/






























































































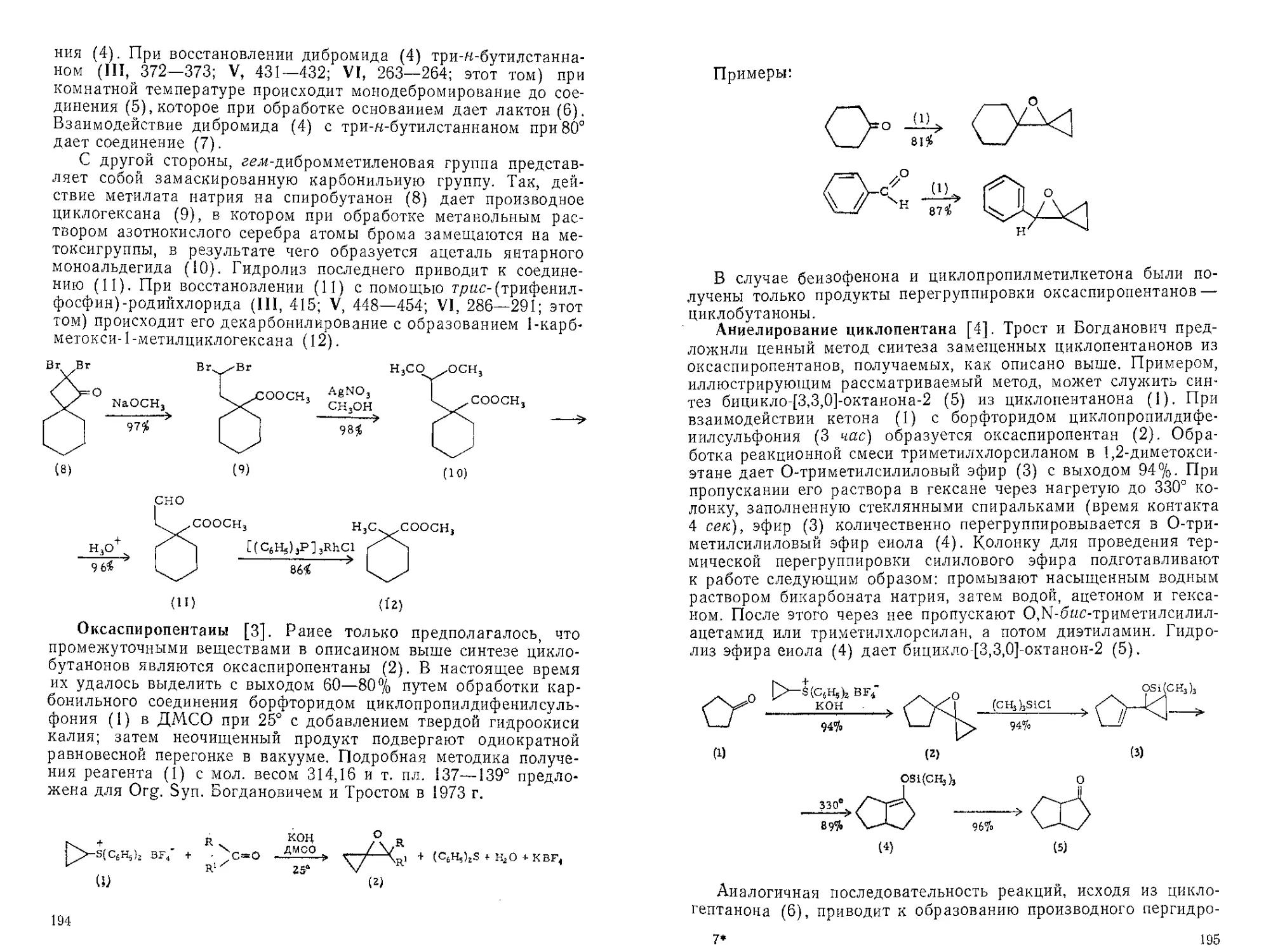








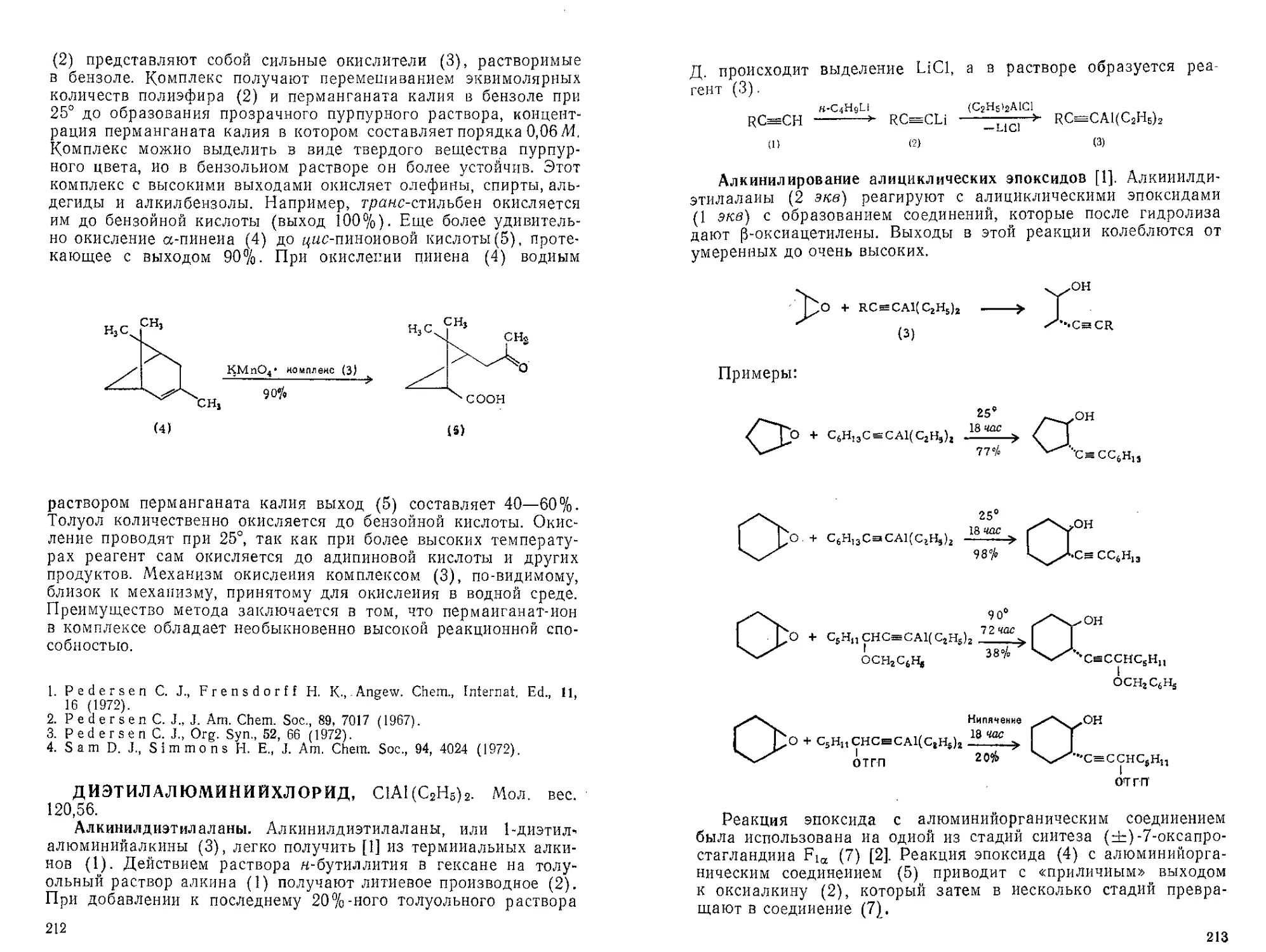











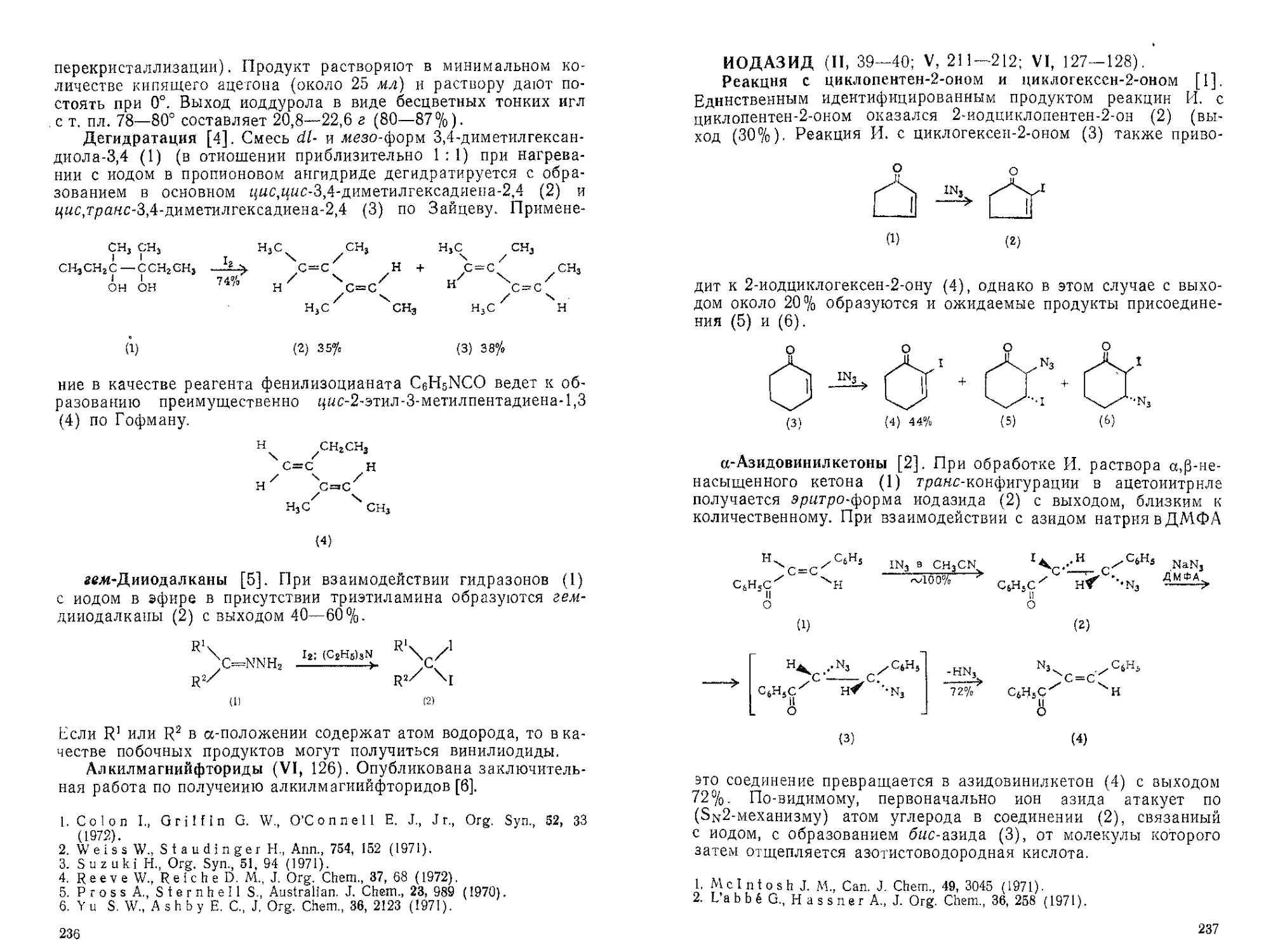














































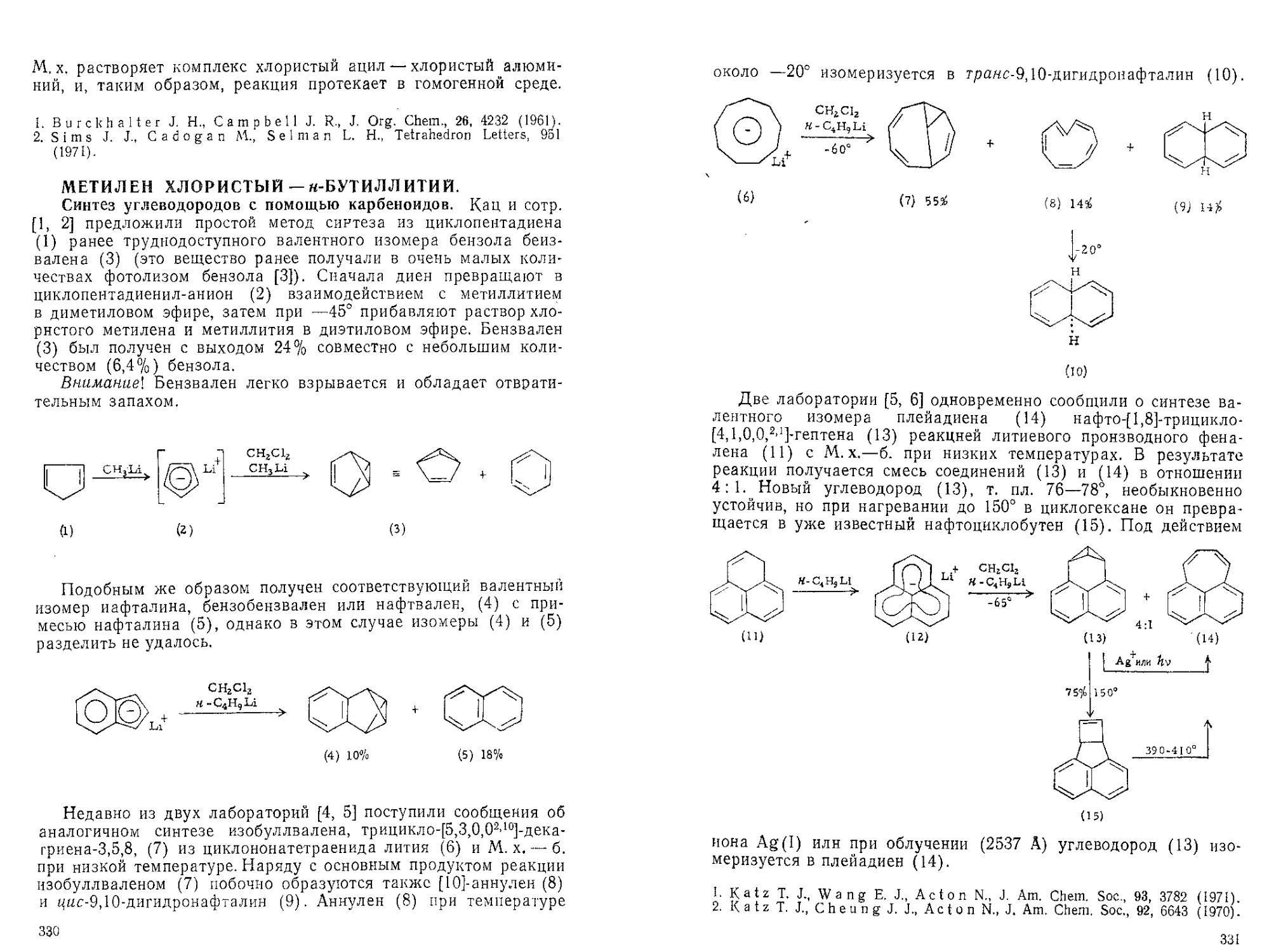


























































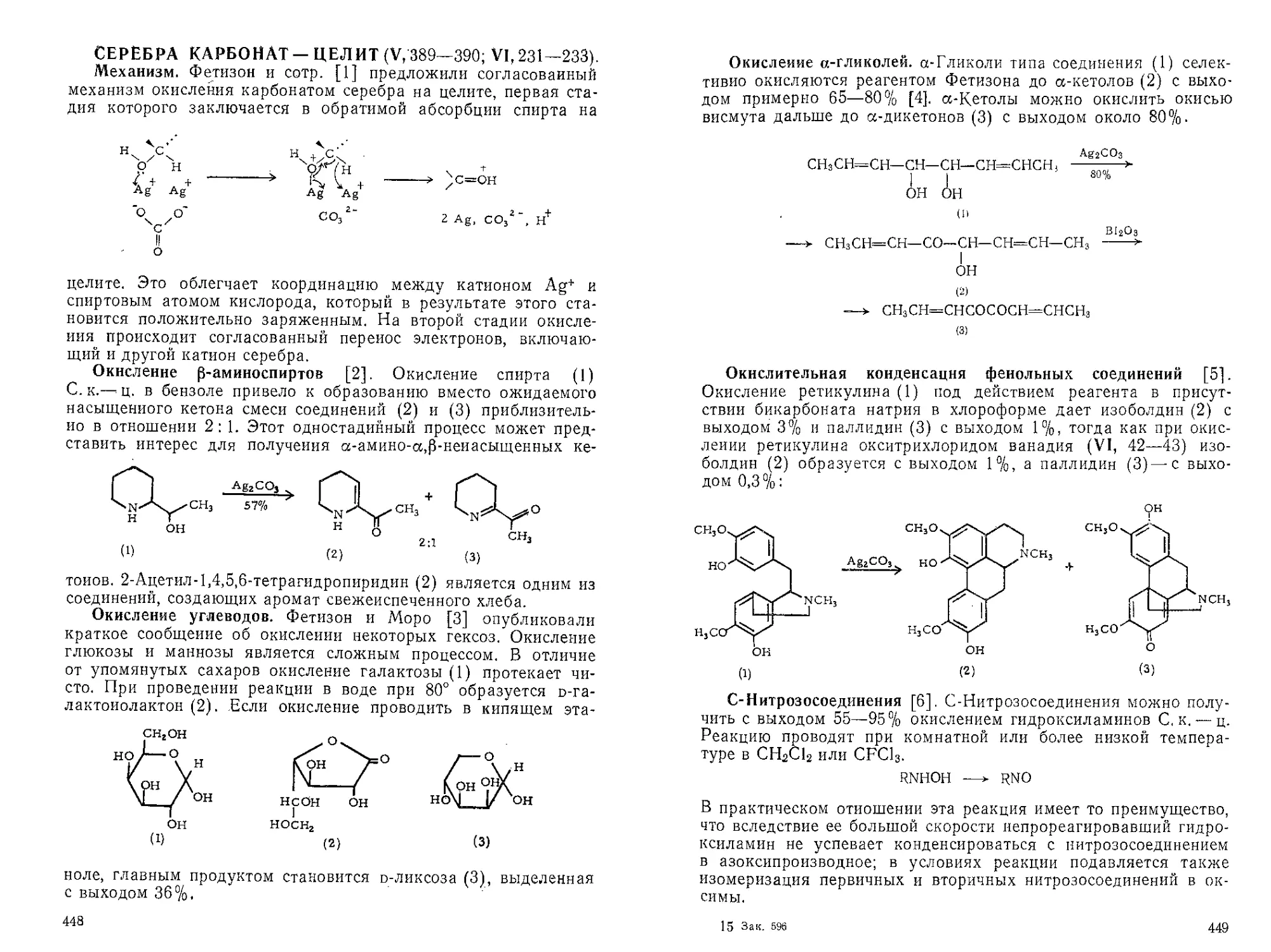













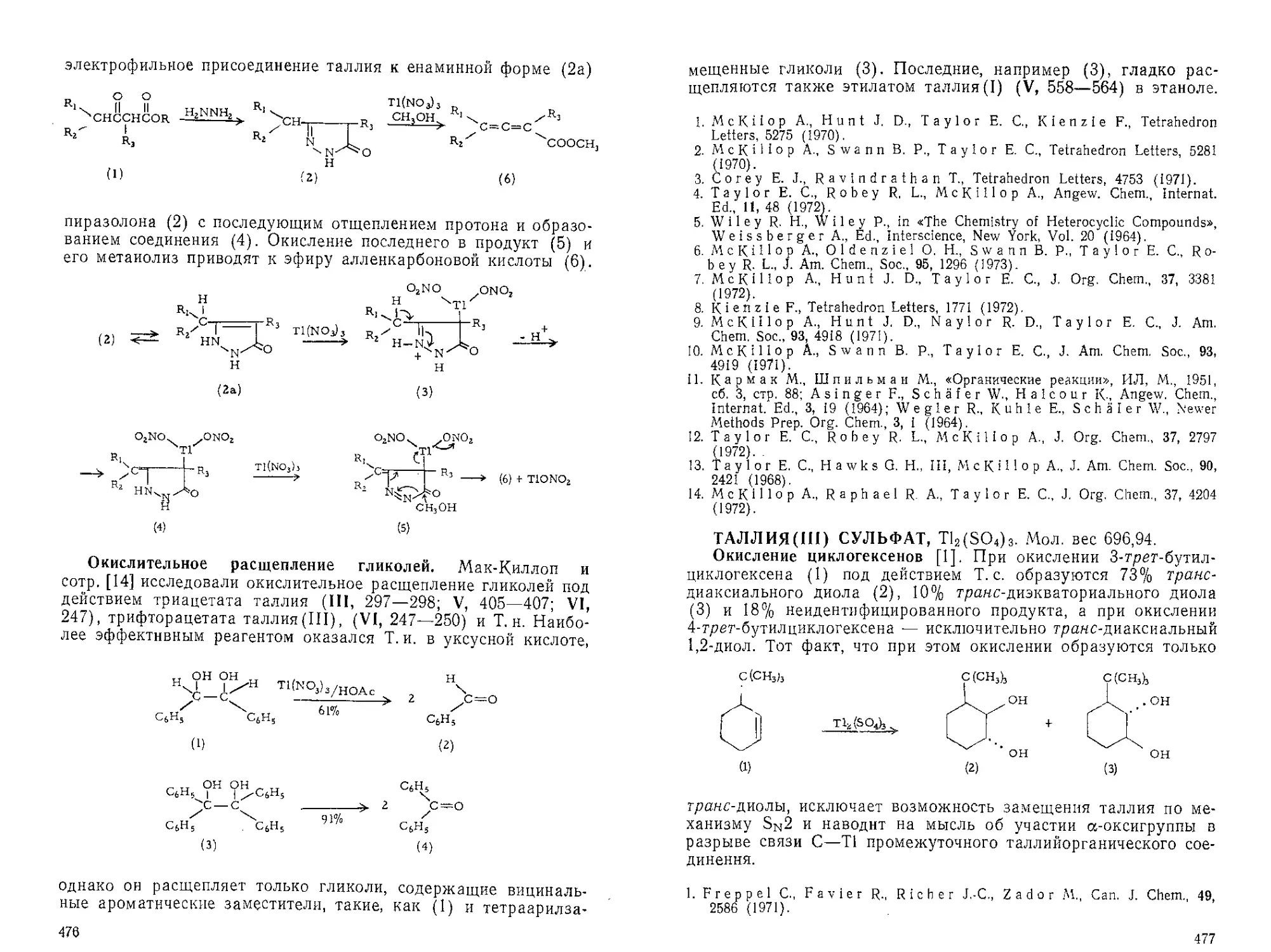





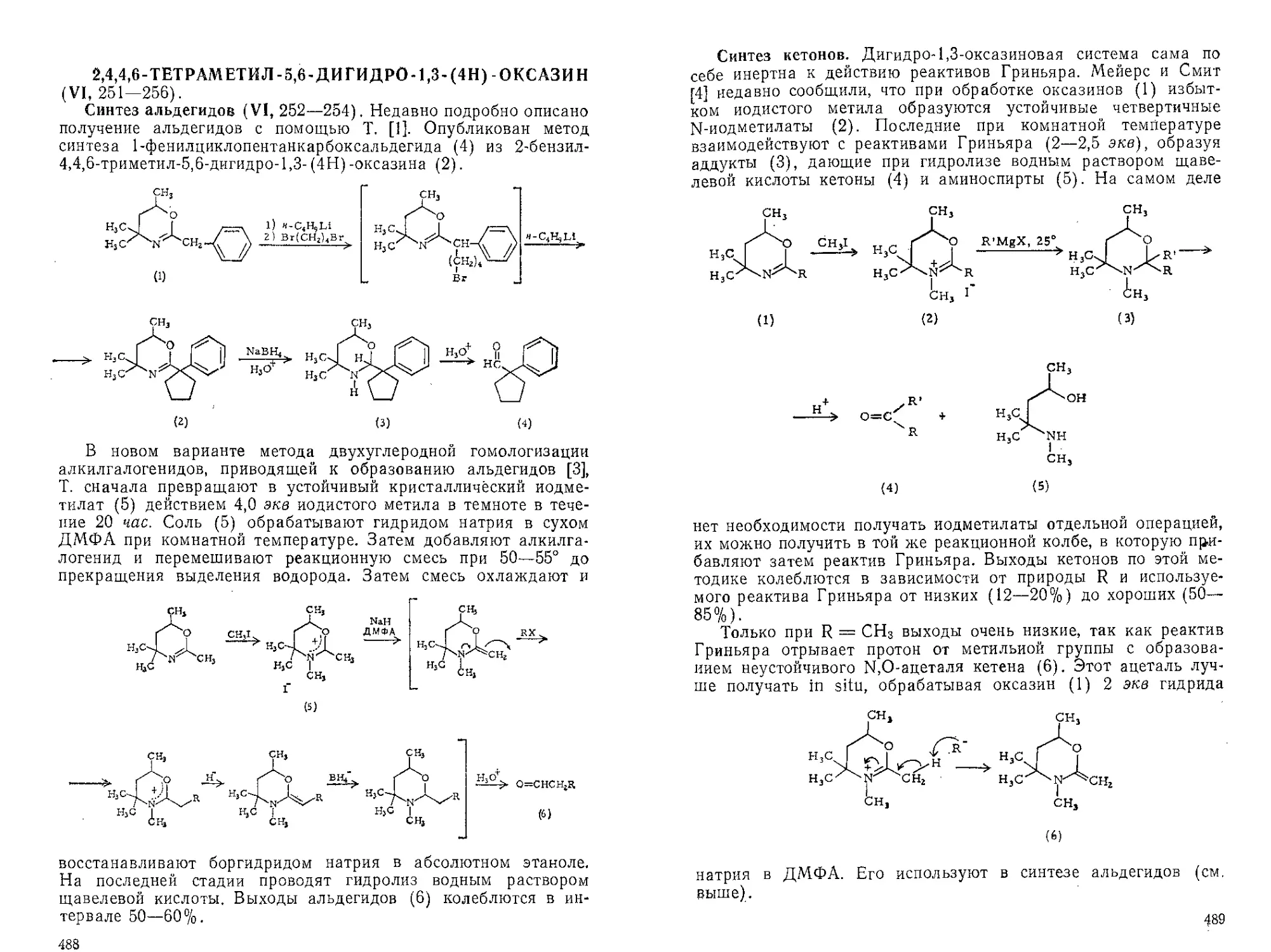

























































































































Текст
Reagents for
Organic Synthesis
VOLUME 4
MARY FIESER
Research Fellow in Chemistry
Harvard University
LOUIS F. FIESER
Sheldon Emery Professor of Organic Chemistry, Emeritus
Harvard University
A WILEY —INTERSCIENCE PUBLICATION
JOHN WILEY & SONS
NEW YORK • LONDON • SYDNEY • TORONTO
1974
Л. ФИЗЕР, М. ФИЗЕР
Реагенты
для органического
синтеза
ТОМ VII
ПЕРЕВОД С АНГЛИЙСКОГО
канд. хим. наук Т. П. ТОЛСТОЙ
ПОД РЕДАКЦИЕЙ
академика И. Л. КНУНЯНЦА
МОСКВА 1978
ИЗДАТЕЛЬСТВО «М И Р»
УДК 661.7/54-41
Книга является очередным томом широко известного совет-
скому читателю справочного пособия по органическому синтезу
(Физер Л., Физер И., Реагенты для органического синтеза, т. I—
VI, пер. с англ., М., 1970—1971, 1970). Данный том содержит
350 ссылок на реагенты, описанные в предыдущих томах;
297 реагентов рассматриваются впервые. Он существенно допол-
няет уже изданные тома в плане как расширения спектра реа-
гентов, так и раскрытия новых свойств уже известных, реаген-
тов.
Книга предназначена для химиков-органиков, а также для
физико-химиков, биохимиков, фармакологов, химиков-технологов.
Редакция литературы по химии
Copyright © 1974, by John Wiley & Sons, Inc. All
rights reserved. Authorized translation from English
20504-107 jn7_77 language edition published by John Wiley & Sons, Inc.
041 (01)-77 © Перевод на русский язык, «Мир», 1978
ПРЕДИСЛОВИЕ АВТОРОВ
Настоящий том охватывает литературу по реагентам, опу-
бликованную в основном в 1970—1972 гг„ а также включает
ссылки, относящиеся к первым месяцам 1973 г. В него входят
ссылки иа 297 реагентов, рассмотренных нами впервые, и на
350 реагентов, обсуждавшихся ранее. Мы поражены тем, как
много новых реагентов введено в органический синтез. Инте-
ресно и то, что и старые реагенты, например хлористый алюми-
ний н перманганат калня, нашли новое применение.
Мы благодарны нашим коллегам, приславшим нам допол-
нительный материал или предложившим интересные рефераты.
Мы признательны за помощь проф. Фуксу, проф. Секристу,
д-ру У анол а, д-ру Уингарду, Денхизеру и Уолленбергу, согла-
сившимся прочитать корректуру.
Госпожа Лнтл и госпожа Киркбай печатали рукопись и впи-
сывали формулы.
Д-р Брахвитц и д-р Вейзелла оказали большую помощь при
чтении корректуры. Ксерокопирование было выполнено Белло-
том.
Мы благодарим Исследовательскую корпорацию за постоян-
ную финансовую поддержку.
Кембридж, Массачусетс, М. Физер,
3 июня 1973 г. Л. Физер
Мы весьма сожалеем о задержке публикации этой книги.
Она вызвана различными причинами. Одна из них не зависела
от издателей, так как рукопись печаталась в Англии в период
энергетического кризиса.
ВВЕДЕНИЕ
Указатели. Для большего удобства книга снабжена не толь-
ко предметным указателем, но также н указателем типов реак-
ций или типов соединений, например: ацетилирование, бромиро-
вание, декарбоксилирование, циклоприсоединение, или: ацето-
ниды, дегидробензола предшественники, карбена предшествен-
ники. Внутри каждой такой рубрики перечислены в алфавит-
ном порядке все реагенты, фигурирующие в цитируемых методи-
ках или имеющие отношение к приведенным в указателе типам
соединений, независимо от того, являются ли они реагентами,
катализаторами, растворителями, улавливающими агентами
и т. д. Часто реагент можно отнести соответственно к двум или
более типам. Если реагент нельзя причислить к какому-либо
типу, мы предпочитаем вовсе отказаться от его классификации,
а не приписывать ему искусственно придуманное назначение.
Что касается большой группы реагентов, известных как окисли-
тели, но использующихся также в качестве восстановителей, то,
по-видимому, не может быть и речи ни о какой попытке приве-
сти в указателе типов реакций какие-нибудь подробности отно-
сительно этих общих реакций.
ОСНОВНЫЕ СОКРАЩЕНИЯ
Краткая форма названий журналов
Accounts of Chemical Research
Journal of the American Chemical Society
Analytical Letters
Angewandte Chemie
Angewandte Chemie, International Edition in English
Annalen der Chemie
Annales de chemie (Paris)
Australian Journal of Chemistry
Chemische Berichte (ранее Berichte der deutschen chemischen
Gesellschaft)
Bulletin de la societe chimique de France
Canadian Journal of Chemistry
Carbohydrate Research
Chemical Communications
Chemical and Pharmaceutical Bulletin Japan
Acta Chemica Scandinavica
Chemistry and Industry
Chemical Reviews
Collection of Czechoslovak Chemical Communications
Comptes rendus hebdomadaires des seances de I’academie des
sciences
Gazzetta Chimica Italiana
Helvetica Chimica Acta
Inorganic Synthesis
Journal of Chemical Education
Journal of the Chemical Society (London)
J.C.S. Chemical Communications
Journal of Heterocyclic Chemistry
Journal of Medicinal Chemistry
Journal of Organic Chemistry
Journal of Organometallic Chemistry
Journal fur praktische Chemie
Monatschefte fur Chemie
Organic Syntheses
Organic Syntheses, Collective Volume
Proceedings of the Chemical Society
Records of Chemical Progress
Receuil des travaux chimique des Pays-Bas (The Netherlands)
Сокращения названий реагентов
Ac — ацетил
AcOH BuOH БОК Bz Cathyl Cb ДАБЦО ДДХ дцк Диглим Димсилнатрий ДМА дмсо ДМФА ДМЭ Тлим (моноглим) ГМ.ТФК (гексаметапол)- ДНФ ДНФГ ЕЮН МеОН ммк — уксусная кислота - бутанол — бутилоксикарбонил — бензоил - карбэтокси - карбобензокси - 1,4-диазабицикло-[2,2,2]-октан - 2>3-дихлор-5,6-дициан-1,4-бензохинон - дициклогексилкарбодиимид -диметиловый эфир диэтиленгликоля - метилсульфииилметилиднатрий - диметилацетамид - диметилсульфоксид - диметилформамид - диметоксиэтан - 1,2-диметоксиэтан - гексаметилтриамид фосфорной кис- лоты - 2,4-динитрофторбензол - 2,4-динитрофенилгидразии этанол - метанол — магния метнлкарбонат
Ms NBA NBC Py ПФК ПФЭ ТГФ ТГП ТМЭДА Триглим Тритил Ts TsCl TsOH — мезил, CH3SO2— — N-бромацетамид — N-бромсукнинимид ~ пиридин — полифосфорная кислота — полифосфорной кислоты эфиры — тетрагидрофуран — тетрагидропиранильная группировка — Ы,Ы,^М-тетраметилэтилендиамин — диметиловый эфир триэтиленгликоля -(С6Н5)3С- — ТОЗИЛ, W-CH3C6H4SO2— — тозилхлорид — га-толуолсульфокислота, h-CH3C6H4SO3H
ТТФА Ph PhTh ГЦА — таллия(Ш) трифторацетат — фенил — фталоил — гексанитратоцеррат(ТУ) аммония (це- рий, аммиакат нитрата)
CPU
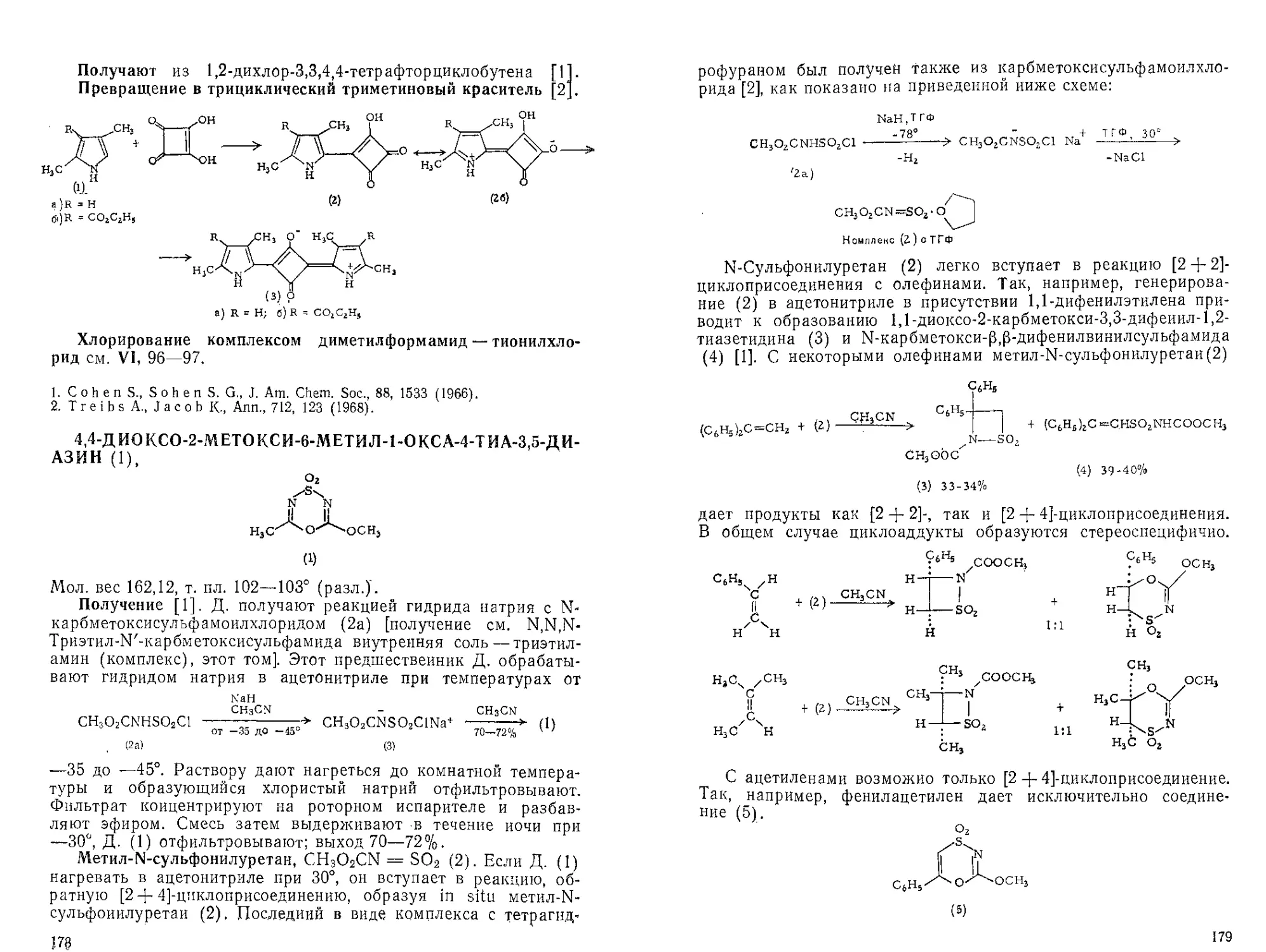
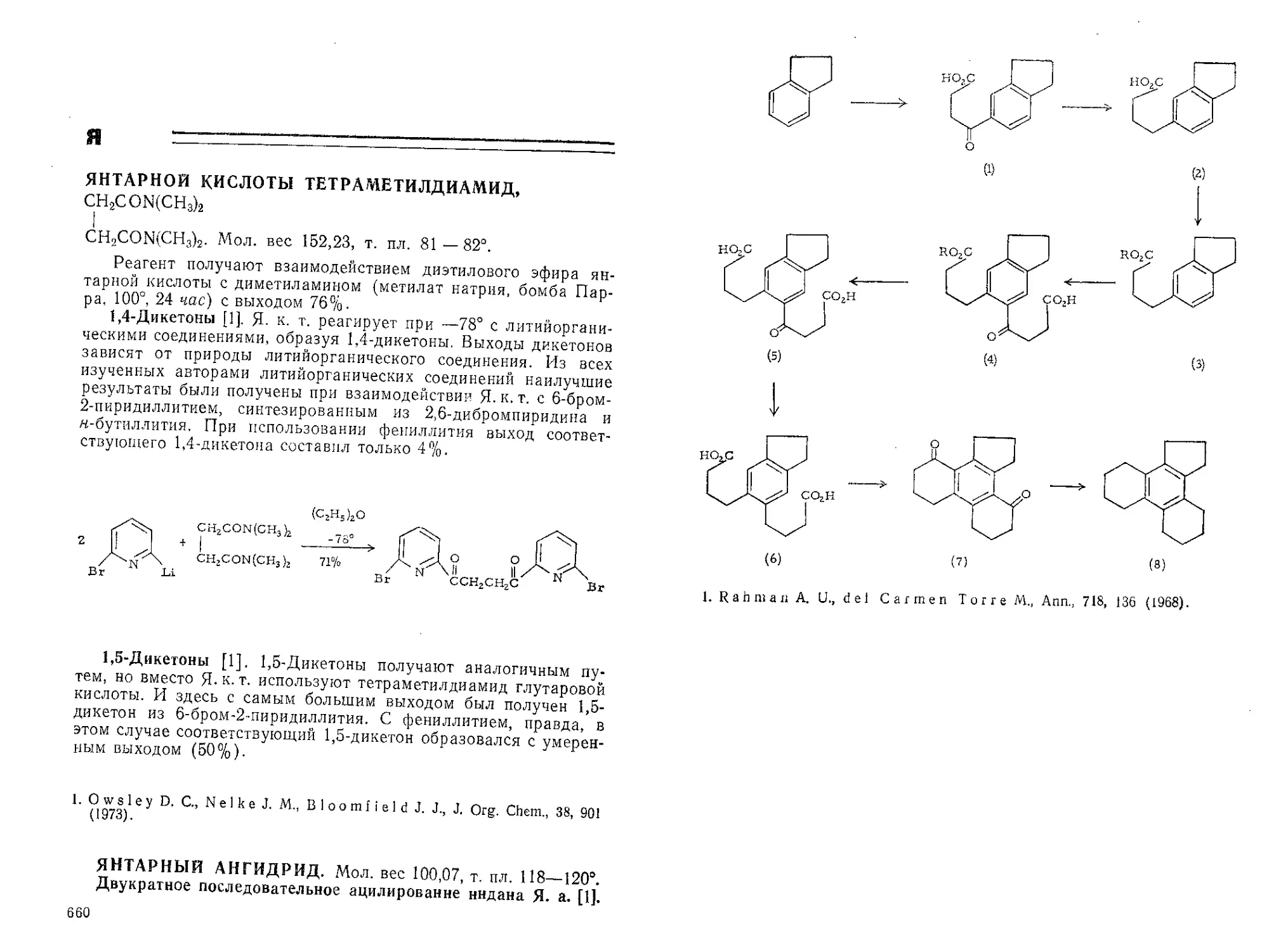
АЗИРИДИН, | /NH. Мол. вес 43,07.
сн/
Получение см. [1].
Реакция с хлороформом [2]:
СНС13, CH3ONa
Бензол
ОСН3
I
N-C-H
I
ОСН3
1. Bestian Н„ Methoden zur Herstellung und
1,3-Alkyleniminen, Houben Weyl, Methoden der
Xl/2.
2. Funke W„ Ann., 725, 15 (1969).
Umwandlung von 1,2- und
Organischen Chemie, Band
АЗОДИКАРБОНОВОЙ КИСЛОТЫ ДИЭТИЛОВЫЙ ЭФИР
(I, 13—15; V, 9—11).
Карбодиимиды [1]. А. к. д. э. (1) реагирует с М,М'-дизаме-
щенными тиомочевинами (2) в ТГФ при комнатной температуре
с образованием (в течение ночи) аддукта состава 1:1 (3) [№,Й2-
дизамещенная 5-(Ы3,Ы4-бцс-карбэтокси)-гидразиноизотиомоче-
вина], обработка которого трифенилфосфииом при комнатной
температуре приводит к соответствующему карбодиимиду (4).
Соединение (3) можно также превратить в карбодиимид (4) ки-
пячением в бензоле или толуоле в течение 8 час. Выходы карбо-
0 0 S
II II II
С2Н5ОС—N=N—СОС2Н5 + RNH— С—NHR' —>
(1) (2)
0 0 О
II II (СвН5)3Р j
С2Н5ОС—N—NH—СОС2Н5 ,------------->- RN=C=NR' + (C2H5OC—NH)2 +
I ) (4) (5)
—> S - + (C6H5)3P=S
I Кипячение ygj
I в толуоле
RN=C—NHR' 1-------(4) + (5) + S
(3)
диимидов в большинстве случаев составляют 80% [!] Пре-
имуществом приведенного метода является также то, что
образующийся бцс-Ы,Ы/-карбэтоксигидразин (5) действием
хлорноватистой кислоты можно снова окислить до А. к. д. э. (1)
с выходом 81—83% [2].
Дегидроабиетиновая кислота. Метиловый эфир дегидроабне-
тиновой кислоты (2) легко получить с 85%-ным выходом де-
гидрированием метилового эфира левопимаровой кислоты (1)
с помощью А. к й. э. в кипящем бензоле [3]:
I. Mltsunobu О., Kato К., К a k е s е F., Tetrahedron Letters, 2473 (1969);
Mitsunobu О,, Kato К., Tom ar i M., Tetrahedron, 26, 5731 (1970).
2. R a b j о h n N., Org. Syn., Coll. Vol., 3, 375 (1955).
3. Mehta G., Kapoor S. K-. Org. Prep. Proc. Int„ 4, 257 (1972).
АЗОТА ЧЕТЫРЕХОКИСЬ (I, 18—24; V, 11 — 12; VI, 5).
Нитрование алкилфенолов [1]. При действии А. ч. на п-кре-
зол (1) в ацетонитриле (25—30°) образуется 2,6-динитро-п-кре-
зол с выходом 66%:
он ОН
(1) (2)
Алкилнитраты [2]. Первичные амины дезаминируются под
действием А. ч. с образованием алкилнитратов. Реакция сильно
зависит от природы растворителя. Лучшими растворителями яв-
ляются ТГФ и другие простые эфиры, способные, как всякие
основания Льюиса, давать комплексы с N2O4. Дезаминирование
происходит с почти полным сохранением конфигурации, что сви-
детельствует об ионном механизме реакции.*
ТГФ, —60°
RNH2 4- NaOj ------> rono2 + N2 + ДО
1. Tao E. V. P„ Christie C. F., Jr., Org. Prep. Proc. Int., 4, 300 (1972).
2. Wudl F„ Lee T. В. K-, J. Am. Chem, Soc., 93, 271 (1971).
АЗОТНАЯ КИСЛОТА (1,28—31; VI, 6—7).
Нитроалканы [1]. Наиболее удобным методом получения
ацилнитратов в лабораторных условиях является взаимодей-
* Образующийся промежуточный карбокатион, по-видимому, несимметрич-
но сольватирован растворителем. — Прим, перев.
12
ствие ангидрида карбоновой кислоты (десятикратный избыток)
с 90%-ной А. к. при 20° (а). Образующуюся при этом карбоно-
вую кислоту можно снова превратить в ангидрид. Удобную мо-
дификацию метода представляет взаимодействие А. к. со сме-
сями карбоновых кислот и уксусного ангидрида (б).
a) (RCO)2O + HNO3 —> RCO2NO2 + RCOOH
б) RCOOH + 2 (СН3СО)2О4-2HNO3 -?=* RCO2NO2+CH3CO2NO3+3CH3COOH
Нагревание полученных ацилнитратов при 270—300° в инерт-
ном растворителе (ацетонитриле или нитроалкане) дает нитро-
270-300°
RCO2NO2 ------► RNO24-CO2
алкаиы с удовлетворительными выходами. Например, из ва-
лероилнитрата был получен 1-нитробутан (56,5%), бутанол-1
(20,0%) и н-бутиловый эфир валериановой кислоты (20,0%).
1. Bachman G. В., Bierman Т. Р., J. Org. Chem., 35, 4229 (1970).
АЛЮМИНИЙ (V, 15—16).
Дегалогенирование [1]. Раствор 1 г ^ис-3,4-дибромгексахлор-
1,2-диметиленциклобутана (1) в 50 мл абсолютного эфира кипя-
тят без доступа влаги в течение 3 час с 2 а алюминиевой фольги.
Полученный раствор фильтруют, промывают водой, высушивают
и удаляют растворитель. Остаток — бесцветные иглы с т. пл.
145—146° (из ацетона)—представляет собой соединение (2):
Аналогично
(з)
(4)
13
Восстановление нитрогрупп [2].
1. Roedig A., DetzerN., Bonse G., Ann., 752, 60 (1971).
2. Christmann O„ Ann., 716, 147 (1968).
АЛЮМИНИЯ БРОМИД (I, 37 — 38; V, 16— 18; VI, 9).
Перегруппировка а-бромэтилдиэтилборана [1]. ct-Бромэтил-
диэтилборан (1) под действием А. б. в сероуглероде при 25°
почти мгновенно изомеризуется в бромидвтор-бутилэтнлбора(2):
сгн5 сгн5 СгН5
знэсн-в-с2н5—А1_в.гз > сн3сн-в-сгн5 > СН3СН-В —сгн5
с 95%
(1) ^А1Вгэ (2)
(а)
Почти такой же эффективностью обладают и некоторые другие
кислоты Льюиса (А1С13, ZnCl2, AgBF4); HgCl2, SnCI4, SbCl3 и
TiCl4 менее эффективны.
1. Brown H. C., Yamamoto Y., 3. C. S. Chem, Comm., 1972, 71.
АЛЮМИНИЯ БРОМИД, in situ.
А. б. можно получить in situ из алюминиевой фольги и брома
(действие иода на алюминиевую фольгу приводит к иодиду алю-
миния).
Обмен галогена в голове моста [1]. Атом галогена, располо-
женный в голове моста, обычно обменивается с трудом. Но под
действием А. б., полученного in situ, эта реакция проходит легко
и быстро. Для замещения галогенов на иод в качестве раство-
рителя используют СНз! или СН212, для замещения на бром —
СН2Вг2 или СНВгз, а на хлор — СНС13 или СС14. Выходы ко-
леблются в пределах 50—90%. Метод использовался для об-
мена атомов галогенов в соединениях (1), (2) и (3).
(1J
(3)
14
I McKinley J. W, Pincock R E.. Scott W B., J. Am, Chem. Soc.,
95, 2030 (1973).
АЛЮМИНИЯ ОКИСЬ (I, 39—41; V, 19; VI, 10—11).
Дегидратация сульфоксидов. При нагревании 2-окиси 1,3-ди-
гидробензо-[с]-тиофена с нейтральной А. о. первой степени ак-
тивности (Woelm) при 120—130° и давлении 25 мм рт. ст. в ап-
парате для возгонки на охлаждаемом «пальце» конденсируется
почти чистый бенз-[с]-тиофен (2), образующийся с выходом 94%.
Нафто-[1,2-с]-тиофен (4) был получен из 2-окиси 1,3-дигидро-
Тропон [2]. При обработке борфторида тропилия (III, 470)
водным раствором азнда натрия образуется азид тропилия (1),
взаимодействие которого с А. о. (Fisher А-540) при интенсивном
перемешивании в течение ночи приводит к тропону-2 с выходом
52%, считая на борфторид тропилия:
' (2)
1. Cava М. Р., Pollack N. М., Marner О. A., Mitchell М. J., J. Org.
Chem. 36, 3932 (1971).
2 McCulIagh L. N„ Wullman D. W., Synthesis, 1972, 422.
АЛЮМИНИЯ ХЛОРИД (I, 41—54; V, 21—24; VI, 11 — 12).
Катализатор реакции Дильса — Альдера (I, 51—52; V, 21 —
22; VI, 12) [1]. Пентадиен-1,3 и 3-бром-4-метилпентен-3-он-2
(бронированная окись мезитила) в присутствии А. х. в качестве
катализатора образуют аддукт Дильса — Альдера (1) с выхо-
дом 87% (ГЖХ). Этот аддукт превращали в р-дамасценон (4)
в три стадии: сначала дегидробромировали его до соединения
(2), которое конденсировали затем с уксусным альдегидом
15
в присутствии М-броммагнезил-Ы-метиланилина (2) в альдоль
(3), а последний уже дегидратировали.
нс
I
НС.
\:н
НзСч ZCH;.
87%
А1С13
40°
ДМФА, 120°
85%
I
сн3
(г) (3} (4)
Из аддукта (5), полученного конденсацией пентадиена-1,3
с окисью мезитила, аналогичным путем был синтезирован б-да-
маскон (6).
Присоединение (1, 48). Реакцией 3-хлорпропионилхлорида
с этиленом в хлористом метилене в присутствии безводного А. х.
[3] можно получить 1,5-дихлорпентанон-З. Продукт реакции пред-
ставляет собой темное масло; выход 93—96 % •
А1С13
С1СН2СН2СОС1 + С2Н4 ----> (С1СН2СН2)2СО
Присоединение хлор ангидридов кислот к олефинам и цикли-
зация по Фриделю — Крафтсу [4]. С целью улучшения сущест-
вующей методики вместо сероуглерода в качестве растворителя
использовали хлористый метилен. Для контроля за ходом реак-
ции время от времени отбирают пробы объемом 2—3 мл, обра-
батывают их холодной водой, органическую фазу отделяют, вы-
сушивают и удаляют растворитель. К концу реакции в ИК-
спектре пробы должна исчезнуть полоса поглощения карбо-
нильной группы хлорангидрида кислоты в области 1786 см-1 и
появиться полоса поглощения карбонильной группы 6-метокси-р-
тетралона при 1701 см-1. Кроме того, к концу реакции исчезают
СН2=СН2
А!С13; СНгС1,
60-68%
16
полосы поглощения в области 1730 и 1661 см~\ Обработка реак-
ционной смесн и перегонка дают 21—24 г (60—68%) 6-метокси-
тетралона-2 с т. кип. 114—11670,2 мм. При хранении в холо-
дильнике продукт затвердевает в бесцветную массу с т. пл.
