/









































































































































































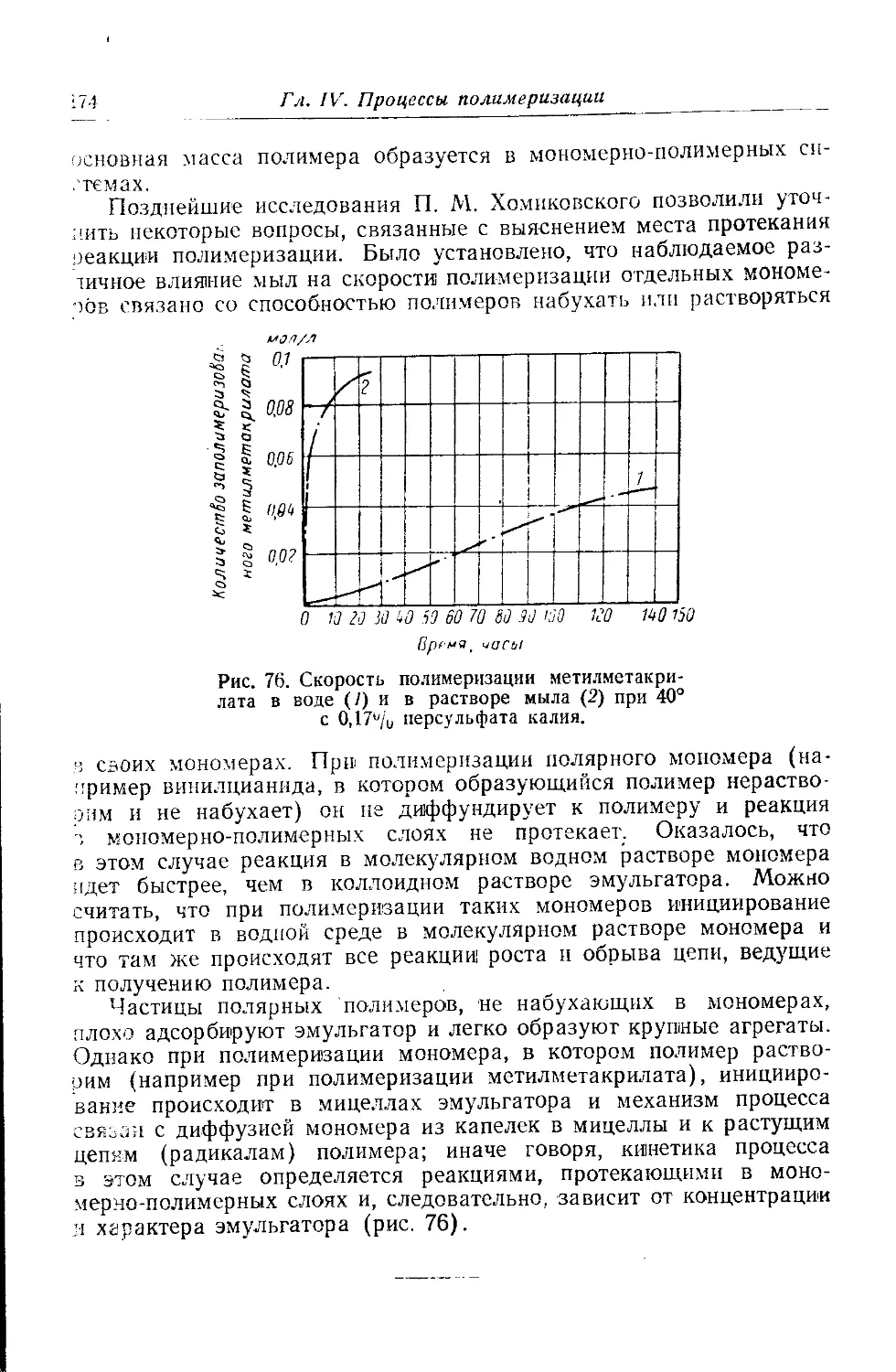







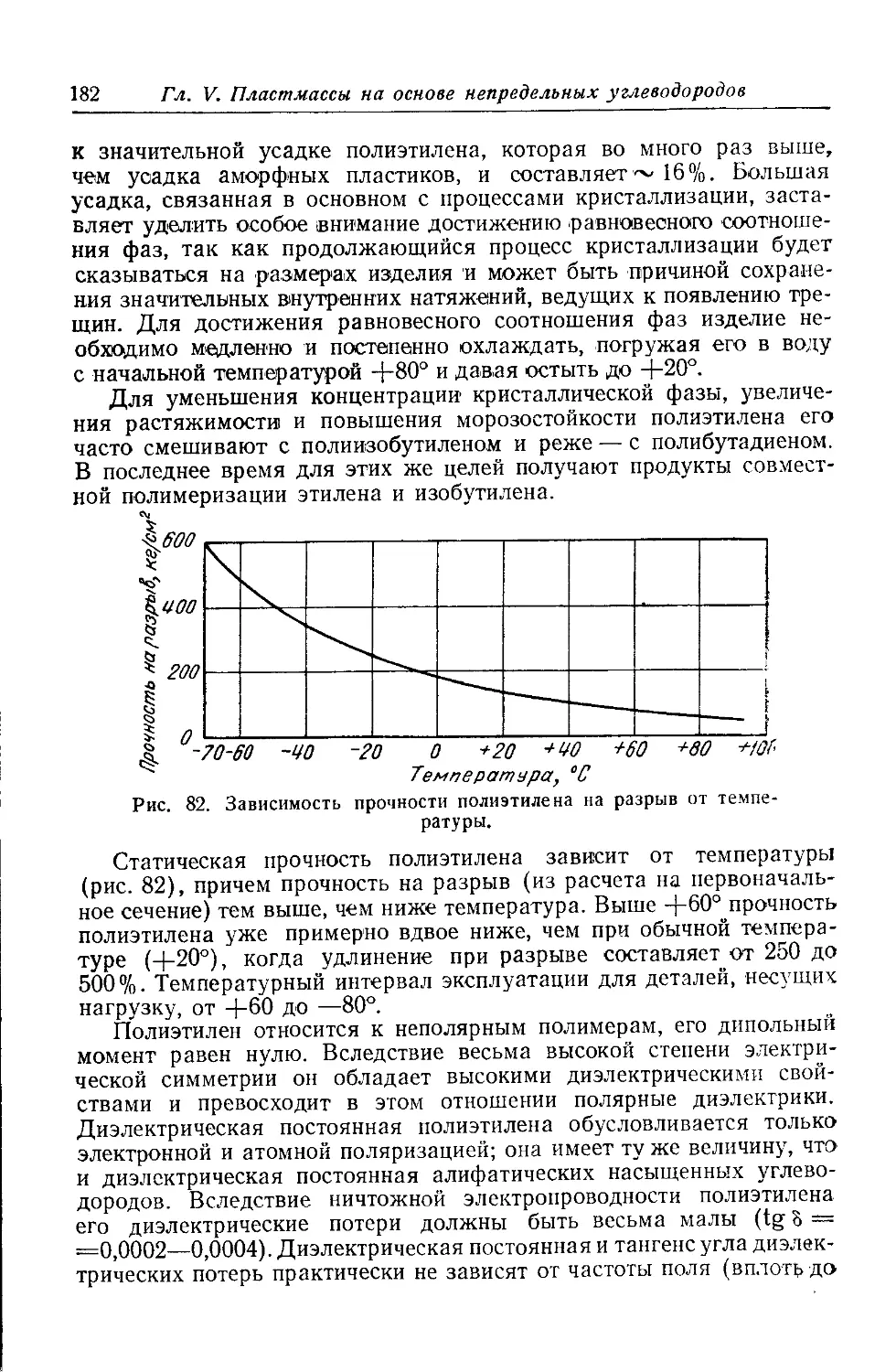



































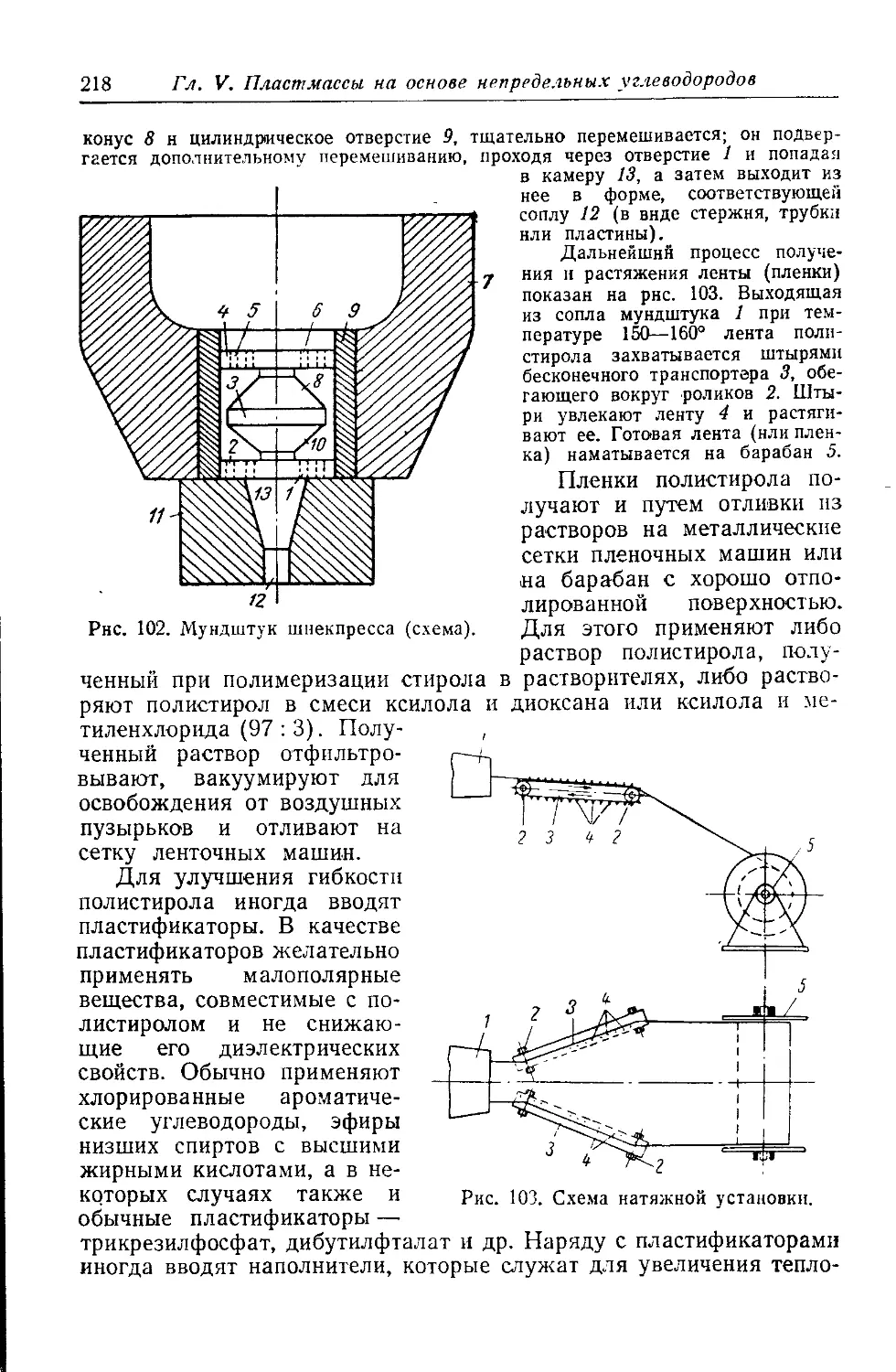















































































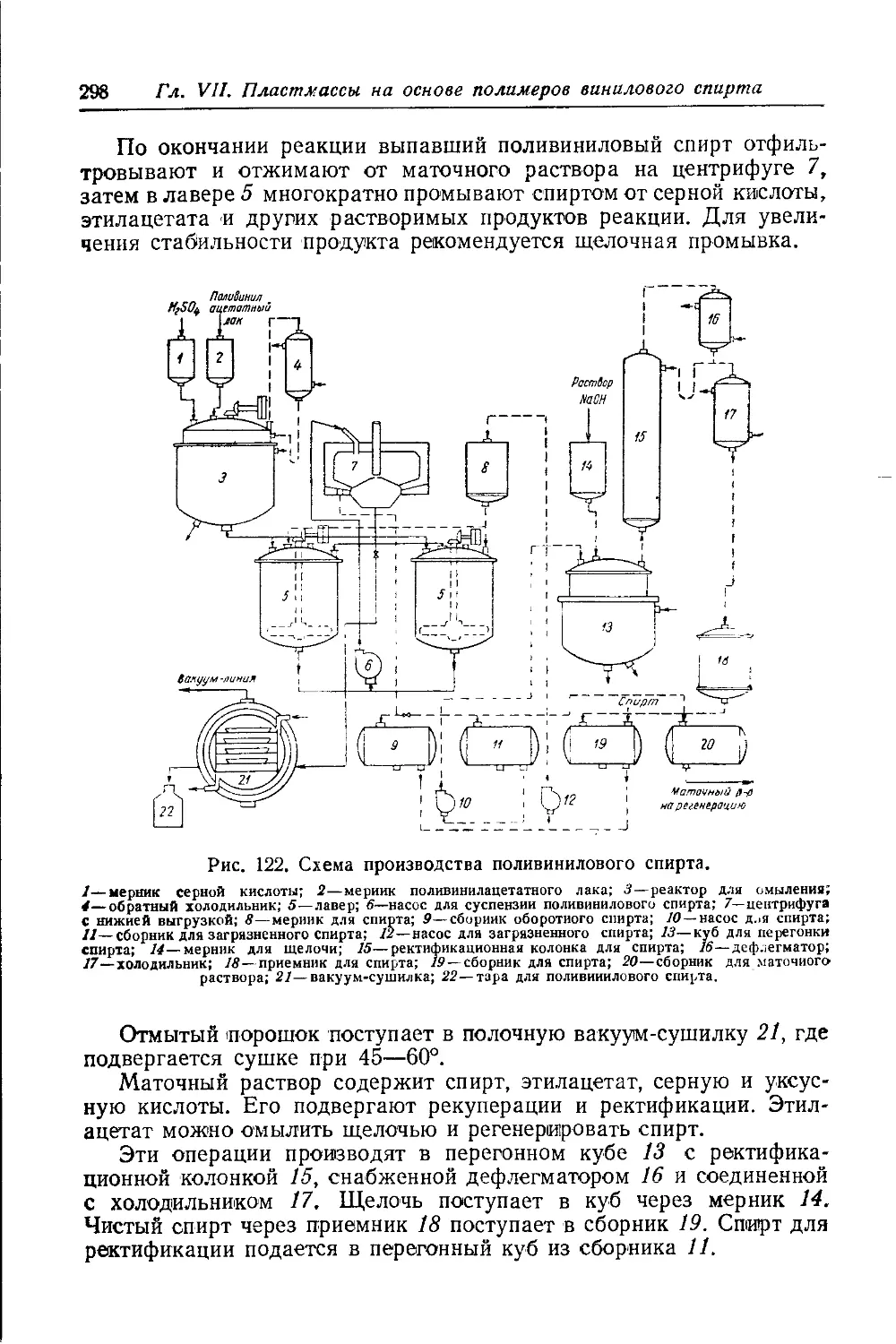
















































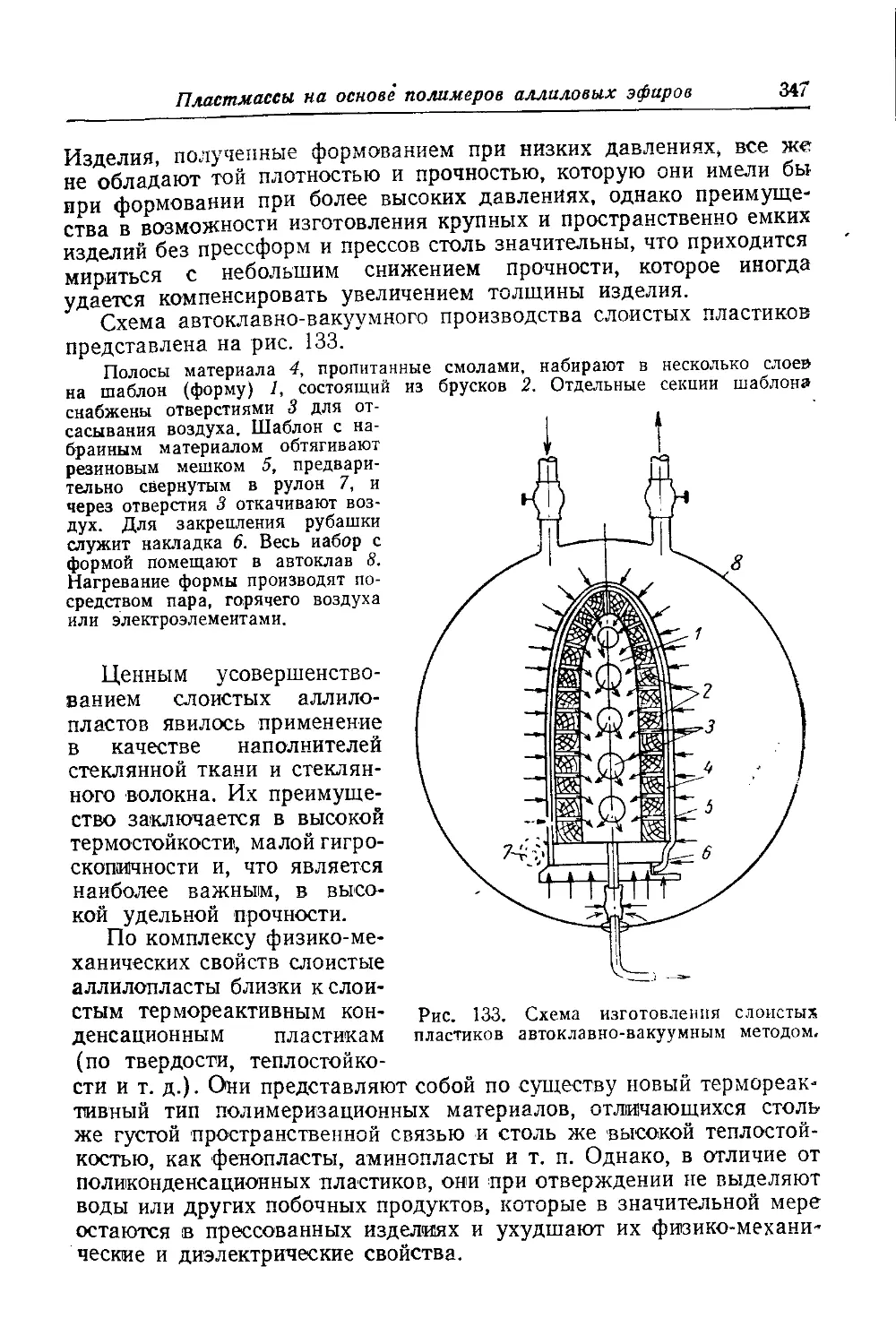








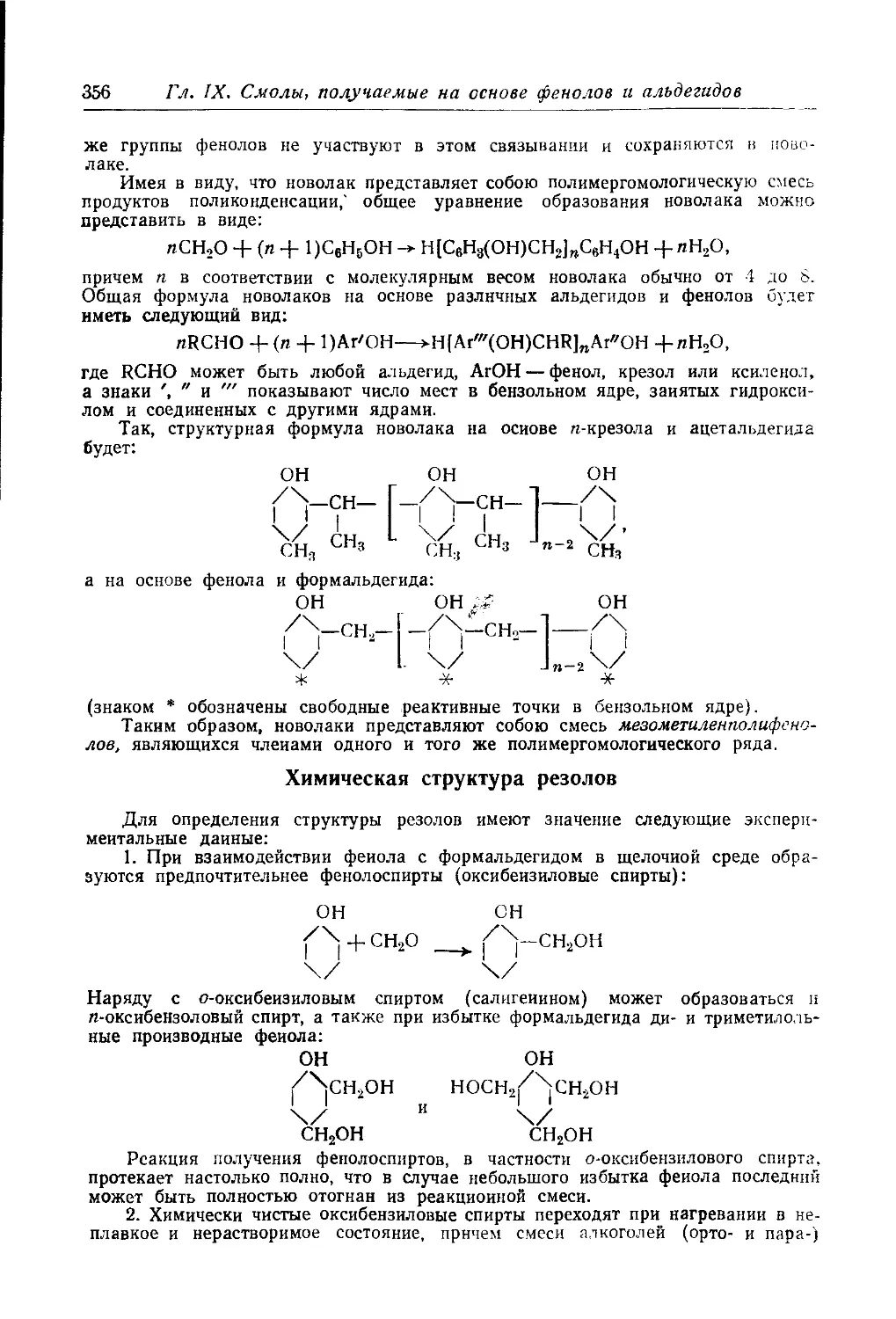






















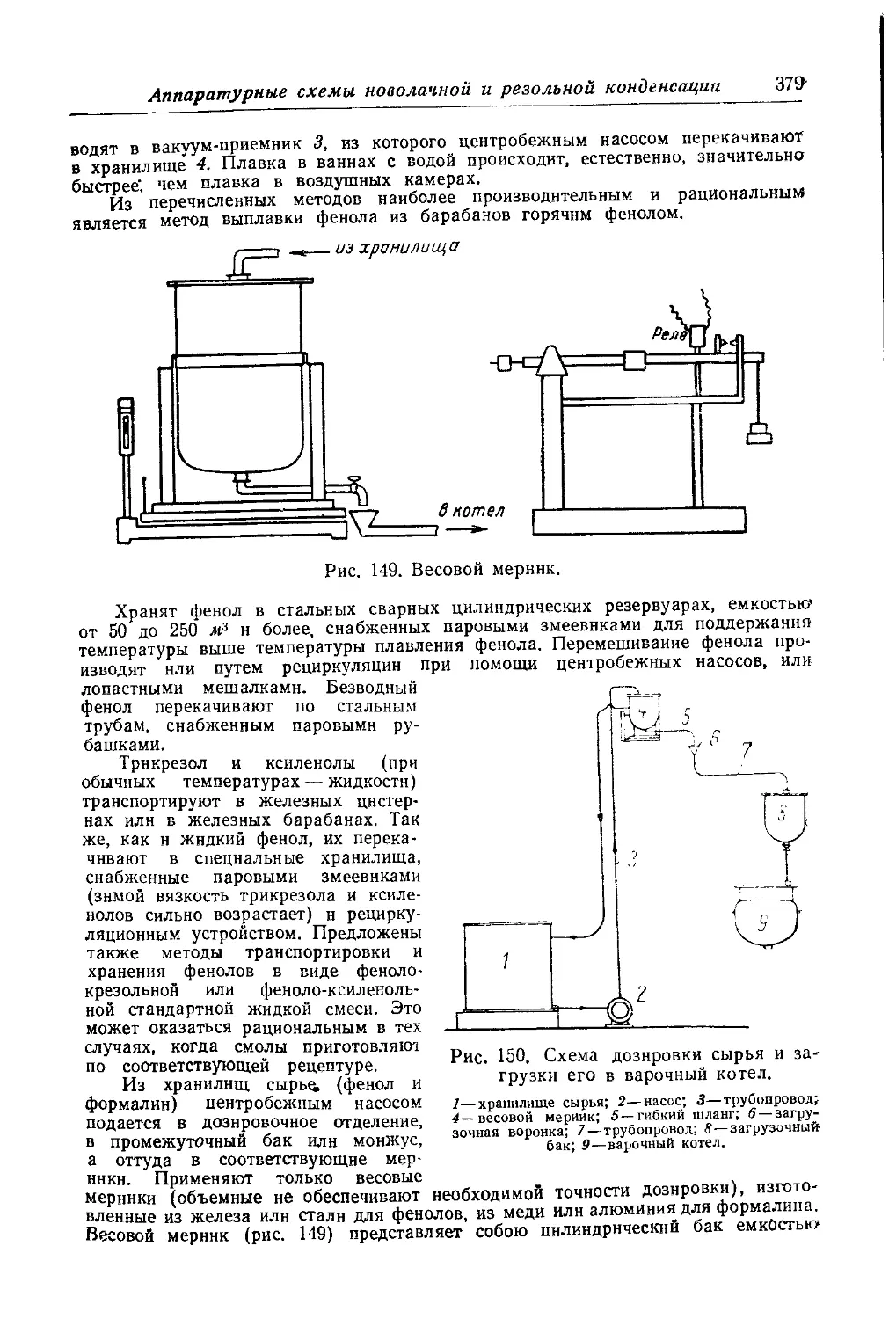









































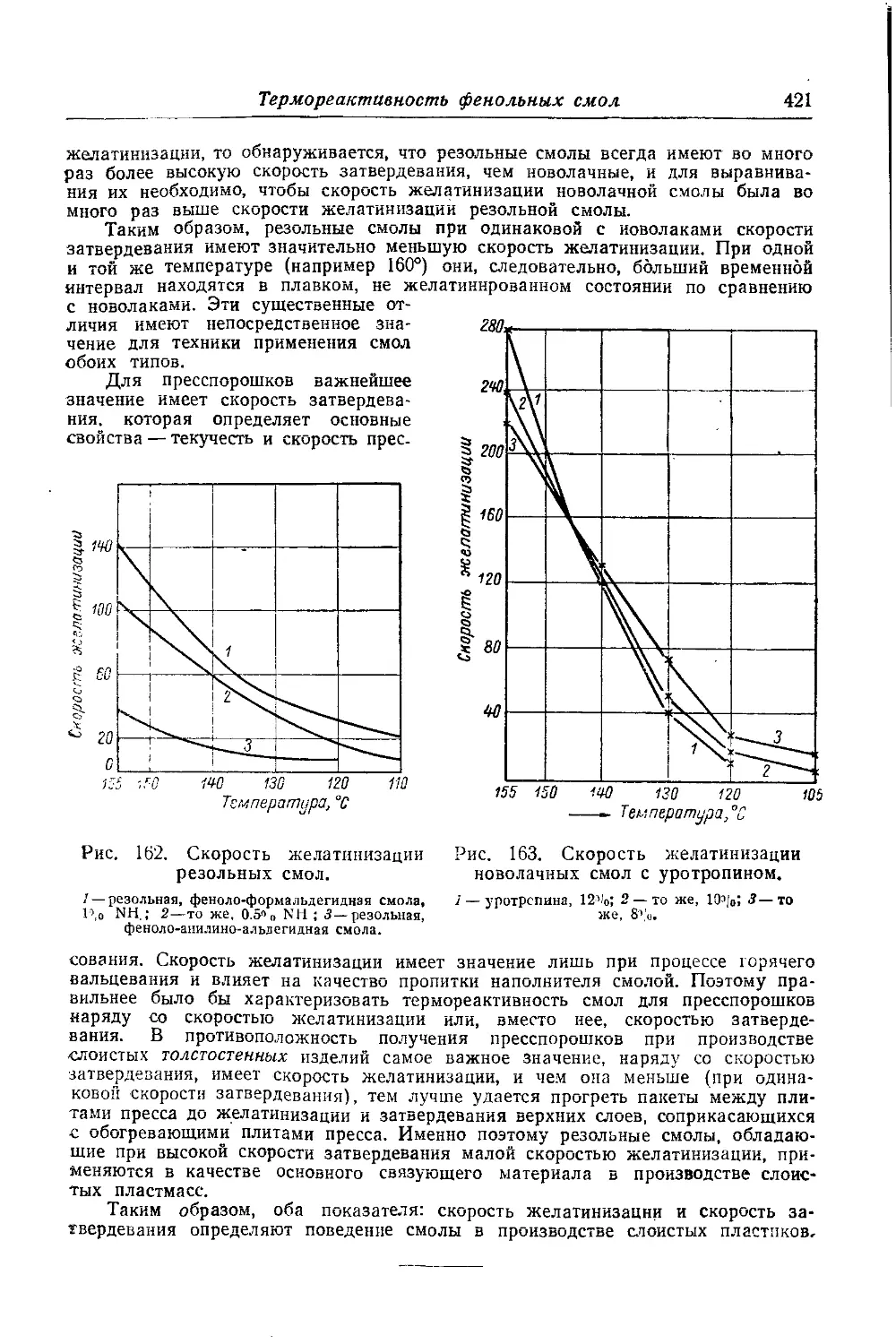








































































































































































































































Текст
СИНТЕТИЧЕСКИХ
ПЛАСТИЧЕСКИХ МАСС
ПОД РЕДАКЦИЕЙ
чл.-корр. АН СССР проф. С. Н. УШАКОВА
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
ХИМИЧЕСКОЙ ЛИТЕРАТУРЫ
ЛЕНИНГРАД • 1954
В книге приведен материал по химии и технологии
синтетических пластических масс. В ней дается подроб-
ная характеристика исходного сырья методов его по-
лучения, описываются технологический процесс про-
изводства различных пластических масс, их применение,
оборудование и технологические схемы процессов.
В книге освещаются вопросы физического и хими-
ческого строения пластических масс и закономерности,
определяющие их технологию и свойства.
Кинга предназначается в качестве руководства для
инженерно-технических работников промышленности
пластических масс и смежных с ней областей, а также
в качестве учебного пособия для студентов химико-тех-
нологических втузов и техникумов.
К ЧИТАТЕЛЮ
Издательство просит присылать Ваши замечания и от-
зывы об этой книге по адресу: Ленинград, Невский, 28,
________ Ленгосхимиздат.
ОГЛАВЛЕНИЕ
Стр.
Предисловие......................................................... 9
Введение
Исторический обзор ................................................ 11
Основные технические свойства, преимущества и недостатки пластиков 17
Применение пластиков...........‘................................... 23
ЧАСТЬ I
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ТЕХНОЛОГИИ ПЛАСТМАСС.
ТЕРМИНОЛОГИЯ И КЛАССИФИКАЦИЯ
Глава I. Химические основы технологии пластмасс.................... 26
Общая характеристика смол.......................................... 26
Процессы образования синтетических смол............................ 28
Полимеризация и поликонденсация.................................... 31
Процессы полимеризации....................................... 31
Ступенчатая полимеризация. Цепная полимеризация. Совместная
полимеризация. Полидисперсность полимеров. Разветвленность
полимеров. Структура звеньев цепи полимера.
Процессы поликонденсации..................................... 62
Глава II. Физические основы технологии пластмасс................... 70
Упругая и пластическая деформации.................................. 70
Упругая деформация.............•............................. 71
Пластическая деформация...................................... 74
Значение временного фактора (процесса релаксации) для низкомолекуляр-
ных веществ ..................................................... 77
Закономерности аморфного состояния низкомолекулярных веществ. 81
Температура стеклования и интервал размягчения низкомолекуляр-
ных смол................................................... 85
Высокоэластическая деформация ..................................... 89
Теория высокоэластической деформации......................... 90
Релаксационный характер высокоэластической деформации......... 102
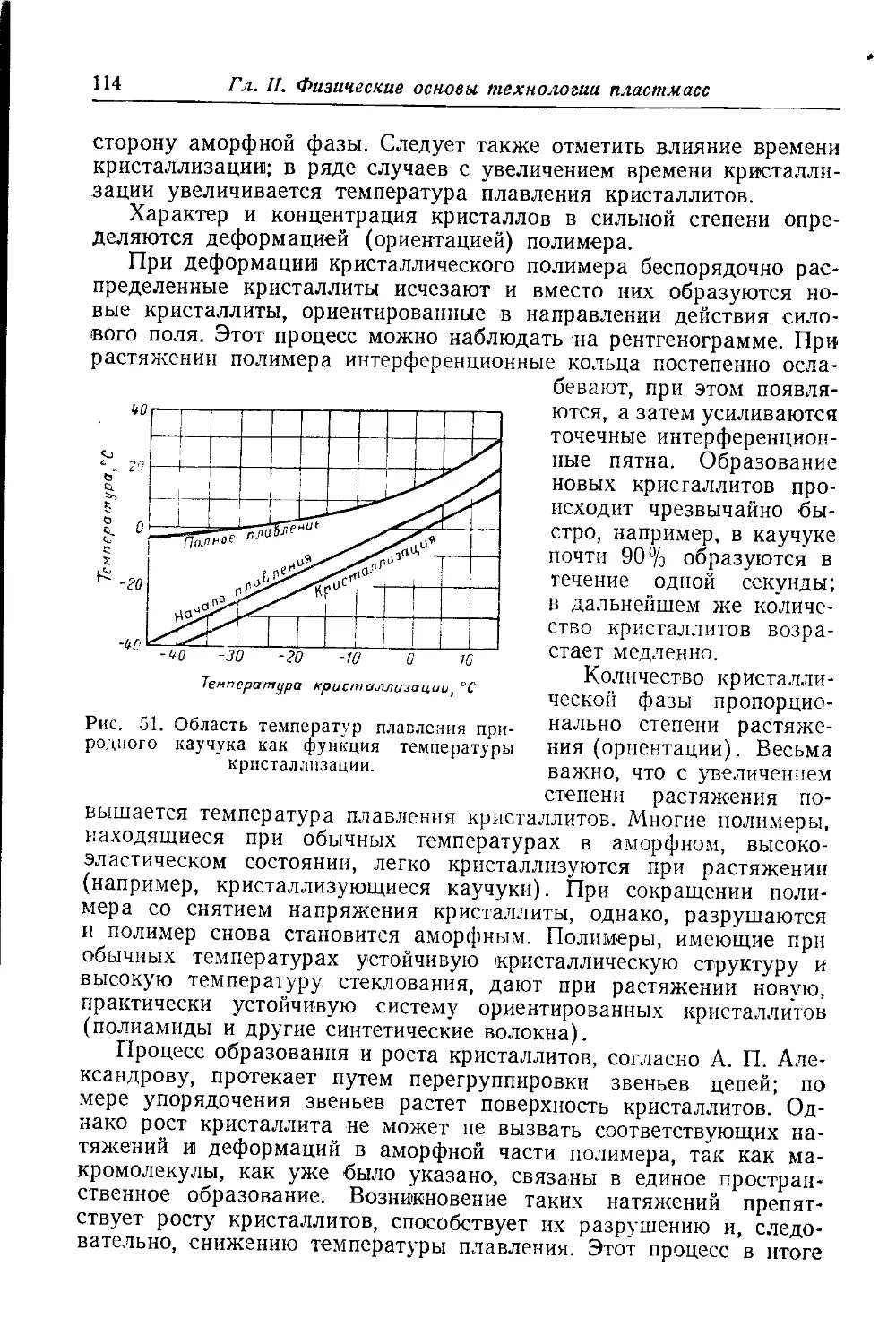
Зависимость температуры стеклования линейных полимеров от раз-
личных факторов. Температура хрупкости ................... 108
.Межмолекулярные силы и фазовая структура полимеров................. 110
Основные особенности в закономерностях кристаллического состоя-
ния полимеров............................................. 112
Способность полимеров к кристаллизации ..................... 116
Теоретическая Pi техническая прочность пластмасс.................. 119
Зависимость технической прочности пластмасс от их структуры....... 126
Пластификация полимеров .......................................... 136
Механическая пластификация (ориентация полимеров)........... 144
г-;
4
Оглавление
Стр.
Глава III. Терминология и классификация пластмасс.................. 147
Терминология пластмасс ............................................ 147
Определение понятия „пластические массы"..................... 148
Обозначение отдельных пластмасс ............................. 14&
Классификация пластмасс............................................ 152
ЧАСТЬ II
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ СИНТЕТИЧЕСКИХ СМОЛ,
ПОЛУЧАЕМЫХ ЦЕПНОЙ ПОЛИМЕРИЗАЦИЕЙ
Глава IV. Процессы полимеризации........................... 159
Технические методы полимеризации........................... 159
Теория эмульсионной полимеризации ......................... 170
Глава V. Пластические массы на основе полимеров непредельных
углеводородов ..................................................... 175
Пластические массы на основе полиэтилена (этиленопласты).............. 175
Получение полиэтилена........................................... 175
Структура полиэтилена .......................................... 178
Свойства полиэтилена ........................................... 179
Применение полиэтилена ......................................... 184
Пластические массы на основе полиизобутилена (изобутиленопласты) . . . 185
Свойства полиизобутилеиа....................................... 19(>
Сополимеры изобутилена...................................... . 192
Применение п переработка полиизобутилена........................ 192
Пластические массы на основе полистирола (стиропласты) ............... 195
Технические методы получения стирола............................ 194
Полимеризация стирола........................................... 20?
Сополимеры стирола ? ? . . ГС ? ГГ '. '. '. 5 : ; ' ~ ' 2Ю~
Структура и свойства полистирола ............................... 212
------Применение полистирола -.—s—s~—— .—— -------------------—2219-
Поропласты из полистирола....................................... 225
Свойства поропластов . . ....................................... 223
Применение поропластов......................................... 223-
Полимеры замещенных стиролов.................................... 224
Полимеры хлорстиролов........................................... 223
Пластические массы на основе поливинилкарбазола (карбазовинипласты) . 227'
Структура и свойства поливинилкарбазола......................... 228
Применение поливинилкарбазола................................... 230
Глава VI. Пластические массы на основе полимеров галоидопроиз-
водных этилена..................................................... 231
Пластические массы на основе полихлорвинила (хлорвинипласты) .... -235
Свойства хлористого винила ..................................... 231
Методы получения хлористого винила.............................. 232
Получение полихлорвинила ....................................... 234
Строение и свойства полихлорвинила ............................. 238
Пластические массы из полихлорвинила, не содержащие пластифи-
каторов .................................................. 24'-.
Хлорвинилоид (винипласт).
Оглавление
5
Стр.
Пластические массы из полихлорвинила, содержащие значительное
количество пластификатора '.............................. 248
Хлорвиниласт (пластикат).
Хлорированный полихлорвинил (перхлорвинил) ................. 253
Сополимеры хлористого винила................................ 254
Пластические массы на основе полихлорвинилидена (хлорвинилиденопласты) 256
Строение полихлорвинилидена ................................ 258
Свойства сополимеров хлористого винилидена.................. 260
Переработка и применение сополимеров хлористого винилидена . . 262
Пластические массы на основе политетрафторэтилена (тетрафторвини-
пласты)....................................................... 263
Строение и свойства политетрафторэтилена.................... 265
Переработка и применение политетрафторэтилена............... 267
Пластические массы на основе политрифторхлорэтилена.............. 267
Глава VII. Пластические массы на основе полимеров винилового
спирта и его производных...................................... 270
Сложные эфиры поливинилового спирта.............................. 270
Поливинилацетат................................................. 270
Синтез винилацетата ........................................ 270
Полимеризация винилацетата ................................. 276
Полимеризация в растворителях. Эмульсионная полимеризация.
Блочная полимеризация.
Строение поливинилацетата .................................. 282
Свойства поливинилацетата .................................. 284
Сополимеры винилацетата..................................... 285
Применение поливинилацетата................................. 285
Лоливинилформиат................................................. 286
Простые эфиры поливинилового спирта.............................. 287
Сополимеры простых виниловых эфиров.............................. 293
Поливиниловый спирт.............................................. 295
Строение и свойства поливинилового спирте .................. 299
Пластификация и применение поливинилового спирта ........... 301
Поливинигацепгали........................... 304
Методы производства поливинилацеталей....................... 306
Строение и свойства поливинилацеталей....................... 310
Промышленные поливинилацетали............................... 312
Поливинилформаль. Поливинилэтаналь. Поливинилбутираль.
Глава VIII. Пластические массы на основе полимеров — производ-
ных этиленкарбоновых кислот и эфиров ненасыщенных спиртов. 317
Пластические массы на основе полимеров производных этиленкарбоновых
кислот (акри[ло]пласты).......................................... 317
Методы получения производных акриловой и метакриловой кислот 317
Эфиры экриловой кислоты. Акрилонитрил. Эфиры метакриловой
кислоты.
Полимеризация акриловых и метакриловых эфиров................. 327
Блочный метод. Эмульсионный метод. Полимеризация в рас-
творителях. Переработка акриловых отходов.
Строение полиакрилатов ....................................... 333
Сополимеры акриловых и метакриловых эфиров.................. 334
Свойства полиакрилатов ....................................... 336
Применение полиакрилатов...................................... 341
Пластические массы на основе полимеров аллиловых эфиров (аллилопласты) 342
Оглавление.
Стр.
ЧАСТЬ III
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ СИНТЕТИЧЕСКИХ СМОЛ,
ПОЛУЧАЕМЫХ ПОЛИКОНДЕНСАЦИЕЙ И СТУПЕНЧАТОЙ
ПОЛИМЕРИЗАЦИЕЙ
Глава IX. Смолы, получаемые на основе фенолов и альдегидов
(Феноло-альдегидные смолы)......................................350
Условия получения новолачных и резольных продуктов конденсации . . . 350
Строение феноло-альдегидных смол..................'.............. 354
Химическая структура новолачных смол....................... 355
Химическая структура резолов............................... 356
Феноло-формальдегидные смолы..................................... 361
Закономерности кислой конденсации и образования новолачных смол 361
Катализаторы. Соотношение компонентов. Влияние условий конден-
сации и сушки. Действие уротропина.
Закономерности щелочной конденсации и образования резольных
смол ...................................................... 370
Катализаторы. Соотношение компонентов. Влияние химического
состава компонентов. Стадии процесса щелочной конденсации,
свойства и применение резольных смол.
Аппаратурные схемы новолачной и резольной конденсации...... 376
Подготовка сырья. Реакционные (варочные) агрегаты. Слив смол.
Утилизация надсмольных вод.
Технологический процесс производства новолачных смол для пресс-
порошков ................................................... 336
Фенольное сырье. Альдегидное сырье. Рецептура и проведение
процесса.
Технологический процесс производства резольных смол........ 390
Производство твердых резольных смол. Производство эмульсион-
ных резольных смол.
Литые продукты резольной конденсации (фенолиты, литые резиты) .... 396
Благородные резиты......................................... 397
Модифицированные феноло-альдегидиые смолы для производства лаков . .—401---
Получение маслорастворимых смол эфиризацией гидроксильных групп
_________новолака................................... . . . . . А 403
Простые эфиры новолака. Сложные эфиры новолака.
Получение маслорастворимых смол путем усложнения молекул
фенольных веществ........................................... 407
Алкилфенольные или 100% фенольные смолы.
Смолы на основе фенолов и заменителей формальдегида.............. 410
Феноло-фурфурольные смолы.................................. 410
Заменители формальдегида, не обладающие явной альдегидной
функцией.................................................... 4 ГА
Феноло-древесная смола (Ф. Д.). Феноло-лигниновые смолы (Ф. Л.).
Феноло-лигниновые смолы на щелочном лигнине (Ф. Щ. Л.).
Использование фенолов дегтей малокалорийных видов топлива ....... 416
Конденсация формальдегида с многоатомными фенолами............... 418
Резорциновые смолы......................................... 418
Термореактивность фенольных смол................................. 420
Глава X. Неслоистые пластические массы иа основе феноло-альде-
гидных смол (фенопласты)....................................... 422
Неслоистые фенопласты (прессматериалы)........................... 423
Технологические методы получения пресспорошков............. 423
Оглавление
7
Стр.
Лаковый метод. Эмульсионный метол. Пропитка наполнителя
смесью смолообразующих компонентов. Вальцовый метод.
Шнековый метод.
Новолачные и резольиые прессматерпалы (пресспорошки).......... 426
Скорость прессования новолачных пресспорошков в зависимости
от содержания уротропина.
Новолачные пресспорошки....................................... 430
Состав новолачных пресспорошков па основе древесной муки
(фенодреволиты). Вальцовый метод производства фенодрево-
лнтов). Шнековый метод производства пресспорошков
Резоль ные пресспорошки (фенодреволиты)....................... 440
Пресспорошки с минеральным наполнителем ...................... 442
Прессматериалы на основе длинноволокнистого наполнителя .... 444
Прессматериалы с целлюлозным волокном (феноцеллолиты,
волокниты). Фенотекстолит (текстолит-крошка). Феноасболиты
(асборезиты).
Определение текучести прессматериалов ........................ 452
Асборезптовые формовочные массы (фаолит)...................... 455
Глава XI. Слоистые пластические массы на основе феноло-альде-
гидных смол (фенопласты)......................................... 461
Слоистые фенопласты (слойматериалы)................................ 461
Смолы для слоистых фенопластов................................ 461
Методы пропитки наполнителя................................... 463
Типы слоистых фенопластов .................................... 463
Фенотекстослой (текстофенолнт, текстолит).................. 464
Ткани
Фенобумослой (картофенолит, бумофенолит, гетинакс)......... 482
Бумага
Слоистые асборезиты (феноасбослой, фепоасботекстослой, асботе-
кстолит, асболпт)........................................ 499
Древесно-слоистые пластики (фенодревослой ДСП, лигнофоль,
дельта-древесина, резитовая фанера, фанерит и др.)..... 502
Собственно фенодревослой. Резитовая фанера
Феностеклослой (стеклолнт, стеклофенолит).................. 507
Глава XII. Пластические массы на основе амиио-альдегидных смол. 514
Пластические массы на основе мочевина- и меламино-формальдегид-
ных (карбамидных) смол (аминопласты)............................. 514
Строение мочевино-формальдегидных и меламино-формальдегидных смол 514
Строение мочевино-формальдегидных смол........................ 514
Строение меламино-формальдегидных смол........................ 519
Методы получения мочевино-формальдегидных смол и прессматериалов
(аминопластов) .................................................. 521
Технологический процесс производства пресспорошков на основе мочевино-
формальдегидных смол и целлюлозы (аминоцеллолиты)................ 525
Приготовление мочевино-формальдегидного раствора. Смешение
раствора с наполнителем и „созревание" массы. Сушка........... 526
Свойства и применение мочевино-формальдегидных пресспорошков .... 532
Слоистые мочевино-формальдегидные прессматериалы (аминобумослой,
ампнотекстослоп и др.).......................................... 533
.Мочевино-формальдегидные смолы для лаков и клеев (модифицированные
смолы) ......................................................... 536
Пластические массы на основе меламино-формальдегидных смол........ 538
Пластические массы на основе анилино-формальдегидных смол (ани-
л ин он ласты)................................................... 545
8
Оглавление
Стр.
Глава XIII. Ионообменные смолы.................................... 55I
Катионообменные смолы ............................................... 555
Анионообменные смолы................................................. 559
Применение ионитов .................................................. 563
Глава XIV. Пластические массы на основе полиэфиров, полиамидов,
полиуретанов и полимочевин ..................................... 565
Полиэфирные и полиамидные смолы (эфиро- и амидопласты)............ 565
Основные закономерности реакций образования полиэфирных и полиамид-
ных смол........................................................ 565
Лаковые смолы и пластические массы на основе полиэфирных смол (эфиро-
пласты)......................................................... 576
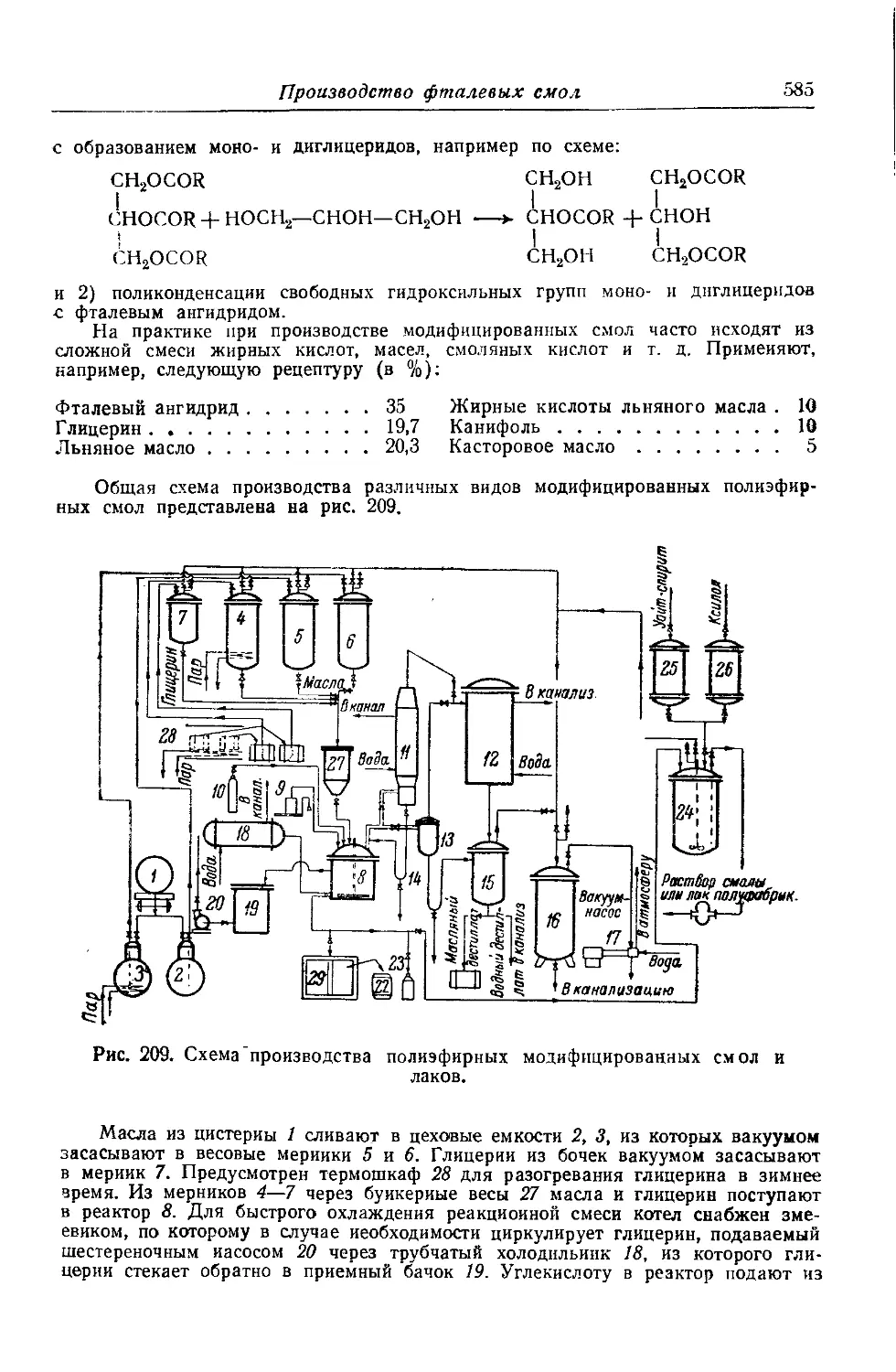
Производство фталевых смол................................... 577
Смолы на основе этиленгликоля и фталевого ангидрида. Смолы
на основе этиленгликоля и терефталевой кислоты. Смолы на
основе глицерина и фталевого ангидрида. Модифицированные
фталевые смолы.
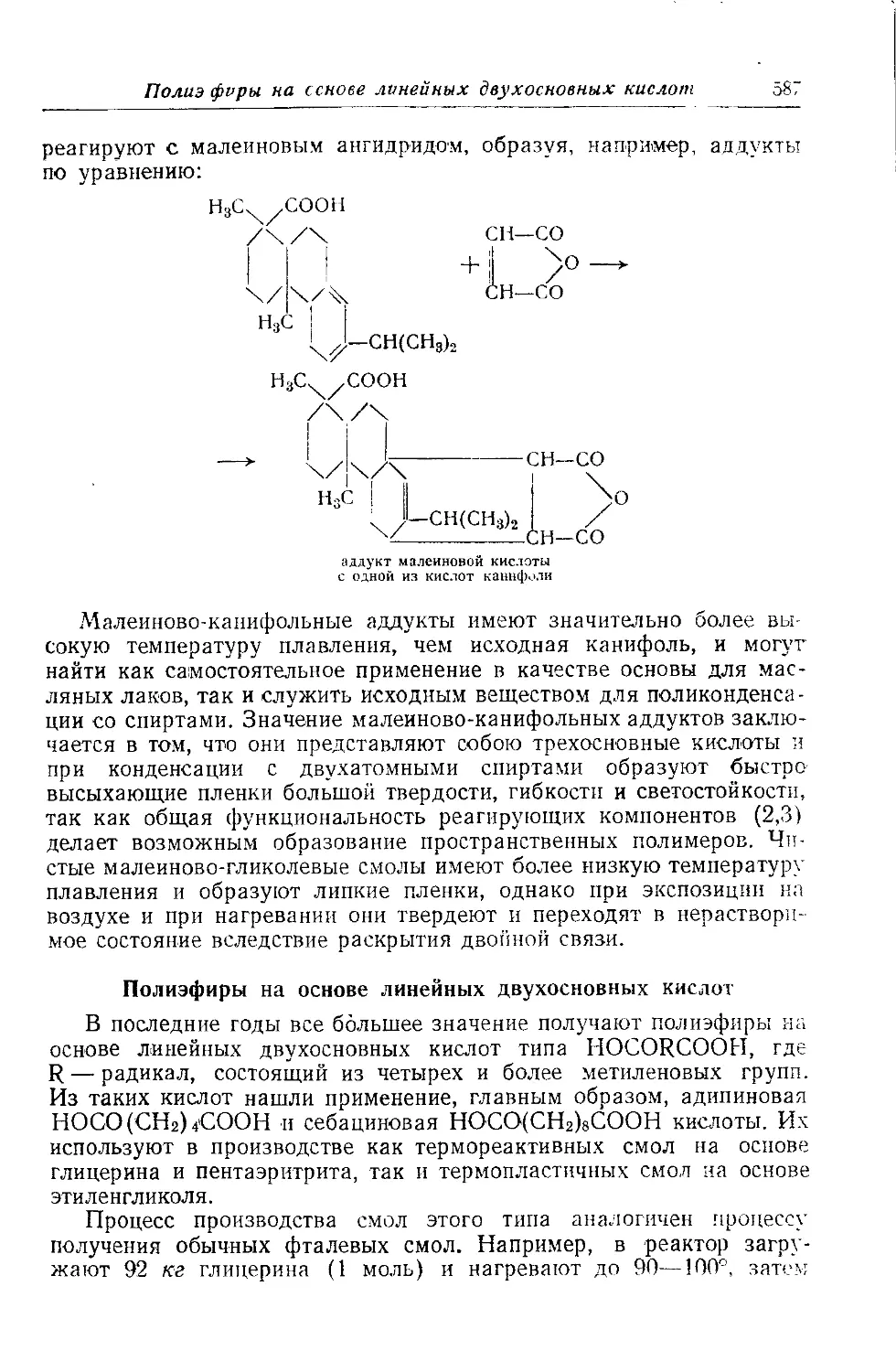
Малеиновые смолы.............................................. 586
Полиэфиры на основе линейных'двухосновных кислот............ 587
Пентаэритритовые смолы........................................ 588
Применение полиэфирных смол................................... 589
Пластические массы на основе синтетических полиамидов (амидоптасты) . 591
Полиамиды, получаемые поликонденсацией диаминов с дикарбоно-
выми кислотами.............................................. 595
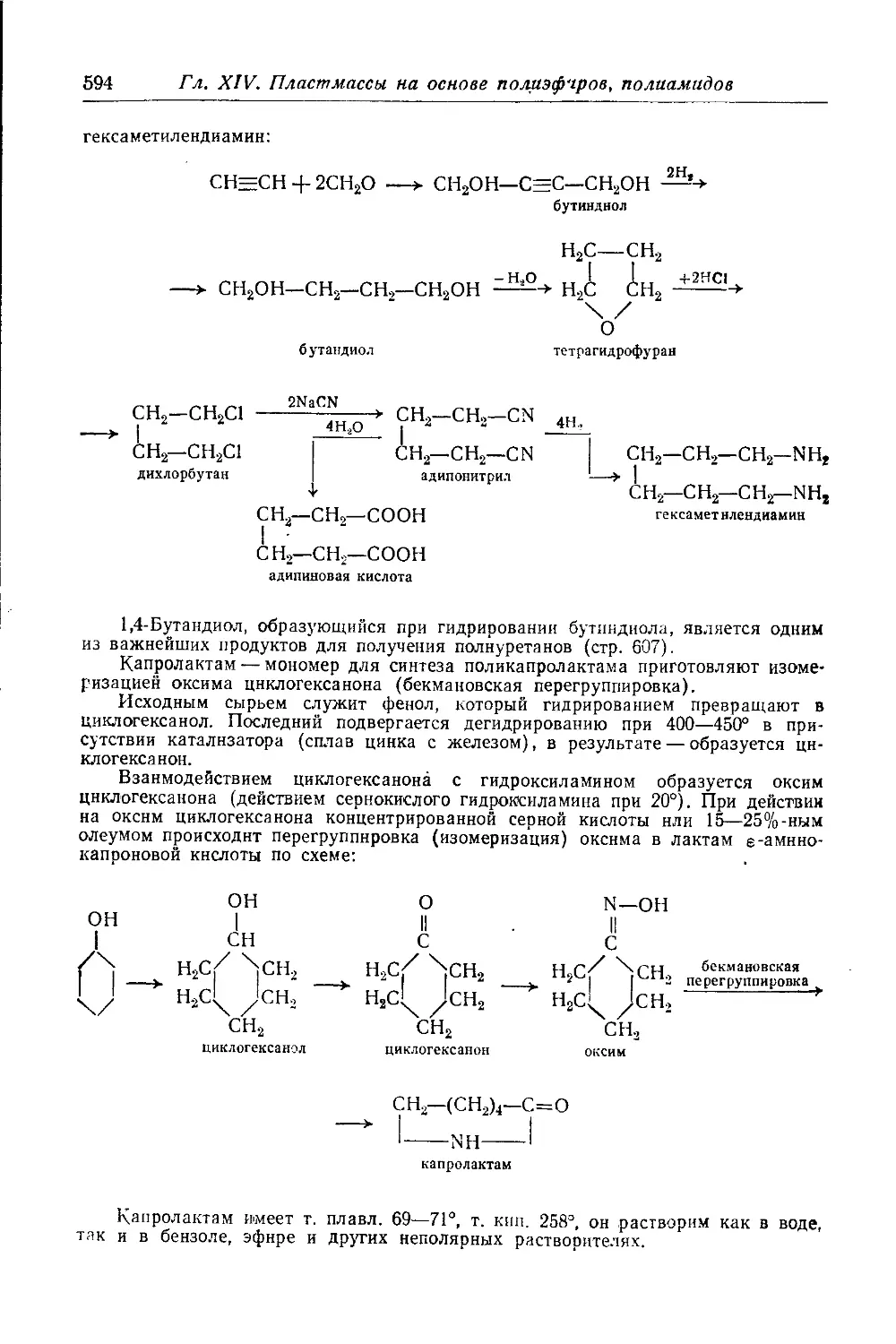
Полиамиды, получаемые полимеризацией циклов—лактамов s-амино-
кислот .................................................... 597
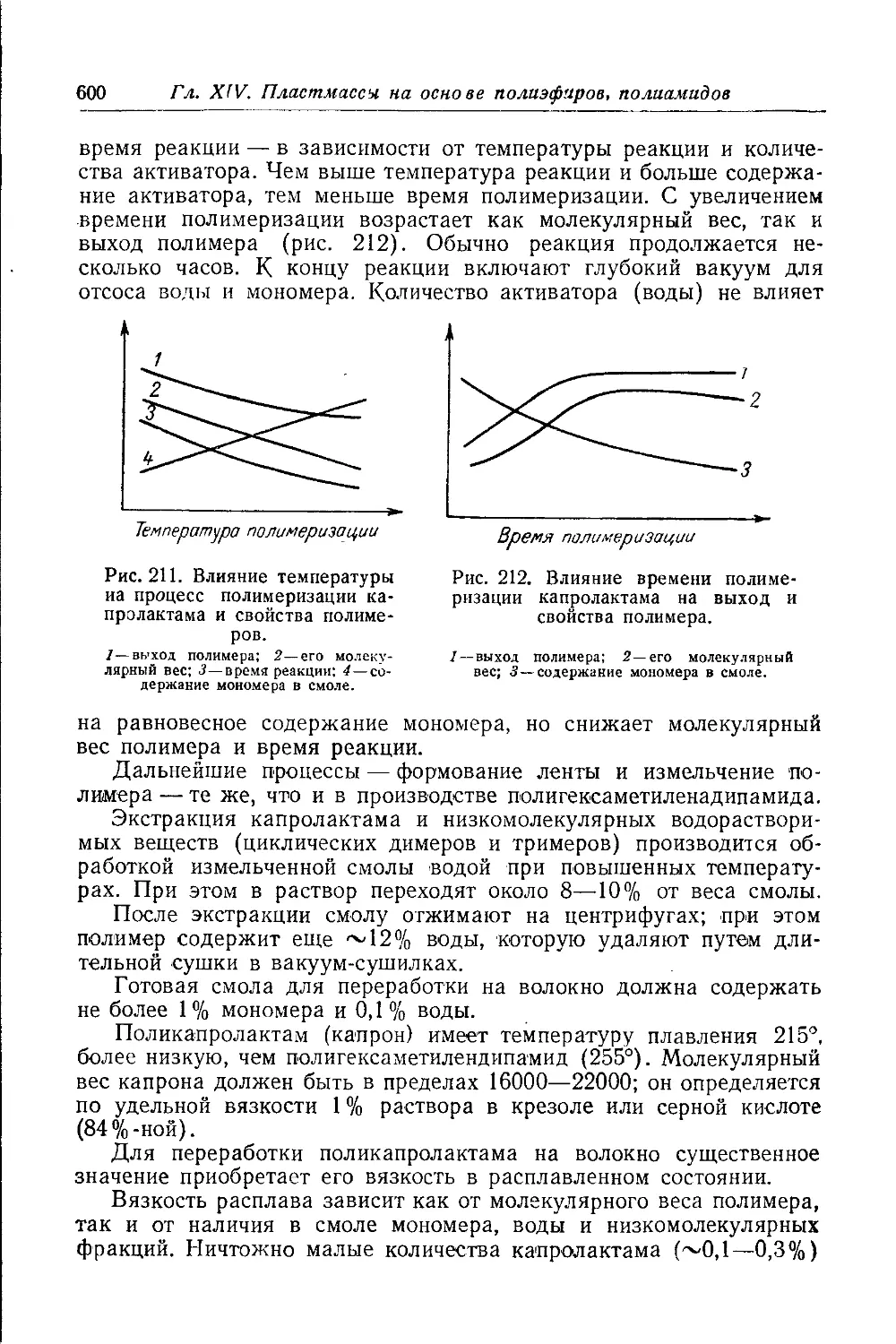
Свойства и переработка полиамидов............................. 601
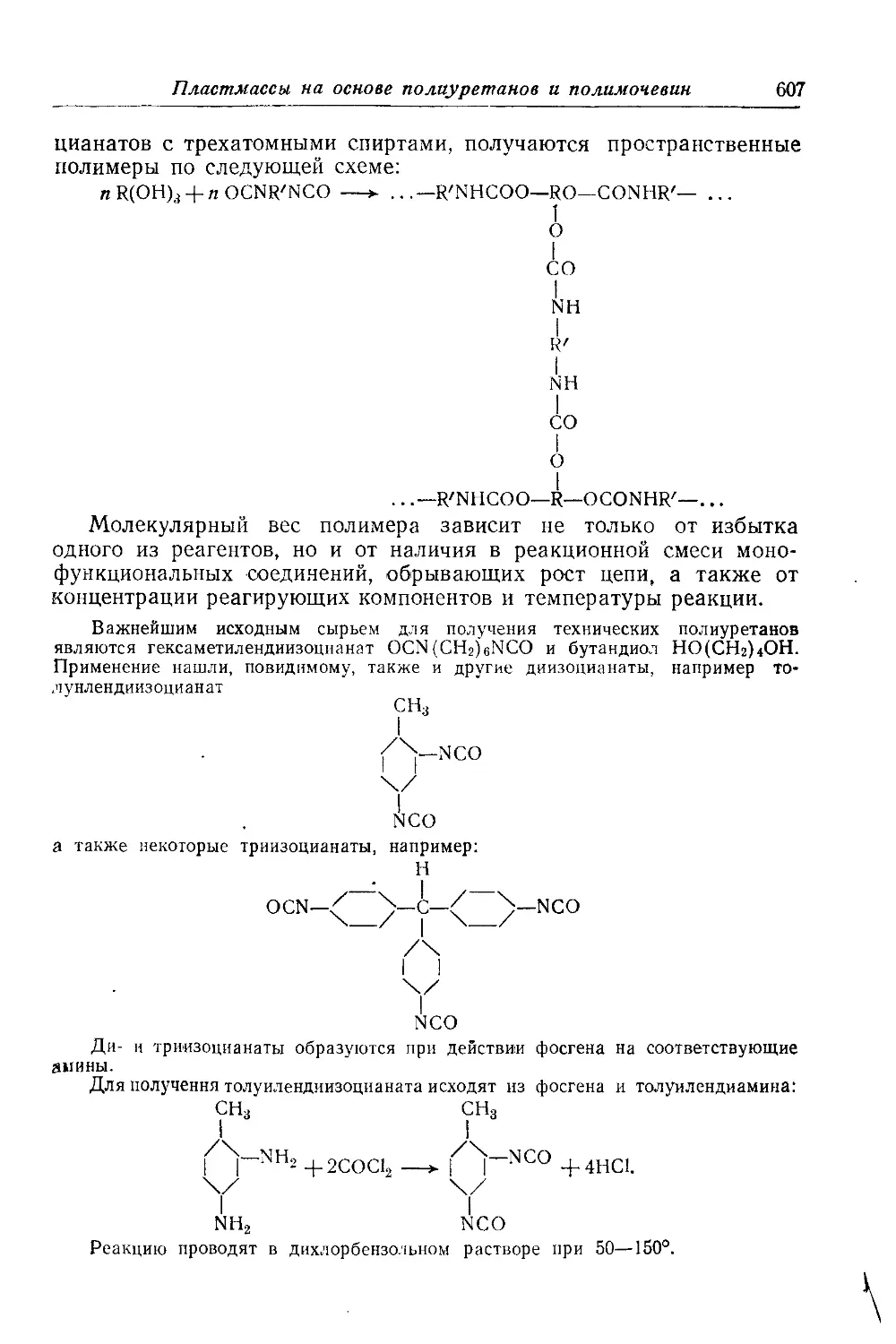
Пластические массы на основе полиуретанов и полимочевин............... W)
Глава XV. Пластические массы на основе кремнийорганических
смол............................................................ 611
Основные типы кремнийорганических смол—.—.—.—.....—. . .—.—.—,—. .—644-
Химические особенности кремнийорганических соединений................ 613
Получение исходных „мономеров".............................. 615
Процессы образования кремнийорганических полимеров '................. 616
Свойства кремнийорганических полимеров............................... 623
Получение пластмасс на основе кремнийорганических полимеров.......... 624
Получение термореактивных полимеров, модифицированных крем-
нийорганическимп соединениями............................... 627
Литература........................................................... 628
Указатель............................................................ 638
1
ПРЕДИСЛОВИЕ
Промышленность синтетических пластических масс за последние
годы достигла больших успехов. Ее рост и развитие тесно связаны
с развитием всего народного хозяйства.
Пластические массы являются не только эффективными замени-
телями цветных металлов, но и материалами, определяющими раз-
решение ряда технических проблем в различных отраслях про-
мышленности.
Директивы XIX съезда партии по пятому пятилетнему плану
развития СССР на 1951 — 1955 гг. обязывают увеличить производ-
ство пластических масс и развивать производство синтетических
материалов — заменителей цветных металлов.
Синтетические пластики получают все большее значение в про-
мышленности, производящей предметы народного потребления. Так,
для развития легкой и пищевой промышленности требуются краси-
вые, прочные пластики, смоляные эмульсии и лаки, упаковочные
пленки, клеящие вещества и т. д.
Технический прогресс промышленности пластмасс основан на
освоении и внедрении успехов советской науки и техники, достиже-
ний новаторов производства, на оснащении промышленности новей-
шими видами оборудования и непрерывном росте инженерно-техни-
ческих и рабочих кадров.
Настоящая книга по своему содержанию в основном отвечает
(в части синтетических пластиков) программе курса технологии
пластических масс для студентов химико-технологических вузов, но
она может быть полезной и для широких кругов инженерно-техни-
ческих работников промышленности пластмасс и смежных с ней
отраслей.
Рассматривая технологию пластических масс как самостоятель-
ную научную дисциплину, автор всемерно старался выдвинуть те
научные принципы, которые должны быть положены в ее основу.
Поэтому в первых главах книги изложены химические и физиче-
ские основы технологии пластмасс. При рассмотрении же вопросов,
относящихся непосредственно к технологии тех или иных видов
пластических масс, возможно ббльшее внимание уделялось харак-
теристике закономерностей реакций, ведущих к получению высоко-
молекулярных соединений, их химическому строению и зависи-
мости комплекса физико-механических и химических свойств поли-
10
Предисловие
Г
меров от строения и химической структуры исходных реагирующих
молекул.
Описание технологии отдельных пластмасс не может быть про-
изведено с одинаковой полнотой, так как технология многих пла-
стиков еще недостаточно освоена и в ряде случаев отсутствуют
литературные данные. Основным же типам пластиков, имеющим
уже оформленные и установившиеся процессы производства и
получившим широкое практическое применение, в книге уделено
наибольшее внимание и приведено подробное описание процессов
их получения.
Глава XIII «Ионообменные смолы» написана кандидатом хими-
ческих наук А. А. Васильевым.
Автор считает своим долгом выразить глубокую благодарность
чл.-корр. АН СССР проф. С. Н. Ушакову за общее научное редак-
тирование, а также за ценные советы, большое внимание и помощь,
которые он оказывал при написании книги.
За все указания читателей на имеющиеся в книге недостатки
автор будет весьма признателен.
ВВЕДЕНИЕ
ИСТОРИЧЕСКИЙ ОБЗОР
Знакомство человека с пластическими свойствами некоторых тел
относится к глубочайшей древности. Так, с незапамятных времен
человеку были известны пластические свойства влажных глин. На-
блюдения древнего человека над кусочками глины, которые после
обжига в присутствии золы теряли свою пластичность и станови-
лись твердыми и водонепроницаемыми, послужили основой для раз-
вития керамического производства.
Керамика является, следовательно, древнейшей пластической
массой. Хотя готовые керамические изделия после обжига и не
обладают свойством пластичности (они тверды и хрупки), однако,
по современным представлениям, керамику следует отнести к пла-
стическим массам, поскольку до обжига, в процессе изготовления
изделий, исходная масса пластична. Развитие керамической про-
мышленности основано именно на использовании пластических
свойств керамической массы.
Наряду с керамикой к древнейшим пластическим массам следует
отнести и стекло, так как изделия из стекла изготовляют исключи-
тельно методом пластической деформации (литье, выдувание, прес-
совка).
Таким образом, еще в древние времена пластические свойства
минеральных веществ использовали по следующим двум напра-
влениям: 1) формуя изделия из глин, пластичных при обычной тем-
пературе, и подвергая эти изделия последующему обжигу (кера-
мика), или же 2) сплавляя при высоких температурах смесь
веществ (песок, мел, зола), не пластичных при обычной темпера-
туре, и используя пластичность сплава при высоких температурах
для литья и формовки изделий (стекол).
В древние времена были известны также и пластические мате-
риалы органического происхождения: асфальты, некоторые виды
ископаемых смол, канифоль, воск и др. Так, древние египтяне, вави-
лоняне, а в дальнейшем римляне и другие народы применяли
асфальт для пропитки тканей и дерева в строительной и гидроизо-
ляционной технике, при выделке архитектурных деталей и т. д.
Однако в течение средних веков использование органических пла-
стических материалов не только не получило дальнейшего развития,
но был даже забыт тот значительный опыт, которым обладали древ-
ние египтяне и вавилоняне.
Введение
Лишь во второй половине XIX и главным образом в XX вв.
использование органических пластических материалов получило
новое развитие и широкое распространение.
История органических пластических материалов может быть
условно разделена на два периода: первый период — вторая поло-
вина XIX в. — характеризуется применением природных и химиче-
ски измененных (модифицированных) смол и высокомолекулярных
веществ — целлюлозы, каучука, белка; второй период — начало
XX в. и до наших дней — характеризуется преимущественным раз-
витием синтеза высокомолекулярных веществ и широким использо-
ванием их в различных отраслях техники.
Наиболее значительным достижением первого периода явилось
открытие процесса вулканизации природного каучука путем нагре-
вания его с небольшим количеством серы (1839 г.). Оно позволило
коренным образом устранить недостатки природного каучука (лип-
кость, текучесть) и получить эластичный и прочный материал
(резины).
Введением больших количеств серы при вулканизации каучука
(20—30%) был получен твердый упругий материал эбонит (1843 г.).
Эбонит является первым типом неплавкого и нерастворимого мате-
риала, типом пластика «пространственной» структуры. В отличие
от сырого каучука и резин, эбонит лишь в весьма незначительной
степени обладает высокоэластическим!! и пластическими свой-
ствами. Его следует считать первым пластиком, полученным путем
химического видоизменения природных полимеров.
В XIX столетии были открыты и другие важнейшие типы моди-
фицированных пластиков. Так, в 1872 г. путем пластификации нит-
роцеллюлозы камфорой был получен целлулоид, который до настоя-
щего времени сохранил промышленное значение. Он явился первым---
пластиком на основе производных целлюлозы и родоначальником
обширного -титпг-целлулоидоподобных материалов, отличающихся —
характерным комплексом механических свойств.
В самом конце XIX в. (1897 г.) был изобретен галалит — первый
пластик на основе химически модифицированных белковых веществ
(казеина).
Широкое применение получили также пластики на основе при-
родных смол и асфальтов.
Таким образом, к концу XIX в. были изобретены и приобрели
промышленное значение почти все типы пластиков на основе важ-
нейших природных высокомолекулярных веществ: каучука, целлю-
лозы и белка.
Во втором периоде — в XX в. — были впервые синтезированы
высокомолекулярные вещества и на их основе создана современная
промышленность пластмасс, синтетического каучука, синтетических
волокон и пленкообразователей.
Принципиальное отличие этого периода заключается, следова-
тельно, в переходе от реакций химических превращений природных
Истерический обзер
13
высокомолекулярных веществ к реакциям синтеза высокомолекуляр-
ных соединений, к получению синтетических полимеров с заданной
конфигурацией и величиной макромолекул.
В последние годы область синтетических пластиков развивается
также в направлении химических превращений внутри цепей макро-
молекул, получаемых синтетическим путем. Так, например, этим пу-
тем были получены различные производные поливинилового и по-
лиаллилового спиртов (эфиры, ацетали и др.).
Реакции, ведущие к получению таких материалов (омыление,
эфиризация, ацеталирование и др.), аналогичны реакциям модифи-
кации природных полимеров. Пластики, получаемые на основе
этих реакций, можно назвать синтетическими модифицированными
пластиками.
Развитие синтеза высокополимеров стало возможным благодаря
успехам органической химии в конце XIX и в XX вв., в особенности
благодаря ряду крупных исследований в области полимеризации и
конденсации.
Для развития химии и техники высокополимеров огромное зна-
чение имели работы творца теории химического строения А. М. Бут-
лерова и других выдающихся русских химиков.
А. М. Бутлерову принадлежат первые исследования в области
полимеризации. Изучая механизм полимеризации непредельных со-
единений, А. М. Бутлеров впервые доказал зависимость способности
вещества к полимеризации от его строения.
Особое значение имеет осуществленный А. М. Бутлеровым син-
тез изобутилена и исследования в области его полимеризации. При
этом Бутлеров впервые получил низшие формы полимеров изобу-
тилена (ди- и тримеры) и учел значение этих простейших форм для
синтеза более высоких полимеров.
В настоящее время изобутилен, синтезированный еще А. М. Бут-
леровым, получил промышленное применение как исходное сырье
для производства полиизобутилена — полимера, имеющего большое
значение в качестве синтетического каучука и каучукоподобных
пластмасс.
Классические исследования превращений йодистого метилена
привели А. М. Бутлерова еще в 1858 г. к открытию формальдегида,
гексаметилентетрамина (уротропина) и основных полимерных форм
формальдегида.
Значение этих работ трудно переоценить. Академик А. Е. Арбу-
зов следующим образом характеризует значение работы Бутлерова
по открытию формальдегида:
<Если И. Н. Зинин получением анилина заложил фундамент анилино-
красочной промышленности, то А. М, Бутлеров открытием формальдегида
и изучением его главнейших свойств как бы впервые открыл дверь в об-
ласть высокомолекулярных соединений, той категории химических веществ,
которым суждено в современной жизни и технике играть совершенно
исключительную по значению роль».
14
Введение
В развитии химии и промышленности органических пластмасс
важнейшую роль сыграла реакция М. Г. Кучерова (1881 г.) по ги-
дратации ацетилена и получению уксусного альдегида — исходного
вещества для нроизводства ряда пластиков. Она явилась и основой
промышленного синтеза сложных и простых виниловых эфиров, при-
меняемых для получения ряда типов современных полимеров
(поливинилацетата и др.).
В этом отношении большое значение имеют работы А. Е. Фавор-
ского, выяснившего механизм изомерных превращений непредель-
ных соединений. Им же разработаны реакции, ведущие к получению
простых виниловых эфиров и соответствующих полимеров.
Химия высокомолекулярных соединений могла выделиться как
важнейшая отрасль науки и техники лишь после того, как были
отброшены различные коллоидо-химические теории «малых ячеек»
и сформулированы современные представления о макромолекулах,
лежащие в основе природных и синтетических полимеров.
В основу развития науки о синтетических полимерах положены
процессы и теории полимеризации и поликонденсации.
Русская наука внесла большой вклад в развитие процессов поли-
меризации. Так еще в 1887 г. В. В. Солонина взаимодействием
аллиловых эфиров с SO2 впервые осуществил реакцию сополимери-
зации.
В 1900 г. И. Л. Кондаков получил диизопропенил и при нагреве
его в присутствии щелочи — каучукоподобный полимер.
В 1909 г. С. В. Лебедев сделал сообщение о способности диви-
нила к полимеризации и о получении каучу ко подобных веществ.
Он обогатил русскую химическую науку своими широко известными
работами по синтезу и полимеризации диеновых соединений, кото-
рые и привели его ж получению синтетического каучука.----------
Основные положения установленных С. В. Лебедевым законо-
мерпостей.известные как «Правила полимеризации диеновых соеди-
нений С. В. Лебедева», имеют важнейшее значение для выбора
условий полимеризации и соответствующей структуры исходного
мономера.
Работы С. В. Лебедева имеют большое значение также и для
теории и техники полимеризации этиленовых соединений и, следо-
вательно, для промышленности пластмасс.
Первыми синтетическими пластиками, получившими промышлен-
ное значение, явились фенопласты (стр. 422 и сл.).
Еще в 1872 г. А. Байер сделал наблюдение, что при взаимодей-
ствии фенолов с альдегидами образуются смолообразные продукты.
Это наблюдение не привело к каким-либо практическим результа-
там, так как ©смоление продуктов реакции в то время рассматри-
вали лишь как серьезную помеху в синтезе индивидуальных низко-
Исторический обзор
15
молекулярных соединений. Потребность промышленности того
времени в новых материалах и синтетических смолах была еще не-
велика — природные продукты и смолы вполне удовлетворяли ее
нужды.
Лишь в конце XIX и в начале XX вв. в связи с развитием
электротехнической промышленности, а также приборе- и аппа-
ратостроения, появилась потребность в материалах с более разно-
образными и совершенными свойствами, чем те, которыми обла-
дали природные материалы и материалы старой техники (каучук,
керамика, дерево, янтарь, шеллак и др.). Эта потребность и напра-
вила работы ученых в сторону более детального и глубокого изу-
чения процессов смолообразования, процессов, целью которых
является получение не кристаллических, а смолообразных продук-
тов. Исследования в этом направлении привели уже в 1902 г. к
получению в полузаводских условиях первой синтетической, спирто-
растворимой смолы на основе конденсации фенола с формальдеги-
дом — лаккаина.
В период 1907—1914 гг. было осуществлено промышленное про-
изводство синтетических твердых нерастворимых и неплавких мате-
риалов на основе феноло-альдегидной конденсации (Л. Бэкеленд,
Г. С. Петров). Таким образом были созданы фенопласты, являю-
щиеся первыми синтетическими пластиками. Впоследствии они, как
известно, получили исключительно большое развитие и в настоящее
время представляют важный конструкционный материал. В 20-х и
30-х годах нашего столетия были изобретены и получили промыш-
ленное значение также и другие классы синтетических поликонден-
сационных пластиков (мочевино-формальдегидные, полиэфирные
и ДР-).
Начиная с 30-х годов, большое промышленное значение приобре-
тают полимеризационные пластики (полистирол, поливинилацетат,
полихлорвинил, полиметилметакрилат и др.), темпы роста произ-
водства которых в этот период значительно превысили темпы роста
производства поликонденсационных пластиков.
Сороковые годы нашего столетия характеризуются весьма быст-
рым развитием промышленности пластмасс всех классов и в осо-
бенности — полимеризационных пластиков. Важное значение полу-
чили новые виды поликонденсационных пластиков: кремнийорга-
нические, полиамидные, полиуретановые и др.
Современная промышленность пластмасс является важнейшей
отраслью народного хозяйства. Она включает большое число различ-
ных технологических процессов и выпускает десятки различных ма-
териалов, получивших применение в самых разнообразных областях
техники.
В настоящее время технологии синтетических пластиков, во-
локна, каучука и пленкообразователей неразрывно связаны друг с
другом и составляют по существу одну техническую область, кото-
16
Введение
рая основывается па данных науки о химии и физике высокополи-
меров.
* * *
Несмотря на замечательные достижения русской химической
науки конца XIX и начала XX вв. и на большой вклад, который
она внесла в химию высокополимеров, царская Россия не имела
промышленности пластмасс. Интересные в практическом отно-
шении открытия русских ученых не получили поддержки и раз-
вития.
Современная мощная отечественная промышленность синтети-
ческих полимеров и пластических масс является у нас детищем со-
ветской эпохи, — эпохи пятилеток. В этот период был организован
ряд научно-исследовательских институтов и лабораторий, развер-
нувших широкие исследования в области пластических масс и вы-
сокополимеров.
Советские ученые обогатили науку ценными исследованиями и
разработали ряд новых пластиков и многие оригинальные техноло-
гические процессы.
С. Н. Ушаков разработал и внедрил ряд новых технологий и ма-
териалов, главным образом в области виниловых пластиков, произ-
водных целлюлозы, фенопластов и др.
Г. С. Петрову принадлежит заслуга изобретения первого синте-
тического пластика — карболита (1912); им проведен ряд крупных
исследований в области фенопластов и других поликонденсацион-
ных смол.
Большое принципиальное значение имеют работы К. А. Андриа-
нова и других советских ученых в области кремнийорганических
соединений. Их работы и привели к открытию нового класса поли-
меров — кремнийорганических.
. Обширные теоретические работы по изучению химической_ струю,
туры полимеров и процессов полимеризации и поликонденсации
проведены С. С. Медведевым, А. А. Ваншейдтом, В. В. Коршаком,
И. П. Лосевым, Б. Н. Рутовским, 3. А. Рогозиным и др.
Важное значение в развитии новой науки о полимерах — физики
полимеров — имеют работы П. П. Кобеко, А. П. Александрова,
В. А. Каргина и др.
За годы пятилеток в СССР были созданы кадры научных работ-
ников, рационализаторов, изобретателей, которые разработали и
освоили в области промышленности пластических масс десятки но-
вых производственных процессов.
Эти достижения советской науки о высокополимерах и советской
промышленности пластмасс служат залогом успешного выполнения
директив XIX съезда партии по пятому пятилетнему плану разви-
тия СССР о дальнейшем увеличении производства пластиче-
ских масс и синтетических материалов — заменителей цветных ме-
таллов.
Основные технические свойства пластиков
17
ОСНОВНЫЕ ТЕХНИЧЕСКИЕ СВОЙСТВА, ПРЕИМУЩЕСТВА И
НЕДОСТАТКИ ПЛАСТИКОВ
Пластические массы за сравнительно короткий период (20—30 лет)
стали одной из значительных отраслей народного хозяйства. Они во
многом определяют как количественное, так и качественное разви-
тие ряда других отраслей промышленности, в частности электротех-
ники, промышленности средств связи, транспорта, приборе- и аппа-
ратостроения и т. д. Рост производства синтетических пластиков
значительно превосходит не только рост производства материалов
старой техники, но также и многих новых материалов, например
производства алюминия, магниевых сплавов и др;
Такое широкое развитие промышленности орга’нических синтети-
ческих материалов связано с присущим пластикам комплексом тех-
нических свойств, исключительным разнообразием в сочетании этих
свойств, а также с эффективнейшими методами переработки пла-
стических масс и наличием неисчерпаемой сырьевой базы для их
производства. Укажем кратко основные преимущества органических
синтетических пластиков.
Малый удельный вес. Удельный вес различных пластиков может
колебаться от 0,9 до 2,2; в среднем пластики в два раза легче алю-
миния, в 5—8 раз легче стали, меди, свинца, бронзы и т. п. Эконо-
мия веса означает экономию энергии и в ряде отраслей (авиа-,
авто- и судостроении, железнодорожном транспорте и др.) имеет
решающее значение. Малый удельный вес определяет некоторые
пластмассы (если исходить из «весовой прочности») как самый
-Арочный материал современной техники (табл. 1).
ТАБЛИЦА 1
Прочность на разрыв металлов и пластмасс
Материал Уд. вес г/сма Предел прочн сти на разрыв к?1с.ч* „Вессвая проч- ность" ^пред. прочн. на разр. \
X уд. вес /
Сталь высоких сортов 8,0 12 800 1 600
Чугун 8,0 1 500 190
Дюраль 2,8 3 900 1 400
Фенотекстослой 1,4 1 500 1 11)0
Фенодревослой 1,4 3 500 2 500
Стеклотекстослой 1,8 3 COO—7 000 1 700—4 000
Ориентированный полихлорвинилиден 1,7 7 000 4 000
Особый класс пластиков представляют собою губчатые, пори-
стые, пенообразные материалы (поропласты) с удельным весом по-
рядка 0,1—0,02 (100—20 кг/м3). Благодаря малому коэффициенту
теплопроводности (0,06—0,015) они получили преимущественное
применение для теплоизоляции. . . ...
2 Зак. 1269. Э. И. Барг > '' т''
«
l.S
Введение
Химическая стойкость. Хорошо известно, в какой мере черные
металлы, а также многие цветные металлы подвержены коррозии
как от действия влажного воздуха, так, в особенности, от различных
агрессивных сред. Для предохранения металлических изделий от
коррозии широко применяют различные методы поверхностной за-
щиты, чаще всего покрытие различными пленками (лаками).
Основные виды пластиков, в отличие от металлов, противостоят
не только действию влажного воздуха, но в широких пределах и
действию различных кислот и щелочей. Изделия из пластиков, как
правило, не требуют каких-либо защитных покрытий, в том числе и
лакировки. Многие из них широко применяются в химическом ма-
шиностроении в качестве антикоррозийного материала.
Некоторые пластики отличаются универсальной химической стой-
костью и превосходят в этом отношении все известные металлы;
у многих химическая стойкость сочетается с высокой термостой-
костью (до 200—300°) и с большой механической прочностью.
К наиболее химически стойким пластикам относятся: политетра-
фторэтилен, полиэтилен, полиизобутилен, фенопласты (последние не
стойки к щелочам), полистирол и полихлорвинил.
Диэлектричность. Все пластмассы являются диэлектриками,
причем некоторые из них — лучшие диэлектрики современной
техники.
Сочетание высоких диэлектрических свойств с механической
прочностью и теплостойкостью позволяет применять пластики в ка-
честве электроизоляционного и основного конструкционного мате-
риала электротехники.
Во многих случаях из металла изготовляют только токонесущие
части электрических аппаратов, а основным конструкционным и изо-
—ляционным материалом являются пластики.------------------------
Особое значение пластики имеют в высокочастотной технике,
ста к как ошг— единственные-совершеннейшие- диэлектрики-для ра-
диосвязи, телевидения, радара и т. п. В этой области наиболее
широко применяют пластики на основе полиэтилена, полистирола,
полидихлорстирола, политетрафторэтилена и других неполярных
полимеров.
Для сильноточной промышленности важнейшими диэлектриками
являются фенопласты, анилинопласты, меламинопласты и другие
термореактивные пластики.
Механическая прочность. Пластические массы представляют со-
бою материалы со сложным и разнообразным комплексом физико-
механических свойств, — от жестких, упругих материалов, напоми-
нающих керамику, дерево, кость, до гибких, растяжимых коже- и
каучукоподобных материалов.
Жесткие типы — пластики (стр. 148) —делятся на три группы:
1) ненаполненные пластики, — обычно различные типы органиче-
ских стекол, литых смол и т. д.; 2) наполненные неслоистым напол-
нителем (прессматериалы); 3) слоистые пластики (слойматериалы).
Основные технические свойства пластиков
19
Последняя группа пластиков относится к наиболее прочным ма-
териалам современной техники, если эту прочность относить к еди-
нице веса (табл. 1).
Пластики могут применяться в широком температурном интер-
вале— от температуры жидкого воздуха до температуры их стекло-
вания (стр. 85), которая характеризует теплостойкость пластика,
температуру его размягчения.
Мягкие типы — эластики (стр. 149)—при рабочих температу-
рах находятся в высокоэластическом состоянии; предельно низкой
температурой их применения является температура стеклования
(морозостойкости); чем она ниже, тем обычно в более широком
диапазоне температур они могут работать. Предельно высокой
является температура текучести, т. е. температура, при которой
наблюдается начало необратимого (пластического) течения.
Некоторые типы пластмасс с микрокристаллической структурой
(стр. ПО) характеризуются сочетанием свойств жестких и мягких
типов и отличаются отсутствием хрупкости при низких температурах
и значительной жесткостью при обычных и повышенных температу-
рах, т. е. они обладают как морозо-, так и теплостойкостью (поли-
этилен, полихлорвинилиден, политетрафторэтилен и т. д.).
Другие полимеры с микрокристаллической структурой в вытяну-
том, ориентированном состоянии представляют собою класс синте-
тических волокон, отличающихся большой устойчивостью кристал-
лической фазы, прочностью, гибкостью, морозостойкостью и высокой
температурой плавления (полиамиды, полиуретаны и др.).
Фрикционные свойства. Многие пластики служат лучшими анти-
фрикционными материалами. Они отличаются низким коэффициен-
том трения и весьма малым износом. Многие типы пластиков не
требуют смазки при использовании их в виде подшипников («само-
смазывающиеся» материалы); для других же в качестве смазки
может служить вода или водные эмульсии.
Лучшим антифрикционным материалом являются слоистые
фенопласты (на основе ткани, дерева и стеклоткани). Они широко
применяются для изготовления подшипников металлургических ста-
нов, для производства шестерен, роликов и т. п. Для работы при
высоких температурах и в агрессивных средах большое значение
приобретают подшипники из политетрафторэтилена.
Другие типы пластиков обладают при сухом трении высокими
фрикционными свойствами и весьма малым износом и их применяют
поэтому в качестве тормозного материала. Лучшим фрикционным
материалом являются фенопласты с асбестовым наполнителем.
Оптические свойства. Многие виды пластиков по праву носят
название органических стекол; они обладают комплексом оптиче-
ских и механических свойств, который делает их исключительно
ценным материалом как для оптической промышленности, так и для
аппарате- и машиностроения.
20
Введение
Органические стекла отличаются высокой прозрачностью и бес-
цветностью, но могут легко быть окрашены минеральными и орга-
ническими красителями; они пропускают лучи света в широком
диапазоне волн, в частности, ультрафиолетовую часть спектра, при-
чем в этом отношении превосходят в десятки раз обычные стекла
^прозрачность для ультрафиолетового света до 90—99%).
Органические стекла имеют чаще всего коэффициент преломле-
ния в пределах 1,5—1,6 (табл. 2); однако имеются теоретические
предпосылки для получения стекол с более высоким коэффициентом
преломления (до 2 и выше).
ТАБЛИЦА 2
Оптические свойства прозрачных пластиков
Полимер Уд. вес г/см ~ Коэффициент преломления Характеристика старения Прозрачность, о)О
первоначальная через год
Полиметилметакрилат .... 1,19 1,49 94 93
Аллиловые смолы 1,32 1 ,а0 92 91
Полистирол Сополимер хлорвинила с ви- 1,0 о 1,00 90 90
нилацегатом 1,34 1,52 83 83
Ацетилцеллюлоза 1,30 1,49 87 83
Ацегобутирагцеллюлоза . . . 1,20 1,47 89 89
Фенольная смола 1,32 1,о0 85 —
Алмаз З.о 1 2,43 100 100
Оконное стекло 2,о0 1,52 Старение и гень слабое
Наряду с высокими оптическими свойствами органические стекла
отличаются также высокой прочностью и способностью к формова-
нию. Пластики, применяемые для оптических целей, представляют
собою либо твердые, упругие материалы, либо гибкие, эластичные
пленки. Первые нашли самостоятельное применение в качестве ор-
ганического стекла, вторые — для склейки силикатных стекол и
получения безосколочного стекла («триплекс»).
Внешний вид. Пластики и изделия из них имеют твердую, бле-
стящую поверхность, которая не меняется в обычных атмосферных
условиях. Изделия из пластиков поэтому не нуждаются в лакировке
или полировке, а также в поверхностной окраске, так как их окра-
шивают в массе, в процессе производства; при желании могут быть
получены любые расцветки — многоцветные имитации натуральных
камней, черепахи, перламутра и др.
Методы переработки. Главное преимущество пластических
масс — возможность бесстружечного, бесстаночного изготовления
из них изделий при помощи разнообразных пластицирующих про-
цессов (простого литья, литья под давлением, прессования, выда-
Основные технические свойства пластиков
21
вливания и т. д.). Развитие пластицируюших процессов привело
к изобретению ряда автоматических машин (пресс-автоматов, авто-
матов для литья под давлением и т. д.), позволяющих изготовлять
в час сотни деталей сложной конфигурации.
Преимущество методов пластической переработки заключается
в значительном уменьшении расхода материала при изготовлении
деталей (нет отходов от стружки), в возможности применять менее
квалифицированную рабочую силу, в значительном снижении коли-
чества станочных- и человеко-часов, затрачиваемых на изготовление
деталей, в уменьшении общего числа станков и в экономии электро-
энергии.
Методы пластичной переработки позволяют с успехом исполь-
зовать пластики взамен других материалов даже в тех случаях,
когда они значительно дороже заменяемых материалов, так как
готовые изделия из пластиков получаются все же более дешевыми.
Таким образом, пластичность-—важнейшее техническое свойство
пластиков, именно благодаря этому свойству они нашли такое ши-
рокое применение.
Неограниченность и доступность сырьевой базы. Синтетические
пластики получают путем химических превращений на основе реак-
ций поликонденсации и полимеризации, исходя из простейших хи-
мических веществ, которые, в свою очередь, получают из столь до-
ступных видов сырья, как уголь, известь, воздух, нефть и т. п.; для
модифицированных природных пластиков сырьем служат целлю-
лоза. белковые отходы, асфальты, битумы и др.
Для производства пластмасс используют и отходы лесотехниче-
ской промышленности, а также сельскохозяйственной, пищевой,
нефтяной, угольной и др.
Потенциальная сырьевая база для синтетических пластмасс
имеется в любой стране, и проблема получения исходных материа-
лов сводится, в основном, к проблеме энергии.
Промышленность пластмасс в настоящее время превратилась
в важнейшего потребителя основных химикалиев органического
синтеза.
♦ *
*
Наряду с перечисленными преимуществами, пластики обладают
и некоторыми недостатками, ограничивающими их применение.
Основной недостаток — низкий «потолок» теплостойкости (от 70
до 200°); только некоторые типы пластиков (кремнийорганические.
политетрафторэтилен) могут работать при несколько более высоких
температурах (до 300—350°).
Плохая теплопроводность пластиков (0,2—0,6 ккал/м • час • °C по
сравнению с 45 для стали, 10—40 для чугуна и 330 для меди), ко-
торая во многих случаях является их преимуществом, в некоторых
областях применения, где необходимо отведение тепла, — суще-
ственный недостаток.
22
Введение
Пластики по сравнению со стеклом, керамикой и металлами
имеют малую поверхностную твердость, что ограничивает во мно-
гих случаях их применение. Большой коэффициент термического
расширения (25-— 120 - 10~6 для пластиков и 11 • 10~6 для стали) и
малая теплопроводность пластиков обусловливают значительные
остаточные внутренние натяжения, которые могут быть причи-
ной появления трещин в изделиях в процессе эксплуатации
при резких изменениях температур; эти напряжения особенна
значительны в тех местах, где запрессована металлическая
арматура.
Даже жесткие типы органических пластиков в гораздо большей
степени, чем это наблюдается для керамики, стекла и металлов, об-
ладают медленно развивающимся пластическим течением — пол-
зучестью, причем эта ползучесть сильно увеличивается с темпера-
турой, что, естественно, должно быть учтено при конструировании
изделий.
Изделия из пластиков, имеющих линейную макромолекулярную
структуру, подвержены также медленным процессам самопроиз-
вольной деформации. Чем ближе температуры эксплуатации или
хранения изделия к температуре стеклования связующего, тем
быстрее изделие деформируется. В основе таких явлений лежат
релаксационные процессы, обусловленные высокоэластической де-
формацией при формовании изделий. В известной мере можно
значительно ослабить эти нежелательные явления путем формо-
вания изделий при более высоких температурах, лежащих в об-
ласти пластического течения.
Существенным недостатком некоторых пластиков является «ста-
рение», которое выражается как в медленно протекающих процес-
сах—окисления;—влагопоглощения,—снижения—поверхностной твер-
дости, потемнения, так и в самопроизвольном разрушении изделий.
Было показано, что на изделиях из ненаполненных хрупких
смол (например полистирола и др.) уже в процессе формования
образуются поверхностные микротрещины, которые в дальнейшем
под воздействием значительных внутренних напряжений приводят
изделие к преждевременному разрушению.
Эти нежелательные явления можно существенно уменьшить,
если применять правильные методы конструирования изделий и
соответствующий режим формования (прессования, литья под да-
влением и т. д.).
Чем глубже изучаются свойства пластмасс, тем в большей сте-
пени выявляются их технические преимущества, ведущие к новым
областям применения, однако одновременно вскрываются и их не-
достатки.
Современный этап развития промышленности пластмасс харак-
теризуется синтезом материалов с заранее заданными свойствами.
Все чаще конструктор не ограничивается «подбором» материала,
со стандартными свойствами, а требует создания новых материалов
Применение пластиков
23
с нужным ему комплексом свойств. Современное состояние техно-
логии пластмасс позволяет удовлетворить эти требования, так как
эта технология основана на фундаментальных достижениях химии
и физики высокополимеров.
ПРИМЕНЕНИЕ ПЛАСТИКОВ
Применение пластиков непосредственно связано с комплексом
их основных свойств. Дать перечень всех областей их применения
почти невозможно — значительно легче указать, в каких случаях
пластики неприменимы.
Пластики не могут быть применены во всех случаях, когда они
подвергаются постоянному действию температуры выше 200—300°,
когда требуются проводящие ток материалы с удельным объемным
сопротивлением ниже 106— 107 2 -см или когда требуется высокая
твердость (выше 3—4 по Моссу) и металлическая теплопровод-
ность.
Если первоначально пластические массы производили и при-
меняли главным образом как заменители цветных металлов, эбо-
нита и дорогих экзотических материалов (слоновой кости, черепахи,
перламутра и др.), то теперь они являются самостоятельным кон-
струкционным материалом и в значительной степени не могут
быть заменены какими-либо природными материалами и метал-
лами.
Стремление улучшить качество изделий, повысить их техниче-
скую и экономическую эффективность и привело к широкому при-
менению пластиков в ряде важнейших отраслей промышленности.
Общее машиностроение. В этой области пластики нашли эффек-
тивное применение для производства вкладышей подшипников для
блюмингов, металлургических станов, станков различных систем,
для изготовления шестерен, шкивов, тормозных колодок и т. п.
Автостроение. В автомашинах среднего класса свыше 200 дета-
лей изготовляют из пластмасс, в том числе магнето, штурвал,
шестерню распределительного вала, аккумуляторные баки и др.
Принципиально разрешен вопрос об изготовлении из пластмасс
всего кузова легкового автомобиля. Такой автомобиль будет легче
металлического примерно на 400—500 кг.
Авиастроение. Наряду с второстепенными деталями (корпуса
приборов, сидение пилота, шкивы) в авиастроении из пластиков
изготовляют все остекление и бронеостекление, а также детали
радиосвязи, пропеллеры, баки для горючего и др. В современной
авиационной технике стремятся к изготовлению цельнопластических
самолетов, в которых по существу только мотор будет сделан
из металла. Изготовление корпусов самолетов из пластмасс мето-
дом формования при низких давлениях (стр. 340) позволяет полу-
чать высокообтекаемые самолеты и решает проблему «миллиона
24
Введение
заклепок», которая связана с производством металлических само-
летов.
Судостроение. В этой области пластики нашли различное при-
менение в зависимости от характера и тоннажа судов. В крупном
судостроении пластики в большом количестве применяют для изго-
товления деталей приборов, штурвалов, в качестве облицовочного,
а также тепло- и звукоизоляционного материала и т. п. В производ-
стве малых судов, так называемого «москитного флота» — шлюпок,
катеров, самоходных и несамоходных судов пластические массы
применяют в качестве основного конструкционного материала, из
которого методом формования при низких давлениях изготовляют
корпуса судов.
Электротехника. Одной из первых отраслей машиностроения,
начавших применять пластмассы, была электротехника.
Различные слоистые пластики на основе термореактивных смол
являются важнейшим конструкционным и изоляционным материа-
лом современной сильноточной электротехники.
В современной слаботочной электропромышленности металл
в ряде случаев применяют лишь в качестве токопроводящих про-
водов, клемм и т. д., тогда как все остальные детали изготавливают
из пластиков.
Развитие промышленности современных средств связи, осно-
ванных на применении ультравысоких частот, стало возможным
лишь в результате освоения новых, более совершенных типов пла-
стиков, отличающихся, в частности, весьма малыми диэлектриче-
скими потерями в широком диапазоне частот и температур.
Можно полагать, что широкие перспективы развития, открытые
перед высокочастотной промышленностью, в свою очередь, несо-
миеино,приведут к разработке, открытию и освоению новых, более
совершенных типов пластиков.
----В последние годы большинство типов кабелей изготовляют с обо-
лочкой из пластмасс, которые заменяют одновременно свинец,
пряжу и каучук. При этом применение пластиков значительно по-
вышает электроизоляцию кабеля, снижает его вес и объем.
Химическая промышленность. В этой области пластики все
в большей степени находят применение в качестве химически стой-
ких конструкционных материалов и антикоррозийных покрытий.
Развитие промышленности пластиков привело к созданию совре-
менной химической аппаратуры из пластмасс любых конфигураций
и габаритов.
Применение химических аппаратов из пластиков взамен метал-
лов, в частности из пластиков, стойких по отношению к действию
соляной и разбавленной серной кислот, позволило во многих слу-
чаях радикально изменить химические процессы (например перейти
от серной кислоты к соляной) и таким образом усовершенствовать
и сделать рентабельным и возможным синтез ряда химических
веществ.
Применение пластиков
Оптическая промышленность. Многие оптические изделия (очки,
лупы, фотоаппараты, бинокли и др.) в настоящее время полностью
изготовляют из пластиков. Быстрота изготовления и многочислен-
ность выпускаемых изделий из пластиков в несколько раз понизили
стоимость биноклей и очков.
Органические стекла стали теперь серьезным конкурентом сили-
катных не только при остеклении самолетов, в производстве дально-
меров и других сложных оптических систем, но и при изготовлении
призм, линз и зеркал для ответственных астрономических приборов
(телескопов).
ЧАСТЬ I
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ТЕХНОЛОГИИ ПЛАСТМАСС.
ТЕРМИНОЛОГИЯ И КЛАССИФИКАЦИЯ
ГЛАВА I
ХИМИЧЕСКИЕ ОСНОВЫ ТЕХНОЛОГИИ ПЛАСТМАСС
ОБЩАЯ ХАРАКТЕРИСТИКА СМОЛ
Смолами первоначально называли только природные, аморфные
органические вещества, твердые или вязко-жидкие, большей частью
растворимые в органических растворителях и нерастворимые в воде.
С развитием производства синтетических смол термин «смола»
стали применять в более широком смысле, и теперь им обозначают
органические вещества в устойчивом аморфностеклообразном со-
стоянии. Основные характеристики смол следующие:
I. Смолы имеют аморфную стеклообразную структуру и яв-
ляются органическими стеклами. Некоторые, внешне аморфные
смолы имеют частично микрокристаллическую структуру.
II. Смолы всегда состоят из смеси молекул, обычно изомеров,
полимергомологов или близких по структуре молекул, образующих
лиофильный твердый раствор. Как коллоидам, смолам свойственна
структура геля или изогеля. Некоторые смолы (например, асфальты,
пеки) отличаются многофазной структурой и представляют собою
сложную дисперсную систему.
III. В отличие от аморфного состояния индивидуальных химиче-
ски* веществ, так иязывяемых «переохлажденных жидкостей»,___
аморфное состояние смол можно рассматривать как практически
стабильное и равновесное (стр. 83). ------
IV. Смолы состоят из сравнительно больших, сложных и неоди-
наковых по величине молекул; большей частью они относятся
к высокомолекулярным веществам. Средний молекулярный вес
является их основным и важнейшим показателем.
По структуре макромолекул различают два класса смол:
1. Смолы со сферической, (глобулярной) структурой.
2. Смолы с нитевидной (линейной) структурой.
Каждый из обоих классов смол, в свою очередь, делится на
плавкие и растворимые смолы, состоящие из молекул, связанных
межмолекулярными связями, и на неплавкие и нерастворимые,
представляющие собою молекулярные цепи или сферические
частицы, соединенные валентными связями. Для этих смол само
понятие «молекула» теряет свой обычный смысл.
К первому классу относятся главным образом низкомолекуляр-
ные смолы. Они состоят из небольших сферических молекул
(мол. вес 500—5000), связанных межмолекулярными силами при-
Общая характеристика смол 27
тяжения, характеризуются низкой температурой плавления, малой
прочностью и хрупкостью; растворы этих смол имеют нормальную
вязкость, которая не находится в прямой зависимости от их моле-
кулярного веса. Выше температуры стеклования эти смолы пока-
зывают пластическое течение; выше температуры плавления они
превращаются в жидкости с нормальным течением.
К смолам первого класса относится большинство природных
смол (канифоль и др.), пеки, асфальты, многие синтетические кон-
денсационные смолы, в том числе термореактивные (феноло-альде-
гидные и др.), в растворимом и плавком состоянии, в стадии А
(стр. 352). Некоторые из них (например термореактивные смолы)
способны к дальнейшим превращениям, которые приводят к замене
межмолекулярных сил валентными связями. В результате глобу-
лярные молекулы образуют гигантскую сферическую «молекулу» —
пространственный полимер.
Замена межмолекулярных связей валентными обусловливает
нерастворимость и неплавкость смол, значительно увеличивает их
прочность (от 10 до 100 раз) и затрудняет процессы пластической
деформации. К этим смолам относятся все термореактивные смолы
в стадии С (стр. 353)—феноло-альдегидные (резиты), амино-
формальдегидные, полиэфирные (глифтали) и др.
Ко второму классу в основном относятся линейные высокомоле-
кулярные смолы (мол. вес 5- 103 — 106 и больше).
Линейные цепи больших молекул этих смол могут иметь раз-
личную длину (до десятков тысяч онгстрем, при ширине в несколько
онгстрем), быть более или менее разветвленными, иметь различную
степень симметричности в расположении боковых групп и различ-
ную степень полидисперсности (стр. 55). Цепи переплетены между
собой и образуют структуру, напоминающую войлок.
Большая или меньшая степень упорядоченности структуры смол
второго класса, вплоть до образования микрокристаллической фазы,
зависит от симметричности линейных молекул и от величины меж-
молекулярных сил (стр. 116).
Смолы второго класса обычно плавки и растворимы. Вязкость
их растворов находится в зависимости от длины молекулярной
цепочки, причем растворы обладают аномальной вязкостью до
весьма малых концентраций, а расплавленные смолы не показывают
нормального течения вязкой жидкости (стр. 75).
Выше температуры стеклования эти смолы находятся в своеоб-
разном, характерном для них высокоэластическом состоянии, кото-
рое при дальнейшем повышении температуры может перейти в
пластическое; во многих случаях термическая деструкция насту-
пает раньше пластического течения. Ниже температуры стеклования
эти смолы теряют высокоэластические свойства и переходят в
твердое, упругое состояние.
Ко второму классу относятся также смолы со «сшитыми» це-
пями, структура которых представляет пространственную сетку
28
Гл. I. Химические основы технологии пластмасс
переплетенных цепей, связанных редкими поперечными мости-
ками.
‘ Наличие даже редкой валентной связи между цепями устраняет
плавкость и растворимость смолы. Смолы с «сетчатой» молекуляр-
ной структурой могут при нагреве лишь в большей или меньшей сте-
пени размягчаться, а действие растворителей приводит к набуха-
нию смолы, вследствие проникновения растворителя между «сши-
тыми» цепями. Чем чаще сетка, чем больше количество поперечных
мостиков между цепями, тем смола менее набухает в растворителях
и тем менее размягчается при нагреве.
Сетчатая структура значительно меняет и механические свой-
ства смол; валентные связи между цепями препятствуют смещению
этих цепей и этим затрудняют возможность пластической (необра-
тимой) деформации.
Когда же количество поперечных связей между цепями стано-
вится соизмеримым с количеством звеньев в цепи, образуется про-
странственный полимер, который по своей структуре мало чем
отличается от глобулярных пространственных полимеров в ста-
дии С.
V . Смолы не могут быть перегнаны без разложения даже
в вакууме. В результате перегонки получаются либо продукты
деструкции смол, либо отгоняются низкомолекулярные компоненты
смолы (например масла в асфальтах).
V I. Смолы могут быть непосредственно синтезированы в не-
растворимом состоянии или же вначале они растворимы, но при
дальнейшей термической или химической обработке переходят в не-
растворимое состояние. Растворимые смолы в зависимости от их
молекулярной структуры могут растворяться в неполярных и слабо-
- полярных или в полярных растворителях.- Некоторые смолы раетво-
римы также и в воде.
V H. Все растворы емол но- мерс испарения растворителя пере-
ходят в гель, содержащий большее или меньшее количество оста-
точного растворителя. Практически это выражается в образовании
пленок.
Способность образовывать пленки — важнейшее свойство смол.
Низкомолекулярные смолы образуют твердые, но хрупкие и непроч-
ные пленки, тогда как высокомолекулярные дают более прочные
и гибкие пленки.
VIII. Низкомолекулярные смолы имеют техническое применение
лишь при температурах ниже температуры стеклования; высоко-
молекулярные применяются при температурах как ниже, так и
выше температуры стеклования (в высокоэластическом состоянии).
ПРОЦЕССЫ ОБРАЗОВАНИЯ СИНТЕТИЧЕСКИХ СМОЛ
Синтетические смолы образуются в результате реакций полиме-
ризации и поликонденсации.
Процессы, образования синтетических смол
2®
Основные закономерности, приводящие к образованию высоко-
молекулярных синтетических смол и устанавливающие связь между
строением исходных молекул и свойствами получаемых полимеров,-
могут быть выражены следующими тремя постулатами:
1) высокомолекулярные соединения образуются лишь тогда,,
когда взаимодействующие молекулы полиреактивны;
2) сцепление происходит при контакте между двумя реактив-
ными точками молекул и определяется вероятностью такого кон-
такта;
3) физические свойства образующегося полимера, т. е. его
жесткость, гибкость, отвердеваемость при нагревании и т. п., опре-
деляются относительным размером и формой реагирующих моле-
кул, а также положением в них реактивных точек.
Первый постулат выражает возможность образования полимеров
лишь в случае, если реагирующие молекулы полиреактивны, т. е.
обладают свойством соединяться больше, чем- одной валентной
связью. Реакционная способность молекулы определяется числом
центров (точек) реакции.
Так, молекула этилового спирта монофункциональна, так как
имеет одну реактивную точку (ОН); молекула гликоля бифункцио-
нальна (2ОН); молекула глицерина — трифункциональна и т. д.
Двойная связь, способная к раскрытию, является бифункциональ-
ной группой, к полиреактивным относятся молекулы, имеющие
больше одной реактивной группы.
Допустим, что вещество А, имеющее одну функциональную
группу, реагирует с веществом В, имеющим также одну функцио-
нальную группу (реакция 1,1), с веществом С, имеющим две функ-
циональные группы (реакция 1,2), или с веществом D, имеющим
три функциональные группы (реакция 1,3), и т. д. Во всех этих
случаях в результате реакции можно ожидать образования лишь
простых химических соединений типа А—В, А—С—А или А—D—А,.
.4
функциональные группы которых быстро блокируются, насыщаются
в результате взаимодействия с монофункциональными молекулами.
Однако, если все реагирующие молекулы бифункциональны
(реакция 2,2), то после каждой реакции присоединения получаются
молекулы, имеющие на концах те же две функциональные группы,
что и исходные, и реакция, если не образуются циклы и концевые
группы не блокируются монофункциональными соединениями,
может идти в том же направлении беспрерывно, в пределе до тех
пор, пока из всех реагирующих простых молекул не образуется
одна гигантская линейная молекула, например из х молекул
СН2=СН2 одна макромолекула (—СН2—СН2—).„•
Реакция между бифункциональными молекулами ведет, следо-
вательно, к образованию лишь одномерных макромолекул, главным,
образом, линейного характера.
i() Г.г. I. Химические основы технологии пластмасс
Если реагируют молекулы с более высокой функциональностью
(реакции 2,3; 2,4; 3,3; 3,4 и т. д.), то после каждой реакции
присоединения свободные функциональные группы остаются не толь-
ко на концах, но и внутри цепей молекул; это приводит к реакциям
и между цепями, с образованием трехмерных пространственных
макромолекул (рис. 1). Чем длиннее синтезируемые линейные цепи
молекул, тем при меньшей концентрации три- и тетрафункциональ-
ных соединений происходит пространственная «сшивка» цепей и
а) о-------- 4- -е----о ------- О------О (1,1)
Рис. 1. Схема реакций моно- и полифункциональных молекул,
а)— тип 1,1; 1,2; 1,3; б) — тип 2,2; я) — тип 2,3.
образование нерастворимого полимера. При длине синтезируемых
молекул порядка 10 000 А достаточно ~ 0,01 % тетрафупкциональ-
ного компонента, чтобы получить пространственный полимер. Не-
растворимый полимер получается в том случае, если число молекул
«сшивки» становится соизмеримым с числом макромолекулярных
цепей.
Второй постулат формулирует основу кинетики процессов по-
лимеризации и поликонденсации.
Третий постулат указывает на зависимость свойств образую-
щихся полимеров от структуры исходных молекул, а также пути
к получению полимеров с желаемыми свойствами. Число функцио-
нальных групп реагирующих молекул определяет лишь общую
структуру образующегося полимера (линейную или пространствен-
ную), в то время как структура этих молекул определяет гибкость
возникающих макромолекул, их разветвленность, характер меж-
молекулярных сил и, следовательно, основные физические и меха-
нические свойства образующегося полимера — его твердость, гиб-
кость, эластичность и др.
Процессы, полимеризации
31
ПОЛИМЕРИЗАЦИЯ И ПОЛИКОНДЕНСАЦИЯ
Реакции, на основе которых получаются синтетические полимеры, — полиме-
ризация и поликонденсация — считаются принципиально отличными.
При полимеризации происходит соединение одинаковых или различных мо-
лекул и образующийся полимер имеет молекулярный вес, равный сумме молеку-
лярных весов реагирующих молекул, в то время как при поликонденсации соеди-
нение простых молекул и образование полимера сопровождается выделением
таких простых веществ, как Н2О, НС1 и др. и, следовательно, молекулярный вес
полимера уже не будет равен сумме молекулярных весов исходных молекул.
Поликонденсация представляет собою частный случай реакций замещения,
когда реагирующие молекулы полифункциональны и условия равновсия ие пре-
пятствуют образованию больших молекул.
Процессы полимеризации
Для однородных молекул процесс полимеризации может быть
представлен схемой
nA -> (Д)„,
где А — простая молекула (мономер); и— степень полимеризации.
Различают два принципиально отличных типа полимеризации:
ступенчатую полимеризацию и цепную.
Ступенчатая полимеризация
Ступенчатая полимеризация относится к типу обычных химических реакций,
протекающих путем последовательного контакта реагирующих молекул с обра-
зованием на каждой ступени реакции равновесных, устойчивых и изолируемых
промежуточных соединений. Обычно она протекает по принципу перескока водо-
рода, например:
1, R—СН=СН2 + НС=СН, —> R—СН2—СН2—С=СН2;
! I
2. R—СН,—СН2—С=СН2 + НС=СН2 —►
I I
R R
—> R—СН,—СН,—СН—СН,—С=СН, и т. д.
'I ‘I
R R
Скорость таких реакций определяется температурой и концентрацией ка-
тализаторов (обычно кислот и солей), а средний молекулярный вес получаемого
полимера тем выше, чем выше температура и длительнее процесс реакции.
Рассмотрим различные виды ступенчатой полимеризации.
1. Полимеризация ненасыщенных одинаковых молекул или различных мо-
лекул, каждая из которых способна к полимеризации. Примерами этого типа
реакций могут служить — реакция полимеризации изобутилена, впервые откры-
тая А. М. Бутлеровым, а также полимеризация формальдегида, окиси этилена,
индена, стирола и др.
Полимеризация стирола, изобутилена и других олефинов при высоких тем-
иературах и при действии катализаторов (HoSCU: SnCl2; ZnCl2; A1C1S и др.)
32
Гл. I. Химические основы технологии пластмасс
протекает по ступенчатому механизму, с перемещением подвижного водорода
от одной молекулы к другой и с образованием устойчивых изолируемых про-
межуточных соединений, например:
______ СН, СН3
сн3 ; ,сн3 | |
>С=СН9 4- СН2=С< —> СНз—С—СН=С—СНз ид. д.
СН/ ‘ Х'СН3 I
СН3
Этот механизм реакции приводит лишь к сравнительно низкомолекулярным
продуктам полимеризации, — к смеси димеров, тримеров и т. п. Теоретически
рассчитанная вероятность образования димеров и тримеров свыше 65%, тогда
как вероятность образования декамеров меньше 0,5%.
Смолы, получаемые в результате этого вида ступенчатой полимеризации,
легкоплавки, хрупки и обладают небольшой прочностью, характерной для простых
низкомолекулярных смол. Они не имеют большого промышленного значения.
При полимеризации формальдегида в водной среде реакция также проте-
кает ступенчато по схеме:
СН2=О + Н2О —>- НОСН2ОН
1 I
носн2он + сн2=о —> носн2—о—сн2он
НОСН2— О—СН2ОН + пСН2=О —> НО(—СН2— С— )„4.2Н.
В этом случае важнейшее условие реакции — присутствие воды. Очевидно,
достаточно даже следов воды, так как реакция начинается с образования мети-
ленгликоля (НОСН2ОН), продукт взаимодействия которого с формальдегидом
непрерывно сохраняет свою функциональность.
Образующийся в водной среде полимер — параформ — имеет небольшую
степень полимеризации 8—12; в присутствии серной кислоты степень полимери-
зации полимера может возрастать до 100 и выше.
При полимеризации безводного газообразного формальдегида получаются
высокомолекулярные полимеры, способные вытягиваться в нити, однако они об-
- разуются, повнднмому, уже но схеме цепной полимеризации.-------------
2. Полимеризация неодинаковых молекул, каждая из которых не способна
самостоятельно полимеризоваться, К этому.типу реакций ргносятся_.реакции.по-
лимеризации диизоцианатов с диоксисоединениями, с образованием полиурета-
нов, реакции полимеризации диизоцианатов с диаминами, с образованием полимо-
чевин и т. д. Они также основаны на перемещении подвижного атома водорода
от одной молекулы к другой, содержащей двойную связь, с последующим при-
соединением всего остатка первой молекулы:
RiO—Н + O=C=N— R2 —> Rj—С—СО—NH—R.,.
I t
Ряд последовательных реакций этого типа ведет к получению полимеров, на-
пример полиуретанов, при взаимодействии гликолей с диизоцианатами по схеме:
(х + 1)НС(СН2)ИСН 4-xCCN(CH2)„.NCO —>
—> H[C(CH2)nCCCNH(CH2)mNHCC]xC(CH2)nOH.
3. Полимеризация циклов, образованных в результате взаимодействия
сильных дипольных групп. Примером служит полимеризация капролактама
в присутствии е-аминокапроновой кислоты (образующейся в результате ката-
Процессы полимеризации
33
литического действия воды на капролактам). Эта реакция протекает ступенчато
и может быть представлена следующей схемой:
!.... !
H2N(CH,)5CO : ОН : + HN(CH2)5CO
—> H2N(CH2)5CONH(CH2)5COOH
и т. д. (стр. 598).
Полимеризации вида 2 и 3, в отличие от полимеризации вида 1, приводят
к получению высокомолекулярных продуктов и протекают в иных условиях, чем
реакции вида 1. В основе этих реакций возможно лежит другой механизм. Так
некоторые реакции вида 2 протекают ио ионному цепному механизму, а реакции
вида 3 по еще не выясненному механизму реакций ненасыщенных циклов.
Цепная полимеризация
Открытие цепных процессов — важнейшее достижение в учении
о скоростях химических превращений.
Механизм кинетических цепных реакций был теоретически раз-
работан Н. Н. Семеновым и, как известно, лежит в основе ряда хи-
мических процессов, в частности процессов горения, взрывов и др.
Цепным процессом определяется также механизм большинства
процессов полимеризации полифункциональных молекул. Экспери-
ментальное подтверждение было дано С. С. Медведевым, Штаудин-
гером и другими учеными.
Цепной процесс полимеризации, если обозначить через М — мо-
номер, М' —активированный мономер и через Av —квант энергии,
в общем виде может быть представлен следующими тремя этапами:
1) активация молекул и образование радикалов:
Л1 Av —> м‘;
2) развитие реакции — рост цепи:
Л]-+л} _> м;;
3) обрыв цепи:
п+г
Первый этап цепной реакции — активация мономера и образова-
ние свободных радикалов в случае полимеризации ненасыщенных
соединений сводится к раскрытию двойных связей валентно-ненасы-
щенных молекул мономера. Этот процесс не может протекать само-
произвольно, так как требует преодоления значительного энергети-
ческого барьера.
гХктивация мономеров может происходить следующими путями.
1. Путем поглощения кванта света при фотохимической реакции
CH2=CHR-|-ftv —> —СН2—CHR—.
Ьак. 1269. Э. И. Барг
34 Гл. I. Химические основы, технологии пластмасс
2. В результате соударения двух молекул под влиянием тепло-
вой энергии
2CH2=CHR —> —СН2—CHR—СН2—CHR—
3. Путем присоединения катализатора, например BF3, и созда-
ния комплексного свободного радикала
CH2=CHR4-BF3 —> BF3. СН2—CHR—
4. Взаимодействием мономера с имеющимися свободными ради-
калами (полученными при распаде инициаторов)
CH2=CHR4-CeH5----> CeH5—СН2—CHR—
В технике при проведении полимеризации активацию мономеров
чаще всего осуществляют с помощью катализаторов и инициаторов,
поэтому различают каталитическую и инициированную полимери-
зации. Ч
Общий механизм действия катализаторов основан на значитель-
ном понижении энергии активации. В качестве катализаторов поли-
меризации применяют различные кислоты, минеральные соли, осно-
вания, щелочные и щелочноземельные металлы (Na, BF3, SnCl4
и т. д.).
Катализаторы не входят в состав образующихся полимеров и
лишь участвуют в промежуточных этапах полимеризации, обра-
зуя нестойкие промежуточные комплексы, легко распадающиеся
в процессе дальнейшей реакции.
В отличие от катализаторов, инициаторы по окончании процесса
остаются в составе полимера. Их активирующее действие заклю-
чается в распаде или в диссоциации с образованием свободных
радикалов, которые соединяются с мономером, превращая его,
в свою очередь, в свободный радикал. Таким образом начинается
второй этап процесса полимеризации — рост цепи полимера. В ка-
честве инициаторов нашли применение главным образом различные
перекиси (органические и неорганические) и соли перкислот.
Однако подразделение на каталитическую и инициированную
недостаточно полно характеризует различия процессов цепной поли-
меризации. Существенное значение имеет не столько характер
активирующего вещества (катализатора или инициатора), сколько
определяемый им механизм активации молекул. В соответствии
с характером последнего различают: 1) радикальную (иницииро-
ванную) и 2) ионную (каталитическую) цепные полимеризации.
Многие мономеры (акриловые и метакриловые эфиры, хлори-
стый винил, хлористый винилиден и др.) полимеризуются только
по механизму цепной радикальной полимеризации. С другой сто-
роны, некоторые мономеры полимеризуются предпочтительно или
только по ионному механизму (винилкарбазол, простые виниловые
эфиры и др.).
Процессы по.шмер-лзац'ш
35
Радикальная (инициированная) цепная полимеризация
Радикальная полимеризация характеризуется образованием ра-
дикалов (мономеров, в которых имеется трехвалентный атом угле-
рода), сохраняющих свою радикальную природу на всем протяже-
нии роста цепи вплоть до ее обрыва. Поэтому основные закономер-
ности роста цепи определяются поведением трехвалентного атома
углерода.
В первом этапе цепной полимеризации, как уже было указано,
активация мономера не может протекать самопроизвольно из-за
сравнительно высокой энергии активации. Последняя значительно
снижается (до 500 кал/моль) вследствие образования свободных
радикалов и соединения их с мономерными молекулами. Этим
обеспечивается самопроизвольный рост цепи полимера.
Образование свободных радикалов может быть обусловлено раз-
личными причинами (действием света, тепла и т. д.); однако основ-
ное значение имеет влияние инициаторов. Так, обычно употребляе-
мая в качестве инициатора перекись бензоила (СеН5СОО)г рас-
падается на два радикала:
(с6н5соо)2 —► с6н; + со2 + с6н5соо-.
Аналогично распадаются и другие инициаторы (например, пере-
кись водорода Н2О2 ->2ОН’).
Распад инициаторов, в частности перекиси бензоила, как пока-
зал С. С. Медведев, в свою очередь, ускоряется присутствием реак-
ционноспособных мономеров. При этом акту возбуждения пред-
шествует образование начального комплекса мономера с инициато-
ром и уже при реакции этого комплекса с другими молекулами
мономера возникают начальные активные центры, обладающие
свойствами свободных радикалов:
1. RCOOOCOR + СН2=СН —> [(RCOOOCOR) + (СН2=СН)];
| Начальный комплекс |
Rt Ri
2. [(RCOOOCOR) + (СН2=СН)] + СН,=СН —>
Ri Ri
—> RCH2—CH- + RCOOCH-2—СН- + со2,
R1 R1
где R — СВН5; Ri — любой радикал.
Наличие осколков инициаторов в полимерах, полученных путем
радикальной полимеризации, было доказано С. С. Медведевым
и другими исследователями на примере полиметилметакрилата,
полистирола, полихлорвинила и др.
Для радикальной полимеризации характерна линейная зависи-
мость скорости образования активных центров от квадратного
корня концентрации инициатора; скорость образования активных
3S;
36
Гл. I. Химические ссновы u'c.xuo.iotuu пластмасс
центров определяет скорость конверсии мономера и, следовательно,
выход полимера (рис. 2).
Второй этап цепной полимеризации (стр. 33) — процесс роста
цепи полимера, в отличие от первого этапа — активации молекул,
сопровождается непрерывным выделением энергии и протекает
весьма быстро и самопроизвольно. При этом в процессе полимери-
зации происходит непрерывная
регенерация свободных валент-
ностей на концах растущей
цепи и непрерывное накопле-
ние радикалов (рис. 3).
ВремЯ' мин.
Рис. 3. Кривые полимеризации I и на-
копления радикалов 2 при полимериза-
ции хлоропрена под влиянием перекиси
бензола.
Рис. 2. Выхо! полибутатиена в за-
висимости от квадратного корня
концентрации инициатора (пе-
рекиси водорода).
Относительно малая энергия активаций " определяй "большую
скорость роста цепи, — во многих случаях цепь образуется практи-
чески мгновенно.
Следовательно, реакция роста цепи, если бы не существовали
причины ее обрыва, продолжалась бы до наступления полного
равновесия. Последнее лежит в области максимальной стабильности
и может быть теоретически достигнуто лишь тогда, когда все моно-
меры соединяются в один полимер:
л(СН2=СН)
I
R
—> (—СН?-СН—)„.
R
Третий этап цепной реакции — обрыв цепи — осуществляется
лишь вследствие насыщения свободных валентностей радикалов.
Это может быть достигнуто столкновением растущей цепи поли-
мера с другой растущей цепью или с активной молекулой моно-
мера, столкновением с молекулами растворителя или, наконец,
столкновением со стенками сосуда. Влияние относительной поверх-
ности реакционного сосуда на скорость реакции и на величину
Процессы полимеризации
37
молекулярного веса полимера является характерной особенностью
радикального процесса полимеризации.
При обрыве реакции в результате взаимодействия двух расту-
щих цепей могут быть два случая:
1) соединение двух растущих цепей монорадикалов или расту-
щей цепи с активной молекулой мономера, с образованием одной
неактивной макромолекулы:
ксн—СН.,— + ксн—сн,—
КСН—СН,— СН,—снк
R R
Растущие цепи
(К — присоединившийся остаток инициатора);
2) переход атома водорода от одной реагирующей цепи к дру-
гой; в результате прекращается рост обеих цепей с образованием
двух неактивных макромолекул (диспропорционирование):
I
КСН-СН2— + ксн—СН2-----► КС=СН2+ ксн—сн3.
При столкновении с неактивной молекулой или с молекулами
растворителя возможен процесс переноса подвижным атомом энер-
гии растущей цепи на неактивные и насыщенные молекулы (про-
цесс переноса цепей). В этом случае растущая цепь теряет свою
активность и становится неактивным полимером; в то же время
образуется новый радикал, дающий начало роста новой цепи:
1. ксн—СН,-(- СС14
КСН—СН,С1 Н--СС13,
2. —СС13 + СН=СН2 —► С13С—СН—сн,-
Насыщение цепи может быть также вызвано столкновением
с активированными молекулами мономера или полимера, способ-
ными выделять водород; этот водород и насыщает активные группы
растущих цепей:
КСН—СН2---h ксн—СН---- ксн—СН—р КСН—СЩ.
' I II I
R R R R
В этом случае может произойти и разветвление второй расту-
щей цепи.
38
Гл. I. Химические основы. технологии пластмасс
В некоторых случаях, в особенности если длина растущей
цепи еще невелика, насыщение свободных валентностей может
происходить путем образования циклов:
—СН,—С Н—СН,- СН-СН,—СН—
' I I I
R R R
R
СН—СН2
—> НгС^ 'cH— R.
Хсн—СН2
R
Скорости обрыва цепей также велики, однако они значительно
меньше, чем скорости роста радикалов.
Характерной особенностью цепной полимеризации является то
обстоятельство, что средняя степень полимеризации (молекулярный
вес полимера) определяется не закономерностями химического
равновесия, как это, например, имеет место в реакциях поликонден-
сации и ступенчатой полимеризации, а лишь кинетикой процесса,
т. е. относительной скоростью каждого этапа цепной реакции.
Процесс полимеризации может быть изучен путем определения
двух основных величин: 1) скорости исчезновения мономера (ско-
рости конверсии) в различных стадиях процесса в зависимости от
температуры и концентрации инициатора и 2) средней степени
полимеризации в зависимости от условий, указанных в п. 1.
Важнейшая характеристика цепной полимеризации — устано-
вленная опытом зависимость конверсии мономера и независимость
среднего молекулярного веса полимера от времени полимеризации
. (в широких пределах- каждог-а-втапа-процесса полимеризации).--
Так как конечная длина цепи получается весьма быстро .(пра-
ктически мгновенно^ после образования активной молекулы, то
с течением времени меняется в основном степень конверсии.
Вместе с тем, на различных этапах процесса полимеризации
в зависимости от изменения в реакционной среде концентрации
мономера длина образующейся макромолекулы может быть раз-
личной. Это обусловлено тем, что с изменением концентрации
мономера меняются условия взаимодействия растущих радикалов
как с мономером, так и между собой.
Средняя степень полимеризации, так же как и скорость про-
цесса исчезновения мономера, зависит от температуры, количества
и характера инициатора. При этом скорость конверсии увеличи-
вается с повышением температуры и с увеличением количества
инициатора, тогда как средняя степень полимеризации при этом
уменьшается. В первом приближении можно считать, что факторы,
ускоряющие процесс реакции, обусловливают уменьшение средней
степени полимеризации, за исключением действия эмульгаторов при
эмульсионной полимеризации (стр. 164).
Процессы полимеризации
39
Средняя степень полимеризации (Р) зависит от соотношения
скорости роста образовавшегося радикала (Уроот) и скорости
обрыва цепи (У об? )
р _ ^Р»ст
^Обр ’
т. е. определяется количеством элементарных актов присоединения
мономера к радикалу до его обрыва (насыщения).
Температурная зависимость Р связана с влиянием температуры
на соотношение скоростей Уроот и УОбР. __
Если на оси ординат нанести логарифм Р, а на оси абсцисс
обратные абсолютные температуры (рис. 4), то графически эта
зависимость выразится в виде прямой; это указывает на обратную
экспоненциальную зависимость Р от
температуры.
Скорость конверсии определяется
скоростью первичного акта — обра-
зования радикалов. Так как можно
считать, что на каждую макромо-
лекулу приходится одна активиро-
ванная молекула (один радикал), то
скорость активации Va должна рав-
няться общей скорости конверсии
Vrohb, деленной на среднюю степень
полимеризации Р:
у
т, г конв
Рис. 4. Зависимость 1g средней
степени полимеризации Р от об-
ратной температуры-^г.
Таким образом, скорость конвер-
сии мономера (У конв) пропорцио-
нальна температуре и количеству
инициатора. Средняя же степень полимеризации (Р) обратно про-
порциональна скорости конверсии (рис. 5), следовательно, прибли-
женно имеет место закономерность:
Р Уконв = const.
Кинетика процесса полимеризации по цепному механизму опре-
деляется таким образом соотношением скоростей активации мо-
лекул, роста и обрыва цепей. Для большинства мономеров реак-
ция начинается не сразу, а лишь после некоторого периода —
периода индукции, который длится иногда несколько часов. Это
может быть объяснено наличием в мономере следов ингибиторов.
Способность к полимеризации естественно определяется строе-
нием полифункциональных молекул.
В этом отношении существует весьма широкая градация, — от
молекул, которые полимеризуются на холоду и весьма быстро
40
Гл. I. Химические основы технологии пластмасс
(например, акролеин), до молекул, которые практически не полиме-
ризуются или полимеризуются лишь при высоких температурах и
давлениях.
Очевидно, что факторы, облегчающие раскрытие двойной связи
ненасыщенных молекул при столкновении их друг с другом или
с активированными молекулами (радикалами), обусловливают
склонность мономера к полимеризации.
3000-
I 2500-
I 2000-
1500-
1000-
500-
0 0,05 0,10 0,15 0,20 0,25
Концентрация инициатора,0/»
Рис. 5. Зависимость скорости конверсии Уконв
и средней степени полимеризации Р от концентрации
инициатора (перекиси бензоила) для полистирола.
Известно, что симметричные ненасыщенные молекулы или нс
полимеризуются или же полимеризуются значительно труднее, чем
несимметричные. Поэтому введение заместителя в молекулу эти-
лена значительно увеличивает его способность к полимеризации,
притом в тем большей - степени, “чем- - менее - симметричной стано-
вится молекула при замещении.
Введение же заместителей, не нарушающих симметричность
молекулы, как правило, делает ее совсем неспособной к полимери-
зации. Так, введение в молекулу этилена С6Н5, С1, Вг и др. при-
водит к таким легко полимеризующимся соединениям^ как
C6HsCH=CH2, С1СН=СН2, ВгСН—СН2; введение двух заместите-
лей у одного атома углерода еще больше усиливает асимметричность
молекулы и, следовательно, еще больше увеличивает склонность
к полимеризации; это видно на примере таких соединений, как
С12С=СН2; (СН3)2С=СН2 и др. Если оба заместителя расположены
симметрично у обоих атомов, то молекула уже неспособна к поли-
меризации; так СбН5СН=СНС6Н5; С1СН=СНС1; ВгСН=СНВг
ит. п. — не полимеризуются.
С физической точки зрения степень симметричности молекулы
определяется степенью ее полярности и может быть выражена ве-
личиной дипольного момента молекулы.
В полярных молекулах центры положительных и отрицательных
зарядов смещены, причем тем больше, чем менее симметричны
Процессы полимеризации
41
молекулы. Схематически неполярные молекулы можно изобразить
<•' о о
в виде СН2=СН2, а полярные СН3—СН=СН2.
Поляризация двойной связи, обусловливаемая введением заме-
стителей, невидимому, разрыхляет эту связь, снижает энергию
активации. Известное влияние на механизм взаимодействия поляр-
ных молекул должен оказывать фактор электростатического при-
тяжения диполей, дающий направленность в сочетании мономеров
в цепи, например, по схеме:
СН2=СН + СН,=СН —> —СН2—СН—СН,—СН— И т. д.
XX XX
Однако фактор полярности сам по себе все же не может с доста-
точной полнотой объяснить все многообразие склонности молекул
к полимеризации.
Прежде всего существует значительная градация в склонности
к полимеризации между неполярными молекулами. Некоторые из
них сравнительно легко полимеризуются (CF2—CF2), другие значи-
тельно труднее (СН2=СН2) и третьи, представляющие большин-
ство, невидимому совсем не полимеризуются (СС12=СС12). Кроме
того, известны случаи, когда усиление полярности не только не уве-
личивает склонности к полимеризации, а, наоборот, уменьшает или
совсем устраняет ее; так, СбН5СН=СН2 хорошо полимеризуется,
а (СбН5)2С=СН^ совсем не полимеризуется, несмотря на большую
асимметричность и полярность.
Для объяснения вышеизложенного В. В. Коршак указал на
влияние пространственных затруднений при полимеризации, связан-
ных с таким стереохимическим фактором, как объем замещающих
атомов и групп. Действительно, вполне вероятно, что на первом
этапе реакции — активации молекул — большое влияние может ока-
зать экранирующий эффект заместителей, пропорциональный их
объему и количеству. Чем больше объем и количество заместителей,
тем больше затруднений для контакта реагирующих молекул
с теми атомами, между которыми образуется новая связь. Поэтому
можно ожидать, что чем менее громоздки замещающие группы, тем
при большем числе заместителей молекула будет способна к поли-
меризации. Так, фторпроизводные этилена способны к полимериза-
ции при всех степенях замещения, в то время как среди производ-
ных, содержащих фенильный радикал, значительно более громозд-
кий, чем фтор, к полимеризации способен только монозамещенный
этилен.
Однако и стереохимические представления не объясняют с до-
статочной полнотой все разнообразие в склонности молекул к поли-
меризации. Так, CF2=CF2 значительно легче полимеризуется, чем
СН2=СН2, несмотря на то, что радиус фтора значительно больше,
чем радиус водорода.
42
Гл. I. Химические сснсвы технологии пластмасс
Необходимо отметить, что до настоящего времени нет еще
вполне надежного критерия для суждения о склонности молекул
к полимеризации. Возможно поэтому, что наблюдаемые отклонения
от закономерностей связаны с наложением двух или нескольких
механизмов, определяющих склонность ненасыщенных молекул
к полимеризации.
Для практической оценки влияния заместителей на увеличение
склонности производных этилена к полимеризации (при наличии
одного заместителя) можно составить следующий ряд:
H<R<OR<C6H5<RCOO<C1<J,
где R — алкил.
Инициаторы, ингибиторы и регуляторы
Скорость полимеризации и длина растущей цепи полимера
определяются количеством активных центров — радикалов, обра-
зующихся при распаде инициаторов.
Для понимания процессов, протекающих при радикальной цеп-
ной полимеризации, необходимо иметь в виду, что радикалы, воз-
никающие при распаде перекисей, способны не только возбуждать
полимеризацию, образуя активные молекулы полимера, но и обры-
вать ее, присоединяясь к растущей полимерной цепи. Двойной
механизм действия инициаторов — наиболее существенное отличие
их от обычных катализаторов.
Наряду с инициаторами известны вещества, которые не уско-
ряют, а замедляют процесс полимеризации; такие вещества назы-
вают ингибиторами. К ингибиторам относятся фенолы, хиноны,
амины, некоторые металлы (медь). - Ингибиторы- играют важную
роль в технике производства и переработки мономеров, так как
предохраняют нх от преждевременной полимеризации.
Механизм действия ингибиторов основан на разрушении (вос-
становлении) перекисных соединений, образующихся в мономере
под влиянием кислорода воздуха. Так, гидрохинон, восстанавливая
перекись, превращается в хинон. Действие ингибиторов заклю-
чается, однако, не только в восстановлении перекисей в мономере,
но и во взаимодействии с образующимися радикалами и растущими
цепями. Ингибиторы обрывают рост цепей и превращают радикалы
в неактивные молекулы. В этом случае ингибиторы, так же как и
инициаторы, входят в состав цепи и могут быть обнаружены в поли-
мере (Б. А. Догадкин, А. Л. Клебанский и др.).
Особое место среди веществ, влияющих на характер полимери-
зации, занимает атмосферный кислород. В ряде случаев он
является весьма эффективным инициатором, тогда как для некото-
рых веществ служит ингибитором. В других случаях влияние кис-
лорода зависит и от его количества: в малых количествах он дей-
ствует как инициатор, в больших — как ингибитор.
Процессы полимеризации
43-
Было доказано, что термическая полимеризация стирола при
полном отсутствии кислорода протекает крайне медленно, тогда как
следы кислорода уже значительно ускоряют реакцию. В то же
время при полимеризации стирола и других мономеров в присут-
ствии несколько больших количеств кислорода — он является инги-
битором. В этом случае кислород реагирует преимущественно со
свободными стирольными радикалами, образуя прочную полимер-
ную перекись состава
(—СН—СН2—О—С—)д..
От ингибиторов следует отличать Так называемые регуляторы —
вещества, механизм действия которых основан на переносе или
передаче цепи реакции. Введение в реакцию регуляторов обеспечи-
вает возможность регулирования средней длины цепей полимера
при сохранении средней скорости полимеризации. Среди регулято-
ров особое значение получили меркаптаны (амилмеркаптан, доде-
цилмеркаптан), а также четыреххлористый углерод, дихлорэтан
и др.
Передача цепи под действием регуляторов происходит при столк-
новении радикалов растущей цепочки полимера с молекулами регу-
лятора. В случае, например, четыреххлористого углерода или
меркаптана от регулятора отрывается атом хлора или, соответ-
ственно, водорода, которые, с одной стороны, присоединяясь
к растущему радикалу, делают его неактивным и прекращают его
рост, а с другой, — превращают регулятор в радикал; последний
начинает рост новой цепи. Естественно, что такой процесс должен
в итоге привести к снижению среднего молекулярного веса
полимера.
Подобно инициаторам, регуляторы входят в состав цепи поли-
мера, так как каждая молекула регулятора, служившая началом
роста новой цепи, обусловливает образование одной молекулы
полимера.
Наличие остатков регуляторов в цепях полимеров было экспе-
риментально доказано советскими учеными.
Обычные растворители также можно рассматривать как регуля-
торы процесса полимеризации. Механизм их действия сводится
к различным процессам, среди которых существенны активация
молекул мономеров при их столкновении с молекулами раствори-
теля, обрыв роста цепи, а также процессы передачи молекуле
растворителя энергии растущей цепи, что ведет к уменьшению
средней степени полимеризации.
Закономерности влияния растворителя очень сложны и зависят
от структуры растворителя. Наиболее вероятно, что активация мо-
лекул мономера происходит при их взаимном столкновении, а не
при столкновении с молекулами растворителя, что, естественно,.
И
Гл. f. Химические основы техно гогии пластмасс
по мере уменьшения концентрации мономера, снижает скорость
реакции. С другой стороны, возможно, что процессы насыщения
(обрыва) растущей цепи происходят быстрее при столкновении
с молекулами растворителя, а это ведет к уменьшению средней
степени полимеризации.
Практически действие растворителей выражается как в сниже-
нии скорости полимеризации (скорости конверсии), так и в умень-
шении средней степени полимеризации.
При этом растворители действуют специфически: снижение как
скорости, так и средней степени полимеризации различно для
каждого растворителя.
Ионная (каталитическая) полимеризация
Как уже было указано, цепная полимеризация может протекать как по
радикальному, так и по ионному механизму.
Раскрытие двойной связи мономера при ионной полимеризации обусловли-
вается ее поляризацией (смещением совместных электронов к одному из атомов
углерода, соединенных двойной связью).
Ионный механизм цепной полимеризации определяется действием катализа-
торов, которые, в отличие от инициаторов, создают с мономером лишь крайне
неустойчивый комплекс и поэтому в состав полимера не входят. Однако не-
которые катализаторы ионной полимеризации, повидимому, также входят в со-
став полимера; тем не менее, они определяют ионный механизм полимеризации.
Такие катализаторы, как шелочные металлы (натрий, литий), трехфтористый
бор, хлористый алюминий и др., в определенных условиях вызывают цепную
полимеризацию, ведущую к получению высокополимеров. Свободные кислоты
в большей мере определяют ступенчатый характер полимеризации.
Однако необходимо указать, что закономерности ионной (каталитической)
полимеризации изучены еще недостаточно и в ряде случаев механизм процессов
еще не выяснен.
Ионная цепная полимеризация, так же как радикальная, протекает по трем
послёдоватёльным этапам? 'активация мблёкул)Трост' цепи и обрыв ее.
Первичный акт — активация молекул в некоторых случаях (например, под
влиянием безводных галогенидов, металлов и т. п.) может происходить при
весьма низких температурах (—100°), что указывает на резкое снижение энер-
гии активации катализатором. Значительно меньшая, по сравнению с радикаль-
ной полимеризацией (в два и более раза), энергия активации весьма харак-
терна для ионной цепной полимеризации.
Повидимому, основное действие катализатора сводится к первоначальному
образованию промежуточного комплекса по схеме:
МХ„ + СН2=СН —> МХ„.СН2—сн,
где М -• -металл, X--галоид, п— число атомов галоида.
Дальнейший рост цепи может происходить по различным механизмам в за-
висимости от характера мономера и катализатора. Так, под влиянием кислот он
Происходит по так называемому карбониевому механизму, когда растущий ком-
плекс сопержит на конце трехвалентный атом углерода:
е ®
мх„ • сн2—сн—...— сн2—сн.
R R
Процессы полимеризации
По такому механизму, невидимому, протекает полимеризация стирола в при-
сутствии SnCU и НС1. По данным С. С. Медведева, скорость полимеризации при
атом пропорциональна концентрации ЪпСц. Активация и рост цепи в этом про-
цессе могут быть представлены схемой:
SnCl4 + НС1 —> S11CI4 • НС1 —> Н + Snct- а;
© ©
н + СН2=СН —> СН3—СН;
I I
С6Н5 С6Н6
СН,—СН + СН.—СН •—> СН,—СН—СН,—СН и Т. д.
I ‘I 1’1
С6н5 С6Н3 СвН5 СВН5
М. Ф. Шостаковский полагает, что активация виниловых эфиров происхо-
дит путем образования оксониевого соединения за счет эфирного кислорода:
СН,=СН—O-f-HX—> ГСН,=СН—СН1®
I I X®
R R 1
Многие катализаторы (BF3, TiCl4 и др.) действуют лишь в случае наличия
в мономере небольшого количества так называемых активаторов (воды, спирта
и других соединений с подвижным водородом). В этом случае катализатор
первоначально образует комплекс с активатором;
МХ„ + HR —> МХ„ HR,
который при взаимодействии с мономером приводит к образованию активных
молекул по схеме:
сн3 сн3
МХ„ HR + СН2=С —> MX„R + СН3-С®
СН3 СН3
Рост цепи в этом случае идет по схеме:
СН3 СН3 СН3 СН3
СН,—с® + СН,=С —> СНУ—С—СН,—С® и т. д.
I "I
сн3 СН3 СН3 сн3
При полимеризации диеновых соединений в присутствии щелочных метал-
лов действует механизм непрерывного передвижения катализатора к концам
растущей цепи:
2Na + СН,=СН—СН=СН2 —> NaCH2—СН=СН—CH2Na
NaCH,—СН=СН—CH2Na 4- СН,=СН—СН=СН2 —►
—> NaCH2—СН=СН—СН2—СН2—СН=СН—CH2Na и т. д.
Обрыв цепи по этому механизму может быть осуществлен образованием не-
активных молекул в результате отщепления катализатора от концов растущих
цепей.
46
Гл. Л Химические основы технологии пластмасе
Характерное отличие кинетики ионной полимеризации от радикальной, как
это доказал С. С. Медведев с сотр., заключается в линейном изменении
скорости полимеризации в зависимости от концентрации катализатора; скорость
же радикальной (инициированной) полимеризации пропорциональна квадратному
корню концентрации инициатора. Однако основные зависимости ионной поли-
меризации (например, молекулярного веса полимера от температуры, кон-
центрации катализатора и т. д.) имеют в основном тот же характер, что и при
радикальной полимеризации. Процесс передачи цепи, повиднмому, играет мень-
шую роль, чем при радикальной полимеризации, и основные зависимости опре-
деляются лишь распадом образующихся комплексов с освобождением катали-
затора в диссоциированном состоянии.
Совместная полимеризация
Образование полимера может происходить путем цепной поли-
меризации не только одинаковых, но и разнородных полифункцио-
нал ьных молекул по схеме:
пА + тВ —> ... — А — В — А— В — ...
Такую полимеризацию называют совместной полимеризацией,
или сополимеризацией.
Следует отметить, что образующийся при сополимеризации поли-
мер лишь очень редко имеет такое правильное чередование элемен-
тов. обоих мономеров, какое показано на схеме, и соотношение
элементов обоих мономеров в цепи обычно также не соответствует
соотношению мономеров в реакционной смеси.
Сополимеризация была впервые осуществлена еще в 1887 г.
русским химиком В. А. Солонина, который при действии SO2 на
аллиловые эфиры получил высокомолекулярные сульфоны. Этим
он открыл не только процесс сополимеризации, но также и особый
тип сополимеризации, так- называемую истинную гетерополимери-
зацию (стр. 48).
И. Л. Кондаковым в 1912 г. впервые был описан дивиниловый
тип сополимеризации. Он получил каучукоподобные массы сополи-
меризацией бутадиена и диметилбутаднена.
Процессы сополимеризации изучались также рядом других рус-
ских и советских ученых: И. И. Остромысленским, Б. Н. Рутовским,
С. С. Медведевым, С. Н. Ушаковым, А. А. Ваншейдтом и др.
Сополимеризация получила весьма большое значение в совре-
менной технике пластмасс и синтетических каучуков. Ряд важней-
ших технических продуктов мог быть получен только на основе
сополимеризации. Широко известны сополимеры хлористого винила
с хлористым винилиденом и винилацетатом, сополимеры акриловых
эфиров, стирола, винилкарбазола и др.
Бутадиенстирольные и бутадиенакрилонитрильные типы каучу-
ков получили большое промышленное значение. С каждым годом
увеличивается число технически важных сополимеров. Изучение
теории и техники процессов сополимеризации имеет исключительно
актуальное и важное значение.
Процессы полимеризации
47
Сополимеризация имеет своей целью создать материал с ком-
плексом свойств, которым не обладает полимер каждого из двух
или трех мономеров в отдельности. В результате сополимери-
зации нарушается правильность структуры макромолекул, что за-
трудняет процессы кристаллизации. Сополимеры являются обычно
аморфными веществами. .
Часто сополимеризацией достигают снижения температуры
стеклования, увеличения гибкости и эластичности материала; в
этом случае эффект сополимеризации подобен эффекту пластифи-
кации; такую сополимеризацию называют внутренней пластифи-
кацией (стр. 137).
Сополимеризация имеет своей целью в некоторых случаях по-
вышение теплостойкости, твердости, получение неплавких и нерас-
творимых материалов. В этом случае сополимеризация приводит
к образованию пространственных полимеров путем «сшивки» линей-
ных макромолекул.
Процессы сополимеризации можно разделить на два основных
типа:
I. Сополимеризация моновинильных (бифункциональных) со-
единений.
II. Сополимеризация моновинильных соединений с дивиниль-
ными (тетрафункциональными).
I. Сополимеризация моновинильных соединений. Не все поли-
меризующиеся соединения способны к сополимеризации. В ряде
случаев вместо ожидаемого сополимера образуется смесь индиви-
дуальных полимеров или же полимер только одного из мономеров.
Так, стирол и винилацетат в отдельности хорошо полимеризуются
по радикальному механизму. При совместной же полимеризации
этих веществ вначале получается только полистирол, и лишь когда
стирол практически исчезает из реакционной смеси образуется вто-
рой полимер — поливинилацетат, т. е. образуется не сополимер,
а в основном смесь полимеров.
С другой стороны, сополимеризация может происходить и между
ненасыщенными соединениями, из которых каждое в отдельности
не полимеризуется.
Таков, например, случай сополимеризации стильбена и малеи-
нового ангидрида в бензольном растворе. Каждый из этих компо-
нентов в отдельности не способен полимеризоваться, при совмест-
ном же их нагревании образуется сополимер со строго чередую-
щимися мономерами в соотношении 1:1.
лСН=СН + отСН=СН —> ... —СН—СН—СН—СН—СН—СН—СН—СН— ...
II I 1 I I I I I I I 1
/\ /\ со со /\ /\ со со /\ /\ со со
III \/ ||||\/||||\/
о
о
о
48
Гл. I. Химические основы технологии пластмасс
По такому же типу сополимеризации образуются сополимеры
олефинов с SO2. При этом реакция протекает по схеме:
О О
II II
СН=СН, + SC, —> ...—СН—СН,—S—СН—СН,—S—СН—...
I I 11 I II I
R R О R О R
Такой вид сополимеризации, когда каждый мономер в отдель-
ности не способен к полимеризации, а образующийся сополимер
состоит из правильно чередующихся исходных мономеров (в соот-
ношении 1:1, вне зависимости от соотношения мономеров в реак-
ционной смеси) называют истинной гетерополимеризацией.
Известен также тип обычной гетерополимеризации, когда в со-
вместную полимеризацию вступают два мономера, из которых
только один не склонен к самостоятельной полимеризации, а вто-
рой является легко полимеризующимся. Примером может служить
сополимеризация стирола (легко полимеризуется) с малеиновым
ангидридом (в отдельности не полимеризуется):
/iCH=CH2-j-mCH=CH —> ...—СН—СН2—СН—СН—СН,—СН—СН—СН—...
I II I I I “ I I I
/\ СО СО /\ СО СО /\ СО СО
При обычной гегерополимеризации в образующемся полимере
обычно нет строгого чередования в расположении мономеров, однако
мономер не полимеризующегося компонента (малеиновый ангидрид)
непременно должен чередоваться с мономером полимеризующегося
компонента (стирола), т. е. сополимер может иметь структуру:
.СН—СН,—СН—СН—СН,—СН—сн,-
I----г--Г-------- I------
/\ СО СО /\
СН—СН—СН—СН,—СН
Как уже указано, состав сополимера редко соответствует соот-
ношению исходных мономеров в первоначальной смеси; однако
известны случаи, когда при любом соотношении в исходной смеси
образующийся сополимер по составу точно соответствует соотноше-
нию мономеров в исходной смеси. Так при сополимеризации хлор-
винилидена с метилакрилатом, исходя из соответствующего соот-
ношения этих мономеров в исходной смеси, можно получить сопо-
лимер с заранее заданным соотношением обоих мономеров в цепи.
Такой вид сополимеризации получил название азеотропической
сополимеризации (аналогия с перегонкой нераздельно кипящей
жидкости).
Особенности сополимеризации находят объяснение в химической
структуре реагирующих мономеров. Активность различных моно-
меров по отношению к данному радикалу растущей цепи опреде-
ляется степенью полярности, а также стереохимическими факторами.
Процессы полимеризации
49
Многие наблюдаемые явления сополимеризации легко объяс-
нить, если считать, что активность мономеров при сополимеризации,
так же как и при полимеризации, зависит от числа, размеров и по-
ложения заместителей.
Влияние этих факторов объясняется тем, что большое число
громоздких заместителей «экранирует» двойную связь и затрудняет
приближение второго мономера; поэтому такие соединения с че-
тырьмя громоздкими группами, как тетрафенилэтилен, тетраиодэти-
лен, не полимеризуются и не вступают в сополимеризацию, в то
время как тетрафторэтилен, имеющий такое же число заместителей,
но значительно меньших размеров, полимеризуется и легко всту-
пает в реакцию сополимеризации.
Однако только стереохимическими факторами, так же как в слу-
чае полимеризации, невозможно объяснить все многообразие раз-
личий в активностях мономеров при сополимеризации, в частности
мономеров с одним заместителем, где пространственные затрудне-
ния не имеют существенного значения. Так же, кай и при полиме-
ризации, существенным фактором, влияющим на активность моно-
меров при сополимеризации, является степень их полярности.
Однако влияние степени полярности при сополимеризации изучено
менее, чем при полимеризации. Для некоторых реакций наблюда-
лось, что мономеры, в которых нет сопряжения со связью С=С
этилена (винилацетат, аллилацетат и др.), обладают меньшей
активностью, чем мономеры, у которых имеется сопряжение с двой-
ной связью (бутадиен, стирол и др.).
II. Сополимеризация моновинильных соединений с дивиниль-
ными. В результате сополимеризации этого типа получаются в за-
висимости от структуры дивинильного мономера или линейный
ненасыщенный сополимер, или же пространственный, трехмерный
сополимер, где макромолекулы «сшиты» поперечными связями
тетрафункционального (дивинильного) соединения.
Примером получения ненасыщенного линейного сополимера
может служить реакция сополимеризации бутадиена со стиролом,
при которой в сополимере остается ненасыщенная связь:
сн2=сн—сн=сн2 + сн2=сн —>
—► ... — сн2—сн=сн—сн2— сн2—сн-сн2— сн=сн—сн2— сн2— сн—...
Получаемые стабильные ненасыщенные сополимеры обычно
подвергаются дальнейшей термической обработке или воздействию
серы или других «вулканизующих» веществ, посредством которых
4 Злк. 1269 Э. И, Барг
50
Гл. I. Химические основы технологии пластмасс
раскрывается двойная связь в сополимере и «сшиваются» макро-
молекулы. Этим путем линейные ненасыщенные полимеры обра-
зуют макромолекулярную трехмерную сетку — переходят в про-
странственный полимер.
Чем больше ненасыщенных связей в сополимере, тем в боль-
шей степени можно «сшить» макромолекулярные цепи. Регулируя
концентрацию дивинильного мономера (например бутадиена),
можно получать сополимер с желаемой степенью ненасыщенности
и, следовательно, после предельной «вулканизации» с заданной
структурой макромолекулярной сетки — с определенным расстоя-
нием между узлами такой сетки.
Примером непосредственного образования пространственного
сополимера может служить реакция сополимеризации стирола
с л-дивинилбензолом
X СН=СН.>+ у СН=СН, —>
сн=сн2
стирол л-дивинилбензол
.СН—СН2—СН—СН2—СН—СН2—СН—.
В этом случае обе ванильные группы дивинилбензола реагируют
с равной или близкой активностью, что, естественно, приводит
к непосредственной «сшивке» образующихся цепей в процессе сопо-
лимеризации. При этом на определенной стадии реакции проис-
ходит желатинизация — образуется нерастворимый пространствен-
ный агрегат (гельфракция). Таким образом, пространственные сопо-
лимеры могут состоять из двух фракций; линейных макромолекул
(зольфракция) и погруженной в их среде пространственной макро-
молекулярной сетки (гельфракция). Концентрация гельфракции
определяется различными условиями: отношением реагирующих
мономеров, степенью полимеризации и относительной активностью
обоих мономеров и радикалов.
Процессы полимеризации
51
Зависимость желатинизации от этих условий определяется урав-
нением:
Ра? \ В >
где: х — доля полимеризованных винильных групп к моменту жела-
п А
типизации; Р — средняя степень полимеризации; — отношение
молярных концентраций моновинильного мономера А к дивиниль-
ному В; <х = —-----относительная активность радикала А (моно-
КЛ'А
винильного) к обоим мономерам (Кл-В — константа скорости реак-
ции радикала А' с мономером В; К А-А— то же радикала А' с мо-
номером Л).
Таким образом, чем больше длина образующихся цепей (Р),
чем больше концентрация дивинильной компоненты, тем раньше
произойдет желатинизация. С другой стороны, чем больше длина
образующихся цепей, тем при меньшей концентрации дивинильного
мономера произойдет образование пространственного полимера.
Эту зависимость легко понять, так как для того, чтобы сшить все
цепи, достаточно в среднем меньше одной молекулы тетрафункцио-
•нального соединения («сшивки») на макромолекулу согласно
уравнению:
p = N— 1,
где: р — число молекул «сшивки»; Af — число макромолекул.
Так, если средняя степень полимеризации Р = 10 000 и если
а= 1, то уже при 0,01 мол. % концентрации «сшивки» все макро-
молекулы будут соединены валентной связью и весь сополимер
будет представлять собою один пространственный нерастворимый
макроагрегат. Естественно, что в этом случае все цепи связаны
в среднем лишь одной поперечной валентной связью. При дальней-
шем увеличении концентрации «сшивки» цепи связываются боль-
шим числом связей, образуются замкнутые петли между макро-
молекулами и конструируется сеточная структура; частота сетки
(расстояние между узлами) будет определяться концентрацией
сшивки. Таким образом, если образуются очень длинные цепи, то ни-
чтожная концентрация дивинильного мономера (~0,01 мол. %) при-
водит к образованию пространственного нерастворимого сополимера.
Теория сополимеризации
Сополимеризация является более общим случаем процессов образования
макроцепей, чем обычная полимеризация. Последнюю следует рассматривать
как частный случай сополимеризации.
Кинетика реакции сополимеризации значительно более сложна, чем кинетика
реакции полимеризации, и поведение смеси разнородных мономеров отлично от
поведения однородных мопомеров.
4*
52
Гл. I. Химические основы технологии пластмасс
В случае полимеризации активируется только один тип мономеров я растет
только один тип радикалов, в результате взаимодействия их с одним типом
мономеров. Это может быть представлено схемой:
+ Я > мпг
где: Мп—радикал растущей цепи; М— мономер; /С—константа скорости.
При бинарной сополимеризации имеются два типа мономеров (например,
Mi и М2) и не одни тип радикалов, взаимодействующих при своем росте с ка-
ждым нз мономеров, а два типа растущих радикалов, отличающихся характе-
ром концевой группы (например Alj —радикал, имеющий в качестве концевой
группы мономер ЛЛ, и М2—радикал, имеющий в качестве концевой группы
мономер Afj). Естественно, что растущий радикал М j при взаимодействии с мо-
номером М2 превращается в радикал М2, а растущий радикал М'2 при взаимо-
действии с мономером Mi превращается в радикал ATj. Таким образом, расту-
щая цепь при взаимодействии с мономерами обоих типов образует все время
новые типы радикалов, отличающихся характером концевой группы. При таком
взаимодействии радикалов с мономерами кинетика роста цепи будет, следова-
тельно, определяться несколькими константами скоростей, так как активность
каждого из радикалов со «своим» или «чужим» мономером неравноценна.
Если принять, что Mi и Л12 — молярные концентрации обоих мономеров,
М и ЛГ— молярные концентрации радикалов с концевыми группами соответ-
ствующих мономеров, — константы скоростей реакций и С® — концентрация
инициатора, то процесс сополимеризации может быть описан следующими уравне-
ниями (принимаем, что обрыв происходит только путем соединения активных
цепей).
Возникновение активных центров Скорости процессов
(радикалов)
1. С® 4-Afi —М[;
к'
2. С®+М2 —т-> М-у К'я„С®М.г
Рост цепи макромолекулы
3. М1 -1- Мt —L-> М^; KiMiMi
4. Mi + /И2 —М'2; К2М[М.2
5. Mi 4- М2 М'2; К3М'2М„
6. М'2 +М± М[; к,м;мх
Обрыв цепи
~ 1 .г ^обрыв /. М± -f- Мг -* неактивный полимер; /Собрыв М"
о 1 iz’ АОбрЫВ о. > неактивный полимер; <бРыв м[М2
„ ... , ... обрыв 9. Л12-[-Л12 > неактивный полимер; рыв М2
В основу теории сополимеризации положены некоторые допущения, которые
в значительной мере соответствуют экспериментальным данным.
Процессы полимеризации
53
Так делается допущение, что на скорость роста цепи не влияет коэффи-
циент полимеризации растущей цепи, что. следовательно, две реакции:
Н
R—(—СН2—CHX)J00—СН2—С’ + сн,=снх
X
н
R—(—СН2—СНХ)1000—сн2—с + сн2=снх
протекают с одинаковой скоростью.
Допускают, что тип радикала и его поведение полностью определяются
только характером крайнего мономерного остатка и не зависят от структуры
и числа других звеньев.
Принимают также, что химический состав сополимера определяется исклю-
чительно конкуренцией между различными реакциями роста цепей, хотя моле-
кулярный вес и распределение молекулярных весов определяются наряду со ско-
ростью роста цепей также реакциями активации и обрыва цепей. Это обстоя-
тельство позволяет связать простым уравнением соотношения между составом
сополимера и составом реагирующих мономеров.
Поэтому из девяти уравнений, описывающих активацию, рост и обрыв
цепей, только четыре уравнения роста цепи (3—6) определяют состав сополи-
мера. Естественно также, что расход мономера на активацию и обрыв цепей
крайне незначителен по сравнению с расходом на рост цепей.
Согласно четырем уравнениям кинетики роста цепи, особенности со-
вместной полимеризации можно объяснить, если допустить, что большинство
мономеров более склонно взаимодействовать с «чужим», а не со «своим»
радикалом, т. е. что Ki < Ki и Л'з < Kt. Это было установлено опытным
путем.
С этой точки зрения механизм гетеросополимернзации заключается в том.
что один из мономеров практически не взаимодействует со «своим» радикалом
<Kt = O), но с большой скоростью взаимодействует с «чужим», а второй моно-
мер взаимодействует как со «своим», так и с «чужим» мономерами. Такое явле-
ние наблюдается при полимеризации малеинового ангидрида со стиролом. Моно-
мер малеинового ангидрида может войти в цепь растущего полимера лишь
в том случае, если он взаимодействует с радикалом стирола. При этом об-
разуется радикал малеинового ангидрида, который не может взаимодействовать
со «своим» мономером и взаимодействует лишь с «чужим» — с мономером сти-
рола. Это определяет такую структуру сополимера, в которой звенья малеино-
вого ангидрида непременно чередуются со звеньями стирола, но внутри цепи
звенья стирола могут находиться и рядом. Такая структура и характерна для
полимеров, образующихся прн гетерополимернзации (стр. 48).
При истинной идеальной гетерополимеризации, например в случае реакции
олефинов с SO2, оба мономера не реагируют со «своими» радикалами, но ка-
ждый реагирует с «чужим» радикалом (Д1 =0; Кз = 0).
Невозможность совместной полимеризации винилацетата со стиролом можно
объяснить тем, что в этом случае один радикал (стирола) значительно более
склонен взаимодействовать со «своим» мономером, чем с «чужим», а второй
радикал (винилацетата), наоборот, — с «чужим» мономером. В итоге образуется
цепь, почти исключительно состоящая из стирольных мономеров.
На основании изложенного можно прийти к выводу, что решающее значение
для понимания механизма и характера сополимеризации имеет относительная
54
Гл. J. Химические ссисвы технологии пластмасс
реакционная способность радикалов к «своим» и «чужим» мономерам, которая
может быть выражена отношением констант скоростей реакции:
где: Г| — относительная активность радикала и г2 — относительная актив-
ность радикала AJ.j.
Чем меньше скорость взаимодействия радикалов со «своими» мономерами
по сравнению со скоростью их взаимодействия с «чужими», тем меньше значе-
ния rt и г2. Последние для данной системы являются величиной постоянной и не
зависят от температуры и количества инициатора.
Исходя из скорости исчезновения каждого мономера в процессе сополимери-
зации, было выведено основное уравнение сополимеризации в дифференциальной
форме
rf/W, _ Mi riMi + M.2
dM-2 ~~ Мг'г^+М1 ’
где Afi и Af2— молярные концентрации обоих мономеров.
Левая часть этого уравнения выражает состав сополимера, полученного в на-
чальной стадии сополимеризации, в виде отношения прореагировавших компонен-
тов; в правой — даны концентрации непрореагировавших компонентов и кон-
станты Г\ И Г2-
Дифференциальное уравнение сополимеризации может быть применено лишь
в том случае, если сополимеры выделяются при очень малых степенях превра-
щения.
Из уравнения сополимеризации следует, что если л или г2 близки нулю,
то происходит гетерополимеризация, если же обе константы (и л и г2) близки
нулю, то происходит истинная гетерополимеризация, так как один или оба ра-
дикала со значительно меньшей скоростью реагируют со «своим» мономером, чем
с «чужим».
Если г\ = Г2 = 1, то из дифференциального уравнения сополимеризации сле-
дует, что
dM} Mi
dM., М2 ’
Этот случай соответствует азеотропическому сополимеру, состав которого
отвечает составу исходной смеси при любом соотношении компонентов этой
смеси. Однако, как уже было указано, такое отношение констант сополимери-
зации л и г2 не является характерным. Обычно состав сополимера отличается
от состава смеси, причем для каждого момента реакции соотношения различны,
что определяет неравенства
г1=#г2¥= 1-
Для оценки констант сополимеризации л и г2 произвольно выбирают
в двух опытах Afi и Af2 (молекулярные концентрации обоих мономеров) и пре
весьма малых степенях превращения непосредственно из эксперимента аналити-
dMi
чески определяют принимая, что отношение скоростей присоединения двух
мономеров равно отношению обоих мономеров в полученном сополимере. Из
уравнения сополимеризации тогда можно найти л и г2. Если повторить эти
определения в ряде опытов, то получаются значения для л и г2, отличающиеся
друг от друга из-за экспериментальных ошибок. Вследствие этого были пред-
ложены различные методы, чтобы найти оптимальные значения для л и г2.
Если степени превращения столь значительны, что происходит заметное из-
менение концентрации непрореагировавших мономеров (например больше
Процессы полимеризации
55
2_3%), то в этом случае применяют уравнение сополимеризации в его инте-
гральной форме
1g _ 1 ig (1-P)-W^2
r p ё(1-р)-м?и
Г'^ М° (1— p)-M. ML, ’
lg —J J- 1g-—----C—-
(1 — p)-M0JM°,
J_J.
Где: p = ---—молекулярные концентрации обоих мономеров в на-
1 г 2
чальной стадии процесса; Mi и Л12— молекулярные концентрации непрореаги-
ровавших мономеров после окончания полимеризации.
Для расчета констант rt и г2 по интегральной форме уравнения сополиме-
ризации определяют молярные концентрации обоих мономеров в начале и в мо-
мент прекращения полимеризации.
Соответствующие значения для Mi и Л12 и и Л12 подставляют в урав-
нение и получают значения г2 для некоторых произвольных значений р.
Из уравнения для р вычисляют соответствующие значения для и. Далее
строится график в координатной системе л (абсцисса) — г2 (ордината); при
этом получается практически прямая линия. Решение для л и г2 находится
в точке пересечения двух прямых, соответствующих двум различным опытам.
При трех опытах пересечение трех прямых дает небольшой треугольник, вели-
чина площади которого позволяет оценить возможные ошибки в значениях
Л и г2.
Пол идисперсно с т ь полимеров
В отличие от некоторых природных полимеров, например бел-
ков, состоящих из макромолекул одинакового строения и одинако-
вой величины и являющихся поэтому монодисперсными, синтетиче-
ские полимеры никогда не состоят из молекул с одинаковой сте-
пенью полимеризации. В процессе их образования всегда получается
смесь молекул, подобных по строению, но различной длины, т. е.
смесь полимергомологов. Такие полимеры, состоящие из макро-
молекул одинакового строения, но различной степени полимериза-
ции, называют полидисперсными (полидисперсными по размерам). *
Степень полидисперсности оказывает влияние на физические и
механические свойства полимеров. Однако характер этого влияния
и его количественные зависимости еще не установлены с достаточ-
ной определенностью. Можно лишь утверждать, что наличие
низкомолекулярных фракций (от мономера и до полимеров со
степенью полимеризации ~100) снижает температуру стеклова-
ния и теплостойкость полимеров и увеличивает их пластические
свойства.
Степень полидисперсности непосредственно можно определить
различными методами, из которых наибольшее значение в лабора-
торной практике получили методы фракционного осаждения и фрак-
ционного растворения. Последние основаны на закономерном изме-
нении свойств молекул по мере увеличения их молекулярного веса.
* Полимеры, состоящие из макромолекул, различающихся как по размерам,
так и по структуре, называют полидисперсными по структуре.
56
Гл. I. Химические основы. технологии пластмасс
Из других методов определения полидисперсности следует ука-
зать на хроматографирование (основанное на различной адсорб-
ционной способности молекул в зависимости от их величины),
ультрацентрифугирование, определение скорости диффузии, а также
на изучение рассеяния света и сравнение молекулярных весов,
определенных различными методами (стр. 59—60).
Рис. 6. Кривые фракционирования полиизобутилеиа со средней степенью
полимеризации 550.
/—интегральная кривая распределения; 2—дифференциальная кривая распределения.
Полидисперсность выражают при помощи кривой распределе-
ния. Для этого экспериментальные значения отдельных фракций
наносят на график в системе координат: степень полимеризации
(или мол. вес) — весовая доля фракции. Через полученные сту-
пеньки (каждой фракции соответствует ступенька) проводят инте-
гральную кривую распределения. Такая кривая показывает весовые
доли полимергомологов со степенью полимеризации от Р до Р dP
в данном полимере.
По интегральной кривой можно построить в координатах Р*~^
дифференциальную кривую распределения, выражающую скорость
Процессы полимеризации
57
возрастания количества с увеличением степени полимеризации
(рис. 6).
Дифференциальные кривые имеют резко выраженный максимум
и позволяют более наглядно, чем интегральные кривые, сравнивать
степень полидисперсности полимеров. На рис. 7 показаны кривые
распределения молекулярных весов для нитроцеллюлозы и поли-
стирола. Как видно из характера дифференциальных кривых,
Степень полимеризации
Рис. 7. Распределение по степени полимеризации.
/ — нитроцеллюлоза сл средней степенью полимеризации 860;
2— полистирол со_сиедней степенью полимеризации 800.
нитроцеллюлоза обладает значительно меньшей степенью полидис-
персности, чем стирол.
Полидисперсность до известной степени удается регулировать
за счет условий реакции полимеризации, однако она определяется
и химической структурой мономера.
Разветвленность полимеров
Структура полимера характеризуется не только степенью полидисперсности
и порядком сочетания звеньев мономера в цепи (в случае сополимеризации), но
также формой и степенью разветвленности цепи макромолекул. Последняя яв-
ляется существенным фактором, определяющим физико-механические свойства
полимеров. Линейные полимеры можно разделить на три группы: 1) полимеры
с прямой цепью, не содержащей боковых групп; 2) полимеры с прямой цепью,
содержащей боковые группы; 3) разветвленные полимеры (группы 1 и 2).
Характер боковых групп в макромолекулах определяется преимущественно
структурой реагирующих молекул, а иногда условиями полимеризации, развет-
вленность же полимеров — главным образом условиями полимеризации. Суще-
ственное значение имеют длина боковых групп, их полярность, концентрация и
симметричность расположения в звене.
Длина боковых групп полимера определяется длиной замещающих ради-
калов в реагирующих мономерах типа CH2=CHR; CH2=CRRi и т. д. Чем они
длиннее, тем больше расстояние между цепями, тем меньше вследствие этого
межмолекулярные силы и тем до известного предела «мягче», пластичнее поли-
мер, тем ниже его температура стеклования. Уменьшение длины боковых групп
в результате изомерии увеличивает «твердость» полимеров.
58
Гл. Л Химические сснсвы технологии пластмасс
Температура хрупкости,
Количестбо углеродных атомов
б бОКПе-'й идпи
Рис. 8. Зависимость температуры
хрупкости в гомологическом ряду
полиакрилатов от количества угле-
родных атомов (СН,-групп) в бо-
ковой цепи полимера.
Однако снижение температуры стеклования полимеров в зависимости от
длины боковых групп в некоторых случаях проходит через минимум, после ко-
торого увеличение длины боковых групп уже усиливает жесткость и повышает
температуру хрупкости полимера. Это может быть показано на примере поли-
меров эфиров акриловой кислоты (рнс. 8).
Повидимому увеличение длины боковых групп усиливает торможение при
цепи и по достижении определенной длины
обусловливаемые ими стерические затруд-
нения начинают превосходить эффект сни-
жения межмолекулярных сил притяжения.
Результирующий эффект влияния длины бо-
ковых групп на снижение температуры сте-
клования полимеров определяется, таким
образом, одновременным действием проти-
воположных факторов — фактора сниже-
ния межмолекулярных сил, увеличивающего
подвижность цепей, и фактора, увеличи-
вающего стерические трудности при враще-
нии звеньев.
Полимеры с боковыми группами могут
быть получены и при полимеризации таких
линейных, неполярных мономеров, как эти-
лен, бутадиен и др. Образование линей-
ной цепи, не содержащей или же содер-
жащей боковые группы, зависит от условий
реакции (температуры, катализатора и др.).
Так реакции, ведущие к образованию
технических полибутадиенов, протекают в
зависимости от условий реакции преимущественно в направлении 1,4 или 1,2. В пер-
вом случае образуется линейный полимер без боковых групп, имеющий структуру:
NaCH2—СН=СН—СН2—СН2—СН=СН—СН2—...
Во втором случае образуются цепи с боковыми группами по схеме:
2Na + CH2=CH—СН=СН2 —> NaCH,—CHNa;
СН=СН,
NaCH2— CHNa-j-CH2=CH—СН=СН2 —>
CH=CH,
—>- NaCH.>—СН—СН,—CHNa и т. д.
I “I
сн=сн, сн=сн.
Разветвленными называются полимеры, в которых преобладают или на-
ходятся в значительном количестве цепи, имеющие большое число ответвле-
ний, соизмеримых по длине с главной цепью и содержащих, следовательно, де-
сятки и сотни звеньев.
Процесс образования разветвленных полимеров, в котором наряду с основ-
ным направлением роста цепи развиваются и другие направления, происходит
преимущественно по механизму отрыва водорода свободным радикалом по схеме:
R R R
I I I
1. ...—СН—СН,—СН—СН..—... 4-КСН,—сн— —>
! <
R R R
—> ксн,—сн.. + • •.—сн—сн2—с—сн2—...
Процессы полимеризации
591
R R R
2. ...—СН—СН2—С—СН3—...+—СН2—СН— —>
' R R R
---> ...—СН—СН2—С—СН2—СН2—СН—... и т. д
С н.,—С HR-
Разветвление полимеров может происходить и при взаимодействии радика-
лов с неактивными цепями полимера. В этом случае при отрыве водорода от
какого-либо звена неактивного полимера осуществляется реакция переноса цепи,
неактивный полимер становится растущим радикалом, а радикал — неактивной
молекулой. Так как вероятнее всего, что активная точка в полимере не находится
в крайнем звене, то рост такого полимера приводит к образованию разветвлен-
ных полимеров:
RCH—СН3—СН—CH2—СН—СН3+ RCH—СН,—... —>
XXX X
—> RCH—СНг—СН—СН—СН—сн3+ RCH—СН3.
I ! ' I
XXX X
Эти реакции позволяют «прививать» полимеру, растворенному в мономере,,
звенья мономера, причем не только звенья «своего» мономера, но и «чужого»,
например звенья стирола к полиметилметакрилату. Такие полимеры называются
привитыми. Образование их представляет собою своеобразный тип сополимериза-
ции, основанной на «прививке» чужого мономера к внутренним звеньям полимера.
Структура привитого полимера может быть изображена следующей схемой:
—Л—А—А—А— А-/1-А—
I
В-В—В-В-
где А — звенья полимера, В — привитых мономеров.
Представление о степени полидисперсности и разветвленности полимера
можно получить, если сравнить его средний молекулярный вес, найденный па
двум принципиально отличным методам определения:
1) определением молекулярного веса в результате деления веса частиц на их
число, как числовой средней величины М,, которую можно выразить уравнением
11 ’
где Л1— молекулярный вес частиц, состоящих из х звеньев; —число частиц,,
состоящих из х звеньев;
2) определением молекулярного веса в результате деления веса частиц на
вес средней частицы, как весовой средней величины М в:
ЕМ® У,.
К первой группе методов относятся химические, криоскопические, эбуллио-
скопические и осмотические, ко второй — метод определения молекулярного веса
по вязкости, а также оптический метод, основанный на определении рассеяния
света растворами высокомолекулярных соединений.
Кроме этих принципиально различных групп методов определения молеку-
лярного веса полимеров непосредственное представление о полидисперсности
«о
Гл. I. Химические основы технологии пластмасс
ТАБЛИЦА 3
АТ,, и Л1В для системы, состоящей
из молекул с мол. весом
100 000 и 1000
99 1 99 000 50 000 1,98
50 50 50 500 1 980 25,50
полимера, о величине, форме и количестве молекул дают методы: седимента-
ционный (с применением ультрацентрифуги) и диффузионный.
Существенное отличие в показателе среднего молекулярного веса, опре-
деляемого различными методами, объясняется полидисперсностью полимеров.
Если бы полимеры были монодисперсны-
ми, т. е. если бы молекулы имели оди-
наковую величину и одинаковый вес, то
все методы определения молекулярного
веса давали бы идентичные результаты
и Л1Ч = Л1в. Однако с появлением поли-
дисперсности обнаруживается разрыв
между числовой средней величиной Л4Ч
и весовой средней величиной Мъ; чем
выше степень полидисперсиости, тем
больше Л1В по сравнению с Мч.
На практике степень полидисперс-
ности чаще всего устанавливают путем
параллельного определения молекуляр-
ного веса вискозиметрическим (влияние
весового процентного соотношения рас-
творенных частиц) и осмометрическим
(влияние числа частиц) методами. Чем
больше расхождение между получен-
Л1в
ными результатами, чем больше показатель -уу , тем, следовательно, больше
степень полидисперсности полимера.
Если в полимере содержание молекул Л12 с низким молекулярным весом по
сравнению с содержанием молекул All с высоким молекулярным весом—не-
велико, то и определенные по тому или другому методу величины среднего
/VI в
молекулярного веса существенно не отличаются, и отношение yj- также не-
велико. Однако расхождение в найденных средних молекулярных весах и со-
М И
отношение -у становится весьма значительным, если количество низкомолеку-
лярной фракции возрастает (табл. 3).
Когда же при испытании узких фракций полимера, в которых полидисперс-
ность не может быть значительной, наблюдается расхождение между значе-
ниями средних молекулярных весов, определенных вискозиметрическим и осмо-
метрическим методами, то это указывает на разветвленность полимера.
Структура звеньев цепи полимера
Структура цепи полимера зависит от порядка присоединения
мономеров. С этой точки зрения различают три типа соединений —
два правильных и один неправильный.
Рассмотрим полимер, образующийся по схеме:
иСН2=СН ----> (...—СН.—СН—...)„.
R R
* Согласно Флори, Мч : Л1в= 1 : (1 +р), где р— степень завершенности
реакции. При полной завершенности (р = 1) А1,,: А1В = 1 : 2.
Процессы полимеризации
61
Цепи такого полимера могут образоваться по следующим типам
соединений:
...—СН,—СН—СН,—СН—СН,—СН—...
I “I ‘I
R R R
..СН2— СН— СН—СН,—СН2— СН—СН—СН,—...
II II
R R R R
.,.— СН,—СН—СН2—СН—СН—СН2—СН—СН,—СН,—СН—...
Illi I
R R R R R
ш
Первый тип — правильный — и построен по принципу 1,3 — «го-
лова к хвосту»; «конец» («хвост») молекулы мономера, как видно
из схемы I, присоединяется к «началу» («голове») другой молекулы
мономера. Второй тип (схема II)—также правильный — и по-
строен по принципу 1,2 — «голова к голове, хвост к хвосту». Третий
же тип соединения (схема III) — неправильный, смешанный.
Определение структуры цепи полимера может быть произведено
различными методами, чаще всего путем определения строения про-
дуктов пиролиза углеродных цепей, а также действием иодистого
калия на полимеры, содержащие галоид в боковой цепи. В послед-
нем случае, если галоид находится при соседних атомах углерода
(по типу 1,2), происходит выделение иода и образование непре-
дельного соединения по схеме:
... —СН,—СН—СН—СН,—... + —..—СН,—СН2—СН—СН—....
I I I I
Cl Cl J J
—> J2+ ... — СН2— СН=СН—СН,—...
Если же выделения иода не происходит, полимер имеет струк-
туру типа 1,3.
В технике часто применяют реакции замещения, омыления, эфи-
ризации и ацетилирования боковых групп макромолекул полимера.
В тех случаях, когда каждая реактивная группа боковой цепи
может реагировать самостоятельно, вне зависимости от соседней
группы, реакция теоретически, а часто и практически, может идти до
конца, до равновесного состояния.
Однако, когда реагировать должны одновременно две боковые
группы цепи полимера, построенного по типу 1,3 — «голова
к хвосту», например при ацетилировании поливинилового спирта,
полное замещение всех гидроксильных групп невозможно и по ста-
62
Гл. I. Химические основы технологии пластмасс
диетическим закономерностям часть гидроксильных групп в поли-
мере всегда может остаться «без пары»:
..—СН.,—СН—СН,—СН—СН2—СН—СН2—... 4-СН3С^
I I I ХН
он он он
—> ...—сн,—сн—сн,—сн—сн,—сн—сн,—сн—сн—сн—.. .
I ‘I “I ‘I "I
о—сн—о он о—сн—о
СН, СН3
Было доказано, что по статистической вероятности при таком
пипе реакций (к ним относятся также и реакции отщепления галои-
дов при действии цинка на галоидсодержащие полимеры и ряд дру-
гих) около 14% групп не смогут вступить в реакцию. Это подтвер-
ждается и на практике. Так реакция дегалоидирования полихлор-
винила действием цинка и реакция ацеталирования поливинилового
спирта никогда не идут до конца (максимум до 85—86%).
В том случае, однако, когда полимер имеет структуру 1,2 («го-
лова к голове») имеется возможность для вступления в реакцию
всех сближенных пар боковых групп и в то же время малая вероят-
ность реакции за счет групп, отдаленных друг от друга на расстоя-
ние нескольких углеродных атомов.
Таким образом, структура полимера, т. е. порядок соединения
мономеров в его цепи, обусловливает кинетику многих реакций за-
мещения, их характер и предел и, следовательно, характер боковых
функциональных групп полимера после реакции.
Процессы поликонденсации
Поликонденсация (стр. 31) представляет собою процесс обра-
зования макромолекул в результате конденсации полиреактивных
(полифункциональных) молекул.
Типичным примером поликонденсации является реакция типа 2,2,
например:
лНОСО—t— COGH 4-пНО—R,—ОН ±2^
нс[—се—се—о—к,—о—]„н +2 («—1)н2с.
В этом случае в реакции участвуют молекулы двух типов
(спирта и кислоты), обладающие одинаковым количеством функ-
циональных групп. Такой тип реакции называют гетерополиконден-
сацией. Когда же в реакции участвуют молекулы одного типа (на-
пример молекулы оксикислоты типа HOR—СООН), процесс назы-
вают гомополиконденсацией-.
?• П,О(СН,)3.СООН Н[О(СН,)сСО];1ОН + (п — 1)Н,С.
Поликонденсация, в отличие от цепной полимеризации, является
ступенчатым процессом. На каждой ступени реакции образуются
Процессы поликонденсации
63
промежуточные, стабильные продукты, которые могут быть изоли-
рованы.
Процессы поликонденсации протекают по тем же законам, что
и процессы низкомолекулярной конденсации: рост цепи происходит
путем последовательного взаимодействия одной молекулы с другой.
Полученный продукт взаимодействует со следующей молекулой
и т. д. Наряду с этим происходят реакции соединения между обра-
зовавшимися цепями, однако вероятность таких реакций умень-
шается по мере увеличения молекулярного веса цепей и возраста-
ния вязкости среды.
Процесс поликонденсации не является самопроизвольным про-
цессом и требует энергии извне. Рост цепи макромолекул происхо-
дит медленно и скорость процесса зависит от температуры, от
скорости удаления отщепляющихся низкомолекулярных продуктов
реакции, а также от количества и характера катализатора (обычно
от концентрации водородных ионов).
Процесс поликонденсации так же, как и обычная конденсация,
характеризуется величиной константы равновесия К, определяющей
во сколько раз прямая реакция протекает быстрее обратной. Раз-
личают два типа реакций поликонденсации.
К первому типу относятся реакции поликонденсации, для кото-
рых характерны малые значения —10. Они определяют боль-
шую чувствительность реакций к присутствию в реакционной среде
побочных низкомолекулярных продуктов реакции (воды). Равно-
весное состояние в таких реакциях наступает быстро и рост молекул
прекращается, если не происходит удаления побочных продуктов
реакции. Если же эти побочные продукты удаляются из сферы
реакции, прямая реакция может идти дальше. При идеальном уда-
лении побочных продуктов равновесное состояние, в пределе, насту-
пит лишь тогда, когда все реагирующие молекулы соединятся
в одну гигантскую молекулу (в случае гетерополиконденсации —
при условии отсутствия избытка одного ив реагирующих компо-
нентов) .
К этим реакциям, ведущим к образованию в цепи •—СО—О—;
—NH—СО— связей относятся взаимодействия полифункциональ-
ных спиртов, кислот, аминов и т. д.
Ко второму типу реакций поликонденсации принадлежат реак-
ции, которые практически протекают в одном направлении. Их
равновесное состояние настолько сдвинуто в сторону прямой реак-
ции, что заметно не влияет на кинетику процесса. Эти реакции ха-
рактеризуются весьма большим значением К (в 1000—10 000 раз
больше, чем для первого класса), вследствие чего они могут проте-
кать в водной среде даже тогда, когда побочным продуктом реакции
является вода. Степень поликонденсации получающихся при этом
смол (величина образующегося макромолекулярного комплекса)
практически мало зависит от условий равновесия. К этому классу
64
Гл. I. Химические основы технологии пластмасс
I I
относятся реакции, при которых образуются связи —С—С— или
—С—N—С—, например, в соединениях, получаемых поликонденса-
I i I
цней фенола и формальдегида.
Средняя степень поликонденсации Р пропорциональна кон-
Рис. 10. Зависимость средней степени по-
ликонденсации Р от величины константы
равновесия К при различном числе мо-
лей воды ян>0 в системе.
станте К и обратно пропор-
циональна пн 0 — количеству
имеющихся в системе молей
воды (побочных продуктов реак-
ции):
г "н3о
На_рис. 9 показана зависи-
мость Р от /?я о, на рис. 10 —
от величины К при различных
значениях п. Из рис. 10 сле-
дует, что средняя степень по-
ликонденсации зависит от К
и ли,о’ причем влияние
wh,o на Р обусловлено вели-
чиной К.
Так для полиэфиров ~ 4
и Р стремится к нулю при
пПа0 порядка 0,3—0,5. В то же
время для соединений с угле-
родной связью (—С—С—1, на-
I I
пример для феноло-формальде-
гидных смол К— 10 000 и Р
достигает значительной величи-
ны даже если лн 0 = 50, т. е.
если реакция происходит в вод-
ной среде.
Таким образом, процессы
поликонденсации, ведущие к
образованию карбоцепных поликонденсатов с очень большим
Процессы поликонденсации
65
значением К можно рассматривать как реакции, практически проте-
кающие в одну сторону.
В реакциях бифункциональных молекул средняя степень поли-
конденсации зависит также и от наличия избытка молекул одного
из реагирующих веществ, способных блокировать концевые группы
растущей цепи. Например, в нижеследующей реакции при избытке
спирта:
НО^ОН + EOCOR2COOH —>- HORjOCORjCOOH 4- Н2О
EORjOCORjCOOH + HORjOH —> HORiOCORaCOCRjOH + H2O
образовавшееся соединение уже не может реагировать с молеку-
лами одного из веществ (спирта), и если исчерпаны молекулы вто-
рого вещества (кислоты), реакция роста цепи приостанавливается.
Следовательно, чем больше избыток молекул одного из реагирую-
щих веществ, тем скорее концевые группы растущей цепи будут
блокированы молекулами, находящимися в избытке, и, следова-
тельно, тем меньше будет средняя длина цепи, т. е. меньше Р.
При точно эквимолекулярных соотношениях реагирующих ве-
ществ и при полном удалении воды из сферы реакции рост поли-
мера теоретически должен продолжаться до установления равно-
весия, которое в пределе может быть достигнуто при бесконечно
больших молекулярных весах полимера.
Зависимость молекулярного веса полимера от избытка одного из
компонентов детально изучали В. В. Коршак и С. Р. Рафиков. Они
вывели следующую формулу для
теоретического подсчета молеку-
лярного веса полимера в зависи-
мости от молекулярного процента
избытка одного из реагирующих
веществ:
ч
где: М — молекулярный вес поли-
мера; q — молекулярный процент
избытка одного из реагирующих
веществ; Ма — молекулярный вес
вещества, взятого в избытке; М6 —
молекулярный вес второго веще-
ства; z — молекулярный вес вы-
деляющегося продукта (воды).
С помощью этой формулы были
подсчитаны предельно возможные
средние молекулярные веса поли-
ТАБЛИЦА 4
Зависимость молекулярного веса
полимера из гексаметилендиамина
и адипиновой кислоты от избытка
последней
Избыток адипиновой кислоты мол. О/о Число сегментов в цепн Молекулярный вес полимера М
100 3 372
50 5 598
20 11 1276
10 21 2406
2 101 11446
1,0 201 22476
0,2 1001 113146
0,02 10001 1130146
0,002 100001 11306146
амидов в зависимости от избытка адипиновой кислоты (табл. 4).
Из табл. 4 следует, что для практически достижимых моле-
кулярных весов (20 000—40 000) избыток одного из компонентов не
должен превышать 1 мол. %.
5 Зак. 1269. Э. И. Бар:
66
Гл. I. Химические основы технологии пластмасс
При приближении к эквимолекулярным соотношениям реакция
становится чрезвычайно чувствительной к ничтожным изменениям
в соотношении компонентов. Так, согласно расчетным данным, из-
менение избытка адипиновой кислоты на 0,09 мол. % может при-
вести в этих условиях к изменению молекулярного веса полиамида
в 10 раз. Поэтому при весьма малых избытках одного из реагирую-
щих веществ (меньше 0,1 мол. %) неизбежен большой разброс
в величине образующихся молекул и, следовательно, большая поли-
дисперсность полимера. Значительный же избыток (больше 1 мол. %)
способствует образованию полимеров, в которых преобладают цепи
более равномерной длины, т. е. уменьшает полидисперсность
полимера.
Подобно избытку одного из бифункциональных компонентов,
влияет присутствие в реакционной среде монофункциональных ком-
понентов, например, одноатомного спирта или одноосновной кис-
лоты. Ничтожные количества таких веществ, блокируя концевые
группы реагирующих молекул, снижают молекулярный вес поли-
мера в такой же степени, как и избыток одного из бифункциональ-
ных компонентов. В технике при производстве полиэфирных и дру-
гих смол широко пользуются добавками монофункциональных
компонентов (смоляных, жирных кислот, моноаминов и др.) для ре-
гулирования молекулярного веса и стабилизации молекул полиме-
ров. Действие монофункциональных соединений можно представить
следующей схемой:
НО(СН2)ПОСО(СН2),„СОО ... (СН2)„ОН+ 2RCOOH —►
—> RCOO(CH2)„OCO(CH2)OTCOO ... (CH2)„OCOR.
Процесс поликонденсации приводит к различным продуктам
в зависимости от строения реагирующих молекул.
При конденсации бифункциональных циклических или развет-
вленных молекул малой величины (например, фенолов, мочевины,
формальдегида, фталевого ангидрида и др.) получаются низкомоле-
кулярные смолы глобулярной структуры.
Процесс поликонденсации более высокофункциональных моле-
кул того же строения (реакции типа 2,3\ 2,4-, 3,3 и т. д.) приводит
к образованию трехмерных пространственных полимеров гло-
булярной структуры. Обычно в технике этот процесс проис-
ходит последовательно. Сначала получают плавкие и раствори-
мые низкомолекулярные смолы, которые при обычной темпе-
ратуре медленно, а при высокой температуре быстрее переходят
в неплавкие и нерастворимые смолы, т. е. в пространственные
полимеры.
Скорость процесса образования трехмерных молекул опреде-
ляется концентрацией остаточных функциональных групп в низко-
молекулярных смолах.
Процессы поликонденсации
67
Так в молекуле резольной смолы (стр. 358), строение которой
может быть представлено схемой:
СН,ОН
ОН |
ОН
имеющиеся свободные метилольные группы способны к дальнейшей
конденсации с водородными атомами бензольных ядер, образуя
макромолекулу с пространственной валентной связью бензольных
колец:
Если реагирующие бифункциональные молекулы имеют линей-
ную форму и если длина молекул обоих компонентов или хотя бы
одного из них превосходит минимальную длину примерно в 5—
6 углеродных атомов, можно ожидать получения высокомоле-
кулярного линейного полимера типа полиамида или некоторых
полиэфиров.
При поликонденсации линейных молекул (линейных спиртов,
кислот, аминов) одновременно протекают два процесса: процесс
образования замкнутых колец (лактамов, лактонов) и процесс, ве-
дущий к получению линейных полимеров. Скорость каждого про-
цесса и, следовательно, соотношение образующихся продуктов кон-
денсации зависит от ряда факторов и, в первую очередь, от числа
углеродных атомов в первичном продукте присоединения. Если
число атомов равно пяти, образуется почти исключительно цикли-
ческий продукт; наличие шести и семи атомов углерода также
обусловливает вероятность образования низкомолекулярных ци-
клических соединений; лишь при числе атомов углерода больше
5*
68
Гл. I. Химические основы технологии пластмасс
8—10 (при нормальных условиях) образуется только линейный
полимер.
Циклизация протекает значительно легче в процессах гомополи-
конденсации. Направление реакции при гомополиконденсации [на-
пример молекул строения NH2(CH2)„COOH или НО(СН2),г СООН]
определяется величиной я, т. е. числом СН2-групп.
Если п < 5, то образуется только лактам, если п > 5, образуется
только линейный полимер, если п = 5, образуется линейный поли-
мер, но имеется вероятность и образования циклов:
NH2 (СН2)лСО-ЫН(СН2)лСО-NH(СН2)„ (СН2)...
Линейный полимер
Примером влияния формы реагирующих молекул на структуру
и молекулярный вес полимера может служить различие в характере
полимеров, образующихся при взаимодействии о- и и-фталевых
кислот с этиленгликолем.
о-Фталевая кислота с гликолем образует ответвленный низкомо-
лекулярный полимер с аморфной глобулярной структурой, имеющий
низкую температуру плавления:
НОСО СООН
+ НО(СН2)2ОН —>
—СН,—СН2 СН,—СН2 СН,—СН, СН,—.
I I I I I I
о о о о о о
I I i I I I
со со со со со со
Терефталевая (я-фталевая) кислота образует линейный высоко-
молекулярный полимер с кристаллической структурой, имеющий
высокую температуру плавления;
НОСО—^>-СООН + НО(СН2)2ОН —►
—>- НО(СН2),ОСО-/ СОО(СН2),ССО— / ССО—...
Длина реагирующих линейных молекул определяет не только
кинетику и направление реакции поликонденсации, но и основные
физические свойства образующегося полимера, так как от длины
Процессы поликонденсации
69
исходных молекул зависит расстояние между дипольными группами
(—NH—СО—; —О—СО— и др.) в макромолекуле. Чем длиннее
цепь реагирующих молекул, тем реже расположение диполей
в макромолекулярной цепи и тем меньше их влияние на метилено-
вые звенья цепи и на величину межмолекулярных сил притяжения.
Удлинение расстояния между диполями повышает, таким обра-
зом, подвижность звеньев (—СН2—СН2—), а это отражается на
увеличении гибкости и эластичности полимера, снижает его темпе-
ратуру плавления. В общем с удлинением цепи реагирующих мо-
лекул увеличиваются эластические свойства полимеров.
С уменьшением длины реагирующих линейных молекул плот-
ность расположения диполей повышается и, следовательно, воз-
растают силы, стабилизующие внутреннюю структуру молекулы,
усиливаются межмолекулярные силы притяжения — все это ведет
к жесткости и повышению температуры плавления полимеров.
