основная
цепь к-рых содержит чередующиеся двойные и орди-
ординарные углерод-углеродные связи (см. Ацетилена
полимеры, Ацетиленовых соединений полимеры, Поли-
Полимеры с системой сопряжения).
ПОЛИВИНИЛ И ДЕНФТОРИД — см. Винилиденфто-
рида полимеры.
ПОЛИВИНИЛИДЕНХЛОРИД — см. Винилиденхло-
рида полимеры.
ПОЛИВИНИЛИДЕНХЛОРИДНЫЕ ПЛЕНКИ [ро-
ly(vinylidene chloride) films, Polyvinylidenchlorid-
filme, films de chlorure de polyvinylidene]. Для изго-
изготовления пленок обычно используют сополимеры винил-
идеихлорида G5—90%) с винилхлорндом, т. к. гомопо-
лимер винилиденхлорида плохо перерабатывается
в пленку из-за хрупкости и низкой термостабильности.
При содержании винилиденхлорида до 70% сополиме-
сополимеры имеют аморфную структуру, выше 70% — кри-
кристаллическую. У сополимеров с содержанием выше
75% винилиденхлорида резко падает растворимость
и повышается темп-pa размягчения. Напр., аморфный
сополимер с содержанием 75% винилиденхлорида раз-
размягчается при 110 СС, 80% — при 160 °С и 90% — при
190 °С.
Обычно композиция для производства П. п. содержит
не менее 90% сополимера. Кроме того, в ее состав вхо-
входят пластификаторы, стабилизаторы и иногда краси-
красители. Сополимеры винилиденхлорида с винилхлоридом
размягчаются при темн-рах, близких к темп-рам раз-
разложения, что создает большие трудности при их пере-
переработке. Введение в сополимер пластификатора умень-
уменьшает вязкость расплава и позволяет экструдировать
его при более низкой темп-ре, что гарантирует от раз-
разложения сополимера. Кроме того, пластификатор сни-
785
ПОЛИВИНИЛИДЕНХЛОРИДНЫЕ ПЛЕНКИ
786
жает хрупкость и модуль упругости пленок, а также
повышает их морозостойкость. Пластификаторов, спо-
способных придавать материалу все эти свойства одновре-
одновременно, не существует. Поэтому в композицию для про-
производства П. п. обычно вводят два или более различных
пластификаторов.
В качестве стабилизаторов используют динатрневую
соль этилендиаминуксусной к-ты, бутил га л лат, эпо-
ксидированные масла, оловосодержащие соединения,
стеараты Са, Cd, Pb и др. При изготовлении П. п. для
упаковки пищевых продуктов и медикаментов выбор
пластификаторов и стабилизаторов строго ограничен
требованиями санитарно-гигиеннч. контроля.
Для окрашивания П. п. в сополимер вводят 0,1 —
0,2% (по массе) индигокармина (натриевая соль дисуль-
фокислоты индиго), тартразина (натриевая соль произ-
производного сульфофенилазопнразона) и др. Для П. п.
технич. назначения можно применять анилиновые
и фталоцианиновые красители.
Ниже приведены типичные композиции для получе-
получения упаковочных П. п.: 1) сополимер (80% винилиден-
хлорида) — 93,5% , метилэтилглнколят — 2,5% , трет-
бутилфенилсалицилат — 2,5%, эпоксиднрованное сое-
соевое масло — 1,5%; 2) сополимер G5% винилиденхло-
рида) — 92%, тетранатрийпирофосфат — 0,3 —1,75%,
дпбутилсебацинат — 2—6% , дифенилмоноортоксилил-
фосфат — 2—4% .
П. и. получают экструзией с раздувом пленочного
рукава (см. Пленки полимерные). Материал экструди-
руется через кольцевую щель головки при 150—180 СС
в виде тонкостенной трубки, к-рую для предотвраще-
предотвращения кристаллизации сополимера быстро охлаждают
в ванне водой при 10—20 СС или водными растворами со-
солей с темп-рой 2—7 °С. Затем трубку из аморфного со-
сополимера раздувают воздухом иод давлением 7 ки/м2
@,07 кгс/см2), что приводит к ориентации пленки в по-
поперечном направлении. Образовавшийся пленочный
рукав диаметром ок. 30 см растягивают в продольном
направлении тянущими валками, складывают и нама-
наматывают в рулоны. Для предотвращения слипания пле-
пленочного рукава внутрь него иногда вводят эпоксиди-
рованное масло или этиленгликоль. Описанный спо-
способ производства дает возможность получить аморфную
или частично кристаллич. П. и.
Для получения П. п. со значительной степенью кри-
кристалличности разработан след. процесс (рисунок).
Экструдированную трубку зажимают в нескольких
местах валками и при нагревании раздувают воздухом,
Схема установки для производства термостабилизированной
поливинилиденхлоридной пленки: I — экструдер с кольце-
кольцевой головкой; 2 — охлаждающая ванна; 3 — тянущие вал-
валки; 4 — туннельная печь; 5 — рулон пленки; 6,7 — пленоч-
пленочные пузыри.
подаваемым на каждом участке отдельно специальными
шприцами (на рис. не показаны). После первого пленоч-
пленочного пузыря образуется П. п. такого же тина, как в пре-
предыдущем методе. Затем аморфная пленка в виде вто-
второго пленочного пузыря попадает в туннельную печь,
где поддерживается оптимальная для кристаллизации
темп-pa. Степень кристалличности П. п. зависит от про-
продолжительности термообработки, к-рая в свою очередь
определяется длиной туннельной печи (скорость движе-
движения пленки в агрегате постоянна). Основные парамет-
параметры процесса не отличаются от описанных выше, однако
давление воздуха во втором пленочном пузыре ниже,
чем в первом. Кроме того, частота вращения двух по-
последних пар тянущих валков выше, чем двух первых,
благодаря чему достигается ориентация пленки в про-
продольном направлении и предотвращается ее усадка при
нагревании в туннельной печи.
Аморфные П. п. подвергаются усадке, к-рая начи-
начинается при 50—60 °С и достигает максимума G0%)
при 80 'С. Напряжение усадки при 80 С составляет
1,3—1,5 Мн/м2 A3 — 15 кгс/см2). Кристаллич. П. п.
практически не усаживаются вплоть до темп-ры плав-
плавления A20 °С). Поэтому с увеличением степени кристал-
кристалличности темп-pa, при к-рой достигается максималь-
максимальная усадка, возрастает, в то время как значение .макси-
.максимальной усадки надает. Напр., при 100 СС усадка П. п.
составляет 30% .
П. п. прочны, эластичны, мало проницаемы для паров
и газов, практически не горючи. Они устойчивы к дей-
действию к-т и щелочей (кроме NH4OH), спиртов, масел и
большинства органич. растворителей (при комнатной
темп-ре). С повышением темп-ры растворимость П. п.
в органич. растворителях растет. Так, при 60 °С они
растворимы в тетрагидрофуране и циклогексаноне,
а при 100—110 СС также в хлорбензоле. П. п. прозрач-
прозрачны для УФ-излучения и видимого света. Ниже приве-
приведены нек-рые свойства П. п.:
Плотность, г/см3 1,60 — 1,75
Прочность при растяжении, Мн/м-
(кгс/см2) 40-100
D00 — 100 0)
Относительное удлинение, % 3 5 — 100
Морозостойкость, °С от —15 до -30
Водопоглощение за 24 ч, % 0,1 — 0,2
Показатель паропроницаемозти за 4 8 ч, г/м2
(г/дм2) 2 — 4@,02 — 0,04)
Коофф. газопроницаемости, м2/(сеп • н/м2)
[см2/(сек- кгс/см2)]
но азоту @,04 —0,06)-10-21
[@,4 — 0,6)-10~13
по кислороду @,12 —0,14)-10-"
[A,2—1,4) • 10-131
по углекислому газу @,4—0,6) • 10~21
[D,0-6,0). 10~13]
Пром-сть выпускает также двухслойные (с целлофа-
целлофаном или алюминиевой фольгой) и трехслойные (с поли-
полиэтиленом и полиэтилентерефталатом) пленочные мате-
материалы, в к-рые входит П. п. Сополимер винилидеихло-
рида с винплхлоридом экструдируют на целлофан или
алюминиевую фольгу через плоскощелевую головку,
плотно прикатывают роликами и немедленно охлаж-
охлаждают. Кристаллизацию сополимера осуществляют при
прогреве дублированного материала без вытяжки.
Трехслойную пленку производят экструзией через
трехщелевую головку.
П. п. хорошо свариваются при помощи токов высокой
частоты и ультразвука. Пригодны также термоконтакт-
термоконтактная, термоимпульсная и др. методы сварки, однако
они дают худшие результаты (см. Сварка). В связи
с узким интервалом между темн-рами размягчения и
разложения сополимеров винилиденхлорида с винил-
хлоридом необходим тщательный контроль режима
сварки.
Печатание на П. п. текстов и рисунков не вызывает
затруднений. Наиболее экономичный способ нолигра-
фич. оформления изделий из П. п.— флексограф-
ская печать с использованием высокопроизводитель-
высокопроизводительных ротационных машин. Для нанесения на П. и. ху-
художественных многоцветных рисунков применяют глу-
глубокую печать красками на летучих органич. раствори-
растворителях (см. Печать на полимерах).
П. п. применяют гл. обр. в пищевой пром-сти в виде
оболочек для колбас, а также для упаковки ветчины,
сыра, хлебобулочных, кондитерских и др. изделий.
787
ПОЛИВИНИЛИДЕНЦИАНИД
788
Мясные и рыбные колбасы упаковывают в рукавную
пленку толщиной 0,04—0,05 мм. Усадку пленки осу-
осуществляют при нагревании до 80—100 °С в течение
2—5 сек (более термостойкую П. п. для усадочной
упаковки не применяют). Минимальная кислородопро-
ницаемость П. п. обеспечивает длительную защиту
упакованных продуктов от окисления, а низкая па-
ропроницаемость — от высыхания. Плотная усадоч-
усадочная упаковка придает товарам привлекательный внеш-
внешний вид.
Для упаковки жиросодержащих продуктов рекомен-
рекомендуется применять пакеты из П. п., к-рые вкладывают
в бумажную тару для защиты от УФ-излучения, вызы-
вызывающего прогоркание жира.
П. п. и многослойные пленочные материалы на их
основе применяют также для вакуумной упаковки пи-
пищевых продуктов сублимационной сушки, фармацев-
тич. препаратов, микробиологич. сред и др.
П. п. выпускаются за рубежом под след. фирменными
названиями: саран, крайовак S (США), в е -
стан (ФРГ), курехалон (Япония) и др.
Промышленный выпуск П. п. был впервые осущест-
осуществлен в США в 1939.
Лит.: Такахаси Г., Пленки из полимеров, пер. с
япон., Л., 1971; Гордон Г. Я., Хлористый винилиден и
его сополимеры, М., 1957; Гуль В. Е., БеляцкаяО. Н.,
Пленочные полимерные материалы для упаковки пищевых
продуктов, М., 1968; Encyclopedia of polymer science and tech-
technology, v. 2, N. Y.— [a. o.], 1965, p. 345; v. 14, N. Y.—
[a. o.l, 1971, p. 540. Я. Г. Муравин, С.В.Генелъ.
ПОЛИВИНИЛИДЕНЦИАНИД — см. Винилиденциа-
нида полимеры.
ПОЛИВИНИЛИМИДАЗОЛЫ — см. Винилимидазо-
лов пол им ер ы.
ПОЛИВИНИЛИМИДЫ — см. Винилимидов поли-
полимеры.
ПОЛИВИНИЛКАРБАЗОЛ — см. Винилкарбазош по-
полимеры.
ПОЛИВИНИЛКЕТОНЫ — см. Винилкетонов по-
полимеры.
ПОЛИВИНИЛНАФТАЛИН — см. Винилнафталина
полимеры.
ПОЛИВИНИЛОВЫЕ ЭФИРЫ — см. Виниловых
эфиров простых полимеры, Виниловых эфиров сложных
полимеры.
ПОЛИВИНИЛОВЫЙ СПИРТ fpoly(vinyl alcohol),
Polyvinylalkohol, alcool polyvinylique] — полимер об-
общей ф-лы
[-сн2-сн-]п
он
Получение. П. с. нельзя синтезировать полимериза-
полимеризацией винилового спирта, т. к. последний в момент по-
получения изомеризуется в ацетальдегид или окись
этилена.
Наиболее распространенный способ получения
П. с— гидролиз или алкоголиз полимеров сложных
виниловых эфиров. В пром-сти П. с. получают алкого-
лизом, гл. обр. метанолизом, поливинилацетата (ката-
(катализатор — к-та или щелочь):
[-CH2-CH-]n -f- СНзОН -* [-СН2-СН-]П + СН3ОСОСН3
ОСОСНз ОН
В полученном П. с. обычно содержатся остаточные аце-
ацетатные группы, количество к-рых в зависимости от ус-
условий процесса может изменяться от 0,05 до 5,0% (по
массе).
Алкоголиз — автокаталитич. реакция, абсолютная
скорость ее увеличивается до степени превращения
ок. 40%. Энергия активации щелочного алкоголиза
54,47 кдж/молъ A3 ккал/молъ), кислотного 55,3 кдж/молъ
A3,2 ккал/молъ).
Обычно для осуществления алкоголиза используют
15—35%-ный метанольный р-р поливинилацетата и
10—20%-ный водный, водно-метанольный или мета-
метанольный р-р катализатора; реакционную смесь интен-
интенсивно перемешивают. Темп-ру реакции в зависимости от
вида аппаратуры варьируют в пределах 30—60 °С.
При омылении ок. 40% вини л ацетатных звеньев проис-
происходит переход полимера из растворимого в жидкой
органич. среде в нерастворимое состояние («точка ге-
геля»). Для получения полимера в виде мелкодисперсного
порошка или зерен используют аппараты с эффектив-
эффективными перемешивающими устройствами: реакторы ем-
емкостного типа с мешалками, шнековые агрегаты, аппа-
аппараты с фрезерно-шестеренными или кулачковыми ме-
мешалками и др.
Так, технологич. схема процесса алкоголиза, осу-
осуществляемого по непрерывной схеме в двухшнековом
горизонтальном агрегате, снабженном для обогрева
водяной рубашкой, заключается в следующем: 25%-ный
метанольный р-р поливинилацетата и 20% -ный водно-
метанольный р-р NaOH (из расчета 20 моль щелочи на
100 моль винил ацетатных звеньев) подаются на шнеки,
вращающиеся навстречу друг другу. Темп-pa реакции
48 °С; время полного омыления поливинилацетата
в П. с. 2,0—2,5 мин. Выходящий из агрегата продукт
представляет собой суспензию П. с. @,05—0,2% оста-
остаточных ацетатных групп) в маточном р-ре, к-рый со-
состоит из метанола, метил ацетата и ацетата натрия,
образующегося вследствие омыления едким натром
метилацетата. Суспензия поступает в центрифугу пе-
периодического, полунепрерывного или непрерывного
действия либо в отжимной шнек. Отжатый от маточного
р-ра П. с, содержащий 30—60% органич. летучих, су-
сушат в различных сушильных устройствах при переме-
перемешивании F0—80 °С, атмосферное давление или вакуум
200—600 мм рт. ст., 1 мм рт. ст. = 133,322 н/м2).
Готовый П. с, представляющий собой порошок или
зерна размером 3—5 мм, содержит 5—8% ацетата
натрия.
Для уменьшения содержания ацетата натрия П. с.
перед сушкой промывают метанолом (или этанолом).
Так, в результате последовательных трех промывок
П. с. пятикратным (по массе) количеством метанола
при 40 °С в течение 1 ч каждая количество ацетата
натрия снижается от 7,0 до 0,4—0,5%. Др. способ
уменьшения содержания ацетата натрия в П. с, ис-
используемом для получения его водных р-ров в тех
производствах, в к-рых невозможно применение мета-
метанола или этанола, заключается в промывке готового
высушенного П. с. холодной водой при комнатной
темп-ре в течение 0,5—1,0 ч. Перед промывкой полимер
должен быть подвергнут обработке с целью уменьше-
уменьшения степени набухания его в воде. Существует несколь-
несколько способов такой обработки, из к-рых наиболее эф-
эффективны след.: 1) прогрев высушенного полимера
в течение 2—4 ч при 100—120 °С; 2) обработка высу-
высушенного полимера острым паром с последующим досу-
досушиванием; 3) введение в отжатый от маточного р-ра
П. с. 10—20% (от массы П. с.) воды (распылением при
перемешивании сырого порошка, зерен) с последую-
последующей сушкой полимера при 120 °С.
При алкоголизе поливинилацетата в присутствии
серной, соляной или хлорной к-т получают П. с. без
примеси солей. Однако при применении серной к-ты
нек-рая часть гидроксильных групп П. с. взаимодейст-
взаимодействует с серной к-той с образованием кислых сернокислых
эфиров, к-рые могут отщеплять H2SO4, способствующую
деструкции П. с. При применении соляной к-ты обра-
образуются П. с, окрашенные в светло-желтый цвет. При-
Применение хлорной к-ты затруднено в связи с ее взрыв-
взрывчатостью.
В зависимости от катализатора, использованного при
омылении поливинилацетата, П. с. может содержать
различные трудноудаляемые примеси. Принципиаль-
Принципиальный смысл всех операций по стабилизации П. с, полу-
789
ПОЛИВИНИЛОВЫЙ СПИРТ
790
ченного при омылении в присутствии к-т, сводится
к нейтрализации содержащихся в нем (адсорбирован-
(адсорбированных или химически связанных) кислотных групп путем
введения перед отжимом в суспензию П. с. р-ров ще-
щелочи в количествах, необходимых для получения нейт-
нейтральных или слабощелочных р-ров полимера. П. с,
полученный при омылении в присутствии щелочи, со-
содержит ацетат натрия, приводящий к окрашиванию по-
полимера при прогреве; такой П. с. перед отжимом от
маточного р-ра стабилизируют A12(SO4K, вводя его в ко-
количествах, близких к стехиометрическим.
Методы получения сополимеров винилового спирта
и винилацетата, содержащих 10—30% (по массе)
остаточных ацетатных групп (т. наз. с о л ь в а р ы,
или с о в и о л ы), принципиально не отличаются от
методов синтеза П. с, за исключением того, что исполь-
используют меньшее количество катализатора (щелочи или
кислоты) и вводят специальные добавки для прекраще-
прекращения реакции омыления в ее заключительной стадии.
Напр., для получения сополимера с содержанием ок.
12% (по массе) ацетатных групп в 15—20%-ный мета-
нольный р-р поливинилацетата при интенсивном пе-
перемешивании вводят —10%-ный метанольный р-р
NaOH (из расчета 0,4 моль NaOH на 100 моль винил-
ацетатных звеньев). До фазового перехода полимера в не-
нерастворимое состояние процесс ведут при 28—30 °С.
Затем темп-ру повышают до 56—58 °С в течение 15—
20 мин и загружают Н3РО4 в стехиометрич. количестве
к исходному NaOH. После загрузки Н3РО4 смесь пере-
перемешивают при 56—58 °С еще в течение 1 ч. Отжим сопо-
сополимера от маточного р-ра, сушка и промывка полиме-
полимера метанолом осуществляются так же, как при син-
синтезе П. с. Для стабилизации сополимеров рекоменду-
рекомендуется вводить небольшие количества фосфорноватистой
кислоты.
В лабораторных условиях синтезированы изотактич.
и синдиотактич. П. с. Первый получают преимуществен-
преимущественно омылением полимеров простых виниловых эфиров,
второй — гидролизным разложением полидивинилаце-
талей.
Ведутся исследования по прямому синтезу П. с.
ионной изомеризационной полимеризацией ацетальде-
гида под давлением в несколько сотен Мн/м2, или не-
несколько тысяч кгс/см2, однако получаемый по этому
способу продукт имеет молекулярную массу всего лишь
около 1000.
Показано также, что П. с. образуется из ацетальде-
гида при темп-pax на 1—2 °С выше темп-ры плавления
мономера в присутствии высокодисперсных щелоч-
щелочных металлов (Na, К) или магния. Реакцию осу-
осуществляют совместным осаждением паров ацетальде-
гида и металла на поверхность, охлажденную жидким
азотом. Инициатором служит продукт взаимодействия
мономера с металлом.
Свойства. П. с.— твердый полимер белого цвета,
без вкуса и запаха; нетоксичен; содержит микрокри-
сталлич. образования. П. с. может кристаллизоваться
при термообработке в интервале 80—225 °С, достигая
степени кристалличности 68%; при кристаллизации
из р-ров многоатомных спиртов могут образовываться
пластинчатые монокристаллы и сферолиты. Макромоле-
Макромолекулы обычного П. с. содержат 1,0—2,5% звеньев, при-
присоединенных по типу «голова к голове», и имеют атак-
тич. строение.
Большая часть гидроксильных групп П. с. связана
водородными связями. Так, при комнатной темп-ре
в связанном состоянии находится ок. 70% гидроксиль-
гидроксильных групп. Практически полное разрушение водород-
водородных связей наступает при 150 °С. Ввиду наличия боль-
большого числа водородных связей П. с. растворяется лишь
в горячей воде (при темп-ре 80—100 °С) при перемеши-
перемешивании в течение 2—4 ч. Водные р-ры П. с. нестабильны
при хранении: через несколько часов после приготов-
приготовления начинается гелеобразование. Для придания та-
такому р-ру первоначальных свойств его следует, пере-
перемешивая, прогреть при 80—90 °С в течение 0,5—1,5 ч.
С увеличением в П. с. количества остаточных ацетат-
ацетатных групп от 5 до 30% (по массе) в связи с уменьше-
уменьшением плотности упаковки макромолекул скорость раст-
растворения полимера повышается, а темп-pa растворения
понижается. П. с, содержащий 8 —10% остаточных
ацетатных групп, уже растворяется в воде при комнат-
комнатной темн-ре. Растворы П. с. с увеличением содержания
в нем ацетатных групп становятся более стабильными,
а при содержании более —16% остаточных ацетатных
групп гель вообще не образуется.
Основным и единственным для П. с. растворителем
на практике служит вода. П. с. растворим также в ди-
метилформамиде и многоатомных спиртах; устойчив
к действию масел, жиров, алифатич. и ароматич. угле-
углеводородов. Мол. масса П. с. в зависимости от способа
получения лежит в пределах 5000—1 000 000. Зависи-
Зависимость между средневязкостной мол. массой М и ха-
рактеристич. вязкостью [ц] П. с. в воде при 20 °С вы-
выражается соотношением:
AW_
АР
Молекулярно-массовое распределение П. с. опреде-
определяется условиями получения исходного поливинил-
поливинилацетата (рис.).
П. с. устойчив к дейст-
действию разб. к-т и щелочей.
Термич., световая, окис-
окислительная и др. виды де-
деструкции начинаются с
дегидратации П. с, соп-
сопровождающейся образова-
образованием двойных, простых
эфирных и др. связей. Об-
Образование изолированных
двойных связей может
привести к ослаблению
взаимодействия между уг-
углерод-углеродными атома-
атомами и углерод-водородными
атомами а-метиленовой
Молекулярно-массовое рас-
распределение П. с, получен-
полученного из поливинилацетата,
синтезированного в среде
1000 2000 3000 4000 5000
Степень полимеризации
метилового спирта при степени превращения мономера
около 50% (а) и при практически полном превращении
мономера (б).
группы и к разрыву цепи. Для стабилизации обра-
образующихся радикалов наиболее эффективными инги-
ингибиторами служат фенолы. Хорошо отмытый от
ацетата натрия или к-т и стабилизированный П. с.
достаточно устойчив к воздействию тепла и света,
поэтому введения специальных противостарителей при
его переработке и хранении, как правило, не требуется.
Ниже приведены нек-рые свойства П. с:
Плотность при 20° С, г/см3 1.20 — 1,30
Показатель преломления п 1,49 — 1,53
Темп-pa, °С
стеклования 8 5
плавления 220-232
деструкции ок. 23 0
Темп-рный коэфф. линейного расширения
@-45 °С), °C-i G-12). Ю-5
Прочность при растяжении B0 °С, мол.
масса 80 000—200 000), Мн/м2 (кгс/см2) 63 — 120
F30-1200)
Относительное удлинение, % 0—3
Газопроницаемость по водороду,
м3/(м • сек • н/м*) 7,49-Ю*
мл/см • сек • мм рт. cm 10~u
791
ПОЛИВИНИЛПИРИДИНЫ
792
Химич. свойства П. с. определяются гл. обр. нали-
наличием гидроксильных групп (содержание их в П. с.
составляет 38,64% по массе). П. с. вступает в реакции,
типичные для многоатомных спиртов. Он способен,
напр., образовывать сложные и простые эфиры, реаги-
реагировать с металлич. натрием, альдегидами и кетонами,
образовывать при низких теми-pax (до 10 °С) в водной
среде ксантогенат с GS2 и NaOH. Наибольшее значение
в практич. отношении имеет реакция П. с. с альдегида-
альдегидами и кетонами,. приводящая к образованию соответст-
соответственно ацеталей и кеталей (см. Ацетали поливинилового
спирта).
П. с. сравнительно легко может образовывать эфиры
с двухосновными карбоновыми к-тами. В зависимости
от природы к-ты и условий реакции могут получаться
растворимые кислые эфиры (I) или нерастворимые сши-
сшитые полимеры (II):
ш
I
сн-сг
i
он
~сн2-
II
ОСО(СН2)
1
ОСО(СН
-сн-сн,-
1
он
nGOOH
2)псо
СНО
1
Реакцию образования кислых эфиров используют на
практике для придания морозостойкости водным дис-
дисперсиям полимеров, при синтезе к-рых в качестве за-
защитного коллоида был применен П. с. Реакцию до бо-
более глубокой степени этерификации П. с. применяют
для его сшивания. П. с. может взаимодействовать с ми-
минеральными к-тами с образованием сложных эфиров.
На концах макромолекул П. с. содержатся преимуще-
преимущественно карбонильные и карбоксильные группы. При
относительно высокой мол. массе концевые группы не
оказывают большого влияния на свойства П. с; при
мол. массе до 1000 это влияние существенно. Полимер
низкой мол. массы можно получить, если обычный П. с.
обработать йодной к-той НЮ4. Образовавшийся при
этом П. с. будет содержать большое число альдегидных
групп, легко ацеталирующих гидроксильные группы
др. макромолекул, что приводит к разветвлению или
сшиванию полимера.
Содержание карбонильных групп в макромолекулах
обычного П. с. не превышает 0,01% (молярная концент-
концентрация), и на большинство свойств полимера эти группы
не оказывают большого влияния. Однако в зависимости
от условий обработки П. с. карбонильные группы могут
ускорять процесс образования двойных связей и, сле-
следовательно, его окрашивание. В еще меньшем коли-
количестве в макромолекулах П. с. содержатся группы
—СН=СН—СН=СН—СО —. Принято считать, что в
макромолекулах обычного П. с. длинные боковые це-
цепи отсутствуют. Во всяком случае их число, прихо-
приходящееся на 1000 звеньев винилового спирта, не превы-
превышает 1; эти ответвления не могут оказывать большого
влияния на свойства П. с.
Анализ П. с. сводится к определению летучих,
содержания золы, ацетата натрия, остаточных ацетат-
ацетатных групп, растворимости, характеристич. вязкости,
вязкости 4%-ного водного р-ра и, в ряде случаев, его
прозрачности.
Применение. П. с. применяют для формования воло-
волокон (см. Поливинилспиртовые волокна), для произ-ва
поливинилацеталей (см. Ацетали поливинилового спир-
спирта), шлихтования основ пряжи и аппретирования тка-
тканей, в качестве защитного коллоида для эмульгирова-
эмульгирования мономеров и стабилизации водных дисперсий поли-
полимеров, как загуститель различных водных р-ров и ла-
тексов, в качестве связующего при изготовлении литье-
литьевых форм для цветных и черных металлов, для изготов-
изготовления водорастворимых пленок, консервирования до-
донорской кожи, изготовления печатных плат в радиотех-
нич. пром-сти и клише в полиграфия, нром-сти. Спе-
Специальные марки тщательно очищенного ннзкомолеку-
лярного П. с. применяют в качестве плазмозаменителя
при переливании крови, а также для изготовления ле-
лекарственного препарата «иодинол» (см. Полимеры
в медицине).
В 1972 мировое производство П. с. составляло
400 000 т.
П. с. впервые синтезирован В. О. Германом и В. Ге-
нелем в 1924 в Германии.
Лит.: Ушаков С. Н., Поливиниловый спирт и его'
производные, т. 1 — 2, М.— Л., 1960; Д ж е и л Ф. X., Поли-
Полимерные монокристаллы, пер. с англ., Л., 1968; Анализ полиме-
ризационных пластмасс, 2 изд., Л., 1967; Н и к о л а е в А. Ф.,
Синтетические полимеры и пластические массы на их основе,
2 изд., М,— Л., 1966; Pritchard J. G., Polyvinylalcohol,
L., 1970; Encyclopedia of polymer science and technology, v. 14
N. Y.— [a. o.], 1971, p. 149. С. С. Мпацаканов.
ПОЛИВМНИЛПИРИДИНЫ — см. Винилпиридина
полимеры.
ПОЛИВИНИЛПИРРОЛИДОН — см. Вииилпирроли-
дона полимеры.
ПОЛИВИНИЛ СПИРТОВЫЕ ВОЛОКНА [ poly (v inyl
alcohol) fibres, Polyvinylalkoholfasern, fibres d'alcohol
polyvinylique] — синтетич. волокна, формуемые из
поливинилового спирта. Поливинилацетат, из к-рого
получают используемый для формования волокна по-
поливиниловый спирт (ПВС), синтезируют обычно ради-
радикальной полимеризацией винилацетата в метаноле.
Для получения малоразветвленного полимера со срав-
сравнительно узким молекулярно-массовым распределе-
распределением процесс ведут до конверсии не более 55—65% за
один проход (см. также Винилацетата полимеры).
Из поливинилацетата ПВС образуется в результате
алкоголиза. Ниже приведены основные требования,
к-рые предъявляются к ПВС как к сырью для произ-
производства волокон:
Степень полимеризации 1200 — 1700
Содержание ацетатных групп, % .... ^0,2
Содержание примесей, %
ацетат натрия
железо
летучие фракции
низкомолекулярные фракции, отмы-
отмываемые водой при 20 СС <3
Степень набухания в воде при 2 0 °С, % <!150 — 2 00
Вид набухшего полимера Мелкие, не слипа-
слипающиеся кусочки
Вязкость 15%-ного р-ра в воде при 5 0 °С,
н-сек/м2 (пз) 2 — 4 B0-40)
Прозрачность 4%-ного р-ра в воде, % ^90
Растворимость в воде при 95 ^С, % . . >99,9%
Число гелеобразных частиц в 1 см3 15%-
ного р-ра в воде (по методу фильтра-
фильтрации на сетке) ^2 — 3
Производство
Получение прядильного раствора. Получение волокон
из ПВС возможно как из р-ров по мокрому или сухо-
сухому методу, так и из пластифицированного водой поли-
полимера по сухому методу. Большинство видов П. в.
формуется по мокрому методу в солевых ваннах.
До растворения ПВС промывается от ацетата натрия
и низкомолекулярных фракций водой при 15—20 QG
и модуле ванны (отношение массы ПВС к массе воды)
от 1 : 10 до 1 : 20. Промывка ведется двукратно в ба-
баках с мешалками или на сетчатом транспортере (про-
(противотоком). После промывки полимер отжимается
в центрифуге или на каландрах. Для выравнивания
влажности его желательно кондиционировать в тече-
течение 12 — 24 ч.
Набухший полимер растворяют в вертикальных
аппаратах с мешалками и рубашками, обогреваемыми
паром, в обессоленной или умягченной воде с темп-рой
95—98 °С в течение 4—8 ч. В процессе растворения до^-
6-1 О
<0,003
793
ПОЛИВИНИЛСПИРТОВЫЕ ВОЛОКНА
794
бавляют поверхностно-активные вещества (для повы-
повышения гомогенности р-ра, гашения иены и облегчения
последующего процесса формования). В конце раство-
растворения рН р-ра доводится до 4—G. Из аппаратов для
растворения прядильный р-р перекачивается в смеси-
смесители, где перемешивается не менее трех партий для
выравнивания состава. После этого р-р дважды фильт-
фильтруют на рамных фильтр-прессах (через ткань) и подают
в баки, где в течение 12—18 ч при атмосферном давлении
осуществляют деаэрацию.
Все процессы обработки прядильного р-ра произво-
производят при темн-ре не ниже 80 °С во избежание его жела-
тпнизации. Поэтому все оборудование и трубопроводы
обогревают водой с температурой 95—98 °С. Раствор,
поступающий на формование, имеет концентрацию
15-16%.
Кроме периодич. схехмы растворения, предложена
также непрерывная, однако она пока не нашла прак-
тич. применения.
При формовании волокон по сухому методу прядиль-
прядильный р-р готовят по той же схеме, что и в предыдущем
случае; концентрация р-ра составляет 30—45%. Воз-
Возможна и др. технологич. схема, при к-рой ПВС после
промывки и набухания в воде уплотняется и расплавля-
расплавляется в шнековом экструдере, затем двукратно фильт-
фильтруется и поступает на формование.
Формование волокон.ПВС может осаждаться из его
водных р-ров с применением водно-солевых или орга-
нич. ванн. Хорошие осгцители — сульфаты натрия
и аммония, ацетон, спирты и др. Обычно формование
ведется в осадительной ванне, содержащей р-р Na2SO4
(концентрация 400—420 г/л), при рН 4—5 и темп-ре
43—45 °С. Длина пути нити в ванне 150—200 см, ско-
скорость движения 7—12 м/мин. Такой длительный процесс
формования необходим из-за медленного осаждения
полимера. Поэтому наиболее рациональная схема —
вертикальное формование в трубках. Осадительная
ванна, разбавленная в результате формования водой,
для регенерации подвергается выпарке под вакуумом.
Свежесформованное волокно подвергается пласти-
фикационной вытяжке (в 3—4 раза) в ванне, содержа-
содержащей 200—400 г/л Na2SO4, при 70—80 °С. Вытяжка обыч-
обычно ведется в две ступени.
После вытягивания волокно промывается от суль-
сульфата натрия водой с темп-рой 10—20 °С. Хотя волокно
на этой стадии имеет еще низкую водостойкость, во
время промывки под натяжением оно не теряет своих
прочностных свойств при контакте с водой. Иногда
волокно после промывки подвергается авиважной об-
обработке для облегчения последующей термовытяжки.
При получении штапельного волокна формование
ведется на фильерах с 4800—15 000 отверстиями, при
получении непрерывных нитей — с 30—1200 отвер-
отверстиями. После прядильной машины волокна собираются
в общий жгут и все дальнейшие обработки производят-
производятся в жгуте.
При производстве нитей их обработка ведется либо
на машине с системой отдельных рабочих органов для
каждой нити, либо в виде ленты из нескольких десятков
параллельных нитей, обрабатываемых на одной системе
рабочих органов.
После промывки и отжима волокно высушивается
под натяжением на вальцах в среде горячего воздуха
или в сушилках с электро- или газовым обогревом. Суш-
Сушка производится в мягком режиме при темп-ре воздуха
не выше 70—100°Сво избежание растворения волокна
в содержащейся в нем воде.
В производстве П. в. по сухому методу формование
ведется в шахте в среде горячего воздуха. Этот метод
пригоден только для получения волокон большой тол-
толщины @,5—0,7 текс после термовытяжки). Надмоле-
Надмолекулярная структура волокон, формуемых по сухому
методу, характеризуется наличием фибриллярных об-
образований большого размера, что в сочетании с боль-
большой толщиной волокон обусловливает их жесткость
и невысокие усталостные свойства. В значительной
мере лишены этих недостатков волокна мокрого
метода формования, к-рые имеют структуру с мень-
меньшим размером фибриллярных образований и мень-
меньшую толщину.
Термическая вытяжка и термообработка волокон.
Для получения различных по свойствам П. в. их после
сушки подвергают различной обработке. Волокна,
к-рые должны иметь повышенную прочность, подвер-
подвергают термич. вытяжке в среде горячего воздуха при
230—260 °С. Степень вытяжки при получении упроч-
упрочненных штапельных волокон составляет 1,5 — 2,5, при
получении технич. нитей — 3—5.
Другая важная операция — термообработка сопро-
сопровождается релаксационными и кристаллизационными
процессами, в результате чего волокно приобретает
равновесную молекулярную структуру. Термообработ-
Термообработка проводится при 220—250 °С в течение 0,3—2 мин.
В зависимости от длительности процесса получают во-
волокна с различной водостойкостью.
Без термообработки затруднена дальнейшая химич.
обработка волокон, т. к. нетермообработанное волокно
набухает, изменяет структуру и резко снижает меха-
нич. свойства под действием воды и водных р-ров.
Аппаратурное оформление процесса термообработки
зависит от вида формуемого волокна. Жгут для полу-
получения штапельного волокна обрабатывается на роликах
с непараллельными осями E—8 переходов), где проис-
происходят последовательно сушка, термовытяжка, термооб-
термообработка и охлаждение волокна. Обогрев камер — га-
газовый или электрический. При получении высокомо-
высокомодульных нитей термовытяжку и термообработку осу-
осуществляют одновременно, в одних и тех же камерах.
После указанных операций штапельное волокно
м. б. получено с водостойкостью от 60 до 95 °С (в зави-
зависимости от темп-ры и времени термообработки). Высо-
Высокомодульные технич. нити имеют водостойкость до
110 °С благодаря равномерному прогреву волокон и
наиболее полному протеканию релаксационных и кри-
кристаллизационных процессов при термовытяжке и тер-
термообработке.
Химическая обработка и заключительные операции
производства волокон. После термообработки П. в.
имеют степень кристалличности ок. 60—75% . С целью
дальнейшего повышения водостойкости волокно м. б.
подвергнуто сшиванию бифункциональными соедине-
соединениями, реагирующими с гидроксильными группами;
другой путь увеличения водостойкости — блокирова-
блокирование свободных гидроксильных групп макромолекул
ПВС более гидрофобными группами. Придание большей
водостойкости обычно необходимо для штапельных
волокон, имеющих менее упорядоченную структуру.
Для получения волокон, стойких даже при длитель-
длительном кипячении в воде, их чаще всего ацеталируют фор-
формальдегидом (иногда — бензальдегидом). Процесс про-
проводят при 65—70 °С в р-ре, содержащем 3—4% фор-
формальдегида, 15—20% серной к-ты (катализатор) и
15—20% сульфата натрия (для уменьшения набуха-
набухания волокон); длительность процесса 25—40 мин.
После ацеталирования волокно промывают умягчен-
умягченной водой и подвергают авиважной обработке для при-
придания ему необходимых текстильных свойств (мягкого
грифа, необходимого коэфф. трения). При получении
штапельных волокон ацеталирование жгута произво-
производится обычно в аппарате U-образного типа, промыв-
промывка — в проходном агрегате. Жгут для придания ему
извитости подвергается гофрировке, режется на шта-
пельки нужной длины, обрабатывается авиважным
р-ром, сушится в конвективной сушилке. Полученное
волокно упаковывается в кипы. Возможен также выпуск
волокна в жгуте.
795
ПОЛИВИНИЛСПИРТОВЫЕ ВОЛОКНА
796
Технич. нить ацеталируется на бобинах в герметич-
герметичных аппаратах, где она затем промывается и обрабаты-
обрабатывается авиважным р-ром. После сушки нить перематы-
перематывают на конич. бобины. Однако большая часть технич.
нитей и все высокомодульные нити не ацеталируют,
а сразу после термич. операций перематывают на конич.
бобины для отправки потребителю.
Формальдегид — наиболее токсичное вещество, ис-
используемое в производстве П. в. Однако его выделение
в производственное помещение и в атмосферу практи-
практически не происходит вследствие достаточной герметич-
герметичности оборудования. Формальдегид, попадающий
в сточные воды, легко разрушается в процессе био-
биоочистки.
Свойства и применение
П. в., в зависимости от их вида и условий получения,
могут иметь различные механич. свойства. Как прави-
правило, они обладают высокой прочностью, высокой устой-
устойчивостью к истиранию и изгибам. Благодаря большому
количеству полярных гидроксильных групп в макро-
макромолекуле ПВС м. б. получено волокно с наибольшей
среди др. синтетич. волокон гигроскопичностью. Вы-
Высокая реакционная способность гидроксильных групп
обеспечивает удовлетворительную окрашиваемость
П. в. красителями, применяемыми для крашения цел-
целлюлозных волокон. По этой же причине волокна из
ПВС обладают хорошей адгезией к пластикам и резине
и легко поддаются химич. модификации.
Карбоцепная структура с высокой химич. регуляр-
регулярностью обеспечивает отличную устойчивость П. в.
к действию света (по этому показателю П. в., наравне
с полиакрилонитрильными волокнами, превосходят
все остальные синтетич. волокна), микроорганизмов,
пота, а также хорошую хемостойкость ко многим реа-
реагентам (к-там, щелочам, окислителям умеренных кон-
концентраций). Волокна из ПВС особенно устойчивы к ма-
малополярным растворителям и нефтепродуктам.
Штапельные волокна. В табл. 1 приведены основ-
основные показатели штапельных П. в., ацеталированных
формальдегидом. Перерабатывают такие волокна по
различным схемам как в чистом виде, так и в смеси
с хлопком, шерстью, льном или др. химич. волокнами.
Их применяют при получении одежных, бельевых, ру-
рубашечных, занавесочных и др. тканей и трикотажа.
Таблица 1. Основные свойства штапельных поливинилспиртовых
волокон
Показатели
Толщина, текс
Прочность, гс/текс
Относительное удлинение, % ....
Сохранение прочности в мокром состоя-
состоянии, %
Устойчивость к двойным изгибам, тыс.
циклов
Длина резки, мм
для хлопчатобумажной пром-сти . .
для шерстяной пром-сти
для бумажной пром-сти
Влагопоглощение, %
Обычное
волокно
0,11-0,55
30 — 40
20-27
75-85
200-1200
35-40
60-100
4-7
3,5-5
Упрочнен-
Упрочненное волок-
волокно
0,11-0,22
40-55
17-25
80 — 90
200—1200
3,5—5
Изделия из смеси хлопка или вискозного штапель-
штапельного волокна с П. в. имеют в 1,5—3 раза более длитель-
длительный срок службы, чем чистохлопковые или чистовис-
козные. Высокая гигиеничность, носкость и устойчи-
устойчивость к химич. реагентам позволяют получать из П. в.
ткани (иногда в смеси с др. волокнами) для высокока-
высококачественной спецодежды и форменной одежды рабочих
многих профессий, школьников и др.
Благодаря тому что П. в. не подвергаются гниению
и действию пота и в то же время обладают хорошей
гигроскопичностью и износостойкостью, их используют
в обувной пром-сти для производства как верха тек-
текстильной обуви, так и особенно подкладки. Волокна
из ПВС — единственные среди химич. волокон, к-рые
не ухудшают свойлачиваемости шерсти при получении
сукон, фетров, войлоков. Поэтому их применение для
получения указанных изделий как бытового, так и
технич. назначения весьма перспективно.
Благодаря высокой устойчивости к светопогоде и
действию микроорганизмов, ограниченной набухае-
мости во влажных условиях и высоким механич. свой-
свойствам П. в.— наилучший среди всех синтетич. волокон
материал для изготовления парусины, брезентов, ту-
туристского и спортивного снаряжения. Волокно из
ПВС для этих целей используют в чистом виде или
в смеси с лубяными и хлопковыми волокнами.
Штапельные волокна из ПВС применяют также для
изготовления неответственных канатов и рыболовных
снастей. Особенно целесообразно использование изде-
изделий, содержащих П. в., в условиях влажного и тропич.
климата.
Хорошая хемостойкость П. в. позволяет изготовлять
на их основе ткани и нетканые изделия, применяемые
в качестве фильтровальных материалов и полупрони-
полупроницаемых перегородок для химически агрессивных сред.
Штапельные П. в. используют также для армирования
пластиков, упрочнения бумаги и нек-рых др. изделий.
В результате модификации П. в. получены ионообмен-
ионообменные волокна, а также волокна различного медицинского
назначения (см., напр., Медицинские нити).
Нити. На основе ПВС, в зависимости от условий тер-
термич. вытяжки и термообработки, м. б. получено не-
несколько видов технич. нитей, основные свойства к-рых
приведены в табл. 2.
Как уже отмечалось, технич. нити из ПВС в боль-
большинстве случаев (за исключением нитей, к-рые постоян-
постоянно эксплуатируют во влажных условиях) не ацетали-
ацеталируют, т. к. они обладают достаточной водостойкостью.
Высокомодульные нити имеют настолько упорядочен-
упорядоченную структуру, что и без ацеталирования выдерживают
кратковременное кипячение в воде или действие многих
др. вызывающих набухание реагентов.
Нити из ПВС используются для армирования тран-
транспортерных лент, шлангов, приводных ремней, мем-
мембран и др. резино-технич. изделий. Достоинства таких
нитей как арматуры определяются их высоким модулем
упругости и малой ползучестью, особенно по сравне-
сравнению с полиамидными нитями. Высокая адгезия П. в.
к резине позволяет упростить технологию (не нужна
сложная адгезионная обработка волокон) и повысить
эксплуатационные свойства резино-технич. изделий.
Таблица 2. Основные
ПОКЯ'ЧЯТРЛИ
Л- л- \J X 1СЛ О СЛ X f <vl XL
Толщина, текс . . .
Прочность, гс/текс
Относительное удли-
удлинение, %
Модуль упругости,
Мн/м2 (кгс/мм2) . .
Сохранение прочно-
прочности в мокром сос-
состоянии, %
Усадка при 150°С, %
Плотность, г/см3 . .
Влагопоглощение, %
свойства технических поливинилспиртовых
нитей
Упрочнен-
Упрочненные нити
29-160
60-70
8-12
8000 — 15000
(800-1500)
75—85
1,5—2,5
1,31
3-4
Высокопроч-
Высокопрочные нити
29-93
70-80
6-9
15000 — 25000
A500 — 2500)
80-90
1,5-2
1 ,32
3-4
Высокомо-
Высокомодульные нити
5 5-93
75-105*
3-5
30000-60000
C000-6000)
85-95
1-1,5
1,32
2,5-3,5
* Прочность одиночного волокна может достигать 125
гс/текс.
Технич. нити мокрого метода формования обладают
более высокими усталостными свойствами, чем нити
сухого метода формования, и их применение в произ-
производстве шин более перспективно.
797
ПОЛИВИНИЛФОРМАЛЬ
798
Высокомодульные нити из ПВС благодаря низкой
плотности, высокой адгезии ко многим связующим,
прочности и высокому модулю упругости являются
прекрасными армирующими наполнителями для пла-
пластиков. Наилучшие результаты получаются в произ-
производстве пластиков на основе эпоксидных, фенольных,
эгюкси-фенольных связующих. Новые материалы полу-
получены также при армировании высокомодульными нитя-
нитями из ПВС полиолефинов и др. термопластов. Для уп-
упрочнения пластиков можно использовать также карбо-
низованные П. в. (т. н. углеродные нити). См. также
Орган о воло книт.
Канаты, тросы, рыболовные снасти из ПВС-нитей
отличаются повышенной механич. жесткостью.
Кроме технич. ПВС-нитей, производятся также нити
для изделий широкого потребления (напр., для пла-
плащевых и бельевых тканей).
Водорастворимые волокна. В зависимости от условий
сушки и термич. обработки м. б. получены водораст-
водорастворимые П. в. с различной водостойкостью. Ниже пред-
представлены свойства таких волокон:
Толщина, тепе
штапельного волокна 0,1—0,5
нити с 16-160
Прочность, гс/текс 25 — 40
Относительное удлинение, %
Водостойкость, %
тип 65 •
тип 75
тип 85
тип 95
Длина резки (для штапельного волокна), мм
для текстильной пром-сти
для бумажной пром-сти
Влагопоглощение, %
18-25
60-70
70—80
80-90
90-98
36-120
4-7
4 — 5
Водорастворимые штапельные П. в. применяют в ка-
качестве вспомогательного (удаляемого) компонента в сме-
смесях с др. волокнами при получении ажурных изделий,
тонких тканей, пористых структур, а также в производ-
производстве водорастворимой ткани-основы, используемой
при получении гипюра (взамен ткани из натурального
шелка).
При введении 7 —15% водорастворимых П. в. в бу-
бумажную массу облегчается процесс отлива бумаги,
а на стадии сушки это волокно склеивает целлюлозные
(базовые) волокна в бумаге. Таким методом произво-
производится бумага и картон для очистки воздуха, фильтра-
фильтрации моторных топлив, масел, гидрожидкостей и др.
П. в. используют также в производстве бумаги из
синтетических волокон, нетканых изделий и высокопроч-
высокопрочных бумажных изделий для однократного пользова-
пользования (белье, салфетки, медицинские изделия).
Промышленное производство П. в. было впервые
освоено в 1950 в Японии, где сейчас выпускается не-
несколькими фирмами под названиями в и н и л о н,
куралон, мьюлон, Кремона и др. П. в.
производятся также в СССР (вино л), КНДР (в и-
н а л о н), КНР и в др. странах.
В 1971 выпуск П. в. во всем мире превысил 100 тыс. т.
Лит.: ПерепелкинК. Е., Хим. волокна, № 1, 39
A962); Сакурада И., Хим. и технол. полимеров, № 10,
80 A964); О s и g i Т., в кн.: Man-made fibres, v. 3, N. Y.,
1968; ПерепелкинК. Е., Пугач Б. М., в сб.: Хи-
Химические волокна, М., 1968, с. 66, 96; П е р е п е л к и н К. Е.
[и др.], Хим. волокна, № 1, 20 A967); Лелинков О. С,
УтевскийЛ. Е., ПерепелкинК. Е., там же, № 6,
с. 45 A969); Перепелкин К. Е., Перепелки-
н а М. Д., Текстильная пром-сть, № 6, 20 A963); № 8, 27, 95
A969); Перепелкин К. Е..Поливинилспиртовые волокна,
в кн.: Карбоцепные синтетические волокна, под ред. К. Е. Пе-
репелкина, М., 1973, с. 165; Новые химические волокна техни-
технического назначения, М.—Л., 1973, с. 89. К. Е. Перепелкин.
ПОЛИВИНИЛСТЕАРАТ — см. Винилстеарата по-
полимеры.
ПОЛИВИНИЛСУЛЬФОФТОРИД — см.
фофторида полимеры.
Винилсулъ-
ПОЛИВИНИЛСУЛЬФОХЛОРИД — см. Винилсулъ-
фохлорида полимеры.
ПОЛИВИНИЛТОЛУОЛ — см. Стирола производ-
производных полимеры.
ПОЛИВИНИЛФЛУОРЕН — см. Винилфлуорена поли-
полимеры.
ПОЛИВИНИЛФОРМАЛЬ [poly(vinyl formal), Po-
lyvinylformal, formal polyvinylique] — карбоцепной
полимер, ацеталь поливинилового спирта и формаль-
формальдегида общей ф-лы [—GH—СН2—GH—СН2 — ]п.
I I
о — сн2—о
Свойства. П.—аморфный полимер белого цвета. Сте-
Степень полимеризации П. составляет 350—500. Нек-рые
свойства технич. П. призедены ниже:
Плотность при 20 °С, г/см3 1,24
Показатель преломления п . 1,1
Температура, °С
стеклования . . . . с „ . .
текучести
Теплостойкость, °С
по Вика
по Мартенсу
Темп-рный коэфф. линейного расширения,
°С~1
Верхний предел рабочих теми-р, °С
при кратковременной эксплуатации . „
при длительной эксплуатации ....
Водопоглощение, %
Влагопроницаемость,
кг/(м-сек-н/м2)
г/(см• ч• мм рт. ст.)
Прочность, Мн/м2 (кгс/см2)
при растяжении . . .
при статич. изгибе.
Модуль упругости при изгибе, Мн/м2
(кгс/см2)
Ударная вязкость, кдж/м2, или кгс • см/см2
Относительное удлинение, %
Уд. электрич. сопротивление
поверхностное, Том(ом)
объемное, Том-м (ом-см)
Диэлектрич. проницаемость
при 1 пгц „
при 1 Мгц „
Тангенс угла диэлектрич. потерь
при 1 пгц
при 1 Мгц
Электрич. прочность, кв/мм
85-95
150 — 170
115 — 120
90-95
5,4- 10~5
120
60-80
0,5—3,0
2,7- Ю-14
1,3- Ю-7
60-70
F00-700)
100-130
A000 — 1300)
4000
D0 000)
15 — 30
5-11
4 • 10*D • 10ie)
3- 102C- 101в)
4,5
3,3
0,01
0,02
20-26
П. растворим в уксусной к-те, хлороформе, крезоле,
диоксане, дихлорэтане, фурфуроле, нитробензоле, пи-
пиридине, тетрахлорэтане, смеси D0 : 60) этанола с толу-
толуолом и др.; нерастворим в спиртах жирного ряда,
эфирах и углеводородах. Растворимость П. ухудшается
с уменьшением содержания винилацетатных звеньев
(одновременно увеличиваются термостабильность и
прочность полимера). При концентрации этих звеньев
2—3% П. растворяется лишь в бензиловом спирте, фено-
феноле, крезоле. П. совмещается с резольными смолами и
реагирует с ними при нагревании с образованием не-
нерастворимых полимеров. Он устойчив к действию разб.
щелочей. Выше 150 °С П. подвергается термоокисли-
термоокислительной деструкции с выделением формальдегида и
воды. Полимер стабилизируют нек-рыми аминами, мо-
мочевиной или ее производными.
Содержание формальных групп в П. определяют по-
лярографич. методом.
Получение. В пром-сти П. получают из поливинил-
ацетата (гомогенный способ) или из поливинилового
спирта (гетерогенный способ). Поливинилацетат раст-
растворяют в 60—80% -ном водном р-ре уксусной к-ты и на-
нагревают при 70—80 °С в присутствии формальдегида
и серной к-ты (как катализатора). Нагревание прекра-
прекращают, когда гидролизуется 92% ацетатных групп и
ацеталируется ок. 90% образовавшихся гидроксильных
групп. Катализатор нейтрализуют ацетатом аммония
и добавляют воду для выделения П., к-рый затем мно-
799
ПОЛИВИНИЛФТОРИД
800
гократно промывают водой, стабилизируют и сушат. П.
содержит (молярная концентрация) 10—8% гидро-
ксильных, 10—8% ацетатных и 80—84% формальных
групп.
Ацеталирование поливинилового спирта осущест-
осуществляют в водной среде при 90—95 °С в присутствии кис-
кислого катализатора, напр. HG1. Образующийся полимер
осаждается из р-ра; его отфильтровывают, многократ-
многократно промывают водой, стабилизируют и сушат. Содержа-
Содержание винил ацетатных звеньев в П. не превышает 2—3%.
Применение. П. применяют гл. обр. для изготовления
электроизоляционных лаков (см. Поливинилацепгалъ-
ные лаки и эмали) и клеев.
В состав клеев входят П. и феноло-формальдегидная
смола резольного типа; клеи применяют для склеива-
склеивания металлов, стеклотекстолита, дерева и резины (см.
Феиоло-формалъдегидные клеи).
При смешении П. с синтетич. каучуком в соотноше-
соотношениях (95—70) : E—30) повышаются ударная вязкость
(с надрезом) и модуль упругости (но сравнению с ана-
аналогичными показателями П.). Из таких смесей можно
формовать изделия для машиностроения и электротех-
электротехники. Композиции на основе П. можно перерабатывать
методами экструзии и литья под давлением.
За рубежом П. выпускается под след. торговыми на-
названиями: ф о р м в а р (США), формадур, м о-
в и таль F, п йодоформ F (ФРГ), р е в и л ь
F, р о в и н а л ь F (Франция) и др.
Впервые П. синтезирован В. О. Германом и В. Гене-
лем в 1927.
Лит.: Ушакове. Н., Поливиниловый спирт и его
производные, т. 1—2, М.— Л., 1960; Николаев А. Ф.,
Синтетические полимеры и пластические массы на их основе,
2 изд., М.— Л., 1966; Справочник по пластическим массам,
под ред. М. И. Гарбара [и др.], т. 1, М., 1967; X у в и н к Р.,
Ставерман А. [сост.], Химия и технология полимеров,
пер. с нем., т. 2, ч. 1, М.— Л., 1965; Ethylene and its industrial
derivatives, ed. S. A. Miller, L., 1969, p. 1041.
M. Э. Розеиберг.
ПОЛИВИНИЛФТОРИД — см. Винилфторида по-
полимеры.
ПОЛИВИНИЛФУРАН — см. Винилфурана поли-
полимеры.
ПОЛИВИНИЛХЛОРИД — см. Винилхлорида поли-
полимеры.
ПОЛИВИНИЛХЛОРИДНЫЕ ВОЛОКНА [poly (vi-
(vinyl chloride) fibres, Polyvinylchloridfasern, fibres de
chlorure de polyvinyle] — синтетич. волокна, формуе-
формуемые из поливинилхлорида или из сополимеров винил-
хлорида.
Сырье. Для получения П. в. помимо гомополимера
винилхлорида, используют: хлорированный поливи-
нилхлорид, содержащий 63—64% G1 (т. наз. перхлор-
виниловая смола); сополимер 13—20% винилхлорида
с 80—85% винилиденхлорида и 0—3% акрилонитрила;
сополимеры винилхлорида с винилацетатом или акрило-
нитрилом; смеси поливинилхлорида с производными
целлюлозы (напр., с ацетилцеллюлозой) и хлорирован-
хлорированным поливинилхлоридом, содержащим 70—72% G1.
Волокнообразующий поливинилхлорид получают
суспензионной или блочной полимеризацией. Он дол-
должен иметь мол. массу 80 000—100 000. Особый интерес
представляют гомополимеры повышенной степени син-
диотактичности, полученные различными методами по-
полимеризации, в основном при низких (минус 20—40 °С)
темп-pax. Из них производят наиболее теплостойкие
и прочные волокна.
Перхлорвиниловую смолу, пригодную для формова-
формования волокон, получают при хлорировании поливинил-
поливинилхлорида, растворенного в тетрахлорэтане. Содержа-
Содержание хлора в смоле должно быть 63—64%, мол. масса
60 000—80 000.
Волокнообразующие сополимеры винилхлорида с ви-
винилацетатом (до 15% винилацетата) или акрилонитри-
акрилонитрилом (до 40% акрилонитрила), полученные эмульсион-
эмульсионной полимеризацией, должны иметь мол. массу соот-
соответственно ок. 30 000 и ок. 100 000, сополимер вини-
винилиденхлорида с винилхлоридом — 20 000—30 000.
Подробно о получении и свойствах полимеров на
основе винилхлорида см. Винилхлорида полимеры,
Винилхлорида сополимеры, П ерхлорвинилоеые смолы.
Получение. П. в. в основном формуют из конц. р-ров
полимеров по сухому или мокрому способу. Применяют
также способ экструзии (см. Формование химических
волокон).
При формовании волокон из гомополимера по су-
сухому способу используют ~30%-ные р-ры
в смесях ацетона с сероуглеродом или бензолом (соот-
(соотношение растворителей в смеси 1 : 1 но массе). Эти
р-ры при нормальных условиях находятся в гелеобраз-
ном состоянии. Поэтому для транспортировки по тру-
трубопроводам, фильтрации и продавливания через филье-
фильеру их подогревают до 50—90 °С; вязкость подогретых
р-ров 100 н-сек/м2 A000 пз). В прядильные р-ры вводят
светостабилизаторы (обычно производные оксибензо-
фенонов — 0,5—1,5% от массы полимера), а при кра-
крашении волокон в массе — органические пигменты
илиацетонорастворимые красители B—3% от массы
полимера).
Для удаления растворителя через шахту прядильной
машины продувают горячий воздух. Образующуюся
газовоздушную смесь, концентрация паров раствори-
растворителя в к-рой меньше нижнего предела взрывоопасных
концентраций, подают на рекуперацию. Во Франции
реализован более экономичный процесс, исключающий
подачу воздуха в шахту. При этом растворитель испа-
испаряется в верхней обогреваемой зоне шахты, а концент-
концентрируется в нижней интенсивно охлаждаемой зоне.
Сформованное волокно вытягивают в 5 — 7 раз при
темп-pax выше 90 СС, обрабатывают антистатиками и
подвергают кручению и сновке (при производстве
нитей) или гофрированию и резке (при производстве
штапельного волокна). Для получения нитей применяют
фильеры с количеством отверстий от 40 до 100, для по-
получения штапельного волокна — с 200—400 отверстия-
отверстиями. Скорость формования 400—600 м/мин.
По сухому способу получают также нити из ацетоно-
ацетоновых р-ров сополимера винилхлорида с винилацетатом
или акрилонитрилом. Концентрация сополимеров в пря-
прядильном р-ре 26—30%, вязкость р-ров 50—70 н-сек/м2
E00—700 пз).
При формовании по мокрому способу при-
применяют: 1) 24—28%-ные р-ры гомополимера в диметил-
формамиде или 12—16%-ные в тетрагидрофуране;
2) 16—22%-ные р-ры гомополимера повышенной сте-
степени синдиотактичности в циклогексаноне или 14 —
20%-ные в диметилформамиде; 3) 24—28%-ные р-ры
перхлорвиниловой смолы или сополимеров винилхло-
винилхлорида с винилацетатом в ацетоне. Вязкость р-ров, по-
поступающих на формование по мокрому способу, 10—
30 п-сек/м2 A00 — 300 пз).
Полимеры растворяют, как правило, при темп-рах
выше темп-ры стеклования полимера. Охлаждение
р-ров ниже 60—80 СС приводит к их гелеобразованию,
сопровождающемуся резким возрастанием вязкости.
В прядильные р-ры вводят 1 — 2% (от массы полимера)
термостабилизатора, а также пигменты и светостабили-
светостабилизаторы. При растворении в диметилформамиде, кроме
того, необходимо добавлять к-ты или комплексообра-
зователи, связывающие продукты гидролиза раство-
растворителя (амины), являющиеся активными катализато-
катализаторами деструкции поливинилхлорида. Растворение нер-
хлорвиниловой смолы и сополимера винилхлорида
с винилацетатом или акрилонитрилом в ацетоне про-
проводят при нормальных теми-рах.
Осадительными ваннами при формовании П. в. из
р-ров в диметилформамиде, ацетоне и тетрагидрофуране
служат смеси растворителей с водой; при формовании
801
ПОЛИВИНИЛХЛОРИДНЫЕ ВОЛОКНА
802
Физические свойства поливинилхлоридных волокон
Показатели
Атактич. поливинилхлорид
волокно
без термо-
термофиксации
волокно
термофик-
сирован-
ное (под
натяже-
натяжением)
волокно
термофик-
сирован-
ное (в сво-
свободном
состоя-
состоянии)
Поливи-
нилхло-
нилхлорид повы-
повышенной
стереоре-
гулярнос-
ти
Перхлор-
винило-
вая
смола
Смесь по-
поливинил-
хлорида и
хлориро-
хлорированного
поливи-
нилхлори-
даG0-72%
хлора)
Сополи-
Сополимер ви-
нилхло-
рида с ви-
винил аце-
ацетатом
Сополи-
Сополимер вини-
лиден-
хлорида
с винил-
хлоридом
Толщина, текс
Диаметр, мкм
Прочность при растяжении, гс/текс . .
Относительное удлинение, %
Прочность в петле, % от прочности
при растяжении
Темп-pa начала усадки, °С
Усадка в кипящей воде за 30 мин, %
Благопоглощение (при относительной
влажности воздуха 65% и 2 0 °С),% .
0,15-0,5
6 — 10
22—30
15-25
60 — 70
75-80
45-55
0,15-0,5
6-10
20-25
15-30
40-50
80-85
35-40
0
0,2 — 0,6
7-12
10 — 12
120-180
90-95
95 — 105
0
0
0,15-0,5
6—10
30 — 35
40
60 — 70
110-130
0
0,15-0,5
5,6-10
14 — 17
25-45
40-60
80 — 90
20-25
0
0,2-0,6
7-10
15-18
40-60
90-95
110
0
0
0, 15 — 0,4
3,5-8
10-12
100 — 120
85 — 90
65-67
55-65
0,5
500 — 1000
20—22
15-25
из р-ров в циклогексаноне — смесь растворителя, воды
и спирта (обычно этилового).
Жгут свежесформованного волокна, содержащего
остатки растворителя, вытягивают при нормальной
темп-ре в 2—8 раз. Затем волокно отмывают от раство-
растворителя, вытягивают в 1,2—3,0 раза при повышенных
темп-pax, сушат, гофрируют, режут на штапельки и
обрабатывают антистатич. препаратами.
По мокрому способу получают в основном штапель-
штапельные волокна (число отверстий в фильере до 50 000,
скорость формования 5—20 м/мин). Нити этим спосо-
способом в пром-сти производят только из перхлорвиниловои
смолы (число отверстий фильеры от 40 до 100, скорость
формования 25—60 м/мин).
При экструз ионном способе формо-
формования полимер, переведенный в результате нагрева
в пластичное состояние, продавливают через фильеру
шнеком. Этим способом получают моноволокно и щети-
щетину, а в опытном масштабе нити.
Волокна из сополимеров винилиденхлорида с винил-
хлоридом получают только экструзионным способом.
Темп-pa нагрева сополимера в зоне шнека составляет
130—170 °С, давление перед фильерой — 15—25 Мн/м2
A50—250 кгс/см2). В сополимер вводят стабилизаторы,
напр, феноксипропиленоксид A — 2% от массы полиме-
полимера). Быстрая кристаллизация сополимера при темп-рах
выше температуры стеклования затрудняет вытяжку
волокон. Поэтому волокна после формования быстро
охлаждают («закаливают»), а затем, для достижения
необходимой прочности, проводят холодную вытяж-
вытяжку уже закаленного волокна. Синтез высокоэффектив-
высокоэффективных термостабилизаторов и усовершенствование кон-
конструкции экструдеров могут привести к тому, что экс-
трузионный способ станет основным для производст-
производства П. в.
Обычно П. в. окрашивают в массе по общепринятой
схеме (см. Крашение химических волокон в массе). Кроме
того, П. в. можно окрашивать с поверхности, применяя
дисперсные красители (азокрасители, производные ант-
рахинона или нафтохинона), а также кубовые красители
в форме кубозолей. Для получения глубоких окрасок
процесс ведут при повышенной темп-ре (~100°С),
применяя вещества, разрыхляющие структуру по-
полимера (см. Крашение волокон).
Свойства. П. в. обладают высокой химпч. стойкостью
(особенно волокна из гомополимера), очень низкой
тепло- и электропроводностью, негорючи, устойчивы
к действию микроорганизмов. Для П. в., не подверг-
подвергнутых термофиксации, характерна высокая усадка,
достигающая для волокон из атактич. гомополимера
в кипящей воде 55% (от первоначальной длины). Тер-
Термофиксация П. в. при темп-pax на 20 — 60 °С выше
темп-ры стеклования значительно уменьшает усадку.
Физич. показатели П. в. зависят в основном от вида
исходного сырья и характера последующей обработки
(таблица).
Применение. П. в. в чистом виде применяют для про-
производства фильтровальных и негорючих драпировочных
тканей, спецодежды, нетканых изделий, технич. вой-
войлока, а также различных теплоизоляционных материа-
материалов, используемых при низких темп-pax. Способность
П. в. накапливать высокий отрицательный электроста-
тич. заряд используют для изготовления из них ле-
лечебного белья. В смесях с другими волокнами П. в.
часто применяют для достижения «эффекта усадочно-
сти». Из таких смесей изготовляют ткани повышенной
плотности, рельефные ткани, ковры, искусственную
кожу, замшу, пушистые трикотажные изделия. Волок-
Волокна из гомополимера повышенной синдиотактичности
и из смесей поливинилхлорида с нек-рыми полимера-
полимерами (ацетилцеллюлозой, хлорированным поливинил-
хлоридом с содержанием хлора 70—72%) после термо-
термофиксации не усаживаются даже при темп-рах 100 —
130 °С и используются для изготовления широкого
ассортимента изделий.
В 1971 объем мирового производства П. в. составил
1,5—2% от общего производства синтетич. волокон. Де-
Дешевизна и доступность исходных полимеров, разрабо-
разработанные в последние годы экономичные и безопасные
методы формования (в частности, из р-ров в диметил-
формамиде), создание П. в. с повышенной теплостой-
теплостойкостью (напр., из полимера повышенной стереорегу-
лярности и смесей полимеров), по-видимому, обусловят
дальнейшее увеличение как абсолютного объема произ-
производства П. в., так и их доли в общем выпуске синтетич.
волокон.
П. в. выпускают в различных странах под след. тор-
торговыми названиями: ПВХ-волокио (СССР),
ровиль, ф и б р о в и л ь (Франция), тевирон
(Япония), м о в н л ь (Италия) — из гомополимера;
хлорин (СССР), пивиацид (ГДР) — из пер-
перхлорвиниловои смолы; ТПВХ-волокно (СССР),
л е а в ин (Италия) — из поливинилхлорида повы-
повышенной синдиотактнчпости; ацетохлорин
(СССР) —из смеси поливинилхлорида с ацетилцеллю-
ацетилцеллюлозой; к л е в и л ь (Франция) — из смеси поливинил-
поливинилхлорида с хлорированным продуктом (содержание хлора
до 70—72%); в и н ь о н (США) — из сополимеров
винилхлорида с винилацетатом или акрилонитрилом;
саран (США), с о в и д е н (СССР) — из сополимера
винилхлорида с винилиденхлоридом.
Промышленное производство П. в. было впервые
освоено в 1936 в Германии (из перхлорвиниловои смо-
смолы) и в 1938 в США (из сополимеров винилхлорида).
Промышленный выпуск волокон из гомополимера впер-
впервые освоен в 1950 во Франции.
Лит.: Ф и х м а н В. Д., Поливинилхлоридные волокна,
в кн.: Карбоцепные синтетические волокна, под ред. К.Е.Пе-
803
ПОЛИВИНИЛХЛОРИДНЫЕ ПЛАСТМАССЫ
804
репелкина, М., 1973; Геллер Б. Э., Химия и технология
хлоринового волокна, М., 1958; Монкрифф Р. У., Хими-
Химические волокна, пер. с англ., М., 1964; Koch P. А., Тех-
tilveredlung, № 6, 318 A968). В. Д. Фихман.
ПОЛИВИНИЛХЛОРИДНЫЕ ПЛАСТМАССЫ [ ро
ly(vinyl chloride) plastics, Polyvinylchloridplaste, pla-
stiques de chlorure de polyvinyl]. К этой группе обычно
относят материалы на основе поливинилхлорида (ПВХ)
или сополимеров, содержащих более 50% винилхлори-
да. П. п. составляют ок. 25% общего мирового произ-
производства полимерных материалов. Широкое распростра-
распространение этих пластмасс и быстрый рост их производства
(ок. 20% в год) обусловлены сравнительно низкой стои-
стоимостью, хорошими физико-механич. и электрич. свой-
свойствами, химич. стойкостью, способностью к модифика-
модификации свойств (при введении различных добавок), а также
возможностью получения из них материалов и изделий
практически всеми известными способами переработки
термопластов (см. Переработка пластических масс).
Состав и свойства. В П. п. для повышения их ста-
стабильности (особенно термостабильности) и улучшения
перерабатываемое™, а также с целью модификации
свойств материала вводят значительные количества
различных ингредиентов — пластификаторы, термо- и
светостабилизаторы, наполнители, красители, моди-
модифицирующие полимеры (модификаторы"!, смазки, добав-
добавки, придающие специальные свойства (напр., фунги-
фунгициды, антипирены, антистатики, осветлители). Ассор-
Ассортимент этих ингредиентов очень широк. Выбор их опре-
определяется назначением материала, условиями его экс-
эксплуатации и переработки, стоимостью и пр.
Выбор полимера обусловлен, как правило, пред-
предполагаемым методом переработки и уровнем требуемых
свойств пластмассы. При этом определяющими фак-
факторами являются тип полимера, его мол. масса, разме-
размеры и морфология частиц порошка (см. Винилхлорида
полимеры). Для пластмасс, перерабатываемых каланд-
рованием, экструзией, литьем под давлением и др.
методами, применяют обычно суспензионный или блоч-
блочный ПВХ, хотя в отдельных случаях используют и
эмульсионный полимер.
Мол. масса полимера определяет уровень физико-
механич. свойств конечного продукта и возможность
его переработки. Чем выше мол. масса, тем выше проч-
прочностные свойства пластмассы, но тем сложнее перера-
переработка. Морфология частиц порошка имеет особенно
большое значение для переработки пластмасс, содержа-
содержащих пластификаторы. Для получения пластмасс высо-
высокого качества желательно применение пористого ПВХ
с морфологически однородными зернами. Для получе-
получения пластизолей (см. Пасты полимерные) применяют
обычно эмульсионный или т. наз. микросуспензионный
ПВХ со специальными характеристиками. Для облег-
облегчения переработки, особенно жестких П. п., при их
приготовлении применяют (самостоятельно или как
добавки к ПВХ) сополимеры винилхлорида с винилаце-
татом, пропиленом или акриловыми мономерами. Со-
Содержание сомономера не превышает 10—15%.
Пластификаторы вводят в П. и. с целью
получения материалов, мягких при обычных темп-рах
и обладающих хорошими свойствами при низких
темп-pax. Небольшие количества пластификаторов вво-
вводят в П. п. для облегчения их переработки. В зависи-
зависимости от растворяющей способности по отношению
к ПВХ пластификаторы можно разделить на первич-
первичные и вторичные. К первичным относят пластификато-
пластификаторы, хорошо совмещающиеся с ПВХ и сохраняющие эту
совместимость в условиях эксплуатации материала:
сложные эфиры фталевой, себациновой, адипиновой
и др. двухосновных к-т, а также ряд сложных эфиров
фосфорной к-ты и др. Их вводят в количестве 5—50%
от массы ПВХ (в зависимости от требуемых свойств
П. п.). Вторичные пластификаторы (хлорированные
парафины и нек-рые высококипящие ароматич. углево-
углеводороды) ограниченно совместимы с ПВХ; их самостоя-
самостоятельно не применяют, а заменяют ими часть первичных
пластификаторов (в зависимости от свойств последних
до 50%) с целью снижения стоимости материала или
придания ему специальных свойств.
В пластмассах, к-рые должны обладать стойкостью
по отношению к экстрагирующим средам, применяют
полимерные пластификаторы. Такие пластификаторы,
как эпоксиалкилфталаты, эпоксиалкилстеараты, обла-
обладают стабилизирующим действием. Слабый стабилизи-
стабилизирующий эффект оказывает также ряд обычных пласти-
пластификаторов. В нек-рых спец. случаях используют пла-
пластификаторы, способные к полимеризации, напр, поли-
гликольдиметакрилаты. При этом в П. п. перед перера-
переработкой вводят инициаторы полимеризации. Пластмас-
Пластмассы, содержащие пластификаторы, часто наз. пласти-
катами.
Термостабилизаторы вводят в пласт-
пластмассы на основе ПВХ с целью снижения интенсивности
деструкции полимера при переработке, в результате
к-рой материал окрашивается; при глубоком разложе-
разложении у него ухудшаются свойства. Для сохранения экс-
эксплуатационных свойств материала введение термоста-
термостабилизаторов обязательно только в тех случаях, если
материал предназначен для использования при высо-
высоких темп-pax или подвергается дополнительной тепло-
тепловой обработке, напр, при формовании, сварке.
Применяемые для ПВХ стабилизаторы можно раз-
разделить на след. группы: а) свинцовые, б) оловооргани-
ческие, в) барий-кадмий-цинковые, г) кальций-цин-
кальций-цинковые. Помимо этих основных стабилизаторов, приме-
применяют также вторичные — эпоксисоединения, фосфиты,
соединения фенольного типа.
Светостабилизаторы (обычно производ-
производные бензофенона, бензотриазола и др.) вводят в мате-
материалы, предназначенные для эксплуатации вне поме-
помещений. В основном их действие сводится к поглощению
активной части излучения и экранированию полимера.
Помимо стабилизаторов, применяемых для защиты
полимера, в пластмассы на основе ПВХ вводят а н-
тиоксиданты, основная роль к-рых — защита
пластификаторов и модификаторов от окисления при
высоких темп-рах.
Количество стабилизатора зависит от его эффектив-
эффективности и от нек-рых побочных воздействий, к-рые он
может оказывать на свойства П. п. Основные стаби-
стабилизаторы вводят в количества 1—3% (реже 5%). Иног-
Иногда вводят большие количества стабилизаторов; в этих
случаях они выполняют роль и наполнителей. Для
придания П. п. определенного комплекса свойств ис-
используют смеси стабилизаторов. Вторичные стабили-
стабилизаторы, светостабилизаторы и антиоксиданты добав-
добавляют в значительно меньших количествах, чем основ-
основные стабилизаторы.
Введение наполнителей в П. п. обусловлено
гл. обр. стремлением к снижению их стоимости, а также
возможностью придания им различных свойств (напр.»
непрозрачности, светостойкости, увеличения электрич.
сопротивления, повышения твердости и др.). Выбор на-
наполнителя зависит от требований, предъявляемых
к свойствам материала и его стоимости. При этом не-
необходимо учитывать, что зависимость нек-рых свойств
пластмасс от содержания наполнителя проходит через
максимум (напр., прочность, электрич. проводимость)»
а изменение др. происходит равномерно.
В качестве наполнителей применяют каолин, тальк,
асбест, слюду, мел, диатомовую землю, сульфат бария
и др. В ряде случаев поверхность наполнителей пред-
предварительно обрабатывают веществами, улучшающими
взаимодействие их с полимером. Количество наполни-
наполнителя в П. п. может колебаться от 2—3 до 100% и более
(от массы ПВХ) в зависимости от природы наполнителя
и назначения П. п.
805
ПОЛИВИНИЛХЛОРИДНЫЕ ПЛАСТМАССЫ
806
Растворимые в полимере органич. красители
применяются сравнительно редко для окрашивания
П. п., гл. обр. прозрачных. Их основной недостаток —
невысокая стойкость и миграция. Наиболее употреби-
употребительны минеральные и органич. пигменты (см.
Красители). Помимо спектральных характеристик,
термо- и светостойкости, существенно важна дисперс-
дисперсность пигментов, от к-рой зависит равномерность окра-
окрашивания материала.
Модификаторы добавляют к ПВХ (в коли-
количестве 10—15%) с целью улучшения его перерабатывае-
мости и повышения ударной вязкости. В качестве моди-
модификаторов ПВХ применяют акриловые сополимеры
(обычно тройные сополимеры акрилонитрила с бута-
бутадиеном и стиролом), хлорированный полиэтилен и
каучуки.
Наилучшие свойства П. п. достигаются при опреде-
определенной фазовой морфологии смеси (оптимальной дис-
дисперсности модификатора, степени агрегирования его
частиц и т. д.) и оптимальной прочности связи между
частицами модификатора и ПВХ. Если показатель пре-
преломления модификатора равен показателю преломления
ПВХ или достаточно близок к этому значению, то по-
получают прозрачные пластмассы, обладающие высокой
ударной вязкостью. Несмотря на нек-рое снижение
физико-механич. свойств и теплостойкости, модифици-
модифицированные пластмассы находят широкое применение
благодаря пониженной хрупкости при обычных и низ-
низких темп-рах.
Смазки — необходимый компонент всех жестких
(не содержащих пластификатора) П. п. В ряде случаев
смазки вводят также и в пластифицированные мате-
материалы. Смазками могут служить пизкомолекулярные
или полимерные вещества, плохо совмещающиеся или
несовмещающиеся с ПВХ. В зависимости от степени
совместимости с ПВХ смазки обычно подразделяют на
внешние и внутренние, хотя это деление в известной
мере условно. Внешние смазки (напр., парафины, вос-
воска, низкомолекулярный полиэтилен) выделяются из
расплава на поверхность раздела расплав — стенки
перерабатывающего оборудования, уменьшая внешнее
трение. Внутренние смазки (моноэфиры глицерина,
стеараты металлов и др. мыла) остаются в расплаве;
распределяясь между элементами надмолекулярной
структуры полимера, они оказывают влияние на вяз-
вязкость расплава и распределение скоростей течения по
профилю канала. Для достижения максимального эф-
эффекта часто используют комбинации различных смазок.
Смазки эффективны в малых концентрациях, их содер-
содержание обычно не превышает 1% , но, тем не менее, они
оказывают заметное влияние на физико-механич. свой-
свойства материала.
В П. п., предназначенные для эксплуатации в спе-
специальных условиях, помимо перечисленных выше ин-
ингредиентов, часто вводят дополнительные добавки.
П. п., как и многие др. полимерные материалы, под-
подвержены воздействию бактерий и грибков. Для предот-
предотвращения этого воздействия используют фунги-
фунгициды: неорганич. соединения мышьяка и меди, оло-
воорганич. соединения, меркаптаны и четвертичные ам-
аммониевые соединения. Общее содержание их в пласт-
пластмассе может колебаться от 0,1 до 5%.
ПВХ — негорючий полимер благодаря высокому
содержанию в нем хлора. Однако при добавлении
25% пластификатора полимер становится горючим. В
связи с этим в пластифицированные материалы иног-
иногда добавляют (в количестве 1—3%) антипире-
н ы — соединения фосфора (обычно сложные эфиры
фосфорной кислоты), трехокись сурьмы (см. Анти-
пирены).
Для предотвращения накопления электростатич. за-
зарядов на поверхности изделий из П. п. используют
антистатики: производные четвертичных аммо-
аммониевых оснований, длинноцепочечные алифатич. амины
и амиды в количестве от 0,5 до 1,5%. Поскольку анти-
антистатики по химич. природе сходны со смазками и могут
обладать смазывающим действием, содержание этих
двух компонентов материала должно быть скоррек-
скорректировано. См. также Антистатики.
Для придания прозрачным П. п. привлекательного
вида, в частности для компенсации желтоватого оттен-
оттенка, в них вводят так наз. осветлители, напр, тинопал,
белофор (в очень малых количествах — от 0,001 до
0,05%, т. к. они ускоряют световое старение пласт-
пластмассы). Действие осветлителей основано на том, что
они люминесцируют голубоватым светом при возбуж-
возбуждении ультрафиолетовой частью спектра.
Получение. П. п. представляют собой сложные мно-
многокомпонентные системы с весьма разнообразными ви-
видами взаимодействий между ингредиентами. В значи-
значительной мере свойства пластмассы зависят от условий
ее получения. Независимо от того, в какой конечной
форме получают пластмассу, начальными стадиями
технологич. процесса являются смешение и гомогени-
гомогенизация. Цель этих операций — приведение всех компо-
компонентов смеси в мелкодисперсное состояние, обеспечи-
обеспечивающее получение изделий и материалов нужного каче-
качества при последующей переработке. Различают два
основных типа процессов смешения и гомогенизации.
К первому можно отнести смешивание и гомогенизацию
в расплавленном состоянии. В этом случае предвари-
предварительно грубо смешанные компоненты перетираются и пе-
перемешиваются в тяжелых смесителях интенсивного
действия, напр, на вальцах, в смесителях типа Бенбери
или в мощных компаундирующих экструдерах. В слу-
случае смесей, не содержащих трудно перерабатываемых
добавок, для компаундирования применяют обычные
экструдеры. Темп-pa массы при этом повышается, гл.
обр. вследствие внутреннего трения. Расплавленная
масса после этого поступает на формование изделий или
на гранулирование. Этот метод наиболее пригоден для
получения П. п. с высоким содержанием наполнителей
или модификаторов, а также для переработки отходов
пластмасс на основе ПВХ.
Второй метод, получивший название метода «сухих
смесей», заключается в добавлении жидких и твердых
добавок к ПВХ и перемешиванию их в легких быстро-
быстроходных или тихоходных смесителях до получения легко
сыпучей порошкообразной смеси, в к-рой ингредиенты
распределены достаточно равномерно для последую-
последующей переработки обычными методами. Этот способ
предъявляет ряд, специфич. требований к сырью, осо-
особенно к ПВХ. Преимущество этого метода в том, что
в процессе приготовления смеси полимер подвергается
значительно меньшему термич. воздействию, чем при
компаундировании расплава. Методом сухого смеше-
смешения перерабатывают смеси, не содержащие больших
количеств наполнителей и др. трудно перерабатывае-
перерабатываемых добавок. В этом случае ПВХ должен быть доста-
достаточно пористым, чтобы обеспечить полное поглощение
пластификатора и др. жидких добавок. Желательно,
чтобы частицы полимера были крупными (сыпучесть
смеси должна быть высокой). Существуют также спе-
специальные приемы агломерирования порошкообразной
смеси для обеспечения высокой сыпучести и повыше-
повышения насыпной массы смеси. См. также Смесители, Сме-
Смешение.
При получении П. п. пластизольным методом (см
Пасты полимерные) смесь ингредиентов с ПВХ пасто-
пастообразной консистенции заливают в форму, наносят нг
какую-либо основу или пропитывают ею ткань, а за-
затем путем нагревания и последующего охлаждения
переводят в монолитное состояние, в к-ром реализуется
требуемый комплекс свойств. К этому методу, в извест-
известной степени, близок способ получения пористых П. п.
(см. Пенополивинилхлорид).
807
ПОЛИВИНИЛХЛОРИДНЫЕ ПЛЕНКИ
Переработка. Порошкообразные или гранулирован-
гранулированные П. п. можно перерабатывать на всех видах совре-
современного перерабатывающего оборудования. Один из
наиболее важных методов переработки П. п.— каланд-
рование. Его можно осуществлять при довольно высо-
высоких значениях вязкости расплава. Этим методом обычно
перерабатывают полимеры высокой мол. массы, высо-
конаполненные композиции как пластифицированные,
так и непластифицированные.
Многие П. п. перерабатывают экструзией (на одно-
шнековых, двухшнековых и многошнековых экстру-
дерах). Для переработки непластифицированных мате-
материалов предпочтительно использовать экструдеры с ва-
вакуумным отсосом. Для эффективной переработки при-
применяют шнеки специальной конструкции, приспособ-
приспособленные для мягких или жестких пластмасс. Все более
широкое признание для производства полых объемных
изделий из П. п. получает метод экструзии с выду-
выдуванием.
Литье под давлением также применимо для получе-
получения изделий из П. п. В этом случае обычно применяют
машины шнекового типа, причем рекомендуют специ-
специальные шнеки. Листовые материалы, полученные ка-
ландрованием или экструзией, легко перерабатывают
в изделия методами вакуумирования, пневмоформова-
ния и штампования.
Применение. Одна из наиболее важных областей ис-
использования П. п.— производство электроизоляцион-
электроизоляционных материалов (см. Пластикат), на к-рое расходу-
расходуется 10—20% общего производства ПВХ.
П. п. широко применяются в строительстве для изго-
изготовления деталей окон и дверей, плинтусов, покрытий
для полов, водосточных желобов и труб, элементов сан-
технич. оборудования (см. Полимеры в строительстве).
Для этих целей расходуется 10—25% общего количест-
количества ПВХ.
В количестве 7—10% общего производства ПВХ рас-
расходуется на изготовление одежды, обуви (см., напр.,
Кожа искусственная). Значительные количества П. п.
применяются для производства различных деталей
в автомобильной, вагоностроительной и самолетострои-
самолетостроительной пром-сти. Ок. 5% ПВХ расходуется на изго-
изготовление граммпластинок, не менее 5% — на изго-
изготовление различных упаковочных материалов и тары,
в том числе для пищевых продуктов (см. Полимеры
в пищевой промышленности). П. п. применяются также
в химическом машиностроении (см. Полимеры в ма-
машиностроении, Винипласт), в медицине (см. Полиме-
Полимеры в медицине), в сельском хозяйстве, особенно в ви-
виде пленок (см. Полимеры в сельском и водном хозяйст-
хозяйстве, Поливинилхлоридные пленки), для изготовления
игрушек (см. Пасты полимерные), спортивных принад-
принадлежностей, канцелярских товаров, товаров домашне-
домашнего обихода.
Лит.: X у в и н к Р., Ставерман А. [сост.], Химия
и технология полимеров, пер. с нем., т. 2, ч. 1, М., 1965; М и н-
скер К. С., Федосеева Г. Т., Деструкция и стабили-
стабилизация ПВХ, М., 1972; Голдинг Б., Химия и технология
полимерных материалов, пер. с англ., М., 1963.
Б. П. Штаркман.
ПОЛИВИНИЛХЛОРИДНЫЕ ПЛЕНКИ [ poly (vinyl
chloride) films, Polyvinylchloridfilme, films de chlorure
de polyvinyle].
П. п. подразделяют на пленки общетехнического на-
назначения и пленки для пищевой и медицинской про-
промышленности, к к-рым предъявляют повышенные то-
токсикологические требования.
Состав. В композицию, предназначенную для полу-
получения пленок, кроме полимера и стабилизатора, могут
входить пластификаторы, пигменты, смазки и некото-
некоторые др. добавки. Содержание и химическая природа
отдельных ингредиентов, входящих в такие компози-
композиции, в основном те же, что и в винипласте и пласти-
пластикате. В зависимости от содержания пластификатора
различают пленки жесткие @—5% пластификатора),
полужесткие E—15%) и мягкие (более 15%). Мягкие
пленки в СССР наз. пленочным пластикатом,
жесткие — пленочным винипластом. В
производстве пленочного пластиката применяют сус-
суспензионный поливинилхлорид с мол. массой 60 000—
75 000, в производстве пленочного винипласта — с мол.
массой ок. 50 000. Для получения пленок используют
также саран — сополимеры винилхлорида с винили-
денхлоридом, содержащие 75—90% последнего (см.
Поливинилиденхлоридные пленки). Выбор стабилизирую-
стабилизирующей системы зависит от назначения пленочного мате-
материала. Для пластифицированных пленок применяют
стабилизаторы на основе соединений бария и кадмия
в сочетании с эпоксидными стабилизаторами-пластифи-
стабилизаторами-пластификаторами, органич. фосфитами и добавками для умень-
уменьшения фотохимич. деструкции (напр., производными
бензофенона или бензтриазола); возможно применение
карбоксилатов диалкилолова в сочетании с солями
жирных к-т бария и кадмия. В жесткие пленки, помимо
этих стабилизирующих систем, вводят меркаптиды
и тиогликоляты диалкилолова и основные свинцовые
соли (стеараты, сульфаты и фосфиты). В производстве
нетоксичных П. п. используют кальций-цинковые стаби-
стабилизаторы в комбинации с эпоксидированным соевым
маслом и низкотоксичными добавками для уменьшения
фотохимич. деструкции (подробнее о составе компо-
композиций на основе поливинилхлорида см. Поливинилхло-
Поливинилхлоридные пластмассы).
Производство. Наиболее распространенные техноло-
гич. методы переработки поливинилхлорида в плен-
пленки — вальцево-каландровый и экструзионный (см.
Пленки полимерные). В обоих случаях первой опера-
операцией является смешение полимера с др. компонентами
композиции в смесителе любого типа в течение 25 —
60 мин (в зависимости от конструкции смесителя и ре-
рецептуры). Порядок введения компонентов и температур-
температурные условия смешения определяются рецептурой и ско-
скоростью поглощения пластификатора полимером.
При вальцево-каландровом спосо-
б е обработка смеси на фрикционных вальцах (при
160—170 °С для винипласта и 155 —185 °С для пласти-
пластиката) предназначена для придания ей гомогенности и
пластичности. При каландровании вальцованной массы
из нее удаляется воздух, материал уплотняется и полу-
получаются листы или непрерывная лента заданной толщи-
толщины и ширины. Этот процесс осуществляют на 3—5-вал-
ковых каландрах; температура первого валка обычно
155 °С, последнего — 170 °С при переработке вини-
винипласта или соответственно 155 и 185 °С при переработ-
переработке пластиката. Правильный выбор и постоянство тем-
температур переработки необходимы во избежание раз-
нотолщинности пленки. Каландрированная пленка
имеет преимущественную ориентацию макромолекул
вдоль полотна.
При экструз ионном способе в зависи-
зависимости от конструкции головки экструдера П. п. полу-
получают в виде: 1) тонкостенной трубы, к-рую затем разду-
раздувают с образованием пленочного рукава (рукавный ме-
метод), и 2) пленочного полотна, к-рое охлаждают на ме-
таллич. барабане или в водяной ванне (плоскощелевой
метод). Рукавным методом получают очень тонкие плен-
пленки (толщина несколько мкм). Температурный режим
экструзии зависит от состава композиции и конструк-
конструкции головки. Темп-ру на выходе из головки поддержи-
поддерживают обычно в пределах от 155 до 185 °С. В ряде случа-
случаев для получения П. п. с заданными физико-механич. и
физико-химич. свойствами применяют дополнительные
технологические операции — вытяжку пленки, ее тер-
мич. обработку, дублирование с бумагой, тканями или
др. пленками, нанесение на поверхность пленки спе-
специальных композиций, придающих ей липкость, гид-
рофильноетъ или др. свойства.
809
ПОЛИГЕКСАМЕТИЛЕНАДИПИНАМИД
810
Для специальных целей находят применение плен-
пленки, получаемые методом полива из раство-
растворов поливинилхлорида в тетрагидрофуране или цик-
логексаноне.
Свойства и переработка. П. п. обладают комплексом
ценных свойств: химической стойкостью, высокими
электроизоляционными свойствами, малой теплопро-
теплопроводностью, ограниченной горючестью, низкой влаго-
проницаемостью, хорошей атмосферостойкостью и мик-
робиологич. устойчивостью. Светопрозрачные пленки
пропускают до 90% коротковолнового излучения и 10%
длинноволнового. Поэтому они более удобны для уст-
устройства теплиц, чем силикатное стекло, не пропускаю-
пропускающее ультрафиолетового излучения.
П. п. перерабатывают в изделия методами вакуум-
и пневмоформования, сваривают токами высокой ча-
частоты и склеивают при повышенных темп-pax. В табли-
таблице приведены нек-рые свойства П. п.
Свойства поливинилхлоридных пленок
Показатели
Плотность, г/см3
Прочность при растяжении, Мн/м2
(кгс/см2)
Относительное удлинение, % ....
Водопоглощение за 24 ч при 20 °С, %
Уд. поверхностное электрич. сопро-
сопротивление, Том (ом)
Уд. объемное электрич. сопротивле-
сопротивление, Том-м (ом-см)
Тангенс угла диэлектрич. потерь
при 5 0 гц
Диэлектрич. проницаемость при 5 0г^
Электрич. прочность, Мв/м, или кв/мм
Светопрозрачность (для прозрачных
пленок), %
Жесткая
поливинил-
хлоридная
пленка
1,37-1,45
45D50)
30-50
0,1-0,5
100A О14)
10A015)
0,01-0,05
3,2-4
15-35
до 80
Пластифи-
Пластифицированная
поливинил-
хлоридная
пленка
1,2-1,6
14-28
A40—280)
150 — 300
0,01-1
A012—10")
0,1
4,2-4,5
6-12
90
Применение. Относительная дешевизна, доступность
сырья и возможность изменения свойств в широких
пределах способствуют постоянному увеличению про-
производства П. п. В США, Японии и Великобритании
ок. 30% производимого поливинилхлорида использует-
используется для получения пленок.
П. п. применяют в качестве обычной и термоусадоч-
термоусадочной упаковки пищевых продуктов и промышленных
товаров широкого потребления, а также в качестве та-
тары для хранения и транспортировки различных жид-
жидкостей. В медицине пластифицированные П. п. исполь-
используют для покрытия аппаратуры, соприкасающейся
с кровью, при изготовлении тары для хранения крови
и повязок, применяемых при пластич. операциях, и во
многих др. случаях.
В сельском хозяйстве П. п. используют для устрой-
устройства теплиц, в к-рых легко поддерживать благоприят-
благоприятный для растений темп-рный режим, высокую относитель-
относительную влажность и освещенность. В качестве материала
для теплиц П. п. более долговечны (срок их службы до
3 лет), чем полиэтиленовые пленки, и, кроме того,
в отличие от последних они не подвергаются необрати-
необратимому загрязнению. Использование П. п. для силосо-
силосования сочных кормов, упаковки удобрений, изготовле-
изготовления надувных переносных складов, устройства проти-
вофильтрационных экранов на оросительных каналах
и для других нужд позволяет значительно снизить се-
себестоимость сельскохозяйственной продукции (см. так-
также Полимеры в сельском и водном хозяйстве). П. п. ис-
используют в строительстве жилых и общественных зда-
зданий. Водостойкие пленки применяют для гидроизоля-
гидроизоляции кровель, фундаментов, пароизоляции, в теплоизо-
теплоизоляционных прокладках и вкладышах, для защитных
укрытий строящихся объектов и др. П. п. могут слу-
служить заменителями стекла.
С помощью П. п. можно производить консервацию
машин и механизмов для защиты от коррозии. Дубли-
Дублированные с тканями П. п. могут заменить брезент при
укрытии крупногабаритных изделий, строящихся соо-
сооружений, стоящих в доках судов и т. д.
Транспортерные ленты, имеющие вместо резиновой
обкладки пленочные покрытия из поливинилхлорида,
более стойки к атмосферным влияниям и воздействию
агрессивных сред, обладают высокой износоустойчи-
износоустойчивостью и прочностью на раздир. П. п. удобны для изго-
изготовления средств индивидуальной защиты, поскольку
они не поглощают радиоактивные загрязнения и хоро-
хорошо очищаются от них, не пропускают токсичных ве-
веществ и свинцовой пыли.
Лит.: Козлов П. В., Брагинский Г. И., Хи-
Химия и технология полимерных пленок, М., 1965; Т а к а х а-
с и Г., Пленки из полимеров, пер. с япон., Л., 1971; Encyclo-
Encyclopedia of polymer science and technology, N. Y.— [a. o.L v. 2,
1965, p. 353; v. 14, 1971, p. 438, 441.
Ю. В. Овчинников, Г. Т. Федосеева, В. Ф. Сливаева.
ПОЛИВИНИЛЦИКЛОАЛКАНЫ — см.
алканов полимеры.
ПОЛИВИНИЛЦИКЛОГЕКСАН - см.
алканов полимеры.
ПОЛИВИНИЛЦИКЛОПЕНТАН— см. Винилциклоал-
канов полимеры.
ПОЛИВИНИЛЦИКЛОПРОПАН — см. Винилцикло-
алканов полимеры.
ПОЛИГЕКСАМЕТИЛЕНАДИПИНАМИД, поли-
полиамид- 6,6 [poly(hexamethylene adipamide), Poly-
hexamethylenadipamid, polyhexamethylene adipamide] —
линейный алифатич. полиамид
Винилцикло-
Винилцикло-
Свойства. П.— твердый роговидный кристаллич. по-
полимер белого цвета, без запаха; мол. масса составляет
15 000—25 000. В обычных растворителях (напр., спир-
спиртах, сложных эфирах, кетонах, алифатич. и ароматич.
углеводородах) П. нерастворим; растворяется в конц.
H2SO4, уксусной и муравьиной к-тах, фторированных
спиртах и фенолах. При нагревании к-ты (напр., сер-
серная, соляная, муравьиная) вызывают гидролиз П. По-
Полимер устойчив к действию масел, разб. и конц. р-ров
щелочей. При темп-pax выше 350 °С П. разлагается
с выделением газообразных продуктов: окиси и двуоки-
двуокиси углерода, аммиака. П. сильно поглощает влагу (по-
(поглощение воды при насыщении составляет 9—10%).
П.— самозатухающий полимер. Он обладает высокой
прочностью, абразивостойкостью и значительно более
высокой термостойкостью, чем большинство др. али-
алифатич. полиамидов. При низкой влажности П.— хоро-
хороший электроизоляционный материал. Ниже приведены
нек-рые свойства П.:
Плотность при 20 °С, г/см3 1,14
Показатель преломления ng 1,532
Темп-pa, °С
плавления 264
размягчения 250
начала деформации
под нагрузкой 1 ,85Мн/м2, или
18,5 кгс/см2 75
под нагрузкой 0,46 Мн/м2, или
4,6 кгс/см2 200
хрупкдсти от 25 до—30
Теплостойкость по Вика, °С 220 — 230
Теплостойкость по Мартенсу, °С 55 — 60
Уд. теплоемкость, кдж/(кг-К) [ккал/(кг-°С)] 1,68—2,1
[0,4-0,5]
Темп-рный коэфф. линейного расшире-
расширения, °С~1 1-Ю
Теплопроводность, вт/(см • К)
[кал/(см-сек-°С)] 0,25[6-10~*J
Усадка при литье, % 1,5
Прочность*, Мн/м2 (кгс/см2)
при растяжении • 80(800)
при сжатии 46D60)
при изгибе 100A000)
811
ПОЛИГЕКСАМЕТИЛЕНСЕБАЦИНАМИД
812
Модуль упругости при растяжении,
Мн/м2(кгс/см2) 3000C0 000)
Относительное удлинение *,% 80—100
Твердость по Бринеллю *, Мн/м2(кгс/см2) . . . 67 — 70
F70-700)
Электрич. прочность, Мв/м, или кв/мм .... 470
Уд. объемное электрич. сопротивление,
ом-см A —4) -1 0~13
Диэлектрич. проницаемость при 60 гцB3сС) 4,6
Тангенс угла диэлектрич. потерь при 60 га
B5 °С) 0,04
* Для неориентированных образцов.
Получение. П. может быть получен поликонденсацией
на границе раздела двух фаз или в расплаве гексаме-
тилендиамина и адипиновой к-ты (в нервом случае ис-
используют дихлорангидрид к-ты). В пром-сти П. полу-
получают поликонденсацией в расплаве соли гексамети-
лендиамина и адипиновой к-ты (т.1 н; соли А Г,
или соли найлона-6,6). Использование соли АГ
(вместо отдельно взятых компонентов) обеспечивает
в процессе поликонденсации строгое соблюдение экви-
молярного соотношения между диамином и дикарбоно-
вой к-той и возможность получения П. необходимой
мол. массы:
п H2N(GH2N NH2 + п НООС(СН2L СООН ->
[-HN(CH2NNHCO(CH2LGO-]n+ 2n Н2О
Г
n H
LOOC(GH2LCOO
Соль АГ — кристаллич. порошок белого цвета, без
запаха; т. пл. 190—191 °С; растворима при комнатной
темп-ре в воде, при нагревании —в метиловом и этило-
этиловом спиртах; нерастворима в эфире. Соль АГ получают
смешением 60—80% -ного р-ра гексаметилендиамина
и 20%-ного р-ра адипиновой к-ты в кипящем спирте.
Выпавшую соль из охлажденной до комнатной темп-ры
смеси отфильтровывают центрифугированием, промы-
промывают спиртом и сушат. В пром-сти соль АГ часто полу-
получают смешением стехиометрич. количеств адипиновой
к-ты и гексаметилен*диамина в водных р-рах. В этом
случае исходные реагенты должны обладать высокой
степенью чистоты, т. к. полученный водный р-р соли
непосредственно используют для получения П.
Поликонденсацию соли АГ проводят в стальных авто-
автоклавах емкостью 1 — 6 м3 в атмосфере инертного газа
(азота) по периодич. схеме. Водный р-р соли 50—
60%-ной концентрации в присутствии регулятора мол.
массы (напр., уксусной или адипиновой к-ты в количе-
количестве 0,2—0,5% от массы соли) нагревают сначала до
220 °С (при этом давление достигает ~1,8 Мн/м2, или
—18 кгс/см2), а через 1—2 ч темп-ру повышают до
270—280 °С и выдерживают при этой темп-ре и дав-
давлении — 1,8 МнГм2 в течение 1 — 1,5 ч. Затем в те-
течение 1 ч давление снижают до атмосферного, по-
после чего (спустя —30 мин) опять повышают до
— 1,8 Мн/м1. Циклы поликонденсации при повышен-
повышенном и атмосферном давлении повторяют несколько раз,
что обеспечивает перемешивание расплава парами во-
воды, вскипающей при снижении давления. Иногда с це-
целью получения П. более высокой мол. массы (—25 000)
на последнем этапе поликонденсации применяют ва-
вакуум. Полученный П. выдавливают из автоклава сжа-
сжатым азотом в виде ленты на барабан. Ленту после ох-
охлаждения измельчают в крошку.
Переработка и применение. П. перерабатывают ли-
литьем, литьем под давлением, экструзией и прессова-
прессованием. Детали из П. можно сваривать (тепловой свар-
сваркой или токами высокой частоты) или склеивать р-рами
этого же полимера в многоатомных фенолах или му-
муравьиной к-те. Применяют П. гл. обр. для изготовления
волокон. См. также Полиамидные волокна, Полиамид-
Полиамидные пленки.
П. производится в различных странах под след. на-
названиями: анид (СССР); н а й л о н-6,6, зай-
т ел- 101 и з а й т е л-105 (США); маранил, лу-
рон, сутрон, брулон (Великобритания);
перлон Т, игамид А, энтернамид
(ФРГ). По объему производства П. занимает первое
место среди др. полиамидов.
Впервые П. получен У. X. Карозерсом в 1931.
Лит.: Коршак В. В., Фрунзе Т. М., Синтетиче-
Синтетические гетероцепные полиамиды, М., 1962, с. 429; Волокна из
синтетических полимеров, под ред. Р. Хилла, пер. с англ.,
М., 1957, с. 115; Н и к о л а е в А. Ф., Синтетические поли-
полимеры и пластические массы на их основе, 2 изд., М.— Л., 1966,
с. 600; Флойд Д. Е., Полиамиды, пер. с англ., М., 1960.
В.В.Курашев.
ПОЛИГЕКСАМЕТИЛЕНСЕБАЦИНАМИД, и о
л и а м и д-6,10 [poly(hexamethylene sebacamide), Po-
lyhexamethylensebazamid, polyhexamethylene sebac-
sebacamide] — линейный алифатич. полиамид
[-HN(CH2NNHCO(CH2)8CO-]n
Свойства. П.— твердый роговидный кристаллич. по-
полимер белого цвета, без запаха; мол. масса обычно со-
составляет — 20 000. П. растворим в конц. минеральных
к-тах, напр. H2SO4, муравьиной и уксусной к-тах, в фе-
фенолах и фторированных спиртах; устойчив к действию
алифатич. и ароматич. углеводородов, спиртов, кето-
нов, масел, разб. и конц. р-ров щелочей; при нагрева-
нагревании гидролизуется к-тами. П. менее гигроскопичен,
чем полигексаметиленадипинамид (поглощение воды
при насыщении составляет 3,5%). П.— самозатухаю-
самозатухающий полимер. Он характеризуется хорошими механич.
и диэлектрич. свойствами, абразивостойкостью и устой-
устойчивостью к истиранию. Ниже приведены нек-рые свой-
свойства П.:
Плотность при 20 °С, г/см3 1,09-1,11
Темп-pa, °С
плавления 213—220
начала деформации
под нагрузкой 1,85 Мн/м2
A8,5 кгс/см*) 55
под нагрузкой 0,46 Мн/м2
D,6 кгс/см2) ,... 160
Теплостойкость, °С
по Вика • 195 — 205
по Мартенсу 6 0
Уд. теплоемкость, кдж/(кг-К) [ккал(кг¦ °C)J l,68[0,4]
Теплопроводность, вт/(М'К)
{кал/(см-сек-0 С)] 0,21[5.10]
Темп-рный коэфф. линейного расшире-
расширения,0^1 15-10
Прочность, Мн/м2 (кгс/см2)
при растяжении 4 5 — 60
D50-600)
при сжатии с... 70 — 90
G00-900)
при изгибе 70 — 90
G00-900)
Модуль упругости при растяжении, Мн/м2
(кгс/см2) 2110B1 100)
Ударная вязкость, кдж/м2, или кгс-см/см2 . . 100-120
Относительное удлинение, % 100 — 150
Твердость по Бринеллю, Мн/м2 кгс/см2). . . . 100-150
A000-1500)
Уд. объемное электрич. сопротивление,
ом-см 4,5 -К)4
Тангенс угла диэлектрич. потерь 0,025—0,030
Получение. В промышленности П. получают поли-
поликонденсацией в расплаве соли гексаметилендиамина
и себациновой кислоты (так называемая соль С Г,
или соль найлона-6,10):
nH2N(CH?)eNH2 + п НООС(СН2)8СООН -»
Гн3Й(СН2)в NH3
L ООС(СН2)8СОб
t [-NH(CH2NNHCO(CH2)eCO-]n +2n H2O
Соль СГ —кристаллич. порошок белого цвета, без за-
запаха; т. пл. 170 °С; она плохо растворима при комнат-
комнатной темп-ре в воде; при нагревании растворяется в ме-
метиловом и этиловом спиртах, нерастворима в эфире.
Получение соли СГ и ее поликонденсацию осуществляют
аналогично получению и поликонденсации соли АГ
(см. Полигексаметиленадипинамид).
813
полигликолид
814
Переработка и применение. П. перерабатывают ли-
литьем под давлением, экструзией и прессованием. Литье-
Литьевые детали из П. могут быть сварены (тепловой сваркой
или токами высокой частоты) либо склеены р-рами этого
же полимера в многоатомных фенолах или в муравьи-
муравьиной к-те. П. применяют для изготовления изделий,
характеризующихся хорошими механическими и анти-
антифрикционными свойствами (в машиностроении, при-
приборостроении, авиационной, электротехнической и др.
отраслях промышленности), изделий, стойких к дейст-
действию щелочей, масел и углеводородов, а также воло-
волокон и пленок. См. также Полиамидные волокна, По-
Полиамидные пленки.
П. производится в ряде стран под след. названиями:
полиамид- 6,8 (СССР), найлон- 6,10, з а й-
тел-31, зайтел-33 (США), перлон Н,
энтернамид С (ФРГ). По масштабам производст-
производства П. значительно уступает др. полиамидам из-за отно-
относительно высокой стоимости себациновой к-ты, полу-
получаемой из касторового масла. П. впервые получен
У. X. Карозерсом в 1936.
Лит.: Коршак В. В., Фрунзе Т. М., Синтетиче-
Синтетические гетероцепные полиамиды, М., 1962, с. 438, Nesme] a-
п о w А. [а. о.], Chem. Technik, 9, № 3, 139 A957).
В. В. Курашев.
ПОЛИГИДРАЗИДЫ (polyhydrazides, Polyhydrazide,
polyhydrazides) — линейные полимеры, содержащие
в основной цепи макромолекулы гидразидные груп-
группировки —CONHNHOC— или —CONHNH —. П. мож-
можно рассматривать как полиамиды, полученные из не-
неорганического диамина (гидразина) и дикарбоновых
кислот.
П. получают поликонденсацией по след. схемам:
1. Из дигидразидов дикарбоновых к-т или гидразин-
гидрата (NH2—NH2H2O) и дихлорангидридов дикар-
дикарбоновых к-т, напр.:
-НС1
C1OC-R-COC1 + H2NHNOC-R'-CONHNH2 >
_> [_ОС—R-CONHNHOC-R'—CONHNH—]n
где R и R' — алифатич. или ароматич. радикалы. Ре-
Реакцию осуществляют в р-ре, напр, в гексаметилфосфор-
амиде @—5 °С), N-метилпирролидоне (—20 °С) или
]Ч,]М-диметилацетамиде (—20 °С). Алифатич. П. в про-
процессе синтеза выпадают из реакционной смеси (ввиду
нерастворимости данных о их мол. массе нет). Мол.
масса ароматич. П. сравнительно высока (приведенная
вязкость Т|пр> 1 дл/г).
2. Из дифениловых эфиров дикарбоновых к-т и ди-
дигидразидов дикарбоновых к-т или безводного гидра-
гидразина, напр.:
-Свн5он
CeH5OOC-R-COOCeH5 + H2NHNOC-R'-CONHNH2 >
_» [-OC-R-CONHNHOC-R'-CONHNH-]n
где R и R' — алифатич. радикалы. Реакцию проводят
в ]Ч,1Ч-диметилформамиде при 110—150 °С. В этом
случае образуются П. сравнительно низкой мол. массы.
3. Из дигидразинов и дифениловых эфиров дикарбо-
дикарбоновых к-т:
-С6Н5ОН
CeH5OOC-R-COOCeH5 + H2NHN-(CH2N-NHNH2 >
-> [_OC-R-CONHNH-(CH2)e-NHNH-]n
где R — алифатич. или ароматич. радикалы. Реакцию
осуществляют в ]Ч,1Ч-диметилформамиде при 100 °С или
в расплаве при 120—130 °С.
П., как правило,— аморфные вещества белого цвета.
Наиболее подробно изучены ароматич. П. Они кри-
кристаллизуются при кипячении в ]Ч,1Ч-диметилацетамиде,
диметилсульфоксиде и др. растворителях, способных
разрушать водородные межмолекулярные связи; кри-
кристаллизация наблюдается также при ориентационной
вытяжке пленок и волокон. Аморфные ароматич. П.
растворяются в диметилсульфоксиде, тетраметилен-
сульфоне, гексаметилфосфорамиде, г^,]Ч-диметилацет-
амиде. Их температуры размягчения лежат в интервале
300—350 °С. При темп-pax выше 250 °С они отщепля-
отщепляют воду с образованием поли-1,3,4-оксадиазолов.