33,5-35°.
Отщепление бензильных групп [5]. При кипячении растворов
5,5-дибензилдитиогидантоина (1) и 4,4-дибензил-2,5-бпс-(метил-
тио)-4Н-имидазола (2) в ароматическом углеводороде в присут-
ствии А. х. от атома углерода гетероцикла отщепляется одна из
бензильных групп и атакует молекулу растворителя:
С6н5снг
н S
с1Н, с4н,нгс-у-^
3
с6нЕсн2 SCH3
с6н5нгс-Р
SCH
(2)
+ с6щснгс6н5
Перегруппировка М,М-дигалогенамииов [6]. Обработка N,N-
дихлор-трис-(я-бутил)-метиламина (2), полученного реакцией
трис- (н-бутнл)-метиламина (1) с гипохлоритом кальция [7], А. х.
в хлористом метилене при —30° и последующий кислотный гид-
ролиз дают с высоким выходом ди-н-бутилкетон (3) и н-бутил-
амин (4). Предполагают, что механизм реакции состоит в ми-
грации алкила со своей электронной парой к электронодефицит-
ному атому азота.
Са(ОС1)2 А1С13 г +
(«-Bu)3CNH3 - (k-Bu)3CNC12 ------(w-Bu)3CNCl
Уи—Уо zo 1
Ц) (2)
+ + СГ Н3О+
—> (w-Bu)2CNCl-«-Bu ----------*- (я-Ви)2С—N-h-Bu ------->
tl CI
—> («-Bu),CNH-«-Bu-l —> (h-Bu)2C=O + h-BuNH2
Ah J
(3) 95 % {4)
17
При проведении реакции с М,М-дихлораминоапокамфаном
(5) помимо ожидаемого продукта (6) в результате р-расщепле-
ния образуются еще три соединения —(7), (8) и (9) [8]:
Получение диамантаиа (VI, 9) [9]. Изомеризацию тетра-
гидробнснора-S (гидрированный димер норборнадиена) в диаман-
там можно осуществить с выходом 82% (считая на выделенный
продукт) кипячением с А. х. в хлористом метилене в тече-
ние 12 час. При этом побочно образуются 1- и 4-хлордиаман-
таны.
Полистирол — А.х. [10]. А.х. образует устойчивый к действию
воды комплекс с сополимером стирола и дивинилбензола (1,8%).
Согласно типовой методике, его получают обработкой гранул
сополимера (59—100 меш, 31,0 г) сероуглеродом, а затем без-
водным А.х. (7,5 а). Смесь кипятят при перемешивании в те-
чение 40 мин, затем избыток А1С13 разлагают осторожным
добавлением воды и снова перемешивают до тех пор, пока пер-
воначальный оранжевый цвет не перейдет в светло-желтый.
Комплекс отфильтровывают, промывают водой, затем последо-
вательно эфиром, ацетоном, горячим изопропанолом, снова эфи-
ром и, наконец, высушивают в вакуумном сушильном шкафу
в течение 18 час. При хранении на воздухе комплекс не теряет
своих свойств более года. Безводный А. х. можно снять с поли-
мера с помощью набухания комплекса в различных раствори-
телях (бензоле, гексане, сероуглероде).
А. х. на полимерном носителе использовали в синтезе простых
эфиров, получающихся в этих условиях с довольно высокими
выходами. При действии этого комплекса на дициклопропилкар-
бииол (1) образуется бис- (дициклопропилметиловый) эфир (2)
с выходом 81%. Можно получить и смешанные простые эфиры,
если вводить в реакцию два различных спирта.
(1) (2)
18
Новый метод, в частности, полезен в тех случаях, когда ис-
ходный карбинол чувствителен к А. х.
Расщепление эфирной связи (I, 49—51; VI, 11). Приведем
еще один пример этой реакции. Нагревание триметилового эфира
флорацетофенона (1) с А.х. при 110° приводит к диметиловому
эфиру (2) [1]. Аналогично, при деметилировании полиметокси-
флавона отщепляется преимущественно метоксигруппа в поло-
жении 5 и образуется соответствующий 5-оксифлавон [12].
сосн3
СОСН3
А1С1з, 110°
------------>
Это было использовано в синтезе некоторых природных флаво-
нов, например генкванина (5) [13]:
1) HCl-AcOH
2) AlClj, 100°
-------L---->
(5}
На одной из стадий полного синтеза агликона противо-
опухолевого антибиотика дауномицинона (8) Хо и сотр. [14]
действием А. х. при комнатной температуре осуществили селек-
тивное расщепление эфирной связи триметилового эфира дау-
номнцинона (6) с образованием 4-О-деметил-7-О-метилдауноми-
цпнона (7):
19
1. А у у а г К. S., С о о k s о n R. С., К a g i D. A., J. С. S. Chem. Comm., 1973,
161.
2. Nielsen A. T., Gibbons C., Zimmerman C. A., J. Am. Chem.
Soc., 73, 4696 (1951).
3. Owen G. R., Reese С. B., Org. Syn., submitted 1972,
4. Sims J. J., Selman L. H., Cadogan M., Org. Syn., 51, 109 (1971).
5. M а г к о v i t s - К о r n 1 s R., Nyitrai J., Lemper t K.f Chem. Ber., 104,
3080 (1971).
6. Kling T. A., White R. E., Kovacic P„ J. Am. Chem. Soc., 94, 7416
(1972).
7. Kovacic P., Chaudhary S, S., Org. Syn., 48, 4 (1968).
8. Fisher R. D., Bogard T. D., Kovacic P., J. Am. Chem. Soc., 94,
7599 (1972).
9. Courtney T., Johnston D. E., Me Ker vey M. A., Rooney J. J.,
J. C. S. Perkin I, 1972, 2691.
10. Neck er s D. C., Kooistra D. A., Green G. W., J. Am. Chem. Soc.,
94, 9284 (1972).
11. von Kostanecki St., Tambor J., Ber., 32, 2260 (1899).
12. Ven ka tar a man K-, Bharadwaj G. K-, Curr. ScL, 2, 50 (1933);
Gulati К. С., V enk a ta г a man K-> J- Chem. Soc., 1936, 267.
13. Mahal H. S., Venkataraman K-, J. Chem. Soc., 1936, 569.
14. Wong С. M. Schwenk R., Popien D., Ho T.-L., Can. J. Chem., 5l,
46 (1973).
АМИДИНЫ БИЦИКЛИЧЕСКИЕ.
Опубликован обзор «Бициклические амидины — реагенты
в органическом синтезе» [1].
ДБУ
I, 240-242; V, 91;
V, 88—89; этот том
этот том
Эти реагенты очень удобны для дегидрогалогенирования не-
стойких органических соединений в мягких условиях. Например,
20
обработка галогенсодержащего соединения (1) ДБН приводит
к образованию ацетата витамина А (2) в результате гладкого
элиминирования галогеноводорода, не сопровождающегося пере-
группировками, которые часто наблюдаются в ряду производ-
ных витамина А, О степени превращения ацетата (1) в ацетат
(2) СН2-ОАс
(2) можно судить по изменению коэффициента экстинкции
в УФ-спектре. Использование этого параметра позволило пока-
зать, что восемь других оснований менее эффективны, чем ДБН.
С помощью ДБН из 4-бром-1,2-эпокси-1,2,3,4-тетрагидронаф-
талина (3) впервые удалось синтезировать 1,2-окись нафтали-
на (4):
Продукт кристаллизуется в виде бесцветных игл с выходом
83%. Температуру плавления этого вещества точно определить
не удалось, поскольку оно быстро перегруппировывается
в а-нафтол.
Реакцией соединения (5) с ДБН был осуществлен и первый
успешный синтез чрезвычайно нестабильного стибина — стиба-
бензола (6):
С1
(5)
ДБН
(6}
Селективность действия ДБН хорошо иллюстрируется реакцией
дегидробромирования дибромида (7). Реакция последнего
с трет-бутилатом калия приводит к соединению (8), а с ДБН
образуется вещество (9).
В случае целого ряда простых бромалканов (см. табл.) вы-
ходы алкенов, полученных с применением ДБУ, в некоторых слу-
чаях почти вдвое выше выходов продуктов, получаемых при
действии ДБН.
Таблица
Сравнительное дегидрогалогенирование с помощью ДБН и ДБУ
Выход, %
Реакция
с ДБН е ДБУ
НгС— СН,—СН,—СН— сн2—СН2—СНз —>
I
Вг
—> НаС—СН2—СН=СН—СН2—СН,—СН3
НаС—СН—СН2—(СН2)3—СН3 —>
Вг
—> Н3С—СН=СН—(СН2)3—СНзН-Н2С=СН—СН2—(СН2)Э—СНз
4:1
н3с—сн—сна—(сн3)4—сн3 —>
^г
—> Н3С—СН=СН—(СН,)4—СНз+Н,С=СН—СН,—(СШ4—СН3
4:1
60 91
36 78
40 84
Элиминирование сульфокислот. В молекулы органических со-
единений двойные связи можно ввести путем отщепления суль-
фокислот от соответствующих сложных эфиров последних. Ре-
акция протекает гладко под действием ДБН или ДБУ. Напри-
мер, обработка З-тозилокснгексадиина-1,5 (10) избытком ДБН
в эфире при комнатной температуре (1 час) приводит к образо-
22
ванню смеси (40 : 60) цис- и транс-гексен-З-диинов-1,5 (11) с вы-
ходом 70% [2]:
ДБН
нс-:С—СН2—СН—С-.СН -------> НС=-С—СН—СН— С—СН
OTs
(10) (1b
Оба циклических амидина катализируют следующие реакции:
Получение илидов фосфора
Элиминирование азотистой кислоты
Стерический контроль в реакциях элиминирования
Альдольная конденсация
Перегруппировки
Реакции с арилизоциаиатами
Получение хлорангндридов карбоновых кислот
Синтезы макромолекул
1. О е d i g е г Н., Moller F., Е i t е г К., Svnthesis, 1972, 591.
2. Barton Т. J., Martz М. D., Zika R." G„ J. Org. Chem., 37, 552 (1972).
АМИД КАЛИЯ (I, 54—57; V, 24; VI, 13—14).
Замена фенольного гидроксила на аминогруппу [1]. Росси и
Беннет предложили общий метод превращения фенолов в ани-
лины. Сначала реакцией с диэтилхлорфосфатом (I, 435—436;
ArOH + NaOH + (С2Н5О)2РОС1 ——> АгОРО(ОС2Н5)2
VI, 119—120) в присутствии NaOH фенол превращают в арил-
диэтилфосфат, затем действуют А. к, и металлическим калием
в жидком аммиаке и с хорошим выходом получают анилины.
Описанная реакция, по-видимому, не применима к нитро- или
галогеизамещеиным фенолам.
Синтез фенантридина [2]. При взаимодействии галогензаме-
щеииого анила (1) с А. к. в жидком аммиаке образуется фенан-
тридин (2) с выходом >90%. Реакция, вероятно, протекает че-
рез стадию образования дегидробензола. Циклизации не мешает
наличие в молекуле анила алкильных, алкокси-, диалкиламино-,
циано-, карбокси-, карбонильных групп и галогена. Однако с ни-
тропроизводными циклизация не идет.
3,3-Диметоксициклопропен (3). Бауком и Батлер [3] предло-
жили сравнительно простой метод синтеза 3,3-диметоксицикло-
23
пропена (3) из 2,3-дихлорпропена (1), Последний действием
NBC н метанола в присутствии серной кислоты (катализатор)
переводят в 1-бром-3-хлор-2,2-диметоксипропан (2), который при
обработке А. к. в жидком аммиаке циклизуется в продукт (3).
С1
।
СН2 = ССН2С1
ИБО, СН3ОН
Н?О+
33-40%
(1)
ОСН3
I
ВгСН2ССН2С1
осн3
KNH2, NHj
----------->
40-50%
(3)
(2)
1. Rossi R. А., В unnett J. F., J. Org. Chem., 37, 3570 (1972).
2. К е s s а г S. V., G о р а 1 R., S i п g h М., Tetrahedron, 29, 167 (1973); К е s-
s а г S. V., Р а 1 D., Singh М., Tetrahedron, 29, 177 (1973).
3. Baucom К. В., Butler G. В., J. Org. Chem., 37, 1730 (1972).
АМИД НАТРИЯ (I, 58—66; V, 24—26).
N-Алкиланилины [1]. При действии А.и. на бромбензолы,
замещенные в .мета-положении группами, обладающими —/-эф-
фектом [ОСН3, Cl, N(CH3)2], образуется замещеииый дегидро-
беизол, который реагирует с первичными алифатическими
аминами с образованием соответствующего Ы-алкил-тиета-заме-
h2nh
щениого анилина с выходом 68—85%. При этом образуется
менее 5% орто-изомера.
1, Biehl Е. R., Р a t г i z i R., Reeves Р. С., J. Org. Chem., 36, 3252 (1971).
АМИД НАТРИЯ — трет-БУТИЛАТ НАТРИЯ.
Дегидробромирование. До сих пор предполагалось, что р-эли-
мииирование НХ, приводящее к образованию олефинов, осуще-
ствляется через переходное состояние, обладающее анти-конфи-
гурацией. В недавнем обзоре Зихера [1] по бимолекулярному
элиминированию отмечалось, что наряду с анти- может происхо-
дить и сан-элиминирование особенно в тех случаях, когда эли-
минированию подвергаются ониевые основания.
Основание
Основа ние
н J
анти сип
24
Французские химики [2] сообщили, что при действии на
транс- 1,2-дибром циклогексан (1) «комплексным основанием»
А. н.— б, и, образуется 36% циклогексена (2) и 60% 1-бромцик-
логексена (3). При действии каждого основания по отдельности
в основном возвращается исходный дибромид. Циклогексен (2)
образуется, очевидно, в результате ожидаемого анти-элиминиро-
вания; образование 1-бромциклогексена (3) возможно только
путем снн-элиминнрования.
NaNHa
NaOC(CH3)3
(U (2) 36% (3) 60%
t. Sichef J., Angew, Chem., Internal Ed,, 11, 200 (1972).
2. Caubere P., Coudert G., J. C. S. Chem. Comm., 1972, 1289.
трет-АМИЛА ГИДРОПЕРЕКИСЬ, CH3CH2C(CH3)2. Мол.
I
вес 104,15, т. кип. 26°/3,5 мм. ООН
Получение [1]. А. г. получают из трет-амилгидросульфата и
концентрированной перекиси водорода при 0°.
Оксазиридины [2]. Окисление оснований Шиффа (1) с по-
мощью А. г. в бензольном растворе, катализируемое гексакарбо-
нилом молибдена (V, 307; VI, 178—179; этот том) или M0CI5,
дает обычно с высокими выходами оксазиридины (2):
j О
R \ А. г. —Мо(СО)а R \ / \
4C=N—-R3 ----——------> ^С—N—R3
(I)
N-Окиси [2]. А. г. вступает в реакцию с азотсодержащими
ароматическими гетероциклами, образуя с высокими выходами
N-окиси (90—100%). Исключение составляют 2,2'-дипиридил н
о-фенантролин, которые при действии А. г. не окисляются.
Нитрамины [2]. Продолжительная обработка нитрозаминов
(1) смесью А. г. — M0CI5 приводит к нитраминам (2) с выходом
около 80%:
RL R‘
J,N— NO —> NO2
R2/ R2/
(П (2)
1. Alii as N. A., Surgenor D, M., J. Am. Chem. Soc., 68, 643 (1946).
2. Tolstikov G. A., J e m 11 e v U. M., J 11 r j e v V. P., G e r s h a n 0 v F. B.,
R a 11 kov S. R., Tetrahedron Letters, 2807 (1971).
трет-АМИЛ-8-ХИНОЛ ИЛ КАРБОНАТ. Мол. вес 259,3, т. пл.
46—46,5°.
См. трет-Бутил-8-хинол ил карбонат.
25
л-АМИНОАЦЕТОФЕНОН, n-CH3COC6H4NH2. Мол вес 135,16,
т. пл. 104—106°.
Арилирование по Меервейну. В [1] описана подробная мето-
дика диазотирования А. в смеси ацетона и 48%-ной бромисто-
водородной кислоты водным раствором нитрита натрия, а также
реакция Меервейна [2] полученной соли диазония с акриловой
кислотой в присутствии бромистой меди (промытой ацетоном до
бесцветных вытяжек и высушенной) с образованием м-ацетил-
а-бромгидрокоричной кислоты (выход 56—59%) в виде бесцвет-
ных игл с т. пл. 159—160°,
NaNO2 + СН2=СНСОгН
я-СН3СОС6ВДН2 —-—> «-CH3COCeH4N^NBr- ' р >
НВг СцВг, НВг
—> гг-СН3СОСеН4СН2СНСО2Н+N2
I
Вг
1. С 1 е 1 a n d G. Н., Org. Syn., 51, 1 (1971).
2. Рондестведт X. С. «Органические реакции», «Мир», М„ 1965, сб. 11,
стр. 199.
4- АМИНО-3 - ГИДРАЗИНО-5-МЕРКАПТО-1,2,4,-ТРИАЗОЛ
(1). Мол. вес 146,19, т. пл. 231—233° (с разл.).
Получение. Подробная методика получения этого триазола
приведена в «Organic Syntheses» [1].
nh2
I
2 CS л.
I
NH,
3 h2nnh2- h,o
nh.
HS nh-nh2
57-65^ V + H*S + 4NH3 +ЗЩО
(П
Реакция с альдегидами. A. (1) взаимодействует только с аль-
дегидами, давая 6-меркапто-5-триазоло- (4,3-5) -сплгм-тегразииы
(2) с окраской от пурпурной до красной. К 100—200 мг реаген-
та (1), растворенного приблизительно в 2 мл 1 и. гидроокиси
натрия, прибавляют одну каплю альдегида. На воздухе в резуль-
тате окисления через несколько минут появляется интенсивная
окраска [2].
+ ясно ->
Возду^
(0 (2)
26
1. Jacobsen N. W., Dickinson R. G., Org. Syn., submitted 1971,
2. Dickinson R. G., Jacobsen N. W., Chem. Comm., 1970, 1719.
2-ЛМИНОТИЛЗОЛ (1). Мол. вес 100,14, т. пл. 90—92°.
2-Аминотиазол (1) с эфирами пропиоловой кислоты образует
2-оксо-7Н-тиазо-[3,2-а]-пиримидии (2а) и небольшие количества
моноциклических аддуктов состава 1 : 1 и 1 : 2 [(3) и (4)]. В слу-
чае эфира ацетилендикарбоновой кислоты образуется 5-карбме-
токсипроизводиое соединения 2а (26) [1].
1. R е i m 1 i n g e r H., Chem. Ber., 104, 2232 (1971).
Дегидрирование [2]. Действие грет-бутилата калия и 3 же А.
в бензоле при комнатной температуре на аддукты Дильса — Аль-
дера из 1,3-диенов и нитрилов цитраконовой и мезаконовой кис-
лот (1) приводит к образованию замещенных бензонитрилов (2).
27
Это, по-видимому, первый зафиксированный пример дегидриро-
вания карбанионов под действием хиноиа.
А, при этом превращается, очевидно, в калиевую соль хингид-
рона антрацена.
1. Fleser L. F,, Org. Expts., 2nd Ed., Heath D. C. and Co., Boston, 1968,
199___203
2. Vaughan W. R., Simonson D. R., J. Org. Chem., 38, 566 (1973).
АСКАРИТ (90% NaOH на асбесте).
Дегидроцианирование динитрилов [1]. Динитрил циклобу-
тан-1,2-дикарбоиовой кислоты (1) под действием гидроокиси на-
трия при 190—225° и давлении 1 мм рт, ст. превращается в ни-
трил циклобутен-1-карбоновой кислоты (2). Наиболее высокие вы-
ходы (59%) получают с помощью А. Метод применим, очевидно,
+ NaOH
(1)
+ NaCN +. Н2О
(2)
только для дегидроцианирования вицинальных динитрилов.
1. Gale D. М., С h е г к о f s к у S. С., J. Org. Chem., 38, 475 (1973).
АЦЕТИЛЕНДИКАРБОНОВОЙ КИСЛОТЫ ДИМЕТИЛ 0-
ВЫЙ ЭФИР (I, 78—79; V, 28; VI, 19—20).
Реакция с Д3-оксазолиноном [1].
СН3)гСН .О
N
снгснгсосн3
III
с
I
согсн3
2 05-210°
--------
-СОг
Реакция с я-диазокетонамн [2]. А. к. д. э. реагирует с 2-ди-
азобутаионом-3 (1) с образованием замещенного N-ацетилпира-
зола (2). При взаимодействии реагента с 2-диазоциклопентано-
ном (3) образуется диметиловый эфир 4,5-дигидро-7(6Н)-оксо-
28
пиридо-[1,5-а]-пиразол-2,3-дикарбоновой кислоты (4).
cz
I +
нэс ''о
CCOOCHj
ССООСН}
Реакция с З-амино-1,2,4-триазолом [3].
1,2,4-триазол (2) в кипящем этаноле дают
(4) и (5):
А. к. д, э. и 3-амино-
смесь продуктов (3),
(4)
Ароматическое аннелирование [4]. Реакция перекиси бензо-
ила (1 моль) с большим избытком А. к. д. э. (10 молей) при 80°
приводит к тетраметиловому эфиру нафталии-1,2,3,4-тетракар-
боновой кислоты (0,5 моля/моль перекиси). В реакции промежу-
точно образуются, видимо, фенильные радикалы, поскольку вме-
сто перекиси можно использовать и другие источники фениль-
ных радикалов (N-нитрозоацетанилид или смесь ацетанилида
с амилиитритом). Предполагается, что реакция протекает сле-
дующим образом: фенильный радикал атакует молекулу
А. к. д. э. с образованием замещенного стирильного радикала, ко-
торый в свою очередь присоединяется ко второй молекуле
А. к. д, э. и, наконец, реакция завершается циклизацией, как по-
казано на приведенной схеме:
29
'Н^СООССеССООСН;
GOOCH,
I
H3COOCC=CCO0CHj
ас.,
^C-COO CH,
^C-COOCH,
C^ ’
I
COOCH3
Бензциклобутадиен в качестве интермедиата [5]. транс-3,4-
Ди-(феиилэтинил)-1,2,3,4-тетраметилциклобутен-1 (1) реагирует
с А. к, д. э. в бензоле в атмосфере азота с образованием димети-
лового эфира 1,2-дифенил-5,6,7,8-тетраметилиафталин-3,4-дикар-
боновой кислоты (2) с выходом 85°/о- По-видимому, эта необыч-
ная реакция протекает через промежуточные соединения (а) —
(в):
Реакция с енаминами и родственными соединениями. Брэн-
нон и сотр. [6] обнаружили, что енамины из ациклических аль-
дегидов и кетонов дают с А. к. д. э. соединения, представляющие
собой продукты перегруппировки цнклобутенов, образовавшихся
первоначально в результате 1,2-циклоприсоединения. Например,
при кипячении М,М-днметилизобутениламина (1) с А. к, д. э.
в эфире образуется диметиловый эфир 2-диметиламинометилен-
зо
3-изопропилиденяитарной кислоты (2) с выходом 49%:
сн,
г_с ССООСЩ
; V + in
H-с ССООСНз
N(CH3)2
NfCHjh
С(СНэ)г
C-COOCHj
49*Й> C-COOCHj
CHN(CH3)j
Промежуточный циклобутен удалось выделять с хорошим вы-
ходом из реакции N-(циклогексен-1-ил-1)-пирролидина (3)
с А. к. д. э. в эфире при 25—35°. Полученный циклобутен (4) тер-
мически неустойчив и при нагревании иа кипящей водяной бане
(11 час) перегруппировывается с расширением цикла в соедине-
ние (5), правда, с низким выходом. Аналогичные продукты были
(3)
соосн,
с
ill _________
COOCHj
COOCHj
COOCHj
получены с хорошим выходом непосредственно из енаминов
циклопеитаноиа, циклогепта нон а и циклооктанона.
При взаимодействии 1-метилиндола (6) с А, к. д. э. в кипя-
щем ацетонитриле в качестве основного продукта образуется дн-
метиловый эфир 1-метил бенз-[/]-азепии-3,4-дикарбоиовой кис-
лоты (8) [7], Очевидно, гетероциклический атом азота и двойная
связь в положении 2,3 действуют как енаминовая система, и
в реакции промежуточно образуется циклобутен (7),
соосн,
соосн,
I
СН,
(8)
Плинингер и Вилд [8], получившие 2-этоксибензазепин (11)
из 2-этокси-1-метилиидол а (9) и А. к.д.э., также полагали, что
реакция протекает с промежуточным образованием циклобу-
теиа (10):
Леман [9] недавно выделил циклобутеновый интермедиат (13)
из реакции 1-метил-1,4-дигидрохинолииа (12) с А. к. д. э. в аце-
тонитриле в атмосфере азота. При кипячении в сухом бензоле
в течение 8 час аддукт (13) перегруппировывается в 1-метил-
3,4-дикарбметокси-1,6-дигидробеиз-[^]-азоцин (14) с выходом
77,9%.
Синтез азулена [10]. Реакция 6-(2-диметиламиновинил)-фуль-
вена (1) [11] с А. к. д. э. (2) приводит к образованию 11% дн-
метилового эфира азулеи-4,5-днкарбоновой кислоты (3) и ~20%
диметилового эфира диметиламиномалеиновон кислоты (4):
+ СН3ОСОС=ССООСН3
n(ch3)2
(1) (2)
[2,2]-л-Циклофаны. Реакция Дильса—Альдера гексатетра-
ена-1,2,4,5 (1, диалленил) с А. к. д. э. приводит к [2,2]-«-цикло-
фану (3), являющемуся продуктом димеризации первоначально
образующегося 2,3-бмс-(каРбметокси)-хиЯодйметана-1,4 (2) [12].
Реакция с фенилгидразонами альдегидов и кетонов [13]. При
взаимодействии феиилгидразона бензальдегида (1) с А. к. д. э.
образуются соединения (2), (3) н (4).
Фенилгидразон ацетофенона (5) дает пиразол (3) с выходом
57%.
с6н.
C IT
нх Н
соосн,
1
с
150\ С‘ЯЧ>АМ'''С‘Я®
соосн,
CHjOOc.A__/ ,н
Н^-COOCHj
(2) 3%
СООСНз
С*Н5 N ДДНз
Y I* +
HjCOOC COOCHj
(3) 16%
I '''СООСН3
COOCHj
И) 5%
/С6н5
СХ N +
i Н
CHj
(5)
СООСН,
I
с
соос нэ
<31
57%
1, Steglich W., Gruber Р., Н einin ger Н. U.f Kneidl F., Chem,
Ber., 104, 3816 (1971).
2, К a t n e r A. S., J. Org. Chem., 38, 825 (1973).
3. Reimlinger H., Jacquier R., Daunis J., Chem. Ber., 104, 2702
(1971).
4. В a i g г i e B. D., Cadogan J, 1. G., Cook J., Sharp J. T., J. C. S,
Chem. Comm., 1972, 1318.
5. Muller E., Huth A., Tetrahedron Letters, 1035 (1972); 4359 (1972).
6. Brannock К. C., Burpitt R. D., G о о d 1 e 11 V. W., Thweatt J. G.,
J. Org. Chem., 28, 1464 (1963).
7. Acheson R. M., В r i d s о n J. N., Cameron T. S., J. C. S. Perkin I,
1972, 968.
8. P 1 i e n i n g e r H., W i 1 d D., Chem. Ber., 99, 3070 (1966).
in л *7 h m a п P. G., Tetrahedron Letters, 4863 (1972),
10. Alder R. W., Whittaker G., Chem. Comm., 1971, 776.
2 Зак. 59S пъ
11. Hainer К. et al., Angew. Chem., 75, 35 (1963); Bredereck H., Ё f f e rt-
b e r g e r F., Z e у f a n g D., Angew. Chem., 77, 219 (1965).
12. Hopf H., Angew. Chem., Internal. Ed., 11, 419 (1972).
13. Saxena M. K., Gudi M. N., George M. V., Tetrahedron, 29, 101
(1973).
1-АЦЕТИЛ-1-МЕТИЛ ГИДРАЗИН, CH3N(Ac)NH2. Мол. вес
88,11, т. кип. 103°/8 мм, т. пл. 16°.
Реагент удобнее всего получать ацетилированием метилгид-
разина уксусным ангидридом в пиридине (выход 76%) [1].
1-Алкил-2-метилгидразины [2]. С альдегидами или кетонами
А. образует, обычно с количественным выходом, ацетилметил-
гидразоны, которые восстановлением NaBH4 и последующим
ЬГаВЩ
CH3N(Ac)NH2 + ЧС=О —> CH3N(Ac)N=C(' ----------->
rZ ^r2
н3о+ Л
—> CH3N(Ac)NHCH -------CH3NHNHCH
^Ra Z?2
гидролизом превращают в 1-алкил-2-метилгидразииы (выход
40-80%).
1. Condon F. Е., J. Org. Chem., 37, 3608 (1972).
2. С о n d о n F. Е., J. Org. Chem., 37, 3615 (1972).
АЦЕТИЛСЕРНАЯ КИСЛОТА.
В т. V на стр. 404 предлагается исправить формулу сульфо-
уксусной кислоты: HOOCCH2SO2OH. Это предложение нуж-
дается в уточнении: соединение, о котором идет речь в т. III, 291,
иа самом деле является ацетилсерной кислотой.
Сулъфоуксусную кислоту лучше всего получать действием
сульфита натрия на натриевую соль монохлоруксусной кислоты
[1].
1. Gilbert Е. Е., Sulfonation and Related Reactions, Interscience, New York,
1965, p. 277.
О s
II II
N-АЦЕТИЛТИОМОЧЕВИНА, CH3CNHC—NH2. Мол. вес
118,16, т. пл. 165—169°.
А. (1) получают из тиоцианата натрия (III, 341—342) и хло-
ристого ацетила с последующей обработкой водным аммиа-
ком [1]:
ГЧН3 ц и
-НС1 II II
NaSCN + СН3СОС1 —> CH3CNHC—NH3
(i)
34
Синтез меркаптанов [1]. При нагревании А. (1) с галогенал-
килами в этаноле в течение 24 час образуются меркаптаны с вы-
ходом 30—75%. Вторым продуктом является ацетилмочевина
(3). Промежуточно образуется галогеигидрат 1-ацетил-2-алкил-
2-тиопсевдомочевины (а). Эти тиопсевдомочевнны можно выде-
лить, если растворителем служит ацетонитрил. Выходы меркап-
танов высоки при использовании первичных галогеналкилов и
несколько ниже в случае вторичных. При взаимодействии А. (1)
Г О SR 1 Г О ОН 1
II I + с2н5он || || I
(D + RX —► Lch3cnhc—nh2x_J ———> Lrs~+ ch3cnhcnh2J —>
4 ' —С2Н5Л
(a) (6)
О о
II II
—> RSH+CH3CNHCNH2
(2) (3)
с трет-бутил бромидом меркаптан ие образуется. Вместо А.
можно использовать беизоилтиомочевину, одиако при этом вы-
ходы меркаптанов более низкие.
1. Klayman D. L, Shine R. J., Bower J. D., J. Org. Chem., 37, 1532
(1972).
АЦЕТИЛ-п-ТОЛУОЛ СУЛЬФОНАТ (V, 29—30).
Опубликованы методы получения и реакции А. [I].
1. Karger М. Н., Mazur Y., J. Org. Chem., 36, 528, 532, 540 (1971).
АЦЕТОФЕНОН, С6Н5СОСН3. Т. кип. 202°/749 мм рт. ст.
Фотоизомеризация бицикло-[2,2,1]-гептадиена-2,5 (1) в при-
сутствии А. в качестве сенсибилизатора служит методом полу-
чения квадрициклана (2). В реакцию вводят 180 г соедине-
ния (1) в 1 л эфира в стандартном иммерсионном фотохимичв-
(1) (2)
ском реакторе (550W), снабженном мешалкой и обратным холо-
дильником, в атмосфере азота в присутствии 8 г А., играющего
роль фотосенсибилизатора; продолжительность реакции 36—
48 час; выход 70—80%.
1. S m i t h C. D., Org. Syn„ 51, 133 (1971).
2*
БАРИЯ ГИДРООКИСЬ, Ва(ОН)2. Мол. вес 171,38.
й-Бромзамещенные кислоты и й-бромзамещенные сложные
эфиры. Стоттер и Хилл [1] предложили новый метод синтеза
ct-бромкарбоновых кислот и их эфиров из замещенных ацетоук-
сусных эфиров (1). Натриевое производное ацетоуксусного
эфира алкилируют в ТГФ первичным иодистым или бромистым
алкилом, получая эфир (2). Если образовавшийся эфир (2) рас-
творить в ТГФ, перевести в енолят натрия (NaH), а затем об-
работать его раствором брома в хлористом метилене при 0°, то
CH3COCH2COOR' —>
NaH
ВГ2
CH3COCHCCW —>
R
Вг
.—> CH3CoicOOR'
R
(3)
(2)
/.Спирт.
Ва(ОН)2
-------> RCHCOOR'
ir
(4)
бромирование направляется исключительно в сс-положение, Ке-
тонное расщепление эфира (3) осуществляют действием 1 г-экв
гидроокиси бария (высушенной в вакууме при 125° до постоян-
ного веса) в абсолютном этаноле при температуре ие выше 0°
в течение 30 мин. Применение избытка гидроокиси бария во
влажном спирте дает плохие результаты.
трет-Бутиловые эфиры дезацетилируют кипячением в бен-
золе, содержащем каталитические количества п-толуолсульфо-
кислоты.
Джонсон и сотр. [2] дезацетилировали а-алкил-а-хлор-р-ди-
кетопы действием гидроокиси бария в 95%-ном этаноле прн 0°.
Дикетон (5), например, легко превращается в хлоркетои (6).
36
1 S t о 11 e r P. L., H111 K. A., Tetrahedron Letters, 4067 (1972) ,
2* Johnson W. S., Wiedhaup K-, Brady S. F., Olson G. L., J. Am.
Chem. Soc,, 90, 5277 (1968); Johnson W. S., Ll T., Faulkner D. J.,
C a m p b e 11 S. F„ J. Am. Chem. Soc., 90, 6225 (1968).
БЕНЗИЛТРИЭТИЛАММОНИЙХЛОРИД (VI, 22).
Дихлоркарбен. Польские химики [I] предложили новый ме-
тод геиерирования дихлоркарбена (или карбеиоидных частиц)
реакцией олефина с хлороформом в присутствии 50%-ного вод-
ного раствора NaOH и каталитических количеств Б. Таким ме-
тодом, например, из циклогексена был получен дихлорноркаран
с выходом 72%. В отсутствие катализатора выход составлял
лишь 0,5% [2].
Б. играет роль катализатора фазового переноса. Хлорид ам-
мония растворяется в водной фазе, имеющей щелочную реак-
цию, и переходит в гидроокись беизилтриэтиламмония, раство-
римую в органической фазе. Ион ОН- гидроокиси реагирует
с хлороформом, давая дихлоркарбен; при этом регенерируется Б.
Немецкие химики [3] с успехом применили этот метод для
синтеза дихлорциклопропанов из таких олефинов, которые при
взаимодействии с дихлоркарбеном, генерированным из хлоро-
форма под действием трет-бутилата калия, или вообще не об-
разовывали дихлорциклопропанов или давали их с ничтожными
выходами. Аналогичным образом они генерировали и дибром-
карбен. Циклопропены из ацетиленов удалось получить лишь
с небольшими выходами, что связано с рядом побочных реак-
ций.
Данный метод был использован японскими химиками [4] для
генерирования дихлоркарбена, который затем вводили в реак-
цию с адамантаном (1), приводящую к 1-дихлорметиладаман-
тану (2); выход составлял 54% (91% в расчете на прореагиро-
вавший адамантан). Это первый пример реакции внедрения ди-
хлоркарбена в насыщенный углеводород. Следует отметить
также высокую степень избирательности внедрения CCI2 в по-
ложение в голове моста. Замещение только одного положения,
происходит, по-видимому, в результате замедления реакции под
влиянием дихлорметильного заместителя.
Те же японские химики [5] нашли, что при реакции с дихлор-
карбеиом, генерированным данным методом, спирты с высокими
37
выходами превращаются в хлориды. Например, 1-оксиадаман-
тан превращается в 1-хлорадамантаи с выходом 94%, а бензи-
ловый спирт — в бензилхлорид с выходом 90%. Реакция проте-
кает преимущественно с сохранением конфигурации (S^Q.
Реакция дихлоркарбена с диамантаном (3) не столь избира-
тельна, как в случае адамантана. Здесь почти количественно об-
разуется смесь 1- и 4-дихлорметилдиамантанов (4) н (5) в от-
ношении 1,7: 1. Следует подчеркнуть, что положение 4 диаман-
тана пространственно менее затруднено, чем положение 1 [6].
Много лет назад Гофман [7] открыл реакцию первичных ами-
нов с хлороформом в присутствии сильного основания, в резуль-
тате которой образуются изонитрилы. Выходы последних в этой
реакции низкие, и она применялась редко. Недавно Вебер и Го-
кель [8] сообщили, что если дихлоркарбен генерировать по ме-
тоду Монкошн н Вавжиневича [1], то изонитрилы можно полу-
чить с выходом 40—60%.
R—NH2 + : СС12 —> [R—NH2—СС1з —> R—N=CHC1] —> R—NC
Реагент Монкошн оказался наиболее подходящим и в реак-
ции дихлоркарбена с диенами, с его помощью бис-аддукты
можно получить с удовлетворительными выходами [9].
38
Индийские химики [10] сообщили, что еще более эффективно
генерируют дихлоркарбен другие катионные агенты, например
цетилтрнметиламмоиийхлорид или индийское моющее средство,
выпускаемое в продажу под маркой «цетримид». Старкс [11] для
той же цели применял «трикаприлметиламмонийхлорид», ал-
кильные группы которого представляют собой смесь неразвет-
вленных углеводородных радикалов С8—Cig.
Израильские химики [12] синтезировали некоторые стерины
с цнклопропановой системой в боковой цепи. Недавно было об-
наружено, что этот структурный фрагмент встречается в неко-
торых стеринах природного происхождения [13]. Взаимодействие
десмостерилацетата (6) с дихлоркарбеиом, генерированным по
(в)
методу Монкошн н Вавжиневича, привело к аддукту (7) с 50%’*
ным выходом. Последний действием системы литий — трет-бута-
нол — тетрагидрофуран (II, 153—155) восстанавливался в Д3-
24,25-метиленхолестенол-Зр (8) с выходом 70%.
Джоши и сотр. [14] удалось выделить аддукт (9) фенантрена
с дихлоркарбеиом, генерированным в присутствии цетилтриме-
тнламмонийхлорида. При температуре плавления (140°) аддукт
перегруппировывается в 6-хлордибензо-[а,с]-тропилинхлорид (10).
39
Фторнодкарбен. Немецкие химики [15] генерировали фтор-
иодкарбен следующим образом:
СН13
СНП2
2 Nal/
CHFBrj
NaOH, HjO
[ (СгНДИСНгС6Н5:)С1~
СНгС12
Реакция карбена с олефинами приводит к образованию 1-фтор-
1-иодциклопропанов с выходом 20—60%.
Циклогексилиденкарбен [16]. При обработке охлаждаемого
до температуры от —10 до —5° раствора 1-(N-нитрозоацетил-
амииометнл)-циклогексанола (1) в смеси пеитаиа и циклогек-
сена 50%-ным раствором NaOH в присутствии тетра алкил ам-
монийхлорида (использовался метилтрикаприламмонийхлорид,
«Аликвот» 336) образуется соединение (2) с 80%-иым выходом:
Алкилирование кетонов. Кетоны гладко алкилируются в «-по-
ложение галогенал килами в присутствии 50%-кого раствора гид-
роокиси натрия и каталитических количеств Б. [17]. Каталитиче-
ский эффект соли особенно ощутим в случае галогеналкилов
с пониженной реакционной способностью. Например, при реак-
ции метилбензилкетоиа с н-бутилбромидом без катализатора вы-
ход З-феиилгептанона-2 составляет лишь 5%, а в присутствии
катализатора выход продукта повышается до 90%. Самые высо-
кие выходы получены при алкилировании кетонов с ароматиче-
ским заместителем в сс-СНг-группе.
Нитрилы глицидных кислот [18]. Эти нитрилы можно полу-
чить с хорошим выходом реакцией кетонов с хлорацетонитрилом
в водном NaOH в присутствии Б. в качестве катализатора:
R\ R\
д. C1CH3CN —> ,С--СН—CN
V
40
Окисление перманганатом с помощью катализа фазового пе-
реноса. Старкс [11] сообщил, что окисление терминальных алке-
нов до карбоновых кислот, содержащих на I атом углерода
меньше, можно осуществить с высоким выходом действием ней-
трального водного перманганата калия в присутствии неболь-
шого количества четвертичной аммониевой соли. Децен-1 окис-
ляют до нонановой кислоты по следующей общей методике.
К энергично перемешиваемой смеси бензола (50 мл), «трикап-
рил метил аммоннйхлорнда» (0,01 моля) и раствора перманга-
ната калия (0,8 моля) в воде (100 мл) прибавляют олефин
(0,2 моля) с такой скоростью, чтобы температура реакционной
смеси не превышала 40—50° (экзотермическая реакция). По
окончании введения олефина реакционную смесь перемешивают
еще 0,5 час. Избыток перманганата разлагают сульфитом на-
трия, реакционную смесь отфильтровывают от МпО2 и подкис-
ляют разбавленной НО. Бензольный слой отделяют и встряхи-
вают с 10%-иым раствором NaOH. Щелочную вытяжку промы-
вают эфиром и затем подкисляют НО. Карбоновую кислоту
экстрагируют эфиром; выход нонановой кислоты составляет
91°/о- Октен-1 окисляют подобным же образом до гептановой
кислоты с почти количественным выходом.
Вебер и Шеферд [19] применили эту новую методику для
окисления алкенов с внутренней двойной связью до цщ?-1,2-гли-
колей с выходами около 50%. Как правило, эта реакция проте-
кает с низкими выходами, поэтому в качестве окислителя обычно
использовали токсичную дорогую четырехокись осмня (111,40—
46) или ацетат серебра и нод во влажной уксусной кислоте
(IV, 19).
Иллюстрацией нового метода может служить окисление цис-
циклооктена. К раствору октена (0,1 моля) в хлористом мети-
лене прибавляют 40%-ный водный раствор гидроокиси натрия
и 1 г Б. Смесь охлаждают до 0° и в течение 2 час небольшими
порциями при энергичном перемешивании вносят КМпСЦ
(0,1 моля). Смесь оставляют на ночь при 0°; выпавшую МпО2
растворяют пропусканием SO2. ц«с-Циклооктаи диол-1,2 экстра-
гируют эфиром. После кристаллизации выход его составляет
50%. В отсутствие катализатора фазового переноса диол обра-
зуется с выходом 7% [20].
Окисление транс-цнклододецеиа в транс-циклододеканди-
ол-1,2 по новой методике протекает также с выходом 50%.
Однако если гликоль хорошо растворим в водной фазе, то н
этим методом он получается с низким выходом. Так, циклогек-
сен окисляется в ц«с-цнклогександиол-1,2 только с 15%-иым
выходом и при этом образуется значительное количество адипи-
новой кислоты.
I. Makosza М., Wa wr zуniеw1 сz М., Tetrahedron Letters, 4659 (1969);
М а к о s z а М., Org. Syn., submitted 1971.
?• D о er i n g W. v. E.( Hollman A. K.( J. Am. Chem. Soo., 76, 6162 (1954).
41
3. D e h m 1 о w E. V., S c h & n e f e 1 d J., Ann, 744, 42 (1971).
4. T a b u s h i 1., Yoshida Z., Takahashi N., J. Am. Chem. Soc., 92,
6670 (1970).
5. T a b u s h i I.f Yoshida Z., Takahashi N., J. Am. Chem Soc., 93, 1820
(1971).
6. Ta bush i I., Ao yam a Y., Takahashi N., Gund T. M., Schley-
er P. v. R., Tetrahedron Letters, 107 (1973).
7. Hofmann A. W., Ann., 144, 114 (1867).
8. Weher W. P., Goke! G. W., Tetrahedron Letters, 1637 (1972).
9. Deh m 1 о w E. V., Tetrahedron, 28, 175 (1972).
10. J о s h i G. C., Singh N., P a n d e L. M., Tetrahedron Letters, 1461
(1972).
11. S t a г к s С. M., J. Am. Chem. Soc., 93, 195 (1971).
12. Ik an R., Markus A., Goldschmidt Z., J. C. S. Perkin I, 1972, 2423.
13. Hale R. L., Led er eq J., Tursch B., Dj eras st C„ Gross R. A.,
Weinheimer A. J., Gupta K., Scheuer P. J., J. Am. Chem. Soc., 92,
2179 (1970); Ling N. C., Hale R. L., Djerassi C., J. Am. Chem.
Soc., 92, 5281 (1970); Schmitz F. J., P a 11 a b h i r a m a n T., J. Am.
Chem. Soc., 92, 6073 (1970).
14. J о s h i G. C., Singh N., P a n d e L. M., Synthesis, f972, 317.
15. Wey erst ahi P., Mathias R., Blume G., Tetrahedron Letters, 611
(1973).
16. Newman M. S„ Din Z., J. Org. Chem., 38, 547 (1973).
17. Jonczyk A., Serafin B., Moksza M., Tetrahedron Letters, 1351
(1971).
18. Jonczyk A., Fedorynskf M., Makosza M., Tetrahedron Letters,
2395 (1972).
19. Weber W. P., Shepherd J. P., Tetrahedron Letters, 4907 (1972).
20. С о p e A. C., Fenton S. W., Spencer C. F., J. Am. Chem. Soc., 74,
5884 (1952).
1,3,2,-БЕНЗОДИОКСАБОРОЛ (пирокатехинбораи)
О)
Мол. вес 119,92, т. кип. 76—77°/100 мм.
Б. (1) получают с выходом 80% реакцией пирокатехина с бо-
раном в ТГФ [1]. Борол почти количественно реагирует с олефи-
нами при 100°, образуя соответствующие 2-алкил-1,3,2-бензоди-
оксаборолы (2), которые быстро гидролизуются до соответ-
ствующих алкилборных кислот (3):
ВН3, ТГФ , 0°
80%
Олефин
100°
------->
ВВ(ОН)г
(1) (2) (з)
Единственное неудобство метода заключается в том, что ре-
акцию с низкокипящими олефинами приходится проводить в за^
паянной ампуле [1]. Для таких случаев Браун рекомендует дру-
гой путь синтеза 2-алкил-1,3,2-бензодиоксаборолов (2), основан-
ный на легко протекающем обмене радикалов между триалкил^
42
боранами и о-фениленборатом [2] и приводящий к требуемым
бородам (2) почти с количественным выходом:
(2)
Гидроборирование алкинов. Б. (1) взаимодействует с алки-
нами стереоспецифично и региоселективно, давая продукты мо-
ногидроборнрования — 2-алкенил- 1,3,2-бензодиоксаборолы (2)
почти с количественным выходом. Гидроборирование протекает
по механизму ^«г-присоединения, причем атом бора атакует ме-
нее экранированный из атомов углерода тройной связи. Обра-
а CFTjCOOfr
(1) + RC = CRi-Z£_> RC-CR' 100°. i--
(2) (3)
1
Н2Ог
ч,ОН~
RCHaCOR’
(4)
зовавшиеся эфиры борной кислоты (2) легко гидролизуются до
соответствующих алкенилборных кислот RCH —С (RZ)B (ОН)2.
Протолиз эфиров (2) дает соответствующие ^мс-олефины (3),
а их окисление перекисью водорода в щелочной среде прн 25—
30° в течение 2 час приводит к альдегидам или кетонам (4) [3].
2-Алкенил-1,3,2-бензодиоксаборолы (2) с ацетатом ртути(П)
прн 0° почти количественно образуют алкенилмеркурацетаты [4].
Hg(OAc)2
(2) -----> RC=C—R'
н HgOAc
1. Brown Н. С., Gupta S. К., J. Am. Chem. Soc., 93, 1816 (1971).
2- Thomas L. H., J. Chem. Soc., 1946, 820.
3. Brown H. C., Gupta S. K., J. Am. Chem. Soc., 94, 4370 (1972).
C., Gupta S. K., Brown H. C., J. Am. Chem. Soc., 94, 4371
43
БЕНЗОИЛА ПЕРЕКИСЬ (1, 98—100).
Свободнорадикальная циклизация [1]. Соединения с двойной
связью в 6-положении циклизуются в алициклические соединения
под действием перекисей, генерирующих свободные углерод-
содержащие радикалы. Этим методом из тозилата трпяс-гексен-
4-ола-1 (1) можно получить этиловый эфир 1-циан-2-метилцик-
логексанкарбоновой кислоты (3). Тозилат конденсируется с эти-
0) о о
11 II
С6Н5СООСС6Н5
циклоген сан
88%
ловым эфиром циануксусной кислоты (гидрид натрия, ДМФА),
образуя этиловый эфир тра«с-2-цианоктен-6-овой-1 кислоты (2),
который под действием инициатора радикальной реакции, пере-
киси бензоила, превращается в этиловый эфир 1-циан-2-метил-
циклогексанкарбоновой кислоты (3) с выходом 88%. При ис-
пользовании перекиси трет-бутила эфир (3) получается лишь с
выходом 68% [2].
а-Метилен-у-бутиролактоны. Оуриссон и сотр. [3] описали
метод превращения трйис-сочлененных сс-метил-у-бутиролакто-
нов в а-метилен-у-бутиролактоны. Например, лактон (1), полу-
ченный в несколько стадий из а-сантонина, обрабатывают три-
фенилметиллитием в диметоксиэтане (ДМЭ) и затем Б. п.
в ДМЭ. Лактон (2) образуется с выходом около 50%. Пироли-
зом этого лактона при ~450° получают а-метилен-у-бутиролак-
тон [(+)-арбускулин-В] (3) с выходом 35%.
44
О превращении грс-сочлененных лактонов в а-метнлен-у-бу-
тиролактоны см. 1,2-Дибромэтан, этот том.
1. Julia М., S u г z и г J. М., Katz L., Bull. soc. chim, France, 1964, 1109;
Julia M., Maumy M., Bull. soc. chim. France, 1969, 2415, 2427;
J u 1 i a M., Accts. Chem. Res., 4, 386 (1971).
2. Julia M., Maumy M., Org. Syn., submitted 1973.
3. Greene A. E., Muller J. C., Ourisson G., Tetrahedron Letters, 3375
(1972).
БЕНЗОИНА ЕНОЛЬНОЙ ФОРМЫ ЦИКЛИЧЕСКИЙ КАР-
БОН АТ (дифенилэтиленкарбонат) (1). q о . Мол. вес
о
(0
238,23, т. пл. 75—76°.
Получение [1]. Карбонат (1) получают с 65—70%-иым выхо-
дом взаимодействием при 5° бензоина (1,0 же) и фосгена
(1,1 же) в бензоле в присутствии свежеперегнанного Ы,Л-диме-
тиланилина. После перемешивания в течение ~ 12 час при ком-
натной температуре солянокислую соль амина отфильтровывают,
а фильтрат кипятят в течение 3 час для циклизации образовав-
шегося первоначально эфира хлоругольной кислоты.
Защита первичной аминогруппы. Шихан и Гузиек [1] пред-
ложили этот реагент для защиты первичной аминогруппы, в ре-
зультате замещения обоих атомов водорода которой образуется
высокостабильная 4,5-дифенил-А4-оксазолинон-2-овая система
(3) [2]. Эти производные получают действием карбоната (1) на
первичный амин или тетраметиламмонневую соль аминокислоты
з ДМФА. После перемешивания в течение 0,5 час смесь подкис-
ляют (НС1) и образовавшуюся смесь диастереомерных оксиок-
сазолидинонов (2) экстрагируют этилацетатом. Смесь (2) де-
гидратируют с помощью трифторуксуснон кислоты (ТФК) до
оксазолиноновых производных (3). Эти производные устойчивы
к действию водных растворов оснований, кипящего спиртового
раствора гидразина, спиртового раствора хлористого водорода,
45
бромистого водорода в уксусной кислоте, кипящей ТФК и без-
водного фтористого водорода.
+ RNHj
- —-А> Cf,H5-CCH(C6H5)OCNHR
ТФН
(3)
Защитную группу можно удалить восстановлением — илн ка-
талитическим гидрогенолизом над палладием (количественно),
или натрием в жидком аммиаке (выход 75—85%). Кроме того,
ее можно отщепить окислением избытком ж-хлорнадбензойной
кислоты с последующим гидролизом (70%)- Простые оксазоли-
нондипептиды получают при использовании хлоргидрата 1-этил-
3- (З'-диметиламинопропил) -карбодиимида (IV, 253—254). Сво-
бодные дипептиды получают гидролизом. Ни в процессе получе-
Восстановление
Восстановление
-----------—> RNH2 + СОг + СьН5СНгСНгС6Н,
, Окисление
(3)—------------->
нго
+ СОг + CjHsCOCOC^Hj
ния оксазолиноновых производных, нн в их реакциях, ни прн
удалении защитной группы рацемизации не наблюдалось.