Если в процессе поликонденсации наряду с компонентами, кото-
рые по своей структуре способны образовать линейный полимер,
участвуют также линейные молекулы с функциональностью выше 2,
то образуются линейные сшитые пространственные поликонденса-
ционные полимеры, во многом напоминающие сшитые линейные
полимеры, полученные методом полимеризации. Такие полимеры
могут быть получены, например, при конденсации себациновой кис-
лоты с линейными многоатомными спиртами, при конденсации
гексаметилендиизоцианатов с трехатомными спиртами и т. д. Для
них характерна линейная структура макромолекул, но с более или
менее густой «сшивкой» между цепями. Однако, если молекулы
обоих компонентов не имеют линейной структуры с соответствую-
щей минимальной длиной цепи и один из компонентов имеет функ-
циональность свыше 2, то в результате реакции поликонденсации
образуются пространственные полимеры не с линейной (сетчатой),
а с глобулярной структурой. Это наблюдается при конденсации
фталевого ангидрида с глицерином (при получении глифталей,
стр. 582). В соответствии со структурой обоих типов пространствен-
ных полимеров находятся и их физико-механические свойства: ли-
нейные полимеры с пространственной структурой обладают большей
эластичностью и растяжимостью, а глобулярные пространственные
полимеры — большей твердостью и хрупкостью.
ГЛАВА 11
ФИЗИЧЕСКИЕ ОСНОВЫ ТЕХНОЛОГИИ ПЛАСТМАСС
УПРУГАЯ И ПЛАСТИЧЕСКАЯ ДЕФОРМАЦИИ
Если на твердое тело действует внешняя сила, то тело меняет
свою форму (деформируется). Все виды деформации могут быть
сведены к следующим трем процессам:
I. Частицы (атомы, молекулы) под влиянием внешней силы
(растяжения) удаляются друг от друга на расстояние, пропорцио-
нальное величине приложенного напряжения, при этом не меняется
существовавший порядок в расположении частиц. Следствием та-
кого процесса является увеличение объема при деформации..
Удаление частиц от равновесного состояния осуществляется
лишь в пределах действия межчастичного притяжения и связано
с увеличением внутренней энергии тела как вследствие деформации
валентных углов, так и натяжения валентных и межмолекулярных
связей. При снятии внешнего напряжения деформация исчезает
практически мгновенно (со скоростью звука) вследствие освобо-
дившейся внутренней энергии. Такая деформация является упругой-
она предшествует всегда всякой другой деформации твердого
тела.
2. Частицы, не удаляясь друг от друга, перегруппировываются,
причем полностью меняется существовавший порядок в расположе-
нии частиц. Деформация, вызванная этим процессом, не сопрово-
ждается изменением внутренней энергии твердого тела, — после
снятия напряжения она не исчезает и деформированное тело остается
без изменения. Такая деформация называется пластической. Де-
формация обычных твердых тел (кристаллических, аморфных), со-
стоящих из изотропных, шарообразных частиц (молекул, атомов),
складывается или только из упругой деформации, или из упругой и
пластической.
3. Частицы, состоящие из анизотропных длинных молекул, со-
держащих большое число звеньев, способных изгибаться и вра-
щаться под влиянием деформирующей силы, меняют свою форму,
однако сдвига частиц в целом не происходит. Деформация опреде-
ляется не изменением междучастичного расстояния (как в случае
упругой деформации) и не перегруппировкой частиц (как в случае
пластической деформации), а лишь изменением равновесной кон-
фигурации длинных молекул, способных к вращательным движе-
ниям. Такая деформация является высокоэластической. Так же, как
и упругая деформация, она является обратимой и исчезает после
Упругая деформация
71
снятия напряжения. Исчезновение деформации происходит не
в фазе с напряжением, поэтому она имеет характер «запаздываю-
щей» деформации.
Упругая деформация
Упругая деформация определяется изменением междучастичного расстояния
в пределах действия межчастичных сил притяжения.
Для того, чтобы получить представление о межчастичных силах, дей-
ствующих в твердом теле, и их зависимости от междучастичных расстояний,
следует рассмотреть условие равновесного состояния, которое определяется
взаимодействием сил притяжения и ....
Оба вида сил изменяются с
расстоянием г по особым законо-
мерностям:
V — -EL
г притяж р 1
Fm
Ет
Резу ль тирующие
силы
Силы отталкивания
Г
I / Силы притяжения
X
Резу ль тирующая
энергия
Отталкивание
Притяжение
Рис. 11. Энергия взаимодействия Е и силы
притяжения между частицами F как функ-
ция междучастичного расстояния.
Е
где: а и b — константы; п — по-
казатель степени (для ионных
кристаллов п может иметь величи-
ну от 7 до 11).
Таким образом, силы отталки-
вания действуют по сравнению с
силами притяжения иа весьма
малых расстояних и резко умень-
шаются с увеличением этих рас-
стояний.
Наглядно это представлено на
рис. 11, где силы F и энергия
взаимодействия Е представлены
как функция междучастичного
расстояния г.
Результирующая сила притя-
жения растет с уменьшением ме-
ждучастичного расстояния, дости-
гая максимума F,„ при расстоянии
гт\ при дальнейшем сближении
начинают значительно сказываться
силы отталкивания, и при г0 они
становятся равными силам притяжения, что и является условием равновесия и
устойчивости системы. Дальнейшее сближение частиц требует преодоления резко
возрастающих сил отталкивания, что может быть осуществлено приложением
внешнего напряжения сжатия. При увеличении расстояния превалируют уже
силы притяжения; увеличение же расстояния может быть достигнуто лишь за
счет приложения внешнего усилия — растяжения. Таким образом, изменение
междучастичного расстояния в любом направлении вызывает силы, стремя-
щиеся восстановить это расстояние. Чтобы разрушить систему, удалить частицы
на бесконечное расстояние друг от друга, нужно приложить усилие, большее,
чем Fm,— максимальные силы притяжения.
Следовательно, Рт характеризует теоретическую прочность системы, которая
при расчете на площадь сечения твердого тела составляет десятки и сотни
тысяч килограммов на квадратный сантиметр (стр. 119—121).
72
Гл. П. Физические основы технологии пластмасс
Энергия взаимодействия частиц может быть найдена интегрированием кри-
вой сил.
Как видно из графика энергии, равновесное состояние системы, соответ-
ствующее междучастичному расстоянию г приходится на минимуме кривой.
Работа, необходимая для удаления частиц на бесконечное расстояние, равна,
следовательно, £—глубине минимума потенциальной энергии при г0 («по-
тенциальной яме»).
В этих «углублениях» при температурах выше абсолютного нуля частицы
находятся в непрерывных гармонических колебаниях. Амплитуда этих колебаний
определяется величиной кинетической энергии — температурой.
Если она становится равной величине потенциального углубления Ет—ча-
стицы могут преодолеть потенциальный барьер и занять новые равновесные по-
ложения. В случае идеально кристаллического тела считают, что оно предста-
вляет собою ряд равных потенциальных углублений (рис. 12), в каждом из
а=3,55 А
Рис. 12. Потенциальные углубления вдоль прямой линии
кристаллической решетки.
О
а—расстояние между минимумами; для решетки алмаза оно равно 3,55 А.
-Г
которых находится элементарная частица. Такой ряд характеризуется равным
потоком энергии, энергетической равноценностью всех частиц, что обусловливает
для них наличие определенной термодинамически фиксированной точки плавле-
ния— температуры «распада» кристаллической решетки. Такая равноценность
потенциалов не может быть при-
нята для реальных твердых тел, в
особенности для аморфных, где
в силу условий затвердевания
частицы «фиксированы» с нерав-
ными потенциальными углубле-
ниями (рис. 13).
Для идеальных твердых тел
упругая деформация является рав-
новесным состоянием, вызванным
Рис. 13. Потенциальные углубления вдоль
прямой линии в аморфном теле.
изменением междучастичного рас-
стояния вследствие приложения внешнего напряжения. Такая деформация не
Должна зависеть от времени действия напряжения и быть полностью упругой,
т. е. после снятия напряжения деформация должна полностью исчезнуть вслед-
ствие взаимодействия межчастичных сил.
Соотношение между упругой деформацией и напряжением определяется, как
известно, законом Гука:
Д/ _ а
£ ’
(1)
где: I — длина образца; — удлинение при растяжении; а — напряжение;
Е — константа (модуль упругости, модуль Юнга).
Графически эта зависимость определяется прямой, проходящей через на-
чало координат.
Упругая деформация
73
Каждому напряжению, даже самому малому, соответствует упругая дефор-
мация, величина которой определяется значением константы Е — тангенсом угла
наклона прямой.
Модуль упругости
£= —
Д/
(2)
является материальной константой и важнейшей характеристикой твердого тела.
Он выражает способность материала упруго сопротивляться деформации; чем
больше Е, тем меньше при одинаковых напряжениях деформация.
Физически модуль упругости Е представляет то напряжение, которое следо-
вало бы приложить к телу, чтобы упруго растянуть его вдвое; это вытекает из
формулы (2), когда Д' = I, т. е. когда удлинение становится равным длине об-
разца, что однако неосуществимо, так как упругое удлинение реализуется лишь
на весьма малую величину (~1°/о), после чего или достигается предел проч-
ности материала, или же развивается необратимая, пластическая деформация.
Упругая деформация характеризуется большими значениями модуля упру-
гости (порядка 5-Ю4 — 5-Ю6 кг/см2) и, следовательно, малыми деформациями.
Модуль упругости находится в определенной зависимости от величины меж-
молекулярных сил притяжения, т. е. от теоретической прочности твердого тела
(стр. 120), а именно, Е = 0,1 сыако , где сыакв—напряжение, соответствующее
теоретической прочности тела.
При упругой деформации, как следствие изменения междучастичных рас-
стояний, происходит изменение объема твердого тела, — в случае растяжения
происходит увеличение объема. Это находит . — '
свое выражение в величине коэффициента
Пуассона, который всегда меньше 0,5.
При упругом растяжении (как следствие
увеличения объема)—тело охлаждается, а
при исчезновении деформации — нагревается.
Наконец, деформация в области упругих
напряжений устанавливается в фазе с на-
пряжением практически мгновенно (со ско-
ростью звука).
Существенной характеристикой твердого
тела является предел упругости, — то наиболь-
шее напряжение, которое сопровождается еще
упругой, обратимой деформацией.
Отношение предела упругости к пределу рис. 14. Кривая хрупкого (7) и
прочности характеризует поведение твердого пластического (2) разрушений,
тела при деформации.
Если исходить из обычных условий деформации, то в этом отношении твер-
дые тела можно разделить на две группы:
1. Твердые тела, которые разрушаются при напряжениях ниже предела
упругости, т. е. у которых предел прочности ниже предела упругости.
Диаграмма деформации таких тел может быть представлена прямой
(рис. 14), величина деформации крайне мала (~1%) и определяется величиной
модуля ’ упругости. Такие тела называются обычно хрупкими, телами (стекло,
керамика, чугун, некоторые пластики и т. д.).
2. Твердые тела, которые разрушаются после достижения предела упру-
гости, т. е. у которых предел прочности выше предела упругости. Диаграмма
растяжения таких тел имеет начальный линейный характер, который выше пре-
дела упругости переходит в нелинейный, обычно дугообразный, заканчиваю-
щийся пределом прочности и точкой разрушения.
Деформация выше предела упругости может иметь значительную величину,
причем, естественно, вся деформация, соответствующая нелинейной части кри-
вой, является необратимой, пластической.
Такие тела называются обычно упруго-вязкими, упруго-пластичными (сталь,
бронза, многие пластмассы).
74
Гл. И. Физические сснсвы технологии пластмасс
Однако, если исходить из различных условий деформации и состояния тела,
то «хрупкость» не может считаться материальной характеристикой тела, а ско-
рее определяется условиями деформации и разрушения.
Правильнее поэтому говорить не о хрупких и упруго-вязких телах, а об
условиях хрупкого и упруго-вязкого разрушения твердого тела.
Хрупкое разрушение наступает во всех случаях, когда создаются условия
для преждевременного разрушения твердого тела (до достижения предела упру-
гости), а также в тех случаях, когда скорость роста напряжения превосходит
скорость развития пластической деформации, — скорость перегруппировки эле-
ментарных частиц.
Поэтому одно и то же тело, например стекло, является хрупким, если
это обычное, объемное стекло, и упруго-вязким в виде стеклянных нитей. Бла-
годаря концентрации микротрещин (стр. 124), техническая прочность обычного
стекла крайне низка и разрушение происходит хрупко, не достигая предела
упругости. Однако в стеклянных нитях отсутствуют в значительной мере мнкро-
трещины, техническая прочность во много раз превосходит прочность обычного
стекла и при деформации достигается предел упругости, т. е. стеклянные нити
ведут себя как упруго-вязкое тело.
Основные закономерности упругой деформации были изложены примени-
тельно к идеально-упругому телу. Эти закономерности характеризуются равно-
весным характером упругой деформации, отсутствием какого-либо влияния фак-
тора времени — времени воздействия напряжения.
Поведение реальных тел характеризуется неравновесным состоянием п
влиянием фактора времени даже в области напряжений ниже предела упру-
гости. Как предел упругости, так и область напр/тжений, где действует законо-
мерность Гука, определяются в значительной мере временным фактором. Чем
длительнее время воздействия напряжения, тем ниже предел упругости, тем при
более низких напряжениях происходят необратимые перегруппировки частиц
макроскопически обусловливающие пластическую деформацию.
Такие процессы, протекающие во времени, должны приводить к снижению
напряжения при заданной величине деформации, т. е. к релаксации напря-
жения.
Пластическая деформация
Пластическая, необратимая деформация твердого тела определяется пере-
группировкой, перемещением частиц, полным изменением порядка в расположе-
нии частиц, без измеиення междучастичных расстояний.
Такая перегруппировка ведет к изменению формы тела, к деформации, без
какого-либо изменения внутренней энергии. Поэтому отсутствуют и силы, кото-
рые могли бы привести частицы в первоначальное состояние.
Пластическая деформация не сопровождается изменением объема, следова-
тельно, коэффициент Пуассона равен 0,5.
Пластическая деформация протекает во времени, скорость деформации
определяется величиной внутреннего трения, — вязкостью.
Тепловой эффект пластической деформации выражается в превращении под-
водимой энергии в тепло.
В твердых телах пластическому течению предшествует упругая часть де-
формации.
Пластическая деформация, принципиально, может наблюдаться у всех твер-
дых тел, если временные условия деформации и техническая прочность тела
позволяют достигнуть предел упругости.
Если отличия пластической деформации от упругой деформации идеально
твердого тела носят принципиальный характер и определяются, в первую оче-
редь, равновесным характером упругой деформации, ее независимостью от вре-
менного фактора, то отличия пластической деформации от вязкого течения
жидкости принципиально не существенны.
Пластическая деформация
75*
Для получения качественной характеристики пластического течения твердого
тела и вязкого течения жидкости целесообразно исходить из закономерностей
напряжения сдвига.
Для упругого тела величина обратимого сдвига у пропорциональна напря-
жению сдвига т:
Т=|. (3)
где g — модуль сдвига.
Для жидкостей любое малое напряжение сдвига вызывает градиент скорости
течения и их поведение определяется законом Ньютона:
dv т
dy ~ V’
(4)
dv
где: -т-
— градиент
скорости;
'П — материальная
константа, характеризующая
вязкость.
Зависимость скорости деформации от напряжения сдвига т выражается,
следовательно, прямой, проходящей через начало координат (рис. 15) с на-
клоном 1/т).
Рис. 15. График течения
ньютоновской жидкости.
Рис. 16. График течения жид-
кости с аномальной вязкостью.
Таким образом, жидкости характеризуются наличием равновесного соотно-
шения между скоростью течения и напряжением, в то время как упругие
тела — соотношением между величиной деформации и напряжением. Как закон
Ньютона для жидкостей, так и закон Гука для упругих тел действительны для
идеальных тел, для которых отсутствуют какие-либо временные зависимости.
В отличие от так называемых «ньютоновских» жидкостей, практически под-
чиняющихся в широком интервале напряжений закону пропорциональности ско-
рости течения напряжению сдвига, некоторые жидкости, в особенности по-
строенные из асимметричных цепных молекул, не подчиняются закону Ньютона.
Их поведение может быть представлено кривыми, изображенными на рис. 16,
t
где, как видно, отношение меняется с изменением значения т, и, следова-
тельно, т) не может быть рассматриваема как материальная константа.
Зависимость скорости течения от напряжения выражается для них, следо-
вательно, формулой
Д =^Е = _1
dy Т|8
(5>
76 Гл. И. Физические основы технологии пластмасс
где —коэффициент, не идентичный с константой -л, а п больше, иногда же
меньше единицы.
Таким образом, хотя напряжение и создает для таких веществ стационар-
ное течение, скорость его, одиако, не пропорциональна напряжению. Для су-
ждения о скорости течения в зависимости от напряжения недостаточно знать
одни какой-либо параметр (например т; для ньютоновских жидкостей), а не-
обходимо знание всей кривой течения.
Вместе с тем между обоими типами веществ имеется та общая законо-
мерность, что кривая скорости течения приходится на начало координат. Та-
ким образом, даже самое малое напряжение сдвига вызывает как у «ньютонов-
ских», так и у «неньютоновских» жидкостей определенный градиент течения.
Рис. 18. График течения пластич-
ных тел; f—предельное напря-
жение сдвига.
рис. 17. График течения идеально-
пластичного тела; f—предельное
напряжение сдвига.
В отличие от этих веществ, обычно называемых «жидкостями», тела, об-
ладающие упруго-вязкими свойствами, требуют для преодоления упругих сил за-
траты части напряжения. В этом случае до некоторого предела напряжения f,
предельного напряжения сдвига, наблюдается упругая деформация, характе-
ризующаяся смещением пропорционально напряжению, и лишь после достиже-
ния этого напряжения может начаться течение вещества.
На рис. 17 и 18 показана зависимость скорости деформации от напряжения
сдвига для упруго-вязких тел.
Вещество, которое следует зависимости, показанной на рис. 17, называется
идеально-пластичным телом.. Эта зависимость выражается следующим уравне-
нием:
D =
dv __ 1
dy ~ т]
О' —f)>
(6)
где ,* — предельное напряжение сдвига, или предел текучести.
До напряжения f вещество не течет и ведет себя как упругое тело; выше f
устанавливается такая стационарная скорость течения, которая пропорцио-
нальна не т, а (т—/) ив этих пределах подчиняется закону Ньютона.
Однако значительное большинство «пластичных» тел не подчиняется линей-
ной зависимости течения от ? —f, а обнаруживает заметную кривизну графика
течения (рис. 18) и по аналогии с течением неньютоновских жидкостей под-
чиняется зависимости:
rfv 1 .
=----------(т — f}n
dy т]® J'
(7)
Таким образом, отличием упруго-вязких или «пластичных» тел от жидкостей
служит наличие на графике течения параметра f — предельного напряжения
Значение временнбго фактора для низкомолек. веществ
сдвига. Чем выше напряжение, соответствующее f, тем больше свойства веще-
ства соответствуют твердому телу, чем меньше значение Д тем тело «пла-
стичнее», «мягче» и приближается по своим свойствам к жидкости.
Таким образом, можно считать, что жидкости характеризуются значением
f = 0, т. е. любое малое напряжение сдвига вызывает течение, тогда как твер-
дые тела характеризуются значением f > 0 и для начала течения требуется
преодолеть f — величину предельного напряжения сдвига.
Однако такое разграничение веществ на жидкости и твердые тела дей-
ствительно лишь постольку, поскольку не учитывается фактор времени — времени
воздействия напряжения.
Вследствие того, что величина предельного напряжения сдвига и само на-
личие его определяется временными условиями и условиями испытания (чув-
ствительностью методов, фиксирующих наличие необратимых структурных из-
менений и величину деформации) принципиальная грань между жидко-вяз-
кими и упруго-вязкими веществами не может быть установлена.
ЗНАЧЕНИЕ ВРЕМЕННОГО ФАКТОРА (ПРОЦЕССА РЕЛАКСАЦИИ)
ДЛЯ НИЗКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Из классических законов механики твердого и жидкого тела —
законов Гука и Ньютона — следует, что каждому приложенному на-
пряжению соответствует либо определенная деформация (закон
Гука), либо определенная скорость деформации (закон Ньютона).
В обоих случаях равновесное состояние устанавливается «мгно-
венно» и фактор времени не имеет значения. Эти законы, однако,
относятся к поведению идеальных тел.
Все реальные тела тем и отличаются от идеальных, что для них
характер деформации и напряжений на любом участке кривой де-
формации зависит от времени действия напряжения.
Значение временнбго фактора и поведение в этом отношении
реальных тел определяется отсутствием абсолютной упорядочен-
ности в расположении элементарных частиц и, следовательно, не-
равноценностью их в энергетическом отношении. Это имеет место
как для аморфных, так и для кристаллических тел.
С другой стороны, даже в том случае, когда средняя кинетиче-
ская энергия элементарных частиц (атомов, молекул) мала для
преодоления действующего между ними потенциального барьера,
в теле всегда найдется некоторое число частиц, кинетическая энер-
гия которых может быть сколь угодно большой.
Иными словами, имеется вероятность, что любая из элементар-
ных частиц тела может приобрести энергию, которая позволит ей
преодолеть потенциальный барьер и, следовательно, оставить свое
старое место между частицами и занять новое положение равнове-
сия в окружении других частиц.
Вероятность такого процесса определяется уравнением
_ п
W=Ae kT , (8)
где: U — величина потенциального барьера; А — константа;
k — константа Больцмана; Т — абсолютная температура.
78
Гл. П. Физические основы технологии пластмасс
Приложение любого малого внешнего напряжения должно вы-
звать направленный процесс перегруппировки частиц, т. е. опреде-
ленную скорость деформации (течения).
Время, необходимое для перехода частиц из первоначального
равновесного состояния в новое состояние равновесия, называется
.временем релаксации.
Время релаксации (X) должно быть в обратной зависимости от
вероятности процесса перегруппировки частиц и определяться урав-
нением
к^Ае^. (9)
Поведение упруго-вязких тел может быть описано соотноше-
нием «внешнего времени» (времени действия напряжения) и «вну-
треннего времени» (времени релаксации).
Если внешнее время равно или больше внутреннего времени,
наблюдаются текучие, жидкие, свойства вещества, если оно
меньше, — упругие, твердые.
Общая феноменологическая схема, учитывающая влияние вре-
менного фактора и одновременное наличие упругих и вязких
свойств в твердых телах, была предложена Максвеллом еще
в 1867 г.
Если рассматривать временную зависимость между напряже-
нием сдвига и деформацией для идеально-упругого тела, не обла-
дающего текучестью, то действительно уравнение
^- = 0-^-
dt и dt '
(Ю)
где: t — время; т — напряжение сдвига; G — модуль сдвига;
Y — деформация.
Если, однако, тело наряду с упругими обладает и вязкими (пла-
стическими) свойствами, напряжение сдвига будет стремиться
уменьшиться (релаксировать) со скоростью, зависящей от ее соб-
ственной величины; тогда уравнение (10) примет вид:
где X — константа (время релаксации).
Если тело деформировано на постоянную величину, т. е.
7 = const, то ~ = 0 и уравнение (11) примет вид:
dt т
dt /. ’
(12)
т. е. напряжение будет снижаться (релаксировать) прямо пропор-
ционально величине напряжения т и обратно пропорционально вре-
мени релаксации X.
Значение временнбго фактора для низкомолек. веществ
79
Остаточное напряжение т, после t времени действия напряже-
ния может быть найдено интегрированием уравнения (12), что
дает:
f
= (13)
где: — напряжение при t = 0.
Таким образом, время релаксации X представляет время, необ-
ходимое для того, чтобы напряжение при постоянной деформации
„ 1
образца уменьшилось до — первоначального значения.
Если же действующее напряжение является постоянным, т. е.
т = const, то деформация упруго-вязкого тела будет со временем
увеличиваться; тогда ^ = 0и уравнение (11) примет вид:
G dt~ \ или dt~G'K' (14)
Интегрируя уравнение (14), получим деформацию ft при вре-
мени t\
(15)
где Уо — деформация при t — 0.
Это уравнение показывает, что при действии постоянного напря-
жения т деформация со временем растет со скоростью, пропорцио-
нальной напряжению и обратно пропорциональной произведению
времени релаксации на модуль сдвига.
Если X очень велико и, следовательно, у--> 0, уравнение (11)
переходит в уравнение идеально-упругого тела (10), в котором на-
пряжение не зависит от времени.
Уравнение (14) может быть рассматриваемо как видоизменен-
ная форма уравнения (4) ньютоновского закона течения нормаль-
ной жидкости, в котором место коэффициента вязкости ч) занимает
произведение GX и, следовательно:
т. е. время релаксации пропорционально iq.
Из этого уравнения следует также, что если X очень мало, то
скорвсть деформации становится очень большой и стремится к бес-
конечности; если же X очень велико, то течение практически пре-
кращается.
Поэтому закономерно рассматривать вязкие жидкости как
аморфные тела с весьма малым временем релаксации. Они будут
выявлять упругие свойства твердых тел, если время действия на-
пряжения будет меньше времени релаксации.
80
Гл. II. Физические основы технологии пластмасс
Применение звуковых и ультразвуковых частот для исследова-
ния механических свойств жидкостей высокой вязкости (~ 500 пуаз
и больше) позволило наблюдать проявление в них упругих свойств.
При наложении электрических полей высокой частоты (106—109 герц}
можно установить наличие упругих деформаций в жидкостях более
низкой вязкости.
Механическое поведение реальных тел, обладающих наряду с упругими
также и вязкими свойствами, может быть наглядно представлено максвеллов-
ским элементом, состоящим из пружины и поршня, включенных
последовательно (рис. 19).
Упругие свойства вещества характеризуются пружиной, мгно-
венная деформация которой определяется величиной
т
а вяз
и
кие свойства — движением поршня со скоростью, равной ~. При
наложении напряжения пружина мгновенно растягивается, а пор-
шень начинает двигаться с определенной скоростью. Если через
t секунд напряжение снято, то пружина полностью сократится.
Рис. 19. Ма-
ксвелловский
элемент (ме-
ханическая
модель для
упруго-вяз-
кого тела).
ио сдвиг поршня на величину t останется постоянным. Если
время действия напряжения, крайне мало по сравнению с X =
(временем релаксации), то вся деформация определяется только
деформацией пружины и полностью исчезает при снятии на-
пряжения. Если, однако, время действия напряжения t велике
7 г
по сравнению с —, то течение, равное t, значительно перекры-
вает упругую деформацию.
В такой же мере эта модель иллюстрирует и релаксацик>
напряжения при постоянной величине деформации, если считать,
что растянутая пружина действует на поршень и по мере'
его выдвижения имеет возможность сокращаться, что при-
водит к снижению напряжения при растяжении модели на постоянную ве-
личину.
Рис. 20. Зависимость деформации от времени*при постоянном напряжении.
•у —деформация; t — время; —время включения напряжения; — время снятия напряжении
На рис. 20 показана зависимость деформации от времени для идеально
твердого тела (/), идеальной жидкости (II), для максвелловского элемента (III).
Максвелловская модель качественно иллюстрирует поведение реальных тел,
состоящих лишь из сравнительно простых и однородных частиц.
Закономерности аморфного состояния низкомол. веществ
81
Для понимания деформации и релаксации аморфных тел с более сложной
” — -------------» ----- ------- ----------модель запаздывающей
молекулярной структурой может служить механическая
упругой деформации (элемент Фойгта).
Запаздывающая упругость может быть представ-
лена моделью, состоящей из пружины и поршня, вклю-
ченных параллельно (рис. 21). В этом случае вязкость
(поршень) играет роль замедляющего сопротивления
установлению упругого разновесия, в то время как
в максвелловском элементе она определяет лишь не-
обратимое добавочное смещение, которое следует при-
бавить к упругому удлинению. На рис. 20 (IV) пока-
зана кривая зависимости деформации от времени для
запаздывающей упругой реакции, если напряжение
включено в момент ta и выключено в момент Л. Прин-
ципиальное отличие обоих элементов отражает также
отличия в молекулярной структуре. Для запаздываю-
щей упругой реакции типичным механизмом деформа-
£
Рис. 21. Механическая
модель для запазды-
вающей упругости
(элемент Фойгта).
сложной смеси веществ
ции являются процессы распрямления и сокращения
макроцепей, т. е. она иллюстрирует главным образом
деформацию и релаксацию линейных полимеров
(стр. 102). Вместе с тем она часто наблюдается и
в системах с малыми молекулами, но состоящими из
(окислевные битумы и Др.).
Закономерности аморфного состояния низкомолекулярных
веществ
С точки зрения механики твердое тело, обладающее пластиче-
скими (упруго-вязкими) свойствами, отличается от вязкой жидкости
наличием предельного напряжения сдвига, вне зависимости от
структуры этого тела — аморфной или кристаллической.
Однако с точки зрения физики твердого тела — к истинно твер-
дым относят лишь тела с высокой внутренней упорядоченностью,
т. е. кристаллические. Твердые же тела стеклообразной, аморфной
структуры считают жидкостями с большой вязкостью, или пере-
охлажденными жидкостями. Аморфное тело, действительно, тем и
отличается от кристаллического, что в нем сохраняется структура,
соответствующая более высоким температурам (выше температуры
плавления), т. е. структура жидкости.
Переход тела из жидкого состояния в твердое осуществляется
путем ослабления разбрасывающего действия теплового движения,
противодействующего силам взаимного сцепления молекул. В опре-
деленный момент силовые поля молекул становятся равными раз-
брасывающему тепловому движению и в жидкости начинают про-
являться ориентационные участки (зародыши кристаллизации), ко-
торые затем быстро растут.
Склонность тела к кристаллизации можно охарактеризовать
скоростями образования и роста зародып/ей кристаллов. Оба про-
цесса зависят как от величины сил сцепления, действующих между
частицами (ионами, атомами, молекулами), так и от их подвиж-
ности. С увеличением размеров частиц уменьшается их подвиж-
6 Зап. 1269. Э. И. Барг
82
Гл. II. Физические основы технологии пластмасс
ность, вследствие чего уменьшается и скорость кристаллизации.
Последняя в такой же степени уменьшается с увеличением вязкости
и, следовательно, с понижением температуры.
На рис. 22 представлена общая схема кристаллообразования. За
пунктирной линией скорость кристаллообразования близка нулю;
возможность кристаллообразования из-за быстрого увеличения вяз-
кости в связи с понижением температуры ограничена узким темпе-
ратурным интервалом; если
v
Рис. 22. Схема кристаллообразования.
/—скорость образования зародышей;
2—скорость роста кристаллов; 3—увели-'
чение вязкости.
этот интервал быстро перейден (на-
пример при быстром охлаждении),
то кристаллизация становится не-
возможной и тело переходит в твер-
дое состояние, сохраняя структуру
жидкости.
Кристаллическое тело представ-
ляет собою наиболее плотно упако-
ванную и упорядоченную систему
частиц, характеризующуюся мини-
мальным значением потенциальной
энергии, практической равноцен-
ностью потенциальных барьеров,
стабильным и равновесным состоя-
нием. Следовательно, для кристал-
лического состояния (по сравне-
характерно меньшее теплосодержа-
НИЮ с любым другим)
кие.
Разность в теплосодержании кристаллического и аморфного тела
(при температурах, близких к температуре кристаллизации) необ-
ходимо, в первую очередь, отнести за счет выделения скрытой
теплоты кристаллизации. Поэтому самопроизвольный переход кри-
сталлического тела в аморфное невозможен.
С этой точки зрения переход жидкого тела в твердое, минуя
фазу кристаллизации, необходимо рассматривать как фиксацию со-
стояния с более высоким термодинамическим потенциалом. По-
этому в первом приближении представляется закономерным аморф-
ные тела рассматривать как переохлажденные жидкости, как
системы с избытком внутренней энергии (по сравнению с кристал-
лическими телами), как неравновесные системы, которые могут
самопроизвольно переходить в более равновесное — кристалличе-
ское состояние.
Принципиально все тела способны к переохлаждению и к пере-
ходу в твердое аморфное состояние. Однако для некоторых тел
разность удельных термодинамических потенциалов обеих фаз (для
краткости будем их называть кристаллизационными потенциалами)
столь велика, что переохлажденное состояние является крайне не-
устойчивым (например для металлов). В то же время для других
тел кристаллизационный потенциал крайне низок, вследствие чего
они имеют столь большую склонность к переохлаждению, что их
Закономерности аморфного состояния низкомол. веществ
83
трудно, а иногда практически и невозможно, получить в кристалли-
ческом состоянии (например глицерин и др.).
Процесс перехода в стеклообразно-аморфное состояние и устой-
чивость этого состояния определяются не только термодинамиче-
скими, но и молекулярно-кинетическими факторами.
Учет влияния молекулярно-кинетических факторов заставляет
отказаться от рассмотрения стеклообразных веществ только как
переохлажденных жидкостей, как неустойчивых, неравновесных си-
стем, и во многом объясняет высокую устойчивость ряда стекло-
образных синтетических и природных веществ.
С точки зрения молекулярно-кинетических представлений важ-
нейшим процессом, развивающимся антагонистически по отноше-
нию к процессу кристаллизации, является процесс ассоциации
(агрегации), т. е. процесс образования крупных комплексов вслед-
ствие действия межмолекулярных сил. Чем выше полярность и
сложнее структура молекул, тем в большей степени процесс агре-
гации конкурирует с процессом кристаллизации и тем вероятнее
направление процесса в сторону перехода в стеклообразное, аморф-
ное состояние.
Степень ассоциации является статистической величиной и опре-
деляется температурой; повышение температуры сдвигает кривую
динамического равновесия в сторону преобладания индивидуальных
моЛекул, понижение температуры — в сторону преобладания ком-
плексов. Эти процессы в такой же степени, как и процессы кристал-
лизации, зависят от строения молекул. При быстром охлаждении
фиксируется молекулярная структура, соответствующая более вы-
сокой температуре (т. е. структура с меньшей степенью агрегации),
и лишь со временем (которое зависит от степени переохлаждения)
достигается молекулярная структура, соответствующая равновес-
ному состоянию для данной температуры (т. е. с большей степенью
агрегации).
Самопроизвольный переход аморфных тел в кристаллическое
состояние теоретически возможен; он становится, однако, крайне
мало вероятным в тех случаях, когда к аморфному состоянию при-
водят в основном не внешние воздействия (быстрый переход зоны
кристаллизации), а молекулярно-кинетические процессы (агрегация
и усложнение структуры молекул), конкурирующие с процессами
кристаллизации.
Достаточно указать, что многие природные смолы (янтарь, ко-
пал и др.) находятся в устойчивом стеклообразном состоянии еще
со времен эоценового периода и что до сих пор не известны случаи
кристаллизации низкомолекулярных смол и стекол при температу-
рах ниже точки стеклования.
Практическую стабильность аморфных тел можно было бы
объяснить как малым термодинамическим потенциалом, так и вы-
сокой вязкостью, обусловливающей длительность перехода в равно-
весное (кристаллическое) состояние. Однако вряд ли эти объясне-
г.
84
Гл. !!. Физические основы технологии пластмасс
ния удовлетворительны для устойчивых природных или синте-
тических аморфных веществ, получивших широкое практическое
применение. Ближайшее рассмотрение таких систем позволяет
прийти к заключению, что и с чисто термодинамической точки зре-
ния не при всех условиях и температурах запас энергии аморфных
тел должен быть выше, чем у кристаллических.
И, действи-
меныпее теплосодержание кристаллических тел является следствием
скрытой ~
Эти соображения были высказаны Б. Л. Блюмбергом.
тельио,
выделения
теплоты
Теплосодержание
Рис. 23. Изменение теплосо-
держания при кристаллическом
и аморфно-стеклообразном за-
стывании.
кристаллизации. Одиако теплосодержание твер-
дого тела определяется также и величиной
его теплоемкости, различной для жидкого, кри-
сталлического и стеклообразного состояний.
На рис. 23 представлено изменение тепло-
содержания при кристаллическом и аморфно-
стеклообразном застывании. Кривая 1—2—3—4
характеризует выделение тепла при кристал-
лизации: отрезок 1—2 — при охлаждении жид-
кости, отрезок 2—3 — при температуре кри-
сталлизации (Ткр) и показывает выделение
скрытой теплоты кристаллизации, а отрезок
3—4 — уменьшение теплосодержания пропор-
ционально охлаждению кристаллического тела,
т Так как теплоемкость кристаллического тела
меньше теплоемкости жидкости, то при охла-
ждении на одинаковое число градусов кри-
сталл отдает меньше тепла, поэтому отрезок
3—4 имеет меньший наклон к оси температур,
чем отрезок 1—2.
Иной вид имеют кривые 1—5—6 и 1—5—4,
характеризующие выделение тепла при про-
цессе стеклообразоваиия. В этом случае
жидкое состояние сохраняется до более низкой
температуры — температуры стеклования (То) и на отрезке 1—2—5 жидкость
успевает «выбросить» значительно больше тепла, чем на отрезке 1—2 кривой кри-
сталлизации. Таким образом, при некоторой температуре Т разность в теплосодер-
жании кристаллического (кривая 1—2—3—4) и стеклообразного (кривая
1—2—5—6) тела уже значительно меньше, чем можно было бы ожидать, судя
по выделившейся скрытой теплоте кристаллизации. Однако теплоемкость стекло-
образного тела может быть больше теплоемкости кристаллического; поэтому воз-
можна кривая 1—2—5—4, показывающая, что при некоторой температуре То
теплосодержание обеих форм твердого тела станет одинаковым, что соответствует
истинному термодинамическому равновесию кристаллической и стеклообразной
фаз. Таким образом, при температурах значительно ниже температуры стеклова-
ния стеклообразное состояние и с энергетической точки зрения можно рассматри-
вать как закономерное, равновесное состояние.
Можно считать, что процессы ассоциации, происходящие при
стеклообразном застывании, также способны в значительной сте-
пени компенсировать избыток внутренней энергии (получающийся
вследствие отсутствия выделения скрытой теплоты кристаллизации),
так как они протекают с уменьшением внутренней энергии тела.
Изложенные выше соображения дают известное основание счи-
тать аморфную стеклообразную форму многих тел самостоятельные
агрегатным состоянием.
То и интервал размягчения низкомолекулярных смол 85
Стеклообразное аморфное состояние весьма распространено
в природе; важнейшие типы синтетических полимеров, играющих
важную роль в современной технике, также относятся к аморфным,
стеклообразным материалам.
Органические вещества со стабильной аморфно-стеклообразной
структурой называются смолами (стр. 26).
По структуре смолы могут быть разделены на два основных
класса: 1) низкомолекулярные и 2) высокомолекулярные.
Температура стеклования и интервал размягчения
низкомолекулярных смол
Хаотический комплекс молекул, характерный для аморфных тел,
обусловливает важнейшее отличие аморфных тел от кристалли-
ческих: отсутствие фазовых переходов и равновесного сосущество-
вания фаз при определенной термодинамически фиксированной
температуре. Аморфные тела остаются гомогенными при всех тем-
пературах.
Если нагревать кристаллическое тело, то при определенной тем-
пературе (температура плавления) с точностью свыше 0,01° все
Рис. 24. Изменение свойств Е
кристаллического тела с тем-
пературой Т.
Рис. 25. Изменение свойств
аморфного тела в интервале
размягчения 7"тек—Ге.
свойства тела скачкообразно меняются (рис. 24); это соответ-
ствует фазовому превращению, изменению структуры тела, пере-
ходу от дальнего порядка твердого состояния к ближнему порядку
жидкого состояния. Такое скачкообразное изменение свойств объяс-
няется практической равноценностью в энергетическом отношении
всех элементарных частиц, составляющих пространственную ре-
шетку кристаллического тела.
В отличие от кристаллических, аморфные тела характеризуются
постепенным изменением свойств в зависимости от температуры,
отсутствием фазового перехода. Вместе с тем в определенном тем-
86
Гл. II. Физические основы технологии пластмасс
пературном интервале наблюдается нелинейная закономерность
изменения свойств.
Если проследить зависимость какого-либо свойства Е (напри-
мер, объема V) низкомолекулярного аморфного тела от темпера-
туры, то получим кривую Е, представляющую две прямые, соеди-
ненные искривленным участком (рис. 25). Точку первого изгиба Те
называют температурой стеклования, — она характеризует измене-
ние линейных закономерностей твердого тела при переходе его в-
вязкопластическое состояние. Точку второго изгиба Ттск называют
температурой текучести, — она характеризует переход линейной за-
кономерности свойств жидкого тела в закономерность вязко-пла-
стичного тела.
Интервал Ттек — Тс называют интервалом размягчения, интер-
валом пластичности. В этом интервале, как видно из рис. 25,
резко меняются термодинамические показатели аморфного тела:
удельный объем, теплоемкость, диэлектрическая проницаемость
и др. Наглядно это видно на зависимостях первой и второй произ-
водных Е от температуры. Так изменение температурного коэффи-
циента расширения будет происходить по кривой , где на
участке 2 две прямые параллельные оси абсцисс показывают изгиб.
Соответственно закономерности второй производной, характеризую-
„ .. d'-’E
щие ускорение в изменении свойств, представлены кривой
имеющей максимум в Тщ>. Таким образом, в интервале размягче-
ния (ГтеК —Го) имеет место ускорение в изменениях свойств, до-
ходящее до максимума в точке Тщ,. Точку Tai. называют точкой
превращения.
Интервал размягчения (Гток — Т„) отражает молекулярные
процессы, происходящие в аморфном теле. До температуры Т тс,.
в нем наблюдаются закономерности жидкого состояния, связанные
со свободным вращательным и поступательным движением моле-
кул. Так, понижение температуры до Гтек вызывает равномерное
уменьшение объема, характерное для жидкого тела и объясняемое
уменьшением междучастичных расстояний и изменением структуры
жидкости, так как каждой температуре соответствует структура,
связанная с установлением ближнего порядка, соответствующего
минимуму потенциальной энергии. Закономерности жидкого со-
стояния прекращаются при Г ток ; свободное вращение всех молекул
уже не может происходить вследствие их сближения и недостатка
свободного пространства; часть молекул может совершать лишь ко-
лебательные движения около фиксированных в пространстве поло-
жений равновесия; чем ниже температура, тем меньше молекул со
свободным вращением и тем больше число закрепленных, фиксиро-
ванных молекул. Нарастанием количества закрепленных молекул
среди свободно вращающихся объясняется нелинейный характер
изменения объема в интервале размягчения.
Та и интервал размягчения низкомолекулярных смол
87
Ниже Тс молекулы закреплены, прекращается возможность из-
менения структуры тела и уменьшение объема происходит только
за счет уменьшения междучастичных расстояний. Это обстоятель-
ство приводит к линейному характеру зависимости свойств от тем-
пературы и к уменьшению наклона кривой по сравнению с накло-
ном при температурах выше TWK.
Таким образом, интервал размягчения характеризуется одно-
временным наличием как свободно вращающихся, так и фиксиро-
ванных молекул и закономерностями, отличными как от жидкого,
так и твердого состояния.
Гомогенность аморфного тела во всем температурном интервале
позволяет характеризовать точки Тс, Ттек и интервал размягчения
величиной и интервалом вязкости этого тела, не рассматривая
структурных изменений, возможных выше и ниже То.
Так для всех низкомолекулярных аморфных тел (смолы, стекла)
~ Лек ~ Ю7 пуаз; следо-
Рис. 26. Зависимость Тс
от скорости нагрева.
/—при медленном подъеме
температуры; 2 — при более
быстром подъеме температуры.
вязкость в точке Та ~ 1013 пуаз, а в точке
вательно, в интервале размягчения вязкость
находится в пределах 1013—107 пуаз.
То обстоятельство, что в пределах интер-
вала размягчения вязкость различных ве-
ществ имеет соизмеримые значения (при
одинаковых методах и скоростях измерения)
свидетельствует, что этот интервал, а так-
же То и Г тек характеризуются определен-
ной скоростью молекулярных перегруппиро-
вок и близкими значениями времен рела-
ксации. Мы можем поэтому считать ТЛ тем-
пературой, при которой время релаксации
(время, необходимое для перегруппировки
частиц) становится равным времени наблю-
дения, времени действия напряжения.
Температура стеклования, хотя и не яв-
ляется точкой фазового перехода и равно-
весного сосуществования фаз, однако она
имеет столь же важное техническое значение для характеристики
аморфного тела, как и температура плавления кристаллических тел.
Релаксационный характер 7’0 определяет ее зависимость от ско-
рости изменения температуры. Чем быстрее происходит охлажде-
ние тела, тем выше То (рис. 26).
Эта зависимость действительна для скоростей, при которых успе-
вает устанавливаться тепловое равновесие, но при которых струк-
тура тела, соответствующая более низкой температуре, не успевает
определиться. Поэтому и более быстрое установление равновесия
при различных температурах дает более высокую точку стеклова-
ния, чем более медленное.
Значения температур стеклования, полученные различными ме-
тодами, не могут быть поэтому сравнимы. Они в значительной
88
Гл. II. Физические основы технологии пластмасс
степени определяются скоростью подъема температуры, величиной и
формой образца, величиной напряжения, скоростью роста напряже-
ния и другими релаксационными факторами. Температура стекло-
вания, если сравнивать при одних и тех же методах определения,
тем выше, чем больше межмолекулярные силы, чем выше поляр-
ность смолы.
Для всех низкомолекулярных тел величина То определяет тем-
пературу размягчения, теплостойкость материала, температуру,
выше которой материал не может быть применен.
Столь же важной технической характеристикой аморфных тел
является величина интервала размягчения, или интервала пластич-
ности (Лек— Л).
Для индивидуальных (переохлажденных) низкомолекулярных
органических и неорганических аморфных веществ, а также и для
низкомолекулярных смол интервал размягчения сравнительно мал
и находится примерно в пределах 20—50°.
Аморфные тела с малым интервалом размягчения (20—50°) от-
носят к так называемым нормальным стеклам (смолам).
Ниже То такие смолы представляют собою хрупкие тела, разру-
шающиеся до достижения предела упругости. График деформации та-
ких смол ниже Л состоит только из линейной упругой составляющей.
При достижении Тв они показывают обычную диаграмму растя-
жения упруго-вязких тел, которая состоит из линейного участка,
соответствующего упругой деформации, и нелинейной части, соот-
ветствующей пластической деформации.
Выше Гтек они показывают график зависимости напряжения
сдвига от температуры, соответствующий нормальным жидкостям
(рис. 15, стр. 75).
Аморфные тела с более сложной и неоднородной структурой ха-
рактеризуются значительно большим интервалом размягчения
(50—150°). Такие тела относят к анормальным стеклам.
Нормальными смолами являются низкомолекулярные смолы гло-
булярной структуры (например, канифоль, пеки, новолачные смолы
и др.); анормальными — главным образом высокомолекулярные
смолы (полистиролы, полиакрилаты, поливинилацетаты, окисленные
битумы, окисленные масла и др.). *
Величина интервала размягчения определяется также внутрен-
ней структурой аморфного тела. Чем однороднее по составу и строе-
нию элементарные частицы, составляющие аморфное тело, тем
меньше интервал размягчения; чем они более различны и
сложны, — тем он больше. Наличие смеси веществ, составляющих
сложную коллоидную систему, а также незначительное количество
структурообразующих трехмерных молекул значительно увеличи-
вает интервал размягчения.
* Интервал размягчения (TreR —То ) линейных полимеров определяется дли-
ной линейных макромолекул (стр. 103).
Высокоэластическая деформация
89
ВЫСОКОЭЛАСТИЧЕСКАЯ ДЕФОРМАЦИЯ
Общая деформация кристаллических и низкомолекулярных
аморфных тел складывается из деформаций: обратимой, упругой
(гуковской) и необратимой, пластической.
Однако не всякая обратимая деформация подчиняется устано-
вленным выше закономерностям обычной гуковской упругости.
На рис. 27 приведены кривые растяжения стали и каучука. Они
показывают, что упругая деформация стали принципиально отли-
чается от упругой деформа-
ции каучука.
Упругая деформация ста-
ли составляет ~0,1 % и рез-
ко определена пределом те-
кучести при напряжении
~25 кг/мм2-, выше этого
предела имеет место пла-
стическая деформация; мо-
дуль упругости стали
2 • 106 кг/см2.
Упругая деформация
каучука—-1000% (и выше);
соответственно модуль упру-
гости имеет величину в
пределах от 2-10 до
2 • 102 кг/см2, т. е. он мень-
ше модуля упругости стали
в 105—104 раза. Такая «кау-
чуковая» упругость прояв-
1000
900-
800-
700-
600-
§ 500-
| W0-
| зоо-
200-
100-
/Каучук
Сталь
I ।-1----1--гг !-----1—
0 5 10 15 20 25 30 35
Напряжение, нг/ммг
Рис. 27. Кривые растяжения стали и кау-
чука.
ляется в определенных тем-
пературных пределах у всех веществ с большими несимметричными
молекулами, в частности у всех линейных полимеров. Этот особый
вид упругой деформации получил название высокоэластической
в отличие от упругой (гуковской).
Высокоэластическая деформация отличается от упругой не
только величиной, но и особым механизмом; последний так же, как
и механизм пластической деформации, характеризуется отсутствием
изменения междучастичных расстояний и, следовательно, постоян-
ством удельного объема при растяжении, что выражается величиной
коэффициента Пуассона, равного 0,5.
Для упругой деформации коэффициент Пуассона меньше 0,5 и
тем меньше, чем более выражены упругие свойства материала.
Из постоянства удельного объема при деформации вытекает, что
высокоэластическая деформация, подобно пластическому течению,
не вызывает изменения внутренней энергии.
Тепловой эффект при высокоэластической деформации противо-
положен по знаку тепловому эффекту при • упругой деформации.
90
Гл. II. Физические основы технологии пластмасс
Так, каучук при растяжении разогревается, а при сжатии охла-
ждается. Если находящийся в растянутом состоянии каучук нагре-
вать, то напряжение и, соответственно, модуль упругости увеличи-
ваются пропорционально абсолютной температуре.
Наконец, высокоэластическая деформация, в отличие от упругой,
проявляется не в фазе с напряжением, а требует определенного
времени для своего развития, т. е. протекает во времени.
Отличия закономерностей упругой и высокоэластической дефор-
маций приведены в табл. 5.
ТАБЛИЦА 5
Сравнение закономерностей упругой и высокоэластической деформаций
Показатели Упругая деформация Высокоэластическая деформация
Величина удлинения в упру- го-эластической области 0' /0 0,1—1 до 1000 и выше
Тепловой эффект:
при растяжении . . при сжатии .... Коэффициент Пуассона . Удельный объём при растя- жении Модуль упругости, кг/см? Модуль при повышении тем- пературы . Деформация Охлаждение Разогревание < 0,5 Увеличивается 104—2 - 10в Уменьшается Устанавливается в фазе с напряжением Разогревание Охлаждение 0,5 Не изменяется 90—200 Увеличивается Отстает от напряжения
Зависимость деформации от температуры Мало зависит Зависит
Высокоэластическая деформация, в отличие от упругой, про-
является лишь в определенном температурном интервале. По ана-
логии с интервалом пластичности (интервалом размягчения) низко-
молекулярных аморфных тел этот интервал может быть назван тем-
пературным интервалом высокоэластичности. Его нижний предел —
температура стеклования Та, ниже которой высокоэластическан
деформация исчезает и остается обычная упругая деформация.
Верхним пределом является 1\ек —точка перехода в пластиче-
ское состояние. Эта точка менее ясно выражена, чем Те, так как
вязкое течение может развиваться одновременно с высокоэластиче-
ской деформацией.
Теория высокоэластической деформации
Принципиальные отличия закономерностей высокоэластической
деформации от упругой должны быть основаны на совершенно осо-
бом ее механизме. Если причиной обратимой деформации (вслед-
Теория высокоэластическсй деформации
91
ствие постоянства удельного объема при деформации) не мо-
жет служить накопление потенциальной энергии, необходимо прийти
к выводу, что причиной может быть только изменение кинетической
энергии.
Известно, что упругостью обладают не только твердые тела, но
и сжатые газы. Однако присущие твердым телам и газам виды
упругости основаны на совершенно различных механизмах. Упру-
гость твердых тел является следствием противодействия элементар-
ных частиц изменению их взаимного расположения (следствием
изменения внутренней энергии), тогда как упругость газа, деформа-
ция и совершаемая им работа — результат изменения кинетической
энергии движения молекул газа. То, что увеличение напряжения
каучука, растянутого на постоянную величину, пропорционально
абсолютной температуре, указывает на аналогию с увеличением
пропорционально абсолютной температуре давления газа в постоян-
ном объеме.
Соответственно для каучука и газа аналогичны и параметры де-
формации. Так: 1) упругая деформация газа (сжатие) так же, как
и упругая деформация каучука (растяжение), может выражаться
многими сотнями процентов, тогда как кристаллические тела упруго
деформируются лишь до 1%; 2) модуль упругости газа в сотни ты-
сяч и миллионы раз меньше модуля упругости кристаллических тел,
но близок к модулю упругости каучука (модуль газа 0,01 кг/мм2,
каучука 0,02—1,0 кг/мм2, стали— ~ 20 000 кг/.и.и2).
Так как в идеальном газе отсутствуют силы взаимодействия
между молекулами, единственной причиной упругости газа может
быть только тепловое движение молекул. Упругость газа очевидно
определяется числом ударов молекул о стенку сосуда в единицу
времени, что, в свою очередь, зависит от числа молекул в единице
объема и скорости их поступательного движения, т. е. от темпе-
ратуры.
Таким образом, упругость газа обусловлена высокой подвиж-
ностью его молекул и не прекращающимся стремлением их к
расширению занимаемого объема. Это соответствует переходу
в наиболее вероятное состояние — состояние наибольшей энтро-
пии.
В такой же мере, применяя первый и второй закон термодина-
мики, можно показать, что внутренняя энергия изотермически растя-
нутого каучука меняется так же мало, как и внутренняя энергия
идеального газа при изотермическом увеличении или уменьшении
его объема. В обоих случаях, однако, меняется энтропия.
На основе такой аналогии механизма упругости каучука с ме-
ханизмом упругости газа разработана кинетическая теория высоко-
эластичности (Марк, Гут, Кун). Учитывая, что высокая подвиж-
ность молекул газа является причиной его упругих сил, кинетиче-
ская теория объясняет упругость высокоэластичных полимеров,
высокой степенью гибкости их макромолекул.
92
Гл. II. Физические основы технологии пластмасс
Все полимеры, которые могут находиться в высокоэластическом
состоянии, построены из нитевидных линейных макромолекул,
большей частью из длинных цепей с углеродной связью. Введенные
Рис. 28. Схема свободного
вращения атомов углерода
вокруг простой С—С связи.
Штаудингером представления о вытяну-
той, жесткой, палочкообразной форме та-
ких длинных молекул в свете многочислен-
ных экспериментальных данных (напри-
мер, рентгеновского анализа, адсорбции
линейных полимеров на жидких поверх-
ностях и поведения полимеров в раство-
ре и др.) оказались неправильными.
Представления Штаудингера не могут
быть также увязаны с высокоэластиче-
скими свойствами полимеров. Поэтому
высокоэластические свойства полимеров
старались объяснить внутренней подвиж-
ностью макромолекул, их гибкостью, спо-
собностью изгибаться и менять свою кон-
фигурацию.
Кинетическая теория высокоэластичности, исходя из гибкости
макромолекул, допускает совершенно свободное вращение угле-
родных атомов макромолекулы вокруг каждой простой химической
связи С—С (двойная
связь не участвует во вра-
щении) с сохранением ве-
личины валентного угла
между атомами, т. е. сво-
бодную вращаемость зве-
ньев линейных полимеров.
Представление о вра-
щении звеньев макромо-
лекулы можно получить
из рис. 28. Если С! и С2 —
первые два атома угле-
рода, то третий атом С3
лежит на периметре осно-
вания конуса, образован-
ного вращением С3 во-
Рис. 29. Схема цепной молекулы полимера.
/—вероятная форма молекулы; //—полностью выпрям-
ленная цепь.
круг связи Ci—С2, причем угол CiC2C3 есть валентный угол <? и ра-
вен 109,5°. Четвертый атом С4 лежит аналогично на периметре осно-
вания конуса, образующая которого С3С4 движется вокруг первого
конуса и т. д. В результате такого свободного вращения звеньев мо-
жет получиться беспорядочно запутанная форма молекул, в которой
расстояние между концами цепи будет составлять лишь часть пол-
ной, максимальной (рис. 29) длины молекулы, величина которой
L = r>.aKc -= 1 — 1) sin У ,
(16)
Теория высокоэласпшческой деформации
9>
где: п — число звеньев; I — длина звена С—С =1,54 А • ф —. В и
лентный угол = 109,5°.
Эта максимальная длина цепи (гмак„) может встретиться лишь
при одной форме молекулы, когда все связи лежат в одной пло-
скости (транс-цепь). Во всех других случаях длина цепи будет
Меньше Г макс .
Представим себе изолированную молекулу (например в рас-
творе), допустив совершенно свободную вращаемость звеньев цепи
под влиянием термического воздействия (с сохранением величины
валентного угла). Такая молекула будет принимать большое число
различных конфигураций, из которых состояние максимальной рас-
прямленности молекулы (Гмако) и состояние такого клубка, когда
расстояние между концами молекулы будет приближаться к нулю,
статистически мало вероятны.
Такую макромолекулу, состоящую из большого числа звеньев,
можно рассматривать как систему со многими степенями свободы
и с внешней формой, отвечающей наиболее вероятному ее со-
стоянию.
Если расстояние между концами макромолекулы фиксировано,
то каждому такому расстоянию может соответствовать болыпое
число конфигураций. Иначе говоря, достигнуть такого расстояния
между концами цепи можно большим числом способов. Чем рас-
стояние меньше максимально возможного, тем большим числом
способов его можно достигнуть. Естественно, что состояние макси-
мальной длины цепи может быть реализовано только одним спо-
собом.
Распределение конфигураций отдельной изолированной макромолекулы ко
расстоянию между концами цепи г может быть вычислено по теории вероятности
и дается следующей формулой:
ГТ • ,1 n •/, 3 Zl-eosaV1
\V,..,r2dr = 3\/ — '• e2nla (1-H-osJ (1/i
r - n 12/° \1-|-cos a/
где: n — число звеньев; I — длина звена; a =-
В этой формуле №(,.) является искомой
термодинамической вероятностью определен-
ного состояния макромолекулы, которая может
быть представлена кривой распределения кон-
фигураций цепей по расстоянию между их
концами (рис. 30). Из рис. 30 видно, что наи-
большее число конфигураций соответствует
примерно средней степени изогнутости цепей.
Такая конфигурация цепей и отвечает термо-
динамически наиболее вероятному (соответ-
ствующему наибольшему числу случаев) со-
стоянию молекулы.
2~— ср (ср— валентный угол)..
Рис. 30. Статистическое распре-
деление расстояний между кон-
цами цепи линейной макро-
молекулы.
Статистические расчеты для последовательно устанавливаемы:-;
координат расположения каждого звена по отношению к предыду-
Гл. II. Физические основы технологии плчстмасс
щему приводят к средней квадратичной длине свернутой молекулы
p = №2H1COSa
1 — COS a
где: rz — средняя квадратичная длина молекулы; N — число звеньев
в макромолекуле; I — расстояние между двумя углеродными ато-
мами; a = 2?т — ,9, где — валентный угол.
Как видно, г пропорционален квадратному корню из числа
звеньев цепи, что и следовало ожидать на основании постулата
о свободной вращаемости звеньев.
Если свернутую молекулу растянуть приложением внешней
силы, она придет в менее вероятное состояние; поэтому после сня-
тия внешней силы молекула будет стремиться, согласно второму за-
кону термодинамики, перейти к статистически наиболее вероятной
длине, т. е. принять ту статистически среднюю форму изогнутости,
которую она имела до растяжения.
Статистическая физика дает зависимость энтропии системы, на-
ходящейся в состоянии равновесия, от термодинамической вероят-
ности состояния этой системы:
S = £lnlC (19)
где: W — термодинамическая вероятность; k — константа Больц-
мана; S — энтропия системы.
Это выражение позволяет определить энтропию отдельной цепи
и объяснить ее упругость стремлением макромолекулы перейти в со-
стояние с наибольшей энтропией (процесс этот происходит само-
произвольно).
Поэтому, для того чтобы вытянуть цепь длиной г до длины
г 4- dr, необходимо совершить работу, отвечающую изменению
(уменьшению) энтропии
Sr - Sr+dr = k [in HZ,. - In №r+dr). (20)
Кинетическая теория высокоэластичности не учитывает какого-либо
взаимодействия между цепями и считает, что энтропия всей системы
(всей совокупности цепей) равна сумме энтропий отдельных цепей
и что упругие силы при деформации полимера определяются изме-
нением энтропий всего количества молекул в полимере.
Естественно, что допущение отсутствия реально существующего
взаимодействия между цепями может характеризовать поведение
лишь «идеального» каучукоподобного полимера, аналогично тому,
как допущение отсутствия какого-либо взаимодействия между мо-
лекулами характеризует поведение «идеального» газа.
Подобно тому, как в идеальных газах внутренняя энергия не
зависит от объема, при идеальной высокоэластической деформации
внутренняя энергия тела не зависит от степени растяжения.
Из сказанного следует, что имеется параллель между давле-
нием р газа, стремящегося к увеличению объема V для перехода
в состояние высшей энтропии
'>= + (svV (2|)
Теория высокоэластической деформации
95
и напряжением а молекул высокоэластического полимера, стремя-
щихся к сокращению длины цепи, также характеризующимся повы-
шением энтропии:
А т
(22)
Таким образом, кинетическая теория рассматривает силы, стре-
мящиеся при снятии напряжения вернуть полимеры в первоначаль-
ное состояние, как силы, обусловленные уменьшением энтропии.
Иначе говоря, интенсивность теплового движения звеньев молекулы
определяет возвратную силу растянутого высокоэластического по-
лимера.
Количественно поведение растянутых полимеров в высокоэла-
стическом состоянии (как и поведение газов под давлением) опре-
деляется их свободной энергией F, которая в случае простого растя-
жения находится как
(23)
где: L — длина растянутого полимера; U — внутренняя энергия;
S — энтропия; Т — абсолютная температура.
Работа внешней силы [ при удлинении dl выражается диффе-
ренциалом F и, следовательно:
Эго уравнение определяет возможность существования двух ви-
дов упругих сил: упругости вследствие изменения (увеличения)
внутренней энергии и упругости вследствие изменения (уменьше-
ния) энтропии.
В случае идеальной высокоэластической деформации отсут-
\(dU\ „1 Л
ствует изменение потенциальной энергии ( 37г = 0 и работа
внешних сил / = — вся Расх°ДУется ПРИ изотермическом
растяжении полимеров только на образование теплоты, отдаваемой
холодильнику: [dl = 8Q.
Кинетическая теория высокоэластической деформации, как по-
казал Кун, дает возможность вывести зависимость между молеку-
лярным весом полимера и модулем упругости в высокоэластиче-
ском состоянии:
’ <25)
где: р — плотность; 7? — газовая постоянная; М — молекулярный
вес; Еэл — модуль высокоэластичности; Т — абсолютная темпера-
тура.
96
Гл Г. Физические основы технологии пластмасс
Отсюда:
М = —=— (2b)
• с
Таким образом чем больше звеньев в линейной цепи, тем меньше-
будет Еэл-
Кинетическая теория в ее первоначальном выражении качественно пра-
вильно передавала механизм высокоэластичности. Она допускает, однако, про-
извольный постулат о том, что энтропия реального полимера равна сумме энтро-
пий всех цепей и не учитывает межмолекулярных сил взаимодействия.
Дальнейшее развитие кинетической теории идет в направлении расчета вв-
одной изолированной цепи, а целой сетки молекул, цепи которых соединены
редкими поперечными связями, как это имеет место в слегка вулканизованной
резине.
Исходя из поведения тел, имеющих строение такой сетки, Уолл допустил,,
что длина каждого ее отрезка меняется при растяжении подобно изменению
длины самого тела, и после соответствующих статистических расчетов нашел
изменение энтропии элементарной сетки при деформации; последнее позволило
ему определить зависимость напряжения s от деформации:
7? Г II
С = т^-Р I;—п I, (27)
М° \со Г]
где: .W®—мол. вес отрезка цепи между двумя связями (узлами) сетки:
Р — плотность; /о — первоначальная длина; Z — конечная длина.
Эта зависимость довольно близко совпадает с экспериментальными данными
как для растяжения, так и для сжатия высокоэластичных тел.
Зависимость Е от ЛТ® для сетчатых полимеров выражается по Куну урав-
нением
Принципиальные возражения против основного постулата кинетической тео-
рии о полной свободе вращения звеньев вокруг связи С—С были впервые вы-
двинуты советскими учеными С. Е. Бреслером и Я. И. Френкелем. Они также
исходят из наличия в полимерах согнутых, гибких цепей, но отрицают полную
свободу вращения звеньев даже в изолированной единичной цепи вследствие
существования определенного и сравнительно значительного потенциального
барьера.
Рассматривая экспериментальные данные о теплоемкости этана, пропана и
других гомологов, Бреслер и Френкель пришли к заключению, что устойчивой
является граяс-конфигурация вытянутой жесткой цепи, тогда как ^ис-конфигу-
рация соответствует максимуму значения потенциальной энергии (рис. 31). По-
этому для того, чтобы крутильные колебания около равновесной траяс-конфигу-
рации сменились вращением при переходе в ^“с-конфигурацию, необходимо
преодолеть потенциальный барьер, равный избытку энергии чис-конФигУРаиин
по сравнению с траяс-конфигурацией.
Исходя из наличия внутримолекулярного потенциального барьера, Бреслер
и Френкель пришли к выводу, что цепь должна вести себя как система шар-
нирно связанных стержней, способных к крутильным колебаниям, но не способ-
ных к вращательным движениям. Цепи с малым числом звеньев при этом
остаются в основном жесткими и прямыми, тогда как цепи с большим коли-
чеством звеньев могут под влиянием теплового движения переходить в сильно
изогнутые конфигурации путем складывания последовательных малых поворо-
тов отдельных звеньев. Большое число таких поворотов дает в сумме эффект
Теория высокоэластической деформации
97
значительного изгиба, вследствие чего статистическая длина изогнутой моле-
кулы, т. е. среднее квадратичное расстояние между ее концами, будет отли-
чаться от расстояния для предельно вытянутой молекулы, и тем больше, чем
больше число звеньев в цепн. Средняя
величина квадратичного расстояния ме-
жду концами цепи полимера в случае
наличия потенциального барьера будет
равна:
72— у/2 2у0(1 4-CQS а)
kT(1 — COS а) ’
(29)
ба-
где — величина потенциального
рьера (3—3,5 ккал/моль).
На основании этой формулы была
рассчитана зависимость средней квадра-
тичной длины (г‘~) изолированной моле-
кулы от числа звеньев. Полученные дан-
ные приведены в табл. 6. Для сравнения
в этой таблице приведены данные, вы-
численные по Куну на основании теории
свободного вращения звеньев.
Наличие потенциального барьера
приводит к представлениям о жестких,
Рис. 31. Транс-
и чае-конфигурации
алифатической цепи.
траяс-конфигурация; II—цис-конфигура-
ция.
вытянутых молекулах, если число звеньев мало, и о сильно изогнутых молеку-
лах, если число звеньев велико. Однако и при большом числе звеньев степень
ТАБЛИЦА 6
Зависимость
эффективной длины
молекулы от числа
звеньев N
Число звеньев АГ /Р8 1
По Бресле- ру и Френ- келю По Куну
16 14 5,7
20 15,5 6,3
100 38 15,5
1000 ПО 45
более стабильной является
гибкости и изогнутости цепей значительно меньше и,
соответственно, эффективная длина молекул значи-
тельно больше, чем это допускает теория свободного
вращения звеньев.
Существенная поправка, внесенная Бреслером и
Френкелем к основному постулату кинетической тео-
рии, основанная на учете действительно существую-
щего в изолированной молекуле потенциального ба-
рьера, имеет большое принципиальное значение и
является новым плодотворным этапом в развитии
теории высокоэластичности.
М. В. Волькенштейн предложил поворот-
но-изомерную теорию линейных полимеров.
Как было показано исследованиями термодина-
мических свойств простых химических соединений,
внутреннее вращение вокруг С—С-связей всегда в
той нли иной степени заторможено, что объясняет-
ся отталкиванием «привесков», например атомов во-
дорода в линейных углеводородах. По этой причине
вытянутая транс-конфигурация цепи, так как в этом
случае «привески» к углеводородным атомам удалены на наибольшее расстоя-
ние.
Следовательно, для того чтобы молекула совершила поворот вокруг С—С-
связи опа должна преодолеть потенциальный барьер, равный, например, дли
этана 2700 кал/моль. Естественно, что чем ниже температура, тем более затор-
моженным является это вращение, при этом все повороты на 120° приводят
к однозначным результатам и энергетически эквивалентным конфигурациям.
Однако, если молекула лишена осевой симметрии, то внутреннее вращение
приводит к появлению энергетически неэквивалентных, неравнозначных конфи-
гураций, так называемых поворотных изомеров. Так молекулы 1,2-дихлорэтана
Зак. 1269. Э. И. Барг
98
Гл. II. Физические основы технологии пластмасс
могут существовать как в транс-конфнгурацнн, так и в двух изогнутых неравно-
значных конфигурациях, — в виде повторных изомеров. На рис. 32 показаны их
схематические изображения — проекции молекулы на плоскость, перпендику-
лярную к центральной С—С-связи 1,2-дихлорэтана, вокруг которой происходит
внутреннее вращение.
Скорость взаимного превращения поворотных изомеров имеет порядок вели-
чин 10~8 — 10~10 сек., следовательно, такие изомеры не могут быть химически вы-
делены. В этом их основное отличие от всех других видов нзомерни. Их свой-
ства и строение могут быть, однако, изучены методами молекулярной оптики,
С1 С1 С1
1
Рис. 32. Поворотные изомеры 1,2-дихлорэтана.
/ - транс-; II. ///—изогнутые изомеры.
спектроскопии и т. д. Явление поворотной изомерии было открыто методом ко-
лебательных спектров комбинационного рассеяния.
При изоэнергетнческом растяжении цепочки количество транс- и изогнутых
изомеров остается прежним, но они перераспределяются по цепочке так, что ее
длина увеличивается. Так как такое распределение обладает меньшей вероят-
ностью, то цепочка, стремясь вернуться в наиболее вероятное состояние, про-
являет конфигурационную (энтропийную) упругость.
Разности между энергиями поворотных изомеров (а также высоты потен-
циальных барьеров) могут быть подсчитаны, исходя из полуэмпирического вы-
ражения для энергии взаимодействия групп, не связанных между собой валент-
ными связями. Подобными расчетами установлено, что разности между энер-
гиями поворотных изомеров для таких полимеров, как полиизобутнлен, нату-
ральный каучук, метилкаучук, полихлоропрен и т. п., весьма малы. Оказывается,
в частности, что вследствие симметрии связи, соседней с двойной, вращение
вокруг нее можно считать практически свободным.
Представления о поворотных изомерах позволяют не только объяснять вы-
сокоэластические свойства полимеров, но по величине разности в энергиях по-
воротных изомеров получить оценку «гибкости» изолированной цепи.
Таким образом, М. В. Волькенштейн рассматривает линейную
макромолекулу не как случайно закрученную цепочку, а как свое-
образную «смесь поворотных изомеров». Он считает, что в цепочке
макромолекул осуществимы не любые повороты вокруг отдельных
звеньев, а лишь повороты на определенные углы, приводящие к по-
явлению относительно устойчивых поворотных изомеров. Гибкость
цепочки и, следовательно, важнейшие свойства полимеров опреде-
ляются разностями энергий поворотных изомеров. Только от раз-
ности между энергиями поворотных изомеров, а не от величины
потенциальных барьеров зависит, следовательно, и средняя квадра-
Теория высокоэластической деформации
99
тичная длина цепочки. Чем меньше разность между энергиями по-
воротных изомеров, тем меньше средняя квадратичная длина це-
почки, тем выше ее гибкость.
Поворотные изомеры по своей энергии отличаются от наиболее
устойчивого транс-изомера, поэтому их «концентрация» убывает с
уменьшением температуры, что приводит к увеличению длины и
уменьшению гибкости цепочки (так как при низких температурах
в основном существует транс-изомер).
Вопрос о степени гибкости полимерной цепочки в конденсиро-
ванной фазе является одним из основных при оценке высокоэласти-
ческих свойств полимера.
Обычно представление о гибкости цепей получают по величине
температуры стеклования полимера (чем она ниже, тем больше
гибкость цепей). Однако Г., зависит не только от величины внутри-
молекулярных тормозящих потенциалов, но и от сил взаимодей-
ствия между макромолекулами. Поэтому «мягкий» полимер (низ-
кая Т,,) может состоять из жестких цепей, если межмолекулярное
взаимодействие очень мало, и, наоборот, «твердый» полимер (вы-
сокая Тс) может состоять из гибких цепей, если межмолекулярные
силы велики.
Характеристику гибкости цепей полимеров в конденсированной
фазе дает величина высокоэластического обратимого удлинения.
Легко видеть, что максимально возможная величина обратимого
удлинения определяется отношением длины полностью распрямлен-
ной молекулы к ее эффективной длине в равновесном состоянии.
Однако средняя квадратичная длина цепей в равновесном со-
стоянии (при одинаковых степенях полимеризации N) определяется
характером тормозящего потенциала, степенью гибкости цепи. Наи-
меньшее значение эффективной длины молекул в равновесном
состоянии, соответствующее г =—NI (ур. 18) и, следовательно,
наибольшее обратимое удлинение полимера предполагает свобод-
ное вращение цепей. Наличие внутримолекулярных тормозящих по-
тенциалов увеличивает эффективную длину цепи и уменьшает, сле-
довательно, возможную степень обратимого удлинения (и тем в
большей степени, чем выше потенциал).
До последнего времени не были опубликованы соответствующие
исследования о предельной величине высокоэластической деформа-
ции полимеров. Известно, что слегка вулканизованные природные
и синтетические каучуки можно обратимо растянуть на 1000—
2000%. В большинстве случаев реализуемые обратимые удлинения
полимеров имеют величину значительно меньшую, чем теоретиче-
ски возможные, если исходить из свободного вращения звеньев.
I I
Так, для насыщенных полимеров типа —С—С — максимальная
I !
длина цепи составляет 1,25 N (в онгстремах), длина свернутой
цепи в равновесном состоянии 2,50 А/1/5 и, следовательно, соотно-
7*
100
Гл. 11. Физические основы технологии пластмасс
шение между обеими величинами составляет 0,50 Л^2, где W —
число звеньев в цепи (степень полимеризации).
В этом случае, например, полиизобутилен с мол. весом 560 000
(jV = 10 000) теоретически может быть обратимо растянут в 100 раз,
однако практически достигнуты обратимые удлинения полиизобути-
лена лишь в 10—20 раз.
Если можно было бы достигнуть обратимых удлинений, которые
соответствовали бы соотношению 0,5 №/г, то это явилось бы дока-
зательством максимально-возможной гибкости цепи полимера —
доказательством незаторможенного вращения звеньев.
В последнее время Э. И. Баргом с сотрудниками на примере
поливинилацетата было показано, что могут быть достигнуты об-
ратимые удлинения, не только соответствующие теоретическим ве-
личинам, но и значительно их превосходящие.
Это должно указывать на высокую гибкость цепей поливинил-
ацетата, на отсутствие внутримолекулярных тормозящих потенциа-
лов. Более высокие, чем теоретически возможные удлинения, могут
быть объяснены, если учесть взаимное влияние переплетенных моле-
кул. Повидимому, влияние этого фактора должно сказываться на
увеличении длины кинетической единицы, которая будет состоять
как бы из многих молекул, сшитых в длину, что равносильно увели-
чению степени полимеризации N. Это, естественно, должно привести
к меньшей эффективной длине молекул в равновесном состоянии и,
следовательно, к большему обратимому удлинению, чем это до-
пускается теорией, которая построена на представлениях о поведе-
нии изолированных молекул.
Как уже было указано, аналогия между поведением молекул газа и
звеньев полимерной цепочки хотя и оправдывается с термодинамической точки
зрения, однако не может быть принята с точки зрения кинетики процесса, так
как звенья полимера действуют в конденсированной фазе. С этой точки зре-
ния качественная характеристика полимера в высокоэластическом состоянии мо-
жет быть лучше представлена сравнением поведения звеньев его в цепи с по-
ведением молекул в жидкостях.
В жидкости молекулы не распределены так беспорядочно, как в газе.
Вследствие действия межмолекулярных сил они принимают равновесное по-
ложение, которое определяется расстоянием от ближайших соседних молекул.
Под влиянием внешнего напряжения молекулы жидкости слегка удаляются от
их равновесного положения; однако уже ничтожные усилия достаточны для
того, чтобы через практически бесконечно малое время молекулы освободились
от напряжения, соскальзывая друг относительно друга и занимая новые поло-
жения равновесия с первоначальными расстояниями.
В такой же мере отдельные звенья, составляющие макромолекулу поли-
мера, в высокоэластическом состоянии обладают вращаемостью вокруг каждой
простой С—С-связн и прн наложении внешнего напряжения ведут себя по-
добно молекулам жидкости: для освобождения от внешнего напряжения, стре-
мящегося удалить их от состояния равновесия, они поворачиваются, занимая
новое положение равновесия.
В отличие от молекул жидкости, каждое звено макромолекулы в двух точ-
ках связано химической связью с соседними звеньями; поэтому последние не мо-
гут быть удалены друг от друга, не могут, как молекулы жидкости, свободно
скользить относительно звеньев соседних молекул. Валентная связь меж д’.
Теория высокоэ.гастической деформации
101
звеньями не позволяет релаксировать напряжение до нуля (как в жидкости), так
как новое положение звеньев не равноценно первоначальному. При изменении
положения звеньев меняется форма всей молекулы и уменьшается энтропия.
Следовательно, для высокоэластического состояния характерна столь же высо-
кая подвижность звеньев, как подвижность молекул в жидкостях, но в сочета-
нии с наличием сильных валентных связей между звеньями, т. е. должны суще-
ствовать слабые связи наряду с сильными.
Действительно, экспериментально было доказано, что подвижность звеньев
высокоэластических полимеров аналогична подвижности молекул в жидкости.
Большая подвижность звеньев в высокоэластическом состоянии сочетается
с неподвижностью самих макромолекул. Для того, чтобы началось пластическое
течение, т. е. чтобы макромолекулы в целом могли участвовать в перегруппи-
ровке, необходима энергия, достаточная для одновременного или последова-
тельного акта перегруппировки всех звеньев молекулы или значительных участ-
ков цепи (сегментов); последнее же может быть достигнуто лишь при более
высоких температурах.
Пластическое течение может быть практически полностью устра-
нено при образовании редких поперечных химических связей между
цепями макромолекул, т. е. при создании сетчатой, пространствен-
ной структуры. Такие сильные, но редкие химические связи между
цепями затрудняют соскальзывание и перемещение макромолекулы
в целом, но не мешают вращению звеньев, т. е. развитию высоко-
эластической деформации. Подобную сетчатую структуру мы имеем,
например, в слегка вулканизованном каучуке (в резине), где атомы
серы поперечно связывают цепи макромолекул, в сополимерах
стирола с дивинилбензолом (стр. 50), где цепи полистирола свя-
зываются мостиками из дивинилбензола, и в других аналогичных
сополимерах.
Наличие в полимере редких поперечных связей между цепями
хотя существенно и не отражается на самом характере высокоэла-
стической деформации, однако температура стеклования такого
полимера несколько повышается. Если же количество поперечных
химических связей между молекулами велико и соизмеримо с чис-
лом звеньев, то будут затруднены не только пластическая, но и вы-
сокоэластическая деформации. Такая структура пространственно свя-
зывает не только все цепи молекул, но и значительное большинство
звеньев в одну сферическую молекулу макроскопических размеров.
Тепловое движение звеньев и макромолекул можно себе пред-
ставить, если исходить из понятий макро- и микроброунского дви-
жения элементарных частиц. Тепловое движение и перемещение
молекул, происходящее в обычных жидкостях, представляет собою
макроброуновское движение, в то время как тепловые вращения
звеньев без перемещения самой молекулы можно обозначить как
микроброуновское движение.
В высокомолекулярных соединениях в интервале высокоэластич-
ности тепловое движение выражается в интенсивном микроброунов-
ском движении, в то время как макроброуновское движение
полностью отсутствует.
При температуре стеклования прекращается также и микро-
броуновское движение и исчезает высокоэластическая часть дефор-
102
Гл. II. Физические основы технологии пластмасс
мации. При высоких температурах, наряду с микроброуновским
движением, развивается и макроброуновское движение — перемеще-
ние макромолекул в целом, что ведет к пластическому течению
полимера.
Релаксационный характер высокоэластической деформации
Как уже было указано, высокоэластическая деформация огра-
ничена определенным температурным интервалом — интервалом
в ысокоэл астичности.
Аморфные линейные полимеры могут находиться в трех состоя-
ниях: стеклообразном (ниже Та), высокоэластическом (выше Т„),
и пластическом (выше Ттск ). Эти состояния можно определить по
кривой деформации в зависимости от температуры при постоянном
напряжении (рис. 33). До Т„ деформация крайне мала и линейно
растет с температурой (упругая де-
формация) ; в точке стеклования раз-
вивается высокоэластическая дефор-
мация, которая на определенном
участке достигает максимума разви-
тия и уже практически не изменяет-
ся с температурой; в точке Ттвк де-
формация по мере увеличения тем-
пературы снова резко возрастает,
что указывает на преобладание в
основном необратимого пластиче-
ского течения.
Рис. 33. Зависимость деформации
от температуры при постоянное,
нагрузке.
А — стеклообразное, Б—высокоэластиче -
ское, В — пластическое состояния.
1—упругая и высокоэластическая деформа-
ция; 2—остаточная деформация.
Таким образом, общая деформа-
ция полимеров в широком темпера-
турном интервале складывается из
всех трех видов деформаций, од-
нако преобладание каждого вида
определяется температурой и време-
нем воздействия напряжения.
Величина интервала высокоэластической деформации пропор-
циональна степени полимеризации и зависит от структуры полимера.
Зависимость величины интервала высокоэластичностн от сте-
пени полимеризации полимера объясняется зависимостью от
степени полимеризации, в то время как Тс (после достижения опре-
деленной величины) уже не зависит от длины молекулы.
Последнее легко понять, так как тепловое движение звеньев
цепи в малой степени может зависеть от всей длины цепи, в то
время как для передвижения макромолекул требуется тем больше
энергии, чем длиннее цепь полимера.
На рис. 34 показана зависимость температурного интервала вы-
сокоэластичности (Лек —Т<,) от логарифма степени полимериза-
ции N. Как видно из рио. 34, при небольших степенях полимериза-
Релаксационный характер высокоэластической деформации
103
7>Z7J------(
0 100 200 30?
Ттек~Тс, °C
Рис. 34. Зависимость темпера-
турного интервала высокоэла-
стичности (7’т.к— 7’с) от лога-
рифма степени полимериза-
ции N.
ции 20—50) интервал Тгек — То равен нулю, т. е. высокоэла-
стическая деформация практически отсутствует и То характеризует
температуру проявления пластической деформации, перемещения
молекул как таковых. Лишь при более высоких N начинают про-
являться высокоэластические свойства и Т', становится отличной
от Ттек , характеризуя температуру начала вращения звеньев при
неподвижности молекул как таковых.
Наряду с интервалом высокоэластичности (Tres —Та) самым
важным и наиболее характерным показателем свойств линейных
полимеров является температура сте-
клования. Она представляет ту темпе-
ратуру, при которой тепловая энергия
при заданных временных условиях уже
не может преодолеть силы, удержи-
вающие звенья цепей в неподвижном
(колебательном) состоянии. Чем боль-
ше эти силы, тем выше п температура
стеклования.
Возможны два вида сил, которые
необходимо преодолеть для того, что-
бы полимер перешел в высокоэласти-
ческое состояние: межмолекулярные
силы, действующие между звеньями
соседних макромолекул, и внутримоле-
кулярные силы, препятствующие изменению формы цепных моле-
кул, т. е. силы взаимодействия между соседними атомами и груп-
пами атомов в самой макромолекуле.
Исходя из этих соображений, можно допустить также и два раз-
личных механизма стеклования полимеров: один из них сводится
к достижению температуры, при которой невозможно преодолеть
взаимодействие между звеньями соседних цепей, другой — к дости-
жению температуры, при которой невозможно преодолеть внутри-
молекулярный потенциальный барьер самой цепи, препятствующий
изменению конфигурации макромолекул.
Наличие в полимерах внутримолекулярного и межмолекуляр-
ного взаимодействий определяет зависимость высокоэластической
деформации от времени, ее релаксационный характер. Основные
закономерности, вытекающие из релаксационного характера высоко-
эластической деформации, были установлены П. П. Кобеко,
А. П. Александровым, В. А. Каргиным и др.
П. П. Кобеко впервые показал, что предел высокоэластической
деформации при постоянном напряжении не зависит или мало за-
висит от температуры, тогда как скорость установления равновес-
ной деформации находится в экспоненциальной зависимости от тем-
пературы.
На рис. 35 представлена временная зависимость деформации
резины в результате наложения постоянного закручивающего
104
Гл. II. Физические основы технологии пластмасс
момента при различных температурах. Легко видеть, что деформа-
ция, достигнутая при высоких температурах за короткое время, бу-
дет достигнута при низких температурах за более длительный про-
межуток времени.
Таким образом, при низких температурах (ниже Т,.) выеокоэла-
стическая деформация по существу не исчезает, но для проявления
ее требуется большее время наблюдения.
Время, сен.
Рис. 35. Кривые установления вы-
сокоэластической деформации резины
при различных температурах.
Рис. 36. Зависимость высокоэласти-
ческой деформации от температуры
при различном времени действия на-
пряжен ия > t2 > t3.
Зависимость высокоэластической деформации от температуры
(при t = const.) имеет такой же экспоненциальный характер, как
и изменение вязкости про-
стых аморфных тел в интер-
вале размягчения (стр. 86).
На рис. 36 показана за-
висимость высокоэластиче-
ской деформации от темпера-
туры. Кривые имеют три
участка; из них нижний вы-
ражает чисто упругий про-
цесс, когда высокоэластиче-
ская деформация не успела
развиться; верхний — полное
развитие высокоэластиче-
ской деформации и сред-
ний — лишь частичное раз-
витие высокоэластической де-
Рис. 37. Зависимость деформации от тем-
пературы для мягкого вулканизата при
различных частотах напряжения
7—1 пер/мин; 2—10 пер/мин; 3—100 пер/мин;
4—1000 пер/мин.
формации, когда она не до-
шла до равновесного состояния. Из кривых рис. 36, соответствующих
различному времени воздействия напряжения, видно, что чем больше
время, тем больше деформация при одной и той же температуре, тем
более низкой температуре соответствует температура стеклования.
Релаксационный характер высокоэласттеской деформации 105
Отставание по фазе от приложенного напряжения обусловли-
вает большую зависимость эластической деформации от частоты
напряжения. Чем больше частота, тем в меньшей степени при оди-
наковых температурах может проявиться высокоэластическая де-
формация. Из кривых на рис. 37 видно, что при одной и той же
температуре материал, эластичный при низкой частоте, является
жестким при высокой частоте, так как при больших частотах (при
меньших временах действия напряжения) высокоэластнческая де-
формация не успевает развиваться и проявляется лишь упругая
часть деформации.
—|----1--1 1 Г~Т—I-------1 о
-ЬО -20 0 20 Ы 60 80 -М С
Рис. 38. Температурная зависимость логарифма
времени релаксации к для различных полимеров.
У —резина из природного каучука с 3°/0 серы; 2—полихлоро-
прен; 3—эбонит; 4—полиметилметакрилат с 30э/о пластифика-
тора; 5—то же с 10\о; £—чистый полиметилметакрилат.
В общем температурная и частотная зависимость деформации
зысокоэластических тел определяется соотношением между време-
нем релаксации и временем действия силы.
На рис. 38 показана зависимость логарифма времени релакса-
ции от температуры для различных полимеров. Как видно из гра-
фика, она является приблизительно экспоненциальной.
Так как увеличение частоты должно вызвать тот же эффект, что
и снижение температуры, всегда можно установить такую частоту,
когда модуль высокоэластичности Е эл достигает величины модуля
упругости Е}ПР при одной и той же температуре.
Температура стеклования полимеров зависит, наряду с частотным факто-
ром, также и от напряжения.
В определенных пределах, как было показано Э. И. Баргом с сотр., Тс яв-
ляется линейной функцией напряжения (рис. 39).
Эта зависимость может быть объяснена влиянием напряжения на энергию
активации молекулярных перегруппировок и тем, что в первом приближении
уменьшение энергии активации происходит по линейному закону с ростом на-
пряжения.
106
Гл. II. Физические основы технологии пластмасс
В этом случае зависимость времени релаксации т от температуры Т и на-
пряжения з будет иметь вид:
ЕГ—ост
-с = . (30)
где: U— энергия активации, а и
Учитывая, что при всех Тс
— константы для данного полимера.
время релаксации полимера должно оставаться
постоянным, легко прийти к зависимости
Г1= 7’о-^ГЛ, (31)
где: Го — температура стеклования при нуле-
вом напряжении, а Г: — при напряжении
Таким образом, величина -р Т„ предста-
вляет собою тангенсы углов наклонов
Общая деформация полимера в ши-
Напряжение кг[сн?
Рис. 39. Зависимость температу-
ры стеклования от напряжения.
1 — поливинилацетат; 2—полистирол
(пластифицированный); 3—поливинил-
бутираль.
роком температурном интервале вклю-
чает как высокоэластическую дефор-
мацию, так и упругую и пластическую.
Упругая деформация не замещается
высокоэластической, а существует на-
ряду с ней и может быть выявлена при
тех температурах, при которых ско-
рость высокоэластичсской деформации становится малой, т. е. при
температурах ниже Гк
Пластическая деформация проявляется с достаточной скоростью
лишь при температурах значительно выше Та и тем выше, чем
больше степень полимеризации полимера.
Развитие высокоэластической деформации во времени может
быть выражено следующим уравнением:
Лл = Ллоо(1-еХ)> (32)
где: Дэл со — высокоэластическая деформация при t = оо , т. е. в
равновесном состоянии; ДЭл—деформация, которая достигнута
при времени t; X — постоянная времени, характеризующая скорость
установления деформации (время релаксации).
Так как Д= где а — напряжение и Е — модуль, то общее
уравнение деформации:
Лл =
а
£
у пр
а
(33)
в котором последний член выражает пластическую деформацию
(>1 — вязкость).
Релаксационный характер высокоэластической деформации 107
Таким образом, суммарная деформация выражается двумя мо-
дулями: модулем упругости Ет и модулем высокоэластичности
£.Э.Т .
Механико-деформационное и релаксационное поведение полимеров можно
изобразить моделью (рис. 40), где G; представляет упругую (гуковскую) дефор-
мацию, устанавливающуюся мгновенно с иапряже-
)нием; G2 и т)2 характеризуют запаздывающую упру-
гость (стр. 81) высокоэластической деформации,
а Лз — пластическое течение.
Если к полимеру приложено напряжение, то
сначала произойдет мгновенная упругая деформа-
х
ция, равная tv- (где —напряжение сдвига, Gi—-
Рис. 41. Зависимость деформации (-j) от време-
ни для линейного полимера (согласно модели
рис. 40).
— время включения напряжения;
I, — время снятия напряжения.
Рис. 40. Механическая мо-
дель деформации поли-
меров: —мгновенная
упругость, G2rh—заиазды-
вающая упругость и
д.—пластичное течение.
де-
модуль сдвига), после чего начнет медленно развиваться запаздывающая упру-
гость (высокоэластическая деформация), связанная с распрямлением цепей по-
х
лимера. Предел этой деформации определяется 7.— , где G2— модуль высоко-
U2
эластичности. Чем меньше т]9, тем быстрее будет установлена равновесная
х
формация 7-. Одновременно развивается пластическое течение, величина
торога равна —t.
Пз
Если напряжение снято, то мгновенно исчезает упругая деформация
ко-
на
величину тг , после чего медленно будет уменьшаться высокоэластическая
Ml ^2
X
н останется без изменения пластическая часть — t. Наглядно это представлена
на рис. 41.
Гл. И. Физические основы технологии пластмасс
Зависимость температуры стеклования линейных полимеров
от различных факторов. Температура хрупкости
Переход полимеров из стеклообразного состояния в высокоэла-
стическое происходит в некоторой области температур, характери-
зуемой условно температурой стеклования (То). А. П. Александров
определяет Тп как температуру, ниже которой равновесие в ближ-
нем порядке не успевает устана-
вливаться, вследствие чего струк-
тура застеклованного состояния
при понижении температуры
остается неизменной.
Отсутствие фазовых перехо-
дов при затвердевании полимеров
Hiiv. 42. Схематический график
зависимости объема от темпе-
ратуры.
Рпс. 43. Зависимость деформации
от температуры для поливипилбу-
тираля при постоянном напряже-
нии (s = 30 кгсм1).
заставляет искать в застеклованном состоянии закономерности,
аналогичные с наблюдаемыми выше Т„.
Эти положения, естественно, определяют зависимость Та от
условий и метода испытаний (скорость подъема температуры, ско-
рость приложения напряжения), от величины напряжения и т. д.
Температура стеклования может быть определена:
а) по точке перегиба на кривой зависимости, некоторых термо-
динамических свойств полимеров (например, объема, теплоемкости
и др.) от температуры (рис. 42); б) по максимуму диэлектрических
потерь — при этом, в соответствии с релаксационным характером
данной зависимости, максимум диэлектрических потерь с увеличе-
нием частоты будет закономерно сдвигаться к более высоким тем-
пературам; в) по кривым зависимости деформации от температуры
при постоянном напряжении (рис. 43).
Наряду с температурой стеклования, свойства и структуру по-
лимера характеризует еще температура хрупкости (Тхр). Так на-
Зависимость То от различных факторов
109
Рис. 44. Зависимость то и.ритурт:
хрупкости полихлорвинилидена
(пластифицированного) от толщи-
ны образца.
зывают температуру, при которой образец полимера разрушается
при практически мгновенной деформации его на заданную вели-
чину.
Температура хрупкости зависит от скорости и величины задан-
ной деформации: чем выше скорость и больше величина заданной
деформации, тем выше температура хрупкости. Она зависит также
от формы, в частности, от толщины образца и от степени ориента-
ции (вытяжки) полимера. Чем тоньше образец и чем выше степень
ориентации, тем ниже температура
хрупкости (рис. 44).
Физический смысл температуры
хрупкости заключается в том, что
определяется температура, при ко-
торой уже не успевают произойти
перегруппировки, способные рела-
ксировать напряжения при заданных
скоростях воздействия и величине
деформации.
Основным различием между Т<.
и 7хр является их различная зави-
симость от степени полимериза-
ции N.
Температура стеклования уже
при N ~ 100 достигает практически
постоянного значения; температура
хрупкости, наоборот, резко увеличи-
вается в области малых степеней по-
лимеризации, но достигает постоян-
ной величины при больших молеку-
лярных весах.
Как видно из рис. 45, оба показателя резко различаются при
малых молекулярных весах, причем снижается, а Т х), резко воз-
растает по мере снижения молекулярного веса; однако у поли-
меров с высокими молекулярными весами оба показателя сбли-
жаются.
Именно благодаря различной зависимости Та и 7\г от величины
молекулярного веса полимеров, параллельное определение этих по-
казателей имеет большое техническое значение и косвенным обра-
зом характеризует степень полимеризации. Чем больше расхожде-
ние между Го и ТУ1, тем ниже молекулярный вес полимера. Зави-
симость состояния и свойств полимеров от степени полимеризации
может быть наглядно представлена диаграммой, приведенной на
рис. 46.
Как видно из рис. 46, ниже Т,, полимер на протяжении всех
значений молекулярного веса (ось абсцисс) находится в твердом,
хрупком (застеклованном) состоянии, причем по мере увеличения
молекулярного веса температура стеклования сначала монотонно
ио
Гл. II. Физические основы технологии пластмасс
возрастает и достигает постоянной величины. Выше Т>, полимер
может находиться в различном состоянии в зависимости от вели-
Рпс. 45. Зависимость величины Тс (кривая 1)
его мо-
чины молекулярного веса;
с увеличением молекуляр-
ного веса он переходит от
жидкого в упруго-вязкое
и, наконец, в высокоэла-
стическое состояние.
Между То и 7хР поли-
мер с малым молекуляр-
ным весом представляет
собою или вязко-пластич-
ное тело (если время дей-
ствия напряжения больше
времени релаксации) или
л 7’1р (кривая 2) полиизобутилена от
лекулярного веса.
Молекулярный вес (шкала
логарифмическая)
Рис. 46. Диаграмма физического со-
стояния полимера в зависимости от
молекулярного веса.
.4 — упруго-вязкое состояние; Б — жидкость,
-хрупкая при быстрых деформациях; В—твер-
дый пластик.4
1 — вязкое состояние; 2 — упруго-вязкое;
3— высокоэластнческое.
хрупкое твердое тело
(если время действия на-
пряжения меньше време-
ни релаксации); соответ-
ственно в этом же интервале по-
лимер с большим молекулярным
весом представляет или высоко-
эластичное или твердое тело.
Таким образом, в интервале
между Тс и Ту,, особое влияние
и особое значение имеет фактор
времени, однако процессы дефор-
мации и выше Лр и ниже Т„ так-
же существенно определяются ре-
лаксационным механизмом, хотя
и не в такой степени, как между
7), и Тхр.
МЕЖМОЛЕКУЛЯРНЫЕ СИЛЫ И
ФАЗОВАЯ СТРУКТУРА
ПОЛИМЕРОВ
Высокомолекулярные веще-
ства чаще всего состоят из хаоти-
чески скомкованных, в беспорядке
спутанных и переплетенных ли-
нейных молекул и показывают ха-
рактерную жидкостную рентгенограмму аморфных тел (рис. 47).
Такими полимерами, например, являются поливинилацетат, поли-
стирол, некоторые синтетические каучуки и др. Однако некоторые
полимеры (полиамиды, полиэтилен и др.), несмотря на внешний
аморфный характер, имеют большую степень внутренней упорядо-
Межмолекулярные силы и фазовая структура полимеров
111
Рис.
47. Рентгеногргмма
аморфного полимера.
свойств, характерных
рентгеновых лучей и
характерную для кри-
бристаллическая
область
А мор ср на я
область
Рис. 48. Схематическое изобра-
жение расположения цепей
макромолекул в кристалличе-
ских и аморфных областях.
ченности и действительно обладают рядом
для кристаллов: они вызывают дифракцию
быстрых электронов и дают рентгенограмму,
сталлических тел. При растяжении они
показывают рентгенограмму ориентиро-
ванных кристаллитов (рентгенограмму во-
локна), а также двойное лучепреломление
и ясно выраженную анизотропию физиче-
ских и механических свойств.
Однако отличие кристаллических по-
лимеров от обычных кристаллических тел
состоит в том, что они не имеют хорошо
образованных плоскостей раздела или рез-
ких симметричных граней, характерных
для индивидуальных кристаллов, и не
обладают точными фазовыми переходами.
Они состоят из субмикроскопических кри-
сталлитов.
Кристаллиты полимеров имеют про-
стую форму параллельных пучков, палочкообразную форму или
форму плоских сеток (ленточную). Форма и размеры кристаллитов
никогда не бывают строго определен-
ными, даже в одном п том же поли-
мере. Размеры кристаллитов могут ко-
лебаться в широких пределах — от 20
до 10 000 А.
Характерной особенностью кристал-
лических полимеров является постоян-
ное наличие в них аморфной фазы на-
ряду с кристаллической; при этом обе
фазы не отделены друг от друга и вза-
имное соотношение их может меняться
в широких пределах под влиянием ме-
ханических или термических воздей-
ствий или с течением времени. Так как
размеры кристаллитов значительно
меньше обычной длины цепочных моле-
кул, следует предположить, что в кри-
сталлических полимерах одна и та же
цепь макромолекул может переходить
через различные области упорядочен-
ности, т. е. через кристаллическую и
аморфную фазы (рис. 48).
Существование в кристаллических
полимерах двух фаз, неразрывно свя-
занных между собой прохождением одних и тех же макромолекул,
делает невозможным рассмотрение их независимо друг от друга.
112
Гл. 11. Физические основы технологии пластмасс
Нужно считать, что в этом случае имеется равновесное состояние
между фазами и что любые тепловые или механические изменения
в одной области должны вызывать соответствующие изменения
в другой.
Установить наличие кристаллической фазы в полимерах и в осо-
бенности дать количественные показатели кристалличности пред-
ставляется во многих случаях весьма затруднительным.
В сущности прямым доказательством кристалличности полиме-
ров является тепловой эффект кристаллизации и уменьшение
объема при постоянной температуре (температурекристаллизации),
отражающие изменение внутренней энергии и междучастичных
расстояний при фазовых переходах.
Рентгено- и электронноструктурный анализ также могут дать
прямое доказательство существования в полимерах кристалличе-
ской фазы. Дифракция рентгеновых волн и быстрых электронов
дает обычные характерные интерференционные кольца, однако они
сильно размыты широкими диффузными кольцами, которые тем
шире, чем больше аморфной фазы в полимере. При малом содер-
жании кристаллической фазы в полимере рентгенограмма часто не
позволяет заключить о наличии кристалличности.
В. А. Каргин на примере целлюлозы и ее производных показал,
что наличие характерной рентгенограммы само по себе еще не мо-
жет служить доказательством межмолекулярной кристалличности,
так как рентгеноструктура может отражать и внутримолекулярную
ориентировку цепей полимера.
Однако при сравнении полученных рентгенограмм с соответ-
ствующими электронограммамп и изучении рентгеноструктуры г
широком интервале температур можно получить правильную ин-
терпретацию рентгенограммы и составить соответствующее сужде-
ние о фазовой структуре полимера.
Основные особенности в закономерностях кристаллического
состояния полимеров
Кристаллическим полимерам вследствие наличия в них двух
фаз — аморфной и кристаллической, находящихся в состоянии
равновесия, свойственны две важнейшие константы, определяющие
весь комплекс их физических и механических свойств: температура
стеклования аморфной фазы и температура плавления кристалли-
ческой фазы. На рис. 49 показана зависимость относительного
объема природного каучука от температуры. Верхняя кривая 1 ха-
рактеризует аморфное состояние полимера, нижняя 2 — кристалли-
ческое. На кривых видно, что перегиб при — 70° соответствует
точке стеклования, причем она одинакова для обеих кривых. Таким
образом, температура стеклования не зависит от наличия кристал-
лической фазы, однако изгиб кривой, характеризующий темпера-
туру стеклования, меньше в кристаллическом полимере, чем в
аморфном. Другой перегиб при 4-20° (на кривой 2) характеризует
Закономерности кристаллического состояния полимеров
113
температуру (интервал) плавления кристаллической фазы. Нали-
чие интервала плавления, а не фиксированной точки фазового пе-
рехода, является характерным отличием кристаллических полиме-
ров.
Температура плавления кристаллических полимеров значительно
ниже, чем этого можно было бы ожидать, исходя из обычных за-
висимостей температуры пла-
вления от молекулярного веса
в полимергомологическом ряду.
Так, точка плавления поли-
этилена, содержащего 1000
СН2-групп, лежит на 20° ниже,
чем этого следовало ожидать
по известной зависимости тем-
пературы плавления парафинов
от их молекулярного веса. Те-
плоты плавления кристалличе-
ской фазы полимеров имеют
также аномально низкие зна-
чения.
Важнейшим отличием кри-
сталлических полимеров от
Рис. 49. Относительные объемы аморф-
ного и частично кристаллического кау-
чука как функция температуры.
1—аморфный каучук; 2—кристаллический каучук.
обычных кристаллических тел является установленная опытом зави-
симость температуры плавления от температуры кристаллизации.
Именно в этой, совершенно несвойственной обычным кристаллам
закономерности, можно ви-
деть все своеобразие кри-
сталличности полимеров.
Полимеры могут кристал-
лизоваться (рис. 50) в ши-
роком температурном интер-
вале (внутри которого обыч-
но лежит температура макси-
мальной скорости полимери-
зации) и чем выше темпера-
тура кристаллизации, тем
Рис. 50. Скорость кристаллизации каучука зыше и темПература плавле-
как функция температуры. г , Г1,
r ния кристаллитов (рис. 51).
Аморфный полимер можно последовательно кристаллизовать
при различных температурах; при этом получится ряд точек плав-
ления с постепенным переходом полимера из одной степени кри-
сталличности в другую и, наконец, в аморфное состояние.
Концентрация кристаллитов в полимере также определяется
температурой. Для каждой температуры устанавливается равновес-
ное состояние между кристаллической и аморфной фазами. С повы-
шением температуры количество кристаллической фазы значи-
тельно снижается и равновесие соответственно передвигается в
8 Зак. 1269. Э. И. Барг
114
Гл. И. Физические основы технологии пластмасс
Температура кристаллизации, °C
Рис. 51. Область температур плавления при-
родного каучука как функция температуры
кристаллизации.
сторону аморфной фазы. Следует также отметить влияние времени
кристаллизации; в ряде случаев с увеличением времени кристалли-
зации увеличивается температура плавления кристаллитов.
Характер и концентрация кристаллов в сильной степени опре-
деляются деформацией (ориентацией) полимера.
При деформации кристаллического полимера беспорядочно рас-
пределенные кристаллиты исчезают и вместо них образуются но-
вые кристаллиты, ориентированные в направлении действия сило-
вого поля. Этот процесс можно наблюдать на рентгенограмме. При
растяжении полимера интерференционные кольца постепенно осла-
бевают, при этом появля-
ются, а затем усиливаются
точечные интерференцион-
ные пятна. Образование
новых кристаллитов про-
исходит чрезвычайно бы-
стро, например, в каучуке
почти 90% образуются в
течение одной секунды;
в дальнейшем же количе-
ство кристаллитов возра-
стает медленно.
Количество кристалли-
ческой фазы пропорцио-
нально степени растяже-
ния (ориентации). Весьма
важно, что с увеличением
степени растяжения по-
вышается температура плавления кристаллитов. Многие полимеры,
находящиеся при обычных температурах в аморфном, высоко-
эластическом состоянии, легко кристаллизуются при растяжении
(например, кристаллизующиеся каучуки). При сокращении поли-
мера со снятием напряжения кристаллиты, однако, разрушаются
и полимер снова становится аморфным. Полимеры, имеющие при
обычных температурах устойчивую кристаллическую структуру и
высокую температуру стеклования, дают при растяжении новую,
практически устойчивую систему ориентированных кристаллитов
(полиамиды и другие синтетические волокна).
Процесс образования и роста кристаллитов, согласно А. П. Але-
ксандрову, протекает путем перегруппировки звеньев цепей; по
мере упорядочения звеньев растет поверхность кристаллитов. Од-
нако рост кристаллита не может не вызвать соответствующих на-
тяжений и деформаций в аморфной части полимера, так как ма-
кромолекулы, как уже было указано, связаны в единое простран-
ственное образование. Возникновение таких натяжений препят-
ствует росту кристаллитов, способствует их разрушению и, следо-
вательно, снижению температуры плавления. Этот процесс в итоге
Закономерности кристаллического состояния полимеров
115
приводит к установлению равновесного состояния между фазами.
Если с повышением температуры часть кристаллической фазы рас-
плавится, произойдет уменьшение натяжений в аморфной фазе, а
это немедленно приведет к образованию и росту новых кристалли-
тов с более высокой температурой плавления.
На основе рассмотренного механизма удается объяснить все
аномалии высокополимерной кристалличности, в том числе суще-
ствование интервала температуры плавления, зависимость темпе-
ратуры плавления от температуры кристаллизации и др.
Кристаллизация во всех случаях приводит к повышению жест-
кости, к снижению высокоэластических свойств полимера.
Вместе с тем комплекс характерных свойств кристаллических
полимеров определяется как концентрацией кристаллической фазы,
так и тем состоянием, в котором находится в них аморфная фаза.
Аморфная фаза кристаллических полимеров может находиться либо
в застеклованном состоянии (ниже Т„), либо в высокоэластическом
(выше Тп). В первом случае кристаллический полимер будет твер-
дым и хрупким, во втором — гибким и упругим.
Подводя итог вышесказанному, все полимеры с точки зрения
состояния внутренней упорядоченности можно разделить на сле-
дующие три группы:
1. Полимеры аморфной структуры. Эти полимеры дают рент-
генограмму, характерную для жидкостного состояния, не изме-
няющуюся в широком температурном интервале (выше Та) и
при растяжении полимера. Последнее вызывает лишь ориентацию
макромолекул, что, естественно, приводит к значительному увели-
чению прочности полимера и к анизотропии его свойств.
Такая ориентация остается практически устойчивой ниже тем-
пературы стеклования и исчезает с повышением температуры; по-
лимер при этом сокращается.
К аморфным полимерам относятся: полистрол, поливинилаце-
тат, полиметилметакрилат, различные сополимеры и др.
2. Полимеры с малой концентрацией и низкой температурой
плавления кристаллической фазы и низкой температурой стекло-
вания (—40° и ниже). Эти полимеры находятся, как правило,
при обычных температурах в аморфном, высокоэластическом со-
стоянии.
Однако в определенном температурном интервале выше Т а
они способны кристаллизоваться.
Подвергаясь растяжению, они быстро кристаллизуются, — кон-
центрация кристаллической фазы может достигать 75% и больше
(в зависимости от степени растяжения). При снятии напряжения
и релаксации кристаллиты быстро плавятся и исчезают. Кристал-
лическая фаза плавится при температурах, близких к обычным тем-
пературам (от + 5 до 4-40°).
К этой группе полимеров относится природный каучук, полиизо-
бутилен, полихлоропрен и др.
8*
116
Гл. II. Физические основы технологии пластмасс
3. Полимеры с большой концентрацией и высокой температурой
плавления кристаллической фазы. В этих полимерах значительно
преобладает кристаллическая фаза. Их скорость кристаллизации
очень велика, а аморфное состояние крайне неустойчиво. Темпера-
тура плавления кристаллической фазы обычно выше 100°. Растя-
жение возможно при температуре ниже температуры плавления
кристаллической фазы, но выше температуры стеклования аморф-
ной фазы. Растяжение вызывает появление ориентированных кри-
сталлитов, которые в зависимости от структуры цепи, от симмет-
ричности расположения диполей вдоль цепи могут иметь различ-
ную степень устойчивости.
К полимерам с высокой степенью упорядоченности относятся:
полиэтилен, политетрафторэтилен, полиамиды, линейные поли-
эфиры, полиуретаны, полимочевины и др.
Способность полимеров к кристаллизации
Степень упорядоченности и способность полимера переходить в
кристаллическое состояние зависят от двух основных факторов: от
величины межмолекулярных сил притяжения и от конфигурации
цепей, т. е. от степени их симметричности и гибкости.
Межмолекулярные силы определяются, в основном, характе-
ром и расположением замещенных групп вдоль цепи макромоле-
кул. Приближенно они могут быть разделены на неполярные и
легко поляризующиеся, или полярные группы. К межмолекулярным
силам относят водородную связь, наличие которой было доказано
для некоторых полимеров (например, для поливинилового спирта,
для полиамидов и др.).
Наряду с межмолекулярным взаимодействием, существенное
влияние на способность полимера к кристаллизации оказывает
взаимодействие групп внутри цепи макромолекулы, которое и опре-
деляет ее гибкость — величину ее тормозящего потенциала. По
сравнению с энергией валентных связей углеродных атомов в са-
мой цепи (60—120 ккал/моль) величина энергии межмолекуляр-
ного притяжения (1—6 ккал! моль) невелика, однако суммарная ве-
личина этой энергии, благодаря огромному количеству звеньев,
образующих цепь, значительно превосходит энергию валентных
связей, действующих в самой цепи. Поэтому температуру стекло-
вания и другие основные физико-механические свойства полимеров,
в том числе во многих случаях и их способность к кристаллиза-
ции, в первую очередь определяет именно величина межмолекуляр-
ных сил.
В табл. 7 приведены энергии молекулярного притяжения для
различных групп.
Для характеристики межмолекулярного взаимодействия раз-
личных полимеров могут служить величины энергии притяжения,
рассчитанные на единицу длины цепи полимера. В табл. 8 приве-
Способность полимеров к кристаллизации
117
дены примерные величины удельных притяжений различных поли-
меров. рассчитанные на длину цепи в 5 А.
ТАБЛИЦА 7
Величины энергии молекулярного притяжения для различных групп
Группы Энергия притяжения кал!моль Группы Энергия притяжения кал!моль
—сн3 1 1780 —Вг 4300
>сн2 f —J 5040
—СИ,— 1 >сн— J —no2 7200
990 —SH 4250
—conh2 13200
—он 7250 —CONH— 16200
—О— 1630
>со 4270 К
—сно —ссон 4700 8970 о- 1 -о 1 п:
—СООСНз 5600
—соос,н5 6230 К
—NH,, 3530 (водородная связь) 4500 — 6000
—F 2060
—С1 3400
ТАБЛИЦА 8
Молекулярные притяжения различных полимеров, рассчитанные
на длину цепи в 5 А
Полимер Группы, обусловливающие силы притяжения Молекулярное притя- жение на цепь длиной в 5 А с координацион- ным числом 4 (каждая цепь окружена 4 дру- гими), кая!моль ГС
Полиэтилен >сн2 1000 — 80
Полибутадиеи —СН=СН—;>СН2 1100 — 60
Полиизобутилен —СН3; >СН2 1200 — 70
Каучук (полиизопропен) —СН=С(СН3)—; >СН2 1300 — 73
Полихлорвинил > СН2; > СНС1 2600 4-70
Поливииилацетат >СН2; —ОСОСН3 3200 4-30
Полистирол —CgHj; > СН2 4000 4-80
Поливиниловый спирт . . > СН2; > СНОН 4200 4-85
Полиамиды > СН,; —CONH—; Н 5800
Целлюлоза —ОН; —О— 6200
Фиброин шелка >CHR; —CONH—; Н 9800
Как видно из табл. 8, полимеры с малым удельным молекуляр-
ным притяжением (порядка 1000—2000 кал/моль) имеют крайне
низкую температуру стеклования и находятся, следовательно, при
118
Гл. II. Физические основы технологии пластмасс
'обычных и низких температурах в высокоэластическом состоянии.
За исключением полиэтилена эта группа полимеров по характеру
своих механико-деформационных свойств относится к каучукам.
Полиэтилен, если судить по Т <. его аморфной фазы, был бы
«идеальным» каучуком, однако он имеет устойчивую кристалличе-
скую структуру, что в значительной степени снижает высокоэласти-
ческие свойства и придает ему свойства эластичного пластика, а не
каучука.
Полимеры, в макромолекулах которых имеются полярные боко-
вые группы или бензольные ядра, обусловливающие значительно
большее удельное молекулярное притяжение (порядка 2000—
4000 кал/моль), не обладают при обычной температуре высокоэла-
стическими свойствами; последние проявляются, однако, при более
высоких температурах (70—150°). Большее взаимодействие между
цепями в этих полимерах и придает им тот комплекс физико-меха-
нических свойств, на основании которых такого рода полимеры
относят к группе пластиков.
Наконец, полимеры, обладающие весьма высоким удельным мо-
лекулярным притяжением — свыше 5000 кал)моль, — чаще всего
обладают кристаллической структурой с высокой температурой
плавления и дают при растяжении устойчивую ориентационную
кристалличность. К такого рода полимерам относятся природные и
синтетические волокна (целлюлоза, фиброин шелка, полиамиды,
полиуретаны, полимочевина и др.).
С термодинамической точки зрения способность к кристаллизации, выражен-
ная величиной температуры плавления кристаллов, зависит от изменения во
время плавления как потенциальной энергии ДГ/, так и энтропии ДУ
ill
^=5 (34)
"’ж °кр ’
где: U-At и t/кр — потенциальная энергия на 1 моль вещества соответственно
в расплавленном и кристаллическом состояниях; Уж н Укр— энтропия на 1 моль
вещества в расплавленном и кристаллическом состояниях.
При одинаковом изменении потенциальной энергии д(7 температура пла-
вления кристаллов зависит от разности в энтропии дУ, причем, чем эта раз-
ность меньше, тем выше температура плавления кристаллов, тем сильнее моле-
кулярная связь в кристаллической решетке. Разность в энтропии расплавленного
и кристаллического состояний будет тем меньше, чем симметричнее построена
макромолекула; в этом случае, чтобы «впрыгнуть» в кристаллическую решетку,
макромолекула не должна значительно менять форму, по сравнению с той,
которую она имела в расплавленном (аморфном) состоянии. Очевидно, что на-
личие сильно разветвленных боковых групп и их несимметричное расположение
вдоль цепи должны мешать макромолекулам принять то правильное располо-
жение, которое характерно для кристаллической решетки. Естественно, что чем
менее симметрична структура макромолекулы, тем больше должны быть меж-
молекулярные силы, чтобы такие молекулы могли «впрыгнуть» в кристалли
ческую решетку.
Зависимость кристаллизации от обоих факторов — изменения энергии и
энтропии во время плавления — объясняет кристалличность полиэтилена и аморф-
ность многих полимеров с более высокой энергией молекулярного притя-
жения.
Теоретическая и техническая прочность пластмасс
119
Способность к кристаллизации в значительной мере определяет
все существо различий между тремя основными типами полиме-
ров — каучуками, пластиками и волокнами.
Для получения «каучуков» — полимеров, способных к высоко-
“I эластической деформации при возможно низких температурах, необ-
ходимы условия, препятствующие закреплению цепей макромолекул
как силами, действующими в кристаллической решетке, так и меж-
молекулярными силами связи. Иначе говоря, макромолекулам кау-
чуков должны быть свойственны малые межмолекулярные силы и
несимметричная линейная структура.
С другой стороны, для получения волокон, т. е. полимеров с по-
стоянной ориентационно-кристаллической структурой, необходима
высокая удельная энергия молекулярного притяжения (свыше
5000 кал/моль), а макромолекулы должны быть симметричными, с
цепями, не содержащими боковых групп.
В этом отношении пластики занимают среднее положение. По-
лимеры, которые относят к классу пластиков, при обычной темпе-
ратуре не обладают достаточно выявленной высокоэластичностью
(в отличие от каучуков), но и не имеют постоянной ориентационно-
кристаллической структуры, свойственной волокнам.
Таким образом, три названные типа высокополимеров не имеют
принципиальных признаков различия. Их отличают по различной
степени гибкости макромолекул, величине межмолекулярных сил
притяжения и по их способности к закреплению в кристаллической
решетке. Большие колебания в величине этих показателей обусло-
вливает и наличие большого количества материалов, занимаю-
щих промежуточное положение между основными типами поли-
меров.
ТЕОРЕТИЧЕСКАЯ И ТЕХНИЧЕСКАЯ ПРОЧНОСТЬ' ПЛАСТМАСС
Механическая прочность твердых тел определяется силами вза-
имодействия между частицами: причем в телах с гетерополярными
кристаллическими решетками — силами взаимодействия между
ионами, в веществах с ковалентными связями (например, в алмазе
или в пространственных полимерах) — силами взаимодействия
между атомами и. наконец, в таких телах, как молекулярные кри-
сталлы, линейные полимеры или низкомолекулярные смолы — си-
лами взаимодействия между молекулами.
Для расчета максимального (теоретического) значения меха-
нической прочности твердых тел исходят из зависимости потен-
циальной энергии Е системы от расстояния г между взаимодей-
ствующими центрами (или от сил F, получаемых дифференцирова-
нием Е по г), а также из равенства сил притяжения и отталкива-
ния в состоянии равновесия (рис. 11, стр. 71).
Максимальная прочность At.™ реализуется при удалении вза-
имодействующих частиц на расстояние ГмаВ0, выше которого
120 Гл. И. Физические основы технологии пластмасс
нарушается равновесие в твердом теле и F начинает падать при
увеличении г, что приводит к разрыву.
Поэтому соответствующее значение F при г«ЛК„определяет макси-
мальное (теоретическое) значение прочности и FMaK0 = FT.
Расчет позволяет лишь приближенно оценить значение Ft, так
как приходится принимать некоторые допущения, сильно сказываю-
щиеся на получаемых результатах.
Однако энергия взаимодействия так сильно зависит от харак-
тера химических сил (валентных, ван-дер-ваальсовских), что даже
при грубом исчислении можно получить приближенное значение
Ft, характеризующее тот или иной механизм взаимодействия.
Для расчета Ft можно исходить из уравнения Морзе для зави-
симости потенциальной энергии Е взаимодействующих частиц от
междучастичного расстояния г
Е = De~2b — 2De~b (35)
где: D — энергия диссоциации связи; г0 — расстояние между взаимо-
действующими частицами в состоянии равновесия; b — постоянная.
Значение b можно вычислить из эмпирических данных, получен-
ных Фоксом и Мартином для связей С—С, С=С и С=С:
Для перехода от энергии к. результирующей силе производят
дифференцирование Е по г
F = d-^ = — 2bDe~2b 4- 2bDe~b (37)
Если в это уравнение вставить для междучастичного расстоя-
ния значение гМако, которое может быть получено при = 0, то
после соответствующих преобразований получаем простое значение
ДЛЯ Fмако -
^ма«о = = ~2 (38)
или для С—С связей:
Таким образом, для расчета теоретической прочности нужно
знать энергию диссоциации D и константу Ь, которая приближенно
для С—С связей может быть определена из значений г0.
Для более грубого представления о величине теоретической
прочности можно исходить из величины модуля упругости при воз-
можно низких температурах.
Теоретические расчеты приводят к соотношению Ft=0,1Ep,
где Ер — экспериментальное значение модуля упругости.
Теоретическая и техническая прочность пластмасс
121
Для многих однородных веществ с изотропной структурой тео-
ретические и экспериментальные значения модуля упругости прак-
тически совпадают, однако значения теоретической прочности в де-
сятки и сотни раз (иногда в тысячи раз) превосходят значения тех-
нической (экспериментальной) прочности.
Вместе с тем для некоторых полимеров, в частности для пространственных
полимеров типа феноло-альдегидных резитов, аминопластов и др., теоретиче-
ские значения модуля упругости значительно (в десятки раз) выше экспери-
ментальных, если считать, что модуль обусловлен деформацией валентных
С—С связей в пространственной решетке. Это различие в расчетных и экспе-
риментальных модулях П. П. Кобеко относит за счет иного рода сил, обусло-
вливающих механические свойства резитов, а именно, он считает, что в
пространственных полимерах эти силы являются не валентными, а диспер-
сионными и, следовательно, феноло-альдегидные резиты следует рассматри-
вать не как единую макромолекулу, а как конгломерат пространственных обра-
зований, скрепленных друг с другом дисперсионными связями. Однако вряд ли
можно считать эти выводы достаточно обоснованными.
Соотношение Ft= 0,1 Ер действительно для температур, близких к абсолют-
ному нулю, в то время как значения Ер для фенопластов, аминопластов и др.
измерены при обычных температурах; необходимо также учитывать, что техни-
ческие продукты никогда не свободны от низкомолекулярных примесей (на-
пример, «свободный фенол»). Кроме того, мы никогда не имеем идеальной про-
странственной сетки, а имеем сетку с весьма значительными дефектами (стр. 361).
Все эти обстоятельства, а не характер химических сил, повидимому, объяс-
няют наблюдаемый разрыв между теоретическим и экспериментальным значениями
модуля упругости для фенопластов.
В табл. 9' приведены теоретические и экспериментальные значе-
ния модулей упругости и механической прочности некоторых ве-
ществ.
Теоретическая прочность определяется характером сил, дей-
ствующих между частицами вещества, которые могут быть валент-
ными или межмолекулярными. В последнем случае естественно, что
как теоретическая, так и техническая прочность оказываются зна-
чительно ниже. Так, например, теоретическая прочность феноло-
формальдегддной смолы в стадии С составляет 4300 кг/мм?, что
соответствует силам разрыва валентных углеродных связей сфе-
рической макромолекулы, а в стадии А — 39 кг/мм2, что соответ-
ствует преодолению лишь межмолекулярных сил связи; в таком
же примерно соотношении находится и техническая прочность
этой смолы в обеих стадиях (4—8 кг!мм2 и 1 — 5 -10—2 кг/мм2).
Значительный разрыв между расчетными и экспериментальными
данными наблюдается лишь в отношении прочности материалов;
что же касается таких показателей, как энергия диссоциации, пла-
вления или испарения твердых тел, то для них расчетные и экспери-
ментальные данные хорошо совпадают.
Чтобы объяснить это явление, необходимо рассмотреть зави-
симость прочности тела от габаритов и характера поверхности, а также
указать на разброс в показателях прочности, получаемый при
ТАБЛИЦА 9
Теоретические и экспериментальные значения для модулей упругости и прочности различных материалов
Вещество Модуль упругости, «г/.#-и3 Механическая прочность, кг/мм1 Отношение теоретической прочности к эксперимен- тальной
теоретическое значение эк сие римеитальное значение теоретическое значение экспериментальное значение
Каменная соль 2000—4000 4 000 200—400 0,4 500—1000
Стекло обычное (толщиной > 1 мм) Тонкие стеклянные нити, травленные в фтористоводородной кислоте — 8 000 800—2200 2—4 200—1000
— 8 000 800—2200 400 2—5
Асбестовые нити — 10 700 —, 365 3
Слюда — 15 000—18 000 — 135—300 5—10
Стальная проволока — 20000 —1 200 10
Лен 11 000 И 000 1000 170 6
Шелк Феноло-формальдегидная смола: ^CONH ~ 27 000 (валентная связь) £('.0ХЯ . . . CON И — — 2700 (межмолекулярная связь) 10 000 2500 80—100 10
стадия С (резит) £'с„с = зо 000 1 000 4300 7—8 400
стадия А (резол) — 500 >39 1,5 - 10-а—Ю-1 400
Полиметилметакрилат (500 400—600 ~30 6—7 4
Полихлорвинил ООО -100— IKK) ->-30 6--7 4
Гл. II. Физические основы технологии пластмасс
Теоретическая и техническая прочность пластмасс
125
определении хрупкой прочности твердых тел. Разброс, как из-
вестно, всегда наблюдается, в той или иной степени, при испы-
тании большого числа образцов одинаковой формы, размеров и
состава.
Влияние так называемого «габаритного» фактора, т. е. разме-
ров и формы испытуемых образцов, на величину удельной проч-
ности материала широко известно, однако оно отрицается класси-
ческими положениями! теории прочности.
Изучением прочности волокон, стеклянных и кварцевых нитей
(С. Н. Журков) было установлено закономерное увеличение проч-
ности с уменьшением диаме-
тра (рис. 52). Оказалось, что
нити с диаметром порядка
одного или нескольких микро-
нов имеют прочность, близкую
к теоретической.
Большое влияние на проч-
Рис. 52. Зависимость прочности стеклян
пых нитей от диаметра.
ность твердого тела имеет со-
стояние его поверхности. Это
может быть проиллюстриро-
вано классическими опытами
акад. А. Ф. Ио'ффе при изуче-
нии прочности каменной соли.
А. Ф. Иоффе показал, что если
разрыв кристалла каменной
соли производить в воде, проч-
ность его увеличивается в де-
сятки и сотни раз, приближаясь к теоретической, — до 200 кг/мм?
(эффект Иоффе). При растяжении в этих условиях достигается
предел текучести и кристалл соли может быть подвергнут значи-
тельной пластической деформации.
Опыты С. Н. Журкова по разрыву стеклянных нитей, подверг-
нутых травлению плавиковой кислотой, показали значительное уве-
личение прочности после растворения поверхностного слоя.
Таким образом, техническая прочность значительно увеличи-
вается и может приближаться к теоретической при переходе
«объемного» твердого тела в состояние, близкое к двухмерному —
в форму тонких пленок и нитей, а также при растворении поверх-
ностного слоя в процессе растяжения.
Для объяснения расхождений между значениями теоретической и техниче-
ской прочности тел и вышеуказанных закономерностей были выдвинуты раз-
личные теории.
Общепринятой в настоящее время является статистическая теория распре-
деления внутренних и внешних дефектов (трещин) в твердом теле, разработан-
ная А. Ф. Иоффе, А. П. Александровым, С. Н. Журковым и др.
Эта теория исходит из положения о действии механизма, концентрирующего^
среднее напряжение, приложенное к телу, на площадь во много раз меньшую,,
чем площадь сечения тела (в сотни и тысячи раз).
124
Гл. П. Физические основы технологии пластмасс
Для объяснения действия такого механизма принимают, что внутри реаль-
ных тел так же, как на поверхности, имеются дефекты в виде микротрещин,
диаметром от нескольких десятков до нескольких тысяч и более онгстрем. Такое
предположение теоретически не является невозможным; гораздо труднее было
бы представить, что реальные тела являются абсолютно «сплошными», если при-
нять во внимание стерические трудности при уплотнении молекул в результате
уменьшения разбрасывающего действия температуры. При наложении внешнего
усилия возникающие напряжения концентрируются на краях микротрещнн и
достигают там величины теоретической прочности. Это приводит к быстрому
развитию, прорастанию трещин и к преждевременному разрушению тела (при
сравнительно небольших напряжениях, если относить их к площади сечения
всего образца).
Перенапряжения, концентрирующиеся на краях трещин, достигают макси-
мума (равного теоретической прочности) при тем меньших средних напря-
жениях F (из расчета на всю площадь сечения тела), чем длиннее и уже тре-
щина. Если трещина имеет эллиптическую форму с полуосями а и b и напря-
жение перпендикулярно большой оси а, то
f-’макс = F 0 + v) ’ ИЛИ /'мавс = KF’ (40)
т. е. напряжение на краях трещины тем больше среднего напряжения в теле,
чем больше отношение а к Ь, чем уже и длиннее трещина. «Опасность» такой
трещины будет в этом случае выражаться коэффициентом К, который указы-
вает, во сколько раз напряжение на краях трещины выше среднего напряжения
во всем сечении тела. Для круговой трещины, где а = Ь, коэффициент К =3.
Разрушение твердого тела начинается из развития самой «опасной» микро-
трещины (имеющей наибольшее К).
Развитие микротрепшн протекает во времени, н чем менее «опасна» тре-
щина, тем позже в процессе разрыва она начинает расти.
В каждом теле имеется, строго говоря, одна наиболее «опасная» трещина
и ее развитие и степень ее «опасности» определяют техническую прочность тела.
При последовательном уменьшении радиуса сечения тела уменьшается ве-
роятность наличия в нем наиболее «опасной» трещины. Это приводит к стати-
стическому распределению, к статистической зависимости самой опасной тре-
щины от диаметра сечения (толщины) тела и, следовательно к такому статисти-
ческому распределению прочностей образцов, при котором средняя величина
прочности непрерывно возрастает пропорционально уменьшению толщины об-
разца. С этой точки зрения эффект упрочнения стеклянных нитей при уменьше-
нии их толщины представляет собой статистическое явление и объясняется мень-
шей вероятностью наличия микротрещин в тонких нитях по сравнению с толстыми.
Статистическая теория приводит к наблюдаемой на практике
обратной зависимости прочности от толщины нити. Прочность до-
стигает теоретического значения при определенной минимальной
толщине нити.
Последовательный характер разрыва хрупкого тела опреде-
ляется первоначальным достижением предельного напряжения,
равного теоретической прочности у наиболее «опасной» трещины,
и последующим прорастанием этой трещины.
Если по пути прорастания такой наиболее «опасной» трещины
нет каких-либо других трещин (менее «опасных») или не суще-
ствуют какие-либо посторонние включения, могущие приостановить
прорастание трещины, а также если нет условий для релаксации
Теоретическая и техническая прочность пластмасс
125-
напряжения, прорастание трещины идет с увеличивающейся ско-
ростью и разрыв тела практически происходит мгновенно. Плоскость
разрыва в таком случае представляет гладкую, блестящую, «зер-
кальную» поверхность.
Наоборот, если на пути прорастания самой «опасной» трещины
встречаются другие менее «опасные» трещины или же посторонние
включения, первоначальная трещина сливается с менее «опасными»
трещинами или упирается в посторонние включения; в обоих слу-
чаях первоначальный рост трещины
прекращается, но может быть во-
зобновлен, если будут достигнуты
более высокие напряжения. Поверх-
ность разрыва в этом случае имеет
чешуйчатый неровный характер, так
как разрыв протекает при много-
численных последовательных соеди-
нениях линий прорастания боль-
шого числа трещин. Действительно,
различный характер поверхности
разрыва наблюдается у стекол и
смол — обычно одновременно зер-
кальный и чешуйчатый, а в пределе
только чешуйчатый или только зер-
Гиперболы А. П. Александрова.
кальный.
Экспериментально было доказано,
что чем больше зеркальная зона по сравнению с чешуйчатой, тем при
меньшем среднем напряжении происходит разрыв хрупкого тела.
Последовательный характер разрыва хрупкого- тела получил
экспериментальное и наглядное подтверждение в работах А. П. Але-
ксандрова. Изучая под микроскопом поверхность разрыва полиме-
тилметакрилата, он наблюдал в конце зеркальной зоны ряд пра-
вильно очерченных гипербол, обращенных головной частью к цен-
тру зеркальной зоны (рис. 53). Чешуйчатая поверхность разрыва
получается благодаря накладыванию одной на другую большого
числа гипербол. А. П. Александров дает следующее объяснение ме-
ханизму образования гипербол: из центра зеркальной зоны растет
круговая трещина; по мере увеличения в образце среднего напря-
жения из других неоднородностей также начинают расти круговые
трещины. Когда первоначальная круговая трещина доходит до тре-
щины других неоднородностей, в образце происходит «скол», выра-
жающийся в образовании чешуйчатой поверхности. Тот факт, что
линия соединения круговых трещин представляет собою не прямую,,
а гиперболу, наглядно указывает на последовательный механизм
развития трещин. Чем позже, по сравнению с первоначальной, на-
чинают расти вторичные трещины, тем уже гипербола, так как если
бы обе трещины начали развиваться одновременно и с одинаковой,
скоростью, то линия встречи была бы прямой.
126
Гл. И. Физические основы технологии пластмасс
Таким образом, относительные размеры зеркальной зоны и ха-
рактер наблюдаемых гипербол могут служить показателем «опас-
ности» первоначально развивающейся трещины по сравнению с
другими трещинами, последова-
тельно вступающими во взаимо-
действие с первой.
Техническая прочность твер-
дых тел является функцией вре-
мени действия напряжения. Чем
больше время, тем при более низ-
ких напряжениях происходит раз-
рушение тела. Зависимость проч-
ности от логарифма времени мо-
жет быть выражена на определен-
ном участке прямой, согласно
уравнению
= (41)
где: т — время до разрыва; а —
прочность, Л и а — константы.
Чем выше напряжение, тем,
следовательно, быстрее дости-
гается разрушение тела. На
Рис. 54. Зависимость'времени т (сек.)
от момента приложения нагрузки до
момента появления видимых трещин,
от величины напряжения а (кг/мм).
У —полистирол; 2— полихлорвинил; 3—поли. ПИС. 54 Приведена ЗАВИСИМОСТЬ
метилметакрилат 4-9г0 дибутилфталата. к .
долговечности (логарифма вре-
мени до разрушения) от величины напряжения для ряда полимеров.
Подобная же зависимость наблюдается и от температуры, а
также от степени пластификации полимеров.
ЗАВИСИМОСТЬ ТЕХНИЧЕСКОЙ ПРОЧНОСТИ ПЛАСТМАСС ОТ ИХ
СТРУКТУРЫ
Пластические массы подразделяются на два вида материалов,
отличающихся по своей структуре: материалы макроскопически
однородные, состоящие только из аморфного, стеклообразного
органического связующего (литые резиты, целлулоид, органическое
стекло и др.), и материалы макроскопически неоднородные, состоя-
щие из грубо диспергированных веществ, так называемых напол-
нителей, распределенных в непрерывной фазе — органическом
пленкообразующем веществе.
Наполнителями могут быть твердые, жидкие и газообразные
вещества. Наибольшее значение имеют твердые наполнители, ко-
торые, в свою очередь, делятся на порошкообразные и волокнистые
наполнители.
По характеру распределения в непрерывной фазе (в смоле) и
.по структуре наполнителя различают слоистые и неслоистые пласт-
Зависимость прочности пластмасс от структуры
127
массы. В слоистых пластмассах обычно применяют наполнитель в
виде листов бумаги, тканей и т. п.
Наполнитель увеличивает прочность пластмасс, а также твер-
дость, теплостойкость, огнестойкость, облегчает переработку и
уменьшает стоимость.
В пластмассах с однородной структурой, не содержащих напол-
нителя и состоящих только из смолы, прочность обусловлена си-
лами взаимодействия элементарных частиц (когезией). При этом
имеется существенное отличие
молекул с глобулярной, сфе-
рической структурой, и смо-
лами, построенными из ли-
нейных макромолекул.
Механическая прочность
смол со сферическими (гло-
булярными) молекулами оп-
ределяется характером меж-
частичных связей, кото-
рые необходимо преодолеть
для разрушения тела. Эти
связи могут быть ван-дер-
ваальсовскими у низкомоле-
кулярных, плавких и раство-
между смолами, построенными из
римых смол или же химиче- р11С. Зависимость прочности на разрыв
скими, валентными — у про- от степени полимеризации.
странственных сферических
полимеров. Как уже было указано, техническая прочность последних
в десятки и сотни раз выше, чем у плавких и растворимых низкомо-
лекулярных смол, и находится до известной степени в соответствии
с разницей в теоретической прочности обоих типов смол, опреде-
ляемой разницей в энергиях разрыва валентных и межмолекуляр-
ных связей.
В табл. 10 показана приближенная зависимость некоторых
свойств линейных полимеров от элементов структуры макромоле-
кулярной цепи. Как видно из таблицы, эта зависимость для некото-
рых элементов структуры точно не установлена-.
Весьма характерна для всех линейных полимеров зависимость
прочности полимера от степени полимеризации (рис. 55).
Как видно из кривой, до некоторой минимальной степени поли-
меризации (40—60) прочность настолько незначительна, что соизме-
рима с прочностью низкомолекулярных смол глобулярной струк-
туры. Затем до степени полимеризации 250—400 наблюдается ее
возрастание, после чего кривая изгибается и при больших сте-
пенях полимеризации прочность уже не зависит от длины мо-
лекул.
Такая зависимость может быть объяснена
полимера. Когда полимер имеет сравнительно
различием в механизме разрыва
короткие молекулы, разрыв его
ТАБЛИЦА 10
Связь между некоторыми свойствами и структурными элементами линейных полимеров
Свойства Структурная xajактсристика макромолекул
Степень полимеризации Разветвленность Гибке,сть цепи Наличие диполей Наличие мостиков между цепями Кристалли- зуемость Ориентация
Температура стеклова- ния аморфной фазы (Го) До известного предела увеличивается, в дальнейшем мало за- висит Увеличивается Уменьшается Увеличивается Увеличивается Не зависит Не зависит
Температура плавления кристалли ческой фазы Увеличивается, но не пропорционально сте- пени полимеризации Уменьшается спо- собность к кри- сталлизации Невидимому уменьшается Увеличивается, а также увеличи- вается способ- ность к кристал- лизации Уменьшается спо- с бность к кри- сталлизации — Увеличи- вается
Модуль упругости и модуль высокоэла- стичности . До известного предела увеличивается, затем не зависит; модуль высокоэластичности уменьшается с уве- личением степени по- лимеризации Модуль высокоэла- стичности повы- шается Уменьшаются Увеличиваются Увеличиваются Увеличи- ваются Увеличи- ваются
Предел прочности на разрыв До известного предела увеличивается; в дальнейшем мало за- висит Уменьшается Уменьшается Увеличивается Увеличивается Увеличи- вается Увеличи- вается
Удлинение при разрыве Увеличивается Уменьшается Увеличивается Уменьшается Уменьшается Умень- шается Умень- шается
Прочность на удар Увеличивается Уменьшается Увеличивается Уменьшается Уменьшается Умень- шается Увеличи- вается
Твердость Увеличивается Уменьшается Уменьшается Увеличивается Увеличивается Увеличи- вается
Гл. II. Физические основы технологии пластмасс
Зависимость прочности пластмасс от структуры
129
связан преимущественно с соскальзыванием (перемещением) молекул друг
относительно друга, подобно, например, тому, как это имеет место при разрыве
шерстяных нитей (межмолекулярный механизм разрыва). При этом необходимо
преодоление межмолекулярных сил, действующих между цепями молекул. Эти
силы растут пропорционально возрастанию длины молекул, и на определенном
отрезке кривой (рис. 55) их ростом и может быть объяснено увеличение прочности
пропорционально степени полимеризации.
Дальнейшее увеличение длины цепей все в большей мере приводит ко вто-
рому механизму разрыва цепей — к разрыву валентных С—С связей цепочки
(внутримолекулярный механизм разрыва). При достижении определенной длины
молекул действует уже только этот механизм и уже не может наблюдаться за-
висимость прочности от длины цепи, так как энергия разрыва валентных связей
не зависит от длины молекулы. Этим объясняетси появление пологого участка
кривой при высоких степенях полимеризации.
Н. В. Михайлов и В. А. Каргин произвели энергетический подсчет разрыва
целлюлозных волокон по обоим механизмам: с разрывом свизей С—С и С—О
(внутримолекулярный механизм разрыва) и с преодолением межмолекуляр-
иых связей ОН. . .ОН в результате соскальзывания цепей (межмолекулярный
механизм разрыва). Считая, что степень полимеризации равна 300 и энергия
разрыва связей С—С и С—О 70 ккал/моль, а энергия разрыва связей
ОН. . .ОН 7,2 ккал/моль и полагая, что средние расстояния, при которых
реализуется разрыв для связей С—С и С—О, равны 2,5 А, а для связей
ОН. . .ОН 4 А, эти авторы рассчитали, что энергия внутримолекулярного
разрыва равна 520 кг/мм2, а энергия межмолекулярного разрыва равна
14 000 кг/мм2.
Таким образом, при степени полимеризации, равной 300, вероятность раз-
рыва целлюлозы по механизму сдвига цепей крайне незначительна и, очевидно,
имеет место внутримолекулярный механизм разрыва цепей.
Прочность пластмасс с неоднородной структурой, содержащих наполнители,
обусловлена не только силами когезии, но также и дополнительными силами,
действующими между связующим и наполнителем. Эти силы могут быть рас-
сматриваемы как силы адгезии — силы прилипания.
Усиливающее действие наполнителей следует связать с тем, что в их при-
сутствии молекулы связующего переходят из неупорядоченного в ориентиро-
ванное состояние тонких пленок.
Как известно, на поверхности раздела двух фаз полирные анизотропные мо-
лекулы жидкости типа СН3(СН2) ,tCOOH располагаются не беспорядочно,
а ориентируются полярными СООН-группами к'поверхности твердого тела (ме-
талла, стекла) и образуют на ней адсорбционный слой перпендикулярно ориен-
тированных молекул. Наличие перпендикулярно ориентированного молекуляр-
ного слоя было доказано рентгенографически, а также с помощью электронной
дифракции при исследовании пленок жирных кислот на стеклянных пластинках.
Минимальная толщина такого слоя равна длине одной адсорбированной це-
почечной молекулы. Однако ориентирующее действие силового поля твердого
тела распространяется также и иа молекулы, расположенные за этим слоем,
ослабевая по мере удаления от поверхности твердого тела. Поэтому за преде-
лами мономолекулярного слоя молекулы также располагаются перпендикулярно
вслед за первым слоем, но только своими неактивными концами они обращены
к неактивным концам молекул первого слоя (рис. 56).
Расстояние, на которое простирается ориентирующее влияние твердого тела,
зависит от степени полярности его молекул и молекул жидкости; повидимому,
это влияние является еще заметным на расстоянии до 0,1 р. и более. При
наличии молекул одинаковой структуры, но различной длины, в первую оче-
редь образуется слой из наиболее коротких и подвижных молекул, способных
быстрее ориентироваться.
Соответственно наполненные пластмассы можно представить как систему,
состоящую из непрерывной фазы (смолы), ориентированной и фиксированной
в виде тонких пленок (слоев) на поверхности прерывной фазы (твердого на-
9 Зак. 1269. Э. И. Барг
130
Гл. II. Физические основы, технологии пластмасс
полнителя), или как слоистую систему из чередующихся слоев твердого тела
и ориентированных, адсорбционных слоев (пленок) смолы.
Из рис. 56 видно, как слой перпендикулярно ориентированных цепей мо-
лекул на поверхности твердого тела (наполнителя) постепенно переходит
в неупорядоченную область. Наибольшая неупорядоченность, очевидно, будет
иметь место по середине слоя между двумя поверхностями наполнителя. Чем
тоньше этот слой, тем меньше будет область хаотически расположенных мо-
лекул.
Образование адсорбционных ориентированных слоев обусловливает и сма-
зывающее действие масел. Снижение коэффициента трения при добавке поляр-
ных веществ к смазочным маслам объяс-
няется фиксацией мономолекулярного слоя
поляряых молекул на поверхности металла.
Коэффициент трения смазочного веще-
ства тем меньше, чем полярнее линейные
молекулы и чем они длиннее, т. е. чем проч-
нее мономолекулярный слой фиксирован на
поверхности металла и чем толще бимоле-
кулярная прослойка перпендикулярно ориен-
тированных молекул между плоскостями
скольжения твердого тела.
Если адсорбционный слой между по-
верхностями твердого тела теряет свою по-
движность (например, когда в связи с пони-
жением температуры или благодаря процес-
сам полимеризации смазка «замерзает») —
смазка переходит в склейку. Прочность
последней тем выше, чем полярнее моле-
Рис. 56. Схематическое изобра-
жение расположения полярных мо-
лекул между двумя поверхностями
твердого тела.
кулы и, следовательно, чем сильнее они
прикреплены своими полярными концами
к поверхности твердого тела. До опреде-
ленного предела прочность склейки тем
выше, чем длиннее линейные молекулы,
чем толще бимолекулярный слой ориенти-
рованных молекул по сравнению с общей
толщиной слоя между плоскостями твердого
Кружками обозначены полярные концы тела’
молекул, штрихами—неполярные. Таким образом, имеется далеко идущая
аналогия между требованиями к идеаль-
ной смазке и к идеальной склейке. Хорошая смазка, обусловливающая ми-
нимальный коэффициент трения, при затвердевании (при температуре ниже Те
должна переходить в хорошую склейку. Прочность склейки (адгезии) есть
в идеальном случае прочность отрыва адсорбционного слоя от поверхности твер-
дого тела и теоретически равна прочности межмолекулярного сцепления (около
3000 кг/см?). Эта прочность часто значительно больше технической прочности
самого склеивающего вещества, благодаря чему часто наблюдается не отрыв
смолы от поверхности твердого тела, а разрыв по склейке.
Чем толще слой склейки, тем в большей мере ее прочность обусловливается
наличием. и развитием неоднородностей и определяется технической прочностью
самого связующего. Однако прочность склейки должна линейно возрастать
с уменьшением толщины слоя, так как при этом все в большей степени будет
расти влияние ориентирующего действия силового поля твердого тела и ориен-
тированная часть склейки будет увеличиваться за счет уменьшения неупорядочен-
ной части. Максимум прочности такой склейки будет достигнут только тогда,
когда пленка станет бимолекулярной, предельно ориентированной. В этом состоя-
нии прочность склейки должна достигнуть теоретической величины, равной вели-
чине силы, необходимой для разрыва ориентированных слоев молекул или для
отрыва их от поверхности твердого тела.
Зависимость прочности пластмасс от структуры
131
Все сказанное непосредственно приложимо к объяснению уси-
ливающего действия наполнителя. В идеальном случае наполнен-
ные пластмассы можно рассматривать как систему адсорбирован-
ных тонких пленок (слоев) смолы, фиксированных между поверх-
ностями наполнителя и удерживаемых поверхностными силами
сцепления.
Согласно этому представлению, прочность пластмасс должна
линейно расти с увеличением активной поверхности наполнителя
вплоть до максимума, соответствующего предельно ориентирован-
ному бимолекулярному слою связующего. Однако эксперименталь-
ные данные показывают, что лишь до определенного предела за-
полнения наблюдается прямая зависимость прочности от количе-
ства наполнителя (от величины поверхности раздела); выше этого
предела обычно наблюдается резкое снижение прочности (рис. 57).
Рис. 57. Зависимость прочности пекоасбослоя (асбо-
пеколита) от содержания наполнителя (асбеста).
Это обстоятельство может быть объяснено технической трудностью
равномерного распределения малого объема связующего на огром-
ной поверхности наполнителя.
Усиливающее действие наполнителя тем больше, чем меньше
«объемная» прочность самого связующего, чем меньше когезия.
При оптимальной степени заполнения техническая прочность
пластмасс на основе различных по прочности смол в значительной
степени уравнивается и в пределе — в случае оптимальной ориентации
наполнителя — приближается к теоретической. Естественно, что
чем больше расхождение между технической и теоретической проч-
ностью смолы, тем эффективнее усиливающее действие наполни-
теля.
Так, прочность на разрыв каменноугольного пека — 10 кг/см2,
феноло-альдегидной смолы в стадии С 500—800 кг! см2. При эф-
фективном их заполнении слоистым ориентированным наполните-
лем (асбест) прочность пластмассы на основе пека (пекоасбослой)
повышается до 2000 кг/см2, а прочность пластмассы на основе
9*
132
Гл. II. Физические основы технологии пластмасс
резита (феноасбослой) —до 3000 кг/см2 (рис. 58). Таким образом,
усиливающее действие наполнителя для пека выражается в увели-
чении прочности в 200 раз, а для резита всего в 5—6 раз.
Закономерности влияния наполнителей на прочность пластмасс
можно объяснить более полно, исходя из физико-механических
Содержание напалнителя(асЬеста) °'-
Рис. 58. Зависимость прочности плас-
тика от степени заполнения.
2—-на основе феноло-альдегиднсй смолы
(феноасбослой,; 2—на основе каменноуголь-
ного пека (пекоасбослой).
представлений, в частности, из
статистической теории распреде-
ления внутренних дефектов (ми-
кротрещин) в твердом теле.
Усиливающее действие напол-
нителей может быть связано с из-
менением условий перенапряже-
ний на краях микротрещин, а
именно, с релаксацией напряже-
ний и перераспределением их на
большее число центров прораста-
ния микротрещин, что, естествен-
но, должно увеличить среднее на-
пряжение, ведущее к разрушению
тела.
Влияние наполнителей с этой
точки зрения становится понят-
ным, если представить себе, что
микротрещина, развиваясь, может
«упереться» в наполнитель и, сле-
довательно, ее дальнейшее разви-
тие требует увеличения напряже-
ния. Чем больше количество на-
полнителя, тем больше создается
препятствий на пути развития
трещин. Таким образом, происхо-
дит торможение процесса разви-
тия трещин и среднее напряже-
ние, необходимое для разруше-
ния массы, должно быть больше, т. е. масса в присутствии напол-
ните чя становится прочнее. Согласно этим воззрениям и в соот-
ветствии с практическими наблюдениями, большое значение
имеет анизотропность наполнителя, его волокнистая и слоистая
структуры.
Олнако на основе механических представлений возможно и дру-
гое объяснение механизма усиливающего действия наполнителя, во
многом аналогичное с физико-химической теорией перевода смол в
присутствии наполнителей в пленочное состояние. Согласно стати-
стической теории распределения микротрещин, прочность тела
увеличивается пропорционально уменьшению толщины и может
быть доведена до теоретической при предельно малом диаметре
1 М).
Зависимость прочности пластмасс от структуры.
133
Диспергирующее действие наполнителя является тем механиз-
мом, который обусловливает перевод «объемной» смолы в систему
тонких пленок и, следовательно, уже одним этим, вне зависимости
от степени полярности и величины поверхностных сил прилипания,
увеличивает прочность связующего.
Такой механизм усиливающего действия наполнителя получает
особую наглядность в слоистых пластиках, представляющих слои-
сто ориентированные волокна, листы или ткани, пропитанные смо-
лами. Наполнитель диспергирует смолу в систему тонких парал-
лельно ориентированных пленок, прочность которых возрастает
пропорционально уменьшению их толщины, что, в свою очередь,
определяется эффективной поверхностью наполнителя в единице
объема пластмассы. Ориентированное состояние наполнителя и
пленок связующего значительно увеличивает прочность благодаря
уменьшению возможности развития микротрещин.
Слоистые пластики представляют собою весьма совершенную,
упорядоченную систему наполненных пластиков. Это позволяет в
наибольшей степени устранить причины, снижающие техническую
прочность объемных тел, и в случае достижения предельной одно-
родности и высоких степеней заполнения (предельно малой тол-
щины слоев) получать материалы с прочностью, близкой к теорети-
ческой.
Принципиально иной механизм усиливающего действия наполнителей наблю-
дается в полимерах, находящихся в высокоэластическом состоянии (например
з каучуках), для которых внесение наполнителя в ряде случаев не увеличивает
Рис. 59. Работа разрыва полимера
(заштрихованная площадь).
Рис. 60. Работа А (заштрихованная
площадь) разрыва двух полимеров
(Z и 2) с равными разрывными на-
пряжением и удлинением.
статическую прочность, а при расчете на действительное сечение поверхности
разрыва — даже снижает ее.
Теория усиливающегося действия наполнителей на каучукоподобиые полимеры
была разработана акад. П. А. Ребиндером с сотрудниками. Он определял уси-
ливающее действие наполнителей мерой добавочной работы, затрачиваемой на
разрушение наполненных полимеров.
Для перевода объемной смолы в пленочное состояние и создания большой по-
верхности раздела необходимо произвести работу преодоления сил поверхностного
134
Гл. II. Физические основы технологии пластмасс
натяжения смолы. Эта работа идет на увеличение активной поверхности
смолы, что является причиной добавочной работы, необходимой для разрушения
наполненных масс.
Работа разрыва определяется площадью, образуемой кривой растяжения
(рис. 59), и равна:
Lb
А = j* a dL,
(42)
где: А — работа разрыва, кг • см; L/,— длина образца после разрыва; £о — на-
чальная длина образца; а — напряжение; L — соответствующее растяжение.
Следует отметить, что два материала, требующие одинакового разрывного
напряжения и дающие равные удлинения, могут обнаруживать различную ра-
боту разрыва (рис. 60)
Работу разрыва полимера, отнесенную к единице объема, называют энер-
гией упругости (кг - см/см3). Увеличение энергии упругости можно принять за
Рис. 61. Увеличение энергии упругости
каучука в зависимости от количества
наполнителя.
основную характеристику усиливаю-
щего действия наполнителей для по-
лимеров, находящихся в высокоэла-
стическом состоянии.
Работа разрыва н энергия упру-
гости не находятся в прямой связи с
силами когезии, с энергией валентных
или межмолекулярных связей, но за-
висят от способности полимера к об-
ратимым и необратимым перегруппи-
ровкам.
Высокополимеры в температур-
ном интервале высокоэластичности
обладают весьма большой энергией
упругости, превышающей в сотни и
тысячи раз энергию упругости других
материалов (стали, чугуна, дерева и
т. д.).
Выражая усиливающее действие
наполнителя увеличением энергии
упругости ЛА, можно заметить, что последнее идет параллельно увеличению ко-
личества наполнителя лишь до определенной концентрации и по достижении ма-
ксимума— падает (рис. 61). Кроме того, не все наполнители обладают одинако-
вым усиливающим действием. Это становится понятным, если учесть их раз-
личную степень дисперсности и лиофильности. На рис. 62 показано усиливаю-
щее действие различных наполнителей (увеличение энергии упругости ЛА)
в зависимости от их концентрации.
Наполнители, не увеличивающие энергию упругости полимеров, называют
инертными или неактивными, в противоположность активным наполнителям,
увеличивающим энергию упругости. Однако принципиальной границы между
ними ие существует. Если усиливающее действие (М) отнести к остающейся
в смеси части каучука, то и неактивные наполнители будут показывать значи-
тельное усиливающее действие (рис. 63).
В высокоэластическом состоянии различные линейные полимеры
(каучуки) обладают различной способностью усиливаться напол-
нителями. Как указал А. П. Александров, полимеры, кристаллизую-
щиеся при растяжении (например природный каучук), усиливаются
наполнителями в меньшей степени, так как они уже в процессе
растяжения в достаточной степени загружаются образующимися
Зависимость прочности пластмасс от структуры.
135
кристаллитами, которые и выполняют роль дисперсной фазы — на-
полнителя; те же высокополимеры, которые при растяжении не
кристаллизуются и, следовательно, не «самозагружаются», в зна-
чительно большей степени усиливаются наполнителями (например
некристаллические синтетические каучуки).
Энергия упругости,нгм/см3
Количество наполнителя
Рис. 63. Схема зависимости энергии упру-
гости от количества наполнителя.
Обсзначения те же, что на рис. 62.
О 5 10 ?5 То 25 30
Наполнитепь3 %
Рис. 62. Усиливающее действие
различных наполнителей для вул-
канизованного каучука.
1—магнезия: 2—газовая сажа; 3—каолин;
4— барит.
Рассмотренные теории усиливающего действия наполнителя
позволяют сделать ряд важных выводов:
1. Механическая прочность пластмасс теоретически должна воз-
растать пропорционально количеству и дисперсности наполнителя.
2. Теоретически предельным является такое количество напол-
нителя, которое обусловливает создание бимолекулярной пленки
связующего между поверхностями наполнителя.
3. Пределом усиливающего действия наполнителя является до-
стижение теоретической прочности связующего. Эта прочность прак-
тически может быть достигнута, если толщина пленок связующего
максимально однородна и будет иметь порядок нескольких микрон.
4. Усиливающее действие наполнителей особенно значительно
для низкомолекулярных смол глобулярной структуры и в тех слу-
чаях, когда затруднены процессы релаксации напряжений (в тем-
пературном интервале ниже Те).
5. Усиливающее действие наполнителей снижается с увеличе-
нием степени полимеризации смол и с увеличением сил когезии,
а также при замене межмолекулярных (ван-дер-ваальсовских)
связей химическими, валентными.
6. В высокоэластическом состоянии (для линейных полимеров)
усиливающее действие наполнителей обусловлено работой, затрачи-
ваемой на увеличение поверхности раздела смолы, и выражается
увеличением энергии упругости из расчета на остаточную в массе
смолу.
1.36
Гл. II. Физические основы технологии пластмасс
ТАБЛИЦА 11
Температура стеклования
системы ацетилцеллюлоза—
трикрезилфосфат
Трикрезилфосфат к ацетилцеллюлозе °0 т °C
0 + 60
20 0
40 — 30
80 — 50
ПЛАСТИФИКАЦИЯ ПОЛИМЕРОВ
Для уменьшения в полимерах межмолекулярных сил притяже-
ния, для увеличения их гибкости и растяжимости широко приме-
няют так называемые пластификаторы-, последние представляют со-
бою обычно низкомолекулярные, высококипящие жидкости, совме-
стимые с полимерами; значительно реже они являются твердыми
кристаллическими телами.
Роль пластификатора сводится к снижению температуры стекло-
вания полимера, что осуществляется взаимодействием молекул
пластификатора с макромолекулами полимера.
Взаимодействуя с пластификаторами, полимеры вначале по-
глощают эти обычно низкомолекулярные жидкости, увеличиваясь
в объеме и весе, т. е. набухают.
Процесс набухания является специ-
фическим для полимеров и неизвестен
для низкомолекулярных веществ. Он
основан на проникновении небольших
молекул растворителя между звенья-
ми макромолекул полимера и ведет
к раздвижению звеньев, а затем и
цепей макромолекул. Первая стадия
набухания связана с образованием
мономолекулярного слоя растворителя
вокруг цепи полимера, с нарушением
связей между звеньями и молекулами
полимера и созданием новых связей
между молекулами полимера и молекулами растворителя. Есте-
ственно, что нарушение связей между молекулами полимера, экра-
нирование дипольных групп и раздвижение цепей полимеров
должно значительно уменьшить силы когезии между макромоле-
кулами и звеньями и, следовательно, снизить температуру стекло-
вания полимера.
В табл. 11 показано изменение температуры стеклования ацетил-
целлюлозы в зависимости от содержания пластификатора.
Однако пластификатор, уменьшая в полимере межмолекулярные
силы и снижая его вязкость, обусловленную наличием сильных
межмолекулярных связей, не может влиять на жесткость самой
цепи, так как эта жесткость обусловлена существованием и ха-
рактером внутримолекулярного потенциального барьера, препят-
ствующего изменению формы цепной молекулы. Практически это
сказывается в том, что хотя внесением пластификатора и можно
значительно снизить температуру стеклования полимера, обладаю-
щего жесткими цепями (например эфиров целлюлозы), при этом,
однако, не происходит превращения пластика в настоящий «каучук»;
последний характеризуется не только низкой температурой стекло-
вания, но и высокой скоростью релаксации. Таким образом, пласти-
Пластификация полимеров
137
ТАБЛИЦА 12
Зависимость температуры
стеклования сополимеров от
соотношения количеств
стирола и бутадиена
Молекулярные соотношения
стирола и бутадиена
1 : 0 (чистый полистирол)
2:1...................
1:1...................
1:2...................
но и большой
Те
°C
4-80
— 30
— 45
— 65
фикатор, снижая межмолекулярное сопротивление, не меняет, од-
нако, кинетических свойств самой макромолекулы.
Изменения потенциального барьера самих макромолекул можно
достигнуть, прибегая к так называемой внутренней пластификации.
Под. этим часто применяемым в технике термином понимают про-
цессы сополимеризации, а также процессы эфиризации и заме-
щения полярных групп макромолекул, ведущие не только к умень-
шению межмолекулярной когезии, но и
к нарушению симметрии макромолекул
и снижению их внутреннего потен-
циального барьера, т. е. к изменению
кинетических свойств макромолекул.
В табл. 12 показана зависимость
температуры стеклования от соотноше-
ния компонентов при сополимеризации
стирола и бутадиена. В отличие от си-
стемы трикрезилфосфат —• ацетилцел-
люлоза (табл. 11), где также дости-
гаются весьма низкие То, система со-
полимеров стирол — бутадиен приво-
дит к получению продуктов, характе-
ризующихся не только низким значением
релаксации. Эти продукты являются синтетическими каучуками.
Такое различие объясняется глубоким изменением кинетических
свойств макромолекул сополимеров.
Отличие внутренней пластификации от действия .пластификато-
ров заключается также в том, что пластификаторы, снижая темпе-
ратуру стеклования полимеров, одновременно снижают и темпера-
туру вязкого течения (Ттек ). При этом после определенной концен-
трации пластификатора Лек снижается быстрее, чем То, что
приводит к уменьшению температурного интервала высокоэластич-
ности (Утек—Го) и к получению полимеров с вязким течением
уже при обычной температуре. Такое действие пластификаторов,
ведущее преимущественно к усилению пластических свойств поли-
мера, называют ложной пластификацией. Переход от истинной
пластификации к ложной зависит не только от количества пласти-
фикатора, но и от его структуры и совместимости с полимером.
В отличие от действия пластификаторов, внутренняя пластифи-
кация не. снижает температуру вязкого течения, если только моле-
кулярный вес сополимера достаточно велик и существенно не от-
личается от молекулярного веса полимера. Техническое преимуще-
ство внутренней пластификации заключается также в том, что
пластификаторы обладают заметной упругостью пара и, следова-
тельно, непрерывно улетучиваются, вследствие чего увеличивается
жесткость полимера и снижаются со временем эластичность и мо-
розостойкость, в то время как при процессах внутренней пластифика-
ции высокоэластические свойства полимера остаются неизменными.
138
Гл. II. Физические основы технологии пластмасс
Вопрос о механизме действия пластификаторов еще не решен.
Если это действие заключается в раздвижении цепей макромолекул,
то оно должно быть аналогично действию температуры. С каче-
ственной стороны это соответствует действительности, однако рас-
чет показывает, что если бы пластификаторы проявляли себя
только в этом отношении, то вызываемое ими снижение темпера-
туры стеклования было бы в несколько раз более значительным,
чем это наблюдается на практике. Различие это объясняется, ко-
нечно, тем, что пластификаторы не только раздвигают цепи поли-
мера, но и взаимодействуют с ними; при этом образуются новые
связи — между полимером и пластификатором.
Первые работы, посвященные исследованию механизма пласти-
фикации, принадлежат С. Н. Журкову. Исследуя влияние большого
числа полярных и неполярных растворителей на снижение темпера-
туры стеклования различных полимеров, он пришел к выводу, что
снижение температуры стеклования неполярных полимеров при их
взаимодействии с неполярными растворителями пропорционально
весовому количеству поглощенного растворителя, следовательно,
если исходить из одного и того же молярного соотношения рас-
творителя и полимера, то температура стеклования снижается про-
порционально молекулярному весу растворителя.
Совершенно другие закономерности Журков наблюдал при дей-
ствии полярных растворителей на полярные полимеры. В этом
случае оказалось, что величина снижения температуры стеклова-
ния (Д Т) пропорциональна не весовому количеству сорбированного
растворителя, а числу его молекул, удерживаемых полимером,
вне зависимости от формы и размеров молекул, т. е.
ДГ=АГ© = Л>7’ (43)
где: дТ— снижение (депрессия) температуры стеклования поли-
мера; С — концентрация; М — молекулярный вес; К — коэффи-
циент, не зависящий от природы растворителя; п — число сорбиро-
ванных молекул.
Эта закономерность в снижении температуры стеклования ана-
логична закономерности, выражаемой уравнением Рауля для де-
прессии температуры кристаллизации раствора от числа молекул
растворенного вещества, хотя Та —не фазовый переход полимера.
На рис. 64 представлена найденная Журковым зависимость сни-
жения температуры стеклования полиметилметакрилата от числа
молекул сорбированного растворителя, которое пропорционально от-
ношению концентрации к мол. весу В случае полярных пласти-
фикаторов и полярных полимеров эта зависимость для различных
пластификаторов выражается прямой линией.
На рис. 65 представлена найденная тем же автором зависимость
коэффициента К от числа углеродных атомов в молекуле раствори-
теля для неполярного полимера (полиизобутилена) при сорбции
Пластификация полимеров
139
неполярных растворителей (бутан, гексан и др.). Как видно из
рисунка, в этом случае имеется линейная зависимость между дли-
ной молекул и снижением температуры стеклования, иными сло-
вами, в случае неполярных полимеров и неполярных растворите-
лей дГ зависит не только от количества сорбированных молекул,
но и от их размеров и формы.
Объясняя аналогию зависимости ДТ = Кп с законом Рауля,
Журков принимает, что полярные полимеры в застеклованном со-
стоянии представляют собою пространственную сетку, узлы которой
Рис. 65. Зависимость коэффи-
циента К от числа углеродных
атомов в молекуле неполярного
растворителя.
Рис. 64. Зависимость снижения Тс по-
лиметилметакрилата (ДТ) от числа моле-
кул сорбированных растворителей I — I.
С—концентрация; М—молекулярный вес раство-
рителя; кружки относятся к различным раство-
рителям (ацетофенон, ацетон, и др.).
составлены взаимодействующими диполями, расположенными вдоль
цепей. При сорбции полярного растворителя каждая его молекула
взаимодействует только с одной полярной группой полимера, кото-
рая при этом «блокируется» и уже выключается из взаимодействия
с соседней полярной группой полимера; это и приводит к снижению
температуры стеклования. Таким образом, последнее должно быть
пропорционально только количеству молекул и не должно^ зависеть
от их величины и формы.
В неполярных полимерах стеклообразное состояние объясняется
межмолекулярным взаимодействием равноценных с энергетической
точки зрения атомов и групп. Поэтому снижение температуры
стеклования зависит не только от количества сорбированных моле-
кул, но и от их размеров, т. е. практически от веса абсорбированной
жидкости.
С. Н. Журков показал также, что неполярные пластификаторы
с линейными молекулами более эффективны, чем с циклическими
при одинаковом молекулярном весе. Таким образом, для неполяр-
140
Гл. II. Фазические основы, технологии пластмасс
них полимеров значение имеют не только размеры молекул пласти-
фикатора, но и форма молекул.
Теория Журкова не может быть принята без существенных ого-
ворок. Она совершенно правильно трактует закономерности, наблю-
даемые при действии неполярных пластификаторов на неполярные
полимеры, однако, по нашему мнению, упрощенно трактует ме-
§
ез
I
I
Рис. 66. Зависимость Г(. поливи-
нилбутираля от числа углеродных
атомов z в цепи линейных моле-
кул пластификатора.
7—диэтилсвый эфир щавелевой кислоты:
?—дибутиловый эфир щавелевой кислоты;
‘—дибутиловый эфир янтарной кислоты;
/—дибутиловый эфир адипинов й кислоты;
5—дигексиловый эфир янтарной кислоты;
о — дигексил.,вый эфир адипиновой кисло-
ты; 7—дигексиловый эфир себаниновой
кислоты: а-диоктиловый эфир себацино-
вой кислоты.
ханизм действия полярных пласти-
фикаторов, так как не учитывает
межмолекулярного взаимодействия
за счет всей молекулы в целом. Если
пластификатор совместим с полиме-
ром, то его действие заключается в
экранировании дипольных групп и в
снижении энергии межмолекуляр-
ного притяжения за счет действия не
только дипольных групп молекул
пластификатора, но и всей массы мо-
лекул. Механизм действия «числа
молекул», вне зависимости от их раз-
меров, мог бы быть понятным, если
бы наблюдалась депрессия темпера-
туры фазового перехода, а не сни-
жение температуры стеклования.
Пластификаторы, применяемые в
современной технике, представляют
вещества, молекулы которых имеют
сравнительно большое число угле-
родных атомов (до 30 и выше) и
значительную длину (до 59А); по-
этому трудно себе представить,
чтобы во взаимодействии с полимером участвовала лишь полярная
группа, а вся остальная часть молекулы оставалась вне взаимодей-
ствия и не влияла на температуру стеклования полимера.
Э. И. Барг и Н. Н. Мельтева изучили зависимость температуры
стеклования поливинилбутираля от длины цепи пластификатора,
применяя гомологический ряд линейных эфиров общей структуры
СН3(СН2)„ОСО(СН2)ОТСОО(СН2)„СН3.
Была получена линейная зависимость температуры стеклования
от длины молекулы пластификатора (от числа углеродных атомов)
(рис. 66); пластификатор вводился в одинаковых молекулярных от-
ношениях (5 мол. %).
В. А. Каргин с сотрудниками показал на примере полихлор-
винила и большого числа полярных пластификаторов, что сниже-
ние Та происходит пропорционально объему внесенной жидкости,
следовательно, зависит не только от числа, но и от величины моле-
кул пластификатора.
Пластификация полимеров
141
Снижение 7*о пропорционально длине линейных молекул (при
введении их в эквимолекулярных отношениях) подчинено такой
же закономерности, как и снижение ?'< пропорционально напряже-
нию о (стр. 106).
Влияние обоих факторов определяется закономерностью линей-
ного снижения энергии активации высокоэластической деформа-
ции U и, следовательно, снижения времени релаксации т как от
концентрации пластификатора с, так и от роста напряжения о:
U— аа— Ъс
1 = Той КТ , (44)
где т0, а и b — константы данного полимера.
Таким образом, между а и с существует однозначная зависи-
мость. Так, для случая поливинилбутираля увеличение напряжения
при растяжении пленки на 150 кг/см2 (с 50 до 200) эквивалентно
снижению То внесением 5 мол. % диэтилоксалата.
Подобная же зависимость существует и при внутренней пласти-
фикации. Как было показано П. П. Кобеко, снижение Т(, полиме-
ров гомологического ряда метакриловых эфиров находится в ли-
нейной зависимости от длины боковой цепи (от числа углеродных
атомов в спиртовом радикале).
Таким образом, для полярных полимеров в основном характерна
та же закономерность, что и для неполярных: снижение темпера-
туры стеклования зависит не только от числа, но и от величины и
формы сорбированных молекул пластификатора и для линейных
пластификаторов — от числа атомов цепи, от длины молекулы.
Из сказанного следует, что механизм пластификации как поляр-
ных, так и< неполярных полимеров, повидимому, заключается
в образовании мономолекулярного слоя абсорбируемого полимером
пластификатора вокруг звеньев макромолекул полимера; чем длин-
нее молекулы пластификатора, тем большая часть звеньев макро-
молекулы полимера оказывается изолированной от влияния сосед-
них макромолекул полимера и тем больше снижение температуры
стеклования.
В качестве пластификаторов в технике применяют лишь высоко-
кипящие, малолетучие вещества, совмещающиеся с полимерами.
Поэтому, кроме эффективности пластификатора, определяемого по
величине снижения температуры стеклования (на единицу объема
или веса пластификатора), техническое значение имеют степень
совместимости пластификатора с полимером и степень летучести
пластификатора.
При проникновении в полимер молекулы растворителя либо
распределяются среди цепей макромолекул полимера, взаимодей-
ствуя с их звеньями, т. е. образуя истинный раствор, характеризую-
щийся достижением равновесного состояния, либо растворитель
коллоидно диспергируется в полимере, образуя эмульсию. Во втором
142
Гл. II. Физические основы технологии пластмасс
случае частицы эмульгированного растворителя постепенно могут
сливаться в большие капли, которые выступают на поверхности
полимера; пластификатор при этом как бы «выпотевает». Этот
процесс протекает во времени; поэтому «выпотевание» пласти-
фикатора наступает обычно не сразу после его диспергирования
в полимере, а через некоторое, иногда очень продолжительное,
время.
Во многих случаях для каждого конкретного полимера известны
совместимые пластификаторы, т. е. такие, которые молекулярно
распределяются в полимере, и пластификаторы несовместимые, —
образующие в полимере эмульсию. Однако пластификаторы, обра-
зующие истинные растворы, могут неограниченно или лишь ограни-
ченно совмещаться с полимером. В последнем случае при данной
температуре истинные растворы пластификаторов с полимерами
образуются лишь при строго определенных соотношениях. При до-
бавлении пластификатора при данной температуре выше этого со-
отношения он выступает в виде эмульсии.
«Выпотевание» пластификатора усиливается при низких тем-
пературах в связи с уменьшением его растворимости с пониже-
нием температуры; с течением времени после выделения избытка
пластификатора достигается равновесное состояние и дальнейшее
«выпотевание» прекращается.
Растворение пластификаторов в полимерах чаще всего подчи-
няется общим закономерностям, действительным для смешиваю-
щихся жидкостей. Эти закономерности определяются как силами
взаимодействия между молекулами пластификатора и полимера,
приводящими к энергетическим изменениям системы, так и силами
диффузии, выражающимися в изменении энтропии при смешении.
Растворимость полимеров тесно связана с гибкостью их цепей.
Гибкая молекула может расположиться в растворителе большим
числом способов, что, естественно, ведет к увеличению диффузии и
энтропии. С другой стороны, энергия связи жестких цепей друг
с другом очень велика и может быть выше связи цепей с молеку-
лами растворителя; в этом случае полимеры становятся нераствори-
мыми или ограниченно растворимыми даже в растворителях с та-
кой же полярностью, как и сам полимер. Такими мало раствори-
мыми полимерами являются целлюлоза, политетрафторэтилен,
полихлорвинил и др.
Однако пластификаторами не обязательно должны быть только
вещества, в которых полимер растворяется; достаточно, если поли-
мер лишь ограниченно набухает в пластификаторе. Чтобы пласти-
фикатор мог продиффундировать в полимер, необходимо, чтобы
энергия взаимодействия между молекулами полимера и пласти-
фикатора была близка к энергии взаимодействия между цепями
макромолекул полимера, т. е. степени полярности полимера и
пластификатора были бы примерно одинаковыми. Поэтому поляр-
ные пластификаторы легче совмещаются с полярными поли-
Пластификация полимеров
143
мерами, а неполярные пластификаторы — с неполярными поли-
мерами.
Пластификаторы должны иметь низкую температуру стеклова-
ния. Температура стеклования пластификатора обычно является
предельной Та полимера после максимальной степени пластифи-
кации.
Все пластификаторы можно разделить на два класса:
1) кристаллические или легко кристаллизующиеся и практически
не стеклующиеся;
2) легко стеклующиеся и практически не кристаллизующиеся.
Рис. 67. Изменение механических
свойств полимера в зависимости от
содержания пластификатора.
Содержание пластификатора,%
Рис. 68. Зависимость объемного эле-
ктрического сопротивления ре поли-
хлорвинила от содержания пласти-
фикатора (дибутилфталата).
К первому классу относятся эфиры линейных дикарбоновых
кислот (себацинаты, адипинаты и др.), ко второму — эфиры цикли-
ческих кислот (фталаты и др.).
Возможность применения кристаллизующихся пластификато-
ров основана на торможении процессов кристаллизации при взаимо-
действии молекул пластификатора с молекулами полимера.
Однако после определенной концентрации пластификатора, когда
все связи полимер — пластификатор уже насыщены, образуются
связи пластификатор — пластификатор, что приводит к образова-
нию кристаллов внутри аморфной массы полимера. В этом случае
с увеличением концентрации пластификатора увеличивается жест-
кость вещества и понижается его «морозостойкость», так как тем-
пература плавления кристаллов пластификатора обычно значи-
тельно выше температуры стеклования полимера.
Несмотря на то, что основная цель введения пластификаторов
заключается в снижении температуры стеклования и температуры
хрупкости полимера, определенное значение имеет также изменение
144
Гл. II. Физические основы технологии пластмасс
других свойств полимеров. На рис. 67 показано изменение некото-
рых механических свойств полимера введением пластификатора.
С увеличением содержания пластификатора статическая прочность
полимера (растяжение, сжатие) падает, тогда как прочность на
удар и способность к удлинению сильно увеличиваются; значи-
тельно снижается (по экспоненциальной кривой) также и модуль
упругости.
Введение пластификатора заметно снижает диэлектрические
свойства полимера (рис. 68). Это обусловлено двумя факторами —
увеличением подвижности диполей полимера в связи со снижением
температуры стеклования и внесением новых диполей, если приме-
няется полярный пластификатор.
Механическая пластификация (ориентация полимеров)
Для достижения большей прочности, гибкости и морозостойкости (снижения
Гхр) полимеров производят так называемую механическую пластификацию.
Практически ее осуществляют путем односторонней или двухсторонней вытяжки
полимера при температуре выше температуры стеклования и охлаждения в рас-
тянутом состоянии. Прн этом происходит ориентация и распрямление макромо-
Температура, °C
Рис. 69. Зависимость времени ре-
лаксации вытянутой полистироль-
иой пленки от температуры.
лекул; при охлаждении возникающая струк-
тура полимера фиксируется.
Для всех типов полимеров при механи-
ческой пластификации характерно увеличе-
ние прочности пропорционально степени вы-
тяжки. При этом односторонняя вытяжка
(вытяжка в одном направлении) увеличи-
вает прочность только в том направлении, в
каком она производится, и соответственно
снижает прочность в перпендикулярном на-
правлении. Двухсторонняя вытяжка увели-
чивает прочность в обоих направлениях,
однако при этом соответственно умень-
шается возможная степень вытяжки.
Вытяжка аморфных и кристаллических
полимеров приводит к различным структур-
ным изменениям и различным изменениям
свойств.
Вытяжка аморфных полимеров в боль-
шинстве случаев вызывает лишь разверты-
вание макроцепей и является процессом
высокоэластической деформации; существен-
ного перемещения макроцепей в целом при этом не происходит. Полимер пере-
ходит в неравновесное, напряженное состояние; устойчивость достигнутой степени
вытяжки будет определяться наличием больших времен релаксации (практически
бесконечных), что может иметь место лишь при температурах значительно ниже
температуры стеклования.
Этим объясняется, что механической пластификации могут подвергаться
только те технические аморфные полимеры, у которых температура стеклова-
ния лежит сравнительно высоко — не ниже 60—70°. Только у таких полимеров
время релаксации при обычных температурах будет практически бесконечным.
На рис. 69 показана зависимость времени релаксации ориентированной по-
листирольной пленки от температуры. Как видно из рисунка, при температуре
ниже +70° время релаксации стремится к бесконечности и состояние вытянутой
пленки может считаться практически устойчивым.
Механическая пластификация
145
Однако чем выше температура, при которой происходит процесс вытяжки и
деформации, тем значительнее процессы необратимой перегруппировки цепей на-
ряду с процессами высокоэластической деформации, т. е. тем значительнее об-
ласть равновесной ориентации по сравнению с областью неравновесной, высоко-
эластической деформации.
При изготовлении изделий из пластмасс методом литья под давлением,
методом прессования или выдавливания (экструзии) необходимо иметь в виду,
что в зависимости от температуры переработки форма, даваемая материалу
при этих процессах, может иметь или преимущественно равновесный, пластиче-
ский характер (при достаточно высоких температурах) или неравновесный,
высокоэластический характер (при недостаточна высоких температурах).
В отличие от обычной пластификации, механическая пластификация не
изменяет температуры стеклования полимеров, однако, значительно снижает
температуру хрупкости.
В табл. 13 приведены данные, характеризующие увеличение прочности и
гибкости полистирольиой пленки при вытяжке.
ТАБЛИЦА 13
Изменение механических свойств полистирольной пленки при вытяжке
Механические свойства | Нерастянутая пленка Растянутая плетка
Предел прочности на разрыв, «г;'с.ч2| 250—280 900—1000
Удлинение при разрыве, % .... i Число двойных перегибов до разру- 1 0—1 Ст нескольких сот до нескольких тысяч в за- висимости от толщины пленки
шеиия [ 0
Таким образом, вытяжка аморфных полимеров значительно увеличивает их
прочность, способность к удлинению и гибкость. При этом увеличивается
также их морозостойкость. Снижение температуры хрупкости происходит почти
пропорционально степени вытяжки; это дает возможность на основе ориентиро-
ванных полимеров получать морозоустойчивые оболочки, шланги, трубки, без
внесения пластификаторов, которые уменьшают статическую прочность и ухуд-
шают диэлектрические свойства полимеров.
Иной характер имеет вытяжка кристаллических полимеров. Она происходит
в температурном интервале, ограниченном температурой стеклования аморфной
фазы и температурой плавления кристаллической фазы полимера. Возможность
вытяжки кристаллических полимеров связана с наличием в них аморфной фазы.
При вытяжке кристаллических полимеров происходит выпрямление тех
частей макромолекул, которые проходят через аморфную область. Одновременно
беспорядочно расположенные кристаллиты разрушаются (плавятся) и обра-
зуются новые кристаллиты, расположенные параллельно плоскости растяжения.
Увеличение прочности и гибкости обусловливается как процессами ориентации
и распрямления макромолекул в аморфной области, так и (в особенности) про-
цессами ориентации кристаллитов. Наложение обоих типов процессов в значи-
тельно большей степени увеличивает прочность, чем это наблюдается при
деформации аморфных полимеров.
Характерным отличием кристаллических полимеров является их практиче-
ская устойчивость в ориентированном состоянии при температурах выше темпе-
ратуры стеклования аморфной фазы. Это объясняется закрепляющим действием
ю Зак. 1269. Э. И. Барг
146 Гл. II. Физические основы технологии пластмасс '
кристаллической решетки и значительным увеличением кристаллической фазы
при растяжении.
Чем больше в полимере содержание кристаллической фазы и чем выше
температура стеклования и температура плавления кристаллитов, тем устойчи-
вее будут ориентированные полимеры. Поэтому на практике не все кристалличе-
ские полимеры могут быть подвергнуты стабильной вытяжке, так как не все
дают в достаточной мере устойчивую ориентацию (к последним, например, отно-
сятся полиэтилен, полиизобутилен и Др.). Однако кристаллические полимеры
с высокой температурой стеклования и плавления и большим содержанием
кристаллической фазы дают практически устойчивую ориентацию в широком
интервале температур. Такие ориентированные кристаллические полимеры соста-
вляют класс синтетических и природных волокон.
Таким образом, ориентированное состояние кристаллических полимеров
характеризуется напряженным состоянием тех частей макромолекул, которые
проходят через аморфную область, и устойчивым, равновесным состоянием тех
их частей, которые закреплены в кристаллической решетке; практическая устой-
чивость полимера является результирующей величиной взаимодействия обеих
фаз.
ГЛАВА III
ТЕРМИНОЛОГИЯ и КЛАССИФИКАЦИЯ ПЛАСТМАСС
Большое значение, которое приобрели пластические массы во
всех областях техники и промышленности, естественно, вызвало не-
обходимость упорядочения их наименований, создания рациональ-
ной терминологии, а также научной классификации.
В капиталистических странах специфика частного предпринима-
тельства привела к безграничному, хаотическому росту числа фир-
менных названий пластмасс. В результате, часто одинаковые как
по химическому составу, так и по свойствам материалы имеют не-
сколько названий даже в одной и той же стране. В некоторых
капиталистических странах делались попытки создания рациональ-
ной, смысловой терминологии и научной классификации пластмасс,
однако они не увенчались успехом.
В 1937 г., на основе предложенного Э. И. Баргом проекта,
был утвержден советский стандарт «Классификация, терминология
и типизация пластмасс» — 2725/' явился не только пер-
вым советским, но и первым в мире стандартом в этой области.
В дальнейшем с развитием науки, техники и промышленности
полимеров и пластических масс некоторые положения этого совет-
ского стандарта оказалось необходимым дополнить или изменить.
В связи с этим Э. И. Баргом совместно с А. С. Файнштейном была
разработана новая классификация и терминология пластмасс, кото-
рая была принята и утверждена в 1951 г. новым Общесоюзным
стандартом: «Пластические массы органического происхождения.
Классификация, термины и обозначения» (ГОСТ 5752—51).
В соответствии с новым стандартом и будут ниже рассмотрены
основы советской терминологии и классификации пластмасс. Од-
нако в дальнейшем, наряду с новыми техническими терминами, бу-
дут приведены и те из старых, произвольных терминов, которые
получили широкое распространение в технической и научной лите-
ратуре.
ТЕРМИНОЛОГИЯ ПЛАСТМАСС
Терминология пластмасс может быть разделена на: 1) опреде-
ление ряда понятий, в том числе понятия «пластические массы», и
2) обозначение отдельных пластмасс.
10*
148
Гл. III. Терминология и классификация пластмасс
Определение понятия «пластические массы»
Пластическими массами могут называться все материалы, изго-
товляемые или перерабатываемые в изделия методами пластической
деформации (прессование, литье под давлением, вытяжка, штам-
повка и т. д.), которые, следовательно, обладают пластическими
свойствами в условиях переработки, но сами по себе в обычных
условиях, в виде готовых материалов или изделий, представляют
твердые, упругие тела, не обладающие при обычных (рабочих) на-
пряжениях и температурах пластическими свойствами.
В такой формулировке почти все известные нам конструкцион-
ные материалы могут относиться к пластическим массам. Однако
из понятия пластические массы легко выделить весь класс метал-
лов, имея в виду резкое отличие металлов от других материалов по
ряду важнейших физических и технических свойств (теплопровод-
ность, электропроводность и т. п.). В связи с этим мы будем назы-
вать пластическими массами все неметаллические материалы,
перерабатываемые, в основном, пластическими методами. Они
могут быть разделены на неорганические (минеральные) и органи-
ческие пластические массы.
Органические пластические массы, или, как их называют, карбо-
пласты объединяют весьма разнообразные по своей структуре и по
своим свойствам группы материалов. В двадцатых и тридцатых го-
дах текущего столетия органические пластические массы обычно
противопоставляли природному каучуку и по старому советскому
стандарту пластмассами назывались «органические материалы,
обладающие пластическими свойствами и не содержащие кау-
чука».
Огромный прогресс в изучении высокополимеров за последние
годы, синтез большого количества полимеров с каучуковыми свой-
ствами (синтетические каучуки), а также полимеров, обладающих
комплексом промежуточных свойств между каучуками и пласти-
ками, и принципиальное тождество химической структуры этих
полимеров сделали невозможным какое-либо принципиальное раз-
граничение понятий «пластмасса» и «каучук».
Таким образом, по современным воззрениям карбопласты пред-
ставляют собою единую в широком значении группу синтетических
и модифицированных природных полимеров, объединенных законо-
мерностями, связанными с макромолекулярным строением их моле-
кул, к которой относятся как пластмассы, так и каучуки.
Вместе с тем, по физическим свойствам карбопласты должны
быть разделены на пластики и эластики.
Пластики представляют собою твердые, упругие тела с высоким
модулем упругости (выше 104 кг/см2) и малой растяжимостью
(меньше 25%). Рабочие температурные условия пластиков лежат
всегда ниже температуры стеклования полимера, которая является
предельной температурой теплостойкости материала.
Обозначение отдельных пластмасс
149
Температуры ниже температуры стеклования, вплоть до самых
низких (включая температуру жидкого воздуха), являются об-
ластью рабочих температур пластиков.
Эластики представляют собою эластичные и мягкие материалы,
с малым модулем упругости (ниже 104 кг/см?) и высокой растяжи-
мостью (от 2,5 до 2000%). Температура стеклования эластиков
лежит в пределах обычных и низких температур. Они в большей
или меньшей степени обладают свойствами высокоэластической
деформации при обычмых температурах. Эластики «работают» при
температурах, лежащих всегда выше точки стеклования, которая
является предельной температурой морозостойкости. Чем ниже тем-
пература стеклования эластика, тем до более низких температур он
может «работать». Верхняя рабочая температура зависит от тем-
пературы пластического течения материала и для многих пластиков
недостаточно точно установлена.
Эластики, в свою очередь, разделяют на резина(каучука)пласты
и эластопласты.
Резинопласты представляют собою по комплексу свойств кау-
чуки. Они характеризуются модулем упругости менее 102 кг)см?,
высокой растяжимостью, большой скоростью релаксации при
весьма низких температурах (ниже —40°).
Эластопласты — эластики с более высоким модулем упругости
(> 102< Ю4 кг/с.и2), поддающиеся значительным деформациям при
обычных температурах и не ниже •—20°, причем значительная часть
деформации после снятия напряжения длительно сохраняется (при
нагреве остаточная часть деформации может полностью или боль-
шей частью исчезнуть).
Естественно, что многие материалы занимают промежуточное
положение между пластиками и эластиками и между эласто-
пластами и резинопластами, однако подавляющее большинство ма-
териалов может быть легко отнесено к одному из этих классов.
Круг органических материалов, который в технике относят
к пластическим массам и который лежит в основе промышленности
пластмасс, может быть представлен пластиками и частью эластиков,
а именно эластопластами. Такое объединение не основано на науч-
ных принципах, а подчинено сложившимся промышленным формам.
Обозначение отдельных пластмасс
Советская терминология пластмасс построена не на произволь-
ных, а на смысловых терминах.
Пластические массы прежде всего различаются по химическому
характеру связующего (смолы, полимера). Однако на основе од-
ного и того же связующего в зависимости от характера и струк-
туры наполнителя, а также технологий производства могут быть
получены различные материалы, весьма отличающиеся по ком-
плексу своих свойств.
150
Гл. III. Терминология и классификация пластмасс
Таким образом, для идентификации пластических материалов
необходимо, кроме характера связующего, указать на характер
и структуру наполнителя (если он применяется) и на метод
пластицирующего процесса. Поэтому советская терминология преду-
сматривает как обобщающие термины, дающие обозначение класса
пластмасс в зависимости от характера связующего, так и уточняю-
щие термины, определяющие характер конкретного пластика. Ниже
даны некоторые термины, принятые новым советским стандартом.
Наименование пластических масс, состоящих из связующего без
наполнителя или с наполнителем, складывается из обозначения
химического характера связующего, обозначения наполнителя (при
наличии его) с соответствующим окончанием, указывающим на
структуру и метод переработки пластических масс.
Обозначение химического характера связующего
Фено — продукты феноло-альдегидной конденсацнн.
Амино, меламина, анилина — соответствующие продукты амино-альдегидной
(карбамидной) конденсации.
Полиэфиро — общее обозначение продуктов конденсации многоосновных кис-
лот с многоатомными ецдртами.
Целло — общее обозначение для производных целлюлозы.
Нитро, ацето, этил, бензил и др. — для соответствующих эфиров целлюлозы.
Протвино, казеино, альбумина — белковые продукты.
Битумино, пеко, асфальта — битумные, пековые, асфальтовые продукты.
Вини(ло)—общее обозначение для продуктов полимеризации производных
этилена.
Хлорвини, хлорвинилидено, ацетовини — обзначение продуктов полимери-
зации хлорвинила, хлорвинилидена, винилацетата.
Стиро(ло), карбазовини — продукты полимеризации стирола и винилкарба-
зола.
Акри(ло), метакри(ло) — продукты полимеризации эфиров акриловой »
метакриловой кислот.
Ацеталь(вини)— общее обозначение для ацеталей поливинилового спирта.
Сили(ко)— продукты поликонденсации кремнийоргаиических соединений.
Амидо — продукты поликонденсации аминов с кислотами.
Обозначение наполнителя
Асбо — асбестовое волокно, асбестовая бумага и картон или асбестовая
ткань.
Стекло — стеклянное волокно или стеклянная ткань.
Текста — текстильный (тканевый) на основе целлюлозы.
Древо — опилки, древесная масса, древесный шпон.
Бумо — бумажный, на основе целлюлозы или древесной массы.
Целло — древесная целлюлоза.
Общее обозначение пластмасс
Общее обозначение отдельных групп пла^масс, отличающихся
химическим характером связующего, складывается из обозначения
химического состава связующего с окончанием — «пласт», вне за-
висимости от характера наполнителя, метода переработки, струк-
туры и свойств пластмасс.
Например:
Фенопласт — общее обозначение для пластмасс на основе феноло-альдегид-
ных смол, вне зависимости от наполнителя, переработки и т. д.
Обозначение отдельных пластмасс
151
Аминопласт — то же, на основе амино-формальдегидных смол.
Полиэфиропласт — то же, на основе смол, получаемых поликонденсацией
многоосновных кислот с многоатомными спиртами.
Целлопласт — то же, на основе эфиров и производных целлюлозы.
Винипласт — то же, на основе продуктов полимеризации производных
этилена.
Наименование пластмасс в зависимости от структуры и основного метода
переработки в изделия
Все пластмассы, перерабатываемые в изделия методом простого
литья, литья под давлением, прессованием, формованием из бесфор-
менной массы неслоистой структуры (пресспорошки, крошка и т. д.)
и состоящие из связующего без наполнителя или с наполнителем,
обозначаются составным наименованием, указывающим на характер
связующего вещества с окончанием — лит-, обозначение наполни-
теля ставится внутри слова, после обозначения связующего.
Например:
Фенолит — литые или прессованные материалы на основе феноло-альдегид-
ной смолы, не содержащие наполнителя.
Фенодреволит—то же с наполнителем древесная мука.
Феноасболит — то же с асбестовым наполнителем.
Аминолит 1 — пластмассы на основе аминопластов без
Аминоцеллолит > наполнителя или с различными наполни-
Меламинодреволит J телями.
Винилит — общее обозначение для прессматериалов и материалов, полу-
чаемых методом литья на основе виниловых полимеров (хлорвинилит, стиролит,
метакрилит и др.).
Целлолит — прессматериалы на основе эфиров целлюлозы.
Все слоистые материалы (слойматериалы), перерабатываемые
в изделия прессованием, формованием, намоткой и состоящие из
связующего и слоистого наполнителя, обозначаются наименованием
связующего и наполнителя с окончанием слой.
Например:
Фенотекстослой — слоистые материалы на основе ткани и феноло-альдегид-
ной смолы (соответственно: аминотекстослой, метакрилотекстослой).
Фенобумослой, аминобумослой— то же, на основе бумаги и соответствую-
щего связующего.
Твердые, гибкие, целлулоидоподобные материалы с удлинением
при разрыве от 5 до 25—30%, состоящие только из связующего
без наполнителя, получаемые пластикацией на вальцах или в ме-
шателе в присутствии пластификаторов и растворителей или без
них, с последующим выдавливанием (экструзией) или каландриро-
ванием и прессованием листов, или путем прессования блоков с по-
следующей строжкой и сушкой листов, или без сушки, изготовляе-
мые в виде пластин, плит, стержней, труб и т. д., обозначаются
наименованием химического вещества с окончанием — лоид.
152
Гл. Ш. Терминология и классификация пластмасс
Например:
Винилоид — общее обозначение для целлулоидоподобных материалов на
основе виниловых полимеров; хлорвинилоид—на основе полихлорвинила.
Нитро(целло)лоид— целлулоид — на основе нитроцеллюлозы.
Ацето(целло)лоид и т. д.—то же, на основе ацетилцеллюлозы.
Мягкие и растяжимые материалы (эластопласты), состоящие
только из связующего, без наполнителя, получаемые в виде пленок,
оболочек, шлангов и т. д. методами испарения растворителей,
коагуляции связующего, каландрирования или промежуточного
вальцевания, с последующей прессовкой, а также методом экструзии,
обозначают названием связующего с окончанием — эласт или ласт.
Например:
Виниласт — общее обозначение эластомеров на основе виниловых полимеров
[хлорвинил аст, акриласт, бути(раль)внниласт].
Целласт — то же, на основе производных целлюлозы.
Для специального обозначения пленочного материала, вне зави-
симости от степени мягкости и растяжимости пленки, после обозна-
чения связующего ставится окончание — пленка.
Материалы с малым удельным весом (0,03—0,3), имеющие по-
ристую, ячеистую структуру, получаемые из синтетических поли-
меров или из модифицированных природных полимеров, обозна-
чаются общим наименованием — поропласты.
Отдельные типы поропластов обозначаются наименованием
химического вещества с окончанием — поропласт или — пор.
Например:
Фенопоропласт (фенопор), аминопоропласт (аминопор), стиропоропласт (сти-
ропор) и т. д.
Синтетические и искусственные материалы в виде твердых или
жидких смол, водных дисперсий смол, органозолей, паст, различных
пластичных композиций с наполнителями, применяемые в качестве
клея для склейки различных конструкционных материалов, готовых
конструкций, а также для нанесения защитных покрывных эмалей
и для пропитки различных веществ, обозначаются общим наимено-
ванием пластоклей, а отдельные типы пластоклеев — наименова-
нием химического вещества с окончанием — клей.
Например:
Феноклей, меламиноклей и т. д.
Для формовочных асбестовых материалов сохраняются суще-
ствующие обозначения (фаолит, асбовинил).
Если связующее является сополимером, то к наименованию пла-
стика добавляется буква С (например стиролит-С).
КЛАССИФИКАЦИЯ ПЛАСТМАСС
Стремление к систематизации высокополимеров и пластмасс
диктуется как интересами промышленности, так и элементарными
требованиями развивающейся науки о синтетических органических
материалах.
Классификация пластмасс
153
Основные формы этой систематизации выражаются в класси-
фикации и типизации. Между обоими понятиями — классификация
и типизация — имеется существенное и принципиальное отличие:
классификация представляет логичное, основанное на научных или
технических принципах распределение полимеров и пластмасс на
их основе, типизация же — объединение в типы, группы материа-
лов, соответствующих строго определенному, гарантийному ком-
плексу свойств.
Классификация высокомолекулярных соединений может суще-
ственно отличаться от классификации технических полимеров, пла-
стических масс. Вместе с тем, часто не проводились разграниче-
ния классификаций, относящихся к полимерам, как химическим
соединениям, от классификаций технических материалов на их
основе.
Химическая классификация высокомолекулярных соединений
была предложена А. А. Ваншейдтом и В. В. Коршаком.
Они положили в основу классификации принцип атомарного
строения основной цепи полимера, с дальнейшим подразделением
по химическому характеру их боковых групп.
Так все полимеры делятся на карбоцепные, основная цепь кото-
рых состоит только из углеродных атомов
и гетероцепные, основная цепь которых содержит, наряду с углерод-
ными, также и другие атомы, например:
II II
—С—О—С или — С—N—С— .
Карбоцепные полимеры разделяются на насыщенные и ненасы-
щенные:
III 1111
—С—С—С— и —С—С=С—С—
Ill I I
В дальнейшем класс карбоцепных полимеров делится на группы
в зависимости от характера и количества замещающих групп
в основной цепи: алкил, арил, гидроксил, галоид и т. д.; например
группа моноалкилзамещенные
—СН2—СН—СН2— (где R —алкил)
R
или дигалоидозамещенные
X
|
—СН2—С—СН2—- (где X — галоид).
X
154
Гл. Ilf. Терминология и классификация пластмасс
Класс гетероцепных соединений делится на группы по характеру
гетероатома в основной цепи; например группа кислородсодержа-
щих полимеров:
—О—СН2— О—СН2—
или группа азотсодержащих полимеров:
—N—-СН2—N—СН2—
R R
В основу классификации пластических масс могут быть поло-
жены различные принципы: химическая структура полимеров
(смолы), химические процессы, ведущие к образованию полимера
(природные, полимеризационные, поликонденсационные пластики),
отношение к температуре (термопласты, реактопласты), характер
пластицирующего процесса и макроструктура полимера (литые
пластики, слоистые, формовочные и т. д.).
Так в основу классификации пластмасс, согласно старому со-
( СТ 27 \
ветскому стандарту (— 2725; ’ °ыл положен принцип ха-
рактера пластицирующего процесса. Все пластмассы были разде-
лены на семь классов: продукты литые, прессовочные, формовоч-
ные, слоистые, получаемые путем высыхания лиогелей и т. д.
В основу нового стандарта классификации пластмасс положена
химическая структура связующего (смолы), однако, при этом перво-
начальное деление на классы не исходит из атомарной структуры
.макроцепей полимеров. Это связано с тем, что атомарная структура
основной цепи макромолекулы не всегда определяет наиболее суще-
ственное различие в физических и технических свойствах соответ-
ствующих классов полимеров и, следовательно, пластмасс.
Так, целлюлоза и глифтали, относящиеся к одной группе гетеро-
цепных полимеров, содержащих кислород в основной цепи, значи-
тельно больше отличаются по комплексу свойств, чем фенопласты
и аминопласты, принадлежащие к двум различным классам: карбо-
цепных (фенопласты) и гетероцепных (аминопласты) полимеров.
Целесообразно и необходимо поэтому, чтобы правильный сам
по себе принцип систематизации пластмасс по. химической струк-
туре основной цепи и характеру побочных групп полимеров был
принят для подразделения внутри классов, объединяющих пластики
на основе более общих принципов, определяющих их важнейшие
физические и технические отличия. Для такой классификации, по
нашему мнению, должны быть учтены основные процессы, ведущие
к образованию полимера или пластика.
Исходя из этого, в новом стандарте технической классификации
пластмасс (ГОСТ 5752—51) все пластмассы предварительно разде-
лены на четыре класса:
Класс А. Пластические массы на основе высокомолекулярных
соединений, получаемых цепной полимеризацией.
Классификация пластмасс
155-
Класс Б. Пластические массы на основе высокомолекулярных
соединений, получаемых поликонденсацией и ступенчатой полиме-
ризацией.
Класс В. Пластические массы на основе химически модифици-
рованных природных полимеров.
Класс Г. Пластические массы на основе природных и нефтяных
асфальтов и смол, получаемых при пирогенетической деструкции
различных органических веществ.
В зависимости же от химической структуры связующего классы
делятся на группы и виды. Так, класс А составляют 9 групп,
в основу которых положен принцип деления полимеров по хими-
ческой структуре боковых цепей макромолекул. Группы, в свою
очередь, делятся на виды, в которых приведены конкретные типы
пластиков, а также их техническое обозначение.
В табл. 14 дана в сокращенном виде классификация пластмасс
в соответствии с ГОСТ 5752—51.