При действии на ароматич. П. оснований (рН 10 — 12)
гидразидные группы способны переходить в енольную
форму
NH—NH
N—N
С
I I
с-
0 0 ОН ОН
к-рая с солями Ni2+, Ca2+, Ag+, Pb2+ образует полимер-
полимерные хелаты. К действию сильных минеральных к-т
ароматич. П. неустойчивы.
При обработке П. анилином в полифосфорной к-те
при 200 °С в течение 140 ч образуются поли- D-фенил)-
1,2,4-триазолы (см. Политриазолы).
При взаимодействии ароматич. П. с пятисернистым
фосфором в среде кипящего пиридина происходит по-
постепенное замещение кислорода серой с образованием
полиоксатиагидразида и политиагидразида. Политиа-
гщуэазиды плохо растворяются, термически неустойчи-
неустойчивы и легко превращаются в политиадиазолы.
Формованием из р-ров ароматич. П. можно получать
прочные эластичные пленки и волокна. Напр., из
сополигидразида регулярного строения на основе изо-
и терефталевой к-т получено волокно с прочностью
6 гс/текс и относительным удлинением 14%. П. ис-
используют в качестве форполимеров при получении поли-
1,3,4-оксадиазолов и изделий из них (см. Полиокса-
диазолы).
Алифатич. П. на основе дигидразидов и дихлоран-
дихлорангидридов дикарбоновых к-т легко превращаются в поли-
1,3,4-оксадиазолы уже при 150 °С, а при 200—260 °С
циклодегидратация заканчивается за 2—4 ч. Алифатич.
низкомолекулярные П. из дифениловых эфиров и ди-
дигидразидов дикарбоновых к-т плавятся при 285 —
290 °С. В расплаве при 290—300 °С одновременно про-
происходят рост основной полимерной цепи и циклодегид-
циклодегидратация, приводящие к образованию поли-1,3,4-окса-
диазолов сравнительно высокой мол. массы. Алифатич.
П. на основе дигидразинов и дифениловых эфиров
дикарбоновых к-т не способны циклизоваться; они
растворяются в гексаметилфосфорамиде, этиленхлор-
гидрине и частично в 1Ч,]Ч-диметилформамиде и хлоро-
хлороформе. Эти П. очень гигроскопичны (расплываются на
воздухе).
Лит.: Фрейзер А. Г., Высокотермостойкие полимеры,
М., 1971; Коршак В. В., Термостойкие полимеры, М.
1969; Лиг., СтоффиД., Н е в и л л К., Новые линей-
линейные полимеры, пер. с англ., М., 1972. Д.Р.Тур.
ПОЛИГЛИКОЛИД (polyglycolide, Polyglykolid,
polyglycolide).
Гликолид (Г.) — внутренний циклический эфир (ди-
лактон) гликолевой кислоты, 2,5-дикето-1,4-диоксан;
кристаллическое вещество белого цвета;
т. пл. 84—85 °С, сублимируется при 80— н г'^СО
84°С (при 133, 322 н/м1, или 1 мм рт. ст.). 21 i
Г. растворим в бензоле, этилацетате и др. oc\q^ch2
органических растворителях; полимери-
зуется и сополимеризуется с рядом виниловых и гете-
гетероциклических мономеров.
Г. получают вакуумной перегонкой гликолевой к-ты
с последующей термической деполимеризацией об-
образующегося циклического олигоэфира гликолевой кис-
кислоты; очищают многократной перекристаллизацией
из этилацетата.
Полигликолид [—ОСН2СО—1^ — линейный слож-
сложный полиэфир; кристаллич. вещество белого цвета;
плотность 1,707 г/см3 B5 °С), т. пл. 227—230 °С, ха-
рактеристич. вязкость [г|] = 1 —10 дл/г. П. нераство-
нерастворим в воде и обычных органич. растворителях; раство-
растворение при нагревании в л*-крезоле и смеси A0 : 7 по
массе) фенола и трихлорфенола сопровождается дест-
деструкцией; П. нетоксичен и в контакте с тканями орга-
815
ПОЛИГРАФИЧЕСКИЕ КРАСКИ
низма не вызывает повышения темп-ры тела; гидроли-
гидролитически устойчив в слабокислых и слабоосновных сре-
средах (в организме гидролизуется с постоянной скоро-
скоростью); легко образует волокна из расплава, имеющие
прочность до 550 Мн/м2 E5 кгс/мм2) и относительное
удлинение 15—20% ; устойчив при нагревании до 240 °С.
В пром-сти П. получают катионной полимеризацией
Г. в массе при 230 °С; катализатор — SbF3; длитель-
длительность процесса ок. 1 ч; выход количественный.
П. применяют взамен кетгута для изготовления хи-
рургич. рассасывающихся нитей, выпускаемых в США
под названием дексон. П. и продукты его биодест-
биодеструкции не вызывают характерной для кетгута тканевой
реакции, а также не влияют на биохимич. показатели
мочи и крови. Нити из П. легко завязываются в эластич-
эластичные, прочные, нерастягивающиеся хирургич. узлы,
не ослабляются при погружении, напр., в консерви-
консервирующие р-ры. Нити стерилизуют обычно у-излучением
(доза ок. 3 Мрад). П., разлагаясь до СО2 и Н2О, пол-
полностью выводится из организма с мочой и выдыхаемым
воздухом.
Время полного рассасывания нитей из П. ~4 мес,
причем основная масса рассасывается за два мес.
Прочность нитей уменьшается во времени по линей-
линейному закону.
Время рассасывания П. можно регулировать, вводя
в мономерную композицию на стадии полимеризации
нек-рое количество (обычно 5—10%, молярная концен-
концентрация) лактида (см. Полилактид). Сополимер более
медленно рассасывается в организме, обладает лучшей
растворимостью по сравнению с П. и поэтому легче
очищается и перерабатывается.
Впервые высокомолекулярный волокнообразующий
П. был синтезирован Бэком в 1952.
Лит.: Ghu]o К. [а. о.], Macromol. Ghem., 100, 262 A967);
Morgan M. N., Brit. Med. J., 2, № 3, 308 A969); К u s H.,
Koziowska, Polimery w medycynie, 2, № 4, 375 A972).
В.С.Лившиц.
ПОЛИГРАФИЧЕСКИЕ КРАСКИ (printing inks,
Druckfarben, encres d'imprimerie) — краски, приме-
применяемые в полиграфич. пром-сти для различных способов
печатания. П. к. наносят слоем ок. 2 мкм, чаще всего
на поверхность бумаги, представляющей собой капил-
капиллярно-пористый материал. В этом состоит специфика
применения П. к. (краски всех др. видов наносят слоя-
слоями значительно большей толщины на хорошо прогрун-
тованную невпитывающую подложку). Кроме того, на
основе многих П. к. получают прозрачные покрытия,
тогда как главное назначение др. красок — образо-
образование непрозрачного (укрывистого) покрытия. П. к.
закрепляются на поверхности бумаги в результате
впитывания в ее поры и капилляры растворителя, со-
содержащегося в краске. При этом пленкообразующее
остается на поверхности бумаги, формируя прочную
эластичную пленку, к-рая закрепляет пигмент.
Органич. растворитель может оставаться в красочной
пленке, а также в порах и капиллярах бумаги продол-
продолжительное время (напр., при использовании типограф-
типографских и офсетных красок без принудительного их высу-
высушивания) или почти полностью удаляться сразу же
после нанесения краски (напр., при использовании
красок глубокой печати).
Классификация. В зависимости от методов печати
П. к. подразделяют на типографские, офсетные, фото-
типные, для глубокой печати, эластографские (флексо-
графские), этмографские (трафаретные) и электрограф-
электрографские, различающиеся в основном скоростью и харак-
характером закрепления на бумаге или на другой подложке.
Кроме того, П. к. в зависимости от особенностей изда-
изданий, для к-рых они предназначены, классифицируют
на газетные, книжные, журнальные, репродукционные
и др., а в зависимости от вида печатных машин — на
полиграфические краски для ротационных и плоско-
плоскопечатных машин.
В типографской печати П. к. наносятся вали-
валиками на поверхность рельефной формы и затем под давлением
печатного цилиндра переходят на бумагу. В офсетной
печати применяют плоские металлич. печатные формы,
к-рые состоят из печатающих элементов, воспринимающих
краску, и т. наз. пробельных элементов, смачивающихся водой.
При печати форму сначала увлажняют водой, а потом накаты-
накатывают на нее краску. Затем краска переходит с формы на рези-
резиновый (т. наз. офсетный) цилиндр, с к-рого под давлением пе-
печатного цилиндра или второго офсетного цилиндра, печатаю-
печатающего оборотную сторону бумажной ленты, переносится на бу-
бумагу. Фототипная печать подобна офсетной. Отли-
Отличие состоит в том, что применяют желатиновую плоскую печат-
печатную форму, изготовляемую фотомеханич. методом. Кроме того,
краска с формы переходит на бумагу непосредственно под дав-
давлением печатного цилиндра. О др. способах печати см. Печать
на полимерах.
Состав. П. к.— коллоидные системы, образованные
из пигментов и пленкообразующих веществ. Исклю-
Исключение — нек-рые эластографские П. к., представляю-
представляющие собой растворы органич. красителей, и электро-
электрографские краски — порошкообразные материалы. Кро-
Кроме пигментов и пленкообразующих, в состав П. к. мо-
могут входить сиккативы, наполнители и т. наз. подцвет-
подцветки (вещества, позволяющие в максимальной степени
выявить основной цвет краски).
В качестве пленкообразующих для П. к.
обычно применяют различные полимеры или олиго-
меры. Феполо-альдегидные смолы на основе п-трет-
бутилфенола, дифенилолпропана и формальдегида, мо-
модифицированные канифолью и пентаэритритом или гли-
глицерином, используют для получения П. к., образую-
образующих твердые и глянцевые пленки. Алкидные смолы,
например на основе пентаэритрита, изофталевой ки-
кислоты и подсолнечного масла, хорошо смачивающие
пигменты, применяют для изготовления высокодис-
высокодисперсных типографских и офсетных красок, образую-
образующих глянцевые покрытия.
Канифольно-малеиновые аддукты, этерифицирован-
ные пентаэритритом, служат для изготовления П. к.,
быстро закрепляющихся на подложке вследствие изби-
избирательного впитывания; пентаэритритовый и глицери-
глицериновый эфиры канифоли (см. Смолы природные) — для
изготовления обычных типографских и офсетных П. к.
Из циклизованного натурального каучука (см. Цик-
Циклизация каучуков) получают быстрозакреиляющиеся
типографские и офсетные краски, а также краски глу-
глубокой печати. Резинаты цинка и кальция применяют
в сочетании с циклизованным каучуком при изготовле-
изготовлении красок глубокой печати. Лаковый битум (см. Би-
Битумные лаки) используют для изготовления черных ти-
типографских и офсетных П. к. и красок темных тонов
для глубокой печати.
Наиболее часто для изготовления П. к. применяют
органич. пигменты (фталоцианиновые, азопигмен-
ты), а также лаки (лаковые пигменты), напр, из
трифенилметановых органич. красителей. Из неорга-
неорганических пигментов используют ферроцианиды Fe,
сульфохроматы (кроны) и молибдаты РЬ, а также TiO2,
ZnO и др. В качестве черного пигмента применяют
сажу, гл. обр. газовую канальную или ее окисленную
разновидность. Для получения П. к., образующих
прозрачные покрытия, применяют пигменты и пленко-
пленкообразующие с совпадающими или близкими показате-
показателями преломления. Наполнителями в П. к.
служат А1(ОНK, BaSO4 и др., подцветкой —
фиолетовые органич. пигменты или фосфорновольфра-
мовомолибденовые соли растворимых органич. краси-
красителей, а также масло- и смолорастворимые красители
соответствующих цветов (см. также Пигменты лако-
лакокрасочных материалов, Красители, Наполнители лако-
лакокрасочных материалов).
В качестве растворителей для пленкооб-
пленкообразующих применяют толуол, бензин, скипидар, уайт-
спирит, керосин и различные минеральные масла. Эти
же вещества используют и для разбавления готовых
красок (см. также Растворители лакокрасочных мате-
817
ПОЛИ-2,6-ДИМЕТИЛ-п-ФЕНИЛЕНОКСИД
818
риалов). Сиккативами служат нафтенаты, рези-
резинаты и линолеаты Со, РЬ и Mg.
Состав П. к. зависит от методов печати, назначения
издания и применяемого печатного оборудования.
Типографские черные газетные ротационные краски
состоят из след. компонентов (в % по массе): газовая
канальная сажа — 10, р-р индулина в олеиновой к-те
(подцветка) — 13, р-р лакового битума в машинно-сма-
машинно-смазочном масле — 77. Аналогичный состав имеют и типо-
типографские черные ротационные книжные краски. Одна-
Однако они содержат (для увеличения вязкости) большее
количество пигмента и подцветки.
Примерный состав цветных офсетных и типографских
красок (в % по массе): органич. пигмент — 20, феноло-
формальдегидная смола — 25, алкидная смола — 15,
трансформаторное масло — 20, керосиновая фракция
нефти (темп-pa кипения 240—290 °С) — 15, сиккатив —2,
стеарат алюминия — 3.
Состав красок для глубокой печати (в % по массе):
органич. пигмент — 15, BaSO4 (наполнитель) — 5,
т. наз. бензиновый или толуольный лак — 80. Бензи-
Бензиновый лак представляет собой р-р смеси резинатов
Zn и Са с циклизованным натуральным каучуком в бен-
бензине, толуольный — смесь феноло-формальдегидной
смолы и толуола в соотношении 1 : 1 (по массе).
Получение. Основная операция в технологии изготов-
изготовления П. к.— дезагрегирование пигментов в пленкооб-
пленкообразующих. При этом, в зависимости от назначения
П. к., используют различные методы и оборудование.
При изготовлении типографских и офсетных цветных
красок пигмент перемешивают с пленкообразующим
в мощных лопастных замесочных машинах планетар-
планетарного типа (частота вращения мешалки ок. 500 об/мин).
Смеси выдерживают 10 ч в покое для лучшего смачива-
смачивания пигментов пленкообразующим, а затем перетирают
на трехвалковых краскотерочных машинах. Получен-
Полученные пасты различных пигментов (т. наз. робюры) сме-
смешивают в нужной пропорции между собой и с др. до-
добавками, подвергают термообработке в течение 10 ч
при 90 °С и расфасовывают в банки.
Газетные ротационные и др. маловязкие П. к. гото-
готовят сначала в виде конц. полуфабрикатов, а затем
разбавляют пленкообразующим или растворителем.
Краски глубокой печати получают, используя шаровые
или шариковые (аттриторы) мельницы, а также ультра-
ультразвуковые установки. Типографские и офсетные краски
на основе алкидных смол, содержащие такие пигменты,
как ферроцианиды Fe, готовят методом «отбивки воды».
Подробно о методах получения красок и применяемом
оборудовании см. Краски.
В готовых П. к. определяют цвет, вязкость, эластич-
эластичность, тиксотропные свойства, степень перетира, ско-
скорость закрепления (пленкообразования) на бумаге, све-
то- и водостойкость и др. (о методиках определения нек-
рых свойств см. Испытания лакокрасочных материалов
и покрытий). Иногда П. к. испытывают на небольших
лабораторных устройствах, моделирующих процесс
печатания (толщина слоя краски, давление при печати,
скорость печатания, темп-pa). При этом используют
приборы институтов графич. техники — «ИГТ» (Ни-
(Нидерланды) и Фогра или Прюфбау (ФРГ).
Свойства. Для офсетных, типографских и нек-рых
других П. к. характерны вязкости аномалия и ясно вы-
выраженные тиксотропные свойства. Реологич. свойства
маловязких красок описываются простейшим законом
Ньютона. Структурная вязкость и эластичность опре-
определяют печатные свойства красок, т. е. их способность
раскатываться валиками, накатываться на поверхность
печатной формы и переноситься под давлением печат-
печатного цилиндра на поверхность бумаги.
Расширение производства и совершенствование П. к.
неразрывно связаны с развитием книгоиздательского
дела и полиграфич. пром-сти. В результате значительно-
значительного роста цветной печати, особенно выпускаемой офсет-
офсетным способом, увеличивается производство П. к. для
трех- и четырехкрасочного печатания, быстро закреп-
закрепляющихся красок и др. В СССР в 1973 было изготовлено
ок. 27 000 т П. к.
Лит.: КозаровицкийЛ. А., Бумага и краска
в процессе печатания, М., 1965; Березин Б. И., Печат-
Печатные краски, М., 1961; его же, Материаловедение полигра-
полиграфического производства, М., 1972; Т ю р и к о в Д. А., Л я-
лина Э. Э., Кудрявцев Б. Б., Печатные краски, М.,
1971; Т а т и е в Д. П., Художественно-живописные и печат-
печатные краски, М., 1969; Askew F. A. [ed.], Printing ink manua-
le, 2 ed., Gamb., 1969; A p p s E. A., Inks for the minor printing
processes and specialized application, L., 1966. Б. И. Березин.
ПОЛИ-2,6-ДИМЕТИЛ -w-ФЕНИЛЕНОКСИД [poly-
B,6-dimethyl-p-phenylene oxide), Poly-2,6-dimethyl-jp-
phenylenoxid, oxyde de poly-2,6-dimetnyl-p-phenyle-
ne] — простой ароматич. полиэфир линейного строения.
СН,
SCH3
Получение. Наиболее распространенный метод по-
получения П.— дегидрополиконденсация 2,6-диметилфе-
нола или га-галоген-2,6-диметилфенола. В первом слу-
случае процесс проводят в инертном органич. растворите-
растворителе; окислителем служит кислород вместе с катализа-
катализатором, в качестве к-рого используют комплексы солей
металлов переменной валентности с алифатич. аминами,
пиридином или диметилформамидом, а также МпО4,
РЬО2 и AgO.
Для получения П. в пром-сти СССР в качестве ка-
катализатора используют комплекс формиата меди с пири-
пиридином (оптимальная концентрация 0,01—0,02 молъ/л);
процесс проводят в смеси растворителей метанол—то-
метанол—толуол при 28—32 °С в течение 40—60 мин. При увели-
увеличении продолжительности синтеза могут образоваться
сшитые П.
В США в качестве катализатора применяют комплекс
Си2С12 с пиридином (молярное соотношение Си2С12:
пиридин равно 1 : 100); темп-pa процесса 40—60 °С.
Увеличение указанного соотношения, использование
катализатора в молярной концентрации менее 1% от
количества мономера, а также повышение темп-ры
реакции способствуют протеканию побочной реакции —
образованию 3,5,3',5'-тетраметилдифенохинона.
При дегидрополиконденсации гс-галоген-2,6-диметил-
фенола в качестве окислителя используют феррицианид
калия в бензольно-щелочной гетерогенной среде или
СиС12 в пиридине; кроме того, можно применять пер-
персульфаты, ди-трет-бутилперекись, РЬО2 и др. Процесс
проводят при комнатной темп-ре.
П. выпускают под названиями а р и л о к с (СССР),
Р.Р.О. и но рил (США), причем два последних в виде
прозрачных или полупрозрачных, бесцветных или ок-
окрашенных в различные цвета гранул, ненаполненных
или наполненных 20 и 30% (по массе) короткорезаного
стекловолокна. Норил производят в реакторах (см.
рис.), содержащих фиксированный слой молекулярных
сит, через к-рые циркулирует 2,6-диметилфенол, ката-
катализатор и ароматич. углеводород, напр, толуол, в про-
противотоке с кислородом, что обеспечивает высокую ско-
скорость поликонденсации, предупреждает взаимодей-
взаимодействие выделяющейся воды A моль на 1 моль мономера)
с катализатором и способствует проведению процесса
в изотермич. условиях (дегидрополиконденсацию, ви-
видимо, проводят по периодич. схеме). После обработки
разб. к-той реакционная смесь отделяется от твердого
осадка и центрифугируется для отделения органич. ча-
части от водной, полимер в осадителе осаждается мета-
метанолом из ароматич. растворителя, образовавшаяся
819
ПОЛИ-2,6-ДИМЕТИЛ-п-ФЕНИЛЕНОКСИД
820
суспензия полимера фильтруется и высушенный поли-
полимер (порошок) поступает на упаковку.
Метанол
Тяжелая фаза
на обработиу
отходов
Полимер
Схема производства поли-2,6-диметил-п-фениленоксида мар-
марки «норил»: 1 — реактор; 2 — насос; 3 — смеситель; 4 —
фильтр; 5 — центрифуга; 6 — осадитель; 7 — вращающийся
вакуум-фильтр; 8 — вращающаяся сушилка
Периодич. технологич. схема производства арилокса
включает следующие стадии: приготовление реакцион-
реакционного р-ра, дегидрополиконденсация 2,6-диметилфенола,
выделение П., очистка его от низкомолекулярных
фракций, дифенохинона и остатков катализатора, суш-
сушка П., регенерация растворителя.
Свойства. П.— твердое самозатухающее вещество
белого цвета; мол. масса 30—700 тыс. Зависимость
между характеристич. вязкостью [г\] и мол. массой
выражается ур-нием: [г\] = 4,83- 10-2М°>64±0>02 (хло-
(хлороформ, 20°С). П., полученный из 2,6-диметилфенола,
аморфен, а из гс-галоген-2,6-диметилфенола частично
кристалличен. Нагреванием в таком растворителе, как
хлористый метилен, трихлорэтилен, циклогексан, этил-
ацетат, ацетон или а-пинен, аморфный П. можно пре-
превратить в кристаллический (размеры моноклинной ре-
решетки П.: а = 11,7А, В = 10,5 А, с = 16,9А, а = 97°).
Темп-pa стеклования аморфного П. 230—250 °С, темп-ра
плавления кристаллич. П. 260 СС (плотность расплава
0,958 г/мл). При нагревании П. на воздухе потеря мас-
массы начинается при 200 °С; после кратковременного
прогрева на воздухе при 200 °С П. теряет растворимость
вследствие частичной кристаллизации и сшивания.
Окисление протекает в первую очередь по СН3-группе
с отщеплением или последовательным превращением
ее в оксиметиленовую, альдегидную и затем в карбок-
карбоксильную; возможна также конденсация вновь образо-
Свойства поли-2,6-диметил-м-фениленоксида отечественной и зарубежных марок
Показатели
Арилокс*
(СССР)
Р.Р.О.
(США)
Норил (США)
Норил, наполненн-ый стекловолокном
2 0 % стекловолок
на (норил-2)
;- 30%стекловолок-
на (норил-3)
Плотность, г/см9
Водопоглощение, %
равновесное E0%, 40 °С)
при погружении в воду D0 °С). .
Деформационная теплостойкость при
1,7 Мн/мг, или 17 кгс/см2, °С
Темп-рный коэфф. линейного расшире-
расширения (от —1,1 до 83 °С), СС~1
Усадка при формовании, см/см ....
Прочность D0 °С), Мн/мг (кгс/см2)
при растяжении
при сдвиге
при статич. изгибе
Относительное удлинение D 0 °С), %
Модуль упругости D 0 °С), Мн/м2
(кгс/см2)
при растяжении
при изгибе
Напряжение при сжатии D0 °С)
и 10 %-ной деформации, Мн/мг
(кгс/см2)
Деформация под нагрузкой
12,8 Мн/мК 128 кгс/см2), 68 °С, %
Деформация при ползучести [нагрузка
12,8 Мн/м2 A28 кгс/см2), 300 ч,
40 °С], %
Ударная вязкость по Изоду D0 СС),
кдж/м2, или кгс см/см2
с надрезом
без надреза
Твердость по Роквеллу
Предел выносливости после 2-Ю6
циклов, Мн/м2 (кгс/см2)
Диэлектрич. проницаемость при
5 0 %-ной относительной влажности
воздуха
40 °С, 60 гц
40 СС, 1 Мгц
94 °С, 1 Мгц
Тангенс угла диэлектрич. потерь при
5 0 %-ной относительной влажности
воздуха
40 °С, 60 гц
94 СС, 1 Мгц
Уд. объемное электрич. сопротивление,
сухой, 40 СС, ом-см
Диэлектрич. прочность (кратковремен-
(кратковременная), в/мм
Дугостойкость (вольфрамовый элект-
электрод), сек
1,06
0,06
0,11
80-83 (800-830
12-18
40-7000
90
2,6
2,5
2-Ю17—1 ,5-Ю17
1800
1,06
0,03
0,10
194
2,9-Ю-5
0,007-0,009
74G40)
70G00)
107A070)
20—40
2,5-1 03B ,51 О4)
2,4-103B,4-104)
106A060)
0,1
0,5
5,8
40
119
7,7G7)
2,58
2,58
2,55
0,00035
0,0004
1018
500
(образец 0,3 еж)
7 5
1 ,06
0,07
0,14
147
3,3-10-3
0,005 — 0,007
61,5F15)
68@80)
87,5(875)
20-30
2,3-103B,3-104)
2, 3-103B ,3-Ю4)
106A060)
0,3
0,75
8,65
40
119
16A60)
2,64
2,64
2,63
0,0004
0,0006
1017
550
(образец 0,3 еж)
75
1,21
0,03
0,14
158
2,0-10~5
0,002
92,5(925)
66,5F65)
131A310)
4-6
5 ,9-103E, 9-1 О4)
4,75-1 03D ,75-1 О4;
113A130)
0,2
0,33
7,7
10
106
1,27
0,03
0,12
173
1,4-10-5
0,001
109A090)
69F90)
141A410)
4-6
,5-103(8, 5- Ю4)
, 5-1()зF,5-104)
115A150)
0, 12
0,2
9,1
10
108
32C20)
2,93
2,92
2,90
0,0009
0,0012
1017
1020
образец 0,08 см)
120
* В пром-сти выпускается двух марок, различающихся в основном по ударной вязкости,— арилокс1 D 0 кдж/м*, или
кгс -см/см2), арилокс II G000 кдж/м2, или кгс -см/см2).
821
ПОЛИДОДЕКАНАМИД
822
вавшихся групп с ароматич. ядрами цепи, что приводит
к получению лестничных или сшитых структур. Окис-
Окисление СН3-групп до карбоксильных, хлорирование до
хлорметиленовых, а также превращение в четвертичные
аммониевые или тетраметилсилановые уменьшают тер-
термостойкость П.
П. растворим в хлорированных и ароматич. углево-
углеводородах, диоксане, тетрагидрофуране и апротонных
диполярных растворителях, напр, в диметилформамиде,
диметилсульфоксиде; устойчив к действию кипящей во-
воды, перегретого пара, разбавленной и концентрирован-
концентрированной минеральных к-т, щелочей и перекисей, радиоак-
радиоактивного излучения. Микроорганизмы на поверхности
П. не развиваются.
Промышленные марки П. мол. массы 25—30 тыс.
обладают свойствами, характерными, напр., для поли-
поликарбонатов] норил имеет лучшую по сравнению с
с Р. Р. О. адгезию к металлам. П. мол. массы ниже
2-Ю4 характеризуются низкими прочностными показа-
показателями; с увеличением мол. массы выше 3-Ю4 ухуд-
ухудшаются реологич. свойства П. В таблице приведены
нек-рые свойства П.
Переработка и применение. П. перерабатывают лить-
литьем под давлением при темп-ре расплава 320—340 °С
и темп-ре формы 120—150 °С; экструзией при 240—
300 °С; пленки можно каландровать, а также формовать
поливом из 10%-ных р-ров в бензоле, толуоле и др.
Свойства полидиметилфениленоксида не изменяются по-
после многократного повторного формования на литье-
литьевых машинах.
П. применяют в электротехнике (электроизоляцион-
(электроизоляционные детали и корпуса электромоторов), электронике,
радиотехнике (детали высокочастотной изоляции радар-
радарных установок, печатные схемы), сантехнике (корпуса
вентилей и элементы водомерного оборудования) и хи-
хирургии (рукоятки мединструментов, детали протезов,
трансплантанты и др.). Из П. изготовляют также кор-
корпуса химич. насосов и турбин, бытовые товары (напр.,
детали стиральных машин), типографские металлизи-
металлизированные матрицы, а также изоляционные и защитные
лакокрасочные материалы.
Благодаря более низкой стоимости П. можно приме-
применять вместо поликарбонатов, полиакрилатов и особенно
полимеров фторированных углеводородов. Кроме того,
возможно их применение в качестве термореактивных
смол низкотемпературного отверждения, термостойких
пенопластов, ионообменных смол.
Первое промышленное производство П. освоено в США
в 1962.
Лит.: Ли Г., С т о ф ф и Д., Невилл К., Новые
линейные полимеры, пер. с англ., М., 1972; Paris A., Ind.
Ghim., 54, № 597, 91 A967); Vollmert В., Saatwe-
ber D., Angew. makromol. Chemie, 2, 114 A968); Con-
1 e у R. Т., J. Macromol. Sci., 1 A, № 1, 81 A967); Новые гюли-
конденсационные полимеры. Сб. переводов и обзоров из ино-
иностранной периодической литературы, под ред. 3. А. Роговина
и П. М. Валецкого, М., 1969; Гуров А., Зеленецкий
А., Хим. и технол. полимеров, № И, 115 A965); Тел ешо в Э.,
Праведников А., Пласт, массы, № 2,3 A973); Ю д-
к и н Б. И. [и др.], там же, с. 41. А. Н. Зеленецкий.
ПОЛИДИСУЛЬФИДЫ — см. Полису лъфидные кау-
чуки.
ПОЛИДОДЕКАНАМИД, полиамид-12 (poly-
dodecanamide, Polydodekanamid, polydodecanamide).
со-Додекалактам (Д.), л актам со-аминододекановой,
или аминолауриновой, кислоты HN(GH2)UGO. Д.— кри-
I |
сталлическое вещество белого цвета; т. пл. 151 —
153 °С, т. кип. 182 °С при 2 мм рт. ст. A мм рт. ст.=
= 133,322 и/м2)\ теплота плавления 9,4 кдж/молъ B,2
ккал/молъ); хорошо растворим в спирте, бензоле, аце-
ацетоне, плохо — в воде.
Додекалактам полимеризуется с большим трудом,
чем е-капролактам; способен сополимеризоваться с
другими лактамами, например с е-капролактамом, ка-
приллактамом.
Д. синтезируют из дивинила по след. схеме:
ЗСН2=СН-СН = СН2
5О-6О°С |(С2Н5JА1С1+ TiCl4
ОН
Циклододекатриен
150°С Н2
Г^ I ,°2
Ч/ ^У
I
¦ О
Циклододекан
INOC1
hn(ch2)i|Co
Выделяют Д. последовательно экстракцией, кри-
кристаллизацией и ректификацией. Для получения чистого
Д. р-ры его обрабатывают ионообменными смолами.
Полидодеканамид [ — HN(GH2)UGO—]п (П.).
Свойства. П.— твердый роговидный продукт бе-
белого цвета, прозрачный в тонком слое, без запаха.
Мол. масса П., выпускаемого в пром-сти, 15—35 тыс.,
степень кристалличности 40—70% (в зависимости от
условий переработки). Зависимость между характе-
ристич. вязкостью [ц] и мол. массой М выражается
ур-нием: [г\] = 15,5- 10-4Л/0»70 (ж-крезол, 25 °С). П.
устойчив к действию большинства органич. растворите-
растворителей, масел, жиров, разб. к-т и в противоположность
всем др. полиамидам не растворяется в муравьиной
к-те; растворяется в сильнополярных растворителях,
напр, в конц. H2SO4, крезоле, фенолах, хлорированных
и фторированных спиртах. Максимальное водопогло-
щение П. 1,7% (в 6 раз меньше, чем у поли-?-капро-
амида). П. биологически безвреден. Подобно др. поли-
полиамидам он характеризуется высокой износостойкостью,
низким коэфф. трения, хорошими электроизоляцион-
электроизоляционными и физико-механич. свойствами. Ниже приведены
свойства П., выпускаемого в пром-сти:
Плотность, г/см3 1,02
Теми-pa плавления, °С 178—180
Теплостойкость, °С
по Вика 140
по Мартенсу 4 5
Деформационная теплостойкость, °С
при напряжении 0,46 Мн/м2
D,6 кгс/см*) 140
при напряжении 1,85 Мн/м2
A8,5 кгс/см2) 42 — 45
Темп-pa хрупкости, °С от - 70 до —90
Теплопроводность,
вт/ (ж-К) 0,24
ккал/(м-час- °С) 0,21
Темп-рный коэфф. линейного расширения,
°С~1 1,25-10-*
Ударная вязкость, кдж/м2, или тс-см/см2
без надреза 90 — 100
с надрезом 5—8 , 5
Прочность, Мн/мЦкгс/см-):
при растяжении 50E00)
при сжатии 60F00)
при изгибе 65F50)
Модуль упругости при растяжении,
Мн/м2 (кгс/см2) 1200 — 1600
A2000-16000)
Относительное удлинение, % 25 0—300
Твердость по Бринеллю, Мн/м2 (кгс/см2) 7 5G 50)
Уд. объемное электрич. сопротивление,
Том-м (ом-м) 10 — 60
(Ы0'5-6-101&)
Уд. поверхностное электрич. сопротивле-
сопротивление, Том(ом): 1-Ю3—2-Ю3
(Ы01&-2-1015)
Диэлектрич. проницаемость при 1 Мгц . . 3,0 — 3,5
Тангенс угла диэлектрич. потерь при
1 Мгц 0,02—0,03
Электрич. прочность, Мв/м, или кв/мм 22—25
823
ПОЛИИЗОБУТИЛЕН
824
Коэфф. трения по стали 0,28 — 0,30
Абразивный износ при нагрузке Юн
A кгс), мм3/м 0,7—0,8
Деформация при ползучести (растяжение
в течение 100 ч при напряжении
20 Мн/м2, или 200 кгс/см2), % 4,0 — 4,2
Водопоглощение, механич. и электрич. свойства за-
зависят от содержания влаги в П., но в меньшей степени,
чем у поли-е-капроамида (см. Капролактама полимеры).
Так, прочность при изгибе после пребывания П. в воде
и на воздухе 65%-ной относительной влажности умень-
уменьшается соответственно на 10—15% и на ~9% (у поли-е-
капроамида соответственно на 70—72% и ~57%).
Электрич. свойства этих двух полиамидов в сухом
состоянии практически равны, однако после пребы-
пребывания в воде или в средах с повышенной влажностью
более высокие электрические характеристики сохра-
сохраняются у П.; напр., уд. объемное электрич. сопротивле-
сопротивление П. уменьшается до 0,1 —1,0 Том-м(\01^—10ыом-см),
у поли-е-капроамида — до 1 • 10~5—1 • 10~4 Том-м
(\0*—\№ ом-см).
П. обладает всеми химич. свойствами, характерными
для полиамидов. Он легко окисляется под действием
тепла и света; на воздухе расплав П. окрашивается
в коричневый цвет. Для повышения срока службы в П.
вводят (в процессе синтеза или переработки) антиокси-
данты и светостабилизаторы — обычно амины или фе-
фенолы, а также йодистый калий или ацетат меди.
Для повышения теплостойкости и снижения коэфф.
трения в П. вводят различные наполнители, напр,
тальк, графит, дисульфид молибдена и стекловолокно.
Для повышения ударной вязкости П. совмещают с пла-
пластификатором, в качестве к-рого используют сульф-
сульфамидные производные. При введении 12 — 14% пластифи-
пластификатора в П. ударная вязкость (с надрезом) возрастает
до 40 кдж/м2, или кгс см/см2.
Получение. В пром-сти П. получают гидроли-
тич. полимеризацией со-додекалактама в присутствии
воды и к-т @,01 — 1,0%), напр, ортофосфорной, борной,
адишшовой, декандикарбоновой и др., вызывающих
гидролиз лактамного кольца:
HN(CH2),,CO
H2N(CH2)nCOOH
(A7-1)HN(CH2)uCO
— [-HN(CH2),,CO-],7
Процесс осуществляют в две стадии. Для раскрытия
цикла, ввиду плохой растворимости Д. в воде даже
при высоких темп-pax, реакцию проводят при 270—
300 СС и давлении 0,5—6,0 Мн/м2 E—60 кгс/см2) в те-
течение 4—8 ч. Образующаяся при этом со-аминододека-
новая к-та превращается в низкомолекулярный П.
Для получения П. высокой мол. массы из реакционной
смеси необходимо полнее удалять воду. Поэтому на
второй стадии реакцию проводят при атмосферном дав-
давлении или даже в вакууме (в расплаве или твердой фа-
фазе). Расплав П. выдавливают инертным газом (содер-
(содержание кислорода в нем не более 0,001%) из реактора
через фильеру в ванну с холодной водой. После ох-
охлаждения П. измельчают, гранулы сушат при 80 °С
в вакууме 20—100 мм pm. cm. A мм рпг. ст.= 133,322
н/м2) до содержания влаги 0,1—0,2%, охлаждают и
упаковывают в герметичную тару. Полученный П. со-
содержит 1—2% Д. и низкомолекулярных продуктов,
к-рые почти не растворимы в воде; их, как правило,
не удаляют из П.
Перспективен метод получения П. анионной полиме-
полимеризацией Д. в присутствии щелочных катализаторов и
сокатализаторов. Процесс проводят в углеводородах для
получения порошкообразного П. или в массе при
темп-ре несколько выше темп-ры плавления мономера.
В последнем случае Д. можно полимеризовать непосред-
непосредственно в формах и получать крупногабаритные изде-
изделия различного профиля массой до нескольких сот кг.
Переработка и применение. П. с уд.
вязкостью г|уД = 0,65—0,8 перерабатывают на литье-
литьевых машинах, снабженных самозапирающимися соп-
соплами; полимер с г|уД0,8—1,1 — на экструдерах и с т]уд
выше 1,1 — на вакуумформовочных машинах. Темп-ра
переработки 240—270 °С, усадка П. 0,7—1,1%. Содер-
Содержание влаги в П. с г|уД выше 0,8 не должно превышать
0,1%. Для увеличения прочностных и деформацион-
деформационных характеристик изделия из П. следует подвергать
термообработке при 150—160 °С, однако эластичность
П. при этом снижается. П. можно подвергать механич.
обработке, сварке, склеивать фенольными или эпоксид-
эпоксидными клеями, окрашивать в различные цвета.
П. применяют как антифрикционный и конструк-
конструкционный материал для изготовления вкладышей под-
подшипников, деталей точных измерительных приборов
(поплавки водомеров, детали газовых счетчиков и др.).
Из П. изготавливают также проточные части турбин
турбобуров, рабочие колеса скваженных погружных
электронасосов, комплектующие детали автомобилей
(напр., втулки, держатели проводов, фильтры, трубки),
каркасы катушек, изоляторы для телефонов и оболочки
для кабелей. Порошок A00—200 мк) П. используют для
нанесения покрытий по металлу методом напыления.
Из П. формуют также пленки (см. Полиамидные
пленки) для упаковки в пищевой и фармацевтич.
пром-сти и волокна (см. Полиамидные волокна).
П. производится в различных странах под назва-
названиями: П-12 (СССР), вестамид (ФРГ), г р и л-
а м и д (Швейцария), рильсан А (Франция).
Лит.: G r i e h I W., Ruestem D., Ind. Eng. Chem.
62, № 3, 16 A970); Лущейкин Г. А., Доброхото-
Доброхотова M. К., ТрещелинаН. К., Пласт, массы, № И, 59
A971); Доброхотова М. К. [и др.], там же, № 1 A973);
М о р о з о в А. Г. [и др.], там же, № 8, 72 A972); Kunstoff
Berater, 12, № 10, A967). М. К. Доброхотова.
ПОЛИИЗОБУТИЛЕН — см. Изобутилена полимеры.
ПОЛИИЗОПРЕН — см. Изопрена полимеры, Изо-
преновые каучуки.
П0ЛИИМИДА30ЛЫ (polyimidazoles, Polyimidazole,
polyimidazoles) — полимеры, содержащие в основной
4 3
НС—N42
цепи имидазольный цикл: II «!
НС"—NH
5 1
П. сравнительно мало изучены; более известны и важны
полибензимидазолы.
Известны два основных типа П.: поли-[2,5-(га-фени-
лен)-2',5'-арилен-4,4'-дифенил]диимидазолы, напр. (I),
и N-замещенные элементоорганич. П. (II, их строение
изображают двумя способами):
3 = Zn, Cu, Co, BR2,SnR3 (R-алкил)
Мол. масса I достигает 3000—6000; они растворимы
в муравьиной к-те и амидных растворителях, напр, ди-
метилформамиде; термостойки до 320 °С на воздухе (по
данным термогравиметрич. анализа).
В II связи между обоими атомами азота, по-види-
по-видимому, выравнены. При Э = Zn и Со II — порошкооб-
порошкообразные продукты, не растворимые в органич. раство-
растворителях; т. пл. выше 360 °С; термостойкость в инертной
атмосфере до 500—575 °С (по данным термогравиметрич.
825
ПОЛИИМИДНЫЕ ПЛЕНКИ
826
анализа). При Э = В(С2Н5J II — эластомер; мол. мас-
масса 6000—12 000; т. стекл. —12 °С. Потеря массы при
нагревании на воздухе до —370° составляет 5%.
Лит.: KorshakV. V., TeplyakovM. M., J. Мас-
romol. Sci. (Revs), С 5, № 2, 409 A971); К г i e g В., М а-
necke G., Macromol. Ghem., 108, 210 A967). М.М.Тепляков.
ПОЛИИМИДНЫЕ ВОЛОКНА — см. Термостойкие
волокна.
ПОЛИИМИДНЫЕ КЛЕИ (polyimide adhesives, Poly-
imidklebstoffe, colles de polyimide) — композиции на
основе полиамидокислот общей ф-лы
ноос
-NH-OC/
\R/
R
CO-NH-R'
ЧСООН
L
к-рые при отверждении превращаются в полиимиды.
Полиамидокислоты получают в результате взаимодей-
взаимодействия диангидридов тетракарбоновых к-т с диаминами
(табл.). Эти полимеры хорошо растворимы в полярных
растворителях — диметилацетамиде, диметилформами-
де, диметилсульфоксиде, пиридине и др.
Состав и свойства. П. к. применяют в виде жидких
или сухих композиций. Жидкий клей готовят раство-
растворением полиамидокислоты в одном из указанных выше
растворителей. П. к. этого типа представляют собой
прозрачные вязкие системы, чувствительные к дейст-
действию влаги и повышенных темп-р. При темп-ре ок. 0 °С
в отсутствие влаги такой клей можно хранить от не-
нескольких месяцев до одного года.
Сухие П. к. выпускают: 1) в виде пленки, к-рую гото-
готовят поливом полиамидокислоты; такой клей содер-
содержит фактически только один компонент — твердый фор-
полимер и 2) в виде стеклоткани с нанесенной на ней
полиамидокислотой; такую композицию готовят 8 —
10-кратной пропиткой ткани р-ром полиамидокислоты
очень низкой концентрации (р-ры этих полимеров даже
небольшой концентрации ИхМеют очень высокую вяз-
вязкость). В промежутках между каждой операцией про-
пропитки материал подвергается сушке путем последо-
последовательного нагрева: при 100 °С — 1 ч, при 150, 200
и 250 °С — по 15 мин, при 300 °С — 5 мин. Содержание
полиамидокислоты в композиции составляет 80% (от
массы ткани). Сухие П. к. можно хранить при обычных
условиях длительное время.
Технология склеивания. Металлич. поверхности пе-
перед склеиванием подвергают специальной обработке.
Изделия из нержавеющих сталей обезжиривают в три-
хлорэтиленс, промывают в горячей и холодной воде,
высушивают на воздухе при 95° С в течение 15 мин и
выдерживают в течение 2 мин в нагретой до 85 °С смеси
к-т G мае. ч. соляной, 17 мае. ч. ортофосфорной и 12
мае. ч. фтористоводородной). Затем изделия промывают
в дистиллированной воде до нейтральной реакции и
высушивают на воздухе при 95 °С. Титановые изделия
обезжиривают в хлорированных растворителях и ме-
тилзтшшетоне, промывают в воде и сушат на воздухе
при 95 °С в течение 15 мин. Затем изделия выдерживают
несколько мин в смеси, содержащей 270 мае. ч. дистил-
дистиллированной воды, 9 мае. ч. фтористого натрия, 4 мае. ч.
окиси хрома и 30 мае. ч. конц. серной к-ты, промы-
промывают в дистиллированной воде и сушат на воздухе при
95 °С в течение 15 мин.
Жидкие П. к. наносят на склеиваемые поверхности
клетью, а сухие закладывают между склеиваемыми де-
деталями. Склеивание осуществляют при нагревании от
180 до 450 °С и давлении порядка 1,4—1,5 Мн/м2
A4—15 кгс/см2) в течение нескольких ч. В этих условиях
происходит термич. циклизация полиамидокислот и пре-
превращение их в неплавкие и нерастворимые полиимиды.
Свойства клеевых соединений. Отличительными свой-
свойствами клеевых соединений, полученных с применением
П. к., являются высокая стойкость к термич., термо-
окислителыюй и радиационной деструкции, влаго-
и хпмстойкость. По электрич. свойствам отвержден-
ные П. к. относятся к среднечастотным диэлектрикам;
диэлектрич. проницаемость этих материалов C,0—3,5)
мало зависит от частоты электрич. колебаний и темп-ры;
уд. объемное электрич. сопротивление при комнатной
темп-ре составляет 103—104 Том-м A017—1018 ом-см),
при 200 °С—1 Том-м A014 ом-см); тангенс угла
диэлектрич. потерь — A —1,5)-10~3. Клеевые соедине-
соединения на основе П. к. работоспособны в течение сотен ч
при темп-ре до 300 °С. Прочностные характеристики
клеевых соединений на основе П. к. представлены
в таблице.
Прочность при сдвиге клеевого соединения нержавеющей
стали при использовании полиимидных клеев различного
состава
Мономеры, использованные для
синтеза полиамидокислот
м-Фенилендиамин
Диангидрид 3,3', 4,4' -бензофенонтет-
ракарбоновой к-ты
м -Фенилвн диамин
Диангидрид 3,3',4,4'-бензофенонтет-
ракарбоновой к-ты
п-Аминоацетанилид
м -Фенилен диамин
Диангидрид 3,3', 4,4' -бензофенонтет-
ракарбоновой к-ты
Анилин
ж-Фенилендиамин
Диангидрид 3,3', 4,4' -бензофенонтет-
ракарбоновой к-ты
Анилин
Фталевый ангидрид
ж-Фенилендиамин
Диангидрид 3,3',4, 4'-бензофенонтетра-
карбоновой к-ты
п-Аминоацетанилид .
Фталевый ангидри
Диангидрид 3,3', 4,4' -бензофенонтетра-
карбоновой к-ты
п,п-Диаминодиэтиловый эфир
Количест-
Количество компо-
компонента,
мае. ч.
100
100
96
100
4
96
100
4
98
98
2
2,2
98
98
2
2
100
100
Прочность
при сдвиге,
Мн/м*
{кгс/см2)
13,4A34)
23, 1B31)
15,6A56)
14,9A49)
12,7A27)
1,5A5)
Применение. Благодаря комплексу высоких тепло-
физич., электроизоляционных и механич. свойств П. к.
находят применение в космич. технике, в приборо-
приборостроении, в авиационной и электротехнич. промышлен-
промышленности для склеивания практически всех металлов
и нек-рых типов керамики.
П. к. выпускают под следующими названиями: С П-1
(СССР), п а й р ML, а м о к о AI (США).
Лит.: Encyclopedia of polymer science and technology,
v. 11, N. Y.— [a. o.l, 1969, p. 247; Хрулев В. М., Син-
Синтетические клеи и мастики, М., 1970. См. также лит. при ст. По-
Полиимиды. А.Б.Давыдов.
ПОЛИИМИДНЫЕ ПЛЕНКИ (polyimide films, Poly-
imidfilme, films de polyimide).
Производство П. п.— обычно двухстадийный про-
процесс. На первой стадии получают пленку поливом
р-ра полиамидокислоты (промежуточный продукт при
синтезе полиимидов) в ]Ч,]>Г-диметилацетамиде, N,N'-
диметилформамиде или др. амидах на бесконечную лен-
ленту или полированный металлич. барабан поливочной
машины. Наиболее широко используют полиамидо-
кислоту на основе пиромеллитового диангидрида и 4,4'-
диаминодифенилоксида. Образовавшуюся пленку вы-
высушивают с принудительной циркуляцией сухого азота
при 100 °С (не снимая с подложки). Пленка из поли-
полиамидокислоты слабо окрашена в желтый цвет, обла-
обладает высокой прочностью при растяжении [120 Мн/м2
A200 кгс/см2)} и относительным удлинением при разры-
разрыве ок. 80%. Однако эти свойства не стабильны во вре-
времени, т. к. полиамидокислота гидролизуется влагой
воздуха.
827
ПОЛИИМИДНЫЕ ПОКРЫТИЯ
828
На второй стадии пленку из полиамидокислоты под-
подвергают химич. или термич. циклизации. Выделение
воды при циклизации затрудняет получение толстых
пленок, т. к. в этом случае одновременно с циклиза-
циклизацией полимер интенсивно гидролизуется.
При химич. циклизации под действием аминов (чаще
всего пиридина) эффективное связывание выделяющей-
выделяющейся воды достигается применением дегидратирующих
агентов (ангидридов монокарбоновых к-т). По одному
из вариантов пленку из полиамидокислоты выдержи-
выдерживают 24 ч в смеси пиридин — уксусный ангидрид при
комнатной темп-ре, затем промывают в течение 2 ч
в диоксане при той же темп-ре, нагревают ее на воздухе
1 ч при 130 °С и 1 мин при 380 °С. Мягкие условия хи-
химич. циклизации почти полностью предотвращают де-
деструкцию полиамидокислоты, что позволяет получить
полимер с высокой мол. массой.
Термич. циклизации подвергают только очень тон-
тонкие пленки, обычно не толще 200 мкм.
При термообработке полиамидокислотной пленки,
закрепленной на раме, подложке или др. способом,
происходит ее двухосная ориентация вследствие усад-
усадки материала. Такая П. п. обладает лучшими физико-
механич. показателями, чем пленка, термообработан-
ная в свободном состоянии.
В зависимости от строения и способа циклизации
П. п. могут приобретать различный цвет — от бледно-
желтого до темно-красного. П. п. сохраняют эластич-
эластичность даже при темп-ре жидкого гелия. Напр., при
темп-ре 4 К их можно наматывать на стержень диамет-
диаметром 6 мм, и при этом они не ломаются. При 50%-ной
влажности окружающей среды П. п. поглощают влагу
в 6 раз быстрее, чем полиэтилентерефталатные плен-
пленки. Вместе с тем П. п. сохраняют 90% исходной проч-
прочности и 75% удлинения после кипячения в воде
15 су т. П. п. становятся хрупкими после пребыва-
пребывания на солнечном свету в течение 6 мес, однако их
радиационная стойкость выше стойкости др. полимер-
полимерных пленок.
По данным термогравиметрического анализа, поли-
полиамидные пленки устойчивы на воздухе до температур
порядка 400 °С.
Ниже приведены нек-рые свойства пленки на основе
полипиромеллитимида, получаемого из пиромеллито-
вого диангидрида и 4,4'-диаминодифенилоксида:
Плотность, г/см3 1,42
Прочность при растяженнии, Мн/м2
(кгс/см2) 160—180
A600-1800)
Модуль упругости, Мн/м2 [кгс/см2] B8 —30)-102
[B8-30)-10«]
Относительное удлинение, % .
Уд. объемное электрич. сопротивле-
сопротивление, Том-м (ом-см)
при 20 °С
70-80
средний слой которой состоит из полиимида, а наруж-
наружные — из сополимера гексафторпропилена и тетра-
фторэтилена. Эти пленки обладают повышенной из-
износостойкостью, хорошей электрич. прочностью, их
можно сваривать при нагревании до 300 °С.
За рубежом в опытно-промышленных масштабах вы-
выпускают термопластичные и растворимые полиимиды,
пленки из к-рых изготовляют поливом из р-ра или экст-
экструзией. Свойства этих пленок аналогичны свойствам
П. п., описанных выше. Кроме того, в США освоено
производство пленок на основе полиимидоэфиров и
полиимидоамидов. Ниже приведены нек-рые свойства
пленки на основе полиимидоэфира:
Прочность при растяжении, Мн/м2
(кгс/см2)
при 20 °С 10 5A0 50)
при 200 °С 44D40)
Относительное удлинение, % 14 — 23
Модуль упругости, Мн/м2 (кгс/см-) .... 3100C1 000)
Тангенс угла диэлектрич. потерь
при 5 0 гц 0,006
Диэлектрич. проницаемость при 1 кгц ... 3,1
Пленки на основе полиимидоэфиров обладают хоро-
хорошим комплексом физико-механич. и электроизоляци-
электроизоляционных свойств, но значительно меньшей термостабиль-
термостабильностью, чем П. п.; например, срок их службы при 240°С
составляет только 1000 ч. Однако гидролитическая
устойчивость полиимидоэфирных пленок выше, чем по-
лиимидных.
Впервые промышленное производство П. п. было
освоено в США в 1961 (фирменное название кап-
тон Н). В СССР П. п. выпускают под названием
«пленка ПМ».
Лит.: Справочник по пластическим массам, под ред.
М. И.Гарбара [и др.], т. 2, М., 1969, с. 317; Такахаси Г.,
Пленки из полимеров, пер. с япон., Л., 1971.
См. также лит. при ст. Полиимиды. А.А .Пешехопов.
ПОЛИИМИДНЫЕ ПОКРЫТИЯ — см. Термостой-
Термостойкие лакокрасочные покрытия.
ПОЛИИМИДОАМИДЫ (polyimide-amides, Polyimid-
amide, polyimide-amides) — полимеры, содержащие в ос-
основной цепи макромолекулы амидные (—СО— NH—) и
имидные (~qo/N— J группы. П. получают полицикло-
конденсацией в две стадии (на первой синтезируют по-
лиамидокислоту А, на второй осуществляют ее цикло-
дегидратацию):
1. Трикарбоновых к-т или их производных с первич-
первичными диаминами, напр.:
n Cl-OC— ]
/ССК
О + п
—OC-r'
102-10з
A01в-1017)
0,1-1
при 200 °С A О13—1 О14)
Тангенс угла диэлектрич. потерь
при 50 гц 0,0025
Электрич. прочность, кв/мм 100—275
Срок службы П. п. на воздухе при 250 °С — 10 лет,
при 275 °С — 1,5 года, при 300 °С — 3 мес, при 350 °С —
6 сут и при 400 °С — 12 ч.
П. п. применяют в качестве прокладочной и обмо-
обмоточной изоляции для электрических машин, в качестве
изоляции для конденсаторов и кабелей, длительно ра-
работающих при температурах до 250 °С, для изготовле-
изготовления гибких печатных радио- и электронных схем, для
мембран топливных насосов и др. Металлизированная
П. п. применялась в качестве наружной теплоизоля-
теплоизоляции американских лунных кораблей и костюмов кос-
космонавтов.
Выпускается также двухслойная пленка из полиими-
полиимида и политетрафторэтилена и трехслойная пленка,
СО
-HN-oc—r; nnr
ч
COHNR'NH— i
COOH J
A*
2. Диангидридов тетракарбоновых к-т, первиччых
диаминов и хлорангидридов дикарбоновых к-т:
nO
чсо/
O + 2/7HoNR'NH2 + n C1OCR"COC1
2 2
— ОС—R—COHN—R— NH—OCR"CO—NH-R — NH— -
—N
COOH
^coN
N-R-NH-OCR"CO—HN-R-
3. Диангидридов тетракарбоновых к-т с первичными
диаминами, в молекуле к-рых содержатся амидные
группы, или первичных диаминов с дикарбоновыми
кислотами, содержащими в молекуле имидные груп-
группы, например:
829
ПОЛ НИМИ ДОЭФ ИРЫ
830
n H2NV^\ .NH-ОС
-HN\^\^-NH-OC
NH2 CO CO
+ n O( X )
чсо' чсо/
NH—OC4 /CO—
HOOCX
X CO
;
хо
ссг
4. Дигидразидов дикарбоновых к-т с диангидридами
тетракарбоновых к-т:
хох .с
п О R
О + л H2N—HN-OC-Rr—CO-NH—NH.
—ОСЧ .COHN-HN-OC
R
7 чсоон
СО.
R'—CO—NH-NH-
—N
R NHNCORCONH-
ЧСО/
П. получают преимущественно по первому методу.
Основными мономерами для синтеза П. являются: ди-
диамины — бензидин, м- и ?г-фенилендиамины, 4,4'-диами-
нодифенилоксид, 4,4'-диаминодифенилметан, 3,3'- и
4,4'-диаминобензанилиды; диангидриды пиромеллито-
вой, 3,3',4,4'-дифенилоксидтетракарбоновой, 3,3', 4,4'-
бензофенонтетракарбоновой к-т, 4-хлорформилфтале-
вый ангидрид (хлорангидрид тримеллитового ангид-
ангидрида).
Синтез полиамидокислот (А) осуществляют
при темп-pax от—10 до 20°С в р-ре ]Ч,К-диметилформ-
амида, ]Ч,]Ч-диметилацетамида, N-метилпирролидона
и др. Полиамидокислоты растворимы в указанных
растворителях, а также в смесях их с алифатич. кето-
нами, сложными эфирами или ароматич. углеводоро-
углеводородами. Такие полиамидокислоты гидролитически более
устойчивы, чем полиамидокислоты, образующиеся на
первой стадии синтеза полиимидов. Тем не менее, как
правило, из р-ра А не выделяют. Циклодегидратацию,
к-рую проводят после формования изделия, осущест-
осуществляют при комнатной темп-ре под действием химич.
агентов (напр., смеси пиридина и уксусного ангидрида)
или нагреванием в течение нескольких ч при 200—
250 °С. При этом концентрация р-ров полиамидокислот,
применяемых, напр., в качестве лаков, составляет
32%, а вязкость таких р-ров при 25 °С в диметилацет-
амиде, диметилформамиде, диметилсульфоксиде и ме-
тилпирролидоне соответственно 1,4, 0,4, 2,0 и 6,3
п-сек/м2 A4, 4, 20 и 63 газ).
Наиболее интересны ароматич. П.—твердые аморф-
аморфные вещества светло-желтого цвета; плотность при
20 °С 1,4г/сж3. П. растворимы в диметилформамиде,
диметилацетамиде с добавкой LiCl, в конц. H2SO4,
нерастворимы в воде. П. отличаются высокой тепло-
и термостойкостью (темп-pa стеклования 250—275 °С,
потеря массы П. за 1 мес при 210 °С составляет 1%).
С увеличением содержания в П. амидных группировок
улучшается эластичность полимера, но ухудшается
термостойкость.
Полиамидокислоты применяют для приготовления
лакокрасочных материалов, из к-рых получают на-
гревостойкие электроизоляционные покрытия, а также
в качестве связующих для стеклопластиков, к-рые прес-
прессуют при темп-pax до 200 °С, что выгодно отличает их
от др. высокотермостойких связующих (напр., полиими-
полиимидов и полибензимидазолов). Срок службы полиамидо-
имидных эмалей при 240 СС достигает 20 000 ч, а стек-
стеклопластики на основе П. можно длительно эксплуати-
эксплуатировать при 260 °С.
П. выпускают в промышленном масштабе в США под
названием амоко AI полимер.
Лит.: ЛиГ., Стоффи Д., Невилл К., Новые
линейные полимеры, пер. с англ., М., 1972. Я.С.Выгодский.
ПОЛИИМИДОЭФИРЫ (polyimide-esters, Polyimid-
ester, polyimide-esters) — гетероцепные полимеры, со-
содержащие в основной цепи макромолекулы сложно-
эфирные (—СО —О—) и имидные (~qq/N—J груп-
группы. В пром-сти П. получают:
1. Полициклоконденсацией диангидридов тетракарбо-
тетракарбоновых к-т, содержащих сложноэфирные связи, с пер-
первичными диаминами в две стадии (на первой синтези-
синтезируют полиэфироамидокислоту А, на второй осущест-
осуществляют ее циклодегидратацию):
n O'
^R—COO-R—OOC-R^
+ n H2N-R—NH2-
Г — ОС—R-COO-R — ООС— R-COHN—r"— NH —
7 ч
7
R
соон
R—COO—R—ООС—
]
in
Ihooc
—N
Синтез полиэфироамидокислот прово-
проводят при комнатной темп-ре в р-ре 1Ч,К-диметилформ-
амида, ]М,]Ч-диметилацетамида, N-метилпирролидона
или др. полярных растворителей, в к-рых А раствори-
растворимы. Таким способом получают А с приведенной вяз-
вязкостью 0,5%-ного р-ра в диметилформамиде не менее
1,7 дл/г. Циклодегидратацию, к-рую проводят после
формования изделия из А, а также при отверждении
лака или клея, осуществляют при 200—250 °С или
при комнатной темп-ре под действием химич. агентов
(напр., смеси пиридина и уксусного ангидрида).
2. Поликонденсацией дикарбоновых к-т или диолов,
содержащих имидные группы, соответственно с диола-
ми или дикарбоновыми к-тами, напр.:
/? НООС
хх>
со'
СООН + n HOROH
COORO—
Процесс проводят в расплаве или р-ре при темп-ре
выше 200 СС в присутствии катализаторов, напр, ук-
уксуснокислого цинка или триоксида сурьмы.
Наиболее интересны ароматич. П. (из ароматич.
первичных диаминов, напр. 4,4'-диаминодифенилового
эфира, бензидина, л«-фенилендиамина, 4,4'-диами-
нодифенилметана, и таких диангидридов, как продукт
взаимодействия тримеллитового ангидрида и диацетата
гидрохинона). П.— твердые аморфные вещества белого
или светло-желтого цвета, легко кристаллизующиеся
при нагревании выше темп-ры стеклования B40—
270 °С) до высоких степеней кристалличности (85—
90%); не растворяются в воде и большинстве органич.
растворителей. Они обладают высокой тепло- и термо-
термостойкостью (остаются гибкими после выдерживания
при 240 °С в течение 750 ч или при 325 °С в течение
100 ч). По термоокислительной стабильности П., как
правило, уступают ароматич. полиимидам, но превос-
превосходят ароматич. полиэфиры (полиарилаты). Ниже
приведены свойства пленок из П.:
Прочность при растяжении, Мн/м2 (кгс/см2)
при 20 °С
при 200 °С
Относительное удлинение, %
при 20 °С
при 200 °С
120-150
A200 — 1500)
50 — 60
E00-600)
15-20
40-65
831
полиимиды
832
Модуль упругости при растяжении
B0 °С), Мн/м* [кгс/сж2]
Уд. объемное электрич. сопротивление,
Том-м [ом-см]
Тангенс угла диэлектрич. потерь при
1 Мгц
Диэлектрич. проницаемость при 1 Мгц
C0-55)-102
[C0 —55I033
A,2—3,5)-1 О2
A235I01]
0,005
3 , 0
Из полиэфироамидокислот изготовляют лакокрасоч-
лакокрасочные материалы, а также клей, термич. обработка к-рых
приводит к конечному продукту — полиимидоэфиру.
Покрытия из П. характеризуются нагревостойкостью
и обладают электроизоляционными свойствами. Из П.
производят также пленки.
Лит.: Ли Г., Стоффи Д., Невилл К., Новые ли-
линейные полимеры, пер. с англ., М., 1972; Власова К. Н.,
Танунина П. М., Палладина Т. И., Пласт, массы,
№ 2, 25 A969); А д р о в а Н. А. [и др.], Высокомол. соед.,
А15, № 1, 153 A973). Я.С.Выгодский.
ПОЛИИМИДЫ (polyimides, Polyimide, polyimi-
des) — полимеры, содержащие в основной или боковой
цепи макромолекулы циклич. имидную группу I. К П.,
у к-рых имидная группа расположена в
боковой цепи, относятся, напр., N-винил-
имидов полимеры и имидов непредельных
кислот полимеры. В данной статье рас-
рассмотрены только П. с имидными группа-
и ми в основной цепи. По строению П. мо-
могут быть алифатическими, алициклическими или аро-
ароматическими, по структуре — линейными или трех-
трехмерными. Наибольшее практич. применение получи-
получили ароматич. линейные П. благодаря ценным физико-
механич. свойствам, не изменяющимся в течение дли-
длительного времени в широком темп-рном интервале
(от—200 до 300 °С).
Получение. П. получают полициклоконденсацией,
радикальной полимеризацией (или сополимеризацией),
по реакциям Дильса — Альдера и 1,3-диполярного
циклоприсоединения. Наибольшее значение имеют П.
общей ф-лы:
/с\
О
-N
N-R-
II
О
II
О
где Q
О- , —СО—, —
или отсутствует)
(Y= —Н, —Hal, —СН3 , —ОСН3 идр ;
—о—, — сн2—, — s-, -so2—, N:(cf3) ,
и пи отсутствует!
Такие П. получают полицпклокондеисацией ароматич.
тетракарбоновых к-т или их производных (диангидрп-
дов, кислых диэфиров) с алифатич. или ароматич. ди-
диаминами.
П., содержащие в основной цепи алифатич. звенья,
получают, как правило, полициклоконденсацией в рас-
расплаве солей алифатич. диаминов с алифатич. или аро-
ароматич. тетракарбоновыми к-тами или их кислыми ди-
эфирами при 110—140 СС. Чтобы завершить реакцию,
реакционную массу дополнительно нагревают в течение
нескольких часов при 250—300 °С. Для достижения
высокой мол. массы образующиеся П. в условиях реак-
реакции должны находиться в расплавленном состоянии.
Наиболее высокомолекулярные П., способные к пере-
переработке, получаются по этому методу из пиромеллитово-
го диангидрида и алифатич. диаминов, содержащих
не менее 9 атомов углерода в линейной цепи или не ме-
менее 7 при наличии разветвлений. Диамины с меньшим
числом атомов С в молекуле дают неплавкие и нераст-
нерастворимые полипиромеллитимиды.
Плавкие формуемые П. высокой мол. массы получе-
получены также из алифатич. диаминов с более короткой угле-
углеводородной цепью, но при использовании производных
тетракарбоновых к-т след. строения:
НООС
Т 1
СО ОН
R'= —Н
>
-сн.
Полициклоконденсация в расплаве не применима для
получения высокоплавких высокомолекулярных П.
(особенно полипиромеллитимидов) на основе ароматич.
диаминов.
Алифатич. П. синтезируют также высокотемператур-
высокотемпературной полициклоконденсацией в органич. растворителях
в одну или две стадии.
Ароматич. П. получают обычно полициклоконденса-
полициклоконденсацией диангидридов ароматич. тетракарбоновых к-т
и ароматич. диаминов в р-ре в одну или две стадии.
Из диангидридов тетракарбоновых к-т наибольшее
практич. применение получили диангидриды пиромел-
литовой, 3,3',4,4'-бензофенонтетракарбоновой, 3,3',
4,4'-дифенилоксидтетракарбоновой и 3,3',4,4'-дифе-
нилтетракарбоновой к-т. Из диаминов чаще всего при-
применяют л*-и ?г-фенилендиамины. бензидин, 4,4'-диамино-
дифенилоксид, 4,4'-диаминодифенилсульфон. Ввиду до-
доступности диангидрида пиромеллитовой к-ты первыми
были получены и наиболее полно изучены полипи-
полипиромеллитимиды, в частности выпускаемый
в пром-сти полидифениленоксидпиромеллитимид.
При синтезе ароматич. П. в две стадии на первой по-
получают п о л и а м и д о к и с л о т у (I):
п О,
/с\ /с\
О + п H2NRNH9
-N Q N-R-
II II
О О
НООСЧ /СО—NHR—
Q
—HN—ОС^ ЧСООН
I
Для этого к р-ру ароматич. диамина в полярном раство-
растворителе (напр., в 1Ч,]\Т-Д11метилформамиде, ]Ч,]М-диметнл-
ацетамиде, N-метилпирролидоне или пиридине) при 15—
35 °С и перемешивании прибавляют оквимолярнос ко-
количество порошкообразного диангидрида тетракар-
боновой к-ты. По мере прибавления диангидрида вяз-
вязкость р-ра увеличивается (до нескольких единиц
н-сек/м2, или до нескольких десятков пз). С измене-
833
полиимиды
834
нием порядка прибавления реагентов молекулярная
масса образующейся полнамидокислоты уменьшается
вследствие протекания побочных реакций диангидри-
да с растворителем или растущей полимерной цепью.
Для образования полиамидокислоты существенное
значение имеют природа реагирующих веществ, чисто-
чистота мономеров и растворителя, темп-pa (при проведении
процесса выше 50 СС в большинстве случаев не удается
достичь высокой мол. массы); менее существенна кон-
концентрация мономеров (обычно концентрация образую-
образующейся полнамидокнелоты 15—25% но массе). Р-ры
полиамндокнелот используют непосредственно для пе-
переработки в изделия; для получения пресспорошков
полиампдокнелоты выделяют из р-ра осаждением в
большой избыток осаднтеля, однако при этом полимер
сильно деструктируется, т. к. карбоксильные группы
в о/шго-положении к амидной связи сильно катализи-
катализируют ее распад.
П о л и а м и д о к и с л оты — волокнистые веще-
вещества белого цвета, растворимые в растворителях амид-
ного типа, днметилсульфоксиде и в смесях их с бензо-
бензолом, ксилолом, циклогексаноном. Мол. массы полиами-
докислот составляют: Мп = 15 000—55 000, Mw =
= 25 000—270 000. Р-ры полиамидокислот при комнат-
комнатной и повышенных темн-рах нестабильны из-за само-
самопроизвольной деструкции, поэтому хранят их при
темн-рах не выше 0 СС. Более устойчивы в р-ре соли
иолиамидокислот и третичных аминов. Высокой устой-
устойчивостью как в р-ре, так и в чистом виде обладают
нолиамидоэфиры (III), получаемые полициклоконден-
сацией дихлорангидридов кислых диэфиров тетракар-
боновых к-т и первичных диаминов в Й,]Ч-диметилаце-
тамиде или N-метилнирролидоне при 5 —15 °С, напр.:
сюс.
COOR
I и + H2NRNH2 —
rooc^sAcoci ~HC1
R - —CH3 , —C2H5 или др алкил
На второй стадии полициклоконденсации осущест-
осуществляют имндизацию иолиамидокислот или их производ-
производных как таковых (в виде порошка) или в виде изделий
(пленки, волокна). Термич. имидизацию проводят
ступенчатым нагреванием полиамидокислоты от 200 °С
до 300—400 СС в вакууме или в инертной атмосфере.
Этим способом получают только тонкие пленки, т. к.
при изготовлении пленок большой толщины выделяю-
выделяющаяся при циклизации вода вызывает деструкцию
полимера и нарушает монолитность пленок.
Химич. нмидизация заключается в обработке поли-
полиамидокислоты дегидратирующими агентами (напр., ан-
ангидридами или хлорангндридами к-т, карбодиимидами)
в присутствии катализатора (третичный амин, напр.
пиридин, триэтиламин, изохинолин) в среде органич.
растворителей. Напр., пленку полиамидокислоты вы-
выдерживают при 20 СС в ванне, содержащей р-р уксус-
уксусного ангидрида и пиридина E0 : 50) в бензоле; спустя
24 ч пленку вынимают, высушивают при 20 С на воз-
воздухе, а затем прогревают в течение 1 ч при 200 — 300 °С
для достижения максимально возможной степени ими-
дпзации. Благодаря отсутствию деструктивных про-
процессов при химич. циклизации получаются П. более
высокой мол. массы, чем при термической; этим спо-
способом получают и более толстые пленки. Физико-химич.
свойства получаемых ароматич. П. зависят от степени
циклизации.
О степени превращения нолиамидокислоты в П. су-
судят по уменьшению в ИК-спектрах полимеров интен-
интенсивности полос поглощения, характерных для амидных
связей A550, 1680, 3260 см~1), карбоксильных групп
A710 см'1), и по появлению и интенсивности полос по-
ГЛО1ЦСНИЯ, характерных для имидного цикла G20, 1380,
1735, 1780 см~1). Хотя по данным ИК-спектров макси-
максимальная степень циклизации достигается после про-
прогрева полиамидокислоты при 200 — 220 °С в течение
90—120 мин, для получения П. с оптимальными фпзи-
ко-хпмич. свойствами требуется дополнительный про-
прогрев при 300—400 °С в течение нескольких часов.
При синтезе П. полициклоконденсацией тетракарбо-
новых кислот или их производных с диизоцианатамп,
например:
о
— N~CO
OCNRNCO
О
\
СО—О
\
СО
\ /
О—СО
\
\
c=o
CO—N—R —
-со2
Полиимид
на первой стадии при 40 — 50 °С, иногда в присутствии
катализаторов (напр., триэтиламина, диметилфенил-
амина, бензойной или акриловой к-ты), образуется
полиацилоксиимид (IV), превращающийся в П. при
дальнейшем нагревании сначала при 80—140 СС и за-
затем при 250—300° С. Полиацилоксиимнд более стабилен,
чем полиамидокислота, но имеет низкую молекуляр-
молекулярную массу.
Ввиду сложности синтеза П. в две стадии, разраба-
разрабатываются одностадийные методы, имеющие особое зна-
значение для получения раство-
растворимых и плавких П. Синтез
—r'oh^ высокомолекулярных раство-
*- Подиимид рИМЫХ л содержащих объем-
объемные циклические группы, а
также ряда др. П. осуществлен
полициклоконденсацией экви-
молярных количеств днан гид-
гидрида тетракарбоновой кислоты и соответствующего
диамина [например, анилинфталеина, 9,9-бмс-D-амино-
фенил)флуорена] в одну стадию при 190 — 210 СС в раст-
растворе (например, в нитробензоле, бензонитриле, сульфо-
лане) в токе инертного газа. Т. к. этот процесс об-
ратим, для получения П. высокой мол. массы особое
значение приобретает полнота удаления или связывания
выделяющегося низкомолекулярного продукта реак-
реакции (воды). В ряде случаев получаемый р-р П. может
быть непосредственно использован как связующее,
а также для изготовления лакокрасочных материалов,
пленок и волокон.
Полициклоконденсацией дипмида ниромеллитовой
к-ты с различными алифатич. и алкилароматич. дигало-
генидами в среде диметилацетамида при 70 —140 °С
в присутствии каталитич. количеств третичных аминов
(напр., триэтиламина, пиридина) или поташа получены
полипиромеллитимиды низкой мол. массы. В одну ста-
стадию проводят также полициклоконденсацию диангид-
рида 2,3,5,6-пиридинтетракарбоновой к-ты с ароматич.
диаминами в полифосфорной к-те при темп-ре ниже
180 °С.
Заменой части бифункционального мономера в исход-
исходных веществах мономером с числом функциональных
групп в молекуле больше двух синтезированы сшитые
П. Так, термореактивные П. синтезируют на основе
бензофенонтетракарбоновой к-ты и различных арома-
ароматич. диаминов в смеси с 2,4-диаминоацетанилидом и
3,5-диаминобензойной к-той. В макромолекулу обра-
образующегося форполпмера входят фрагменты А и В (см.
кол. 835), содержащие соответственно ацетампнную и
карбоксильную группы.
835
полиимиды
836
NHCOCH3
соон
При темп-pax выше 200 СС форполимер непосредствен-
непосредственно в изделии переходит в неплавкое и нерастворимое
состояние в результате реакции по ацетамишюй и кар-
карбоксильной группам. Такие термореактивные П. при-
применяют в качестве высокотермостойких связующих в
производстве стеклопластиков.
Свойства. Алифатич. П.— твердые, легко кристал-
кристаллизующиеся вещества белого или желтого цвета (в за-
зависимости от строения тетракарбоновой к-ты и диа-
диамина); могут быть получены высокой молекулярной
массы.
Алифатич. полшшромеллитимиды на основе диами-
диаминов, содержащих менее 7 атомов С в молекуле, харак-
характеризуются высокими темп-рами плавления, лежащи-
лежащими выше их темп-р разложения, и не растворяются
в известных органич. растворителях. Поэтому из та-
таких П. обычными методами не удается получить изделий
с хорошими физико-химич. свойствами. Полигшромел-
литимиды алифатич. диаминов с более длинной нор-
нормальной (не менее 9 атомов С) или разветвленной (не
менее 7) углеводородной цепью, а также П. др. тетра-
карбоновых к-т размягчаются при темп-pax ниже
300 еС; такие П. хорошо перерабатываются прессова-
прессованием или литьем под давлением, для них характерна
весьма низкая темп-pa стеклования A00—200 СС).
В аморфном состоянии такие П. хорошо растворимы
в крезоле, с^лим-тетрахлорэтане, хлороформе, нераст-
нерастворимы в диметилацетамиде, диметилформамиде, аце-
ацетоне, бензоле и др. Из р-ров этих П. можно формовать
пленки (прочность при растяжении пленки П. дифе-
нилоксидтетракарбоновой к-ты и октаметилендиами-
на 50 Мн/м2, или 500 кгс/см2, относительное удлине-
удлинение 65%).
Ароматич. П.— твердые негорючие вещества пре-
преимущественно аморфной структуры; цвет зависит от
химич. строения исходных веществ (напр., полидифе-
ниленоксидпиромеллитимид — золотистого цвета, по-
лидифениленпиромеллитимид — темно-красного, а П.
дифенилоксидтетракарбоновой к-ты и анилинфталеи-
на — белого цвета). Ароматич. П. отличаются от боль-
большинства органич. полимеров весьма высокой плотно-
плотностью A,35—1,48 г/см3). Мол. масса (MW)U. 50 000—
150 000.
Ряд свойств П. обусловлен существованием сильного
межмолекулярного взаимодействия, связанного с на-
наличием в основных цепях имидных циклов и бензоль-
бензольных колец. Так, для ароматич. П. характерны высокие
темп-ры переходов: темп-ры размягчения выше 200 °С
и темп-ры плавления выше 400 СС. Многие П. хорошо
кристаллизуются. Большинство ароматич. П. не раст-
растворяется в органич. растворителях и инертно к дейст-
действию масел, а также почти не изменяется при действии
разб. к-т. Аморфные П. с боковыми метильными, мето-
ксильными группами или с объемными циклич. груп-
группировками (фталидной, флуореновой, антроновой, фтал-
имидной) растворяются вплоть до высоких концентра-
концентрации A0% и выше) в 1Ч,1Ч-диметилформамиде, N,N-ah-
метилацетамиде, метиленхлориде, диоксане, симм-тет-
рахлорэтане, крезоле, нитробензоле. В конц. кислотах,
таких, как дымящая азотная или серная, особенно при
нагревании, ароматич. П. растворяются, однако раст-
растворение сопровождается деструкцией.
Под действием щелочей и перегретого пара ароматич.
П. гидролизуются. П. несколько более чувствительны
к воде, чем большинство др. линейных гетероцепных
полимеров. Напр., водопоглощение пленкой кантон Н
(см. Полиимидные пленки) при относительной влажно-
влажности 50% происходит в 6 раз быстрее, чем полиэтилен-
терефталатной пленкой. Вместе с тем у пленки сохра-
сохраняется 75% начального удлинения и 90% исходной
ударной вязкости после кипячения в воде в течение
15 сут. П. (на основе пиромеллитового ангидрида и
4,4'-диаминодифенилоксида) после кипячения в воде
содержит 3% воды; при этом прочность при растяже-
растяжении уменьшается в 2 раза. П. отличаются высокой ус-
устойчивостью к действию озона: после выдержки в те-
течение 3700 ч на воздухе, содержащем 2% озона, проч-
прочность при растяжении пленки кантон Н уменьшается
в 2 раза. Пленка становится хрупкой после облуче-
облучения солнечным светом в течение 6 лес. П. деструкти-
руются под действием гидразингидрата.
Изотропные пленки из ароматич. П. обладают высо-
высоким модулем упругости [при комнатной темп-ре 3-Ю3—
10-103 Мн/м2 C0-Ю3 —100-Ю3 кгс/см2)], к-рый может
быть дополнительно увеличен в результате ориентацп-
онной вытяжки пленки. При ориентации волокон уве-
увеличение модуля упругости еще значительнее. Если
ориентация пленки или волокна сопровождается силь-
сильным упорядочением структуры и кристаллизацией, мо-
модуль упругости может достигать значений 3-Ю4—
4-Ю4 Мн/м2 C0-Ю4—40-Ю4 кгс/см2), характерных для
неорганич. стекол и металлов.
Изделия из ароматич. П. характеризуются высокой
стабильностью размеров и низкой ползучестью при
высоких темп-pax. Прочности при растяжении при
комнатной темп-ре неориентированных пленок арома-
ароматич. П. достаточно высоких мол. масс в зависимости
от химич. строения исходных веществ составляют
100—300 Мн/м2 A000—3000 кгс/см2), а относительное
удлинение 10—150%. При наличии в диангидридном
или диаминном компоненте атомов кислорода или серы
температура стеклования П. составляет 200 — 250 °С.
В высокоэластическом состоянии такие П. ведут се-
себя как слабосшитые каучуки. При этом у П. сохра-
сохраняются достаточно высокие прочностные свойства (см.
таблицу).
П. являются среднечастотными диэлектриками; ди-
электрич. проницаемость лежит в области 3 — 3,5 и
мало зависит от частоты и темп-ры. Уд. объемное эле-
ктрич. сопротивление составляет 103 —104 Том- м
(Ю17—Ю18 ом-см) при 20 °С и 1 Том-м A014 ом-см)
при 200 °С; выше 200 °С зависимость удельного объ-
объемного электрического сопротивления от температу-
температуры определяется температурой размягчения. У боль-
большинства ароматич. П. тангенс угла диэлектрических
потерь при 20 °С равен A —1,5)-10~3; его величина
незначительно зависит от частоты и температуры в
интервале 50—230 СС.
П. характеризуются высокой термостойкостью и
устойчивостью к действию у-лучей, быстрых электронов
и нейтронов. Наиболее высокой термостойкостью обла-
обладают П., макромолекулы к-рых содержат только аро-
ароматич. кольца и имидные циклы. Потери в массе у та-
таких П. при 500 °С в вакууме не превышают 1 — 1,5%
(по данным термогравиметрич. анализа). П. на основе
диангидрида 3,3',4,4'-дифенилтетракарбоновой к-ты и
ароматич. диаминов устойчивы при нагревании до
450—500 °С. При наличии в макромолекулах между бен-
бензольными кольцами атома кислорода или серы потери
в массе при этой же темп-ре достигают ок. 7%. П.,
содержащие в цепи метиленовые группы, разлагаются
при 260—300 СС.
Ароматич. П. устойчивы в условиях длительного нзо-
термич. нагревания при темп-pax до 350 °С. Еще более
термостойки П., не содержащие в макромолекуле ато-
837
полиимиды
838
Свойства ароматических полиимидов общей формулы
—N^
О
II
\
N-NT
N-R-
Строение полиимида
Q
То же
ХГ<Х
То же
То же
тех
R
-о-о
То же
-О-о-О-
-очн>
XT
-о-о
-оо-
-ок>~о-
0Q0
Прочность при рас-
растяжении, Мн/м2
(кгс/см2)
20 СС
200 B000)
250 B500)
180 A800)
200 B000)
140 A400)
180 A800)
140 A400)
150 A500)
100 A000)
400 ^С
100 A000)
60 F00)
50 E00)
50 E00)
-
80 (800)*
—
-
-
Относитель-
Относительное удлине-
удлинение, %
20 (
2
20
80
120
30
100
40
100
6
| 400 "С
15
80
120
150
-
120*
-
-
-
Модуль упругости,
Мн/м* (кгс/см2)
20 "С
8-103
(80-Ю3)
7-103
G0-Ю3)
3,5-1 О3
C5-103)
3,2-Ю3
C2-Ю3)
2,9-Ю3
B9-1 О3)
3,2-1 О3
C2-Ю3)
3-Ю3
C0-Ю3)
2,8-103
B8-Ю3)
4,5-Юз
D5-Ю3)
400 °С
1,2-103
A2-1 О3)
0 , 8-1 О3
(8-Ю3)
0,6-Ю3
@-1 О3)
0,5-1 О3
E-1 О3)
-
1 ,7-103
A7-1 О3)
-
-
-
Темп-ра
размягче-
размягчения, °с
Не разм.
То же
» »
» »
290 D90)**
270
260
200 D20)**
370
* При 200 СС. ** В скобках приведена темп-pa плавления закристаллизованного П.
мов водорода, с отрыва к-рых при высоких темп-рах
начинается деструкция. Так, П. на основе диангидрида
пиразиытетракарбоновой к-ты и диампнотиадиазола не
изменялся при нагревании при 400 °С в течение 25 ч,
в то время как аналогичный П. на основе пиромелли-
тового диангидрида в этих же условиях полностью де-
структировался. Пленки П., содержащих в макромо-
макромолекулах бензимидазольные, бензоксазольные или окса-
диазольные звенья (такие П. получены полициклокон-
денсацией соответствующих диаминов с диангидридами
пиромеллитовой или 3,3,4,4'-бензофенонтетракарбо-
ыовой к-ты), после нагревания на воздухе при 325 СС
в течение 332 ч теряли в массе 4 —18%, но оставались
прозрачными и достаточно гибкими.
Полиимид (в СССР марка ДФО) характеризуется
след. показателями:
Теплостойкость по Вика, °С 265
Прочность при растяжении, Мн/м2
(кгс/см2)
при 20°С 120 A200)
при 200°С 40-50 D00 — 500)
Относительное удлинение при 2 0 0°С,% . 20 — 30
Модуль упругости, Мн/м2 {кгс/см2)
при 20°С 3200 C2 000)
при 200°С 1500 A5 000)
Стеклопластики на основе термореактивного полиимид-
вого связующего (на основе 3,3',4,4'-бензофенонтетра-
карбоновой к-ты и ж-фенилендиамина) после 100 ч на-
нагревания при 315 СС обладают в 2 раза большей проч-
прочностью при растяжении, чем стеклопластик па основе
линейного П. Деструкция ароматич. П. при 500—
600 °С в вакууме и на воздухе происходит в результате
распада имидных циклов с выделением СО, СО2 и обра-
образованием карбонизованного азотсодержащего остатка,
обладающего полупроводниковыми свойствами (при
деструкции П. в вакууме).
Для П. характерны низкий коэфф. трения @,05—0,17
по стали), весьма высокая для органических полиме-
полимеров теплопроводность 150—180 вт/(м-К) [125 — 150
/(^С)]
()
Переработка и применение. Монолитные изделия из
неплавких полиппромеллитимидов получают по техно-
технологии, общей с порошковой металлургией. Сформован-
Сформованные заготовки подвергают механич. обработке. Формо-
Формование стеклопластиков с использованием разб. р-ров
связующего осуществляют в эластичном мешке посред-
посредством выдержки изделия в нем при 150 —170 СС (и не-
небольшом разрежении) в течение 4 ч и затем в течение
10 ч при 300—315 °С (мешок одноразового использо-
использования). Стеклопластик с более высоким содержанием
связующего получают прессованием: сначала иод дав-
давлением 0,07—0,175 Мн/м2 @,7 — 1,75 кгс/см2) при 100—
120 °С выдерживают в течение 1 ч, затем 1 — 2 мин при
300—315 °С, после чего давление повышают до 1,5 — 2,0
Мн/м2 A5 — 20 кгс/см1) и выдерживают в течение 30 мин.
Стеклопластики изготавливают также намоткой стекло-
стекловолокон, пропитанных связующим, на нагретый до
65 °С сердечник с последующей выдержкой при 150 —
175 СС в течение 4 ч и затем при 300 — 315 °С в течение
10 ч. Полиимид (марка ДФО) перерабатывается литьем
под давлением и прессованием при 380—390 С.
839
ПОЛИ-Е-КАПРОАМИД
840
Па основе ароматич. П. получают все виды технич.
материалов, предназначенных для длительной падеж-
падежной эксплуатации при теми-рах 250—300 °С. В полу-
полупромышленных н промышленных масштабах выпус-
выпускаются электроизоляционная полиимидпая пленка,
эмаль для обмоточных проводов, заливочные компаун-
компаунды, связующие, пластмассы, пепонласты, волокна (см.
Термостойкие волокна), клеи (см. Полиимидпые клеи),
лакокрасочные материалы (см. Термостойкие лакокрасоч-
лакокрасочные покрытия). Ароматич. П. рекомендованы в США
для применения в самолетах и космич. двигателях.
Монолитные изделия из ароматич. П. используются
в качестве высокотеилостойкях антифрикционных са-
мосмазывающпхся материалов, прокладок и конструк-
конструкционных изделий.
Впервые высокомолекулярный ароматич. П. был син-
синтезирован Одвардсом и Робинсоном в 1955. Первый син-
синтез П. из 4-амипофталевой к-ты осуществлен Бод-
жертом Т. М. и Реншоу Р. Р. в 1908.
Лит.: Полинмиды — новый класс термостойких полимеров,
Л., 19E8; Коршак В. В., Термостойкие полимеры, М.,
1909; К о т о н М. М., ЖПХ, 42, в. 8, 1841 A969); Куд-
Кудряв цо в Г. И., Термостойкие полимеры и волокна на их ос-
основе. Хим. волокна, Л» 4, 23 A969); Ли Г., С т о ф ф и Д.,
Невилл К., Новые линейные полимеры, нер. с англ., М.,
1972; К а р д а ш И. Е., ТелешовЭ. Н., Синтез, свойства
и применение высокотермостойких гетероциклических полиме-
полимеров, М., 1971 (Итоги науки. Серия Химия, 27); Фрей-
Фрейзер А. Г., Высокотермостойкие полимеры, пер. с англ., М.,
1971. М. М. Нотон.
П0ЛИ-8-КАПР0АМИД — см. Капролактама поли-
полимеры.
ПОЛИКАПРОЛАКТАМ — см. Капролактама поли-
полимеры.
ПОДИКАРБОДИИМИДЫ (polycarbodiimides, Poly-
karbodiimide, polycarbodiimides) — полимеры общей
ф-лы [— R — N = С = N—}п, где R — бифункцио-
бифункциональный алифатич. или ароматич. радикал.
П. обычно получают с количественным выходом по-
ликонденсацней ди- и полиизоциаиатов в р-ре при 25 —
250 °С и атмосферном давлении в присутствии фосфор-
органич. катализаторов (фосфиноксидов, фосфоленов,
фосфоланов или др.), напр.:
— R—
R— N=C=N— R—
Растворителями служат, гл. обр., ароматич. углеводо-
углеводороды или их смеси с полярными органич. соединениями,
напр. смесь хлорбензола с диметилсульфоксидом.
В зависимости от функциональности изоцианата
образуются линейные, разветвленные или сшитые П.
Варьируя время и темп-ру реакции, можно получать
линейные П. с концевыми группами — N^C = O или
без них.
П.— продукты от белого до рыжевато-коричневого
цвета, нерастворимые в органич. растворителях и воде.
Мета стабильные р-ры П. можно получить посредством
прекращения реакции до выпадения образующегося
П. из р-ра (особенно в присутствии полярных раствори-
растворителей, напр. диметилсульфокенда). П. плавятся, как
правило, выше 200 СС и характеризуются высокой
вязкостью расплава. Для ИК-сиектров П. характерна
полоса поглощения в области 4,70—4,80 мкм (колеба-
(колебания группы —N = C=N —).
Наиболее интересны ароматич. П., обладающие вы-
высокими механич. и дизлектрич. свойствами. Так, плен-
пленки из полн-3,3'-диметокси-4,4'-дифениленкарбодшшида
имеют модуль упру-
оснз гости 8300 Мн/м2
=C==N_ (83 000 кгс/см2); отпо-
\п сительное удлинение
6,2%; прочность нри
растяжении 280 Мн/м2 B800 кгс/см2)', диолектрич. про-
проницаемость при 102 —105 гц 3,4 B5 °С); 3,8 A70 °С); тан-
тангене угла диэлектрич. потерь 0,002—0,003 B5 °С);
уд. объемное ;>лектрич. сопротивление выше 1-1014од
B5 °С), 2,2-1011 ом B20 °С). Поли-2,4-толуиленкар-
бодиимид при 100 °С ведет себя
как эластомер. П., полученные /N=C=N—
ноликопденсацпей полиэфиров, со- [__/ \_рН
держащих концевые изоцпанатные [ \ / 3
группы, также являются эласто-
эластомерами. Поликарбодиимид, синтезированный поли-
коыденсацпей толуилендиизоцианата [катализатор —
2,4,6-??г/шс-(диалканоламино)-С-триазин] является же-
жестким пенопластом; он обладает высокими диэлектри-
диэлектрическими свойствами, в пламени не горит и не выде-
выделяет дыма.
Пленки из многих П. устойчивы к действию кипящих
водных р-ров к-т A0%-ной H2SO4), щелочей C н. р-ра
NaOH) и оснований A0%-ного этаноламина). Однако
П. в виде тонкодисперсных порошков или метастабиль-
ных р-ров взаимодействуют с этими реагентами и др.
соединениями, содержащими подвижные атомы водо-
водорода, напр. с водой, сероводородом, спиртами, аминами
и к-тами, с образованием соответственно полимочевины,
политиомочевипы, иоли-0-алкилизомочевины, полигу-
аиидина и полиацплмочевины.
Методом полива из расплавов или р-ров П. можно
получать пленки. П. могут служить стабилизаторами
полимеров, содержащих сложноэфирные группы. Защит-
Защитное действие поликарбодиимидов заключается в пре-
преимущественном взаимодействии групп —N = C=N —
с геиелотами, выделяющимися при деструкции полиэфи-
полиэфиров, что предохраняет полимер от автокаталитичес-
автокаталитического разложения.
Лит.: Б о ч а р о в Б. В., Усп. хим., 34, № 3, 488 A965);
Kurzer P., Douraghi-Zadeh К., Chem. Revs 67
№ 2, 107 A967); Windemuth E., Kunststoffe, 57, № 5,
337 A967). Д. Г. Вальковский.
ПОЛИКАРБОНАТЫ (polycarbonates, Polykarbonate,
polycarbonates).
Содержание:
Получение 84 0
Свойства 84 5
Переработка и применение 849
Поликарбонаты — сложные полиэфиры угольной к-ты
и диоксисоединений. Общая формула поликарбона-
поликарбонатов [ — О — R — О—СО — О — R — ]л. В зависимости от
природы R поликарбонаты м. б. алифатич., жпрно-
ароматич. и ароматическими, в зависимости от струк-
структуры макромолекулы — линейными, разветвленными
и трехмерными. Наибольший интерес представляют
линейные ароматич. П. благодаря определенному
комплексу физико-механич. показателей.
Получение. Для синтеза П. используют ароматич.
дноксисоединения — двухатомные фенолы типа бис-
D-оксифепил)алканов
л'
где в=-сн2-; -сн(сн3)-; -С(сн.,J-; -сн(С0н5)-;
-С(СН3) (СвН3)-; _/~Л_/~~\_ ; -О- и др.;
\—/ \==/
R'=-H; —С1; -Вг; —СН3; СН2=СН-СН2-
В пром-сти применяют гл. обр. 2,2-бмс-D-оксифенил)-
проиан (наз. также дианом, дифеянлолпропаном, бис-
фенолом А). Вторым компонентом служит фосген
(дихлорангидрнд угольной к-ты) или диарилкарбонаты
(напр., дифенилкарбонат).
В нром-сти П. получают фосгенированпем диокси-
соедииений газообразным фосгеном в среде пиридина
841
ПОЛИ КАРБОНАТЫ
842
или р-ром фосгена в инертном органич. растворителе
(напр., в хлористом метилене) на границе раздела фаз
(межфазной поликоиделсацией), а также пере;)терифи-
кацией диарилкарбонатов ароматич. дноксиеоедине-
ниями.
Ф о с г е н п р о в а н и е в растворе. Про-
Процесс осуществляют в отсутствие воды в среде пиридина
(см. Поликопденсация в растворе), к-рый служит одно-
одновременно катализатором и акцептором хлористого
водорода (по периодич. схеме):
п НО
сн,
сн
сн
Кроме фосгена, можно использовать дихлоруголь-
ные эфиры ароматич. диокслсоединений, а также три-
хлорметиловый эфир хлоругольноп к-ты (дифосген),
бггс-трихлорметилкарбонаты ароматич. диоксисоеди-
нсний.
В реакции используют избыток сильной щелочл
(NaOH, КОН), т. к. реакционная среда должна иметь
рН > 10. Процесс экзотермичен, поэтому необходимо
охлаждение реакционной смеси (оптимальная тсмп-ра
реакции 20—25 °С). При повышении тсмп-ры возра-
возрастает скорость гндро-
НС1
Реакция протекает при комнатной (пли более низкой)
темн-ре с высокой скоростью, что позволяет органи-
организовать процесс и но непрерывной схеме. Необходимое
условие получения П. с высокой мол. массой и опти-
оптимальными свойствами — растворимость образующегося
П. в пиридине. Т. к. в процессе реакции выделяется
2 моль хлоргидрата пиридина на 1 моль ароматич.
диоксисоединения, необходимо использовать большой
избыток пиридина (на 1 моль фосгена не менее 2 моль
пиридина), чтобы реакция протекала в жидкой фазе.
Для растворения полученного П. в реактор вводят
также инертный растворитель (метиленхлорид, бен-
бензол, толуол и др.) в таком количестве, чтобы после
отделения пиридина и хлоргидрата пиридина оставался
вязкий р-р П. в этом растворителе, что облегчает выде-
выделение и очистку П.
К полученному вязкому р-ру П. после отмывки
хлоргидрата пиридина прибавляют при перемешива-
перемешивании осадитель (напр., гептан, петролейный афир),
отфильтровывают- образовавшийся тонкодисперсный
белый осадок, к-рый сушат при 120 °С в вакууме.
Смесь растворителя и осадителя, полученную после
фильтрования, высушивают, разделяют перегонкой
и возвращают в процесс. Водную фазу, содержащую
хлоргидрат пиридина и растворитель, нейтрализуют
эквимолярным количеством каустич. соды. Из полу-
полученной смеси сначала отгоняют растворитель, а затем
азеотроп пиридина с водой, к-рый разделяют. Пири-
Пиридин вторично перегоняют до требуемой чистоты и воз-
возвращают в процесс.
Описанный процесс можно использовать для полу-
получения всех П., растворимых в инертном растворителе.
Метод применяют обычно для синтеза большинства
П. на основе производных б^с-D-оксифенил)метана,
а также таких диоксисоединений, к-рые претерпевают
химич. изменения в щелочных средах или неустойчивы
при нагревании до высоких темп-р B80—300 °С). Мол.
масса образующегося П. F0 000—80 000) в основном
зависит от темп-ры реакции, количества пиридина,
скорости прибавления фосгена, присутствия агентов
обрыва цепи (монофенолов).
Преимущество данного метода заключается в том,
что ноликонденсация протекает в гомогенной жидкой
фазе при комнатной или более низкой темп-ре; недо-
недостатки — использование дорогостоящего пиридина,
растворителя и осадителя, к-рыс необходимо регене-
регенерировать.
Фосгенирование на границе раз-
раздела фаз. При получении ароматич. П. этим ме-
методом используют водные щелочные р-ры ароматич.
циоксисоединений и р-р фосгена в инертном органич.
растворителе:
та NaO
р р
лиза фосгена и бис-
фенольных эфиров
хлоругольноп к-ты,
что приводит к сни-
снижению мол. массы и
выхода П. Полимер, обладающий лучшим комплек-
комплексом свойств, получается при использовании в ка-
качестве инертного растворителя метиленхлорида или
хлороформа, в к-рых растворим и фосген, н обра-
образующийся П. Т. к. щелочные соли ароматич. диокси-
диоксисоединений легко окисляются в щелочной среде, в реак-
реакционную смесь рекомендуется вводить восстановители
(дитионат натрия, сульфит натрия и др.)-
Процесс осуществляют путем непрерывного пере-
перемешивания реакционной смеси в реакторе (см. рис.)
при комнатной (или более низкой) темп-ре. В резуль-
Раетвор , Н3О
щелочи
в метиленхлориде
Дифенилолпропан
Раствор
NaCl I
Схема получения П. фосгенированием бисфенола А методом
межфазной поликонденсации: 1 — реактор, снабженный
мешалкой; 2 -— флорентийский сосуд; 3 — аппарат для
промывки; 4 — флорентийский сосуд для отделения р-ра П.
в СН2Сь от водной фазы; 5 — сушилка.
тате образуется р-р П. незначительной вязкости. К нему
добавляют катализатор (тршзтиламин), и перемешива-
перемешивание продолжают до получения вязкого р-ра П., к-рый
разбавляют хлористым метиленом, доводя вязкость
до желаемой величины. Органич. фазу отделяют, П.
промывают водой до полного удаления солей и щелочи.
Для удаления электролитов и части воды р-ры П. мож-
можно также охлаждать ниже 0 °С, а затем отфильтровывать
соль и лед. Р-ры П. в органич. растворителе, предвари-
предварительно высушенные, напр, азеотропной отгонкой или
осушающими агентами, можно использовать непо-
непосредственно для получения пленок или волокон. Не-
Необходимая вязкость р-ра достигается отгонкой избыт-
843
ПОЛИКАРБОНАТЫ
ка растворителя. Из р-ра П. выделяют осаждением не-
растворптелем (напр., гептаном); образовавшийся мелко-
мелкодисперсный порошок отфильтровывают и сушат. Метод
выделения осаждением может быть применен только
для хорошо кристаллизующихся П. Полимеры с низкой
способностью к кристаллизации при добавлении оса-
дителя образуют стойкие эмульсии, из к-рых выделить
П. очень трудно.
Для получения П. высокой мол. массы в качестве
катализаторов м. б. использованы третичные амины
или четвертичные аммониевые основания, способные
претерпевать перегруппировку Стивенса и превра-
превращаться в третичные амины, а также четвертичные фос-
фониевые,, арсониевые и третичные сульфониевые сое-
соединения. Оптимальное количество катализатора опре-
определяется реакционной способностью исходных диокси-
соединений (напр., 2— 3% от количества исходного
бисфенола).
Триэтиламин (или др. третичный амин) образует
с дифенолом или гидроксильной группой олнгомера
комплекс, к-рый переходит в органич. фазу:
Ангидридоэфнрная группа угольной к-ты стабилизиру-
стабилизируется, превращаясь в эфирную группу с элиминирова-
элиминированием СО2:
-j + С02
О
ОЦ + N(C2H5K
..N(C2H5
Нуклеофильная атака фосгена комплексом приводит
к разрушению последнего и образованию повой связи
за счет свободной пары электронов кислородного атома
дифенола но схемам:
Образующийся хлоргидрат третичного амина пере-
переходит в водную фазу, где под действием щелочи превра-
превращается в основание, способное к дальнейшим превра-
превращениям:
[HN+(C2H3K] CI- + NaOH -> N(G2H5K + NaCl + H2O
Таким образом, в присутствии третичного амина частич-
частичный гидролиз хлорформиатных групп не приводит
к прекращению роста полимерной цепи, а способствует
образованию более высокомолекулярного П.
При содержании катали-
\ затора, меньшем оптималь-
5), ного> группы_0С00Н об-
образуются с более высокой
скоростью, чем группы — OCOONHR3 взаимодействуют
с хлорформиатными. Это приводит к накоплению в си-
системе групп —ОСООН. После израсходования всех
хлорформиатных групп на обоих концах макромоле-
ci-c—ci
/II
o-hn(c2h5K —R-s,
ci—с—ci
4
y
8+1 I
"Ж
Приведенный выше механизм действует на началь-
начальной стадии реакции образования П. При более глу-
глубоких степенях превращения роль катализатора сво-
сводится к следующему: в ходе образования поликарбо-
поликарбоната происходит частичный гидролиз концевых хлор-
хлорформиатных групп и превращение их в менее активные
группы —ОСООН, к-рые очень медленно взаимо-
взаимодействуют с группами NaOAr— и значительно легче
с катализатором (третичным амином), находящимся
в гидратной или основной форме:
~0C0-/)-R
II \—/
О
0C00H + HONHR.3
OCOONHRo
о
Образующаяся соль взаимодействует с негидролнзовав-
шимися хлорформиатными группами др. полимерной
или олнгомерной молекулы:
—ОСО-</ )-R—\ /—
\_/ * \=
OCOONHR, + C1C— O—
—R
О о
R-/~Vo-c-ci + (hn(c2h5K|ci
\=/
II
о
HN(C2H5K C1
кул будут находиться нереакционноспособные группы
— ОСООН. При большом избытке катализатора уско-
ускоряется гидролиз хлорформиатных групп, в результате
чего повышается основность среды, и, следовательно,
группы —ОСООН будут образовываться быстрее, чем
расходоваться в реакциях с катализатором и хлорфор-
хлорформиатными группами. Гидролиз хлорформиатных кон-
концевых групп в макромолекулах П., полученных в при-
присутствии третичных аминов, протекает достаточно
быстро. При получении П. в отсутствие третичного
амина для завершения гидролиза необходима допол-
дополнительная выдержка реакционной смеси в щелочной
среде.
• При использовании в качестве катализаторов тре-
третичных аминов и четвертичных аммониевых оснований
межфазную поликонденсацию можно проводить по не-
непрерывному методу.
Преимущество данного процесса — низкая темп-ра
реакции B5 °С), применение только одного органич.
растворителя и возможность получения П. высокой
молекулярной массы (до 200 000); недостатки — необ-
необходимость промывания р-ра полиме-
полимера очень большим количеством воды
для полного удаления из него элек-
электролитов и использование для этого
смесителей с сигмаобразными лопа-
—осо—
о
HC1 и Р е э т е р и ф и к а ц и е й по-
лучают ароматич. П., температура
845
ПОЛИКАРБОНАТЫ
846
плавления к-рых ниже их темн-ры разложения. По-
Поскольку нереэтерификация диалкилкарбонатов в при-
присутствии катализаторов щелочного характера проте-
протекает медленно даже при темп-ре выше 200 °С и при
этом дпалкилкарбонат и образующийся П. частично
разлагаются, в пром-сти в качестве эфиров угольной
к-ты используют диарилкарбонаты (напр., дифенил-
карбонат):
Переэтерификация — обратимая реакция, поэтому для
получения высокомолекулярных П. с хорошим выхо-
выходом необходимо удалять из реакционной смеси обра-
образующийся фенол. Процесс проводят в расплаве (см.
Ноликопденсация в расплаве) но периодич. схеме в две
стадии в присутствии в качестве катализаторов гидро-
гидроокисей или гидридов щелочных металлов, окисей или
органич. соединений алюминия, титана и цинка в ко-
количестве 0,0001—0,1% от массы образующегося П.;
дифенилкарбонат берут в избытке для получения
макромолекул с концевыми фенилкарбонатными груп-
группами. Реакцию осуществляют в автоклавах; первую
стадию при 150 — 200 °С и остаточном давлении
2,67 кн/м1, или 20 мм рт. ст. (при этом выделяется
и удаляется из реактора основная масса фенола и обра-
образуется П. низкой мол. массы); вторую стадию при 250—
280 СС и остаточном давлении 0,13—0,26 кн/м2, или 1 —
2 мм рт, ст. В результате образуется очень вязкий
расплав, к-рый при охлаждении превращается в свет-
светлый, прозрачный, упругий полимер (мол. масса
20 000 — 50 000).
Преимущество данного метода — отсутствие раство-
растворителя, что позволяет получить сухой П., пригодный
для непосредственного гранулирования. Благодаря
низкому содержанию примесей электролитов такой П.
обладает высокими термостойкостью и диэлектрич.
свойствами.
Недостатки процесса — необходимость проведения
переэтерификации при высокой темп-ре и глубоком
вакууме и невозможность получения П. с мол. мас-
массой выше 50 000 вследствие значительного повыше-
повышения вязкости расплава и протекания побочных реак-
реакций химич. деструкции.
Из смесей диоксисоединений, взятых в различных
соотношениях, можно получать рассмотренными выше
методами сополикарбонаты, или смешанные П. Свойст-
Свойства таких П. зависят от соотношения и химич. строения
исходных дифенолов. Для синтеза блоксополикарбо-
натов сначала получают методом межфазной поликон-
поликонденсации из различных двухатомных фенолов низко-
низкомолекулярные П. с концевыми реакционноспособными
группами — О — GOG1 (в виде р-ров в инертных раст-
растворителях). Затем различные р-ры смешивают и добав-
добавляют катализаторы (третичные амины), в результате
чего получают высокомолекулярные блоксополимеры,
состав, длину фрагментов и мол. массу к-рых можно
регулировать, изменяя мол. массу исходных олигомер-
ных П. и их соотношение. По сравнению со смесями
соответствующих гомополикарбонатов блоксополикар-
бонаты обладают более высокими темп-рами плавления
и модулем упругости.
Свойства. Ароматич. П.— высокоплавкие самозату-
самозатухающие термопластичные полимеры белого цвета.
Способность П. к кристаллизации, форма кристаллич.
образований и степень кристалличности A0 — 40%)
определяются строением исходного дифенола. Так,
кристаллизация П. затруднена, если у центрально-
центрального атома углерода дифенола
содержатся несимметричные ..—* | *—ч
или большие по объему замес- НО—f /~с—\ у
тителп R и R'. П. на осно- \==/ I, \—/
ве бисфенола А кристаллизу- Й
ется весьма медленно даже при выдерживании в
оптимальных для кристаллизации условиях. П. ха-
характеризуются сильным межцегшым взаимодействием,
обусловленным высокой полярностью карбонатных
групп.
Темп-ры плавления и стеклования П. зависят от при-
природы исходного дифенола; большое влияние оказывает
наличие заместителей в ароматич. ядрах дифенола
(табл. 1). Так, при введении атомов галогена в арома-
ароматич. ядра производных бггс-D-оксифенил)метана в 3,3'-
и 5,5'-положения темп-ры плавления и стеклования
Таблица 1. Влияние заместителей в ароматических ядрах
производных ог^-D-оксифенил)метана на темп-ры
плавления и стеклования поликарбонатов
Исходные дифенолы
но-/~
С1
\
но-(
>
С1
_/~
С!
\
яо7~
У-
С1
н°-С
С1
у
но—(
У
С1
Вг
Вг
CI
V-
//
но-/~
но \Z
н
н
ж
=/ 1 N.
н
>$¦
V-C-f
сн3
УН
сн3
СНз
л СНз/-
/ ? V
СН3 "
сн,
¦х 1 г—
ж
СН3
сн3
Л I у
СНз
СНз
С1
)-он
CI
>он
С1
/
Л-он
-Г
С1
N-OH
С1.
-У •
Л—он
С1
Вг
/
\—он
Вг
С1
\—он
СНз
\—он
Д—он
Темп-ра
плавления,
°с
223 — 225
234—239
250—260
260—270
220—230
250-260
240—260
190-210
150-170
210—220
Темп-ра
стеклова-
стеклования, °С
147
-—
171
163
149
180
157
147
95
140
847
ПОЛИКАРБОНАТЫ
848
соответствующих П. повышаются. Если оба ароматич.
ядра содержат атом галогена или алкильную группу
в о/j иго-положении к гидроксильной группе, темп-ры
плавления и стеклования П. понижаются; это пониже-
понижение незначительно при наличии заместителя только
в одном фенильном радикале.
Темп-ры плавления и стеклования П. на основе
дпфеыола, содержащего симметричные алкильные или
арильные заместители в ядре, невысоки вследствие не-
невысокой мол. массы таких П. Полимеры высокой мол.
массы на основе указанных дифенолов получить не уда-
удается, т. к. последние с трудом реагируют с производ-
производными угольной к-ты.
Зависимость между мол. массой М и характеристич.
вязкостью [г]] р-ра П. в различных растворителях
выражается ур-ннем Марка — Хувинка [г]] = К-Ма\
значения К и а приведены в табл. 2.
Таблица 2. Константы К и а в уравнении Марка — Хувинка для
определения мол. массы фракций П. на основе бисфенола А
Раствори-
Растворитель
Метилен-
хлорид
То же
»
»
»
»>
Хлороформ
Диоксан
Тетрагид-
рофуран
То же
»
Метод определе-
определения мол. массы
Седиментация ^
Диффузия (
CBeTopaeceHimeJ
То же
Оемометрия
То же
Светорассеяние
То же
Седиментация ^
1
Диффузия |
Светорассеяние)
Область мол.
масс
8-103—2 , 7 • 105
1 , 3- Ю4—8-Ю4
2,5 -1 О4— 1 0 -1 О4
2,2-104-20-Ю4
1 ,5-104-6-104
1.1 04-40-1 О4
8-103-27-104
1
1
1
0
2
5
3
,11
,11
,23
,92
,77
,54
,99
ri
• ю-2
•10~2
•К)
•К)
-ю-1
• ю-2
• ю-2
0
0
0
0
0
0
0
ее
,82
,82
,83
,87
,50
,67
,70
Растворимость П. зависит от их структуры и межмо-
межмолекулярного взаимодействия. Быстро кристаллизую-
кристаллизующиеся П. [напр., на основе гидрохинона, б^с-D-окси-
фенил)метана, 1,2-бггс-D-оксифеннл)отана] практически
нерастворимы в обычных органич. растворителях. П.