1. Sheehan J. С., Guziec F. S., Jr., J. Am. Chem. Soc., 94, 6561 (1972).
2. Filler R., Advan. Heterocyclic Chem., 4, 103 (1965).
БЕНЗОЛСУЛЬФОНИЛАЗИД (VI, 23—24).
Реакции. Поправка к стр. 24, строки 9—10 сверху. С норбор-
неном (1) реагент образует дкзо-азнриднн (2) [1]. Норборна-
46
диен (3) реагирует аномально, давая продукт внедрения азота
в цикл с предположительным строением (4) [2].
CtH5SOjN3
-N,
(2)
1 Franz J. E., О such C., Dietrich M. W., J. Org. Chem., 29, 2922
’ (1964).
2 . tranz J. E, Osuch C., Chem. Ind., 1964, 2058.
бмс-(БЕНЗОНИТРИЛ)ПАЛЛАДИЙ(П)ДИХЛОРИД,
(C6H5CN)2PdCl2. Мол. вес 357,85.
2-Замещениые бензофураиы [1], При обработке натриевой
соли 2-аллилфенола (1), полученной из 2-аллилфенола и мети-
лата натрия, 1 г-экв металлоорганического реагента в кипящем
бензоле (3 час) образуется 2-метилбензофуран (2) с выходом
(2)
31 %. 2-Бензилбензофуран был получен аналогичным образом
с 53%-ным выходом из 2-циннамилфенола.
Этим же методом нз 1-аллил-2-нафтола (3) получали 2-ме-
тил-[2,1-Ь]-нафтофуран (4):
(C6H5CN)2PdC12
42%
1- Но sok a wa Т., Maeda К„ Koga Moritani I., Tetrahedron Let-
ters, 739 (1973/,
БЕРЧА ВОССТАНОВЛЕНИЕ (I, 108—110; V, 33—35; VI,25).
Обзоры. Берч и Субба Рао [1] опубликовали обзор по вос-
становлению растворами металлов в жидком аммиаке. Кайзер
[2] рассмотрел общие черты и различия методов восстановления
по Берчу и литием в низших аминах по Бенкесеру. Автор при-
шел к заключению, что в сравнении с восстановителем Берча
восстановитель Бенкесера более сильный, но менее селективный.
Восстановление двойной связи в стероидах. На последней
стадии полного синтеза (±)-2,3,4-триметоксиэстратриен-1,3,5(10)-
ола-17{3 [2] осуществляют стереоспецифическое восстановление
стероидной двойной связи в соединении (1) натрием в жидком
аммиаке, Первоначально соединение (1) восстанавливали 4 г-ат
натрия в смеси жидкого аммиака и анилина в течение 30 мин,
но оказалось, что в этих условиях продукт реакции теряет 3-мет-
оксигруппу. Впоследствии авторы нашли, что если взять
2,5 г-атом натрия и ограничить время реакции 10 мин [3], то же-
лаемое восстановление проходит с выходом 85%.
Восстановление малоновых эфиров металлами. Днметиловый
эфир диметилмалоновой кислоты (1) восстанавливается на-
трием, диспергированным в ксилоле, в присутствии триметил-
хлорсилана (ТМХС) до метилтриметилсилилацеталя диметил-
кетена (2) [4]:
Na —ксилол
ТМХС
(СН3)2С(СООСНз)2 ———
с/ %
(1)
—> (СН3)2С=С(ОСНз) OSi(CH3)3 + СО + CH3OSi(CH3)3.
(2)
Поскольку ацеталь (2) легко гидролизуется до эфира (3),
такой двухстадийный процесс представляет собой удобный ме-
тод декарбоксилирования.
(2) + СН3ОН —> (CH3)2CHCOOCH3 + CH3OSi(CH3)3
(3)
В результате восстановления эфира (1)4 г-экв натрия в жид-
ком аммиаке и последующей обработки ТМХС образуется пять
48
веществ, наиболее интересным из которых является 3,3-диметил-
1,2-бис-(триметилсилокси)-циклопропан (4) [5]:
1) 4 Na-NH3
(1} 2> ™ХС >
25%
OSi(CH3)3
Н3С
3 '-Н
м
1. Birch A. J., Subb a RaO G., Advan. Org. Chem., Methods and Results,
8, 1 (1972).
2. Kaiser E. M., Synthesis, 1972, 391.
3. Rao P. N., Jacob E. J., Axelrod L. R., J. Chem. Soc. (C), 1971, 2855.
4. К u о Y.-N., Chen F., Ainsworth C., Bloomfield J. J., Chem. Comm.,
1971, 136.
5. Chen F., Ainsworth C., J. Am. Chem. Soc., 94, 4037 (1972).
9-БОРАБИЦИКЛО43,3,1]-НОНАН (9-ББН) (V, 35—36;
VI, 25—30),
Реагент представляет собой белый кристаллический порошок
с т. пл. 150—152°. Его можно хранить при комнатной темпера-
туре в атмосфере сухого N2. При работе с ним следует свести
к минимуму контакт реагента с кислородом и влагой воздуха.
Внимание! Алкилбораны могут быть пирофорны.
БОРА ТРИБРОМИД (I, 110—111; V, 37—38; VI, 30—31).
Гексабромциклопентадиен (V, 279). Опубликована улучшен-
ная методика получения гексабромциклопентадиена (2) по Уесту
обменной реакцией гексахлорциклопентадиена (1) с Б. т. в при-
сутствии трехбромистого алюминия и брома [1]:
+ ВВгз
А1Вг3,
78%
Вг
Вг
(О (2)
1. Ungefug G. A., Roberts С. W., J. Org. Chem., 38, 153 (1973).
БОРА трис-(ТРИФТОРАЦЕТАТ), B(OOCCF3)3. Мол. вес
349,88.
Получение [1, 2]. Б. т. получают из ВВгз и CF3COOH в CH2CI2
при 0°. Образующийся реагент выпадает в осадок; растворитель
3CF3COOH 4-ВВгз —> B(OOCCF3)3 + ЗНВг
удаляют в вакууме при 20°.
Удаление защитных групп в синтезе пептидов [2]. Раствор
Б. т. в трифторуксусной кислоте уже при 0° отщепляет различ-
ные N-защитные группы, снимаемые кислотами (СЬ, БОК и др.),
49
а также удаляет нитро-, тозил- или n-метоксибензильные груп-
пы, применяемые для защиты функциональных групп в боковой
цепи.
Таким образом, новый реагент по своему действию подобен
жидкой HF.
1. Gerrard W., LappertM. F., Schafferman R,, J. Chem. Soc., 1958,
3648.
2. Pless J., Bauer W., Angew. Chem., Internal. Ed., 12, 147 (1973).
БОРА ТРИФТОРИДА ДИ-н-БУТИЛЭФИРАТ,
BF3—O(CH2CH2CH2CH3)2. Мол. вес 198,04.
Формилциклопропаи. При дегидратации смеси цис и транс-
циклобутанднолов-1,2 (1) под действием Б. т. д. при 230° обра-
зуется формилциклопропан (2) с выходом 65—80% [1], дегидра-
тация этого же диола (1) л-толуолсульфокислотой приводит
к несколько меньшему выходу (66%) альдегида (2) [2]:
Г—1ОН
I__1он.
BF3- О(С4Н9)г
65-80%
—с но
(2)
I. Barnier J. Р., Champion J.. С о n i a J. М., Org. Syn., submitted 1972.
2. Conia J. M, Barnier J. P., tetrahedron Letters, 4981 (1971).
БОРА ТРИФТОРИДА ЭФИРАТ (I, 113—116; V, 38—40;
VI, 33).
/J-Глюкопиранозиды [1]. (З-Глюкопиранозиды можно получать
с хорошим выходом взаимодействием агликона с 2,3,4,6-тетра-О-
ацетил-^-п-глюкопиранозой (1) в присутствии Б. т. э. в 1,2-ди-
хлорэтане при температуре —20°. В глюкозиде сохраняется ис-
(1)
ходная ^-конфигурация глюкозной части молекулы, но конфигу-
рация гидроксильных групп агликона часто нарушается, так как
реакция протекает через стадию образования карбониевого иона.
Расщепление трет-БОК-производиых пептидов [2]. трет-Бу-
тилоксикарбонильную защитную группу обычно снимают дей-
ствием трифторуксусной кислоты или хлористого водорода. Как
недавно сообщалось, для этой цели можно использовать Б. т. э.
в ледяной уксусной кислоте или в смесях уксусной кислоты
с хлороформом. Реакция легко протекает при комнатной те^*
пературё, необходима лишь защита от влаги. Новый метод Имеет
преимущество в тех случаях, когда нежелателен растворитель
с сильно выраженными кислотными свойствами.
Внутримолекулярное присоединение диазокетонов к олефи-
нам. Эрман и Стоун [3] впервые осуществили присоединение ди-
азокетона к олефину, катализируемое кислотами. Реакция на-
шла применение в синтезе бициклических кетонов, особенно
COCHNa
(1) (2) 30% (3) 3%
в ряду сесквитерпенов. Так, при обработке Б. т. э. в течение
3 час при 0—27° диазокетон (1) дает кетоны (2) и (3) с выхо-
дами соответственно около 30 и 3%. Таким образом, новая реак-
ция оказалась проще по сравнению с обычно применяемым
методом термической циклизации в присутствии меди до цикло-
пропилтрициклического кетона и последующего его расщепле-
ния в присутствии кислоты до бициклического кетона.
Этерификация. Б. т. э. в сочетании с большим избытком
спирта является эффективным этерифицирующим агентом для
4-аминобензойной кислоты [4]. Эта смесь особенно удобна для
этерификации ненасыщенных карбоновых кислот [5].
Спирт в присутствии Б. т. э. легко этерифицирует также гете-
роциклические карбоновые кислоты [6].
Циклизация костанолида [7]. Обработка костанолида (1)
BF3- (C2Hs)2O при комнатной температуре в течение 1 —10 мин
приводит к образованию продуктов (2), (3) и (4) с выходами
U) (З^экзо-изомер (4)
(3), ЭЯЙО-изомер
соответственно 28, 37 и 1,5%. Интересно отметить, что в продук-
тах реакции оказалось соединение (4)—4а-оксициклокостано-
лид. Этот сесквитерпен, как полагают, мог появиться в резуль-
тате катализируемого ферментами присоединения воды к (1).
О-Тетрагидропираиил-2-овые производные стероидов. В ре-
зультате реакции тестостерона с 2,3-дигидропираном, катализи-
51
руемой моногидратом n-толуолсульфо кислоты, Отт и сотр. [8]
получили О-тетрагидропиранил-2-овое производное тестостерона.
Для проведения реакции потребовались три недели; выход со-
ставил 59% Если в качестве катализатора использовать Б. т. э„
выход повышается до 67%, а время реакции сокращается до
10 час [9].
1. Kuhn М, von Wartburg A.t Helv. Chim. Acta, 51, 1631 (1968),
2. H i s k e у R. G., Beacham L. M. (Ill), M a 11 V. G., Smith J. N.,
Williams E. B.f Jr., Thomas A, M., Wolters E, T., J. Org. Chem.,
36, 488 (1971).
3. Erm an W. F., Stone L. C., J. Am. Chem. Soc., 93, 2821 (1971).
4. Kadaba P. K., Carr M., Tribo M., Triplett J., Glasser A. C.,
J. Pharm. Sci., 58, 1422 (1969).
5. Kadaba P. K-, Synthesis, 1971, 316.
6. К a d a b a P. K., Synthesis, 1972, 628.
7. Jain T. C., McCloskey J. E,, Tetrahedron Letters, 1415 (1971).
8. 011 A. C., Murray M. F., Pederson R. L., J. Am. Chem. Soc,, 74, 1239
(1952).
9. Al per H., Dinkes L., Synthesis, 1972, 81.
БОРА ТРИФТОРИД—ТРИФТОРУКСУСНЫЙ АНГИДРИД.
Реакция Померанца — Фрича. По методу Померанца — Фри-
ча [1] изохннолины получают циклизацией ацеталей сс-бензаль-
аминоальдегидов (1) в присутствии конц. серной кислоты.
(1)
В одной из недавних работ сообщалось, что использование
Б. т. — Т. а. [2] повышает выход изохинолинов. Аналогичным же
образом реагент применяли для синтеза N-замещенных индолов
и бензтиофена.
1. G en s 1 er W. J., Org. React., 6, 191 (1951).
2. Bevis М. J., Forbes E. J., Naik N. N., Uff В. C., Tetrahedron, 27,
1253 (1971).
БОРА ТРИХЛОРИД (I, 116—118; V, 41; VI, 33—34).
Расщепление эфиров пространственно затрудненных карбоно-
вых кислот [1]. Эфиры пространственно затрудненных кислот
легко расщепляются при обработке Б. т. в хлористом метилене.
Например, при 0° реагент расщепляет метиловый эфир О-метил-
подокарповой кислоты (1) до О-метилподокарповой кислоты
с 90%-ным выходом. Простая эфнрная связь при этом не за-
трагивается. Аналогично из метиловых эфиров адамаитан-1-кар-
52
боновой (2) и 2,4,6-триметилбензойной кислоты с высоким вы-
ходом получаются соответствующие кислоты.
Деметилирование (V, 41). Описано селективное деметилиро-
вание метоксигруппы, находящейся в орто-положении к карбо-
нильной группе. Однако Бартон и сотр. [2] в случае некоторых
полиоксибензолов наблюдали избирательное деметилирование
метоксигруппы, находящейся в /гара-положении к карбонильной
группе. Например, единственным продуктом реакции соедине-
ния (1) с Б, т. в хлористом метилене при —80° оказалось веще-
ство (2).
(1) 702 мг
(2J 507 мг
О-Деметиленирование [3]. В определенных условиях Б. т.
способен отщеплять метилендиоксигруппу, не затрагивая мето-
ксигруппы. Так, производное изохинолииа (1) можно превратить
в диоксипроизводное (2) с выходом 78% взаимодействием с Б. т.
в хлористом метилене при 4°. Трехбромистый бор в этих усло-
виях реагирует неизбирательно.
(Б (2)
53
1. M a n c h a n d P. S., Chem. Comm., 1971, 66?-
2. Barton D. H. R., В о u 1 d L., Clive D. L. J., Magnus P. D., Hase T.,
J. Chem. Soc. (C), 1971, 2204.
3. T e i t e 1 S., O’B r i e n J., В г о s s i A., J. Org. Chem., 37, 3368 (1972).
БОРНАЯ КИСЛОТА (1,118—122; V, 42; VI, 34—35).
О-Ацилирование фенолов [1]. Прямое О-ацилирование фено-
лов катализируется смесью борной н серной кислот. Например,
фениловый эфир бензойной кислоты (1) можно получить азео-
тропной перегонкой толуольного раствора фенола и бензойной
кислоты в присутствии каталитических количеств борной и сер-
ной кислот.
о о
1. Lawrence W. W., Jr., Tetrahedron Letters, 3453 (1971).
БРОМ (VI, 35).
Циклобутаидион-1,2 [1]. Все попытки получить циклобутан-
дион-1,2 окислением легкодоступного сс-оксициклобутанона по-
терпели неудачу. Хайне получил этот дикетон бромированием
1,2-бис- (триметилсилокси) -циклобутена-1 (1):
^OSi(CH3)3
(В
OSi(CH3)3
+ 2 (CH3)3SiBr
К смеси 46 г вещества (1) и 50 мл абсолютного изопентана
в течение 20 мин при перемешивании и охлаждении до —70°
прибавляют раствор 32 г брома в 50 мл изопентана. При нагре-
вании светло-желтого раствора до —10° начинают выделяться
желтые кристаллы с т. пл. 67—68°; выход 11,7 г (70%).
Этот метод одновременно предложили н французские хи-
мики [2].
OSi(CH3)3
OSi(CH3)3
Br
OSi(CH3)3
Br
OSi(CH3)3
Homh.
.темп.
30°
200 .«.и
(2)
(a)
Они проводили реакцию сначала при —20°, затем при ком-
натной температуре и, наконец, при 30° в вакууме. Реакцию
можно контролировать с помощью ПК- и ЯМР-спектров. Выход
дикетона (2) близок к количественному.
54
1. Heine H.-G., Chem. Ber., 104, 2869 (1971).
2. С о n i a J. M., D e n i s J. M., Tetrahedron Letters, 2845 (1971).
N-БРОМАЦЕТАМИД (NBA) (I, 125—126; V, 44).
Реакция с олефинами. Воль [1] в 1919 г., упомянув в своей
статье реакцию тетраметилэтилена с NBA, высказал предполо-
жение, что эта реакция представляет собой аллильное бромиро-
вание. Вольф и Аванг [2], однако, установили, что в реакции
NBA с олефинами аллильного бромирования не происходит, а
образуется новый класс соединений — И-бромацетимино-2-бром-
алкиловые эфиры. Так, кипячение циклогексена (1) с NBA в
СС14, 8 0°
+ 2 CHjCONHBr Лм
перекись
XC=NBr
+ CH3CONH3
1
СН<
четыреххлористом углероде при УФ-облучении приводит в основ-
ном к образованию Г4-бромацетимино-2-бромциклогексилового
эфира (2) с выходом 36%. В тех же условиях из тетраметил-
этилена (3) получается соединение (4).
СНдСОКНВг |
(СНз)гС=С(СНз)2 (СНз)зС—С(СН3)2
H3CC==NBr
(4)
Реакция, по-вндимому, протекает в две стадии: через свобод-
норадикальное аутобромирование NBA с образованием N.N-ди-
бромацетамида и нонное присоединение последнего по двойной
связи.
Буфалин. На заключительных стадиях синтеза ацетилбуфа-
лина (3) 14-дегидроацетилбуфалнн (1) обрабатывают NBA
в водном ацетоне, а затем восстанавливают образовавшийся
бромгидрин (2) никелем Ренея в хлористом метилене. Ацетиль-
ОН Вг
55
N1 Йене я
Na
32^, считая на (I)
ную группу в соединении (3) гидролизуют соляной кислотой
в водном метаноле (20°, 48 час, выход 68%) [3].
1. Woh 1 А., Вег., 52, 51 (1919).
2. Wolfe S., Awang D. V. С., Can. J. Chem., 1971, 1384.
3. Sondheim er F., Wife R. L., Tetrahedron Letters, 765 (1973).
N-БРОМАЦЕТАМИД — ФТОРИСТЫЙ ВОДОРОД (I, 126;
v, 44).
Присоединение фтористого брома к ацетиленам [1]. Фтори-
стый бром присоединяется к алкинам-1 согласно правилу Мар-
ковникова:
СНз(СН2)3О=СН
CH3(CH2)3CF=CHBr
с=сн
CF=CHBr
Прн этом происходит преимущественно трбшс-присоединение.
Электроноакцепторные группы затрудняют реакцию: так,
СНзОС(С1?з)= CII и CH3OC(CF3)2C = CC1 вообще в нее не
вступают. Диметиловый эфир ацетилендикарбоновой кислоты
дает неидентифицируемые продукты.
1. Dear R. Е. A., J. Org. Chem., 35, 1703 (1970).
БРОМИСТОВОДОРОДНАЯ КИСЛОТА (I, 128—131; V, 45—
46; VI, 35—36).
Тризамещениые олефины [1]. При взаимодействии с 48%-ной
Б. к. при 0° в течение 1 час оксиран (1) превращается в (Е)-
5-бром-2-метилпентен-2-ол-1 (2а) с выходом 81% (чистого),
А сн:
н О'
(1)
(Za^X = Вг
(2б^Х я I
а при обработке (1) смесью иодида натрия с ацетатом натрия
в смеси уксусной и пропионовой кислот (IV, 89—90) при —18°
в течение 30 мин образуется соединение (26). Оксиран (1) по-
лучали реакцией метилциклопропилкетона и диметилсульфоний-
метилида (1,339—341; V, 139—141; VI, 91; этот том) в ДМСО
при —5° в течение 20 мин и при 25° в течение 1 час.
Изомерный оксиран (3) в эту реакцию не вступает, однако
с безводным бромидом цинка (V, 547—548) в эфире при 0° в те-
чение 3 час он образует (Е)-5-бром-З-метил пентен-2-ол-1 (4)
с выходом 73% (чистый):
Следует отметить, что 2- и 3-метилбутен-2-олы-1 являются
концевыми группами цепи изопреноидов.
Деметилирование [2]. Деметилирование 4,5-диметоксибензо-
циклобутендиона-1,2 (1) было с успехом осуществлено дейст-
вием 48°/о-ной Б. к. Этот дикетон является бензологом квадрат-
ной кислоты. Он дает положительную пробу с хлорным же-
лезом.
сн3о
сн3о
(2)
1. Nakamura Н., Yamamoto Н., Nozakl Н., Tetrahedron Letters, 111
(1973).
2. McOmie J. F. W., Perry D. FL, J. C. S. Chem. Comm., 1973, 248.
БРОМИСТЫЙ ВОДОРОД
Получение циклобутаноиов реакцией расширения цикла 1-ви-
ннлциклопропанолов [1]. 1-Винилциклопропанол, полученный
кипячением соединения (1) в ТГФ с винилмагнийбромндом, при
взаимодействии с избытком НВг в хлористом метилене пере-
группировывается в 2-метилцикло бутанов (3). Перегруппировка,
вероятно, включает промежуточную стадию образования ка-
тиона циклопропилкарбония. Расширение цикла происходит так-
же при нагревании (приблизительно до 100°) [2].
но осгн5
/\ 64%
(1}
67
В циклопропйноле (2) расширение цикла происходит под
действием и других электрофильных агентов, например при об-
работке надбензойной кислотой в эфире он превращается в 2-ок-
симетилциклобутанон.
1. Wasserman Н. И., С о с h о у R. Е., Baird М. S., J. Am. Chem. Soc.,
91, 2375 (1969).
2. S a 1 a u n J. R., С о n i a J. M., Tetrahedron Letters, 2849 (1972).
БРОМИСТЫЙ ВОДОРОД БЕЗВОДНЫЙ (I, 131 — 132).
Перегруппировка. Под действием безводного Б. в. в диметил-
формамиде при 130° 3-метоксиэстратриен-1,3,5( 10) -он-17 (1)
в течение 6 час с хорошим выходом перегруппировывается
в 3-окси-17р-метил-14р-гонапентаен-1,3,5(10),6,8 (2) [1]:
Присоединение (1,4 и 1,6) к 4-метнл-о-бензохинону [2].
Хлористый водород присоединяется аналогично.
1. Hilscher J.-C., Chem. Вег., 104, 2341 (1971).
2. Н о г n е г L., В u г g er Т., Ann., 708, 105 (1967).
БРОМИСТЫЙ ВОДОРОД (I, 131 — 132)— ТРИФТОРУК-
СУСНАЯ КИСЛОТА (V, 459—460).
Рассматривается скорость отщепления кислотами различной
концентрации важнейших N- и С-защитных групп пептидов в ук-
сусной и трифторуксусной кислотах [1].
1. Losse G., Zeidler D., Grieshaber T., Ann., 715, 196 (1968).
N-БРОМПОЛИМАЛЕИНИМИД (МБПМИ),
—сн—сн—
I I
58
Полималеинимид получают свободнорадикальной полимери-
зацией малеинимида в присутствии дивинилбензола (2,5—5%)
в качестве сшивающего агента. ЫБПМИ образуется при броми-
ровании полимера в водном растворе гидроокиси натрия.
Бромирование [1]. Бромирование кумола (1) с помощью
ЫБПМИ в кипящем четыреххлористом углероде (перекись бен-
зоила) дает смесь трех продуктов:
В отличие от этого применение N-бромсукцинимида приводит
к образованию соединений (5) и (6), причем в присутствии
большого избытка NEC дибромид (6) становится основным про-
дуктом реакции. Различие в действии NBIIMH и NEC наблю-
далось также и в случае других алкилбензолов. Бромирование
NEC в полярном растворителе сходно с действием ЫБПМИ
в четыреххлористом углероде. Качальский относит необычные
реакции NBIIMH за счет его полимерной структуры. Ср. с N-
Хлорполималеинимидом, этот том.
1. Yaroslavsky С., Patchornik A., Katchalski Е., Tetrahedron
Letters, 3629 (1970).
N-БРОМСУКЦИНИМИД (I, 134—137; V, 47—49; VI, 36—
37).
Бромноватистая кислота, HOBr (I, 136). Эриксон и Ким [1]
получали бромноватистую кислоту in situ из NBC н воды. С ме-
тнленциклобутаном (1) она образует 1-(бромметил)-циклобута-
нол (2) примерно 90%-иой чистоты с 78%-ным выходом, т. е.
реакция региоспецифична.
ЫБС,Н,О
--------->
78^
(2)
69
Японские химики [2] сообщили, что с бромноватистой кисло-
той, генерированной из NBC и воды, производное 4Л,5Л-дидегид-
роадеиозина (3) в качестве основного продукта дает соедине-
ние (4). Образование последнего является первым примером
N,^-циклизации, сопровождающейся одновременным расщепле-
нием кольца азотистого основания пуринового нуклеозида.
(5) 12$
Селективное эпоксидирование. Ван Тамелен и Курфи [3] от-
мечали, что NEC в водном полярном растворителе (1,2-димет-
оксиэтане) генерирует бромноватистую кислоту, которая про-
являет некоторую избирательность в реакциях с соединениями
с двойной связью. Этот метод был использован для селектив-
ного эпоксидирования концевой двойной связи в фарнезилаце-
тате (1) с образованием 10,11-эпоксифарнезилацетата (4) [4].
Все подробности превращения описал Ханцлик [5]. Под дей-
ствием NEC во влажном трет-бутаноле фарнезилацетат (смесь
транс, транс- и цис, транс-изомеров или чистый транс, транс-
фарнезилацетат) дает бромгидрин (2) с выходом около 65%,
ео
который при обработке К2СО3 в метаноле переходит в эпоксид
(3). Ацетилирование последнего приводит к 10,11-эпоксифарне-
зилацетату (4) с общим выходом около 60%.
Этот метод, лежащий в основе биогенеза, был использован
в синтезе ди- и тритерпенов [6], а также в синтезе ювенильного
гормона Cecropia [7].
Аллильное окисление (VI, 36—37). Как сообщал Томсон [12],
единственным подходящим растворителем для аллильного окис-
ления является диоксан, причем реакция идет с высоким выхо-
дом только в тех случаях, когда двойная связь пространственно
сильно затруднена. Например, 4,4-диметилхолестерилацетат дает
7-оксопроизводное с количественным выходом, а холестерилаце-
тат— 7-оксохолестерилацетат с выходом 80%. Холестен-1, холе-
стен-2 и холестен-3, напротив, образуют лишь бромгидрнны, ди-
бромнды и продукты их дальнейших превращений, но не дают
продуктов аллильного окисления. Применение карбоната каль-
ция не обязательно.
Окисление ацетиленов [8]. Прибавление к раствору дифенил-
ацетилена в ДМСО 2 же NEC приводит к образованию бензнла
с 98%-ным выходом. Реакцию катализирует только NEC [эле-
ментарный бром и дибромид бромгидрата пирролидона-2
NEC (ДМСО)
98$ >
(VI, 211) оказались неэффективными,] а в качестве раствори-
теля пригоден лишь ДМСО.
Синтез караханаенона [9]. Ионная реакция NEC (в СС14 при
комнатной температуре) с третичными спиртами, содержащими
двойную связь в у-положении, приводит к образованию а-бром-
тетрагидрофуранов. Именно эту реакцию применили на одной
из стадий синтеза составной части масла хмеля — караханаено-
на (4), исходя из линалоола (1). Реакция линалоола с NEC
дает 2-метил-2-винил-5-(1-бром-1-метилэтил) -тетрагидрофуран
(2) с выходом 85%, дегидрогалогенирование которого под дей-
ствием коллидина при 110° приводит к аллилвиниловому эфиру
(1)
NBC, ССЦ, 20s
---------J--->
85%
Коллидин , П0ъ
----------’---->
(П
сн3
S2C*
(общий
считая на (1)
61
(3), тотчас же подвергающемуся [3,3]-сигматропной перегруппи-
ровке в караханаенон (4); общий выход 62%, считая на (1).
Реакция с триалкилборанами. В 1971 г. Лэйн и Браун [10]
предложили изящный метод синтеза сильно разветвленных спир-
тов, исходя из триалкилборанов. Например, бромирование три-
этилборана (1) бромом в присутствии воды при УФ-облучении,
а затем окисление перекисью водорода в щелочной среде дает
З-метилпентанол-З (2) с 88%-ным выходом. Реакция протекает
сн3сн2 сн3сн2
2Bra, hv | Н2О2, ОН" I
(С2н5)3в > СН3СВ(ОН)2 -------------> СН3С-ОН
СН3СН2 СН3СН2
(1) (2)
в несколько стадий. Первая стадия — свободнорадикальное бро-
мирование триалкилборана в «-положение с образованием
а-бромтриалкилборана (а), который в присутствии воды гладко
перегруппировывается в диалкилборную кислоту (б). Кислота
в свою очередь тоже бромируется в «-положение (в) и в присут-
ствии воды перегруппировывается в алкилборную кислоту (г).
Окисление последней приводит к спирту (2).
Вг2) A-v
(С2Н5)3В -------> СН3СНВ(С2
Вг
(О (а)
С2Н5
I Н2О
—> сн3с—в—с2н5 —-
I 1
Вг ОН J
(В)
сгн3
Н2О | Вг2, hv
Н5)2 —> CH3CHBC2HS -------------
—НВг ।
он
(б)
ад
I Н2О2. ОН"
Н3С—С—В(ОН)2 ------------> (2)
C2HS
(г)
Для успешного проведения а-бромирования и перегруппировки
и подавления полибромирования бром необходимо прибавлять
медленно. Брауи и Ямамото [11] сообщили, что применение
NBC в присутствии воды ведет к увеличению выхода реакции
(1)--*-(2) до 97%- Увеличение выхода связано с тем, что при
взаимодействии NBC с бромистым водородом, образующимся
в процессе реакции, бром, как известно, выделяется в малых
+ НВг
концентрациях. Удовлетворительным источником брома может
служить также бромат натрия (II, 388—389).
62
Другие примерь!:
CH2CH2Ctf3
(»-С,Н,),В 2 NSC> СН,(СНг),С(СНг),СН,
in
5" Пропилнона нол— 5
I. Erickson К. L., Kim К., J. Org. Chem., 36, 2915 (1971).
2. Sasaki Т., Min a mo to К., Hat tori К., J. Am. Chem. Soc., 95, 1350
(1973).
3. v a n Tamelen E. E., Curphey T. J., Tetrahedron Letters, 121 (1962).
4. van Tamelen E. E„ Storni A., Hessler E. J., Schwartz M.,
J. Am. Chem. Soc., 85, 3295 (1963).
5. H a n z 11 к R. P., Org. Syn., submitted 1973.
6. v a n Tamelen E. E., Accts. Chem. Res., 1. Ill (1968); van Tame-
len E. E. et al., J. Am. Chem. Soc., 94, 8225, 8228, 8229 (1972).
7. Corey E. J., Katzenellenbogen J. A., Gilman N. W., Ro-
man S. A, Erickson B. W., J. Am. Chem. Soc., 90, 5618 (1968);
van Tamelen E. E., McCormick J. P., J, Am. Chem. Soc., 92, 737
(1970).
8. Wolfe S., Pilgrim W. R., Garrard T. F., Chamberlain P., Can.
J. Chem., 49, 1099 (1971).
9. Demo le E., E n g gl s t P„ Helv. Chim. Acta, 54, 456 (1971).
10. Lane C. F., Brown H. C., J. Am. Chem. Soc., 93, 1025 (1971).
11. Brown H. C., Yamamoto Y., Synthesis, 1972, 699.
12. Thomson J. В., частное сообщение.
БРОМЦИАН (I, 138—141; V, 52—53).
транс-Олефины. Цвейфель и сотр. [1] предложили удобный
метод синтеза транс-ди- и транс-тризамещенных олефинов из ал-
кинов. Сначала алкины гидроборируют диалкилборанами в ТГФ
до диалкилвинилборанов. После удаления ТГФ к остатку при 0°
добавляют хлористый метилен и Б. (или иодциан), перемеши-
вают 2 час, затем добавляют гидроокись натрия, а олефин
экстрагируют пентаном и очищают перегонкой. Выходы транс-
олефинов колеблются в пределах 60—75%.
Вт
R..-C —С---B-CSN =
/ L ।
Н R1 R
R.. .-н
63
1. Zweifel G., Fisher R. P., Snow J. T,. Whitney С. C. J. Am. Chem.
Soc., 94, 6560 (1972).
БУТАДИЕНА-1,3 1,2-ОКИСЬ, CH2=CHCH-CH2. Мол. вес
О
70,09, т. кип. 65—66°.
Гомологизация триалкилбораиами [1]. В присутствии кисло-
рода или других инициаторов свободнорадикальных реакций
•триалкилбораны и Б. о. вступают в реакцию четырехуглеродной
гомологизации, давая 4-алкилбутен-2-олы-1, например
1) ог
2) н2о
(С3Н&)3В + СН2=СНСН—СН2 С2Н5СН3СН=СНСН2ОН + (C2HS)3BOH
\ 68 %
о
(избыток) 89% (транс-)
Наиболее подходящими растворителями для реакции служат
бензол и эфир.
1. Suzuki A., Miyaura N., It oh М., Brown Н. С., Holland G. W.,
Negishi E., J. Am. Chem. Soc., 93, 2792 (1971).
я-БУТИЛАМИН, СНз(СН2)зМН2. Мол. вес 73,14, т. кип. 78°,
п™ 1,4015.
Арилацетилены. При взаимодействии Б. в эфире при комнат-
ной температуре с 5,5-дизамещенными 3-нитрозооксазолидоиа-
ми-2 (1) (получение см. II, 447—448), у которых хотя бы один
из заместителей при С5 — арильная группа, с выходом, близким
к количественному, образуются арилацетилены [1, 2]. Этот метод
использовали также для получения 2-этинилтиофена [3].
\no
CH3(CH2)3NH2 RjC = CR2 + СО2 4- Н2О 4- Na
99-100%
(1) Rt или R2 =С6Н5
О других превращениях соединений (1) см. VI, 151 —152.
1. Hogan Н. Р., S е eha fer J., J. Org. Chem., 37, 4466 (1972).
2. Newman M. S., Lee L. F., J. Org. Chem., 37, 4468 (1972).
3. Patrick T. B., Disher J. M., Probst W. J., J. Org. Chem., 37, 4467
(1972).
трет-БУТИЛAT КАЛИЯ (I, 147—168; V, 56—66; VI, 38—40).
64
Дегидробромирование. Чабаттопи и сотр. [1] разработали
следующий путь синтеза борфторида три-трет-бутилциклопропе-
нилия. Сначала синтезируют по Гриньяру динеопентил кетон (1)
(см. приведенную ниже схему), переводят его в а,а'-дибромпро-
изводное (2), которое подвергают затем двойному дегидробро-
мированию с помощью Б, к. Образовавшийся циклопропенон (3)
далее вводят в реакцию с раствором трет-бутиллития в пентане.
Реакционную смесь разлагают водой, пентановый слой промы-
вают, сушат и упаривают на роторном испарителе. Оставшееся
бледно-желтое масло растворяют в эфире и обрабатывают при
о
(CHjjCCHjMgCi +(СН.),ССНгСОС1 ------------» MgClj + (CHjJjCCHsCCHzCfCHsJj
78-77%
Cl)
о
(СН^ССЩССН^СН,), + 2 Вг, —иг.->
85%
(1)
ВгОВг
I II I ,
(СН,),С6НССНС(СНз), + 2 НВг
(2)
Вг О Вг
I II I
{СН^ССНС СНС{СН,Ь + 2 (сносок —------------>
79“83% (сн,),
(2) (3)
° ) (CH,),CU__________
Я 2) Н2О &
3)HBF(, АсгО, 68-79% /С\
(СН, fee с (СН,), (СНз ),С С (СН,),
(э) (4)
охлаждении до 0° и интенсивном перемешивании магнитной ме-
шалкой свежеприготовленным 10%-ным раствором борфтористо-
водородной кислоты в уксусном ангидриде (приготовлен в ат-
мосфере азота при —40°). После 20-минутного перемешивания
получившейся суспензии белый осадок отсасывают на пористом
стеклянном фильтре со средним размером пор и тщательно про-
мывают тремя порциями эфира по 75 мл каждая. Продукт рас-
творяют в минимальном количестве кипящего ацетона (около
300 мл) и охлаждают раствор в холодильнике примерно до
—25°. При этом выпадает 15,3 г борфторида три-трет-бутилцик-
лопропенилия в виде белых игл, который отфильтровывают и
промывают эфиром. Концентрирование маточных растворов дает
еще две порции чистого вещества. Общий выход составляет 24—
28 г (68—79%).
Дегидрохлорирование. Дегидрохлорирование 1,6,7,7-тетра-
хлор-2,5-дифенилбицикло-[4,1,0]-гептена-3 (1) небольшим избыт-
ком Б. к. в ТГФ приводит к 7,7-дихлор-2,5-дифенилбензоцикло-
3 Зак. 596
65
пропену (2) с выходом около 80%. Обработка соединения (1)
гидроокисью калия в метаноле дает ортоэфир (3), который об-
разуется, по-видимому, через стадию циклопропена (2) в ре-
зультате сольволитического расщепления трехчленного цикла
[2]. Дегидрохлорирование бензобициклогептена (4) действием
CI
(4)
Б. к. в ТГФ ведет к образованию 1,1-дихлор-2,7-дифен илцикло-
пропа-[^]-нафталииа (5); в этом случае выход составляет лишь
25% [3].
2,3-Дихлорнорборнен-2 (8) синтезировали в три стадии [4].
Взаимодействием циклопентадиена (IV, 200—201) и трихлорэти-
лена по реакции Дильса — Альдера получали 5,5,6-трихлорнор-
борнен-2 (6), который затем гидрировали 5% Pd/C до трихлор-
норборнана (7). Дегидрохлорирование последнего с помощью
Б. к. в тре'г-бутаноле давало дихлориорборнен (8) с невысоким
общим выходом в основном из-за того, что образовавшийся на
+ CTCH=.-GC1,
19 0°
?2-24^>
Н2
Pd/C
84
(6)
66
первой стадии трихлорнорборнен (6) вновь реагирует с цикло-
пентадиеном по реакции Дильса — Альдера.
Шоберт и Ханак [5] описали простой метод синтеза циклопро-
пилацетилена (11) из метилциклопропилкетона (9). Взаимодей-
ствием с очищенным пятихлористым фосфором [6] в четыреххло-
ристом углероде кетон переводят в дихлорпроизводное (10), ко-
торое затем дегидрогалогенируют с помощью Б. к. в ДМСО.
Хлор-, бром- и иодциклооктатетраены [7]. ^ис-7,8-Дигалоген-
циклооктатриены-1,3,5, полученные хлорированием или бромиро-
ванием циклооктатетраена при —60° в хлористом метилене, де-
гидрогалогенируют in situ действием Б. к. при —45°; выходы
бром- и хлорциклооктатетраеиов составляют 85 и 74—83% со-
ответственно. Иодциклооктатетраен можно получить из цикло-
октатетраениллития и иода. Описаны удобные методы получе-
ния метокси-, фенокси-, ацетокси-, метил- и фенилциклоокта-
тетраенов.
Циклопропилкетоиы [8]. Циклопропилкетоны образуются о
выходом 42—59% при кипячении солей 3-ацилоксипропилфос-
фония с Б. к. в трет-бутаноле. Например, бромид 3-ацетоксипро-
пилтрифеиилфосфония (1), полученный с выходом 97% из бро-
мида 3-бромпропилтрифенилфосфоиия и ацетата натрия в смеси
ацетон—вода (4:1), превращается в метилциклопропилкетон
(2) с выходом 49%. Авторы предлагают следующую схему реак-
ции:
3*
67
С> + КОС(СН3)3 п о
CH3COCH2CHzCH2P(C6H5)3Br' -Д°ЗЯН^> СН3СОСН2СН2СНР(С6Н5)3
(1) L
о
(G6HS)3P ^Сн2
сн-сн2
I
сн3с=о
о
(ОД^Р^ ^СН2
СН3ССН—СИ,
II
о J
СН3ССН -----снг
(2)
СН3С = О
+ (С6Н5)3РО
Бензоциклопропен [9]. Предложен удобный двухстадийный
синтез бензоциклопропена (3). На первой стадии из циклогекса-
диена-1,4 (1) и дихлоркарбена (генерированного из хлороформа
и Б. к.) получают 7,7-дихлорбицикло-[4,1,0]-гептен-3 (2) с выхо-
дом 41—44%, который обрабатывают Б. к. в ДМСО сначала
(П
wpem-BuOK
jj СНСЧ
' 41-44$
трет- ВиОК
С1 ДМСО
с1 37$
(3)
при 15—20°, затем при комнатной температуре в течение 25 мин.
Бензоциклопропен (3) образуется с 37%-ным выходом в резуль-
тате катализируемой основанием реакции дегидрохлорирова-
ния — изомеризации.
Винилалкилиденцихлопропаиы [10]. Винилалкилидеицикло-
пропаны можно получить дегидрохлорированием геж-дихлорцик-
лопропанов с помощью Б. к. в ДМСО. Простейшим примером
сн.
трет- ВиОК
ДМСО
-НС1
-НС1
сн2сн3
---~>
62$
(21
68
служит образование винилметиленциклопропана (2) из 1,1-ди-
хлор-2-этил-З-метилциклопропана (1), вероятно, через стадии,
приведенные на схеме в квадратных скобках.
Бутатриены [11]. При взаимодействии олефинов и дихлор-
карбена с последующей обработкой Б. к. в трет-бутаноле (или
с метилатом натрия в ДМСО) образуются бутатриены с выхо-
дами в пределах 50—90%:
R1 R2C =С HCHR3 R4 —R1 R2 с — СНС HR3 R4--
с
с/ ХС1
R1 R2с = С =С = СR3R4
9-Оксабицикло-[ЗД1]-ионеи~1 (3) [12]. Этот алкен с двойной
связью в голове моста синтезируют гидроборированием — окис-
лением циклооктадиена-1,5 до цис-циклооктандиола-1,5, кото-
рый далее окисляют реагентом Джонса до 1-окси-9-оксаби-
цикло-[3,3,1]-нонана (1) с общим выходом 49%. Попытки полу-
чить производные соединения (1), как правило, приводили
к производным оксикетона (4), однако в результате реакции со-
единения (1) с метансульфохлоридом в присутствии триэтил-
амина с хорошим выходом образуется мезилат (2). Реакция по-
(4)
следнего с Б. к. в трет-бутаноле при 80° приводит к элиминиро-
ванию метансульфокислоты и образованию соединения (3)
с 32%-ным выходом.
Изомеризация непредельных соединений (I, 151 —153; V, 56—
57; VI, 39). Реакция металлилхлорида (1) с амидом натрия в
кипящем диоксане или ди-я-бутиловом эфире приводит к обра-
зованию смеси метиленциклопропана (2) и 1-метилциклопро-
пена (3). Обработка этой смеси Б. к. в ДМСО дает метилен-
циклопропаи 98,3—98,6%-ной чистоты (ГЖХ) [13].
ХН3
нгс=с
СН2С1
(1)
NaNHz
-NH3, -NaCl
75%
(2)
(зр
/дмсо
(2)
70%
ЙО
На второй стадии синтеза [18]-аннулена [14] 35 г неочищен-
ного циклооктадекагексаина (4), полученного на первой стадии,
растворяют в 800 мл бензола в 2-литровой круглодониой колбе,
снабженной обратным холодильником с хлоркальциевой труб-
кой, и нагревают до кипения на водяной бане. Затем прибавляют
раствор Б. к., приготовленный кипячением 44 г калия в 1 л су-
хого трет-бутилового спирта в атмосфере азота до полного рас-
творения металла, и кипятят реакционную смесь в течение
30 мин\ за это время три тройные связи изомеризуются в диено-
вые группировки. Обработка реакционной смеси весьма тру-
доемка и включает хроматографические методы.
(сндсок
(СНДСОН
Миграция метиленовой группы [15]. Обработка сульфониевой
соли (1) Б. к. в 1,2-диметоксиэтане приводит к внутримолеку-
лярной миграции метиленовой группы с образованием цикло-
пропильного соединения (2) с выходом >95%:
Окись циклопентена и окись циклогексена [16]. При взаимо-
действии тра/<с-2-оксициклопентилмеркурхлорида (1а) и транс-
2-оксициклогексилмеркурхлорида (16) с Б. к. в диглиме с высо-
ким выходом образуются окиси циклопентена (2а) и циклогек-
сена (26):
он
JL ,.нес1 кос(снэ)3
' ] 90-120° „
----(снг)п 70-95%
(1)
а) И = 1
б) П = 2
(СНг)д
(2)
al Д = 1
б) П = 2
70
Соответствующее циклогептилы-юе производное (1s), (п = 3)
превращается в основном в циклогептанои (вы.ход 70%).
^-Оксидитиокоричиые кислоты [17]. Б. к.— наиболее под-
ходящее основание для синтеза p-оксидитиокоричных кислот (1)
реакцией кетонов с CS2.
сзг
АгССН,
Н
о
КОС(СНЭ)3 _
. ArCCHac-sK+ KQC№4
II II
О S
/S н о+
---> А г С СН =С --------->
4 S" 60-90%
2К+_
Нх ZSH
,c-cv
ArC S
ЧО-Н--'
Действием бисульфата тетра-я-бутнламмония в присутствии
NaOCH3 р-оксидитиокоричные кислоты (1) можно перевести
в аммониевые соли; алкилирование полученных солей приводит
к эфирам дитиокислот (2) с высоким выходом. Меркаптали (3)
получают с выходом 70—88% взаимодействием эфиров (2)
с этилатом таллия (V, 558—564) и галогеналкилом. Если один
(О
(«-Bu)4NOH‘
Нх zsS(^-Bli)4
АгС^ ---RX >
ХОН J 50-9 5%
Нч XSR
^С —
Ar —С S
хо-н-'‘
(2)
TIOC2HS
(2)-----
70-88%
нс-сн,
СН3— HCU 4S Нагревание
нс=с -•- —>
Ar-Сб XSCHj
О
СН = СНг
н3с-нсч /SCH:
нс-с
/
Аг-С^ S
О
(5)
из заместителей у атома серы в меркаптале (3)— аллильная или
пропаргильная группировка, то наблюдается кляйзеновская пе-
регруппировка (4)->(5).
Расщепление нуклеозидов [18]. Действие Б. к. в диоксане
позволяет отщепить пуриновые и пиримидиновые основания от
производных аденозина, цитидина п уридина.
71
I. C i a b a 11 о n i J., Nathan E. C., Feiring A. E., К о c i e n s к i P, J.,
Org. Syn., submitted 1971.
2. Hal ton В., M i I s о m P. J., Chem. Conun., 1971, 814.
3. Browne A. R., Halton B., J. C. S. Chem. Comm., 1972, 1341.
4. NagendrappaG., Org. Syn., submitted 1973.
'5. Schoberth W., Hanack M., Synthesis, 1972, 703.
6. Отт Э., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 547.
7. G a steiger J,, Gream G. Е., Huis gen R., Konz W. E. Chem. Ber.,
104, 2412 (1971).
8. Schweizer E. E.. Greasy W. S., J. Org. Chem,, 36, 2379 (1971).
9. В i 11 u p s W. E„ Blakeney A. J., Chow W. Y., Chem. Comm., 1971,
1461; procedure submitted to Org. Syn., 1972.
10. Billups W. E., Shields T. C„ Chow W. Y., Deno N. C., J. Org.
Chem., 37, 3676 (1972).
11. Kajigaeshi S., Kuroda N,, Matsumoto G., Wada E., Naga-
sh i m a A., Tetrahedron Letters, 4887 (1971).
12. Quinn С. B., Wiseman J. R., J. Am. Chem. Soc., 95, 1342 (1973).
13. Koster R., Arora S., Binger P., Synthesis, 322 (1971).
14. Stoke! K, S о n d h e i m e r F., Org. Syn., submitted 1971.
15. Matthews R. S., Meteyer T. E., Chem. Comm., 1971, 1576.
16. Kretchmer R. A., Conrad R. A., Mihelich E. D., J. Org. Chem.,
38, 1251 (1973).
17. Larsson F. С. V., Lawesson S.-О., Tetrahedron, 28, 5341 (1972).
18. Fol Iman FL, Tetrahedron Letters, 397 (1973).
грет-БУТИЛГИПОХЛОРИТ (I, 169—174; V, 67; VI, 40).
Хлорирование амииохинонов [I]. 2-Аминохиноны-1,4 (1) се-
лективно хлорируются в течение нескольких минут 1 же Б.
в эфирном или бензольном растворе при комнатной температуре.
о
о
В)
(СНЭ)3СОС1
8 0-9 6%
О
о
Хлорирование происходит исключительно в положение, соседнее
с аминогруппой. Хиноны, не содержащие аминогрупп, в эту ре-
акцию не вступают.
Азоалкаиы. Тимберлак и сотр. [2] рекомендуют следующий
метод синтеза азоалканов (4) из ГфГФ-диалкилсульфамидов (1)
(о получении см. [3]). Суммарные выходы составляют 35—85%.
пентан
NaH/эфир - -----------
RNHSO.NHR --------------> RNHSO2NRNa+ ЮН3)зСОС1
(1)
RN---NR —> RN=NR
t3) (4)
Реакция с 1 ;Тд^и)Кс.о-2-диазо-1,2-дип1дрофеналеном [4].
6-Метоксилирование производных пенициллина. Окисление
ангидропенициллина (1) действием Б. в метаноле в присутствии
пербората натрия (II, 421) в качестве буфера приводит к обра-
зованию 6-метоксипроизводного (2) с почти количественным вы-
ходом [5]. Атом серы тиазолидина можно защитить превраще-
нием в сульфон или сульфоксид.
сн?он
NaBO}
(г)
6-Метоксипенициллины можно получить и другим'путем [6].
При взаимодействии соединения (3) с метилатом лития в ТГФ
при —80° образуется анион амида (в), который после обработки
Б., а затем уксусной кислотой дает метоксипроизводное (4)
с выходом 70%.
73
Ij (CH3)3COC1/CH5OH
2) HOAc
70%
О CH,
(4)
Этот же метод применим для 7а~метоксилирования цефало-
споринов. Так, из соединения (5) получают метоксипроизводное
(6) с выходом 73%.
cooch2c6h4no2
(5) (6)
1. Moore И. W., С a j i р е G., Synthesis, 1973, 49.
2. Timberlake J. W., Hodges M. L., В e 11 e r t о n K., Synthesis, 1972,
632.
3. Ohme R., Preuschhoff H., Ann., 713, 74 (1968); Stowell J. C.,
J. Org. Chem., 32, 2360 (1967).
4. Regitz M., Adolph H.-G., Ann., 723, 47 (1969).
5. Baldwin J. E., Urban F. J., Cooper R. D. G., Jose F. L., J. Am.
Chem. Soc., 95, 2401 (1973); см. также, Firestone R. A., Christen-
sen B. G., J. Org. Chem., 28, 1437 (1973).
6. Koppel G. A., Koehler R. E„ J. Am. Chem. Soc., 95, 2403 (1973).
«-БУТИЛЛИТИЙ (I, 174—175, V, 68—71).
Присоединение к альдегидам и кетонам [1]. При добавлении
раствора карбонильного соединения в гексане или эфире к ли-
тиевому реагенту при —78° с максимальными выходами обра-
зуются продукты присоединения Б. к альдегидам и кетонам и
лишь следы продуктов восстановления или енолизации. Неено-
лизующиеся альдегиды и кетоны дают спирты с количествен-
ным выходом. При проведении реакции с трет-бутиллитием (I,
74
- R\ /OLi -
C
- R'/ \i-Bu-
С=0 + «-BuLi
H2O
75-90%
R/ /ОН
C
RZ// ^w-Bu
175—176) в тех же условиях выходы спиртов несколько сни-
жаются.
Десульфуризация эписульфидов. Десульфуризация эписуль-
фидов цис- и транс-бутенов-2 под действием Б. происходит с пол-
ным сохранением конфигурации с образованием соответствую-
щих цис- и транс-бутенов-2. Трост [2] высказал предположение,
что образуется промежуточное сульфурановое соединение (а).
Десульфуризация эписульфидов с помощью нонакарбонила
железа (II, 5—6; V, 202—203; VI, 123) не столь стереоспеци-
фнчна; потерю стереоспецифичности (2—6%) можно отнести за
счет одновременной изомеризации олефнна.
«^-Ненасыщенные сульфоны [3]. Б, в ТГФ при —78° генери-
рует из эфира сульфонометилфосфоновой кислоты (1) анион, ко-
торый реагирует с алифатическими и ароматическими альдеги-
дами и кетонами, давая алкилиденсульфоны (2), выход 70—97%.