ТАБЛИЦА 14
Классификация пластических масс
I № группы | Группа № вида Вид
химическое наименование техническое наименование
Класс А. Пластические массы на основе высокомолекулярных соединений,,
получаемых цепной полимеризацией
1 Полимеры этилена (этиленопласт) 1 2 Полиэтилен То же Этиленолоид Этиленолит
2 Полимеры галоидо- 1 Поливинилхлорид Хлорвииилит
производных этилена (хлор- 2 То же Хлорвиииласт
винипласт, хлор- 3 V Хлорвинилоид
винилидено- пласт; тетра- фторвинипласт 4 Сополимеры хлорвинила с винилацетатом Хлорвинилит-С
и др.) 5 То же, с эфирами акрило- Хлорвинилит-С
вой и метакриловой ки-
слот
6 Поливинилиденхлорид Хлорвинилиденолит
7 Сополимеры хлорвинили-1 Хлорвинилиденолит-С
дена с хлористым винилом) Хлорвинилиденоласт-С
8 Политетрафторэтилен Тетрафторвинилит
9 Политрифторхлорэтилен Трифторхлорвинили-
3 Полимеры алкил- 1 Полиизобутилеи Изобутиленоласт
производных этилена (изобу- тилеиопласт)
156
Гл. III. Терминология и классификация пластмасс
Продолжение
з i Группа Вид
Мд S химическое наименование техническое наименование
4 5 •в 7 /8 '•0 Полимеры арил- этилеиов (стиро- пласт) Полимеры произ- водных винил- амина (винил- карбазопласт) Полимеры вини- лового спирта и их производные (этенолопласт, ацетальвини- пласт и др.) Полимеры винил- кетонов Полимеры эфиров этиленкарбоно- вых кислот (акрипласт, мет- акрипласт) Полимеры эфиров аллилового спирта с много- основными ки- 1 слотами (алли- ; лопласт) 1 2 3 4 1 2 1 2 3 4 (5 7 1 1 2 3 4 5 6 7 8 9 1 о Полистирол То же Полидихлорстирол Поливинилкарбазол Сополимеры винилкарба- зола и стирола Поливиниловый спирт Поливинилэтиловый эфир Поливинилацетат Формаль поливинилового спирта То же Ацеталь поливинилового спирта Бутираль поливинилового спирта Полиметилизопропенил- кетон Полиметилметакрилат То же Сополимеры метилмет- акрилата со стиролом Полибутилметакрилат Сополимеры бутилмет- акрилата с эфирами акри- ловой кислоты Полиметилакрилат Сополимеры метилакри- лата с эфирами мет- акриловой кислоты Полиаллилметакрилат Сополимеры аллилмет- акрилата п метилмет- акрилата Полидиаллилсебацин ат Полидиаллилфталат СтиролИТ Стиропленка Стиропор Дихлорстиролит Винилкарбазовинилит Винилкарбазовини- лит-С Этенолоид Винипластоклей Ацетовииипленка Формальвиниплеика Формальвииилит Этанальвинилит Бутиральвииипленка Бутиральвиниклей Пропенилвинилит Метакрилит Метакритекстослой Метакрилит-С Метакрипленка Метакриласт Метакриласт-С Акрипленка Акритекстослой Акриласт-С Метакрилит Метакрилит-С Аллплтекстослой
Классификация пластмасс
15Г
Продолжение
3 g Г руппа CS К Вид
S м
g химическое наименование техническое наименование
Класс Б. Пластические массы на основе высокомолекулярных соединений,
получаемых поликонденсацией и ступенчатой полимеризацией
1 Пластические массы на основе феноло-альдегид- ных смол (фено-' пласт) 1 2 3 4 5 6 7 8 9 10 11 Феноло-формальдегидные смолы Феноло-, крезоло-, ксиле- ноло-формальдегидные смолы Феноло-фурфурольные смолы i Феноло-анилино-формаль- дегидные смолы Феноло-лигниновые смолы То же в V в в в » . Фенолит Фенодреволит Феноцеллолит Феноасболит Фаолит Фенотекстослой Фенобумослой Феноасбослой Фенодревослой Феностеклослой Фенопор
2 Пластические массы на основе резорцино-форм- альдегидных смол (резорци- нопласт) 1 2 3 Резорцино-формальдегид- ные смолы То же 1, » Резорциноасболи г Резорциностеклослой Резорцинопластоклей
3 4 Пластические массы на основе амидо- и амино- формальдегид- ных смол (амино- пласт, меламино- пласт, анилино- пласт) Пластические массы на основе полиэфиров 1 2 3 4 5 6 7 8 9 1 2 3 Мочевино-формальдегид- ные смолы То же В в V 9 Меламино-формальдегид- ные смолы То же Анилино-формальдегидные смолы То же Глицерофталат То же в » Аминоцеллолит Аминолит Аминотекстослой Аминобумослой Аминопор Меламинодреволит Меламиностеклослой Анилинолит Анилинобумослой Г лифтальслюдослой Глифтальасболит Глифтальстеклослой
5 Пластические массы на основе полиамидов (амидопласт) 1 Смолы на основе гексаме- тилендиамина и адипино- вой кислоты; гексамети- лендиамина и себацино- вой кислоты; поликапро- лактама и др. i Амидолит Амидолоид 1
1-58
Гл. 1П. Терминология и классификация пластмасс
Продолжение
№ групп | Группа № вида 1 Вид
химическое наименование техническое наименование
6 Пластические массы на основе полиуретанов и полимочевТш (уретанопласт) 1 2 Смолы на основе гексаме- тилендиизоцианата и бу- тандиола Смолы на основе гекса- ме тиле ндиизоциа на та и глицерина; гексаметилен- диизоцианата и диами- нов и т. д. Уретаполит Уретанолоид
7 Пластические массы на основе кремнийоргани- ческих полиме- ров (снлипласт) 1 Смолы на основе продуктов гидролиза диалкилди- хлорсиланов, моноалкил- трихлорсиланов в смеси с триалкилмонохлорси- ланами и т. д. Силиласт Силиасболит Силислюдолит Силислюдослой
Класс В. Пластические массы на основе природных химически-модифицирован-
ных полимеров
1 Пластические 1 Метилцеллюлоза Этилцеллюлоза Метилцеллопленка
массы на основе 2 Этилцеллолит
простых эфиров 3 То же Этилцеллолоид
целлюлозы (цел- 4 Этилцеллопленка
лопласт) 5 Бензилцеллюлоза Бензилцеллолит
? Пластические I Нитроцеллюлоза Нитроцеллолоид
массы на основе 2 Нитроцеллюлоза Нитроцеллопленка
сложных эфи- 3 То же Нитроцеллолит
ров целлюлозы 4 Ацетилцеллюлоза Ацетоцеллолоид
(целлопласт) 5 То же Ацетоцеллолит
6 Ацетоцеллопленка
7 Ацетобутиратцеллюлоза Ацетобутиратцеллолоид
3 Пластические 1 На основе казеина (молоч- Протеинолоид
массы на основе ного, растительного)
белковых ве- 2 На основе альбумина Протеинолит
ществ (протеино- пласт)
Класс Г. Пластические массы на основе природных и нефтяных асфальтов,
а также смол, получаемых деструкцией различных органических веществ
1 Пластические 1
массы на основе
природных и 2
нефтяных ас-
фальтов и раз- 3
личных пеков
(битуминопласт)
Сплавы асфальтов с асфаль-
титами
Сплавы пеков с асфаль-
тами
Сплавы на основе асфаль-
тов и высыхающих масел
Битуминоцеллолит
Пекоасбослой
Битуминолит
Часть II
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ СИНТЕТИЧЕСКИХ СМОЛ,
ПОЛУЧАЕМЫХ ЦЕПНОЙ ПОЛИМЕРИЗАЦИЕЙ
ГЛАВА IV
ПРОЦЕССЫ ПОЛИМЕРИЗАЦИИ
ТЕХНИЧЕСКИЕ МЕТОДЫ ПОЛИМЕРИЗАЦИИ
Синтетические смолы, получаемые методом цепной полимериза-
ции, промышленное значение приобрели значительно позже, чем
поликонденсационные. Многие из важнейших полимеризационных
смол были синтезированы всего 5—10 лет тому назад.
Развитие науки и техники в области полимеризационных смол в
настоящее время происходит столь интенсивно, что почти каждый
год появляются новые полимеризационные пластики.
В современной технике, в частности в электротехнике, в технике
высоких частот и в приборостроении полимеризационные пластики
получили выдающееся значение.
Производство полимеризационных смол складывается из двух
процессов: 1) синтеза мономеров, индивидуальных ненасыщенных
веществ, способных к полимеризации, и 2) полимеризации мономе-
ров с образованием полимеров — синтетических смол.
В некоторых случаях на заводах пластмасс осуществляют оба
процесса; иногда же мономеры производят на других заводах, —
тогда процесс получения синтетической смолы сводится лишь к про-
цессу полимеризации.
Современная техника полимеризации предъявляет большие тре-
бования к чистоте исходного сырья — мономеров. Наличие даже
незначительных примесей отражается как на кинетике процесса
полимеризации, так и на физических свойствах и структуре полиме-
ров. Чистота мономера приобретает особое значение, если полимер
предназначен для применения в качестве диэлектрика.
В зависимости от характера и назначения полимеров техниче-
ские условия на исходное сырье обычно предусматривают содержа-
ние мономера от 98,5 до 99,99%.
Мономеры часто содержат ингибиторы — вещества, добавляемые
для предупреждения преждевременной полимеризации мономеров
при хранении; эти вещества перед полимеризацией должны быть
удалены промывкой или дестилляцией мономеров.
В качестве ингибиторов применяют фенолы (гидрохинон, пиро-
галлол), соли меди, сернистые соединения и др. Действие этих ве-
ществ (стр. 42) основано главным образом на восстановлении
образующихся в мономере перекисей, которые, распадаясь, превра-
щаются в радикалы — центры роста цепи.
160
Гл. IV. Процессы полимеризации
Возможность образования в мономерах перекисей обусловлена
действием атмосферного кислорода, который способствует ини-
циированию процесса полимеризации. Однако часто кислород мо-
жет проявить себя и как ингибитор, так как он стимулирует взаимо-
действие растущих радикалов, ведущее к обрыву цепи и получению
неактивных молекул. Учет влияния атмосферного кислорода имеет
исключительно большое значение, так как многие процессы поли-
меризации могут привести к технически годным продуктам лишь
при тщательно контролируемых дозах кислорода.
Из всех известных методов полимеризации главное применение
в технике получили методы, основанные на внесении инициаторов
и катализаторов, а также методы чисто термической полимериза-
ции. Другие способы инициирования процессов полимеризации
(действие света, электрических разрядов, ультразвука и др.) не
нашли пока значительного технического применения.
Техническая полимеризация в присутствии инициаторов обычно
происходит в жидкой фазе, т. е. в среде жидкого мономера или
в растворителях и эмульсиях как при атмосферном, так и при вы-
соких давлениях (в автоклавах, если мономеры при температурах,
полимеризации — газы).
Полимеризация в газовой фазе имеет небольшое техническое
применение, лишь иногда ее используют для получения низкомоле-
кулярных продуктов.
Разнообразные технические приемы полимеризации ненасыщен-
ных соединений могут быть разделены на следующие три основных
метода:
1) полимеризация чистого мономера в жидкой фазе (блочный
метод полимеризации);
2) полимеризация в растворителях;
3) полимеризация в водных эмульсиях.
Эти методы будут подробно рассматриваться применительно
к отдельным производствам полимеров; здесь же приведены лишь
основные закономерности каждого метода.
Полимеризация чистого мономера может быть осуществлена пу-
тем заливки смеси мономера с инициатором (или чистого моно-
мера) в формы; иногда одновременно добавляют пластификатор и
краситель. Залитые формы нагревают при строго регулируемой
температуре, обычно в термошкафах. В качестве инициатора при-
меняют чаще всего перекись бензоила. Количество инициатора и
температура определяют степень полимеризации полимера и кинетику
процесса. Если требуется, в полимеризующуюся смесь вносят регу-
ляторы (меркаптаны и другие вещества), которые позволяют полу-
чать полимеры в более узких пределах заданных молекулярных весов.
По окончании полимеризации твердый полимер может иметь
форму блока, пластины, стержня, трубки, а также форму неслож-
ных заготовок для дальнейшей механической обработки в готовые
изделия.
Технические методы полимеризации
161
Основным затруднением, с которым приходится встречаться при
блочной полимеризации, является трудность отвода значительного
количества тепла, образующегося в процессе экзотермической реак-
ции. Вследствие плохой теплопроводности блоков происходят мест-
ные перегревы, в результате чего скорость конверсии и степень
полимеризации в различных местах блока неодинаковы (меньшая
степень полимеризации в середине блока, большая — в поверхност-
ных слоях). Естественно, что
эти факторы определяют и
различие физико-механиче-
ских свойств блока в раз-
личных точках сечения и воз-
никновение в нем внутрен-
них натяжений. Чем толще
блок и чем больше его
размеры, тем труднее по-
лучить технически годный
образец, без вздутий и тре-
щин.
При изготовлении мето-
дом блочной полимеризации
пластин, труб, стержней,
различных заготовок и про-
филей необходимо иметь
ТАБЛИЦА 15
Удельные веса мономеров и полимеров,
расчетная и наблюдаемая усадки
некоторых полимеров
Мономер Уд. вес. моно- мера Уд. вес поли- мера Расчетная усадка, Наблюдаемая усадка, °(о
Стирол 0,907 1,06 14,7 14,5
Метилметакрилат 0,940 1,19 20,1 21,2
Винилацетат . . . 0,939 1,19 20,t 20,9
Акрилонитрил . . 0,797 1,17 31,0 31,0
Диаллилфталат . . 1,12 1,27 11,8 11,8
Винилкарбазол 1,11 1,20 6,9 7,5
в виду, что в процессе полимеризации значительно увеличивается
удельный вес вещества, что приводит к соответствующему измене-
нию объема и определяет величину усадки. Чем больше усадка,
тем больше в изделиях остаточных внутренних напряжений, веду-
щих к появлению, со временем, трещин, тем меньше точность от-
ливки изделия.
Процент усадки можно рассчитать, исходя из соотношения
удельных весов мономера и полимера. Получаемые при этом дан-
ные в большинстве случаев хорошо совпадают с наблюдаемой усад-
кой (табл. 15).
Значительным достижением явились непрерывные методы поли-
меризации чистого мономера, так называемая непрерывная блочная
полимеризация. Она осуществляется в башнях, состоящих из не-
скольких секций. Каждая секция башни имеет самостоятельный
обогрев и постоянную температуру. Мономер обычно поступает для
предварительной полимеризации в кубы, откуда частично полиме-
ризованный жидкий продукт непрерывно подается в верхнюю сек-
цию башни и по мере полимеризации проходит ряд секций. Гото-
вый полимер в расплавленном состоянии выдавливается в виде
ленты или стержня из нижней секции башни. Подробнее эти схемы
будут рассмотрены в дальнейшем. Преимущество непрерывной по-
лимеризации чистого мономера по сравнению с периодической по-
лимеризацией в блоках заключается как в большей производитель-
11 Зак. 1269. Э. И. Барг
162
Гл. IV. Процессы полимеризации
мости, автоматизации процесса и улучшении условий труда, так и
в большей стандартности получаемого продукта, в более легкой
возможности регулировки молекулярного веса полимера.
Однако непрерывная полимеризация, естественно, не может
быть применена в тех случаях, когда конечный продукт (полимер)
должен иметь форму блоков, листов и различшых заготовок, полу-
чаемых простым литьем в формах, без прессования.
Полимеризация в растворителях может быть проведена по
двум вариантам.
По первому варианту применяют растворитель, в котором рас-
творимы как мономер, так и образующийся полимер. В этом слу-
чае конечный продукт представляет собою раствор полимера в рас-
творителе (лак) и может быть применен непосредственно в качестве
лака или полимер может быть выделен путем отгонки растворителя,
осаждением водой или какой-либо другой жидкостью, в которой он
нерастворим.
По второму варианту полимеризацию проводят в таком раство-
рителе, в котором растворим только мономер. В этом случае обра-
зующийся полимер непрерывно осаждается из раствора в виде тон-
кой суспензии и может быть отделен центрифугированием, филь-
трацией с последующими промывкой и сушкой'
Кроме мономера и растворителя реакционная смесь содержит
обычно растворимый в мономере инициатор, иногда регулятор сте-
пени полимеризации и, если необходимо, пластификатор.
Механизм полимеризации в растворителях определяется:
1) взаимодействием растущих цепей (радикалов) с молекулами
растворителя, которое ведет к реакциям передачи цепи и, следова-
тельно, к уменьшению среднего молекулярного веса полимера,
2) снижением скорости реакции вследствие уменьшения концен-
трации мономера. Реакции передачи цепи связаны с химической
структурой растворителя; кинетика процесса и средний молекуляр-
ный вес полимера зависят таким образом от структуры и концен-
трации растворителя.
Иногда для регулирования среднего молекулярного веса поли-
мера применяют смесь растворителей.
Полимеризацию в растворителях обычно проводят при темпера-
туре кипения смеси, при энергичном перемешивании в течение
сравнительно длительного времени (обычно 12—36 часов).
Благодаря равномерному распределению температуры, а также
процессам «передачи цепей» образуются полимеры с меньшей по-
лидисперсностью, чем при блочной полимеризации.
В частности, полимеризация по первому варианту (полимер рас-
творим в растворителе) приводит к меньшей полидисперсности, чем
по второму, когда образующийся с значительно более высоким мо-
лекулярным весом полимер осаждается из растворителя.
Степень полимеризации зависит от температуры, количества
инициатора и, кроме того, ог характера растворителя и концентра-
Технические методы полимеризации
163
ции мономера в смеси. Чем выше концентрация мономера, тем
выше и степень полимеризации полимера. Влияние характера рас-
творителя на степень полимеризации и на кинетику процесса
подробно будет рассмотрено при описании процессов получения
отдельных полимеров.
Полимеризация в водных эмульсиях является наиболее распро-
страненным, а для некоторых полимеров почти единственным тех-
ническим методом и имеет ряд существенных преимуществ перед
другими методами. Полимеризация по этому методу протекает
весьма быстро, полимеры получаются с большим молекулярным
весом, обычно в виде тонкодисперсного порошка, а также в
виде стойкой водной суспензии (эмульсии), так называемого ла-
текса.
Метод эмульсионной полимеризации основан на диспергирова-
нии мономера в воде в присутствии эмульгаторов. Роль эмульга-
тора заключается в понижении поверхностного натяжения на гра-
нице капля мономера — вода; последнее существенно для облегче-
ния диспергирования и для образования прочных адсорбционных
слоев вокруг частицы мономера.
Реакционные смеси обычно состоят из сравнительно боль-
шого числа компонентов: жидкого (или сжиженного) мономера,
воды, эмульгатора, инициатора, регуляторов (pH среды, по-
верхностного натяжения, степени полимеризации и разветвленности
полимера).
Регуляторами pH среды служат вещества с буферными свой-
ствами (цинкацетат и др.); применение их необходимо потому, что
pH непосредственно влияет на скорость реакции, так как водорас-
творимые перекиси с повышением pH разлагаются, кроме того, ве-
личина pH значительно влияет на стабилыность эмульсии.
Регуляторами поверхностного натяжения обычно служат одно-
атомные алифатические спирты (аллиловый, гексиловый), добавле-
нием которых можно уменьшить поверхностное натяжение и таким
образом регулировать размер капель мономера в эмульсии.
Регуляторами степени полимеризации являются различные мер-
каптаны, четыреххлористый углерод и другие вещества, ускоряющие
реакции передачи цепей.
Кинетика процесса и степень полимеризации образующегося
полимера при эмульсионной полимеризации определяются в основ-
ном температурой, количеством инициатора, количеством и характе-
ром эмульгатора, а также скоростью и способом механического
перемешивания. От последних зависит дисперсность системы и до
известной степени ее стабильность и скорость реакции, которая
определяется также и величиной диспергированных частиц.
При полимеризации в эмульсиях обычно применяют температуру
несколько более низкую, чем температура кипения воды (часто
60—70°), а в некоторых случаях — близкую к температуре кипения
(90—95°) или к температуре замерзания воды (5—10°).
н*
164
Гл. IV. Процессы полимеризации
Углеводородная фаза
tj 8 8 Поверхность
§ § раздела
водная фаза
Рис. 70. Строение адсорб-
ционного слоя на поверхно-
сти раздела двух жидкостей.
Содержание мономера в системе может варьировать в широких
пределах. Обычно применяют концентрации от 30 до 60% мономера.
Продолжительность полимеризации в эмульсиях значительно
меньше, чем в растворителях, а именно от 1,5 до 6 часов.
В отличие от растворов, эмульсии и суспензии в термодинами-
ческом отношении не являются равновесными системами и имеют
тенденцию к самопроизвольному разрушению — к слиянию диспер-
гированных частиц. Основные факторы, определяющие стабильность
системы, — характер и количество эмульгатора. Механизм стаби-
лизации эмульсии заключается в образовании (в зависимости от
характера эмульгатора) моно- или полимолекулярных защитных
адсорбционных слоев поверхностно
активных веществ вокруг диспергиро-
ванных частиц мономера.
Наиболее существенное отличие
эмульсионной полимеризации — зави-
симость скорости и степени полимери-
зации от характера и количества эмуль-
гатора.
Эмульгаторы, применяемые для
эмульсионной полимеризации, могут
быть разделены на три группы.
Первая группа включает коллоид-
ные поверхностно-активные вещества,
молекулы которых состоят из длинных
углеводородных цепей, имеющих на конце полярную, гидрофиль-
ную группу. К этим веществам относятся: мыла — соли жирных
кислот, соли органических сульфокислот и др. Их молекулы своими
гидрофобными (углеводородными) концами ориентируются пер-
пендикулярно к поверхности капель мономера, а полярными груп-
пами — к дисперсионной среде — воде, образуя «частокол» из мо-
лекул вокруг капель мономера. Именно такой «частокол» из моле-
кул эмульгатора предохраняет частицы мономера от слипания
(рис. 70). Эмульгаторы этой группы даже при малых концентра-
циях (~0,1%) образуют прочные адсорбционные слои, обладаю-
щие, как показал акад. Ребиндер, гелеобразной структурой и
значительной упругостью. Эти слои выдерживают обычные на-
пряжения при диспергировании, если, конечно, сила, действующая
при столкновениях частиц, не превышает предельного напряжения
сдвига.
Эмульгаторы первой группы применяют в присутствии водо-
растворимых инициаторов для получения дисперсий типа латекса.
Вторая группа эмульгаторов — высокомолекулярные, водорас-
творимые вещества, например, поливиниловый спирт, полиакрило-
вая кислота и др. Их применяют главным образом для получения
грубодисперсных систем в больших концентрациях (3—5%), в при-
сутствии инициатора, растворимого в мономере.
Технические методы полимеризации
165
Третья группа — твердые эмульгаторы — представляет высоко-
дисперсные гидрофильные порошки, частицы которых вследствие
избирательного смачивания прилипают к поверхности раздела ка-
пель со стороны дисперсионной среды и создают твердую брони-
рующую оболочку. К эмульгаторам этой группы относятся дисперс-
ные глины, тальк, окислы металлов (алюминия, магния). Эти
вещества, так же как и высокомолекулярные эмульгаторы, приме-
няют для получения крупных капель (0,1—5 мм).
Различают два основных типа эмульсионной полимеризации:
1) с применением водорастворимых инициаторов и 2) с примене-
нием инициаторов, нерастворимых в воде, но растворимых в моно-
мере; такую полимеризацию называют бисерной, или капельной.
В качестве инициаторов эмульсионной полимеризации первого
типа применяют водорастворимые перекиси, обычно перекись водо-
рода, которая легко распадается на радикалы ОН'. Наряду с пере-
кисью водорода широкое применение нашли надсернистые соли и
пербораты, которые служат как инициаторами, так и (полностью
или частично) эмульгаторами.
Важнейшими эмульгаторами для эмульсионной полимеризации
первого типа являются соли жирных кислот (мыла) и соответ-
ствующие им по структуре синтетические моющие вещества и сма-
чиватели.
Мыла относятся к коллоидным электролитам, — они могут рас-
творяться в воде молекулярно или коллоидно, образуя мицеллы.
Молекулярная растворимость мыл в воде очень незначительна; так,
для пальмитата натрия при 60° она равна 30 - 10~* мол/л-, при
избытке мыла образуется коллоидный, мицеллярный раствор. Обра-
зование мицелл и.их структура, а также критическая концентрация
мыла, ведущая к образованию мицелл в процессе эмульсионной
полимеризации, имеют существенное значение. Опытным путем
была установлена значительно большая растворимость мономеров
в растворах мыла по сравнению с их растворимостью в чистой воде,
а также особое расположение молекул мономеров в мицеллах мыла
(стр. 172). В этом дополнительное значение применения мыл при
эмульсионной полимеризации.
Как уже было указано, характерными особенностями эмульсион-
ной полимеризации являются большая скорость реакций и образо-
вание полимеров с высокими молекулярными весами, а также поли-
меров в тонкодисперсном состоянии. Было установлено, что вели-
чина частиц, образующихся при эмульсионной полимеризации,
в десятки и сотни раз меньше диаметра капель соответствующих
мономеров в эмульсии.
В результате полимеризации жидкая дисперсная фаза (мономер)
переходит в твердую (полимер), которая, адсорбируя мыло, обра-
зует стойкую суспензию. Однако, если образующийся полимер при
этом находится в высокоэластическом состоянии, его нельзя при
столь высокой дисперсности полностью отнести к твердым телам,
166
Гл. IV. Процессы полимеризации
тем более, что часто такие дисперсные частицы представляют ча-
стицы полимера, окруженные молекулярным слоем эмульгатора.
В этом случае и образуется латекс — дисперсный полимер в высо-
коэластическом состоянии, который по своему характеру напоми-
нает скорее эмульсию, чем суспензию. Получаемая таким образом
дисперсия может, как уже было указано, найти самостоятельное
применение, например, для пропитки тканей, кожи, для внесения в
состав бумажной массы и т. д. Она имеет ряд преимуществ перед
лаковым раствором — малую вязкость и высокую концентрацию по-
лимера, приготовление ее не требует расхода дорогих растворите-
лей, обычно огнеопасных и токсичных.
В некоторых случаях в состав реакционной смеси вносят пла-
стификатор, в котором дисперсные частицы твердого полимера на-
бухают и вся система также приобретает консистенцию латекса.
При получении жестких полимеров с высокой температурой
стеклования чаще всего прибегают к осаждению суспензии. Оса-
ждение производят путем внесения электролитов, снижающих pH
среды и разрушающих адсорбированные слои дисперсной фазы.
После коагуляции суспензию фильтруют и тщательно отмывают от
эмульгаторов и электролитов.
Недостатком эмульсионного метода полимеризации является
трудность полного удаления электролитов после осаждения; послед-
нее в ряде случаев (например при применении того или иного поли-
мера в качестве диэлектрика) имеет исключительно большое зна-
чение.
Второй тип эмульсионной полимеризации, так называемая
бисерная, или капельная полимеризация, получил техническое при-
менение для изготовления ряда полимеров. Как было уже ука-
зано (стр. 165), принципиальное отличие этого типа полимери-
зации — применение инициаторов, растворимых в мономере, но
нерастворимых в воде. Инициатор, обычно перекись бензоила, рас-
творяют в мономере, после чего полученный раствор диспергируют
в воде.
При бисерной полимеризации применяют не мыла, а эмульга-
торы второй и третьей групп (стр. 164, 165), т. е. такие высокополи-
мерные гидрофильные коллоиды, как полиакриловая кислота, по-
ливиниловый спирт, причем в относительно больших концентрациях
(3—5%). В зависимости от концентрации и условий перемешивания
эти эмульгаторы способны создавать и предохранять от коагуляции
сравнительно большие капли мономера (0,1—-5 мм).
Процесс капельной полимеризации протекает под действием
инициатора внутри крупных капель мономера. Получаемый про-
дукт легко и самопроизвольно осаждается, без внесения электро-
литов. Отмывка высокомолекулярного эмульгатора происходит
сравнительно легко вследствие малой удельной поверхности круп-
ных гранул и слабых сил адсорбции.
Бисерная полимеризация протекает в основном по закономер-
Технические меп оды полимеризации
167
ностям блочной полимеризации. Каждая капля в этом случае пред-
ставляет как бы «микроблок»; при этом «микроблоки» находятся
в условиях постоянной температуры и хорошего отвода тепла. Зна-
чительно большая скорость бисерной полимеризации по сравнению
с блочной заставляет предполагать, что значение имеют также реак-
ции, протекающие в поверхностных слоях, на границе мономер —
эмульгатор. Чем выше дисперсность мономера, тем в большей мере
бисерная полимеризация по своему характеру будет сближаться
с обычной эмульсионной полимеризацией.
Техника эмульсионной полимеризации получила свое дальней-
шее развитие с применением так называемого окислительно-восста-
новительного цикла.
При полимеризации под влиянием окислительно-восстановитель-
ных систем, кроме обычных инициаторов перекисного типа (перекись
водорода, перекись бензоила и др.), добавляют восстановители.
Действие восстановителей сводится к увеличению скорости ини-
циирования, что приводит к значительному увеличению скорости
реакции (выхода полимера).
Увеличение скорости реакции введением восстановителей при-
обретает особое значение в тех случаях, когда условия получения
полимера с высокими свойствами требуют проведения процесса
полимеризации при низких температурах. Так, например, известно,
что полимеризация бутадиена или сополимеризация бутадиена со
стиролом при низких температурах (4-10° и ниже) приводят пре-
имущественно к реакциям типа 1,4 (стр. 58) и к получению поли-
меров с более высокими техническими свойствами. Реакция типа
1,2, ведущая к получению полимеров с более низкими свойствами,
при этом подавляется. Однако снижение температуры полимериза-
ции, благоприятствующее получению полимеров с более высокими
свойствами, значительно уменьшает скорость инициирования и, сле-
довательно, увеличивает время полимеризации. В этом случае при-
менение окислительно-восстановительного метода эмульсионной
полимеризации во много раз увеличивает скорость реакции и обес-
печивает возможность проведения процесса при низких темпера-
турах.
Активирующее действие восстановителей в процессах радикаль-
ной полимеризации было показано Е. М. Чилкиной и С. С. Медве-
девым еще в 1939 г. на примере реакций, протекающих под
влиянием гидроперекисей в присутствии ароматических аминокис-
лот. В дальнейшем было предложено большое количество такого
рода ускоряющих полимеризацию восстановителей, среди которых
особое значение приобрели соли металлов с переменной валент-
ностью (Fe11, Си1 и др.), а также многие органические и неоргани-
ческие восстановители (глюкоза, левулеза, сорбоза и другие сахара,
сульфит, гидросульфит и т. п.). Часто применяют одновременно
два различных восстановителя, например сахар и соль закиси
железа.
168
Гл. IV. Процессы полимеризации
Реакции, инициированные под влиянием окислительно-восстано-
вительных систем, заключаются в том, что при взаимодействии
перекиси с восстановителем образуются радикалы. Так, например,
в системе, состоящей из перекиси водорода, мономера и ионов
двухвалентного железа, имеют место следующие реакции:
H»O.,4-Fe + + —> Fe + + +-f-Oir-f-ОК-
ОН' -f- Л4 (мономер) —> ОНЛГ
онлг+м —> онл<2 —> о нм;,
OH’4-Fe + + —► OH-4-Fe+ + +
онм;, 4-м- —> OHMn+1
Так как образование радикалов ОН’ сопровождается образо-
ванием равного количества ионов ОН-, этот процесс может идти
лишь в полярных средах и эффективнее всего — в водной среде, при
эмульсионной полимеризации.
Процессы, протекающие по окислительно-восстановительному
механизму между мономером, органической перекисью, железным
и органическим восстановителями, можно выразить подобной же
схемой:
(RCOO)24-Fe + + —► RCOO'4-RCCO-4-Fe*- + + (1)
RCCO’4-Fe’-+ —> RCOO-4-Fe^ + + (2)
RCOO’ 4- M —► RCCOM’ (3)
RCUOM’4-M —► RCOOMg —► RCOOM’„ (4)
RCOOM;, 4- (RCCO)2 —► RCOOMn4- CO2 4- RCOO’ (5)
Fe + + +
органический „ , ,
—--------------> Fer +
восстановитель
(6)
Реакция (1) представляет образование из перекиси при ее
взаимодействии с ионом закисного железа одного свободного ради-
кала и одного иона кислоты, который связывается в виде соли.
Реакция (2) ведет к разложению образовавшегося радикала ионом
закисного железа — с образованием иона кислоты, который так же
может быть связан в виде соли. Реакция (2) привела бы к полному
восстановлению перекиси железом, если бы этому не препятство-
вали другие реакции. Так, свободный радикал может взаимодей-
ствовать с мономером, образуя радикалы растущей цепи по реак-
циям (3) и (4); при этом радикалы растущей цепи уже не спо-
собны к восстановлению по типу реакции (2), так как они не могут
образовать стабильных солей с железом. Реакция (5) подобна
реакциям передачи цепи и ведет к образованию неактивного поли-
мера и нового радикала, т. е. перекись при полимеризации может
играть роль не только инициатора, но и регулятора процесса.
Реакция (6) выражает действие присутствующих органических вос-
становителей на ионы окисного железа, которые переходят при этом
вновь в ионы закисного железа.
Технические методы, полимеризации
169
Таким образом, наличие в системе ионов закисного железа ведет
к образованию свободных радикалов и к ускорению реакции поли-’
меризации; однако очевидно, что для успешного течения этого
процесса необходимо ограничить возможность реакции (2); огра-
ничение может быть достигнуто регулированием во время протека-
ния процесса концентрации ионов закисного железа по отношению
к концентрации мономера, например, вводя их в малых количествах.
Тогда реакция (3) будет преобладать над реакцией (2).
Процесс эмульсионной полимеризации по этой схеме идет
таким образом, что ионы Fe++, растворенные в углеводородной фазе
в виде стеарата железа, ускоряют разложение перекиси бензоила
на свободные радикалы; при этом они окисляются до ионов Fe+++ и
переходят в водную фазу, где добавленной к воде солью пирофос-
форной кислоты связываются в пирофосфатный комплекс.
Затем по реакции (6) образующиеся ионы окиси железа Fe+4+
восстанавливаются растворимыми в воде органическими восстано-
вителями (например сорбозой) и переходят уже в виде Fe++ в угле-
водородную фазу, где снова вступают в реакцию. Этим заканчи-
вается цикл окислительно-восстановительного процесса.
Этот процесс, однако, имеет циклический характер лишь в при-
сутствии восстановителей как перекиси, так и ионов Fe444', а также
и в присутствии таких эмульгаторов, которые с ионами закисного
железа дают соединения, растворимые в углеводородной фазе. Если
стеарат натрия заменить другим эмульгатором, не дающим с же-
лезом соединения, растворимого в углеводородной фазе, то цепная
реакция нарушается.
В ряде случаев полимеризация в окислительно-восстановитель-
ных системах протекает с отрицательным температурным коэффи-
циентом, т. е. выход полимера увеличивается с понижением тем-
пературы. Это объясняется тем, что при высоких температурах
происходит быстрый расход перекиси, после чего процесс прекра-
щается; в результате выход полимера снижается.
Таким образом, окислительно-восстановительные системы при
эмульсионной полимеризации позволяют значительно снизить тем-
пературу процесса без снижения скорости полимеризации.
Окислительно-восстановительную полимеризацию проводят в ще-
лочной среде (pH 10).
В качестве примера можно привести нижеследующий состав
реакционной смеси (в частях) при сополимеризации бутадиена со
•стиролом по окислительно-восстановительному методу:
Мономеров...................................20
Воды.......................................4<)
Перекиси бензоила ....................... <1,1
Железо-аммиачных квасцов (Ретт) 0,1
Пирофосфата натрия (регулятор pH)......... 0.1
Стеарата натрия (эмульгатор)................ I
Сорбозы ..................................
170
Гл. IV. Процессы, полимеризации
ТЕОРИЯ ЭМУЛЬСИОННОЙ ПОЛИМЕРИЗАЦИИ
Несмотря на то, что техника эмульсионной полимеризации
нашла- очень большое применение, теория и механизм этого метола
до последнего времени не получили достаточного развития.
Основным вопросом теории эмульсионной полимеризации
является вопрос о местах реакции в эмульсионной системе и о роли
каждого места этой системы. Существенное отличие эмульсионной!
полимеризации — нахождение инициатора не в мономере, а в вод-
ной среде. Соответственно возникает необходимость выяснения, где
протекают реакции образования радикалов, роста и обрыва цепи.
Теоретически мыслимы следующие места реакции: 1) в капле
мономера, 2) в водной среде (в молекулярном растворе мономера
в воде), 3) в мицеллах эмульгатора и 4) на поверхности раздела
капля мономера — вода.
Согласно первоначальным воззрениям, развитым еще в конце
тридцатых годов машего столетия, реакция протекает в водной
среде. Растворенные в воде молекулы мономера (имеющиеся
в воде даже при практически нерастворимом мономере) взаимодей-
ствуют с молекулами инициатора, образуя зародыши полимериза-
ции (радикалы), которые растут за счет реакции с другими моле-
кулами мономера. По мере образования нерастворимого в воде
полимера в раствор переходят новые молекулы мономера, и равно-
весное состояние в растворе похдерживается до тех пор, пока не
будут исчерпаны молекулы мономера в каплях, которые выполняют,
следовательно, роль резервуара мономера. Реакция инициирования
вследствие больших размеров в эмульсионных системах поверх-
ности раздела может также проходить на границе раздела в погра-
ничных адсорбционных слоях, куда могут диффундировать моле-
кулы мономера.
В современных теориях эмульсионной полимеризации учиты-
вается возможность протекания реакции как в водном слое, так и
в пограничных слоях, однако в них особое значение придается роли
эмульгатора. Современные представления основаны на ряде эмпи-
рически установленных закономерностей, а именно:
1. Мономер, как правило, значительно больше растворим в вод-
ных растворах мыла, чем в воде.
2. Скорость реакции и степень полимеризации зависят от ха-
рактера и количества эмульгатора.
3. Скорость полимеризации зависит от размеров диспергирован-
ных капель мономера. Чем выше степеш, диспергирования, тем
больше скорость полимеризации (рис. 71).
4. Размер частиц образующегося полимера значительно меньше,
чем размер капель диспергированного мономера (средний радиус
капель в синтетических латексах 0,5—5 и, а средний размер частиц
полимера 0.01—0,5 ,ч).
Теория эмульсионной полимеризации
5. В процессе полимеризации происходит закономерное умень-
шение среднего радиуса частиц осаждаемого полимера.
Чем выше первоначальный радиус эмульгированных капель, тем
значительнее уменьшение частиц со временем. На рис. 72 предста-
Рис. 71. Скорость полимеризации
стирола в эмульсиях с различным
начальным размером частиц (;л).
Рис. 72. Уменьшение среднего размера
частиц с увеличением времени эмульси-
онной полимеризации для трех образцов
стирола с различным начальным ра-
диусом капель.
влево уменьшение среднего размера частиц полимера в зависимост.:
от времени полимеризации и от исходных размеров частиц дисперс-
ной фазы (мономера).
6. Было эксперименталыно показано, что мономер диффундирует
из капель в мицеллы мыла и к частицам полимера, а также
непосредственно к частицам полимера, если мицеллы мыла отсут-
ствуют.
7. Было также показано, что в процессе полимеризации кол-
лоидно-растворенное (мицеллярное) мыло постепенно исчезает н
адсорбируется на поверхности образующегося полимера благо-
даря более высокой степени дисперсности и большей удельной
поверхности полимера по сравнению с поверхностью капелек
мономера.
Все перечисленные закономерности, естественно, не могут быть
объяснены на основе только старых представлений, согласно кото-
рым роль эмульгатора ни в какой мере не связана с кинетикой г.
механизмом полимеризации. Из приведенных экспериментальных
данных, естественно, вытекает, что механизм эмульсионной поли-
меризации неразрывно связан, наряду с другими факторами, с
механизмом действия эмульгатора. В этом, собственно говоря, за-
Ц72
Гл. IV. Процессы полимеризации
ключается основной прогресс развитии представлений о механизме
эмульсионной полимеризации.
Действие эмульгатора
образовывать
Вода—
Рис. 73. Попе-
речное сечение
пластинчатой
мицеллы. Рас-
стояние d не-
сколько больше
двоенной дли-
ны молекулы
мыла.
(мыла) объясняется его способностью
мицеллы после достижения критической
концентрации, которая крайне низка
и колеблется в пределах 10-4—
10-2 молей/л.
Повидимому, существуют два вида
мицелл: 1) круглые, ионные, и 2) пла-
стинчатые, нейтральные или слабо за-
ряженные. Пластинчатые мицеллы со-
стоят из двойного слоя молекул мыла
и являются более упорядоченной и ори-
ентированной системой по сравнению
с ионными, круглыми мицеллами.
Экспериментально присутствие пла-
стинчатых мицелл обнаружено по
интерференционным кольцам на рент-
генограммах растворов мыл. Рентге-
новские измерения, однако, показали,
что расстояние d, отвечающее повто-
ряющейся структурной единице, боль-
ше, чем удвоенная длина молекулы
мыла, и что это может быть объяснено
нахождением между полярными груп-
пами слоя воды (рис. 73).
При эмульгировании мономера про-
исходят два процесса: равновесное рас-
творение мономера в мицеллах мыла
(образование мицеллярного раствора)
и образование эмульсий (эмульсион-
ное диспергирование). Реакционная
таким образом из двух видов дисперсных частиц:
в растворе
— Вода—
О о о о
Мономер
6
~ Воде
Рис. 74. По-
перечное се-
чение мицел-
лы мыла
с включен-
ным в нее
мономером.
смесь состоит
мицелл коллоидного электролита, содержащих мономер, и капель
эмульсии, стабилизированных эмульгатором.
Сущность мицеллярного растворения состоит во включении мо-
лекул мономера внутрь мицелл мыла между неполярными концами
его молекул (рис. 74).
Было установлено, что расстояние между слоями молекул мыла
в мицеллах значительно увеличивается с повышением концентрации
мономера в системе и что для ненасыщенных растворов существует
зависимость
Ml = км,
где: с/ — расстояние между слоями молекул в мицеллах; М — кон-
центрация мономера (углеводорода); К. — константа, зависящая от
характера мономера.
Теория эмульсионной полимеризации
173
Коллоидная (мицеллярнзя) растворимость различных мономе*
ров в мылах в зависимости от их характера может достигать зна-
чительных размеров; так, в 3% растворе олеата натрия раство-
ряются 6,8% стирола, 9% изопрена и 17,8% нитрила акриловой
кислоты. Чем больше полярность мономера, тем выше его коллоид-
ная растворимость.
Кроме распределения мономера внутри мицелл мыла и в отдель-
ных стабилизованных капельках, имеет место также диффузия
мономера из капелек эмульсии в мицеллы
мыла. Скорость диффузии увеличивается с
уменьшением размера капель, с повышением
температуры среды и концентрации мыла.
Таким образом, основная масса мономера
в эмульсионной системе находится в виде
большого количества стабилизированных капе-
лек диаметром 1 —10 ц; из этих капелек моно-
мер диффундирует в мицеллы мыла, внутри ко-
торых он распределяется, образуя слои толщи-
ной в несколько десятков онгстрем. Общая
схема такой мицеллы представлена на рис. 75.
А. П. Юрженко считает, что полимеризация
первоначально протекает в пластинчатых ми-
целлах эмульгатора, в которые по мере хода
реакции диффундирует мономер из диспер-
гированных и стабилизированных капелек.
Образующиеся молекулы полимера, достигая
определенной величины, становятся уже не-
растворимыми в мицеллах мыла; поэтому они
переходят из мицелл в водную фазу. К этим
молекулам диффундирует мономер из капель.
Такие полимерно-мономерные частицы являют-
ся местами дальнейшего роста цепи и образо-
вания основной массы полимера.
Представления А. П. Юрженко о диффузии
мономера из капель в мицеллы мыла, а затем
Вода
-----Мономер----
—-----Вода-------
Рис. 75. Общая схема
пластинчатой мицеллы
эмульгатора, содер-
жащей мономер.
к молекулам полимера, извергаемым после достижения известного
предела роста цепи из мицелл в воду, — весьма существенно. Они
позволяют объяснить почти все наблюдаемые при эмульсионной
полимеризации закономерности, в частности значительную разницу
между величиной частиц полимера и размером капелек, а также
зависимость скорости полимеризации от концентрации и ха-
рактера эмульгатора и др. Таким образом, очевидно, что в каплях
эмульсии не происходит инициирования и что капли эти следует
рассматривать лишь как хранилища мономера. Инициирование
в известных условиях может происходить в водной среде, при
этом роль его возрастает по мере адсорбции мыла на поверх-
ности полимера. Однако при достаточной концентрации мыла
74
Гл. IV. Процессы полимеризации
основная масса полимера образуется в мономерно-полимерных си-
стемах.
Позднейшие исследования П. М. Хомиковского позволили уточ-
нить некоторые вопросы, связанные с выяснением места протекания
реакции полимеризации. Было установлено, что наблюдаемое раз-
шчное влияние мыл на скорости полимеризации отдельных мономе-
ров связано со способностью полимеров набухать или растворяться
Рис. 76. Скорость полимеризации метилметакри-
лата в воде (/) и в растворе мыла (2) при 40°
с 0,17% персульфата калия.
з своих мономерах. При полимеризации полярного мономера (на-
пример винилцианида, в котором образующийся полимер нераство-
рим и не набухает) он не диффундирует к полимеру и реакция
: мономерно-полимерных слоях не протекает. Оказалось, что
в этом случае реакция в молекулярном водном растворе мономера
идет быстрее, чем в коллоидном растворе эмульгатора. Можно
считать, что при полимеризации таких мономеров инициирование
происходит в водной среде в молекулярном растворе мономера и
что там же происходят все реакции роста и обрыва цепи, ведущие
к получению полимера.
Частицы полярных полимеров, не набухающих в мономерах,
плохо адсорбируют эмульгатор и легко образуют крупные агрегаты.
Однако при полимеризации мономера, в котором полимер раство-
рим (например при полимеризации метилметакрилата), иницииро-
вание происходит в мицеллах эмульгатора и механизм процесса
связан с диффузией мономера из капелек в мицеллы и к растущим
цепям (радикалам) полимера; иначе говоря, кинетика процесса
в этом случае определяется реакциями, протекающими в моно-
мерно-полимерных слоях и, следовательно, зависит от концентрации
и характера эмульгатора (рис. 76).
ГЛАВА I'
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИМЕРОВ
НЕПРЕДЕЛЬНЫХ УГЛЕВОДОРОДОВ
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИЭТИЛЕНА
(Этиленопласты)
Полимер этилена — полиэтилен — представляет собою высоко-
молекулярный парафин ...—СН2—СН2—СН2—.... в основном нор-
мального, линейного строения. По структуре цепи—эго наиболее
простой полимер, производными которого являются полимеры дру-
гих классов. Так, почти все полимеры на основе моновинильных
соединений можно рассматривать как производные полиэтилена,
в котором один или несколько атомов водорода замещены другими
атомами или группами.
Полиэтилен — сравнительно новый пластик; он был синтезиро-
ван в 1939 г. и получил техническое применение лишь в годы вто-
рой мировой войны.
Исходным веществом для получения полиэтилена служит эти-
лен— газ с т. плавл.— 169°, т. кип.— 103,8° и уд. в. 0,570 (при
темп. кип.). Он является дешевым и доступным сырьем, получаемым
главным образом при пиролизе и крекинге нефти.
Наряду с ацетиленом, этилен широко.применяют для получения
ряда важнейших мономеров (хлорвинила, стирола, хлорвинилидена,
акриловых эфиров и др.) однако, несмотря на сравнительную до-
ступность и дешевизну сырья, на пути непосредственного получения
из этилена высокомолекулярного продукта возникли большие труд-
ности; последние объясняются главным образом значительной
инертностью этилена в раскрытии двойной связи. Как уже было
показано (стр. 40), симметричное строение и отсутствие дипольных
групп в моновинильных соединениях определяет их малую склон-
ность к полимеризации.
Получение полиэтилена
Полимеры этилена были впервые получены еще в 1884 г. рус-
ским ученым Г. Г. Густавсоном при полимеризации этилена в при-
сутствии бромистого алюминия. Подобные же, в основном низко-
молекулярные парафины, были позднее получены по реакции
Е. И. Орлова при каталитическом гидрировании окиси углерода,
а также в ряде других реакций.
В настоящее время из этилена могут быть получены продукты
различного молекулярного веса и различной консистенции (от бен-
зинов и смазочных масел до твердых парафинов); образование их
176
Гл. V. Пластмассы на основе непредельных углеводородов
зависит от процессов, ведущих к ступенчатой или цепной полиме-
ризации. Эти процессы, в свою очередь, определяются условиями
полимеризации, характером катализатора (инициатора), давлением
и температурой.
Чтобы направить реакцию в сторону получения высокомолеку-
лярных твердых продуктов (технического полиэтилена), т. е. в сто-
рону цепной реакции полимеризации, необходимо применять высо-
кие давления и проводить реакцию в присутствии инициаторов
(кислорода, перекиси бензоила и др.).
Ускорение процессов полимеризации и получение более высоко-
молекулярных продуктов при применении высокого давления на-
блюдается во многих реакциях полимеризации. Особенно же на-
глядно влияние высокого давления при полимеризации этилена. При
нормальных давлениях в присутствии катализаторов этилен поли-
меризуется в жидкие углеводороды. Так, при полимеризации эти-
лена в присутствии кобальта, осажденного на активированном угле
при 200°, образуется в основном бутилен. При более высоких тем-
пературах (350—500°) в присутствии катализаторов образуются
жидкие углеводороды с молекулярным весом до 200. Повышение
давления до 30—50 ат при применении катализаторов типа хлори-
стого алюминия или фтористого бора приводит к получению поли-
меров с молекулярным весом до 1000, которые могут быть
использованы как , высококачественные смазочные масла. При-
менение давления до 300 ат дает возможность получать парафино-
подобные продукты с молекулярным весом 2000—3000. Однако
лишь при давлениях порядка 1200—2500 ат характер реакции по-
лимеризации резко меняется. Она протекает при этом по цеп-
ному механизму с большой скоростью и приводит к получению
твердого, высокомолекулярного, технически ценного продукта —
полиэтилена.
Технический полиэтилен получается двух типов: сравнительно
низкомолекулярный (мол. вес 2000—3000) и высокомолекулярный
(мол. вес 15 000—35 000).
Низкомолекулярный полиэтилен, известный в Германии под на-
званием луполен N, получается полимеризацией этилена в авто-
клавах под давлением 200—300 ат. Полимеризацию проводят в
растворителе (метаноле); инициатором процесса полимеризации
служит перекись бензоила. Перед началом процесса путем вакууми-
рования из системы тщательно удаляют воздух (атмосферный кис-
лород). Получающийся полимер нерастворим в метаноле и оса-
ждается по мере образования.
Высокомолекулярный полиэтилен, известный в Германии под
маркой луполен Н, а в Англии как политен, получается полимериза-
цией этилена при давлениях 1500—2500 ат и при температурах на
300—100° ниже температуры взрыва, в присутствии строго контро-
лируемых количеств кислорода, являющегося инициатором. Полиме-
ризацию ведут без растворителей, так как присутствие растворите-
Получение полиэтилена
177
лей приводит к обрыву реакции полимеризации и к образованию
низкомолекулярных продуктов.
Наибольшие трудности в проведении процесса, протекающего
при высоких давлениях и температурах, заключаются в его техни-
ческом, аппаратурном оформлении. Полимеризация может проис-
ходить в автоклавах из нержавеющей стали, снабженных при-
способлением для перемешивания массы или во вращающихся
автоклавах, в которых перемешивание производится шарами. Для
непрерывных процессов применяется трубчатка с непрерывной пода-
чей под давлением этилена и с непрерывным выходом полиэтилена.
Технический этилен в зависимости от происхождения и метода
синтеза содержит различные примеси и может быть получен раз-
личной концентрации.
Для целей полимеризации он должен быть высококонцентриро-
ван и тщательно очищен от примесей, в особенности от ацетилена.
Так, в Германии поступавший на полимеризацию газ имел сле-
дующий состав (в %): этилена 98; этана 1—2; азота 0,5—1 и аце-
тилена 0,1—0,3. Перед поступлением на полимеризацию к нему до-
бавляют строго нормированное количество кислорода (0,05—0,1%).
Схема процесса получения полиэтилена по непрерывному методу
представлена на рис. 77.
Рис. 77. Схема процесса получения полиэтилена.
/—насос; 2— газовый счетчик; 3—компрессор на 300 к?(см*\ 4, 6— смазкоотделители;
5—компрессор на 2500 л-г/сл3; 7—реактор; 8—тцисмник высокого давления; 9— при-
емник низкого давления; 10— ловушка полиэтилена; 11—скруббер для ^промывки
этилена; 12— сборник с раствором щелочи.
Готовая газовая смесь из газгольдера с помощью насоса 1 подается через
газовый счетчик 2 в компрессор 3. Из компрессора газ, сжатый до 300 кг/см2,
12 Зак. 1269. Э. И. Варг
178
Гл. V. Пластмассы на основе непредельных углеводородов
через смазкоотделитель 4 поступает в дополнительный компрессор 5, где сжатие
доводится до рабочих давлений (1200—2500 кг/см"). Под этим давлением через
смазкоотделитель 6 газ поступает в трубчатый реактор 7.
Реактор представляет собою змеевик, первая часть которого состоит из
труб с внутренним диаметром в 10 мм, а вторая — из труб с диаметром
16—24 мм. Трубы первой части снабжены рубашкой для подогрева газа, в кото-
рой циркулирует вода, нагретая на 200°; трубы второй части имеют рубашку,
в которой циркулирует вода, нагретая на 100°; с помощью последней проис-
ходит отвод избытка тепла, образующегося при полимеризации. В первой части
змеевика происходит в основном лишь подогрев газа до 180—200°, во второй —
основной экзотермический процесс полимеризации. Из реактора полимер вместе
с непрореагировавшим этиленом через редукционный вентиль перепускается
в приемник высокого давления 8, а из последнего — в приемник 9, где давление
снижается до атмосферного и этилен отводится через ловушку 10 на промывку
в скруббер 11. Полиэтилен из приемника 9 вытекает в формы, в которых и за-
стывает.
В газ перед его поступлением в скруббер вводят дополнительное количе-
ство кислорода, так как в процессе полимеризации кислород расходуется. После
промывки щелочным раствором, подаваемом в скруббер из сборника 12 (для
удаления альдегидов), газ возвращается в газгольдер и снова поступает на
компримирование. За один цикл процент превращения составляет 8—15%, сум-
марное превращение 93—98%.
Молекулярный вес образующегося полиэтилена определяется
условиями полимеризации; при понижении температуры, уменьше-
нии содержания кислорода и увеличении давления молекулярный
вес полиэтилена повышается; однако соответственно уменьшается и
процент превращения.
Структура полиэтилена
Молекулы полиэтилена имеют плоскую зигзагообразную струк-
туру обычной парафиновой цепи. На концах полиэтиленовой цепи
имеются двойные связи, наличие которых доказывается спектром
поглощения, а также реакцией присоединения иода. Для молекул
характерна линейная неразветвленная структура с весьма редкими
боковыми метильными группами; при полимеризации они обра-
зуются по схеме:
сн2=сн., + сн, + сн,=сн2 —> ...—СН.,—СН,-СН—СН.—СН—.
II I
СН., сн3
Наличие метильных групп в полиэтилене было доказано и коли-
чественно определено путем спектрографического анализа (инфра-
красная спектроскопия). В соответствующих условиях полимериза-
ции были получены продукты, содержащие одну метильную группу
на каждые 8, 15, 40, 200 и 400 углеродных атомов. В технических
полиэтиленах одна метильная группа приходится на 15—25 угле-
родных атомов. Кроме боковых метильных групп в цепи полиэти-
лена содержится незначительное число атомов кислорода. Цепи
полиэтилена в зависимости от условий полимеризации могут иметь
большую или меньшую степень разветвленности.
В обычных температурных условиях полиэтилен состоит при-
мерно на 75% из кристаллической фазы и на 25% из аморфной.
Свойства полиэтилена
179
.Панные рентгенографического анализа показывают, что элемен-
тарная ячейка кристаллита полиэтилена имеет орторомбическое
строение и содержит ’четыре СН2-группы; период идентичности
вдоль цепи 2,534 А; ближайшее расстояние между атомами раз-
О о
личных цепей 4,3 А; размеры кристаллической ячейки: а — 7,4 А,
Ь — 4,93 А, с — 2,534 А (период идентичности).
Свойства полиэтилена
Полиэтилен представляет собою твердый, желтовато-белый ро-
говидный продукт и является одним из самых легких полимеров
(уд. вес 0,92—0,95).
Физические свойства полиэтилена определяются его химической
структурой. Линейный характер макромолекулы, ее высокая сте-
пень симметричности обусловливают весьма малые (меньше, чем
в каком-либо другом полимере) межмолекулярные силы, высокую
гибкость цепи и высокую подвижность звеньев. Поэтому в аморф-
ном состоянии полиэтилен имеет весьма низкую температуру стекло-
вания ~ —80°, ниже, чем у какого-либо другого «каучука». Если бы
его аморфное состояние было устойчивым, он был бы самым морозо-
стойким каучуком.
Однако аморфное состояние полиэтилена является крайне не-
устойчивым и он легко и быстро кристаллизуется (хотя никогда не
содержит 100 % кристаллической фазы). Способность полиэтилена
к кристаллизации, несмотря на малую величину межмолекулярных
сил притяжения, объясняется высокой симметричностью его макро-
цепи. Естественно, что участки цепей, между которыми действуют
силы кристаллической решетки, уже не способны к вращению, они
жестко скреплены и составляют основной каркас полимера. Участки
цепей, образующие неупорядоченную (аморфную) часть полимера,
сохраняют при этом способность к вращательным движеньям
звеньев, т. е. к высокоэластической деформации.
Наличие аморфной фазы, температура стеклования которой
весьма низка, позволяет полиэтилену сохранять известную степень
гибкости и эластичности до весьма низких температур, т. е. придает
ему высокую морозостойкость (до —80°). С другой стороны, значи-
тельное содержание кристаллической фазы, температура плавления
которой находится в пределе ПО—115°, определяет его жесткость
(по сравнению с «каучуками»), характеризующуюся более высоким
модулем эластичности, весьма малой скоростью релаксации и прак-
тической теплостойкостью до ~80. Полиэтилен, следовательно, —
типичный эластопласт.
Сочетание жесткости и твердости при обычных температурах с
высокой морозостойкостью является самым ценным и важным в
характеристике механических свойств полиэтилена.
Физические свойства полиэтилена зависят от степени его полиме-
ризации. На рис. 78 показана зависимость температуры плавления
12*
180
Гл. V. Пластмассы на основе непредельных углеводородов
от молекулярного веса полимера. Низкомолекулярный полиэтилен
(мол. вес 2000—3000) имеет температуру плавления 95—105е
и представляет собою воскоподобное вещество, легкорастворимое
на холоду в парафиновых и ароматических углеводородах. Высоко-
молекулярный полиэтилен (мол. вес 15 000—35 000) имеет темпе-
ратуру плавления ПО—115°; он нерастворим на холоду почти ни
в одном из известных растворителей и лишь при высоких темпера-
турах (+80° и выше) за-
М олекулярный Sec t тысячи
Рис. 78. Молекулярный вес и температура
метно растворяется в че-
тыреххлористом углероде,
трихлорэтилене, толуоле,
бензоле и т. п.
Предел прочности на
разрыв полиэтилена зави-
сит от его молекулярного
веса (от 100 кг/см2 для
низкомолекулярного до
150—200 кг/см2 для вы-
сокомолекулярного поли-
мера).
Так же, как и все кри-
сталлические полимеры,
полиэтилен можно под-
вергнуть холодной вытяж-
ке при температуре ниже
плавления технических полиэтиленов. температуры плавления
кристаллитов. Она про-
исходит, повидимому, за счет аморфной фазы, находящейся в вы-
сокоэластическом состоянии. В процессе растяжения образуются
новые ориентированные кристаллиты вдоль оси вытяжки.
Растяжение полиэтилена увеличивает его прочность примерно в
десять раз (до 2500 кг/см2). Кристаллическая структура и высокая
прочность ориентированных нитей из полиэтилена должны были
привести к широкому его использованию в качестве синтетического
волокна, однако в этой области он нашел сравнительно ограничен-
ное применение (см. ниже), главным образом из-за недостаточно
высокой температуры плавления (110°), весьма низких температур
стеклования и резкого снижения концентрации кристаллической
фазы с температурой. Как видно из рис. 79, еще значительно ниже
температуры плавления (выше 60°) наблюдается резкое уменьше-
ние кристаллической фазы и, следовательно, увеличение аморфной;
это, естественно, должно усиливать процессы релаксации вытяну-
того и ориентированного полимера.
Как все кристаллические полимеры, полиэтилен плавится в
узком интервале (3—5°), однако уже при температуре на 15—20°
ниже температуры плавления концентрация кристаллической фазы
полиэтилена настолько уменьшается, что его можно подвергать
Свойства полиэтилена
181
вытяжке, формовке и некоторым другим пластицирующим процес-
сам. Выше температуры плавления полиэтилен переходит в пласти-
ческую, но не жидкую массу, которую можно перерабатывать ме-
тодом экструзии, вальцевания, литья под давлением и т. д. Воз-
можность применения этих методов переработки связана с сравни-
Рис. 79. Зависимость содержания
кристаллической фазы от темпе-
ратуры (в полиэтилене).
Рис. 80. Зависимость вязкости
технического полиэтилена (7) и
парафинов (2) от температуры.
тельно высокой вязкостью расплавленного этилена. На рис. 80
показана сравнительная вязкость технических полиэтиленов и обыч-
ных парафинов в зависимости от тем-
пературы.
Нетрудно заметить, что и выше
температуры плавления высокомоле-
кулярные полиэтилены имеют вяз-
кость в 106—Ю7 раз более высокую,
чем расплавленные парафины.
Перевод полиэтилена в аморфное
состояние можно осуществить быст-
рым охлаждением расплава. Аморф-
ный полимер мягок, каучукоподобен
и при обычных температурах имеет
вязкость примерно того же порядка,
что и при температуре плавления.
Переход его в кристаллическое со-
стояние происходит непрерывно, но
равновесное соотношение фаз насту-
Рис. 81. Зависимость уд. веса по-
лиэтилена от температуры.
пает очень медленно. Этот переход связан с увеличением плотности.
На рис. 81 показана зависимость удельного веса полиэтилена от тем-
пературы и, косвенно, — от содержания в нем кристаллической фазы.
Увеличение плотности с изменением фазовой структуры приводит
182
Гл. V. Пластмассы на основе непредельных углеводородов
к значительной усадке полиэтилена, которая во много раз выше,
чем усадка аморфных пластиков, и составляет~ 16%. Большая
усадка, связанная в основном с процессами кристаллизации, заста-
вляет уделить особое внимание достижению равновесного соотноше-
ния фаз, так как продолжающийся процесс кристаллизации будет
сказываться на размерах изделия и может быть причиной сохране-
ния значительных внутренних натяжений, ведущих к появлению тре-
щин. Для достижения равновесного соотношения фаз изделие не-
обходимо медленно и постепенно охлаждать, погружая его в воду
с начальной температурой 4-80° и давая остыть до +20°.
Для уменьшения концентрации кристаллической фазы, увеличе-
ния растяжимости и повышения морозостойкости полиэтилена его
часто смешивают с полиизобутиленом и реже — с полибутадиеном.
В последнее время для этих же целей получают продукты совмест-
Рис. 82. Зависимость прочности полиэтилена на разрыв от темпе
ратуры.
Статическая прочность полиэтилена зависит от температуры
(рис. 82), причем прочность на разрыв (из расчета на первоначаль-
ное сечение) тем выше, чем ниже температура. Выше +60° прочность
полиэтилена уже примерно вдвое ниже, чем при обычной темпера-
туре (4-20°), когда удлинение при разрыве составляет от 250 до
500%. Температурный интервал эксплуатации для деталей, несущих
нагрузку, от 4-60 до —80°.
Полиэтилен относится к неполярным полимерам, его дипольный
момент равен нулю. Вследствие весьма высокой степени электри-
ческой симметрии он обладает высокими диэлектрическими свой-
ствами и превосходит в этом отношении полярные диэлектрики.
Диэлектрическая постоянная полиэтилена обусловливается только
электронной и атомной поляризацией; она имеет ту же величину, что
и диэлектрическая постоянная алифатических насыщенных углево-
дородов. Вследствие ничтожной электропроводности полиэтилена
его диэлектрические потери должны быть весьма малы (tg 5 ~
—0,0002—0,0004). Диэлектрическая постоянная и тангенс угла диэлек-
трических потерь практически не зависят от частоты поля (вплоть до
Свойства полиэтилена
183
109 гц) и в значительной мере от температуры. Однако необходимо
учитывать, что в техническом полиэтилене посторонние включения
и продукты окисления снижают его диэлектрические показатели.
Полиэтилен является лучшим диэлектриком для высокочастот-
ной техники, отличаясь в интервале от —80 до -4-60° наименьшей
изменяемостью диэлектрических свойств от температуры.
При нагревании без доступа воздуха полиэтилен устойчив до 290°.
Деструкция полиэтилена наступает при более высоких темпера-
турах, причем образуются жидкие маслянистые, а также газообраз-
ные продукты, но мономер (этилен) — не образуется.
При нагревании полиэтилена в присутствии воздуха уже при
120° наступает его окисление, которое постепенно ведет к попереч-
ной «сшивке» макромолекул с образованием пространственных,
полностью нерастворимых полимеров. В процессе окисления в по-
лиэтилене образуются различные группы, предположительно гидро-
ксильные и кетонные, а также увеличивается количество ненасы-
щенных связей.
При термической обработке полиэтилена (вальцевание, экстру-
зия и т. п.) также происходят деструкция и окисление, ведущие к
снижению механических и, в особенности, диэлектрических свойств
полиэтилена. Ниже показана зависимость тангенса угла диэлектри-
ческих потерь полиэтилена от времени вальцевания при 160°:
tgo
до вальцевания .... 30-10 ° после 200 мин......... 232- 10г>
после 100 мин.........114- 10~5 после 400 мин......... 600- 10“5
Поэтому при процессах переработки к полимеру часто добав-
ляют антиокислители (стабилизаторы), что, естественно, имеет
также большое значение и для сохранения его свойств.
Полиэтилен при температурах до 60—80° обладает высокой
стойкостью по отношению к действию почти всех кислот, включая
фтористоводородную, а также щелочей.
Свойства технического полиэтилена:
Уд. вес, г'слС................................. 0,92—0,95
Температура плавления, °C........................ 108—115
Температура хрупкости, °C:
низкомолекулярный.............................. —15°
высокомолекулярный....................... —68,5°
Теплостойкость, °C................................ 50—60°
Предел прочности на разрыв, кг/см3............ 110—135
Удлинение, %..................................... 300—500
Предел прочности на изгиб, кг-см-................ 115—160
Водостойкость (привес в воде), с/о.............. 0,0—0,01
Коэффициент преломления света, к®................... 1,52
Газопроницаемость, г/час • смя-мм рт. ст.......1,5—0,7-10
Уд. теплоемкость, кал/г - °C.................. 0,5—0,68
Коэффициент теплового расширения.............. 7—8,3 • 10
Теплопроводность, кал/см • сек • °C............... 7-10~4
Пробивное электрическое сопротивление, кв/мм . . 24—40
Уд. объемное электрическое сопротивление, Q-см . Ю17
184
Гл. V. Пластмассы на основе непредельных углеводородов
Диэлектрическая постоянная при:
60 гц...........................................
КР........................................
10е.......................................
2,3—2,4
2,3
2,3
Тангенс угла диэлектрических потерь при:
60 гц
10®,
10«
5-103
2—5 • 10-*
2—5 • 10 ,
2-5-10
30-1О-4
Применение полиэтилена
Сочетание весьма высоких технических свойств, широкая и доступная
сырьевая база и освоение производства в больших промышленных установках
открывают исключительно широкие области применения полиэтилена.
Полиэтилен легко перерабатывается почти всеми известными методами
пластицирующей обработки, однако наибольшее значение имеют методы валь-
цевания, каландрирования и экструзии (продавливания), позволяющие получать
• трубы, шланги, оболочки, пленки и
Рис. 83. Аппарат для распыления полн-
t этилена через пламя.
др. Перерабатывают полиэтилен и ме-
тодом литья под давлением, а также
методом прессования. Однако при этом
необходимо учитывать узкий интервал
плавления (3—5°) и сравнительно ме-
дленный процесс «отверждения» —
кристаллизации полиэтилена и свя-
занную с этим значительную усадку
материала.
Полиэтилен поддается сварке, при-
чем прочность получаемого шва не
уступает прочности материала. Он так-
же хорошо обрабатывается на токар-
ных и фрезерных станках. В распла-
вленном состоянии полиэтилен можно
перерабатывать методом выдувания
(сконструированы машины для выду-
вания бутылок).
Широкое применение получают
методы нанесения расплавленного по-
лиэтилена на различные поверхности
путем распыления его в жидком го
/—приемник для порошка; 2—пыльная камера;
«3—пистолет; 4—поверхность покрываемого пред-
мета; 5 — канал пистолета; 6 — баллон с воздухом.
рячем состоянии (огневое распыле-
ние). Для этой цели сконструированы
специальные аппараты. Схема работы
аппарата представлена на рис. 83.
Порошок полиэтилена помещают в воронкообразный приемник и перемешивают
воздушным вибратором. Частицы порошка увлекаются воздухом в пыльную
камеру наверху питательной воронки. Из пыльной камеры порошок и воздух
всасываются через гибкий шланг в пистолет, который и выбрасывает порошок
через пламя на поверхность покрываемого предмета. Проходя через пламя
с большой скоростью, порошок полиэтилена успевает лишь плавиться и, попа-
дая на горячую металлическую поверхность, прочно прилипает в виде пленки.
Регулирование количества полиэтилена, выходящего из пистолета (толщины
пленки), производят с помощью воздуха, поступающего через обводный канал
пистолета.
Методами экструзии, вальцевания й каландрирования из ^полиэтилена полу-
чают для различных целей пленки толшниой от 0.1 до 0,005 лм.
Пластические массы на основе полиизобутилена
185
Наиболее технически важная область использования полиэтилена — промыш-
ленность средств связи, так как он является незаменимым материалом для
изоляции высокочастотных кабелей в радиотехнике, радиолокации, телевидении,
телемеханике, телеграфной и телефонной связи. Широко применяется полиэти-
лен для изготовления морского кабеля, а пленки из полиэтилена—в производ-
стве конденсаторов.
Весьма перспективным является применение полиэтилена в химической
промышленности как в качестве самостоятельного конструкционного материала
(трубы, шланги, детали арматуры), так и различных защитных антикорро-
зийных пленок, футеровочных пластин, химической лабораторной посуды
и тары.
Большое промышленное значение приобретают полиэтиленовые пленки для
упаковки фармацевтических препаратов, пищевых продуктов; они совершенно
не гигроскопичны, безвредны, ие имеют запаха и стойки по отношению к хими-
ческим реагентам. Полиэтилен частично используется в виде волокна для изоля-
ции кабелей, для изготовления фильтр-полотиа, сеток, ткани для обнвки
•мебели и др.
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИИЗОБУТИЛЕНА
(Изобутиленопласты)
Полиизобутилен представляет высокомолекулярный продукт по-
лимеризации изобутилена. В зависимости от условий полимериза-
ции получаются полимеры с различным молекулярным весом — от
вязких жидкостей до твердых эластичных материалов.
Изобутилен был синтезирован в 1868 г. А. М. Бутлеровым (путем
дегидратации третичного бутилового спирта). Первые исследования
по полимеризации изобутилена были опубликованы А. М. Бутлеро-
вым и В. Горяйновым в 1873 г. Они получили и исследовали лишь
низшие (димерные, тримерные и т. п.) формы полиизобутилена,
однако правильно указали структуру полимера. Катализаторы, ко-
торыми они пользовались (фтористый бор, хлористый алюминий и
др.), применяют и в современной технике для производства высоко-
молекулярного полиизобутилена.
Основоположник промышленного синтеза каучука С. В. Лебе-
дев продолжил исследования, начатые Бутлеровым, и изучал поли-
меризацию изобутилена под действием флоридина. Он изолировал
низшие формы полиизобутилена вплоть до декамера и установил, что
в отсутствие мономера низшие формы не способны к дальнейшей
полимеризации. При низких температурах (около —105°) Лебедеву
удалось получить продукты с молекулярным весом 7 000—8 000.
Преждевременная смерть помешала ему закончить начатые изы-
скания по получению высокомолекулярных форм полиизобутилена.
Дальнейшие работы были направлены на изыскание условий,
ведущих к цепному механизму полимеризации и к получению вы-
сокомолекулярного продукта. Было найдено, что в присутствии
группы галоидных соединений, указанных еще А. М. Бутлеровым
(фтористый бор, хлористый алюминий и др.), и при низких
температурах (—80, —100°) реакция протекает по цепному ион-
ному механизму с образованием высокомолекулярных полимеров
186
Гл. V. Пластмассы на основе непредельных углеводородов
(с молекулярным весом 100 000—500 000 и выше). Техническая за-
дача получения высокомолекулярного полиизобутилена была решена
в 1938—1939 гг.
Изобутилен СН2=С(СН3)2 представляет бесцветный газ с тем-
пературой кипения —6,9° (760 мм), температурой замерзания
—140,8°, уд. весом (при —10°) 0,631 и показателем преломления
(при —25°) 1,3796.
Под давлением 1—3 ат изобутилен можно хранить и перево-
зить в баллонах.
В промышленных условиях его получают пиролизом бутан-бути-
леновых фракций, являющихся побочными продуктами крекинга
нефти, а также дегидратацией изобутилового спирта:
СН3—СН—СН.ОН СНз— с=сн,.
СНз СНз
Для получения высокомолекулярного полиизобутилена реакцию
проводят при низких температурах (~—100°) в присутствии рас-
творителей, применяя в качестве катализаторов различные галоид-
ные соединения (BF3, А1С13, TiCU). Катализаторы этого типа в
весьма большой степени ускоряют реакцию. По своей активности
они могут быть расположены в следующий ряд:
BF3, А1С13, А1Вг3, TiCl4, Т1Вг4, ВС13, BBr3, SnCl4.
BF3 ускоряет реакцию очень сильно. При внесении 0,5% ката-
лизатора скорость полимеризации изобутилена столь велика, что
граничит со скоростью взрыва. Эта реакция является одной из
самых быстрых в химии высокополимеров. Она сопровождается
выделением большого количества тепла (~ 10 000 кал/моль), что
вызывает значительные трудности в проведении процесса, так как
требуется быстро отводить выделяющееся тепло. Именно поэтому
технически полимеризацию проводят в растворителях. Чем актив-
нее катализатор, тем меньше его концентрация и тем меньше
должно быть содержание мономера в реакционной среде (табл. 16).
ТАБЛИЦА 16
Полимеризация изобутилена под действием различных катализаторов
Катализатор Количество катализатора °;о Содержание изо- бутилена в реак- ционной смеси °; о Время полимеризации Выход полимера °, о Молекулярный вес
BF3 0,05 10 Секунды 100 120 000—150000
А1Вг3 0,05 20 1—5 мин. 70—90 120000—150 000
Т1С14 0,12—0,25 30 20-70 „ 35—50 100 000—130 000
Т1Вг4 1,0—1,5 30-50 12—18час. 30—50 70 000— 90000
SnCl4 1,5—4,5 50 17-50 „ 10—18 12 000— 25 000
Пластические массы на основе полиизобутилена
187
Полимеризация изобутилена в присутствии BF3 и других ката-
лизаторов протекает с большой скоростью лишь тогда, когда в реак-
ционной среде в незначительных количествах присутствуют соедине-
ния, имеющие протонный водород и действующие как активаторы.
При полимеризации с BF3 таким активатором обычно служит третич-
ный изобутиловый спирт, присутствующий в техническом изобутилене
в виде незначительной примеси (~ 1 %). При полимеризации с TiCL»
активатором служат следы воды, вносимые с влажным воздухом.
Механизм действия активаторов можно представить следующей
схемой:
BF3 4- RH —> F3B • RH
СН3 СН3
I е I
F3B • RH 4- СН2=С —> F3B • R 4- СН3— С®
СН3 СН3
сн3 сн3 сн3 сн3
I ® I I I &
СН3—С 4- СН,=С —> СН3—С—СН,—С и т. д.
I । I I
сн3 сн3 сн3 сн3
Повидимому, действие активаторов специфично для различный
катализаторов. Так, в присутствии BF3 вода не только не активи-
рует, а замедляет процесс полимеризации изобутилена.
Ингибиторами в этом процессе, т. е. агентами, препятствую-
щими процессу полимеризации, являются сера, меркаптаны, серо-
водород, хлористый водород и др.
К особенностям реакции полимеризации изобутилена следует
отнести влияние примесей низкомолекулярных изобутиленов, напри-
мер, ди- и триизобутилена, присутствие которых сильно снижает
средний молекулярный вес полимера. Такое же действие оказывает
н. бутилен, который, участвуя в построении цепи, ограничивает
среднюю степень полимери-
зации полимера. Молекуляр-
ный вес получаемого поли-
мера зависит и от темпера-
туры реакции: чем она ниже,
тем молекулярный вес выше
(табл. 17).
Степень полимеризации
полиизобутилена зависит
также от характера раство-
рителя (его полярности) и
концентрации изобутилена.
Кривая зависимости молекул
лена характеризуется максимумом, лежащим обычно в пределах
20—40% (рис. 84).
ТАБЛИЦА 17
Полимеризация изобутилена с BF3 пр»
различных температурах
Темпера- тура, °C Мол. вес Темпера- тура, °C Мол. вес
— 10 10 000 — 80 80 000
—25 13 000 — 90 120 000
—45 25 000 —105 220 000
веса от концентрации изобути-
188
Г.1. V. Пластмассы на основе непредельных углеводородов
В качестве растворителя чаще всего применяют жидкий этилен.
Технологический процесс производства полиизобутилена во мно-
гом определяется методом смешения мономера с катализатором
«ввиду чрезвычайной, скорости
Рис. 84. Зависимость мол. веса по-
лиизобутилена от концентрации
изобутилена при полимеризации.
реакции), а также условиями, обеспе-
чивающими быстрый отвод тепла
реакции (так как высокомолекуляр-
ный продукт получается лишь при
весьма низких температурах).
Отвод тепла может происходить
за счет наружного охлаждения реак-
тора или за счет скрытой теплоты
испарения растворителя в самом ре-
акторе.
Последний метод, повидимому,
чаще всего применяется. В этом слу-
чае растворитель должен иметь тем-
пературу кипения, близкую к темпе-
ратуре полимеризации мономера
(около —100°).
Процесс получения полиизобути-
лена по одному из вариантов может
схемой (рис. 85).
быть представлен следующей
Полииэобутилен
Рис. 85. Схема получения полиизобу.тилена.
Жидкий этилен, охлажденный до —40°, под давлением посту-
пает в теплообменник 1, где он дополнительно охлаждается газо-
образным этиленом, идущим из испарителя 2, после чего
дросселируется в испарителе 2 до 1 ат. Здесь этилен охлаждается
за счет частичного испарения до температуры кипения (—104), при
этом газообразный этилен поступает в теплообменник 1, а жидкий
Пластические массы, на основе полиизобутилена
18У
этилен в дозер 3, где он охлаждает змеевик, по которому протекает
жидкий изобутилен.
При выходе из дозера жидкий изобутилен смешивается в тру-
бопроводе с жидким этиленом и смесь направляется в полимериза-
тор на движущийся транспортер 5. Туда же поступает из прием-
ника 4 раствор фтористого бора в этилене.
Реакция полимеризации изобутилена завершается практически
мгновенно при смешении с катализатором, при этом теплота реак-
ции поглощается испарением жидкого этилена, чем достигается по-
стоянство температуры полимеризации.
Рис. 86. Схема строения полиизобутилена.
© — атомы углерода; О—атомы водорода.
Для разрушения оставшегося в полимере катализатора и для
предохранения от деполимеризации при повышении температуры
на ленту транспортера подается из дозера 6 стабилизатор (раствор
третичнобутилфенилсульфида).
В дальнейшем для более полного удаления газа полимер посту-
пает в обогреваемый смеситель 7, а оттуда для охлаждения на
стеллажи 8, после чего охлажденный полимер прессуют в прес-
сах 9.
Из полимеризатора 5 этилен, содержащий некоторое количестве
фтористого бора, поступает для нейтрализации на колонну 10, а
оттуда на ректификацию, после чего направляется в этиленовый
цикл.
Полиизобутилен имеет, повидимому, структуру «голова к хво-
сту» (рис. 86). Такой вывод сделан на основании вычисления
периода идентичности по рентгенограмме растянутого полимера. Пе-
риод идентичности равен 18,63 А, что соответствует трем изобута-
190
Гл. V. Пластмассы на основе непредельных углеводородов
новым группам. Однако при пиролизе полиизобутилена образуются
соединения, которые по строению соответствуют структуре «голова
к голове»; возможно, что во время пиролиза происходит перегруп-
пировка.
Свойства полиизобутилена
Полиизобутилен представляет собою каучукоподобный эластич-
ный мягкий материал. В нерастянутом состоянии полиизобутилен
имеет, повидимому, аморфную структуру, но при растяжении
он легко кристаллизуется и дает четкую точечную рентгено-
структуру ориентированных кристаллитов. Кристаллическая фаза
в растянутом полиизобутилене плавится при 50—70°. При сокраще-
нии полимера (после снятия нагрузки) основная часть кристаллитов
разрушается и исчезает. Техническое применение получили, глав-
ным образом, высокомолекулярные твердые полимеры со средним
молекулярным весом от
ТАБЛИЦА 18 ЮОООО до 500 000. Поли-
Зависимость механических свойств полиизобутилена от молекулярного веса меры с молекулярным весом ниже 50 000 представляют
Средний моле- кулярный вес Временное сопротивление разрыву кг[см* Удлипенине при разрыве '\о 4 С 9 С С удлинение после 24 ч. э,'о Твердость по TIJopy собою вязкие жидкости. Чем выше молекулярный вес полимера, тем выше его прочность на разрыв, тем больше высокоэласти-
ЮОООО 200 000 Зав> пол 20 60 1CHMOCT имериз: более 1000 более 1000 ь Ттек — Та 1ЦИИ полии 20—50 около 4 ТАБЛИ от степе зобутиле 27 35 ЦА 19 ‘ИИ на ческая, обратимая деформа- ция и тем выше темпера- тура перехода в пластиче- ское состояние (точка Ттек, стр. 103). В табл. 18 пока- зана зависимость прочности полимера от степени полиме- ризации, а в табл. 19 — зависимость (по данным В. А. Каргина) интервала
Молекулярный вее Степень полимеризации Лгек -Г,
6-103 7-11OI 5-8105 1 610е 3 • 5107 склонен к кр1 107 1270 10-400 28-600 62 • 500 металлизации пластического течения (J тек — 30 Та ) полиизобутилена от ве- 80 личины его молекулярного 23Q веса. 280 Полиизобутилен имеет очень низкую температуру стеклования (—74°). Он мало и в интервале от —74° до 100—150° на-
ходится в высокоэластическом состоянии; поэтому полиизобутилен
по комплексу его механических свойств следует рассматривать как
«каучук». Однако в отличие от основной группы «каучуков» (поли-
Свойства полиизобутилена
191
бутадиен, полиизопрен и т. д.) полиизобутилен не способен- к реак-
циям вулканизации («сшивки» макромолекул), гак как является на-
сыщенным полимером. Наряду с полиэтиленом полиизобутилен
один из самых легких пластиков, его удельный вес 0,91—0,93.
Высокомолекулярные полиизобутилены (например с молекуляр-
ным весом 200 000 и выше) эластичные и прочные материалы. При
растяжении они дают удлинение свыше 2000% и их прочность в пе-
ресчете на действительное (а не на первоначальное) сечение 600—
1200 кг!см2.
Полиизобутилен отличается высокой химической стойкостью и
водостойкостью. При обычных температурах он устойчив к воздей-
ствию почти всех кислот, в том числе азотной, а в некоторых усло-
виях •— и к действию царской водки; по отношению к щелочам и
галогенам полиизобутилен также устойчив.
Высокая химическая стойкость полиизобутилена, значительно
превосходящая стойкость обычных «каучуков», имеет своей причи-
ной насыщенный характер его макромолекулы. Полиизобутилен от-
носится к слабополярным полимерам, что определяет его высокие
диэлектрические свойства, в частности малую зависимость диэлек-
трической постоянной и тангенса угла диэлектрических потерь от
температуры и частоты. В отношении химической стойкости и ди-
электрических свойств полиизобутилен, а также его смеси с поли-
этиленом, полистиролом и его сополимеры уступают только поли-
этилену и политетрафторэтилену.
При действии кислорода в присутствии ультрафиолетовых лу-
чей и при длительном воздействии солнечных лучей полиизобути-
лен окисляется и разлагается с образованием маслянистых продук-
тов деструкции. Введение активных наполнителей (сажи, графита,
талька и др.), а также некоторых смол (полиэтилена, каучука, фе-
ноло-альдегидных смол и др.) улучшает его стойкость к свету и к
.атмосферному кислороду, уменьшает текучесть и увеличивает
•прочность и жесткость композиции. Количество активных наполни-
телей может доходить до 90% ко всей массе; техническое примене-
ние получил главным образом.наполненный полиизобутилен.
Полиизобутилен сравнительно легко растворяется в ароматиче-
ских углеводородах, в сероуглероде, в хлорированных углеводоро-
дах. Он нерастворим в спиртах, в кетонах, в сложных эфирах и
других полярных растворителях. Вследствие высокой вязкости рас-
творов в качестве лака применяют лишь низкомолекулярный поли-
изобутилен.
Как типичный эластик полиизобутилен «работает» в температур-
ном интервале, в котором сохраняются его высокоэластические свой-
ства (практически от —60 до +60°). При более высоких темпера-
турах (80—120°) и длительных нагрузках медленно развивается
пластическое течение и тем быстрее, чем ниже молекулярный вес
полимера. __ .
192 Гл. V. Пластмассы на основе непредельных углеводородов
Приводим некоторые свойства полиизобутилена:
Уд. вес, г[смЛ........................................ 0,91—0,93
Температура стеклования, ............................. —74°
Температура хрупкости, °C............................. —50°
Предел прочности на разрыв, кг^м-..................... 20—135
Удлинение, °/0 ....................................... 1000—2000
Водостойкость (привес в воде), % ..................... 0—0,05
Пробивное электрическое сопротивление, кв/мм.......... 23—25
Уд. объемное электрическое сопротивление, Ы-с.ч .... 1015—1016
Диэлектрическая постоянная при 10в гц................. 2,3—2,2
Тангенс угла диэлектрических потерь при 106 гц ... . 0,0004—0,0008
Сополимеры изобутилена
Важнейшими техническими сополимерами изобутилена являются
продукты сополимеризации с диеновыми соединениями, главным
образом с бутадиеном и изопреном. Цель сополимеризации — вве-
дение в состав цепи ненасыщенных групп, которые в процессе вул-
канизации (взаимодействием с серой или другими соединениями>
могли бы служить местом образования валентной связи («сшивки»)
между цепями полимера.
Для получения мягких резин достаточно внесения лишь 2—5%
диенового мономера. Образующийся при этом сополимер изобути-
лена после вулканизации является практически насыщенным поли-
мером, в отличие от природного и синтетических диеновых каучу-
ков, и обладает, соответственно, более высокой химической стой-
костью и диэлектрическими свойствами. Сополимер изобутилена с
небольшим количеством бутадиена, известный под названием поли-
изобутиленовый каучук, или бутилкаучук, приобрел большое про-
мышленное значение.
Изобутилен образует также сополимеры с моновинильными со-
единениями, с этиленом, акрилонитрилом, полистиролом, винилхло-
ридом, сложными и простыми виниловыми эфирами и др. В зави-
симости от характера винильного соединения и его соотношения с
изобутиленом получаются сополимеры с различными свойствами,
нашедшие, однако, лишь ограниченное применение для ряда спе-
циальных назначений.
Применение и переработка полиизобутилена
Полиизобутилен нашел применение в ряде областей техники. В электротех-
нике его применяют для изоляции проводов и кабелей, часто в смеси с природным
или синтетическим каучуком; в промышленности пластмасс — в качестве добавки
К полиэтилену.
Важнейшей областью применения высокомолекулярного полиизобутилена
(не сополимера) является химическая промышленность. Полиизобутилен пред-
ставляет ценный антикоррозийный материал и применяется в виде обкла-
дочных листов, прокладочных уплотнительных материалов и отчасти в виде
Пластические массы на основе полистирола
193
антикоррозийных защитных пленок, наносимых лаковым методом или распыле-
нием. Для этого высоконаполненные смеси, состоящие из полиизобутилеиа,
сажи и графита, а также талька, асбеста и других наполнителей (содер-
жание наполнителей обычно составляет 60—70% и выше), подвергают горя-
чему смешению в лопастном мешателе при 130—150° в течение 20—30 мин. Из ме-
шателя горячую массу переносят на горячие фрикционные вальцы, где происходит
гомогенизация при 80—90° в течение 5 мин., после чего массу переносят на вторые
вальцы без фрикции, где при температуре 50—60° происходит прокатка и листо-
образование; после этого на горизонтальных или вертикальных каландрах
(стр. 244) при 20° путем многократного пропускания (отжима) при постепенном
уменьшении зазора получают листы желательной толщины. Если хотят получить
шланги, трубки, оболочки, массу после мешателя или первых нальцов загру-
жают в экструзионную машину (шнекпресс), в которой при различных зонах
обогрева происходит гомогенизация ее и выдавливание через мундштук в виде
беспрерывной ленты нли трубки (рис. 102, стр. 218).
Для получения прокладочных и уплотнительных пластин применяют волок-
нистые наполнители, например асбест, асбест в смеси с тальком, каолином
и т. д. Так, смесь, состоящую из полиизобутилена, асбеста и талька, смеши-
вают в мешателе при 130—150°, после чего переносят на смесительные фрик-
ционные вальцы, где при 60—70° про-
исходит дальнейший процесс гомогениза-
ции массы. После этого смесь загружают
в прессформы и при 160—190° при
медленном поджатии плит в течение не-
скольких минут производят прессование
пластин. После горячей выдержки охла-
ждают под давлением.
Процесс смешения и в особенности
вальцевания полиизобутилена следует
вести при строго нормированных темпе-
ратуре и времени. При вальцевании
всегда происходит деструкция полимера
н уменьшение его молекулярного веса.
Деструкция тем больше, чем ниже тем-
пература и длительнее процесс вальце-
вания, так как с уменьшением темпера-
туры возрастает вязкость массы и быст-
Ародолжительмагть вальце-
вания, мин.
Рис. 87. Изменение молекулярного
веса полиизобутилеиа в зависимости
от времени и температуры вальце-
вания.
рее происходит разрыв макромолекул от
механического трения (рис. 87).
Обкладочные листы, шланги и про-
кладочные пластины из полиизобутилеиа
отличаются удовлетворительной проч-
ностью и высокой химической стойкостью.
Их применяют для внешней н внутренней защиты металлических труб, для
внутренней обкладки реакторов, железнодорожных цистерн, кнслотохранилищ.
Их укрепляют при помощи специальных клеев и замазок, а также соединяя
методом сварки. Прокладочные пластины применяют также н качестве прокла-
дочных материалов для фундаментов, при облицовке полов, сооружений, тон-
нелей, для создания различных видов водонепроницаемой изоляции.
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИСТИРОЛА
(Стиропласты)
Полимер стирола — полистирол — является одним из наиболее
давно известных и наиболее полно изученных полимеризационных
пластиков. Он широко известен своими выдающимися свойствами:
13 Зак. 1269. Э. И. Барг
194
Гл. V. Пластмассы, на основе непредельных углеводородов
почти абсолютной водостойкостью, высокой химической стойкостью,
весьма совершенными диэлектрическими свойствами и прозрач-
ностью.
Стирол впервые был получен в 1831 г. Бенастром при сухой пе-
регонке живицы (смолы) styrax. Уже в 1839 г. стало известно, что
при хранении стирол переходит в твердое вещество, причем перво-
начально этот переход связывали с окислением стирола. В 1878 г.
русский ученый А. Кракау впервые изучал полимеризацию стирола
под действием металлического натрия; он установил, что в то время
как металлический натрий способствует процессу полимеризации,
другие вещества замедляют его. Более детально процесс полимери-
зации стирола изучал И. И. Остромысленский, который впервые
выяснил действие на него ультрафиолетовых лучей, а также уста-
новил структуру образующегося полимера.
Еще в начале настоящего столетия были изучены многие свой-
ства полистирола и предложено применение его для различных це-
лей. В начале двадцатых годов полистирол в ограниченном коли-
честве применяли в слаботочной электропромышленности как в
виде лака, так и в виде пластин, блоков и т. п.
Первые попытки технического применения полистирола натал-
кивались на значительные трудности, связанные с его быстрым
старением, самопроизвольным разрушением, хрупкостью.
Лишь постепенно, по мере преодоления ряда трудностей, свя-
занных с получением чистого стирола и освоением современных
методов полимеризации, удалось получить полимер со стандарт-
ными и стабильными свойствами; это обеспечило широкое приме-
нение полистирола не только в электротехнике, но и в других от-
раслях промышленности.
Последние 10 лет характеризуются новым развитием исследова-
тельских работ, идущих в направлении синтеза и технического
получения производных стирола и их полимеров (галоидо- и алкил-
производных полистирола). Эти исследования уже привели к полу-
чению ряда технически важных продуктов, отличающихся в част-
ности большей теплостойкостью.
Вторым направлением работ были исследования в области син-
теза сополимеров стирола как с моновинильными, так и в особен-
ности с дивинильными соединениями. Эти работы привели к полу-
чению ряда важных пластиков и нового вида стирол-бутадиенового
синтетического каучука, который является одним из основных ви-
дов синтетических каучуков.
Технические методы получения стирола
Стирол С6Н5СН=СН2 (винилбензол, или фенилэтилен) предста-
вляет собою бесцветную жидкость со слабым характерным запа-
хом. Температура кипения его 145,2° (760 мм), температура замер-
зания— 30,6°, показатель преломления nf" 1,5469, плотность при
Технические методы, получения стирола
195
20° — 0,9060. Он нерастворим в воде, растворим в бензоле, четы-
реххлористом углероде, этиловом эфире и др.
В небольших количествах стирол содержится в дегтях, получае-
мых при сухой перегонке битуминозного угля, при крекинге нефти
и при пиролизе некоторых органических веществ. Вместе с другими
ненасыщенными углеводородами — кумароном, инденом, циклопен-
тадиеном и др. он содержится (0,2—1%) в легкой фракции каменно-
угольного дегтя, в так называемой сольвент-нафте, кипящей в пре-
делах 160—180°.
В последние годы значительным источником сырья для получе-
ния стирола являются так называемые ксилольно-стирольные фрак-
ции, получаемые при пиролизе нефти при высокой температуре
(1400—1500°), а также при специальном крекинге нефти при низ-
ких температурах (600—700°) в атмосфере азота и в присутствии
медных катализаторов. Последним методом можно получить до
5—7% стирола (считая на нефть).
Из большого числа известных методов синтеза стирола промыш-
ленное применение получили лишь немногие, преимущественно те,
в которых промежуточным продуктом является этилбензол, а сы-
рьем — этилен и бензол.
Следует, однако, упомянуть и о некоторых методах, не нашед-
ших еще промышленного применения, но являющихся перспектив-
ными или же используемых для лабораторных целей.
Одним из давно известных в органической химии путей полу-
чения стирола является пиролиз коричной кислоты, протекающий
с выделением СО2 по уравнению:
С6Н5СН=СНСООН —► С6НбСН=СН2 + сст.
Этим методом в 1890 г. был впервые получен синтетический сти-
рол. Основное препятствие к его промышленному применению —
сложный и дорогой еще синтез коричной кислоты из бензальдегида
и уксусной кислоты, а также сравнительно малые выходы стирола.
В последние годы этот метод был улучшен и выход стирола дове-
ден до 70%.
Другим методом синтеза стирола является дегидратация а- и
р-фенилэтиловых спиртов. Ниже (стр. 198) мы подробнее остано-
вимся на методе дегидратации а-фенилэтилового спирта (фенил-
метилкарбинола), нашедшем промышленное применение.
Дегидратацию Р-фенилэтилового спирта производят путем на-
гревания его с расплавленной щелочью при 220°; реакция проте-
кает почти количественно по уравнению:
СвНбСН2СН2ОН С6Н6СН=СН2.
Этот метод может получить промышленное применение, так как
£-фенилэтиловый спирт, ранее мало доступный, в настоящее время
легко получается конденсацией окиси этилена с бензолом в присут-
13
196
Гл. V. Пластмассы на основе непредельных углеводородов
ствии хлористого алюминия. По этому методу можно получать наи-
более чистый стирол.
Стирол может быть также получен из ацетофенона путем его
восстановления в а-фенилэтиловый спирт (фенилметилкарбинол),
либо водородом in statu nascendi, например, при нагревании в кис-
лой среде с амальгамой цинка, либо восстановлением изопропила-
том алюминия в присутствии изопропилового спирта, с последую-
щей дегидратацией а-фенилэтилового спирта в стирол по уравнению:
С6НБСОСН3 С6Н5СН(ОН)СН3;
С6НБСН(ОН)СН3 С6НБСН=СН,.
Необходимо указать на принципиальную возможность синтеза
стирола непосредственным действием ацетилена на бензол по урав-
нению:
СбН6 + СН=СН —► С6НБСН=СН3.
Реакция протекает в газовой фазе при пропускании паров бен-
зола и ацетилена через железные трубки, нагретые до 800—1000°;
выход стирола составляет 5—10%. Лучшие выходы (до 40%) мо-
гут быть получены при пропускании смеси паров бензола и ацети-
лена над А1С1з и ZnCl2, осажденными на инертном носителе при
температурах до 150°. Этот метод — безусловно весьма перспекти-
вен и в принципе наиболее прост. Однако малые выходы и большой
расход катализатора делают его пока нерентабельным.
В настоящее время основным промышленным методом является
синтез стирола из этилбензола.
Этилбензол — СвНзСНгСНз в промышленных условиях полу-
чают в основном двумя путями: 1) из бензола и хлористого этила
и 2) из бензола и этилена.
В первом случае реакция протекает в присутствии хлористого
алюминия по уравнению:
С6Н6 + СН3СН2С1 С6Н5СН2СН3+НС1.
Однако наряду с этилбензолом по этому методу получаются в
большом количестве гомологи этилбензола (полиэтилбензолы). Вы-
ход этилбензола можно увеличить, если процесс вести при большом
избытке бензола (50—60% против теории и выше) и применять в
качестве катализатора алюминий в виде порошка или стружек, пре-
вращаемый в хлористый алюминий пропусканием хлористого водо-
рода. Хлористый этил вводят в нагретый до 70° бензол, содержа-
щий 1% катализатора (от количества бензола); реакция протекает
в течение 5 часов; в конце температуру поднимают до 90° и считают
реакцию завершенной при практическом прекращении выделения
НС1. В этих условиях общий выход этилбензола ~ 75%, а выход
гомологов ~ 20%; последние, однако, также могут быть превра-
щены в этилбензол при кипячении с большим избытком бензола в
присутствии 10% хлористого алюминия. Общий выход этилбензола
Технические методы, получения стирола
197
может, следовательно, достигать 90% и более. После окончания
реакции смесь подвергают промывке и ректификации; отгоняют
бензол и другие побочные продукты и отбирают фракцию 135—
155°, которая в основном содержит этилбензол.
Второй способ синтеза этилбензола — из бензола и этилена —
является основным в промышленном производстве стирола.
Реакция протекает по уравнению:
С6Нв + СН2=СН2 —> С6Н5СН2СН3.
Ее ведут, главным образом, жидкофазным методом, путем про-
пускания этилена через бензол при 85° и давлении около 1 ат в при-
сутствии хлористого алюминия. Наряду с этилбензолом получаются
в количестве 25% более высокоэтилированные гомологи, а также
происходит некоторое осмоление продукта.
Продукты реакции после удаления катализатора подвергают
ректификации. Вначале отгоняют непрореагировавший бензол, ко-
торый направляется в реактор на этилирование, затем этилбензол.
Оставшиеся в кубе полиэтилированные бензолы частично также на-
правляются в реактор для повышения выхода этилбензола.
Выход этилбензола при циклическом процессе составляет около
90% теоретического.
Превращение этилбензола в стирол может быть осуществлено
в основном двумя путями: 1) через а-хлорэтилбензол и 2) непо-
средственным дегидрированием этилбензола.
1. Получение стирола из этилбензола через а-хлорэтилбензол.
Этот метод, в свою очередь, подразделяется на два варианта:
1) омыление хлорэтилбензола с образованием фенилметилкарби-
яола и с последующей дегидратацией его в стирол и 2) непосред-
ственное получение стирола из хлороэтилбензола путем отщепле-
ния хлористого водорода.
Первый вариант получения стирола из хлорэтилбензола был
разработан в СССР Ю. С. Залькиндом, И. П. Берковичем и
М. К. Амусиным.
Перегнанный и промытый этилбензол подвергают прямому хло-
рированию газообразным хлором при 70° в присутствии PCU, ко-
торый способствует введению хлора в боковую цепь:
С6Н5СН2СН3 С6Н5СНС1СН8 + НС1.
В этих условиях а-хлорэтилбензол является главным продуктом;
если же в техническом этилбензоле присутствует бензол, то обра-
зуются также р-хлорэтилбензол, полихлорэтилбензолы и хлорбен-
зол.
Реакцию ведут до 60—70%, после чего этилбензол и другие по-
бочные продукты отгоняют под вакуумом, причем а-хлорэтилб1ен-
зол остается в котле. Выход 40—42% от теории.
Следующая операция — омыление а-хлорэтилбензола в фенил-
метилкарбинол. Для этого а-хлорэтилбензол обрабатывают раство-
198
Гл. V. Пластмассы на основе непредельных углеводородов
ром соды при 70—100° в присутствии эмульгаторов (мыла) в те-
чение 4—6 часов:
С6Н5СНС1СН3 —ОН-> С6Н5СН(ОН)СН3.
Скорость реакции зависит от концентрации соды: оптимальная
концентрация отвечает 10% избытку. Процесс контролируют по вы-
делению СО2. Выход карбинола ~ 90%.
Последней операцией является дегидратация фенилметилкарби-
нола в стирол в присутствии кислых водоотнимающих средств:
С6Н5СН(ОН)СН3 свн5сн=сн2.
Для этого карбинол нагревают при 150—200° с 3% бисульфата
калия. Образующийся стирол отгоняют из реактора с водяным па-
ром. Сырой продукт подсушивают и подвергают ректификации.
Вследствие осмоления части карбинола и стирола выход последнего
составляет ~ 80 %. Процесс дегидратации значительно облегчается
введением около 1 % тетрахлорфталевой кислоты.
Более эффективным, повидимому, является процесс дегидрата-
ции фенилметилкарбинола в паровой фазе, разработанный
А. А. Ваншейдтом и В. М. Зельцер. Этот процесс может быть осу-
ществлен с хорошими выходами при пропускании паров карбинола
через катализатор, состоящий из А12О3 при температуре 350—400°.
Пары, выходящие из реактора, конденсируются в холодильнике,
и конденсат, поступающий в приемник, состоит из двух слоев —
водного и углеводородного; последний содержит стирол, непрореа-
гировавший карбинол и некоторые продукты распада.
В этих условиях выход стирола достигает 85%.
Второй вариант получения стирола (через а-хлорэтилбензол пу-
тем непосредственного отщепления от последнего НС1) протекает
по уравнению:
С6Н5СНС1СН3 СвН5СН=СН2.
Отщепление НС1 можно осуществить путем нагревания а-хлор-
этилбензола с некоторыми органическими основаниями, например,,
с избытком хинолина или пиридина. Однако этот метод требует
большой чистоты хлорэтилбензола и значительного расхода орга-
нического основания; при этом потери этилбензола существенно не
уменьшаются. Более перспективно каталитическое отщепление HCI
путем пропускания а-хлорэтилбензола через нагретые поверхност-
но-активные вещества (активированный уголь, окись кальция, бо-
кситы и др.).
Метод получения стирола и а-хлорэтилбензола путем непосред-
ственного отщепления НС1 не имеет существенных преимуществ и
не нашел технического применения.
Рассмотренные процессы получения стирола слишком сложны;
они состоят из многих операций, требуют большого расхода хими-
калий и дают малый выход стирола — около 60—65% в расчете на
этилбензол.
Технические методы получения стирола
199
2. Получение стирола непосредственным дегидрированием этил-
бензола. Этот процесс вследствие большой простоты наиболее
эффективен и в настоящее время является основным методом про-
изводства синтетического стирола. Реакция дегидрирования
с6н6сн2сн3 — -> С6Н5СН=СН2
эндотермична и обычно проводится при высоких температурах и
низких парциальных давлениях этилбензола.
Для уменьшения парциального давления этилбензола применяют
вакуум, разбавление инертными газами и чаще всего разбавление
водяным паром.
Н. Д. Зелинский с сотрудниками проводил дегидрирование па-
ров этилбензола при 650° в присутствии медно-хромовых катализа-
торов, а также в присутствии азота или углекислого газа в качестве
разбавителей. В этих условиях выход стирола достигает 50—55%.
Ю. С. Залькинд с ’сотрудниками проводил дегидрирование
при 650° над 7п-А120з (1 :9) при 10—13 мм остаточного давления
и получил стирол с выходом до 80%.
В процессе, разработанном А. А. Ваншейдтом, пары этилбензола
либо предварительно пропускают через подогреватель, либо не-
посредственно в реактор (контактный аппарат), нагретый до
650—700° и заполненный силикагелем, активированным углем, на
поверхности которого осажден катализатор, или же другим мелко-
дисперсным катализатором; контактированные пары направляют
в холодильник, где они конденсируются. Выход стирола ~ 50%;
конденсат, наряду со стиролом, содержит примерно такое же коли-
чество этилбензола, а также немного толуола и ряд высококипящих
продуктов.
Выход стирола и продолжительность службы катализатора,
а следовательно, и продолжительность непрерывного, циклического
процесса дегидрирования этилбензола определяются в первую оче-
редь составом катализатора, его дисперсностью, характером носи-
теля, на котором диспергирован катализатор, а также соотношением
между водяным паром и этилбензолом.
В Германии применяли в качестве катализатора окись цинка
(77,4%), активированную калием; реакцию проводили в труб-
чатке при молярных соотношениях водяного пара к этилбензолу.
Температуру реакции поддерживали над свежим катализатором
в 580° и постепенно, по мере уменьшения активности катализатора,
ее поднимали до 610°.
В США применяют в качестве катализатора щелочные окислы
трехвалентного железа (83%) с небольшим количеством окиси
хрома; 1 кг этилбензола смешивают с 2,6 кг водяного пара,
температуру смеси в реакторе поддерживают 630°, а температуру
выходящих паров 565°. Отходящие пары содержат водород, окись
углерода и углекислоту, метан и этан (за счет взаимодействия пара
с коксом, образовавшимся в результате пиролиза).
200 Гл. V. Пластмассы на основе непредельных углеводородов
' Выход стирола при возврате непрореагировавшего этилбензола
составляет ~90% от теории. Процесс продолжается непрерывно
много месяцев.
Жидкие углеводороды после отделения воды направляют в ряд
непрерывно действующих перегонных аппаратов, где сначала уда-
ляют бензол и толуол, затем этилбензол, который возвращается
в аппарат для дегидрирования, после чего отгоняют стирол.
Выделение стирола
из сырца связано с пре-
одолением значитель-
ных трудностей, заклю-
чающихся в близости
точек кипения этилбен-
зола и стирола. Ректи-
фикацию ведут на ва-
куумных колонках вы-
сотой свыше 18 м, при
остаточном давлении
ниже 50 мм. Колонку
готовят из меди, так
как последняя замедля-
ет процесс полимериза-
ции стирола; кроме то-
го, с этой же целью при
разгонке в стирил вно-
сят ингибиторы, напри-
мер, 0,1% гидрохино-
на 1 % серы и др. Таким путем можно получить стирол с чистотой
98—99%; повторная ректификация может дать очень чистый стирол.
Учитывая потери при очистке сырья и повторное использование этил-
бензола, общий выход стирола составляет ~80%.
К чистоте стирола современная техника предъявляет весьма
высокие требования, так как это определяет стандартность процесса
полимеризации и технические свойства полимера. Продукт, идущий
для полимеризации, должен содержать не менее 99% стирола,
причем часто требуется еще более чистый стирол — до 99,9%.
Из примесей к стиролу особое значение имеет п-дивинилбензол;
он получается при дегидрировании л-диэтилбензола, который может
находиться в виде примеси в этилбензоле:
Непрореагировавшии
Бензол бензол
Этилен I * л" } Этилбензол
3 р д рованив
На Вегидри-
Т То
1— ’ Полиэтилиро-
ванные бензолы
Этилбензол
1
толуол
у Лар
9
Вода
Стирол
Остаток
Рис. 88. Схема получения стирола.
1 — реактор для этилирования бензола; 2 — нейтрализатор:
j, 4, 7, 8, 9 — реактификационные колонны; 5— аппарат для
дегидрирования этилбензола; 6 ~ конденсатор.
сн.—сн3
сн=сн.
- 2Н.
сн.—сн3
сн=сн.
Полимеризация стирола
201
Достаточно самых ничтожных количеств примесей (~ 0,01 %)
дивинилбензола в стироле, чтобы при полимеризации образовался
неплавкий и нерастворимый полимер сетчатой структуры, т. е. тех-
нически негодный продукт (стр. 51). Это объясняется тем, что
п-дивинилбензол, благодаря наличию в нем двух способных к поли-
меризации двойных связей, склонен с большой легкостью «сшивать»
цепи полимера в пространственную макромолекулу (стр. 50). Кроме
того, п-дивинилбензол, полимеризуясь в процессе дегидрирования,
дает неплавкие продукты, загрязняющие аппаратуру и уменьшаю-
щие активность катализатора. Поэтому тщательная очистка этил-
бензола от диэтилбензола является крайне необходимой.
На рис. 88 приводится общая схема получения стирола из бен-
зола и этилена.
Полимеризация стирола
Стирол полимеризуется сравнительно легко, в особенности
по цепному механизму в присутствии инициаторов, что, повидимому,
должно быть объяснено наличием фенильной группы и, следова-
тельно, перегрузкой одного из атомов в этиленовой связи.
Полимеры стирола могут быть получены как цепной, так и сту-
пенчатой полимеризацией, однако техническое значение имеют
только высокомолекулярные продукты, получаемые на основе цеп-
ной полимеризации.
Полимеризация стирола по цепному механизму протекает под
влиянием инициаторов (инициированная полимеризация) или под
воздействием только температуры в присутствии инертных газов
(термическая полимеризация).
Термическую полимеризацию в присутствии воздуха нельзя счи-
тать чисто термическим процессом, так как кислород вызывает
образование перекисей, являющихся зародышами полимеризации.
В то же время атмосферный кислород при полимеризации стирола
действует не только как инициатор полимеризации, но и как инги-
битор, так как он реагирует и со свободными радикалами стирола,
образуя устойчивую полимерную перекись (стр. 43). Как видно из
рис. 89, влияние воздуха сказывается в некотором ускорении про-
цесса полимеризации в начальной стадии и в замедлении — в по-
следующих.
Термическую полимеризацию стирола в отсутствие воздуха осу-
ществляют при непрерывных башенных методах с применением вы-
соких температур (150—200°). Широко применяется также поли-
меризация стирола в присутствии инициаторов, которые значительно
увеличивают скорость полимеризации. Применение инициаторов
(органических и неорганических перекисей) позволяет вести поли-
меризацию при низких температурах в присутствии растворителей
пли в водной эмульсии.
Скорость и степень полимеризации стирола, а также выход поли-
202 Гл. V. Пластмассы на основе непредельных углеводородов
мера определяются закономерностями, общими для всех полимеров,
получаемых на основе цепного механизма. Чем выше температура
полимеризации, тем ниже молекулярный вес образующегося поли-
мера (рис. 90) и тем быстрее протекает процесс. Такая же зависи-
мость наблюдается и в отношении количества инициатора.
Рис. 89. Кинетика полимеризации стиро-
ла при различных температурах и газо-
вых средах.
Для полимеризации стиро-
ла могут быть применены все
три основных варианта — поли-
меризация чистого мономера,
полимеризация в растворителе
и эмульсионная полимериза-
ция. Так называемый блочный
метод с заливкой мономера в
формы и полимеризацию в рас-
творителях применяют сравни-
Температура полимеризации,°C
Рис. 90. Кривая зависимости мол.
веса полистирола от температуры
полимеризации.
* /—воздух; II—азот.
тельно редко. Наибольшее распространение получили непрерывные
методы блочной полимеризации чистого мономера и эмульсионный
метод.
При блочной полимеризации чистый стирол заливают в формы,
и с течением времени, обычно при нагревании, в присутствии воз-
духа или инертного газа (N2, СО2), в присутствии инициаторов или
без них, стирол постепенно густеет и превращается в блок — твер-
дый, прозрачный материал, имеющий форму сосуда, в котором про-
водился процесс.
Метод полимеризации чистого мономера в формах позволяет в ла-
бораторных условиях получать полистирол с весьма высоким моле-
кулярным весом, недостижимым при других методах, если полиме-
ризация проводится при низких температурах (30—40°) без внесе-
ния инициаторов в течение очень длительного времени.
Полимеризация стирола
203.
Техническая полимеризация стирола в формах обычно происхо-
дит при повышенных температурах (60—90°) и в присутствии
инициаторов (перекиси бензоила).
В этих условиях получают полимеры
со сравнительно невысоким молеку-
лярным весом, однако процесс про-
ходит значительно быстрее (12—
24 часа) и полистирол получается
с более стандартными техническими
свойствами.
Полимеризация чистого стирола
в формах имеет ряд существенных
недостатков, связанных с высокой
вязкостью и плохой теплопровод-
ностью образующегося блока. Бла-
Поперечное сечение блока
Рис. 91. Схема распределения тем-
пературы (7) и степени полиме-
ризации (2) в различных точках,
сечения блока.
годаря значительной экзотермично-
сти процесса и плохой теплопровод-
ности полимера внутри блока обра-
зуются зоны местного перегрева.
Неравномерность распределения тем-
пературы в блоке определяет различную скорость и, следовательно,.
различную степень полимеризации в отдельных его частях — мень-
шую в центре и большую на поверхности (рис. 91), которая
является причиной различной усадки блока и неизбежно приводит
к внутренним натяжениям, ведущим к появлению со временем тре-
щин в блоке.
При блочном методе полимеризации никогда не происходит пол-
ного превращения мономера, наличие же в блоке остаточного моно-
мера сильно снижает теплостойкость полистирола и является основ-
ной причиной его старения; последнее проявляется в помутнении
полимера и в образовании в нем трещин.
Вместе с тем блочный метод дает возможность без прессования
изготовлять прозрачные пластины, бруски и блоки. Последнее важно
при получении полистирола для некоторых специальных назначе-
ний, например для изготовления деталей в высокочастотной про-
мышленности.
Такие литые пластины и изделия имеют преимущество перед
прессованными (из порошка или гранул) в большей технической
теплостойкости, так как при достижении температуры стеклования
(~ 80°) они лишь несколько размягчаются, но не разрушаются и не
распадаются на кусочки, из которых они были запрессованы (что
в той или иной степени имеет место в прессизделиях).
Многие из вышеуказанных недостатков блочной полимеризации
могут быть уменьшены, если отливать плоские блоки, т. е. пластины
небольшой толщины, в которых условия теплопередачи значительно
лучше, чем в толстых блоках.
204 Гл. V. Пластмассы на основе непредельных углеводородов
Блочную полимеризацию обычно проводят путем предваритель-
ного смешения стирола с перекисью бензола (0,1—0,5%) и за-
ливки смеси в стеклянные формы. Эти формы нагревают в термо-
шкафах при 50—100° до образования твердого продукта. Время
полимеризации и молекулярный вес зависят как от температуры и ко-
личества инициатора, так в некоторой степени и от толщины блока.
Как уже было указано, кроме стационарных методов обычной
блочной полимеризации, для полимеризации чистого стирола особое
значение приобрели непрерывные методы.
Непрерывные методы полимеризации стирола позволяют полу-
чить продукт практически свободный от мономера с более высоким
молекулярным весом, чем это достижимо стационарными методами
блочной полимеризации в формах. Они позволяют автоматизировать
управление процессом, обеспечивают стандартность — постоянство
свойств полимера, улучшают условия труда, повышают производи-
тельность аппаратуры, значительно удешевляют продукт.
Непрерывную полимеризацию проводят в непрерывно действую-
щих аппаратах (башнях).
Аппараты (башни) для непрерывной полимеризации стирола
(жидких мономеров) построены по принципу «идеального вытесне-
ния». Сущность их работы заключается в том, что непрерывно по-
ступающие в аппарат исходные компоненты реакции по мере их
перемещения в аппарате вступают во взаимодействие с образова-
нием новых соединений, которые непрерывно вытесняются, не
смешиваясь с вновь поступающей реакционной смесью. Исключение
смешения поступающих компонентов реакции с ее промежуточными
и конечными продуктами позволяет получать однородный и стан-
дартный продукт. Чтобы устранить смешение и обеспечить необхо-
димую полноту реакции, следует соблюдать определенное соотно-
шение между временем реакции и высотой аппарата, а также опре-
деленную скорость реакции.
Основная аппаратура непрерывной блочной полимеризации стирола (рис. 92)
состоит из полимеризационной башни и двух реакторов предварительной поли-
меризации (форполимеризаторов) 4. Свежеперегнаиный стирол, содержащий не
менее 99,5% мономера и менее 0,03% дивинилбензола, * перекачивается центро-
бежным насосом из цехового хранилища в один из двух реакторов, причем
предусматривается также и предварительная фильтрация стирола от случайных
загрязнений. Реакторы представляют собою алюминиевые аппараты емкостью
около 2 л3. Они снабжены лопастными мешалками, делающими 50—60 об/мин.,
и змеевиками, в которых циркулирует вода с температурой 74—78°. Вода
уносит часть тепла, образующегося в результате экзотермической реакции;
* Содержание в мономере свыше 0,03% дивинилбензола приводит к получению
полностью нерастворимого продукта или же способствует осаждению неплавкой
смолы.
Полимеризация стирола
205-
таким обпазом достигается
Полимеризация происходит в
жается 55—65 часов до по-
лучения раствора с содер-
жанием 33—36% полимера
(содержание полимера опре-
деляют по показателю пре-
ломления). По окончании
предварительной полимери-
зации раствор поступает в
полимеризационную башню
для дальнейшей полимери-
зации. Башня имеет шесть
секций. В первой и вто-
рой секциях поддерживает-
ся температура 100—110°,
в третьей 150°, в четвертой и
в пятой 180°, в шестой
200°.
Каждая секция обогре-
вается индивидуально. Обо-
грев осуществляется паро-
вой рубашкой (первая и
вторая секции), паровой ру-
башкой со змеевиками (тре-
тья, четвертая и пятая
секции) и электрическим обо-
гревом (шестая секция).
Пары, выделяющиеся из
башни, отводятся в хо-
лодильник, а конденсат не-
постоянная температура в реакторе 80 + 2‘.
атмосфере азота без инициаторов и продол-
Рис. 92. Схема непрерывной (башенной) полнме-
ступает обратно н реактор
предварительной полимери-
зации.
В нижней, конусной
части башни находится
расплавленный полистирол,
практически свободный от
ризации стирола.
1 — паровая рубашка 2 —электрический обогреватель; 3—
шнекпресс; 4— реакторы предварительной полимеризации Z
5— холодильник; 6 — конвейер; 7— мельница.
мономера. Он непрерывной струей с температурой 200—220° подается на
шнекпресс 3, из которого выдавливается в виде непрерывного стержня, наре-
зается вращающимся ножом на короткие отрезки и уносится конвейером, на
котором охлаждается и затем попадает на мельницу для размола. Получаемый
таким образом гранулированный полистирол имеет мол. вес 150 000 (по центри-
фугальному методу). Средняя продолжительность пребывания в колонне
25—30 час. Производительность установки около 1 т/сутки, причем в каждый
реактор подаются 22,3 кг стирола в час.
Непрерывную блочную полимеризацию чистого стирола можно производить
с применением барабанной сушилки вместо полимеризационной башни по схеме,
изображенной на рис. 93. Из реакторов предварительной полимеризации реак-
ционная смесь с содержанием ~35% полимера поступает на барабаны (вальцы)
из хромированной стали (диаметр —- 50 см, длина — 900 сл), делающие
1,5—2 об/мин. и обогреваемые паром под давлением 14 ат. Полимеризация
на барабанах осуществляется в вакууме (остат. давл. 10—15 мм), который под-
держивается в сушильной камере трехступенчатым эжектором с конденсацией
пара. Готовый полимер срезается с барабанов специальными ножами, попадает
в тележки и после’охлаждения измельчается на ножевых мельницах. Выделяю-
щийся из сушильной камеры мономер конденсируется и поступает на ректифи-
кацию. Этот метод (по сравнению с башенным) дает возможность получать поли-
стирол с более высоким молекулярным весом (300 000—400 000), чем башенный.
206
Гл. V. Пластмассы на основе непредельных углеводородов
однако не обеспечивает в достаточной мере стандартности полимера и возможно-
сти регулирования его молекулярного веса. Производительность установки, со-
стоящей из двух реакторов и одной барабанной сушилки, ~350 кг/сутки.
При получении полистирола с низким молекулярным весом (—50000), кото-
рый находит применение для производства электроизоляционных лаков, можно
значительно упростить схему процесса, вводя мономер непосредственно в по-
лимеризационную башню, не применяя реакторы предварительной полиме-
Стирол
Рис. 93. Схема непрерывной полимеризации стирола
с применением барабанной сушилки.
1 — реакторы предварительной полимеризации; 2—барабаны (вальцы);
3 — тележки; 4, 5,6—холодильники; 7—сборник; 8— вакуумный эжектор.
ризации. Более низкий молекулярный вес в этом случае будет обусловлен более
высокой начальной температурой полимеризации стирола (100—110° в башне
вместо 82° в реакторах предварительной полимеризации) и более быстрым прохо-
ждением процесса полимеризации.
Полимеризация стирола в растворителях не получила сколько-
нибудь значительного применения. Это объясняется тем, что рас-
творители замедляют процесс полимеризации, причем замедляющее
действие возрастает по мере увеличения молекулярного веса рас-
творителей (как ароматических, так и жирного ряда). Раствори-
тели кроме того способствуют образованию более низкомолекуляр-
ных продуктов, молекулярный вес которых тем ниже, чем меньше
концентрация стирола (рис. 94). Значительные трудности встре-
чаются также и при удалении растворителя из полимера. Эта
операция может быть произведена различными методами, например
осаждением раствора в нерастворителях — петролейном эфире,
бензине и др., отгонкой растворителя при нагревании и перемеши-
вании или распылением раствора при помощи перегретого пара.
Однако все эти методы не дают возможности полностью удалить
растворитель, между тем наличие даже следов его крайне вредно
отражается на основных свойствах полистирола (в частности на
теплостойкости).
Полимеризация стирола
207
К преимуществам метода полимеризации в растворителях необ-
ходимо отнести возможность получения продукта с меньшей поли-
дисперсностью, не содержащего мономера. Благодаря равномер-
ному распределению температуры и меньшей вязкости среды, про-
цесс в этих условиях поддается более эффективному контролю, чем
при блочной полимеризации. Однако указанные недостатки ме-
тода, а также неизбежный расход рас-
творителей делают его малоэффектив-
ным.
Водно-эмульсионный метод полиме-
ризации стирола получил весьма широ-
кое применение. На 1 часть стирола
обычно берут 2—3 части воды. В каче-
стве эмульгатора применяют соли жир-
ных кислот (мыла), сульфированные
высшие спирты жирного ряда, соли
сульфокислот (например соль изопро-
пилнафталинсульфокислоты), а также
соли сульфокислот высоко кипящих па-
рафиновых углеводородов Ci3—Gie, по-
лучаемых при производстве синтетиче-
ского бензина. В качестве инициаторов
применяют главным образом водорас-
творимые перекиси (перекись водорода,
персульфат аммония или калия). Над-
сернокислые соли служат не только
Рис. 94. Зависимость молеку-
лярного веса полистирола от
концентрации мономера.
хорошими инициаторами, но могут в значительной мере заменять
эмульгаторы.
Обычный технологический процесс получения полистирола
эмульсионным методом складывается из следующих операций:
1) удаления ингибитора из стирола; 2) полимеризации стирола;
3) осаждения полимера; 4) отделения маточного раствора и про-
мывки полимера; 5) центрифугирования; 6) сушки; 7) измельчения,
просева и тарирования.
1) Удаление ингибитора производят путем промывки сти-
рола раствором едкой щелочи. В эмалированный чугунный реактор,
снабженный мешалкой, самотеком поступает дестиллированная
вода.
Через люк вносят едкий натр и после перемешивания в течение
получаса из мерного бачка самотеком или посредством вакуума
в реактор подают стирол. На 100 ч. стирола дают 50 ч. воды и
5 ч. едкого натра. После перемешивания в течение часа мешалку
останавливают и в продолжение 1,5—2 час. отстаивают раствор
щелочи. После этого нижний слой, состоящий из водного рас-
твора щелочи, сливают, а стирол с помощью вакуума перекачивают
в цеховые хранилища. Стирол не должен содержать свободной
щелочи.
208
Гл. V. Пластмассы на основе непредельных углеводородов
2) Полимеризация стирола. В качестве примера можно при-
вести следующий рецепт (в частях) полимеризационной смеси:
Стирол..................... 100
Вода дестиллированная . . . 300
Эмульгатор.................1—3
Едкий натр................... 0,2
Инициатор (надсернокислый.-
калий) ....................0,25—0,50
Полимеризацию ведут в эмалированном чугунном реакторе
(рис. 95) со сферическим днищем, обычно емкостью в 3—5 .и3,
снабженном паровой рубашкой, лопастной или рамной мешалкой,
делающей 50—75 об/мин. и работающей от мотора через редуктор.
Рис. 95. Реактор для эмульсион-
ной полимеризации стирола.
Рис. 96. Аппарат для осаж-
дения полистирола.
Реактор снабжен обратным холодильником; в крышке его имеются
штуцеры для подачи воды и стирола, для вакуумной линии и люк
для загрузки эмульгатора и инициатора.
Для проведения процесса в реактор через мерник самотеком
поступает дестиллированная вода с температурой около 50°, затем
включают мешалку и через люк загружают эмульгатор и едкий
натр. После перемешивания в течение 20—30 мин. в реактор из
мерника самотеком поступает стирол и после 15-минутного переме-
шивания через люк вносят инициатор (надсернокислый калий, взму-
ченный в воде). После внесения всех компонентов в рубашку реак-
тора дают пар, включают обратный холодильник и в течение
1,5—2 час. производят нагревание смеси до 65—70°. Дальнейший
Полимеризация стирола
209
подъем температуры до §5—95° происходит, главным образом, за
счет экзотермической реакции полимеризации. В этом интервале
температур реакция продолжается еще 2—3 часа. Конец реакции
определяют по содержанию мономера (должно быть менее 0,5%).
По окончании реакции содержимое реактора с помощью вакуума
или сжатого воздуха перекачивают в промежуточный бак.
3) Осаждение полимера. По окончании полимеризации реак-
ционная смесь представляет собою мелкодисперсную, стойкую
суспензию — латекс, для коагуляции которой необходимо разру-
шить эмульгатор. Это может быть достигнуто увеличением кислот-
ности среды до pH = 6—5,5 путем внесения в реакционную смесь
соляной или серной кислоты или дающих кислую реакцию солей
(например, сернокислого алюминия и др.). Для удаления остатков
мономера реакционную смесь продувают острым паром.
Коагуляцию производят в отдельных аппаратах (рис. 96) из
нержавеющей стали или эмалированного чугуна, снабженных ме-
шалкой и паровым барботером. В аппарат подают воду и кислоту
(или квасцы, взмученные в воде), а затем после перемешивания
медленной струей — реакционную смесь. После подачи всей реак-
ционной смеси производят продувку острым паром и при интенсив-
ном перемешивании доводят смесь до температуры 75—85°. После
перемешивания в течение 1,5—2 часов масса разделяется на два
слоя: нижний содержит осажденный полимер, верхний состоит из
водного раствора.
4) Отделение маточного раствора и промывку полимера произ-
водят в лаверах, куда передавливают массу из аппарата для оса-
ждения. Лаверы представляют собою цилиндрические сосуды из
нержавеющей стали или из алюминия с коническим ложным дни-
щем и рамной мешалкой. В крышке лавера имеются штуцеры для
подачи полистирола и воды и боковой штуцер для спуска маточ-
ника через ловушку. Вслед за полистиролом в лаверы подают
горячую воду и после 15-минутного перемешивания мешалку
останавливают. Полимер оседает, и маточный раствор спускают
через ловушку в канализацию. Такую промывку повторяют
3—5 раз.
5) Центрифугирование. Для отжима полистирола от воды всю
массу после последней промывки подают на центрифугу. Центри-
фуга из нержавеющей стали с числом оборотов 1000—1200
обычно имеет диаметр корзины 900—ЮООлл и объем 0,15—0,20 л3.
Отжим вместе с подачей длятся 30—40 мин. Влажность отжатого
полистирола 60—70%. Его выгружают в мешки и передают на
сушку.
6) Сушка полистирола может быть произведена различными
методами: стационарными в полочных вакуум-сушилках или (не-
прерывно) в сушильных барабанах. Стационарные методы сушки
полистирола пока наиболее употребительны. Влажный поли-
стирол раскладывают слоем 5—6 см на алюминиемые противни,
14 Зак. 1269. Э. И. Барг
210
Гл. V. Пластмассы на основе непредельных углеводородов
которые вставляют в вакуум-термошкафы, обогреваемые паром.
Температура в сушилках должна быть в пределах 60—70°, вакуум
100—200 мм остаточного давления. Время от времени сушилки не-
обходимо открывать и перемешивать массу на противнях. Сушка
продолжается 6—8 час. до остаточной влажности не более 0,5%.
7) Измельчение и просеивание. Высушенный полистирол со-
держит небольшие комки и должен быть подвергнут измельчению.
Измельчение можно производить в шаровых мельницах или (не-
прерывно) на дезинтеграторах. В последнем случае необходимо
следить, чтобы температура кожуха и вращающегося диска дезин-
тегратора не поднималась выше 50—60°. Измельченный полистирол
просеивают через сетку № 30—40 и тарируют в мешки.
Степень полимеризации полистирола, полученного эмульсион-
ным методом, и скорость процесса определяются количеством ини-
циатора, характером и количеством эмульгатора. От характера
эмульгатора зависит также технологический процесс отмывки поли-
стирола. Применение в качестве эмульгатора натриевых солей
сульфокислот высших парафиновых дестиллятов, содержащих
13—18 атомов углерода, обусловливает легкую отмывку полимера
водой и высокую степень полимеризации.
Приведенная технология может быть широко модифицирована
в отношении рецептуры, типа применяемых аппаратов, режима
и т. д.; здесь она приведена как примерная.
Эмульсионный метод имеет следующие преимущества: 1) про-
цесс полимеризации идет быстрее, чем в гомогенной среде или
в растворителях, и легко поддается регулированию и контролю;
2) полистирол, несмотря на значительную скорость полимеризации,
получается с тем средним молекулярным весом (100 000—200 000),
который необходим для дальнейшей его переработки (для экструзии
и литья под давлением), не содержит мономера (остаточный моно-
мер легко отгоняется с паром) и получается в виде тонкодисперс-
ного порошка, удобного для дальнейшей переработки (путем валь-
цевания или таблетирования).
При производстве полистирола необходимо учитывать токсич-
ность, взрыво- и огнеопасность стирола, который так же токсичен,
как и толуол, и при концентрациях свыше 0,2 мг/'л может вызывать
раздражение кожи, слизистых оболочек глаз и органов дыхания.
Поэтому при работе со стиролом необходимо применять защитные
приспособления и прежде всего обеспечить хорошую вытяжную и
проточную вентиляцию, а также принять меры к герметизации
аппаратов и хранилищ. Полистирол физиологически совершенно без-
вреден, но его пыль может образовать с воздухом взрывоопасные
смеси.
Сополимеры стирола
Как мы уже указали, значительное количество стирола нашло
применение в виде сополимеров с дивинильными соединениями для
производства синтетических каучуков. Таким, например, является
Сополимеры стирола
211
советский каучук СКС, представляющий сополимер бутадиена и
стирола.
Введение стирольных групп в макромолекулу полибутадиена
значительно увеличивает механические показатели полимера и в то
же время мало повышает его температуру стеклования, если содер-
жание стирола в сополимере не превосходит 20—30%
При сополимеризации стирола с дивинилбензолом, диаллило-
выми эфирами, с эфирами малеиновой кислоты и другими тетра-
функциональными соединениями можно непосредственно получить
неплавкие и нерастворимые продукты. Чем выше степень полиме-
ризации, тем при меньшей концентрации четырехфункциональной
компоненты достигается нерастворимость сополимера.
Так, при степени полимеризации 10 000 достаточно 0,01 мол. %
дивинилбензола для получения нерастворимого полистирола.
Техническое значение таких сополимеров со сшитыми структу-
рами пока еще невелико, так как, будучи неплавкими и нераство-
римыми, они не поддаются переработке
обычными методами (литью под давле-
нием, прессованию и др.). Однако
блочным методом сополимеризации
можно получать пластины, стержни,
блоки и т. п., которые обладают более
высокой теплостойкостью и из которых
детали могут быть изготовлены меха-
нической обработкой.
В табл. 20 приведена зависимость
температуры стеклования полистирола
от содержания дивинилбензола.
Значительно бдльшее техническое
значение имеют сополимеры стирола с
моновинильными соединениями, в част-
ности с неполярными или малополяр-
ТАБЛИЦА 20
Зависимость температуры
стеклования полистирола от
содержания дивинилбензола
Содержание дивинилбен- зола в сополи- мере стирол- дивинилбензол п/о Среднее число мономерных ! молекул сти- рола между поперечными связями । Температура стеклования полистирола •г.
0 0,6 0,8 1,0 1,5 172 101 92 58 87 89,5 92 94,5 97
ними, которые не снижают высоких диэлектрических свойств про-
дукта и в то же время значительно увеличивают его теплостой-
кость. Так большое значение имеет получение сополимера, стойкого
к кипящей воде.
Важнейшим продуктом является сополимер стирола с винил-
карбазолом. Теплостойкость такого сополимера лежит между
теплостойкостью полистирола (80°) и поливинилкарбазола (150°);
в зависимости от соотношения обоих мономеров можно получить
сополимеры, стойкие к кипящей воде, с теплостойкостью между
110—130°, которые в отличие от поливинилкарбазола (стр. 229)
хорошо перерабатываются методами экструзии и литья под
давлением.
Техническое значение имеют также продукты сополимеризации
2,5-дихлорстирола со стиролом, дающие полимеры с большей тепло-
стойкостью, чем полистирол.
12
Гл. V. Пластмассы на основе непредельных углеводородов
С. Н. Ушаков с сотрудниками изучил сополимеры стирола
с n-хлорстиролом и показал, что степень полимеризации мало за-
висит от соотношения компонентов, но скорость реакции увеличи-
Время полимеризации, часы
Рис. 97. Сополимеризация стирола
с л-хлорстиролом при 100° без ка-
тализатора.
/—стирол и я-хлорстирол 10: 90; 2—то же
30 : 70; 3 — то же £0:50; 4—то же 70:30;
5 —то же 90: 10; 6—стирол.
вается с повышением содержания
в реакционной смеси п-хлорсти-
рола (рис. 97).
Значительное увеличение теп-
лостойкости может быть достигну-
то при сополимеризации стирола с
акрилонитрилом и с нитрилом фу-
маровой кислоты, однако вслед-
ствие полярного характера этих
мономеров значительно снижают-
ся диэлектрические свойства поли-
мера. В табл. 21 показана зависи-
мость теплостойкости сополимера
стирола и нитрила фумаровой
кислоты от содержания нитрила.
Техническое значение получили
ТАБЛИЦА 21
Зависимость тепло-
стойкости сополимера
от содержания нитрила
Содержание нитрила фумаровой кислоты °!о Теплостой- кость сополимера °C
0 76—78
2,5 84
5,5 94
14 107
21 124
30 140
сополимеры стирола с хлор-
винилом, с хлорвинилиденом, а также с метилметакрилатом. По-
следний сополимер (с содержанием 10—30% стирола) широко при-
меняется в виде порошка, перерабатывае-
мого путем литья под давлением.
Следует указать на возможность гетеро-
полимеризации стирола. Гетерополимером
является, например, продукт сополимериза-
ции стирола с малеиновым ангидридом,
имеющий структуру:
... - СН2—СН—СН—СН—СН2—СН—СН—СН—СН2-...
Ill III
/\ СО со /\ со со
I i \ / I I \ /
\/ о \/ о
Работы по использованию подобных со-
полимеров, а также сополимеров стирола с
малеиновыми эфирами ведутся в последние
годы. Так, при сополимеризации стирола
с гликолевыми эфирами малеиновой кислоты образуются линейные
и плавкие продукты, способные, однако, к образованию простран-
ственных структур и к переходу в неплавкое состояние в процессе
прессования при 180—200°.
Структура и свойства полистирола
Технические свойства полистирола определяются в основном
условиями и методами полимеризации. От условий полимеризации
зависят: 1) степень полимеризации; 2) наличие мономера в поли-
Структура и свойства полистирола
213
мере; 3) степень полидисперсности и разветвленности цепи; 4) чи-
стота продукта (загрязнение эмульгатором и др.).
В технике различают «блочный» и «эмульсионный» полистирол.
Блочный (получаемый непрерывными методами) отличается боль-
шой чистотой, не содержит электролитов (эмульгаторов, солей)
и инициаторов. Его диэлектрические свойства поэтому значительно
выше, чем у «эмульсионного» полистирола.
Молекулярный вес полистирола, получаемого методом блочной
полимеризации, может варьировать в весьма широких пределах —
от 20 000 до 800 000 и выше. Технические продукты блочной поли-
меризации имеют обычно молекулярный вес от 50 000 (применяются
в качестве лаков) до 200 000—300 000 (применяются для прессова-
ния плит, для литья под давлением). Полимеры, получаемые эмуль-
сионной полимеризацией, обычно имеют молекулярный вес от 70 000
до 200000. Эмульсионный метод позволяет получать продукты и зна-
чительно большего молекулярного веса, однако такие продукты труд-
нее перерабатывать методом прессования и литья под давлением.
Макромолекулы полистирола характеризуются значительной раз-
ветвленностью и полидисперсностью, на что указывает расхождение
в значениях молекулярных весов, определенных вискозиметриче-
ским методом и методами осмотическим и центрифугальным.
Структура полистирола изучена различными методами, в част-
ности методом термической деструкции; оказалось, что он имеет
строение 1,3 («голова к хвосту») (стр. 60).
Подтверждением такого строения служит наличие в продуктах
сухой перегонки полистиролов:
1.3-дифенилбутена
сн2—сн2—с=сн2
1,3-дифенилпропана
СН2—СН2—СН.,
1,3,5-трифенилпентана
сн2—сн2—сн—сн2—сн2
На рис. 98 показана схема строения молекулы полистирола.
Полистирол имеет аморфную структуру. При растяжении проис-
ходит ориентация и распрямление его макромолекул, что приводит
к значительному увеличению прочности в направлении вытяжки;
214 Гл. V. Пластмассы на основе непредельных углеводородов
однако рентгеноструктура ориентированных пленок не дает указа-
ния на образование кристаллов. Таким образом, жидкостная
структура полистирола сохраняется как в обычном, так и в ориен-
тированном состояниях.
Физико-механические и диэлектрические свойства полистирола
обусловлены слабой полярностью его макромолекул (дипольный
момент ~ 0,37 • 10-18). По комплексу диэлектрических свойств поли-
стирол относится к наиб1олее совершенным диэлектрикам и уступает
лишь таким неполярным полимерам, как полиэтилен и политетра-
фторэтилен. Важнейшие диэлектрические показатели полистирола,
имеющие значение для высокочастотной техники, — диэлектрическая
постоянная и тангенс угла диэлектрических потерь — имеют весьма
низкие значения и в широких пределах не зависят от температуры
и частоты поля.
Рис. 98. Схема строения молекулы полистирола.
• —атома углерода: О—атома водорода.
Полистирол обладает практически абсолютной водостойкостью
(даже после месяца нахождения в воде вес образцов не увеличи-
вается), высокой химической стойкостью как к кислотам (даже
к фтористоводородной), так и к щелочам.
Полистирол бесцветен и отличается прозрачностью, пропуская
до 90% лучей видимого света, растворим в ароматических угле-
водородах, во многих эфирах (бутилацетате, этилгликольацетате
и др.), нерастворим в спиртах и в бензине.
При обычной температуре полистирол представляет собою твер-
дое, упругое тело с уд. весом 1,05. Лишь при 80—90° начинают
проявляться его высокоэластические свойства. Температура стекло-
вания — около 80°, теплостойкость по Мартенсу ниже (70—80°).
Теплостойкость полистирола, как это было показано А. П. Але-
ксандровым, не зависит от его молекулярного веса (при достиже-
нии определенной величины степени полимеризации), но сильно
снижается при увеличении содержания мономера (рис. 99). Эта
Структура и свойства полистирола
215
закономерность, как и в случае других полимеров, объясняется тем,
что температура размягчения связана лишь с подвижностью звеньев
макромолекул, а не цепей в целом и обусловлена только характе-
ром связи между звеньями молекул. При температурах ниже тем-
пературы стеклования и при обычных скоростях нагрузки полисти-
рол ведет себя как твердое, упругое тело.
Механические свойства полистирола зависят в определенном
пределе от степени его полимеризации. Низкомолекулярные поли-
меры чрезвычайно хрупки и имеют
малую прочность. По мере увели-
чения степени полимеризации уве-
личивается прочность, растяжи-
мость и уменьшается хрупкость
полимера. Однако выше опре-
деленной степени полимериза-
ции (соответствующей примерно
мол. весу 100 000) механические
свойства уже мало изменяются.
Низкомолекулярные полистиролы
выше температуры размягчения в
сравнительно узком температур-
ном интервале легко переходят в
Рис. 99. Зависимость температуры
размягчения полистирола от процент-
ного содержания мономера (стирола).
вязкую жидкость; высокомолеку-
лярные полистиролы выше темпе-
ратуры стеклования переходят в
высокоэластическое (каучукопо-
добное) состояние, которое сохраняется в пределах широкого тем-
пературного интервала (от 80 до 150° и выше).
При высоких температурах происходит деполимеризация поли-
стирола, причем чем выше его молекулярный вес, тем при меньшей
температуре наблюдается деструкция цепей. В присутствии воздуха
процессы деструкции ускоряются. Среднемолекулярный полистирол
деполимеризуется при 300° с выделением мономера и других про-
дуктов (трифенилбензола, дифенилбензола и др.). Высокомолеку-
лярный полистирол (мол. вес 200 000 и выше) деполимеризуется
при 250—300°.
Полистирол легко перерабатывается методами прессования,
литья под давлением, экструзии и выдувания. Некоторое применение
находит и механическая обработка пластин и блоков из полисти-
рола, в частности для линз и некоторых деталей электротехники.
Прессование имеет лишь ограниченное применение, так как поли-
стирол после горячего прессования требует охлаждения под давле-
нием. Применяется прессование блоков ио пресспорошка с после-
дующей строжкой этих блоков на строгальных машинах для
получения листов и пленок.
Основным методом переработки полистирола, отличающегося
высокой текучестью, является литье под давлением.
216
Гл. V. Пластмассы на основе, непредельных углеводородов
Для этого применяют полистирол в виде порошка или гранул. Литье ведут
при 180—230° и при удельном давлении 500—2000 кг/см.'1.
Литье под давлением основано на принципе доведения массы в обогревае-
мых цилиндрах до пластического состояния и выдавливания в гнездо закрытой
прессформы через специальные литниковые каналы. Прессформу не обогревают,
Рис. 100. Схема расположения изделий, литника и лит-
никовых каналов.
Г— литник; 2—литниковые каналы; 3—отлитые изделия.
а большей частью охлаждают. Залитая масса под давлением заполняет свобод-
ное пространство прессформы, охлаждается и превращается в твердое изделие,
которое выталкивают из гнезда наружу. Этим путем изготовляются тонкостенные
изделия (0,5—3 мм) сложной конфигурации сравнительно небольшой величины
(до 100—300 г). На рис. 100 показана схема расположения отлитых изделий,
литника и литниковых каналов. Схема рабочей части машины для литья под
давлением приведена на рис. 101. Материал из бункера 1 поступает в обогре-
7 в
Рис. 101. Схема рабочей части машины для литья под давлением.
Г—бункер; 2—цилиндр; 3. 8—элементы обогрева; 4—литниковый канал; 5—прессформа;
6 —сопло; 7—сердечник; 9—плунжер
ваемый цилиндр 2, откуда с помощью плунжера 9 через сопло 6 продавли-
вается сквозь литниковый канал 4 в прессформу 5. Обогрев машины произво-
дится элементам» 3 и 8.
Современные машины для литья под давлением полностью автоматизиро-
ваны. Они состоят из двух основных частей: 1) устройства для выдавливания
пластической массы через сопло в прессформу и 2) устройства для открывания,
закрывания и герметизации прессформы. Первая часть, в свою очередь, состоит
из трех основных частей и ряда регулирующих устройств: 1) бункера с регуля-
Структура и свойства полистирола
217
тором подачи порошка в цилиндр машины; 2) обогреваемого цилиндра с голов-
кой и 3) плунжера, создающего давление.
Обогрев цилиндра производится либо непосредственно электрическими эле-
ментами, либо через нагреваемое ими масло, циркулирующее в рубашке
цилиндра. Для более равномерного нагрева материала близ сопла устанавли-
вается сердечник 7, облегчающий нагревание материала, так как последний
в этом случае должен проходить по тонким каналам. Чтобы материал не при-
липал к плунжеру и, следовательно, не двигался вместе с плунжером при его
обратном ходе, в задней части рабочего цилиндра имеются каналы для охла-
ждения. Подвод прессформы к соплу машины, зажатие ее и раскрытие произ-
водятся при помощи плунжера, приводимого в движение гидравлическим или
рычажным механизмом.
ТАБЛИЦА 22
Зависимость удлинения и числа переменных
изгибов пленки полистирола от вытяжки
Наряду с литьем под давлением широко применяется метод
переработки полистирола выдавливанием на шнекпрессах (путем
экструзии или шприцевания). Метод этот дает возможность произ-
водить пленки, прутки, трубки и пластины; в частности, он нашел
большое применение для получения полистироловых пленок (стиро-
пленки).
Для изготовления
стиропленки выходя-
щую из мундштука
шнекпресса трубку в
горячем состоянии
('>' 140—160°) растяги-
вают сжатым воздухом
и после охлаждения и
отверждения разрезают
в продольном напра-
влении. Для этого при-
меняют также и метод обычной механической вытяжки ленты после
выхода ее из мундштука. Вытяжку часто ведут одновременно в про-
Пленка полистирола толщиной 0,1 мм Предел прочности на разрыв KZjMM* Удлине- ние °,о Число переменных изгибов
Без вытяжки .... 2,5—2,8 1 1
С вытяжкой (~4ОО°/о) 9,2—10 5 1000—2000
дольном и поперечном направлениях, достигая равномерного уве-
личения прочности пленки в обоих направлениях, однако возможная
степень вытяжки при этом уменьшается вдвое. Методом экструзии
с одновременной вытяжкой удается получать весьма тонкие поли-
стирольные пленки (толщиной 0,1—0,01 мм) и нити (~ 0,01 мм).
Удельная прочность ориентированной пленки зависит от степени ее
вытяжки. Одновременно с увеличением прочности на растяжение
увеличивается и внутренняя гибкость пленки, в частности прочность
на переменный изгиб, как это видно из табл. 22.
Для изготовления пленки и других изделий методом выдавливания на
шиекпрессах существенное значение имеет устройство мундштука шнекового
пресса. Схема обычных шнекпрессов показана на рис. 172 (стр. 439), а схема
устройства мундштука шнекпресса—на рис. 102. В обогревательном цилиндре
пресса 7 закрепляется мундштук 11 с соплом 12, из которого выходит форму-
емая масса. Мундштук 11 имеет патрон 6, состоящий из конических деталей 8
и 10, связанных цилиндрическими пластинками 2, 3, 4. В пластинке 4 имеется
отверстие 5. Пластинка 3 — меньшего диаметра, поэтому вокруг нее образуется
цилиндрическое отверстие 9. Пластинка 2 имеет отверстие 1. Разогретый пла-
стичный полистирол продавливается через отверстие патрона 5, затем, обтекая
218 Гл. V. Пластмассы на основе непредельных углеводородов
конус 8 н цилиндрическое отверстие 9, тщательно перемешивается; он подвер-
гается дополнительному перемешиванию, проходя через отверстие 1 и попадая
Рнс. 102. Мундштук шнекпресса (схема).
раствор полистирола, полу-
ченный при полимеризации стирола в растворителях, либо раство-
ряют полистирол в смеси ксилола и диоксана или ксилола и ме-
тиленхлорида (97: 3). Полу-
ченный раствор отфильтро-
вывают, вакуумируют для
освобождения от воздушных
пузырьков и отливают на
сетку ленточных машин.
Для улучшения гибкости
полистирола иногда вводят
пластификаторы. В качестве
пластификаторов желательно
применять малополярные
вещества, совместимые с по-
листиролом и не снижаю-
щие его диэлектрических
свойств. Обычно применяют
хлорированные ароматиче-
ские углеводороды, эфиры
низших спиртов с высшими
жирными кислотами, а в не-
которых случаях также и
обычные пластификаторы —
трикрезилфосфат, дибутилфталат и др. Наряду с пластификаторами
в камеру 13, а затем выходит из
нее в форме, соответствующей
соплу 12 (в виде стержня, трубки
нли пластины).
Дальнейший процесс получе-
ния и растяжения ленты (пленки)
показан на рнс. 103. Выходящая
из сопла мундштука 1 при тем-
пературе 150—160° лента поли-
стирола захватывается штырями
бесконечного транспортера 3, обе-
гающего вокруг роликов 2. Шты-
ри увлекают ленту 4 и растяги-
вают ее. Готовая лента (нли плен-
ка) наматывается на барабан 5.
Пленки полистирола по-
лучают и путем отливки из
растворов на металлические
сетки пленочных машин или
на барабан с хорошо отпо-
лированной поверхностью.
Для этого применяют либо
иногда вводят наполнители, которые служат для увеличения тепло-
Применение полистирола
219
стойкости, твердости и статической прочности полистирола, однако
при этом увеличивается и его хрупкость. В общем, как правило,
наполнители усиливают те свойства, которые ослабляются введе-
нием пластификаторов. В качестве наполнителей полистирола при-
меняют минеральные порошки, главным образом кварцевый поро-
шок (при получении материалов для электротехники), а также си-
ликагель, углекислый кальций (мраморная мука), слюдяной порошок,
тальк, окись цинка и т. п.
Для получения прессматериалов на основе полистирола с напол-
нителями, красителями, пластификаторами и т. д. исходные ингре-
диенты предварительно смешивают в мешателе или в шаровой мель,
нице, затем обрабатывают на смесительных фрикционных вальцах
при 80—120°. Полученную массу размалывают в шаровой мельнице
или в дезинтеграторах и просеивают через сито 40—80 меш.
Свойства полистирольных прессматериалов (ненаполненных и
наполненных) сильно варьируют в зависимости от молекулярного
веса полистирола, метода полимеризации, характера пластифика-
тора и наполнителя.
В СССР выпускают полистирол различных марок, например:
«масса для литья под давлением», являющаяся композицией эмуль-
сионного полистирола с наполнителем, пластификатором и красите-
лем; «полистирол эмульсионный» — для получения литьевого
материала методом горячего вальцевания и методом таблетирова-
ния с последующим прогревом, а также плиты и трубки из поли-
стирола, получаемые горячим прессованием и экструзией блочного
полистирола. Показатели свойств полистирольных прессматериалов
следующие:
Уд. вес, г/с.и3 .................................
Коэффициент преломления света, п~£...............
Предел прочности на разрыв, кг; см-...............
Предел прочности на изгиб, кг{см3.................
Уд. ударная вязкость, кг-см!см3...................
Уд. ударная вязкость с надрезом, кг • см/см3 . . .
Твердость по Бринелю, кг/мм3......................
Теплостойкость по Мартенсу, °C.................
Водостойкость (привес за 24 часа), %...........
Теплопроводность, калием • сек • °C............
Коэффициент теплового расширения...............
Уд. объемное электрическое сопротивление, Q-cm . .
Уд. поверхностное электрическое сопротивление, Й .
Диэлектрическая постоянная при 106 гц..........
Тангенс угла диэлектрических потерь при 10е гц .
1,05—1,65
1,59—1,67
300—5(Ю
400—800
5—15
1,0—2,5
12—21
65—85
0—0,05
1,9—3,810-4
6—П .10-->
5 • 1015—5 10'7
1015—1017
2,2—2,8
0,0002—0,001
Применение полистирола
Полистирол получил весьма широкое применение и является одним из наи-
более распространенных полимеризационных пластиков. Высокие диэлектриче-
ские свойства, водостойкость и химстойкость, прозрачность и бесцветность, спо-
собность легко перерабатываться методом литья под давлением и экструзии
220
Гл. V. Пластмассы на основе непредельных углеводородов
обеспечили ему широкие области распространения в различных отраслях техники
и для производства предметов широкого потребления.
Важнейшей областью использования полистирола является промышленность
средств связи и высокочастотная электротехника. Так полистирол применяют
для производства радиотехнических деталей, пленок для высокочастотных
конденсаторов для изоляции высокочастотных кабелей и т. п. Из поли-
стирола изготовляют основания для конденсаторов, основания под про-
ходные и опорные изоляторы, антенны, ламповые панели, каркасы катушек,
лабораторную химическую посуду (воронки, фильтры, стаканы и др.). В част-
ности, полистирол является лучшим материалом для производства сосудов для
хранения фтористоводородной кислоты. Он пригоден для производства аптечной
тары, тары для пищевой промышленности и др. Из полистирола делают про-
зрачные баки для кислотных аккумуляторов, кислотопроводы, различные детали
химаппаратуры; в полиграфической промышленности полистирол служит для
изготовления пробельного материала и шрифта (взамен свинца).
В различных отраслях техники находят применение полистирольные лаки
и компаунды. Так в радиотехнике — покрывные пропиточные компаунды транс-
форматоров, лаки для катушек индуктивности; в химической промышленности
полистирольные лаки применяют для борьбы с коррозией. Следует, однако, ука-
зать, что в ряде случаев они обладают недостаточной адгезионной способностью.
Полистирольным лаком пропитывают бумагу, ткань, дерево, мрамор, шифер
и т. д.; иногда их пропитывают стиролом с последующей полимеризацией.
Новый вид слоистых материалов получают путем прессования ткаии и
волокна, изготовленного из полистирола. Прессование волокна и ткаии из поли-
стирола производят на плиточных прессах при 120—150°, т. е. в температурном
интервале высокоэластичности (Те — Ттек). В этих условиях получают монолит-
ный, прозрачный материал, сохраняющий, однако, слоистую структуру ориенти-
рованных нитей. Такие материалы обладают прочностью класса слоистых пласти-
ков. В отличие от обычных слоистых пластиков (стр. 461), имеющих гетеро-
генную структуру (смола и наполнитель), пластики этого типа гомогенны и
состоят только из полимера. Получаемые материалы обладают различной плот-
ностью, зависящей от степени плавления нигей и удельного давления при
прессовании.
Широкое распространение получили полистирольные пленки (стиропленка,
стирофлекс), получаемые методом экструзии с последующей вытяжкой
(в 5—10 раз). Эти пленки применяют в различных отраслях техники, главным
образом в радиотехнике, а также для упаковки пищевых продуктов, фармацев-
тических препаратов и др.
Благодаря высокой прозрачности, полистирол применяют для остекления
общественных зданий, для производства оптических деталей и прозрачных
моделей. Из полистирола в галантерейной промышленности изготовляют пуго-
вицы, гребни, ручки для щеток и т. д„ граммофонные пластинки, а также про-
тезы, в частности — искусственные челюсти и зубы.
Наряду с вышеуказанными достоинствами полистирольные пластики обла-
дают и существенными недостатками, которые ограничивают их применение и
заставляют искать пути к улучшению их свойств.
Одни из основных недостатков — низкая теплостойкость (~80°), которая
существенно ограничивает область применения полистирола.
В частности, полистирол не противостоит кипячению в воде, что ограничи-
вает возможность его применения в электротехнике, в производстве медицин-
ских приборов, для изделий, применяемых в пищевой промышленности, для изде-
лий народного потребления и т. д.
Более важным недостатком является его большая хрупкость, низкие показа-
тели удельной ударной вязкости, высокая чувствительность к надрезу. Работами
советских ученых (В. Р. Регель и др.) было показано, что изделия из полисти-
рола могут самопроизвольно разрушаться вследствие внутренних напряжений.
Разрушение происходит вследствие непрерывного развития и роста внутренних и
знешних микротрещин (стр. 124). Рост микротрещин происходит под действием
Поропласты из полистирола.
221
внутренних напряжений, которые вследствие высокой хрупкости материала не
могут релаксировать. Эти недостатки могут быть отчасти устранены при над-
лежащей конструкции изделия и прессформы.
Поропласты из полистирола
Полистирол применяют для производства легковесных пористых
пластиков (поропластов или пенопластов), состоящих из замкну-
тых ячеек, наполненных воздухом или каким-либо другим газом
(азотом). Вследствие замкнутой пористости такие пластики не впи-
тывают влаги; они отличаются очень малой плотностью.
Следует указать, что поропласты изготовляют не только из поли-
стирола, но и из ряда других полимеров — полихлорвинила, поли-
винилформаля, ацетилцеллюлозы, мочевино-формальдегидных и
феноло-формальдегидных смол, полиуретанов, полиэфиров, алли-
ловых смол и др. В принципе поропласты можно изготовлять из
любых полимеров и смол, обладающих достаточной текучестью
в процессе переработки при совместном действии температуры и
давления. Они могут быть изготовлены различными методами,
большинство которых основано на внесении газа в состав пластич-
ной композиции полимера. Известны следующие технологические
методы, ведущие к получению поропластов:
1. Вязкую пластическую массу вспенивают путем механиче-
ского перемешивания и при таком ее состоянии проводят процесс
отверждения. Твердая вспененная масса сохраняет пористую
структуру.
2. В пластичной, вязкой массе под давлением растворяют газ;
после снятия давления масса вспенивается за счет расширения
газа. Отверждение фиксирует пористую структуру.
3. В массу вводят водорастворимые твердые вещества, которые
после формования растворяют и вымывают из массы.
4. Волокнистую рыхлую массу пропитывают связующим и от-
верждают при небольшом давлении, — получается легкий пластик
волокнистой структуры. В некоторых случаях для этого применяют
термопластические волокна, обычно из полистирола, в виде рыхлого
мата. Система закрытых пор при этом образуется за счет расплавле-
ния рыхлой, пластичной массы.
5. Пластики в виде листов бумаги или ткани, пропитанных свя-
зующим, а также в виде пленок, гофрируют на специальных
вальцах, а затем формуют (склеивают) в изделия с низкой плот-
ностью (трубы, полые стержни, сетки и др.). Изготовленные мате-
риалы обладают малой плотностью, жесткостью и прочностью.
6. В состав композиции вводят вещества, которые при опреде-
ленных температурах (при т. плавл. полимера порядка 100—150°)
разлагаются с образованием газа (N2 или СО2); такие вещества
называют порофорами. Выделяющийся газ вспенивает массу, в ко-
торой при отверждении фиксируется пористая структура.
222
Гл. И Пластмассы на основе непредельных углеводородов
Из всех перечисленных методов наибольшее применение получил
последний — изготовление поропластов с помощью порофоров.
В качестве порофоров можно применять различные вещества:
двууглекислый натрий, углекислый аммоний и др.; однако наиболее
эффективными порофорами оказались органические вещества типа
азосоединений, имеющие группу —N=N—, которые при определен-
ной температуре распадаются с выделением N2; последний обусло-
вливает мельчайшую пористость пластика. Из ряда испытанных
порообразователей техническое применение получили лишь немно-
гие. В качестве примера укажем на следующие соединения:
1) азоизобутиродинитрил
сн3 сн3
СНз-С—N=N—С—СН3
C=N C=N
(т. плавл. 103—104°; т. разл. 120°); разложение с выделением азота
протекает по схеме:
СН3 сн3 СН3 СН3
II II
сн3—с—N=N—С—СН3 —> СН3—С----С—CH3 + N.;;
II I I ‘
C=N C=N C=N C=N
2) азобензол CeH5N = NCeH5 (т. плавл. 68°; т. разл. 100—110°);
3) азодикарбамид NH2CON = NCONH2 (т. разл. 180—200°);
4) азотолуол CH3C6H4N = NCgHiCHa (т. разл. 55°).
Процесс получения поропластов с помощью порофоров состоит
из следующих операций.
Тонкоизмельченный полимер смешивают в шаровой мельнице
с твердым измельченным порофором примерно в соотношении
70—90 ч. полимера на 30—10 ч. порофора. Полученную гомогенную
смесь загружают в прессформу и прессуют при удельном давлении
150—300 кг/см2, медленно поднимая температуру до точки разло-
жения порофора. Обычно эта температура значительно выше тем-
пературы размягчения полимера. Полимер плавится (размягчается)
и образует пластическую, гомогенную массу, причем противодавле-
ние образовавшегося внутри пластичной вязкой массы газа уравно-
вешивается внешним давлением пресса. Когда образование газа
заканчивается, прессформу под давлением охлаждают до комнат-
ной температуры (20—25°).
По охлаждении изделие вынимают из формы. При комнатной
температуре полимер имеет достаточную твердость и прочность (по-
рядка 300—500 кг/сл12); поэтому он удерживает газ внутри замкну-
тых пор и имеет плотность, близкую к его нормальной плотности.
Для превращения полимера в поропласт его подвергают мед-
ленному нагреванию. Этот этап является решающим в производ-
стве поропластов, так как материал в результате постепенного раз-
мягчения и расширения газа должен во много раз увеличиться
Применение поропластов
223
в объеме (до 10—15 ряз). Скорость подъемз температуры нс
должна превышать 8—10° в час, а при достижении температуры
размягчения полимера 3—5° в час. Нагрев производят в калори-
ферных сушилках при принудительном движении воздуха. По окон-
чании прогрева и расширения материала температуру в сушилке
медленно снижают до 20—30°.
Свойства поропластов
Свойства поропластов зависят как от химической структуры
полимера и его физических свойств, так и от плотности самого
поропласта.
Поропласты могут быть получены с весьма широкими пределами
плотности (от 8 до 250 кг/м3). Напомним, что плотность пробки,
наиболее легкого из известных естественных материалов,
200—250 кг)м2. Соответственно, поропласты имеют и наиболее низ-
кую по сравнению со всеми известными теплоизоляционными мате-
риалами теплопроводность (табл. 23), которая зависит от плотности
и от размера ячеек. Чем мень-
ше размеры ячеек при одной и
той же плотности, тем ниже
теплопроводность.
Прочность поропластов опре-
деляется их плотностью и
структурой, а также химиче-
ским составом полимера. Чем
меньше плотность, тем соответ-
ственно ниже прочность поро-
пласта. Очевидно, что проч-
ность поропласта должна быть
характеризована его удельной
прочностью из расчета на еди-
ТАБЛИЦА 23
Коэффициент теплопроводности
различных материалов
Материал Коэффициент теплопровод- ности кал/м^час^С
Шлаковое волокно . . . 0,073
Стеклянное волокно . . 0,064
Пробка 0,038
Поропласты (в зависи- мости от состава и плотности) 0,06—0,015
ницу веса материала.
Поропласт на основе полистирола имеет ряд преимуществ перед
другими поропластами. Эти преимущества заключаются, главным
образом, в высокой водостойкости, прочности (из расчета на еди-
ницу веса), а также в высоких диэлектрических свойствах. Свойства
полистирольного поропласта характеризуются следующими дан-
ными:
Плотность, кг/лг3............................. 15—10Э
Предел прочности на сжатие, кг]слР............ 2—65
Предел прочности на разрыв, кг/см2............ 2,5—25
Наибольшая рабочая температура, °C............ —|- 80
Применение поропластов
Весьма малая плотность (в некоторых случаях в десять—пятнадцать раз
меньше плотности пробки), химическая стойкость и водостойкость, негорючесть
или малая горючесть и сравнительно хорошие механические свойства позволяют
применять поропласты из различных полимеров в самых разнообразных обла-
стях: в строительной технике, машиностроении, судостроении и т. д. Их
224
Г.1. V. Пластмассы на основе непредельных углеводородов
применяют в качестве теплоизоляционного и конструкционного материала, про-
межуточного слоя в сочетания с жесткими пластиками для производства легких и
прочных конструкций. Широко применяют полистирольный поропласт для изо-
ляции рефрижераторов в домашних и промышленных холодильниках, для
изоляции термосовых котлов и автоклавов, а также как заполнитель для пере-
городок перекрытий, в качестве звуко- и теплоизоляционного материала.
В радиочастотной технике поропласты из полистирола применяют в каче-
стве конструкционного электроизоляционного материала.
Полимеры замещенных стиролов
Как уже было указано, недостаточная теплостойкость полисти-
рола (65—85° по Мартенсу) значительно ограничивает область его
применения, в частности в электротехнике. Естественно поэтому
стремление получить полимер, являющийся таким же совершенным
диэлектриком, как полистирол, и так же хорошо поддающийся
обработке методом литья под давлением, но со значительно более
высокой теплостойкостью. Основным направлением для достижения
этой цели является введение заместителей в ядро стирола, в част-
ности галоидов. Работы по синтезу галоидостиролов и по получе-
нию более теплостойких «полистиролов» особенно широко проводи-
лись в последние годы.
Введение заместителей в кольцо стирола увеличивает асимме-
тричность его молекулы и, следовательно, не уменьшает, а уве-
личивает его полимеризационную способность. Однако введение
одного заместителя, естественно, приводит к ухудшению диэлек-
трических свойств полимера, так как молекула последнего приобре-
тает больший дипольный момент. Если же ввести в кольцо два оди-
наковых заместителя в пара-положение друг к другу, то суммарный
дополнительный дипольный момент молекулы будет равен нулю и
макромолекула в целом остается неполярной (слабополярной)
в той же мере, как и сам полистирол. * Вместе с тем эффект усиле-
ния межмолекулярных сил, благодаря взаимодействию введенных
полярных групп, а также вследствие утяжеления ядра, приводящего
к торможению вращения сегментов, сохраняется; а это приводит
к более высокой теплостойкости полимера. Примером может слу-
жить полимер 2,5-дихлорстирола, имеющий столь же малый ди-
польный момент, как и полистирол:
... — СН2—СН—СН2— СН— ...
A-ci А-С1
С1—\/ С1—\/
Все замещенные производные стирола можно разделить на
три группы — производные, полученные замещениями: 1) в цепи,
2) в ядре, 3) в ядре и в цепи.
Техническое значение имеет вторая группа. Стиролы, замещен-
ные в ядре, обычно полимеризуются так же легко, как стирол, или
* Диполи характеризуются векторной величиной, поэтому два равных диполя,
введенные в пара-положение, дают суммарный дипольный момент, равный нулю.
Полимеры хлорстиролоа
225
даже значительно быстрее и дают полимеры с высокой степенью
полимеризации. По характеру заместителя замещенные в ядре
стиролы также могут быть разделены на ряд групп: 1) алкилсти-
ролы и арилстиролы, 2) алкоксистиролы и арилоксистиролы и 3) га-
лоидостиролы. Из них техническое значение получили полимеры
галоидостиролов, главным образом хлорстиролы.
Полимеры хлорстиролов
В настоящее время синтезированы и изучены о-, м- и п-хлорсти-
ролы и все шесть изомерных дихлорстиролов (2,3-; 2,4-; 2,5-; 2,6-;
3,4- и 3,5-). Как мы уже указали, особый интерес представляет
2,5-дихлорстирол, полимер которого обладает столь же высокими
диэлектрическими свойствами, как и полимер стирола.
С. Н. Ушаков с сотрудниками синтезировал о-, м- и п-хлорсти-
ролы и изучил условия их полимеризации, степень полимеризации
образующихся полимеров и технические свойства последних.
Для синтеза .и-хлорстирола исходят из лг-нитробензальдегида;
действием хлористого олова в кислой среде его превращают
в л«-аминобензальдегид, который, по Зандмайеру, переходит в ^-хлор-
бензальдегид. Последний по реакции Гриньяра через магний-орга-
ническое соединение переводят в метил-л*-хлорфенилкарбинол, де-
гидратацией которого в присутствии KHSO4 получают ж-хлорсти-
рол. Все эти процессы могут быть представлены схемой:
сно сно
л«-Хлорстирол — жидкость с т. кип. 51°, d-° =1,11, п"" 1,5682.
Полиметахлорстирол—прозрачный пластик с температурой размяг-
чения свыше 90°, обладает высокими диэлектрическими свойствами,
хотя в этом отношении он несколько и уступает полистиролу.
о-Хлорстирол был синтезирован, исходя из о-толуидина, заме-
щением аминогруппы хлором, окислением метильной группы до
альдегидной и превращением образовавшегося 2-хлорбензальдегида
по реакции Гриньяра в метил-о-хлорфенилкарбинол с последующим
отщеплением от этого соединения молекулы воды:
СН3 СН3 | | /СН сно сн—сн3 сн=сн2 1 1 i
0-NH2 (j-C1 0 ci CHMgj ' /X—Cl -ню ' C1 \/ KKSO,"* 1^/1
n-Хлорстирол был из п-толуидина. синтезирован аналогичным образом, исходя
15 Зак. 1269. Э. И. Барг
226
Гл. V. Пластмассы на основе непредельных углеводородов
Из дихлорстиролов применение в технике нашел главным обра-
зом уже упомянутый 2,5-дихлорстирол.
Для получения 2,5-дихлорстирола можно исходить из этил-
/г-дихлорбензола и вести синтез по схеме:
/ОН
СН2-СН3 СНС1—сн3 сн—сна сн=сн2
/\_С1 С12 /\—С1 омыление С1 ~нэ° , С1
С1 \/ —* С1\/ С1 \/ С1 \/
или СН=СН,
I
-НС1 С1
С1 \/
Полимеризацию дихлорстиролов ведут аналогично полимериза-
ции стирола. Ее осуществляют в присутствии перекисных ка-
тализаторов либо в блоке, либо в эмульсиях или в растворите-
лях. Дихлорстиролы имеют более высокую полимеризационную
способность, чем стиролы. Полимеризация 2,5-дихлорстирола при
4-70° продолжается 3—5 часов, причем идет настолько полно, что
в образующемся полимере мономер практически отсутствует
(остается 0,1—0,5% мономера).
Поли-2,5-дихлорстирол представляет собою прозрачный пластик
с уд. весом 1,38—1,40. Теплостойкость его значительно выше тепло-
стойкости полистирола (по Мартенсу она равна 115°), и он вы-
держивает кипячение в горячей воде. Диэлектрические свойства
этого пластика столь же совершенны, как и полистирола, но воз-
можная область его применения вследствие высокой теплостой-
кости значительно шире. Свойства поли-2,5-дихлорстирола могут
быть представлены следующими данными:
Уд. вес, г1см?..................................... 1,38—1,40
Теплостойкость по Мартенсу, °C.............. 115
Предел прочности на изгиб, кг! см-......... 600
Предел прочности на разрыв, кг]см^.......... 450
Уд. ударная вязкость, кг-CMfcM'i............ 6—12
Водостойкость (привес за 24 часа), % • . . . . 0
Уд. объемное электрическое сопротивление Q-см 101в—1017
Диэлектрическая постоянная при 106 . 2,6
Тангенс угла диэлектрических потерь при 10е гц 0,0002—0,0004
Полидихлорстирол может быть переработан методом литья под
давлением, методом экструзии (вытяжкой), он так же хорошо обра-
батывается и на станках.
Основное применение полидихлорстирол нашел в качестве тепло-
стойкого диэлектрика в производстве высокочастотных изоляцион-
ных деталей для радиотехники; его применяют также для изгото-
вления медицинских инструментов (в этом случае важна его
стойкость при кипячении в воде), для приготовления химически стой-
кого лака, применяемого для покрытия деталей, работающих в актив-
Пластические массы на основе поливинилкарбазола
227
ных средах при повышенных температурах, а также в оптике —
для производства линз с высоким коэффициентом преломления.
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИВИНИЛКАРБАЗОЛА
(Карбазовинипласты)
Поливинилкарбазол является полимером N-винилкарбазола, ко-
торый получается при действии ацетилена на карбазол в присут-
ствии щелочи:
|/Ч| |/Х| + сн=сн —> f4! |/Х|
сн=сн2
Карбазол (дибензопиррол) относится к ряду производных пир-
рола; вместе с пирролом и индолом (бензопирролом) он находится
в каменноугольном дегте и может быть выделен из антраценовой
фракции; при перегонке сырого антрацена над едким кали карбазол
остается в виде карбазолкалия, а при обработке последнего кисло-
тами выделяется в чистом виде. Карбазол представляет собою кри-
сталлическое вещество с т. плавл. 238° и т. кип. 355°.
Винилирование карбазола ацетиленом производят в автоклаве
под давлением в присутствии растворителя; в качестве катализа-
тора применяют КОН и ZnO.
В стальной автоклав загружают 100 ч. гексагидроксилола (рас-
творитель), 100 ч. карбазола, катализатор (3,2 ч. КОН + 1 ч. ZnO)
и после вытеснения воздуха азотом под давлением нагнетают 16 ч.
ацетилена. Процесс ведут при 180° в течение 3—4 час.; к концу
температура поднимается до 260°. Реакционную смесь отмывают
от непрореагировавшего карбазола 5% раствором КОН. После
отделения щелочного раствора и отмывки остатка от едкого
кали очищают полученный винилкарбазол путем дробной пере-
гонки с последующей перекристаллизацией из метанола. Вы-
ход — около 75 %.
Синтез винилкарбазола может быть осуществлен также и паро-
фазным методом путем пропускания смеси паров карбазола и аце-
тилена над катализатором — натронной известью; однако этот ме-
тод встречает значительные трудности вследствие малой упругости
паров карбазола.
N-винилкарбазол представляет собою белое кристаллическое
вещество с т. плавл. 64° и т. кип. 140—150° (при 1 мм рт. ст.).
В совершенно чистом виде он весьма быстро полимеризуется (на-
пример, при +80° за 20 мин.). Однако технический продукт, содер-
жащий некоторое количество примесей фенольного характера, слу-
жащих ингибиторами, полимеризуется медленно, например при
+80° за 20—30 час.
15*
228
Гл V. Пластмассы на основе непредельных углеводородов
Реакция полимеризации имеет цепной характер и может проте-
кать как по радикальному, так и по ионному механизму.
Техническое применение получила полимеризация N-винил-
карбазола в водносуспендированном состоянии в присутствии ката-
лизаторов — щелочных окислителей (по ионному механизму). Так
по одному из вариантов в стальной реактор емкостью 2300 л, сна-
бженный мешалкой, паровой рубашкой и герметически закрываю-
щейся крышкой, загружают 750 кг N-винилкарбазола, 112 кг едкого
натра (в виде 50% водного раствора), 14 кг двухромовокислого
натрия и около 600 л воды. Смесь в реакторе нагревают в течение
70 час., повышая температуру со 120 до 180°, при этом внутреннее
давление в реакторе достигает 18 ат. Для предотвращения агломе-
ризации полимера в качестве эмульгатора вносят около 0,4 % натрие-
вой соли бутилнафталинсульфокислоты.
После окончания реакции содержимое из реактора переводят
в разделитель, отсасывают маточную жидкость и полимер измель-
чают на вальцах с водой. Для удаления мономера, который может
находиться в полимере в количестве 5—10%, размолотый полимер
многократно экстрагируют сначала водой при 70—80°, потом не-
сколько раз кипящим метанолом в реакторе с обратным холодиль-
ником в течение 3—5 час., а потом снова водой при 80—90°; затем
полимер отжимают на центрифугах. Сушку производят горячим
воздухом на ленточных сушилках при ПО—115°.
Полимеризация по радикальному механизму в присутствии ини-
циаторов может быть произведена эмульсионным методом. Для
этого в стальном реакторе, снабженном паровой рубашкой, быстро-
ходной пропеллерной мешалкой и обратным холодильником,
эмульгируют 100 ч. N-винилкарбазола в 500 ч. воды, содержащей
в растворе 2 ч. эмульгатора (натриевой соли бутилнафталинсульфо-
кислоты), 1 ч. ализаринового масла и немного аммиака.
Эмульгирование происходит при работающей мешалке (темпе-
ратура 95—98°), при этом медленно вносят 3 ч. 30% перекиси водо-
рода. В течение процесса эмульгирования необходимо следить за
сохранением слабой щелочности среды. По окончании полимериза-
ции эмульсию коагулируют прибавлением небольшого количества
кислоты, фильтруют, полимер отмывают горячей водой от электро-
литов и экстрагируют этиловым спиртом.
Структура и свойства поливинилкарбазола
Поливинилкарбазол представляет собою высокомолекулярный
линейный полимер, построенный, повидимому, преимущественно по
типу «голова к хвосту»:
... —сн2—сн—сн2—сн—сн,—...
I I
N N
/\/\/\ /\/\/\
Структура и свэйстгва полаванилкарбазола
229
В отличие 01 полистирола поливинилкарбазол — кристаллический
полимер и при растяжении показывает четкую рентгенограмму
ориентированных кристаллитов. Кристалличность поливинилкарба-
зола объясняется, повидимому, его малой степенью разветвления и,
следовательно, более симметричной структурой. Благодаря наличию
в цепи больших громоздких колец, препятствующих вращению
звеньев, температура стеклования поливинилкарбазола значительно
выше, чем у полистирола (~150°). Отличаясь высокой теплостой-
костью, он одновременно, будучи мало полярным полимером, обла-
дает высокой гидрофобностью, химической стойкостью и высокими
диэлектрическими свойствами, лишь незначительно уступающими
полистиролу. Так же, как у полистирола, диэлектрические свойства
поливинилкарбазола мало зависят от частоты поля и темпера-
туры. Вследствие более высокой теплостойкости он может быть
использован в более широком температурном интервале, чем поли-
стирол.
Поливинилкарбазол получается серого цвета, поэтому обычно
его применяют в окрашенном виде. Он растворим в кетонах, эфи-
рах, ароматических и хлорированных углеводородах.
По комплексу механических свойств поливинилкарбазол усту-
пает полистиролу, в особенности по показателю удельной ударной
вязкости (2—4 кг/см2). Вследствие высокой теплостойкости поли-
винилкарбазол перерабатывается в значительно более жестких
условиях, чем полистирол, главным образом методом прессования.
Для литья под давлением необходимы более высокие температуры
(270—290°) и более высокие удельные давления (до 2000 кг/см2).
Из-за хрупкости поливинилкарбазол часто применяют в смеси с на-
полнителями (кварцевая мука, асбест, слюда и т. д.). Изделия, при-
готовленные из поливинилкарбазола методом литья под давлением,
имеют ясно выраженную волокнистую структуру, ориентированную
в направлении течения массы.
В последние годы из нитей поливинилкарбазола удалось полу-
чить волокнистую массу, которую запрессовывают при малых
давлениях и при температурах несколько ниже температуры пла-
вления; этим путем получают слоистые массы с переменной плот-
ностью.
Винилкарбазол имеет весьма важное значение в качестве ком-
понента при получении сополимеров. Наибольшее техническое зна-
чение получили его сополимеры со стиролом и частично с акрило-
нитрилом. При сополимеризации винилкарбазола с небольшим
количеством стирола образуется сополимер, хотя и с несколько
сниженной теплостойкостью, однако значительно лучше подвергаю-
щийся переработке. В табл. 24 приведены свойства поливинилкар-
базола и сополимеров винилкарбазола со стиролом в тех соотно-
шениях, которые получили техническое применение. Все эти поли-
меры выдерживают кипячение с водой и устойчивы по отношению
к действию кислот и щелочей.
230
Гл. V. Пластмассы на основе непредельных углеводородов
ТАБЛИЦА 24
Свойства поливинилкарбазола и сополимеров винилкарбазола
со стиролом
Свойства Поливинил- карбазол Сополимер с 15° о стирола Сополимер с ЗО^/о стирола
Уд. вес, г/см3 Теплостойкость по Мартенсу, °C . . 1,2
150 125 100
Температура переработки (литья под давлением), °C 270 250 230
Предел прочности на изгиб, кг/см2. 1000 900 800
Уд. ударная вязкость, кг см]см2 . Твердость по Бринелю, кг/мм2 . . . 3 2,4 4
14 12 12
Водостойкость (привес), мгЦООсм2 . 10 10 10
Уд. теплоемкость (0—100 °C) кал[г- °C 0,3 0,3 0,3
Коэффициент теплового расширения, °C 4 -10-5 4-Ю'5 4- Ю-5”
Пробивное электрическое сопроти- вление, кв/мм 25 25 25
Уд. поверхностное сопротивление, 2 1013 1013 1О’«
Диэлектрическая постоянная при 800 гц . • 2,8 3,0 2,8—2,7
Тангенс угла диэлектрических потерь при 10* гц 0,0008—0,001 0,001 0,0006—0,001
Применение поливинилкарбазола
Особое значение поливинилкарбазол приобрел для изготовления изделий,,
стойких к кипящей воде и к воздействию температур выше 100°. Его приме-
няют в электротехнике в качестве заменителя слюды при изготовлении мика-
нитов и в качестве заменителя асбеста, обладающего более высокими диэле-
ктрическими свойствами и большей водостойкостью, чем асбест. Основное назна-
чение поливинилкарбазола и его сополимеров — изготовление деталей высоко-
частотной электротехники, где все в большей мере требуется высокая тепло-
стойкость материалов наряду с высокими диэлектрическими свойствами; боль-
шое значение приобретает также стойкость поливинилкарбазола по отношению
к действию электрических разрядов.
В химической промышленности поливинилкарбазол применяют для произ-
водства деталей химаппаратуры, стойкой при температурах до 120° к действию
щелочей, кислот (в том числе фтористоводородной) и фтористых соединений.
Сополимеры винилкарбазола с акрилонитрилом нашли применение в каче-
стве легкого и гигиенического заменителя типографского металла.
ГЛАВА VI
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИМЕРОВ
ГАЛОИДОПРОИЗВОДНЫХ ЭТИЛЕНА
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИХЛОРВИНИЛА
(Хлорвинипласты)
Хлористый винил (винилхлорид) был синтезирован еще в 1835 г.
(Реньо) и уже тогда было замечено, что при действии света он
образует полимер в виде аморфного порошка. Однако в продол-
жение последующих десятилетий полихлорвинил не находил ка-
кого-либо применения; не производились и существенные работы
по изучению полимеризации хлористого винила и по переработке
его полимера.
В 1912 г. И. И. Остромысленский впервые опубликовал ряд ра-
бот по фотополимеризации хлористого винила и по фракциониро-
ванию его полимера методом дробного растворения и осаждения.
Оказалось, что полихлорвинил растворим лишь в ограниченном
числа растворителей, имеет высокую температуру плавления, близ-
кую к температуре разложения, и плохо совмещается с пласти-
фикаторами; поэтому практическое использование этого поли-
мера встретило ряд трудностей. Лишь в конце двадцатых и
в начале тридцатых годов нашего столетия, когда был освоен ме-
тод горячей пластификации хлористого винила и были получены
его сополимеры с винилацетатом и другими мономерами, полимери-
зация хлористого винила приобрела промышленное и техническое
значение.
Развитие современной технологии хлорвинипластов, а также
применение их в различных областях техники связаны с научными
работами ряда советских ученых: П. И. Павловича, И. П. Лосева,
Б. Н. Рутовского, В. О. Седлиса и др.
Свойства хлористого винила
Хлористый винил СН2 = СНС1 при комнатной температуре и
при обычном давлении представляет собою бесцветный газ
с приятным эфирным запахом, т. кип. —13,9°+ 0,1°, т. плавл.
—159,7° + 0,1°. Критическое давление хлористого винила равно
52,2 ат, критическая температура 142°, уд. вес при —12,96° = 0,9692.
Он растворяется в обычных растворителях: ацетоне, этиловом
спирте, ароматических и алифатических углеводородах.
232 Гл. Vr. Пластмассы на основе полимеров галопдопроизв. этилена
Методы получения хлористого винила
В промышленности хлористый винил получают двумя путями:
1) присоединением хлористого водорода к ацетилену:
СН=СН + HCI —> СН,=СНС1 +477 кал-,
2) отщеплением хлористого водорода от дихлорэтана, который,
в свою очередь, получают хлорированием этилена:
-НС1
СН2=СН, + С1, —> С'СН.2— СН,С1 --> СН2=СНС1.
Первый путь получения хлористого винила — присоединение НС!
к ацетилену — может быть осуществлен жидкофазным или паро-
фазным методами.
Современные варианты жидкофазного метода основаны на про-
ведении реакции в присутствии специальных катализаторов. Про-
цесс осуществляют пропусканием ацетилена через перемешиваемый
раствор концентрированной соляной кислоты и катализатора (полу-
хлористая медь и хлористый аммоний). Для повышения активности
катализатора к раствору добавляют хлористую медь в порошке,
хлористый кальций. Реакцию ведут при 20—25° в реакторе, снаб-
женном мешалкой и штуцерами для впуска и выхода ацетилена,
для ввода НС1 (по мере его связывания), для загрузки и выгрузки
катализаторной смеси. Вся система должна быть изолирована от
доступа кислорода и заполнена азотом. Образующийся хлористый
винил уносится из системы вместе с ацетиленом и парами воды и
проходит специальную рекуперационную систему, состоящую из
обычного холодильника для отделения паров воды и из холодиль-
ника глубокого охлаждения (—60°) для отделения хлористого ви-
нила. Не вошедший в реакцию ацетилен направляется обратно
в реактор, куда поступает также и НС1 для возмещения расхода
хлористого водорода, вступающего в реакцию. Сырой, жидкий хло-
ристый винил для отделения от растворенного в нем ацетилена под-
вергают перегонке на колонке.
Непрерывный парофазный метод ведут каталитически при по-
вышенных температурах (140—200°). Существенное значение
в этом процессе имеет характер катализатора и носителя (трегера).
В качестве последнего обычно применяют активированный газовый
уголь, который пропитывают водным раствором хлористой ртути
или хлористого бария и тщательно просушивают. Носитель и ка-
тализатор берут в соотношении 12: 1. Газовая смесь из ацетилена
и хлористого водорода (с 10—15% избытком НС1), предвари-
тельно тщательно просушенная, проходит через трубку подогрева-
теля и направляется через конвертор, заполненный активированным
углем с нанесенным на него катализатором. Выход хлористого ви-
нила определяется скоростью прохождения газовой смеси, количе-
ством катализатора и температурой; чем ниже скорость и выше
температура,—тем он больше. Выходящая из конвертора газовая
Методы получения хлористого винила
233
смесь проходит рекуперационную систему с охлаждением смеси до
—40°; при этом хлористый винил и вышекипящие примеси полно-
стью сжижаются, а ацетилен и НС1 возвращаются в реактор.
Жидкие продукты реакции поступают на ректификацию, при
этом хлористый винил уходит из верхней части колонны, а более
высококипяшие вещества собираются в нижней части.
Второй путь получения хлористого винила — отщепление НС1
от дихлорэтана — также может быть осуществлен различными ме-
тодами, в частности действием на дихлорэтан спиртового раствора
щелочи или каталитическим пиролизом его при высоких темпе-
ратурах.
По первому методу в реактор из нержавеющей стали, снабжен-
ный обогревающей рубашкой, мешалкой и обратным холодильни-
ком, загружают едкий натр, спирт и дихлорэтан в соотношениях
0,5 ч. едкого натра и 0,5 ч. спирта на 1 ч. дихлорэтана. Реакция
протекает по уравнению:
СН2С'—СН Cl + NaOH —> СН2=СНС1 + NaCl + Н2О.
Для ее ускорения рекомендуют применять избыток спирта, так
как избыток щелочи ведет к образованию в небольшом количестве
ацетилена. В качестве побочного продукта реакции образуется
также и этиленгликоль.
Значительный расход едкого натра, спирта и громоздкость аппа-
ратуры привели к необходимости разработки второго метода —
каталитического пиролиза дихлорэтана.
Процесс осуществляют пропусканием паров дихлорэтана через
нагретую (240—350°) контактную трубку, изготовленную из железа
или из керамики и наполненную активированным углем. Контакти-
рованные пары проходят затем конденсационные и рекупера-
ционные установки, а избыточный дихлорэтан возвращается в
реактор.
В современной технике выпускают очень чистый технический
хлористый винил (98,5—99,5%). В качестве примеси последний мо-
жет содержать ацетилен, но не должен содержать щелочи, так как она,
в свою очередь, способствует образованию ацетилена и повышению,
таким образом, взрывоопасности продукта.
Хлористый винил хранят и перевозят в стальных резервуарах
(баллонах) под давлением. Вентили и все части, соприкасающиеся
с хлористым винилом, не должны содержать сплавов меди. Без
ингибиторов хлористый винил можно хранить лишь при низких тем-
пературах (—40° и ниже); в отсутствие же кислорода и под давле-
нием азота его можно хранить и при комнатной температуре.
Обычно к хлористому винилу в качестве ингибиторов добавляют
гидрохинон, катехин и др., причем для достижения необходи-
мого эффекта достаточно 1 ч. ингибитора на 10 000 ч. хлористого
винила.
234 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
Получение полихлорвинила
Хлористый винил легко полимеризуется под влиянием света
(фотополимеризация), тепла и различных инициаторов (органиче-
ских и неорганических перекисей). Полимеризация носит цепной,
радикальный характер. В технике основное значение приобрела
инициированная полимеризация жидкого хлористого винила.
Особенностью полимеризации хлористого винила является то,
что образующийся полимер нерастворим в мономере, вследствие
чего в процессе полимеризации выпадает из него. Это, естественно,
исключает возможность применения блочного метода полимериза-
ции. Полихлорвинил получают поэтому полимеризацией в раство-
рителях или водно-эмульсионными методами.
Полимеризация в растворителях может быть проведена в двух
вариантах: 1) в растворителе (спирте), в котором нерастворим
полимер, или 2) в растворителе (дихлорэтан, ацетон и др.), в кото-
ром растворимы как мономер, так и полимер. В первом случае поли-
мер по мере течения реакции осаждается в виде тонкого порошка, во
втором — остается в гомогенном растворе (лак). Последний приме-
няют либо как таковой, либо выделяют из него полимер, осаждая
водой. Выделение полимера может быть произведено и путем от-
гонки растворителя. В качестве инициаторов полимеризации при-
меняют обычно перекись бензоила. Реакцию проводят под давле-
нием в автоклавах при 35—45°. Автоклавы и вся арматура к ним
должны быть тщательно освинцованы, иметь паровую рубашку для
обогрева и охлаждения и мешалку для перемешивания реакцион-
ной смеси. Сначала в автоклав вносят растворитель, а затем из
баллонов жидкий хлористый винил. Скорость полимеризации зави-
сит от температуры, концентрации инициатора, характера и концен-
трации растворителя. Степень полимеризации, наряду с ее обратной
зависимостью от скорости полимеризации, определяется характером
растворителя и его концентрацией. Полихлорвинил, полученный при
полимеризации в гомогенной среде (в растворителе в виде лака),
обладает лучшей растворимостью, меньшим молекулярным весом
и, повидимому, меньшей полидисперсностью, чем полимер, полу-
ченный полимеризацией в растворителях, в которых он нерас-
творим.
Полимеризация в растворителях из-за длительности процесса
и расхода растворителей не нашла широкого применения.
Основным методом технической полимеризации хлористого ви-
нила является водно-эмульсионный метод. На практике его осуще-
ствляют либо обычным, прерывным методом полимеризации, либо
непрерывным, башенным методом.
В первом случае полимеризацию проводят в вертикальных футе-
рованных свинцом автоклавах периодического действия емкостью от
400—800 л до нескольких куб. метров, рассчитанных на рабочее
давление до 10 аг и снабженных мешалкой, рубашкой для обогрева
Получение полихлорвинила 235
паром или горячей водой, а также для охлаждения холодной водой.
Применяют и горизонтальные автоклавы большой емкости (да
10—15 м3), футерованные свинцом или выполненные из кислото-
упорной стали. Они рассчитаны на внутреннее давление до 15 ат,
имеют рубашку для обогрева и охлаждения и вращаются для пере-
мешивания реакционной смеси на бандажах со скоростью1
10—15 об/мин. Реакционная смесь состоит из дестиллированной
воды (или очищенной при помощи двойного ионного обмена), эмуль-
гатора, стабилизатора, регулятора кислотности среды (pH), регуля-
тора поверхностного натяжения, инициаторов и мономера.
В качестве эмульгаторов применяют различные мыла, напри-
мер ализариновое, триэтаноламиновое, натриевую соль изобутил-
сульфонафталиновой кислоты (некаль), а также натриевые соли
кислот, получаемых окислением синтетических парафинов С12—Cie<
Эмульгаторы вводят в количестве от 0,1 до 0,5% от веса мономера.
С увеличением количества эмульгатора увеличивается дисперсность
частиц полимера. Концентрация эмульгатора существенно влияет
на скорость реакции и средний молекулярный вес. В качестве
инициаторов применяют водорастворимые перекиси (перекись водо-
рода, персульфат калия и др.).
Стабилизаторы применяют чаще всего, если реакция протекает
с инициаторами, растворимыми в мономере (перекись бензоила) t
с получением водносуспензионной смолы. В качестве стабилизато-
ров вводят желатин, поливиниловый спирт, полиакриловую кислоту,,
растворимые эфиры целлюлозы — оксиэтилцеллюлозу, оксиметил-
этилцеллюлозу и др. (в количестве от 0,5 до 2%).
В качестве буферных регуляторов pH среды применяют 0,5—2%
буферных солей (пирофосфат натрия, фосфорную кислоту, уксусно-
кислый натрий и др.). pH среды влияет на скорость реакции: чем
выше pH, тем энергичнее происходит разложение перекиси на ради-
калы, тем быстрее протекает реакция.
Для уменьшения поверхностного натяжения среды вводят
спирты — амиловый, гексиловый и др. в количестве 0,1—0,5%.
Уменьшение поверхностного натяжения увеличивает дисперсность
частиц полимера. Существенное значение имеют температура, давле-
ние, концентрация мономера, продолжительность реакции, характер
и концентрация эмульгаторов (стабилизаторов), а также характер
и концентрация инициаторов.
Для получения устойчивых тонких дисперсий типа латекса в ка-
честве инициатора применяют водорастворимые перекиси (пере-
кись водорода и др.). Часто берут равные части хлористого винила
и воды. С повышением концентрации мономера и давления (обычно
реакцию ведут при 5—7 ат) увеличивается как скорость реакции,,
так и молекулярный вес полимера, однако экзотермичность реакции
и необходимость отвода тепла ставят предел концентрации моно-
мера.
236 Гл. Vr. Пластмассы на основе полимеров галоидопроизв. этилена
Так по одной из рецептур в реактор загружают 1000 л хлори-
стого винила, 1000 л воды, 27 кг смеси жирных кислот, 1 л NaOH
(50%), 0,7 л Н3РО4 (63%) и 1,2 л Н2О2.
Технологический процесс состоит из следующих операций. В
эмульсионном баке предварительно подготавливают водную эмуль-
сионную смесь, которую затем переводят в автоклав и при охла-
ждении пуском в рубашку холодной воды загружают из резер-
вуаров сжиженный хлористый винил, все время тщательно пере-
мешивая смесь. По окончании загрузки в рубашку автоклава дают
горячую воду, поддерживая температуру в пределах 40—50°; при
этом давление в рубашке автоклава достигает 5—7 ат. Период ин-
дукции — обычно несколько часов, после чего начинается экзотер-
мическая реакция полимеризации, которая быстро ведет к подъему
давления в автоклаве. В это время необходим ввод холодной воды
в рубашку автоклава и тщательное регулирование температуры.
Степень полимеризации и дисперсность полихлорвинила в основ-
ном определяются температурой смеси в реакторе, поэтому точный
режим температуры имеет самое существенное значение. Отклоне-
ния от заданных температур и давлений не должны превышать со-
ответственно +2° и +0,2 ат. Несоблюдение температурного режима
может привести к резкому повышению температуры, ведущему к
спеканию массы в виде сплошного куска («козла»), или к образо-
ванию грубодисперсных частиц («гороха»), а иногда и к разложе-
нию массы.
По окончании реакции и спада давления практически почти весь
мономер оказывается заполимеризованным. В зависимости от со-
става эмульсионной смеси (характера эмульгатора) полимер может
•быть получен либо в виде тонкого дисперсного порошка, который
затем легко осаждается, фильтруется и сушится в полочных ва-
куум-сушилках, либо в виде стойкой эмульсии типа латекса, кото-
рая может быть осаждена лишь внесением электролитов и пони-
жением pH среды. Полихлорвиниловые латексы имеют также и
самостоятельное применение для пропитки и поверхностной отделки
тканей, кожи, бумаги и т. д.
В последние годы в промышленных условиях применяют неко-
торые новые методы выделения полимера из эмульсии, в основе
которых лежат следующие операции: 1) эмульсию пропускают че-
рез непрерывно работающую барабанную сушилку; 2) полимер вы-
деляют непрерывным процессом сушки распыляемой эмульсии и
3) эмульсию осаждают центрифугированием с последующей суш-
кой полимера в полочных вакуум-сушилках.
Более эффективными являются непрерывные методы эмуль-
сионной полимеризации хлористого винила, которые нашли ши-
рокое применение. Аппаратура состоит из одного или из двух
последовательно включенных полимеризационных автоклавов, пред-
ставляющих собою вертикальные котлы диаметром 1500—1800 мм
И высотой 7000—8000 мм, рассчитанные на давление до 10 ат. Вну-
Получение полихлорвинила
237
три атоклавы облицовывают стеклом; они снабжены рубашкой для'
нагревания и охлаждения. Нагревание производят горячей водой
(50—60°), охлаждение — путем циркуляции в рубашке рассола с
температурой — 20° или .воды с температурой 15°. Регулируя коли-
чество охлаждающей смеси, можно довольно точно поддерживать-
температуру в реакторе и с большей скоростью отводить вы-
деляющееся при реакции тепло. В верхней части реакторы сна-
бжены лопастными мешалками, которые приводятся в движение от
индивидуального мотора через редуктор с переменным числом (от'
0 до 120) оборотов. Мотор и редуктор смонтированы на крышке
автоклава. Во избежание загрязнения реакционной смеси смазоч-
ными маслами, которые действуют как ингибиторы, сальники всех
насосов, подающих сырье в автоклав, а также сальник вала ме-
шалки набивают асбестом, пропитанным специальным составом.
Схема работы такого автоклава показана на рис. 104. Раствор инициатора
давлением азота подают в расходный мерник 1, из которого насосом 2 нагне-
тают в автоклав 6. Хлористый винил и раствор эмульгатора проходят автома-
тические кольцевые весы 4, соединенные с самопишущими и регулирующими
Рис. 104. Схема непрерывной эмульсионной по-
лимеризации хлористого винила
1—мерник; 2—плунжерный насос; 3— регулирующие клапаны;
4— кольцевые весы; 5 — контрольно-измерительные приборы;
6—автоклав; 7 — горизонтальный фильтр; 3 — центробежный
насис; 9— паро-водосмеситель; 10— обратный клапан.
приборами 5, которые управляют мембранами, устанавливающими клапаны <?.
Подачу воды (или охлаждающего рассола) в рубашку автоклава производят
центробежным насосом 8. Полимер в виде тонкой устойчивой эмульсии (латекса>
238 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
непрерывно из автоклава вытекает снизу и, пройдя дроссельный вентиль, посту-
пает на горизонтальный фильтр 7, где фильтруется от крупных частиц поли-
мера. Полученный латекс поступает на коагуляцию, промывку, фильтрацию и
сушку. Производительность автоклава ~25 кг полимера иа 1 м3 полезного
объема в час.
При проведении полимеризации в двух последовательно включенных авто-
,клавах реакционная смесь вначале проходит первый автоклав, в котором
в основном завершается конверсия; затем смесь подают в верхнюю часть вто-
рого автоклава. Первый автоклав, в котором в наибольшей степени выделяется
тепло реакции, охлаждают рассолом с температурой —20°, второй автоклав —
водой.
При первоначальной загрузке автоклавы заполняют на 70% раствором
эмульгатора и катализатора (в соотношениях 17:1), после чего добавляют
1000 л хлористого винила и, пуская в рубашку горячую воду, нагревают реак-
циоиную смесь до 45—55°. В момент подъема температуры, которая указывает
на начало реакции полимеризации, включают охлаждение. Процесс полимериза-
ции контролируют определением удельного веса реакционной смесн. Когда он
достигает 1,070, то непрерывной загрузкой компонентов реакционной смеси и
.отбором снизу равного по объему количества латекса процесс постепенно пере-
водят на непрерывный. Когда же удельный вес реакционной смеси становится
равным 1,100, то непрерывная нагрузка и отбор латекса достигают наибольших
размеров; при этом автоклав работает непрерывно в течение 10—12 суток, после
чего его останавливают и очищают от прилипшего к стенкам полимера.
Непрерывный метод эмульсионной полимеризации позволяет
регулировать степень полимеризации как изменением температуры
в автоклавах (при постоянном соотношении компонентов), так и
количества и характера инициатора. Степень стабильности и дис-
персности эмульсии регулируется изменением состава реакционной
смеси.
Строение и свойства полихлорвинила
На основании химических, а также рентгенографических и опти-
ческих методов исследования установлено, что полихлорвинил имеет
структуру 1,3 по принципу «голова к хвосту». Схема строения мо-
лекулы полихлорвинила показана на рис. 105.
Рис. 105. Схема строения молекулы полихлорвинила.
• — атомы углерода; О —атомы водорода; QC1—атомы хлора
Полихлорвинил обычно выпускают в виде белого аморфного
порошка; его уд. вес ~1,4, показатель преломления 1,544, темпера-
•тура стеклования высокая (-f-85°). Температура пластического те-
Строение и свойства полихлорвинила
239
чения полихлорвинила близка к температуре разложения, а часто
даже выше ее. Уже при температурах выше 140° он заметно разла-
гается с выделением НС1, причем образующаяся кислота каталити-
чески ускоряет разложение полимера; последнее ускоряется также
солями железа, цинка и в меньшей степени солями меди. Разложе-
ние легко определить по изменению цвета и потемнению поли-
мера.
При более высоких температурах и при сухой перегонке проис-
ходит деструкция полихлорвинила с выделением НС1 и образова-
нием смеси различных низкомолекулярных продуктов; мономер при
деструкции не образуется.
Температура разложения является самым важным техническим
показателем полихлорвинила.
Важное значение при переработке полихлорвиниловых смол
имеет также и показатель так называемой термостабильности смолы.
За термостабильность принимают время (в минутах) нагрева смолы
в стандартных условиях при постоянной температуре (165 или 175°)
до появления первых признаков выделения НС1.
Чем ниже температура разложения, тем меньше и термостабиль-
ность; выделение НС1 тем больше, чем меньше молекулярный вес
полихлорвинила, что, повидимому, объясняется большей реакцион-
ной способностью атомов внутри цепи.
Так как выделяющийся НС1 каталитически ускоряет процесс
разложения смолы, то внесением мелкодисперсных веществ, спо-
собных связывать выделяющийся хлористый водород, можно суще-
ственно повысить температуру разложения, повысить термостабиль-
ность полимера.
Вещества, вводимые в состав полихлорвиниловых композиций
для связывания выделяющегося НС1 при термической обработке
(вальцевание, прессование) и повышающие их термостабильность,
называются стабилизаторами.
В качестве стабилизаторов применяют различные вещества основ-
ного характера, неорганические соединения (Na2CO3, PbCO3, Na3PO4
и др.), органические аминокислоты, металлические мыла и др.
По степени эффективности (по повышению термостабильности
и температуры разложения) стабилизаторы можно расположить в
следующий ряд: свинцовые белила > углекислый свинец стеарат
свинца свинцовый глет свинцовый сурик сода > стеарат ба-
рия стеарат кальция и т. д.
Как видно, наиболее эффективными стабилизаторами являются
свинцовые соединения, которые поэтому чаще всего применяются,
несмотря на токсичность свинца.
Полихлорвинил — полимер со значительной полидисперностью.
Степень полимеризации для различных фракций одной и той же
смолы может находиться в пределах от 100 до 2000. Содержание
высокомолекулярных фракций в полимере имеет существенное зна-
чение для его физико-механических свойств.
240 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
Для получения прочных и эластичных материалов фракции со
средней степенью полимеризации 1000 и более должны составлять
большую часть полимера (~70%).
Действие высоких температур (как при переработке, так и в
условиях эксплуатации), ультрафиолетового облучения, электриче-
ских разрядов и т. д. приводят к утрате растворимости полихлор-
винила и к переходу его в трехмерный полимер. Сшивка цепей
происходит, невидимому, вследствие отщепления хлора в двух со-
седних цепях:
—СН2—СН—СН.,—СН—СН2—
I
С1
С1
I
—сн2—сн—сн2—сн—сн2—
Промышленные марки полихлорвинила характеризуются пока-
зателем вязкости 1% раствора полимера в дихлорэтане, выражен-
ном в сантипуазах; косвенно он характеризует величину молекуляр-
ного веса полихлорвинила.