на основе бисфенола А растворяется или набухает
в большинстве органич. растворителей, за исключе-
исключением алифатич. и циклоалифатич. углеводородов,
одно- и многоатомных спиртов (кроме метанола),
растительных и животных жиров, масел. Основные
растворители П.— метиленхлорид, хлороформ, 1,1,2-
трихлорэтан, 1,1, 2,2-тетрахлорэтан. П. устойчивы
к действию водных р-ров неорганич. и органич. к-т,
солей и окислителей, слабых щелочей (карбонат и би-
бикарбонат натрия). Амины, гидроокись аммония и р-ры
сильных щелочей вызывают гидролитич. деструкцию
(омыление) П. Стойкость П. к гидролизу повышается
при введении в ароматич. ядра атомов хлора, брома
или метильных групп в opmo-положение к сложноэфир-
ной группе, а также при блокировании концевых
фенольных оксигрунп.
П. на основе 2,2-б^с-D-оксифенил)пронана не изме-
изменяется при нагревании в расплавленном состоянии
при 300 °С в течение многих часов и кратковременном
нагревании при 320 °С; выше 330 °С начинается деструк-
деструкция, сопровождающаяся изменением цвета до бурого.
Присутствие небольшого количества воды вызывает
гидролитич. деструкцию П., поэтому перед переработ-
переработкой его необходимо высушить до содержания влаги
не более 0,01%. Вязкость расплава П. в интервале
темп-р 240—300 °С (темп-ры переработки) изменяется
от 10 000 до 1000 н-сеп/м2 (от 100 000 до 10 000 пз),
оставаясь высокой по сравнению с вязкостью распла-
расплавов др. термопластов.
С увеличением однородности полимера по мол. массе
вязкость уменьшается.
П. обладают высокими прочностными свойствами.
Прочность сохраняется при наложении быстродейст-
быстродействующих внешних усилии; относительное удлинение
изменяется почти линейно с увеличением напряжения
вплоть до высоких значений последнего (такая зави-
зависимость свойственна большинству металлов). Изделия
из П. характеризуются высокой стабильностью разме-
размеров при переработке, их склонность к ползучести
очень мала. Благодаря застекловываиию формован-
формованного образца, к-рое препятствует кристаллизации,
и высокой темп-ре стеклования, прочностные свойства
П. изменяются незначительно в очень широком интер-
интервале температур (от —150 до 200 °С). Механич. свойства
изделий из литьевых марок П. на основе бисфенола А
при комнатной темп-ре остаются неизменными неогра-
неограниченное время вследствие незначительной склоннос-
склонности этого П. к кристаллизации; прочность при растя-
растяжении неориентированной аморфной пленки пони-
понижается только на 40% при повышении температуры
от 20 до 140 °С.
П. обладают хорошими диолектрич. свойствами как
при комнатной, так и при повышенной темп-pax, зави-
зависящими от степени кристалличности и ориентации
пленок. Кристаллизация уменьшает диэлектрич.
потери в интервале темп-р от —120 до 200 С. Ниже
приведены физико-механич. и электрич. свойства литье-
литьевых промышленных марок П. на основе 2,2-б^с-D-
оксифенил)пропана (мол. масса 32 000 — 35 000):
Плотность при 20°С, г/см* 1,2
20
Показатель преломления п^ 1,56 — 1,65
Темп-pa плавления, °С 22 0-^27 0
Прочность, Мн/м2 (кгс/см2)
при растяжении 56 — 78 E60—780)
при сжатии 77 — 95 G70 — 950)
при статич. изгибе 77 — 120 G70 — 1200)
Относительное удлинение, % 50-110
Твердость по Бринеллю, Мн/м2
(кгс/мм2) 150-160 A5-16)
Ударная вязь-ость, кдж/м2, или
кгс-см/см2 250 — 5 00
Усадка при литье, % 0,5-0,8
Теплостойкость по Вика, °С 15 0—165
Деформационная теплостойкость, °С
при нагрузке 1,87 Мн/м2
A8,7 кгс/см2) 138
при нагрузке 0,4 6 Мн/м2
D , 6 кгс/см2) 143
Уд. теплоемкость, кдж/(кг-К)
[ккал/(кг^С)] 1,17 [0,28]
Морозостойкость, °С „ ниже —100°
Водогюглощение, %
за 24 ч 0,1-0,15
за 168 ч 0,2 —0 ,35
равновесное B 5°С, 60%-ная
относительная влажность) ... 0,2
Уд. поверхностное электрич. сопротив-
сопротивление, Том (ом) 1 , 6 • 1 О3—8-Ю3
A,6- 101а—
— 8-1 О15)
Уд. объемное электрич. сопротивление,
Том-м [ом-см] A,5 — 6,1)• 102
[1 ,5-10lt5-6, 1-101С]
Диэлектрич. проницаемость
при 5 0 гц 3,17
при 1 Мгц 2,9 6
при 1010 гц 2,74
Электрич. прочность, Мв/м, или к«/мм 15,5 — 21,0
Дугостойкость, сек 10 —11*
Тангенс угла диэлектрич. потерь
при 5 0 гц 0,0009
при 1 Мгц 0,007-0,008
П. на основе бисфенола А — оптически прозрачный
материал. Так, прозрачность пленки из П. толщиной
100 мкм, отлитой из р-ра, в видимой части спект-
спектра колеблется от 85 до 90% , мало изменяясь при уве-
увеличении толщины пленки до 2 мм.
П. обладают низкой гигроскопичностью; количество
поглощаемой влаги зависит от степени кристаллич-
кристалличности полимера, темп-ры и парциального давления
водяного пара. Поглощение такого количества воды
не вызывает изменения размеров образца, что имеет
существенное значение для применения изделий, из П.
849
ПОЛИКАРБОНАТЫ
850
Паропрошщаемость П. сильно зависит от толщины
пленки и степени ее ориентации.
П. достаточно устойчивы к действию УФ-света
и излучений высокой энергии. При облучении пленки,
отлитой из р-ра, кварцевой лампой без фильтра в те-
течение 72 ч при 45 °С не наблюдается изменения окраски
и физич. свойств полимера. Стойкость П- к атмосфер-
атмосферным воздействиям (влаги, света, воздуха) при различ-
различных теми-pax так велика, что во многих случаях можно
обойтись без их стабилизации.
* П. устойчивы к действию микроорганизмов, гумн-
новых к-т, содержащихся в почве, к плесени; инертны
к водным р-рам мыл, детергентов, отбеливающих ве-
веществ, фотографич. химикатов, дезинфицирующих
средств, природных и синтетич. красителей, пигмен-
пигментов, ко всем видам пищевых продуктов. П. биологически
неактивны и при экстракции не дают продуктов, к-рые
бы обладали биологич. активностью. Они хорошо
совмещаются с др. полимерами (полиолефинами, поли-
полиэфирами, виниловыми полимерами). Такие компози-
композиции обладают повышенной ударопрочностыо, проч-
прочностью на изгиб и др.
П. можно модифицировать для придания им спе-
цифич. свойств. Так, для повышения темп-ры размяг-
размягчения и понижения горючести в основную цепь макро-
макромолекул вводят фрагменты различных днкарбоновых
к-т (напр., терефталевой, изофталевой, фенилфосфор-
ной, фенилфосфиновой), для чего используют их ди-
хлорангидриды. Для повышения относительного удли-
удлинения, термич. и термоокислительной устойчивости,
улучшения релаксационных свойств при сохранении
прочностных характеристик в макромолекулы П. вво-
вводят силоксановые звенья (замещением части фосгена
органоднгалогенсилоксанами). П., содержащие в цепи
мочевинные, амидные, нитрильные и уретановые
группы, прозрачны, обладают достаточно высокими
темп-рами размягчения, стойкостью к щелочному
гидролизу, высокими механич. прочностью и диэлект-
рич. показателями.
Недостатком П. на основе уретанобисфенолов типа
CNH-R-NHCO
b h
является невысокая термостабильность (ароматнч.
эфиры карбамнновой к-ты выше 150 СС разлагаются
на изоцианаты и ароматич. оксисоединения). П.,
содержащие эпоксидные или аллильные группы в фе-
нильных ядрах остатков бисфенолов, способны отверж-
даться с образованием продуктов, обладающих высо-
высокой гидролитич. устойчивостью и повышенными меха-
механич. показателями. П. трехмерной структуры можно
получать также из «обычных» П. действием на них
ионизирующих излучений, нагреванием на воздухе,
проведением синтеза с использованием полифункцио-
полифункциональных фенолов и др.
Переработка и применение. Из большинства арома-
ароматич. П. промышленное применение нашли гл. обр. го-
мополикарбонат и нек-рые смешанные П. на основе
бисфенола А, т. к. последний необходимой степени
чистоты получают из дешевого и доступного сырья.
Применение ароматических П. на основе других двух-
двухатомных фенолов ограничено из-за высокой стоимости
исходных дифенолов, хотя по некоторым свойствам та-
такие П. превосходят полимеры, полученные на основе
бисфенола А.
П. перерабатывают всеми широко распространенны-
распространенными методами, используемыми для переработки термо-
термопластов. Пленки из П. формуют гл. обр. из р-ров
в метиленхлориде. Оптически прозрачные пленки тол-
толщиной до 250 мкм формуют из р-ров П. на основе бис-
фенола А с мол. массой 75 000—90 000, толщиной
> 300 мкм — из р-ров смешанных П. Пленки и пла-
пластины большей толщины лучше формовать из расплава
методом экструзии через плоско-щелевую головку
(см. Органическое стекло).
Очень тонкие пленки (толщиной < 6 мкм) ие сни-
снимают с подложки до момента использования (напр.,
для намотки в конденсаторах). Тонкие пленки можно
получить формованием П. из расплава с последующим
раздувом (см. Пленки полимерные). Растворимые аро-
ароматич. П. применяют для получения лекарственных
препаратов пролонгированного действия (активные
ингредиенты покрывают тонкой пленкой П.), а также
в качестве связующего при изготовлении светонро-
нускающих композиций для электрофотографии. Плен-
Пленки служат для изоляции магнитных сердечников слож-
сложной конфигурации. Пленки из П. можно подвергать
металлизации в вакууме.
Литьем под давлением перерабатывают П. со средней
мол. массой 32 000—35 000 и содержанием влаги
не более 0,01% во избежание деструкции в процессе
переработки B40—330 СС). Для предотвращения дест-
деструкции ароматич. П. при повышенных темп-pax ис-
используют стабилизаторы [силикаты свинца пли цинка,
оловоорганпч. соединения, днарнлстаннатокспды, пол-
полные эфиры фосфористой к-ты и ароматич. оксисоеди-
нений, напр. три-B-трет-бутил-5-метилфенил)фосфит].
Для снижения содержания влаги П. сушат перед
переработкой в течение нескольких ч при 120 С в ва-
вакуум-сушилке. Необходимо также тщательно удалять
растворитель (метиленхлорид), т. к. при темн-рах
переработки он разлагается с образованием хлористого
водорода, вызывающего коррозию аппаратуры. Гра-
Гранулированный П. на основе бисфенола А можно пере-
перерабатывать на любых промышленных литьевых маши-
машинах, темн-ра нагревательного цилиндра может быть
доведена до 350 °С. Крупногабаритные изделия полу-
получают на литьевых машинах с червячной пластикацией
[давление 70—220 Мн/м2 G00—2200 кгс/см'2)\.
Изделия из П. на основе бисфенола А, полученные
литьем под давлением, применяют главным образом в
электротехнике для изготовления ограничителей,
защитных экранов для кинескопов телевизоров, плит
для печатных схем, каркасов для катушек, штепсель-
штепсельных вилок, клеммовых панелей, телефонных аппара-
аппаратов, электротехнической арматуры и др. П. применя-
применяют также для производства прецизионных деталей
инструментов и контролирующих приборов, пишу-
пишущих машинок, вычислительных машин и вентилято-
вентиляторов, деталей машин, перерабатывающих молоко, ку-
кухонной утвари, деталей холодильников, калориферов,
посуды и др.
В медицинской пром-сти П. используют для изготов-
изготовления чашек Петри, фильтров для крови, корпусов
бормашин, зубных протезов, к-рые обладают хорошей
стабильностью размеров, хорошими механич. свойст-
свойствами, легко ремонтируются. П. используют для изго-
изготовления чертежного оборудования, логарифмпч. ли-
линеек, защитных шлемов, шахтерских касок. В авто-
автомобильной промышленности из П. изготовляют про-
прозрачные крышки и колпаки сигнальных ламп, линзы
для сигнальных фонарей, распределители, защитные
решетки.
Методом экструзии при 240—300 "С из П. произво-
производят стержни, листы, профилированные изделия, трубы
и шланги. Вследствие высокой механич. прочности
и незначительной текучести из П. можно изготавливать
трубы с меньшей толщиной стенок, чем из др. поли-
полимерных материалов.
Методом экструзии с раздувом можно изготавливать
пустотелые изделия (напр., бутылки), используемые
для хранения фармацевтич. и косметич. препаратов,
пищевых продуктов.
851
ПОЛИКАРБОРАНЫ
852
П. можно перерабатывать также прессованием, т. к.
благодаря высокой темп-ре стеклования изделия из фор-
формы можно вынимать уже при 120 —130 СС. Прессова-
Прессованием получают прозрачные панели с гладкой поверх-
поверхностью. П. можно точить, сверлить, фрезеровать, пи-
пилить, резать ножницами, штамповать, шлифовать, по-
полировать и вырезать но шаблону; соединять детали
из П. друг с другом или с деталями из др. материа-
материалов гвоздями и заклейками.
Благодаря малой хладотекучести, а также хорошей
пластичности, малой упругости расплава, жесткости,
высокому модулю упругости и высокой теплостойкости
П. можно перерабатывать методами холодного формо-
формования, характерными для металлообрабатывающей
пром-сти (штамповка, прокат, клепка, вытяжка). Грам-
Грампластинки, изготовленные штампованием дисков из П.,
хорошо воспроизводят звук, стабильны при перепадах
темп-р, практически не бьются.
Изделпя из П. можно сваривать горячим воздухом
с использованием сварочного прутка из того же ма-
материала или склеивать при помощи растворителей или
агентов, вызывающих набухание П. (дихлорэтан, бен-
бензол, стирол, специальные клеи). На поверхность изде-
изделий из П. хорошо наносятся лаки, полиграфич. краски.
Для получения иен из П. обычные порообразователи
применять нельзя, т. к. они могут вызвать деструкцию
полимера в процессе вспенивания. Парообразователями
для П. служат мономерные или полимерные соединения,
содержащие группы смешанных ангидридов карбоно-
вых к-т и угольной к-ты, напр, смешанный ангидрид
изофталевой к-ты и метилового эфира угольной к-ты,
выделяющие при повышенных темп-pax двуокись угле-
углерода:
О о
с-о-с-о-сн,
С-О-С-О-СН,
II 'I
О О
с-о-сн.
+ 2 СО
—о—сн3
При нагревании смеси такого порообразователя и аро-
матич. П. выше его темп-ры размягчения происходит
вспенивание массы в результате выделения двуокиси
углерода; образующиеся при этом сложные эфиры
(или полиэфиры) совмещаются с П. По др. способу
получения пен расплавленный П. смешивают под дав-
давлением с инертным растворителем с соответствующей
теми-рой кипения или инертным газом и дают смеси
остыть, поддерживая в системе определенное давление.
Если затем смесь нагреть в открытых формах выше
темп-ры размягчения П., произойдет вспенивание
массы в результате испарения растворителя или рас-
расширения растворенного газа. Так, при смешивании
П. с 15 — 25% (концентрация по массе) циклогексана
при 230 —240 °С и давлении 2 Мн/м2 {20 кг с/см2) и по-
последующем охлаждении массы иод давлением получают
бесцветные, прозрачные куски материала, к-рые при
нагревании до 150 °С превращаются в мелкопористую
пену с плотностью 0,05—0,25 г/см*. Пены из П. харак-
характеризуются высокой термостабильностью, хорошими
механич. свойствами, низкой теплопроводностью,
устойчивостью к старению.
Многие П. и смешанные П. растворяются в раство-
растворителях, применяемых в лакокрасочной пром-сти,
поэтому их можно использовать в качестве связующих
при изготовлении лаков, обеспечивающих свето- и
погодостойкие покрытия с глянцевой поверхностью,
высокой эластичностью, отличной влагостойкостью,
малым водопоглощением и хорошими электрич. свойст-
свойствами, сохраняющимися при высоких темн-рах. Лаки
на основе П. обладают адгезией к металлич. поверхно-
поверхностям. Порошкообразные П. используют для покрытия
металлич. деталей напылением.
Ароматич. П. хорошо совмещаются со многими пла-
пластификаторами, но несовместимы с нек-рыми др. поли-
полимерными связующими, что ограничивает их применение.
Продукты полимераналогичных превращений П. нахо-
находят применение как ионообменные полимеры.
П. на основе бисфенола А производят в различных
странах иод след. названиями: д и ф л о н (СССР);
м а к р о л о н (ФРГ); лексан, мерлон (США);
пепла ii т, ю п и л о н, т о у ф л о н (Япония); б и-
с т а н (ПНР).
Общее производство П. в США, ФРГ и Японии в 1975
должно составить 100 000 т.
Лит.: Ш н е л л Г., Химия и физика поликарбонатов,
пер. с англ., М., 1967; Encyclopedia of polymer science and
technology, v. 10, N. Y. — [a. o.], 1969; Смирнова
О. В., Ерофеева СБ., Поликарбонаты, М., [в печати];
Johnson К., Polycarbonates recent developments, Chem.
Process Rev., № 47, 1970; Poliwe.glany, Warszawa, 1971.
О. В. Смирнова.
ПОЛИКАРБОРАНЫ, карборансодержа-
щ и е полимеры (polycarboranes, Polykarborane,
polycarboranes) — полимеры, содержащие в основной
или боковой цепи макромолекулы карборановые груп-
группировки. Известны карборансодержащие полисилокса-
ны, полиэфиры, полиамиды, полиуглеводороды, поли-
гидразиды, полиоксадиазолы, полиимиды, полибенз-
имидазолы, полимочевины, полиуретаны, полифор-
полиформа ли и др. Наиболее полно описаны первые три класса.
о-, д*- и п-Карбораны-12 — кристаллич. вещества
белого цвета, плавящиеся соответственно при темп-рах
ок. 320 °С, 262—263 °С и 259—261 °С. Рентгенострук-
турными исследованиями показано, что молекулы
карборанов-12 имеют структуру икосаэдра, в к-ром
все связи близки по длине (напр., для о-карборана
0,168^0,002 им) и форма существенно не искажена:
о - Карооран
п - КарЬоран
I — углерод, Q— бор, • — водород
Обычно карбораны-12 изображают графически след.
образом:
вюню
О- Карборан—12
л/—Карборан—12
/7 — Кгрборзн—12
о-Карборан получают взаимодействием декаборана
В10Н14 и ацетилена в присутствии оснований Льюиса.
Нагреванием о-карборана при 450—600 СС получают
ж-карборан, из к-рого при 630 — 700 СС образуется
д-карборан (в виде равновесной смеси с ж-карбораном).
Для синтеза П. применяют в основном производные
карборанов (таблица). При введении карборановых
группировок в макромолекулы различных классов
полимеров у последних, как правило, повышается
растворимость и возрастает термостойкость.
Карборансодержащие по лиси л океаны. Из этого клас-
класса полимеров наиболее полно исследованы поли-jt-
карбораниленсилоксаны, гл. обр. полиалкилсилоксаны
с ж-карборановыми группами в основной цепи. Полу-
Получают их в основном каталитич. поликонденсацией
б^с-(метоксидиметилсилил)-ж-карборана с бг
853
ПОЛИКАРБОРАНЫ
854
диметилсилил)-л1-карбораном, алкилхлорсиланами или
алкилхлорсилоксанами в присутствии FeGl3
CH3O(CH3JSiCB10Hl0CSi(CH3JOCH3
з СН3 СН
сн.
(CH3JSiClCB10Hl0CSi(CH3JCl
-св
или гидролитич. поликонденсацией карборансодержа-
щих силоксанов, таких, как
CISi (CH3JOSi (CH3J CB10H10CSi (CH3JOSi (СН3J Cl
и
CISi (CH3J [OSi (СН3J]2 СВ10Н10С [Si (СНзШзЗКСНзШ
Поли-л-карбораниленсилоксаны содержат от од-
одной до пяти силоксановых групп между ^ двумя
л-карборанилышми группировками. Первый член
ряда карборансодержащих полидиметилсилоксанов
[—Si(CH3JGB10H10CSi(GH3JO — ]n — кристаллич. поли-
полимер белого цвета, хорошо растворимый в органич.
растворителях; мол. масса 16 500; т. пл. 238 °С. Ос-
Остальные члены ряда представляют собой растворимые
в органич. растворителях вязкие жидкости или элас-
эластомеры с темп-рами стеклования ~ —70 СС; мол.
масса 10 000—20 000. Эти полимеры характеризуются
устойчивостью на воздухе при 300 СС. При частичном
замещении метильных групп в карборансодержащих
полидиметилсилоксанах на фенильные термостойкость
полимеров возрастает на -~ 50 °С.
Карборансодержащие полиэфиры получают поликон-
поликонденсацией в расплаве, высокотемпературной или акцеп-
торно-каталитич. поликонденсацией в р-ре.
о-Карборансодержащие полиэфиры общей ф-лы:
—ОСН2С— CCH2OCORCO—
синтезируют из бмс-(оксиметил)-о-карборана и алифа-
алифатич. или ароматич. дикарбоновых к-т или их произ-
производных. Полиэфиры, у к-рых R — алкилен, представ-
представляют собой вязкие жидкости или хрупкие твердые в-ва
светло-желтого или коричневого цвета; мол. масса до
5000; т. размягч. до 100 °С. Они аморфны, раствори-
растворимы в ароматич. углеводородах, хлоруглеводородах,аце-
хлоруглеводородах,ацетоне и нерастворимы в алифатич. углеводородах, спир-
спиртах, воде. При замене СН2-групп в макромолекулах этих
полиэфиров на CF2-rpynnbi или о-карборановой группы
на „%-карборановую темп-ры размягчения полимеров
снижаются на 10—30 °С. о-Карборансодержащие поли-
полиэфиры, у к-рых R — арилен, представляют собой
твердые вещества белого цвета; т. размягч. 90—220 °С.
Они* обладают очень упорядоченной структурой и
поэтому не растворяются даже в серной к-те. И в этом
случае при замене о-карборановой группы на ж-кар-
борановую температура размягчения полимеров по-
понижается.
ж-Карборансодержащие полиэфиры общей форму-
формулы [ —COCCB10H10CCOO — R—О—]п, где R — арилен,
синтезируют из дихлорангидрида л-карборандикарбо-
новой к-ты и бисфенолов. Это — белые порошкообраз-
порошкообразные или волокнистые вещества; мол. масса 3000—
20 000; т. размягч. 225—300 °С. Они хорошо раствори-
растворимы в тетрагидрофуране, хлорированных углеводородах,
трикрезоле и растворителях амидного типа; при нагре-
нагревании на воздухе начинают разлагаться ок. 300 С,
при 800'С образуется прочный коксовый остаток
(~ 8О°о от первоначальной массы); при кипячении
в воде медленно гидролизуются. При замене ж-карбо-
рановой группы на л-карборановую темп-ры размяг-
размягчения полиэфиров возрастают на 40—100 °С, масса
коксового остатка, образующегося при 800 °С, умень-
уменьшается на 10—20% и появляется устойчивость к депст-
вию кипящей воды.
Полиэфиры общей формулы
[—COC6H4R'CGH4COORO—]n, где R'— о-кар-
боранилен, ж-карборанилен, R — арилен,
получают из дихлорангндридов 1,2- или 1,7-бмс-
D-карбоксифенил)карборана и бисфенолов. Это волок-
Мономеры, применяемые для синтеза
полимеров
карборансодержащнх
Название
мономера
R'
R
Производные о - к а р б о р а
Винил-о-карборан
Изопропенил-о-
карборан . . .
Аллил-о-карборан
Метил-о -карбора-
ниленметил-
акрилат ....
бис-(Оксиметил)-о-
карборан . . .
бис-D-Оксифе-
нил)-о-карборан
бис-( 4 -Карбокси-
фенил)-о-карбо-
ран
Дихлорангидрид
бис-D-карбок-
сифенил)-о-кар-
борана
RC CR'
\О/
СН2=СН Н
СНО=С(СН,)
СН2=СНСН2
СН2=СНСООСН2
СН2ОН
С6Н4ОН
С6Н4СООН
СвН4СОС1
н
н
СНз
СН2ОН
С6Н4ОН
С6Н4СООН
С6Н4СОС1
Темп-ра
плавления, °С
A мм рт-ст.—
= 133, 322
и'м2)
на
78-79
46,7-47,7
63-65
135*@,5 мм
рт. ст.)
303-304
190-191
370-371
178-179
Производные д-карборана
R Cj Jj iq H j qLj Ja
1,7-Метилвинил-
м-карборан . .
1,7 -Метила ллил-
„ч-карборан . .
Изопропенил-лг-
карборан . .
бис-(Оксиметил)-
лг-карборан
лг-Карборанди-
карбоновая к-та
Дихлорангидрид
м-карборанди-
карбоновой к-ты
бис-D-Карбонси-
фенил)-л1-карбо-
ран
Дихлорангидрид
бис-D-карбок-
сифенил)-л1-
карборана . . .
Дигидразид бис-
D-карбоксифе-
нил)-л1-нарбора-
на
СН2=СН
СН2=СНСН2
СН2=С(СН3)
СН2ОН
соон
сосл
С6Н4СООН
С3Н4СОС1
CeH4CONHNH2
СН3
СН3
н
СН2ОН
соон
СОС1
С6Н4СООН
С6Н4СОС1
C6H4CONHNH,
75-76
142—143*
A3 мм рт.
ст.)
46-47
194-196
202—204
86* @,5 мм
рт. ст.)
301-302
160-161
243-245
Производные п-карборана
п-Карборанди-
карбоноваяк-та
Дихлорангидрид
п-карборанди-
карбоновой к-ты
СООН
СОС1
соон
СОС1
173-174
102 , 5 — 10 4 , 5
* Темп-pa кипения в °С.
нистые или порошкообразные вещества белого цвета;
мол. масса 20 000—120 000; т. размягч. 200—360 °С.
Они хорошо растворимы в большинстве органич. раст-
855
ПОЛИКОНДЕНСАЦИЯ
856
ворителей и способны образовывать из р-ров бесцвет-
бесцветные прозрачные пленки с прочностью при растяжении
50—110 Мн/м2 E00—1100 кгс/см2) и относительным
удлинением 4—23% . При нагревании на воздухе такие
полиэфиры начинают разлагаться выше 400 °С, при
900 С образуется коксовый остаток E0—90% от пер-
первоначальной массы). Эти полимеры можно перерабаты-
перерабатывать в изделия прессованием.
Карборансодержащие полиамиды получают необра-
необратимой низкотемпературной поликонденсацией в р-ре
(в тетрагидрофуране или апротонных диполярных
растворителях).
Полиамиды общейф-лы[— COCCB10H10CCOi4HRNH—]n,
где R — арнлен, получают из дихлорангидрида ле-кар-
борандикарбоновой к-ты и ароматич. диаминов. Это
волокнистые аморфные вещества белого цвета; мол.
масса 7000—50 000. Они хорошо растворимы в тет-
тетрагидрофуране и растворителях амидиого типа, обра-
образуют прозрачные пленки, характеризующиеся проч-
прочностью при растяжении 50—90 Мн/м2 E00—900 кгс/см2)
и хорошей адгезией к стеклу и металлам, устойчивостью
к гидролизу при 100 СС. Эти полиамиды не размяг-
размягчаются, не изменяются при нагревании на воздухе
до 200 — 250 °С, почти не теряют в массе при нагрева-
нагревании в инертной атмосфере до 1000 °С, т. к. образуется
прочное термостойкое вещество. При замене м-карбо-
рановой группы на /г-карборановую растворимость
:)тих полимеров ухудшается и повышается устойчи-
устойчивость к термоокислительной деструкции. /г-Карборан-
содержащие полиамиды не изменяются при нагревании
на воздухе до 350—400 °С.
Полиамиды общей формулы
[_COC6H4R'C6H4CONHRNH —]п, где R' — о-карбо-
ранилен или ж-карборанилен, R — арилен, синтези-
синтезируют из дихлорангидридов 1,2- или 1,7-бггс-D-карбо-
ксифенил)карборана и ароматич. диаминов. Они пред-
представляют собой волокнистые в-ва белого цвета; мол.
масса 70 000—80 000; т. размягч. 280—390 °С; хорошо
растворимы в ряде органич. растворителей и образу-
образуют из них прочные пленки (прочность при растяже-
растяжении до 120 Мн/м2, или до 1200 кгс/см2, относительное
удлинение до 30%). Такие полиамиды можно перера-
перерабатывать прессованием.
Карборансодержащие полиамиды с большим выхо-
выходом образуют коксовый остаток при нагревании до 800—
900 °С; основным компонентом газообразных продуктов
деструкции является водород.
Применение поликарборанов. Из П. практич. при-
применение пока нашли только поли-ж-карбораниленси-
локсаны, выпускаемые в США иод названием дек-
сил. Метилзамещенные поли-л^-карбораниленсилок-
саны используют в качестве подвижной фазы при хро-
матографич. разделении таких высококипящих соеди-
соединений, как углеводороды, силиконовые масла, поли-
фениленовые :)фиры, триглицериды.
Имеются сведения, что карборансодержащие поли-
полимеры можно применять для изготовления ракетного
топлива, в качестве абляционностойких материалов
и связующих для армированных пластиков.
Лит.: Grimes S., Carboranes, N. Y.— L., 1970; Про-
Прогресс полимерной химии 1968, М., 1969, с. 321; S с h г о е-
d е г Н., Inorg. Macromol. Chem. Revs, 1, 45 A970); H e-
y inp; Т., в кн.: Progress in boron chemistry, v. 2, Oxf., 1970,
p. 119; Williams R. E., Pure Appl. Chem., 29, № 4,
p. 569 A972). H. И. Бекасова, П. М. Валецкий.
ПОЛИКОНДЕНСАЦИЯ (polycondensation, Polykon-
densation, polycondensation).
Содержание:
Классификация и терминология 856
Мономеры 857
Основные стадии и сопутствующие процессы 859
Кинетика 861
Зависимость молекулярной массы от некоторых
факторов 863
Заключение 865
Классификация и терминология
П. обычно определяют как процесс образования
полимеров из би- или полифункциональных соедине-
соединений, сопровождающийся выделением побочного низ-
низкомолекулярного вещества (воды, спирта, галогено-
водорода и др.). Типичный пример П.— синтез слож-
сложного полиэфира из гликоля и дикарбоновой к-ты:
п HOROH + п HOOCR'COOH—>[-- OROOCR'CO-]^ -f 2n II2O
Процесс наз. г о м о п о л и к о н д е н с а ц и с й,
если в нем участвует минимально возможное для дан-
данного случая число типов мономеров. Чаще всего ото
число равно 2, как в приведенном выше примере,
однако может быть и единицей, напр. при синтезе
полиамида из а-аминокислоты:
nH2NRCOOH ^ [-HNRCO-]n + ?*Н2О
Если в П., помимо мономеров, необходимых для
данной реакции, участвует по крайней мере еще один
мономер, процесс наз. со пол и конденсацией
(см. также Сополикондеисация). По такой реакции
образуются, например, т. наз. смешанные полиамиды
из двух или более диаминов и дикарбоновой к-ты:
2nHOOCRCOOH -f nH,NR'NH2 -f- ?iH2NR"NH_, i-
±5 [-HNR'NHCORCONHR"NHCORCO--]n -f 4?iH2O
П., в к-рой участвуют только бифункциональные
молекулы, приводит к образованию линейных макро-
макромолекул. Такой процесс наз. линейной поли-
поликонденсацией. Если же в П. участвуют моле-
молекулы с тремя или большим числом функциональных
групп, образуются трехмерные структуры; соответст-
соответствующий процесс наз. трехмерной п о л и к о н-
д е н с а ц и е й. Двухступенчатая реакция, в к-рой
образовавшийся на первой стадии продукт линейной
П. подвергается внутримолекулярной циклизации,
наз. п о л и ц и к л о к о н д е н с а ц и е й. Подоб-
Подобный процесс осуществляется, напр., при синтезе иоли-
имидов (см. также Полициклоконденсация).
В соответствии с термодинамич. характеристиками
реакции различают обратимую (равновес-
(равновесную) и необратимую (неравновес-
(неравновесную) поликонденсацию (об этой терми-
терминологии см. стр. 859).
П. осуществляют как в гомогенных жидких системах
(см. Поликоидеисация в расплаве, Поликонденсация
в растворе), так и в гетерогенных (см. Межфазная
поликонденсация). Известен также способ проведения
П. в твердой фазе (см. Твердофазная поликонденсация).
В самом общем виде схему П. можно записать след.
образом:
Mt + Mj^Mt + i A)
где М^ Mj и Mi + j — соответственно г-, /- и (/ +/)-
меры, причем i и / могут быть любым числом, в том
числе и единицей. Из этой схемы следует, что при П.
реагирующие группы разных молекул мономеров вна-
вначале взаимодействуют друг с другом и получаются
димеры; последние при взаимодействии с мономером
или димером образуют соответственно тримеры или
тетрамеры и т. д. Так, по мерс протекания процесса,
образуются все более длинные цени. В отличие от это-
этого, при полимеризации к активному концу г-мера
присоединяются только молекулы мономера, т. е. про-
процесс протекает по схеме:
Л/. + М-> Mi+1 B)
Многие из реакций, идущих по схеме A), сопровож-
сопровождаются выделением низкомолекулярных продуктов,
на чем и основано приведенное выше традиционное
определение П.
Вместе с тем в последние годы все большее значение
приобретают процессы, к-рые не сопровождаются
выделением низкомолекулярных соединений, но идут
857
ПОЛИКОНДЕНСАЦИЯ
858
по схеме A). Таким образом осуществляется, напр.,
синтез полиуретанов из гликолей и диизоцианатов или
образование полиамидов из диаминов и недоокиси
углерода по реакции
и H2NRNH, л. п О=С=С=--С=О —> [—HNRNHCOGH2CO-]n
С др. стороны, открыт и изучен ряд реакций, идущих
но схеме B), но с выделением низкомолекулярных
веществ (см., напр., ТЯ-Карбоксиаигидридов а-амино-
кислот полимеризация, Диазосоединений полимериза-
полимеризация). Все :>то обусловливает целесообразность дать
новое определение П. (как и полимеризации), основан-
основанное не на внешних проявлениях процесса, а на специ-
специфике роста цепи. Такое определение можно сформу-
сформулировать следующим образом: и о л и к о н д е н-
с а ц и я — процесс синтеза полиме-
полимеров из б и - или пол и функционал ь-
н ы х соединений, в к - р о м рост мак-
макромолекул происходит путем х и-
м и ч. взаимодействия молекул м о-
номеров друг с другом и с к - м е р а-
м и, накапливающимися в ходе
реакции, а также молекул п - м еров
м е ж д у с о б о и.
Мономеры
Мономерами при П. могут быть любые соединения,
содержащие в молекулах не менее двух функциональ-
функциональных групп. Как уже отмечалось, П. бифункциональ-
бифункциональных соединений приводит к образованию линейных
полимеров, П. иолифункциональных — разветвленных
и сетчатых полимеров. При всем их многообразии
бифункциональные мономеры можно разделить на 3 ос-
основные класса.
1. Мономеры, содержащие одинаковые функциональ-
функциональные группы, не способные в условиях данной реакции
взаимодействовать между собой. К ним относятся,
напр., диамины, дикарбоновые к-ты и их производные
и др. Полимер в этом случае образуется путем взаимо-
взаимодействия друг с другом молекул различных мономеров
этого типа, содержащих соответственно различные
и способные взаимодействовать друг с другом функ-
функционал ьные группы. Примером может служить П.
диаминов с дихлорангидридами:
п H2NR'NH2 + n G1COR"GOC1 ->
-> [HNR'NHGOR"GO-]n + 2n HG1
Подобные процессы часто наз. гетерофунк-
ц и о н а л ь н о й П., или гетерополикон-
д е н с а ц и е й.
2. Мономеры, содержащие различные функциональ-
функциональные группы, способные взаимодействовать друг с дру-
другом. К мономерам этого типа относятся оксикислоты,
аминокислоты и др. Образование полимера происходит
нутем взаимодействия различных функциональных
групп одного и того же мономера, напр.:
?/H2NRCOOH ±5 [- RGONH—]n + пН2О
3. Мономеры, содержащие одинаковые и способные
в условиях реакции взаимодействовать друг с другом
функциональные группы, напр, гликоли. Примером
П. таких мономеров может служить образование про-
простых полиэфиров:
п HOROH *± [-OR-]n + ™Н2О
Изменением условий П. можно изменить характер
функциональности мономеров. Так, м. б. подобраны ус-
условия, когда гликоли будут реагировать лишь как мо-
мономеры класса 1. Так. обр., при отнесении мономеров
к какому-либо классу следует учитывать конкретные
условия П.
Хпмнч. природа реагирующих групп поликонден-
сациоиных мономеров м. б. весьма разнообразной.
К числу наиболее распространенных относятся моно-
мономеры со стабильными валентнонасыщенными функцио-
функциональными группами, такими как амино-, окси-, кар-
бокси-, карбонильная группа и др. (табл. 1).
Таблица 1. Функциональные группы поли конденсационных
мономеров, участвующие в синтезе полимеров различных классов
Функциональные
группы
Примеры мономерных пар
Простые полиэфиры
-ОН
-он
—он
-он
-OR
-ОН
—ОМе
-С1
-ОН
-NH2
—NH2
-NH,
-nh;
-G=N
-NH,
—OH'
-NH,
-OH
—Cl
I Гликоль + гликоль
[ Гликоль -j- дихлоралкан
Сложные полиэфиры
-COOH
-COOR
-COOH
-COC1
—COC1
OCOMe
-C=C=O
—COOH
-COC1
-COOR
-C=C=O
-OH
—OCOC1
Гликоль -}- дикарбоновая к-та
Гликоль -f- диэфир дикарбоновой к-ты
Эфир гликоля + дикарботювая к-та
Гликоль -J- дихлорангидрид дикарбо-
дикарбоновой к-ты
Алкоголят -f- дихлорангидрид дикар-
дикарбоновой к-ты
Дихлоралкан + соль дикарбоновой
к-ты
Гликоль -J- недокись углерода
Полиамиды
Диамин + дикарбоиовая к-та
Диамин + дихлорангидрид дикароо-
новой к-ты
Диамин -f- диэфир дикарбоповой к-ты
Диамин -{- недокись углерода
Динитрилы -г диолы (третичные)
Диамин -f бис-хлорформиат
Гликоль + диизоцианат
Диамин + бициклич. карбонат
о
\
с
-СН-СН,
I I
О
/
с
II
о
Полиацетали
—ОН | — СН=О | Гликоль + диальдегид
Полишиффовые основания
—NH2 I —СН=О | Диамин + диальдегид
П о л и а н г и д р и д ы
I — ООСОСНд I Смешанные диангидриды дикарбоно-
—OOCOLH3| | вои и уксусной к-т
Координационные полимеры
о о
II II
—С—СН,—С—СН3
сн3
-С
/
—Me
сн
Тетракетон -\- ацетилаце-
тонат металла
-С1
-сн,
|-NH2
-NH,
-NH2
f-NH2
i-OH
—NHNH2
—SiOH
—SiOR
СН3
Пол и углевод о роды
—С1 гг-Дихлорбензол
—СН3 ?1-Ксилол и подобные соединения
—СН2ОН Ф^нол + метилольные производные
фенола
П о л и б е н з и м и д а з о л ы
—COOR I Тетраамины + диэфиры дикарбоновых
| к-т
Полиамиды
(— СООН I Диамин + производные тетракарбо-
(—ОСОСНз I новых к-т (напр., диангидриды)
П о л и б е н з о к с а з о л ы
—СОС1 I бис-о-Аминофенолы + дихлорангид-
I риды дикарбоновых к-т
Полипиразол ы
—COCH2COR | Дигидразид -f- тетракетоп
П о л и с и л о к с а н ы
—SiOH
—SiOOCCH3
Гидроксисилан
R-Оксисиланы -j- ацетилсиликаты
859
ПОЛИКОНДЕНСАЦИЯ
860
Из др. типов реагирующих групп поликонденса-
поликонденсационных мономеров следует отметить группы радикаль-
радикальной природы. Они очень реакционноспособны, что
определяет особенности П. с их участием, напр, поли-
полирекомбинации, нек-рых случаев дегидрополиконденса-
ции, поликонденсации по Вюрцу — Фиттигу.
Основные стадии и сопутствующие процессы
Рост макромолекул при П. может осуществляться
путем самых разнообразных химич. реакций. Необхо-
Необходимо лишь, чтобы в результате взаимодействия функ-
функциональных групп образовывалась химич. связь между
несущими их фрагментами.
Константы иоликонденсационного равновесия К для
П., протекающей без выделения и с выделением побоч-
побочных продуктов, записывается соответственно след.
образом:
(безРа3меРная)
К = [А] [В]
где [А] и [В] — равновесные концентрации функцио-
функциональных групп, не вступивших в реакцию, [X] — рав-
равновесная концентрация образовавшихся в результате
П. связей, [Z] — концентрация побочного продукта
в реакционной системе.
Если в условиях процесса степень завершенности
П. и средняя длина макромолекул лимитируются рав-
равновесными концентрациями реагентов и продуктов
реакции, то П. называют равновесной. Обычно
это характерно для процессов с К ~ 10—102. Если
К ^ 103, то степень завершенности П. и средняя мол.
масса полимера лимитируются обычно не термодинами-
термодинамическими, а кинетич. факторами. Такую П. наз. н е-
равновесной. Поскольку в данном случае
смысл, вкладываемый в указанные термины, отличается
от принятого в классич. химич. термодинамике, а ве-
величина К характеризует обратимость процесса, рав-
равновесную П. удобнее называть обратимой, а не-
неравновесную — необратимой.
Значения констант равновесия для нек-рых практи-
практически важных случаев П. приведены в табл. 2.
Таблица 2. Значение констант равновесия для некоторых
случаев поликонденсации
Тип поликонденсации
Н е о б р а т
ГГолиотерификация
Полиамидирование
То же
Мономеры
имые реакции
Фенол 4- бензоилхлорид (мо-
(модельная реакция)
Тетраамины -f диангидриды
ароматич. тетракарбоно-
вых к-т
Диамины + дихлорангидри-
ды ароматич. дикарбоно-
вых к-т
Константа
равнове-
равновесия К
4260
Ю15—1 О25
>1015
Обратимые реакции
Полиэтерификация
Полиамидирование
Пол икоорд и нация
Пеытаметиленгликоль -f ади-
пиновая к-та
о)-Аминоундекановая к-та
Ацетилацетонат бериллия и
4,4 '-бис-(ацетоацетилфе-
нил)оксид
6,0
8,9
4,0
Особенность роста полимерной цепи обратимой П.—
наличие процессов, обратных росту, т. е. реакций
полимера с низкомолекулярным выделяющимся про-
продуктом. При необратимой П. этого не происходит,
а наблюдающееся часто на практике уменьшение мол.
массы полимера в присутствии низкомолекулярного
продукта происходит при этом по иному пути, наир.
в результате реакции с мономерами.
Сопутствующие П. процессы, в результате к-рых
реагирующие группы хмономеров, олигомеров и поли-
полимерных цепей расходуются не по основному пути и
утрачивают способность участвовать в реакции роста,
можно разделить на химические и физические. К пер-
первым из них относятся:
а. Взаимодействие функциональной группы бн- или
полифункционального мономера или олигомера с моно-
монофункциональным .соединением, аналогичным второму
мономеру, присутствующим в качестве нрпмссп или
специально введенной добавки, напр.:
^CICORCONHR" + HG1
ClOCR'COCl + H2NR"<
Nr"NHCOR'CONHR" + 2HG1
(Здесь и далее R — любой радикал, в том числе фраг-
фрагмент полимерной цепи).
б. Химич. превращение функциональных групп
вследствие протекания побочных реакций, иногда
с участием активных посторонних примесей, наир.
декарбоксилирование концевых карбоксильных групп
при повышенных темп-pax или гидролиз хлорангидрид-
ных групп:
HOOC-R-COOH
R-C0C1 + HoO -> R-COOH 4- HC1
Физич. Ихммобилизация реагирующих групп моно-
мономеров и олигомеров чаще всего происходит при выпа-
выпадении (самопроизвольном выделении) полимера из реак-
реакционной среды в ходе синтеза, особенно если это сопро-
сопровождается кристаллизацией полимера, а также при
резком повышении вязкости системы и образовании
полимера сетчатой структуры.
В реальных условиях П. часто осложняется одновре-
одновременно протекающими побочными реакциями циклиза-
циклизации, в к-рые могут вступать как исходные мономеры,
так и конечный продукт (полимер). Из такого рода
процессов следует отметить внутримолекулярную цик-
циклизацию, когда друг с другом взаимодействуют реак-
ционноспособные группы одной молекулы, напр.:
СН2-СНг
HNX \
NH2(CH2 M СООН -» ! /СН2
"~ осч /
ХртТ ПТТ /
Li±l2 — L-xl2
Ниже приведен пример межмолекулярной циклизации:
СН2-СН2
НОСН2СН2ОН + С2Н8СОО-СООС2НБ «± О
О + 2С2Н5ОН
ЧС-С '
II II
о о
Особой склонностью к внутримолекулярной цикли-
циклизации отличаются соединения или пары мономеров,
дающие 5 —7-членные продукты циклизации. Однако
при П. могут образоваться и циклы из 20—40 атомов.
Так, в продуктах синтеза полиэтилентерефталата вы-
выделено соединение след. строения:
СОО — СН2СН2ООС— / \—
СОО —СН2СН2ООС
-о-
соо
сн
сн2
соо
ВнутрИхМолекулярную циклизацию можно в значи-
значительной степени предотвратить увеличением концент-
концентрации мономеров и уменьшением темп-ры процесса
(см. также Поликонденсация в растворе).
861
ПОЛИКОНДЕНСАЦИЯ
862
Другими распространенными побочными реакциями
при П., особенно проводимой при высоких темн-рах,
являются деструктивные процессы (напр., обменные
реакции), приводящие к разрыву макромолекул обра-
образовавшегося полимера.
Конкуренция (по скоростям) стадий роста и побоч-
побочных реакций определяет выход, мол. массу и молеку-
лярно-массовое распределение поликонденсационного
полимера.
Во многих случаях П. стадии роста цепи предшест-
предшествует образование в молекулах мономера реакционно-
способных групп. Так, при взаимодействии с формаль-
формальдегидом вначале образуются метилолпроизводные фе-
фенола:
он он он
сн2о
сн2он
сн2о
СН2ОН
к-рые, собственно, и участвуют в П.:
ОН
СНоОН
,СН2ОН
СН2ОН
4- Н2О
и т.д.
Стадия образования функциональных групп оказы-
оказывает существенное влияние на кинетику и закономер-
закономерности П. только в том случае, если ее скорость меньше,
чем скорость роста цепи. Это наблюдается тогда,
когда в стадии роста участвуют очень активные функ-
функциональные группы, напр, радикальной природы.
Кинетика
Для П. характерно исчезновение мономера уже
на сравнительно ранних стадиях процесса, когда мол.
масса полимера очень мала. Дальнейший рост цепи
происходит при практически полном отсутствии моно-
мономера в системе (при полимеризации на всех стадиях
наряду с высокомолекулярным полимером присутствует
мономер). При количественном анализе кинетики П.
обычно используют принцип, согласно к-рому актив-
активность реагирующих групп не зависит от длины присое-
присоединенной к ним части макромолекулы (принцип Фло-
ри). Это позволяет значительно сократить количество
одновременно решаемых кинетич. ур-ннй (иногда
до одного). Известен, однако, ряд случаев, когда
принцип Флори не соблюдается, напр, для П., приво-
приводящей к получению полимеров с сопряженными свя-
связями:
п H2N
— NH2 4-
Н
\
С
н
[ = СН -<
,_CH=N-/~\-N=]/7
2пН2О
При рассмотрении кинетики П. обычно пользуются
понятием «глубина превращения» (х), т. е. доля про-
прореагировавших к данному моменту времени функцио-
функциональных групп. Отметим, что при полимеризации иод
глубиной превращения понимают долю прореагировав-
прореагировавшего мономера. При П. доля прореагировавшего моно-
мономера х меньше х.
Рассмотрим кинетич. ур-ния для П. мономеров, взя-
взятых в эквимолярных количествах. Ур-ние для измене-
изменения концентрации функциональных групп запишется:
dIB1=M A, fB] = *[AP
dt
dt
где к — константа скорости реакции.
После интегрирования и введения начальной концент-
концентрации функциональных групп [Ао] и глубины превра-
превращения х получим:
Это ур-нне справедливо для необратимых процессов
и для начальных стадий обратимых процессов, когда
можно пренебречь обратной реакцией. Для ряда про-
простейших случаев это ур-ние м. б. преобразовано:
где п — среднечнсленная степень полимеризации (поли-
конденсацип), т. е. среднее число звеньев в макромоле-
макромолекуле. Подход к выводу кинетич. ур-ний др. случаев П.
аналогичен изложенному.
Принцип Флори позволяет также изучать нек-рые
особенности П. (напр., подбирать эффективные ката-
катализаторы и растворители для проведения процессов,
грубо оценивать реакционную способность разных
классов мономеров) на реакциях модельных монофунк-
монофункциональных соединений. Это значительно упрощает
исследование кинетики П. В тех случаях, когда прин-
принцип Флори не выполняется, П. описывается более
сложными кинетич. ур-ниями, для решения к-рых
целесообразно использовать электронные вычисли-
вычислительные машины.
Кинетические и др. характеристики обратимой и
необратимой П. сильно различаются. Обратимые про-
процессы характеризуются малыми скоростями [констан-
[константа скорости к равна 10~3—10~5 л/(моль-сек)], срав-
сравнительно большими значениями энергии активации
[83,8—167,6 кдж/молъ B0—40 ккал/молъ)]; они могут
быть как экзо-, так и эндотермическими.
Для большинства случаев необратимой П. характер-
характерны высокие скорости [к до 105 л/(молъ-сек)], низкие
значения энергии активации [8,38—41,9 кдж/молъ
B—10 ккал/молъ)]; эти процессы, как правило, сильно
экзотермичны.
Для обратимой П. предельная в данных условиях
мол. масса определяется термодинамич. факторами,
а именно значением константы поликонденсацнонного
равновесия. Так, для гомополиконденсации характерно
след. соотношение:
/
к
[Z]
Из этого соотношения следует, что для получения
высокомолекулярных продуктов процесс следует про-
проводить в условиях, когда в системе не накапливает-
накапливается низкомолекулярный продукт, т. е. в вакууме, при
высоких температурах, в тонком слое реагирующих
веществ.
Для необратимой П. степень полимеризации практи-
практически никогда не достигает значения, определяемого
константой поликонденсационного равновесия. Для
таких процессов мол. масса образующегося полимера
определяется конкуренцией реакций роста с парал-
параллельно протекающими процессами, приводящими
к дезактивации реагирующих групп. Т. обр., в этом
случае мол. масса определяется не термодинамиче-
термодинамическими, а кцнетич. факторами. Возможны и промежу-
промежуточные случаи, когда влияние термодинамических
и кинетич. факторов на предельную мол. массу поли-
полимера соизмеримо.
Молекулярно-массовое распределение полимера
в большинстве простейших случаев описывается
ур-нием:
где Рп — доля гс-меров, х — глубина превращения. См.
также Молекулярно-массовое распределение.
В гетерогенных системах кинетика П. осложняется
рядом явлении, связанных с наличием границы раздела
863
ПОЛИКОНДЕНСАЦИЯ
между фазами. Это — распределение реагентов между
фазами, поверхностная активность реагентов (адсорби-
руомость), различные процессы массоиереноса (напр.,
диффузия мономеров через слой образовавшегося поли-
полимера) н др. Лимитирующей стадией П. в таких систе-
системах м. б. не только химич. взаимодействие функцио-
функциональных групп мономеров (как в гомогенных систе-
системах), но и различные процессы переноса реагентов
и продуктов.
В зависимости от того, какая стадия является лими-
лимитирующей, реакционная зона может находиться в раз-
различных частях гетерогенной системы (табл. 3). При
Таблица 3. Некоторые характеристики поликоиденсацни
в гетерогенных двухфазных системах
Область
(режим)
протека-
протекания
Внутрен-
Внутренняя кине-
кинетическая
Внутрен-
Внутренняя диф-
диффузионная
Внешняя
кинетиче-
кинетическая
Внешняя
диффузи-
диффузионная!
Лимитирующая
стадия
Основные фак-
факторы, определя-
определяющие стадию
роста
Хим. взаимо-
взаимодействие моно-
мономеров и олиго-
олигомеров
Диффузия мо-
мономеров внутри
реакционной зо-
зоны через слой
полимера
Химич. взаи-
взаимодействие мо-
мономеров на по-
поверхности раз-
раздела
Диффузия мо-
мономеров из объ-
объема нереакцион-
нереакционной фазы к по-
поверхности раз-
раздела
Реакционная
способность мо-
мономеров; коэфф.
распределения
мономеров меж-
между фазами
Коэфф. диф-
диффузии мономе-
мономеров через слой
продукта; коэф-
коэффициент распре-
распределения мономе-
мономеров между фа-
фазами; величина
поверхности
раздела
Реакционная
способность мо-
мономеров; по-
поверхностная ак-
активность моно-
мономеров
Коэффициент
переноса моно-
мономеров; величина
поверхности
раздела
Местонахож-
Местонахождение реакци-
реакционной зоны
Полный объем
реакционной
фазы
Часть объема
реакционной
фазы
Полная* по-
поверхность раз-
раздела (концент-
(концентрация мономе-
мономеров на ней рав-
равновесна)
Часть поверх-
поверхности раздела
(концентрация
мономеров на
ней неравновес-
неравновесна)
рассмотрении кинетики П. в таких системах следует
учитывать, что действующие концентрации мономеров
(концентрации в реакционной зоне) отличаются от ис-
исходных объемных концентраций. Именно поэтому наи-
наиболее характерные зависимости мол. массы от нек-рых
факторов, наблюдаемые при П. в гетерогенных систе-
системах, оказываются иными, чем нри П. в гомогенных
системах.
Зависимость молекулярной массы от некоторых
факторов
Для П. характерна, как уже отмечалось, след. зави-
зависимость мол. массы образующегося полимера от г л у-
б и н ы проведения процесса: исчезно-
исчезновение мономера на ранних стадиях, а затем увеличение
мол. массы нри небольшом изменении глубины процесса
в области более чем 95%-ного превращения. Прибли-
Приближенно эта зависимость выражается ур-нием, кото-
которое достаточно хорошо описывает экспериментальные
данные:
п^ 1/A—я).
Из этого ур-ния следует, что высокомолекулярный
продукт м. б. получен лишь нри значительной глубине
процесса. Так, полимер с д = 200 образуется нри завер-
завершенности процесса на 99,5%. В ряде особых случаев
(когда, напр., реакционная способность групп моно-
мономеров и олигомеров меняется) приведенное ур-ние
не будет справедливо, однако качественный характер
зависимости мол. массы (резкое увеличение п при х-Л)
сохранится.
П. в гетерогенных системах может привести к обра-
образованию высокомолекулярного полимера при глубине
процесса, много меньшей 100% (в расчете на общее
исходное количество реагентов). Это связано с влия-
влиянием на П. процессов переноса мономеров в таких
системах.
Необходимым условием образования высокомолеку-
высокомолекулярных полимеров нри линейной П. является эквива-
эквивалентность реагирующих между собой исходных соеди-
соединений (правило эквивалентности функциональных
групп). Может быть несколько причин, приводящих
к нарушению этого правила: в реакцию вводят неэкви-
неэквивалентные количества исходных веществ; в реакционной
смеси присутствуют монофункциональные соединения,
к-рые взаимодействуют с исходными веществами; функ-
функциональные группы мономеров претерпевают химич.
изменение, превращаясь в группы, нереакционноспособ-
ные в условиях П.
В наиболее распространенном случае П. (реагируют
два различных мономера класса 1) мол. масса поли-
полимеров существенно зависит от м о л я р н о г о соот-
соотношения взятых мономеров. При избыт-
избытке одного из исходных веществ, т. е. при неэквивалент-
неэквивалентности количеств мономеров, П. протекает до тех пор,
пока мономер, присутствующий в меньшем количестве,
полностью вступит в реакцию. В этот момент все обра-
образовавшиеся макромолекулы будут иметь на концах
цепи одинаковые функциональные группы (такие же,
как у избыточного исходного соединения), что приве-
приведет к остановке процесса П.
Мол. массу полимера, образующегося в указанных
условиях при х — > 1, находят из ур-ния:
где q — относительное количество мономера, взятого
в избытке (q > 1).
Для обеспечения эквивалентности между двумя моно-
мономерами класса 1 иногда используют специальные прие-
приемы. Так, нри получении полиамидов вместо индиви-
индивидуальных соединений применяют стехиометрич. соль
диамина и дикарбоновой к-ты.
Для необратимой П. эквивалентность исходных ве-
веществ в ряде случаев достигается след. способом:
в реакционный объем, где находится один из мономе-
мономеров, медленно вводят другой мономер. Если скорость
дозирования этого исходного соединения значительно
меньше той скорости, с к-рой он вступает в реакцию,
все его функциональные группы прореагируют, прежде
чем будет введена новая порция. В какой-то момент
времени количество мономера, постепенно вводимого
в реакционную систему, станет равным количеству
первого мономера, т. е. будет соблюдено правило
эквивалентности функциональных групп. Дальнейшее
поступление мономера, взятого в избытке, не влияет на
молекулярную массу полимера, т. к. все функциональ-
функциональные группы первого мономера уже вступили в ре-
реакцию.
При П. в гетерогенных системах можно получить
полимер с большой мол. массой и при неэквивалентном
количестве исходных соединений, введенных в реак-
реакционный объем. Однако это лишь кажущееся наруше-
нарушение правила эквивалентности функциональных групп,
т. к. в зоне реакции эквимолярное соотношение моно-
мономеров будет соблюдаться.
Общей для поликонденсационных процессов явля-
является резкая зависимость средней мол. массы полимера
от количества в системе монофунк-
монофункционального соединения аналогичной
химич. природы: мол. масса резко уменьшается при
увеличении содержания такого соединения. Для обра-
865
ПОЛИКОНДЕНСАЦИЯ В РАСТВОРЕ
866
тимой П. эта зависимость выражается приближенным
соотношением:
п^1/р
где р — количество монофункционального соединения
(мольные доли). Монофункциональные соединения мо-
могут специально вводиться в реакцию для регулиро-
регулирования мол. массы образующегося полимера, попадать
в сферу реакции в виде примеси к основным реагентам
и растворителю или образовываться в ходе П.
вследствие понижения функциональности исходных
веществ. Чем больше монофункциональных соединений
содержится в реакционной смеси, тем на более ранней
стадии П. прекращается рост мол. массы полимера.
Т. обр., из рассмотренной зависимости мол. массы
полимера от количества монофункционального соедине-
соединения следует, что для получения высокомолекулярных
продуктов необходимо применять высокочистые ве-
вещества. Если же требуется ограничить мол. массу
полимера, вводят точно дозируемое количество моно-
монофункционального соединения, называемого в этом
случае регулятором мол. массы.
Заключение
Процессы П. играют важйую роль в природе и тех-
технике. П. или подобные ей процессы лежат в основе
биосинтеза наиболее важных биополимеров — белков,
нуклеиновых кислот, целлюлозы и др.
Поликонденсацией в 1909 был получен первый про-
промышленный синтетич. олигомер — феноло-формальде-
гидная смола. Теперь П. широко используется в про-
промышленности для получения полиэфиров (полиэтилен-
терефталата, поликарбонатов, алкидных смол), поли-
полиамидов, нек-рых кремнийорганич. полимеров, многих
термореактивных смол на основе формальдегида (мо-
чевино-формальдегйдных, феноло-формальдегидных
и др.)- В 1965—70 П. приобрела большое значение
в связи с организацией промышленного производства
ряда новых, в том числе термостойких, полимеров
(полиарилатов, ароматич. полиамидов, полипиромел-
литимидов, полифениленоксидов, полисульфонов и др.).
Лит,: Соколов Л. Б., Поликонденсационный метод
синтеза полимеров, М., 1966; Коршак В. В., Виногра-
Виноградова С. В., Равновесная поликонденсация, М., 1968;
Step-growth polymerization, ed., D. H. Solomon, N. Y., 1972;
Морган П. У., Поликонденсационные процессы синтеза^
полимеров, пер. с англ., М., 1970; Коршак В. В., Вино-
гр адова С. В., Неравновесная поликонденсация, М., 1972;
Оудиан Дж., Основы химии полимеров, пер. с англ., М.,
1974. Л. Б. Соколов.
ПОЛИКОНДЕНСАЦИЯ В РАСПЛАВЕ (melt poly
condensation, Schmelzpolykondensation, polycondensa-
tion en fusion) — способ проведения поликонденсации
в отсутствие растворителя или разбавителя; образую-
образующийся в этом процессе полимер находится в расплав-
расплавленном состоянии. Для осуществления П. в р. смесь
мономеров или олигомеров длительно нагревают при
темп-ре, на 10—20 °С превышающей темп-ру плавления
(размягчения) образующегося полимера, т. е. обычно
при 200—400 °С. Во избежание окисления мономеров
и термоокислительной деструкции полимера процесс
вначале проводят в токе инертного газа (часто осу-
осушенного), а затем, для удаления побочных продуктов
реакции, под вакуумом.
П. в р. обладает рядом преимуществ по сравнению
с другими способами проведения поликонденсации:
1) обеспечивает возможность применения мономеров
с пониженной реакционной способностью (по сравнению
с мономерами, участвующими в поликонденсации
в р-ре или на границе раздела фаз); 2) отличается срав-
сравнительной простотой технологич. схемы; 3) дает высо-
высокий выход полимера; 4) позволяет получать полимеры
высокой чистоты; 5) обеспечивает возможность непо-
непосредственного использования полученного расплава
полимера для формования волокон и пленок. К недо-
недостаткам метода П. в р. относятся: необходимость
использования термически устойчивых мономеров, дли-
длительность процесса и необходимость его проведения
при высоких темп-рах.
Как уже отмечалось, П. в р. протекает обычно мед-
медленно, т. к. этот способ используют чаще всего для
поликонденсации мономеров с низкой реакционной спо-
способностью. Нек-рому ускорению процесса способствуют
повышение темп-ры, допустимое в пределах, определяе-
определяемых термич. стабильностью мономеров, олигомеров
и образующегося полимера, и применение катализато-
катализаторов (солей, окислов и гидратов окислов Fe, Pb, Al
и др., а также карбоновых к-т).
В большинстве ноликонденсационных процессов для
получения полимеров с высокой мол. массой необхо-
необходимо тщательно соблюдать стехиометрич. соотношение
(эквивалентность) мономеров. При П. в р. нарушение
этого соотношения может происходить по след. при-
причинам: 1) разложение функциональных групп одного
из мономеров, интенсивдо протекающее при высоких
теми pax (напр., декарбоксилирование или окисление);
2) унос части более летучего мономера током инертного
газа или его испарение (возгонка) в вакууме. Для того
чтобы избежать этого, в начальный период процесса
поддерживают минимально необходимую для начала
реакции темп-ру и повышают ее только после того, как
основная масса мономеров превращается в олигомеры.
Вязкость расплавов большинства полимеров доволь-
довольно высока [напр., вязкость расплава полиэтилентере-
фталата при 280 °С достигает 100 н-сек/м2 A000 пз)\.
Поэтому взаимное перемещение макромолекул на за-
заключительных стадиях П. в р. затруднено, и скорость
процесса определяется не столько реакционной способ-
способностью функциональных групп, сколько подвижностью
концов макромолекул.
В связи с большой длительностью и высокой темп-рой
П. в р. образовавшиеся макромолекулы постоянно
участвуют в обменных реакциях. Эта их способность
используется для синтеза соцолимеров, напр, сплавле-
сплавлением полиамидов и полиэфиров получают полиамидо-
эфиры.
,П. в р. применяют в лабораторной практике для син-
синтеза полиамидов, полиэфиров сложных (в том числе
полиарилатов), полиимидов и др. полимеров с гетеро-
циклами в основной цепи. Наиболее часто процесс про-
проводят в конденсационной пробирке; для получения
высокомолекулярных полимеров завершающую стадию
осуществляют в молекулярном кубе.
П. в р.— практически единственный промышленный
способ синтеза алифатич. полиамидов и полиэфиров
(напр., полиамида-6,6 и полиэтилентерефталата). Ее
осуществляют как по периодической, так и по непрерыв-
непрерывной схеме. В первом случае процесс проводят в стан-
стандартном автоклаве, а готовый полимер выдавливают
из него азотом через обогреваемый вентиль. Непрерыв-
Непрерывный процесс проводят в U-образном, L-образном или
трубчатом аппарате, разделенном на независимые
температурные зоны. Последнюю зону аппарата снаб-
снабжают щнековой мешалкой, к-рая обеспечивает тщатель-
тщательное перемешивание расплава и выдавливает его через
фильеру в виде моноволокна, жгута или пленки. Труб-
Трубчатый аппарат не нуждается в мешалке, т, к. процесс
в нем происходит в тонком слое. Расплав в этом случае
выдавливается из последней зоны аппарата шестеренча-
шестеренчатым насосом.
Лит.: СеренсонУ., КемпбелТ., Препаративные
методы химии полимеров, пер. с англ., М., 1963.
См. также лит. при ст. Поликондепсация. Я.С.Выгодский.
ПОЛИКОНДЕНСАЦИЯ В РАСТВОРЕ (solution
polycondensation, Losungspolykondensation, polycon-
densation en solution) — способ проведения поликон-
поликонденсации, при к-ром мономеры находятся в одной жид-
жидкой фазе в растворенном состоянии.
Известны следующие варианты П. в р.: 1) мономеры
и полимер растворимы в реакционной среде; 2) моно-
867
ПОЛИКОНДЕНСАЦИЯ В РАСТВОРЕ
меры и полимер частично растворимы в реакционной
среде; 3) мономеры полностью растворимы в реакци-
реакционной среде, тогда как образующийся полимер выпа-
выпадает из р-ра; 4) мономеры частично (до 50%) раство-
растворимы в реакционной среде, в к-рой полностью раство-
растворим образующийся полимер. Наиболее благоприятные
условия для получения полимера с высокой мол. мас-
массой создаются в том случае, когда мономеры и полимер
полностью растворимы в реакционной среде. Однако
для многих реакционных систем трудно подобрать та-
такой растворитель. Использование в качестве реакцион-
реакционной среды смеси двух или более растворителей позво-
позволяет в нек-рых случаях добиться полного растворения
как мономеров, так и полимера. Это связано с синер-
гич. действием растворителей, когда растворимость
вещества в смеси значительно выше, чем в каждом
из ее компонентов в отдельности.
При проведении П. в р. полимер может образовы-
образовывать пересыщенные термодинамически неустойчивые
(метастабильные) р-ры. В этом случае после выделения
или выпадения полимера из реакционной среды заново
растворить его в данном растворителе уже нельзя.
Время жизни метастабильных р-ров м. б. достаточно
большим и увеличиваться с уменьшением концентра-
концентрации р-ра. После выпадения из р-ра кристаллич. поли-
полимера, не способного набухать в реакционной среде,
дальнейший рост макромолекул прекращается, в то
время как в выпавшем в осадок аморфном полиме-
полимере, способном к набуханию, поликонденсация продол-
продолжается.
П. в р. обладает рядом особенностей, отличающих
ее от других способов проведения поликонденсации:
1) возможно осуществление процесса в относительно
мягких условиях, что особенно существенно при син-
синтезе высокоплавких полимеров, когда высокая темп-ра
реакции в расплаве может вызвать деструкцию моно-
мономеров и полимера; 2) растворитель часто выполняет
функции катализатора реакции; 3) облегчен вывод
из сферы реакции низкомолекулярного продукта;
4) обеспечивается хорошая теплопередача, что осо-
особенно важно для экзотермич. реакций; 5) мономеры
смешиваются быстро, что ведет к уменьшению доли
побочных реакций и способствует соблюдению правила
эквивалентности функциональных групп; 6) получен-
полученные в результате П. в р. р-ры полимеров можно непо-
непосредственно использовать для изготовления волокон
и пленок.
Закономерности поликонденсации в растворе
Основные закономерности П. в р. во многом идентич-
идентичны общим закономерностям поликонденсации (напр.,
влияние на процесс соотношения мономеров, колич.
монофункциональных добавок, темп-ры реакции и др.),
хотя в ряде случаев строение растворителя оказывает
существенное влияние на полученные результаты.
В любой реакционной системе растворитель в большей
или меньшей степени влияет на величину мол. массы
образующегося полимера и способствует формированию
той или иной его структуры.
Влияние природы растворителя на П. в р. обуслов-
обусловлено сольватацией исходных веществ, промежуточных
соединений и продуктов реакции. Сольватация м. б.
как физической (электростатической), так и химической
или специфической (донорно-акцепторное взаимодейст-
взаимодействие, образование водородных связей и др.). Поскольку
поликонденсация протекает через ряд промежуточных
элементарных стадий, на каждой из которых дейст-
действие растворителя может быть различным, наблюдаемое
влияние растворителя на П. в р. является суммарным
эффектом.
При поликонденсации одних и тех же мономеров
в различных растворителях реакционная способность
мономеров, скорость процесса, а следовательно,
и величина мол. массы образующихся полимеров обыч-
обычно плохо коррелируются с диэлектрич. проницаемостью
растворителя. Это объясняется значительным вкладом
специфич. сольватации в общий эффект среды. Специ-
фич. взаимодействие тесно связано с катализом поли-
поликонденсации, когда растворитель, помимо обычных
функций, может играть роль катализатора. Напр.,
пиридин, используемый в качестве растворителя при
синтезе поликарбонатов, выполняет также роль ката-
катализатора, образуя реакционноспособный комплекс-
с фосгеном:
Быстро
СОС12 + 2C5H5N « х COOJ'CgHc), • 2СГ
CO(N+C5H5)- 2СГ+ НО
2C5H5N -НС1
Кроме того, пиридин играет роль акцептора образую-
образующегося в ходе реакции хлористого водорода. В данном
случае связывание низкомолекулярного продукта по-
поликонденсации акцептором не является необходимым
условием получения полимера с высокой мол. массой,
т. к. взаимодействие фосгена с бисфенолом — практи-
практически необратимый процесс (константа равновесия
равна 103—104). Иная картина наблюдается при взаи-
взаимодействии дихлорангидридов дикарбоновых к-т с диа-
диаминами. Реакционная способность мономеров вполне
достаточна для того, чтобы взаимодействие происхо-
происходило без катализатора при достаточно низкой темп-ре,
но применение акцептора хлористого водорода совер-
совершенно необходимо; в противном случае хлористый
водород взаимодействует с аминогруппами, превращая
диамины в малореакционноспособные соли. Поэтому
при полиамидировании в качестве растворителей при-
применяют органич. соединения, обладающие свойствами
оснований. Нек-рые из них (напр., 1Ч,1Ч-диметилацет-
амид, гексаметилфосфортриамид, N-метилпирролидон
и др.) являются одновременно катализаторами поли-
поликонденсации. П. в р. указанного типа предложено
называть акцепторно-каталитической
поликонденсацией.
Растворитель может способствовать протеканию одно-
одновременно с поликонденсацией и нежелательных про-
процессов: обменных реакций и реакций дезактивирования
и блокирования функциональных групп. Напр., при
поликонденсации в среде N-замещенных амидов воз-
возможна побочная реакция дихлорангидридов дикарбо-
дикарбоновых к-т с растворителем. Способность растворителя
участвовать в побочных реакциях м. б. оценена по теп-
тепловому эффекту побочной реакции термохимич. мето-
методом. Известны многочисленные примеры дезактиви-
дезактивирования или блокирования функциональных групп
при П. в р. за счет примесей (напр., влаги), к-рые
могут содержаться в растворителе. Отрицательное
влияние примесей в значительной мере определяется
реакционной способностью мономеров. Так, при поли-
полиамидировании дихлорангидридов дикарбоновых к-т
предъявляются очень жесткие требования к отсутствию
влаги в растворителе. Применение дифторангидридов
тех же к-т позволяет существенно снизить эти требо-
требования.
Особое место среди побочных реакций при П. в р.
занимает процесс образования циклов в результате
циклизации мономеров или деструкции полимеров. Рав-
деструкция
новесие линейный полимер _
полимеризация
макроцикл,
869
ПОЛИКОНДЕНСАЦИЯ В РАСТВОРЕ
870
по-видимому, является характерной особенностью П.
в р., приводящей к образованию гетероцепных полиме-
полимеров. Циклообразование облегчается по мере уменьше-
уменьшения концентрации реакционной системы и при нек-ром
разбавлении может стать доминирующей реакцией.
Подбирая соответствующие растворители, можно
повлиять на степень упорядоченности полимерных
цепей, конформацию и конфигурацию макромолекул
и др. Так, при синтезе полиарилатов в растворителе,
к-рый хорошо растворяет мономеры, но плохо —
полимер, происходит сворачивание образующихся мак-
макромолекул в глобулы. Напротив, синтез полиарилатов
в растворителе, к-рый хорошо растворяет образующийся
полимер, приводит к возникновению фибриллярных
макромолекул, что обусловливает лучший комплекс
физико-механич. свойств полимера.
Выпадение полимера из реакционной среды во время
синтеза часто приводит к упорядочению его структуры.
Степень кристалличности полимеров, находящихся
после синтеза в р-ре, во многом зависит от скорости
и темп-ры осаждения, природы осадите ля, его взаимо-
взаимодействия с растворителем и т. д.
На практике выбор растворителя для
поликонденсации обычно осуществляют эмпирически
несколькими способами. Первый заключается в непо-
непосредственном проведении поликонденсации в ряде
растворителей с тем, чтобы затем выбрать тот из них,
в к-ром процесс протекал с наибольшей скоростью
и сопровождался образованием полимера с высокой мол.
массой. Однако при таком способе оценки растворите-
растворителей исследователь наблюдает суммарный эффект всех
протекающих в реакционной среде превращений.
По второму способу первоначально синтезируют
полимер (безразлично каким методом), а затем оцени-
оценивают его взаимодействие с различными растворителями
по абсорбции жидкости, набухаемости полимера и т. д.
Кроме того, существует косвенный способ подбора
и классификации растворителей, заключающийся
в изучении растворимости низкомолекулярных соеди-
соединений, моделирующих полимер. Напр., для полимеров
диметилпиперазина и терефталевой к-ты такими моде-
моделями являются:
.НзС
ю
СН3
н3с
Этот способ применим лишь к ограниченной группе
полимеров.
Технология и аппаратурное оформление
Обратимая (равновесная) поликонденсация. Техно-
логич. особенности проведения обратимой П. в р. за-
заключаются в следующем: 1) низкомолекулярный про-
продукт реакции удаляют, как правило, путем отгонки
из сферы реакции; 2) применяют растворители, темп-ра
кипения к-рых выше темп-ры кипения низкомолеку-
низкомолекулярных продуктов реакции; 3) в ряде случаев исполь-
используют растворители, дающие азеотроп с низкомолеку-
низкомолекулярным продуктом реакции, темп-pa кипения к-рого
ниже темп-ры кипения применяемого растворителя;
при этом низкомолекулярный продукт удаляется в виде
азеотропа (такой процесс наз. азеотропной
поликонденсацией); 4) применяют раство-
растворители, в к-рых хорошо растворяются образующиеся
полимеры и плохо — низкомолекулярные продукты
реакции; 5) процесс осуществляют при 100—200°С и
постоянном притоке свежего растворителя взамен отог-
отогнанного вместе с низкомолекулярным продуктом.
Существует разновидность П. в р.— т. н. осади-
тельная поликонденсация, при к-рой
равновесие реакции сдвигается в сторону образования
полимера вследствие его выпадения из р-ра.
Обратимая П. в р.— широко распространенный
лабораторный метод синтеза полимеров. Аппаратурное
оформление этого метода, как правило, несложное:
колба Вюрца, снабженная прямым (иногда и обрат-
обратным) холодильником и приспособлениями для ввода
растворителя и инертного газа или подключения
к вакуумной линии. Для проведения азеотропной поли-
поликонденсации требуется колонка специальной конст-
конструкции.
В пром-сти обратимая П. в р. применяется редко.
Так, первая стадия промышленного производства
ненасыщенных полиэфиров, алкидных смол и полиэти-
лентерефталата представляет собой разновидность
П. в р., когда растворителем служит один из мономеров
(напр., гликоль), взятый в избытке. Процесс проводят
в автоклаве с мешалкой, снабженном обратным холо-
холодильником для конденсации высококипящих реагентов
реакционной смеси и прямым холодильником для уда-
удаления паров воды (гликоля), а также приспособлением
для ввода инертного газа. Реактор обогревается высо-
высокотемпературным теплоносителем.
Необратимая (неравновесная) поликонденсация. Не-
Необратимую П. в р. обычно подразделяют на высоко-
высокотемпературную поликонденсацию
(процесс проводится при темп-pax выше 100 °С) и
низкотемпературную поликонден-
поликонденсацию (ниже 100 °С). При высокотемпературной ре-
реакции необходимо медленное (иногда — ступенчатое)
нагревание реакционной смеси до рабочей темп-ры,
т. к. быстрый ее подъем может привести к потере части
мономера (например, вследствие возгонки) и нару-
нарушению эквимолярного соотношения мономеров. Для
предотвращения окислительной деструкции реаген-
реагентов процесс проводят в токе инертного газа (азот,
аргон).
Осуществление низкотемпературной П. в р., как
правило, не требует принятия специальных мер пред-
предосторожности. Известно несколько особых разновидно-
разновидностей низкотемпературной необратимой П. в р. Одна
из них — эмульсионная поликонден-
поликонденсация, при проведении к-рой образование полимера
происходит в объеме одной из фаз двухфазной гетеро-
гетерогенной системы, напр, синтез ароматич. полиамидов
из ароматич. диаминов и дихлорангидридов дикарбо-
новых к-т в эмульсии органич. растворителей в воде.
В этом случае дихлорангидриды растворяют в органич.
фазе, а диамины — в водной. Применение высалива-
телей приводит к почти полному переходу диамина
в органич. фазу, в к-рой и происходит поликонденса-
поликонденсация. Нейтрализацию выделяющегося хлористого водо-
водорода осуществляют в водной фазе карбонатами или
щелочами. Реакция протекает обычно с большой ско-
скоростью и заканчивается за несколько мин.
Др. разновидность низкотемпературной необратимой
П. в р.— межфазная поликонденсация в том ее вариан-
варианте, когда процесс образования полимера протекает в ор-
органич. фазе.
Необратимая П. в р.— широкораспространенный
лабораторный метод синтеза полимеров. Аппаратур-
Аппаратурное оформление его проще, чем обратимой П. в р., т. к.
отпадает необходимость в приспособлениях для отгонки
низкомолекулярного продукта реакции и возвращения
растворителя в зону реакции. Необратимую П. в р.
проводят в конденсационной пробирке, снабженной
приспособлением для ввода инертного газа и газообраз-
газообразного мономера (напр., фосгена). В нек-рых случаях
необратимую П. в р. можно осуществить в круглодон-
871
поликоординация
872
ной колбе с обратным холодильником (если применя-
применяется метиленхлорид или др. низкокипящие раствори-
растворители) и приспособлением для ввода инертного газа.
В пром-сти необратимую П. в р. используют в про-
из-ве поликарбонатов, полиарилатов, нек-рых поли-
полиамидов и др. Для этой цели применяют стандартный
автоклав с мешалкой и обратным холодильником, снаб-
снабженный приспособлением для ввода инертного газа и,
при необходимости, газообразного мономера. Автоклав
снабжен рубашкой, к-рую иногда используют для обо-
обогревания или охлаждения (напр., при получении поли-
поликарбоната автоклав охлаждают водой).
П. в р.— перспективный лабораторный и промыш-
промышленный метод синтеза полимеров.
Лит.: Органические растворители, под ред. Я. М. Варшав-
Варшавского, пер. с англ., М., 1958.
См. также лит. при ст. Поликонденсация. В. А. Васн'ев.
ПОЛИКООРДИНАЦИЯ — см. Координационные
полимеры.
ПОЛИп-КСИЛИЛЕНЫ (poly-p-xylylenes, Poly-p-
й
xylylene,1 poly-p-xylylenes)
общей ф-лы:
— CR
— линейные полимеры
2 I
An
где R = Н, алкил, галоген, ацетил, алкокси- или
цианогруппа. Известны также П., в1 макромолекулах
к-рых атомы водорода алкиленовой группы замещены
на алкил, фтор или цианогруппу. Наибольшее значе-
значение имеют производимые в пром-сти поли-га-ксилилен
и поли(монохлар-гс-ксилилен).
Получение. В пром-сти поли-и-ксилилен (R == Н)
получают из гс-ксилола. Процесс протекает по след.
схеме:
СН3-( V-СНз
СН;
сн,
СИ
,-сн2-( У-с
IV
-сн2-/Л-сн2-1
L \=/ Jn
Пиролизом га-ксилола (I) при 950 °С в присутствии
водяного пара сначала синтезируют ди-га-ксилилен
(II); выход 15%. Последний выделяют охлаждением
газов пиролиза в бензоле или толуоле и затем подвер-
подвергают пиролизу при 600 °С в вакууме менее 133,322 н/м2
(менее 1 мм рт. ст.).
Пары образующегося при
этом га-ксилилена (III)
вступают в реакцию по-
полирекомбинации при осаж-
осаждении их на какую-либо
поверхность (при получе-
получении пленок) либо при
поглощении органич. ра-
растворителем или водой
(при получении порош-
филя. Для этого изделия помещают в конденсацион-
конденсационную камеру, непосредственно соединенную с пиро-
литич. трубкой.
Аналогичным методом (только при более высоких
тёмп-рах) из замещенных д-ксилолов можно получать
замещенные П. В пром-сти реализован способ получе-
получения цоли(монохлор-д-ксилилена) (R = С1). При пиро-
пиролизе ди-д-ксилиленов, замещенных в одном бензольном
кольце, раздельно (при разных темп-pax осаждения)
образуются замещенный и незамещенный П.
В лаборатории П. получают также по реакции Вюрца
из бромистого или хлористого га-ксилилена, разложе-
разложением гидроокиси метйлбензилтриметиламмония или
по реакции Фриделя — Крафтса из дигалогеналкилов
и бензрла (см. Полиалкиленарилены). Однако всеми
этими методами получаются полимеры низкой мол.
массы и менее регулярного строения, чем описанным
выше методом.
Свойства. Поли-га-ксилилен — бесцветное кристал-
лич. вещество. Полимер, полученный пиролизом ди-
n-ксилилена, имеет строго линейное строение; мол.
масса ок. 500 000. При темп-pax выше 200 °С он раст-
растворим в хлорированном дифениле и бензилбензоате,
из к-рых при 160 °С осаждается в виде геля. Для
поли-га-ксилилена характерны две кристаллич. формы
с темп-рой перехода второго рода 220—260 °С (по раз-
разным лит. источникам). На воздухе при темп-pax выше
170 °С поли-гс-ксилилен окисляется по метиленовой
группе с образованием гидроперекисей, при распаде
к-рых образуются ароматич. к-ты. В результате декар-
боксилирования этих к-т получаются низкомолекуляр-
низкомолекулярные ароматич. углеводороды.
Термич. распад полй-гс-ксилилена начинается при
350 °С с разрыва связей, образовавшихся в результате
побочных реакций при получении полимера, и далее
протекает по цепному механизму.
Большинство замещенных П.— также кристаллич.
вещества; их темп-ры плавления и стеклования, раство-
растворимость и физико-механич. свойства зависят от при-
природы заместителей (см. табл. 1 и 2). Поли(монохлор-дг-
ксилилен) и поли(монобром-тг-ксилилен) растворимы
в а-хлорнафталине при 150 °С; алкилзамещенные П.—
в хлоралканах при 20 °С.
У всех П. хорошо сохраняются линейные размеры
при изменении относительной влажности воздуха
в интервале 35—95% при 40 °С. Газопроницаемость
пленок из П. сравнима или меньше, чем у пленок
из поливинилиденхлорида, полиэтилентерефталата,
полиэтилена, политетрафторэтилена и полистирола.
Термич. и термоокислительный распад замещенных
П. во многом аналогичен распаду поли-гс-ксилилена.
П. сохраняют хорошую гибкость даже при криогенных
темп-рах.
Недостаток П.— низкая устойчивость к продолжи-
продолжительному воздействию солнечного света.
Таблица 1. Свойства поли-я-ксилилена и его аналогов, замещенных в бензольном кольце
СН2
сн.
CHfQ=CH2
Показатели
ков); выход полимера ко-
количественный.
Метод м. б. использован
для нанесения пленок по-
ли-гс-ксилилена (толщиной
от 50 нм до 250 мкм) на
изделия различного про-
Тёмп-ра плавления кри-
кристаллитов, °С
Темп-pa стеклования, °С
Прочность при растяже-
растяжении при 20 °С, Мн/м2
(кгс/см*)
Модуль упругости при
растяжении, Мн/м2
(кгс/см2)
при 20 °С . . . .
лри 200 °С . . .
Поли-п-
ксилилен
400
60-70
63 F30)
2500
B5 000)
175 '
A750)
Поли(мо-
нохлор-п-
ксилилен)
280-300
80—100
90 (900)
2800
B8000)
175
A750)
Поли(мо-
нобром-
п-ксили-
лен)
270
80
56 E60)
2800
B8000)
140
A400)
Поли(ди-
хлор-п-
ксили-
Лен)
Поли(мо-
ноциан-
п-ксили-
лен)
300
НО
40 D00)
2800 •
B8000)
175
A750)
270
90
60 F00)
3000
C0000)
140
A400)
Йоли(мо-
нометил-п-
ксилилен)
200-210
50^60
70 G00)
2800
B8000)
G0)
Поли-
(ЭТИЛ-П-
ксилилен)
160-170
25
80 (800)
1200
A2000)
0,7
G)
873
ПОЛИМЕРАНАЛОГИЧНЫЕ ИЗВРАЩЕНИЯ
874
Таблица 2. Некоторые свойства поли-п-ксилилена и поли(моно-
хлор-п-ксилилена)
Показатели
Плотность при 20°С, г/см3
Показатель преломления,
ПТ\
Интервал рабочих темп-р,
сс
\j
в вакууме
на воздухе
Диэлектрич. проницае-
проницаемость при 106 гц . . . .
Тангенс угла диэлектрич.
потерь при 106 гц ...
Электрич. прочность,
К€/М, ИЛИ в /ММ
Уд. поверхностное элект-
электрич. сопротивление при
23°Си 50%-нойотносит.
влажности воздуха, ом
Поли-тг-ксилилен
(парилен N)
1,103-1,120
1,669
от —60 до 220
от —60 до 100
2,65
0,0006
26 000
10"
Поли(монохлор-?г-
ксилилен)
(парилен С)
1,289
1,629
от —80 до 220
от —80 до 120
2,9
0,0128
15 000
1014
Переработка и применение. Поли-га-ксилилен и поли-
(монохлор-д-ксилилен) применяют в основном в виде
электроизоляционных или защитных пленок для кон-
конденсаторов, для покрытия внутренней поверхности
контейнеров, предназначенных для хранения жидко-
жидкостей, растворяющих полиэтилен, а также гидроокиси
натрия, бихромата калия и щелочных металлов (плен-
(пленки наносят в вакууме по описанной выше технологии).
Из тонких пленок изготавливают окна для оптич.
приборов. П. применяют также для приготовления кле-
клеев и герметиков.
Из поли-га-ксилилена и поли(монохлор-га-ксилилена)
можно получать изделия прессованием, однако после
хранения поли-га-ксилилен становится трудно формуе-
формуемым, видимо вследствие перехода в высокоплавкую
кристаллич. форму. Для более легкого перевода в вяз-
котекучее состояние к нему добавляют эквимолекуляр-
эквимолекулярное количество триалкилфосфина или триалкилфос-
фита. Товарными продуктами являются либо димеры,
либо пленки полимеров, нанесенные на подложки;
порошки пока не нашли практич. применения.
Первое пром. произ-во П. освоено в США в 1962,
где его производят под торговыми марками пари-
парилен N (поли-га-ксилилен) и парилен С [поли-
(монохлор-д-ксилилен)].
Лит.: Ли Г., Стоффи Д., Невилл К., Новые
линейные полимеры, пер. с англ., М., 1972; Gorham W. F.,
J. Polymer. Sci., pt. A-l, 4, JSfe 12, 3027 A966); Kubo S.,
Wunderlich В., J. Polymer. Sci., Polym. Phys., 10, № 10,
1949 A972); Chem.Eng., 72, №7, 48 A965); Jel linek H.H.,
Ron el S., J. Polymer. Sci., pt. A-l, 9, 2605 A971)
A. H. Зеленецкий.
ПОЛИЛАКТИД (polylactide, Polylaktid, polylactide).
Лактид (Л.) — внутренний циклич. эфир (дилактон)
молочной к-ты, 2,5-дикето-3,6-диметил-1,4-диоксан. Л.
может существовать в виде оптически активных R(—)-и
8-(+)-форм и двух неактивных —
?д НС^СО рацемич. и мезо-форм. П. получа-
3 or гн—гн ют из более доступного S-(-f)-Л.—
О кристаллического вещества бело-
белого цвета, легко растворимого в
органич. растворителях; т. пл. 96 °С; т. кип. 148—
150 °С при 1,6 кн/м2 A2 мм рт. ст.); уд. вращение
20
М 300° (бензол). Л. гидролизуется водой до молочной
кислоты, сополимеризуется с лактонами. Получают
S-(+)-JI. вакуумной перегонкой S-(—)-молочной к-ты
с последующей термич. деполимеризацией образующе-
образующегося циклич. олигоэфира молочной к-ты в присутствии
цинковой пыли; очищают многократной кристаллиза-
кристаллизацией из этил ацетата.
Полилактид — оптически активный линейный поли-
полиэфир [—ОСН(СН3)—СО—]п; кристаллич. вещество бе-
белого цвета; т. пл. 176 °С, т. стекл. 110—115 °С, [а]"
— 165° (бензол), —283° (метиленхлорид); характеристич.
вязкость [г]] = 0,50—2,0 дл/г (бензол, 25 °С). П. раст-
растворим в обычных органич. растворителях, легко обра-
образует пленки из р-ров, напр, в бензоле, хлороформе,
диоксане и бутилацетате, а также волокна из расплава
или р-ра (прочность при растяжении волокон ок.
250 Мн/м2, или 25 кгс/мм2, относительное удлинение
ок. 25%). П. не гидролизуется даже в кипящей воде;
0,01 н. р-ром NaOH при 25 °С за 50 ч гидролизуется на
50%; устойчив при нагревании до 290 °С. Он нетокси-
нетоксичен и не вызывает тканевой реакции отторжения,
а продукты его биодеструкции не накапливаются в жиз-
жизненно важных органах.
П. получают ионной полимеризацией в массе или
р-ре S-(-j-)-JI. в присутствии 0,001—0,5% (по массе)
(C2H5JZn,(C2H5KAl,SnCl4, К2СО3,КОНили др. катали-
катализаторов.
П. может быть применен в восстановительной хирур-
хирургии для изготовления кровеносных сосудов. В качестве
рассасывающихся хирургич. нитей запатентованы мате-
материалы на основе сополимеров S-(+)-JI. с гликолидом,
содержащих 5—10% (молярная концентрация) Л. и
обладающих лучшей растворимостью, чем полигликолид.
Впервые высокомолекулярный П. был синтезирован
в ФРГ Кляйне в 1954.
Лит.: К 1 е i n e J., Kleine H-, Makromol. Chem.,
30, 23 A959); К u 1 k a r n i R. К. [а. о.], Arch. Surg., 93, 839
A966); D ittrich W., SchulzR. C, Angew. Makromol.
Chem., 15, 109 A971); Kulkarni R. K. [a. o.], J. Biomed.
Mater. Res., 5, JSft 3, 169 A971); De SantisP., Kovacs
A. J., Biopolymers, 6; 299 A968). • В. С. Лившиц.
ПОЛИМАЛЕИНИМИД — см. Имидов непредельных
кислот полимеры.
ПОЛИМЕРАНАЛОГИЧНЫЕ ПРЕВРАЩЕНИЯ (poly-
meranalogous reactions, polymeranaloge Reaktionen,
reactions polymeranalogues) — химич. реакции макро-
макромолекул с низкомолекулярными соединениями, в про-
процессе к-рых изменяется природа связанных с основной
цепью функциональных групп, но сохраняется длина
и строение скелета основной цепи. В результате П. п.
фрагменты низкомолекулярного реагента (атомы или
группы атомов) входят в состав образующегося поли-
полимера.
Для многих П. п. характерно образование одной но-
новой (функциональной) группы в результате превраще-
превращения лишь одной исходной группы, напр, при гидролизе
поливинилацетата
+Н2О
[~сн2—сн--] > [_сн2-сн-1
I n -СНзСООН | п
ОСОСНз ОН
илд метилировании полиакриловой к-ты диазометаном
+CH2N2
[-СН2-СН-1 > [-СН2-СН-1
I n -N3 | п
СООН СООСНз
К этой же группе П. п. относятся разнообразные реак-
реакции замещения в полиолефинах. и в бензольных ядрах
поливинилароматич. соединений.
К П. п. относят также реакции, приводящие к обра-
образованию в макромолекуле циклич. структур в резуль-
результате взаимодействия двух исходных функциональных
групп, напр, ацеталирование поливинилового спирта:
—СН2—СН-СН2—СН-СН2-СН-СН2-СН^ + п RC^ ~>
он он он он
-» ^СН2—СН—СН2-СН-СН2-СН—СН2—СН Ь пН2О.
о о о о
СН
I
R
R
875
ПОЛИМЕРАЦАЛОГИЧНЫЕ ПРЕВРАЩЕНИЯ
876
Отнесение таких реакций к П. п. в значительной сте-
степени условно, т. к. нельзя утверждать, что при цикли-
циклизации (или при обратной реакции, связанной с раскры-
раскрытием циклов) сохраняется строение основной цепи по-
полимера; правда, при этом строение основной цепи не
изменяется столь существенно, как, напр., в реакциях
присоединения по двойным связям 1,4-полидиенов.
П. п., протекающие с образованием циклич. структур,
сходны с внутримолекулярными превращениями, одна-
однако в последнем случае в состав циклов не входят фраг-
фрагменты участвовавших в реакции низкомолекулярных
соединений.
Использование П. п. открывает широкие возможности
химич. модификации полимеров и получения новых по-
полимерных материалов, в частности таких, к-рые трудно
или невозможно синтезировать другим путем (см. также
Модификация химическая). Так, поливиниловый спирт
получают с помощью П. п.— гидролизом поливинило-
поливиниловых эфиров, дебензилированием поливинилбензилового
эфира. Изотактическую полиметакриловую кислоту не
удается получить с помощью стереоспецифической
полимеризации метакриловой к-ты, однако этого мож-
можно достичь полным гидролизом изотактического поли-
метилметакрилата.
Продукты П. п. при степенях конверсии, отличных
от 100%, представляют собой сополимеры, построен-
построенные из непрореагировавших и прореагировавших зве-
звеньев. Распределение звеньев в цепи таких сополиме-
сополимеров может значительно отличаться от распределения
звеньев в продуктах сополимеризации соответствую-
соответствующих мономеров. Следовательно, при одинаковом сред-
среднем составе свойства модифицированных путем П. п.
полимеров и продуктов сополимеризации м. б. сущест-
существенно различными.
Ряд П. п. уже нашел промышленное применение.
Такие реакции происходят, например, при получении в
промышленности простых и сложных эфиров целлюлозы,
поливинилового спирта, хлорирование полиэтилена
и поливинилхлорида. П. п. широко используются
также в практике лабораторных исследований. Так,
проведенная в свое время Штаудингером серия П. п.
NaOH (CH3COJO
[-СН2-СН-]П > [_СН,-СН-]П > [-СН.-СН-],,
ОН
ОСОСНз ОН ОСОСНз
послужила одним из наиболее убедительных доказа-
доказательств цепной природы макромолекул.
Основные закономерности. П. п.— обычные органич.
реакции, однако полимерная природа реагента вносит
существенный вклад в закономерности этих процессов.
Прежде всего, П. п. часто не доходят до конца вследст-
вследствие значительного изменения растворимости полимера
с глубиной конверсии — частично превращенный поли-
полимер выпадает в осадок, «уводя» непрореагировавшие
группы из сферы реакции.
При ацеталировании поливинилового спирта ОН-
группы, у к-рых обе соседние гидроксильные группы
уже прореагировали, не могут участвовать в образо-
образовании циклич. структур (хотя и могут взаимодейство-
взаимодействовать с альдегидом, образуя полуацетали). Если ацета-
лирование протекает по закону случая, то по заверше-
завершении процесса в макромолекулах остается —13% изо-
изолированных ОН-групп.
Исследование реакционной способности макромоле-
макромолекул в П. п. опирается на принцип Флор и,
постулирующий независимость реакционной способ-
способности функциональной группы от длины цепи, с кото-
которой эта группа связана. Этот принцип дает возмож-
возможность количественно описывать кинетику П. п. для об-
образцов полимеров, состоящих из макромолекул раз-
различной степени полимеризации. Более того, во мно-
многих случаях функциональные группы в макромолеку-
макромолекулах не отличаются по реакционной способности от
соответствующих низкомолекулярных аналогов. Это
справедливо, напр., для гидролиза поли-1\~-винил-
пирролидона и ]Ч-изопропил-2-метилпирролидона, для
распада гидроперекисных групп, содержащихся в ата-
ктическом полипропилене и гидроперекиси трет-
бутила.
Однако уже накоплен большой экспериментальный
материал, показывающий, что для многих П. п. реак-
реакционные способности макромолекул и их низкомоле-
низкомолекулярных аналогов существенно различны, и П. п.
характеризуются весьма специфич. особенностями,
обусловленными длинноцепочечной природой реагента.
Эти особенности проявляются при проведении П. п. не
только в конденсированной фазе, но и в разб. р-рах,
когда макромолекулы находятся в квазиизолированном
состоянии. В таких р-рах макромолекулы принимают
форму статистич. клубков. Если молекулы растворите-
растворителя и низкомолекулярного реагента значительно отли-
отличаются по полярности от макромолекулы или ее фраг-
фрагментов, то вследствие эффектов избирательной сольва-
сольватации и сорбции они могут неравномерно распреде-
распределяться между клубком и остальной частью р-ра. Такой
клубок можно рассматривать как микрореактор, в к-ром
реакционная среда и локальные концентрации реаген-
реагентов будут существенно иными, чем для р-ра в целом.
Уже по этой причине измеряемые константы скорости
П. п. могут заметно отличаться от таковых для низко-
низкомолекулярных аналогов.
Кроме того, связывание функциональных групп в еди-
единую гибкую макромолекулярную цепь приводит к из-
изменению таких определяющих реакционную способ-
способность факторов, как пространственные эффекты, элект-
электронные взаимодействия и участие соседних групп в ста-
стабилизации промежуточного соединения или переход-
переходного состояния (анхимерное содействие). Если же в
П. п. участвуют заряженные реагенты, на реакцион-
реакционной способности полиионов весьма специфич. образом
сказывается электростатич. взаимодействие.
Электростатические эффекты. Вы-
Высокая локальная концентрация зарядов в полиионах
прежде всего проявляется в том, что полимерные к-ты
и основания при одинаковых значениях рН ионизованы
значительно слабее, чем соответствующие однооснов-
одноосновные к-ты или основания. С этим связаны различия
в кинетике реакций частично ионизованных полимеров
и их низкомолекулярных аналогов как с незаряжен-
незаряженными, так и с заряженными агентами. Так, с уменьше-
уменьшением рН скорость кватернизацииполи-4-винилпиридина
алкилгалогенидами понижается гораздо медленнее,
чем в случае 4-пиколина.
Изменение степени ионизации полииона может вли-
влиять в определенных условиях на его сольватацию
низкомолекулярным незаряженным реагентом, что,
в свою очередь, отразится на кинетике П. п. Напри-
Например, при взаимодействии карбоксилат-анионных групп
полиметакриловой кислоты с а-бромацетамидом наблю-
наблюдаемое уменьшение константы скорости с увеличени-
увеличением степени ионизации полимера объясняют именно
снижением локальной концентрации а-бромацетамида
в клубке.
Особенно ярко электростатич. эффекты проявляются
при взаимодействии полиионов с заряженными реаген-
реагентами. Если карбоксилат-анионы полиметакриловой
кислоты вытесняют Вг~ из а-бромацетамида, то в слу-
случае бромацетат-иона такая реакция не идет вслед-
вследствие сильного отталкивания последнего полианио-
полианионом. Электростатическое отталкивание сильно прояв-
проявляется при щелочном гидролизе пектинов: констан-
константа скорости реакции быстро снижается с глубиной
превращения.
Электростатич. притяжение способствует ускорению
П. п. Так, реакция а-бромуксусной к-ты с частично
ионизованным поли-4-винилпиридином:
877
ПОЛИМЕРАНАЛОГИЧНЫБ ПРЕВРАЩЕНИЯ
878
~СН2— СН—СН2— СН— СН2—СН~
—СН—СНо—СН—СН2—СН~
BrCH2COO
ускоряется благодаря взаимному притяжению ионизо-
ионизованных пиридиниевых групп и бромацетат-ионов.
Влияние электростатич. взаимодействий на кине-
кинетику П. п. может быть описано количественно. Так, при
щелочном гидролизе пектинов наблюдаемая при сте-
степени ионизации полииона а константа скорости реакции
второго порядка ~к описывается ур-нием:
где к0 — константа скорости гидролиза эфирных групп
электронейтрального пектина; AG^(a) — свободная
электрич. энергия активации, необходимая для преодо-
преодоления электростатич. отталкивания, создаваемого за-
зарядом полианиона, при приближении ионов ~ОН
к эфирной группе полимера; к — константа Больцмана;
Т _ абсолютная темп-ра. Полагают, что для этой ре-
реакции AG^(a) равна свободной электрич. энергии ио-
ионизации АОдЛ(а). Такое допущение представляется
оправданным, т. к. для образования переходного со-
состояния ионы ~ОН должны подойти достаточно близко
к основной цепи полимера:
I
c=o
о
R
он
I I *
С=О НО-С—0й
о о5
R
В др. случаях, очевидно, AGJ^ (a) не обязательно рав-
равна AG^ (а). Напр., заряд полииона влияет на иониза-
ионизационное равновесие поли-4-винилпиридина значитель-
значительно сильнее, чем на скорость его реакции с бромацетат-
ионом, т. е. AG^(a)<AG^(a).
Общую картину взаимодействия заряженных реаген-
реагентов с полиионами представляют на основе двух раз-
различных моделей. В одной из них принимается, что
внешний реагент взаимодействует со сферич. клуб-
клубком, на поверхности к-рого равномерно распределен
заряд полииона; в другой — что заряды локализованы
и реагент испытывает электростатич. воздействие лишь
тех заряженных групп, к-рые являются ближайшими
соседями атакуемой функциональной группы полиме-
полимера. Остается, однако, не ясным, каковы условия реали-
реализации для каждой из этих моделей. Так, титрование
полиоснований удовлетворительно описывается при
учете взаимодействия с ближайшими соседями, тогда
как для поликислот такого согласия, как правило, не
наблюдается.
Эффект соседних звеньев. Ближайшие
соседние звенья могут влиять на реакционную способ-
способность функциональных групп в П. п. не только путем
электростатич. взаимодействия с низкомолекулярным
агентом, но и по др. механизмам. При хлорировании
полиэтилена в р-ре реакционная способность метиле-
новых групп в ходе процесса уменьшается в результате
отрицательного индукционного влияния образующихся
соседних хлорметиновых групп, к-рое передается по
углеродному скелету макромолекулы. Прореагировав-
Прореагировавшие звенья замедляют также кватернизацию поли-4-
винил пиридина ал кил- и бензилгалогенидами, что свя-
связано, вероятно, с пространственными затруднениями.
Значительные ускоряющие эффекты соседних зве-
звеньев обнаружены при гидролизе полиакрилатсхв и по-
лиметакрилатов. Сополимер га-нитрофенилметакрилата
с акриловой к-той гидролизуется в ~106 раз быстрее,
чем гс-нитрофенилтримети л ацетат. Причиной столь силь-
сильного ускорения является анхимерное содействие сосед-
соседних карбоксильных групп, стабилизирующее 6-член-
ный цикл в переходном состоянии:
> \-» v-»rlft UH"™CHo's/
Н20
СН,
~CH2—C—CH2—CH—CH2~
Если образование устойчивых 6- или 5-членных цик-
циклов невозможно, анхимерное содействие не реализуется.
Поэтому при гидролизе поли-Й-винилпирролидона об-
образующиеся в ходе П. п. карбоксильные группы не
ускоряют реакцию непрореагировавших соседних зве-
звеньев. Не наблюдается ускорение реакции и при гидро-
гидролизе сополимера этилакрилата с бутадиеном, где обра-
образующиеся карбоксильные группы отделены от эфирных
звеньями бутадиена (тогда как гидролиз гомополимера
этилакрилата протекает с ускорением). Обнаружены
также ускоряющие эффекты соседних гидроксильных
групп в гидролизе поливинилацетата и поливинил-
ацеталя.
Конфигурационные эффекты. Взаи-
Взаимодействие соседних звеньев существенным образом
зависит от их взаимного пространственного располо-
расположения. Естественно поэтому, что конфигурация макро-
макромолекулы может оказывать значительное влияние на
кинетику и механизм П. п. Так, изотактические поли-
метилметакрилат и полиметилакрилат гидролизуются
обычно с большей скоростью, чем атактические и ешь
диотактические полимеры. При гидролизе поливинила-
поливинилацетата и поливинилацеталя более реакционноспособ-»
ными оказались синдиотактич. образцы. Вклад конфи-»
гурационных эффектов не сводится, однако, лишь к вза-
взаимодействию соседних прореагировавших и непрореаги-
непрореагировавших звеньев. Так, в условиях, когда реакция
протекает под влиянием внешнего агента, константы
скорости кислотного гидролиза метилметакрилатных
звеньев в изо-, гетеро- и синдиотактич. триадах поли-
метилметакрилата относятся как 43 : 2,7 : 1. Этот ре-
результат, вероятно, объясняется различной доступностью
сложноэфирных групп в триадах разного типа для ата-
атаки катализирующих гидролиз ионов Н+.
Конформационные эффекты. Реак-
Реакционная способность функциональных групп при П. п.
879
ПОЛИМЕРАИАЛОГЙЧНЫЕ ПРЕВРАЩЕНИЯ
в сильной степени связана также с конформационными
характеристиками макромолекул. Прежде всего, при
конформационных переходах изменяется доступность
функциональных групп для атаки внешних агентов.
Напр., константа скорости дейтеро-водородного обме-
обмена по связи N — Н в поли-1Ч-винилацетамиде в 20 раз
меньше, чем константа скорости такого обмена для
N-метилацетамида, а энергии активации обмена прак-
практически одинаковы для полимера и низкомолекуляр-
низкомолекулярного модельного соединения. Для объяснения этого
факта предполагается, что макромолекула претерпе-
претерпевает быстрые конформационные переходы, при этом
амидогруппа ~95% времени экранирована близлежа-
близлежащим участком цепи от атаки молекул D2O; когда же
атака возможна, амидогруппа в полимере не отличается
по реакционной способности от низкомолекулярного
аналога. Экранирование функциональных групп осо-
особенно резко выражено в случае стабилизированных
специфич. конформаций, свойственных глобулярным
белкам. Так, тиольные группы яичного альбумина вос-
восстанавливают порфиридин лишь после денатурации
белка.
Благодаря гибкости полимерной цепи на реакцион-
реакционную способность функциональных групп могут влиять
не только ближайшие соседи, но и удаленные (вдоль
цени) звенья. Так, в тройном сополимере акрилами-
да с небольшими количествами ]Ч-D-пиколил)акрила-
мида и м-нитрофенилового эфира N-акрилилглицина
или д-нитрофенилового эфира N-акрилил-б-аминокап-
роновой к-ты гидролиз сложноэфирных групп проте-
протекает в результате внутримолекулярного катализа
пиридиновыми остатками. Вероятность взаимодействия
катализатора и субстрата убывает с увеличением рас-
расстояния между ними. Из теоретич. представлений о
конформациях гибких макромолекул в хороших раст-
растворителях следует, что скорость гидролиза эфирных
групп должна быть пропорциональна X (X — число
звеньев, разделяющих эфирный и пиридиновый остат-
остатки). Это предсказание теории хорошо согласуется с экс-
экспериментом.
Надмолекулярные эффекты. В гетеро-
гетерогенных системах, когда полимер находится в конден-
конденсированном состоянии, кинетика П. п., а также харак-
характер продуктов реакции в значительной мере опреде-
определяется морфологическими особенностями полимерного
образца.
С увеличением степени упорядоченности надмолеку-
надмолекулярных структур затрудняется доступ внешнего реа-
реагента к функциональным - группам макромолекул и,
соответственно, уменьшается скорость П. п. Так, при
хлорировании полиэтилена в твердой фазе аморфные
участки хлорируются быстрее, чем кристаллические,
поэтому линейный полиэтилен превращается медлен-
медленнее, чем разветвленный. Скорость окисления полиэти-
полиэтилена уменьшается при предварительной ориентации
образцов. Напротив, увеличение доступности функцио-
функциональных групп в результате мерсеризации приводит
к ускорению П. п. целлюлозных волокон.
Естественно, что при одинаковой средней степени
превращения продукт гетерогенной реакции будет зна-
значительно более неоднородным по составу, чем получен-
полученный в р-ре. Следовательно, эти продукты могут разли-
различаться по физич. и химич. свойствам, что и наблюда-
наблюдалось, напр., для образцов полиэтилена, хлорирован-
хлорированных в р-ре и в суспензии.
Следует заметить, что обусловленная межмолекуляр-
межмолекулярным взаимодействием склонность гибких макромоле-
макромолекул к агрегации в р-ре может сказаться и на кинетике
П. п. в гомогенных условиях.
Рассмотренные выше факторы могут существенным
образом влиять не только на кинетику П. п., но и на
такие фундаментальные характеристики продуктов ре-
реакции, как распределение прореагировавших и непро-
реагировавших звеньев в цепи и композиционная неод-
неоднородность. Напр., сильные ускоряющие эффекты со-
соседних групп должны приводить к блочному распреде-
распределению звеньев и увеличению композиционной неодно-
неоднородности полимера. Следует признать, однако, что де-
детальный механизм действия специфических для П. п.
факторов, оказывающих столь существенное влияние
на процесс, крайне слабо исследован как в экспери-
экспериментальном, так и в теоретич. аспектах.
Количественное описание. Задача количественного
описания П. п. с учетом всех присущих этим процессам
особенностей является весьма сложной. Для построе-
построения общей теории П. п. необходимо изучить действие
каждого из перечисленных выше факторов в отдель-
отдельности. В 60-е годы XX в. была начата разработка тео-
теории эффекта соседних звеньев.
Рассматривается след. модель П. п. Звенья А пре-
превращаются в звенья В по необратимой реакции первого
порядка (относительно А), и реакционная способность А
зависит только от природы ближайших соседей., Пусть
No, Мц N2 — доли А в центре триад ААА, ААВ (и ВАА)
и ВАВ, а кОч ки к2 — соответствующие константы ско-
скорости. Задача теории — описать кинетику реакции,
а также распределение звеньев в цепи и композицион-
композиционную неоднородность продуктов П. п. как функции
Ki ^1» ^2 п времени (или степени превращения).
Кинетика П. п. с эффектом соседа описывается
ур-нием:
2 (ftt- ftp) ,.
Р {А} = *-*«* [2 (V- *i) e *° J e(fts ~ 2fti) ' X.
X exp {2 (ftp-ft t) e-ftoM
2 (ftt-
X e
X exp
'j
(h2 -ft0-
hA
dt + C]
A)
где P[A} —доля А в полимере в момент времени t,
С — константа интегрирования.