9 1) h-BliLI, ТГФ, -78е
// 7) R1R2C=O
(C3H5O)2PCH.,SO3R------------------> R’R-sC=CHSO2R
(I), R = CH3, п-С1С6Щ (2)
Ранее в этой реакции, называемой реакцией Хорнера — Вит-
тита [4], использовали гидрид или метилат натрия и область ее
применения ограничивалась лишь синтезом арилиденсульфонов.
О реакции «^-ненасыщенных сульфонов с диалкилмедьли-
тием см. Диметилмедьлитий в этом томе.
1,3-Дегидроадамантан [5]. Реакция 1,3-дибромадамантана (1)
с Б. в гексаметаполе приводит к образованию 1,3-дегидроада-.
маитана (2), который можно выделить из реакционной смесн
методом препаративной ГЖХ или возгонкой. Два атома угле-
к -Bu Li
ГМТФК
(2)
75
рода циклопропильного кольца, находящиеся в голове моста,
«инвертированы», т. е. все соединенные с ними атомы располо-
жены внутри полусферы, описанной вокруг каждого из атомов
в голове моста. Углерод-углеродная связь в положении 1,3 цик-
лопропановой системы необычайно длинная (1,64 А), и именно
поэтому энергия напряжения в 1,3-дегидроадамантане не осо-
бенно высока.
Этим же методом, исходя из 1,3,5-трибромадамантана, ка-
надские авторы [6] получили разнообразные 5-замещенные
1,3-дегидроадамантаны.
Избирательное металлирование метилпиридииов и метилхи-
иолинов [7]. Взаимодействие 2,4,6-триметилпиридина (1) с Б.
в смеси эфир — гексан приводит к образованию литиевого про-
изводного по 2-метильной группе (2), которое метилируется
иодистым метилом до диметилэтилпиридина (3). Металлирова-
ние амидом калия или натрия в жидком аммиаке или диизопро-
пиламидом лития в смеси эфир — гексан ведет к производным
щелочных металлов по 4-метильной группе (4), которые метили-
руются иодистым метилом до диметилэтилпиридина (5). По-
скольку метильная группа в положении 4 обладает более кис-
лым характером, чем в положении 2, металлирование колли-
дина (1) с образованием металлического производного (4) не
является неожиданностью. Кайзер приписал неожиданное на-
правление реакции коллидина (1) с Б., приводящее к образова-
нию изомера (2), координации литиевого катиона по азоту
пиридинового цикла, в связи с чем обладающая основным ха-
рактером бутильная группа оказывается в непосредственной
близости от метила в положении 2. В той же работе сообщается
об избирательном металлировании метилированных хинолинов.
Металлические производные (2) и (4), помимо галогеналкилов,
реагируют и с другими электрофильными агентами, в частности
с альдегидами и кетонами.
0)
(5)
се, /3-Ненасыщенные альдегиды. Кори и Терашима [8] разра-
ботали простой метод синтеза «.^-ненасыщенных альдегидов из
пропаргиловых спиртов. Например, октин-2-ол-1 переводят обыч-
ным путем в тетрагидропираниловый эфир (1) и металлируют
его Б. (1,05же) в ТГФ при —25° в атмосфере аргона в течение
2,5 час. Образовавшееся литиевое производное (2) разлагают
смесью метанола со льдом в присутствии небольшого количества
карбоната калия, получая смесь аллена (3) и исходного ацети-
лена (1) в отношении 70 : 30. При обработке этой смеси смесью
н-BtiLl ;
С5НцС^ССН2ОТГП ----------> [СаНнС^ССНОТПЖ1 ---------------
(И (2)
|снгон—н2о
НОАс, Н2О.
ТГФ
С5НцСН=СНСНО -с------------ С5НцСН=С==СНОТГП-[-(1) --------
(4), транс+цис (3)
|n-TsOH
С5Н11Ч уН
хс--с/
ц/ ^сно
(5)
уксусной кислоты с водой и ТГФ (в объемном отношении
1:1:2) при 40—45° в течение 4 час проходит гладкий селектив-
ный гидролиз эфира енола (3) с образованием смеси транс- и
цпс-октен-2-алей-1 (4) в отношении 76:24. Эта смесь под дей-
ствием 0,02 экв n-толуолсульфокислоты в хлористом метилене
количественно изомеризуется в транс-альдегид (5).
При очистке октеналей можно легко выделить исходный аце-
тилен (1) и вновь ввести его в реакцию, так что потери веще-
ства в этом синтезе лишь чисто механические.
I. Buhler J. D., J. Org. Chem., 38, 904 (1973).
2. Trost В. M., Ziman S. D., J. Org. Chem., 38, 932 (1973).
3. Posner G. H., Brunelle D. J., J. Org. Chem., 37, 3547 (1972).
4. P о p о f f I. C., Dever J. L., Leader G. R., J. Org. Chem., 34, 1128
(1969); Shahak I., Almog J., Synthesis, 1969, 170; 145 (1970).
5. Pincock R. E., Schmidt J., Scott W. B., Torupka E. T., Can. J.
Chem., 50, 3958 (1972).
6. Scott W. B., Pincock R. E., J. Am. Chem. Soc., 95, 2040 (1973).
7. Kaiser E. M., В art ling G. J., Thomas W. R., Nichols S. B„
Nash D. R., J. Org. Chem., 38, 71 (1973).
8. Corey E. J., Terashima S., Tetrahedron Letters, 1815 (1972).
трет-БУТИЛЛИТИЯ КОМПЛЕКС C N,N,N',N'-TETPAME-
ТИЛЭТИЛЕНДИАМИНОМ.
Ж
См, Н,Н,Н/,Н/-Тетраметилэтилендиамин, V, 411; VI, 256; этот
том.
7/К7-БУТИЛМАГНИЙХЛОРИД, (CH3)3CMgCI, см. I, 227—
238.
Внутримолекулярная альдольная конденсация. На одной из
стадий синтеза предшественника эпиаллогибберовой кислоты
Хаузу и corp. [1] удалось осуществить с помощью Б. направлен-
ную внутримолекулярную альдольную конденсацию благодаря
промежуточному образованию ковалентного алкоголята магния.
Взаимодействие соединения' (1) с 2 же Б. в ТГФ и диметокси-
этане (ДМЭ) н последующий гидролиз водной уксусной кисло-
той привели к образованию альдоля (3) с 96%-ным выходом.
Применение метилмагнийбромида снижает выход альдоля (3)
до 45% • Продукт альдольной конденсации (3) гидролизуется
в исходный (1) водным раствором бикарбоната натрия.
1. House Н. О., Mel il Io D. G., Sauter F. J., J. Org. Chem., 38, 741
(1973).
«-БУТИЛМЕРКАПТАН (V, 71— 72).
«-Метилирование кетона. На одной из стадий полного синте-
за логанина (1) Бюхи с сотр. [1] метилировали кетон (2) по
методу Айрланда и Маршалла (V, 71—72) обработкой метил-
формиатом в присутствии основания. Последующее действие то-
зилхлорида и Б. приводит к образованию смеси трех н-бутил-
тиометиленкетонов (3), (4) и (5), в которой, к счастью, преобла-
78
дает требуемый изомер (5). Гидрогенолиз последнего никелем
Ренея дает смесь эпимерных метилкетонов, но в ней обычно пре-
валирует кетон (6). Под действием оснований этот кетон цели-
ком превращается в требуемый метилкетон (7), термодинами-
чески более устойчивый.
Геминальное алкилирование кетонов. Коутс и Соуэрби [2]
предложили новый метод региоселективного геминального ал-
килирования кетонов, заключающийся в восстановлении н-бу-
тилтиометиленового производного кетона литием в аммиаке с об-
разованием метилзамещенного енолят-аниона, который алкили-
руют in situ. Кетон, например циклогексанон (1), конденсируют
с этилформиатом, а затем реакцией с Б. (V, 71—72) превращают
в н-бутилтиометиленовое производное (2). Полученное произ-
водное восстанавливают избытком лития в жидком аммиаке при
—33° в присутствии 2 экв донора протонов (обычно используют
воду, чтобы избежать полиалкилирования). При алкилировании
образующегося енолята лития большим избытком йодистого
79
метила получается 2,2-диметилциклогексанон (3) с 83%-ным вы-
о
01) СгН5ОСНО, NaH
2) TsOH
(1)
Li-NH3 .
2НгО-(СгН;)гО.
(4) 2%
ходом и следы 2,2,6-триметилциклогексанона (4). Алкилирова-
ние лучше всего проходит в случае первичных галогеналкилов
или тозилатов.
Те же химики нашли, что 2-н-бутилтиометиленциклогексанон
(2) вступает в реакцию двойного сопряженного присоединения
с диметилмедьлитием в эфире при 0°, давая после гидролиза
2-изопропилциклогексанон (5) с выходом более 95%.
(СН3)гСиЫ
>95$
(5)
1. В п с h i G., Carlson J. A., Powell J. E., Jr., T i e t z e L.-F., J. Am.,
Chem. Soc., 95, 540 (1973).
2. Coates R. M., Sowerby R. L., J. Am. Chem. Soc., 93, 1027 (1971)'.
1-трег-БУТИЛОКСИКАРБОНИЛТРИАЗОЛ-1,2Д
О
— \ П
N-C-OC(CH3)3
Мол. вес 169,18, т. пл. 40°.
Реагент получают [1] с выходом 80% реакцией 1-триметил-
силилтриазола-1,2,4 [2] с азидом трет-бутилового эфира уголь-
ной кислоты (IV, 6; V, 471—473; этот том):
I А и \ II
N-Si(CH3)3 + (CH3)3COCN3 ----> N —С —ОС(СН3)3 + N,Si(CH3)3
8 0% LztY
80
N-трет-Бутилоксикарбоииламииокислоты [1] Аминокислоты в
форме солей с гидроокисью бензилтриметиламмония (тритон В)
реагируют с Б. (1) в ДМСО или ДМФА, в течение нескольких
часов образуя БОК-аминокислоты с выходом 80—90%.
БОК-Аминокислоты можно также получить, правда с не-
сколько меньшими выходами, реакцией бензилтриметил аммоние-
вых солей аминокислот с трет-бутил фенил карбонатом (2)
(IV, 6—7) в присутствии 1 мол. же триазола-1,2.4.
(CH3)3Co4oCflHs
(2)
1 BramG., Tetrahedron Letters, 469 (1973).
2 . Birkolfer L., Richter P., Ritter A., Chem. Ber., 93, 2804 (1960).
трет-БУТИЛ-8-ХИНОЛ ИЛ КАРБОНАТ. Мол. вес 245,3, т. пл.
69,5—72°.
Получение. Реагент получают из 8-оксихинолина и трет-бу-
тилхлорформиата в присутствии триэтиламина в смеси тетра-
гидрофуран — эфир при температуре от —60 до —25° (выход
72%). Использование этого реагента существенно облегчает
получение ^-защищенного лизина.
Описано также получение нескольких других БОК-г-амино-
кислот [1].
1. Rzeszotarska В., W i е j a k S,, Ann., 716, 216 (1968).
трет-БУТИЛХРОМАТ (I, 178; V, 73; VI, 40).
Окисление (4-)-карена-3 [1]. Для окисления ( + )-карена-3
(1) в (~)-карен-3-он-5 (2) вместо перманганата или хромового
ангидрида гораздо лучше использовать Б. При этом помимо ке-
тона (2) наблюдается образование побочных продуктов и среди
них соединений (3) и (4) в заметных количествах. Основные
(2) 50%
(4) 11,7%
продукты реакции легко разделить методом колоночной хрома-
тографии.
Р- Б., Cocker W., Grayson D. Н., J. Chem. Soc. (C), 1971,
81
(СН3)3СЧ
трег-БУТИЛЦИАНКЕТЕН, V=C=O. Мол. вес 123,15.
NC'
Получение [1], Б. (2) получают с почти количественным вы-
ходом термическим расщеплением 2,5-диазидо-3,6-ди-трет-бутил-
1,4-бензохинона (1) [2] в кипящем бензоле. Этот кетен необык-
(П
Нагревание
”">95^
2
(СНз)3С<
^С = С
NC/
(2)
новенно устойчив; после хранения его бензольного раствора при
комнатной температуре в течение восьми дней было отмечено
лишь незначительное разложение.
[22]-Циклоприсоединение. Моор и Уейлер [1] отмечают,
Что трет-бутил ци анкетен необычайно активен в реакциях цик-
лоприсоединения. Так, например, с циклогексеном (3) он обра-
зует циклобутанон (4) с выходом 63% (на выделенный про-
дукт). Следует отметить, что фенилцианкетен не вступает в реак-
цию с циклогексеном [3].
2L
6 з%'
Б. стереоспецифически реагирует с цис- и тра«с-циклоокте-
нами, давая циклобутаноны (5) и (6) соответственно [4]:
(6)
С циклическим алленом — циклононадиеном-1,2 (7) Б. при
комнатной температуре легко образует смесь диастереомерных
82
циклобутанонов (8) и (9) в отношении 3 : 2:
(7) (8) 312 (9)
При введении в реакцию частично расщепленного аллена (7)
оба продукта (8) и (9) обладают заметной оптической актив-
ностью.
. Б. вступает также в реакцию циклоприсоединения с ацетиле-
нами, давая производные циклобутен-2-она-1 с выходом 40—
80% [5]. Например, с фенилацетиленом в бензоле при комнатной
температуре он образует циклобутенон (10). При этом полу-
чается только один из двух возможных продуктов циклоприсо-
единения, и, следовательно, реакция региоспецифична. Струк-
тура кетона (10) была установлена на основании данных спек-
С6Н5СеСН
троскопии. Реакция Б. с трет-бутилфенилацетиленом также ре-
гиоспецифична и приводит только к циклобутенону (11).
СбН^СС(с.н;)з
(2)
1.3-Ди-трет-бутил-1,3-дициаиаллен [6]. Действие 0,01 же
триэтиламина на Б. в бензольном растворе вызывает медленную
димеризацию (8—12 час) его в р-лактон (2), структура кото-
рого была установлена на основании как спектральных, так и
химических свойств этого соединения. При обработке лактона
(2) 0,1 экв триэтиламина при комнатной температуре выделяется
Двуокись углерода и образуется 1,3-ди-грег-бутил-1,3-дицианал-
83
лен (3) с выходом 68%. Последний можно получить непосред-
ственно из Б., действуя 0,1 же триэтиламииа (выход 50%) .
С(СН3)з
ооо
NG (1)
50% (QH^N
С(СН3)3
С(сн3^
С(СН3)3 ^С(СН3)3
с=с=с *
(C^N
мс/ \n 68^
(3)
1. Moore Н. W., W е у 1 е г W., Jr., J. Am. Chem, Soc., 92, 4132 (1970).
2. Moore H, W., Weyler W-, Jr., J. Am. Chem. Soc., 93, 2812 (1971).
3. DeSeims R. C., Tetrahedron Letters, И79 (1969).
4. Weyler W., Jr., Byrd L. R., Caserio M. C., Moore H. W., J. Am,
Chem. Soc., 94, 1027 (1972).
5. Gheorghiu M. D., Drgghici C., St^nescu L., Avram M., Tetra-
hedron Letters, 9 (1973).
6. Moore H. W„ Duncan W. G., J. Org. Chem., 38, 156 (1973).
в
ВИНИЛМАГНИЙХЛОРИД, CH2 = CHMgCl.
Получение (I, 231). Normant Н., J. Org. Chem., 22,
1602 (1957).
1,6-Дикетоны [1]. В. реагирует с жирными кислотами или их
эфирами в присутствии хлористой меди в качестве катализа-
тора, образуя обычно с довольно низким выходом (5—55%)
1,6-дикетоиы. «-Валериановая кислота (1) в условиях этой ре-
акции дает н-тетрадекандион-5,10 (2) с выходом 53%.
1) Си2С1г, ТГФ
2) NH4C1
СНз(СН,)3СООН + CH2==CHMgCl ---------—-------
53%
(I)
—> СН3(СН2)3ССН2СН2СН2СН2С(СН2)3СН3
4 о
(2)
Реакция с карбоновыми кислотами [2]. С бензойной кислотой
в ТГФ В. образует 5-оксо-5-фенилпентен-1, а с «-валериановой
СоНбСООН + 2CHa=CHMgCl —С6Н5ССН2СН2СН=СН2
85 % п
кислотой в отсутствие катализатора дает смесь н-бутилдивинил-
карбинола и 5-оксононена-1,
сн=сн2
СН3(СН2).,С—СН=СН2 + СН3(СН,)3ССН>СН2СН=СН2
'А
он о
35% 30%
1. Watanabe S., Suga К., Fujita Т., Takahashi У., Can. J. Chem.,
50, 2786 (1972).
2. Watanabe S., Suga К., Yamaguchi Y., J. Appl. Chem. Biotechnol.,
22, 43 (1972).
ВИСМУТА ТРИАЦЕТАТ, Bi(OCOCH3)3. Мол. вес 322,13.
Получение [1]. Окись висмута (50 а) нагревают с уксусной
кислотой (300 мл) и уксусным ангидридом (35 мл) при 140—
150° до растворения (1,5—2 час). По охлаждении В. т. выде-
ляется в виде блестящих бесцветных пластинок (выход 90%).
Реакция с аминами и спиртами [2]. При взаимодействии В. т.
с аминами [реакция (1)] и со спиртами [реакция (2)] при ~150°
образуются соответственно N- и О-ацетилпроизводные. И в том
и в другом случае выходы колеблются в пределах 30—80%; вто-
рым продуктом реакции (неорганическим) является висмутил-
ацетат.
/R /Н
I) Bi(OAc)3 4- HlZ —> CH3CON^ + Bi(O)OCOCH3 + СНзСООН
^R1 XR*
2) Bi(OAc)3 + HOR —> CH3COOR + Bi(O)OCOCH3 + СНзСООН
1. Rigby W., J. Chem. Soc., 1951, 794.
2. Reese A. L., McMartin K., Miller D., Wickham P. P., J, Org.
Chem., 38, 764 (1973).
ВИТТИГА РЕАКЦИЯ.
Механизм. Смит и Триппетт [1] привели доказательства того,
что в реакции Виттига не получается винилфосфониевой соли
(а), промежуточное образование которой предполагал Швейцер
Но\
CRj
(а)
[2]. Таким образом, по-видимому, правилен принятый в настоя-
щее время механизм реакции, включающий промежуточное об-
разование четырехчленного цикла.
(С6Н5)зР=СНП 4- R*2C=O
» (с6ндр-снк--------*
б-CR'j
.___> (CfcH^P-CHR ---~>rch=cr'2 + (C6H№0
o-cr*2
Синтез Виттига иа полимерном носителе. Две группы иссле-
дователей [3, 4] сообщили о синтезе Виттига, осуществленном на
полимерном носителе. Сополимер получают из стирола н фосфи-
на (1) с небольшой добавкой 1,4-дивинилбензола в качестве
сшивающего агента. Алкилирование и обработка смолы основа-
нием приводят к полимеру, содержащему илидные группы (2).
При добавлении карбонильного соединения происходит реакция
86
Виттига.
О) (2)
1. Smith D. J. H., T г 1 p p e 11 S., Chem. Comm., 1972, 191.
2. Schweizer E. E., Crouse О. M., Minami T., Wehman A. T.,
Chem. Comm,, 1971, 1000.
3. Camps F., Castells J-, Font J„ Vela F., Tetrahedron Letters, 1715
(1971).
4. McKinley S. V., R a к s h у s J. W., J r., Chem. Comm., 1972, 134.
ВОЛЬФРАМА ГЕКСАХЛОРИД, WC16. Мол. вес 396,66.
Дезоксигенирование. Шарплес и сотр. [1] получили из В. г.
ряд галогенидов вольфрама более низкой валентности:
ТГФ
WC16 4- 2RLi ---> Реагент I
ТГФ
WCle-bSRLi -----> Реагент II
ТГФ
WCIo + 4RLi ----> Реагент III
ТГФ
WCls + 2Li (диспергированный) --> Реагент IV
130°
WC16 + 3L1I --------> Реагент V
в вакууме
130°
WCle + ^Lil --------Реагент VI
в вакууме
Под действием этих реагентов (особенно мягко под дей-
ствием реагента I) происходит восстановительное дезоксигени-
рование двух молекул альдегида или кетона с образованием оле-
финов. Обработкой избытком реагента I бензальдегид, напри-
мер, можно превратить в стильбен с выходом 70—76%.
Реагент I
2С6Н5СНО ----- > с6н5сн=снсен5
7U —7Ь%
В типовом эксперименте к 10 мл ТГФ, охлажденного до —78°,
прибавляют сначала WC16 (0,8 .ммоля), а затем н-бутиллитнй
(1,6 ммоля). Смеси дают нагреться до комнатной температуры
и после этого вводят карбонильное соединение (0,2 лгмоля). По
прошествии 6 час реакцию прерывают добавлением 20%-иого
раствора NaOH и олефин экстрагируют эфиром.
Наиболее высокие выходы были получены в случае аромати-
ческих альдегидов и кетонов; проведение этой реакции, напри-
мер с бутаноном-2, дает олефин лишь с 10%-ным выходом.
87
В описанной реакции сдваивания реагент I более эффективен,
чем другие вольфрамовые реагенты (II—VI).
Новые вольфрамовые реагенты (I—VI) оказались эффектив-
ными и в реакции дезоксигенирования эпоксидов в исходные
олефины. Для этой цели наиболее пригодны самые дешевые реа-
генты V и VI; реагенты I—III, с другой стороны, хорошо про-
явили себя в реакциях с небольшими и средними загрузками.
В типичном эксперименте к 420 мл ТГФ, охлажденного до —62°,
прибавляют сначала WC16 (0,15 моля), а после этого при той же
температуре — медленно «-бутиллитий (0,30 моля). Смеси дают
нагреться до комнатной температуры и затем вводят эпоксид
(0,08 моля). Сразу начинается быстрая экзотермическая реак-
ция восстановления. Через 0,5 час реакционную смесь выливают
в водно-щелочной раствор тартрата натрия и олефин экстраги-
руют гексаном.
Примеры:
Реагенты I, IV, VI
Окись траяс-циклододецеиа ---+ граяс-Циклододецеп
97—98%
95% транс
Реагент I
Окись ци с -цикл од о децена —. — > ^ас-Цнклододецен
70 — 90%
94-98% цас
Реагент I
Окись тоаяс-стильбена ----> траяс-Стильбен
86% н
100% транс
Восстановление ациклических ди- и тризамещенных эпокси-
дов протекает не вполне стереоселективно.
Селективное восстановление диэпоксида стигмастерилацетата
до 22,23-эпоксистигмастерилацетата можно осуществить с 83 %-
ным выходом при использовании небольшого количества реа-
гента I (0,3 час, комнатная температура).
См. также Калия гексахлоровольфрамат(IV) в этом томе.
Диспропорционирование олефинов. В. г. в сочетании с
С2Н5А1С12 [2] или с м-бутиллитием [3] в качестве катализаторов
применяют для диспропорционирования олефинов. Было пока-
зано, что эффективным сокатализатором является также алюмо-
гидрид лития, преимущество которого состоит в том, что он лег-
ко доступен и гораздо более устойчив на воздухе [4]. Так, геп-
тен-3 при взаимодействии с WC16—LiAlH4 в хлорбензоле в тече-
ние 3 час дает смесь гептена-3 (39%), октеиа-4 (23%) и гексена-3
(18,5%). В этой реакции также образуются нонен, пентен и
бутен с общим выходом 10%.
1. Sharpless К. В., Umbreit М. A., Nieh М. Т., Flood Т. С., J. Ат.
Chem. Soc., 94, 6538 (1972).
2. Calderon N., Judy W. A., О f s t e a d E. A., Scott K. W., Wood J. P.,
J. Am. Chem. Soc., 90, 4132 (1968).
3. Menapace H. R., Wang J. L., J. Org. Chem., 33, 3749 (1968).
4. Matlin S. A., Sammes P. G., J. C. S. Chem. Comm., 1973, 174.
г
ГАЗЫ ИНЕРТНЫЕ (I, 187).
Аргон. В методике, описанной в Org, Syn., диалкилирование
1,3-дитиана с помощью 1-бром-З-хлорпропана (первая стадия в
синтезе циклобутанона) рекомендуется проводить в атмосфере
аргона, если он доступен, вследствие его большей плотности по
сравнению с азотом [1]:
1) И-С4Щ,Ы
2) Cl(CHz)3Br
з) «-с4н9и
н,о
HgCIa
1. SeebachD., BeckA. K-, Org1- Syn., 51, 76 (1971).
ГАЛЛИЯ ОКИСЬ, Ga2O3. Мол. вес 187,44.
Г. о. катализирует обмен винильных и аллильных атомов во-
дорода на дейтерий с высокой степенью селективности. Под
действием Г. о. происходит взаимное превращение цис- и транс-
изомеров бутенов и пентенов при относительно низких темпера-
турах (110°).
1. Charlton F. В., Quinn Н. A., Rooney J. J., J. С. S. Chem. Comm.,
1973 , 231.
ГЕКСАМЕТИЛЕНТЕТРАМИН (уротропин) (I, 189—193;
V, 77—78).
Реакция Даффа (I, 192—193). Обзор [1]. Согласно класси-
ческой методике, активированные к электрофильному замеще-
нию ароматические соединения действием Г. в присутствии гли-
церинборной кислоты превращаются в формильные производные
обычно с невысокими выходами. Смит [2] нашел, что большое
число ароматических соединений, включая незамещенные угле-
водороды, при кипячении с Г. в грифторуксусной кислоте
(82—90°) превращаются в имины, которые при гидролизе дают
альдегиды:
о CF3cooH
2) н2о
ArH + C6H]2N4 ------> АгСНО
Даже бензол можно превратить в бензальдегид с выходом
32% (в запаянной ампуле при 125—150°). Высокие выходы
89
наблюдаются в случае активированных ароматических соедине-
ний; так, 2,6-ксиленол (1) превращается в 3,5-диметил-4-окси-
бензальдегид (2) с выходом 95%. Замещение направляется пре-
имущественно в «ара-положение к заместителю и при наличии
свободных орто-положений. Толуол превращается в «-толуило-
вый (выход 50%) и о-толуиловый (выход 11%) альдегиды.
Реакция Соммле. Ньюмен и Ханг [3] предложили усовершен-
ствованный метод синтеза З-метилнафталин-2-карбоновой кис-
лоты (4). Раствор 2,3-диметилнафталина (1) в CCU обраба-
тывают NEC (в присутствии перекиси бензоила), получая смесь
продуктов с преобладанием 2-бромметил-З-метилнафталииа (2).
После удаления растворителя и обработки остатка Г. в водной
уксусной кислоте выход альдегида (3) составляет 78%. На по-
следней стадии альдегид (3) окисляют щелочным раствором
азотнокислого серебра. Общий выход кислоты (4), считая иа (1),
составляет 69%.
1. Ferguson L. N., Chem. Rev,, 38, 230 (1946).
2. Smith W. E., J. Org. Chem,, 37, 3972 (1972).
3. Newman M, S., kiting W. M., Org. Prep. Proc. Inh, 4, 227 (1972).
ГЕКСАМЕТИЛТРИАМИД ФОСФОРИСТОЙ КИСЛОТЫ
(I, 193—194; V, 78).
Оксираны. С помощью реагента Марка при соблюдении опре-
деленных предосторожностей из диальдегида (1) был получен
90
с 75%-ным выходом неустойчивый оксиран (2) [1]. Ранее сооб-
щалось, что для этого случая данный метод непригоден [2].
Диметиламиды карбоновых кислот [3]. Ангидриды карбоно-
вых кислот при нагревании с избытком Г. ф. к. при 150—170° пре-
вращаются в диметиламиды карбоновых кислот (выход 40—
80%);
R—С
R—С
\)(СН,)2
1. Goh S. Н., Н а г v е у R. G., J. Am. Chem, Soc., 95, 242 (1973).
2. Newman М.. S., Blum S., J. Am. Chem. Soc., 86, 5598 (1964).
3. Schindlbauer H., Fischer S., Synthesis, 1972, 634.
ГЕКСАМЕТИЛТРИАМИД ФОСФОРНОЙ КИСЛОТЫ (гек-
саметапол, ГМТФК (I, 195; V, 78—81; VI, 45—48).
Дегидратация спиртов [1]. Первичные и вторичные спирты
дегидратируются под действием ГМТФК при температуре 220—
240°. Первичные спирты дают с умеренными выходами алкены-1
и 1-диметил аминоалканы, вторичные спирты превращаются
в олефины (без образования диметиламиноалканов).
Дегидратацией третичных спиртов под действием ГМТФК
при повышенных температурах можно получить стерически за-
трудненные олефины. Таким способом из карбинола (1) полу-
чают олефин (2) с выходом 77%, Описанный метод, конкури-
рующий с методом пиролиза сложных эфиров, применяют обыч-
j'per-C.pic. /ОН
/С\
грег-С4Нв' ^СН2С3Н7-уз<9
(1)
ГМТФК
77%
ypeT’-C.jHgx хС3Н7-изо
/С=С\
(2)
но в тех случаях, когда эфиры труднодоступны [2].
Нитрилы [3]. Амиды алифатических и ароматических карбо-
новых кислот дегидратируются ГМТФК при 220—240°, образуя
с хорошим выходом нитрилы, Предполагается, что реакция.
91
протекает через промежуточное соединение диамидофосф атного
типа (а).
ГМТФН
---------->
-HN(CHj)z
о
II
R-C=N + НО-Р-ЩСНД
Щснд
(г)
Амидииы [4]. N-Алкиламиды карбоновых кислот (1) при ки-
пячении в ГМТФК дают N.N-диметиламидины (2) с прекрасным
выходом:
R—с
%
ГМТФН
---------J
-ны(сн3)г
^NR1O
R-С У
(1)
О — Р-—N (С Н3 )2
N(CH3)2
^NR'
R-C
XN(CH3)a
(2)
1,3,2,4-Диазадифосфетидины. При кипячении анилина в
ГМТФК в течение 3—7 час образуется 1,3,2,4-Диазадифосфети-
дин (1) с выходом 76% [5]:
С6Н5ХНг
ГМТФН
------
сьн< ° ,СН3
XN —P-N
НзСх I I ХСН
XN-P—N 3
J \с6Н5
(П
Дегидрогалогенирование [6]. Нормальные первичные гало-
геналкилы при нагревании в ГМТФК при 180—210° гладко де-
гидрогалогенируются и без перегруппировки дают алкены-1
с выходом 60—65%.
Децианирование [7]. Алифатические нитрилы с хорошим вы-
ходом восстанавливаются до углеводородов калием в ГМТФК
и эфире с одновременным отщеплением CN-группы. В случае
третичных нитрилов добавки протонного растворителя (спирта)
не требуется. Выходы колеблются в пределах 60—90%.
к, ГМТФд
RCN ---------> RH + KCN
5-Галогензамещенные рибонуклеозиды [8]. Рибонуклеозиды
можно легко превратить в б'-хлор- и б'-бромпроизводные взаи-
модействием с тионилхлоридом или тионилбромидом соответ-
ственно в присутствии ГМТФК. Таким путем с удовлетворитель-
ными выходами были получены, например, б'-хлор-б'-дезоксици-
92
тидин (1) и 5'-бром-5'-дезоксиаденозин (2):
(2)
Реакция с циклоалканонами [9]. При кипячении циклогекса-
нона в ГМТФК (215—220°) в течение 40 мин в значительных
количествах образуется еиамин — 1-диметиламиноциклогексен.
Если после этого охладить смесь до комнатной температуры, то
енамин можно проалкилировать в той же колбе.
ГМТФН
------>
40 мин
1) RBT
2) Н2О
С увеличением продолжительности кипячения (до 4 час) енамин
разрушается, и главным продуктом реакции становится цикло-
гексилдиметиламин (1) (45,2%), а побочным (выход 4,8%) —
1,2,3,4,5,6,7,8-октагидроакридин (2). Эта неожиданная реакция,
приводящая к образованию октагидрбакрндинов из енаминов
диметиланилина, в настоящее время изучается.
ГМТФН
------->
4 час
N( СН3)2
(1) 45,2^
Восстановление карбонильных соединений [10]. Альдегиды
и кетоны восстанавливаются до соответствующих спиртов ще-
лочными металлами (Li, Na, К) в ГМТФК в присутствии протон-
ных растворителей, таких, как трет-бутанол.
Восстановление ароматических углеводородов. Натрий в
ГМТФК в присутствии ТГФ количественно восстанавливает ан-
трацен до 9,10-дигидроантрацена [11]. Реакция была распростра-
нена на бензантрацен, тетрацен, 9-алкил- и 9,10-диалкилантра-
цены с целью получения соответствующих лгезо-дигидропроиз-
водных. Источником протонов служат добавляемые к ТГФ вода
или метанол [12].
93
Тетрагидропираниловые эфиры w-окси ал кил ацетиленов. По-
чти все половые аттрактанты чешуекрылых представляют собой
ненасыщенные спирты с длинной цепью или их ацетаты. Обычно
их синтезируют через тетрагидропираниловые эфиры типа (2).
Шварц и Уотерс [13] нашли, что эти эфиры можно получить
почти с количественным выходом реакцией ацетиленового соеди-
нения типа (1), растворенного в ТГФ, с 1 г-же «-бутнллития
п K-C4H9L!
2) Х-(СН2)т-СН3
ТГП—О—(СН2)„—с^сн -----------------•>
(1)
—> ТГП—О—(СН2)а— С=С—(СГШ— СНз
(2)
в гексане при —65°. После прибавления всего количества н-бу-
тиллития прикапывают раствор галогеналкила в ГМТФК, следя
за тем, чтобы температура не поднималась выше 25°. По клас-
сической методике эфир (2) получают из алкина (1), используя
амид натрия в жидком аммиаке с последующим алкилирова-
нием, однако некоторые ацетилениды плохо растворяются в жид-
ком аммиаке.
Аттрактанты насекомых легко получить из эфиров (2) путем
восстановления тройной связи до двойной с последующим гид-
ролитическим расщеплением ТГП—О-связи.
Алкилирование натриевой соли иидола [14]. N-Алкилиидолы
можно синтезировать с прекрасными выходами взаимодействием
галогеналкилов с натриевой солью индола, полученной реакцией
индола с гидридом натрия в ГМТФК- При проведении этой ре-
акции в ТГФ побочно образуются 3-алкил- и 1,3-диалкилин-
долы с выходом около 10%. Добавка бензола к ГМТФК также
приводит к указанным продуктам.
Этерификация стерически затрудненных карбоновых кис-
лот [15]. Стерически затрудненные карбоновые кислоты можно
этерифнцировать с высоким выходом алкилированием их калие-
вых солей в смеси спирта с ГМТФК- В типовой методике кис-
лоту, например (1), растворяют в смеси этанол — ГМТФК
(1 : 1), добавляют порошкообразную гидроокись калия и смесь
нагревают при 50° до полного растворения. Затем прибавляют
СНз
CH3(CH2)3i—СООН
(СН2)2СНз
(1)
94
галогеналкил и смесь перемешивают при 50° в течение 30 мин.
После подкисления реакционной смеси полученный эфир экстра-
гируют гексаном. Предложенный метод особенно удобен именно
в случае стерически затрудненных кислот, поскольку пеларго-
новая кислота СН3(СН2)?СООН, например, реагирует гораздо
медленнее. В отсутствие ГМТФК степень превращения значи-
тельно уменьшается. Натриевые соли кислот реагируют обычно
медленнее калиевых.
Количественная этерификация карбоновых кислот [16]. К ра-
створу 10 лмолей мезитойиой кислоты в 25 мл ГМТФК в дели-
тельной воронке прибавляют 20 лшолей гидроокиси натрия
в виде 25%-ного водного раствора. Смесь встряхивают в тече-
ние 5 мин, затем добавляют 40 кмолей йодистого метила и про-
должают встряхивать еще в течение 5 мин. После этого смесь
подкисляют (НС1) и экстрагируют эфиром. Судя по данным
NaOH R'—X
ГМТФК ГМТФК
R-C-OH -------> R-C-ONa -> R-C-OR'
|| 95-100%
ИК-спектроскопии и ГЖХ, метиловый эфир мезитойной кислоты
образуется с выходом 99%.
1. Monson R. S., Tetrahedron Letters, 567 (1971).
2. LomasJ. S., Sagatys D. S., Dubois J.-E., Tetrahedron Letters, 165
(1972).
3. Monson R. S., Priest D. N., Can. J. Chem., 49, 2897 (1971).
4. Pedersen E. B., Vesterager N. O., Lawesson S.-О., Synthesis,
1972, 547.
5. Vesterager N. O.. Dyrnesli R., Pedersen E. B,, Lawes-
son S.-О., Synthesis, 1972, 548.
6. Monson R. S., Chem. Comm., 1971, 113.
7. Cuvigny T., LarchevSque M., Normant H., Compt. rend., 274,
797 (1972).
8. К i k u g a w a K„ 1 c h i п о M., Tetrahedron Letters, 87 (1971)
9. Monson R. S., Priest D. N., Ullrey J. C., Tetrahedron Letters, 929
(1972).
10. Larchev£que M., Cuvigny T., Compt. rend., 272, 794 (1971).
11. Labandibar P„ Lapouyade R., Bouas-Laurent H., Compt.
rend., 269 (C), 701 (1969).
12. Labandibar P., Lapouyade R., Bouas-Laurent H., Compt.
rend., 272 (C), 1257 (1971).
13. Schwarz M., Waters R. M., Synthesis, 1972, 567.
14. R u b o 11 o m G. M., C h a b a 1 a J. C., Synthesis, 1972 , 566.
15. Pfeffer P. E., Foglia T. A., Barr P. A., Schmeltz I., Sil-
ber! L. S., Tetrahedron Letters, 4063 (1972).
16. Shaw J. E., Kuner th D. C., Sherry J, J,, Tetrahedron Letters, 689
(1973).
ГЕКСАНИТРАТОЦЕРРАТ(1¥)АММОНИЯ (ГЦА) (IV 184—
185; V, 541—543; VI, 342—343).
См. Церий, аммиакат нитрата; этот том.
95
зоваиию соответствующего 6-ацетата (2)
с выходом 60—65% [8]:
(О
Окисление тропилидена. Циклогептатриен-^8 окисляется с по-
мощью ПДА с той же скоростью, что и сам циклогептатриен.
Этот факт, следовательно, может свидетельствовать в пользу
того, что ПДА охотнее реагирует с неароматической углерод-
углеродной двойной связью, чем с очень реакционноспособной
углерод-водородной связью [9].
цис- и транс-Стильбены из фенилдиазометана [10]. Фенил-
диазометан в пентане при —5° под действием каталитических
количеств ПДА (0, 03 мол. экв) быстро разлагается с образова-
нием цис- и транс-стильбенов, причем в качестве главного про-
74%
13%
дукта образуется первый из них. Для этого окисления предло-
жен цепной радикальный механизм, однако столь высокий вы-
ход цис-изомера пока не нашел себе объяснения. Сильные элек-
троноакцепторные заместители затрудняют реакцию.
Регенерация кетонов из !,3-дитиоланов и 1,3-дитианов. При
обработке 1,3-дитиоланов или 1,3-дитианов в 75%-ном водном
ацетонитриле ПДА при комнатной температуре в течение 3 мин
с выходами 70—87% образуются исходные карбонильные со-
единения [11].
Этот метод использовали в синтезе цис-жасмона для удале-
ния дитиокетальной группировки в мягких условиях [12]. При
окислении соединения (1) с помощью ГЦА ундекандион-2,5 (2)
образуется с выходом 80%. Применение хлорамина-Т (этот том)
привело к образованию дикетона (2) с выходом 67,3%.
98
1. Т г a h a n о v s к у W. S., Gilmore J. R., Heaton P. C., J, Org. Chem.,
38, 760 (1973).
2. Kurz M.. E., Steele E. S.. V e c h i о R. L., J. C. S. Chem. Commun., 1973,
109.
3. Brener L., McKennis J. S., Pettit R., Org. Syn., submitted 1972.
4. H о T.-L., Hall T. W, Wong С. M., Chem. Ind., 1972, 729.
5. H о T.-L., Synthesis, 1972, 560.
6. H о T.-L., Wong С. M., Synthesis, 1972, 561.
7. Ho T.-L., Ho H. C.. Wong С. M., Synthesis, 1972, 562.
8. P i a t a к D. M.., E 1 c h m e i e r L. S., Chem. Comm., 1971, 772.
9. Muller P,, Kat ten E., Rocek J., J. Am. Chem. Soc., 93, 7114 (1971).
10. Trahanovsky W. S. Robbins M. D., Smick D., J. Am. Chem.
Soc., 93, 2086 (1971).
11. H о T.-L., Ho H. C., Wong С. M., Chem. Comm.. 1972, 791.
12. Ho T.-L., Ho H. C., Wong С. M, Can. J. Chem., 50, 2718 (1972).
ГЕКСАХЛОРЦИ КЛОТРИФОСФАЗАТРИЕН (фосфонитрил-
хлорид) (1) (V, 82). Мол. вес 347,67; т. пл. 112—114°.
С1 С1
Cl^l: :| .CI
ОС. .-'PZ
Cl^ Cl
Продажный препарат можно очистить кристаллизацией из
ц-гептана [1] или перегонкой [2].
Дегидратация амидов с образованием нитрилов [2]. При
нагревании смеси амида и реагента (1) в отношении 3:1 при
температурах выше 100° быстро образуются нитрилы с высоким
выходом (обычно 75—100%). Дегидратацию можно проводить
также кипячением реагентов в растворителе (обычно в хлорбен-
золе). Во многих случаях оптимальное соотношение амида и
реагента (1) составляет 6:1.
Нитрилы из альдоксимов [3]. Оксимы алифатических, арома-
тических и ненасыщенных альдегидов (1 моль) дегидратируются
при действии триэтиламина (3 моля) и Г. (1 моль) в эфире или
ТГФ при комнатной температуре в течение 8—24 час, выходы
составляют 70—95%.
Оксим циклогексанона реагирует с Г., образуя соединение
(1) с выходом 63%:
0)
4*
99
1. Allcock H. R., Ku gel R. L., J. Am. Chem, Soc., 91, 5452 (1969).
2. Gr a ham J. C., Marr D. H., Can. J. Chem., 50, 3857 (1972).
3. Ros ini G., Baccolini G., Cacchi S., J. Org. Cnem., 33, 1060 (1973),
ГЕКСАЭТИЛТРИАМИД ФОСФОРИСТОЙ КИСЛОТЫ
[трис- (диэтиламино)-фосфин] (I, 196; V, 82—83; VI, 50—51).
Частичная десульфуризация дисульфидов (V, 82—83). Харп
и Глисон [1] сообщили, что диалкил-, аралкил- и алициклические
дисульфиды под действием Г. ф. к. быстро десульфуризуются
в соответствующие сульфиды, трис- (Диметиламино)-фосфин об-
ладает такой же эффективностью. Процесс стереоспецифичен,
поскольку у одного из атомов углерода в «-положении относи-
тельно дисульфидной группы происходит обращение конфигура-
ции. Так, при десульфуризации дисульфида (1), г^с-З.б-дикарб-
метокси-1,2-дитиана, с количественным выходом образуется
трсшс-2,5-дикарбметокситиолан (2). Диалогичным образом ди-
сульфид (3) количественно превращается в тиолан (4).
соосн3
количеств*
(2)
н3соос
количеств*
COOCHj
(4)
В реакции с ди-2-бензотиазолнлдисульфидом (5) были полу-
чены промежуточные термически неустойчивые соли фосфония
(6), выделяющиеся в виде масла. При нагревании до 80° они
превращаются в ди-2-беизотиазолилсульфид (7).
Изучение механизма десульфуризации привело к выводу, что
стадией, определяющей скорость реакции, является нуклеофиль-
ное расщепление дисульфидной связи.
100
1. Harpp D. N, Gleason J. G., J. Am. Chem. Soc., 93, 2437 (1971).
ГИДРАЗИН (I, 197—211; V, 84; VI, 51).
Восстановление по Хуаиг-Миилону —- Вольфу — Кижнеру
(IV, 449—450) [1]. Восстановление 3,4,5-триметоксибензальде-
гида:
сно
Ы2Н4, (НОСН2СН2ОСН2)2
87%
ОСН3
В синтезе 3,4-беизпирена из пирена [стадии (1) — (7)] Шлюде
[2] провел восстановление (2а)->(3) по Вольфу — Кижнеру —
Хуаиг-Миилону с выходом 80%, работая с загрузками в 100 г и
используя в качестве растворителя трет-бутанол.
Сукцинилирование пирена, описанное ранее другими авто-
рами, дает кетокислоту (2) с выходом до 95%; этерификация ее
по Фишеру приводит к метиловому эфиру (2а) с 95%-ным вы-
ходом.
Реакция с 1,5-дифеиилпентандииноном [3].
НгМЫНг + C6H5C=C-C-C=CC6HS
О
абс. СаН5ОН
79%
> C6HSC^C
1. Aquila Н, Ann, 721, 220 (1969).
2. S с h 1 u d е Н, Chem. Вег, 104, 3995 (1971),
3. Reimlinger H, Vandewalle J. J. M., Ann, 720, 117 (1968).
ГИДРОКСИЛАМИН-О-СУЛЬФОКИСЛОТА (I, 211—214;
V, 85—86; VI, 51).
Дибензо-1,4-диазепины [1]. 1-Метилдибензо-[b, ^]-1,4-диазе-
пин (2) получают с выходом 71,5% реакцией иодида N-метилак-
ридиння (1) с Г. в абсолютном метаноле, содержащем 30% ам-
миака (3—4 час, комн, температура). По-видимому, реагент
атакует положение 9 иодметилата (1) с последующим образо-
101
ванием азиридинового цикла; в результате расширения цикла
образуется соединение (2).
।
сн3
(1)
H2NO5O3H
NH,, СН,ОН
-------=-->
71, 5%
(2)
Синтез пиррола по Кнорру. Г. взаимодействует с некоторыми
соединениями, содержащими активную метиленовую группу, да-
вая продукты С-аминирования. Так, реакцией Г. с ацетоуксус-
ным эфиром (1) можно в одну стадию получить 2,4-диметил-3,5-
дикарбэтоксипиррол (2) [2]:
nh2
сн3сосн2соос2н5 _..NHz£s°3^ [ снэсоснсоос2н5]
води. K2COj
(1)
HjC
СаЩООС
н
(2)
(I)
1. Н i г о b е М., Ozawa Т., Tetrahedron Letters, 4493 (1971).
2. Tamura Y., Kato S., Ikeda M... Chem. Ind., 1971, 767,
ГЛУТАРОВОЙ И ЯНТАРНОЙ КИСЛОТ ДИНИТРИЛЫ.
МС(СНз)зСМ. Мол. вес 94,12, т. кип. 122—124°/5 мм; 137—
1407Ю мм.
NC(CH2)2CN. Мол. вес, 80,09, т. кип, 265—267°, т. пл. 53—
60°.
Синтез нитрилов. В патентной литературе [1] сообщалось
о превращении кислот в нитрилы реакцией с ацетонитрилом при
высоких температурах (150—300°). Недавно Клейн [2] нашел,
что использование для этой цели динитрилов янтарной, глутаро-
вой или сс-метилглутаровой кислот дает более высокие выходы
нитрилов и позволяет обойтись без автоклава. Реакция катали-
зируется сульфокислотами, а также серной или фосфорной кис-
лотой. Клейн использовал разработанный им метод для синтеза
алифатических динитрилов. Так, например, динитрил 1,10-де-
кандикарбоновой кислоты можно получить с высоким выходом
кипячением соответствующей кислоты с 2 же динитрила янтар-
ной, глутаровой или сс-метилглутаровой кислот в течение 18 час
в присутствии кислотного катализатора.
н
н+
НООС(СЬ^)10СООН + 2 NC(CHj)„CN --7—> NC (СН3 )]0CN + 2 0=7
(СНг)п
1. Loder D. J., пат. США 2377795 (1945); франц, пат. 1525498 (1968).
2. Klein D. A., J. Org. Chem., 36, 3050 (1972),
ДЕГИДРО-N, и'К-ТРИЦИ КЛОГЕКСИЛ ГУАН ИДИ НОГЕК-
САКАРБОНИЛДИЖЕЛЕЗО(О) (1). Мол. вес 583,23
Д. (1) получают в виде красных диамагнитных кристаллов
кипячением дициклогексилкарбодиимида (ДЦК) с Fe2(CO)s в
гептане в течение 3 дней,
Дихлоркарбен [1], При взаимодействии Д. (1) с хлорофор-
мом, содержащим 0,75% этанола, наблюдается отщепление всех
шести СО-групп, приводящее к образованию соли (2) и дихлор-
карбена. Последний в присутствии, например циклогексена, дает
N-C6Hn
C6Hn-N^ ^N-C6Hn CHCS :СС1а + [ FeClJ
(CO)jFe-Fe(COP
дихлорноркаран и смесь изомерных продуктов внедрения ди-
хлоркарбена по С—Н-связям атомов С2 и С3. Таким образом,
генерируемый этим способом дихлоркарбен по своему поведе-
нию отличается от дихлоркарбена, генерированного обычным
путем. В последнем случае реакции внедрения наблюдаются, как
правило, редко.
1. Bremer N. J., Cutcliff е А. В., Fa го па М. F., Chem. Comm., 1970,
932.
1,5-ДИАЗАБИЦИКЛО-[4ДО]-НОНЕН-5 (ДБН) (I, 240—242;
V, 88—89; VII, 20—23).
Дегидрогалогенирование. Было показано, что алкилгексатрие-
ны-1,3,5 высокой степени чистоты легко получить с выходом
30—40% дегидрогалогенированием винилаллилгалогенметанов
под действием ДБН в ДМСО (25—80°). Использование для этой
цели трет-бутилата калия в ДМСО приводит лишь к полимер-
ным продуктам [1].
Вг
| ДБН
R1CH==CHCHCH2C=CHR2 ----> К1СН=СНСН=СНС=СНКг
Из Из
На одной из стадий синтеза сД-мускона (4) из экзальтона (1)
необходимо было провести дегидробромирование соединения (2).
ЮЗ
Реакцию осуществили с помощью ДБН с 70%-ным выходом.
Если для этой цели применять трет-бутилат калия в трет-бута-
ноле или ДМСО, то получается только диоксин (5) [2],
9 1) HOCHzCHaOH Q о О Ю
Н 'Хч 'Чу < kjllj
-СЛ 2) Лсснвг .ДВН.» АсзГТ ХсиС'с-о
(сн,),. (с^)„ 70,1 '(сн1),т'/ )
(снг )12
(1; (2) (3). (41
трет - ВиОК
(5)
Дегидрогалогенирование бромида (6) под действием ДБН
дает смесь изомеров (7) и (8), которые были разделены с по-
мощью газовой хроматографии [3].
(С3НГ)2С(СН2)3СО2СН?
Вт
сн,снгсн2
>С(СН2)3СОгСН3 + (С3Нт)2С=СНСНгСН2СО2СН3
сн3сн2сн
(71
(8)
Окись трифенилфосфирена (2) [4]. Этот потенциально аро-
матический гетероцикл был синтезирован действием ДБН на
бензольный раствор бис- (сс-бромбензил)-фенилфосфинокснда (1),
полученного бромированием дибензилфенилфосфиноксида. Стайл
и сотр. [4] полагают, что превращение соединения (1) в про-
дукт (2) протекает через стадию 1,3-отщепления НВг с проме-
жуточным образованием окиси фосфирана, которая в свою
очередь претерпевает 1,2-отщепление бромистого водорода. Об-
разовавшийся при этом фосфациклопропен (2) легко гидроли-
зуется щелочью до 1,2-дифенилвинилфеннлфосфиновой кислоты
(3). Пиролиз фосфациклопропена (2) при 120° н давлении
104
10'5— Ю-6 мм рт. ст.' приводит к дифенилацетилену (4).
ДБН
-НВг
CfeH5 C6HS
/С=С\
С6Н5-Р-ОН н
4
о
(2)
о
ф
(С6Н5СНВг)гРС6Н5
(1)
ДБН
-НВг
(2)
Нагревание
С6Н5С=СС6Н5
(4)
Дегидробромирование и декарбметоксилирование в одну ста-
дию; расщепление метиловых эфиров карбоновых кислот по свя-
зи О—алкил. Майлс и Париш [5] пытались синтезировать лактон
(2) действием ДБН на бромкетон (1), полученный из дитер-
пена — подокарповой кислоты. Ранее в этой реакции в качестве
основания использовали коллидин, но выход лактона (2) был
низким. Однако кипячение (1) и ДБН в о-ксилоле (165°, 6 час)
неожиданно привело к образованию а,р-ненасыщенного кетона
(3) с выходом 93%. ДБН известен как дегидрогалогенирующнй
агент, приведенная же реакция представляет собой первый при-
мер декарбметоксилирования под действием этого реагента. Ре-
акция включает две стадии: расщепление метилового эфира кар-
боновой кислоты по связи О—метил и последующее декарбокси-
лирование. Действительно, ДБН оказался удобным реагентом
для расщепления метиловых эфиров пространственно затруд-
ненных карбоновых кислот. Например, метиловый эфир мезнтой-
ной кислоты превращали в мезитойную кислоту с выходом 90%
кипячением с ДБН в о-ксилоле (165°) в течение 6 час. Метиловый
105
эфир триизопропилуксусной кислоты гидролизуется до соот-
ветствующей кислоты в тех же условиях с выходом 94%, ДБН
также селективно расщепляет сложноэфириую связь в метило-
вом эфире 3[3-ацетокси-А5-этиеновой кислоты (4). Кипячение по-
следнего с 4,0 г-экв ДБН в о-ксилоле дает 27% исходного ве-
щества, 56% ожидаемой ацетоксикислоты, 7% оксикислоты и
5% диена, образовавшегося в результате отщепления ацетатной
группы. Таким образом, ДБН является несколько менее селек-
тивным агентом, чем w-пропилмеркаптид лития (VI, 153), но об-
ладает заметно большей селективностью по сравнению с иоди-
дом лития в кипящем лутидине (II, 197—198).