Полихлорвинил обладает высокой прочностью и теплостой-
костью, что должно быть отнесено за счет сильных дипольных свя-
зей, действующих между соседними макромолекулами. Этому же
обстоятельству следует приписать и малую растворимость поли-
хлорвинила, с трудом растворяющегося в сравнительно ограничен-
ном числе растворителей; образующиеся растворы даже при не-
больших концентрациях имеют часто характер студней. Раствори-
мость уменьшается с увеличением степени полимеризации, причем
водно-эмульсионные полихлорвинилы имеют наименьшую раство-
римость. Низшие полимеры полихлорвинила относительно легко,
растворимы в ацетоне, высшие — лишь в дихлорэтане, хлорбензоле,
тетрагидрофуране и диоксане. С пластификаторами на холоду по-
лихлорвинил также мало совместим; однако при повышенных тем-
пературах набухание идет значительно лучше, в частности, при же-
латинизации смеси полихлорвинила с пластификаторами на горячих
вальцах.
Полихлорвинил, по крайней мере в нерастянутом состоянии,
имеет аморфную структуру. При растяжении он не дает достаточно
четкой рентгенограммы ориентированных кристаллитов; однако
рентгенограмма все же позволяет вычислить период идентичности
полихлорвинила, равный 5,2 А, что соответствует двум звеньям мо-
номера, связанным зигзагообразно; эти звенья, однако, не лежат-
в одной плоскости, в последнем случае период идентичности был
бы равен 2,7.
Вследствие полярности полимера (дипольный момент звена
1,66-10-18) диэлектрическая постоянная и диэлектрические потери
полихлорвинила значительно выше, чем у неполярных и слабопо-
лярных полимеров (полиэтилена, полистирола и др.). Кроме того,
Пластические массы, не содержащие пластификаторов
241
диэлектрические свойства полихлорвинила в значительной мере за-
висят от частоты поля и температуры (рис. 106). Резкое увеличение
диэлектрической постоянной (&) и тангенса угла диэлектрических
потерь (tg 8) наблюдается при достижении температур, близких к
температуре стеклования, когда скорость ориентации диполей стано-
вится соизмеримой со скоростью перемены поля. При значительно
более высоких температурах (120° и выше) на дипольный механизм
Рис. 106. Зависимость е, tg о и (уд. объемное
электрическое сопротивление) полихлорвинила от
температуры.
В СССР полихлорвинил выпускается разных марок, отличаю-
щихся как по характеру инициатора, применяемого при эмульсион-
ной полимеризации (перекись бензоила, персульфат аммония), так
и по величине вязкости 1% раствора в дихлорэтане (от 1,28 до
2,8 сп).
Пластические массы из полихлорвинила можно разделить на
две группы: 1) не содержащие пластификаторов и 2) содержащие
пластификаторы.
Пластические массы из полихлорвинила, не содержащие
пластификаторов
X л ор вини л оид (винипласт)
Введение пластификаторов понижает температуру размягчения
смолы и, следовательно, дает возможность перерабатывать ее при
более низких температурах, что значительно облегчает процесс про-
изводства. Однако пластификаторы, как правило, ухудшают хими-
ческую стойкость и диэлектрические свойства полимера, снижают его
]6 Зак. 1269. Э. И. Барг
242 Гл. VI. Пластмассы на основе полимеров салоидопроазв. этилена
теплостойкость. Кроме того, пластификаторы обладают некоторой ле-
тучестью, которая является причиной «старения» пластиков (увеличе-
ния жесткости со временем), вызываемого потерей пластификатора.
Поэтому для получения материалов высокой теплостойкости и
химической стойкости применяют композиции из полихлорвинила
без пластификаторов.
Для приготовления такого рода пластиков термическую обра-
ботку полихлорвинила (пластикацию, прессование, экструзию и т. д.)
проводят при сравнительно высоких температурах (160—170°); для
этого, естественно, необходимо обеспечить высокую термостабиль-
ность полихлорвиниловых смесей внесением соответствующих ста-
билизаторов. Непластифицированный тепло- и химически стойкий
и упругий полихлорвинил (хлорвинилоид) производят в СССР под
названием винипласт.
Процесс получения винипласта (хлорвинилоида) в основном со-
стоит из двух стадий.
Первая стадия заключается в смешении порошка полимера со
стабилизатором. Стабилизатор (обычно стеарат или силикат свинца
или кальция) добавляют в количестве 2—3%. Смешение произво-
дят в лопастном мешателе или шаровых мельницах в течение 25—
50 мин. Перед смешением полихлорвинил и стабилизатор просеи-
вают через сита (№ 25—30).
Вторая стадия заключается в так называемой термической пла-
стикации, которая сводится к обработке смеси на смесительных
фрикционных вальцах при 160—170° (конструкции вальцов см.
рис. 169, стр. 434). Результатом такой обработки является увели-
чение пластичности полимера (что в некотором отношении ана-
логично действию обычных пластификаторов). Можно считать,
что эффект термической пластикации обусловливается частичной
деполимеризацией макроцепей и их ориентацией в процессе вальце-
вания. Возможно, что при этом имеют место и окислительно-восста-
новительные процессы, также приводящие к увеличению пластич-
ности смеси.
Вальцевание производят при температуре выше температуры
текучести Ттвк (стр. 102), которая для 'полихлорвинила лежит в
пределах 150—160°. Чем выше температура вальцевания, тем быст-
рее протекает процесс образования гомогенной пластичной массы,
однако тем быстрее, естественно, и термическая деструкция смолы.
С другой стороны, более низкая температура (ниже Т^к) при-
водит к механической деструкции полимера (разрыв цепей за
счет механического воздействия при вальцевании), при этом по мере
деструкции Ттек снижается до пределов температуры вальцева-
ния, при которой и образуется гомогенная пластичная масса.
В последнем случае механическая прочность полимера соответ-
ственно снижается.
Вальцевание производят на двух или более парах'вальцов. Коли-
чество вальцов определяется соотношением их производительности
Пластические массы, не содержащие пластификаторов
243
к производительности каландра, куда поступает масса после окон-
чательного вальцевания. Работа вальцов и каландра должна быть
строго синхронизирована как для получения пленки соответствую-
щего качества, так и для устранения излишних потерь массы и про-
стоя оборудования.
Вальцевание происходит на фрикционных вальцах. Оптимальная
фрикция вальцов, обеспечивающая получение гомогенной и плотной
массы без включения пузырьков воздуха, составляет 1,2 (отноше-
ние окружной скорости обоих валков 1 : 1,2); зазор между валками
может быть различен: от 0,5—1 мм в зависимости от характера
смолы. Вальцеванию подвергаются как композиции из свежеприго-
товленной смеси смолы и стабилизатора, так и возвратные отходы.
Вальцевание состоит из двух операций: 1) предварительного и
2) окончательного вальцевания.
Предварительное вальцевание начинается с подачи на вальцы све-
жей композиции вручную совками. Масса вначале лишь слегка пла-
стицируется и в виде кусочков падает через зазор валков. В даль-
нейшем, после двух- и трехкратной повторной подачи на вальцы
масса размягчается и образует сплошное пластичное полотно, кото-
рое несколько раз снимают (подрезают), закатывают в ролик, снова
его подают на вальцы, и, наконец, гомогенную массу снимают и
направляют на окончательное вальцевание. На других вальцах,
в случае необходимости, предварительно провальцовывают возврат-
ные отходы.
Цель окончательного вальцевания — доведение массы до нуж-
ной степени пластичности. На горячие вальцы подают полотна
из свежей массы и из отходов, производят тщательное сме-
шение полотен и обрезают листы соответственно расстоянию между
щеками каландра. После окончательного вальцевания полотно сре-
зают, скатывают в ролик и подают в горячем состоянии в загру-
зочную камеру каландра для получения непрерывной ленты опре-
деленной толщины (калибровка пленки) и для удаления воздуха и
уплотнения массы. Обычно применяют вертикальные каландры, со-
стоящие из ряда валков, расположенных в подшипниках один над
другим (двух-, трех-, четырехвалковые каландры в, зависимости
от числа валков) и имеющие одинаковые линейные скорости вра-
щения (рис. 107). Валки в процессе каландрирования обогреваются
паром или перегретой водой. Зазор между валками весьма точно
устанавливают с помощью специального приводного механизма, —
зазор и определяет толщину листа; ширина его определяется дли-
ной валка. Горячую массу с вальцов загружают непрерывно или
периодически на загрузочную плиту каландра, где она проходит ме-
жду верхним и средним валками, огибает средний валок, проходит
далее в зазор между средним и нижним валком, огибает его и, в
случае трехвалкового каландра, направляется далее через охла-
ждающий валок на приемный стол, где пленка режется на полосы
или листы.
16*
244 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
Температура валков различна, наиболее низкую имеет верхний
(155—160°), наиболее высокую — нижний (165—170°); такой пере-
пад температур облегчает переход пленки от верхнего валка к
нижним.
Каландрированием на трехвалковом каландре получают пленки
с гомогенной поверхностью толщиной от 0,2 до 1 мм. Увеличение
числа горячих валков в каландре и их диаметра позволяет изготов-
лять пленки большей толщины с вполне удовлетворительной поверх-
ностью.
Рис. 107. Трехвалковый каландр.
1 — фундаментальная плита; 2— станина; 3— приводная шестерня;
4 —шестерни; .5 —червячная передача; 6 — мотор; 7 —редуктор.
Пленка, полученная с каландров, имеет ориентацию макромо-
лекул и, следовательно, соответствующее растяжение вдоль напра-
вления каландрирования, что приводит к анизотропии свойств
пленки, к так называемому каландровому эффекту. Последний вы-
ражается в том, что прочность и относительное удлинение у образ-
цов, вырезанных в продольном направлении, выше, чем у образцов,
вырезанных в поперечном направлении; соответственно усадка
пленки при прогреве (релаксация) выражается в сокращении по
длине и в увеличении ширины и толщины пленки.
Предельные значения усадки пленки составляют ~30% сокра-
щения длины и ~12% увеличения ширины.
Листовой винилоид (винипласт) получают горячим прессова-
нием каландрированной пленки.
Нарезанные листы пленки укладывают между стальными нике-
лированными пластинами с таким расчетом, чтобы толщина набора
пленки была на 10—20% больше толщины изготовляемого листа.
Прессование производят на гидравлических многоэтажных пли-
точных прессах (рис. 184, стр. 477). На каждый этаж пресса уклады-
Пластические массы, не содержат,ие пластификаторов
245
вают различное число наборов, — от одного до десяти в зависи-
мости от их толщины и расстояния между плитами пресса.
Прессование производят при двух режимах обогрева: предвари-
тельного с прогревом прессуемых наборов до 80—120° и оконча-
тельного— с нагревом пакетов на 5—10° выше температуры калан-
дрирования (170—175°). Общее время обогрева ~60—90 мин.
Удельное давление из расчета на прессуемую поверхность изде-
лия составляет 10—30 кг/с.и2.
Обогрев производится подачей горячей воды под давлением в
плиты пресса.
По окончании горячего прессования в плиты пресса пускают хо-
лодную (водопроводную) воду и охлаждение ведут до 4-50°. В про-
цессе охлаждения увеличивают удельное давление и доводят его до
50—80 кг/сл!2. Как удельное давление, так и температурный ре-
жим на различных стадиях прессования определяются толщиной
прессуемых пластин и вязкостью исходных полихлорвиниловых
смол.
По окончании прессования пакеты выгружают из пресса, от-
деляют прессованные листы и обрезают кромки (прессовочный
грат) на обрезных станках.
Листы выпускают толщиной от 2 до 20 мм.
Кроме листового материала (пластин, плит) из каландрирован-
ной пленки путем перфорации (вырубания частых небольших от-
верстий) и гофрирования перфорированной пленки изготовляют
сепараторы для аккумуляторов (для разделения анодных и катод-
ных пластин в кислотных аккумуляторах).
Из хлорвинилоида (винипласта) изготовляют трубы различного
внутреннего диаметра (от 2 до 150 мм и больше), а также стержни
различного профиля путем выдавливания (экструзии) пластичной
массы на шнекпрессах или гидравлических горизонтальных прессах
через специальные формы (прессинструмент), оформляющие про-
филь изделия.
Для этого хорошо провальцованную горячую пластичную массу
скатывают в ролик и загружают в загрузочный цилиндр шнек-
пресса.
Давлением поршня или ходом шнека пластичная масса прода-
вливается через прессинструмент, выходит из пресса в виде стержня
или трубы. Скорость выдавливания от 0,7 до 2,0 м в мин. Темпера-
тура массы 170—180°, а матрицы прессинструмента 180—200°,
уд. давление в зависимости от диаметра и профиля изделия от 50
до 500 кг! см2.
Отрезки труб, длиной 2—3 м, в зависимости от диаметра или
откатывают на станках для предотвращения деформации в процессе
охлаждения, или же поддерживают форму трубы вдуванием сжа-
того воздуха; давление ~1—1,5 ат.
Хлорвинилоид представляет собою твердый упругий материал
с сравнительно высоким модулем упругости (~10000 кг/см2),
24<> Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
с высокой удельной ударной вязкостью (~100—200 кг1см2) и срав-
нительно большой статической прочностью (500—800 кг/см2) на рас-
тяжение.
Однако при суждении о механических свойствах винипласта не-
обходимо учитывать их большую зависимость от температуры и от
эффекта надреза (стр. 444—445). Так, удельная ударная вязкость
надрезанных образцов в 5—10 раз ниже гладких; при снижении
температуры с -[-20 до —60° удельная ударная вязкость снижается
примерно в пять раз, в то время как прочность на растяжение уве-
личивается почти вдвое.
Свойства винипласта различны в продольном и поперечном на-
правлениях. При обычных температурах винипласт не находится в
равновесном состоянии; с течением времени он релаксирует, что
приводит к медленно развивающимся процессам деформации, кото-
рые резко ускоряются при нагреве.
Неравномерность нагрева и охлаждения, различие в плотности
и дефекты в слипании (склейки) слоев создают значительные кон-
центрации внутренних напряжений, распределенных неравномерно
и снижающих прочность изделия.
Влияние внутренних напряжений, эффекта надреза, анизотроп-
ности, температуры, релаксации делают необходимым устанавли-
вать большой запас прочности при расчетах габаритов изделий (ог
2 до 4).
Недостаток хлорвинилоида — низкая температура стеклования,
определяющая его техническую теплостойкость (порядка 60—70°).
Изделия из него, если они подвергаются постоянно действующему
напряжению растяжения или сдвига, не могут работать при темпе-
ратурах выше 50—60°.
Хлорвинилоид обладает большой химической стойкостью
(к кислотам и щелочам) и хорошими диэлектрическими свой-
ствами.
Он характеризуется следующими показателями:
Уд. вес, г1слГ..............................
Предел прочности на разрыв, кг/см2...........
Удлинение, %.................................
Предел прочности на изгиб, кг'см2 . ... .
Предел прочности на сжатие, кг/см?............
Модуль упругости, лгг/елг2....................
Уд. ударная вязкость, кг см/см2.............
Уд. ударная вязкость с надрезом, кг см/см2 . .
Твердость по Бринелю, кг/мм2..................
Теплостойкость по Мартенсу, °C................
Теплопроводность, кал)см сек -°C............
Коэффициент линейного расширения..............
Уд. объемное электрическое сопротивление, Q-cm
Уд. поверхностное электрическое сопротивление, й
Пробивное электрическое напряжение, кв/мм
Диэлектрическая постоянная при 10’ гц ....
Тангенс угла диэлектрических потерь при Ю1* гц
1,38—1,40
500—700
10—25
800—1200
800—1600
4000-10000
100—250
10—30
15—18
65—80
3,8—4 • 10-1
0,00006—0,00007
ЮЫ—Ю'5
10t2_i0H
15-35
3,2—3,6
0,04—0,10
Пластические массы, не содержащие пластификаторбв
247
Химическая стойкость:
1) водопоглощение (за 24 часа), г!см?....0,1 • 10~4— 0,3- 10~4;
2) до + 60° стоек к действию НС1 — всех концентраций; H2SO4 — до 90%;
HNO3—до 50%; СН3СООН — до 80%; щелочей — всех концентраций;
3) стоек к бензину, керосину, маслам, глицерину, спиртам и т. д.
Сочетание механической прочности с высокой химической стой-
костью, возможность подвергнуть его различным видам механиче-
ской обработки (формованию, сварке, склеиванию) сделали вини-
пласт одним из ценных конструкционных антикоррозийных
материалов.
Листовой и пленочный винипласт широко применяют для изго-
товления и футеровки электролизных и травильных ванн, для из-
готовления вентиляционных воздухопроводов, резервуаров кислот
и щелочей, сепараторов в аккумуляторах, а также для выделки де-
талей насосов (прокладок, вентилей, клапанов, поршней) и т. д.
Трубы из винипласта служат для транспортировки агрессив-
ных жидкостей и с успехом заменяют свинцовые и эмалированные
трубы.
Для соединения «руб, для создания сложных конструкций из
листов применяют сварку винипласта.
Сварку осуществляют путем одновременного нагрева сваривае-
мых элементов и сварного прутка, изготовленного из мало пласти-
фицированного полихлорвинила. Нагрев доводят до температуры
выше вязко-текучего состояния (180—200°). При этой температуре
с большой скоростью протекают процессы диффузии макроцепей,
ведущие к образованию гомогенной массы. Для сварки скон-
струированы специальные аппараты. Хорошие результаты полу-
чают при применении сварочного пистолета, служащего для нагрева
и направления струи воздуха.
Воздух в сварочный пистолет поступает из компрессора, где он
нагревается до нужной температуры, проходя над каналами с рас-
каленной нихромовой проволокой. Температура воздуха регули-
руется как изменением давления поступающего воздуха, так и из-
менением. силы тока, питающего электрический обогрев.
Струя горячего воздуха из сопла пистолета направляется на
сварочный пруток и свариваемые поверхности; пруток быстро ста-
новится пластичным и прочно схватывается с основной массой.
Прочность шва при правильной сварке составляет 60—80%
прочности основного материала.
Наряду со сваркой применяют склейку тонких листов перхлор-
виниловой смолой (лаком) (стр. 253), а также приклеивание тонких
лент винипласта к другим материалам, в том числе и к металлам.
Можно перерабатывать винипласт методом снятия стружки, свер-
ловкой, распиливанием, фрезеровкой и т. д. Широко пользуются
также горячим прессованием, штамповкой, тиснением и формовкой
как на холоду, так и при небольшом нагреве (60—80°),
248 Гл. Vf. Пласп массы на основе полимеров галоидопроизв. этилена
Винипласт применяют в качестве заменителя целлулоида для
производства гребней, пуговиц и т. д., а в электротехнике — взамен
эбонита.
Из непластифицированного полихлорвинила изготовляют упако-
вочные и изоляционные пленки толщиной 0,01—0,05 мм. Процесс
производства таких пленок состоит в горячем вальцевании поли-
хлорвинила при 160—180° в течение 5—10 мин. и в дальнейшей об-
работке вальцованной смеси на каландрах при той же температуре.
Затем образующаяся пленка в течение 5—10 сек. проходит нагре-
тые до 260—270° вальцы и растягивается на подвижных валках,
нагретых до 120°, до толщины в несколько десятков микронов.
Пластические массы из полихлорвинила, содержащие значительное
количество пластификатора
X л о р в и н и л а с т (пластикат)
Пластифицированный полихлорвинил применяют главным об-
разом для производства мягких материалов, обладающих высоко-
эластическими свойствами при обычных и пониженных температу-
рах.
Об эффективности пластификаторов можно судить по величине
снижения температуры стеклования полимера на единицу веса или
объема внесенного пластификатора. Пропорционально понижению
Тс (повышению морозостойкости) снижается статическая прочность
материалов (из расчета на первоначальное сечение) и увеличи-
вается удлинение; при этом (до известных пределов содержания
пластификаторов) значительно увеличивается удельная работа де-
формации.
В качестве пластификаторов полихлорвинила применяют поляр-
ные высококипящие и малолетучие жидкости: фосфаты (трикрезил-
фосфат, тригексилфосфат и др.), фталаты (дибутилфталат, диоктил-
фталат), эфиры линейных дикарбоновых кислот (дибутилсебацинат,
диоктилсебацинат, дибутиладипинат и др.), а также хлорпроизвод-
ные нафталина, продукты этерификации смесей гликолей с низшими
членами ряда кислот (Ci2—Ci8), полученных окислением синтетиче-
ских парафинов, и т. д.
Эффективность действия пластификатора определяется его хи-
мической структурой. Для получения морозостойких материалов же-
лательно, чтобы сам пластификатор был морозостойким, т. е. имел
низкую температуру стеклования. Пригодность пластификаторов
для получения морозостойких материалов можно определить, в пер-
вом приближении, снятием кривой температурной зависимости их
вязкости. Чем более пологий характер будет иметь кривая, тем эф-
фективнее действие пластификатора. Предельная концентрация
пластификатора в полимере определяется его совместимостью с по-
лимером и склонностью к кристаллизации: чем выше совместимость
Пластические массы из полихлорвинила, содержащие пластификаторы 249
и меньше склонность к кристаллизации, тем больше можно ввести
пластификатора.
В технике широко известно, что часто смесь двух пластифика-
торов более эффективна, чем каждый из пластификаторов в отдель-
ности. В этом отношении интересны наблюдения В. О. Седлиса, ко-
торый нашел, что смесь
из лиофильного пласти-
фикатора и жидкого ве-
щества, в котором по-
лимер не набухает,
дает значительно более
морозостойкий мате-
риал, чем только один
пластификатор. Зави-
симость прочности по-
лихлорвинила от хара-
ктера и количества пла-
стификаторов представ-
лена на рис. 108.
В качестве пласти-
фикаторов для поли-
хлорвинила были пред-
ложены высокомолеку-
лярные вещества, обла-
дающие низкой темпе-
ратурой стеклования и
достаточно высокой по-
Рис. 108. Зависимость прочности на разрыв по-
лихлорвинила от содержания и характера пласти-
фикатора.
1—трикрезилфосфат; 2 —дибутилфталат.
лярностью для получения совместимых и гомогенных продуктов.
Смешение и получение твердого раствора обоих полимеров проис-
ходит на горячих смесительных вальцах. В качестве примера таких
«пластификаторов» могут служить синтетические каучуки — сопо-
лимеры акрилонитрила с бутадиеном. Основное их преимущество —
стабильность материала вследствие практической нелетучести таких
«пластификаторов»..
Механические свойства пластифицированных так же, как и
не пластифицированных полихлорвинилов в значительной мере
определяются степенью полимеризации (вязкостью) исходного
полимера. Чтобы получить материал с одинаковой «мягкостью»
и пластичностью при переработке, нужно по мере повыше-
ния степени полимеризации увеличивать количество пластифи-
катора. $
Технологический процесс производства пластифицированного
полихлорвинила складывается из следующих операций: 1) просеи-
вание, 2) замешивание, 3) созревание, 4) вальцевание и 5) калан-
дрирование или выдавливание (экструзия).
Смолу просеивают через сита от № 10 до № 35 в зависимости
от назначения материала. Стабилизатор обычно просеивают через
2.'>(t Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
сито № 25; наполнители в зависимости от их структуры и харак-
тера — через сита от № 6 до № 20.
Просеянную смолу смешивают в двухлопастном мешателе или в
шаровой мельнице с пластификатором и стабилизатором, а чаете
также и с наполнителем, пигментами и органическими красителями,
Спирте- и жирорастворимые красители предварительно растворяют.
Их вводят при замешивании или же во время вальцевания. Если
смешение происходит в мешателе, то обычно туда сначала вводят
просеянную смолу, а также наполнители и пигменты и после 10—
15 мин. перемешивания — раствор красителя. Через 5—10 мин. до-
бавляют постепенно пластификатор с размешанным в нем стабили-
затором. После введения всех составных частей перемешивание
продолжают от полутора до двух часов, обычно при комнатной тем-
пературе или при 60—70°.
Затем массу выгружают в оцинкованные или алюминиевые бачки
и переводят в камеру с обогревом для «созревания», которое про-
должается 4—8 час. при 65—75°. В процессе «созревания» происхо-
дит перераспределение пластификатора и предварительная желати-
низация (набухание) смолы.
Далее желатинизация продолжается на горячих фрикционных
вальцах. Температура вальцевания определяется содержанием в
смеси пластификатора и вязкостью смолы. Обычно вальцуют при
130—160°.
Массу в виде порошка засыпают на обогреваемые паром вальцы
с первоначальным зазором 0,8—1,2 мм. Соприкасаясь с поверх-
ностью горячих валков, масса плавится и образует первоначальную
пленку (часть массы проходит через зазор и снова подается на
вальцы). В дальнейшем на валках накапливается такой слой пла-
стицируемой массы, который уже полностью не помещается в зазоре
между валками, и часть ее отжимается. При этом вследствие фрик-
ции происходит постепенный сдвиг и перемещение массы. Для луч-
шего перемешивания время от времени производят надрез пленки.
В процессе вальцевания зазор между валками постепенно умень-
шают (до 0,5—0,3 мм), а через 10—15 мин. его снова увеличивают.
Вальцевание продолжается 25—45 мин.
Вальцованная масса поступает для калибровки на каландры
для получения пленочного материала или на шнекпрессы для изго-
товления гибких труб, стержней различных профилей, а также пле-
нок.
В СССР пластифицированный полихлорвинил (хлорвиниласт,
хлорвинипленка) выпускают под названием пластикат (различных
марок) для самого разнообразного применения (ленты и трубки
для кабельных изделий, листы и пленки для товаров широкого по-
требления и различного технического назначения.
Высокопластифицированный полихлорвинил применяют в основ-
ном для изоляции проводов и кабеля (главным образом телефон-
Пластические массы из полихлорвинила, содержащие пластификаторы 251
кого, по также и силового) взамен свинцовой оболочки, резины, бу-
маги и т. п. Для этих целей он должен обладать высокими диэлек-
трическими свойствами, поверхностной твердостью, прочностью и
теплостойкостью (до +60°), а также высокой морозостойкостью,
т. е. способностью сохранять определенную степень гибкости при
низких температурах (—40° и ниже). Комплекс этих свойств дости-
гается сочетанием определенной смеси пластификаторов с напол-
нителями, в качестве которых применяют коллоидно-дисперс-
ные минеральные вещества (коллоидный кремнезем, кизельгур и
ДР-) •
В виде примера можно привести следующий рецепт (в вес. ч.)
для приготовления пластифицированного полихлорвинила:
Полихлорвинила ............................. 100
Смеси пластификаторов (трикрезилфосфат, ди-
октилфталат. хлорированный дифенил и др.) . 40—50
Дисперсного кремнезема......................15—20
Стеарата кальция ........................... 3
Физико-механические и диэлектрические свойства различных
пластикатов, применяемых для изоляции кабеля, могут в широкой
степени варьировать в зависимости от состава и количества приме-
ненных пластификаторов и наполнителей, а также от вязкости по-
лихлорвинила:
Морозостойкость, °C ...........................от —15 до —50
Предел прочности на разрыв, к?[см2.......... 100—250
Удлинение при разрыве, %.................... 100—300
Уд. объемное электрическое сопротивление, П-сл/ 1 - 1013—5-Ю13
Температура разложения, °C.................. 180—220°
Морозостойкость кабельной оболочки может быть значительно
повышена, если в процессе экструзии произвести вытяжку. Для
увеличения морозостойкости на 10—15° достаточна вытяжка на
80—100%. При этом, соответственно, увеличивается и прочность
изделия.
Обкладку кабеля пластикатом производят в шнекпрессах (рис. 172, стр. 439)
со специальной головкой, поставленной обычно под углом 90° к цилиндру ма-
шины (рис. 109).
Внутри головки строго центрируется дорн, который служит для прохожде-
ния провода к мундштуку. Провод проходит через канал дорна с большой ско-
ростью (до 15—20 км)час}. Вследствие значительного трения и возможности
износа дорн делают из высокотвердых сортов стали.
Массу в виде лент или крошки с температурой 120—130° загружают
в камеру шнекпресса, цилиндрическая часть которого нагрета на 130—160°
(паром или горячей водой под давлением), головка до 140—160° и мундштук
до 170—190° (электрообогрев). Строгое постоянство температуры, в частности
в головке и в мундштуке, — основное условие получения оболочки стандартной
толщины и заданных свойств. Подлежащий обкладке металлический провод
пропускают через канал дорна, откуда он выходит через мундштук, уже
обложенный пленкой пластифицированного полихлорвинила. По выходе из
мундштука кабель проходит через охлаждаемый барабан и наматывается
на валик.
252 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
Обычным методом экструзии из пластиката получают шланги и
различные изоляционные трубки.
Пластикат находит чрезвычайно разнообразное применение в раз-
личных отраслях промышленности: для производства обуви (верх и
Рис. 109. Головка шнекпресса для
обкладки кабеля.
/—шнек; 2—камера; 3 — решетка; 4 — спуск-
ной кран; 5—обогрев; 6—установочные болты;
7 -д^рн; S - мундштук; 9- канал для преве-
лики.
подошва), непромокаемых пла-
щей, клеенки, поясных ремней и
т. п. В больших масштабах про-
изводятся тонкие пленки (0,01 мм
и ниже), используемые для упако-
вочных целей, для изоляции про-
водов и т. д.
Широко развиваются методы
переработки пластифицированно-
го полихлорвинила в виде водных
эмульсий и безводных вязких
паст. Водные эмульсии полихлор-
винила применяют для поверх-
ностной обработки кожи, дерева,
бумаги, ткани и т. д.
В последние годы широкое
распространение получают поли-
хлорвиниловые пасты. Их готовят
путем предварительного смешения
тонкодисперсного полихлорвинила
с пластификатором (например
трикрезилфосфатом) в отноше-
ниях от 2 : 1 до 1 : 1. По выходе
из мешателя смесь растирают на
вальцах или размалывают на ша-
ровых мельницах. Принцип при-
менения паст основан на незна-
чительной растворимости и малой скорости желатинизации
полихлорвинила в пластификаторах на холоду и сравнительно хоро-
шей растворимости и быстрой желатинизации — при высоких
температурах. Получаемая паста имеет сравнительно невысокую
вязкость (до 5000 сп), однако обладает большой липкостью и
консистенцией, позволяющей наносить ее в виде тонкого слоя
на ткань, бумагу, кожу, а также на металлические формы. Если
после этого форму или ткань нагреть (при 100—110°), то про-
исходит быстрый процесс желатинизации и образования проч-
ной, эластичной пленки. Этим методом получают перчатки, галоши,
обувь, а также искусственную кожу, плащи и различные ткани,
пропитанные смолой.
Полихлорвинил нашел применение и в качестве поропласта
(стр. 220). Поропласт из полихлорвинила отличается от других
поропластов, в частности — полистирола, своей негорючестью и
теплостойкостью.
Хлорированный полихлорвинил
253
Хлорированный полихлорвинил
(Перхлорвинил)
Если на раствор полихлорвинила действовать хлором, то происходит даль-
нейшее замещение водорода хлором и получаются полимеры с более высоким
содержанием хлора, чем в полихлорвиниле (до 65—68% вместо 56,8 для поли-
хлорвинила) .
Хлорируют полихлорвинил, пропуская через его раствор (—10%) в тетра-
хлорэтане в течение 12—16 час. газообразный хлор при 60—100°. Образую-
щийся хлорированный полимер высаживают из раствора метанолом, осадок
в виде белого порошка отфильтровывают на центрифуге и сушат в полочных
вакуум-сушилках. Реакция хлорирования протекает, невидимому, с замещением
атома водорода при незамещенных атомах углерода цепи, причем, в среднем,
каждый третий незамещенный атом углерода обменивает водород на хлор, что
можно представить примерно следующей схемой:
... — Cri — СН—Г—СН,—СИ—СН2— СН—СНа— СН1 -СН.-СН-+-^- С13 -->
I ’ I I I I 3
Cl L ci Cl Cl Jn_ Cl
3
---> ... — СН,—СН—Г—CHS—СН—СН—СП—СН.,—СН—“] —СН —СН— ... +4- HCI
I III ” I 'I 3
Cl L CI C! Cl C1 j’2_ fl!
3
Перхлорвинил растворяется в значительно большем числе растворителей.
Он хорошо растворим в ацетоне и других кетонах, в хлорбензоле, в тетрахлор-
этане и т. д., обладает лучшей адгезией к металлу, коже, дереву и, что очень
важно, — и к самому полихлорвинилу. В технике это используют для покрытия
металлических изделий полихлорвиниловыми пластиками. Для этого на металл
наносят пленку перхлорвинила, а сверху прессованием — полихлорвинил.
Перхлорвинил, однако, несколько менее термостоек и при нагревании легче
отщепляет хлор, чем полихлорвинил.
Основное применение хлорированный полихлорвинил получил в производ-
стве антикоррозийных лаков. Для этих целей применяют либо чистую смолу,
либо в смеси с пластификаторами, с глифталевыми и другими смолами и т. д.
Такого рода пленки отличаются эластичностью, твердостью, хорошими адгезион-
ными свойствами, кислота- и щелочестойкостью, стойкостью по отношению
к бензину и маслам.
Перхлорвинил входит в состав различных красок, — его используют, в част-
ности, для наружной окраски зданий, а также и для производства эле-
ктроизоляционных и упаковочных пленок.
Все большее значение хлорированный полихлорвинил приобретает для про-
изводства волокна. Для этого применяют перхлорвинил со степенью полимери-
зации ~250—300 и с содержанием 63—64% хлора; смолу растворяют обычно
в ацетоне (~25-% раствор), последовательно фильтруют через слои различ-
ных хлопчатобумажных тканей (бязь, вата, батист и др.) и после охлажде-
ния до +20° пускают на прядение. Прядильный раствор подается из бака
по трубопроводу в прядильные машины. Его продавливают через фильеры
в стеклянный цилиндр, где циркулирует водный раствор ацетона. Тонкие струйки,
выходящие нз фильер, осаждаются и постепенно затвердевают в волокна. По
выходе из цилиндра нить наматывается на бобину, проходит в дальнейшем ряд
натяжных валиков и вытягивается в 4—6 раз. Чем выше степень вытяжки, тем,
естественно, выше прочность волокна. После вытяжки нити подвергают круче-
нию и перематывают на катушки.
Высокая химическая стойкость (по отношению к серной, соляной и другим
кислотам, а также к щелочам, маслам, бензину и т. д.), прочность, хорошие
диэлектрические свойства и влагостойкость — преимущества перхлорвинилового
254 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
волокна. Недостатком является размягчение и релаксация растянутого волокна
при 70—80°.
Из перхлорвинилового волокна изготовляют фильтровальные ткани, ткани
для спецодежды в химической промышленности, ленты для транспортеров, сети,
канаты, щетки и др.
Сополимеры хлористого винила
Наряду с большими преимуществами (химическая стойкость,
прочность, теплостойкость), у полихлорвинила отмечается ряд су-
щественных недостатков: 1) малая текучесть, вследствие которой
он не может перерабатываться методом литья под давлением;
2) растворимость лишь в ограниченном числе растворителей; 3) пло-
хие адгезионные свойства; 4) недостаточная термостабильность и
др. Использование пластификаторов для облегчения переработки
полихлорвинила и улучшения его пластичности не во всех случаях
целесообразно, так как пластификаторы ухудшают стабильность и
химстойкость полимера.
Естественно, что решение этой задачи искали в процессах сопо-
лимеризации. Введение в цепь полимера второго компонента, как
известно, может снизить межмолекулярные силы взаимодействия,
нарушить симметричность расположения диполей. С этой точки
зрения сополимеризацию можно рассматривать как процесс струк-
турной пластификации.
Первым и важнейшим техническим сополимером хлористого ви-
нила является продукт его сополимеризации с винилацетатом. Оба
мономера легко образуют совместные полимеры.
В начале процесса сополимеризации несколько быстрее идет
реакция между мономерными молекулами хлористого винила, чем
между хлористым винилом и винилацетатом, и полимер, образую-
щийся в начале процесса, содержит несколько больший процент
хлора, чем исходная смесь. К концу процесса сополимеризации со-
став исходной смеси и состав полимера почти одинаковы. Таким об-
разом, готовый полимер все же должен содержать цепи с различным
соотношением реагирующих компонентов. Следовательно, в этом
случае полимер полидисперсен не только по длине макромолеку-
лярных цепей, но и по их структуре. Фракционирование полимера
дает возможность обнаружить оба вида полидисперсности.
Техническое применение получили сополимеры с преобладаю-
щим содержанием хлористого винила. Однако чем выше в сополи-
мере содержание ацетильных групп, тем лучше он растворим, тем
ниже его температура стеклования и тем он эластичнее. Варьируя
соотношение компонентов, а также степень полимеризации, можно
получить сополимеры с большим разнообразием свойств, приме-
няемые для различных назначений (табл. 25).
Процесс сополимеризации хлористого винила с винилацетатом
может быть осуществлен обычными методами как в растворителях,
так и в эмульсиях.
Сополимеры хлористого винила
255
Обычно сополимеризацию ведут в таких растворителях, которые
растворяют оба мономера, но в которых образующийся сополимер
нерастворим (например, в гексане, пентане и т. п.) и высаживается
из растворителя в виде тонкого дисперсного порошка, который за-
тем легко может быть отфильтрован, отмыт и высушен. Иногда же
сополимеризацию ведут в таких растворителях, как ацетон, из ко-
торого сополимер осаждают затем спиртом.
ТАБЛИЦА 25
Назначение сополимерэв хлористого вини на с винилацетатом
в зависимости от их состава и молекулярного веса
Содержание хлори- стого винила, о/о Молекулярный вес Назначение
65—70 4 000— 6 000 Покрытие тканей совместно с ннтроцсл-
люлозой
86—87 8 500— 9 500 Изготовление различных пленок
86—87 9 600—10 500 Изготовление патефонных пластинок,
покрытие полов
85—88 12 000—13 000 Для прессовочных композиций
88—90 15 000—16 000 Изготовление листового материала
85—90 20 000—23 000 Получение синтетического волокна
95 20 000—23 000 Покрытие проводов
Сополимеры хлористого винила с винилацетатом получили более
разнообразное применение, чем полихлорвинил. Оли выгодно отли-
чаются от полихлорвинила возможностью переработки методом
литья под давлением. Прессматериалы из сополимеров обычно не
содержат пластификатора или содержат его лишь в небольшом
количестве (до 10%).
Отличаясь лучшей растворимостью, сополимеры хлористого ви-
нила и винилацетата нашли применение в лаковой технике, для
нанесения пленок на бумагу, ткань и т. д. Особый тип сополимеров,
содержащих до 90% хлористого винила, пластифицированных три-
этил енгликольгексоатом, эфирами себациновой и фталевой кислот,
может быть получен прозрачным, гибким и эластичным.
Сополимеры с более высоким молекулярным весом используют
для производства синтетического волокна, получаемого путем про-
давливания растворов сополимера через фильеры. Сополимеры
применяют для покрытия проводов (для производства кабельной
оболочки); при этом они требуют меньше пластификатора, чем
полихлорвинил. Фракции сополимеров, не содержащие низкомоле-
кулярных частей, применяются в зубопротезной технике.
Комплекс механических свойств сополимеров с содержанием
85—87% хлористого винила несколько выше, чем полихлорвинила
(в частности, сополимеры имеют большее удлинение и большую
256 Гл. VI. Пластмассы на основе голимеров галоидопроизв. этилена
динамическую прочность); однако теплостойкость и химическая
прочность сополимеров несколько ниже. Как правило, сополимеры
для достижения одинаковой степени эластичности требуют введения
значительно меньшего количества пластификаторов, чем полихлор-
винил.
Промышленное применение получили также сополимеры хло-
ристого винила с метилметакрилатом, бутилакрилатом, простыми
виниловыми эфирами, диметил-, диэтил- и диизобутилмалеатами,
а также трехкомпонентные сополимеры. Технические преимущества
этих сополимеров заключаются в основном в большей их ста-
бильности и термостойкости, растворимости и эластичности; они
более прозрачны и легче перерабатываются. В табл. 26 приведены
состав и назначение некоторых из указанных сополимеров.
ТАБЛИЦА 26
Различные сополимеры хлористого винила и их назначение
Состав сополимера, °.'о Назначение
Хлористый винил 80 Диметилмалеат 10 Диэтилмалеат 10 Прозрачные полированные листы, циферблаты и т. п.
Хлористый винил 84 Метилакрилат 16 Оболочки кабеля, шланги и т. п.
Хлористый! винил 80 Метилакрилат 13,5 Диизобутилмалеат 6,5 Аккумуляторные баки, изготовлен- ные литьем под давлением и прес- сованием
Хлористый винил 73 Изобутилвиниловын эфир 27 Для литьевых и прессизцелий
Одним из важнейших сополимеров хлористого винила является
также продукт его сополимеризации с хлористым винилиденом.
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИХЛОРВИНИЛ ИДЕНА
(Хлорвинилиденопласты)
Хлористый винилиден—1,1-дихлорэтилен СН2 = СС12 был впер-
вые получен Реньо свыше ста лет тому назад. Уже тогда было
обнаружено, что при стоянии он образует трудно растворимую бе-
лую массу, выпадающую из мономера. Однако работы по изучению
процесса полимеризации хлористого винилидена были проведены
лишь в последние 15—20 лет; они привели в 1939—1940 гг. к про-
мышленному освоению нового весьма ценного пластика — поли-
хлорвинилидена.
Пластические массы на основе полихлорвинилидена
257
Хлористый винилиден получают путем отщепления хлористого
водорода от трихлорэтана при обработке его при 90° гидратом
окиси кальция:
2СН2С1—СНС12Н-Са(ОН)2 —> 2СН2=СС12 + СаС12 + 2Н2О.
Исходным сырьем является этилен, хлорированием которого по-
лучают дихлорэтан:
СН3=СН3 + С12 —> СН3С1—СН2С!.
Последний затем хлорируют в трихлорэтан:
СН2С1-СН3С1 + С1, —► СН2С1—СНС12 + НС!.
Хлористый винилиден представляет собою бесцветную жидкость
со слабым характерным запахом, с т. кип. 31,7°, т. плавл. —122,1°,
уд. вес d“° — 1,2124 и показателем преломления 1,4244.
Тщательно очищенный мономер, не содержащий кислорода,
полимеризуется очень медленно, однако технический продукт поли-
меризуется относительно быстро, и так как полимер нерастворим в
мономере, то он осаждается в виде порошка.
Чисто термическая полимеризация, без катализаторов, протекает
медленно, в то время как фотополимеризация при действии света
с длиной волны менее 4500 А идет сравнительно быстро.
Хлористый винилиден полимеризуется по механизму цепной
полимеризации как в присутствии инициаторов (перекиси бензоила,
перекиси водорода), так и катализаторов (карбонильные соедине-
ния, кислоты, соли). Однако в технике применяют, главным обра-
зом, обычные инициаторы радикальной полимеризации: перекись
бензоила, перекись водорода и т. п.
Полимеризацию вследствие низкой температуры кипения хло-
ристого винилидена проводят в автоклавах под давлением при
30—60° в присутствии небольшого количества перекиси бензоила
(0,05—0,5%). Техническое применение получила полимеризация
в растворителях и в водной эмульсии. При блочной полимеризации
чистого мономера ввиду нерастворимости полимера в мономере по-
является твердая фаза; уже при 10% конверсии полимер выде-
ляется в виде хлопьевидного осадка, а при дальнейшей полимери-
зации жидкая фаза исчезает и вся масса превращается в пористое
твердое вещество.
Хлористый винилиден образует сополимеры с различными про-
изводными этилена (хлористым винилом, винлацетатом, стиролом,
акрилатами и метакрилатами и др.), а также с диеновыми соедине-
ниями и их производными (бутадиен и др.).
Важнейшее техническое значение имеют сополимеры хлористого
винилидена с хлористым винилом. Процесс сополимеризации ха-
рактеризуется рядом закономерностей. Содержание хлорвинилиде-
новы.х групп в образующемся сополимере обычно превышает содер-
жание хлористого винилидена в исходной смеси мономера. Скорость
17 Зак. 1269. Э. И. Барг
258 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
полимеризации смеси мономеров значительно меньше, чем скорость
полимеризации отдельных мономеров (рис. ПО). В начальной ста-
дии реакции конверсия хлористого винилидена идет быстрее, чем
хлористого винила, тогда как в конечной стадии в реакцию вступает
почти исключительно хлористый винил. Продукт сополимеризации
всегда содержит некоторое количество смеси полимеров каждого из
реагирующих мономеров. Сополимер состоит из макромолекул
с различными боковыми группами и с различной длиной цепей.
Рис. 110. Скорость сополимеризации хлористого ви-
нила с хлористым винилиденом (с 0,5% перекиси бен-
зоила, при 45°, в темноте).
Сополимеры хлористого винилидена с хлористым винилом могут
быть получены в различных соотношениях мономерных групп.
В технике применяют главным образом следующие три типа сопо-
лимеров, которые содержат: 1) менее 20%; 2) 20—40% и 3) более
50% хлорвиниловых групп. Наибольшее техническое значение имеют
сополимеры с содержанием менее 20% хлорвиниловых групп.
Строение полихлорвинилидена
Полихлор винилиден имеет структуру 1,3 («голова к хвосту»)
(рис. 111), при этом пары атомов хлора располагаются попере-
менно на противоположных сторонах цепи; период идентичности
равен 4,7 А, что указывает на некоторое отклонение от плоской
зигзагообразной цепи.
Полихлорвинилиден легко кристаллизуется и содержит преобла-
дающее количество кристаллической фазы. Рентгенограмма поли-
мера в кристаллическом состоянии показывает весьма четкую ли-
нию колец; при вытяжке полимера рентгенограмма принимает вид,
4
Строение полихлорвинилидена
259
характерный для ориентированных кристаллитов, с четкими точеч-
ными сгущениями.
При быстром охлаждении расплавленный полихлорвинилиден
может быть полностью переведен в аморфное состояние. Скорость
рекристаллизации зависит от температуры. При комнатной темпе-
ратуре процесс продолжается несколько дней, при 30° — 20 мин.,
а при 60° — 1 мин.
Рис. 111. Схема строения молекулы полихлорвинилидена.
•—атомы углерода; О —атомы водорода; О С1— атомы хлора.
В аморфном состоянии при обычных температурах полихлорвн-
нилиден мягок и каучукоподобен; температура стеклования аморф-
ной фазы —17°. В этом отношении, он резко отличается от
полихлорвинила, точка стеклования которого -|-80о. Следует пред-
положить, что симметричное замещение обоих водородных атомов
у одного углеродного атома мономерного звена одинаковыми груп-
пами приводит к компенсации влияния диполей и, следовательно,
снижает внутримолекулярный потенциал, увеличивает гибкость и
подвижность звеньев.
В кристаллическом состоянии полихлорвинилиден — твердое,
упругое вещество с температурой плавления 185—200°. Растяже-
ние, вызывая ориентацию кристаллитов, приводит к увеличению
кристаллической фазы полимера, а также к значительному (в не-
сколько раз) увеличению прочности. Растяжение полимера произ-
водят при температурах ниже температуры плавления кристал-
лической фазы, что может быть достигнуто при невысоких
температурах благодаря низкой температуре стеклования аморф-
ной фазы.
Процессы сополимеризации, как и можно было ожидать, умень-
шают содержание кристаллической фазы, и тем больше, чем меньше
в сополимере хлорвинилиденового компонента. Однако большин-
ство технических сополимеров хлористого винилидена, в частности
сополимеры с хлористым винилом, показывает еще четкую кри-
сталлическую рентгенограмму даже при содержании в сополимере
только 70% хлорвинилиденовых групп.
17*
260 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
Полихлорвинилиден имеет примерно ту же степень полярности,
что и полихлорвинил (дипольный момент ~ 1,8 • 10~18). Однако по-
казатели его диэлектрической постоянной и тагенса угла диэлектри-
ческих потерь имеют меньшую зависимость от температуры и ча-
стоты поля, чем у полихлорвинила.
Свойства сополимеров хлористого винилидена
Полихлорвинилиден вследствие высокой температуры размяг-
чения и малой термостабильности получил лишь ограниченное при-
менение, однако сополимеры хлористого винилидена имеют большое
промышленное значение (в частности с хлористым винилом).
Сополимеры хлористого винилидена с хлорвинилом содержат
обычно свыше 85% хлорвинилиденовых групп и обладают кристал-
лической структурой и физико-механическими свойствами, близ-
кими к полихлорвинилидену; их преимущество заключается
в том, что они легче перерабатываются и имеют большую термо-
стабильность. Средний молекулярный вес технических сополимеров
20 000—100 000, уд. вес ~ 1,7; они относятся, следовательно, к наи-
более тяжелым пластикам. Отличительная особенность этих сопо-
лимеров — высокая химическая стойкость. Неорганические кислоты
и щелочи не оказывают на них практически никакого действия, за
исключением серной кислоты концентрации свыше 95%, концентри-
рованного раствора едкого натра и аммиачной воды, вызывающих,
однако, лишь побеление продукта.
Сополимеры с высоким содержанием хлористого винилидена
(свыше 85%) практически противостоят почти всем известным
органическим растворителям, за исключением весьма немногих
(о-дихлорбензол, диоксан, циклогексанон), которые, однако, дей-
ствуют только при повышенных температурах. Промышленные про-
дукты стойки к воздействию алифатических и ароматических угле-
водородов, спиртов, эфиров, кетонов и т. д. Высокая химическая
стойкость полихлорвинилидена и его сополимеров является важ-
нейшим фактором, определяющим их техническое применение. Они
отличаются сравнительно узким интервалом плавления, связанным
с обычным процессом плавления кристаллической фазы.
Выше температуры плавления (185—200°) они переходят в вяз-
кое состояние, однако при этих температурах происходит уже с за-
метной скоростью разложение полимера. При кристаллизации
выделяется тепло — около 3,4 кал на 1 г полимера — и наблю-
дается увеличение удельного веса, твердости, изменение диэлектри-
ческих свойств, увеличение прочности на разрыв и уменьшение удли-
нения при разрыве, увеличение сопротивления действию раствори-
телей; рентгенограмма постепенно приобретает вид, характерный для
кристаллического полимера.
Кинетику процесса и индукционный период кристаллизации
можно довольно точно наблюдать по изменению светопроницае-
Свойства сополимеров хлористого винилидена
261
мости образца, помещенного между скрещенными ппколими. ве-
личина индукционного периода кристаллизации является характер-
ной константой для каждого сополимера. Зависимость индукцион-
ного периода и скорости кристаллизации от температуры довольно
сложна. Наименьшую величину индукционный период имеет
в пределах примерно 50—120°. Выше 120° индукционный период
увеличивается под влиянием относительно высокой термической
энергии, дезориентирующей макромолекулярные цепи. Ниже 50°
увеличение индукционного периода определяется резким нараста-
нием вязкости, которая тормозит кристаллообразование. Темпера-
турная зависимость кристаллизации имеет существенное значение
для технологии переработки полимера.
Молекулярный fee
Рис. 112. Влияние величины молеку-
лярного веса на температуру хруп-
кости полихлорвинилидена, содержа-
щего 9% пластификатора.
Содержание хлорвинила С сополин.
мол. %
Рис. ИЗ. Зависимость температуры
стеклования сополимера хлористого
винилидена с хлористым винилом от
его состава.
В аморфном состоянии полимер обладает каучукоподобными
свойствами, что имеет значение в некоторых стадиях его перера-
ботки.
Переход из аморфного в кристаллическое состояние сопрово-
ждается незначительным изменением плотности полимера (от 1,66
до 1,69) и, следовательно, лишь небольшой усадкой.
В кристаллическом состоянии полимер обладает рядом свойств,
характерных для типичных пластиков: твердостью, жесткостью,
прочностью и теплостойкостью. Механическая прочность, гибкость
и эластичность полимера значительно увеличиваются при растяже-
нии. При максимальном удлинении в 400—600% прочность на рас-
тяжение достигает 4000—7000 кг/см2, что значительно превосходит
прочность других ориентированных полимеров.
262 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
Технические свойства полихлорвинилидена и сополимеров хлори-
стого винилидена во многом определяются степенью полимеризации.
Как видно из рис. 112, с уменьшением степени полимеризации (после
определенной величины) резко возрастает температура хрупкости
полимера, одновременно резко снижается и его прочность.
Полихлорвинилиден имеет высокую удельную ударную вязкость,
высокую морозостойкость и эластичность, которые определяются
сочетанием аморфной фазы, находящейся в высокоэластичном со-
стоянии (точка стеклования —17°), со значительным содержанием
кристаллической фазы, имеющей высокую температуру плавления.
Полимер, таким образом, представляет собою систему, состоящую,
в основном, из жестких и упругих кристаллитов, соединенных, од-
нако, подвижными молекулами, находящимися в непрерывном
микроброуновском движении.
Для технических сополимеров температура стеклования аморф-
ной фазы закономерно повышается с уменьшением содержания
групп хлористого винилидена. Соответственно также уменьшается
и концентрация кристаллической фазы. Это приводит к меньшей
морозостойкости и гибкости сополимеров по сравнению с чистым
полихлорвинилиденом. На рис. ИЗ показана зависимость темпера-
туры стеклования сополимера хлористого винилидена и хлористого
винила от его состава.
Пределы физико-механических свойств сополимеров хлористого
винилидена и хлористого винила могут быть представлены следую-
щими данными:
Уд. вес, г/слс’.............................. 1,6—1,75
Предел прочности на разрыв, кг/см2:
в неориентированном состоянии ............... 400
в ориентированном (400—600°) состоянии . 4000—7000
Удлинение (для ориентированного), % . . . . 25
Предел прочности на изгиб, кг)см2............ 1000—1100
Уд. ударная вязкость, кг-см/см2.............. 100—150
Теплостойкость по Мартенсу, °C............... 70—95®
Температура размягчения, °C.................. 95—135
Уд. объемное электрическое сопротивление, Q-см 6-1013—1 • 101в
Водостойкость (привес в воде за 24 часа), % • 0,01
Теплопроводность, кал/см - сек - °C.......... 2,2 • 10-4
Теплоемкость, кал]г -°C...................... 0,316
Тангенс угла диэлектрических потерь при 106 гц 0,02—0,08
Диэлектрическая постоянная при 106 гц .... 3,5
Переработка и применение сополимеров хлористого винилидена
Пластические массы из сополимеров хлористого винилидена получают
путем пластицирующей обработки их на горячих вальцах со стабилизаторами,
пластификаторами, красителями и наполнителями.
Техника переработки сополимеров требует специальных приемов и специаль-
ного аппаратурного оформления. Это связано с тем, что сополимеры обладают
малой термостабильностью, высокой температурой плавления и двух-
фазной структурой. Для этого в горячих зонах аппаратуры применяют спе-
циальные сплавы, например, магниевые н никелевые, так как железо и медь
Пластические массы на основе политетрафторэтилена
263
ускоряют процесс деструкции сополимеров и выделение хлористого водорода.
Деструкция наблюдается уже при 130°. Особенно важно создать условия для
максимального сокращения времени нагрева полимера, а также снижения тем-
пературы переработки н повышения термостабильности путем внесения пласти-
фикаторов и стабилизаторов.
Основным методом переработки сополимеров хлористого винилидена
является метод выдавливания (экструзии), позволяющий изготовлять нити,
шланги, оболочки, стержни, ленты, пластины, пленки и т. д. По этому методу
смесь надлежащего состава может быть загружена непосредственно в прием-
ную воронку аппарата без предварительного вальцевания. Все возрастающее
техническое значение ориентированных нитей из сополимера, которые при-
меняют для производства тканей, щеток, фильтровального полотна и т. д.,
потребовало производства специальных машин с приспособлениями для прямо-
линейного течения массы и равномерного нагревания наряду с приспособле-
ниями для односторонней и двухсторонней вытяжки получаемых изделий.
Выходящие из шнекпресса нити и шланги имеют аморфную структуру, оии про-
зрачны, мягки и каучукоподобиы, однако при медленном охлаждении в интервале
100—120° они быстро кристаллизуются и приобретают упругость и прочность.
Наряду с экструзией широкое применение для переработки сополимеров
имеет метод прессования, а также литья под давлением. Как известно, обыч-
ный метод прессования является малоэффективным для переработки термо-
пластов, так как он требует охлаждения прессформ после запрессовки; однако
в случае полихлорвинилидена можно обойтись без значительного охлаждения
прессформ, так как процесс отвердевания (кристаллизации) изделия требует
высокой температуры (100—130°) и в этом случае в известной мере аналогичен
отверждению термореактивиых прессмасс (фенопластов и др.) в постоянно на-
гретых прессформах.
Литье под давлением может быть осуществлено с вливанием массы либо
в холодную, либо в нагретую форму. В первом случае получается мягкая
масса с аморфной структурой, которую затем отдельно подвергают процессу
кристаллизации. Во втором случае из формы можно получить твердое, прочное
изделие. Мягкие изделия, получаемые при литье в холодных формах, могут
быть до наступления кристаллизации подвергнуты еще дальнейшей обработке.
Высокоэластичность сополимеров хлористого винилидена в комбинации
с кристалличностью допускает использование и таких методов переработки, как
штамповка, холодная и горячая прокатка, ковка и т. п. Широко применяется
также сварка труб, шлангов и пластин.
В последнее время получили распространение полихлорвииилидеиовые эмуль-
сии, которые применяют для пропитки тканей, бумаги, кожи.
Из сополимеров хлористого вииилидеиа изготовляют трубы для кислото- и
щелочепроводов, различные шланги, фильтровальное полотно, корпуса насосов,
рукоятки, облицовочные плиты для травильных и электролизных ванн и др.
Нити, лента, шнуры, оболочки и т. п. — идут на изготовление ремней кана-
тов, рыболовных сетей, щетины, различных щеток, полотна, обивочного мате-
риала для чемоданов и для сидений вагонов, кресел, для изготовления тен-
нисных ракеток и т. д.
Ткани, полотна, нити и т. п. из полихлорвинилидена отличаются высокой
прочностью на износ, прочностью на разрыв, негорючестью и весьма высокой
химической инертностью (практически) по отношению к действию всех кислот
и щелочей.
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИТЕТРАФТОРЭТИЛЕНА
(Тетрафторвинипласты)
Быстрое развитие химии соединений фтора привело к техниче-
скому синтезу тетрафторэтилена и к получению нового пластика —
политетрафторэтилена.
264 Гл. VI. Пластмассы на основе полимеров галоид опр о из в. этилена
Исходный тетрафторэтилен CF2 = CF2 представляет собою
бесцветный нетоксичный газ, без запаха, с т. кип. —76,3°, т. за-
мерз. —142,5°, критической температурой —33,3°, критическим
давлением 37,8 ат и уд. вес (жидкости) 1,202-- 0,00414 t (от —100
до —40°).
Тетрафторэтилен может быть получен различными методами,
например, исходя из монохлордифторметана, путем пиролиза его
при 400°:
2CHF2C1 —CF2=CF.. + 2HC1,
а также из тетрафтордихлорэтана действием цинка при высоких
температурах:
C1F2C-CF2C1-+-Zn —-> F2C=CF2 -f- ZnCl2.
Несмотря на симметричное строение тетрафторэтилен сравни-
тельно легко (по сравнению с этиленом) полимеризуется. Реакция
полимеризации протекает под умеренным давлением в присутствии
инициаторов (перекиси бензоила, кислорода, перекиси водорода,
персульфатов и др.); она имеет типично цепной радикальный харак-
тер и ведет к получению полимера с высокой степенью полимериза-
ции.
При реакции полимеризации тетрафторэтилена необходимо счи-
таться с возможностью его разложения (на углерод и тетрафтор-
метан), вследствие экзотермичности реакции (~ 25 000 кал! мол) и
малой термодинамической устойчивости тетрафторэтилена.
Реакция может протекать весьма бурно, а иногда и со взрывом,
так как скорость полимеризации резко возрастает с увеличением
давления. Для получения технически годного продукта необходимо,
таким образом, создать условия, обеспечивающие быстрый отвод
тепла и сохранение в процессе полимеризации постоянного темпе-
ратурного режима. Полимеризацию поэтому ведут в эмульсиях.
Реакция проводится в автоклавах, изготовленных из кислотоупор-
ной стали и снабженных мешалкой и рубашкой для нагрева и охла-
ждения смеси.
В автоклав, предварительно продутый азотом, загружают, на-
пример, 160 ч. воды и 0,14 ч. персульфата калия; автоклав
охлаждают пуском в рубашку охладительной смеси, закрывают и
создают в нем разрежение. Затем в автоклав вводят 10—15 ч.
тетрафторэтилена и при постоянном перемешивании доводят темпе-
ратуру до 70—80°; реакция протекает под давлением 40—100 ат.
По окончании реакции реакционную смесь охлаждают, отгоняют
газообразный мономер и содержимое автоклава переводят на
центрифугу, где полимер, полученный в виде белых волокон, филь-
труют, промывают и затем передают на сушку.
Тетрафторэтилен, как и многие мономеры, сравнительно быстро
полимеризуется при хранении. Полимеризация инициируется сле-
дами кислорода воздуха и незначительным количеством перекиси.
В качестве ингибиторов предложены вещества, содержащие сульф-
Строение и свойства политетрафторэтилена
265
гидрильную группу или аминоазот (сероводород, бутилмеркаптан.
аммиак, амины и др.). Так, было обнаружено, что 0,5% бутилмер-
каптана’ задерживает полимеризацию тетрафторэтилена, находяще-
гося под давлением 200 ат в течение нескольких месяцев.
Строение и свойства политетрафторэтилена
Политетрафторэтилен имеет правильную линейную структуру
F F F F
1111
... —С—С—С—С— ...
1111
F F F F
Его макромолекула, несмотря на наличие сильно электроотрица-
тельных атомов вследствие симметричности их расположения, не
имеет неуравновешенных диполей и ее суммарный дипольный мо-
мент равен нулю; таким образом, политетрафторэтилен является
насыщенным неполярным веществом.
Симметричное строение макромолекул, тождественное со строе-
нием полиэтилена, обусловливает его кристаллическое строение.
Однако ввиду большей энергии взаимодействия между атомами
фтора соседних молекул (~ 2000 кал/моль) температура плавле-
ния и концентрация кристаллической фазы должны быть у поли-
тетрафторэтилена значительно выше, чем у полиэтилена.
В аморфном состоянии политетрафторэтилен каучукоподобен.
Получают его в аморфном («закаленном») состоянии быстрым охла-
ждением после расплава. Такой «закаленный» полимер мало устой-
чив и постепенно «твердеет» — переходит в кристаллическое состоя-
ние. Этот переход ускоряется повышением температуры. При кри-
сталлизации, наряду с другими термодинамическими свойствами,
меняется (увеличивается) уд. вес полимера, чем и объясняется его
значительная усадка.
Температура стеклования политетрафторэтилена столь же
низка, как и у полиэтилена (~—80°), однако его температура пла-
вления равна 320—327°, т. е. примерно на 200° выше. Он является
самым высокоплавким из всех кристаллических полимеров.
Политетрафторэтилен характеризуется чрезвычайно широким ин-
тервалом рабочих температур, — практически от температур жид-
кого воздуха до 300°.
Сочетание каучукоподобной аморфной фазы с высокоплавкой
кристаллической обусловливает малую твердость политетрафтор-
этилена, его чрезвычайно низкую температуру хрупкости, гибкость
и некоторую эластичность при растяжении.
Политетрафторэтилен самый тяжелый из известных в настоящее
время технических полимеров (уд. вес 2,2—2,3).
Наиболее характерными свойствами политетрафторэтилена
являются его весьма высокие химстойкость и термостабилыюсть;
266 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
в этом отношении он превосходит все известные полимеры, в том
числе полихлорвинилиден, полиэтилен и др.
В связи с некоторой подвижностью звеньев аморфной фазы
теплостойкость политетрафторэтилена, определяемая показателями
зависимости скорости деформации от температуры в пределах от
комнатных температур до 100—130°, зависит также и от величины
нагрузки; однако этот полимер сохраняет достаточную твердость,
жесткость и высокую прочность вплоть до температуры фазового
перехода (327°). Термостойкость политетрафторэтилена может быть
охарактеризована тем, что после нагревания в течение месяца при
300° его первоначальная прочность снижается лишь на 10—15%;
разлагается же он лишь при температуре выше 400°.
Высокая теплостойкость политетрафторэтилена сочетается
с почти абсолютной химической инертностью. Он практически не-
растворим, на него не действуют минеральные кислоты, в том числе
царская водка и фтористоводородная кислота, щелочи, соли, га-
лоиды и т. д. Лишь щелочные металлы при 200° вызывают деструк-
цию полимера.
Как неполярный полимер политетрафторэтилен является самым
совершенным диэлектриком; его диэлектрическая постоянная и тан-
генс угла диэлектрических потерь столь же малы, как и у поли-
этилена, практически не зависят от частоты поля (в пределах до
108 гц) и постоянны в весьма широком температурном интеовале
(от—160 до+280°).
Механическая прочность политетрафторэтилена зависит от сте-
пени ориентации. В неориентированном состоянии его прочность
на растяжение равна 140—160 кг/см?, тогда как в ориентированном
она 1000—1200 кг/см2.
Свойства политетрафторэтилена могут быть охарактеризованы
следующими данными:
Уд. вес, г/см3.............................. 2,1—2,3
Предел прочности на разрыв, кг/см2.......... 140—315
Удлинение при разрыве, %.................... 360—400
Предел прочности на изгиб, кг/см2.............140 и 1200 (для
ориентированных
пленок)
Предел прочности на сжатие, кг/см2........... 120
Теплостойкость при низкой нагрузке, °C ... . 130
Теплостойкость при высокой нагрузке, °C . . . 20—60
Температура хрупкости, °C.....................от — 80 до—100
Коэффициент расширения (25—60°).............. 5,5 • Ю-э
Водостойкость (привес в воде), °/о........... 0
Показатель преломления, Лд................... 1,37—1,38
Коэффициент трения по стали.................. 0,2—0,3
Уд. объемное электрическое сопротивление, Q-см 1016—1017
Диэлектрическая постоянная в диапазоне от 60
до 10® гц................................... 2,0
Тангенс угла диэлектрических потерь в диапа-
зоне от 60 до 108 гц....................... 0,0002
Пластические массы на основе политрифторхлорэтилена
26.
Переработка и применение политетрафторэтилена
Политетрафторэтилен вследствие высокой вязкости с большим трудом под-
дается обычной горячей пластнцирующей переработке (прессованию, литью пол
давлением, экструзии и т. д.). Для этого не могут быть использованы обычные
литьевые машины стандартного типа. Мало применим и метод экструзии на
обычных машинах.
Длн переработки политетрафторэтилена разработана особая техника. Так,
для некоторых назначений изделия простой формы изготовляют холодным прес-
сованием порошка, после чего заготовку подвергают спеканию в печи при тем-
пературе выше точки плавления. Дальнейшую обработку производят механиче-
ским путем на токарных и фрезерных станках; например, тонкие листы и
пленки можно получать путем строгания блоков и цилиндров.
При переработке на шнекпрессах со специальными приспособлениями при
высоких давлениях и температурах из политетрафторэтилена удается получать
трубки, а также покрывать им провода, при этом скорость продвижения массы
значительно ниже, чем в случае других пластиков.
Важнейшей областью применения политетрафторэтилена является химиче-
ское машиностроение, изготовление химически стойких пластин и прокладок,
деталей химической арматуры (краны, вентили, клапаны и т. п.) для работ при
высоких температурах в смесях концентрированных кислот (азотной, хромовой
и др.).
Благодаря высокому сопротивлению изнашиваемости, а также весьма низ-
кому коэффициенту трения политетрафторэтилен оказался превосходным мате-
риалом для производства подшипников, работающих в присутствии агрессивных
сред. Так как он имеет невысокую поверхностную твердость, то подшипники из
него являются «самосмазывающимися» и почти не нуждаются в специальной
смазке.
В электротехнике политетрафторэтилен ' широко применяют в тех случаях,
когда от материала требуются высокие теплостойкость и электроизолирующие
свойства, в частности для изготовления деталей высокочастотной техники (оболо-
чек высокочастотного кабеля, изоляции катушек, конденсаторов и др.).
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИТРИФТОРХЛОРЭТИЛЕНА
Как уже было указано, политетрафторэтилен является неполяр-
ным полимером с самыми совершенными диэлектрическими свой-
ствами, наиболее высокой температурой плавления, морозостой-
костью, а также с непревзойденной химической стойкостью. Вслед-
ствие этого область его применения была бы весьма широкой, если
бы он к тому же обладал хорошей текучестью и мог легко под-
вергаться обычной пластицирующей переработке в изделия сложной
формы. Стремление повысить текучесть политетрафторэтилена
привело к изучению ряда его сополимеров, с одной стороны, и
к получению подобных полимеров, в которых один из атомов фтора
замещен каким-либо другим атомом. Среди последних техническое
применение нашел политрифторхлорэтилен:
F F F F
1111
—С—С—с—с—....
1111
F Cl F С1
268 Гл. VI. Пластмассы на основе полимеров галоидопроизв. этилена
который образуется при полимеризации трифторхлорэтилена
CF2 — CFC1. Последний, в свою очередь, может быть получен обра-
боткой 1,2,2-трихлор-1,1,2-трифторэтана цинком в спиртовой среде:
С1Р2С—CC1..F -сйПш* F2C=CFC1 + ZnCL.
Трифторхлорэтилен представляет собою при обычной темпера-
туре бесцветный газ, конденсирующийся в жидкость при —27°; его
т. плавл. —147,9°. При хранении в газообразном или жидком со-
стоянии трифторхлорэтилен полимеризуется при комнатной тем-
пературе.
Полимеризация технически может быть осуществлена, напри-
мер, под действием перекиси бензоила (0,1—0,3%) в растворе
четыреххлористого углерода или хлороформа в автоклаве под
давлением при 45—70°. Образующийся полимер нерастворим
в растворителе и осаждается в виде порошка или белых хлопьев.
Реакция может протекать и в водной среде в присутствии пере-
кисных водорастворимых инициаторов, а также перекиси бензоила,
растворенной в мономере.
Высокомолекулярный политрифторхлорэтилен имеет такое же
правильное линейное построение, как и политетрафторэтилен. Он
также обладает кристаллической структурой, однако со значительно
большим содержанием аморфной фазы.
По стойкости к агрессивным средам политрифторхлорэтилен
мало уступает политетрафторэтилену. Так, на него не действуют
серная кислота, царская водка и т. д.
Политрифторхлорэтилен можно применять при температурах
в пределах от —80 до +180° Верхний предел значительно меньше,
чем для политетрафторэтилена. Политрифторхлорэтилен обладает
относительно высокими диэлектрическими свойствами, однако, так
как симметрия макромолекулы нарушена введением атома хлора,
создается диполь, который проявляется при высоких частотах и
температурах, обусловливая некоторую зависимость диэлектриче-
ской постоянной и угла диэлектрических потерь от частоты поля и
температуры.
Политетрафторэтилен может находиться в устойчивом аморф-
ном (закаленном) состоянии в пределах до 50—70°. В этом состоя-
нии он эластичен, мягок и растяжим.
Политрифторхлорэтилен обладает более высокой поверхностной
твердостью, более высоким модулем упругости, большей статиче-
ской прочностью и меньшей растяжимостью, чем политетрафтор-
этилен.
Технические показатели политрифторхлорэтилена следующие:
Ул. вес, г/см2................................ 2,1—2,15
Предел прочности на разрыв, кг/см2.......... 400
Предел прочности на изгиб, кг/см2........... 500—600
Предел прочности на сжатие, кг'см-.............. 250—560
Предел упругости, кг/см'-.................... 150—250
.Модуль упругости при растяжении, кг'см2 ... 15 090—18 000
Пластические массы на основе политрифторхлорэтилена
2G9
1 Бер/lVV » «’ «1^ .......................... 'Az- 1 х>
Водостойкость (привес в воде), %.............. 0,00—0,01
Теплопроводность, кал'.см сек - °C.......... 1,44 • 10 ~4
Коэффициент преломления, .......................... 1,43
Пробивное электрическое сопротивление (0,76мм)
кв! мм,....................................... 48
Уд. объемное электрическое сопротивление, 0,-см 1017—101е
Диэлектрическая постоянная при 106 гц .... 2,5—3
Тангенс }тла диэлектрических потерь при 106 гц 10-3—1,5 • 10~'-’
Преимущесгвом политрифторхлорэтилена по сравнению с поли-
тетрафторэтиленом является его хорошая текучесть при термиче-
ской переработке. Его можно перерабатывать почти всеми извест-
ными пластицирующими процессами на обычных, стандартных
машинах и аппаратах. Методом экструзии из него можно получать
шланги, оболочки, пленки и листы, а также непосредственно покры-
вать провода. Изделия сложной конфигурации могут быть изго-
товлены горячим прессованием и литьем под давлением.
Политрифторхлорэтилен может быть получен в виде неводных
дисперсий типа латекса и использован для нанесения защитных пле-
нок на металлы, с последующей после сушки термической обработ-
кой при 330—360° в течение нескольких минут.
Области возможного применения политрифторхлорэтилена чрез-
вычайно разнообразны. В высокочастотной электротехнике его
применяют для изготовления радиодеталей, для изоляции кабелей,,
изготовления конденсаторов, а также для изоляции моторов и
трансформаторов в тех случаях, когда материал подвергается дей-
ствию высоких температур и корродирующих сред. Однако наибо-
лее эффективным является применение политрифторхлорэтилена
в качестве антикоррозийного конструкционного материала, для из-
готовления деталей насосов, труб, прокладок, подшипников, для
облицовки аппаратов и т. д.
ГЛАВА VII
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИМЕРОВ
ВИНИЛОВОГО СПИРТА И ЕГО ПРОИЗВОДНЫХ
(Сложные и простые эфиры поливинилового спирта, поливиниловый
спирт, поливинилацетали)
Большое промышленное значение имеют полимеры, которые
можно рассматривать как производные поливинилового спирта,
а именно его сложные и простые эфиры и ацетали.
Так как до сих пор получить свободный виниловый спирт не
удалось, то поливиниловый спирт получают не путем полимериза-
ции винилового спирта, а омылением полимеров его сложных эфи-
ров. Поэтому целесообразно вначале рассмотреть те полимеры, из
которых в технике получают поливиниловый спирт (поливинилаце-
тат, поливинилформиат и др.), а затем уже сам поливиниловый
спирт и его ацетали.
Сложные эфиры поливинилового спирта
ПОЛИВИНИЛАЦЕТАТ
Поливинилацетат является важнейшим техническим полимером
среди сложных эфиров поливинилового спирта.
Поливинилацетат — полимер винилацетата СН2=СНОСОСН3 —
сложного эфира уксусной кислоты и неизвестного в свободном виде
винилового спирта СН2=СН—ОН. При попытках получить вини-
ловый спирт, например, присоединением воды к ацетилену (реакция
Кучерова), отщеплением хлористого водорода от этиленхлоргидрина
и др., обычно образуются либо ацетальдегид, либо окись этилена:
СН2-СН2 СН,=СНОН СН3СНО.
О
Виниловый спирт является неустойчивой энольной формой ацет-
альдегида. Устойчивы лишь его сложные и простые эфиры (винил-
ацетат, винилэтиловый эфир и др.), которые получаются не непо-
средственно из винилового спирта, а специальными методами.
Синтез винилацетата
Винилацетат представляет собою бесцветную легкоподвижную
жидкость с характерным эфирным запахом. Он кипит при 75°
(760 мм), замерзает при —84°, имеет коэффициент преломле-
ния 1,3958, вязкость 0,432 сп (20°) и уд. вес 0,9342; выдерживает
Синтез винилацетата
271
сравнительно высокие температуры, но разлагается с выделением
уксусной кислоты около 400°. В воде при 20° растворяются ~2,5%
винилацетата.
Основным сырьем для его получения служат ацетилен и
уксусная кислота. Последнюю, в свою очередь, в промышленных
условиях получают преимущественно из ацетилена. Винилацетат
образуется в результате присоединения уксусной кислоты к ацети-
лену по схеме:
СН=СН + СН3СООН -—> CH2=CHOCOCHs + 28,3 ккал.
Реакция протекает в присутствии катализаторов, которые одно-
временно могут способствовать и взаимодействию винилацетата
с уксусной кислотой с образованием этилидендиацетата:
СН2=СНОСОСНз+СН3СООН -—> CH3CH(OCOCHs)2 + 6,2 ккал.
Впервые винилацетат был синтезирован в 1912 г.; в 1917 г. были
описаны условия его полимеризации, а в 1920 г. поставлено опытное
производство. В Советском Союзе исследования в области синтеза
винилацетата были проведены С. Н. Ушаковым совместно
с Ю. М. Файнштейн, Е. Н. Ростовским, И. А. Арбузовой. Эти
исследования привели к разработке оригинальной технологии син-
теза винилацетата в паровой фазе, осуществленной в Советском
Союзе,
Технологический процесс производства винилацетата ведут двумя
методами: жидкофазным и парофазным.
В основе жидкофазного метода лежит известная реакция гидра-
тации ацетилена по Кучерову: пропускание ацетилена через рас-
творы ртутных солей в разбавленной серной кислоте. Первоначаль-
ным продуктом присоединения воды к ацетилену должен был бы
быть виниловый спирт, который, однако, немедленно изомеризуется
в ацетальдегид путем миграции водорода от кислорода к углероду
(правило А. П. Эльтекова, 1877 г.):
СНееСН + НОН сн2=снон —► сн3—
1 'Н
По типу реакции Кучерова может протекать взаимодействие
ацетилена со всеми веществами, содержащими гидроксильные и
карбоксильные группы. Так как в образующихся при этом продук-
тах присоединения подвижный водород замещен углеводородным
радикалом или остатком органической кислоты, то они предста-
вляют собой стабильные производные винилового спирта. Поэтому
устранение или связывание воды — основная предпосылка устране-
ния образования ацетальдегида при взаимодействии ацетилена
с органическими кислотами.
Жидкофазный метод проводят пропусканием ацетилена через
жидкую уксусную кислоту. Выход винилацетата зависит от ско-
рости пропускания ацетилена, состава катализатора и температуры.
272 Гл. VII. Пластмассы на основе полимеров винилового спирта
Для увеличения выхода необходимо быстрое выведение образую-
щегося винилацетата из сферы реакции, что может быть достигнуто
быстрым пропусканием избытка ацетилена через реакционную
среду. При этом ацетилен увлекает с собой винилацетат, — как
только он образуется. При более длительном соприкосновении с
уксусной кислотой реакция идет в сторону образования этилиден-
диацетата.
Оптимальная температура реакции в значительной степени за-
висит от характера катализатора; в случае применения сернокислых
солей ртути она составляет 60—65°, при более высоких темпера-
турах (выше 70—75°) образуется главным образом этилиден-
диацетат.
Состав катализатора также определяет срок его службы, кине-
тику реакции и направление ее в сторону преимущественного обра-
зования винилацетата или этилидендиацетата. В качестве катали-
заторов могут быть применены ртутные соли серной, ацетилсерной,
фосфорной и других кислот. Они катализируют реакцию в сторону
образования винилацетата только в свежеосажденном тонко-
дисперсном состоянии.
Наиболее эффективным катализатором является ртутная соль
ацетилсерной кислоты, которую получают взаимодействием HgO со
смесью уксусной кислоты и олеума при обычных температурах.
Хорошим катализатором является также фосфорнокислая соль
закиси ртути. Ее приготовляют смешением ортофосфорной кислоты
с HgO в ледяной уксусной кислоте при 80°.
Добавка ВЕз всегда значительно усиливает каталитическое дей-
ствие ртутных солей; при этом уменьшается образование диэфира
и снижается температура реакции. Действие катализатора стано-
вится еще более эффективным, если кроме BF3 прибавить еще HF
в следующих, например, количествах на килограмм уксусной
кислоты:
4 г HgO-f-l —1,5 г BF3-|-0,5 г HF.
Жидкофазный процесс производства винилацетата осуществляют,
пропуская с большой скоростью избыток ацетилена через стальной
реактор из нержавеющей стали, в котором находится смесь ледяной
уксусной кислоты и уксусного ангидрида, содержащая диспергиро-
ванные ртутные соли. Температура смеси 60—65° или 65—75° в за-
висимости от характера катализатора и скорости пропускания аце-
тилена. Образующийся винилацетат, удаляемый током ацетилена,
вместе с другими летучими веществами конденсируется в холодиль-
нике, а ацетилен, проходя сушильные агрегаты, возвращается
в реактор. Когда скорость связывания ацетилена падает ниже
определенного предела, реакцию прерывают и перезаряжают реак-
тор. Последний содержит отработанный катализатор, а также этили-
дендиацетат, уксусную кислоту и уксусный ангидрид. Неоргани-
ческий остаток отфильтровывают и жидкость фракционируют. Этили-
Синтез винилацетата
273
дендиацетат пропусканием его паров над окисью алюминия при
220° может быть превращен в винилацетат.
Жидкофазный процесс включает приготовление и регенерацию
катализатора, рекуперацию и ректификацию отработанной серной
кислоты и этилидендиацетата, а также ректификацию винилацетата.
Жидкофазный метод получения винилацетата не свободен от
ряда существенных недостатков, к которым относятся: дороговизна
катализатора, относительно большой его расход, вредность работы
с ртутными солями, сильное корродирующее действие реагентов на
аппаратуру, применение наряду с уксусной кислотой также и уксус-
ного ангидрида и образование этилидендиацетата.
Более эффективным является непрерывный парофазный метод.
Принцип этого метода заключается в пропускании парогазовой
смеси из ацетилена и уксусной кислоты при высоких температурах
через реактор, заполненный катализатором, осажденным на про-
каленном активированном угле (носитель). Основные показатели
процесса — производительность, процент конверсии, время службы
катализатора и т. д. зависят от состава катализатора и носителя,
температуры в реакторе, концентрации ацетилена в парогазовой
смеси, скорости пропускания смеси через реактор, относительных
количеств катализатора и т. п.
В качестве катализатора применяют уксуснокислые соли цинка
(или кадмия). Активированный, прокаленный газовый уголь пропи-
тывают свежеприготовленным раствором катализатора (цинкаце-
тата), затем отделяют его от раствора и просушивают при 160—170°.
Уголь должен обладать высокой активностью и определенной сте-
пенью дисперсности. Кроме угля в качестве носителя можно
применять силикагель и активированную окись алюминия. До из-
вестного предела процент конверсии уксусной кислоты увеличи-
вается по мере относительного увеличения содержания катализа-
тора, достигая 99% (рис. 114).
Оптимальная температура реакции лежит в пределах 180—220°.
Процент конверсии в большой степени зависит и от температуры
реакции (рис. 115).
Получение винилацетата парофазным методом ведут при из-
бытке ацетилена. Наибольшая степень конверсии достигается,
когда на 8—10 ч. ацетилена берут 1 ч. уксусной кислоты. Однако
на практике выгоднее получать несколько меньший процент кон-
версии, но работать со значительно меньшим избытком ацетилена
(4—5 ч.).
Если вести реакцию при оптимальных температурных условиях,
при оптимальных количествах катализатора и соотношениях компо-
нентов, то степень конверсии уксусной кислоты достигает 95—98%;
такой степени не удается достигнуть при жидкофазном методе.
Большое значение имеет срок службы и активность при-
меняемого катализатора. Как было доказано С. Н. Ушаковым,
Е. Н. Ростовским и И. А. Арбузовой, основные причины, ведущие
18 Зак. 1269. Э. И. Барг
274
Гл. VII. Пластмассы на основе полимеров винилового спирта
к снижению активности катализатора, связаны с процессами поли-
меризации ацетилена и винилацетата в порах активированного
угля, активная поверхность которого уменьшается при закрытии пор
полимерами; кроме того, имеют значение процессы миграции ката-
лизатора и летучесть цинкацетата при температурах реакции.
Относит. к-Ио катализатора
г катализатора
л аиетилена.часы
Рис. 114. Зависимость процента кон-
версии уксусной кислоты от относи-
тельного количества катализатора при
парофазном получении винилацетата.
Рис. 115. Влияние температуры на
процент конверсии уксусной кис-
лоты при парофазпом получении
винилацетата.
Реакция полимеризации ацетилена экзотермична, она приводит
к местному перегреву и разрушению катализатора. Для уменьше-
ния миграции последнего рекомендуется менять направление га-
зового потока через реактор; постепенный подъем температуры
по мере снижения эффективности катализатора повышает его
активность. Срок службы катализатора зависит также от степени
очистки ацетилена и удаления из него фосфор- и мышьяксодержа-
щих соединений.
В промышленном синтезе винилацетата применяют совершенно
безводную уксусную кислоту (для предотвращения реакции гидра-
тации по Кучерову) и ацетилен, предварительно освобожденный от
вредных примесей.
Упрощенная схема процесса (рис. 116) может быть представлена следую-
щими операциями. Ацетилен из генератора 1 поступает в газгольдер 2, из кото-
рого его для очистки и осушки пропускают с помощью вакуум-насоса 4 в ряд
колонок 3; одна из них представляет окислитель, наполненный пемзой, про-
питанной хромовой смесью, другая — поглотитель, наполненный щелочью, и
третья — осушитель, наполненный хлористым кальцием и фосфорным ангидри-
дом. Очищенный и сухой ацетилен собирается в промежуточный буферный
бак 5, ив которого насосом 4а подается в насытитель 7. Последний состоит
из котла и колонки с насадкой. Проходя колонку, ацетилен движется навстречу
уксусной кислоте, которая, поступая из мерника 6, непрерывно орошает на-
Синтез винилацетата
275
садку колонки. Скорость прохождения ацетилена и температура кислоты
в последней должны быть точно отрегулированы, так как соотношение ацетилена
и уксусной кислоты в парогазовой смеси определяется упругостью пара уксус-
ной кислоты при данной температуре. Пары ацетилена, насыщенные уксусной
кислотой, направляются в трубчатый подогреватель 8, наполненный фарфоро-
вой насадкой и обогреваемый маслом (~250), после чего нагретая смесь посту-
пает в реактор (конвертор) 9, представляющий собою так же, как и подогре-
Рис. 116. Схема получения винилацетата.
/ —газогенератор; 2 — газгольдер для ацетилена; 3—колонки для очистки и осушки ацетилена;
4 и 4а — иасосы; 5—буферный бак; 6—мерник для кислоты; 7—испаритель кислоты (иасытитель);
8 — подогреватель; 9— контактный аппарат; 10— ловушка-брызгоуловитель; 11— холодильник; 12—
рекуператор (нагрев конденсата); 13— приемник эфира-сырца; 14— рекуператор эфира-сырца;
15 — дефлегматор; 16 —холодильник; 17 — приемник; 18 — бутыль для готовой продукции.
ватель, ряд трубок, но наполненных катализатором. Трубки с катализатором
обогреваются маслом, и температуру можно регулировать подачей в систему хо-
лодного масла. Контроль за температурой осуществляют с помощью термопар,
вставленных в нескольких местах реактора. Конвертированные газы проходят
ловушку 10 и трехступенчатый холодильник 11, в котором ацетилен в значи-
тельной степени отделяется от винилацетата. Из холодильников винилацетат
проходит ряд трубчатых подогревателей 12, в которых происходит отделение
последних следов ацетилена, и направляется в сборник сырого эфира 13. Сво-
бодный ацетилен, выделяющийся из холодильников и частично из подогрева-
телей, возвращается через осушитель 3 обратно в систему. Иногда применяют
276 Гл. VII. Пластмассы на основе полимеров винилового спирта
метод сорбции различными растворителями, например, ксилолом; в этом слу -
чае газы из конвертора проходят колонку, орошаемую ксилолом, а свободный
ацетилен направляется обратно в реактор.
Для получения чистого винилацетата сырой эфир поступает в ректифика-
ционный аппарат, состоящий из куба с колонкой 14, дефлегматора 15, холо-
дильника 16 и приемника чистого эфира 17. Чистый продукт загружают
в тару 18. Готовый продукт может быть получен с очень высокой степенью
чистоты (до 99,9% винилацетата). В качестве материала аппаратуры при-
меняют алюминий специальных марок, ие содержащий железа, и кислотоупор-
ные стали.
Преимущества парофазного метода получения винилацетата пе-
ред жидкофазным весьма существенны. В парофазном методе при-
меняются более дешевые и невредные катализаторы, имеющие боль-
шие сроки службы; процесс ведут непрерывно и достигают высокого
процента превращения уксусной кислоты. Однако парофазный метод
требует тщательной очистки ацетилена, более высоких температур и
применения ряда дополнительных мер пожарной и взрывобезопас-
ности.
Технический винилацетат должен отвечать следующим показа-
телям:
Уд. вес, г/с.и3 ............................ 0,9335—0,9345
Предел кипения (при 760 мм), °C............. 71,8—73
Кислотность (содержание уксусной кислоты), " 0 не более 0,02
Содержание альдегида, %......................... » » 0,05
Остаток после кипения, °/0........................ „ 0,05
Влажность, " о.................................. „ ,, 0,15
Пары винилацетата ядовиты и действуют на центральную нерв-
ную систему, кроме того раздражают слизистые оболочки и вызы-
вают слезотечение.
Винилацетат полимеризуется при наличии следов перекисей и
его нельзя свыше 24 часов хранить без стабилизатора (в закрытых
сосудах это может привести к взрыву); при пониженной темпера-
туре он может храниться более длительное время. Наиболее эффек-
тивными стабилизаторами являются дифениламин (в количестве
0,01—0,02%), резинаты двухатомных металлов (Си, Zn, Mg) и др.
Перевозят винилацетат в алюминиевых, железных и стальных
цистернах.
Полимеризация винилацетата
В технике полимеризацию винилацетата ведут главным образом
в присутствии инициаторов (органических и неорганических пере-
кисей) .
Применяют три основных метода технической полимеризации:
полимеризацию в растворителях, эмульсионную и блочную.
Полимеризация винилацетата по любому из методов протекает
по закономерностям, свойственным обычной цепной полимеризации:
молекулярный вес полимера уменьшается с увеличением концентра-
Полимеризация винилацетата
277
ции инициатора и с повышением температуры и находится в обрат-
ной зависимости от скорости конверсии. Период индукции и, в из-
вестной мере, кинетика процесса зависят от наличия в винилацетате
примесей, в частности сернистых соединений меди и железа, тор-
мозящих процесс полимеризации. Чем больше содержание таких
активных примесей, тем больше требуется перекисей, так как часть
последних расходуется на исключение их влияния.
Аналитическое определение примесей в винилацетате не всегда
дает достаточно данных для того, чтобы можно было судить о его
поведении при полимеризации при заданном количестве катализа-
тора. На практике лучшие результаты можно получить анализом
кривых зависимости вязкости от времени полимеризации при опре-
деленной температуре и концентрации инициатора. В зависимости
от характера кривых можно рассчитать то количество инициатора,
которое необходимо для обеспечения заданной скорости процесса
полимеризации.
Контроль полимеризации осуществляют определением процента
выхода полимера и его вязкости.
Обычно технические продукты из поливинилацетата различают по
числовым значениям вязкости молярного раствора в сантипуазах.
Полимеризация в растворителях
Метод полимеризации винилацетата в растворителях распро-
странен наиболее широко. Это объясняется характером применения
поливинилацетата, который в значительной мере используют в виде
раствора, например, для получения поливинилового спирта и его
производных, в виде лака для грунтовок, в качестве клея при
производстве искусственной кожи и т. д.
Течение процесса полимеризации винилацетата в растворителях
зависит от характера и концентрации растворителя, температуры и
продолжительности процесса. Условия процесса отвечают обычным
закономерностям цепной полимеризации в растворителях, опреде-
ляемых реакциями обрыва и передачи цепи молекулами раствори-
теля. Из растворителей наибольшей активностью обладает толуол,
наименьшей бензол, что, повидимому, связано с большей актив-
ностью водорода у толуола. Поэтому при полимеризации в толуоле
образуются полимеры с наименьшим молекулярным весом (вяз-
костью), а в бензоле — с наибольшим. Другие растворители в этом
отношении занимают промежуточное положение. Зависимость
выхода поливинилацетата и его вязкости от характера растворителя
может быть представлена следующими данными:
Растворители:
Толуол Ацетон Абсолют- ный спирт Этилацетат Бензол
Выход полимера, % . . Вязкость, сп ..... . 28 2,9 68,5 3 22,6 5,6 89,3 8,2 55 18,2
278 Гл. VII. Пластмассы на основе полимеров винилового спирта
Зависимость молекулярного веса поливинилацетата от концен-
трации растворителя настолько значительна, что, изменяя ее, можно
в широких пределах и с удовлетворительной для многих целей точ-
ностью регулировать степень полимеризации. Чем больше концен-
трация растворителя, тем, естественно, меньше степень полимери-
зации (вязкость) поливинилацетата:
Концентрация растворителя, % . . 40 45 50 55
Вязкость поливинилацетата, сп . . 18,4 12,4 8,8 6,2
Степень полимеризации поливинилацетата в значительной мере
зависит от содержания в мономере ацетальдегида (или других
альдегидов); присутствие альдегидов сильно снижает вязкость
полимера:
Содержание ацетальдегида в винилацетате, % . 0 0,1 0,2 0,6
Вязкость поливинилацетата, сп............. 30 20 15 6
Ацетальдегид или пропионовый альдегид применяют для регу-
лирования вязкости поливинилацетата.
Степень полимеризации поливинилацетата, естественно, можно
регулировать и изменением концентрации инициатора.
В технике стараются выбрать такой растворитель, применение
которого позволяет получать полимеры с заданной степенью поли-
меризации (вязкостью) и который может быть введен в количестве,
достаточном для получения лака с не слишком высокой вязкостью.
Из разнообразных растворителей техническое применение нашли
лишь бензол, этилацетат, этиловый спирт, отчасти метанол и др.
Полимеризацию в растворителях ведут в никелевых или алюми-
ниевых реакторах, снабженных паровой рубашкой, мешалкой и
обратным холодильником. Реакционную смесь составляют из воз-
можно более чистого винилацетата, растворителя, перекиси бен-
зоила и альдегида (для регулирования вязкости). Реакция проте-
кает при температуре слабого кипения растворителя в течение
12—18 часов. Получаемый раствор (лак) находит самостоятельное
применение, но из него можно получать также и чистый поливинил-
ацетат. Для этого после окончания полимеризации раствор пере-
ливают в перегонный куб, в котором действием острого пара произ-
водят отгонку растворителя и остаточного мономера. После этого
смолу в виде тонкой ленты продавливают сжатым воздухом из
щелевидного отверстия на вращающийся барабан или на бесконеч-
ную стальную ленту, где она охлаждается и разрезается на не-
большие полоски.
Полимеризацию винилацетата в растворителях ведут и непре-
рывным процессом по башенному методу с применением смесителей
и аппаратуры периодического действия для завершения полимери-
зации. По этому методу могут быть получены полимеры со сравни-
тельно широким интервалом молекулярных весов. В качестве
растворителя применяется этилацетат, а степень полимеризации
Полимеризация винилацетата
279
регулируется изменением концентрации перекиси бензоила и про-
пионового альдегида.
Аппаратура для непрерывной полимеризации винилацетата (рис. 117) со-
стоит из двух цилиндрических смесителей («мешалок») 1, 2, служащих для
приготовления полимеризационной смеси, двух башен 3, 4, в которых проис-
ходит основной процесс полимеризации, и двух реакторов 5, 6 для последую-
щей полимеризации. Смесители изготовляют из алюминия. В каждый смеситель
Рис. 117. Непрерывная полимеризация винилацетата в растворе.
непрерывно в определенном соотношении поступают компоненты смеси: в пер-
вый — этилацетат и инициатор, во второй — мономер и альдегид. Из обоих
смесителей смесь с помощью отрегулированных с большой точностью насо-
сов 7 непрерывно подается в первую башню. Башни изготовляют из стали;
изнутри они облицованы стеклом или эмалью, имеют диаметр около 0,6 м и
высоту около 9 м. Первая башня для облегчения выхода паров растворителя
имеет расширенную верхнюю секцию и снабжена также мешалкой из нержа-
веющей стали, которая состоит из серии лопастей, чередующихся с каждой
стороны вала, вращающегося со скоростью 40 об/мин. Вторая башня не имеет
мешалки и не расширена в верхней части. Обе башни снабжены рубашками
(для обогрева циркулирующей горячей водой), а также трубчатыми обратными
холодильниками 8, в которых конденсируются пары растворителя и мономера.
Поступающая в верхнюю часть первой башни реакционная смесь проходит до
низа башни, затем поступает в нижнюю часть второй башни и поднимается
вверх до выхода из башни, а оттуда поступает в цилиндрические реакторы,
облицованные внутри стеклом и снабженные мешалкой и рубашкой для горя-
чей воды. Температура обогревающей воды в обеих башнях 80°; такая же
температура поддерживается и в реакторах. Выходящий из второй башни
раствор содержит до 5—8% мономера. После нескольких часов обработки
280
Гл. VII. Пластмассы на основе полимеров винилового спирта
в реакторах при 80° содержание мономера снижается до 1—1,5%. Таким обра-
зом, не во всех стадиях процесс является непрерывным, и башни в данном
случае следует рассматривать как непрерывные аппараты предварительной
полимеризации.
Основные технические трудности описанного процесса заключаются в обра-
зовании и нарастании на стенках башен пленки полимера, которую необходимо
периодически удалять. Облицовка поверхности стали стеклом и эмалью зна-
чительно уменьшает нарастание
ТАБЛИЦА 27 пленки.
Производительность установки и
молекулярные веса поливинилацетатов
в зависимости от состава реакционной
смеси
Состав реакционней смеси Молекулярный вес
14000! 20 000 1 32 000
Винилацетат, кг ... . 782 782 782
Этилацетат, кг .... Пропионовый альдегид 325 325 325
кг Перекись бензоила 35,2 7,85 4,03
(100%), кг Производительность 35,6 14,8 1,56
кг!час 147 102 76,3
Производительность уста-
новки и молекулярный вес
поливинилацетата зависят
от состава реакционной
смеси.
Как уже указывалось, по-
ливинилацетат удается полу-
чать в широком интервале
молекулярных весов путем
регулировки количества аль-
дегида и инициатора. В
табл. 27 приведены молеку-
лярные веса поливинилаце-
тата и производительность
установки в зависимости от
состава реакционной смеси.
Эмульсионная полимеризация
Обычно пользуются двумя вариантами эмульсионной полимери-
зации: 1) с применением водорастворимых инициаторов, что ведет
к получению устойчивой суспензии (эмульсии) и 2) с применением
инициаторов, растворимых в мономере, что дает полимер в виде
гранул («бисерная» полимеризация).
По первому варианту эмульсионную полимеризацию винилаце-
тата ведут в присутствии в качестве эмульгаторов преимущественно
солей жирных кислот и сульфокислот, а также и других мыл, а в ка-
честве инициатора — главным образом перекиси водорода.
Так, согласно одной из рецептур, в реактор вводят: 100 ч. винил-
ацетата, 100—120 ч. воды, 0,5—1,5 ч. перекиси водорода, 0,1—0,5 ч.
олеата калия. Реакционную смесь нагревают при 65—75° и энер-
гично перемешивают в течение 90—120 мин. Продукт полимериза-
ции представляет собой устойчивую эмульаию типа латекса.
Поливинилацетатные эмульсии в значительной мере находят
самостоятельное применение, например для обработки кожи, ткани
и т. п. В тех случаях, когда необходим чистый твердый поливинил-
ацетат, применяют второй вариант эмульсионной полимеризации,
который позволяет получать весьма чистый полимер практически
любого молекулярного веса (то 500 000 и более).
Полимеризация винилацетата
281
По второму варианту применяют инициаторы, растворимые в
винилацетате (обычно перекись бензоила). Раствор инициатора в
мономере диспергируют в водном растворе высокомолекулярных
веществ, например в растворе поливинилового спирта, полиакрило-
вой кислоте и др. Диспергирование производят в реакторах, сна-
бженных быстроходными мешалками обычно пропеллерного типа.
Величина образующихся гранул полимера зависит как от состава
полимеризационной смеси, так и от интенсивности диспергирования.
Внесением небольших количеств нерастворимых щелочных солей —
сульфатов или карбонатов — можно улучшить стабильность поли-
меризующейся дисперсии.
Так, по одной из рецептур в реактор загружают 100 ч. 5%
раствора поливинилового спирта, 100 ч. винилацетата, в котором
растворено 0,5 ч. перекиси бензоила, 0,1 ч. MgCOs, взмученного
в 10 ч. воды. Реакция при 70—90° и энергичном перемешивании про-
должается от 2 до 6 час. Получаемый в результате реакции «бисер»
может иметь различную и строго устанавливаемую степень дисперс-
ности; он легко осаждается, фильтруется и промывается водой.
Блочная полимеризация
Обычный метод блочной полимеризации чистого винилацетата
имеет ограниченное применение. Его используют главным образом
для получения низкомолекулярных полимеров (с мол. весом от 3500
Рис. 118. Схема непрерывной блочной полимериза-
ции винилацетата.
1 и 2~аппараты для приготовления полимеризационной смеси;
3 —полимеризацилиная башня; 4—обратный холодильник;
5—ленточный транспортер; б—плунжерный насос; 7—приспо-
собление для охлаждения поливинилацетата.
то 7500). Более высокомолекулярные продукты получают приме-
нением непрерывного башенного метода полимеризации, подобного
соответствующему методу полимеризации стирола (стр. 205). Од-
нако специфические условия полимеризации винилацетата и физи-
282
Гл. VII. Пластмассы на основе полимеров винилового спирта
ческая структура его полимера потребовали внесения многих изме-
нений в аппаратурную схему (рис. 118).
Полимеризационная алюминиевая башня состоит из цилиндрической части,
снабженной рубашкой для обогрева горячей водой; кроме того, внутрь нее вста-
влен торпедовидный обогреватель, в который также поступает горячая вода.
Нижний конец башни снабжен патрубком, через который на конвейерные сталь-
ные плиты 5 в виде тонкой ленты выгружается конечный продукт. Полимер
сверху интенсивно охлаждается током воздуха. В верхней части башни имеете:)
снабженная мешалкой расширенная секция для облегчения выхода газов и для
перемешивания реакционной смеси со свежим потоком мономера и инициатора,
поступающего из обратного холодильника 4, расположенного в верхней части
башни.
Аппаратура для предварительной подготовки реакционной смеси состоит из
двух алюминиевых цилиндрических сосудов 1 и 2, снабженных лопастными ме-
шалками. Сосуды имеют рубашки для охлаждения водой или внутренние холо-
дильники. В первом сосуде происходит растворение перекиси бензоила в части
мономера, после чего концентрированному раствору дают отстояться с тем,
чтобы вода и другие примеси, содержащиеся в перекиси бензоила, могли
быть отделены от раствора. Мономер с инициатором поступает во второй сосуд
одновременно с мономером, содержащим небольшое количество пропионового
альдегида. Из второго сосуда реакционная смесь дозировочным плунжером
подается в полимеризационную башню. В башне она находится около восьми
часов. Производительность одного агрегата—1,5 т в сутки.
Башенный метод позволяет получать поливинилацетат лишь
в узких пределах молекулярных весов, примерно от 30 000 до 60 000.
Более низкомолекулярные продукты слишком жидки при темпера-
туре полимеризации 70—80°, более высокомолекулярные чрез-
мерно вязки и с трудом передвигаются в башне.
Строение поливинилацетата
Структуру поливинилацетата изучали главным образом по
реакциям образующегося при его омылении поливинилового
спирта (стр. 295), так как было установлено, что длина мо-
лекулы полимера при омылении поливинилацетата практически не
меняется.
До последнего времени все проведенные реакции указывали, что
поливиниловый спирт, а следовательно, и поливинилацетат по-
строены по принципу «голова к хвосту». Если бы поливиниловый
спирт содержал группы а-гликоля (т. е. был бы построен по прин-
ципу «голова к голове»), то он легко окислялся бы таким специфи-
ческим реагентом на а-гликоли, как иодная кислота (ШО4). Од-
нако даже при длительном нагревании (до 30 часов) не удалось
обнаружить взаимодействия иодной кислоты с поливиниловым
спиртом.
Подтверждением структуры «голова к хвосту» служит также
образование щавелевой кислоты при окислении поливинилового
спирта азотной кислотой:
...-4-СН2—СН——СН2—Сн—^-... НО—С—С-СН.
• I ; I ; II II
он сн о о
Строение поливинилацетата
283
Если бы поливиниловый спирт (поливинилацетат) имел струк-
туру «голова к голове», то в результате окисления была бы полу-
чена янтарная кислота:
... J—сн2—сн2—сн2—сн-i—сн—сн2— ... _±° но—с—сн2— сн2—с—сн,
: I I : I II II
он он он о о
Наконец, близкое сходство спектров поглощения поливинилового
спирта и 2,4-пентандиола СН3—СН—СН2—СН—СН3 (рис. 119) f
I I
ОН ОН
а также данные рентгеновского анализа доказывают, что поливи-
ниловый спирт, а следовательно, и поливи-
нилацетат имеют ^-гликолевую структуру.
На рис. 120 представлена схема строе-
ния молекулы поливинилацетата.
Однако в последние годы были получены
данные, которые указывают, что в поливи-
нилацетате и образующемся из него поливи-
ниловом спирте, построенных главным об-
разом по принципу «голова к хвосту», имеют-
ся от 1 до 2% групп, построенных, как
структура «голова к голове» (а-гликоль).
Этот вывод был сделан на основании умень-
шения вязкости поливинилового спирта при
обработке его небольшим количеством иод-
ной кислоты; при этом, после достижения
Рис. 119. Спектры по-
глощения 2,4-пентандио-
ла (7) и поливинилового-
спирта (2).
некоторой степени падения вязкости, последняя стабилизуется (и
от дальнейшего воздействия иодной кислоты больше не падает) -
Рис. 120. Схема строения молекулы поливинилацетата.
> —атомы углерода; О— атомы водорода; О — атомы кислорода
Падение вязкости объясняется деструкцией цепи под действием^
иодной кислоты.
*284 Гл. VII. Пластмассы на основе полимеров винилового спирта
Количество в полимере групп а-гликоля зависит от температуры
полимеризации винилацетата и при проведении ее при температу-
рах от 25 до 110° меняется в пределах 1—2 мол. %.
До сих пор не выяснен вопрос о характере конечных групп в
поливинилацетате. Некоторые исследователи (Марвел и др.) дока-
зывают, что на концах цепей имеются альдегидные группы:
—СН,—СН—CHS— Г—СН—СН,— 1 -сн,—сно.
I I
о—со—сн3 L о—со—сн3 ]„
Это, однако, встречает ряд возражений и во многих случаях не
подтверждается. С. Н. Ушаковым было установлено, что поливини-
ловые спирты не содержат концевых альдегидных групп, а имеют
на конце остатки инициатора, что хорошо согласуется с наличием
некоторого количества групп а-гликоля, могущих образоваться при
смыкании двух растущих цепей поливинилацетата. Новейшие иссле-
дования поливинилового спирта методом спектрального анализа
доказывают наличие в 'нем до 0,4 мол. % карбонильных групп, по-
видимому, кетонных:
...—СН2—СН— ...—СН2—С—СНа—...
I II
ОН о
Поливинилацетат имеет жидкостную, аморфную структуру; рент-
генограмма даже высокоориентированных полимеров не позволяет
обнаружить наличия кристаллитов.
Свойства поливинилацетата
Поливинилацетат представляет собою прозрачный бесцветный
полимер; уд. вес 1,191, коэффициент рефракции 1,4665. Он не-
сколько набухает в воде и легко поддается действию сильных кислот
и щелочей.
При нагревании сдыше 150° поливинилацетат легко деполиме-
ризуется, однако продуктом деполимеризации является не винил-
ацетат, а уксусная кислота.
Поливинилацетат растворяется во многих растворителях — в
спиртах, сложных эфирах, ароматических углеводородах и т. п.
Однако он стоек к действию бензина, керосина и масел. Вязкость
растворов поливинилацетата зависит от характера растворителя;
ацетон дает растворы с наименьшей вязкостью, сложные эфиры
(бутилацетат) — с наибольшей. Сорта поливинилацетата характери-
зуют по его молярной вязкости в бензоле (86 г/л).
В промышленности выпускаются сорта поливинилацетата с вяз-
костью от 2,5 до 40 сп, чаще всего 2,5; 7 и 15. Пропорционально
вязкости увеличивается температура плавления (вязкого течения)
поливинилацетата, тогда как температура стеклования в широких
пределах молярной вязкости остается без изменения.
Применение поливинилацетата
285
Температура стеклования поливинилацетата 28;, поэтому он уже
при обычных температурах обладает заметной скоростью дефор-
мации. Однако такая хладотекучесть поливинилацетата является
только обратимой, высокоэластической деформацией; она быстро
исчезает при нагревании образца в пределах 40—80°. Необратимое
пластическое течение можно наблюдать лишь при более высоких
температурах (120° и выше).
Прочность поливинилацетата зависит от температуры, молеку-
лярного веса и степени растяжения: при —10° предел прочности на
разрыв ~600 кг/с-М2; при +10° 400кг/с.и2 и при +30° 150—200 кг/см2.
Ориентированные пленки поливинилацетата при 5—6-кратном рас-
тяжении имеют прочность 1500—2000 кг/см2.
Чем выше степень полимеризации, тем выше прочность ориен-
тированных пленок поливинилацетата. При весьма больших растя-
жениях (сто раз и больше) высокомолекулярного поливинилацетата
(мол. вес ^> 500 000) прочность может достигать 10 000 кг/см2.
Поливинилацетат обладает значительной адгезией к стеклу,
коже, ткани и т. д. Его адегизионные свойства часто увеличиваются
при внесении пластификаторов.
Сополимеры винилацетата
Винилацетат широко применяют для сополимеризации с дру-
гими соединениями. Техническое значение получили сополимеры
винилацетата с хлористым винилом (стр. 254), акриловым и мет-
акриловыми эфирами (стр. 335).
Применение поливинилацетата
Поливинилацетат применяют главным образом для дальнейшей химической
переработки в поливиниловый спирт и его ацетали.
Самостоятельное применение поливинилацетат получил в производстве'
лаков, где он ценен благодаря высоким адгезионным свойствам, эластичности,
светостойкости, бесцветности. Его применяют в композиции с нитроцеллюлоз-
ными лаками и другими смолами. Высокие адгезионные свойства поливинилаце-
тата дают возможность применять его для производства клеящих составов (для
склейки стекол, кожи, древесины, для наклейки металлической фольги на бумагу
и ткани и т. д.) и в качестве связки для производства абразивных кругов.
В виде водных эмульсий и суспензий, содержащих пластификаторы (трикрезил-
фосфат, дибутилфталат и др.), его применяют для поверхностной обработки
кожи, аппретирования ткани, для обработки бумаги, производства искусствен-
ной кожи и т. д.
В сочетании с другими смолами, например, с глифталем и с ацетилцеллю-
лозой, поливинилацетат применяют для получения граммпластипок. Малая
теплостойкость поливинилацетата, низкая морозостойкость и невысокая водо- н
химическая стойкость препятствуют самостоятельному использованию его для
производства пластмасс и прессизделий. Однако для этих целей большое про-
мышленное значение приобрели сополимеры поливинилацетата.
Для каждого назначения выпускаются специальные марки поливинилаце-
тата, отличающиеся по вязкости. Так, для производства лаков и клеев при-
меняют полимер с молярной вязкостью 1,5—2,5 сп, для аппретуры и обра-
ботки бумаги и кожи 7—15 сп; для переработки на поливиниловый спирт
15—35 сп и т. д.
-286 Гл. VII. Пластмассы на основе полимеров винилового спирта
ПОЛИВИНИЛФОРМИАТ
Винилформиат — СН2=СНОСОН представляет собою жид-
кость с эфирным запахом, т. кип. 46—46,5° и —0,9592, легко
гидролизуется теоретическим количеством воды, образуя ацеталь-
дегид и концентрированную муравьиную кислоту. Реакция гидро-
лиза значительно ускоряется в присутствии кислот (катализатором
является также выделяющаяся при гидролизе муравьиная кислота).
Синтез винилформиата может быть осуществлен как в жидкой,
так и в паровой фазе.
Синтез в жидкой фазе был описан Ю. С. Залькиндом, который
получал винилформиат с выходом в 57—67% путем пропускания
ацетилена в муравьиную кислоту в присутствии фосфорной ртути
и трехфтористого бора. В ряде патентов дан синтез винилформиата
путем взаимодействия виниловых эфиров, кислотный остаток кото-
рых содержит более, чем один атом углерода (винилацетат, винил-
бутират, винилстеарат и др.), с му-
равьиной кислотой в присутствии солей
ртути.
С. Н. Ушаков, И. А. Арбузова и
Е. Н. Ростовский разработали парофаз-
ный метод получения винилформиата
по схеме, в основном аналогичной схе-
ме парофазного получения винилацета-
та. В качестве катализатора они при-
менили цинкацетат (или формиат),
нанесенный на активированный газо-
вый уголь.
По этой схеме хорошо очищенный
и высушенный ацетилен поступает в
испаритель муравьиной кислоты, поме-
щенный в гидротермостат, и затем
Рис. 121. Зависимость конвер-
сии муравьиной кислоты (/)
и уксусной кислоты (//) от тем-
пературы.
проходит зону предварительного нагрева и зону контактирования.
Оптимальная температура в зоне контактирования 180°, т. е. сдви-
нута к более низким температурам, чем при получении винилацетата
(рис. 121). В этих условиях при соответствующих катализаторах
можно получать столь же высокий выход, как и при синтезе винил-
ацетата (95% и выше). Сырой продукт реакции, кроме винилаце-
тата и ацетилена, содержит ацетальдегид (до 5%) и муравьиную
кислоту. Ацетальдегид образуется при взаимодействии винилфор-
миата с водой, которая получается при распаде муравьиной кислоты
по вероятной схеме:
I—> СО +Н2о
а) п НССОН !-> СО2+Н„
!-> СО2 + Н2СО + Н2О;
О) СН2=СНСССН +Н2о —> НССОН 4- СН3СНО.
Простые эфиры поливинилового спирта
287
Техническое оформление процесса получения винилформиата
осложняется корродирующим действием муравьиной кислоты (в жид-
кой фазе) на аппаратуру; процесс поэтому необходимо вести в ке-
рамиковой или эмалированной аппаратуре.
Полимеризация винилформиата может быть проведена как в
растворителях, так и блочным методом. Эмульсионный метод поли-
меризации в этом случае неприменим, так как вода гидролизует и
мономер и полимер. В качестве растворителя может быть приме-
нен ацетон; полимеризация протекает при температуре кипения аце-
тона в течение 8—24 час. Полимеризацию чистого мономера блоч-
ным методом можно проводить в герметически закрытых формах в
присутствии перекиси бензоила при 60—80° в течение 1,5—2,5 час.
Полимеры винилформиата еще мало изучены. Особое значение
имеет легкая омыляемость поливинилформиата при кипячении с
водой (катализатором служит отщепляющаяся муравьиная кис-
лота), с образованием поливинилового спирта, свободного от про-
дуктов, образующихся при действии минеральных кислот и щелочей.
Поливинилформиат имеет аморфную структуру; его температура
стеклования ~40°, он растворим в ацетоне и диоксане, нераство-
рим в спиртах, углеводородах и сложных эфирах.
Простые эфиры поливинилового спирта
/—сн,—сн—сн..—сн—\
I I
\ OR OR Л
Простые эфиры поливинилового спирта получают полимериза-
цией простых виниловых эфиров СН2=СН—OR, где R — алкил или
арил.
Полимеры и сополимеры простых виниловых эфиров получили
значительное применение в последние годы, когда были разрабо-
таны технические методы их синтеза и изучены свойства полимеров
и сополимеров.
Синтез виниловых эфиров был впервые осуществлен в 1870 г.
А. М. Бутлеровым. Взаимодействием бромистого изокротила с эти-
латом натрия он получил р,р-диметилвинилэтиловый эфир по схеме:
СН3, СН3,
)С=СНВг 4- NaOC,H5 —> >С=СН—OC2Hr, + NaBr.
Сн/ ‘ СН/
Реакция Бутлерова применима ко всем винильным соединениям,
содержащим галоид при двойной связи, при их взаимодействии с
алкоголятами. Многие современные методы синтеза виниловых
эфиров имеют своей основой реакцию Бутлерова.
В 1887 г. А. Е. Фаворский открыл реакцию взаимодействия
простейшего, однозамещенного ацетиленового углеводорода — алли-
лена с этиловым спиртом в присутствии щелочи при 170—180°, при
288
Гл. VII. Пластмассы на основе полимеров винилового спирта
этом аллилен присоединяет спирт по тройной связи с образованием
изопропенилэтилового эфира:
СН3—С=СН + С3Н5ОН —> СН2=С-ОС2Н6.
сн3
Развитием этой реакции явилась общая реакция винилирова-
ния — синтеза простых эфиров взаимодействием ацетилена в при-
сутствии щелочи с соединениями, содержащими ОН-группы (спирты,
фенолы, аминоспирты, углеводы) или другие группы с подвижным
водородом по схеме:
120 — 1Я0°
СН=СН + ROH —щелцчь -> СН2=СН—OR.
Эта схема была разработана и подробно изучена А. Е. Фавор-
ским и М. Ф. Шостаковским; она является основной реакцией про-
мышленного синтеза простых виниловых эфиров.
Реакцию винилирования проводят как в жидкой, так и в паро-
вой фазе.
По одному из вариантов жидкого метода в стальной вращаю-
щийся автоклав, снабженный рубашкой для обогрева и охлажде-
ния, загружают соответствующий безводный спирт и ~5% КОН,
затем в автоклав нагнетают под давлением 12—15 ат хорошо очи-
щенный и высушенный ацетилен, предварительно вытеснив ацети-
леном воздух из автоклава.
Реакцию для большинства эфиров ведут при высоких темпера-
турах в пределах 140—160°. Давление в автоклаве, которое необхо-
димо для удержания соответствующего спирта в жидком состоя-
нии, будет зависеть от температуры кипения спирта; в случае низ-
ших спиртов (метиловый, этиловый) давление в автоклаве может
достигать 20—30 ат; для высших спиртов, температура кипения
которых выше температуры реакции (н. первичный гептиловый,
октиловый и т. д.), процесс может протекать и без давления.
Для предохранения от возможного взрыва ацетилена при нагре-
вании под давлением было предложено проводить реакции при
разбавлении ацетилена азотом (2 : 1), а также другими инертными
газами, что, однако, приводит к соответствующему уменьшению
скорости реакции.
Более рациональным является разбавление ацетилена парами
соответствующих реагирующих спиртов и образующихся виниловых
эфиров, как это было предложено М. Ф. Шостаковским.
Во избежание образования взрывчатого ацетиленида меди все
части аппаратуры, которые могут соприкасаться с ацетиленом, не
должны содержать меди.
По окончании реакции, что определяется падением давления,
автоклав охлаждают до комнатной температуры. Если весь спирт
не прореагировал, операцию ввода ацетилена повторяют до тех пор,
пока не прекратится поглощение ацетилена.
Простые эфиры поливинилового спирта
289
После вступления в реакцию всего спирта реакционную смесь
выгружают и подвергают промывке, дестилляции и ректификации.
Парофазный метод состоит в пропускании смеси паров спирта
и ацетилена над контактным катализатором при высоких темпера-
турах.
Так смесь паров этилового спирта и ацетилена пропускают при
400° над активированным углем, насыщенным фосфорной кислотой.
Были предложены также и другие катализаторы, например цинко-
вые и кадмиевые соли органических кислот, нанесенные на акти-
вированный уголь. Простые виниловые эфиры получают также на
основе реакции расщепления ацеталей при пропускании их паров
над контактным катализатором при высоких температурах
CR
СН3—СН —> ch2=chor + roh.
'or
Так, этилвиниловый эфир получают из диэтилацеталя, применяя
в качестве катализатора платину, нанесенную на асбест. Темпера-
тура контактной массы 280—290°. В качестве катализаторов были
предложены также пористый глинозем, окись тория, никель, се-
ребро, палладий и другие металлы в дисперсном состоянии. Темпе-
ратура реакции предлагалась от 100 до 450°.
Для целей полимеризации простые виниловые эфиры не должны
содержать растворенный ацетилен, спирты, альдегиды, ацетали,
воду и другие полярные примеси, понижающие активность катали-
заторов типа Фриделя—Крафтса (AICI3, TiCK и др.).
Тщательная очистка мономеров является поэтому основным
условием для проведения реакции полимеризации.
Простые виниловые эфиры могут быть разделены на две основ-
ные группы:
1) моновиниловые эфиры — производные одноатомных спиртов
(метилового, этилового, бутилового, октилового):
СН3С—СН=СН2 С4Н9О-СН=СН2
С2Н5О—СН=СН2 С8Н17О—СН=СН2;
2) ди- и тривиниловые эфиры — производные двух- и трехатом-
ных спиртов и спиртов высшей атомности, например дивинилглико-
левые эфиры:
СН2—С—СН=СН2
сн2—о—сн=сн2
и тривинилглицериновые эфиры:
сн,—о—сн=сн2
сн—о—сн=сн2
сн2—о—сн=сн2.
19 Зак. 1269. Э. И. Барг
290
Гл. VII. Пластмассы на основе полимеров винилового спирта
Простые виниловые эфиры полимеризуются по механизму, от-
личному от цепного, радикального механизма полимеризации слож-
ных виниловых эфиров, что можно видеть из приведенного в табл. 28
сопоставления отношения
ТАБЛИЦА 28
различных виниловых произ-
водных к обычным факто-
Способность виниловых соединений
к полимеризации под влиянием различ-
ных факторов
рам, вызывающим процесс
цепной (радикальной) по-
лимеризации (+ полиме-
Виниловые соединения
Винилацетат . . .
Хлористый винил
Эфиры акриловых
кислот . . . .
Простые винило-
вые эфиры . .
протекает под действием различных
Фриделя — Крафтса.
ризуется, —не полимеризует-
ся) .
Таким образом, в отли-
чие от сложных виниловых
эфиров простые эфиры не
полимеризуются под влия-
нием тех факторов, которые
вызывают процесс радикаль-
ной полимеризации.
Так, перекиси и кислород
воздуха не активируют мо-
номер и не инициируют по-
лимеризацию. Последняя
кислот и катализаторов типа
Причина различия в механизме полимеризации простых и слож-
ных виниловых эфиров пока еще недостаточно выяснена.
Для объяснения каталитического действия галоидоводородных
кислот и хлоридов металлов на полимеризацию простых виниловых
эфиров М. Ф. Шостаковский предполагает, что первичным актом
является взаимодействие катализатора с кислородом простого не-
предельного эфира и образование промежуточного неустойчивого
комплекса:
СН2=СН—OR-f-HX —► [СН.=СН—ОН] X,
R
где X — галоид.
Этот неустойчивый комплекс в дальнейшем распадается с об-
разованием иона
© е
[СН2=СН—ОН]Х —► [CH3CHOR] х,
R
который дает начало ионному механизму роста цепи (стр. 45).
Полимеризация мономеров может быть проведена в блоке и в
растворе. Почти для всех простых виниловых эфиров в качестве
катализаторов чаще всего применяют SnCI2, AIC13, FeCl2 и BF3.
Простые эфиры поливинилового спирта
291
В технике используют и комбинированные катализаторы, напри-
мер, SnCb и BF3. Натрий не активирует реакцию полимеризации.
Перекиси, повидимому, замедляют процесс полимеризации, если он
протекает под действием катализаторов.
Кинетика процесса и молекулярный вес полимера определяются
характером катализатора, его концентрацией и температурой реак-
ции.
При проведении реакции при обычных или повышенных темпе-
ратурах получают сравнительно низкомолекулярные, бальзамопо-
добные, пластичные и вязкие полимеры. В этом случае катализато-
ром обычно служит SnCl2 или FeCU. С увеличением молекулярного
веса спиртового остатка скорость реакции снижается; полимериза-
ция виниловых эфиров высших спиртов требует большего количе-
ства катализаторов. Вязкость растворов полимеров от метилвинило-
вого эфира до бутилвинилового повышается, но затем, по мере
увеличения спиртового радикала, снижается (при одинаковых усло-
виях полимеризации).
Высокомолекулярные полимеры, твердые и хрупкие или каучуко-
подобные, получают полимеризацией виниловых эфиров при низких
температурах (—40° и ниже). Для этого применяют эфиры развет-
вленной структуры: изобутилвиниловый эфир, виниловые эфиры
диизопропилкарбинола, метилтретично-бутилкарбинола и др. Их
полимеризуют при низкой температуре в среде разбавителей (на-
пример жидкого пропана) в присутствии трехфтористого бора или,
применяя комплексные катализаторы с трехфтористым бором. Чем
меньше скорость реакции при низких температурах, тем выше моле-
кулярный вес полимера.
Полимеры простых виниловых эфиров в зависимости от струк-
туры и молекулярного веса получают самой разнообразной конси-
стенции — от жидких продуктов до твердых, хрупких и каучукопо-
добных. Соответственно и температура стеклования их может на-
ходиться в пределах от —20 (и ниже) до +70° (и выше). Выше Тс
они либо вязкие жидкости, либо высокоэластические, каучукопо-
добные вещества, что определяется степенью полимеризации.
Жидкие полимеры отличаются хорошими адгезионными свой-
ствами (к дереву, металлу, ткани, коже и т. п.); они стойки к рас-
творам щелочей и кислот, чем отличаются от полимеров сложных
виниловых эфиров. Вязкость и консистенция жидких полимеров
мало меняются в широком температурном интервале (от —30 до
+ 150°); они выдерживают и более высокие температуры (200—
250°), но при этом несколько темнеют.
Полимеры моновиниловых эфиров растворимы во всех обыч-
ных растворителях, полярных и неполярных, за исключением
этилового спирта. Полиметилвиниловый эфир растворим в холод-
ной воде, однако осаждается из воды при нагревании. Свойства не-
которых полимеров простых виниловых эфиров приведены в табл 29.
19*
292
Гл. VII. Пластмассы на основе полимеров винилового спирта
Полимеры второй группы виниловых эфиров — дивинилглико-
левых, тривинилглицериновых и т. д., а также виниловых эфиров
ненасыщенных спиртов представляют собою пространственные по-
лимеры, — это хрупкие смо-
лы, неплавкие и нераствори-
мые и лишь набухающие в
растворителях. Некоторые из
них могут быть получены в
растворимом состоянии, но
при нагревании они медлен-
но переходят в нераствори-
мое и неплавкое состояния (в
пространственный полимер).
Структура полимеров про-
стых виниловых эфиров точ-
но еще не установлена. При
исследовании продуктов оки-
сления поливинилэтиловых
эфиров была обнаружена
ТАБЛИЦА 29
Показатели простых поливиниловых
эфиров
Эфиры Уд. вес, г!смъ Коэффициент п реломления Уд. эл. объем- нее сопроти- вление, 'ь.-см Диэлектриче- ская постоян- ная
Поливинилме- тиловый . . 1,045 1,467 5 • 1012 3,5
Поливинил- этиловый . . 0,96 1,454 6-1013 3,0
Поливинилизо- бутиловый . 0,91 1,452 1 • 10’’ 2,2
щавелевая кислота, что дает основание предполагать для этих эфи-
ров структуру 1,3 («голова к хвосту»):
... —СК—СН—СН,—СН— ...
I I
CCgHg СС2Н5
Чтобы получить полимеры простых виниловых эфиров с высо-
кой степенью полимеризации, соответствующей обычным техниче-
ским полимерам (например, поливинилацетату, поливиниловому
спирту и др.), С. Н. Ушаков применил реакцию между щелочными
производными поливинилового спирта и галоидоалкилами и га-
лоидоар ил алкилами:
... —СН2—СН—СН2—СН— ...
ONa ONa
—► —СН2—СН—СН2—СН— ... + NaX.
OR OR
Щелочные производные поливинилового спирта были получены
путем обработки поливинилового спирта водными растворами ед-
кого натра. При обработке таких щелочных производных галоидо-
алкилами, галоидоарилами или диалкилсульфатами были полу-
чены бензиловый, метиловый, этиловый и бутиловый эфиры поли-
винилового спирта различных степеней замещения с относительна
высоким молекулярным весом. Эти продукты еще мало исследо-
ваны и изучены, но несомненно представляют большой интерес и
могут иметь техническое значение.
Сополимеры простых виниловых эфиров
293
Полимеры простых виниловых эфиров в зависимости от их со-
става и свойств применяют для изготовления лакокрасочных ком-
позиций для добавки к смазочным маслам, для обработки кожи и
пропитки ткани при производстве искусственной кожи, для аппре-
турных целей в текстильной промышленности, в качестве водорас-
творимого клея (для изоляционных лент и пластырей), пластифи-
цирующих добавок для понижения температуры стеклования
полимеров, загустителей, в фармацевтической промышленности (за-
менители перувианского бальзама) и т. д.
Особый промышленный интерес представляет поливинилметило-
вый эфир, который, будучи растворим в воде, нашел основное при-
менение для аппретурных целей и для производства клеев.
СОПОЛИМЕРЫ ПРОСТЫХ ВИНИЛОВЫХ ЭФИРОВ
Сополимеризация простых виниловых эфиров может происходить
как между мономерами простых виниловых эфиров, так и между
простыми виниловыми эфирами и другими виниловыми соедине-
ниями.
Сополимеризация между простыми виниловыми эфирами осуще-
ствляется по ионному механизму. М. Ф. Шостаковский с сотрудни-
ками получал сополимеры винилбутилового эфира с винилэтило-
вым и винилизопропиловым эфирами в присутствии хлорного же-
леза в качестве катализатора. Были получены также сополимеры
метилвинилового эфира с октадецилвиниловым эфиром, а также
алкилвиниловые эфиры с дивиниловым эфиром диэтиленгликоля и
бутан-диоля 1,4.
Техническое значение приобрели сополимеры простых виниловых
эфиров с другими виниловыми соединениями — с производными
акриловой кислоты, хлористым винилом и хлористым винилиде-
ном, акрилонитрилом, малеиновым ангидридом и его производными,
винилацетатом и т. д.
Сополимеризация осуществляется по радикальному механизму
действием перекисных соединений в качестве инициаторов.
Так как молекулы простых виниловых эфиров не реагируют друг
с другом в присутствии перекисей, то макромолекула сополимера
может состоять из звеньев другого мономера или из чередующихся
звеньев другого мономера со звеньями простых виниловых эфиров.
Концентрация звеньев простых виниловых эфиров в сополимере не
может поэтому превышать 50%.
Введение простых виниловых эфиров имеет цель увеличить
эластичность и гибкость полимера и служит для «внутренней пла-
стификации» соответствующих полимеров. Так, при сополимери-
зации эфиров акриловой кислоты и акрилонитрила с простыми
виниловыми эфирами получают каучукоподобные сополимеры.
294 Гл. VII. Пластмассы на основе полимеров винилового спирта
В технике применяют сополимеры этилового эфира акриловой
кислоты с 2-хлорэтилвиниловым эфиром. Сополимеризацию прово-
дят, главным образом, эмульсионным методом в присутствии пер-
сульфата аммония в качестве инициатора. Например, реакционную
смесь, содержащую 95 ч. этилового эфира акриловой кислоты,
5 ч. 2-хлорэтилвинилового эфира, 50 ч. воды и 0,06 ч. персульфата
аммония нагревают при интенсивном перемешивании до темпера-
туры кипения в течение часа, после чего остаточные мономеры
отгоняют паром и продукт желатинируют обработкой на горячих
смесительных вальцах. В виде эмульсии выпускают каучукоподоб-
ные сополимеры, получаемые сополимеризацией этилакрилата
(75 ч.) с изобутилвиниловым эфиром (25 г). Применяют также и
сополимеры акрилонитрила. Так, по одной из рецептур методом
бисерной (суспензионной) сополимеризации 70 ч. акрилонитрила,
30 ч. изобутилвинилового эфира, 400 ч. воды, 1 ч. перекиси бен-
зоила и 2 ч. поливинилового спирта нагревают при интенсивном
перемешивании до 70—75° в течение 1—3 час. Сополимер оса-
ждается в виде тонкой суспензии, отделяется от раствора и много-
кратно промывается горячей, а затем холодной водой.
Мягкие, каучукоподобные сополимеры, с относительно большим
содержанием (20—25%) звеньев простых виниловых эфиров, нашли
применение для производства искусственной кожи, отделки текстиль-
ных тканей, в производстве технических резин и т. д.
Для увеличения эластичности и стабильности полихлорвинила
и полихлорвинилидена также были получены их сополимеры с не-
большим содержанием алкилвиниловых эфиров.
Добавка простого винилового эфира замедляет полимериза-
цию хлористого винила и, повидимому, также и хлористого винил-
идена. Содержание звеньев простых виниловых эфиров в сополи-
мерах всегда меньше, чем в реакционной среде.
Сополимеризация проводится эмульсионным методом в авто-
клаве под давлением. Например, в автоклав загружают 100 ч. воды,
1—1,5 ч. эмульгатора (натриевая соль жирной кислоты), 0,5 ч. пер-
сульфата калия, тщательно перемешивают, охлаждают пуском охла-
дительного рассола в рубашку автоклава, загружают после этого
жидкие мономеры (50 ч. хлористого винила и 5 ч. изобутилвинило-
вого эфира), пускают азот под давлением 10—15 ат и проводят
реакцию при 60—65° в течение 8—12 часов.
Получаемый сополимер применяется в качестве лаковой смолы,
обладает хорошей адгезией к металлу, дереву и коже.
Значительный интерес представляют сополимеры малеинового
ангидрида, а также эфиров малеиновой кислоты с простыми вини-
ловыми эфирами.
Оба мономера в отдельности не полимеризуются в присутствии
перекисей, однако они легко образуют сополимеры с правильно че-
редующимися звеньями, так как радикал одного мономера может
Поливиниловый спирт
295
вступать в реакцию лишь с радикалом второго мономера
... —сн—сн—сн2—сн—сн—сн—сн2—сн—...
II III I
СО СО OR СО СО R
Х/ х/
Реакцию проводят в блоке или в растворителях в присутствии
перекиси бензоила в качестве инициатора.
Сополимеры малеинового ангидрида были получены с метил-,
этил-изопропилвиниловыми эфирами.
Сополимеры метилвинилового эфира с малеиновым ангидридом
представляют вязкие, прозрачные водорастворимые жидкости, ко-
торые предложено применять для обработки кожи, для отделки тка-
ней и т. д.
Поливиниловый спирт
Поливиниловый спирт является единственным синтетическим
полимером, который получают не полимеризацией мономера, а пу-
тем превращения других полимерных веществ, — обычно гидроли-
зом его сложных эфиров (поливинилацетата и др.). Поливиниловый
спирт в основном сохраняет структуру и степень полимеризации
исходного поливинилового эфира, омылением которого он был по-
лучен. Было неоднократно доказано, что как при омылении поли-
меров сложных эфиров, ведущих к образованию поливинилового
спирта, так и при обратном процессе — эфиризации поливинило-
вого спирта с получением полимеров сложных эфиров, если эти
процессы проведены в определенных условиях, степень полимериза-
ции, т. е. длина цепи макромолекулы, практически мало меняется.
Такая устойчивость основной макромолекулы к химическим про-
цессам, ведущим к изменениям за счет побочных функциональных
групп полимера, открывает возможность для ряда макромолекуляр-
ных превращений, обеспечивающих получение новых и ценных в
промышленном отношении продуктов.
Технически поливиниловый спирт получают, главным образом,
омылением поливинилацетата в спиртовом растворе. Омыление
можно вести при помощи кислот:
...—СН2—СН — СН2-СН — СН2— ... C^f--»
О—СО—СН3 о—СО—СНз
—► ...- СН2—СН—СН2—СН—СН2 —... + nCH3COOC2H5 + тСН3СООН
он он
или щелочей
... —СН2—СН—СН2---СН-----СН2—... к”онН-»
О—СО—СНз о—СО—СН3
—► ...— СН2—СН—СН2—СН—СН2— ... + nCHsCOOC2H6 + т CHsCOONa.
ОН он
296
Гл. VII. Пластмассы на основе полимеров винилового спирта
При омылении щелочами кислотные остатки первоначально об-
разуют эфиры со спиртом-растворителем (в приведенной схеме об-
разуется этилацетат). Эти эфиры при дальнейшем действии щелочи
омыляются с образованием соли (ацетата) по уравнению:
СН3ССОС2Н6 4- NaOH —> CH3COONa ф- С2Н5ОН.
Процесс омыления поливинилацетата происходит при нагрева-
нии и интенсивном перемешивании раствора. Кислоты и щелочи
обычно берут в количестве меньшем, чем эквимолекулярное. По
мере омыления полимер теряет свою растворимость в спирте и оса-
ждается в зависимости от условий реакции в виде тонкого по-
рошка, нитей или гелеобразной массы. Выпадение из раствора
происходит при замещении около 60% ацетильных групп гидро-
ксильными. При дальнейшем нагревании процесс омыления про-
текает уже гетерогенно до получения почти полностью омыленного
продукта.
Кислоты и щелочи, добавляемые при кислотном и щелочном
омылении, затрудняют отмывку и стабилизацию образующегося по-
ливинилового спирта; кроме того они реагируют с ним с образова-
нием побочных продуктов. Стремление к устранению этих недостат-
ков привело к разработке процесса каталитического алкоголиза
в присутствии абсолютных спиртов при незначительных количествах
щелочи по схеме:
... — СН — СН2 — СН — СН2 — ... —"кой^"» п СН3СООС2Н6 +
О-СО-СНз О-СО-СН3
+... — СН—СН2—СН—СН2— ...
I I
ОН он
Для этого в раствор поливинилацетата, например в абсолютном
метиловом спирте, вносят метилат натрия (из расчета 0,1 —
0,4% щелочи от теоретически необходимого количества для пол-
ного омыления) и реакцию проводят при комнатной температуре
или при ~35°; через несколько часов (6—24) поливиниловый спирт
выделяется в виде геля, если реакция происходила без перемеши-
вания, или в виде аморфных сгустков, нитей и порошка, если реак-
ция происходила при перемешивании.
Регулируя количество катализатора (щелочи) и количество
воды в спирте, можно за определенный промежуток времени до-
стигнуть любой степени омыления. Получаемый поливиниловый
спирт легче поддается очистке и является более стабильным продук-
том, чем при кислотном омылении. Он может быть освобожден от
примесей растворением в воде и осаждением в избытке ацетона.
Вместо метилата натрия могут быть применены безводные едкие
щелочи, в особенности КОН.
Поливиниловый спирт
297
В технике применяют главным образам кислотное омыление и
каталитический щелочной алкоголиз.
Кислотное омыление может быть осуществлено при помощи
ряда кислот: соляной, серной, хлорной и др. Характер кислоты су-
щественно влияет на качество полимера.
Соляную кислоту применяют мало, так как она дает полимер,
окрашенный в желтый цвет (возможно вследствие образования не-
стойких продуктов присоединения).
Наиболее широко пользуются серной кислотой. Она дает свет-
лые полимеры в виде тонкого порошка, который может быть легко
Ьтмыт и просушен. Недостатком при этом является, однако, обра-
зование кислых сернокислых эфиров:
... —СНа—СН ----- СН2—СН— ...
О—SO2OH он
Эти эфиры нестойки и при нагревании легко отщепляют серную
кислоту, которая способствует деструкции полимера.
Были предложены и другие кислоты (фосфорная, хлорная, хлор-
уксусная), которые, повидимому, не образуют нестойких эфиров с
поливиниловым спиртом. Получаемый продукт может оказаться по-
этому более стабильным.
Скорость реакции омыления определяется концентрацией водо-
родных ионов в реакционной среде и температурой. Очень важно,
чтобы полимер получался в виде тонкодисперсного порошка, кото-
рый может быть легко отделен и промыт. На степень дисперсности
и характер агрегатного состояния осаждаемого полимера влияет
величина модуля реакционной среды (отношение количества поли-
мера к количеству растворителя) и интенсивность перемешивания.
Чем больше модуль и чем интенсивнее перемешивание, тем выше
дисперсность порошка.
При сернокислотном омылении применяют примерно следующую
реакционную смесь (вес. ч.):
Поливинилацетата ... 20
Спирта.............100
Серной кислоты .... 1,0—2
Реакцию проводят (рис. 122) в эмалированном реакторе 3 (или
в реакторе из кислотоупорной стали), снабженном паровой рубаш-
кой, обратным холодильником и пропеллерной мешалкой. Раствор
поливинилацетата и спиртовый раствор кислоты поступают само-
теком из весовых мерников 1 и 2. Процесс идет при температуре ки-
пения спиртовой смеси или же при несколько более низкой темпе-
ратуре в течение 8—20 час. (в зависимости от температуры и
количества серной кислоты). После значительного продвижения
реакции продукт начинает осаждаться в виде тонкой суспензии.
Реакцию продолжают до остаточного содержания ацетильных групп
в полимере не более 1,5—2,5%.
298
Гл. VII. Пластмассы на основе полимеров винилового спирта
По окончании реакции выпавший поливиниловый спирт отфиль-
тровывают и отжимают от маточного раствора на центрифуге 7,
затем в лавере 5 многократно промывают спиртом от серной кислоты,
этилацетата -и других растворимых продуктов реакции. Для увели-
чения стабильности продукта рекомендуется щелочная промывка.
Рис. 122. Схема производства поливинилового спирта.
/—мерник серной кислоты; 2—мериик поливинилацетатного лака; 3—реактор для омыления;
4—обратный холодильник; 5—лавер; 6—насос для суспензии поливинилового спирта; 7—центрифуга
С нижней выгрузкой; 8—мериик для спирта; Р—сборник оборотного спирта; 10 — насос д,.я спирта;
//—сборник для загрязненного спирта; 12—насос для загрязненного спирта; 13—куб для перегонки
спирта; 14—мерник для щелочи; 15—ректификационная колонка для спирта; 16—дефлегматор;
17—холодильник; 18—приемник для спирта; 19—сборник для спирта; 20—сборник для маточного
раствора; 21—вакуум-сушилка; 22—тара для поливинилового спирта.
Отмытый Дорошок поступает в полочную вакуум-сушилку 21, где
подвергается сушке при 45—60°.
Маточный раствор содержит спирт, этилацетат, серную и уксус-
ную кислоты. Его подвергают рекуперации и ректификации. Этил-
ацетат можно омылить щелочью и регенерировать спирт.
Эти операции производят в перегонном кубе 13 с ректифика-
ционной колонкой 15, снабженной дефлегматором 16 и соединенной
с холодильником 17. Щелочь поступает в куб через мерник 14.
Чистый спирт через приемник 18 поступает в сборник 19. Спирт для
ректификации подается в перегонный куб из сборника 11.
Строение и свойства поливинилового спирта
299
Строение и свойства поливинилового спирта
Поливиниловый спирт как полимер, содержащий равноценные
вторичные спиртовые группы, характеризуется рядом реакций, свой-
ственных вторичным спиртам. При окислении он образует кетонные
группы; вступает в реакцию с металлическим натрием, образуя по-
ливинилалкоголят, который дает простые поливиниловые эфиры при
взаимодействии с галоидоалкилами (стр. 292) и образует сложные
эфиры. При взаимодействии поливинилового спирта с альдегидами
получаются соответствующие ацетали.
Нетрудно заметить, что поливиниловый спирт ведет себя во мно-
гом подобно целлюлозе, которая также склонна к аналогичным
реакциям, основанным на взаимодействии первичных и вторичных
гидроксильных групп с кислотами и спиртами.
Поливиниловый спирт способен к некоторым реакциям, которые
мало характерны для низкомолекулярных соединений и которые
определяются его макромолекулярным строением. К этим реакциям
относятся, в частности, реакции дегидратации поливинилового
спирта, связанные с различными условиями взаимодействия его
функциональных групп. Так С. Н. Ушаков и Р. К. Гавурина уста-
новили следующие превращения при дегидратации
... —СН—СН2—СН—СН2—СН—СН,— ..
I I I
он он он
1) образование групп внутреннего простого эфира
ОН
...—СН—СН2—СН—СН2—СН—сн2—...,
1—о----------------1
2) образование непредельных соединений
—> ... сн—сн2—сн=сн—СН—сн2—... ,
I I
он он
легко окисляющихся под действием кислорода воздуха с образо-
ванием перекисей:
... — сн—сн,—сн—сн—сн—сн2—..
I III
он о—о он
3) образование продуктов циклизации:
ОН ОН
I I
... — сн—сн,—сн—сн—сн—сн,—...
I I
... —CH2—СН—CH—СН—СН,—СН —...
I I
он он
300
Гл. VH. Пластмассы на основе полимеров винилового спирта
4) сшивка цепей
6
... — сн—сн2—сн—сн,—in—сн2—...
он (J
... —сн—сн2—сн—сн,—сн—...
I I
он о
Дегидратация поливинилового спирта может начаться уже при
-температурах выше 100°; процесс ускоряется в присутствии следов
кислот или щелочей. Дегидратация приводит к глубокому измене-
нию физико-химических свойств поливинилового спирта, которое
выражается обычно в потере или в уменьшении растворимости в
воде, в увеличении хрупкости и жесткости, а также и температуры
стеклования.
Поливиниловый спирт имеет такое же строение макромолеку-
лярной цепи, как и поливинилацетат, в основном структуру 1,3
(«голова к хвосту»).
Степень полимеризации поливинилового спирта, как уже было
указано, практически определяется степенью полимеризации исход-
ного поливинилацетата. Вместе с тем было показано, что при про-
цессах омыления всегда имеет место, хотя и небольшое, снижение
вязкости полимера. Доказано также, что деструкция цепи тем силь-
нее, чем выше температура омыления и чем выше степень полиме-
ризации поливинилацетата. При кислотном омылении деструкция
цепи больше, чем при щелочном.
Можно предположить, что деструкция цепи происходит не за
счет углерод—углеродных связей цепи (С—С), а за счет разрыва
менее стабильных кислородных связей, повидимому, в небольшом
количестве также имеющихся в полимере. Было показано, что после
разрыва таких связей при первом процессе омыления поливинил-
.ацетата дальнейшие реакции реацетилирования и вторичного омы-
ления уже не ведут к существенному снижению вязкости полимера.
Превращение эфирных групп поливинилацетата в гидроксиль-
ные группы поливинилового спирта увеличивает полярность поли-
мера и межмолекулярные силы притяжения, обусловленные, как это
было доказано С. Н. Журковым, силами водородной связи между
гидроксильными группами соседних цепей.
В соответствии со своей химической структурой поливиниловый
спирт растворим в воде, в кислотах (в том числе в фосфорной кис-
лоте), в концентрированных водных растворах многоатомных спир-
тов (глицерине, гликоле, диэтиленгликоле и др.) и нерастворим в
одноатомных спиртах, кетонах (ацетоне), эфирах и т. д. Таким
образом, он является одним из наиболее полярных, гидрофильных
и водорастворимых полимеров.
Пластификация и применение поливинилового спирта
301
Поливиниловый спирт представляет собой белый порошок с
— 1,293, без вкуса и запаха, совершенно нетоксичный. Он ха-
рактеризуется высокой температурой стеклования (80°) и большой
прочностью. В неориентированном состоянии прочность на растя-
жение достигает 500—600 кг/см2-, при 5—6-кратной вытяжке проч-
ность увеличивается до 4000—4500 кг!см2.
В техническом продукте всегда имеется небольшое количество
остаточных эфирных групп (1—5%). Однако в технике получают
специальные полимеры с большим содержанием (20% и более)
эфирных (ацетильных) групп. С увеличением количества эфирных
групп значительно меняются свойства полимера и при 20% содер-
жании таких групп он уже нерастворим в холодной, а при 50% —
и в горячей воде. Такие полимеры, с содержанием 50—80 мол. %
ацетильных групп, имеют преимущество перед поливиниловым
спиртом в водостойкости и в то же время они значительно более
прочны и теплостойки, чем поливинилацетат. Таким образом, на
основе продуктов омыления сложных поливиниловых эфиров можно
получить как поливиниловый спирт, так и ряд полимеров с пере-
менным количеством ацетатных и гидроксильных групп. Полимеры,
содержащие заданное число боковых гидроксильных групп, наряду
с другими группами, можно получать омылением сополимеров
винилацетата с другими винильными соединениями, например, с хло-
ристым винилом, акрилонитролом и др.
Поливиниловый спирт, невидимому, включает некоторое коли-
чество кристаллической фазы (при растяжении выше температуры
стеклования его пленки показывают характерную, точечную рентге-
нограмму волокна). Сам процесс ориентации, в свою очередь, спо-
собствует процессам кристаллизации. Элементарный кристаллит
поливинилового спирта представляет собою орторомбическую
ячейку с размерами: а — 7,82 А; b = 5,60 А и с = 2,52 А (период,
идентичности).
Характер рентгенограммы поливинилового спирта и период
идентичности в 2,52 А указывают на плоскую, зигзагообразную
конфигурацию углеродных цепей с гликолевыми группами в поло-
жении 1,3, расположенными в одной плоскости («голова к хвосту»)
Пластификация и применение поливинилового спирта
Поливиниловый спирт в основном используют для производства поливинил-
ацеталей, однако он находит и самостоятельное применение. Так, им пользуются
главным образом для производства коже- и каучукоподобных изделий, беизо-
стойких шлангов и прокладок, для изготовления приводных ремней, волокна,
шнуров (для медицинских целей), абразивных кругов и т. д,; в виде водо-
растворимого коллоида он служит эмульгатором (в том числе для ведения про-
цессов суспензионной «бисерной» полимеризации), клеем, сгустителем лекарсти-
и т. п.
302
Гл VII. Пластмассы на основе полимеров винилового спирта
В качестве пластика, как правило, применяют пластифицированный поли-
виниловый спирт..
Применяемые в технике пластификаторы поливинилового спирта могут
-быть разделены на: 1) истинные и 2) косвенные пластификаторы; в первых
поливиниловый спирт растворяется, во вторых в безводном состояния не рас-
творяется и весьма мало набухает; при этом косвенные пластификаторы рас-
творимы в воде и в водных растворах, в которых поливиниловый спирт либо
растворяется, либо набухает.
К первой группе пластификаторов относится прежде всего вода.
Хотя вода является почти единственным техническим растворителем поли-
винилового спирта и низкокипящим, летучим веществом, однако ее необходимо
рассматривать и как пластификатор поливинилового спирта. Объясняется это
тем, что изделия из поливинилового спирта всегда содержат сравнительно зна-
чительные количества воды (в соответствии с относительной влажностью воз-
духа), которая резко снижает Тс полимера. При внесении других пластифика-
торов (не летучих) всегда учитывается содержание воды в полимере, которая
также значительно улучшает совместимость многих пластификаторов и пере-
работку (пластикацию) продукта.
К этой же группе могут быть отнесены и различные кислоты; однако прак-
тическое применение получила, повидимому, лишь фосфорная кислота, так как
она мало летуча и не приводит к эфиризации и деструкции полимера.
Опыт с применением фосфорной кислоты показал, что она может считаться
одним из лучших технических пластификаторов поливинилового спирта.
Ко второй группе пластификаторов поливинилового спирта относятся, в пер-
вую очередь, многоатомные спирты — глицерин, гликоль, диэтиленгликоль, три-
этилеигликоль и бутиленгликоль. К этой же группе пластификаторов относятся
некоторые амиды кислот (ацетанилид) и аминогидроксильные соединения, а
также фенолоспирты, диметиломочевина и др.; все они обладают еще меньшей
желатинирующей способностью.
Лучшими пластификаторами поливинилового спирта для промышленных
целей являются: глицерин, фосфорная кислота, бутиленгликоль, смесь глицерина
с бутиленгликолем и триэтиленгликоль.
Комплекс физико-механических свойств пластифицированного полимера
в значительной мере определяется количеством пластификатора. Материалы
х содержанием свыше 65 мол. % пластификатора (плюс равновесная влага)
представляют собою типичные эластопласты и в ряде случаев могут быть при-
менены вместо каучука; при меньшем содержании пластификатора получаются
кожеподобные материалы.
Поливиниловый спирт обладает практически абсолютной стойкостью к дей-
ствию алифатических и ароматических углеводородов (бензин, беизол) и в этом
отношении превосходит все известные пластики. Он поэтому применяется
в качестве бензостойкого каучукоподобного материала для изготовления бензо-
шлангов, бензиноемкостей и т. д.
Большая прочность, эластичность и малая истираемость пластифицирован-
ного поливинилового спирта были широко использованы для производства при-
водных ремней, мешков для прессования пластмасс при низких давлениях
(стр. 340), шнуров, а также ниток для хирургических целей.
Волокно и ткани из поливинилового спирта также находят применение.
Волокна получают пропусканием через фильтры водного раствора поливинило-
вого спирта в осадительную ванну (в водный раствор сернокислого калия).
Влажная нить подвергается вытяжке (300—400%), а иногда (для снижения
растворимости в воде) обработке в течение нескольких минут альдегидом; при
этом на поверхности, нити образуется соответствующий ацеталь (см. ниже).
Ткани из волокна поливинилового спирта производят как непосредственно, так
и в сочетании с волокнами из других полимеров. Их применяют для фильтрации
неполярных жидкостей и т. п.
Получение пластифицированной массы для дальнейшей переработки ее на
пленки, шланги и т. п. производят на горячих смесительных вальцах, на кото-
рые подается поливиниловый спирт, предварительно набухший («созревший»)
Пластификация и применение поливинилового спирта
303
в смеси пластификатора с водой. Вальцованную массу перерабатывают калан-
дрированием на листы и пленки и методом экструзии—на шланги, трубки,
стержни. Методом экструзии могут быть получены и пленки; для этого произво-
дят продольное разрезание выходящего из мундштука шнекпресса с одновре-
менной вытяжкой образующегося листа в одном или в двух направлениях.
Пленки отличаются прочностью и газонепроницаемостью. Они имеют весьма
низкий коэффициент диффузии к различным газам и отличаются высокой стой-
костью по отношению к действию озона.
Существенный недостаток пластиков из поливинилового спирта — их рас-
творимость в воде. Делались попытки устранить этот недостаток поливинилового
спирта, сохранив его важнейшие положительные качества (в частности стой-
кость к бензинам и к другим неполярным растворителям). Для этого изделия
из поливинилового спирта подвергают поверхностной защитной лакировке или
поверхностной химической обработке; можно также производить глубокое хими-
ческое изменение всей массы, например путем сшивкн цепей макромолекул.
Покрытие поверхности изделий защитными пленками, поливинилацетат-
ными, нитроцеллюлозными и др. дает в ряде случаев удовлетворительные
результаты, если, конечно, изделия подвержены лишь случайному воздействию
влаги и от них не требуется стойкости к неполярным растворителям.
Химическое изменение поверхности или превращение всей массы в нераство-
римое состояние может быть достигнуто многими методами: 1) химической
обработкой поверхности изделий альдегидами (например формальдегидом)
с превращением в соответствующие ацетали (см. ниже); 2) нагреванием при
150—160° или при более низкой температуре (110—130°) в присутствии следов
кислот, эффективнее всего—1—5% фосфорной кислоты (нагревание в присут-
ствии кислот вызывает процессы дегидратации и переход полимера в нераство-
римое состояние); 3) получением нерастворимых комплексов с некоторыми
соединениями двух- и трехвалентных металлов, главным образом с растворами
л(—СН2—СН—СН2—СН—СН2—СН— ...) п HOCORCOOH
I I I + ►
ОН ОН ОН п OCHRCHO
—> ..СН—СН2—СН—СН.,—СН—СН2—СН—СН»--------
I 1'1 I
ОН О—СН—о о
I
со
I
R R
I
со
I
О—СН—о о
,.. —СН—СН2—СН—СН»—СН—СН2—СН—СН2—СН— ...
он о
I
со
R
I
СО
I
о
I
... —СН—СН2—СН—СНг—СН—...
I I
ОН он
304
Гл. VII. Пластмассы на основе полимеров винилового спирта
солей бора, борной кислоты, с солями хрома (бихроматами) и медноаммиач-
ными солями (даже незначительное количество этих соединений вызывает при
действии света или при нагревании переход полимера в нерастворимое состоя-
ние) и 4) образованием пространственно-сшитых структур путем проведении реак-
ции эфиризации нагреванием поливинилового спирта с дикарбоновыми кислотами
(щавелевой, адипиновой, себацииовой) и диальдегидами, способными реагиро-
вать с двумя соседними макромолекулами поливинилового спирта по схеме,
представленной на стр. ’303.
Однако вопрос водостойкости изделий из поливинилового спирта еще не
получил успешного разрешения, так как все приведенные методы либо сильно
снижают основные характерные свойства полимера, либо не предотвращают
набухания его в воде.
В заключение необходимо указать и на некоторые менее значительные об-
ласти применения поливинилового спирта. Так, его применяют в текстильной
промышленности для шлихтовки тканей, в кондитерской промышленности для
улучшения всхожести теста, в производстве фруктовых желе и мороженого, для
производства фармацевтических облаток, а также в качестве лекарственного
препарата (против гипертонической болезни). Применяют поливиниловый спирт
и в типографском деле в качестве добавки к типографским краскам, а также для
изготовления светочувствительных слоев при фоторепродукциониых процессах.
Поливинилацетали
Среди производных поливинилового спирта поливинилацетали
являются важнейшими техническими продуктами.
В основе процесса образования поливинилацеталей лежит реак-
ция конденсации поливинилового спирта с карбонильными соедине-
ниями, главным образом с альдегидами, по схеме:
... —СН—СН,—СН—СН2—СН—СН,— ... 4- RCHO —>
I I I
ОН он он
—> ... — сн—СН2—СН—СН,—СН—сн,—...
I I I
он о—сн—о
I
R
Как видно из этой схемы, две гидроксильные группы одной и
той же макромолекулы реагируют с одной молекулой альдегида.
Значительно менее вероятно, что молекула альдегида вступает во
взаимодействие с гидроксильными группами двух соседних макро-
молекул по схеме:
—СН—СН2—СН—Сн2—СН— ..
I I I
он он он
он он он
I I I
... —сн—сн2—сн—сн2—сн—..
... сн—сн2—сн—сн,—сн—...-
+ RCHO ч 1 он 1 о 1 1 ОН
HCR
он О он
I I I
СН—CH2—СН—СН,—СН— ....
Поливинилацетали
305
По последней схеме реакция должна была бы привести к образо-
ванию нерастворимого пространственного полимера, тогда как поли-
винилацетали являются хорошо растворимыми полимерами.
Реакция ацеталирования катализируется водородными ионами,
т. е. теми же катализаторами (H2SO4; НС1 и т. п.), что и процесс
гидролиза поливинилацетата. Вследствие этого представляется воз-
можным проводить в одну операцию (в «одной ванне») как процесс
омыления поливинилацетата, так и процесс ацеталирования обра-
зующегося поливинилового спирта. Эта возможность облегчается
также соотношением скоростей реакций, которое обеспечивает столь
быстрое ацеталирование образующихся гидроксильных групп, что
полимер может оставаться в растворе в течение всего процесса гид-
ролиза и ацеталирования, если, конечно, применяют растворитель,
в котором растворимы как исходный поливинилацетат, так и обра-
зующийся ацеталь. Наряду с однованным методом применяют и
двухванный., заключающийся в ацеталировании изолированного по-
ливинилового спирта, полученного самостоятельным гидролизом
поливинилацетата.
Технические поливинилацетали никогда не содержат только аце-
тальные группы. Наряду с последними в молекулах поливинил-
ацеталей всегда имеются незамещенные гидроксильные и некоторое
количество эфирных (ацетильных) групп, так что схематически по-
ливинилацеталь может быть представлен формулой:
... — СН—С Но— ... —сн—сн.—сн—сн..—...—сн—сн,—...
I I “ I I
ОН О—СН—О О—СО-СНз
I
R
Содержание небольшого количества ацетильных групп (обычно
1,5—2 мол. %) объясняется наличием этих групп в техническом по-
ливиниловом спирте. Естественно, что в принципе можно получать
поливинилацетали, свободные от ацетильных (эфирных) групп.
В противоположность этому количество гидроксильных групп,
содержание которых в техническом продукте обычно выше 15—
20 мол. %, может быть уменьшено лишь до определенного предела.
Статистическая вероятность взаимодействия равноценных гидро-
ксильных групп с карбонильными определяет неизбежность наличия
«одиноких» гидроксильных групп между ацетальными циклами
(стр. 61).
По математическим подсчетам Флори, максимально достижимая
полнота замещения гидроксильных групп составляет 86,47 мол. %;
таким образом, в поливинилацеталях всегда должно быть не менее
13,53 мол. % свободных гидроксильных групп. Следует указать, что
на практике удавалось добиться большего замещения, — повидимому
приведенный математический подсчет, основанный на некоторых
произвольных допущениях, не является абсолютно достоверным;
однако недостижимость 100% ацеталирования остается доказанной.
20 Зак. 1269. Э. И. Барг
306
Гл. VII. Пластмассы на основа полимеров винилового спирта
Скорость ацеталирования зависит от температуры и количества
катализатора (от величины pH); степень же ацеталирования зави-
сит от продолжительности реакции, концентрации реагирующих
компонентов и характера альдегида.
Для достижения высоких степеней ацеталирования применяют
избыток альдегидного компонента (обычно около 1 моля альдегида
на 1 моль —СНг—СН — вместо теоретически рассчитанного 1 моля
ОН
альдегида на 2 моля —СН2—СН—).
ОН
При определенных условиях поливинилацетали образуются почти
со всеми альдегидами — алифатическими и циклическими, предель-
ными и непредельными. Однако, промышленное значение получили
лишь немногие, а именно:
поливинилформальдегидацеталь (поливинилформаль)
—сн»—сн—СН3—СН—СН2— ...+СН2О —►...—сн2—сн—сн2—сн—сн,—
он он о—сн2—b
поливинилацетальдегидацеталь (поливинилэтаналь)
..СН«—СН—СН3—СН—СН, —.. .-f-CHaCHO —► ... — сн,—сн—сн,—сн—сн,- .. .
II II
ОН ОН О—сн—о
I
сн,
поливинилбутир альдегидацеталь (поливинилбутираль)
—сн,—сн—СН,—СН—СН,—...+С3Н7ОН —> —сн—сн—сн,—сн—сн,—
‘1'1’ I I
ОН ОН о—сн—о
I
сн2
I
сн2
СН3
а также смешанные ацетали, образующиеся при одновременном
действии на поливиниловый спирт двух альдегидов, например поли-
винилформальэтаналь, получаемый при действии формальдегида и
ацетальдегида.
Методы производства поливинилацеталей
Поливинилацетали получают двумя различными уже упомяну-
тыми выше методами: 1) совместным, гомогенным процессом ги-
дролиза и ацеталирования, исходя из раствора поливинилацетата
(однованный метод) и 2) гетерогенным ацеталированием, исходя из
водных растворов поливинилового спирта (двухванный метод).
Методы производства поливинилацеталей
307
Однованный метод имеет два варианта, различающиеся в за-
висимости от характера растворителя. По первому варианту приме-
няют растворители, смешивающиеся с водой, например, этиловый
спирт, метиловый спирт и др., по второму — растворители, не сме-
шивающиеся с водой, например, бензол, бутилацетат и др.
По первому варианту однованного метода обычно исходят либо
из спиртового раствора поливинилацетата (при получении поливи-
нилэтаналя и поливинилбутираля), либо из раствора поливинилаце-
тата в уксусной кислоте (при получении поливинилформаля, когда
образующийся ацеталь нерастворим в спирте).
Процесс гидролиза и ацеталирования проводят в эмалирован-
ном реакторе или в реакторе, изготовленном из кислотоупорной
стали, снабженном мешалкой, паровой рубашкой и обратным холо-
дильником. В реактор из весового мерника поступает раствор поли-
винилацетата и при постоянном перемешивании через мерник мед-
ленно вводится спиртовый раствор кислоты (обычно серной 2—3%)
и соответствующий альдегид; последний берут в количестве до од-
ного моля на 1 моль звена поливинилового спирта (от 130 до 200%
от теоретического количества). Процесс ведут при 60—70° при по-
стоянном и энергичном перемешивании в течение 8—16 часов. Об-
разующийся в результате гидролиза поливиниловый спирт, не оса-
ждаясь из раствора, немедленно превращается в соответствующий
ацеталь, растворимый в том же растворителе, в котором был рас-
творен поливинилацетат.
По окончании процесса гомогенная реакционная смесь наряду
с поливинилацеталем содержит этилацетат (если в качестве рас-
творителя был взят этиловый спирт), альдегид, серную и уксусную
кислоты и воду. Выделение из этого раствора чистого полимера мо-
жет быть осуществлено осаждением его в воду. Однако для полу-
чения полимера в мелкораздробленном виде необходимо предвари-
тельно разбавить раствор приблизительно до 10—5% концентрации
полимера и медленной струей при постоянном перемешивании вво-
дить избыток (примерно пятикратный) воды. Осажденный в виде
хлопьев или зерен полимер фильтруют и отжимают на центрифуге.
Существенным недостатком этого варианта является значитель-
ный расход растворителей и трудность их регенерации.
По предложению С. Н. Ушакова, выделение чистого полимера
может быть осуществлено значительно более эффективным «соле-
вым методом». Полученный реакционный раствор ацеталя смеши-
вают в мешателе с большим избытком дисперсной соли (NaCl).
Затем при подогреве и включении вакуума отгоняют практически
всю летучую часть (при работающей мешалке). Оставшуюся
смесь ацеталя с солью многократно обрабатывают водой. Соль
растворяется и отделяется, таким образом, от полимера, который
тщательно промывают и в диспергированном виде переносят с водой
на центрифугу. Отжатый и снова промытый полимер поступает на
сушку. Этот способ дает возможность избежать главных затрудне-
20 s
30s Гл. VII. Пластмассы на основе полимеров винилового спирта
ний, связанных с образованием большого количества сильно разве
денных растворителей, и получать чистый ацеталь в мелкодисперс-
ном состоянии.
По второму варианту однованного метода реакцию ведут обычно
в растворе поливинилацетата в бутилацетате. Получаемый раствор
поливинилацеталя в несмешивающемся с водой растворителе может
быть в мешателе тщательно отмыт водой от серной кислоты и дру-
гих водорастворимых примесей; после этого его освобождают от
растворителя путем отгонки последнего с паром; оставшийся рас-
плавленный ацеталь продавливают шнековым устройством через
мундштук в виде лент, нарезают на штабики и сушат.
Двухванный метод получения поливинилацеталей также может
быть осуществлен по двум вариантам в зависимости от характера
реакционной среды (растворителя).
По первому варианту двухванного метода реакцию ацеталирова-
ния поливинилового спирта ведут в спиртовой среде, т. е. в усло-
виях, в которых он нерастворим. По мере продвижения реакции
ацеталирования диспергированный продукт все в большей мере
переходит в раствор, образуя прозрачный спиртовой раствор поли-
винилацеталя. Ацеталь в дальнейшем может быть выделен осажде-
нием водой. Этот вариант редко применяется, повидимому, из-за
сложных операций осаждения, а также необходимости регенерации
растворителей и отделения побочных продуктов. По существу он не
имеет никаких преимуществ перед обычным однованным методом.
По второму варианту двухванного метода реакцию ацеталиро-
вания ведут в водном растворе поливинилового спирта. По мере
продвижения реакции полимер становится нерастворимым и выпа-
дает из водного раствора. Было доказано, что выпадение полимера
происходит уже при незначительном ацеталировании гидроксиль-
ных групп поливинилового спирта и что дальнейший процесс аце-
талирования идет гетерогенно, т. е. протекает на поверхности по-
лимера. Такой процесс может быть назван водно-гетерогенным
ацетилированием поливинилового спирта. Он был разработан
С. Н. Ушаковым с сотрудниками для всех видов ацеталей поли-
винилового спирта.
Степень ацеталирования зависит от характера и количества
альдегида, его концентрации в реакционной смеси, от времени и
температуры реакции. Реакцию проводят в зависимости от приме-
няемого альдегида при 60° (масляный альдегид), 80° (формальде-
гид) в течение от 8 до 16 часов. Альдегид берут в теоретическом
или несколько большем количестве (если он не летуч), причем
альдегиды должны быть очищены перегонкой. Из кислот применяют
серную, фосфорную, соляную, муравьиную и др.
Технологический процесс производства и рецептура реакцион-
ной смеси должны обеспечивать получение поливинилацеталей с
оптимальной (заданной) степенью ацеталирования и высокой сте-
пенью чистоты и дисперсности.
Методы производства поливинилацеталей
309
Рецептура реакционной смеси определяется техническими тре-
бованиями к полимеру, а также методом и конкретными техниче-
скими условиями ацеталирования. Так, по одной из рецептур для
Масляный альдегид
Рис. 123. Схема производства ацеталей поливинилового спирта.
1—мерник для альдегида (масляный альдегид, ацетальдегид или формалин). 2—мерник для кислоты;
Л'—бункер для поливинилового спирта; 4—мерник для оживленного маточного раствора; 5—реактор
для ацеталирования поливинилового спирта; 6—обратный холодильник; 7—лаверы для промывки
ацеталей: 8—насос для суспензии ацеталей; 9 — центрифуга; 10—сборник маточного раствора;
11—насос для маточного раствора; 12— куб для перегонки маточного раствора; 13—холодильник;
14—приемник к кубу; 15—сборник перегнанного маточного раствора; 16—мерник для альдегида;
17—мерник для кислоты; 18 — мерник для воды; 19— иасос для „оживленного" маточного раствора;
20—куб для перегонки масляного альдегида; 21—холодильник; 22 — приемник масляного альдегида;
23 — сборник для масляного альдегида; 24— насос для масляного альдегида; 25—вакуум-сушилка для
ацеталей; 26— тара для ацеталей.
получения поливинилбутираля 100 ч. поливинилового спирта рас-
творяют в 800 ч. воды (дестиллированной) и при соответствующих
условиях ведения процесса вносят 50 ч. фосфорной кислоты и 85 ч.
масляного альдегида.
Процесс производства поливинилацеталей по гетерогенному
двухванному методу в общих чертах складывается из следующих
операций (рис. 123).
310 Гл. VII. Пластмассы на основе полимеров винилового спирта
1. В эмалированный или изготовленный из кислотоупорной
стали реактор, снабженный паровой рубашкой и обратным
холодильником, загружают через мерник воду и затем при интенсив-
ном перемешивании через люк вводят поливиниловый спирт. Рас-
творение поливинилового спирта происходит при нагревании до
60—80° и энергичном перемешивании, после чего в реактор вно-
сят раствор кислоты и свежеперегнанный альдегид и устанавливают
необходимую температуру. В результате реакции вскоре выпадает
ацеталь; однако, реакцию продолжают еще несколько часов до до-
стижения заданной степени ацеталирования.
2. По окончании реакции выпавший ацеталь (обычно в виде
порошка или волокон) отфильтровывают и подвергают отмывке.
Вода для промывок подается в лаверы, откуда она насосом вместе
с суспендированными частицами полимера подается на центрифугу.
После водных промывок рекомендуется проводить сначала про-
мывку ацеталя стабилизирующими агентами, а под конец холодной
или теплой водой.
3. Промытый и отжатый полимер выгружают из центрифуги и
подают в полочную вакуум-сушилку, где его сушат при темпера-
туре до 60°.
Маточный раствор и промывные воды, содержащие кислоту и
немного альдегида, поступают на рекуперацию, а концентраты на-
правляются обратно в реактор и для первичных промывок.
С. Н. Ушаков с сотрудниками разработал оригинальную схему
получения поливинилацеталей, исходя из поливинилформиата. Прин-
ципиальное преимущество этого процесса основано на том, что по-
ливинилформиат более легко гидролизуется по сравнению с поли-
винилацетатом. Омыление поливинилформиата происходит уже при
нагревании с водой, без катализаторов; в результате реакции вы-
деляется ’муравьиная кислота, которая и катализирует процесс.
Строение и свойства поливинилацеталей
Макромолекулярные цепи поливинилацеталей имеют, есте-
ственно, такую же структуру, как поливинилацетат и поливинило-
вый спирт, т. е. структуру 1,3. Степень полимеризации поливинил-
ацеталей практически также определяется степенью полимеризации
исходного поливинилацетата или поливинилового спирта.
Физические свойства поливинилацеталей определяются харак-
тером альдегида, содержанием ацетальных, ацетильных и гидро-
ксильных групп и степенью полимеризации полимера. Чем дальше
расположен в гомологическом ряду алифатический альдегид, тем,
соответственно, длиннее боковая цепь полимера и тем, следова-
тельно (как и для всех других полимеров), ниже его температура
стеклования и теплостойкость (рис. 124). По той же зависимости
циклические и разветвленные альдегиды с одним и тем же числом
Строение и свойства поливинилацеталей
311
углеродных атомов, как и алифатические, дают полиацетали с более
высокой температурой стеклования. В общем с переходом к альде-
гидам с более длинной цепью имеет место монотонное увеличение
гибкости и эластичности полимера. Растворимость полимеров в
органических растворителях повышается с увеличением длины цепи
альдегида.
Непредельные альдегиды, как и можно было ожидать, обуслов-
ливают сшивку макромолекулярных цепей и образование нераство-
римых полимеров. Последние пока еще мало изучены и не имеют
технического значения.
Существенное различие в физических свойствах поливинилаце-
талей проявляется в зависимости от химического строения альде-
Рис. 125. Зависимость температуры
размягчения по Вику поливинилбу-
тираля от содержания бутиральных
групп.
Рис. 124. Зависимость температу-
ры плавления алифатических поли-
винилацетелей от числа углерод-
ных атомов в боковой цепи (в аль-
дегиде).
вают усиление гидрофобных свойств поливинилацеталей, тогда как
окси-, алкокси- и аминоальдегиды уменьшают их гидрофобность.
Кроме характера альдегида существенное значение имеет степень
ацеталирования. Чем она выше, тем ниже температура стеклования
(температура размягчения) полимера, тем ниже также статическая
прочность и выше удлинение при разрыве (рис. 125).
С увеличением степени ацеталирования увеличиваются гидро-
фобные свойства полимера, его водостойкость и электроизолирую-
щие свойства, увеличивается растворимость в ароматических угле-
водородах и других слабополярных растворителях.
По характеру и величине сил взаимодействия между макромоле-
кулами ацетальные группы занимают среднее положение между
чрезвычайно сильными гидроксильными и значительно более
312
Гл. VII. Пластмассы на основе полимеров винилового спирта
слабыми ацетильными группами. Характерный комплекс свойств тех-
нических поливинилацеталей определяется именно этим сочетанием
влияния всех трех видов функциональных групп.
Степень полимеризации (вязкость) исходного поливинилацетата
определяет также степень полимеризации поливинилацеталя, так
как процессы гидролиза и ацеталирования мало изменяют длину
цепи макромолекулы.
Степень полимеризации существенно отра-
жается на основных свойствах ацеталей; чем
выше (до определенного предела) степень
полимеризации, тем ниже температура хруп-
кости полимера, тем больше можно увели-
чить его морозостойкость внесением пласти-
фикаторов, тем больше его прочность и
удлинение (табл. 30).
Поливинилацетали имеют, повидимому,
аморфную структуру, по крайней мере это
относится к поливинилбутиралю.
В отличие от многих других полимеров,
поливинилацетали представляют собою груп-
пу полимеров, отличающихся практически
неограниченной возможностью модифика-
ции и изменения свойств. Основные свойства
могут существенно изменяться под влия-
ТАБЛИЦА 30
Зависимость прочности
и удлинения поливи-
нилбутираля от сте-
пени полимеризации
(вязкости) исходного
поливинилацетата
Вязкость СП Предел прочности на разрыв к г/см* Удлине- ние
2,44 307 2,5
11,00 478 4,2
16,00 550 6,1
поливинилацеталей
нием: 1) химического состава альдегида; 2) количества ацетиль-
ных групп в
ния количеств
полимеризации
ацетата.
полимере; 3) степени ацеталирования (соотноше-
ацетальных и гидроксильных групп); 4) степени
и степени полидисперсности исходного поливинил-
Промышленные поливинилацетали
Как уже было указано, промышленное применение нашли лишь
немногие ацетали, а именно: поливинилформаль (формальвини-
пласт, формаль, или формвар), поливинилэтаналь (этанальвини-
пласт, альвар, поливинилэтилаль), поливинилбутираль (бутираль-
винипласт, бутираль, бутвар) и смешанные поливинилацетали. Все
эти ацетали представляют собой весьма ценные пластики, получив-
шие разнообразное применение в современной технике.
Поливинилформаль
Поливинилформаль получают чаще всего двухванным мето-
дом— действием формальдегида (в виде 40% формалина) на вод-
ный раствор поливинилового спирта в присутствии кислого ката-
лизатора. С этой целью берут, например, 4—10 ч. поливинилового
спирта, 3—8 ч. формальдегида, 0,01—0,05 ч. хлористого водорода
(в виде соляной кислоты) и 100 ч. воды.
Промышленные поливинилацетали
313
Поливинилформаль может быть получен и по однованному ме-
тоду действием 40% формалина на 15—20% раствор поливи-
нилацетата в уксусной кислоте в присутствии 2—3% серной кис-
лоты.
Степень ацеталирования технического поливинилформаля 75—
85 мол. %. Наличие в нем ацетальных групп с наиболее короткой
боковой цепью обусловливает наиболее высокую, по сравнению
с другими ацеталями, температуру размягчения. По этой же при-
чине поливинилформаль имеет и наибольшую статическую проч-
ность; он является наиболее жестким и твердым поливинилацеталем.
Свойства поливинилформаля могут быть представлены следующими
показателями:
Уд. вес, г'см'''........................... 1,23
Теплостойкость по Вику, °C..................110—115
Предел прочности на разрыв, кг/см2 .... 600—700
Коэффициент преломления света, п2£° .... 1,5
Водостойкость (привес в воде), °/о......... 13
Диэлектрическая постоянная при 800 гц . . . 3,7—3,2
Поливинилформаль ограниченно или совсем не растворяется в
обычных доступных растворителях; в зависимости от степени ацета-
-лирования он растворим в уксусной и муравьиной кислотах, в
пиридине, диоксане, хлорированных углеводородах, в фенолах, в
60% водном этиловом спирте, а также в смесях спирт — бензол
(30 : 70) и спирт — толуол (40 : 60). Высокая прочность, теплостой-
кость, твердость, хорошие диэлектрические свойства поливинилфор-
маля, а также стойкость его пленок к истиранию сделали этот по-
лимер ценным материалом для производства электроизоляционных
покрытий и, в частности, для эмалировки и покрытия магнитных
проводов в динамомашинах. Для эмалировки проводов формаль
применяют в композиции с резольными феноло- и крезоло-формаль-
дегидными смолами (стр. 353). Введение резольных смол (до 20%),
способных при нагревании реагировать с функциональными груп-
пами ацеталей и к переходу в неплавкое состояние, улучшает теп-
лостойкость поливинилформаля, повышает его нерастворимость и
прочность к истиранию. Применение формаля для эмалировки про-
водов позволяет уменьшить объем и вес электромоторов и повысить
их эффективность. По сравнению с обычными составами, применяе-
мыми для эмалировки и изоляции магнитных проводов, формаль
имеет преимущества в большей стойкости к горячим растворителям,
в более высокой гибкости и прочности к истиранию. Это приводит
к лучшей сохранности изоляции при намотке, хранении и примене-
нии проволоки. Изоляция из формаля отличается также более вы-
сокими диэлектрическими свойствами, которые сравнительно мало
меняются в широком температурном интервале. Изоляция из фор-
маля не требует какой-либо дополнительной защиты в виде хлопча-
тобумажной ткани и т. п. В табл. 31 сопоставлены диэлектрические
314
Гл. VII. Пластмассы на основе полимеров винилового спирта
свойства проволоки, изолированной с помощью формаля и обычно
эмалированной.
ТАБЛИЦА 31
Диэлектрические свойства эмалированной проволоки
и проволоки, изолированной поливинилформалем
Свойства Обычно эмалиро- ванная проволока при Проволока, изолированная формалем при
10® 75° 10’ 75®
Тангенс угла диэлектрических потерь Диэлектрическая постоянная . 0,0085 3,0 0,0366 3,6 0,007 3,6 0,0046 3,4
В сочетании с пластификаторами формаль применяется также и
для производства небольших электротехнических изделий методом
литья под давлением и прессования.
Поливинилэтаналь
Поливинилэтаналь в промышленных условиях получают обыч-
ными методами, исходя из поливинилацетата, как гомогенным спир-
товым, так и гетерогенным водным методами. Реакцию ведут обычно
не с ацетальдегидом, имеющим низкую температуру кипения, а с
паральдегидом.
Поливиниэтаналь имеет более низкую температуру стеклования
и, следовательно, более низкую теплостойкость, чем поливинилфор-
маль. Его прочность ниже, но удлинение при разрыве — выше.
Поливинилэтанали могут быть охарактеризованы следующими
показателями:
Уд. вес, г.'с.и3..........................1,14—1,18
Температура плавления, °C.................140—180
Теплостойкость по Вику, °C................ 85—90
Предел прочности на изгиб, кг^см*......... 550—600
Водостойкость (привес в воде), °/0........ 2
Поливинилэтаналь хорошо растворяется во всех обычных рас-
творителях — в спиртах, кетонах, сложных эфирах, ароматических
углеводородах и т. п. Он нашел применение в качестве лакового
сырья и для производства прессизделий, является высококачествен-
ным заменителем шеллака для политур и лаков, в производстве
граммпластинок и т. д. В виде пресскомпозиций поливинилэтаналь
применяют с добавкой пластификаторов; эти композиции могут быть
переработаны методом литья под давлением.
Промышленные поливинилацетали
315
П о л и в и н и л б v т и р а л ь
Поливинилбутираль получают действием масляного альдегида
на поливиниловый спирт обычными описанными выше методами
производства ацеталей. Так, реакционную смесь из водного рас-
твора поливинилового спирта и свежеперегнанного масляного аль-
дегида в соответствующем молярном соотношении (в зависимости от
требуемой степени замещения) в присутствии 0,01—0,1% НС1
нагревают при интенсивном перемешивании. Образующийся вскоре
нерастворимый дисперсный осадок содержит всего 15—20% бути-
ральных групп и дальнейшая реакция ацеталирования продол-
жается гетерогенно в течение нескольких часов. По окончании
ацеталирования полимер отфильтровывают и подвергают много-
кратной промывке. Технический продукт содержит обычно-
55—75 мол. % бутиральиых групп.
Длинные боковые группы поливинилбутираля обусловливают
сравнительную «мягкость» полимера и низкую температуру стеклова-
ния (~50°). Поливинилбутираль растворим в этиловом и бутило-
вом спиртах, в уксусной кислоте, циклогексаноле, пиридине, в смеси
спирта с бензолом и т. д. Пленки его отличаются высокой прозрач-
ностью и бесцветностью, прочностью и гибкостью. Статическая
прочность поливинилбутираля ниже, чем у других технических аце-
талей, но зато удлинение при разрыве и динамическая прочность
значительно выше. Свойства технических бутиралей могут быть
охарактеризованы следующими показателями:
Уд. вес, г/см2. . . . :.....................1,11—1,14
Температура размягчения, °C.................60—65
Температура стеклования, °C................. —50
Предел прочности на разрыв, кг/см2 .... 400—500
Коэффициент преломления, ...................... 1,489
Водостойкость (привес в воде), °/о.......... —4
Техническое применение получили главным образом пластифи-
цированные поливинилбутирали, содержащие обычно от 15 до 35%
пластификатора. В качестве пластификаторов применяют дибутил-
себацинат, диоктилсебацинат, диоктифталат и др. Введением
пластификаторов можно значительно снизить температуру стеклова-
ния бутираля и обеспечить пленкам высокую гибкость и морозо-
стойкость (до —60° и ниже), с сохранением при этом значительной
прочности (прочность на разрыв технических пластифицированных
бутиралей находится в пределах 250—350 кг! см2, при удлинении
при разрыве — 400% и выше).
Поливинилбутираль применяют главным образом для изготовле-
ния прозрачных и бесцветных пленок для склейки силикатных сте-
кол при производстве безосколочного стекла. Преимущество поли-
винилбутиральных пленок перед пленками из других полимеров
заключается в их высокой прозрачности, светостойкости, прочности
316 Гл. VII. Пластмассы на основе полимеров винилового спирта
п гибкости, морозостойкости и высоких адгезионных свойствах
к стеклу и к металлу. Обычно прочность склейки поливинилбути-
раля со стеклом превосходит прочность технических стекол. В на-
стоящее время он безусловно является лучшим промежуточным ма-
териалом для склейки стекол.
Из поливинлибутираля методом выдавливания (экструзии) изго-
товляются шланги, прутки и т. п.; ряд изделий изготовляется мето-
дом литья под давлением.
Особое значение приобрели конструкционные клеи из поливи-
нилбутираля. Высокие адгезионные свойства этого полимера
значительно усиливаются при добавках к нему термореактивных
(феноло-альдегидных, меламиновых и др.) продуктов конденсации.
Получаемые таким образом клеи имеют большое промышленное
значение, так как они весьма прочно склеивают разнообразные ма-
териалы — металлы, пластмассы, дерево, силикатные мате-
риалы и др.
Поливинилбутираль с термореактивными добавками широко
применяется и для изготовления непромокаемых плащей, наки-
док и т. п.
ГЛАВА VIII
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИМЕРОВ
ПРОИЗВОДНЫХ ЭТИЛЕНКАРБОНОВЫХ КИСЛОТ И ЭФИРОВ
НЕНАСЫЩЕННЫХ СПИРТОВ
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИМЕРОВ ПРОИЗВОДНЫХ.
ЭТИЛЕНКАРБОНОВЫХ КИСЛОТ
(Акри[ло]пласты)
Смолы, известные под названием полиакрилатов, акрилопластов.
акрилоидов и т. д., представляют собою полимеры производных
акриловой и метакриловой (а-метилакриловой) кислот — их слож-
ных эфиров, нитрилов, амидов и др., а также продукты сополиме-
ризации акриловых производных друг с другом и с другими нена-
сыщенными соединениями.
Полиакрилаты — обширный и разнообразный класс полимериза-
ционных пластиков, получивший большое техническое значение. Все
они бесцветные, светостойкие и прозрачные смолы; некоторые из
них представляют собою твердые, упругие стекла, получившие ши-
рокое применение в качестве органического стекла, другие — более
мягкие, каучукоподобные и даже воскообразные тела.
Методы получения производных акриловой и метакриловой кислот
Эфиры акриловой кислоты
Акриловая кислота СНг = СН — СООН представляет собой бес-
цветную жидкость с резким кислотным запахом; 1,4224, уд.
вес 1,062 (при 16°), т. плавл. 13°, т. кип. 141°.
Акриловая кислота, не содержащая перекисей, в атмосфере
азота не полимеризуется даже при 180°. Кислород в небольших
количествах ускоряет полимеризацию.
В присутствии органических или неорганических перекисей акри-
ловая кислота легко полимеризуется при температурах ниже 100°
как в блоке, так в растворителях и в эмульсиях.
Полимеры акриловой кислоты (так же, как метакриловой,
стр. 322) имеют лишь ограниченное промышленное значение по срав-
нению с полимерами ее производных (эфиров, нитрилов и др.), по-
этому технический синтез обычно направлен на непосредственное
замещение (эфиризацию) образующейся в результате реакции
акриловой кислоты с получением ее производных.
Известны разнообразные методы получения акриловой кислоты
и ее эфиров, однако техническое применение имеют лишь немногие.
Так важнейшим методом является этиленхлоргидриновый синтез.
Он заключается в конденсации этиленхлоргидрина с цианистым
318 Гл. VIII. Пластмассы на основе полимеров произв. эпшленкарб. кислот
натрием или синильной кислотой, с образованием этиленцианги-
дрина, который путем гидролиза и дегидратации превращают в
акриловую кислоту
СН2(ОН)СН2С1 + NaCN —> CH2(OH)CH2CN + NaCl;
CH2(OH)CH2CN ^2--^+н^-0-э- CH2(OH)CH2COOH4-NH4HSO4;
CH2(OH)CH2COOH CH2=CHCOOH;
или путем алкоголиза и дегидратации — в эфир акриловой кислоты
CH2(OH)CH2CN ±g°H+H*s?‘ -> CHa(OH)CH2COOR 4-NH4HSO4;
CH3(OH)CH2COOR CH2=CH • COOR.
В производственных условиях омыление, эфиризацию и дегидра-
тацию производят в одну операцию и в одном аппарате.
Для получения циангидрина тщательно измельченный цианистый
натрий нагревают с хлоргидрином
Рпс. 126. Непрерывная схема получе-
ния метилакрилата.
в течение пяти часов при 50°. По
окончании реакции хлористый на-
трий отфильтровывают, а филь-
трат перегоняют. Выход циангид-
рина 75—85%. При применении
HCN процесс идет с лучшими вы-
ходами.
Для проведения процессов омы-
ления, эфиризации и дегидратации
смесь, состоящую, например, из
1 ч. циангидрина и 2 ч. серной кис-
лоты и воды (или спирта), нагре-
вают с обратным холодильником
в течение одного часа; при этом
образуется акриловая кислота или
ее эфир, которые отгоняют в ва-
кууме.
Реакцию омыления, во избежа-
ние преждевременной полимери-
зации и дегидратации, ведут в
присутствии ингибитора (гидро-
хинона), а перегонку эфира — в
медных колонках.
Производство акриловых эфи-
ров (например, метилового) мо-
жет быть осуществлено непрерыв-
ным методом.
Аппаратурная схема состоит из реактора 1 (рис. 126), соединенного с ко-
лонкой 2, дефлегматором 3 и отводной трубкой 4. Реактор снабжен паровой
рубашкой, выложен внутри свинцом и имеет постоянную температуру в пре-
Методы получения производных акриловой и метакриловой кислот 319
делах 140—160°. В него непрерывно поступает метанол, который испаряется и
•образующиеся пары заполняют всю колонку. Колонка имеет около 65 см в диа-
метре и 8 -и в длину, изготовлена из железа, футерована каучуком, выложена
фарфоровыми плитками и заполнена керамиковыми кольцами. Сверху колонки
навстречу парам метанола с постоянной скоростью поступает серная кислота
и этиленциангидрин. В колонке протекают основные реакции и образуется ме-
тилакрилат, в небольшом количестве диметиловый эфир и формальдегид. Все
этн вещества в парообразном состоянии вместе с парами воды и метаиола про-
ходят при 78° дефлегматор, который задерживает серную кислоту, этиленциан-
гидрин и частично воду и направляются по трубке 4 в котел 5, наполненный
горячей водой (95°). Трубки дефлегматора медные, а дно свинцовое. Котел 5,
в который попадают пары из дефлегматора, соединен с колонкой 6, подобной
первой колонке и заполненной керамиковыми кольцами. Вода с температурой 75°
непрерывно поступает сверху колонки. Метанол отмывают в котле 5 и промыв-
ные воды, содержащие 3—5% метанола, непрерывно выпускают через сливной
штуцер.
Колонка 6 соединена с дефлегматором 7. Сверху в нее поступает ингибитор
(метиленовый синий). Дестиллат, выходящий из дефлегматора, состоит на 80%
из метилакрилата, а также из воды, метанола и метилового эфира.
Конденсат из дефлегматора 7 поступает в отдельный бак 8, в котором он
отделяется от воды и метанола, собирающихся на дне бака, и сливается через
переливную трубку с содержанием 93—94% метилакрилата. Дестиллат и конден-
сат, хранящиеся в отдельных сборниках, поступают на дальнейшие операции
промывки и дестилляцни.
Кроме этиленхлоргидринового метода синтеза акриловой кис-
лоты техническое значение имеют и некоторые другие методы.
Так, необходимый для синтеза этиленциангидрин может быть
получен также, исходя из окиси этилена и синильной кислоты:
СН2—CH, + HCN —> CH2(OH)CH2CN.
Техническое значение имеют также методы синтеза акриловых
эфиров из Р- и а-оксипропионовых кислот.
Для синтеза из fi-оксипропионовой (гидракриловой) кислоты
предварительно получают гидракриловый альдегид, который обра-
зуется при взаимодействии формальдегида и ацетальдегида:
СН2О + СН3СНО —> СН2(ОН)СН2СНО.
гидракриловый альдегид
Альдегид окисляют воздухом в присутствии марганцовых или
кобальтовых солей и получают £>-оксипропионовую кислоту:
СН2(ОН)СН2СНО СНа(ОН)СН2СООН.
Путем эфиризации кислоты с одновременной дегидратацией
образуются эфиры акриловой кислоты:
СН2(ОН)СН2СООН CH,=CHCOOR.
Значительно ббльшее значение имеет метод синтеза акриловых
эфиров через з.-оксипропионовую кислоту: для синтеза применяют
техническую молочную кислоту.
320 Гл. VII'. Пластмассы на основе полимеров произв. эталенкаро. кислот
Молочную кислоту подвергают эфиризации путем нагревания
самой кислоты или ее солей с соответствующими спиртами в при-
сутствии серной кислоты:
СН3СН(ОН)СООН СН - CH(OH)COOR.
Полученный эфир ацетилируют уксусным ангидридом или уксус-
ной кислотой в присутствии серной кислоты:
CH3CH(OH)COOR CH.CH(OCOCH3)COOR.
Наконец, в результате нагревания ацетилированного продукта
до 450—500° происходит отщепление уксусной кислоты и образова-
ние акрилового эфира:
CH3CH(OCOCH3)COOR ~-с-НаС—он -» CH2=CHCOOR.
Метод синтеза акриловых эфиров из молочной кислоты даег
хороший выход ацетилированного продукта — 75%, а реакция от-
щепления уксусной кислоты протекает с выходами до 90%. Выход
зависит от характера спиртового остатка: с увеличением молекуляр-
ного веса спиртового радикала выход при пиролизе значительно
снижается.
В настоящее время техническое значение приобретает весьма
перспективный синтез акриловых эфиров из ацетилена, окиси угле-
рода и спирта по схеме:
СН = СН + СО + ROH —> ch2=chcoor.
Процесс протекает при обычном давлении. Его особенностью
является применение карбонила никеля Ni(CO)4 в качестве ката-
лизатора и окиси углерода.
Карбонилникель в виде раствора в соляной кислоте и спирт
вносят в реактор и перемешивают, после чего смесь насыщают
ацетиленом. При этом эфир акриловой кислоты получается почти
с 100% выходом. Можно вначале насытить спирт ацетиленом и
в раствор медленно впускать раствор Ni(CO)4. Наконец, можно
вести процесс с одновременной подачей карбонила никеля и ацети-
лена.
Для получения акриловых эфиров высших спиртов (например
октилового и др.) применяют реакцию переэфиризации, нагревая
низшие эфиры (например метиловый эфир акриловой кислоты)
с высшими спиртами и отгоняя образующийся низший спирт
CH2=CHCOOCH3 + ROH — -> CH2=CH-COOR + CH3OH,
где R — радикал высшего спирта.
Методы получения производных акриловой и метакриловой кислот 321
Акриловая кислота, так же как и метакриловая, применяется
для синтеза водорастворимых полимеров и для получения некото-
рых сополимеров.
Свойства некоторых эфиров акриловой кислоты приведены
в табл. 32; зависимость удельного веса и коэффициента преломле-
ния от числа углеродных
атомов в спиртовом ради-
кале видна из рис. 127.
Акрилонитрил
Акрилонитрил CH2=CHCN
в последние годы приобрел
большое промышленное зна-
чение главным образом для
получения одного из видов
синтетических каучуков —
сополимера с бутадиеном, —
а также ряда других тех-
нических сополимеров.
Акрилонитрил кипит при
77,3° (760 мм), т. плавл. —83°,
преломления 1,3911.
Получают акрилонитрил различными методами, чаще всего де-
гидратацией этиленииангидрина
ТАБЛИЦА 32
Свойства некоторых эфиров акриловой
кислоты
Эфир Т. кип. °C {ат\ Давле- ние, •H.1Z Уд. вес. d*>° Коэффи- циент преломле- НИЯ
Метиловый 80 760 0,9535 1,4040
Этиловый 43 103 0,9234 1,4038
Пропиловый 44 40 0,9078 1,4130
Бутиловый 35 8 0,8998 1.4190
Гексиловый 40 1,0 0,8882 1,4285
Г ептиловый 57 1,0 0,8846 1,4311
Октиловый 57 0,05 0,8810 1,4360
уд. вес при 20° - - 0,8060, показатель
синтезом из ацетилена и синиль-
ной кислоты.
Дегидратацию этиленцнан-
гидрина
CH2(OH)CH2CN ch2=chcn
проводят в жидкой или паро-
вой фазе в присутствии самых
разнообразных катализаторов,
причем дегидратацию в жид-
кой фазе ведут в присутствии
инертных растворителей (ди-
фенилэтан, минеральные мас-
ла) при температуре 250—255°,
применяя в качестве катализа-
тора фосфаты аммония, гидрат
окиси кальция и др. При де-
гидратации же в паровой фазе
лары этлленциангидрина пропускают над активированной окисью
аммония.
Из ацетилена и синильной кислоты акрилонитрил получают по
реакции
9,94 -
.-^2 -
0,90
0,86
1,^5 § §
oj г.
______________1,0-05 |
О 4 8 12 16
Число углеродных атомоЗ 8
спиртовом радикале
Рис. 127. Зависимость уд. веса и коэф-
фициента преломления акриловых эфи-
ров от числа углеродных атомов в спир-
товом радикале.
1—ул. вес; 2—коэффициент преломлении.
и
НС = СН 4- HCN — > CH2=CHCN.
21 Зак. 1269. Э. И. Барг
322 Гл. VIII. Пластмассы, на основе полимеров этиленкарб. кислот
Реакцию проводят как в жидкой, так и в паровой фазе. Реак-
ция в жидкой фазе катализируется водным раствором полухло-
ристой меди, содержащим хлористый аммоний и соляную кислоту.
Парофазным методом акрилонитрил предлагали получать про-
пусканием смеси равных объемов ацетилена и синильной кислоты
при 400—500° над цианидами щелочных металлов, нанесенных на
активированную окись алюминия, древесный уголь или другие
носители.
Эфиры метакриловой кислоты
Метакриловая (а-метилакриловая) кислота СН2 = С(СН3)СООг1
представляет собою жидкость с т. плавл. 15°, т. кип. 161°, уд. ве-
сом 1,015, обладающую менее острым запахом, чем акриловая
кислота.
Метакриловая кислота, ее эфиры и производные (нитрил, амид)
могут быть синтезированы различными методами, из которых неко-
торые аналогичны методам получения эфиров акриловой кислоты.
Это, в частности, относится к ацетонциангидриновому методу, кото-
рый нашел наибольшее техническое применение и весьма близок
к методу получения акриловых эфиров из этиленциангидрина.
Исходным сырьем для ацетонциангидринового синтеза служит
ацетон, синильная кислота или ее соли, серная кислота и соответ-
ствующие спирты.
Первой стадией синтеза является получение ацетонциангидрина
действием на ацетон синильной кислоты в присутствии щелочных
катализаторов (едкой щелочи, углекислых солей, пиридина, хино-
лина и др.) или же цианистого натрия или калия в присутствии
бисульфита натрия пли серной кислоты:
СН3 СНз
I I
СН3—C + HCN —> СНз—С—CN.
II I
о он
ацетонциангидрин
Ацетонциангидрин — бесцветная жидкость без запаха, кипит
при 82° (при 23 мм), уд. вес 0,932, растворим в воде, в спиртах,
эфирах и других растворителях.
Вторая стадия синтеза включает процессы дегидратации, омы-
ления и эфиризации, которые проводят последовательно в одном и
том же реакторе и в результате получают эфир метакриловой
кислоты.
Превращение ацетонциангидрина в эфиры метакриловой кис-
лоты осуществляют действием на него серной кислоты в спиртовой
среде (в водной среде получается метакриловая кислота); процесс
Методы получения производных акриловой и метакриловой кислот 323
протекает таким образом, что вначале образуется амид метакрило-
вой кислоты по схеме:
СН,; СН;
СНз—С—C = N CHs-t-C=NH
I I I
OH OH OSO3H
CH3 CH;i
—> CH2=C—C=NH — -» CH,=C—C—NH».
I II
OSOjH о
амид метакриловой кислоты
В дальнейшем амид подвергается омылению и эфиризации:
СН.; сн3 сн:.
CH,=ti—с—nh-> — -> сн2=ё—с—он — -> cH,=i—с—or.
А ’ " 1
ООО
эфир метакриловой кислоты
Из всех эфиров метакриловой кислоты в технике наиболее ши-
роко применяют метиловый эфир. Поэтому следует несколько по-
дробнее остановиться на процессе его получения по ацетонциан-
гидриновому методу.
Исходным сырьем являются технический (98%) ацетонциангид-
рин, метанол и серная кислота. На 1 вес. ч. ацетонциангидрина бе-
рут примерно 2 ч. серной кислоты, 0,6—0,7 ч. метанола и примерно
столько же воды. Процесс по одному из вариантов проводят в че-
тыре стадии.
1. В эмалированный стальной реактор, снабженный мешалкой
и рубашкой для водяного охлаждения, загружают серную кислоту;
затем при перемешивании вводят 98% ацетоциангидрин и в ка-
честве ингибитора 0,2% к ацетоциангидрину — танина. Циан-
гидрин вливают в охлажденную серную кислоту с такой ско-
ростью, чтобы поддерживать температуру не выше 70—80°.
2. По прибавлении всего циангидрина смесь переливают во вто-
рой выложенный свинцом стальной реактор, снабженный мешалкой
и паровой рубашкой. Температуру смеси при постоянном переме-
шивании поддерживают около 125° в течение 20 мин. В этих усло-
виях продукт присоединения серной кислоты к ацетонциангидрину
превращается в сернокислую соль амида метакриловой кислоты.
К концу реакции смесь охлаждают до 90° и выливают в большой
приемник, выложенный свинцом, в который собирают несколько
последовательных партий.
Превращение сернокислой соли амида метакриловой кислоты
в эфир проводят в отдельном реакторе. Последний выложен свин-
цом или керамическими плитками, снабжен мешалкой из крем-
21*
324 Гл. VIII. Пластмассы на основе полимеров этиленкарб. кислот
Ряс. 128. Аппарат для ректификации .ме-
тилметакрилата.
/—куб; 2—колонка; 3—дефлегматор; 4 —холо-
дильник;^, 6, 7—сборники.
нистого чугуна, паровой ру-
башкой и обратным холодиль-
ником. В него загружают мета-
нол, воду и метакриламид; по-
следний добавляют с такой ско-
ростью, чтобы температура не
поднималась выше 90°. После
внесения всего метакриламида
реакция продолжается 2—3
часа.
Образовавшийся метилмет-
акрилат выделяют из реакци-
онной смеси перегонкой с па-
ром. Сырой продукт содержит
метанол и метакриловую кис-
лоту, для удаления которых
его несколько раз промывают
водой в специальных промыва-
телях, представляющих собою
обычно медные сосуды с лопа-
стной мешалкой. Затем эфир
сливают в сборник, откуда он
поступает в куб ректифика-
ционного аппарата.
Ректификационный аппарат
(рис. 128) состоит из куба, ко-
лонны, дефлегматора, холо-
дильника и сборников для чи-
стого эфира и для азеотропа.
Основная аппаратура должна
быть медной (для торможения
полимеризации). Ректифика-
цию отмытого эфира произво-
дят в присутствии гидрохинона
с отбором фракции, перегоняю-
щейся при 100—102°, которая
представляет собой технически
чистый метилметакрилат.
Ниже этой температуры пе-
регоняется фракция с меньшим
содержанием эфира, предста-
вляющая собой азеотроп, со-
бирающийся в отдельный сбор-
ник.
Другой вариант синтеза метилметакрилата — проведение про-
цесса «в одном котле», г. е. в одном аппарате. Схема такого про-
цесса приведена на рис. 129.
Методы получения производных акриловой и метакриловой кислот 325
В стальной освинцованный реактор 1, соединенный с холодиль-
ником 6, через мерник 2 подают серную кислоту. При охлаждении
смеси (пуском воды в рубашку реактора) и работающей мешалке
через мерник 3 постепенно загружают ацетонциангидрин; после
этого в рубашку реактора пропускают пар и, подняв температуру
до 100—130°, выдерживают смесь при этой температуре необходи-
мое время. Затем из мерника 4 в реактор вводят воду и метанол,
Рис. 129. Схема получения метилметакрилата.
7 — реактор; 2, 3, 4, 5—мерники; 6—холодильник; 7—бак; 8—промыватель; 9— сборник;
10 —15 — ректификационные аппара гы.
а также раствор гидрохинона. После перемешивания в течение
60—90 мин. при 70—90° образуется метилметакрилат, который при
переключении холодильника 6 на «прямой» перегоняется в медный
бак 7 в виде сырого эфира. Дальнейшие операции по промывке и
ректификации аналогичны операциям предыдущего метода. Сырой
эфир поступает в промыватель 8, затем в сборник промытого
эфира 9, из которого направляется в ректификационные аппа-
раты 10—15.