Распределение звеньев в цепи продук-
продуктов П. п. описывается в терминах вероятностей после-
последовательностей из п звеньев Р{А^ Вп_,-} при любом за-
заданном расположении i звеньев А и (п — i) звеньев В
на п местах последовательности. Точное решение зада-
задачи основано на том, что P{Ah B?w} можно выразить
через вероятности последовательностей из одних
АР{Ад} и последовательностей из А и X P{Ah, Xm_ft},
где X обозначает место, к-рое может занимать либо А,
либо В. Напр., Р{АВАВ} = (Р{АХА) — Р{АХА2}) —
— (Р{А3} — Р{А4}) P{Aq] находят из ур-ния A) при
q = 1 и из ур-ния B) при q>2\
P{kq}=
exp {2
*0-*i) [* - i I kOt
B)
Сложнее найти вероятности ^{A^, Xm_fe). Для
простейшей из них Р{АХА} приходится решать на
ЭВМ систему из 4 дифференциальных ур-ний. С увели-
увеличением числа звеньев В в последовательности {А,,
Вп_/} растет число необходимых для расчета вспо-
вспомогательных вероятностей P{Ak, Xm_fe}, соответ-
соответственно возрастает число ур-ний и сложность решения.
Поэтому предложены приближенные методы описания
распределения звеньев, при этом степень приближения
оценивалась путем сопоставления приближенных реше-
решений с точным.
Наилучшие результаты дает следующий метод. По-
Последовательность {А/, Вп_.-} разбивают на отрезки
типа {Ад} и {ABjA}. Вероятности P{AQ} находят по
881
ПОЛИМЕРБЕТОН
882
ур-нию B), а вероятности P{ABjA} по ур-нию C):
dP {ABjA}
dz
= *j,i iV0 + 2 A - b5il) P {ABMA} ky -
k' A — '
X
C)
AV2
- 2P {АВ,-А} [Ay +.*' A - Y)l +
i-2
X
Здесь к = V/cq, A:' = /г2/&0, т =
6 = 1 при / = 1 и б = 0 при y Ф 1. iV0, iV1? iV2 и y легко
выражаются через Р{А}, Р{А2}, Р{А3} и, следователь-
следовательно, рассчитываются по ур-ниям A) и B). Вероятность
Р{А,-, Вп_;} находят как произведение вероятностей
P{Aq} и P{ABjA}.
В случае замедляющих эффектов соседних звеньев
A:0>к1>к2) хорошие результаты дают марковские при-
приближения. В частности, используя марковское при-
приближение первого порядка, P{Ah Bn_f-} можно
представить как функцию двух независимых условных
вероятностей Раа — вероятность найти звено А спра-
справа от А, и Ръа — вероятность найти А справа от В.
Раа и РЬа легко отыскать, решив систему:
dPaa/dz = Раа [(к' - 2к) - Раа B - Раа) (к' -2к + 1)]
dPbJdz = Pba
р2
аа
1 - Р
+ 2кРаа+ к' A - Раа)
q,a
D)
Для к о м п о з и ц и онной неоднород-
неоднородности продуктов П. п. строгого аналитич. решения
пока не найдено. Аналогом точного решения могут
служить результаты исследования композиционной
неоднородности методом статистич. испытаний (метод
Монте-Карло). На основании хорошего согласия с ре-
результатами метода Монте-Карло рекомендуется след.
приближенный аналитич. метод расчета.
Предполагается, что композиционная неоднород-
неоднородность описывается функцией нормального распределе-
распределения. Дисперсия этого распределения Dn рассчитыва-
рассчитывается — в одномарковском приближении — по ф-ле:
Mm Dn/n =
^-Раа + РЬаУ
E)
где п — длина отрезков, на к-рые разбивается беско-
бесконечная цепь при вычислении Dn. Следует подчеркнуть,
что предположение о применимости соотношений мар-
марковской цепи первогр порядка относится только к рас-
расчету Dnln в заданный момент времени, вся же предысто-
предыстория образца определяется истинными закономерностя-
закономерностями П. п. с эффектом соседа. Поэтому Раа и РЬа нель-
нельзя рассчитывать по ур-нию D), для этого используют
соотношения Раа = Р{А2}/Р{А}, РЬа = {Р{А} —
— Р{А2})/A — Р{А}), а Р{А} и Р{А2} находят по
ур-ниям A) и B).
Разработанные методы теоретич. описания П. п.
с эффектом соседа можно использовать для установле-
установления механизма процесса, а также для расчета «таких
характеристик распределения звеньев и композицион-
композиционной неоднородности продуктов реакции, к-рые не уда-
удается найти экспериментально.
Лит.: Химические реакции полимеров, под ред. Е. Феттеса,
пер. С англ., т. 1—2, М., 1967; Моравец Г., Макромоле-
Макромолекулы в растворе, пер. с англ., М., 1967, гл. 9; ЙлатэН.А.,
Литманович А. Д., Высокомол. соед., А14, № 11, 2503
A972); Litmanovich A. D. [а. о.], Units distribution
in the products of the polymeranalogous reactions, в кн.: IUPAG
Intern. Conference: Chemical Transformations of Polymers, v. 3,
Bratislava, 1971, P56. А. Д. Литманович, В. А. Агасандян.
ПОЛИМЕРБЕТОН (resin concrete, Plastbeton, beton
de resine) — бетон на основе органич. высокомолеку-
высокомолекулярного связующего (вяжущего). Связующим в П. слу-
служат преимущественно термореактивные смолы. Раз-
Разработан П. на основе фурановых (получаемых гл. обр.
из фурфурольно-ацетонового мономера — мономера
ФА), фенольных, ненасыщенных полиэфирных, эпок-
эпоксидных смол. Иногда для изготовления П. применяют
термопластичные продукты, напр, кумароно-индено-
вые смолы. В качестве наполнителей (заполнителей)
в П. вводят гранитный или андезитовый щебень, квар-
кварцевый песок и др. Размер частиц наполнителя состав-
составляет 0,1—40 мм, влажность — не более 5%. Соотно-
Соотношение в П. связующее : грубодисперсный наполнитель
может изменяться в пределах от 1 : 3 до 1 : 20 (по мас-
массе). Если используют связующие, к-рые отверждаются
кислыми отвердителями (напр., мономер ФА), содержа-
содержание карбонатов в наполнителях ограничивают (в пре-
пределах 0,5—1%), т. к. взаимодействие карбонатов с от-
вердителямн обусловливает значительное газовыделе-
газовыделение, приводящее к понижению плотности и прочности
П. Содержание отвердителя в композиции составляет
2—30% от массы связующего.
Помимо грубодисперсных наполнителей, в компози-
композицию вводят 10—50% (от массы связующего) различных
веществ, улучшающих технологич. свойства, а также
эксплуатационные показатели П.: 1) тонкодисперсные
наполнители (графит, сажу, фарфоровую муку, барит
и др.), повышающие прочность, модуль упругости, а
в нек-рых случаях и химстойкость П.; 2) пластифика-
пластификаторы (дибутилфталат, синтетич. каучуки), способст-
способствующие повышению эластичности изделий из П.; 3) ра-
растворители и разбавители (например, фурфурол в
фурановые смолы, толуол или ацетон в эпоксидные
смолы), повышающие пластичность композиции и об-
облегчающие ее формование; 4) порообразователи и дру-
другие добавки.
Типичный состав композиции (в % по массе): ще-
щебень — 52, речной песок — 29, молотый кварц — 7,
мономер ФА — 10, бензолсульфокислота — 2.
При изготовлении]!, компоненты тщатель-
тщательно перемешивают в обычном лопастном или шнековом
смесителе или в вибросмесителе (отвердитель загружа-
загружают в последнюю очередь). Процесс пожароопасен. Изде-
Изделия из П. формуют методами свободного литья или виб-
виброформования. Композицию выдерживают в формах
сначала 1 сут при 18 — 25 °С, а затем 10—30 ч при
80—120 °С до полного отверждения связующего. В тех
случаях, когда композицию не подвергают термообра-
термообработке, прочность изделий из П. повышается в тече-
течение 1—3 мес после их изготовления.
Свойства П. определяются типом и количеством
связующего и наполнителя, а также степенью отверж-
отверждения связующего. Прочность П. при сжатии, изгибе
и растяжении находится в пределах 50—120, 12—40
и 6—20 Мн/м2 соответственно A Мн/м2ъ10 кгс/см2),
ударная вязкость — в пределах 10—20 кдж/м2, или
кгс-см/см2. Ползучесть П. зависит в основном от типа
связующего, степени его отверждения, а также от усло-
условий нагружения. При длительном действии нагрузки,
не превышающей 50% от разрушающей, деформация
образцов П. на основе мономера ФА прекращается че-
через 240 сут нагружения. В интервале темп-р 20—70 °С
образцы II. на основе эпоксидных смол характеризуют-
характеризуются незатухающей ползучестью.
Теплостойкость П. на основе различных связующих
следующая (в °С): фурановые смолы — 150—200, эпок-
эпоксидные — 80—120, полиэфирные — 70—100, феноль-
ные — 120—180. Температурный коэфф. линейного
расширения П. в 2—6 раз превышает этот показатель
для стали и обычного бетона; при повышении темп-ры
от —40- до 60 СС он изменяется от 20 -Ю^ до
бО-Ю^. Теплопроводность П. на основе мономера
883
ПОЛИМЕР ГОМОЛОГИ
ФА меньше, чем у гранита и стали, соответственно
в 10 и 100 раз. П. обладают высокой стойкостью к дей-
действию химич. реагентов (таблица).
Химическая стойкость полимербетона и обычного бетона
в различных средах (по 10 -балльной шкале)
Вид бетона
Фурановый . . .
Эпоксидный . . .
Полиэфирный . .
Фенольный . . .
На основе порт-
портландцемента . .
Кисло-
Кислоты
10
9
8-9
9-10
1
Окис-
Окислители
2
3
6-7
3-4
1
Щело-
Щелочи
9
8
3-4
5-7
9
Соли
10
10
8-10
10
5
Раст-
вори-
ворители
8
6-7
4-5
7
5-7
Масла
и неф-
тепро-
тепродукты
8
9
7-9
8
5-6
Водопоглощение плотного П. составляет 0,2—1,5%
(за 30 сут). П. морозостоек: после 100 циклов замора-
замораживания и оттаивания масса фуранового П. уменьшает-
уменьшается на 0,1—0,2%, а его прочность снижается лишь на
5—8% (заметное снижение прочности наблюдается
после 300 циклов). П., особенно на основе полиэфирных
и эпоксидных смол, обладают хорошей адгезией ко мно-
многим материалам; прочность связи при испытании П.
на отрыв изменяется в пределах 2—10 Мн/м2 B0—
100 кгс/см2). Для П., содержащих связующие, к-рые от-
верждаются к-тами, характерна низкая адгезия к порт-
ландцементному бетону. Для повышения адгезии такой
бетон перед нанесением на него П. кислотного отверж-
отверждения покрывают кислотостойким материалом.
П. широко применяют для покрытия полов
в производственных помещениях с агрессивными сре-
средами, покрытия мостов и дорог, подвергающихся воз-
воздействию интенсивных нагрузок, а также для декора-
декоративной отделки различных сооружений. Из П. изготов-
изготовляют тюбинги, шахтную крепь, трубы. Армированный
металлом П. (сталеполимербетон) перспективен как
высокопрочный материал, к-рый м. б. использован
в конструкциях, контактирующих с агрессивными сре-
средами. Применение П. в строительных конструкциях
ограничивается в нек-рых случаях его ползучестью
при низких темп-pax и горючестью.
О свойствах бетонов, изготовляемых на основе ком-
композиций неорганических вяжущих веществ и органи-
органических высокомолекулярных связующих, см. Полимер-
цемент.
Лит.: Химически стойкие мастики, замазки и бетоны на ос-
основе термореактивных смол, М., 1968; Соломатов В. И.,
Полимер цементные бетоны и пластбетоны, М., 1967; Синтети-
Синтетические смолы в строительстве, Киев, 1969; Скупин Л.$
Полимерные растворы и пластбетоны, пер. с чеш., М., 1967;
Сталеполимербетонные строительные конструкции, М., 1972.
Ю. С. Черкинский.
ПОЛИМЕРГОМОЛОГИ (polymer-homologues, Poly-
merhomologe, homologues polymeres) — полимеры, мак-
макромолекулы к-рых построены из одинаковых звеньев и
различаются только степенью полимеризации. Поня-
Понятие о П. тесно связано с понятием о молекулярно-массо-
вом распределении полимеров.
ПОЛИМЕРИЗАЦИЯ (addition polymerization, Poly-
Polymerisation, polymerisation).
Содержание:
Терминология и классификация 883
Мономеры 884
Некоторые особые случаи 885
Кинетика 886
Механизм 88 9
Способы проведения 890
Заключение 892
Терминология и классификация. Полимеризация —
процесс получения высокомолекулярных веществ, при
к-ром макромолекула образуется путем последователь-
последовательного присоединения молекул одного или нескольких
низкомолекулярных веществ (мономеров) к растущему
активному центру.
По числу участвующих в П. мономеров различают
гомополимеризацию (один мономер) и с о-
полимеризацию (два и более).
В зависимости от природы активного центра, веду-
ведущего цепь, и от механизма акта роста цепи различают:
1) радикальную П., в к-рой активным центром
является свободный радикал, а акт роста является
гомолитич. реакцией (см. Радикальная полимеризация)
и 2) ионную П., при к-рой активные центры яв-
являются ионами или поляризованными молекулами,
а раскрытие двойной связи (или цикла) происходит ге-
теролитически (см. Ионная полимеризация). Соответст-
Соответственно, растущие макромолекулы в радикальной П.
представляют собой мак ро радикалы, а в ионной —
макроионы. В свою очередь, ионная П. подразделяется
на анионную, если концевой атом растущей цепи не-
несет полный или частичный отрицательный заряд, и
катионную, если этот атом заряжен положительно
(см. Анионная полимеризация, Катионная полимери-
полимеризация).
Активные центры ионной П. редко являются свобод-
свободными ионами; обычно в состав активного центра на-
наряду с растущим концом цепи входит противоположно
заряженный компонент (п р о т и в о и о н). Во многих
случаях противоион принимает непосредственное уча-
участие в актах роста цепи, образуя с присоединяющейся
молекулой мономера координационный комплекс или
циклич. переходное состояние. Такую П. наз. коор-
координационно-ионной. Благодаря регули-
регулирующему действию противоиона при координационно-
ионной П. часто образуются стереорегулярные полиме-
полимеры, т. е. полимеры с высокой степенью упорядочен-
упорядоченности пространственного строения (см. Координацион-
Координационно-ионная полимеризация, Стере о специфическая поли-
полимеризация).
До последнего времени два основных способа полу-
получения высокомолекулярных веществ — П. и поликон-
поликонденсацию — различали по их стехиометрии. К первому
случаю относили процессы, в к-рых полимер был един-
единственным продуктом реакции, а ко второму — процес-
процессы, сопровождающиеся выделением низкомолекулярных
веществ (воды, аммиака и т. п.). Однако в результате
интенсивных исследований механизма реакций синтеза
высокомолекулярных соединений стало ясно, что прин-
принципиальное различие между П. и поликонденсацией
лежит не в составе образующихся продуктов, а в ме-
механизме этих процессов. П.— особый тип цепных про-
процессов, в к-ром развитие кинетич. цепи сопровождается
ростом материальной цепи макромолекулы, в то время
как поликонденсация представляет собой совокупность
бимолекулярных реакций, кинетически не связанных
друг с другом. Поэтому, напр., реакции образования
полимеров из диазосоединений или N-карбоксиангид-
ридов а-аминокислот являются полимеризацией, хотя
они и сопровождаются выделением соответственно азо-
азота или двуокиси углерода (см. Диазосоединений поли-
полимеризация, N-Карбоксиангидридов а-аминокислот по-
полимеризация), а синтез полиуретанов из диизоциана-
тов и диолов — поликонденсацией, хотя в этом процес-
процессе и не выделяется никаких низкомолекулярных про-
продуктов.
Мономеры. Подавляющее большинство мономеров
принадлежит к одному из след. двух классов:
1. Мономеры, полимеризующиеся вследствие раскры-
раскрытия кратных связей С=С, С = С, С=О, G = N и др.
(олефины, диеновые и ацетиленовые углеводороды, аль-
альдегиды, нитрилы и др.). П. мономеров, принадлежа-
принадлежащих к этому классу, может быть описана следующей
общей схемой:
п А=В-
[-А-В-]п
A)
885
ПОЛИМЕРИЗАЦИЯ
886
2. Мономеры, полимеризующиеся вследствие раскры-
раскрытия циклич. группировок, напр, окиси олефинов, цик-
лич. эфиры, лактамы, лактоны и др. гетероциклич.
соединения. В этом случае П. протекает по схеме:
ГА-В1
L с J
> [-А-В-С-1
B)
Как правило, мономеры представляют собой вполне
устойчивые вещества; самопроизвольная П.— редкое
исключение. Поэтому способность данного мономера
к П., наряду с термодинамич. факторами (условие убы-
убыли свободной энергии — см. Термодинамика полиме-
полимеризации), определяется также и кинетическими, т. е.
наличием подходящего катализатора, выбором условий
и т. д. Эти две группы факторов далеко не всегда взаи-
взаимосвязаны. Напр., этилен из числа винильных соедине-
соединений имеет наибольшую теплоту П. и, следовательно,
находится в наиболее благоприятных термодинамич.
условиях (изменение энтропии при П. большинства
винильных мономеров почти одинаково), однако до
открытия Циглера — Натта катализаторов он считал-
считался весьма инертным мономером, малоактивным в ради-
радикальной и неактивным в ионной П. В то же время изобу-
тилен, теплота П. к-рого вдвое меньше, полимеризуется
с почти взрывной скоростью даже при —180 °С.
Некоторые особые случаи. П. большинства мономе-
мономеров происходит по ур-ниям A) и B), т. е. состав и струк-
структура мономерного звена в макромолекуле соответствует
составу и строению исходного мономера (за исключе-
исключением, конечно, размыкающейся в ходе процесса связи).
Однако известен ряд примеров, в к-рых образующиеся
при П. мономерные звенья отличаются от исходного
мономера по структуре, а иногда и по составу. Боль-
Большинство из известных случаев такой «необычной» П.
можно отнести к одному из след. типов: образование
новых связей внутри мономерного звена, сдвиг одного
или группы атомов во время акта роста, П. с выделе-
выделением низкомолекулярных веществ.
Наиболее частым случаем образования но-
новых связей внутри мономерного
звена является П. несопряженных диенов путем
чередования межмолекулярных и внутримолекуляр-
внутримолекулярных актов роста с образованием циклич. мономерных
звеньев, напр.:
Н2с
сн2
сн2
* II II * II
~СН2+СН СН->^СН2—CHj^CH СН -*
II II
СН2СН2 СН2 СН2
сн2 сн2
Межмолекулярный акт роста
СН2
/\*
->~СН2-СН2-СН СН
сн2 сн2
Ун;.
Внутримолекулярный акт роста
В результате такого процесса возможно замыкание не
только низших E—6-членных) циклов, но и высших
A0—15-членных), хотя, конечно, со значительно
меньшим выходом. Интересным случаем меж-внутри-
молекулярной П. является П. мономеров, в к-рых не-
несопряженные двойные связи разделены функциональ-
функциональными группами (дивинилацеталей, диаллиловых эфи-
ров, диаллиловых аммониевых солей и т. п.). В этом
случае образуются полимеры с гетероциклич. мономер-
мономерными звеньями, напр.:
СН2 СН2 СН2
II II /\
п СН СН->[-СН2-СН СН-1
II I ! п
о-сн-о о о
R \/
СН
R
К этому же типу относится и т. наз. трансаннулярная
П., т. е. П. циклич. ненасыщенных мономеров с замы-
замыканием дополнительной связи внутри цикла, напр. П.
1,4-диметиленциклогексана
—СН
или 1,5 циклооктадиена
-О-
При П. со сдвигом одного или группы
атомов во время акта роста чаще всего
происходит перемещение водорода в виде гидрид-иона
или протона, напр, при катионной П. нек-рых развет-
разветвленных а-олефинов
н
. I
R + СН2=СН— С-СН3
сн3
+ I
R —СН2 —СН —С—СН3
сн3
сн3 сн3
*- R — СН2— СН2—С - R— -СН2—СН2— С —
| L I J n
сн3 сн3
при анионной П. акриламида и нек-рых др. амидов
R-+ сн2=»сн—с— nh2
О
R—СН2-СН—C-N
о н
- -CH2-CH2—CONH-
или при П. пропилена на нек-рых комплексных ката-
катализаторах.
Известны также случаи миграции и более тяже-
тяжелых групп, например хлор-иона или метильной груп-
группы. Подробно об этом см. Изомеризационная полиме-
полимеризация.
По существу процессы этого типа — предельный
случай характерной для большинства ионных систем
тенденции к изомеризации активных центров в более
устойчивое состояние, при к-ром скорость изомериза-
изомеризации становится равной или даже превышает скорость
роста цепи.
Пока известны лишь единичные примеры реакций П.,
сопровождающихся выделением
визко молекулярных веществ, напр.
диазосоединений полимеризация, протекающая с выде-
выделением азота, N-карбоксиангидридов а-аминокислот
полимеризация, при к-рой выделяется двуокись угле-
углерода.
Кинетика. В процессе П. можно выделить несколько
основных стадий, т. наз. элементарных актов: иниции-
инициирование полимеризации, рост цепи, обрыв цепи, пере-
передача цепи.
Инициирование — это превращение неболь-
небольшой доли молекул мономера в активные центры, спо-
способные присоединять к себе новые молекулы мономера.
Для создания активных центров в систему вводят спе-
специальные вещества — инициаторы (см. Инициирова-
Инициирование полимеризации) или катализаторы полимеризации,
либо подвергают ее действию ионизирующего излуче-
887
ПОЛИМЕРИЗАЦИЯ
888
ния (см. Радиационная полимеризация), света (см. Фото-
Фотополимеризация) или электрич. тбка.
Рост цепи состоит из ряда многократно повто-
повторяющихся однотипных реакций присоединения моле-
молекул мономера (М) к активному центру (М*):
М*
М -> М*
М*
М -> М* . . . . М* + М -> М*
C)
В результате роста цепи исходный низкомолекулярный
активный центр вырастает в макромолекулу.
Реакция роста цепи в принципе является обратимой;
наряду с присоединением мономера к активному цент-
центру (реакция 3) может происходить и его отщепление:
м* + 1-»м*+м (За)
Поэтому в равновесии с активными центрами всегда
остается нек-рое количество мономера. Равновесная
концентрация мономера определяется термодинамич.
характеристиками полимеризующейся системы и по-
поэтому не зависит от механизма процесса (подробнее см.
Термодинамика полимеризации).
Обрыв цепи — это гибель активного центра
в результате его реакции с другим активным центром,
каким-либо посторонним веществом Z или вследствие
мономолекулярной перестройки:
М* 4- Z
ИнеРтные
продукты
D)
В результате передачи цепи активный центр
с растущей макромолекулы переходит на какую-либо
другую частицу X, начинающую рост новой макромоле-
макромолекулы:
М* + X -> Мп + X*
X* +М-> ХМ* и т. д.
E)
В зависимости от того, какой из компонентов полиме-
ризационной системы выступает в роли передатчика
цепи, различают передачу цепи на мономер, раст-
растворитель, полимер и т. д. В нек-рых случаях при пе-
передаче цепи образуется устойчивое соединение, не
присоединяющее к себе мономер. Такая реакция, ки-
кинетически эквивалентная обрыву, наз. и н г и б и р о-
в а н и е м, а вызывающее ее вещество — ингибитором
(см. Ингибирование полимеризации). Реакции D) и E)
ведут к прекращению роста макромолекулы, поэтому
их иногда объединяют под общим названием реак-
реакций ограничения роста цепи.
В отсутствие передачи цепи длина кинетич. цепи
процесса (т. е. число молекул мономера, прореагиро-
прореагировавших с активным центром от момента его появления
до гибели) равна длине молекулярной цепи (т. е. числу
звеньев в образующейся макромолекуле). При наличии
реакций передачи длина кинетич. цепи превышает
длину молекулярной. Иными словами, каждый акт
инициирования приводит к образованию одной макро-
макромолекулы (если нет передачи цепи) или нескольких (ес-
(если такие реакции есть).
Элементарные акты П. могут, в свою очередь, состоять
из нескольких простых реакций. Химизм элементарных
актов в конкретной полимеризующейся системе опреде-
определяется природой ее компонентов и условиями проведе-
проведения процесса.
В ряде реальных полимеризационных систем процесс
состоит только из стадий инициирования и роста цепи
(наличие этих двух стадий обязательно для всех про-
процессов полимеризации). В таких системах рост макро-
макромолекулы продолжается в течение всего процесса и во-
возобновляется при введении новых порций мономера
(см. Живущие полимеры). Предельные размеры обра-
образующихся в таких системах макромолекул определя-
определяются в конечном счете количеством вступившего в ре-
реакцию мономера, приходящимся на одну растущую
макромолекулу.
В других случаях процесс включает также различные
реакции обрыва и передачи цепи. В этих системах
время роста каждой отдельной макромолекулы, как
правило, значительно меньше продолжительности про-
процесса в целом, и размер макромолекулы (ее степень
полимеризации Р) определяется отношением скорости
роста цепи ур к сумме скоростей всех процессов огра-
ограничения роста цепи, т. е. обрыва voqp и передачи vueX):
Р = ¦
F)
"пер
Т. о., вводя в систему передатчики цепи, можно по
желанию снижать молекулярную массу полимера (см.
Регуляторы молекулярной массы). Если в системе
присутствуют эффективные передатчики цепи в боль-
больших количествах, то образуются только низкомоле-
низкомолекулярные вещества; в этом случае процесс называет-
называется теломеризацией.
Поскольку в реакцию обрыва или передачи цепи
может с некоторой вероятностью вступить растущий
активный центр любой длины, степень полимеризации
и мол. масса полимера являются статистич. величина-
величинами. Характер распределения макромолекул по разме-
размерам определяется механизмом процесса и в принципе
м. б. вычислен, если известна кинетич. схема процесса
(см. Молекулярно-массовое распределение).
Кинетич. ур-ния П., т. е. ур-ния, связывающие ско-
скорость процесса с концентрациями основных компонен-
компонентов, могут принимать самый разнообразный вид в за-
зависимости от механизма конкретных процессов. Но об-
общий принцип их вывода во всех случаях одинаков и
основан на небольшом числе упрощающих допущений.
Важнейшим из них является предположение, что реак-
реакционная способность растущих полимерных цепей не
зависит от их длины, если последняя превышает нек-рый
предел C—4 звена). Обоснованность такого предполо-
предположения для реакции роста цепи подтверждается тем, что
практически во всех экспериментально исследованных
случаях для кинетич. описания П. достаточно учесть
влияние последнего или, в немногих случаях, предпос-
предпоследнего звена растущей цепи. Что касается обрыва
цепи, то зависимость его скорости от длины растущей
макромолекулы в определенных условиях (напр., в вы-
высоковязких средах) представляется более вероятной,
однако в большинстве случаев этой зависимостью пре-
пренебрегают.
При образовании достаточно больших макромолекул
скорость П., т. е. скорость расходования мономера,
равна скорости роста цепи. Таким образом:
где п — число растущих цепей, [М] — текущая кон-
концентрация мономера, /ср — константа скорости роста
цепи.
Для простейшего случая живущих полимеров, где
скорость инициирования равна или выше скорости ро-
роста цепи, а реакции обрыва отсутствуют, концентрация
растущих макромолекул п равна исходной концентра-
концентрации инициатора 10, т. е.:
~
р 'о [Щ (8)
(поэтому системы с живущими полимерами наиболее
удобны для экспериментального определения А'р, см.
Анионная полимеризация).
В других случаях, когда время жизни растущих це-
цепей мало по сравнению с общим временем развития
процесса, для исключения величины п из ур-ния G)
используют т. наз. принцип стационар-
889
ПОЛИМЕРИЗАЦИЯ
89U
н о с т и, т. е. считают, что концентрация растущих
цепей не изменяется во времени или, что то же, что ско-
скорости инициирования и обрыва цепей равны:
Г = »«-"**= 0 (9)
Очевидно, что принцип стационарности применим лишь
для такой фазы реакции, в к-рой скорость изменения
концентрации активных центров мала по сравнению со
скоростями их образования и исчезновения. Для не-
неустановившихся процессов, в к-рых происходит быст-
быстрое нарастание или спад концентрации активных цент-
центров, он неприменим.
Использование принципа стационарности для случая
бимолекулярного обрыва (t'06p = К™2) дает:
"~\~1 (Ю)
Для мономолекулярного обрыва (uqq^ = коп):
К
(И)
"ин
ь0
В свою очередь, иИН зависит от концентрации ини-
инициирующего агента (часто пропорциональна ей), а так-
также может зависеть от концентрации мономера и т. д.
Ур-ния типа A0) и A1) удобнее всего применять
к начальной стадии процесса, когда все входящие в них
величины, в том числе и концентрацию мономера,
можно считать постоянными. Однако, поскольку при
их выводе не вводились никакие ограничения (кроме
предположений о независимости &р и к0 от длины цепи,
принципа стационарности и вида обрыва), эти ур-ния
универсальны и их применимость не зависит от кон-
конкретного механизма процесса, глубины полимеризации
и др. факторов до тех пор, пока указанные предположе-
предположения остаются справедливыми. Однако следует учесть,
что входящие в ур-ния A0) и A1) величины (иИН, к0, /ср)
могут сами зависеть, напр., от глубины превращения,
и поэтому применение этих ур-ний с начальными зна-
значениями констант для глубокой П. может приводить
к ошибочным заключениям.
В случае сополимеризации кинетич. схема усложня-
усложняется, т. к. увеличивается число возможных актов ини-
инициирования, роста и обрыва цепи. Напр., при сополи-
меризации двух мономеров А и В надо учитывать по
меньшей мере 4 акта роста цепи:
A2)
А
^А* + А > - А*
B
-Ь В > '
-В* + А
> ~> А*
A3)
A4)
^в* + в — ->~в* A5)
(А* и В* — активные центры, соответствующие моно-
мономерам А и В).
Обработка кинетич. схемы сополимеризации произ-
производится с помощью тех же методов, что и гомополимери-
зации. В случае сополимеризации наряду с кинетич.
ур-ниями можно вывести и ур-ния, связывающие со-
состав образующегося сополимера с концентрациями
мономеров в системе.
Механизм. Суммарная скорость ионной П., особенно
при комнатной и более низких темп-pax, часто бывает
выше скорости радикальной П. того же мономера. Это
связано прежде всего с тем, что стационарная концент-
рация активных центров в ионных системах, как пра-
правило, значительно выше, чем в радикальных. Сравни-
Сравнивать же реакционную способность макрорадикалов
и ионных активных центров трудно. Дело в том, что
в большинстве ионных Систем в актах роста цепи одно-
одновременно участвует несколько форм активных центров
с различной реакционной способностью (полярные
молекулы, ионные пары, свободные ионы), поэтому на-
наблюдаемое значение константы скорости роста &р,
полученное, напр., из ур-ния (8), часто оказывается
усредненной величиной, зависящей, в частности, от
концентрации активных центров.
Определение истинных величин А*р для индивидуаль-
индивидуальных активных центров возможно лишь при сочетании
кинетич. методов с определением числа различных
форм активных центров с помощью независимых физи-
ко-химич. измерений, что проделано лишь в единичных
случаях. Поэтому, несмотря на большое число работ
по ионной П., количественно она изучена значительно
меньше, чем радикальная.
Наибольший интерес, естественно, представляет срав-
сравнение активности радикалов и свободных ионов. В тех,
к сожалению пока еще единичных, случаях, когда уда-
удалось оценить реакционную способность свободных
макроионов, они оказались значительно более актив-
активными, чем радикалы (табл. 1).
Таблица 1. Константы скорости роста цепи на свободных
радикалах и макроионах
Мономер
Тип актив-
активного
центра
Стирол .
То же .
а-Метилсти-
рол ....
То же ...
Радикал
Анион
Катион
Анион
Катион
Условия полимери-
полимеризации
В массе, 25 °С
В тетрагидрофура-
не, 25 °С
В массе, 15 °С
В тетрагидрофура-
не, 25 °С
В массе, 25 °С
kP, лЦсек-моль)
52
6,5.104-1,3.10*
3,5-10°*
830
3,(М0в*
* Радиационная П., оценочные данные.
Установление механизма процесса, особенно при ис-
исследовании новых катализаторов или новых мономеров,
часто представляет собой трудную задачу. В специаль-
специальной литературе можно встретить ряд рекомендаций по
этому поводу, включающих как общие соображения
(рассмотрение химич. природы инициатора и мономера,
влияние среды), так и конкретные «рецепты» (напр.,
порядок реакции по инициатору, состав сополимеров,
образующихся из нек-рых пар мономеров); иногда ука-
указывают «индикаторные» мономеры, якобы способные
полимеризоваться только по одному механизму или по
разным механизмам образующие полимеры разной
структуры и т. д.
Однако ни одна из подобных проб не является и не
м. б. однозначной. В лучшем случае они могут служить
более или менее полезными рабочими инструментами,
в худшем — при некритич. применении — привести
к серьезным ошибкам. В частности, надо иметь в виду,
что большинство мономеров (если их П. термодинами-
термодинамически разрешена) способно к П. по любому механизму,
и если мономер считается неспособным к П. по какому-
либо механизму, то это м. б. следствием лишь отсутст-
отсутствия подходящих инициаторов. Поэтому надежные вы-
выводы можно сделать лишь на основании всестороннего
изучения процесса, включающего как исследование ки-
кинетики и свойств полимеров, так и физико-химич. ис-
исследование механизма процесса и природы активных
центров.
Способы проведения. П. м. б. осуществлена различ-
различными способами, различающимися по агрегатному со-
состоянию полимеризуемой системы. Выбор способа про-
891
ПОЛИМЕРИЗАЦИЯ
892
ведения П. определяется задачами, к-рые ставятся при
осуществлении процесса, требованиями, предъявляе-
предъявляемыми к получаемому продукту, природой компонентов
полимеризационной системы, технологич. требованиями
и т. д. (табл. 2).
Таблица 2. Промышленные способы получения полимеров
Полимер
Полиэтилен
Полипропилен
Сополимер этилена с пропи-
пропиленом
Полистирол
Поливинилхлорид
Полибутадиен
Полиизопрен
Бутадиен-стирольные сополи-
сополимеры
Сополимеры бутадиена с акри-
лонитрилом
Полимер АБС (акрилонитрил-
бутадиен-стирол)
Полиизобутилен
Фторопласты
Полиакрилаты
Полиацетали
Поливинилацетат
Способ полимеризации
В мас-
массе
В рас-
растворе *
+
Эмуль-
Эмульсионная
1
Суспен-
Суспензионная
+
* Включая гетерогенную полимеризацию.
Простейшим случаем является П. жидкого мономера
в отсутствие растворителя (полимеризация в массе).
Если реакцию доводят до полного превращения моно-
мономера, то полимер получают в виде монолитного блока,
имеющего форму сосуда, в к-рый был залит мономер.
Преимуществами этого способа являются возможность
использования блоков полимера без последующей пе-
переработки и отсутствие стадии отделения от раствори-
растворителя. Основной недостаток — сложность отвода выде-
выделяющегося при реакции тепла, особенно при высокой
вязкости системы.
Другой распространенный способ гомогенной жидко-
фазной П.— полимеризация в растворе. В этом случае
проблема теплоотвода по сравнению с полимеризацией
в массе решается относительно легко. Для нек-рых
целей (при получении клеев, лаков, связующих для
слоистых пластиков) полученный р-р полимера можно
использовать непосредственно, для других — требу-
требуется выделить дюлимер и освободить его от растворите-
растворителя, что является одним из основных технологич. недо-
недостатков этого процесса. П. в массе и в р-ре можно осу-
осуществлять под действием инициаторов как радикаль-
радикальной, так и ионной природы.
Технически важным видоизменением предыдущего
способа является гетерогенная полимеризация раство-
растворенного мономера под действием различных диспергиро-
диспергированных или гранулированных твердых катализаторов.
При этом образующийся полимер может либо находить-
находиться в растворе, либо осаждаться на частицах катали-
катализатора. Основные преимущества этого способа те же,
что и предыдущего; к числу недостатков прибавляет-
прибавляется необходимость удаления из полимера остатков
катализатора.
В связи с тем, что наиболее дешевым и распростра-
распространенным технич. растворителем является вода, большое
промышленное распространение получила П. в вод-
водных эмульсиях и суспензиях. В первом случае моно-
мономер диспергируют в воде в присутствии диспергирую-
диспергирующих веществ (эмульгаторов), а для инициирования ис-
используют обычно водорастворимые инициаторы (чаще
всего — окислительно-восстановительные системы, эф-
эффективные при низких темп-pax). Эмульсионная поли-
полимеризация, в механизме к-рой существенная роль при-
принадлежит адсорбционным слоям эмульгатора на по-
поверхности мономер-полимерных частиц, отличается
рядом специфич. особенностей. В частности, этот про-
процесс позволяет осуществлять П. с высокой скоростью
при низких темп-pax и, кроме того, получать продукт
большой мол. массы.
При суспензионной полимеризации мономер диспер-
диспергируют в виде капель размером 1 мм—1 мкм и полиме-
ризуют под действием растворимого в мономере ини-
инициатора; процесс, протекающий в каждой отдельной
капле, можно рассматривать как П. в массе.
При эмульсионной П. полимер получается в виде ла-
латекса, при суспензионной — в виде крошки. Недо-
Недостатками обоих процессов являются образование боль-
больших количеств сильно загрязненных сточных вод,
а также необходимость удаления из полимера эмуль-
эмульгаторов и стабилизаторов. Процессы эмульсионной и
суспензионной П., связанные с применением воды,
наиболее характерны для радикальных процессов.
В связи с быстрым развитием ионной и координацион-
координационно-ионной П., позволяющей в ряде случаев получать
из тех же мономеров полимеры с более регулярной
структурой и с более высокими эксплуатационными
качествами, намечается тенденция к замене эмульси-
эмульсионных процессов ионной полимеризацией в р-ре. С дру-
другой стороны, появляются ионные каталитич. системы,
напр., катализаторы на основе металлов VIII группы,
способные инициировать П. в водных средах.
Интенсивно исследуется твердофазная полимериза-
полимеризация, при к-рой охлажденные ниже темп-ры плавления
мономеры или их р-ры полимеризуют под действием
ионизирующего излучения. При твердофазном процессе
в ряде случаев образуются полимеры с лучшими свойст-
свойствами, чем при полимеризации в р-ре или расплаве (с бо-
более регулярной структурой, более высокой мол. массой
и т. п.); этот процесс, по-видимому, может получить тех-
технич. применение для П. мономеров, являющихся при
комнатной темп-ре твердыми веществами.
Наконец, необходимо упомянуть еще о т. наз. газо-
газофазной полимеризации. Очевидно, что осуществление
П. целиком в газовой фазе практически невозможно,
т. к. из-за ничтожно малой растворимости полимера
в парах своего мономера растущие цепи уже при до-
достижении относительно небольших размеров выпадают
в виде твердой фазы. Газофазной П. обычно называют
процессы, в к-рых мономер находится в газообразном
состоянии. Инициирование может происходить либо
в газовой фазе (под действием у-излучения или парооб-
парообразного инициатора), либо на поверхности твердого
катализатора. Сама же П. (рост цепи) происходит на
поверхности или в объеме твердой фазы полимера.
Заключение. Явление полимеризации было открыто
еще в середине XIX в., практически одновременно с вы-
выделением первых способных к П. мономеров (стирола,
изопрена, метакриловой кислоты и др.). Однако сущ-
сущность П. как процесса образования истинных хими-
химических связей между молекулами мономера была поня-
понята лишь в 20—30-х гг. XX в., когда установились пра-
правильные представления о природе высокомолекуляр-
высокомолекулярных веществ.
На долю полимеров, получаемых методом полимери-
полимеризации, сейчас приходится около 3/4: общего мирового
выпуска полимеров. Поэтому не удивительно, что
пром-сть полимеризационных процессов составляет
сейчас одну из наиболее мощных и, вероятно, наиболее
быстро растущую отрасль промышленности органиче-
органического синтеза.
До середины 50-х гг. XX в. господствующее место
в пром-сти занимали радикальные процессы. Ионные
процессы (получение полибутадиена под действием
натрия, синтез бутилкаучука) были единичными иск-
исключениями и, казалось, не имели большой перспекти-
перспективы. Однако после открытия новых высокоэффективных
стереоспецифич. катализаторов началось широкое
893
ПОЛИМЕРИЗАЦИЯ В МАССЕ
894
внедрение в пром-сть ионных и координационно-ион-
координационно-ионных процессов, характеризующихся более высокой
производительностью, высокой стереорегулирующей
способностью и позволяющих более гибко контролиро-
контролировать свойства получаемых продуктов. В настоящее вре-
время полимеры, получаемые методами координационно-
ионной полимеризации (полиэтилен высокой плотности,
полипропилен и их сополимеры, стереорегулярные
полиизопрен и полибутадиен и т. д.), составляют
10—15% от общей массы полимеризационных материа-
материалов, причем уд. вес ионных процессов в выпуске по-
полимеров непрерывно возрастает.
Лит.: Багдасарьян X. С, Теория радикальной
полимеризации, 2 изд., М., 1966; БреслерС.Е., Еруса-
лимский Б. Л., Физика и химия макромолекул, М.— Л.,
1965; ФренкельС. Я., Введение в статистическую теорию
полимеризации, М.— Л., 1965; Г е й л о р д Н., М а р к Г. Ф.,
Линейные и стереорегулярные полимеры, пер. с англ., М.,
1962; Катионная полимеризация, под ред. П. Плеша, пер.
с англ., М., 1966; Химия и технология полимеров, пер. с нем.,
т. 1, т. 2, ч. 1—2, М.—Л., 1965; Шварц М., Анионная
полимеризация, пер. с англ., М., 1971; О у д и а н Д ж., Осно-
Основы химии полимеров, пер. с англ., М., 1974; Encyclopedia of
polymer science and technology, v. 1—15, N. Y., 1964—71.
А. А. Арест-Якубович.
ПОЛИМЕРИЗАЦИЯ В МАССЕ, полимериза-
полимеризация в блоке (mass polymerization, Massenpoly-
merisation, polymerisation en masse).
Содержание:
Особенности кинетики процесса 893
Расчет вязкости системы 894
Аппаратурное оформление процесса 895
Выбор типа реактора и режима процесса . ... 897
Технологический расчет реактора 898
Математическое моделирование процесса . ... 900
Преимущества и недостатки метода 901
Полимеризация в массе — способ проведения поли-
полимеризации, при к-ром исходный мономер находится
в жидкой фазе в неразбавленном виде. Механизм П.
в м. может быть различным — радикальным, ионным
или координационно-ионным в зависимости от природы
возбудителя процесса и мономера. Реакционная систе-
система м. б. гомогенной (полимер растворим в мономере,
в конце процесса система представляет собой конц. р-р
или расплав); гетерогенной (полимер образует отдель-
отдельную жидкую или твердую фазу). Помимо мономера и
возбудителя полимеризации, система может включать
различные добавки, регуляторы и стабилизаторы.
Обычно в результате П. в м. образуются продукты ли-
линейной или разветвленной структуры. Образование
трехмерных (сшитых) структур при полимеризации
многофункциональных мономеров и олигомеров (см.так-
(см.также Трехмерная полимеризация) часто рассматривают
как особый тип полимеризационного процесса. Однако
по основным кинетич. закономерностям его следует
отнести к П. в м.
Особенности кинетики процесса
П. в м. получила широкое распространение
в пром-сти, где в большинстве случаев процесс ведут до
глубоких степеней превращения мономера. Для П. в м.
характерна аномальная зависимость скорости процес-
процесса и мол. массы полимера от глубины превращения
мономера. Это явление в радикальной полимеризации
получило название гель-эффект. Оно в различной сте-
степени присуще всем мономерам и в наибольшей степени
тем, к-рые являются «плохими» (в термодинамич. смыс-
смысле) растворителями для полимера или вообще не раст-
растворяют его. Изменение кинетич. констант полимериза-
полимеризации обусловлено значительным увеличением в ходе
процесса вязкости среды из-за накопления полимера и
диффузионных затруднений перемещению активных
центров и молекул мономера. Вязкость большинства
жидких мономеров составляет 0,005—0,010 н-сек/м2
@,05—0,10 пз), вязкость конц. р-ров и расплавов по-
полимера в условиях П. в м. может достигать 103—
105 н-сек/м2 A04—106 пз). Столь значительное изменение
свойств среды оказывает заметное влияние на условия
массообмена и теплопередачи при П. в м.
Отклонение кинетики процесса радикальной полиме-
полимеризации от «нормальной» зависит от темп-ры и обычно
уменьшается с увеличением последней, однако во всех
случаях гель-эффект необходимо учитывать при кине-
кинетич. расчетах с самого начала процесса. Наиболее
чувствительными к изменению вязкости системы яв-
являются константа скорости реакции обрыва цепи (к0^)
соединением и диспропорционированием радикалов
и эффективность инициирования / (см. Клетки эффект).
Экспериментально установлено, что &об при глубоких
степенях превращения для нек-рых систем может
уменьшаться в 100 раз, а эффективная энергия актива-
активации обрыва цепи возрастать с 8,4 до 50,4 кдж/молъ
(с 2 до 12 ккал/моль). Значение / в ряде изученных систем
(включая образование сшитых структур) уменьшалось
в 3—10 раз.
Предельная глубина превращения мономера опреде-
определяется природой и концентрацией возбудителя про-
процесса, темп-рой и структурой образующегося продукта.
Если темп-pa близка к темп-ре стеклования полимера,
П. в м. затормаживается при относительно низких
глубинах превращения.
Для кинетич. описания П. в м. в реальных реакто-
реакторах необходимо для каждой конкретной системы опре-
определить константы скорости элементарных актов поли-
полимеризации (инициирования, роста, передачи и обрыва
цепи) и установить корреляцию вязкости системы и глу-
глубины превращения мономера при расчетных темп-рах
процесса. Для упрощения часто принимают, что кон-
константы скорости роста &р и передачи цепи на мономер
ки не зависят от вязкости. Тогда значения k0Q и / можно
определить по данным о скорости полимеризации и сред-
нечисловой степени полимеризации при данной глубине
превращения. На примере полимеризации стирола
в массе при вязкости до 10 н-сек/м2 A00 пз) показано,
что данный метод дает хорошее совпадение с экспери-
экспериментом при расчете молекулярно-массового распреде-
распределения (ММР) полимера. Для исследования кинетики
П. в м. используют дилатометры (для гомогенных си-
систем) или гравиметрич. метод (для гетерогенных).
Расчет вязкости системы
Вязкость р-ров и расплавов полимеров сложным об-
образом зависит от ММР и концентрации полимера.
Поэтому для каждой конкретной системы необходимо
экспериментально исследовать эту зависимость в изу-
изучаемом диапазоне темп-р. Нек-рые эмпирич. зависи-
зависимости этого типа приводятся в литературе.
Для ньютоновского течения р-ров и расплавов (при
малых напряжениях и скоростях сдвига) теория пред-
предсказывает зависимость вязкости только от среднемас-
совой мол. массы Mw и от концентрации С полимера:
7j = KMws'bC*>*-5>°
где тг] — вязкость в пз A пз = 0,1 н-сек/м2), К — кон-
константа, зависящая от природы системы и от темп-ры.
Эта зависимость применима при значениях Mw и С
больше нек-рых критических.
При синтезе полимеров методом П. в м. в промышлен-
промышленных условиях обычно вязкость р-ров и расплавов зави-
зависит от скорости сдвига (неньютоновское течение при
больших скоростях и напряжениях сдвига). В этом
случае эффективная (неньютоновская) вязкость слож-
сложным образом зависит от ММР и концентрации и не м. б.
рассчитана теоретически.
Связь между вязкостью и темп-рой выражается экспо-
экспоненциальным законом
_ехр ga
R.T
895
ПОЛИМЕРИЗАЦИЯ В МАССЕ
896
где Еа — энергия активации вязкого течения. Эта ве-
величина сложным образом зависит от концентрации
полимера, ММР и скорости сдвига. Она постоянна
в узком интервале темп-р и может колебаться от 21
до 125 кдж/моль (от 5 до 30 ккал/молъ).
Для гетерогенных систем эффективная вязкость сус-
суспензии т(с в н-сек/м2 (пз) м. б. определена по ур-нию:
* = Т'ж A-Соб)а
гДе ^ж — вязкость жидкого мономера в н-сек/м21 (пз),
Coq — объемная доля твердой фазы, а — эмпирич, ко-
коэффициент, зависящий от формы частиц полимера (для
сферических частиц его значение минимально и состав-
составляет 2,5).
Аппаратурное оформление процесса
В лабораторной практике П. в м. обычно проводят
в ампулах или (при небольших значениях вязкости)
в аппаратах с мешалкой. Если реакционная система
гомогенна, полимер выделяют, добавляя осадитель.
Реакторные устройства, используемые для проведе-
проведения П. в м. в пром-сти, можно разделить на 4 основные
группы:
1. Реакторы смешения (или каскад про-
проточных реакторов этого типа) — разнообразные по кон-
конструкции вертикальные и горизонтальные емкостные
аппараты, оборудованные различными перемешиваю-
перемешивающими устройствами (лопастными, турбинными, лен-
ленточными, дисковыми мешалками и др.), рассчитанными
на высоковязкие среды. Единичный реактор может
работать в периодич. или непрерывном режиме. Тепло-
съем в аппаратах этого типа осуществляется через ру-
рубашку и (или) внутренние змеевики, полости в мешал-
мешалке, где циркулирует теплоноситель.
При увеличении размеров реактора отношение его
поверхности к объему уменьшается, что создает допол-
дополнительные трудности для отвода тепла. В нек-рых
случаях для увеличения теплосъема и сохранения теп-
теплового баланса процесса полимеризацию ведут при ки-
кипении мономера, регулируя соответствующим обрааом
давление (разрежение) в аппарате. Избыточное телло
реакции расходуется на испарение части мономера,
к-рый затем может возвращаться в цикл. Подобный
прием используют, напр., при получении ударопроч-
ударопрочного полистирола.
Интервал давлений, применяемых при П. в м.,—
от вакуума 0,05—0,02 Мн/м2 @,5—0,2 кгс/см2) при по-
полимеризации стирола с частичным испарением мономе-
мономера до 200—270 Мн/м2 B000—2700 кгс/см2) при полиме-
полимеризации этилена по радикальному механизму в кон-
конденсированной фазе. Последний процесс иногда рас-
рассматривают как газофазную полимеризацию, однако
правильнее ее отнести к П. в м. При давлениях выше
180 Мн/м2 A800 кгс/см2) реакционная масса представ-
представляет собой гомогенную среду, плотность этилена в этих
условиях ок. 0,5 г/см3.
В реакторах смешения получают продукт относитель-
относительно однородного состава и поддерживают темп-рный
режим, близкий к изотермическому, на каждой ступе-
ступени каскада. Применение аппаратов этого типа ограни-
ограничено верхним пределом вязкости 10—102 н-сек/м2
A02—Ю3 пз).
2. Реакторы вытеснения — аппараты
трубчатого и колонного типов, шнековые устройства.
Теплосъем осуществляется через рубашку, в колоннах—
с помощью встроенных поверхностей. Характерно на-
наличие градиента темп-р и концентраций по длине ре-
реактора. По сравнению с единичным реактором смешения
здесь достигают более высоких степеней превращения
мономера.
3. Комбинированные установки,
включающие последовательно соединенные реакторы
первого и второго типов. В этом случае удается обеспе-
обеспечить наиболее гибкое управление процессом, что осо-
особенно важно при получении различных марок продук-
продуктов путем регулирования режима синтеза.
4. Полимеризацию иные формы. К это-
этому типу относятся различные периодически и непрерыв-
непрерывно действующие устройства, в к-рых. не происходит
перемешивания реагентов и продукт получается непо-
непосредственно в виде готового изделия (см., напр., Орга-
Органическое стекло). Стадии окончательного формования
(дополимеризации) часто предшествует полученце жид-
жидкого форполимера в реакторе периодич. действия с ме-
мешалкой. При получении покрытий из олигоэфиракри-
латов полимеризация в тонком слое ведется непрерывно
с высокой скоростью при использовании, напр., фото-
фотоинициирования.
При проведении П. в м. технологи сталкиваются
с рядом трудностей. Напр., большие затруднения вызы-
вызывает образование пленки полимера на поверхности
аппаратуры, что приводит к резкому снижению коэфф.
теплоотдачи. Для каждой конкретной системы задача
решается эмпирич. путем за счет подбора материала
реактора или специального покрытия, поверхности
аппаратуры (полимеризация винилхлорида), примене-
применения пульсирующего режима давления (полимеризация
этилена в трубчатом реакторе), использования специаль-
специальных перемешивающих устройств для непрерывного уда-
удаления пленки со стенок.
Важная технич. задача — удаление незаполимеризо-
вавшегося мономера из суспензии или расплава поли-
полимера. Относительно просто эта задача решается в тех
случаях, когда полимеризация ведется при высоком
давлении (полиэтилен). Благодаря резкому снижению
давления в разделителе и' газообразному состоянию
мономера при обычных условиях он полностью ис-
испаряется из расплава полимера. В других процессах
для удаления мономера используют специальные уст-
устройства (шнеки, вакуум-камеры), работающие под ва-
вакуумом.
Упругость паров мономера над конц. р-ром полиме-
полимера заметно понижается с ростом концентрации полиме-
полимера согласно зависимости,
P/P0=f(CM)
где Ро — давление паров чистого мономера, Р — дав-
давление паров над р-ром, См — концентрация мономера.
Для каждой конкретной системы эта зависимость опре-
определяется эмпирически. Напр., для р-ра стирола в поли-
полистироле она имеет вид:
-?- = exp [In См + A - С„) + х A - СМ)Ц
где % = 0,4. Используя зависимости подобного типа,
можно рассчитать темп-ру и вакуум, необходимые для
достижения требуемой очистки полимера от мономера.
Удаление мономера в реальных условиях затруднено
из-за низкой интенсивности темплообмена и большого
диффузионного сопротивления расплава полимера.
В производствах среднетоннажного масштаба (сополи-
(сополимеры стирола, поливинилхлорид, полиформальдегид)
для этой цели широко применяют вакуум-экструдеры.
Для расчета параметров подобного процесса предложе-
предложена ф-ла:
С =С„оехр (--?-)
где Сщ — начальное содержание мбномера в расплаве
полимера, Сы — конечное содержание мономера,
w = (t — I) cos ср, т — время пребывания материала
в шнеке, t — шаг шнека, I — толщина слоя полимера,
w — ширина канала, ф — угол подъема винтовой ли-
линии канала.
В крупнотоннажных производствах применяют спе-
специальные .вакуум-камеры, где массообмен осущест-
897
ПОЛИМЕРИЗАЦИЯ В МАССЕ
898
вляется в тонком слое полимерного расплава, пропу-
пропущенного под давлением через трубки или пластины и
попадающего затем в расширительную камеру.
Выбор типа реактора и режима процесса
Подходящий реактор и режим процесса (периоди-
(периодический или непрерывный) выбирают в результате сов-
совместного рассмотрения след. вопросов: а) влияние па-
параметров процесса (скорости потоков, распределения
времен пребывания, градиента темп-р и концентраций
реагентов, давления) на кинетику полимеризации и
характеристику продукта в реакторах различного типа;
б) условия регулирования параметров процесса в ре-
реакторах различного типа с учетом физич. и теплофизич.
свойств среды; в) сравнение экономич. показателей
процесса.
Переход от периодич. режима полимеризации к не-
непрерывному связан с изменением ММР продукта и
нек-рым уменьшением степени превращения мономера,
а также с усложнением аппаратуры. Однако непрерыв-
непрерывный процесс позволяет получать продукты более одно-
однородные по свойствам, применять эффективные методы
автоматизации и контроля, повышать производитель-
производительность при тех же объемах реакторов. Поэтому для ре-
реакторов первых 3 типов (см. выше) явно наметилась
тенденция к переходу на непрерывные режимы и созда-
созданию высокопроизводительных, полностью автоматизи-
автоматизированных агрегатов большой мощности.
Сложность выбора оптимального типа реактора свя-
связана со многими причинами: недостаточным знанием
макрокинетики процессов, допустимостью изменений
свойств продуктов в достаточно широком диапазоне,
недостатком данных по связи структуры полимеров с их
свойствами. Это приводит к тому, что для каждого кон-
конкретного процесса существует несколько вариантов
аппаратурного оформления, обеспечивающих пример-
примерно равный экономич. эффект при сопоставимом качестве
продуктов.
Так, в производстве полиэтилена при высоком давле-
давлении используют трубчатые реакторы и автоклавы с ме-
мешалками (см. Этилена полимеры). В первом случае
большая длительность пребывания реакционной массы
в аппарате, наличие градиента темп-р по длине реак-
реактора и большая поверхность теплообмена обеспечивают
увеличение степени конверсии мономера при более
широком ММР продукта. Во втором случае теплосъем
через стенку практически равен нулю, и реактор рабо-
работает в автотермич. режиме с очень малыми временами
удерживания реакционной массы. Стационарность до-
достигается вследствие непрерывного подвода холодного
этилена. Благодаря малому градиенту темп-р и кон-
концентраций получают продукт с более узким ММР. Одна-
Однако свойства продуктов, получаемых в обоих случаях,
достаточно близки друг к другу (за счет превалирую-
превалирующего влияния разветвленности макромолекул и плот-
плотности, к-рые мало зависят от типа реактора, на техно-
логич. и физико-химич. свойства полиэтилена по срав-
сравнению с влиянием ММР).
Сказанное выше справедливо и применительно к про-
процессам получения полистирола и сополимеров стирола
методом П. в м., где применяют каскад реакторов сме-
смешения или комбинированный процесс в неизотермич.
условиях (с контролируемым или адиабатич. подъемом
темп-ры на завершающей стадии процесса). Из анализа
кинетики следует, что при непрерывном режиме поли-
полимеризации в каскаде реакторов со ступенчатым подъе-
подъемом темп-ры на каждой ступени каскада будет полу-
получаться продукт с мультимодальным ММР. Более плав-
плавное изменение температуры в реакторах колонного
типа приводит к получению продуктов с относительно
широким унимодальным ММР (см. также Стирола по-
полимеры). Оба типа продуктов достаточно близки друг
к другу по технологическим и физико-химическим
Свойствам, что оправдывает параллельное развитие
обоих типов процесса.
Применение периодич. режимов при полимеризации
в форме обусловлено необходимостью получать твердые
продукты с конечной глубиной превращения, близкой
к 100%. Этого достигают за счет относительно большо-
большого времени полимеризации. Однако, если необходимо
получить продукт в виде тонкого слоя (покрытия), то
процесс удается осуществить непрерывно и с высокой
скоростью. Так, фотоинициированная полимеризация
акрилатов с образованием сшитого полимера протекает
за несколько сек.
Технологический расчет реактора
Технологич. расчет реактора для П. в м. включает
составление материального и теплового балансов, рас-
расчет молекулярной структуры продукта на основании
кинетич. данных, а также расчет нек-рых епецифич. тех-
технологич. параметров процесса (величины частиц поли-
полимера, уд. поверхности, скорости налипания пленки по-
полимера на стенки аппаратуры и др.).
Объем, конечная глубина превращения мономера и
производительность реактора рассчитываются по кине-
кинетич. данным (на основании установленных зависимостей
скорости реакции от концентрации реагентов и темп-ры).
Оптимальная глубина превращения мономера зависит от
кинетич. констант процесса, теплоты полимеризации,
теплофизич. и гидродинамич. свойств среды, а также
от экономич, факторов. При ионной полимеризации
и сополимеризации гетероциклов методом П. в м.
(капролактам, триоксан с окисью этилена или с диоксо-
ланом) низкие теплоты полимеризации и высокие ско-
скорости процесса позволяют вести реакцию почти до
100%-ной конверсии. В то же время при полимеризации
этилена тепловой эффект и скорость реакции столь ве-
велики, а возможности теплоотвода при высоком давле-
давлении столь ограничены, что глубина превращения моно-
мономера за один проход этилена через реактор не превы-
превышает 12—20%.
При разработке технологии полимеризации и сопо-
сополимеризации стирола долгое время стремились достиг-
достигнуть глубины конверсии, близкой к 100% , т. к. в этом
случае необходимо получать продукты с предельно низ-
низким содержанием мономера. Увеличение продолжи-
продолжительности процесса на последней стадии, когда кон-
концентрация мономера минимальна, приводила к сниже-
снижению производительности. Более выгодным оказалось
вести процесс с высокой скоростью до конверсии поряд-
порядка 75—95% , а непрореагировавший мономер удалять из
продукта под вакуумом.
Среднюю степень полимеризации, ММР и степень
разветвленности (или прививки в случае сополимери-
сополимеризации) можно рассчитать на основе известной кинетич.
схемы процесса (см. также Радикальная полимериза-
полимеризация, Молекулярно-массовое распределение).
Для расчета теплопередачи в процессе П. в м. исполь-
используют эмпирич. зависимости для реакторов различного
типа в критериальной форме.
В трубчатых реакторах на начальном участке режим
течения мономера турбулентный и коэфф. теплоотдачи
а можно рассчитать по ур-нию Крауссольда:
Nu = 0,024 Re0»8 Pr°>35
где Nu—критерий Нуссельта Nu = ad/h d — диаметр,
X — коэффициент теплопроводности; Re — критерий
Рейнольдса Re = Wd/ч, W — скорость, v = —-, ;х —
коэффициент внутр. вязкости, р — плотность; Рг —
критерий Прандтля Рг = v/a, a — коэффициент тем-
температуропроводности.
При высоких степенях конверсии мономера режим
течения среды становится ламинарным. При условии
стабилизации гидродинамического ттотпкя пягппогглттл
899
ПОЛИМЕРИЗАЦИЯ В МАССЕ
900
ние скоростей по сечению трубы описывается пара-
параболическим законом Пуазейля:
где Wo — скорость в центре трубы, R — радиус трубы,
Wr — скорость в точке на расстоянии г от центра
трубы.
Для неньютоновских сред в широком интервале зна-
значений критерия Пекле Ре = Wd/a при 1<Ре -^ > 50,
где L — длина, критерий Нуссельта определяется по
эмпирич. ф-лам, к-рые приводятся в специальной лите-
литературе. Отсутствие надежных расчетных данных по
теплообмену в высоковязких средах приводит к тому,
что расчеты коэфф. теплопередачи для реакторов в ос-
основном базируются на экспериментальных данных,
получаемых при исследовании моделей и опытных
реакторов.
Более подробно изучен теплообмен в реакторах сме-
смешения. Для маловязких сред при турбулентном режиме
перемешивания коэфф. теплоотдачи можно определить
по ф-ле:
Nu = К Re0'67 Рг1/з (rjr^yM
где г]ст — вязкость у стенки реактора, К — коэффи-
коэффициент, зависящий от конструкции мешалки и геомет-
геометрии аппарата.
Для высоковязких неньютоновских сред применяют
аналогичные ф-лы; показатели степени зависят от типа
перемешивающего устройства.
Для расчета коэфф. теплоотдачи суспензии (гетеро-
(гетерогенная П. в м.) предложена след. зависимость:
ж
где ас — коэфф. теплоотдачи суспензии, аж — коэфф.
теплоотдачи жидкой фазы в данном режиме перемеши-
перемешивания, СОб— объемная доля твердых частиц, а — коэф-
коэффициент, зависящий от формы и плотности частиц; для
рыхлых частиц а = 10—15.
При расчете теплового баланса реактора важное
значение имеют вопросы динамики, устойчивости теп-
теплового режима процесса и др. При П. в м. возможности
регулирования теплового ре-
режима весьма ограничены (по
сравнению с полимеризацией в
р-ре или в суспензии) и опас-
опасность теплового взрыва вслед-
вследствие автотермич. разогрева
весьма велика.
Наглядно устойчивость вы-
выбранного температурного ре-
График зависимости теплоотвода (J)
и тепловыделения B) от темпера-
температуры процесса: QM — тепловой эф-
эффект, соответствующий максималь-
максимальной степени превращения; точки
М', М" иМ'" отвечают стационар-
стационарным состояниям работы реактора.
VV Т"
Температура Т
жима процесса можно оценить при графич. решении
ур-ний материального и теплового балансов реактора.
Скорость теплоотвода через стенку линейно возрастает
с увеличением темп-ры (см. рисунок). Зависимость теп-
тепловыделения от темп-ры имеет более сложный ха-
характер, т. к. скорость реакции возрастает экспонен-
экспоненциально. Точке М"' соответствует предельная конвер-
конверсия мономера. Возможные побочные реакции, напр, раз-
разложение мономера, в приведенной на рисунке схеме не
учтены.
Стационарный режим процесса будет достигнут в том
случае, если скорости теплоотвода и тепловыделения
будут равны (точки пересечения кривых на рисунке).
В общем случае это может произойти при 3 различных
температурах: Т', T",Tfn. Состояние, отвечающее точ-
точке М'\ неустойчиво, поскольку при незначительном
увеличении темп-ры количество выделившегося тепла
резко возрастет. Анализ этой схемы позволяет с учетом
всех ограничений выбрать режим устойчивой работы
реактора.
Математическое моделирование процесса
Нек-рые процессы П. в м. и прежде всего полимериза-
полимеризация этилена и стирола представляют большой интерес
как объекты математич. моделирования. Обычно при
разработке нового технологич. процесса полимериза-
полимеризации центр тяжести исследований приходится на опыт-
опытные установки различного масштаба, где исследуются
различные режимы работы, проверяется влияние типа
реактора и его размеров. Переход к промышленному
производству осуществляется путем последовательной
проверки процесса (и свойств соответствующего про-
продукта) на установках все более крупного масштаба.
Очевидно, что такой метод моделирования процесса
связан с большими затратами времени и средств.
Теоретически возможно заменить физич. эксперимент
на установках математич. расчетами на ЭВМ. Для это-
этого необходимо построить математич. модель процесса,
к-рая представляет собой систему дифференц. ур-ний,
описывающих полную кинетич. схему реакции, ур-ния
материального и теплового балансов реактора, гидро-
гидродинамику всех реакционных потоков и характеристику
продукта.
Тип реактора обычно учитывается в виде моделей
идеального смешения (реактор, в к-ром все
реагенты мгновенно и равномерно перемешиваются по
всему объему; отсутствует градиент темп-р) и иде-
идеального вытеснения (в любом поперечном
сечении, нормальном к движению жидкости, объемная
скорость и свойства жидкости однородны; диффузия
пренебрежимо мала).
Реальные реакторы не могут быть описаны с помо-
помощью идеальных моделей из-за наличия различных от-
отклонений: температурных градиентов, диффузии и т. д.
Поэтому при моделировании используют различные
усложненные модели. Так, для описания процесса те-
течения в трубчатом аппарате используют «диффузион-
«диффузионную» модель или модель «последовательно соединенных
аппаратов идеального смешения». В первом случае
учитывают отклонение от идеальности, вызванное диф-
диффузией в направлении движения потока и в попереч-
поперечном (радиальном) направлении, к-рая описывается за-
законом Фика:
дС дЮ
dt ~ дх2
где D — коэфф. молекулярной диффузии, различный
для продольного и радиального направления, х — ли-
линейный размер. Вторая модель рассматривает дискрет-
дискретное изменение концентрации реагентов при переходе
от одного элементарного объема (реактора смешения)
к другому и особенно удобна при моделировании про-
процесса с помощью ЭВМ.
Реакторы смешения описывают с помощью «смешан-
«смешанных» моделей. В этом случае реальный аппарат рас-
рассматривают как совокупность нескольких взаимосвя-
взаимосвязанных моделей течения потока. При построении по-
подобной модели используют след. типы течения среды:
поток идеального смешения, байпасный поток (часть
жидкости, к-рая движется как бы параллельно реакто-
реактору), застойная зона и др. Удельный вес каждой этой
модели в суммарной определяют методом последова-
последовательного приближения, используя динамич. характе-
характеристику реактора — кривую распределения времен
пребывания.
901
ПОЛИМЕРИЗАЦИЯ В РАСТВОРЕ
902
Математич. моделирование позволяет для нек-рых
полимеризационных процессов с хорошо изученной ки-
кинетикой и механизмом достаточно точно рассчитать
ММР и конверсию мономера для выбранного тина ре-
реактора.
Достоинства и недостатки метода
Широкое использование в пром-сти П. в м. обуслов-
обусловлено несколькими причинами: а) высокой степенью
чистоты получаемых полимеров (отсутствуют загрязне-
загрязнения, привносимые растворителями или диспергирую-
диспергирующими агентами при полимеризации в р-ре и эмульсии);
б) отсутствием стадии обработки полимера с целью
удаления полимеризационной среды; в) отсутствием
стадии сушки продукта; г) наибольшими потенциаль-
потенциальными возможностями для интенсификации процесса
(концентрация мономера максимальна, следовательно,
полимер с заданной молекулярной массой можно по-
получать при наибольших температурах и концентраци-
концентрациях возбудителя процесса); д) возможностью использо-
использовать в большинстве случаев непрерывные режимы по-
полимеризации, что обеспечивает высокую производи-
производительность метода.
В то же время при П. в м. ограничены возможности
регулирования мол. структуры полимера путем моди-
модификации свойств реакционной среды. Значительную
трудность представляет задача равномерного распреде-
распределения возбудителя полимеризации и других гетероген-
гетерогенных добавок.
Трудности, связанные с плохим тепло- и массообме-
ном в высоковязких средах, в большинстве случаев пре-
преодолимы за счет использования неизотермич. режимов
и повышенных темп-р. В связи с этим П. в м. обычно
применяют в тех случаях, когда допустимы относитель-
относительно широкие ММР продуктов.
П. в м. более экономична, чем полимеризация в р-ре
и эмульсии. Этим объясняются попытки использовать
ее в пром-сти в еще больших масштабах. Ок. 75% ми-
мирового производства полиэтилена получают П. в м.
под высоким давлением. Постоянно растет доля П. в м.
в процессах синтеза полистирола и сополимеров сти-
стирола. Этим способом в основном получают полиметил-
метакрилат, поликапролактам, полиформальдегид (из
триоксана). В то же время в производстве поливинил-
хлорида жесткие требования к температурным услови-
условиям процесса в значительной мере ограничивают при-
применение П. в м.; этим способом получают только неко-
некоторые марки поливинилхлорида. Полимеризация в мас-
масле ограниченно применима в производстве эластоме-
эластомеров, где также необходима тонкая регулировка темпе-
температуры процесса.
Лит.: ГладышевГ. П., Г и б о в К. М., Полимери-
Полимеризация при глубоких степенях превращения и методы ее иссле-
исследования, Алма-Ата, 1968; North A. M., Diffusion, control
of homogeneous free-radical reactions, в сб.: Progress in high
polymers, v. 2, L., 1968, p. 95; Денбиг К. Г., Теория хи-
химических реакторов, пер. с англ., М., 1968; Л е в е н-
шпиль Д., Инженерное оформление химических процессов,
пер. с англ., М., 1969; Штербачек 3., Тауск П.,
Перемешивание в химической промышленности, пер. с чеш.,
Л., 1963. С. А. Вольфсон.
ПОЛИМЕРИЗАЦИЯ В РАСТВОРЕ (solution poly-
polymerization, Losungspolymerisation, polymerisation en
solution) — способ проведения полимеризации, при
к-ром исходный мономер находится в жидкой фазе в ра-
растворенном виде. Реакционная система м. б. гомогенной
(образующийся полимер растворим в реакционной сре-
среде) или гетерогенной [полимер и (или) возбудитель по-
полимеризации образуют отдельную твердую или жид-
жидкую фазу]. Мономер растворяют в реакционной среде
заранее или подают в реакционное устройство непо-
непосредственно из газовой фазы. В последнем случае он
растворяется в реакционной среде, где находится
возбудитель полимеризации, и одновременно вступает
в реакцию.
Механизм П. в р. может быть радикальным, ион-
ионным или координационно-ионным. Температурный ин-
интервал процесса определяется областью, в к-рой моно-
мономер и растворитель существуют в жидком состоянии.
Повышение температуры процесса достигается также
увеличением давления, т. е. повышением точки ки-
кипения раствора.
Особенности кинетики процесса. Гомогенная П. в р.
описывается классич. ур-ниями кинетики. При гетеро-
гетерогенной П. в р. возможны диффузионные затруднения
для поступления молекул мономера к активным цент-
центрам. Частным случаем гетерогенной является гетеро-
фазная П. в р., когда рост цепей происходит одновре-
одновременно в р-ре и осажденном полимере.
Диффузионные процессы играют значительную роль
и в том случае, когда газообразный мономер под
нек-рым избыточным давлением поступает в реактор,
где находится р-р или суспензия катализатора в инерт-
инертном растворителе. Избыточное давление м. б. самым
различным — от десятых долей кн/м2 (нескольких
мм рт. ст.), как при полимеризации формальдегида,
до нескольких Мн/м2 (десятков кгс/см2), как при поли-
полимеризации этилена и пропилена.
В зависимости от соотношения скоростей подвода
мономера, его растворения и полимеризации процесс
может протекать в кинетической, диффузионной или
переходной области, что существенно влияет на кине-
тич. закономерности процесса и мол. массу продукта.
В процессах, где самой медленной стадией является
диффузия мономера, общая скорость при стационарном
режиме описывается след. ур-нием:
^ = P(c?-cM) = MCM]sm!/ A)
где v — скорость процесса, моль - л~г • сек~г, Р — эффек-
эффективный коэфф. массопередачи, молъ-м~2-сек~г, С^ —
растворимость мономера в жидкой фазе, моль-л'1,
Сш — текущая концентрация мономера в р-ре, моль-л'1,
к — эффективная константа скорости полимеризации,
А* — концентрация активных центров в р-ре, моль-л'1,
х, у — порядок реакции по мономеру и возбудителю
полимеризации соответственно.
Если в A) считать концентрацию активных центров
постоянной, обозначить /с[<4*]^ через &* и принять, что
х = 1, то после преобразования A) получаем, что на-
наблюдаемая скорость полимеризации равна:
" = ^С? = *ЭффС? B)
Ф-ла B) принимает простой вид в двух предельных
случаях, соответствующих протеканию процесса в ки-
кинетической и диффузионной областях. При /с*<^|3 полу-
получаем, что &Эфф ~ к* и См = Cjj. В последнем случае
скорость наблюдаемого процесса всецело определяется
истинной кинетикой П. в р. и не зависит от условий
диффузии. То же следует сказать о молекулярной мас-
массе и молекулярно-массовом распределении (ММР)
продукта.
В др. предельном случае к*> Р и кЭфф — J3; при этом
^м^^м и скорость наблюдаемого процесса всецело
определяется скоростью диффузии. В диффузионной
области скорость П. в р. всегда имеет первый порядок
по мономеру, энергия активации реакции равна энер-
энергии активации диффузии; активность и концентрация
возбудителя полимеризации влияют на скорость про-
процесса незначительно.
В общем случае ионной полимеризации в диффузи-
диффузионной области молекулярная масса полимера опреде-
определяется механизмом передачи цепи на примеси (или
регуляторы). Если передачи цепи нет, то переход про-
процесса из кинетической области в диффузионную бу-
будет приводить к снижению молекулярной массы про-
продукта из-за снижения См.
903
ПОЛИМЕРИЗАЦИЯ В РАСТВОРЕ
904
Для перевода процесса в кинетич. область необхо-
необходимо повысить скорость диффузии мономера по срав-
сравнению со скоростью полимеризации. Этого достигают,
повышая давление газообразного мономера, увеличивая
поверхность контакта газа с жидкой фазой (т. е. эффек-
эффективность перемешивания), уменьшая темп-ру (т. е.
понижая скорость реакции в большей степени, чем ско-
скорость диффузии), снижая концентрацию возбудителя
полимеризации. Кинетич. области процесса соответст-
соответствуют условия, когда скорость полимеризации зависит от
концентрации возбудителя и не зависит от эффектив-
эффективности перемешивания при варьировании этих перемен-
переменных в нек-ром интервале значений.
Если в жидкую фазу из газообразной поступает не-
несколько компонентов (сомономеры при сополимериза-
ции, регуляторы роста цепи), то может оказаться, что
реакционная система находится в кинетич. области от-
относительно одного компонента и в диффузионной отно-
относительно другого.
Аппаратурное оформление процесса. В лаборатор-
лабораторной практике П. в р. проводят в дилатометрах, ампу-
ампулах, колбах и т. д. При использовании «вещественных»
возбудителей полимеризации необходимо тщательно пе-
перемешивать реакционную смесь. В пром-сти П. в р.
проводят в вертикальных и горизонтальных емкостных
аппаратах, оборудованных перемешивающими устрой-
устройствами различного типа (мешалками, насосами, шне-
шнеками и др.). Реже используют аппараты трубчатого
или колонного типа, работающие по принципу вытесне-
вытеснения. Периодич. процессы осуществляют обычно в еди-
единичных реакторах объемом до нескольких м3, часто в не-
изотермич. (переменных) темп-рных режимах. Для
ведения непрерывных процессов используют каскады
последовательно соединенных аппаратов, работающих
при одинаковых или различных темп-pax. Такая тех-
нологич. схема обусловлена малыми степенями превра-
превращения реагентов в одном реакторе смешения непрерыв-
непрерывного действия.
П. в р. с использованием газообразных мономеров
осуществляют в реакторах смешения полупериодич.
или непрерывного действия. В первом случае в реак-
реактор периодически загружают растворитель и возбуди-
возбудитель полимеризации, а мономер подают непрерывно
с постоянной скоростью до получения расчетного коли-
количества продукта, после чего процесс прерывают.
Тепловой режим в реакторе регулируют, используя
теплообмен через рубашку аппарата, встроенные зме-
змеевики или специальные поверхности, в к-рых циркули-
циркулирует теплоноситель. Более эффективный способ под-
поддержания изотермич. режима — отвод тепла за счет
испарения части растворителя и (или) мономера из ре-
реакционной зоны (т. наз. автотермический режим про-
процесса). Таким способом ведут П. в р. этилена, пропиле-
пропилена, изобутилена, характеризующиеся большими теп-
тепловыми эффектами. Темп-ру процесса в этом случае
можно регулировать, подбирая растворитель с опреде-
определенной темп-рой кипения (используют, напр., смеси
растворителей) или соответствующим образом регули-
регулируя давление в реакторе. Другой распространенный
способ поддержания автотермического режима в ре-
реакторе непрерывного типа — использование тепла
реакции на подогрев исходной смеси в реакторе сме-
смешения.
Образовавшийся продукт отделяют от реакционной
среды (там, где это требуется) обычными для химич.
технологии методами. При гетерогенной П. в р.— это
фильтрация, центрифугирование, реже отгонка раст-
растворителя под вакуумом или перегонка с острым па-
паром. Следы катализатора и растворителя полимера
удаляют из полимера путем промывки легколетучими
растворителями или водой. Порошкообразные продук-
продукты сушат в сушилках различного типа (вакуумных,
в псевдоожиженном слое и др.).
Как правило, продукты, полученные в результате
ионной П. в р., содержат сорбированные или химиче-
химически связанные катализаторы, к-рые отрицательно влия-
влияют на физико-химич. свойства полимеров. Поэтому ката-
катализатор, содержащийся в продукте, разлагают или нейт-
нейтрализуют с помощью какого-либо химич. агента и за-
затем отделяют путем промывки водой или растворите-
растворителем. Такая обработка полимеров требует значительных
затрат и повышает себестоимость продукции. Возмож-
Возможный способ преодоления этих трудностей — подбор
высокоактивных катализаторов, которые можно при-
применять в столь малых концентрациях, что они, ос-
оставаясь в продукте, не окажут влияния на свойства
полимеров.
Расчет реактора. При расчете реакторов смешения,
предназначенных для проведения П. в р., обычно
используют модель аппарата «идеального смешения».
В первом приближении это означает, что молекулы,
подведенные ко входу в реактор, в след. момент с рав-
равной вероятностью могут оказаться в любой точке реак-
реакционного объема. Отсюда следует, что состав смеси
на входе в реактор претерпевает мгновенное измене-
изменение и на выходе из реактора состав такой же, как и во
всем объеме. В реальных полимеризационных реакто-
реакторах при малой вязкости среды и интенсивном переме-
перемешивании отклонения от идеальной модели невелики.
Критерием «идеальности» для реактора непрерывного
действия является распределение элементов среды
по временам пребывания (динамич. характеристика
реактора). Для модели «идеального смешения» распре-
распределение по временам пребывания представляет собой
экспоненциальную зависимость:
Х = Хое-*'Г C)
где X — концентрация индикатора (нейтральное ве-
вещество, используемое для анализа режима работы ре-
реактора) на выходе из реактора, Хо — начальная кон-
концентрация индикатора, введенного в реактор практи-
практически мгновенно (т. наз. импульсный ввод), t — время,
соответствующее X, т — среднее время пребывания
в реакторе (т = V/G, где V — объем жидкой фазы в ре-
реакторе, G — объемная скорость движения потока через
реактор при стационарном режиме).
Изменяя режим работы перемешивающего устройст-
устройства, объем жидкой фазы, конструкцию аппарата, во
многих случаях можно добиться удовлетворительного
приближения к режиму идеального смешения. Это по-
позволяет с достаточной точностью при кинетич. расчете
пользоваться значением среднего времени пребывания
реакционной массы в аппарате. Существуют прибли-
приближенные методы, позволяющие описать «неидеальный»
аппарат с помощью набора идеальных моделей, рабо-
работающих последовательно и параллельно. Прямой рас-
расчет кинетики процесса на основании кривой распреде-
распределения по временам пребывания для реакций не нуле-
нулевого н не первого порядков — неопределенная задача,
если не определен характер массообмена между эле-
элементарными объемами внутри реактора.
Для кинетич. расчета проточного реактора смеше-
смешения необходимо располагать ур-ниями брутто-кинетики
процесса (ур-ниями скорости и средней степени полиме-
полимеризации как функциями выхода полимера или концент-
концентрации мономера). Для расчета ММР продукта необхо-
необходимо иметь более полную кинетич. информацию о меха-
механизме процесса (см. Молекулярно-массовое распределе-
распределение). Расчет производительности реактора основан на
решении ур-ний материального баланса. Для стацио-
стационарного режима:
GM0 = GMX + VW D)
где Мо и М1 — соответственно молярная концентрация
мономера на входе и выходе из реактора, W — ско-
скорость расхода мономера, определяемая кинетич.
905
ПОЛИМЕРИЗАЦИЯ В РАСТВОРЕ
906
ур-нием. Аналогичное ур-ние материального баланса
можно составить для возбудителя полимеризации.
Третье ур-ние можно получить для продукта, однако
оно будет зависимым. Поскольку в результате полиме-
полимеризации плотность реакционной смеси р изменяется,
часто удобно ур-ние материального баланса выразить
через скорость превращения. Для мономера имеем:
mG0
Ро
E)
= Go — Мх + mVW
Pi
где ро и Pi — соответственно начальная и конечная
плотность среды, т — мол. масса мономера, Go — ско-
скорость движения потока на входе в реактор. Совместное
решение ур-ний материального баланса для мономера,
возбудителя полимеризации и соответствующих кине-
тич. ур-ний скорости расхода этих реагентов позволя-
позволяет при заданных значениях скорости подачи и объ-
объема реактора рассчитать его производительность и на-
наоборот.
Для реакции первого порядка, скорость к-рой равна
dM/dt — кэффМ, стационарная концентрация М1 (из
ф-лы 4) определяется выражением:
Чем больше т, тем ниже Мъ тем больше выход поли-
полимера, но ниже скорость превращения и, следовательно,
производительность реактора.
Для расчета теплового режима реактора (см. Поли-
Полимеризация в массе) решается ур-ние теплового баланса.
Скорость выделения тепла определяется тепловым
эффектом А# и скоростью реакции:
— = — AHVW
dt
G)
Подставляя Мх из ф-лы F) и учитывая зависимость
скорости П. в р. от темп-ры, получаем
02*
dt
-E/RT
VAH
(8)
При т = const ddu/dt растет с увеличением темп-ры
до тех пор, пока длительность реакции не станет мень-
меньше т.
Для высоковязких сред нужно учитывать также теп-
тепло, образующееся в результате перехода механич.
энергии в тепловую при перемешивании среды (тепло
дессипации). Эффективное управление тепловым ре-
режимом реактора возможно тогда, когда производная
от скорости отвода тепла по температуре выше анало-
аналогичной производной для скорости выделения тепла.
Это необходимое условие устойчивости режима рабо-
работы аппарата.
При непрерывном режиме П. в р. каскад последова-
последовательно соединенных реакторов смешения имеет мень-
меньший суммарный объем, чем одноступенчатый реактор,
при одинаковой производительности и степени прев-
превращения. Для расчета каскада реакторов смешения
составляют материальный баланс каждой ступени. Су-
Существуют специальные графич. методы расчета числа
ступеней каскада изотермич. реакторов смешения для
достижения заданной производительности при извест-
известном объеме единичного реактора. В общем случае при-
применяют численные решения, используя ЭВМ. Хотя эко-
номич. эффект (выигрыш в капитальных затратах) бу-
будет снижаться при очень большом числе ступеней, на
практике для медленных процессов П. в р. используют
каскады из 20 и более реакторов смешения (напр.,
при синтезе каучуков).
Принципиальное отличие расчета полимеризацион-
ного реактора от расчета обычного химич. реактора
состоит в необходимости учета мол. массы, ММР и
структурных параметров образующегося продукта.
Мол. масса продукта в одноступенчатом реакторе сме-
смешения проточного типа определяется стационарной
концентрацией реагентов согласно ур-нию Р =
= f([M],[A*]) для конкретного механизма П. в р.
и темп-рой. Для каскада реакторов смешения Р полу-
получают путем усреднения выходов и мол. масс для всех
ступеней каскада. ММР полимера рассчитывают на
основании кинетич. модели П. в р. с учетом элементар-
элементарных стадий. Для одноступенчатого реактора смешения
механизм, включающий быстрое инициирование и
медленный обрыв цепи, будет приводить к получению
полимера с относительно широким ММР (большому
значению отношения Mw/Mn). Наоборот, медленное
инициирование и быстрый обрыв цепи будут приводить
к относительно узкому ММР. При образовании живущих
полимеров в реакторе периодич. действия получают
продукт с отношением Mw/Mn близким к единице.
Расчет процесса для реактора смешения проточного
типа дает MJМп= 2. Сужения ММР можно достичь,
применяя каскад последовательно соединенных реак-
реакторов смешения. В этом случае Му/Мп =1-\——, где
п — число реакторов в каскаде.
При кинетич. расчете реактора смешения периодич.
действия выход продукта определяют, интегрируя по
времени ур-ние скорости реакции, а конечную мол.
массу — интегрируя ур-ние зависимости степени поли-
полимеризации от времени (или выхода продукта). Часто
в периодич. процессах темп-ру повышают по мере рас-
расходования мономера, чтобы скомпенсировать снижение
скорости реакции. В этом случае вышеуказанные
ур-ния решают совместно с ур-ниями зависимости выхо-
выхода и мол. массы от темп-ры.
Расчет реактора вытеснения непрерывного действия
(трубчатого или колонного типа) также основан на
интегрировании по времени (или по длине реактора,
что то же самое) ур-ний скорости реакции, средней
степени полимеризации совместно с ур-ниями тепло-
теплового баланса (ур-ниями скорости выделения и отво-
отвода тепла). Модель «идеального вытеснения» предпола-
предполагает в первом приближении допущение, что среда в
трубе движется подобно поршню. Перемешивание раз-
различных элементов среды в направлении движения по-
потока отсутствует. Реальные аппараты описываются с
помощью более сложных моделей, учитывающих ради-
радиальную и осевую диффузию и т. д. (см. Полимеризация
в массе).
При расчете реактора смешения, в к-ром происхо-
происходит растворение газообразного мономера и его П. в р.,
суммарная скорость превращения мономера будет опре-
определяться скоростями 3 различных процессов: подвода
газообразного мономера на границу раздела жидкость —
газ, диффузии и полимеризации. Расчет режима ра-
работы реактора в кинетич. области ведется так же,
как указано выше. При работе в диффузионной области
следует различать 2 случая: 1) весь мономер, поступаю-
поступающий в реактор, растворяется и реагирует; 2) часть
мономера не успевает раствориться и поступает в
рецикл. Скорость полимеризации и производительность
реактора в последнем случае будут определяться ско-
скоростью диффузии.
Применение метода, его достоинства и недостатки.
П. в р. широко используют в лабораторной практике
для изучения кинетич. закономерностей и механизма
полимеризации. При этом процесс ведут в разб. р-рах
мономеров до небольших глубин превращения, чтобы
избежать усложняющего влияния высоких концентра-
концентраций полимера на кинетику процесса (см. также Гель-
эффект, Полимеризация в массе). Влияние природы ра-
растворителя на механизм процесса зависит от природы
возбудителя полимеризации и мономера. Обычно вы-
выбирают растворители, химически нейтральные по отно-
отношению к мономеру, однако их физико-химич. свой-
свойства могут в значительной степени влиять на кине-
907
ПОЛИМЕРИЗАЦИЯ В ЭМУЛЬСИИ
•908
тику процесса, ММР и структуру получаемых полиме-
полимеров. Реакционная система может содержать различные
модифицирующие добавки, влияющие на активность
возбудителя процесса, мономера или на физич. свойства
полимера.
В пром-сти способ П. в р. имеет преимущество по
сравнению с др. методами полимеризации (в массе,
эмульсии, суспензии) с точки зрения легкости управ-
управления процессом. При П. в р. можно осуществлять
тонкую регулировку темп-ры реакции вплоть до глубо-
глубоких степеней превращения мономера, регулировать мол.
массу продукта в широких пределах. Возможный интер-
интервал изменения параметров процесса и свойств реак-
реакционной системы при П. в р. значительно шире, чем
при др. методах полимеризации.
В то же время разбавление мономера растворите-
растворителем приводит к снижению скорости реакции и уменьше-
уменьшению мол. массы продукта (вследствие передачи цепи
на растворитель и на примеси, содержащиеся в нем).
К недостаткам процесса относится также необходимость
дополнительных затрат на подготовку растворителя,
отделение и регенерацию полимеризационной среды,
промывку и сушку продукта.
При радикальном инициировании и крупнотоннаж-
крупнотоннажном производстве полимеров способ П. в р. экономиче-
экономически наименее выгоден по сравнению с полимеризацией в
массе и в дисперсных водных системах. При средне- и
малотоннажном производстве П. в р. используют там,
где конечный продукт применяют в виде р-ра (лак,
клей, связующее в производстве пластмасс) или в тех
случаях, когда др. методы не позволяют получать про-
продукты требуемой структуры (нек-рые полиакрилаты,
поливинил ацетат, пенополистирол, политетрафторэти-
политетрафторэтилен и др.).
Значительно более важную роль П. в р. играет при
осуществлении ионных и координационно-ионных про-
процессов. Это важнейший и практически единственный
промышленный способ проведения полимеризации на
гетерогенных каталитич. системах (в частности, на ка-
катализаторах типаЦиглера—Натта). При гомогенном ка-
катализе полимеризации высокая активность .каталитич.
систем позволяет осуществлять промышленные процес-
процессы с достаточно высокой скоростью и в разб. р-рах моно-
мономеров. Возможность эффективного контроля параметров
процесса обеспечивает способу П. в р. ив этом случае
преимущества по сравнению с полимеризацией в мас-
массе и неводных дисперсиях (синтез бутилкаучука и
бутадиен-стиролъных каучуков на литийорганич. ката-
катализаторах, полимеризация этилена и пропилена на
растворимых каталитич. системах, полимеризация изо-
бутилена и др.). Полимеризация в массе технически
целесообразна при низких значениях теплового эффек-
эффекта (напр., при получении полимера из триоксана, капро-
лактама и др. малонапряженных гетероциклов).
Лит.: X у в и н к Р., Ставерман А. [сост.], Химия
и технология полимеров, пер. с нем., т. 1—2, М.— Л., 1965—
1966; Крамере X., Вестертерп К., Химические
реакторы, пер. с англ., М., 1967; Кристаллические полиоле-
фины, под ред. Р. А. Раффа и К. В. Дака, пер. с англ., т. 1,М.,
1970; Берлин А. А., Вольфсон С. А., Кинетический
метод в синтезе полимеров, М., 1973. С. А. Вольфсон.
ПОЛИМЕРИЗАЦИЯ В ЭМУЛЬСИИ — см. Эмуль-
Эмульсионная полимеризация.
ПОЛИМЕРЦЕМЕНТ (polymer cement, Polymerze-
ment, ciment resineux) — материал на основе компози-
композиционного связующего, включающего органич. полимер
и неорганич. вяжущее вещество. Органич. компонентом
П. служат водные дисперсии поливини л ацетата, нату-
натуральный и синтетич. (бутадиен-стирольный, хлоропре-
новый) латексы, водорастворимые эпоксидные, поли-
полиэфирные, фурановые и карбамидные смолы, эфиры цел-
целлюлозы и др. В качестве неорганич. вяжущих исполь-
используют портландцемент, глиноземистый и магнезиальный
цемент, жидкое стекло, гипс, известь.
Различают полимерцементные бетоны, строительные
р-ры и отделочные составы. При получении бетонов и
р-ров используют грубодисперсные наполнители (за-
(заполнители): для бетонов — обычно песок и щебень, для
р-ров — песок. Наполнителями в отделочных составах
служат тонкодисперсные материалы — мел, каменная
мука, молотый песок, асбест, тальк. Для получения
цветных отделочных составов применяют органич. и
неорганич. пигменты (см. Красители).
П. получают, смешивая цемент и наполнитель с вод-
водной дисперсией полимера в обычных или вибросмеси-
вибросмесителях (см. Смесители). Иногда П. приготовляют смеше-
смешением цемента, воды и мономера (напр., метилметакрила-
та, акриловой к-ты). Режим твердения П. определяется
видом материала или изделия. Так, бетоны в течение
первых 3—5 сут выдерживают во влажной среде (поли-
(поливают водой или хранят под слоем влажных опилок),
а затем 14—42 сут при нормальных условиях. Отделоч-
Отделочные составы твердеют на воздухе при обычных темп-рах
в течение 1—2 сут, при использовании сушки ИК-лу-
чами — в течение 10—30 мин. В отдельных случаях до-
допустимо твердение П. при 80 °С и относительной влаж-
влажности воздуха 100%; продолжительность процесса 10—
15 ч.
Для предохранения водных дисперсий полимеров от
коагуляции в результате взаимодействия с цементом
применяют стабилизаторы, обеспечивающие т. наз. вре-
временную A0—30 мин после смешения всех компонентов)
или постоянную (вплоть до схватывания цемента) ста-
стабильность системы. В первом случае используют поташ,
соду, жидкое стекло, фосфаты Na и К A—5% от
массы полимера), во втором — комплексный стабили-
стабилизатор E—10%), представляющий собой смесь неионо-
генного поверхностно-активного вещества (например,
продукт ОП-7) и амфотерного гидрофильного кол-
коллоида (казеинат аммония, гидролизованный костный
клей и др.).
Выбор органич. компонента П. определяется в
первую очередь рН водной суспензии вяжущего. Так,
в средах с рН > 7 (портландцемент, глиноземистый
цемент) наиболее пригодны стабилизированные водные
дисперсии термопластичных полимеров, а также термо-
реактивные олигомеры (напр., резорцино-формальде-
гидные или эпоксидные смолы) или мономеры (напр.,
фуриловый спирт), отверждающиеся в щелочной среде
в присутствии соответствующих отвердителей. В тех
случаях, когда неорганич. вяжущее имеет рН < 7
(напр., магнезиальный цемент, полуводный гипс), чаще
всего применяют олигомеры, отверждающиеся в кислой
среде (напр., карбамидные или феноло-формальдегид-
ные смолы); иногда эти олигомеры отверждаются в ука-
указанных условиях без отвердителя.
Свойства материалов и изделий на основе П. зави-
зависят от вида и соотношения органич. и неорганич.
компонентов связующего, типа заполнителя, соотноше-
соотношения (по массе) между водой и цементом (т. наз. водо-
вяжущего отношения), метода приготовления компози-
композиции, режима формования изделия и условий его тверде-
твердения. Для П., содержащих водорастворимые полимеры,
оптимум свойств достигается при отношении массы по-
полимера к массе неорганич. вяжущего вещества, равном
0,005—0,02; для П., получаемых на основе водных дис-
дисперсий полимеров,— при 0,05—0,2.
Прочность полимерцементного бетона при сжатии
составляет обычно 30—50 Мн/м2 C00—500 кгс/см2), в
отдельных случаях — до 100 Мн/м2 A000 кгс/см2);
прочность при изгибе — 8—21 Мн/м2 (80—210 кгс/см2).
Прочность П. на основе латексов каучука меньше, чем
у П. на основе дисперсий поливинилацетата: напр.,
прочность сцепления при сдвиге составляет соответ-
соответственно 3 и 8 Мн/м2 C0—80 кгс/см2). В случае при-
применения бутадиен-стирольных латексов прочность П.
возрастает с увеличением содержания стирола в сопо-
909
ПОЛИМЕРЫ В АВИАСТРОЕНИИ
910
лимере. П. на основе латексов выдерживает 300 цик-
циклов замораживания и оттаивания. Он стоек в водных
и в слабокислых средах (в отличие от поливинилаце-
татного П.)- Усадка и набухание бетонов на основе
П. больше, чем у обычных бетонов. Введение полиме-
полимера в несколько раз повышает стойкость бетона к удару
и снижает модуль его упругости.
П. применяют гл. обр. для отделочных работ. В
механосборочных цехах, в радио- и электротехнич.
производстве из материалов на основе П. устраивают
сплошные полы (см. Покрытия для полов). Строитель-
Строительные р-ры на основе П. используют для приклеивания
керамич. плитки, выравнивания бетонных поверхно-
поверхностей, заделки стыков между бетонными конструкцион-
конструкционными элементами. Для наружной и внутренней отделки
зданий применяют полимерцементные краски и шпат-
шпатлевки. П. используют также для целей гидро- и масло-
изоляции, защиты стальной арматуры в легких и си-
силикатных бетонах от коррозии, а также при изготов-
изготовлении частей железобетонных конструкций, подверга-
подвергающихся растяжению.
Лит.: ЧеркинскийЮ. С, Полимерцементный бетон,
М. 1960; О h a m a Y., Study on properties and mix proportio-
proportioning of polymer-modified mortars for buildings, Tokyo, 1973 (на
япон. яз.). См. также лит. при ст. Полимербетон.
Ю. С. Черкинский.
ПОЛИМЕРЫ (polymers, Polymere, polymeres) —
высокомолекулярные соединения (В. с), молекулы
к-рых (макромолекулы) состоят из большого числа
одинаковых группировок, соединенных химич.связями.
Часто, однако, П. называют также и В. с, цепи к-рых
состоят из различных нерегулярно повторяющихся
групп, напр, статистические синтетич. сополимеры,
биополимеры (белки, нуклеиновые кислоты). См. также
Высокомолекулярные соединения, Макромолекула.
ПОЛИМЕРЫ В АВИАСТРОЕНИИ (polymers in
aircraft construction, Polymere im Flugzeugbau, poly-
polymeres dans construction aeronautique).
Содержание:
Введение 909
Реактопласты 910
Термопласты 912
Пено- и сотопласты 913
Резины 913
Герметики 914
Клеи 915
Лакокрасочные материалы 915
Введение. Полимерные материалы стали играть за-
заметную роль в авиастроении в 50-е гг., когда поя-
появились планеры и легкие самолеты, изготовленные
почти полностью из стеклопластика. Достигнутое бла-
благодаря этому значительное уменьшение массы самоле-
самолетов (до 50%) позволило существенно увеличить их по-
полезную нагрузку и дальность полетов. Интерес к са-
самолетам, изготовленным целиком из пластмасс, вызван
также их минимальной доступностью для обнаружения
радиолокаторами.
Целесообразность применения полимерных материа-
материалов в авиастроении обусловлена их легкостью, ва-
риабильностью состава и строения и, следовательно,
широким диапазоном технич. свойств. За период
1940—70 число авиационных деталей из полимерных
материалов увеличилось от ~25 до 10 000.
В тяжелых самолетах доля полимеров среди др.
конструкционных материалов сравнительно невелика.
Напр., в дозвуковых пассажирских самолетах использу-
используется в среднем 2% композиционных материалов на осно-
основе реактопластов, 4% термопластов и 1% резин (в
расчете на массу летательного аппарата). Еще меньше
их доля в сверхзвуковых самолетах. Все же благодаря
применению полимерных материалов масса основных
элементов самолетов указанных типов уменьшается на
10-40%.
Наибольший прогресс в использовании полимерных
материалов достигнут при создании легких самолетов
и вертолетов. Тенденция к все более широкому их при-
применению характерна также для производства ракет и
космич. аппаратов, в которых масса деталей из поли-
полимерных материалов может составлять ~ 50% общей мас-
массы аппарата.
Реактопласты. Широкое применение в авиастроении
армированных пластиков обусловлено прежде всего
их высокой уд. прочностью, а также термостойкостью.
Первые попытки применить стеклопластик вместо ме-
металла в конструкции передней части авиационных
реактивных двигателей, детали к-рых подвержены
длительному воздействию темп-р от 100 СС до 300 °С,
относятся к началу 50-х гг. Первоначальные разработ-
разработки ограничивались газотурбинными двигателями
самолетов вертикального взлета и посадки, для к-рых
увеличение тяговооруженности (отношение тяги к мас-
массе) особенно важно. Согласно расчетам, при замене
металла на углеродо- и боропластик тяговооруженность
подъемных авиационных двигателей удастся повысить
до 4 кн/кг D00 кгс/кг). Значительный эффект м. б. полу-
получен и в маршевых реактивных двигателях.
Наибольшее внимание уделяется применению арми-
армированных пластиков при разработке таких силовых
агрегатов, как оперение, крыло, фюзеляж. К 1980 долю
пластиков в дозвуковых тяжелых самолетах предпо-
предполагается довести до 25% (по массе).
Перспективны для авиастроения прессматериалы,
наполненные нитевидными монокристаллами («усами»)
графита, сапфира, карбидов кремния и бора, обла-
обладающими очень высокой прочностью и жесткостью (см.
Наполнители пластмасс). Изделия из таких материа-
материалов могут успешно сочетать функции несущих силовых
элементов и тепловой защиты. Напр., фирма «Филко»
(США) использовала фенопласты, наполненные «уса-
«усами» сапфира, для изготовления стенок камеры сгора-
сгорания и сопла ракеты, работающей на топливе N2O4—
гидразин.
Разрабатывается технология получения композиций
с регулируемым расположением «усов». Изучаются
возможности применения изотропных прессматериалов,
поскольку даже при неориентированном расположении
«усов», напр, в фенопластах, отмечается увеличение
прочности на 20—50%. Из изотропных материалов
можно изготовлять точным литьем детали небольших
двигателей, гироскопов и др.
По объему использования в силовых элементах ле-
летательных аппаратов первое место среди армирован-
армированных пластиков принадлежит стеклопластикам. Напр., в
конструкции самолета F-3 фирмы «Мак-Донне л» (США)
из общей массы деталей из этих материалов, равной
5040 кг, на долю эпоксидных пластиков приходится
2900 кг, фенольных —1360 кг, полиэфирных (диаллил-
фталатных) —72 кг, полиимидных —9 кг. Одна из при-
причин широкого применения эпоксидных пластиков, по-
помимо их высокой прочности,— возможность изготов-
изготовлять из них детали при сравнительно небольшом давле-
давлении. Благодаря этому из таких материалов методом
прессования можно получать не только небольшие
изделия — лопатки компрессоров, кронштергаы, крыш-
крышки лючков и др., но и крупногабаритные элементы —
створки контейнеров, колеса, каркасы рулей, обтекате-
обтекатели, панели крыльев и фюзеляжа.
Способом намотки из стеклопластиков в США изго-
изготовляют корпуса ракет типа «земля-воздух», стратегич.
баллистич. ракеты «Минитмен» (из стеклопластика вы-
выполнена их третья ступень) и «Поларис». В резуль-
результате применения стеклопластика вместо легирован-
легированных сталей для изготовления корпусов обеих ступеней
ракеты «Поларис А-2» радиус ее действия почти удво-
удвоился. При изготовлении корпусов сверхзвуковых само-
самолетов «Боинг-747» используют намотку стеклоровницы,
пропитанной полиимидом. Известно также, что методом
намотки стеклолентой изготовляют монококовые (без
911
ПОЛИМЕРЫ В АВИАСТРОЕНИИ
912
силового набора стрингеров и шпангоутов) хвостовые
балки вертолетов.
Литьевые стекловолокниты успешно используют
при изготовлении таких ответственных элементов авиа-
авиационных конструкций, как вертолетные колеса и соеди-
соединительные дуги в статоре компрессора газотурбинного
двигателя.
Существенное препятствие для реализации высокой
уд. прочности стеклопластиков в авиационных конст-
конструкциях — низкая (по сравнению с металлами) уд. же-
жесткость. С расширением производства углеродо- и
боропластиков, обладающих более высокой уд. жестко-
жесткостью, стеклопластики заметно уступают свои позиции,
особенно в производстве сверхзвуковых самолетов.
Например, из угле род опластое в США созданы тормоз-
тормозные щитки предкрылков, стабилизаторы (самолеты F-5
и «Скайхок А-4»), крыло сверхзвукового беспилотного
самолета-мишени BQM-34F, створки купола для уборки
основного шасси (CF-14), наружные панели обшивки,
лонжероны, узлы крепления и стойки шасси для др.
самолетов. В Великобритании углеродопласты приме-
применяют в производстве поводков управления циклич.
шагом (WO-13), трансмиссионных валов («Васп/Скоут»),
монококовых хвостовых балок и др. деталей вертоле-
вертолетов, для к-рых жесткость является одной из определяю-
определяющих характеристик.
Аналогичные конструкции создаются из боропласти-
боропластиков, а иногда из их комбинации с углеродопласта-
ми. По данным фирмы «Боинг», такая комбинация
позволяет создавать несущие винты вертолетов, жест-
жесткость к-рых выше, чем у алюминиевых и стальных, со-
соответственно в 6—8 и 2—3 раза. Уменьшение массы
деталей во всех названных выше случаях применения
углеродо- и боропластиков находится в пределах
15—50%. По предварительным оценкам, в случае ши-
широкого использования боропластиков удастся снизить
массу вертолетов на 35%, военно-транспортных само-
самолетов — на 22% и самолетов вертикального взлета и
посадки — на 21%. Наиболее перспективными высоко-
высокомодульными армированными пластиками считаются
углеродопласты, стоимость к-рых к 1980 должна быть
в 3—5 раз меньше, чем боропластиков. При этом само-
самолеты, в которых углеродопласты найдут широкое при-
применение, будут дешевле изготовленных целиком из
металла.
Высокие демпфирующие свойства армированных пла-
пластиков обусловили, в частности, применение углеродо-
пластов для изготовления втулки несущего винта
вертолета «Сен Кинг» (Великобритания), стекло- и
боропластика — для изготовления вертолетных колес
и стоек. Элементы летательных аппаратов из этих
материалов характеризуются более высокой выносли-
выносливостью в условиях вибрационного нагружения, чем их
металлич. прототипы. Так, направляющий аппарат для
вертолетных газотурбинных двигателей фирмы «Вэрко
пластике» (США), изготовленный из эпоксидного стек-
стеклопластика, не разрушается после 30 млн. циклов
испытаний на электромагнитном вибраторе, тогда как
алюминиевый аппарат не выдерживает 1 млн. циклов.
Усталостная выносливость боропластиков еще выше:
при 1 млн. циклов испытаний разрушающее напряжение
у них в 1,5 раза выше, чем у той же конструкции из
стеклопластика.
На крыльях одного из самолетов фирмы «Боинг»
устанавливают предкрылки переменной кривизны с
обшивкой из стеклопластиков и сотовым заполнителем.
Раскрываясь при взлете и посадке самолета, предкрылки
принимают благодаря гибкости полимерного материала
необходимый аэродинамич. контур. Это свойство пла-
пластиков обусловливает лучшую динамич. устойчивость и
управляемость, а также более высокую надежность
бесшарнирных несущих винтов. Удачным сочетанием
уд. прочности, демпфирующих и нек-рых др. свойств
характеризуются также пластики, армированные
синтетич. волокнами (см. Органоволокниты).
Решающее значение при выборе полимерных мате-
материалов для внешних элементов обшивки самолета,
нагревающихся из-за трения о воздух и торможения
потока, имеет термостойкость. Перспективными термо-
термостойкими связующими для армированных пластиков,
помимо модификаций фенольных и циклоалифатич.
эпоксидных смол, являются полибензимидазолы. Ком-
Композиции на основе карбонизованных полимеров, со-
содержащие асбестовые и углеродные волокна (см.
Углеродопласты) и выдерживающие темп-ры 800 СС
и выше, используют при изготовлении тормозных ди-
дисков на авиационных колесах.
Широкое применение получили теплозащитные (абля-
ционностойкие) покрытия из реактопластов, в частно-
частности из фенопластов. Нек-рые из этих материалов спо-
способны длительное время находиться в контакте с откры-
открытым пламенем, темп-pa к-рого м. б. выше 5000 СС
(см. также Абляция).
Основной недостаток высокодисперсных наполните-
наполнителей (особенно порошкообразных), применяемых в теп-
теплозащитных материалах,— унос их газообразными про-
продуктами деструкции еще до того, как они выполнят
свою основную функцию. Поэтому в прессматериалах
для защитных покрытий порошкообразные наполнители
используют только в сочетании с коротковолокнисты-
ми. Напр., асбофенопласты с добавкой высокодисперс-
высокодисперсных порошков бора и графита сохраняют свою струк-
структуру в атмосфере сгорающего ракетного топлива при
темп-ре до 1100 °С в течение 0,5 ч. Такие комбини-
комбинированные материалы применяют для облицовки выхлоп-
выхлопных труб и экранов ракетных двигателей.
Нанесение теплозащитных покрытий на поверхность
летательных аппаратов осуществляется различными
способами в зависимости от типа материала: приклеи-
приклеиванием, послойной выкладкой, напылением и др. Теп-
Тепловая защита может составлять значительную часть
массы летательного аппарата (напр., 20—40% от мас-
массы ракеты без топлива).
Прессматериалы применяют в производстве подвес-
подвесных топливных баков. Изготовление баков из асбо-
текстолита и стекловолокнита методом центробежного
формования позволяет существенно снизить их стои-
стоимость. Высокими фрикционными характеристиками
асбоволокнита обусловлено его применение для коло-
колодок, накладок и дисков в тормозных устройствах само-
самолетов.
Технологич., механич. и колористич. свойства по-
полимерных материалов открывают также широкие пер-
перспективы для их использования при создании интерье-
интерьера кабины, салона и вспомогательных помещений ле-
летательных аппаратов. Распространенный отделочный
материал — декоративный бумажно-слоистый пластик.
Термопласты. Из термопластов, используемых в са-
самолетостроении, в наименее благоприятных эксплуа-
эксплуатационных условиях (большие механич. и тепловые на-
нагрузки) находятся элементы остекления (фонари,
блистеры, иллюминаторы и др.)» к-рые изготовляют
обычно из полиметилметакрилата, обладающего высо-
высокой светопрозрачностью, низкой плотностью и способ-
способностью легко формоваться (см. Органическое стекло).
Возможность повышения прочности и теплостойкости
органич. стекол (выше 110—140 °С), напр, с помощью
армирующих волокон, ограничивается тем, что для со-
сохранения прозрачности стекол компоненты, входящие
в их состав, должны иметь близкие показатели прелом-
преломления. Проблема повышения теплостойкости органич.
стекол не м. б. также решена применением многослой-
многослойных стекол (триплексов) из полиакрилатов (см. Стекло
многослойное). Предпринимаются попытки использовать
для изготовления внутренних слоев триплексов поли-
поликарбонат.
913
ПОЛИМЕРЫ В АВИАСТРОЕНИИ
914
Трудности подбора органич. стекол связаны с высо-
высокими требованиями, предъявляемыми не только к их
теплостойкости, но также и к абразиво-, огне- и пти-
цестойкости. Эти требования, особенно последнее,
вынуждают иногда отказываться от применения
даже достаточно теплостойких органич. стекол в поль-
пользу минеральных.
Абляционностойкие антенные обтекатели изготовляют
из фторопластов, наполненных керамич. волокнами.
Из этих же материалов, стойких к маслам, охлаж-
охлаждающим жидкостям, электролитам и др. агрессивным
средам, изготовляют трубы, фланцы, фитинги, эле-
элементы насосов, уплотнители и др. Предполагают,
что армирование полиэтилена «усами» карбида крем-
кремния, исключительно стойкого к действию плавиковой
к-ты, позволит изготовлять из него трубы и др. де-
детали для нек-рых ракетных двигателей. Однако при-
применение термопластов ограничено их ползучестью.
В частности, уплотнители из фторопласта нельзя
использовать в тех случаях, когда конструкция узла
крепления не исключает ползучесть материала.
Термопласты используют также при декоративной
отделке интерьеров самолетов. В частности, широ-
широкое применение для этой цели находят разнообразные
материалы на основе поливинилхлорида (см. Винил-
хлорида полимеры) — искусственная кожа, пленки и др.