(4)
3,6-Дегидрооксепин (3). Действие ДБН на эритро-З-окси-4-
д-толуолсульфонилоксигексадиин-1,5 в эфире приводит к смеси
цис- и тра«с-1,2-диэтиннл оксиранов (2а) и (26) с выходом 53%
[6]. Пиролиз (2а) или (26) в атмосфере азота (400°, время кон-
такта — 20 сек) дает 3,6-дегидрооксепин (3)—циклическую 8л-
электронную систему. Оксепин (3) чрезвычайно чувствителен
к кислороду.
Оксепин (3) может подвергаться парциальному гидрирова-
нию при —65° над восстановленной окисью платины до 3-оксаби-
106
цикло-[3,2,0]-гептадиепа-1,4 (4), который более стабилен, чем его
предшественник (3) [7].
Стибабензол. ДБН использовали также для превращения
1,4-дигидро-1-хлорстибабензола (1) в стибабензол (2) [8]:
1. Spangler С. W., Е i с h е п R., Silver К., Butzlaff В.. J. Org. Chem.,
36, 1695 (1971).
2. Mookherjee В. D., Patel R. R., Ledig W. O.t J. Org. Chem., 36,
4124 (1971).
3. Eiter K.., Truscheit E., Boness M,, Ann., 709, 29 (1967).
4, Koos E. W., Vander Koot J. P., Green E. E.. Stille J. K., Chem.
Comm., 1972, 1085.
5. Miles D. H., Parish E. J., Tetrahedron Letters, 3987 (1972).
6. V о 11 h a г d t К. P. C., Bergman R. G-, J. Am Chem. Soc., 94, 8950
(1972).
7. Bergman R. G., V о 11 h a r d t К,. P. C., J. C. S. Chem. Comm., 1973, 214.
8. Ashe A. J., HI, J. Am. Chem. Soc., 93, 6690 (1971).
1,4-ДИАЗАБИЦИКЛО-[2,2,2]-ОКТАН (ДАБЦО) (III, 458;
V, 89—90).
Дезалкилирование четвертичных аммониевых солей [1]. Об-
работка четвертичной аммониевой соли (1 жмоль) ДАБЦО
(2 ммоля) в кипящем этаноле или ДМФА приводит к отщепле-
нию третичного амнна с выходом 50—90 %
+ -
r4nx +
R3N +
К
1. Но Т.-L.. Synthesis., 1972, 702.
1,5-ДИАЗАБИЦИКЛО-[5,4,0]-УНДЕЦЕН-5 (ДБУ) (V.91,;
VII, 20—23).
Расщепление метиловых эфиров карбоновых кислот по связи
О—алкил [1]. Нагревание бромкетона (1) с 2 г-экв ДБУ в
о-ксилоле при 165° в течение 5 час приводит к образованию со-
единения (2) с выходом 92%. Реакция с метиловым эфиром
подокарповой кислоты (3) в тех же условиях с высоким выхо-
дом дает кислоту (4). Аналогичным путем гидролизуется сильно
стерически затрудненный метиловый эфир мезитойной кислоты;
выход кислоты 94,6%.
10/
1. Parish E. J., Miles D. H, J. Org. Chem., 38, 1223 (1973).
ДИАЗОАЦЕТАЛЬДЕГИД (V, 91—92; VI, 52),
Получение. Новый метод получения Д. см. Уксусномуравьи-
ный ангидрид в этом томе.
ДИАЗОМЕТАН (I, 242—248; V, 92—95; VI, 52).
Присоединение по кратным С—N-связям см. [1].
Реакция с сопряженными диинами и енинами [2].
Синтез п-дизамещенных тетрафенилбицикло-[3,1,0]-гексенонов
[3].
108
Присоединение к ди-трет-бутилхинонам [4].
а-Хлорсульфоксиды [5]. Дизометан гладко реагирует с суль-
фннилхлоридами (получение см. [6]), образуя с высоким выхо-
дом хлорметнлсульфоксиды:
Н3СЖ + RS(O)C1 H,CCIS(O)R
В реакции можно использовать и другие диазоалканы, однако
они дают несколько меньшие выходы.
Синтез диазоацетофенона [7]. При медленном прибавлении
хлорангидрида кислоты к 2 же Д. выделяется хлористый водо-
род [реакция (1)], вступающий далее в реакцию (2). При обрат-
С6НЁСОС1 + CH2N2 —> C6H5COCHNo + НС1 (1)
CH3N2 + HCI —> CH3C1 + N2 (2)
C8HECOCHN2 + HC1 C6H5COCH2C1 + N2 (3)
ном порядке введения реагентов и использовании только 1 моля
Д- получившийся диазокетон реагирует с хлористым водородом
по реакции (3) с образованием а-хлоркетона. В приведенном
здесь методе, разработанном Ньюменом и Билом [8], для связы-
вания хлористого водорода применяется триэтиламин и, следо-
1 ла
вательно, в этом случае необходим только 1 же. Д. Однако ука-
занная модифицированная методика приводит к получению
лишь ароматических а-диазокетонов; хлорангидриды алнфати-
.ческих кислот дают смесь продуктов.
С6Н5СОС1 + CH2N, + (C2H5)3N —> CBHSCOCHN2 + (С2Н5)3ЙНСГ
I. Hoberg H, Ann. 707, 147 (1967).
2. Reimlinger H., Van dew a lie J. J. M., van Overstraeten A.,
Ann., 720, 124 (1968).
3, Durr H., Heitkamper P., Ann., 716, 212 (1968).
4. .R u n d e 1 W., Kastner P., Ann,, 737, 87 (1970).
5. Ven i er C. G., Hsieh Н.-Ы,, Barager H. J., Ill, J. Org. Chem., 38, 17
(1973).
6. Douglass I. B., Norton R. V., J- Org, Chem., 33, 2104 (1968).
7. В r i d s о n J. N., H о о z J., Org. Syn., submitted 1971.
8. Newman M. S., Beal P., Ill, J. Am. Chem. Soc., 71, 1506 (1949).
ДИАЗОПРОПИОНОВОЙ КИСЛОТЫ МЕТИЛОВЫЙ ЭФИР,
n2
Н3С—С—СООСН3. Мол. вес 114,11, т. кип. 52°/18 мм, желтого
цвета.
Получение [1]. Реагент можно получить несколькими спосо-
бами:
nh2
сн3снсоосн.
Нагревание
HCl/NaNO:
Метилкарбметоксикарбен [2]. Алкилкарбены, как правило,
не могут выступать в качестве предшественников циклопропа-
нов, так как в синглетном состоянии они быстро претерпевают
гидридный сдвиг. Преимущественным направлением реакции
этих активных частиц является поэтому внедрение по связи уг-
лерод—водород. Например, при облучении Д. к. м. э. (1) в от-
сутствие сенсибилизатора в качестве главного продукта обра-
110
зуется метилакрилат. Однако при сенсибилизированном бензо-
феноном фотолизе Д. к. м. э. возникает триплетный метилкарб-
метоксикарбен (2), который в присутствии изобутилена (3) дает
/сх
сн/ соосн,
сенсибилизатор
сх
СООСН3
/СН3
(3)НгС=С^
сн3
(1) (2)
+ НгС=СНСООСНз
(5)
(4)
производное циклопропана (4) с выходом 72% и только 3% ме-
тилакрилата (5). Фотосенсибилизированное разложение соеди-
нения (1) в присутствии цис- и тцаяс-бутена-2 приводит к соот-
ветствующим аддуктам с выходами 50 и 46% соответственно.
Фотосенсибилизированное облучение диазоалкана (6) в при-
сутствии изобутилена дает в качестве главного продукта соеди-
нение (7), но лишь с 33%-ным выходом.
'соосн3
COOCHj
+ НС^С—ССОСИ,
(9) 3%
I. Sohn М. В., Jones М., Jr., Hendrick М. Е., Rand о R. R., von
Е. Doering W., Tetrahedron Letters, 53 (1972).
2. Sohn M. В.. Jones М., Jr., J. Am. Chem, Soc., 94, 8280 (1972).
диазоуксусной кислоты АЗИД,
о
i! . -
N—N—CHCN=N“N
(1).
Мол. вес 111,07, т. кип. (взрывоопасно) 20—2Г/0,2 мм, т. пл.
7—8°.
Получение, Д. к. а. получают диазотированием гидразида
глицина [1].
Присоединение к диметиловому эфиру ацетилендикарбоновой
кислоты приводит к моноазиду диметилового эфира пиразол-
111
3,4,5-трикарбоновой кислоты:
СН3ОгС
(1) + снэогс-с=с-согсн3
COjC Hj
CON,
1. Neunhoeffer Н., Сипу G., Franke W. К., Ann., 713, 96 (1968).
ДИАЗОУКСУСНЫЙ ЭФИР (I, 249—252, V, 96—98; VI,
54-55).
Катализируемое ацетатом меди (II) присоединение Д. э.
к комплексам меди (II) с производными порфирина дает воз-
можность идентифицировать винильные группы в последних, ра-
ботая с миллиграммами вещества [1].
Д. э. в присутствии днэтиламина реагирует с N-метилизати-
ном и в отсутствие растворителя, в течение 2—3 недель образуя
при комнатной температуре кристаллический аддукт (2) [2]:
Эфиры алкинилуксусных кислот. Как сообщали Хуз и Лей-
тон [3], триалкинилбораны (2) легко получить действием эфирата
трифторнда бора в ТГФ при —20° на ацетиленид лития (1),
приготовленный из монозамещенного ацетилена и w-бутнл лития.
Последующее взаимодействие борана (2) с Д. э. дает после гид-
ролиза эфиры алкинилуксусных кислот (3) с выходами около
80—90% (считая на выделенный продукт).
ТГФ
3RC^CLi + BF. O(C2HS)2 (RCsC)3B
(О (2)
1} ТГФ, -20°
2) Н2О
(2) + N2CHCOOC2He ----------> RC=CCH2COOC2H5
(3)
Рассматриваемая реакция открывает также простой и удоб-
ный путь к эфирам у-кетокислот, так как гидратация сложных
112
эфиров алкинилуксусных кислот, промотируемая ионами рту-
ти (II), протекает региоспецнфично с образованием эфиров у-ке-
токислот (4):
Hg2+, н2о
(3) -------> RCOCH2CH2COOC2H5
(4)
Реакция с диалкилхлорборанами [4]. Реагент вступает в ре-
акцию двухуглеродной гомологизации с диалкилхлорборанами
[5] при —78°, образуя эфиры алкилуксусных кислот с выходами
90—95%. Браун и сотрудники предполагают, что реакция вклю-
чает координацию диазосоединения и борана [уравнение (1)],
отщепление азота и миграцию алкильной группы [уравнение (2)]
или хлора [уравиеиие (3)] и, наконец, [уравнение (4)] метано-
лнз (II) и (III).
R
L
1) R2BC1 + N2CHCOOC2H5 —> R—в—снсоос2нЁ
I I
Cl n2
I
2) R—B^~CHCOOC2HS ► RBCHCOOC2H5 + N2
ii ci
II
3) R—B—CHCOOC2H5 —RBCHCOOC2Hs + n2
Cl N2 Cl
III
R
CH3OH I
4) II или III --> RBCHCOOC2H5-4-HC1
icH3
|ch3oh
RB(OCH3)2 + RCH2COOC2Hs
1. Budzikiewicz H., Taraz K-, Ann., 737, 128 (1970).
2. E i s t e r t В., Borggrefe G., Ann., 718, 142 (1968).
3. Hooz J,, Layton R. B., Can. J. Chem., 1972, 1105.
4. Brown H. C., Midland M. M., Levy A. B., J. Am. Chem. Soc,, 94,
3662 (1972).
5. Brown H. C., Midland M. M., Levy A. B.t J. Am. Chem. Soc., 94,
2114 (1972).
ДИАЛКОКСИКАРБОНИЯ БОРФТОРИДЫ, (RO)2CHBFL
См. Днэтоксикарбония борфторид, этот том.
Борх [1] синтезировал Д. б. с выходом около 85% реакцией
ортомуравьнных эфиров с эфиратом трехфтористого бора в хло-
1 I ч
ристом метилене при —30° (VI, 301 *). Они представляют собой
низкоплавкие крнсталлическне вещества. Согласно данным
Борха, Д. б. являются более активными алкилирующими аген-
тами, чем борфториды триалкилоксония.
Перегруппировка Стивенса — отщепление. Бокельхайде и Ан-
дерсон [2] сообщили о синтезе [2,2]-метапарациклофандиена-1,9
(5). Ключевой стадией этого синтеза является замена сульфид-
ной группировки двойной углерод-углеродной связью, происхо-
дящая в результате перегруппировки Стивенса с последующим
элиминированием. При взаимодействии 2,11-дитиа-[3,3]-метапа-
(1) (2)
* Поправка. Эти
ния. — Прим, персе.
соли были ошибочно названы солями диалкилкарбО'
114
рациклофана (1) с борфторидом днметоксикарбония почти с ко-
личественным выходом образуется соль дисульфония (2). Под
действием гидрида натрия в ДМСО эта соль подвергается пере-
группировке Стивенса [3], давая смесь стереоизомеров (3) с вы-
ходом 67%. Дальнейшее алкилирование этого продукта приво-
дит к смеси изомеров (4) с выходом 87%. На последней стадии
синтеза происходит отщепление диметилсульфида под действием
н-бутиллития, протекающее с 50%-ным выходом.
В сущности, тот же метод был использован и для превраще-
ния соединения (6) в [2,2]-метациклофандиен-1,9 (7). В этом
случае на конечной стадии образуется смесь продукта (7) и его
валентного таутомера 15,'16-диметнлдигидропирена (8). При на-
гревании или хроматографировании на силикагеле смесь пре-
вращается целиком в соединение (8) [4]; другой способ синтеза
(8) см. V, 538—539.
I. В orch R. F., J. Org. Chem., 34, 627 (1969).
2. Boekelheide V., Anderson P. H., Tetrahedron
3. Ингольд К., «Теоретические основы органической
1973, стр. 649.
4. Mitchell R. Н., Boekelheide V., Tetrahedron
Letters, 1207 (1970).
химии», «Мир», М.,
Letters, 1197 (1970).
ДИАЦЕТИЛА И ТРИМЕТИЛФОСФИТА АДДУКТ (2,2,2-
триметокси-4,5-диметил-1,3-диоксафосфолен) (1) (V, 98—99).
Фосфорилирование [1]. Действие фосгена на реагент (1)
при 0° приводит к соединению (2), которое нагреванием при 120°
в течение 2 час превращается в смесь диастереомерных цикли-
ческих ацилфосфатов (За) и (36) и ендиолфосфата (4). Изо-
меры (За) и (36) в принципе могут быть разделены, но для фос-
форилирования спиртов пригодна и нх равновесная смесь
(30 : 70). Например, изомеры (3) чрезвычайно быстро реагируют
со спиртами с раскрытием цикла и декарбоксилированием, об-
разуя производные ацетоина типа (5) [алкилметил-(1-метил-2-
кетопропил)-фосфаты]. Последние очень легко гидролизуются до
115
эфиров фосфорной кислоты (6). Реакционная способность ацил-
фосфатов (3) по отношению к гидрокснлсодержащнм соедине-
ниям выше, чем у любого из ранее известных фосфорорганиче-
ских соединений, включая соединение (4).
RQH НзСхг/нок нго
н3ссо ор-оснэ
о
(5)
OR
1
НОР-ОСН,
II
о
(6)
Ацилфосфат (3) также метилирует пиридин по азоту с об-
разованием соли (7). При обработке последней борфторидом
триметилоксония регенерируется соединение (3). Взаимодей-
ствие соли (7) со спиртами или фенолами приводит к новой
соли (8), которая быстро гидролизуется до эфира фосфорной
кислоты (9).
OR
I
НОР-ОН
II
о
(8)
(9)
I, Ramirez F., Glaser S., Stern P„ Gillespie P. D,, U g i I., Angew.
Chem., Internal. Ed.. 12, 66 (1973).
ДИБЕНЗИЛ КЕТОН (1, 256).
Конденсация с фенилциклобутендионом [1].
1. Ried W, Kunkl W, Ann., 717, 54 (1968).
ДИБОРАН (I, 258—268; V, 100—102; VI, 56—58).
Д. образует сравнительно стабильные комплексы с ТГФ
и ди метил сульфидом. ВН3-ТГФ следует хранить при 0°;
ВН3- (CH3)2S можно хранить при комнатной температуре.
116
Оптически активные амины [1]. Ацилирование оксима, на-
пример 2-оксимино-1-фенилпропана (1), хлорангидридом хн-
ральной кислоты, например хлорангидридом 5-( + )-2-фенилмас-
ляной кислоты, приводит к эфиру окснма (3), который при вос-
становлении Д. дает смесь оптически активных аминов (4а) и
(46) с небольшим преобладанием S-энантиомера (4а); оптиче-
ский выход около 6 %.
^он
?г 5 Л
+ С1ОС*>С-С6Н6----> N Н
I II'
Н .С
C6HSCH2 сн3
(2) В)
NH2 jsJHj
С6Н5СНг*С-"СН3 + Н3С*-С-СНгС6Н5
н н
(4а) (46)
Восстановление амидов в амины [2]. Амиды, N-алкиламиды
и особенно П,]М-диалкиламиды кислот быстро и почти количе-
ственно восстанавливаются Д. до соответствующих аминов. Ре-
акцию проводят при 25° при получении третичных аминов и при
кипячении в случае первичных и вторичных аминов. Метод не-
применим к ненасыщенным амидам, так как Д. легко присоеди-
няется по двойной связи. Однако на другие заместители, та-
? ВНз
тгф
(Ar)RCNR2 яч > (Ar)RCH,NR2
од "У / уд
R' = Н или алкил
кие, как нитро- и сложноэфирная группы и галогены, Д. ие дей-
ствует.
1,З-Днолы. Клейн и Медлик [3] сообщили о новом методе
получения 1,3-диолов гидроборированием аллиллитиевых произ-
водных. Так, аллилбензол сначала металлируют н-бутиллитием
в эфире; после стояния в течение ~ 12 час реакционную смесь
прибавляют к охлажденному раствору Д. в ТГФ. Последующее
окисление водным щелочным раствором перекиси водорода при-
водит к образованию 1-фенил-пропандиола-1,3, который выде-
ляют в виде диацетата.
I) H-BuLl
2) BaHg; H2O2, “ОН
cgh3ch2ch=ch2 ---------—--------> СбН5СНСН2СН2ОН
Об h j
он
1 17
1,2-Днолы [4]. Гидроборирование енолятов натрия или лития
с последующим окислением приводит к образованию 1,2-диолов.
Например, реакция Д. (3 моля) в ТГФ при охлаждении на ледя-
ной бане с натрнйенолятом циклогексанона, полученным нагре-
ванием кетона с гидридом натрия в диметоксиэтане, и после-
дующее окисление щелочным раствором перекиси водорода дает
транс-циклогександиол-1,2 (1) с выходом 45%. Взаимодей-
ствие Д. с натрнйенолятом 4-метилциклогексанона приводит
П в2н6
2) Нгрг, ОН
4 5%
к смеси транс-диолов (2) и (3) в отношении 3:2 (суммарный
выход 55%).
Обработка 2-метилциклогексанона трифенйлметиллитием
дает кинетически контролируемый енолят (4), который при гид-
роборировании-окислении превращается в смесь транс-диолов
(5) и (6) в отношении 42:58 (суммарный выход 70%)- Термо-
динамически более устойчивый енолят (7) образует с Д. транс-
днол (8) с выходом 60%.
(П (8)
Аналогичным путем трнметилсилильные эфиры еиолов пре-
вращаются в 1,2-днолы.
Фенолы [5]. При взаимодействии ртутьорганическнх галоге-
нидов типа ArHgX с Д. в ТГФ по обычной методике гидробори-
118
рования образуются промежуточные борорганические соедине-
ния, которые, не выделяя, окисляют щелочным раствором пере-
киси водорода и с хорошим выходом получают фенолы.
Взаимодействие с реактивами Гриньяра [6]. С реактивами
Грнньяра Д. образует борорганические соединения, которые без
выделения можно окислить до соответствующих спиртов щелоч-
ным раствором перекиси водорода. Наиболее высокие выходы
спиртов получаются в том случае, когда реактив Гриньяра при-
вн + RMgBr
HMgBr+
ОН/Н2О2
ROH
готовлен в присутствии ВН3. Стандартные условия реакции та-
ковы: 10 ммолей галогеналкила взаимодействуют с 10 мг-атом
Mg и 15 лшолями ВНз при кипячении в ТГФ. Образование бор-
органического соединения в основном заканчивается через
45 мин. Область применения реакции ограничена доступностью
реактивов Гриньяра, которые всегда количественно превра-
щаются в борорганические соединения. Рассмотренная реакция
представляет собой простой и удобный метод замещения гало-
гена на гидроксил как в алифатических, так и в ароматических
соединениях.
Примеры:
Бромбензол
jw-Хлориодбензол
1 -Бромбутан
Бензилбромид
Циклогексилбромид
Г Б рома дама итак
- —> Фенол (67%)
— —> л-Хлорфенол (77%)
— > я-Бутанол (73%)
— > Бензиловый спирт (99%)
— > Циклогексанол (67%)
—> 1-Оксиадамантан (48%)
1. Busse г U., Haller R., Tetrahedron Letters, 231 (1973).
2. Brown Н. C., Heim Р., J. Org. Chem., 38, 912 (1973).
3. Klein J., Med 1 i k A., J. Am. Chem. Soc., 93, 6313 (1971).
4. К1 e i n J., Levene R., Dunkel blum E., Tetrahedron Letters, 2845
(1972).
5. Breuer S. W., Leath am M. J., Thorpe F. G,, Chem. Comm., 1971,
1475.
6. Breuer S. W., Bros ter F. A., J. Organometallic Chem., 35, C5 (1972).
1,2-ДИБРОМЭТАН (этилен бромистый) ВгСН2СНзВг. Мол.
вес 187,87, т. пл. 9—10°, т. кип. 132°.
а-Метилен-у-бутиролактоны. Оуриссон и сотр. [1] предло-
жили новый путь синтеза а-метилен-у-бутиролактонов, исходя
из а-метил-у-бутиролактонов. Найденный метод был использо-
ван для превращения лактона (1), полученного в несколько ста-
дий ыз 6-эписантонина, в природный сесквитерпен (—)-фрулла-
нолид. При действии на лактон (1) избытка трифеннлметиллп-
1 10
тня в днметоксиэтане (ДМЭ) в присутствии тетраметилэтнлен-
диамина (ТМЭДА) образуется соответствующий енолят, обра-
ботка которого Д. при 5° дает а-бромлактон (2) с выходом
около 50%. Дегидробромирование последнего избытком 1,5-ди-
азабицикло-[4,3,0]-нонена-5 (ДБН, I, 240—242; V, 88—89) в ки-
пящем толуоле приводит к образованию (—)-фрулланолида (3)
с выходом около 80%. Предложенный метод является, по-вндн-
мому, первым примером использования виц-дигалогенидов для
получения а-галогенлактонов.
Необходимо отметить, что этот метод применим лишь к лак-,
тонам с цис-сочленением колец. В случае траис-сочленеиных
бромлактонов происходит транс-элиминирование НВг с образо-
ванием только эндоциклических олефинов.
Оуриссон и сотр. разработали также метод превращения
транс-сочлененных лактонов в а-метилен-у-бутиролактоны (см.
Бензоила перекись, этот том).
1. Greene А. Е., Muller J.-C., Ourisson G., Tetrahedron Letters, 2489
(1972).
2,6-ДИ-трет-БУТИЛ-а-ДИМЕТИЛАМИНО-п-КРЕЗОЛ (этил-
антиоксидант 703). Мол. вес 263,4, т. пл. 94°, т. кип. 179°/40 мм.
Растворимость при 18°. вес. %
Толуол................ 22 Вода...............
Этанол................ 28 10%-пый NaOH . . . .
< 0,0007
< 0,002
120
Ингибитор окисления натурального и синтетических каучу-
ков, полиолефиновых пластмасс, смол, клеев, нефтетоплива и
восков.
ДИ-трет-БУТИЛ ИМИНОКСИЛ (I, 271; VI, 60).
Механизм гашения триплетного состояния с помощью Д. об-
суждается Шверцелем и Колдуэллом [1].
1. Schwerzel R. Е.. Caldwell R. A., J. Am. Chem. Soc., 95, 1382 (1973).
ДИ-н-БУТИЛМЕДЬЛИТИЙ (V, 121; VI, 60—62).
Синтез кетонов [1].
СН3о2ССН2СН2СОС]
2w-C4H9Li + Cui —> (H-C+H9)sCuLi -+
—> СН3О2ССН2СН2СОС4Нэ-н
Кетоны. Одним из наиболее удобных и общих реагентов для
получения кетонов из хлорангидридов кислот являются медь-
органические соединения благодаря разнообразию их структур-
ных типов, простоте выполнения эксперимента и мягким усло-
виям реакций, а также стереоселективности их действия и се-
лективности по отношению к функциональным группам [2].
I. Posner G. Н., Whitten С. Е., Org. Syn., submitted 1971.
2. Reinheckel H., H a a ge K., Jahnke D., Organometallic Chem. Rev.(A),
4, 55 (1969).
ДИ-трет-БУТИЛ ПИРО КАРБОНАТ,(CH3)3COCO2CO2C (CH3)3.
Мол. вес 209,17, т. пл. 21—22°.
Получение [1]. Д. получают, как показано ниже, из др-трет-
бутилтрикарбоната:
coci2
(СНз)зСОК + СО2 —> (СН3)зСОСО2К —->
ДАБЦО
—со2
—> (СН3)3СОС02С02С02С(СН3)Э —-——> (СН3)3СОСО2СО2С(СН3)з (1)
84—89%
Защита аминогрупп [2]. Д. (1) взаимодействует с эфирами
аминокислот, давая с хорошим выходом N-трет-бутоксикарбо-
121
нилпропзводпые (трет-БОК-производные) (2). Согласно типовой
методике, к суспензии хлоргидрата этилового эфира глицина
в хлороформе приливают водный раствор бикарбоната натрия,
а затем хлористого натрия. После этого добавляют Д. (1) в хло-
0 R'
(I) +R/CHCOOR'/ —> (СН3)3СОС—NhAhCOOR"
nh2
(2)
роформе и кипятят смесь в течение 90 мин. Этиловый эфир трет-
БОК-глицина получается с выходом 89%.
1. Pope В. М., Yamamoto Y., Tarbell D. S., Org. Syn., submitted 1973.
2, Pope В. M., Yamamoto Y., Tarbell D. S., Proc. Nat. Acad. Sci.
U. S. A., 69, 730 (1972).
ДИВИНИЛМЕДЬЛИТИЙ, (CH2~CH)2CuLi. Мол. вес.
124,57.
Получение [1]. Этот металлоорганический реагент получают
взаимодействием раствора иодида меди(1) в динзопропилсуль-
фиде (3,1 мол. эке) с 2,2 М раствором виниллития (1,92 эхе) в
ТГФ при температурах от —25 до —15°. Затем раствор охлаж-
дают до —72° и оставляют до завершения реакции.
Стереоселективное введение ангулярной винильной груп-
пы [1]. Д. применяют для введения винильной группы (даже
ангулярной) в р-положеяие к карбонилу. Так, например, при до-
бавлении енона (1) к раствору 1,3 мол. же Д. в ТГФ при —72°
в аргоне происходит экзотермическая реакция с образованием
единственного p-винилированного кетона (2) с выходом >90%.
Последний был использован для построения характерной для
гиббереллинов карбоциклической системы в синтезе гибберелло-
вых кислот (см. Диметилдихлорсилан, этот том)
Гомосопряженное присоединение к циклопропанам. Кори и
Фукс [2] исследовали реакцию циклопропанов с медьорганиче-
скими соединениями с целью ее применения в синтезе проста-
ноидов. Например, трициклический карбметоксилактон (1) при
взаимодействии с Д. (2,0 же) в эфире при —12° (19 час) дает
122
вииилциклопропанкарбметоксилактон (2). При кипячении по-
следнего в пиридине (II, 197—198) в течение 3 час в присутствии
иодида лития (5 экв) образуется лактон (3) с выходом около
37%.
(з)
Другим примером такого гомосопряженного присоединения
является реакция этилового эфира а-циаициклопропанкарбоно-
вой кислоты (4) с 2 экв Д., приводящая к образованию этило-
вого эфира 2-циангексен-5-овой кислоты (5) с выходом 70%.
I\/CN
СООСгН5
(СНг—CH)2CuLi
— Г"1 >Vj — Xi vj
7 си
CN
соосгн5
(4) (5)
Присоединение успешно осуществлено н с помощью диметил-
медьлития.
Стереоспецифический синтез 1,3-диенов. Кори и сотр. [3]
описали новый метод синтеза 1,3-диенов, основу которого со-
ставляет в высшей степени стереоспецифическое цис-присоеди-
нение алкилмедных реагентов к а,р-ацетиленовым карбониль-
ным соединениям (VI, 76). Так, реакция между метиловым эфи-
ром 4-триметилсилоксинонин-2-овой кислоты (1) и Д. (1,25 же)
в ТГФ, проводимая сначала при —90°, а затем при —78°, дает
чистый цис-аддукт (2) с выходом >90%• При обработке послед-
него метанольным раствором хлористого водорода происходит
отщепление триметилсилильной группы и лактонизация с обра-
зованием (3). Метиловый эфир (1) получали в одну стадию по-
следовательной обработкой метилпропиноата в ТГФ при —78°
123
ц-бутиллитием, гексаналем и триметилхлорсиланом; выход 71%'.
Ч-С5Н11СНО+ НС^ССООСНэ м-СБНиСНС^ССООСН3 —>
(CH3)3SiO
0)
О5ЦСНз)з
т. H-CsHnCH СООСН.
(СН3=СН) 2GuLi^ \с=с<
>90$ H2C=Clf" ХН
(2)
(3)
Присоединение метилового эфира бутин-2-овой кислоты (4)
к винилмеди [получена из 2 же виииллития и 2 же иодида
меди(1) в атмосфере N2 при —78°] дает метиловый эфир 3-метил-
гранс-пентадиен-2,4-овой кислоты (5) с выходом 74%.
сн хсн
СН3С==ССООСН3 CHCu> HzC'Z ''"'COOCHj
74$ CH3
(4) (5)
Сообщалось также об аналогичном стереоспецифическом син-
тезе 1,4-диеиов с использованием аллилмедных реагентов. Так,
реакция 2 же аллилмеди, полученной из 1 же аллилмагнийхло-
рида и 1 же иоднда меди(1) при температурах от —30 до —40°
в течение 2 час и окрашенной в темио-красиый цвет, с метил-
пропиноатом (6) в эфире при —78° в атмосфере N2 приводит
к образованию метилового эфира трацс-гексадиен-2,5-овой кис-
лоты (7):
СН2=СНСНгСи „ „
НС 5SС С ООС Н3-i-------->НгС /С Нг С Н
СН ЮООСНз
(6)
(7)
1. Corey Е. J,, Carney R. L., J. Am. Chem. Soc.. 93, 7318 (1971).
2. Corey E. J,, Fuchs P. L., J. Am. Chem. Soc., 94, 4014 (1972).
3. С о г e у E. J., Kim C. U., Chen R. H. K-, Takeda M„ J. Am. Chem.
Soc., 94, 4395 (1973).
ДИВИНИЛМЕДЬЛИТИЯ КОМПЛЕКС С ТРИ-н-БУТИЛ-
ФОСФИНОМ, (CH2=CH)2CuLiP (ц-С4Н9)3. Мол. вес 302,86.
Получение [1]. Раствор 2 же виниллития в ТГФ медленно
при перемешивании прибавляют к раствору 1 же тетракис-[нол-
(три-н-бутилфосфин)-меди(1)] (V, 220; VI, 134) в ТГФ при —78°
в атмосфере азота. Комплекс имеет сине-черный цвет.
у,6-Ненасыщенные кетоиы. Сопряженное присоединение ком-
плекса к а,р-ненасыщеиным кетонам приводит к образова-
124
нию у,б-ненасыщенных кетоиов с выходом 65—90%. Заслужи-
вает внимания реакция с изофороном (см. ниже), в результате
которой образуется З-винил-3,5,5-триметилциклогексанон. При
взаимодействии изофорона с винилмагнийхлоридом, катализи-
руемом иодидом меди(1), выход продукта 1,4-присоедииеиня со-
ставляет всего лишь 8% [1].
0. 5 час, -78°
—--------------->
65$
СН= СН;
О
II
снг=снссн3
о
0.75 час, -78° II
—— ----'--1—> СНг=СН-СНгСНгССН3
7 0$
1 час , о'
85$
Дивииилмедьлитнй (СН2==СН)2СиЫ, не содержащий фос-
фина, можно получить действием 1 же иодида меди(1) на 2 же
виниллития, но скорость образования этого реагента н скорость
его реакций с еноиами значительно ниже, чем в случае фосфин-
содержащих комплексов.
Подобного рода комплекс использовался в недавно осущест-
вленном синтезе (±)-15-дезоксипростагландина Ej [2]. Реакция
соединения (1) с 2 мол. экв 1-литий-транс-октеиа-1 и 1 же тет-
рак«с-[иод-(три-н-бутилфосфин)-меди(1)] в эфире при 0° приво-
дит после гидролиза защитной тетрагидропиранильной группы
к образованию этилового эфира (±)-15-дезоксипростагландина
Е[ (2) с выходом 60%.
У^^\СНг)6СООСгН5 ) [lCuP(H-C4H,)3 ]+ II (СНДСООСгЩ
| |j z) НОАс, НгО, ТГФ | |
Тгп(У--- 60% но>----
(1) (г)
1. Н о о z J., L а у t о и R. В., Can. J. Chem., 48, 1626 (1970).
2. Sih С. J., Salomon R. G., Price P., Peruzzoti G., Sood R., Chem
Comm., 1972, 240.
ДИИЗОБУТИЛАЛЮМИНИИГИДРИД (I 275—278- V
108—110; VI, 66—67).
гра«с,гранс-1,3-Днены [1]. Гидроалюминирование терминаль-
ных ацетиленов с помощью Д. приводит к образованию терми-
125
нальных винилалаиов, при обработке которых хлористой медью
в ТГФ получаются транс, транс- 1,3-диеиы с выходом около 70%.
Другие изомеры при этом не образуются. Винилаланы, очевидно,
реагируют с хлористой медью, давая соединения винилмеди(1),
которые затем разлагаются с образованием диенов.
2 rc^ch 2
и-Гексан
*4 /Н г R /Н
------> с = с _н
Н А1(«о-С4Н9)г ТГФ ''с = с/
НХ "\
Метод применим также к вииилаланам, полученным из диза-
мещеиных алкинов. Так, гексин-3 превращали в 4,5-диэтил-траяс,
транс-октадиен-3,5 с выходом 71%.
/СгН3
С.Н.С-СС.Н, НА1(...-С.Н,),; С=-КСИС/С=Н- Cud Н/С-СЧС_С/"
МХ ЧА1(Ио.-С4нрг 71% Н5СгХ ХСгНя
Циклопропаны. Алкилциклопропаны (4) можно получить из
1-алкинов (1) по приведенной ниже последовательности реак-
ций [2]:
с.н,с-сн СЛ\С=С/Н СН=Вг<
ц/ \А1(С4Н,)г 2n-Cu
0) (2)
цмс-Присоединение Д. к (1) приводит к алкенил-1-алану (2).
Реакция последнего с бромистым метиленом в присутствии цинк-
медной пары (IV, 214—215) дает циклопропилалаи (3), который
гидролизуется до алкилциклопропаиа (4). Промежуточное со-
единение (3) действием брома или иода можно превратить так-
же в транс- 1-галоген-2-алкилциклопропан (5).
(з)
з Вгг
’ 58%
к / \ .Н
ус----'
(5)
транс-Аллиловые спирты [3]. траяс-Винилаланы (1), полу-
ченные в результате ц«с-присоедннения Д. к 1-алкинам (V, 109),
взаимодействуют с альдегидами или кетонами в эфире или бен-
золе, давая после подкисления соответствующие аллиловые
125
спирты (2) с выходом 30—50%. Новый метод алкенилирования
альдегидов и кетонов удобен в том отношении, что винил аланы
получить легче, чем виниллитий или винилмагнийгалогеииды.
в1
1) ,с = о
R2
RC^CH Rx _ -Н /)Нд°+
1-\ Г1 । in in - пи . i— ) ' *"< II . .
Hz хА1(изо-С4Но)э
(О
R ZH
R1
?г С
НО R2
(2)
Сопряженное восстановление а^-ненасыщенных эпокси-
дов [4]. Прн восстановлении 1,2-оксидо-2-метилбутена-3 (1), по-
лученного из изопрена по методике Ренета и сотр. [5], Д. в ки-
пящем гексане происходит в высокой степени стереоселективиое
сопряженное присоединение его к (1) с образованием спирта
(2), имеющего Z-конфигурацию; Е-изомер (3) является побоч-
ным продуктом. Восстановление 1,2-окиси (1) с помощью Д.
HAI Щвд-С4Н9)2 н г
СН Г1Н 3 \
—————А,—С"
сн2 71% /
7 но-сн2
н
=с
СНЭ
носн2 н
уз=с
Н3с СНз
НА1 (язо-С4Н9)2 Н3С
) ^с-сн
носнг \;нг
(2)
(4)
в ТГФ дает главным образом гомоаллиловый спирт (4), обра-
зующийся в результате прямого присоединения (по Марковни-
кову) гидрид-иона к эпоксиду. Восстановление соединения (1)
алюмогидридом лития приводит исключительно к третичному
аллиловому спирту (5).
Г)
LiAiH4
эфир
шГ
HjC
НО^Р-СН
н2с Хсн2
(5)
ь противоположность этому восстановление 1,2-окиси (1)
кальцием в жидком аммиаке дает почти исключительно Е-изо-
127
мер (3). При использовании металлических лития или натрия
стереоселективность реакции уменьшается.
Распространение этих исследований на другие «^-ненасы-
щенные эпоксиды показало, что направление восстановления
(прямое или сопряженное) зависит от степени экранирования
реакционного центра обеими функциональными группами.
Селективное восстановление [6]. При действии Д. нитрил (1)
восстанавливается до соответствующего альдегида (2) с выхо-
дом 75—85% (ср. 1,277—278):
(О (2)
1. Zweifel G., Miller R. L., J. Am. Chem. Soc., 92, 6678 (1970).
2. Zweifel G., Clark G. M., Whitney С. C., J. Am. Chem. Soc., 93, 1305
(1971).
3. Newman H., Tetrahedron Letters, 4571 (1971).
4. Lenox R. S., Katzenellenbogen J. A., J. Am. Chem. Soc., 95, 957
(1973).
5. Reist E. J., Junga I. G., Baker B. R., J. Org. Chem., 25, 1673 (1960).
6. Stevens R. V., DuPree L. E., Jr., Loewenstein P. L., j. Org.
Chem., 37, 977 (1972).
ДИИЗОПИНОКАМФИЛБОРАН (1,279).
Реагент в действительности представляет собой скорее произ-
водное диборана (1), а не мономер [1]. В Chemical Abstracts этот
реагент называется тетра-3-пинаиилдибораном и ему приписана
формула Р114В2Н2.
(О
Реакции с алленами. Касерио и сотр. [2] сообщили, что в ре-
зультате частичного гидроборирования рацемических 1,3-диме-
тилаллена и 1,3-дифенил аллен а с помощью (+)-Рп4В2Н2 [из
(—)-а-пинена] с умеренным оптическим выходом можно полу-
чить (—)-аллеи. Метод был детально изучен Моором и сотр. [3].
Они нашли, что во всех изученных ими случаях продукт реак-
128
ции обогащен ^-энантиомером. Таким образом, реагент Брауна
пригоден для получения оптически активных алленов, одиако их
оптическая активность невелика и, конечно, часть аллена безвоз-
вратно теряется в ходе реакции.
Асимметрическое гидроборирование. Ключевой стадией асим-
метрического синтеза [4] логанииа (1), важнейшего интерме-
диата в биосинтезе индола и монотерпеновых алкалоидов, яв-
ляется асимметрическое гидроборирование 5-метилциклопента-
O-p-D-Гликоза
диеиа (2). Эту реакцию осуществляли с использованием (+)-
или (—)-Д. Применение (+)-борана приводит к образованию
спирта (За), »d —169°, (—)-бораи дает спирт (36), ац +170°,
причем выходы каждого из спиртов составляли >30%. Ни в од-
ном случае не были обнаружены цис-спирты. Было показано,
Д+)-ЕгВН
>30$
(-)-RzBH^
>30$
(36)
(За) (2)
что оба спирта имеют оптическую чистоту ие ниже 98—99%.
I. Brown Н. С., К 1 е n d е г G. J., Inorg. Chem., I, 204 (1962).
2, Waters W. L., Caserio M. C., Tetrahedron Letters. 5233 (1968); Wa-
ters W. L., Linn W. S., Caserio M. C., J. Am. Chem. Soc., 90, 6741
(1968).
3. Moore W. R., Anderson H. W., Clark S. D., J. Am. Chem. Soc., 95,
835 (1973).
4. Partridge J. J., Chad ha N. K., Uskokovid M. R., J. Am. Chem.
Soc., 95, 532 (1973).
X,N-Д И ИЗОПРОПИЛ ГИДРАЗИН,
(CH3)2CHNHNHCH(CH3)2. Мол. вес 116,20, т. кип. 124—124,5°.
Д. получают каталитическим восстановлением азииа аце-
тона [1].
Защита карбоновых кислот [2]. Д. взаимодействует с произ-
водными карбоновых кислот (ацилхлоридами и смешанными ан-
гидридами) с образованием моио ацил гидр азидов.
5 Зак. 596 129
RCON (CHMe2)NH (СНМе2), устойчивых к действию кислот и
оснований. Кислота регенерируется селективным окислением
гидразида, предпочтительно тетраацетатом свинца. Новый ме-
тод защиты был использован в синтезе пенициллинов.
1. Lochte Н. R., Bailey J. R., Noyes W. A.. J. Am. Chem. Soc., 43, 2597
(1921).
2. Barton D. H. R., G i г i j a v a 11 a b h а п M.., Sammes P. G., J. C. S.
Perkin I, 1972, 929.
ДИИМИД (I, 280—282; V, НО; VI, 67—69).
Выделение [1]. Д. был выделен из продуктов пиролиза ли-
тиевого производного /г-толуолсульфонилгидразина (2) путем
низкотемпературной конденсации (—196°, жидкий Na). Литиевое
производное (2) получают реакцией тозилгидразина с б«с-(три-
метилсилил)-амидом лития [2] в бензоле при комнатной темпе-
ратуре. Пиролиз производного (2) проводят в высоком вакууме
(< 10“4 мм рт. ст.).
TsNHNH
ЬЩЗПСНзЬЪ
-HN[Si(CHah]2
Ts\ /н
/N—NZ
Lr \н
HN=NH
(1) (2) (3)
Восстановление двойных и тройных связей [3]. На последней
стадии стереоселективиого синтеза ц«с-4-метилгексеи-3-ола-1 (2)
происходит восстановление диенола (1). Эту реакцию осущест-
ви (2)
вляли с выходом 74% при использовании большого избытка гид-
разиигидрата и Н2О2 в этаноле. При этом было получено также
небольшое количество полностью восстановленного спирта; ка-
тализ ионами Си2+ увеличивает выход полимерного побочного
продукта. Восстановлением ацетилена (3) под действием Д.
в тех же условиях был получен енол (2) также с хорошим вы-
ходом. Для получения (2) предлагаемый метод удобнее, чем
(3)
HN (2)
71%
гидрирование ацетилена (3) над катализатором Линдлара с по-
следующим восстановлением Д,
130
1 Wiberg N., Bachhuber H., Fischer G., Angew Chem., Internal. Ed.,
’ II, 829 (1972).
2. Wannagut U., Niederpr uni H., Chem. Ben, 94, 1540 (1961).
3. Mori K-, Ohki M., Sato A., Matsui M., Tetrahedron, 28, 3739 (1972).
ДИИМИНОЯНТАРНОИ КИСЛОТЫ ДИНИТРИЛ (диими-
HN^ /CN
ИОСуКЦИНОННТрИЛ), )C—С
NCZ ^NH
Мол. вес 106,09, т. п. 165—166° (разл.), возгоняется при
10071 л-ьп.
Внимание\ Д. к. д. во влажном состоянии или при контакте
с гидроксилсодержащими растворителями выделяет цианистый
водород; реагент вызывает слабое раздражение кожи и слизи-
стой оболочки носа.
Получение [1,2]. Д. к. д. образуется с 96%-ным выходом при
катализируемом основаниями присоединении цианистого водо-
рода к дициану при —40°. Его также можно получить пропуска-
нием хлора в толуольный
—15° (выход 65%).
2HCN + NCCN
(и
4HCN + 2(CH3)3N + С12 —(1) 4-2(CH3)3NHCl
65%
раствор HCN и триметиламина при
Hbk yCN
хс—cz
(C2hsi3n
-------->
96%
Д. к. д. легко и количественно восстанавливается водородом
(Pd/C) до динитрила диамииомалеиновой кислоты, тетрамера
HCN [3]:
™4c-c/CN
мс/ *Ч[Н ДДХ
nh2
CN
(2)
(О
Реакции. При нагревании с водой Д. к. д. гидролизуется до
щавелевой кислоты; контролируемый гидролиз с помощью
TsOH в ТГФ дает оксалилцианид. Это вещество в высшей сте-
пени реакционноспособно и вводится в последующие реакции
2ТзОН V V
Н2° II J1 _ , k
(1) ----—> NCC—CCN 4-2TsO nh;
(2
без выделения. При взаимодействии с метанолом в условиях кис-
лотною или основного катализа Д. к.д. дает диметиловый эфир
Дииминощавелевой кислоты (3) с выходом 79,5%- Он реагирует
131
с аминами, например анилином, образуя дифенилоксамидин (4J.
+ 2 сн3он
79, 57?
+ 2 HCN
(1) (3)
(1) + 2 G6H5NH,-^
h2n .NC6Hs
cz
I
c6h9n^>nh2
(4)
С хлористым ацетилом Д. к. д. образует с очень низким вы-
ходом М,М'-диацетилдииминосукцинонитрил (5). Использование
уксусного ангидрида приводит к соединению (5) с выходом 11%.
NC4
С
1
NG NH
(1)
+ ЛсгО
NC /NCOCHj
____Чсг
CH3OCNZ 4CN
(5)
Прибавление избытка хлора к раствору Д. к. д. в ацетонит-
риле при температурах от —40 до —20° приводит к количест-
венному образованию М,Ь1'-дихлордииминосукцииоиитрила (6):
Ncx ^NCl
(i) + ci2 -sa™» ?
количеств,
NC 4NC1
(Ч
Азотистые гетероциклы [1, 4]. Д. к. д. чрезвычайно широко
используется в синтезе азотистых гетероциклов. Например, ои
вступает в реакции [2 + 4]-циклоприсоедииеиия с олефинами,
содержащими активированную электронодонорными заместите-
лями двойную связь. Так, реакция Д.к.д. (1) с цнс-1,2-диметок-
сиэтиленом (2) в ацетонитриле приводит к образованию 76%
2,3-диметокси-5,6-дициаио-1,2,3,4-тетрагидропиразина (3) с со-
хранением конфигурации.
NC ^NH Н3СО /ОСНЭ гм
HN^ XCN Н Н 76%
(1) (2)
132
При взаимодействии Д, к.д. с фосгеном при —20° в ТГФ с
высоким выходом образуется 4,5-дихлор-4,5-дициаиимидазоли-
дон-2 (4). Этот 1 : 1-аддукт неустойчив, он легко реагирует со
спиртами, давая с хорошим выходом 4,5-дициан-4,5-диалкокси-
имидазолидоны-2 (5). Реакция (4) с аминами дает имидазо-
лов (6).
ci н но н
ci н ио н
(4) (5)
10%
С двухлористой серой Д.к.д. образует 3,4-дицнан-1,2,5-ти-
адиазол (7), который легко гидролизуется до соответствующей
дикарбоновой кислоты (8).
NC
(1) + SC1Z --S
93% NC г/
(7>
HOOCr^N\g
НООС^к'
(8)
Катализируемая серной кислотой реакция Д.к. д. с 2,2-диме-
токсипропаном дает 2,2-диметил-4,5-дициаиизоимидазол (9) с
выходом 80%. Та же реакция, катализируемая щавелевой кис-
лотой, приводит к соединению (9) с выходом 80%.
осн3
(1) 4- сн3ссн3
осн3
(9)
При взаимодействии с самим ацетоном соединение (9) обра-
зуется с низким выходом.
1. В eg land R. W., Cairncross A., Donald D. S., Hart ter D. R.,
Sheppard W. A., Webster 0. W., J. Am. Chem. Soc., 93, 4953 (1971).
2. Webster O. W., H a r 11 e r D, R., В e g 1 a n d R. W., Sheppard W. A„
Cairncross A., J. Org. Chem., 37, 4133 (1972).
3. BredereckH., Schmotzer G., Oehler E., Ann., 600, 81 (1956).
< Be gland R. W., H artier D. R, J. Org. Chem., 37, 4136 (1972).
133
ДИКОБАЛЬТОКТАКАРБОНИЛ (I, 286—291; VI, 69—70).
Реакция с гем-дигалогенидами [1]. При взаимодействии Д.
с активированными гелг-дигалогенидами при 50° в ТГФ или в
бензоле сначала образуется продукт сдваивания, который за-
тем дегалогенируется до «димерного» олефина:
Со2(СО)8
R2CX2 R2C—CR2
Со2(СО)8
------> R2C=CR2
X X
Дихлордифенилметан и 9,9-дибромфлуорен в этой реакции дают
«димерные» олефины с хорошим выходом.
1. Seyferth D., Millar М. D., J. Organometallic Chem., 38, 373 (1972).
2,2'-ДИЛИТИЙДИФЕНИЛ (1).
Получение [1].
Образование атропоизомерных о-гексафениленов [2], При
обработке at-комплекса хрома (2), полученного из 2,2'-дилитий-
дифенила и хлорида хрома (III), галогенидами переходных ме-
таллов (C0CI2, FeCl3) образуются атропоизомеры о-гексафеии-
лена (За) и (36):
(За)
Центросимметричная форма Скрученная форма
Атропоизомеры о-гексафенилена
1. Gilman Н., Kirby J. Е., Kinney С. R., J. Am. Chem. Soc., 51, 2252
(1929); Collette J., McCreer D,, Crawford R., Chubb F., San-
din R. B., J. Am. Chem. Soc., 78, 3819 (1956); W i 11 i g G., Her wig W.,
Chem. Ber., 87, 1511 (1954).
2. Wittig G., Rumpier K.-D., Ann., 751, 1 (1971).
134
4-ДИМГ.ТИЛАМИ НО-1 -трет-БУТИЛ ОКСИКАРБОН ИЛ ПИ-
РИДИНИЯ ХЛОРИД (1). Мол. вес 258,75, т. пл. 50° (разл.),
растворим в воде.
Реагент получают прибавлением 4-диметиламинопиридина
(мол. вес 122,17, т, пл. 108—110°) к избытку трет-бутилового
эфира хлоругольной кислоты [1] в сухом эфире при 0°:
N(CH3)Z И(СНЭ)2
+ С1СОС(СН3)3 —> Г|М сг
О = СОС(СН3)3
(1)
С натриевыми солями аминокислот в водных растворах при
25° Д. х. с хорошими выходами образует трет-бутилоксикарбо-
ниламинокислоты (БОК-амииокислоты) [2].
I. Woodward R. В. et al., J. Am. Chem. Soc., 88, 852 (1966).
2. Guibe-Jampel E., Wakselman M., Chem. Comm., 1971, 267.
1,8-бис-(ДИМЕТИЛАМИНО)-НАФТАЛИН (VI, 71).
Усовершенствованный метод получения [1]. Это основание
получают с выходом 87% при обработке избытком диметил суль-
фата в присутствии гидрида натрия кипящего раствора 1,8-ди-
амиионафталина в ТГФ.
ЫН2 МН2 (СН3)2М М(СН3)2
+ 4 (CH3O)2SO2 + 4-NaH ТГФ > *
+ 4 NaSO3(OC&3) + 4 Н2
1. Quast H., R 1 s I e r W., D 6 11 s c h e r G., Synthesis, 1972, 558.
N,N-ДИМЕТИЛАМИ НОЦИКЛОПРОП ИЛ ФЕНИЛ СУЛЬФ-
ОКСОНИЯ БОРФТОРИД, Мол. вес. 309,15, т. пл. 121 — 122°.