Метиловый эфир метакриловой кислоты — важнейший мономер
группы акриловых эфиров. Он представляет собою бесцветную,,
легко подвижную прозрачную жидкость со специфическим запахом,
т. кип. 100,3° (при 760 мм), т. плавл. —48°; его уд. вес d^a= 0,949
и коэффициент преломления 1,4168.
Технический продукт должен содержать не менее 99,5% метил-
метакрила.
326 Гл. Vir. Пластмассы на основе полимеров этиленкарб. кислот
Для синтеза метакриловых эфиров была предложена также
реакция взаимодействия пропилена с фосгеном, ведущая к получе-
нию хлорангидрида ^-хлоризомасляной кислоты
СН3
СОС1., + СНз—СН=СН2 —> С1СН-—СН—СС1.
II
о
При обработке ^-хлоризобутирилхлорида едкими щелочами
происходит омыление и отщепление хлористого водорода с образо-
ванием метакриловой кислоты. Для получения эфиров метакриловой
кислоты хлорангидрид подвергают эфиризации непосредственным
нагреванием с 2—3-кратным избытком спирта, после чего отщепле-
ние хлористого водорода может быть осуществлено либо примене-
нием органических оснований, либо пропусканием паров эфиризо-
ванного продукта через силикагель или активированный уголь при
высоких температурах (300°), либо обработкой хлорангидрида хлор-
ным железом в жидкой фазе при 98—105°:
СН3 СН3
С1СН2—СН—С—С1 СН2=С—С—OR.
I' I'
О О
Приведенными методами обычно получают метиловый, а также
этиловый и бутиловый эфиры метакриловой кислоты. Высшие
эфиры метакриловой
ТАБЛИЦА 33 кислоты, так же как и Свойства некоторых эфиров метакриловой высшие эфиры акрило кислоты вой кислоты, получают nvTPM прт>рэгЬит<чяттии
Эфиры Т. кип. °C Давле- ние лс.м обычно из метилового эфира. Для этого смесь метилметакрилата, со-
Метиловый Этиловый Пропиловый Изопропиловый .... Бутиловый Изобутиловый (первич- ный) Изобутиловый (третич- ный) Октиловый 100,3 116,5—117 141—143 125 51—52 46—47 67—68 105 760 760 765 760 14 13 20 5 ответствуюшего высше- и,_ го спирта, серной кис- 0,921 лоты, ингибитора и o’88<s инертного растворителя 0,894 (например бензола) на- 0 884 гревают Д° кипения и ’ через дефлегматор Ol- О.887 гоняют образующий- — ся метиловый спирт вместе с растворите- лем.
Свойства некоторых эфиров метакриловой кислоты приведены
в табл. 33.
Полимеризация акриловых и метакриловых эфиров 327
Полимеризация акриловых и метакриловых эфиров
Значительная асимметричность молекул акриловых и метакри-
ловых эфиров определяет их большую склонность к полимеризации.
Полимеризация имеет цепной, радикальный характер и осуще-
ствляется воздействием света, температуры, перекисями и другими
факторами, инициирую-
щими рост свободных ра-
дикалов.
Чисто термическая по-
лимеризация протекает
крайне медленно. Так,
тщательно очищенный ме-
тилметакрилат в отсут-
ствие воздуха и света не
полимеризуется вплоть до
-4-170°. Технические про-
дукты, однако, способны к
полимеризации в отсут- _ ,,,,, ..
r J Рис. 130. Кривые кинетики термической по-
ствие воздуха и света при димеризации метилметакрилата в темноте без
сравнительно невысоких инициатора.
температурах (рис. 130).
В технике, однако, чисто термическую полимеризацию приме-
няют редко; обычно ее проводят в присутствии инициаторов (пере-
киси бензоила и водорастворимых перекисей).
Как и при технической полимеризации многих других виниловых
соединений, применяют три основных метода инициированной поли-
меризации: блочный (полимеризация чистых мономеров), водно-
эмульсионный и полимеризацию в растворителях.
Блочный метод
Блочный метод полимеризации широко применяют главным
образом в производстве полиметилметакрилата; последний при этом
получается в виде прозрачных и бесцветных пластин и блоков
(органическое стекло).
Полиметилметакрилат в виде блочного полимера (органического
стекла) получают тщательным смешением инициатора (перекиси
бензоила) с мономером с последующей заливкой смеси в стеклян-
ные формы.
В некоторых случаях в состав реакционной смеси вносят неболь-
шое количество (5—15%) пластификатора, который должен быть
тщательно перемешан с мономером и инициатором.
Концентрация инициатора и температура полимеризации опре-
деляют как скорость реакции полимеризации, так и молекулярный
вес полимера, причем чем ниже температура полимеризации и
меньше концентрация инициатора, тем выше молекулярный вес по-
лимера, но тем меньше скорость полимеризации.
328 Гл. Vfff. Пластмассы на основе полимеров этиленкарб. кислот
Как и в других случаях блочной полимеризации, основная труд-
ность процесса заключается в регулировке температуры внутри
блока. Вследствие экзотермичности реакции полимеризации и ма-
лой теплопроводности полимера (0,15 ккал/м • час • °C) неизбежны
местные перегревы внутри блока, обусловливающие, в свою оче-
редь, увеличение скорости реакции и, следовательно, резкое повы-
шение температуры; это ведет к испарению мономера и к образо-
ванию вздутий, если внешние слои блока уже достаточно вязки и
не допускают выделения газов.
До известной степени избежать вздутия можно изменением кон-
центрации инициатора и температуры полимеризации.
Чем толще получаемый блок, тем меньше должна быть концен-
трация инициатора, тем медленнее подъем температуры или тем
ниже температура полимеризации.
Однако необходимо при этом учитывать, что выполнение этих
условий приводит к получению полимера с более высоким молеку-
лярным весом.
Необходимо отметить, что местные перегревы, избежать которых
полностью невозможно, неминуемо ведут к внутренним напряже-
ниям в блоке из-за различной степени полимеризации во внутренних
и внешних слоях блока.
Процесс производства органического стекла по одному из ва-
риантов состоит из приготовления и заливки форм, предварительной
и окончательной полимеризации и разъема форм.
Формы обычно изготовляют из полированного зеркального сили-
катного стекла, которое должно быть тщательно промыто в усло-
виях, исключающих попадание пыли.
Для изготовления формы берут два стеклянных листа, на края
одного кладут прокладки из гибкого, эластичного материала, по вы-
соте равные толщине изготовляемого блока, и покрывают эти про-
кладки вторым листом стекла, после чего края обклеивают прочной и
тонкой бумагой, оставляя при этом отверстие для заливки мономера.
Одновременно подготовляют смесь, тщательно перемешивая
мономер, инициатор и пластификатор. Смешение может быть про-
изведено в никелевом котле, снабженном пропеллерной или якор-
ной мешалкой, герметически закрывающейся сферической крышкой,
на которой имеются люк и штуцеры для загрузки мономера,
инициатора и других компонентов. Перемешивание ведут при обыч-
ной температуре в течение 30—60 мин., после чего через сливной
нижний штуцер смесь поступает в весовые мерники, а из мерников
через воронку — в формы.
Полимеризацию проводят путем последовательного прохожде-
ния залитых форм ряда печей с различной температурой. При этом
формы проходят печи при следующем примерно режиме: в первой
печи при 45—55° они находятся 4—6 часов; во второй (55—65°)
8—10 часов и в третьей печи (85—125°) 8 часов.
Полимеризация акриловых и метакриловых эфиров
329
По другому варианту залитые формы устанавливают на рамы,
которые погружают в водяную баню; при этом полимеризация
происходит при 32—42° в течение 36—48 часов до достиже-
ния 93% конверсии. Рамы затем вынимают из ванны, помещают
в термошкаф, выложенный свипцом, и четыре часа нагревают
при 100°.
Вместо заливки форм мономером более рациональной, повиди-
мому, является заливка форм 0,5—1,5% раствором полимера
в мономере, что должно уменьшить образование пузырей в
блоке.
Тепловой и временной режимы полимеризации зависят от кон-
центрации инициатора и от толщины изготовляемых блоков и, сле-
довательно, могут варьировать в известных пределах.
По окончании полимеризации формы погружают в воду, после
чего блоки могут быть легко отделены от силикатных стекол.
Готовые листы направляют на обрезку краев и на поли-
ровку, — они должны быть прозрачными, без пузырей, вздутий и
соответствовать допускам по толщине, а также техническим усло-
виям по физико-механическим свойствам. Полиметилметакрплат-
ные стекла изготовляют различной толщины — от 0,5 до 50 мм
и больше.
Эмульсионный метод
Эмульсионную полимеризацию акрилатов применяют для полу-
чения литьевых и прессовочных порошков, а также стойких водных
дисперсий типа «латекс». Процесс в общем подобен эмульсионной
полимеризации других виниловых мономеров. Воду и эфир берут
в отношении 2—3:1 (модуль). Инициаторами обычно служат
водорастворимые перекиси (перекись водорода, персульфат ам-
мония и другие персульфаты). Как известно, весьма хороший
эффект дают надсернокислые соли, так как они сравнительно легко
отмываются от полимера и позволяют проводить реакцию полиме-
ризации без эмульгатора (или с меньшим его содержанием). В ка-
честве эмульгаторов применяют различные мыла (например, олеино-
вое и кокосовое), сульфированные масла, желатин, некаль и др.
Специальными эмульгаторами для бисерного варианта являются
полимеры акриловой и метакриловой кислот, а также другие водо-
растворимые синтетические полимеры. В состав реакционной смеси
обычно входит пластификатор (дибутил-, диоктилфталат, дибутил-
себацинат и др.), который в зависимости от состава мономера и
назначения полимера может быть внесен в различных соотношениях
(от 5 до 30%).
Для изготовления прессовочных порошков, материала для произ-
водства зубных протезов и для других целей, где требуется жесткий,
330 Г.1. VIII. Пластмассы на основе полимеров этиленкарб. кислот
упругий материал рационально применять бисерный метод эмуль-
сионной полимеризации, получая гранулированный полимер, обычно
полиметилметакрилат или сополимеры со стиролом, с метилакрила-
том и др. При производстве бисерного пли гранулированного поли-
акрилата инициатором служит перекись бензоила, которую рас-
творяют в мономере в количестве несколько большем, чем при
блочной полимеризации (от 0,5 до 1 %); в качестве эмульгато-
ров применяют карбонат магния, а также полиакриловую кислоту,
поливиниловый спирт и другие водорастворимые полимеры. Вели-
чина гранул зависит от концентрации эмульгатора и скорости
перемешивания. Воду и мономер берут в соотношениях 2 : 1 или
3:1. Процесс производства складывается из загрузки сырья в реак-
тор, полимеризации, фильтрации и промывки гранул полимера,
сушки и просеивания.
В никелевый реактор, снабженный паровой рубашкой и мешал-
кой, последовательно загружают из мерников дестиллированную
воду и мономер (или смесь мономеров), затем вручную через шту-
цер вносят эмульгатор. После перемешивания в течение 10—20 мин.
в реактор вносят пластификатор, краситель и инициатор, раство-
ренный <в мономере. В рубашку реактора дают пар и температуру
поднимают до 70—75°. Через 40—60 мин. за счет тепла, выделяю-
щегося в результате полимеризации, температура в реакторе повы-
шается до 80—85°. Температуру можно регулировать подачей воды
или пара в рубашку реактора. Контроль процесса производят опре-
делением содержания мономера. Полимеризация продолжается
2—4 часа.
По окончании полимеризации реакционную смесь переносят
в центрифугу с корзиной из нержавеющей стали, в которой гранулы
полимера легко отделяются и многократно отмываются водой от
эмульгатора.
Отмытый порошок загружают на алюминиевые противни тонким
слоем и сушат в термошкафах при медленном подъеме температуры
в пределах 40—70° в течение 8—12 час. После сушки порошок про-
сеивают и затаривают.
Получаемые этим методом гранулированный полиметилметакри-
лат, а также его сополимеры с метилакрилатом, со стиролом и т. п.
без дальнейшей переработки могут быть применены для изготовле-
ния зубных протезов, для производства лаков и др. Однако для
изготовления прессовочных порошков гранулированный полимер не-
обходимо пропустить через вальцы в течение 3—5 мин. при
170—190°; в процессе этой операции к полиметилметакрилату мо-
гут быть добавлены пластификаторы и красители. Вальцованные
листы измельчают на ударно-крестовой мельнице и просеивают
через сито № 30—40.
Процесс вальцевания, необходимый для получения текучего и
легко таблетируемого прессматериала, снижает однако молекуляр-
ный вес полимера примерно на 25%.
Полимеризация акриловых и метакриловых эфиров
33'.
Физико-механические свойства прессовочных материалов на
основе полимеров метилметакрилата и его сополимеров со стиролом
следующие:
Уд. вес, г[см*.............................. 1,2
Водостойкость (привес в воде), %............0,4—0,05
Теплостойкость по Мартенсу, °C..............60—70
Уд. ударная вязкость, кг - см/см*........... 5—10
Предел прочности на изгиб, кг[см2........... 500—700
Твердость по Бринелю, кг/мм*................15—18
Полиакрилаты нашли широкое применение в виде устойчивых
водных дисперсий типа «латекс».
Они получаются методом эмульсионной полимеризации акрило-
вых эфиров (метил-, этил- или бутилакрилового эфира). Технологи-
ческий процесс складывается из загрузки компонентов в реактор
и проведения реакции полимеризации, после чего дисперсию смоль:
как готовую товарную продукцию сливают в тару. Реак-
ционная смесь состоит из 100 ч. мономера (например метил-
акрилата), 250—300 ч. дестиллированной воды, 0,1—0,2 ч. водорас-
творимого инициатора (персульфата аммония), 1—2 ч. эмульгатора
(аммониевой соли дибутилнафталинсульфокислоты— некаль) и
10—20 ч. пластификатора. Порядок загрузки компонентов и режим
полимеризации примерно те же, что и при полимеризации метакри-
ловых эфиров для получения эмульсионных порошков. Полимери-
зация считается законченной, если содержание мономера в реакторе
не превышает 1—2%.
Полимеризация в растворителях
Полимеризация акриловых и метакриловых эфиров в раствори-
телях не нашла широкого применения.
Как уже было указано (стр. 162), полимеризация может проис-
ходить как в среде, в которой растворимы мономер и полимер, так
и в среде, в которой растворим только мономер. Первым типом рас-
творителей являются ароматические углеводороды (бензол, толуол),
хлорированные углеводороды, сложные эфиры, ацетон, диоксан
и др.; в качестве второго типа служат, главным образом, смеси
воды со спиртами, например смесь воды и метилового спирта.
Полимеризацию акриловых производных в растворителях по
первому методу применяют в тех случаях, когда конечный продукт
полимеризации используют в качестве лака. Концентрация таких
лаков невелика вследствие высокой вязкости полимера; обычно
она не превышает 10—15%. Вязкость лака, определяемая степенью
полимеризации полимера, зависит как от режима полимеризации
(количество инициатора, температура), так и от концентрации и
характера растворителя: чем выше концентрация мономера, тем
больше степень полимеризации. Так, при полимеризации метил-
Гл. VIII. Пластмассы на основе полимеров этиленкарб. кислот
метакрилата в метаноле молекулярный вес полимера-при концен-
трации 5% —26 000; при 10% —33 000 и при 20% —52 000.
С уменьшением концентрации мономера уменьшается также и
скорость полимеризации. В табл. 34 показана зависимость скорости
полимеризации и молекулярного веса полимера от характера рас-
творителя. 1
ТАБЛИЦА 34
Зависимость скорости полимеризации метилметакрилата
(в 20j/0-hom растворе) от характера растворителя
Растворитель Выход полимера, о,'о, через Молекулярный вес полимера в тысячах
24 ч. 48 ч. 72 ч. 225 ч.
Ацетон .... 0 42 73 75 (через 225 час.)
Диоксан . . . 69 84 88 98 35 „ „ „
Бутилацетат . 0 6 40 86 41 „ . „
При полимеризации по второму методу, например в водно-спир-
товых растворах (в 50% растворе метанола), в которых полимер
не растворяется, молекулярный вес полимера и скорость полимери-
зации обычно тем выше, чем
ТАБЛИЦА 35 больше содержание воды в
Зависимость мол. веса полимера и индук-
ционного периода от содержания воды
в спирте и от концентрации мономера
спирте, т. е. чем меньше
растворяющая способность
водно-спиртовой смеси по
Растворитель Концентра- ция мономера °.о Индук- ционный период часы Мол. вес (ТЫСЯЧИ) отношению к полимеру. Индукционный период также зависит от содержания в спирте воды (табл. 35).
Полимеризацию акрило-
50% метанол . О 10 45 21 100 140 вых и метакриловых эфиров
20 18 168 в водно-спиртовой смеси осу-
45°/о метанол . 20 170 75 ществляют при соотношении
30 140 77 спирта к воде от 50 : 50 до
50 96 90 30 : 70 в присутствии 0,5—
1 % перекиси бензоила и при
концентрации мономера от 20 до 40%. Полимеризацию ведут при
55—75° в эмалированном реакторе, снабженном мешалкой, паровой
рубашкой и крышкой с соответствующими штуцерами и люком для
ввода ингредиентов. По мере хода реакции полимер осаждается из
раствора в виде порошка; его отфильтровывают и в реактор вводят
добавочное количество мономера с инициатором. Отфильтрованную
зодно-спиртовую смесь периодически подают обратно в реактор,
который соединен с центрифугой; в последнюю время от времени
через нижний сливной штуцер подается реакционная смесь с оса-
жденным полимером. После фильтрации полимер тщательно
Строение полиакрилатов
333
Рис. 131. Схема непрерывной деполиме-
ризации полиметилметакрилата.
/ — приемная воронка; 2—реактор для деполи-
меризации; 3— парс гая труба; 4—бак с проточной
водой.
промывают свежей водно-спиртовой смесью, затем дестиллирован-
ной водой, сушат на противнях в термошкафах при 40—60°, про-
сеивают и затаривают.
Переработка акриловых отходов
В процессе механической обработки органического стекла (полимети.т-
метакрилата) и, частично, в процессе самого производства полимера получаются
значительные отходы. Одним из методов утилизации отходов является деполи-
меризация полимера в мономер. Полиметилметакрилат деполимеризуется при
температуре выше 300°, с хорошим выходом мономера (более 90%).
Процесс деполимеризации можно вести непрерывным методом. Для этого
отходы органического стекла предварительно раздробляют на мелкие кусочки
(1—3 см) и в таком виде загружают
в приемную воронку аппарата, от-
куда их шнеком непрерывно подают в
реактор (рис. 131), в котором они
продвигаются с постоянной скоростью
к приемнику. Реактор состоит из
стальной трубы 2—3 м длиной И
30—50 см в диаметре, внутри которой
проходит червячный транспортер.
Трубу обогревают электричеством.
Рабочая температура (380—400°)
устанавливается терморегуляторами.
Конец реактора соединен с паровой
трубой, ведущей к холодильнику,
куда направляются пары образующе-
гося мономера, а твердый оста-
ток, состоящий из небольшого коли-
чества углерода и неизменившегося
полимера, поступает в приемник с
проточной водой, которая уносит
углистые частицы, а полимер остается
на дне приемника; его периодически возвращают в приемную вороню
реактора.
Поступающие из реактора пары конденсируются в холодильнике. Конденсат
обрабатывают серной кислотой и разбавляют водой. Эфирный слой отделяют,
промывают, после чего он поступает в ректификационный котел для очистки
мономера. Сернокислотный слой продувают паром для отгонкн мономера;
в дальнейшем дестнллат обрабатывают, как сырой эфир.
Строение полиакрилатов
По некоторым данным полиакриловые эфиры построены по
типу 1,2 («голова к голове»):
СН3 СН3 СН3 СН3
II II
.. ,_СН2—с---С—сн2— сн2—с----с—сн2—сн2—...
COOR ioOR COOR COOR
Такая структура была доказана, например, для полимеров
&.-галоидакрилового эфира; так как галоид почти полностью от-
щепляется цинковой пылью, то это указывает, что атомы его в по-
лимере находятся в положении 1,2.
;34 Гл. VIII. Пластмассы, на основе полимеров этиленкарб. кислот
Однако по другим данным полиакриловые эфиры построены по
типу — «голова к хвосту». Это доказывается, например, тем, что
в продуктах деструкции полиметилакрилата найден эфир метилен-
глутаровой кислоты
сн2—сн2—с=сн2,
I I
с=о с=о
I I
OR OR
который мог образоваться лишь из цепи, построенной по прин-
ципу 1,3.
Таким образом, возможно, что полимеры галоидзамещенных
акриловых эфиров имеют структуру «голова к голове», а полимеры
чистых акрилатов, по крайней мере частично, имеют структуру «го-
лова к хвосту».
Сополимеры акриловых и метакриловых эфиров
Несмотря на большое число акриловых и метакриловых полиме-
ров и на разнообразие их свойств, в технике все большее значение
приобретают продукты сополимеризации акриловых мономеров.
Сополимеры акрилатов можно условно разделить на три группы:
1) сополимеры различных акриловых эфиров, 2) сополимеры с дру-
гими виниловыми соединениями, 3) сополимеры с тетрафункцио-
нальными соединениями, обусловливающими образование про-
странственных полимеров.
Сополимеры первой группы применяют для получения эластич-
ных, мягких материалов, обладающих растяжимостью при обычных
температурах и хорошими адгезионными свойствами. Такими
являются сополимеры метилметакрилата, а также нитрила акрило-
вой кислоты с бутиловым и другими высшими эфирами акриловой
и метакриловой кислот. Они могут быть получены в виде прозрач-
ных мягких листов, используемых в качестве промежуточного слоя
при изготовлении стекла «триплекс», а также литьевых пресс-
порошков, так как чистый полиметилметакрилат трудно пере-
рабатывается литьем под давлением. Широкое применение акрилат-
ные сополимеры находят в виде эмульсий для обработки кожи, для
пропитки тканей и бумаги и для получения слоистых пластиков.
Из сополимеров второй группы большое техническое применение
приобрели сополимеры метакриловых эфиров со стиролом; в част-
ности, метилметакрилата с 10—20% стирола. Сополимеризацию
метилметакрилата со стиролом проводят эмульсионным бисерным
методом, часто в присутствии пластификатора. Получаемый поро-
шок вальцуют на горячих вальцах, после чего листы размалывают
в порошок.
Сополимер обладает значительно более высокой текучестью, чем
полиметакрилат (при 166—180°), и может перерабатываться мето-
Сополимеры акриловых и метакриловых эфиров
335
дом литья под давлением и методом экструзии на стандартных
машинах и автоматах.
Известны сополимеры эфиров акриловой и метакриловой кислот,
а также акрилонитрила с разнообразными виниловыми соедине-
ниями, — с хлористым винилиденом, хлористым винилом, винил-
ацетатом, простыми виниловыми эфирами и др. В ряде случаев
акриловые эфиры применяют в тройных системах, например, при
сополимеризации хлористого винила, стирола и метилакрилата.
Иногда присутствие третьего компонента — акрилового производ-
ного делает возможным течение процесса сополимеризации, который
между двумя компонентами не протекает. Так, образование сополи-
меров стирола с винилацетатом возможно только в присутствии не-
которого количества метилметакрилата.
Большое промышленное значение имеют также сополимеры
третьей группы акриловых производных с тетрафункциональными
соединениями. Они, в свою очередь, могут быть разделены на:
1) сополимеры, в которых остается свободной одна из ненасы-
щенных групп тетрафункционального соединения и которые, следо-
вательно, получаются в виде линейного, растворимого ненасыщен-
ного полимера, способного, однако, к дальнейшим процессам «сшив-
ки» (вулканизации) и образования пространственного полимера, и
2) сополимеры, получаемые непосредственно в процессе полимери-
зации в виде трехмерного, неплавкого и нерастворимого продукта.
Примером первых сополимеров могут служить сополимеры
акрилонитрила с бутадиеном и др., представляющие собою новый
тип вулканизующихся каучуков, имеющих теперь столь большое
техническое значение.
Примером вторых могут служить сополимеры акриловых эфи-
ров с гликолевыми эфирами акриловой и метакриловой кислот,
а также с аллиловыми производными акриловой кислоты; так метил-
акрилат с аллилакрилатом образует трехмерный сополимер по сле-
дующей схеме:
Н2С=СН—СО +Н»С=СН—СО —>
I I
о—сн2—сн=сн2 о—сн3
аллилакрилат метилакрилат
-—>- ...—сн.,—сн—сн2—сн—сн2—сн—сн2—сн—...
I I I I
с=о с=о с=о с=о
i I I I
О—СН3 О О—СН-, о
I I
сн2 сн2
I I
... — сн—сн2—сн—сн2—сн—сн2..
I
с=о
I
О—сн3
336 Гл. VIII. Пластмассы на основе полимеров этиленкарб. кислот
Сополимеры с трехмерной структурой представляют собою про-
зрачные стекла, неплавкие и нерастворимые, с более высокой твер-
достью и теплостойкостью, чем твердость и теплостойкость
полиакриловых эфиров. Чем выше концентрация «сшивки» (тетра-
функциональной компоненты), тем больше твердость и тепло-
стойкость получаемого органического стекла.
Основные свойства пространственных «сшитых» сополимеров
(растворимость, теплостойкость, твердость и др.) определяются, од-
нако, не абсолютным значением концентрации «сшивки», а соотно-
шением между концентрацией «сшивки» и длиной цепи образую-
щихся макромолекул. Чем выше степень полимеризации, тем при
меньшей концентрации «сшивки» образуется пространственный,
нерастворимый сополимер. Теоретически необходимо около одной
молекулы тетрафункционального вещества на каждую цепь поли-
мера для образования трехмерного нерастворимого продукта.
Пространственно сшитые акрилаты не получили большого при-
менения, хотя проблема увеличения теплостойкости и твердости
органических стекол весьма актуальна. Это объясняется суще-
ственным недостатком указанных материалов — их неспособностью
к пластицирующим методам переработки. Получаемые пластины
или стержни могут быть подвергнуты только механической, станоч-
ной обработке.
Пространственные сополимеры, образующиеся непосредственно
в процессе полимеризации, получают только блочным методом.
Сополимеризацию бифункциональных компонентов проводят
теми же методами, что и полимеризацию, а именно: блочным мето-
дом, в эмульсиях или в растворителях. Соотношение реагирующих
мономеров и метод полимеризации должны быть выбраны с учетом
относительной активности каждого мономера, выраженной в соот-
ветствующих значениях констант сополимеризации и желаемого
комплекса физико-механических и химических свойств сополимера.
Свойства полиакрилатов
Свойства полиакрилатов, так же как и всех других полимериза-
ционных пластиков, определяются химическим строением мономера
и условиями полимеризации.
Полиакрилаты имеют аморфную структуру. При растяжении
происходят выпрямление и ориентация макромолекул; однако даже
при весьма значительных растяжениях рентгенограмма не дает воз-
можности обнаружить каких-либо заметных признаков кристалли-
зации. Различные физические константы также не указывают на
наличие фазовых изменений в полимерах.
Так же, как и для всех полимеров аморфной структуры, самая
характерная константа полиакрилатов — температура стеклования.
Важной закономерностью является то, что все полиметакриловые
эфиры имеют более высокую точку стеклования и, следовательно,
более высокую теплостойкость по сравнению с полиакриловыми
Свойства полиакрилатов
3)7
эфирами (при одинаковом спиртовом радикале). Повышение тем-
пературы стеклования с введением метильной группы может быть
объяснено меньшей подвижностью четвертичного углерода, обусло-
вленной несимметричным замещением, в то время как симметричное
замещение обоих водородов одними и теми же группами приводит
к компенсации диполей и к снижению температуры стеклования, что
мы видим при сравнении полихлорвинилидена (Гс —17°) с поли-
хлорвинилом (Т., +80°).
Более высокая теплостойкость метакриловых полимеров имеет
особое значение при применении их в качестве прозрачного кон-
струкционного материала (органического стекла). Более «мягкие»
акриловые полимеры используют главным образом при получении
морозостойких материалов, т. е. материалов, То для которых лежит
в области температур значительно ниже обычных.
Температура стеклования полимеров зависит также и от ха-
рактера спиртового остатка в эфире. В этом отношении имеется
одинаковая зависимость как для полиакрилатов, так и для поли-
метакрилатов: чем длиннее боковая цепь полимера, т. е. чем больше
молекулярный вес и длина цепи спиртового радикала, тем «мягче»
иолимер, тем ниже его температура стеклования (табл. 36). Так
как в данном случае имеет значение длина спиртового остатка, то
эфиры с изоспиртовыми радикалами образуют более твердые поли-
меры, чем с нормальными, с третичными — более твердые, чем с вто-
ричными.
ТАБЛИЦА 36
Зависимость Та полиакриловых и полиметакриловых эфиров
от характера спиртового остатка
Полиметакрилаты
Полиакрилаты
Метил.............
Этил..............
Пропил............
Изопоопил.........
Бутил.............
Первично изобутил .
Третично бутил . . .
Олекл.............
Сктил ............
Циклогексил . . . .
98
50
36
95
16
54
76
— 5
— 15
105
Метил
Этил
Бутил
8
— 20
— 40
Зависимость температуры хрупкости от длины боковой цепи
в гомологическом ряду полиметакрилатов дана на рис. 8 (стр. 58).
Как видно из рис. 8, обратная зависимость температуры хруп-
кости (и температуры стеклования) от длины боковой цепи в гомо-
логическом ряду полиакрилатов действительна лишь до некоторой
22 Зак. 1269. Э. И. Барг
338 Гл. VIII. Пластмассы на основе полимеров этиленкарб. кислот
предельной длины (до 8 углеродных атомов), после чего она резко
возрастает.
Растворимость и вязкость полиакрилатов в значительной мере
зависят также от строения спиртового остатка: с увеличением его
длины полимер приобретает способность растворяться в более ши-
роком ряду растворителей. Полимеры низших эфиров растворимы
в ароматических углеводородах, в сложных эфирах, в хлорирован-
ных углеводородах и т. д. Полимеры высших эфиров растворимы,
кроме того, в парафиновых углеводородах. Вязкость растворов, как
правило, снижается с увеличением молекулярного веса спиртового
остатка, возможно, из-за уменьшения степени полимеризации
(рис. 132).
Полимеры низших и высших эфиров различаются и по отноше-
нию к пластификаторам; полиакрилаты высших спиртов совместимы
Рис. 132. Вязкость растворов поли-
метакриловых эфиров в толуоле.
1 — метиловый; 2—этиле вый; 3—гропилсвый;
4— бутиловый; 5— изобутиловый эфиры.
со значительно большим числом
пластификаторов, чем полиакри-
латы низших спиртов.
Различие между метакриловы-
ми и акриловыми полимерами про-
является и в их химической стой-
кости; метакриловые полимеры
химически более стойки и более
водостойки, чем акриловые, что,
.невидимому, объясняется более
плотной упаковкой макромолекул
полиметилметакрилата.
Они отличаются также отно-
шением к действию температур:
выше температуры разложения
полиметакрилаты деполимери-
зуются с образованием исходного
мономера, тогда как полиакрила-
ты разлагаются в этих условиях в
значительной мере без образова-
ния исходного мономера-
Технические продукты в зависимости от их назначения полу-
чают с различной степенью полимеризации. Наибольшую степень
полимеризации чаще всего имеют органические стекла (полиметил-
метакрилат), получаемые в результате блочной полимеризации
(мол. вес выше 200 000), меньшую — пресспорошки и порошки
для литья под давлением, получаемые эмульсионной полимериза-
цией (мол. вес от 40 000 до 200 000), и наиболее низкую — ла-
тексы и лаки из акриловых эфиров и сополимеров, применяемые
для пропитки бумаги, тканей, для поверхностной обработки кожи
и т. д. С увеличением степени полимеризации повышается тем-
пература плавления (температура пластического течения) поли-
Свойства полиакрилатов
339»
мера, до известного предела улучшаются его механические свой-
ства, в частности, удельная ударная вязкость.
Ценным техническим свойством полиакрилатов является их про-
зрачность и бесцветность, а также способность пропускать ультра-
фиолетовые лучи. Так, полиметилметакрилат пропускает свыше
99% солнечного света и в этом отношении значительно превосхо-
дит силикатные стекла. Преимущество полиакрилатных стекол ста-
новится еще нагляднее, если сравнивать их способность про-
пускать только ультрафиолетовую часть спектра; например, квар-
цевое стекло пропускает 100% ультрафиолетовых лучей, полиме-
тилметакрилатное — 73,5 %, зеркальное силикатное — 3 %, обыч-
ное силикатное — 0,6 %.
Полиметилметакрилат, пожалуй, первый пластик, который на
основании комплекса его свойств может быть назван органическим
стеклом. Преимуществом перед обычным стеклом является более
высокая прочность (в частности к знакопеременным динамическим
нагрузкам), превосходящая в десятки раз обычные стекла.
Однако полиметакрилатные стекла по сравнению с минеральными
имеют меньшую поверхностную твердость, меньшую стойкость
к действию абразивов. Важным преимуществом является их спо-
собность подвергаться обработке как механическим методом (сня-
тие,м стружки), так и методом пластической и высокоэластической
деформации.
Крупные изделия сферической формы изготовляют из листов
органического стекла методом формования, которое рационально
проводить вакуумным методом, впервые предложенным С. Н. Уша-
ковым и получившим применение в технике.
Для этого предварительно нагретые (120—150°) пластичные
листы укладывают и закрепляют по поверхности сферы металличе-
ской формы, в которой имеется отвод к вакууму; при включении
вакуума листы втягиваются внутрь формы и в этом состоянии охла-
ждаются, полированная поверхность изделий при этом сохра-
няется’.
Более мелкие изделия несложной формы могут быть получены
штамповкой заготовок из нагретого листа с последующей формов-
кой в прессформах при-низком давлении или без формовки. Так же
успешно применяют вытяжку и выдувание горячим воздухом.
Трубы и другие полые изделия изготовляют «центробежным» ме-
тодом из вязкой, текучей массы, приготовленной растворением поли-
мера в мономере. Ее вносят в быстровращающуюся полую
цилиндрическую форму, нагретую до ПО—160°. Под влиянием цен-
тробежной силы масса отбрасывается к стенкам формы, где полиме-
ризуется и переходит в твердое состояние. Этим путем можно приго-
товить трубы до 4 л длиной и 500 мм в диаметре.
Полиметилметакрилатные пресспорошки перерабатываются ме-
тодом прессования и литья под давлением значительно труднее и
при более высоких температурах, чем полистирол и некоторые
22*
340 Гл. VIII. Пластмассы на основе полимеров этиленкарб. кислот
другие полимеры. Объясняется это высокой вязкостью его в об-
ласти температур высокоэластического состояния и высокой точ-
кой его пластического (вязкого) течения, обусловленной
большим молекулярным весом полиметилметакрилатных пресс-
материалов.
Вместе с тем для получения изделий, к которым предъявляют
требования высокой стабильности во времени и сохранения формы
и размеров при температурах, близких к температуре стеклования,
необходимо, чтобы при прессовании преобладал процесс необрати-
мого вязкого течения массы, а не обратимой высокоэластической
деформации. В последнем случае форма изделия не находится в
равновесном состоянии и с течением времени, которое экспонен-
циально уменьшается с температурой, изделие деформируется и
получает в итоге ту форму и размеры, которые материал имел до
прессования.
Поэтому переработку полиметилметакрилата так же, как и всех
линейных полимеров, следует вести при более высоких температу-
рах, обеспечивающих пластическое течение материала (~200—220°).
Пресспорошки на основе сополимеров метилметакрилата, в част-
ности со стиролом, имеют более высокую текучесть при переработке,
меньшую температуру вязкого течения и, следовательно, легче пере-
рабатываются методом литья под давлением, который является од-
ним из самых производительных и эффективных методов пере-
работки полимеризационных термопластов.
Полиметилметакрилат лишь незначительно меняет свои свой-
ства с понижением температуры; это один из весьма немногих пла-
стиков, удельная ударная вязкость которого почти не меняется с
понижением температуры и практически стабильна в пределах от
—183° до +60°, хотя модуль упругости и статическая прочность
монотонно повышаются с понижением температуры.
Свойства полиметилметакрилата (техническое органическое
стекло, блочный литой продукт) следующие:
Уд. вес, г/см3........................................ 1,18
Предел прочности на изгиб, кг/см3................. 800—1400
Предел прочности на разрыв, кг[см2:
при — 40 ...................................... 990
, +20 .......................................... 790
Предел прочности на сжатие, кг/см2 ............... 1200—1600
Уд. ударная вязкость, кг-см [см2.................. 1>—20
Модуль упругости, кг[см2...........................32 000—40 000
Твердость по Моссу................................... 2—3
Твердость по Бринелю, кг/мм2........................... 20
Водостойкость (привес в воде при 20° за 24 часа), % 0,17
Теплостойкость по Мартенсу, °C......................... 80
» . Вику, °C......................... 100—120
Теплопроводность, кал[см сек - °C................ 4,4 • 10~*
Коэффициент теплового расширения.................. 8,2- 10-5
Теплоемкость, кал[г-°С............................ 0,343
Применение полиакрилатов
341
Коэффициент преломления, Лд.......................... 1,49
Прозрачность для видимого света (1=767—385), % • 90,99
Среднее пробивное электрическое напряжение, кв/мм 25
Внутреннее электрическое сопротивление, Q......... 1012
Уд. объемное электрическое сопротивление, Q-см . . 2—3- 10та
Диэлектрическая постоянная при 103 гц............. 3—3,6
Тангенс угла диэлектрических потерь при Ю3 гц . . 0,02—0,03
Применение полиакрилатов
Полиметилметакрилат широко применяется в различных областях техники
и быта. Так, им пользуются для остекления самолетов, в качестве предохрани-
тельных стекол для аппаратов и приборов, часового и оптического стекла, пресс-
порошка для изготовления технических изделий и предметов широкого потребле-
ния, для производства протезов (зубы, глаза и др.), изготовления медицин-
ских приборов.
Кроме «жестких» полимеров и сополимеров метилметакрилата, которые при-
меняют в качестве органического стекла и пресскомпозиций, большое значе-
ние имеют также и «мягкие», каучукоподобные полимеры, перерабатываемые
в виде пленок, оболочек, грунтовых составов и т. д. «Мягкие» материалы мо-
гут быть получены на основе полимеров как метакриловых, так и акриловых
эфиров.
Из числа «мягких» полиметакриловых эфиров следует указать на полибу-
тилметакрилат. Он отличается высокими адгезионными свойствами, хорошей
растворимостью в ароматических углеводородах, эластичностью пленок; его
используют в производстве художественных красок, для пропитки мрамора,
керамики и других материалов, как составную часть многих лаков, для обра-
ботки кожи, а также в производстве пленок для пищевой и фармацевтической
промышленности.
Из «мягких» полимеров акриловых эфиров нашли применение полиэтил-,
пропил- и бутил акрилаты, а также сополимеры акриловых эфиров с метилмет-
акрил атом.
«Мягкие» акриловые полимеры получают главным образом методом эмуль-
сионной полимеризации. В зависимости от желаемой степени «мягкости» под-
бирают соответствующие мономеры. Полученные продукты обычно не содержат
пластификаторов.
Гибкие полиакрилатные шланги, трубки и оболочки получают методом
экструзии. Известно применение полиакрилатов в качестве оболочек для кабеля;
преимущество заключается в том, что они не содержат летучего пластификатора,
обладают высокой маслостойкостью, стойкостью к действию атмосферных усло-
вий н озона.
Большое значение приобретают акриловые водные дисперсии типа латекс
для пропитки ткани, дерева, бумаги, обработки кожи и т. д. Они нашли
применение в производстве липких медицинских пластырей, для получения
немнущихся тканей и в качестве композиции при производстве искусственной
кожи. Значительный интерес эти дисперсии представляют для пропитки бумаж-
ной массы в ролле, получения картона, который применяют в производ-
стве искусственной кожи. Они используются также как защитные лаки для
металла и дерева. Благодаря совместимости с нитро- и ацетилцеллюлозой они
входят в состав целлюлозных лаков, увеличивая их адгезию, водостойкость
и стойкость к атмосферным влияниям. Наконец, акриловые дисперсии нашли
применение в строительной технике для придания водонепроницаемости бетону,
в качестве грунтовки при внутренней окраске стен, пропитки пористых строи-
тельных материалов и т. д.
342 Гл. VIII. Пластмассы на основе полимеров этиленкарб. кислот
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИМЕРОВ
АЛЛИЛОВЫХ ЭФИРОВ
(Аллилопласты)
Полимеры аллиловых эфиров («аллиловые смолы») представ-
ляют собою тип термореактивных полимеризационных пластиков.
Впервые аллиловые эфиры были синтезированы в 1855 г.
Н. Н. Зининым, который указал на склонность этих соединений к
полимеризации. Техническое применение аллиловых эфиров стало
возможным лишь в конце тридцатых годов, так как в этот период
аллиловый спирт стал доступным соединением, образующимся в ка-
честве побочного продукта при утилизации кубовых остатков в ле-
сохимическом производстве и при химической переработке газов
крекинга нефти.
Первые сведения о технических продуктах на основе аллиловых
смол относятся к 1942 г.
Для производства аллиловых смол применяют главным образом
диаллиловые эфиры двухосновных кислот (фталевой, адипиновой,
себациновой, малеиновой и др.):
/СООН /СО—О—снг—сн=сн2
R< + 2НОСН2— СН=СН2 —> R<
чсоон хсо—о—сн2—сн=сн2
Ведутся работы по изучению полимеров триаллиловых эфиров
трехосновных кислот, а также моноаллиловых эфиров с ненасыщен-
ными кислотами.
Диаллиловые эфиры представляют собою тетрафункциональные
соединения, содержащие две ненасыщенные группы и способные,
следовательно, к образованию пространственного полимера.
Синтез аллиловых эфиров может быть осуществлен прямой эфи-
ризацией соответствующих кислот аллиловым спиртом при нагре-
вании в присутствии серной кислоты, разбавителей или раствори-
телей, взаимодействием хлористого или бромистого аллила с солями
кислот, реакцией переэфиризации, исходя из других эфиров, а также
взаимодействием ангидридов и галоидангидридов кислот с аллило-
вым спиртом.
Полимеризация аллиловых эфиров протекает сравнительно мед-
ленно, причем можно установить две фазы процесса: вначале обра-
зуется линейный, ненасыщенный полимер, и лишь по достижении
определенной величины конверсии начинается процесс образования
пространственного полимера, связанный с раскрытием вторых не-
насыщенных групп. Так, работами Б. Н. Рутовского на примере
полимеризации триаллиловых эфиров было показано, что желати-
низация и, следовательно, образование пространственного полимера
ироисходят лишь после того, как содержание полимера в полиме-
ризующейся смеси достигает 25—28%.
Пластмассы на основе полимеров аллиловых эфиров
343
Зависимость точки желатинизации от степени полимеризации тетрафункцно-
иальных соединений определяется согласно статистическим расчетам Флори:
— 1
~ 7>—1 ’
где: х — доля полимеризованных винильиых групп при наступлении желати-
низации; Р — средняя степень полимеризации.
Это уравнение, выведенное для соединений, в которых активность обеих
винильиых групп в процессе полимеризации одинакова, связывает наступление
желатинизации с обратной зависимостью от длины образующихся цепей. Однако,
если первая винильная группа вошла в состав цепи, то вторая не имеет той
степени свободы, какую она имела в мономере, и, следовательно, ее активность
всегда будет меньше.
Высокий процент превращения до наступления желатинизации, наблюдаемый
при полимеризации аллиловых эфиров, указывает как на малую длину образую-
щихся цепей (степень полимеризации Р). так и на существенное уменьше-
ние активности второй винильной группы мономера, вошедшего в состав расту-
щей цепи.
Таким образом, общая схема полимеризации складывается из
двух последовательных процессов: образования ненасыщенного ли-
нейного полимера (/) и пространственного насыщенного поли-
мера (2):
... — СН—СН2----СН--------СН2— ...
I
СН2—О—СО сн2—о—со
I I
R R —►
I I
сн2—о—со сн2—о—со
Ан=сн2 сн=сн2
1
... —сн----сн2—
сн2—о—со
I
R
сн2—о—со
... —сн—сн2—...
2
---СН—сн2—...
I
СН2—О—со
I
R
сн2—о—со
. —сн—сн2—,..
Полимеризация аллиловых эфиров инициируется перекисями,
например перекисью бензоила, и проводится обычно при постепенно
возрастающей температуре (от 70 до 120°). В первой стадии обра-
зуется растворимый, сиропообразный продукт, представляющий со-
бой раствор линейного полимера в мономере. Если прервать про-
цесс полимеризации (например охлаждением смеси), то полученная
вязкая жидкость способна долгое время храниться без заметного
увеличения вязкости. Вторая стадия полимеризации — образование
предельно твердого, неразмягчающегося пространственного поли-
мера — идет при дальнейшем нагревании.
344
Гл. VHI. Пластмассы, на основе полимеров этиленкарб. кислот
Протекание реакции полимеризации аллиловых эфиров в две
фазы имеет существенное значение для технического использования
полимеров. Образующиеся в первой фазе сиропообразные жидкие
полимеры используют как для получения литых чистых смол (орга-
нических стекол), так и наполненных прессматериалов.
Для получения органических стекол формы заливают жидкими
полимерами первичной полимеризации, иногда дополнительно вно-
сят инициатор и при соответствующем температурном режиме (100—
120°) в течение многих часов проводят вторую стадию полимериза-
ции. Применение для этих целей жидких полимеров более целесо-
образно, чем мономеров.
Аллиловые полимеры — прозрачные неплавкие неразмягчаю-
щиеся теплостойкие стекла. Преимущество таких стекол (по срав-
нению с полиметилметакрилатными) заключается в их значительно
большей теплостойкости, стойкости к растворителям, поверхностной
твердости, стойкости к абразивным воздействиям, например, к шли-
фующему действию пыли. Однако, несмотря на эти существенные
преимущества, аллиловые полимеры в качестве органических стекол
имеют значительно более ограниченное применение, чем полиметил-
метакрилатные ввиду крайне незначительной способности к пла-
стической или высокоэластической деформации вплоть до темпера-
тур. близких к деструкции (до -}-250°). Это полностью устраняет
возможность их переработки методами формования, прессования
и т. д.
Вместе с тем способность к формованию является весьма цен-
ным свойством органических полиметилакрилатных стекол, позво-
ляющим изготовлять пространственно изогнутые изделия, например,
стеклянные куполы и др.
Таким образом, структура аллиловых смол, характеризующаяся
густой пространственной сеткой, обусловливает как их основные
преимущества — более высокую твердость, теплостойкость, нерас-
творимость, неплавкость, так и их недостатки — неприменимость
пластицирующих методов переработки.
Ниже приведены показатели некоторых свойств аллиловых сте-
кол (для сравнения в скобках даны значения для полиметил-
метакрилатных стекол):
Уд. вес, г/см3.....................................
Предел прочности на разрыв, кг'см-.................
Уд. ударная вязкость, кг • см/слА..................
Модуль упругости при изгибе, кг/см2................
Твердость по Моссу.................................
Теплоемкость, кал]г- °C............................
Теплостойкость по Вику, °C .........................
Теплостойкость (максимально допустимая температура
без нагрузки):
продолжительное действие........................
действие в течение 1 часа ...................
j 31_। 3g
350—450’ (600—800)
5—12(15—20)
30 000—40 000
3-3,5 (2—3)
0,55
150(100-120)
100° 60’)
>150° (~ 100°)
Пластмассы на основе полимеров аллиловых эфиров
345
Водостойкость, °/0 привеса:
за 24 часа в воде.................................. 0,2—0,4
за 7 суток „ „ .............................. 0,5—0,8
Теплопроводность, кал/см сек °C................. 5 • 10-4
Коэффициент теплового расширения.................. 1,07—1,09- 10~*
Прозрачность, для видимого света, %............... 90—92
Значительно большее применение, чем литые аллиловые
смолы, имеют слоистые материалы на основе этих смол (см. также
стр. 469).
Слоистые материалы получают путем пропитки наполнителей
(хлопчатобумажной ткани, древесного шпона, стеклоткани и т. п.)
жидкими, вязкими продуктами первичной полимеризации.
Пропитанная ткань или древесный шпон обладают значитель-
ной липкостью и могут служит материалом для формования путем
последовательного наложения слоев на формуемую модель. По
окончании формования деталь под небольшим давлением подвер-
гают вторичному процессу полимеризации при 90—120° до получе-
ния твердого, неплавкого и нерастворимого продукта — простран-
ственного полимера.
Особенностью технологии формования и «отверждения» алли-
ловых пластиков по сравнению со слоистыми поликонденсацион-
ными пластиками (стр. 478) является то, что процесс протекает без
выделения каких-либо летучих веществ, так как жидкая смола не
содержит растворителя, а процесс полимеризации, в отличие от по-
ликонденсации, не сопровождается выделением воды или других
простых соединений.
Следует указать, что до применения аллиловых смол слоистые
термореактивные пластики изготовлялись только из поликонденса-
ционных смол. В процессе горячего прессования последних про-
текала реакция поликонденсации с выделением воды. Для устране-
ния образования вздутий в прессуемом изделии применяли высокое
давление (~100 кг/см2) до момента возможно полного отвержде-
ния смолы. Высокое же давление при прессовании обусловли-
вало применение стальных прессформ и прессов большой мощ-
ности. Для изготовления изделий больших габаритов (например,
лодок, фюзеляжей самолетов и др.) прессование в стальных
прессформах с помощью прессов является малоэффективным и
часто технически невыполнимым. Таким образом, из слоистых по-
ликонденсационных пластиков могли быть изготовлены лишь изде-
лия ограниченных габаритов. При этом изготовление простран-
ственно емких изделий и изделий сложной конфигурации во многих
случаях было вообще невозможным или требовало сложной пред-
варительной сборки заготовок и выкроек и укладки их в пресс-
форму (стр. 479). Поэтому применение полимеризационных термо-
реактивных аллиловых смол, отверждение которых не сопрово-
ждается выделением каких-либо летучих веществ, позволило формо-
вать слоистые аллилопласты при малых давлениях (1—5 кг/см2),
346 Гл. VIII. Пластмассы на основе полимеров этиленкарб. кислот
достаточных, однако, для склейки соприкасающихся слоев. Суще-
ственное значение при этом имеют липкость и вязкость первичных
продуктов полимеризации, делающие возможным склеивание слоев
наполнителя при обычной температуре в процессе предваритель-
ного формования.
Возможность формования слоистых пластиков при малых давле-
ниях позволила разработать новые методы формования изделий из
слоистых пластиков без применения стальных прессформ и прессов,
а именно автоклавно-вакуумные методы с применением резинового
мешка. Они позволяют изготовлять изделия практически любых
габаритов, а также пространственно сложные изделия глубокой и
обтекаемой формы (например, для самолетных конструкций, для
лодок, кузовов и т. п.).
Основы изготовления изделий методом формования при низких
давлениях крайне просты. Пропитанные вязкими аллиловыми смо-
лами слои ткани или древесного шпона укладывают на внешнюю
или внутреннюю поверхность формы, соответствующей конфигура-
ции изготовляемого изделия, и уплотняют (склеивают) вручную,
пользуясь соответствующими шаблонами. Число слоев в каждой
части формы должно отвечать толщине изделия. Формы изготовляют
из твердых пород дерева, слоистых пластиков (фенодревослой и др.),
цветных металлов, гипса, цемента ит. д. После наложения и склейки
слоев форму помещают в резиновый мешок, из которого затем вы-
качивают воздух (если слои нанесены на внешней поверхности
формы); резиновый мешок при этом прижимает пластичные слои к
форме. Для того, чтобы масса не прилипала к мешку, изделие, со-
прикасающееся с мешком, покрывают целлофаном.
Отверждение изделия ведут без дополнительного применения
внешнего давления или же, наоборот, с применением — путем вне-
сения формы в автоклав, в котором создают давление 2—10 ат.
Обогрев в автоклавах осуществляют сжатым горячим воздухом,
паром или водой, которые непосредственно накачивают в автоклав;
применяют также электрический или паровой обогрев стенок авто-
клава.
Отверждение ведут в течение многих часов при ступенчатом
подъеме температуры от +70° до 120°. По окончании процесса
отверждения изделие охлаждают под давлением в автоклаве, после
чего выгружают из автоклава и снимают резиновый мешок. Полу-
чают прочное изделие с твердой поверхностью.
Если листы массы уложены во внутренней поверхности формы,
давление создается внутри резинового мешка с помощью пара, го-
рячей воды или сжатого воздуха. Давление в этом случае не может
превышать 4—5 ат.
Конструкция крупных изделий, изготовленных методом формо-
вания с помощью резинового мешка, носит иногда название «скор-
лупкой» конструкции. Метод формования «скорлупных» конструк-
ций называют не совсем точно контактным методом прессования.
Пластмассы на основе полимеров аллиловых эфиров
347
Изделия, полученные формованием при низких давлениях, все же
не обладают той плотностью и прочностью, которую они имели бы
при формовании при более высоких давлениях, однако преимуще-
ства в возможности изготовления крупных и пространственно емких
изделий без прессформ и прессов столь значительны, что приходится
мириться с небольшим снижением прочности, которое иногда
удается компенсировать увеличением толщины изделия.
Схема автоклавно-вакуумного производства слоистых пластиков
представлена на рис. 133.
Полосы материала 4, пропитанные смолами, набирают в несколько слоев
на шаблон (форму) 1, состоящий из брусков 2. Отдельные секции шаблона
снабжены отверстиями 3 для от-
сасывания воздуха. Шаблон с на-
бранным материалом обтягивают
резиновым мешком 5, предвари-
тельно свернутым в рулон 7, и
через отверстия 3 откачивают воз-
дух. Для закрепления рубашки
служит накладка 6. Весь набор с
формой помещают в автоклав 8.
Нагревание формы производят по-
средством пара, горячего воздуха
или электроэлемеитами.
Ценным усовершенство-
ванием слоистых аллило-
пластов явилось применение
в качестве наполнителей
стеклянной ткани и стеклян-
ного волокна. Их преимуще-
ство заключается в высокой
термостойкости, малой гигро-
скопичности и, что является
наиболее важным, в высо-
кой удельной прочности.
По комплексу физико-ме-
ханических свойств слоистые
аллилопласты близки к слои-
стым термореактивным КОН- Рис. 133, Схема изготовления слоистых
денсационным пластикам пластиков автоклавно-вакуумным методом,
(по твердости, теплостойко-
сти и т. д.). Они представляют собой по существу новый термореак-
тивный тип полимеризационных материалов, отличающихся столь
же густой пространственной связью и столь же высокой теплостой-
костью, как фенопласты, аминопласты и т. п. Однако, в отличие от
поликонденсационных пластиков, они при отверждении не выделяют
воды или других побочных продуктов, которые в значительной мере
остаются в прессованных изделиях и ухудшают их физико-механи-
ческие и диэлектрические свойства.
.348 Гл. VIII. Пластмассы на основе полимеров этиленкарб. кислот
Механические свойства аллилопластов так же, как и других слои-
стых пластиков, определяются в основном характером и составом
слоистой основы, удельной прочностью элементарного волокна, тол-
щиной ткани, соотношением количества нитей в основе и в утке и,
наконец, содержанием связующего в массе.
Для аллилопластов наибольшая прочность достигается приме-
нением стеклянной ткани.
Прочность в этом случае тем больше, чем тоньше элементарное
волокно, чем тоньше ткань и чем меньше содержание связующего.
Необходимость, однако, получения однородного материала с равно-
мерным распределением связующего на поверхности наполнителя,
а также с определенной степенью пластичности и вязкости мате-
риала в процессе, формования, приводит к оптимальному содержанию
смол в пределах 30—40%.
Средние свойства пластика на основе стеклянной ткани могут
быть представлены следующими показателями:
Предел прочности на разрыв, кг/см2.................. 1800—2500
Предел пропорциональности, кг/см2................... 1400—2300
Модуль упругости при растяжении, кг/см2.............1,2—1,4 • 105
Предел прочности на сжатие, кг/см2 .................. 1600—2200
Предел прочности на срез, кг1см2 ................... 800—1000
Теплостойкость по Мартенсу.......................... 135—150°
Благодаря особому сочетанию их свойств аллиловые полимеры
все в большей степени привлекают к себе внимание исследователей.
Изучаются полимеры на основе разнообразных органических и даже
неорганических кислот. Наличие трех двойных связей обусловливает
более густую пространственную связь и более высокую теплостой-
кость.
Представляют интерес также и триаллиловые эфиры борной кис-
лоты, получаемые при эфиризации борной кислоты с аллиловым
спиртом в присутствии серной кислоты:
/ОН /ОСН2—сн=сн2
В—ОН 4- зноен,—сн=сн, —> в—осн2— сн=сн,
4 он ^осн,—сн=сн2
Можно ожидать, что неорганическая основа такого эфира
должна определить более высокую теплостойкость полимера.
Следует также упомянуть об особом классе аллиловых смол,
получаемых полимеризацией аллиловых эфиров ненасыщенных кис-
лот, содержащих реакционноспособную виниловую группу, напри-
мер, аллиловых эфиров акриловой и метакриловой кислот, а также
эфиров малеиновой кислоты и т. п. Высокая функциональность этих
эфиров дает возможность использовать их в качестве компонента
при сополимеризации с виниловыми соединениями для получения
сшитых виниловых структур.
Пластмассы на основе полимеров аллиловых эфиров
349
Большой интерес представляет предложенное С. Н. Ушаковым
применение для целей гетерополимеризации с другими ненасыщен-
ными соединениями ацеталей аллилового спирта (диаллилформаля,
этилаля, бутираля и др.) - Эти ацетали неспособны к полимеризации
ни по радикальному, ни по ионному механизму, но легко образуют
сополимеры сшитой структуры с полярными мономерами (винил-
ацетатом, акрилатами и др.).
Применение аллиловых эфиров для сополимеризации с монови-
пильными соединениями (акриловыми эфирами и др.) представляет
сравнительно новую область исследования.
В результате сополимеризации могут образоваться нераствори-
мые пространственные сополимеры. Желатинизация может насту-
пить при весьма малой концентрации аллиловых эфиров. Чем
больше степень полимеризации сополимера, чем выше активность
аллиловых эфиров по отношению к радикалу моновинильного мо-
номера, тем при меньшей концентрации произойдет образование
пространственного сополимера.
Изучение систем, в которых концентрация моновинильных
мономеров значительно превосходит (обычно в десятки раз) кон-
центрацию аллиловых эфиров, имеет своей целью увеличение
теплостойкости или поверхностной твердости моновинильных поли-
меров (например полиметилметакрилата) путем создания редкой
пространственной молекулярной сетки. При этом, однако, при-
ходится считаться с уменьшением пластических и высокоэластиче-
ских свойств и, соответственно, с ухудшением условий переработки
(формования) сополимера.
ЧАСТЬ HI
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ СИНТЕТИЧЕСКИХ СМОЛ
ПОЛУЧАЕМЫХ ПОЛИКОНДЕНСАЦИЕЙ И СТУПЕНЧАТОЙ
ПОЛИМЕРИЗАЦИЕЙ
ГЛАВА IX
СМОЛЫ, ПОЛУЧАЕМЫЕ НА ОСНОВЕ ФЕНОЛОВ
И АЛЬДЕГИДОВ
(Феноло-альдегидные смолы)
Феноло-альдегидные смолы и пластмассы на их основе (фено-
пласты) являются первыми синтетическими пластиками и относятся
к одному из важнейших классов пластмасс. Несмотря на значитель-
ное развитие промышленности других синтетических смол (в част-
ности полимеризационных), фенопласты сохраняют свое значение
в современной технике синтетических пластмасс, причем из года в
год наблюдается расширение областей их применения.
Фенопласты — важнейшие заменители цветных и черных метал-
лов; они завоевали ряд областей техники, в которых эффективность
их применения столь велика, что они не могут быть заменены дру-
гими материалами. Некоторые области машиностроения (автопро-
мышленность, электротехника и др.) немыслимы без применения
феноло-альдегидных пластмасс.
УСЛОВИЯ ПОЛУЧЕНИЯ НОВОЛАЧНЫХ И РЕЗОЛьных
ПРОДУКТОВ КОНДЕНСАЦИИ
Феноло-альдегидные смолы получаются при взаимодействии фе-
нолов (собственно фенола, крезолов, ксиленолов и др.) с аль-
дегидами (формальдегидом, ацетальдегидом, фурфуролом, масля-
ным альдегидом, бензальдегидом и др.) в присутствии кислых или
щелочных катализаторов, а при известных условиях — и без них
(в этом случае катализаторами служат соответствующие примеси
в исходном сырье кислоты и основания).
Взаимодействие фенолов с альдегидами представляет собой
реакцию поликонденсации, условием протекания которой является
полифункциональность реагирующих молекул (реакция типа 2,2
или 2,3 и т. д., стр. 29). Реакция поликонденсации — ступенчатая.
На каждой ступени образуются относительно стабильные про-
межуточные продукты, которые, однако, способны к дальнейшему
взаимодействию с получением новых стабильных веществ и
с выделением простых молекул (Н2О, NH3, НС1 и др.).
Поликонденсация фенолов с альдегидами ведет к образованию
Условия получения новолачных и резольных продуктов конденсации 351
прочных карбоцепных связей; реакция характеризуется большим
значением константы равновесия (К ~ 10 000) и, следовательно,
практически протекает в одном направлении — в сторону образова-
ния полимера (стр. 63); выделяющиеся же при реакции продукты
(НгО) незначительно влияют на равновесие и на скорость процесса,
и реакцию можно вести даже в водной среде. Вследствие этого ки-
нетика и степень поликонденсации в большей мере зависят от кон-
центрации катализатора, температуры и времени, чем от удале-
ния побочных продуктов реакции.
Характер и технические свойства получаемого продукта поликон-
денсации определяются, в первую очередь, химической структурою
реагирующих молекул, их величиной, функциональностью и сте-
пенью полярности.
Из одноатомных фенолов только в феноле, л<-крезоле и 1,3,5-кси-
леноле имеются три реактивные точки (*):
zt-крезол о-крезол фенол
лг-крезол
1,3,5-ксиленол 1,2,4-ксилеиол
' Другие фенолы, например, n-крезол, о-крезол, а также
некоторые изомерные ксиленолы — бифункциональны, а 1,2,6- и
1,2,4-ксиленолы — монофункциональны. При этом считают, что ги-
дроксильные группы фенолов не участвуют в реакции поликонден-
сации и что метильные группы не активируют соседние водород-
ные атомы бензольного ядра. Хотя эти предпосылки и не абсолютно
действительны, все же для практических целей можно исходить из
вышеуказанного разделения фенолов на три-, би- и монофункцио-
нальные.
При взаимодействии альдегидов, имеющих реактивность 2,.с фе-
нолами с реактивностью 2 или 3 могут быть получены либо по-
стоянноплавкие и растворимые — термопластичные смолы (при
реактивности 2,2), либо смолы, способные к переходу в неплавкое и
нерастворимое состояние — термореактивные смолы (при реактив-
ности 2,3). В случае недостатка альдегидной компоненты и при
pH < 7 термопластичные смолы получают и при реактивности 2,3.
Следовательно, при взаимодействии формальдегида с о- и «-кре-
золами, а также со всеми ксиленолами, за исключением 1,3,5-кси-
ленола, т. е. при реактивности 2,2 при любых соотношениях компо-
нентов получаются только термопластичные смолы; при взаимодей-
ствии же формальдегида с фенолом, .и-крезолом и 1,3,5-ксиленолоМ
352
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
могут быть получены как термореактивные смолы, так и термопла-
стичные (при недостатке формальдегида и в кислой среде).
Из альдегидов только формальдегид способен при взаимодей-
ствии с трифункциональными фенолами давать термореактивные
смолы; другие альдегиды гомологического ряда формальдегида
по мере увеличения длины молекулярной цепочки все в меньшей
степени и даже совершенно неспособны образовывать простран-
ственные полимеры. Исключением до некоторой степени являются
ненасыщенные альдегиды — фурфурол, акролеин и др.
Если термопластичные смолы реагируют с избытком формаль-
дегида, то они могут перейти в термореактивные смолы (резол,
см. ниже) или непосредственно в пространственный полимер. Однако
эту способность проявляют только те термопластичные смолы, ко-
торые были получены на основе смесей фенолов, содержащих и
трифункциональные фенолы. Смолы, полученные на основе только
бифункциональных фенолов, даже при избытке формальдегида
остаются термопластичными.
Способность формальдегида при взаимодействии с трифункцио-
нальными фенолами образовывать термопластичные или термореак-
тивные смолы зависит от избытка альдегида или фенола и от ха-
рактера катализатора (концентрации водородных ионов). Кислые
катализаторы при недостатке формальдегида способствуют образо-
ванию только термопластичных смол, тогда как щелочные при
весьма широких соотношениях фенола и формальдегида образуют
только термореактивные смолы, причем в случае недостатка форм-
альдегида часть фенола остается растворенной в смоле («свободный
фенол»).
Таким образом, в зависимости от функциональности исходного
фенольного сырья, характера альдегидного компонента, соотноше-
ния альдегида и фенола и характера катализатора образуются два
типа продуктов поликонденсации фенолов с альдегидами: 1) термо-
реактивные смолы, способные при нагревании переходить в неплав-
кое и нерастворимое состояние (в пространственные полимеры), и
2) термопластичные смолы — постоянно плавкие и растворимые, не
отверждающиеся при нагревании.
Термореактивные смолы в исходном плавком и растворимом со-
стоянии называют резолами, или смолой в стадии А. Резолы пред-
ставляют собою нестабильные продукты реакции; в зависимости от
температуры они с большей или меньшей скоростью переходят в
конечное, неплавкое и нерастворимое состояние. Скорость этого
перехода—скорость образования пространственных связей — назы-
вают скоростью отверждения смолы.
Переходу в состояние полного отверждения и нерастворимости
предшествует стадия перехода в промежуточное состояние, которое
характеризуется потерей плавкости и растворимости и наличием
высокоэластического, резинообразного состояния (при нагревании)
и значительной набухаемостью в растворителях. В такой промежу-
Условия получения новолачных и резольных продуктов конденсации 353
точной сталии эти смолы называют резитолами, или смолами в
стадии В.
Конечная стадия поликонденсации характеризуется неплав-
костью и нерастворимостью смолы, неспособностью ее размяг-
чаться при нагревании и набухать в растворителях. В этой ко-
нечной стадии смолы называют резитами, или смолами в ста-
дии С.
Термопластичные смолы известны под общепринятым назва-
нием — новолачные смолы.
Резолы получаются:
1) при взаимодействии альдегидов с трифункциональными фе-
нолами или фенольными смесями, содержащими трифункциональ-
ные фенолы;
2) при конденсации трифункциональных фенолов только с форм-
альдегидом (или с ненасыщенными альдегидами);
3) при соотношениях фенола и формальдегида, близких к экви-
молекулярным, или при избытке формальдегида (в кислой среде).
Новолаки получаются:
1) при взаимодействии альдегидов как с трифункциональными,
так и с бифункциональными фенолами; в случае же применения
трифункциональных фенолов и формальдегида только при избытке
фенольной компоненты;
2) при конденсации фенолов как с формальдегидом, так и с
другими альдегидами;
3) при избытке фенола; в случае применения бифункциональных
фенолов также при избытке формальдегида.
Новолачные смолы получаются в основном при кислых ка-
тализаторах. При избытке формальдегида, однако, кислый ката-
лизатор может привести к получению резитов. Щелочной катализа-
тор обусловливает получение резольных смол при взаимодей-
ствии триреактивных фенолов с формальдегидом даже в том
случае, если в реакционной смеси имеется избыток фенольного ком-
понента.
Весьма важно то обстоятельство, что оба состояния (новолачное
и резольное) могут быть обратимы. Так, например, если новолач-
ную смолу обработать избытком формальдегида, например в виде
гексаметилентетрамина, и заменить кислый катализатор щелочным,
из новолака можно получить резол или непосредственно резит. Эта
способность новолаков переходить в термореактивные смолы при до-
бавлении избытка альдегида весьма эффективно используется в
технике производства фенопластов. Следует, однако, указать, что
такой переход возможен только в том случае, если при производстве
новолака были применены трифункциональные фенолы. С другой
стороны, резолы, а в некоторых условиях даже резиты могут стать
термопластичными (новолачными) смолами при обработке их из-
бытком фенола.
23 Зак. 1269. Э. И. Барг
354
Гл. IX. Смолы, получаемые на основе фенолов и альдегадов
Все приведенные условия могут быть представлены следующей
схемой (рис. 134).
Фенол +
Альдегид
4
Новолак
t 4
При добавлении избытка форм-
альдегида (если новолак полу-
чен на основе трифункциональ-
ных фенолов)
------------------------------->
При добавлении избытка фенола и переводе
щелочной реакции в кислую
Резол
4
Резитол
4
Резит
Рис. 134. Схема предпочтительного влияния отдельных
факторов на образование новолака или резола.
СТРОЕНИЕ ФЕНОЛО-АЛЬДЕГИДНЫХ СМОЛ
Несмотря на то, что феноло-альдегидные смолы производятся
уже более сорока лет, — до последнего, времени их химическая при-
рода и процессы образования не были в достаточной мере устано-
влены.
В продолжение ряда лет выдвигались различные гипотезы и тео-
рии, большинство которых не базировалось на какой-либо суще-
ственной экспериментальной основе. Лишь в конце тридцатых годов
работами А. А. Ваншейдта, Кетнера и др. были положены теорети-
ческие основы строения новолаков и резолов, базирующиеся на
большом экспериментальном материале.
Химическая структура, новолачных смол
35-5
Химическая структура новолачных смол
Для установления механизма получения и химической природы новолач-
ных продуктов поликонденсации имеют значение следующие экспериментальные
данные:
1. При взаимодействии фенола с формальдегидом в кислой среде образуется
4,4'-диоксидифенилметан:
НО-./ \+СН,О + / >-он
НО—:
—сн,—.
и одновременно также 2,4'- и 2,2/-диоксидифенилметан.
Эти соединения были выделены из продуктов кислой конденсации фенолов
с альдегидами. На этом основании Рашиг предположил, что новолак представляет
собою смесь всех трех изомерных диоксидифенилметанов. Однако, как доказал
А. А. -Ваншейдт, даже при десятикратном
избытке фенола по отношению к формальде-
гиду в результате конденсации образуются
не только диоксидифенилметаны, а одно-
временно и более сложные вещества.
2. По мере уменьшения молярного от-
ношения фенола к .формальдегиду уве-
личивается молекулярный вес новолака
(рис. 135).
Как видно из рис. 135, наименьший
молекулярный вес (200), соответствующий
диоксиднфенилметану, не достигается и при
весьма значительном избытке фенола.
3. Новолаки представляют собой поли-
мергомологическую смесь с значительной
полимолекулярностью, т. е. с большим раз-
бросом значений молекулярных весов от-
дельных фракций. Так, при фракциониро-
вании при помощи дробного осаждения во-
дой спиртового раствора новолака с моле-
кулярным весом 640 получается ряд фрак-
ции с мол. вес. от 200 до 1300 (что соот-
Рис. 135. Зависимость молекуляр-
ного веса новолака от молярного
отношения фенола к формальде-
гиду.
веютует содержанию от 2 до 12 феноль-
ных остатков), причем характер вязкости растворов этих фракций указывает на
полнмергомологическую зависимость вязкости от молекулярного веса.
4. Новолачная смола при обработке формальдегидом переходит в неплавкое
и нерастворимое состояние, однако чистый 4,4'-диоксидифенилметан не пере-
ходит в трехмерный полимер.
5. Новолак, полученный на основе о- и n-крезола и других фенолов, в кото-
рых водород какой-либо из реактивных точек (2, 4, 6) замещен неполярной груп-
пой, не переходит при обработке формальдегидом в неплавкое и нерастворимое
состояние.
6. Работами А. А. Ваншейдта доказано, что процентное содержание ОН-групп
в новолаке. вне зависимости от молекулярного веса новолака. практически по-
стоянно и близко к содержанию ОН-групп в феноле. Это может быть объяс-
нено лишь тем, что эти группы не принимают или принимают в весьма неболь-
шой степени участие в процессе поликонденсации. Было также доказано, что
почти весь кислород в новолаках содержится в виде ОН-групп; поэтому нельзя
предполагать, что в новолаках в заметных количествах" имеются вещества
с эфирносвязанным кислородом, а также метилольные группы (т. е. феноло-
спирты).
Все эти экспериментальные данные могут быть легко объяснены, если при-
нять, что процесс образования новолака сводится лишь к связыванию молекул
фенола посредством метиленовых групп за счет ароматических ядер, гидроксильные
356
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
же группы фенолов не участвуют в этом связывании и сохраняются в пово-
лаке.
Имея в виду, что новолак представляет собою полимергомологическую смесь
продуктов поликонденсации,' общее уравнение образования новолака можно
представить в виде:
пСН,0 + (п + 1)С6Н6ОН -> Н[СвН3(ОН)СН2]пСвН4ОН Н-пН2О,
причем п в соответствии с молекулярным весом новолака обычно от 4 до 8.
Общая формула новолаков на основе различных альдегидов и фенолов будет
иметь следующий вид:
nRCHO + (п + 1)Аг'ОН—>H[Arw(OH)CHR]nAr"OH +пН2О,
где RCHO может быть любой альдегид,
а знаки " и показывают число мест
лом и соединенных с другими ядрами.
Так, структурная формула новолака
будет:
АгОН — фенол, крезол или ксиленол,
в бензольном ядре, занятых гидрокси-
ла основе и-крезола и ацетальдегида
ОН
ОН
ОН
а на основе фенола и формальдегида:
Х’
(знаком * обозначены свободные реактивные точки в бензольном ядре).
Таким образом, новолаки представляют собою смесь мезометиленполифено-
лов, являющихся членами одного и того же полимергомологического ряда.
Химическая структура резолов
Для определения структуры резолов имеют значение следующие экспери-
ментальные данные:
1. При взаимодействии фенола с формальдегидом в щелочной среде обра-
зуются предпочтительнее фенолоспирты (оксибеизиловые спирты):
ОН
ОН
Наряду с о-оксибеизиловым спиртом (салигенином) может образоваться и
n-оксибензоловый спирт, а также при избытке формальдегида ди- и триметилоль-
ные производные фенола:
ОН
ОН
^СН2ОН
^СНаОН
НОСНг/^СНаОН
И
СН2ОН
Реакция получения феполоспиртов, в частности о-оксибензилового спирта,
протекает настолько полно, что в случае небольшого избытка фенола последний
может быть полностью отогнан из реакционной смеси.
2. Химически чистые оксибензиловые спирты переходят при нагревании в не-
плавкое и нерастворимое состояние, причем смеси алкоголей (орто- и пара-)
Химическая структура резолов
357
переходят значительно быстрее. Этот процесс связан с реакцией дегидратации
н поликонденсации.
3. о-Оксибензиловый спирт (салигенин) образует в кислой среде смолоподоб-
ную смесь (салиретиновую смолу), которая при нагревании переходит в неплав-
кое состояние. Салиретиновая смола, реагируя с фенолом, образует соедине-
ния с характерной для новолаков структурой. Процесс получения салиретнновых
смол заключается в поликонденсации салигенина, с образованием монометилоль-
ных производных мезометиленполифенолов.
4. При образовании резольной смолы, даже при эквимолекулярных отноше-
ниях фенола к формальдегиду, на 1 моль прореагировавшего феиола может
связываться до 1,5 молей формальдегида.
5. Содержание кислорода в резоле примерно в 1,4 раза превосходит содер-
жание его в новолаке, что указывает не только на сохранение в резоле феноль-
ных ОН-групп, но и на наличие в нем метилольных или других кислородсодер-
жащих групп.
6. При переходе резола в резит при нагревании выделяются свыше 5% кон-
денсационной воды, что может быть объяснено лишь конденсацией метилоль-
ных групп с фенольными ядрами.
7. Замещенные (бифункциональные) фенолы при взаимодействии с форм-
альдегидом не образуют резолов.
8. Мол. вес резолов от 300—400 до 600—800 соответствует небольшому
числу фенольных ядер.
А. А. Ваншейдт установил, что при взаимодействии фенола с формальдеги-
дом в щелочной среде образуется смесь моно- и диалкоголей (а также, вероятно,
и триалкоголи), которые отщепляют воду за счет метилольных групп и водо-
родных атомов бензольных колец, образуя более сложные полифенолы, содержа-
щие метилольные группы, по схеме:
ОН г ОН он он
СН2ОН + [/Х|—СН2ОН |'/Х|—СНг—/4}—СН2ОН
'ЧСН,ОН CH^OH
Таким образом, резол можно рассматривать как смесь метилольных производ-
ных феиола и мезометиленполифенолов, причем формальдегид может быть при-
соединен в о- или «-положения к ОН-группе, и его количество будет определяться
отношением фенола к формальдегиду в исходной среде. Общая формула резола
может быть написана следующим образом:
H[C6H2(OH)(CH2OH)CH2]m[C6H3(OH)CH2]„OH.
Согласно А. А. Ваншейдту, показатель п для резолов может находиться
в пределах от 4 до 10, а число метилольных групп т— от 2 до 5.
Переход резола в резит при нагревании представляет собою процесс сшивки
молекул путем конденсации метилольных групп с атомами водорода бензольных
колец, с выделением соответствующего количества воды.
Общая схема образования резола и резита может быть представлена сле-
дующим образом:
а) образование смесн феноло-спиртов:
он он он он
+ СН2О —> СН2ОН; . ^-CHjOH ;
Х'/ ^CHjjOH 'сН2ОН
ОН
НОСН2—СН2ОН
'сн2он
б) конденсация феноло-спиртов с образованием смеси метилольных произ-
водных полифенолов (резола):
358
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
в) конденсация резольных молекул с образованием пространственного по-
лимера (резита):
Химическая структура резолов
359
Вышеприведенная теория строения и образования резола и резнта хорошо
соответствует основным экспериментальным данным и находится в согласии
с физическими и химическими свойствами продуктов. Так, плавкость резола
и растворимость в спиртах и щелочах объясняются его сравнительно неболь-
шим молекулярным весом и наличием свободных фенольных гидроксильных н
метилольных групп. Эта же схема объясняет и большее содержание кисло-
рода в резоле по сравнению с новолаком. Наконец, она объясняет тот факт, что
если реагирует фенольное производное с замещающими группами в о- или п-по-
ложении (например n-крезол), то резольная смола не может образоваться, а по-
лучается лишь смола с новолачной структурой, что видно из следующей схемы:
ОН ОН ОН
/'Ч| + СН2О —> СН2ОН; НОСН2—СН2ОН —>
Y Y Y
СН3 СН3 СН3
В последние годы (начиная с 1940 г.) появился ряд работ немецких
ских авторов, в которых делались попытки доказать, что отверждение
происходит главным образом за счет образования эфирных связей при
действии метилольных групп путем выделения воды за счет фенольных
товых гидроксилов с образованием резита следующей структуры:
ОН ОН он
и швед-
резолов
взаимо-
и спир-
СН3
Невидимому, такие реакции
щены о- или и-положения,
функциональных фенолов с
НО—>--------СН2--О
Г“
сн,—...
до известной степени могут иметь место, если заме-
однако они мало вероятны при конденсации трн-
избытком формальдегида, когда реакция вероятнее
360
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
всего идет между метилольными группами и атомами водорода бензольных
ядер с выделением воды только за счет метилольных групп и с образованием
метиленовых мостиков.
В последние годы сделаны также попытки объяснить механизм образования
твердеющих смол. Согласно Хулче, смолы, способные к отверждению, должны
иметь стабильные метилольные группы, которые лишь при нагревании могут
реагировать с образованием пространственного полимера. В тех случаях, когда
нет возможности стабилизовать метилольные группы, образуется либо непо-
средственно стадия С (резит), либо новолак — в зависимости от избытка мети-
леновых групп в реакционной среде. Устойчивость метилольных групп зависит
от особого механизма, связанного с образованием шестичленных колец, благо-
даря «протоновым мостикам» между водородом фенольной гидроксильной группы
и кислородом метилольной группы:
В тех случаях, когда образование «протоиовых мостиков» и шестичленных
колец невозможно, например в случае п-окснбензилового спирта, метилольные
группы не могут быть стабильны; они быстро распадаются с образованием
Д жидкий (резол)
А твердый (резол)
В (рвзитол)
С (резит)
Рнс. 136. Схематическое изображение струк-
туры феноло-альдегидных смол в различных
стадиях конденсации (А, В н С).
метиленовых групп или соеди-
нений типа диоксидифенилме-
таиа; при избытке же форм-
альдегида реакция идет сра-
зу до конца, — до образования
резита. От концентрации водо-
родных ионов и от соотноше-
ния альдегида и фенола зави-
сит соотношение о-метнлоль-
ных (устойчивых) производных
и и-метилольных (неустойчи-
вых) производных в продуктах
реакции, что определяет ново-
лачный или резольный харак-
тер смолы.
Вопрос о функционально-
сти замещенных фенолов в
свете ряда новых эксперимен-
тальных данных не может счи-
таться еще достаточно разра-
ботанным. Было, например, до-
казано, что о- и п-алкилфенолы
способны давать смолы, кото-
Q—частицы, закрепленные межмолекулярными'силами;
ф— частицы, закрепленные валентными (молекулярными)
силами; пунктирные кружки—свободно вращающиеся
частицы.
рые, хотя и весьма медленно и
лишь при высоких тем пер ату-
рах (выше 180°), могут, одна-
ко, переходить в неплавкое
и нерастворимое состояние.
Это же явление наблюдалось и для новолаков, полученных при конденсации 0,8 мол.
формальдегида с 1 мол. фенола при более высоких температурах (250—280°).
Эти наблюдения стараются объяснить действием одного из следующих ме-
ханизмов: 1) возможностью активации водорода в .н-положенин при высоких
температурах, если о- или n-положения замещены, и 2) участием гидроксиль-
ной группы фенолов в образовании эфирных связей. Пока иет еще достаточных
данных для того, чтобы установить, в какой мере и при каких условиях дей-
Феноло-формальдегидные смолы.
361
ствует каждый из этих механизмов. Необходимо подчеркнуть, что отверждение
новолаков на основе замещенных фенолов происходит столь медленно и при
столь относительно высоких температурах, что оно не может считаться характер-
ным для реакции резольной и новолачной конденсации. Однако в виде побочных
(вторичных) реакций в особых температурных условиях возможны медленно
протекающие реакции за счет фенольных гидроксилов, а также процессы актива-
ции атомов водорода в .«-положении, которые приводят к потере плавкости и
растворимости новолачиых смол и смол на основе замещенных фенолов.
С коллоидной и физико-химической точек зрения резольные и новолачные
смолы представляют собою изогель, т. е. смесь лиофильных, растворимых и
близких по химической структуре веществ, или, точнее, смешанный изогель. Со-
гласно рентгенографическим, оптическим и вискозиметрическим исследованиям,
резолы и новолаки представляют собою весьма разветвленные, шарообразные
глобулярные комплексы, что объясняется сильными дисперсионными силами при-
тяжения, действующими между бензольными ядрами. При переходе в резит,
т. е. при замене межмолекулярных сил притяжения валентными связями, обра-
зуется в пределе гигантский сферический комплекс шарообразной структуры.
Переход смолы из состояния А в С характеризуется тем, что фиксирован-
ные молекулы постепенно начинают преобладать над молекулами со свободным
поступательным и вращательным движениями (рис. 136).
При образовании резита многие валентные связи не могут быть осущест-
влены из-за пространственных условий и в макромолекулярном комплексе
остаются свободные метилольные группы, которые можно рассматривать как
неплотности, «дырки» в макромолекулярной решетке резита (стр. 358). С другой
стороны, значительные напряжения, которые создаются вследствие неравномер-
ного и последовательного закрепления фенольных ядер, вызывают внутримоле-
кулярные разрывы и образование микротрещии. Наличие таких неоднородностей
на поверхности резита можно непосредственно наблюдать под микроскопом на
тонких пленках (0,01 мм), например после набухания их в ацетоне.
ФЕНОЛО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
Закономерности кислой конденсации и
образования новолачиых смол
Постоянноплавкие феноло-альдегидные смолы — новолаки при-
меняют в основном для производства быстропрессующихся ново-
лачных пресспорошков и пресскомпозиций.
Возможность получения термореактивных пресскомпозиций на
основе термопластичных смол — новолаков основана на взаимодей-
ствии новолака с твердыми производными формальдегида, главным
образом с уротропином (гексаметилентетрамином) при высоких
температурах и давлениях, т. е. в условиях горячего прессования,
в результате которого термопластичная смола (новолак) переходит
в неплавкий нерастворимый продукт (резит). Таким образом, при-
менение новолачиых пресскомпозиций предполагает наличие в них
наряду с новолаком также и твердого производного формальдегида,
способного к взаимодействию с новолаком при высоких темпера-
турах.
Важнейшая характеристика новолаков, применяемых для произ-
водства пресспорошков, — их термореактивность, т. е. скорость их
перехода в резит при взаимодействии с уротропином в определен-
ных температурных условиях. Чем выше термореактивность
362
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
новолака, тем больше скорость прессования пресскомпози-
ции. ;
Термореактивностью, как указано, обладают лишь новолаки, по-
лученные на основе трифункциональных фенолов. Чем выше кон-
центрация этих фенолов в общей фенольной смеси, тем выше и тер-
мореактивность новолака. Из технических фенолов (фенол, крезолы,
ксиленолы) только фенол С6Н5ОН является полностью трифунк-
циональным веществом; крезолы и ксиленолы, представляющие со-
бою смеси изомеров, содержат значительные концентрации бифунк-
циональных соединений.
В такой же мере новолаки, полученные на основе формальде-
гида, обладают большей термореактивностью (при взаимодействии
с уротропином), чем новолаки на основе уксусного, масляного и
других насыщенных альдегидов с открытой цепью. Поэтому наи-
большее применение для пресскомпозиций нашли новолаки на
основе фенола и формальдегида, обладающие наибольшей реактив-
ностью с уротропином. Для производства лаков могут найти приме-
нение также новолаки на основе как бифункциональных фенолов,
так и других альдегидов, кроме формальдегида.
Катализаторы
Реакция поликонденсации и образования новолака катализи-
руется водородными ионами. В тех случаях, когда катализатор не
вносится, реакция катализируется муравьиной кислотой, которая
всегда имеется в техническом формалине. При pH < 7 реакция идет
в сторону образования полиметиленфенолов — новолачной смолы.
Выход смолы, условия равновесия и в значительной мере свойства
новолака, повидимому, не зависят от количества катализатора (pH),
но скорость реакции является линейной функцией концентрации во-
дородных ионов, что почти во всех случаях подтверждается на прак-
тике.
Однако химический характер катализатора сказывается не
только на его катализирующем действии, полностью определяемом
степенью его диссоциации, но влияет и на некоторые технические
свойства смолы. Следует различать катализаторы, удаляемые при
сушке из смолы, и катализаторы, остающиеся в смоле в свободном
или связанном виде. Естественно, что вторые больше влияют на
свойства смолы, чем первые. Катализаторы влияют на цветность
смолы, ее светостойкость, на процессы конденсации, сушки и термо-
обработки.
Наиболее энергичный катализатор — соляная кислота. Концен-
трация НС1 в реакционной среде должна быть от 0,1 до 0,3% (к фе-
нолу) и обусловливается как степенью кислотности (pH) техниче-
ского формалина (количества в нем муравьиной кислоты), так и
устанавливаемыми пределами pH для реакционной смеси (обычно
от 2,2 до 1,8). Реакция новолачной поликонденсации протекает
Закономерное» и образования новолачиых смол
363
со значительным выделением тепла (до 150 ккал на моль фенола),
что может привести к бурному вспениванию и выбросу реакцион-
ной смеси из реактора. Поэтому соляную кислоту рекомендуют
вводить в два—три приема. Большое преимущество этого катали-
затора, — что в процессе сушки смолы соляная кислота в основном
удаляется из реакционной смеси вместе с парами воды. Недостат-
ком же применения соляной кислоты является корродирующее дей-
ствие ее на аппаратуру.
Серную кислоту в качестве катализатора применяют в особых
случаях и сравнительно редко. Она несколько менее энергично ка-
тализирует реакцию, чем соляная кислота. Кроме того, серная кис-
лота остается в смоле; поэтому необходима последующая нейтра-
лизация, приводящая к образованию химически инертных солей
[нейтрализацию производят, добавляя Ва(ОН)2 или Са(ОН)2];
смолы же получаются более темные, чем при применении соляной
кислоты; корродирует аппаратуру; концентрация обычно 0,4—0,6%.
Щавелевая кислота как слабо диссоциированная органическая
кислота действует менее энергично и ее следует брать в соответ-
ственно больших количествах (обычно 1,5—2,5%). При этом процесс
конденсации протекает более спокойно, им легче управлять, но он
более длителен, чем при применении в качестве катализатора соля-
ной кислоты; получаемые новолаки светлее и более светостойки.
Подобно щавелевой кислоте на процесс конденсации влияют и дру-
гие слабодиссоциированные кислоты, например фосфорная. Нейтра-
лизацию этих кислот после конденсации не производят.
Муравьиная кислота всегда присутствует в техническом форма-
лине. Однако ее содержание (~0,1 %) не обеспечивает нужной ско-
рости реакции поликонденсации. Поэтому, если конденсация прово-
дится при атмосферном давлении и при температуре кипения смеси,,
необходимо добавить катализатор (кислоту) до снижения pH реак-
ционной среды от 4,5—3,5 (соответствующей смеси фенола с фор-
малином) до 1,8—2,2.
Если же проводить реакцию под давлением и при более высо-
ких температурах (в автоклавах), то поликонденсация проте-
кает с достаточной скоростью и без внесения других катализаторов
при кислотности среды, соответствующей смеси фенола с форма-
лином. Такой процесс не совсем правильно называют бескатализа-
торной автоклавной конденсацией.
Соляная кислота является основным катализатором для ново-
лаков, применяемых в производстве прессматериалов. Щавелевую
кислоту (реже фосфорную) часто используют для новолаков, иду-
щих для производства абразивов и лаков.
До и после внесения катализатора, а иногда и в процессе реак-
ции необходимо определять и контролировать pH реакционной
среды.
364
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
Соотношение компонентов
При проведении конденсации трифункциональных фенолов с
формальдегидом при pH < 7 в зависимости от соотношения компо-
нентов образуются либо термопластичная смола — новолак, либо
неплавкие, желатинирующиеся продукты.
Новолачные смолы могут быть получены только при избытке
фенольного компонента. С приближением к эквимолекулярным со-
отношениям или при избытке формальдегида образуются неплавкие
продукты.
При одинаковых условиях поликонденсации, чем больше отно-
шение формальдегида к фенолу, тем
ТАБЛИЦА 37
Характеристика новолака в зависимости
от отношения формальдегида к фенолу
Количество граммов формальдегида на 100 г фенола Выход СМОЛЫ, Температура капле- падения по Уббелоде, °C I Скорость желатиниза- ции (переход в ста- дию В при смешении с 10°/'о уротропина | при 150°), сек. Вязкость 50о'о спир- тового раствора, сп Содержание свобод- ного фенола, «/о
24 108,9 97,5 160 83 8,7
26 109,6 103,0 80 136 5,9
28 112 112,0 65 370 4,7
29 Смола переходит в стадию В
выше температура размягче-
ния образующегося новола-
ка и его средний молекуляр-
ный вес (выше вязкость рас-
творов), тем больше термо-
реактивность и выход смолы
по отношению к фенолу, тем
меньше в ней остается не-
прореагировавшего (свобод-
ного) фенола (табл. 37).
На практике максималь-
но возможное отношение
формальдегида к фенолу —
10 молей формальдегида на
12 молей фенола, или 28 г
формальдегида на 100 г фе-
нола. Оптимальным соотно-
шением, дающим возмож-
ность безопасного ведения процесса в заводских условиях и по-
лучения смолы с оптимальными свойствами, является 26,5—27,5 г
формальдегида на 100 г фенола, или приблизительно 7 молей
фенола на 6 молей формальдегида.
Влияние условий конденсации и сушки
Процесс получения новолачных смол включает стадию поликон-
денсации с образованием смолообразных первичных продуктов и
стадию сушки (термообработки) смолы. Для новолачной конденса-
ции характерной является гидрофобность первичных продуктов кон-
денсации, нерастворимость их в реакционной смеси; поэтому стадия
конденсации может быть установлена по резкому разделению
слоев. Однако и после разделения слоев конденсация может про-
должаться до желаемой степени завершения реакции. Было
установлено на обширном экспериментальном материале (А. Д. Со-
колов с сотр., И. П. Михеев и др.), что основные технические пока-
затели новолака определяются условиями конденсации и сушки.
Закономерности образования новолачных смол
365
Чем длительнее процесс поликонденсации, тем полнее связы-
вается фенол и формальдегид, тем выше средний молекулярный вес
новолака, тем больше выход смолы; это и естественно, так как про-
цесс поликонденсации протекает во времени и является ступенча-
тым, последовательным процес-
сом взаимодействия реагирующих
молекул (табл. 38).
Процесс сушки смолы ведут с
целью возможно полного удале-
ния воды и частично непрореаги-
ровавших компонентов (фенола,
формальдегида, соляной кислоты
и др.), а также для углубления и
завершения процессов поликон-
денсации при повышенных темпе-
ратурах для получения смолы с
заданными свойствами. Поэтому
процесс сушки складывается из
1) нагревания до начала кипения
смеси; 2) испарения воды при
постоянной температуре кипения
ботки — подъема температуры вь
ТАБЛИЦА 38
Характеристика новолака в зави-
симости от продолжительности
конденсации
[родолжи- ельность кон- енсации, мин. ыход смолы фенолу, о/о ф S о © Я U 6“ « о н g о m й язкость >/0 спирто- ого раствора, емпература аплепадения о Уббелоде, С
Снм CQ и <->©• CQ ю аз ч» НйВо
55 107,6 6,7 106 102
60 107,8 6,4 107 103
90 107,9 6,0 126 110
150 108,0 5,8 129 111
270 108,2 5,2 163 117
смеси; 3) термической обра-
зе температуры кипения воды
(рис. 137, стр. 366).
От продолжительности термической обработки и ее конечной
температуры зависят те свойства смолы, которые определяются ве-
личиной ее молекулярного веса: вязкость растворов, температура
размягчения и каплепадения, а также содержание свободного фе-
нола. Как уже было указано, эти показатели, в первую очередь, ха-
рактеризуются соотношением реагирующих компонентов. На рис. 138
показаны зависимости температуры каплепадения от конечной тем-
пературы сушки для новолаков с различным отношением формаль-
дегида к фенолу. Как видно из кривых, при этом наблюдается моно-
тонное увеличение температуры каплепадения по мере повышения
конечной температуры сушки, однако наиболее резко эта зависи-
мость проявляется для новолаков с более высоким отношением форм-
альдегида к фенолу. Таким образом, для каждой реакционной
смеси получаются в этом отношении характерные кривые, которые
могут быть положены в основу контроля процесса сушки смолы (в
одном и том же реакторе при одинаковых условиях подъема тем-
пературы) .
Из приведенных кривых (рис. 138) следует, что одна и та же
температура каплепадения новолака может быть достигнута:
1) увеличением отношения формальдегида к фенолу в реакцион-
ной смеси и 2) повышением конечной температуры сушки. Для до-
стижения максимальной «твердости» (температуры каплепадения)
новолака необходимо применять как предельные отношения
формальдегида к фенолу, так и наиболее высокую конечную
366
Гл. IX. Смолы., получаемые на основе фенолов и альдегидов
температуру сушки. Вместе с тем, одинаковая температура каплепа-
дения двух смол с различным отношением формальдегида к фенолу,
достигнутая применением различной
ечнои температуры сушки,
не свидетельствует еще,
что эти смолы одинаковы
по другим своим свой-
ствам, по вязкости рас-
творов и скорости отвер-
ждения (скорости желати-
низации; см. стр. 421).
Значение соотношения
компонентов выражается в
том, что для смолы с боль-
шим отношением форм-
альдегида к фенолу вяз-
кость раствора и скорость
желатинизации будут вы-
ше, чем для смолы с мень-
шим отношением форм-
альдегида к фенолу при
одинаковой температуре
каплепадения.
Как видно из табл. 39,
а также из рис. 139, вяз-
кость новолака хотя и на-
ходится в линейной зави-
симости от отношения
формальдегида к фенолу
в исходной смеси, однако
и при одинаковой вязкости
смолы могут иметь совер-
шенно различную темпе-
ратуру каплепадения, ес-
ли соотношение исходных
Конечная температура сушки, ° с
Рис. 138. Зависимость температуры каплепаде-
ния новолака от конечной температуры сушки
(термообработки) смолы при различных отноше-
ннях (в г) формальдегида к фенолу.
компонентов в реакционной смеси было различным. В такой же сте-
пени это относится и к скорости отверждения смолы (перехода но-
волака в резит), которая в основном зависит от отношения форм-
альдегида к фенолу и в значительно меньшей степени от харак-
тера термической обработки. Таким образом, для получения ново-
лачных смол с максимальной температурой размягчения необхо-
димо: 1) предельное отношение формальдегида к фенолу в реак-
ционной смеси, 2) оптимальное время конденсации и 3) термообра-
ботка после сушки до максимальной температуры.
В том случае, когда желательно получать смолы с высокой тем-
пературой размягчения, Но с малой вязкостью, необходимо рабо-
тать на пониженном отношении формальдегида к фенолу, достигая
высокой конечной температуры сушки (180° и выше). Варьируя
Закономерности о5разования новолачиых смол
367
ТАБЛИЦА 39
Соотношение между температурой
каплепадения и вязкостью дня
смол с различным отношением
формальдегида к фенолу
Температура капле- падения смолы по Уббелоде, ®С Вязкость (в СП) 50\о спиртового раствора смолы с отношением формальдегида к фенолу
24 : 100 26 : 100 28 : 100
96 83 114 —
102 92 1130 | 278
112 109 136 314
123 1 1301 141 —
соотношения компонентов, продолжительность конденсации и конеч-
ную температуру сушки, пользуясь соответствующими кривыми
(рис. 138), можно установить режим конденсации и сушки для ка-
ждого аппарата в отдельности,
обеспечивающий получение смолы
с заранее заданными свойствами.
Характер термообработки и
соотношение компонентов опреде-
ляют содержание в новолаке сво-
бодного фенола. Как было пока-
зано А. Д. Соколовым, зависи-
мость между температурой капле-
падения новолака и содержанием
в нем свободного фенола опреде-
ляется не только соотношением
компонентов, но и методом сушки.
При сушке в открытом котле
(рис. 140) наблюдается обратная
линейная зависимость между тем-
пературой каплепадения и содер-
жанием свободного фенола в смо-
ле. Полученные прямые располо-
жены таким образом, что при рав-
ной температуре каплепадения
большее содержание свободного фенола имеет смола, изготовленная
при наибольшем отношении формальдегида к фенолу. При сушке
в закрытом реакторе или в лабораторной колбе (без вакуума) полу-
Температура каплепадения,ЛС
Рис. 139. Зависимость вязкости от тем-
пературы каплепадения для смол с раз-
личным отношением формальдегида
к фенолу (сушка в колбе).
бодного фенола, в то время как при
личение молекулярного
веса происходит
чаются иные зависимости: не-
смотря на значительные изме-
нения температур каплепаде-
ния, содержание свободного фе-
нола для каждой смолы не из-
меняется, однако оно резко раз-
лично для смол с различным
отношением формальдегида к
фенолу (рис. 141). Таким обра-
зом, увеличение молекулярного
веса новолака (что косвенно
в известной мере определяется
увеличением температуры ка-
плепадения) определяется при
сушке в закрытом реакторе
(колбе) лишь процессами поли-
конденсации, а не отгонкой сво-
сушке в открытых котлах уве-
как за счет процессов
поликонденсации, так и за счет отгонки свободного фенола.
368
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
Минимальное содержание свободного фенола особенно важно
для новолаков, применяемых для лаковых целей. Для получения
таких новолаков работают на предельных количествах формальде-
СоЗержание свободного фенола^
Рис. 140. Зависимость содержания
свободного фенола от температуры
каплепадения для смол с различным
отношением фенола к формальдегиду
(сушка в открытом котле).
5 130
S 120
§
§ 110
^юо
S- 90
§
J 80
. О 2 0- 6 8 10 12
Содержание свободного фенола,%
Рис. 141. Зависимость содержания
свободного фенола от температуры
каплепадения для смол с различным
отношением формальдегида к фено-
лу (сушка в колбе без вакуума или
в закрытом реакторе).
гида и проводят возможно более глубокую конденсацию и термо-
обработку; кроме того, для удаления свободного фенола в процессе
сушки продувают смолу перегретым паром.
Действие уротропина
Новолачные смолы могут быть применены для производства
пресскомпозиций лишь в присутствии уротропина, с которым они
взаимодействуют в процессе горячего прессования (при 160—180°),
с образованием неплавких и нерастворимых продуктов (резита).
Однако механизм реакции новолака и фенола с уротропином еще
нельзя считать достаточно установленным.
Ход реакции конденсации фенола с гексаметилентетрамином
представляли следующей схемой:
ОН
I
2 (/'4| + N(CH2-N=CH2)3 —>
ОН он
I I
—> NH,-CH2— СН,—+ NH(CH,—N=CH2)2
Законоцерностги образования новолачных смол 369
ОН
+ NH(CH2-N=CH2)2
он он
I I
NH2—СНо-^—СН2— + NH2(CH2— N = CH,)
он он он
I I I
2/4j+NH2(CH2-N=CH2)~>nh2-ch2-/'^-ch2-/\+nh3
Эта реакция продолжается до тех пор, пока все метиленовые
группы не соединятся с фенольными ядрами, a NH3 не выделится
в виде побочного продукта. Однако Г. С. Петров экспериментально
показал, что при взаимодействии уротропина с фенолом выделяются
лишь 40—50% всего азота. В такой же степени часть азота остается
в смоле и при взаимодействии уротропина с новолаком как в про-
цессе горячего вальцевания новолачных пресспорошков, так и после
горячего прессования.
В табл. 40 приведен состав новолачных пресскомпозиций на раз-
личных стадиях обработки. Как видно из таблицы, новолачные
смолы после перехода в стадию резита следует рассматривать как
азотсодержащие соединения.
ТАБЛИЦА 40
Состав новолачньх пресскомгогиций на различных стадиях обработки
Стадия обработки Своб(>Д1И..Й фенол “о Уротропин ^0 Свободный аммиак %. Азот °;о
Исходный пресспорошок . 3,20 6,67 — —
После вальцевания при 85—115° 2,40 6,23 0,20
После прогревания таблеток при 180° 2,20 4,00 0,23 4,33
После прессования при 175—180° 0,60 — 2,09 4,20
24 Зак. 1269. Э. И. Барг
370 Гл. IX. Смолы, получаемые на основе фенолод и альдегидов
Закономерности щелочной конденсации и
образования резольных смол
Резольные смолы получаются только при взаимодействии трех-
функциональных фенолов, главным образом с формальдегидом.
Катализаторы
Реакция поликонденсации фенолов с формальдегидом приводит
обычно к образованию резольных смол при pH > 7, т. е.
в присутствии щелочных катализаторов. Щелочные катализаторы
определяют резольный характер смолы не только при избытке
формальдегида, но также и при избытке фенола. Непрореаги-
ровавший фенол остается в смоле в виде свободного фенола.
Важнейшие катализаторы резольной конденсации: NaOH,
Ва(ОН)2, NH4OH, иногда Na2CO3, Na2SO3 и др.
Гидрат окиси натрия представляет собой весьма энергичный
катализатор и применяется, соответственно, в небольших кон-
центрациях (~0,1%). Он обусловливает, по сравнению с дру-
гими катализаторами, большую растворимость первичных продук-
тов поликонденсации в реакционной среде и его используют в тех
случаях, когда важно тормозить процесс коагуляции смолы во
время конденсации и сушки. Он облегчает также условия получе-
ния обезвоженных (так называемых «сухих») жидких резольных
смол. Недостаток такого рода катализатора — необходимость ней-
трализации щелочи, которая остается в смоле. Присутствие свобод-
ной щелочи, естественно, значительно снижает технические свойства
смолы (цветность, водостойкость и, главным образом, диэлектриче-
ские свойства). Нейтрализацию смолы обычно производят в про-
цессе сушки, лучше всего действием органических малодиссоцииро-
ванных кислот (молочной, бензойной, щавелевой).
Гидрат окиси бария Ва(ОН)2-8Н2О является более слабым,
«мягким» катализатором и применяется, соответственно, в больших
концентрациях (1—1,5%). Он легко может быть нейтрализован, на-
пример, пропусканием углекислого газа. «Мягкость» этого катали-
затора позволяет легко управлять процессом конденсации. Присут-
ствие в резоле нерастворимых, инертных и нелетучих бариевых
солей не приводит к ухудшению химической стабильности и
диэлектрических свойств смолы. Было установлено, что присут-
ствие в смоле двухвалентных катионов бария улучшает ее диэлек-
трические свойства.
Резольно-баритовые смолы были разработаны Г. С. Петровым
и используются в производстве электротехнических изоляционных
прессизделий.
Гидрат окиси аммония (обычно применяют аммиачную воду —
25%)—наиболее распространенный катализатор для получения
резольных омол различного назначения. Он связывается, в первую
очередь, с формальдегидом и образует уротропин. Вместо аммиака
Закономерности образования резольных смол
371
фенолу. Это относительно слабо
Количество катализатора (ИН^ ОН), %
Рис. 142. Зависимость скорости конден-
сации формальдегида с фенолом от кон-
центрации катализатора.
/—35,6°/о формальдегида; 2—33,2^ формальде-
гида.
можно применять и сам уротропин. Аммиак берут в количестве от
0,5 до 3% (обычно ~1,5%) к
действующий катализатор и
позволяющий в такой же мере,'*
как и гидраты окисей двух-
валентных металлов, легко
управлять процессом конден-
сации. Недостатком примене-
ния этого катализатора являет-
ся образование вздутий в
прессизделиях при горячем
прессовании, что объясняется
выделением в процессе отвер-
ждения смолы (наряду с кон-
денсационной водой) и газооб-
разного аммиака. Резольно-ам-
миачные смолы широко приме-
няются в производстве слои-
стых масс и пресспорошков.
Соотношение компо-
нентов
Несмотря на то, что при ще-
лочной конденсации резольные
смолы получаются и при избытке фенола и что влияние соотношения
компонентов не имеет
ТАБЛИЦА 41
Характеристика резола
от отношения фенола к
в зависимости
формальдегиду
Молекулярное отно- шение фенола к форм- альдегиду Выход смолы по от- ношению к фенолу, о'(. Температура капле- падения по Уббелоде °C Скорость желатини- зации при 150®, сек. 1 Вязкость бО’.'о спирто- вого раствора, сп Содержание свобод- ного фенола, о;о
5:4 112 42 160 23,0 24,3
5:5 118 50 98 39,5 16,8
5:6 122 65 100 42,0 15,5
5:7 126 66 96 42,5 14,8
табл. 41. Скорость
такого значения, как при новолачнои конден-
сации, все же на практике
для большинства- назначений
установлен сравнительно уз-
кий диапазон отношений фе-
нола к формальдегиду, близ-
ких к эквимолекулярным. В
общем, с увеличением количе-
ства формальдегида свойства
смолы меняются в том же на-
правлении, как и при ново-
лачной конденсации, т. е. по-
вышается температура капле-
падения смолы, вязкость, ско-
рость отверждения (желати-
низации) , увеличивается вы-
ход смолы по отношению к
фенолу и уменьшается содер-
жание свободного фенола,
конденсации пропорциональна
как это видно из
содержанию в реакционной смеси как катализатора, так и формаль-
дегида (рис. 142).
24*
372 Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
Резольные смолы для пресспорошков и слоистых пластмасс
получают обычно при эквимолекулярных отношениях формальде-
гида и фенола или при небольшом избытке формальдегида, напри-
мер, на 6 молей фенола берут 7 молей формальдегида. Соотношения
компонентов часто определяются также характером катализатора и
техническим назначением резола. Резольные смолы на аммиачном
катализаторе изготовляют при соотношении 36—38 вес. ч. формаль-
дегида на 100 вес. ч. фенола; в особых случаях при других катали-
заторах (едкий ‘натр) количество формальдегида может достигать
42 вес. ч. или может быть снижено до 30—32 вес. ч. Значительный
избыток формальдегида (от 2 до 3 молей на 1 моль фенола) при-
меняют при производстве так называемых «литых смол» (стр. 396).
Влияние химического с о ста в а к омпонеито в
От характера фенола, участвующего в образовании резольной
смолы, зависят; 1) скорость самого процесса поликонденсации (ско-
рость связывания альдегида и образования резольной смолы) и
2) термореактивность смолы (скорость
Время конденсации, мин.
Рис. 143. Скорость смолообра-
зования в зависимости от ха-
рактера фенольного сырья.
/ — лг-крезэл; 2/ — технический три-
крезол; 111 — крезольная смесь; IV—
фенол.
перехода резола при нагревании в ре-
зитольное состояние или в резит). Наи-
большее значение имеет концентрация
в фенольном сырье трехфункциональ-
ных компонентов.
Фенол имеет три реактивные точки;
он энергично реагирует с формальде-
гидом в процессе конденсации и об-
разует резольные смолы с наиболее
высокой термореактивностью.
лгКрезол, подобно фенолу, образует
смолы со столь же высокой термореак-
ТИВНОСТЬЮ.
Однако скорость образования резо-
ла (скорость смолообразования) зна-
чительно выше у крезолов, чем у фе-
нола (рис. 143). Это, возможно,
объясняется активирующим действием
СНз-группы на соседние водородные
атомы бензольного кольца.
Таким образом, скорость первичной стадии конденсации {ско-
рость смолообразования) не находится в прямой зависимости от
функциональности изомеров, а определяется также строением мо-
лекул фенольного сырья, в частности расположением метильных
групп, как это видно из рис. 144. Однако термореактивность резола
в основном определяется только концентрацией в фенольном сырье
трехфункциональных компонентов. Поэтому смолы на основе фе-
нола и чистого .м-крезола имеют наибольшую и примерно одинако-
Закономерности образования резольных смол
373
вую скорость отверждения, в то время как скорость отверждения
резолов на основе трикрезола и технических ксиленолов умень-
шается по мере снижения в них концентрации трехфункциональных
фенолов — л/-крезола в трикрезоле и 1,3,5-ксиленола в техническом
ксиленоле (рис. 145).
Так как м-крезол реагирует с формальдегидом в 4—6 раз быст-
рее, чем о- и н-крезолы, то при применении трикрезола в первую
очередь с формальдегидом связывается ж-крезол. Конденсацию
Рис. 144. Относительная реактивность фенола и его гомологов (числа внутри
бензольного кольца показывают относительную скорость конденсации (смоло-
образования) по сравнению с фенолом; * — реактивные точки.
с трикрезолом можно проводить таким образом, чтобы в реак-
цию вошел только м-крезол; остальные не вступившие в реак-
цию крезолы смогут быть затем отогнаны. Однако этот метод не
нашел применения как вследствие сложности процесса, так и по-
тому, что некоторые свойства резольной крезоло-формальдегидной
смолы, в первую очередь механические и электрические, могут быть
улучшены, если в смоле остаются менее полярные (на основе
n-крезола) и термоплавкие компоненты, действующие как пластифи-
каторы при отверждении смолы. Этим объясняется большая эла-
стичность и лучшие диэлектрические свойства пленок крезольных
смол резольной конденсации по сравнению с фенольными.
В общем трикрезол с 60 и даже с 40% л«-крезола в процессе об-
разования первичных продуктов конденсации (резольной смолы)
обладает большей реактивностью с формальдегидом, чем фенол,
но полученная на его основе смола уступает фенольной по термо-
реактивности и скорости отверждения при 150—160°.
374
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
Ценность для резольной конденсации технических ксиленолов,
высших фенолов и различных кислых фракций малокалорийных
видов топлива (стр. 416) в основном определяется, как уже было
Рис. 145. Скорость 'отверждения (выра-
женная снижением растворимости смолы
при нагревании) резольных смол на основе
трикрезола и фенола.
указано, содержанием в них
трехфункциональных фено-
лов (фенола, At-крезола и
1,3,5-ксиленола).
Многоатомные фенолы
реагируют с формальдегидом
более интенсивно, чем одно-
атомные, в том случае, если
в бензольном кольце остают-
ся свободными три реактив-
ные точки. Так, реакция смо-
лообразования на основе ре-
зорцина протекает на холоду
и часто требует охлажде-
ния. Как уже было ука-
зано, формальдегид является
единственным из альдегидов
его гомологического ряда,
который приводит к образо-
ванию резольных смол. При-
меняют обычно 40% форма-
15 % метанола, который оставляют в
ТАБЛИЦА 42
Влияние содержания в формалине
метанола на свойства смол
ГСодержание мета- нола в формалине, о/о Выход СМОЛЫ, о/о Температура капле- падения по Уббелоде °C Время желатинизации (155°) (переход смолы в стадию В), сек. Вязкость 50% спирто- । вого раствора, сп Содержание свобод- ного фенола,
20 120,7 38 190 18,2 22,0
15 120,3 43 175 22,2 18,80
10 121,0 51 120 30,1 16,30
4 121,9 61 100 93,0 14,7
лин, содержащий от 7 до
формалине для предохранения
от образования параформа — твердого полимера формальдегида.
Применение получил также 30% формалин, который практически
не содержит метанола (~1—
2%), так как при меньшей
концентрации формальдеги-
да скорость образования па-
роформа очень мала. Нали-
чие метанола в формалине
уменьшает выход смолы
и ухудшает ее основные
показатели (табл. 42). Кро-
ме того, сам метанол не ис-
пользуется.
Поэтому конденсацию це-
лесообразно проводить на
безметанольном формалине.
Для этого можно применять
30% формалин, однако бо-
лее низкая концентрация его
требует большей кубатуры реактора и приводит к меньшей скорости
реакции конденсации. Наиболее эффективно применение 40% без-
Закономерности образования резольных смол
375
метанольного формалина, что, однако, возможно лишь в случае,
если производство формалина непосредственно приближено к заво-
дам фенольных смол (параформ выделяется лишь при длительных
хранениях высокопроцентного формалина).
Стадии процесса щелочной конденсации,
свойства и применение резольных смол
В отличие от кислой конденсации, отдельные стадии процесса
щелочной конденсации не могут быть установлены с достаточной
определенностью. Это объясняется меньшей экзотермичностью (80—
100 ккал/моль) щелочной конденсации по сравнению с кислой
и большей растворимостью и гидрофильностью начальных про-
дуктов конденсации (феноло-спиртов) в реакционной смеси (ще-
лочи). Вследствие этого расслоение смеси и коагуляция смолы не
происходят так отчетливо, как при кислой конденсации, и в некото-
рых случаях расслоение смеси и отделение воды не наблюдаются.
Можно все же различить следующие стадии процесса: 1) нагре-
вание до начала экзотермической реакции (55—75°); 2) экзотерми-
ческую реакцию; 3) расслоение и коагуляцию смолы; 4) обезвожи-
вание (сушка смолы).
В зависимости от соотношения компонентов, характера катали-
затора и режима сушки, конечный продукт конденсации может быть
жидким (но практически свободным от воды) или твердым. Полу-
чение твердых (хрупких при обычной температуре) резольных смол
значительно сложнее, чем вязко-жидких, вследствие склонности этих
смол при более глубокой конденсации и сушке переходить в рези-
тольное состояние. Твердые резольные смолы легче получить, если
реакционная смесь содержит некоторое количество анилина (~10%
к фенолу). Обычно твердые резолы имеют температуру каплепаде-
ния по Уббелоде в пределах 70—85°.
Жидкие (безводные) резольные смолы довольно широко приме-
няют для пропитки ткани, волокна и получения формовочных масс.
Во многих случаях непосредственно применяют водные конден-
саты («эмульсионные» смолы), получаемые после окончания про-
цесса конденсации и слива надсмольных вод. В этом случае сушку
смолы производят после смешения конденсата с наполнителем.
Твердые резольные смолы могут быть получены в стандартных
условиях. Их преимущества: стабильность свойств, меньшее содер-
жание свободного фенола, более высокие диэлектрические и хими-
ческие свойства. Они отличаются от твердых новолачных смол как
более иизкой температурой плавления, так обычно и большим со-
держанием свободного фенола. Последнее зависит от соотно-
шения компонентов, характера и количества катализатора,
глубины конденсации и продолжительности сушки. Обычно твер-
дые резолы содержат до 8—12% свободного резола, жидкие 20%
и выше. Небольшое содержание свободного фенола в резоле в
376 Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
некоторых случаях, невидимому, желательно, так как это улучшает
плавкость и текучесть смолы, а также гибкость пленок после отвер-
ждения, Однако значительное количество свободного фенола крайне
ТАБЛИЦА 43
Влияние продотжи-
тепьности термообра-
ботки на физико-хими-
ческие свойства твер-
дого резола
0,5 37 169 12.4
1 52,5 137 10.9
2 61 100 8,4
3 68 85 7,5
нежелательно, так как уменьшает скорость
отверждения и ухудшает физико-химиче-
ские свойства пресскомпозиций. В табл. 43
показана зависимость некоторых свойств
твердого резола от продолжительности суш-
ки (считая с момента, когда содержание
влаги в смоле достигает 3—4 %) - Сушка ре-
зольной смолы (термообработка) проводит-
ся в вакууме и при температуре не выше
90—105°. '
В отличие от новолачных смол, которые
могут храниться длительное время без из-
менения свойств, резольные смолы (даже
твердые) уже при обычной температуре за-
метно теряют текучесть, плавкость и рас-
творимость, увеличивают вязкость раство-
ров, т. е. их свойства при хранении изме-
няются в направлении образования про-
странственных, сетчатых полимеров и резол постепенно переходит
в резитольное состояние.
Термореактивность резольных смол при высоких температурах
(150—180°) меньше, чем у новолаков в смеси с уротропином (т. е.
меньше скорость перехода из стадии А в стадию С). Однако при
невысоких температурах (до 120°) резол значительно быстрее пе-
реходит в стадию В, чем новолачные смолы в смеси с оптимальным
количеством уротропина.
Аппаратурные схемы новолачной и резольной конденсации
Аппаратура для новолачной и резольной конденсации состоит
из хранилищ сырья (фенола, трикрезола, ксиленола, формалина
и др.), насосных установок, прирельсовых хранилищ, линий передач
из хранилища в цеховые приемники, плавителя фенола, дозировоч-
ных и реакционных аппаратов, устройств для слива смолы, охла-
ждения ее и размола.
Подготовка сырья
Формалин обычно поступает на завод (рис. 146) в алюминиевых железно-
дорожных цистернах /, из которых поршневым вакуум-насосом 4 его засасывают
в промежуточный бачок 2, а оттуда центробежным насосом 6 подают по трубам
в хранилище 5. Хранилище для формалина представляет алюминиевую цистерну,
емкостью 50—200 м3, снабженную поплавком для указания уровня жидкости.
Аппаратурные схемы новолачной а резольной конденсации 377
Применяют также резервуары, имеющие форму усеченного конуса, емкостью
20—50 л£3, изготовленные из парафинированного дерева.
Хранилища формалина должны быть устроены так, чтобы по возможности
предотвратить выпадание твердого параформа, что легче происходит при низких
температурах н при отсутствии перемешивания или циркуляции. Поэтому храни-
лища должны находиться либо в утепленном помещении (зимой), либо иметь
внутри паровые змеевики. Желательно создание системы непрерывной или эпи-
зодической циркуляции формалина или же частый спуск его в расходную ли-
нию. Рекомендуется также производить перемешивание лопастными мешалками,
которыми должны быть снабжены хранилища. Часто применяют сжатый воз-
дух, однако при этом несколько снижается концентрация формалина (прибли-
зительно на 5%).
Рис. 146. Схема перекачки формалина на сливном пункте.
I —железнодорожная цистерна; 2—промежуточный бачок; 3—буферный бачок;
4— паршневэй вакуум-насос; 5 — хранилище; 6 — центробежный насос.
Целесообразно иметь по крайней мере два хранилища: одно под загрузкой и
второе — расходное. Из хранилища формалин центробежным насосом перекачи-
вают в отделение подготовки сырья.
Фенол поступает либо в кристаллическом безводном, либо в жидком виде,
с содержанием 7—12% воды. В последнем случае он прибывает в железных
цистернах и его вакуум-насосом подают в промежуточные бачки, из которых
уже центробежным насосом — в хранилища. Твердый, кристаллический фенол
обычно прибывает в железных, оцинкованных барабанах, емкостью по 100 кг,
и идет в плавитель. Кристаллический фенол доставляют также в железно-
дорожных или автомобильных цистернах, снабженных паровыми змеевиками
для плавки фенола.
Фенол можно перевозить также и в необогреваемых но хорошо изолирован-
ных цистернах. В этом случае залитый горячий фенол, нагретый до ~100°,
сохраняет температуру жидкого состояния (~45°) в течение 5—6 суток летом
и 3—4 суток зимой.
Плавление фенола из барабанов может быть произведено различными ме-
тодами: при нагревании паром, горячим фенолом или путем помещения бара-
банов в обогреваемые камеры или ванны с горячей водой.
По первому методу — плавки острым паром — получают водный жидкий
фенол; этот метод может быть применен в тех случаях, когда рационально при-
менять водный фенол. Барабаны с фенолом поступают в плавильную камеру и
их устанавливают таким образом, чтобы отверстия в барабанах приходились
над соплами паропровода (давление 2—3 аг). Фенол, смешанный с конденса-
том и нагретый до 50—60°, стекает в приемник, из которого его перекачивают
в хранилище.
По второму методу — плавки горячим фенолом (рис. 147)—барабаны /
ставят на тележку 2 таким образом, чтобы их отверстия в иижием дне
установились над соплами фенолопровода, укрепленного на тележке. Тележку
378
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
вводят в камеру 3 и фенолопровод прикрепляют к фланцу распределительной ко-
лонки горячего (80—100°) фенола. Жидкий феиол центробежным насосом по-
дают из хранилища 4 в камеру 3, где он проходит через систему паровых труб.
Рис. 147. Схема установки для выплавки фенола из барабанов расплавленным
фенолом.
1—барабаны с фенолом; 2—вагонетка; 3—камера; 4—хранилища фенола.
уложенных под тележкой, и через сопла под давлением поступает в барабаны,
расплавляя в них фенол и стекая в сборник, выложенный паровыми змееви-
ками; из сборника фенол перекачивают обратно в хранилище.
По третьему методу, как уже указано, барабаны разогревают в камерах
или в ваннах с водой. Общая схема процесса представлена на рис. 148. В обо-
Рис. 148. Схема установки для выплавки фенола из барабанов в камере
нли ванне.
1—барабаны с фенолом; 2—обогреваемая камера нли ванна с горячей водой; 3—вакуум-
приемник; 4—хранилища фенола; 5 —блок, движущийся по монорельсу.
греваемую змеевиками камеру или в ванну с горячей водой 2 посредством
блока 5, движущегося по монорельсу, устанавливают барабаны 1 отверстием
вверх. Расплавленный фенол из барабанов с помощью гибкого шланга пере-
Аппаратурные схемы новолачной и резольной конденсации
379
водят в вакуум-приемник 3, из которого центробежным насосом перекачивают
в хранилище 4. Плавка в ваннах с водой происходит, естественно, значительно
быстрее’, чем плавка в воздушных камерах.
Из перечисленных методов наиболее производительным и рациональным
является метод выплавки фенола из барабанов горячим фенолом.
Хранят фенол в стальных сварных цилиндрических резервуарах, емкостью
от 50 до 250 м3 н более, снабженных паровыми змеевиками для поддержания
температуры выше температуры плавления фенола. Перемешивание фенола про-
изводят нли путем рециркуляции при помощи центробежных насосов, или
лопастными мешалками. Безводный
фенол перекачивают по стальным
трубам, снабженным паровыми ру-
башками.
Трнкрезол и ксиленолы (при
обычных температурах — жидкости)
транспортируют в железных цистер-
нах илн в железных барабанах. Так
же, как н жидкий фенол, их перека-
чивают в специальные хранилища,
снабженные паровыми змеевиками
(зимой вязкость трикрезола и ксиле-
нолов сильно возрастает) н рецирку-
ляционным устройством. Предложены
также методы транспортировки и
хранения фенолов в виде феноло-
крезольнон или феноло-ксиленоль-
ной стандартной жидкой смеси. Это
может оказаться рациональным в тех
случаях, когда смолы приготовляют
по соответствующей рецептуре.
Из хранилищ сырье, (фенол и
формалин) центробежным насосом
подается в дозировочное отделение,
в промежуточный бак илн монжус,
а оттуда в соответствующие мер-
ники. Применяют только весовые
мерннки (объемные не обеспечивают необходимой точности дозировки), изгото-
вленные из железа илн стали для фенолов, из меди илн алюминия для формалина.
Весовой мерннк (рис. 149) представляет собою цилиндрический бак емкостью
Рис. 150, Схема дозировки сырья и за-
грузки его в варочный котел.
1—хранилище сырья; 2—насос; 3—трубопровод;
4—весовой мериик; 5—гибкий шланг; 6—загру-
зочная воронка; 7—трубопровод; 8—загрузочный
бак; 9—варочный котел.
380 Гл. IX. Смолы., получаемые на основе фенолов а альдегидов
1—3 л<3, установленный вертикально на весах. Весы должны быть снабжены
электрическим реле, выключающим насос,
Рис. 151. Автоклав.
1—корпус; 2—крышка; 3—мешалка; 4—паровая
рубашка; 5—сливнлй штуцер; сальник; 7—фла-
нец; 8— гильза для термометра; 9—манивакуумметр;
10 —штуцер загрузочного отверстия; 11—штуцер
для предохранительное клапана; 12—вакуумная
линия; 13— линия сжатого воздуха; /-/—линия, со-
единяющая автоклав с атмосферой; 15— штуцер
для входящей паровой трубы; 16—штуцер для
выхода конденсата; 17—штуцер входа воды; 18—
штуцер для выхода воды.
подающий сырье, по достижении не-
обходимого веса. Залив мерника про-
изводят по трубе, опущенной в от-
верстие в крышке мерника.
Отвешенные материалы из мер-
ника с помощью гибкого шланга
переливают через специальную за-
грузочную воронку в загрузочный
бак, в котором должна храниться го-
товая навеска для варки смолы. Из
загрузочного бака сырье самоте-
ком поступает в варочные котлы
(рис. 150).
Реакционные (варочные)
агрегаты
Основным методом совре-
менного производства смол яв-
ляется прерывный метод, за-
ключающийся в изготовлении
отдельных партий смол путем
проведения самостоятельных
законченных циклов загрузки,
конденсации, сушки и слива
смолы. Различные схемы непре-
рывных методов не получили
технического применения.
Для производства смол слу-
жат как аппараты, в кото-
рых процесс конденсации про-
ходит при высоких давлениях и
температурах (автоклавы), так
и аппараты (реакторы), рабо-
тающие при атмосферном и по-
ниженном давлении (варочные
котлы). Последние имеют наи-
большее значение.
Автоклавы (рис. 151) пред-
ставляют собою толстостенные
стационарные сосуды из нержа-
веющей стали или фосфористой
бронзы со сферическим дни-
щем и крышкой, снабженные
паровой рубашкой, на которой
расположены штуцеры для
ввода пара, выхода конденсата и охлаждения автоклава во-
дой. Слив смолы производят через отверстие, закрываемое про-
бочным краном и расположенное в центре сферического днища. Для
перемешивания реакционной смеси в автоклаве имеется вертикаль-
Аппаратурные схемы новолачной и резольной конденсации
381
пая, якорного типа мешалка. Крышка автоклава соединена (бол-
тами) с корпусом и уплотнена асбестовым шнуром; на ней имеются
отверстия (штуцеры) для загрузки сырья, установки манометра,
термометра и присоединения вакуум-линии и линии сжатого
воздуха, а также предохранительного клапана. Емкость автоклавов
500—1000 л.
В автоклавах происходит лишь процесс конденсации. Сушку
смолы производят в отдельных сушильных агрегатах.
Варочные и сушильные котлы (реакторы) служат основными
аппаратами для изготовления новолачных и резольных смол. Их
применяют для проведения процесса либо по одноаппаратному, либо
по двухаппаратному методу.
Наибольшее распространение нашел одноаппаратный метод,
который осуществляют в реакторе, приспособленном для проведе-
ния в одну операцию всего цикла получения готовой смолы (кон-
денсацию, сушку и термообработку).
Варочно-сушильный котел (моноаппарат) представляет собою (рис. 152)
обычно стальной или никелевый сварной цилиндр с полусферической, иногда
плоской крышкой и сферическим днищем. Он снабжен паровой рубашкой,
Рис. 152. Варочный котел.
7—корпус котла; 2—крышка; 5—мешалка; 4— мотор; 5—редуктор; 6—смотровые фонари; 7 — шту-
цер для предохранительного клапана; 8 — штуцер для загрузки; 9— штуцер для паромера; 10 — шту-
цер для трехходового крана; //—штуцер для взятия проб; 12 — см» тр> вой люк; 13 — смотровое
стекло; 14 — штуцер для пароотв< да к конденсатору; 15— штуцер для линии от конденсатора; /5 —
штуцер входящей паровой линии; V — штуцер выводи, й паровой линии; 18— штуцер входящей
водяной линии; 19— штуцер выводной ВОДЯНцЙ линии.
рассчитанной на рабочее давление до 15 аг, высота ее составляет 0,4—0,6 вы-
соты цилиндрической части котла. В рубашку может быть пущена и вода
для охлаждения. Аппарат снабжен вертикальной мешалкой, которая должна
382 Гл. /X. Смолы, получаемые на основе фенолов и альдегидов
быть рассчитана на перемешивание вязкой смолы. Обычно применяют ме-
шалку якорного типа с чйслом оборотов 30—50 в мин., приводимую в дви-
жение через редуктор от электромотора; последний часто располагают на
крышке котла. В ннжней, сферической части котла имеется штуцер для слнва
смолы, снабженный пробочным краном или специальным клапанным затвором
Хрис. 153). На крышке аппарата имеются два смотровых фонаря (диаметр ~
150 мм), люк для чистки котла, иногда для
внесения катализатора. Кроме того, на крыш-
ке и на цилиндрической части котла распо-
ложены штуцеры для подачи сырья, отвода
паров в холодильник н стока конденсата,
для гильз термометра и мановакуумметра,
взятия пробы, предохранительного клапана,
пуска воды в реактор в случае аварии и т. д.
(см. рис. 152). Рубашка реактора должна
иметь два штуцера—-для ввода пара и слива
воды, штуцер для ввода воды и спуска кон-
денсатора и штуцер для предохранительного
клапана.
Отношение диаметра варочно-сушильного
котла к его высоте — от 1:1,2 до 1:1. Ем-
кость такого аппарата может быть различной
в зависимости от назначения: для новолачиых
смол можно применять большие котлы емко-
стью от 3 до 10 л3, для резольных смол более
рациональными являются котлы емкостью от
1,5 до 3 м3
Аппарат соединен с конденсатором — труб-
чатым поверхностным холодильником, кото-
Рис. 153. Клапанный затвор.
/—дно варочного котла; 2— дно паре-
бой рубашки; 3—сальниковое устрой-
ство; 4—корпус клапанного затвора;
•5—клапан; 6— штуцер для слива.
рый может работать как прямой и как
обратный. В некоторых котлах имеются два
отдельных холодильника (прямой и обрат-
ный). Холодильник (прямой) соединен с дву-
мя сборниками для конденсата (надсмольных
вод), которые, в свою очередь, приключены
к вакуум-насосу. Холодильник состоит из стальной обечайки и медных тон-
ких трубок с поверхностью охлаждения от 10 до 25 м2 на 1 м3 котла;
емкость приемников 0,4—1 .ч3. Для удобства монтажа, чистки н ремонта рацио-
нально ставить холодильник наклонно под углом 20—30°. Схема установки
варочного котла, конденсатора и сборника приведена на рис. 154.
Одноаппаратная система производства смол является в настоя-
щее время наиболее рациональной как для резолов, так и для ново-
лаков. Преимущества одноаппаратного метода — однотипность
всего оборудования и минимум операций — столь значительны, что
они искупают некоторые его недостатки. Последние заключаются в
необходимости испарения всей содержащейся в реакционной смеси
воды и в применении более усложненных и конструктивно менее
эффективных для каждого этапа процесса аппаратов, чем это было
бы необходимо и, возможно, если вести оба процесса — конденса-
цию и сушку — в отдельных аппаратах.
Двухаппаратный метод, сущность которого заключается в про-
ведении поликонденсации и сушки смолы в отдельных реакторах,
имеет сравнительно небольшое применение.
Аппаратурные схемы новолачной и резольной конденсации
333
По одному из вариантов этого метода конденсацию проводят
в варочном агрегате, затем сливают реакционную смесь в отстой-
ник, отделяют надсмольные воды и после этого реакционную смесь
подают в сушильный агрегат. Этот вариант двухаппаратного ме-
тода оказался нерациональным вследствие большого числа опера-
ций, громоздкости аппаратуры, больших потерь сырья в надсмоль-
ных водах и т. д.
Рис. 154. Схема реакционного агрегата для производства смол.
/--реактор; 2—холодильник; 3—приемник конденсата; 4—смотровые фонари.
Обычно после окончания процесса конденсации все содержимое
в котле перекачивают непосредственно в сушильный агрегат, по кон-
струкции близкий к варочно-сушильному моноаппарату.
Существенным преимуществом двухаппаратного метода является
возможность ведения каждой стадии процесса в таком аппара-
турном оформлении, которое в наибольшей степени соответствует
условиям проводимой операции.
Так, при отдельном проведении операции конденсации рацио-
нально конструировать варочный агрегат с облегченной паровой
рубашкой и с облегченного типа мешалкой, так как конденсация
производится при сравнительно невысоких давлениях пара
(0,5—1 ат) и реакционная смесь обладает малой вязкостью; не
нужна также и вся система для вакуума и для конденсации над-
смольных вод. Реактор должен иметь малую поверхность испарения;
отношение высоты котла к его диаметру может быть большим
(примерно 2:1).
384 Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
Двухаппаратную схему применяют главным образом для полу-
чения новолачиых и только иногда резольных смол. Она имеет ряд
вариантов.
1 вариант. Варочный аппарат по емкости примерно равен су-
шильному, при этом вся его загрузка может быть перенесена в один
сушильный аппарат. Это наиболее распространенный вариант для
производства новолачиых смол.
2 вариант. Сушильный аппарат по емкости значительно меньше
варочного и состоит из небольших плоских котлов с диаметром
в 3—4 раза превышающим высоту и с большим отношением по-
верхности обогревающей рубашки к объему, что обеспечивает
быстрое испарение воды и быструю сушку. Этот вариант был пред-
ложен для производства резольных смол, так как вследствие термо-
реактивности смолы очень важно, чтобы процесс сушки был прове-
ден как можно быстрее и чтобы можно было быстрее произвести
слив смолы.
3 вариант. Сушильный аппарат по емкости равен нескольким
варочным аппаратам. Конденсацию производят в нескольких аппа-
ратах обычной емкости (3—5 лг3), а содержимое всех котлов пере-
качивают в один большой сушильный аппарат емкостью 10—20 м3.
Этот метод применим только для производства новолачиых смол,
выдерживающих длительное нагревание при высоких температурах.
Преимущество его заключается в получении однородных по ка-
честву больших партий смол.
Слив смол
По окончании сушки смолу из котла сливают. Операции слива, охлаждения
и дробления твердых при обычных температурах смол до сих пор недостаточно
механизированы. Однако были
Рис. 155. Противень для слива смол.
сделаны многие попытки ра-
ционализации и механизации
этого процесса.
Большое применение полу-
чил метод слива смолы в на-
бор железных противней, уста-
новленных один над другим
по семь—десять штук. Против-
ни (рис. 155) имеют конические
стенки высотой 30—40 мм
для слива резольных смол и
80—120 мм для слива ново-
лачных смол. Противни (за
исключением самого нижнего)
снабжены переливными труба-
ми. Расплавленная смола из
варочного котла по сливной
трубе заполняет верхний про-
тивень и затем через перелив-
ную трубу попадает во второй нижележащий, далее в третий и т. д. до тех
пор, пока весь набор противней не будет заполнен; после этого подвижную
часть сливной трубы направляют во второй набор противней. Перед спуском
Аппарап урные схемы новолачной и резольной конденсации
385
смолы сливную трубу необходимо прогреть пуском пара в ее паровую рубашку.
Слив смолы рационально проводить в отдельной вентиляционной камере, в ко-
торой на съемной площадке устанавливают тележки с противнями и где также
происходит охлаждение смолы после слива (рис. 156). Твердую смолу обычно
впучиую выколачивают из противней на решетку, где раздробляют молотком на
вручную выколачивают из противней на решетку,
небольшие куски; последние из-под решетки
попадают в бункер.
По другому более рациональному вариан-
ту смолу сливают из сушильных агрегатов в
закрытые, снабженные вытяжной вентиляцией
камеры с полом, выложенным метлахскими
плитками. Смола растекается тонким слоем по
полу камеры, застывает н легко может быть
раздроблена в небольшие тонкие кусочки.
И. П. Михеев, Н. Ф. Бесхадарный и
П. 3. Ли предложили способ слива расплавлен-
ной смолы путем распыления ее во время
слива сжатым воздухом: последний подается
под падающую струю смолы через специальное
сопло. При этом падающие тонкие нити рас-
пыленной смолы охлаждаются в токе воздуха
и собираются на дне сливной камеры, где по-
падают на горизонтальный шнек, дополнитель-
но измельчаются и выносятся из нее.
Были предложены также методы одно-
временного проведения процессов сушки, слива
и измельчания смолы путем распыления сжа-
тым воздухом. Смолу после конденсации рас-
пыляют Г~ "
высоты 20—30-метровой
на дно
личные зоны обогрева: в верхней части 70—80°,
в средней 40—50° и в нижней — холодная
зона. Влажный воздух уносится вентилятором
или насосом. Сухая твердая смола собирается
в виде порошка на дне башни и выносится оттуда шнеком. Этот метод не нашел
еще применения вследствие сложности установки.
1
Пар
6
Конденсат.
на мельчайшие капельки, которые с
. "i башни падают вниз
ее. В башне предусмотрены раз-
Рис. 156. Схема слипа човолач-
иой смолы.
/—ваточный котел; 2 —сливная труба
с паровым обогревом (рубашкой); 3 -
подвижная часть сливной трубы; 4 —
противни; 5— вентиляционным шкаф;
6 •- съемная плошадка; 7— патрубок
для присоединения трубы, идущей к
вентилятору.
Утилизация надсмольных вод
В процессе сушки феноло-формальдегидных смол в виде отхода образуются
отгоны, так называемые надсмольные воды, содержащие 2—3% фенола, 1—5%
формальдегида и весь метиловый спирт, имевшийся в исходном формалине
(4—8%). Примерно на одну тонну новолачной смолы получаются около 600 кг,
а на тонну резольной смолы — около 900 кг надсмольных вод.
Для утилизации надсмольных вод предложены различные методы, главней-
шие из которых основаны на:
1) экстракции феиола путем обработки надсмольных вод растворителями
(трикрезилфосфатом и др.), после чего они поступают на формалиновые заводы
для отделения метанола дестилляцией и использования как низкоконцентриро-
ванных формалиновых вод в качестве поглотителя в производстве формалина;
2) утилизации фенола и формалина путем дополнительной конденсации
надс.мольной воды с избытком серной кислоты (кислый метод) или щелочп
(щелочной метод), при предварительном доведении отношения формальдегида
к фенолу до необходимого (примерно 28:100). В этих условиях может быть
достигнуто практически полное освобождение надсмольной воды от фенола, с об-
разованием соответствующих смол низкого качества, которые, однако, могут
быть применены как добавки к стандартным смолам. После обесфеноливания
25 Зак. 1269. Э. И. Барг
386 Гл, IX. Смолы, получаемые на основе фенолов и альдегидов
надсмольные воды могут быть подвергнуты ректификации для регенерации ме-
танола.
Однако все эти методы утилизации надсмольных вод недостаточно рацио-
нальны. Применяя щелочное или кислотное обесфеноливание иадсмольных вод,
стремятся, главным образом, использовать метанол и настолько освободить над-
смольные воды от фенола, чтобы их можно было спустить в общую канализа-
цию без заражения водоемов (рек, озер).
Технологический процесс производства новолачных смол
для пресспорошков
Фенольное сырье
При производстве новолачных смол для быстропрессующихся
пресспорошков с наивысшими физико-механическими свойствами
применяют фенол — безводный или водный.
Другие виды фенольного сырья (крезолы, ксиленолы) не обеспе-
чивают должного качества новолачных смол для пресскомпозиций,
поэтому чаще всего приходится их смешивать с фенолом.
Применение водного фенола (с содержанием 10—15% воды)
имеет определенное преимущество, так как водный фенол при обыч-
ных температурах — жидкость и поэтому более удобен для транспор-
тировки. Заводская практика показала, что небольшие добавки воды
к реакционной смеси не только не снижают, но даже увеличивают
скорость реакции и уменьшают содержание свободного фенола в го-
товой смоле; это необходимо отнести за счет снижения концентра-
ции метанола в реакционной смеси. Однако при применении 30%
безметанольного формалина не рекомендуется брать водный
фенол, так как дальнейшее разбавление реакционной смеси ведет
к снижению скорости реакции и выходов смолы.
Кроме фенола для производства новолачных смол применяют:
1) смесь фенола и трикрезола (~ 50 : 50), 2) смесь фенола и ксиле-
нола (~ 60 : 40), 3) торфяные и буроугольные фенолы (стр. 416).
Альдегидное сырье
При получении новолачных пресспорошков в основном поль-
зуются формальдегидом в виде 40% формалина, содержащего ме-
танол, реже в виде безметанольного 30%-ного. Из других альде-
гидов практическое применение для этой цели нашел фурфурол,
продукты конденсации которого будут рассмотрены отдельно
(стр. 410).
Рецептура и проведение процесса
Для производства новолаков для пресскомпозиций (пресспорош-
ков) оптимальное соотношение 26—27 ч. формальдегида на 100 ч.
Производство новолачных смол для пресспорошков
387
фенольного сырья. Катализатором служит техническая соляная кис-
лота (37%).
Процесс получения (конденсация, сушка и термообработка) ти-
пичной смолы для пресспорошков в варочно-сушильном аппарате
(емкость 3—5 м3, коэффициент заполнения ~ 0,8) складывается из
следующих операций: 1) загрузки фенола и формалина; 2) переме-
шивания и взятия пробы для определения pH; 3) загрузки первой
порции катализатора, перемешивания и взятия пробы для опреде-
ления pH; 4) нагревания смеси до 75°; 5) нагревания до 95° за счет
тепла экзотермической реакции; 6) выдержки при 95° и внесения
второй порции катализатора; 7) завершения реакции; 8) охла-
ждения до температуры сушки (в вакууме, 700 мм); 9) сушки
смолы; 10) термообработки; 11) загрузки жирных кислот и пере-
мешивания; 12) слива смолы.
Фенольное сырье и формалин (учитывая анализы партий на со-
держание в них формальдегида, воды и т. п.) отвешивают в весовых
мерниках (каждый вид сырья в отдельности) и самотеком через
промежуточный загрузочный бак перепускают в реактор. Соляную
кислоту дозируют мерным сосудом и вносят непосредственно в реак-
тор через загрузочную воронку. Количество соляной кислоты дают
с таким расчетом, чтобы pH среды находился в пределах 1,6—2,3
(в зависимости от характера фенольного сырья). После загрузки
фенола и формалина включают холодильник на обратный; пере-
мешивают 10 мин. и берут пробу на определение pH среды.
Если pH соответствует требованиям, в рубашку реактора пускают
пар под давлением 1,0—1,5 ат и при работающей мешалке смесь
нагревают в течение 35—45 мин. до 70—75°. Затем подачу пара в
рубашку реактора прекращают и дальнейший подъем температуры
до кипения происходит за счет тепла экзотермической реакции. При
температуре смеси 85—90° останавливают мешалку и для предот-
вращения бурного кипения в рубашку реактора пускают воду, при-
чем через несколько минут после начала равномерного кипения воду
выключают, а через 15—20 мин. снова включают мешалку и вносят
строго определенное количество (0,06 ч. на 100 ч. фенола) соляной
кислоты. По истечении 10—15 мин. в рубашку реактора дают пар
для поддержания кипения смеси. Общая длительность периода кипе-
ния 60—90 мин. Затем берут пробы для определения удельного
веса конденсата, и если удельный вес укладывается в соот-
ветствующие нормы (от 1,17 до 1,2 в зависимости от характера
фенольного сырья) процесс конденсации заканчивают. Кроме
пробы на удельный вес берут пробы на поведение смолы при
нагревании ее на плитке при 200°. В этих условиях смола не должна
желатинизироваться (переходить в стадию В), в противном слу-
чае конденсат не может быть подвергнут сушке и его необходимо
слить.
По окончании конденсации смолу переводят на сушку: конден-
388 Гл. IX. Смолы, получаемые на основе фенолов ti альдегидов
сатор переключают с обратного на прямой и постепенно, во избе-
жание вспенивания, аппарат соединяют с вакуумом 600—700 мм.
Для поддержания интенсивного кипения и для термообработки
смолы в рубашку дают пар давлением 5—8 ат. По отгонке
основной массы воды температура в реакторе начинает повышаться.
Время, часы
Рис. 157. Температурная кривая варки
(конденсации) новолачной смоль: (по
двухаппаратной схеме).
АБ — загрузка сырья и перемешивание с катали-
затором; БВ — нагревание до кипения; ВГ — период
кипения; ГД—охлаждение под вакуумом; ДЕ—
перегрузка смолы на сушку; EKJI—hk\ иод осмот-
ра аппарата и начала следующей загрузки.
Рис. 158. Температурная кривая
сушки новолачной смолы.
ОД—сушка при постоянной температуре;
АБ —период подъема температуры; БВ — пе-
риод .максимальной температуры; ВГ—вы-
грузка смолы иосмотр котла; ГДЕ — загрузка
котла.
Если требуется, то в начале сушки в смолу вводят краситель.
Сушку и термообработку заканчивают, когда взятая проба
смолы имеет заданную температуру каплепадения по Уббелоде
(в пределах 95—105°). Затем давление пара в рубашке сни-
жают примерно на половину и в реактор вводят олеиновую кис-
лоту или другие жирные кислоты, применяемые в качестве «смаз-
ки» для пресспорошков (для устранения прилипания к пресс-
форме в процессе прессования). После 15—20 мин. перемешивания
смолу сливают.
Контроль за ходом конденсации, сушки и термообработки про-
изводят с помощью самопишущих термо- и вакуумографов. Режим
процесса ведут по заранее установленным кривым температуры и
вакуума. На рис. 157 и 158 представлены кривые конденсации и
сушки новолачной смолы. Выход смолы к фенолу составляет
105—110%. Производительность на 1 м3 объема реактора
1,15—1,35 т/сутки.
Производство новолачных смол для пресспорошков
389
Новолачная смола Для пресспорошков должна соответывоиать
следующим условиям:
Температура каплепадения по Уббелэде, °C . . 95—105°
Вязкость 50% спиртового раствора смолы, сп. не более 130
Время желатинизации с 10% уротропина при
150°, сек....................................... 40—50
Содержание свободного фенола, %............. 6—9
Описанный процесс получения новолака является типичным. Од-
нако на практике применяются и различные его видоизменения. Ре-
жим типичного процесса характеризуется тем, что конденсацию
смеси фенола и формалина ведут при атмосферном давлении,
а сушку и термообработку конденсата — под вакуумом. При другом
варианте процесса конденсацию осуществляют под вакуумом,
а сушку и термообработку — при атмосферном давлении. Приме-
нение вакуума на стадии конденсации целесообразно для котлов
большой емкости. Температура кипения смеси при этом снижается.
Конденсация под вакуумом проходит более плавно и без резкого
подъема температуры. Сушка и термообработка при атмосферном
давлении дают возможность точно регулировать конечную темпера-
туру термообработки и, следовательно, температуру каплепадения
новолака; однако процесс при этом протекает медленнее.
В настоящее время выпускают новолачные смолы различных
марок, используемые для производства пресспорошков, отличаю-
щихся по вязкости растворов (среднему молекулярному весу), со-
держанию свободного фенола, скорости желатинизации и др., что
существенно сказывается на условиях переработки и физико-меха-
нических свойствах пресскомпозиций, изготовляемых на их основе
(скорость прессования, текучесть, теплостойкость, диэлектрические
свойства).
Указанные различия в свойствах новолачных смол определяются
главным образом характером фенольного сырья: чем меньше кон-
центрация трехфункциональных компонентов, тем ниже теплостой-
кость, водостойкость и скорость прессования новолачного пресс-
порошка.
В последние годы находят применение новолачные смолы на
основе комплексной конденсации фенола, анилина и формальдегида
в присутствии кислого катализатора. Такие смолы применяют для
получения новолачных пресспорошков с более высокими диэлектри-
ческими свойствами.
Особый тип новолака, характеризующийся высокой температу-
рой каплепадения (130—150°) и малым содержанием свободного
фенола (не более 5%), применяют в виде тонкоизмельченного по-
рошка (так называемого пульвер-бакелита) в качестве связую-
щего вещества для производства наждачных кругов и других ви-
дов абразивов.
390
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
Смолы со столь высокой температурой плавления образуются
при максимально допустимых соотношениях формальдегида к фе-
нолу и при высокой конечной температуре сушки (180—200°); обо-
грев ведут при давлении пара в рубашке реактора 10—12 ат (без
вакуума) или же в специальных котлах с газовым обогревом.
Новолак лишь в ограниченном количестве применяют для произ-
водства спиртовых лаков. Для этих целей требуется отсутствие сво-
бодного фенола и возможно более высокая «твердость» новолака
(температура каплепадения).
Новолак для производства лаков выпускают в СССР под назва-
нием идитол. Он может быть изготовлен на основе обычной рецеп-
туры с солянокислым катализатором. Лучшие результаты полу-
чаются при применении в качестве катализаторов малодиссоцииро-
ванных кислот (щавелевой, фосфорной). Смолы на основе этих
катализаторов являются более светлыми и отличаются от обычного
новолака лучшей светостойкостью. В рецептуре указывается не-
сколько большее содержание формальдегида (27—27,5 ч. на 100 ч.
фенола), большее количество (1,5—3%) кислоты (щавелевой), не-
обходимое для достижения pH в пределах 1,5—1,7. Процесс ха-
рактеризуется большей длительностью конденсации (3—6 час. при
85—95°) и введением продувки паром в процессе сушки смолы.
Первую стадию сушки, когда количество образующихся паров еще
велико, проводят без продувки паром, а иногда и без вакуума.
Когда же основное количество воды уже отогнано, включают
вакуум и усиленно пропускают острый пар для отгонки фенола, од-
нако с таким расчетом, чтобы вакуум значительно не снижался.
Этим методом можно практически полностью обесфенолить ново-
лак. Если содержание свободного фенола снижается медленно,
вводят добавочное количество формальдегида. Процесс термообра-
ботки ведут до получения смолы с содержанием свободного фенола
не более 0,1% и с температурой каплепадения в пределах
110—120°.
Технологический процесс производства резольных смол
В соответствии с вышеизложенными (стр. 370) зависимо-
стями резольной конденсации для получения резольных смол
в качестве фенольного сырья применяют трехфункциональные
фенолы или смеси фенолов, содержащие значительное количе-
ство трехфункциональных соединений (фенола, ж-крезола, 1,3,5-кси-
ленола).
Достижением последних лет является широкое использование
комплексных феноло-анилино-формальдегидных резольных смол,
отличающихся более совершенными диэлектрическими свойствами
и более высокой водостойкостью.
В качестве фенольного сырья для феноло-формальдегидных ре-
золов применяют: фенол, трикрезол, фенольные и феноло-крезольные
Производство резольных смол
391
фракции (содержащие смеси фенола, крезолов и ксиленолов) и
ксиленолы.
Альдегидным компонентом в производстве резольных смол прак-
тически является исключительно формальдегид.
Катализатором чаще всего служит аммиачная вода
(25% NH4OH), едкий натр, а также едкий барий. Иногда ще-
лочной катализатор во второй стадии процесса конденсации пол-
ностью или частично нейтрализуют кислотами (фосфорной, молоч-
ной и др.).
Для производства феноло-анилино-формальдегидных смол в ка-
честве фенольного компонента применяют только фенол.
Для примера можно привести несколько рецептур резольных
смол (в вес. ч.):
1) Фенол.............100
Формальдегид ... 37
Аммиак............ 1,5—2
2) Трикрезол.........100
Формальдегид . . . 37—38,5
Едкий барий .... 2
3) Фенол................50
Трикрезол.............50
Формальдегид..........45
Едкий натр...........0,4
Аммиак...............4,0
Фосфорная кислота . . 0,71
В рецептуре феноло-анилиновых смол отношение анилина к фе-
нолу может варьировать в широких пределах — от 10 до 100 ч. (и
более) анилина на 100 ч. фенола, например (в вес. ч.):
Фенол................100
Анилин.............. 50
Формальдегид.........36
Аммиак.............. 1,5
Резольные смолы выпускают в виде жидких (не содержащих
воду), твердых (с температурой каплепадения 60—85°) и так назы-
ваемых эмульсионных смол, представляющих собою конденсат,
частично, отделенный от надсмольных вод. Для каждого из этих
типов применяют соответствующие рецептуру и технологию.
Производство твердых резольных смол
Твердые резольные смолы изготовляют, главным образом, для
производства слоистых пластмасс, а также пресспорошков и во-
локнистых прессмасс. Они могут быть получены конденсацией фе-
нола и формальдегида, а также комплексной конденсацией фенола,
анилина и формальдегида. Конденсацию и сушку, как правило, про-
водят в варочно-сушильном аппарате (моноаппарате).
Основные стадии процесса получения твердых резольных смол
в общем те же, что и при производстве новолачных смол, за.
исключением термообработки, которая либо полностью отсутствует,
либо имеет подчиненное значение. Вследствие меньшей экзо-
термичности процесса конденсации катализатор обычно вносят
392
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
в атмосферу
Сырье
в приемник
надсмольных
вод
Слив лама
в rnapy
Рис. 159. Схема
для получения
установки
резольной
смолы и лака.
/- варочный котел; 2--конден-
сатор; 3 — атмосферная труба:
/ — аварийный приемник; 5 —сбор-
ники конденсата; б—трехходовые
краны; 7 —мерник для спирта;
сборник для лака; 9 - смотро-
вой фонарь.
в один прием. В зависимости от характера исходного сырья,
рецептуры и технических требований к смоле процесс смолообразо-
вания может варьировать в определенных пределах. Ниже приво-
дится схема установки и некоторые технологические нормативы
получения твердых резольных смол.
В варочно-сушильный аппарат 1 (рис. 159) при температуре не
выше 35—40° загружают из мерников сначала фенольное сырье,
которое тщательно перемешивают, затем
формалин и через люк вносят катализа-
тор (обычно аммиачную воду). После за-
грузки сырья и катализатора конденса-
тор 2 переключают на обратный, в холо-
дильник пускают воду, всю систему со-
единяют с атмосферой (<?, 4), в рубашку
реактора дают пар (1,0—1,5 ат) и дово-
дят температуру реакционной смеси до
65—75°. После этого пуск пара прекра-
щают и дальнейший подъем температуры
до 90—95° (обычно до кипения) происхо-
дит за счет тепла экзотермической реак-
ции. При этой температуре реакцию про-
должают определенное, строго контроли-
руемое время, примерно 25—45 мин. Вви-
ду того, что не всегда можно точно уста-
новить время начала кипения, необходимо,
строго руководствоваться заданными кри-
выми термографа. На практике руко-
водствуются также определением темпе-
ратуры помутнения смеси (из-за выпаде-
ния кристаллических феноло-спиртов):
чем глубже прошла реакция поликонден-
сации, тем при более высокой темпера-
туре наступает помутнение смеси. Обычно
концом конденсации считают момент начала помутнения при темпе-
ратуре кипения. Затем постепенно (во избежание переброса) вклю-
чают вакуум, который должен быть по возможности глубоким (не
ниже 700 ми) и приступают к сушке смолы.
При проведении комплексной конденсации фенола, анилина и
формальдегида экзотермическая реакция может начаться уже при
смешении реакционной смеси с катализатором, при этом темпера-
тура в реакторе может достигнуть 50—60°. В этом случае пар
(0,5—1 ат) в рубашку реактора дают через 15—20 мин. после ин-
тенсивного перемешивания и доводят температуру реакционной
смеси до 90—95°.
Сушка резольной смолы — операция весьма ответственная
ввиду возможности быстрого повышения вязкости смолы и желати-
низации — перехода смолы в резитольное состояние. Поэтому тем-
Производство резольных смол
393
пература в смоле должна повышаться равномерно и не более, чем
до 95—105°; при достижении 90—95° берут пробу для определения
температуры каплепадения и скорости желатинизации смолы. В не-
которых случаях, если температура в котле уже доходит до 90—95°,
а скорость желатинизации еще не достигла установленной величины,
можно внести в котел небольшое количество воды (2—5% от веса
фенола и анилина) и продолжать реакцию до получения необходи-
мых показателей.
По окончании сушки смолу сливают. Слив смолы может быть
затруднен высокой вязкостью и склонностью смолы к желатиниза-
ции; поэтому производить его необходимо как можно быстрее. Про-
цесс слива облегчается, если в котле предусмотрены специальные
затворы (стр. 382) и применено
давление (в котле) сжатым воз-
духом.
Смолу сливают в противни
слоем не более 30—40 мм, так как
при заливке более толстого слоя
она вследствие медленного охла-
ждения может перейти в рези-
тольное состояние. Некоторые
анилино-фенольные смолы склон-
ны в процессе слива к самовоз-
горанию, в связи с чем необходи-
мо принять соответствующие меры
предосторожности, например сли-
вать смолу в специальные ту-
шильники.
Рис. 160. Температурная кривая по-
лучения резольной смолы.
АБ - нагревание до кипения: БВ —период ки-
пения; ВГ— перевод на сушку под вакуумом:
ГД—сушка при постоянной температуре;
ДЕ— период конца сушки; ЕК—растворение:
КЛ — выгрузка лака; ЛМ — осмотр котла и на-
чало новой загрузки.
Условия резольной конденса-
ции по сравнению с новолачной
характеризуются большей гидро-
фильностью начальных продуктов
конденсации (из-за которой нельзя
с достаточной определенностью наблюдать конец процесса и разде-
ление слоев), а также склонностью смолы к переходу в желатиниро-
ванное (резитольное) состояние в процессе сушки. Все это опреде-
ляет необходимость тщательного контроля процессов конденсации
и сушки. На практике придерживаются графика по определенным,
заданным кривым термографа и вакуумографа (рис. 160), а также
тщательно наблюдают за состоянием реакционной смеси в котле, за
температурами как в смоле, так и в газовой среде котла, которые
тем больше расходятся, чем меньше осталось воды в смоле. Особое
значение приобретает контроль физических свойств смолы (скорость
желатинизации, температура каплепадения, прозрачность смолы по-
сле охлаждения и др.); проверка скорости желатинизации в опреде-
ленной стадии сушки должна производиться систематически, на-
пример каждые 10 мин.
394 Гл. IX. Смолы, получаемые на основе фенолов а альдегидов
Для многих назначений твердый резол применяют в виде спир-
тового раствора (лака), обычно 50—60% концентрации.
Для получения лака спирт вводят в котёл немедленно после того,
как анализ смолы устанавливает ее соответствие техническим
условиям.
Для этого спирт из спиртового мерника 7 (рис. 159) подают
в котел 1, причем предварительно закрывают вакуумную линию,
выключают пар в рубашке и переключают конденсатор 2 на обрат-
ный. Растворение смолы происходит при непрерывно работающей
мешалке в течение 40—60 мин., после чего лак сливают в сбор-
ник 8.
Выход резольной смолы ПО—130% к фенолу (или к фенолу
и анилину). Производительность 1 лР реактора 0,9—1,3 т/сутки.
Свойства твердых резольных смол могут значительно варьиро-
вать в зависимости от рецептуры, процесса конденсации и сушки.
Твердые резолы характеризуются обычно следующими показа-
телями.
Температура каплепадения по Уббелоде, °C........ 60—85
Скорость желатинизации при 150э, сек............ 60—180
Содержание свободного фенола и анилина, % .... 5—12
Содержание влаги, %...............................не более 2—4
Производство эмульсионных резольных смол
Эмульсионные смолы представляют собою вязкие водные конденсаты после
отстаивания и отделения надсмольных вод или после частичного испарения воды.
Их применяют для пропитки волокнистых наполнителей: древесной муки, целлю-
лозы, ткани. Рациональность их применения (по сравнению с твердыми резоль-
ными смолами) заключается в том, что не требуется производить сушку смолы,
а также расходовать спирт для получения спиртового резольного лака. Недоста-
ток таких смол — их малая стабильность, нестандартность свойств и более вы-
сокое содержание в них свободного фенола и низкомолекулярных метилольных
продуктов конденсации.
Среди эмульсионных резольных смол особое значение приобрели разрабо-
танные Г. С. Петровым крезоло-баритовые смолы, отличающиеся более высо-
кими диэлектрическими свойствами. В качестве катализатора для получения
такого рода смол применяют гидрат окиси бария Ва(ОН)2 • 8Н2О, который хо-
рошо растворяется в горячей воде.
Наряду с крезоло-баритовыми эмульсионными смолами изготовляются и
феноло-баритовые, а также феноло- и крезоло-аммиачные смолы по следующим
(примерным) рецептурам (в вес. ч.):
I п
Фенол..............100
Формалин (37%) .... 100
Аммиак.............. 2,5
Трикрезол............100
Формалин (37%) . . . 100
Ва(ОН)2............... 2
В качестве примера приведем процесс получения эмульсионной крезоло-
баритовой смолы.
В реактор загружают крезол и формалин и затем через люк вводят предва-
рительно смешанный с равным количеством воды протертый через сито едкий
барий. Затем в рубашку реактора пускают пар (~0,5 ат) и реакционную смесь
при работающей мешалке и обратном холодильнике нагревают в течение
Производство резольных смол
395
20—25 мин. до 55—60°, после чего пар выключают, дают температуре подняться
до 75—80° за счет тепла экзотермической реакции (в случае резкого подъема
температуры в рубашку реактора пускают воду). При этой температуре смесь
выдерживают до тех пор, пока вязкость ее не достигнет 180—250 сп. Контроль
вязкости производят через каждые 10—15 мин. Потом реакционную смесь охла-
ждают до 30—35° и сжатым воздухом передавливают в отстойник, в котором
происходит отделение надсмольных вод от смолы (в течение 2—6 час.). После
отстаивания готовую смолу сливают в железные бочки.
Вязкость готовой смолы 350—500 сп, влажность ~25%, содержание свобод-
ного фенола — до 18%. Выход смолы (из расчета на сухое) 105—110% (к фенолу).
По другому варианту конденсацию проводят под вакуумом со включенным
обратным холодильником. Реакционную смесь быстро нагревают до кипения
и через 15—20 мин. включают вакуум, так что в дальнейшем кипение смеси
продолжается при 60—65°. Благодаря низкой температуре кипения нарастание
вязкости происходит более медленно. По достижении соответствующей вязкости
смолу охлаждают и перекачивают в отстойник.
Физико-механические и диэлектрические свойства резольных
смол, как было указано выше, определяются, в первую очередь,
характером фенольного сырья. Чем выше концентрация трехфунк-
циональных фенолов, тем выше скорость прессования, теплостой-
кость, твердость и прочность прессматериалов, изготовленных на
основе резольных смол. Поэтому резольные смолы на основе фе-
нола обладают наилучшими показателями этих свойств. С другой
стороны, для диэлектрических свойств и водостойкости имеет зна-
чение степень полярности исходных фенолов; чем выше полярность
последних, тем ниже диэлектрические свойства и водостойкость
смол. Поэтому смолы на основе трикрезолов имеют лучшие диэлек-
трические свойства и лучшую водостойкость, чем смолы на основе
фенола, так как полярность фенола выше полярности крезолов.
Введение анилина в комплексную конденсацию в еще большей сте-
пени снижает полярность смолы и резко улучшает диэлектрические
свойства. Поэтому для многих назначений преимущество имеют
крезольные и анилино-фенольиые смолы, хотя они и уступают фе-
нольным смолам по теплостойкости и скорости затвердевания. На-
личие в трикрезоле бифункциональных фенолов (п- и о-крезолов)
снижает скорость затвердевания смолы, однако создает более благо-
приятные условия для релаксации напряжений, что повышает эла-
стичность пленок из этих смол. Как уже было указано, резольные
смолы отличаются от новолачных (в смеси с уротропином) меньшей
скоростью затвердевания (перехода в резит) при высоких темпе-
ратурах (150—160°), но они скорее, чем новолачные смолы, пере-
ходят в резитольное состояние при низких температурах (80—120°).
Это является преимуществом резольных смол при производстве
слоистых пластмасс, для которых нежелательна чрезмерная ско-
рость затвердевания (стр. 421), однако малая скорость затвердева-
ния затрудняет их использование для производства быстро прессую-
щихся пресспорошков. Резольные смолы применяют поэтому преиму-
щественно для производства слоистых пластиков и таких марок
пресспорошков, к которым предъявляют высокие требования в отно-
-396
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
тении диэлектрических свойств и водостойкости, а также в произ-
водство специальных формовочных масс типа фаолит (етр. 455)
и антикоррозионных лаков и замазок. Прессматериалы на основе
резольных смол значительно превосходят по водостойкости, хим-
стойкости и диэлектрическим свойствам соответствующие мате-
риалы на основе новолачиых смол (с уротропином).
ЛИТЫЕ ПРОДУКТЫ РЕЗОЛЬНОЙ КОНДЕНСАЦИИ
(Фенолиты, литые резиты, литой карболит, неолейкорит)
Особый тип резольных смол служит для получения твердых
упругих, неплавких и нерастворимых органических стекол, так на-
зываемых литых смол (литых резитов). Литыми эти смолы назы-
вают потому, что их получают путем простого литья. Жидкую смолу
в стадии резола заливают в формы, где ее отверждают путем дли-
тельного нагре-вания. Литые резиты получаются в виде пластин,
блоков, стержней и различных заготовок. Изделия из литых резитов
большей частью производят путем механической, станочной обра-
ботки заготовок.
Литые резиты были первыми пластическими массами на основе
резольных смол. Они были получены Бэкелендом в 1907 г. при кон-
денсации эквимолекулярных количеств фенола с формальдегидом
в присутствии щелочного катализатора. Отверждение резольных
смол производили под давлением в специальных герметических
аппаратах «бакелизаторах». Последние были снабжены паровой
рубашкой, в которую давали пар под давлением 3—5 ат. Внутри
бакелизатора на полках ставили формы с залитой жидкой, резоль-
ной смолой, после чего в бакелизаторе создавали избыточное давле-
ние углекислотой примерно на 2—3 ат, превышавшее давление пара
в рубашке. Благодаря давлению отвержденные смолы получались
без вздутий и пузырей. Полученные таким образом смолы
(«бакелиты») имели приятный янтарный цвет, но хрупки и не-
прочны; они нашли применение лишь для производства галанте-
рейных изделий.
Крупным достижением в производстве литых резитов явился
пластик «карболит», изобретенный Г. С. Петровым в 1912 г. В от-
личие от других литых резитов карболит, обладающий высокими
диэлектрическими свойствами, получил, главным образом, техниче-
ское применение, в частности в электротехнике.
Для производства карболита по одному из вариантов берут 6 мол. фенола
или трикрезола на 7 мол. формальдегида. В качестве катализатора применяют
уксуснокислый цинк, который в процессе реакции гидролитически распадается на
гидрат окиси цинка и уксусную кислоту. Реакцию конденсации ведут в две или
з три фазы, со ступенчатым введением формалина.
Так, фенол (или крезол), около 70% всего формалина и 0,5% (к фенолу)
уксуснокислого цинка загружают в реактор и тщательно перемешивают, после
„Благородные" резиты
397
чего в рубашку реактора дают пар (2 ат}, смесь доводят до кипения и выдер-
живают при кипении в течение 2 часов. После этого смолу быстро охлаждают
до 60—65°, включают вакуум и производят отгонку основной массы воды. Затем
вносят вторую порцию формалина (~20%), а также краситель; массу тщательно
перемешивают, снова включают вакуум и при 65—70° производят отгонку основ-
ного количества воды; после этого смолу охлаждают до 30° и смешивают с неф-
тяными сульфокислотами («контакт Петрова») и с последней порцией форма-
лина (—10%). На 100 вес. ч. смолы вводят 10—12 вес. ч. контакта. Необходимо
следить, чтобы температура смолы при этом не поднималась выше 25—30°. За-
тем смолу вновь подвергают вакуумированию, после чего разливают в брон-
зовые формы (пластины, блоки и др.). Залитые смолой формы помещают
в термостаты (воздушные или водяные), в которых при постепенном подъеме
температуры до 100° в течение нескольких часов происходит отверждение-
смолы.
Отвержденные заготовки из карболита (стержни, блоки, пластины) выни-
мают из формы. Они представляют собою черного или коричневого цвета упру-
гие твердые материалы, которые отличаются хорошими диэлектрическими свой-
ствами, хорошей теплостойкостью и удовлетворительной для ряда назначений
прочностью. Для улучшения диэлектрических свойств готовые изделия из кар-
болита подвергают в продолжение нескольких часов прогреванию в масле
при 125—150°.
Литой карболит (табл. 44, стр. 401) нашел применение в основ-
ном для производства высоковольтных изоляторов и других элек-
тротехнических деталей, а также различных деталей технического
назначения.
С развитием производства прессматериалов значение литых ре-
зитов значительно уменьшилось и сейчас литой карболит произ-
водят в небольшом количестве для специальных целей.
«Благородные» резиты
В дальнейшем производство литых резитов пошло по пути полу-
чения так называемых «благородных» литых смол, отличающихся
более высокой динамической прочностью, с одной стороны, и боль-
шей светостойкостью, светлой окраской и даже бесцветностью,
с другой. Цвет же обычных резольных смол и литых резитов жел-
тый, со временем переходящий в коричневый. Такие смолы могут
служить заменой слоновой кости, причем без добавочной окраски
и без внесения красителей они имеют присущий последней харак-
терный цвет. Они могут служить также заменой янтаря, черепахи,
перламутра и т. п. Как установили А. А. Ваншейдт и О. Н. Симо-
нова, причиной окраски фенольных смол в различных стадиях
отверждения является окисление свободного фенола и продуктов
первичной конденсации. В результате окисления этих соединений,
которые сами по себе не содержат хромофорных групп и, следова-
тельно, не могут поглощать лучи видимой части спектра, обра-
зуются, вероятно, соединения хиноидной структуры.
J93 Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
Так 4,4'-диоксидифенилметан можно рассматривать как лейко-
соединение красителя аналогично бензаурину и, возможно, что
именно этот темный краситель образуется при окислении по схеме
НО-/ СН3-/ ОН /U О=/==/=СН—/ он.
Чтобы, по возможности, устранить образование в резите хиноид-
ных соединений и получать светлые и нетемнеющие смолы, необхо-
димо обесфенолить смолу, создавать максимально густую
«сшивку» фенольных ядер метиленовыми мостиками и устра-
нять все условия, облегчающие окисление фенола или диокси-
дифенилметана. Для этого: 1) применяют значительный избы-
ток формальдегида (2—2,5 мол. на 1 мол. фенола); 2) используют
перегнанный фенол и формальдегид и исключают железную и свин-
цовую аппаратуры, так как железо и свинец сами окрашивают
смолу и являются катализаторами окислительных процессов; 3) уда-
ляют воздух из сферы реакции, заменяя его азотом или углекисло-
той; 4) вводят в реакционную смесь восстановители (гидрохинон) и
антиокислители (оксикислоты, например молочную); 5) проводят
реакцию конденсации при низкотемпературном режиме, так как
при низкой температуре окисление происходит медленно; б) в про-
цессе конденсации меняют щелочной катализатор, нейтрализуя ще-
лочь небольшим избытком кислоты (лучше всего оксикислотой, на-
пример молочной, которая одновременно является и антиокислите-
лем). Смена катализатора и создание к концу процесса кислой
среды необходимы для обесцвечивания красителей типа бенз-
аурина.
Для получения литого резита большой прочности и упругости
необходимо, во-первых, создать наиболее плотную пространствен-
ную валентную связь между фенольными ядрами, что достигается
введением значительного избытка формальдегида, и, во-вторых, со-
здать коллоидно-дисперсную, ячеистую структуру резита, что может
быть достигнуто диспергированием в нем воды, глицерина и других
жидких веществ, которые, будучи диспергированы в твердой смоле,
так же содействуют упрочнению, как и твердые наполнители.
Диспергирования воды достигают специальным процессом сушки и
отверждения резита, при котором часть воды (5—20%) остается
коллоидно распределенной в смоле.
В СССР выпускают пластик неолейкорит, представляющий со-
бою тип «благородного» резита. Процесс производства неолейко-
рита был разработан С. Н. Ушаковым и А. Д. Соколовым.
Технологический процесс производства «благородных» литых ре-
зитов состоит из следующих операций: конденсации, сушки и ней-
трализации смолы, проводимых в варочно-сушильном аппарате,
слива и разлива смолы в формы, отверждения залитой в формы
Благородные' резаты.
399
смолы путем длительного нагрева в термостатах, разъема форм и
механической обработки и полировки полученных заготовок. Рецеп-
тура (в вес. ч.):
Фенол (перегнанный) . 100
Формалин (37%) .... 202
Едкий натр............. 3
Молочная кислота ... 16,8
на 1 мол. фенола
2,4 мол. формаль-
дегида
Процесс конденсации и сушки ведут в никелевом вакуум-аппа-
рате (моноаппарате), снабженном рубашкой, в которой циркулирует
вода, подогреваемая уложенными внутри нее паровыми змеевиками.
Реактор снабжен якорной мешал-
кой, прямым и обратным холо-
дильниками, пеноотделителем, ин-
дивидуальным вакуум-насосом,
мерниками для фенола и форма-
лина (рис. 161).
Фенол, формалин и едкий натр
загружают в аппарат, перемеши-
вают, пускают пар в змеевики и
воду в рубашку котла, включают
обратный холодильник и подни-
мают температуру в котле в тече-
ние 40—45 мин. до 60—65°, а
воды в рубашке до около 85—90°.
Реакцию в этих условиях ведут
от 2 до 2,5 час. После этого
постепенно (во избежание пере-
броса смолы в холодильник)
включают вакуум и в течение
30—40 мин. доводят его до 600—
650 мм; холодильник при этом пе-
реключают на прямой. По исте-
чении некоторого времени сушки
в реакционную смесь вносят мо-
лочную кислоту с избытком около 0,5% для нейтрализации щелочи,
затем сушку смолы продолжают еще 2—3 часа. Окончание сушки
устанавливают анализом пробы, а также наблюдением за тем-
пературой в котле, выделением конденсата и т. д. Анализ произ-
водят, определяя твердость и внешний вид охлажденной пробы
или вязкость смолы. Весь процесс конденсации и сушки длится
8—10 час.
Готовая смола содержит еще некоторое количество воды; в та-
ком виде ее сливают в деревянные бачки, а из них после охлажде-
ния или еще в горячем виде разливают в свинцовые или в стеклян-
ные формы. Отверждение смолы производят путем длительного
(от 3 до 5 суток) нагревания (во избежание вздутий) в воздушных,
Рис. 161. Схема получения литых
резитов.
/- вакуум-аппарат; 2—обратный холодильник;
• - прямой холодильник; 4, 6, 7—мерники;
5 приемник для конденсата; 8—формы;
9 —полимеризатор; 10— вакуум-насос.
400
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
водяных или масляных термостатах; температуру поднимают посте-
пенно от 60 до 80°, например:
2 суток ........... 60—62°
2 „ 70-72°
1 сутки............80°
По окончании процесса отверждения отлитые заготовки (блоки,
пластины, стержни и т. п.) вынимают из форм; при этом свинцовые
формы часто из-за механических повреждений не могут быть более
использованы и поступают на переплавку или перештамповку.
Так как неолейкорит (без красителей) имеет цвет слоновой
кости, то во многих случаях он и применяется вместо слоновой!
кости, например, при производстве биллиардных шаров, черенков для
ножей и вилок, различных поделочных и галантерейных изделий.
Неолейкорит можно получать и окрашенным в различные цвета.
Для этого в смолу под конец или после сушки при тщательном пере-
мешивании вносят краситель, который должен быть светостойким,
стойким к формалину, кислотам и к повышенным температурам.
Применяют почти исключительно органические спирторастворимые
красители.
В состав смол обычно вводят 0,005—0,03% спиртового раствора
красителя в зависимости от характера последнего. Могут быть
использованы как основные, так и кислотные красители, хотя, пови-
димому, — ни один из типов органических красителей не удовле-
творяет полностью всем предъявляемым требованиям. Для окраски
феноло-формальдегидпых смол применение нашли: аурамин, литоль-
красный, родамин 6Ж, ксиленовый голубой, сафранин, основной
бирюзовый и др. Кроме того, непрозрачные смолы окрашивают и
минеральными пигментами; последние обладают более высокой
термоустойчивостыо и светостойкостью.
Окрашивание смолы производят следующим образом. После сушки ее
разливают в небольшие медные луженые котелки емкостью в 30—5б л, сна-
бженные паровой рубашкой и лопастной мешалкой; когда температура в смоле
достигнет 70—85°, при работающей мешалке медленно вносят спиртовый рас-
твор красителя. После 15—25 мин. перемешивания смолу разливают в формы.
Для получения окраски под мрамор ряд партий смол, окрашенных в различ-
ные цвета, в том числе и неокрашенную смолу, сливают поочередно в одну
форму, после предварительного охлаждения до 40—50° (для увеличения вязкости
и уменьшения скорости диффузии смешиваемых смол). Формы после этого под-
вергают термической обработке.
В некоторых случаях получают прозрачные сорта неолейкорита.
Для этого в состав реакционной среды вводят, например, глицерин
(10—15%), который в резите частично заменяет диспергированную
воду. Непрозрачность литого резита является следствием наличия
диспергированной в нем воды; введение глицерина облегчает усло-
вия стабилизации и коллоидного диспергирования жидкой фазы и
таким образом способствует улучшению прозрачности резита. Пре-
имущества глицерина (перед водой) — малая упругость его паров,
обусловливающая большую стабильность материала. До некоторой
Феноло-альдегидные смолы для производен-ва лаков
401
степени глицерин можно рассматривать и как «пластификатор» в ре-
шайте, так как, повидимому, он способствует увеличению «гибкости»
материала, уменьшению хрупкости и динамической прочности.
Наличие в неолейкорите дисперсной воды снижает его электриче-
ские свойства, теплостойкость и химическую стойкость и является
причиной коробления и деформации. Поэтому, чем лучше высушена
смола перед заливкой в формы, тем лучше физико-химические и ди-
электрические свойства неолейкорита; однако удельная ударная
вязкость и упругие свойства, зависящие от коллоидно-дисперсной
структуры, при этом снижаются. Литые резиты лишь относительно
светостойки, так как очень трудно получить смолу, не содержащую
фенола и низкомолекулярных продуктов конденсации, способных к
окислению.
Неолейкорит отличается светлой окраской, хорошей механиче-
ской прочностью, в частности прочностью к динамическим напряже-
ниям, и высокой упругостью, поэтому его применяют для производ-
ства биллиардных шаров, деталей мебели, разнообразных поделоч-
ных деталей взамен слоновой кости и т. д.
Литые резиты имеют также и техническое применение. Из них
изготовляют крупные изоляторы, детали нефтяных врубовых машин,
крышки и детали различных приборов и т. д.
Сравнительные свойства литых резитов приведены в табл. 44.
ТАБЛИЦА 44
Свойства литых резитов
Показатели Литой карболит Неолейкорит
Уд. вес, г[см* 1,17—1,18 1,25
Теплостойкость по Мартенсу, °C . . . 125—108 70—100
Уд. ударная вязкость, Кь-слцсм- . . . 2-6 10-20
Предел прочности на изгиб, кг[см" . . 400—650 800-1000
Зьердость по Ьринелю, /сг/Л'.И' . . . 20—30 20—30
Уд. поверхностное электрическое со- противление, L 10'-’—1013 Юн—Ю’з
Уд. объемное электрическое сопротив- ление, У-С.1Г 10”—10’2 1010—1011
Пробивное электрическое сопротивле- ние, кв/мм 10—14 8—12
МОДИФИЦИРОВАННЫЕ ФЕНОЛО-АЛЬДЕГИДНЫЕ СМОЛЫ ДЛЯ
ПРОИЗВОДСТВА ЛАКОВ
Уже первые новолачные смолы нашли применение для производ-
ства спиртовых лаков взамен шеллака — важнейшей природной
спирторастворимой смолы.
26 Зак. 1269. Э. И. Барг
402
Гл. IX. Смолы, получаемые на основе фенолов а альдегидов
Возможность применения синтетических смол для получения ла-
ков во многом определяется степенью их полярности, от которой’
зависит их растворимость в соответствующих растворителях, совме-
стимость с маслами, водостойкость, погодоустойчивость и т. д. Так,
новолак обладает недостаточной полярностью, чтобы растворяться
в воде, в буре и соде, однако его полярность слишком велика для
того, чтобы он мог растворяться в неполярных растворителях
(бензоле, бензине и др.) и совмещаться с маслами. Он раство-
рим в спирте, ацетоне и других органических полярных раство-
рителях.
Чтобы феноло-альдегидные смолы могли совмещаться с маслами
и получить применение в качестве основы масляных лаков, они
должны обладать значительно меньшей полярностью, чем новолак.
С другой стороны, для многих назначений (для производства поли-
тур, туши и др.) требуются смолы, способные, подобно шеллаку,
диспергироваться в слабых щелочных растворах, например в буре и
соде. В этом случае необходимы смолы с большей полярностью, чем
обычный новолак.
Модификация новолачных смол для использования в производ-
стве лаков сводится, главным образом, к уменьшению полярности
новолака с целью сделать его растворимым в неполярных раствори-
телях и совместимым с маслами, т. е. пригодным для производства
масляных лаков.
Полярность новолака определяется наличием ОН-групп в бен-
зольных ядрах, связанных метиленовыми мостиками:
он он он
I I I
iQj—сн,-^—сн2-(0
При одном и том же значении и расположении диполя степень
полярности молекулы характеризуется отношением веса полярной
группы (гидроксила) к весу всей молекулы; чем это отношение
меньше, тем слабее полярность. Кроме того, полярность — величина
векторная, являющаяся результатом геометрического сложения
векторов диполей; это, естественно, приводит к неравноценности
орто-, мета- и паразамещенных производных бензольного кольца и
к уменьшению полярности при переходе от орто- к парапроизвод-
ным. Поэтому при переходе в производстве новолака от фенола к
его замещенным (фенол — крезол — ксиленол и т. д.), в особенно-
сти к паразамещенным, а также при переходе от формальдегида
к его высшим гомологам (ацетальдегид — масляный альдегид
и т. д.) и при применении более сложных паразамещенных фенолов,
можно настолько уменьшить полярность новолака, что он будет
растворяться в неполярных растворителях и совмещаться с мас-
лами. ' . Г. -
Получение маслорасггворимых смол эфарчзацпей новолака
403
Таким образом, для модификации новолака с целью уменьшения
его полярности и получения маслорастворимых смол могут быть
применены следующие методы: 1) блокирование, замещение поляр-
ных (гидроксильных) групп путем получения простых и сложных
эфиров новолака и 2) усложнение молекул новолака путем введе-
ния больших неполярных групп в параположении.
Для модификации новолаков с целью получения более полярных
смол можно исходить из соединений, содержащих две гидроксиль-
ные группы (например резорцин), или из соединений, содержащих
че гидроксильную, а более полярную — карбоксильную группу.
В последнем случае образуются поликонденсаты, растворимые
в слабых щелочах, буре, соде и т. п.
Получение маслорастворимых смол эфиризацией
гидроксильных групп новолака
Простые эфиры новолака
Простые эфиры новолака могут быть получены действием алкил-
хлоридов на щелочные производные (феноляты) новолака по схеме:
• • + «NaOH
•+пН2О
• + nRCl •—►
Феноляты новолака получают растворением новолака в водной
щелочи. Реакцию эфиризации проводят путем внесения хлоридов в
горячий раствор фенолята, причем она завершается быстро и про-
стой эфир новолака выпадает в осадок.
По этой схеме получали маслорастворимые смолы действием на
човолак м-толуолсульфохлоридом, являющимся отходом сахарино-
вого производства.
Превращение новолака в простой эфир приводит к следующему
лзменению его свойств: 1) уменьшается полярность смолы, вслед-
26*
404 Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
ствие чего опа становится растворимой в неполярных растворите-
лях, в том числе и в маслах; 2) улучшается ее светостойкость,
так как в ней отсутствуют фенол и фенольные группы; 3) несколько
повышается температура размягчения смолы (на 10—15°).
Сложные эфиры новолака
Сложные эфиры новолака, в частности эфиры новолака со смо-
ляными кислотами канифоли, а также с жирными кислотами со-
вместно с канифолью получили большое промышленное значение.
Смоляные кислоты канифоли представляют смесь нескольких изомерных
кислот, имеющих циклическое строение и содержащих одну карбоксильную
группу и одну или две ненасыщенные связи. Из них наибольшее значение имеют
абиетиновая и левопимаровая кислоты. Под абиетиновой кислотой часто под-
разумевают все виды кислот канифоли.
Были выделены и изучены пять смоляных кислот канифоли, а именно:
абиетиновая
декстрапимаровая
неоабиетиновая
изодекстрапимаров ая
Левопимаровая кислота, имеющая так же, как и абиетиновая, две нена-
сыщенные группы внутри колец и одинаковые боковые группы, переходит
в абиетиновую кислоту при нагревании канифоли свыше 150°. Можно считать,
что при реакциях, проходящих при высоких температурах (свыше 150°), лево-
пимаровая кислота, как таковая, уже не реагирует, так как полностью изомери-
зуется в абиетиновую. Левопимаровую кислоту можно отделить от других кис-
лот, в частности от абиетиновой, при действии на нее малеиновым ангидридом,
С последним она реагирует уже при обычных температурах, образуя продукты
присоединения (аддукты, стр. 588), в то время как абиетиновая кислота реаги-
рует с малеиновым ангидридом лишь при 150° и выше. Неоабиетиновая кислота
также сравнительно легко изомеризуется в абиетиновую. Эти три кислоты харак-
теризуются сопряженной диеновой группировкой, которая отсутствует в осталь-
ных двух кислотах.
Получение маслорасп>ворамы.х смол эфаразацаей новолака
405
Реакции, протекающие при взаимодействии канифоли с новола-
ком или с другими продуктами феноло-альдегидной конденсации,
еще недостаточно изучены. Их обычно рассматривают как реакции
эфиризации гидроксильных групп новолака кислотными группами
смоляных кислот:
ОН ОН OCOR OCOR
I I I I
---СН2—------------И rzRCOOH—>----/\-СН2—/Х|-----1- лН2О
(COR — остаток смоляных кислот), а образующиеся продукты счи-
тают сложными эфирами новолака и смоляных кислот. Однако в
последние годы появились работы, указывающие на возможность
взаимодействия продуктов феноло-альдегидной конденсации со смо-
ляными кислотами и без участия карбоксильных групп этих кислот.
Такие реакции могут быть осуществлены между о-метилольными
производными феноло-альдегидной конденсации и двойными свя-
зями смоляных кислот. Согласно предложенной схеме, о-метилоль-
ные производные при высоких температурах переходят в промежу-
точную хинонметидную форму и далее реагируют со смоляными кис-
лотами по месту двойной связи с образованием продуктов присоеди-
нения, имеющих структуру бензопирановых (хроменовых) колец:
хинометид ненасыщенная производное
кислота бензопирана
Так, при взаимодействии о-феноло-спиртов с абиетиновой кисло-
ой получаются продукты присоединения следующего строения:
406
Гл. IX. Смолы, получаемые на основе фенолов и альдегидов
Возможность образования таких продуктов присоединения при
наличии в смолах феноло-альдегидной конденсации о-метилоль-
ных производных (в случае, когда конденсация была проведена
в щелочной среде) не исключает и реакции эфиризации новолаков
смоляными кислотами. По всей вероятности протекают оба процесса,
и преобладание какого-либо из них определяется условиями реак-
ции (характером исходных реагирующих веществ, pH среды
и др.).
Возможны два технических метода получения фенольных смол,
модифицированных смоляными кислотами канифоли: 1) метод не-
посредственного сплавления новолака с канифолью — или с выде-
ленной из нее абиетиновой кислотой и 2) метод совместной конден-
сации фенола с формальдегидом в присутствии смоляных кислот
канифоли, которые в стадии конденсации ведут себя как кислый
катализатор; после конденсации и сушки смолу подвергают терми-
ческой обработке и тогда происходит эфиризация новолака кисло-
тами канифоли. Этот процесс протекает при более высоких темпе-
ратурах. Избыток абиетиновой кислоты может быть эфиризован
глицерином, пентаэритритом или нейтрализован окисью магния или
кальция с образованием резинатов. Поэтому полученные таким
образом смолы нельзя рассматривать как чистые эфиры новолаков.
Наряду с процессами эфиризации известную роль играют и взаимо-
действие формальдегида с ненасыщенными группами канифоли,
а также процессы переэфиризации в присутствии глицерина. Масло-
растворимые смолы (сложные эфиры новолака и канифоли) из-
вестны в СССР под названием искусственных копалов. Технология
искусственных копалов была разработана С. Н. Ушаковым и
В. М. Зельцер.
Производство искусственных копалов рационально проводить по
первому методу — непосредственного сплавления новолака с кани-
фолью. Его преимущество — в применении стандартных твердых,
смол; при этом процессе обычно не требуется дополнительная эфи-
ризация глицерином. Целесообразно применять крезольно-формаль-
де1*идные новолаки и новолаки на основе торфяных фенолов
(стр. 418), так как они обладают меньшей полярностью. По темпера-
туре плавления новолак должен быть близок к идитолу (стр. 390) и
содержать не более 0,1% свободного крезола. Технологический про-
цесс получения искусственного копала по этому методу складывается
из следующих операций: смешения и сплавления новолака с раз-
дробленной канифолью, продувки полученной смолы перегретым
паром, сливания, охлаждения и дробления полученной смолы.
Эфиризация новолака непосредственным сплавлением с ка-
нифолью является основной операцией. Ее ведут при 280—300°
в течение длительного времени (до 24 час.). Для устранения
окислительных процессов через реакционную смесь пропускают
углекислый газ. По окончании сплавления для удаления лету-
чих продуктов разложения новолака и канифоли (канифоль-
Получение маслорастворимых смол усложнением молекул фенолов 407
ное масло) производят продувку расплавленной смолы перегретым
паром.
Аппарат (эфиризатор) для сплавления представляет собою
стальной котел (обычно диаметром 700—900 мм, высотой 1200—
1500 мм), снабженный крышкой с отводной трубкой к холодиль-
нику. Применяют котлы с выносной, огневой или газовой топкой;
можно вести нагревание перегретой водой по системе фредеркинга.
В этом случае горячая вода под высоким давлением циркулирует в
трубах, расположенных на поверхности цилиндрической и сфериче-
ской части котла и залитых свинцом, а нагрев воды происходит от-
дельно от котла огневой топкой в специальной трубчатке.
Соотношение новолака к канифоли может варьировать в широ-
ких пределах. Лучшие результаты получаются при 200 ч. канифоли
на 100 ч. новолака.
Искусственные копалы обладают столь малой полярностью, что
растворяются в неполярных растворителях и хорошо совмещаются
с маслами. Они отличаются также большей твердостью, более вы-
сокой температурой плавления (130—140°) и лучшей светостой-
костью и погодоустойчивостью, чем новолачные смолы. Искусствен-
ные копалы должны иметь малое кислотное число (20—40) г
которое характеризует их неполярность и химическую устой-
чивость.
Искусственные копалы нашли применение в качестве заме-
нителя природных копалов — основного и важнейшего сырья
в производстве масляных лаков и линолеума.
Получение маслорастворимых смол путем усложнения
молекул фенольных веществ
Алкилфенольные или 100% фенольные
смолы
Снижение полярности и маслорастворимость фенольной смолы
могут быть достигнуты и при сохранении в новолаке гидроксильных
групп, если в «-положение исходного фенола введен какой-либо
сложный неполярный заместитель. К такого рода смолам относятся,
например, продукты поликонденсации формальдегида с п-третично-
алкилфенолами (например, с n-третичноизобутил- или п-третично-
изоамилфенолом), а также с циклогексил-, метилциклогексил- и
первично изобутилфенолом, н. гексил-, н. гептил-, н. октил- и дру-
гими алкилфенолами с длинными цепями.
Эти смолы получили название 100% фенольные смолы, так
как они содержат исключительно фенольные смолы и не содержат
других смол, например канифоли.
Реакция образования алкилфенольных смол может быть выра-
408 Гл. IX. Смолы, по гучлечче на основе фенолов и альдегидов
жена обычной схемой образования новолака:
Такие смолы могут быть только новолачными, термопластич-
ными, что определяется бифункциональностью «-замещенного фе-
нола; малая полярность и маслорастворимость — влиянием слож-
ного неполярного радикала в пара-положении к гидроксильной
группе. Применяемые для получения алкилфенольных смол п-тре-
тичноизоамил- и изобутилфенолы могут быть синтезированы различ-
ными путями, например действием на фенол в присутствии хлори-
стого алюминия ненасыщенных углеводородов, изобутилена или
изоамилена.
СН3 СН3
С=СН2 V-он Н3С—С—/ он,
I
СН, СН.;
а также соответствующих галоидных алкилов, в свою очередь полу-
чаемых действием хлористого водорода на третичные спирты:
СН3 СН3
Н3С—С—ОН Н3С—С—CI
I [
СНз СНз
СН3 СН3
Н3С—С—Cl-ф/ ОН—> Н3С—С—/ OH4-HCI.
СНз СН.
Алкилфенольные смолы были изучены А. А. Ваншейдтом с сотруд-
никами. Они получали алкилфенолы путем взаимодействия 1 ч. фе-
нола и 1,2 ч. алкилхлорида, в присутствии хлористого алюминия, в
течение 4—6 часов при 60—90°. По окончании выделения хлористого
водорода алкилфенол перегоняли с водяным паром и сушили.
Конденсация алкилфенолов с формальдегидом происходит при
избытке формальдегида (2 мол. формальдегида на 1 мол. фенола).
Избыток формальдегида не может вызвать перехода смолы на
основе бифункционального фенола в неплавкое состояние и в то же
время обеспечивает более полное связывание алкилфенола. Реакция
может идти в присутствии как кислого, так и щелочного катализа-
тора. Обычно применяют щелочную конденсацию (с едким натром),
так как реакция с кислотой идет более медленно и не полно.
Так, 100 вес. ч. zz-третичноизобутилфенола, 100—120 вес. ч.
07% формалина и 1% едкого натра (к фенолу) нагревают при 100°
Получение маслорастворпмых смол усложнением молекул фенолов 409
з течение 6—12 час. при интенсивном перемешивании и пропускании
углекислоты, поступающей через барботер; после этого щелочь ней-
трализуют соляной кислотой и смолу сушат под вакуумом при
120—140°. Получаются твердые, хрупкие смолы желто-лимонного
цвета с температурой размягчения ПО—115°, растворимые в бен-
зине, бензоле, в высыхающих маслах. Температура плавления
алкилфенольных смол значительно повышается при длительном на-
гревании, но они остаются, однако, плавкими и растворимыми.
Дешевые маслорастворимые смолы типа «100%-ных» могут быть
-получены при замене индивидуальных алкилфенолов соответствую-
щими фракциями сланцевой смолы, содержащими смеси замещен-
ных фенолов.
Для получения алкилфенольных смол, наряду с обычным мето-
дом конденсации алкилфенолов с формальдегидом, было предло-
жено применять алкилирование новолаков, например при помощи
олефинов в присутствии НС1О4.
Алкилфенольные смолы являются весьма ценным сырьем для
масляных лаков. Они обладают рядом преимуществ перед другими
смолами, в частности перед эфиризованными новолаками. Алкил-
фенольные смолы: 1) не желтеют, весьма погодоустойчивы и прак-
тически не стареют, что объясняется отсутствием в них свободных
смоляных кислот, которые обычно имеются в искусственных копа-
лах; 2) обладают хорошими механическими свойствами, гибкостью
пленок, хорошей адгезией, твердостью и эластичностью; 3) весьма
мало газопроницаемы. Последнее является важным преимуществом
этих смол, благодаря чему они нашли применение в качестве лаков
для аэростатов и других целей.
В последнее время приобретают значение термореактивные
маслорастворимые смолы. Основы строения их являются феноло-
спирты, т. е. те же соединения, которые служат основой обычных
резольных смол. Для получения такого рода смол реакцию конден-
сации фенола с формальдегидом в присутствии щелочи ведут лишь
до образования феноло-спиртов. В этой стадии реакцию обрывают
( первичные продукты реакции — феноло-спирты — частично эфири-
зуют спиртами (бутиловым или амиловым):
он
\
+зсн3о—>
он
НОН2С—1/\—СН2ОН
сн.он
он
1
НОН2С—j/\-CH3—о— С4Н9
Y
СН..ОН
+с4наон ,
410 Гл. IX. Смолы, получаемые на основе фенолов а альдегидов
Такие смолы становятся растворимыми в маслах, однако они
сохраняют способность при нагревании переходить в неплавкое со-
стояние, если некоторая часть феноло-спиртов не подверглась эфи-
ризации.
Феноло-спирты или их эфиры с высшими спиртами реагируют
также с ненасыщенными жирными кислотами и маслами, образуя
так называемые пластифицированные резолы, способные к отвер-
ждению за счет как ненасыщенных связей жирных кислот, так и ме-
тилольных групп резола. При этом ненасыщенные жирные кислоты
одной из двойных связей взаимодействуют с феноло-спиртом, обра-
зуя путем выделения воды соединения следующего типа:
—ОН
НОН2С—
—СН,ОН
сн—сн=сн—(сн2)5—соон
+ II
СН—СН=СН—(СН2)6-СН3
СН—СН=СН—(СН2)6—СООН
СН—СН=СН—(СН2)5—СН3
XCH2Z
СМОЛЫ НА ОСНОВЕ ФЕНОЛОВ И ЗАМЕНИТЕЛЕЙ
ФОРМАЛЬДЕГИДА
Формальдегид до настоящего времени является наиболее важ-
ным и часто единственным альдегидом, применяемым для производ-
ства фенопластов. Однако были проведены обстоятельные работы
по изучению возможности применения и других альдегидов, из ко-
торых большое практическое значение получил лишь фурфурол.
Феноло-фурфурольиые смолы
СН—СН
11 11
СН С—С<^ является весьма реакционноспособным
'о/
Фурфурол
соединением. Как альдегид он вступает в поликонденсацию с фе-
нолами, а как производное фурана, в гетероциклической группи-
ровке которого имеется система сопряженных двойных связей, — он
склонен к полимеризации.
Реакция полимеризации, ведущая к желатинизации фурфурола,
ускоряется воздействием' крепких кислот. По этой причине поли-
конденсация фурфурола с фенолами в присутствии сильных кислот
может закончиться образованием желатинированных и неплавких
Феноло-фурфурольные смолы
411
смол. На практике поэтому конденсацию чаще всего ведут в щелоч-
ной среде, обычно в присутствии углекислых щелочей. В реакцию
вводят, например, на 1 мол. фенола 0,75—0,90 мол. фурфурола, —
получаются новолачные смолы со сравнительно высокой температу-
рой плавления. При большем количестве фурфурола в результате
щелочной конденсации получаются смолы, способные при высоких
температурах (180°) переходить в неплавкое состояние. Возможно,
что реактивные точки бензольного кольца не участвуют в этом
процессе, и переход в неплавкое состояние основан на раскрытии
сопряженных связей фуранового кольца, с образованием попереч-
ных пространственных связей.
Феноло-фурфурольные смолы можно получать, проведя конден-
сацию под давлением в автоклаве. Так, в автоклав (рис. 151, стр. 380)
загружают: 100 ч. фенола, 80 ч. фурфурола и 0,5—0,75 ч. едкого
натра. После загрузки сырья производят интенсивное перемешивание
сжатым воздухом, автоклав герметически закрывают, пускают ме-
шалку и дают в рубашку автоклава пар (5—6 ат). Нагревание ве-
дут до тех пор, пока давление внутри автоклава не достигнет 4,5—
5,5 ат, затем пар выключают и дальнейший подъем температуры
в автоклаве и, следовательно, повышение давления происходят за
счет экзотермической реакции. Давление постепенно поднимается до-
10—12 ат, причем для регулирования давления в рубашку автоклава
периодически пускают воду. При 8—10 ат (~170—185°) реакцию
продолжают в течение 40—60 мин.; в случае же падения давления
в рубашку снова дают пар. Потом автоклав охлаждают. Когда
давление снижается до 1 —1,5 ат, смолу сливают в промежуточный
сборник или в сушильный агрегат. Сушку производят в вакуум-
сушильных агрегатах, при постепенном подъеме температуры в смоле
до 125—135°. Процесс заканчивается при получении смолы с тем-
пературой размягчения по Кремер-Сарнову 80—85°; метод капле-
падения по Уббелоде не дает для этих смол однозначных результа-
тов, вследствие высокой вязкости расплавленных смол.
Полученные смолы могут содержать до 10% свободного фенола;
их скорость желатинизации (с 10% уротропина) 60—120 сек. при
150° и 40—60 сек. при 180°.
Для получения термореактивных (резольных) смол Г. С. Петров
предлагал проводить комплексную конденсацию фенола с формаль-
дегидом и фурфуролом в две стадии: в первой стадии в присутствии
кислого катализатора с фурфуролом и во второй стадии — с форм-
альдегидом в присутствии щелочного катализатора.
Фурфурольные смолы имеют некоторые преимущества перед феноло-форм-
альдегидными: онн лучше пропитывают наполнитель и дают прессизделия более-
однородного цвета и лучшего внешнего вида. Важнейшим отличием этих смол
является их особое поведение при различных температурах обработки и прес-
сования. Так, феноло-фурфурольные смолы резольного, а также новолачного
типа в смеси с уротропином проходят обычные стадии отверждения В и С
с иной скоростью и в ином температурном интервале.
412
Гл. IX. Смолы., получаемые на основе фенолов и альдегидов
Значительно более сложные комплексы феноло-фурфурольных продуктов кон-
денсации (по сравнению с феноло-формальдегидными) взаимодействуют с обра-
зованием сшитых молекул лишь при более высоких температурах; вследствие
этого и каучукоподобная, нетекучая стадия В достигается только при более вы-
соких температурах и смола в значительном временном и температурном интер-
вале (130—150°) сохраняет свою высокую подвижность. При 180—200° потен-
циально-реактивная смола быстро переходит в стадию С, повидимому, также
в результате полимеризации, за счет ненасыщенных связей фурфурола. Этот
переход в стадию резита при высоких температурах (180—200°) происходит
с большей быстротой, чем в случае феноло-формальдегидных смол.
Поэтому кривая зависимости вязкости от температуры для феноло-фурфу-
рольных смол по сравнению с феноло-формальдегидными имеет иной характер:
она более пологая при низких температурах (130—150°) и более крутая при вы-
соких (180—200°). Вышеуказанные температурные зависимости фе.чоло-фурфу-
рольных смол более благоприятны для переработки пресскомпозиций на основе
этих смол методом литья под давлением (пресслитья), так как такой метод
требует более длительного сохранения подвижности массы в тигле машины
при температурах текучести композиции (~150°) и быстрого отверждения массы
в прессформе при 180—200°.
Преимущества фурфурольных смол заключаются также в большей теку-
чести пресспорошков на их основе и способности лучше заполнять прессформу;
•они дают меньший процент брака по показателям цветности и неоднородности
прессованных изделий и большую производительность пресса при работе при
высоких температурах (180—200°). Их преимущество особенно выявляется при
прессовании больших изделий сложного профиля, когда требуется более высо-
кая подвижность массы и сохранение ее текучести в процессе прессования до
момента заполнения прессформы и оформления изделия.
Заменители формальдегида, не обладающие явной
альдегидной функцией
Смолы могут быть получены при конденсации фенола с древеси-
ной или с какой-либо из трех ее основных составных частей—цел-
люлозой, лигнином и пентозанами.