Пено- и сотопласты. Эти материалы благодаря их
низкой плотности, а также звукопоглощающим и тепло-
теплоизоляционным свойствам используют в качестве запол-
заполнителей высоконагруженных трехслойных авиацион-
авиационных конструкций. Пенопласты изготовляют из компо-
композиций фенольных смол с каучуками, полистирола,
эластифицированного поливинилхлорида (см. Пено-
фенопласты, Пенополистирол, Пенополивинилхлорид).
При использовании последнего достигается высокий
коэфф. звукопоглощения (~0,9 при 1 кгц). В трехслой-
трехслойных конструкциях широко применяют также пенополи-
пенополиуретаны. В этом случае собранные панели заполняют
через технологич. отверстия жидкой смесью исходных
продуктов, к-рая вспенивается под действием газов, вы-
выделяющихся в результате реакции между компонента-
компонентами, образуя пенопласт. Иногда для повышения прочно-
прочности и жесткости пенопласт армируют волокнами
(обычно стеклянными).
Сотопласты успешно конкурируют с пенопластами
в производстве трехслойных силовых конструкций.
Одна из причин этого — возможность достижения более
высоких показателей уд. прочности при сжатии. Напр.,
на самолете «Боинг-747» ок. 6300 м2 площади панелей
заполнено сотами из синтетич. бумаги на основе аро-
матич. полиамида («номекс») с кажущейся плотностью
0,045 г/см3. В конструкции того же самолета используют
большое количество стеклосотопласта на полиимидном
связующем, к-рый сохраняет работоспособность до
300 °С. Помимо обеспечения жесткости и радиопрозрач-
радиопрозрачности (в антенных обтекателях), стеклосотопласты по-
повышают безопасность полетов, уменьшая вероятность
поражения летательных аппаратов молнией.
Пено- и сотопласты вытесняют ребра жесткости во
всех силовых конструкциях проектируемых самолетов
(исключение — топливные отсеки, применение ребер
жесткости в к-рых позволяет увеличить долю внутрен-
внутреннего полезного объема).
Резины. Из резин изготовляют шины, амортизато-
амортизаторы, рукава, мягкие топливные баки, разнообразные
профилированные монолитные шнуры, к-рыми гермети-
герметизируют люки, окна, двери кабин и др. В производ-
производстве авиашин используют гл. обр. резины из нату-
натурального и синтетич. изопреновых каучуков и кордные
ткани (см. Кордные нити и ткани) из полиамидных во-
волокон (см. также Шины, Резино-технические изделия).
При изготовлении поверхностных нагревателей и
антиобледенителей, для экранирования деталей элек-
электро- и радиоаппаратуры, а также в производстве
шлангов и покрышек, к-рые не должны накапливать за-
зарядов статич. электричества, применяют токопроводя-
щие резины, наполненные ацетиленовой сажей. Из мас-
лостойких резин изготовляют топливные баки, в
том числе взрывобезопасные. В последние помещают
специальный пенополиуретан, занимающий 3—5% по-
полезного объема бака и препятствующий взрывной вол-
волне от разорвавшергся пули достигнуть критической
скорости.
В элементах авиационных конструкций больших раз-
размеров и невысокой жесткости используют резиновые
абляционностойкие теплозащитные покрытия, не раз-
разрушающиеся при деформации конструкции. Перспек-
Перспективный материал для таких покрытий — резины на
основе кремнийорганич. каучуков, в том числе напол-
наполненные полыми микросферами (см. Пластики с полым
наполнителем), волокнами или сотами. Бутилкаучук,
вулканизованный фенолоформальдегидными: смолами,
может стать заменителем фторопласта в усовершенст-
усовершенствованных вытеснительных емкостях систем подачи
жидких компонентов топлива при низких темп-рах.
Использование резин для сверхзвуковых самолетов
и космич. кораблей ограничено из-за высоких требо-
требований к тепло- и морозостойкости материалов, а так-
также к их стойкости в условиях действия радиации и
в вакууме. Однако общее потребление каучуков в авиа-
авиастроении не уменьшается. Напр., в США к началу
70-х гг. объем использования только кремнийорганич.
каучуков, включая фторсилоксановые, достиг 25%
от их общего выпуска в стране.
Герметики. Применение герметизирующих составов
для поверхностной и внутришовнои герметизации авиа-
авиационных конструкций позволило увеличить высоту по-
полета до 10 км и более, а также значительно усовер-
усовершенствовать самолеты — повысить их надежность, умень-
уменьшить массу, увеличить дальность полета. Эффективная
герметизация кабин, топливных отсеков, воздухово-
воздуховодов, клепаных и др. соединений оказалась практически
возможной с появлением эластичных герметиков на
основе каучуков. Невысыхающие пластичные замазки,
напр, на основе полисульфидных каучуков, использо-
использовавшиеся для герметизации кабин высотных самолетов
после второй мировой войны, в современных самолетах
применяют лишь в неответственных соединениях. Вмес-
Вместо них используют вулканизующиеся герметики, обес-
обеспечивающие большую теплостойкость соединительного
шва и его меньшую чувствительность к перепадам дав-
давлений. Требованиям сверхзвуковой авиации отвечают
герметики на основе кремнийорганич. каучуков, при-
применяемые для поверхностной герметизации.
Большие и малодоступные поверхности, напр, топ-
топливные отсеки, герметизируют вулканизующимися при
обычной темп-ре составами на основе полисульфидных
каучуков. Такая герметизация позволяет размещать
топливо непосредственно в отсеках фюзеляжа и
крыльев, благодаря чему исключается применение
резиновых топливных баков. Количество топлива на
самолете м. б. при прочих равных условиях увеличено
на 30—40% . Для сверхзвуковых самолетов с относитель-
относительно малыми толщинами крыльев такие герметизирован-
герметизированные отсеки — единственно возможные емкости для
топлива.
Недостаток герметиков на основе каучуков — низ-
низкую адгезию ко многим конструкционным материа-
материалам — устраняют модификацией составов, напр, изо-
цианатами или эпоксидными смолами. Распространено
также применение клеевых подслоев, обладающих оди-
одинаково высокой адгезией как к герметизируемому
материалу, так и к герметику. Герметики с хорошими
адгезионными свойствами, напр, на основе полиизобу-
тилена, феноло-формальдегидных и эпоксидных смол,
полиуретанов, используют только при герметизации
915
ПОЛИМЕРЫ В АВИАСТРОЕНИИ
916
малонагруженных узлов, т. к. эти герметики значитель-
значительно изменяют форму и размеры под действием деформа-
деформаций и при колебаниях темп-ры.
Клеи. Основное назначение клеев в авиастроении —
сборка самих самолетных конструкций. Наиболее ши-
широко для этой цели применяют термореактивные клеи
на основе эпоксидных, полиэфирных и фенольных смол,
полиуретанов и их модификаций (см. Клеи синтетиче-
синтетические). Применение клеев для крепления обшивок фюзе-
фюзеляжа, крыла, стабилизатора и др. элементов со стрин-
стрингерами и шпангоутами, пено- и сотозаполнителями
обусловлено тем, что клеевые соединения, обеспечи-
обеспечивая необходимую герметичность, более равномерно,
чем заклепочные, болтовые или сварные, распределяют
напряжения. Кроме того, склеивание осуществляют
по более простой технологии и при значительно более
низких темп-pax, чем сварку. Клеевая пленка выпол-
выполняет одновременно роль демпфера, способствующего га-
гашению вибрации. Благодаря применению клеев для
сборки отсеков вертолетных лопастей и крепления их
на лонжероне ресурс лопастей увеличился до 1,5—
2 тыс. ч. Известны также примеры использования
клеев в производстве ракет, космич. кораблей и спут-
спутников.
Чисто клеевые соединения наиболее часто встреча-
встречаются в сочленениях, работающих на сжатие и сдвиг, а
также в малоответственных сочленениях, работающих
на отрыв. В ответственных элементах авиационных кон-
конструкций иногда создают клеезаклепочные и клеебол-
товые соединения.
Применение клеев ограничивается в нек-рых случаях
длительностью сборки. Поэтому при разработке
новых клеев значительное внимание уделяется выпу-
выпуску их в форме, пригодной для быстрого нанесения и
формирования клеевого соединения. Напр., при креп-
креплении обшивок к сотовым заполнителям в трехслойных
конструкциях применяют пленочные клеи. Они пред-
представляют собой сухие пленки, к-рые плавятся и отверж-
даются при нагревании. Высокая производительность
склеивания этим способом, простота и равномерность
нанесения пленки обусловили, в свою очередь, новые
конструкторские разработки. В частности, при соз-
создании самолета «Боинг-737» использованы двухслой-
двухслойные алюминиевые панели, склеенные пленкой, выпол-
выполняющей одновременно функции вибропоглотителя и
защитного средства при химич. фрезеровании панелей
на точно заданную глубину. Склеивание в этом случае
происходит при прокатке панелей, тогда как обыч-
обычная технология предусматривает применение струб-
струбцин, вакуумных мешков, автоклавов и др. оснастки.
Формирование клеевых соединений удается значи-
значительно ускорить при использовании клеев-расплавов.
В зависимости от требуемой теплостойкости исполь-
используют различные термопласты, чаще всего полиэтилен
и политетрафторэтилен, к-рые наносят на соединяемые
поверхности в виде порошка, пленки или волокон.
После кратковременного нагревания под давлением
(обычно несколько кн/м2, или гс/см2) до темп-ры плазле-
ния полимера и последующего охлаждения давление
снимают. Такие соединения характеризуются высокой
прочностью при сдвиге. Напр., при использовании
полиэтилена в конструкциях из алюминиезых сплавов
она равна 14,5—29 Мн/м2 A45—290 кгс/см2).
Лакокрасочные материалы. Ассортимент лакокрасоч-
лакокрасочных материалов для авиастроения насчитывает более
200 наименований. Антикоррозионные лакокрасочные
покрытия (см. Защитные лакокрасочные покрытия)
защищают самолеты от атмосферных воздействий и
агрессивных сред (топлив, масел и др.). Коррозия
обшивки самолета особенно опасна потому, что толщи-
толщина ее составляет всего 0,6—1,5 мм. Важное требование
к антикоррозионным покрытиям — эффективная за-
защита при малой толщине (при больших толщинах мо-
может значительно увеличиться масса самолета). Наилуч-
Наилучшими эксплуатационными показателями характеризу-
характеризуются полиакриловые покрытия толщиной 15—20 мкм
(см. Полиакриловые лаки и эмали). Такие покрытия ти-
типичны для самолетов со скоростью до М2,2. Дальнейшее
повышение скорости влечет за собой необходимость по-
повышать теплостойкость покрытий, а также создавать
новые покрытия для защиты металлов, вытесняющих
алюминиевые сплавы. Предполагают, что для самоле-
самолетов со скоростями полетов до МЗ доминирующую роль
будут играть покрытия на основе кремнийорганич.
полимеров (см. Кремнийорганические лаки и эмали),
срок службы к-рых при 315 °С составляет 10 000 ч.
Для скоростей порядка М4 наиболее перспективны,
по-видимому, полиимидные покрытия, получаемые на-
напылением порошкообразных композиций.
Лакокрасочные материалы на основе пластифици-
пластифицированных полиуретанов, эфиров целлюлозы (см. По-
лиуретановые лаки и эмали, Эфироцеллюлозные лаки и
эмали) и нек-рых др. пленкообразующих обеспечивают
морозостойкость от —40 до —60 °С, полиакриловые —
до —100 °С.
Внешние поверхности самолетов подвергаются
интенсивной эрозии, особенно в результате соударения
с каплями дождя при полетах на больших скоростях.
Интенсивные поиски эрозионностойких лакокрасочных
покрытий связаны как с опасностью обнажения защи-
защищаемой поверхности, так и со стремлением сохранить
ровную поверхность, поскольку появление неровности,
даже равной 0,01 (отношение глубины неровности к
ее длине), приводит к снижению скорости самолета
примерно на 1%.
Нанесение лакокрасочных покрытий на тонколисто-
тонколистовую обшивку самолета может в 20—25 раз снизить ее
вибрацию. Это обусловило поиски эффективных виб-
рогасящих материалов, способных работать в широком
диапазоне частот B0—3000 гц) и темп-р (от —50 до
—300 °С). Перспективными считают покрытия на осно-
основе теплостойких каучуков, напр, карбораниленсилок-
сановых.
Защита внутренних поверхностей самолетов не менее
сложна, чем защита внешних, т. к. конденсирующаяся
на них влага может не испаряться в течение длитель-
длительного времени. Кроме того, повторное окрашивание
труднодоступной внутренней поверхности осуществля-
осуществляется лишь через —10 лет. Чаще всего на внутренние
поверхности наносят пассивирующие грунтовки на
основе комбинации эпоксидных смол и полиамидов,
пигментированные хроматами стронция или бария.
Терморегулирующие лакокрасочные покрытия устра-
устраняют перегрев обшивки самолетов, напр, под действием
аэродинамич. потока и интенсивного радиационного
облучения (в стратосфере). Такие покрытия должны
характеризоваться высокими коэфф. отражения и излу-
излучения. Коэфф. отражения покрытий для космич. аппа-
аппаратов должен быть не менее 0,9. Этому требованию удов-
удовлетворяют, напр., кремнийорганич. полимеры, на-
наполненные окислами цинка, титана, циркония. Для сни-
снижения темп-ры внутри жилого отсека космич. станции
«Скайлэб» внешние элементы тепловой защиты были
выполнены из полимерной пленки, металлизированной
алюминием и золотом.
Терморегулирующие покрытия, к-рые наносят на
внутренние поверхности летательных аппаратов, долж-
должны защищать кабины экипажа и пассажирский салон от
переохлаждения. Коэфф. излучения в этом случае дол-
должен быть не более 0,4. Покрытия с такими свойствами
получают из эпоксидно-полиамидных композиций,
наполненных чешуйчатым алюминием. В результате ста-
старения коэфф. излучения покрытий повышается до 0,7.
Используют также светопоглощающие (для окраски
внутренних поверхностей, оптич. приборов и др.) и
светящиеся лакокрасочные покрытия (для шкал прибо-
917
ПОЛИМЕРЫ В АВТОМОБИЛЕСТРОЕНИИ
918
ров, стрелок указателей, деталей управления и др.).
Лакокрасочные материалы применяют для создания раз-
разнообразных маскировочных покрытий (под цвет снега,
зелени, ночного неба и др.)- Подбирая состав ком-
компонентов, добиваются эффекта слияния контура воен-
военных самолетов с окружающей средой при визуаль-
визуальном и оптич. наблюдении.
В отделке интерьеров самолетов важную роль играют
декоративные лакокрасочные покрытия.
Лит.: ВольмирА. С, Павленко В. Ф., Поно-
Пономаре в А. Т., Механика полимеров, № 1, 105 A972); Приме-
Применение конструкционных пластмасс в производстве летательных
аппаратов, под ред. А. Л. Абибова, М., 1971; Павлен-
Павленко В. Ф., Силовые установки летательных аппаратов верти-
вертикального взлета и посадки, М., 1972; Булатов Г. а., Пе-
Пенополиуретаны и их применение на летательных аппаратах,
М., 1970; Пригода Б. А., Кокунько В. С, Обтека-
Обтекатели антенн летательных аппаратов, М., 1070; Scow A. L.,
SAMPE Journal, 8, № 2, 25 A972); Peterson G. P.,
AIAA Paper, № 367, 1, A971); WetterR., Kunststoffe, 10,
№ 10, 756 A970); Johnson Z. P., Rubber World, 161,
№ 6, 79 A970); Encyclopedia of polymer science and techno-
technology, v. 1, N. Y.— [a. o.], 1964, p. 568. Г. С. Головкин.
ПОЛИМЕРЫ В АВТОМОБИЛЕСТРОЕНИИ (polymers
in motor-car construction, Polymereim Kraftwagenbau, po-
Потребление пластмасс в зару- lymeres dans l'autoconstru-
бежном автомобилестроении ction). Надежность работы
(в кг*) автомобиля, его долговеч-
ность, комфорт при езде и
безопасность движения м. б.
обеспечены только при ус-
ловии применения полимер-
ных материалов — пласт-
пластмасс, резин, лаков и кра-
СОК И др.
Пластмассы. Потребление
пластмасс в автомобиле-
строении непрерывно воз-
растает (см. таблицу).
* В среднем на 1 автомобиль. Ниже на примере авт0.
мобиля «Фиат-125» показан расход различных пласт-
пластмасс на одну машину (в кг):
Страна
США . . .
Велико-
Великобритания
ФРГ . . .
Япония . .
Франция
Италия . .
1965
13,6
1Й.6
14,0
—
15,0
17,0
1970
45,4
30,4
30,4
30,0
40,0
40,0
1975
(прог-
(прогноз)
80-100
45-50
45-50
40—45
45-50
45-50
Поливинилхлорид
Полиуретаны ....
Полистирол и сопо-
сополимеры стирола. .
Полиэтилен
Фенопласты
19,5
6,0
6,0
4,4
О j
Полипропилен . . .
Полиакрилаты . . .
Этролы
Полиамиды
Прочие пластики . .
1,4
1,3
1 0
о; 6
0,5
Из пластмасс изготовляют кузова и кабины автомо-
автомобилей и их отдельные крупногабаритные детали, раз-
разнообразные малогабаритные детали конструкционного
и декоративного назначения, тепло- и звукоизоляцион-
звукоизоляционные детали и др. Благодаря применению пластмасс
улучшается внешний вид автомобиля, уменьшается его
масса, снижается шум при езде, совершенствуется
конструктивное оформление деталей, увеличивается
срок их службы и уменьшается трудоемкость изготов-
изготовления. Замена металлов и др. традиционных материа-
материалов пластмассами при изготовлении деталей сложной
конфигурации дает значительный технико-экономич.
эффект, т. к. многие детали из пластмасс м. б. получены
на автоматизированных установках с минимальными
отходами перерабатываемого материала.
Особенно большую перспективу имеет применение
пластмасс для изготовления кабин и кузовов и их
крупногабаритных деталей, т. к. на долю кузова при-
приходится ок. половины массы автомобиля и ~40% сто-
стоимости. Кузова из коррозионностойких пластмасс бо-
более надежны и долговечны в эксплуатации, чем метал-
металлические G0% автомобилей с металлич. кузовами не
выдерживают 10-летнего срока эксплуатации из-за
коррозии металла), а их ремонт дешевле и проще.
При изготовлении кабин и кузовов автомобиля
наиболее широкое применение находят полиэфирные
стеклопластики и слоистые пластики на основе фе-
нольных смол и тканей из растительных волокон (фе-
нотекстолиты). Методом горячего прессования из стек-
стеклопластика изготовляют, напр., кузов легкового авто-
автомобиля «Корвет» (США), к-рый монтируют из отдель-
отдельно формуемых панелей, а также капот и оперение грузо-
грузового автомобиля «Форд» серии L. Стеклопластик исполь-
используют также для изготовления кабины грузового авто-
автомобиля «Фаун» (ФРГ) и кузова легкового спортивного
автомобиля «ВМС» модели 1100 (Великобритания) ме-
методом контактного формования. В ГДР выпускается
легковой автомобиль «Трабант» с кузовом из фенотек-
столита, к-рый монтируют из панелей, получаемых
прессованием (см. Прессование реактопластов). Как
правило, отдельные детали кузова крепятся на метал-
металлич. каркасе. Ведутся также работы по созданию кабин
и кузовов без такого каркаса.
Для изготовления кузовов применяют также сополи-
сополимер АБС (см. Стирола сополимеры) и жесткие пенопо-
пенополиуретаны. Напр., кузов автомобиля «Диана-6-Мек-
сари» (Франция) состоит из 11 деталей, получаемых
вакуумформованием сополимера АБС. В ФРГ имеются
опытные образцы легкового кабриолета «YAK» (масса
65 кг) из пенополиуретана.
Несмотря на отмеченные выше преимущества пласт-
пластмасс перед металлами, они не получили еще широко-
широкого распространения в производстве крупногабаритных
деталей автомобиля, гл. обр. из-за недостаточной
жесткости (низкого модуля Юнга) и сравнительно невы-
невысокой атмосферостойкости, напр, у сополимера АБС.
Наиболее широко пластмассы применяют в производ-
производстве деталей внутренней отделки салона автомобиля,
особенно его передней части. При изготовлении деко-
декоративных деталей пластмассы окрашивают в массе или
металлизируют. На наружные видовые детали металл
наносят трудоемким, но позволяющим получать более
износостойкие покрытия гальванич. способом, на
внутренние детали — вакуумным способом (см. Ме-
Металлизация пластмасс).
Из пластмасс изготовляют детали двигателя, транс-
трансмиссии, шасси. При использовании пластмасс в под-
подшипниках скольжения уменьшается трудоемкость обслу-
обслуживания автомобиля, т. к. подшипники с вкладышами
из пластмассы и консистентной смазкой, к-рую закла-
закладывают во время сборки, не требуют периодич. смазки
при пробеге автомобиля до 80—100 тыс. км.
Ниже приведены примеры использования пласт-
пластмасс в производстве малогабаритных комплектующих
деталей автомобиля.
Из п о л и в и н и л х л о р и д а изготовляют шлан-
шланги для омывателя ветрового стекла, сильфоны, изоля-
изоляцию электропроводов, мягкие ручки, кнопки, канты,
прошвы и др. Для звукоизоляции, защиты днища кузо-
кузова от коррозии, герметизации сварных швов внутри
кузова, препятствующей проникновению воды и пыли,
уплотнения желобка водослива, склеивания фильтрую-
фильтрующих элементов масляных фильтров с верхней и нижней
картонными крышками, изготовления прокладок воз-
воздушного фильтра и др. широко используют поливи-
нилхлоридные пласт и золи (см. Пасты полимер-
полимерные). Поливинилхлоридными пленками отделывают
потолок, сиденья, дверную и боковую обшивку салона.
Вследствие повышения требований к безопасности
при езде большое внимание уделяют отделке салона
эластичными пенополиуретанами. При за-
замене традиционных пружинных сидений подушками
из этого пенопласта повышается боковая устойчивость
сиденья, комфорт, надежность опоры и благодаря это-
этому уменьшается утомляемость водителя при длитель-
длительных поездках. Производство подушек из пенополиуре-
пенополиуретана м. б. автоматизировано. Из полужесткого пенопо-
пенополиуретана изготовляют стойки ветрового стекла, щит-
щитки приборов, подлокотники, внутренние дверные пане-
панели, противосолнечный козырек и др.; из монолит-
монолитных полиуретанов — подшипники скольже-
919
ПОЛИМЕРЫ В МАШИНОСТРОЕНИИ
920
ния рулевого управления, подвески, ремни привода
распределительного вала, амортизатор рулевого меха-
механизма.
Сополимер АБС используют в производстве
вентиляционных решеток, картера системы охлажде-
охлаждения, колпаков колес, щитка приборов, дверных карма-
карманов, чехлов для сидений, перчаточного ящика авто-
автомобиля «BMW». Этот сополимер используют также
для облицовки радиатора, вентиляционных отверстий,
эмблем.
Нек-рые зарубежные фирмы («Дженерал моторе»—
США, «Фиат»— Италия, «Тайота»— Япония) устанав-
устанавливают на автомобилях решетки радиаторов из сополи-
сополимера АБС, хорошо окрашиваемого в массе (эти детали
изготовляют также из наполненных стекловолокном
полиамидов и полипропилена). Трудоемкость их изго-
изготовления из пластмасс в 4—5 раз меньше, чем из метал-
металла. Решетки радиаторов из пластмассы, устанавли-
устанавливаемые на машинах США, металлизируют гальванич.
способом, на европейских — окрашивают в массе; в
последнем случае повышается безопасность при езде
вследствие уменьшения бликов.
Полипропилен используют для изготовле-
изготовления вентиляционных трубопроводов, лопастей венти-
вентиляторов, педалей акселератора, а также для облицов-
облицовки дверей; из этого полимера изготовляют ручки, крюч-
крючки и др.
Полиметилметакрилат — основной по-
полимер для изготовления деталей внутрисалонного осве-
освещения, защитных колпаков фонарей заднего света.
Пластмассы на основе ацетобутирата цел-
целлюлозы используют для облицовки рулевого коле-
колеса, изготовления кнопок управления, а также раз-
разнообразных декоративных деталей. Из полиами-
полиамидов изготовляют лопасти вентиляторов, подшипники,
топливопроводы, направляющие сидений, детали
дверных замков, из полиэтилена — топливные
баки (емкостью до 100 д), уплотнительные прокладки,
облицовку дверей, багажников, из поликарбо-
поликарбонатов — крышку ступицы колеса, внутренние осве-
осветители, изоляторы и крышки, облицовку репродукто-
репродукторов, плафоны.
Политетрафторэтилен применяют для
изготовления втулок подшипников скольжения, ф е-
нопласты — для электроизоляционных деталей
системы зажигания и др. Из полиэфирного
стеклопластика, помимо крупногабаритных
деталей, изготовляют картер системы отопления и за-
защитные трубы.
Резины. К важнейшим и наиболее материалоемким
резиновым изделиям для автомобилестроения относят-
относятся шины (подробно об их типах и конструкции см.
Шины). Большое значение в этой отрасли пром-сти
имеют также многочисленные резино-технические изде-
изделия, от качества к-рых во многом зависит надежность
работы автомобиля.
Наряду с резинами на основе натурального, бута-
диен-стирольного, бутадиен-нитрильного, хлоропрено-
вого, нек-рых бутадиеновых каучуков (см. Каучук на-
натуральный, Каучуки синтетические), к-рые издавна
используют в автомобилестроении, большое значение
приобрели резины из новых каучуков специального
назначения. Так, из фторсодержащих каучуков изго-
изготовляют уплотнители, эксплуатируемые при темп-рах
до 200 °С, из кремнийорганических каучуков — уплот-
уплотнители и манжеты, работающие в контакте с консистент-
консистентными смазками при темп-pax от —50 до 180 °С, а также
амортизирующие и теплоизоляционные материалы,
напр, пористые уплотнители.
Значительное распространение в автомобилестрое-
автомобилестроении получили масло-, свето- и озоностойкие акрилат-
ные каучуки, из к-рых изготовляют манжеты, диафраг-
диафрагмы, радиаторные рукава и др. Из атмосфере- и химстой-
ких этилен-пропиленовых каучуков получают губчатые
и монолитные оконные и дверные прокладки, манжеты
для тормозных систем, шланги радиаторов, пневматич.
амортизаторы, детали рессор и др., из высокопроч-
высокопрочных и износостойких уретановых каучуков — вклады-
вкладыши рулевых тяг, крестовины карданных валов, подушки
амортизаторов, диафрагмы тормозов и др. Весьма пер-
перспективны для применения в производстве уплотнитель-
ных автомобильных деталей эпихлоргидриновые каучу-
каучуки (см. Эпоксидные каучуки), превосходящие бутадиен-
нитрильные по маслостойкости, а акрилатные —
также и по свето- и озоностойкости.
Помимо твердых каучуков, в производстве нек-рых
автомобильных деталей применяют латексы. Напр., из
бутадиен-стирольных латексов изготовляют губчатые
подушки сидений (см. Губчатые резины). Малоответст-
Малоответственные изделия, напр, коврики для салонов автомоби-
автомобиля, изготовляют из регенерата резины (см. Регенерация
резины).
Получили распространение резино-тканевые изделия,
напр, приводные и вентиляторные ремни, с полиамид-
полиамидными и высокопрочными вискозными волокнами, при-
применение к-рых позволило существенно повысить экс-
эксплуатационные свойства изделий.
Лакокрасочные материалы. Эти материалы, приме-
применяемые для грунтования и окончательной отделки
металлич. поверхностей, должны образовывать покры-
покрытия, к-рые надежно защищают металл от коррозии (см.
Защитные лакокрасочные покрытия), обладают высокой
твердостью, эластичностью, ударопрочностью, термо-
и износостойкостью. Особенно большой интерес для
автомобилестроения представляют полиакриловые
эмали (см. Полиакриловые лаки и эмали), в том числе
пигментированные металлич. порошками различных
цветов, придающими покрытиям красивый металлич.
блеск, а также полиуретановые эмали, образующие
атмосферостойкие покрытия (см. Полиуретановые лаки
и эмали).
Для нанесения лакокрасочных материалов в автомо-
автомобилестроении особенно широко используют метод пнев-
пневматич. распыления, а также окунание и обливание.
Водорастворимые лакокрасочные материалы (см. Во-
доразбавляемые грунтовки и эмали) наносят методом
электроосаждения (о методах нанесения см. Лакокра-
Лакокрасочные покрытия).
Большую перспективу для автомобилестроения имеет
получение защитных и декоративных покрытий методом
напыления с применением порошковых красок.
Лит.: М а с и н а М. А., Алексеев В. Н., М о т о-
вилин Г. В., Автомобильные материалы, М., 1971; М а-
лышевГ. А., ЕзерскийА. Н., Основы проектирования
и производства деталей из пластмасс в автомобилестроении,
М., 1963; Кауч. и рез., № 6, 52 A971); Экспресс-информация.
Автомобилестроение, № 7, 14 A970); № 40, 29 A973); № 43,
10 A973); Automobilwirtschaft, № 5, 295, 298 A971); Kimst-
stoffe, jsfi 10, 738 A970); Automotive News, № 8, 24 A969);
J. P. D. (Italia) № 20026, с 13, A970); Kunststofftechnik, ii,
№ 9, 395 A970); Des News, 2, № 26, 25 A971); Rubb. World,
156, № 2, 107 A969); Автомобильная промышленность, № И,
41 A969); там же, № 11, 41 A970). В. М. Ильин.
ПОЛИМЕРЫ В МАШИНОСТРОЕНИИ (polymers
in mashine-building, Polymere im Maschinenbau, poly-
meres dans construction des mashines). Полимеры зани-
занимают одно из ведущих мест среди конструкционных
материалов для машиностроения. Так, потребление
пластмасс в этой отрасли становится соизмеримым (в
единицах объема) с потреблением стали. Непрерывно
возрастает также применение лакокрасочных материа-
материалов, синтетич. волокон, клеев, резины и др.
Целесообразность использования полимеров в маши-
машиностроении определяется, прежде всего, возможностью
удешевления продукции. При этом улучшаются также
важнейшие технико-экономич. параметры машин —
уменьшается масса, повышаются долговечность, надеж-
надежность и др. В результате внедрения полимеров высво-
921
ПОЛИМЕРЫ В МАШИНОСТРОЕНИИ
922
бождаются ресурсы металла, а благодаря уменьшению
отходов при переработке существенно повышается
коэфф. использования материалов (средние значения
коэфф. использования пластмасс примерно в 2 раза вы-
выше, чем для металлов).
Основные достоинства полимерных конструкцион-
конструкционных материалов — высокая уд. прочность (отноше-
(отношение прочности к плотности), хим- и износостойкость,
хорошие диэлектрич. характеристики. Свойства этих
материалов можно варьировать в широких пределах
модификацией полимеров или совмещением их с раз-
различными ингредиентами. В частности, при введении в
полимеры соответствующих наполнителей (см., напр.,
Наполнители пластмасс) можно получать фрикцион-
фрикционные и антифрикционные материалы, а также материа-
материалы с токопроводящими, магнитными и др. специаль-
специальными свойствами.
К недостаткам полимерных материалов относятся
склонность к старению и деформированию под нагруз-
нагрузкой (ползучесть), зависимость прочностных характе-
характеристик от режимов нагружения (темп-pa, время), срав-
сравнительно невысокая теплостойкость, относительно боль-
большой температурный коэфф. линейного расширения,
изменение размеров при воздействии на материал влаги
или агрессивных сред.
Из пластических масс изготовляют обширный ассор-
ассортимент деталей и узлов машин, а также технологич.
оснастку различного назначения (см. таблицу).
Основные области применения пластмасс в машиностроении
Виды деталей, узлов машин
и технологич. оснастки
Зубчатые и червячные колеса
Шкивы, маховички, рукоятки, кнопки
Ролики, катки, бегуны
Подшипники скольжения
Направляющие станков
Детали подшипников качения
Тормозные колодки, накладки
Трубы, детали арматуры, фильтры мас-
масляных и водных систем
Рабочие органы вентиляторов, насосов и
гидромашин
Уплотнения
Кожухи, корпуса, крышки, резервуары
Детали приборов и автоматов точной ме-
механики
Болты, гайки, шайбы
Пружины, рессоры, кулачковые механиз-
механизмы, клапаны
Крупногабаритные элементы конструк-
конструкций, емкости, лотки и др
Электроизоляционные детали, панели,
щитки, корпуса приборов
Светопропускающие оптич. детали (лин-
(линзы, смотровые стекла и др.)
Копиры, контрольные шаблоны
Холоднолистовые штампы
Литейные модели
Условные номера
пластмасс
1, 6, 10-16
1, 4, 13-16
1, 5, 6, 11, 16
1-3, 6, 8-16
1, 9, 15
1, 11, 12
13, 14, 16
2, 5, 6, 11, 17
1, 2, 5, 6, 10, И, 17
1 — 3, 5, 6, 10
2, 4-8, 11, 13, 17
1, 2,
1, 2,
1, 5,
2, 5,
1-17
2, 4,
2, 5,
8, 9,
7-9,
5, 6, 10 — 14
4, 5, 6, 10-14
6, И, 12, 15, 17
7, 17
6-8, 11
6, 9
13, 17
13, 17
Примечание: 1—полиамиды, 2—полиэтилен, З^фторо-
пласты, 4 — аминопласты, 5 — поливинилхлорид, 6 — полипро-
полипропилен, 7 — полистирол, 8 — полиакрилаты, 9 — эпоксипласты,
10 — пентапласты, 11—поликарбонаты, 12 — полиформальдегид,
13—фенопласты, 14 — волокниты, 15—текстолит, 16—древесные
пластики, 17 — стеклопластики.
Ниже рассматриваются примеры использования по-
полимерных материалов в производстве деталей обще-
общемашиностроительного назначения (подшипники, зуб-
зубчатые колеса, ремни, шкивы и др.). О специфике при-
применения этих материалов в различных отраслях маши-
машиностроения См. Полимеры в авиастроении, Полимеры в
автомобилестроении, Полимеры в пищевой промыш-
промышленности, Полимеры в судостроении, Полимеры в элект-
электротехнике, Полимеры на железнодорожном транспорте.
Для изготовления подшипников сколь-
скольжения используют разнообразные материалы, обла-
обладающие большой износостойкостью и низким коэфф.
трения (см. Антифрикционные полимерные материалы),
а также теплостойкостью, стабильностью размеров в
условиях эксплуатации и длительным сроком службы
при больших значениях несущей способности (произве-
(произведения допустимых нагрузки и скорости скольжения).
Износостойкость, несущая способность и др. свойства
подшипниковых материалов резко повышаются при вве-
введении в них наполнителей (при наполнении фторопла-
ста-4 скрытокристаллич. графитом износостойкость
возрастает в 1000 раз). Подшипники из графитонапол-
ненного фторопласта-4 могут работать без смазки, а
также в агрессивных средах (см. Графитопласты).
Основные требования к пластмассам для зубча-
зубчатых колес — высокие контактная прочность
и сопротивление изгибу, износостойкость, демпфирую-
демпфирующая способность, динамич. выносливость, стабиль-
стабильность размеров. При использовании пластмасс, удовлет-
удовлетворяющих этим требованиям, повышается долговеч-
долговечность колес, в среднем в 1,5 раза снижается уровень шу-
шума, уменьшается чувствительность передачи к наличию
смазки, снижаются требования к точности изготовле-
изготовления колеса. Однако единичный зуб из полиамида со стан-
стандартным контуром по статич. прочности уступает зубьям
из алюминия, улучшенной или закаленной стали соот-
соответственно в 1,4, 3—5 и 7 раз. Деформация зубьев из
пластмассы достигает десятых долей мм, а размеры кон-
контактной площадки становятся соизмеримыми с разме-
размером зуба. Все же благодаря новым технологич. и кон-
конструктивным решениям удалось расширить области
применения зубчатых колес из пластмасс, увеличить
их несущую способность, повысить кинематич. точность,
износостойкость и др. Армирование колес из пластмасс
металлом (из него изготовляют ступицы, диск, венец
и др. элементы) позволяет наиболее эффективно исполь-
использовать достоинства обоих материалов.
Пластмассы все более широко используют вместо
нержавеющих сталей и др. материалов в волновых пе-
передачах, отличающихся компактностью и большими
передаточными отношениями (например, от 64 : 1 до
320 : 1), а также для изготовления звездочек в цеп-
цепных передачах.
Плоские, клиновые и зубчатые ремни из пласт-
пластмасс (полиамидов, поливинилхлорида), а также из ре-
резины (см. Резино-технические изделия) м. б. использова-
использованы для передачи даже значительных мощностей. В отли-
отличие от ремней из традиционных материалов, ремни из
полимерных материалов можно эксплуатировать в
агрессивных средах без применения натяжных роли-
роликов. Многослойные ремни шириной 10—1200 мм, арми-
армированные синтетич. волокнами, м. б. использованы для
передачи мощностей до 3600 кет при скоростях 50 —
80 м/сек.
Применение в ременных передачах прочных и изно-
износостойких шкивов из пластмасс, характеризую-
характеризующихся малой плотностью, высоким коэфф. сцепления
с ремнем, стабильностью размеров, позволяет умень-
уменьшить инерционные силы, увеличить срок службы рем-
ремней, сократить мощность, потребляемую станком, а в
нек-рых случаях повысить тяговую способность пере-
передачи. Использование полимерных материалов для фу-
теровок блоков и барабанов подъемных устройств повы-
повышает коррозионную стойкость этих деталей и увеличи-
увеличивает долговечность канатов.
Использование труб из полимерных материалов
вместо металлических приводит к упрощению их монта-
монтажа вследствие снижения массы, уменьшению гидрав-
лич. потерь и расхода мощности на транспортировку
материалов, увеличению пропускной способности труб,
повышению срока службы (особенно в агрессивных сре-
средах, в земле и воде) и стойкости к гидравлич. удару.
Применение прозрачных труб позволяет, кроме того,
визуально наблюдать за движением продукта. О тру-
трубах из полимерных материалов см. также Полимеры в
сельском и водном хозяйстве, Полимеры в строительстве.
923
ПОЛИМЕРЫ В МЕДИЦИНЕ
924
Основным материалом для уплотнительных
прокладок, к-рые, помимо высокой износо- и
теплостойкости, должны обладать эластичностью, а
также стойкостью в различных агрессивных средах,
служат резины на основе хлоропренового, бутадиен-
нитрильного, кремнийорганич., фторсодержащих и
др. каучуков специального назначения (см. Каучуки
синтетические, Резино-технические изделия). Для
уплотнения подвижных соединений или соединений,
к-рые подвергаются действию высоких давлений, ис-
используют обычно уплотнители из пластмасс.
Полимерные материалы применяют для фикса-
фиксации резьбовых соединений, осуществ-
осуществляемой различными способами: использованием гаек
из пластмасс, нарезку на к-рых создают при ввинчива-
ввинчивании в них металлич. болтов, применением шайб и вкла-
вкладышей из пластмасс, а также с помощью быстроотверж-
дающихся компаундов (см. Компаунды полимерные).
Эти способы фиксации обеспечивают повышение срока
службы резьбовых соединений, выполняющих одновре-
одновременно функции уплотнительных элементов.
Эпоксидные и акрилатные компаунды применяют в
качестве универсальных компенсато-
компенсаторов погрешностей при сборке узлов машин
и приборов. Благодаря их использованию процесс сбор-
сборки (напр., редукторов) сводится к установке деталей
с требуемой точностью и заливке компаундом простран-
пространства между сопрягаемыми деталями. Заполняя зазоры,
компаунд компенсирует все погрешности обработки и
сборки деталей. Применение компенсаторов позволяет
на 2—3 класса расширить допуски на изготовление
поверхностей, снизить себестоимость обработки дета-
деталей, уменьшить трудоемкость их сборки. Заданная точ-
точность замыкающего звена сборочных размерных цепей
м. б. обеспечена за одну выверку.
С помощью клеев (см. Клеи синтетические) удалось
создать сборные зубчатые колеса из металлов и пласт-
пластмасс, упростить сборку узлов подшипников, удешевить
ремонт машин, повысить их надежность. Напр., в
результате применения направляющих с приклеен-
приклеенными накладками из антифрикционных материалов
повысились эксплуатационные свойства станков и упро-
упростился их ремонт. Использование синтетич. клеев при
изготовлении магнитных плит привело к улучшению их
электроизоляционных свойств.
Технологическая оснастка из пласт-
пластмасс (кондукторы для сверления деталей, шаблоны
для контроля деталей сложной конфигурации, штампы,
приспособления для разметки и др.) легче, дешевле,
проще в изготовлении, чем аналогичная металлическая.
Эксплуатационные свойства такой оснастки повышают-
повышаются при ее армировании металлами, применением в ка-
качестве наполнителей металлич. волокон или металли-
металлизацией рабочих поверхностей (см. Металлизация пласт-
пластмасс). Из пластмасс изготовляют различную литей-
литейную оснастку. Так, в пром-сти широко используют
метод литья деталей по выжигаемым моделям из пено-
полистирола', из фенопластов изготовляют формовоч-
формовочные смеси, оболочковые формы и стержни. Полимер-
Полимерные материалы служат также связующим в абразивном
инструменте (напр., при изготовлении термо- и во-
водостойких шлифовальных шкурок).
Важное народнохозяйственное значение имеет при-
применение лакокрасочных и др. полимерных материалов
для антикоррозионной защиты металлич. конструкций
при их сооружении, транспортировке, консервации и
эксплуатации, а также для декоративной отделки и при-
придания специальных свойств (электроизоляционных,
антифрикционных и др.). Объем потребления таких ма-
материалов составляет —30% общего потребления поли-
полимерных материалов в машиностроении. См. Лакокра-
Лакокрасочные покрытия, Антикоррозионные полимерные покры-
покрытия, Защитные лакокрасочные покрытия, Напыление.
Лит.: К о г а н А. М., Р а х л и н И. В., Экономика про-
производства изделий из пластмасс, М., 1974; А л ь ш и ц И. Я.,
Анисимов Н. Ф., Благов Б. Н., Проектирование
деталей из пластмасс, М., 1969; Б о к и н М. Н., Ц ы п л а-
к о в О. Г., Расчет и конструирование деталей из пластмасс,
М.— Л., 1966; Воробьев Ю. А., БежелуковаЕ. Ф.,
Допуски и посадки деталей из пластмасс, М., 1964; Основы
конструирования изделий из пластмасс, под ред. Э. Бэра пер.
с англ., М., 1970; КестельманН. Я., Кестель-
м а н В. Н., Номограммы по расчету и конструированию пласт-
пластмассовых деталей машин, М., 1970; Полимеры в машинах, М.,
1968; Суровяк В., Худзиньски М., Применение
пластмасс в машиностроении, пер. с польск., М., 1965; Бегид-
жанова А. Я., Крейндлин Л. М., Применение пласт-
пластмасс в тракторном машиностроении, М., 1970; ГенельС. В.,
Кестельман Н. Я., Кестельман В. Н., Полимер-
Полимерные материалы в пищевом машиностроении, 2 изд., М., 1969;
Мирзоев Р. Г., Пластмассовые детали машин и приборов,
2 изд., Л., 1971; БелыйВ. А., С в и р и д е н о к А. И.,
Щербакове. В., Зубчатые передачи из пластмасс, Минск,
1965; Кестельман В. Н., Короб А. Д., Пластмассо-
Пластмассовые шкивы и клиноременные передачи, М., 1968; Пластмассы
в подшипниках скольжения, М., 1965; Поликарбонат в машино-
машиностроении, М., 1972; Иванов Ю.М., Пластмассовая техно-
технологическая оснастка к станкам, М., 1964; Ш т у р м а н А. А.,
Холоднотвердеющие акриловые пластмассы в инструменталь-
инструментальном производстве, М., 1964; Р а х л и н И. В., Экономика при-
применения пластмасс в машиностроении, М., 19 73.
В. А. Белый, В. Н. Кестельман^
И. В. Рахлин, А. И. Свириденок.
ПОЛИМЕРЫ В МЕДИЦИНЕ (polymers in medicine,
Polymere in Medizin, polymeres dans medicine).
Содержание:
Особенности применения полимерных материалов
в медицине 924
Полимеры медико-технического назначения ... 925
Полимеры, используемые в восстановительной
хирургии 926
Сердечно-сосудистая хирургия 927
Хирургия внутренних органов и тканей . . 928
Травматология и ортопедия 928
Офтальмология 929
Стоматология и челюстно-лицевое протези-
протезирование 929
Полимеры, используемые в функциональных
узлах хирургических аппаратов 930
Полимеры направленного биологического дей-
действия 930
Крове- и плазмозаменители 931
Пролонгаторы 932
Полимерные лекарственные вещества .... 933
Вспомогательные вещества для создания различ-
различных лекарственных форм 934
Особенности применения полимерных материалов
в медицине
Полимерные материалы, находящиеся в контакте с
биологич. средами живого организма, могут раство-
растворяться в этих средах без изменения мол. массы или
подвергаться биодеструкции по след. основным меха-
механизмам: 1) гидролиз с образованием макромолекуляр-
ных осколков и мономерных продуктов; 2) каталитич.
гидролиз под влиянием ферментов; 3) фагоцитарное
разрушение (защитная клеточная реакция организма
на инородное тело). В реальных условиях скорость
биодеструкции, по-видимому, обусловлена суммарным
воздействием указанных факторов.
Биологич. активность полимерных материалов
связана с образованием продуктов биодеструкции, а
также с присутствием в полимерах остаточных моно-
мономеров и добавок (пластификаторов, стабилизаторов,
красителей, наполнителей, эмульгаторов, инициато-
инициаторов и др.).
Среди многочисленных проблем санитарно-химич.
исследований особое значение имеют следующие: 1)
выявление токсикологич. опасности полимерных ма-
материалов на основании качественного и количественно-
количественного определения состава низкомолекулярных продуктов;
2) изучение закономерностей миграции примесей из
полимеров в зависимости от их химич. природы и сред
живого организма; 3) исследование процессов метабо-
метаболизма, изменений функциональных систем организма,
путей выведения из него продуктов биодеструкции.
См. также Санитарно-гигиеническая характеристика
полимерных материалов.
925
ПОЛИМЕРЫ В МЕДИЦИНЕ
921
Особое значение имеет токсикологич. оценка поли-
полимерных материалов, применяемых в медицине в усло-
условиях непосредственного контакта с живым организмом.
Необходимость тщательной токсикологич. оценки поли-
полимеров, даже обладающих высокой химич. стойкостью и
инертностью, связана с тем, что процессы их перера-
переработки часто осуществляются при темп-pax, близких или
превосходящих начальные темп-ры разложения этих
полимеров (табл. 1). Продукты термической и термо-
термоокислительной деструкции могут присутствовать в ма-
материале в сорбированном виде и оказывать токсич.
воздействие на организм, к-рое непосредственно не
связано с химич. природой и структурой исходного
полимера.
Имплантация в организм животных ряда поли-
полимерных материалов, не обладающих общетоксич. дей-
действием, может приводить к возникновению злокачест-
злокачественных опухолей. Так, через 6—8 мес после имплан-
имплантации в различные оргдны крыс гладких пластинок из
полиэтилена, поливинилхлорида, фторопласта, поли-
акрилатов, полиамидов, кремнийорганич. каучука и
др. наблюдалось возникновение злокачественных опу-
опухолей. Однако такое бластоматозное действие наблюда-
наблюдалось лишь на мелких животных (крысы, мыши, хомяки,
морские свинки), причем аналогичным образом в
этих условиях проявляли себя такие инертные мате-
материалы, как стекло, благородные металлы. Установлено
Таблица 1. Допустимые темп-ры переработки полимерных
материалов при производстве изделий медицинского
назначения
Наименование
материала
Полиамиды
Поливинилхлорид ....
Полиметилметакрилат
Полипропилен
Полиорганосилоксаны
Полистирол
Политетрафторэтилен
Полиэтилен
Темп-pa, °С
разло-
разложения
(началь-
(начальная)
150
150
300
280
260
250
300
100
перера-
переработки
(макси-
(максимальная)
280
160
225
260
210
205
375
120
250
Способ
переработки
Прядение
Вальцевание,
сварка
Сварка
Литье под дав-
давлением
Прессование
Литье под дав-
давлением
Спекание
Вальцевание
Литье под дав-
давлением
также, что имплантация полимеров в виде порошка
или перфорированных пластин не вызывает образова-
образования опухолей и оказывает слабый бластоматозный
эффект. Большинство исследователей считает, что бла-
стомогенное действие биоинертных полимеров обуслов-
обусловлено не их химич. природой, а механич. длительным
раздражением стенок соединительнотканной капсулы,
возникающей вокруг имплантированного материала, и
нарушением нормального обмена в ней.
Полимеры медико-технического назначения
Применение полимеров для изготовления изделий
медицинской техники позволяет осуществлять серийный
выпуск инструментов, предметов ухода за больными,
специальной посуды и различных видов упаковок для
лекарств, обладающих рядом преимуществ перед ана-
аналогичными изделиями из металлов и стекла: экономич-
экономичностью, в ряде случаев — повышенной стойкостью к
воздействию различных сред, возможностью выпуска
изделий разового использования и др. Основными тре-
требованиями, предъявляемыми к полимерам и материа-
материалам на их основе, используемым в производстве изде-
изделий медицинской техники, являются: необходимый
комплекс физико-механич. свойств, зависящий от конк-
конкретного назначения материала; повышенная химич
стойкость, обусловливающая стабильность изделий по;
воздействием жидких сред, в том числе стерилизующие
жидкостей; минимальное содержание низкомолекуляр
ных примесей, стабилизаторов, катализаторов и др
Таблица 2. Ассортимент и области применения полимерных
материалов медико-технического назначения
Наименование
материала
Полиэтилен вы-
высокой плотно-
плотности
Полиэтилен
низкой плот-
плотности
Полиамиды
Поликарбонат
Фторопласт-4
Пластикат
Полистирол
Ацетобутират-
целлюлозный
и ацетилцел-
люлозный эт-
этролы
Полипропилен
нестабилизи-
рованный
Области применения
Детали медицинских приборов и инструмен-
инструментов, предметы ухода за больными, лабора-
лабораторное оборудование, футляры-стерилизато-
футляры-стерилизаторы, пробирки, пипетки и др.
Мягкие емкости различного назначения, сое
динительные трубки, шприц-тюбики, про
тезно-ортопедич. изделия, бачки для гамма-
глобулина
Детали медицинских приборов и инструмен-
инструментов, воронки, трубки., оправы очков, канюл!
переходные
Протезно-ортопедич. изделия
Медицинские инструменты и их детали, зонды
катетеры, канюли", емкости различного на-
назначения, лабораторная посуда, предметы
ухода за больными
Эластичные медицинские инструменты—кате-
инструменты—катетеры, бужи пищеводные, трахеотомич. труб-
трубки, системы для взятия и переливания
крови, клеенка
Шприцы разового использования, чашки
Петри, футляры, упаковка для лекарствен-
лекарственных препаратов
Оправы коррегирующих очков, линзы защит-
защитных и солнцезащитных очков
Детали медицинских приборов и аппаратов
технологич. добавок; отсутствие запаха; способность
выдерживать тепловую стерилизацию (в том числе ав-
токлавирование) и радиационную стерилизацию; ста-
стабильность состава жидких медицинских препаратов,
находящихся в контакте с- полимерным материалом, и
др. В табл. 2 приведен ассортимент и области приме-
применения полимерных материалов, разрешенных к приме-
применению для изготовления изделий медицинского на-
назначения.
Полимеры, используемые в восстановительной
хирургии
Полимерные материалы, применяемые в восстанови-
восстановительной хирургии, предназначены для постоянной иле
временной замены пораженных или утраченных тканек
и органов живого организма. Требованиями, предъяв-
предъявляемыми к таким полимерам, являются физиологич,
безвредность, отсутствие токсичности и канцероген-
канцерогенных свойств, минимальное раздражающее действие на
контактирующие с полимером ткани и др. Кроме того,
конкретные области применения полимеров при про-
протезировании тканей и органов предъявляют разно-
разнообразные и жесткие требования по комплексу физико-
химич. и механич. свойств.
Биа инертные полимеры предназначены
для длительного обеспечения функционирования ор-
органов и тканей. Такие полимеры должны обладай
высокой устойчивостью к воздействию сред организ-
организма, не изменять своих первоначальных характерис-
характеристик при многократных деформациях, допускать тепло-
тепловую, радиационную и химическую стерилизующую об-
обработку.
Биоассимилируемые поли мерь
используют для временного обеспечения функциониро-
функционирования органа на период регенерации тканей. Биоас-
Биоассимилируемые материалы должны обладать способ-
способностью растворяться или деструктироваться под влия-
влиянием жидких сред с образованием нетоксичных продук-
927
ПОЛИМЕРЫ В МЕДИЦИНЕ
928
тов, ассимилируемых тканями, с последующим выведе-
выведением их из организма. Скорость превращения твердых
биоассимилируемых полимеров в жидкие продукты
под влиянием биологич. среды должна соответствовать
скорости регенерации тканей организма и составлять
от нескольких недель при протезировании мягких тка-
тканей до нескольких месяцев при протезировании кост-
костных тканей. В табл. 3 приведен ассортимент полимер-
полимерных материалов, используемых для внутреннего проте-
протезирования.
Таблица 3. Ассортимент и области применения полимеров,
используемых для внутреннего протезирования и создания
функциональных узлов «искусственных органов»
Наименование
материала
Полиэтилен
низкой плот-
плотности
Поликапролак-
там (капрон)
Поликарбонат
Политетрафтор-
Политетрафторэтилен (фторо-
пласт-4)
Полипропилен
Полиэтиленте-
рефталат
(лавсан)
Полиметилмета-
крилат
Кремнийорга-
нич. каучук
Цианакрилат-
ный клей
Области применения
Изделия, контактирующие с тканями орга-
организма
Протезно-ортопедич. изделия, хирургич. ни-
нити, изделия, контактирующие с тканями
организма
Корпуса и детали искусственных желудочков
и стимуляторов сердца
Протезы сосудов, клапанов сердца, фетр для
реконструктивных операций на сердце
Детали искусственных клапанов сердца, про-
протезы сосудов
Изделия для внутреннего протезирования и
восстановительной хирургии — сетки, нити,
протезы кровеносных сосудов, ленты для
пластики связок и сухожилий
Изделия для кератопротезирования, детали
аппаратов «искусственная почка», «серд-
«сердце — легкие» и др.
Изделия для внутреннего протезирования,
детали аппаратов искусственных органов
Бесшовное соединение тканей организма при
хирургич. операциях
Сердечно-сосудистая хирургия. Использование поли-
полимеров в этой области хирургии связано в первую оче-
очередь с протезированием клапанов сердца и сосудов. С
этой целью в клинич. практике используют след. поли-
полимерные материалы: для протезирования сосудов — во-
волокна из фторированных полиолефинов (фторлон), по-
полипропилена, полиэфирные волокна (лавсан); для кла-
клапанов сердца — кремнийорганические (силиконовые)
каучуки, полипропилен, волокна из фторлона. В экспе-
экспериментальных моделях искусственного сердца широко
используют поликарбонат. При нек-рых реконструк-
реконструктивных операциях на сердце применяют войлок различ-
различной плотности из фторлона (см. также Медицинские
нити).
Помимо общехирургич. требований к материалам,
применяемым для протезирования сердца и сосудов,
предъявляются и епецифич. требования: они не долж-
должны вызывать гемолиз (разрушение) крови и образова-
образование тромбов.
Ряд полимеров, таких как полиамиды, полистирол,
способствуют тромбообразованию. Лавсан, политетра-
политетрафторэтилен, полиэтилен, полиуретаны не влияют на
процесс тромбообразования, а нек-рые из полимеров
даже задерживают образование тромбов (кремнийорга-
нич. каучук, поливинилпирролидон и др.)- Большое
влияние на скорость тромбообразования оказывает
состояние поверхности полимерного материала. Име-
Имеются данные о влиянии на интенсивность тромбообразо-
тромбообразования электрич. потенциала поверхности материала,
а также его смачиваемости.
Проводятся работы по приданию антитромбогенно-
сти различным группам полимеров. Установлено, что
эффект тромбообразования можно подавить путем нане-
нанесения на поверхность имплантируемых материалов
коллоидного графита, обработкой стиролсульфокисло-
той, этиленимином, гепарином и др. (табл. 4).
Таблица 4. Влияние химической природы полимеров и обработ-
обработки их поверхности гепарином на время свертывания крови
Наименование
материала
Полистирол
Полиэтилен
Поливинилхлорид
Целлофан
Натуральный каучук
Бутадиен-винилпи-
ридиновый каучук
Этилен-пропиленовый
каучук
Кремнийорганичес-
кий фторсодержа-
щий каучук ....
Кремнийорганич. ка-
каучук
Время свертывания
крови, мин
необрабо-
необработанная по-
поверхность
9
11
12
6
10
12
5
18
15
обрабо-
обработанная
поверх-
поверхность
1440
1440
40
60
60
60
60
60
60
Содержание гемогло-
гемоглобина через 4 ч, мг%
необрабо-
необработанная по-
поверхность
16
25
10
600
13
37
15
15
5
обрабо-
обработанная
поверх-
поверхность
50
200
14
600
75
20
20
40
Хирургия внутренних органов и тканей. Хотя опера-
операции на легких, пищеводе, кишечнике, мочевыводящих
путях и др. с применением полимерных материалов
сравнительно многочисленны, большинство из них все
еще носит характер экспериментальных работ, и лишь
сравнительно небольшой круг материалов нашел ши-
широкое клинич. применение. К таким материалам в пер-
первую очередь следует отнести клеящие композиции на
основе эфиров цианакриловой кислоты (см. Полиакри-
Полиакриловые клеи). Соединение тканей при различных хирур-
хирургич. операциях с помощью клея — значительный шаг в
совершенствовании медицинских методик, т. к. обеспе-
обеспечивает герметичность соединения, возможность резкого
сокращения количества накладываемых швов и даже
бесшовного соединения, ускорение операций и сокра-
сокращение времени заживления ран.
Большое количество операций на диафрагме, при
лечении грыж, замещении дефектов тканей брюшной
стенки, закрытии дефектов пищевода и др. осуществ-
осуществляется с применением сетчатых материалов из капроно-
капронового волокна, полиэфирных волокон, волокон из поли-
полипропилена и фторлона. Имеются сообщения об успеш-
успешном протезировании желчных протоков, мочеточников
с помощью трубок из полиэтилена, пластифицирован-
пластифицированного поливинилхлорида, кремнийорганич. каучуков.
Однако ряд исследователей отмечает, что применение
протезов из указанных материалов дает лишь времен-
временный положительный эффект, т. к. в большинстве случаев
наблюдается «инкрустация» протезов солями, приводя-
приводящая к последующей их закупорке.
Весьма актуальная проблема хирургии легких —¦
восстановительные операции на трахеях, бронхах, а
также операции, связанные с необходимостью заполне-
заполнения послеоперационных полостей. Помимо клеев,
при этих операциях могут широко использоваться вспе-
вспененные и гелеобразные композиции на основе биоинерт-
биоинертных и биосовместимых полимеров. Имеются данные о
положительном опыте применения полиорганосилокса-
нов (монолитных и вспененных) для пломбирования
послеоперационных полостей, восстановления формы
грудной железы и др.
Травматология и ортопедия. Для создания различ-
различных изделий внешнего протезирования (протезов ко-
конечностей, ортопедич. вкладок, туторов и др.) широ-
широкое применение находят полиэтилен, поливинилхлорид,
стеклопластики, жесткие и эластичные пенопласты.
Применение полимеров для указанных целей позволяет
резко облегчить протезы, улучшить их функциональ-
функциональные, гигиенич. свойства и внешний вид.
Широкое развитие получили работы по созданию по-
полимеров для внутреннего протезирования суставов,
участков костей, сухожильных и мышечных связок.
929
ПОЛИМЕРЫ В МЕДИЦИНЕ
930
Имеется положительный опыт применения полиэтиле-
полиэтилена высокого и низкого давления, полиметилметакрилата
и поликарбоната для изготовления протезов коленных
и бедренных суставов. Установлена целесообразность
применения комбинированных протезов, в к-рых наря-
наряду с металлич. деталями используют части из полиоле-
финов. Полимеры с низким коэфф. трения можно нано-
наносить на поверхность металлич. протезов суставов для
улучшения их функциональных свойств.
Для замены сухожилий и связок применяют лавсано-
лавсановые ленты. Закрытие дефектов черепа осуществляют с
помощью пастообразных, отверждающихся без нагре-
нагревания композиций на основе акриловых полимеров и
сополимеров.
Актуальная проблема травматологии — создание раз-
различных соединительных элементов (штифтов, скоб) из
биосовместимых полимеров. Это позволит отказаться
от операций по извлечению этих элементов после за-
завершения регенерации костных тканей. Важной зада-
задачей является также разработка клеевых композиций,
обеспечивающих высокую прочность склеивания кост-
костных тканей.
Офтальмология. К. полимерным материалам, приме-
применяемым в офтальмологии, помимо общехирургических
требований, предъявляются требования по прозрачно-
прозрачности, смачиваемости поверхности слезной жидкостью,
устойчивости к воздействию жидких сред конъюнктивы
глаза и др.
Для создания искусственных хрусталиков и радуж-
радужной оболочки глаза применяют акриловые полимеры,
для искусственного стекловидного тела — гелеобраз-
ные кремнийорганич. композиции. Большой лечебный
эффект при офтальмологич. операциях достигается при
применении цианакрилатных клеев.
Стоматология и челюстно-лицевое протезирование.
Успехи, достигнутые в проведении стоматологич. опе-
операций, имеющих массовый характер, во многом связа-
связаны с широким внедрением в стоматологич. практику по-
полимеров, к-рые используют как в виде вспомогательных
материалов, так и непосредственно для создания сто-
стоматологич. протезов, изготовления искусственных зу-
Таблица 5. Характеристики отечественных полимерных
материалов стоматологического назначения
Наимено-
Наименование
материала
АКР-7
АКР-15
(Этакрил) ММА
Акреал
Протак-
рил
ЭГМАС-12
Норакрил-
-65
Состав*
порошок
жидкость
Эмульсион-
Эмульсионный окрашен-
окрашенный и пласти-
пластифицированный
ПММА, замут-
замутненный ZnO или
TiO2
Сополимер
метил-
акрилата,этил-
метакрилата
Суспензион-
Суспензионный окрашен-
окрашенный ПММА, за-
замутненный ZnO
или TiO2
Смесь ПММА
с перекисью
бензоила и ди-
дисульфан амином
Смесь ПВХ и
TiO2
ПММА, может
быть окрашен
в 6 цветов
Метилметакри-
лат, содержащий
стабилизаторы —
гидрохинон и ди-
фенилолпропан
Смесь метилово-
метилового и этилового
эфиров метакри-
ловой к-ты
ММА с добав-
добавкой метилолмета-
криламида и ста-
стабилизатора
ММА с актива-
активатором— диметил-
паратолуидином
Дибутилфталат
№1-ММА с
гидрохиноном и
диметилпарато-
луидином,
№2—ММАсме-
такриловой к-той
и гидрохиноном
Область приме-
применения
Изготовлениеба-
зисов, мостовид-
ных протезов, ис-
искусственных зу-
зубов, шин, лице-
лицевых протезов
Изготовление
базисов, съемных
протезов, искус-
искусственных зубов
Изготовление
базисов
Перебазировка
и починка съем-
съемных протезов
Изготовление
защитных боксер-
ких шин
Пломбирование
зубов, восстанов-
восстановление углов и
краев фронталь-
фронтальных зубов
* ПММА — полиметилметакрилат,
рид, ММА — метилметакрилат.
ПВХ — поливинилхло-
бов, ортодонтич. аппаратов, челюстно-лицевых проте-
протезов и др. В СССР выпускается широкий ассортимент
синтетич. материалов стоматологич. назначения
(табл. 5), основу большинства из к-рых составляют
полимеры и сополимеры акрилового ряда. Проводятся
работы по изысканию новых полимеров для стоматоло-
стоматологич. целей. Имеются сообщения об использовании для
челюстно-лицевого протезирования (исправление лице-
лицевых дефектов, изготовление искусственных ушных ра-
раковин и т. п.) полипропилена, поликарбоната, крем-
кремнийорганич. каучуков, пластифицированного поливи-
нилхлорида и др.
Полимеры, используемые в функциональных узлах
хирургических аппаратов
В отечественной пром-сти и за рубежом разработа-
разработаны многочисленные аппараты, выполняющие роль
отдельных органов или являющиеся средствами под-
поддержания функционирования систем человеческого
Таблица 6. Полупериоды переноса некоторых соединений
через различные полимерные мембраны
Соединение
Мочевина
Глицин
L-Аланин
Саркозин
Д,Ь-Серин
Креатинин . . . .
Мочевая к-та . . .
Аскорбиновая к-та
Глюкоза
Лимонная к-та . .
Тиаминхлорид . .
Сахароза
Иолупериод переноса, мин
пленка из
полиуре-
полиуретана на
основе по-
лиокси-
этилен-
гликоля
168
624
417
869
510
420
276
1056
258
490
1700
медноам-
миачная
целлюлоз-
целлюлозная
пленка
99
150
171
189
227
223
471
254
318
218
386
386
полиэти-
лентере-
фталатная
пленка
57
183
201
262
249
150
240
142
298
191
167
412
организма. К таким аппаратам относятся различные
аппараты искусственного кровообращения (АИК), пе-
ритониального диализа (АИП), вживляемые стимуля-
стимуляторы сердца и др. органов и т. п. К полимерам, исполь-
используемым в этих аппаратах, предъявляют те же жесткие
требования, что и к материалам, предназначенным для
внутреннего протезирования. Полимерные мембраны,
выполняющие в АИК и АИП роль основного функцио-
функционального узла, должны обладать селективной пропу-
скаемостью по отношению к компонентам крови, вы-
высокой эффективностью диализа, достаточной механич.
прочностью, оказывать наименьшее травмирующее
действие на кровь.
Целлофановые пленки ранее широко применялись для
указанных целей, но в настоящее время уже не удов-
удовлетворяют требованиям, к-рые предъявляются к мате-
материалам, предназначенным для создания портативных и
высокоэффективных диализаторов, АИК и др. аппара-
аппаратов. Мембраны нового типа получают путем модифи-
модификации пленок из целлофана, используют мембраны из
кремнийорганич. полимеров, модифицированных поли-
олефинов, блоксополимеров полиоксиэтиленгликоля с
полиэтилентерефталатом, полиуретановых эластомеров
и др. (табл. 6).
Полимеры направленного биологического действия
Роль полимеров в медицине, в фармакологич. аспек-
аспекте, пока относительно невелика. В лечебной практике
их используют мало. К веществам, вводимым в орга-
организм, тем более к таким, к-рые должны в растворен-
растворенном виде попасть в кровь, лимфу, межклеточные и кле-
клеточные полости и могут достигнуть любой части тела,
931
ПОЛИМЕРЫ В МЕДИЦИНЕ
932
любого его рецептора, предъявляются, естественно,
очень жесткие требования. До начала широкого исполь-
использования полимеров в фармакологии должно быть изу-
изучено множество вопросов, связанных с взаимодействием
полимер — организм. Однако потенциальные воз-
возможности получения положительных эффектов от при-
применения полимеров в этой области весьма велики и
поэтому экспериментальные (на животных) и клинич.
исследования приобретают все больший размах.
Полимеры применены как фармакологические (тера-
(терапевтические) препараты в виде лекарств или компонен-
компонентов лекарственных форм и композиций. Наиболее общим
свойством таких полимеров является их растворимость
(рассасываемость) в воде, водно-солевых или в био-
биологических (желудочный и кишечный соки, лимфа,
плазма) средах.
Перевод лекарственных соединений в полимерное со-
состояние позволяет: на более длительное время задер-
задержать лекарство в организме, т. е. пролонгировать его
действие; селективно направить в определенные орга-
органы или ткани; получить такие лекарственные формы ве-
веществ, в к-рых ранее они не могли применяться, напр,
нерастворимые вещества перевести в растворимые или
наоборот; инъекционные препараты превратить в пе-
роральные, а применявшиеся в виде порошков или таб-
таблеток — в инъекционные (ампульные).
В ряде случаев биологич. действие (сохранение или
повышение кровяного давления, дезинтоксикация,
интерфероногенное, противовирусное, антикоагуля-
ционное действие) проявляется синтетич. макромолеку-
макромолекулой, в структуру к-рой не введено никаких низкомоле-
низкомолекулярных фармакологич. веществ. Явно выраженным
терапевтич. действием обладают, напр., поливинилпир-
ролидон, карбоксилатные сополимеры, сульфовинол,
сульфодекстран, N-окись поливинилпиридина.
Наиболее широкие масштабы имеет применение во-
водорастворимых высокомолекулярных веществ в каче-
качестве крове- или плазмозаменителей.
Крове- и плазмозаменители (синонимы — инфузион-
ные среды, кровезамещающие, плазмозамещающие
р-ры). Введенные в кровяное русло кровезамещающие
жидкости (водные р-ры высокомолекулярных ве-
веществ) должны временно выполнять роль крови как
своеобразного «жидкого органа». Отсюда вытекают
особые требования к полимерам-кровезаменителям: дли-
длительно удерживаться в кровяном русле, для чего мол.
масса полимера должна быть достаточно высокой; пол-
полностью выводиться из организма или вступать в обмен
веществ; обладать постоянными физико-химич. свойст-
свойствами (осмотич. давлением, вязкостью и др.), близкими
по значению соответствующим показателям плазмы кро-
крови; не вызывать гемолиза (распада) или агглютинации
(склеивания) эритроцитов; не быть анафилактогенны-
ми, не вызывать сенсибилизации организма при пов-
повторном введении; быть нетоксичными, непирогенными,
легко стерилизоваться и выдерживать достаточно
длительные сроки хранения.
Основные функции кровезаменителей: заполнение
кровяного русла, обеспечивающее поддержание посто-
постоянного давления в' нем; удаление из организма ток-
токсичных веществ различного происхождения; перенос
питательных энергетич. веществ. Кровезаменители по
выполняемым ими лечебным функциям делят на три
главные группы: 1) противошоковые; 2) дезинтоксика-
ционные и 3) препараты парентерального питания. Со-
Соответственно различаются и нек-рые требования к
полимерным веществам. В качестве препаратов проти-
противошокового действия можно использовать полимеры с
достаточно высокой мол. массой (оптимально 30 000—
60 000), что обеспечивает длительное пребывание поли-
полимера в организме для восстановления гемодинамики.
Дезинтоксикаторы эффективны при сравнительно низ-
низкой мол. массе A0 000—20 000), т. к. они должны бы-
быстро выводиться из организма, унося токсичные веще-
вещества. Для препаратов третьей группы этот показатель
не регламентируется, т. к. они в организме расщепля-
расщепляются и ассимилируются (усваиваются).
Кровезаменители противошоко-
противошокового действия. Наиболее широко используют
для получения таких кровезаменителей плазму натив-
ной крови, декстран, поливинилнирролидон (см.
Винилпирролидона полимеры) и желатину. Из них
готовятся след. препараты: полиглюкин —
6%-ный солевой р-р продукта частичного гидролиза
соляной к-той нативного декстрана, синтезируемого
определенным штаммом бактерий (наиболее эффективна
фракция с мол. м. 55 000 ^ 15 000); гемовинил —
3,5%-ный солевой р-р фракции поливинилпирролидо-
на с мол. м. 30 000—40 000; желатиноль —
8%-ный р-р частично гидролизованной желатины,
в его состав входят различные полипептиды с мол. мас-
массой от 5000 и выше; раствор Б К- 8— получают из
гетерогенных белков, специально обработанных с
целью лишения их антигенных свойств; за рубежом
широко применяют препарат г е м а ц е л, получаемый
путем гидролиза и последующего ресинтеза пептидных
цепей желатины (мол. масса ок. 35 000).
Кровезаменители для дезинтокси-
дезинтоксикации. Дезинтоксикационный эффект, или свойст-
свойство р-ров полимеров выводить из организма токсины
бактериального и иного происхождения, обусловлива-
обусловливается способностью макромолекул сорбировать или свя-
связывать в комплексы вещества различной природы.
Наиболее эффективными препаратами являются: г е-
м о д е з — 6%-ный р-р низкомолекулярного поливи-
нилпирролидона с мол. м. 12 000—27 000 (до 80% пре-
препарата выводится почками в течение первых 4 ч); поли-
поливиниловый спирт с мол. м. 10 000; реополиглю-
к и н — низкомолекулярные фракции гидролизата
декстрана с мол. массой ок. 35 000.
Все кровезаменители готовят на физиологич. р-ре
с доведением рН до 5 — 7. В качестве др. компонентов
кровезаменителей, приближающих их по свойствам к
крови (достижение изотоничности и изоионичности)
и обуславливающих дополнительный лечебный эффект,
применяют глюкозу, лактат натрия, соли Na, К, Са,
Mg и др.
В экспериментах на животных и в клиниках в каче-
качестве кровезаменителей испытывается ряд др. пре-
препаратов на основе синтетических и природных полиме-
полимеров: гидроксиэтилкрахмал —6%-ный р-р частично гид-
ролизованного и обработанного окисью этилена крах-
крахмала (по терапевтич. действию и побочным реакциям
этот препарат близок декстрану); метилцеллюлоза —
2%-ный солевой р-р натриевой соли карбоксиметил-
целлюлозы с мол. м. 30 000—70 000; сополимеры окиси
этилена с окисью пропилена; р-ры левана (биосинте-
тич. препарат полифруктозы), гуммиарабика (мол. м.
2000), пектинов (мол. м. 4000—6000), фракций гидро-
гидропектина яблок, амилопектина и др.
Ведутся широкие исследования по синтезу поли-
полимерных кровезаменителей, к-рые, кроме вышеперечис-
вышеперечисленных основных свойств, обладали бы способностью к
переносу кислорода и углекислого газа, функциями ле-
лечебных препаратов направленного действия.
Кровезаменители для паренте-
парентерального питания представляют собой про-
продукты полного или частичного расщепления белков.
Пролонгаторы. Действие ряда лекарственных ве-
веществ можно продлить, если их вводить в р-ре вместе с
полимерами. В качестве таких р-ров используют крове-
кровезаменители (полиглюкин, поливинилпирролидон, поли-
поливиниловый спирт и др.). Чем выше мол. масса полиме-
полимера и его концентрация, тем длительнее действуют такие
препараты. При этом улучшается растворимость и
снижаются токсичность и побочные действия лекарст-
933
ПОЛИМЕРЫ В МЕДИЦИНЕ
934
венных веществ. Эффект пролонгации и уменьшение
токсичности обусловлены тем, что чекарственные веще-
вещества более или менее прочно связываются с полимерами,
затрудняется диффузия лекарства из места введения.
Большие молекулы (мол. масса более 50 000) с трудом
или вовсе не проходят через биологич. барьеры и более
длительно находятся в крови, лимфе или межклеточ-
межклеточной жидкости. По-видимому, постепенно отщепляясь и
достигая соответствующего рецептора, лекарствен-
лекарственное вещество проявляет свое действие.
Применение пролонгированных лекарств позволяет
уменьшить число приемов или инъекций; увеличить
вводимые дозы без увеличения токсичности и в то же
время уменьшить общее количество используемого ле-
лекарственного средства; уменьшить или устранить коле-
колебания концентрации активного вещества, неизбежные
при периодич. приемах обычных препаратов. В смеси с
полимерами более длительным действием обладают,
напр., антибиотики, инсулин, новокаин и др. Нек-рые
из препаратов такого типа уже используются в практи-
практике, др. проходят проверку на животных и в клинике.
Пролонгирующее действие полимеров м. б. усилено,
если использовать полимеры, имеющие функциональ-
функциональные группы; в этом случае могут образовываться более
прочные соединения типа комплексов или солей. Для
их получения используют поливинилпирролидон,
крахмал, декстран, поливиниловый спирт, полиэти-
ленгликоль и сополимеры. Наиболее известны комплек-
комплексы полимеров с иодом, к-рые обладают высокой бакте-
бактерицидной активностью. Их применяют как в виде вод-
водных р-ров, так и в виде гелей, пленок, нитей. Препарат
и о д и н о л — 1%-ный водный р-р йодного комплекса
поливинилового спирта, нашел широкое применение в
медицине и ветеринарии. В качестве антисептиков пред-
предложены йодные комплексы поливинилпирролидона.
Описано применение комплексов железа и декстрана
(для лечения анемии), кобальта и декстрана, производ-
производных полиэтиленоксида и различных лекарственных
препаратов.
Полимерные лекарственные вещества. Как уже отме-
отмечалось, биологич. активностью, свойствами терапевти-
терапевтически действующих препаратов обладает и ряд полиме-
полимеров, в структуре к-рых нет специально введенных ле-
лекарственных соединений. К таким полимерам можно
относить и плазмозаменители, поскольку они также
осуществляют лечебные функции (лечение шока, ожо-
ожоговой болезни и др.). Сульфированный поливиниловый
спирт м. б. использован как антикоагулянт крови —
заменитель гепарина. В качестве препаратов, нейтра-
нейтрализующих антикоагуляционное действие гепарина,
используют полимерные четвертичные соли. Широкое
практич. применение нашел препарат «полибрен»—
продукт взаимодействия тетраметилгексаметилендиами-
на с триметилендибромидом. Известно применение
поли-Г^-окисей винилпиридина (см. Винилпиридина
полимеры) для лечения силикозов. Имеются сведения
о синтезе длительно действующих полимерных веществ,
обладающих защитным действием при облучении.
Полимеры и сополимеры с кислотными функциональ-
функциональными группами эффективны в борьбе с вирусными за-
заболеваниями. В этом случае действие полимеров заклю-
заключается как в стимулировании выработки в организме
особого защитного вещества белковой природы —
интерферона, так и в непосредственной инактивации
вирусов. В качестве таких противовирусных и интер-
фероногенных препаратов испытываются полимеры
и сополимеры ненасыщенных карбоновых и сульфоно-
вых к-т, малеинового ангидрида и др. Наибольшей
интерфероногенной активностью обладают специфич-
специфичные комплексы породных полимеров — полинуклеоти-
дов (полигуаниловой, полицитидиловой к-т и др.),
получение к-рых возможно путем ферментативного и
химич. синтезов или их комбинацией.
Широкие возможности модификации известных и
получения новых лекарственных соединений представ-
представляют методы присоединения к полимерам терапевтич.
средств с помощью химич. связей, а также полимери-
полимеризация или поликонденсация соответствующих произ-
производных. При этом получаются фармакологически или
биологически активные препараты, специфика дей-
действия к-рых определяется макромолекулярной приро-
природой вещества: мол. массой, конформацией, прочностью
связей в основной полимерной цепи или в боковых
активных группах по отношению к гидролитическому
или ферментативному расщеплению.
Для синтеза полимерных лекарственных препаратов
методом полимер аналогичных превращений можно
использовать практически любые водорастворимые
полимеры с функциональными группами (альдегидны-
(альдегидными, кислотными, аминными и т. п.), напр, карбоцеиные
поликислоты (метакриловую, акриловую), сополимеры
винилпирролидона или винилового спирта, окислен-
окисленные или модифицированные иным образом декстраны,
крахмал, целлюлозу и т. д. Описано применение в ка-
качестве лекарственных веществ, присоединяемых к поли-
полимерам, антибиотиков, гормонов, ферментов, салицила-
тов, анестетиков, алкалоидов, противотуберкулезных
и противоопухолевых препаратов, витаминов и др.
Поведение полимерных лекарственных соединений в
организме, их эффективность, специфичность действия
и возможности применения изучаются пока в основном
в экспериментах на животных.
Полимер, используемый в качестве лекарства, напр,
плазмозаменителя или терапевтич. препарата, остается
в организме более или менее продолжительное время и
в конце концов должен выводиться в неизменном или
деструктированном виде. Полимеры с мол. массой до
12 000 выводятся практически нацело за несколько
часов.
Вспомогательные вещества для создания различных
лекарственных форм
Все более важное значение синтетич. полимеры при-
приобретают в создании новых лекарственных форм уже
известных терапевтич. средств и в качестве заменителей
восков, жиров и масел. Полимеры используют как без-
безжировые основы паст, мазей и пластырей,
а также для стабилизации р-ров, эмульсий, суспензий.
Требования к полимерам в отношении их физиологич.
активности в этих случаях менее спеиифичны, посколь-
поскольку практически все большие полимерные молекулы не
проникают через кожные покровы и клеточные мембра-
мембраны. Основными из применяемых для этих целей поли-
полимеров являются полиэтиленоксид (см. Окиси этилена
полимеры), поливиниловый спирт, поливинилпирроли-
поливинилпирролидон. В экспериментальных и поисковых работах исполь-
используют также ряд производных целлюлозы, гомо- и сопо-
сополимеры акриламида, винилпирролидона, винилового
спирта, этиленоксида и др.
Низкомолекулярные формы полиэтиленоксида (мол.
м. 4000—10 000) используют как заменители жировых
основ и вазелина. Преимущества их в том, что они раст-
растворяются в воде, обеспечивают хороший контакт
введенных в их состав лекарственных веществ с кожей,
слизистой или раневой поверхностью, и лекарства при
этом легко всасываются; при наружном применении
такие мази, в отличие от вазелиновых, образуют эла-
эластичную «кожицу», а затем легко смываются водой или
отдираются. В состав мазей вводят лекарственные
(гл. обр. против кожных заболеваний), дезинфицирую-
дезинфицирующие или бактерицидные вещества. Такие мази не про-
прогоркают и могут храниться длительное время. Эффек-
Эффективно применение их для массажа, а также для смазки
медицинских инструментов.
Поливиниловый спирт (ПВС) применяется в качестве
основы водорастворимых мазей при лечении кожных
935
ПОЛИМЕРЫ В ПИЩЕВОЙ ПРОМЫШЛЕННОСТИ
936
заболеваний. Широко используется действие ПВС
как защитного коллоида и поверхностно-активного
вещества для стабилизации р-ров, суспензий и эмуль-
эмульсий. В гормонотерапии, напр, при лечении диабета,
ПВС используют для создания устойчивых суспензий,
содержащих инсулин в тонкодисперсном состоянии,
допускающем инъекции. Такие препараты обладают
длительным и ровным действием. Известно примене-
применение ПВС для создания кровеостанавливающих средств
(порошки на основе ПВС и хлорного железа, р-ры ПВС
с добавкой сахара и мочевины), а также ряда др. лечеб-
лечебных препаратов, включая пероральные и инъекционные
формы, пленки, р-ры для пропитки марли и т. д.
Перспективной формой использования ПВС является
создание на его основе гелей (студней) с включенны-
включенными в их состав лекарственными веществами. Гели могут
содержать сшивающие агенты, образующие (в зависи-
зависимости от целей и способа использования) более или
менее прочные связи между молекулами ПВС. В част-
частности, м. б. использованы борная к-та, бура, конго
красный, иод и др. Темп-pa плавления гелей может
регулироваться соотношением ингредиентов, а также
концентрацией и вязкостью исходного ПВС. Гели пла-
плавятся в интервале темп-р 50—70 °С и застывают при
30—45 °С. Скорость их рассасывания в организме мож-
можно регулировать, используя полимеры (ПВС, его про-
производные, сополимеры винилового спирта) различной
мол. массы, а также меняя условия обработки поли-
полимерных композиций. По консистенции и плотности ге-
гели м. б. мягкие или плотные (хрящевидные).
Поливинилпирролидон с успехом применяют в ка-
качестве основы различных мазей, кремов, косметических
жидкостей и лекарств для кожи. В отличие от ПВС
и полиэтиленоксида, он растворим не только в воде,
но и в ряде органич. жидкостей, что бывает целе-
целесообразно использовать при приготовлении некоторых
препаратов.
В качестве покрытий и составных частей табле-
таблеток используют гомополимеры, композиции (сме-
(смеси) полимеров и сополимеров, обеспечивающие требуе-
требуемые свойства по проницаемости (размерам пор), раст-
растворимости, рассасываемости в различных средах, адге-
адгезионным и др. показателям. Нек-рые лекарственные
вещества должны быть защищены от инактивации или
разрушения содержимым желудка, чтобы их действие
проявилось после всасывания в том или ином отделе
кишечного тракта. Важным является и регулирование
скорости всасывания лекарства. В качестве полиме-
полимеров, растворимых в желудке, можно использовать: по-
ливинилпиридин; поливинилалкилпиридины; ацетат
и диэтиламиноацетат целлюлозы, бензиламинометил-
целлюлозу; статистич. и привитые сополимеры поливи-
ниламина, поливинилацетата, поливинилацеталей, эфи-
ров целлюлозы и др.
В качестве соединений, не растворимых в желудке,
но растворимых в кишечнике, применяют полимеры со
свободными карбоксильными группами и их производ-
производные: производные целлюлозы (ацетилфталилцеллюлоза
и ее аммонийная соль), сополимеры малеиновой, акри-
акриловой и метакриловой к-т.
В ряде случаев нужны соединения, обладающие спо-
способностью растворяться (с различной скоростью) как
в щелочной, так и в кислотной среде, но не растворяю-
растворяющиеся в нейтральной среде. В качестве таких веществ
используют тройные сополимеры, состоящие, напр.,
из звеньев винилпиридина (или алкилвинилпиридина),
акриловой к-ты и какого-либо винильного мономера,
служащего для регулирования гидрофобности макро-
макромолекул. Таблетки с использованием пористых ионо-
ионообменных смол применяют для пролонгации действия
нек-рых лекарств, вводимых перорально. Разработаны
методы создания таблеток с двух- и многослойными по-
полимерными покрытиями.
Расширяется использование полимеров для создания
оболочек капсул, в к-рые заключаются лекарствен-
лекарственные вещества. Ранее такие оболочки (напр., из желати-
желатины) создавались только для лекарств перорального
применения. В последние годы разработаны способы
получения микрокапсул таких размеров (несколько
мкм в диаметре), что их суспензии можно вводить
инъекционно. Помещенные внутри микрокапсул белки,
ферменты, суспендированные вещества не выходят за
их пределы, но могут реагировать с проникающими
внутрь оболочек капсул низкомолекулярными соеди-
соединениями и осуществлять обменные процессы как в аппа-
аппаратах (напр., искусственная почка), так и в организме
(детоксикация, изменение баланса ионов или молекул
и др.). Делаются попытки заключения в микрокапсу-
микрокапсулы гемоглобина и создания искусственных эритроцитов.
См. также Микрокапсулирование.
Известно применение ПВС и его сополимеров, дек-
страна, полиуретанов и производных целлюлозы для
создания гемостатических (кровеостанавливающих)
средств, применяемых в виде пористых материалов
(губок), порошков, пленок, р-ров для пропитки марли
и т. п. В качестве композиций для пластырей исполь-
используют бутилированные, ацетилированные или формили-
рованные полимеры и сополимеры ПВС (напр., сополи-
сополимеры с хлорвинилацетатом) и ряд др. сополимеров. Ве-
Ведутся исследования по применению полимеров (напр.,
гомо- и сополимеров винилпирролидона, окиси этилена,
винилового спирта) для консервации трансплантатов
(в том числе мозговой ткани, крови).
Лит.: Полимеры в медицине. [Сб. ст.], пер. с англ., под ред.
Н. А. Платэ, М., 1969; П е т р о в с к и й Б. В., Соловь-
Соловьев Г. М., Шумаков В. И., Протезирование клапанов
сердца, М., 1966; Рабинович И. М., Применение поли-
полимеров в медицине, Л., 1972; Токсикология высокомолекуляр-
высокомолекулярных материалов и химического сырья для их синтеза. Сб.,
под ред. С. Л. Данишевского, М.— Л., 1966; Справочник
по кровезаменителям и препаратам крови, М., 1969; Уша-
Ушаков С. Н., Синтетические полимеры лекарственного назна-
назначения, Л., 1962; 3-й Симпозиум по физиологически активным
синтетическим полимерам и макромолекулярным моделям
биополимеров. Тезисы докладов, Рига, 1971; Всесоюзный сим-
симпозиум «Синтетические полимеры медицинского назначения».
Тезисы докладов, Ташкент, 1973; XXIII International congress
of pure and applyed chemistry, Boston, 1971.
А. Б. Давыдов, В. А. Нропачев.
ПОЛИМЕРЫ В ПИЩЕВОЙ ПРОМЫШЛЕННОСТИ
(polymers.in food industry, Polymere in Nahrungsmittel-
industrie, polymeres dans industrie alimentaire).
Содержание:
Конструкционные материалы и покрытия в пище-
пищевом машиностроении 937
Таро-упаковочные материалы , 938
Уплотнительные пасты 941
Консервные лаки и эмали 942
Иониты ., 942
Данная статья посвящена рассмотрению проблем,
связанных с применением в пищевой пром-сти только
синтетич. полимеров.
Полимерные материалы, используемые в пищевой
пром-сти, должны соответствовать комплексу опреде-
определенных санитарно-гигиенич. требований, обусловлен-
обусловленных контактом этих материалов с продуктами питания:
не изменять органолептич. свойств продуктов (вкус,
запах, цвет и др.), не содержать компонентов (в част-
частности, токсичных), к-рые могут экстрагироваться
пищевыми средами или реагировать с ними.
Обязательное условие применения полимерных ма-
материалов в пищевой пром-сти — разрешение органов
санитарного надзора, к-рое выдается на основании
комплекса испытаний, включающих оценку органо-
органолептич. свойств, а также санитарно-химич. и токсико-
логич. 'исследования полимеров и отдельных ингре-
ингредиентов, входящих в состав композиционных материа-
материалов и изделий. При санитарно-химич. исследованиях
определяются концентрация и состав соединений,
937
ПОЛИМЕРЫ В ПИЩЕВОЙ ПРОМЫШЛЕННОСТИ
938
мигрирующих из материала в пищевую среду в усло-
условиях, приближающихся к эксплуатационным. При
токсикологич. исследованиях выявляется действие ис-
испытуемых материалов или вытяжек из них на организм
теплокровных животных. Подробно см. Санитарно-
гигиеническая характеристика.
По степени пригодности для контакта с продуктами
питания полимерные материалы делят обычно на след.
группы: 1) допущенные без ограничений; 2) допущен-
допущенные для контакта с нек-рыми продуктами при опреде-
определенных условиях; 3) не допущенные из-за токсичности
или изменения состава при соприкосновении с пище-
пищевыми средами.
Конструкционные материалы и покрытия в пищевом
машиностроении. Пищевое машиностроение относится
к числу крупных потребителей полимерных материалов;
их применение в этой отрасли пром-сти обусловливает
значительный технико-экономич. эффект. Так, при
транспортировке зерна вместо металлич. шнеков ис-
используют шнеки с рабочей поверхностью, покрытой
полиуретаном, поликапролактамом, политетрафтор-
политетрафторэтиленом (фторопластом-4). Благодаря уменьшению
коэфф. трения зерна о поверхность шнека производи-
производительность при транспортировке повышается в среднем
на 25% и, кроме того, зерно значительно меньше по-
повреждается. В рыбоперерабатывающей, консервной,
молочной пром-сти и др. широко распространены транс-
транспортерные ленты, звенья к-рых изготовляют из срав-
сравнительно легких и коррозионностойких полиамидов
или полиэтилена высокой плотности (см. Этилена
полимеры), а также подшипники из фторопласта-4 и
полиамидов. Смазкой таких подшипников может слу-
служить вода, благодаря чему удается сохранить вкусо-
вкусовые качества и питательную ценность пищевых про-
продуктов.
Для контакта с наиболее агрессивными пищевыми
средами перспективна аппаратура из стеклопластиков,
плакированная химстойкими термопластами, напр, по-
лиолефинами, винипластом, фторопластами.
Из «пищевых» резин изготовляют прокладки молоч-
молочных пастеризаторов, аппаратов и машин винодельче-
винодельческой, пивоваренной, консервной пром-сти, уплотни-
тельные рукава, транспортерные ленты, приводные
ремни; из теплостойких резин на основе кремнийорга-
нических каучуков и бутадиен-нитрилъных каучуков —
прокладки для сушильных агрегатов, ультрапастери-
ультрапастеризаторов и др. аппаратов, в к-рых жидкие пищевые
среды стерилизуют при 120—130 °С и выше.
В узлах автоматов, предназначенных для изготов-
изготовления бумажных пакетов с полиэтиленовым покрытием,
заполнения их молоком и герметизации сваркой, уста-
устанавливают амортизаторы с прижимными роликами из
резин на основе фторсодержащих каучуков или кремний-
органич. каучуков, наполненных фторопластом-4. Про-
Продолжительность эксплуатации деталей из этих резин
в 7—8 раз больше, чем, например, деталей из бута-
диеннитрильного каучука. Эффективно применение
резин высокой твердости и износостойкости из ком-
композиций бутадиен-стирольного и натурального кау-
каучуков при изготовлении дек станков для шелуше-
шелушения зерна.
Использование клеев синтетических, напр, поливи-
поливинил ацетатной дисперсии, вместо натуральных дает
возможность изготовлять бумажную тару улучшенного
качества на высокопроизводительных автоматах.
Важное значение в пищевой пром-сти имеют поли-
полимерные антиадгезионные покрытия, к-рые наносят на
металлич. конструкции, чтобы предотвратить прили-
прилипание сырья и полуфабрикатов к поверхностям обору-
оборудования. Антиадгезионными свойствами обладают
нек-рые лакокрасочные покрытия, напр, получаемые
при нанесении суспензий фторопластов, феноло-фор-
мальдегидных лаков, модифицированных поливинилбу-
тиралем и наполненных порошком фторопласта-4 (см.
Феноло-алъдегидные лаки и эмали), кремнийорганич.
лаков, напр, полиметилфенилсилоксановых, модифици-
модифицированных эпоксидными смолами (см. Кремнийоргани-
ческие лаки и эмали). Помимо лакокрасочных, распро-
распространены также антиадгезионные покрытия, получае-
получаемые при нанесении порошков модифицированных фто-
фторопластов, поливинилбутираля, полиэтилена высокой
плотности методом напыления. Иногда металлич. де-
детали обкладывают листовыми или пленочными поли-
полимерными материалами, а ткани для транспортерных
лент обрабатывают кремнийорганическими жидкостями,
гл. обр. полиэтилгидросилоксановыми.
Металлич. инвентарь или оборудование с антиадге-
антиадгезионными покрытиями (напр., транспортерные ленты,
формы) используют в тесторазделочных линиях хлебо-
хлебопекарных предприятий, при транспортировке фарша,
выпечке мясных хлебов и ветчины, замораживании
пельменей, в механизированных линиях изготовления
кулинарных изделий на рыбоконсервных предприятиях,
выпечке пшеничного и ржаного хлеба и др. Антиадге-
Антиадгезионные покрытия наносят также на внутреннюю по-
поверхность бункеров, из к-рых сыпучие и пастообраз-
пастообразные продукты поступают в расфасовочные и укупороч-
укупорочные автоматы. При этом повышаются срок службы и
производительность оборудования. Кроме того, благо-
благодаря исключению операций очистки и смазки деталей
сокращается расход пищевых жиров на смазку, улуч-
улучшаются санитарное состояние цехов и качество про-
продукции.
С целью антикоррозионной защиты емкостей для со-
солевого и дрожжевого р-ров в хлебопекарном производ-
производстве, ковшей вертикальных элеваторов и лотков зер-
зерновых сепараторов на мукомольных предприятиях и
др. оборудования применяют порошкообразные поли-
олефины, к-рые наносят напылением. Поверхность обо-
оборудования, эксплуатируемого в наиболее агрессивных
средах, защищают покрытиями из политрифторхлорэти-
лена (фторопласта-3) и сополимеров тетрафторэтилена.
Хорошей адгезией к металлич. поверхностям, меха-
нич. прочностью и химстойкостыо обладают антикор-
антикоррозионные покрытия, получаемые при нанесении эпок-
эпоксидной смолы, наполненной минеральными наполни-
наполнителями. После отверждения и промывки кислотными и
щелочными р-рами покрытия могут контактировать со
многими пищевыми средами. Их используют с целью
защиты внутренних поверхностей больших емкостей
для вина, спирта, плодовых соков, цистерн для молоч-
молочных продуктов, аппаратов хлебопекарной пром-сти
и дрожжевого производства, бункеров расфасовочных
автоматов, силосов для бестарного хранения муки,
деталей рыбоперерабатывающих машин, эксплуати-
эксплуатируемых в морских условиях.
Декоративно-защитные покрытия пищевого оборудо-
оборудования получают с использованием поливинилбути-
ральных и полиэтиленовых порошковых красок, наноси-
наносимых напылением, а также разнообразных лакокрасоч-
лакокрасочных материалов (см. Декоративные лакокрасочные по-
покрытия). На пищевое оборудование не наносят, как
правило, рельефных покрытий, т. к. их стерилизация
вызывает затруднения.
Таро-упаковочные материалы. Для упаковки пище-
пищевых продуктов используют одно- и многослойные
пленки (см. Пленки полимерные)', комбинированные
материалы на основе бумаги, пропитанной различными
полимерными композициями, а также на основе алю-
алюминиевой фольги или бумаги с полимерными покрытия-
покрытиями; полужесткую и жесткую тару (бутыли, флаконы,
стаканы, тубы). Такую тару изготовляют методами
вакуумформования, пневмоформования, литья под дав-
давлением, экструзии с раздувом и др. и используют гл.
обр. для упаковки жидких и пастообразных продуктов
на высокопроизводительных линиях, объединяющих
939
ПОЛИМЕРЫ В ПИЩЕВОЙ ПРОМЫШЛЕННОСТИ
940
в одном потоке изготовление самой тары, заполнение
ее продуктом, герметизацию и товарную отделку.
Особый вид упаковки, защищающей некоторые про-
продукты от потери необходимой влаги (усушки) и дей-
действия микроорганизмов, — полимерные покрытия, по-
получаемые из расплавов, содержащих парафин, поли-
полиэтилен и полиизобутилен, из водных р-ров поливини-
поливинилового спирта, спиртового раствора поливинилбутираля,
водных дисперсий поливини л ацетата, сополимера ви-
нилиденхлорида с винилхлоридом и др.
Требования к упаковочным материалам определяют-
определяются видом продуктов, условиями их обработки, хране-
хранения и транспортировки. Так, для обеспечения герме-
герметичности упаковки и ее стойкости к ударным нагруз-
нагрузкам необходимы материалы, обладающие достаточной
механич. прочностью и эластичностью; для упаковки
гигроскопичных продуктов '— влагонепроницаемые ма-
материалы; для упаковки в вакууме или в атмосфере
инертного газа — газонепроницаемые. В отдельных
случаях, напр, при упаковке биологически активных
продуктов, используют материалы с селективной газо-
газопроницаемостью. Нек-рые материалы должны быть не-
непроницаемы для пахучих веществ и жиров, обладать
достаточной морозостойкостью и стойкостью к старе-
старению, обеспечивающими сохранность как самой упаков-
упаковки, так и пищевых продуктов в различных условиях.
Для использования на расфасовочных автоматах необ-
необходимы материалы, обладающие способностью свари-
свариваться, а в нек-рых случаях также и достаточной жест-
жесткостью, чтобы сохранять форму упаковки после ее
заполнения продуктом. Многие пленочные материалы
должны усаживаться, плотно облегая продукты слож-
сложной конфигурации, быть пригодными для нанесения
красочной печати (см. Печать на полимерах), а также
прозрачными для возможности визуального контроля
содержимого.
Для упаковки охлажденного мяса, к-рое
необходимо предохранять от изменения цвета (обуслов-
(обусловленного разрушением миоглобина при отсутствии до-
доступа кислорода) и др. органолептич. свойств, а также
от действия бактерий наиболее пригоден целлофан
с наружным лаковым покрытием. Для этой цели ис-
используют также нелакированный целлофан, пленки
из поливинилхлорида, сополимера винилиденхлорида
с винилхлоридом (саран), полиэтилена, полистирола,
гидрохлорида каучука. Срок хранения мяса в поли-
полимерной упаковке 2—3 сут при 0 °С и 1,5 сут при 6 °С.
См. также Гидратцеллюлозные пленки, Поливинилхло-
ридные пленки, Поливинилиденхлоридные пленки, По-
лиолефиновые пленки, Полистиролъные пленки, Гидро-
Гидрохлорид каучуковые пленки. Соленое мясо, пред-
предназначенное для длительного хранения, расфасовывают
и упаковывают (напр., в США) на высокопроизводи-
высокопроизводительных автоматах в вакууме или в атмосфере инерт-
инертного газа. В качестве упаковочных материалов, к-рые
должны защищать продукт от проникновения кислоро-
кислорода и влаги, а также от действия света применяют много-
многослойные пленки целлофан — полиэтилен, полиэтилен-
терефталат — полиэтилен, полиамид — полиэтилен
(см. также Полиэтилентерефталатные пленки, По-
Полиамидные пленки), саран — поливинилхлорид — саран
и целлофан — фольга — полиэтилен. Используют так-
также пленки из поликарбоната, полиуретана или поли-
поливинилового спирта в сочетании со сваривающейся
(обычно полиэтиленовой). пленкой.
При упаковке мяса, прошедшего тепло-
тепловую обработку (обычно после предваритель-
предварительного посола), используют те же пленки, что и для со-
соленого мяса. Для мясных консервов, подлежащих
стерилизации, особенно пригоден комбинированный
материал полиэтилентерефталат — фольга — полиэти-
полиэтилен, а также др. комбинированные материалы со слоя-
слоями фольги.
Наиболее компактный и удобный упаковочный мате-
материал для замороженной птицы — усадочные плен-
пленки типа саран, а также гидрохлоридкаучуковые и по-
полиэтиленовые пленки. Иногда птицу перед упаковкой
помещают для поглощения влаги на лотки из пенопо-
листирола или пенополивинилхлорида. При получении
колбасных и сосисочных оболочек
используют преимущественно целлофан, а также длин-
длинноволокнистую изотропную бумагу, пропитанную вис-
вискозой, пленки типа саран и поливинилепиртовые.
При упаковке натурального сыра, к-рый необ-
необходимо защищать от проникновения кислорода, бакте-
бактерий, влаги, применяют целлофан с двухсторонним
влагостойким покрытием, чаще всего из пленок типа
саран или гидрохлоридкаучуковых. В такой упаков-
упаковке сыр можно хранить до 3 сут. Более длительное
хранение E—8 сут) возможно в многослойных плен-
пленках целлофан — саран — полиэтилен (пригодны для
упаковки в вакууме), полиэтилентерефталат — са-
саран — полиэтилен, полиамид — саран — полиэтилен
или полипропилен — полиэтилен — саран — поли-
полиэтилен.
Плавленый сыр (он более стабилен, чем натураль-
натуральный, т. к. подвергается пастеризации при изготовле-
изготовлении) упаковывают в фольгу с лаковым покрытием на
основе сополимера винилхлорида с винилацетатом,
в комбинированный материал бумага — фольга — по-
полиэтилен, в пленку из поливинилхлорида или типа са-
саран. Сыр м. б. также расфасован в целлофан, покрытый
микровоском, в состав к-рого входят церезин и низко-
низкомолекулярный полиэтилен (в этом случае расплавлен-
расплавленный сыр заливают в пакеты, к-рые затем герметизи-
герметизируют сваркой). Широко распространена упаковка
плавленого сыра и в жесткую тару, напр, в ста-
стаканы из ударопрочного полистирола (см. Стирола по-
полимеры).
Свежую рыбу упаковывают в сетки или в пакеты
из полиэтилена (иногда в пакеты вкладывают лотки
из пенополистирола, поглощающие жидкость). Для
упаковки мороженой рыбы используют обычно целло-
целлофан, полиэтилен или картонные коробки, покрытые
изнутри слоем полиэтилена или сополимера винилиден-
винилиденхлорида с винилхлоридом. Упаковка копченой рыбы
должна обеспечивать сохранность запаха продукта,
предохранять его от проникновения посторонних за-
запахов, а также защищать от бактерий. Обычно такую
рыбу упаковывают в вакууме в многослойные пленки
целлофан — полиэтилен, полиэтилентерефталат — цел-
целлофан — полиэтилен, а также в пленку из полиами-
да-12 (в последнем случае рыбу можно разогревать,
не снимая упаковки). Соленую рыбу часто упаковы-
упаковывают в деревянные бочки или ящики с вкладышами
из полиэтилена или пластифицированного поливинил-
поливинилхлорида, к-рые герметизируют сваркой. Рыбу в рас-
рассоле расфасовывают также в банки из полиэтилена
низкой плотности и герметизируют с помощью крышек
из этого же полимера.
Гигроскопичные кондитерские изделия
(напр., карамель, ирисы) упаковывают во влагонепро-
влагонепроницаемые пленки, гл. обр. полипропиленовые, а также
в комбинированные материалы полиэтилен — фольга.
Применение целлофана в этом случае ограничивается
тем, что при контакте с гигроскопичным продуктом
он становится хрупким. Использование нек-рых пле-
пленок вызывает затруднения при упаковке на высокопро-
высокопроизводительных автоматах вследствие возникновения
зарядов статич. электричества.
Менее гигроскопичные кондитерские изделия (напр.г
желейные конфеты, мармелад, марципаны) можно упа-
упаковывать в паропроницаемые пленки из нелакирован-
нелакированного целлофана или ацетата целлюлозы (см. Эфиро-
целлюлозные пленки). При необходимости длительного
хранения практически негигроскопичных шоколадных
941
ПОЛИМЕРЫ В ПИЩЕВОЙ ПРОМЫШЛЕННОСТИ
942
изделий используют комбинированный материал поли-
этилентерефталат — фольга — полиэтилен. Такая упа-
упаковка предотвращает образование плесени, а также
миграцию жира и сахара на поверхность шоколадного
изделия при повышенных темп-рах.
Хлебобулочные изделия упаковывают
гл. обр. в полиэтиленовые или полипропиленовые плен-
пленки с целью защиты от черствения, образования пле-
плесени, а также соблюдения правил гигиены. При необ-
необходимости упаковку герметизируют на специальных
автоматах с помощью зажимов из пластмассы или
металла. Особый интерес представляет применение
пленки из полиамида-12, в к-рой можно хранить за-
замороженное тесто, а также выпекать хлеб. После сте-
стерилизации хлеб может сохраняться в такой упаковке
до 7 мес.
Для упаковки свежих овощей и фрук-
фруктов большое значение имеют материалы с селектив-
селективной газопроницаемостью (напр., более проницаемые
для СО2 и менее — для О2), обеспечивающие возмож-
возможность длительного хранения этих продуктов. Наиболее
пригодны для них пленки из кремнийорганич. каучу-
ков, а также гидрохлоридкаучуковые. Используют,
кроме того, пленки из полиэтилена (в том числе перфо-
перфорированные), из пластифицированного поливинилхло-
рида, ацетата целлюлозы. См. также Полимеры в сель-
сельском и водном хозяйстве.
Молоко, кисломолочные продук-
продукты, с о к и упаковывают в бумагу, покрытую изнутри
полиэтиленом, а снаружи — парафином. Для стерили-
стерилизуемых жидких продуктов используют также бумагу,
покрытую полипропиленом, и комбинированный мате-
материал бумага — фольга — полипропилен.
Для упаковки полуфабрикатов и кон-
концентратов применяют обычно комбинированные
материалы с промежуточным слоем фольги, напр, по-
лиэтилентерефталат — фольга — полиэтилен, бума-
бумага — фольга — полиэтилен. Используют также одно-
однослойные пленки, напр, поливинилспиртовые или по-
полиэтиленовые, многослойные пленки саран — целло-
целлофан — саран, а также бумагу с нанесенным из водной
дисперсии покрытием на основе сополимера винили-
денхлорида с винилхлоридом. Иногда, напр, при упа-
упаковке кофе, в картонные коробки помещают вклады-
вкладыши, напр, из пленок типа саран. Полимерная упаков-
упаковка надежно защищает концентраты и полуфабрикаты
от проникновения влаги, измельчения и прогоркания.
В нек-рых случаях в пищевые концентраты для повы-
повышения их стабильности при хранении вводят микро-
капсулированные (защищенные оболочкой, к-рая раз-
разрушается только при повышенных темп-pax или др.
воздействиях на продукт) жиры и душистые вещества
(см. Микрокапсулирование).
Уплотнительные пасты. Эти материалы, представ-
представляющие собой 40—98%-ные дисперсии или р-ры поли-
полимерных композиций, имеют важное значение при упа-
упаковке пищевых продуктов в металлическую, стеклян-
стеклянную или др. тару. Пасты наносят с помощью высоко-
высокопроизводительных автоматов на металлич. крышки,
к-рые затем подвергают термообработке в течение
0,5—1 мин при 100—240 °С (в зависимости от состава
пасты и ее назначения). Основой паст могут служить
синтетич. латексы (напр., бутадиен-стирольные — см.
Латексы синтетические), натуральный каучук, поли-
винилхлорид, композиции полиэтилена высокой плот-
плотности с полиизобутиленом. Они содержат обычно пла-
пластификаторы, наполнители, эмульгаторы, стабилиза-
стабилизаторы и др. ингредиенты. Напр., широко распростра-
распространенная паста для герметизации металлич. колпачков
(кроненпробок), используемых при укупорке бутылок
с безалкогольными напитками, состоит из примерно
равных (по массе) количеств поливинил хлорида, ди-
октилфталата и сульфата бария. В пасты, к-тэыми гею-
метизируют стерилизуемую продукцию, вводят от-
вердители или вулканизующие вещества. Отвержден-
ные (вулканизованные) материалы можно использо-
использовать при ~ 125 °С.
Консервные лаки и эмали. Назначение этих материа-
материалов — защита металлич. консервной тары от корро-
коррозии. Важнейшие требования к покрытиям на их осно-
основе — высокая устойчивость к действию различных кон-
консервных сред, сохранение свойств в условиях стерили-
стерилизации, способность выдерживать механич. воздействия
при изготовлении тары и хорошая адгезия к ее по-
верхности. Наиболее перспективны эпокси-фенольные
консервные лаки, а также белковоустойчивые эмали,
к-рые получают введением в эти лаки окиси цинка.
См. также Консервные лаки и эмали.
Иониты. При обработке свежего коровьего молока
катионообменными смолами получают т. наз. ионит-
ное молоко, к-рое характеризуется уменьшенным со-
содержанием кальция и более равномерным распределе-
распределением частиц казеина. Такое молоко пригодно даже для
кормления грудных детей. При использовании анионо-
обменных смол в производстве сгущенного молока сни-
снижается его кислотность и в 3—4 раза ускоряется про-
процесс сгущения. С помощью ионитов удаляют катионы
железа, меди и марганца из промывной воды, приме-
применяемой в производстве сливочного масла; при этом
уменьшается окисляемость молочного жира.
В производстве сахара иониты служат для осветле-
осветления свекловичного сока, ускорения операции очистки
соков второй сатурации. Благодаря их использованию
выход сахара повышается на 5—10%. В винодельче-
винодельческой пром-сти с помощью ионитов очищают вино и
сусло от солей кальция, калия, железа и тяжелых
металлов, уменьшают кислотность вин, осветляют их
и повышают стабильность. Нек-рые катиониты, способ-
способствующие ускорению биохимич. процессов, используют
для сокращения сроков старения вин.
В консервной пром-сти иониты применяют для ста-
стабилизации и уменьшения кислотности плодово-ягодных
соков, очистки маточных р-ров в производстве лимон-
лимонной, молочной, винной к-т, а также для очистки ук-
уксусной, щавелевой и муравьиной к-т от катионов ни-
никеля, железа, кобальта, магния. В производстве кон-
кондитерских изделий и в хлебопечении иониты служат
для очистки патоки и дрожжей, в пивоварении —
для осветления пива, в производстве фруктовых вод —
для удаления избытка к-ты.
Перспективы применения полимеров в пищевой
пром-сти связаны, в первую очередь, с увеличением
объемов их использования для упаковки. Это обуслов-
обусловлено созданием высокопроизводительного расфасовоч-
но-упаковочного оборудования и расширением сети
магазинов самообслуживания, торгующих гл. обр.
расфасованными продуктами. Большое значение в
связи с этим приобретает создание полимерных мате-
материалов, дифференцированных применительно к свой-
свойствам определенных пищевых продуктов, характеру
их потребления, способу реализации, особенностям
хранения и транспортировки.
Важные направления в области усовершенствования
оборудования для пищевой пром-сти — разработка
эффективных средств антикоррозионной защиты ме-
металлич. поверхностей, а также применение в деталях
оборудования тепло- и морозостойких полимеров с
целью интенсификации технологич. процессов и со-
сохранения питательной ценности продуктов при их
переработке.