о
c6H5s
n(ch3)2
(1)
Получение. Реагент можно получить из фенил-3-хлорпропил-
сульфида [1] или из циклопропилфенилсулъфида [2].
Спиросоединения [3]. Реакция Д. б. с гидридом натрия в
ДМСО приводит к циклопропилиду (2), который при взаимо-
действии с кетонами дает спиросоединения. Так, илид (2) обра-
135
зует с окисью мезитила (3) 1-ацетил-2,2-диметилспиропентан (4)',
который при 70° перегруппировывается в 1-ацетонил-1-изопро-
пеиилциклопропан (5).
о ДМСО
(СН3)2С=СНСОСН3 + С6Н5-3=<И 2Q1W„ >
N(CH3)a Ш
Реакция и лида (2) с циклогексаноном дает неустойчивый
диспироэпоксид (6), который в процессе препаративной ГЖХ
перегруппировывается в соединение (7).
(6) (?)
I. Zimmerman Н. Е., Thyagarajan В, S., J, Am, Chem, Soc,, 82, 2505
(1960).
2. Truce W. E., Hollister K. R., Lindy L. B., Parr J. E., J, Org.
Chem., 33, 43 (1968),
3. Johnson C. R., Katekar G. F., Huxol R. F., Janiga E. R., J. Am.
Chem. Soc., 93, 3771 (1971).
ДИМЕТИЛАЦЕТАМИД (I, 305—306; V, 115).
Винилирование карбазола (эффект растворителя) [1]. Най-
дено, что ДМ.А, ДМСО и М-метил-2-пирролидон являются са-
мыми подходящими растворителями для получения винил кар-
базола (2) из N-металлических производных карбазола (1), где
М —Li, Na, К, и ацетилена.
(1),М^Ы, Na, К
HCSCH
1. Sandler S, R.f Chem, Ind., 1973, 134.
133
ДИМЕТИЛАЦЕТАМИДА ДИМЕТИЛ АЦЕТАЛЬ,
СНз
(CH3)2NC(OCH3):. Мол. вес 133,19.
Получение Д.д. описал Меервейн [1].
Реакция с вицинальными rfuc-диолами [2]. Д. д. (2) взаимен
действует с вицинальными цме-диолами, такими, как метил-0-D-
рибофуранозид (1), в 1,1,1-трихлорэтане, образуя ацеталь (3)
с выходом более 80%. Это производное устойчиво в отсутствие
влаги, но количественно
нон2с осн,
W
он он
(1)
НОН2С ОСН,
сн3 II
(CH^N-cjoCHab (2),
сн3он, н2о V-м
V
/с\
(СНЦДГ .сн,
(3)
НОНгС о РСН3
(3)
НОАс>
НО од с
НОН2С о осн3
* У
АсО ОН
(4) (5)
гидролизуется при кипячении в 3%-ном водном метаноле (реак-
цию можно проводить также при комнатной температуре). Д.д.
(2), следовательно, может применяться для защиты вициналь-
ных диолов. Прн pH 4—5 в 95 %-ной водной уксусной кислоте
ацеталь (3) расщепляется, давая смесь приблизительно равных
количеств 2- и 3-моноацетатов (4) и (5).
Подобным же образом можно использовать диметилацеталь
КЫ-диметилбензамида.
Перегруппировка Клайзенаамиды [3]. Д4-Холестеиол-3£
с т. пл. 130,5—131° можно получить по методу Бергсталлера и
Нордина [4]; заниженная т. пл. получаемого таким путем про-
дукта свидетельствует о загрязнении его некоторым количеством
5-а-оксиизомера. В круглодоиную колбу объемом 50 ж л, снаб-
137
женную магнитной мешалкой, ротор которой покрыт тефлоном,
и обратным холодильником, соединенным с трубкой для ввода
газа, помещают 0,97 г (2,5 ямоля) Д4-холестенола-3{3 и 30 мл
о-ксилола и перемешивают смесь до полного растворения.
К полученному раствору добавляют 1,67 г (12,5 лсмоля) Д.д.
Колбу заполняют аргоном, нагревают ее содержимое на песча-
ной бане до кипения и далее кипятят при избыточном давлении
аргона и интенсивном перемешивании в течение 65 час. После
охлаждения летучие вещества отгоняют иа роторном испарите-
ле, остаток высушивают в вакууме (0,1 мм рт. ст.) в течение
1 час, при этом получают 1,2 г желтого масла, которое хрома-
тографически разделяют на 60 г силикагеля, элюируя эфиром.
Элюирование 200 мл эфира дает смесь холестадиенов, которую
выбрасывают; дальнейшим элюированием еще 500 мл эфира
выделяют 0,74 г 5р-М,М-диметилкарбоксамидометилхолестена-3
в виде прозрачного бесцветного масла, которое при растирании
с ацетоном дает 0,74 г (65%) амида в виде белых пластинок
с т. пл. 128—129,5°.
Реакция с аллиловыми спиртами (I, 306—307) [5]. Кипяче-
ние транс-пентен-З-ола-2 (1) с Д. д, илн с его синтетическим
эквивалентом 1-метокси-1-диметиламиноэтиленом [6] в ксилоле
в течение 17 час дает Ы,М-диметиламид З-метил-транс-гексен-4-
овой кислоты (2) с выходом 80%. При проведении этой реакции
с (S)- (—)-транс-пентен-З-олом-2 амид (2) образуется более чем
90%-ной оптической чистоты и с обращением конфигурации. Со-
ответственно высокая асимметрическая индукция наблюдалась
N(CH3)a
СН3С(ОСН3)3
CHa=CN(CH3)2
осн3
во?£ > н3с
С)
в реакции оптически активного спирта (1) с триэтиловым эфи-
ром ортоуксусиой кислоты (VI, 202—204), в результате которой
образовался этиловый эфир (3).
, сн3с(ос2н5)3 сн3соосгн5
(3)'
I. М.еегwеin Н., Florian W., Schon N., Stopp G., Ann., 641, 1
(1961).
2. Hanessian S., M о г a 11 о g 1 u E., Tetrahedron Letters, 813 (1971); Can.
J. Chem., 50, 233 (1972).
3. Ireland R. E,, Dawson D. J., Org. Syn., submitted 1971.
138
4. Bnrgstahler A. W„ Nor din 1, C., J. Am. Chem. Soc., 83, 198 (1961).
5. Hill R. K., Soman R., Sa wad a S., J. Org. Chem., 37, 3737 (1972).
6. В re derec к H., Effenberger F., Simchen G., Chem. Ben, 36, 1350
(1963).
бис-(3,6-ДИМЕТИЛ)-БОРЕПАН (1). Мол. вес. 274,12.
Получение [1]. Этот диалкилборан (1) получают с выходом
84% при кипячении 2,5-диметилгексадиена-1,5 с бораном в ТГФ
в течение 1 час. бис- (3,5-Днметил)-боринам (2) был получен
н,с
1) ТГФ , 0°
2) кипячение
84$
аналогичным образом из 2,4-диметилпентадиена-1,4.
(2)
Гидроборирование [1]. Оба б«с-борациклана (1) и (2) яв-
ляются высокоселективными агентами, позволяющими количе-
ственно проводить гидроборирование олефинов в спирты против
правила Марковникова. Они сравнимы с 9-борабицикло-[3,3,1]-
ионаном, 9-ББН (V, 35—36; VI, 25—30; этот том).
Синтез вторичных спиртов из алкинов-1 [2]. Дигидробориро-
вание терминального алкина при комнатной температуре дейст-
вием Д. (1) или 9-ББН приводит к образованию 1,1-диборил-
алкана (3). Обработка последнего при 0—5° 1 экв метиллития
в эфире дает продукт (4), тотчас же превращающийся в соеди-
нение (5). При добавлении к нему 100%-ного избытка алкилга-
логенида образуется вторичный алкилборан (6), который окис-
ляют щелочным раствором перекиси водорода до вторичных
спиртов (7) с 70—85%-ным выходом.
RCssCH + (1)
I
-CH
I.
— В
RCH2-CH
CH3LR
—~~г—
КСНг
L? CH3
(4)
в J
139
R~CHS-CH-B^;)
t U >-R’^ RCH^CHR1 RCH,CHR’
V \ I
В OEI
RCH,CH=B I !f \
u+
(5) (6) <7)
1. Brown H, C., Negishi E., J. Organometallic Chem., 28, Cl (1971).
2. Zweifel G., Fisher R, P,, Horng A,, Synthesis, 1973, 37.
Д ИМЕТИЛ БРОМСУЛ ЬФОНИЯ БРОМИД, (CH3)2SBr2.
Мол. вес 221,97, оранжевые кристаллы, т. пл. 81—82° (разл.).
Получение. Реагент получают взаимодействием диметилсуль-
фида с бромом. В темноте без доступа влаги его можно хранить
в течение нескольких недель при комнатной температуре.
Алкилбромиды [1]. Д. б. реагирует со спиртами (в избытке)
при ~80° в течение 4—5 час, образуя алкилбромиды с выходом
40—80%. Реакция протекает с обращением конфигурации. Так,
(СН3)гЗВг2 + ROH —> [(CH3)2SOR]Br- —> RBr + (CH3)2SO
(-р)-втор-октаиол при взаимодействии с Д. б. дает (—) -втор-
октилбромид (выход 70%, оптический выход 91%). См. также
стр. 159—160 этого тома.
1. Furukawa Н., Inoue Т., Aida Т., Оае S., J. С. S, Chem. Comm.,
1973, 212.
2,3-ДИМЕТИЛ БУТИЛ-2-БОРАН (тексилбораи), ThBH2.
(I, 307—308; V, 115—117).
Селективное восстановление. Браун и сотр. [ 1 ] подробно
изучили восстановление большого числа органических соедине-
ний тексилбораном и свои результаты сравнили с результатами,
полученными при использовании диборана (ВН3) и «днизоамил-
борана» (Sia2BH, I, 313—316; V, 123; VI, 84—86). Все три вос-
становителя обладают сходными характеристиками, за исклю-
чением лишь незначительных различий. Так, ThBH2 и Sia2BH
при восстановлении кетоиов проявляют меиьшую стереоселек-
тивиость по сравнению с ВН3. При восстановлении эфиров их
реакционная способность уменьшается в ряду ВН3 > ThBH2 >
7> Sia2BH.
Миогоуглеродиая гомологизация олефинов [2]. В новом об-
щем методе синтеза кетонов ThBH2 реагирует с эквимолярным
количеством олефина, образуя тексилмоноалкилборан (1). До-
бавление к (1) со-алкенилалкилацетата приводит к тексилди-
ал кил борану (2), карбонилирование которого с последующим
140
окислением дает гомологизованный кетон (3). Выходы колеб-
сн3 сн3 сн3 сн3 „
HLиД — Д — И НгС^СН-(СНг)п-ОАС >
сн3 сн3 Анэ Дн3
(1)
<fH3 < сн3 ZR 1 со/нго о
нс_< СН3 ( i:—в' 5н3 (СН3)^_|_^ОАс i) ЩОг/NaOAc „ Д ч » ac-lcH^a-OAc
(2)
(3)
лютея в интервале 60—75%. Суммарный процесс может быть
представлен в следующем виде:
СН3СНгСН2СН^СН2 + СН2=СН(СН3)эОАс + СО —>
О
СН3(СНг)4С(СН2)пОАс
Синтез транс-дизамещенных олефинов. Браун и сотр. [3] со-
общили о новом стереоселективиом синтезе транс-дизамещен-
ных олефинов (4) с использованием в качестве промежуточных
соединений тексилмоиоалкилбораиов (1). Их получают, дейст-
вуя на олефин ThBH2 в атмосфере азота при —25° в течение
1 час. Затем добавляют 1-бромалкнн-1 и по истечении 1 час
образующийся тексилалкил-1 -бромалкенил- 1-бораи (2) обраба-
тывают метилатом натрия при 25° в течение 1 час. Промежуточ-
ное соединение (3) гидролизуют кипячением с изомасляной кис-
лотой (1 час). Выходы составляют 80—95%.
сн, сн3
I I
н-с—с—вн2
I I
сн3 сн3
сн3 СНз
н—с—с—в
I I
СН3СН3
cHjchs
S5 Н-С—С — В
I I хн
СН3 снз
О)
Br-Q~CR2, —25°
СН3СН3 /R1
Н—С—Д—в Н
I I \ /
СН3СН3 с = с
Вг \г
(2)
NaOCH,, 2 5'
R1
СНз СН3 \ 'н КЗО-С3Н7СООН „1 „
___. „ I j, z кипячение х
---> Н-С—С—В^ R2 --------------> С=СХ
СН3 СН3 ОСНэ Н/ (4) R*
(3)
141
Таким путем, например, из транс-гексепа-З и 1-бромгексина-1
был получен с выходом 78% (выделенного) rpa/ic-4-этилде-
ден-5:
сн3сн2сн2
сн3се2сн^ zh
/с^с\
Н (СНг)зСНз
(5)
I. Brown Н. С,, Heim Р„ Yoon N. М„ J. Org. Chem., 37, 2942 (1972).
2. N е g i s h i Е., В г о w n Н. C., Synthesis, 1972, 196.
3. N e g i s h i E., Katz J.-J., Brown H. C., Synthesis, 1972, 555.
C(CH3)3
ДИ MET И Л-трег-БУ T И ЛХ ЛОРС ИЛАН, (CH3)2S iCl. Мол.
вес. 151,73, т. пл. 92,5°, т. кип. 125°/733 мм.
Получение. К 12И раствору диметилднхлорсилана (1,15эюз)
в пентане при 0° в атмосфере азота при перемешивании прибав-
ляют по каплям раствор трет-бутиллития в пентане. Темпера-
туру поддерживают при 0° в течение 1,5 час и затем при 25° в
течение 48 час. Перегонкой при атмосферном давлении полу-
чают чистый препарат (т. кип. 125°) с выходом 70% [1]. См.
также получение реагента, описанное Соммером и Тайлером [2].
Защита гидроксильных групп [1]. Триметилсилиловые эфиры
вследствие повышенной чувствительности к сольволизу в про-
тонсодержащих средах не нашли широкого применения в син-
тетической практике. Диметнл-трет-бутилсилоксильная группа,
однако, почти в 104 раз устойчивее триметилсилоксильной груп-
пы. В ТГФ в присутствии избытка пиридина Д. лишь медленно
реагирует со спиртами, но если в качестве катализатора исполь-
зовать имидазол (II, 27—30; V, 208), а в качестве растворите-
ля— ДМФА, то диметнл-трет-бутилсилиловые эфиры образуют-
ся в мягких условиях с высокими выходами. В этом случае си-
лил ирующим агентом является, вероятно, N-диметил-трег-бу-
тилсилилимидазол, Полученные эфиры устойчивы к действию
водных и спиртовых растворов оснований, к гидрогенолизу
(Н2—Pd) и к восстановлению в мягких условиях (Zn—СН3ОН).
Диметил-трег-бутил силильная группа является эквивалентом
тетрагндропираиильиой, однако обладает тем преимуществом,
что не имеет хирального центра.
Диметил-грег-бутилсилиловые эфиры расщепляются смесью
уксусной кислоты с водой (2: 1) при 25° или под действием фто-
рида тетра-н-бутиламмония (см. этот том) в ТГФ при 25°.
Эти новые эфиры были широко изучены в ходе исследова-
ний, посвященных синтезу простагландинов и представляют осо-
бую ценность именно в этой области химии.
U2
трет-Бутилдиметилсилильная группа рекомендуется [2, 3] так-
же для защиты 5'-первичных гидроксильных групп нуклеозидов.
Например, реакция тимидина (1) с Д. в ДМФА в присутствии
имидазола (2,5 мол. экв) приводит к образованию простого
эфира (2) с 60%-ным выходом.
он он он он
(!) (2)
Эта группа устойчива к действию слабых оснований (15%'
ный NH4OH в этаноле) и сохраняется в обычных условиях
фосфорилирования. Она легко снимается 80%-ной уксусной кис-
лотой (паровая баня, 15 мин) или фторидом тетр а-н-бути л ам-
мония (22°, 30 мин).
1, Corey Е. J., Venkateswarlu A., J. Am. Chem. Soc., 94, 6190 (1972).
2. Sommer L. H., Tyler L. J., J. Am. Chem. Soc., 76, 1030 (1954).
3. Ogilvie K. !<., Iwacha D. J., Tetrahedron Letters, 317 (19/3).
ДИМЕТИЛДИХЛОРСИЛАН, (CH3)2SiCl2 (VI, 74). Мол, вес
129,07, т. кип. 70,3°.
Восстановительная циклизация с образованием пинакона [1].
Ключевой стадией нового метода синтеза карбоциклической си-
стемы, характерной для гибберелловых кислот, является вос-
становительная циклизация кетоальдегида (1) в пинакон (2).
Эту циклизацию осуществили с выходом 75% при добавлении
соединения (1) к смеси амальгамы магния (7,5 эле) и Д. (2экв)
в сухом ТГФ при 25° с последующим щелочным десилилирова-
иием. Продукт (2) получается в виде смеси цис- и транс-диолов
в отношении 80:20. Использование Д. оказалось решающим
для успешного проведения циклизации, так как без него полу-
чалась сложная смесь продуктов. Ср. с применением триметил-
143
хлорсилана в родственных реакциях (III, 390—391; V, 439—442;
VI, 276—278; этот том).
1. Corey Е. J., Carney R. L., J. Am. Chem. Soc., 93, 7318 (1971).
ДИМЕТИЛМЕДЬЛИТИЙ (V, 120—122; VI, 74—81).
Сопряженное присоединение. Опубликован обзор [1] реакций
сопряженного присоединения медьорганических соединений.
Сопряженное присоединение Д. к 3,4,5,6,7,8-гексагидроиафта-
лин-1 (2Н)-он-7-карбоновой кислоте (1) дает смесь трех стерео-
изомерных 4й-метил-3,4,4а,5,6,7,8,8а-октагидронафталин-1 (2Н)-
он-7-карбоновых кислот (2), (3) и (4) в отношении 4 : 3 : 2. Реак-
ция нашла применение в синтезе сесквитерпена р’-эйдесмола,
проходящем без стадии аннелироваиия по Робинсону [2].
Реагент использовали [3] также в полном стереоселектнвном
синтезе рацемической формы трициклического сесквитерпена сей-
челлена (8), одного из компонентов, содержащихся в незначи-
тельных количествах в масле пачули [4]. В качестве исходного
соединения использовали кетон Виланда — Мишлера (5), кото-
рый превращали в соединение (6) селективным восстановлением
боргидридом натрия с последующим превращением спирта в
144
тетрагидропираниловый эфир. Последний обрабатывали Д. при
—25°, а затем ацетилировали ацетил хлоридом. Из полученного
таким путем соединения (7) синтезировали в 10 стадий сей-
челлен.
Реакция Д. с а,^-ненасыщенным кетоном (9) в эфире при 0°
приводит в основном к ожидаемому 1,4-аддукту (10) с выходом
55%, однако при этом побочно образуются два продукта рас-
щепления циклопропанового кольца: 39% (11) и 6% (12). Про-
дукт 1,4-присоединения содержит главным образом транс-изо-
мер [5]. Расщепление циклопропанового кольца происходит, по-
видимому, лишь при наличии в молекуле кетона сопряженной
с карбонильной группой двойной связи. Так, бицикло-[4,1,0]-геп-
танон-2 (13) не вступает в реакцию с Д. [6].
о
СН3)гСuL.|_|е реагируют
(13)
Сопряженное присоединение Д. к 2-метил-б-кетооктагидро-
изохинолину (14) дает исключительно 2,Ю-диметил-6-кетодека-
гидроизохинолин (15) с ipc-сочлеиением ядер [7]:
Сопряженное присоединение к сопряженным диенонам. Мар-
шалл и сотр. [8], исследовавшие взаимодействие Д. с циклогек-
садиенонами, пришли к заключению, что соотношение 1,4- и
1,6-присоединения определяется пространственными факторами.
Последняя из приведенных ниже реакций является примером
того случая, когда ангулярное метилирование практически не
происходит.
145
Примеры:
(3) 14)
Циклизация [9]. Реакция иодсодержащего эфира (1) с Д.
помимо ожидаемого метилированного соединения [(2) выход
30%] дает еще три продукта—(3), (4) и (5)—в отношении
50:45:5. Особый интерес представляет соединение (3), посколь-
ку формально его образование можно рассматривать как ре-
зультат внутримолекулярного нуклеофильного присоединения
винильной группировки по двойной связи с последующей ал-
лильной перегруппировкой.
(1) (2)
сн3
'V/CHjCH
CsCCHzOH
(4) (5)
Ufi
Сесквитерпены группы эйдалена. В методах синтеза сескви-
терпенов группы эйдалена для построения бициклической си-
стемы обычно применяют реакцию аннелирования по Робинсо-
ну. Однако реакция аннелирования часто протекает с низкими
выходами и не всегда стереоспецифично. Хафмен и Моул [10]
недавно сообщили о новом стереоселективном синтезе. 8-Меток-
ситетралин-2-карбоновую кислоту (I) восстанавливают по Бер-
чу, затем реакционную смесь обрабатывают кислотой. При этом
13) 3 изомера
(4)
образуется смесь четырех соединений, из которых методом хро-
матографии можно выделить ненасыщенную кетокислоту (2)
с выходом 49%. При введении в нее ангулярной метильной
группы реакцией с Д. образуются примерно в равных количе-
ствах три изомера кетокислоты (3) с почти количественным вы-
ходом. Следующую стадию, реакцию Виттига с метилентрифе-
нилфосфораном, проводят со смесью изомеров (3), поскольку
эта реакция как с цис-, так и с транс-декалоном-! приводит к
образованию продуктов преимущественно с тронс-сочленением
колец. В данном случае в результате реакции образуется смесь
трех кислот примерно в отношении 1 :3:6. Этерификация, взаи-
модействие с раствором метилата натрия в метаноле и гидролиз
дают желаемую 23-кислоту (4) с выходом 75%, считая на
смесь (3). Кислоту (4) превращали в (3-эйдесмол, а последний
в свою очередь — в криптомеридиол и неомеридиол.
Пространственно затрудненные кетоны. Д. (и родственные
реагенты) вступает в реакцию диспропорционирования с таки-
ми а-бромкетонами, как, например, (1), образуя стерически за-
трудненные кетоны, например, такие, как (2), Можно использо-
вать даже третичные купраты лития [11].
(CH3)aCuLl «30-Р1Ч
«зо-Рг—СНСО—«зо-Ви ---------'СНСО—изо-Вц
Н8с/
Вг
(1)
(2)
147
Получение фосфатов енолов [12]. Д. в эфирном растворе
используют для введения ангулярной метильной группы в Д4-хо-
лестенон-3 (1) с одновременным превращением его в соответст-
вующий енолят лития (2), который затем реакцией с диэтил-
хлорфосфатом в присутствии триэтиламина превращают в фос-
фат енола (3). Взаимодействие последнего с литием в этил-
амине и последующая обработка трет-бутанолом приводят к
образованию 5-метил-Д3-копростена (4).
Реакция с «^-ненасыщенными сульфонами [13]. Альдегиды
и кетоны можно превратить в аеж-диалкилалканы (3) следую-
щим образом. Сначала из альдегидов и кетонов получают
а,^-ненасыщенные сульфоны (1) (см. н-Бутиллитий, этот том),
обрабатывают их диалкилмедьлитиевым реагентом и получают
с хорошим выходом алкиларилсульфоны (2). Гидрогенолиз по-
следних проводят 6%-ной амальгамой натрия в кипящем эта-
ноле [14].
R\
^C=CHSO2Ar
R2Z
1) Д2СиЫ/(СгНБ)2О, 0°
2) Н+
(1), Аг = л-С1СеН4
СН2 SO2Ar
R2
55-95%
6% Na —Hg
C2H5OH
55—100%
Получение метилкетонов из хлорангидридов карбоновых кис-
лот (VI, 80). Познер и сотр. [15] нашли, что метод синтеза кето-
нов из хлорангидридов кислот и медьорганических реагентов
применим и в случае хлорангидридов кислот, содержащих иод-,
циан-, ацил- и карбалкоксигруппы. Эта реакция, однако, имеет
некоторые ограничения. Альдегидная группа реагирует с диал-
(С2Н5)йО
— 78°
R'COCl + R2CuLi -----> R'COR
148
килмедьлитием даже при температуре —90°. Поэтому для до-
стижения оптимальных выходов целевого продукта требуются
три эквивалента R2CuLi. Другим недостатком рассматриваемой
реакции является то обстоятельство, что литийоргаиические реа-
генты обычно менее доступны, чем магнийорганические.
Реакция с бензилхлоридами. Познер и Брюнель [16] приво-
дят следующие примеры реакции Д. с бензилхлоридами в ди-
этиловом эфире:
(CHgJjCuLi
CSHECHC12 ----------->- CSH5CH(CH3)2 + CeH5CH~CHCfiH6
сн3 <Lh3
40% 40%
(СНз)гСиы
СеН5СНС1 -----~~----С6НЕСН(СН3)2 + С6Н5СН-СНС6Н5
I II
СН3 СН3 СН3
40% 40%
(CH3)2CuLi
СеН5СН2С1 -------—> СвНьСН3СН3
80%
(СН3)2СиЫ
(СеНЕ)2СС12 ~ (CsH5)2C=C(C6H5)2
81%
Реакция с моноокисями 1,3- н 1,4-цнклогексадненов. Д. вза-
имодействует с 3,4-эпоксициклогексеном-1 (1), образуя продукт
нуклеофильного раскрытия эпоксидного цикла (2) и продукт
сопряженного присоединения (3), обладающие т’рйнс-конфигу-
рацией [17]:
(1) (2) 35% (3) 42% (4) 23%
Аналогичные результаты были получены Виландом и Джон-
соном [18].
С другой стороны, с моноокисью циклогексадиеиа-1,4 (5) Д,
дает исключительно продукт транс-р а скрытия оксиранового
кольца (6) [17]:
(5)
(снз)гСцД^
149
Реакция с винилбромидами, Реакция замещения атома бро-
ма в винильном положении при действии Д. протекает с сохра-
нением конфигурации [19]. Так, r/шнс-а-бромкоричная кислота
(1) взаимодействует с Д., образуя после обработки подкислен-
ной водой транс-а-метилкоричную кислоту (2), Такое поведе-
ние алкилиденбромидов прямо противоположно поведению на-
сыщенных галогеналкилов в реакции с диалкилмедьлитиевымн
реагентами, где наблюдается обращение конфигурации [20].
СйН5. ,Вг
с=с<
Н/' ^соои
( С Н;) 3 С И1Л
СЙН; СН3
о—сд
и/ ^соон
(1)
(2)
1. Posner G. Н., Org. Reac., 19, 1 (1972).
2. Н u f f m a n J. W., M. о 1 е М. L., J. Org. Chem., 37, 13 (1972).
3. Piers Е., de Waal W., Britton R. W., J. Am. Chem. Soc., 93, 5113
(1971).
4. Wolff G., О u r i s s о n G., Tetrahedron, 25, 4903 (1969).
5. Marshall J. A., Ruden R. A., J. Org. Chem., 37, 659 (1972).
6. Marshall J. A., Ruden R. A., Tetrahedron Letters, 2875 (1971).
7. Sicsic S., L u о n g - T h i N.-T., Tetrahedron Letters, 169 (1973).
8. Marshall J. A., Ruden R. A., H i г s c h L. К., P h 1111 p p e M., Tetra-
hedron Letters, 3795 (1971).
9. Katzenellenbogen J. A., Corey E. J., J. Org. Chem., 37, 1441
(1972).
10. Huffman J. W„ Mole M. L., Tetrahedron Letters, 501 (1971).
11. Dubois J.-E., Lion C., Moulineau C., Tetrahedron Letters, 177
(1971).
12. Muchmore D. C., Org. Syn., 52, 109 (1972).
13. Posner G. H., Brunelle D. J., Tetrahedron Letters, 935 (1973).
14. Dabby R. E., Kenyon J., Mason R. F., J. Chem. Soc., 1952, 4881.
15. Posner G. H., Whitten С. E., McFarland P. E., J. Am. Chem. Soc.,
94, 5106 (1972).
16. Posner G. H., Brunelle D. J„ Tetrahedron Letters, 293 (1972).
17. Staroscik J., Rickborn B„ J. Am. Chem. Soc., 93, 3046 (1971).
18. Wieland D. M., Johnson C. R., J. Am. Chem. Soc., 93, 3047 (1971).
19. К 1 e 1 n J., L e v e n e R., J. Am. Chem. Soc., 94, 2520 (1972).
20. W h i t e s i d e s G. M., San F i 11 i p p о J., Jr., Casey С. P., Pa-
nek E. J., J. Am. Chem. Soc., 89, 5302 (1967); White si des G. M.,
Fischer W. F., Jr., San Fillip po J., Jr., В a she R. W.,
House H. O., J, Am. Chem. Soc., 91, 4871 (1969).
N, N-ДИМЕТИЛМЕТИЛЕНАММОНИЯ ИОДИД,
(CH3)2N=CH2r. Мол. вес 185,01, разл. около 240°.
Соль легко получить реакцией триметиламина с иодистым ме-
тиленом в смеси диоксана с этанолом. Нагревание первоначаль-
но образующегося продукта (1) при ~50° в течение примерно
12л-ш« в сульфолане приводит к Д. и. (2) с выходом 81% [1].
+ ISO0, —СН31 +
(CH3)3N+CH2I2 —> (СНЭ)3ИСН21Г ------------------—---->- (CH3)2N=CH2r
рУ 70 ТО
(1) (2)
150
Метиленаммониевая соль (2)' — чрезвычайно активный реа-
гент Манниха. Ее использовали для введения метильной группы
в ядро коррина, При перемешивании раствора коррина (3) в
хлористом метилене с Д. н. (2) образуется чистое 15-(диметил-
амино)-метилпроизводное (4) с выходом 85%, Полученное про-
изводное (4) превращали в 15-метилкоррин (5) каталитическим
гидрированием [1],
(з), к = н
(4), Rs CH2-N(CH3)2
(5), R = СН3
Региоспецифичный синтез оснований Маиииха. Хуз и Бридсон
[2] описали новый региоспецифичный (об определении термина
«регио» см, [3]) синтез некоторых оснований Манниха, Сначала
реакцией триалкилборана с а-диазокетоном в ТГФ получают
эфир енола и диалкилборной кислоты (1) [4]. По прекращении
выделения азота прибавляют Д, и, (2) в ДМСО и после гидро-
лиза реакционной смеси получают основание Манниха (3) с вы-
ходом 85—100%. Для обеспечения высоких выходов в качестве
сорастворителя следует использовать ДМСО,
R'
R3B + R'COCHN2 —R2BO(L=CHR 4- (CH3)2N=CH2r —>
(1) (2)
—> R'CCHR
ii!
OCH2N(CH3)2
О)
Например, последовательная обработка триэтилборана ди-
азометил-н-пропилкетоном (ТГФ), а затем Д. и, (1,15 экв в
ДМСО) приводит к образованию 3-димети л аминометилгепта-
нона-4 (4) с 91%-ным выходом,
Д. и.
(С2Н5)3В + CH3(CH2)2COCHN2 —> —СН3(СН2)2СОСНСН2СН3
У1 |
CH2N(CH3)2
(4)
Эфиры енолов и борной кислоты являются промежуточными
продуктами также при взаимодействии борорганических соеди-
нений с а,^-ненасыщенными кетонами и альдегидами. Так, реак-
ция между триэтилбораном и З-метилбутен-З-оном-2 (5) с по-
1Э1
следующим добавлением Д. и. в ДМСО приводит к образова-
нию основания Маиииха (6) с выходом 87%.
, Н2Сх хСН,
(С2Н5)3В + v
А
о чсн3
(5)
1. Schreiber J., М a a g Н., Hashimoto N., Eschenmoser A., An-
gew. Chem., Internal. Ed., 10, 330 (1971).
2. Hooz J„ Bridson J. N., J. Am. Chem. Soc., 95, 602 (1973),
3. H a s s ne r A., J. Org. Chem., 33, 2684 (1968).
4. Hooz J., Bridson J. N„ Can. J. Chem., 50, 2387 (1972); см. приведенные
в работе ссылки.
М,1Ч-ДИМЕТИЛМЕТИЛ ЕНАММОНИЯ ТРИФТОРДЦЕТДТ
(VI, 81—82).
Опубликована заключительная статья французских исследо-
вателей [1].
1. Aho nd А„ Cave A, Kan-Fan С., Potler Р., Bull. soc. chim. France,
1970, 2707.
бис-(1,2-ДИМЕТИЛ ПРОПИЛ-1)-БОРДН («диизоамил бо-
ран»), Sia2BH (1,313—316; V, 123; VI, 84—86).
Восстановление ацетиленов (I, 314) [1]. На последней стадии
синтеза полового аттрактанта насекомых ацетата цис-додецен-8-
ола-1 (2) [2] проводят восстановление ацетилена (1). Эту реак-
цию осуществляют с помощью Sia2BH и затем обработкой пе-
1) (Sla)2BH
диглим
2) НОАс
3) Н2О3
СН3(СН2)2С=С(СН2)6СН2ОСОСНз —-—>
бои
(1)
н н
I 1
— •> СН8(СН2)2С=С(СН2)вСН2ОСОСН3
(2)
рекисью водорода удаляют борсодержащие
иие (2), содержащее >98% ^пс-изомера,
дом 66%.
примеси; соедине-
получают с выхо-
1. Н о 1 a n G., O’Keefe D. F., Tetrahedron Letters, 673 (1973).
2. R о е1 о 1s W. L., Comeau A., Selle R., Nature, 224, 723 (1969).
ДИМЕТИЛ-М-СУКЦИНИЛСУЛЬФОНИЯ ХЛОРИД,
о
£^N-s(CH3)aci'‘. Мол. вес 163,67, т. пл. 70—72°.
о
162
Вильсмайер и Шпрюгель [1] получили этот комплекс с 85 % -
ным выходом реакцией N-хлорсукцинимида с диметилсульфи-
дом в хлористом метилене при 0°. Было показано, что он яв-
ляется промежуточным продуктом при получении хлорметилме-
тилсульфида из N-хлорсукцииимида и диметилсульфида:
(сн3)г5 +
\ *
N-S(CH3)jCl
> CH3SCI4C1 +
Окисление первичных и вторичных спиртов до карбонильных
соединений. Кори и Ким [2] нашли, что комплекс N-хлорсукцин-
имида с диметилсульфидом в некоторых отношениях превосхо-
дит комплекс диметилсульфида с хлором (этот том), также
применяемый для окисления первичных и вторичных спиртов,
В реакции с ним не образуется свободный хлористый водород,
и поэтому выходы карбонильных соединений, как правило, бы-
вают выше. Метод иллюстрируется примером окисления 4~трет~
бутилциклогексанола (2) до 4-трет-бутилциклогексанона (4),
Комплекс (1) получают добавлением диметилсульфида
(4,1 жмоля) к перемешиваемому раствору NXC (3,0 жмоля) в
толуоле при 0° в атмосфере аргона. Смесь охлаждают до —25°
и прибавляют к ней по каплям раствор 4-трет- бутил циклогек-
санола (2,0 лмоля, смесь цис- и транс-изомеров) в толуоле. Ре-
акционную смесь перемешивают еще 2 час при —25°, затем до-
бавляют по каплям раствор триэтиламина (Зл/моля) в толуоле.
Кетон (4) получают почти с количественным выходом. Как и
при окислении комплексом диметилсульфида с хлором, в рас-
сматриваемой реакции промежуточно образуется сульфоксоние-
(3)
[(CxHshN
О
3 + (CHj)iS + (ОД^МНСГ
С(СНЭ)3
(4)
153
вый комплекс (3). По аналогичной методике окисляют окта-
нол-1 до октаналя-1 с выходом 96%.
Новый метод окисления имеет, однако, одно серьезное огра-
ничение. Аллиловый и бензиловый спирты в этих условиях не
окисляются, вместо этого наблюдается замещение гидроксила
хлором. В полярной среде может протекать и другая реакция.
Так, если в качестве растворителя использовать смесь хлористо-
го метилена с диметилсульфоксидом, то в заметных количествах
образуется хлор метил метил сульфид.
Кори и Ким [3] использовали Д. х. в улучшенном методе син-
теза простагландинов. Так, с его участием оксикислоту (5) окис-
ляли до кетокислоты (6) с выходом >90%. Раньше это окис-
ление проводили реактивом Джонса при —20° и получали ке-
токислоту (6) с выходом около 70%, Согласно новой методике,
карбоксильную группу кислоты (5) сначала защищают in situ,
превращая ее в изопропилдиметилсилиловый эфир. Для этого
кислоту (5) в толуоле обрабатывают изопропилдиметилсилил-
хлоридом в присутствии триэтиламина. При добавлении к ре-
акционной смеси окислителя образуется силиловый эфир кис-
лоты (6), который затем гидролизуют разбавленной соляной
кислотой и выделяют кетокислоту (6).
В той же работе описано окисление окснлактона (7) до аль-
дегида (8) комплексом метилфенилсульфида (тиоанизола) с
хлором состава 1 : 1 в системе растворителей CCU—CH2CI2.
CI2-CHsSC6H5
СС1^-СН2С1г
9 3$
(7}?к =П-С6н5едсо
(8)
Превращение аллиловых и бензиловых спиртов в галогениды
[4]. Как уже было сказано выше, аллиловые н бензиловые спир-
ты не окисляются под действием Д. х., вместо этого они превра-
щаются в хлориды. Кори н сотр. [4] недавно предложили вос-
пользоваться этим приемом для получения с высоким выходом
аллил- или бензилхлоридов. Для этого достаточно оставить
154
промежуточное сульфоксониевое производное, соответствующее
(3) (см. выше), свободно разлагаться в хлористом метилене
без добавления третичного амина. Например, аллиловый или
бензиловый спирт обрабатывают Д.х. в хлористом метилене,
как описано выше, и затем выдерживают реакционную смесь
при —25° в течение 4 час. В случае бензгидрола [(С6Н5)2СНОН]
бензгидрилхлорид можно получить с выходом >95%.
о
[^N-S(CH3hCl Ж RjRjCHOH
(1)
+
(CHjJjjSOCHRjRj
сГ
(2)
-25е, 4 час
RiRjCHCl + (CHPjSO
(3)
В тех же условиях циклогексен-2-ол-1 превращается в 3-хлор-
циклогексен тоже с выходом, превышающим 95%.
С помощью комплекса N-бромсукцинимида и диметилсуль-
фида состава 1 : 1 аллиловые и бензиловые спирты аналогич-
ным путем превращаются в соответствующие бромиды с 80~
90%-ным выходом. Гераниол, например, превращается в гера-
нилбромид с выходом 82%.
Насыщенные первичные и вторичные алифатические и али-
циклические спирты в тех же условиях почти не вступают в ре-
акцию. Наглядным примером избирательности этой реакции
может служить превращение 2-3-метилпентен-2-диола-1,5 (4)
под действием комплекса (1) в хлористом метилене (недолго
при —20°, а затем в течение 1 час при 0°) в аллильный моно-
хлорид, выделенный с выходом 87%.
Н3СХ /Н н3сх уН
%=cz . У.и 'C=cz
HOCH2HSCZ ^СН2ОН 87% НОСН2Н2с/ \CH2C1
(4) (5)
1. V i 1 s m a i er E., Sprugel W., Ann., 747, 151 (1971).
2. Corey E. J., Kim C, U., J. Am. Chem, Soc., 94, 7586 (1972).
3. Corey E. J., Kim C. U„ J. Org. Chem., 38, 1233 (1973).
4. Corey E. J., Kim C. U., Takeda M,, Tetrahedron Letters, 4339 (1972).
ДИМЕТИЛСУЛЬФАТ (1,316—318).
Для метилирования фенолов химики, имеющие дело с объек-
тами растительного происхождения, обычно используют метод,
который заключается в продолжительном кипячении фенола
с Д, в присутствии карбоната калия в ацетоне. Если в феноле
155
при этом содержится спиртовая группа, то под действием ди-
метилкарбоната, образующегося при взаимодействии Д. с К2СО3,
она может превратиться в соответствующий карбонат.
(CH3)2SO4 + K2CO3 —> (СН3)2СО3
(СН3)2СО3 + ROH —> R—О— СООСН3 + СН3ОН
Так, например, было доказано, что одним из продуктов метили-
рования лаккаииовой кислоты В (I) является соединение (II) [1].
Высказанное предположение о пути образования последнего бы-
ло подтверждено кипячением ^-фенилэтаиола с Д. и карбона-
том калия в ацетоне в течение 36 час, в результате которого
был получен р-феиилэтилметилкарбонат с выходом 45%. На-
гревание р-фенилэтанола с диметилкарбонатом при 100° в тече-
ние 48 час привело к тому же продукту с выходом 90%.
Этерификация. Д. широко применяется для О-метилирования
фенолов и спиртов, но для этерификации карбоновых кислот
его использовали всего несколько раз [2} — он не привлек осо-
бого внимания. На самом деле Д.—удобный реагент для этой
реакции, особенно для этерификации пространственно затруд-
ненных кислот, таких, как (1) [3]. Кислоту (1) превращали в ме-
тиловый эфир с выходом 95% двумя способами. Концентриро-
ванный водный NaOH (1,1 моля) приливают при комнатной
температуре к хорошо перемешиваемой смеси кислоты (1)
(1 моль) и Д. в диоксане и кипятят реакционную смесь в тече-
ние 0,5 час для завершения реакции. Другой способ: раствор
кислоты (1) и Д. в ацетоне кипятят в течение 3 час в присут-
ствии К2СО3 (10%-ный избыток).
Описанный метод с успехом применяли для этерификации
2,6-бнс-трифтор метил бензойной и 2,6-динитробензойной кислот.
Этерификацию можно осуществить в тех же условиях и диэтил-
сульфатом [4].
156
I. Pandhare E. D, Rama Rao A. V., Shaikh I. N-. Venkatara-
man K., Tetrahedron Letters, 2437 (1967),
2. Stoermer R., Ber., 44, 637 (1911); Riiber C. N., Ber., 48, 823 (1915);
U 11 у о 11 G. E., Taylor H. W,, Dawson N., J. Am. Chem. Soc., 70, 542
3. Grundy J-, James B. G., Pattenden G., Tetrahedron Letters, 757
(1972).
4. HallasG., Hep worth J. D., Chem. Ind., 1972, 691.
ДИМЕТИЛСУЛЬФАТ-КАЛИЯ КАРБОНАТ (1,318).
Под действием этих реагентов в кипящем ацетоне карбоно-
вые кислоты с высоким выходом превращаются в соответствую-
щие метиловые эфиры [1]. Метод удобен для этерификации про-
странственно затрудненных кислот [2]. В тех же условиях тро-
полон превращается в 2-метокситропои с выходом 96% [3].
1. Р а 11 е г М„ Bergthaller Р., Monatsh, 99, 103 (1968).
2. G г u n d у J., James В. G., Pattenden G„ Tetrahedron Letters. 757
(1972).
3. Evans D. H„ Greenwald R. B., Org. Prep. Proc. Int., 4, 75 (1972).
ДИМЕТИЛСУЛЬФИД (V, 123—124).
орто-Алкилнрованне ароматических аминов. Гасман и Груэтц-
махер [I] описали простую методику специфического орто-мети-
лирования анилинов, протекающего с высоким выходом. Анилин
(1) обрабатывают 1 же трет-бутилгипохлорита (I, 169—174; V,
67; VI, 40) или N-хлорсукцинимида (IV, 155; V, 520—521; VI,
37, 353) в хлористом метилене при —65°. Затем добавляют при
той же температуре Д. (Зэке) и по истечении 40 жш раствор
метилата натрия (1,2 же) в метаноле. При этом с хорошими
выходами (около 50—90%)' получаются 2-тиометоксиметилани-
лины (2). Десульфуризация с помощью никеля Ренея дает про-
изводные о-толуидина (3) с выходом 60—88%.
Процесс не ограничивается только метилированием. Так,
проводимая по той же методике реакция анилина (1) с тетра-
метиленсульфидом с последующей десульфуризацией над нике-
лем Ренея приводит к образованию o-w-бутиланилина (5) с до-
157
статочмо высоким общим выходом.
---т>
62$
(I) (4) (5)
аМИ2
снгснгснгсн3
Синтез индолов. Гасман и ван Берген [2] использовали мо-
дификацию приведенного выше метода для синтеза 2-замещен-
ных индолов из анилинов. Анилин, как указано выше, обраба-
тывают хлорирующим агентом при —65°, а затем при той же
температуре эквивалентным количеством метилтиопропанона-2
[3] и добавляют эквивалентное количество основания (обычно
триэтиламина). После обычной обработки реакционной смеси
получают производное индола (2) с выходом 60—70%, Тиоме-
тильную группу удаляют с помощью никеля Ренея (выход
>70%) • Кетосульфиды можно варьировать; так, использование
метилфенацилсульфида [СН35СН2С( = О)СбН5] [4] приводит к
образованию 2-фенилиндолов.
1) (СН})3СОС1
г} ch3schec(=o)ch3>
3) (СЕН5)Щ
(2) (3)
Незамещенный индол (или индолы с заместителями в аро-
матическом кольце) тоже можно синтезировать только что опи-
санным методом [5], если кетосульфид заменить метилтиоацет-
альдегидом [CH3SCH2(C=O)H] или соответствующим диметил-
кеталем [6]. Обработка анилина (1) реагентами, указанными в
уравнении, дает соединение (4) с выходом 57%. Непродолжи-
тельная обработка последнего соляной кислотой приводит к тио-
метилиидолу (5) с выходом 97%.
1) (СНЩСОС1
1. Gassman Р. G., Gruetzmacher G., J. Am. Chem. Soc., 95, 588
(1973).
2. Gassman P. G., van Bergen T. J., J. Am. Chem. Soc., 95, 590 (1973).
3. В r a d sher С. K., Brown F. C., Grantham R. J., J. Am. Chem. Soc.
76, 114 (1954).
158
4. Prelog V., Hahn V., BrauchH H.T Beverman H. C., Helv. Chim.
Acta, 27, 1209 (1944).
5. Gassman P. G., van В e r g e n T. J., J. Am. Chem. Soc., 95, 591 (1973),
6, Wick E. H., Y a m a n i s h i T„ Wertheimer H. С., H о f f Y. E., Proc-
tor В. E., Goldblith S. A., J. Agr. Food. Chem., 9, 289 (1961).
ДИМЕТИЛСУЛЬФИД — БОРАН, (CH3)2S-BH3. Мол. вес
75,98, т. пл. от —40 до —38°. Жидкость, устойчивая при комнат-
ной температуре в атмосфере N2.
Д.—б. получают, конденсируя равные объемы газов (СНзДЙ
и В2Н6 в тензиметре [1] и нагревая затем смесь до комнатной
температуры [2].
Диборан неустойчив при комнатной температуре, и его обыч-
но получают in situ. Адамс и сотр. [3] нашли, что Д.— б. являет-
ся ценным заменителем диборана. Он, правда, менее реакцион-
носпособен, чем диборан, но все же его можно использовать в
большинстве методик гидроборирования и восстановления.
1. Burg А. В., Schlesinger Н. J., J. Am. Chem, Soc., 59, 785 (1937).
2. Burg А. В., Wagner R. I., J. Am. Chem. Soc., 76, 3307 (1954).
3. Braun L. M., Braun R. A., Crissman H. R., Opperman M.,
Adams R. M., J. Org. Chem., 36, 2388 (1971).
ДИМЕТИЛСУЛЬФИД — ХЛОР, (CH3)2S+C1C1“ Мол.
вес 133,05.
Окисление первичных и вторичных спиртов до карбонильных
соединений [I]. Действие на раствор диметилсульфида в четы-
реххлористом углероде при 0° раствора 1 же хлора в том же
растворителе приводит к быстрому образованию частично вы-
падающего в осадок комплекса (1). Кори и Ким использовали
этот комплекс для окисления первичных и вторичных спиртов.
Согласно типовой методике, комплекс охлаждают до —25° (су-
хой лед — ССЦ), добавляют циклогексилкарбинол и перемеши-
{ снгон
(ch3rs + с1г секу (CH3)2s+cicr------>
(П
(3) 80$
159
вают при этой температуре в течение 2 час. Образующийся
сульфоксониевый комплекс (2) действием 2 же триэтиламина
превращают в циклогексилкарбоксальдегид (3) (выход 80%)
и диметилсульфид.
Это окисление можно провести с большим выходом (93%),
если использовать комплекс N-хлорсукцииимида с диметилсуль-
фидом (этот том).
См. также Диметилсульфоксид — хлор, этот том.
1. Corey Е. J., Kim С. U., J. Am. Chem. Soc., 94, 7586 (1972).
' ДИМЕТИЛСУЛЬФОКСИД (I, 319—335; V, 124—130;
VI, 86—89).
ЭФФЕКТЫ РАСТВОРИТЕЛЯ
Дегидробромированне. Гримшоу и сотр. [1] иашли, что де-
гидробромирование p-бромнорпиноиа (3), приводящее к образо-
ванию аповербенона [(4), 6,6-диметилнорпинен-3-он-2], под дей-
ствием обычно употребляемых оснований протекает с большим
трудом. В этих условиях [3-изомер (3) лишь превращается в
смесь а- и [3-изомеров (2) и (3). В данном случае две причины
вызывают затруднение реакции элиминирования. Во-первых, ще-
лочная среда не способствует закреплению транс-диаксиальиой
конфигурации уходящих групп. Во-вторых, гел-г-диметильная
группировка затрудняет доступ основания к реакционному цент-
ру. Та же реакция, проведенная при 140—150° в диметилформа-
миде или в диметилацетамиде в присутствии карбоната и бро-
мида лития (II, 199—202; V, 256), приводит преимущественно
к образованию трудноразделимой смеси соединений (2) и (4).
Использование ДМСО, содержащего соли лития, позволило осу-
ществить желаемое дегидробромирование соединения (3) до
(4) с 77%-ным выходом. Высокая эффективность ДМСО в ка-
честве растворителя объясняется тем, что он сольватирует пре-
(2)
(3)
(+) -U)
имущественно катионы, предоставляя таким образом бромид-
иону возможность выступать в качестве сильного основания
Льюиса.
РЕАКЦИИ
Дегидратация (I, 325; V, 127). Селективную дегидратацию
2,4-ди-(2-окси-2-пропил)-циклогексена-1 (1) до диена (2) мож-
но осуществить нагреванием диола в ДМСО при 130° [2]. Реак-
ция, по-видимому, катализируется кислотами, поскольку добав-
ление 1,5-диазабицикло-[4,3,0]-нонеяа-5 (I, 240—242) ее пол-
160
Было замечено, что в этом случае трудно
ностью ингибирует.
(П
(2) 80% 20%
воспроизводится и дегидратация нагреванием диола с обрабо-
танной пиридином окисью алюминия [3].
Реакции с оксиранами (эпоксидами) [4]. ДМСО реагирует
с эпоксидами (1) в присутствии сильных кислот [использовали
2,4,6-тринитробензолсульфокислоту (ТНБС)], образуя с хоро-
шим выходом алкоксисульфониевые соли (2). Поскольку при
гидролизе алкоксисульфониевых солей атака реагента направ-
ляется исключительно по атому серы, эта реакция позволяет
осуществить стереоспецифический синтез 1,2-гликолей. Так, на-
пример, в результате реакции цис- и трлнс-9,10-эпоксистеарино-
вых кислот с ДМСО—ТНБС и последующего гидролиза обра-
зуются соответственно трео- и эритро-§, 10-диоксистеариновые
Н
НА ?
R' I
^С-ОН А
С-OS+(CH3)2
*А
(1)
ЙЛНИЛ или C6HS
R1 = Н или алнил
(2)
кислоты с выходами 65—70%. Таким образом, при взаимодейг-
ствии ДМСО с циклическими окисями наблюдается полное об-
ращение конфигурации.
Окисление эфиров 4-ннтробензолсульфокнслоты [5]. Окисле-
ние первичных спиртов до альдегидов с помощью ДМСО тре-
бует температур около 100° или добавления кислот или ионов
тяжелых металлов. По новой методике спирт действием 4-нит-
робензолсульфохлорида и пиридина сначала превращают в
эфир 4-нитробензолсульфокислоты, а затем полученный эфир
окисляют ДМСО в присутствии бикарбоната натрия. Реакцию
проводят при комнатной температуре при перемешивании в те-
чение около 100—200 час.
RCH2OH
O2n
—SO2OCH2R
|каНСО3
(CH3)2S=O
--------->
NaHCOs
RCH2OS(CH3)2 + O2N
RCHO + S(CH3)2 + NaHjCOj
6 Зак. 596
161
Окисление тиокарбонильных соединений [6]. ТиоКарбониль-
ные соединения окисляются до карбонильных под действием
ДМСО в присутствии кислотных катализаторов (серная кисло-
та, TsOH, трифторуксусная кислота) или трифторида бора.
R\
V=S 4- (CH3)2SO
Rz/
R\
V=O+ (CH3)2S 4- S
RZ//
Выходы колеблются в пределах 65—90%, Методика окисления
применима также к селенкарбонильным соединениям.