Первые сведения о конденсации фенола с целлюлозой в кислой
•среде и о получении смолообразных продуктов относятся еще к
1909 г. Можно полагать, что реакция в этом случае идет по пути
гидролиза целлюлозы с образованием глюкозы, вступающей затем
в конденсацию с фенолом.
Были также проведены работы по конденсации пентозансодер-
жащих веществ (колосья, лузга) с фенолами в присутствии крепких
кислот. Образование смол, повидимому, происходит в результате
гидролиза пентозанов с образованием фурфурола и конденсации
последнего с фенолом.
Технология феноло-лигниновых и феноло-древесных смол впер-
вые была разработана в Советском Союзе в результате работ
С. Н. Ушакова, Е. Н. Фрейдберга, И. И. Матвеева, а также
И. П. Лосева с сотрудниками.
Ф е н о л о - д р е в е с н а я смола (Ф. Д.)
При взаимодействии древесины с фенолом в присутствии серной
кислоты получаются плавкие смолы новолачного характера. Химизм
Заменители формальдегида, не облад. альдегидной функцией 413-
реакции', невидимому, очень сложен, так как реагируют продукты
деструкции и гидролиза различных веществ (целлюлозы, пентоза-
нов, лигнина).
Для получения феноло-древесных смол берут примерно 100 ч.
фенола, 60 ч. сосновых опилок и 3—5 ч. серной кислоты. Распла-
вленный фенол загружают в реактор из кислотоупорной стали, в ру-
башку которого пускают пар (1—1,5 ат), вносят серную кислоту и
после 10 мин. перемешивания при продолжающей работать мешалке
начинают загружать древесные опилки. Вначале вносят 30—40% от
всего количества опилок (в течение 15—25 мин.), через час еще1
30%, а через 1,5—2 часа остальное количество. По окончании за-
грузки давление пара поднимают до 1,5—2 ат и при НО—115° ве-
дут конденсацию с обратным холодильником еще 3—4 часа; затем
приступают к сушке смолы, для чего давление пара в рубашке'
доводят до 6 ат и переключают холодильник на прямой. Когда
сушка закончена, в котел вносят водный раствор извести (для ней-
трализации кислоты), перемешивают и сушат в течение часа, после’
чего готовую смолу сливают.
Реактор представляет собою обычный варочно-сушильный агре-
гат, изготовленный из кислотостойкой стали.
Получаемая новолачного типа смола имеет температуру капле--
падения 90—110°, окрашена в коричневый цвет, содержит до 15%
свободного фенола и при смешении с 15% уротропина скорость ее'
отверждения (при 150°) 40—60 сек. Выход смолы 130—140%
к фенолу; это примерно на 30% больше выхода обычных феноло-
формальдегидных смол. Таким образом, производство феноло-дре-
весных смол по сравнению с феноло-формальдегидными дает эконо-
мию всего формальдегида и около 30% фенола.
Прессматериалы, получаемые на основе феноло-древесной смолы,,
уступают однако по водостойкости, текучести и механической проч-
ности нормальным новолачным прессматериалам (на основе феноло-
формальдегидных смол).
Феноло-лигниновые смолы (Ф. Л.)
Лигнин является составной частью древесины и спутником цел--
люлозы.
При техническом получении целлюлозы лигнин удаляют путем
обработки древесины реагентами, разрушающими лигнин, но не дей-
ствующими на целлюлозу. В процессе образования целлюлозы по
натронному или сульфатному методу из сульфатных щелоков может
быть выделен так называемый щелочной лигнин, который в значи-
тельной степени деструктирован; при производстве целлюлозы по*
сульфитному методу лигнин остается в растворе сульфитных ще-
локов.
При осахаривании древесины минеральными кислотами целлю-
лоза гидролизуется до глюкозы, в то время как лигнин остается1
414 Гл. ГХ. Смолы, получаемые на основе фенолов и альдегидов
мало измененным. Таким образом, лигнин получают в громадных
количествах как в виде значительно деструктированного щелочного
лигнина и лигнина сульфитных щелоков, так и в виде малодеструк-
тированного так называемого кислотного, гидролизного.
Химический состав лигнина еще точно не известен. Установленным является
лишь наличие в нем метоксильных и гидроксильных групп и ароматический
характер его строения. Молекулы лигнина не имеют нитевидной, линейной струк-
туры, подобно целлюлозе, а представляют собою глобулярные комплексы; воз-
можно, что лигнин в древесине представляет собою трехмерную макромолекуляр-
ную структуру, чем и объясняется его нерастворимость.
Ароматический характер лигнина вытекает как из его оптических свойств,
так и из характера продуктов его пирогенетического разложения и ряда дру-
гих реакций; свободные гидроксильные группы лигнина имеют фенольный ха-
рактер, а метоксильные — характер метилированных фенольных гидроксилов.
Однако необходимо подчеркнуть, что с достаточной определенностью не вы-
яснен не только химический состав лнгнина, но до сих пор не удалось даже
с достаточной четкостью провести отграничение его от других сопутствующих
ему одеревеневших клеток и инкрустов древесины.
В 1897 г. Бюлер для получения целлюлозы подверг древесину обработке
фенолом; при этом нецеллюлозиая часть оказалась растворенной в феноле. До
1915 г. исследователи считали, что процесс извлечения лигнина фенолом есть
процесс растворения, пока не было установлено, что фенол взаимодействует
с лигнином, причем реакция эта ускоряется кислотами и другими катализаторами.
Было доказано, что при взаимодействии фенола с лигнином на каждую
молекулу фенола выделяется одна молекула воды и что гидроксильные группы
фенола, повидимому, в большинстве своем остаются без изменения, а реагируют
лишь водородные группы (атомы) бензольного ядра фенола с гидроксильными
-группами лигнина по схеме:
\/ \/
он + \ /—он —\ у—ОН
водородные и гидро- структурные элементы
ксильные группы молекулы феноло-лнгиина
лигиииа
Гидроксильные группы фенола могут частично образовывать также и эфир-
ную связь. Исходя из этих положений и принимая состав молекулы лигнина
>C47H55Oi9 и что 1 мол. лигнина связывает 4 мол. фенола, Браунс и Гибберт
предложили следующую формулу для феноло-лигнина:
НО—
НО—
но—
но—
/—°—
—осн3
—осн3
—осн3
осн3
осн3
С42Н32О6
Из четырех молекул фенола 1 молекула связывается эфирной связью,
а 3 молекулы — путем конденсации гидроксильных групп лигнина с активными
водородными атомами ядра фенола.
Предложенной структуре феноло-лигнина соответствует растворимость его
,з щелочах и полярных растворителях (в спирте, ацетоне и др.).
Заменители формальдегида, не облад. альдегидной функцией
♦15
По современным воззрениям феноло-лигнин представляет слож-
ную смесь полимергомологов и аналогов и является продуктом
соединения фенола с деполимеризованными, деструктированными
частями лигнина. Чистый феноло-лигнин — полностью освобожден-
ный от фенола — неплавкий продукт. Технический продукт конден-
сации всегда представляет собою твердый раствор феноло-лигнина
в феноле и такой раствор является плавкой смолой. Таким образом,
наличие «свободного» фенола в смоле является не недостатком (как
в случае обычных новолаков), а в определенных пределах необхо-
димым условием получения технически годного продукта — плавкой
смолы новолачного характера. Повидимому, такое же значение
имеет свободный фенол и для смолы Ф. Д.
Отверждение феноло-лигниновой смолы происходит в результате
дальнейшей конденсации с формальдегидом или с уротропином.
Повидимому, в процессе отверждения важное значение приобретает
фенол, молекулы которого выполняют роль промежуточного соеди-
нительного звена между метиленовыми группами уротропина и фе-
ноло-лигнина.
Для получения феноло-лигниновых смол применяют примерно
следующую рецептуру: на 100 ч. фенола берут от 80 до 140 ч. гидро-
лизного лигнина (из расчета на сухой) и 3—4 ч. серной кислоты.
В варочно-сушильный аппарат загружают фенол, затем 2/з количества серной
кислоты и смесь при постоянном перемешивании нагревают до 100—105°; после
этого вносят 25% всего лигнина небольшими порциями в течение нескольких
минут. По мере внесения лигнина вязкость в котле сильно возрастает и необ-
ходимо наблюдать за нагрузкой мотора мешалки. После внесения первой пор-
ции лигнина (25%) температуру в котле поднимают до ПО—120° (при этом по
мере растворения лигиина вязкость в котле падает) и через полчаса загружают
вторую порцию — около 10% от общего количества лигнина; затем через каж-
дые четверть часа загружают такие же порции, пока не будет загружен весь
лигнин, после чего вводят остаток (!/з) серной кислоты и реакцию продолжают
при 110—120° в течение 60—90 мин. Затем холодильник переключают на обрат-
ный, температуру в реакторе поднимают до 120—140° и производят сушку смолы
до тех пор, пока температура каплепадения смолы не достигнет 120—130°. По
окончании сушкн для нейтрализации смолы в котел вводят водный раствор
извести, а затем — после испарения воды — олеиновую кислоту (2,5% от веса
смолы); смолу перемешивают и сливают.
Готовая феноло-лигниновая смола содержит 12—16% свободного
фенола; при 150° скорость желатинизации такой смолы с 10% уро-
тропина 50—60 сек., температура каплепадения 120—140°.
По основным свойствам феноло-лигниновая смола близка к ново-
лачным феноло-формальдегидным смолам. Физико-механические
свойства пресспорошков, получаемых на ее основе, почти не усту-
пают обычным новолачным пресспорошкам, в частности по скорости
прессования. Некоторый недостаток феноло-лигниновых смол —
большая вязкость в расплавленном состоянии (что обусловливает
худшую пропитку наполнителя и требует более высокой темпера-
туры при вальцевании), а также некоторая хрупкость при механи-
ческой обработке изделий. Важным преимуществом, однако,
416
Гл. IX. Смолы, получаемые на основе фенолов а альдегидов
является высокий выход этих смол (до 200% к фенолу); эк.
дает значительную экономию фенола (кроме экономии формаль-
дегида).
Феноло-лигниновые смолы на щелочном
лигнине (Ф. Щ. Л.)
Щелочной лигнин, который образуется в виде отхода при натронном или
сульфатном методе получения целлюлозы, отличается значительно большей реак
ционной способностью, чем гидролизный лигнин.
С. Н. Ушаков установил, что щелочной лигнин непосредственно способен
реагировать с уротропином в присутствии фенола примерно в такой же сте-
пени, как и феноло-лигнин, получаемый на основе гидролизного лигнина. Фенол
выполняет в этой реакции роль промежуточного звена по схеме:
Oil
лигнин—СН2——СН2—лигнин.
Это позволяет значительно снизить содержание фенола (примерно дс
20—30% к лигнину), причем отпадает необходимость в проведении процесса
конденсации фенола с лигнином, а можно ограничиться лишь приготовлением
сплава из щелочного лигнина, фенола и уротропина примерно по следующей
рецептуре (в вес. ч.)
Лигнин................. 300
Фенол.................. 100
Уротропин ..........12—15.
Выход смолы (сплава)—до 400% к весу фенола. Путем непосредственногс
смешения древесной муки с перечисленными составными частями н последую-
щей вальцовки смеси можно получать пресспорошки, обладающие хорошими,
механическими свойствами, но недостаточно высокой водостойкостью.
ИСПОЛЬЗОВАНИЕ ФЕНОЛОВ ДЕГТЕЙ МАЛОКАЛОРИЙНЫХ ВИДОВ
ТОПЛИВА
Дегти малокалорийных видов топлива (торф, бурый уголь, сла-
нец и др.) могут быть использованы для дальнейшей переработки
на жидкое горючее, смазочные масла и т. д. лишь после предва-
рительного выделения из них фенолов. Последние могут быть при-
менены для получения синтетических смол.
Дегти малокалорийных топлив содержат значительно большее
количество фенолов, чем коксовальные каменноугольные. Так
в торфяной газогенераторной смоле находятся (в %):
Кристаллический фенол................. 1,5
Трикрезол ............................ 4,8
Ксиленолы и высшие фенолы ............ 5,2
Всего . . . 11,5.
В буроугольном газогенераторном дегте содержится ~ 10%,
а в коксовальном 15—20% фенолов.
Использование фенолов дегтей малокалорийных топлив
417
В сланцевых дегтях содержание кислой фракции (фенолов)
также достигает 15—20%.
Большая часть фенолов, содержащихся в дегтях малокалорий-
ных топлив, относится к так называемым высшим фенолам, выде-
лить которые весьма трудно. Поэтому применение их может идти
по пути как извлечения части индивидуальных фенолов, так и
использования всей фенольной фракции посредством выделения
смеси всех фенолов и комплексной ее конденсации с формальде-
гидом.
В последнем случае, например, буроугольный деготь разгоняют и отби-
рают фракцию 210—280°, которая наиболее богата фенолами. Эту фракцию об-
рабатывают раствором едкого иатра, водный щелочной раствор фенолов осво-
бождают от эмульгированных углеводородов путем обработки острым паром и
затем разлагают 20%-раствором соляной кислоты. Выделившиеся фенолы про-
мывают горячей водой от кислоты, после чего они могут быть перегнаны под
вакуумом.
Полученные таким образом фенолы конденсируют с формальдегидом в ще-
лочной среде; при этом образуются коричневые и черные резольные смолы,
свойства которых в сильной степени зависят от химического состава фенолов.
С. Н. Ушаков и Г. С. Бродский получали пресспорошки и слоистые пластики на
основе смол, образовавшихся в результате комплексной конденсации фенолов
кислых фракций буроугольных дегтей. Ввиду недостаточно высоких технических
свойств полученных материалов, метод этот мало применим.
Более рациональное использование фенолов буроугольного и
торфяного дегтя может быть достигнуто, если предварительно про-
извести разгонку дегтя, например, на три фракции
I фракция.......... 180—205°
II ................ 205—230°
III ............... 230—280°.
Из этих фракций фенолы извлекают путем обработки водным
10% раствором щелочи. Водную часть, содержащую фенолы
в виде фенолятов, отделяют от смоляной части и подвергают очистке
от увлеченного масла (продувкой перегретым паром, промывкой
бензолом). После удаления масла феноляты разлагают кислотой,
фенолы промывают от кислоты и перегоняют под вакуумом или
с перегретым паром.
Первая фракция (< 10%) состоит из фенола и частично крезо-
лов, вторая фракция (30—40%) —из крезолов, ксиленолов и этил-
фенолов, а третья (около 50%)—частично из ксиленолов, но в
основном — из высших продуктов кислого характера. Во всех
фракциях имеются примеси карбоновых кислот, присутствие кото-
рых вредно отражается на процессе конденсации с формальде-
гидом; поэтому они должны быть удалены, что может быть до-
стигнуто промывкой фенолов 10% раствором соды.
Фенолы первой фракции близки по составу к так называемым
фенольным фракциям каменноугольного дегтя и могут быть,
следовательно, использованы в обычном процессе феноло-альде-
гидной конденсации для получения новолачиых и резольных смол.
27 Зак. 1269. Э. И. Барг
418
Гл. fX. Смолы, получаемые на основе фенолов и альдегидов
Фенолы второй фракции содержат в основном ксиленолы и частично
крезолы.
Ввиду небольшой концентрации в них трехфункциональных
соединений их используют для производства как резольных, так и
новолачных смол в смеси с фенолом, реже с трикрезолом. Для
производства слоистых пластиков они могут быть применены в ряде
случаев и без добавки других фенолов.
Фенолы третьей фракции характеризуются весьма малым содер-
жанием трехфункциональных фенолов; поэтому они могут найти
применение только для производства лаковых смол, например
искусственных копалов.
КОНДЕНСАЦИЯ ФОРМАЛЬДЕГИДА С МНОГОАТОМНЫМИ ФЕНОЛАМИ
Резорциновые смолы
Из трех изомерных двухатомных фенолов (о-, м- и п-диоксибен-
золов) только резорцин (лг-диоксибензол) имеет три реактивных
водорода бензольного ядра (табл. 45) и при конденсации с форм-
ТАБЛИЦА 45 альдегидом образует про-
Свойства изомерных диоксибензопов
Формула (-Х- — реактив- ные ТОЧКИ) Название Т. плавл. °C Т. кип. °C
он I
< /—ОН он Резорцин по 277
*п* Г идрохи- 169 25 (при
он он НОН 75 мм)
\ *|Z ''j-—он * Пирокате- хин 105 245
странственные продукты по-
ликонденсации.
Однако наличие второй
гидроксильной группы акти-
вирует водороды в .w-поло-
жениях (для о- и п-диокси-
бензолов). Поэтому смолы,
полученные на основе гидро-
хинона (n-диоксибензола) и
пирокатехина (о-диоксибен-
зола), также в некоторой
степени способны при высо-
ких температурах к медлен-
ным переходам в неплавкое
состояние. Кроме того, нали-
чие второй гидроксильной
группы у резорцина еще бо-
лее увеличивает подвиж-
ность водородов в о- и «-по-
ложениях и, кроме того, де-
лает вероятным образование
кислородных эфирных мо-
стиков между бензольными
ядрами.
Благодаря этому резорцин более энергично взаимодействует с форм-
альдегидом, чем фенол, и реакция может с достаточной скоростью
протекать на холоду и без внесения катализаторов. В технике из
всех изомерных диоксибензолов для производства термореактивных
Резорциновые смолы
419
смол применение получил только резорцин. Высокая реактивность
последнего делает более целесообразным применение твердых поли-
меров формальдегида — параформа. Так на 1 мол. резорцина берут
’.G мол. альдегида, причем для предупреждения преждевременного
и самопроизвольного перехода образующейся смолы в резит реко-
мендуется параформ вводить частями. Вначале вводят 'Д мол. пара-
форма и получают смолу новолачного типа с большим содержанием
свободного резорцина, которую уже непосредственно при приме-
нении переводят в резит добавлением остальной части параформа.
Процесс конденсации по этому методу представляет собою сплавле-
ние твердых и безводных веществ. Так, 55 ч. резорцина и 8 ч. па-
раформа смешивают в порошкообразном виде и нагревают в от-
крытом реакторе на водяной бане при 60—70°. Реакция протекает
очень быстро и приводит к образованию плавкой и растворимой
смолы, которая при дальнейшем нагревании с 10 ч. параформа пере-
ходит в термореактивную смолу, а затем в резит.
Смолы получают также при конденсации резорцина с техническим
формалином. Так, 100 ч. резорцина растворяют в 90 ч. 40% форма-
лина и раствор (без катализатора) медленно нагревают при пере-
мешивании до 65—70°. При этой температуре, поддерживаемой по-
стоянным охлаждением, реакцию продолжают час, после чего смолу
высушивают.
Прессовочные массы можно получать при непосредственном
вальцевании при 70—75° смеси резорцина и параформа с напол-
нителем (например с древесной мукой), после чего вальцованную
массу размалывают и прессуют в горячих прессформах.
Слоистые массы приготовляют путем пропитки листов бумаги
или ткани водным раствором уротропина, сушки листов и вторичной
пропитки водным раствором резорцина. После сушки листы при
горячем прессовании образуют твердый, неплавкий пластик. Реакция
поликонденсации с образованием пространственного полимера в этих
случаях протекает в основном в процессе горячего прессования.
Г. С. Петров получал водноэмульсионные смолы резольного типа
при конденсации без катализатора 1 мол. резорцина с 1 мол. форм-
альдегида в водной среде при 60—70°. Для предупреждения жела-
тинизации эмульсию разбавляли спиртом. Такие смолы могут быть
применены для пропитки слоистого наполнителя в производстве
слоистых пластмасс.
Резорциновые смолы в отвержденном состоянии обладают зна-
чительно большей теплостойкостью и твердостью, чем феноло-альде-
гидные смолы, и их применяют поэтому для изготовления деталей
нагревательных приборов. Они характеризуются также хорошими
электрическими свойствами и, в отличие от обычных резитов, не
образуют науглероженных поверхностных плёнок при электриче-
ских разрядах.
Благодаря большой реактивности и способности переходить
на холоду в резиновое состояние из резорциновых конденсатов
27’
420
Гл. IX. Смолы, получаемые на основе фенолов а альдегидов
изготовляют водные дисперсии, замазки и клеи, отвердевающие без
нагревания и в нейтральной среде. Резорциновые клеи отличаются
хорошей адгезией и теплостойкостью.
Применяют резорциновые смолы и в зубоврачебном деле в ка-
честве материала для зубных пломб, так как, в противоположность
фенольным смолам, они не токсичны и затвердевают на холоду.
Наконец, на основе резорциновых смол изготовляют прессмате-
риалы для производства изделий крупных габаритов, отверждаемых
при низких давлениях и низких температурах.
Применяют также смолы, получаемые комплексной конденсацией
резорцина, фенола с формальдегидом; такие смолы дешевле, чем
резорциновые, и обладают большей термостойкостью и твердостью,
чем феноло-формальдегидные. Смесь резорцина с параформом мо-
жет служить хорошим отверждающим агентом для новолачиых
смол вместо уротропина.
ТЕРМОРЕАКТИВНОСТЬ ФЕНОЛЬНЫХ СМОЛ
Из всех свойств термореактивных смол важнейшее значение для техники
пластмасс имеет характер термореактивности — скорость перехода в простран-
ственный полимер — в резнтол и резит.
Общая скорость образования пространственных полимеров складывается из:
1) скорости конденсации и получения смолообразных термоплавкнх продуктов
(резола или новолака), 2) скорости желатинизации и перехода в резнтольное
состояние, для которого характерны высокоэластические каучукоподобные свой-
ства, нерастворимость, но набухаемость в растворителях, и 3) скорости перехода
в конечное, твердое, неплавкое состояние (в резит).
Как установлено многими экспериментальными данными, скорости реакций
каждого из указанных этапов не зависят друг от друга; могут быть получены
смолы быстро желатинизирующиеся, но медленно переходящие затем в стадию
резита, и, наоборот, — медленно желатинизирующиеся, но быстро переходящие
в конечное, твердое состояние. Поэтому, говоря вообще о «скорости полимери-
зации» (поликонденсации), т. е. о скорости превращения смолы, нужно всегда
указать, о какой скорости идет речь—скорости ли перехода в резнтольное или
в резитовое состояние. Для большей ясности мы предлагаем называть скоростью
желатинизации — скорость перехода смолы из стадии .4 в стадию В, а скоростью
затвердевания — скорость перехода из стадии А в стадию С.
При изучении температурной зависимости скорости желатинизации и ско-
рости затвердевания отмечают существенное отличие в поведении резольных и
иоволачных смол (в смеси с уротропином). Это отличие наблюдается, например,
при рассмотрении кривых на рис. 162 и 163, где приведена зависимость ско-
рости желатинизации смол обоих типов от температуры. Резольные смолы при
высоких температурах (150—160°) имеют меньшую скорость желатинизации,
чем новолачные, однако у них менее резко выражена температурная зави-
симость, и при низких температурах (80—100°) их скорость желатинизации уже
выше, чем новолачиых. Таким образом, новолаки (с уротропином) представляют
собою смолы, быстро желатинизирующиеся при высоких температурах (150—160°)
и малореактивные при низких (до 120°); в то же время скорость желатинизации
резолов постепенно повышается с температурой: она меньше, чем у новолаков
при высоких температурах н значительно выше — при низких.
Характерное отличие резольных н новолачиых смол наблюдается и в ско-
рости их затвердевания. Если сравнить скорость затвердевания обоих типов
смол, имеющих при высоких температурах (150—160°) одинаковую скорость
Термореактивность фенольных смол
421
желатинизации, то обнаруживается, что резольные смолы всегда имеют во много
раз более высокую скорость затвердевания, чем новолачные, и для выравнива-
ния их необходимо, чтобы скорость желатинизации новолачной смолы была во
много раз выше скорости желатинизации резольной смолы.
Таким образом, резольные смолы при одинаковой с иоволаками скорости
затвердевания имеют значительно меньшую скорость желатинизации. При одной
и той же температуре (например 160°) они, следовательно, больший временной
интервал находятся в плавком, не желатинированном состоянии по сравнению
с новолаками. Эти существенные от-
личия имеют непосредственное зна-
чение для техники применения смол
обоих типов.
Для пресспорошков важнейшее
значение имеет скорость затвердева-
ния. которая определяет основные
свойства — текучесть и скорость прес.
Тсмпература, °C
Рис. 163. Скорость желатинизации
новолачных смол с уротропином.
7— уротропина, 12л10; 2 — то же, 19i:oi 3—то
же, в1,'!).
Рис. 162. Скорость желатинизации
резольных смол.
/ — резольная, феноло-формальдегидная смола,
F,o NH.; 2—то же. 0.5% NH ; 5—резольная,
феноло-анилино-альдегидная смола.
сования. Скорость желатинизации имеет значение лишь при процессе горячего
вальцевания и влияет на качество пропитки наполнителя смолой. Поэтому пра-
вильнее было бы характеризовать термореактивность смол для пресспорошков
наряду со скоростью желатинизации или, вместо нее, скоростью затверде-
вания. В противоположность получения пресспорошков при производстве
слоистых толстостенных изделий самое важное значение, наряду со скоростью
затвердевания, имеет скорость желатинизации, и чем она меньше (при одина-
ковой скорости затвердевания), тем лучше удается прогреть пакеты между пли-
тами пресса до желатинизации и затвердевания верхних слоев, соприкасающихся
с обогревающими плитами пресса. Именно поэтому резольные смолы, обладаю-
щие при высокой скорости затвердевания малой скоростью желатинизации, при-
меняются в качестве основного связующего материала в производстве слоис-
тых пластмасс.
Таким образом, оба показателя: скорость желатинизации и скорость за-
твердевания определяют поведение смолы в производстве слоистых пластиков.
ГЛЛПЛ Л’
НЕСЛОИСТЫЕ ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ
ФЕНОЛО-АЛЬДЕГИДНЫХ СМОЛ (ФЕНОПЛАСТЫ)
Пластические массы на основе феноло-альдегидных смол (фено-
пласты) делятся на: 1) литые (ненаполненные) материалы и
2) прессовочные (формовочные) материалы.
Литые материалы (литые резиты) представляют собою отвер-
жденные ненаполненные смолы, получаемые литьем в формах; из-
делия из них изготовляют в основном механической, станочной
обработкой (стр. 396).
Важнейшими фенопластами являются прессовочные материалы
(не менее 90% всех фенопластов).
Прессовочные (формовочные) материалы представляют собою,
в основном, композиции из смол (связующего) и наполнителя.
Как правило, система из смолы и наполнителя прочнее самой
смолы.
Наполнителями называют вещества, диспергированные в смоле
и химически к ней инертные. Условием усиливающего действия на-
полнителя является, однако, наличие таких остаточных сил сродства
между обоими компонентами, которые были бы достаточны для
смачивания наполнителя смолой. Таким образом, лиофильность
обоих компонентов — первое и основное условие для возможности
применения наполнителя. В этом случае физико-химические про-
цессы, связанные с развитием поверхностных сил прилипания (ад-
гезии), с диспергированием объемной смолы в систему тонких пле-
нок и с рядом других факторов, приводят к значительному упроч-
нению материала.
Наполнитель в сильной степени обусловливает и другие физико-
химические свойства материала — его водостойкость и химстой-
кость, диэлектрические свойства, теплостойкость, твердость и т. д.
Существенное значение имеет не только химическая, но и физиче-
ская структура наполнителя, его анизотропность, дисперсность,
ориентация, а также концентрация в материале. В первом
приближении, без учета ряда факторов, можно считать, что усили-
вающее действие наполнителя пропорционально активной (смачи-
ваемой) поверхности его на единицу объема смолы.
Введение наполнителей, которые значительно дешевле синтети-
ческих смол, снижает также и стоимость массы.
Композиции из смолы и наполнителя позволяют широко исполь-
зовать наиболее совершенные методы переработки в изделия — го-
рячее прессование, литье под давлением и формование. Развитие
Методы получения пресспорошков
423
этих методов определило во многом эффективность применения
прессовочных фенопластов; применение же наполнителей дало воз-
можность значительно снизить усадку фенопластов и изготовлять
изделия с высокой точностью размеров.
Прессовочные фенопласты делят на два класса: 1) неслоистые
(прессматериалы) и 2) слоистые (слойматериалы).
НЕСЛОИСТЫЕ ФЕНОПЛАСТЫ
(Прессматериалы)
По структуре наполнителя различают порошкообразные (прессо-
вочные порошки) и волокнистые (волокниты) прессматериалы.
Прессовочные порошки (пресспорошки) состоят из феноло-альде-
гидной смолы и тонкодисперсного, порошкообразного наполнителя
(древесная мука, тонкоразмолотый линтер, микроасбест, кизельгур,
каолин и т. д.).
Волокниты содержат обычно длинноволокнистый, свойлоченный
наполнитель (хлопок, асбест, стеклянное волокно и др.).
По характеру смолы прессматериалы делят на резальные и ново-
лачные.
Технологические методы получения пресспорошков
Пресспорошки получают либо так называемыми «мокрыми» ме-
тодами (лаковый и эмульсионный, пропитка смолообразующими
компонентами и др.), либо «сухими» (вальцовый, шнековый и др.).
Несмотря на то, что в производстве некоторых видов прессмате-
риалов мокрые методы пока еще не могут быть заменены сухими,
развитие производства прессматериалов идет по пути все более ши-
рокого внедрения сухих методов.
Основной недостаток существующих мокрых методов — необхо-
димость большого числа немеханизированных операций, что неиз-
бежно приводит к соприкосновению рабочих с парами фенола и
формальдегида. Кроме того, по этому методу приходится выпускать
мелкие партии продукта, — последний получается нестандартным.
Ниже дана краткая характеристика основных методов производ-
ства пресспорошков.
Лаковый метод
Сущность лакового метода состоит в пропитке наполнителя
(обычно волокнистого) спиртовым раствором твердой резольной
смолы.
Спиртовый раствор смолы — лак — получают в варочно-сушиль-
ном аппарате непосредственно после окончания варки и сушки
смолы (стр. 394). Обычно применяют лак с концентрацией смолы
55—65%.
424
Гл. X. Фенопласты неслоастые
Пропитку наполнителя производят в двухлопастном вакуум-ме-
шателе (рис. 173, стр. 447). В мешатель вводят спиртовый раствор
смолы, краситель и минеральный наполнитель; после 20—30-минут-
ного перемешивания постепенно (в продолжение 30 мин.) при-
бавляют волокнистый наполнитель и перемешивание продолжают
еще 60—90 мин. при обычной температуре либо при слабом подо-
гревании (40—50°). После смешения массу выгружают и переводят
на сушку в полочный вакуум-сушильный шкаф. Сушку ведут при
60—70° и при возможно более глубоком вакууме. Слой массы на
противнях должен быть толщиной не более 35—50 мм. В процессе
сушки необходимо периодически разрыхлять и перемешивать массу.
Испаряющийся спирт может быть использован, если вакуум-сушилка
снабжена конденсационной установкой. Однако и в этом случае
допустимы потери спирта до 15%. Более равномерную сушку и
большую производительность обеспечивают сушилки непрерывного
действия (рис. 196, стр. 530). По окончании сушки массу измель-
чают, просеивают н тарируют.
Лаковый метод применяют для получения прессмасс на основе
резольных смол и волокнистых наполнителей (целлюлоза, асбест).
Этот метод имеет ряд достоинств: дает хорошую пропитку напол-
нителя, возможность применять предварительно высушенные жид-
кие и твердые смолы; недостатками его являются (кроме расхода
спирта) получение нестандартного готового продукта из-за неравно-
мерности сушки в полочных вакуум-сушилках и невозможность уда-
ления остатков спирта, вследствие его высокой лиофильности
к смоле, комкования и слипания материала.
Эмульсионный метод
По эмульсионному методу пропитку наполнителя производят
водно-эмульсионными резольными смолами (стр. 394).
Резольная эмульсионная смола поступает в двухлопастный ва-
куум-мешатель и при небольшом подогреве (для снижения вязкости)
к ней постепенно прибавляют наполнитель (древесную муку, лин-
тер, асбест и др.). По окончании перемешивания массу выгружают
из мешателя и подвергают сушке, обычно на противнях в вакуум-
сушильных шкафах. Вследствие меньшей лиофильности удаление
остатков воды происходит легче, чем удаление спирта, несмотря на
его более высокую упругость пара.
Сравнивая лаковый и эмульсионный методы, можно прийти
к заключению, что эмульсионный имеет преимущества в отсутствии
расхода спирта, операций по сушке смолы, а также в более совер-
шенном удалении воды при сушке пропитанного материала.
Существенные недостатки эмульсионного метода заключаются в
применении нестандартных по вязкости конденсатов, склонных
к желатинизации и содержащих значительные количества свобод-
ного фенола и низкомолекулярных соединений.
Методы получения пресспорошков
425
Преимущества лакового метода — применение стандартных ре-
зольных смол.
Пропитка наполнителя смесью
смо л оо бразу ю щ их компонентов
Для лучшей пропитки волокнистых наполнителей был применен
метод пропитки не смолами, а исходными смолообразующими ком-
понентами, с последующим проведением процесса конденсации «на
волокне».
При осуществлении этого метода в двухлопастный вакуум-меша-
тель вносят фенол, формалин, катализатор и наполнитель (целлю-
лозу и др.) и при перемешивании нагревают до 65—85° в течение
1,5—2,5 час. В процессе конденсации смола образуется не только
на поверхности, но и внутри клеток целлюлозного наполнителя. По
окончании конденсации включают вакуум-мешатель и частично
переключают на сушку (до остаточной влажности 7—8%), после
чего массу выгружают и сушат в полочных вакуум-сушилках (до
остаточной влажности ~ 3% ).
Этот метод обеспечивает совершенную по равномерности про-
питку наполнителя связующим. Однако он имеет и крупные недо-
статки. Один из них — невозможность достаточно точно контроли-
ровать процесс конденсации и сушки; другой — повышенный расход
фенола и формалина, так как в этом случае конденсация не идет
до конца, даже в присутствии избытка катализатора. Метод кон-
денсации на волокне известен в СССР как неоконденситовый метод.
Он был детально разработан И. П. Эдельштейном.
Вальцовый метод
В процессе получения пресспорошков по вальцовому методу
применяют твердые смолы, в основном новолачные, но также и ре-
зольные. Метод состоит в предварительном измельчении и смеше-
нии твердых компонентов, входящих в состав прессматериала
(смола, древесная мука, уротропин и др.), и в последующем горячем
вальцевании и дроблении массы.
При вальцевании смола плавится и пропитывает наполнитель под
давлением. Пропитка под давлением необходима вследствие боль-
шой вязкости расплавленных смол; одновременно происходит хоро-
шее перемешивание, гомогенизация массы и испарение остаточной
влаги. Регулируя, время и температуру смешения, можно легко до-
вести пресспорошок до желаемой степени текучести, так как в
процессе вальцевания смола частично переходит в нетекучую ста-
дию В.
После вальцевания массу подвергают дроблению и измельчению.
Вальцовый метод имеет следующие существенные преимущества:
исходными продуктами являются стандартные твердые смолы;
426
Гл. X. Фенопласты неслоистые
значительно сокращена продолжительность процесса изготовле-
ния пресспорошков; применяется компактное оборудование с вы-
сокой производительностью, позволяющее механизировать техноло-
гический процесс; кроме того, он допускает регулирование
процесса и установление пофазного контроля, а также изготовле-
ние пресспорошков с заданной текучестью и с малым удельным
объемом.
По одному из вариантов (стр. 438) все стадии процесса (в том
числе и вальцевание) могут быть проведены непрерывно.
К недостаткам его следует отнести следующее: 1) метод в основ-
ном пригоден при применении порошкообразных наполнителей,
тогда как при длинноволокнистых наполнителях он менее рациона-
лен из-за недостаточной гомогенизации смеси и уменьшения проч-
ности массы, вследствие истирания волокна при вальцевании;
2) использование этого метода при применении резольных тверды?;
смол требует некоторой осторожности, причем в этом случае труднее
получить порошок с заданной текучестью; 3) не всегда достигается
вполне удовлетворительное распределение уротропина в массе;
4) процессы сухого смешения и горячего вальцевания, дробления и
размола массы требуют соблюдения специальных санитарно-гигие-
нических мер.
Несмотря на перечисленные недостатки вальцовый метод счи-
тается в настоящее время основным в технике производства ново-
лачных, а также и резольных пресспорошков.
Шнековый метод
Сущность этого нового метода состоит в предварительном раз-
моле и смешении твердых компонентов (твердых смол, древесной
муки, уротропина и др.) и в непрерывном проведении процесса го-
рячего смешения и пластикации через шнекпресс (стр. 438).
Преимущество этого метода — проведение производства пресспо-
рошков в виде непрерывного процесса.
Новолачные и резольные прессматериалы (пресспорошки)
Новолачные пресспорошки состоят из новолачной твердой смолы,
уротропина, красителя, смазки (жирные кислоты), древесной муки,
иногда окиси кальция и минерального наполнителя (каолин, тяже-
лый шпат и др.).
В состав резольных пресспорошков входят резольиая смола,
краситель, смазка, минеральный наполнитель, древесная мука, окись
кальция, иногда небольшое количество уротропина (1—3%).
Жирные кислоты и органический краситель вносят непосред-
ственно в котел после сушки смолы. Жирные кислоты добавляют
для устранения прилипания массы к стенкам прессформы в про-
цессе прессования.
Новолачные и резольные прессматериалы
42/
Уротропин добавляют для связывания молекул новолачной
смолы, состоящих из полиметиленфенольных ядер, метиленовыми
мостиками по месту активных точек фенольных ядер для перевода-
смолы в пространственный полимер (резит).
Небольшое количество уротропина добавляется к резольным
пресспорошкам с целью увеличения их термореактивности.
Окись кальция также до некоторой степени служит ускорителем
реакции отверждения; добавление ее улучшает теплостойкость
пресспорошков. Предполагают, что известь в процессе горячего
прессования связывает гидроксильные группы фенольных ядер с об -
разованием фенолятов кальция:
---( \-О—Са—О—/ ----
Основные отличия в свойствах новолачных и резольных пресс-
порошков сводятся к следующему: 1) новолачные пресспорошки
значительно более термореактивны при температуре прессования
(160—180°) и соответственно прессуются с большей скоростью,
чем резольные (время прессования новолачных пресспорошков
40—20 сек. на 1 мм толщины изделия, резольных 60—40 сек.);
2) соответственно этому новолачные прессматериалы имеют боль-
шую теплостойкость, чем резольные; 3) дают более прочные изде-
лия; 4) водостойкость, химстойкость и диэлектрические свойства
выше у резольных прессматериалов (в том числе и пресспорошков).
Следует указать на некоторые преимущества в оформлении тех-
нологического процесса получения новолачных пресспорошков, свя-
занные с тем, что твердая новолачная смола является более стабиль-
ным и стандартным материалом, чем жидкие, эмульсионные и даже
твердые резолы, и что новолачные прессматериалы могут длительно
храниться при обычных температурах без существенного изменения
их свойств, в то время как резольные — со временем изменяют свои
свойства вследствие дальнейших процессов поликонденсации.
Ввиду этого новолачные пресспорошки применяют для самого
разнообразного назначения, в то время как резольные — в основ-
ном для производства специальных деталей, для которых особое
значение имеют диэлектрические показатели или требуются высокие
водостойкость, химстойкость и стойкость по отношению к действию
атмосферных влияний. Однако резольные смолы чаще всего приме-
няют для производства прессматериалов на основе длинноволок-
нистых наполнителей.
Основным и важнейшим наполнителем для пресспорошков
является древесная мука. Высокая степень дисперсности и волок-
нистая структура древесной муки обусловливают прочность, теку-
честь, хорошую внешнюю поверхность, малый удельный вес и высо-
кие диэлектрические свойства прессматериала. Недостатки древес-
ной муки — гигроскопичность и большая, чем у минеральных
428
Гл. X. Фенопласты неслоистые
наполнителей (например у асбеста), чувствительность к термиче-
ским воздействиям. Основные требования, предъявляемые к дре-
весной муке, идущей в производство пресспорошков, относятся к ее
дисперсности и влажности: она должна проходить через сито № 30,
а степень допускаемой влажности зависит от метода приготовления'
пресспорошка; так, для вальцового метода допустима влажность не
свыше 3%.
Скорость прессования новолачиых
пресспорошков в зависимости от содержания
уротропина
Изделия из феноло-альдегидных пресспорошков изготовляют ме-
тодом горячего прессования в постоянно нагретых прессформах, из
которых их вынимают в горячем состоянии.
Продолжительность процесса прессования складывается из:
1) времени прогревания прессматериала в прессформе до темпера-
туры прессования (160—180°), 2) времени, необходимого для за-
полнения всей прессформы массой, перешедшей в пластичное, теку-
чее состояние, зависящего от скорости хода плунжера пресса и
конструкции прессформы и изделия и, наконец, 3) длительности хи-
мических процессов отверждения смолы. Последний фактор, являясь
функцией химического состава и свойств пресспорошка, в свою оче-
редь, зависит от термореактивности применяемой смолы, от содер-
жания в пресспорошке уротропина, от режима производства (напри-
мер, от длительности, температуры и других условий вальцевания)
и от предварительного подогревания пресспорошков перед прессо-
ванием. Последнее сокращает время прессования вследствие уда-
ления ’ влаги и частичной полимеризации смолы (переход в ре-
зитол).
Обычная выдержка прессматериала в прессформе (~40—60 сек.
на 1 мм толщины стенок) соответствует тому минимальному вре-
мени, которое необходимо, чтобы изделие после снятия с формы
в горячем состоянии не имело вздутий и деформаций; это дости-
гается лишь тогда, когда поверхностный слой изделия достаточно
тверд, чтобы выдержать внутреннее давление паров воды и ам-
миака. Такое состояние прессматериала далеко не совпадает с его
полным отверждением, т. е. с конечной стадией поликонденсации,
с переходом в твердое при температуре прессования состояние.
Практически для большинства изделий из фенопластов — это до-
пустимо.
Зависимость многих свойств прессматериалов (например диэле-
ктрических) от времени прессования проходит через максимум. Это
объясняется тем, что в процессе прессования, с одной стороны, уве-
личивается концентрация валентных пространственных связей, что
улучшает показатели прочности, теплостойкости и т. д., а с другой,
увеличивается выделение конденсационной воды, которая при
Новолачные и резольные прессмапгериалы
429
высоком давлении при прессовании не может быть удалена из
изделия и снижает диэлектрические свойства, водостойкость и хим-
стойкость.
Поэтому оптимум времени прессования лежит значительно ниже
времени, необходимого для полного отверждения материала. Когда
же требуется более высокая степень отверждения, применяют
термическую обработку изделий вне пресса. В этом случае
конденсационная вода имеет возможность испариться через поры
наполнителя.
Скорость отверждения и теплостойкость новолачных пресспорош-
ков зависят от количества
торый реагирует как со сво-
бодным фенолом, содержа-
щимся в новолаке (до 9 % ),
так и с молекулами ново-
лака.
Количество же уротропи-
на, необходимого для дости-
жения максимальной скоро-
сти прессования, зависит как
от содержания свободного
фенола в смоле, так и от
химического состава ново-
лака, определяемого отноше-
нием формальдегида к фе-
нолу в исходной смеси, про-
должительностью конденса-
ции и термической обработ-
ки смолы. Количество уро-
тропина зависит также от
того, насколько полно про-
исходит смешение и рас-
пределение уротропина в
введенного в смесь уротропина, ко-
Рис. 164. Зависимость теплостойкости и во-
достойкости пресспорошка от содержа-
ния (%) уротропина в новолачной смоле.
1— теплостойкость; 2— водостойкость.
пресспорошке, так как часть уротропина, которая вследствие
несовершенства распределения не находится в непосредствен-
ном контакте со смолой, практически не влияет на скорость от-
верждения. Чем более совершенен метод смешения, тем меньше
уротропина необходимо для достижения одинаковой скорости от-
верждения.
Следует иметь также в виду, что если недостаток уротропина
уменьшает теплостойкость и скорость прессования пресспорошка, то
его избыток, не увеличивая теплостойкости и скорости отверждения,
значительно ухудшает водостойкость прессматериала, его диэле-
ктрические свойства и в процессе прессования ведет к образованию
в изделиях вздутий (рис. 164). Оптимальное количество уротро-
пина— от 10 до 15% по отношению к смоле (в зависимости от ха-
рактера смолы и методов смешения).
.130
Гл. X. Фенопласты неслоистые
Новолачные пресспорошки
Состав новолачиых пресспорошков наоснове
древесной муки (фенодреволиты)
Новолак для изготовления пресспорошков должен удовлетворять
примерно следующим условиям:
Температура каплепадения по Уббелоде, °C ... . 95—105
Свободного фенола, %........................... 6—9
Время желатинизации с 10% уротропина при
150°, сек......................:............... 40-50
Вязкость 50% спиртовых растворов, сп....... 100—130
Основное различие в рецептурах новолачных пресспорошков
(фенодреволитов) заключается в содержании наполнителя, коли-
чество которого может варьировать от 60 до 40, реже от 70
до 30%. Наполнителем в
Upei&n прочности на изгиб,кг/см2
Рис. 165. Зависимость прочности на изгиб
новолачного пресспорошка от содержания
древесной муки.
основном является древес-
ная мука и в небольшом ко-
личестве также и минераль-
ные вещества — каолин, тя-
желый шпат, мумия, охра
(последние служат одновре-
менно и красителями), из-
весть и др.
Количество древесной му-
ки определяет основные фи-
зико-механические свойства
пресспорошка — текучесть
при прессовании, водостой-
кость, механическую проч-
ность. С увеличением количе-
ства наполнителя прочность
увеличивается (рис. 165),
однако лишь до определен-
ного предела, выше которого она снижается; объясняется снижение
прочности увеличением неоднородности порошка вследствие ухуд-
шения условий перемешивания. Текучесть и водостойкость с увели-
чением количества наполнителя (древесной муки) снижаются.
Новолачные пресспорошки можно разделить на две группы:
1) пресспорошки с оптимальным наполнением и 2) высоконапол-
ненные.
Пресспорошки первой группы содержат от 40 до 50% древесной
муки и, в свою очередь, подразделяются на ряд марок по химиче-
скому характеру новолачной смолы (например, феноло-формальде-
гидные, феноло-ксиленоло-формальдегидные, феноло-фурфурольные
Новолачные пресспорошки
431
н др.)- В качестве примера можно привести типичные рецептуры
(в %) новолачных пресспорошков с оптимальным наполнением:
Новолак.....................43,5 или 47,0
Древесная мука............. 43,5 45,5
Уротропин................... 6,8 5,0
Стеарин....................... — 0,5
Известь..................... 1,0 1,0
Минеральный наполнитель . . 4,2 —
Краситель................... 1,0 1,0
Высоконаполненные пресспорошки содержат от 60 до 70% дре-
весной муки. Они характеризуются пониженной водостойкостью и
текучестью, а также, чаще всего, и пониженной механической
прочностью.
Рис. 166. Дпобилка Блека.
Вальцовый метод производства
ф е н о д р е в о л и т о в
Процесс производства новолачных пресспорошков по вальцовому
методу складывается из следующих операций:- дробление и размол
смолы; размол и смешение со-
ставных частей; вальцевание сме-
си; дробление и размол вальцо-
ванной массы; укрупнение партий
и тарирование.
Измельчение смолы от сравни-
тельно крупных кусков, получен-
ных после выколачивания смолы
из противней, до необходимой тон-
кости помола, соответствующей
ситу № 30—40, производят в две
или три последовательные стадии
размола на различных агрегатах.
Для первоначального, грубого
дробления смолы может быть
применена, например, дробилка
типа Блека, схематически изображенная на рис. 166. Существен-
ными частями ее являются две рифленые щеки А и Б; первая не-
подвижна, а вторая, подвижная, качается на горизонтальной оси и
при движении дробит подаваемую сверху смолу; при этом полу-
чаются главным образом частицы размером 5—10 мм и более.
Средняя степень помола с получением частиц несколько меньших
размеров может быть достигнута на ударно-крестовых дробилках,
на вальцах, бегунах и т. д. И, наконец, тонкий помол смолы про-
изводят в шаровых мельницах и на ударноцентробежных дисковых
мельницах.
Процесс размола смолы осуществляют на дробильно-размоль-
ном узле, состоящем обычно из ударно-крестовой дробилки (для
432
Гл. X. Фенопласты неслоистые
среднего помола) и дисковой мельницы (для тонкого помола), со-
единенных между собою ковшевым элеватором.
Ударно-крестовая дробилка состоит из цилиндрического кожуха,
диаметром 800—1000 мм, внутри которого на быстро вращающемся
(1000—1200 об/мин.) валу укреплены 4 крыла. Основная и боковые
поверхности кожуха снабжены неподвижными ребрами, а нижняя
часть представляет собою решетку.
Дисковая мельница состоит из
щается (1000—1200 об/мин.) диск,
Рис. 1С7. Схема установки ударно-кресго-
вой мельницы с воздушной сепарацией
1 — мельница; 2—вентилятор; 3— конический воздуш-
ный сепаратор; 4—в .здуховод; 5 — циклон; 5— ру-
кавный фильтр; 7—питатель.
кожуха, внутри которого вра-
диаметром 600—650 мм, укреп-
ленный на стальном валу.
Диск снабжен тремя рядами
концентрически расположен-
ных кулачков, вращающих-
ся между кулачками непо-
движной боковой крышки
кожуха.
Смолу небольшими пор-
циями вручную загружают в
бункер крестовой мельницы;
после дробления она посту-
пает в приемник, из кото-
рого норией подается на ди-
сковую мельницу, а затем
поступает в приемный бун-
кер. Из бункера смолу вруч-
ную загружают в тару.
Тонкость помола смол
определяют номером сита,
через которое проходят
95—98% всего продукта. Обычно применяют сита № 30—40. Про-
изводительность дисковых мельниц 1200—1500 кг/час.
Более тонкий размол можно получить на мельницах ударного
типа с воздушной сепарацией, где проходящий ток воздуха обеспе-
чивает лучший отвод тепла и большую однородность помола. Схема
ударно-крестовой мельницы с воздушной сепарацией представлена
на рис. 167.
Вентилятор 2 создает разрежение в коническом воздушном сепараторе 3
мельницы 1. Смола увлекается с потоком воздуха и поступает в сепаратор, а от-
туда в циклон 5. Избыток воздуха выбрасывается через фильтр 6. Изменением
сечения прохода в верхней части сепаратора можно регулировать тонкость по-
мола, так как при этом меняется скорость воздуха. Крупные частицы, увлекае-
мые потоком воздуха, теряют скорость в верхней части сепаратора и возвра-
щаются на дополнительное измельчение через питатель 7.
Размол смолы производят также на узлах, состоящих из кони-
ческой зубчатой дробилки, соединенной норией с шаровой мельни-
цей. Схема такого узла, включающего выгрузку размолотой смолы
Новолачные пресспорошки
433
сжатым воздухом через воздушный сепаратор на механические
сита, представлена на рис. 168.
Смолу загружают в приемную воронку зубчатой мельницы для предвари-
тельного размола 5, откуда элеватором 2 через приемную воронку 1 она по-
дается в хранилище 3, а затем через затвор 4—в шаровую мельницу 8. Смола
определенной степени размола уносится током сжатого воздуха из мельницы и
попадает через передаточную трубку 9 и воздушный сепаратор 11 на механи-
ческие сита 12, 13, 14.
Рис. 1С8. Схема измельчения смол в шаровой мельнице.
1— приемная воронка; 2— элеватор; 3 — хранилище; 4— затвор; 5 —зуб-
чатая мельница для смолы; 6—передаточный вал к мотору;
7 — впуск сжатого воздуха; 8 — шаровая мельница; 9—передаточная
труба; 10—мешки для фильтрации воздуха; 11—воздушный сепара-
тор; 12, 13, 14—сита; 15— сборник для грубого помола; 16— барабан
для готового продукта.
Вторую стадию производства новолачных пресспорошков — сме-
шение размолотой смолы со всеми компонентами, входящими в со-
став пресспорошка, — обычно проводят в лопастных мешателях или
шаровых мельницах, в которых наряду с перемешиванием осуще-
ствляется и дальнейшее измельчение массы.
Шаровые мельницы, применяемые для смешения (или размола)
смол, футерованы диабазом или фарфором и снабжены диабазо-
выми или фарфоровыми шарами; это исключает загрязнение мате-
риала, а также предохраняет от взрыва органической пыли с воз-
духом, так как таким образом исключается возможность искрообра-
зования. Число оборотов шаровых мельниц должно соответствовать
оптимальному отношению
где: D — внутренний диаметр мельницы; п — число оборотов.
Введение составных частей и размол в шаровой мельнице прово-
дят в две фазы; вначале ведут загрузку и размол всех составных
28 Зак. 1269. Э. И. Барг
434
Гл. X. Фенопласты, неслоистые
частей, кроме древесной муки: смолы, уротропина, минерального
наполнителя, красителя. Продолжительность смешения и размола
30 мин. Затем загружают древесную муку и смешение продолжают
еще 30 мин.
После размола смесь
выгружают из шаровой
мельницы в приемный бун-
кер, из которого она по-
ступает на вальцевание.
Третью стадию произ-
водства новолачных пресс-
порошков — вальцева-
ние — проводят на сме-
сительных (фрикционных)
обогреваемых вальцах.
Благодаря различной ско-
рости вращения валков,
происходит сминание и
перемешивание расплав-
ленной массы, пропитка
наполнителя под давле-
нием и некоторое его исти-
рание, а также дополни-
тельная сушка и процес-
сы взаимодействия смолы
с уротропином, т. е. даль-
нейшие процессы поликон-
денсации и частичного пе-
ревода смолы в нераство-
римое состояние.
Вальцы (рис. 169)
представляют собою два
полых цилиндрических
валка, вращающихся в
подшипниках, укреплен-
ных на двух чугунных ста-
нинах; наружный диаметр
валков 400—600 мм, дли-
на 800—1200 мм. Внутри
валков через коллектор
подается пар для обогрева
и вода для охлаждения (в
зависимости от режима
работы). Вальцы имеют
ограничительные щеки из
чугуна или текстолита для регулирования рабочей длины валков,
которая определяет размеры снимаемого с вальцов листа, нож для
Новолачные пресспорошки
435
надрезания и снятия вальцуемой массы (перед передним рабочим
валком), приспособление для регулирования зазора между валками
и аварийный трос для автоматической остановки вальцов. Один из
валков — рабочий — приводится в движение от мотора через ре-
дуктор и пару стальных цилиндрических шестерен; второй валок —
холостой — приводится в движение от рабочего валка через пару
шестерен. Валки вращаются с различной скоростью; например, ра-
бочий со скоростью 18 об/мин., холостой— 14 об/мин.; обычно отно-
шение скоростей (коэффициент фрикции) от 1,10 до 1,30. Величина
фрикции оказывает существенное влияние на режим работы валь-
цов и на качество получаемого прессматериала.
Для каждой укрупненной партии смолы предварительно устана-
вливают режим вальцевания, т. е. температуру каждого из валков и
размеры зазора. *
Температура рабочего валка варьирует в пределах 80—130°,
холостого 60—100°. Можно также работать с «обратным» режимом,
т. е. с более холодным рабочим валком. Продолжительность валь-
цевания от 30 сек. до 2—3 мин. (для резольных смол до 5 мин.). За-
зор между валками, определяющий толщину листа, устанавливают
от 3 до 7 мм. Загрузка для одной операции составляет примерно
8—12 кг, часовая производительность вальцов 200—350 кг.
Перед засыпкой смеси на вальцы дают «затравку» — кусок го-
рячей, вальцованной массы (0,5—1 кг) и затем смесь совком вруч-
ную распределяют равномерно по всей длине валков; она размяг-
чается, плавится и прилипает к горячему валку, но потом посте-
пенно переходит на передний валок и образует на нем равномерный
слой. Когда такой слой образуется, на нем производят несколько
надрезов ножом для лучшего перемешивания и продолжают валь-
цевание. Затем массу срезают в виде целого листа, который в раз-
вернутом виде кладут на плиты, охлаждаемые водой. От темпера-
туры валков, времени вальцевания и величины зазора зависят теку-
честь пресспорошка и скорость его прессования; поэтому весьма
важно соблюдать установленный режим процесса вальцевания,
а также выдерживать определенную температуру листа при снятии
его с вальцов (она не должна быть выше НО—115°).
В процессе вальцевания резко меняется нагрузка мотора, причем
по ней можно судить о протекании процесса вальцевания. Нагрузка
мотора наиболее высока непосредственно после засыпки порошка,
когда масса еще холодная и смола не расплавилась. В дальнейшем
идет процесс плавления смолы и нагрузка резко падает, а затем
постепенно снова возрастает в связи с частичным отверждением
смолы, обусловливающим постепенное возрастание вязкости
(рис. 170). Такой характер распределения энергии при вальцева-
нии позволяет применять реле для автоматического включения ножа
* Температуру поверхности валков регулируют посредством вентилей,
пуском пара и воды; может быть применено также и автоматическое регулиро-
вание системой обогрева перегретой водой и охлаждения холодной водой.
28*
436
Гл. X. Фенопласты неслоастые
рами оил, расположенных на
Рис. 170. Распределение энергии
в процессе вальцевания.
АБ—нагрузка мотора при холостом ходе;
БВ— увеличение наг1узки щи засыпке
смолы; В — Ж—нагрузка при переме-
шивании; ЖЗ—снятие листа.
и среза листа при достижении определенной вязкости массы,
соответственно при достижении определенной нагрузки мотора.
В следующей стадии производства новолачных пресспорошков
охлажденные листы вальцованной массы размалывают на дро-
бильно-размольном узле, состоящем обычно из зубчатой дробилки
для грубого помола и мельницы деэинтеграторного типа, соеди-
ненных норией. В этой мельнице измельчение происходит под уда-
вращающемся диске и на неподвиж-
ной откидной стенке мельничной ка-
меры. Степень размола регулирует-
ся соответствующими ситами, вста-
вленными в нижней части стенки
мельничной камеры. Провальцован-
ную массу загружают вручную в
зубчатую дробилку, а готовый поро-
шок из приемного бункера мельницы
тонкого помола шнеком выгружает-
ся в тару. Измельчение провальцо-
ванной композиции ведут с расчетом
получения порошка средней ди-
сперсности (с величиной зерен
1—3 мм) и с минимальным содер-
жанием пыли.
Для стандартизации порошка от-
дельные партии загружают в сме-
сительный барабан, имеющий вид
горизонтального цилиндра, объемом
от 5 до 10 м3, вращающийся на двух
бандажах, катящихся по опорным
роликам. На внутренних стенках барабана укреплены винтовые
полосы и перегородки. Барабан снабжен шнеком для загрузки
и выгрузки материала. Его приводят во вращение мотором
через редуктор, и он делает от 6—10 об/мин. Норией барабан соеди-
нен с загрузочным бункером и разгрузочным шкафом. Полученную
партию порошка после анализа тарируют в бочки или мешки.
Производство пресспорошков вальцовым методом может быть
осуществлено на компактной установке с поточным процессом
(рис. 171).
Смолу вручную вносят на зубчатую дробилку 1; через ковшевой элеватор 2
она попадает в бункер 3, из которого самотеком поступает на мельницу тонкого
помола 4. Из мельницы смола поступает в бункер 5, а из него через автомати-
ческие весы 6 в смеситель 7 (лопастной мешатель или шаровую мельницу).
Древесная мука пневмотранспортом подается в бункер 8, из которого самотеком
через автоматические весы 9 поступает в смеситель 7, куда вручную вносят
краситель, уротропин и другие составные части. Воздух из бункеров уда-
ляется через фильтры 10, а оседающая в них древесная мука возвращается
в бункер 8. Из смесителя 7 порошок через бункер 12 подается на вальцы 13.
Листы массы с вальцов переносят на конвейер 15, где они охлаждаются. Охла-
Новолачные пресспорошка
437
жденные листы подвергаются размолу на зубчатой дробилке 16, и полученный
продукт грубого помола пневмотранспортом (или норией) направляется через
циклон 19 в бункер 20, а из него в мельницу 21 дезинтеграторного типа для
тонкого помола и далее в бункер 22. Из бункера порошок поступает в смеси-
тель 23 (для получения укрупненных партий) и далее на фасовочную машину 24
для фасовки.
Рис. 171. Схема поточного метода производства прессматериалов.
1 — зубчатая дробилка; 2—ковшовый элеватор; 5—цеховой бункер смолы; 4—мельница тонкого
помола; 5 — бункер для измельченной см?лы; 6, 9—автоматические весы; 7—смеситель; 8 — цехо-
вой бункер древесной муки; 10 — матерчатый фильтр; 11 — шнек для древесной муки; 12—
бункеры; 13—вальцы; 15—охлаждаемый транспортер; 16— зубчатая дробилка; 17—транспортер;
18—вентилятор; 19—циклон; 20, 22— бункеры; 21 — мельница дезинтеграторного типа; 23— смеситель;
21—фасовочная машина.
По другому варианту рационализация процесса и ускорение
вальцевания достигаются введением системы спаренных вальцов.
Скорость вальцевания пропорциональна температуре, однако при-
менение температур выше 120—130° невозможно, так как холодная
масса плохо пристает и распределяется на очень горячих (~ 150°)
вальцах. При работе же на спаренных вальцах листообразование
происходит на первых вальцах, температуру которых поддерживают
в пределах 90—100°. При такой температуре смола легче всего
прилипает к валкам и быстро образуется лист, который автома-
тическим включением ножа срезается и попадает на вторые вальцы,
стоящие к первым под углом в 90° и нагретые уже на 140—150°; на
них в течение весьма короткого времени завершается термическая
обработка и доведение массы до желаемой степени текучести. Такая
система позволяет проводить оба основных процесса вальцевания
438
Гл. X. Фенопласты неслоистые
(листообразование и термообработку) при оптимальных термиче-
ских условиях и ускорить общий процесс вальцевания.
В последнее время освоен метод непрерывного вальцевания. Для
этого применяют вальцы с более длинными валками (1800—2200 мм).
Поступающая из бункера готовая смесь проходит через дозатор, рас-
положенный над вальцами и с постоянной скоростью попадает на
середину вальцов. По мере вальцевания пластичная масса передви-
гается от середины к краям вальцов. Над серединой переднего рабо-
чего валка на штанге симметрично установлены два ножа, которые
подрезают и переворачивают пластичную массу; по краям рабочего
валка укреплены два дисковых ножа, которые непрерывно отделяют
в виде ленточек уже готовый материал. Срезаемые полоски в даль-
нейшем попадают на ленточный конвейер, где они охлаждаются при
интенсивной продувке воздуха и далее поступают на размол.
Шнековый метод производства
пресспорошков
Этот метод позволяет в более совершенном виде оформить про-
цесс получения пресспорошков в виде поточного, непрерывного
цикла.
Предварительные процессы (размол смолы, подготовка напол-
нителей, смешение ингредиентов), ведущие к получению порошко-
образной смеси, одинаковы для шнекового и вальцового метода.
Различие заключается лишь в процессе пластикации (горячего сме-
шения), который осуществляется при шнековом методе непрерывно
через шнекпресс.
Конструкция обычного червячного пресса (шнекпресса) показана на рис. 172.
На фундаментальной плнте 1 установлена станина, на которой расположен
цилиндр 2 машины.
Внутри цилиндра вращается червяк (шнек) 3, состоящий из рабочей и хво-
стовой частей.
На хвостовой части насажена большая приводная шестерня 14, прн помощи
которой шнек получает вращение от малой приводной шестерни, сидящей на
валу 16. Вал 16 помещен в коробке передач 15, в которой находится и второй
вал 17, соединенный с валом электромоторов 19. В зависимости от зацепления
соответствующих шестерен вал 16 может вращаться с различными скоростями,
что дает возможность менять скорость вращения шнека.
Смесь для пластикации поступает в цилиндр 2 через загрузочную воронку 5
и шнеком 3 проталкивается к головке 4\ в последней имеется мундштук 6. Для
обогрева цилиндр и головка машины снабжены рубашками 8, в которые через
распределительную коробку 7 поступают пар или холодная вода. Подача регу-
лируется вентилями распределительной коробки. Для охлаждения и нагрева
шнека внутрь его полости по трубке 9 подается вода или пар.
С целью пластикации массы, идущей для получения пресспорош-
ков, применяют особые конструкции шнекпресса или вносят соот-
ветствующие изменения в обычные пресса. Так, на валу машины
укрепляют стальные пластины для грубого измельчения пластици-
рованной массы; последняя выходит из машины в грубоизмельчен-
ном виде и поступает для охлаждения на транспортер.
Рис. 172. Червячный пресс (шнекпресс) для непрерывной пластикации пресспорошков.
У —фундаментальная плита; 2—цилиндр; 3- рабочая часть червяка шнека:; 4-гсл<вка; 5—загрузочная воронка; «—мундштук: 7—распре-
делительная головка- S— рубашка; 9-трубе провод для подачи пара и воды внутрь полости червяка; 10—трубопровод для отвода конден-
сатаИиеЛ отработанной воды: ZZ—уплотнительная втулка; 12, 13-мыш™»*-, //-большаяшестерня; /5-коробка передач; 16, /7-валы
коробки передач; 18— соединительная муфта; 19—электромотор; 20—шестерни коробки передач, 21 фрикционная передача.
440
Гл. X. Фенопласты неслоистые
Процесс непрерывной пластикации происходит следующим
образом.
Порошок через дозировочное устройство непрерывно и равно-
мерно подается в загрузочную камеру, а из нее через загрузочную
воронку в корпус шнекмашины; здесь он уплотняется, разогре-
вается от трения, перемешивается и увлекается шнеком к зонам бо-
лее высокого обогрева, где и происходят основные процессы пласти-
кации и термообработки; после этого масса грубо измельчается
ножами, укрепленными в корпусе машины, и выдавливается. Кор-
пус шнекмашины имеет три зоны обогрева: первая — в загрузочной
части с температурой 40—60°, поддерживаемой циркуляцией холод-
ной воды внутри полого вала; вторая — в средней части шнека
с температурой 80—100°, поддерживаемой циркуляцией в каналах
корпуса горячей воды или пара, и третья — в участке измельчаю-
щих ножей — с температурой 120—130°, поддерживаемой электро-
нагревателем.
Пластицированная масса, выходящая в измельченном виде из
шнекпресса, непрерывно поступает на транспортер, на котором
охлаждается и направляется на размол, просеивание и т. д.
Непрерывные методы пластикации имеют значительные техниче-
ские преимущества. Основные из них — экономия рабочей силы,
лучший контроль процесса и качества продукции, меньшая вред-
ность производства, экономия расхода электроэнергии, пара,
воды и т. д.
В табл. 46 приведены сравнительные технические характеристики
некоторых методов получения пресспорошков.
ТАБЛИЦА 46
Характеристика различных методов получения пресспорошков
Показатели Периодическое вальцевание Непрерывное вальцевание Шнековый метод
Производительность аппарата, кг)час . 350 400—500 90—100
Кубатура здания, м^/т год Расход электроэнергии (по установочной 0,5 0,16 0,31
мощности), квт-ч!т 228 178 190
Расход пара, т)т 1,5 0,23 0,6
Расход воды, мя/т 40 2,5 1,5
Затрата рабочего времени, нел.иас1т . 2,9 0,6 1,8
Резольные пресспорошки (фенодреволиты)
Производство резольных пресспорошков на основе древесной
муки в принципе мало чем отличается от производства новолачных.
Так же, как и новолачные пресспорошки, резольные изготовляют
сухими методами смешения, в частности, вальцовым методом.
Вместе с тем, имеются и некоторые .трудности в применении этого
Резольные пресспорошки
441
ТАБЛИЦА 47
Технологические нормативы фенодреволитов и свойства стандартных
отпрессованных образцов (на основе новолачных и резольных смол)
! Пресспорошки
Показатели I i------------------
; новолачный резольный
Уд. объем пресспорошка, сж’/г .... 2,0-2,9 2,0—2,9
Влажность (летучие), % 2—3 ! 1,0-2,0
Текучесть по Рашигу: (стр. 453). . . . :
низкая, мм 30—70 ' 30—70
средняя, мм 70—120 I 70—120
высокая, мм 120—180 120—180
Условия прессования: предварительное подогревание . . пресспорошка:
температура, СС 180±10 130-140
время, мин оптимальная температура прессо- 3-6 6-15
вания после предварительного по-
догревания, °C 180±5 160—180
время выдержки под прессом на
мм. толщины изделия, сек 20—45 60—80
оптимальное уд. давление при прес-
совании, кг/см2 200-500 300—500
Уд. вес прессматериала, г'см2 1,35—1,4 1,4—1,6
Водостойкость (привес стандартных 0,25—0,35
брусков за 24 часа в воде), °/0 ... 0,10-0,25
Теплостойкость по Мартенсу, °C ... . 110—125 100—125
Усадка, % 0,6—1 0,6—1,0
Уд. ударная вязкость, кг см/'см2 . . . 4—7 4—6,0
,. „ „ с надрезом,* . .
кг слг/см2 . . 1,8—2,2 1,6—2,0
Предел прочности на изгиб, кг/см2 . . 500-800 500—600
,, „ „ разрыв, „ . . . 300-500 250—350
.. „ сжатие 1 400—1 800 1 400—1 600
Твердость по Брипелю, кг/м.к2 .... Модуль упругости, кг/см2 30—35 30—35
70 000—90 000 70 000-90 000
Теплоемкость, ккал'°С 0,32-0.38 0,35—0,36
Теплопроводность, «кдд/.и • чдс/°С . . . 0,18—0,20 0,18-0,20
Коэффициент термического линейного 4,5—5,4
расширения 10~5 4,3-5,3
Уд. поверхностное электрическое со- 1 • 109—1 • юн 5. ЮЧ—Ю’4
противление, Q
Уд. объемное электрическое сопротив- 1 • 101—1 • юн .5.1012—10»
ление, Q-см
Пробивное электрическое сопротив- !
ление, кв',мм । 7—12 10—15
Диэлектрическая постоянная 1 7—10 | 6-7
Тангенс угла для электрических потерь 0,4—0,8 0,06—0.10
при 50 гц
при 106 гц — 0,01—0.02
* Значение влияния надреза см. стр. 444.
442
Гл. X. Фенопласты неслоистые
метода, связанные со значительно большей термореактивностью ре-
зольных смол в пределах 80—110°, т. е.—-тех температур, при
которых происходит горячее вальцевание, а также с затруднениями
при производстве твердых, хрупких резолов. Поэтому резольные
пресспорошки изготовляют также и мокрыми методами (лаковым
и эмульсионным).
В состав резольных пресспорошков входят резольные смолы,
древесная мука, минеральные наполнители, известь, красители и
уротропин; последний вводят в небольшом количестве для увели-
чения скорости отверждения пресспорошка. Рецептура различных
марок резольных пресспорошков обычно варьирует в следующих
пределах (в вес. ч.):
Резольная смола...........40—50
Древесная мука............50—40
Минеральный наполнитель . . 0—10
Уротропин.............. 0—2
Известь................ 3—0
Краситель..............1,5—0.
Резольные пресспорошки выпускают различных марок, в зави-
симости от химического характера связующего (феноло-формальде-
гидные, крезоло-формальдегидные, анилино-феноло-формальдегид-
ные), состава и количества наполнителя.
Пресспорошки на основе анилино-феноло-формальдегидных
твердых резолов отличаются более высокой водостойкостью и более
высокими диэлектрическими свойствами, чем резольные пресспо-
рошки на основе феноло- и крезоло-формальдегидных смол. Более
высокая водостойкость и более высокие диэлектрические свойства
являются общей характерной особенностью резольных прессмате-
риалов по сравнению с новолачными. Их применяют поэтому, глав-
ным образом, для производства электротехнических прессизделий.
Однако по механической прочности, теплостойкости и по скорости
прессования резольные пресспорошки уступают новолачным.
Сравнительная характеристика новолачных и резольных пресс-
порошков приведена в табл. 47. Фенодреволиты выпускают в СССР
следующих марок:
новолачные: монолит, К-18-2, К-15-2, К-17-2, К-101-201 и др.
резольные: К-21-22, К-211-2, К-214-2 и др.
Пресспорошки с минеральным наполнителем
Кроме пресспорошков, содержащих в качестве основного напол-
нителя древесную муку, имеются различные марки пресспорошков,
в которых древесная мука либо в значительной мере, либо пол-
ностью заменена минеральными порошкообразными наполнителями
(слюдой, асбестом, графитом, каолином, кремнеземом и др.).
Замена древесной муки минеральным порошкообразным напол-
нителем, естественно, снижает механическую прочность массы, но
значительно увеличивает ее теплостойкость, термостойкость и
водостойкость. Однако во многих случаях такие наполнители при-
Пресспорошки с минеральным наполнителем
443
меняют для придания массе также и ряда специальных свойств.
Так, слюду и кварцевую муку вводят для увеличения электрического
сопротивления материала, а графит — для получения пластика,
обладающего весьма малым электрическим сопротивлением (по-
рядка ~ Ю7 ом) и по существу являющегося полупроводником.
Применение асбеста низших сортов служит для увеличения термо-
стойкости, прочности при истирании и т. д.
Пресспорошки с минеральным наполнителем изготовляют как
на основе резольных (если массу предназначают для целей элек-
троизоляции), так и новолачных смол.
Учитывая больший удельный вес минеральных наполнителей?
их вводят в состав массы в количестве от 60 до 75%. Обычно на-
полнитель состоит из двух или даже трех компонентов. Так, при-
меняют массу следующего состава (%):
Резольная смола .... 35—40 Древесная мука .... 10—15
Слюда............. 40—35 Кварцевая............15—10
Пресспорошки с минеральным наполнителем изготовляют сухими
методами — вальцовым или шнековым.
Вальцевание проводят при режиме, отличающемся от вальце-
вания пресспорошков на основе древесной муки. Применяют мень-
ший коэффициент фрикции вальцов (1 : 1,1), допускают больший
зазор между валками (7—12 мм), более низкие температуры
(60—90° для резольных смол) и соответственно более длительное
время вальцевания (4—8 мин.).
Основное применение минеральные пресспорошки получили для
производства термо- и водостойких изделий электротехнического
назначения.
Применяя резольные феноло-анилино-формальдегидные смолы
в качестве связующего и кварцевую муку и слюду в качестве напол-
нителя, можно получать прессматериалы высокой термостойкости,
водостойкости и высокого электрического сопротивления, однако
ТАБЛИЦА 48
Технические свойства резольных пресспорошков с минеральным
наполнителем
А — наполнитель — кварц и слюда; Б — кварц слюда и древесная мука (1О°/0У
Показатели А Б
Уд. вес, г)смъ 1,8—1,9 1,6—1,8
Уд. ударная вязкость, кг см[см? . . . 2,5—3,5 4,0—5,0
Предел прочности на изгиб, кг: см1 . . 500—550 550—600
Водостойкость (привес в воде), % . . 0,06—0,02 0,10-0,06
Теплостойкость по Мартенсу, °C . . . 150—250 150—180
Уд. объемное электрическое сопротив- ление, Q-CM 10й—5- 1014 10’3—5 • 10”'-
444
Г л. X. Фенопласты неслоистые
лакие материалы будут непрочными и хрупкими (по сравнению
с фенодреволитами).
Чтобы несколько уменьшить хрупкость и в то же время сохра-
нить основные преимущества минеральных пресспорошков, вводят
небольшое количество (5—15%) древесной муки.
В последнему случае улучшения механических показателей можно
достигнуть лишь за счет ухудшения водо- и термостойкости массы.
В табл. 48 приведены свойства двух типов минеральных резоль-
ных пресспорошков: А — содержащих только минеральный напол-
нитель (кварц, слюда) и Б — содержащих те же наполнители, но
также и 10% древесной муки.
Прессматериалы на основе длинноволокнистого наполнителя
Основной недостаток древесной муки как наполнителя — боль-
шая чувствительность к действию высоких температур и гигроскопич-
ность древесных волокон, что связано с самой химической природой
.древесины. Предельная температура, до которой можно длительно
нагревать фенодреволиты без существенного ущерба для их свойств,
100—120°.
Кроме того, фенодреволиты, имеющие показатели удельной
ударной вязкости 5—8 кг • см/см2, являются хрупкими и не могут
быть применены для ответственных деталей приборо- и машино-
строения, несущих значительные динамические напряжения.
Статическая прочность (~ 350 кг/см2 при растяжении) также
недостаточна.
Однако показатели динамической и статической прочности, полу-
ченные при обычных методах испытания на «гладких» стандартных
образцах (120X15X10 мм), не характеризуют прочность мате-
риала в изделиях, имеющих сложную конфигурацию и резкие
изменения площади сечения, например острые края (углы), выемки,
наплывы, бортики, ребра, отверстия и т. д. Для расчета прочности
таких изделий конструктор должен исходить не из показателей
удельной прочности, полученных на «гладких» образцах, а из пока-
зателей прочности образцов, имеющих выемку «надрез».
Наличие надреза приводит для многих типов пластмасс к рез-
кому снижению удельной прочности (особенно удельной ударной
вязкости). Отношение показателей удельной прочности, полученных
на гладких образцах, к показателям прочности на образцах с над-
резом является коэффициентом надреза, который характеризует
степень его опасности, меру снижения прочности материала при рез-
ком изменении его сечения.
Чем больше коэффициент надреза, тем более хрупким является
изделие в условиях эксплуатации при равных показателях проч-
ности массы, полученных на гладких образцах.
Объясняется эффект надреза концентрацией внутренних напря-
жений в местах резкого изменения сечения при отсутствии условий
Прессматериалы на основе длинноволокнистого наполнителя 445
для их релаксации. Естественно, что чем больше скорость роста на-
пряжения, тем меньше возможность для релаксаций перенапряже-
ний, концентрирующихся в основании надреза, поэтому эффект
надреза в наибольшей степени наблюдается при ударных напряже-
ниях в показателях удельной ударной вязкости.
Эффект надреза лишь в малой степени зависит от химической
структуры связующего (в твердом упругом его состоянии), но
в весьма большой степени от наполнителя, его структуры и коли-
чества.
Наибольшее снижение прочности при «надрезе» показывают
пластики на основе связующего без наполнителя, например литые
резиты, полистирол, полиметилметакрилат, хлорвинилоид (вини-
пласт) и др.
Наполнитель, следовательно, во всех случаях уменьшает коэф-
фициент надреза. Степень приближения прочности надрезанных об-
разцов к прочности гладких зависит от структуры наполнителя.
Мелкодисперсный, порошкообразный наполнитель (каолин, камен-
ная мука и др.) показывает наибольший, в то время как наполни-
тель волокнистой анизотропной структуры обусловливает тем мень-
ший коэффициент надреза, чем длиннее волокно, чем более свойло-
ченную структуру имеет материал, чем более ориентированы в нем
волокна.
Эффект надреза в значительной мере и часто полностью исче-
зает в пластмассах слоистой структуры, где наполнитель (листы
бумаги, ткани и др.) создает строго ориентированную слоистую
систему.
Снижение опасности надреза с увеличением длины волокна и
степени ориентации объясняется улучшением условий для релакса-
ции напряжений.
Как видно из табл. 49 (стр. 446), коэффициент надреза для
фенодреволитов составляет 3,3, т. е. наличие «надреза» более, чем
в три раза снижает показатель прочности гладких образцов.
В этом заключается один из наиболее существенных недостат-
ков фенодреволитов. Для устранения хрупкости фенодреволитов
необходимо не только увеличить показатель прочности на глад-
ких образцах, но, главным образом, значительно снизить коэффи-
циент надреза.
Так, снижение коэффициента надреза вдвое (при одинаковой
прочности на гладких образцах) означает (для конструктора) со-
ответственное увеличение технической прочности изделия.
Для увеличения эффективной удельной ударной прочности мате-
риала в качестве наполнителей для фенопластов вводят длинно-
волокнистый наполнитель, — органический (целлюлоза) или мине-
ральный (асбест, стекловолокно). Такие материалы получили на-
звание волокнитов. Они превосходят по удельной ударной вяз-
кости фенодреволиты (~ 12—20 кг см/см2 — на гладких образцах)
и имеют значительно более низкий коэффициент надреза (~ 1,5—1,3),
446
Гл. X. Фенопласты неслоасты?
чю примерно соответствует увеличению технической прочности из-
делий в 4—8 раз (в 2—4 раза за счет показателя прочности и в два
раза за счет более низкого коэффициента надреза).
Недостатком волокнитов является их более низкая текучесть
при прессовании (по сравнению с фенодреволитами). Это обстоя-
тельство ограничивает их применение для прессования тонкостен-
ных изделий сложного и глубокого профиля.
ТАБЛИЦА 49
Удельная ударная вязкость пластиков с различным наполнителем,
определенная на гладких образцах и образцах с надрезом,
и коэффициент надреза
Пластики 1 Уд. ударная вязкость, К?* см!см? Коэффициент надреза
I образцы ' гладкие образцы с надрезом
Без наполнителя i i
Фенолит (литые резиты) 12 1,2 10
Полистирол . . 1 10 0,8 15
Полихлорвинил (винипласт) .... . . : 100—200 15—20 6,6—10
Наполненные 1
Фенодреволит 6 1,8 3,3
Аминоцеллолит (аминопласт) . . . . . | 8 2,2 3,6
Феноцеллолит (волокнит) 15 11 1,37
Фенопласт с абестовым шнуром . . 25 25 1,00
Фенотекстослой . . । 35 32 1,01
Фенобумослой . . 1 20 16 1,24
Фенолиты на основе длинноволокнистых наполнителей не
могут быть изготовлены обычными сухими методами (валь-
цовым, шнековым), так как они неизбежно приводят к исти-
ранию и уменьшению длины волокна. Вследствие этого приме-
няют, главным образом, мокрые методы (водно-эмульсионный и,
отчасти, лаковый), и в качестве связующего чаще всего — резоль-
ные смолы.
Пропитку волокнистого наполнителя раствором или эмульсией
смолы производят в двухлопастном вакуум-мешателе. На рис. 173
показана чаша мешателя с расположенными в ней Z-образными
лопастями. Она снабжена рубашкой для обогревания паром или
горячей водой. Лопасти мешателя вращаются в противоположные
стороны с соотношением числа оборотов 2 : 3; изменение вращения
лопастей производят переключением электромотора. Мешатель
снабжен массивной крышкой, соединенной через патрубок с вакуум-
насосом. Выгрузку из мешателя производят откидыванием чаши;
Прессматериалы на основе длинноволокнистою наполнителя 447
Рис. 173. Чаша двухлопастного ва-
куум-мешателя.
прессматериалы на основе мине-
смолы), а также включают вакуум для удаления части растворителя
для этого имеется цепной привод, вал которого расположен на
колоннах позади мешателя. В зависимости от направления дви-
жения лопастей различают пере-
мешивание массы «внутрь» и
«в развал». Перемешивание обыч-
но происходит с вращением ло-
пастей «внутрь», однако время
от времени направление движе-
ния лопастей на несколько ми-
нут меняют «в развал», что обес-
печивает лучшую гомогенизацию
массы.
При перемешивании вязких
длинноволокнистых масс необхо-
димо иметь в виду возможность
наматывания волокна на лопасти
мешалки, что приводит либо к
остановке мешателя, либо к не-
полному смешению массы. Чем
длиннее волокно, тем труднее про-
изводить операцию его пропитки
в мешателе.
Длинноволокнистые прессма-
териалы подразделяют на две
группы: 1) прессматериалы на
основе целлюлозного волокна и 2)
рального наполнителя (асбест).
Прессматериалы с целлюлозным волокном
(Феноцеллолиты, волокниты)
Этот вид прессматериалов изготовляют чаще всего на основе
резольной смолы и длинноволокнистого целлюлозного наполнителя
(линтер различных сортов, очесы хлопка, льняное волокно, пенька
и др.). Иногда в состав массы вводят небольшое количество мине-
рального наполнителя (каолин, графит и др.).
Производство феноцеллолитов обычно ведут по лаковому или
эмульсионному методу. Так, в мешатель, в соответствии с рецепту-
рой, загружают водно-эмульсионную резольную смолу, смесь жир-
ных кислот (смазка), иногда аммиачную воду или около 1% Уро-
тропина и перемешивают в течение 15—20 мин. После этого неболь-
шими порциями в течение 15—25 мин. при работе лопастей
мешателя «внутрь» вносят линтер. Когда введено все количество
линтера, перемешивание продолжают еще 60—90 мин., при этом
каждые 15—20 мин. меняют направление движения лопастей. Пере-
мешивание производят при обычной температуре, без вакуума, од-
нако иногда смесь подогревают до 45—60° (для снижения вязкости
448
Гл. X. Фенопласты неслоистые
(воды). После перемешивания массу выгружают на противни и
распределяют слоем толщиной не более 30 мм. Противни затем по-
ступают в вакуум-сушилку. Сушку производят при 50—60° при ва-
кууме 500—700 мм до остаточной влажности ~3%. В процессе
сушки необходимо периодически разрыхлять и перемешивать массу
в противнях и контролировать влажность и текучесть ее.
Рецептура получаемых таким образом прессмасс может широко
варьировать в зависимости от характера и длины целлюлозного во-
локна, а также характера смолы и от других факторов. Типичные
рецептуры содержат следующие основные компоненты (в %):
Смола эмульсионная (в перерасчете на сухую
смолу) ...............................40—50
Слеиновая кислота ........................1,0—1,5
Линтер....................................48—43
Аммиачная вода............................0,2—1,0
Минеральный наполнитель (каолин)..........10—5.
В мешателе целесообразно пропитывать линтер лишь низших и
средних сортов.
Когда же применяют более длинноволокнистые материалы, про-
питка в мешателе не дает хороших результатов вследствие истира-
ния волокна в процессе перемешивания. Для этих целей были раз-
работаны специальные методы пропитки: суспензионная пропитка
в ролле (стр. 492), пропитка феноло-спиртами (стр. 472), а также
исходными смолообразующими веществами и др. Применяют также
пропитку под давлением. Для этого волокно в закрытых формах
заливают дисперсией смолы и прессуют. Получается таблетка из
волокна, пропитанного смолой. Разрыхлением и повторным прессо-
ванием удается пропитать волокно без снижения его средней длины
(способ С. Н, Ушакова).
Волокниты (феноцеллолиты), имея преимущество в механиче-
ской прочности, уступают, однако, фенодреволитам по водостой-
кости, диэлектрическим свойствам и показателю текучести прессма-
териала. Чем более длинноволокнистым является наполнитель, тем
меньше текучесть прессматериала. Ввиду этого прессование изде-
лий требует применения более высоких давлений, а в некоторых
случаях вообще невозможно оформить из волокнита изделие слож-
ной, глубокой конфигурации при обычной (~2 мм) толщине стенок.
В СССР феноцеллолиты выпускают под маркой волокнит.
Свойства волокнита приведены в табл. 50.
Феноцеллолиты нашли применение в качестве материала горя-
чего прессования для изготовления технических изделий, к которым
предъявляют требования высокой механической прочности (в част-
ности к ударным напряжениям). Их применяют для производства
деталей в приборо- и машиностроении (например, для производ-
ства футляров, корпусов и крышек аппаратов, шестерен, роликов,,
маховиков и др.), а также различных изделий строительной тех-
ники (дверные ручки, различная арматура и т. д.).
29 Зак. 1269. Э. И. Бар:
ТАБЛИЦА 50
Физико-механические свойства неслоистых фенопластов
Показатели Смола без наполнителя (литые резиты) Смолы с наполнителями
древесной мукой фенодрев лит (новолачные и резольные пресспорошки) хлепковой’ целлюлоз' й фсноцеллолит (волокиит) кусочками хлспчатс- бумажней ткани фенотскстолит (текстслит- -крошка) асбестом фсноасболит (асборезиты) асбесте вым шнуром ф<ноасботе- кстолит
Уд. вес, г'см2 Уд. ударная вязкость, кг-см!см2 Уд. ударная вязкость с надрезом, кг см 1см2 Предел прочности на изгиб, кг/см2 „ ,, „ сжатие, „ „ „ „ разрыв, „ Твердость по Бринелю, кг/мм2 . . . Модуль упругости, кг/см2 Водостойкость (привес в воде за 24 часа), % Теплостойкость по Мартенсу, °C . . Жаростойкость по Шрамму, класс . Уд. поверхностное электрическое сопротивление, Q Уд. объемное электрическое сопро- тивление, Р-слг Электрическая прочность, кв/мм . . Тангенс угла диэлектрических потерь при 50 гц 1,3 5—15 0,8—1,2 500—800 350—450 25—35 50 000—70 000 0,01—0,05 70-110 2—3 10™—10>2 10”—10™ 1,35—1,45 4-8 1,8—2,2 500- 800 1 400—1 800 250—500 30 70 000—90 000 0,1-0,30 110-125 2—3 10™—10’« Ю’О—10™ 7—15 0,8-0,06 1,35-1,45 9-15 5-8 600-800 1 200—1 600 350- С 00 25 70 000— 80 000 0,3-0,5 110—125 2—3 109—10П 109—10И 7-9 0,4—0,9 1,4 12—25 10—20 800—1 000 1 400—2 000 450—600 0,4-0,6 120—135 3 109—10’1 109—10” 7—15 1,7—1,85 10—25 6-15 600—1 000 800—1 400 30-40 150 000—250 000 0,4—1,0 200—250 4 107—10™ 107—юн 1-3 0,6—0,9 1,7—1,8 20-25 20-25 600—1000 1000—1400 400—600 30—40 0,4—0,8 200—250 4 107—10» 107—юн 1—3
Прессматериалы на основе длинноволокнистого наполнителя
450
Гл. X. Фенопласты неслоистые
Фенотекстолит (текстолит-крошка)
Этот прессматериал представляет собой прессованные кусочки
ткани (1—2 см), пропитанной резольной смолой. Их получают вы-
рубкой из листов пропитанной ткани или из ее отходов. Пропитка
ткани резольными смолами составляет часть технологии слоистого
пластика на основе фенотекстослоя (текстолита, стр. 464).
Кусочки сухой ткани, пропитанные резольными смолами, содер-
жат 45—50% смолы.
Изделия из фенотекстолита изготовляют прессованием при 155—
165° с выдержкой 60—90 секунд на 1 мм.
Текучесть текстолита-крошки ниже, чем волокнитов; чем
меньше размеры кусочков ткани, тем больше текучесть. Тонко-
стенные глубокие изделия не удается прессовать из текстолита-
крошки.
Фенотекстолит, а также феноасботекстолиты имеют самый боль-
шой показатель удельной ударной вязкости (20—25 кг/см2) из всех
неслоистых прессматериалов и почти не показывает эффекта над-
реза. Статическая прочность (разрыв, изгиб) также выше, чем
у других неслоистых материалов. Его широко применяют в машино-
строении (для изготовления подшипников, шестерней, шкивов
и т. д.), часто наравне или в комбинации с фенотекстослоем, т. е.
с тем же материалом слоистой структуры. Свойства фенотексто-
лита в основном зависят от удельной прочности ткани и от вели-
чины кусочков «крошки», а также от содержания смолы (табл. 50).
Феноасболиты (асборезиты)
*
Применяемый для изготовления этих прессматериалов наполнитель —
асбест — представляет собою минерал с волокнистой структурой. Ои является
линейным высокополимером следующего строения:
I I I
---Si—O-Si—О—Si--
I I I
ООО
I I I
Катион Катион :
О О ;
---81—О—Si—О—Si--
' I I
: О О
Прессматериалы на основе длинноволокнистого наполнителя
451
Основные цепи полимера состоят из весьма прочных силоксановых связей
i i
—Si—О—Si—, в то время как между цепями, повидимому, имеются более
III i
слабые связи типа —Si—О— катион —О—Si—, которые могут быть преодо-
I I
лены механическим воздействием (например при распушке в ролле).
Асбестовое волокно легко расщепляется в продольном направлении. Удель-
ная прочность его очень велика и зависит от диаметра волокна. Чем тоньше
волокно, тем выше его прочность. При толщине волокна в 5—10 ц прочность
достигает 300 кг/мм2 и выше.
Термические свойства асбеста определяются его составом, наличием в нем
химически связанной, абсорбционной и так называемой внешней воды. При на-
гревании до 105° — удаляется внешняя, от НО до 360° — абсорбционная, около
800° — химически связанная вода и, наконец, около 1500° происходит плавле-
ние асбеста.
Асбесты обладают большой удельной поверхностью н высокой абсорбцион-
ной способностью.
Различают два типа асбеста: змеевиковый или хризотиловый и роговообмаи-
ковый.
Хризотиловый асбест представляет собою водный силикат магния
3MgO • 2SiO2 • 2Н2О.
Роговообманковый асбест известен в нескольких разновидностях, в частно-
сти антофилит (MgFe) О • SiO2 и крокидолит или синий капский асбест
Na2O • Ре20з • 2FeO • 6SiO2.
Хризотиловый асбест нестоек к действию кислот; роговообманковые сорта
асбестов кислотостойки. По механической прочности хризотиловый асбест зна-
чительно прочнее роговообманковых сортов асбеста.
В СССР имеются богатейшие залежи хризотилового асбеста на Урале. Из
кислотоупорных сортов в СССР разрабатывают антофилит.
По текстуре асбесты делят на три марки: 1) «О» — жесткая текстура,
2) «И» — мягкая, 3) «Г» — полужесткая.
Текстура асбеста в основном зависит от метода и характера его обработки
и распушки.
По длине волокна и содержанию пыли технические асбесты делятся на
шесть (кроме текстильных) сортов (I—VI).
Высокая механическая прочность и термостойкость асбеста сде-
лали его ценным наполнителем для различных типов пластмасс.
Феноасболиты отличаются высокой термостойкостью и теплостой-
костью.
Механические свойства асборезитов определяются сортностью и
структурой асбеста. Чем выше сорт асбеста, чем длиннее волокна,
тем прочнее получаемые пластики. Асбесты жесткой текстуры дают
более прочный материал, чем асбесты мягкой текстуры.
Прочность асборезитов определяется также методом распушки
асбеста, степенью повреждения и деформации волокна в процессе
смешения и содержанием асбеста в массе.
Чем выше степень распушки для одного и того же сорта асбеста
в массе, тем ниже прочность, но выше водостойкость и текучесть
массы. Во всех случаях, когда распушка приводит не к продоль-
ному расщеплению волокна, а к поперечному, к уменьшению длины,
прочность массы снижается. В такой же степени снижается проч-
ность с увеличением времени перемешивания массы, которое также
29*
452
Гл. X. Фенопласты неслоистые
приводит к истиранию и уменьшению длины волокна. Поэтому
весьма важно, чтобы в процессе предварительной обработки асбеста
(распушки) и смешения (пропитки) со смолой не происходило бы
истирания и поперечного разрыва волокон.
Ввиду этого, как правило, применяют различные мокрые ме-
тоды смешения: пропитку асбестового волокна в мешателе вод-
ными эмульсиями и спиртовыми растворами смол, а также феноло-
спиртами, пропитку в ролле распушенного в воде волокна суспен-
зиями и эмульсиями смол, пропитку эмульсиями смол под
давлением в закрытых формах и другие методы. При примене-
нии длинноволокнистого асбеста (сорт № 1 и 2) перемешивание ас-
беста в мешателе приводит к резкому снижению прочности из-за из-
мельчения волокна; в этом случае рекомендуется применять методы
пропитки под давлением.
В качестве смол применяют чаще всего резольные смолы. Фено-
асболиты, в зависимости от длины волокна и метода смешения, мо-
гут быть получены с весьма широким диапазоном показателей проч-
ности, например с удельной ударной вязкостью от 6—8 до 20—
25 кг • см/см2.
Однако все асборезиты отличаются высокой термо- и теплостой-
костью (теплостойкость по Мартенсу 200—250°), высокими фрик-
ционными свойствами при сухом трении (коэффициент трения
0,3—0,35) и малой истираемостью.
Асборезиты применяют для производства тормозных колодок,
изготовления высоковольтных и низковольтных коллекторов, за-
прессовки электроизолирующего слоя деталей, для которых
необходимы повышенные механические свойства и термостой-
кость, а также для деталей приборостроения, к которым предъ-
являются требования высокой механической прочности и термо-
стойкости.
В СССР асборезиты выпускают под марками К-6, К-3,
К-6-Б и др.
Феноасботекстолит. Асборезиты приготовляют также на основе
кусочков асбестовой ткани или асбестового шнура, пропитанных ре-
зольными смолами.
Прессматериалы на основе асбестовой ткани отличаются более
высокой удельной ударной вязкостью и, практически, полным от-
сутствием влияния надреза.
В табл. 50 приведены пределы свойств различных фенолитов
в зависимости от характера наполнителя, в том числе и различных
асборезитов.
Определение текучести прессматериалов
Текучесть — важнейший показатель прессовочных свойств пресс-
материалов при переработке их в изделия. Под текучестью (пла-
стичностью) прессматериалов подразумевают способность заполнять
прессформу в условиях переработки.
Определение текучести пресспорошков
453
Текучесть является функцией применяемого при прессовании да-
вления Р, температуры Т и времени t прессования.
Большинство методов определения текучести основано на прес-
совании образца при постоянных давлении, температуре и вре-
мени, причем показателем пластичности (текучести) в этом случае
является величина деформации I, обыч-
но длина пути, который проходит масса -4--
в прессформе.
Рис. 174. Прессформа для определения
текучести.
Рис. 175. Форма и максималь-
ная длина призматического
стержня, характеризующего те-
кучесть пресспорошка.
Применяются и другие методы, основанные на постоянной вели-
чине деформации I при постоянных температуре Т и давлении прес-
сования Р; при этом определяется время I, при котором достигается
заданная степень деформации, или же давление Р при постоянном
времени, необходимое для достижения этой степени деформации.
Широкое применение получил метод определения текучести,
основанный на измерении длины пути I, который проходит масса
в прессформе при постоянных давлении, температуре и времени
прессования (метод Рашига).
В стальную обойму (рис. 174) высотой в 250 см вставляют со-
стоящий из двух половин стальной конус, в котором имеется приз-
матический канал с постепенно уменьшающимся поперечным сече-
454
Гл. X. Фенопласты неслоистые
нием. Таблетированный на холоду порошок (7,5 г) закладывают
в цилиндрическую часть канала (диаметр 30 мм), причем форму
предварительно нагревают до 160°. Таблетку прессуют при давлении
300 кг!см2 в течение 3 мин. После разъема прессформы определяют
длину призматического стерженька (рис. 175) в миллиметрах, кото-
рая и служит показателем текучести порошка.
Для фенопластов зависимость текучести от температуры очень
сложна. Под действием температуры смола в стадии А плавится,
Температура прессования, °C
Рис. 176. Зависимость текучести пресс-
порошка от температуры.
становится вязкой, и чем выше
температура, тем большую теку-
честь приобретает масса. Если бы
под воздействием температуры не
происходили структурные измене-
ния в массе, то текучесть была бы
в широких пределах пропорцио-
нальна температуре прессования.
Однако на первоначальный
процесс плавления массы немед-
ленно накладывается второй про-
цесс — процесс поликонденсации,
с образованием сшитых, простран-
ственных структур, который резко
повышает вязкость массы и в
итоге полностью устраняет текучесть. Чем выше температура, тем
быстрее протекают процессы сшивки молекул и тем меньше остается
времени для развития пластической деформации, которую мы усло-
вно определяем как «текучесть». Суммарный эффект обоих процес-
сов выражается длиной стержня, характеризующей подвижность
(текучесть) массы, зависимость которой от температуры имеет ха-
рактерный максимум (рис. 176).
Текучесть пресспорошка зависит от ряда факторов: соотношения
смолы и наполнителя, термореактивности смолы, влажности пресс-
порошка, структуры наполнителя, степени размола, а также от сте-
пени предварительного подогревания порошка перед прессованием,
характера поверхности прессформы и т. д. Чем больше в массе со-
держится наполнителя, чем длиннее волокно, чем более оно свойло-
чено и чем выше термореактивность смолы, тем ниже текучесть
массы. Увеличение влажности порошка увеличивает текучесть, од-
нако при этом, естественно, ухудшаются физико-химические свой-
ства материала. Предварительный подогрев нетаблетироваиного
порошка в термошкафах снижает текучесть, так как вследствие
плохой теплопроводности пресспорошок прогревается неравномерно.
Однако, если производить более быстрый и равномерный подогрев
таблетированного порошка токами высокой частоты (при этом тепло
развивается внутри таблеток), то вследствие равной интенсивности
теплового воздействия на все части таблетки текучесть увеличи-
вается.
Асборезитовые формовочные массы
455
Асборезитовые формовочные массы
(Фаолит)
Уже в первые годы производства резольных смол и прессмате-
риалов из фенопластов были сделаны попытки применения их в ка-
честве химически стойких антикоррозийных замазок, поверхностных
защитных покрытий и т. д. Естественно, что вскоре возникла мысль
об использовании высокой химической стойкости резитов для из-
готовления из них как конструкционных химических материалов,
так и готовых конструкций — химаппаратуры.
Для решения этой задачи необходимо было, во-первых, найти
наполнитель, который был бы достаточно химически стойким, термо-
стойким и в то же время, чтобы он обеспечивал высокую прочность
изделия; во-вторых, разработать рациональный метод производства
крупных изделий из фенопластов без прессования в формах при
высоких давлениях. Изготовление изделий путем горячего прессо-
вания эффективно лишь для изделий сравнительно небольших га-
баритов и при условии их массового стандартного производства.
Исследовательская мысль была поэтому направлена на изыскание
других путей для решения этой задачи. Она была удовлетворительно
решена применением в качестве наполнителя кислотосюйких сортов
асбеста, а также графита, кремнезема и разработкой метода ста-
ночного или ручного формования массы без высоких давлений, по-
добно формованию керамических изделий.
Сущность соответствующей технологии заключается в пропитке
кислотостойких сортов асбеста жидкой, безводной резольной смо-
лой в мешателе, осторожном вальцевании массы с получением пла-
стичных при обычной температуре листов, из которых формовкой,
пользуясь шаблонами, деревянными моделями и формами, можно
изготовлять крупные изделия сложной конфигурации. Полученные
таким образом изделия подвергают в дальнейшем термической обра-
ботке в термошкафах для перевода смолы в резитовое состояние.
При этом пластичная масса превращается в твердый, нераствори-
мый, неплавкий и прочный материал с высокой химической стой-
костью (резит).
Формовочные резитовые материалы и химаппаратура на их
основе, известные под названием «хавег» (Германия), выпущены
различных марок, отличающихся по стойкости к химическим реа-
гентам и по составу наполнителя и связующего (табл. 51).
Химическая стойкость формовочных масс определяется химиче-
ским составом как смолы, так и наполнителя. «Хавег-41» изгото-
вляется из феноло-альдегидной смолы и кислотоупорных сортов
асбеста. Его стойкость к действию кислот определяется кислото-
стойкостью резита и асбеста; присутствие в резите свободных гидро-
ксильных групп обусловливает его нестойкость к щелочам. Нестой-
кость по отношению к фтористоводородной кислоте связана с не-
стойкостью к этой кислоте асбеста. Поэтому в «хавег-43» асбест
456
Гл. X. Фенопласты неслоистые
заменен графитом и этим объясняется его стойкость к фтористо-
водородной кислоте. «Хавег-ВС (50)» стоек к действию едких щело-
чей, что, повидимому, достигнуто модификацией резольной смолы
с блокированием свободных гидроксильных групп в резите.
ТАБЛИЦА 51
Различные марки формовочных резитовых материалов „хавег“
„Хавег-41“ „Хавег-43“ „Хавег-ВС (50)“
стоек к действию: не стоек к действию: стоек к действию: стоек к действию:
H2SO4 (60%), НС1 всех концентраций; молочной, муравьи- ной, уксусной кис- лот; неполярных растворителей; со- лей анилина; хло- ристых солей и т. д. Конц. H2SO4; HNO3; хромовой кислоты, едких щелочей, пири- дина, анилина, фтористоводо- родной кислоты Фтористоводорол ной кислоты в остальном ве- дет себя, как „хавег-41“ Едких щелочей (NaCH; КОН)
В СССР кислотостойкий формовочный асборезит изготовляют
под названием фаолит. Технология производства фаолита была раз-
работана А. Д. Соколовым и М. К. Амусиным.
Процесс производства фаолита складывается из получения ре-
зольной жидкой смолы, замешивания ее с асбестом, вальцевания
массы, переработки массы (каландрирования, шприцевания и из-
готовления изделий) и термообработки (отверждения) изделий из
фаолита.
Резольную жидкую безводную смолу получают конденсацией
фенола с формальдегидом в молярных отношениях 5 : 6 в присут-
ствии щелочного катализатора (обычно аммиака). В полученной
смоле допускается содержание не более 16% свободного фенола;
скорость ее желатинизации при 155° от 1 до 3 мин.
Для производства фаолита применяют в основном кислотостой-
кий антофилитовый асбест высших сортов (№ 1 и № 2); раствори-
мость антофилитового асбеста в соляной кислоте при кипячении не
должна быть выше 15%.
Наряду с антофилит-асбестом в фаолит вводят небольшое коли-
чество хризотил-асбеста (5—10%).
Введение некислотостойкого хризотил-асбеста преследует цель
улучшения механических свойств фаолита, так как хризотиловый
асбест значительно прочнее антофилитового. Содержание хризоти-
лового асбеста снижает, однако, кислотостойкость фаолита.
Соотношение смолы и наполнителя в основном зависит от на-
значения фаолита и метода его переработки. Оно может варьиро-
Асборезитовые формовочные массы
457
вать в сравнительно больших пределах, например, от 100 до
150 вес. ч.; асбеста (антофилитовый + хризотиловый) на 100 вес. ч.
резольной смолы.
Жидкую резольную смолу загружают в двухлопастный мета-
тель, снабженный паровой рубашкой; затем к смоле небольшими
порциями в течение 10—15 мин. добавляют асбест и в рубашку ме-
шателя дают пар с таким расчетом, чтобы температура массы была
в пределах 30—60°. Перемешивание при этой температуре продол-
жают в течение 30—60 мин. после прибавления всего асбеста, меняя
при этом несколько раз направление вращения лопастей. По окон-
чании перемешивания массу выгружают, измельчают на валках на
небольшие кусочки и раскладывают на стеллажи.
Следующая операция — вальцевание массы на обогреваемых
фрикционных вальцах. Температура валков ~40—70°. До начала
вальцевания устанавливают минимальный зазор между валками и
по мере вальцевания его увеличивают до тех пор, пока весь избыток
массы между валками не исчезнет; после этого массу срезают
в виде листа. Длина и ширина листа определяются длиной и диа-
метром рабочей части валков. После вальцевания листы охлаждают
до 20—30°, посыпают тальком и пропускают несколько раз через ка-
ландр (рис. 107, стр. 244), причем каждый раз зазор между валками
каландра равномерно уменьшают, пока не будет достигнута задан-
ная толщина листа. После каландрирования листы обрезают, вновь
посыпают тальком, и в таком виде они поступают на изготовление
изделий.
Из различных изделий из фаолита, наибольшее значение имеют
химически стойкие трубы (кислотопроводы), в частности, для со-
ляной кислоты.
Трубы изготовляются методом выдавливания (экструзии) через
шнекпресс (рис. 172, стр. 439).
Для этого получаемую после вальцевания горячую массу в виде
листов грубо измельчают и загружают в загрузочную воронку шнек-
пресса. В случае необходимости массу предварительно подогревают
в термошкафе до 40—60°. К корпусу машины привинчивают сталь-
ную головку с рубашкой для обогревания. Головка, в свою очередь,
соединена с конусом, переходящим в съемный мундштук, состоящий
из стакана и стержня. Перед пуском пресса устанавливают стакан
и стержень мундштука необходимых размеров, центрируют мунд-
штук и температуру корпуса шнекпресса и мундштука регулируют
в пределах 50—70°.
Выходящую из шнека трубу принимают на металлический пусто-
телый дорн, который изнутри охлаждается водой. Один конец дорна
имеет нарезку, при помощи которой он привинчивается к мундштуку,
другой конец укладывается на специальную переносную подставку.
Диаметр дорна на 2—3 мм меньше внутреннего диаметра изгото-
вляемой трубы, которая должна скользить по дорну, не давая скла-
док. Когда труба требуемой длины получена, машину останавли-
458
Гл. X. Фенопласты неслоистые
вают, массу у мундштука срезают, вывертывают дорн и снимают
с него трубу, затем снова вставляют в мундштук.
Химические аппараты (гидролизные ванны, дестилляционные
колонки, мешалки, резервуары и т. п.) изготовляют из каландриро-
ванных пластичных листов массы. Фаолитовые листы накладывают
на соответствующие деревянные каркасы или гипсовые формы, при-
чем края крепят «внахлестку» и прижимают шаблонами; при этом
они слипаются друг с другом.
Трубы и изделия вместе с каркасом подвергают термической
обработке в так называемой полимеризационной камере; обогре-
вают ее паровыми батареями, а для циркуляции воздуха (для охла-
ждения) в ней имеются вентиляторы.
Общая длительность термической обработки от 30 до 50 час.
Обработку ведут, постепенно поднимая температуру по установлен-
ному режиму от 60 до 130°; после этого изделия медленно охла-
ждают в камере до 60° и выгружают. Так, аппараты и детали, изго-
товляемые из сырых фаолитовых листов, проходят отверждение
в полимеризационной камере по следующему режиму:
При 60—70°................... 6 час.
70—80°................... 5 „
80—90°................... 4 „
90—100°.................. 3 „
100—110°.................. 4 ,
110—120°.................... 5 „
120—130°.................... 3 „
Всего ... 30
Иногда готовые изделия лакируют резольным лаком и снова
прогревают для отверждения лака.
Часто химаппаратуру изготовляют из отвержденных фаолитовых
плит или же изготовляют части аппарата, которые отверждают и
затем склеивают специальной замазкой, затвердевающей на холоду
(фаолитовая замазка). Последняя представляет собою пластичную
композицию из резольной смолы, дисперсного кремнезема и коротко-
волокнистого асбеста, к которой перед применением добавляют
«ускоритель», представляющий собою раствор серной кислоты
в спирте.
Из фаолитовой массы можно получать изделия и обычным
методом горячего прессования, так изготовляют, например, химиче-
скую арматуру: краны, вентили, маховики и т. д.
Фаолит выпускают- различных марок, отличающихся составом
и количеством наполнителя (асбест, графит, кремнезем).
Фаолит представляет собою кислотостойкий и теплостойкий ма-
териал, который успешно применяется для производства труб и
аппаратов химической промышленности, однако его механическая
прочность недостаточно высока и ниже, чем у прессованных фено-
пластов (волокнитов и др.).
Асборезитовые формовочные массы.
459
Свойства фаолита характеризуются следующими показателями -
Уд. вес, г!смъ..................................
Уд. ударная вязкость, «г-слг/слг2...............
Предел прочности на изгиб, кг/см2...............
, „ , сжатие, „ .............
. „ „ разрыв, „ .............
» » срез, » .............
Теплостойкость по Мартенсу, °C..................
Коэффициент линейного термического расшире-
ния ............................................
Водостойкость (привес в воде за 24 часа), °/0 . .
Привес или потери в весе при действии соляной
кислоты в течение 24 часов, °/0,................
1,6—1,8
2—6
400—800
500—1000
175—350
175—400
150—200
17-20- 10-®
0,05—0,5
1,25
(не более)
Фаолит с асбестовым наполнителем при обычных или повышен-
ных температурах (120—150°) стоек почти ко всем кислотам, за
исключением концентрированной серной (концентрации свыше 60%)
и окисляющих кислот (азотной, хромовой); кроме того, он не стоек
и к действию щелочей. Следует указать, что фаолит противостоит
действию неполярных органических растворителей (нефтепродукты,
ароматические и хлорированные углеводороды и др.).
-Для получения формовочных резитов, стойких к действию щело-
чей, применяют модифицированные резольные смолы, в которых
блокированы свободные фенольные гидроксилы. Для этого готовят
резольную смолу с избытком формальдегида и в присутствии такого
количества едкого натра, при котором значительная часть феноль-
ных групп резола превращается в фенолятные. Щелочной раствор
смолы обрабатывают алкил- или арилалкилгалогенидом, например,
хлористым бензилом; при этом образуется бензиловый эфир резола,
выпадающий из раствора по схеме:
ONa ONa
--------------------+С6Н5СН2С1 —+
СН2 СН2ОН
Н5С6Н2СО
ОСН2С6Н5
Реакцию ведут на неполное замещение, так как при значи-
тельном замещении активность водорода в орто- и параположениях
460
Гл. X. Фенопласты неслоистые
настолько уменьшается, что при термической обработке смола уже
не может переходить в пространственный полимер. На основе бен-
зилированной смолы можно получать материалы в значительной
мере стойкие к действию разбавленных едких щелочей.
Сравнивая фаолит, как новый конструкционный химически стой-
кий материал, с известными и распространенными материалами
химического машиностроения, можно отметить следующие его
преимущества и недостатки. По сравнению с кислотоупорной кера-
микой он имеет преимущество в меньшем удельном весе (примерно
на 50% легче) и в значительно большей статической и динамиче-
ской прочности (~4—6 раз), меньшей чувствительности к резким
изменениям температуры. Фаолит, стойкий против действия фто-
ристоводородной кислоты, естественно, не может быть заменен ни-
какими силикатными материалами.
Однако меньший температурный интервал химстойкости яв-
ляется его основным недостатком по сравнению с керамикой.
Преимущества фаолпгга по сравнению с цветными и черными
металлами очевидны, так как за исключением некоторых специаль-
ных марок кислотоупорной стали (хром-никелевых и хром-никель-
молибденовых) они не стойки к соляной, разбавленной серной,
уксусной и другим кислотам. Однако значительно большая проч-
ность металлов и их термостойкость являются их преимуществом
перед фаолитом. Преимущество же фаолита — его малая теплопро-
водность: однако во многих случаях это является и недостат-
ком (там, где требуется быстрый отвод тепла). Температурные
пределы химической стойкости этого материала невелики. Необхо-
димо иметь в виду, что температура 120—150° предельная для
стойкости фаолита к действию химических реагентов. Существенной
является относительная простота изготовления крупных изделий из
фаолита и несложность технологического процесса. Его недоста-
ток — трудность механизации этого процесса.
Фаолит применяют в химической промышленности в качестве
одного из важных антикоррозийных конструкционных материалов.
Из него изготовляют трубы, различную химическую арматуру,
ванны, колонки и т. п. Он является эффективным заменителем
цветных металлов (свинца, бронзы), кислотоупорных сплавов
и керамики. Его стойкость к действию соляной кислоты имеет особо
•большое значение, так как во многих случаях из-за корродирую-
щего действия соляной кислоты химические процессы приходилось
вести на серной кислоте, хотя это делало их менее эффективными.
Новые полимеризационные химически стойкие материалы (вини-
пласт и др., стр. 241) также широко применяют в качестве кон-
струкционного материала для химической промышленности; однако
•большим преимуществом фаолита является его значительно более
высокая теплостойкость.
ГЛАВА XI
СЛОИСТЫЕ ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ
ФЕНОЛО-АЛЬДЕГИДНЫХ СМОЛ (ФЕНОПЛАСТЫ)
СЛОИСТЫЕ ФЕНОПЛАСТЫ
(Слойматериалы)
Слоистыми пластмассами называют материалы, имеющие ясне1
выраженную слоистую структуру. Наполнитель в этих массах имеет
ориентированную поверхность, развитую в двух направлениях (на-
пример листы бумаги, ткань, шпон). Так как наполнитель в слои-
стых массах представляет собой уже скрепленную, свойлоченную
или переплетенную структуру, то текучесть слоистых масс при прес-
совании весьма ограничена; поэтому из слоистых масс методом
горячего прессования можно изготовлять по преимуществу плоские
материалы (пластины, плиты, блоки, стержни, бруски). Путем
намотки с последующей прессовкой или без нее могут быть изго-
товлены также стержни, трубы, цилиндры. Однако в последнее
время стали известны методы, дающие возможность изготовлять из
слоистых масс крупные изделия сложной конфигурации без помощи
пресса, например по способу «резинового мешка» под небольшим
давлением (стр. 346). Этот метод наряду с другими открывает ши-
рокие перспективы для применения слоистых пластиков.
Механическая прочность и физико-химические свойства слоистых
пластмасс зависят от характера и количества смолы в массе (от
толщины смоляной пленки между листами), характера 'слоистого
наполнителя и толщины его листов, а также от его удельной проч-
ности по обоим направлениям, наконец, от метода пропитки, сте-
пени отверждения при прессовании и от термической обработки
после прессования.
Смолы для слоистых фенопластов
Характер феноло-альдепидной смолы в значительной мере опре-
деляет физико-химические и диэлектрические свойства слоистых
фенопластов.
Для получения слоистых фенопластов применяют резольные
смолы.
Кроме преимуществ в отношении водостойкости и диэлектриче-
ских свойств, резольные смолы обладают меньшей термореактив-
ностью (меньшей скоростью желатинизации, стр. 420) при темпе-
ратурах прессования (150—160°) по сравнению с новолачными.
Поэтому они в процессе прессования сохраняют некоторую
пластичность в наружных слоях материала до прогревания внутрен-
них слоев, а это важно для сохранения монолитности прессуемого
материала.
-462
Гл. XF. Фенопласты слоистые
Термореактивность смолы определяется не только общим вре-
менем отверждения (достижения стадии С), но и временным ходом
отверждения смолы, который схематически может быть представлен
кривыми 1 и 2 на рис. 177. При
тучшие результаты дают смолы,
изготовлении слоистых пластиков
ход отверждения которых выра-
жен кривой 2. Обычно приме-
няют резольные смолы со сред-
ней скоростью желатинизации
в пределах 70—100 сек.
Для производства слоистых
фенопластов наряду с феноло-
формальдегидными резолами
широко применяют крезоло-
и ксиленоло-формальдегидные
резолы. Так, значительная
часть технического трикрезола
идет для производства слои-
стых пластиков. Это объяс-
няется тем, что резольные смо-
лы на основе крезолов и ксиле-
нолов менее полярны и изгото-
вляемые из них пластики имеют
более высокую водостойкость
и лучшие диэлектрические
свойства. Кроме того, крезоль-
рис. 177. Зависимость перехода смолы
в нерастворимое состояние от времени
нагревания при 150°.
1—феноло-альдегидная резольная смола; 2—фе-
ноло-анилино-альдегидная резальная смола.
ные смолы обладают меньшей термореактивностью при темпера-
туре прессования, что также благоприятствует процессу изготовле-
ния слоистых пластиков.
Для производства бумажных фенопластов уже давно исполь-
зуют твердые резолы, полученные с добавкой в реакционную смесь
6—10% анилина к фенолу. В последнее время находят применение
феноло-анилино-формальдегидные твердые резолы, полученные с со-
держанием в реакционной смеси 30—50 и более процентов анилина,
что позволяет получать пластики с еще более высокими диэлектри-
ческими свойствами. Диэлектрические свойства таких пластиков,
естественно, тем выше, чем выше содержание анилина в смоле, т. е.
чем меньше фенольных гидроксильных групп в резите. Однако
термореактивность и теплостойкость пластика тем ниже, чем больше
содержание анилина.
Введение в состав реакционной смеси анилина улучшает также
условия получения твердых, хрупких резолов, имеющих в производ-
стве слоистых пластиков значительные преимущества перед эмуль-
сионными смолами. Эти преимущества: большая стабильность твер-
дых резолов, возможность применения их для ряда новых техноло-
гических процессов (пропитка наполнителя сухой расплавленной
.смолой, рольно-суспензионный метод и др.), а также более высоких
Типы слоистых фенопластов
463
технических свойств этих смол (меньшее содержание свободного
фенола, более высокая степень поликонденсации).
Однако наряду с твердыми резолами для производства слоистых
пластиков широко применяют жидкие и водно-эмульсионные резолы
и феноло-спирты. Вообще же характер, рецептура и свойства
резола, применяемого для получения слоистых пластиков, опреде-
ляются техническим назначением слоистого материала.
Твердые резольные смолы для производства слоистых пластиков
должны иметь температуру каплепадения по Уббелоде не ниже 65°,
содержать не более 12% свободного фенола и скорость желатини-
зации должна быть не менее 70 сек. и не более 3 мин.
Этим условиям удовлетворяют феноло- и крезоло-формальдегид-
ные аммиачные смолы, а также феноло-анилино-формальдегидные.
Методы пропитки наполнителя
Вследствие высокой вязкости смолы (как в расплавленном со-
стоянии, так и в виде растворов и эмульсий) и огромной действую-
щей поверхности волокнистого наполнителя по сравнению с неболь-
шим объемом смолы, совершенная и равномерная пропитка на-
полнителя встречает значительные трудности. Вместе с тем, равно-
мерность толщины смоляного слоя на поверхности наполнителя
имеет важнейшее значение для технических показателей слоистого
пластика. Таким образом, весьма существенно оформление техно-
логического процесса пропитки листов. Известны следующие ме-
тоды пропитки: 1) спиртовыми растворами резольной смолы (ла-
ковый метод); 2) водными конденсатами смолы (эмульсионный
метод); 3) расплавленной смолой под давлением; 4) феноло-спир-
тами; 5) смолообразующими реагентами (фенол, формалин, ката-
лизатор) путем конденсации их на волокне с последующим получе-
нием из этого волокна листов; 6) пропитка волокна водными
суспензиями или эмульсиями смол в ролле с получением листов
на папп-машине (рольный метод).
Типы слоистых фенопластов
Слоистые резиты являются наиболее прочными органическими
материалами. По комплексу механических свойств они прибли-
жаются к цветным и черным металлам и даже значительно превос-
ходят их, если отнести прочность к единице веса (так называемой
«весовой прочности», стр. 17). По назначению слоистые резиты
прежде всего конструкционные материалы современного машино-
строения, электротехники, строительной техники и т. д.
Слоистые фенопласты по характеру наполнителя разделяют
на: 1) фснотекстослой (текстолит), 2) фенобумослой (картонфено-
лит, бумофенолит, гетинакс), 3) феноасбослой (асболит), 4) фено-
древослой (древофенолит, лигнофоль, дельтадревесина, 5) фено-
стеклослой (стеклолит).
464
Гл. Xf. Фенопласты, слоистые
Фенотекстослой
(Текстофенолит, текстолит)
Фенотекстослой представляет собой прессованный слоистый пла-
стик, получаемый из листов органической (целлюлозной) ткани,
пропитанной резольной смолой.
Ткани
Свойства фенотекстослой в значительной мере зависят от свойств
и характера ткани.
Ткани подразделяют по составу, по типу плетения и по тол-
щине.
По составу органические ткани делят на хлопчатобумажные,
льняные и др. Основное применение получили хлопчатобумажные
ткани. Льняные ткани более прочны, однако из-за дороговизны их
применяют редко. В последнее время начинают применять ткани
из синтетических волокон, например из капрона, из полихлорвини-
лидена, политерефталата и др. Фенотекстослой на основе синтети-
ческих тканей имеет более высокую влаго-
стойкость и более высокие диэлектриче-
ские св°йства. Эти материалы, однако,
еще мало изучены.
По характеру плетения различают
ткани с простой формой переплетения
основных и уточных нитей, так называе-
Рис. 178. Киперное плете- мое тафтяное плетение, где нити основы и
ние ткани. утка расположены перпендикулярно друг
к другу (примерами являются батист,
миткаль и др.), и киперное (рис. 178) плетение (примером может
служить саржа). Кроме того, различают диагональные ткани, где
при сохранении простого, тафтяного переплетения нити утка и
основы расположены под углом в 45°. Прочность тканей киперного
плетения выше, чем тафтяного, так как при растяжении работают
обе нити, в то время как при тафтяном плетении — только нити
основы или утка. По этой же причине прочность диагоналевых тка-
ней несколько меньше, чем киперных, но выше простых тафтяных.
В производстве фенотекстослой обычно применяют ткани про-
стого плетения и диагоналевые ткани.
В зависимости от условий, в которых работает фенотекстослой,
от характера распределения в нем внешних напряжений требуются
ткани с различным характером плетения.
Для большинства назначений, в частности для производства
деталей с равномерным распределением напряжения в различных
сечениях, требуются ткани с возможно близкой прочностью по утку
и по основе.
Однако в особых случаях, в частности для деталей, работаю-
щих при одностороннем растяжении, применяют ткани, состоящие
Фенотекспюслой
465
главным образом, из одной основы (так называемые кордовые).
Ткани не должны иметь узелков, утолщений и других дефектов,
должны изготовляться из сурового волокна и не быть отбелены.
так как отбелка снижает их
прочность и термостойкость.
Шлихтование нитей и аппре-
тирование тканей снижают
их впитывающую способ-
ность, ухудшают распреде-
ление смолы внутри волок-
на, что приводит к умень-
шению прочности материала.
Поэтому шлихтованные и
аппретированные ткани ре-
комендуется «расшлихто-
вать» путем водной вы-
варки.
По толщине различают
легкие ткани (до 150 г/м2),
средние (до 300 г/м2) и тя-
желые (свыше 300 г/м2).
В табл. 52 приведены на-
именования и свойства неко-
ТАБЛИЦА 52
Ткани для производства
фенотекстослоя
Ткань
Разрывная прочность полоски 250 X 50 мм кг Уд. проч- ность при пересчете на единицу веса кг/г
« се
св а
о а о а
X о X о
G>
О >1
Башмачная . . . 400 114 93 23 19
Палаточная . . . 257,5 72,2 64,4 22,3 20
Фильтрполотно 242 62,9 49,4 20,7 16,5
Бязь 173,2 39,2 38,4 18,5 —
Диагональ .... 122,2 83 45,8 54 30
Рогожка тонкая . 180 66 46,1 29 20
торых хлопчатобумажных тканей, применяемых в производстве
фенотекстослоя.
Как видно из табл. 52, некоторые ткани значительно разли-
чаются в прочности по основе
ТАБЛИЦА 53
Впитывающая способность
различных тканей
Миткаль . . . 114,2 0,22 55
Бязь 173,2 0,35 50,5
Палаточная . . 303,5 0,50 41,0
Башмачная . . 483,0 0,70 40,0
и по утку. Удельные прочности
таких тканей, как бязь, палаточ-
ная, башмачная, довольно близки,
причем тяжелые ткани имеют не-
сколько большую удельную проч-
ность, чем легкие. Однако по-
следние равномернее и полнее
пропитываются смолой и способ-
ны впитывать ее относительно
большее количество (табл. 53).
Прочность пластика в конеч-
ном счете зависит от количества
слоев на единицу толщины пресс-
материала, и в этом отношении
преимущество имеют материалы
на основе тонких тканей. Только
более высокая цена и большая
стоимость процесса производства
текстослоя из этих тканей ограничивает их применение.
Весьма существенным является то обстоятельство, что ткань
после пропитки резольной смолой приобретает большую удельную
30 Зак. 1269. Э. И. Барг
466
Гл. XI. Фенопласты слоистые
прочность. При этом было выяснено (П. 3. Ли), что 1) прочность
ткани возрастает лишь до 40—50% содержания в ней смолы и
2) упрочнение ткани обратно пропорционально ее толщине.
Пропитку смолой производят чаще всего лаковым, а также
эмульсионным и спирто-эмульсионным методами. Некоторое при-
менение имеет горячий метод пропитки ткани расплавленной смолой
под давлением, который будет описан при изготовлении фенобумо-
слоя (стр. 490).
Производство фенотекстослоя (текстолита) по лаковому
и эмульсионному методам
Производство фенотекстослоя по лаковому методу складывается
из получения лака, пропитки и сушки ткани в пропиточных маши-
нах, разрезания ее, дополнительной сушки в вакуум-сушилках
и прессования.
Лак получают в реакторах непосредственно по окончании про-
цесса сушки резольной смолы, путем растворения последней в
спирте (стр. 394). Перед прибавлением спирта смолу проверяют на
соответствие техническим условиям по скорости желатинизации,
температуре плавления и вязкости. Лак должен иметь по возмож-
ности высокую концентрацию, при вязкости в пределах примерно
500—1200 сп. Обычно работают с лаком, концентрация которого
не менее 55%. Применение менее концентрированных растворов
не оправдывается с экономической точки зрения и не приводит
к улучшению качества фенотекстослоя. Применение для пропитки
ткани более концентрированных и вязких лаков стало возможным
благодаря использованию системы отжимных валков в пропиточной
машине.
Технологический процесс пропитки и сушки ткани
Пропитку и сушку ткани производят на пропиточно-сушильных
машинах. Для этого чаще всего применяют машины вертикальные
шахтного типа, реже горизонтальные, которые описаны на стр. 485
Типичные вертикальные пропиточно-сушильные машины состоят
из следующих основных частей: ванны для пропитки, вертикальной
сушильной камеры (шахты), воздушно-вентиляционной системы и
тягового аппарата.
На рис. 179 представлена конструкция пропиточной машины,
рассчитанной на одновременную пропитку и сушку двух полотнищ
ткани, сматываемых из двух рулонов.
Ткань со съемных валиков 2, снабженных тормозным устройством, проходя
направляющий валик 6, подается с двух сторон в ванну машины 4. В ванне
расположены валики 7, дающие направление движению. По выходе из ванны
пропитанная ткань проходит отжимные валики Sue помощью валиков 9
Рис. 179. Вертикальная пропиточная машина.
-'-станина; 2 -валики для непропитанной ткани; 3—валики для пропитанной ткани; 4 ванна для
- J < он и смол; 5 —сушильная шахта; ij, 7, 9, Л7 —направляющие валики; 8— отжимные валики;
1 натяжные валики; /_ коробка скоростей: 13— валик для заправки ткани в машину.
468
Гл. XI. Фенопласты слоистые
подается в вертикальную сушильную шахту 5, расположенную над ванной. Вы-
сушенная в шахте ткань проходит валики 10, натяжные валики 11 и наматы-
вается на съемные валики 3 в рулоны. В период прохождения между вали-
ками 10 и съемным валиком 3 ткань должна охладиться до такой температуры,
чтобы слон в рулоне не могли слипаться (конечная температура зависит от ха-
рактера смолы и количества остаточного растворителя). Вся система (съемные
валики, отжимные и др.) приводится в движение от электромотора посредством
цепных приводов и коробки скоростей 12, которая позволяет менять число обо-
ротов электромотора и, следовательно, в случае необходимости и скорости дви-
жения ткани в машине (от 50 до 300 и более .и/час).
Ванна (емкостью около 0,5 м3), в которой производится про-
питка, имеет рубашку для подогревания лака или эмульсии смолы
(пуском горячей воды). Подогрев (до 40—50°) применяют в тех
случаях, когда необходимо снизить вязкость раствора. Для поддер-
жания в ванне постоянной концентрации лака или эмульсии реко-
мендуется во время работы пропиточной машины непрерывная или
периодическая циркуляция раствора между ванной и сборником
лака. Это осуществляют с помощью шестеренчатого насоса. Реко-
мендуется также фильтрация лака, отбираемого из ванны. По-
стоянство уровня в ванне достигается посредством переливной
трубки. Время, в течение которого ткань должна находиться
в ванне, зависит от ее толщины и вязкости лака.
Практика показывает, что достаточно пребывания ткани в ванне
от 20 до 45 сек.; более длительные сроки, повидимому, не сказы-
ваются существенно на степени ее пропитки.
Проходя через ванну, ткань в значительной мере увлекает на
своей поверхности вязкий раствор смолы. Для точного регулирова-
ния содержания смолы в ткани, а также для достижения равно-
мерной пропитки и удаления избытка смолы над ванной располо-
жены отжимные гумированные валики, между которыми и проходит
пропитанная ткань. Содержание смолы в ткани регулируют точной
установкой степени отжима. Из отжимных валиков ткань выходит,
имея равномерное, заданное распределение смолы; при этом проис-
ходит не только распределение смолы на поверхности, но и глубокая
пропитка волокна. Сушильная часть машины, в которую направ-
ляется ткань после отжимных валиков, представляет собою вер-
тикальную, прямоугольную шахту с внутренними паровыми труб-
чатыми радиаторами, расположенными вдоль стенок шахты. Кон-
структивно она оформлена в виде железного каркаса, с внутренней
стороны покрытого листовым железом, которое изолировано асбе-
стовой облицовкой. Она имеет высоту около 12—13 м и поперечную
площадь ~ 4 м2. Площадь сечения шахты должна быть мини-
мально возможной, что необходимо для сокращения потерь тепла
и расхода вдуваемого воздуха.
Шахту обогревают с помощью паровых труб и продувкой горя-
чего воздуха.
Греющие трубы располагают в несколько рядов вдоль оси
шахты; ряды труб состоят из отдельных секций, каждая из которых
Фенотекстослой
469
самостоятельно включена в паровую сеть. Тепловой режим в отдель-
ных зонах шахты может быть регулирован путем отключения
отдельных секций или изменением давления и количества поступаю-
щего пара.
Температура поверхности труб 160—190°, давление пара
~ 8—10 ат. Наиболее низкая температура внизу шахты; повышаясь
по мере движения сукна, она наиболее высокая на выходе ткани.
Постепенное повышение температуры в шахте достигается увеличе-
нием поверхности нагрева труб от начала сушильной шахты к концу
ее. Первый ряд труб располагают па высоте 1,5—2 м от начала
шахты.
В шахту при помощи центробежного вентилятора через после-
довательно соединенные калориферы подводят горячий воздух.
Температурный режим в шахте и скорость движения воздуха
определяют скорость сушки ткани. Однако температурный интервал
и скорость подъема температуры ограничены
условиями, обеспечивающими качественные
показатели пропитанной ткани и взрыво-
безопасные концентрации растворителя.
Так, при быстром подъеме температуры или
быстрой подаче горячего воздуха и при тем-
пературах свыше 140° могут образоваться
вздутья и произойти быстрое отверждение
смолы с поверхности; последнее препят-
ствует диффузии растворителя из внутрен-
них слоев ткани к ее поверхности.
В обычных пропиточно-сушильных ма-
шинах температурный режим характеризует-
ся следующим распределением температур-
ных зон в шахте: низ 60—80°, середина
80—100° и верх 100—130°. Постоянство тем-
Время, мин.
и
скоро-
180. Кривые сушки
„дук“ при 130°
Рис.
ткани
при различных
стях v движения воздуха.
1—v = l м(сек\ 2—т>=2 м]сек\
3—г?=4 м!сек.
пературы и непрерывный приток тепла обес-
печиваются конвекцией и лучеизлучением
тепла от радиаторов и циркуляцией горя-
чего воздуха.
Изучением оптимальных температурных
условий для пропиточных машин занимался
А. Н. Левин. Он нашел, что лучшие условия сушки тяжелой и лег-
кой ткани могут быть достигнуты при более высокой средней темпе-
ратуре сушки и при меньшем температурном перепаде, а именно, при
120° в нижней части шахты (начиная с 2—3 метров выше входа
ткани) и 140° на выходе ткани. Было также установлено, что луч-
шие результаты могут быть достигнуты при скорости воздуха 4 м/сек
(рис. 180). В этих условиях продолжительность сушки в шахте
для тяжелой ткани около 3,5 мин., а для легкой — около 2 мин.
Движение ткани через шахту происходит однократно по прин-
ципу противотока: .наиболее горячий воздух встречает наиболее
470
Гл. XI. Фенопласты слоистые
сухую ткань. Обычно предусматривают рециркуляцию воздуха с ре-
куперацией спирта с расчетом устранения в шахте взрывоопасных
концентраций (57 г этилового спирта на 1 кг абсолютно сухого
воздуха).
Пропиточно-сушильные машины должны быть снабжены взрыво-
безопасными моторами и выключателями. Конструкции фрикцион-
ного привода, редукционных и шестереночных передач должны
исключать возможность образования искры. Улучшение качества
пропитанной ткани, увеличение производительности машин и повы-
шение коэффициента использования тепла достигают применением
сушки инфракрасными лампами. В последние годы такими лам-
пами оборудуют сушильные камеры (шахты) пропиточных машин.
Инфракрасные лучи способны более глубоко проникать в лаковую
пленку и этим обеспечивают более равномерный обогрев по тол-
щине пленки, что приводит к более быстрому испарению спирта.
При применении инфракрасного обогрева можно значительно
уменьшить количество подаваемого горячего воздуха или же уве-
личить скорость движения ткани.
Процесс термической обработки ткани в сушильной части про-
питочной машины преследует цель не только сушки (от
спирта или воды), но и углубление реакции поликонденсации смолы
на ткани, с частичным переводом резола в резитол.
Скорость, с которой должна продвигаться ткань, зависит от ее
толщины, концентрации лака, содержания в ткани смолы, темпе-
ратурного режима в шахте и температуры размягчения смолы.
От скорости движения ткани, в свою очередь, зависит производи-
тельность машины. Более тонкие ткани, естественно, допускают
большую скорость их движения, однако производительность ма-
шины в весовом отношении при этом не увеличивается, а умень-
шается (табл. 55). С другой стороны, чем выше концентрация лака
или чем меньше содержание смолы в ткани и чем выше температура
размягчения смолы, тем больше может быть скорость прохождения
ткани и выше производительность машины.
Сушку в пропиточно-сушильной машине доводят обычно
до содержания 4—8% летучих, считая на смолу (или 2—4%, считая
на пропитанную ткань). При этом для уменьшения чрезмерного
вытекания из пропитанной ткани смолы при прессовании плит
небольшую часть смолы (5—20%) необходимо перевести в рези-
тольное (нерастворимое) состояние.
Минимальное время сушки определяется необходимостью су-
шить «до отлипа», т. е. до такого состояния, при котором нама-
тываемая в рулоны ткань при 30—40° не слипалась бы; это, есте-
ственно, — основное условие нормальной работы машины. Однако
липкость и мягкость пропитанной ткани определяются не только
содержанием в ней остаточного растворителя, но и температурой
размягчения смолы. Поэтому, чем выше эта температура и чем
меньше в смоле летучих, тем быстрее может быть произведена
Фенотекстослой
471
сушка «до отлипа», тем с большей скоростью может двигаться
ткань, т. е. тем больше производительность машины. При заданных
температурах время нахождения ткани в шахтной сушилке лими-
тируется скоростью перехода смолы в нерастворимое состояние.
В табл. 54 показано изменение растворимости крезоло-формальде-
гидного резола в зависимости от температуры и времени сушки.
ТАБЛИЦА 54
Зависимость растворимости крезоло- формальдегидного резола от температуры и времени сушки
Время Температура, ’С
80 100 110 120 130 150 170
Нерастворимая часть смолы, %
10 мин. 20 „ 40 „ 1 час 9 3 ”, 4 „ 0 0 0 0 0 2,7 й’з 0 0 3,1 30,6 59,4 65,3 71,4 0 0 40,7 61,3 82,6 86,7 91,0 6,8 34,4 65,4 74,7 88,8 93,3 95,4 46,0 74,8 90,4 98,3 100 100 100 78,0 97,2 100 100 100 100 100 100 100 100 100 100 100 100
Скорость движения ткани в шахте устанавливают в зависимо-
сти от температурного режима, содержания летучих и смолы в
ткани, температуры размягчения смолы, толщины ткани и от вре-
мени, необходимого для частичного перевода смолы в резитольное
состояние. Она обычно варьирует е пределах от 150 до 700 пог. м/час;
соответственно ткань находится в сушилке от 2 до 5 мин.
ТАБЛИЦА 55
Технические показатели работы вертикальных пропиточных машин
Показатель Единицы измерения Величина показателя
Производительность машины по тяжелой ткани пог. м/час 412
т/час 0,323
То же, по легкой ткани пог. м/час 720
т/час 0,175
Длина хода ткани пог. м 12
Время сушки тяжелой ткани мин. 3,5
Уд. производительность на 1 ж3 сушильного пространства для тяжелой ткани пог. м/м3час 68,8
Расход пара на 1 пг пропитанной тяжелой ткани т/т 0,625
Расход электроэнергии на 1 т пропитанной тяжелой ткани квт-ч/т 14,5
472
Гл. XI. Фенопласты слоистые
В табл. 55 приведены некоторые технические показатели работы
вертикальных пропиточных машин.
Контроль производства осуществляют путем анализа лака (или
эмульсии смолы) и анализа пропитанной ткани. Определяют вяз-
кость лака, а также его концентрацию по удельному весу. Пробы
для анализа берут периодически из пропиточной ванны. Готовую
ткань анализируют на содержание смолы, летучих и нерастворимой
части смолы.
Пропитка ткани феноло-спиртами
А. А. Ваншейдт, А. А. Васильев и Р. И. Груз, разработавшие
процесс получения феноло-спиртов, применили их для пропитки
волокнистого, а также слоистого наполнителя при производстве
слоистых пластиков.
Феноло-спирты представляют собою простейшие продукты при-
соединения фенолов к формальдегиду в щелочной среде (стр. 357).
Они могут образоваться при отношении 1,1—1,4 молей формаль-
дегида к 1 молю фенола в присутствии 1,5—3% едкого натра или
гидрата окиси кальция в качестве катализатора. Реакцию проводят
при 50° в течение 6—12 час. Образовавшиеся таким образом
продукты — маловязкие жидкости, содержащие 50—60% феноло-
спиртов. Даже при длительном хранении не происходит значитель-
ного увеличения их вязкости; они могут быть разбавлены водой
в любых отношениях, причем вязкость растворов практически мало
отличается от вязкости воды. Феноло-спирты могут быть получены
и в безводном состоянии в результате отгонки воды под вакуумом
при низких температурах. Безводные феноло-спирты также пред-
ставляют собою сравнительно маловязкие жидкости, способные
легко пропитывать волокнистые наполнители.
Малая вязкость феноло-спиртов является их преимуществом
перед растворами резольных смол; она дает возможность значи-
тельно легче и более совершенно пропитать волокнистый наполни-
тель, в том числе ткань и бумагу. Кроме того, при применении
феноло-спиртов отпадает необходимость в расходе растворителя.
В процессе сушки и термообработки пропитанной ткани в пре-
делах 70—80° феноло-спирты в результате поликонденсации, про-
текающей на волокне, переходят в смолообразные продукты,
в основном идентичные резольным продуктам конденсации. При
горячем прессовании пропитанных феноло-спиртами и термически
обработанных волокон ткани или бумаги получаются резитовые
твердые и неплавкие продукты.
Технологический процесс производства фенотекстослоя на основе
феноло-спиртов складывается таким образом из следующих опера-
ций: получения феноло-спирта, пропитки и сушки ткани на про-
Фенотекстослой
473
ниточно-сушильных машинах, дополнительной сушки и термической
обработки в термостатах и, наконец, прессования на этажных прес-
сах. В общем он во многом аналогичен процессу производства
фенотекстослоя по лаковому методу.
Содержание смолы, о|0 Удельная ударная вязкость, : tC2*CMjCM* Предел, прочность на изгиб, кг!см* Предел прочности на разрыв,
60 23 1094 659
53,3 27,3 1378 786
51,2 34,2 1462 882
47,5 35,6 1410 799
45,1 46,4 1434 908
40,0 49,5 1825 1238
Зависимость свойств фенотекстослоя от соотношения
и характера компонентов
Механическая прочность фенотекстослоя (как и всех слоистых пластиков)
определяется удельной прочностью слоистого наполнителя (ткаии), его толщи-
ной и соотношением между наполнителем и смолой. При одном и том же содер-
жании смолы в пластике прочность тем больше, чем тоньше ткань. Зависимость
прочности фенотекстослоя от содержания cmoj
приведена в табл. 56. Как видно из таблицы,
прочность слоистого пластика тем выше, чем
меньше в нем смолы.
Обратная зависимость прочности от содер-
жания смолы может быть объяснена диспер-
гирующим действием наполнителя, который
переводит «объемную» смолу в систему ориен-
тированных тонких пленок. Чем больше со-
держание наполнителя, тем тоньше элементар-
ные слои смоляных пленок.
Известно, что техническая прочность уве-
личивается с уменьшением толщины образца и
в пределе (при толщинах порядка нескольких
микрон) может достигать теоретической проч-
ности тела (стр. 122—125).
Следовательно, можно ожидать, что с
уменьшением концентрации смолы прочность
пластика будет возрастать за счет увеличения
прочности пленок смолы. Можно теоретиче-
ски рассчитать зависимость прочности пла-
стика от содержания наполнителя (смолы);
такая зависимость в первом приближении
должна быть линейной, при этом прочность
пластика будет значительно выше как прочно-
сти наполнителя (ткани), так и смолы (резита).
На рис. 181 показаны теоретическая и экспериментальная кривые зависимости
прочности от содержания смолы. Из рисунка видно, что экспериментальная, кри-
вая, в отличие от теоретической, имеет резко выраженный максимум, который для
различных слоистых пластиков обычно находится в пределах ~30% содержания
смолы. Причина этого максимума — неравномерность распределения смолы при
увеличении содержания наполнителя; чем равномернее пропитка, тем при мень-
шем содержании смолы лежит этот максимум.
На практике, однако, содержание смолы невозможно уменьшить ниже опре-
деленной величины, так как это приводит к ухудшению водостойкости мате-
риала и к необходимости применения чрезмерно высоких давлений при прессо-
вании. Поэтому устанавливают оптимальное содержание смолы, соответствую-
щее заданным свойствам и назначению материала. Для большинства назначе-
ний выпускают слоистые материалы, содержащие 30—50% смолы. Свыше 50%
смолы содержат лишь материалы, к которым предъявляют повышенные требо-
вания в отношении химстойкости и водостойкости.
Механические свойства текстослоя различны в продольном и поперечном на-
правлениях. Это связано с тем, что прочность ткани по основе всегда пре-
вышает, а иногда весьма значительно (примерно на 50—100%), прочность
по утку.
(от толщины смоляного слоя)
ТАБЛИЦА 56
Зависимость прочности фе-
нотекстослоя от содержания
в нем смолы
Гл. X!. Фенопласты слоистые
Свойства текстослоя неодинаковы при действии напряжения вдоль слоев или
перпендикулярно к ним. Прочность на сжатие, изгиб, электрическая проч-
ность, водостойкость значительно выше в перпендикулярном направлении
к слоям, прочность же на растяжение, естественно, выше, если оно напра-
влено вдоль слоев наполнителя. Поэтому не безразлично, в каком направлении
расположены слои в конструкциях. Необходимо, чтобы в изделиях соответствую-
щие напряжения, возникающие в эксплуатационных условиях, действовали бы
в направлении максимальной прочности ма-
Рис. 181. Зависимость прочности
слоистого пластика от содержа-
ния смолы.
1— теоретическая кривая; 2 - эксперимен-
тальная кривая.
териала.
Физико-механические свойства слоисто-
го пластика определяются во многом проч-
ностью склейки его слоев. Последнюю
можно характеризовать величиной показа-
теля прочности на раскалывание в на-
правлении параллельном слоям. От проч-
ности склейки зависит способность мате-
риала к механической обработке, влаго-
поглощение, коэффициент допускаемой на-
грузки и т. д.
Снижение прочности на раскалыва-
ние чаще всего объясняется несоблюде-
нием должного режима производства про-
питочной ткани и режима прессова-
ния. Более высокая влажность пропи-
танной ткани, «пересушка» (уменьше-
ние содержания растворимых) и недо-
статочное содержание смолы — основные
причины снижения прочности склейки
слоев.
Уменьшение содержания смолы и
усиление этим материала допускается
лишь до того предела, пока это не вы-
зывает уменьшения прочности склейки
слоев.
Фенотекстослой имеет более высокую прочность на скалывание, чем дру-
гие слоистые фенопласты. При равном содержании смолы она зависит от типа
ткани. Тканн с преобладанием нитей основы (кордовые, частично ткани
кпперного плетения) дают значительно более прочный материал (прочность
на растяжение до 2000 кг/см2), однако только в продольном направлении,
при соответствующем снижении прочности в поперечном направлении (до
400—600 кг/сл«2).
Физико-химические свойства фенотекстослоя — водостойкость, химическая
стойкость, а также, косвенно, и диэлектрические свойства зависят от степени
защиты гидрофильного целлюлозного наполнителя (ткани) гидрофобным свя-
зующим (резитом). Водостойкость снижается по экспоненциальной зависимости
с уменьшением содержания смолы.
Слоистые фенопласты отличаются от всех типов неслоистых значительно
более высокой прочностью и меньшим эффектом надреза (стр. 446). Учитывая
влияние надреза, удельная ударная вязкость фенотекстослоя в 10—20 раз выше
фенодреволитов. Его прочность можно сравнить с прочностью твердых пород
дерева, цветных и черных металлов (алюминий, бронза, чугун и др.). По «весо-
вой» прочности (стр. 17) он приближается к прочности стали. Техническое
значение фенотекстослоя часто определяется не только прочностью, но и рядом
других характерных и ценных свойств. Так он обладает малым коэффициентом
трения (0,05—0,01) и малой истираемостью, и поэтому успешно применяется
в производстве подшипников. При умеренных напряжениях износ фенотексто-
слоя ниже, чем у цветных металлов, чугуна и закаленной стали. Очень важное
качество фенотекстослоя — высокая способность поглощать вибрационную энер-
гию (вдвое больше, чем дерево, и во много раз больше, чем металлы). На
Фенотекстослой
475
рис. 182 приведена диаграмма, показывающая способность гасить вибрации, воз-
никающие на лопастях пропеллера, изготовленного из стали и текстослоя.
видно из резонансных кривых, эта способность
у текстослоя значительно выше, чем у стали.
Статическая прочность фенотекстослоя моно-
тонно снижается при переходе к высоким
температурам, так, например, прочность на
растяжение падает от 1100 кг/см? при +20°
до 450 кг/см2 при 100°. Удельная ударная вяз-
кость показывает обратную зависимость: она
непрерывно снижается с понижением темпера-
туры и ее максимум находится при 100—120°.
Длительные действия высоких температур
(—120) приводят к резкому снижению проч-
ности, однако длительный нагрев при 60—80°
сопровождается увеличением прочности (если
последняя испытывается при +20°). Можно счи-
тать, что фенотекстослой может длительно ра-
ботать прн температурах 90—105°.
Изучение поведения фенотекстослоя при
знакопеременных нагрузках, а также изучение
скорости деформации при ее длительном воз-
действии дают основание считать, что фено-
текстослой может нести постоянную нагрузку
от 50 до 60% предела прочности (различ-
ную в зависимости от характера напряжения).
Рис. 182. Резонансные кривы
для стали и текстолита.
Ниже приведены пределы показателей свойств фенотекстослоя;
Уд. вес, г/см3............................‘ 1,3—1.4
Теплостойкость по Мартенсу, °C............ 125—150
Жаростойкость по Шрамму, класс............ 2
Водостойкость (привес стандартного бруска в
воде за 24 часа), %....................... 0,4—1,5
Уд. ударная вязкость, кг • см/см2......... 30—60
Предел прочности на изгиб, кг[см2......... 800—1800
„ „ „ сжатие „ ........ 2000—3000
„ „ „ разрыв „ ........ 600—1600
„ ,, „ скалывание, кгфм2 . . . 250—350
Модуль упругости при растяжении, „ . . . 50 000—90 000
Твердость по Брннелю, кг/мм2.............. 30—45
Коэффициент трения (при нагрузке в 100 кг) 0,015—0,02
Уд. теплоемкость, кал[г • °C.............. 0,3—0,4
Теплопроводность, ккал'м- час. °C......... 0,12
Коэффициент теплового расширения.......... 20- 10'6—30- 10_®
Уд. объемное электрическое сопротивление,
‘-’-слг................................... щи—1(Я
Уд. поверхностное электрическое сопротив-
ление, 2.................................. 109 —101-
Электрическое пробивное напряжение, KejMM 8—20
Изготовление плит из фенотекстослоя
Из пропитанной и высушенной ткани прессуют илн плиты и пластины, или
фигурные изделия (шестерни, подшипники и др.); ограниченное применение по-
лучили прессование и намотка труб. Процесс изготовления плит складывается
из укладки и резки ткани, сборки пакетов, дополнительной сушки, горячего
прессования, обрезки кромки; в некоторых случаях применяют термо-
обработку плит.
476
Гл. XI. Фенопласты слоистые
Листы из пропитанной ткаии укладывают и разрезают в пакеты на специаль-
ном столе (рис. 183) с четырьмя стойками. Ткань из рулона пропускают через
валик, укладывают между стойками стола и закрепляют на сгибах при помощи
металлических прутьев. Длина и ширина ткани должны соответствовать длине
и ширине прессуемых плит.
Рис. 183. Стол для укладки заготовок.
Количество листов в пакете зависит от заданной толщины прессуемой плиты,
причем для каждого рода ткани, с учетом содержания в ней смолы, имеется
установленный наборный коэффициент — количество листов на 1 мм прессуемой
детали.
Перед прессованием заготовки во многих случаях сушат в полочных ва-
куум-сушилках при 60—90° в течение 1—3 час. Предварительная сушка улуч-
шает диэлектрические свойства и водостойкость материала. В виду гигроскопич-
ности пропитанной ткани вакуумную подсушку производят вне зависимости от
степени сушки в пропиточно-сушильной машине. После сушки заготовки
немедленно прессуют; для этого и.х кладут между стальными никелированными
пластинами, предварительно смазанными олеиновой кислотой. В зависимости от
толщины прессуемых плит, одну или несколько таких заготовок с прокладочными
стальными листами кладут между обогревающими плитами пресса.
Для прессования применяют многоэтажные гидравлические прессы обычно
с нижним давлением, мощностью от 500 до 3000 т и более, с плитами размером
от 500X500 до 1500ХЮ00 мм и более (рис. 184); гидравлическое давление
з цилиндре такого пресса обычно 200—300 кг/см2.
Пресса столь большой мощности получают питание рабочей жидкости от
насосно-аккумуляторных станций. Целесообразно, в связи с большим расходом
рабочей жидкости, применять предварительное заполнение рабочих цилиндров
жидкостью низкого давления (~50 кг/см2), после чего давать высокое давление
I~-300 кг/см2).
Между верхней рамой и нижней плитой пресса помещается обычно от
S до 12 плит, расположенных параллельно одна над другой. Они обогреваются
паром, подаваемым в них через медиые или резиновые шланги; расстояние
между плитами от 200 до 400 мм.
Фенотекстослой
477
После укладки пакетов в плиты пресса дают пар (6—8 ат) и медленно
включают давление, большей частью ступенчато, сначала низкое, затем высо-
кое. Под влиянием температуры и давления смола плавится, листы становятся
мягкими и пластичными; при этом смола частично вытекает из заготовок, не-
одновременно происходит и отверждение
ее, которое начинается с внешних слоев
прессуемого материала и постепенно дохо-
дит до середины. После выдержки при на-
гревании до возможно полного отверждения
материала пар выключают и в плиты пресса
пускают холодную воду до полного охла-
ждения (40—50°) пресс-материала; после
этого прессование прекращают, пресс раз-
гружают и отпрессованные плиты отделяют
от металлических прокладочных листов.
Плиты и пластины направляют для обрезки
кромок на дисковую пилу.
Оптимальная температура прессования
фенотекстослоя 155—165°. При меньшей
температуре требуется значительно более
длительное время прессования,а более вы-
сокая — ведет к быстрому отверждению
внешних слоев, примыкающих к обогре-
вающим плитам пресса, в то время как
внутренние слои еще не перешли в пла-
стическое состояние; вследствие этого умень-
шается прочность склейки слоев.
Фенотекстослой обычно прессуют при уд.
давлении 70—90, реже при 100—150 кг/см2.
Высокое давление при прессовании
слоистых пластиков необходимо прежде
всего для обеспечения возможно более
тесного контакта между связующим со-
прикасающихся слоев для выявления по-
верхностных сил прилипания. Естественно,
что чем тоньше пленка связующего (чем
меньше смолы в ткани), тем более вы-
сокое давление требуется для контакта
слоев. Однако высокое давление при прес-
совании слоистых фенопластов необходимо
и при наличии избытка смолы; оно пре-
пятствует образованию вздутий и расслое-
нию под влиянием внутреннего давления
паров, образующихся как за счет влажно-
сти прессуемого материала, так и вследствие
выделения паров конденсационной воды.
Некоторые экспериментальные данные поз-
воляют считать, что в пределах опоеделен-
ных давлений водостойкость, модуль эла-
стичности И предел текучести получаемого
слойматериала возрастают с увеличением
давления при прессовании (рис. 185); это,
невидимому, связано с увеличением плот-
ности материала. Прочность материала
на скалывание обычно тем выше, чем
Рис, 184. Плиточный 10-этажныи
гидравлический пресс мощностью
2500 т с размерами плит
1250X2500 мм
Г—рабочий цилиндр; 2—рабочий плуижс;;
подвижной стол; 4—плита; 5—колонн,::
6—архитрав; 7—вспомогательный цилиндр;
5—вспомогательный плунжер; Р—рейка.
больше давление при прессовании.
Время, необходимое для прессования, складывается из времени на: 1) про-
гревание материала до температуры прессования (150—165°), 2) завершение
реакции поликонденсации (отверждения) и 3) охлаждение прессматериала под
давлением (до 40—50°).
4.8
Гл. XL Фенопласты слоистые
Время, затрачиваемое на прогревание материала, находящегося между обо-
гревающими плитами пресса, по опытным данным, около 1 мин. иа 1 зглг толщины
прессуемой плиты (или нескольких плит, уложенных в один просвет пресса).
Время для охлаждения материала в зависимости от температуры охлаждаю-
щей (водопроводной) воды составляет
0,8—1,2 мин. на 1 мм толщины прес-
суемых плит.
Время на отверждение (переход в
стадию С) в зависимости от состава ре-
зольной смолы и ряда других факторов
(глубины конденсации и сушки пропи-
танной ткани) составляет 5—10 минут.
Естественно ожидать, что время
отверждения не должно зависеть от тол-
щины прессуемых изделий.
В этом случае суммарное время
прессования / должно было бы выра-
жаться уравнением:
t = ad + bd с,
где а — время для прогрева 1 мм толщи-
ны изделия до 150—160°; b — то же для
охлаждения; d — толщина прессуемых
плит, уложенных в один просвет пресса;
с — время на отверждение.
Вместе с тем, практически устано-
влено, что время для полного отвержде-
ния прессматериала зависит и от толщи-
ны прессуемых изделий; для слоистых
фенопластов оно составляет в среднем
от 3 до 5 мин. на 1 мм, а для фено-
текстослоя 4—5 мии. на 1 мм толщины
прессуемого изделия. Зависимость вре-
мени отверждения от толщины плит
объясняется условиями теплопередачи
от поверхностных слоев в глубь материа-
ла, различными условиями отверждения
Рис. 185. Зависимость свойств фено-
текстослоя от уд. давления при прес-
совании.
1— водопоглощенне, 2 — прочность на рас-
тяжение. 102 к г 1см*, 3—модуль упругости X 104,
к?/см*; 4—уд. вес, г,'сма', 5 — уд. ударная вяз-
кость, 10 кг-см/см--
поверхностных и внутренних слоев, и условиями диффузии и распределения влаги
и конденсационной воды внутри материала в процессе прессования.
Таким образом, общее время прессования слоистых фенопластов составляет,
примерно, 5—7 мин. на 1 мм суммарной толщины материала между плитами
пресса (для фенотекстослоя 6—7 мин.).
Значительный прогресс в технологии прессования слоистых пластиков мо-
жет быть достигнут применением обогрева токами высокой частоты. Последний
основан на превращении части энергии высокочастотного поля в тепло благо-
даря работе, затрачиваемой на вращение диполей. Коэффициент превращения
энергии высокочастотного поля в тепловую пропорционален полярности прессуе-
мого материала (коэффициенту диэлектрических потерь), а также частоте поля
и квадрату напряжения. Он выше в начальной стадии прессования и умень-
шается по мере завершения отверждения материала. Применение обогрева
прессуемых изделий токами высокой частоты коренным образом меняет характер
распределения влаги, тепла и внутренних напряжений в материале в процессе
прессования.
Основным техническим преимуществом обогрева токами высокой частоты
является равномерное образование и распределение тепла внутри прессуемого
материала, в то время как при обычном обогреве через греющие плиты пресса
тепло распределяется крайне неравномерно, следовательно неравномерно проис-
ходит и отверждение материала (весьма быстро отверждаются внешние слои,
крайне медленно внутренние). Неравномерность отверждения приводит к рез-
Фенотекстослой
479
кому нарастанию внутренних напряжений, к короблению и расслоению массы.
Выделяющаяся конденсационная вода не может из внутренних слоев диффунди-
ровать из-за отверждения внешних слоев. В этих условиях для получения тех-
нически годных материалов необходимо применять высокие давления, противо-
стоящие упругости давления паров, низкие температуры прессования и длитель-
ное время выдержки. Однако и при этом внутренние напряжения в материале не
могут быть сняты, что в значительной мере снижает его физико-механические
свойства. Естественно, что обогрев токами высокой частоты, определяющий
равномерное отверждение материала, должен устранить основные недостатки
обычного горячего прессования (высокое давление, длительную выдержку и др.).
Экспериментально было показано, что применение токов высокой частоты
имеет следующие преимущества: 1) процесс нагрева и отверждения происходит
по толщине материала равномерно и с одинаковой скоростью; 21 разогревание
происходит быстрее и допустимо применение более высоких температур; 3) зна-
чительно сокращается время (отверждения) выдержки (~10—30 мин.), которое
мало зависит от толщины прессуемого пакета; 4) ввиду равномерного отвер-
ждения создаются благоприятные условия для удаления влаги и снятия вну-
тренних напряжений, что позволяет вести прессование при значительно меньшем
уд. давлении (20—30 кг/см2); 5) вследствие уменьшения внутренних напряжений
материал может быть вынут из пресса при более высоких температурах, или без
охлаждения; 6) материал получается значительно более однородным, увеличи-
вается его прочность на раскалывание, улучшаются диэлектрические свойства и
другие показатели.
Высокочастотное нагревание особо эффективно при прессовании толстостен-
ных изделий (из материала, обладающего значительной полярностью). Оно, в
частности, с успехом может быть использовано при прессовании всех видов фено-
пластов, аминопластов и др.
Высокочастотный обогрев не получил еще большого применения при прессо-
вании слоистых пластиков. Это объясняется многими трудностями, стоящими на
пути его внедрения. Он требует применения высокочастотного генератора вы-
сокой мощности (~150 кет для плит пресса 1000X 1000 мм); коэффициент ис-
пользуемой энергии крайне низок, в ряде случаев происходят перегревы и вы-
жигание материала; не разрешен вопрос эффективного охлаждения. Поэтому
применяют высокочастотный прогрев пакетов из пропитанного материала
(~120°) с последующим прессованием на этажных прессах обычного обогрева.
В этом случае требуется генератор значительно меньшей мощности.
После прессования желательно, а в некоторых случаях необходимо, подвер-
гать изделия термической обработке, с целью углубления процесса поликонденса-
ции смолы, снятия внутренних напряжений в материале и удаления из пластика
в процессе медленного прогревания влажности. Термическая обработка значи-
тельно улучшает как механические, так и диэлектрические свойства пластика. Ее
обычно производят в масляных или воздушных термостатах при ступенчатом
подъеме температуры от 60 до 130° в течение 6—12 час. Применяют также метод
длительного прогревания материала между греющими плитами при низком давле-
нии (2—5 кг/см2).
Прессование фигурных деталей из фенотекстослоя
Листы ткани, пропитанные резольной смолой, имеют, естественно, лишь
крайне ограниченную текучесть при прессовании. Она осуществляется, главным
образом, за счет способности тканей к относительному удлинению при растя-
жении как вследствие удлинения нитей, образующих ткань, так и вследствие, их
перестройки (распрямления нитей основы и скручивания нитей утка при течении
смолы). Так относительное удлинение бязи составляет 12% (как по основе, так
и по утку), при этом 6% за счет удлинения нитей и 6% за счет их перестройки.
480
Гл. XI. Фенопласты слоистые
Способность тканей к относительному удлинению используют для непо-
средственного прессования плоских и несложной формы изделий из набора про-
питанных, параллельно уложенных листов.
Эту же способность используют и для формования изделий из отвержденных
прессованных листов фенотекстослоя по так называемому методу последую-
щего формования и методами сгибания листов. Для этого из прессованных листов
фенотекстослоя вырезают заготовки с учетом формы и габаритов изделия. За-
готовки возможно быстро нагревают (в термостатах, между горячими металли-
ческими плитками, в ванне с расплавленным легкоплавким сплавом, а также
токами высокой частоты и т. д.) и быстро укладывают на горячую поверхность
формы, изготовляя изделия методом штамповки, прессования, вытяжки и различ-
ными методами сгибания. Температура разогрева материала (130—160°) зависит
от его состава и толщины, причем верхний ее предел ограничен возможностью
появления вздутий, а нижний определяется степенью отверждения листов.
Метод основан на использовании способности не полностью отвержденного
резнта к явлениям остаточной деформации при высоких температурах (выше
его «теплостойкости»). Поэтому применяют листы или не полностью отвержден-
ные или содержащие термопластичные смолы (или высококипящие «пластифи-
каторы») наряду с резольными смолами. Этими методами изготовляют лишь
плоские изделия несложной конфигурации.
Фигурные изделия более сложной формы, как правило, изготовляют методом
горячего прессования из заранее заготовленного набора пропитанных листов
ткани. Необходимо до внесения массы в прессформу собрать из соответствую-
щих выкроек (сегментов и др.) н набора прессованных таблеток заготовку, соот-
ветствующую форме прессуемой детали. При этом надо, чтобы места, где кон-
центрируются наибольшие напряжения, имели строго слоистую структуру и на-
пряжение было бы направлено в сторону наибольшей прочности материала,
т. е. в направлении основы н перпендикулярно слоям в случае, например, на-
пряжения сжатия.
Расположение слоев должно быть сделано таким образом, чтобы по воз-
можности полностью устранить скалывающие напряжения. Менее ответственные
части изделия могут быть изготовлены без сохранения строго слоистой струк-
туры и даже из кусочков пропитанной ткани. Процесс получения фигурных
деталей начинается, таким образом, с выяснения направления действующих на-
пряжений и с разработки формы и размеров выкроек для производства таких
деталей.
В качестве примера опишем один из вариантов процесса производства за-
готовок шестерен распределительного вала для автомашин. Наиболее ответ-
ственной частью шестерни является обод, который должен быть изготовлен из
материала строго слоистой структуры. Процесс складывается из ряда операций,
а именно: высечки сегментов, рубки ткани (приготовления крошки), набора
обода, предварительной прессовки обода, профилирования шайб, предваритель-
ного (холодного) прессования всей заготовки и окончательного (горячего) прес-
сования в нагретых прессформах.
Производство сегментов осуществляют на специальных автоматах или на
штамповочных механических прессах, а также на ручных винтовых прессах.
Сегменты для формования обода набирают на диске в специальном цилиндре,
диаметр которого соответствует диаметру заготовки шестерни; после этого нх
прогревают до 60—70° и прессуют при удельном давлении 300 кг/см-.
Среднюю часть шестерни (кроме обода) прессуют из крошки, причем
с обеих сторон накладывают профилированные шайбы из пропитанной ткани.
Шайбы вырубают из нескольких предварительно нагретых лент при помощи
профилировочно-просечного штампа. Крошку рубят из отходов при раскрое
тканн специальным штампом (размер крошки 2X2 см). Из подпрессованного
обода, крошки и шайб, предварительно взвешенных, составляют заготовку, ко-
торую таблетируют при подогревании до 70°. Полученную таблетку закладывают
в прессформу и прессуют на этажных прессах (по одной или по две прессформы
на каждом этаже) при удельном давлении 250—300 кг[см? и температуре
Феноггекстослой
481
150—160° в течение 25 мин. По окончании выдержки прессформу охлаждают на
специальных прессах прн давлении 25—30 кг/см2; полученные шестерни под-
вергают термической обработке в масляной ванне с постепенным подъемом
температуры до 120—150° и выдержкой при этой температуре в течение 8—12 час.
Изготовление фигурных детален из слоистых фенопластов полу-
чило принципиально новое развитие с использованием техники
прессования при низких давлениях методом «резинового мешка»
(стр. 346). Как уже было указано, этот метод дает возможность
изготовлять изделия практически любых габаритов и сложной, глу-
бокой формы с сохранением слоистой структуры без применения
прессов и стабильных прессформ. С наибольшей эффективностью
он применим к слоистым пластикам на основе полимеризационных
термореактивных смол (например типа аллиловых), способных к от-
верждению без выделения воды, которая, естественно, создает не-
желательное внутреннее давление в слоях пластика, нарушая их
связь и уменьшая плотность материала.
Тем не менее, метод прессования при низких давлениях приме-
няется и для получения изделий из фенопластов и других термо-
реактивных поликонденсационных пластиков. Эффективность этого
метода, позволяющего, как уже указывалось, непосредственно из-
готовлять изделия больших размеров и сложной формы, столь ве-
лика, что сознательно пренебрегают некоторым снижением физико-
механических показателей изделий (главным образом, водостойко-
сти, модуля упругости, прочности на скалывание и т. д.) вследствие
выделения конденсационной воды.
Для более эффективного использования этого метода получают
модифицированные смолы, так называемые «смолы для низкого
давления», которые при нагревании частично полимеризуются и
меньше выделяют конденсационной воды. Такие смолы готовят на
основе конденсации фенолов, альдегидов и ненасыщенных компо-
нентов полиэфиров. Метод «резинового мешка» был также усовер-
шенствован применением смол с соответствующей скоростью от-
верждения и уточнением режима отверждения (времени, давления,
температуры); это сделало возможной диффузию значительной
части конденсационной влаги до периода отверждения слоев, без
существенного нарушения связи между слоями.
Применение фенотекстослоя
Фенотекстослой — один из важных материалов современного машинострое-
ния. Из большого числа областей применения фенотекстослоя укажем прежде
всего на изготовление из него шестерен различного назначения (в частности,
когда требуется бесшумность хода, например автомобильных) и вкладышей под-
шипников.
Подшипники из фенотекстослоя получили большое распространение в черной
и цветной металлургии для шеек прокатных, отжимных и других металлургиче-
ских станов. Целесообразность применения фенотекстослоя для изготовления
подшипников определяется его высокими антифрикционными свойствами и малой
истираемостью. По некоторым данным замена баббитовых подшипников фено-
31 Зак. 1269. Э. И. Барг
482
Гл. XI. Фенопласты слоистые
текстослойными дает до 30% экономии в энергии. Важным преимуществом при
этом является сохранность шейки вала, которая предохраняется от царапин и
практически не снашивается. Срок службы текстослойных подшипников также
выше, чем бронзовых и других металлических; кроме того, в качестве смазки для
текстослойных подшипников применяют воду или водные эмульсии, что умень-
шает расход смазочных масел.
Следует, однако, указать, что текстослойные вкладыши для подшипников
из-за их малой теплопроводности плохо отводят тепло и не могут работать на
больших скоростях. Теплопроводность фенотекстослоя в 500—1000 раз меньше,
чем теплопроводность обычных подшипников из металла, а коэффициент их
теплового расширения значительно выше (для фенотекстослоя а = 5 • 10-5; для
бронзы а = 1,75 • 10-5).
Скорость вращения вала в подшипниках из текстослоя поэтому рекомен-
дуется в пределах 0,1—10 м!сек при удельном давлении 30—300 кг'см-.
Для снижения коэффициента трения и истираемости фенотекстослоя, а также
увеличения его теплопроводности в состав лака, применяемого для пропитки
ткани, рекомендуется вводить дисперсный графит в количестве от 2 до 10%.
Есть также указания на значительно большее содержание графита в подшипни-
ках (вплоть до 30%). По данным автора, содержание графита до 20% не
снижает механических свойств слоистых резитов, первые же 10% графита зна-
чительно их повышают.
Кроме использования для изготовления шестерен и подшипников, фено-
текстослой применяют в качестве конструкционного материала для производства
разнообразнейших деталей машиностроительной техники. Из него изготовляют
шкивы, колеса, пропеллеры, гребные винты, втулки, прокладки, кольца и другие
конструкционные детали, а в последнее время — цветные и узорчатые пластины,
служащие для облицовки стен, в качестве панелей, для изготовления ме-
бели и т. ц.
Удовлетворительные электроизоляционные показатели некоторых сортов
фенотекстослоя обусловили применение его в электротехнике — для изготовления
распределительных щитов и монтажных панелей.
Большое распространение получили так называемые «намоточные» тексто-
слойные изделия, например, трубы, втулки, стержни. Намотку трубчатых слои-
стых пластиков производят на специальных намоточных станках (стр. 489).
Фенобумослой
(Картофенолит, бумофенолит, гетинакс)
Фенобумослой представляет собой слоистый прессованный ре-
зит, состоящий из листов целлюлозной бумаги, пропитанной резоль-
ной смолой. Это один из первых слоистых резитов, получивший
основное применение в электротехнике (в качестве электроизоля-
ционного и конструкционного материала), а последние годы и в дру-
гих отраслях техники.
Существуют два принципиально отличных метода изготовления
фенобумослоя. По первому методу наполнитель (специальных сортов
бумага) пропитывают либо резольным лаком (лаковый метод), либо
расплавленной смолой под давлением (сухой метод). По второму
методу резольную смолу смешивают с бумажной массой в процессе
изготовления самой бумаги. Этот метод известен под названием
рольного метода производства бумажно-слоистых пластиков.
Фенобумослой
483
Бумаги для фенобумослоя
Состав и свойства применяемых бумаг в значительной мере опре-
деляют технические качества бумажно-резитовых пластиков.
Наибольшей теплостойкостью (термостойкостью) обладают бу-
маги, содержащие только целлюлозное волокно; различные инкру-
стирующие вещества, древесное волокно и др. ухудшают ее проч-
ность и термостабильность.
Чем меньше кислотность бумаги, тем выше ее термостабиль-
ность и диэлектрические свойства.
Назначение же бумаг в производстве различных сортов фено-
бумослоя зависит от их состава.
Для производства электротехнического материала применяют
почти исключительно бумагу из сульфатной целлюлозы. Она обла-
дает меньшей кислотностью (дает водную вытяжку с наиболее
высоким pH), более высокой термостойкостью (меньшей потерей
прочности при нагревании до 120°) и большей прочностью, чем
сульфитная целлюлоза.
Фенобумослой для декоративных целей изготовляют из суль-
фитной целлюлозы или из смеси сульфитной и хлопковой целлю-
лоз. Для производства более дешевых сортов строительного мате-
риала применяют бумаги из древесной массы или из смеси древес-
ной массы и сульфитной целлюлозы.
Прочность бумаги не одинакова в продольном и поперечном на-
правлениях: в продольном (по длине) она в 2—2,5 раза больше,
чем в поперечном, что объясняется большей ориентацией волокон в
процессе отлива на сеточной машине в направлении движения
массы.
Бумаги различаются по- впитывающей способности, которая за-
висит главным образом от характера размола массы в ролле,
а также от состава волокна, степени его очистки и отбелки.
Для разных сортов фенобумослоя требуются бумаги с различ-
ной впитывающей способностью.
Так для изготовления плиточного материала применяют более
впитывающую, так называемую пропиточную бумагу, а для произ-
водства намоточных изделий (труб, цилиндров и др.) — менее впи-
тывающую, так называемую намоточную бумагу.
Поскольку фенобумослой в значительной мере применяется для
электротехнических целей, большое значение имеют электроизоля-
ционные свойства бумаги. Они зависят от ее состава, плотности
и влажности. Наиболее стабильными свойствами обладают суль-
фатные бумаги.
Бумаги в абсолютно сухом состоянии являются хорошими ди-
электриками; так, удельное объемное электрическое сопротивление
31*
484
Гл. Х!. Фенопласты слоистые
сухой бумаги равно 1013 Q-см, тангенс угла диэлектрических по-
терь ~ 0,001. Однако технические бумаги с обычной влажностью
порядка 6—7% имеют показатели диэлектрических свойств на не-
сколько порядков ниже, а именно удельное объемное сопротивление
~ 1О9 42-си, тангенс угла диэлектрических потерь —0,1.
Было показано, что логарифм электрического сопротивления
бумаг находится в обратной линейной зависимости от относитель-
ной влажности воздуха, в котором хранятся бумаги. Ввиду столь
резко выраженной зависимости диэлектрических свойств от влаж-
ности важно соблюдать надлежащий режим хранения и сушки
бумаги. Электрическая прочность находится в линейной зависи-
мости от плотности бумаги. Так, бумага с удельным весом 0,6 г! см3
имеет электрическую прочность 8 кв/мм, а с удельным весом
1,0 г/см?—16 кв!мм. Электрическая прочность бумаги находится
в зависимости от относительной влажности воздуха, т. е. от влаго-
содержания в бумаге. Пробивное напряжение резко возрастает при
снижении относительной влажности воздуха от 60% до 20%.
Бумаги различаются по толщине: чем она тоньше и выше ее
удельная прочность, тем прочнее фенобумослой.
Важным показателем качества бумаги является ее термостой-
кость, определяемая уменьшением прочности на излом при нагреве
в течение 10 час. при 120°.
В технике производства фенобумослоя различают два основных
типа бумаг:
1) пропиточные — для производства листовых и плиточных
прессматериалов;
2) намоточные—для производства труб и цилиндров методом
намотки.
В табл. 57 приводятся некоторые свойства сульфатной бумаги
для электротехнического фенобумослоя.
ТАБЛИЦА 57
Свойства сульфатной бумаги для производства электротехнического
фенобумослоя
Свойства Пропиточная бумага Намоточная бумага
Состав волокна: небеленой сульфатной цел- люлозы, и/о 100 юо
г* ntiTiT- -а. Толщина листов, мм 0,10—0,13 0,05—0,1
Уд. вес, г/см3 0,55—0,60 0,75—0,80
Содержание золы, % до 1 —
Разрывной груз на ширину 15 мм, в про- дольном направлении, кг 5—6 45—15-
Фенобумослой
485
Продолжение
Свойства
Пропиточная
бумага
Намоточная
бумага
Удлинение при разрыве, % 1,8—2 —
Впитывающая способность по Клемму — в
течение 5 мин. подъем воды на бумаге, мм 25—40 5—10
Число двойных перегибов в продольном на-
правлении 200—300 300—2000
Стношение разрывного груза в продольном
и поперечном направлениях 1 ,Ь—2 •—
Уменьшение числа двойных перегибов после
нагрева при 120° в течение десяти часов, % 20—25 8—10
Электрическая прочность, кв}мм 5 —
pH водной вытяжки 7—8,5 7—8,5
Смолы для фенобумослой должны в основном удовлетворять
тем же условиям, что и для фенотекстослоя. При получении эле-
ктротехнических сортов фенобумослоя предпочтение отдают крезоло-
и феноло-крезоло-формальдегидным резольным смолам. Чем выше
содержание крезола в крезоло-фенольной смеси и .м-крезола в три-
крезоле и чем ниже содержание о-крезола, тем выше диэлектриче-
ские свойства получаемого материала.
Значительно более высокие показатели в отношении диэлектри-
ческих свойств могут быть достигнуты при пользовании феноло-
анилино-формальдегидными твердыми резолами.
Как правило, применяют твердые резолы и лишь значительно
реже водные конденсаты (эмульсионные смолы, см. стр. 394).
Двухсторонняя пропитка бумаги и получение
прессованного фенобумослоя
Технология двухсторонней пропитки бумаги ст зтовыми раство-
рами резольных смол (или водными эмульсиями) и применяемая
при этом аппаратура в основном такие же, что и при про-
питке ткани. '
Из-за малой прочности бумаги пропитку производят не на верти-
кальных, а на горизонтальных пропиточно-сушильных машинах.
Горизонтальные машины по существу состоят из тех же элементов,
что и вертикальные: из ванны для пропитки, сушильной камеры
(горизонтальной), системы обогревающих труб, воздушно-вентиля-
ционной системы, тягового аппарата и т. д.
Бумага, сматываемая с рулона 1 (рис. 186), через направляю-
щие валики 2, 6 и валик 4 поступает в ванну 3, в которой цирку-
486
Гл. XI. Фенопласты слоистые
лирует раствор смолы, и оттуда через отжимные гуммированные
валики 5 направляется в сушильную камеру 7 длиной 12—20 м,
опираясь на поддерживающие валики 8.
В сушильной камере бумага проходит над паровыми трубами 9,
протягивание ее через пропиточную машину с постоянной ско-
ростью осуществляется ведущими валиками 10, имеющими меха-
нический привод; после же выхода из сушильной камеры она про-
ходит через валики 11, регулирующие равномерность натяжения, и
наматывается на рулон 12.
Рис. 186. Схема горизонтальной пропиточной машины.
У—рулон бумаги; 2, 6—направляющие валики; 3—пропиточная ванна; 4—валик
пропиточной ванны; 5—отжимные валики; 7 —сушильная камера; 8— поддерживаю-
щие валики; Р —паровые трубы; 10 — ведущие валики; 11 — регулирующие валики;
/2—рулон пропитани й бумаги; 13—вытяжная труба.
Горячий воздух, подаваемый вентилятором, поступает в сушиль-
ную камеру в большей своей части у выхода бумаги из машины,
часть воздуха распределяется по зонам сушилки. Воздух, насыщен-
ный парами, удаляется на рекуперацию спирта у места входа
бумаги в сушилку через вытяжную трубу 13.
Температурный режим в сушилке регулируется по принципу
противотока; наи 5олее горячая зона находится у выхода бумаги из
машины. Темпе] атура у выхода из машины вследствие чувстви-
тельности бумаг [ к повышенным температурам не должна быть
выше 120°; скорссть же пропитки бумаги в основном зависит от кон-
струкции и теплового режима сушильной части машины; обычно
она значительно больше скорости пропитки ткани 'и составляет
от 400 до 1000 м/час.
Так же, как и при производстве фенотекстослоя, целесообразно
работать с концентрированными лаками (55—60%), применяя от-
жимные валики и регулируя содержание смолы в бумаге степенью
отжима. Однако это возможно лишь при применении прочных суль-
фатных бумаг, со сравнительно ограниченной впитывающей способ-
ностью и при работе на спиртовых растворах смол. При применении
менее прочных бумаг (например, на основе сульфитной целлюлозы,
Фенобумослой
487
древесно-целлюлозных масс) использование отжимных валиков де-
лается затруднительным. Для регулирования содержания смолы
в бумаге в этом случае устанавливают соответствующую концентра-
цию лака или соответствующую скорость прохождения бумаги че-
рез ванну. Чем больше скорость, тем выше содержание смолы, так
как при большей скорости большее количество смолы увлекается
бумагой. Соответственно увеличение скорости движения бумаги
дает тот же эффект, что и увеличение концентрации лака. Так, для
получения бумаг с одинаковым содержанием смолы при скорости
1,2 м/мин требуется лак с уд. весом 0,940, а при скорости
2,5 м/мин — лак с уд. весом 0,930.
При применении водных эмульсий и бумаг с высокой впиты-
вающей способностью, обладающих малой прочностью в мокром
состоянии, предусматривают специальный движущийся транспортер,
поддерживающий бумагу после выхода из пропиточной машины.
Сушку листов в пропиточной машине доводят до остаточной
влажности (летучих) ~4—8% (к смоле). При этом 10—20% смолы
переводят в нерастворимое состояние (резитол).
Пропитанную бумагу из рулонов нарезают на листы на рота-
ционной бумагорезательной машине, складывают в стопки и перед
прессованием подвергают дополнительной сушке в полочных ва-
куумных сушилках. Температуру в сушилке поддерживают около
60—90°; время сушки зависит от содержания летучих в бумаге
и колеблется от 0,5 до 4 час. По окончании сушки бумага должна
немедленно поступить на прессовку.
Для этого просушенные листы нужного формата укладывают в
пакеты. Прессование пластин и плит бумослоя производят тем же
методом на многоэтажных прессах, как и прессование фенотексто-
слоя: при 150—160° и удельном давлении 80—150 кг/см2. Мате-
риал выдерживают под прессом при этой температуре из расчета
4—5 мин. на 1 мм толщины материала; время выдержки отсчиты-
вают с момента достижения температуры прессования внутри плит
150—160°. По окончании выдержки материал охлаждают под да-
влением. Изделия особого назначения после прессования подвер-
гают дополнительной термической обработке путем ступенчатого
прогревания в масле или в воздушных термостатах.
Прессованные сорта фенобумослоя содержат в зависимости от
назначения от 30 до 55% смолы. Материалы электроизоляционного
назначения содержат 45—55% смолы, а материалы в основном кон-
струкционного назначения 30—45%.
Односторонняя пропитка (лакировка) бумаги
и получение намоточного фенобумослоя
Для производства намоточных изделий (труб, цилиндров) при-
меняют более плотную и менее впитывающую бумагу из сульфат-
ной целлюлозы (так называемую намоточную).
488
Гл. XI. Фенопласты слоистые
Бумагу односторонне пропитывают (лакируют) спиртовыми рас-
творами резольных смол.
Для производства лакированных бумаг применяют специальные
лакировально-сушильные машины, которые отличаются от пропи-
точно-сушильных главным^ образом устройством для односторонней
лакировки бумаги.
Лакировально-сушильная машина обычно состоит из системы
вращающихся валиков (от одного до трех), расположенных один
над другим. На рис. 187 приведена схема лакировального устрой-
ства, состоящая из трех валиков. Нижний валик на половину диа-
метра погружен в ванну с лаком; при вращении он соприкасается
Рис. 187. Схема лакирозки бумаги.
/—бумага (рулен); 2—тормозное устройства; 3—ванна с лаком;
4 — лакировочный валик: 5—натяжной валик; 6 — направляющие
валики; 7—вытяжная труба; 8 - электрообогрев; 9—лакированная
бумага.
со вторым валиком, вращающимся вне ванны, а последний —
с третьим лакировочным (барабаном). Бумага поступает с рулона
с тормозным устройством, проходит направляющий валик и обо-
гревающий вал для подсушки (на схеме не показано), касается
лакировочного валика, смачиваемого лаком, и лакируется с одной
стороны. Окружная скорость вращения лакировочного валика
больше скорости движения бумаги, благодаря чему лак снимается
бумагой. После этого бумага через натяжные и направляющие
валики поступает в сушильную часть машины и по выходе из нее
наматывается в рулон. Толщину слоя на бумаге регулируют, из-
меняя расстояния между первым и вторым валиком.
Применяют также систему, состоящую только из двух валиков —
нижнего, частично погруженного в лак, и верхнего, лакировочного,
имеющих принудительное вращение, — при вращении нижний ва-
лик захватывает лак из ванны и непосредственно передает его
Фенобумослой.
489
верхнему, соприкасающемуся с бумагой. Толщину пленки регули-
руют величиной зазора между валиками.
Лакировальные машины конструируют и с одним валиком, по-
груженным частично в лак и непосредственно соприкасающимся
с движущейся бумагой. Количество смолы, наносимой на бумагу,
регулируют степенью погружения валика в ванну, а также посред-
ством вспомогательного прижимного валика, расположенного парал-
лельно лакировочному.
Сушильная камера обогревается паровыми змеевиками и продув-
кой горячего воздуха, а также инфракрасными лампами. Темпера-
тура внутри сушильной камеры в нача-
ле сушки 70°, а в самой горячей зоне
ПО—130°; в конце машины она пони-
жается до 60—70°. Скорость движе-
ния бумаги больше, чем в пропиточ-
ных машинах, и достигает 500—
1500 м/час.
В отличие от пропитанной бумаги,
внутренние слои лакированной очень
мало пропитаны смолой, в то же
время на поверхности бумаги имеется
слой чистой смолы; содержание по-
следней в бумаге также значительно
меньше (20—35%). Даже после горя-
чей намотки или прессования смола не
полностью пропитывает бумагу. В ре-
зультате прессованный материал имеет
более выраженную (чем материал из
пропитанной бумаги) слоистую струк-
туру с последовательным чередова-
нием слоев. Такая структура материала
определяет его более высокие механи-
ческие и электрические свойства (в су-
хом состоянии), но приводит к значи-
тельному снижению водостойкости. При этом плоскость, параллель-
ная слоям (торцовая часть) материала, наименее устойчива к дей-
ствию влаги. Вследствие этого для уменьшения гигроскопичности
материала торцовые части намоточных изделий или даже всю их
поверхность тщательно покрывают резольным лаком.
Намотку труб и цилиндров производят на трехвалковых намо-
точных станках.
Основными конструкционными элементами станка (рис. 188)
являются два горизонтальных вала, обогреваемые паром или эле-
ктричеством (температура 150—160°), вращающиеся в одну и ту же
сторону. На эти валы кладут цилиндрическую оправку, диаметр
которой соответствует внутреннему диаметру изготовляемой трубы
1
Рис. 188. Схема устройства на-
моточ.юго ста.тка.
1—прижимной вал; 2— изготовляе-
мая т|.уба; 3 —съемный вал (оправка);
4, 5 — нагревательные валы; 6 — тор-
мозное устр<йство; 7—рулон лакиро-
ванной бумаги.
490
Гл. XI. Фенопласты слоистые
(цилиндра). Третий вал, прижимной, опускают на оправку в про-
цессе намотки трубы.
Рулон односторонне лакированной бумаги укрепляют на стой-
ках намоточного станка и заправляют таким образом, чтобы бу-
мага, обогнув направляющие и тормозные валики, а также вал
для предварительного подогрева бумаги (на схеме не показано),
соприкасалась бы нелакированной стороной с передним валом 4
и наматывалась бы на оправку 3, которая прижимается верхним
валом 1.
Оправка вращается либо самостоятельно от привода (для из-
делий диаметром до 100—150 мм), или же от привода вращаются
нагревательные валы 4, 5, а оправка получает вращение уже от
них (для изделий диаметром больше 100—150 мм). При принуди-
тельном вращении оправки нагревательные валы вращаются от
трения наматываемого на оправку изделия.
Давление верхнего прижимного вала составляет 10—15 кг на
1 см длины.
Поддерживая температуру нагревательных валов около 150—
160°, достигают плавления смолы и перехода ее в резнтольное и ча-
стично в резитное состояние за время намотки трубы. Плотность
получаемой трубы зависит от содержания смолы, температуры и
скорости вращения валов и давления прижимного вала.
Бумагу наматывают на оправку до тех пор, пока не получится
труба нужной толщины, затем ее обрезают и оправку снимают
со станка.
Оправки, снятые со станка после намотки, направляют для тер-
мической обработки в стационарные камерного типа печи, в кото-
рых с помощью системы паровых радиаторов и нагнетания горя-
чего воздуха поддерживают постоянную температуру в пределах
ПО—130°. Время термической обработки в зависимости от тол-
щины изделий и температуры колеблется от 2 до 24 час. и более.
После термической обработки трубы или цилиндры снимают
с оправок посредством кабестана (волочильного станка). Длина
труб ограничена, естественно, длиной валов намоточного станка
(обычно 1,5—3 м). Диаметр труб зависит от диаметра применяе-
мых оправок. Для нужд электротехнической промышленности изго-
товляют трубы с внутренним диаметром от 1 до 100 мм и более
и цилиндры диаметром до 2000—3000 мм.
Пропитка бумаги расплавленной смолой
Для производства фенобумослоя широко применяют пропитку
бумаги расплавленной смолой. Практическая целесообразность
этого метода заключается в экономии растворителя (спирта),
а также в получении материала, почти не содержащего лету-
чих веществ.
Пропитку производят на станке (рис. 189), состоящем из вала
для рулона с бумагой, ряда натяжных валиков и обогревательной
Фенобумослой
491
плиты, над которой проходит пропитываемая бумага. На чугунной
станине расположены нагревательные валы, а над ними — прижим-
ной вал (соединенный системой рычагов со стойкой), который может
быть поднят или опущен. Для создания необходимого давления и
для его регулировки служит передвижной груз, закрепленный на
плече коромысла, перемеще-
нием которого изменяется да-
вление прижимного вала. Бума-
га с рулона проходит натяжные
валы и передвигается над обо-
греваемой плитой, а из дозато-
ра, помещенного над плитой, на
нее поступает порошок резоль-
ной смолы, который плавится.
Бумага вместе с расплавленной
смолой направляется на нагре-
вательные валы, где под давле-
нием верхнего прижимного ва-
ла смола пропитывает бумагу,
при этом избыток смолы отжи-
мается, образуя бортик между
прижимным и нагревательными
валами. Количество смолы в
Рис. 189. Схема станка для пропитки бу-
маги расплавленной смолой.
/—рулон с бумагой; 2—порошок смолы; 3 — обо-
гревательная плита; 4—нагревательные валы;
5—прижимной вал; 6—передвижной груз; 7— про-
питанная бумага.
бумаге регулируется исключи- •
тельно силой отжима верхнего вала, т. е. переставлением груза на
плече коромысла. В дальнейшем пропитанная бумага проходит
через ряд направляющих валов и наматывается на рулон.
Этот метод имеет наряду с преимуществами и существенные не-
достатки. Важнейший из них — неравномерное распределение смолы
по ширине бумаги. К применяемой при работе по этому методу
смоле предъявляются весьма строгие требования в отношении тем-
пературы плавления, хрупкости и скорости отверждения. Плавле-
ние смолы сопровождается выделением вредных летучих веществ
и требует усиленной местной вентиляции.
Рольный метод производства фенобумослоя
Описанные методы получения резитовых бумажных пластиков-
основаны на двух самостоятельных процессах: производства бу-
маги и ее пропитки. Характер и качество пропитки в этих случаях
зависят как от свойств самой бумаги (ее прочности и впитывающей
способности), так и от условий пропитки. Однако во всех случаях
пропитки готовых листов на поверхности удерживается более тол-
стый слой смолы, чем внутри волокон. Задача равномерного
распределения смолы в бумаге решается применением рольного
метода, который объединяет процесс производства бумаги с процес-
сом пропитки, путем введения смолы в состав бумажной массы..
Гл. XI. Фенопласты слоистые
При этом элементарные бумажные волокна пропитываются смолой
еще до свойлочивания их в листы.
Рольный метод производства слоистых пластмасс имеет также
ряд экономических преимуществ и дает возможность получения но-
вых видов слоистых пластиков. Он был разработан впервые в СССР
(Э. И. Баргом, А. К. Садиковой).
Производство фенобумослоя по рольному методу складывается
из следующих операций: 1) получения водных дисперсий резоль-
ных смол (эмульсий, водных щелочных растворов, суспензий);
2) размола волокна в ролле; 3) смешения в ролле дисперсий резоль-
ных смол с разработанным (размолотым) волокном и осаждения
смолы на волокне; 4) получения бумаги или картона из резольно-
целлюлозной массы на бумажных и папочных машинах; 5) сушки
листов; 6) прессования фенобумослоя.
Разработка бумажной массы в ролле
Рис. 190. Массный ролл.
чгво
-Ъ= ISO об/мин
Технология производства слоистых пластмасс рольным методом в значи-
тельной мере аналогична технологии производства бумаги и картона.
Процесс производства бума-
ги начинается с размола целлю-
лозного волокна, который про-
изводят в специальных аппара-
тах — массных роллах.
Ролл состоит из бетонной,
чугунной или деревянной ван-
ны овальной формы (рис. 190).
Основной частью аппарата
является размалывающее при-
способление, которое состоит из
ножевого барабана Б, вращаю-
щегося внутри ванны А, и из
расположенной под барабаном
гребенки Г, составленной из
стальных ножей и укреплен-
ной в чугунном ящике. На по-
верхности барабана по его об-
разующим также установлен
ряд стальных планок-ножей, ко-
торые имеют толщину 8—10 мм,
ширину 130—150 мм и длину
900—1050 леи. Они должны вы-
ступать из укрепляющих их де-
ревянных планок приблизи-
тельно на 50 мм. Ножи, равно-
мерно распределенные, отстоят
друг от друга на 50—60 мм.
Гребенка состоит из 10—12 стальных планок-ножей, укрепленных в чугунном
ящике при помощи деревянных планок-клиньев, причем гораздо чаще, чем ножи
у барабана (на расстоянии всего 5—8 мм). Ножи гребенки устанавливают под
углом в 1,5° к ножам барабана, что улучшает распушку волокна.
Фенобумослой
493-
Бэл баоабанз установлен на подвижных подшипниках, которые могут Сьыь
подняты или опущены при помощи червячной передачи и, следовательно, зазор
между ножами барабана и гребенки может изменяться. Дно ванны ролла, где
вращается барабан, круто поднимается, образуя так называемую горку Д с по-
верхностью, концентричной поверхности барабана. Расстояние от ножей до горки
не должно превышать 20 мм.
Размол целлюлозы в ролле производят в водной среде. В ролл заливают
воду, а затем в него вносят по частям целлюлозное волокно (всего 4—7%). При
вращении барабана волокно уносится жидкостью и, попадая между ножами ба-
рабана и гребенки, размалывается. В зависимости от степени присадки разма-
лывающего барабана (расстояния ножей барабана от гребенки) получают раз-
личный характер помола — жирный или тощий (садкий). В первом случае ножи
барабана не столько рубят волокно, сколько расщепляют его и разминают. Раз-
рыв происходит по длине волокна, причем волокна расщепляются на весьма
тонкие элементарные волоконца толщиной в 10—5 ц, при одновременном значи-
тельном ослизнении и набухании волокна. Во втором — преобладает в основном
рубка волокна в поперечном направлении. Отдельные пучки волокна становятся
при этом более короткими, но расщепление по ширине волокна происходит лишь
в малой степени. На практике не удается вести процесс таким образом, чтобы
достигался только один вид размола волокна; обычно довольствуются преобла-
данием одного или другого вида (жирного или тощего). Время размола может
быть различным, в зависимости от желаемой степени помола (от 3 до 8 час.).
Степень помола обычно определяют аппаратом Шоппер — Риглера и выра-
жают в градусах Шоппер — Риглера (Ш. — Р.). Чем выше число градусов по-
Ш. — Р., тем больше степень размола (для целлюлозных волокон обычно в пре-
делах 40—65 градусов).
В процессе размола целлюлозное волокно сильно набухает и все содержи-
мое в ролле представляет собой полужидкую набухшую массу. Целлюлоза из
твердого состояния переходит в полупластичное, частично студнеобразное. Раз-
мол сообщает целлюлозе коллоидные свойства набухшего геля; одновременно
значительно увеличивается реагирующая поверхность волокна.
Когда волокно размолото до установленной степени, в ролл вводят водные
дисперсии смол и осаждают на волокне. Смола удерживается на поверхности
волокна благодаря силам сцепления и взаимодействия двух набухших гелей —
волокна и смоляной дисперсии.
Методы осмоления
Получение бумажно-смоляных масс или осмоление волокна может быть
осуществлено двумя основными методами; 1) смешением размолотой целлюлозы
с водными эмульсиями резольных смол или со щелочными растворами смол;
последние затем тщательно перемешивают в ролле с разработанным волокном
и осаждают путем разрушения эмульсии кислотами (или сернокислым алюми-
нием); 2) смешением размолотой целлюлозы с водными суспензиями твердых
резольных смол; последние после перемешивания в ролле с разработанным во-
локном также осаждают внесением соответствующих электролитов. Ниже мы
рассмотрим второй, так называемый суспензионный (или рольно-суспензионный)
метод, который имеет то важное преимущество перед методом, основанным на
применении эмульсий и растворов, что в ием исходным сырьем являются ста-
бильные твердые смолы.
Процесс получения фенобумослоя по рольно-суспензионному методу
Первая стадия в производстве фенобумослоя по рольно-суспензионному ме-
тоду — получение водной суспензии резольной смолы.
Суспензии представляют собою неравновесную систему диспергированных
твердых частиц в жидкости. Стойкость суспензии определяется рядом факторов.
.494
Гл. XL Фенопласты слоистые
а именно: степенью полярности обеих фаз, характером и концентрацией эмуль-
гатора, степенью дисперсности и др. В суспензии можно различать два вида
входящей в ее состав воды: свободную и связанную (или адсорбционную).
В технических суспензиях смол адсорбционная вода, которая практически ие
выделяется при хранении (нли, точнее, выделяется очень медленно), может соста-
влять до 100% (к весу смолы). Количество адсорбционной воды пропорцио-
нально степени диспергирования смолы.
Стойкость суспензии в технике определяется следующими факторами: 1) сте-
пенью гидрофобности смолы (чем более гидрофобна смола, тем устойчивее
суспензия); 2) характеров и концентрацией эмульгатора; 3) pH среды и 4) сте-
пенью дисперсности. Стойкость суспензии значительно увеличивается, если часть
фенола, входящего в состав смолы, заменить анилином; это объясняется умень-
шением полярности (и гидрофильности) смолы. Поэтому рациональнее всего
применять твердые анилимо-фенольные резолы.
Смола должна быть хрупкой при 30—35° и не слипаться при сухом размоле
в шаровой мельнице.
В качестве эмульгаторов применяют либо мыла, либо минеральные дисперс-
ные вещества (каолин, кизельгур), либо оба вида эмульгаторов одновременно.
Мыла добавляют в количестве до 1%, минеральные вещества — до 10% и более
(последние служат одновременно и наполнителями).
Суспензия может быть получена путем размола твердой резольной смолы
с водой в шаровых мельницах. Смолу вносят в шаровую мельницу кусками раз-
мером не более 3—5 см. Одновременно в шаровую мельницу загружают ми-
неральный эмульгатор (наполнитель) — каолии или кизельгур. Вначале произво-
дят сухой помол смеси без воды; по истечении примерно двух часов в шаровую
мельницу пускают воду в таком количестве, чтобы получить суспензию, содержа-
щую 50—55% воды. Вместе с водой вводят органический эмульгатор (мыло)
и размол продолжают еще 4—6 час., до получения суспензии, проходящей
на 100% через сито № 80.
Готовую суспензию через сливное отверстие шаровой мельницы (с сеткой,
предохраняющей от выпадания шаров) спускают по жолобу в промежуточный
бак, снабженный мешалкой, из которого через весовой мерник насосом подают
в ролл, где суспензию в течение 15—25 мин. перемешивают с разработанным во-
локном до образования равномерной по окраске массы; после этого для разру-
шения органического эмульгатора и осаждения суспензии на волокне вводят
сернокислый алюминий; pH среды при этом 6,5—5,5.
После тщательного перемешивания массу разбавляют водой (в 2—3 раза)
и спускают в мешалку.
Мешалка состоит нз железного или бетонного резервуара, внутри которого
проходит вал; на валу закреплены лопасти, вращением которых достигается не-
прерывное перемешивание. В передней части резерзуара на том же валу закре-
плено черпальное колесо, несущее по окружности ряд ковшей, которые захваты-
вают массу, поднимают ее и выливают в жолоб, по которому масса через песоч-
ницу и узлоловитель попадает в корыто сетчатого цилиндра папп-машины. Как
в жолобе, так и в сетчатом цилиндре массу разбавляют водой до концентра-
ции 1—0,5%.
На папп-машине происходит процесс листообразования — обезвоживание,
.свойлочивание, ориентация пропитанного смолой волокна (образование эле-
ментарного слоя) и навивка слоев на форматном вале с образованием
картона.
Папп-машина — простая круглосеточная машина для производства ручного
картона — состоит из двух основных частей: сгустительной н прессовой. Через
обе части, соединяя их, движется бесконечное съемное сукно. В железобетонной
ванне сгустительной части папп-машины расположен сетчатый цилиндр, гауч-вал
с нажимным приспособлением и трубка для промывки сетчатого цилиндра. Сет-
чатый цилиндр имеет стальную ось, на которой укреплен каркас, изготовлен-
ный из продольных латунных прутьев, проходящих через опорные кольца; на
каркас натянута редкая латунная сетка, сверху покрытая второй частой бронзовой
Фенобумослой
495
сеткой № 40—60. Прессовая часть состоит из: форматного барабана, опорного
вала, направляющих валиков, трепалки, трубок для промывки сукна и валков
для отжима влаги из сукна. Обе эти части приводятся в движение
сукном, транспортирующим отфильтрованный слой массы (элементарный слой)
от сетчатого цилиндра к форматному барабану; движение сукна происходит от
вращения опорного вала.
Схема движения бесконечного сукна показана на рис. 191. При вращении
сетчатого цилиндра, вследствие разницы в уровнях воды внутри и вне цилиндра.
Рис. 191. Схема движения сукна на папп-машине.
/—сетчатый цилиндр; 2~ванна; 3 — гауч-вал; 4—натяжные
валики; 5—форматный барабан; 6 — опорный вал; 7—вакуум-
ная коробка.
вода процеживается сквозь сетку в его внутреннюю часть, причем иа сетке
остается слой массы (элементарный слой). Толщина элементарного слоя тем
больше, чем выше концентрация массы, чем мельче сетка (чем выше ее но-
мер) и чем больше скорость вращения сетчатого цилиндра. Обычно толщина
элементарного слоя может быть получена в пределах от 0,1 до 0,3 мм (из рас-
счета на сухое).