Лит.: Гуль В. Е., Беляцкая О. Н., Пленочные
полимерные материалы для упаковки пищевых продуктов, М.,
1968; Г е н е л ь С. В., КестельманН. Я., К е с т е л ь-
ман В. Н., Полимерные материалы в пищевом машинострое-
ТТ Т/Г Т/Г О T/Гптт TV/Г A ClQ Cl • A ¦¦-» -»-г •>.•>. ~ ~ * ¦*-» •*• *
943
ПОЛИМЕРЫ В РАДИОЭЛЕКТРОНИКЕ
944
производствах и способы защиты, 2 изд., М., 1965; Знамен-
Знаменский Н. Н., Полимерные материалы в молочной промыш-
промышленности, 2 изд., М., 1967; Ларионов В. В., Применение
пластмасс в рыбной промышленности, М., 1965; Лепил-
к и н А. Н., НоздринС. И., С о к о л М. Г., Пенопластиче-
ские массы в мясной и молочной промышленности, М., 1968;
Применение полимерных материалов в консервной промыш-
промышленности, М., 1971; Ткачев Н. И., Полимеры в хлебопе-
хлебопекарной, кондитерской, макаронной и дрожжевой промышлен-
промышленности, 2 изд., М., 1970; Применение синтетических материалов
в винодельческой промышленности, М., 1966; Шишки-
наН. Н., Назаров А. С, Применение полимерных пле-
пленок для упаковки мясопродуктов, М., 1965; Корень-
ков Г. Л. [и др.], Хим. пром-сть за рубежом, в. 1 (85), 3
A970); Аксенова Т. В., там же, в. 8 (92), 51 A970);
ШевченкоМ. Г., ГенельС. В., Феофанов В. Д.,
Гигиенические требования к полимерным материалам, приме-
применяемым в пищевой промышленности, М., 1972; М о г g a r e i-
d g e К., в кн.: Encyclopedia of polymer science and technology,
v. 7, N. Y.— [a. o.], 1967, p. 219; Kunststoffe, № 10, 784
A970). С. В. Генелъ.
ПОЛИМЕРЫ В РАДИОЭЛЕКТРОНИКЕ (polymers
in radioelectronics, Polymere in Radioelektronik, poly-
meres dans radioelectronique). В радиоэлектронной ап-
аппаратуре (РЭА) полимерные материалы применяют
гл. обр. как электроизоляционные и конструкционные.
Кроме того, в РЭА используют нек-рые полимерные
материалы специального назначения, напр, магнито-
диэлектрики, полупроводники.
Применение полимерных материалов позволяет зна-
значительно уменьшить объем и массу РЭА, расширить
диапазон допустимых внешних воздействий, способ-
способствует внедрению в производство РЭА прогрессивных
технологич. процессов. Большой экономич. эффект,
к-рый дает использование полимеров, обусловлен сни-
снижением стоимости РЭА, увеличением ее долговечности
и уменьшением расходов на обслуживание. Так, гер-
герметизация РЭА компаундами (см. Компаунды полимер-
полимерные) позволяет экономить металлич. крепежные эле-
элементы, применять блочный принцип конструирования
и ремонта РЭА, дает возможность отказаться от ис-
использования металлич. герметизирующих кожухов.
Благодаря применению компаундов во многом решает-
решается проблема электрич. изолированного вывода через
металл и уменьшается трудоемкость изготовления
изделий.
Заливка РЭА компаундами — конструктивный спо-
способ миниатюризации и создания влагостойких типовых
конструкций в бескорпусном исполнении. Благодаря
применению микромодулей, особенно перспективных
в портативной переносной РЭА, ракетах, искусствен-
искусственных спутниках Земли, значительно уменьшаются раз-
размеры аппаратуры по сравнению с ее размерами при
традиционном (уплотненном) монтаже. Так, напр.,
размеры ЭВМ, источников питания и радиостанций
уменьшаются соответственно в 17, 2 и 1,8 раза.
При использовании микромодулей в радиоаппара-
радиоаппаратуре связи масса приемо-передающих станций умень-
уменьшается в 3 раза, а их объем — в 20 раз; масса и объем
устройства для уплотнения каналов с временным
разделением -— соответственно в 25 и 35 раз. Приме-
Применение тонких полимерных пленок, обладающих луч-
лучшими, чем бумага, электрическими характеристиками,
привело к уменьшению размеров конденсаторов в
2—4 раза.
Печатный монтаж на слоистых пластиках или плен-
пленках полнее удовлетворяет требованиям миниатюриза-
миниатюризации и высокой надежности, чем классический прово-
проволочный монтаж. Наряду с выигрышем в объеме (при-
(примерно в 7—10 раз) и в массе РЭА печатные платы обес-
обеспечивают хороший доступ к элементам схем, облегчают
стандартизацию конструкций РЭА и позволяют авто-
автоматизировать ее производство.
Еще более высокую плотность размещения элемен-
элементов (до 200 в 1 см3) и большую потенциальную надеж-
надежность (в среднем на 1 порядок), чем схемы, выполнен-
выполненные методом уплотненного монтажа, и микромодули,
имеют тонкопленочные схемы РЭА. Все эти важные
достижения в радиоэлектронике тесно связаны с при-
применением новых полимерных материалов и техноло-
технологич. процессов.
Новая область применения полимерных материалов
в РЭА — микроминиатюрные голографич. запоминаю-
запоминающие устройства для вычислительной техники. Один
из наиболее перспективных материалов для этого —
поливинилкарбазол (см. Винилкарбазола полимеры),
применение к-рого позволило получить запоминающее
устройство с высокой чувствительностью и большой
разрешающей способностью. Максимальная емкость
устройств, в к-рых применены полимерные материалы,
на 2—3 порядка больше, а среднее время выборки на
7 порядков меньше, чем у запоминающих устройств
на магнитных лентах.
Полимерные материалы применяют не только во
вновь разрабатываемой РЭА. Для нек-рых видов се-
серийной РЭА они оказались полноценными замените-
заменителями остродефицитных цветных металлов, сплавов и
др. материалов и, кроме того, способствовали усо-
усовершенствованию конструкции изделий. Например,
корпуса интегральных схем и полупроводниковых
приборов из пластмасс не уступают по надежности
керамическим, но значительно дешевле и легче по-
последних.
Требования к полимерным материалам, применяе-
применяемым в производстве радиоэлектронной аппаратуры.
8 большинстве случаев полимерные материалы при-
применяют в РЭА, работающей при сравнительно невысо-
невысоких напряжениях в слаботочных высокочастотных и
импульсных цепях. В этих случаях к электрич. проч-
прочности (#Пр) предъявляют менее жесткие, а к уд. по-
поверхностному (ps) и уд. объемному (pv) электрич.
сопротивлению, тангенсу угла диэлектрич. потерь tg б
и диэлектрич. проницаемости е — более жесткие тре-
требования. Материалы, предназначенные для примене-
применения при высоком напряжении (в высокочастотной ап-
аппаратуре и, особенно, в высоковольтных устройствах),
должны иметь малые значения е и tg 6 и высокие зна-
значения pv и ps, т. к. в противном случае мощность,
рассеиваемая в материале, может достигнуть недопус-
недопустимо больших значений. Если полимерный материал
используют в колебательном контуре, большие диэлек-
диэлектрич. потери нежелательны в связи с увеличением ак-
активного сопротивления, а следовательно, и значения
затухания контура. К материалам, работающим в вы-
высоковольтных устройствах, предъявляют также тре-
требование высокой jE'np-
Параметры РЭА в большой степени зависят от меха-
нич. свойств полимерных материалов: прочности при
различных нагружениях, в том числе вибропрочности,
а также твердости, эластичности и др.
Важное требование к заливочным и пропиточным ком-
компаундам, особенно применяемым для РЭА в миниатюр-
миниатюрном и микроминиатюрном исполнении,— высокая теп-
теплопроводность. Доступный и широко применяемый
метод повышения этой характеристики — введение
теплопроводных наполнителей, напр, порошков ме-
металлов и их окислов, нитрида бора, молотого кварца,
талька и др.
К компаундам, помимо необходимых влагозащитных
и электроизоляционных свойств, предъявляют также
след. требования: отсутствие химич. взаимодействия
с др. материалами и деталями; достаточно низкая
темп-pa стеклования и небольшие внутренние напря-
напряжения (при больших напряжениях возможен обрыв
тонких проводов намоточных изделий, обратимое или
необратимое изменение параметров герметизирован-
герметизированных деталей, растрескивание и отслоение компаундов
от стенок кожуха). Во многих случаях требуются
также малая усадка компаунда, его хорошая адгезия
и близость температурных коэфф. линейного расшире-
расширения компаунда и материала герметизируемой детали.
945
ПОЛИМЕРЫ В РАДИОЭЛЕКТРОНИКЕ
946
Иногда большая усадка компаунда играет и полезную
роль, напр, в случае герметизации выводов с помощью
компаунда, обладающего плохой адгезией к материалу
детали.
Нек-рые материалы для РЭА (компаунды, прессма-
териалы), применяемые для ремонта контактов реле,
контактных колец и выводов, должны обладать по-
повышенной электрической проводимостью. Способы
улучшения этого показателя такие же, как при по-
повышении теплопроводности; наилучшие результаты
получают при наполнении полимерных материалов
порошками серебра или графита (см. Металлонапол-
ненные полимеры, Электропроводящие полимерные ма-
материалы).
Микроминиатюризация и связанная с ней высокая
плотность монтажа предъявляют к герметизирующим
материалам особые требования — обеспечение надеж-
надежной изоляции между элементами при малых изоляцион-
изоляционных расстояниях; сохранение функциональной точ-
точности аппаратуры; обеспечение защиты сложных эле-
элементов, чувствительных к механич. нагрузкам. К по-
полимерным материалам для РЭА все чаще предъявляет-
предъявляется требование высокой нагревостойкости. Однако это
необходимо далеко не всегда (напр., для схем с полу-
полупроводниковыми приборами выполнение этого требо-
требования не обязательно).
Особенности технологии применения полимерных ма-
материалов в производстве радиоэлектронной аппара-
аппаратуры. Основные методы герметизации РЭА — заливка
(получение монолитной, или литой, изоляции — см.
Литье компаундов) или пропитка компаундом (лаком)
с пониженной вязкостью. На одном или нескольких
герметизированных пропиткой элементах РЭА создают
дополнительный слой герметизирующего материала,
погружая изделие в жидкий компаунд или помещая
его в форму, в к-рой на изделии получают слой, напр,
термопласта, методом литья под давлением. При по-
погружении для образования слоя повышенной толщи-
толщины применяют компаунды, обладающие тиксотропны-
ми свойствами.
Прогрессивные приемы герметизации намоточных
изделий — струйная (капельная) пропитка, а также
совмещение этого процесса с намоткой. Перспективен
способ герметизации РЭА напылением в псевдоожи-
женном слое порошкового полимерного материала.
Заливка и напыление обеспечивают лучшую защиту
РЭА от воздействия влаги, чем др. способы гермети-
герметизации.
Отвержденные заливочные компаунды или клеевые
соединения, а также покрытия, получаемые напыле-
напылением, образуют, как правило, монолитные системы,
к-рые с трудом поддаются ремонту, а чаще вообще не
пригодны для ремонта. Это учитывают при конструи-
конструировании РЭА, напр, собирают аппаратуру из отдель-
отдельных герметизированных блоков. Такой блок в случае
выхода его из строя заменяют новым. Затраты на эту
операцию частично или полностью компенсируются
экономией, получаемой от повышения надежности за-
залитого блока и блочной РЭА в целом. Кроме того,
замена блока м. б., в отличие от ремонта, выполнена
оператором невысокой квалификации.
При низких темп-pax литая изоляция может рас-
растрескиваться (особенно в случае деталей с острыми уг-
углами). Проблемы предупреждения растрескивания ре-
решаются путем создания деталей соответствующих кон-
конструкций, применения демпфирующих прокладок, пра-
правильного выбора компаундов и модифицирующих до-
добавок к ним.
В нек-рых случаях герметизация может вызвать
повышение массы РЭА, а герметизирующие материалы
оказаться несовместимыми с другими, применяемыми
в изделии, напр. с материалами для изоляции прово-
проводов, клеями и др. Наконец, свойства полимерных
материалов менее стабильны, чем свойства керамики,
слюды и др. неорганич. электроизоляционных мате-
материалов, а также металлов.
Основная причина старения полимерных материалов
в РЭА — повышенная темп-pa. При этом основные
электроизоляционные свойства полимеров изменяются
незначительно или даже в желательном направлении:
повышается pv, снижаются е и tg 6. Более опасно для
РЭА изменение при старении физико-механич. свойств
полимера, сопровождающееся увеличением внутрен-
внутренних напряжений в материале, снижением его относи-
относительного удлинения. Это приводит к растрескиванию
изоляции, в результате чего ухудшаются ее влагоза-
влагозащитные свойства и уменьшается электрич. прочность.
Факторы, ускоряющие старение полимерных материа-
материалов в РЭА,— одновременное действие повышенной
темп-ры, влаги, радиации, а также озона, образующе-
образующегося при ионизации воздуха в высоковольтных устрой-
устройствах.
Виды полимерных материалов, применяемых в про-
производстве радиоэлектронной аппаратуры. Ассортимент
полимерных материалов, применяемых в РЭА, весьма
разнообразен. Наряду с компаундами широко исполь-
используют также лакокрасочные материалы, клеи, пено-
пласты, пленки, слоистые пластики, прессматериалы
и др.
Применение в РЭА заливочных и пропиточных
эпоксидных компаундов, обладающих ма-
малой усадкой, отличной адгезией к герметизируемым
поверхностям, хорошими влагозащитными и электро-
электроизоляционными свойствами, обусловило переход от
крупногабаритных высоковольтных конструкций с ме-
мета л л ич. корпусами, жидким диэлектриком и керамич.
изоляторами к бескорпусной конструкции РЭА с ли-
литой изоляцией. Такие конструкции проще, меньше по
габаритам, легче, могут храниться в течение длитель-
длительного времени, стойки в тропич. климате. Применение
некоронирующих изоляторов из эпоксидных смол с
введенными в их тело заземленными экранами обеспе-
обеспечивает равномерное распределение напряженности но-
ноля. Эпоксидные компаунды используют также в про-
производстве малогабаритных высоковольтных трансфор-
трансформаторов, дросселей, герметичных токоподводов, к-рые
могут эксплуатироваться иод высоким давлением,
компактных блоков аппаратуры и др. Темп-pa дли-
длительной эксплуатации этих компаундов не превышает
в большинстве случаев 130 —150 °С.
Морозостойкость, высокая нагрево- и влагостой-
влагостойкость, стойкость к тепловым ударам, малая зависи-
зависимость электрич. характеристик от темп-ры, хорошие
технологич. свойства обусловили широкое применение
в РЭА кремнийорганических компа-
компаундов. Последние могут длительно работать при
темп-pax от —80 до 260 °С. Способность этих компаун-
компаундов отверждаться при комнатной темп-ре используют
при герметизации РЭА, не допускающей нагрева. Пло-
Плохая адгезия компаундов препятствует их применению
в нек-рых конструкциях; известные же методы исполь-
использования адгезионных подслоев усложняют техноло!ию
герметизации.
Эластичные полиуретановые компаун-
д ы морозостойки, имеют хорошую адгезию к метал-
металлам и пластмассам. Применение этих материалов це-
целесообразно для герметизации элементов, чувстви-
чувствительных к внутренним напряжениям, напр, трансфор-
трансформаторов с пермаллоевыми или ферритовыми сердечни-
сердечниками. Способность отверждаться при темп-pax не выше
60—80 °С делает эти компаунды пригодными для залив-
заливки схем с полупроводниковыми приборами, селеновых
выпрямителей и др.
В тех случаях, когда требуются материалы с малыми
диэлектрич. потерями, часто применяют компаун-
компаунды на основе полистирола, а также со-
947
ПОЛИМЕРЫ В РАДИОЭЛЕКТРОНИКЕ
948
полимеров с высоким содержанием стирола. Такие
материалы работают, как правило, при темп-pax ниже
100—120 °С, имеют большую усадку и большой темпе-
температурный коэфф. линейного расширения, подвержены
растрескиванию при термоударах. Это, а также нек-рые
технологич. трудности ограничивают применение по-
полистирол ьных компаундов.
Пропиточные и заливочные компаунды на ос-
основе метакрилатов характеризуются эластич-
эластичностью в широком диапазоне темп-р, малыми внут-
внутренними напряжениями и высокой стойкостью к тер-
термоударам. Их применяют для герметизации блоков
РЭА, работающих в условиях высокой влажности.
Эти компаунды могут полимеризоваться при темп-рах
до 70 °С и удобны для герметизации блоков с полупро-
полупроводниковыми приборами и деталями, чувствительными
к воздействию внутренних напряжений. Длительно
допускаемая рабочая темп-pa метакрилатных компаун-
компаундов не превышает 120 °С.
Важное значение в производстве РЭА имеют л а-
кокрасочные материалы на основе эпок-
эпоксидных, алкидных, полиэфирных смол, кремнийорга-
нич. полимеров, нитроцеллюлозы и др. пленкообразую-
пленкообразующих веществ. Эти материалы выполняют в РЭА элек-
электроизоляционные, антикоррозионные и декоративные
функции. Лаки и эмали применяют для пропитки на-
намоточных изделий, волокнистой и пористой изоляции
с целью повышения их электрич. и механич. прочности,
теплопроводности и влагостойкости, для образования
на поверхности лакируемых деталей прочной, гладкой
и влагостойкой пленки, увеличивающей поверхностное
сопротивление утечки и напряжение поверхностного
разряда.
Среди разнообразных синтетич. клеев (см. Клеи
синтетические) особое место в производстве РЭА зани-
занимают эпоксидные клеи, к-рые отличаются высокими
адгезионными свойствами, хим-, плесене- и влагостой-
влагостойкостью. Сравнительно небольшая вязкость таких клеев
позволяет заполнять узкие полости и достигать высо-
высокой прочности и герметичности клеевого соединения.
Эпоксидные клеи не требуют, как правило, давления
при склеивании; многие из них м. б. отверждены при
комнатной температуре. Применение эпоксидных и др.
клеев упрощает технологию изготовления РЭА. Заме-
Замена пайки и сварки склеиванием обусловливает мень-
меньшее коробление и др. деформации РЭА (это особен-
особенно важно в производстве прецизионных приборов), а
также устраняет опасность возникновения гальвани-
гальванических пар.
Применение в производстве РЭА пенопластов
обусловлено их малой объемной массой, низкими зна-
значениями е и tg S, повышенной радиопрозрачностью.
Благодаря этому пенопласты применяют в обтекателях
антенн радиолокационных станций, для герметизации
и изолирования деталей и схем РЭА. Вспенивающиеся
компаунды применяют вместо компаундов, образую-
образующих монолитные герметизирующие слои, для заливки
РЭА в тех случаях, когда необходимо уменьшить ее мас-
массу и снизить стоимость.
Для заливки схем с печатным монтажом и модулей
широко применяют пенополиуретаны с объемной мас-
массой 0,032—0,32 г/см3. Основные недостатки этих мате-
материалов — ухудшение электроизоляционных свойств
при длительном увлажнении, относительно невысокая
стойкость к тепловому старению. Помимо пенополиуре-
пенополиуретанов, в РЭА применяют пенофенопласты, пеноэпокси-
ды, пенополиорганосилоксаны, пенополистирол. Для
всех пенопластов характерна низкая теплопровод-
теплопроводность.
Разнообразные пленочные материалы
толщиной 2—200 мкм используют как прокладочный
материал в производстве трансформаторов, конденса-
конденсаторов и электромеханич. устройств, а также для изо-
изолирования проводов и кабелей, изготовления гибких
плат печатного монтажа. Для этих целей применяют
полиэтиленовую, поливинилхлоридную, полистироль-
ную, полиэтилентерефталатную, фторопластовую плен-
пленки (см. Пленки полимерные). Значительный интерес
для РЭА, работающей в широком диапазоне темп-р
(от —190 до 250 °С), а также в условиях вакуума и
действия радиации, представляют полиимидные плен-
пленки, имеющие малую усадку при старении. Их приме-
применяют для изготовления изоляционных прокладок,
основы печатных схем, магнитных лент и др.
В ремонтных и монтажных работах используют плен-
пленки из поливинилхлорида или полиэтилена с клеящим
подслоем. Из этих же полимеров, подвергнутых облу-
облучению, изготовляют тонкостенные трубки, колпачки,
муфты и наконечники, к-рые при нагревании до опреде-
определенной темп-ры дают значительную усадку. Такими
изделиями герметизируют и упрочняют места пайки,
стыки кабелей и др.
Слоистые пластики и волокниты
широко применяют как конструкционные и изоляцион-
изоляционные материалы для изготовления щитов, панелей, плат
и др. деталей. Очень велика роль слоистых пластиков,
особенно стеклотекстолита, как материала подложки
плат печатного монтажа толщиной 0,25—3 мм (жесткие
платы) или 0,25—0,1 мм и менее (гибкие платы). В ус-
условиях повышенной влажности и действия механич.
нагрузок наиболее пригодны эпоксидные стеклопласти-
стеклопластики, стоимость к-рых относительно высока.
Пропитанные электроизоляционными лаками волок-
волокнистые материалы, напр, лакошелк, применяют как
прокладки в обмотках. Трубки из стекловолокна,
синтетич. или хлопчатобумажных волокон служат
изоляторами проводов при монтаже электроаппара-
электроаппаратуры. Такие трубки должны быть износостойки, вы-
выдерживать перегибы на 90°, а также воздействие нек-рых
растворителей, полимерных заливочных и пропиточ-
пропиточных материалов и влаги.
Большое значение в РЭА приобретают также б у-
мага и картоны, изготовленные из нагрево-
стойких синтетич. волокон, напр, из ароматич. полиа-
полиамидов (см. Бумага из синтетических волокон).
В производстве РЭА, особенно изделий с малыми
изоляционными расстояниями между токоведущими
элементами, широко применяют к р е м н и й о р-
ганические гидрофобизаторы (см. Ги-
дрофобизаторы), к-рые предотвращают появление то-
коведущих мостиков.
В значительном количестве в производстве РЭА ис-
используют прессовочные и литьевые
пластмассы для изготовления установочных
деталей, изоляторов, механизмов управления, корпу-
корпусов приборов, а также для опрессовки элементов схем
(конденсаторов, сопротивлений и др.). Основные кри-
критерии при выборе этих материалов — нагревостойкость
и частота электромагнитного поля, в к-ром они должны
эксплуатироваться. В высокочастотных цепях приме-
применяют преимущественно термопласты с пониженными
диэлектрич. потерями (полистирол, полиэтилен, поли-
полипропилен, фторопласты), а также кремнийорганич.
полимеры; в низкочастотных — гл. обр. пластмассы
на основе термореактивных смол (полиэфирных, эпок-
эпоксидных, феноло-формальдегидных и др.).
Лит.: Б е л е в ц е в А. Т., Технология производства
радиоаппаратуры, 2 изд., М., 1971; Богородиц-
кийН. П., Пасынков В. В., Материалы радиоэлектрон-
радиоэлектронной техники, М., 1969; В а с и л ь е в А. В., Микроминиатюри-
Микроминиатюризация военной электронной аппаратуры, М., 1969; ВолкМ.,
Леффордж Ж., Стетсон Р., Герметизация электро-
электротехнической и радиоэлектронной аппаратуры, пер. с англ.,
М.— Л., 1966; МайофисИ. М., Химия диэлектриков, М.,
1970; Харпер Ч., Заливка электронного оборудования
синтетическими смолами, пер. с англ., М.— Л., 1964; Чер-
Черняк К. И., Неметаллические материалы в судовой электро-
и радиотехнической аппаратуре. Справочник, 2 изд., Л., 1970.
А. К. Варденбург, А. И. Галушко.
949
ПОЛИМЕРЫ В СЕЛЬСКОМ И ВОДНОМ ХОЗЯЙСТВЕ
950
ПОЛИМЕРЫ В СЕЛЬСКОМ И ВОДНОМ ХОЗЯЙ-
ХОЗЯЙСТВЕ (polymers in agriculture and melioration, Poly-
mere in Landwirtschaft und Melioration, polymeres
dans agriculture et amelioration).
Содержание:
Растениеводство 949
Земледелие 953
Упаковка и хранение сельскохозяйственной
продукции, химикалий и гербицидов 954
Животноводство 954
Мелиорация и сельскохозяйственное водоснаб-
водоснабжение 955
Применение полимерных материалов в производстве
сельскохозяйственных продуктов, животноводстве,
при орошении и осушении земель, водоснабжении и
обводнении пастбищ — одно из важнейших направле-
направлений научно-технич. прогресса в этих отраслях народ-
народного хозяйства. Тенденция ко все более широкому
использованию полимерных материалов характерна
для всех стран с развитым сельским хозяйством.
Растениеводство. В этой отрасли сельскохозяйствен-
сельскохозяйственного производства полимерные материалы используют
в строительстве культивационных сооружений, для
мульчирования почвы, дражирования семян и др.
Применение полимеров в строительстве
культивационных сооружений поз-
позволяет значительно уменьшить число несущих деталей
и благодаря этому снизить капиталовложения.Из поли-
полимерных материалов м. б. построены сооружения сферич.
формы с минимальным числом непрозрачных элементов,
в т. ч. такие, к-рые с применением стекла построить
невозможно (напр., воздухоопорные конструкции).
В строительстве культивационных сооружений
используют пленки из полиэтилена, пластифицирован-
пластифицированного поливинилхлорида, полиамидов (см. Пленки поли-
полимерные), а также жесткие материалы из полиэфирных
стеклопластиков, непластифицированного поливинил-
поливинилхлорида (винипласта) и др.
Значительная прозрачность полиэтиленовой пленки
в ИК-области спектра E—15 мкм — см. таблицу) обус-
обусловливает понижение темп-ры внутри культивационно-
культивационного сооружения в ночное время (при конденсации влаги
на внутренней поверхности пленки степень охлаждения
резко уменьшается). Полиэтиленовые пленки больше,
чем др. пленочные материалы, рассеивают проникаю-
проникающую радиацию.
Прозрачность нек-рых материалов в различных областях
солнечного спектра (%)
Материал*
Полиэтиленовая пленка
стабилизированная . - .
Поливинилхлоридная
пленка
Ацетобутиратцеллюлозная
пленка
Полиамидная пленка . . .
Стеклопластик
Стекло
Длина волны, мкм
0,295—
0,400
26
22
30
73
4
46
0,400-
0,750
80
88
85
87
80
83
0,750-
2,000
80
88
85
88
85
85
5-15
80
10
5
30
2
0
* Толщина пленок всех типов 0,1 мм, стеклопластика —
1,5—2,0 мм, стекла — 2,5—3,0 мм.
Гидрофобность поверхности полиэтиленовой пленки
(угол смачивания 83°) и накопление на ней значитель-
значительного количества статич. электричества приводят к ее
необратимому запылению и, следовательно, к умень-
уменьшению прозрачности (на 20% и более в течение севона).
Этот недостаток устраняют при помощи антистатиков,
к-рые вводят в полимер или наносят на поверхность
пленки. Гидрофилизация поверхности полиэтиленовой
пленки при одновременном улучшении ее атмосферостой-
кости достигается облучением пленки, содержащей фо-
фотосенсибилизаторы, УФ-лучами; проницаемость самой
пленки для УФ-лучей при этом не изменяется. Для
районов с высокими летними темп-рами перспективны
металлизированные (напр., алюминием) пленки с вы-
высокой отражательной способностью, благодаря чему
уменьшается опасность перегрева растений в укры-
укрытии. См. также Полиэтиленовые пленки.
Поливинилхлоридные пленки более прозрачны, чем
полиэтиленовые, в видимой области спектра. При
запылении они м. б. легко очищены водой. Вследствие
меньшей проницаемости для теплового излучения в
укрытиях из этой пленки лучше сохраняется тепло.
Стабилизированные пленки могут эксплуатироваться не
менее 8—10 мес, т. е. в течение 2—3 сезонов. В Японии
используются также пленки из пластифицированного
поливинилхлорида, покрытые специальными состава-
составами, препятствующими вымыванию пластификатора.
Поверхность такой пленки длительно сохраняет гидро-
фильность и не запыляется. См. также Поливинилхло-
Поливинилхлоридные пленки.
Высокая прозрачность полиамидных пленок в види-
видимой части солнечного спектра, малая проницаемость
для ИК-облучения и гидрофильность их поверхности
создают благоприятные микроклиматич. условия для
развития растений. Урожайность растений и качество
урожая в укрытиях из этих пленок обычно выше,
чем в укрытиях из других пленочных материалов.
Однако, несмотря на отмеченные достоинства, поли-
полиамидные пленки имеют в растениеводстве ограниченное
применение из-за их склонности к значительному водо-
поглощению и изменению размеров при эксплуатации.
Поэтому такие пленки используют гл. обр. при соору-
сооружении малогабаритных укрытий (см. ниже) с подвиж-
подвижными натягивающими устройствами. См. также По-
Полиамидные пленки.
Перспективны для сельского хозяйства пленки из
сополимера этилена с винилацетатом (е в а ф и л ь м,
нипофлекс GF — Япония). Они атмосферостой-
ки, мало запыляются, имеют высокую прозрачность в
видимой части солнечного спектра, мало проницаемы
для УФ- и ИК-лучей, эластичны и морозостойки. Боль-
Большой интерес как ограждающий материал могут предста-
представить пленки из ацетобутирата целлюлозы (см. также
Эфироцеллюлозные пленки) и прозрачные, прочные,
хим- и термостойкие полиэтилентерефталатные плен-
пленки, применение к-рых ограничено их относительно
высокой стоимостью.
Проводятся работы по использованию цветных пле-
пленок, что может в ряде случаев оказывать благоприят-
благоприятное влияние на процессы обмена веществ в растениях.
Напр., салат, выращенный под цветными пленками,
отличается от выращенного в укрытиях из прозрачной
пленки повышенным содержанием углеводов и общего
азота. Разрабатываются также пленки, отражающие
ИК-радиацию; в укрытиях из этих пленок создается
более равномерный температурный режим.
Довольно значительное распространение в строитель-
строительстве самонесущих культивационных сооружений (без
опорных элементов) получили жесткие материалы на
основе непластифицированного поливинилхлорида
и полиэфирных стеклопластиков. Последний отлича-
отличается хорошей прозрачностью для видимой части сол-
солнечного спектра и почти полной непроницаемостью
для дальних ИК-лучей. Вследствие способности этого
материала рассеивать солнечные лучи он может стать
одним из эффективных средств борьбы с перегревом
растений в культивационных сооружениях, эксплуати-
эксплуатируемых в южных районах. Благодаря высокой атмо-
сферостойкости стеклопластика срок его службы может
достигать 3—5 лет.
Пленочные полимерные материалы широко исполь-
используют для строительства обычных многоблочных теп-
951
ПОЛИМЕРЫ В СЕЛЬСКОМ И ВОДНОМ ХОЗЯЙСТВЕ
952
лиц (типа стеклянных). Получили также распростране-
распространение теплицы арочной формы (крупногабаритные тон-
тоннели) шириной ок. 4 м, длиной 25—30 м и высотой 1,7 —
2,0 м. Опорами для них служат прутковое железо,
профилированный алюминий, стальные трубы, дерево.
Используются блочные теплицы с арочной кровлей,
а также теплицы башенного типа из жестких полимер-
полимерных материалов. Южнее 60° сев. шир. теплицы из поли-
полимерных материалов могут эксплуатироваться круг-
круглый год.
Для создания малогабаритных укрытий (тоннелей
высотой 0,7—1,2 м и шириной 1,0—1,5 м) применяют
тонкие полимерные пленки E0—80 мкм). Опорами для
них служат дуги из жестких полимерных прутков
(напр., поливинилхлоридных), деревянные планки или
металлич. проволока диаметром 4—6 мм.
Для воздухоопорных теплиц, дешевизна сооружения
и эксплуатации к-рых удачно сочетается с технич.
возможностями оптимизации режимов выращивания
растений, применяют пленки толщиной не менее 0,12—
0,15 мм. Для поддержания формы внутри теплицы со-
создают небольшое избыточное давление [выше наруж-
наружного на 98—118 н/м2 A0—12 мм вод. ст.)] путем постоян-
постоянного поддува воздуха, подаваемого вентилятором.
Разработана также блочная ветроустойчивая теплица
с плоской кровлей, к-рая может работать и без подду-
поддува воздуха. Существуют надувные теплицы таких раз-
размеров, что в них можно применять обычные машины
для обработки почвы.
Увеличение срока службы культивационных соору-
сооружений из полимерных материалов и повышение их вет-
ветроустойчивости достигаются при использовании пле-
пленок, армированных стекловолокном или синтетич.
волокнами, напр, полиэфирными.
Из-за большей герметичности культивационных со-
сооружений (по сравнению со стеклянными теплицами) в
них возможны сильные перегревы в дневные часы и
резкое понижение темп-ры воздуха в ночные. Поэтому
сооружения, эксплуатируемые в континентальном кли-
климате, должны иметь устройства для обогрева воздуха.
Для борьбы с перегревами в малогабаритных тоннелях
целесообразно при возделывании отдельных культур,
напр, земляники, применять перфорированные пленки.
При возделывании нек-рых культур, напр, томатов, в
теплицах или в малогабаритных тоннелях устраивают
принудительное вентилирование.
На микроклимат в сооружениях из полимерных мате-
материалов влияет также характер конденсата на внутрен-
внутренней поверхности пленки. Напр., на полиэтиленовой
пленке, не содержащей антистатиков, образуется кон-
конденсат в виде мелких сферич. капель, на поливинил-
хлоридной — плоские капли, имеющие тенденцию сли-
сливаться в один сплошной слой. В последнем случае в
укрытие проникает больше солнечной радиации, в том
числе и прямой.
В теплицах из полимерных материалов выращивают
гл. обр. огурцы и томаты, к-рые созревают в них в
среднем на 25—30 дней раньше, чем в открытом грун-
грунте. В таких укрытиях можно выращивать овощи, тре-
требующие для своего развития большого количества теп-
тепла, в тех районах, где их возделывание в открытом
грунте вообще невозможно. В ранневесенний период в
теплицах выращивают также салат, редис, лук, в позд-
неосенний их часто используют для выращивания
цветов.
Теплицы и малогабаритные туннели из полимерных
материалов широко применяют при выращивании зем-
земляники. В условиях Ленинградской области эта культу-
культура созревает в пленочных теплицах на 35 дней раньше,
чем в открытом грунте, а прибавка урожая составляет
ок. 20%. Ягоды тепличных растений отличаются повы-
повышенным содержанием Сахаров и уменьшенной кислот-
кислотностью. Выращивание земляники в малогабаритных
укрытиях из поливинилхлоридной пленки особенно
распространено в Японии, где такими укрытиями заня-
занята х/з площадей под эту культуру. Эффективным ока-
оказалось использование перфорированной пленки с
отверстиями диаметром 27—29 мм, через к-рые могут
свободно пролетать пчелы.
В ФРГ под перфорированной пленкой выращивают
салат, цветную капусту, кольраби, морковь, петрушку
и др. овощи. Такую пленку используют и для кратко-
кратковременного (на 10—20 дней) укрытия растений, что
позволяет после высадки в открытый грунт обеспечить
их ускоренное развитие (на 8—10 дней).
В полиэтиленовых теплицах и малогабаритных тон-
тоннелях создаются благоприятные условия (высокая
влажность, рассеянная радиация) для укоренения че-
черенков винограда. На Украине в пленочных теплицах
выращивают бахчевые культуры (арбузы, дыни). В Ве-
Великобритании, Франции, ФРГ дыни возделывают как
в теплицах, так и в малогабаритных укрытиях. Наи-
Наиболее пригодный материал для этой цели — пленка,
мало рассеивающая солнечную радиацию, напр, поли-
винилхлоридная или из сополимера этилена с винилаце-
татом. Важная проблема плодоводства — защита
нек-рых культур, напр, цитрусовых, от неблагоприят-
неблагоприятных зимних условий — решается также применением
укрытий из полимерных пленок, пропускающих ок.
30% видимого света и отражающих максимально воз-
возможное количество ИК-лучей. В таких укрытиях
темп-pa растений в сильные морозы бывает на 10 —
12 °С выше, чем в открытом грунте. Укрытия из черной
полиэтиленовой пленки используют в Великобритании
для выращивания шампиньонов, урожай к-рых дости-
достигает при этом 16 кг/ж2.
Пригодные для посадки сеянцы хвойных деревьев
под укрытиями из полиэтиленовой пленки можно полу-
получить за 1 год вместо обычно необходимых 2 лет. При
этом стоимость таких сеянцев в ~ 2 раза ниже тех,
к-рые выращены в открытом грунте. В пленочных тон-
тоннелях нек-рые сорта нарциссов начинают цвести на
Черноморском побережье Кавказа на 19—30 дней рань-
раньше, чем в открытом грунте.
При возделывании растений как в открытом, так и
в защищенном грунте почву покрывают различными ма-
материалами. Этот агротехнич. прием, наз. мульчи-
мульчированием почвы, существенно улучшает гид-
ротермич. условия корнеобитаемого слоя почвы, спо-
способствует более раннему прогреванию ее весной, устра-
устраняет образование почвенной корки, приводит к значи-
значительной экономии труда на борьбу с сорняками. Наибо-
Наиболее распространенный полимерный материал для муль-
мульчирования — полиэтиленовые пленки.
Разрабатывается способ мульчирования почвы в теп-
теплицах заполненными водой пленочными рукавами
(толщина водяного слоя ок. 0,2 ж), к-рые укладывают
между рядами растений. В дневное время вода в рука-
рукаве хорошо прогревается, а ночью отдает тепло. Мульчи-
Мульчирование в открытом грунте с использованием гл. обр.
прозрачных пленок толщиной 30—40 мкм распростра-
распространено при выращивании дынь, спаржи, томатов, арбузов,
огурцов и др. Для земляники применяют преимущест-
преимущественно черную пленку, толщина к-рой при однолетней
культуре составляет 30 мкм, при двухлетней — 40—
50 мкм и более. Мульчирование весьма эффективно и
при выращивании винограда. Черная пленочная муль-
мульча на плодовых культурах улучшает не только водно-
физич. свойства почвы и подавляет прорастание сорня-
сорняков. Она служит также хорошим средством защиты кор-
корневой системы от первых сильных морозов (при отсут-
отсутствии снегового покрова).
При использовании мульчирующих пленок ускоряет-
ускоряется развитие многих цветочных культур, напр, флок-
флоксов, гладиолусов; одновременно с этим заметно уве-
увеличивается количество соцветий.
953
ПОЛИМЕРЫ В СЕЛЬСКОМ И ВОДНОМ ХОЗЯЙСТВЕ
954
Черной полиэтиленовой пленкой покрывают посадоч-
посадочные клубни раннего картофеля. Пленку расстилают
специальными машинами и после прорастания ботвы
делают на ней прорези. При этом молодые клубни обра-
образуются на две недели раньше, чем в случае неукры-
неукрытых растений, а урожай повышается на 30—50%.
Ведутся работы по созданию мульчирующих пленок,
разрушающихся при соприкосновении с почвенной вла-
влагой, а также под действием солнечной радиации. Для
этой цели применяют, в частности, полиэтиленовую
пленку, покрытую сверху слоем крафт-бумаги, а так-
также пленку из полибутенов (см. Бутенов полимеры).
Благодаря использованию таких материалов исклю-
исключается операция уборки пленки, а почва не засоряет-
засоряется ее отходами.
Водорастворимые пленки на основе поливинилового
спирта используют для посева семян. При этом семена
закладывают между слоями пленки и помещают в поч-
почву. Скорость прорастания семян регулируется водо-
растворимостью пленки. При таком способе посева иде-
идеально соблюдаются глубина и равномерность распреде-
распределения семян по площади.
С целью борьбы с сорняками в пленку, предназна-
предназначенную для мульчирования, в отдельных случаях до-
добавляют гербициды, растворяющиеся под действием
влаги, к-рая конденсируется на поверхности пленки,
обращенной к земле.
Дражирование семян (обволакивание их
искусственной оболочкой с целью придания округлой
формы и увеличения размеров) позволяет благодаря
лучшей сыпучести семян высевать их на нужные глу-
глубину и расстояние друг от друга. Это способствует
ускоренному развитию растений и повышению качест-
качества корнеплодов. Пленки широко используют для
обвязки прививок при вегетативном размножении пло-
плодовых растений. Благодаря этому значительно сокра-
сокращаются затраты труда, улучшается приживаемость
глазков, выход саженцев повышается на 30%. Для
обвязки прививок наиболее пригодна пленка из неста-
билизированного поливинилхлорида, к-рая быстро
стареет и сама отпадает от прививок через 1,5—2 мес.
Стволы плодовых деревьев защищают поливинилхло-
ридными пленками, содержащими репелленты. За рубе-
рубежом в полиэтиленовых пленках транспортируют ле-
лесопосадочный материал, что обеспечивает его хорошую
сохранность. При хранении подвойной лозы широко
применяют полиамидную пленку, к-рая позволяет избе-
избежать контакта лозы с песком, землей и опилками и
исключить т. обр. заражение черенков пятнистым не-
некрозом. При этом повышается также выход саженцев.
Поливинилацетатная дисперсия служит ценным ком-
компонентом побелок стволов деревьев, к-рые защищают их
от неблагоприятных климатич. условий, напр, солнеч-
солнечных ожогов. Для защиты растений от града и птиц при-
применяют сети из тонких полиакрилонитрилъних волокон.
Земледелие. Полимерные структурообразователи
(гидролизованный полиакрил онитрил, полиакрил амид,
сополимер метакриловой к-ты с метакриламидом), при-
применяемые в количестве 0,05—0,1% от массы сухой поч-
почвы, обусловливают слипание ее мельчайших частиц,
что способствует восстановлению макроструктурного
состава и резкому улучшению водно-воздушных усло-
условий в почве.
Хороший эффект достигается при внесении смеси
полиакриламида с поверхностно-активными вещества-
веществами (напр., диметиламмонийхлоридом) в поверхност-
поверхностный слой почвы толщиной 2—3 см. Этот слой защищает
почву от образования вредной для влаго- и газообмена
поверхностной корки, аккумулирует осадки, умень-
уменьшает испарение влаги, благоприятно действует на био-
логич. процессы в почве. При использовании поверх-
поверхностно-активного вещества резко сокращается расход
структурообразователя.
Большой интерес представляет применение поли-
полимерных структурообразователей для защиты поверхно-
поверхности почвы от эрозии. С этой целью используют, в част-
частности, бутадиен-стирольные латексы (см. Латексы син-
синтетические), образующие пленки, к-рые обладают хо-
хорошей адгезией к частицам почвы и стойкостью к ста-
старению. Применение латексов в количестве 100—
150 кг/га обеспечивает устойчивость почвы к ветровой
эрозии при скорости ветра до 25 м/сек.
Новые возможности для улучшения водно-воздушных
свойств почвы открывает использование пенопластов,
напр, на основе мочевино-формальдегидных смол (см.
Мипора), к-рые м. б. получены непосредственно в поле-
полевых условиях одновременным распылением всех входя-
входящих в их состав компонентов. Эти материалы, отвер-
ждающиеся с образованием рыхлых хлопьев, исполь-
используют как в открытом, так и в защищенном грунте. При
выращивании растений они могут также служить заме-
заменителями корнеобитаемого слоя почвы. Мочевино-форм-
альдегидные пенопласты содержат до 70% открытых
пор, к-рые могут заполняться водой, почти полностью
доступной для растений. Эти материалы могут слу-
служить также источником азота для растений. При оро-
орошении полей они способствуют более длительному
удерживанию влаги в почве.
За рубежом в земледелии широко используют также
пенополистирол с закрытыми порами, к-рый особенно
эффективен на переувлажненных глинистых почвах с
плохим воздушным режимом. Этот пенопласт не пог-
поглощает воду, не подвержен воздействию микроорганиз-
микроорганизмов и может использоваться в течение нескольких лет.
Упаковка и хранение сельскохозяйственной продук-
продукции, химикалий и гербицидов. Для этой цели широко
применяют гидрохлоридкаучуковые пленки, а также
водорастворимые пленки на основе поливинилового
спирта (последние — для хранения токсичных, пачкаю-
пачкающих или пылевидных материалов).
Разработан способ длительного хранения корнепло-
корнеплодов, напр, моркови, в открытых или перфорированных
полиэтиленовых мешках емкостью до 35 кг. При этом
естественная убыль моркови в 1,5—2 раза меньше, чем
при ее хранении в песке или в деревянных ящиках. В
пленочных емкостях длительно сохраняются аромат,
вкус и цвет продуктов соления и квашения. Свежие
огурцы, упакованные под вакуумом в полиэтиленовую
пленку толщиной 50 мкм, могут храниться при обыч-
обычной темп-ре в течение 2 недель.
Для хранения яблок используют герметизированные
надувные камеры/ покрытые изнутри пенополиурета-
пенополиуретаном. В газовой среде,содержащей 92% азота, 3% кисло-
кислорода и 5% углекислого газа, срок хранения достигает
10 мес. Мочевино-формальдегидные пенопласты приме-
применяют для хранения моркови непосредственно в поле.
Пена толщиной 2,5 см, к-рая отверждается в течение
1 ч после нанесения ее на корнеплоды, обеспечивает
хорошую защиту от морозов всю зиму.
Животноводство. Использование полимерных пле-
пленок, гл. обр. полиэтиленовых, для силосования кормов
обеспечивает более высокое качество силосной массы.
Пленки применяют как при наземном способе силосова-
силосования, так и для перекрытия полубашен и траншей перед
укрытием силоса слоями соломы и земли. При этом слой
гниения силоса уменьшается от 15 до 2—3 см, создается
лучшая герметичность при его закладке, снижаются
затраты труда. Силосная масса, укрытая пленкой,
может храниться в течение длительного времени. При
использовании пленок для силосования сырья с влаж-
влажностью не более 75% почти полностью устраняются
отходы силоса, а скрытые потери сухого вещества вслед-
вследствие угара составляют всего 4—10%. Качество сило-
силоса при этом улучшается: в нем увеличивается содержа-
содержание молочной кислоты и уменьшается содержание ма-
масляной.
955
ПОЛИМЕРЫ В СЕЛЬСКОМ И ВОДНОМ ХОЗЯЙСТВЕ
956
Полимеры успешно используют в ветеринарии в каче-
качестве компонентов лечебных препаратов и профилактич.
средств. Такие препараты, как «синий иод» (йодный
комплекс поливинилового спирта), поливинилпирроли-
дон нетоксичны, обладают широким антимикробным
спектром действия. Перспективно применение полиме-
полимеров в качестве пролонгаторов действия некоторых ле-
лечебных средств; например, карбоксиметилцеллюло-
за — эффективный пролонгатор действия акарицидных
препаратов.
Мелиорация и сельскохозяйственное водоснабжение.
Полимерные материалы применяют при орошении
для предотвращения потерь воды на фильтрацию из оро-
оросительных каналов и водоемов, при изготовлении труб
различного назначения, а также в строительстве водо-
водохозяйственных сооружений.
Основные материалы для противофильтрационных
экранов, используемые в СССР,— пленки из полиэтиле-
полиэтилена низкой плотности, стабилизированного газовой ка-
канальной сажей A,5—2,0% в расчете на массу полиме-
полимера), и из пластифицированного поливинилхлорида.
Толщина этих пленок обычно 0,2—0,4 мм, а ширина
должна быть максимально возможной для уменьшения
числа швов в экране. Полиэтиленовые экраны более
эластичны и морозостойки, чем поливини л хлоридные
(последние становятся жесткими в результате постепен-
постепенного улетучивания пластификатора). Преимущество
поливинилхлоридных пленок — лучшее сопротивление
прокалыванию. За рубежом для устройства экранов
используют также пленки из каучуков, напр, хлоропре-
нового, бутилкаучука, а также иолиизобутилена.
Устройство пленочных экранов возможно во всех
грунтах, тогда как строительство бетонных экранов
на просадочных грунтах обычно не рекомендуется.
При использовании пленок сокращаются сроки строи-
строительства сооружений, уменьшается их стоимость, резко
снижаются транспортные расходы благодаря уменьше-
уменьшению массы строительных материалов.
Пленочные экраны м. б. постоянными (с защитным
слоем из грунта или бетона) или поверхностными (без
защитного слоя). Последние используют и для защиты
оросительных каналов от размывания водой. Незащи-
Незащищенные экраны разрушаются обычно после 2—3 лет
эксплуатации. Расчетный срок службы защищенных
окранов из пленок толщиной не менее 0,2 мм для южных
районов СССР 25—30 лет, для северных — 35—40 лет.
Стоимость пленочных экранов с защитным слоем из
грунта в 1,5—3 раза ниже стоимости монолитной бе-
бетонной облицовки и почти в 5 раз меньше стоимости
сборной железобетонной. Производительность при
строительстве пленочных экранов в 4—8 раз выше,
чем при ручном способе бетонирования каналов, и
практически равна ей при бетонировании с помощью
бетоноукладочных машин.
Противофильтрационные пленочные экраны соору-
сооружают на различных водоемах и водохранилищах, их
используют для экранирования низконапорных земля-
земляных дамб и плотин, для гидроизоляции оползневых отко-
откосов. Они служат также для охраны водных источников
от загрязнения сточными водами.
Из полиэтилена и из поливинилхлорида изготовляют
трубы для закрытых оросительных и коллекторно-дре-
нажных систем, а также для временных трубопроводов,
к-рые укладывают на поверхности земли. Относитель-
Относительная легкость труб из пластмасс по сравнению с этими
изделиями из традиционных материалов (стали, чугу-
чугуна, асбестоцемента) позволяет снизить стоимость строи-
строительства протяженных оросительных трубопроводов.
Это обусловлено сокращением трудовых затрат на
строительно-монтажные работы, а также транспорт-
транспортных расходов и объема земляных работ, поскольку
трубопроводы из полимерных материалов можно мон-
монтировать на бровке траншей.
Трубы из пластмасс можно укладывать в агрессивных
средах, а также в подвижных грунтах, где укладка труб
из традиционных материалов требует значительных до-
дополнительных затрат. Кроме того, трубы из пластмасс
меньше истираются наносами, имеют лучшие пропуск-
пропускную способность и гидравлич. характеристики. Поли-
Полиэтиленовые трубы более стойки, чем поливинилхлорид-
ные, к гидравлич. удару. При размораживании они
восстанавливают свое первоначальное поперечное се-
сечение.
Трубы из полимерных материалов диаметром 100 мм
и выше, наиболее широко используемые в оросительных
системах, пока еще дороже труб из традиционных мате-
материалов. Однако отмеченные выше преимущества труб
из полимерных материалов обусловливают целесообраз-
целесообразность их применения при строительстве закрытых
оросительных систем.
В коллекторно-дренажных системах обычно исполь-
используют гладкостенные трубы (диаметр до 200 мм) из
полиэтилена высокой плотности с круглой перфора-
перфорацией C—5 мм). Применение для этих целей гофрирован-
гофрированных труб позволяет укладывать их при высоком стоя-
стоянии грунтовых вод и в оплывающих грунтах без пред-
предварительного рытья траншей (бестраншейная укладка
специальными дреноукладчиками). Производительность
при таком способе укладки примерно в 10 раз больше,
чем при обычном траншейном, а укладка труб обходит-
обходится примерно в 3—4 раза дешевле.
В поливной технике полимерные материалы исполь-
используют взамен металлов, древесины и др., а в отдельных
случаях и в сочетании с ними. Этим обеспечиваются
сравнительно малая масса конструкций поливного обо-
оборудования, легкость их сборки, разборки и транспорти-
транспортировки. Разработаны конструкции поливных трубопро-
трубопроводов, сифонов-водовыпусков, регулирующих щитов,
водосливов, гидротехнич. лотков и деталей насосов
из стеклопластиков, а также труб диаметром до 800 м,
изготовляемых из полиэтиленовых лент способом на-
намотки, и др. Из полиэтилена высокой плотности,
полипропилена, поликапролактама и др. изготовляют
детали дождевальных аппаратов. При поливах по бо-
бороздам и полосам широко применяют гибкие трубопро-
трубопроводы из капроновой ткани, пропитанной полиизобути-
леном. Производительность при поливе увеличивается
при этом в 2,5 раза. В поливных устройствах широко
используют также гибкие напорные шланги из армиро-
армированного найлоном поливинилхлорида диаметром 100 —
500 мм, выдерживающие напоры до 3,6 Мн1м2
C6 кгс/см2).
При облицовках оросительных каналов свежеуложен-
ный монолитный бетон защищают лаком этиноль (см.
Дивинилацетиленовые лаки и эмали). Благодаря этому
в значительной степени предотвращается испарение
влаги из бетона в начальный период его твердения и
бетон приобретает необходимую прочность за более
короткое время.
Для герметизации деформационных швов в сборных
и монолитных бетонных и железобетонных облицовках
оросительных каналов, лотках и др. гидротехнич. со-
сооружениях используют герметизирующие составы,
напр, на основе тиокола или бутилкаучука. При замене
металлич. листов или древесины полимер бетоном на
основе мономера ФА трудоемкость облицовки поверхно-
поверхности гидросооружений, работающих в зоне интенсивного
истирания или кавитационной эрозии, уменьшается
на 30—35%, а себестоимость — на ~20%.
Для закрытого дренажа — наиболее совершенного
способа осушения земель — применяют гл. обр.
трубы из полиэтилена высокой плотности.
Опыт СССР свидетельствует о том, что применение
дренажных труб из пластмасс вместо гончарных для
строительства осушительных систем в устойчивых ми-
минеральных грунтах позволяет повысить производитель-
957
ПОЛИМЕРЫ В СТРОИТЕЛЬСТВЕ
958
ность дренажных машин на 25%, сократить транспорт-
транспортные расходы в 4—6 раз и снизить затраты ручного
труда в 2,5—4 раза. Длинные плети дренажных труб
из пластмасс можно укладывать в траншеи без пред-
предварительного осушения массива. При укладке дрена-
дренажа из пластмасс в глубоких торфяниках, плывунах и
др. грунтах с неустойчивыми дном и стенками траншей
отпадает необходимость в тяжелом ручном труде, про-
производительность почти в 2 раза выше, чем при укладке
гончарного, а стоимость строительства в нек-рых райо-
районах (напр., в республиках Прибалтики, БССР) снижает-
снижается на 10-15%.
В мелиоративной практике используют гофрирован-
гофрированные и гладкостенные трубы. Дренажная вода поступает
в них через отверстия щелевой или круглой формы.
Гладкостенные трубы из пластмасс имеют более ровную,
чем гончарные, внутреннюю поверхность, к-рая не сма-
смачивается водой, а число стыков в них сводится к мини-
минимуму. Поэтому пропускная способность труб из пласт-
пластмасс выше, чем у гончарных, что позволяет значи-
значительно уменьшать их диаметры и "уклоны.
Наиболее экономичны по расходу материала и стои-
стоимости гофрированные трубы, имеющие высокую проч-
прочность в поперечном сечении при минимальной толщи-
толщине стенок и одновременно большую продольную гиб-
гибкость, что позволяет транспортировать их в бухтах
небольших диаметров. За рубежом наибольшее распро-
распространение получили гофрированные трубы из непласти-
фицированного поливинилхлорида, в СССР — из более
ударопрочного и морозостойкого полиэтилена высокой
плотности.
В нек-рых случаях, прежде всего в глубоких торфя-
торфяниках, свободных от пней и кустарника, дренаж из
пластмассы можно укладывать при помощи специаль-
специальных дреноукладчиков без траншей.
Для сочленения и наращивания полимерных дренаж-
дренажных труб используют соединительную арматуру (муф-
(муфты, переходы, тройники, уголки) из полиэтилена низ-
низкой плотности. Тройники из этого полимера применяют
также для соединения гончарных дренажных коллек-
коллекторных труб с трубами из пластмассы или с гончарны-
гончарными дренами-осушителями. Все это позволяет значитель-
значительно повысить производительность труда при строитель-
строительстве дренажных систем. Перспективны для изготовле-
изготовления осушительных дренажных систем наполненные ком-
композиции на основе полиэтилена и непластифицирован-
ного поливинилхлорида, а также пористые материалы.
Для крепления откосов осушительных каналов в не-
неустойчивых грунтах применяют пористые плиты из
химстойкого полимербетона на основе мономера ФА,
эпоксидных или кумароно-ннденовых смол.
При сельскохозяйственном водо-
водоснабжении и обводнении пастбищ
используют гл. обр. трубы из полиэтилена высокой
плотности; применение труб из поливинилхлорида
ограничено из-за склонности полимера к деструкции с
выделением НС1. Наиболее важное свойство полиэтиле-
полиэтилена, определяющее целесообразность использования его
в водоснабжении,— морозостойкость: трубы из поли-
полиэтилена сохраняют гибкость при темп-pax до —60 °С.
Кроме того, в трубах из полимерных материалов вода
замерзает в 3—4 раза медленнее, чем в стальных, а при
многократном замерзании и оттаивании воды трубы не
разрушаются.
Трубы стойки к коррозии и не подвержены зараста-
зарастанию. Поэтому их пропускная способность и качество
воды в них при эксплуатации не изменяются. Срок
службы труб из полиэтилена достигает 50 лет, т. е. выше
срока службы стальных труб без защитных покрытий
почти в 10 раз. Стоимость строительства трубопроводов
из полиэтиленовых труб диаметром 100 мм (без учета
стоимости самих труб) меньше стоимости строительства
стальных, асбестоцементных и чугунных трубопроводов
соответственно в 1,5; 2 и 2,5 раза. Возможность строи-
строительства полимерных трубопроводов небольших диа-
диаметров из разворачиваемых бухт позволяет уменьшить
объемы земляных работ и сократить число стыков,
что упрощает и удешевляет монтажные работы. При
требуемых для поселковых водопроводов сравнительно
небольших напорах, не превышающих в большинстве
случаев 0,4 Мн/м2 ( 4 кгс/см2), применение металлич.
труб не оправдано технически и экономически. Поли-
Полиэтиленовые трубы соединяют между собой сваркой
или с помощью муфт, поливинилхлоридные трубы —
склеиванием. Стоимость контактной сварки полиэтиле-
полиэтиленовых труб в 3 раза ниже стоимости сварки стальных.
Снижение затрат труда на транспортировку, монтаж
и эксплуатацию обусловили целесообразность приме-
применения труб из пластмасс для обводнения пастбищ.
Расширяется также применение труб из полиэтилена
диаметром 200 мм и более для буровых артезианских
скважин.
Лит.: Колясева В. А., Пащенко Т. Е., Ро-
Рожа некая О. Д., Микроклимат культивационных соору-
сооружений с пленочными покрытиями, Л., 1966; Мировая практика
применения синтетической пленки в овощеводстве, обзор
ВИНТИСХ, М., 1968; К о т о в и ч И. Н., П а щ е н к о Т. Е.|
Применение полимерных материалов в плодоводстве, Л., 1968;
Применение синтетических пленок в лесном хозяйстве, М.'
1969; Бауман X., Пластопоника, Л., 1970; Г о н ч а-
р у к Н. С, Полимеры в овощеводстве, М., 1971; Пути регу-
регулирования почвенных условий жизни растений, под ред.
И. Б. Ревута, Л., 1971; Ж е м о й ц А. А., В а щ е н к о С. Ф.,
Технология возделывания овощей в защищенном грунте, М.,
1972; 4th International colloquium on plastics in agriculture'
June, Paris, 1970; Plastics in agriculture, 5th international col-
colloquium, 5-11 June, Budapest, 1972; M у р а ш к о А. И., Пласт-
Пластмассовый дренаж, Минск, 1969; Полимерные материалы в вод-
водном хозяйстве, М., 1971; Ел шин И. М., Пластбетон, К., 1967;
Сокольская В. В., Гидротехническое строительство,
№ И, 18 A969); Баркан И. Л., Новиковский В. Э.
Гидротехника и мелиорация, № 5, 117 A969); S v i k 1 i s
P. В., Novikowsky V. E., Soviet experience on the
use of synthetic materials in irrigation and drainage, в кн.: 8th
Congress on irrigation and drainage, Varna, 1972.
И. Л. Баркан, В. Э. Новиковский, Т.Е. Пащенко.
ПОЛИМЕРЫ В СТРОИТЕЛЬСТВЕ (polymers in
building, Polymere im Bauwesen, polymeres dans con-
construction).
Содержание:
Введение 958
Материалы и изделия для покрытия полов 95 9
Отделочные и конструкционно-отделочные мате-
материалы 900
Мастики и клеи 962
Теплоизоляционные материалы и акустич. изделия 963
Гидроизоляционные, кровельные и антикоррозион-
антикоррозионные материалы 963
Санитарно-технич. трубы и оборудование „ . . . . 964
Элементы зданий и сооружений 964
Введение. Строительство относится к наиболее круп-
крупным потребителям синтетич. полимеров, их примене-
применение в этой отрасли народного хозяйства непрерывно
возрастает. Так, если в 1960 мировое потребление
пластмасс в строительстве составило 12,4% от общего
объема их производства, то в 1965 и 1970— соответст-
соответственно 20,8 и 25,1%. При увеличении мирового произ-
производства пластмасс за период 1960—1970 примерно в
4 раза объем их потребления в строительстве возрос в
8 раз. Это обусловлено не только уникальными физико-
механич. свойствами полимеров, но также и их ценны-
ценными архитектурно-строительными характеристиками.
Важнейшие проблемы современного индустриально-
индустриального строительства — сокращение сроков возведения
зданий и их максимальное облегчение, т. к. приме-
применение тяжелых материалов сопряжено с большими тран-
транспортными расходами и необходимостью использо-
использования тяжелой механизации. Основные преимущества
полимеров перед др. строительными материалами —
легкость и большая уд. прочность. Благодаря этому
м. б. существенно уменьшена масса строительных кон-
конструкций; напр., многоэтажное крупнопанельное зда-
здание с железобетонным каркасом и ограждающими сте-
959
ПОЛИМЕРЫ В СТРОИТЕЛЬСТВЕ
960
нами, перегородками и встроенным оборудованием из
полимерных материалов легче кирпичного в 3—4 раза.
Области применения полимеров в строительстве
чрезвычайно разнообразны. Известны примеры опытно-
опытного строительства зданий целиком из полимерных ма-
материалов (от конструктивных элементов до оборудо-
оборудования помещений). Однако такие здания еще не по-
получили широкого распространения из-за относительно
высокой стоимости полимеров и отсутствия приемле-
приемлемых для массового строительства оптимальных архи-
архитектурных, конструктивных и технологич. решений.
Накопленный опыт и технико-экономич. подсчеты по-
показывают, что значительный эффект в строительстве
м. б. получен при сочетании полимерных материалов
с железобетоном, металлом, стеклом, асбоцементом.
Наиболее широко полимеры используют для покры-
покрытия полов и др. отделочных работ, герметизации, гидро-
и теплоизоляции, а также для производства труб и др.
санитарно-технического оборудования. Из них изготов-
изготовляют, кроме того, стеновые панели и перегородки, эле-
элементы кровельных покрытий (в том числе светопроз-
рачных), оконные переплеты, двери, объемные сани-
тарно-технич. кабины, пневматические (надувные)
строительные конструкции, домики для туристов,
оленеводов, полярников, павильоны, киоски и др.
Общие требования, к-рые предъявляют к полимер-
полимерным материалам независимо от области их примене-
применения,— возможность использования при современных
индустриальных методах строительства, простота
обработки и экономич. эффективность. Последнему
требованию отвечают материалы, к-рые характери-
характеризуются относительно малой приведенной стоимостью,
учитывающей единовременные затраты в строительст-
строительстве (стоимость материала и производства работ по его
применению), капиталовложения в промышленное про-
производство материала, его долговечность и средне-
среднегодовые эксплуатационные расходы.
Общее архитектурно-строительное требование к
отделочным и конструкционно-отделочным материа-
материалам — достаточный ассортимент и соответствие необхо-
необходимым физико-механич. показателям и эстетич. (ху-
(художественно-декоративным) характеристикам. Мате-
Материалы для ограждающих конструкций должны обладать
высокой механич. прочностью, атмосферостойкостью,
низким коэфф. теплопроводности, а светопрозрач-
ные ограждения, кроме того,— высоким коэфф. све-
топропускания. Материалы для интерьера должны
удовлетворять специальным санитарно-гигиенич. тре-
требованиям (отсутствие токсичных летучих веществ и за-
зарядов статич. электричества; последнее требование
предъявляют к материалам, к-рые при эксплуатации
подвергаются трению).
Материалы и изделия для покрытия полов. Эти ма-
материалы изготовляют в виде рулонов, плиток, а также
мастичных и полимерцементных составов для бесшов-
бесшовных (монолитных) покрытий. Важнейшие эксплуа-
тационно-технич. требования к ним — высокая изно-
износостойкость, небольшая деформируемость под нагруз-
нагрузкой, сохранение цвета, а для рулонных и плиточных
материалов, кроме того, стабильность линейных разме-
размеров. См. также Покрытия для полов.
Экономич. эффективность применения полимерных
покрытий полов вместо традиционных (деревянных и
др.) обусловлена уменьшением трудоемкости работ
(более чем на 50%), значительным сокращением рас-
расхода древесины (при замене в одном 80-квартирном
доме дощатых полов на полы из синтетич. материалов
сохраняется ок. 300 м3 пиломатериалов), уменьшением
капиталовложений на организацию производства мате-
материалов и снижением единовременных затрат на устрой-
устройство полов.
Рулонными материалами покрывают
полы в помещениях жилых, общественных и нек-рых
промышленных зданий. В условиях полносборного
индустриального строительства с железобетонными меж-
междуэтажными перекрытиями весьма эффективно при-
применение свариваемых между собой поливинилхлорид-
ных рулонных материалов на тепло- и звукоизолирую-
звукоизолирующей подоснове, а также ворсовых ковровых материалов
из синтетич. волокон, к-рые укладывают непосредствен-
непосредственно на железобетонную панель перекрытия, обеспечивая
т. обр. необходимую звукоизоляцию без устройства
трудоемкой и дорогой многослойной тепло- и звукоизо-
звукоизолирующей подготовки под иолы. В жилых зданиях
поливинилхлоридные и алкидные рулонные материалы
настилают во всех помещениях. В общественных зда-
зданиях их не рекомендуется использовать в помещениях
с интенсивными пешеходными потоками. Резиновые и
коллоксилиновые материалы не применяют обычно в
детских учреждениях (сады, ясли) и жилых комна-
комнатах. В промышленных зданиях рулонные материалы
настилают в бытовых и административных помещениях,
а также в таких производственных цехах, где отсут-
отсутствуют интенсивное движение и воздействие на иол
ударных нагрузок, жиров, масел, абразивных мате-
материалов.
Достоинства плиточных изделий для ио-
иолов — простота приклейки к основаниям и ремонта,
хорошие эксплуатационные свойства, возможность
создания иолов разнообразного рисунка. На производ-
производство плиток расходуется меньше полимеров, чем на ру-
рулонные материалы, т. к. их изготовляют из более на-
наполненных композиций. Однако укладка плиток более
трудоемка, чем настилка рулонных материалов, а чис-
число швов в покрытии в несколько раз больше.
Плитки из поливинилхлорида применяют преимуще-
преимущественно для устройства полов в помещениях обществен-
общественных зданий, а также в кухнях, санитарных узлах и под-
подсобных помещениях жилых домов. В общественных зда-
зданиях укладывают, кроме того, плитки из резины, а
также из синтетич. волокон. Плитки из фенолита
и полимер вето на предназначены гл. обр. для устрой-
устройства полов в цехах, лабораториях и подсобных поме-
помещениях промышленных зданий. В подсобных поме-
помещениях зданий различного назначения (кроме поме-
помещений с повышенной влажностью) укладывают плитки
из кумароно-инденовых смол.
Для получения бесшовных покрытий
применяют мастичные составы, напр, на основе диспер-
дисперсий поливини л ацетата, а также составы на основе поли-
мерцемента, получаемые, напр., из дисперсий поливи-
нилацетата или бутадиен-стирольных латексов (см.
Латексы синтетические). Мастичные покрытия (тол-
(толщина 3—5 мм) применяют обычно в помещениях обще-
общественных и производственных зданий, в к-рых полы
подвергаются слабым механич. воздействиям. Составы
на основе полиэфирных и эпоксидных смол используют
в помещениях общественных (кроме лечебно-профи-
лактич. и детских) и промышленных зданий (кроме
зданий пищевой промышленности) с интенсивным дви-
движением и умеренными механическими воздействиями
на полы.
Из полимерцементных составов устраивают полы
(толщина 15—20 мм) в помещениях общественных
и производственных зданий, где на полы воздействуют
умеренные механич. нагрузки; поливинилацетатные со-
составы пригодны для полов с сухим режимом эксплуа-
эксплуатации. Бесшовные полы могут быть окрашены в любые
цвета, а полы из полимерцементного бетона, кроме то-
того, содержать мелкие включения цветного природно-
природного камня.
Отделочные и конструкционно-отделочные материа-
материалы. Для отделочных работ широко используют рулон-
рулонные материалы — поливинилхлоридные и полиэтиле-
полиэтиленовые пленки (см. Поливинилхлоридные пленки, По-
лиолефиновые пленки), а также влагостойкие бумажные
961
ПОЛИМЕРЫ В СТРОИТЕЛЬСТВЕ
962
обои. Непрозрачные поливинилхлоридные пленки,
окрашенные в массе в любые цвета, м. б. покрыты кле-
клеевым слоем или иметь подоснову — бумажную, ткане-
тканевую, эластичную звукопоглощающую (напр., из пено-
пенополиуретана). На пленки м. б. нанесен печатный рису-
рисунок (см. Печать на полимерах); их поверхность м. б.
гладкой или рельефной. Пленки без подосновы и без
клеевого слоя иногда бывают прозрачными или полу-
полупрозрачными, благодаря чему их можно использовать в
качестве светопропускающих материалов.
Пленки с клеевым слоем применяют для отделки
встроенной мебели, дверных полотнищ, стен и пере-
перегородок в помещениях с повышенными гигиенич. тре-
требованиями (больницы, детские учреждения и др.)
и в санитарных узлах квартир; пленки на бумажной
подоснове — преимущественно для оклейки стен и пе-
перегородок в помещениях жилых и общественных зда-
зданий. Пленки на тканевой подоснове служат для отдел-
отделки стен и устройства раздвижных перегородок в поме-
помещениях общественных зданий; пленками на звукопогло-
звукопоглощающей эластичной подоснове отделывают помещения
с повышенными акустич. требованиями. Пленки без
подосновы используют гл. обр. для устройства занавесей
и отделки помещений с повышенной влажностью. Из
полупрозрачных пленок устраивают подвесные светя-
светящиеся потолки; для этой цели м. б. также использованы
пленочные материалы, армированные стекловолокном.
Бумажные обои с прозрачным пленочным покрытием из
поливинилхлорида или полиэтилена выдерживают мно-
многократное мытье теплой водой с мылом. Срок службы
обоев с покрытием из поливинилацетатной дисперсии
в 2—3 раза больше срока службы обычных. В ворсовых
обоях, бумажная основа к-рых покрыта короткими син-
тетич. волокнами, сочетаются высокие декоративные
характеристики с звукопоглощающей способностью.
Группа конструкционно-отделочных материалов
включает различные листовые и плиточные материалы.
Декоративный бумажно-слоистый пластик, к-рый мо-
может иметь зеркальную или матовую поверхность, лю-
любой цвет и рисунок, используют для облицовки стен,
панелей, перегородок помещений общественных зда-
зданий, для изготовления дверных полотнищ и др. Не-
Непрозрачный листовой жесткий поливинилхлорид (ви-
(винипласт) применяют для облицовки панелей, перего-
перегородок, дверных полотнищ и др.; прозрачный бесцвет-
бесцветный и полупрозрачный тонированный — для заполне-
заполнения световых проемов и устройства верхнего освеще-
освещения. Ударопрочный и атмосферостойкий материал на
основе поливинилхлорида, модифицированного хлори-
хлорированным полиэтиленом (см. Полиолефины хлорирован-
хлорированные), используют для наружной облицовки трехслой-
трехслойных навесных панелей, фасадов зданий, ограждений
балконов, устройства светопрозрачной кровли, заполне-
заполнения стеновых проемов, изготовления окон и дверей.
Для внутренней облицовки стен и панелей помеще-
помещений с повышенными гигиенич. требованиями (продо-
(продовольственные магазины, предприятия общественного
питания, лабораторные помещения, кухни, санитар-
санитарные узлы) применяют плитки из полистирола (см.
Стирола полимеры). Для устройства потолков, изго-
изготовления дверных полотнищ, перегородок, стенных
шкафов и др. встроенной мебели м. б. использованы
древесно-стружечные плиты, облицованные синтетич.
пленкой, шпоном из ценных пород дерева, декоратив-
декоративным бумажно-слоистым пластиком, а также покрытые
лакокрасочными материалами. Древесно-волокнистыми
плитами с полимерными покрытиями облицовывают
стены и встроенную мебель; их используют также для
устройства стеновых панелей в кухнях и санузлах жи-
жилых зданий, в лабораториях, магазинах, медицинских
учреждениях. См. также Древесные пластики.
Из древесно-слоистых пластиков изготовляют стено-
стеновые трехслойные панели. Бакелизированная фанера
служит гл. обр. для внутренних облицовок в обще-
общественных зданиях, а также в качестве наружного слоя
щитовых дверей. Эти материалы имеют гладкую, на-
напоминающую лакированную, поверхность с хорошо
просматриваемой текстурой древесного шпона.
Непрозрачные листовые стеклопластики образуют ли-
лицевые поверхности слоистых (преимущественно навес-
навесных) панелей. При введении пигментов м. б. получены
материалы с необходимыми декоративными характери-
характеристиками. Бесцветные или окрашенные в массе прозрач-
прозрачные или полупрозрачные стеклопластики в виде пло-
плоских или волнистых листов применяют для ограждения
лестниц и балконов, заполнения проемов, устройства
перегородок, просвечивающих кровель, световых фона-
фонарей, плафонов искусственного освещения, навесов и ко-
козырьков над входами. Листы прозрачного волнистого
стеклопластика в сочетании с непрозрачными волнисты-
волнистыми асбоцементными или стальными листами служат для
устройства верхнего освещения в скатных кровельных
покрытиях зданий. Их используют и для устройства
светопрозрачных панелей бокового освещения.
Полиметилметакрилат (органическое стекло) служит
для заполнения оконных и дверных проемов, устрой-
устройства прозрачных перегородок, лестничных и балконных
ограждений, акустич. экранов и др. Его используют
также для изготовления световых плафонов, ламп лю-
люминесцентного освещения. Сферич.,конич. ицилиндрич.'
колпаки из органич. стекла применяют в качестве све-
световых фонарей на плоских кровлях общественных и
промышленных зданий. При введении красителей полу-
получают цветные прозрачные, полупрозрачные или непроз-
непрозрачные разновидности этого материала.
Важное значение при отделке зданий, в частности в
полносборном строительстве, имеют профильные
(погонажные) изделия — плинтусы, поруч-
поручни для лестниц, балконов, накладки на проступи лест-
лестниц, наличники, нащельники, профили для крепления
и обработки швов листовых и рулонных облицовочных
материалов и обработки стыков в крупнопанельных зда-
зданиях, изготовляемые гл. обр. из поливинилхлорида.
Из этого же полимера, а также из полиуретанов и резин
на основе синтетич. каучуков изготовляют эластичные
уплотняющие профили (ленты, полосы, жгуты и др.)
для окон, дверей, витрин и стыков в крупнопанель-
крупнопанельных зданиях.
Одно из ведущих мест среди отделочных полимерных
материалов занимают лакокрасочные мате-
материалы. Наиболее перспективны среди них краски на
основе синтетич. пленкообразующих — алкидных смол
(напр., алкидно-стирольные), продуктов совмещения
каучуков с кумароно-инденовыми смолами (т. наз. ку-
мароно-каучуковые), хлорированных каучуков, пер-
хлорвиниловых и эпоксидных смол, полиуретанов, дис-
дисперсий поливинил ацетата, полимерцемента и др. См.
также Лаки и эмали, Краски, Строительные краски,
Лакокрасочные покрытия.
Мастики и клеи. Мастики, применяемые для приклеи-
приклеивания покрытий полов и погонажных изделий, а также
для крепления отделочных материалов, должны обла-
обладать хорошей адгезией как к этим материалам, так и
к материалам основания, био- и термостойкостью (до
50—60 °С), быть нетоксичными. Они должны, кроме то-
того, легко наноситься тонким слоем и склеивать без спе-
специальной «пригрузки». В наибольшей степени этим тре-
требованиям отвечают мастики на основе продуктов совме-
совмещения кумароно-инденовых смол с каучуками, хлоро-
пренового каучука, эпоксидных смол.
Мастики на основе полисульфидных каучуков,
бутилкаучука, полиизобутилена, кремнийорганич. и
хлоропреновых каучуков используют для герметиза-
герметизации стыков между панелями полносборных зданий,
для заделки швов в витринах, окнах, фонарях верх-
верхнего света и др. См. также Герметизирующие составы.
963
ПОЛИМЕРЫ В СТРОИТЕЛЬСТВЕ
964
Для склеивания строительных конструкций (из асбо-
асбоцемента, алюминия, стали, пластмасс, дерева) как друг
с другом, так и с пенопластами и др. теплоизолирую-
теплоизолирующими материалами применяют эпоксидные, феноло-
и мочевино-формальдегидные, полиэфирные и резино-
резиновые клеи (см. Клеи синтетические). Наиболее прочные
соединения получают при использовании эпоксидных
клеев, к-рые применяют в мостостроении для склейки
стыков между сборными железобетонными элементами.
Это обеспечивает высокие темпы сборки конструкций
в любых климатич. условиях, уменьшает трудоемкость
и стоимость монтажных работ. По прочностным и дефор-
деформационным свойствам такие сборные конструкции не
уступают сооружениям из монолитного железобетона.
Для склеивания древесины чаще всего применяют
феноло- и мочевино-формальдегидные клеи. При сборке
деревянных конструкций, не предназначенных для
эксплуатации во влажной среде, широко используют
казеиновые клеи (см. Клеи природные).
Теплоизоляционные материалы и акустические изде-
изделия. К весьма эффективным тепло- и звукоизоляцион-
звукоизоляционным материалам относятся пенопласты с объемной мас-
массой 15 —100 кг/м3— пенополистирол, пенополиформалъ-
дегид, пенополиуретаны, пенополивинилхлорид, к-рые
применяют, как правило, в качестве внутреннего теп-
теплоизолирующего слоя в трехслойных легких панелях
ограждающих конструкций зданий. Пропиткой мине-
минеральной (в частности, стеклянной) ваты феноло- или
мочевино-формальдегидными смолами, а также синте-
тич. латексами изготовляют тепло- и звукоизоля-
звукоизоляционные маты и плиты (жесткие и полужесткие) с
объемной массой 25—150 кг/м3 и теплостойкостью не
ниже 150 °С.
Покрытые белым фактурным слоем перфорированные
минераловатные плиты используют для звукопогло-
звукопоглощающей облицовки потолков и верхних (недоступных
для механич. повреждений) участков стен.
К акустич. изделиям относятся, кроме того, листы из
перфорированных пластмасс (.винипласта, органич.
стекла, полиэфирного стеклопластика), к-рые приме-
применяют в сочетании со звукопоглощающим слоем минера-
ловатного материала.
Гидроизоляционные, кровельные и антикоррозион-
антикоррозионные материалы. В качестве гидроизоляционных мате-
материалов применяют полипропиленовые, полиэтиленовые,
полиизобутиленовые пленки, а также пленки из бу-
тилкаучука и модифицированного синтетич. каучуком
(эластифицированного) поливинилхлорида. Пленки и
пленкообразующие составы для кровельных покрытий
изготовляют из поливинилхлорида, полиолефинов и
полиэфирных смол. Жесткий поливинилхлорид, в том
числе армированный тканью, стеклопластики, органич.
стекло, а также армированные поливинилхлоридные и
полиэтиленовые пленки используют для устройства
светопропускающей кровли при перекрытии парников,
спортивных площадок, отдельных участков торговых
улиц.
Для кровельных и гидроизоляционных работ при-
применяют также материалы на основе битумов: рубероид,
в том числе армированный стекловолокном (для кров-
кровли), гидроизол (пропитанный битумом асбестовый
картон) и бризол (рулонный материал из измельченной
отработанной резины, битума и добавок асбеста).
Весьма перспективно устройство кровли из полимер-
полимерных мастик или модифицированных полимерами биту-
битумов нанесением их на защищаемую поверхность на-
напылением (иногда с одновременным распылением ар-
армирующего стекловолокна). Такой способ дает эконо-
экономию трудовых затрат и капиталовложений, посколь-
поскольку исключается стадия производства рулонных ма-
материалов.
Для антикоррозионной защиты строительных кон-
конструкций служат замазки на основе феноло-формаль-
дегидных смол, листовой полиизобутилен, наполнен-
наполненные песком строительные растворы на основе эпоксид-
эпоксидных или фурановых смол, а также композиции на
основе хлорсульфированного полиэтилена (см. Поли-
Полиэтилен хлорсульфированный).
Санитарно-технические трубы и оборудование. Тру-
Трубы из пластмасс получили значительное распростране-
распространение в системах водоснабжения и канализации. Важ-
Важнейшие их достоинства — коррозионная стойкость и
небольшая масса. Трубы м. б. надежно соединены друг
с другом при помощи специальных фасонных частей
(изготовляемых обычно из того же материала, что и са-
сами трубы), а также сваркой. Напорные трубы изготов-
изготовляют из полиэтилена, винипласта, полипропилена,
безнапорные канализационные трубы — из полиэтиле-
полиэтилена высокой плотности и винипласта, дренажные —
из полиэтилена высокой плотности. Из листов вини-
винипласта м. б. изготовлены вентиляционные короба, а
также трубы и желоба наружных водостоков. Пласт-
Пластмассы, окрашенные в различные цвета, используют
также для изготовления ванн, моек, умывальников,
сифонов, кранов, смесителей и др. санитарно-технич.
оборудования.
Элементы зданий и сооружений. В многоэтажных кар-
каркасных зданиях широкое распространение получили
легкие, как правило трехслойные, навесные панели,
к-рые состоят из двух листов, образующих наружные
(несущие) слои, и внутреннего (заполняющего) тепло-
и звукоизолирующего слоя. Материалом для наруж-
наружного слоя, наряду с алюминием, сталью, асбоцементом,
закаленным и армированным стеклом, может служить
стеклопластик, древесный пластик, винипласт, а также
поливинилхлорид, модифицированный хлорирован-
хлорированным полиэтиленом. Внутренний слой панелей изготов-
изготовляют как из пенопластов (полистирольных, поли-
уретановых и др.), так и из волнистых листов стекло-
стеклопластика. Этот слой м. б. выполнен в виде т. наз. сото-
сотовых конструкций, изготовляемых из крафт-бумаги, про-
пропитанной термореактивными смолами (см. Сотоплас-
ты), или в виде ячеистой конструкции, пустоты к-рой
заполнены минеральной ватой или пенопластом.
Полимерные материалы используют для изготовления
оконных переплетов. При строительстве зданий в теп-
теплых безлесных районах применяют полые переплеты
из полиэфирного стеклопластика или жесткого поли-
поливинилхлорида. Для более холодных районов более при-
пригодны деревянные переплеты с тонким ударопрочным
и атмосферостойким покрытием на основе поливинил-
поливинилхлорида.
Из стеклопластика изготовляют кровельные панели,
зонтичные конструкции, объемные санитарно-технич.
блоки.
Для создания пневматич. конструкций — промыш-
промышленных и складских зданий, культурно-бытовых зда-
зданий временного типа (выставочные павильоны, спортза-
спортзалы, туристич. палатки), оранжерей с покрытиями из
светопропускающих пленок, временных сооружений
на строительных площадках — используют ткани из
синтетич. волокон или стеклоткани с воздухонепрони-
воздухонепроницаемыми покрытиями из поливинилхлорида или рези-
резины (см. Прорезиненные ткани), а также полиэтилено-
полиэтиленовые, полиэфирные и поливинилхлоридные пленки,
армированные синтетич. волокном. О применении поли-
полимерных материалов для изготовления внутренних пере-
перегородок, дверей, светопрозрачных фонарей см. выше в
разделе «Отделочные и конструкционно-отделочные
материалы».
С применением полимеров в значительной степени
связан прогресс в строительной практике и архитекту-
архитектуре. Внедрение новых полимерных строительных мате-
материалов способствует разработке эффективных конст-
965
ПОЛИМЕРЫ В СУДОСТРОЕНИИ
966
рукций, развитию индустриальных методов их про-
производства, созданию красивых, прочных и экономич-
экономичных зданий. Наиболее перспективны в строительстве
трехслойные ограждающие панели, покрытия для по-
полов на тепло- и звукоизолирующей подоснове и др.
многослойные изделия и материалы, в к-рых возмож-
возможно комбинирование полимеров, выполняющих в кон-
конструкции свои особые функции. Сочетание полимер-
полимерных материалов со стеклом и легкими металлами так-
также позволит создавать новые конструктивные узлы,
детали и элементы зданий.
Лит.: Каталог отделочных материалов и изделий, под ред.
М. П. Макотинского, раздел 1 — Пластмассы, раздел 2 — Кра-
Краски и лаки, М.> 1961—62; Якубовский Э., НитшФ.,
Пластмассы во внутренней отделке зданий, пер. с нем., М.,
1963; МакотинскийМ.П., Новые отделочные материалы
в строительстве, М., 1967; Пластмассы в строительстве, пер.
с чеш., М., 1969; Строительные нормы и правила, ч. 1, раздел В,
гл< 15 — Материалы и изделия на основе полимеров (СН и П.
1 _ В, 15—69), М., 1970; Губенко А. Б., Строительные
конструкции с применением пластмасс, М., 1970; Полуя-
Полуянов А. Ф., Стройматериалы, № 4, 11 A971); Симпозиум
по применению пластмасс и каучуков в строительстве. Сб.
материалов и информации постоянной комиссии СЭВ по строи-
строительству (Интерстройинформация), М., 1971; Макотин-
с к и й М. П., Новые отделочные материалы, М., 1972; К 1 е i-
nickeW.E., В i k a 1 е s N. М., в кн.: Encyclopedia of poly-
polymer science and technology, v. 2, N. Y.— [a. o.J, 1965, p. 614.
M. П. Макотинский.
ПОЛИМЕРЫ В СУДОСТРОЕНИИ (polymers in
shipbuilding, Polymere im Schiffsbau, polymeres dans
construction de navires).
Содержание:
Введение 965
Материалы для изготовления корпусов судов и
корпусных конструкций 965
Уплотнительные материалы 967
Клеи 968
Материалы для палубных мастичных покрытий 969
Защитные и декоративные покрытия, отделочные
материалы 970
Материалы, используемые в судовом машино-
машиностроении 972
Введение. Полимерные материалы начали применять
в судостроении в 30-х гг. В 1936 в Германии на лин-
линкоре «Тирпиц» были впервые установлены поливинил-
хлоридные трубопроводы для питьевой, технич.,
охлаждающей и забортной воды, а несколько лет спустя
армированный асбестом прессматериал стали применять
для изготовления мебели и переборок кают на траль-
тральщиках и миноносцах.
Современная судостроительная пром-сть — один из
крупнейших потребителей синтетич. полимерных ма-
материалов, причем области их применения очень раз-
разнообразны, а перспективы использования практически
неограниченны. Полимеры применяют для изготовле-
изготовления корпусов судов и корпусных конструкций, в произ-
производстве деталей судовых механизмов, приборов и
аппаратуры, для окраски судов, отделки помещений
и их тепло-, звуко- и виброизоляции и др. Благодаря
использованию полимерных материалов значительно
улучшаются технич. и эксплуатационные характери-
характеристики судов, повышаются их надежность и долго-
долговечность, сокращается продолжительность и снижает-
снижается трудоемкость постройки.
Материалы для изготовления корпусов судов и кор-
корпусных конструкций. Среди конструкционных поли-
полимерных материалов наибольшее значение в судострое-
судостроении имеют стеклопластики. Это объясняется их высо-
высокими механич. свойствами, достаточно хорошей тер-
термостойкостью, устойчивостью к коррозии и к старению,
сравнительной легкостью переработки в изделия самых
сложных форм, простотой и удобством эксплуатации и
ремонта. В США, напр., в 1958 из этих материалов
было построено 80 тыс., в 1963 — 214 тыс., в 1972—
700 тыс. судов. В 1969 в мировом судостроении было
использовано около 100 тыс. т стеклопластиков; к
1980 их потребление в этой отрасли пром-сти должно
возрасти примерно в 4 раза. Данные о росте потребле-
потребления стеклопластиков в судостроительной пром-сти
США и ФРГ приведены в табл. 1.
Таблица
Год
1966
1967
1968
1969
1970
1. Потребление стеклопластиков в
США и ФРГ
США
всего,
тыс. т
219
275
353
440
525
из них в судо-
судостроении, %
20,7
25,4
25,9
24,7
15,4
судостроении
ФРГ
всего,
тыс. т
36
38
53
72
95
из них в судо-
судостроении, %
4
7
7
6
7
Таблица 2. Сравнительные
характеристики траулеров,
построенных из различных
материалов
Для изготовления корпусов судов и корпусных кон-
конструкций используют гл. обр. полиэфирные стеклопла-
стеклопластики холодного отверждения. В тех случаях, когда тре-
требуются материалы с особо высокими прочностными
характеристиками (напр., для корпусов глубоководных
аппаратов), применяют эпоксидные стеклопластики.
Использование в судостроении стеклопластиков на
основе феноло-альдегидных, карбамидных, фурановых
и кремнийорганич. смол затруднено из-за необходимо-
необходимости формования изделий при высоких темп-рах
(>170 °С) и давлениях [2,5 — 10 Мн/м2 B5 — 100 кгс/см-)].
Одно из наиболее важных преимуществ стеклопла-
стеклопластиков перед традиционными судостроительными ма-
материалами (деревом, сталью, алюминиево-магниевыми
сплавами) — высокая уд. прочность (см. Армированные
пластики), благодаря чему м. б. уменьшена масса судо-
судовых конструкций (табл. 2).
Корпуса и корпусные
конструкции из стеклопла-
стеклопластиков изготовляют гл. обр.
тремя методами: 1) кон-
контактное формование; 2) на-
напыление; 3) прессование.
Основное достоинство м е-
тода контактного
формования, наибо-
наиболее распространенного в су-
судостроении,— простота. По-
Помимо форм (матриц), он не
требует практически ника-
никакой сложной оснастки, оборудования и приспособле-
приспособлений. Поскольку все операции выполняются вручную,
производительность этого метода невысока, а условия
труда тяжелы. Однако он позволяет изготовлять кор-
корпуса сравнительно крупных судов (длиной до 50 м и
водоизмещением до 300 т). Имеются также разработки
для постройки судов длиной более 60 м и водоизме-
водоизмещением до 1000 т.
Механизированный метод напыления бо-
более прогрессивен. Его применяют при изготовлении
корпусов мелких судов или их отдельных деталей.
Для производства корпусов более крупных судов этот
метод непригоден вследствие меньшей прочности стек-
стеклопластика, чем в случае ручного контактного формо-
формования.
Наиболее прогрессивный метод формования корпу-
корпусов судов — прессование, в к-ром все операции,
кроме укладки стеклоткани в форму и заливки связую-
связующего, механизированы. Метод более производителен,
чем описанные выше, но его применение ограничива-
ограничивается размерами прессовой оснастки и мощностью обо-
оборудования. О методах формования см. также Стекло-
Стеклопластики.
Корпуса крупных судов изготовляют преимуществен-
преимущественно из монолитного стеклопластика. Трехслойные кон-
конструкции, в к-рых в качестве заполнителя между дву-
Материал
Дерево ....
Сталь
Стеклопла-
Стеклопластик ....
Грузо-
вмести-
вместимость,
м3
170
187
195
Масса
порож-
порожнего су-
судна, т
232
233
170
967
ПОЛИМЕРЫ В СУДОСТРОЕНИИ
968
мя слоями стеклопластика применяют пенопласты и
сотопласты, при изготовлении таких судов себя не
оправдали. Стремление получить максимальный выиг-
выигрыш в массе обусловило использование трехслойных
конструкций в судах на подводных крыльях и на воз-
воздушной подушке. Такие конструкции широко приме-
применяют для изготовления переборок, секций палуб, над-
надстроек и др. деталей (они легче монолитных из стекло-
стеклопластика на 20%, а металлических — в 1,5 раза). Из
трехслойных конструкций, заполненных пенополиуре-
пенополиуретаном, изготовляют также спасательные шлюпки. Этот
заполнитель, помимо облегчения корпуса, обеспечивает
необходимую непотопляемость судна. Введение в свя-
связующее для стеклопластика антипиренов или исполь-
использование самозатухающих связующих позволяет изго-
изготовлять спасательные шлюпки для танкерного флота,
к-рые могут преодолевать водные пространства с горя-
горящими на поверхности нефтепродуктами (см. также
Огнестойкость).
Несмотря на сравнительно высокие цены на исход-
исходное сырье, стоимость судна с корпусом из стеклопла-
стеклопластика практически равна стоимости деревянного. Это
обусловлено меньшей трудоемкостью изготовления
корпуса из пластмассы, а также возможностью исполь-
использования малоквалифицированной рабочей силы.
Эксплуатационные расходы на содержание корпусов
судов из пластмассы составляют не более 25% от
расходов на содержание деревянных; они также зна-
значительно ниже расходов на содержание металлич. кор-
корпусов. Низкие эксплуатационные расходы — одна из
причин резкого роста применения стеклопластиков для
изготовления корпусов рыбодобывающих и промысло-
промысловых судов.
Помимо корпусов судов, из армированных пласт-
пластмасс изготовляют также переборки, надстройки и руле-
рулевые рубки, мачты, дейдвудные трубы, люковые закры-
закрытия дверей, оборудование помещений и др. детали.
Стеклопластик — немагнитный материал, и поэтому
его с успехом применяют для постройки тральщиков и
др. военных кораблей. Напр., в США из этого ма-
материала в 1947—65 было построено более 3000 военно-
морских судов различного назначения.
Большие перспективы имеет использование стекло-
стеклопластиков для изготовления корпусов глубоководных
аппаратов и подводных лодок. Напр., из высокопроч-
высокопрочных легированных сталей м. б. изготовлены корпуса
обитаемых глубоководных аппаратов, предназначен-
предназначенных для погружения на глубину до 3000 м, из алюми-
алюминиевых сплавов — до 4500 м, из титановых сплавов —
до 6000 м, из стеклопластиков — до 9000 м. Такое пре-
преимущество стеклопластиков перед др. конструкцион-
конструкционными материалами обусловлено прежде всего их вы-
высокой уд. прочностью. Внутренние (прочные) детали
этих аппаратов из стеклопластика изготовляют ме-
методами прессования или намотки на специальных
станках. Секции цилиндрич. корпусов крупных аппа-
аппаратов получают намоткой на специальные разборные
оправки. Наружные (легкие) корпуса изготовляют из
полиэфирных стеклопластиков методом контактного
формования.
Весьма перспективные материалы для производства
глубоководных аппаратов — пластики, армированные
углеродными или борными волокнами. Широкое приме-
применение этих волокон ограничивается их высокой стои-
стоимостью. Однако, по данным исследователей США, эпок-
эпоксидный слоистый пластик, армированный углеродными
волокнами (см. Углеродопласты), более перспективен
для изготовления корпусов глубоководных аппаратов,
чем стеклопластик, вследствие более высоких модуля
упругости и усталостной выносливости и меньшей
плотности.
Уплотнительные материалы. Эти материалы приме-
применяют в судостроении для тепло-, звуко- и виброизоля-
виброизоляции, для герметизации разъемов и соединении деталей и
механизмов и многих др. целей. Независимо от назначе-
назначения все уплотнительные материалы должны обладать
влаго- и огнестойкостью, устойчивостью к действию
биологич. факторов и к перепадам темп-р, отвечать тре-
требованиям пожарной безопасности, не выделять при
эксплуатации токсичных веществ и иметь хорошие тех-
нологич. свойства.
Теплоизоляционные материалы
на основе синтетич. полимеров (табл. 3) превосходят
по большинству показателей пробковую изоляцию:
они имеют меньшие теплопроводность, влагопоглоще-
ние и объемную массу, обладают большей огнестой-
огнестойкостью. Помимо своей основной функции, эти материа-
материалы обеспечивают звукоизоляцию. Иногда для
звукоизоляции применяют также губчатые резины на
основе различных каучуков.
Для виброизоляции, особенно необходимой в
судах на подводных крыльях и на воздушной подушке,
отдельные судовые конструкции покрывают вибропо-
глощающими пластмассами на основе полибутилметак-
рилата или поливинилхлорида. Главные и вспомога-
вспомогательные двигатели, приборы и судовую аппаратуру
устанавливают на виброизолирующих резиновых амор-
амортизаторах.
Герметики на основе синтетич. полимеров, спо-
способные отверждаться при обычных темп-pax с образо-
образованием прочных и эластичных соединений, устойчивых
к действию агрессивных сред, применяют для уплот-
уплотнения разъемов и стыков различных судостроитель-
судостроительных деталей. Наиболее широко для этих целей ис-
используют составы на основе жидких полисульфидных
каучуков (тиоколов), а также обладающие повышенной
водостойкостью составы на основе композиций тиоко-
тиокола с фенольными смолами (см. также Герметизирую-
Герметизирующие составы).
В 50-х гг. в судостроительной пром-сти многих стран
получили распространение одноупаковочные составы
на основе полиэфирных смол, отверждающиеся в малых
зазорах без доступа воздуха и предназначенные для
уплотнения деталей механизмов, приборов и аппаратов,
а также для стопорения деталей, имеющих резьбо-
резьбовые соединения. В последнем случае составы исполь-
используют вместо установки стопоров, шайб, контргаек,
шплинтов; их применяют также для склеивания де-
деталей из различных металлов, пластмасс, дерева, стек-
стекла, резины.
Для получения разнообразных прокладочных
материалов применяют гл. обр. резины на осно-
основе различных синтетич. каучуков. Так, уплотнитель-
уплотнительные прокладки для дверей, иллюминаторов, крышек
люков, конусов гребных валов в местах насадки греб-
гребных винтов и др. аналогичных деталей изготовляют из
кислото- и щелочестойких резин (напр., на основе
хлоропреновых каучуков). Для уплотнительных деталей
(манжет, колец, воротников, прокладок) масляных и
топливных систем применяют масло- и бензостойкие
резины на основе бутадиен-нитрилъных каучуков и
полисулъфидных каучуков, для деталей механизмов,
приборов и устройств, эксплуатируемых при низких
темп-pax (напр., на открытых палубах или в рефриже-
рефрижераторных камерах) — морозостойкие резины из бута-
бутадиеновых каучуков и бутадиен-стиролъных каучуков.
Теплостойкие резины из кремнийорганических каучуков
и фторсодержащих каучуков, а также из бутилкаучука
используют для изготовления деталей, работающих при
повышенных температурах (прокладки теплых ящиков,
трубки для подачи горячей воды и газов, уплотнения
осветительной и сигнальной аппаратуры, эжекторы,
теплообменные аппараты, эластичные муфты и др.).
Клеи. Для склеивания деталей из пластмасс, при-
приклеивания различных материалов к металлам и склеи-
склеивания металлов друг с другом широко используют
969 полимеры в судостроении 970
Таблица 3. Основные характеристики теплоизоляционных материалов, применяемых в судостроении
Материал
Маты из штапельного ка-
капронового волокна
Плиточный пенополиви-
нилхлорид
Напыляемый пенополи-
пенополиуретан на основе полиизо-
цианата и фосфорсодержа-
фосфорсодержащего олигоэфира
Плиточный суспензион-
суспензионный иенополистирол
Плиты из сплава феноло-
формальдегидной и фурфу-
фурфурол-ацетоновой смол
Пробка спрессованная
в плитах
Объем-
Объемная
масса,
кг /ж3
60
85-195
30-50
25-40
70-100
240
Коэфф. теп-
лопроводно-
лопроводности, вт/(м-К)
0,052
[0,045]
0,058
[0,050]
0,041
[0,035]
0,052
[0,045]
0,058
[0,050]
0,058
[0,050]
Возго-
рае-
раемость
К*
2,1
1 ,63-
1 ,75
0,83
2,1
0,58
4,0
Темп-
ра
экс-
плуа-
плуатации,
СС
60
60
80
60
40
100
Влаго-
погло-
щение
за 24 ч,
% (по
массе)
9,0
2 ,5**
3,6
4,0
2,8
12,0
Изолируемые детали
Корпуса и др. поверхности малых судов, холод-
холодные трубопроводы, вентиляционные каналы (за ис-
исключением охлаждаемых камер и трюмов)
Борта, наружные переборки, палубы и их риббан-
ды (только во вспомогательных помещениях)
Борта наружных переборок и палуб в жилых и
вентилируемых служебных помещениях (кроме пи-
пищеблоков и провизионных кладовых на всех судах,
за исключением пассажирских)
Борта, переборки, палубы в вентилируемых поме-
помещениях, трубопроводы и вентиляционные каналы
(кроме пищеблоков на всех судах, за исключением
пассажирских)
Борта, переборки, палубы и риббанды (во всех по-
помещениях)
Жилые, служебные и др. помещения
* К = дв/фь где qB и
ния. ** При 120 °С.
— соответственно количество тепла, выделенное образцом и подведенное к образцу для его сжига-
эпоксидные клеи, а также эпоксидно-полиамидные клеи,
к-рые обладают большой жизнеспособностью и отверж-
даются с помощью нетоксичного низкомолекулярного
полиамида; при этом получают эластичные клеевые
соединения. Для клеесварных соединений применяют
полибутилметакрилатныи клей холодного отверждения
(см. Полиакриловые клеи), позволяющий производить
точечную сварку сразу после нанесения и образующий
вибро- и ударопрочный, а также химстойкий эластич-
эластичный шов.
Для приклеивания теплоизоляционных материалов
используют латексные клеи, а также полиамидные
клеи (напр., дифенилолкапролактамовые), не вызываю-
вызывающие коррозии металлов. Последние наиболее удобны
в применении, т. к. не содержат растворителей и отверж-
даются в короткие сроки. Нетоксичные и не вызываю-
вызывающие коррозии металлов поливинилацетатные клеи
используют вместо декстринового и казеинового для
приклеивания отделочных материалов. Для этой же
цели пригодны клеи на основе композиции хлорирован-
хлорированного полихлоропрена (хлорнаирит) и феноло-формаль-
дегидной смолы. В судостроении широко применяют
также клеи на основе мочевино-формальдегидной смолы
и модифицированного фенольной смолой поливинил-
бутираля. В нек-рых случаях клеи выполняют одновре-
одновременно функции герметиков. См. также Клеи синтетиче-
синтетические, Склеивание.
Материалы для палубных мастичных покрытий.
Покрытия из мастичных материалов должны устранять
скользкость открытых металлич. палуб в любую пого-
погоду, предохранять палубы от коррозии, обладать хоро-
хорошей адгезией к защищаемой поверхности, атмосферо-
стойкостью (в том числе в тропич. условиях), стойко-
стойкостью к действию морской воды, а также нефтестойко-
стью. Эти свойства мастик должны сохраняться в ин-
интервале темп-р от — 40 до 80 °С. Мастичные материалы
наносят обычно в местах интенсивного хождения;
иногда их используют для выравнивания палуб перед
наклейкой линолеума. Нанесение мастичных материа-
материалов на железобетонные палубы предотвращает образо-
образование трещин в железобетоне. В отдельных случаях
мастики выполняют роль теплоизоляционных или
огнезащитных материалов.
Для покрытий, устраняющих скользкость палуб, при-
применяют мастики на основе эпоксидной смолы, пласти-
пластифицированной низкомолекулярным бутадиен-нитриль-
ным каучуком, на основе алкидной (глифталевой) грун-
грунтовки, пластифицированной хлорированным поливи-
нилхлоридом (см. Пер хлор винило вые смолы), а также
полимерцементные составы (напр., на основе бутадиен-
стирольного латекса; см. Полимер цемент). Последние
м. б. использованы и для выравнивания палуб. Необхо-
Необходимые свойства мастик достигаются введением в них
наполнителей, абразивов и др. ингредиентов.
Защитные и декоративные покрытия, отделочные
материалы. Особое место в судостроении занимает
проблема защиты конструкций (гл. обр. металлических)
от воздействия морской воды, влажного воздуха, на-
насыщенного частицами морской воды, солнечной радиа-
радиации. Защитные материалы должны выдерживать рез-
резкие колебания темп-ры, связанные с плаванием судов в
различных широтах и в разное время года, противо-
противостоять действию живых организмов, обитающих в
морской воде и в атмосфере, а также агрессивных про-
продуктов, перевозимых на судне.
Для указанных целей в судостроении широко при-
применяют лакокрасочные материалы на
основе синтетич. продуктов. Используемые в судострое-
судостроении грунтовки служат не только для получения ниж-
нижних слоев лакокрасочных покрытий, но и как самостоя-
самостоятельный материал для защиты изделий от внешних
воздействий в период межоперационного хранения
(напр., стальные изделия защищают грунтовками на ос-
основе алкидно-стирольного лака или эпоксидных смол).
Нижними слоями покрытий по стали и алюминиево-
магниевым сплавам служат фосфатирующие грунтов-
грунтовки на основе поливинилбутираля. На внутренние по-
поверхности цистерн для питьевой воды, резервуаров
для винных продуктов, изготовляемых из стали и алю-
алюминиевых сплавов, наносят нетоксичные хим- и водо-
водостойкие грунтовки на основе сополимера винилхло-
рида с винилиденхлоридом. Из алкидных лаков гото-
971
ПОЛИМЕРЫ В СУДОСТРОЕНИИ
972:
вят грунтовки для стальных и деревянных поверхно-
поверхностей, эксплуатируемых в сухих помещениях.
Для окраски наружной обшивки корпусов судов, под-
подводных металлич. поверхностей, деревянных изделий,
цистерн для хранения воды используют краски на осно-
основе лака этиноль (см. Дивинилацетиленовые лаки и
эмали). Эти же краски, а также краски на основе эпок-
эпоксидных смол применяют при получении антикоррозион-
антикоррозионных и нефтестойких покрытий цистерн для хранения
нефтепродуктов и для окраски аккумуляторных поме-
помещений, подводной части корпуса, грузовых трюмов
и другого.
Особенно жесткие требования предъявляются к по-
покрытиям, защищающим район переменной ватерли-
ватерлинии судна. В наибольшей степени этим требованиям
отвечают покрытия на основе поливинилхлорида, со-
сополимера винилхлорида с винилиденхлоридом, алкид-
ных, феноло-формальдегидных и перхлорвиниловых
смол.
Для защиты подводной части судов применяют кра-
краски, к-рые образуют водо- и коррозионностойкие по-
покрытия, устойчивые, кроме того, к обрастанию живот-
животными и растительными морскими организмами (см. Не-
обрастающие лакокрасочные покрытия). Антикорро-
Антикоррозионные покрытия корпусов судов из алюминиевых
сплавов получают с применением эпоксидных красок;
стальные корпуса защищают дивинилацетиленовыми
красками пли красками на основе сополимера винил-
хлорида с винилацетатом.
Наибольшее распространение для окрашивания на-
наружных судовых поверхностей (надводного борта, над-
надстроек, рубок и др. открытых конструкций) получили
эмали на основе пентафталевого лака, краски на ос-
основе поливинилхлорида, смесей перхлорвиниловой
и алкидной смол, а также сополимеров винилхлорида
с винилацетатом.
Внутренние поверхности судовых помещений окра-
окрашивают материалами, к-рые обеспечивают пожарную
безопасность и отвечают требованиям эстетики и гигие-
гигиены. Этому комплексу требований в наибольшей сте-
степени отвечают эмульсионные краски на основе бута-
диен-стирольного латекса и пластифицированной по-
поливинил ацетатной дисперсии, а также пентафталевые
эмали.
Трюмы машинно-котельных отделений, внутренние
поверхности балластных и топливных цистерн, грузо-
грузовые танки нефтеналивных судов окрашивают красками
на основе сополимера винилхлорида с винилиденхло-
винилиденхлоридом, эпоксидной смолы, сополимера частично омылен-
омыленного винилацетата с винилхлоридом, к-рые образуют
нефтестойкие покрытия. Для окраски внутренних по-
поверхностей цистерн, в к-рых хранят горячее масло
(до 200 °С), используют краски на основе феноло-
формальдегидного (бакелитового) лака. Поверхности,
к-рые подвергаются воздействию агрессивных р-ров,
защищают гл. обр. красками для химстойких покрытий
на основе эпоксидных смол и сополимера винилхлори-
винилхлорида с винилиденхлоридом. Последние, обладающие наи-
наименьшей токсичностью, используют для защиты ци-
цистерн, в к-рых хранят пресную воду и перевозят хи-
химически активные пищевые продукты, напр, поварен-
поваренную соль. См. также Лаки и эмали, Краски.
Для внутренней отделки судов все более широко при-
применяют разнообразные пленочные и др.
отделочные материалы на синтетич. осно-
основе, вытесттяющие такие дефицитные природные продук-
продукты, как древесина, кожа, хлопчатобумажные, шелковые
и шерстяные ткани, растительные масла. При этом
улучшается качество отделки и сокращается трудоем-
трудоемкость работ.
Для покрытия металлич. палуб внутри судовых по-
помещений используют рулонные материалы (линолеум)
на основе алкидных (глифталевых) смол, а также по-
ливинилхлоридные на тканевой основе и трудновос-
пламеняющиеся безосновные (см. Покрытия для полов).
При облицовке щитов из фанеры или негорючего ма-
материала на основе минеральных волокон (нептунит,
маринит, асбосилит и др.), используемых для отделки
кают, изготовления переборок и зашивки подволоков
кают, широко применяют декоративный бумажно-слои-
бумажно-слоистый пластик. Из этого же материала получают трех-
трехслойные щиты с легким заполнителем, применяемые для
изготовления судовой мебели и каютных переборок.
Детали судовых помещений, облицованные бумажно-
слоистым пластиком, не требуют последующей трудоем-
трудоемкой окраски.
Переборки жилых судовых помещений отделывают
мягким рулонным отделочным материалом пави-
павинол, представляющим собой поливинил хлоридную
пленку на тканевой основе. Для сокращения трудоем-
трудоемкости отделочных работ выпускают павинол с заранее
нанесенным клеевым слоем. Мягкую судовую мебель
отделывают кожей искусственной и эластичным пено-
пенополиуретаном. Жесткие пенополиуретан и пенополисти-
рол, а также пенополивинилхлорид используют в качест-
качестве легкого заполнителя в трехслойных щитах, приме-
применяемых при изготовлении щитовой судовой мебели и
конструкций переборок. Из пластмасс на основе поли-
метилметакрилата, перхлорвиниловой смолы и поливи-
поливинилхлорида изготовляют детали судовой мебели. Такая
мебель проста в изготовлении, дешевле деревянной
и щитовой и, кроме того, трудновоспламеняема. Из
поливинилхлорида получают профили, к-рые исполь-
используют для декоративной заделки стыков щитовых мате-
материалов, изготовления поручней и ступеней трапов,,
облицовки кромок щитовой мебели, плинтусов и др.
отделочных деталей. Способность этого полимера хо-
хорошо окрашиваться позволяет создавать изделия, гар-
гармонирующие но цветовым оттенкам с интерьером.
Материалы, используемые в судовом машинострое-
машиностроении. Полимерные материалы нашли применение и при
изготовлении ответственных судостроительных деталей,,
эксплуатируемых в условиях воздействия агрессивных
сред, больших колебаний темп-р, тропич. влажности
и др. Напр., взамен дорогостоящего тропич. дерева
бакаута, служившего единственным материалом для
изготовления вкладышей дейдвудных подшипников, с
успехом применяют резину, текстолит, капролон и др.
полимерные материалы. Наиболее полно требованиям
судостроения отвечает капролон, из к-рого изготовляют
разнообразные машиностроительные детали — дейд-
вудные втулки, вкладыши подшипников рулевых
устройств и насосов, детали опорных подшипников ва-
лопроводов и сальниковых уплотнений, шестерни, обте-
обтекатели и др.
С применением стеклотекстолитов на основе эпок-
эпоксидных и феноло-формальдегидных смол, обладающих
высокой прочностью и коррозионной стойкостью, изго-
изготовляют гребные винты диаметром до 3 м (как цельные,
так и со съемными лопастями из этого прессматериала),
к-рые легче и дешевле латунных, а также менее тру-
трудоемки в изготовлении. Из стекловолокнитов на ос-
основе феноло-формальдегидных смол, модифицирован-
модифицированных др. полимерами, методами прямого или литьево-
литьевого прессования изготовляют маховики арматуры,
(включая паровую), блоки и коуши для пеньковых ка-
канатов, ненагруженные шестерни ручных приводов,
переборочные стаканы, детали иллюминаторов, изоли-
изолирующие звенья такелажа и многие другие судострои-
судостроительные детали.
Лит.: Архангельский Б. А., Пластические массы,
Л., 1961; Михайлов М. В., Ткаченко В. С, Судо-
Судостроение за рубежом, № 10, 62 A967); Современные глубоко-
глубоководные аппараты, под общ. ред. А. К. Сборовского и А. В.
Кирсанова, ДО.], 1967 (ЦНИИТЭИС); Прохоров Б. Ф.,
Шпитальникова Л. И., Современное судостроение.