Реакции с эфирами 6,7-дизамещенных 4-хлорхинолин-З-кар-
боновых кислот [7]. При нагревании в течение 2 час с ДМСО
при 100° хинолины этого типа (I) превращаются в соответст-
вующие 4-оксихинолины (2). Однако реакцию не удается осу-
ществить с 2,4-дииитрохлорбензолом, а также с этиловыми эфи-
рами 2-хлорбензойной или 2-хлор-5-нитробензойной кислоты.
С!
0)
R] = алнонсил , алкил
Н2 =алионсил
R3 => Н, COOR
1. Grimshaw J., Grimshaw J, Т., June] а Н. R., J. С. S. Perkin I,
1972, 50.
2. Wharton Р. S., Sundin С, Е., Johnson D, W., Kluender Н. С.,
J. Org. Chem., 37, 34 (1972).
3. v о n R u d 1 о f f E., Can. J. Chem., 39, 1860 (1961).
4. Khuddus M. A., S w e r n D., Tetrahedron Letters, 411 (1971).
5. Snyder С. H., Gendler P. L., Chang H.-H., Synthesis, 1971, 655.
6. MikolajczykM., LuczakJ., Chem. Inm, 1972, 76.
7. Harris N. D., Synthesis, 1972, 625.
ДИМЕТИЛСУЛЬФОКСИДА ПРОИЗВОДНЫЕ, а) МЕТИЛ-
СУЛЬФИНИЛМЕТИЛИДНАТРИЙ (димсилнатрий) (I, 335—
339; V, 134—139; VI, 89—90).
Октаметилнафталнн [I]. Недавно опубликована заключитель-
ная работа по синтезу октаметилнафталина (V, 137, ссылка
[Не]). Под действием димсилнатрия мостиковое соединение (3)
почти с количественным выходом превращается в соединение (5),
В качестве второго продукта реакции идентифицирована изо-
масляная кислота. Другие основания (метилат натрия в мета-
ноле, гидрид натрия) оказались в этой реакции неэффектив-
ными.
162
ЬМетокси-4,5; 10,11-бис-(тетраметилен)-6,8-бис-дегидро- [13]-
аниуленил-анион (2). Ле Гофф и Зондхеймер [2] использовали
димсилнатрий в ДМСО для отщепления водорода от соедине-
ния (1) с целью получения 13-членной карбоциклической систе-
мы (2) ЯА^Р-спеКтР соединения (2) подтверждает ароматич-
ность этой системы (она содержит 4л+ 2 л-электронов).
CH3SOCH2Na+
дмсо
----:-------->
(2)
Получение простых эфиров [3]. Обработка вторичных и тре-
тичных спиртов димсилнатрием в ДМСО с последующим алки-
лированием диалкилсульфатом дает простые эфиры с выходом
60—90%. Наличие в молекуле спирта сложноэфирных и третич-
ных аминогрупп, а также двойных связей не мешает проведе-
нию этой реакции. Однако таким путем не удается проалкили-
ровать а- и ^-оксикетоны.
Синтез олефинов [4]. Согласно новой методике синтеза оле-
финов, неенолизующиеся альдегиды и кетоны, например бензо-
фенон, обрабатывают димсиллитием в ТГФ. К полученному
раствору при —80° добавляют о-фениленхлорфосфит (V, 491).
CH3SOCI-I2Li+ (о-С6Н4О2)Р—С1
(СеН5)2С=О--------------> CH3SOCH,C(C6H5)2 -----------—------>
—‘LlGi
Li
Г CH3SOCH2C(C6Hs)r
[(o-CeHjO^PO
СН2=С(С6Н5)2 + (0-СйН4О2)Р—S—сн3
II
о о
Реакционной смеси дают нагреться до комнатной температуры,
после чего кипятят ее в течение 1 час. 1,1-Дифенилэтилен по-
лучается этим методом с выходом 91%. Енолизующиеся карбо-
нильные соединения дают низкие выходы олефинов.
Реакция Чугаева (VI, 89—90). Мерлинг и сотр. [5] получили
ксантогенаты с хорошим выходом, обрабатывая спирты эквива-
лентным количеством димсилнатрия в ДМСО, а затем сероугле-
родом и алкилирующим агентом. Температура разложения
ксантогенатов понижается при проведении пиролиза в ДМСО.
6*
163
Циклизация. На одной из стадий синтеза (±) -р-копаена (3)
Кори и Уатт [6] с помощью этого реагента осуществили цикли-
зацию соединения (1) в трициклический кетой (2). Использова-
ние для этой цели трет-бутилата калия или 1,5-диазабицикло-
[4,3,0]-нон ев а-5 ие привело к успеху.
1. Н а г t Н., О k и А„ J. Org. Chem., 37, 4269 (1972).
2. L е Goff Е., Sondheimer F„ Angew. Chem., Internal. Ed., 11, 926
(1972).
3. S j 6 b e r g B., S j 6 b e r g K, Acta Chem. Scand., 26, 275 (1972).
4. Ruwaj ima I., U c h 1 d a M., Tetrahedron Letters, 649 (1972).
5. Meurling P., Sjoberg K„ SJoberg B., Acta Chem. Scand., 26, 279
(1972).
6. С о г e у E. J., W a 11 D. S., J. Am. Chem. Soc., 95, 2303 (1973).
б) ДИМЕТИЛСУЛЬФОНИЙМЕТИЛИД (1,339—341- V, 139—
141; VI, 91).
Синтезы фуранов [1]. При реакции н-бутилтиометиленовых
производных (см. w-Бутилмеркаптан, V, 71—72; этот том) кето-
нов с Д. с хорошим выходом образуются 3- или 3,4-замещенные
фураны. (Д. получали взаимодействием w-бутиллития в гексане
с борфторидом триметилсульфония [2] в диметоксиэтане. Иодид
триметилсульфония для этой цели непригоден.)
II
R1 СН,
1 )Формилирование
2) Я-С4Н95Н
1) (СН3)23=СН2
2)HgSO4
В реакции с Д. соединения (а) и (б), по-видимому, являются
промежуточными. Соединения типа (б) были обнаружены спек-
тральным путем.
(б)
164
Примеры:
В большинстве случаев образовавшееся фурановое соединение
загрязнено непрореагировавшим н-бутилтиометиленовым произ-
водным кетоиа (около 20—30%).
Этой реакцией из кетона (1) синтезирован природный фу-
рилпериллеи (2):
о _
н3с а н3с /Г"'0
и3с н3с/
(0 (2) .
N-Метилирование индолов [3]. Индолы можно селективно ме-
тилировать по азоту действием Д. в ТГФ при комнатной темпе-
ратуре:
I, Gar st М. A., Spencer Т. A., J. Am. Chem. Soc., 95, 250 (1973).
2. Teichmann H., H i 1 g e t a g G., Chem. Ber., 96, 1454 (1963).
3. Bravo P., Gaudiano G., Umani-Ronchj A., Gazz. Chim. Ital., 100,
652 (1970).
В) ДИМЕТИЛСУЛЬФОКСОНИЙМЕТИЛИД (I, 341—345;
V, 141 — 143; VI, 91—93).
Поправка (I, 343). Формула соединения (5) должна быть:
165
Адамантан-2-карбоновая кислота [1]. Предложен новый удоб-
ный метод синтеза адамантан-2-карбоновой кислоты (4). Ада-
мантаном (1) обрабатывают Д. и иа образующийся эпоксид
2-метиленадамаитана (2) действуют эфиратом трифторида бо-
ра [2], что вызывает его перегруппировку в нестабильный аль-
дегид (3). Окисление альдегида реагентом Джойса дает кисло-
ту (4) с общим выходом около 70% в расчете на (1).
79%, считая на (2)
4,5-Бензогомотропон [3]. 4,5-Бензогомотропон (2) получали
реакцией 4,5-беизотропоиа (1) с Д. в ТГФ. Выход продукта
реакции (2) не приведен. Попытки дальнейшей гомологизации
соединения (2) оказались безуспешными.
Эпоксиолефины [4]. Эпоксиолефины можно получить с пре-
красным выходом, приливая по каплям раствор соответствую-
щего альдегида в ДМСО к Д. При этом побочно образуются
также полимерные продукты конденсации альдольного типа.
В результате реакции получается неразделимая смесь диасте-
реомерных эпоксидов. Ниже в качестве примера приведена ре-
акция цитронеллаля (1) с Д., приводящая к образованию
166
8,9-эпокси-2,б-днметнлнонена-2 (2а)' и (26)
1-Окись тиабензола [5]. Реакция 1,3-дизамещенных пропино-
нов-2, таких, как 1,3-дифенилпропин-2-он-1, с. Д. в ДМСО при
16,5° дает 1-окись 1-метил-3,5-дизамещенного тиабензола. В слу-
чае соединения (1) продуктом реакции является 1-окись 1-ме-
тил-3,5-дифенилтиабензола (3), получающаяся с выходом 76%.
Если реакцию проводить в среде ТГФ—ДМСО при —8°, то
можно выделить промежуточное соединение . (2), которое обра-
зуется, по-видимому, в результате присоединения Д. к соедине-
нию (1) по реакции Михаэля.
b
о о Ji
II ф О /С\
С6Н3С=СССйН£ + (CH3)2S=CH2 > Ф ^-С=С С6Н5 ________________
(CH3)2S=CH ХН
(2) 63%
„—-----------
76%, считая на (1)
I. F£rca$iu D., Synthesis, 1972, 615.
2. House Н. О., J. Am. Chem. Soc., 77, 3075 (1955).
3. Sugimura Y., Soma N., Rishi da Y., Tetrahedron Letters, 91 (1971).
4. Clark В. C., Jr., Goldsmith D. J., Org. Prep. Proc. Int., 4, 113 (1972).
5. Hortmann A. G., Harris R. L., J. Am. Chem. Soc., 93, 2471 (1971).
ДИМЕТИЛ СУЛЬФОКСИД — N-БРОМ СУКЦИНИМИД
(ДМСО —NBC).
Метиленацетали [1]. Метиленацетали получают с хорошим
выходом реакцией спиртов н днолов с ДМСО и NBC или N-хлор-
сукцинимидом. Например, раствор циклогексанола (1) в сухом
ДМСО, содержащем 1—2 мол. же NBC, нагревают при 50° при
перемешивании в течение 12 час. После обычной обработки
167
Получают ди-(циклогексилокси)-метан (2) с выходом 86%’.
Другие примеры:
Оба атома водорода метиленовой группы отщепляются от
ДМСО: если реакцию проводить в ДМСО-й6, получающиеся
ацетали содержат СО2-фрагмент.
I. Hanesslan S., Yang-Chung G., Lava И ее P., Pernet A. G.,
J. Am. Chem. Soc., 94, 8929 (1972).
ДИМЕТИЛСУЛЬФОКСИД - СЕРЫ ТРЕХОКИСЬ (I, 334;
V, 133—134).
Д. — с. т., или реагент Парика и Деринга, с успехом приме-
няли для окисления иорборнадиеиол-7-железотрикарбонила (1)
до иорборнадиенои-7-железотрикарбонила (2) [1]. Использование
смеси трехокись хрома — пиридин или окисление по Пфитц-
неру — Моффату приводит к фрагментации до бензальдегида и
бензола.
Fe(COj3
(2)
Гоу и Харвею [2] удалось с удовлетворительным выходом
окислить zjwc-диол (3) до соответствующего о-хиноиа (4) дейст-
вием пиридиисульфотриоксида в смеси ДМСО и триэтиламина.
Применение для той же цели сочетания ДМСО—уксусный ан-
гидрид (I, 330; V, 131 —133; VI, 88) дает прекрасные выходы
хинонов, однако эта реакция очень капризна. Другие реагенты
168
вовсе непригодны. Следует отметить, что окисление ароматиче-
ских 1,2-диолов такого типа до соответствующих 1,2-диоиов
осложняется из-за наблюдающихся в этих случаях относительно
легкой дегидратации и окислительного расщепления С—С-связи.
Окисление производных углеводов. При взаимодействии
частично ацетилированных углеводов с Д.—с. т. происходит ие
только окисление, ио и элиминирование элементов уксусной кис-
лоты, что открывает путь к получению с высокими выходами
новых ненасыщенных углеводов [3]. Самопроизвольное элими-
нирование ^-ацетоксигруппы, по-видимому, следует за стадией
окисления. Оксигруппы, защищенные бензильной или триметил-
силильной группировкой, не элиминируются.
Примеры:
1. L a n dеsbег g J. М., Sieczkowski J., J. Am. Chem. Soc., 93, 972
(1971).
2. Goh S. H„ Harvey R. G., J. Am. Chem. Soc., 95, 242 (1973).
3. Cree G. Mackie D. W., P e r 1 i n A. S., Can. J. Chem., 47, 511 (1969).
ДИМЕТИЛСУЛЬФОКСИД —УКСУСНЫЙ АНГИДРИД
(l> 330; V, 131 — 133; VI, 88).
|69
Полипоровая кислота (1) под действием ДМСО-Ас2О (2:1)
при 60° в течение 15 мин превращается с 95%-ным выходом в
дилактон пульвиновой кислоты (2) [1]. Это окисление осуще-
о
о . о
U) (2)
ствлено также in vivo [2]. Для ознакомления с предложенным
механизмом этого окисления, а также с рассмотрением биосин-
тетического аспекта реакции следует обратиться к оригинальной
статье.
1. Wikholm R. J., Moore Н. W., J. Am. Chem. Soc., 94, 6152 (1972).
2. Ma as s W. S. G., N e ish A. C„ Can. J. Bot., 45, 59 (1967).
ДИМЕТИЛ СУЛЬФОКСИД “ ХЛОР, (CH3)2SO — Cl2.
Окисление первичных и вторичных спиртов [1]. Прибавление
ДМСО к раствору хлора в хлористом метилене, охлажденному
до —45°, приводит к образованию неустойчивого комплекса,
строение которого, вероятно, можно представить формулой (1).
СН3. ,0
(CH3)3SO + C13 —г X СГ
-15 сн/ \ci+
(1)
Полученный комплекс, как и комплекс диметилсульфида с хло-
ром (этот том), окисляет первичные и вторичные спирты до кар-
бонильных соединений. Реакцию проводят приблизительно со
100%-иым избытком комплекса при —45° (2,4 час) с последую-
щим добавлением избытка триэтнламина. Выходы колеблются
в пределах 94—98%,
О
(CH3)2SO-С1д 11+
R*R2CHOH -----—-> R!R2CH—OS(CH3)2C1
— 45
I NfCjHslg
I от —45 до —10°
О
R’CR2 + (C2H5)3NHC1 + (CH3)2SO
Д.—x. имеет один недостаток: он быстро реагирует с олефи-
нами с образованием виц-дихлоридов, так, например, циклоок-
тен легко превращается в 1,2-дихлорциклооктан. Поэтому для
окисления ненасыщенных спиртов реагент непригоден.
I. С от е у Е, J,, Kim С. U., Tetrahedron Letters, 919 (1973).
170
2,4-ДИМЕТИЛТИАЗОЛ,
Мол. вес. 113,18, т, кип.
144—1457719 мм.
Синтез альдегидов [I]. Д., обработанный последовательно
н-бутиллитием в сухом ТГФ и хлористым бензилом, превра-
щается в 2-(2-фенилэтил)-4-метилтиазол (2) с выходом 94%.
Полученный продукт алкилируют борфторидом триметилоксо-
ния и восстанавливают боргидридом натрия. Образующийся
при этом с высоким выходом 2-(2-фенилэтил)-3,4-диметилтиазо-
лидин (3) гидролизуют в присутствии хлориой ртути в качестве
катализатора в 3-фенилпропаналь (4); выход последнего 60%.
1) Н-ВиЫ
г) с6н5снгс^
34%
СН; С^Н5
1) (СН3)О+ВКД so2
2) NaBH4
91, 5%
<9 (2)
(3)
HgCb, Н,О „
' 60% > C6HSCH2CH2CHO
И)
Рассмотренная последовательность реакций представляет со-
бой путь синтеза альдегидов, равноценный методу с использова-
нием 2,4,4,6-тетраметил-5,6-дигидро-1,3(4Н)-оксазина (VI, 252—
254; этот том), предложенному Лйейерсом, однако имеющий то
преимущество, что не требует для своего осуществления сильно-
кислой среды.
I. Altman L. J., Richheimer S. L., Tetrahedron Letters, 4709 (1971).
ДИМЕТИЛТИОКАРБАМОИЛХЛОРИД (V, 145—146).
Фенолы—► тиофенолы. Опубликована проверенная методика
получения иафталин-2-тиола из р-нафтола [1].
I. NewmanM. S., Неtzе 1 F. W., Org. Syn., 51, 139 (1971).
ДИМЕТИЛ ФОРМАМИД (I, 346—351; V, 146—147; VI, 95).
Аминоантрахиноны [1]. Реакция 1-хлорантрахинона (1) с
ДМФА (кипячение 24 час) дает 1-метиламиноантрахинои (2) в
качестве главного продукта реакции и небольшое количество
1-диметиламиноантрахинона (3). Из 2-хлорантрахиноиа и
ДМФА в этих условиях образуется только 2-диметиламиноаи-
171
трахинон.
В реакции кроме ДМФА могут участвовать и соединения об-
щей формулы R1CONR2R3, где R1 = Н или алкил, R2 и R3 = Н,
алкилы или арилы, но не R2 — R3 = Н [2]. Особенно высокие
выходы замещенных амииоантрахинонов наблюдаются при вза-
имодействии 1-хлорантрахиноиа с М,Ы-диэтилформамидом, N-ме-
тилформамидом и ЫДЧ-диметилацетамидом.
Деметилирование кватернизованных азотистых гетероциклов
[3]. Йодиды N-метилпиридиния, N-метилхинолиния и N-метил-
изохинолиния при нагревании в ДМФА в течение 6—95 час де-
метилируются с высокими выходами.
1. Lord W. М., Peters А. Т., J. Chem. Soc. (С), 1968, 783.
2. L о г d W. М., Peters А. Т., Chem. Ind., 1973, 227.
3. Anmann D,, D e a d у L. W., J. C. S. Chem. Comm., 1973, 32.
ДИМЕТИЛФОРМАМИДА ДИМЕТИЛАЦЕТАЛЬ (см. Ди-
метилформамида диэтил ацеталь, I, 351—352; V, 147; VI, 95—96).
Синтез олефинов. Иствуд и сотр. [1] предложили новый ме-
тод превращения вицинальных диолов в олефины. Например,
(±)-1,2-дифенилэтандиол-1,2 (1) при нагревании с Д.д. дает
2-диметиламиио-т'/?анс-4,5-дифеиил-1,3-диоксолаи (2). Нагрева-
ние полученного диоксолана с уксусным ангидридом при 165—
180° приводит к образованию транс-стильбена (3) с выходом
80%. Подобным же образом жезо-1,2-дифенилэтандиол-1,2 пре-
вращается в ^нс-стильбен (75%), загрязненный следами транс-
изомера, если элиминирование проводится при 90°. траис-Стиль-
(1)
(СНэ)гЫСН(ОСНд)
н с6ня
+ НО А с + AcN(CH3)z + со2
С6Н3 Н
(ЗУ
172
бен становится основным продуктом реакции при повышении
температуры элиминирования до 150°. Реакция, по-видимому,
стереоспецифична, но образующийся олефин в присутствии кис-
лот может изомеризоваться.
Алкилирование меркаптанов [2]. При кипячении Д. д. с гете-
роциклическими меркаптопроизводиыми в бензоле или ацето-
нитриле с прекрасными выходами образуются соответствующие
алкилтиопроизводные:
RSH + (CH3)3N—СН(ОСН3)а —> RSCH3 + СН3ОН + (CH3)2NCHO
Например, при кипячении в бензоле в течение 1 час 2-меркап-
топиримидин и Д. д. дают 2-метилтиопиримидин с выходом 86%.
Хромон. Удобный метод получения хромона (4) [3] основан
на реакции 2-оксиацетофенона (I) с Д.д. (2) в кипящем кси-
лоле с одновременной отгонкой образующегося метанола. По-
лучающийся енамииокетои (3) под действием водного раствора
серной кислоты при 100° циклизуется в хромон.
сн3
н3со
^CH-N(CH,)a
Н3ССГ
120°
80%
(3) (4)
1. East wood F. W., Harrington R. I., J о s a n J. S., P u r a J. L.,
Tetrahedron Letters, 5223 (1970).
2. Hol у A., Tetrahedron Letters, 585 (1972).
3. F о h 1 i s c h B., Chem. Ber., 104, 348 (1971).
ДИМЕТИЛФОРМАМИД - ТИОНИЛХЛОРИД (I, 354—357;
VI, 96—97).
Изонитрилы [1]. Изонитрнлы можно получить с выходом 70—
95% дегидратацией формамидов в диметилформамидном рас-
творе хлорида хлордиметилформиминия, приготовленого in situ
— н2о
RNHCHO ---> RN=C:
из ДМФА и хлористого тионила. Для получения удовлетвори-
тельных выходов изонитрилов существенную роль играет низ-
173
кая температура (—50°) и добавление карбоната натрия (для
нейтрализации образующейся соляной кислоты).
2-Диметиламинобензо-1,3-диоксол [2]. Реакция пирокатехина
(1) с хлоридом хлордиметилформиминия в хлористом метилене
(40°) приводит к хлориду иминия (2), который под действием
триэтиламина в хлористом метилене (40°) циклизуется в диок-
сол (3). Превращение пирокатехина (1) в диоксол (3) можно
сг
н +
^C=N(CH3)2
сг
(2)
(C2H5)3N
79% (общий)
(3)
осуществить в одну стадию, правда с несколько меньшим выхо-
дом (69%), взаимодействием (1) с Д.—т. в присутствии три-
этиламина в хлористом метилене.
1. Walborsky Н. М., Nlznik G. Е., J. Org. Chem., 37, 187 (1972),
2. Ege G., Frey H. 0., Tetrahedron Letters, 4217 (1972).
ДИМЕТИЛФОСФИТ (I, 360).
Этерификация [1]. При нагревании холестерина (1)в течение
нескольких часов в Д. получается главным образом холестерил-
метилфосфит (2), Одиако в присутствии и-TsOH образуется
преимущественно 3^-метокси-Д5-холестен (3) (выход около60%).
Этим методом холестанол превращают в Зр-метоксихолестан
с выходом 60—70%.
Дифенилфосфит использовали лишь для этерификации Д5-3->
оксистероидов (в этом случае, вероятно, промежуточно обра-
зуется гомоаллильиый катион). Холестерин, например, при об-
работке дифеннлфосфитом в условиях кислотного катализа
дает Зр-фенокси-Д5-холестен с выходом 50%.
174
Реакция с N,N/-б«c-бeнзoлcyльфo-n-xинoндииминoм [2].
NSOaC^Hfi
+ НР(О)(ОСИз)2 -----J
50?!
мзадня
C6H5SO2NH
осн3
OCHS
I. к ashman Y, J. Org. Chem, 37, 912 (1972).
2. Mustafa A, Sidky M. M, Zayed S. M. A. D, Zayed M. F, Ann,
716, 198 (1968).
3,5-ДИМЕТИЛ-4-ХЛОРМЕТИЛИЗОКСАЗОЛ (I,361;V, 148—
149).
Синтез пиридинов. Опубликована заключительная работа по
синтезу пиридинов через 4-(3-оксоалкил) -изоксазолы (V, 148—
149).
1. Stork G, Ohashi М, Kamachl Н, K^klsawa Н, J. Org. Chem,
36, 2784 (1971).
2,4-ДИМЕТОКСИБЕНЗИЛАМИН, (CH3O)2C6H3CH2NH2.
Мол. вес 167,20. Хлоргидрат, мол. вес 203,45, т. пл. 185—186°.
Получение. Д. получают с выходом 86% восстановлением
2,4-диметоксибензальдоксима бис- (2-метоксиэтокси) -алюмогид-
ридом натрия (VI, 189—190).
Защита глутамина и аспарагина [1]. Для защиты амидных
групп в остатках аспарагина и глутамина в пептидном синтезе
рекомендуется 2,4-диметоксибензильная группа (ДМБ). Произ-
водные прекрасно кристаллизуются, а ДМБ-группу можно уда-
лить действием трифторуксусной кислоты или безводного фто-
ристого водорода.
I. Р i е 11 а Р. G, Cavallo Р, Marshall G. R, J. Org. Chem, 36, 3966
(1971).
2-(2,2-ДИМЕТОКСИЭТИЛ )-1,3-ДИТИАН,
CH(OCHj)2
Мол. вес 192,34.
Д. получают [1] взаимодействием пропандитиола-1,3 (1 же)
С бис-ди метил ацеталем малонового диальдегида по методике,
175
аналогичной методике синтеза 1,3-дитиаиа [2].
HS(CH2)3SH + снгСсн(осн3)2]
(I)
Дигидро-у-пироны и дигидрофураноны-3 [1]. По новому ме-
тоду синтеза дигидро-у-пиронов (5) и дигидрофуранонов-3 (8)
д. (1) действием «-бутиллития в ТГФ при —304 превращают
в 2-ЛИТИЙ-2- (2/,2/-диметоксиэтил) -1,3-дитиан (2). При взаимо-
действии последнего с эпоксидом образуется продукт (3), кото-
рый при кипячении в ТГФ или бензоле в присутствии п-толуол-
сульфокислоты циклизуется в ацеталь (4). Дигидро-у-пирои (5)
получают затем удалением тиоацетальной группы из соедине-
ния (4) с последующим элиминированием метанола (и-TsOH
в кипящем водном ТГФ). Дигидрофураноны-3 (8) получают
аналогичным путем из литиевого производного (2), но в реак-
ции с ним вместо эпоксида участвует кетон. Общие выходы
в этой реакции лишь умеренные, что объясняется главным об-
разом трудностью удаления тиоацетальной группировки. Опи-
санные методы гидролиза дают низкие выходы; в некоторых
случаях использование NEC повышает выходы (см. этот том).
I. Sher F.( I si do г J. L,, Taneja H. R., Carlson R. M., Tetrahedron
Letters, 577 (1973).
2. С о г e у E. J., Seebach D., Org. Syn., 50, 72 (1970).
J 76
1,2-ДИОКСИ-З-БРОМПРОПАН (а-бромгидрин),
ОН
HOCH2CHCH2Br (1). Мол. вес 155,01, т. кип. 106—110°/4 мм.
Д. получают [1] из эпибромгидрина.
О
/ \ n-TsOH; Н2О
Н2С--СНСН2Вг ----> (1)
Защита карбонильных групп [2]. При кипячении Д. (1) с аль-
дегидами или кетонами в бензоле (катализатор — n-TsOH) с
прекрасными выходами образуются бромметилэтиленкетали (2).
Rl + <СНгВг Zn-
"с=О + (1)-' СНз°Н > R’COR2 + СНг=СНСН2ОН
R2' °х/
Карбонильную группу можно регенерировать с высоким выхо-
дом 12-часовым кипячением (2) с активированной цинковой
пылью в метаноле в атмосфере аргона. Цинковую пыль активи-
руют кратковременной обработкой уксусной кислотой, а затем
промывают метанолом.
Следует отметить, что для удаления кетальных защитных
групп обычно необходимы кислотные катализаторы.
1, Winstein S., Goodman L., J. Am. Chem. Soc., 76, 4368 (1954).
2. Corey E. J., Ruden R. A., J. Org. Chem., 38, 834 (1973).
4,4'-ДИОКСИ-2,6,2/,6'-геграк«с-(трет-БУТИЛ)-ДИФЕНИЛ-
METAH («этил антиоксидант 702»). Мол. вес 424,6, т. пл. 154°,
Т. КИП, ZDU /IU ММ.
3
Не растворим
Не растворим
Растворимость при 20°, вес. %
Толуол................... 32 Изооктан.........
Бензол................... 20 Вода.............
Этанол...................3,8 10%-ный NaOH . .
Ингибитор окисления натурального и синтетических каучу-
ков, полиолефиновых пластиков, смол, клеев, нефтетоплива и
восков.
3,4-ДИОКСИЦИКЛОБУТЕН-3-ДИОН-1,2 (квадратная кис-
лота), ГТ Мол. вес 114,06, т. пл. >300°, рК22,2.
0^
177
Получают из 1,2-дихлор-3,3,4,4-тетрафторциклобутена [11.
Превращение в трициклический триметиновый краситель [2].
6i)R = СОгС2Н,
a) R = Н; б) R = СОгС2И,
Хлорирование комплексом диметилформамид — тионилхло-
рид см. VI, 96—97.
1. CohenS., S о h е п S. G., J. Am. Chem. Soc., 88, 1533 (1966).
2. Т г е i b s A., Jacob К., Ann., 712, 123 (1968).
4,4-ДИОКСО-2-МЕТОКСИ-6-МЕТИЛ-1-ОКСА-4-ТИА-3,5-ДИ-
АЗИН (1),
°2
N N
А 1
НЭС'^Х' О'-'^-оС нэ
(1)
Мол. вес 162,12, т. пл. 102—103° (разл.)’.
Получение [1]. Д. получают реакцией гидрида натрия с N-
карбметоксисульфамоилхлоридом (2а) [получение см, N,N,N-
Триэтил-Ы'-карбметоксисульфамида внутренняя соль — триэтил-
амин (комплекс), этот том]. Этот предшественник Д, обрабаты-
вают гидридом натрия в ацетонитриле при температурах от
КаН
СНзСМ - CH3CK
CmOiCNHSOjCl * CH,O2CNSO2ClNa* > (I)
(2 a) (3)
—35 до —45°. Раствору дают нагреться до комнатной темпера-
туры и образующийся хлористый натрий отфильтровывают.
Фильтрат концентрируют на роторном испарителе и разбав-
ляют эфиром. Смесь затем выдерживают в течение ночи при
—30°, Д. (1) отфильтровывают; выход 70—72%.
Метил-ГС-сульфонилуретан, CH3O2CN = SOa (2). Если Д. (1)
нагревать в ацетонитриле при 30°, он вступает в реакцию, об-
ратную [2 -% 4]-циклоприсоединению, образуя in situ метил-N-
сульфоиилуретаи (2). Последний в виде комплекса с тетрагид*
17$
рофураном был получен также из карбметоксисульфамоилхло-
рида [2], как показано на приведенной ниже схеме:
Ь’аНДГФ
-78° ~ + ТГФ 30°
CH3O2CNHSOZC1 ------------> CH3O3CNSOZC1 Na ----------->
-H2 -NaCl
'2а)
ch3o2cn=so2- О
Н омпленс (2 ) с ТГФ
N-Сульфонилуретан (2) легко вступает в реакцию [2 +21-
циклоприсоединения с олефинами. Так, например, генерирова-
ние (2) в ацетонитриле в присутствии 1,1-дифенилэтилена при-
водит к образованию 1,1-диоксо-2-карбметокси-3,3-дифеиил-1,2-
тиазетидина (3) и Ы-карбметокси-р,р-дифенилвинилсульфамида
(4) [1]. С некоторыми олефинами метил-Ы-сульфонилуретаи(2)
(ед)гс=снг + (г)
с6нв
С6Н5----“I
> I + (С6Н6)гС>=С.Н5ОгЫНСООСНэ
so,
CH3QOCX
(4) 39-40%
(3) 33-34%
дает продукты как [2 + 2]-, так и [2 + 4]-циклоприсоединения.
В общем случае циклоаддукты образуются стереоспецифичио.
?бН5 гссюсн3
C6HS ZH Н—т N
ЧС , CH3CN I
£ + (2)-----* Н-1— во.
HZ чн н
снэ
С ацетиленами возможно только [24]-циклоприсоедииение.
Так, например, фенилацетилен дает исключительно соедине-
ние (5).
о2
X Л
О^^ОСН3
(5)
179
С некоторыми гетероатомными л-системами метил-Ы-суль-
фонилуретан (2) вступает в реакцию [2 -Г 2]-циклоприсоедине-
ния, давая промежуточные соединения, которые самопроизволь-
но отщепляют трехокись серы. В целом это превращение можно
рассматривать как «квазиреакцию» Виттига. Так, например, ди-
фенилциклопропеион (6) при взаимодействии с уретаном (2) в
ТГФ при 30° дает с количественным выходом соединение (7).
ХСООСН3
((>)
ТГФ
зо% -so3
ноличеств,
(7)
1. В u г g е s s Е. М., W i 11 i a m s W. M., Org. Syn., submitted 1973.
2. Burgess E. M., Williams W. M.t J. Am. Chem. Soc.; 94, 4386 (1972).
1,3-ДИТИАН (V, 157—164; VI, 100—102). Ссылка [9] (V, 161):
S e e b a c h D., В e с k A. K.., Org. Syn. 51, 76 (1971).
Окислительный гидролиз производных 2-ацил-1,3-дитиана [1].
Опубликована работа, посвященная исследованию гидролиза
производных 2-ацил-1,3-дитиаиа в присутствии хлорной ртути
(HgCl2). Однако и в этих условиях они гидролизуются медлен-
но. В таких случаях рекомендуется использовать N-галогеисук-
цинимиды. Окислительный гидролиз производных Д. лучше все-
го проводить с помощью N-бромсукцинимида или N-хлорсукци-
нимида в присутствии азотнокислого серебра. N-Галогенсукци-
нимиды можно использовать и при гидролизе 2-ацил-1,3-дитиа-
нов до 1,2-дикарбонильиых соединений.
Окисление альдегидов до сложных эфиров и кислот [2]. Об-
работка 2-литий-2-алкил-1,3-дитианов (1) ди метилдисульфидом
в ТГФ дает с выходом около 90% соответствующие ортотиофор-
миаты (2). Их с высоким выходом превращают в сложные эфи-
ры (3) кипячением в водном спирте в присутствии хлорной рту-
ти и окиси ртути в качестве катализаторов. Для превращения
тиоформиатов (2) в кислоты их кипятят в водном ацетоне в те-
чение 24 час с хлорной ртутью и окисью ртути (катализаторы).
Выходы в этом случае колеблются в пределах 40—65%.
Данный метод удобен в случае альдегидов, чувствительных
к действию обычных окислителей.
Синтез р,у-ненасыщеиных альдегидов [3]. При взаимодей-
ствии аллильного бромида (1) с Д. с высоким выходом обра-
180
зуется бромид сульфонил (2), обработка которого «-бутилли-
тием в ТГФ при —78° дает илид (3). Последний при нагревании
реакционной смеси до 20° перегруппировывается в 2-замещен-
иый 1,3-дитиан (4). Гидролиз соединения (4) обычными мето-
дами приводит к р,у-ненасыщенному альдегиду (5).
ь-Стрептоза [4]. 2-Литий-1,3-ДИТиан был использован для
синтеза сахаров разветвленного строения с альдегидными или
кетоиными группами в боковой цепи. Приведенная ниже после-
довательность реакций иллюстрирует синтез ь-стрептозы (4) из
3-улозы (1). Успешное осуществление всего синтеза зависит от
(1)
89%^
(4)
стадии десульфуризации [(2)—>(3)], которую необходимо про-
водить в тщательно контролируемых условиях.
181
I. Corey Ё. J., Erickson E. W., J. Org. Chem., 36, 3553 (1971).
2. Ellison R. A., Woessner W. D., Williams С. C., J. Org. Chem., 37,
2757 (1972).
3. H unt E,, Ly thgoe B., Chem. Comm., 1972, 757.
4. Paulsen H., Sinn well V., Stadler P., Angew. Chem., Internal. Ed.,
11, 149 (1972).
1,3-дИТИЕНИЯ БОРФТОРИД (1), Мол. вес
T ВГ."
206,05, т. пл. 188—189° (разл.). f1)
Получение [1]. Д. б. (1) получают с выходом 92% в виде
желтого кристаллического вещества кипячением 1,3-дитиана (V,
157—164; VI, 100—102; этот том) с трифеиилметилборфторидом
(III, 399—401; этот том) в сухом хлористом метилене в течение
30 мин. Затем следует удаление растворителя при пониженном
давлении, растирание с охлажденным эфиром и высушивание
в вакууме. Д.б. может храниться без доступа влаги при —20°
в течение нескольких месяцев.
/13-Циклопентеноны [1]. Реагент (1) взаимодействует с 1,3-
диенами (2) при 0—25° по реакции, аналогичной диеновому
синтезу Дильса — Альдера, с образованием аддуктов (3) с вы-
сокими выходами. Реакцию можно проводить в сухом хлори-
стом метилене либо, если диен в нем плохо растворим, в смеси
сухих хлористого метилена и нитрометана (2: 1), При действии
1 же «-бутиллития сначала при —78°, а затем при температу-
рах от —78 до 25° аддукты (3) перегруппировываются с высо-
ким выходом в производные винилциклопропаиа (4), которые
при нагревании в бензольном растворе (200°) в свою очередь
перегруппировываются в тиокеталн циклопеитеноиов (5). Тио-
кетали легко гидролизуются до соответствующих кетонов (6)
в смеси ацетон — вода (8:1) при 25° в присутствии избытка
хлориой ртути и карбоната кальция. Выходы на некоторых ста-
182
днях достигают 90—95%. В целом итог реакции формально
эквивалентен 1,4-прнсоединеиию окиси углерода к 1,3-диенам —
процессу, который, вероятно, неосуществим.
1. Corey Е. J., Wil insky S. W., J. Am. Chem. Soc., 94, 8932 (1972).
дИТИОУГОЛЬНОЙ КИСЛОТЫ N-ЦИАНИМИДА ДИКА-
ЛИЕВАЯ СОЛЬ, NCN=C^ (1). Мол. вес 194,37.
^SK
Реагент получают [1] реакцией цианамида с сероуглеродом
н последующим добавлением гидроокиси калия (выход 86%):
c2hsoh /SK
NCNH2 + CS3 + 2КОН ——> NCN=C
86A \SK,
З-Галоген-1,2,4-тиадиазолы. При обработке хлористым суль-
фурилом соединение (1) подвергается окислительной циклиза-
ции с образованием 1,2,4-тиадиазола (2) с выходом 80—
100% [2]:
(О
CIS
5ОгС1г
80-100%
(2)
Перед окислительной циклизацией реагент (1) можно пре-
вратить в моиоалкилироваиный продукт (3) [I, 3]:
1)
RX MSK
------» NCN=C
XSR
SO2C12
Cl
(3) (4)
1. W i 11 e n b г о о k L. S., Smith G. L., Timmons R. J., J. Org, Chem., 38,
465 (1973).
2. Thaler W. A., McDivitt J. R., J. Org. Chem., 36, 14 (1971).
3. Timmons R. J., WittenbrookL. S., J. Org. Chem., 32, 1566 (1967).
ДИТИОЦИАН (родан) (1,379—380).
Улучшенный метод получения [1]. Д. можно получить с выхо-
дом 85—90% реакцией тиоцианата натрия и газообразного хло-
ра в системе двух иесмешивающихся растворителей (вода и то-
луол), Д., образующийся в водном слое, экстрагируется толуо-
лом. Раствор Д. можно хранить при пониженных температурах.
1 • W е 1 с h е г R. Р., С u t г ц f е 11 о Р. R„ J. Org. Chem., 37, 4478 (1972).
183
ДИФЕНИЛДИАЗОМЕТАН (1,385—387)'.
Присоединение к циклооктину [1]:
сйн5
CN2
C6HS^
I. Wittig G., Hutchison J. J., Ann., 741, 79 (1970).
ДИФЕНИЛДИ-(1,1,1,3, 3,3-ГЕКСАФТОР-2-ФЕНИЛ-2-ПРО-
ПОКСИ)-СУЛЬФУРАН (1). Мол. вес 672,53, т. пл. 107-109°.
Получение [1, 2]. Наиболее удобным и почти количественным
методом получения Д. (1) является обработка хлором эфирного
раствора калиевого алкоголята гексафтор-2-фенилпропанола-2
[(RfOK), полученного реакцией спирта с металлическим калием]
и дифенилсульфида при —78°. Выделившийся хлористый калий
отфильтровывают, эфир испаряют в вакууме. Белый кристалли-
ческий остаток представляет собой Д. (1), который можно пере-
кристаллизовать из пентана. Улучшенная методика приведена
в работе [2]. В новой методике вместо хлора рекомендуется при-
менять бром. Общий выход Д. составляет 65—75%. Гексафтор-
2-фенилпропанол-2 можно получить по методу [3]. При проведе-
нии синтеза следует полностью исключить попадание влаги, так
как сульфуран легко гидролизуется до дифенилсульфоксида и
RpOH. В отличие от большинства сульфуранов Д. (1) устойчив
CF3
(C6H5)2S 4-2С6Н5СОК
CF3
С12
-2КС1
RfOK
ceH5
I
CF3—C-CF3
I
О
t
C6H5—S—C6HS
0
1
CF3—C—CF3
CeH3
(0
при комнатной температуре, если он защищен от действия вла-
ги. Он медленно разлагается при комнатной температуре толь-
ко в растворе.
Дегидратация спиртов [4]. Наблюдавшийся авторами
быстрый обмен алкоксильиых лигандов Д. (1) с добавленными
спиртами навел их на мысль, что этот реагент можно использо-
вать в качестве дегидратирующего агента. И в самом деле,
трет-бутиловый спирт дегидратируется Д. (1) в хлороформе при
184
—50° в течение нескольких секунд, давая изобутилен с количе-
ственным выходом. Дегидратация, по-видимому, протекает че-
рез образование (путем обмена лигандов) сульфурана (2) с по-
следующим отщеплением р-протона.
ОС(СН3)з CFs
(СН3)3СОН4-(1) - (CeHs^S 4- СеНаСОН ОС(СРз)2С6Н5 cf3 (2) (RpOH)
(Сен5) i3SO + (СН3)2С=СН2 4- rfoh
Д, (1) дегидратирует даже трициклопропилкарбинол (3) до
олефина (4); использование в этом случае серной кислоты или
пятиокиси фосфора не дает олефина вовсе. Для выделения оле-
фина (4) кислый RpOH удаляют с помощью гидроокиси нат-
рия, хлороформ отгоняют при атмосферном давлении, а (4) от-
деляют от менее летучего дифенилсульфоксида — однократной
равновесной перегонкой в высоком вакууме при 10-2 мм.
320/0
(3) (4J
Третичные спирты уже при комнатной температуре мгновенно
дегидратируются Д. (1). Большинство вторичных спиртов также
быстро дегидратируется при комнатной температуре. Имеются
доказательства, что предпочтительным является тра/щ-диакси-
альное расположение уходящих групп. Так, например, цис-4-
трет-бутилциклогексанол превращается в 4-трет-бутилциклогек-
сен по меньшей мере в 150 раз быстрее, чем трсшс-изомер. Пер-
вичные спирты (ROH) обычно не дегидратируются, но при —50°
быстро реагируют с Д. (1), образуя с количественным выходом
эфиры Ri?OR; дегидратация происходит только в том случае,
если p-протон первичного спирта достаточно кислый.
Расщепление замещенных амидов [5]. Реакция Д. (1) с неко-
торыми замещенными амидами даже при комнатной темпера-
туре приводит к легкому расщеплению амидной связи. Напри-
мер, обработка бензанилида 0,5 моля реагента (1) в ДМ.ФА при
4Г в течение 3 мин дает иминосульфуран (2), выделенный с вы-
ходом 72%, и эфир бензойной кислоты (3). Эта реакция является
185
общей, но на скорость ее сильно влияют пространственные отно-
шения, определяемые объемом как N-алкильной, так и ацильной
групп.
С6Н5
CF3—A—CF3
1
О
С6Н5—S—СаН5 + CeH6CONHCeHB —>
О
CF3-A-CF3
АаНд
(1)
CF3
(CeH5)2S=NC6Hs + C6H5CO2C(CF3)2C5H5 + С6Н5- (!он
(2) |
CF3
(3), cshscoorf rfoh
Эпоксиды. Мартин и сотр. [6] на заседании Американского
химического общества доложили об одностадийном превраще-
нии гликолей в эпоксиды с помощью Д. (1). Реакция протекает
с высоким выходом в случае диолов, геометрия которых разре-
шает плоское т/цшс-расположеиие гидроксильных групп.
1. Martin J. С., Arhart R. J., J. Am. Chem. Soc., 93, 2341 (1971),
2. Martin J. C., Arhart R. J., Franz J, A., Perozzi E. F., Kap-
lan L. J., submitted to Org. Syn. (1972),
3. F a r a h B. S., G i 1 b e r t E. E., S i b i 1 i a J. P.( J. Org. Chem., 30, 998
(1965).
4. Martin J. C., Arhart R. J., J, Am. Chem, Soc., 93, 4327 (1971).
5. Franz J. A., Martin J. C., J. Am. Chem, Soc., 95, 2017 (1973).
6. A r h a r t R. J., Franz J. A., Martin J. С., A. C. S. National Meeting,
Washington, Sept, 1971, Abstract ORGN 175.
1,3-ДИФЕНИЛИ30БЕН30ФУРАН (1,388—390; V, 164).
Получение. Поттс и Эллиот [1] разработали улучшенный ме-
тод превращения о-дибеизоилбензола (1) в Д. (4). Восстанов-
ление о-дибензоилбензола (1) избытком боргидрида натрия с
последующей обработкой уксусным ангидридом при нагревании
дает Д. (4) с выходом около 70%. Выделение промежуточно
образующегося спирта (2) не обязательно.
сос6н5
\>^-.СОС6Н5
NaBH*
(2)
(0
186
При кипячении о-дибензоилбензола (1) с P2S5 — Ру в тече-
ние 3 час можно получить с выходом 74% дифенилизобензотио-
фен.
Циклопентин. Виттиг и Тейн [2] использовали Д. в качестве
улавливающего агента, чтобы наглядно доказать образование
циклопентина:
1. Potts к. т., Elliott A. J., Org. Prep. Proc. Int., 4, 269 (1972).
2. W 111 i g G„ H e у n J„ Ann. 726, 59 (1969).
ДИФЕНИЛИОДОНИЙ-2-КАРБОКСИЛАТА МОНОГИДРАТ
(4) T. пл. 220—222° (разл.).
Д. m,, впервые описанный Ле Гоффом [1], можно получить
[2, 3] с выходом 72—79% окислением о-иодбеизойной кислоты
персульфатом калия в серной кислоте сначала до соли иодония
(2), которая затем реагирует с бензолом по реакции типа Фри-
деля-Крафтса с образованием соли дифенилиодония (3), Ней-
187
трализация последней гидроокисью аммония приводит к полу-
чению внутренней соли (4), кристаллизующейся из воды в виде
моногидрата.
Генерирование дегидробензола для реакции Дильса — Аль-
дера. Методика, приведенная в Org. Syn. [2]. а) Дифенилиодо-
иий-2-карбоксилат. В трехгорлую колбу емкостью 2 л, снабжен-
ную механической мешалкой с закругленной тефлоновой ло-
пастью, работающей с небольшим числом оборотов, делительной
воронкой на 250 мл и термометром до 100°, закрепленном на
штативе таким образом, чтобы была видна часть шкалы (15—
25°), помещают 20 г (0,081 моля) тонкоизмельченной о-иодбен-
зойной кислоты и 26 а (0,096 моля) тщательно растертого пер-
сульфата калия. Колбу охлаждают в бане со льдом и добав-
ляют в нее 80 мл заранее охлажденной во льду концентриро-
ванной серной кислоты. Не включая мешалки, колбу в течение
4—5 мин вручную вращают в бане со льдом, чтобы ее содержи-
мое превратилось в однородную суспензию и чтобы контролиро-
вать экзотермическую начальную стадию реакции. Затем пре-
кращают охлаждение и отмечают время. Реакционная смесь
слегка вспенивается и несколько раз последовательно изменяет
свою окраску.
Колбу помещают в баню с ацетоном и через 20 мин после
начала окисления добавлением раздробленного сухого льда до-
водят температуру ацетона в баие до 10°; перемешивают содер-
жимое колбы при этой температуре в течение 2—3 мин. Затем
приливают 20 мл ие содержащего тиофена бензола (17,6 г
0,226 моля) и перемешивают смесь при 10° еще в течение 1 час.
Если реагенты смешивать при комнатной температуре, то тем-
пература поднимается примерно на 7°. Реакцию можно довести
до конца при 50° в течение 5 мин или при комнатной темпера-
туре в течение 20 мин, однако в обоих этих случаях продукт реак-
ции загрязнен значительным количеством коричневого пигмента.
Температуру бани понижают до —15° добавлением сухого
Льда и при этой температуре и энергичном перемешивании при-
ливают в колбу 190 мл предварительно охлажденной льдом ди-
стиллированной воды для осаждения бисульфата дифенилиодо-
ний-2-карбоновой кислоты, выпадающего в смеси с бисульфатом
калия. После этого приливают в колбу 400 мл заранее охлаж-
денного льдом хлористого метилена, а затем при перемешива-
нии и охлаждении (температура бани — 15°) 230 мл также пред-
варительно охлажденного во льду 25%-ного раствора гидро-
окиси аммония с такой скоростью, чтобы температура реакци-
онной смеси держалась в пределах 15—25° (это займет прибли-
зительно 10 мин), после чего водный слой должен иметь ще-
лочную реакцию по индикаторной бумаге (pH 9). Мешалку и
термометр вынимают и смывают водой и хлористым метиленом.
Большую часть водного слоя декантируют в колбу Эрленмейера
на 500 мл, а содержимое реакционной колбы, со стенок которой
188
смывают и остатки вещества, переносят в делительную воронку
на 500 мл. Нижний слой — раствор Д. м. в хлористом метилене
бледного рыжевато-коричневого цвета сливают в колбу Эрлен-
мейера емкостью 1 л, а объединенный с декантированным ранее
водный слой экстрагируют двумя порциями по 100 мл хлористо-
го метилена. Вытяжки, объединенные с основной порцией рас-
твора Д. м. в хлористом метилене, высушивают иад безводным
сульфатом натрия, фильтруют в колбу Эрленмейера емкостью
1 л и выпаривают фильтрат на кипящей водяной бане досуха.
Остаток в виде сухой сероватой лепешки вынимают из колбы,
разбивают шпателем из нержавеющей стали и переносят основ-
ную часть продукта на бумагу. Вещество, прилипшее к стенкам
колбы и шпателю, растворяют в кипящем хлористом метилене,
переносят раствор во взвешенную колбу на 500 мл и снова вы-
паривают досуха. Затем добавляют в колбу основную массу
вещества и объединенный твердый продукт доводят до постоян-
ного веса нагреванием на паровой бане в вакууме водоструй-
ного насоса. Выход неочищенного продукта 23,3—24,4 г.
В колбу вливают кипящую воду (275 мл) и растворяют про-
дукт при кипячении. К слегка охлажденному раствору осторож-
но добавляют 0,4 г активированного угля (иорита). Раствор
снова нагревают до кипения, фильтруют и оставляют для кри-
сталлизации на ночь при 0°. Выделившийся в виде бесцветных
призм Д. м. отфильтровывают, высушивают на воздухе при ком-
натной температуре до постоянного веса. Выход продукта с
т.пл. 220—222° (разл.) составляет 20—22 г (72—79%).
б) 1,2,3,4-Тетрафенилнафталин. В круглодониую колбу ем-
костью 100 мл, снабженную 11-сантиметровым водяным обрат-
ным холодильником (модель Асе Glass № 8244), помещают
60 мл диэтилбензола (смесь мета- и гшра-изомеров) и нагре-
вают колбу в вытяжном шкафу открытым пламенем микрого-
релки до тех пор, пока жидкость не начнет конденсироваться в
холодильнике. Если конденсат в верхней части холодильника
будет мутным, то эту часть конденсата удаляют палочкой (или
толстой проволокой) с ватным тампоном на конце. Тот же прием
используют позже для удаления выкипающей в начале реакции
гидратной воды. Если ее не удалить, она скапывает обратно в
колбу и мгновенно испаряется с сильным шипением и разбрыз-
гиванием содержимого колбы. Горелку убирают и вносят в кол-
бу 10 г (0,026 моля) тетрафенилциклопентадиенона и 11,8 г
(0,035 моля) Д. м. Смесь нагревают на микрогорелке с такой
скоростью, чтобы не прекращалось интенсивное выделение газа
при слабом кипении жидкости в колбе. Кристаллизационная во-
да выделяется за 8—10 , мин. Затем колбу снабжают обычным
обратным холодильником и продолжают нагревание. Через
30 мин па дне колбы еще видно значительное количество нерас-
творившегося Д. м. В течение следующих 5 мин цвет реакцион-
ной смеси изменяется иа ярко-красный, а еще через 1—2 мин
189
раствор приобретает светло-яитарную окраску. Кипячение про-
должают до полного растворения твердого остатка (еще 10—
15 мин). Затем обратный холодильник заменяют нисходящим и
отгоняют 55 мл жидкости (диэтилбензол и иодбензол, т. кип,
188°). Остаток охлаждают и растворяют в 25 мл диоксана. Рас-
твор переносят в колбу Эрленмейера емкостью 125 мл, разбав-
ляют 25 мл 95%-ного этанола, нагревают до кипения и посте-
пенно добавляют к нему воду (6—7 мл) до тех пор, пока вы-
падающие при этом блестящие кристаллы не перестанут рас-
творяться при дальнейшем кипячении раствора. После этого
смесь оставляют кристаллизоваться сначала при комнатной тем-
пературе, а затем в течение нескольких часов при 0°, Осадок
отфильтровывают, из маточника при дальнейшем стоянии вы-
падает вторая, небольшая порция кристаллов (0,3 г, т, пл. на
1° ниже). Основной продукт вначале плавится в интервале 196—
199°, при охлаждении затвердевает и снова плавится четко при
203—204°. Общий выход продукта 9,2—10,2 г (82—90%).