С вращающегося сетчатого цилиндра слой массы принимается съемным
бесконечным сукном, прижимаемым к поверхности сетчатого цилиндра гауч-
валом; при этом слой массы в значительной степени отжимается и обезвожи-
вается. Дальше сукно через вакуумную коробку и по ряду направляющих ва-
лов проходит между опорным валом и форматным барабаном. При этом слой
массы с сукна переходит на форматный барабан и непрерывно наматывается
на него до получения слоя заданной толщины. По мере прохождения через
гауч-вал, вакуумную коробку и опорный вал вода отжимается и масса обезво-
живается до концентрации 45—55%. По образовании на форматном барабане
слоя массы заданной толщины последний разрезают ножом по вертикальным и
горизонтальным желобкам форматного барабана; при этом одновременно сни-
маются четыре листа. Полученные листы подвергают дальнейшему обезвожива-
нию путем отжима на прессах; затем они поступают в канальную сушилку, где
высушиваются при ° 40—60° до остаточной влажности 3—5%. Более глубокая
сушка до остаточной влажности не более 0,5—1 % происходит в вакуум-сушилках
непосредственно перед прессованием.
Полученные листы характеризуются двумя важнейшими показателями: 1) со-
держанием смолы в массе и 2) толщиной элементарного слоя на сетчатом
цилиндре.
Соотношение между смолой и целлюлозой в получаемых листах всегда
меньше, чем в ролле. Это объясняется тем, что в процессе листообразоваиия
(образования элементарного слоя) дисперсная смола в большем количестве про-
ходит сквозь сетку и теряется в промывных водах, чем целлюлозное волокно.
Эти потери как волокна, так и смолы, могут в значительной мере быть снижены
496
Гл. XI. Фенопласты слоистые
применением соответствующих отстойников и замкнутого цикла оборотных вод.
для разбавления массы в ролле, мешалке и в корыте сетчатого цилиндра
Чем больше концентрация смолы в массе, чем тоньше элементарный слой,
тем значительнее относительные потери смолы по сравнению с целлюлозой. По-
этому рольный метод получения слоистых пластиков может быть рационально’
применен лишь при условии использования оборотных вод.
Толщина элементарного слоя больше, чем какой-либо другой фактор, опре-
деляет основные показатели процесса и свойства фенобумослоя. Чем тоньше эле-
ментарный слон, тем прочнее материал, однако тем больше и потери смолы
в промывных водах.
Бумажно-резитовые пластики, изготовленные по рольно-суспензионному ме-
тоду, более монолитны и однородны, чем соответствующие ма1ериалы, изгото-
вленные по лаковому методу; они более водостойки и обладают лучшими физико-
механическими свойствами.
Преимуществом рольного метода является то, что при его применении пол-
ностью отпадает весь процесс пропитки бумаги и не требуется расхода спирта.
По рольному методу можно в принципе получать бумаги и, соответственно,
прессматериалы, содержащие любые количества смолы (от нескольких до-
50—60%). При этом смола равномерно распределяется по всей массе и содер-
жание ее в бумаге можно легко регулировать. Возможность производства роль-
ным методом слоисто-бумажных пластиков с малым содержанием смолы
(~10%) особенно большое значение получила в последние годы в связи с ши-
роким применением фенобумослоя в качестве доступного строительного мате-
риала. Наконец, рольный метод дает возможность получать как бумагу, так и
картой различной толщины, что облегчает технику прессования и в ряде случаев
устраняет необходимость предварительного таблетирования материала.
Рольный метод имеет большое значение для получения слоистых масс на
основе не только целлюлозы, но и асбеста, и не только с резольной смолой,
но и с любыми вяжущими веществами (искусственные смолы, битумы, эфиры,
целлюлозы) или с любой комбинацией вяжущих веществ. Благодаря рольному
методу удалось получить ряд новых материалов с высокими техническими пока-
зателями (слоистые асборезиты, асбопекослой и др.).
Свойства фенобумослоя
Свойства фенобумослоя определяются: 1) характером волокна
(целлюлозы), его происхождением, прочностью, длиной и техниче-
ским процессом его получения; 2) характером и степенью разра-
ботки волокна в ролле (жирный или тощий помол), толщиной листа
(элементарного слоя), прочностью бумаги в продольном и попереч-
ном направлениях; 3) химическим характером резольной смолы;
4) соотношением количества смолы и волокна; 5) структурой мате-
риала (из слоев пропитанной или лакированной бумаги); 6) режи-
мом прессования и термообработки после прессования; 7) остаточ-
ной влажностью бумаги перед прессованием.
Механическая прочность фенобумослоя тем выше, чем больше-
удельная прочность листов бумаги, чем тоньше листы (элементар-
ные слои) и, до известного предела, чем меньше содержание смолы
в массе. Однако прочность на раскалывание в широких пределах,
содержания смолы (до 45—55%) тем больше, чем меньше напол-
нителя. Водостойкость находится в экспоненциальной зависимости
от содержания смолы.
Диэлектрические свойства фенобумослоя тем выше, чем менее
полярна применяемая смола и чем меньше кислотность бумаг.
Фенобумослой 497
Значительно сложнее зависимость диэлектрических-свойств фено-
бумослоя от содержания в нем целлюлозы, т. е. от соотношения
смолы и наполнителя. Диэлектрические свойства абсолютно сухой
целлюлозы или целлюлозы с содержанием ~1% влаги значительно
выше, чем резита, однако они резко снижаются при достижении
целлюлозой равновесной влажности (~4—6%). Ввиду этого на-
значение смоляного слоя — защищать целлюлозные волокна от про-
никновения влаги. Однако это не может быть полностью достигнуто
даже при избытке смолы, хотя все же смоляные пленки значительно
снижают скорость диффузии воды и, следовательно, сильно удли-
няют время достижения целлюлозой равновесной влажности.
Поэтому с повышением содержания смолы диэлектрические
свойства увеличиваются в связи с уменьшением скорости диффузии
влаги, однако по достижении концентрации 45—50% дальнейшее
увеличение содержания смолы уже практически не влияет на ско-
рость диффузии воды, но приводит к снижению диэлектрических
свойств из-за более низких, по сравнению с абсолютно сухой целлю-
лозой, диэлектрических свойств резита.
Таким образом, зависимость диэлектрических свойств фенобумо-
слоя от содержания смолы выражается наличием максимума в пре-
делах 45—55% концентрации смолы. Вместе с тем диэлектрические
свойства фенобумослоя, — вне зависимости от соотношения компо-
нентов, должны с большей или меньшей скоростью снижаться
в процессе эксплуатации. Это снижение обусловливается постепен-
ным увеличением влагосодержания в материале.
Улучшения диэлектрических свойств можно достигнуть вакуум-
ной сушкой пропитанных листов перед прессованием.
Термообработка фенобумослоя после прессования также при-
водит к улучшению диэлектрических свойств в связи с удалением
конденсационной влаги. Для уменьшения скорости диффузии воды
и сохранения достигнутых в процессе прессования и термообработки
высоких диэлектрических свойств поверхность изделий из фено-
бумослоя покрывают резольными лаками (в частности с торцовой
стороны) с последующей термообработкой. Для стабильного же
улучшения диэлектрических свойств фенобумослоя следует приме-
нять менее полярные смолы (анилино-феноло-формальдегидные).
В общем бумажно-резитовый пластик с высокими диэлектриче-
скими свойствами получить сравнительно легко, но весьма трудно
сохранить эти свойства в процессе эксплуатации, особенно во влаж-
ной атмосфере. Поэтому и наблюдается своеобразное «старение»
фенобумослоя, связанное со снижением главным образом его диэле-
ктрических свойств. Зависимость механико-деформационных свойств
фенобумослоя от различных факторов (температуры, времени дей-
ствия напряжения и др.) имеет тот же характер, что и у фенотексто-
слоя.
Фенобумослой выпускают в качестве электроизоляционного и
конструкционного материала для высоковольтной электротехники и
32 Зак. 1269. Э. И. Барг
498
Гл, XI. Фенопласты слоистые
в качестве конструкционного материала для других отраслей про-
мышленности. В первом случае к нему предъявляют требования
высокой водостойкости и прочности на электропробой, что дости-
гают большим содержанием смолы (40—50%), во втором случае
основным требованием является высокая механическая прочность,
которая обеспечивается меньшим содержанием смолы (35—40%).
В СССР фенобумослоя для электротехнических целей выпускают
различных марок под названием гетинакс.
Ниже приведены пределы показателей некоторых свойств фено-
бумослоя:
Удельный вес, г/сж3............................ 1,3—1,4
Предел прочности:
при растяжении, кг>см-........................... 800—1400
при изгибе, hz'icaT............................. 1000—1600
при раскалывании, кг! см~ .................... 150—250
Модуль упругости при растяжении, кг/см? .... 70000—100000
Удельная ударная вязкость, кг-смфм*............ 15—25
Твердость по Бринелю, кг) мм*.................. 25—30
Водостойкость (привес в воде за 24 часа бруска
120 X 15 X Ю мм), %............................. 1,0—2,0
Теплостойкость по Мартенсу, °C................. 150—160
Удельное поверхностное электрическое сопротивле-
ние, 2.......................................... 1010—Ю12
Удельное объемное электрическое сопротивление,
2-сж............................................ 1010—1012
Тангенс угла диэлектрических потерь при 50 гц . . 0,1—0,05
Диэлектрическая проницаемость, 50 гц........... 5—8
Средняя пробивная напряженность перпендику-
лярно слоям (d = 6—10 мм), кв!мм................ 15—25
По сравнению с фенотекстослоем фенобумослой имеет более вы-
сокую теплостойкость (по Мартенсу), лучшие диэлектрические свой-
ства и, как правило, большую прочность к статическим напряже-
ниям (разрыв, изгиб); однако он значительно уступает фенотексто-
слою по прочности к динамическим напряжениям (удар), по проч-
ности на раскалывание и по водостойкости.
Применение фенобумослоя
Слоистые бумажные фенопласты являются важнейшим электроизоляционным
и конструкционным материалом электротехники. Фенобумослой наиболее широко
применяют в промышленности, выпускающей электромашины и электроаппараты
сильных токов, в производстве высоковольтных трансформаторов, конденсато-
ров для рентгеновских установок, бушингов, гасильных камер для масляных вы-
ключателей, изоляторов, электрощитов и т. д.
Применяют фенобумослой и в различных отраслях общего приборо- и ма-
шиностроения для изготовления шестеренок и роликов в качестве прокладоч-
ного и установочного материала, для изготовления цилиндров, корпусов и кры-
шек аппаратов, а также в мебельном производстве.
Бумажные фенопласты в виде пластин и плит используют для внутренней
облицовки зданий, при этом на поверхности пластин запрессовывают тонкую
пленку из бумаги, пропитанной бесцветными и светостойкими смолами (напри-
мер мочевино-формальдегидными).
Слоистые асборезиты
499
Специальные сорта слоисто-бумажных фенопластов с малым содержанием
смолы (~10%), полученные путем ввода дисперсий смол в состав бумажной
массы, применяют для производства деталей сборных домов, перегородок, пли-
ток для пола и т. д.
Слоистые асборезиты
(Феноасбослой,. феноасботекстослой, асботекстолит, асболит)
Феноасбослой представляет собою слоистый пластик на основе
асбестовой бумаги и резольной смолы.
Из-за малой прочности асбестовой бумаги (картона) и пло-
хой ее впитывающей способности, применение обычных «мокрых»
методов пропитки (лаковый, эмульсионный) для получения фено-
асбослоя встречает значительные трудности. По тем же причинам
затруднительна и пропитка расплавленной смолой. Вследствие этого
слоистые асборезиты на основе пропитанной асбестовой бумаги
(картона) изготовляют редко. Большее, но все же ограниченное
применение получили слоистые резиты на основе асбестовой ткани,
которая ввиду большей прочности позволяет производить про-
питку обычными «мокрыми» методами.
Пропитку асбестовой ткани производят на обычных вертикаль-
ных или горизонтальных пропиточных машинах спиртовыми рас-
творами или водными дисперсиями резольной смолы. Пропитка
асбестовой ткани происходит труднее, чем хлопчатобумажной.
Это объясняется отсутствием внутренних капилляров в асбестовом
волокне. I
Асбестовая ткань обычно содержит некоторое количество хлоп-
чатобумажных волокон, что повышает ее прочность, улучшает усло-
вия пропитки, но ухудшает термостойкость. Поэтому асбестовая
ткань не должна содержать более 10—15% хлопковых волокон.
Обычно ее применяют толщиной до 1,6 мм, весом до 1 кг/л/2; проч-
ность ткани по основе 60—80, по утку 25—40 кг/см2.
Слоистые асборезиты на основе ткани имеют более высокую ди-
намическую прочность, по сравнению с бумажными, однако из-за
дефицитности и высокой стоимости текстильных сортов асбеста
они получили лишь весьма ограниченное применение. Их изго-
товляют для деталей, к которым наряду с высокой термостойкостью
и теплостойкостью или же с высокими фрикционными свойствами
предъявляют требования высокой прочности к динамическим напря-
жениям.
Вместе с тем, обычные, нетекстильные и не высшие сорта хризо-
тилового асбеста (сорта III, IV, V, см. стр. 451) являются отно-
сительно доступным материалом и могут быть широко использованы
для изготовления слоистых бумажных асборезитов, если применять
достаточно эффективные методы пропитки или, точнее, методы вве-
дения смолы в состав асбестовой бумаги. Хорошие результаты,
39*
500
Гл. XI. Фенопласты слоистые
в частности, дает рольно-суспензионный метод получения слоисто-
бумажных пластиков.
Рольно-суспензионный метод получения феноасбослоя
Асбест легко набухает в воде и может быть расщеплен на тон-
чайшие волокна. В набухшем расщепленном (распушенном) со-
стоянии он обладает высокими адсорбционными свойствами и спо-
собен удерживать смолу в дисперсном состоянии. Чем выше степень
распушки асбеста, тем тоньше его волокна и тем выше их удельная
прочность. Эти свойства асбеста обусловливают эффективность
применения рольно-суспензионного метода для получения слоистых
асборезитов.
В качестве волокнистого наполнителя применяют хризотиловый
асбест жесткой текстуры — III, IV и V сортов (стр. 451). Чем
выше сортность асбеста, чем длин-
нее и чище его волокно, тем,
естественно, будет прочнее пла-
стик. В качестве связующего при-
меняют твердые, хрупкие резолы.
Общий принцип производства
асбобумажных фенопластов по
рольному методу во многом ана-
логичен производству целлюлоз-
Рис. 192. Схема производства феноасбослоя рольно-суспензионным методом.
1—бегуны; 2—ролл; 3— шаровая мельница; 4—воронка; 5—бак; 6—мешалка; 7—папп-ма-
шина; 8—пресс; 9—туннельная сушилка; 10—вакуум-сушилка; 11—каландр.
которое отличие имеется в процессе распушки асбеста, который
складывается из двух операций: 1) распушки до ролла и 2) рас-
пушки в ролле.
Распушка асбеста до ролла дает возможность освободить его от
имеющейся в нем породы (не волокна), а также в результате раз-
давливания и растирания обеспечивает продольное расщепление во-
локон. Эту операцию обычно производят на бегунах. В чашу бегу-
нов загружают 15—25 кг асбеста, увлажняют его равным количе-
ством воды и обрабатывают в течение 5—15 мин.
После обработки на бегунах производят вторую операцию —
распушку асбеста с водой в ролле. Концентрация асбеста может
Слоистые асборезиты
501
варьировать в пределах от 4 до 10% в зависимости от текстуры и
сортности асбеста. Она должна быть тем меньше, чем жестче асбест
и чем выше его сортность (стр. 451). Распушку производят до до-
стижения 88—90° по Шопер — Риглеру, причем размол должен быть
жирным. Время достижения такого размола от 30 до 90 мин. в за-
висимости от сортности асбеста: чем выше сорт, тем длительнее
время размола.
Схема производства феноасбослоя представлена на рис. 192.
Асбест, предварительно распушенный на бегунах 1, вводят в ролл 2. По
•окончании распушки асбеста в ролле к нему добавляют водную суспензию ре-
зольной смолы, полученную в шаровой мельнице 3. Из шаровой мельницы сус-
пензию через воронку 4 спускают в бак 5, из которого ее можно перекачивать
и ролл. После смешения массу из ролла спускают в метальный чан 6, в кото-
ром разбавляют водой до 2—Зп/а концентрации. Из чана смесь с помощью чер-
пального колеса направляется на папп-машину 7, на которой происходит обра-
зование листов (картона). Листы, снятые с папп-машины, отжимают на прессе8,
сушат сначала в туннельной сушилке 9 до 2—3% остаточной влажности н не-
посредственно перед прессованием дополнительно в вакуум-сушилках 10. Если
листы коробятся, рекомендуется перед прессованием пропустить их для выпря-
мления и уплотнения через каландр 11; после этого они поступают на гидравли-
ческие прессы (в схеме не показаны) для горячего прессования.
Свойства феноасбослоя
Механическая прочность феноасбослоя находится в линейной
обратной зависимости от содержания в нем смолы (рис. 193), а
также от текстуры и сортности асбеста:
чем длиннее волокно и чем оно менее
повреждено, тем выше прочность пла-
стика. Как правило, из жестких асбестов
получают более прочные материалы, чем
из мягких. Содержание смолы в фено-
асбослое обычно находится в пределах от
30 до 40%.
Феноасбослой может быть получен с
весьма высокой статической прочностью,
превосходящей таковую фенобумослоя
и фенотекстослоя. Он характеризуется
значительно более высокими теплостой-
костью и термостойкостью. Высокий
коэффициент трения (от 0,250 до 0,350),
малая истираемость и высокая тепло- и
Рис. 193. Зависимость проч-
ности на изгиб феноасбо-
слоя от содержания смолы.
термостойкость определяют эффектив-
ность применения феноасбослоя в каче-
стве фрикционного материала (для про-
изводства тормозных колодок).
Диэлектрические свойства феноасбослоя, в частности его эле-
ктрическая прочность, значительно (в 5—40 раз) ниже, чем у слои-
стых фенопластов на основе целлюлозного волокна.
502
Гл. XT. Фенопласты слоистые
Удельный вес асбестовых пластиков значительно выше, чем пла-
стиков на основе целлюлозы (1,8 вместо 1,4).
Свойства феноасбослоя, полученного рольно-суспензионным ме-
тодом, приведены в табл. 58. Для сравнения в ней даны также и
свойства феноасботекстослоя — слоистого пластика на основе асбе-
стовой ткани. Последний, как видно из таблицы, обладает более
высокой динамической, но меньшей статической прочностью и водо-
стойкостью, чем бумажные асборезиты, получаемые рольно-суспен-
зионным методом.
ТАБЛИЦА 58
Свойства феноасбослоя и феноасботекстослоя (асботекстолнта)
Показатели Феноасбослой Феноасботексто- слой
Уд. вес, г/см3 Теплостойкость по Мартенсу, °C 1,8 1,7
180—250 150—250
Жаростойкость по Шрамму, класс 3 —
Водостойкость (привес в воде за 24 часа бруска 120 X 15 X Ю мм), % 0,05—0,01 0,4—0,6
Предел прочности на изгиб, кгГм'1 1600—2500 800—1500
Предел прочности на сжатие, к?!см~ .... 2500—3000 —
Твердость по Бринелю, кг/мм^ 60—65 —
Уд. ударная вязкость, кг • см/см3 20—30 25—40
Средняя пробивная напряженность, квмм . 2—5 0,6—1
i
Основное применение асборезиты получили в качестве фрикцион-
ных материалов для производства тормозных лент, колодок и
дисков сцепления, а также в различных отраслях машинострое-
ния для производства деталей, к которым наряду с высокой меха-
нической прочностью предъявляют требования высокой тепло-
и термостойкости.
Древесно-слоистые пластики
(Фенодревослой ДСП, лигнофоль, дельта-древесина, резитовая
фанера, фанерит и др.)
Фенодревослой представляет собою слоистый резитовый мате-
риал на основе прессованных слоев древесины (шпона), пропитан-
ных резольной смолой.
В зависимости от содержания смолы в материале и применяе-
мого при прессовании удельного давления получаются материалы
различной плотности; при этом различают два типа древесно-
слоистых пластиков: 1) собственно фенодревослой, представляю-
щий собою монолитный слоистый пластик, по свойствам мало на-
поминающий дерево, с уд. весом выше 1,25 (~1,4) и 2) материалы
типа фанеры (резитовая фанера), в которых альбуминовый или
казеиновый клей заменен пленками резольной смолы со сравни-
Древесно-слоистые пластики
503
тельно небольшим содержанием смолы и с уд. весом меньше 1,25
(обычно в пределах 0,8—1,1). Кроме этих основных типов древес-
ных пластиков известны материалы, занимающие промежуточное
положение; вообще же возможны разнообразные переходные мате-
риалы от резитовой фанеры с удельным весом около 1,0 до наи-
более плотного материала с удельным весом до 1,4.
Собственно фенодревослой
Древесно-слоистые пластики с плотностью свыше 1,25 получают
путем пропитки березового шпона спиртовыми растворами или вод-
ными эмульсиями резольных смол, а также водными растворами
смол и фенолоспиртами.
Березовый шпон должен иметь толщину в пределах 0,24—0,6 мм
и соответствовать по своим свойствам ГОСТ 99—41; он дол-
жен, в частности, содержать возможно меньшее число различных
дефектов, как то: прорости, свилеватости, косослоя, завит-
ков и т. д.
Применяемые для пропитки растворы резольных смол или
эмульсии должны иметь невысокую вязкость (~20 сл); концентра-
ция смолы в них обычно 35—45%.
Пропитку шпона производят в глубоких прямоугольных ван-
нах, снабженных паровой рубашкой или паровыми змеевиками.
Температуру лака в ванне обычно поддерживают в пределах
20—25°; если же применяют вязкие эмульсии, температура может
быть поднята до 40—50°.
Для пропитки шпон укладывают в пакеты толщиной 5 мм, ко-
торые ставят вертикально в железную корзину с сетчатым дном и
перекладывают металлическими сетками. Корзину с пакетами по-
гружают на 40—60 мин. в ванну, после чего вынимают и поддер-
живают над ванной в течение 20—30 мин. для стекания лака.
После этого листы вынимают из корзины и подвергают сушке.
Однако пропиткой шпона в ваннах не всегда достигают полноты и
однородности. Поэтому применяют также пропитку в герметичных
котлах, наполненных лаком, попеременно создавая в них вакуум и
давление.
Обработку шпона производят также жидкими резольными смо-
лами, получаемыми с едким натром в качестве катализатора,
а также феноло-спиртами. Содержание смолы в шпоне регули-
руют, подбирая концентрации и вязкость растворов смолы.
Сушку производят либо в полочных вакуум-сушилках камер-
ного типа, либо в сушилках непрерывного действия с принудитель-
ной циркуляцией горячего воздуха.
В полочных сушилках пропитанные листы укладывают в стопки
на полки, причем между листами кладут деревянные прокладки
толщиной 1,5—2 см, и сушат при 60—70° и остаточном давлении
150—200 мм от 30 до 90 мин.
504
Гл. XI. Фенопласты слоистые
В сушилках непрерывного действия пропитанные листы, перело-
женные деревянными прокладками, укладывают на вагонетки, на
которых они поступают в шахтную сушилку. Последняя имеет
обычно три зоны обогрева (70, 90 и 120°) и работает, как правило,
по принципу противотока: горячий воздух нагнетается в третью
зону и выходит у первой зоны. Вагонетки последовательно прохо-
дят все зоны обогрева. Сушку продолжают 40—60 мин. до оста-
точной влажности материала ~5%.
Высушенные листы прессуют в пластины или плиты на много-
этажных прессах. В современном производстве фенодревослой
пользуются мощными прессами (10 000 т и более), позволяющими
получать плиты длиной в 5—10 м и шириной в 1—1,5 м. Для обеспе-
чения равномерного давления на всей прессуемой поверхности ра-
ционально применять многоплунжерные прессы. Прессование ведут
при 150—160°.
Удельное давление при прессовании применяют в зависимости
от содержания смолы в материале, его толщины и назначения.
Материалы с содержанием смолы выше 40% прессуют при
60—80 кг/см2; при 20—30% прессуют при 100—150 кг/см2. Плиты
прессуют при более высоком давлении, чем пластины. С увеличением
давления при прессовании увеличивается удельный вес, прочность
на раскалывание, предел прочности на растяжение и водостойкость.
Улучшение этих показателей происходит лишь до достижения опре-
деленного предела давления, после чего они уже мало зависят от
давления. Этот предел тем ниже, чем больше смолы содержится в
прессуемом материале. Давление повышают ступенчато: сначала
дают 20—25 кг/см2, а через 15—20 мин. полное.
Прессование ведут из расчета выдержки при нагреве 3—4 мни.
на 1 мм толщины пакета между плитами, считая с момента дости-
жения заданной температуры в середине пакета.
По окончании выдержки пар выключают, пускают воду в плиты
пресса и разгружают последний при охлаждении прессуемых паке-
тов до 40—60°.
Так же, как и для других слоистых фенопластов, применение
высокочастотного обогрева в процессе прессования, а также вы-
сокочастотного подогревания пакетов перед прессованием улучшает
свойства фенодревослоя, а также дает возможность применять более
низкие давления и снизить время прессования за счет сокращения
времени выдержки и охлаждения.
Из древесного шпона, пропитанного жидкими резольными смо-
лами, а также специальными модифицированными, термореактив-
ными смолами, изготовляют изделия больших габаритов формова-
нием при низких давлениях методом резинового мешка (стр. 346).
Этим методом из фенодревослоя изготовляют лодки, катера, части
самолетов и т. д.
Древесно-слоистые пластики
505
Свойства фенодревослоя зависят от качества шпона, его тол-
щины, состава резольной смолы и метода укладки листов шпона
в прессуемом материале.
Чем тоньше применяемый шпон, тем выше показатели прочности
прессматериала, в том числе прочности на раскалывание (при оди-
наковом содержании смолы).
Содержание смолы в фенодревослое может варьировать в срав-
нительно широких пределах от 20 до 45%, — чаще всего оно
25—30%. Чем оно больше, тем выше водостойкость прессматериала,
тем выше его плотность и прочность на раскалывание (до опреде-
ленного предела содержания смолы); однако прочность на разрыв и
па изгиб с увеличением содержания смолы снижается.
Свойства древесно-слоистых пластиков в значительной мере
определяются характером расположения в них волокон древесины.
Если -все листы шпона уложены в одном и том же порядке, то
свойства материала по взаимно перпендикулярным направлениям
(вдоль и поперек волокон) не одинаковы, при этом прочность
вдоль волокон значительно выше, чем поперек. Для получения ма-
териала с меньшей анизотропностью свойств прибегают к чередо-
ванию в укладке слоев при составлении пакетов перед прессова-
нием; в некоторых случаях применяют последовательный сдвиг
слоев на определенный угол. Так каждые 10—20 листов шпона
с одинаковым направлением волокон перемежают с одним пере-
крестным листом под углом 90° либо листы укладывают так, что в
смежных слоях волокна располагаются во взаимно-перпендикуляр-
ном (перекрестном) направлении, или же звездообразно (ра-
диально), т. е. так, что в каждом последующем слое волокна дре-
весины смещены на угол в 30°.
Свойства древесно-слоистых пластиков (ДСП) различных ма-
рок, выпускаемых в СССР, могут быть выражены следующими по-
казателями:
Уд. вес, г/см*................................... 1,3—1,4
Теплостойкость по Мартенсу, °C...................... 150—170
Водостойкость (привес в воде за 24 часа бруска
120Х15ХЮ мм), %.................................. 0,8—5
Предел прочности на разрыв вдоль волокон, кг'с.’Д 1500—4500
Предел прочности на сжатие вдоль волокон, кг/см* 1200—2000
Предел прочности на изгиб, кг)см^................ 1000—4000
Предел прочности на раскалывание по плоскости
склейки, кг/сл?.................................. 130—180
Твердость по Бринелю, кг! мм-.................... 22—35
Модуль упругости, кг'см1........................ 100 000—300000
Уд. ударная вязкость, кг • см/см-................ 30—100
Фенодревослой отличается от других слоистых пластиков (на
основе ткани, бумаги, асбестовой ткани) более высокой прочностью
на растяжение и изгиб, более высокой удельной ударной вяз-
костью, однако он уступает фенотекстослою по прочности на рас-
калывание, на сжатие, а также по водостойкости.
506
Гл. XI. Фенопласты слоистые
Фенодревослой широко применяют в различных областях машиностроения и
строительной техники как взамен цветных и черных металлов, так и для произ-
водства деталей, изготовляемых из фенотекстослоя (подшипники, винты,
шестерни, ролики и др.).
Древесно-слоистые пластики находят большое применение для производства
труб, корпусов насосов, винтов, деталей разборных домов, внутренних и внешних
облицовочных материалов, цельноформованных лодок и малых судов, деталей
автомобилей и самолетов и т. д. Возможность получении изделий из фенодре-
вослоя при низких давлениях без применения прессов, широкая доступность
древесины и высокая механическая прочность фенодревослоя, превышающая
прочность стали (если исходить из расчета «весовой» прочности), обеспечили
большое техническое значение фенодревослоя и возможность широкого приме-
нения его в будущем в различных областях техники.
Резитовая фанера
Большое значение, главным образом в авиапромышленности и
в судостроении, имеет резитовая фанера (бакелизированная фа-
нера, фанерит) — материал, в котором листы шпона, в отличие от
фенодревослоя, не пропитаны, а склеены смолой.
Нанесение слоя жидкой смолы (эмульсии или раствора) на
листы шпона производят на лакировальных вальцах. Для этого
листы шпона однократно пропускают через вальцы, в которых
обычно нижний валок частично погружен в раствор смолы. Коли-
чество смолы, наносимое па шпон, регулируют, изменяя расстоя-
ние между валками.
Такое нанесение пленок резольной смолы на древесный шпон
встречает некоторые затруднения; поэтому производство резитовой
фанеры осуществляют также и методом запрессовки между двумя
листами шпона специальных бумажно-резольных пленок. Послед-
ние изготовляют путем пропитки бумажной пленки резольным
спиртовым лаком на обычных пропиточных или лакировальных
машинах; они имеют толщину от 0,05 до 0,1 мм. Роль такого
рода промежуточной резольной пленки может играть и пропитан-
ный резольным лаком лист шпона, который прокладывают между
двумя непропитанными листами.
Применяют также нанесение на шпон водной резольной сус-
пензии, с последующей сушкой и горячей прессовкой. Прес-
совку производят при 150—160° и удельном давлении от 15 до
25 кг/см2 без последующего охлаждения под давлением.
По сравнению с обычными фанерами на основе белковых (ка-
зеиновых, альбуминовых) клеев, резитовая фанера имеет преиму-
щество большей прочности, меньшей горючести и большей водо-
стойкости. Резитовые фанеры, в отличие от обычных, совершенно
не подвержены гниению.
Резитовая фанера естественно уступает фенодревослою как по
механической прочности (в частности, по показателю прочности на
раскалывание, по твердости), так и, в особенности, по водостойкости.
Феностеклослой
501
Свойства резитовой фанеры могут быть представлены следую-
щими показателями:
Уд. вес, г[см*................................... 1,00—1,05
Предел прочности на разрыв, кг1см^............... 1000—1500
Предел прочности на изгиб, кг/см*................ 1500—2000
Предел прочности на сжатие, кг/см*............... 1000—1500
Уд. ударная вязкость, кг • см/см*................ 40—100
Модуль упругости, KtjcM*......................... 200 000—220 000
Водостойкость (привес в воде за 24 часа), % . . . 10—15
Производство резитовых древесных пластиков включает также
пропитку растворами резольных смол или смолообразующими ком-
понентами дерева или изделий и деталей из него.
Пропитку производят в автоклавах под давлением 10—15 ат
при 150—180° в течение нескольких часов. В автоклав загружают
смесь фенола, формальдегида и катализатора или низкомолекуляр-
ные продукты конденсации (феноло-спирты). После пропитки
дерево подвергают сушке в сушилках или между обогреваемыми
плитами пресса при низких давлениях (~5 кг/см2). При этом про-
исходит и дополнительная полимеризация веществ, взятых для
пропитки.
Полученные таким образом материалы отличаются значительно
большей твердостью, монолитностью, прочностью, водостойкостью
и химической стойкостью по сравнению с необработанным дере-
вом и во многих случаях находят ценное применение.
Феностеклослой
(Стеклолит, стеклофенолит)
В последние годы большое техническое значение приобретают
слоистые пластики на основе стеклянного волокна и стеклянной
ткани.
Стекло — наиболее древний искусственный материал; его цен-
ными качествами являются прозрачность и изотропность свойств,
но оно хрупко и мало прочно. Вместе с тем, на основе стекла по-
лучают слоистые пластики, относящиеся к наиболее прочным (при
расчете на единицу веса) материалам. Последнее объясняется тем,,
что при изготовлении их применяют не трехмерное, объемное стекло,,
а практически одномерное — стеклянные нити (стеклянное волок-
но). Было доказано, что прочность стеклянных нитей так же, как
и в случае других волокон, находится в обратной зависимости от
их диаметра и что при достаточно малом диаметре их удельная
прочность может быть в десятки и сотни раз выше прочности объ-
емного стекла. Так, прочность на разрыв объемного стекла (тол-
щина образца ~10 мм) 3—5 кг[мм2, тогда как прочность нити
jO8
Гл. XI. Фенопласты слоистые
толщиной в 20 у. — 60 кг/мм2, а толщиной в 3 у- — 200 кг/лш2 (см.
,шс. 52, стр. 123).
В отличие от природных волокон (целлюлоза, асбест), стеклян-
ные волокна могут быть изготовлены любой длины и тонкости,
что очень важно для получения на их основе прочных пластмасс
с заданными свойствами. Стеклянное волокно имеет и ряд других
преимуществ: оно мало гигроскопично, обладает высокой химиче-
ской прочностью, термостойкостью и огнестойкостью.
Термостойкость стеклянного волокна значительно превышает тер-
мостойкость не только органических волокон (хлопок, шелк), но и
минеральных (асбест). Если термостойкость характеризовать
температурой, при которой волокно теряет 50% своей первоначаль-
ной прочности после месяца нагрева, то для стеклянного волокна
она равна 260°, для асбеста 175°, для хлопчатобумажной ткани 125°
и для природного шелка 80°.
Свойства стеклянного волокна определяются также его химиче-
ским составом. Так диэлектрические свойства, влагостойкость и
механическая прочность у малощелочного стекла выше, чем
у щелочного.
Существенно различие этих стекол и в степени снижения проч-
ности от действия температуры и влажности: малощелочное стекло
менее чувствительно в этом отношении, чем щелочное. Так в интер-
вале от 40 до 160° удельное объемное электрическое сопротивление
снижается для малощелочного стекла от 1014 до 5 • 1013 ом см, в то
время как у щелочного от 1013 до 9- 109 ом • см.
Поэтому для получения феностеклослоя для электротехниче-
ских целей применяют малощелочное волокно.
Для увеличения водостойкости слоистых стеклопластиков, а
также для предохранения волокна от потери прочности при хране-
нии К- А. Андрианов предложил метод гидрофобизации минераль-
ных волокон путем обработки их органохлорсиланами (стр. 616).
В этом случае алкилхлорсиланы образуют на поверхности стекла
хремнийорганическую пленку сетчатой структуры:
I I
: R о о
1111
...— О—Si—О—Si—О—Si—О—Si ...
II XR I
R О R О
I I I
. ..-O Si-O—Si—O—Si-...
I । I
R О R
Она образуется в результате реакции
сацпи при взаимодействии с молекулами
гидролиза и поликонден-
адсорбированной воды на
Фенос/геклослой
.50?
поверхности стекла. В такой сетке гидрофобные органические ра-
дикалы направлены наружу, а гидрофильные >Si—О— связаны
О
I
со стеклом, в результате поверхность стекла защищена от действия
влаги «частоколом» из гидрофобных радикалов.
Материалы, получаемые на основе таких волокон, более водо-
стойки, характеризуются практически стабильной прочностью при
хранении в воде или влажной атмосфере, в то время как стекло-
слойматериалы с необработанным волокном снижают свою проч-
ность при действии влаги.
Наибольшую прочность стеклянные нити имеют непосредственно
после изготовления, после вытяжки их из расплава. Как показал
С. Н. Журков, высокая первоначальная прочность («сверхпроч-
ность») мало стабильна и резко снижается от действия незначи-
тельных механических напряжений, а также влажного воздуха.
В этом случае стеклянные нити переходят из малостабильного со-
стояния сверхпрочности в более стабильное состояние обычной
прочности. Однако и в этом состоянии, хотя и в меньшей степени,
прочность нитей непрерывно снижается под влиянием влаги, темпе-
ратуры и различных механических воздействий. Следовательно хра-
нение и переработка нитей обусловливают непрерывное и значитель-
ное снижение прочности.
В частности это наблюдается в процессе изготовления ткани
из стекловолокна вследствие скручивания, скольжения и перепле-
тения нитей.
Так прочность нити толщиной в 20 у непосредственно из рас-
плава составляет 80 кг/мм2 («сверхпрочность»), после некоторого
хранения она снижается до 60 кг/мм2 (стабильная прочность) и
после переработки и выделки ткани имеет прочность 25—35 кг/лш2.
Таким образом, процесс изготовления ткани значительно сни-
жает (примерно в два-три раза) прочность нитей. Из этого сле-
дует, что можно было бы получить более прочный слоистый пла-
стик, если пропитку стеклянных нитей производить непосред-
ственно после получения их из расплава и если слоистый пластик
изготовлять не из ткани, а из параллельно уложенных нитей.
При этом нити в минимальной степени могли бы быть подвергнуты
действию механических напряжений и влажности и сохраняли бы в
значительной мере свою первоначальную прочность.
Таким образом, стеклянные пластики могут быть изготовлены
двумя методами:
1. Пропиткой стеклянной ткани. В этом случае процесс произ-
водства волокна и стеклянной ткани не связан с процессом
пропитки.
510
Гл. XL Фенопласты слоистые
2. Пропиткой стеклянного волокна при его получении из рас-
плава. В этом случае оба процесса — получение стеклянного
волокна и его пропитка — неразрывно связаны друг с другом.
Получение феностеклослоя путем пропитки стеклянной
ткани растворами или дисперсиями резольных смол
Этот метод получил основное промышленное применение.
Преимущества его: хорошо освоенный, сравнительно простой произ-
водственный процесс и удобство применения отдельных, не связан-
ных друг с другом операций: производства стекловолокна и ткани
и пластмасс (пропитка, прессование) на их основе.
Что касается достигаемой прочности по этому методу, то она
весьма значительна и превосходит прочность слойматериалов на
основе хлопчатобумажной ткани.
Применяют или ткани, состоящие только из стеклянных нитей
или же в сочетании с хлопчатобумажными (например, основа из
стеклянных нитей, уток из хлопчатобумажных, или наоборот —
уток из стеклянных нитей, а основа — из хлопчатобумажных).
Введением хлопчатобумажных нитей стремятся улучшить процесс
дропитки, увеличить эластичность и гибкость ткани.
Прочность пластика будет тем выше, чем больше удельная
прочность ткани и чем она тоньше. Однако удельная ударная вяз-
кость может быть увеличена применением более толстой ткани (при
равной ее удельной прочности).
При использовании стеклянной ткани с хлопчатобумажной осно-
вой (или утком) действие рабочего напряжения при растяжении
изделия должно быть в направлении стеклянных нитей. В тех слу-
чаях, когда хотят увеличить прочность пластика на растяжение
в одном направлении, берут ткань с большим числам нитей по
основе, чем по утку.
Стеклянные ткани выпускают нескольких сортов, различаю-
щихся по толщине, прочности, удлинению, размерам и т. д. Тол-
щина тканей обычно находится в пределах от 0,10 до 0,25 мм,
вес 1 м2 от 100 до 350 г, разрывное усилие (полоски 25 X Ю0 мм)
20—150 кг (по основе) и удлинение при разрыве 2—2,5%.
Пропитку стеклянных нитей производят на обычных вертикаль-
ных, реже горизонтальных пропиточно-сушильных машинах, при-
меняя спиртовые растворы резольных смол или водные эмульсии.
Концентрация смолы и ее вязкость вследствие очень гладкой и
не впитывающей поверхности стеклянной ткани может быть выше,
чем при пропитке хлопчатобумажной ткани и бумаги.
Содержание смолы в пластике зависит от его назначения: для
конструкционных материалов оно составляет 30—35%, для мате-
риалов электротехнического назначения 40—45%.
Феностеклослой
511
Прессование производят на обычных этажных прессах. Мате-
риал в виде пакетов укладывают в просветы пресса на плиты,
предварительно нагретые до 80°. Вначале дают давление
10—15 кг/см2 и после плавления смолы и ее желатинизации
(обрыва нитей выступающей смолы) температуру поднимают до
160—165° и увеличивают давление до 60 кг/см2. Выдержка при
достижении этой температуры составляет 5 мин. на 1 мм тол-
щины пакетов между плитами пресса. После выдержки охла-
ждают (до -j—40°) и пресс разгружают. Готовые изделия рекомен-
дуется дополнительно термически обрабатывать в печах при 100—
120° в течение нескольких часов.
В табл. 59 приведены показатели некоторых свойств фено-
стеклослоя различного назначения и в зависимссти от характера
волокна.
ТАБЛИЦА 59
Показатели некоторых свойств феностеклослой различного
назначения
Конструкционного назначения
Показатели на основе на основе стеклянного и Электротехни-
стеклянного волокна хлопчато- бумажного назначення
волокна
Уд. вес, г/см2 Водостойкость (привес в воде за 24 часа 1,7—1,85 1,55—1,65 1,65—1,85
для листов толщиной 4 мм), % . . . 3,0 5,0 2
Теплостойкость по Мартенсу, °C ... Предел прочности на разрыв, кг/см2; 200—225 175—185 180—200
по основе 2300—2700 1100-1500 900—1300
по утку 1500—2000 800—1000 —
Удельная ударная вязкость, кг см/см2:
по основе 45—55 100—150 35—50
по утку • 35—45 60—80 —
Сопротивление раскалыванию, кг/см2 . Удельное поверхностное электрическое — — 225—250 1012—5 - ю!2
сопротивление. 12 Удельное объемное электрическое со- — —
противление, Q-cm — —— 1012—5 • 10-
Диэлектрическая постоянная при 50 гц Электрическая прочность при 90° для — — 7—8,5
листов толщиной в 2 мм, кв/мм . . — — 12—15
Промышленность выпускает слоистые стеклянные пластики
различных марок, различающихся не только толщиной волокна и
ткани, содержанием смолы, но и содержанием в материале цел-
люлозных волокон и ткани.
Целлюлозное волокно снижает статическую прочность, водо-
стойкость, термостойкость, однако увеличивает показатель удель-
ной ударной вязкости.
512
Гл. Xf. Фенопласты слоистые
Получение слоистых пластиков путем пропитки стеклянного
волокна в процессе его производства
Этот метод был предложен А. К, Буровым и М.
Неклюдовой для различных видов связующего.
Стекло плавят в тигле, имеющем ряд фильер
мм). Жидкое стекло, выходящее из фильер,
В. Клаасен-
(диаметром
вытягивается
в тонкую нить (~ 20—10 ц) и не-
прерывно наматывается на вра-
щающийся барабан со скоростью
400—500 об/мин. Барабан в про-
цессе вращения медленно про-
двигается (со скоростью 5—
10 мм/мин.) вдоль горизонтальной
оси пока нить не покроет плотным
слоем всю его поверхность, после
чего он движется в обратном
направлении. Этим путем на по-
верхности барабана образуются
плотные слои стеклянных нитей.
Одновременно путем распыления
или другим методом слои из ни-
тей покрывают смолой, которая
их склеивает и предохраняет от
действия влаги.
Когда на барабане образует-
ся материал нужной толщины
(1—2 мм), его разрезают вдоль
оси барабана и получают тон-
кую пластинку, так называе-
мый «шпон». Из этих пластинок
путем склеивания или прессова-
ния при небольших давлениях
(~ 10 кг!см2) изготовляют пла-
стины или плиты. Не рекомен-
давлениях (выше 20 кг/см2), так
lo.nuj.una стеклянных нитей,у
Рис. 194. Зависимость толщины плен-
ки связующего слоистого пластика от
диаметра волокна при различной кон-
центрации волокна.
дуется прессование при высоких
как снижается прочность нитей.
После прессования плиты подвергают термообработке.
Получаемые этим методом стекломатериалы имеют значительно
более высокую прочность, чем на основе стеклоткани (до 5000—
7000 кг/см2), что объясняется меньшим повреждением волокна в
процессе производства.
Высокую прочность (по сравнению с пластиком на основе стекло-
ткани) нужно отнести также за счет того, что такой пластик состоит
только из нитей основы и, следовательно, все нити в нем участвуют
в растяжении, в то время как в пластике на основе ткани или уточ-
ные нити или нити основы в растяжении не участвуют.
Феностеклослой
513
В поперечном направлении прочность такого пластика значи-
тельно ниже, чем пластиков на основе стеклоткани, так как при этом
нити не участвуют в растяжении и прочность обусловлена лишь
прочностью пленок смолы.
Слоистые стекломатериалы получают на основе различных
термореактивных смол как поликонденсационных (феноло-альде-
гидных, меламино-формальдегидных, кремнийорганических и др.),
так и полимеризационных (полиаллиловых эфиров и др.).
Характер смолы определяет многие физические свойства стекло-
пластика — водостойкость, электроизоляционные свойства, тепло-
стойкость и другие. Прочность материала зависит как от удельной
прочности стеклянных нитей, так и от характера смолы.
Смола должна прежде всего обладать хорошей адгезией
к стеклу. Гидрофобные пленки (каучук, битумы и др.) не прили-
пают к стеклу и в этом случае не могут служить связующим
материалом. Феноло-формальдегидные, мочевино-формальдегид-
ные, сложно-эфирные и другие смолы, для которых характерно
наличие полярных групп, хорошо смачивают стекло и обладают
хорошей адгезией к стеклу. Затем необходимо, чтобы пленка
смолы выдерживала то максимальное напряжение, какое выдержи-
вает стеклянное волокно и обладала большим удлинением, чем
стеклянные нити. В этом случае прочность материала будет
в основном ограничена лишь удельной прочностью стеклянного
волокна. В противном случае разрушение материала начнется
с разрушения связующего.
Как известно, прочность смоляной пленки определяется как ее.
молекулярной структурой, так и толщиной. Чем тоньше смоляные
пленки, действующие между нитями и слоями наполнителя, тем они
прочнее. Толщина смоляной пленки зависит от соотношения волокна
и смолы в материале. Чем больше относительное содержание во-
локна, тем, в среднем, тоньше пленки связующего (рис. 194). Так
можно рассчитать, что при содержании в материале 20% волокна
(диаметром 12 у) средняя толщина пленки смолы будет 13,5 у,
при 50% этого же волокна — 4,5, а при 70% —всего 1,55 у.
Стеклопластики получают большое применение в различных
отраслях машиностроения в качестве конструкционного и электро-
технического материала.
Изделия из стеклопластиков особенно рационально изготовлять
методом прессования при низких давлениях. Таким путем получают
крупные, пространственно изогнутые детали, применяемые в раз-
личных отраслях техники.
33 Зак. 1269. Э. И. Барг
ГЛАВА XII
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ
АМИНО-АЛЬДЕГИДНЫХ СМОЛ
Пластические массы на основе мочевино- и меламино-
формальдегидных (карбамидных) смол
(Аминопласты)
Смолообразные продукты конденсации формальдегида с моче-
виной и ее производными — тиомочевиной, дициандиамидом, мела-
мином и др. — относятся к термореактивным смолам, способны
переходить из плавкого и растворимого состояния (стадия А) в про-
странственный полимер — неплавкий и нерастворимый (стадия С).
Эта способность обусловлена полпфункциональным характером реа-
гирующих молекул.
Техническое значение получили главным образом мочевино- и
меламино-формальдегидные смолы и пластмассы на их основе.
По комплексу основных физико-механических свойств и по струк-
туре промежуточных и конечных продуктов поликонденсации амино-
пласты имеют много общего с фенопластами. Технологические про-
цессы переработки, а в значительной мере также и основные обла-
сти применения этих пластиков во многом сходны.
Существенное преимущество аминопластов перед фенопла-
стами — их бесцветность и светостойкость, отсутствие запаха и
выделения при действии воды отравляющих веществ.
Техническое значение получили главным образом мочевино- и
меламино-формальдегидные смоты и пластмассы на их основе.
СТРОЕНИЕ МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫХ И
МЕЛАМИНО-ФОРМАЛЬДЕГИДНЫХ СМОЛ
Строение мочевино-формальдегидных смол
Химическое строение мочевино-формальдегидных смол менее изучено, чем
феноло-форм альдегидных, в особенности это относится к промежуточным и
конечным продуктам поликонденсапии.
Это объясняется тем, что изучение мочевино-форм альдегидной конденсации
было начато значительно позже, чем феноло-формальдегидной, а также тем, что
опа связана с более сложными процессами, приводящими уже на ранней стадии
конденсации к образованию желатинирующихся, нерастворимых продуктов.
Теория мочевино-формальдегидной конденсации исходит из ряда экспери-
ментальных данных.
Установлено, что в водной среде, в зависимости от определенных условий,
главным образом от pH среды и соотношения компонентов, а также от темпера-
туры, мочевина и формальдегид образуют различные продукты: 1) кристаллические
водорастворимые вещества, 2) нерастворимые аморфные — выпадающие из рас-
творов в виде порошка или геля н, наконец, 3) смолообразиые растворимые
вещества.
Мочевино-формальдегидные смолы
515
Наиболее полно изучены кристаллические продукты. Они образуются в ней-
тральной или слабощелочной среде при умеренных температурах в результате
взаимодействия одной или двух молекул формальдегида с молекулой мочевины и
представляют собой моно- и диметилмочевину:
NH, NH—СН2ОН
СО + сн,о —► со
nh2 \н.
•пли
NH, NH—СН,ОН
СО +2СН.0 —> СО
\н, NH—СН2ОН
Монометилолмочевипа имеет т. плавл. 111—113°, диметилолмочевина 121—126°;
они растворимы в воде, в метиловом и этиловом спиртах, нерастворимы в
эфире.
Более высокометилольные продукты не удавалось изолировать. Лишь в по-
следнее время получены три- и тетраметилолмочевины, которые легко образуют
циклические продукты:
/NH,
СО +4СН2О —>
^NH,
ZCH,CH
/ ХСН,ОН
co
\ /СН..ОН
N<
ХСН,ОН
В какой степени достоверна структура этих соединений, а также какова их
роль в процессе образования более сложных молекулярных комплексов, — пока
решить еще невозможно.
Диметилолмочевина и монометилолмочевина, в особенности в смеси с моче-
виной, прн нагревании в присутствии кислот переходят в неплавкое нераствори-
мое смолообразное состояние.
Реакции между первичными метилольными производными мочевины воз-
можны в следующих направлениях:
1) образование эфирных связей
,NH—СН2ОН 2 СО : xnh2 -NH—СН,ОН 2 СО — XNH—СН,ОН /NH—СН,—О—СН3—NH СО /СО xnh2 h2n / N Н—С Н,—О—С Н2—N Н СО /СО; XNH—СН.,—О-СН,—NH
516
Гл. XII. Аминопласты
2) образование, в- дальнейшем, метиленовых связей с выделением СН2О
/NH—СН2— О—СН2—NH /NH—СН2— NH
СО /СО—> СО /СО+2СН2О.
XNH—СН2—О—СН2—NH XNH—СН2—NH
В присутствии мочевины метилольные производные могут реагировать с об-
разованием метиленовых мостиков
/.NH—СН2ОН /NH2 /NH—СН2—NH
СО + СО —► СО /СО + Н2О.
XNH2 XNH2 ''4NH2 HaN
Возможно, что эти реакции приводят также к образованию метилол-мети-
леновых соединений типа
NH—СН2—NH
I I
СО СО н
I I
NH—СН2— N—СН2ОН
NH—СН2—N—СН2ОН
СО СО
I I
NH—СН2—N—СН2ОН
и в итоге к образованию полимерного
приписывают следующую структуру:
продукта, которому некоторые авторы
Такай структура, однако, является упрощенной, так как она не учитывает
возможного наличия эфирных связей, свободных метилольных групп, а также нс
дает наглядного представления о «сшивке» макромолекул.
При конденсации в сильнокислой среде (pH < 3) и при высоких температу-
рах выпадает аморфный нерастворимый порошок метиленмочевины. Образова-
ние метилеимочевины представляли себе как результат непосредственной реакции
конденсации мочевины с формальдегидом, протекающей с выделением воды и
с образованием ненасыщенного мономера:
/NH2
СО 4- СН2О
XNH2
zN=CH2 yN=CH2
со »
xnh2 xn=ch2
Так как метиленмочевииа трудно растворима, неплавка и имеет аморфную
структуру, ее рассматривали как полимерное соедииеиие
- /N=CH2
СО
- XNH,
Однако еще не выяснено, в какой мере действительно имеют место процессы
полимеризации до сих пор невыделенных мономеров метиленмочевины.
Известно, что при нагревании метилольных производных мочевины выде-
ляется вода и формальдегид и образуются аморфные продукты, идентичные
с метиленмочевинами, получаемыми в сильнокислей среде при взаимодействии
мочевины с формальдегидом.
На основании аналитических данных трудно решить, имеет ли место в рас-
сматриваемых условиях процесс поликонденсации или процесс полимеризации,
ведущий к образованию трехмерных макромолекул, так как процессу полимери-
Мочевино-формалъдегадные смолы
517
зации должна предшествовать реакция поликонденсации с выделением воды:
однако характер кинетики процесса, например образования неплавких смол при
горячем прессовании композиций из смеси моно- и диметилолмочевины, позволяет
считать, что по крайней мере основным процессом, ведущим к конечному со-
стоянию смолы, является поликонденсация.
По механизму протекающих реакций метилолмочевииы напоминают феноло-
спирты, превращение которых из стадии резола в стадию резита протекает как
процесс поликонденсации. При реакциях метилолмочевииы в присутствии мо-
чевины линейные и впоследствии разветвленные цепи образуются за счет взаимо-
действия гидроксилов метилольных групп с водородами амидных групп; при
этом так же, как и при конденсации феноло-спиртов, образуются в основном ме-
тиленовые мостики и частично эфирные связи как внутри цепи, так и между цепями.
Поэтому метиленмочевину следует рассматривать ие как продукт полимери-
зации ненасыщенных мономеров, которые с достаточной достоверностью не вы-
делены, а как продукт поликонденсации моно- и диметилолмочевины, которые
первично образуются присоединением мочевины к формальдегиду.
А. А. Ваишейдт рассматривает метиленмочевины, которые образуются в
сильнокислой среде, как смесь полимергомологов общего полимергомологиче-
ского ряда (мезометиленполимочевин);
Н[—NHCONHCH,—]„NHCONH2.
Реакция протекает по общей схеме:
лСН3О + (л + 1)CO(NH2)2 —► H[-NHCONHCH2—]„NHCONH2 + «H2O.
Свойства членов полимергомологического ряда Н[—NHCONHCH2]„NHCONH2
(мезометиленполимочевин) определяются показателем п, причем с увеличением
этого показателя повышается температура размягчения, уменьшается раствори-
мость и увеличивается вязкость полимергомологов.
При нагревании растворов мочевины в формалине в слабокислой среде
(pH = 4—6) образуются смолообразные гидрофильные продукты, не выделяю-
щиеся обычно из раствора и переходящие после обезвоживания и нагревания в
неплавкое и нерастворимое состояние, подобно переходу резола из стадии А
в стадию С. В этом случае смолообразный продукт в растворимом состоянии
представляет собою конгломерат молекул, содержащих кроме метиленовых не-
которое количество (30—40%) свободных метилальных групп; структуру таких
молекул можно выразить примерно следующей формулой:
Н[—NH—СО—N--------СН,—МН—СО—NH—СН2—J^-OH.
I
СН2ОН
Переход в пространственный полимер («сшивка» молекул) осуществляется
взаимодействием боковых метилольных групп цепи как с водородами NH-rpynn,
так и с метилольными группами соседних цепей с образованием эфирных
связен:
----NH—СО—N—СН2—N—СО—NH—СН2—N—СО—...
Ан
Grig
1Н
Gr*2
...— N—СН2— N—СО—МН—СН2— N—СО—...
518
Гл. XII. Аминопллсты
Таким образом, наличие в метилол-метилен-мочевинных соединениях реак-
ционноспособных имино- и метилольных групп делает возможным дальней-
ший процесс поликонденсации с образованием трехмерных полимеров. От сте-
пени замещения водородов аминогрупп, плотности поперечных связей, образуе-
мых метиленовыми и эфирными группами, зависят нерастворимость, неплавкость,
механическая и химическая стойкость и прочность продукта.
Образующийся пространственный полимер содержит тем меньше свобод-
ных метилольных групп, чем дальше протекала реакция поликонденсации.
Однако в технических смолах некоторые метилольные группы из-за стерических
трудностей остаются невступнвшими в реакцию даже при длительных сроках и
высоких температурах поликонденсации, образуя неплотности — «дырки» в поли-
мере, что ослабляет жесткость и прочность пространственной решетки. Чем
больше остается таких свободных метилольных групп и чем больше эфирных
связей находится в макромолекулярной решетке, тем больше вероятность выде-
ления формальдегида (а также воды) при дальнейших процессах поликонден-
сации, например, при термической обработке готовых изделий или при кипячении
их в воде. На практике такое выделение формальдегида обычно происходит
в случае мочевино-формальдегидных пластиков. Водопоглощаемость амино-
пластов также должна быть отнесена за счет наличия в отверждаемом полимере
свободных метилольных групп.
Таким образом, пространственная макромолекулярная решетка аминопластов,
может быть представлена следующей схемой:
..—NH—СН,—N—СН2—О—СН,—N—СН2- -NH .— N—
СО СО СО 1 СН,
Н2С—О—СН,—N N—СН,- -N—СН,—О—СН — N
1 1 1 —N СН, СН, „дырка14 4 ! !
1 1 " I СН, N СН N ! “ у. | ' | :НОСН,—NH СО
. . N—: СН,ОН; СО СО СО
CO-N-H.C-?! СП, - ?<!—СП,— -О—СП_—N—СН,
Принципиально иная схема строения мочевино-формальдегидных смол была
предложена Марвелем. Согласно его взглядам, мочевина ведет себя в этом про-
цессе как амид аминокислоты. Вначале она дает метиленимин:
R—NH:+CH,O —> R—N—СН,-|-Н._О,
(где R представляет CONH2), который переходит в тример с образованием цикли-
ческого трнметилентриаминового кольца:
R
N
Нос' сн.,
3R—N=CH„—> | '
R—N N—R
\н.
При этом свободные NHs-группы R (радикала — СОЫЩ) реагируют с форм-
альдегидом, образуя метилен-бис-амидные связи между кольцами. Образующийся
Строение меламино-формальдегидных смол
519
в результате пространственный полимер имеет строение, изобилующее попереч-
ными связями, которое может быть представлено следующей схемой:
N Н-------С Н ._>---N Н
СО СО
I I
N N
IkX Хн, Хн,
Il' I I '
..СН2—NH—СО—N N—CO—NH N—СО—N N—СО—NH—СП,—...
XX СН, | XX,
I ‘ СН2
СН, СО—NH I СН2
...—NH—СО—N N----СО—NH HN—CO-N N—СО—NH—CI.
II I i
Н2С сн, Н2С сн,
XX XX
I I
Конечная стадия процесса поликонденсации заканчивается образованием
сильно разветвленной, пространственной макромолекулы шарообразной струк-
туры.
Эту схему пока можно рассматривать лишь как научную гипотезу, так как
циклические триметилентриаминовые кольца, которые здесь считаются основой
структуры конечных продуктов реакции, до сих пор не были изолированы.
Строение меламино-формальдегидных смол
Меламин относится к триазинам (2,4,6-триамино-1,3,5-триазин или триамчд
циануровой кислоты). При гидролизе меламин образует циануровую кислоту.
Предполагают существование меламина в двух формах: (I) и (II)
NH,
I
С
И I
H2N—С С—NH,
XX
NH
II
С
HN Хн
HN=C C=NH
NH
I
аминоформа
II
иминоформа
Более устойчивой и более вероятной
ского исследования является форма I.
Меламин в промышленных условиях
формой по данным рентгенографиче-
получают из дицианидиамида путем
520
Гл. XII. Аминопласты
нагревания его в автоклаве при 120—20б° в присутствии аммиака, а иногда и
растворителей, при этом реакция циклизации протекает по схеме
NH2
I
C=NH
I
NH
CN
NH2
C
NZ
II I
H.,N—С C—NH.,
Меламин представляет собою белые призматические кристаллы, мало раство-
римые в воде (5% прн 100°), растворимые в глицерине и пиридине; растворы
имеют слабощелочной характер, с кислотами дают соли, применяемые для его
анализа; т. пл. 354°, уд. вес (25°) — 1,573.
Поликонденсация меламина с формальдегидом менее изучена, чем мочевино-
формальдегидная, однако, повидимому, механизм реакции конденсации и прин-
ципиальная схема строения смол подобны схемам для мочевино-формальдегид-
ных продуктов конденсации.
Так же, как и при поликонденсации мочевины с формальдегидом, перво-
начально образуются метилольные производные меламина. Однако отличитель-
ным является то, что в этом случае, при наличии соответствующего избытка
формальдегида, могут реагировать все шесть водородов амидных групп меламина
с образованием гексаметилолмеламина (а также ди-, три-, тетра- и пентамети-
ло.к'роизводных), по схеме:
ПОСН,—N—CILOII
С
NH2
С
/ \
НОН2С N N СН,ОН
I II I I
+ 6СН2О —>N—С С—N
нон2с сн2он
меламин
Метилольные продукты в дальнейшем (в кислой среде) реагируют друг
с другом и с меламином, образуя метиленовые и, частично, эфирные мостики
между кольцами:
R—NH—СН2ОН + H2N—R —> R—NH—СН2—HN—R + Н2О,
R—NH—СН2ОН + НОСН2—HN— R —> R—NH—СН2—О—СН2—HN—R.
Согласно работам В. С. Киселева, гексаметилолмеламин получается при
взаимодействии меламина с двойным избытком формальдегида (12 молей форм-
альдегида на 1 моль меламина) в нейтральной среде (pH = 7—7,5) при уме-
ренных температурах (60—80°); пеитаметилолмеламин образуется в этих же
условиях, если берут 8 молей формальдегида на 1 моль меламина. Дальней-
шие процессы, ведущие к образованию пространственных полимеров, протекают
в слабокислой среде (pH = 4,5—5) и при температурах 80—90°.
Более высокая функциональность меламина по сравнению с мочевиной
обусловливает большую плотность метиленовых межмолекулярных связей и, сле-
довательно, большую прочность, теплостойкость и гидрофобность конечного про-
дукта поликонденсации. Строение этого полимера, по аналогии с мочезино-
формальдегидными смолами, можно себе представить в виде пространственного
конгломерата колец, соединенных по всем направлениям группами —СН2— и
Получение мочевино-форм альдегидных смол и прессматериалов 521
- CHi—О—CHs— с сохранением в отдельных местах разрывов («дырок»),
вследствие наличия иепрореагировавших метилольных групп:
NH
МЕТОДЫ ПОЛУЧЕНИЯ МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫХ СМОЛ И
ПРЕССМАТЕРИАЛОВ (АМИНОПЛАСТОВ)
Основным затруднением, с которым встречались исследователи,
изучавшие процессы образования и отверждения литых мочевино-
формальдегидных смол, являлась преждевременная желатинизация
смолы при конденсации, которая, естественно, затрудняла сушку
смолы и приводила к образованию пузырчатых масс при отвержде-
нии.
Желатинизация объясняется исключительной чувствительностью
процесса поликонденсации к изменению кислотности среды; кроме
того, известное значение имеют соотношение компонентов, темпера-
турные условия и т. д. Поэтому прозрачные твердые и технически
годные смолы были получены лишь когда были установлены зави-
симости процесса конденсации от вышеперечисленных факторов и,
в первую очередь, от степени кислотности среды.
Так было установлено, что реакцию конденсации целесообразно
проводить в две фазы, вводя во второй фазе процесса добавочное
количество мочевины для связывания формальдегида. По этому
методу в начальной стадии конденсаций вводят 1 моль мочевины
на 2 моля формальдегида; в результате связывается только 1,6 моля
522
Гл. XII. Аминопласты
формальдегида, а 0,4 моля остаются свободными. Во второй ста-
дии добавляют мочевину для связывания свободного формальде-
гида, доводя молярное соотношение мочевины и формальдегида
до 1 : 1,5.
Изучение кинетики реакции поликонденсации позволило для
каждой стадии процесса установить оптимальные значения pH. Так,
для того, чтобы реакция начала протекать с нужной скоростью необ-
ходима слабокислая среда (pH в пределах 4,5—5,0). Однако для
устранения в дальнейшем чрезмерного нарастания вязкости сле-
дует снизить степень кислотности и поддерживать ее в пределах
pH = 5,5—6,5. В стадии сушки смолы, во избежание желатиниза-
ции, необходима практически нейтральная реакция (pH в пределах
6,5—7,5) и, наконец, в последней стадии поликонденсации для
отверждения смолы (перевода ее в стадию С) рекомендуется снова
увеличить кислотность до pH = 3,5—4,5.
Для понимания сущности этого процесса следует иметь в виду,
что технический формалин вследствие наличия в нем некоторого
количества муравьиной кислоты имеет кислую реакцию (pH =
2,5—3,5) и что при процессе конденсации количество муравьиной
кислоты может увеличиваться за счет окисления кислородом воз-
духа, а также по реакции окисления—восстановления:
2СН,О + Н.О —> СН;!ОН + нсоон,
которая может протекать и в кислой среде.
Для устранения нарастания кислотности в процессе конденсации
и поддержания ее в пределах pH = 5,5—6,5 в реакционную массу
рекомендуется вводить ряд щелочных солей слабых кислот (на-
пример ацетат натрия, фосфат натрия и др.), обладающих буфер-
ным действием. Буферные катализаторы регулируют pH среды
в процессе конденсации.
Для устранения желатинизации в процессе сушки смолы введе-
нием соды устанавливают нейтральную реакцию (pH = 6,5—7,5).
Последняя стадия поликонденсации — отверждение смолы (переход
в стадию С) — может быть ускорена повышением кислотности
среды до pH = 3,5—-4,5, что достигается внесением твердых орга-
нических кислот или продуктов их конденсации с многоатомными
спиртами, жирных кислот, кислых солей и т. д.
Первоначальная (исходная) кислотность среды определяет на-
чальное соотношение применяемых компонентов; так, при конден-
сации в слабокислой среде (pH ~ 4,5—5,5) на 1 моль мочевины
берут 2 моля формальдегида; при более кислой среде (pH —
2,5—3,5) для предупреждения желатинизации рекомендуется брать
избыток формальдегида (2,5—3 моля и более); чтобы связать избы-
ток формальдегида во второй фазе конденсации вводят мочевину
или тиомочевину, дициандиамид, фенол и др.
Этими методами получали как литые, ненаполненные смолы, так
и прессматериалы с целлюлозным наполнителем. Литые смолы
Получение мочевино-формальдегидных смол и ирессматерьалов 523
несмотря на бесцветность, прозрачность и твердость, не нашли
в дальнейшем широкого применения из-за склонности к образова-
нию трещин.
Современное назначение мочевино-формальдегидных смол — по-
лучение наполненных прессматериалов, главным образом на основе
целлюлозного наполнителя (аминоцеллолитов). Для этого получали
вышеуказанным методом конденсации в присутствии буферных
катализаторов необезвоженные смолы («сиропы»), которыми пропи-
тывали наполнитель. В дальнейшем масса подвергалась сушке и
горячему прессованию.
Метод производства неслоистых прессматериалов (аминоцелло-
литов) путем пропитки их водным раствором смол в дальнейшем
получил лишь ограниченное распространение как из-за сложности
процесса получения «сиропов» с заданными показателями, так и
из-за трудности контроля степени кислотности и вязкости среды.
Этот метод сохранил, однако, свое значение для производства слои-
стых аминопластов (стр. 534).
Существенное усовершенствование процесса получения пресс-
композиций на основе мочевино-формальдегидных смол было вне-
сено А. А. Ваншейдтом и Краусом.
Было показано, что слабощелочной или нейтральный раствор
реакционной смеси, полученный в результате прибавления уротро-
пина при нагревании и даже на холоду, по мере развития реакции
поликонденсации становится кислым, что способствует образованию
смолообразных продуктов.
В дальнейшем А. А. Ваншейдт установил, что во всех случаях,
когда нагревают уротропин с мочевиной и формалином, раствор
с течением времени приобретает ту кислотность, которую он имел
до прибавления уротропина, независимо от количества последнего,
и что при достижении первоначальной кислотности (если она была
в пределах pH = 3—3,5) сравнительно быстро происходит желати-
низация смолы (табл. 60).
Увеличение кислотности среды до величины pH, которую раствор
имел перед введением уротропина, может быть объяснено взаимо-
действием уротропина с формальдегидом и мочевиной с образова-
нием продуктов нейтрального характера.
Таким образом, наличие в реакционной среде уротропина при-
водит к непрерывному изменению pH среды; скорость этого про-
цесса и момент наступления желатинизации будет определяться
концентрацией уротропина, температурой и первоначальной кислот-
ностью среды.
Следовательно уротропин, являясь регулятором кислотности ре-
акционной массы, тем самым регулирует скорость реакции смолооб-
разования, которая ускоряется (возбуждается) водородными ионами.
Первоначальную кислотность среды (до введения уротропина)
можно, по желанию, понизить едкой щелочью или содой.
Таким образом, путем установления определенного pH реакцион-
Гл. ХИ. Аминопласты
ной среды как до, так и после введения уротропина можно регулиро-
вать течение процесса конденсации, заранее установив зависимость
скорости конденсации и предела желатинизации от первоначальной
кислотности среды, количества вводимого уротропина и темпера-
туры.
ТАБЛИЦА 60
Зависимость pH среды от количества уротропина и времени нагревания
при получении мочевино-формальдегидных смол.
(pH среды до введения уротропина 3,8)
Количество уротропина в “(о к весу мочевины
1 3 5 10 15
pH среды до нагрева (немед- ленно после введения уро- тропина) 5,0 6,2 6,8 7,8 7,8
pH среды после 5 мин. нагрев. Желати- низация — 3,8 6,2 7,6
То же 10 „ » Желати- низация Желати- низация 4,4 6,4
15 , 3,8 5,4
20 » Желати- низация 4,2
. 25 „ » 3,8
» 32 и „ Желати- низация
Желатинизация реакционной смеси наступает, когда количество
метилен-мочевинных производных значительно превосходит (при-
мерно в четыре раза) количество первоначально образующихся
метилольных производных мочевины; при этом, чем больше кислот-
ность среды, тем быстрее происходит накопление метилен-мочевин-
ных производных.
В связи с регулирующим действием уротропина на кислотность
реакционной смеси и, следовательно, на скорость реакции, пред-
ставляется возможным проводить реакцию как при непродолжитель-
ном нагревании, так и на холоду. В последнем случае перво-
начально образующиеся кристаллические метилольные производные
мочевины параллельно с процессом связывания уротропина и уве-
личением активной кислотности среды будут переходить в более
вязкие и желатинизирующиеся продукты — метиленпроизводные
мочевины. Если пропитать наполнитель, например волокно, рас-
твором формалина, мочевины и уротропина, то уже на холоду, по-
степенно, образуется пленка коагулированной смолы. Этот процесс
может быть значительно ускорен повышением температуры, умень-
шением количества уротропина или повышением первоначальной
кислотности среды (до введения уротропина).
Для ускорения отверждения смолы при горячем прессовании
необходимо в состав реакционной смеси вводить ускорители.
Производство мочевино-формальдегидных пресспорошков 525-
Ускорителями обычно являются твердые органические кислоты
(щавелевая и др.), которые должны иметь температуру плавления,
лежащую в пределах температур горячего прессования пластика.
В этом случае жидкая смола взаимодействует с расплавленной кис-
лотой, что ведет к резкому повышению кислотности и к увеличе-
нию скорости отверждения и, следовательно, скорости прессования..
Количество ускорителей должно соответствовать достижению вы-
сокой скорости отверждения смолы при горячем прессовании, од-
нако при этом необходимо иметь в виду, что слишком большое
количество ускорителей может снизить текучесть прессматериала
при прессовании.
На основе вышеприведенных положений был разработан про-
цесс производства аминопластов (см. ниже). Процесс этот основан
на получении на холоду или при слабом нагреве конденсацион-
ного раствора, состоящего из мочевины, формалина, уротро-
пина и щавелевой кислоты, в котором преобладают метилоль-
ные производные мочевины и которым пропитывают волокнистый
наполнитель.
После пропитки масса «созревает», т. е. в определенных темпе-
ратурных условиях протекают процессы связывания уротропина и
формальдегида и желатинизации массы на волокне. По окончании
«созревания» массу подвергают сушке, размолу и просеиванию.
При прессовании пресспорошка в горячих прессформах щавелевая
кислота взаимодействует с расплавленной смолой, ускоряя процесс
отверждения смолы.
К мочевино-формальдегидным продуктам реакции относят и
конденсаты тиомочевины с формальдегидом. Тиомочевина приме-
няется либо самостоятельно, либо, чаще всего, в смеси с мочевиной..
Тиомочевину рекомендуют вводить, вместо мочевины, во второй
стадии процесса конденсации, для связывания избытка альдегида.
Тиомочевина, внесенная вместе с мочевиной, позволяет уменьшить
начальное молярное отношение альдегида к мочевине до 1,5 молей
на 1 моль мочевины; считают, что добавка тиомочевины улучшает
водостойкость смолы и пресскомпозиций, увеличивает скорость
отверждения, а также улучшает условия обезвоживания смолы.
Однако применение тиомочевины усиливает коррозию аппаратуры,
которую в этом случае приходится готовить из спецсталей.
ТЕХНОЛОГИЧЕСКИЙ ПРОЦЕСС ПРОИЗВОДСТВА ПРЕССПОРОШКОВ НА
ОСНОВЕ МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫХ СМОЛ И ЦЕЛЛЮЛОЗЫ
(Аминоцеллолиты)
Производство пресспорошков на основе мочевино-формальдегид-
ных смол может быть проведено по двум методам.
Первый метод основан на предварительном получении водного
конденсата смолы, так называемого «сиропа», с последующей про-
питкой этим конденсатом наполнителя — волокна.
526
Гл. XII. Аминопласты
Второй метод — более совершенный и широко распространенный
в промышленности — основан на пропитке наполнителя (волокна)
исходными смолообразующими реагентами.
Первый метод в настоящее время применяется уже редко,
однако получение водных конденсатов (сиропов) мочевино-формаль-
дегидных смол имеет значение для пропитки ткани, бумаги и дру-
гих слоистых наполнителей.
Ниже приводится описание производства пресспорошков по вто-
рому методу, разработанному А. А. Ваншейдтом, 3. И. Наумовой
и др. Он заключается в пропитке наполнителя смесью моно- и ди-
метилолмочевины, регулировании кислотности реакционной смеси
и, следовательно, скорости реакции в процессе поликонденсации дей-
ствием уротропина. Этот метод может быть выполнен в двух вариан-
тах: с промежуточным «созреванием» массы после пропитки напол-
нителя реакционной смесью и без «созревания», с непосредственной
сушкой массы после пропитки в мешателе.
По первому варианту производство пресспорошков складывается
из следующих стадий: 1) приготовления мочевино-формальдегидного
раствора; 2) смешения раствора с волокном и другими компонен-
тами (краситель, пигменты; соли жирных кислот и др.); 3) «созре-
вания» массы; 4) сушки массы; 5) измельчения; 6) укрупнения
партий; 7) просеивания и упаковки.
В состав мочевино-формальдегидного раствора входит мочевина,
формалин, уротропин, щавелевая кислота. Молярные отношения
мочевины к формальдегиду берут обычно в пределах 1 : 1,5— 1,6.
Примерный состав реакционной смеси (в вес. ч.):
.Мочевина........ 100 Уротропин.............5—8
Формалин (37р0) . . . 200—225 Щавелевая кислота . . . 0,5—1
Приготовление мочевино-формальдегидного
раствора
Формалин из заводского хранилища центробежным насосом по-
дают в цеховой напорный бак, откуда он самотеком поступает
в весовой мерник и затем в реактор. Применяют реакторы обычной
конструкции, желательно эмалированные или алюминиевые. Ру-
башка для обогревания может быть крайне облегченной, рассчитан-
ной на давление до одной атмосферы, мешалка и мотор также
должны быть рассчитаны на перемешивание маловязкой среды;
применяют мешалки как якорного, рамного типа, так и пропеллер-
ные. Реактор должен быть снабжен обратным холодильником, рас-
считанным на малую упругость паров в реакторе и нагрев реакцион-
ной смеси не выше 40°. Обогревание реактора производят водой,
реже паром.
Формалин, имеющий pH -= 2,8 — 3,5, подогревают в реакторе
до 30—35°, затем приливают постепенно при перемешивании водный
раствор уротропина, после чего среда становится почти нейтральной
Производство мочевино-формальдегидных порошков 527
(pH — 7,0—7,5). После, перемешивания в течение 15—20 мин. по-
степенно вводят мочевину, а после растворения мочевины — водный
раствор щавелевой кислоты. Введением щавелевой кислоты pH
реакционной смеси лишь незначительно снижается и находится
в пределах 6,5—7,5.
Конденсацию ведут в течение 60—90 мин., считая с момента
ввода уротропина, при 25—30° и прерывают при содержании в реак-
ционной среде 10—12% формальдегида..
По мере протекания конденсации pH постепенно снижается. При
более высокой температуре (40—45°) может начаться экзотермиче-
ская реакция, резкое снижение pH и образование неплавких про-
дуктов; при pH = 5,0—5,5 могут получаться твердые нерастворимые
продукты, которые весьма нежелательны.
Контроль процесса сводится главным образом к определе-
нию pH; реже определяют содержание формальдегида.
Количество уротропина, необходимое для достижения задан-
ного pH, устанавливается лабораторией в зависимости от pH фор-
малина.
После окончания перемешивания раствор спускают через ниж-
ний штуцер для фильтрования через путч-фильтр или же сначала
в отстойник для отстаивания, после чего фильтруют через путч-
фильтр.
Отфильтрованный раствор центробежным насосом подают в ве-
совой мерник, расположенный над мешателем. Раствор не должен
храниться до замешивания более 3—5 час., во избежание желати-
низации и выпадения кристаллических продуктов реакции, могущих
набить трубопроводы.
Смешение раствора с наполнителем и
«созревание» массы
В качестве основного наполнителя применяют древесную отбе-
ленную сульфитную целлюлозу. Целлюлоза, поставляемая в виде
листов, предварительно измельчается на небольшие кусочки
(0,5—2 см); измельчение ведут обычно на волчках.
Кроме целлюлозы в состав массы вводят стеарат цинка нли
кальция (в качестве смазки), органический краситель и минераль-
ные пигменты. Например, на 100 вес. ч. раствора берут 25—35 ч.
сульфитной измельченной целлюлозы, 0,3—0,5 ч. стеарата цинка,
от 3 до 10 ч. минеральных пигментов (литопон и др.) и от 0,1 до
0,3 ч. органических красителей.
Смешение раствора с наполнителем производят в двухлопастном
вакуум-мешателе, внутренние части и лопасти которого должны
быть футерованы никелем или алюминием, чтобы устранить воз-
можность окрашивания массы продуктами коррозии металла.
Раствор поступает в.мешатель из весового мерника через сетку;
затем при работающих лопастях постепенно (в течение 15—20 мин.)
528
Гл. XII. Аминопласты
добавляют целлюлозу, после чего массу перемешивают еще
15—20 мин. По окончании перемешивания в мешатель вносят стеа-
рат цинка, пигменты и красители и перемешивают еще в течение
часа.
Перемешивание ведут при обычной температуре.
После смешения масса содержит до 50% воды и значительные
количества свободного формальдегида. Ее выгружают в алюминие-
вые или эмалированные цилиндрические тушильники, снабженные
крышкой диаметром ~400 мм; толщина слоя массы не должна
превышать 400—500 мм. Массу в тушильниках перевозят в отдель-
ную камеру для «созревания».
Созревание массы в тушильниках ведут при комнатной темпера-
туре. Необходимо следить, чтобы температура массы не превышала
40—50°, для чего массу, во избежание местного перегрева, необ-
ходимо регулярно (примерно раз в два часа) перемешивать. В про-
цессе «созревания» происходит связывание свободного формальде-
гида, увеличение кислотности среды и образование смолообразных
продуктов на волокне.
Конец «созревания» определяют по содержанию свободного фор-
мальдегида в растворе, отжатом от целлюлозы; оно не должно быть
более 4%.
Сушка
Сушку производят либо в стационарных камерных сушилках
периодического действия, либо в сушилках непрерывного действия.
В первом случае массу после созревания перекладывают из
сушильников на алюминиевые противни слоем ~20 мм, ставят на
тележки и направляют в камерные сушилки полочного типа с ре-
циркуляцией воздуха (рис. 195). Обогревание ведут нагнетанием
горячего воздуха (80—90°) вентилятором из калориферов, а также
с помощью системы паровых змеевиков. Выходящий из сушилки
воздух имеет температуру 60—70°. Сушку считают законченной,
если остаточная влажность материала не превосходит 3%. Продол-
жительность сушки обычно ~ 4 часа. Важно не «пересушить» массу,
т. е. чтобы процесс поликонденсации не привел к уменьшению теку-
чести прессматериала. Поэтому наряду с определением остаточной
влажности процесс контролируют и определением текучести пресс-
материала (стр. 452).
Высушенный материал измельчают на дисковых или шаровых
мельницах, футерованных фарфором. Применяют также предвари-
тельное измельчение на ударно-крестовых мельницах, с последую-
щим измельчением на шаровых.
Краситель, пигменты и ускоритель (щавелевая кислота) можно
вносить непосредственно в шаровые мельницы. В этом случае
упрощается процесс перемешивания в мешателе и для многих ком-
Производство мочевино-формальдегидных пресспорошков
529
позиций получаются более яркие цвета. Измельченный материал
просеивают через вибрационные сита в 900—1200 отв/см2.
Для получения больших и стандартных партий измельченный
пресспорошок загружают в смесительный барабан, где укрупнен-
ную партию перемешивают в течение 40—60 мин., затем порошок
просеивают.
По второму варианту рассматриваемого метода (стр. 526) з сме-
сителе происходят не только пропитка и механическое перемешива-
ние, но и дальнейшая конденсация и смолообразование на волокне,
Рис. 195. Схема камерной (сушилки.
1—вентилятор; 2—калорифер; 3—камеры; 4—воздуховод; 5 —
подводка воздуха; 6—вывод воздуха; 7—вагонетки со стеллажами.
т. е. частично те процессы, которые по первому варианту протекают
при «созревании» массы. В этом случае перемешивание проводят не
на холоду, а при 50—60° в продолжение 90—120 мин., после чего
масса может непосредственно поступать на сушку. Отсутствие про-
межуточной операции «созревания» значительно облегчает произ-
водство пресспорошков п дает возможность введения непрерывных,
поточных методов.
Условием введения элементов непрерывного, поточного произ-
водства пресспорошков является применение непрерывной сушки
(взамен стационарной, периодической), которую осуществляют
в ленточных и турбинных сушилках. Непрерывные методы сушки
более производительны, связаны с меньшими затратами рабочей
силы, меньшим расходом пара и позволяют получать пресспорошки
более высокого качества.
На рис. 196 приведена схема ленточной сушилки. Последняя
состоит из камеры, в которой расположены одна над другой
34 Зак. 1269. Э. И. Барг
530
Гл. ХИ. Аминопласты
бесконечные ленты, изготовленные из стальных сеток. Мате-
риал, поступающий из бункера питателя 1, распределяется равно-
мерным слоем по первой (верхней) ленте 5 и движется вместе
с лентой до противоположного конца камеры, где ссыпается на
Рис. 196. Схема ленточной сушилки.
1—бункер питателя; 2—бункер для сухого материала; 3—калориферы; 4— паровые
змеевики; 5—бесконечная лента.
нижнюю ленту и движется с ней в противоположном направлении'
вдоль камеры, затем ссыпается на третью ленту и т. д. С последней
ленты материал поступает в бункер 2 для высушенного продукта.
Обогрев сушилки производится нагнетанием горячего воздуха, по-
ступающего из калориферов 3, а также системой паровых змееви-
ков 4, расположенных внутри камеры. Движение массы и воздуха
осуществляется по принципу противотока, как это показано на схеме.
При отсутствии операции «созревания» массы и при применении
непрерывных методов сушки все производство пресспорошков может
быть в значительной степени оформлено как непрерывный процесс
(рис. 197).
В реактор 3 через напорный бачок 1 и весовой мерник 2 поступает форма-
лин; через бункер 5 подается мочевина; остальные компоненты вносят вручную
через загрузочный люк. Готовый раствор из реактора поступает в отстойник 6
и через фильтр 7 подается центробежным насосом в весовой мерник 8, а от-
туда в двухлопастный вакуум-мешатель .9, в который вручную загружают цел-
люлозу и другие компоненты. Из мешателя влажную массу выгружают в бун-
кер 10; через питатель 11 и ленточный транспортер 12 она поступает в загру-
зочную воронку сушилки непрерывного действия 13. Из сушилки материал
пневмотранспортом передается на измельчение. На схеме показан также процесс
промежуточного горячего вальцевания массы, который применяют при производ-
стве мочевино-меламиновых и меламиновых прессматериалов (стр. 541). В этом
Рис. 197. Схема производства карбамидных пресспорошков.
/—напорный бак; 2, <9 -весовые мерники; 3—реакционный аппарат; 4 — обратный холодильник; 5, 23— весовые бункеры;
б —отстойник; 7 —фильтр; 9—двухлопастный вакуум-мешатель; 10—бункер; 11—питатель; 12—ленточный транспортер;
/3—сушилка непрерывного действия; //—шнек; 15—бункер с питателем; 16—вальцы; 17—транспортер; 18 — вальцовая дробилка;
19 — вентилятор; 20-циклон; 21, 25-дозировочные бункеры; 22—молотковая дробилка; 24— шаровая мельница; 2о —сме-
ситель; 27—сито; 23—теплообменник.
532
Гл. ХИ. Аминопласты
случае масса из сушилки шнеком 14 подается в бункер 15, а из него на горя-
чие вальцы непрерывного действия (стр. 438) 16. Вальцованная масса подается
транспортером 17 на дробильные вальцы 18, и размолотый материал пневмо-
транспортом переносится через циклон 20 в дозировочный бункер 21, а из него
в молотковую дробилку 22, откуда материал через весовые бункеры 23 посту-
пает на тонкое измельчение и окраску в шаровые мельницы 24. Затем материал
поступает через дозировочные бункеры в смеситель для укрупнения партий, а из
смесителя — на непрерывно работающие сита 27 и на упаковку.
Следует обратить особое внимание на организацию процесса
окраски аминопластов. Обычно выпускают порошки нескольких
(4—8) стандартных цветов. Наилучшим методом, обеспечивающим
чистоту окраски, явилась бы организация специальной производ-
ственной линии для порошка каждого цвета. Поэтому для укрупне-
ния размеров агрегатов целесообразно проводить окраску в шаро-
вых мельницах и начать разделение производственного потока
с.' размольного отделения. В крайнем случае группируют в одной
линии производство 3—4 порошков, близких по тону окраски.
СВОЙСТВА И ПРИМЕНЕНИЕ МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫХ
ПРЕССПОРОШКОВ
Качество аминопластов в значительной мере определяется цве-
том материала, равномерностью окраски и его стойкостью по отно-
шению к действию света, влаги и горячей воды.
По цветности мочевино-формальдегидные порошки делятся на
непрозрачные и прозрачные (просвечивающие). Непрозрачные, как
правило, содержат минеральные пигменты (сернокислый барий.,
литопон и др.).
Пресспорошок обычно удовлетворяет следующим условиям:
1) влажность не более 3%; 2) текучесть по Рашигу 50—180 мм
(р зависимости от класса порошка); 3) дисперсность—порошок
должен проходить через сита 900 отв/см2; 4) диск, прессованный
при 140—150° и выдержке 1—1,5 мин/мм должен быть ровного
цвета, без посторонних включений, вздутий и т. д.
Мочевино-формальдегидные пресспорошки следует хранить в су-
хом и холодном месте, в герметически закрытой таре или в плот-
ных многослойных бумажных мешках.
Не рекомендуется длительное хранение, так как при этом непре-
рывно уменьшается текучесть пресспорошка.
Прессование аминопластов производят при 140—150° и удель-
ном давлении 250—350 кг/см2.
Аминопласты более, чем фенопласты чувствительны при прессо-
вании к колебаниям давления и температуры, а также к неравно-
мерности обогрева. Строгое соблюдение времени выдержки под
давлением, постоянства и равномерности обогрева и давления —
основные условия для получения высококачественных изделий из
аминопластов. Поэтому прессование аминоцеллолитов рекомендуется.
Слоистые мочевино-формальдегидные прессматериалы
533
производить на прессах с автоматической регулировкой давления,
времени и температуры прессования.
В СССР аминоцеллолиты на основе мочевино-формальдегидных
смол выпускают под названием аминопласт.
Ниже приведены показатели физических и механических свойств
аминопластов:
Уд. вес, г/смъ..................................1,45—1,55
Предел прочности на изгиб, кг/CAfi.............. 600—900
Предел прочности на растяжение, кг/см"*......... 350—400
Предел прочности на сжатие, кг/см2.............. 1500—2000
Удельная ударная вязкость, кг-см/см-............ 6—8
То же с надрезом, кг-см/слР..................... 1,8—2,6
Теплостойкость по Мартенсу, °C.................. 100—120
Водостойкость (привес в воде за 24 часа бруска
120 X 15 X 10 мм), %..........................0,35—1,5
Удельное объемное электрическое сопротивление,
Q см............................................ 10”—10й
Удельное поверхностное электрическое сопротивле-
ние, й.......................................... 1010—1011
Тангенс угла диэлектрических потерь при 50 гц . 0,02—0,1
Диэлектрическая постоянная при 50 гц........... 5—7
Электрическое пробивное напряжение, кв!мм . . . 10—15
Аминопласты стойки к действию спирта, бензина, ацетона,
хлороформа и др.
По водостойкости они значительно уступают фенодреволитам.
При кипячении в воде они поглощают до 4% воды за 30 мин; кроме
того, выделяют при этом формальдегид.
При длительном действии повышенной температуры аминопла-
сты стойки до 80°, при кратковременном — до ПО—120°. Более
высокие температуры вызывают уменьшение прочности и измене-
ние веса.
Пресспорошки на основе мочевино-формальдегидных смол получили приме-
нение главным образом для изготовления изделий широкого потребления. Из-
готовляют из них только такие технические детали, к которым не предъявляют
требований высокой водостойкости и высоких диэлектрических свойств; так .из
мочевино-формальдегидных пластиков делают корпуса и ручки телефона, штеп-
селя, выключатели и т. д. Из них изготовляют также дверные ручки, облицо-
вочные плитки, плафоны, абажуры, лампы и т. д. Широко применяют эти
пластики для изготовления различной посуды, однако не рекомендуется изго-
товлять из них посуду для горячих жидкостей (тарелки, стаканы и т. д.).
СЛОИСТЫЕ МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ ПРЕССМАТЕРИАЛЫ
(Аминобумослой, аминотекстослой и др.)
Технологический процесс производства слоистых аминопластов
в принципе мало чем отличается 'от производства слоистых фено-
пластов. Он складывается из получения связующего (раствора) и
пропитки слоистого наполнителя (бумаги или ткани).
В качестве связующего применяют водный конденсат смолы
(«сироп»), получаемый по первому методу (стр. 525), т. е. путем
534
Гл. XII. Аминопласты
конденсации при нагревании реагирующих компонентов в присут-
ствии буферных солей до получения прозрачного и немутнеющего
раствора.
Реакцию проводят в обычных котлах, применяемых для варки
феноло-альдегидных смол, изготовленных из эмалированного чугуна,
алюминия или кислотоупорной стали, снабженных паровой рубаш-
кой, якорной мешалкой, прямым и обратным холодильником.
Формалин поступает в реактор самотеком из алюминиевого
мерника; к нему добавляют такое количество щелочной соли (аце-
тата натрия), чтобы pH был в пределах 5—5,5, после чего в котел
постепенно вносят мочевину из расчета 1 моль мочевины на 2 моля
формальдегида. Реакционную смесь при постоянном перемешива-
нии и включенном обратном холодильнике нагревают пуском пара
в рубашку реактора до 85—90°; при этой температуре ведут реак-
цию в течение 60—90 мин. до получения прозрачного, немутнею-
щего раствора. Конец реакции контролируется взятием пробы на
прозрачность. Если реакция смолообразования не закончена и
в смеси имеются еще низкомолекулярные соединения (моно- и диме-
тилолмочевины), последние при охлаждении смеси выкристаллизо-
вываются и взятая проба быстро мутнеет. Отсутствие мути — при-
знак окончания процесса смолообразования.
Для связывания избытка формальдегида и доведения молярного
соотношения формальдегид — мочевина до 1,5:1 вводят доба-
вочное количество мочевины (25—30% от первоначального) и после
15—20 мин. перемешивания раствор нейтрализуют содой до
pH = 6,5—7,5.
В состав реакционной смеси входят также твердые органические
кислоты (0,5% щавелевой) для ускорения прессования.
Готовый раствор сливают в промежуточный сборник, откуда он
поступает в ванны пропиточных машин.
Следует указать и на ряд других типов смол, применяемых для
производства как слоистых пластиков, так и пресспорошков. Они
получаются введением в состав реакционной смеси некоторого коли-
чества тиомочевины (взамен части мочевины) и применением других
регуляторов кислотности, например, углекислого магния, аммиака
и др.
Ниже приведены некоторые рецептуры (в кг) таких смол:
1 11 111
Мочевина.................... 90 НО 120
Тиомочевина................. 40 20 —
Формалин (ЗО°/о)........... 300 300 300
Аммиак (25%)............... 4,5 4,5 4,5
MgCO3 .................... 0,13 0,13 0,13
Слоистые аминопласты получаются пропиткой бумаги или тка-
ни сиропом смолы на пропиточно-сушильных машинах.
Пропитку бумаги производят на горизонтальных пропиточно-
сушильных машинах, а пропитку ткани как на горизонтальных, так
и на вертикальных машинах.
Слоистые мочевино-формальдегидные прессматериалы.
535
Для производства бумажно-слоистых пластиков (аминобумослоя)
применяют специальные сорта наполненных окрашенных бумаг,
изготовленных из сульфитной целлюлозы, отличающихся высокой
плотностью и ограниченной впитывающей способностью.
Ввиду малой прочности влажной бумаги, она после выхода из
пропиточной ванны передвигается по сушильной части машины при
помощи специального транспортера.
Режим сушки значительно отличается от режима сушки бумаги,
пропитанной фенольными смолами. Это объясняется как различ-
ным составом раствора, так и различной прочностью применяе-
мых бумаг. Вследствие малой прочности бумаги, увлажненной
водным раствором смолы, необходима более интенсивная сушка ее
по выходе из ванны. Для этого горячий воздух, поступающий из
калориферов, частично вводят к месту входа бумаги в сушилку.
С другой стороны, конечная температура и скорость сушки должны
быть несколько меньше, чем для фенопластов, в среднем ПО—120°
при скорости 3—5 м!мин.
Необходимо указать, что режим сушки должен быть с особой
тщательностью регламентирован.
Количество вводимой в бумагу смолы регулируется концентра-
цией раствора последней, а иногда и температурой пропитки; отжим-
ные валики из-за малой прочности бумаги не применяют.
Прессование слоистых аминопластов производят на многоэтаж-
ных прессах между никелированными стальными пластинами при
140—145° и при удельном давлении 80—150 кг/см2 — для бумаж-
ных и тканевых материалов или 50—100 кг/см2 — для материалов
на основе древесного шпона.
Слоистые аминопласты на основе бумаги, ткани, дерева и стекло-
ткани имеют примерно ту же механическую прочность, что и соот-
ветствующие фенопласты, однако они значительно уступают им по
водостойкости, химической стойкости и теплостойкости. Их основ-
ное преимущество — светостойкость, окрашиваемость в яркие тона,
отсутствие запаха, даже при повышенных температурах.
Слоистые аминопласты применяют в качестве строительно-обли-
цовочного материала. Из них изготовляют также мебель, освети-
тельную арматуру и т. д.
Обычно слоистые пластики, используемые в качестве облицовоч-
ного материала, имеют только внешние тонкие слои из аминопла-
стов, промежуточный же слой состоит из слоистых фенопластов,
асбоцемента, слоистых пековых масс и др. Изготовление такого
комбинированного пластика производят при одновременном про-
цессе горячего прессования пакета бумаг, внешние слои которых
пропитаны мочевино-формальдегидными, а внутренние — феноль-
ными или другими смолами. Толщина лицевого слоя 0,1—0,5 мм.
Внутренний слой из фенопластов, будучи в меньшей мере гигро-
скопичным, в некоторой степени устраняет опасность короб-
ления.
536
Гл. ХП. Аминопласты
Из чистых мочевино-формальдегидных смол изготовляют пори-
стые пластики (аминопоропласт). По сравнению с поропластами на
основе термопластов, например полистирола (стр. 220), аминопоро-
пласт имеет преимущество в термостойкости: он не размягчается и
не плавится. Его широко применяют в качестве теплоизоляционного
промежуточного слоя в различных конструкциях.
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ ДЛЯ ЛАКОВ И КЛЕЕВ
(МОДИФИЦИРОВАННЫЕ СМОЛЫ)
Мочевино-формальдегидные смолы применяют в качестве клеев, лаков и
для пропитки ткани. В текстильной промышленности их используют для прида-
ния несминаемости хлопчатобумажным, льняным и шерстяным тканям. Для
этого волокна хлопка или ткань предварительно обрабатывают слабым раство-
ром щелочи и после тщательной промывки пропитывают водным раствором
(конденсатом) карбамидной смолы с последующим, после сушки, пропусканием
ткани через горячие каландры. После такой обработки ткань обладает несми-
наемостыо и более низкой влаго.юглощаемостью; однако такие ткани часто об-
ладают некоторой жесткостью.
В качестве склеивающего вещества мочевино-формальдегидные смолы при-
меняют либо в виде сухого порошка, растворимого в воде, либо в виде сиропо-
образного раствора. Обычно жидкие смолы для отверждения требуют нагрева;
твердые же смолы содержат твердый ускоритель, делающий возможным отвер-
ждение склейки на холоду. Можно и в жидкие клеевые смолы перед самым при-
менением вводить кислые соли пли кислоты (например молочную) и добиться
таким образом отверждения склейки на холоду. Клеевые смолы применяют для
склейки деревянных деталей, а также фарфора, металла и т. д. Эти смолы пред-
ставляют интерес для получения фанеры путем склейки шпона без горячего
прессования, однако водостойкость таких фанер уступает материалам, получен-
ным при горячем прессовании.
Мочевино-формальдегидные смолы, полученные в водных растворах ни в
одной из стадий конденсации не растворимы в спиртах. Для получения менее
гидрофильных смол и смол, растворимых в спиртах, производят эфиризацию
свободных метилольных групп спиртами. Имеются два метода получения таких
модифицированных смол: 1) сначала получают диметилолмочевину (или смесь
моно- и диметилолмочевины), которую затем эфиризируют спиртом; 2) конден-
сацию мочевины с формалином ведут в присутствии спирта.
Чаще всего для эфиризации применяют бутиловый, а также гексиловый и
октиловый спирты, глицерин и гликоль. Смолы, образующиеся при эфиризации
высшими спиртами (например октиловым и др.), способны растворяться как в
спиртах, так и в маслах.
При эфиризации метилольных групп по схеме
HOCH;NHCONHCH.OH + HOC4Ha —> HOCH2NHCONHCH2OC4H<),
каждая молекула одноатомного спирта блокирует одну функциональную (ме-
тилольную) группу; при этом, следовательно, соответственно уменьшается термо-
реактивность смолы. Очевидно, что для получения термореактивных модифи-
цированных смол необходимо, чтобы все же оставалось значительное количество
свободных метилольных групп, соответствующее средней функциональности
больше двух. Чем выше функциональность реагирующих молекул, т. е. чем
больше возможная концентрация метилольных групп, тем, следовательно, боль-
шее количество спиртовых групп можно ввести, сохраняя заданную степень
термореактивности смолы.
Технические свойства модифицированных смол определяются таким образом
соотношением между количеством эфиризированных н свободных метилольных
групп в смоле, а также характером введенных спиртовых групп.
Мочевино-формальдегидные смолы для лаков и клеев
537
По данным Г. С. Петрова, химические процессы, ведущие к получению
модифицированных мочевино-формальдегидных смол, складываются из: 1) обра-
зования в нейтральной среде диметилолмочевины; 2) эфиризации диметилолмо-
чевины бутиловым спиртом с образованием диэфира и 3) дальнейшего процесса
поликонденсации при нагревании в слабокислой среде. В условиях последнего
этапа происходит частичное отщепление молекул спирта и за счет восстановлен-
ных функциональных групп идет процесс смолообразования. Отверждение свя-
зано также с дальнейшим отшеплением молекул спирта, в результате чего обра-
зуется трехмерный полямер. Процесс образования и отверждения спиртораство-
римой смеси можно изобразить следующей схемой:
/NH2 /NH—СН2ОН
1) СО +2СН2О —> СО
'4nh2 ^nh—сн2он
/NH—СН2ОН
2) СО
^NH—СН2ОН
3)
/NH—СН2ОС4Н9
со
ZNH—СН2ОС4Н9
+ 2С'Н»?НЧ. qq
\nH—СН2ОС4Н9
-С4Н„ОН^
^NH—СН2ОС4Н9
/NH—СН2ОС4Н9
СО
\nh—СН2ОН
NH—СН2ОС4Н9
со
—> ...—N----------СН2-
/NH—СН2ОС4Н9
СО
XNH—СН2ОН
/NH—СН2ОН
+ СО —►
\мН,
NH—СН2ОС4Н9 NH—..
СО СО
— n--------СН2— N—СН2—N—...
СО
NH—СН2ОН
Малая эластичность, а также несовместимость со многими другими смо-
лами и ограниченная растворимость смол, модифицированных спиртами, заста-
вили исследователей искать новые пути модификации. Лучшая растворимость и
большая гибкость пленок были достигнуты модификацией аминопластов поли-
эфирными смолами или проведением эфиризации в присутствии реагирующих
компонентов мочевино-формальдегидной и полиэфирной конденсации (стр. 589).
С другой стороны, мочевино-формальдегидные смолы (или промежуточные про-
дукты мочевино-формальдегидной конденсации), в свою очередь, находят при-
менение в качестве модификаторов полиэфирных смол и служат, например, уско-
рителями отверждения этих смол.
Для этой цели, например, приготовляют сначала полиэфирную смолу, кото-
рую затем нагревают с некоторым количеством диметилолмочевины в присут-
ствии бутилового спирта или моноэтилового эфира гликоля. Полученные таким
путем лаки, содержащие, например, 80% полиэфирной и 20% карбамидной
смолы, образуют эластичные твердые пленки, способные отверждаться в течение
30 мин. при 150°.
Полиэфиры из себациновой и адипиновой кислот с гликолями могут служить
в качестве пластификаторов для мочевино-формальдегидных смол. Смолы, моди-
фицированные октиловым спиртом, легко совмещаются с полиэфирными смолами,
модифицированными высыхающими маслами, а также с эфирами канифоли и
высыхающими маслами.
538
Гл. XII. Аминопласты
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ МЕЛАМИНО-ФОРМАЛЬДЕГИДНЫХ
СМОЛ
Значительным достижением в области аминопластов явилось
производство и применение смол и пластиков на основе продуктов
поликонденсации меламина с формальдегидом.
Меламиновая конденсация, в отличие от мочевинной, характери-
зуется большей гидрофильностью первоначальных продуктов, что
обусловлено более высокой концентрацией полярных метилольных
групп в меламине.
Большая гидрофильность смолообразных продуктов конденсации
устраняет опасность преждевременной желатинизации и делает про-
цесс конденсации и сушки менее чувствительным к изменению
температуры и pH среды. Это позволяет вести процесс конденсации
и обезвоживания смолы в более широком интервале температур и
значений pH.
С другой стороны, большая функциональность меламиновых
продуктов конденсации определяет большую концентрацию валент-
ных связей в пространственной макромолекуле и, следовательно,
бблыпую гидрофобность и прочность отвержденного пластика
(в стадии С).
Соотношение компонентов в рецептурах меламиновых смол
можно варьировать в значительно более широких пределах, чем б
мочевинных.
При получении смол для пластмасс чаще всего берут 3 моля
формальдегида на 1 моль меламина, для модифицированных лако-
вых смол (стр. 544) — от 6 до 12 молей формальдегида, получая
при этом изолируемые высокометилольные меламины (тетра-, пента-
и гексаметилолмеламины). Реакцию ведут в нейтральной, слабокис-
лой и слабощелочной среде.
В нейтральной и слабощелочной среде (pH = 7 — 7,5) обра-
зуются устойчивые метилольные производные, а в слабокислой
(pH = 5,5 — 6,5) происходит поликонденсация метилольных произ-
водных с образованием смолообразных продуктов.
Существенное значение имеет температура реакции. До 40—50е
меламин плохо растворим в формалине и реакция протекает гетеро-
генно и сравнительно медленно. Выше 60° меламин легко растворим,
и реакция резко ускоряется с температурой.
Обычно реакцию ведут в пределах 70—100° от 20 мин. до 2 час.,
при pH = 5,5 — 6,5. На количество присоединенного формальдегида
не оказывает влияние pH среды. Конечное содержание формальде-
гида определяется соотношением формальдегида к меламину в реак-
ционной среде. Так, для получения пентаметилолмеламина вводят
8 молей, а для получения гексаметилолмелаимина — 12 молей форм-
альдегида на 1 моль меламина. Соответственно значительные
количества формальдегида остаются не связанными. От pH среды
Пластмассы на основе меламано-формалъдегидных смол
53$
зависит не скорость связывания формальдегида, а скорость
дальнейшей реакции поликонденсации метилольных производных:
при pH > 7 она мала и метилолмеламины сравнительно устойчивы,
при pH < 7 скорость велика и образующиеся метилольные произ-
водные легко реагируют и образуют смолообразные высокомолеку-
лярные продукты. Поэтому кинетика смолообразования (а не только
связывания формальдегида) характеризуется как температурой,
соотношением компонентов, так и pH среды.
Для изучения кинетики образования метилольных производных
определяли скорость связывания формальдегида при различных
температурах и pH среды. Было установлено (В. С. Киселев и
М. Ф. Сорокин), что при отношении формальдегида к меламину
6 : 1 реакция близка ко второму порядку; константа скорости реак-
ции в широких пределах мало зависит от pH среды.
Однако при уменьшении отношения формальдегид — меламин
константы скорости начинают отличаться, и при отношении 3 : 1
реакция имеет .мономолекулярный характер (время половинного
превращения не зависит от начальной концентрации). Было также
установлено, что энергия активации для реакций присоединения
меламина к формальдегиду остается постоянной в широких пре-
делах pH и, следовательно, реакция не катализуется водородными
ионами. Однако водородные ионы весьма сильно катализируют
реакцию взаимодействия метилольных производных, приводящую
к образованию смолообразных продуктов.
Присоединение меламина к формальдегиду идет с различной
скоростью, в зависимости от степени замещения. Образование три-
метилольного производного протекает с большой скоростью, реак-
ция практически необратима (константа скорости прямой реакции
в десятки раз больше обратной) и сопровождается выделением
тепла; дальнейшее же взаимодействие с формальдегидом идет мед-
ленно, реакция обратима (константа скорости обратной реакции в не-
сколько раз больше константы скорости прямой реакции) и проте-
кает с поглощением тепла. Поэтому она доходит до конца (с обра-
зованием гексметилолмеламина) лишь при избытке формаль-
дегида и при высоких температурах.
Кинетику процесса реакции при отношении 3 моля формаль-
дегида на 1 моль меламина изучали А. А. Ваншейдт и 3. К. Нау-
мова, определяя при разных температурах изменение концентрации
формальдегида (рис. 198). Было показано, что если реакцию про-
водить при 90°, то через 3—5 мин. нагревания раствор уже не мут-
неет при охлаждении; однако, если нагревание продолжают дольше,
то раствор снова начинает мутнеть. С другой стороны, если реак-
цию проводить при более низкой температуре (50°), то раствор
с самого начала мутнеет при охлаждении и остается мутным даже
после длительного нагревания.
Это объясняется тем, что при низких температурах образуются
кристаллические метилольные производные меламина, выпадающие
-540
Гл. ХИ. Аминопласты
при охлаждении из раствора, даже если реакция продолжалась
более длительное время. С другой стороны, при более высоких тем-
пературах с самого начала образуются более высокомолекулярные,
некристаллизующиеся, но еще водорастворимые соединения, кото-
рые, однако, при дальнейшем нагревании переходят в смолообраз-
ные, гидрофобные продукты, выпадающие из раствора при
охлаждении.
Для получения конденсата, годного для пропитки волокнистого
наполнителя, оптимальным является проведение реакции при 70°
Время нагревания, мин.
Рис. 198. Зависимость скорости свя-
зывания формальдегида при взаимо-
действии его с меламином при раз-
личных температурах.
в течение 30—40 мин. (при pH —
= 5,5—6,5). В этих условиях полу-
чаются некристаллизующиеся при
охлаждении конденсаты, причем
процесс конденсации еще не дохо-
дит до желатинизации реакцион-
ной смеси и образования водоне-
растворимых продуктов.
Для нейтрализации техниче-
ского формалина (pH = 2,8—3,5)
и получения слабокислой реакции
(pH = 5,5—6,5) в реакционную
смесь вводят едкий натр или
уротропин; едкий натр в боль-
шей степени замедляет процесс
конденсации, чем уротропин (при
равном pH).
Так же, как и при конденсации
мочевино-формальдегидных смол,
введение твердых органических
кислот ускоряет отверждение смолы при горячем прессовании.
Оптимальной является, например, добавка 0,3% щавелевой кис-
лоты, тогда как добавление 1 % этой кислоты уже значительно сни-
жает текучесть пресспорошка.
Меламино-формальдегидные пресспорошки получают как с цел-
люлозным, так и с древесным и минеральным наполнителями.
Технологический процесс производства меламино-формальдегид-
ных пресспорошков складывается из следующих операций: 1) варки
водного конденсата смолы; 2) смешения конденсата с наполните-
лем — целлюлозой или древесной мукой, ускорителем и красителем;
3) сушки массы; 4) вальцевания; 5) размола, просева и укрупнения
партий.
Производство меламиновых прессматериалов может быть офор-
млено по схеме, идентичной с механизированными и в значительной
степени непрерывными процессами производства аминопластов
(рис. 197, стр. 531).
В реактор из весового мерника загружают формалин из расчета
3 моля формальдегида на 1 моль меламина, после чего вно-
сят 0,3—0,5% щавелевой кислоты и уротропин, с доведением pH
Пластмассы на основе меламино-формальдегидных смол 545-
среды до 5,5—6,5. После перемешивания в течение 15—20 мин. через-
весовой бункер вводят меламин. Затем включают обратный конден-
сатор, в рубашку реактора пускают пар и реакцию проводят при
70° в течение 35—45 мин. до получения немутнеющего при охла-
ждении раствора. Полученный раствор сливают в промежуточный
сборник и через нутч-фильтр центробежным насосом подают в ве-
совой мерник. Из мерника раствор поступает в мешатель, куда
загружают также наполнитель, краситель, пигменты и стеарат
кальция.
Количество наполнителя по отношению к связующему обычно
варьируют в следующих пределах: 60—80% для целлюлозы..
80—100% для древесной муки и 120—150% для минерального на-
полнителя, считая на меламин.
Смешение производят при обычной температуре в течение 60—
90 мин., после чего массу выгружают из мешателя в бункер, откуда
ленточным транспортером ее подают в сушилку непрерывного дей-
ствия (стр. 531), а из сушилки шнеком — в бункер с питателем,
расположенным над вальцами. Ввиду дополнительного процесса
горячего вальцевания массу сушат лишь до 5—7% остаточной
влажности. 1, „1й
Вальцевание массы производят при различных режимах в зави-
симости от характера наполнителя. Оно преследует цель улучшить
однородность и пропитку материала и уменьшить удельный объем
пресспорошка. При применении древесной муки массу вальцуют
при 80—90° в течение 2—3 мин. Вальцованные листы транспортером
подаются на дробильные вальцы, после чего по обычной, уже-
описанной схеме (стр. 531) их подвергают непрерывному процессу
измельчения, просева и т. д.
Наряду с чистыми меламино-формальдегидными прессматериа-
лами производят прессматериалы на основе комплексной мочевино-
меламиновой конденсации с формальдегидом. Обычно берут соот-
ношения мочевины и меламина 1 : 1 или 1 : 2. Так, применяют сле-
дующую рецептуру (в вес. ч.):
Меламин................126
Мочевина.............. 60
Формалин (30%).........350
Прессматериалы с целлюлозным наполнителем на основе этих,
смол используют для изготовления посуды, стойкой к горячим жид-
костям, что, как было указано, невозможно при применении моче-
вино-формальдегидных пластиков.
Меламиновые пресскомпозиций прессуют при 145—165°, при-
чем скорость их прессования примерно та же, что и для резольных
пресспорошков, т. е. 40—60 сек. на 1 мм толщины изделия. Обычно
для прессования применяют подогретые при 100—110° в течение
5 мин. таблетки, которые прессуют при удельном давлении 200—-
—350 кг]см2.
.542
Гл. ХГ. Аминопласты
Меламино-формальдегидные пресспорошки отличаются высо-
кими физико-механическими, диэлектрическими и химическими
свойствами, значительно превосходящими свойства мочевино-фор-
мальдегидных прессматериалов. По многим показателям меламино-
формальдегидные пресспорошки превосходят резольные и новолач-
ные пресскомпозиции. Их значительным преимуществом является
высокая водостойкость, теплостойкость, прочность и стойкость к дей-
ствию электрических разрядов (дугостойкость).
Свойства меламиновых пресскомпозиций в сильной степени
определяются характером наполнителя. Материалы с высокой тер-
мостойкостью (250°), теплостойкостью и стойкостью по отношению
к действию электрических разрядов получаются при применении спе-
циальных минеральных наполнителей (слюды, дисперсного кремне-
зема и др.), иногда в смеси с целлюлозным волокном или с асбестом.
В табл. 61 приведены сравнительные показатели свойств мела-
миновых, мочевинных и фенольных прессматериалов на основе дре-
весной целлюлозы.
ТАБЛИЦА 61
Свойства прессматериалов на основе целлюлозы и различных смол
Свойства Смола
мочевино- формальде- гидная меламино- формальде- гидная фенол о-форм- альдегидная резол)
Возможность окрашивания Неограни- Неограни- Ограни-
ценная ценная ценная
Температура прессования (чаще всего применяемая), °C 145—150 150—170 160-180
Время прессования, сек!мм 60—90 45—60 45—60
Родопоглощение при 20° за 24 часа, % 0,75—2,0 0,3-0,6 0,4—1,0
'Го же за 7 суток, лгг/100 см2 800 270 200—750
То же в кипящей воде за 10 мин., % • 3,4 0,4 0,4—1,0
Дугостойкость Хорошая Хорошая Плохая
Теплостойкость по Мартенсу, °C . . . 120 135—160 125—150
Предел прочности на разрыв, кг]см'2 400—600 500—600 400—600
Предел прочности на изгиб, кг/см2 . . 600—700 800—900 700—800
Уд. ударная вязкость, кг -см/см2 . . . 6—10 6—10 6—10
Диэлектрическая постоянная при 106 гц 6,5—7,5 6,5—7,5 4,5—6
Уд. поверхностное электрическое со- противление, 2 ’ Уд. объемное электрическое сопроти- вление, Q-см Ю»—10’0 10’0—5-104 1010—10”
5-10»—5-10’0 10’1—5 • Ю’з 5-Ю’о-о-ЮП
Пробивное электрическое сопротивле- ние, кв/мм 10—15 12—18 9—18
Обладая высокими техническими показателями, меламиновые
пресскомпозиции бесцветны, светостойки, нетоксичны и хорошо
«окрашиваются.
Пластмассы на основ? меламино-формальдегидных смол
543
Доступность сырьевой базы и высокие технические показатели
создают 'благоприятные предпосылки для развития производства
этих пластиков, главным образом за счет мочевино-формальдегид-
ных, а также феноло-формальдегидных.
Слоистые меламино-формальдегидные пластики получают пу-
тем пропитки ‘бумаги или ткани водными конденсатами («сиро-
пами») смол. Пропитку обычно производят на горизонтальных про-
питочносушильных агрегатах.
Конструкция машин, условия транспортировки бумаги в сушиль-
ной части машины, подача горячего воздуха и тепловой обмен в су-
шильной части, скорость прохождения бумаги должны соответство-
вать условиям пропитки и сушки слоистых мочевино-формальдегид-
ных пластиков.
Для получения слоистых пластиков на основе стеклоткани поль-
зуются как обычными методами пропитки на пропиточно-сушильных
агрегатах, так и нанесением смолы на стеклоткань путем распыле-
ния водных растворов смол или другими методами поверхностной
обработки. В этом случае целесообразно исходить из порошков су-
хой водорастворимой смолы, содержащей твердый ускоритель (ща-
велевую кислоту). Такие порошки имеют преимущество в стабиль-
ности перед обычно применяемыми «сиропами».
Водные растворы порошков содержат ~50% смолы, имеют
pH = 5,5—6,0 и сравнительно невысокую вязкость (~50—100 сп).
Распыление производят сжатым воздухом через ряд распылитель-
ных аппаратов, наносящих на стеклоткань поверхностный слой
смолы нужной толщины. Затем непрерывные полотна ткани, сматы-
ваемые из рулона, проходят различные зоны горизонтальных су-
шильных камер, где при 120—140° происходит сушка смолы.
Слоистые меламиновые пластики, в особенности пластики на
основе стеклоткани и стекловолокна, приобретают из года в год все
большее техническое значение. Изготовляют также слоистые стекло-
материалы на основе смесей меламиновых и фенольных смол, а так-
же меламиновых смол с рядом полимеризационных смол, в част-
ности с производными поливинилового спирта. Эти материалы, об-
ладающие большой прочностью, водо- и теплостойкостью, приме-
няют для производства крупных изделий, изготовляемых методом
прессования при низких давлениях (при помощи резинового мешка,
стр. 346).
Слоистые меламиновые пластики на основе древесного шпона,
целлюлозной ткани и бумаги применяют для производства облицо-
вочных материалов, строительно-архитектурных деталей и т. д.
В сильноточной электротехнике из меламиновых прессматериалов
на основе стеклоткани, а также на основе обычных бумаг изгото-
вляют изоляцию, стойкую по отношению к действию электрических
разрядов.
Лаковые (модифицированные) смолы на основе меламина вследствие их
прозрачности, термостойкости и хороших диэлектрических свойств, широко при-
544
Гл. XII. Аминопласты
Рис. 199. Зависимость прочности бу-
маги от содержания в ней мелами-
новой смолы.
1—прочность в сухом состоянии; 2—то же,
в мокром состоянии.
меняют в электротехнической и в других отраслях промышленности. Модифици -
рованные смолы получают эфиризацией метилольных групп, имеющихся в смоле,,
спиртами (бутиловый, октиловый), а также путем сочетания с полиэфирными
смолами, модифицированными высыхающими маслами.
Возможность образования высокометилольных производных меламина (пента-
и гексаметилолмеламина) делает меламиновые продукты конденсации особенно
ценными для модификации и позволяет
получать спирторастворимые, а в некото-
рых случаях и маслорастворимые смолы
высокой термореактивности.
Методы получения модифицирован-
ных меламиновых смол изучались
В. С. Киселевым. Он нашел, что опти-
мальные условия могут быть достигнуты
при эфиризации пентаметилолмеламинов
бутиловым спиртом.
Процесс состоит из следующих эта-
пов: 1) приготовления пентаметилолмел-
амина; 2) эфиризации метилолмеламина
бутанолом; 3) отделения смолы от вод-
ного слоя; 4) промывки и сушки смолы.
В реактор из нержавеющей стали
загружают формалин, вводят раствор
едкого натра до pH = 7—7,5, пропу-
скают пар в рубашку, включают обрат-
ный холодильник и доводят температуру
смесн -до 80°. Затем при перемешива-
нии загружают постепенно меламин из-
расчета 1 моль меламина на 8 молей
формальдегида.
Получение метилолмеламинов считают законченным, когда в реакцию войдут
60% всего количества формальдегида.
После получения метилолмеламинов в реактор вводят бутанол (из расчета
4—8 молей иа 1 моль меламина) с растворенным в нем фталевым ангидридом
или жирными кислотами в качестве катализаторов (3—4% от веса меламина).
Температуру реакции поднимают до 85—90°. В этих условиях протекает процесс
смолообразования в спиртовом слое, куда диффундируют метилолмеламины.
Реакция продолжается до расслоения. После этого водный слой сливают, спирто-
смоляной слой заливают горячей водой, перемешивают, отделяют воду и сливают.
Промытая смола должна иметь кислотное число не более 2—5. Затем вклю-
чают вакуум и сушат смолу до возможно полного удаления воды. Конец сушки
определяют по прекращению помутнения пробы дестиллата с ксилолом.
Реакция смолообразования идет в спиртовой среде лишь в том случае, если
в ней растворены кислоты, нерастворимые в воде; чем больше концентрация кис-
лот, тем быстрее идет реакция эфиризации и смолообразования. В зависимости
от отношения спирта к меламину получают смолы с различной термореактив-
ностью: чем больше спирта входит в реакцию, тем меньше термореактивность,
но тем выше растворимость смолы и гибкость пленки.
Модификацию смолы можно производить и многоатомными спиртами — гли-
церином, пентаэритритом и др. В этом случае образуются смолы, которые в даль-
нейшем могут взаимодействовать с многоосиовными кислотами, в частности,
с адипиновой и себацииовой кислотами в смеси с жирными кислотами. При этом-
протекают реакции переэфиризации и поликонденсации полиэфирного характера,
ведущие к получению эластичных и твердых смол.
Так же, как н мочевинные, меламиновые смолы применяют как пропитываю-
щие и клеящие композиции. В качестве клея наряду с меламиновыми смолам®
применяют также продукты комплексной конденсации формальдегида с мелами-
ном и мочевиной, а также с фенолами.
Пластмассы на основе анилино-формальдегидных смол
545
Меламиновые смолы используют для получения водостойких бу-
маг, в частности для повышения их прочности во влажном со-
стоянии. Для этого меламиновые смолы вводят в виде водных дис-
персий в состав бумажной массы и в дальнейшем перерабатывают
обычными для технологии бумаги методами; к целлюлозе, напри-
мер, добавляют от 4 до 8% меламиновой смолы. Чем больше содер-
жание смолы, тем выше прочность бумаги во влажном состоянии
(рис. 199).
Пластические массы на основе анилино-формальдегидных смол
(Анилинопласты)
При взаимодействии анилина с формальдегидом в зависимости
от pH среды получаются смолообразные продукты с различной
структурой и различными свойствами. Некоторые из этих смол по-
лучили большое техническое значение и привлекают все в большей
степени внимание исследователей. Работы последнего времени,
в частности Г. С. Петрова, А. К. Андрианова, И. П. Лосева и др.,
позволили выяснить основные закономерности синтеза и строение
анилино-формальдегидных смол.
При взаимодействии анилина с формальдегидом в щелочной
среде образуются только растворимые низкомолекулярные, не смо-
листые продукты, состоящие, согласно Дроздову, из метиленди-
фениламина:
C6H6NFL
2C6H5NH.,+ СН2О —> >сн2 + н2о,
c6h5nh/
который перегруппировывается в диаминодифенилметан:
N Н2—СВН4—СНс—С6Н4—N Н2.
При взаимодействии анилина с формальдегидом в эквимолеку-
лярных соотношениях в нейтральной или слабокислой среде обра-
зуется метиленанилин:
C6H5NH2 + CH2O —> C6H6N=CH,4-I-rO.
Эта реакция, вероятно, идет через образование метилолани-
лина:
C6H5NH£ 4-СН2О —> CbH5NHCH2OH —> C6H5N=CH2+H2O.
Метиленанилин, а также метилоланилин в чистом виде не вы-
делены. Получающийся белый кристаллический порошок с т. плавл.
143° представляет собой тример метиленанилина, которому припи-
35 Зак. 1269. Э. И. Бзрг
546
Гл. ХП. Анилинопласты
сывают гетероциклическое строение и который известен под назва-
нием — ангидроформальдегиданилин:
СвН5
zn—сн2
н,с ^N-CeH5.
XN—СН2
С6Н3
При нагревании с кислотами ангидроформальдегиданилин обра-
зует низкоплавкие смолы желто-красного цвета, которые являются
полимерами метиленанилина и которые, повидимому, имеют сле-
дующую структуру:
x(CeHsN=CH2)3 —> .N—СН2— N—СН2—N— СН2— ...
Доказательством, что в основе этих продуктов лежит метилен-
анилин, служит реакция восстановления, приводящая к образова-
нию монометиланилина:
C6H5N = CH2 —C6H5NHCH3.
Характер и свойства таких смол до известной степени зависят
от характера и количества применяемых органических кислот, тем-
пературы и времени нагревания. Эти продукты в общем напоминают
новолачные смолы, но они значительно менее полярны, обладают
высокими диэлектрическими свойствами и щелочестойкостью. Од-
нако, вследствие хрупкости и низкой температуры плавления, они
не нашли применения для производства пластмасс.
Особый тип анилиновых смол был получен К. А. Андриановым с сотрудни-
ками при конденсации алкилзамещеиных анилина с формальдегидом. Необходи-
мые при этом алкилзамещенные анилина образуются конденсацией анилина
с алкилхлоридами.
Получаемые смолы дают эластичные растяжимые пленки. При этом, чем
длиннее цепочка алкильного радикала, тем эластичнее смола. Еще более гибкие
смолы получаются при конденсации с формальдегидом анилидов насыщенных
и ненасыщенных жирных кислот.
Для практического осуществления процесса исходят из ангидроформальде-
гиданилина, который при 230—240° подвергают конденсации с жирными кис-
лотами. *
Реакцию взаимодействия метиленанилина с жирными кислотами можно пред-
ставить следующим уравнением:
N=CH, NHCOR
А " А
। | 4-RCOOH —► ! | где R — радикал жирной кислоты.
СН2ОН
Пластмассы на основе анилино-формалъдегидных смол 547
Метиленанилин присоединяет жирную кислоту по месту аминогруппы, а ме-
тиленовая группа мигрирует при этом в параположение, образуй параметилоль-
ное производное, которое способно к дальнейшим процессам конденсации, пови-
днмому, по следующей схеме:
2
NHCOR
NHCOR
_ I
RCONH—/ \—СН2—
Y
СН2ОН
В результате процессов поликонденсации и полимеризации за счет ненасы-
щенных радикалов жирных кислот образуются эластичные, гибкие, твердые и
неплавкие смолы. Эластичность и гибкость смол объясняется главным образом
введением радикала жирных кислот в макромолекулу смолы.
Эти смолы отличаются также высокими диэлектрическими свойствами н
водостойкостью. Лаки на основе этих смол не нуждаются в пластификаторах,
так как наличие радикала жирной кислоты обусловливает «внутреннюю пласти-
фикацию», гибкость и эластичность пленки. Смолы с высоким содержанием жир-
ных кислот растворимы в бензоле, уайт-спирите, в дихлорэтане и других непо-
лярных растворителях. Они могут быть применены в качестве электроизоляцион-
ных лаков и связующего для производства гибких слоистых пластиков.
При реакции анилина с избытком формальдегида (~ 1,5 молей
на 1 моль анилина) в сильнокислой среде (pH < 4) образуются
•высокомолекулярные смолы линейной структуры, обладающие ха-
рактерным комплексом физико-механических свойств: высокой тем-
пературой размягчения, прочностью и способностью к своеобраз-
ному процессу отверждения, напоминающему переход резолов в
резиты.
Реакция взаимодействия анилина с формальдегидом в сильнокис-
лой среде проходит через промежуточное образование и-аминобензи-
лового спирта, переходящего в ангидроаминобензиловый спирт.
л-аминобензиловый
спирт
(т. плавл. 65°)
ангидроаминобензиловый
спирт
(т. плавл. 214—216°)
При нагревании ангидроаминобензиловый спирт образует ли:
нейный полимер следующей структуры:
^>-СН2- NH-/ \-CH2-NH-<^ )-CH2-NH-/-^>-СН2-...
Линейные полимеры получаются в результате присоединения ме-
тиленовых групп к NH-группам, что определяет основную нераз-
ветвленную группировку —CH2NH—... и положение бензольных
колец не в боковой, а в основной цепи.
35*
548
Гл. XII. Анилинопласты
При избытке формальдегида в 2 и более моля на 1 моль ани-
лина в сильнокислой среде формальдегид реагирует с водородами
бензольного кольца, образуя с метилольными группами производные
не только в пара-, но и в ортоположении.
NH2
—СН2ОН
СН2ОН
Наличие небольшого числа диметилольных соединений приводит
к «сшивке» линейных цепей с образованием пространственного по-
лимера следующей структуры:
Такие неплавкие, но размягчающиеся и нерастворимые полимеры
были впервые получены А. М. Настюковым, когда он применял
большой избыток формальдегида.
Процесс поликонденсации протекает в две фазы: в первой фазе
образуются линейные, растворимые и плавкие полимеры, содержа-
щие некоторое количество свободных метилольных групп; во вто-
рой — при длительном нагревании и высоких температурах мед-
ленно вступают в реакцию боковые метилольные группы, образуя
пространственную «сшивку» цепей. Этот процесс отверждения про-
исходит значительно медленнее, чем отверждение резолов и, пови-
димому, дальше стадии В не идет.
Техника горячего прессования таких «сетчатых» — неплавких,
но размягчающихся полимеров, содержащих однако и некоторое
количество свободных линейных молекул, в значительной степени
основана на использовании как пластичных, так и высокоэластиче-
ских свойств полимера. При этом удается получать изделия
с практически стабильной формой и с удовлетворительными свой-
ствами. Сохранение формы таких изделий наблюдается, однако,
лишь до 80—90°, так как выше этих температур релаксация приво-
дит к быстрой деформации.
Пластмассы на основе анилино-формальдегидных смол
549
Линейные полимеры анилино-формальдегидной конденсации
(анилинопласты типа «совенит») получают при конденсации 1 моля
анилина с 1,5 молей формальдегида в присутствии избытка соля-
ной кислоты по следующей схеме (рис. 200).
В реактор 1 из кислотоупорной стали, снабженный якорной
мешалкой, рубашкой для охлаждения смеси и обратным конденса-
тором, загружают, из мерника анилин (100 ч.); при энергичном пере-
Рис. 200. Схема получения анилино-формальдегидной
смолы.
/ — реактор; 2—смеситель; 3— нутч-фильтр; 4—вакуум-сушилка.
мешивании и охлаждении (путем пуска воды в рубашку) реактора
медленно вносят соляную кислоту (уд. вес 1,28) примерно в таком
же количестве, как и анилин. Для снижения теплового эффекта в
реактор вводят также 300—500 ч. воды. После тщательного переме-
шивания при 30—40° медленно из мерника загружают формалин
(~120 ч., 37%). Реакцию продолжают при указанной температуре
в течение 60—90 мин. Температуру регулируют пуском воды в ру-
башку реактора. По окончании реакции смолу выгружают в смеси-
тель 2, где нейтрализуют кислоту 25 % раствором щелочи. При этом
смола выделяется в ваде дисперсного тонкого порошка, который
в дальнейшем отделяют на нутч-фильтре 3. Смолу отфильтровы-
вают и тщательно промывают водой от хлористого натрия и следов
кислоты. Лучшие результаты, повидимому, могут быть достигнуты,
если Отмывку производить при размоле смолы в шаровой мельнице.
После промывки смола поступает в. стационарную вакуум-су-
шилку 4, где ее сушат при 60—70° в вакууме (600—700 мм), затем
измельчают в шаровых мельницах и просеивают через сито в
900 огв/см2. Анилинопласт перерабатывают в виде чистых смол без
наполнителей.
550
Гл. XII. Анилинопласты
Порошок смолы представляет собой готовый прессматериал. Его
прессуют без наполнителя в горячих прессформах при 165—185° и
при давлении от 300 до 800 к.г]см2. Порошок не отличается хорошей
текучестью и не может быть переработан методом литья под давле-
нием. Переработка же методом горячего прессования требует по-
следующего охлаждения прессформы, так как смола при темпера-
туре свыше 90—100° находится в высокоэластичном состоянии.
Частичная замена анилина толуидином по некоторым данным на-
столько улучшает текучесть, что позволяет перерабатывать смолу
методом литья под давлением. Анилинопласты (не содержащие на-
полнителя) характеризуются следующими показателями:
Уд. вес, г^С-М’.............................. 1,20—1,2 а
Предел прочности на изгиб, кг/см?............ 1000—1200
Уд. ударная вязкость, кг • см/см*............ 15—25
Уд. ударная вязкость с надрезом, кг-см/см^... 3—5
Теплостойкость по Мартенсу, °C.................. 90—110
Водостойкость (привес в воде за 24 часа при 20°), и/о 0,1—0,15
Электрическая прочность, кв/лм............... 15—30
Уд. объемное электрическое сопротивление, ’-2-с. и . . Ю13—5-1014
Диэлектрическая постоянная при 60 гц......... 3—4
Тангенс угла диэлектрических потерь при 60 гц . . . 0,002—0,01
То же, при 10й гц .................. 0,0015—0,006
Прессовочные смолы — анилинолиты отличаются прочностью, водостойкостью
и хорошими диэлектрическими свойствами. Эти свойства обусловлены линей-
ной структурой макромолекулы, не содержащей боковых групп в цепи и поляр-
ных групп в бензольном кольце.
Существенный недостаток этих смол — низкая, по сравнению с фенопластами,
теплостойкость (~90°) и малая эффективность их переработки (в основном
только горячим прессованием, при этом требуется последующее охлаждение).
Анилиновые смолы применяют также для изготовления бумажно-слоистых
пластмасс. Последние получают рольно-суспензионным методом (стр. 492), т. е.
пропиткой в ролле бумажной массы водными дисперсиями (суспензией) анилино-
вых смол, с последующим формованием листов на папп-машине или на бумажной
машине. Полученные таким образом бумажно-целлюлозные листы с содержанием
45—55% смолы прессуют при 160—170° и при удельном давлении 100—150 кг!см?.
Анилинобумослой представляет собой прочный и гибкий пластик, отличаю-
щийся высокими диэлектрическими свойствами; по сравнению с слоистыми фено-
пластами он имеет преимущество в дугостойкости, однако уступает им по
теплостойкости.
Слоистые бумажноанилиновые пластики (анилинобумослой) характери-
зуются следующими показателями:
Уд. вес, г/см3.......................................... 1,4
Предел прочности на изгиб, кг-см-.................... 1300—1800
Уд. ударная вязкость, кг-слг/слг3........................ 25
Теплостойкость по Мартенсу, °C...................... 115—125
Уд. объемное электрическое сопротивление, (->-см . ,5-Ю12—1014
Электрическая прочность, кв/мм........................15 (при 90°)
ГЛАВА Х!П
ИОНООБМЕННЫЕ СМОЛЫ*
Применение синтетических смол в качестве ионообменных ма-
териалов является новой, чрезвычайно своеобразной и многообе-
щающей областью. По сравнению с другими ионообменными мате-
риалами синтетические смолы характеризуются особенно ценными
свойствами.
Под ионообменными материалами — ионитами (ионообменни-
ками, обменно-ионными сорбентами) подразумевают твердые, прак-
тически нерастворимые в воде и в других растворителях вещества,
минеральные или органические, способные обменивать содержа-
щиеся в них катионы или анионы на различные другие катионы или
анионы, присутствующие в растворе, с которым иониты приводятся
в соприкосновение.
Первой по времени областью применения ионитов было умяг-
чение (пермутирование) котловой воды. В настоящее время ион-
ный обмен далеко перешагнул за первоначальные рамки применен
ния и используется в самых разнообразных областях промышленной
практики и исследовательских работ, связанных с удалением, выде-
лением и концентрированием различных ионов.
Иониты делятся на два класса: катиониты и аниониты. Катио-
ниты — вещества кислотного характера, способные обменивать ионы
водорода на ионы металлов, присутствующие в растворе. Соответ-
ственно аниониты — вещества основного характера, способные
к обмену анионов. Например:
2HRI + CaSO4 CaRI2H-HSO4
или
2RnOH 4-H,SO4 R"SO4 + 2H2O,
где R1 и R11 — сложные минеральные или органические остатки,,
в данном случае играющие роль аниона или катиона.
Обмен ионов происходит в строго эквивалентных количествах.
После насыщения ионита до определенной степени или до пре-
дела теми или иными ионами производят его «регенерацию». Катио-
ниты регенерируются кислотами, аниониты — разбавленными рас-
творами едких или углекислых щелочей. После промывки ионитов
* Глава написана А. А. Васильевым.
552
Гл. ХИГ. Ионообменные смолы
водой цикл поглощения может быть начат заново и повторен та-
ким образом многократно.
Насыщенные теми или иными катионами катиониты или насы-
щенные анионами аниониты способны заменять эти ионы другими,
содержащимися в растворе, с которыми иониты приводятся в со-
прикосновение, например:
HR1-!- NaCi NaR' + HCl,
2NaRr4- CaSO4 CaR* + Na2SO4j
или
RnOH4-HCl RnC14-H,O,
2RnC14-Na2SO4 R"SO4 + 2NaCl.
Основы метода избирательной сорбции, нашедшего впоследствии
широкое применение для очистки, концентрирования и разделения
веществ и известного под названием хроматографического, были
разработаны около 50 лет назад русским ученым М. С. Цветом.
Хроматографический метод анализа, одной из разновидностей
которого является метод разделения ионов, основан на различной
сорбируемости отдельных компонентов анализируемой смеси ве-
ществ различными сорбентами из различных растворителей. При
пропускании раствора анализируемой смеси через колонку сорбента
она разделяется на отдельные компоненты, располагающиеся в виде
зон. Хроматографический метод анализа исключительно чувствите-
лен и позволяет разделять смеси чрезвычайно сложного состава,
содержащие очень близкие по химическим свойствам вещества.
Сорбционный метод разделения ионов получил плодотворное
развитие в работах Е. Н. Гапона, Б. П. Никольского, С. Е. Брес-
лера и др.
Синтез ионообменных смол рассматривается как новый этап по-
лучения и применения ионитов. Преимущества синтетических смол
по сравнению с ионитами других типов (алюмосиликатами, природ-
ными и сульфированными углями и др.) заключаются в их значи-
тельно более высокой механической прочности и химической устой-
чивости, а также в высокой обменной емкости. Чрезвычайно важно
то обстоятельство, что наряду с катионообменными появились также
и анионообменные смолы, пригодные для промышленного примене-
ния, тогда как известные ранее аниониты не имели никакого прак-
тического значения.
Первые ионообменные смолы были синтезированы в 1935 г. на
основе многоатомных фенолов и формальдегида (катиониты) и аро-
матических аминов и формальдегида (аниониты).
Ионообменные смолы должны быть нерастворимы в водных рас-
творах или в других жидких средах, с которыми они приводятся
в соприкосновение; они должны быть в достаточной мере
Ионообменные смолы
553
устойчивы к действию химических реагентов, нагреванию и обладать
хорошей механической прочностью.
Для того чтобы смола обладала способностью к ионному об-
мену, необходимо присутствие в ней активных групп, придающих
ей кислый или основной характер.
В сущности, уже обычные феноло-формальдегидные и анилино-
формальдегидные смолы в конечной стадии их образования (ста-
дия С) удовлетворяют перечисленным выше требованиям. Однако
активные группы, содержащиеся в таких смолах (фенольные
гидроксилы или ароматические аминогруппы), являются очень сла-
быми; обычно промышленные ионообменные смолы содержат более
сильные группы.
Группы, характерные для катионитов: —SO3H (в ароматиче-
ском ядре или в боковой цепи), —СООН, —ОН (фенольные).
Группы, характерные для анионитов: —NHa, =NH, sN (являю-
щиеся остатками ароматических или алифатических аминов).
Смола, содержащая сульфогруппы, обладает более сильно выра-
женными кислотными свойствами по сравнению со смолой, содер-
жащей карбоксильные группы, а тем более со смолой, содержащей
одни лишь фенольные гидроксилы. Аналогичным образом смолы,
содержащие одинаковые аминогруппы (например, =МН-группы),
обладают более сильно выраженными основными свойствами в тех
случаях, когда эти группы входят в состав алифатических, а не аро-
матических аминов.
С накоплением активных групп повышается обменная способ-
ность смол, но одновременно увеличивается их растворимость
в воде или способность к набуханию. С увеличением числа попе-
речных связей в структуре ионообменных смол, напротив, раство-
римость и способность к набуханию понижаются.
В случае синтетических смол обмен ионов совершается во всем
объеме и ионы, содержащиеся в растворе, могут свободно прони-
кать сквозь структурную решетку смолы.
Иониты обычно применяют в виде зерен размером 0,25—2,0 мм.
Чем меньше размер зерен ионита, тем быстрее совершается про-
цесс обмена, однако в тех случаях, когда ионит служит фильтрую-
щим слоем, применение зерен размером менее 0,25 мм вызывает
резкое увеличение сопротивления слоя.
По количеству извлекаемых из раствора ионов, отнесенному
к единице веса или объема ионита, судят об обменной емкости (об-
менной способности) того или иного ионита. Обменная емкость
ионитов может быть определена в статических и динамических
условиях.
В статических условиях можно производить определение как
максимальной, так и равновесной обменной емкости ионитов. О по-
следней судят по результатам титрования раствора той или иной
исходной концентрации, находившегося в продолжение определен-
ного промежутка времени в соприкосновении с ионитом до уста-
554
Гл. XIII. Ионообменные смолы
новления равновесия. Обменная емкость, определенная в таких
условиях, в значительной мере зависит от структуры ионита и ха-
рактера содержащихся в нем активных групп, а также от природы
обменивающихся ионов, концентрации раствора, соотношения
между твердой и жидкой фазами и pH среды, в которой происходит
ионный обмен. При повышении pH среды емкость катионитов воз-
растает, тогда как емкость анионитов понижается; напротив, при
понижении pH среды обменная емкость катионитов понижается,
а анионитов повышается.
У ионитов, содержащих какой-нибудь определенный вид актив-
ных групп, обменная емкость мало зависит от значения pH среды
в том случае, если эти иониты обладают свойствами сильных кис-
лот или оснований.
Иониты, обладающие свойствами слабых кислот или оснований,
постепенно изменяют свою обменную емкость с изменением pH
среды.
Обменная емкость ионитов, содержащих различные активные
группы, может значительно различаться для высших и низших зна-
чений pH среды.
Так, например, у сульфофенольных катионитов, содержащих
наряду с сильнокислотными сульфогруппами слабокислотные фе-
нольные гидроксилы, последние будут принимать участие в реак-
циях обмена только при высоких значениях pH; поэтому обменная
емкость таких смол при высших и низших значениях pH заметно
различается.
В ряде случаев полная обменная емкость ионита может быть
определена путем действия в течение более или менее продолжи-
тельного промежутка времени на катионит избытка щелочи, на
анионит — избытка кислоты с последующим обратным титрова-
нием.
При определении динамической обменной емкости ионит служит
фильтрующей средой, через которую с определенной скоростью про-
пускают раствор той или иной концентрации до тех пор, пока
в фильтрате не будет обнаружен при помощи того или иного спо-
соба «проскок» поглощаемого иона. Таким путем устанавливается
«рабочая» обменная емкость ионита.
Если продолжить пропускание раствора через слой ионита, то
наступит момент, когда концентрации растворов — исходного и вы-
текающего из фильтра — сравняются. Это дает возможность вы-
числить полную обменную емкость ионита.
Динамическая емкость ионита зависит от тех же факторов, что
и статическая. Кроме того, она зависит и от размера зерен ионита
и толщины его слоя, а также от скорости фильтрации раствора.
Сопоставление обменной емкости до проскока и полной обмен-
ной емкости ионита в динамических условиях дает возможность
охарактеризовать кинетику ионного обмена для определенных ви-
дов ионитов.
Катионообменные смолы
555»
Очень удобным способом определения полной обменной емкости
ионитов является способ потенциометрического титрования, позво-
ляющий судить об обменной емкости ионитов при различных значе-
ниях pH и о наличии тех или иных активных групп в составе
ионитов.
Обменная емкость ионитов может быть выражена в различных
единицах: миллиграмм-эквивалентах (мг-экв), % СаО, тонно-граду-
сах (т-град) и т. д. и отнесена либо к единице веса, либо к единице-
объема ионита. В настоящее время статическую обменную емкость
принято выражать в миллиграмм-эквивалентах на грамм сухого
ионита.
катионообменные смолы
В Советском Союзе широкое развитие изысканий синтетических
смол началось с тридцатых годов нашего столетия.
В настоящее время широко известны многие советские катионо-
обменные смолы, например: эспатит-l, катиониты марок МСФ, СБС,
КУ-2 и многие другие.
Катиониты могут содержать однотипные или разнотипные актив-
ные группы, например лишь одни 8О3Н-группы или одновременно
SO3H- и фенольные ОН-группы.
Соответственно этому различают монофункциональные и поли-
функциональные катиониты.
В зависимости от характера кислотных групп катиониты обла-
дают свойствами сильных или слабых кислот. Нолифункциональные
катиониты (например, так называемые, сульфофенольные катио-
ниты) могут объединять в себе и те и другие свойства.
При синтезе ионообменных смол либо исходят из мономерных
соединений, уже содержащих активные группы, либо вводят эти
группы в уже готовые смолы. И тот, и другой путь, в частности, при-
меняют при получении феноло-альдегидных катионитов, имеющих
большое распространение среди синтетических катионообменных
смол.
Феноло-альдегидные ионообменные смолы обычно содержат
в качестве активных групп сульфо- и иногда — карбоксильные
группы. Строение этих смол так же, как и ионитов других типов,,
освещено до настоящего времени очень мало.
По мнению ряда исследователей, продукты, получающиеся при
введении сульфогрупп в готовые феноло-альдегидные смолы, харак-
теризуются случайной, не поддающейся стандартизации структу-
рой.
Такие смолы обладают высокой набухаемостью в воде и низ-
кой химической стойкостью. Сульфирование смол в стадии С не-
избежно связано с их деструкцией; получаемые катиониты имеют
низкую емкость. При сульфировании смол в стадии А сульфогруппы
занимают о- и «-положения фенольных ядер, что является препят-
556
Гл. ХШ. Ионообменные смолы.
ствием для дальнейших процессов поликонденсации (для образо-
вания трехмерных молекул).
Обычно при получении сульфофенольных катионитов предвари-
тельно подвергают сульфированию фенол, и затем уже проводят
конденсацию полученного сульфопроизводного с формальде-
гидом.
И. П. Лосев и Е. Б. Тростянская считают, что твердые водостой-
кие продукты, обладающие стандартными свойствами, могут быть
получены только на основе п-сульфопроизводного фенола. Сказан-
ное относится также и к другим фенолам: л-крезолу, резорцину,
флороглюцину и др.
Элементарной ячейкой продукта, полученного при поликонденса-
ции п-фенолсульфокислоты и формальдегида, можно считать звено:
ОН
/X СНг-
R—\/ R
SO;H
где R—Н, ОН или СН,.
Те же авторы указывают на малую термическую стабильность
фенолсульфокислот и необходимость тщательного соблюдения тем-
пературного режима конденсации. Уже при температурах выше
80° наблюдается заметное отщепление сульфогрупп.
К сульфофенольным катионитам принадлежат советские ионо-
обменные смолы эспатит-1, МСФ, ПФСК. Эспатит-1 представляет
собой черный зернистый продукт с величиной зерен 0,3—2,0 мм,
обладающий достаточной механической прочностью. Эспатит-1 тер-
мически устойчив при температуре до 90—95° и химически стоек
к разбавленным кислотам. Его насыпной вес 0,7 т/м3; коэффициент
набухания 1,6—2,0; влажность товарного продукта 50%.
В качестве примера ниже приводится описание способа полу-
чения сульфофенольного катионита, известного под названием
катионит ПФСК. Его получают в две стадии: 1) сульфирование фе-
нола; 2) конденсация сульфопроизводных фенола с формальдеги-
дом.
1 вес. ч. фенола и 1 вес. ч. концентрированной серной кислоты
нагревают в реакторе при периодическом перемешивании при
140+ 10°. После десятичасового нагревания прибавляют еще 10%
от взятого ранее количества серной кислоты и нагревание продол-
жают еще четыре часа. По окончании сульфирования продукту не
дают выделиться в твердом виде или загустеть, а переливают в ва-
рочный аппарат, где обычно разбавляют водой (0,5 вес. ч. на
Катионообменные смолы
557'
1 вес. ч. продукта сульфирования) во избежание бурной экзотермиче-
ской реакции с формальдегидом. После полного растворения про-
дукта сульфирования в воде вводят формалин, — обычно в количе
стве, несколько большем 0,5 вес. ч. на 1 вес. ч. продукта сульфиро-
вания (т. е. несколько больше 1 моля формальдегида на 1 молы
фенола). Смесь подогревают до 70—80°; пдд этой температуре про-
текает конденсация, завершающаяся образованием геля. Гель из-
влекают и выдерживают в отдельном аппарате в течение 4—5 ча-
сов при температуре около 100°. Полученный твердый, неплавкий и
нерастворимый продукт подвергают дроблению, промыванию от из-
бытка серной кислоты, сушке при температуре не выше 50—60°,
окончательному дроблению и просеиванию для отбора фракции
0,5—2,0 мм. Готовый продукт представляет собою черный или тем-
нобурый зернистый материал, имеющий насыпной вес 0,76 т/м\,
истинный удельный вес 1,0—1,2 и влажность, обычно, 15—20%.
Рабочая обменная емкость катионита — порядка 450 мг-экв1л.
Среди катионообменных смол феноло-альдегидного типа, вы-
пускаемых за рубежом, известны, в частности, германские вофатиты,
и американские амберлиты.
Так вофатит Р содержит сульфогруппы как в бензольных ядрах,,
так и в боковых цепях. Его изготовляют в две стадии. В первой ста-
дии получают смолу при взаимодействии фенола и формальдегида
в присутствии сульфита натрия. При этом вначале идет реакция:
ОН ОН
( + СН2О + NazSOj —> -j- NaOH.
CH2SO3Na
Затем при действии избытка формальдегида получается жела-
тинообразная масса, которую подвергают термообработке, дробле-
нию и просеиванию.
Во второй стадии смолу обрабатывают концентрированной сер-
ной кислотой. При этом, очевидно, протекают дальнейшие процессы
поликонденсации и дополнительное обогащение смолы сульфогруп-
пами в бензольных ядрах.
По сравнению с другими вофатитами, вофатит Р —при сравни-
тельно невысокой обменной емкости — отличается большей устойчи-
востью по отношению к щелочным средам при повышенных темпе-
ратурах (до 90°).
Вофатит К. получают из бензальдегид-2,4-дисульфокислоты, ре-
зорцина и формальдегида также в две стадии.
В первой стадии производят конденсацию бензальдегид-2,4-ди-
.558
Гл. Х’П. Ионообменные смолы
сульфокислоты с избытком резорцина в кислом водном растворе;
при этом начальная реакция протекает по схеме:
Во второй стадии полученный продукт конденсируют с формаль-
дегидом в щелочном растворе; при этом образуется гель.
За последние годы все большее значение начинают приобретать
слабокислотные катиониты, содержащие карбоксильные группы и
обладающие способностью к избирательному извлечению ионов из
растворов.
Карбоксильные катиониты могут применяться для умягчения вод
с высокой карбонатной жесткостью.
К числу феноло-альдегидных катионообменных смол, содержа-
щих в ароматических ядрах, наряду с фенольными гидроксильными
группами, также карбоксильные группы, — принадлежит вофатит С.
Для изготовления вофатита С применяют оезорциловую (3,5 ди-
оксибензойную) кислоту, фенол и формалин. Резорциловую кислоту
и фенол конденсируют в слабощелочной среде с формальдегидом,
после чего полученный гель подвергают термообработке, дроблению,
-сушке и рассеву.
Возможно, что строение вофатита С следующее:
он
Апионообменные смолы
559
В настоящее время достигнуты определенные успехи в области
получения катионообменных смол новых типов, в том числе поли-
меризационных смол. Появились катиониты, содержащие один опре-
деленный вид активных групп (монофункциональные катиониты),
например, полимеры сульфированных углеводородов, содержащие
сульфогруппы в ароматических ядрах. Этот вид смол очень удобен
при изучении и при практическом использовании. Различные актив-
ные группы, содержащиеся в одном катионите, как это, например,
имеет место для сульфофенольных катионитов, могут обладать раз-
личной скоростью ионного обмена. Обменная емкость такого катио-
нита сильно меняется с изменением pH и т. д.
В литературе, появившейся в последние годы, имеются указания
о получении катионитов на основе сополимеров стирола, а также
метакриловой кислоты и описания их свойств. В первом случае
активными группами являются сульфогруппы, во втором — карбо-
ксильные группы. Компонентом, необходимым для образования мо-
лекул трехмерной структуры, служит дивинилбензол (стр. 50).
Сополимеризацию стирола и дивинилбензола проводят при 80°
в продолжение 18 час. Затем сополимер подвергают сульфированию
концентрированной серной кислотой при 100° в продолжение 8 час.
в присутствии 1% (от сополимера) сернокислого серебра в каче-
стве катализатора. Полистирольные катиониты (дауэкс-50 и др.)
устойчивы как к кислотам, так и к щелочам и окисляющим агентам.
По другому способу товарную метакриловую кислоту (после по-
вторной перегонки в вакууме) подвергают сополимеризации с ди-
винилбензолом (10% от основного компонента) в присутствии 1%
перекиси бензоила. Полимеризацию проводят в запаянных трубках
при 60° в продолжение 24 час. Затем полимер обрабатывают
2н. раствором едкого натра для удаления растворимых продуктов,
промывают и сушат.
К числу монофункциональных катионитов, содержащих сульфо-
группы, относятся советские ионообменные смолы СБС, КУ-2 и др.
Катионит КУ-2 обладает высокой обменной емкостью, практически
не меняющейся в широком интервале pH, и устойчивостью по от-
ношению к кислотам и щелочам даже при температурах до 100°.
АНИОНООБМЕННЫЕ СМОЛЫ
Анионообменные смолы не имеют практически пригодных анало-
гов среди неорганических или естественных органических ионитов.
Довольно продолжительное время было известно лишь незначи-
тельное число анионообменных смол. Следует отметить, что в на-
стоящее время число известных анионитов приближается к числу
известных катионитов.
Разработаны способы получения анионитов, обладающих как
слабо-, так и сильноосновными свойствами и высокой обменной
емкостью.
560
Гл. XIII. Ионообменные смолы
При получении анионообменных смол для конденсации с форм-
альдегидом могут быть применены разнообразные амины, при-
надлежащие как к ароматическому (анилин, л-фенилендпамин), так
и к жирному ряду (мочевина, меламин, гуанидин). Некоторые ис-
следователи считают, что высокоактивные анионообменные смолы
могут быть получены только в том случае, если в качестве исход-
ных соединений применяют амины с сильно выраженными основ-
ными свойствами. Однако можно считать доказанным, что актив-
ность анионитов в значительно большей степени определяется
направлением реакции конденсации аминов с альдегидами и харак-
тером расположения активных групп в структуре анионита, чем вы-
бором исходного сырья. Анилино- и мочевиноформальдегидные
аниониты обладают низкой обменной емкостью, в основном из-за
того, что в процессе поликонденсации исходные аминогруппы не
сохраняются, а в зависимости от условий конденсации образуются
смолы, обладающие структурой, характерной для вторичных или
третичных аминов:
или-
или
...— N—CH,-N—СН,—N—СН,—...
io io io
I I I
...— N—CH2—N—CH2— N—CH2—...
При переходе первичных аминов во вторичные и особенно в тре-
тичные происходит резкое понижение основных свойств аминов.
Было доказано, что, в зависимости от условий конденсации (pH
среды, количественных соотношений компонентов и т. д.), можно,
исходя из относительно слабых оснований (мочевины, меламина),,
получать аниониты, обладающие высокой обменной емкостью.
Анаонообменные смолы
561
Основные свойства некоторых аминосмол можно повысить пу-
тем обработки их галоидалкилами и сульфалкилами в щелочной
среде. В этом случае имеет место образование соединений типа чет-
вертичных аммониевых оснований.
Высокоактивные анионитовые смолы можно получать путем
полимеризации ненасыщенных четвертичных аммониевых оснований.
Анионообменные смолы, обладающие высокой обменной ем-
костью и химической устойчивостью, могут быть получены на основе
диаминов и полиаминов (иминов) жирного ряда.
В качестве примера синтеза анионообменной смолы на основе
ароматического амина и формальдегида ниже дано описание спо-
соба получения так называемой аминосмолы, синтезированной
б 1938 г. Ф. Г. Прохоровым, К- А. Янковским и Ф. А. Куткиным.
Раствор .м-фенилендиамина (8%) загружают в реактор, в который
при размешивании подают серную кислоту уд. вес. 1,83 (3 моля
на 1 моль .м-фенилендиамина). Смесь подогревают до 95°, после
чего подогрев выключают. Затем при работающей мешалке равно-
мерной струей вводят формалин (3 моля на 1 моль .м-фениленди-
амина). Через 10—12 мин. после прибавления формалина мешалку
выключают и образовавшийся гель выдерживают в аппарате в про-
должение четырех часов. Затем аппарат заполняют водой, вклю-
чают мешалку и через 3—5 минут разбитую на кусочки смолу сжа-
тым воздухом передавливают на нутч-фильтр, работающий под
вакуумом. Отмытую до слабокислой реакции смолу подвергают
сушке на деревянных противнях в сушилке при 130—150°. Сухую
смолу дробят и просеивают для отбора фракции 0,3—1,5 мм. Вы-
ход недробленой смолы 105—110% от л£-фенилендиамина. Готовая
смола представляет черный блестящий зернистый материал с на-
сыпным весом 0,65—0,75 т/л<3. Рабочая обменная емкость амино-
смолы порядка 150 мг-экв/л по НС1 и 250 мг-экв/л по H2SO4.
Вследствие малой стабильности и невысокой обменной емкости
аминосмола утратила свое значение.
В настоящее время известно значительное количество советских
анионообменных смол (марки: эспатит ТМ, МГ, ММГ-1, Н-0, ПЭ-9,
ЭДЭ-10 и многие другие), отличающихся значительным разнообра-
зием свойств, позволяющих успешно применять эти смолы в отдель-
ных, специальных случаях. Большинство упомянутых смол по своим
качествам, в частности по величине обменной емкости, значительно
лучше аминосмолы.
Ряд анионообменных смол был синтезирован на основе л-фени-
лендиамина, мочевины, гуанидина, меламина, цианамида и т. д.
Большим достижением советских исследователей в области син-
теза анионитов является получение анионообменных смол, отличаю-
щихся сильно основными свойствами. Применение таких анионитов
позволяет удалять из обессоливаемой воды не только сильно-, но и
слабодиссоциированные кислоты (H2SiO3). При эксплуатации паро-
вых котлов очень большое практическое значение имеет удаление
36 Зак. 1269. Э. И. Барг
662
Гл. XIII. Ионообменные смолы
из воды кремнекислоты. До последнего времени обескремнивание
воды путем непосредственной сорбции H2S1O3 при помощи аниони-
тов было невозможно вследствие слабоосновных свойств известных
анионообменных смол.
Из зарубежных анионообменных смол следует упомянуть вофа-
тит М, амберлит JR-4B, амберлит JRA-4W, дауэкс-1, дауэкс-2 и др.
Вофатит М получают при поликонденсации л-фенилендиамина,
полиэтилендиамина НгНСгНДЫНСгН^ННг и формальдегида. При-
готовляют смесь 5 вес. ч. воды, 4 вес. ч. л-фенилендиамина,
2,25 вес. ч. концентрированной соляной кислоты и 2,1 вес. ч.
льда. Перемешивание ведут до растворения лг-фенилендиамина.
Затем прибавляют 0,6 вес. ч. полиэтилендиамина, 1,5 вес. ч. соляной
кислоты и 1,5 вес. ч. льда. При последующем прибавлении 1,5 вес. ч.
воды температура смеси повышается до 8—11°. Тогда вводят
8,25 вес. ч. 30% формалина, охлажденного путем прибавления
1,5 вес. ч. льда; при этом температура смеси быстро поднимается
до 65°. Через несколько часов образовавшуюся смолу извлекают
из аппарата и подвергают сушке при 80—90э. Затем ее измельчают
и промывают.
К числу успехов, достигнутых за последние годы в области син-
теза ионообменных смол, следует отнести и синтез ионитов селек-
тивного действия (А. С. Смирнов, В. А. Клячко и др.). Избира-
тельное извлечение ионообменными смолами некоторых ионов из
растворов наблюдается в том случае, если в ионит при его синтезе,
кроме обычных активных групп, вводят вещества, способные к обра-
зованию комплексных соединений с ионами, подлежащими избира-
тельному извлечению.
ТАБЛИЦА 62
Значения полной обменной емкости ионитов
Катионит Полная обменная емкость (мг~экв/г) при рп Анионит Полная обменная емкость (мг-экв/г) при pH
3 5 7 7 1
Сульфоуголь 1,18 1,77 2,0 тм 0,28 4,45
Кь-4 0,16 1,08 4,3 Н-0 1,42 4,49
Эспатит I (КУ-1) . . . 2,6 2,75 2,88 ММГ-1 2,42 4,61
СБС 3,08 3,29 3,43 ПЭ-9 0,84 5,3
КУ-2 4,7 4,7 4,75 ЭДЭ-10 1,51 7,79
Из работ последнего периода в области синтеза ионито»
нужно отметить исследования А. Б. Даванкова и сотрудников,
синтезировавших неизвестные до настоящего времени амфотерные
ионообменные смолы, содержащие как кислотные, так и основные
Применение ионитов
563
группы. Амфотерные иониггы могут быть получены как из кислот,
так и из органических оснований и представляют практически инте-
рес при разделении аминокислот и амфотерных элементов.
В табл. 62 приводятся величины полной обменной емкости иони-
тов некоторых отечественных марок при различных значениях pH.
ПРИМЕНЕНИЕ ИОНИТОВ
Иониты с каждым годом находят все более широкое применение, причем не-
прерывно увеличивается область их использования в энергетической, химической,
пищевой, фармацевтической, металлургической и других отраслях промышлен-
ности. Ниже приводятся некоторые примеры.
Иониты могут быть широко использованы для извлечения металлов из при-
родных вод, из производственных растворов, из сточных вод и для разделения
металлов. Самой старой областью применения ионитов в этом направлении
является умягчение воды. Способ катионирования позволяет практически пол-
ностью удалять из воды, идущей для питания паровых котлов, катионы кальция
и магния.
В ряде случаев подлежащие извлечению металлы содержатся в растворах
в виде положительно или отрицательно заряженных комплексных ионов и могут
быть удалены при помощи катионитов или анионитов. Иногда для извлечения
металлов специально прибегают к образованию комплексных ионов. Например,
в виде комплексных анионов могут быть извлечены: серебро (из отработанных
фиксажей и промывных вод фото-кинофабрик) и золото (из сточных вод копи-
ровальных цехов).
Анионообменные смолы могут быть применены для извлечения кислот, со-
держащихся в растворах различного происхождения (уксусной и щавелевой
кислот Из сточных вод газогенераторных станций, работающих на торфе или
дровах, муравьиной кислоты из формалина и т. д.).
Ряд производственных процессов требует полного удаления как катионов,
так и анионов из перерабатываемых растворов, т. е. деминерализации (обессо-
ливания) последних. В этом случае требуется комбинированная обработка рас-
творов с применением агрегата, состоящего из катионитового и анионитового
фильтров.
Для паровых котлов высокого давления необходима вода, приближающаяся
в ряде случаев по составу к дестиллироваиной. Полная деминерализация воды
достигается последовательным фильтрованием ее через катионитовый, а затем
через аниоиитовый фильтр. Очищенная от солей вода успешно выдерживает
сравнение с дестиллироваиной водой.
В производстве свекловичного сахара можно успешно применять деминера-
лизацию диффузионного сока; таким путем исключаются операции дефекации и
сатурации и значительно повышается выход сахара, который по своему качеству
ие отличается от рафинада. Аналогичным образом можно вести удаление катио-
нов и анионов из фруктовых, овощных и виноградных соков, благодаря чему
иониты могут быть широко использованы в консервной промышленности.
Иониты применяют для разделения аминокислот методом ионообменной
хроматографии, для извлечения и разделения витаминов и т. п. веществ.
Употребляются иониты и в производстве химически чистых реактивов.
В настоящее время иониты достаточно широко применяют в аналитической
практике как в области качественного, так и количественного анализа при ана-
лизе воды, металлов и сплавов, пищевых продуктов и т. д. Достигаемая точ-
ность определений сравнима с точностью спектрографического или полярографи-
ческого методов.
Хроматографический ионообменный метод анализа позволяет ввести значи-
тельные упрощения в трудоемкие и длительные аналитические операции и пред-
ставляет возможность широкого выбора различных путей для выделения ионов
из сложных смесей.
36*
564 Гл. ХШ. Ионообменные смолы
В последние годы опубликован ряд работ по применению ионитов для исклю-
чительно тонких разделений ионов, до настоящего времени не достижимых при
помощи иных методов; так было осуществлено разделение редкоземельных элемен-
тов, извлечение и разделение продуктов распада радиоактивных катионов и т. п.
Наконец, иониты, в том числе ионообменные смолы, могут быть использованы
в качестве катализаторов, для чего их предварительно насыщают теми или иными
ионами, катализирующими процесс. Так катиониты можно использовать в каче-
стве катализаторов для реакций эфиризацин, гидролиза эфиров, дегидратации
спиртов, при инверсии сахарозы н т. д.
ГЛАВА XIV
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ ПОЛИЭФИРОВ,
ПОЛИАМИДОВ, ПОЛИУРЕТАНОВ И ПОЛИМОЧЕВИН
Полиэфирные и полиамидные смолы
(Эфиропласты и амидопласты)
Полиэфирные смолы получают поликонденсацией двухосновных
кислот и многоатомных спиртов, а полиамидные — поликонденса-
цией двухосновных кислот и диаминов, («-аминокислот, а также
ступенчатой полимеризацией лактамов аминокислот.
Основные реакции образования, строение полимеров, а также
(в значительной степени) исходное сырье и частично аппаратурное
оформление процесса производства близки для обоих типов поли-
меров. Поэтому представляется целесообразным совместно рассма-
тривать некоторые закономерности, ведущие к образованию обоих
типов смол, получивших большое промышленное значение в произ-
водстве лаков, синтетических волокон и пластмасс.
Полиэфирные смолы наиболее важны как синтетические пленко-
образователи. По объему производства они занимают второе место
среди синтетических смол.
Полиамидные смолы получили применение в качестве первых
синтетических волокон. По темпу роста производства они значи-
тельно превосходят полиэфирные. Синтезированные в промышлен-
ных условиях всего 10 лет тому назад, они в настоящее время про-
изводятся десятками тысяч тонн.
Как полиэфирные, так и полиамидные смолы получают все воз-
растающее значение для производства пластиков, прессовочных и
слоистых материалов.
ОСНОВНЫЕ ЗАКОНОМЕРНОСТИ РЕАКЦИЙ ОБРАЗОВАНИЯ
ПОЛИЭФИРНЫХ И ПОЛИАМИДНЫХ СМОЛ
В отличие от реакций поликонденсации, ведущих к образованию
карбоцепных полимеров (например реакции фенола с альдеги-
дами), которые характеризуются большим значением константы
равновесия (К ~ 10 000) и для которых установление равновесия
практически не играет роли (стр. 63), реакции поликонденсации,
ведущие к образованию полиэфирных и полиамидных смол, харак-
теризуются небольшим значением константы равновесия. Поэтому
на последние основное влияние оказывают условия, сдвигающие
равновесие в сторону образования полимера с возможно высоким
566
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
молекулярным весом. Так, процесс поликонденсации гексаметилен-
диамина с адипиновой кислотой является обратимым:
х [HjN(CHJ,NH2] + X [HOCO(CH,),COОН] ч—Г
HOCO(CH,)<CO[NH(CH3)sNHCO(CHa).CO]a;_2NH(CH2),NH,-f-(x- 1)Н.О.
Если из сферы реакции удалять образующуюся воду, равновесие
можно сдвинуть вправо.
Однако вследствие высокой вязкости и гидрофильности поли-
конденсата полное удаление воды практически недостижимо.
Нарастание длины образующейся цепи имеет ступенчатый харак-
тер, реакция протекает медленно и. практически, без заметного
выделения тепла. Скорость реакции зависит от температуры реак-
ционной среды и поэтому ее проводят при нагревании.
Рис. 201. Зависимость сте-
пени конденсации этилен-
гликоля с адипиновой кисло-
той от времени при различ-
ных температурах.
Рис. 202. Зависимость кон-
станты скорости поликон-
денсации от температуры.
Процесс поликонденсации, ведущий к образованию полиамидов
и полиэфиров, как это доказали А. И. Коган и В. В. Коршак, про-
текает по бимолекулярному механизму. Константа скорости этого
процесса зависит от молярной концентрации функциональных групп
(гидроксильных, карбоксильных и т. п.), но не от длины цепи расту-
щего полимера.
Зависимость степени поликонденсации от времени при различ-
ных температурах выражается расходящимся пучком прямых
(рис. 201). Зависимость же константы скорости реакции поликон-
денсации от температуры в области широкого интервала темпера-
тур подчиняется уравнению Аррениуса и может быть представлена
прямой (рис. 202).
Скорость реакции и степень поликонденсации, естественно, зави-
Закономерности реакций образов, полиэфирн. и полиамидных смол 567
сят от скорости удаления побочных продуктов реакции, в данном
случае воды. Чем быстрее и полнее удаляется вода, тем больше
скорость реакции, достигаемая степень ее завершенности и молеку-
лярный вес полимера.
Образующийся продукт поликонденсации на каждой стадии
реакции представляет собою полидисперсную смесь молекул различ-
ной величины, причем в состав смолы всегда входят небольшое ко-
личество исходных реагирующих веществ и побочные продукты
реакции.
Характерной закономерностью таких процессов поликонденсации
является зависимость степени полидисперсности продукта не только
от характера реагирующих молекул,
но, в значительной степени, и от их
соотношения, причем, чем больше из-
быток одного типа реагирующих мо-
лекул, тем меньше полидисперсность
(см. ниже).
Полидисперсность продукта регули-
руется также непрестанно протекаю-
щими, наряду с основными процес-
сами роста полимера, процессами его
деструкции (стр. 576). К этим по-
следним молекулы полимера тем бо-
лее чувствительны, чем выше их
степень поликонденсации. Это обстоя-
ло/?. бес в тысяча»
’ Рис. 203. Дифференциальная
кривая распределения полиами-
.. да по молекулярным весам.
Рис. 204. Дифференциальная
кривая распределения по моле-
кулярным весам сополимера
винилацетата и хлористого ви-
нила.
тельство создает внутренний механизм,
регулирующий в известных пределах
как среднюю степень полимеризации,
так и степень полидисперсности продук-
та, причем чем больше возможность
протекания процессов деструкции, тем
сильнее сказывается их нивелирующее
действие на характер полидисперсно-
сти. В итоге, когда достигается равно-
весное состояние, это приводит к воз-
можно малой степени полидисперсно-
сти. Поэтому степень полидисперсно-
сти полиэфиров и полиамидов сравни-
тельно мала.
На рис. 203 и 204 для сравнения
приведены дифференциальные кривые распределения по молекуляр-
ным весам полиамида и сополимера винилацетата и хлористого ви-
нила. Как видно из этих рисунков, полиамиды состоят преимуще-
ственно из однородных по величине молекул (что выражается резким
максимумом кривой распределения), в то время как для полимери-
зационных смол большей частью характерны пологие, сильно раз-
мытые кривые распределения.
568
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
Как показал В. В. Коршак, коэффициент полидисперсности для
полиэфиров по мере протекания процесса поликонденсации непре-
рывно снижается и при достижении равновесного состояния имеет
минимальную величину.
Таким образом, для получения продуктов с наименьшей поли-
дисперсностью процесс поликонденсации необходимо доводить до
возможно большей степени завершенности.
Процессы поликонденсации линейных молекул теоретически
могли бы привести к линейным полимерам с бесконечно высокими
молекулярными весами, если растущие молекулы сохраняли бы
свою бифункциональность в течение всего процесса реакции.
В действительности, однако, ряд факторов приводит к тому, что
цепи становятся неактивными при, сравнительно, небольшой степени
поликонденсации. К таким факторам относятся процессы деструк-
ции цепи, протекающие наряду с реакциями поликонденсации, а
также наличие даже незначительного избытка одного из компонен-
тов реакции, что приводит к изменению функциональности (к блоки-
ровке функциональных групп полимера) и, следовательно, к пре-
кращению роста полимера. Наконец, даже крайне незначительные
количества воды, которые всегда остаются в продуктах реакции
из-за их высокой вязкости, приводят преждевременно к равновесию
и к прекращению роста полимера.
Эти причины, а также все увеличивающаяся вязкость продукта
и уменьшение концентрации реагирующих групп обусловливают
практическую невозможность достигнуть молекулярного веса поли-
мера выше 30 000.
Поэтому предпосылки, необходимые для получения высоких
ртепеней поликонденсации, должны включать возможно более со-
вершенное удаление из сферы реакции образующейся воды и мини-
мальный избыток одного из реагирующих веществ.
Расчеты В. В. Крршака и С. Р. Рафикова показывают, что при
поликонденсации гексаметилендиамина с адипиновой кислотой
избыток одного из компонентов в 1 % дает теоретический мол. вес
11446, а при избытке в 0,1%—мол. вес 137206, что уже значи-
тельно выходит за пределы достижимых на практике величин моле-
кулярного веса.
В том же направлении, как и избыток одного из бифункцио-
нальных компонентов, действует прибавление монофункционального
компонента — одноосновной кислоты или одноатомного спирта,
влияние которых основано на блокировании функциональных групп
растущей цепи. Одноосновные компоненты (например, абиетиновую
кислоту и др.) поэтому широко применяют для регулирования про-
цесса поликонденсации, для модификации свойств полимера и
в качестве стабилизаторов, препятствующих дальнейшему процессу
поликонденсации.
Свойства и структура продуктов поликонденсации определяются
строением реагирующих молекул.
Закономерности реакций образов, полиэфирн. и полиамидных смол 56°
Если реагирующие молекулы имеют разветвленное или цикличе-
ское строение (например, фталевый ангидрид, пентаэритрит и др.).,
то получится полимер с аморфной, глобулярной структурой, хруп-
кий, твердый или вязко-жидкий с низкой температурой плавления,
•подобный обычным низкомолекулярным смолам (например, новола-
кам). Если функциональность таких молекул выше двух, то на опре-
деленной стадии поликонденсации получатся неплавкие и нераство-
римые пространственные полимеры, которые, однако, будут состоять
из тех же глобулярных частиц, но связанных валентными связями.
Объясняется это сильно разветвленным характером поликонденсата
и легкостью образования циклов с двумя, тремя и т. д. элементар-
ными группами эфира.
Так, при взаимодействии фталевого ангидрида с глицерином,
возможно на определенной стадии процесса образование кольчатой
молекулы, согласно уравнению:
НО[СО—С6Н4—СО—С—СН2—СНСН—СН2О]2Н —>
сн2—ос—с—с6н4—со—о—сн2
I \
—> НОСН />СНСН.
СН2—СС—О—С6Н4—СО—О—СН2
Наряду с образованием циклов получаются и сильно разветвлен-
ные молекулы, обусловливающие в целом глобулярное строение по-
лимера.
Если реагирующие молекулы имеют линейную структуру (линей-
ные дикарбоновые кислоты, линейные диамины), то могут быть
получены как глобулярные, так и линейные полимеры (стр. 67)..
Преобладание той или иной структуры зависит от длины реагирую-
щих молекул. В частности, если число атомов, входящих в состав
основного звена, находится в пределах 5—7, то вероятность обра-
зования циклов особенно велика, так как, согласно теории напря-
жений Байера, такие циклы образуются с минимальной деформа-
цией валентных углов. Циклизация происходит легче при процессах
гомополиконденсации (например, амино- и оксикарбоновых кислот);
однако и в этом случае решающее значение имеют симметричность
структуры молекулы и число углеродных атомов в прямой цепи.
Возможность развития реакции по обоим направлениям является
одним из существенных факторов, определяющих структуру поли-
мера.
При взаимодействии дикарбоновых кислот и диаминов линейные
полимеры образуются, если в реагирующих молекулах число мети-
леновых групп не менее четырех. Если число метиленовых групп
менее четырех, образуются устойчивые циклы, типа
СО—NH
r/ >R2.
СС—NH
570
Гл. XIV. Пластмассы, на основе полиэфиров, полиамидов
При наличии четырех метиленовых групп могут образоваться и ли-
нейные полимеры и циклы (40—60% циклов); при наличии пяти
метиленовых групп образуются на 80—90% линейные полимеры и
на 10—20% циклические продукты. При большем числе метилено-
вых групп получаются только линейные полимеры. Образующиеся
первоначально циклы могут иметь различную степень устойчивости.
Во многих случаях они способны в дальнейшем участвовать в по-
строении цепи полимера.
Полиэфиры и полиамиды различаются по степени разветвлен-
ности молекул и по величине боковых цепей.
Только линейные макромолекулы, не имеющие боковых групп
в цепи или имеющие лишь редкие и небольшие группы (например
СНз-группу), могут кристаллизоваться. Кристаллические поликон-
денсационные полимеры имеют высокую прочность в ориентирован-
ном состоянии и комплекс свойств, характерный для полимеров типа
волокон. Они могут быть получены только, если реагируют линей-
ные, симметричные, в основном бифункциональные молекулы с чис-
лом метиленовых групп между диполями не меньше четырех или же
молекулы, состоящие из бензольного кольца с симметрично (в п-по-
ложении) расположенными функциональными группами.
Влияния структуры реагирующих молекул на свойства полимера
можно, например, наблюдать при получении продуктов поликонден-
сации гликоля с орто- и терефталевой кислотой (стр. 68).
Физико-механические свойства полимеров определяются также
концентрацией и степенью полярности диполей в цепи, точ-
нее — размером отрезка метиленовых групп, расположенного
между диполями макромолекул, значением х и у в общей
формуле:
...— (СНа^СО—NH(CH2)yNH—СО—...
Чем полярнее боковые группы и чем выше концентрация их
в цепи, тем, естественно, жестче полимер, тем выше его температура
плавления, и, наоборот, — полимер тем эластичнее, чем больше
неполярный отрезок между диполями.
Эти же факторы определяют и способность полимера к кри-
сталлизации, так как чем больше промежуток между диполями
(до установленного предела), тем подвижнее — СН2 — звенья и тем
легче они укладываются в кристаллическую решетку. Однако термо-
стабильность кристаллитов при этом уменьшается, так как силы,
действующие в кристаллической решетке, уменьшаются с удлине-
нием неактивной части молекулы.
Таким образом, удлинение промежутка между диполями, ослаб-
ляя межмолекулярные силы, увеличивает подвижность звеньев, что
делает полимер растяжимее, эластичнее и уменьшает его темпера-
туру размягчения. Однако эти же причины одновременно облегчают
процесс кристаллизации, что также приводит к жесткой структуре
Закономерности реакций образов, полиэфирн. и полиамидных смол 571
Рис. 205. Рентгенограмма ориен-
тированного полигексаметилен-
адипамида.
с высокой температурой плавления кристаллической фазы. Зависи-
мость концентрации кристаллической фазы полиамида от длины от-
резка между диполями характеризуется поэтому максимумом, ле-
жащим в интервале 6—8 углеродных атомов между диполями.
Таким образом, способность полиэфиров и полиамидов к кри-
сталлизации и образование при растяжении гибкого и прочного
полимера, обладающего свойствами «волокна», определяются: 1) ли-
нейным строением макромолекулы и отсутствием боковых цепей, за
исключением небольших редко расположенных групп (СН3 — и др.);
2) подвижностью звеньев, расположен-
ных между полярными группами, кото-
рая появляется при некоторой мини-
мальной длине цепи между ними;
3) наличием сильных, симметрично
расположенных диполей в цепи
(—СО—HN—; —О—СО—), обусло-
вливающих высокие межмолекулярные
силы притяжения.
Так же, как и все кристаллические
полимеры, кристаллические полиэфиры
и полиамиды всегда содержат боль-
шее или меньшее количество аморф-
ной фазы. При температуре ниже тем-
пературы плавления кристаллической
и выше температуры стеклования
аморфной фаз (между которыми
имеется значительный температурный
интервал) они поддаются растяжению.
При этом образуется устойчивая ориентация кристаллитов, показы-
вающая характерную рентгенограмму концентрических сгустков,
так называемую рентгенограмму волокна (рис. 205). Ориентация
кристаллитов приводит к значительному увеличению механической
прочности в направлении растяжения. Ориентированные таким
образом полиамиды и полиэфиры имеют кривую растяжения, ха-
рактерную для упругих тел, подобную кривой растяжения природ-
ных волокон. Ориентация кристаллитов (в отличие от кристалличе-
ских каучуков) не исчезает со снятием нагрузки, а сохраняется
вплоть до весьма высоких температур (150° и выше), что
объясняется высокой температурой плавления, значительной кон-
центрацией кристаллической фазы, а также температурой стеклова-
ния аморфной фазы, лежащей в пределах положительных темпера-
тур (40—100°).
Среди различных путей модификации свойств полиамидов и поли-
эфиров особое значение имеет блокировка части диполей в цепи,
например путем замещения подвижного водорода в —СО—NH-груп-
пах введением в состав реагирующих диаминов и дикарбоновых
кислот некоторого количества алкилзамещенных диаминов. Блоки-
572 Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
ровка части диполей в макромолекулярной цепи приводит к сниже-
нию межмолекулярных сил притяжения, повышению высокоэласти-
ческих свойств полимера, а в некоторых случаях к устранению воз-
можности кристаллизации (стр. 118).
Закономерности, ведущие к образованию трехмерных полиами-
дов, определяются показателем средней функциональности реаги-
рующих молекул, если он больше двух.
Чем выше средняя функциональность реагирующих молекул, тем
при меньшей степени завершенности реакции, при меньшем исполь-
зовании функциональных групп произойдет желатинизация — обра-
зование неплавкого, нерастворимого пространственного полимера.
Кроме того, чем выше функциональность реагирующих молекул, тем
выше концентрация пространственных связей в полимере в конечной
стадии его отверждения и тем больше его твердость, термостойкость
и прочность.
Характер и структура пространственного полимера в такой же
мере, как и линейного, определяются структурой реагирующих
молекул. Если реагируют циклические и несимметричные соедине-
ния, например фталевая кислота с пентаэригритом, то получаются
жесткие и разветвленные полимеры, состоящие из глобулярных
комплексов, соединенных густой пространственной связью. Если же
реагируют бифункциональные линейные молекулы (например
НОСО(СН2)вСООН, ЫН2(СН2)бМН2 и т. п.) в смеси с небольшим
количеством три- функциональных соединений (триамины, глице-
рин и др.), то образуются пространственные «сетки» со сравни-
тельно редкой «сшивкой» между цепями. Естественно, что физико-
механические свойства обоих типов пространственных полимеров
весьма отличны. В последнем случае получаются более эластичные,
гибкие, хотя и неплавкие и нерастворимые пленки.
Образование пространственных «сшивок» и переход смолы
в гелеобразное состояние может начаться задолго до исчезновения
всех реагирующих (мономерных) молекул и завершения реакции
эфиризации.
Если, рассматривая образование полиэфира, обозначить че-
рез / — количество функциональных групп молекулы и через No —
количество исходных мономерных молекул, то выражение Nof пред-
ставляет общее количество функциональных групп в реакционной
среде. Вступлением в реакцию мономерной молекулы связываются
две функциональные группы; поэтому, считая, что по окончании
реакции получаются N молекул полиэфира, общее количество остав-
шихся после реакции функциональных групп можно определить вы-
ражением: 2GV0--M,
а отношение оставшихся в реакционной смеси реакционноспособных
групп к исходным
где р — коэффициент глубины реакции эфиризации.
Закономерности реакций образов, полиэфирн. и полиамидных смол 573
Если обозначить через х степень поликонденсации, равную ,
и произвести соответствующее преобразование, то получится най-
денное Карозерсом выражение для р:
При высоких значениях х можно выражением —, пренебречь;
тогда будет получено приближенное значение
Если оба реагирующие компонента имеют две функциональные
группы, то коэффициент глубины эфиризации будет приближаться
к единице, что и имеет место, например, в случае поликонденсации
фталевого ангидрида с гликолем, при которой степень эфиризации
может быть доведена почти до 100%.
Однако при взаимодействии фталевого ангидрида с глицерином,
взятых в эквивалентных отношениях, на 3 моля фталевого анги-
дрида и 2 моля глицерина имеются 12 функциональных групп;
поэтому
12 2
/=у = 2-4; /> = -=0,83,
т. е. степень эфиризации может быть доведена лишь до 83%, после
чего должна наступить желатинизация массы (переход в простран-
ственный полимер); это почти соответствует экспериментальным
данным.
Таким образом, реакции, ведущие к образованию трехмерных
полимеров, имеют ту существенную особенность, что желатинизация
и образование трехмера должны наступить задолго до завершения
реакции (до использования всех реакционноспособных групп) и тем
раньше, чем выше функциональность реагирующих молекул.
Уменьшение концентрации трифункциональных молекул или
введение в реакционную смесь монофункциональных снижает f
и, следовательно, ведет к увеличению р, т. е. степени завершен-
ности реакции эфиризации до наступления желатинизации. Для
этой цели в технике производства полиэфирных смол широко приме-
няют сложные и большие молекулы ненасыщенных одноосновных кис-
лот— смоляные кислоты, а также ненасыщенные жирные кислоты и
их эфиры — глицериды, которые вводят в состав реакционной
смеси из дикарбоновых кислот и многоатомных спиртов. Смоляные
и жирные кислоты способны не только к процессам поликонден-
сации, в которых они ведут себя как монофункциональные соедине-
ния, но, в некоторой степени, и к процессам полимеризации (непо-
средственно или посредством вступающего в реакцию кислорода).
Было экспериментально установлено, что реактивность этих соеди-
нений не соответствует, однако, «молекулярной» функциональности
и что их поведение не отвечает закономерностям, действительным
574 Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
для малых функциональных молекул. Влияние ненасыщенных жир-
ных кислот на образование трехмерного полимера и вообще актив-
ность функциональных групп реагирующих молекул, по А. Я. Дрин-
бергу, определяется величиной удельной функциональности, которая
может быть выражена уравнением:
где: д — удельная функциональность; / — молекулярная функцио-
нальность; М — молекулярный вес.
Таким образом, А. Я. Дринберг отличает молекулярную функ-
циональность, выражающую число реакционноспособных групп в
молекуле, от удельной функциональности, выражающей активность
молекулы на единицу молекулярного веса (или длины молекулы).
Кинетика процесса поликонденсации и характер полимера опреде-
ляются, согласно А. Я. Дринбергу, не столько молекулярной, сколько
удельной функциональностью реагирующих молекул (табл. 63).
ТАБЛИЦА 63.
Значение молекулярной и удельной (в скобках) функциональностей
для кислых моноэ^иров
Кислота Спирт
диэтилен* ГЛИКиЛЬ этилен- гликоль глицерин пента- эритрит маннит
Себациновая . . . 2 (0,67) 2 (0,81) 3 (1,08) 4 (1,24) 6 (1,61)
Фталевая .... 2 (0,/8) 2 Щ,Уа) 3 (1,2а) 4 (1,42) 6 (1,81)
Адипиновая . . . 2 (0,8а) 2 (1,08) 3 (1,3о) 4 (1,61) 6 (1,90)
Трикарбаллиловая 3 (1,10) 3 (1,44) 4 (1,о0) 5 (1,4) 7 —
Образование трехмерных полимеров, если реагируют жирные
ненасыщенные кислоты, может произойти лишь в том случае, когда,
значение удельной функциональности достигает определенной вели-
чины. Из этого непосредственно следует, что чем длиннее цепь
линейной молекулы кислоты, тем больше должно быть в ней функ-
циональных групп (двойных связей) для образования простран-
ственного полимера или же тем больше должна быть функциональ-
ность второго компонента — многоатомного спирта.
Удельная функциональность не учитывает, однако, неравноцен-
ности отдельных функциональных групп в молекуле (положение
двойных связей, различие первичных и вторичных гидроксилов
и т. д.) и может быть поэтому рассматриваема лишь как потен-
циальная функциональность, а не как используемая. Очевидно, что
для учета этой неравноценности в формулу удельной функциональ-
ности необходимо ввести еще один коэффициент, который дает
величину используемой функциональности. Таким коэффициентом,.
Закономерности реакций образов, полизфирн. и полиамидных смол 575>
по А. Я. Дринбергу, является р— коэффициент полноты реакции, и-
тогда используемая удельная функциональность будет:
г /И ’
где: 8 — используемая удельная функциональность; Д — удельная
функциональность; / — молекулярная функциональность; М — моле-
кулярный вес.
Процессы, ведущие к образованию полиэфирных смол, модифи-
цированных маслами (триглицеридами), включают реакции пере-
эфиризации, так называемого алкоголиза и ацидолиза; при этом-
происходит вытеснение связанных в сложном эфире (масле) остат-
ков спирта и замещение их остатками другого спирта (алкоголиз)
или же — вытеснение остатков кислот остатками другой кислоты
(ацидолиз). Этими процессами пользуются для введения жирных
кислот в состав молекул полиэфирных смол.
Так, при алкоголизе триглицеридов глицерином получаются
сложные эфиры жирных кислот, содержащие свободные гидро-
ксильные группы, способные к поликонденсации с двухосновными,
кислотами:
CH2CCCR! СН2СН CH-jCCCRi CHaOCOR3
Illi
CHOCCR2 4- СНОН —> CHCCORj, + CHCH
Illi
CHCCCRa CH2OH CH2CH CH2CH.
При ацидолизе триглицеридов, например фталевой кислотой,,
образуются свободные карбоксильные группы, способные к поли-
конденсации с многоатомными спиртами:
CH.OCCRt CH2CCORi
I .СООН 1
CHCCCR2+C6hZ —> CHCCORa + R3COCH.
I XCOOH I
CH2CCOR3 CHaOCCC6H4COOH
Характерной особенностью полиэфирной конденсации, как пока-
зали работы В. В. Коршака с сотрудниками, является одновремен-
ное действие процессов роста и деструкции цепи. Оба процесса на
различных стадиях поликонденсации и роста цепи протекают с раз-
личной скоростью.
Рост цепи осуществляется путем ряда последовательных эле-
ментарных реакций взаимодействия растущего полимера с моно-
мерами. С меньшей скоростью вследствие малой подвижности мо-
лекул и малой вероятности взаимодействия этот процесс может про-
текать между растущими цепями. С увеличением молекулярного веса
рост цепи замедляется.
Деструкция цепи происходит при взаимодействии растущего
полимера с реагирующими мономерными молекулами, причем, чем
длиннее цепь полимера, тем вероятнее ее деструкция.
576
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
Процессы деструкции цепи полиамидов и полиэфиров под дей-
ствием свободных кислот В. В. Коршак назвал ацидолизом, под
воздействием диаминов — аминолизом и под действием спирта —
алкоголизом. *
Так ацидолиз полиамида действием адипиновой кислоты можно
представить следующей схемой:
H2N(CH2)GNH[CO(CH2)4CONH(CH2)6NH]/,COOH + 2НОСО(СН2)4СООН —>
-» HOCO(CH2)4CONH(CH2)eNH[CO(CH,)4CONH(CH2)6NH]m_1CO(CH2)4COOH-f-
4-HOCO(CH2)4CONH(CH2)6NH[CO(CH2)4CONH(CH2)eNH]rt_1CO(CH2)4COOH ,
где р = т 4- п.
Образовавшиеся продукты ацидолиза являются карбоновыми
кислотами.
Аналогично при действии на полиамидную цепь диамина проис-
ходит разрыв цепи с образованием осколков молекул, в которых
полностью отсутствуют свободные карбоксильные группы.
Средний молекулярный вес продуктов ацидолиза и аминолиза
полиамидов тем меньше, чем больше избыток кислоты или диамина.
Как было уже указано, такого рода процессы являются автомати-
чески действующими регуляторами среднего молекулярного веса
полимера и определяют сравнительно высокую степень гомогенности
поликонденсационных продуктов и их малую полвдисперсность.
ЛАКОВЫЕ СМОЛЫ И ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ
ПОЛИЭФИРНЫХ СМОЛ
(Эфиропласты)
Первые смолообразные продукты этого типа были получены
еще в 1847 г. Берцелиусом при конденсации винной кислоты с гли-
церином. Однако лишь в начале настоящего столетия (1901 г.)
были получены технически годные смолы на основе фталевого
ангидрида и глицерина и предложены различные варианты их
использования.
Практическое применение глицеро-фталевых смол оказалось воз-
можным после того, как было осуществлено производство дешевого
фталевого ангидрида методом каталитического окисления нафта-
лина (1916 г.). Первые глицеро-фталевые смолы были выпущены
в 1920—22 гг., модифицированные'—в 1927—31 гг., а полиэфиры на
основе себациновой и адипиновой кислот — в 1934 г.
Полиэфирные смолы применяют главным образом в качестве
лаковых смол и лишь небольшую их часть — для производства
пластмасс, главным образом электроизоляционного материала
миканит.
* Следует поэтому различать термины ацидолиз и алкоголиз, выражающие
процессы переэфиризаиии триглицеридов (стр. 575), от тех же терминов, выра-
жающих процессы деструкции цепи полимеров.
Лаковые смолы и пластмассы на основе полиэфирных смол
577
Полиэфирные смолы можно разделить на термоплавкие, не способ-
ные к отверждению, и термореактивные, способные к отверждению.
Последние, в свою очередь, подразделяются на отверждающиеся
только под действием температуры и отверждающиеся под
действием как температуры, так и окислительно-полимеризационных
процессов.
По структуре полимера полиэфирные смолы делят на смолы
глобулярной и линейной структур.
Различают также: 1) чистые полиэфиры, получаемые на основе
поликонденсации лишь полифункциональных кислот и спиртов;
2) полиэфиры, модифицированные смоляными или жирными кисло-
тами.
С точки зрения химического состава различают следующие виды
полиэфиров: глицеро-фталевые; гликолево-фталевые, гликолево-те-
рефталевые, пентаэритритовые (на основе фталевого ангидрида и
других кислот); смолы на основе малеинового ангидрида, а также
малеинового ангидрида со смоляными кислотами и различными
спиртами; смолы на основе линейных двухосновных кислот — себа-
циновой, адипиновой и др., с многоатомными спиртами; смолы, мо-
дифицированные значительным количеством кислот высыхающих
масел и другими соединениями.
Производство фталевых смол
Смолы на основе этиленгликоля и фталевого
ангидрида
Так как и этиленгликоль и фталевый ангидрид являются бифунк-
циональными соединениями, при их взаимодействии получаются
постоянноплавкие и растворимые смолы.
Реакция протекает таким образом, что сначала быстро обра-
зуются кислые эфиры:
Г““ + НОСН^СН!ОН _ Л-со-о-сн^с^он+^о.
I—СООН 122 I J—СООН
которые в дальнейшем уже медленно реагируют друг с другом,
с гликолем и с кислотой, образуя в итоге сложные, несимметричные
молекулы сравнительно малой степени поликонденсации. Конечным
продуктам с некоторым приближением можно приписать следующее
строение:
.—сн,—сн, сн2—сн, сн,—сн, сн,—..
I ‘ I I ‘ I I I
о о о о о о
Illi II
со со со со со со
37 Зяк. 1269. э. И. Барг
578 Гл. XIV. Пластмассы, на основе полиэфиров, полиамидов
Достигаемая полнота эфиризации и степень поликонденсации
зависят от степени удаления реакционной воды, избытка одного из
компонентов, температуры и времени реакции.
Реакцию ведут продолжительное время при высоких температу-
рах (240—300°), не опасаясь желатинизации смолы и перехода
в неплавкое состояние; при этом достигается высокая степень эфири-
зации и получаются продукты с весьма низким кислотным числом
(до 20—15). Для устранения окисления и потемнения смолы реак-
цию ведут в атмосфере азота или углекислого газа.
Варку смолы проводят в стационарных реакторах, изготовленных
из алюминия или из нержавеющей стали емкостью от 0,5 до 2 лг.
Реакторы имеют пропеллерную или якорную мешалку, сферические
крышку и днище, люк для загрузки фталевого ангидрида, штуцеры
для загрузки этиленгликоля (или других жидких компонентов), для
термометра, для взятия пробы. Реакторы соединены с уловительной
установкой. Обогрев производится топочными газами, отходящими
от индивидуальных топок или от центральной топки для несколь-
ких реакторов. Регулирование обогрева котла проводится с по-
мощью шибера на газоходе.
Обогрев топочными газами, естественно, имеет ряд недостатков,
среди которых важнейшим является трудность регулирования в этих
условиях температуры. Обогрев может осуществляться также
электричеством, паром высокого давления (50—70 ат) или перегре-
той водой. Каждый из этих способов имеет наряду с преимуще-
ствами и существенные недостатки, связанные, например, с услож-
нением аппаратуры (при применении пара и перегретой воды) или
с малым коэффициентом теплопередачи (при применении электро-
обогрева). В последние годы в качестве теплоносителя стали при-
менять пары динила— дифенильной смеси, состоящей из 26,5% ди-
фенила СеНб—С6Н5 и 73,5% дифенилоксида (CcHshO, кипящей при
нормальном давлении при 255—258° и застывающей при 12,3°. Пре-
имущество применения этого способа заключается в возможности
нагревания аппарата путем циркуляции паров теплоносителя по
трубам и в рубашке реактора при нормальном давлении и даже
под вакуумом (при температуре ниже 258°). Однако при работе
с динилом все аппараты и трубы должны быть тщательно гермети-
зированы, чтобы устранить потери и не ухудшить условий труда
из-за некоторой токсичности теплоносителя.
В случае обогрева дифенильной смесью реактор должен быть
снабжен рубашкой, через которую поступают пары динила; для
охлаждения реактора через ту же рубашку пропускают жидкую
дифенильную смесь.
Существенное значение имеет система уловительной установки,
назначение которой улавливать и по возможности возвращать в
реактор летучий фталевый ангидрид, частично этиленгликоль, и обес-
печить возможно более полное удаление реакционной воды и воды,
содержащейся в спиртах. Для этого котлы, обогреваемые топочными
Производство фталевых смол
579
газами, сна’бжены вытяжными трубами с искусственной тягой
(в виде паровой эжекции) и скрубберами для улавливания фтале-
вого ангидрида.
Применяют также систему уловительной установки, предста-
вленную на рис. 206.
Реактор 1 с рубашкой для обогрева дифенильной смесью соеди-
нен с обратным конденсатором 2, охлаждаемым водой, который,
в свою очередь, соединен с прямым змеевиковым конденсатором 3,
вакуум-приемником 4, охлаждаемым водой, и ловушкой 5. Система
может работать под вакуумом, создаваемым вакуум-насосом 7,
перед которым установлен вакуум-приемник 6. Эта установка
переключением трехходового крана у ловушки позволяет либо
отводить из реактора конденсат, либо частично возвращать его
в реактор.
Обычно исходят из эквимолекулярных отношений реагентов или
же берут небольшой избыток фталевого ангидрида, учитывая потери
последнего из-за его летучести. Так, в реактор загружают
60—62 части гликоля и нагревают до 80—110°, после чего вводят
155—180 частей фталевого ангидрида. Когда фталевый ангидрид
.г5
580
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
растворится, температуру медленно поднимают до 200—220°. Реак-
ция вначале идет очень быстро и только после того как степень
эфиризации достигнет 60—65% (после образования кислых эфиров),
скорость реакции падает. Процесс в первой стадии необходимо
вести с учетом летучести ангидрида, а также гликоля с парами
конденсационной воды. Вследствие увеличения вязкости среды и
уменьшения концентрации исходных реагирующих молекул скорость
реакции постепенно сни-
жается. Поэтому в послед-
ней стадии реакции темпе-
ратуру поднимают до 260—
300° и процесс заканчивают
при снижении кислотного
числа до 25—15. Контроль
производства осуществляют
определением кислотного
числа и вязкости, а иногда
и температуры плавления
продукта. Уменьшение кис-
лотного числа сопровождает -
Рис. 207. Изменение вязкости 1 и кислот-
ного числа 2 при конденсации гликоля
с фталевым ангидридом.
ся постепенным увеличением
вязкости смолы (рис. 207).
По окончании реакции
смолу охлаждают в реакторе
до 150—180° и сливают на алюминиевые противни, если она засты-
вает в твердую массу, или в барабан, если при обычной темпера-
туре она имеет вязкую консистенцию.
Когда нужно получить лак, смолу перекачивают в смеситель, где
ее обрабатывают соответствующими растворителями.
Готовый лак выгружают в бочки или бидоны. Гликолево-фтале-
вые смолы растворимы в ацетоне, циклогексане, спирто-бензоле.
Они совместимы с эфирами целлюлозы, отличаются хорошей водо-
стойкостью и хорошими диэлектрическими свойствами. По консистен-
ции они весьма разнородны — от мягких, воскоподобных до твердых
и хрупких смол. Молекулярный вес от 600 до 1500.
Смолы на основе этиленгликоля и терефталевой
кислоты
(Политерефталаты)
При поликонденсации гликоля с терефталевой кислотой
НОСО —— СООН, получаемой окислением n-ксилола, об-
разуются высокомолекулярные линейные полимеры, обладающие со-
вершенно иными свойствами, чем смолы, получаемые при конденса-
ции гликоля с о-фталевой кислотой (фталевым ангидридом).
Производство фталевых смол
581
Реакция протекает по схеме:
НОСН2— СН2ОНН-НОСО—СООН —►
—► НОСН2—СН2ОСО—/ соосн2—СН2ОСО—/ СОО—...
Уже давно было известно, что терефталевая кислота при кон-
денсации с гликолями образует смолы с высокой вязкостью раство-
ров и высокой температурой плавления. Это различие смол, полу-
чаемых при конденсации о- и n-фталевых кислот с гликолем, сле-
дует отнести за счет различия в структурах реагирующих кислот.
Симметричная структура терефталевой кислоты приводит к обра-
зованию линейных полимеров, не содержащих боковых групп. Вы-
сокая концентрация симметрично расположенных диполей
(—СО—О—) обусловливает кристалличность полимера. Полимеры
содержат 65—75% кристаллической фазы; они имеют температуру
плавления 220—240°, температуру стеклования ~ 60—80°. Молеку-
лярный вес полимеров 12 000—20 000.
Вытянутые на холоду (выше температуры стеклования) пленки
и нити показывают рентгенограмму ориентированных кристаллитов.
Прочность при растяжении (~400—500%) увеличивается до десяти
раз и достигает 3500—4500 кг/см2.
Политерефталат отличается сравнительно высокими диэлектри-
ческими свойствами (е ~3—4; tgS ~ 0,005—0,01, р„10и—1015 Q-cm),
однако они резко снижаются при достижении температуры стекло-
вания.
Полигликольтерефталат, выпущенный под названием терилен,
был получен в 1947—1948 гг.
Технологический процесс производства политерефталата и про-
цесс его переработки подобен, в основном, процессу получения поли-
амидов.
Полиэфир получают при строго эквимолекулярных отношениях
гликоля и терефталевой кислоты.
Процесс ведут в автоклаве, обычно без растворителей и катали-
заторов; в первой стадии при 180—200° он проходит под давлением,
в дальнейшем температуру поднимают до 280—300°, включают глу-
бокий вакуум и нагрев при перемешивании продолжают в течение
4—6 часов, до получения смолы с кислотным числом в пределах
10—15. Реакция проходит в токе азота, тщательно очищенного от
кислорода.
Политерефталат получил применение в качестве синтетического
волокна, а также как пластик, перерабатываемый экструзией, валь-
цеванием, прессованием. Из него изготовляют пленки, пластины,
прокладки, трубки и т. д.
Смолы на основе глицерина и фталевого
ангидрида
Глицеро-фталевые смолы, широко известные под названием гли-
фтали, являются важнейшими полиэфирными смолами как для
582
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
изготовления лаков и пластмасс. Они обладают термореактив-
ностыо и способны переходить в неплавкое и нерастворимое со-
стояние.