Трехслойные конструкции в судостроении и других отрас-
отраслях техники, [Л.], 1970; Пашкеев В. Л., Пласт, массы».
973
ПОЛИМЕРЫ В ЭЛЕКТРОТЕХНИКЕ
974
№ 6, 21 A972); Scharping H. D., Kunststoffe, № 10,
747 A970); Winans R., в кн.: Encyclopedia of polymer
science and technology, v. 8, N. Y.— [a. o.], 1068, p. 405.
В. Л. Пашкеев.
ПОЛИМЕРЫ В ЭЛЕКТРОТЕХНИКЕ (polymers in
electrotechnics, Polymere in Elektrotechnik, polyme-
res dans electrotechnique).
Содержание:
Введение 973
Низковольтные и тяговые машины 974
Высоковольтные машины 0 76
Трансформаторы и коммутационная аппаратура 977
Силовые, конденсаторы 978
Электрические провода и кабели 978
Введение. Полимеры являются основой или обяза-
обязательными компонентами практически всех элементов
изоляции электрич. машин, аппаратов и кабельных
изделии. Их широко применяют также для защиты
изоляции от механич. воздействий и агрессивных сред,
для изготовления конструкционных и полупроводя-
полупроводящих материалов, клеящих составов и др. Использова-
Использование полимеров обусловливает возможность создания
электрооборудования с высокими технико-экономиче-
технико-экономическими характеристиками и повышенной эксплуата-
эксплуатационной надежностью и создает предпосылки для комп-
комплексной механизации технологических процессов его
производства.
Требования, предъявляемые к полимерным материа-
материалам электротехнич. назначения, разнообразны. Так,
материалы, применяемые в производстве электрич.
машин и аппаратов, должны иметь достаточно высокие
электрич. прочность и уд. сопротивление во всем интер-
интервале рабочих темп-р, а материалы для высоковольтно-
высоковольтного оборудования, кроме того,— малые диэлектрич. по-
потери и стойкость к коронному разряду (см. также
Диэлектрические свойства). Эти свойства должны соче-
сочетаться с удовлетворительными механич. характеристи-
характеристиками (прочностью, износостойкостью и др.), т. к. при
изготовлении и эксплуатации электрич. машин и аппа-
аппаратов полимерные материалы могут подвергаться раз-
разрушающим воздействиям. Важное значение для элект-
электротехники имеют теплофизич. свойства полимерных ма-
материалов: они должны иметь высокие теплостойкость
и теплопроводность, а также нагревостойкость, т. е.
длительно, в течение нескольких лет, выдерживать
воздействие повышенных темп-р без существенного
изменения электрич. и др. практически важных ха-
характеристик. Электрич. характеристики полимерных
материалов не должны существенно ухудшаться в
условиях высокой влажности, а в нек-рых случаях
и в агрессивных средах.
Электротехнич. пром-сть — один из крупнейших
потребителей полимеров. Напр., их потребление в
электротехнич. и электронной пром-сти США в 1970
составило 1147 тыс. т (~13% общего объема производ-
производства полимеров в этой стране), а к 1975 должно увели-
увеличиться до 2100 тыс. т. Среди полимеров, к-рые исполь-
используют в электротехнике, наиболее значительная доля в
общем объеме их потребления принадлежит материа-
материалам, применяемым для изоляции проводов и кабелей—
полнолефннам C7%) и поливинилхлориду C3%), а так-
также фенольным смолам A0%); см. Олефинов полимеры,
Винилхлорида полимеры, Феноло-альдегидные смолы.
Однако научно-технич. прогресс в этой отрасли пром-сти
определяют полимеры, объем потребления к-рых отно-
относительно невысок,— эпоксидные смолы, кремнийорга-
нические полимеры, ароматич. полиамиды, полиимиды,
полиэфирные смолы и др. Благодаря применению этих
полимеров существенно повышается нагревостойкость
изоляции и уменьшается ее толщина, что отвечает глав-
главной тенденции развития электромашиностроения —
увеличению уд. мощности при одновременном повыше-
повышении надежности электрооборудования.
Низковольтные и тяговые машины. Применение син-
тетич. полимеров в производстве асинхронных
двигателей мощностью 0,6—100 кет позволило
значительно уменьшить массу и габариты изделий,
напр, массу двигателей мощностью 1 кет — почти в
2 раза. Благодаря применению начиная с 50-х гг. па-
пазовой изоляции на основе полиэтилентерефталатной
пленки (вместо электрокартона и хлопчатобумажной ла-
коткани) толщина изоляции уменьшилась почти в два
раза, а ее нагревостойкость повысилась от 105 °С
(класс А) до 120 °С (класс Е). Разработанные в 60-е гг.
пленочные и волокнистые материалы, лаки и пропиточ-
пропиточные составы на основе таких полимеров, как, напр., аро-
ароматич. полиамиды, полиэфирные и эпоксидные смолы,
в к-рых повышенная нагрево- и влагостойкость соче-
сочетается с хорошими электрич. и механич. характеристи-
характеристиками, позволили создать системы изоляции с нагрево-
стойкостью 130 °С (класс В) и 155 °С (класс F). Сущест-
Существенное достоинство этих полимерных материалов —
возможность использования в механизированных про-
процессах изготовления электродвигателей.
Для пазовой изоляции статоров электродвигателей,
помимо полиэтилентерефталатной пленки толщиной
0,20—0,25 мм, применяют комбинированный материал
на основе более тонкой полиэтилентерефталатной плен-
пленки с подложкой из бумаги на основе полиэфирного во-
волокна (см. Бумага из синтетических волокон). Выпу-
Выпускаемая в Японии пленка «Q-филм», не уступающая
полиэтилентерефталатной по механич. свойствам, рас-
рассчитана на длительную работу при 150 °С. Применение
этой пленки для пазовой изоляции весьма перспектив-
перспективно, поскольку нагревостойкие полиимидные пленки до-
дороги и изготовляются в узком интервале толщин. Поэ-
Поэтому в асинхронных электродвигателях их используют
лишь в сочетании с бумагой из ароматич. полиамида или
стеклянной тканью для систем изоляции, рассчитанных
на рабочие темп-ры 180 °С (класс Н) и выше (класс С).
В качестве материала для пазовой изоляции с нагре-
востойкостью, соответствующей классам F и Н, широко
применяют бумагу на основе волокна из ароматич.
полиамидов (номекс, США), в к-рой удачно соче-
сочетаются высокие механич., электрич. и технологич. ха-
характеристики. Однако в тех случаях, когда пазовая
изоляция класса F должна вследствие особенностей тех-
технологич. процесса обладать высоким сопротивлением
надрыву, предпочитают применять комбинированный
материал на основе бумаги номекс и полиэтилентерефта-
полиэтилентерефталатной пленки. Пленочные и волокнистые материалы из
нагревостойких полимеров постепенно вытесняют тра-
традиционные материалы на основе щипаной слюды или
слюдяной бумаги.
Применение в 50-е гг. стеклослюдяных изоляцион-
изоляционных материалов на кремнийорганич. связующих (см.
Кремнийорганические лаки и эмали) в электродвигате-
электродвигателях, предназначенных для тяжелых условий эксплуа-
эксплуатации (напр., врубовых, морских, крановых), позволи-
позволило почти на 20% повысить мощность машины при тех
же габаритах и одновременно значительно увеличить
сроки их службы. При замене кремнийорганич. изоля-
изоляции на полиимидные пленки или бумагу из ароматич.
полиамидов толщина изоляции уменьшается примерно
на 30% и снижается трудоемкость обмоточно-изолиро-
вочных работ.
Для изоляции обмоток сердечников малогабаритных
электродвигателей применяют высокопроизводитель-
высокопроизводительный способ напыления порошкообразных композиций
на основе эпоксидных смол. Преимущества изоляции,
полученной таким способом, перед изоляцией обыч-
обычного типа — большая однородность и более высокая
теплопроводность. Кроме того, при использовании
способа напыления могут быть уменьшены размеры
электродвигателей и сокращен расход изоляционных
материалов.
975
ПОЛИМЕРЫ В ЭЛЕКТРОТЕХНИКЕ
976
Полимерные материалы, аналогичные применяемым
для пазовой изоляции, используют также для между-
междуфазной изоляции обмоток, а в нек-рых случаях и для
изготовления пазовых крышек, к-рые закрепляют обмот-
обмотки в пазах статора. Для этой же цели применяют пазо-
пазовые клинья, изготовляемые из профильных стекло-
стеклопластиков на основе полиэфирных (класс В) или эпок-
сидно-фенольных (класс F) смол, а также из слоистых
пластиков — гетипакса и стеклотекстолита. Изоля-
Изоляцию выводных концов и мест соединений катушек обмот-
обмотки в асинхронных электродвигателях осуществляют,
как правило, с помощью гибких трубок из стеклочул-
ка, покрытого, напр., эпоксидно-полиэфирным лаком
(см. Эпоксидные лаки и эмали), а также трубок из рези-
резины на основе кремнийорганического каучука, к-рые м. б.
армированы стеклочулком.
Выбор лака для пропитки обмоток определяется
требованиями к нагревостойкости изоляции. Так, для
изоляции класса В используют меламино- и феноло-
алкидные лаки (см. Алкидние лаки и эмали), класса
F — эпоксидно-полиэфирные и полиэфирно-цианурат-
ные, класса Н — кремнийорганические лаки. Наряду
с лаками, содержащими органич. растворители, для
пропитки обмоток все более широко применяют быст-
роотверждающиеся составы без растворителей, обеспе-
обеспечивающие возможность механизации пропитки и сушки
обмоток и включения этих операций в общий непрерыв-
непрерывный технологич. поток. При использовании составов
без растворителей на основе полиэфирных или эпоксид-
эпоксидных смол весь процесс пропитки статорных обмоток
электродвигателей средних габаритов, включая отверж-
отверждение состава, занимает 15—20 мин вместо 20—24 ч,
необходимых при пропитке лаками.
В машинах постоянного тока мощ-
мощностью выше 10 кет ив тяговых машинах
для тепловозов и электровозов, якорные обмотки к-рых
выполняются в виде жестких катушек, для систем изоля-
изоляции классов В и F используют пропитанные эпоксидно-
полиэфирным лаком ленты из слюдяной бумаги с
подложками из стеклоткани и полиэтилентерефталатной
пленки. В тяговых машинах нек-рых типов применяют
ленты из слюдяной бумаги со связующим на основе
кремнийорганич. каучука. В этом случае обмотки не
пропитывают, а промазывают кремнийорганич. гер-
герметизирующими составами. Такая изоляция сохра-
сохраняет эластичность и стабильные электрич. характери-
характеристики в условиях длительной эксплуатации при высо-
высоких темп-рах.
Высокие значения уд. мощности достигаются при изо-
изоляции якорных обмоток несколькими слоями ленты из
полиимидной пленки с наружным слоем из бумаги на
основе ароматич. полиамида и последующей пропит-
пропитке обмоток кремнийорганич. лаком. Толщина такой изо-
изоляции на 20—50% меньше, чем традиционной стекло-
слюдяной, а срок ее службы значительно больше. По-
лиимидную пленку вследствие ее высокой стоимости
применяют только при изготовлении электрич. машин
с тяжелыми условиями эксплуатации.
Наиболее широко используемый способ изоляции
обмоток возбуждения машин постоянного тока, для
к-рых применяют провода со стекловолокнистой изоля-
изоляцией или «голую» медь,— пропитка эпоксидным ком-
компаундом (см. Компаунды полимерные) с применением
вакуума и давления. Такой способ обеспечивает высо-
высокую механич. прочность, влаго- и нагревостойкость
изоляции, а также существенное снижение перегревов
обмоток благодаря высокой теплопроводности изоля-
изоляции. В результате внедрения этого способа в производ-
производство катушек возбуждения тяговых двигателей расход
меди уменьшился на 25%, трудоемкость процесса —
почти в два раза.
Большой экономич. эффект и повышение надежности
при бандажировке якорей и роторов достигнуты в ре-
результате замены стальнои проволоки на пропитанные
лаком (для изоляции класса В — эпоксидным, класса
F — эпоксидно-полиэфирным) нетканые бандажные
ленты из однонаправленных стеклонитей. Такие лен-
ленты имеют высокую механич. прочность и упругость,
а их хорошие электроизоляционные свойства позволяют
исключить применение подбандажной изоляции.
В производстве коллекторов машин постоянного
тока получила распространение изоляция из прессма-
териалов на основе новолачных и резольных феноло-
формальдегидных смол, а также из смеси асбо-каучу-
ковой композиции с пресспорошком на основе мелами-
но-формалъдегидной смолы. При использовании этих
материалов значительно снижается трудоемкость изго-
изготовления коллекторов и уменьшается вероятность ра-
радиального смещения коллекторных пластин при экс-
эксплуатации.
Высоковольтные машины. Одно из существенных тре-
требований, предъявляемых к изоляции обмоток высоко-
высоковольтных электродвигателей и генераторов,— стойкость
к коронному разряду. Изоляционные материалы
только на полимерной основе, обеспечивающие надеж-
надежность и достаточно высокие технико-экономич. показа-
показатели машин, еще не созданы. В наибольшей степени
требованию высокой короностойкости отвечают мате-
материалы из щипаной слюды или слюдяной бумаги и тер-
термореактивных связующих и пропиточных составов на
основе эпоксидных смол. По сравнению с широко при-
применявшейся ранее изоляцией из микаленты на битум-
но-масляном лаке (см. Битумные лаки и эмали), пропи-
пропитанной битумным компаундом (класс нагревостойко-
нагревостойкости А), такая изоляция более нагревостойка (классы
В и F), монолитна, не склонна к размягчению и, следо-
следовательно, к «миграции» из пазов, имеет более высокую
электрич. прочность (как кратковременную, так и при
длительном воздействии электрич. напряжения) и луч-
лучшие механич. свойства. Благодаря этому толщина
изоляции м. б. уменьшена на 15—35% , а мощность ма-
машин при тех же габаритах увеличена на 20%.
При изоляции токоведущих стержней обмоток лента-
лентами из слюдяной бумаги или щипаной слюды применяют
два способа:
1. Наложение лент, содержащих не более 5% свя-
связующего («сухие» ленты), и их последующая пропитка
жидким компаундом (этот способ иногда используют
и для изоляции якорных обмоток низковольтных ма-
машин постоянного тока);
2. Наложение лент, содержащих 30—35% связующе-
связующего, без последующей их пропитки.
Известно много разновидностей изоляции первого
типа. Напр., фирма «Вестингауз» (США) в качестве
пропиточного состава использует жидкую эпоксидную
смолу DER-330, отверждаемую гексагидрофталевым
ангидридом. Для уменьшения вязкости и улучшения
пропитывающей способности компаунд разбавляют сти-
стиролом (—30% в расчете на смолу). Пропитку осу-
осуществляют при 20—25 °С и давлении 0,3 Мн/м2
C кгс/см2). В СССР для пропитки изоляции на основе
«сухих» лент применяют эпоксидные компаунды, не
содержащие токсичного стирола (изоляция монолит).
Основной компонент компаундов—низковязкая эпоксид-
эпоксидная смола с высокой жизнеспособностью (напр., типа
ЭД-22). Отвердителем служит обычно изометилгидро-
фталевый ангидрид (для получения изоляции с повышен-
повышенной эластичностью часть этого отвердителя заменяют,
напр., на полиангидрид азелаиновой к-ты), ускори-
ускорителем — триэтаноламин. Обмотки пропитывают ком-
компаундом при 40—60 °С с применением вакуума и давле-
давления и отверждают компаунд в изоляции при 150 °С.
К изоляции второго типа относятся, напр., системы
ВЭС и слюдотерм (СССР). При изготовлении изоля-
изоляции В Э С применяют компаунд на основе эпоксидной
смолы ЭД-16 и олигоэфиракрилата, содержащий в ка-
977
ПОЛИМЕРЫ В ЭЛЕКТРОТЕХНИКЕ
978
честве отвердителя ^с-3,6-эндометилен-1,2,3,6-тетра-
гидрофталевый ангидрид (эндик-ангидрид). Связую-
Связующее не содержит растворителя и поэтому после наложе-
наложения лент на стержни изоляцию не вакуумируют, а
опрессовывают в нагретых прессформах. В изоляции
слюдотерм в качестве связующего используют
компаунды на основе эпоксидной смолы ЭД-16, к-рую
отверждают новолачной феноло-формальдегидной смо-
смолой. В зарубежной практике применяют связующее
на основе высокомолекулярных эпоксидных смол,
отверждаемых карбоксилсодержащими полиэфирами.
В этом случае получают материал с более высокой эла-
эластичностью при повышенных температурах. Однако в
такие компаунды для понижения их вязкости необ-
необходимо вводить растворители, а затем изоляцию ваку-
умировать.
За рубежом ведутся опытные работы по применению
в высоковольтных генераторах бумаги из волокна на
основе ароматич. полиамидов, наполненной измельчен-
измельченной слюдой (н о м е к с М, США).
За рубежом выпускаются электродвигатели, рас-
рассчитанные на напряжение 6 кв, и небольшие генераторы
с обмотками, изолированными самосклеивающимися
лентами из радиационного вулканизата кремнийорга-
нич. каучука. Такая изоляция обладает высокой нагре-
во- и короностойкостью, но имеет сравнительно невы-
невысокую механич. прочность, повышенную толщину и
склонна к «миграции» из пазов вследствие высокого тем-
температурного коэфф. линейного расширения. Поэтому
ее используют только в таких машинах, к-рые эксплуа-
эксплуатируются в условиях воздействия воды и агрессивных
химич. сред.
Трансформаторы и коммутационная аппаратура. В
производстве высоковольтных силовых трансформаторов
и коммутационной аппаратуры полимеры выполняют
гл. обр. функции связующих для намоточных изделий
(электроизоляционных цилиндров, несущих обмотки,
и трубок). Детали, предназначенные для крепления
токоведущих частей, изготовляют из слоистых пласти-
пластиков. В масляных трансформаторах и др. аппаратах при-
применяют гл. обр. гетинакс, текстолит и древесные пла-
пластики на основе феноло- и крезоло-формальдегидных
смол. В сухих высоковольтных трансформаторах с изо-
изоляцией класса Вив воздушных выключателях исполь-
используют цилиндры из эпоксидных стеклопластиков. При
изготовлении сухих трансформаторов, рассчитанных
на рабочие темп-ры 180—200 °С (классы Н и С), в ка-
качестве связующих для слоистых пластиков используют
кремнийорганич. полимеры. Нек-рые зарубежные фир-
фирмы для междукатушечной и междуслойной изоляции
сухих трансформаторов класса Н применяют бумагу
из волокна на основе ароматич. полиамидов. Обмот-
Обмотки масляных и сухих трансформаторов с изоляцией
класса В пропитывают меламино-алкидными, обмотки
сухих трансформаторов с изоляцией класса Н — крем-
кремнийорганич. лаками.
Благодаря замене традиционной фарфоровой изоля-
изоляции на литую изоляцию из эпоксидных смол масса
опорных трансформаторов тока, эксплуатируемых при
напряжении 10 кв, уменьшилась более чем в 3 раза, а
расход черных и цветных металлов — соответственно
более чем в 5 и 2 раза. Для этой цели используют так-
также наполненные пылевидным кварцем эпоксидные ком-
компаунды на основе диановой смолы ЭД-8, отверждаемые
фталевым ангидридом. Такая изоляция непригодна
для эксплуатации в условиях наружной установки
из-за недостаточной трекингостойкости. Большей стой-
стойкостью в этих условиях характеризуются компаунды
на основе циклоалифатич. эпоксидных смол.
Широкое применение для изоляции катушек конст-
конструкционных и изоляционных деталей низковольтной
аппаратуры (плат, каркасов, колодок и др.) находят
прессматериалы на основе феноло-крезоло-формальде-
гидных смол. В нек-рых случаях конструкционные де-
детали изготовляют из полиамидов, полифениленоксида,
поликарбоната. Для деталей, к-рые должны обладать
повышенной дугостойкостью (напр., камеры контакто-
контакторов), используют прессматериалы на основе мелами-
но-формальдегидной смолы, а также асбо-каучуковые
композиции. В тех случаях, когда дугостойкость долж-
должна сочетаться с высокой нагревостойкостью, применяют
кремнийорганич. прессматериалы.
Силовые конденсаторы. В качестве диэлектриков
силовых конденсаторов, применяемых для повышения
уд. мощности электроустановок и пропускной способ-
способности линий электропередач, эффективны тонкие по-
полимерные пленки (8—20 мкм), обладающие высокими
диэлектрич. характеристиками (тангенс угла диэлект-
рич. потерь не более 5-10~4 в интервале темп-р от
—50 до 100 °С, электрич. прочность 100—150 Мв/м,
или кв/мм). К механич. прочности, а также к влагостой-
влагостойкости пленок не предъявляют высоких требований,
поскольку конденсаторы герметизированы.
Характеристики конденсатора существенно улуч-
улучшаются при использовании комбинированного диэлект-
диэлектрика, состоящего из полипропиленовой пленки (см.
Полиолефиновые пленки) и пропитанной жидким дифе-
нилхлоридом конденсаторной бумаги, к-рая выполняет
гл. обр. функции фитиля, обеспечивающего хорошую
пропитку конденсаторов. Применение такого диэлект-
диэлектрика позволяет примерно в 1,5 раза повысить уд. мощ-
мощность конденсаторов при одновременном снижении
их перегревов. Благодаря этому оказалось возможным
изготовление мощных конденсаторов, эксплуатируе-
эксплуатируемых при частоте 500—20 000 гц без водяного охлаж-
охлаждения.
Интересен также опыт применения полистиролъ-
ных пленок с пропиткой нефтяным маслом при изготов-
изготовлении конденсаторов для бесконтактных шахтных
электровозов, а также комбинированного диэлек-
диэлектрика с полиэтилентерефталатной пленкой в импуль-
импульсных силовых конденсаторах и конденсаторах по-
постоянного тока. Еще более перспективно применение
чисто пленочного диэлектрика, однако для этого не-
необходима разработка специальных пропиточных соста-
составов, хорошо смачивающих полиэтилентерефталатную
пленку.
Электрические провода и кабели. Специфич. требо-
требования к изоляции обмоточных проводов —
стойкость к истиранию, тепловым ударам, перегрузкам
по току, к действию пропиточных лаков, эластичность
и пригодность для механизированной намотки. В наи-
наибольшей степени перечисленным требованиям отвечают
эмалированные провода (см. также Электроизоляцион-
Электроизоляционные лакокрасочные покрытия).
Первое место по объему выпуска во всех странах
занимают эмалированные обмоточные провода классов
нагревостойкости В и F с изоляцией на основе полиэти-
лентерефталатных лаков и их модификаций, что обусло-
обусловлено повышением температур эксплуатации электро-
электрооборудования. Эта изоляция обладает достаточной ме-
механической прочностью, позволяющей использовать
провода при механизированной намотке. Модифика-
Модификация полиэтилентерефталата полиамидами, полиими-
дами, циануратами способствует повышению нагрево-
нагревостойкости изоляции, ее стойкости к тепловым ударам
и механич. прочности.
Провода класса А изолируют поливинилацетальны-
ми лаками (см. Поливинилацеталъные лаки и эмали),
пленки к-рых обладают высокой электрич. и механич.
прочностью, стойкостью к тепловым ударам и очень
высокой износостойкостью. Специфич. свойства пленок
на основе поливинилформаля — стойкость к действию
горячего трансформаторного масла и фреона. Иногда
поверх поливинилацетальной изоляции накладывают
дополнительное термопластичное покрытие, напр, на
979
ПОЛИМЕРЫ В ЭЛЕКТРОТЕХНИКЕ
980
основе поливинилацетата (см. Винилацетата полиме-
полимеры). В этом случае стадия пропитки обмоток м. б.
исключена, что особенно важно при изготовлении ка-
катушек приборов и аппаратов.
Покрытия класса Е, для к-рых применяют полиуре-
тановые лаки (см. Полиуретановые лаки и эмали),
обладают высокими изоляционными характеристиками,
стойкостью к тепловым ударам и действию раствори-
растворителей. Кроме того, провода с такой изоляцией можно
лудить, погружая их в олово или в припой, без приме-
применения специальных флюсов и без предварительной ме-
ханич. зачистки. Износостойкость полиуретановой
изоляции несколько ниже, чем поливинилацетальной.
Выше 150 °С полиуретановое покрытие размягчается.
За рубежом используют также полиуретановую изоля-
изоляцию, защищенную дополнительным термопластичным
покрытием на основе поливинилацетата.
Для изоляции эмалированных проводов классов Н
и С применяют полиимиды и их модификации. Такая
изоляция может длительно работать при 220—240 °С,
обладает исключительной стойкостью к тепловым уда-
ударам. Износостойкость полиимидных покрытий ниже,
чем полиэтилентерефталатных. Наилучшей износостой-
износостойкостью, превосходящей износостойкость поливинил-
ацетальных покрытий, обладают покрытия на основе
полиимидоамидов, пригодные для длительной эксплуа-
эксплуатации при 200 °С.
Для машин с особенно тяжелыми условиями эксплуа-
эксплуатации или повышенной надежности применяют обмоточ-
обмоточные провода со стекловолокнистой изоляцией, пропи-
пропитанной алкидными, эпоксидными или кремнийорга-
нич. лаками. Провода для высоковольтных электрич.
машин изолируют полиэтилентерефталатными пленка-
пленками. Для нагревостойких систем изоляции большой
интерес представляют полиимидные пленки.
В шахтных двигателях, тяговых двигателях маги-
магистральных электровозов, а также двигателях, работаю-
работающих в условиях радиационного облучения, применяют
провода с изоляцией из полиимидной пленки, покры-
покрытой сополимером тетрафторэтилена с гексафторпропиле-
ном. При этом повышается коэфф. заполнения паза, а
допустимая темп-pa эксплуатации машин возрастает
до 200—220 °С. Изоляция обеспечивает также герме-
герметизацию обмоток вследствие прочного соединения слоев
пленки при нагревании. Несмотря на то, что такая изо-
изоляция дороже стекловолокнистой, ее применение дает
значительный экономич. эффект благодаря увеличе-
увеличению срока эксплуатации машин.
При изготовлении обмоточных проводов для погруж-
погружных электродвигателей насосов, применяемых при до-
добыче нефти, руды, а также при выкачивании воды из
артезианских скважин, используют гл. обр. пластикат
повышенной твердости. Рабочая темп-pa таких проводов
70 °С. При использовании в качестве изоляционного
материала полиэтилена высокой плотности (см. Эти-
Этилена полимеры) рабочая темп-pa двигателей, в частно-
частности водопогружных, составляет 90 °С. Для рабочих
темп-р 180—200 °С м. б. использованы провода с лен-
ленточной изоляцией из фторопласта, а также из полиимид-
полиимидной пленки с покрытием из фторопласта.
Применение полимеров для изоляции силовых
кабелей, предназначенных для передачи электро-
электроэнергии больших мощностей, позволяет отказаться от
использования металлич. оболочек и облегчает их мон-
монтаж и эксплуатацию. Основной полимерный материал
для изоляции силовых кабелей, рассчитанных на напря-
напряжение переменного тока до 6 кв,— пластикат. Такие
кабели стойки к перегрузкам, т. к. их рабочая темп-ра
65—70 °С, а темп-pa размягчения пластиката 150—
160 °С. Пластикат самозатухает, имеет достаточно
высокие электрич. прочность и светостойкость, однако
большие диэлектрич. потери и высокая диэлектрич.
проницаемость не позволяют использовать его при
напряжениях выше 6 кв (в отдельных случаях — не вы-
выше 10 кв). Изоляционным материалом для кабелей, рас-
рассчитанных на напряжение 10—69 кв, служит полиэти-
полиэтилен, превосходящий поливинилхлорид по электрич.
свойствам и влагостойкости.
При введении в полиэтилен антипиреиов (напри-
(например, хлорпарафина и трехокиси сурьмы) получают са-
самозатухающий материал. Нагревостойкость изоляции
повышается при сшивании полиэтилена, напр, орга-
нич. перекисями: длительная рабочая темп-pa кабе-
кабелей с такой изоляцией 85—90 °С, при токах короткого
замыкания — до 220 °С. Выпускаются опытные партии
кабелей с полиэтиленовой изоляцией, рассчитанной
на напряжение 110—138 кв; ведутся исследовательские
работы с целью создания кабелей, пригодных для
эксплуатации при напряжении 220—230 кв.
При монтаже силовых кабелей с изоляцией из пласт-
пластмасс, а также с традиционной изоляцией из бумаги,
пропитанной минеральным маслом или масло-канифоль-
масло-канифольным составом, в качестве материала для заливки ко-
кожухов муфт используют эпоксидные компаунды, обла-
обладающие малой усадкой при отверждении.
Полиэтилен и пластикат — основные изоляционные
материалы для кабелей связи. Целесообразность
применения этих материалов вместо традиционной бу-
бумажной изоляции обусловлена их лучшими механич.
свойствами, что особенно важно при скручивании в ка-
кабель большого числа жил, а также влагостойкостью,
позволяющей отказаться от применения оболочек из
дефицитного свинца. Кабели с изоляцией из пласт-
пластмасс технологичны, пригодны для прокладки в земле,
воде, для подвески по стенам зданий и опорам. Темпера-
Температурный диапазон их эксплуатации от —40 до 60 °С.
Для кабелей местной связи широко применяют пори-
пористый полиэтилен (см. Пенополиолефины), диэлектрич.
проницаемость к-рого примерно в 1,5 раза меньше, чем
у монолитного. При его использовании м. б. снижена
рабочая емкость цепей при сохранении габаритов или
при той же емкости уменьшена толщина изоляции.
Применение пористой изоляции из полиэтилена с
одновременной заменой кабелей симметричной кон-
конструкции на коаксиальные перспективно для маги-
магистральных кабелей связи. Монолитный и пористый
полиэтилен — наиболее распространенные материалы
и для изоляции радиочастотных кабелей. В тех случаях,
когда их рабочие темп-ры превышают 70 °С, применяют
фторсодержащие полимеры, в частности политетрафтор-
политетрафторэтилен, в виде лент. В радиочастотных кабелях нек-рых
конструкций используют колпачковую изоляцию, изго-
изготовляемую из полистирола (см. Стирола полимеры),
полиэтилена или фторопластов литьем под давлением.
Защитные оболочки радиочастотных кабелей изготов-
изготовляют, как правило, из пластиката. Изоляционным ма-
материалом для кабелей дальней связи чаще всего слу-
служит полистирол.
В установочных проводах, предназначенных для
прокладки в осветительных и силовых сетях (обычно с
напряжением 380 в), применявшаяся ранее резиновая
изоляция почти полностью заменена на поливинил-
хлоридную. Провода с такой изоляцией не нуждаются
в защитной оболочке, поскольку пластикат стоек к
солнечной радиации и к механич. нагрузкам.
Для изоляции монтажных проводов и
кабелей, используемых при соединении различной
электрич. аппаратуры и при монтаже схем, применяют
пластикат, полиэтилен и, в нек-рых случаях, резины
на основе смеси бутадиенового или бутадиен-стироль-
ного каучука с натуральным. Для монтажных проводов
повышенной нагревостоикости используют изоляцию на
основе фторопластов. Гибкие провода для выводных
концов электрич. машин изолируют резинами на основе
бутилкаучука, кремнийорганических каучуков (в т. ч.
фторсилоксановых), а также политетрафторэтиленом.
«981
ПОЛИМЕРЫ НА ЖЕЛЕЗНОДОРОЖНОМ ТРАНСПОРТЕ
982
Перспективный материал для монтажных проводов —
полиэтилен, подвергнутый радиационному сшиванию.
Рабочая темп-pa такой изоляции достигает 150 °С при
продолжительности эксплуатации 10 000 ч.
Во многих случаях монтаж аппаратуры существенно
упрощается при использовании ленточных проводов
и кабелей, в к-рых расположенные в одной плоскости
токопроводящие жилы опрессовывают полиэтиленом
или пластикатом или обкладывают склеенными вдоль
лентами из полиэтилентерефталата.
Наиболее распространенный изоляционный материал
для кабелей специального назначе-
назначения — резина. Напр., шланговую оболочку кабелей,
предназначенных для питания погружных электрона-
электронасосов, добывающих нефть и рассчитанных на напряже-
напряжения 1 и 1,5 кв, рабочую темп-ру 90 °С и давление пласто-
пластовой жидкости до 10 Мн/м2 A00 кгс/см2), изготовляют из
негорючей масло- и бензостойкой резины на основе
бутадиен-нитрилъного каучука.
Резиной изолируют также кабели для питания элект-
электробуров, рассчитанные на напряжения до 3 кв. При
эксплуатации кабелей в нефтяных скважинах с повы-
повышенным содержанием газов предпочитают изоляцию
из полиэтилена, обладающего низкой газопроницае-
газопроницаемостью.
В изоляции геофизич. кабелей, используемых в
геологоразведочных работах, поверх резиновой изоля-
изоляции жил накладывают оболочку из резины на основе
зслоропренового каучука. Иногда эти кабели изолируют
также полиэтиленом или фторопластами.
Изоляцию и оболочку шланговых прово-
проводов и кабелей, используемых для присоединения
передвижных механизмов и рассчитанных на напряже-
напряжения до 3 кв, изготовляют из резин на основе смесей бу-
бутадиенового или бутадиен-стирольного каучука с нату-
натуральным или синтетич. изопреновым. Кабели, предназ-
предназначенные для эксплуатации при более высоких напря-
напряжениях, изолируют резинами, к-рые наряду с высокими
электроизоляционными свойствами должны обладать
также стойкостью к озонному старению. Чаще всего
для этого используют резины на основе бутилкаучу-
ка с малой непредельностью или этилен-пропилено-
вого каучука.
Лит.: Андрианов К. А., Высокомолекулярные сое-
соединения для электрической изоляции, М., 1961; Петраш-
к о А. И., ШахновичМ. И., Синтетические полимеры
в электрической изоляции, М., 1968; Забырина К. И.,
Развитие работ в области синтеза и исследования полимеров
м создания электроизоляционных материалов на их основе,
в сб.: Электротехническая промышленность, серия «Электро-
«Электротехнические материалы», вып. 11 —12, М., 1972; О с и -
пова Л. В., Кореньков Г. Л., Современное состояние
и тенденции в потреблении синтетических смол и пластмасс
в США, Хим. пром-сть за рубежом, в. 8 A04), 3 A971); Encyclo-
Encyclopedia of polymer science and technology, v. 5, N. Y.— [a. o.],
1966, p. 482; Kunststoffe, № 10, 708 A970).
В. В. Скипетр ов, Н. В. Александр ов, И. Б. Пешков.
ПОЛИМЕРЫ НА ЖЕЛЕЗНОДОРОЖНОМ ТРАНСПОР-
ТРАНСПОРТЕ (polymers in railway transport, Polymere im
Eisenbahntransport, polymeres dans transport de chemin
«de fer).
Содержание:
Введение 981
Пассажирские вагоны 982
Грузовые вагоны 984
Локомотивы и моторные вагоны электропоездов 986
Устройство пути и контактной сети 987
Железнодорожная телемеханика, автоматика и
связь 988
Введение. Первые опыты использования полимерных
материалов на ж.-д. транспорте относятся к началу XX
в., когда подвижной состав стали оборудовать автотормо-
автотормозами, нормальная эксплуатация к-рых оказалась воз-
возможной только при условии применения резиновых
шлангов и уплотняющих прокладок. Начавшаяся
jB 30-е гг. интенсивная электрификация железных до-
дорог и постепенный ввод в эксплуатацию пассажирских
вагонов с электрич. освещением привели к расширению
использования резин и пластмасс для электроизоляции.
Создание более совершенного подвижного состава
в послевоенные годы и интенсивное оборудование же-
железных дорог системами автоматич. сигнализации, бло-
блокировки и централизации управления движением обус-
обусловили дальнейший рост потребления полимерных ма-
материалов во всех областях ж.-д. техники. Число из-
изделий и деталей ж.-д. оборудования, изготовляемых
из этих материалов, насчитывает несколько тысяч на-
наименований.
Различные пластмассы и резины используют для
изготовления конструкционных деталей, воспринимаю-
воспринимающих механич. нагрузки и одновременно изолирующих
электрич. ток; фрикционных, антифрикционных, ан-
антикоррозионных, уплотняющих и амортизационных
деталей, а также в качестве гидро-, термо- и звукоизо-
звукоизоляционных, декоративных и конструкционных мате-
материалов в экипажной части вагонов и локомотивов.
Большие потери металла вследствие коррозии ж.-д.
оборудования в условиях эксплуатации обусловливают
широкое применение лакокрасочных материалов, гл.
обр. на основе алкидных смол, для антикоррозионной
защиты (см. Алкидные лаки и эмали, Защитные лако-
лакокрасочные покрытия).
За 1960—70 потребление пластмасс, резин и лако-
лакокрасочных материалов на отечественном ж.-д. транс-
транспорте (без учета использования этих материалов
в строительстве нового подвижного состава) возросло
соответственно в 2,2; 5,2 и 2 раза.
Пассажирские вагоны. В строительстве вагонов
полимеры начали применять в 30-х гг. для изготовления
деталей общего электротехнич. назначения. В после-
послевоенный период получили распространение детали
из полимерных материалов конструкционного назна-
назначения. Так, из древесно-слоистого пластика на основе
феноло-формальдегидной смолы в пассажирских ваго-
вагонах некоторых типов изготовляют решетки, защищаю-
защищающие окна у продольных верхних пассажирских мест,
подоконные и боковые столики, детали санитарно-тех-
нич. оборудования. Внешний вид и эксплуатационные
свойства этих деталей лучше, срок службы в 2—3 раза
больше, а стоимость в 1,5—2 раза ниже, чем аналогич-
аналогичных деталей из твердых пород древесины.
Из волокнита, древеснопрессовой крошки, аминоплас-
тов и пресспорошков др. марок изготовляют детали
потолочных вентиляторов и плафонов электрич. осве-
освещения, корпуса светильников, коробки стоп-кранов,
дверные ручки и десятки др. деталей внутривагонного
оборудования. Использование этих деталей из поли-
полимерных материалов экономически целесообразно из-за
меньшей трудоемкости и стоимости их изготовления.
В ряде случаев применение полимеров приводит
к сокращению расхода цветного металла и нек-рому
уменьшению массы оборудования.
Профилированные и формовые резиновые детали
на основе бутадиен-стиролъных каучуков применяют
для уплотнения и герметизации окон, дверей, нек-рых
сопряжений конструкций, деталей систем вентиляции,
водоснабжения и др. узлов пассажирских вагонов. Од-
Одновременно такие детали амортизируют удары, погло-
поглощают вибрацию и шум. В приборах и устройствах
тормозной системы используют манжеты, воротники,
прокладки из морозостойких (до —55°С) резин на осно-
основе бутадиен-стирольного каучука и маслостойких ре-
резин из бутадиен-нитрилъных каучуков.
Способность к большим упругим деформациям, га-
гашению ударов и вибрации обусловливает все большее
применение резин в сопряжениях, к-рые являются
источником шума в вагонах, и в амортизирующих
устройствах подвижного состава. Значительная часть
пассажирских вагонов в скоростных поездах многих
983
ПОЛИМЕРЫ НА ЖЕЛЕЗНОДОРОЖНОМ ТРАНСПОРТЕ
984
стран оборудована рессорным подвешиванием с амор-
амортизаторами, представляющими собой баллонные или
диафрагменные конструкции, в к-рых рабочей средой
служит воздух, заключенный в многослойную резино-
резинотканевую оболочку. Относительно широкое распрост-
распространение на дорогах ряда стран получили вагонные
колеса, в конструкцию к-рых входят резиновые элемен-
элементы, а также рессоры из резины или с резино-металлич.
элементами.
При строительстве отдельных серий вагонов внед-
внедряются ударопоглощающие аппараты автосцепки,
снабженные резино-металлич. амортизирующими эле-
элементами. Эти аппараты легче, имеют лучшие динамич.
характеристики и значительно более долговечны, чем
металлические.
Тормозные колодки из полимерных материалов ши-
широко используют в подвижном составе железных дорог
многих стран (напр., в СССР — из стереорегулярного
бутадиенового каучука, наполненного смесью асбеста,
сажи и др. компонентов). Износостойкость таких ко-
колодок в несколько раз больше, а масса втрое меньше,
чем чугунных; их коэфф. трения меньше зависит от
темп-ры и скорости движения поезда. Колодки из поли-
полимерных материалов применяют также в вагонах поез-
поездов метрополитена, где недопустимо образование пыли
от износа чугунных колодок, проводящей электрич. ток,
а также в вагонах, оборудованных дисковыми тормозами.
В узлах трения, работающих при кратковременных
эпизодич. нагрузках и малых скоростях скольжения,
наряду с металлич. втулками применяют втулки из ио-
ликапролактама. Такие втулки, работая без смазки,
меньше изнашиваются (срок их службы в два раза
больше) и не вызывают износа и повреждений сопря-
сопрягающихся с ними стальных деталей. Кроме того, се-
себестоимость их вдвое меньше. Срок службы горизон-
горизонтальных и вертикальных скользунов из пластмассы
в опорах кузова вагона на тележках в 3—4 раза вы-
выше, чем стальных и чугунных, а себестоимость и тру-
трудоемкость изготовления соответственно в 1,5 и 10 раз
меньше. Использование «сухарей» буксовых гасителей
колебаний, выполненных из пластмасс (вместо закален-
закаленной стали 45), в конструкции фрикционных аморти-
амортизаторов подвески кузова вагона повышает срок службы
всего узла в 2—3 раза.
Водопроводные трубы в системах холодного водо-
водоснабжения можно изготовлять из полиэтилена высокой
плотности, фитинги и арматуру — из полиэтилена и
поликапролактама, баки для воды, располагающие-
располагающиеся над санузлами,— из полиэфирного стеклопластика.
Достоинства системы — уменьшение массы вагона,
удлинение срока службы, облегчение монтажа и ремон-
ремонта, возможность применения труб меньшего диаметра
из-за более низкого коэфф. трения воды, чем в чугун-
чугунных и стальных трубах.
Для усовершенствования термоизоляции цельноме-
таллич. вагонов вместо мипоры, имеющей низкую ме-
ханич. прочность и очень высокое водопоглощение,
успешно внедряется пенополистирол.
Применение пластиката для внутренней отделки
пассажирских вагонов повышает их комфортабельность,
снижает стоимость и ремонтные расходы. Декоратив-
Декоративный тисненый пластикат, дублированный с хлопчато-
хлопчатобумажной тканью, применяют для покрытия стен,
внутренних перегородок, дверей, потолков вагонов.
Для покрытия полов используют алкидный или рези-
резиновый линолеум (см. Покрытия для полов), для обивки
сидений и спинок кресел и диванов — кожу искусст-
искусственную на основе поливинилхлорида. Стены и потол-
потолки отделывают также декоративным бумажно-слоистым
пластиком, к-рый наклеивают на деревянные столяр-
столярные плиты или древесно-стружечные панели внутрен-
внутренней обшивки стен, перегородок и потолков (в вагонах
нек-рых типов — на алюминиевые листы).
Применение полимерных материалов для отделки
пассажирских вагонов привело к резкому сокращению
расхода благородных сортов древесины и полному
вытеснению линолеума на основе растительных масел.
Такая отделка несравненно менее трудоемка, срок служ-
службы ее больше, гигиенич. условия при ее использовании
лучше и, кроме того, облегчена санитарная обработка
вагонов.
Для изготовления сидений и спинок кресел исполь-
используют эластичные пенополиуретаны и губчатые резины
из латекса. Себестоимость пенополиуретановых матра-
матрацев для мягких вагонов в четыре раза меньше, чем
пружинных, а трудоемкость их изготовления втрое
меньше. Благодаря применению таких матрацев масса
вагонов уменьшилась более чем на 1 т. Пенополиуре-
тановые диваны и матрацы гигиеничнее обычных, соз-
создают большие удобства пассажирам. Перспективно-
использование пенополиуретана и в качестве тепло-
и звукоизоляционного материала, наносимого на сте-
стены, полы и потолки вагона методом напыления.
За рубежом в большом количестве, не превышающем,
однако, масштабы широкого экспериментирования,
эксплуатируются вагоны, в к-рых стены, полы, потолки,
двери, внутреннее оборудование, кабины санузлов,
крыши и даже целиком кузова выполнены из стекло-
стеклопластиков на основе полиэфирных и эпоксидных смол
холодного отверждения. Стеклопластики используют в
виде листовых материалов, крупногабаритных элемен-
элементов, а также как оболочки для трехслойных конструк-
конструкций типа «сэндвич». Заполнителем в таких конструк-
конструкциях служат пенопласты, гл. обр. пенополиуретан. В
результате использования стеклопластика уменьшает-
уменьшается масса вагона, снижается трудоемкость его изготов-
изготовления, увеличивается срок службы. В нек-рых вагонах
полы санузлов изготовляют из полиэфирного стекло-
стеклопластика. Срок службы таких полов в 5 раз больше, чем
обычных бетонных с покрытием из метлахской плитки.
Благодаря лучшей гидроизоляции снижается коррозия
кузова вагона, что приводит к сокращению ремонт-
ремонтных расходов в 10 раз.
Эксплуатируются вагоны, в к-рых внутренние сте-
стены, перегородки между купе, полы, багажные и пас-
пассажирские полки изготовляют не из дерева, а из трех-
трехслойных панелей (оболочка — сверхтвердые древесно-
древесноволокнистые плиты или фанера, заполнители — пено-
поливинилхлорид, пенополистирол или пенополиуре-
пенополиуретан). Лицевые стороны панелей покрыты пластикатом.
Срок службы трехслойных панелей вдвое больше, чем
столярных плит, масса вагона снижается на 3 т, затра-
затраты на ремонт уменьшаются втрое.
Получают распространение декоративные и антикор-
антикоррозионные покрытия по металлич. деталям (стальные
рычаги кресел, кронштейны багажных полок, оконные
поручни и др.), к-рые получают нанесением порошко-
порошковых красок (гл. обр. на основе поливинилбутираля)
методом напыления в псевдоожиженном слое. Эти по-
покрытия дешевле гальванических, а их нанесение менее
трудоемко, особенно на детали сложной конфигурации,
полирование к-рых затруднено.
В ряде стран для снижения в вагонах уровня шу-
шума и вибрации внутренние поверхности металлич.
обшивки и др. элементов кузова покрывают антикорро-
антикоррозионными составами на основе эпоксидных смол,
полиуретанов или их модификаций, наполненными
асбестом, пробкой и др. материалами.
Грузовые вагоны. В этих вагонах широко используют
детали из резин на основе бутадиен-нитрильного и
стереорегулярного бутадиенового каучуков: 1) тормоз-
тормозные колодки, применение к-рых позволяет повысить
скорость и безопасность движения грузовых поездов;
2) «пылевые» шайбы в буксах, на осях грузовых ваго-
вагонов для уплотнения и защиты подшипников скольже-
скольжения от грязи и пыли: использование резиновых шайб
985
ПОЛИМЕРЫ НА ЖЕЛЕЗНОДОРОЖНОМ ТРАНСПОРТЕ
986
вместо войлочных уменьшает доступ в буксу пыли и
абразивных частиц, снижает расход и потери смазки,
улучшает режим работы подшипников и значительно
сокращает расход шерсти; 3) прокладки на крышках
смотровых люков букс с подшипниками скольжения,
благодаря к-рым устраняется потеря смазки; 4) детали
клапанов, приборов и устройств в цистернах всех типов.
На вагонах с подшипниками качения начинают
применять буксы с резиновыми элементами между
опорными поверхностями букс и роликовыми подшип-
подшипниками. Такие элементы, амортизируя радиальные и
осевые динамич. нагрузки и равномерно распределяя
их на ролики, улучшают режим работы подшипников.
Это более чем в 2 раза увеличивает срок службы под-
подшипников, исключает случаи их аварийного выхода
из строя и удлиняет сроки эксплуатации др. деталей
вагона.
Гуммирование стальных котлов цистерн для перевоз-
перевозки соляной к-ты устраняет коррозию котлов и исклю-
исключает необходимость перевозки кислоты в стеклянных
бутылях.
В новых отечественных рефрижераторных вагонах
полы покрывают резиновым линолеумом или изготов-
изготовляют из стеклопластика. Такие полы более долговечны,
а трудоемкость их сборки и ремонта меньше, чем дере-
деревянных, покрытых оцинкованным железом; благодаря
лучшей гидроизоляции дольше сохраняется находя-
находящийся под ними термоизоляционный материал (напр.,
мипора), уменьшается коррозия кузова и ходовых
частей вагона. Напольные решетки отечественных
изотермич. вагонов, к-рые изготовляют из древесно-
слоистого пластика и применяют вместо оцинкован-
оцинкованных железных, отличаются большей легкостью и проч-
прочностью. В нек-рых грузовых вагонах из пластмасс вы-
выполняют также крышки верхних и вентиляционные
решетки боковых загрузочных люков.
За рубежом эксплуатируют рефрижераторные и изо-
изотермич. вагоны, кузова к-рых выполнены целиком
из стеклопластиков. Стены, полы и потолки таких ва-
вагонов изготовлены из трехслойных конструкционных
панелей типа «сэндвич», заполненных в большинстве
случаев пенополиуретаном. Последний используют
также в качестве термоизоляционного материала реф-
рефрижераторов др. конструкций.
Весьма эффективным м. б. использование стекло-
стеклопластиков для изготовления элементов конструкций
кузовов вагонов. При этом возможно снижение массы
вагонов с одновременным сокращением эксплуатацион-
эксплуатационных расходов благодаря удлинению срока сменяемости
отдельных деталей и элементов конструкций. Вагоны
с крышами различной конструкции из полиэфирных
и эпоксидных стеклопластиков широко применяют
во многих странах.
Перспективно изготовление стен и полов из деревян-
деревянных досок или панелей с наружным слоем, работающим
на растяжение, из стеклопластика толщиной 0,5—1 мм.
Это позволит уменьшить массу вагона на 0,7 т, увели-
увеличить срок службы стен и пола в 2—3 раза, сократить
на 90% поступление их в текущий ремонт и, кроме того,
уменьшить благодаря повышению плотности полов и
стен потерю сыпучих грузов.
Контейнеры из полимерных материалов, достаточ-
достаточно широко эксплуатируемые за рубежом, надежны в
эксплуатации, легче обычных стальных и деревянных
и не уступают им по прочности. Жесткие контейнеры
изготовляют чаще всего из стеклопластиков или из их
комбинации с др. материалами (древесноволокнистыми
плитами, фанерой, древесиной, алюминием, сталью и
др.). Эластичные контейнеры цилиндрич. формы, зани-
занимающие в незагруженном состоянии в 8—10 раз мень-
меньший объем, чем в загруженном, снабженные гермети-
герметически закрывающимися загрузочными люками, изготов-
изготовляют из нескольких слоев дублированной прочной тка-
ткани с двухсторонним покрытием из резин на основе
хлоропренового или бутадиен-нитрильного каучука.
В опытных масштабах за рубежом эксплуатируются
железнодорожные цистерны, котлы к-рых изготовляют
из полиэфирных и эпоксидных стеклопластиков.
Локомотивы и моторные вагоны электропоездов.
В этих видах подвижного состава широко применяют
фенопласты для изготовления сотен деталей обычно-
обычного электроизоляционного назначения в аппаратуре
электрич. сетей, а также общетехнич. деталей (ру-
(рукояток управления, поручней, клеммных колодок,
кнопок и др.).
Многочисленные уплотнительные, гидро- и элект-
электроизоляционные детали приборов, аппаратов и
устройств изготовляют из резин и пластиката; детали,
работающие в условиях сосредоточенного и циклич.
нагружения, вибрации, периодич. трения и служащие
одновременно для электроизоляции (кулачковые шай-
шайбы и панели контроллеров управления, детали реле
времени и обратного тока и др.) — из армированных
фенопластов. Из этих материалов выполняют также
разнообразные детали устройств контактирующей аппа-
аппаратуры, быстродействующих выключателей, релейных
приборов, электромагнитных контакторов, реверсоров
и мн. др.
В механизмах тормозных и пневматич. устройств,
в топливных насосах и в системах маслоподачи при-
применяют резиновые прокладки, диафрагмы, манжеты,
воротники и уплотнители, выполняющие одновремен-
одновременно функции амортизаторов. Масло- и бензостойкие
детали, к-рые должны сохранять эластичность до —55 °С,
изготовляют гл. обр. из резин на основе бутадиен-нит-
бутадиен-нитрильного каучука; теплостойкие детали — из резин
на основе кремнийорганических каучуков и фторсодер-
жащих каучуков.
На электровозах переменного тока применяют по-
полиэтиленовые трубопроводы жидкостного охлаждения
игнитронов. Это позволяет уменьшить массу оборудо-
оборудования и дает экономию труб из нержавеющей стали.
Применение полиэтиленовых труб для пневматич.
электроизоляционных шлангов в устройстве пантогра-
пантографов и быстродействующих выключателей повышает на-
надежность работы и увеличивает срок службы деталей.
Из пластиков на основе кремнийорганических полиме-
полимеров, наполненных асбестом, изготовляют дугогаситель-
ные камеры нек-рых контакторов.
Для деталей локомотивного оборудования, работаю-
работающих при высоких механич. нагрузках, колебаниях
темп-ры от —50 до 50 СС (иногда до 150 СС), а также под
током высокого напряжения, вместо стали с изоляцией
из фарфора и слюдяных материалов используют стек-
стеклопластик. Благодаря этому исключаются аварии,
связанные с разрушением изоляции, уменьшаются
ремонтные и эксплуатационные расходы, снижаются
трудоемкость изготовления и себестоимость деталей,
к-рые м. б. отпрессованы целиком из стеклопластика.
Перспективно применение полиэтилентерефталатау
полиимидов и модифицированного полиимидом поли-
этилентерефталата для изоляции обмоток якорей тя-
тяговых электродвигателей локомотивов (см. также
Полимеры в электротехнике).
В конструкции буксовых узлов локомотивов исполь-
используют резино-металлич. амортизирующие элементы,
к-рые обеспечивают лучшую амортизацию и гашение
колебаний, чем стальные рессоры. Амортизация эки-
экипажной части нек-рых электро- и тепловозов осуществ-
осуществляется с помощью резиновых амортизаторов централь-
центральной подвески кузова.
Втулочные резино-металлич. элементы (сайлент-бло-
ки) в электровозах и тепловозах выполняют роль амор-
амортизаторов гибкого привода от тягового двигателя к оси
колесной пары. Они улучшают режим работы дви-
двигателя, смягчая переход с одной скорости на другую.
987
ПОЛИМЕРЫ НА ЖЕЛЕЗНОДОРОЖНОМ ТРАНСПОРТЕ
988
Применение сайлент-блоков в приводах от валиков
буксовых поводков упростило конструкцию приводов
и их эксплуатацию. На локомотивах нек-рых типов
применяют буксы, в к-рых подшипники качения снаб-
снабжены резиновыми амортизаторами осевых усилий.
Внедрение их, улучшив режим работы подшипников,
позволило увеличить межремонтный пробег локомоти-
локомотивов и повысить их скорость.
На электровозах нек-рых типов устанавливают
выполненные из стеклопластика кожухи зубчатых пе-
передач и крупногабаритные детали механизма песко-
подачи. Это, улучшив конструкцию узлов, повысило
надежность их работы и обеспечило снижение массы
неподрессоренных частей локомотива. В отечественных
опытных тепловозах и моторных вагонах электросекций
головная часть кузова, включая кабину машиниста,
выполнена из пластика на основе полиэфирной смолы
холодного отверждения, армированного стеклотканью
и стекломатом. Трудоемкость изготовления этих элемен-
элементов кузова локомотива ниже обычной.
Применение антифрикционных осевых опорных ди-
дисков из пластика на основе феноло-формальдегидной
смолы, наполненной древесной крошкой, повышает срок
службы этих узлов и дает до 400 кг годовой экономии
бронзы на каждом электровозе. Этот же пластик исполь-
используют для изготовления наличников буксовых направ-
направляющих электроподвижного состава, а древесно-слоис-
тый пластик — для деталей опор кузова моторного ваго-
вагона электропоездов на тележку. Применение этих и др.
антифрикционных деталей из полимерных материалов,
работающих эпизодически и с незначительными скоро-
скоростями относительного перемещения, снижает трудоем-
трудоемкость изготовления деталей, а благодаря увеличению
срока службы уменьшает также и эксплуатационные
расходы.
Устройство пути и контактной сети. Необходимость
применения полимерных материалов в верхнем строении
пути вызвана оборудованием железных дорог автоматич.
блокировкой, условия работы к-рой требуют электрич.
изоляции рельсов каждого блокучастка. Задача ослож-
осложняется высокими механич. напряжениями, к-рые возни-
возникают на стыках рельсов. Решение ее оказалось воз-
возможным в результате разработки конструкции т. наз.
изолирующего стыка, в к-ром накладки, скрепляющие
концы рельсов, были выполнены из высокопрочного
пластика (первоначально — из феноло-формальдегид-
ного древесно-слоистого пластика).
Рост перевозок грузов и скорости поездов, повышение
нагрузки на оси привели к необходимости создания
изолирующего стыка, оборудованного стальными,
воспринимающими механич. нагрузки накладками,
изолированными от рельсов профилированными про-
прокладками из полиэтилена высокой плотности. Втулки
и прокладки из этого же полимера применяют и для изо-
изоляции крепежных болтов в конструкции стыка. Такие
стыки в 1,5 раза дешевле, чем стыки с накладками из
древесно-слоистого пластика (последние сохранились
только на метрополитене и на малонагруженных участ-
участках наземных дорог). Срок службы новых изолирую-
изолирующих стыков в 2 раза больше, чем прежних. Перспектив-
Перспективное направление дальнейшего усовершенствования
стыков — применение монолитных, клееных, клее-бол-
товых и др. конструкций с деталями и элементами из
эпоксидного стеклопластика.
Для амортизации динамич. взаимодействий колесных
пар подвижного состава с рельсами и предотвращения,
т. обр., быстрого износа и разрушения рельсов и непод-
неподрессоренных частей подвижного состава во всех конст-
конструкциях промежуточного скрепления рельсов с железо-
железобетонными шпалами применяют прокладки из полимер-
полимерных материалов — резины, кордоволокнита (см. Во-
локнит), полиэтилена высокой плотности. Эти проклад-
прокладки осуществляют одновременно и электрич. изоляцию
рельсов от железнодорожного полотна. Для предотвра-
предотвращения утечки тока от рельсов по крепежным болтам в
каждом узле скреплений рельсов со шпалами устанав-
устанавливают изолирующие втулки из фенопластов и более
совершенные — из стеклопластика.
Перспективно применение резиновых или полиэти-
полиэтиленовых прокладок и на пути, уложенном на деревян-
деревянных шпалах. Прокладка, уложенная на шпалу в узле
скрепления рельса под металлич. подкладкой, пре-
предохраняя древесину шпалы от износа, смятия и раз-
разрушения этой подкладкой, увеличивает срок службы
шпалы на несколько лет. Возможно и эффективно
также локальное упрочнение древесины шпал в зоне
прикрепителей рельсового скрепления путем заливки
специальной композиции (например, полиэфирной,
эпоксидной) холодного отверждения в шурупное
или костыльное отверстие шпалы. При этом прочность
скрепления повышается в несколько раз, т. к. увели-
увеличивается сопротивление древесины усилиям смятия
и вырыва.
За рубежом для укрепления железнодорожного по-
полотна на водонепроницаемых грунтах начинают успеш-
успешно применять гидроизоляционные пленки из поливи-
нилхлорида, синтетич. каучуков и др., а также
дренажные устройства из систем сетчатых поливинил-
хлоридных или полиэтиленовых труб, заполненных
пенополистиролом. С этой же целью и для предотвра-
предотвращения «пучинных» деформаций земляного полотна при
промерзании грунтов начинают использовать тепло-
теплоизоляционные слои из пенополистирола или из поли-
мерцементного бетона на его основе с плотностью 0,5—
0,6 г/сж3, на к-рые укладывают верхнее строение пути
из железобетонных рельсовых плит. Проводятся опыты
по применению полимерных связующих для повышения
монолитности и стабилизации щебеночной балластной
призмы с целью улучшения ее демпфирующих характе-
характеристик, повышения прочности, долговечности и, сле-
следовательно, увеличения межремонтных сроков эксплуа-
эксплуатации.
В конструкции нек-рых изоляторов, применяемых
в устройствах контактной сети, получили распростра-
распространение изолирующие элементы из стеклопластиков (вме-
(вместо фарфоровых), что позволило значительно умень-
уменьшить размеры и массу конструкции, повысить надеж-
надежность ее работы и срок службы. В малогабаритных изо-
изоляторах, к-рые эксплуатируются на электрифицирован-
электрифицированных дорогах постоянного тока с напряжением 3000 в,
изолирующие элементы выполнены в виде брусков из
стеклопластика с лакокрасочным покрытием, напр,
на основе кремнийорганич. лака. Благодаря этому м. б.
сняты ограничения скорости движения поездов в зоне
секционных изоляторов. На электрифицированных до-
дорогах переменного тока с напряжением 27 кв успешно
^применяют секционные изоляторы с изолирующими эле-
^ментами-скользунами, непосредственно по к-рым сколь-
скользит токоприемник пантографа. Эти элементы изготов-
изготовляют из однонаправленного стеклопластикового прут-
прутка с защитными чехлами в виде втулок из фторопласта,
полиэтилена, стеклопластика на основе кремнийорга-
кремнийорганич. полимера или керамики. Перспективно примене-
применение изолирующих элементов из стеклопластика и для
устройств фиксаторов, тяг, консолей, разъединителей,
струновых зажимов подвески контактного провода и др.
Применение таких элементов значительно повышает
надежность контактной сети, безопасность ее эксплуа-
эксплуатации и обслуживания и облегчает конструкцию.
Железнодорожная телемеханика, автоматика и связь.
Создание на железных дорогах систем автоматич.
блокировки и сигнализации, а также централизации
управления движением оказалось возможным только
благодаря применению многочисленных деталей из по-
полимерных материалов в электронных, радио- и электро-
технич. приборах и устройствах, пультах управления
989
ПОЛИМЕРЫ С СИСТЕМОЙ СОПРЯЖЕНИЯ
990
диспетчера, механизмах автоматич. стрелочных пере-
переводов, кабельных ящиках контактных устройств свето-
светофоров. Большинство этих деталей одновременно с изо-
изоляцией электрич. тока выполняет функции конструк-
конструкционных элементов, воспринимающих сосредоточенные
динамич. и статич. нагрузки. В результате использо-
использования поливинилхлорида, полиэтилена, резин и др.
для получения защитных покрытий и изоляции сиг-
нально-блокировочного кабеля, проводов и жил зна-
значительно снизился расход свинца для этих целей.
Из этих же материалов изготовляют гибкие шланги,
к-рые широко используют при монтаже многих автома-
автоматич. и телемеханич. устройств. В обмотках и изоляции
токопроводящих контуров и индивидуальных катушек
приборов применяют также различные электроизоля-
электроизоляционные лаки (см. Электроизоляционные лакокрасочные
покрытия).
Фенопласты незаменимы при изготовлении панелей
и плит приборов, клеммных колодок, электроизоляцион-
электроизоляционных прокладок, шайб и втулок токопроводящих бол-
болтов и др. деталей, насчитывающих несколько сотен
наименований. Для электроизоляционных деталей си-
систем телемеханики, автоматики и связи перспективны
полиолефины и др. термопласты, а также новые термо-
термореактивные материалы, для несущих конструкций
пультов, шкафов и аппаратов — слоистые пластики и
стеклопластики.
На реле нек-рых типов вместо металлич. защитных
колпаков успешно применяют колпаки из прозрач-
прозрачного сополимера метилметакрилата, стирола и акри-
лонитрила (МСН) и из негорючего прозрачного этрола.
Это привело к улучшению качества реле, упрощению
их монтажа и снижению трудоемкости изготовления
колпаков на 30—35% .
Перспективно применение красного, желтого и зеле-
зеленого МСН при изготовлении светофильтров для локомо-
локомотивных светофоров, буферных сигнальных фонарей ло-
локомотивов и ручных сигнально-осветительных фонарей.
Такие светофильтры превосходят по эксплуатацион-
эксплуатационным характеристикам светофильтры из силикатного
стекла вследствие более высокой ударной вязкости и
меньшего коэфф. светопоглощения в видимой области
спектра. Они проще в изготовлении и в несколько
раз дешевле силикатных. См. также Полимеры в радио-
радиоэлектронике, Полимеры в электротехнике.
Лит.: Исследование свойств конструкционных полимерных
материалов, применяемых на железнодорожном транспорте,
М 1966- Исследование полимеров для железнодорожного
транспорта, М., 1968; Ситковский И. П., Полимерные
материалы и их применение в железнодорожной технике, М-,
1968; Рыжова 3. А., Генкин С. М., Ситков-
Ситковский И. П. Использование эпоксидных смол при ремонте
подвижного состава, М., 1968; Полимерные материалы в конст-
конструкции подвижного состава, М., 1969; Антифрикционные поли-
полимерные материалы в узлах трения подвижного состава, М., 1970;
Новые полимерные материалы на транспорте, М., 1971; Поли-
Полимерные материалы в элементах подвижного состава и пути,
М., 1973; Ситковский И. П., МаевскийВ. И.,
Полимерные материалы на зарубежных железных дорогах, М.,
1973 И. П. Ситковский.
ПОЛИМЕРЫ С СИСТЕМОЙ СОПРЯЖЕНИЯ (po-
(polymers with a conjugation system, Polymere mit konju-
giertem System, polymeres a systeme conjugue).
Содержание:
Введение 989
Специфика реакций получения. Структура макро-
макромолекул 991
Физические свойства J^JJ
Химические свойства 99J
Перспективы практического использования . . .1002
Введение
Полимеры с системой сопряжения (П.) — соедине-
соединения, содержащие в основных цепях макромолекул до-
достаточно длинные непрерывные последовательности,
в к-рых каждая одинарная связь находится между
двумя кратными связями или между кратной связью
и атомами с неподеленными парами /^-электронов, атом-
атомными зарядами или с?-электронными вакансиями.
Поскольку строение сопряженных систем нельзя
описать структурами, построенными путем комбина-
комбинации двухцентровых связей, рассматривают много-
многоцентровые связи, образующиеся в результате перекры-
перекрывания волновых функций я-электронов кратных свя-
связей и обусловливающие вследствие этого делокализа-
цию я-электронов и проявление коллективного элект-
электронного взаимодействия по цепи сопряжения. При вве-
введении в цепь сопряжения гетероатомов или ионов изме-
»*
10
II6
10 20
30
40
it г
О
10 20
30
40
S 8
S 4
Число
10 20 30 40
= электронов, участвующих в сопряжении
Зависимость сродства к электрону, потенциала ионизации
и энергетич. щели полисопряженных углеводородов от числа
я-электронов, участвующих в сопряжении: 1 — полиены,
2 — полиацены, 3 — поликумулены, 4 — полиины, 5 —
поли-п-фенилены, 6 — поли-ж-фенилены.
няется распределение я-электронной плотности. Де-
локализация электронов по цепи сопряжения сопро-
сопровождается выигрышем энергии, наз. энергией
сопряжения, что приводит к уменьшению внут-
внутренней энергии системы, разности между энергиями
основного и возбужденного состояний и потенциалов
ионизации, а также к возрастанию электронной по-
поляризуемости и сродства молекулы к электрону (см.
рисунок).
Критерием, позволяющим оценить делокализацию
я-электронов и тип электронного взаимодействия, к-рое
определяет сопряжение в основных цепях молекул,
может служить изменение энергетич. щели (АЕ) с уве-
увеличением числа я-электронов (Nn), участвующих в
сопряжении (АЕ — энергия перехода я-электрона из
основного в низшее возбужденное состояние).
По характеру зависимости АЕ от Nn ( см. рисунок),
а также на основании результатов исследования коле-
колебательных спектров и квантово-механич. расчетов
сопряженных систем различают три группы полисопря-
полисопряженных систем: а) АЕ убывает весьма быстро и при опре-
определенном значении Nn может достигать величины,
близкой к кТ (к — постоянная Больцмана, Т — абсо-
абсолютная темп-pa); такая закономерность характерна для
кумуленов, аценов и цианиновых красителей; б) АЕ
991
ПОЛИМЕРЫ С СИСТЕМОЙ СОПРЯЖЕНИЯ
992
уменьшается довольно медленно для первых членов го-
мологич. ряда и далее стремится к пределу, как это
наблюдается для полиенов, полиинов, поли-д-фениле-
нов; в) увеличение Nk не приводит к заметному умень-
уменьшению АЕ, что характерно для поли-ж-фениленов и ря-
ряда ангулярно-аннелированных ароматических углево-
углеводородов.
П. обладают специфич. физич. и химич. свойствами,
резко отличающими их от полимеров, у к-рых отсутству-
отсутствует делокализация я-электронов. В связи с этим целесо-
целесообразно деление П. на 6 типов, отражающее зависимость
их свойств от химич. структуры макромолекул и при-
природы сопряжения в основных цепях (см. также табл. 1):
1. П. с я,я-электронным взаимодействием (я,я-соп-
ряжение).
2. П. с взаимодействием я-электронов и неподе лен-
ленных р-электронов (/?,я-сопряжение). К этому типу при-
принадлежат соединения, содержащие в цепи сопряжения
гетероатомы N, О и S с неподеленными /^-электронами.
3. П., характеризующиеся взаимодействием я-элект-
ронов кратных связей или ароматич. циклов с вакант-
вакантными с/-орбиталями атомов таких элементов, как Si,
Р, Ge, Sn, Fe и др. (я,с?-сопряжение).
4. П., характеризующиеся взаимодействием р-элект-
ронов с вакантными d-орбиталями (/^-сопряжение).
В П. этого типа основная цепь содержит связи, образую-
образующиеся при взаимодействии /^-электронов элементов
2-го периода периодич. системы с вакантными с?-орбита-
лями элементов 3-го и более высоких периодов.
5. П., в к-рых реализуется взаимодействие я- и р-
электроно-в с вакантными d-орбиталями (я,/^-сопря-
(я,/^-сопряжение). В молекулах полимеров этого типа содержатся
гетероатомы с неподеленными /^-электронами, кратные
связи и такие элементы, как Fe, Gu.
6. П., характеризующиеся взаимодействием я- или
р-электронов с заряженными атомами, свободными спи-
спинами или /^-электронными вакансиями цепи полисопря-
полисопряжения. В цепи полисопряжения таких П. содержатся
поляризованные центры или стабилизированные кар-
бониевые ионы, заряженные гетероатомы, свободные
спины или свободные /?-электронные вакансии.
Специфика реакций получения.
Структура макромолекул
П. могут быть получены полимеризацией (раскры-
(раскрытие тройных связей С = С и С = N или размыкание
азотсодержащих ароматич. гетероциклов), поликонден-
поликонденсацией, а также полимер аналогичными превращениями
(см. табл. 1, а также Лестничные полимеры, Ацетилена
полимеры, Нитрилов полимеры, Полиазапорфины, По-
лифениленоксиды, Полифениленсулъфиды, Полифосфазе-
ны, Ферроцена полимеры).
Специфич. метод синтеза П. основан на т. наз. по-
полисопряженных реакциях, в к-рых получение мономера
и полимера не разделяют, а осуществляют одновремен-
одновременно, напр.:
-2НВг
RCHBrCH2Br > RC==CH -> [ -C(R)=CH-]n
В большинстве случаев этим методом получают хи-
химически неоднородные полимерные продукты, т. к.
в результате протекания побочных реакций образуются
мономеры, олигомеры и полимеры, различающиеся по
структуре и свойствам.
Изучение кинетики полимеризации ацетилена, фе-
нилацетилена, циклогексилацетилена, пропаргилово-
го спирта, ацетонитрила и пропионитрила указывает
на мономолекулярный механизм обрыва и независи-
независимость мол. массы от темп-ры, концентрации инициато-
инициатора и мощности дозы. Обрыв цепи при невысоких степе-
степенях полимеризации (мол. масса не превышает обычно
5—8 тыс.) характерен для всех синтезированных П.,
независимо от их химич. природы и метода получения.
Реакционноспособный центр непосредственно связан
с системой сопряжения, поэтому на его активность в
реакции должны влиять все факторы, определяющие
степень делокализации и подвижность я-электронов.
Внутримолекулярное взаимодействие неспаренного
электрона растущего макрорадикала (радикальная по-
полимеризация), заряда макроиона (ионная полимериза-
полимеризация) или электронов концевой функциональной группы
(иоликонденсация) с я-электронами приводит в конеч-
конечном итоге к инактивации реакционноспособного центра.
Кроме того, при росте цепи сопряжения снижаются
потенциалы ионизации и возрастает сродство к элект-
электрону образующихся макромолекул (см. рисунок), что
создает благоприятные условия для образования
межмолекулярных я-комплексов. Такие ассоциаты бо-
более прочны, чем аналогичные комплексы мономерных
гомологов, и, по-видимому, в значительной степени
определяют специфику формирования и свойств П.
При нарушении непрерывности цепи сопряжения
рост цепи может происходить практически без инакти-
инактивации реакционноспособного центра. Напр., при по-
полимеризации производных ацетилена, содержащих под-
подвижные атомы водорода в а-положении к тройной связи,
цепь сопряжения нарушается в результате изомериза-
изомеризации части звеньев макромолекулы:
СН
I
(т + п) C =
Г—с—cJ—Рс—сн-1
L | J/77 L | J/7
R R
В результате образуются высокомолекулярные слабо-
окрашенные продукты.
Термодинамич. возможность полимеризации опре-
определяется изменением изобарно-изотермич. потенциала
системы (AZ):
AZ = АН — TAS
где АН и AS — изменение соответственно энтальпии и
энтропии системы. При формировании П. уменьшение
энтальпии определяется не только разностью теплот
образования изолированных связей в полимере и моно-
мономере, к-рая вносит основной вклад в энтальпию поли-
полимеризации виниловых мономеров, но и существенным
вкладом энергии сопряжения и межмолекулярного
взаимодействия в конечном продукте (напр., тепловой
эффект я-комплексообразования). Жесткость цепей П.
обусловливает уменьшение энтропии полимеризации.
С развитием цепи полисопряжения, приводящим к
уменьшению длины одинарных связей, возрастает
взаимное отталкивание электронных оболочек атомов,
составляющих основную цепь и боковые группы макро-
макромолекулы. При этом уменьшение энтальпии и энергии
перехода я-электрона из основного в низшее воз-
возбужденное состояние становится все менее интенсив-
интенсивным и в конце концов достигает такого предела, когда
дальнейшее развитие копланарной цепи сопряжения
будет энергетически невыгодным. Вследствие этого
сопряженные фрагменты макромолекул П. стремятся
расположиться по отношению друг к другу под углом
0° < в < 90°. Т. обр., длинные цепи сопряжения с
копланарным расположением кратных связей тер-
термодинамически и кинетически невыгодны. Следователь-
Следовательно, макромолекулы П. не являются плоскими и для
них возможны различные пространственные конформа-
ции, в к-рых обменное взаимодействие я-электронов
уменьшается с увеличением угла в. Поэтому длина це-
цепи сопряжения, определенная физич. методами, соот-
соответствует плоской модели с меньшим числом я-связей,
чем в сопряженном фрагменте макромолекулы П.
Для оценки длины цепи сопряжения введено поня-
понятие «эффективное сопряжение пк», или
«блок сопряжения», к-рое условно выражает-
выражается числом копланарно расположенных сопряженных
993
ПОЛИМЕРЫ С СИСТЕМОЙ СОПРЯЖЕНИЯ
994
Таблица 1. Методы синтеза, структура и систематика некоторых полимеров с системой
сопряжения
Шифр
поли-
полимера
Общая ф-ла и название
Методы синтеза
Тип 1
I
II
III
IV
V
VI
VII
VIII
R
Г-С=С-]П
1
R'
Поливинилены (полиены, полиметины)
[-С^С-]И
Полиацетилен
R
[-С=С=С=С-]П
R
Поликумулен
Г i i . .
[^Г"СГ].
Полициклопентадиен
[~Аг-]п
Полиарилен
R R
| 1
[(-C=CH-)c-Ar-(-CH=C-)m-Ar'-]n
Полиариленвинилены
[-G = G-Ar-GsG-]n
Полиариленацетилены
R
•т
Пг
Г Rl R
. \ Лп \
R R
лиацены
Полимеризация ацетилена; по реакции
Виттига из
[(СвН5)зРСН2СН=СНСН2Р(СвН5)з] • 2Вг-
и ОНС(СН=СНKСНО;
пегидрогалогенирование поливинилхло-
рида или поливинилбромида; отщепле-
отщепление воды или спирта от поливинилового
спирта или его эфиров; отщепление ук-
уксусной к-ты от поливинилацетата; поли-
полимеризация монозамещенных ацетиленов;
гомополиконденсация GH3G(O)R; полисо-
полисопряженные реакции; полимеризация ди-
замещенных ацетиленов
Дегидрополиконденсация ацетилена
Твердофазная полимеризация
RC=C-C=CR
Полимеризация циклопентадиена
Дегидрополиконденсация ароматич. со-
соединений; по реакции Фриделя —Крафтса;
окислительное дегидрирование полицик-
логекгена
Взаимодействие соответствующих диаль-
дегидов с бис-фосфониевыми солями (ре-
(реакция Виттига) или с компонентами, со-
содержащими активную метиленовую груп-
группу (реакция Кневенагеля)
Дегидрополиконденсация
НС^С-Аг-С=СН
Полимеризация
RC=C-C=CR
Т и п 2
IX
X
XI
свн5
[-C=N-]n
Полибензонитрил
[-GH=CH-GH=GH-GH=N-]n
Полипиридин (линейный)
[=NwCH=CH-CH=I
Полихинолин (линейный)
Полимеризация 2,4,6-трифенил-силш-
триазина
Полимеризация пиридина
Полимеризация хинолина
элементов структуры, имею-
имеющих электронный спектр по-
поглощения или люминесценции,
аналогичный по положению
максимума спектру соответст-
соответствующего полимерного гомоло-
гомолога. Величина п* зависит от ус-
условий синтеза и мало изменя-
изменяется с увеличением мол. мас-
массы полимергомологов. П. оди-
одинаковой химич. природы могут
различаться пространственной
структурой макромолекул. По-
Поэтому для таких полимеров
характерно проявление изоме-
изомерии, обусловленной простран-
пространственным расположением фраг-
фрагментов цепи полисопряжения
(изомерия полисо-
полисопряжения).
Следовательно, в отличие от
полимеров, не содержащих
блоков сопряжения, комплекс
свойств П. определяется не
только молекулярно-массовым
распределением, но и строе-
строением и длиной блоков сопря-
сопряжения, распределением их по
длинам и характером фрагмен-
фрагментов, разделяющих эти блоки.
Конформационные превраще-
превращения в П. сопровождаются изме-
изменением свойств самих макро-
макромолекул. Так, практически
идентичные по мол. массе, со-
составу настроению, но различа-
различающиеся по пк полифенилвини-
лены (I, R = С6Н5, R' = H), по-
полученные термич. и каталитич.
полимеризацией фенилацети-
лена, различаются но электрон-
электронным спектрам поглощения,
люминесценции и реакционной
способности.
При формировании твердой
фазы возможны как конформа-
конформационные превращения, способ-
способствующие выводу сопряжен-
сопряженных фрагментов макромоле-
макромолекулы из копланарности и тем
самым уменьшению эффектив-
эффективного сопряжения, так и про-
противоположные явления. Воз-
Возникновению копланарных
структур благоприятствует,
как показано на примере по-
липропиоловой к-ты (I, R =
= СООН, R' = Н), подавление
диссоциации полярных групп
и образование внутримолеку-
внутримолекулярных водородных и ионных
связей. При создании условий,
способствующих ассоциации,
плотной упаковке и усилению
межмолекулярного я-элек-
тронного обменного взаимо-
взаимодействия между блоками соп-
сопряжения (как в случае блок-
сополимеров, состоящих из
жестких полисопряженных и
гибких насыщенных фрагмен-
фрагментов), комплекс присущих П.
свойств (напр., парамагнетизм,
995
ПОЛИМЕРЫ С СИСТЕМОЙ СОПРЯЖЕНИЯ
996
Продолжение табл. 1
Шифр
поли-
полимера
Общая ф-ла и название
XII
I I
R R
Полиазины
XIII [=C-R'-C=N-R'/-N=In
I I
R R
Полишиффовы основания
XIV
г О
Полиариленаминохиноны
XV
[n
Полиазоарилены
XVI
Ji=CH—R— с
Поливиниленпиридины
XVII
Лестничный полипиридин (Х = СН);
парацианоген (X = N)
XVIII
Н Н
Полиариленпиразолы
XIX
Полифениленизоксазолы (X = GH);
полифениленоксадиазолы (X = N)
XX
X
Полииндиго
Методы синтеза
Поликонденсация дикарбонильных со-
соединений с гидразином; разложение бис-
диазониевых солей
Поликонденсация дикарбонильных со-
соединений с диаминами
Дегидрополиконденсация ароматич. ди-
диаминов с хинонами;реакция ди-или тет-
рагалогенхинонов с ароматич. диамина-
диаминами
Дегидрополиконденсация ароматич. ди-
диаминов; разложение бис-диазониевых со-
солей ароматич. аминов; азосочетание бис-
диазониевых солей с фенолами и арома-
ароматич. аминами
Поликонденсация 2,6-лутидина с аро-
ароматич. диальдегидами или диацетилом,
гомополиконденсация 2-метил-6-пири-
динальдегида
Окислительное дегидрирование лестнич-
лестничного полиакрилонитрила (Х=СН); поли-
полимеризация CNCN или поликонденсация
(CONH2J (X = N)
Циклополиприсоединениебис-диазоалка-
+- - +
нов N=NCH—Ar—CHN=N к диацетиле-
нам НС==С—Аг-С=СН
Циклополиприсоединение окисей дини-
трилов
—C=N-
к n-диэтинилбензолу (Х =
рефталонитрилу (X = N)
или к те-
Дегидрополиконденсация дииндоксила
сдвиг максимума в электрон-
электронных спектрах поглощения и
люминесценции в длинновол-
длинноволновую область) проявляется в
значительно большей степени.
Физические свойства
Наиболее общее свойство П.,
отличающее их от полимеров,
не содержащих блоков сопря-
сопряжения,— наличие парамагнит-
парамагнитных центров (ПМЦ). Спектр
ЭПР таких полимеров обычно
представляет собой узкий (ши-
(ширина 0,5—15 редко до 50 эрстед)
одиночный сигнал с ^-факто-
^-фактором, близким к g-фактору
свободного электрона {g ^
2,00). Концентрация несиарен-
ных электронов составляет
1016 —1021 спин/г, или 1 спин
на 10 —106 молекул. Темп-рная
зависимость интенсивности сиг-
сигнала, как правило, соответст-
соответствует закону Кюри (см. Элект-
Электронный парамагнитный резо-
резонанс, Полупроводники поли-
полимерные). Форма линии, интен-
интенсивность и ширина сигнала
могут зависеть от характера
предварительной обработки
образца, темп-ры измерения и
наличия адсорбированных га-
газов или др. добавок. Наиболее
удовлетворительное объясне-
объяснение основных особенностей па-
парамагнетизма П. дают след.
две гипотезы.
Одна из них, предложенная
в результате исследования ком-
комплексов с переносом заряда,
предполагает, что наблюдае-
наблюдаемые ПМЦ представляют собой
устойчивые ион-радикалы, воз-
возникающие при переносе эле-
электрона между макромолекула-
макромолекулами с развитой системой л-со-
пряжения или при захвате за-
заряда дефектами кристалла.
Согласно др. гипотезе, ПМЦ
возникают вследствие локаль-
локального распаривания л-связей в
макромолекулах П. с наиболее
длинными фрагментами поли-
полисопряжения. При этом прини-
принимается, что сингл ет-триплет-
ный переход может осущест-
осуществляться при тепловом возбуж-
возбуждении, а образующийся три-
триплет способен превращаться в
стабильный бирадикал. Воз-
Возможность таких переходов
обусловлена малым значением
\Е в ассоциированных фрак-
фракциях полимер гомологов с
длинными цепями сопряжения,
нарушением копланарности в
таких системах и выигрышем
энергии сопряжения при де-
локализации распаренного
спина по системе сопряжения.
Взаимодействие ПМЦ с ок-
окружающими макромолекулами
997
ПОЛИМЕРЫ С СИСТЕМОЙ СОПРЯЖЕНИЯ
998
Продолжение табл. 1
Шифр
поли-
полимера
Общая ф-ла и название .
Методы синтеза
Тип 3
XXI
[_Эл—СН=СН—R—СН=СН—]„
Полиэлементовинилены
XXII
Полиферроценилен
XXIII
г аг- Ar\Fc/l
L4Ai—N=N— Агх J/7
Полиазоариленферроцен
Полиприсоединение К2ЭлН2 к
НС=С—R—СееСЩЭл = Si,Ge,Sn)
Взаимодействие бром-и 1,1-дибромфер-
роцена с медью; термин, распад поли-
меркурферроцена; реакция литийферро-
цена с СоС12
Реакция ферроцена (Fc) с бис-диазоние-
выми солями ароматич. диаминов
Тип 4
XXIV G1
[-N=P-]n
I
G1
Полифосфонитрилхлорид
Полимеризация фосфонитр ил хлорида
Тип 5
XXV
N С С=—N
II V I
II Л
N С C=N
Металлсодержащий полифталоцианин
Полимеризация тетрацианбензола и др.
тетрациансодержащих мономеров; Me—
=Gu, Fe (см. Координационные полиме-
полимеры)
Тип 6
XXVI
сг
Полипиридинийхлорид
XXVII
[ОН.
Полиилиц
XXVIII
Заряженный полимер IV
Полимеризация 4-хлорпиридина
Дегидрохлорирование
ридинийхлорида
поли-4-метилпи-
Взаимодействие полимера IV с сильны-
сильными протонными к-тами и к-тами Льюиса
сопровождается явлениями,
для которых предложено общее
название «эффект локаль-
локальной активации». Сущ-
Сущность этих явлений заключа-
заключается в активирующем действии
ПМЦ на реакционную способ-
способность и физико-химические
свойства комплексующихся
с ними диамагнитных моле-
молекул. Активирующее влияние
ПМЦ проявляется: в увели-
увеличении скорости низкотемпе-
низкотемпературного пиролиза ароматич.
углеводородов и изменении
состава продуктов реакции; в
повышении реакционной спо-
способности полисопряженных си-
систем по отношению к стабиль-
стабильным радикалам; при изомери-
изомеризации непредельных соедине-
соединений; в инициировании П. по-
полимеризации электроноакцеп-
торных соединений и их блок-
сополимеризации с соедине-
соединениями, содержащими делока-
делокализованные л-электроны; в из-
изменении ингибирующей актив-
активности полисопряженных си-
систем в зависимости от длины
цепи сопряжения и количества
ПМЦ; в изменении ряда физич.
и электрич. характеристик
(флуоресценции, времени спа-
спада фототока, электропровод-
электропроводности и др.).
С увеличением числа звень-
звеньев в цепи линейно-аннелиро-
ванных и /ге/ж-конденсирован-
ных ароматич. углеводородов
с ненарушенной из-за враще-
вращения фрагментов вокруг оди-
одинарных связей симметрией мо-
молекул (напр., ацены, га-фени-
лены) возрастают темп-ры
плавления и теплоты субли-
сублимации, уменьшается раствори-
растворимость (табл. 2, 3). При откло-
отклонении от линейного располо-
расположения ядер (м- и о-фенилены) и
при введении объемных заме-
заместителей темп-ры плавления
резко понижаются, повышает-
повышается растворимость. Положение
и интенсивность максимумов
поглощения аценов и фениле-
нов в электронных спектрах,
обусловленные различиями
между энергиями невозбуж-
невозбужденного и возбужденного со-
состояний, м. б. объяснены толь-
только эффектом сопряжения. В
соответствии с изложенным
выше, П., способные к образо-
образованию прочных межмолеку-
межмолекулярных я-комплексов, явля-
являются обычно твердыми окра-
окрашенными соединениями с вы-
высокими темп-рами плавления,
часто превышающими их
темп-ры разложения, и плохой
растворимостью; они ассоции-
ассоциированы в р-рах. Нарушение
999
ПОЛИМЕРЫ С СИСТЕМОЙ СОПРЯЖЕНИЯ
1000
расположения сопряженных циклов, наличие объем-
объемных боковых заместителей, обусловливающих стерич.
препятствия, отсутствие структурного порядка в цепи
сопряжения нарушают закономерное уменьшение потен-
потенциалов ионизации и возрастание сродства к элект-
электрону с увеличением длины цепи сопряжения. В резуль-
результате уменьшается плотность упаковки макромолекул
П., они становятся менее термостабильными, но более
растворимыми.
Таблица 2. Зависимость некоторых свойств углеводородов
аценового ряда* от длины цепи сопряжения
Число
циклов
Темп-
ра
плав-
плавления,
°С
80
218
357
388-
-398
ДН субли-
сублимации,
кдж/(молъ -
• К) [ккал/
(моль • °G)J
AS субли-
сублимации,
дж/(молъ-К)
{калКмолъ
71.2 [17]
96.3 [23J
117,2 [28]
Темп-pa суб-
сублимации
(при давле-
давлении
1,33 к/д2,
или 10~2
мм рт. ст.),
СС
171,
175
161,3 [39]
,7 [41]
,8 [42]
27
87
186
Раствори-
Растворимость в
СНС13
Легко
1,77 г в
100 мл
Плохо
Оч. плохо
* См. полимеры VIII в табл. 1 (R = Н).
Таблица 3. Зависимость некоторых физических параметров
и характера электронных спектров полиариленов от длины
цепи сопряжения
Число
циклов
3
4
5
6
Число
циклов
3
4
5
6
Поли-п-фенилены
Темп-pa плав-
плавления, °С
210
320
395
475
545
Хмакс нм
280,0
300,0
310,0
317,0
?макс'10
25,0
39,0
62,5
56,0
Поли-.м-фени лены
Темп-pa плав-
плавления, °С
85
85-86
112
147-148
129-131
макс'
нм
251,5
247
249
248
248
?макс*10~3
44,0
61
79,5
105
142
Растворимость
в толуоле при
20 °С, г/л
8,5
0,22
0,1
0,01
Поли-о-фенилены
Темп-ра
плавле-
плавления, °С
57
118
216-217
312—320
макс'
нм
233
Выражен
не явно
То же
Примечание. X—длина волны; е—интенсивность погло-
поглощения.
П. обладают высокоселективными каталитич. свой-
свойствами. Так, они инициируют полимеризацию элект-
роноакцепторных мономеров, причем скорость реакции
и степень превращения резко повышаются с увеличе-
увеличением содержания ПМЦ в полимере. Обнаружен также
автокаталитич. эффект, заключающийся в изменении
скорости полимеризации при получении П. по мере
накопления полимера или при введении в реакционную
смесь заранее полученного полимера. Установлена
каталитич. активность П. в реакциях разложения
гидразина, перекиси водорода, закиси азота, мура-
муравьиной к-ты, окисления, гидрирования, дегидрирова-
дегидрирования, дегидратации и изомеризации спиртов и углево-
углеводородов и др. Обнаружена корреляция между катали-
каталитич. активностью и парамагнитными свойствами П.
(см. также Полупроводники полимерные).
Химические свойства
При галогенировании полиалкилвиниленов (I, R =
= Н, w-C3H7, м-С4Н9, IT = Н; см. табл. 1) наряду с
присоединением галогена по двойным связям происхо-
происходит замещение атомов водорода. Бромирование проте-
протекает труднее, чем хлорирование (это согласуется с
меньшей электрофильностью брома). При бромирова-
нии интенсивнее происходит замещение. Хлорирование
может сопровождаться частичной циклизацией полиме-
полимера. Поливинилены с электроноакцепторными заме-
заместителями (I, R = GOOH, COOR, Cl, R' = Н), а также
полифенилвинилены (I, R = С6Н5, R' = Н, С6Н5)
практически не галогенируются. Атомы галогена при-
присоединяются по концам наиболее длинных блоков
сопряжения, а также образуют с полимерами устойчи-
устойчивые в обычных условиях донорноакцепторные комплек-
комплексы. Бромирование линейных и блок-лестничных поли-
полимеров на основе дифенилбутадиина, содержащих звенья
I (R = C6H5, R' = -C=CC6H5) и VIII (R = С6Н5), сопро-
сопровождается внутримолекулярной циклизацией линейных
участков с присоединением брома по концам образовав-
образовавшихся лестничных фрагментов.
Скорость присоединения озона к полифенилвиниле-
ну (I, R = С6Н5, R' = Н) примерно на два порядка
меньше, чем к его мономерным и высокомолекулярным
аналогам с изолированными двойными связями. Это
связано, по-видимому, с выигрышем энергии, достигае-
достигаемым при сопряжении.
При нитровании I(R = С6Н5, R' = Н, С6Н5) и поли-
дифенилбутадиина в звено каждого полимера вступают
две нитрогруппы. В случае I(R = C6H5, R' = Н) за-
замещение протекает как в основной цепи, так и в боко-
боковых фенильных заместителях, в случае I(R = R' =
= С6Н5) и полидифенилбутадиина, не имеющих атомов
водорода в основной цепи,— только в боковых группах.
После нитрования сигнал ЭПР I(R = R' = С6Н5) и
полидифенилбутадиина остается без изменения, а в
спектрах ЭПР I(R = C6H5, R' = Н) появляется сверх-
сверхтонкая структура, обусловленная расщеплением неспа-
ренных электронов на ядре 14N.
Поливинилены с электронодонорными заместителя-
заместителями (I, R = «-С3Н7, н-С4Н9, R'= H) гидрируются в
более жестких условиях, чем каротин, образуя бесцвет-
бесцветные насыщенные продукты. Поливинилены с электро-
электроноакцепторными заместителями (I, R = Cl, COOH,R' =
= Н) практически не гидрируются. При гидрировании
I(R = С6Н5, R' = H) и полидифенилбутадиина умень-
уменьшается концентрация ПМЦ и образуется смесь пол-
полностью гидрированных и исходных макромолекул.
Полимеры с сопряженными связями C=N (X —
XIII, XVII) под действием таких нуклеофильных
реагентов, как гидразин и фенилгидразин, деструкти-
руются по связи G = N. Глубина гидразинолиза зави-
зависит от структуры полимера и условий процесса. Эта
реакция может служить аналитич. методом установле-
установления структуры соответствующих П.
Специфич. особенность парамагнитных полиэлект-
полиэлектролитов, способных к ионному и (или) электронному
обмену, помимо их термич., химич., радиационной
стойкости и электропроводности, заключается в том,
что их обменная способность благодаря трансформи-
трансформирующему влиянию цепи полисопряжения в значитель-
значительной степени зависит от состава и структуры основной
цепи и боковых групп макромолекулы. В аценовом
ряду (VIII, R = Н) окислительный потенциал умень-
уменьшается с увеличением мол. массы и, следовательно,
длины цепи сопряжения.
При введении электроноакцепторных SO3H-rpynn
в полиариленхиноны полимер становится способным
наряду с электронным к ионному обмену, при этом
увеличивается его окислительный потенциал. Более
слабые кислотные свойства полипропиоловой к-ты
(I, R = СООН, R' = Н) с преимущественно транс-
трансоидной конфигурацией полиеновой цепи (карбо-
(карбоксильные группы расположены по одну сторону полие-
полиеновой цепи), чем полипропиоловой к-ты с транс-цп-
1001
ПОЛИМЕТАКРОЛЕИН
1002
соидной или tyitc-трансоидной конфигурацией ^карбо-
^карбоксильные группы расположены по разные стороны це-
цепи), объясняются возможной делокализацией заряда
продиссоциировавших карбоксильных групп по цепи
сопряжения и, следовательно, ослаблением влияния
электростатич. поля на диссоциацию.
Реакционная способность П. по отношению к ради-
радикалам определяется не общим числом делокализован-
ных я-электронов, а средним эффективным сопря-
сопряжением, связанным с молекулярной и надмолекуляр-
надмолекулярной структурами полимера, а также содержащимися
в полимерах ПМЦ (эффект локальной активации). В
реакциях П. с радикалами резко повышается роль по-
полярного переходного состояния, реализующегося, ве-
вероятно, вследствие повышения электронодонорных
свойств субстрата, активированного ПМЦ. В связи с
этим П. могут служить ингибиторами деструкции ряда
низкомолекулярных и полимерных соединений (не
только линейных, но и пространственно-сетчатых).
Регулируя состав, молекулярно-массовое распреде-
распределение и содержание ПМЦ, можно изменять эффектив-
эффективность этих ингибиторов при умеренных и высоких тем-
температурах.
Растворимые в мономерах или олигомерах П. тор-
тормозят окислительную деструкцию при сравнительно
высоких темп-pax и не препятствуют радикальной
полимеризации при умеренных темп-рах (80—100 °С).
Ингибирующую активность проявляют, по-видимому,
П., возникающие в насыщенных высокомолекулярных
соединениях в результате протекания реакций внутри-
внутримолекулярного отщепления и циклизации, а также П.,
образующиеся в условиях ингибированного окисления
из обычных антиоксидантов (напр., фенолов и аминов),
содержащих подвижные атомы водорода. П. обладают
повышенной абляционной стойкостью и являются
ингибиторами абляционного разрушения.
Повышенная термостойкость — одно из характерных
свойств П., обусловленное снижением внутренней
энергии системы при образовании цепи полисопряже-
полисопряжения. П., приведенные в табл. 1 (см. также Лестнич-
Лестничные полимеры, Полибензимидазолы, Полихиноксалины)
выдерживают без заметной деструкции нагревание в
инертной атмосфере до 450—500 °С, на воздухе — до
300 °С. Своеобразие процессов термич. превращения
П. заключается прежде всего в том, что кинетич.
кривые, характеризующие зависимость потери массы
во времени, достигают максимума при каждой данной
темп-ре.
Такой ступенчатый характер кривых термодеструк-
термодеструкции в широком темп-рном интервале связан, по-види-
по-видимому, с протеканием конкурирующих реакций, обус-
обусловливающих, с одной стороны, рост цепи сопряжения в
образующихся при деструкции полиароматич. струк-
структурах, а с другой — разрыв слабых связей. Послед-
Последними, вероятнее всего, являются места стыков блоков
сопряжения и связи боковых заместителей, не сопря-
сопряженных с основной цепью. Первый процесс ответствен
за повышение термодинамич. стабильности системы,
второй — за образование летучих продуктов. При
каждой заданной темп-ре реализуется состояние, в
к-ром завершаются конкурирующие реакции, но на-
накапливаются ПМЦ, способные катализировать даль-
дальнейшие превращения при повышении темп-ры.
На термич. и термоокислительную стабильность
П. оказывают влияние характер основной цепи сопря-
сопряжения, наличие разветвлений и тип заместителя. Не-
Незамещенные поливинилены с открытой цепью легко
окисляются кислородом воздуха; при наличии алкиль- |
ных и особенно арильных заместителей в основной
цепи устойчивость поливиниленов к действию кисло- |
рода повышается. В зависимости от структуры полие-
новой цепи кислород может присоединяться как в
положение 1,4, так и 1,2. :
Перспективы практического использования
Ввиду трудностей, связанных с получением раство-
растворимых или плавких достаточно высокомолекулярных
П., резко ограничивается возможность их переработки
в различные полимерные материалы и изделия. В свя-
связи с этим актуально решение следующих проблем:
1. Синтез полимеров и сополимеров с развитой систе-
системой сопряжения, содержащих боковые ответвления
или объемные группы, препятствующие плотной упаков-
упаковке макромолекул, а следовательно, образованию проч-
прочных ассоциатов и раннему обрыву цепи при формиро-
формировании П. Сюда относится синтез растворимых и плав-
плавких полиариленов (полиантрацен, сополимеры бензола
с нафталином и с о- и гс-терфенилом, фенилированные
полифенилены), полигетероциклич. соединений и др.
2. Получение П., содержащих электронообменные груп-
группы, при помощи к-рых можно осуществлять переход
растворимой восстановленной формы в нерастворимую
окисленную (полимеры с хинонными, индигоидными
или индофениновыми группами в цепи сопряжения).
3. Синтез растворимых высокомолекулярных соедине-
соединений цепной структуры с нарушенным сопряжением зве-
звеньев, способных при последующей обработке в резуль-
результате внутримолекулярной циклизации превращаться
в полимеры с сопряженными фрагментами (см., напр.,
Лестничные полимеры).
4. Синтез блок- и привитых сополимеров, макромо-
макромолекулы к-рых включают фрагменты с многоцентровой
и двухцентровой связью. Такие в-ва должны сочетать
свойства участвующих в их образовании фрагментов.
П., содержащие хинонные, хинониминные, индиго-
идные и др. группы в цепи сопряжения (см. Окисли-
Окислительно-восстановительные полимеры), сочетают свой-
свойственную П. термостабильность и радиационную стой-
стойкость с электрич. проводимостью и способностью к
электронному обмену, что открывает новые возможно-
возможности применения этих полимеров для дегидрирования
и переноса протонов, моделирования действия биока-
биокатализаторов, аккумулирования электрич. энергии и др.
П. имеют различную окраску и м. б. использованы
в самых разнообразных областях: как окрашенные
пластмассы и аппреты, пигменты, сенсибилизаторы
внутреннего фотоэффекта в высокоомных полупровод-
полупроводниках и фотографич. слоях.
Ввиду низкой летучести и высокой термостабиль-
термостабильности П. можно использовать в качестве антиокси-
антиоксидантов многих промышленных полимеров.
В электрич., фотоэлектрич. и оптич. характеристи-
характеристиках П. заложены потенциальные возможности создания
материалов, к-рые могут найти широкое применение
в различных областях энергетики и радиоэлектроники.
Лит.: Берлин А. А., Хим. пром-сть, № 5, 23 C75),
№ 6, 6 D44) A960), № 12, 23 (881) A962); его же, Высо-
комол.соед., А13, № 2, 276 A971); Б е р л и н А. А., Чер-
кашинМ.И., там же, А13, М 10, 2298 A971); Вег-
1 i n A. A., J. Macromol. Sci., A 5, № 7, 1187 A971); Пень-
ковский В. В., Усп. хим., 33, в. 10, 1232 A964); Ду-
Дулов А. А., там же, 35, № 10, 1853 A966); П о п 0 в Е. М.,
Коган Г. А., там же, 37, в. 2, 256 A968); Ш у с т о р о-
вичЕ.М., Электронное строение полимерных молекул с крат-
кратными связями в основной цепи, М., 1967; Органические полу-
полупроводники, 2 изд., М., 1968; Богуславский Л. И.,
Ванников А. В., Органические полупроводники и био-
биополимеры, М., 1968; Паушкин Я. М., В и ш н я к о-
ваТ. П., Лунин А. Ф., Н и з о в а С. А., Органические
полимерные полупроводники, М., 1971; Химия полисопря-
полисопряженных систем, М., 1972. А. А. Берлин, М. Г.Чаусер.
ПОЛИМЕТАКРИЛАТЫ — см. Метакрилатов по-
полимеры.
ПОЛИМЕТАКРИЛОВАЯ КИСЛОТА — см, Мет-
акриловой кислоты полимеры.
ПОЛИМЕТАКРИЛОНИТРИЛ — см. Метакрилонит-
рила полимеры.
ПОЛИМЕТАКРОЛЕИН — см, ' Метакролеина поли-
полимеры.
1003
ПОЛИМЕТИЛБУТЕН
1004
ПОЛИМЕТИЛБУТЕН — см. З-Метилбутена-1 поли-
полимеры.
ПОЛИМЕТИЛЕН — см. Этилена полимеры.
ПОЛИМЕТИЛЕНОКСИД, полиформальде-
полиформальдегид [poly(methylene oxide), Polymethylenoxid, оху-
de de polymethylene].
Мономеры
Формальдегид СН2=О (Ф.) — бесцветный газ с рез-
резким запахом. Нек-рые его свойства приведены в таблице.
Ф. хорошо растворим в воде и спиртах, в к-рых присут-
присутствует соответственно в виде моно- и полимергидратов
НО(СН2О)ПН и гемиформаля RO(CH2O)nH. Доля сво-
свободного Ф. в р-ре весьма низка; константа равновесия в
водном р-ре при 0 °С составляет 10~4. Теплота рас-
растворения в воде и спиртах ок. 63 кдж/молъ (ок.
15 ккал/молъ). Газообразный Ф. плохо растворим
в большинстве органич. растворителей; при комнат-
комнатной темп-ре образует истинные р-ры в ароматич. угле-
углеводородах [концентрация ок. 1 кмолъ/м3 (молъ/л)],
циклогексане, тетрагидрофуране и др. Жидкий моно-
мономерный Ф. при темп-ре ниже —19 °С хорошо смешивает-
смешивается с ароматич. углеводородами, эфирами, нек-рыми
галогенпроизводными углеводородами. Ф. самопроиз-
самопроизвольно полимеризуется при хранении; следы влаги,
спиртов, к-т и др. полярных соединений ускоряют
этот процесс. Тщательно очищенный от этих примесей
жидкий Ф. или его р-р удается сохранять при —80 СС
в течение лишь нескольких дней. Газообразный Ф. не
полимеризуется при темп-pax выше 119 °С (давление
101,3 кн1м2, или 760 мм рт. ст.); выше этой теми-ры
интенсивно протекает реакция Канниццаро:
2СН2О + Н2О -» СН3ОН + HGOOH
Поэтому в технике Ф. используют непосредственно
после получения. Трубопроводы, арматуру и все
устройства, соприкасающиеся с Ф., нагревают до
ЮО—120 °С для предотвращения образования на их
поверхности твердого полимера. Ф. самовоспламеня-
самовоспламеняется в смесях с воздухом при теми-ре порядка 250 °С.
Он обладает сильным слезоточивым действием; предель-
предельно допустимая концентрация в воздухе 5 мг/м*.
Основной способ получения Ф. в пром-сти — окис-
окисление метанола кислородом воздуха в присутствии
различных катализаторов. Так, в присутствии сереб-
серебряного или медного катализатора образуется парога-
парогазовая смесь, содержащая Ф., метанол, воду, водо-
водород, окись углерода и др. Ф. и метанол поглощают
из этой смеси в скрубберах, орошаемых водой. Товар-
Товарным продуктом является формалин — водный р-р Ф.
Технич. формалин содержит 37% (по массе) Ф. и 4—
12% метанола, к-рый служит стабилизатором. В
нек-рых случаях метанол специально вводят в форма-
формалин. При охлаждении ниже 10 °С из формалина выпа-
выпадают твердые низкомолекулярные полимергидраты.
При использовании в качестве катализаторов оки-
окисей железа и молибдена получают формалин, практи-
практически не содержащий метанола. Устойчивость такого
формалина сильно зависит от концентрации Ф. и
темп-ры. Конц. р-ры формалина (выше 40% Ф.) устой-
устойчивы только при темн-рах выше 50 °С. Формалин ки-
кипит при 102 СС (давление 101 кн/м2, или 760 мм рт. ст.);
при этом происходит разложение гидратов и выделяет-
выделяется свободный Ф. и вода. При перегонке формалина
в вакууме формальдегидом обогащается кубовый оста-
остаток, при перегонке под давлением — дистиллят. Ф.
выделяют из конц. р-ра формалина E0—55% Ф.),
не содержащего метанола, одноступенчатой или двух-
двухступенчатой парциальной конденсацией; выход Ф.
25—30% от его содержания в формалине. Выделенный
таким образом Ф. содержит ок. 1,5% воды.
Ф. получают также пиролизом твердых низкомоле-
низкомолекулярных полимеров Ф. (см. ниже) — параформа,
триоксана или а-полиоксиметилена; этим методом полу-
получается более чистый мономер. При нагревании до 150 —
180 °С эти полимеры практически полностью разла-
разлагаются на мономерный Ф., содержание воды в к-ром
0,5—1,0%.
Для получения высокомолекулярного пластичного
полиформальдегида необходимо, чтобы содержание
примесей (Н2О, СН3ОН, НСООН и др.) в мономере не
превышало 0,05—0,10%. Ф. очищают частичной по-
полимеризацией его при низких темп-pax (ниже 0 СС) на
твердой поверхности, сорбентах (напр., цеолитах или
катионитах) или парциальной конденсацией примесей
в специальных растворителях (гемиформалях).
уо-сн2Ч
Триоксан Н2С<( ^>О — бесцветное кристаллич.
Ч)—СН/
вещество с характерным запахом; нек-рые свойства
его приведены в таблице. С водой триоксан образует
азеотропную смесь, кипящую при 91 °С (содержание
триоксана 70%); хорошо растворим во многих органич.
соединениях и сам используется как растворитель.
Дипольный момент в бензоле 2,18/) (ID = 3,33364X
ХЮ~30 к-м); диэлектрич. проницаемость 3,2 B0 СС) и
8,0 G0 °С). Триоксан устойчив во всех агрегатных
состояниях, а также к действию щелочей; гидролизует-
ся под действием минеральных к-т. Триоксан, содер-
содержащий менее 1% воды, спонтанно полимеризуется при
плавлении и кристаллизации. Ингибиторы полимери-
полимеризации — амины.
Свойства формальдегида и триоксана
Свойства
Плотнопть, г/см3 ......
Темп-pa, °С
плавления
кипения ... ...
Теплота, кдж/молъ
(кпал/молъ)
плавления . . . . с .
испарения
полимеризации* . .
сгорания
Пределы взрывоопасных
концентраций в смесях с
воздухом, % (по объему)
Формальдегид
0,8153 (-20 °С)
-118
— 19
—
23,3E,57)
69,9 A6,7)
561,4A34,1)
7-72
Триоксан
1,17F3 ГС)
61-62
114-115
14,6C,43)
41 (9,8)
62,93A5,03)
1520 C62,7)
3,6-28,7
* При полимеризации газообразного мономера с образова-
образованием кристаллич. полимера.
Триоксан получают нагреванием 50—55%-ного
р-ра формалина, не содержащего метанола, с 2% (по
массе) H2SO4. При 100 °С равновесное содержание
триоксана в реакционной массе не более 6% . Процесс
проводят в кубе ректификационной колонны, из верх-
верхней части к-рой отбирают дистиллят, по соста'ву близ-
близкий к азеотропу триоксан — вода. Триоксан выделяют
экстракцией бензолом, хлороформом и др. растворите-
растворителями или кристаллизацией при 0 °С. Для получения
высокомолекулярного полиформальдегида используют
триоксан, содержащий менее 0,01% Н2О, СН3ОН,
НСООН и др. примесей. Очищают триоксан ректифи-
ректификацией с последующей перегонкой над твердой щелочью.
6-СН2-О
I I
Тетраоксан СН2 СН2 — бесцветное кристаллич.
I I
О—СН2—О
вещество; темп-pa плавления 112 °С; легко возгоняется.
Свойства мало изучены. М. б. использован для получе-
получения высокомолекулярного П.
Гомополимеры
Полиметиленоксиды (П.) — линейные полимеры
полиацетальной структуры [—СН2 — О—]п; кристал-
кристаллич. вещества белого цвета со слабым запахом Ф.
1005
ЛОЛИМЕТИЛЕНОКСИД
1006
Низкомолекулярные П.— параформ (степень
полимеризации 8—12; содержание Ф. 90—96% , осталь-
остальное — вода) и т. наз. а-п олиоксиметилен
(степень полимеризации ок. 100; содержание Ф. 99,5%).
Оба продукта плавятся с разложением соответственно в
интервалах 120—150 и 170—180 °С; степень кристал-
кристалличности близка к 100% . При растворении в воде оба
полимера разлагаются и гидролизуются (параформ —
при нагревании, а-п олиоксиметилен — при
кипячении с добавкой щелочи). Полимеры легко разла-
разлагаются также при нагревании. К-ты и щелочи ускоряют
разложение. Эти полимеры используют обычно в каче-
качестве источника Ф. в органич. синтезе и при произ-ве
различных полимеров на основе формальдегида.
Параформ получают в технике концентрированием
формалина под вакуумом до образования твердого
остатка в кубе. Полученный продукт размалывают и
высушивают. а-Полиоксиметилен синтезируют дейст-
действием щелочи на конц. р-ры Ф., не содержащие метано-
метанола. Твердый продукт отделяют от формалина, промы-
промывают водой для удаления щелочи и высушивают под
вакуумом.