Так как глицерин имеет три функциональные группы, то можно
ожидать, что еще задолго до завершения процесса эфиризации
может начаться образование пространственных связей и желатини-
зация смолы в реакторе, чего, конечно, нельзя допускать. Теорети-
чески желатинизация может наступить, когда степень завершенно-
сти реакции (степень эфиризации) достигнет 84%, поэтому реакцию
прерывают до завершения эфиризации, когда реакционная смесь
еще содержит значительное количество свободных гидроксильных и
карбоксильных групп. Возможность желатинизации смолы в про-
цессе реакции определяет и режим процесса производства.
Реакция поликонденсации фталевого ангидрида с глицерином
в первоначальной стадии протекает так же, как и с гликолем, т. е.
с образованием кислых эфиров, причем кислотное число в течение
первых же минут нагрева (165—180°) резко снижается; дальнейшее
же снижение кислотного числа происходит медленно. Некоторые
считают, что в первую очередь при температуре до 180° реагируют
а-гидроксильные группы глицерина и лишь потом (при температуре
выше 180°) в реакцию вступают ^-гидроксилы, которые при темпе-
ратуре около 260° становятся настолько активными, что вызывают
образование пространственного полимера.
Согласно Кинли, первоначально образуется смесь моно- и дигли-
церидов, с эфиризацией а-гидроксильных групп:
/\_со
I I \о-f-НОСН2—СН—СН.ОН—> соосн —сн—сн,он
со I I
он I J—соон он
/\_со -
| | /О2НОСН2—СН—СН2ОН —> СООСН2— СН—СН..ОН
х'/ со он он
—соосн,—сн—сн,он
Образовавшиеся кислые эфиры могут реагировать друг с другом
по схеме: НОСО СООСН.—СНОН—СН,ОН
НОСО СООСН,—СНОН—СН2ОСО соон
НОСО СООСН2СНОНСН2ОСО СОО СН2 СНОН СН2О СО СООН
Производство фталевых смол 583
В результате последующих реакций конденсации этих соедине-
ний друг с другом, а также и с исходными реагирующими моле-
кулами получается смесь сравнительно низкомолекулярных продук-
тов, образующая первоначальную плавкую смолу, которая содержит
реакционноспособные группы ОН и СООН и, подобно резолам, при
дальнейшем нагревании переходит через промежуточную стадию В
в стадию С — неплавкое состояние. Превращение смолы может
иметь место лишь при условии, если в процессе дальнейшей тер-
мической обработки реагируют полностью или частично Р-гидро-
ксилы глицерина. Отверждение глифталей происходит значительно
медленнее, чем отверждение резолов и требует длительного нагре-
вания при 150—180°. Это обстоятельство значительно ограничивает
их применение для изготовления прессизделий.
Технологический процесс производства глифталей требует бо-
лее осторожного нагрева и более тщательного контроля процесса
конденсации, чем при производстве гликолевых смол.
Обычно исходят из эквивалентных отношений реагирующих ве-
ществ. На 2 моля глицерина берут 3 моля фталевого ангидрида.
Процесс складывается из трех стадий: 1) растворения ангидрида в
глицерине; 2) образования первичных кислых эфиров и 3) заклю-
чительной стадии образования плавкой и растворимой смолы. Схема
процесса в основном аналогична с процессом получения модифи-
цированных смол (см. рис. 209, стр. 585).
В реактор загружают глицерин и нагревают до 110—120°; после
этого при работающей мешалке постепенно загружают фталевый
ангидрид. Когда ангидрид растворится, температуру медленно под-
нимают до 150—160°. При этой температуре резко снижается
кислотное число смеси и происходит образование кислых эфиров.
После того как падение кислотного числа замедлится, температуру
постепенно поднимают до 210—240°. Дальнейший процесс ведут при
тщательном контроле температуры плавления, кислотного числа и
растворимости смолы в смеси спирта с бензолом. По достижении
соответствующих показателей смолу сливают в противни. Готовая
смола в плавком и растворимом состоянии имеет кислотное число
в пределах 70—120 и т. плавл. (по Уббелоде) 90—120°.
По другому варианту в реактор загружают весь глицерин и при-
мерно 60—65% всего фталевого ангидрида и смесь выдерживают
при 200° в течение 1,5—2 час. до получения смолы с кислотным
числом 20—25, после чего загружают остаток фталевого ангидрида
и реакцию ведут в течение 2—3 час. при 160—165° до получения
продукта с кислотным числом 100—140.
Модифицированные фталевые смолы
Чистые глицеро-фталевые смолы часто не могут быть применены в качестве
лаков из-за хрупкости пленок, малой водостойкости и погодоустойчивости,
обусловленных большим кислотным числом. Они также нерастворимы в масле.
Путем модификации с маслами, со смоляными и жирными кислотами получают
584 Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
эластичные, маслорастворимые смолы с весьма низким кислотным числом, хоро-
шей водо- и погодоустойчивостью.
Реакцию образования полиэфиров, модифицированных непредельными жир-
ными кислотами, можно представить в первой фазе уравнением:
СО
I /° +
\/~С°
СН2ОН
(кюн +RCOOH —>
!
СН2ОН
ch2ocor
(^НОН
СН-ОСО—СООН
(R — непредельный радикал жирной кислоты).
Жирные ненасыщенные кислоты ведут себя в процессе поликонденсации в
значительной степени как монофункциональные соединения, в которых реагирует
карбоксильная группа. Поэтому введение их в состав реакционной смеси при-
водит к уменьшению активной функциональности реа-
Рис. 208. Зависимость
скорости смолообразова-
ния модифицированного
глифталя от функцио-
нальности масла.
гирующих молекул, к снижению реактивности поли-
мера. Это уменьшает опасность преждевременной
желатинизации смеси и позволяет достигать более
высокой степени эфиризации.
Конечный продукт поликонденсации представляет
собою поэтому эфиризованный полимер, обладающий
лишь небольшим количеством свободных функцио-
нальных групп, которому, в первом приближении, мо-
жет быть приписано строение:
сн,— 1 1 4 сн- -СН, сн2— сн- -сн, 1 2 сн2- 1 -СН-.
О О о О о он о 1 о о
io со со СО со со со со
ч / R / / R
цесс <высыхания» лаковых
Скорость процесса смолообразования, а также ха-
рактер и свойства модифицированной смолы сильно
зависят от степени ненасыщенности (функционально-
сти) жирных кислот и их количества (рис. 208). Чем
выше степень ненасыщенности кислоты, тем быстрее
протекает как реакция смолообразования, так и про-
пленок в результате термического воздействия и про-
цессов окислительной полимеризации.
Модифицированные смолы могут быть получены следующими методами:
1) поликонденсацией глицерина или полиглицеринов с фталевым ангидридом и
жирными кислотами; 2) синтезом на основе переэфиризованных масел — поли-
конденсацией фталевого ангидрида с моно- и диглицеридами; 3) нагреванием
с маслами полиэфирной глицеро-фталевой смолы, предварительно полученной
с небольшим содержанием жирных кислот; 4) модификацией как жирными, так
и смоляными кислотами и маслами.
Процесс получения модифицированных маслами глицеро-фталевых смол со-
стоит из двух стадий: 1) переэфиризации масел (триглицеридов) глицерином,
Производство фталевых смол
585
с образованием моно- и диглицеридов, например по схеме:
ch2ocor сн2он ch2ocor 1 2 1
CHOCOR + НОСН2—СНОП—СН2ОН — ch2ocor -> CHOCOR + СПОН сн2он ch2ocor
и 2) поликонденсации свободных гидроксильных групп моно- и диглицеридов
с фталевым ангидридом.
На практике при производстве модифицированных смол часто исходят из
сложной смеси жирных кислот, масел, смоляных кислот и т. д. Применяют,
например, следующую рецептуру (в %);
Фталевый ангидрид...........35
Глицерин....................19,7
Льняное масло................20,3
Жирные кислоты льняного масла . 10
Канифоль........................10
Касторовое масло ............... 5
Общая схема производства различных видов модифицированных полиэфир-
ных смол представлена на рис. 209.
Рис. 209. Схема производства полиэфирных модифицированных смол и
лаков.
Масла из цистерны 1 сливают в цеховые емкости 2, 3, из которых вакуумом
засасывают в весовые мерники 5 и 6. Глицерин из бочек вакуумом засасывают
в мериик 7. Предусмотрен термошкаф 28 для разогревания глицерина в зимнее
время. Из мерников 4—7 через бункерные весы 27 масла и глицерин поступают
в реактор 8. Для быстрого охлаждения реакционной смеси котел снабжен зме-
евиком, по которому в случае необходимости циркулирует глицерин, подаваемый
шестереночным насосом 20 через трубчатый холодильник 18, из которого гли-
церин стекает обратно в приемный бачок 19. Углекислоту в реактор подают из
586
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
баллона 10. Твердые компоненты (фталевый ангидрид и др.) взвешивают на ве-
сах 9 и загружают вручную через люк реактора. Пары, выделяющиеся при
варке смолы, проходят через систему труб для улавливания фталевого ангид-
рида, а затем в дефлегматор 11, в котором при помощи паровой рубашки под-
держивают заданную температуру. Конденсат через сифон 14 стекает обратно
в реактор, а неконденсированныс пары поступают в трубчатый холодиль-
ник 12, из которого жидкость через ловушку 13 стекает в приемник 15. Пары
могут также непосредственно поступать в холодильник через ловушку 16.
Вакуум в системе создается при помощи вакуум-насоса 17, соединенного с ре-
сивером 16. Готовую смолу либо сливают в бидоны 23, либо подают в шкаф 29
для розлива в противни. Из последних смолу в виде кусков загружают
в тару 22. '4
В случае изготовления лака смолу засасывают вакуумом в смеситель 24,
в который после охлаждения смолы по трубопроводам через мерники 25—25
поступают растворители, обычно смесь ксилола и лакового бензина. После рас-
творения готовый лак фильтруют или центрифугируют и разливают в тару.
Малеиновые смолы
Малеиновый ангидрид широко применяется в производстве поли-
эфирных и модифицированных смол для получения лаков.
Так как малеиновый ангидрид содержит двойную связь, то
молекулу малеиновой кислоты в реакциях конденсации необходимо
Продолжи т>ельносгт реаииии,
часы
Рис. 210. Изменение кислотного
числа при конденсации малеи-
нового ангидрида с диэтиле.i-
гликолем.
рассматривать как потенциально тетра-
функциональную. При конденсации
малеинового ангидрида с этиленглико-
лем можно достичь при соответствую-
щем ведении процесса значительной
полноты эфиризации и малых кислот-
ных чисел смолы, однако все же не
исключена возможность желатиниза-
ции за счет двойных связей ангидрида
(рис. 210). При конденсации малеи-
нового ангидрида с глицерином и
спиртами высшей атомности желатини-
зация наступает значительно раньше,
чем при конденсации фталевого анги-
дрида. Получаемые смолы имеют более
высокое кислотное число и большую
концентрацию гидроксильных групп.
Они обладают способностью в тонких
пленках и без добавок высыхающего
масла постепенно на воздухе переходить в неплавкое состояние
(высыхать).
Чистые глицеро-малеиновые смолы применяют редко, чаще по-
лучают модифицированные глицеро-малеиновые, в частности моди-
фицированные канифолью и жирными -маслами.
Кислоты, входящие в состав канифоли, при 150—160° легко
Полиэ фирм, на сснове линейных двухосновных кислот 587
реагируют с малеиновым ангидридом, образуя, например, аддукты
по уравнению:
аддукт малеиновой кислоты
с одной из кислот канифоли
Малеиново-канифольные аддукты имеют значительно более вы-
сокую температуру плавления, чем исходная канифоль, и могут
найти как самостоятельное применение в качестве основы для мас-
ляных лаков, так и служить исходным веществом для поликонденса-
ции со спиртами. Значение малеиново-канифольных аддуктов заклю-
чается в том, что они представляют собою трехосновные кислоты и
при конденсации с двухатомными спиртами образуют быстро
высыхающие пленки большой твердости, гибкости и светостойкости,
так как общая функциональность реагирующих компонентов (2,3)
делает возможным образование пространственных полимеров. Чи-
стые малеиново-гликолевые смолы имеют более низкую температуру
плавления и образуют липкие пленки, однако при экспозиции на
воздухе и при нагревании они твердеют и переходят в нераствори-
мое состояние вследствие раскрытия двойной связи.
Полиэфиры на основе линейных двухосновных кислот
В последние годы все большее значение получают полиэфиры на
основе линейных двухосновных кислот типа HOCORCOOH, где
R— радикал, состоящий из четырех и более метиленовых групп.
Из таких кислот нашли применение, главным образом, адипиновая
НОСО(СН2)гСООН и себациновая НОСО(СН2)8СООН кислоты. Их
используют в производстве как термореактивных смол на основе
глицерина и пентаэритрита, так и термопластичных смол на основе
этиленгликоля.
Процесс производства смол этого типа аналогичен процессу
получения обычных фталевых смол. Например, в реактор загру-
жают 92 кг глицерина (1 моль) и нагревают до 90—100°, затем
588
Гл. XIV. Пластмассы, на основе полиэфиров, полиамидов
постепенно вносят 219 кг (1,5 моля) адипиновой кислоты и темпе-
ратуру поднимают до 180—190°; после того как кислотное число
перестает резко снижаться, температуру поднимают до 240—260°
и реакцию ведут до получения смолы с заданными кислотным чис-
лом и температурой размягчения.
В связи с меньшей удельной функциональностью линейных кис-
лот (адипиновой, себациновой) образование пространственного по-
лимера (желатинизация) наступает при более высоких степенях
эфиризации. Вследствие этого реакцию линейных кислот с глицери-
ном можно вести при более высоких температурах и более дли-
тельно, достигая больших степеней эфиризации, чем при взаимо-
действии глицерина с фталевым ангидридом. Получаемые смолы
имеют малый молекулярный вес (~ 1000) и низкую температуру
плавления. Чем больше число углеродных атомов в кислоте, тем
эластичнее пленки, тем ниже температура размягчения. Несмотря на
малый молекулярный вес, смолы этого типа обладают заметными
высокоэластическими свойствами, гибкостью и пластичностью. При
поликонденсации линейных двухосновных кислот с гликолями обра-
зуются постоянноплавкие и растворимые смолы, при взаимодействии
же с глицерином, пентаэритритом и др. — термореактивные смолы,
переходящие при нагревании в нерастворимое состояние.
Смолы на основе линейных двухосновных кислот применяют
главным образом в качестве пластификаторов для целлюлозных
лаков и для модификации мочевино-формальдегидных смол
(стр. 538;.
Пентаэритритовые смолы
Значительным достижением в области производства полиэфир-
ных смол явилось применение пентаэритрита в качестве спиртового
компонента при конденсации с двухосновными кислотами.
Ведущие работы в области синтеза пентаэритритовых смол при-
надлежат А. Я. Дринбергу, В. С. Киселеву и их сотрудникам.
Как четырехатомный спирт с равноценными первичными спирто-
выми группами пентаэритрит энергичнее глицерина реагирует с двух-
основными кислотами, при этом желатинизация смолы теоретически
может наступить уже при достижении 50% эфиризации. Поэтому
чистые пентаэритрито-фталевые смолы имеют ограниченное приме-
нение, так как их можно получить лишь с малой степенью эфириза-
ции и они содержат значительное количество не вступивших в реак-
цию кислот и спирта.
Однако более высокая функциональность пентаэритрита, по
сравнению с глицерином, является большим преимуществом при
получении модифицированных пентаэритритовых смол.
Она позволяет, без ущерба для реактивности смолы, вводить
значительно большие количества масел, а также заменять высыхаю-
щие масла полувысыхающими и даже невысыхающими. Полученные
Применение полиэфирных смол
589
таким образом смолы обладают более высокой термореактив-
ностью, чем глицеро-фталевые при введении того же количества
масла одинаковой функциональности, или такой же термореактив-
ностью при введении большего количества масел или масел более
низкой функциональности. Возможность же введения бдлыпего ко-
личества масла или жирных кислот позволяет получать более гиб-
кие, эластичные пленки.
Так, А. Я. Дринберг получал маслорастворимые смолы при кон-
денсации: 6,4 мол. пентаэритрита с 4 мол. фталевого ангидрида и
2 мол. жирных кислот хлопкового масла. Реакцию проводили при
190—220° до образования смолы с кислотным числом 13—27.
Количественное соотношение реагентов можно варьировать
в широких пределах, однако для получения нейтральных смол
с высокими физико-механическими свойствами необходимо, чтобы
сумма молей всех взятых кислот соответствовала или была бы не-
сколько больше числа молей спирта.
Пентаэритритовые смолы обладают большей совместимостью
с эфирами целлюлозы с меламино- и мочевино-формальдегидными
смолами, чем глицериновые.
Большое применение получил метод, заключающийся в переэфи-
ризации масел пентаэритритом в присутствии щелочных катализато-
ров с последующей эфиризацией полученных продуктов дикарбоно-
быми кислотами. На основе этого метода были получены искусствен-
ные олифы, так называемые пентоли. Производство пентолей состоит
в переэфиризации обычно невысыхающих масел (например хлопко-
вого) пентаэритритом или в поликонденсации пентаэритрита
непосредственно с жирными кислотами. В последнем случае полу-
чаются более реактивные (полимеризующиеся) олифы, так как в
них отсутствуют остатки менее функционального спирта — глице-
рина. В зависимости от функциональности масла или жирных кис-
лот образуются олифы с различной способностью к высыханию. В
общем переэфиризация или эфиризация пентаэритритом переводит
менее высыхающие масла в более высыхающие.
Применение полиэфирных смол
Полиэфирные смолы применяют главным образом для производства изоля-
ционных, масляных и спиртовых лаков, в качестве важнейшего компонента при
получении эфирно-целлюлозных лаков, в качестве пластификаторов н для моди-
фикации феноло-альдегидных, мочевино- и меламино-формальдегидных лаков.
Сравнительно меньшее, но все же важное значение они имеют для производ-
ства замазок, клеев, цементов, пластмасс и синтетического волокна.
Полиэфирные смолы, используемые для производства лаков, могут быть раз-
делены на три группы: 1) пластифицирующие смолы для эфироцеллюлозных и
других лаков; 2) высыхающие смолы, способные при самостоятельном примене-
нии давать сравнительно быстросохнущие лаки и полимеризоваться при экспози-
ции на воздухе или при термообработке; 3) твердые высокоплавкие смолы, глав-
ным образом на основе малеиново-канифольных продуктов конденсации, в ком-
7Ю Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
позиции с феноло-альдегидными и карбамидными смолами для производства
спиртовых н масляных паков.
В качестве добавок к эфироцеллюлозным лакам полиэфирные смолы имеют
то преимущество перед обычными пластификаторами, что они не «выпотевают»,
химически более устойчивы и являются пленкообразующими веществами.
Твердые малеиново-канифольные смолы различаются температурой размяг-
чения (в пределах от 120 до 150°).
Обычно лаковые полиэфирные смолы имеют небольшое кислотное число
(от 20 до 40), растворимы в скипидаре, ароматических углеводородах, в этил-
ацетате, совмещаются с маслами и могут быть разбавлены лигроином и бензи-
ном. Некоторые сорта смол растворимы в спиртах и находят применение в ка-
честве заменителя шеллака.
Лаковые полиэфирные смолы отличаются хорошей адгезионной способностью,
гибкостью и эластичностью образуемых ими пленок, светостойкостью и во мно-
гих случаях большой твердостью, а также высокой стойкостью к атмосферным
зоздействиям.
Глицеро-фталевые смолы — ценный материал в производстве цемента для
.цоколей электроламп.
Важное значение чистые глицеро-фталевые смолы (глифтали)
имеют для производства слюдяной изоляции — так называемых
миканитов. Высокая клеящая способность, теплостойкость, неплав-
кость и сравнительно высокие диэлектрические свойства сделали
глифтали во многих случаях незаменимыми в высоковольтной эле-
ктротехнике. Весьма ценным для указанного назначения является
их дугостойкость, стойкость против ползучести тока.
Миканит на глифталевой основе имеет значительно более высо-
кие показатели диэлектрических свойств, чем на основе резольных
смол или шеллака (который ранее применяли для производства
миканита), а также более высокую термостойкость и прочность.
Так, глифталевый миканит по объемному и поверхностному эле-
ктрическому сопротивлению в 4—6 раз превосходит соответствую-
щий миканит на основе шеллака и в 10—15 раз — миканит на
основе резола; в несколько меньшей степени он превосходит эти
материалы по электрической прочности и диэлектрическим потерям.
Процесс производства глифталевого миканита .складывается из
следующих операций; покрытия и склеивания слюдяных пластинок
раствором смолы; высушивания в вакуумной сушилке при 80—90°
с последующей сушкой при 180—200° до частичного перехода смолы
в стадию В; прессования склеенных листов при 170—180° и охла-
ждения. Кроме этих операций иногда производят последующую
термообработку при 150—180° до полного отверждения.
В последние годы глицеро-фталевые смолы применяют для про-
изводства конструкционных электроизоляционных материалов на
основе стеклоткани, что весьма важно для повышения рабочих тем-
ператур электрических машин и увеличения их мощностей. Такого
рода стеклянные изоляции могут работать при 150° и выше.
Более широкого применения прессизделия из глифталей не по-
лучили, вследствие весьма малой скорости их отверждения. Различ-
ные приемы, которые были предложены для ускорения прессования
глифталей, не дали еще положительных результатов.
Пластмассы на основе синтетических полиамидов
591
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ СИНТЕТИЧЕСКИХ ПОЛИАМИДОВ
(Амидопласты)
Важнейшее достижение синтетической химии последних лет —-
промышленное производство высокомолекулярных полиамидов. На
основе этих смол впервые удалось получить синтетическое волокно,
которое по комплексу своих свойств близко к таким важным бел-
ковым волокнам, как шелк и шерсть, а в некоторых отношениях
даже превосходит их.
Хотя полиамидные смолы впервые были синтезированы еще
в 1899 г., промышленное производство их началось лишь в 1940 г.
Оно основано на исследованиях В. Карозерса, который разработал
научные и технические основы производства полиамидов. Работы
большого теоретического и практического значения в этой области
принадлежат В. В. Коршаку, 3. А. Роговину, И. Л. Кнунянцу и др.
В промышленных условиях полиамидные смолы получают на
основе следующих реакций:
1) поликонденсации диаминов с дикарбоновыми кислотами
(стр. 525):
H2NRNH2 + HOCOR1COOH —>
—> H2NRNH[COR1CONHRNH]j.COR1COOH + (2х + 1)Н2О
(7? и Ri — не менее 4 метиленовых групп);
2) аутоконденсацип «-аминокислот:
H2NRCOOH —> H2N[RCONH]a.RCOOH + (x+l)H2O
(R — не менее 5 метиленовых групп);
3) полимеризации циклов—лактамов г-аминокислот (стр. 597).
Из других реакций, также приводящих к получению полиамидов,
но имеющих меньшее практическое значение, следует отметить:
4) конденсацию диаминов с динитрилами в присутствии воды:
NCRCN+H2NR1NH2 HOCORCO[NHRjNHCORCO]3,NHR1NH2+(2x+2)NH3
или конденсацию аминонитрилов в присутствии воды:
IUNRCN H2NRCO(NHRCO)a.NHRCOOH + (х + 2)NH3;
5) конденсацию двухосновных кислот с диизоцианатами:
HOCORCOOH + OCNRjNCO —>
—>- HOCORCO[’SHR,NHCORCO]^NHRtNCO + (2х + 1)СО,;
6) конденсацию циклических имидов с диаминами:
СО
R\ /NH + H2NR!NH2 —> HaNCORCOfNHRtNHCORCOJa, NHRiNH2 + x NH3;
^CO
592
Гл. XIV. Пластмассы, на основе полиэфиров, полиамидов
7) конденсацию мононитрилов двухосновных кислот с диаминами:
NCRCOOH + H^RjNHj —► NCRCO[NHR1NHCORCO]a,NHR1NH2-(-
+ xNH3 + H2O.
По структуре цепи различают следующие два типа полиамидов;
1. ..NH(CH2)xNHCO(CH2)!/CONH(CH2)a.NHCO(CH2)j/—..
получаемые поликонденсацией диаминов с дикарбоновыми кисло-
тами, и
2. .МН(СН2),СОШ(СН2)ЖСОНН(СН2)Ж—..
получаемые поликонденсацией а>-аминокислот и полимеризацией их
лактамов.
Несмотря на кажущееся сходство обеих структур, в них имеется
существенное отличие: в полимерах первого типа макромолекулы
построены таким образом, что чередующееся число метиленовых
групп между диполями может быть различно, при этом последова-
тельно меняется и порядок расположения дипольных групп, в то
время как полимеры второго типа построены по принципу последо-
вательного присоединения одной и той же мономерной структурной
единицы.
Полиамиды получают как на основе поликонденсации (главным
образом диаминов с дикарбоновыми кислотами, а также “-амино-
кисл от), так и на основе ступенчатой полимеризации лактамов
е-аминокислот. Оба процесса могут привести к одной и той же
структуре макромолекулярной цепи, например:
поликон-
NH2(CH2)6COOH -?^дция
СН2— (СН^—со
!---NH---1
поли-
меризация
Н^ЦСНЖСОГШЫСНгЬСООН.
Основными видами сырья для производства полиамидов являются адипино-
вая кислота, гексаметилендиамин и капролактам.
Исходным сырьем для получения адипиновой кислоты и гексаметилен-
диамина служит фенол (получаемый в основном синтетически из бензола).
Реакции протекают по следующим схемам:
ОН
фенол
он
Хн
СИ
н2с/ ХСН2
Hgci^ Jch2
сн2
циклогексанол
о
II
с
Н2с/ ХСН,
H2Cl\ Jch2
сн2
циклогексанон
с</°
I хон
с^о
СН, I хон
\ /СН2
ХСН2
адипин вая
кислота
Пластмассы на основе синтетических полиамидов
593
0 0
НОСО(СН2)4СООН —’-> NH4OCO(CH2)4COONH4 ^5-» H2NC(CH2)4CNH2 —
адипиновая кислота аммонийная соль диамид
-2^^. NC(CH2)4CN —* H2NCH2(CH2)4CH2NH2.
адипонитрил
гексаметилендиамин
Для получения адипиновой кислоты фенол гидрируют при 135—150° и давле-
нии 15—20 аг в присутствии никеля в качестве катализатора и получают цикло-
гексанол, который окисляется 65—68% азотной кислотой при 45—60° через цикло-
гексанон в адипиновую кислоту.
Гексаметилендиамин образуется при пропускании паров адипиновой кислоты
и аммиака над катализатором (фосфат бария и др.) при 320—380°, при этом
образуется адипонитрил (динитрил адипиновой кислоты); в качестве промежу-
точного продукта реакции образуется диамид, который при высокой температуре
над катализатором дегидрируется. Адипонитрил восстанавливается при 160° под
давлением 20 ат в гексаметилендиамин.
Ввиду дефицитности фенола техническое применение получили методы про-
изводства адипиновой кислоты и гексаметилендиамина из фурфурола, через
фуран, тетрагидрофуран, дихлорбутан и адипонитрил по схеме:
НС---СН о НС СН 2Нг Н2С СН2 2НС1 Н2С СН2 2NaCN
II II. —со/* I II * I I -нао”* I
нс с—сн=о нс СН Н2С сн2 2 Н2С сн2
V ci ci
фурфурол фуран тетрагидро- дихлорбутан
фуран
СН2—СН2
Ан2 сн2
CN CN
адипонитрил
4Н2 । 4Н2О
сн2—сн2
сн2 сн2
СН2 СН2
I I
nh2 nh2
гексаметилен-
диамии
сн2—сн,
I I
сн2 сн2
ХОН \он
адипиновая
кислота
Адипиновая кислота и гексаметилендиамии могут быть получены также из
ацетилена и формальдегида. При конденсации ацетилена с двумя молекулами
формальдегида в присутствии ацетиленидов меди и других тяжелых металлов
образуется бутиндиол, который при гидрировании переходит в 1,4-бутандиол.
Последний при дегидратации дает тетрагидрофуран, который через дихлорбутан
и адипонитрил по вышеуказанной схеме переходит в адипиновую кислоту и
38 Зак. 1269. Э. И. Барг
594
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
гексаметилендиамин:
СН=СН + 2СН2О —> СН2ОН—С=С—СН2ОН —>
бутинднол
н2с—сн2
сн2он—сн2—сн2—сн2он н2с сн2
О бутандиол тетрагидрофуран
СН2—СН2С1 —СНа—СН,—CN
СН2—СН2С1 дихлорбутан СН2—СН2—CN I СН2—СН2—СН2—NH, адипонитрил —> 1 4- СН2—СН2—СН2— NHj сн2—СН2—СООН гексаметнлендиамин СН2—СН,—СООН адипиновая кислота
1,4-Бутандиол, образующийся при гидрировании бутиндиола, является одним
из важнейших продуктов для получения полиуретанов (стр. 607).
Капролактам — мономер для синтеза поликапролактама приготовляют изоме-
ризацией оксима циклогексанона (бекмановская перегруппировка).
Исходным сырьем служит фенол, который гидрированием превращают в
циклогексанол. Последний подвергается дегидрированию при 400—450° в при-
сутствии катализатора (сплав цинка с железом), в результате — образуется ци-
клогексанон.
Взаимодействием циклогексанона с гидроксиламином образуется оксим
циклогексанона (действием сернокислого гидроксиламина при 20°). При действии
на оксим циклогексанона концентрированной серной кислоты нли 15—25%-ным
олеумом происходит перегруппировка (изомеризация) оксима в лактам е-амнно-
капроновой кислоты по схеме:
ОН
I
СН
Н2с/ \сн2
н2с!^ Jch2
сн2
циклогексанон
циклогексанол
N—ОН
II
С
Н2с/ ''сн,
н2с^ Jch,
сн2
бекмановская
перегруппировка
СН2—(СН2)4—с=о
I----NH-----1
капролактам
Капролактам имеет т. плавл. 69—71°, т. кип. 258°, он растворим как в воде,
так и в бензоле, эфире и других неполярных растворителях.
Полиамиды, получ. поликонд. диаминов с дикарбонов, кислотами 595
Полиамиды, получаемые поликонденсацией диаминов
с дикарбоновыми кислотами
Технологический процесс поликонденсации дикарбоновых кислот
с диаминами основан на учете следующих закономерностей:
1) чем полнее удаляется из сферы реакции образующаяся вода,
тем выше молекулярный вес полимера;
2) чем выше температура реакции и длительность процесса, тем
в определенных пределах выше молекулярный вес полимера и сте-
пень превращения реагирующих веществ;
3) чем выше степень эквимолекулярности (чем меньше избыток
одного из реагентов), тем выше молекулярный вес полимера.
Реакция получения технических полиамидов обычно протекает
без растворителей и катализаторов.
Важнейшим техническим полимером этого класса является по-
лигексаметиленадипамид, получаемый поликонденсацией гексаме-
тилендиамина и адипиновой кислоты. Смолы этого типа применяют
для получения волокна, известного под названием найлон.
Процесс производства полигексаметиленадипамида складывается
из двух этапов:
1. Получение соли гексаметилендиамина и адипиновой кислоты
(соли АГ). Для этого оба компонента растворяют в метаноле; об-
разующаяся соль нерастворима в метаноле и выпадает в виде бе-
лого кристаллического порошка (т. плавл. 190—191°):
H2N(CH2)eNH2 + НОСО(СН2)4СООН —> H2N(CH2)6NH3OCO(CH2)4COOH.
Соль очищают от избытка реагирующих веществ перекристалли-
зацией из метанола. После очистки и сушки она является исход-
ным продуктом для получения полиамида. Применение соли обеспе-
чивает высокую степень эквимолекулярности, которая практически
недостижима при использовании смесей диамина и дикарбоновых
кислот.
2. Поликонденсация соли и получение полиамида. Процесс про-
водится в автоклавах при высоких температурах (275—280°).
Конечная температура в автоклавах должна быть несколько выше
температуры плавления полиамида (250—255°), что необходимо
для быстрой выгрузки расплавленной смолы.
Обогрев автоклава осуществляют электричеством или жидкими
теплоносителями или их парами, циркулирующими в рубашке авто-
клава. Обогрев парами обеспечивает более равномерное распреде-
ление температуры во всем объеме автоклава и более точную регу-
лировку температуры (в пределах 1—3°), чем обогрев электриче-
ством. Применяют или водяной пар высокого давления (50 ат и
выше) или пары динила (эвтектической смеси дифенила и дифенил-
оксида) при нормальном давлении.
Материал и конструкция паровой рубашки автоклава опре-
деляются характером теплоносителя. При применении динила
38*
596
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
рубашка не рассчитывается на давление выше нормального, однако
необходимо предусмотреть тщательную герметизацию.
Нагревание соли в автоклаве, весь процесс поликонденсации и
дальнейшие операции с расплавленной смолой осуществляют в ат-
мосфере азота, тщательно очищенного от кислорода (содержание
кислорода не более 0,01%). Необходимость проведения реакции
в .утствии азота объясняется тем, что при высоких температу-
рах происходит частичное окисление полимера, что приводит:
1) к изменению цвета (потемнению) смолы, крайне нежелатель-
ному, если полимер применяется для производства волокна, 2) к ча-
стичному образованию сетчатой структуры, «сшивки» макромолекул,
что уменьшает плавкость смолы и затрудняет дальнейшую пере-
работку полимера при производстве волокна (засорение фильер, за-
труднение при вытягивании нити и др.).
В состав реакционной смеси входят так называемые «стабили-
заторы», представляющие собою моноамины или одноосновные кис-
лоты. В процессе поликонденсации они блокируют концевые группы
растущей цепи полимера и делают эту цепь неактивной. Этим дости-
гают как регулирования молекулярного веса полимера, так и его
стабилизации, т. е. прекращения дальнейшего роста при плавле-
нии и нагревании. Последнее обстоятельство крайне важно при
переработке полимера, осуществляемого при высоких температурах.
Поэтому строго нормированные количества «стабилизаторов» иг-
рают весьма существенную роль в технологии производства поли-
амидов.
Для получения высокомолекулярного продукта большое значе-
ние приобретает скорость удаления из автоклава воды, выделяю-
щейся при реакции поликонденсации. Удаления воды достигают или
включением высокого вакуума (1—2 мм) или непрерывным про-
пусканием тока очищенного азота.
Реакция поликонденсации идет практически до конца, равно-
весное содержание «мономеров» составляет меньше одного про-
цента. Это является большим преимуществом по сравнению с поли-
меризацией капролактама (стр. 597, 599), где конечный продукт
реакции содержит значительные количества мономера.
Таким образом, процесс поликонденсации проходит путем нагре-
вания в автоклаве смеси соли АГ со «стабилизаторами» в атмосфере
азота. Нагревание производят первоначально при 220—230° в те-
чение 2—4 час., после чего постепенно температуру поднимают до
270—275° и выдерживают при этой температуре 1,5—3 часа до за-
вершения реакции. Во второй стадии процесса включают глубокий
вакуум или пропускают ток очищенного азота.
Контроль поликонденсации может быть проведен определением
электропроводности, кислотного числа, вязкости расплава и молеку-
лярного веса.
После завершения процесса поликонденсации полимер выдавли-
вают сжатым азотом (1,5—2 ат) из автоклава через широкую щель
Полиамиды, получ. полимеризац. циклов — лактамов е-аминокислот 597
в виде непрерывной ленты, которую пропускают через бассейн
с проточной водой для быстрого охлаждения и предотвращения
окисления и наматывают на барабан.
Ленту затем дробят на специальном рубильном станке на мел-
кую «крошку». Весьма важно, чтобы размеры кусочков смолы были
по возможности одинаковыми. Для удаления воды «крошку» сушат
в вакуум-сушилках при 60—80° до остаточной влаги 0,05%, что до-
стигается в течение 1 часа, так как влага находится только на по-
верхности смолы. Высушенный «продукт» является исходным по-
лимером для дальнейшей переработки на волокно и на изделия,
изготовляемые прессованием, литьем под давлением и т. д.
Полиамиды, получаемые полимеризацией циклов — лактамов
е-аминокислот
В промышленных условиях получают полимеры капролактама
CO(CH2)5NH.
1________L
Полимеризация лактамов представляет собою ступенчатый про-
цесс и характеризуется следующими закономерностями: 1) чем
выше температура реакции и длительнее время реакции, тем выше
молекулярный вес полимера; 2) для каждой температуры устана-
вливается равновесие между количеством полимера и мономера;
чем выше температура (до определенного предела), тем при боль-
шем отношении полимера к мономеру устанавливается равновес-
ное состояние; 3) активаторами реакции являются вода, органиче-
ские кислоты, металлический натрий, соли и др., в то время как
кислород и перекиси существенно не влияют на скорость реакции и
на молекулярный вес полимера.
Действие воды в качестве активатора при полимеризации ка-
пролактама может быть выражено, согласно 3. А. Роговину, сле-
дующей схемой: >
HN(CH2)5CO H2N(CH2)6COOH.
I I
HOCO(CH2)6NH2 + CO(CH2)5NH —> HOCO(CH»)5NHCO(CH,)6NH2
I I
i ;
HOCO(CH?)6NHCO(CH2)6NH2 + CO(CH2)6NH —►
—► HOCO(CH2)6NHCO(CH2)5NHCO(CH2)6NH2 и t. д.
Образующаяся в результате взаимодействия с водой аминокис-
лота реагирует своей аминогруппой со следующей молекулой капро-
лактама; продукт присоединения имеет линейную структуру и содер-
жит на конце аминогруппу, которая, в свою очередь, реагирует с
капролактамом, и т. д.
598
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
Имеется, однако, вероятность, что наряду с механизмом ступен-
чатой полимеризации действует и механизм поликонденсации.
В этом случае образующиеся в результате действия воды молекулы
аминокислоты реагируют друг с другом, выделяющаяся при этом
вода непрерывно превращает новые молекулы лактама в молекулы
аминокислоты и т. д. по схеме:
2CO(CH2)5NH + 2Н2О —> 2HOCO(CH2)5NH2 —>
HOCO(CH2)5NH2 + HOCO(CH2)5NH2 —> HOCO(CH2)5NHCO(CH2)5NH24-H2O.
Повидимому, поликонденсация образующихся аминокислот про-
текает значительно медленнее полимеризации капролактама.
Количество воды, необходимое для активации процесса поли-
меризации, составляет несколько процентов (1—3%). Степень поли-
меризации тем ниже, чем больше введено воды. Эта зависимость
в некоторой степени аналогична влиянию инициаторов в процессах
радикальной полимеризации, хотя все остальные закономерности
образования полиамидов из лактамов соответствуют в основном ме-
ханизму ступенчатой полимеризации.
Активаторами реакции полимеризации капролактама могут слу-
жить также моно- и дикарбоновые кислоты. Реакция, вероятно, идет
по следующей схеме:
-J . I
RCOOH4-HN(CH2)5CO —> RCONH(CH2)5COOH и т. д.
Капролактам полимеризуется с чрезвычайной быстротой в при-
сутствии металлического натрия (0,2—0,5%) или натриевой соли
лактама. Вначале металлический натрий нагревают с лактамом при
100° до прекращения выделения водорода:
2CO(CH2)5NH + 2Na —> 2CO(CH2)5NNa ф-Н2.
I I I I
После этого температуру поднимают до 230—250°. Реакция образо-
вания полимера заканчивается в течение нескольких минут.
Механизм реакции полимеризации лактамов нельзя считать
в достаточной степени установленным. Повидимому он в известной
мере различен для каждого вида «активатора». Возможно, что про-
цессы полимеризации циклов имеют свои закономерности и про-
ходят по особому механизму ступенчатой полимеризации. Реакция
полимеризации в присутствии натрия имеет, повидимому, цепной
характер.
Технологические параметры и аппаратурное оформление про-
цесса производства поликапролактама (капрона) в основном такие
же, как при получении поликонденсационных полиамидов на
основе диаминов и дикарбоновых кислот (соли АГ). Вместе с тем
Полиамиды, получ. полимериз. циклов—лактамов е-аминокислот 599
имеются и существенные отличия. Эти отличия обусловлены введе-
нием в состав реакционной массы воды в качестве активатора
процесса и наличием в конечном продукте реакции (в смоле) зна-
чительных количеств мономера и низкомолекулярных фракций. По-
лимеризация в присутствии воды делает необходимым ведение про-
цесса полимеризации под высоким давлением (15—20 ат), что не
обязательно для процесса поликонденсации соли АГ.
Автоклав должен быть рассчитан также и на глубокий вакуум,
который желательно включать в последнюю стадию процесса по-
лимеризации для отсоса воды и мономера.
Наличие в смоле в равновесном состоянии значительного коли-
чества мономера объясняется обратимым характером реакции по-
лимеризации капролактама:
n[HN(CH2)6CO] ...—HN(CH2)6COHN(CH2)6COHN(CH2)6CO—...,
при этом чем выше температура реакции (начиная с 220°), тем
больше равновесие сдвигается влево (в сторону мономера), что в
итоге приводит к увеличению содержания мономера в смоле.
Так при температуре полимеризации 230° смола содержит 3—4%
мономера, а при 290° 10—12%.
Содержание в смоле мономера и водорастворимых низкомоле-
кулярных фракций уменьшает прочность смолы и затрудняет ее
переработку на волокно. Поэтому необходимо удалять их из смолы,
что вызывает ряд дополнительных операций, которые не требуются
в процессе производства поликонденсационных полиамидов.
Эти дополнительные операции заключаются в экстракции моно-
мера горячей водой .из измельченной смолы («крошки»), в отжиме и
сушке смолы, а также выпаривании больших количеств воды (до
50 кг на 1 кг смолы) для регенерации капролактама.
Таким образом, процесс производства поликапролактама скла-
дывается из следующих операций: 1) плавления капролактама в
плавителе, смешения с активатором и стабилизатором и загрузки
в автоклав; 2) полимеризации капролактама в автоклаве; 3) вы-
грузки и формовании ленты; 4) измельчения ленты; 5) экстракции
мономера; 6) отжима и сушки смолы.
Плавление капролактама и смешение его с активатором (вода)
и стабилизатором (уксусная, салициловая, адипиновая и другие
кислоты) ведут в отдельном аппарате — плавителе в токе азота,
тщательно очищенного от кислорода. Полимеризацию реакционной
смеси проводят в автоклаве при 240—270° и давлении 15—20 ат.
Минимальная конечная температура реакции должна быть выше
температуры плавления полимера (215°).
Оптимальную температуру устанавливают, учитывая влияние
последней как на скорость процесса реакции, выход полимера,
так и на скорость образования мономера (рис. 211), а оптимальное
600
Гл. XfV. Пластмассы, на основе полиэфиров, полиамидов
время реакции — в зависимости от температуры реакции и количе-
ства активатора. Чем выше температура реакции и больше содержа-
ние активатора, тем меньше время полимеризации. С увеличением
времени полимеризации возрастает как молекулярный вес, так и
выход полимера (рис. 212). Обычно реакция продолжается не-
сколько часов. К концу реакции включают глубокий вакуум для
отсоса воды и мономера. Количество активатора (воды) не влияет
Температура полимеризации
Рис. 211. Влияние температуры
иа процесс полимеризации ка-
пролактама и свойства полиме-
ров.
1—выход полимера; 2—его молеку-
лярный вес; 3—время реакции; 4—со-
держание мономера в смоле.
Рис. 212. Влияние времени полиме-
ризации капролактама на выход и
свойства полимера.
1—выход полимера; 2— его молекулярный
вес; 3—содержание мономера в смоле.
на равновесное содержание мономера, но снижает молекулярный
вес полимера и время реакции.
Дальнейшие процессы — формование ленты и измельчение по-
лимера — те же, что и в производстве полигексаметиленадипамида.
Экстракция капролактама и низкомолекулярных водораствори-
мых веществ (циклических димеров и тримеров) производится об-
работкой измельченной смолы водой при повышенных температу-
рах. При этом в раствор переходят около 8—-10% от веса смолы.
После экстракции смолу отжимают на центрифугах; при этом
полимер содержит еще ~12% воды, которую удаляют путем дли-
тельной -сушки в вакуум-сушилках.
Готовая смола для переработки на волокно должна содержать
не более 1% мономера и 0,1% воды.
Поликапролактам (капрон) имеет температуру плавления 215°,
более низкую, чем полигексаметилендипамид (255°). Молекулярный
вес капрона должен быть в пределах 16000—22000; он определяется
по удельной вязкости 1 % раствора в крезоле или серной кислоте
(84 %-ной).
Для переработки поликапролактама на волокно существенное
значение приобретает его вязкость в расплавленном состоянии.
Вязкость расплава зависит как от молекулярного веса полимера,
так и от наличия в смоле мономера, воды и низкомолекулярных
фракций. Ничтожно малые количества капролактама (~0,1—0,3%)
Свойства и переработка пол иамидов
601
снижают в несколько раз вязкость расплава. Поэтому строгий
контроль концентрации этих примесей обязателен для получения
смолы с заданной вязкостью расплава.
Свойства и переработка полиамидов
Полиамиды, полученные по механизму полимеризации или поли-
конденсации, представляют собою твердые высокоплавкие смолы
микрокристаллического строения.
Интервал плавления обычно нахо-
дится в пределах 3—5°, что косвенно
свидетельствует о высокой концен-
трации кристаллической фазы и ма-
лой полидисперсности полимера.
Молекулярный вес технических
полиамидов находится в пределах
8000—25000. Несмотря на сравни-
тельно небольшую степень полиме-
ризации, они представляют собою
эластичные, прочные, высокоплавкие
материалы (т. плавл. 180—250°).
Прочность полиамидов в не-
растянутом состоянии ~ 400—
450 кг/см2, при вытяжке в 350—
500% она возрастает примерно в
10 раз (до 4000—4500 кг/см2); соот-
Рис. 213. Диаграмма растяжения
полиамидных смол в зависимости
от ориентации.
ветственно удлинение при растяжении резко снижается (рис. 213).
Ориентированные нити не релаксируют вплоть до температур,
близких к температуре плавления.
Прочность полиамидов определяется действием водородной
связи между цепями, наличием сильных диполей, высокой концен-
трацией кристаллической фазы и ориентацией кристаллитов (в рас-
тянутом состоянии). Вытяжка полимеров имеет большое техниче-
ское значение (в процессе получения волокна и пластиков), она
производится при температурах, значительно более низких, чем
температура плавления. В большинстве случаев изделия из поли-
амидов в той или иной степени ориентированы.
Температура плавления смолы тем ниже, чем длиннее исходные
реагирующие молекулы, т. е. чем больше метиленовых групп распо-
ложено между диполями. Вместе со снижением температуры пла-
вления увеличивается растяжимость, эластичность полимера и
облегчаются условия переработки. Полимеры с четным числом угле-
родных атомов между диполями имеют большую температуру пла-
вления, чем полимеры с ближайшим нечетным числом. Приведенные
в табл. 64 данные характеризуют зависимость температуры плавле-
ния полимера от числа углеродных групп в реагирующих
молекулах.
602 Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
Различие в температурах плавления полимеров с четным и не-
четным числом углеродных атомов в звене может быть объяснено
расположением диполей в зигзагообразных цепях. Наибольшее число
связей за счет взаимодействия дипольных групп соседних цепей
(—NH—СО—) может образоваться лишь в случае четного
числа СН2-групп в кислоте и амине;
в случае же нечетного — взаимодей-
ствует лишь половина соседних ди-
полей. Это относится также и к во-
дородным связям, которые могут
устанавливаться между NH- и СО-
группами соседних цепей (рис. 214).
Все это приводит, естественно, к бо-
лее высоким температурам плавле-
ния «четных» полимеров.
ТАБЛИЦА 64
Температуры плавления (°C) полиами-
дов в зависимости от числа атомов
углерода в образующих их
компонентах
нсвые кислоты С. св С, с, ^8 Св С10 с„ с„
С6 278 223 250 226 235 204 230 209
с7 233 183 202 196
С8 250 202 217 — 203 — — — —
с9 223 178 185 — 197 163 194 168 ——
Сю 239 195 209 187 — 175 — — 172
Сц 208 173
Если в молекуле полимера водо-
род в NH-группах полностью или ча-
Рис. 214. Схема расположения це-
пей при четных и нечетных чис-
лах углеродных атомов в исход-
ных молекулах.
1—кислота «четная» и амин «четный». Все
NH и СО принимают участие в образовании
водородной связи. Температура плавления
высокая. 2— кислота «нечетна.», амин «чет-
ный». Половина всех NH- и СО-групп при-
нимают участие в образевании водородной
связи. Температура плавления низкая.
стично замещен метильными, этиль-
ными или другими радикалами, то
пропорционально замещению и дли-
не радикала снижается температура
плавления кристаллической фазы,
или же способность к кристаллиза-
ции настолько снижается, что поли-
мер остается в аморфном состоянии.
Для получения алкилзамещенных полиамидов в состав реаги-
рующих молекул вводят также алкилированные ю-аминокислоты
алкилкапролактамы или алкилзамещенные диамины. Образующаяся
цепь имеет тогда следующую структуру;
..CO(CH2)a.CON(CH2)2ZNHCO(CH2)a.CON(CH2)2ZNHCO(CH2)a.CONH—...
R R
Свойства и переработка полиамидов
603
Зависимость температуры плавления полиамида от процента за-
мещения приведена в табл. 65.
Полиамидные смолы нерастворимы в обычных растворителях,
но хорошо растворяются в горячей муравьиной и уксусной кислотах
и в фенолах. Они относительно стойки к щелочам, но чувствительны
к кислотам, под воздействием которых легко гидролизуются; прак-
тически они негорючи и весьма трудно вос-
пламеняются.
Полиамиды широко применяются в каче-
стве пластиков для производства различ-
ных изделий. Для этих целей пользуются
почти всеми методами пластицирующей пе-
реработки.
Так полиамиды могут быть переработаны
методом простого литья; при температуре
лишь на несколько градусов выше темпера-
туры плавления их можно заливать в метал-
лические или гипсовые формы.
Изделия из полиамидов могут быть
изготовлены и методом прессования и литья
под давлением. При этом необходимо счи-
таться с малым интервалом плавления поли-
амидов и быстрым переходом их в жидкое
состояние; эта их особенность делает необ-
ТАБЛИЦА 65
Температура плавлений
N-метилзамещенного
полипекаметиленсе-
бацамида в зависи-
мости от содержания
метильных групп
Содержание СН-групп °, о Температура плавления полиамида, °C
0 196
20 183
50 152
80 108
100 63
ходимым введение соответствующих изменений в конструкции пресс-
форм и автоматов для литья под давлением, а именно, — уплотнений
и запоров для предохранения массы от вытекания.
Применение получили также методы переработки полиамидных
смол вальцеванием, каландрированием, вытяжкой, штамповкой и
даже выдуванием. Для этих целей рациональнее, однако, применять
модифицированные замещенные полиамиды, отличающиеся боль-
шим интервалом плавления, например смолы, получаемые конден-
сацией декаметиленмонометилдиамина с себациновой кислотой.
Полиамиды перерабатывают также на шнек-машинах для полз -
чения труб, стержней, лент и т. п. И в этом случае смолы с боль-
шим интервалом размягчения значительно легче поддаются процессу
обработки. Процесс обработки на вальцах, каландрах и в особенно-
сти на шнек-машинах обычно сочетается с процессом вытяжки, ко-
торая при этих методах обработки может доходить от 50 до 500%.
Вытяжка может быть произведена в одном направлении (например,
для труб, брусков, нитей) или в двух (например, для лент и пла-
стин). Вытяжка в двух взаимноперпендикулярных направлениях
обусловливает повышенную прочность как в продольном, так и в
поперечном направлениях, но общий процент вытяжки при этом со-
ответственно уменьшается.
Основное применение полиамиды имеют в производстве
волокна — капрон, получаемого полимеризацией капролактама, и
604
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
найлон, получаемого поликонденсацией гексаметилендиамида с ади-
пиновой кислотой.
Для изготовления волокна полимер в виде «хлопьев» («крошки»)
загружают в закрытые алюминиевые или стальные аппараты, сна-
бженные расплавительной решеткой. В этих аппаратах при 260—
270° в атмосфере азота производят плавление полимера; расплав
продавливают через фильеры, образующиеся волокна охлаждают
в шахте и наматывают на бобины.
Агрегаты для расплавления и прядения нитей состоят из: за-
грузочного бункера; дозирующего приспособления; аппарата, сна-
бженного плавительной решеткой с электрическим обогревом;
прядильного устройства, состоящего из насоса, фильтра и фильер,
через которые продавливается расплав; воз-
душной шахты высотой 4—6 м, через кото-
рую проходит охлаждаемое волокно; прием-
ного механизма для накручивания нити, а
также для ее растяжения и кручения.
Образующиеся нити проходят через на-
тяжные приспособления и наматываются на
бобины, после чего их обрабатывают на
прядильных машинах для получения пряжи.
Для увеличения прочности и гибкости пря-
жу подвергают холодной вытяжке (в 4—6
раз), что достигается протяжкой ткани на
специальных машинах между валками, вра-
щающимися с различной скоростью. Вы-
Удлинение, %
Рис. 215. Диаграмма рас-
тяжения полиамидного
волокна в сухом и мок-
ром состояниях.
расплава значительно
тяжку можно производить и непосредственно
при получении нитей, перед обработкой на
прядильных машинах для получения пряжи.
Скорость прядения полиамидных нитей из
выше (раз в десять), чем скорость пряде-
ния других искусственных волокон, обычно получаемых из рас-
твора.
Полиамидное волокно является более прочным, чем природные
волокна, и по внешнему виду напоминает шелк.
В отличие от шелка, полиамидное волокно имеет меньшую
гигроскопичность и большую прочность во влажном состоянии
/рис. 215).
Однако более низкая гигроскопичность полиамидных волокон
не является их преимуществом, так как она обусловливает меньшую
скорость сорбции и десорбции влаги (пота) и этим ухудшает гигие-
нические свойства ткани.
При удлинении до 4% полиамидное волокно полностью упруго
и возвращается к исходной длине, в то время как шелк в этих усло-
виях дает значительную остаточную деформацию. Прочность поли-
амидного волокна достигает 40—60 кг!мм2, тогда как природный
шелк имеет прочность до 35 кг/мм2.
Пластмассы на основе полиуретанов и полимочевин 605-
Из полиамидного волокна изготовляют чулки, ткани, трикотаж,,
технические ткани (нити, шнуры, канаты, сети), а также щетины,
например для платяных, зубных и других щеток.
Полиамидные смолы обладают также хорошими диэлектриче-
-скими свойствами, благодаря чему их применяют для производства
изоляционной оболочки для кабеля, заменяя одновременно ткане-
вую, бумажную, каучуковую изоляцию и свинцовую оболочку.
Некоторое и более ограниченное применение полиамиды нахо-
дят в качестве лаков, эмалей, заливочных и уплотняющих масс.
Кроме того их используют для пропитки ткани, бумаги, для произ-
водства приводных ремней, шлангов, уплотняющих пластин, клеевг
замазок и т. д.
Пластические массы на основе полиуретанов и полимочевин
Реакция образования уретанов — эфиров карбаминовых кислот,
основанная на взаимодействии между изоцианатом и спиртом, была
открыта Вюрцем еще в 1848 г.
RN=C=O4-HOR' —> RNHCOOR'.
Обычно уретаном называют этиловый эфир карбаминовой кис-
лоты NH2COOC2H5.
Конденсация уретана с формальдегидом в кислой среде, а также с мочеви-
ной и формальдегидом приводит к образованию светлых, низкомолекулярных
продуктов типа мочевино-формальдегидных смол.
В качестве промежуточного продукта в сильнокислой среде образуется мети-
ленуретан, который, повидимому, является полимерным веществом:
NH2COOC2H54-CH2O —> (CH2=NCOOCiH5)a,
В нейтральной илн слабокислой среде при этом образуются метилольные
производные, конденсацией которых можно получить светлые, плавкие и термо-
реактивные смолы. Эти смолы, однако, не нашли сколько-нибудь значительного
применения.
Практическое значение получили реакции полиизоцианатов
с многоатомными спиртами или с диаминами, ведущие к образова-
нию новых типов линейных высокополимеров — полиуретанов и
полимочевин.
Полиуретаны образуются при полимеризации диизоцианатов с
многоатомными спиртами по следующей схеме;
HOROH 4- OCNR'NCO —> HOROCONHR'NCO
I t
HOROCONHR'NCO + HOROH —> HOROCONHR'NHCOOROH н т. д,
t I
или вообще:
x HOROH + x OCNR'NCO —> (—OROCONHR'NHCO—) x.
606
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
Полимочевины получают полимеризацией диизоцианатов и ди-
лминов по аналогичной схеме:
H2NRNH2 + OCNR'NCO —> HjjNRNHCONHR'NCO
I t
H2NRNHCONHR'NCO + H2NRNH2—> H2NRNHCONHR'NHCONHRNH2 И t. д
дли вообще
xNH2RNH2 + xOCNR'NCO —► (—NHRNHCONHR'NHCO—)x.
Известны также реакции взаимодействия диизоциаиатов с ди-
карбоновыми кислотами, однако они приводят к образованию поли-
амидов (стр. 591).
Если 7? и R.' — линейные радикалы с числом углеродных атомов
ле менее четырех, естественно, можно ожидать образования высоко-
молекулярных линейных полимеров.
Процессы образования полиуретанов и, возможно, полимочевин
представляют собою реакцию ступенчатой полимеризации, которая
Рис. 216. Влияние соотношения компонен-
тов на вязкость полимера при взаимодей-
ствии бутандиола с гексаметилендиизоциа-
натом.
/—в 95»,1о HsSO4; 2—в крезоле.
основана на миграции по-
движного водорода гидро-
ксильной или аминогруппы
к атому азота изоциановой
группировки. На каждом
этапе образуются изолируе-
мые, способные к дальней-
шей полимеризации молеку-
лы. Реакция протекает без
катализаторов с большим
выделением тепла и сравни-
тельно с большой скоростью
при невысоких температурах
и даже на холоду. Влияние
избытка одного из компо-
нентов должно сказываться
на величине молекулярного
веса в такой же степени,
как и при реакциях дикарбоновых кислот с полиспиртами или
с диаминами; это экспериментально подтверждено в работах
Л. А. Стрепихеева, А. А. Артемьева и Я. А. Шмидта при изучении
реакции взаимодействия бутандиола с гексаметилендиизоцианатом
(рис. 216).
В случае реакции типа 2, 3, например при взаимодействии диизо-
Пластмассы на основе полиуретанов и полимочевин
607
цианатов с трехатомными спиртами, получаются пространственные
полимеры по следующей схеме:
n R(OH)3 + л OCNR'NCO —► ..R'NHCOO—RO—CONHR'— ...
1
О
CO
NH
R'
NH
CO
О
...—R'NHCOO—R—OCONHR'—...
Молекулярный вес полимера зависит не только от избытка
одного из реагентов, но и от наличия в реакционной смеси моно-
функциональных соединений, обрывающих рост цепи, а также от
концентрации реагирующих компонентов и температуры реакции.
Важнейшим исходным сырьем для получения технических полиуретанов
являются гексаметилендиизоцианат OCN(CH2)6NCO и бутандиол НО(СНг)4ОН.
Применение нашли, повидимому, также и другие диизоцианаты, например то-
лунлендиизоцианат
СН3
Q-NCO
NCO
а также некоторые триизоцианаты, например:
Н
I I
Y
NCO
Ди- и триизоцианаты образуются при действии фосгена на соответствующие
амины.
Для получения толуилендиизоцианата исходят из фосгена и толуилендиамина:
сн3 сн3
NH2 +2СОС12 —► Y~NCO _|_4НС!-
Y Y
NH2 NCO
Реакцию проводят в дихлорбензольном растворе при 50—150°.
608
Гл. XIV. Пластмассы на основе полиэфиров, полиамидов
Гексаметилендиизоцианат получают действием фосгена на гексаметиленди-
амин в дихлорбензольном растворе. Реакция протекает в две стадии, с проме-
жуточным образованием карбамата:
. H2N(CH2)cNH2 + CO2 —> H3N(CH2)6NHCOO
H3N(CH2)eNHCOO + 2COCL, —>- OCN(CH2)6NCO + 4HC1 + CO,,.
I I
1,4-Бутандиол получают из ацетилена и формальдегида через бутиндиол
(стр. 594).
Реакцию получения полиуретановых смол из гексаметилендиизо-
цианата и 1,4-бутандиола проводят в присутствии растворителей
(смесь хлорбензола и дихлорбензола) при кипении смеси.
В стальной реактор, снабженный обратным холодильником, за-
гружают смесь растворителей, в которую затем добавляют 1 моль
бутандиола и после нагревания до 60° 1 моль гексаметилендиизоциа-
ната. Затем температуру доводят до кипения смеси и в холодильник
пускают воду. Реакция завершается после 4—5 час. кипения и поли-
мер осаждается в виде порошка или хлопьев. Продукт реакции от-
фильтровывают и растворитель отгоняют острым паром, после чего
полимер высушивают под вакуумом при 65°.
В зависимости от исходных диизоцианатов и полиспиртов, а так-
же от характера растворителя и его концентрации скорость реакции
может быть различной. В некоторых случаях реакция с большой
скоростью протекает уже при обычных температурах.
Значительно более активно протекают процессы образования
полимочевин. Если приливать разбавленный раствор диамина к рас-
твору дийзоцианата при обычной температуре или при охла-
ждении, то практически мгновенно, со скоростью, близкой к ион-
ным реакциям, выпадает осадок полимочевины. Естественно, что
при столь высокой скорости реакции существенную роль играет ско-
рость загрузки компонентов, например, скорость загрузки диамина
в диизоцианат, так как этим определяется сохранение условий
эквимолекулярности. Было установлено, что особую роль играет
характер среды, в которой протекает реакция; высокомолекуляр-
ные полимеры получаются лишь в среде хлорбензола. Реакция ве-
роятно протекает по цепному механизму.
Свойства полиуретановых и полимочевинных смол обусловлены
теми же закономерностями, что и свойства полиэфирных и поли-
амидных смол. Чем больше число углеродных атомов между
полярными функциональными группами, тем ниже температура пла-
вления, тем выше растяжимость, эластичность, а также раствори-
мость полимера (табл. 66).
Так же, как и в случае полиамидных смол (стр. 601), темпера-
тура плавления полиуретанов с четным числом углеродных атомов
в исходном изоцианате выше, чем полиуретанов на основе изо-
цианата с нечетным числом углеродных атомов (рис. 217).
Пластмассы на основе полиуретанов и поламочевин
609
Модификация свойств, в частности улучшение растворимости,
может быть достигнута применением алкил- или арилпроизводны.х
диизоцианатов.
Улучшение растворимости имеет весьма-существенное значение,
так как многие «чистые» полиуретаны и полимочевины не раство-
ряются почти ни в каких растворителях, за
исключением смеси фенола и воды (90 : 10).
Значительное улучшение растворимости
может быть достигнуто также, если прово-
дить совместную полимеризацию диизоциа-
ната с двумя или тремя гликолями. Напри-
мер, при совместной полимеризации 1,4-бу-
тандиизоцианата с 1,4-бутандиолом и 1,6-
гександиолом получается полимер, полно-
стью растворимый в хлорированных угле-
водородах и совмещающийся с пластифика-
торами.
Полиуретановые смолы по комплексу
своих свойств весьма близки к полиамидам.
Так же, как и полиамиды, они представляют
собою чаще всего высоко-плавкие линейные
кристаллические полимеры, способные при
вытяжке давать ориентированно-кристал-
лическую структуру — структуру волокна.
ТАБЛИЦА 66
Температура плавле-
ния различных поли-
уретанов в зависимо-
сти от числа углерод-
ных атомов в исходных
молекулах
Число атомов углерода Темпера- тура плавления °C
В ДИИЗы- цианате в гликоле
4 4 190
4 (i 180
4 10 170
6 3 167
6 5 159
6 9 147
12 12 128
Полиуретаны при одинаковом числе СНг-групп имеют более
Число атомоЁ углерода
в изоцианате
Рис. 217. Температура плавле-
ния полиуретанов в зависимо-
сти от числа атомов углерода
в исходном изоцианате.
приведены сравнительные
низкую температуру плавления, чем
полиамиды, а также более высокую
водостойкость и кислотность.
Более низкая температура плавле-
ния облегчает условия пластицирую-
щей переработки полиуретанов (валь-
цевание, прессование и т. д.), а также
условия формования волокон. Мень-
шая гигроскопичность и большая кис-
лотостойкость являются крупным пре-
имуществом полиуретанов, так как
полиуретановое волокно, в отличие от
полиамидного, применяется почти ис-
ключительно для производства техни-
ческих тканей. По механической проч-
ности полиуретаны несколько уступают
полиамидам. Они, однако, их превос-
ходят по морозостойкости. В табл. 67
показатели свойств среднеориентирован-
ных полиамида и полиуретана.
Полиуретаны и полимочевины так же, как и полиамиды, пере-
рабатывают на волокно (вытяжка порядка 400—600%), на пла-
39 Зак 1269. Э. И. Барг
610 Гл. IV. Пластмассы на основе полиэфиров, полиамид ов
станы, шланги, трубы (вытяжка 100—200%) или на прессизделия
(ориентация 50—100%).
Волокно из полиуретанов применяют преимущественно для про-
изводства щетины, сетей, фильтровального полотна, электроизоля-
ционной ткани и т. д. Метод производства волокна аналогичен ме-
тоду производства волокна из полиамидов.
ТАБЛИЦА 67
Сравнительные свойства среднеориентированных
полиуретана и полиамида
Свойства Полиуретан Полиамид
Уд. вес, г/см3 Температура плавления, °C . . . Твердость по Бринелю, кг/мм2 . 1,21 1,14—1,15
180 210—255
7,5 6—10
Предел прочности на изгиб, кг/см2 600—800 800—1000
Предел прочности на растяже- ние, кг>см2 ......... 870 1050
То же, в поперечном направле- нии, кусм* . . . Теплостойкость по Мартенсу, °C 400 860
45 65
Морозостойкость, °C —30 —10
Водопог.тссцение, °/о (объемн.) 2 8
Полиуретановые смолы применяют в качестве прессовочных и
литьевых композиций как в чистом виде, так и в смеси с наполни-
телями (древесной мукой и др.) и пластификаторами. Широко рас-
пространены, в частности, пресскомпозиции на основе термореактив-
ных смол, полученных полимеризацией триизоцианатов с гликолями
или диазоцианатов с многоатомными спиртами (глицерин, пентаэри-
трит) , причем «сшивка» молекул 'может происходить в процессе го-
рячего прессования, которое ведут при 140—160° с выдержкой от
3 до 10 мин. на 1 мм толщины.
По тепло- и термостойкости термореактивные полиуретаны усту-
пают фенопластам.
Из полиуретанов и полимочевин изготовляют антикоррозийные
лаки; их применяют для склейки фанеры, пропитки тканей, получе-
ния газонепроницаемых защитных покрытий и т. п.; большое значе-
ние приобретают полиуретановые и полимочевинные лаки и для
производства искусственной кожи.
ГЛАВА XV
ПЛАСТИЧЕСКИЕ МАССЫ НА ОСНОВЕ
КРЕМНИЙОРГАНИЧЕСКИХ СМОЛ
ОСНОВНЫЕ ТИПЫ КРЕМНИЙОРГАНИЧЕСКИХ СМОЛ
До последнего времени были известны два класса природных
и синтетических (искусственных) полимеров: 1) органические по-
лимеры (смолы), для основной цепи макромолекул которых харак-
терны углерод-углеродные —С—С—С—, углерод-кислородные
—С—О—С—О—, углерод-азотные —С—N—С—N— и другие по-
добные связи и 2) минеральные полимеры, не содержащие углерода;
важнейшими представителями последних являются силикатные по-
лимеры (стекла), для основной цепи макромолекул которых харак-
терна кремний —кислородная (силоксановая) связь —Si—О—Si—О.
Основное отличие в свойствах полимеров обоих классов следую-
щее: органические полимеры неустойчивы в атмосферных условиях
при температуре выше 300—400°, так как при этом происходит де-
струкция полимера, в то время как минеральные (силикатные)
полимеры, как правило, устойчивы и не деструктируются при тем-
пературе выше 400—500°.
Термостойкость полимера, как известно, определяется главным
образом величиной энергии связи атомов. Высокая термостойкость
силикатных полимеров объясняется более высокой энергией связи
кремния с кислородом в силоксановых группах —Si—О—Si—
(89,3 ккал/моль] по сравнению с энергией связи между атомами
углерода (58,6 ккал/моль], между атомами углерода и кислорода
или углерода и азота.
«Смолы» и «стекла» отличаются также и физико-механическими
свойствами. Хотя в этом отношении зависимость свойств от строе-
ния полимера очень сложна, все же можно установить, что наличие
только силоксановых групп, характерных для силикатных полиме-
ров, — обусловливает твердость и жесткость, в то время как угле-
родные группы, характерные для органических полимеров, способны
создавать во многих случаях гибкие, пластичные и ?ысокоэластиче-
ские вещества.
В техническом отношении крайне ценными оказались бы такие
материалы, которые, наряду с комплексом физико-механических
свойств, характерных для органических полимерных веществ (эла-
стичность и др.), обладали бы такой термо- и химической стой-
костью, какой отличаются силикаты. Структура таких материалов
должна была бы, естественно, характеризоваться содержанием эле-
ментов, типичных для обоих вышеназванных классов полимеров.
39*
612 Гл. XV. Пластмассы на основе кремнийорганических смол
Например, одной из возможных форм является такая структураТ
при которой основная цепь содержит силоксановые связи, а боко-
вые группы — углеродные связи алкильных или арильных радика-
лов.
R R R
... — L—О—L-О—к—О—...
R R R
Такие кремнийорганические полимеры в настоящее время полу-
чены; они представляют новый класс полимеров, сочетающий основ-
ные структуры органических и силикатных полимеров и их свойства.
Ведущая роль в развитии химии и технологии кремнийоргани-
ческих соединений и, в частности, кремнийорганических полимеров,
принадлежит русским ученым.
Принципиальные различия в химии углерода и кремния были
установлены еще Д. И. Менделеевым; он также впервые выдвинул
гипотезу о полимерном строении двуокиси кремния и обратил вни-
мание на склонность кислородсодержащих соединений кремния
к полимеризации (поликонденсации).
А. М. Бутлеров создал химическую теорию гидролиза и поли-
конденсации эфиров и других производных кремневой кислоты.
Ему принадлежат высказанные еще в 70 годах прошлого столетия
следующие, имеющие актуальное значение слова:
«Распространение принципа полимерии на область минеральной химии де-
лает не только понятным состав, строение и законы образования соединений, но
и позволяет предвидеть новые соединения» (подчеркнуто нами. Э. Б.).
Работы Д. И. Менделеева и А. М.. Бутлерова послужили осно-
вой современных воззрений на процессы гидролиза и поликонденса-
ции и определили направление в работах по синтезу кремнийорга-
нических соединений.
Кремнийорганические соединения получили промышленное зна-
чение лишь после того, как были установлены основные закономер-
ности, ведущие к получению высокомолекулярных кремнийоргани-
ческих соединений и были разработаны технические методы произ-
водства кремнийорганических полимеров. Эта задача впервые была
разрешена К. А. Андриановым в 1935—1937 гг.
Таким образом, приоритет в области синтеза кремнийорганиче-
ских полимеров, представляющих собою одно из крупнейших дости-
жений химии и технологии высокополимеров, принадлежит Совет-
скому Союзу.
Технические кремнийорганические полимеры построены главным
образом на основе силоксановых связей в главной цепи и органиче-
ских радикалов в боковых цепях; они имеют линейную 1, цикличе-
Химические особенности кремнийорганических соединений
613
скую 2, а также разветвленную пространственную структуру 3:
R R
R R R / \
I I I 0 0
...—Si—О—Si—О—Si—... R | | ,R
III >Si Si<
R R R Rz| I XR
i
I I
О О
''s/
r\
2
R R R ’
...— Si—O—Si—O—Si—O—Si—...
R I R R
О . . .
R | О R
...—Si—O—Si—O—i—O—Si—...
R R ! R
О
R R | R
...—Si—O—Si—O—Si—O—Si—...
Jill
О R R R
3
В этих схемах R — насыщенные
жирного или ароматического ряда.
и ненасыщенные радикалы
ХИМИЧЕСКИЕ ОСОБЕННОСТИ КРЕМНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИИ
Химия кремнийорганических соединений основана, главным образом, на
закономерностях химии кремния. Кремний обладает большой склонностью к об-
разованию кислородных связей — силоксановых групп и содержащих их слож-
ных полимерных соединений. Б этом сродстве кремния к кислороду, в спо-
собности создавать с ним связи более прочные, чем с любыми другими элемен-
тами, заключается основное отличие химических реакций кремниевых соединений
от углеродных.
Несмотря на то, что кремний и углерод имеют одинаковую валентность и
дают многие аналогичные реакции н соединения, закономерности химии кремния
существенно отличаются от закономерностей химии углерода.
С точки зрения способности к образованию высокополимеров основным от-
личием соединений кремния является непрочность и нестабильность кремний —
кремниевой связи. Энергия связи —Si—Si— меньше энергии связи кремния
614
Гл. XV. Пластмассы на основе кремнийорганических смол
с другими элементами (табл. 68). Связь эта легко гидролизуется в присутствии
воды и гидроксильных ионов:
...—SI—St—... 4-2Н..0 —»...— SiOH 4-HOSt—... 4-Н.
11 'l 1 '
и с увеличением числа звеньев —Si—Si— малоустойчива и декструктируется. Так,
высшим насыщенным кремнийводородом, который удалось синтезировать и изо-
лировать, был гексасилан СбНц.
Существенным отличием кремния, имеющим непосредственное отношение
к образованию высокополимеров, является то, что он неспособен образовывать
устойчивые двойные и тройные связи ни с одним из элементов. Поэтому неиз-
вестны соединения кремния, аналогичные альдегидам, кетонам, оксимам, нитри-
лам, а также ненасыщенным углеводородам. По этой причине кремниевые по-
лимеры не образуются путем процессов полимеризации и получаются лишь по
реакциям поликонденсации.
Существенно для химии кремнийорганических полимеров то, что кремний
образует прочные связи с углеродом. Связь атомов кремния с алкилами или
с арилами можно считать не менее прочной, чем связь этих радикалов с угле-
родными атомами.
ТАБЛИЦА 68
Энергия связи кремния с некоторыми химическими элементами
Связь Энергия связи ккал/моль Связь Энергия связи хкал!моль
1 — Si — Si = । 42,5 = Si — и 75,0
= Si — с — 57,6 SSi —О — 89,3
— С _ с = 58,6 = Si — Cl 85,8
U Si — F 143,0
Функциональной группой, определяющей процессы поликонденсации кремний-
органических соединений, ведущие к образованию силоксановых связей, служит
гидроксильная группа в соединениях типа R3S1OH (силанолы), R3Si(OH)2
(силандиолы), RSi(OH)3 (силантриолы). Хотя некоторые из этих соединений и
подобны соответствующим спиртам, однако по характеру реакций они суще-
ственно от них отличаются. Так, при дегидратации третичные спирты RjCOH
могут переходить в непредельные соединения, например
(СН3)3СОН (СН3)2С = СН2,
тогда как соответствующие силанолы R3S1OH образуют при этом прочные сило-
ксановые связи с участием двух молекул:
R3SiOH + HOSiR3 R3Si—О—SiR3.
Значительно большее сродство к кислороду атомов кремния по сравнению
с атомами углерода определяет основное отлнчие и в поведении ОН-групп,
связанных с кремнием или углеродом. Способность силанолов к поликонденсации
и к образованию силоксановых связей столь велика, что многие из них не могут
быть изолированы.
Реакционная способность ОН-групп, связанных с кремнием, легкость, с ко-
торой они вступают в реакцию конденсации, зависят также и от величины
арилов или алкилов, присоединенных к кремнию. Чем более громоздкими
являются эти радикалы, тем больше устойчивость силанола, т. е. тем меньше его
реакционная способность. Так, триметилсиланол (CH3)3SiOH заметно конден-
сируется при температуре его кипения (98°) с образованием гексаметилди-
силоксана:
(CH3)3SiOH + HOSi(CH3)3 (CH3)3Si—О—Si(CH3)3,
в то время как трнэтилспланол перегоняется без изменения при 150°.
Получение исходных «мономеров»
615
Силандиолы с небольшими углеродными радикалами, например диметил-
силандиолы (СНз)251(ОН)2, столь энергично взаимодействуют с образованием
силоксановых связей, что ни они, ни их димеры не были изолированы; лишь
дифеиилсиландиол является относительно стойким кристаллическим веществом;
в еще меньшей степени стабильны силантриолы.
Существенное отличие наблюдается и в характере — Si—Cl-связей. Галоиды,
образуя прочные соединения с кремнием, одновременно увеличивают стабиль-
ность силанов. Сродство кремния к галоидам значительно больше, чем к водо-
роду, поэтому галоидосиланы, в отличие от соответствующих галоидоуглеродных
соединений, не восстанавливаются при гидрировании. Однако хлор крайне легко
реагирует с подвижными атомами водорода, связанными с кислородом, азотом
и т. п.; при этом образуются соответствующие силанолы, эфиры и т. п.:
=si—ci 4- нон —> =si—он + нс1
=Si—Cl 4-НО—Si= —»- =Si—О—Si= 4- HC1
=Si-Cl 4- HOR —► =Si—OR 4- HC1
=si—ci 4- hnr2 —> =si—nr2 4- hci.
Механизм, ведущий к образованию высокополимеров, построенных из
звеньев
R R
...—1м—О—S>i—...
R R
(в настоящее время синтезированы полимеры, макромолекулы которых содержат
более, чем 10 000 таких звеньев), основан на высокой реакционной способности
силанолов и большой прочности силоксановых связей. Эта же прочность опре-
деляет и термостойкость кремнийоргаиических полимеров, которая тем больше,
чем выше концентрация силоксановых связей в полимере.
Получение исходных «мономеров»
Исходными веществами для получения кремнийоргаиических полимеров слу-
жат соединения общей формулы R,;SiX4-n, где X = С1 или OR. Эти вещества
могут быть разделены на две группы: 1) алкил- или арилхлоренланы (RSiClg;
R2SiCl2; RaSiCl) и 2) алкил- или арилзамещеииые ортокремневой кислоты
[RSi(OR)3; R2Sii(OR)2; R2SiOR],
Органохлорсиланы получаются в промышленных условиях из четыреххлори-
стого кремния (SiCl4) и алкил- нли арилхлоридов по реакции Гриньяра, а также
другими методами. Четыреххлористый кремний получается прямым хлорирова-
нием ферросилиция.
По реакции Гриньяра процесс, обычно, включает стадию приготовления
грииьяровского реактива, исходя из магния и алкил(арил)хлоридов, и стадию
последовательного его взаимодействия с SiCI4 по схеме:
С2Н5С1 4- Mg —> C2H5MgCl
SiCl4 4- C,H5MgCl —> C,H5SiCl3 4- MgCl2
C2H6SiCl3 4- C2H5MgCl —> (C2H;)2SiCl2 4- MgCl2
или:
SiCl44-C6HBMgC14-2C2H5MgCl —> (C2H5)2C6H5SiCl 4-3MgCl2.
Реакция проводится обычно в растворителе действием эфирного раствора
магнийорганического соединения на SiCl4. В зависимости от молекулярных соот-
ношений взятых веществ образуются моно-, ди- и триалкил (арил) хлорсиланы,
причем в сыром продукте присутствует некоторое количество гомологов замеще-
ния. Тщательным фракционированием получают чистые продукты.
616 Гл. XV. Пластмассы, на основе кремнийорганических смол
Органохлорсиланы могут быть получены также и прямым синтезом — путем
пропускания паров хлоридов через нагретую смесь кремния и катализатора (медь,
серебро). Реакция идет по уравнению:
2CH3C1 + Si —нагрев- -» (CHs),SiCl2.
* 1 катализатор ' *
Эфиры ортокремневой кислоты получаются, согласно Д. И. Менделееву, дей-
ствием абсолютного спирта на четыреххлористый кремний:
SiCl4 + 4С2Н5ОН —> Si(OC2H6)4 ф- 4HCI.
Замещенные эфиры ортокремневой кислоты получаются в промышленных
условиях из алкилхлоридов и эфиров ортокремневой кислоты по реакции
Гриньяра:
CHgCl + Mg —► CH3MgCl
Si(OC2H5)4 + CH3MgCl —> CH3Si(OC2H5)3 + C2H6OMgCl
илн непосредственным действием па металлический магний смесью алкилхлорида
н эфира ортокремневой кислоты:
Mg + СН8С1 + Si(OC2H5)4 —► CH3Si(OC2H5)3 + C2H5OMgCi.
ПРОЦЕССЫ ОБРАЗОВАНИЯ КРЕМНИЙОРГАНИЧЕСКИХ ПОЛИМЕРОВ
Реакции поликонденсации силанолов, например силандиолов, ве-
дущие к получению кремнийорганических полимеров, по схеме:
R R
п R2Si(OH)2 —> НО—Si—О--------Si—О
R L R
R
—Si—ОН + (« — !) Н2О
”"2 R
характеризуются в основном обычными закономерностями гетеро-
цепной поликонденсации.
Эти реакции имеют характер ступенчатого процесса и кинетика
их определяется температурой, катализатором (если его применяют)
и скоростью удаления воды из сферы реакции. Молекулярный вес
полимера, в случае применения бифункциональных силанолов (си-
ландиолов) тем больше, чем полнее удалена вода, чем выше темпе-
ратура процесса и чем длительнее время реакции (если не обра-
зуются циклические соединения).
При взаимодействии бифункциональных силанолов R2Si(OH)2
можно ожидать получения как линейных, так и циклических поли-
меров. Если реакция протекает с образованием линейного полимера,
то при совершенном удалении реакционной воды из сферы реакции,
можно получать полимеры с весьма большим молекулярным весом.
Когда же с бифункциональными силанолами реагируют моно-
функциональные R3SiOH, последние, блокируя концевые функцио-
нальные группы растущей цепи, ограничивают, естественно, молеку-
лярный вес полимера и останавливают рост цепи по схеме:
R R R R
2RaSiOH -j- п R2Si(OH),—>- R—Si-О—L—О—Si— —О—Si—R.
I I I „ I
R R R J" R
Процессы образования кремнийорганических полимеров
617
Чем больше отношение монофункциональных силанолов к би-
функциональным, тем меньше будет средняя длина цепи полимера.
Введением заданного количества монофункциональных соединений
можно получать полимеры с определенным интервалом степени по-
ликонденсации.
Если, однако, в реакции участвуют трифункциональные соеди-
нения (силантриолы) или бифункциональные с некоторым количе-
ством трифункциональных или тетрафункциональных, то, есте-
ственно, можно ожидать образования неплавкого и нераствори-
мого пространственного полимера по схеме:
п R2Si(OH)2 -f- т RSi(OH)3 —►
I
R О R R
I I I I
.. .—Si—О -Si—О—Si—О—Si—О—...
R R О R
R R | R
...li—О—Si—О—Si—О—Si—О—...
1111
R О R R
R R R
....—Si—О—Si—О—Si—О—Si—...
R R R О R
... .—О—Si—О -Si—О— Si—...
I I
О R
Чем больше в реакционной среде отношение три- и тетрафунк-
циональных соединений к бифункциональным, тем раньше насту-
пает «желатинизация» смеси, тем выше термореактивность смолы
(если она получена в плавком и растворимом состоянии), а также
термостойкость, твердость и жесткость конечного продукта поли-
конденсации.
Технические кремнийорганические полимеры получают, однако,
не взаимодействием предварительно изолированных силанолов,
а непосредственно в процессе гидролиза алкил- или арилхлорсила-
нов или алкил- или арилзамещенных эфиров ортокремневой кислоты
и поликонденсации образующихся из них силанолов. Естественно, что
в этом случае некоторые закономерности, ведущие к получению вы-
сокополимеров, отличны от закономерностей для реакций между
индивидуальными силанолами.
Наблюдения над процессами поликонденсации, протекающими
при гидролизе эфиров ортокремневой кислоты, были сделаны еще
618
Гл. XV. Пластмассы на основе кремнийоргаиических смол
в прошлом столетии. Так, было известно, что при получении эти-
лового эфира ортокрем новой кислоты по реакции
SiCI4 4- 4С2Н5ОН —> Si(OC,H5)4 + 4НС1
образуются также побочные продукты с более высокой точкой ки-
пения, при этом чем выше влажность спирта, тем в большей мере
образуются более высокомолекулярные продукты.
Это объясняется последовательно протекающими процессами
омыления эфира и конденсации, ведущими к получению поли-
меров по схеме:
2Si(OC2H5)4 4- 2Н2О —> 2HOSi(OC2H5)3 4- 2С2Н5ОН
(C2H5O)3SiOH 4- HOSi(OC2H5)3 —> (C,H5O)3Si—О—81(ОС2Н5)з4-Н,О
2[(C2HsO)gSi—О—Si(C2H5O)3]4-2H,O —>
—> 2[(OC.,H5)3Si—О—Si(OC2H5)2OH] 4-2С,Н5ОН
ОС2НБ ОС2НГ)
(GHjOJgSi—О—S1—OH 4~ НО—Si—О—Si(OC2H5)3 —>
ОС2Н5 ОС2Н5
ОС..Н5 ОС2Н5
I * I
—> (C2H5O)3Si—О—Si— О—Si— О—Si(OC2H5)3 и т. д.
OC2HS ОС2Н5
и вообще:
...—Si...— ;
ООО
л Si(OC2H8)44-4яН.О —> ...—Si—О—Si—О—Si—... —> (SiO2Xr
III
ООО
i I I
.. .—Si—О—Si—О—Si—...
Конечным продуктом омыления и конденсации органических
эфиров ортокремневой кислоты в присутствии избытка воды яв-
ляется неорганический полимер (SiO2).T.
Для получения, однако, устойчивых, с неомыляемыми радика-
лами кремнийоргаиических полимеров необходимо, чтобы наряду с
силоксановыми связями в главной цепи существовала стабильная
I I
кремнийуглеродная связь —Si—С— в боковой цепи. Такие поли-
меры получают ступенчатым омылением алкил- и арилзамещенных
эфиров ортокремневой кислоты, например С2Н581(ОС2Н5)з, или же,
Процессы образования кремнийорганических полимеров 619
соответственно, замешенных хлорсиланов, например CsHsSiCU, с по-
следующей конденсацией за счет образующихся гидроксильных
групп. Процесс гидролиза связан с поглощением, а процесс конден-
сации — с выделением воды, которая, в свою очередь, служит для
омыления по схеме:
1. п RSi(OC2H5)a -(- п Н2О —> п RSl(OC2Hs)2OH -р п С2НзОИ
ОС2Н5 ОС2Н5
2. п RSi(OC„H Д.ОН —> R—SI—O-Ji—R + ~ Н2О.
1 I I 2
ОС2Н5 ос.,н5
Выделяющаяся вода действует на следующие молекулы эфира,
и так происходит до тех пор, пока вся вода не будет химически
связана, после чего, естественно, прекращаются и омыление и поли-
конденсация.
К. А. Андрианов установил зависимость между количеством
воды и степенью поликонденсации моноалкилзамещенных эфиров:
п — т ’
где: А — средняя степень поликонденсации (число атомов кремния
Рис. 218. Выделение спирта при
омылении этилтриэтоксимоно-
силава.
взятой в реакцию воды в молях;
п — число молей алкилзамещенно-
го эфира ортокремневой кислоты.
Так, если омыление 1 моля ди-
этилдиэтоксимоносилана произво-
Рис. 219. Изменение вязкости
при омылении этилтриэтокси-
моносилана.
дится 0,75 мол. воды, то степень поликонденсации А = } = 4;
соответственно при 0,83 мол. воды А = 6 и т. д
Скорость и степень гидролиза и поликонденсации алкилзаме-
щенных эфиров ортокремневой кислоты зависят от температуры и
времени реакции, а также от степени замещения и длины цепи
радикала.
<520
Гл. XV. Пластмасы на основе кремнийорганических смол
С увеличением молекулярного веса образующихся полимеров
скорость поликонденсации и, соответственно, скорость гидролиза
уменьшаются.
На рис. 218 и 219 приведены зависимости процесса омыления
(% выделения спирта) и поликонденсации (изменение вязкости)
этилтриэтоксимоносилана от времени. Скорость омыления этоксиль-
ных групп уменьшается также с введением второго радикала; она
зависит от длины цепи радикала; с увеличением молекулярного веса
Рис. 220. Омыление алкилтри-
этоксимоиосиланов.
1—C,HiaSi 'Оа.Н,)„; 2—C,H,SifOC,H,K;
3— C.HjSKOCjHj).,; Si(OC,H^4.
Время, часы
Рис. 221. Зависимость молекуляр-
ного веса от продолжительности
конденсации диэтилдиэтокси моно-
силана (С2Н6)2 Si(OC2H6)2 и коли-
чества воды.
1—1,0 моль Н,О; 2—то же, 1,5 моля;
3—то же, 2,0 моля; 4—3,0 моля.
радикала уменьшается скорость омыления (рис. 220). Зависимость
среднего молекулярного веса от количества воды и времени кон-
денсации диэтилдиэтоксимоносилана показана на рис. 221.
В процессе поликонденсации бифункциональных соединений, как
уже указывалось, могут образоваться не только линейные, но и ци-
клические продукты, которые получаются внутримолекулярной кон-
денсацией полидиалкилсилоксандиолов по схеме:
СН3 СНз СН8 СНз СНз
НО—Si—О—Si—О—Si—О—Si—О—^i—ОН —►
I I I I I
СНз СНз СНз СН8 СНз
НзС СН. Н3С СНз
Процессы образования кремнийорганических полимеров 621
В отличие от циклических продуктов с углеродными связями
в кольце, в которых, как известно, циклы более чем с шестью-семью
членами неустойчивы, силоксановые связи образуют устойчивые
кольца, содержащие значительно большее число членов в кольце.
Можно с большой вероятностью считать, что между обеими
формами полимера в зависимости от условий реакций и характера
среды (катализатора, температуры, количества монофункциональ-
ных соединений, воды и т. д.) устанавливается равновесие, которое
можно сдвинуть в сторону получения практически лишь одной
формы. Линейные полимеры преобладают, когда гидролиз и поли-
конденсация происходят в нейтральной среде при недостатке воды
и в присутствии монофункциональных соединений.
Стабильность обеих форм полимеров различна по отношению*
к температуре и катализаторам. При обычных температурах линей-
ные формы стабильны по отношению к кислому катализатору, в то
время как в этих условиях происходит перестройка циклических
форм с превращением их в линейные полимеры. Так, смесь гекса-
метилдисилоксана и циклических полидиметилсилоксанов под дей-
ствием серной кислоты на холоду превращается почти полностыо-
в линейные полидиметилсилоксаны. Такая «перестройка» цикличе-
ских молекул под действием серной кислоты получила техническое
применение для регулирования структуры полимера.
Омыление и конденсацию замещенных эфиров ортокремневой
кислоты производят при нагревании в водном спирте, причем кон-
центрация спирта должна соответствовать количеству воды, необхо-
димому для достижения заданного молекулярного веса полимера.
Концентрация спирта, однако, лимитируется его растворяющей спо-
собностью по отношению к полимеру и не должна быть менее 50%.
Конденсацию проводят в реакторе с обратным холодильником,
мешалкой и паровой рубашкой при нагревании до кипения в тече-
ние от 2 до 12 час. в зависимости от состава эфира. По мере про-
текания реакции увеличивается вязкость спиртового раствора, при-
чем на определенной стадии процесса получаются (после испарения
растворителя) пластичные или твердые пленки.
Контроль процесса можно вести путем определения вязкости,,
содержания этоксильных групп, коэффициента рефракции, а также
пробой на получение пленки после испарения спирта. По оконча-
нии реакции спирт, если необходимо, отгоняют под вакуумом и
смолу сливают. Для ряда назначений может найти применение спир-
товый раствор смолы.
Структура и характер кремнийорганических полимеров опре-
деляются функциональностью реагирующих молекул, средней сте-
пенью поликонденсации и характером органического радикала.
Постоянноплавкие и растворимые полимеры, естественно, по-
лучаются лишь в том случае, когда среди реагирующих веществ не
содержится трифункщюнальных соединений. С другой стороны, и
при наличии в реакционной смеси трифункциональных соединений.
622 Гл. XV. Пластмассы на осносе кремнийорганических смол
процесс поликонденсации можно вести таким образом, чтобы пре-
рывать его, когда прореагировала лишь часть функциональных
групп с образованием плавкого и растворимого продукта. Такие
промежуточные продукты могут реагировать в дальнейшем
при нагревании или в процессе горячего прессования после
смешения их с наполнителями, образуя твердые, неплавкие мате-
риалы.
Для получения линейных кремнийорганических полимеров вы-
сокого молекулярного веса (свыше 100 000) необходимо тщательно
освобождать бифункциональные соединения, например (CH3)2SiCl2,
от монофункциональных, например (CH3)3SiCl, так как содержание
последних даже в количестве 1 % делает теоретически невозможным
достижение степени поликонденсации выше 200. В такой же мере
необходимо тщательно удалять примеси и трифункциональных со-
единений, наличие которых ведет к разветвлению полимеров и
к созданию пространственных структур.
Чем больше молекулярный вес линейных кремнийорганических
полимеров, тем выше
ТАБЛИЦА 69
Молекулярный вес
различных фракций
полисилоксаиового
каучука
Вес фрак**
Фрак- ций по от-
ции ношению
к полимеру
Мол. вес
1 17 28-10**
2 14 15-106
3 18 6,1 • 106
4 27 2,9 • 10в
их высокоэластические свойства, прочность
и температура плавления. Низшие члены
полимергомологического ряда линейных
кремнийорганических полимеров предста-
вляют собою жидкости; по мере увеличения
молекулярного веса наблюдается переход
к вязким, пластичным телам и, наконец,
к твердым, эластичным, каучукоподобным
полимерам (табл. 69).
Влияние величины (длины) органиче-
ских радикалов сказывается, в первую оче-
редь, в уменьшении межмолекулярных сил
притяжения с возрастанием длины цепи
алкилов, что, естественно, при одном и том
же молекулярном весе приводит к увеличе-
нию «мягкости» и гибкости полимера. С воз-
растанием длины алкилов уменьшается также термостойкость по-
лимера и возрастает его «органический» характер. В образовании
макромолекул полимеров могут участвовать кремнийорганические
•соединения, содержащие не только линейные и насыщенные
радикалы, но также разветвленные и ненасыщенные радикалы;
такими соединениями, например, являются изобутилтриэтоксимоно-
силан, аллилтриэтоксимоносилан и т. п.
Радикалы изостроения, в частности третичные радикалы, есте-
ственно, обусловливают меньшую «мягкость» полимера (более вы-
сокую Тс), чем соответствующие им линейные радикалы нормаль-
ного строения.
Если радикал является непредельным, то образование высоко-
молекулярного соединения может идти одновременно как за счет
процессов поликонденсации, так и за счет процессов полимеризации.
Свойства кремнийоргаиических полимеров 623
связанных с раскрытием двойной связи непредельного радикала,
например: .сн2—СН—СН2—СН—...
СН, СН,
I I
ОС2НБ Н5с,о—Si---О---Si—ОС,Н3 .
2СН,=СН—СН,—Si— ОН —► ОС,Н5 ОС,Н3
I "
ос,н5
Дальнейшие реакции гидролиза, конденсации и полимеризации
должны, естественно, привести к образованию полимера с про-
странственной «сшивкой» цепей.
СВОЙСТВА КРЕМНИЙОРГАИИЧЕСКИХ ПОЛИМЕРОВ
Кремнийорганические полимеры — вещества различного агрегат-
ного состояния. Они могут быть жидкими, твердыми и хрупкими,
а также высокоэластическими, каучукоподобными.
Наиболее характерное _ отличие кремнийоргаиических полиме-
ров — их более высокая термостойкость по сравнению с органиче-
скими полимерами.
Неполярные боковые группы и симметричность макромолекул
кремнийорганического полимера определяют его большую гидро-
фобность и высокие диэлектрические свойства.
Гидрофобность кремнийоргаиических полимеров — одно из важ-
нейших преимуществ, определивших ряд областей технического при-
менения этих полимеров.
Кремнийорганические полимеры более совершенные диэле-
ктрики, чем другие поликонденсационные пластики (фенопласты,
глифтали, аминопласты и др.).
Макромолекулярная структура таких полимеров характери-
зуется малой величиной межмолекулярных сил притяжения, что,
естественно, определяет низкую температуру стеклования и малую
прочность линейных полимеров.
Наличие жидких кремнийоргаиических полимеров со сравни-
тельно большим молекулярным весом (1000 и выше) объясняется
малыми межмолекулярными силами притяжения. Лишь при высо-
ких степенях поликонденсации (соответствующих мол. весу ЮОООО
и более), благодаря переплетению молекул с длинными цепями и
созданию «войлочной» структуры, линейный полимер приобретает
характер высокоэластического твердого тела с низкой температурой
стеклования (—60°).
В отличие от линейных высокополимеров пространственные по-
лимеры представляют собою твердые, хрупкие смолы, — тем более
твердые, чем выше концентрация пространственных связей.
Рентгенографические исследования показывают, что кремний-
органические полимеры имеют аморфную структуру и что кристал-
лическая структура отсутствует даже в ориентированных пленках.
624
Гл. XV. Пласп массы на основе кремнийорганических смол
ПОЛУЧЕНИЕ ПЛАСТМАСС НА ОСНОВЕ КРЕМНИЙОРГАНИЧЕСКИХ
ПОЛИМЕРОВ
В зависимости от средней функциональности реагирующих ком-
понентов могут быть получены различные кремнийорганические
полимеры. При гидролизе и поликонденсации чистых бифункцио-
нальных соединениях типа R2SiCl2 получают высокомолекулярные
линейные полимеры (типа каучуков). Если реагирует смесь моно- и
бифункциональных соединений R2SiCl2 4- RsSiCl, то легче всего по-
лучаются полимерные жидкости; если же реагируют более высоко-
функциональные соединения, например R2SiCl2 -j- RSiCh, то полу-
чаются твердые пространственные полимеры (пластики).
Пластические массы получают на основе термореактивных крем-
нийорганических полимеров. Как уже было указано, они могут
быть получены, если среди реагирующих компонентов имеются
также и трифункциональные соединения. Степень реактив-
ности этих смол может быть выражена соотношением углеводород-
ных радикалов и кремния R/Si. Образование термореактивных смол
возможно лишь в том случае, когда R/Si < 2; чем меньше
это отношение, тем более термореактивным будет полимер, тем
выше будет его термостойкость, твердость и прочность в конечном
состоянии.
Применяют два технических метода получения термореактивных
пространственных полимеров: 1) метод совместного гидролиза и
конденсации бифункциональных и более высокофункциональных
соединений, например, диметилдихлорсилана и метилтрихлорсила на
(а также четыреххлористого кремния) и 2) метод гидролиза и кон-
денсации бифункциональных соединений, например, диметилдихлор-
силана, с последующим окислением полученного продукта до до-
стижения желаемого соотношения метильных радикалов и атомов
кремния (R/Si).
По первому методу в первоначальных продуктах поликонденса-
ции сохраняется некоторое количество функциональных групп, ко-
торые в дальнейшем участвуют в процессе «сшивки» макромолекул;—
в результате образуется неплавкий пространственный полимер.
По этому методу гидролиз и конденсацию следует проводить
в таком растворителе (этиловый эфир и некоторые кетоны), кото-
рые смешиваются с исходными веществами и водой и растворяют
получаемый в результате реакции полимер; без соответствующего
растворителя могут образоваться желатинирующиеся, нераствори-
мые продукты.
Концентрация реагирующих молекул в растворе должна быть
тем ниже, чем меньше соотношение R/Si, т. е. чем больше содержа-
ние CHaSiCU.
Чтобы предотвратить получение нерастворимого продукта, не-
обходимо проводить реакцию при низкой температуре в холодном
Пластмассы на основе кремнийорганических полимеров
625
растворе. В этих условиях можно получить плавкую и растворимую
смолу, которая при дальнейшем нагревании переходит в нераство-
римый и неплавкий полимер.
По второму методу образующиеся термопластичные смолы пере-
водят окислением в термореактивные. Окисление алкильных или
арильных групп для снижения соотношения R/Si и освобождения
дополнительных функциональных групп производят продувкой через
смолу воздуха при 200—300° (для окисления алкильных групп) или
при 170—180° (для окисления арильных групп). Этот процесс окис-
ления органических радикалов может быть ускорен прибавлением
МпО2 или НС1.
Полученная по второму методу плавкая смола способна при
дальнейшей термической обработке переходить в пространственный,
неплавкий полимер.
Свойства термореактивных смол определяются как соотноше-
нием R/Si, так и химической структурой радикалов.
Метилполисилоксаны с отношением CH3/Si от 1 до 1,3 —твер-
дые, хрупкие смолы. Чем больше это отношение, тем мягче поли-
мер, однако тем при более высоких температурах или тем длитель-
нее происходит отверждение. Фенилполисилоксаны при C6Hs/Si
~ 1—1,3) вязкие жидкости, превращающиеся в твердые, хрупкие
смолы, отличающиеся весьма высокой термостойкостью (500—600°).
Термореактивные органополисилоксановые смолы можно сме-
шивать с порошковыми наполнителями или пропитывать ими волок-
нистый или слоистый наполнитель (стеклоткань, слюду, бумагу,
хлопчатобумажную ткань, асбест) и подвергать горячему прессова-
нию. Таким образом, получают жесткие, твердые и неплавкие пла-
стики. Термореактивность этих пресскомпозиций и, следовательно,
скорость прессования определяются концентрацией трифункциональ-
ных силанолов и длиной органических радикалов в реагирующих
молекулах. Чем больше R/Si и чем длиннее радикал, тем ниже ско-
рость прессования. Скорость отверждения можно увеличить введе-
нием катализаторов — различных дегидратирующих веществ, спо-
собных связывать выделяющуюся реакционную воду; такими ката-
лизаторами являются: олеат свинца, окиси металлов, эфиры борной
кислоты, серная кислота, силикагель и др. Так, прибавление 1%
олеата свинца уменьшает время отверждения от нескольких часов
до нескольких минут.
Пресскомпозиции на основе органополисилоксановых смол обыч-
но прессуют при 170—195° (при удельном давлении 150—300 кг/см-)
в течение от 15 мин. до 3 час. в зависимости от термореактивности
и толщины материала.
Из различных видов органополнсилоксановых материалов наи-
большее техническое значение имеют слоистые пластики, в част-
ности на основе стеклоткани (силистеклослой) и на основе слюды
(миканиты) и асбеста. Такого рода слоистые пластики содержат
обычно от 25 до 35% связующего.
40 Зак 1269 Э И. Барг
626
Гл. XV. Пластмассы на основе кремнийорганических смол
Полисилоксановые термореактивные пластики с минеральным
наполнителем имеют значительно более высокую термостойкость, чем
соответствующие материалы на основе органических термореактив-
ных смол (например фенопластов).
Высокие диэлектрические свойства полисилоксановых слоистых
пластиков и малая зависимость этих свойств от температуры (до
200..250°) делают полисилоксановые пластики особо ценным эле-
ктроизоляционным материалом.
На рис. 222 приведены сравнительные кривые зависимости
удельного объемного сопротивления от температуры для стеклослоя
Температура, °C
Рис. 222. Уд. объемное эле-
ктрическое сопротивление р,,
стеклослоя, изготовленного
на основе полисилоксано-
вой (7) и феноло-альдегид-
ной смолы (2).
0 50 100 150 200 250
Температура^ °C
Рис. 223. Изменение tg о и диэлектри-
ческой постоянной е стеклотекстослоя,
изготовленного на алкилполисилоксано-
вой смоле в зависимости от темпера-
туры.
на основе гюлисилоксановой и феноло-альдегидной смол; на рис. 223
показано изменение тангенса угла диэлектрических потерь и ди-
электрической постоянной стеклослоя на основе полисилоксановой
смолы в зависимости от температуры.
Как видно из приведенных кривых, полисилоксановые пластики
практически устойчивы до 250° и значительно превосходят по термо-
стойкости соответствующие фенопласты.
Применение полисилоксановых пластиков в качестве изоляцион-
ных материалов и смол для покрытия проводов дает возможность
значительно повысить допустимые рабочие температуры нагрева
моторов, динамомашин и т. д. Известно, что недостаточная термо-
стойкость органической изоляции является лимитирующим факто-
ром для увеличения мощности моторов. Применение более термо-
стойкой полисилоксановой изоляции взамен обычной позволяет
изготовлять моторы вдвое меньше по объему и весу при одной и той
же мощности. При значительных перегревах полисилоксановых изо-
ляционных материалов происходит окисление органических радика-
Пластмассы на основе кремнийорганических полимеров
V27
лов и выделение кремнезема. Последний — хороший диэлектрик и
не приводит к пробою, в то время как органические смолы, в част-
ности фенопласты, в аналогичных условиях выделяют углеродные
частицы, служащие проводником тока.
Прессматериалы на основе полисилоксановых смол, в частности
слоистые на основе стеклоткани и асбеста, получают большое значе-
ние и в других областях техники и в первую очередь там, где требуется
высокая термостойкость, стойкость к химическим воздействиям,
действию перегретого пара, влажности и т. д. По комплексу ме-
ханических свойств слоистые полисилоксановые пластики мало
уступают пластикам на основе органических термореактивных смол.
Получение термореактивных полимеров модифицированных
кремнийорганическими соединениями
Г. С. Петров и А. И. Крешков применяли кремнийорганические соединения
для модификации ряда термореактивных смол.
Получение таких смол основано на совместной поликонденсации промежу-
точных (первичных) продуктов конденсации феноло-альдегидных, мочевиио-аль-
дегидных и других смол с эфирами ортокремневой кислоты или с алкил- или
арилзамещенными галоидопроизводными силана, или полимерными кремнийорга-
ническими соединениями.
Процесс такого рода совместной поликонденсации, например с продуктами
мочевпно-формальдегидной конденсации, можно представить следующей схемой:
...—СН2—N—СО—N—СН2ОН + lOCJ l3) ,,Si СзН5°Н
-с.н,он
...—СН2—N—СО—N—CH2OSi(OC2H5)a,_t и т. д.
Механизм этой реакции подтверждается выделением на всех стадиях реакций
этилового спирта и уменьшением числа метилольных групп.
Присутствие этоксильных групп значительно повышает реакционную способ’
пость смолы (функциональность), так как при дальнейших процессах конденсации
происходит омыление этоксильных групп и конденсация продукта гидролиза по
схеме:
(RO)rcSi(OC2H5)// + НОН —> (RO)a!Si(OCiH5)2/_lOH + С2Н5ОН
(RO^SiCOCoHB^-iOH + HO(C2H5O)?/_1Si(OR)J;
-^5-* (RC)J,Si(OC2H5)?/_t—О—(C2H5O)?/_tSi(OR).r и т. д.
Полученные смолы содержат силоксановые связи, что определяет их более
высокую термостойкость, водостойкость и лучшие диэлектрические свойства по
сравнению с обычными конденсационными смолами.
40*
ЛИТЕРАТУРА
Общая
А. Я. Дринберг. Технология пленкообразующих веществ. Госхимиздат,
1948.
В. В. Корш а к. Химия высокомолекулярных соединений. АН СССР, 1950.
В. В. К о р ш а к. Общие методы синтеза высокомолекулярных соединений.
Сборник «Методы высокомолекулярной органической химии», т. I, АН СССР, 1953.
И. П. Лосев, Г. С. Петров. Введение в химию искусственных смол и
пластмасс. Госхимйздат, 1938.
И. П. Лосев, Г. С. Петров. Химия искусственных смол. Госхимиздат, 1951.
Мономеры. Сборник статей № 1. ИЛ, 1951.
Мономеры. Сборник статей № 2. ИЛ, 1953.
Г. С. Петров, Б. Н. Рутовский, И. П. Лосев. Технология синтети-
ческих смол и пластических масс. Госхимиздат, 1947.
И. Шайбер. Химия и технология искусственных смол. Госхимиздат, 1949.
К введению
Б. А. Архангельский и Б. С. Вайсгант. Пластические массы. Лен-
издат, 1950.
Э. И. Бар г. Пластические массы и их применение. Госхимтехиздат, 1933.
Э. И. Барг. Современные пластики и их конструктивно-технологические
возможности в применении к аппарате- и приборостроению. Сборник «Техноло-
гичность конструкций в машиностроении». Машгиз, 1950.
К. Б. Пиотровский. Значение работ отечественных ученых в области
изучения процессов полимеризации. Сборник «Химия и физико-химия высокомо-
лекулярных соединений». АН СССР, 1952.
К. Б. Пиотровский. Работы русских химиков в области синтеза каучука.
Высокомолекулярные соединения, 11, I (1951).
К главе I
Н. С. Акулов. Теория цепных процессов. Гостехиздаг, 1951.
Г. А. Ал фр ей и др. Сополимеризация. ИЛ, 1953.
А. М. Бутлеров. Избранные работы по органической химии, стр. 67,
211—223, 290—293; 332, 335, 381. АН СССР, 1951.
В. В. К о р ш а к, С. В. Рафиков. Синтез и исследования высокомолеку-
лярных соединений. АН СССР, 1949.
А. И. Коган. Смолы из многоосновных кислот и многоатомных спиртов.
ГОНТИ, 1938.
П. Пауэрс. Синтетические смолы и каучуки. Госхимиздат, 1948.
Н. Н. Семенов. Цепные реакции. ОНТИ, 1933.
М. Ф. Шостаковскнй. Простые виниловые эфиры. АН СССР, 1952.
Г. Шт ауд ингер. Высокомолекулярные органические соединения. Хим-
теорет, 1935.
К. Эллис. Химия синтетических смол, т. I, 1938; т. II, 1940. Госхимиздат.
Литература
629
А. Абкин. С. Медведев, Л. Гиндин. Совместная полимеризация вп-
нилцианида и бутадиена. Труды III Конференции по высокомолекулярным соеди-
нениям. АН СССР, 1949.
А. Н. Ба х. Современное состояние учения о процессах медленного окисле-
ния и активации кислорода. Успехи химии, 3, 177 (1934).
X. С. Багдасарьян. О связи между строением виниловых соединений и
их способностью к полимеризации. Сборник «Исследования в области высокомо-
лекулярных соединений, стр. 346. АН СССР, 1949.
А. А.. В а н ш е й д т. Механизм образования и строения феноло-альдегидных
смол. Труды I и II Конференции по высокомолекулярным соединениям. АН
СССР, 1945.
А. А. В а н ш е й д т и Р. И. Груз. К вопросу о влиянии свободных ради-
калов типа трифенилметила на полимеризацию стирола. Сборник «Химия и
«физико-химия высокомолекулярных соединений». АН СССР, 1952.
В. В. Воеводский и В. Н. Кондратьев. Радикалы в цепных реак-
циях. Успехи химии, 19, 673 (1950).
А. Р. Гантмахер, С. С. Медведев. Каталитическая полимеризация
ненасыщенных соединений. Сборник «Исследования в области высокомолекуляр-
ных соединений». АН СССР, 1949.
А. Я. Д р и н б е р г. Теория преимущественного влияния атомности спирто-
вого остатка полимеров эфиров полифункциональных кислот. Труды ЛТИ, XIV.
Госхимиздат, 1, 1947.
С. Каменская, С. Медведев. Кинетика полимеризации винилацетата
в присутствии перекиси бензоила в бензольном растворе. ЖФХ, 14, 922 (1940).
В. В. К о р ш а к, В. В. Голубев, Г. С. К о л е с н и к о в. О значении соот-
ношения исходных веществ в процессе линейной поликонденсации. Сборник
«Исследования в области высокомолекулярных соединений». АН СССР, 1949.
В. В. Корш ак, С. Р. Рафиков, В. А. Замятина. О механизме линей-
ной поликонденсации. Сборник «Исследования в области высокомолекулярных
соединений». АН СССР, 1949.
В. В. Корш а к, С. В. Рогожин. О причинах остановки роста цепи при
тюлиэтерификации. Сборник «Химия и физико-химия высокомолекулярных соеди-
нений». АН СССР, 1952.
С. В. Лебедев. Двуэтнлеиовые углеводороды. ЖРФХО, 45, 1249 (1913).
С. С. Медведев. Свободные радикалы в процессах полимеризации.
Труды 1 и II Конференции по высокомолекулярным соединениям. АН СССР, 1945.
С. С. М. е д в е д е в, О. К о р и ц к а я, Е. А л е к с е е в а. Свободные радикалы
в процессах полимеризации, ЖФХ, 17, 391 (1943).
Р. С. Муромова. Влияние химического строения на свойства линейных
конденсационных полимеров. Высокомолекулярные соединения, 10, 28 (1950).
Б. Н. Р у т о в с к и й, А. М. Шур*. Сополимеризация. Высокомолекулярные
соединения, 6, 1 (1947).
Н. Н. Семенов. Цепные реакции в химии. Успехи химии, 20, 1 (1951).
В. Солонина. ЖРФХО, 30, 826 (1898).
С. Н. Ушаков, Р. К. Г а в у р и н а, X. В. Ц у б и н а. О дегидратации по-
ливинилового спирта. Сборник «Исследования в области высокомолекулярных со-
единений». АН СССР, 1949.
С. Н. Ушаков, С. П. М и ц е н г е н д л е р. Г. А. Ш т р а й х м а н. Современ-
ное состояние теории сополимеризации. Успехи химии, 19, 265 (1950).
М. Ф. Ш оста ко в с ки й, В. И. Микентьев и В. А. Серебрянни-
кова. Каталитическая ионная сополимеризация винилалкиловых эфиров. Сбор-
ник «Химия и физико-химия высокомолекулярных соединений». АН СССР, 1952..
К главе II
А. П. Александров и Н. С. Журков. Явления хрупкого разрыва.
ГТТИ, 1933.
Т. А л ф р е й. Механические свойства высокополимеров. ИЛ, 1952.
630
Литература
Б. Я- Блюмберг. Введение в физическую химию стекла. Госхимиздат, 1940.
Б. А. Дог ад кин. Химия и физика каучука. Госхимиздат, 1947.
М. В. Классен-Неклюдова Пластические свойства и прочность кри-
сталлов. ГТТИ, 1933.
П. П. Кобеко. Физико-химические свойства диэлектриков. Госхимиздат, 1934.
П. П. Кобеко. Аморфные вещества. АН СССР, 1952.
П. В. Козлов. Физико-химия эфироцеллюлозных пленок. Госхимиздат, 1948.
С. М. Липатов. Высокополимерные соединения. АН БССР, 4943.
Г. Марк. Физика и химия целлюлозы. Химтеорет, 1935.
В. Б. Маргаритов. Физико-химия каучука и резины. Госхимиздат, 1941.
А. А. Тагер. Растворы высокомолекулярных соединений. Госхимиздат, 1951.
Г. Т а м м а н. Стеклообразное состояние. ОНТИ, 1935.
Л. Трелоар. Физика упругости каучука. ИЛ, 1953.
Я. И. Френкель. Теория твердых и жидких тел. ГТТИ, 1934.
Я. И. Френкель. Кинетическая теория жидкости. АН СССР, 1947.
Н. А. Ю з е ф о в и ч, Е. В. К У в ш и н с к и и. Изучение механических свойств
полимеров в области размягчения. ЖТФ, 23, 8 (1953).
А. П. Александров. Кристаллизация полимеров и прочность кристалли-
зующихся полимеров. Труды I и II Конференции по высокомолекулярным соеди-
нениям. АН СССР, 1945
А. П. Александров. Морозостойкость высокомолекулярных соединений.
Там же.
А. П. А л е к с а н д р о в и Ю. С. Л а з у р к и н. Температура размягчения по-
лимеров. ДАН СССР, 43, 396 (1944).
А. П. Александров и Ю. С. Л а з у р к и н. Прочность аморфных и кри-
сталлизующихся каучукоподобных полимеров. ДАН СССР, 45, 308 (1944).
Э. И. Барг. Влияние поверхностных сил прилипания на механическую проч-
ность пластмасс. Пластические массы, 2, 29 (1935).
Э. И. Барг. Физико-химические процессы смешиваемости смол. Сборник
«Пластмассы», № 1 (1935).
Э. И. Б а р г, Н. Н. Мельтева. К структуре поливинилового спирта. ДАН
СССР, 92, 2 (1953).
Э. И. Барг, Н. Н. М е л ь т е в а, Д. М. Спитковский. Влияние напря-
жения иа температуру стеклования полимеров. ДАН СССР, 85, 5 (1952).
Э. И. Барг, Д. М. Спитковский и Н. Н. Мельтева. К вопросу об
эластических удлинениях полимеров. ДАН СССР, 84, 2 (1952).
Э. И. Барг и В. Н. X о р ь к о в а. Характеристика пластичных свойств
асфальтов и пеков. Сборник «Пластмассы», № 2 (1937).
Г. М. Б а р т е н е в. О стекловании высокополимерпых тел при периодической
деформации. ДАН СССР, 69, 373 (1949).
С. Е. Бреслер, Я. И. Френкель. О характере теплового движения длин-
ных органических цепей и о причинах эластических свойств каучука. ЖЭТФ, 9,
1094 (1939).
Р. Ф. Б о й е р и Р. С. Спенсер. Переходы второго рода в каучуках и
других полимерах. «Химия больших молекул», № 2, Госхимиздат (1948).
В. М. В о л ь к е и ш т е й и, О. Б. Птицын. Статистическая физика линей-
ной полимерной цепочки. Успехи физических наук, 49, 4 (1953).
Г. И. Гуревич, П. П. Кобеко. К методике механических испытаний вул-
канизатов каучука и пластмасс. ЖТФ, 9, 14, 1267 (1939).
Э. Г у т, Г. Д ж е м с, Г. М а р к. Кинетическая теория эластичности каучука
«Химия больших молекул», № 1, Госхимиздат, 1948.
П. А. Д о г а д к и н, Г. М. Бартенев, М. М.Р е з н и к о в с к и й. Исследо-
вание роли межмолекулярных сил в механизме высокоэластической дефоомации.
Коллоид, журн. 11, 5, 314 (1949).
С. Н. Журков. Эффект повышенной прочности тонких нитей. ЖТФ, 4, 9,
1940 (1934).
С. Н. Журков. Влияние объемной сорбции па механические свойства по-
лимеров. ДАН СССР, 17, 3 (1945).
Литература 631
С. Н, Журков. Исследование механизма отвердевания полимеров. Сборник
Труды I и II Конференции по высокополимерным соединениям. АН СССР, 1945.
В. А. Каргин, Ю. М. Айалинский. Влияние объемной концентрации
пластификатора на температуру стеклования пластиката. ДАН СССР. 73, 967
(1950).
В. А. К а р г и н, Г. А. С л о н и м с к и й. Об определении молекулярного веса
линейных полимеров по их механическим свойствам. ЖФХ. 23, 363 (1949).
В. А. К а р г и н и Г. А. Слонимский. О деформации аморфно-жидких
линейных полимеров. ДАН СССР, 62, 239 (1948).
В. А. Каргин, Т. И. С о г о л о в а. К вопросу о трех физических состояниях
аморфно-жидких линейных полимеров. ЖФХ, 23, 530, 540, 551 (1949).
П. П. Кобе к о, Е. В. К У в ш и н с к и й, Г. И. Гуревич. Исследование
аморфного состояния, эластичность аморфных тел. АН СССР, серия физич. 6,
3, 329 (1937).
П. П. К о б е к о и Н. М. К у м ш а ц к а я. Диэлектрические свойства гомоло-
гического ряда полиэфиров метакриловой кислоты. Сборник, посвященный 70-ле-
тию акад. А. Ф. Иоффе. АН СССР, 1950.
П. П. К о бе к о, Г. П. Михайлов, 3. И. Новикова. Зависимость ди-
электрической постоянной совместных полимеров от температуры. ЖТФ, 19, 1,
116 (1949).
Ю. С. Л а з у р к и н, Р. Л. Фогельсон. О природе больших деформаций
высокомолекулярных веществ в стеклообразном состоянии. ЖТФ. 21, 3, 267
(1951).
Лоуренс и Вуд. Явления кристаллизации в натуральных и синтетиче-
ских каучуках. «Химия больших молекул», № 2. Госхимиздат (1948).
В. Б. М а р г а р и т о в и П. А. Р е б и н д е р. Проблема активности наполни-
телей. Журнал резиновой промышленности, 11, 991 (1935); 12, 1091 (1935); 3,
270 (1936); 5, 552 (1936).
И. А. Майгельдинов. Сравнительная характеристика технических мето-
дов определения теплостойкости полимеров. Химия и физико-химия полимеров.
АН СССР, 1952.
В. Р. Регель. О механизме хрупкого разрушения пластмасс. ЖТФ, 21,
287 (1951).
В. О. Се дли с. Пластификация и морозостойкость. Труды I и II Конферен-
ции по высокомолекулярным соединениям. АН СССР, 1945.
К главе III
Э. И. Бар г. Классификация и типизация пластических масс. Пластические
массы № 2. 1 (1935).
В. В. К о р ш а к. Вопросы систематики в химии высокополимерных соедине-
ний. Исследования в области высокомолекулярных соединений. АН СССР, 1949.
В. В. Корш а к. Химия высокомолекулярных соединений, гл. IX. (Номенкла-
тура и классификация высокомолекулярных соединений). АН СССР, 1950.
А. М. На ст юков. Введение в курс технической химии пластмасс, гл. II.
ОНТИ, Госхимиздат, 1934.
Пластические массы органического происхождения. Классификация, термины
и обозначения. ГОСТ 5752—51.
Терминология, классификация и типизация пластмасс. Стандарт Главоргхим-
прома — 2725 •
К главе IV
Э. В а л ь к о. Химические основы текстильной технологии (гл. Коллоидная
химия мыл). Гизлегпром, 1940.
С. С. Воюцкий. Успехи в области производства и применения синтетиче-
ских латексов и каучукоподобных дисперсий. Высокомолекулярные соединения, 2,
632
Литература
С. С. В о ю ц к и й, Л. Зайцева. Сопряженная растворимость и ее значение
в эмульсионной полимеризации. Успехи химии, 16, 69 (1947).
В. А. Д е р е в и ц к а я, А. А. С т р е п и х е е в. О роли окислительно-восстано-
вительных реакций в процессе полимеризации винильных соединений. Высокомо-
лекулярные соединения, 7, 14 (1948).
Исследования по физико-химии технических суспензий. Сборник работ под
редацией А. П. Ребиндера. Госхимтехиздат, 1933.
А. Н: Левин и Т. Л. Фабрикант. О скорости полимеризации хлорви-
нила в эмульсиях. ЖХП № 2, 16 (1947).
С. Л. Талмуд, Р. М. Гольдина, В. Я- А л д и к у ш к и н. О механизме
полимеризации бутадиена в водных эмульсиях. Труды III Конференции по высо-
комолекулярным соединениям, стр. 77. АН СССР, 1948.
П. М. X о м и к о в с к и й. Теория эмульсионной полимеризации. Высокомоле-
кулярные соединения, 7, 11 (1948); 8, 1 (1949).
П. М. Хомиковский, Е. В. Заболотская, С. С. Медведев. Поли-
меризация винилцианида и метилметакрилата в растворах мыл и в эмульсиях.
«Исследования в области высокомолекулярных соединений», АН СССР, 1949.
П. М. Хомиковский, С. С. Медведев. Механизм эмульсионной поли-
меризации. ЖФХ, 22, 1027 (1948).
А. И. Юрженко. Исследование различных типов перекисей в качестве ини-
циаторов эмульсионной полимеризации. Труды III Конференции по высокомоле-
кулярным соединениям, стр. 107. АН СССР, 1948.
А. И. Юрженко. Физико-химические исследования в области полимериза-
ции углеводородов в эмульсиях. ЖОХ, 16, 1171 (1946).
А. И. Юрженко и С. М. Минц. Растворимость полимеризующихся угле-
водородов в водных растворах эмульгаторов. ДАН СССР, 47, 106 (1945).
А. И. Юрженко. Механизм образования синтетических латексов. Труды
Конференции по высокомолекулярным соединениям. АН СССР, 1948.
К главе V
II. Ш. Пик. Прессовочные и поделочные пластические массы. Госхимиздат,
1951.
А. Н. Решетов, Е. И. Макарова. Полиизобутилены и применение их
в технике. Госхимиздат, 1952.
Н. В. Шорыгина. Стирол. Госхимиздат, 1950.
Н. П. Богородицкий, И. Д. Фридберг. Пластмассы из полистирола.
Сборник Пластические массы № 3. ГОНТИ, 1939.
А. И. Д и н ц е с, И. П. Л о с е в. Полиэтилен и его галоидопроизводные. Ус-
пехи химии, 20, 4, 430 (1951).
А. Я. Д р и н б е р г. Превращение полиэтилена в трехмерный полимер. ЖПХ,
15, 1 (1952).
Ю. С. Зал ьк инд, И. П. Беркович, М. К. А м усни. Синтез стирола.
Пластические массы, 1 (1943).
Н. Д. Зелинский, Г. М. Марукяи, О. К. Богданова. Получение
стирола каталитической дегидратацией этилбензола. ЖПХ, 14, 161 (1941).
А. Н. Левин, Б. Н. Рутовский. Непрерывная полимеризация производ-
ных винилового ряда. Высокомолекулярные соединения, 11, 9 (1951).
И. П. Лосев, А. И. Динцес. Полимеризация этилена и тетрафторэтилена
при высоких давлениях. Высокомолекулярные соединения, 11, 1 (1951).
С. Н. Ушаков. О синтезе хлорстирола. ЖОХ, 14, 121 (1944).
С. Н. У ш а к о в. О сополимеризации хлорстпролов со стиролом и метилмета-
крилатом. ЖПХ, 17, 1—2, 52 (1944).
И. П. Шо р ы г и н, Н. В. Шорыгина. Изучение способности к полимери-
зации замещенных стиролов в зависимости от их строения. ЖОХ, 9, 845
(1939).
Литература- 633
К главе VI
А. С. Гуляев. Изделия из полихлорвиниловой смолы. Госместпром, 1947.
Технология заменителей кожи (под род. И. П. Павлович), т. II, Гизлег-
пром, 1944.
С. В. Щуп, к ий, В. С. Пуркин. Винипласт. Госхимиздат, 1953.
А. А. Берлин, Н. И. Ден кер. Изменение химических и механических
свойств хлорированного полихлорвинила при повышенных температурах. ЖПХ,
24, 3 (1951).
И. Л Кнунянц. Полимеризация фторолефинов. Успехи химии, 20, 419
(1951).
А. Н. Левин, Б. Н. Рутовский. Непрерывная полимеризация произ-
водных винилового ряда. Высокомолекулярные соединения, 11, 9 (1951).
А. Н. Л е в и н, Т. Л. Ф а б р и к а н т. О скорости полимеризации хлористого
винила в эмульсиях. ЖПХ, № 2, 16 (1947).
И. П. Лосев, С. М. Ж и в у х и н. Совместная полимеризация винилхлорида
с ненасыщенными эфирами.
И. П. Павлович. Полимеризация хлористого винила в растворах и эмуль-
сиях. ЖПХ, 14, 551 (1941).
И. П. Павлович. Получение хлористого винила. Сборник работ по синте-
тическим смолам. Госхимиздат, 1948.
И. П. Павлович. Получение винилхлорида и винилиденхлорида путем пи-
ролиза дихлорэтана и трихлорэтана и их сополимеризация. Труды I и II Кон-
ференции по высокомолекулярным соединениям. АН СССР, 1945.
И. П. Павлович. Сополимеры хлористого винила с хлористым винилиде-
ном. Труды III Конференции по высокомолекулярным соединениям. АН СССР,
1948.
В. О. С ед лис. Пластификация и морозостойкость. Сборник трудов по син-
тетическим смолам. Госхимиздат, 1948.
Химия фтора. Сборник 1 и 2 под ред. И. Л. Кнунянц. ИЛ, 1949.
К главе VII
Е. Ф. Гросс, Я. И. Рыски и. Инфракрасный спектр поливинилового спирта
в интервале размягчения и водородная связь. Сборник, посвященный 70-летию
акад. А. Ф. Иоффе. АН СССР, 1950.
Е. К. П о д г о р о д е ц к и й. Тенхология производства пленок из высокомоле-
кулярных соединений. Искусство, 1953.
М. Ф. Ш о с т а к о в с к и й. Простые виниловые эфиры. АН СССР, 1952.
Э. И. Барг. К структуре поливинилового спирта, ДАН СССР, 92, 2 (1953).
Г. И. Д и с т л е р, 3. Г. П и иске р. Электронографическое исследование пле-
нок поливинилового спирта. ЖФХ, 23, 11 (1949).
И. Джонс. Поливиниловый спирт. Высокомолекулярные соединения, 4, 68
(1945).
В. А. Каргин, И. Я. Петров. Рентгенографическое исследование поли-
винилового спирта и поливинилхлорида. ЖФХ, 3, 16 (1951).
Б. Н. Рутовский, А. П. Даванков. Полимеризация винилацетата в
эмульсиях. ЖХП, № 2—3, 36 (1944).
С. Н. Ушаков. Поливинилацетат как основа для получения поливинилфор-
маля. ЖПХ, 19, 853 (1941).
С. Н. Ушаков. Новые пути синтеза поливинилового спирта и его производ-
ных. Сборник трудов по синтетическим смолам. Госхимиздат, 1947.
С. Н. У ш а к о в, М. М. Б и и к и н а, А. В. Е г о р о в а, И. А. Б р о й т м а н.
Полимеризация винилацетата. ЖПХ, 13, 1208 (1940).
С. Н. Ушаков, Д. И. Маневич, В. Г. Круть ко. О синтезе поливи-
нилбутираля. Сборник трудов по синтетическим смолам. Госхимиздат, 1947.
С. Н. Ушаков, Е. М. Ростовский, Б. А. Арбузова. Синтез винил-
ацетата в паровой фазе. ЖПХ, 13, 1629 (1940).
634 Литература
С. И. У ш а к о в, IO М. Ф a й н ш т е и и. О полимеризации винилацетата.
Сборник Пластические массы. Химтеорет, 1937.
А. Е. Фаворский, М. Ф. Ш о с т а к о в с к и й. К вопросу о простых вини-
ловых эфирах. ЖОХ, 18, 1 (1943).
К главе VIII
А. Я. Д р и и б е р г, Б. М. Фундыл ер, К. Г. 3 и н г е р. О полимерах алли-
ловых эфиров. Химия и физико-химия высокомолекулярных соединений. АН
СССР, 1952.
Д. А. К а р д а ш е в. Аллиловые смолы. Высокомолекулярные соединения, 5.
1946.
П. П. К обе ко, Н. М. Кум шацкая. Диэлектрические свойства гомологи-
ческого ряда метакриловой кислоты. Сборник, посвященный 70-летию акад.
А. Ф. Иоффе. АН СССР, 1950.
3. А. Роговин, А. А. Ц а п л и н а. Влияние вязкости среды на полимери-
зацию метилметакрилата. ЖПХ, 20, 875, 887 (1947).
Б. Н. Р у т о в с к и й, Е. А. Голышева. Поверхностное натяжение метил-
метакрилата при полимеризации. ЖФХ, 21, 4, 509 (1947).
Б. Н. Р у т о в с к и й, Н. С. Л е з н е в. Аллиловые эфиры трехосновных кис-
лот и их полимеризация. ЖПХ, 8, 9 (1949).
И. П. Хомиковский. Полимеризация метилметакрилата в воде и в рас-
творах мыл. ДАН СССР, 60, 615 (1948).
К главам IX, X и XI
Б. А. Архангельский. Пластические массы в судостроении. Гос. издат.
обор, пром., 1940.
П. А. Архангельский, И. Н. Ш у с т л е р. Древесная мука и ее произ-
водство. Гостехиздат, 1933.
И. А. Егоров, М. И. Назаров. Фаолит, Госхимиздат, 1946.
А. А. Козловский. Производство искусственных смол и материалов на их
основе. КОГИЗ, 1941.
П. Козлов. Слоистые пластические материалы и их применение в машино-
строении. Гостехиздат, УССР, 1935.
А. Н. Левин, Н. Ф. Бесходарный. Оборудование заводов пластических
масс. Госхимиздат, 1951.
Я. М. Мейтин. Пластические массы. Гостехиздат, УССР, 1949.
П. П. Михеев. Производство феноло-альдегидных смол. Госхимиздат, 1946.
Г. С. Петров. Искусственные смолы и пластмассы. ОНТИ, 1937.
И. Ш. П и к. Прессовочные и поделочные пластические материалы. Госхпм-
издат, 1951.
Э. И. Бар г. Рольно-суспензионный метод производства пластмасс и новые
типы материалов. Труды ЛТП, № 11, Госхимиздат, 1944.
А. К. Б у р о в, М. В. К л а с с е н - Н е к л ю д о в а. Исследования возможно-
сти получения высокопрочных анизотропных материалов на основе тонких cie-
кляиных нитей. ЖТФ, 15, 407 (1945).
А. А. Ваншейдт. Механизм образования и строение феноло-альдегидных
смол. Труды I и II Конференции по высокомолекулярным соединениям. АН СССР.
1945.
А. А. Ваншейдт, А. А. Васильев. Феноло-спирты как заменители тер-
мореактнвных феноло-альдегидных смол в производстве пластмасс. ЖПХ, 19, 1
(1946).
А. А. Ваншейдт, Р. И. Груз. О получении феноло-спиртов пз фенола и
параформа. ЖПХ, 21, 5 (1948).
А. А. Ваншейдт, А. Итенберг, Т. Андреева. Строение феноло-аль-
дегидных смол. Сборник Пластические массы № 2, Химтеорет (1937); № 3 (1939).
Литература
635
А. А. Ваншейд т. А. Итенберг, Ц. Ерлыков а, Г. С и м о и о в. Строе-
ние феноло-альдегидных смол. /Куриал Пластические массы, № 3, 17 (1934).
А. А. В а н ш е й д т, О. Н. Симонова. Получение бесцветных смол из фе-
нола и формальдегида. Сборник Пластические массы, № 3, ОНТИ, 1939.
И. П. Лосев, В. Каминский. Об использовании лигнина в промышлен-
ности пластмасс. Лесохимическая промышленность, X? 4, 4 и № 7, 12 (1939).
И. А. Майгельдинов, П. Н. Щербак. Электрическая прочность фено-
пластов. Сборник Пластические массы, № 2, Химтеорет, 1937.
Г. С. Петров, Е. Я. Голышева. О конденсации фенола с параформом.
ПОХ, 6, 3 (1939).
Г. С. Петров, Р. С. Крыловская. О конденсации фенола с гексамети-
лентетрамином. ЖХП, 12, 7 (1935).
Г. С. Петров, Т. М. Луковенко. Получение новолаков и резолов кон-
денсацией фенола с твердыми полимерами, ПОХ, 3, 702 (1937).
Г. С. Петров, Р. Г. Р и ш и н а. Конденсация фенола и крезола с форм-
альдегидом в присутствии гидратов окиси бария. Пластические массы, 4, 1
(1934).
А. Д. Соколов. Новая искусственная смола — неолейкорит. Пластические
массы, 2, 4 (1932).
А. Д. Соколов, С. И. Кирилова, А. В. Купфер. Исследования ново-
лачных смол, Сборник Пластические массы, № 3, ГОНТИ, 1939.
С. Н. Ушаков, Г. С. Бродский. Пластические массы на основе буро-
угольных фенолов. Пластические массы, 1, 12 (1934).
С. Н. Ушаков, В. М. Зельцер. О синтезе маслорастворимых смол типа
альбертолей. Сборник Пластические массы, Химтеорет, 1937.
С. Н. Ушаков, И. М. Матвеев. Применение гидролизного лигнина в про-
мышленности пластмасс. Лесохимическая промышленность, 1 (1939).
С. Н. Ушаков, Е. Н. Фрейдберг. Пластические массы на основе про-
дуктов конденсации углеводов с фенолом. Сборник Пластические массы,
Химтеорет 1937.
С. Н. Ушаков, Е. Н. Фрейдберг. Пластические массы на основе про-
дуктов конденсации фенола с масляным альдегидом. Сборник Пластические
массы ГОНТИ, 1939.
К главе XII
А. И. Лазарев, М. Ф. Сорокин. Синтетические смолы для лаков. Гос-
химиздат, 1953.
Г. С. Петров. Карбамидные смолы и прессовочные композиции. ГУУЗ
НКХП, 1940.
К- А. Андрианов и Грибанова. Новые эластичные анилино-формаль-
дегидные смолы. Сборник трудов по синтетическим смолам и пластическим мас-
сам. Госхимиздат, 1947.
А. А. Ваншейдт, 3. К. Наумова, Н. И. Сорокина. О конденсации
меламина с формальдегидом. .ЖПХ, 20, 3 (1947).
А. А. В а н ш е й дт, 3. К Наумова. О действии воды на аминопласты.
ЖПХ. 21, 6 (1948).
А. А. Ваншейдт, 3. К. Наумова. О смолах и прессовочных порошках
из мочевины и формальдегида. Сборник Пластические массы. ГОНТИ, 1939.
Г. С. Петров. Анилино-альдегидные смолы и их техническое применение.
Сборник трудов по синтетическим смолам и пластическим массам. Госхимиздат,
1947.
/\ главе XIII
И. Э. Апельцин, В. А. Кляч ко, Ю. Ю. Лурье, А. А. Смирнов
Иониты и их применение. Стандартгиз, 1949.
636
Литература
Ионный обмен. Сборник статей. Перевод под редакцией проф. К. В. Ч м у-
това. ИЛ, 1951.
Р. К у и и и, Р. Майерс. Ионообменные смолы. Перевод под редакцией
проф. Г. С. Петрова. ИЛ, 1952.
А. Б. Д а в а н к о в. О связи между строением некоторых амииоформальде-
гидных смол и нх способностью к ионному обмену. Труды всесоюзного совеща-
ния по хроматографии 21—24 ноября 1950 г. АН СССР, 1952.
И. П. Лосев, Е. Б. Т р о с т я н с к а я, А. С. Т е в л и н а. Катиоиообменные
смоляные сорбенты. Там же.
Ф. Г. Прохоров, М. Г. Корнеева. Характеристика основных органиче-
ских катионитов. Известия ВТИ нм. Ф. Э. Дзержинского, 6, 1 (1947).
Д. И. Рябчиков. Применение ионообменного хроматографического метода
в аналитической химии. Труды Всесоюзного совещания по хроматографии 21—24
ноября 1950 г. АН СССР, 1952.
Д. И. Рябчиков, М. М. С е и я в и н, К. В. Филиппов а. Сравнительная
характеристика некоторых ионообменивающих веществ Журнал аналитической
химии, 7, 135 (1952); 8, 220 (1953).
Д. И. Рябчиков, Е. А. Терентьева. Иониты и их применение. Успехи
химии, 19, 220 (1950).
К главе XIV
В. И. Иванов. Молекулы гиганты. АН СССР, 1951.
А. И. Коган. Смолы из многоатомных спиртов и миогоосновиых кислот.
ГОНТИ НКТП, 1938.
3. А. Роговин. Химия и технология искусственных волокон. Гизлегпром,
1952.
А. Я. Д р и н б е р г. Теория преимущественного влияния атомности спиртового
статна полимеров эфиров полифункциоиальных кислот. Труды ЛТИ, 14, 3 (1947).
А. Я. Дрииберг, В. М. Кобецкая, А. И. Волкова. Спиртораствори-
мые алкидные смолы на основе пентаэритрита. Там же, стр. 54.
А. Я. Дрииберг, Б. М. Фундылер. Полиэфиры малеиновой кислоты
гликоля и пентаэритрита. Там же, стр. 75.
В. В. К о р ш а к, С. Р. Рафиков. О продуктах поликонденсации некоторых
дикарбоновых кислот и диаминов. ЖОХ, 13, 974 (1944).
В. В. Корш а к, С. В. Рогожин. О причинах остановки роста цепи при
полиэтерификации. Химия и физнко-химия высокомолекулярных соединений. АН
СССР, 1952.
А. И. Лазарев, М. Ф. Сорокин. Синтетические смолы нз многоатомных
спиртов и многоосиовных кислот. Высокомолекулярные соединения, 9, 1 (1949).
А. Б. П а к ш в е р. Получение и свойства полиамидных смол. Высокомолеку-
лярные соедииення, 9, 14 (1949).
Г. С. Петров, К. Н. Власова. Конденсация пентаэритрита с малеино-
вым ангидридом. Химия и физико-химия высокомолекулярных соединений АН
СССР, 1952.
С. Р. Рафиков, В. В. К о р ш а к, Л. Н. П и н к и и а. О влиянии соотноше-
ния компонентов на рост цепи. ЖОХ, 13, 1003 (1944).
А. А. Стрепихеев, А. А. Артемьев, Я- И. Шмидт. Синтез полиурета-
нов. Исследования в области высокомолекулярных соединений. АН СССР, 1949.
Ю. А. Стрепихеев, И. А. Грибова и др. О реакции сополимеризации
динзоцианатов с гликолями. Химия и физико-химня высокомолекулярных соеди-
нений. АН СССР, 1952.
К главе XV
К- А. А и д р и а н о в. Кремнииорганические полимерные соединения Госэнерго-
издат, 1946.
Литература 637
К. А. Андрианов, М. В. Соболевский. Высокомолекулярные кремний-
органические соединения. Оборонгиз, 1949.
А. М. Б у т л е р о в. О полисоединениях в минеральной химии. Спб., 1879.
Б. Н. Долгов. Химия кремнийоргаиических соединений. Госхимиздат, 1933.
Н. А. Евстропьев, Н. А. Торопов. Химия кремния и физическая хи-
мия силикатов. Промстройиздат, 1950.
А. И. Крешков. Кремнийорганические соединения в технике. Промстрой-
издат, 1950.
Д. И. Менделеев. Основы химии, изд. 2-е, ч. II, ст. 688, Спб., 1873.
К. А. Андрианов, А. А. Туанов и др. Кремнийорганические соедине-
ния. Успехи химии, 18, 1 (1949).
К. А. Андрианов, Б. М. Б р е й т м а н. Кремнийорганические полимерные
продукты из фенилсилантрихлорида и дифенилсиландихлорида. ЖОХ, 17, 1522
(1947).
К. А. Андрианов, О. Грибанова. Исследования в области алкил- и
арилзамещенных ортоэфиров кремневой кислоты. ЖОХ, 8, 558 (1938).
АЛФАВИТНЫЙ
Адгезия 129, 422
Адипиновая кислота 587, 592—594
---ди а мид 593
Адипонитрил 593 и сл.
Акриласт 152, 156
Акриловая кислота, получение 317
— — эфиры 317 н сл.
—------свойства 321
— — — синтез из оксипропионовых
кислот, 319 н сл.
Акриловые отходы, переработка 333
Акрилонитрил 161, 321 н сл.
Акрипласты 156, 317
Акрилопласты см. Акрипласты
Акрипленка 156
Акритекстослой 156
Активаторы при полимеризации 45
—------капролактама 597
Активация мономеров 33 и сл., 44
Александров А. П. ЮЗ. 108, 114, 123,
125, 134, 214
Алкоголиз каталитический 296 и сл.
— полиамидов и полиэфиров 576
Аллилакрилат 335
Аллилопласты 342
— слоистые, применение 347
--- свойства 348
Аллилтекстослой 156
Аллиловые пластики, «отверждение»
345 и сл.
— полимеры 348
— — свойства 314
— смолы 342
— — литые 345
— — структура 344
— стекла, свойства 314 и сл.
- - эфиры 342
— — полимеризация 342—311
— — полимеры 342
Алюмосиликаты 552
Амберлит(ы) 557, 562
Амидолит 157
Амндолоид 157
Амидоплает(ы) 157, 565, 591 и сл.
«-Аминобензиловый спирт 547
УКАЗАТЕЛЬ
Аминобумослой 151, 157, 533
— применение 535
— производство 533 и сл.
Амннолиз полиамидов и полиэфиров
576
Аминолнт 151, 157
Аминопласт(ы) 121, 151, 154, 157, 514,
533
— методы получения 521
— прессование 535
Аминопор 152, 157
Аминопоропласт (ы) 152, 157, 536
Аминосмола, синтез 561
Аминотекстослой 157, 533 и сл.
— производство 533
Аминоцеллолиты 151. 157. 523
Аморфное состояние низкомолекуляр-
ных веществ 81 и сл.
— тело 81
Аморфные полимеры см. Полимеры
Амусин М. К. 197, 456
Ангидроаминобензиловый спирт 547
Ангидроформальдегиданилин 546
Андрианов К. А. 16, 508, 545 и сл.,
612, 619
Анилинобумослой 157, 550
— применение, свойства 550
Анилинолит 157
— свойства 550
Анилинопласты 157, 545, 548, 550
— типа «совенпт» 549
Аниониты 551
— анилино-формальдегидные 560
— мочевино-формальдегидные 560
Анионообменные смолы 561
--- получение 559 и сл.
Арбузов А. Е. 13
Арбузова И. А. 271, 274, 286
Артемьев .4. А. 606
Асбовинил 152
Асболит 499
Асборезиты, применение 452
- - свойства 450 и сл.
— слоистые 499
— формовочные массы 455
Алфавитный указатель
639
Асботекстолит 499, 502
Ацетилирование 305 и сл.. 308
Ацетальвпнипласт 156
Ацетилцеллюлоза 158
— температура стеклования 136
Ацетобутиратцеллолоид 158
Ацетобутиратцеллюлоза 158
Ацетовинипленка 156
Ацетолоид 152
Ацетонциангидрин 322
Ацетоцеллолит 158
Ацетоцеллолоид 152, 158
Анетопеллопленка 158
Ацидолиз полиамидов и полиэфиров
576
Бутандиол 594
Бутвар 312
Бутилакрилат 341
Бутилкаучук 192
Бутилнафталинсульфокислота (на-
триевая соль), эмульгатор 228
Бутиндиол 594
Бутиральвиниклей 156
Бутиральвнниласт 152
Бутиральвиниплеика 156
Бутлеров А. М. 13, 31, 185, 287, 612
Бюлер 414
Бэкеленд Л. 15, 396
Байер 14, 569
Бакелит 396
Барг Э. И. 100, 105, 140, 147, 492
Бенастр 194
Бензилцеллолит 158
Бензилцеллюлоза 158
Беркович И. П. 197
Берцелиус 576
Бесхадарный Н. Ф. 385
Бисерная полимеризация 165, 169, 281
Битуминолит 158
Битуминопласт 158
Битуминоцеллолит 158
Бифункциональные молекулы 29
«Благородные» резиты 397 и сл.
Блочная полимеризация 160 и сл.,
202—205, 213, 281, 327
Блюмберг Б. Л. 84
Браунс 414
Бреслер С. Е. 96, 97, 552
Бродский Г. С. 417
Бумага для фенобумослоя 483 и сл.
— двухсторонняя пропитка 485
— односторонняя пропитка (лакиров-
ка) 487
— пропитка расплавленной смолой
490
Бумаги намоточные 484
— пропиточные 484
— сульфатные, свойства для произ-
водства фенобумослоя 484 и сл.
Бумажная масса, разработка в ролле
492, 493
Бумажно-анилиновые пластики см.
Анилинобумослой
Бумажно-слоистые пластики, рольный
метод производства 482
Бумажно-смоляная масса, получение
осмоленнем 493
Бумофенолит 482
Буров А. Л'. 512
Ваншейдт А. А. 16, 46, 153, 198 и сл.,
354 и сл., 357, 397, 40в, 472, 517,
523, 526, 539
Варочные агрегаты см. Реакционные
агрегаты
Варочный котел (схема) 381
Варочно-сушильный аппарат 381 и сл.
Васильев А. А. 10, 472, 551
Весовая прочность 17, 463
Винилацетат 161
— полимеризация бисерная 281
----блочная 281 и сл.
-----в растворителях 277
----— — башенная 278 и сл.
----------непрерывная 278 и сл.
— — эмульсионная 280 и сл.
— преимущества парофазного мето-
да перед жидкофазным 276
— производство парофазным методом
271
----жидкофазным методом 271
— свойства 270
— синтез 270 и сл.
— технические показатели 276
— ядовитое действие паров 276
Винилит 151
N-Вииилкарбазол 161
— компонент при получении сополи-
меров 229
— синтез, свойства 226—230
— сополимеры со стиролом 229
Винилкарбазолит 156
Винилкарбазопласт 156
Виниловые эфиры см. Простые вини-
ловые эфиры
Винилформиат, полимеризация 287
— свойства 286
— синтез в жидкой и в паровой фазе
286
Винилхлорид см. Хлористый винил
Винипласт 151 и сл.. 241
— вальцевание 242 и сл.
640
Алфавитный указатель
Винипласт, заменитель целлулоида
248
— листовой, применение 247
— механические свойства 246
— пленочный, применение 247
— получение 242
— прессование 244, 245
— сварка 247
— склейка тонких листов перхлор-
виниловой смолой 247
Винипластоклей 156
Волокниты 423, 445, 447
— свойства 448
Волькенштейн М. В. 97 и сл.
Вофатит(ы) 557, 558, 562
Временной фактор, значение для
низкомолекулярных тел 77 и сл.
Время релаксации 78, 106, ПО, 141,
144
----- температурная зависимость 105
Высокоэластическая деформация 89 и
сл.; см. также Деформация вы-
сокоэластическая
Вязкое течение жидкости 75
Габаритный фактор 123
Гавурина Р. К. 299
Галалит 12
Гапон Е. Н. 552
Гексаметилендиамин, синтез 593 и сл.
Гексаметилендиизоцианат, исходное
сырье для получения полиурета-
нов 607
Гексаметилолмеламин 538 и сл., 544
Гель-фракция 50
Гетерополиконденсация 62
Гетерополимеризапия истинная 46, 48,
54
— обычная 48, 54, 212
Гетинакс 482, 498; см. Фенобумослой
Гибберт 414
Гидракриловый альдегид 319
Гидрофобизация минеральных стекол
(волокон) 508
Гиперболы А. И. Александрова 125
Глифтали 27, 154, 581; см. также
Смолы глицеро-фталевые
Глифтальасболит 157
Глифтальслюдослой 157
Глифтальстеклослой 157
Глицерофталат 157; см. также Глиф-
тали
Гомополиконденсация 62
Горяйнов В. 185
Градусы Шоппера-Риглера (Ш.—Р.)
493
Груз Р. И. 472
Густавсон Г. Г. 175
Гут 81
Даванков А. Б. 562
Дауэкс 559, 562
Дельта-древесина 502
Деформация высокоэластическая 70,
89 и сл., 145
---- закономерности 96
----зависимость от температуры
104 и сл.
--------от частоты напряжения 105
---- механизм 89
----релаксационный характер 102
и сл.
---- теория 90 и сл.
----тепловой эффект 89
— зависимость от времени напряже- .
ии я 80
-----от температуры 108
— общая аморфных низкомолекуляр-
ных тел 89
— — зависимость от времени 107
------- от температуры 102
----кристаллических тел 89
---- механическая модель 107
----пластическая 70, 74 и сл.
---- скорость 75
— упругая 70, 71 и сл., 89 и сл.
— — запаздывающая 81
— — идеальных тел 74, 78
-------твердых тел 72
----реальных тел 74
Диаллилфталат 161
Дибутиладипииат 140
Дибутилоксалат 140
Дибутилсукцинат 140
Дибутилфталат 143
Дигексиладипинат 110
Дигексилсебацинат 140
Дигексилсукцинат 140
Диизобутилмалеат 256
р,^-Диметилвииилэтиловыи эфир 287
Диметилмалеат 256
Диметилолмочевина, свойства 515 и
сл.
Динил 578
Диоксибеизолы, свойства 418
Диоктилсебацинат 140
Дихлорбутан 593 и сл.
2,5-Дихлорстирол, получение 225 и сл.
— полимеризация 226
— применение 226
Дихлорстиролит 156
Диэтилмалеат 256
Диэтилоксалат 140
Догадкин- Б. ,4. 42
Древесно-слоистые пластики (ДСП)
502; см. также Фенодревослой,
Резитовая фанера
---- — свойства 505
Алфавитный указатель
641
Дринберг А. Я. 574 и сл., 588 и сл.
Дробилка Блека 431
Желатинизация пространственных со-
полимеров 50 и сл., 420 и сл.
Жидкость ньютоновская 75 и сл.
Журков С. Н. 123, 138, 300, 509
Закон Гука 72, 75, 77
— Ньютона 75, 77
Закономерности кислой конденсации
и образования новолачных смол
361
— реакций образования полиэфир-
ных и полиамидных смол 565—
576
— щелочной конденсации и образо-
вания резольных смол 370
Залькинд Ю. С. 197, 199, 286
Зандмайер 225
Зелинский Н. Д. 199
Зельцер В. М. 198, 406
Занин Н. Н. 13, 342
Золь-фракция 50
Идеально-пластичное тело 76
Идитол 390
Изобутилвиниловый эфир 256
Изобутилен, полимеризация 186 и сл.
----ингибиторы 187
— свойства 186
— сополимеры 192
Изобутиленоласт 155
Изобутиленопласты 155, 185; см.
также Полиизобутилен
Изогель 361
Ингибиторы 42 и сл.
— при полимеризации 159
Инициаторы 34, 42 и сл.
Интервал высокоэластичности 90, 137
— — зависимость от степени полиме-
ризации 102 и сл.
— размягчения 86
---- низкомолекулярных смол 85
и сл.
---- линейных полимеров 88
— пластичности 86
Иониты, классификация 551
— • обменная емкость 553 и сл.
— ---Динамическая 554
-------полная 554, 562
---- способность 553
— применение 551, 563 н сл.
— селективного действия 562
Ионообменные материалы см. Ионнты
Иоффе А. Ф. 123
Искусственные копалы, производ-
ство 406 и сл.
Каландр трехвалковый 244
Каландрирование винипласта 243 и
сл.
Каландровый эффект 244
Капельная полимеризация см. Би-
серная полимеризация
Капролактам 594
Капрон 464, 598, 603
Карбазовинипласты 227; см. также
Поливиннлкарбазол
Карбазол 227
— винилирование ацетиленом 227
— свойства 227
Карболит литой 16
— — производство 396
---- свойства 401
Карбопласты 148
Каргин В. Л. 16, 103, 112, 129. 140,
190
Карозерс В. 5ТЗ, 591, 595
Картофенолиг 463, 482; см. также
Фенобумослой
Катализаторы 34
— комбинированные при полимери-
зации простых виниловых эфи-
ров 290 и сл.
— новолачной конденсации 362
— резольной конденсации 370 и сл.
Катионит ПФСК, получение 556
Катионит СБС 555
Катионит СБС, К-2, 555
Катиониты 551
Катнонообменные смолы 555 и сл.
Каучук бутадненнитрильный 46
— бутадиенстирольный 46
— изобутиленовый 192
— природный 112, 115, 134
---- вулканизация 12
— синтетический 148
Каучукопласты 149
Кетнер 364
Кинли 364
Киселев В. С. 520, 539, 544, 588
Кислород, влияние на полимеризацию
42 и сл.
Клаасен-Неклюдова М. В. 512
Классификация пластмасс 146, 152—
158
----ГОСТ 5752-51 154—158
Клебанский А. Л. 42
Клячко В. А. 562
Кнунянц И. Л. 591
Кобеко П. П. 103, 121, 141
Коган А. И. 566
Когезия, 129, 131
Кондаков И. Л. 14, 46
Кормак В. В. 16, 41. 65, 153, 566,
568. 575 п сл., 591
41 Зак. 1269. 3 II. Барг
642
Алфавитный указатель
Коэффициент Пуассона 73
Кракау А. 194
Краус 523
Кремнийорганические полимеры, полу-
чение исходных «мономеров» 615
----- процессы образования 616—620
— — свойства 623
•----структура 621—623
— смолы, основные типы 611
— — химические особенности 613—
615
Крешков А. И. 627
Кристаллизационные потенциалы 82
Кристаллизация полимеров 118—119
-----тепловой эффект 112
Кристаллиты полимеров 111
— ориентированные 116
— процесс образования и роста 114
Кристаллические полимеры 111 и сл.,
145
Кристаллическое состояние 82
-----изменение свойств с темпера-
турой 85
Кун 81, 95 и сл.
Куткин Ф. А. 561
Кучеров М. Г. 14
Лакировально-сушильная машина
488—490
Лаккаин 15
Лаковые смолы мочевино-формальте-
гидные 536 и сл.
•----на основе меламина, примене-
ние 543 и сл.
---------—--------полиэфирных смол 576,
589
Латекс 329, 331
Лебедев С. В. 14, 185
Левин Л. И. 469
Лезнев И. С. 348
Ли. П. 3. 385, 466
Лигнин 413 и сл.
— химический состав 414
Лигнофоль 502
Литой карболит; см. Карболит литой
Литые продукты резольной конденса-
ции 396
— резиты 396, 401, 422
Лосев И. П. 16, 231, 412, 545, 556
Луполен 176
Макробрауновское движение 101
Макромолекулы, вращение звеньев
цепи 92. 93, 96
— средняя квадратичная длина 94
— схема строения цепи 92
Максвелл 78
Малеиново-канифольные аддукты 587
Марвел 284, 518
Марк 81
Мартин 120
Маслорастворимые смолы, получение
407
Массный ролл 492
Матвеев И. И. 412
Медведев С. С. 16, 33, 35, -15 и сл.,
96, 167
Междучастичные расстояния, зависи-
мость прочности 71
Межмолекулярное взаимодействие 116
и сл.
Межмолекулярные силы ПО и сл.
Меламин 519 и сл.
— поликонденсация с формальдеги-
дом 520
Меламинодреволит 151, 157
Меламиноклей 152, 544
Меламинопласт 157, 539
Меламиностеклослой 157, 544
Меламино-формальдегидные пресспо-
рошки, производство 540
— смолы 157, 519, 538, 545
----модифицированные 544
Мельтева II. Н. 140
Менделеев Д. И. 612, 616
Метакриламид 324
Метакриласт 156
Метакрилит 156
^Метакриловая кислота, амид 323
---- свойства 322
----эфиры и производные, получе-
ние 322
— — — свойства 326
----ацетонцизигидриновый синтез
322
Метакрипласт 156; см. также Поли-
метилметакрилат
Метакрипленка 156
Метакритекстослой 156
Метилакрилат 256, 335
— схема непрерывного получения 318
а-Метилакрнловая кислота см. Мет-
акриловая кислота
Метиленанилии 545—547
Метиленимин 518
Метнленмочевина, свойства 516 и сл.
Метилметакрилат 161, 323 и сл
— полимеризация 174, 327 и сл.
— синтез 323 и сл.
----«в одном котле» 324
— схема получения 325
Метиловый эфир метакриловой кис-
лоты см. Метилметакрилат
Метилоланилии 545
Метилолмеламины. получение 544
— реакция смолообразования 544
Алфавитный указатель
Мегилпелисилоксань 625
Метилцеллопленка 158
Метилцеллюлоза 158
Миканит 576, 590
Микробрауновское движение 101
Михайлов Н. В. 129
Михеев И. П. 364, 385
Мицеллярный раствор, образование
172
Модифицированные смолы 536 и сл.,
583 и сл.
Модуль высокоэластичности 95, 105,
107, 128
— упругости 72 и сл. 105, 107, 120—
"123, 128
— Юнга 72
Молекулярная функциональность 574
Молекулярный вес полимеров, опре-
деление 59
Моноаппарат см. Варочно-сушильный
аппарат
Монометилолмочевииа, свойства 515
и сл.
Монофункциональные молекулы 29
Морзе 120
Мочевино-формальдегидные смолы
514—519
— — методы получения 521 и сл.
— — модифицированные для лаков и
клеев 536 и сл.
— — прессматериалы, методы полу-
чения 521 и сл.
-----пресспорошки, применение,
свойства 532
----- производство 525 и сл.
-----слоистые, производство 533
Мыла как эмульгаторы 165
Надсмольные воды, утилизация 385
Найлон 595, 604
Наполнители 126
— активные 134
— волокнистые 126
— диспергирующее действие 133
— длинноволокнистые 444 и сл.
— инертные (неактивные) 134
— минеральные 445, 450 и сл.
— обозначение 150
— органические (целлюлоза) 445, 447
— порошкообразные 126
— усиливающее действие 129—135
------- на высокоэластические поли
меры 133
Напряжение сдвига идеально упру-
гого тела 75, 78
Настюков А. М. 548
Наумова 3. И. 526, 539
Неоконденситовый метод 425
Неолейкорит 396, 398—401
Неслоистые пластмассы на основе
феноло-альдегндных смол 422
— фенопласты, свойства 449
Никольский Б. П. 552
Нитролоид 152, 158
Нит.роцеллолит 158
Нитроцеллолоид 152, 158
Нитроцеллопленка 158
Нитроцеллюлоза 158
— степень полидисперсности 57
-----размягчения 366
Новолак(и) 353, 355, 361
— для пресспорошков, характеристи-
ка 361
— процесс производства 386—388
— температура каплепадения 365
и сл.
----- размягчения 366
— термореактивность 361 и сл.
Новолачная конденсация, аппаратур-
ная схема 376
-----подготовка сырья 376 и сл.
Новолачные пресскомпозиций, состав
на различных стадиях обработки
369
— прессматериалы 425—429
— пресспорошки 430—440
-----вальцовый метод производства
431, 438
— — поточный метод производства
437
-----различные методы получения
440
----- рецептура 431
— — свойства 427
----- скорость прессования 428 и сл.
----- состав 426
-----на основе древесной муки 430
----шнековый метод производства
438
-----шнекпресс для непрерывной
пластикации 438—440
----- условия изготовления 430
Новолачные смолы 353
-----для пресспорошков, рецептура
386 и сл.
-----------сырье альдегидное 386
-----------сырье фенольное 386
----------- технологический процесс
производства 386, 387
— — модификация 402 н сл.
----- получение 353
— — применение для производства
пресскомпозиций в присутствии
уротропина 368
----- свойства 389
— — химическая структура 355, 356
644
Алфавитный указатель
Окислительно-восстановительный цикл
при эмульсионной полимериза-
ции 167 и сл.
Органические стекла, 126, 327, 338 и
сл., 344
Органохлорсиланы, получение 615
Ориентация полимеров 144
Орлов Е. И. 175
Остромысленский И. И. 46, 194, 231
Отталкивание частиц 71
Павлович П. И. 231
Параформ 32, 374 и сл., 377
Пек каменноугольный 132
Пекоасбослой 131 и сл., 158
Пенопласты см. Поропласты
Пентаметилолмеламин 538, 544
Пентаэритрит 544, 569
Пентолн 589
Перекись бензоила 35, 160
Перекиси, образование в мономерах
160
Переохлажденные жидкости 82
Перхлорвинил 253
Петров Г. С. 15 и сл., 369 и сл., 396,
411, 419, 537, 545, 627
Пластикат см. Полихлорвинил пла-
стифицированный
Пластики 148
— древеснослоистые см. Фенодрево
слой
— общая характеристика 18
— полимеризационные 159
— применение 23—25
— слоистые на основе стеклянного
волокна и стеклянной ткани, тех-
ническое значение 507
--- получение пропиткой стеклян-
ного волокна в процессе его про-
изводства 512 и сл.
— характеристика, свойства, приме-
нение см. Пластмассы
Пластинчатая мицелла эмульгатора,
схема 173
Пластификаторы 136
— «выпотевание» 142
— высокомолекулярные для поли-
хлорвинила 249
— кристаллические (кристаллизую-
щиеся) 143
— летучесть 141
— механизм действия 136, 138
— неполярные 139 и сл.
— несовместимые 142
— полярные 140
— совместимость с полимером 141,142
— стеклующиеся 143
— температура стеклования 143
Пластификация внутренняя 47, 137
—• ложная 137
— механизм 108—141
— механическая 144 — 146
— полимеров 136—146
Пластические массы см. Пластмассы
Пластическое течение 100
--- твердого тела 75
Пластмассы, диэлектрические свой-
ства 18
— из полихлорвинила, не содержа-
щие пластификаторов 241
— литые 154
— классификация, ГОСТ 5752-51
154—158
— методы переработки 20
— механическая прочность 18
— на основе амидо-формальдегидных
смол 157
--------амино-альдегидных смол 514
-------амино-формальдегидных
смол 157
--------анилнно-формальдегидных
смол, значение 545
---------- белковых веществ 12, 158
—-------кремнийорганических поли-
меров 158
_ — .--------свойства 625
------------- получение 624
---------- смол 611
--------меламино-формальдегидных
смол 514, 538 и сл.
-------------прессматериалы 54С
и сл.
--------мочевино-формальдегидных
смол 514
--------нефтяных асфальтов 158
--------полиамидов 157, 591—594
-------- поливинилкарбазола 227
-------- полиизобутилена 185
—------полимеров аллиловых эфи-
ров 342
---------- винилового спирта и его
производных 270 и сл.
----------галоидопроизводных эти-
лена 231
----------непредельных углеводо-
родов 175
----------производных этиленкар-
боновых кислот 317
--------полимочевин 605—610
— —• — полистирола 193 и сл.
-------- политетрафторэтилена 263
-------- полнтрифторхлорэтилена
— — — полиуретанов 605—610
----------и полимочевин 158
Алфавитный указатель
645
Пластмассы на основе полихлорви-
нила 231
-------- полихлорвинилидена 256
-------- полиэтилена 175
--------полиэфирных смол 157, 576
-----------полиамидов, полиурета-
нов и полимочевин 565
—-------природных асфальтов 158
—-------простых эфиров целлюлозы
158
------- — различных пеков 158
--------сложных эфиров целлюлозы
158
--------смол и асфальтов 12
--------резорцино-формальдегидных
смол 157, 419 и сл.
--------синтетических полиамидов
591—594
-----------— смол, получаемых поли-
конденсацией и ступенчатой по-
лимеризацией 350
--------феноло-альдегидных смол
157; см. также Фенопласты
--------целлюлозы 12
—-------эфиров ненасыщенных спир-
тов 317
— наполненные 129
— недостатки технических свойств
21—23
— не слоистые 126
— неорганические (минеральные) 148
— обозначение 149—152
— определение понятия 148—149
— оптические, свойства 19
— органические 148
— природные (органические) 11
— поликонденсационные 15
— полимеризационные 15
— прессовочные 154
— производство из полиэфирных
смол 576
— прочность 119 и сл.
---зависимость от степени запол-
нения 132
-------- от поверхности наполните-
ля 131
-------- от структуры 126 и сл.
— синтетические модифицированные
13
— слоистые 126, 154
— содержащие значительное коли-
чество пластификаторов 248
— терминология 147—152
— технические свойства 17 и сл.
— типизация 153
— удельный вес 17
— физические основы технологии 70
и сл.
Пластмассы, формовочные 154
— фрикционные, свойства 19
— химическая стойкость 18
— химические основы технологии 26
и сл.
Пластоклей 152
Поворотные изомеры 97
Полиакрнлаты 331, 338, 341
— применение 341
— свойства 317, 336 и сл.
— строение 333 и сл.
— технические продукты и изделия
338 и сл.
— техническое значение 317
Полиалкилметакрилаты 141
Полналлилметакрилаты 156
Полиамидное волокно, получение 604
Полиамидные нити 604
— смолы, образование 566
---применение 565
Полиамиды 116 и сл„ 601, 608 и сл.,
610
— методы переработки 603—605
— получаемые поликонденсацией ди-
аминов с дикарбоновыми кисло-
тами 595—597
---полимеризацией циклов — лак-
тамов g-аминокислот 597—600
— получение 592
— свойства 601 и сл., 609 и сл.
Полибутадиен 117
Полибутилакрилат 341
Полибутилметакрилат 156
Поливинилацетали 270, 304
— методы производства 306—308
— образование 304
— однованный и двухванный методы
получения 305
— промышленные 312
— процесс производства 308:—310
— рецептура реакционной смеси 309
— свойства 310 и сл.
— скорость ацеталирования 306
— степень ацеталирования 308
--- полимеризации 312
— строение 310
— технический комплекс свойств 312
Поливинилацетат 106, ПО, 115, 117,
156, 270
— применение 285
— процесс омыления 296 и сл.
— реакция Кучерова 270 и ел., 274
— свойства 284 и сл.
— спектры поглощения 2,4-пентади-
ола и поливинилового спирта 283
— строение 282
— схема строения молекулы 283
Алфавитный указатель
Поливинилбутираль 106, 108, 110, 315
— методы получения 315
— пластифицированный, применение
315
— применение 315 и сл.
— свойства 315
Поливииилиденхлорид 155. 256
Поливинилкарбазол 156, 227 и сл.
— применение 230
— свойства 228 и сл.
— структура 228
Поливиниловый спирт 117, 156, 270,
295 и сл.
-----область применения 301 и сл.
-----пластификация 301 и сл.
-----применяемые в технике пласти-
фикаторы 302
— — получение 295—298
----- свойства 301
----— — степень полимеризации 300
строение 299
-----схема производства 298
—----------ацеталей 309
Поливинилформаль, показатели
свойств 313
— способы получения 312 и сл.
— степень ацеталирования 313
Поливинилформиат 286
— свойства 287
Поливинилхлорид 155; см. также По-
лихлорвинил
Поливинилэтаналь 314
Поливинилэтиловый эфир 156
Полигексаметиленадипамид 65, 595
Полигликольтерефталат 581
Полидекаметиленсебацамид 603
Полидисперсность полимеров 55
-----методы определения 56
Полидихлорстирол 156
Полиизобутилен 100, ПО, 115, 117,
155, 185
— переработка 192 и сл.
— получение 188
— применение 192 и сл.
— свойства 190—192
— строение 189
— фракционирование 56
Полиизопрен 117
Поликапролактам см. Капрон
Поликонденсация 31, 63 и сл.
— диаминов с дикарбоновыми кис-
лотами 595 и сл. ___
— зависимость от функциональности
молекул 66 и сл.
— катализаторы 362 и сл.
— константа равновесия 63
— линейных молекул 67
Поликонденсация мочевины и мела-
мина с формальдегидом 514
— образование «сшитых» полимеров
69
— условия образования циклов 67
и сл.
— фенола и формальдегида 64
— фенолов с альдегидами, свойства
продуктов 351
— фталевых кислот с гликолем 68
и сл.
Полимеризация 31 и сл.
— акриловых эфиров 34, 327
блочный метод 327 и сл.
------- — в растворителях 331
--------эмульсионный метод 329 и
сл.
— активаторы 45
— бисерная 165, 169, 281
— блочная 160 и сл., 202—204, 327
— бутадиена 36
— в водных эмульсиях 160, 163
---------- технический метод 163
— в газовой фазе 160
— в жидкой фазе 160
— винилацетата 276
— N-винилкарбазола 228
— виниловых эфиров 45
— влияние растворителей 43
— в растворителях 160, 162, 277
-------- механизм 162
--------эмульгаторы 235
— диеновых соединений 14, 45
— диизоцианатов с диаминами 32,
605, 606
---с диоксисоединениями 32, 605
и сл.
— дихлорстиролов 226
— изобутилена 31, 187
— индена 31
— капролактама 32
— метакриловых эфиров 34, 327
блочный метод 327 и сл.
-------- в растворителях 331
— неполярных молекул 41
— непрерывные методы 161 и сл.
— несимметричных молекул 40
— окиси этилена 31
— полярных молекул 40
— пространственные затруднения 41
— процессы 159
— симметричных молекул 40
— скорость реакции 39 и сл.
— стирола 31, 43, 45, 194
---башенный (непрерывный) ме-
тод 204 и сл.
— — блочная 202 и сл.
Алфавитный указатель
647
Полимеризация стирола водно-
эмульсионная 207
---- в растворителях 206
— ступенчатая 31 и сл.
— технические методы 159
— требования чистоты исходных мо-
номеров 159
— формальдегида 31 и сл.
— фторпроизводных этилена 41
— хлористого винила 34, 234 и сл.
— — винилидена 34, 257
— хлоропрена 36
— цепная 31, 33 н сл.
----инициирования 34—36
— — ионная 34, 44 и сл.
----каталитическая 34, 44 и сл.
---- механизм 33
----радикальная 34—36
— чистого мономера 160—162
— эмульсионная 166, 207, 234 , 280
и сл., 329, 341
---- бисерная (капельная) 165, 169
---- инициаторы 165
----регуляторы 163—166
— — эмульгаторы 164 и сл.
Полимеры азотсодержащие 154
— акриловой кислоты 317, 338
— алкилпроизводных этилена 155
— аллиловых эфиров 342 и сл.
— аморфные 111, 115, 144
— арнлэтиленов 156
— вннилкетонов 156
— винилового спирта и нх производ-
ные 156
— виниловых эфиров, пространствен-
ные 292
— винилформиата 287
— высокоэластические 118
— высокоэластическое обратимое
удлинение 99 н сл.
---- состояние 92
— вытяжка 144 и сл.
— галондозамещенные 153
— галондопронзводных этилена 155
— гетероцепные 153
— зависимость времени релаксации
от температуры 105
свойств от содержания пласти-
фикатора 143
— замещенных стиролов 224
— изобутилена 13
— карбоцепные 153
— каучукоподобные идеальные 94
— кислородсодержащие 154
— кристаллизующиеся 135
— кристаллические 111, 115 н сл.,
118, 145
— линейные, свойства 128
Полимеры метакриловые 338
— молекулярный вес 65
— моноалкнлвиниловых эфиров 291
— монодисперсные 55
— морозостойкость 144 н сл.
— насыщенные 153
— ненасыщенные 153
— неполярные, температура стеклова-
ния 139
— определение молекулярного веса
58 и сп.
— — структуры цепи 61
— ориентация 144
— пластификация 136—146
— полидисперсность 55, 60
— полярные, температура стеклова-
ния 138
— привитые 59
— производных внниламина 156
— пространственные, скорость обра-
зования 420
— простых виниловых эфиров 291
— —-----применение 287
-------------для изготовления ла-
кокрасочных композиций 293
— прочность 126
---зависимость от степени полиме-
ризации 127
— разветвленные 57 и сл.
— — условия образования 58 н сл.
— способность к кристаллизации 116,
118 и сл.
— структура звеньев цепи 60 и сл.
— фазовая структура 110 н сл.
— физическое состояние в зависимо-
сти от молекулярного веса 110
— формальдегида 13, 32
— хлорстиролов 225
— этилена 155
— эфиров этнленкарбоновых кислот
156
Полиметакрнлаты см. Полиметилме-
такрилат
Полиметилакрилат 156
Полнметнлизопропенилкетон 156
Полнметнлметакрилат 115, 122, 125
и сл., 138, 156, 327, 330, 338, 344
— деполимеризация 333
— пресспорошки 338—340
— применение 341
— свойства 338—341
Полнмочевины 116, 608 и сл.
Полисилоксановые пластики для изо-
ляции 626
Полипропилакрилат 341
Полистирол 106, 115, 117, 126, 137,
144 и сл., 156, 193 и сл.
— «блочный» 213
648
Алфавитный указатель
Полистирол зависимость температуры
стеклования от содержания диви-
. нилбензола 211
— измельчение и просеивание 210
— марки 219
— методы сушки 209
— переработка выдавливанием на
шнекпрессах 217
---литьем под давлением 215 и сл.
— пленки 218
— получение см. Стирол, полимери-
зация
---эмульсионный метод, техноло-
гический процесс 207 и сл.
— прессматериалы, свойства 219
— применение 194, 219 и сл.
— свойства 194, 212—215
— степень полидисперсности 57
— структура 213 и сл.
— «эмульсионный» 213, 219
Политен 176
Политерефталат (ы) 464, 580 и сл.
Политетрафторэтилен 116, 155, 265
— переработка и применение 267
— получение 263 и сл.
— строение и свойства 265 и сл.
Политрифторхлорэтилен 267—269
• — технические показатели 268 и сл.
Полиуретаны 116, 609
— образование 605
— свойства 605, 609 и сл.
Полиуретановые смолы см. Полиуре-
таны
Полнфункциональные (полиреактив-
ные) молекулы 29 и сл.
Полихлорвинил 117, 122, 126, 143
— аморфная структура 240
• — непластифицированный 249
— пластифицированный 248 и сл.
пластикат 248, 250 и сл.
------ применение 248
— получение 234
— поропласт 252
— свойства 238—240
— строение 238
— хлорированный 253
Полихлорвиниловые пасты, примене-
ние 252
Полихлорвинилиден 109, 464
— переход в аморфное состояние 259
— свойства 258 и сл.
— строение 258 и сл.
— технические свойства 262 *"
Полихлорвинилиденовые эмульсии 263
Полихлоропрен 115
Полиэтилакрилат 341
Полиэтилбензолы, получение 196 и
сл.
Полиэтилен 116 и сл., 118, 155, 175
— молекулярный вес 178, 180
— получение 175—177
— применение 184 и сл.
— свойства 179 и сл., 184
— структура 178
— технический 176
Полиэфиропласт 151
Полиэфирные смолы 565 и сл.
— — глобулярной структуры 577
---- закономерности образования
565 и сл.
----классификация 577
----линейной структуры 577
----модифицированные смоляными
или жирными кислотами 577
---- на основе линейных двухоснов-
ных кислот 587 н сл.
----применение 576, 589 и сл.
— — термоплавкие 577
— • — термореактивные 577
----чистые 577
Поропласты 152, 221
— из поливинилформаля 221 и др.
— из полистирола 221
---- полихлорвинила 252
— получение 221 и сл.
— применение 221, 223
— свойства 223
Порообразователи, техническое при-
менение 222
Порофоры 221 и сл.
Правило Эльтекова 271
Предел упругости 73
— прочности 73
— текучести 76
Прессматериалы на основе длинно-
волокнистого наполнителя 444—
452
----— меламино- и мочевиноформ-
альдегидных смол 540 и сл.
---------- полимеров метилметакри-
лата, физико-механические свой-
ства 331
-------целлюлозы и различных
смол, свойства 542
— новолачные и резольные 426 и сл.
— с целлюлозным волокном 447
----------пропитка волокна по ме-
тоду С. Н. Ушакова 448
---------- рецептура 448
— текучесть 451—453
Прессование при низких давлениях,
метод «резинового мешка» 346,
481
Пресспорошки мочевино-формальде-
гидные 525 и сл.
— — производство, методы 525 и сл.
Алфавитный указатель
G49
Пресспорошки мочевино-формальде-
гидные, приготовление мо-
чевино-формальдегидиого рас-
твора 526
--------смешение раствора с на-
полнителем и «созревание» мас-
сы 527
—-------сушка 528
-----свойства и применение 532 и сл.
-----схема производства 531
— новолачные 430 и сл.
-----и резольные 426—429
-----производство, вальцовый ме-
тод 431—438
—-------шнековый метод 438
— резольные 440 и сл.
— с минеральным наполнителем
442—444
— феиоло-альдегидные 423 и сл.
— — получение, вальцовый метод
425
—-------лаковый метод 423
-------- пропиткой наполнителя
смолообразующими компонентами
425
—-------текучесть 425, 448
--------шнековый метод 426
--------эмульсионный метод 424
Притяжение частиц 71
Пропеиилвинилит 156
Пропилакрилат 341
Пропиточная машина вертикальная
466—472
Пропиточная машина (сушильная)
горизонтальная 485 и сл.
Простые виниловые эфиры, синтез
• 287 и сл.
-------- структура полимеров 292
— поливиниловые эфиры, показатели
292
— эфиры новолака, получение 403
Протеииолит 158
Протеииолоид 158
Протеинопласт 158
Прохоров Ф. Г. 561
Прочность пластмасс см. Пластмассы
— полимеров см. Полимеры
— стеклянных нитей 123
Пульвер-бакелит 389
Равновесное состояние сил притяже-
ния и отталкивания 71 и сл.
Радикалы мономеров при сополиме-
ризации 52 и сл.
— при цепной полимеризации 33
Разветвленность полимеров 57 и сл.
Разрушение твердого тела, упруго-
вязкое 74
Разрушение твеодого тела (хрупкое)
74
Разрыв цепочки, механизм 129
Растворители, влияние на полимери-
зацию 43
— неполярные 139 и сл.
— полярные 138 и сл.
Рафиков С. Р. 65, 568
Рашиг 355
Реакции обрыва цепи 33, 36, 45
— переноса цепей 37
— роста цепи 33, 36, 44
----— карбониевый механизм 44
Реакционные агрегаты для производ-
ства смол 380—384
Реакция Бутлерова, взаимодействие
бромистого изокротила с этилатом
натрия 287
— винилирования для эфиров в жид-
кой и в паровой фазах 288
— Гриньяра 225, 615
— конденсации фенола с гексамети-
лентетрамином 368 и сл.
— переэфиризации для получения
акриловых эфиров высших спир-
тов 320
— поликонденсации — взаимодействие
фенолов с альдегидами 350 и сл.
----фенолов с формальдегидом в
присутствии щелочных катализа-
торов 370
— Фаворского, взаимодействие алли-
лена с этиловым спиртом 287
Ребиндер П. А. 133, 164
Регель В. Р. 220
Регуляторы 42 и сл.
— полимеризации 160
Резинопласты 149
Резит 122, 372 и сл., 420
— образование и пространственное
строение 358 и сл.
— слоистый см. Фенобумослой
Резитовая файера 502, 506 и сл.
Резитолы 352
Резиты 353, 360
— литые 126
Резол 122, 372, 375 и сл.
— свойства 359
Резолы, получение 353
— химическая структура 356—358
Резольная конденсация, аппаратур-
ная схема 376
Резольные прессматериалы 426—429
— пресспорошки, производство 440
---- свойства 441 и сл.
•— — с минеральными наполнителя-
ми 442 и сл.
----— •— свойства 443
650
Алфавитный указатель
Резольные смолы, образование, влия-
ние химического строения компо-
нентов 372 и сл.
----свойства 375, 395
— — скорость отверждения 374
------- твердые 375, 391 и сл.
----термореактивность 376
---- технологический процесс про-
изводства 390 и сл.
— — установка для получения 392
и сл.
----феноло-анилино-формальдегид-
ные 390 и сл.
---эмульсионные, производство 394
Резорциноасболит 157
Резорцинопласт 157
Резорцинопластоклей 157
Резорциностеклослон 157
Релаксации процесс, значение для
низкомолекулярных тел 77
Релаксация напряжения 74
Реньо 256
Роговин 3. А. 15, 591, 597
Ростовский Е. И. 271, 274, 286
Рутовский Б. И. 16, 46, 231, 342, 348
Садыкова .4. К. 492
Связи межчастичиые 127
Себацнновая кислота 587
Седлис В. О. 231
Семенов Н. Н. 33
Силандиолы 616
Силанолы 616—618, 625
Силантриолы 617
Силиасболит 158
Сил пласт 158
Силипласт 158
Силислюдолит 158
Силислюдослой 158
Симонова О. Н. 397
Скорость конверсии мономера 39 и
сл.
— полимеризации 39
СКС 211
Слив смол 384 и сл.
Сложные эфиры новолака 404
— — поливинилового спирта 270
Слойматериалы 461
Слоистые аминопласты, получение
534
— асборезиты 499
— материалы на основе аллиловых
смол, получение 345
— меламиновые пластики на основе
стеклоткани и стекловолокна, зна-
чение 543
— мочевино-формальдегидные пресс-
материалы, получение 533—535
Слоистые пластики, облицовочный
материал 535
— пластические массы на основе фе-
ноло-альдегидных смол 46!
— термореактивные пластики, свой-
ства 347
— фенопласты, получение 461 и сл.
-----различные типы 463; см. так-
же Фенопласты слоистые
-----смолы 461
Слюдяная изоляция 590
Смирнов А. С. 562
Смола феноло-альдегидная 31 и сл.
—। феноло-лпгниновяя на щелочном
лигииие (Ф. Щ, Л.) 416
-----отверждение 415
— феноло-формальдегидпая 122
Смолообразование, скорость 372
Смолы алкилфенольные 407—409
— амино-формальдегидные 27
— анилино-феиольные 395
— анилино-формальдегидные 157
-----схема пдлучения 549
— анормальные 88
— в стадии А см. Резолы
-----В см. Резитолы
-----— С см. Резиты
— высокомолекулярные 27, 85
— глобулярной структуры 26
— для слоистых фенопластов 461
-----методы пропитки напол-
нителя 463
— г.тицеро-малеиновые 586
-----модификация канифолью 586 и
сл.
— глицеро-фталевые 590
-----модифицированные 583 и сл.'
-----применение 576, 583, 590
— ионообменные, см. Иониты
— ионообменные, см. Иониты
— крезоло-баритовые, получение 394
— кремнийорганические, основные
типы 611
— крезоло-формальдегидные 157
— лаковые полиэфирные 590
— литые 372, 396
— линейной структуры 27, 127
— малеиново-канифольные (твердые)
590
— малеиновые 586 и сл.
— маслорастворимые 403 и сл., 406
— меламино-формальдегпдные 157,
519, 538, 545
— мочевино-формальдегидные 157
----- строение 514
•— на основе альбумина 158
— — — гексаметилендиамина и ади-
пиновой кислоты 157
Алфавитный указатель
651
Смолы на основе гексаметилендиа-
мина и себациновой кислоты 157
--------- гексаметилендиизоцианата и
бутандиола 158
---------- и глицерина 158
----------и диаминов 158
------- глицерина и фталевого ан-
гидрида 581—583
-------молочного казеина 158
------- поликапролактама 157
------- продуктов гидролиза диал-
килдихлорсиланов 158
------- моиоалкилтрихлорсиланов с
триалкилмонохлорсиланами 158
---- растительного казеина 158
----— фенолов и альдегидов 350
— —-------и заменителей формаль-
дегида 410
------- этиленгликоля и фталевого
ангидрида 577—-580
----------и терефталевой кислоты
580 и сл.
— низкомолекулярные 26, 85
— новолачные 353
— нормальные 88
— обозначение химического харак-
тера 150
— общая характеристика 26 и сл.
— органополисилоксановые термо-
реактивные 625
— пентаэритритовые 588 и сл.
— полиамидные 601, 608
----получение 565, 591
— полиуретановые 609
— полиэфирные 27
---- получение 565
— • — применение 589 и сл.
— природные 27
— резорцино-формальдегидные 157
— резорциновые 418—420
— сетчатой структуры 28
— синтетические, получаемые мето-
дом цепной полимеризации 159
процессы образования 28 и сл.
условия образования 29
— со сшитыми цепями 27
— сферической структуры 26, 127
— твердеющие, механизм образова-
ния 360
— термопластичные 351 и сл.; см.
также Смолы новолачные
— термореактивные 27, 351 и сл.
— феноло-альдегидные 27, 350
— — маслорастворимые, получение
403 и сл.
---строение 354
----структура, стадии А, В и С
360
Смолы феноло-аиилино-форма.тьдегид-
ные 157
— феноло-лигнииовые 157
— феноло-формальдегидные 157, 361
и сл.
— феноло-фурфу рольные 157
— фталевые модифицированные
583—586
— эмульсионные 375
Смоляные кислоты 404 и сл.
Совенит 549
Совместная полимеризация 46 и сл..
Соколов А. Д. 364, 367, 398, 456
Солонина В. 14, 46
Сополимеризация 46 и сл.
— азеотропическая 48, 54
— аллиловых эфиров с сернистым
ангидридом 14, 46
— бифункциональных соединений
47
--------с тетрафункциональными 47
— бутадиена со стиролом 49
— влияние полярности маиомеров
49
— • дивиниловый тип 46
— 2,5-дихлорстирола со стиролом,
техническое значение 211
— моновинильных соединений с ди-
винильными -17. 49 и сл.
— • олефинов с сернистым ангидридом
48
— пространственные затруднения 49
— стильбена с малеиновым ангидри-
дом 47
— стирола с малеиновым ангидридом
48
— — с п-дивинилбензолом 50
— теория 51 и сл.
Сополимеры акриловых эфиров
334-336
— а.т.тилметакрилата и метилмет-
акрилата 156
— бутилметакрилата с эфирами
акриловой кислоты 156
— винилацетата 285
— винилбутилового эфира с винил-
этиловым и винилизопропнловым
293
— метакриловых эфиров 334—336
— метилакрилата с эфирами мет-
акриловой кислоты 156
— метилметакрилата со стиролом 156
— полихлорвинилидена, свойства 260
и сл. *
— пространственные 50, 192, 211, 335
и сл.
— простых виниловых эфиров 293
---------- применение 287
052
Алфавитный указатель
Сополимеры простых виниловых эфи-
ров с другими виниловыми со-
единениями 293 и сл.
— стирола 210 и сл.
----- с бутадиеном 137
-----с винилкарбазолом 156, 211, 230
-----с дивинильными соединениями
194
-----с дихлорстиролом 211
----- с метилметакрилатом 212
-----с моновиннльными соединения-
ми 194, 211
-----с хлорвинилом, техническое
значение 212
----- с хлорвинилиденом 212
-----с п-хлорстиролом 212
— «сшитые» см. Сополимеры про-
странственные
— хлорвинила 254 и сл.
-----с винилацетатом 155
— — с метилметакрилатом и др.,
промышленное применение 256
— — с хлорвинилиденом 155
-----с эфирами акриловой кислоты
155
— — — — метакриловой кислоты
155
----- применение в лаковой технике
255
-----техническое применение 254
— хлорвинилидена, переработка 262
-----и хлорвинила, пределы физи-
ко-механических свойств 262
----- применение 262
----- свойства 260
— — техническое значение 257 и сл.
Сорбционный метод разделения
ионов 552
Сорокин М. Ф. 539
Спирт поливиниловый 270
Сплавы асфальтов с асфальтитами
158
— на основе асфальтов и высыхаю-
щих масел 158
— пеков с асфальтами 158
Средняя степень поликонденсации 64
-----полимеризации 38 и сл.
Стабилизаторы полихлорвиниловых
композиций 239
Стекла анормальные 88
— нормальные 88
Стекла полиметилметакрилатпьте 329
Стекло органическое 126, 327, 333, 337
• и сл.
- свойства 507
Стеклолит 507
Стеклообразно-аморфное состояние
83 и сл.
Стеклофенолит 507
Стеклянное волокно 223
---- обработка алкилхлорсиланами
(гидрофобизация) 508
----свойства 508
Стеклянные нити, пропитка 510
— пластики, методы изготовления
509 и сл.
— ткани, прессование 511
---- сорта 510
Стирол 161
— гетерополимеризация 212
— полимеризация 194, 201 и сл.
---- блочная 202—204
----водно-эмульсионная 207
-------- непрерывная 205
----в растворителях 206
----термическая 201
— получение 194—200
— реактор для эмульсионной поли-
меризации 208
— скорость полимеризации в эмуль-
сиях 171
— сополимеры 210 и сл.; см также
Сополимеры стирола
— схема непрерывной полимериза-
ции 205
Стиролит 152, 156
Стиропласт(ы) 156, 193
Стиропленка 156, 217
Стиропор 152, 156
Стиропоропласт 152
Стрепихеев А. А. 606
«Сшивка» цепей при сополимериза-
ции 50
Текстолит см. Производство фено-
текстослоя
Текстолит-крошка см. Фенотекстолит
Текстофенолит 464
Текучесть по Рашигу, определение 452
Температура вязкого течения 137
— плавления кристаллических поли-
меров 112 и сл.
----кристаллической фазы 128
----полимеров, зависимость от рас-
стояния между диполями 69
— стеклования 86, 137, 145
----аморфной фазы 128
Температура стеклования аморфной
фазы кристаллических полимеров
112
---- влияние неполярных раствори-
телей 139
--------межмолекулярных взаимо-
действий 99
--------полярных растворителей 138
и сл.
Алфлвитный указатель
Температура стеклования, влияние
тормозящих потенциалов 99
-----зависимость от длины боковых
групп 57 и сл.
—------------молекулы пластифика-
тора 140 и сл.
— —-------молекулярного веса ПО
-----напряжения 105 и сл.
-----линейных полимеров 103, 108
-----низкомолекулярных смол 85 н
сл.
----- определение 108
-----пластификаторов 143
— текучести 86
— хрупкости 108, 143, 145
— — зависимость от различных фак-
торов 109
-----------длины боковых групп 58.
— — полиакрилатов 58
Теория высокоэластической деформа-
ции 90 и сл.
— кинетическая, высокоэластичности
91 и сл.
— поворотно-изомерная линейных по-
лимеров 97 и сл.
— распределения внутренних дефек-
тов в твердом теле 123 и сл.. 132
— сополимеризация 51 п сл.
— усиливающего действия наполни-
телей, выводы 135
—----------на каучукоподобные по-
лимеры 133
— эмульсионной полимеризации 170
Терефталевая кислота 68, 570, 580
н сл.
Терилен 581
Терминология пластмасс 147—152
Термореактивные полимеры, модифи-
цированные кремнийорганически-
ми соединениями 627
— пресскомпозиций, получение 361
— пространственные полимеры, свой-
ства 624 и сл.
— фенольные' смолы, свойства 420
и сл.
Тетрагидрофуран 593 и сл.
Тетрафторвинилит 155
Тетра фторвинипласты 155, 263
Тетрафторэтилен 263
— получение, свойства 264
Течение пластичных тел 76
— полимеров 102
Типизация пластмасс 153
Ткани диагональные 464
— для фенотекстослоя 464 и сл.
— из синтетических волокон, при-
менение 464
— плетение киперное 464
Ткани, плетение тафтяное 464
— пропитка феноло-спиртами 472
— технологический процесс пропит-
ки н сушки 466 и сл.
— хлопчатобумажные, применение
464 и сл.
Точка превращения 86
Триаллиловые эфиры борной кис-
лоты 348
Трикрезилфосфат 136
Трикрезол, способ транспортировки.
379
Три функциональные молекулы 29
Тростянская Е. Б. 556
Угли природные 552
— сульфированные 552
Уолл 96
Упруго-вязкие тела 73
Упруго-вязкое тело, механическая мо-
дель 80
Упруго-пластичные тела 73
Упругость запаздывающая, механиче-
ская модель 81
Уретан 605
Уретанолит 158
Уретанолоид 158
Уретанопласт 158
Ушаков С. И. 10, 16, 46, 271. 273, 28!,
286, 292, 299, 307 и сл., 310, 339.
349, 398, 403, 412, 417. 448
Фаворский А. Е. 14, 287 и сл.
Файнштейн .1. С. 147
Файнштейн Ю. М. 271
Фанера бакелизированная 506
— резитовая 506
Фанерит 502, 506
Фаолит 152, 157, 455
— применение 457 и сл., 460
— производство 456 и сл.
— свойства 459
.и-Фенилендиамин 561
Фенилметилкарбинол 197 и сл.
Фенилполисилоксаны 625
Феноасболит (ы) 151, 157, 450
Феноасбослой 132, 157, 499
— образование 499
— рольно-суспензионный метод по-
лучения 500 и сл.
— свойства 501 и сл.
Феноасботекстолит 452
Феноасботекстослой 499
— свойства 502
Фенобумослой 151, 157, 482 и сл.
— методы изготовления 482
— намоточный, получение 487
— показатели свойств 498
654
Алфавитный указатель
Фенобумослой, получение по рольно-
суспензионному методу 493—496
— прессованный, получение 485
— применение 498
— производство пропиткой бумаги
расплавленной смолой 490 и сл.
---рольным методом 491 и сл.
— свойства 496—498
Фе.чодреволит(ы) 151, 157,430,440,445
• — вальцовый метод производства 431
— (на основе новолачных и резоль-
ных смол) свойства образцов 441
и сл.
— свойства 441
Фенодревослой 157, 505
— получение 503
— применение 503
— свойства 502—505
Феноклей 152
Фенол 377
— относительная реактивность гомо-
логов 373
— плавка горячим фенолом 377 и сл.
----острым паром 377
— способ хранения 379
— хранилище, устройство 377
Фенолит 151, 157
Фенолиты на основе длинноволокни-
стых наполнителей, метод изго-
товления 446 и сл.
Феноло-альдегидные смолы, литые
материалы 422
— — модифицированные для лаков
401 и сл.
---- получение 350
— — прессовочные материалы 422
---- применение 402
Феноло-древесная смола (Ф. Д.) 412
— — получение 413
Феноло-лигнин 414 н сл.
Феноло-лигниновые смолы (Ф. Л.)
413
Феноло-спирты, свойства 472
Феноло-фурфурольные смолы 410
---- получение 411
Фенолы для производства смол 351
и сл., 373, 386, 390 418 и сл.
— дегтей малокалорийных видов
топлива 416 и сл.
Фенольные смолы, термореактивность
420
Феноляты, получение 403
Фенопласт(ы) 14, 121, 150, 154, 157
422, 461
— бумажные, производство 462
— заменители цветных и черных ме-
таллов 350
Фенопласт(ы) неслоистые 423
------ прессматериалы на основе
длинноволокнистого наполнителя
444—452
---- пропитка наполнителя 423 и
сл., 463
----------неоконденситовый метод
425
— слоистые, свойства 461
---- типы 463
Фенопор см. Фенопоропласт
Фенопоропласт 152, 157
Феностеклослой 157
Фенотекстолит 450
Фенотекстослой 151, 157, 463, 498, 507
и сл.
— зависимость свойств от соотноше-
ния и характера компонентов
473—475
— изготовление плит 475—479
— получение пропиткой стеклянной
ткани растворами или диспер-
сиями резольных смол 510
— прессование фигурных деталей
479—481
— применение в машиностроении
481 и сл.
— производство по лаковому методу
466
----по эмульсионному методу 466
— свойства 464 и сл., 511
Феноцеллолиты 157, 447—450
— применение 448
— производство 447 и сл.
Фиброин шелка 117
Флори 305
Фокс 120
Формалин 376 и сл., 386, 526
Формальвиннлит 156
Формальвинипленка 156
Формальдегид 350, 352 и сл., 386
— заменители, не обладающие явной
альдегидной функцией 412
Фрейдберг Е. Н. 412
Френкель Я- И. 96 и сл.
Фталевые смолы модифицированные
583—586
---- производство 577—586
Функциональность молекул при об-
разовании смол 29
— молекулярная 574
— удельная 574 и сл.
Фуран 593
Фурфурол 593
«Хавег», свойства 455 и сл.
Химические особенности кремний ор-
ганических соединений 613
Алфавитный указатель
655
Хлорвинил см. Хлористый винил
Хлорвиниласт 152, 155, 248 и сл., 259
Хторвинилиденоласт-С 155
Хлорвинилиденолит 155
Хлорвинилиденолит-С 155
Хлорвинилиденопласт(ы) 155, 256
Хлорвинидит 155
Хлорвинилит-С 155
Хлорвинилоид см. Винипласт
Хлорвинипласты 155, 231
Хлорвинилпленка 250
Хлористый винил 231 и сл.
— — башенный метод эмульсионной
полимеризации 236
-----непрерывная эмульсионная по-
лимеризация 237
— — полимеризация 234 и сл.
----- — в растворителях 234
----- получение 232 и сл.
-----применение 235 и сл.
----- свойства 231
-----фотополимеризация 231
----- фракционирование его поли-
мера 231
Хлористый винилиден 256 и сл.
----- полимеризация 257
----- получение 257
— — применение сополимеров 262 и
сл.
----- свойства 257
-----сополимеры с производными
этилена 257
------- переработка 262 и сл.
Хлорстиролы, получение 225
— свойства 225
Хомиковский П. М. 174
Хризотиловый асбест 500
Хрупкие тела, определение 73
Хулче 360
Ивет М. С. 552
Целласт 152
Целлолит 151
Целлопласт 151, 158
Целлулоид 12, 126, 152
Целлюлоза 117, 128, 154
Циангидрин, получение 318
Циклогексанол 592 и сл.
Циклогексанон 592 и сл.
Червячный пресс (шиекпресс) для
непрерывной пластикации пресс-
порошков 439
Чил кина Е. М. 167
Шлаковое волокно 223
Шмидт Я- Л. 606
111 остаковский М. Ф. 45, 288, 293
Штаудингер 33, 92
Щелочная конденсация, соотношение
компонентов 371
— — стадии процесса 375
Щелочной лигнин 416
Эбонит 12
Эдельштейн И. П. 425
Эластики, общая характеристика 19,
148 и сл.
Эластопласты 149
Элемент Максвелла . 80
— Фойгта 81
Эльтеков 271
Эмульгаторы 164 и сл.
Эмульсионная полимеризация 163 и
сл., 166, 168, 170, 202, 236—238.
280 и сл., 329, 341
----с окислительно-восстановитель-
ным циклом 166
---- теория 170
Эмульсионное диспергирование 172
Энергия взаимодействия частиц 71
и сл.
— молекулярного притяжения раз-
личных групп в полимерах 117
— упругости каучука 134
— — полимеров 134
Энтропия цепи макромолекулы 94
Эспатит-1 555 и сл.
Этанальвиннлит 156
Этенолоид 156
Этенолопласт 156
Этилбензол, получение 196
Этилвиниловый эфир, получение из
диэтилацеталя 289
Этилена полимеризация 40 и сл., 175
и сл.
Этилеиолит 155
Этиленолоид 155
Этиленопласты 155, 175
Этиленхлоргидрнновый синтез эфи-
ров акриловой кислоты 317 и сл.
Этиловый эфир карбаминовой кис-
лоты см. Уретан
Этилцеллолит 158
Этилцеллолоид 158
Этилцеллопленка 158
Этилцеллюлоза 158
Эфиропласты 565, 576
Эфир поливинилметиловый, промыш-
ленное значение 293
Эфиры акриловые, производство не-
прерывным методом 318
---- свойства 321
— аллиловые, применение 342
656 Алфавитный указатель
Эфиры ди- и тривиниловыс 289
— метакриловой кислоты 322
— получение 325 и сл.
— свойства 326
— моновиниловые 289
— новолака с смоляными кислотами
канифоли, промышленное значе-
ние 404—406
— производных акриловой кислоты,
методы получения 317—319
--- метакриловой кислоты, методы
получения 317
Эфиры простые виниловые 289
-----механизм полимеризации 290
— — поливинилового спирта 270
---------- получение 287
— сложные поливинилового спирта
270
— тривинилглицериновые 289
Эффект Иоффе 123
Юрженко А. П. 173
Янковский К- А. 561
Редактор 3. Я- Хавин
Техн, редакторы: £. В. Климина, Е. Я. Эрлих
Корректор К. А. Мухина
Сдано в набор 9/П1 1154 г.
Формат бумаги 60/92,
Объем 41 физ. л.
Объем 45,63 уч.-лзд. л.
Тираж Е000 экз.
Подписано к печати 15,-Х Г.,'54 г. 18
М17460.
,, 679-Б6
Индекс —-----— 36—Б—3. -Л
Бар. 24
Цена 24 руб. 30 к.
Зак. 1269.
ЛЕНГОСХИМИЗДАТ, Ленинград, Невский пр., 28.
Министерство культуры СССР. Главное управление полиграфической промышленности.
4-я тип. им. Евг. Соколовой. Ленинград, Измайловский пр., 29.
Отпечатано с готового набора во 2-й типографии Водтраненздата
, ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
4 Лэ Строка Напечатано Должно быть
; ?J8 2 сверху повторных поворотных
149 6 сверху 2,5 25
114 В подписи к рис.98 атома атомы
225 18 сверху переходит переводят
235 ;12 сверху изобутилсульфонафталино- бу ти л нафтал иисул ьфо -
вой кислоты кислоты
240 6 сверху полихлор- хлор-
«3 19 снизу бутан-диоля бутандиола
294 12 сверху (25 г) (25 ч.)
ш 8 сверху этил-изопропилвинило- этил- и изопропил ВИ-
выми НИЛОВЫМИ
303 4 сверху шнекпресса с шнекпресса цилиндра с
306 15 снизу (нижняя
формула) + С3Н7ОН + С3Н7СНО
310 22 снизу реактор и для реактор для
358 3—6 (формул) Все метиленовые группь горизонтального ряда
должны быть соединены с бензольным ядром в
орто-положении к гидроксильной группе
375 2 снизу свободного резола свободного фенола
382 17—18 сверху конденсатора конденсата
405 17 сверху хинонметидную хинометидную
450 16 сверху (20-25 кг/сл2) (20—25 кг-сл/сл2)
502 13 снизу (Фенодревослой ДСП, (Фенодревослой, ДСП, |
515 4 сверху диметилмочевииу диметилолмочевину j
N—СО— HN-СО- । 1 1
519 формула сн2 сн2 >
HN -СО- HN—CO— j
. 519 1 снизу дицианидиамида дициандиамида
.604 1 сверху гексаметилендиамида гексаметилендиамииа ]
- о о О R
613 формула 3 1 1 1 1
2-й ряд ... — O-Si-O— Si—о-... ...-O-Si-O-Si-0-... 1 1
R О 1 R О 1
. Зак« С' » М 1269. Э. И. Барг
л
-