Высокомолекулярный II. Свойства. П. высокой
мол. массы (степень полимеризации выше 1000) —
термопласт. Основная кристаллич. модификация —
гексагональная. Постоянные решетки: а = 0,446 им
D,46 А), с = 1,73 им A7,3 А). В определенных усло-
условиях можно получить орторомбич. модификацию, од-
однако она метастабильна и переходит в гексагональную
выше 60 СС. Степень кристалличности П. 70—100% в
зависимости от метода получения. Нек-рые свойства
высокомолекулярного П. приведены ниже:
Плотность при 25 °С, г/см3 1,43
Темп-pa, °С
плавления 173—180
стеклования —60
Теплота плавления, кдж/молъ (ккал/молъ) 7,45A,78)
Уд. теплоемкость, кдж/(кг-К) [кал/(г-сС)] . . 1,47@,35)
Темп-рный коэфф. линейного расширения, "С 81-10~в
Теплопроводность, вт/(м-К) [ккал/(м>Ч'°С)] 0,23@,2)
Прочность, Мн/м2 (кгс/см2)
при растяжении 68—71
F80-710)
при сжатии A0%) 110—130
A100-1300)
при изгибе 100—120
A000-1200)
Модуль упругости, Гн/м2 (кгс/см2)
при растяжении 2,9 B9-103)
при изгибе 3,5C5-103)
Ударная вязкость, кдж/м2, или кгс-см/см2
без надреза 90—120
с надрезом 7—12
Относительное удлинение, % 15—45
Твердость по Бринеллю, Мн/м2 (те/мм2) . . . 150 — 180
A5-18)
Теплостойкость по Вика, °С 167
Деформационная теплостойкость при напря-
напряжении 1,85 Мн/м2, или 18,5 кгс/см2, °С . . 110
Диэлектрич. проницаемость при 100 гц—
100 пгц 3,7—3,8
Тангенс угла диэлектрич. потерь 0,004
Уд. электрич. сопротивление
объемное, Том-м (ом-см) 6 F-1014)
поверхностное, Том (ом) 2000 B-1015)
Водопоглощение за 24 ч, % 0,2
Практич. ценность представляет П. с молекуляр-
но-массовым распределением (Mw/Mn), близким к 2,
и Мп = C0—80) • 103. С повышением средней степени
полимеризации увеличиваются прочность и эластич-
эластичность П., однако текучесть расплавленного материала
резко понижается. Чем шире молекулярно-массовое
распределение, тем ниже текучесть и выше хрупкость
полимера.
Вязкость р-ра П. измеряют при 150 °С в ]Ч,]Ч-диме-
тилформамиде, а также при 90—100 °С в /г-хлорфеноле
или смеси C:1) тетрахлорэтана с фенолом (концентра-
(концентрация р-ра П. 0,5%). Обычно для предотвращения раз-
разложения полимера при нагревании р-р стабилизуют
добавкой 2% дифениламина или а-пинена. Для П. с Мп,
указанным выше, характеристич. вязкость [г\], опре-
определенная в диметилформамиде, составляет 0,5 —
0,7 дл/г; [\\], определенная в двух др. указанных выше
растворителях, приблизительно в 2 раза больше. За-
Зависимость между Мп и [г\] (в дл/г) р-ра П. в смеси те-
тетрахлорэтана с фенолом выражается ур-нием: [г\] =
= 2,754-Ю-4 Мп°>*°.
При темп-pax ниже 60 °С П. растворяется только в
гексафторацетонгидрате или гексафторацетоноксиме;
выше 100 °С П. ограниченно растворим в фенолах, аро-
матич. аминах, бензиловом спирте, метилендиацетате
и др. Ниже 60 °С П. незначительно набухает (менее
3% по массе) практически во всех нейтральных орга-
органич. веществах. При постоянной эксплуатации П. в
воде набухание составляет ок. 1%. Проницаемость
пленки из П. для водяного пара такая же, как и у пле-
пленок из полистирола и полиметилметакрилата и в 10 раз
больше, чем у пленки из полиэтилена. Проницаемость
П. для органич. жидкостей и газообразных углеводоро-
углеводородов исключительно низка; воздухопроницаемость плен-
пленки П. в 100 раз ниже, чем полиэтиленовой (см. Поли-
Полиэтиленовые пленки).
П. не стоек к действию минеральных к-т и НСООН.
Устойчивость к действию щелочей определяется при-
природой концевых групп в макромолекулах (ацетальные
связи не гидролизуются под действием оснований). П.
горюч (сгорает практически без остатка), не токсичен.
Независимо от метода получения большинство мак-
макромолекул П. содержит концевые группы ОН вслед-
вследствие протекания при полимеризации передачи цепи
на молекулы воды. Такой П. имеет низкую термич.
стабильность: при нагревании в вакууме или инерт-
инертной атмосфере до 100 °С он деполимеризуется с вы-
выделением газообразного Ф. При темп-ре плавления
П. скорость разложения его на воздухе достигает 1%
в 1 мин и более. Поэтому перед переработкой П. под-
подвергают стабилизации, «блокируя» концевые группы
ОН. Наибольший практич. интерес представляет
ацетилирование П. уксусным ангидридом в присутст-
присутствии катализатора основного характера (напр., пириди-
пиридина, ацетата натрия). Реакцию проводят в чистом
ангидриде или инертном растворителе при 140—
150 °С в течение 1—2 ч. Выход П. с «блокированными»
(ацетильными) концевыми группами 90—95% . Модифи-
Модифицировать концевые группы ОН можно также ангидри-
ангидридами и хлорангидридами карбоновых к-т, спиртами,
ацеталями, ортоэфирами, эпоксисоединениями и др.
Однако большинство этих реакций происходит в кислой
среде, что приводит к значительным потерям П. вслед-
вследствие деструкции.
«Блокированный» П. устойчив при нагревании до
250 °С только в отсутствие кислорода; его можно пере-
перерабатывать литьем под давлением и экструзией в
темп-рном интервале 190—240 °С, если расплав П.
обладает хорошими реологич. свойствами. На воздухе
такой П. подвергается термоокислительной деструкции
при темп-pax выше 160 °С с выделением газообразного
Ф. и уменьшением мол. массы. В вязкотекучем состоя-
состоянии A90—240 °С) скорость разложения составляет 0,2 —
0,5% в 1 мин, в результате чего переработка «блокиро-
«блокированного» П. на обычных литьевых машинах оказывает-
оказывается невозможной. Чтобы замедлить термоокислительную
деструкцию, в «блокированный» П. вводят специальные
стабилизирующие добавки для увеличения индукцион-
индукционного периода разложения в условиях переработки до
40—60 мин. Стабилизирующие композиции содержат
антиоксидант, акцептор газообразного Ф. и акцептор
НСООН. Антиоксидантами служат ароматич. амины и
фенолы (напр., ди-р-нафтил-/г-фенилендиамин, дифенил-
дифениламин, алкилзамещенные метиленбисфенолы), акцепто-
1007
ПОЛИМЕТИЛЕНОКСИД
1008
рами Ф.— вещества, содержащие амидные группы
(напр., полиамиды, дициандиамид).
П. характеризуется высокой усталостной прочностью
к динамич. знакопеременным нагрузкам (по этому пока-
показателю П. превосходит др. термопласты, в частности
поликарбонат, хотя уступает ему по прочности к одно-
однократным нагрузкам), стабильностью размеров и низкой
ползучестью при повышенных темп-pax, сохранением
достаточно высокой прочности и жесткости при темп-рах
ок. 100 °С, высокой износостойкостью (уступает толь-
только полиамидам), хорошими фрикционными свойствами.
Получение. Существуют три принципиально
возможных способа получения П.: 1) анионная или ка-
тионная полимеризация безводного мономерного Ф.;
2) катионная полимеризация с раскрытием цикла цик-
лич. олигомеров Ф.— триоксана или тетраоксана; 3)
полимеризация водных или спиртовых р-ров Ф.
При получении полимера из Ф. очищенный газооб-
газообразный мономер непосредственно после выделения по
обогреваемым трубопроводам поступает в реактор, в
к-ром энергично перемешивается инертная реакцион-
реакционная среда (бензин, циклогексан, толуол или др.), со-
содержащая обычно анионный катализатор (напр., ами-
амины, фосфины, соли к-т жирного ряда или др.), к-рый
менее чувствителен к примесям, чем катионный. Про-
Процесс проводят в изотермич. условиях B0—60 °С); тепло
реакции отводят через рубашку реактора. Для регули-
регулирования мол. массы в газообразный Ф. или непосред-
непосредственно в жидкую фазу вводят регуляторы роста цепи
(воду, спирты, к-ты). Полимер нерастворим в реакцион-
реакционной среде и выпадает в виде кристаллич. мелкодис-
мелкодисперсного порошка, к-рый отделяют или ацетилируют
непосредственно (в виде суспензии) в реакционной
среде. Для этого в реактор добавляют уксусный ангид-
ангидрид, катализатор и суспензию нагревают до кипения.
Продукт выделяют, высушивают под вакуумом, вводят
в него стабилизаторы и др. добавки и гранулируют.
П. легко окрашивается в расплаве в различные цвета.
При получении П. из триоксана полимеризацию мож-
можно проводить в газовой (выше 115 °С), жидкой и твер-
твердой (ниже 62 °С) фазах или р-ре в присутствии катион-
ных катализаторов (напр., комплексов BF3, карбоние-
вых солей), а также при облучении ^-лучами (при твер-
твердофазной полимеризации). В зависимости от способа
полимеризации продукт получают в виде порошка или
блока. По свойствам он идентичен продукту полиме-
полимеризации газообразного Ф., однако из-за нек-рых
различий в морфологии кристаллич. структуры гете-
гетерогенное ацетилирование такого П. невозможно. По-
Поэтому полимер растворяют в чистом уксусном ангидри-
ангидриде или в его смеси с растворителем (напр., метиленди-
ацетатом или диметилформамидом) при 150 °С, затем
высаждают и обрабатывают, как описано выше.
При получении П. полимеризацией Ф. в водных
или спиртовых р-рах реакцию проводят в равновес-
равновесных условиях (т. е. в условиях, исключающих образо-
образование низкомолекулярных твердых полимеров); ско-
скорость образования высокомолекулярного П. очень
низка. Степень кристалличности продукта 100%.
Реакция протекает по механизму ступенчатой полиме-
полимеризации. При использовании специальных каталитич.
систем образуется орторомбич. П., к-рый подвергают
этерификации.
Переработка. П. перерабатывают на обычных
литьевых машинах, а также на экструдерах с предпла-
стикацией материала. Соотношение длина шнека:
диаметр в экструдерах должно быть не менее 20 : 1.
Контроль за темп-рой расплавленного материала дол-
должен быть очень точным, чтобы избежать перегрева и
разложения П. Нежелательно оставлять П. в расплав-
расплавленном состоянии более 20—30 мин, т. к. может начать-
начаться разложение материала. Для очистки машин до и
после переработки П. используют полиэтилен.
Так как П. обладает высокой текучестью расплава,
при литье под давлением применяют самозапирающиеся
сопла или сопла малых сечений (см. Литьевые машины).
П. быстро кристаллизуется, поэтому литьевые формы
нагревают в зависимости от толщины и формы изделий
до 60—80 °С. Газообразный Ф. обладает очень резким
запахом даже при минимальных неопасных концентра-
концентрациях, поэтому при переработке П. машины оборудуют
вытяжной вентиляцией, а изделия, вынутые из формы,
охлаждают в воде.
Применение. Более 90% П. используют для
замены цветных металлов и сплавов в машиностроении,
автомобилестроении и др. областях пром-сти. Экономич.
эффект при замене металлич. литья достигается благода-
благодаря тому, что для изделий из пластика не требуется мно-
многостадийная станочная обработка. Т. обр., хотя стои-
стоимость П., даже с учетом низкой плотности, значительно
выше, чем цветных металлов, стоимость изделий из не-
него ниже. Кроме того, во многих случаях срок службы
изделий из П. больше, т. к. они не корродируют.
Литьем из П. изготовляют втулки, зубчатые колеса,
шестерни, пружины, рукоятки, корпуса приборов,
детали переключателей, краны, масло- и бензопроводы,
арматуру для водопроводов и т. д. Детали из П., рабо-
работающие при переменных нагрузках в условиях постоян-
постоянной влажности при повышенных темп-рах (до 100 °С),
обладают лучшими эксплуатационными свойствами,
чем детали из полиамидов, фенопластов и др. конструк-
конструкционных пластмасс. В США в полупромышленном
масштабе организовано производство волокна (для ры-
рыболовных сетей и технич. назначения — см. Полифор-
мальдегидные волокна), труб и контейнеров для аэрозо-
аэрозолей.
Промышленный выпуск ацетилированных гомопо-
лимеров Ф. (д е л ь р и н а) был организован впервые
в США в 1960.
Сополимеры
Ацетилированный П. по стабильности в условиях
переработки уступает др. термопластам, что затрудняет
его применение. Лучшее стабилизирующее влияние
на П. при повышенных темп-рах оказывает введение
небольших количеств сомономеров, содержащих связи
С — С (напр., олефинов, циклич. эфиров и ацеталей).
При статистич. распределении сомономера в макромоле-
макромолекулах П. разрыв ацетальной связи (под действием
кислорода или к-т) приводит к деполимеризации не
всей макромолекулы, как в случае гомополимера, а
только участка цепи, заключенного между двумя свя-
связями С — С. Статистич. распределение звеньев сомо-
сомономера достигается в результате протекания при сопо-
лимеризации передачи цепи с разрывом на полимер.
При этом доля термически неустойчивых конце-
концевых фрагментов макромолекул, имеющих структуру
~ (ОСН2—)п ОН, обратно пропорциональна количе-
количеству введенного сомономера.
В результате введения сомономера кристаллич.
структура П. нарушается и, следовательно, снижаются
темп-pa плавления, жесткость и твердость. Одновре-
Одновременно возрастают эластичность, прочность к ударным
нагрузкам, текучесть. Т. обр., изменяя количество
сомономера, можно варьировать свойства сополимера
в широких пределах. Чтобы получить материал, близ-
близкий по механич. свойствам к гомополимеру, но превос-
превосходящий его по стабильности в условиях переработки,
целесообразно вводить не более 2,5—3,0% сомономера
от массы П.
Свойства сополимера практически не зависят от хи-
мич. природы сомономера. Модуль упругости при ра-
растяжении, твердость, теплостойкость по Вика, темп-ра
деформационной теплостойкости таких сополимеров
лишь на 10—15% ниже, чем у гомополимера. Однако
вследствие повышения стабильности в условиях пере-
1009
ПОЛИМОЧЕВИНЫ
1010
работки и улучшения реологич. свойств это различие
в свойствах практически полностью нивелируется при
переработке на типовом оборудовании, и в большинстве
случаев гомополимер вытесняется сополимером. Плот-
Плотность сополимера 1,41 г/см3\ температура плавления
164—168 °С; ударная вязкость 120 — 140 кдж/м2, или
кгс-см/см2. По фрикционным и электрич. свойствам
сополимер не отличается от гомополимера. Сополимер
более стоек к действию щелочей, солей, органич. к-т.
Сильные минеральные к-ты его разлагают.
Сополимер получают катионной сополимеризацией
Ф. или триоксана с окисью этилена, 1,3-диоксоланом и
др. сомономерами. Наибольшее значение приобрела
сополимеризация триоксана вследствие экономич. и тех-
нологич. преимуществ. При этом сополимеризацию про-
проводят в р-ре или расплаве. В последнем случае исполь-
используют реактор шнекового типа. Сомономер перемешивают
с триоксаном, после чего вводят катализатор (напр.,
комплексы BF3, SnCl4, SbF5). Реакцию проводят в изо-
термич. условиях. Тепловой эффект при полимериза-
полимеризации жидкого триоксана всего ок. 8,4 кдж/молъ (ок.
2 ккал/молъ). Продукт отделяют от непрореагировавше-
го триоксана (к-рый возвращают в цикл) и катализато-
катализатора. Сополимер содержит 5—10% (по массе) концевых
нестабильных сегментов, к-рые разлагают термообра-
термообработкой в экструдере под вакуумом. Сополимер стаби-
стабилизируют и перерабатывают так же, как гомополимер;
по внешнему виду оба продукта одинаковы и одинако-
одинаково также их применение (см. выше).
Анализ тенденций развития производства и потреб-
потребления сополимера показывает, что этот материал обла-
обладает значительными потенциальными возможностями.
Дешевое сырье, получаемое в больших масштабах
из природного газа, и ценные свойства материала
создают предпосылки для дальнейшего развития
произ-ва: Массовое использование сополимера в
промышленности пока ограничено из-за относительно
высокой цены на материал и некоторых трудностей
переработки.
В 1962 в США и ФРГ было впервые совместно
организовано производство сополимера триоксана с
2,5% окиси этилена (фирменные марки соответственно
целкон и хостаформ С). По свойствам этот
сополимер аналогичен дельрину.
Общий выпуск П. в 1972 составил 120 тыс. т.
Лит.: Уокер Д ж., Формальдегид, [пер. с англ.], М.,
1957; Е н и к о л о п я н Н. С, Вольфсон С. А., Химия
и технология полиформальдегида, М., 1968. С. А. Вольфсон.
ПОЛИМЕТИЛМЕТАКРИЛАТ — см. Метилмета-
крилата полимеры.
ПОЛИМБТИЛПЕНТЕН — см. 4-Метилпентена-1 по-
полимеры.
ПОЛИ-а-МЕТИЛСТИРОЛ — см. Стирола произ-
производных полимеры.
ПОЛИМОРФИЗМ полимеров (polymorphism,
Polymorphic, polymorphisme) — существование вещест-
вещества в разных кристаллич. формах (полиморфных моди-
модификациях). П. свойствен большинству кристаллизую-
кристаллизующихся полимеров. Следует различать две категории П:,
одна из к-рых связана с неизменностью конформации
макромолекул в различных кристаллич. модификациях
данного полимера (полиморфизм упаков-
упаковки), а другая — с возможным изменением конформа-
конформации при полиморфном переходе (конформацион-
ный полиморфиз м). К особому случаю П.
можно отнести существование т. наз. газокристаллич.
состояния полимеров, вызванное вращением макромоле-
макромолекул вокруг их длинной оси и возможным смещением
в плоскости, перпендикулярной макромолекулярным
осям, при сохранении взаимной параллельности це-
цепей. К полиморфным модификациям первой катего-
категории можно отнести известные кристаллические фор-
формы полиэтилена, изотактического полипропилена, по-
полиамидов, 1-замещенных 1,4-транс-полибутенов и др.
Конформационный П. характерен для кристаллических
полимеров, макромолекулы которых обладают сравни-
сравнительно большой конформационной свободой. К подоб-
подобным полимерам можно отнести полиметиленоксид, по-
либутен-1, политетрафторэтилен, синдиотактический
полипропилен.
В идеальном случае каждая полиморфная модифика-
модификация должна быть стабильна во вполне определенной
области темп-р и давлений, где все др. возможные
кристаллич. формы данного вещества неустойчивы
(метастабильны). Переход из одной модификации
в другую м. б. обратимым (энантиотропным) или не-
необратимым (монотропным). Переход неустойчивой
модификации в устойчивую монотропен.
Теория устойчивости кристалла позволяет предска-
предсказать теоретически, какая из возможных модификаций
полимера наиболее устойчива в данных условиях. Ста-
Стабильная модификация должна соответствовать мини-
минимуму свободной энергии кристалла F, к-рая склады-
складывается из внутренней энергии кристалла U, зависящей
от плотности упаковки молекул, энтропии S, возра-
возрастающей с увеличением симметрии кристаллической
решетки, и Екол — свободной энергии колебания:
F = U + ?кол - TS
Метастабильные модификации должны обладать ли-
либо меньшей плотностью упаковки, либо низкой сим-
симметрией, либо тем и другим.
При понижении темп-ры роль колебаний и энтропий-
энтропийного фактора в установлении равновесия в решетке
снижается. При Т — 0 К равновесная структура кри-
кристалла определяется минимумом потенциальной энер-
энергии решетки (с точностью до величины эффектов нуле-
нулевых колебаний, влияние к-рых на установление равно-
равновесной структуры кристалла, состоящего из больших
молекул, незначительно). Структура кристалла, отве-
отвечающая минимуму его потенциальной энергии, должна
с большой точностью совпадать со структурой его,
соответствующей абсолютному нулю темп-р. Внутрен-
Внутреннюю энергию кристаллич. вещества можно рассчитать,
используя известные потенциальные функции вида
U = — 2d ~ ~J + В ехР ("" аг^) + ^эл
г, j rij
гдег/j — расстояние между г-м и /-м атомами взаимодей-
взаимодействующих полимерных молекул; Л, 5, а — эмпирич.
константы; Еэл — энергия электростатич. взаимодей-
взаимодействия. В общем случае внутренняя энергия кристалла
зависит от симметрии и параметров кристаллич. решет-
решетки, ориентации макромолекул и их конформации, в
свою очередь определяющих гц в вышеприведенной
ф-ле.
Для выяснения структуры кристалла при абсолют-
абсолютном нуле темп-р следует найти минимум U по всем
параметрам. В случае П. упаковки внутренняя энер-
энергия кристалла является функцией параметров решет-
решетки и ориентации молекул. Если макромолекула обла-
обладает большой конформационной свободой (конформа-
ционный П.), поле кристалла может деформировать
ее. В этом случае энергия кристалла — функция также
параметров макромолекулы, характеризующих ее
конформацию (внутренние углы вращения вокруг оди-
одинарных связей, валентные углы).
Лит.: Уразовский С. С, Молекулярный полимор-
полиморфизм, Киев, 1956; KitajgorodskijA. I., Acta Grystal-
lographica, 18, pt 4, 585 A965); GorradiniP., Avita-
b i 1 e G., European Polymer J., 4, № 3, 385 A968); Теорети-
Теоретические аспекты конформации макромолекул, под ред. А. И. Ки-
Китайгородского (Итоги науки. Высокомолекулярные соедине-
соединения), М., 1970. И. О. Умарова.
ПОЛИМОЧЕВИНЫ, поликарбамиды, по^
лиамиды угольной кислоты (polyureas,
Polyharnstoffe, polyurees), — гетероцепные линейные по-
1011
ПОЛИМОЧЕВИНЫ
1012
лимеры, макромолекулы к-рых содержат в основной це-
цепи мочевинные группы —NH—СО — NH —.
Получение. В основе важнейших методов получения
П. лежит в основном поликонденсация диаминов с сое-
соединениями различных классов.
1. Взаимодействие диаминов с диизоцианатами в
р-ре или на границе раздела двух фаз:
эт H2N-R-NH2 + n OGN-R'-NGO -»
-> [-NH-R-NH-GO-NH-R'-NH-CO-]n
Реакция протекает со значительной скоростью даже
при комнатной темп-ре. Для получения П. с высокой
мол. массой процесс проводят при постепенном повы-
повышении темп-ры до темп-ры кипения растворителя
(ацетон, бензол, хлорбензол, третичные спирты, тет-
рагидрофуран и др.), иногда в присутствии катали-
катализаторов (третичные амины, металлоорганич. соеди-
соединения, вода). Для проведения реакции на границе раз-
раздела фаз диамин или его хлористоводородную соль ра-
растворяют в воде, а диизоцианат — в органич. раствори-
растворителе. Процесс подчиняется всем закономерностям
межфазной поликонденсации.
2. Поликонденсация диаминов с мочевиной или ее
производными в р-ре:
п H2N-R-NH2 + nH,N-CO-NH2 ->
-> [-R-NH-CO-NH-]n + 2nNH3
Реагенты нагревают 10—25 ч при 150—250 °С в кре-
крезоле, феноле или воде при атмосферном давлении
или в вакууме. Осуществление этого способа синтеза
П. осложняется неустойчивостью мочевины при высо-
высоких темп-pax. Однако разложения мочевины можно
избежать, если повышать темп-ру реакции постепенно.
При этом вначале образуются более термостойкие, чем
мочевина, олигомеры, к-рые затем вступают в даль-
дальнейшую поликонденсацию. Такой процесс осуществлен
в Японии в промышленном масштабе для получения П.
из нонаметилендиамина и мочеви" -i.
Реакция диаминов с производными мочевины (соля-
(солями циановой к-ты) приводит к образованию димочевин,
к-рые при взаимодействии с пополнительным количе-
количеством диамина образуют П. Аналогично мочевине во
взаимодействие с диаминами вступает тиомочевина.
3. Поликонденсация диаминов с фосгеном в р-ре
или на границе раздела двух фаз (жидкость — жидкость
или жидкость — газ):
п H2N-R-NH2 + n COC12 -> [-R-NH-CO-NH-]n + 2n HG1
При проведении реакции в органич. растворителе
(тетрагидрофуран, хлорированные углеводороды и др.)
выделяющийся HG1 м. б. связан щелочами или третич-
третичными аминами. Для осуществления межфазной поли-
поликонденсации водно-щелочной р-р диамина сливают с
р-ром фосгена в органич. растворителе, не смешиваю-
смешивающимся с водой.
4. Поликонденсация диаминов с диуретанами в р-ре
при 200—250 °С:
п H2N-R-NH2 + n C2H5OOCNH-R'-NK"OOC2H5 ->
-> [-R-NH-CO-NH-R'-NH-CO-NH-I^ -:- 2n G2H5OH
В качестве растворителей применяют фонол, крезол,
о-оксидифенил.
5. Взаимодействие диаминов с сероокисью углерода
в вакууме. При взаимодействии диамина с сероокисью
углерода в растворителе (толуол, бутанол, гликоли и
др.) образуется соль тиокарбамата, к-рая при повыше-
повышении темп-ры превращается в полимер:
п H2N—R—NH2 + п COS -> +H3N-R—NH—GO—S~ -*
80°G
> [-NH-CO-NH-R-]m + n H2S -*
180—200°G
у [-NH-GO-NH-R-]n + H2N-R-NHa
где т > n.
6. Поликонденсация диаминов с двуокисью углерода
при 100—350 °С и давлении 10 Мн/м2 A00 кгс/см2):
п H2N-R-NH2 + п СО2 -* [ - R-NH-CO-NH-]n + п Н2О
7. Поликонденсация диизоцианатов с водой с после-
последующим взаимодействием их с образовавшимися диа-
диаминами:
nOCN-R- NGO + п Н2О -> п H2N-R-NH2 -f n CO2
п OGN-R-NCO + п H2N-R-NH2 -> [-NH-GO-NH-R]n
8. Взаимодействие с водой при кипячении азидов ди-
карбоновых кислот (пробковая, азелаиновая, себаци-
новая и др.):
п N3OG—R-CON3 + п Н2О -> [-NH—CO-NH—R—]п + п СО2
Два последние способа носят препаративный характер.
Свойства П.—твердые бесцветные или белые кри-
сталлич. или аморфные продукты, к-рые в зависимо-
зависимости от способа получения м. б. порошкообразными,
комкообразными или волокнистыми. Свойства (темп-ра
плавления, растворимость, термостойкость и др.) П.
одного и того же строения существенно зависят от
способа их получения.
В связи с тем, что П. имеют узкое молекулярно-
массовое распределение и в большинстве случаев кри-
сталлич. структуру, они плавятся в узком интервале
темп-р. Темп-pa плавления П. с четным числом атомов
углерода в радикале выше, чем с нечетным. С увели-
увеличением числа метиленовых групп между атомами азота
темп-pa плавления полимеров понижается. Ароматич.
П. плавятся при более высоких темп-pax, чем алифати-
алифатические. Темп-pa плавления П., особенно ароматиче-
ароматических, близка к темп-ре разложения, что затрудняет
и у переработку в изделия. Значения темп-р плавления
П. различного строения лежат в интервале 180—320 °С.
При 300—350 °С начинается интенсивная термоокисли-
термоокислительная деструкция П.
П. растворимы в крезоле, муравьиной и серной к-тах,
диметилформамиде, метилпирролидоне, диметилацет-
амиде с добавкой LiCl. Алкилирование П. по атому
азота и частично в углеводородный радикал снижает
их темп-ру плавления, повышает термостойкость и
превращает в продукты, растворимые в обычных орга-
органических растворителях. В системе растворитель —
полимочевина существует сильное межмолекулярное
взаимодействие. Поэтому при разбавлении растворов
П. их вязкость не уменьшается, а увеличивается.
П. обладают высокой водостойкостью. Водопоглощение
П. колеблется в пределах 0,05—1,70% за 24 ч. Ме-
Механические и электрические свойства П. приведены
в таблице.
Механические и диэлектрические свойства полимочевин
Показатель
Прочность при растяжении,
Мн/м2 (кгс/см2)
Модуль упругости при растя-
растяжении, Мн/м2 (кгс/см2) . . . .
Предел текучести, Мн/м2
(кгс/см2)
Относительное удлинение, %
Ударная вязкость (с надре-
надрезом), кдж/м2, или кгс-см/см2
Уд. объемное электрич. со-
сопротивление, Том • м (ом • см)
Уд. поверхностное электрич.
сопротивление, Гож (ом) . .
Электрич. прочность, Мв/м,
или кв/мм
Полинонаме-
тиленмоче-
вина
66F60)
620-710
F200-7100)
33,5 C35)
215-238
0,82
56 E6X1014)
>1000(>10ls)
>20
760-800
G600-8000)
53E30)
195
4,7
—
>1000 (>1015
>20
Полидекаме-
тиленмоче-
вина
60 F00)
Применение. В течение длительного4^времени П. не
производились пром-стью по нескольким причинам:
1013
ПОЛИНОЗНЫЕ ВОЛОКНА
1014
1) близость темп-р плавления и разложения П. суще-
существенно осложняет их переработку; 2) высокая стои-
стоимость сырья (диамины); 3) преимущества П. [низкая
плотность A,03 —1,25 г/см3) и малое водопоглощение]
по сравнению с их ближайшими конкурентами —
полиамидами не очень велики.
Промышленное производство П. осуществлено толь-
только в Японии (в начале 60-х гг. XX в.), где из П. на
основе нонаметилендиамина и мочевины формуют
волокно (торговое название у р и л о н). Полинона-
метиленмочевина обладает темп-рой гелавления 245 °С
и может перерабатываться обычными для термопластов
методами. У рил он используют для производства ры-
рыболовных сетей и трикотажа.
П. потенциально пригодны для изоляции морского
кабеля. Из него можно изготовлять листовые материа-
материалы, трубы, стержни, пленки, лаки, стабилизаторы
эмульсий. Водо- и щелочерастворимые П., имеющие
в макромолекуле группы —ОН и —СО ОН, м. б. ис-
использованы для антистатич. обработки текстиля, в
электролитич. процессах для облегчения осаждения
тяжелых металлов на электродах, для получения пле-
пленок поливом из водных р-ров.
П. были впервые получены Байером в 1937.
Лит.: Федотова О. Я., Пластмассы, № 12, 23 A970);
Encyclopedia of polymer science and technology, v. 11, N. Y.—
[a. o.], 1969, p. 464. О. Я. Федотова.
ПОЛИНИТРИЛЫ — см. Нитрилов полимеры.
ПОЛИНОЗНЫЕ ВОЛОКНА, хлопкоподоб-
ные волокна (polynosic fibers, Polynosefasern,
fibres polynosiques) — разновидность вискозных воло-
волокон, по свойствам близких к хлопковым. П. в. выраба-
вырабатывают преимущественно в виде штапельных волокон.
Получение. П. в. получают в основном по той
же технологической схеме, что и обычные вискозные
волокна. Их формуют также из вискозы по мокро-
мокрому способу в кислотно-солевых осадительных ваннах.
Однако технологические режимы формования П. в. и
обычных вискозных волокон существенно различа-
различаются. В первом случае создают условия для полу-
получения свежесформованного волокна в гелеобразном
состоянии и с высокой степенью этерификации ксан-
ксантогената целлюлозы, что позволяет подвергать П. в.
значительно большей пластификационной вытяжке,
чем обычные вискозные волокна.
Режимы приготовления прядильных
р-ров для формования вискозных и
полинозных волокон также несколько
различаются. Для производства П. в.
применяют высококачественную цел-
целлюлозу с высоким содержанием а-цел-
люлозы и узким молекулярно-массо-
содержат 15--25 г/л H2SO4, 50—60 г/л Na2SO4 и 0,3—
0,5 г/л ZnSO4. В таких условиях ксантогенат целлюло-
целлюлозы разлагается медленно, а свежесформованное волок-
волокно получается в пластичном состоянии и способно,
как уже отмечалось, подвергаться большой пластифи-
пластификационной вытяжке. Одновременно с ориентацией при
вытягивании пластичного волокна происходит химич.
разложение ксантогената целлюлозы, сопровождаю-
сопровождающееся перестройкой межмолекулярных связей меж-
между целлюлозными цепями. Полученное таким спосо-
способом волокно отличается высокой степенью ориента-
ориентации и однородностью структуры в поперечном сече-
сечении. Скорость формования П. в. обычно не превыша-
превышает 20 м/мин.
Кроме стандартного П. в., известно еще высокопроч-
высокопрочное П. в., к-рое вырабатывается пока только в опытном
масштабе. При его формовании к вискозе или осади-
тельной ванне добавляют небольшое количество фор-
формальдегида. Предполагается, что вследствие образова-
образования промежуточного соединения между ксантогенатом
целлюлозы и формальдегидом разложение ксантогената
целлюлозы замедляется еще в большей степени, чем
при получении обычных П. в., что позволяет значитель-
значительно увеличить пластификационную вытяжку и резко
повысить прочность волокна.
К группе П. в. в нек-рых странах относят также вис-
вискозное волокно, получаемое при использовании в ка-
качестве осадительной ванны конц. растворов серной
к-ты (метод Лилиенфельда). Это волокно обладает вы-
высокой прочностью, низким удлинением и очень вы-
высоким модулем высокоэластичности, т. е. имеет основ-
основные характерные для П. в. свойства. Его вырабаты-
вырабатывают преимущественно в виде филаментной нити.
Наряду с П. в. получило развитие производство
хлопкоподобного вискозного волокна с большим моду-
модулем высокоэластичности во влажном состоянии (ВВМ).
Волокна этого типа получают по технологии, близкой
к получению высокопрочного вискозного кордного
волокна. Однако при формовании ВВМ применяют
осадитегьные ванны с более низкой темп-рой (^-30 °С)
и меньшим содержанием сульфата цинка; скорость
формования составляет 25—30 м/мин. В этих условиях
образуются волокна с относительно большими струк-
структурными элементами, обусловливающими большую
жесткость волокна и более высокий модуль высоко-
Свойства различных вискозных волокон и хлопкового волокна
Показатели
в сухом состоянии
в мокром состоянии
вым распределением. Прядильные р-ры Толщина)
(вискозу) приготовляют в мягких ус-
условиях для предотвращения деструк- прочность^гс/шекс
ции макромолекул. Мерсеризацию, из- " ™— ™
мельчение и ксантогенирование щелоч- Прочность в'петле, гс/текс
ной целлюлозы проводят при темп-pax Относительное удлинение,
не выше 20-22 СС, стадия предсозре- /о в сухом состоянии
вания обычно не проводится (подроб- в мокром состоянии
но об этих стадиях см. Вискоза). Рас- Модуль высокоэластично-
ход сероуглерода при ксантогениро- мн/л12МA^°жл^СТ0ЯНИИ'
вании на 20—25% превышает количе-
количество сероуглерода, применяемого при
получении обычного вискозного волок- Степень использования
на. Прядильные р-ры отличаются вы- прочности волокна в пря-
сокой вязкостью, достигающей 20— же, % .
40 н-сек/м* B00-400 па) при концен- Н?|уханиеВВОдепри2° С'.
трации целлюлозы 5 — 7% .
П. в. формуют по мокрому методу
с использованием осадительной ванны
с низким содержанием к-ты и темп-рой
,15—30 °С. Обычно осадительные ванны
Растворимость
NaOH, %
в 6% -ном
Стандарт-
Стандартное поли-
нозное во-
волокно
0,167-
0,133
36-40
26-34
4-6
8—12
8-12
2700—
5400
B70—540)
40-50
55-70
5
450-600
Высоко-
Высокопрочное
полиноз-
ное волок-
волокно
0,167 —
0,133
50-70
40-58
6-8
5-10
5-10
500-1200
F75-
1600)
55-60
3-5
600-800
ВВМ
0,143-
0,125
36-40
24—30
7,5-9
14-16
17-19
1600-
2400
A60-240)
45-55
65-80
8-10
400-450
Обычное
вискозное
штапель-
штапельное волок-
волокно
0,167
18-23
9-12
5-6
20-25
25-30
550-1000
E5-100)
50-55
88-93
12-18
300-350
Хлопковое
волокно
0,200-
0,143
32-52
34-54
21-23
10-12
13-15
950—2000
(95-200)
65
45
0
2000-
3000
1015
ПОЛИНОЗНЫЕ ВОЛОКНА
1016
эластичности, чем у высокопрочного вискозного корда.
Однако значение модуля в мокром состоянии у ВВМ
ниже, чем у П. в. Оба типа волокон следует отнести
к классу высокомодульных. В нек-рых странах П. в.
и ВВМ наз. модальными волокнами.
Структура и свойства. Специфич. свойства П. в.
определяются особенностями их физич. структуры
(наличием больших надмолекулярных структурных
единиц), характер к-рой зависит гл. обр. от условий
формования. П. в. имеют фибриллярную структуру,
подобную структуре хлопковых волокон. Характерная
особенность П. в.— устойчивость структуры к дей-
действию воды и щелочей, поэтому механич. свойства
этих волокон мало изменяются при обработке щело-
щелочами. Этим же объясняется стабильность формы воло-
волокон и их низкая сминаемость. Структура П. в. отли-
отличается от структуры обычных вискозных волокон на-
наличием многочисленных микропустот, к-рые позволяют
красителям и отделочным препаратам проникать в
глубь волокна.
Для П. в. характерна высокая прочность, низкое
относительное удлинение, высокий начальный модуль
в сухом и мокром состоянии. Эти волокна имеют пони-
пониженную степень набухания в воде, поэтому потеря
их прочности в мокром состоянии незначительна A5 —
20%). В таблице приводятся показатели свойств ПВ
в сравнении со свойствами ВВМ, обычных вискозных
и хлопковых волокон.
Большой модуль высокоэластичности П. в., особен-
особенно в мокром состоянии, является одним из важнейших
положительных свойств, обеспечивающих сохранение
размеров волокон и формы изделий, полученных из
них. Недостатки П. в.— высокая хрупкость и склон-
склонность к фибриллированию. Высокая хрупкость прояв-
проявляется при текстильной переработке и обусловливает
понижение коэфф. использования прочности волокна
в пряже. Прочность волокна в петле из-за хрупкости
невысокая (см. таблицу). В ряде стран проводятся
широкие исследования, направленные на получение
П. в. с пониженной хрупкостью.
Применение. П. в. успешно применяют для изготов-
изготовления широкого ассортимента тканей взамен тонково-
тонковолокнистого хлопка. Благодаря высокой прочности
П. в. в сухом и мокром состоянии, высокому модулю
высокоэластичности, а также возможности подвергать
их мерсеризации полученная из этих волокон пряжа
по свойствам приближается к пряже из гребенного
хлопка. Из П. в. получают тонкие ткани, обладающие
высокой стабильностью формы и размеров, хорошими
эксплуатационными свойствами, приятным внешним
видом, шелковистостью. Высокая устойчивость к дей-
действию р-ров щелочей имеет большое значение, посколь-
поскольку ткани, получаемые из П. в. в смеси с хлопком, под-
подвергаются обработке щелочным реагентом при мерсе-
мерсеризации. П. в. широко используют в смеси с различ-
различными синтетич. волокнами. Из П. в. как в чистом виде,
так и в смеси с др. волокнами вырабатывают сорочеч-
сорочечные, бельевые, плащевые, плательные, костюмные,
декоративные ткани, а также технич. ткани различного
назначения. П. в. применяют, кроме того, для полу-
получения бельевого и спортивного трикотажа.
Наибольшее развитие производство П. в. получило
в Японии (торговые названия гиполан, полино
и п о л и к о т), где в 1971 было выработано около
50 000 т этих волокон. В небольшом объеме полиноз-
ные волокна выпускаются также в США (з а н т р е л),
Великобритании (в и н ц е л) и др. странах Западной
Европы.
Лит.: Николаева Н. С, Конкин А. А., Хим.
волокна, № 5, 5 A962); Николаева Н. С, Ф и н к е л ь-
штейн Т. А., Хлопкоподобные вискозные волокна, М., 1965;
Drisch N., Melliand Textilberichte, №4, 389 A972); Ни-
Николаева Н. С., Могилевский Е. М., Маслен-
Масленников К. Н., веб.: Вискозные штапельные волокна, М.,
1973, с. 56; Б о ч к и н а Б. С. [и др.], там же, с. 120;
Herzog W., Lenzinger Berichte, № 31, 22 A971);
Graham C, Proc. Roy. Austral. Chem. Inst., 39, № 10, 305
A972). E. M. Могилевский, Н. С. Николаева.
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ*
АБС сополимер 194, 891, 919
Авиважная обработка волокон 539
Авкоеет 449
Адипинаты 623, 625, 626
Азеотропная поликонденсация 869
Акреал 929
Акрилан 707
Акриловые клеи — см. Полиакриловые
клеи
Акриловые лаки и эмали — см. Полиакри-
Полиакриловые лаки и эмали
Акрилонитрил, сополимер с метилметак-
рилатом 507
Акулон 727
Акцепторно-каталитическая поликонден-
поликонденсация 868
Алкидно-карбамидные лаки и эмали —
см. Мочевино-формальдегидные лаки и
эмали
Алкидные олифы 476, 477
Алкидные смолы 625
Алкидный лак 27
Алкидный линолеум 685, 687
Алкиларилфосфат 623
Алкиленсульфиды 718
Алкилсульфонаты 665
Алкилхлорсиланы 307
Алкилэпоксистеарат 623
Алкоголиз полимеров 402
Алкокс 430
Аллил-о-карборан 854
Аллилметакрилат 181
Аллил-а-цианакрилат 698
Аллофоам 570
Америпол GB 333
Америт 379
Америте 316
Амилан 727
к-Амил-а-цианакрилат 698
Аминолиз полимеров 402
Аминопласты 921
f-Аминопропилтриэтоксисилан 308
Аминоцел 316
Амоко AI 826
Амоко AI полимер 830
Аморфно-кристаллическое состояние по-
полимеров 236
Аморфные полимеры 234, 321, 518, 520,
523, 524, 547
— набухание 318
Амфион 177
Амфифильные макромолекулы 105
Амфотерные ПАВ 664, 666
Ангидрит 341
Анид 725, 727, 811
Анизотропия полимеров 527
сегмента 112, 124
Анилана 706, 707
Аниониты 440
Анионоактивные ПАВ 665
Анкалит 173
Антикодоны 394
Антикоррозионные материалы 963
Антимикробные перевязочные материалы
156
Антипирены 409
Антипластификация 633
Арабаны 577
Арабева 376
Арабоксиланы 577
Арахиновая кислота 144
Арахне 375 /
Арилокс 818, 819
Арилон 763
Арилхлорсиланы 307
Армированные пластики 238, 239, 529, 910
Армосель 559
* Отсылки, напечатанные курсивом, означают, что соответствующие статьи будут помещены в третьем томе энциклопедии.
Асбест 341, 345, 347
Асимплекс 177
Астрел 360, 763
Атактические полимеры 107
Атмосферостойкие пленки 648
Атмосферостойкость пигментов 603
Ауто-сет 316
Ацетобутиратцеллюлозная пленка 949
Ацетонитрил 380
Ацетохлорин 802
Ацидолиз полимеров 402
Аэролам 559
Аэросил 11, 341, 357
Б
Бакешт 573
Бактериальная ДНК 391
Бартрев 316
Бекурол 316
Белки 105, 119, 132
— синтез 394, 395
Белые пигменты 599
Бензилметакрилат 181, 185
Бензилоктиладипинат 623
Бензойная кислота 681
Бензонитрил 380
Бентонит 341
Бетолит 688
Бизли распределение 291, 294
Бинт эластичный 157
Биоассимилируемые полимеры 926
Биоинертные полимеры 926
Биополимеры 120, 132, 150, 286
Биполярные ионитовые мембраны 174
Бланфикс 341
Ближний конфигурационный порядок 108
Ближний конформационный порядок 111
Блок сопряжения 992
Блоклестничные полимеры 58
Боданил 727
Б оропластики 911
Брулон 812
Бумага 35 0
самокопирующая 255
Буна S 333
Бутадиен, сополимеры с акрилонитрилом
891
Бутадиен-винилпиридиновый каучук 928
Бутадиен-нитрильные каучуки 616
Бутадиен-нитрильные латексы 49, 53, 55
Бутадиеновый латекс 55
Бутадиен-стирольные каучуки
— вулканизаты 339
маслонаполненные 333, 334, 336
эмульсионные 336
Бутадиен-стирольные латексы 49, 51, 53, 55
Бутадиен-стирольные сополимеры 891
Бутацит 783
Бутвар 783
Бутен-1 456
Бутилбензилфталат 623, 625
Бутилизодецилфталат 623
Бутилкаучук, латекс 49, 55
изо-Бутилметакрилат 181, 185
н-Бутилметакрилат 181, 182
mpem-Бутилметакрилат 181, 185
Бутилоктилфталат 623
Бутилолеат 623
Бутилфталилбутилгликолят 623, 625
н-Бутил-а-цианакрилат 698
7-Бутиролактон 36, 37
Бутиронитрил 380
р-Бутоксиэтил-а-цианакрилат 698
В
Вакуумформование 584
6-Валеролактон 36, 37
Ведоил 209
Велвик 546, 613
Верел 707
Версамиды 727
Веслау распределение 291
Вестамид 824
Вестан 787
Вибрафоам 570
Виброизолирующие материалы 968
Виброползучесть полимеров 691
Виналон 797
Винидур 593
Виникорк 559
N-Виниладенин 751
2-Винилбензоксазол 750
2-Винилбензтиазол 7 50
М-Винил-1,2,3-бензтриазол 751
2-Винил-4,5-диметилоксазол 749
2-Винил-4,4-диметил-5-оксазолон 749
N-Винил-З-диметилпиразол 749
Винилиденфторид, сополимер с гексафтор-
пропиленом 407
Винилиденхлорид, сополимеры 649, 651,
802
N-Винилиндол 750
Винилит XYHL 783
Винил-о-карборан 854
8-Винил-2-меркаптобензтиазол 751
Винилон 797
Винилпиразин 750
Винилпиридиновые латексы 49, 53, 55
4-Винилпиримидин 750
2-Винилпиррол 749
2-Винилтиазол 749
]Ч-Винил-1,2,4-триазо л 749
Винилтрихлорсилан 308
Винилфенолы — см. Оксистиролы
Винилхлорид 456
— сополимеры 649, 651, 802
Винипласт 221, 222, 961
пленочный 808
Винипор 556, 558, 559
Винифоль 593
Винихлон 613
Вино л 797
Винцел 1016
Виньон 802
Випласт 546
Вискозные волокна 345, 347, 1013
Витасел 559
Витерит 341
Внутреннее трение 233
Водорастворимые волокна 797
Волокна как наполнители 348
Волокна медноаммиачные 157
полиакрилонитрильные 702
поливинилспиртовые 792
поливинилхлоридные 799
полинозные 1013
химические
модификация 275
отделка 538
Волокниты 921, 948
Волокно В, PRD-49 727
Вольпрюла 707
Вольтекс 376
Вторичная структура 119, 124
Вулкалент А 681
Вулканизационные структуры 120, 123
Вулкапрены 736
Вультекс 40
Вырубка 227
Высокомолекулярные ПАВ 664, 670
Высокотемпературная поликонденсация
870
Высокоэластическое состояние 235
Высокоэластичность полимеров 231
вынужденная 232
Вязкость полимеров 233
растворов и расплавов полимеров
894
Вязкотекучее состояние полимеров 235,
238
Вязкоупругое тело 233
Газонаполненные пластмассы 549
Газофазная полимеризация 892
Галтовка 14, 228
Гаусса распределение 291, 295
Гауссовы клубки 115
а-Гваяцилглицерин
— C-конифериловый эфир 68
Гедамин 316
н-Гексадецилметакрилат 181
н-Гексилметакрилат 181
Гектолеке 40
Гелион 727
Гелларидин 379
Гемацел 932
Гемовинил 932
Гемодез 932
Генетический код 392
Геркулес GN 379
Геркулоид 379
Германийсодержащие полимеры 366
Герметики 914, 968
микрокапсулированные 256
Гетерогенная полимеризация 891
Гетерогенные ионитовые мембраны 171, 173
Гетерополиконденсация 857
Гетероцепные полимеры
— обменные реакции 401
неорганические 367
Гетероциклические полимеры 60
Гетинакс 30, 221, 222, 229
феноло-формальдегидный 193
1017—1018
Гибкость макромолекул 117, 237
Гидроабразивная очистка поверхности 14
Гидроизоляционные материалы 963
2-Гидроксиэтилметакрилат 181
Гидродинамические характеристики мак-
макромолекул 116
Гидролиз полимеров 401
Гидролитическая полимеризация 31
Гидропескоструйная очистка поверх-
поверхности — см. Гидроабразивная очистка
поверхности
Гидросилилирование 304
Гидротропия 675
Гидрофильно-липофильный баланс 676
Гиполан 1016
Гипохромный эффект 389
Гипс 341
Гистоакрил 698
Гликолид 814
Глифталевая олифа 476, 477
Глицерин, триацетат 623
Глицидилметакрилат 181
Глобулы 48, 49, 51, 55, 103, 113, 124, 322
Глобулярные белки 119, 120
Глобулярные структуры 121
Гомогенные ионитовые мембраны 171
Гомополиамиды 728, 736
Гомопо ликонденсация 857
Гомополимеризация 884
Гомополимеры 100, 105, 108
Гомополиолефины 453
Гомоцепные неорганические полимеры
366, 371
Горение полимеров 408
Графит 345, 34 7, 366
Гребневидные полимеры 107, 124
Гризошаум 259
Гриламид 824
Грилон 727
Грунтование 13
Губчатые резины 44
Гука закон 231, 529
д
Дайнел 707
Дакрон 347
Дальний конфигурационный порядок 108
Дальний конформационный порядок 111
Данлоп G-60 40
Двухтяжевые макромолекулы 107
Дегидратированные масла 141
Дедерон 727
Дезоксирибонуклеиновая кислота (ДНК)
111, 150, 151, 382
денатурированная 390
Декаборан 366
Декорт 546
Дексил 855
Дексон 815
Деламин 316
Дельрина 1008
Дендритная структура 322
Деполимеризация 297, 299
Деструктивные реакции полимеров 401,
402
Деструкция полимеров 150, 299
механическая 215
Десульфурация химических волокон 538
Деформационная долговечность 690
Деформационные свойства полимеров
231, 526, 640, 689
Дефо-твердость 642
Дефо-эластичность 642
к-Децилметакрилат 181
Джеон 613
Диакон 209
Диалкилфталат 623, 789
2,4-Диамино-6-винилтриазин 750
Дибенол 546
Дибутилсебацинат 623
Дибутилфталат 623, 625
Дидодецилфталат 623
Диеновые полимеры 108
Диизотактические полимеры 107
Диизобутиладипинат 623
Диизобутилазелаинат 623
Диизодециладипинат 623, 625
Диизодецилфталат 623
Диизооктиладипинат 623
Диизооктилазелаинат 623
Дикаприлфталат 623
2-1Ч,]Ч-Диметиламин-4-винилпиримидин
750
1,3-бис- Диметиламиноизопропилметакри-
лат 751
1019-1020
Диметиламиноэтилметакрилат 181, 751
(8)-2,4-Диметилгексан 493
(8)-2,5-Диметилгептан 493
Диметилдихлорсилан 308
<8)-2,3-Диметилпентан 493
C,3-Диметил-3-пропиолактон 36, 37
Диметилфенилхлорсилан 308
Диметилфталат 623
Диметилхлорсилан 308
(8)-2,6-Диметилоктан 493
Дин 573
Динапор 558, 559
Динонилфталат 623
Диоктилсебацинат 623
Диплазоль 546
Дипольный момент 112, 495
Дисиндиотактические полимеры 107
Дисперсия оптического вращения 485
показателя преломления 500
Дитридецилфталат 623, 625
Дифенилдихлорсилан 308
Дифильные макромолекулы 105, 131
Дифлон 852
Дихлорфенилтрихлорсилан 308
Дициклогексилфталат 623, 625
Диэлектрические свойства полимеров 631
Диэпоксиды 422
Ди-B-этилбутил)азелаинат 623
Ди-B-этилгексил)адипинат 623, 625
Ди-B-этилгексил)азелаинат 623
Ди-B-этилгексил)фталат 623, 625
Диэтилдихлорсилан 308
о,?г-Диэтилтолуолсульфамид 625
Диэтилфталат 623
Диэтилхлорсилан 308
н-Додецилметакрилат 181
Долговечность полимеров 233, 690
Доломит 341
Дорил 710
Дралон 707
Древесная мука 345
Древесные пластики 921
Дуко 3 79
Дуросил 357
Дуфлекс 558, 559
Дютрал 334
Изопропенил-о-карборан 854
Изотактические полимеры 102, 107
Изошаум 259
Имидайт 771
Имплекс 209
Ингибирование полимеризации 887
Инжекционное прессование 63
Инжекционный механизм 85
Инициаторы отверждения 534
Инициирование полимеризации 886
Инсулфоум 5 73
Интерполимерные ионитовые мембраны
171, 172
Интерференционная микроскопия полиме-
полимеров 483
Интрузия 72
Иодинол 933
Ионак 177
Ионизирующие излучения, действие на
полимеры 240
Ионитовые мембраны 171
Иониты 942
Ионная полимеризация 295, 297, 884
— обрыв цепи 406
Ионогенные ПАВ 664, 665
Иономер 649, 651
Ионообменная емкость 443
Ионообменные пленки 648
Ипорка 259, 316
Испытания механические 231
Истмен-910 698
К
El, 5К
Евафильм 950
Желатинизация 544
Желатиноль 932
Желтые пигменты 599
Жесткость макромолекул 113
полимеров 232
«Живущие» полимеры 297, 578
Жидкие ионитов-ые мембраны 171
Жирные кислоты 145
Жиры растительные — см. Масла расти-
растительные
3, И
Загустители 42
Зайтел-31 727, 813
Зайтел-33 813
Зайтел-101 811
Зайтел-105 812
Закись-окись железа 599
Заливка компаундами — см. Литье ком-
компаундов
Зантрел 1016
Зародышеобразователи искусственные
266, 267
Защитные лакокрасочные покрытия 12
Звездообразные полимеры 107
Зеленые пигменты 599
Золь 573
Игамид А 812
Игелит 593
Износостойкость полимеров 234
Изоборнилметакрилат 181
Изобутеналь — см. Метакролеин
Изобутенамид — см. Метакриламид
Изоляционные материалы 973
Изомеризованные масла 141
Изомерия полимеров 107, 109, НО, 111
Изопрен 456
Изопреновые каучуки маслонаполненные
334
Изопреновый каучук, латекс искусствен-
искусственный 49
2-Изопропенил-4,5-диметилоксазол 749
Изопропенил-лг-карборан 854
Кабосил 357
Кадорит 559
Каландрование 584
Каларок 316
Калджитекс 156
Камфолоид 379
Канекалон 707
Каптон-Н S28
тгКаприллактам 31
Капринлактам 31
Капролактам 31
s-Капролактон 36, 37
Капролан 727
Капролон СН 239
Капрон 239, 725, 727, 927, 969
Капроновый корд 239
Карбанионы 99
Карбин 366
Карбкатионы 99
Карбоксилатные латексы 49, 53, 55
бис-D-Карбоксифенил)-лг-карборан 854
бис-D-Карбоксифенил)-о-карборан 854
бис-D-Карбоксифенил)-лг-карборанди-
гидразид 854
бис-D-Карбоксифенил)-л1-карборандихлор-
ангидрид 854
бис-D-Карбоксифенил)-о-карборандихлор-
ангидрид 854
Каобониевые ионы 416
.м-Карборандикарбоновая кислота 854
— дихлорангидрид 854
п-Карборандикарбоновая кислота 854
— дихлорангидрид 854
Карборансодержащие полиамиды 85 5
Карборансодержащие полимеры — см. По-
ликарбораны
Карборансодержащие полиэфиры 853
Карбоциклические полимеры 6 0
Каринекс 567
Карифлекс BR 333
Каром 333
Касторовая олифа 476, 477
Касторовое масло 139
Катализаторы металлоорганические 419
отверждения 534
окиснометаллические 444
Катиониты 44 0, 441
Катионоактивные ПАВ 666
Каучуки
— пластикация 614
— пластификаторы 625
бутадиен-нитрильные 616
бутадиен-стирольные 339
маслонаполненные 333,
334, 336
изопреновые маслонаполненные 334
маслонаполненные 332
наполненные лигнином 340
наполненные синтетическими пласти-
пластиками 338
наполненные сополимерами стирола
338
наполненные термореактивными
смолами 339
натуральный 65, 607, 615, 928
неорганический 368
пипериленовый 604
сажемаслонаполненные 335, 336
хлоропреновые 616, 632
этилен-пропиленовые маслонаполнен-
маслонаполненные 334
Кащмилон 706, 707
Квалитекс 40
Квиана 727
Келтан 334
Кельвина модель 135, 262, 263
Керра эффект 503
Кетгут 156
Киана 727
Кинофотопленки 648
Кислородные индексы 407
Кислоты Льюиса 416
Кларекс 209
Клевиль 802
Клежесель 558, 559
Клеи 915, 968
латексные 46
микрокапсулированные 255
полиакриловые 695
полиамидные 727
полиимидные 825
синтетические 947, 963
Клубок — глобула переход 124
Клупанодоновая кислота 141
Коацервация 250
Кодоны 394
Кожа искусственная 57, 544, 546
Кокосовое масло 139
Коллаген 120
Коллоксилин 377, 379
Колфоам 259
Компаунды полимерные 946, 975, 976
— литье 70
Комплементарности принцип Уотсона —
Крика 386
Комплементарность 151
Компрессионное прессование 83, 84
Конопляное масло 139
Контактное формование 966
Конфигурация звена 106
макромолекулы 103, 106, 108
Конформационные переходы 129, 130
Конформация звена НО
макромолекулы 104, НО, 112, 113, 114
Концевые группы 101
Координационная сфера 113, 115
Координационно-ионная полимеризация
295, 884
Кооперативноети параметр 127
Кооперативности эффекты 113, 114, 116,
119, 128
Корогель 546
Кор-фоум 564
Коттона — Мутона эффект 504
Кохинор 613
Коэффициент вязкости 233
полимеризации — см. Полимериза-
Полимеризации степень
трения 233
Крайовак S 787
Красители
— система на основе красителей 437, 442
микрокапсулированные 256
Красные пигменты 599
Красящая способность пигментов 599
Крафта точки 673
Крезилдифенилфосфат 623
Кремний четыреххлористый 3 08
Кремнийорганические компаунды 946
Кремнийорганические мономеры 302
Кремнийорганический каучук 927, 928
Кремона 797
Крип — см. Ползучесть полимеров
Кристакрил 209
Кристаллизация полимеров 323, 547
Кристаллизующиеся полимеры 518, 519,
523, 524
— набухание 320
Кристаллическое состояние полимеров
236, 527, 631
Кристалличности степень 323, 324
Критическая концентрация мипеллообра-
зования 664
Критическая объемная концентрация пиг-
пигмента 9, 602
Критическая опалесценция 125
Кровезаменители 931
Кровельные материалы 963
Крон молибдатный 599
свинцовый 599
цинковый 599
Крэмера-Лансинга распределение 291
Ксиланы 577
Ксифталевая олифа 476, 477
Кумулены 366
Куралон 797
Курехалон 787
Куртель 706, 707
м
л
Лавсан — см. Полиэфирные волокна,
а также 239, 927
Лавсановая нить 156
Лаки и эмали 9
Лаки масляные 147
меламино-формальдегидные 167
микрокапсулированные 256
мочевино-формальдегидные 310
на основе канифоли 147
¦ на основе копалов 148
перхлорвиниловые 588
полиакриловые 698
поливинилацетальные 779
поливинилбутиральные 780
поливинилформалевые 7 79
Лаковые масла 142
Лакокрасочные материалы 12, 10, 13, 24,
29, 915, 920, 947, 962, 970
— наполнители 340
— пигменты 597
Лакокрасочные покрытия 12
необрастающие 362
по дереву 24
по металлу — см. Защитные ла-
лакокрасочные покрытия (т. 1), Лакокра-
Лакокрасочные покрытия
по пластмассам 28
Лаконаливные машины 21, 26
Лактамы, полимеризация 30
Лактид 873
Лактоны
— полимеризация 33
— полимеры 36
Лаллеманциевое масло 139
Латекс натуральный 39
Латексные изделия 40
Латексные клеи 46
Латексные краски — см. Эмульсионные
краски
Латексы искусственные 48, 56
синтетические 48, 954
Лауринлактам 31
Лауриновая кислота 139, 141
Леавин 802
Лекарственные вещества полимерные 933
Лексан 852
Леофлекс 209
Лестничные полимеры 57, 108, 113, 115
Летилан 156
Лигнин 66, 340
Лигносульфоновые кислоты 69, 70
Ликановая кислота 139, 141
Ликра 1065, 1067
Лилион 727
Линейно-кристаллическая структура
макромолекулы 120
Линейно-упорядоченная структура мак-
макромолекулы 120
Линейные макромолекулы 101, 107, 113
Линейные полимеры 70, 368
Линолевая кислота 140, 141
Линоленовая кислота 140, 141
Линолеум — см. Покрытия для полов
Лиофильные золи 260
Лиофильные коллоиды 261
Лиофобные золи 259
Литература о полимерах 70
Литниковые системы 94
Литопон 147, 599
Литье компаундов 70
¦ под давлением 71, 584
реактопластов 76
резиновых смесей 77
термопластов 79
¦ центробежное — см. Центробежное
литье
Литьевое прессование 83
Литьевые машины 71, 72, 76, 77, 79, 93
для лабораторных и исследова-
исследовательских работ 89
для литья двухцветных изделий
из термопластов 88
для литья реактопластов 89
для литья резиновых смесей 89, 90
для литья термопластов 85
для полимерных материалов 85
Литьевые формы 91, 63, 79
Локфоам 570
Лоркасель 567
Л урон 812
Льняное масло 139
Люколен G 613
Люминесценция 502
Люсайт 209
Магнитооптические явления 504
Макание 545
Маканые изделия — см. Латексные изде-
изделия
Макроионы 99
Макролон 852
Макромолекула 99, 64, 132, 133, 237, 4 96
Макромолекулярная конформация 112,
114, 119
Макропористые ионообменные смолы —
см. Пористые ионообменные смолы
Макрорадикалы 133
Макросферы 617
Максвелла модель 135, 262, 263
Малеинизированные масла 143
Малеиновая кислота 137
— сополимеры 137
Малеиновый ангидрид 136
— полимеры и сополимеры 135
Маливатт 375
Малимо 375
Малиполь 375
Маранил 812
Марка — Хувинка уравнение — см. Вяз-
Вязкость характеристическая (т. 1)
Масла растительные 138
Маслонаполненные каучуки — см. На-
Наполненные каучуки
Маслостойкость полимерных материалов —¦
см. Бензо- и маслостойкость (т. 1)
Масляные краски 145
Масляные лаки и эмали 147
Масс-спектрометрия — см. Масс-спектро-
скопия
Масс-спектроскопия полимеров 149
Масштабное формование 551
Матирующие агенты 10
Матричная полиреакция 150
Матричная РНК 389
Матричный синтез ДНК 386, 395
Медицинские нити 155
Медноаммиачные волокна 157
Международная система единиц 160
Межфазная поликонденсация 164
Межцепной обмен полимеров 403
Меламино-алкидная эмаль 701
Меламино-алкидные лаки и эмали — см.
Меламино-формальдегидные лаки и эмали
Меламино-формальдегидные лаки и эма-
эмали 167
Меламино-формальдегидные смолы 167
Мембраны ионитовые 171
полимерные 930
разделительные — см. Разделитель-
Разделительные мембраны
Мерлон 852
Мерсеризация — см. Вискоза (т. 1)
Метакриламид 178
— полимеры 178
Метакрилатные компаунды 947
Метакрилат-М-З-оксиэтиладенин 752
Метакрилаты 180
— полимеры 180
Метакриловая кислота 186
— метиловый эфир — см. Метилметакрилат
— полимеры 186
Метакрилонитрил 188
— полимеры 188
Метакролеин 189
— полимеры 189
Металлизация пластмасс 191
Металлонаполненные пластики 194
Металлоорганические катализаторы 419
Металлопласт 197
Метальвин 779
Метеорологические оболочки 45
Метилакрилат 200
— полимеры 200
а-Метилакриловая кислота — см. Мета-
Метакриловая кислота
1,7-Метилаллил-лг-карборан 854
6-Метиламино-9-винилпу рины 751
1-Метилбутадиен-1,3 — см. Пиперилен
З-Метилбутен-1 202
— полимеры 202
Метилвинилдихлорсилан 308
Метилвинилпиридиновые каучуки — см.
Винилпиридиновые каучуки (т. 1)
Метилдифенилхлорсилан 308
Метилдихлорсилан 308
З-Метил-5-изопропенилпиразолин 749
Метил-о-карбораниленметилакрилат 854
Метилметакрилат 181, 182, 185, 203
— полимеры 203
Метилоксипропилцеллюлоза 213, 215
Метилоксиэтилцеллюлоза 213, 215
Метилолполиамиды 728, 743
1021 — 1022
4-Метилпентен-1 209
— полимеры 209
2-Метилпропеналь — см. Метакролеин
C-Метил-C-пропиолактон 36
Метилстиролы
— полимеры — см. Стирол, производных
полимеры
Метил-^-трифторпропилдихлорсилан 308
Метилтрихлорсилан 308
Метилфенилдихлорсилан 308
Метилфталилэтилгликолят 625
Метилхлорсиланы 303
Метил(хлорфенил)дихлорсилан 308
Метилцеллюлоза 211
Метил([3-цианэтил)дихлорсилан 308
Метилэтилцеллюлоза 213, 215
Метолоза 215
Метофас 215
Метоцел 215
Механическая деструкция полимеров 215
Механическая обработка пластмасс 219
246
Механические свойства полимеров 230, 525
Механопневмоформование 660
Механохимия полимеров 241
Микробаллоны — см. Микросферы
Микрокапсулирование 247, 936
Микрокапсулированные продукты 255
Микропористые резины — см. Кожа искус-
искусственная (т. 1)
Микросферы 617
Минорные основания 384
Мипора 258
Миристиновая кислота 140, 141
Мицелла 259
Мицеллообразующие ПАВ 664
Мовиль 802
Мовиталь В 783
Мовиталь F 799
Модакриловые волокна 702
Модальные волокна 1015
Модели релаксации механической 262
Модификация структурная полимеров 265
физическая полимеров — см. Моди-
Модификация структурная
химическая полимеров 269
химических волокон 275
Модуль 278
Молекулярная кибернетика 132
Молекулярная масса полимера 283 103
112
Молекулярная рефракция 495
Молекулярно-массовое распределение 286,
Молекулярные мицеллы 121
Мольтопрен 570
Моноволокно 300
Монокристаллы полимеров — см. Кри-
Кристаллическое состояние (т. 1)
Мономер 302, 857, 884
Мономерные звенья 100
Мономеры кремнийорганические 302
Мононить — см. Моноволокно
Монтопор 567
Морозостойкость полимерных материалов
309, 52, 241
Мочевино-алкидные лаки и эмали —• см.
Мочевино-формальдегидные лаки и эмали
Мочевино-формальдегидные лаки и эмали
310
Мочевино-формальдегидные смолы 310
Мусковит 341
«рН-Мускул» 131
Мутагенез 392
Мутность полимеров 498
Мьюлон 797
Мягкость полимеров 232
н
Набухание полимеров 317, 131
Набухания давление 318
Набухания степень 317
Надмолекулярная структура полимеров
320, 120, 237, 268, 518, 526
Наирит — см. Хлоропреновые каучуки
Найлон — см. Полиамидные волокна,
а также 347
BNS-Найлон 727
Найлон-3 725, 727
Найлон-4 725, 727
Найлон-6 727
Найлон-6,6 727, 811
Найлон-6,10 727, 813
Найлон-7 727
1023—1024
Найлон-9 725
Найлон-11 727
Найлон-12 727
Налфилм 177
Наполнение полимеров 325, 238
Наполненные каучуки 332
Наполненные пластмассы 634
Наполнители лакокрасочных материалов
340, 10
пластмасс 344
резин 351
Наполнители-пигменты 343
Напыление 357, 199, 252
Натрий-бутадиеновый каучук — см. Бу-
Бутадиеновые каучуки (т. 1)
Натрозол 449
Натуральный каучук — см. Каучук нату-
натуральный (т. 1)
Неионогенные ПАВ 664, 666
Необрастающие лакокрасочные покрытия
362
Необратимая поликонденсация 857, 859,
870
Неолан 1067
Неопрен — см. Хлоропреновые каучуки
Неорганические полимеры 363
Нептон 177
Неравновесная поликонденсация — см.
Поликонденсация
Нетканые изделия 371
Низкотемпературная поликонденсация 870
Ника темп 593
Никелевый катализатор 446
Ниплон 727
Нипол 333
Нипофлекс GF 950
Нити для изготовления перевязочных
материалов (средств) 155
для изготовления протезов трубчатых
органов 156
для изготовления шовных материа-
материалов 156
для прочих медицинских материалов
157
медицинские 155
поливинилспиртовые 796
Нитраты целлюлозы 377
Нитрилсилоксановые каучуки — см. Крем-
нийорганические каучуки (т. 1)
Нитрилы 380
— полимеры 380
Нитро-алкидно-карбамидный лак 25
Нитро-алкидно-эпоксидный лак 25
N-Нитрозодифениламин 681, 682
Нитролаки — см. Эфироцеллюлозные лаки
и эмали
Нитрон — см. Полиакрилонитрильные
волокна
Нитроцеллюлоза — см. Нитраты целлю-
целлюлозы
Нитроцеллюлозные лаки и эмали — см.
Эфироцеллюлозные лаки и эмали
Новолаки — см. Новолачные смолы
Новолачные смолы 381, 528
Номекс 727
Номекс М 977
Норакрил-65 929
Норил 818, 819
Норфлекс 1054
Нуклеиновые кислоты 381, 105, 120, 135
Нуклеозид 383
Нуклеотиды 383, 392
О
Обменные реакции гетероцепных полиме-
полимеров 401
Обратимые процессы полимеризации и
поликонденсации 297, 857, 859, 869
Обрыв цепи при полимеризации 405, 887
Объемные нити — см. Высокообъемные
нити (т. 1)
Огнестойкость полимеров 407
Озонное растрескивание — см. Озонное
старение
Озонное старение 410
Озоностойкость резин 412
Ойтисиковое масло 139
Окиси органические 413
— полимеризация 413
— полимеры 421
— сополимеры 423
Окислительная поликонденсация — см.
Дегидрополиконденсация (т. 1)
Окислительно-восстановительная ем-
емкость 433, 442
Окислительно-восстановительное иниции-
инициирование полимеризации -*- см. Иниции-
Инициирование полимеризации (т. 1)
Окислительно-восстановительные иониты
433, 438
Окислительно-восстановительные полиме-
полимеры 432
Окиснометаллические катализаторы поли-
полимеризации 444
Окисномолибденовый катализатор 445
Окиснохромовый катализатор 444
Окись пропилена 425
— полимеры 425
Окись этилена 414
— полимеры 427
Оксациклобутаны 414, 421
Оксетаны 421
Оксидирование 17
Оксидированные масла 143
бис-(Оксиметил)-лг-карборан 854
бис-(Оксиметил)-о-карборан 854
Оксираны 421
О ксистиро лы 446
— полимеры 446
бис-D-Оксифенил)-о-карборан 854
бис-D-Оксифенил)метан 846
2,2-бис-D-Оксифенил)пропан 847, 848
Оксицеллюлоза 155
Оксиэтилцеллюлоза 447
Оксоль 476
н-Октадецилметакрилат 181
Октилдециладипинат 623
н-Октилметакрилат 181
Олеиновая кислота 140, 141
Олефины 449, 453
— полимеризация 449
— полимеры 453
Олигоамиды 727
Олигомеризация 461
Олигомеры 457, 101
Олигоэфиракрилаты 468
Олифы 474
Оловоорганические полимеры 477
Омыления число 480
Омыления эквивалент 480
Опалесценция 498
критическая 125
Оптическая активность 503
Оптическая анизотропия 496
Оптическая микроскопия полимеров 481
Оптическая поляризуемость 495, 496
Оптическая чистота 485
Оптически активные полимеры 484
Оптические постоянные макромолекул
496
упруго растянутой сетки 497
Оптические свойства полимеров 494
Органическое стекло 504, 30, 206, 912,
962
Органоалкоксисиланы 306
Органоаминосиланы 306
Органоволокнит 510
Органогетинакс 513
Органозоли 543
Органосилазаны 306
Органоацилоксисиланы 306
Органохлоркремнийгидриды 302
Органохлорсиланы 302, 303, 306, 309
Органоциклосилоксаны 309
Ориентационная вытяжка 516
Ориентированное состояние полимеров
515, 236
Орлон — см. Полиакрилонитрильные во-
волокна, а также 347
Ортоноволаки 528
Ортотропные полимерные материалы 528
Осадительная ванна — см. Формование
химических волокон
Осадительная поликонденсация 870
Осмометрия полимеров 531
Отбелка химических волокон 539
Отвердители 533
Отверждение 535, 473, 533
Отделка покрытий 24, 27
химических волокон 538
п
Павилит 688
Пайр MS 826
Пальмитиновая кислота 140, 141
Пальмитолеиновая кислота 139, 141
Парафины 493
Параформ 1005
Парацианоген 995
Парилен С 873
Парилен N 873
Пасты полимерные 541 , 555
уплотнительные 941
Пачки 546, 322, 323
Пектаты 548
Пектинаты 548
Пектиновые вещества 548
Пектины 548
Пектинэстеразы 548
Пектовая кислота 548
Пеларгонлактам 31
Пенлайт 852
Пеногасит.ели 42
Пеногерметики 562, 564
Пенопласты 549, 562, 564, 913, 947, 954
Пенополивинилхлорид 555, 969
Пенополиолефины 559
Пенополиорганосилоксаны 561
Пенополистирол 564, 954, 969
Пенополиуретаны 567, 918, 947, 969
Пенополиэтилен 560
Пенофенопласты 570
Пеноэласт 556, 559
Пеноэпоксиды 573
Пентадиен-1,3 — см. Пиперилен
Пентапласт 575, 921
Пентафталевая олифа 476, 477
Пентафталевые лаки и эмали — см. Ал-
кидные лаки и эмали (т. 1)
Пентафталевые смолы — см. Алкидные
смолы (т. 1)
Пентозаны 577
Пентон — см. Пентапласт
Первичная структура макромолекулы 102,
106, 119
Передача цепи при полимеризации 578,
887
с разрывом 580
Переработка пластических масс 582
Переэтерификация масел 141
Перилловое масло 139
Перлон — см. Полиамидные волокна
Перлон Н 813
Перлон Т 812
Пермаплекс 177
Персистентная длина макромолекул 112,
124
Перспекс 209
Перхлорвиниловые лаки и эмали 588, 27
Перхлорвиниловые смолы 590, 802
Печать на полимерах 594
Пиатерм 259
Пивиацид 802
Пигменты лакокрасочных материалов
597, 9, 146, 147, 341
Пиолоформ В 783
Пиолоформ F 799
а-Пиперидон 31
Пиперилен 603
— полимеры 603
Пипериленовый каучук — см. Пиперилен,
полимеры
Пиридин 439
— полимер 606
2-Пиридинметакрилат 751
Пироксилин 377, 379
а-Пирролидон 31
Пирроны — см. Поли(ароилен-бис-бенз-
имидазолы), а также 765
Плавления температура 607
Плазмозаменители 931
Пластазот 561
Пластигели 543
Пластизоли — см. Пасты полимерные
Пластикат 609, 926
пленочный 808
Пластикация каучуков 614, 246
пластмасс 617
Пластики — см. Пластические массы
Пластики металлонаполненные 194
с полым наполнителем 617, 549
Пластификаторы 620, 10
Пластификационная вытяжка химически*
волокон 538
Пластификация полимеров 627
Пластические массы 633
металлизированные 191
Пластичность полимеров 232, 640
Пластмассы — см. Пластические массы
Пластограф Брабендера — см. Брабен-
дера пластограф (т. 1)
Пластометры — см. Пласто-эластические
свойства
Пласто-эластические свойства 640
Плейномеры — см. Олигомеры
Плексиглас 209
Плексигум 209
Плексидур 209
Пленка ЛМ 828
Пленка из нитратов целлюлозы 378
Пленки из оксиэтилцеллюлозы 448
из поликарбонатов 849
полиамидные 732
поли^инилиденхлоридные 784
поливинилхлоридные 807
полиимидные 826
полимерные 645, 198, 947, 949, 951,
952
Пленкообразование 11
при микрокапсулировании 247
Пленкообразующие вещества 652, 9, 138
Пленочный винипласт 808
Пленочный пластикат 808
Плюродаты 668
Плюроники 668
Пневмоформование 657
Поверхностно-активные вещества 663, 10,
42, 266
Податливость полимеров 678, 530, 689
Подвулканизация 679
Подсолнечное масло .139
Покровные слои лакокрасочных покрытий
13
Покрытия для полов полимерные 683, 959
Полвинит 613
Ползучесть полимеров 689, 233
Полиазапорфины 691
Полиазины 693, 995
Полиазоариленферроцен 997
Полиазоарилены 995
Полиакриламид — см. Акриламид, поли-
полимеры (т. 1)
По лиакри латный латекс 55
Полиакрилаты — см. Акрилаты, полимеры
(т. 1), а также 81, 82, 507, 891, 921
Полиакриловая кислота — см. Акрило-
Акриловая кислота, полимеры (т. 1)
Полиакриловые клеи 695
Полиакриловые лаки и эмали 698
Полиакрилонитрил — см. Акрилонитрил,
полимеры (т. 1)
Полиакрилонитрильные волокна 702, 345,
347
Полиакролеин — см. Акролеин, полиме-
полимеры (т. 1)
Полиалкиленарилены 707
Полиалкиленарилы — см. Полиалкилен-
Полиалкиленарилены
Полиалкиленгликольмалеинаты 710
Полиалкиленгликольфумараты 710
Полиалкиленсульфиды 718
Полиалкиленсульфоны 721
Полиалломеры 454, 455
Полиамид-6 649, 651, 731, 733, 737, 740
Полиамид-6,6 — см. Полигексаметиленади-
пинамид, а также 607, 733, 740
Полиамид-6,8 30, 813
Полиамид-6,10 — см. Полигексамети-
ленсебацинамид, а также 731, 740
Полиамид-11 733
Полиамид-12 — см. Полидодеканамид,
а также 731, 733
Полиамидирование 737, 859
Полиамидное волокно 345, 347
Полиамидные клеи 727
Полиамидные моноволокна 301
Полиамидные пластмассы 729
Полиамидные пленки 732, 949
Полиамидоарилаты 736
Полиамидокислоты 771, 829, 832, 833
Поли-[бис-(п-амидофенилен)-метиленан-
гидрид] 753
Полиамидоэфиры 735
карборансодержащие 855
Полиамиды 736, 625, 919, 921, 925, 926
— литье под давлением 81, 82
угольной кислоты — см. Полимо-
Полимочевины
Поли-п-аминостирол 437
Полиаминотриазолы — см. Политриазолы
Полиамины 747
Полиангидриды 752
Полиангидризация 752
Полиарилаты 755
Полиариленалкилены — см. Полиалки-
Полиалкиленарилены
Полиариленаминохиноны 995
Полиариленацетилены 993
Полиариленвинилены 993
Полиариленпиразолы 995
Полиариленсульфоны 760
Полиарилены 999
Поли(ароилен-бис-бензимидазолы) 764
Полиаценафтилен — см. Аценафтилен, по-
полимеры (т. 1)
Полиацены 64, 993
Полиацетали 891
Полиацетальдегид — см. Ацетальдегид,
полимеры (т. 1)
Полиацетилен — см. Ацетилен, полимеры
(т. 1), а также 993
Полиацетоксиметилен 783
Поли-7-бензил-Ъ-глутамат 111
Поли-К-бензилиденвиниламин 778
Полибензилметакрилат 182, 184
Полибензимидазолы 767
Полибензимидазопирролоны 765
Полибензоксазиноны 771
Полибензоксазолы 772
Полибензонитрил 993
Полибензтиазолы 775
Полибитиазолы — см. Политиазолы
Полибутадиен — см. Бутадиен, полимеры,
Бутадиеновые каучуки (т. 1), а также
58, 65, 891
Полибутен — см. Бутен, полимеры (т. 1),
а также 457
Поли-1Ч-бутилизоцианат 111
Поли-1Ч-бутилмалеинимид 64
Поли-изо-бутилметакрилат 182, 183, 184
Поли-к-бутилметакрилат 182, 183, 184
Поли-тпретп-бутилметакрилат 182
Поливиниламин 778
Поливинилантрацены — см. Винилантра-
цен, полимеры (т. 1)
Поливинилацетали — см. Ацетали поли-
поливинилового спирта (т. 1)
Поливинилацетальные лаки и эмали 779
Поливинилацетат — см. Винилацетат, по-
полимеры (т. 1), а также 625, 792, 891
Поливинилацетатные краски — см. Эмуль-
Эмульсионные краски
Поливинилацетатный латекс 55
Поливинилбензойная кислота — см. Ви-
нилбензойная кислота, полимеры (т. 1)
Поливинилбромид — см. Винилбромид,
полимеры (т. 1)
Поливинилбутираль 781, 780
Поливинилбутиральные лаки и эмали 780
Поливинилиденхлоридный латекс 55
Поли-4-винил-4', 4 '-бис-(диметиламино)-
трифенилкарбинол 43 7
Поли-4-винил-2' ,5'-диоксидифенил 437
По ЛИ-4-ВИНИЛ-2', 5' -диоксидифенилсульфон
437
ПоливинилB' ,5'-диоксифенил)сульфон 437
Поливинилдифенил — см. Винилдифенил,
полимеры (т. 1)
Поливиниленгликоль 783
Поливиниленкарбонат — см. Виниленкар-
бонат, полимеры (т. 1)
Поливиниленпиридины 995
Поливинилены 993
Поливинилиденфторид — см. Винилиден-
фторид, полимеры (т. 1), а также 407
Поливинилиденхлорид — см. Винилиден-
хлорид, полимеры (т. 1), а также 194,
407, 625
Поливинилиденхлоридные пленки 784
Поливинилиденцианид — см. Винилиден-
цианид, полимеры (т. 1)
Поли-6-винилизатин 437
Поливинилимидазолы — см. Винилимид-
азолы, полимеры (т. 1)
Поливинилимиды — см. Винилимиды, по-
полимеры (т. 1)
Поли-6-винилиндирубин 437, 440
ПолиF-винилиндол-3)-(тионафтен-2)ин-
диго 437
Поливинилкарбазол — см. Винилкарба-
зол, полимеры (т. 1)
Поливинилкетоны — см. Винилкетоны,
полимеры (т. 1)
Поли-2-винил-К-метилпиридон 440
Поливинилнафталин — см. Винилнафта-
лин, полимеры (т. 1)
Поливиниловые эфиры — см. Виниловые
эфиры простые, полимеры, Виниловые
эфиры сложные, полимеры (т. 1)
Поливиниловый спирт 787, 407, 437, 649,
651, 934
Поливинилоны 784
Поливинилпиридины — см. Винилпири-
дин, полимеры (т. 1)
Поливинилпирролидон — см. Винилпир-
ролидон, полимеры (т. 1)
Поливинилспиртовые волокна 792
Поливинилстеарат — см. Винилстеарат,
полимеры (т. 1)
Поливинилсульфофторид — см. Винил-
сульфофторид, полимеры (т. 1)
Поливинилсульфохлорид — см. Винил-
сульфохлорид, полимеры (т. 1)
Поливинилтиол 437
Поливинилтолуол — см. Стирол, произ-
производных полимеры
По ли-2-винилфентиазин 437
Поливинилфлуорен — см. Винилфлуо-
рен, полимеры (т. 1)
Поливинилформалевые лаки 779
Поливинилформаль 798
1025—1026
Поливинилфторид — см. Винилфторид,
полимеры (т. 1), а также 407, 649, 651?
Поливинилфуран — см. Винилфуран, по-
полимеры (т. 1)
Поливинилхлорид — см. Винилхлорид,
полимеры (т. 1), а также 81, 82, 193,
407, 590, 609, 625, 802, 891, 918, 921,
925, 928, 955
Поливинилхлоридные волокна 799
Поливинилхлоридные пластизоли 542
Поливинилхлоридные пластмассы 803
Поливинилхлоридные пленки 807, 649
949
Поливинилхлоридный латекс 49, 55
Поливинилхлоридный линолеум 684, 687
Поливинилциклоалканы — см. Винил-
циклоалканы, полимеры (т. 1)
Поливинилциклогексан — см. Винилцик-
лоалканы, полимеры (т. 1)
Поливинилциклопентан — см. Винилцик-
лоалканы, полимеры (т. 1)
Поливинилциклопропан — см. Винилцик-
лоалканы, полимеры (т. 1)
Полигалактуроназы 548
Поли-н-гексадецилметакрилат 182, 183, 184
Полигексаметиленадипинамид 810, 725,
727, 741
Полигексаметиленсебацинамид 812, 727,
731, 741
Поли-н-гексилметакрилат 182, 184
Полигерман 366
Полигидразиды 813
Полигидрохинонамин 43 7
Полигидрохинондиоксин 437
Полигидрохинонметилен 437
Полигидрохиноноксид 437
Полигидрохинонсульфон 440
Полигидрохинонтиазин 437
Полигидрохинонтиоксин 437
Полигидрохинонфенодитиазин 437
Полигликолид 814, 156
Полиглицидилметакрилат 182
Полиглюкин 932
Полиграфические краски 815, 597
Полидиалкил(арил)станнилены 477
Поли-]Ч-диалкилвиниламины 778
Полидекаметиленмочевина 1012
Поли-н-децилметакрилат 182
Поли-1Ч-диметилвиниламин 778, 779
Полидиметилсилоксан 111
Поли-2,6-диметил-п-фениленоксид 818
Поли-2,5-диоксиксилилен 437
Поли-3,4-диоксистирол 437
Поли-ди(сульфофенилен)гидрохинон 440
Полидифенилбутадиен-1,3 65
Полидифениленгидрохинон 43 7
Поли-4,4' -дифенилфталидангидрид 753
Поли(дихлор-л-ксилилен) 872
Поли-3,3-дихлорметилоксациклобутан 422
Полидихлорфосфазен — см. Полифосфо-
нитрилхлорид
Полидиэтиленгликоль-бис-амилкарбонат
507
Полидодеканамид 821, 727, 731
Поли-к-додецилметакрилат 182, 184
Полиены — см. Поливинилены
Полиизоборнилметакрилат 182
Полиизобутилен — см. Изобутилен, поли-
полимеры (т. 1), а также 55, 891
Поли-К-изобутилмалеинимид 111
Полиизопрен — см. Изопрен, полимеры,
Изопреновые каучуки (т. 1), а также
891
Полиизопрен, гидрохлорид 649
3,4-Полиизопрен 65
1,4-^ис-Полиизопрен
— латекс 55
Полиизоцианаты — см. Изоцианаты, поли-
полимеры (т. 1), а также 124
Полиилид 997
Полиимидазолы 824
Полиимидные волокна — см. Термостой-
Термостойкие волокна
Полиимидные клеи 825
Полиимидные пленки 826, 239
Полиимидоамиды 828
Полиимидоэфиры 830
Полиимиды 831
Полииндиго 437, 995
Полиионы 99
Поли-е-капроамид — см. Капролактам,
полимеры (т. 1), а также 725, 731, 741
Поликапролактам — см. Капролактам,
полимеры (т. 1), а также 239
Поликарбамиды — см. Полимочевины
Поликарбодиимиды 839
1027—1028
Поликарбонаты 840, 81, 82, 221, 222, 507,
649, 651, 919, 921, 926, 927
Поликонденсация 855, 297
в расплаве 865
в растворе 866
межфазная 164
на границе раздела жидкость — газ
166
на границе раздела фаз 254
Поликоординация — см. Координацион-
Координационные полимеры (т. 1), а также 859
Поликот 1016
Поли-п-ксилилены 871
Поликумулен 993
По ли-C-л актам 725
Полилактид 873
Полилактоны 38
Полималеинаты 710
Полималеинимид — см. Имиды непредель-
непредельных кислот, полимеры (т. 1)
Полималеиновая кислота 137
Полималеиновый ангидрид 136
По лимерана логичные превращения 874,
277
Полимербетон 882
Полимергомологи 883
Полимеризации степень 103
Полимеризация
— обрыв цепи 405
— передача цепи 578
в блоке — см. Полимеризация в массе
в массе 893, 891
в растворе 901, 891
в эмульсии — см. Эмульсионная по-
полимеризация
Полимерцемент 907
Полимеры 909
в авиастроении 909
в автомобилестроении 917
в машиностроении 920
в медицине 924
в пищевой промышленности 936
в радиоэлектронике 943
в сельском и водном хозяйстве 949
в строительстве 958
в судостроении 965
в электротехнике 973
на железнодорожном транспорте 981
Полимеры с длинноцепными ветвлениями
107
с короткоцепными ветвлениями 107
¦ со сдвоенной цепью — см. Лестнич-
Лестничные полимеры
с регулярной линейной сеткой —см.
Лестничные полимеры
с системой сопряжения 989
¦ элементов VI группы 366
Полиметакриламид 179
Полиметакрилаты — см. Метакрилаты, по-
полимеры, а также 82
Полиметакриловая кислота — см. Метак-
риловая кислота, полимеры
Полиметакрилонитрил — см. Метакрило-
нитрил, полимеры
Полиметакролеин — см. Метакролеин,
полимеры
Полиметаллиловый спирт 191
Полиметилакрилат 201
Полиметилбутен —см. З-Метилбутен-1, по-
полимеры
Поли-З-метилбутен-1 202
Поли-2-метил-5-винил-]Ч-бутилпиридин-
бромид 111
Поли-(8)-4-метилгексен-1 493
Поли-(8)-5-метилгептен-1 493
Полиметилен — см. Этилен, полимеры
Полиметиленоксид 1003
Полиметилметакрилат — см. Метилметак-
рилат, полимеры, а также 124, 194, 205,
221, 222, 229, 239, 407, 506, 507, 919,
925, 927
Полиметилметакрилат атактический 111
Поли-(8)-6-метилоктен-1 493
Полиметилпентен — см. 4-Метилпентен-1,
полимеры
Поли-(8)-3-метилпентен-1 493
Поли-4-метилпентен-1 209
Поли-а-метилстирол — см. Стирол, про-
производных полимеры
Полиметины — см. Поливинилены
Поли(монобром-п-ксилилен) 872
Поли(монометил-п-ксилилен) 872
Поли(монохлор-?1-ксилилен) 872
Поли(моноциан-п-ксилилен) 872
Полиморфизм полимеров 1009
Полимочевины 1010
Полинафтоиленаминогидрохинон 437
Поли(нафтоилен-бис-бензимидазобензо-
фенантролин) 58
Поли(нафтоилен-бис-бензимидазолы) 765
Полинитрилы — см. Нитрилы, полимеры
Полино 1016
Полинозные волокна 1013
Полинонаметиленмочевина 1012
Полиокс 430
Полиоксациклобутан 422
Полиоксигерман 366
2-Полиоксиметилен 1005
Полиоксистиролы 446
Поли-[бпс-(п-оксифенилен)триметиленан-
гидрид] 75 3
Полиоксиэтиленалкиламиды 667
Полиоксиэтиленовые эфиры 666, 667
Поли-н-октадецилметакрилат 184
Полиоктаметиленангидрид 753
Поли-к-октилметакрилат 182, 183, 184
Поли-ос-олефины 493
Полиорганосилоксаны — см. Кремнийор-
ганические полимеры (т. 1), а также
307, 925
Полиорганостанносилоксаны 478
Полиорганостанноксаны 478
Полиорганостаннометаллоксаны 479
Полипеларгонамид 725
Полипентамер 454, 455
Полипиперилены 604
Полипиразолохинон 440
Полипиридин — см. Пиридин, полимер,
а также 993, 995
Полипиримидин 58
Полипиридинийхлорид 997
Полипирролидонамид 725, 727
Полипорфиразины — см. Полиазапорфины
Полипропилен
— литье под давлением 81, 82
— механическая обработка 221, 222
— надмолекулярная структура 268, 321
— плавления параметры 607
— получение 891
— применение 919, 921, 925, 926, 927
— свойства 456
Полипропиленовые пленки 649, 651
Полипропиленоксид 422, 607
Поли-к-пропилметакрилат 182, 183
Поли-р-пропиолактон 38
Полирование лакокрасочных покрытий 24
пластмасс 227
Поли-]Ч-салицилиденвиниламин 778
Полисар Тактин 333
Полиселен 367
Полисера 367
Полиспирокеталь 58
Полистирол
— кислородный индекс 407
— лакокрасочные покрытия на нем 30
— литье под давлением 80, 81
— механическая обработка 229
— механические характеристики 194, 239,
241
— плавления параметры 607
— пластификаторы 625
— получение 891
— преломления показатель 507
— применение 921, 925, 926, 928
— структурные характеристики 64
Полистирол атактический 111
Полистирольные компаунды 946
Полистирольные пленки 649, 651
Полистирольный латекс 49, 55
Полисульфоны 763
Полителлур 367
Политетрагидрофуран 422
Поли-к-тетрадецилметакрилат 182
Политетраметиленангидрид 753
Политетрафторэтилен
— огнестойкость 407
— применение 919, 925
порошкообразный 345
Политетрафторэтиленовые пленки 649,
651
Политиоиндиго 437
Поли-п-тиолстирол 437
Поли(п-тиометил)стирол 43 7
Полиундеканамид 725, 727, 741
Полиуретановые компаунды 946
Полиуретановые лаки и эмали 27
Полиуретаны 625, 918
Полифениленаминогидрохинон 43 7
Поли-ж-фениленангидрид 7 53
Поли-п-фениленангидрид 753
Полифениленизоксазолы 995
Поли-м-фениленизофталамид 741
Полифениленоксадиазолы 995
Полифениленоксид 407
Поли-п-фенилены 999
Полифенилметакрилат 182
Полиферроценилен 997
Полиформальдегид — см. Полиметилен-
Полиметиленоксид, а также 81, 82, 607, 921
— плавления параметры 607
Полифосфонитрилхлорид 368, 997
Полифосфорная кислота 37 0
Полифталоцианин металлсодержащий
Полифумараты 710
Поли-Й-фурфурилиденвиниламин 778
Полихинолин 993
Полихлоргерман 366
Поли-[3,3-бис-(хлорметил)оксетан] —
см. Пентапласт
Поли(п-хлорметил)стирол 440
Полициклогексилметакрилат 182, 184
Полициклоконденсация 857
Полициклопентадиен 64, 993
Полишиффовы основания 995
Полиэлектролиты 105, 111, 123, 131
Полиэлементовинилены 997
Полиэнантоамид 725, 727, 741
Полиэтерификация 859
Полиэтилен 134
— кислородный индекс 407
— лакокрасочные покрытия на нем 30
— литье под давлением 80, 81
— механическая обработка 221, 222
— механические характеристики 239
— плавления параметры 607
— получение 891
— применение 919, 921, 925, 926, 927, 928
хлорированный 407
хлорсульфированный 55, 270
Полиэтиленгликоли 430
Полиэтиленгликольадипинат 62,3
Полиэтиленгликольсебацинат 623
Полиэтиленовые пленки 649, 651, 949, 952,
955
Полиэтиленовый латекс 55
Полиэтиленоксид — см. Окись этилена,
полимеры, а также 407, 422, 607
Полиэтиленсульфид 720
Полиэтилентерефталат 30, 649, 651
Полиэтилентерефталатные моноволокна
301
Поли-[бис-(этилен)-п-фениленангидрид]
753г
Поли-[бг?с-(этилен)-2,5-фуриленангидрид]
753
Поли(этил-п-ксилилен) 872
Полиэтилметакрилат 182, 183, 184, 507
Полиэфиракрилаты — см. Олигоэфиракри^
латы
Полиэфирмалеинаты 710
Полиэфирная смола 221, 222, 712
Полиэфирное волокно 347
Полиэфирные моноволокна 301
Полиэфирный лак 27
Полиэфироамидокислоты 830
Полиэфирсульфоны 761
Полиэфирфумараты 710
Полиэфиры 622, 623, 625
карборансодержащие 853
Полулестничные полимеры —см. Блоклест-
ничные полимеры
Поляризационная микроскопия полимеров
481
Поляризуемость 495, 49G
Поляроидные пленки 648
Помутнения точки 67 4
Порозаполнители 25
Поролон 570
Поропласты 549
Предел вынужденной высокоэластичности
232
прочности 233
текучести 232
Предпластикатор 86
Предполимеры 459
Преломления показатель 499, 507
Пресс трансферный 83, 84
Прессматериалы 912
Прессование 584
литьевое 83
резиновых смесей 78
Привитые ионитовые мембраны 171, 172
Прозрачность полимеров 498, 949
Прокладочные материалы 968
Проксамины 669
Пролонгаторы 932
Промывка химических волокон 538
Пропенкарбоновая-2 кислота — см. Метак-
риловая кислота
Пропиленоксид — см. Окись пропилена
Пропиленсульфид 718
изо-Пропилметакрилат 181, 185
н-Пропилметакрилат 181, 185
н-Пропил-а-цианакрилат 698
|Ь-Пропиолактон 36, 37
Пропионитрил 380 Сажемаслонаполненные каучуки 335, 336 1029—1030
Пропомины 667 Саженаполненные каучуки 335, 336
Пространственно-сетчатые структуры 120, Салициловая кислота 681, 682
122, 123 Самикон М 613 Сульфан 369
Протакрил 929 Сантогард PVI 681 Сульфидолиз полимеров 403
Протонные кислоты 415 Сантофоум 567 Суспензионная полимеризация 892
Протопектины 548 Санфоум 561 Сутрон 812
Прочностные свойства полимеров 233, 239, Саран 787, 802 Сушка покрытий 22, 23, 26, 27
530, 630 Сверление пластмасс 226 химических волокон 540
Прочность жесткоцепных полимеров 331 Сверхсопряжение 124 Сферолит 321, 323
ориентированных полимеров 526 Светопропускание 498, 499 «Сшивки» между макромолекулами 277
Пуассона распределение 291, 295 Светостойкость пигментов 603 Сшитые макромолекулы 101
Себацинаты 623, 626
Р Сегментальная анизотропия 112, 124 m
Седжицелл 155 'I'
Селемион 177 -*-
Радикальная полимеризация 293, 884 Селлобонд 718
— обрыв цепи 405 Серум 39, 51 Тактичность 102, 108
Радиационная стойкость 240 Серые пигменты 599 Талловое масло 139
Разветвленные макромолекулы 101, 107 Сеточная модель строения полимеров 517 Тальк 341, 345
Размер макромолекул 116 Сетчатые макромолекулы 109 Танга распределение 291
Распыление лакокрасочных материалов Сидонур 316 Тангенс угла механических потерь 281
17, 18, 19, 20, 26 Сизаль 345, 347 Тафтинг-ковры 375
паст полимерных 546 Сиккативы 10 Твердость полимеров 233
Рассеяние света 502 Сикрон 613 Твердофазная полимеризация 892
Растворимость полимеров 116 Силанолы 306 Тевирон 802
Растворители лакокрасочных материалов 10 Силастик 564 Тейнохимический принцип 131
Растрескивание полимеров 239 Силен 357 Текстолит
Растяжение полимера 521 Силикондисульфид 369 — лакокрасочные покрытия по нему 30
Реактопласты 634, 637 Силтег 357 — механич. обработка 222, 229
— литьевое прессование 83 Синдиотактические полимеры 102, 107 — огнестойкость 407
— литье под давлением 76 Синие пигменты 599 Текучесть полимеров 233, 628
— машины для литья 89 Синтактические пены — см. Пластики Теломеризация 461, 579, 888
— механич. обработка 229 с полым наполнителем Теплостойкость полимеров 240
— переработка 585 Синтез белка 394, 395 Тергиталы 668
— применение 910 ДНК 395, 396 Термические свойства ориентированных
Реакторы вытеснения 895, 900 линейных сополимеров 277 полимеров 527
смешения 895, 900, 904 нуклеиновых кислот 396 Термодинамика структурных переходов
Ребиндера эффект 240, 675 привитых сополимеров 275 в макромолекулах 124
Ревертекс Т 40 Скорчинг — см. Подвулканизация Термокаталитическое силилирование 304
Ревертекс стандарт 40 Слюда 341, 345 Термообработка химических волокон 540
Ревиль В 783 Слюдотерм 977 Термопластичный органоволокнит 512
Ревиль F 799 » Совиден 802 Термопласты 584, 634, 636
Ревультекс 40 Совиолы — см. Сольвары — литье под давлением 79
Регенерация окислительно-восстановитель- Соевое масло 139 — машины для литья 859
ных полимеров 440 эпоксидированное 623, 625 — механич. обработка 22
Редокс-иониты 433, 439, 441 Соли АГ 811 _ переработка 585
Редокситы — см. Окислительно-восстано- Солпрен 334 — применение 912
вительные полимеры Сольвары 789 Термореактивный органоволокнит 510
Редокс-полимеры 432, 435, 442, 443 Сольвитерм 593 Терморегулирующие лакокрасочные по-
Редупликация ДНК полуконсервативная Солюбилизация 674 крытия 916
387 Сополиамиды 728 Термостойкость полимеров 241
Резиновая обувь 45 Сополиконденсация 857 Термоэластопласты 237
Резиновые клеи 46 Сополимеризация 884, 889 Тетрагидропиран 414
Резиновые нити 46 Сополимеры 100, 104, 105 Тетрагидрофуран 414
Резиновые перчатки 45, 46 Сорбланит 567 Тетрагидрофурфурилолеат 623
Резиновые смеси Сотопласты 913 н-Тетрадецилметакрилат 181
— литье под давлением 77 Софтлон 561 Тетраоксан 1004
— машины для литья 89, 90 Спандекс 157 Тетроники 669
— подвулканизация 679 Спектрополяриметрия 492 ' Тиираны — см. Алкиленсульфиды
— пласто-эластические свойства 640 Спираль — клубок переход 127, 130 Тиксотропные лаки и эмали 11, 13
Резины Спиральная конформация 111 Тилоза 215
— механич. характеристики 239 Спиральная упорядоченность 120 Тиснение горячее 194
— наполнители 351 Спирополимеры 57, 58, 107 красочной или металлизированной
— озоностойкость 412 Стайкаст 620 пленкой 596
— применение 913, 919, 937 Стальные волокна 347 Токолит 379
Рейон. 347 Старение полимеров 239 Тоуфлон 852
Релаксации механической модели 262 Статистический клубок 103, 111, 113, 114 Транскрипция РНК 387
Релин 686, 687 Стационарности принцип 888 Транслокация 395
Релон 727 Стеариновая кислота 140, 14J Трансляция (белковый синтез) 395
Релсин 727 Стеклование полимеров 628 Транспортная РНК 389
Ренофлекс 593 Стеклообразное состояние полимеров Трансферное прессование — см. Литьевое
Реополиглюкин 932 . 234, 237 прессование
Рестичел 567 Стеклопластик 30, 769, 909, 910, 919, 921, Трансферные цилиндры 85
Рефракция молекулярная 495 949, 962, 965, 966 Траубе — Дюкло правило 671
Рибонуклеиновые кислоты (РНК) 150, 151, Стеклопластик СВАМ 239 Третичная структура 119, 120, 132
382 Стеклотекстолит 229, 272 Трехмерные полимеры 370
Рибосомная РНК 389 Стеклянное волокно 345, 347 Триалкилоксониевые соли 415
Ригизоли 543 Стереорегулярная макромолекула 102 Трибензоатцеллюлоза 111
Рильсан А 824 Стереоизомерия 107, 109 Трибутилфосфат 623
Рильсан 727 Стереон 334 Трикрезилфосфат 623, 625
Рицинолевая кислота 139, 141 Стереорегулярности степень 102 Триметилхлорсилан 308
Ровиль 802 Стереорегулярность 237 Тричитроцеллюлоза 111
Ровиналь В 783 Стереоселективная полимеризация 489 Триоксан 1004
Ровиналь F 799 Стереоспецифические процессы 490 Триоктилтримеллитат 623
Родиа-найлон 727 Стереоэлективная полимеризация 489 Трифенилфосфат 623 625
Родэстер 718 Стилон 727 Трифенилхлорсилан 308
Рост цепи 887, 890 Стирол 456 Трифторацетонитрил 380
Ротационное формование 546 Стирол-бутадиеновый латекс 49, 55 777~тРиФтоРпРопилтРихлоРсилан 308
Роулинсона <?>2-точка 116 Стиролизованные масла 144 Трихлормеламин 681
Рутил 147 Стиромуль 567 Трихлорсилан 308
Руфмейт 567 Стиропор 567 Три-B-этилгексил)фосфат 623
Стиросел 567 Триэтиленгликоль-ди-B-этилбутират) 623
|1 Стирофоум 567 Триэтиленгликоль-ди-B-этилгексоат) 623
\J ?труйное формование 76 Тровидур 593
^Структура макромолекулы 101, 106, 112, Тровипор 558, 559
Сажа белая 341, 351 119, 120, 121, 122, 132 Тропоколлаген 111
.газовая 345 надмолекулярная 518, 526 Трубы из полимерных материалов 956, 957,
как наполнитель резин 351 Структурные переходы 124, 127, 129 964
как пигмент 147, 599 Ступенчатый синтез олигомеров 465 Тунговое масло 139
1031—1032
У, Ф
Углеродные волокна 345, 347
Углеродопласты 911
Ударная вязкость 233
Укрывистость пигментов 598
Ультразвук, облучение растворов полиме-
полимеров 246
Ундекан 727
Ундеканлактам 31
Упаковочные материалы 938, 954
Упаковочные пленки 647
Уплотнительные материалы 967
У плотните льные пасты 941
Упрочнение полимеров 330
эластомеров 330
Упруговязкие полимерные жидкости 233
Упругость полимеров 231
У рак резин 316
Уретановые масла 144
Урилон 1013
Усадка полимеров 640
после литья 80, 82
Усталостная прочность 233
«Усы» 347
Фазовые состояния полимеров 323
Фактисы 627
Фафф 259
Фенекпон 573
Фенилдихлорсилан 308
Фенилметакрилат 181, 185
Фенилтрихлорсилан 308
Фенилхлорсиланы 303
Фенодюр 573
Фенолиз полимеров 402
Фенолит 573
Феноло-формальдегидная смола 239
Феноло-формальдегидный гетинакс 193
Фенопласты 229, 919, 921, 989
Ферроцен 437, 439
Фестопас 316
Фибриллы 524, 525
Фибриллярная структура 322
Фибровиль 802
Фи-ви 558, 559
Фиталак 316
Флори параметр 117
Флори принцип 298, 875
Флори распределение 291, 294, 295
Фоамекс 570
Формадур 779, 799
Формасет 316
Формвар 799
Формекс 779
Формование
— вакуум- 583
контактное 966
масштабное 551
пневмо- 657
ротационное 546
химических волокон 702, 723
Формоустойчивость химических волокон
540
Формтеналь 779
Форполимеры 459
Фосфатирование 16
Фотоупругость полимеров 504
Фракционирование олигомеров 460
Фрезерование пластмасс 226
Фреоны 574
Фрикционные свойства полимеров 233
Фталаты 621, 623, 626
Фталевый ангидрид 681, 682
Фторволокно 347
Фторкаучук 407
Фторопласт-4
— лакокрасочные покрытия по нему 30
— механич. обработка 221, 222, 229
— применение 926, 927
Фторопласты
— получение 891
— применение 921
Фторсодержащий латекс 55
Фтортензиды 664, 670, 676
Фумаровая кислота 137
— сополимеры 137
Фурилметакрилат 181
х, ц, ч, ш
Хайпалон 270
Хайсил 357
Характеристическая вязкость 112
Хемилюминесценция 502
Хи этилен С 561
Хлопковое волокно 345
Хлопковое масло 139
Хлопкоподобные волокна — см. Полиноз-
ные волокна
Хлорин 157, 802
Хлорлигнин 69
Хлорметилдиметилхлорсилан 308
Хлорметилдихлорсилан 308
3,3-бис-(Хлорметил)оксетан 575
а,а-бис-(Хлорметил)-C-пропиолактон 36
Хлорметилтрихлорсилан 308
Хлоропреновые латексы 49, 55
Хлоропреновые каучуки 616, 632
Хлорпарафины 623
Хлорфенилтрихлорсилан 308
Хостаформ С 1009
Хрупкость полимеров 233
Цветность полимеров 501
Целакол 215
Целкон 1009
Целлозайс 449
Целлофан 649, 928
Целлофас 215
Целлюлоза
— ацетат 625
— ацетобутират 507, 625, 649, 919, 949
— ацетопропионат 649
— диацетат 649
— метиловый эфир 211
— монофенилацетат 111
— нитраты 377, 625
— триацетат 649, 651
— тринитрат 378
как наполнитель 345
Цетилоксибензойная кислота
— полифенилметакриловый эфир 184
Циакор резин 316
Цианакрилатные клеи 697, 927
Цианобонд-5000 698
(З-Цианэтилтрихлорсилан 308
Циклогексилметакрилат 181, 185
N-Циклогексилтиофталимид 681, 682
Циклоолефины
— полимеры 454
Циклоолигомеры 466
Циклополибутадиен 65
Циклополиизопрен 65
Циклосилоксаны 306
Черные пигменты 599
Четвертичная структура макромолекул 119
Шейка 520
Шишковского уравнение 671
Шлифование лакокрасочных покрытий 24
пластмасс 227
Шпатлевки 13, 26, 29
Шпредингование 546
Штапельные волокна 157, 795
Шульца распределение 291, 294
Эарекс 558, 559
Эдифас 215
Экзальтация рефракции 495
Эккосток 620
Эккофлоут 620
Эккофоумз 620
Эксонайт 379
Экструзия полимеров 546, 550, 556, 584,808
Эластичность 640
Эластомеры
— упрочнение 330
микрокапсулированные 256
Эластофоум 558, 559
Электрические модели релаксации меха-
механической 264
Электроизоляционные пленки 647
Электроноионообменники 433
Электронообменники 432
Электростатическая коагуляция 254
Элеостеариновая кислота 139, 141
Элмопласт 316
Эмали — см. Лаки и эмали
Эмали масляные 147
меламино-формальдегидные 167
мочевино-формальдегидные 310
перхлорвиниловые 588
полиакриловые 698
поливинила детальные 779
поливинилбутиральные 780
Эмульсионная поликонденсация 870
Эмульсионная полимеризация 52, 891
Энант 727
Энантолактам 31
Энергия связи 364, 371
Энергия сопряжения 990
Энтернамид 812
Энтернамид С 813
Эпоксидированные масла 144
Эпоксидированные соединения421, 623, 625
Эпоксидные клеи 947, 963, 969
Эпоксидные компаунды 946
Эпоксидные смолы 573, 625
Эпоксипласты 921
1,2-Эпоксипропан — см. Окись пропилена
Эпоксиэтан — см. Окись этилена
Эрозионностойкие лакокрасочные покры-
покрытия 916
Эруковая кислота 139, 141
Этакрил 929
Эталоза 449
Этафоум 561
2-Этилгексилдифенилфосфат 625
2-Этилгексилметакрилат 181
2-Этилгексилэпокситаллат 623
Этилдихлорсилан 308
Этилен
— окись 414, 427
блоксополимеры 668
полимеры 427
— сополимер с винилацетатом 649, 651
— сополимер с пропиленом 891
Этиленгликольдиметакрилат 181
Этиленоксид — см. Окись этилена
Этилен-пропиленовые каучуки маслонапол-
ненные 334
Этилен-пропиленовый каучук 928
Этиленсульфид 718
Этилметакрилат 181, 182, 185
Этилтрихлорсилан 308
Этилфталилэтилгликолят 623
Этилхлорсиланы 303
Этилцеллюлоза 625, 649
Этил-а-цианакрилат 698
Этодуомины 667
Этомины 667
Эфирное число 480
Юпилон 852
Энциклопедия Полимеров. Ред. коллегия: В. А. Кабанов
(глав, ред.) [и др.] Т. 2— М., «Советская Энциклопедия», 1974.
(Энциклопедии. Словари. Справочники),
т. 2. Л-И. 1974. 1032 стб. с илл.
Сдано в набор 23/XI 19 73 г. Подписано к печати 20/Ш 1974 г.
Издательство «Советская Энциклопедия». 109817. Москва, Ж-28, Покровский бульвар, д. № 8.
Т-02182. Тираж 3 5 000 экз. Заказ № 2720. Бумага типографская № 1. Формат бумаги 84xlO8Vie- Объем 32,25 физич. п. л.;
54,18 усл. п. л. текста. 93,5 уч.-изд. л. Цена книги 4 р. 40 к.
Московская типография № 2 «Союзполиграфпрома» при Государственном комитете Совета Министров СССР по делам издательств,
полиграфии и книжной торговли, Москва И-85, Проспект Мира, 105.