в) Методика для студентов [3]. Поместите в пробирку раз-
мером 25 X 150 мм 1,0 г Д. м. и 0,1 г тетрафенилциклопентадие-
нона и добавьте 6 мл диметилового эфира триэтиленгликоля
(т, кип, 222°), смывая этим количеством растворителя вещество
со стенок пробирки. Пробирку закрепите вертикально, вставьте
в нее термометр и нагрейте на микрогорелке. Когда температу-
ра достигнет 200°, горелку уберите и отметьте время. Затем пе-
риодически нагревайте пробирку, поддерживая температуру на
уровне 200—205° до тех пор, пока не прекратится выделение
газов (СО + СОй) и пурпурная окраска раствора не перейдет
в светло-желтую. В случае если пурпурная или красная окраска
через 3 мин при 200—205° не исчезнет, добавьте небольшое ко-
личество предшественника дегидробензола и продолжайте на-
гревание до полного растворения твердого вещества. Дайте
желтому раствору охладиться до 90°, а за это время нагрейте
до кипения 6 мл 95%-ного этанола. Перелейте желтый раствор
в колбу Эрленмейера емкостью 25 мл и несколькими порциями
горячего этанола ополосните пробирку. Затем добавьте в колбу
остатки этанола и нагрейте смесь до кипения. Если при этом
блестящие кристаллы выделятся не сразу, добавьте к кипящему
раствору микрокаплями небольшое количество воды до начала
кристаллизации. Оставьте вещество кристаллизоваться сначала
при комнатной температуре, а затем при 0°. Отделите кристал-
лический продукт, промойте его небольшим количеством мета-
нола; выход 0,8—0,9 г. Чистый углеводород плавится сначала
в пределах 196—199°, затем затвердевает и снова плавится при
203—204°.
1. L е Goff Е„ J. Am. Chem. Soc., 84, 3786 (1962).
2. Fieser L. F., Haddadin M. J., Org. Syn., 46, 107 (1966).
3. Fieser L. F., Org. Expts., 2nd Ed., б. C. Heath and Co., Boston, 303—306
(1968).
190
NjN-ДИФЁНИЛ КАРБАМОИЛ ПИРИДИНИЯ ХЛОРИД,
(cfeH5)aNCON^ сг Мол. вес 310,78, т. пл. 105—110°.
Получение [1].
Смешанные ангидриды [2]. Смешанные ангидриды карбоно-
вой н Ы,Ы-дифенилкарбаминовой кислот можно получить обра-
боткой карбоновой кислоты Д. х. и триэтиламином в водном
растворе;
RCOOH + (C6H6)2NCON+ АсГ RCO2C0N(C6Hs)2
__у 60-90%
Ангидриды представляют собой устойчивые кристаллические
продукты.
1. Herzog J., Вег., 40, 1831 (1907).
2. S h е р а г d К. L., Chem. Comm., 1971, 928.
ДИФЕНИЛКЕТЕН (1,393—394; VI, 102—103).
Реакция с тиокарбонилилидами. Пиролиз предшественников
тиокарбонилилидов (1а), (16) в углеводородных растворителях
в присутствии Д. приводит к образованию циклоаддуктов (За)
и (36) с выходами 85—94%. Следовательно, реакция включает
1,3-присоединение транс-тиокарбонилилида по карбонильной
группе кетена с сохранением конфигурации олефинового компо-
нента (супраповерхиостное циклоприсоединение).
(с6н5)гс.
С-R4
XR! rZ
(с6н5)гс=с-оч
85-94#
(3)
a) ..R1 = R4 = трет-Bu? R2 = R3 = н
б) R1 = R4 = СгН3; R2 = R3 = Н
1. Kellogg R, М., J. Org. Chem., 38, 844 (1973).
ДИФЕНИЛ СУЛЬФОНИЙЦИКЛОПРОП ИЛ ИД (VI, 104—
105), Мол. вес 227,34.
Получение [1].
C6HsSC6H5 + 1-СНгСНгСНгС1 —^ggF4------> (С6Н5)г5СНгСНгСНгС1.ВГ4’
87%
, ^н-тг?_^(с>Н5)^ 7 9% О II - + CH^SCH^Na bf4* — > (1)
Спироалкилирование [1], Д. (1) при взаимодействии с а,0-
ненасыщенным кетоном (2) дает спиропентан (3):
При реакции Д. (1) с альдегидами и кетонами образуются спи-
робутаноны:
В ряде случаев реакция высоко стереоселективна [2], Вза-
имодействие 4-тргт-бутилциклогексанона с этим илидом серы и
последующая перегруппировка промежуточно образующегося
оксаспиропентана под действием Eu(fod)g приводят к образо-
ванию с 80%-ным выходом спиробутанона (4), не загрязненно-
го другим изомером. Аналогично из трициклического кетона (5)
192
при использовании в качестве агента перегруппировки перхло-
рата лития был получен исключительно спиробутанон (6).
1) [>=5(0^,)!
2) Eu(fod)3
80$" *
i) £>=5<с6не)я
2) UC1O.
-------------э
78$
Геминальное алкилирование [2]. Трост и Богданович разра-
ботали интересный метод геминального алкилирования через
спиробутаноны, полученные, как описано в предыдущем разде-
ле. Кетон (1) при взаимодействии с Д, дает соединение (2),
бромирование которого Вгз (или лучше бромидом-пербромидом
пиридиния, III, 119—123) приводит к а,<х-дибромкетону (3).
Обработка последнего метанольным раствором метилата нат-
рия приводит к раскрытию цикла с образованием соедине-
7 Зак. 596
193
ния (4). При восстановлении дибромида (4) три-н-бутилстанна-
ном (III, 372—373; V, 431—432; VI, 263—264; этот том) при
комнатной температуре происходит монодебромирование до сое-
динения (5), которое при обработке основанием дает лактон (6).
Взаимодействие дибромида (4) с три-н-бутилстаннаном при 80°
дает соединение (7).
С другой стороны, аелг-дибромметиленовая группа представ-
ляет собой замаскированную карбонильную группу. Так, дей-
ствие метилата натрия на спиробутанон (8) дает производное
циклогексана (9), в котором при обработке метанольным рас-
твором азотнокислого серебра атомы брома замещаются на ме-
токсигруппы, в результате чего образуется ацеталь янтарного
моноальдегида (10). Гидролиз последнего приводит к соедине-
нию (11). При восстановлении (И) с помощью трас-(трифенил -
фосфин)-родийхлорида (III, 415; V, 448—454; VI, 286—291; этот
том) происходит его декарбонилирование с образованием 1-карб-
метокси-1 -метилциклогексана (12).
(fz)
Оксаспиропентаиы [3], Ранее только предполагалось, что
промежуточными веществами в описанном выше синтезе цикло-
бутанонов являются оксаспиропентаны (2), В настоящее время
их удалось выделить с выходом 60—80% путем обработки кар-
бонильного соединения борфторидом циклопропилдифенилсуль-
фония (1) в ДМСО при 25° с добавлением твердой гидроокиси
калия; затем неочищенный продукт подвергают однократной
равновесной перегонке в вакууме. Подробная методика получе-
ния реагента (1) с мол. весом 314,16 и т. пл. 137—139° предло-
жена для Org, Syn. Богдановичем и Тростом в 1973 г.
SfCjHjh вг/ +
R
R1
^С=О
КОД
ДМСО
(2)
+ (C6H5)2S + На 0 + KBF^
25“
194
Примеры:
В случае бензофенона и циклопропилметилкетона были по-
лучены только продукты перегруппировки оксаспиропентанов —
циклобутаноны.
Аниелирование циклопентана [4]. Трост и Богданович пред-
ложили ценный метод синтеза замещенных циклопентанонов из
оксаспиропентанов, получаемых, как описано выше. Примером,
иллюстрирующим рассматриваемый метод, может служить син-
тез бицикло-[3,3,0]-октанона-2 (5) из циклопентанона (1), При
взаимодействии кетона (1) с борфторидом циклопропилдифе-
иилсульфония (3 час) образуется оксаспиропентан (2). Обра-
ботка реакционной смеси триметилхлорсиланом в 1,2-диметокси-
этане дает О-триметилсилиловый эфир (3) с выходом 94%. При
пропускании его раствора в гексане через нагретую до 330° ко-
лонку, заполненную стеклянными спиральками (время контакта
4 сек), эфир (3) количественно перегруппировывается в О-три-
метилсилиловый эфир еиола (4), Колонку для проведения тер-
мической перегруппировки силилового эфира подготавливают
к работе следующим образом: промывают насыщенным водным
раствором бикарбоната натрия, затем водой, ацетоном и гекса-
ном. После этого через нее пропускают О,М-бш>триметилсилил-
ацетамид или триметилхлорсилан, а потом диэтиламин. Гидро-
лиз эфира еиола (4) дает бицикло-[3,3,0]-октанон-2 (5).
Аналогичная последовательность реакций, исходя из цикло-
гептанона (6), приводит к образованию производного пергидро-
7* 195
азуленона (7).
Следует отметить, что промежуточно образующиеся силило-
вые эфиры енола, например (4), можно алкилировать, обрабо-
тав сначала метиллитием в 1,2-диметоксиэтане, а затем гало-
геналкилом. Алкильную группу таким путем можно ввести
исключительно в положение у головы моста.
1. Trost В. М., Bogdanowicz М. J., J. Am. Chem. Soc., 93, 3773 (1971).
2. Trost В. М., Bogdanowicz М. J., J. Am. Chem. Soc,, 95, 2038 (1973).
3. Trost В. М., Bogdanowicz М. J., Tetrahedron Letters, 887 (1972).
4. Trost В. M., Bogdanowicz М. J., J. Am. Chem. Soc., 95, 289 (1973).
О
II
ДИФЕНИЛФОСФИТ, НР(ОСбН5)2. Мол. вес 234,19, т. пл. 12°.
Пептидный синтез. Японские химики [1] сообщили об удоб-
ном методе синтеза пептидов с помощью Д. и третичного амина
(был использован пиридин):
о
II
НР(ОС3Н5)2
TptJT-BOK-NHR’CHCOOH+ NHjR2CHCOOX пиридин >
О
II/он
—> rpe7-BOK-NHR’CHCONHR3CHCOOX+нр' + С6Н5ОН
^ОС6Н5
Этим путем с высокими выходами были получены различные
пептиды высокой оптической чистоты. Метод применим также
для синтеза оптически активных эфиров аминокислот.
1. Y a m a z a k i N., Higashi F., Tetrahedron Letters, 5047 (1972).
ДИФЕНИЛФОСФОРНОЙ КИСЛОТЫ АЗИД, N3PO(OC6H5)2.
Мол. вес 275,20, т. кип. 15770,17 мм.
Реагент получают с выходом >90% обработкой дифенил-
хлорфосфата (1, 400—401; V, 168—169) небольшим избытком
азида натрия в ацетоне при комнатной температуре. Д. к. а.—
устойчивая невзрывающаяся жидкость [1].
Уретаны. Уретаны легко получить кипячением эквимолярной
смеси карбоновой кислоты, триэтиламииа, спирта и Д.к.а. в те-
196
чение 5—25 час. Эта видоизмененная методика реакции Курниу-
са менее трудоемка, чем классическая.
N3PO(oCeI{5>2
RCOOH + R'OH - ц , >' RNHCOOR'
Пептидный синтез. В вышеупомянутом синтезе уретанов в ка-
честве промежуточного соединения образуется азид карбоновой
кислоты, и это обстоятельство побудило японских химиков ис-
следовать возможность применения Д. к. а. в пептидном синтезе.
И действительно, реагент в присутствии основания позволяет
конденсировать с высоким выходом и практически без рацеми-
зации ациламинокислоты или пептиды с эфирами аминокислот
или пептидов. Этот новый метод применим и при наличии в мо-
лекулах реагентов различных функциональных групп.
1. Shiolri Т., Ninomiya К., Yamada S., J. Am. Chem. Soc., 94, 6203
(1972).
ДИХЛОРБОРАН, BHC12, Мол. вес 82,74.
Получение [1]. Д. получают взаимодействием борана
(516 лшолей) в ТГФ со свежеприготовленным раствором три-
хлорида бора (1032 жмоля; I, 116—118; V, 41; VI, 33—34) в
ТГФ при 0°. При 0° реагент устойчив в течение нескольких ме-
сяцев.
2ВС13 + ВНз —> ЗНВСЬ
Дезоксигенирование сульфоксидов до сульфидов [1]. Д. вос-
станавливает алифатические сульфоксиды до сульфидов почти
количественно за несколько минут при 0° в ТГФ. Восстановле-
ние арилсульфоксидов происходит значительно медленнее
(24 час, 25°, избыток BHCh) с более низкими выходами, Слож-
R\
ВНС12
RZ
"R\ +
;s-o-вне ц
_RZ
R\
/S + HOBCI
RZ
ные эфиры, хлор ан гидр иды, нитрилы, нитро- и даже карбониль-
ные группы не восстанавливаются в условиях дезоксигенирова-
ния алифатических сульфоксидов.
I. Brown Н. С., R a v i n d г a n N., Synthesis, 1973, 42,
ДИХЛОРБОРАНА ДИЭТИЛЭФИРАТ, BHCI2 • O(C2H3)2.
Мол. вес 156,86.
Получение [1, 2]. Д.д. получают взаимодействием боргидри-
Да лития с трихлоридом бора (5—10%-ный избыток) в днэти-
ловом эфире при 0°. Его можно хранить в течение нескольких
недель при 0—5°.
L1BH4 + ЗВС1з + 4(С3Н8)2О —> 4ВНС12-O(CeHB)2 + LIC1
197
Синтез алкилдихлорборанов. Д. д. очень медленно реагирует
с олефинами в диэтиловом эфире или ТГФ. Однако при введе-
нии трихлорида бора (кислота Льюиса) уже при 0° начинается
бурная реакция, в результате которой образуются алкилдихлор-
бораны с выходом 80—90%.
ВНС13 • O(C2HS)2 + CH3(CH2)GCH=CH2 4- ВС13 —>
—> СН3(СН2)еСН2ВС12+ВС13-О(С2Н5)2
При добавлении избытка спирта к продуктам описанной вы-
ше реакции могут быть получены эфиры алкилборной кислоты
RB(OR')2[2].
1. Brown Н. С., Tierney Р. A., J. Inorg. Nucl. Chem., 9, 51 (1959).
2. Brown H. C., Ravin dr an N., J. Am. Chem. Soc., 95, 2396 (1973).
2fЗ-ДИХЛОР-5, 6-ДИЦИАНЛ, 4-БЕНЗОХИНОН (ДДХ)
(I, 407—413; V, 178-183; VI, 107—111).
Дегидрирование. Фогель и сотр. [1] приводят методики каж-
дой стадии трехстадийного синтеза трицикло-[4,4,1.0’-^-ундека-
диена-3,8 (В). Нафталин восстанавливают до изотетралина (А),
к которому затем присоединяют дихлоркарбен (Б), и, наконец,
дехлорируют образующийся трициклический продукт (В). Де-
гидрирование получаемого на стадии В трицикла действием
ДДХ в смеси диоксан — уксусная кислота (Г) приводит к обра-
зованию чрезвычайно ненасыщенного бициклического соедине-
ния 1,6-метано-[10]-аннулена с т. пл. 27—28° (выход 85—87%).
Дегидрирование. При кипячении (36 час) с ДДХ в диоксане
дигидродиктамнин (1) почти количественно дегидрируется до
диктамнина (2) — алкалоида с конденсированными фурановым
и хинолиновым циклами. В бензоле, ацетонитриле или метаноле
дегидрирование не идет [2].
Дегидрированием алкалоида (3) с помощью ДДХ или хлор-
анила в бензоле при 80° был получен новый ароматический не-
альтернантный углеводород бензо-[4,5]-циклогепта-[с/е-1,2,3]-на-
фталин (4). Применение для той же цели палладия на угле при
повышенных температурах приводит к образованию смеси флуо-
ресцентных смол [3].
Для установления механизмов главных реакций в химии сте-
роидов и гормонов изучено дегидрирование с помощью ДДХ
эстрогенов и 3-ацетаминоэстратриен-1,3,5 (10)-ола-170 [4].
Дегидрирование жирных кислот нормального строения [5].
Карбанионы жирных кислот, полученные взаимодействием нат-
риевых солей соответствующих кислот с диизопропиламидом
лития (V, 252—253; VI, 149—151; этот том), в смеси ТГФ-гекса-
метапол при обработке ДДХ (кипячение с обратным холодиль-
ником, 3 час) превращаются в соответствующие (Е)-а,р-неиасы-
RCH2CH2COONa —> [RCH2CHCO2]2“Na+Li+ RCH=CHCOOH
шейные кислоты (о2,Е-номенклатуре см. [6]). Выходы состав-
ляют около 30% •
Дегидрирование неоэргостернна [7]. При действии ДДХ
неоэргостерин (1) дегидрируется до 19-норэргостапеитаен-
5,7,9,14,22-ола-Зр (2) с выходом 80%. Следовательно, гидрид-
ион отщепляется исключительно из положения 14а от третич-
ного бензильного углерода. Браун и Тернер [7] обсуждают воз-
можные причины такой избирательности. Одна из них заклю-
199
чается в том, что 14сс-водород фактически перпендикулярен
плоскости ароматического кольца В, что способствует более
эффективному о — л-перекрыванию в переходном состоянии.
Кроме того, образование третичного карбониевого иона у См
сопровождается уменьшением напряжения в трбшс-сочлененных
кольцах С/D. Еще одной причиной избирательности может слу-
жить образование л-комплекса между ароматическим кольцом В
и хиноном.
Дегидрирование карбонильных соединений (V, 178—179).
Пельк и Кодичек [8] дегидрировали эргостерон (1) действием
ДДХ в надежде получить Д1>4'7>22-эргостатетраеион-3 в резуль-
тате 1,2-дегидрироваиия. Вместо этого они выделили
Д4,6,8(14),22_эргостатетраенор1_3 (2). Тот же продукт был получен
при дегидрировании эргостерона (I) кипячением с хлоранилом
в ксилоле. Поскольку этот неожиданный результат мог объяс-
няться наличием двойной связи в положении 7,8 соединения (1),
авторы вслед за тем изучили дегидрирование изоэргостерона (3)
под действием ДДХ, катализируемое n-толуол сульфокислотой,
и получили ожидаемый дМА^-эргостатетраенон-З (4).
200
Окисление кольца В в соединениях ряда подокарповоЙ кисло-
ты [9]. Фенолы ряда подокарповоЙ кислоты под действием ДДХ
в спиртовых растворах при комнатной температуре окисляются
избирательно по кольцу В, Так, окисление метилового эфира
подокарповоЙ кислоты (1) 2,5 же ДДХ в метаноле дает смесь
продуктов (2) и (3); последний из них является основным (вы-
ход 77%). При использовании 2 же ДДХ атаке подвергается
Р) (2) 13% (3) 77%
в основном бензильная метиленовая группа с образованием
7-кетона (2), тогда как действие 3 же приводит к образованию
5,6-дегидро-7-кетона (3) в качестве главного продукта. Насы-
щенный 7-кетои образуется, по-видимому, через промежуточное
хинометановое соединение (4), так как ацетат фенола (1) не
окисляется в 7-кетон,
1. V о g е 1 Е., К. 1 u g W., Breuer A., Org. Syn., submitted 1972.
2. P i о z z i F., Venturella P., В e 11 i n о A., Org. Prep, Proc. Int., 3, 223
(1971).
3. Muller J. F„ Cagniant D., Cagniant P., Bull. soc. chim. France,
1972, 4364.
4. Bodenberger A., Dannenberg H., Chem. Ber., 104, 2389 (1971).
5. C a j n e 11 i G., Cardillo G., R о n c h i A. U.f J. C. S. Chem. Comm., 1973,
94.
6. Blackwood J. L., Gladys C. L., Loening K. L., Petr area A. E.,
Rush J. E., J. Am. Chem. Soc., 90, 509 (1968).
7. Brown W., Turner A. B,, J. Chem. Soc. (C), 1971, 2057.
8. P e I с В., К о d i с e k E., J, Chem. Soc., J971, 859.
9. F i n d 1 a у J. W. A., T u г n e г A. B., J. Chem. Soc. (C), 1971, 547.
ДИХЛОРКАРБЕН.
6-Хлорфульвен [1]. Реакция циклопентадиеиа (1) с Д. (ге-
нерированным из хлороформа действием трет-бутилата калия)
201
приводит к образованию в качестве основного продукта 6-хлор-
фульвена (2) с выходом 10%; при этом образуется также хлор-
бензол (3) с выходом 1—2%.
СНС1Э
ко-mpe/fl-Bu.
• ---------->
1. D’A more М. В., Bergman R. G., Chem. Comm., 1971, 461.
ДИХЛОРКЕТЕН (I, 414; V, 183—184; VI, 111 — 113).
Дихлорметиленциклоалканы [1]. Д., образующийся in situ
в результате дехлорирования цинком трихлорацетилхлорида в
присутствии циклических кетонов, вступает с ними в реакцию
циклоприсоединения с образованием 3,3-дихлороксетанонов-2
(1). Оксетаноны-2 при перегонке в вакууме легко декарбоксили-
руются, образуя дихлорметиленциклоалканы (2) с общим вы-
Награаание
4 0-50%*
ходом 40—50%. Дехлорирование последних до метиленцикло-
алканов (3) можно осуществить с высоким выходом действием
металлического натрия в жидком аммиаке при —78° [2].
1,2-Циклоприсоединенне (I, 414; V, 183—184; VI, 112—113).
Недавно опубликованы два обзора, посвященные 1,2-циклопри-
соединению Д. к олефинам и дненам [3, 4]. Как правило, наи-
лучшие выходы циклобутанонов достигаются в тех случаях, ког-
да дегидрохлорирование дихлорацетил хлорида под действием
триэтиламина проводится в интервале температур 30—50° в
углеводородном растворителе в присутствии избытка кетено-
фила. По своей реакционной способности по отношению к цик-
лопентену кетены располагаются в следующий ряд:
С]2С=О=0 > (С6Н5)2С—0=0 > (СНДС=О=0 > НгС=С=О
202
Джеффе и Молина [5] описали циклоприсоединение Д. к три-
замещенным олефинам. Так, Д. с 1-метилциклогексеном (1) об-
разует дихлорциклобутанон (2), который восстанавливается до
циклобутанона (3) с выходом 59%. Такие же циклобутаноны
получены из 1-метилциклопентена.
1. В г a d у W. Т„ Р a t е 1 A. D., Synthesis, (972, 565.
2 Hoff М. С., Greenlee К- W., В о о г d С. Е., J. Am. Chem. Soc., 73,
' 3329 (1951).
3. В г a d у W. Т., Synthesis, 1971, 415.
4, Ghosez L., Montaigne R., Roussel A., Vanlierde H., Mol-
let P„ Tetrahedron, 27, 615 (1971).
5. J e f f s P. \V., Molina G., J, C. S. Chem. Comm., (973, 3.
/CO
ДИХЛОРМАЛЕИНОВЫЙ АНГИДРИД, cljf Мол.
cic /
CO
вес 166,95.
Изомеризация 1-метоксициклогексадиенов-1,4 в 1-метокси-
циклогексадиены-1,3. 1 -Метоксициклогексадиены-1,4, получаемые
восстановлением анизолов по Берчу, обычно изомеризовали в
более устойчивые циклогексадиены-1,3 действием амида калия
в жидком аммиаке [1]. Тот факт, что 1,4-диены, большей частью
при повышенных температурах, образуют с диенофилами аддук-
ты Дильса — Альдера, соответствующие 1,3-диенам, привел Бер-
ча и Дастура [2] к мысли исследовать потенциальные диенофилы
лишь в качестве катализаторов изомеризации без последующего
введения их в конденсацию по Дильсу — Альдеру. Было най-
дено, что для этой цели подходит Д. а. Например, если несопря-
женный диен (1) осторожно кипятить с Д. а. (0,1%) в течение
2—3 час, то образуется смесь диена (1) и нужного 1,3-диена (2)
в отношении 17:38 (общий выход смеси, содержащей некото-
рое количество полимерных продуктов, 75—85%).
203
Д. а. можно использовать и в качестве катализатора реакции
Дильса — Альдера между 1,4-диеиами и некоторыми диенофи-
лами, что позволяет понизить температуру и сократить время
реакции.
1. В i г с h A. J., S u b b a RaoG., Advan. Org. Chem., 8, 1 (1972).
2. Birch A. J., DasturK. P., Tetrahedron Letters, 4195 (1972).
ДИХЛОРМЕТИЛЕНДИМЕТИЛАММОНИЯ ХЛОРИД,
Ck
^C==N(CH3)2C1 (1). Мол. вес 162,45, т. пл. около
C1Z
180°
(разл.).
Д.х. получают [1] с выходом около 90% хлорированием тет-
раметилтиурамдисульфида [(CHa^NCSaC^NfCHgja, мол. вес
240,43, т. пл. 146—148°] в СН2С12.
Д.х. легко вступает в реакции с соединениями, обладающи-
ми протонной подвижностью. Например, ои реагирует с фено-
лом (2 мол. экв), образуя аммониевую соль (2), которая после
гидролиза дает дифенилкарбоиат (3).
г с,н5он + () С‘Щ °>^(сн,)гХ ""он~> С‘Н,’5с-о
90% С6Н5-О Cl- С6Н5-О
(3)
Реакция Д.х. (1) с 2 молями диметиламина приводит к ди-
хлориду тетр а метили очевины (4), который при обработке суль-
фидом натрия образует тетраметилтиомочевину (5).
Na2S
2(CH3)2NH+(1) —► (CH3)3NCN(CH3)2 ------------> (CH3)2NCN(CH3)2
cf ^Ci s
(4) (5)
При кипячении иммониевой соли (1) (2 моля) с М,Ы-диме-
тиламидами карбоновых кислот [например, (6), 1 моль] в хло-
ристом метилене или хлороформе в течение 1—3 час с высокими
выходами образуются 1,3-дихлормалонилцианииы (7). В при-
сутствии бикарбоната натрия они гидролизуются до амидов за-
мещенной малоновой кислоты [например, (8)] [2].
204
' - и
c6h5ch2c-n(ch3)2
(6)
£ NaHCOj
(H}C)zN-C!!!+y7,N(CH,)i Нг~ >
Cl Cl
Cl"
(7)
C6H5 CON(CH3)a
H CON(CH3)a
(8)
Еще одним примером взаимодействия Д.х. (1) с соедине-
ниями, содержащими метиленовую группу, активированную со-
седним карбонилом, является реакция с циклоалканонами [3].
Так, при кипячении Д.х. (1) с циклоалканонами (9) в хлоро-
о
—
*- — (СНг) л
(9)П=2. 3, 4
(11)
форме образуются хлориды иммония (10), которые при гидро-
лизе дают амиды (11) с общими выходами около 70—85%. Реа-
гент (1) вступает в реакцию с енаминами, содержащими
p-водородный атом (12), с образованием амидохлоридов (13)
с высоким выходом [4]. Их можно превратить в аминопирими-
дииы (14) и аминопиразолы (15), как показано иа приведенной
схеме:
(12)
90-95%
(13)
206
При кипячении Д.х. (1) с N-алкиламидами (16) в хлоро-
форме с высокими выходами (85—95%) образуются хлорфор-
мамндиииевые соли (17) [5]. Эти соли можно гидролизовать
сначала до N-(1-хлоралкеиил)-мочевии (18), а затем до N-ацил-
мочевин (19). При дегидрохлорировании соединений (18) трет-
бутилатом калия образуются бывшие не известными до настоя-
щего времени этииилмочевииы (20).
щ fii /
КСНгСх --------и--->RCH=C +
''NHR1 85-95% N —C=N(CH3)a
I, I
R5 Cl
Ci
(16) (17)
НгО
* щоЩснЩ
RCH=C-N —22 R CH2CONCON(CJ^)»
I I
Ci к J?’
(IS) (19)
(CH3)3COK
ТГФ
^сощснщ
R-C^C-N
Ri
(20)
«Биуреттрихлориды» [6]. Диметилцианамид (1) почти коли-
чественно реагирует с Д.х. в хлористом метилене, давая
Н,Ы,М/,М/-тетраметил-1,3-дихлор-2-азатриметиицианин (2)— «би-
уреттрихлорид». В мягких условиях он гидролизуется до N’.N1-
диметил-№-(диметилкарбамоил)-хлорформамидина (3), который
в более жестких условиях в свою очередь гидролизуется до про-
изводного биурета (4).
(CH3%N'-Cs=N + (СН3)гМ=СС1гСГ C-^clz >
(1)
(СН3)гК ZN ^(СНЩ
•О’ ’V
! 1
Cl Cl
(2)
HjO, NaHCOj
0°
------------> (CHj)2N-C=N — C-N(CH,)a
7!% 1г
(3)
H2O
(CH3)iiN-C-NH-C-N(CHi)i
О О
(4)
Азацианин (2) применяют в синтезе гетероциклов. Напри-
мер, в результате реакции азацианина (2) с фенилгидразином
206
с выходом 93% образуется 1,2,4-триазол (5).
1) (СгНЛИ N
(2) + e6H5NHNHz кон/нго.......(CHjhN_^ y-N(cH3)2
9 3% N—N
4C6HS
(5)
1. Vie he H. G., Janousek Z., Angew. Chem., Internal. Ed., 10, 573 (1971).
2. Janousek Z., Viehe H. G., Angew. Chem., Internal. Ed., 10, 574 (1971).
3. ' Viehe H. G., Janousek Z., D e f r e n n e M.-A., Angew. Chem., Internal.
Ed., 10, 575 (1971).
4. Viehe H. G., van Vyve T„ Janousek Z., Angew. Chem., Internal.
Ed., 11, 916 (1972).
5, Janousek Z., Collard J., Viehe H. G., Angew. Chem., Internal. Ed.,
11, 917 (1972).
6. Janousek Z„ Viehe H. G., Angew. Chem., Internal. Ed., 12, 74 (1973).
ДИХЛОРМЕТИЛЛИТИЙ (I, 416—417; V, 184-185; VI, 113).
Обзор [1].
а-Галогенкетоны [2]. Из бензальдегида (1) можно получить
фенацилхлорид (2) с выходом 72% следующим путем. Альдегид
добавляют к раствору Д. (1,20 же) в ТГФ при —95°, затем
смесь при этой же температуре обрабатывают избытком н-бу-
тиллития. Полученному раствору дают нагреться до 0° и затем
разлагают его разбавленной соляной кислотой. Другие альде-
гиды реагируют аналогично с образованием хлорметилкетонов.
CJhCHLI
С6Н5СНО ----------;
(1)
Н
1
С6НЕССНС12
н
h-BliIJ |
----->- С6Н5С—СС12
О’
CGH5C=CHCI
О" Li+ -
Li+ 2Li+
С6Н5СОСН2С1
(2)
Кетоны также вступают в эту реакцию. Так, циклопентанон
можно превратить в 2-хлорциклогексанон с выходом 64%, а
циклододекаион — в 2-хлорциклотридеканон с выходом 46%.
I. Kobrich G., Angew. Chem., Internal. Ed., 11, 473 (1972).
2. T a g u c h i H., Yamamoto H., Nozaki H., Tetrahedron Letters, 4661
(1972); см. также Villieras J., Bacquet C., Norm a nt J. F., J. Or-
ganometallic Chem., 40, Cl (1972).
ДИХЛОРСИЛАН, H2SiCl2. Д4ол. вес 101,02.
Присоединение к олефинам [1]. Д. присоединяется непосред-
ственно к олефинам с внутренней двойной связью в присутствии
в качестве катализатора платинохлористоводородной кислоты.
При этом с высоким выходом образуются соответствующие ал-
207
килсиланы. Реакцию проводят в автоклаве, который нагревают
иа масляной бане при температуре 130—150° в течение 8—
24 час.
H2sich
H2PtCIe
СН3(СН2)3СН=СНСН3 ——’
О J /О
—> СН3(СН2)3СНСН2СН3 + СН3(СН2)3СН2СНСН3
siHCi2 siHci2
36% 64%
1. Benkeser R. A., Muench W. C., J. Am. Chem. Soc., 95, 285 (1973).
М,1М-ДИХЛ ОРУРЕТАН (V, 187—189).
Обзор [1]. Нил опубликовал обзор, посвященный присоеди-
нению Д. и N-хлоруретана CINHCOOC2H5 к олефинам.
1. N е а 1 е R. S., Synthesis, 1971, 1.
ДИ ЦИАНАЦЕТИЛ ЕН (I, 418—419; V, 190—191).
Реакция Дильса — Альдера с тиофеном. Хелдер и Уинберг
[1] сообщили о первой успешной попытке ввести тиофены в реак-
цию Дильса — Альдера, использовав в качестве диенофила Д.
2,5-Диметилтиофен (1) при нагревании с избытком свежепри-
готовленного Д. в запаянной ампуле при 100° в течение 12 час
дает динитрил 3,6-ди метил фталевой кислоты (2) с выходом
49%. В этой реакции тиофеновое кольцо активируют алкиль-
ные заместители; реакция с незамещенным тиофеном протекает
лишь с выходом 8%.
Несколько позднее Уинберг и Хелдер [2] нашли, что сущест-
венное влияние на реакцию 2,3,4,5-тетраметилтиофена (3) с Д.
в хлористом метилене при комнатной температуре оказывает
катализ хлоридом алюминия (J, 51—53). В результате этой ре-
акции с выходом 57% было получено светло-желтое твердое ве-
щество, которое оказалось соответствующим замещенным тие-
(з)
208
пином (4). При нагревании в течении 7 мин при 300° в присут-
ствии трифенилфосфина этот тиепии количественно превращал-
ся в динитрил тетраметилфталевой кислоты (5).
1. Н е 1 d е г R., W у п b е г g Н„ Tetrahedron Letters, 605 (1972).
2.' W у n b е г gH., Н е 1 d е г R„ Tetrahedron Letters, 3647 (1972).
ДИЦИКЛОГЕКСИЛ БОРАН (VI, 114).
Сопряженные цйс,цй<?-диены [1]. При дигидроборировании
сопряженных диииов, например додекадиина-5,7 (1), под дейст-
вием Д. и последующем протонолизе промежуточного борорга-
нического соединения уксусной кислотой образуются цис,цис-
/i-C4H9C=C—С=СС4Нэ-«
1) (СвНц)2ВН
2} СНзСООН
Н\ zh
«-С4Н9Ч /с=с\
хс=с^ хС4Н9-н
79%
(1)
диены (цис, цис-до декадиен). В случае пространственно затруд-
ненных диинов, таких, как 2,2,7,7-тетра метилоктадиин-3,5, вос-
становление проводят в две стадии. Сначала диин реакцией с
1 же Д. и последующим протоколизом превращают в еиин,
а затем взаимодействием с диизоамилбораном с последующим
протонолизом получают цис,цис-диеи.
1. Z we if el G„ Polston N. L., J. Am. Chem. Soc., 92, 4068 (1970),
ДИЦИКЛОГЕКСИЛ КАРБОДИИМИД (ДЦК), (I, 422—428;
V, 192—193; VI, 114—116).
a,j3-Ненасыщенные и a-циклопропилкетоиы [1]. При обра-
ботке ДЦК в присутствии следов однохлористой меди р-оксике-
тоны претерпевают внутримолекулярную дегидратацию с обра-
зованием а,р-ненасыщенных кетонов. Эта реакция представляет
собой удобный метод получения метиленциклоалканонов:
П= 2-8
_дцн
60-70%
о
у-Оксикетоны в тех
про пи л кетоны:
же условиях превращаются в а-цикло-
пнс6ни
+
NHCtHu.
L Alexandre С., Rouessac F., Bull. soc. chim. France, 1971, 1837.
209
ДИЦИКЛОГЕКСИЛ КАРБОДИИМИДА ИОДМЕТИЛАТ,
Мол. вес 348,27, т. пл. 111—113°.
СбН; IV +
СвНцГ
СН3/
Д. и. получают с выходом 75% при продолжительном нагре-
вании дициклогексилкарбодиимида в иодистом метиле.
Спирты—”иодиды [1]. Алифатические первичные и вторич-
ные спирты реагируют с Д. и. в ТГФ, бензоле или гексане при
35—50°, давая соответствующие иодиды, как правило, с высо-
кими выходами. В реакцию вступают даже стерически затруд-
ненные спирты; правда, иодиды в этом случае образуются с не-
сколько меньшими выходами. Реакция протекает с обращением
конфигурации; например, 3|3-холестанол превращается в За-иод-
холестан с выходом 84%, а холестерин — в ранее неизвестный
Зсс-иод-Д5-холестен (выход 40%).
I. Scheffold R„ Saladin Е., Angew. Chem., Internal. Ed., 11, 229 (1972).
ДИЦИКЛОГЕКСИЛ-18-КРАУН-ЭФИР-6 (2). Внимание]
Токсичен.
Химики фирмы «Дюпон» на протяжении нескольких лет про-
являли интерес к макроциклическим простым полиэфирам (см.
недавний обзор [1]). Ими синтезировано более 60 таких поли-
эфиров, но из иих лучше всего изучены полиэфиры (1) и (2).
Систематические названия этих полиэфиров чрезвычайно гро-
моздки, поэтому их называют «краун-эфирами»; такое название
появилось из рассмотрения их молекулярных моделей. Так, по-
лиэфир (1) известен под названием дибензо-18-краун-эфир-6,
так как в его полиэфирном кольце содержится 18 атомов, и
шесть из иих являются атомами кислорода. Тривиальное назва-
ние полиэфира (2) — дициклогексил-18-краун-эфир-6. Дибензо-
18-крауи-эфир-6 (1) получают в виде белых кристаллов с т. пл.
164° реакцией (3,(3'-ди- (о-оксифенокси)-диэтилового эфира с
(3,(3'-дихлордиэтиловым эфиром в присутствии гидроокиси нат-
рия; выход 80%. Д. (2) получают гидрированием полиэфира (1)
210
Над рутениевым катализатором. При этом образуются два изо-
мера, один из которых плавится при 61—62,5°, а другой при
gg—70°. Для большинства целей можно пользоваться смесью
обоих изомеров [2].
Позже тем же автором была описана улучшенная методика
синтеза Д. [3].
Самым замечательным свойством циклических полиэфиров
является их способность образовывать с солями металлов комп-
лексы, растворимые в органических растворителях. Эти комп-
лексы проявляют повышенную реакционную способность. На-
пример, пространственно затрудненные эфиры 2,4,6-триметил-
бензойной кислоты, которые не омыляются гидроокисью калия
в гидроксилсодержащих растворителях, можно гидролизовать
действием комплекса гидроокиси калия с Д. в ароматических
углеводородах. Активность таких растворов комплекса, вероят-
но, обусловлена присутствием несольватнрованных ионов гидро-
ксила, у которых реакционная способность гораздо выше, чем
у гидроксильных ионов, сольватированных, как обычно. Ниже
приведен еще более эффектный пример использования таких
комплексов.
Окисление перманганатом калия в бензоле. Химики фирмы
«Дюпои» [4] сообщили, что комплексы перманганата калия с Д.
211
(2) представляют собой сильные окислители (3), растворимые
в бензоле. Комплекс получают перемешиванием эквимолярных
количеств полиэфира (2) и перманганата калия в бензоле при
25° до образования прозрачного пурпурного раствора, концент-
рация перманганата калия в котором составляет порядка 0,06 М.
Комплекс можно выделить в виде твердого вещества пурпур-
ного цвета, ио в бензольном растворе он более устойчив. Этот
комплекс с высокими выходами окисляет олефины, спирты,аль-
дегиды и алкилбензолы. Например, транс-стильбен окисляется
им до бензойной кислоты (выход 100%). Еще более удивитель-
но окисление а-пинеиа (4) до ^uc-пиноиовой кислоты (5), проте-
кающее с выходом 90%. При окислении пииена (4) водным
(4)
(в)
раствором перманганата калия выход (5) составляет 40—60%.
Толуол количественно окисляется до бензойной кислоты. Окис-
ление проводят при 25°, так как при более высоких температу-
рах реагент сам окисляется до адипиновой кислоты и других
продуктов. Механизм окисления комплексом (3), по-видимому,
близок к механизму, принятому для окисления в водной среде.
Преимущество метода заключается в том, что пермаиганат-ион
в комплексе обладает необыкновенно высокой реакционной спо-
собностью.
I. Pedersen С. J., Frensdorf! Н. К., Angew. Chem., Internat, Ed., 11,
16 (1972).
2. Pedersen C. J„ J. Am. Chem. Soc., 89, 7017 (1967).
3. P e d e r s e n C. J., Org. Syn., 52, 66 (1972).
4. Sam D. J., Simmons H. E., J. Am. Chem. Soc., 94, 4024 (1972).
ДИЭТИЛАЛЮМИНИЙХЛОРИД, ClAi(C2H5) 2. Мол. вес.
120,56.
Алкинилдиэтилаланы. Алкинилдиэтилаланы, или 1-диэтил-’
алюминийалкины (3), легко получить [1] из терминальных алки-
нов (1). Действием раствора н-бутиллития в гексане на толу-
ольный раствор алкина (1) получают литиевое производное (2).
При добавлении к последнему 20%-ного толуольного раствора
212
Д. происходит выделение LiCl, а в растворе образуется реа-
гент (3).
H-CHHsLi (CjHgtaAlCl
RC=CH ------------> RC^CLi --------^_L1C1 > RC^CA1(C2HE)2
(1) (2) (3)
(2}
Алкинилирование алициклических эпоксидов [1]. Алкииилди-
этилалаиы (2 экв) реагируют с алициклическими эпоксидами
(1 экв) с образованием соединений, которые после гидролиза
дают [3-оксиацетилены. Выходы в этой реакции колеблются от
умеренных до очень высоких.
Примеры:
25'
Zj> 4- C^HnCeCAlfCjHj), >
77 у.
0 25°
О.+ С6Н13СэСА1(СгН9)2 -.8^ >
98#
9 0°
С5НпСНС=СА1(СгН5)2 72ТОС
OCH2C6Hj
а ОН
CsCCHCsHn
ОСНгС6Нд
О + С5НпСНСеСА1(С|Н6)л
ОТГП
Нипячение
18 час
-—
20#
ОН
CsCCHCjHn
ОТГП’
Реакция эпоксида с алюминийорганическим соединением
была использована иа одной из стадий синтеза (±)-7-оксапро-
стагландииа Fia (7) [2]. Реакция эпоксида (4) с алюминийорга-
ническим соединением (5) приводит с «приличным» выходом
к оксиалкину (2), который затем в несколько стадий превра-
щают в соединение (7).
213
ОСНгС6Н5
6сНгС6Нд
А1( СгН5)2С^С(СНг)БСН3
О)
ОСНгС6М5
.-ОН
С = С(СНг)5СН3
ОСНгС6Н5
(4) (6)
Несиольно стадий
йнн
он
7 1
...о(снг)3соон
13 14
'C = C^1S- (снг)4снэ
' с
HOZ х н
Р)
у,6-Ацетиленовые кетоны [3]. ЬДиэтилалюминийалкины (3)
реагируют с сс,(3-неиасыщениыми кетонами (8), образуя после
подкисления продукты 1,4-присоединения (9) с выходами от
умеренных до очень высоких. Успех реакции решающим обра-
зом зависит от условий эксперимента. В качестве растворителя
I II III
3 + —С=СС=О —> RC=CCCHC=O
I
(8) (9)
обычно используют смеси эфира с лигроином. Реакцию прово-
дят при температуре от —15 до 25°. Область применения реак-
ции ограничена кетонами, способными принимать цисоидную
конфигурацию. Кетоны, существующие только в трансоидной
конфигурации (например, циклогексеион-3), реагируют с обра-
зованием продуктов 1,2-присоедииения к ацетиленовому фраг-
менту.
Примеры:
О о
н-С4НэС^САЦС3Н6)2 + СНз=СНССНз —> w-C4HgCsC—СН2СН2ССНз
48%
о С6Н5 о
СаН5С=СА1(С2Н5)2 + C6HSCH=CH(?CH3 —> С6Н5С=С— (1нсн2(!сн3
95%
I. Fried J., Lin С.-H., Ford S. H., Tetrahedron Letters, 1379 (1969).
2. Fried J., Heim S., Eiheredge S. J., Sunder-Plassman P„
Santhanakrishnan T. S., H i mi zu J., Lin С.-H., Chem. Comm.,
1971, 634.
3. H о о z J., L a у to n R. B., J. Am. Chem,, Soc., 93, 7320 (1971),
214
ДИЭТИЛАЛЮМИНИЙЦИАНИД (I, 428—429; V, 193—194).
Методика получения реагента опубликована в Org. Syn.[l].
В отличие от алюминийорганических соединений Д. непирофо-
рен.
(C2Hs)3A1 + HCN —> (C2H5)2A1CN+С2Н6
С U 70
Гидроцианироваиие. Нагата и сотр. [1—4] опубликовали
подробное сообщение о результатах исследования реакций гид-
роциаиирования с помощью Д. (метод Б), а также действием
триалкилалюминия и цианистого водорода (RsAl—HCN, метод
А); обычно использовали триэтилалюминий. (Алкилпроизвод-
иые алюминия весьма пирофорны, обращаться с ними следует
с величайшей осторожностью, все операции проводить в атмо-
сфере инертного газа.) Основное различие между этими двумя
методами заключается в том, что гидроцианирование по мето-
ду А необратимо, и, следовательно, в этом случае можно гово-
рить о кинетическом контроле реакции, тогда как гидроциани-
рование по методу Б обратимо, в силу чего здесь осуществляет-
ся как кинетический, так и термодинамический контроль, т. е.
образование кинетически контролируемого продукта происходит
лишь на ранних стадиях реакции.
В цитированных работах на большом количестве примеров
сравниваются оба метода гидроцианирования.
Рассмотренная реакция является ключевой стадией стерео-
направлеиного полного синтеза сДгиббереллииа А15 (3) [5]. Так,
реакция енона (1) с избытком Д. в хлористом метилене при
комнатной температуре приводит к образованию с выходом 87%
цианкетона с цис-сочленением колец С и D. Следует обметить,
что еион (4) не вступает в реакцию гидроцианироваиия, вероят-
215
но, вследствие ингибирующего влияния кислородсодержащей
группы, соседней с реакционным центром (9а) .
ОАс
(4У
Получение циангидринов из нереакционноспособных карбо-
нильных соединений [6]. Исходное соединение не вступает в ре-
акцию с цианистым водородом.
1. Nagata W., Yoshioka М., Hirai S., J. Am. Chem. Soc., 94, 4635
(1972); Nagata W., Yoshioka M., Org. Syn., 52, 90 (1972).
2. Nagata W., Yoshioka M., Murakami M., J. Am. Chem. Soc., 94,
4644 (1972).
3. Nagata W., Yoshioka M., Murakami M., J. Am. Chem. Soc., 94,
4654 (1972).
4. Nagata W., Yoshioka M., Terasawa T., J. Am. Chem. Soc., 94, 4672
(1972).
5. Nagata W., Wakabayashi T., Na r is a da M., HayaseY., Kama-
ta S., J. Am. Chem. Soc., 93, 5740 (1971).
6. Nagata W., Yoshioka M., Murakami M., Org. Syn., 52, 96 (1972)
ДИЭТИЛАМИН (I, 312; III, 282; IV, 65, 249—250).
Это основание в эфире используют на двух стадиях синтеза
грег-бутилцианкетена для дегидрохлорирования хинондихлори-
Да [1].
216.
HN(CzHsh
О(СгН}):
1. Weyler W., Jr., Duncan W. G„ LlewenM. B., Moore H. W., Org.
Syn., submitted 1972.
ДИЭТИЛИЗОЦИАНМЕТИЛФОСФОНАТ,
(C2H5O)2P(O)CH2N=C (1). Мол. вес 167,14.
Д. (1) получают реакцией
Р(ОС2Н5)з + [(CH3)3NCH2NHCHO]Br' —>
POCI3
N(CsHs)3
—> (C2H6O)2P(O)CH2NHCHO -----> (C2H5O)2P(O)CH2N=C
(1)
Вииилизоциаииды [1]. Взаимодействие литиевой соли Д. с
карбонильными соединениями приводит к образованию вииил-
изоцианидов с выходами 70—85%,
217
M-BuLi Li/
(i)—ГГФ’ ,~LQ°> (czh5o)2p(o)chn=c
xr* Г “1
О=<Д,г OLi Р(О)(ОСгН5)г
--------> R1—C —CH
Нг N=C
70-85%
LiOP(O)(OC2H})2
4-Диэтилфосфоиил-2-оксазолииы [1]. При обработке Д. (1)
примерно 10%-ным раствором цианида натрия в этаноле, а за-
тем альдегидом или кетоном образуются 4-диэтилфосфонил-2-
оксазолины (2) с выходами 70—85%.
NaCN Na+
(1) С*н5°н > (C2H5O)ZP(O) CHN=C
zrI
O=C
^2
NaO Р(О)(ОС2Н5)2
R'-C-^H IF
I, ।
R2 N=C
czH>OH
ai_c---C-P(O)(OC2HS)Z -C2HsONa
M н
P(O)(OC2H5)2
(2)
1-Формиламиио-1-диэтилфосфонилалкены [1]. Обработка
оксазолина (2) (см. выше) трет-бутилатом калия в ТГФ приво-
дит к образованию 1 -формиламино- 1-диэтилфосфоиилалкенов с
выходами 78—92%.
КО-/прет-В и
(2) ТГФ
R4 “ ,pN
С—*cz
yP(O)(OC2Hs)
>
R1^ XNHCHO
R*' "р(О)(ОСгН5)г
Ср. Тозилметилизоиитрил, этот том.
1. Schollkopf U., Schroder R., Tetrahedron Letters, 633 (1973).
ДИЭТИЛ-(1-МЕТИЛТИО)-ЭТИЛФОСФОНАТ,
о
CH3SCHP(OC2H5)2. Мол. вес 202,26, т. кип. 68-70°/0,20 мм.
сн3
218
Реагент получают [1] из диэтил-(метилтиометил)-фосфоната
(VI, 117—118) обработкой его н-бутиллитием в атмосфере арго-
на при —78° с последующим добавлением йодистого метила (вы-
ход 77%).
Д. использовали для превращения кетодиена (1) в сескви-
терпен оциденталол (5) [2]. Реакция Уодсворта — Эммонса
между кетодиеном (1) и Д. (3 эка) в смеси гексаметапола и ди-
метоксиэтана (62°, 1 час) приводит к виниловому тиоэфиру (2).
При гидролизе последнего в водном ацетонитриле в присутствии
хлорной ртути образуется смесь 7а- и 7[3-ацетилдиенов (3) и
(4) в отношении 2,9: 1 с выходом 70%. Эти эпимеры разделяли
методом препаративной хроматографии в толстом слое. Нако-
нец, действием метиллития на 7а-изомер диена (3) получали
(+)-оциденталол (5) с выходом 72%.
I. Corey Е. J., S h u 1 m а и J. I., J. Org. Chem., 35, 777 (1970).
2. Watt D. S., Corey E. J., Tetrahedron Letters, 4651 (1972).
ДИЭТИЛ-(1,1,2-ТРИФТОР-2-ХЛОРЭТИЛ)-АМИН (I, 432—
433; V, 195-196; VI, 119).
219
Алленовые кислоты [1] Реакция 3-ацетата 17а-дифторцик-
лопропеиил-5а-андростаидиола-3р,17р (1) с Д. в сухом хлори-
стом метилене приводит к образованию смеси трех изомерных
продуктов (2)—(4):
Взаимодействие циклопропеионкарбинола (5) с фторамииом
дает фтораигидрид алленовой кислоты (6) с выходом 80%. Об-
работка (6) метилатом натрия в метаноле в течение 16 час при-
водит к соединению (8) через промежуточное образование
сложного эфира алленовой кислоты (7). И наконец, гидролиз
эфира 17,20-еиола (8) соляной кислотой дает с высоким выхо-
дом сложный эфир [3-кетокислоты (9) .
220'
Таким образом, рассмотренная совокупность реакций пред-
ставляет собой метод синтеза как алленовых кислот, так и слож-
ных эфиров [3-кетокислот.
1. Crabbe Р., Carpio Н., Velarde Е., Chem. Comm., 1971, 1028.
ДИЭТИЛХЛОРФОСФАТ (I, 435—436; V, 72).
Аминодегидроксилирование. Росси и Бениет [1] описали
метод получения анилинов из фенолов в две стадии. Сначала
ArOH + (C2HSO)2POCI J-NaOH 8Q_90%» АгОРО(ОС2НБ)2
феиол взаимодействием с Д, в присутствии гидроокиси натрия
превращают в соответствующий диэтилариловый эфир фосфор-
ной кислоты. На второй стадии полученный эфир реагирует с
амидом калия и металлическим калием в жидком аммиаке.
В трех изученных примерах выходы на этой стадии составляли
56—78%.
АгОРО(ОС2Н5)2 + KNHa + К .р 7Й » ArNH2
Рассмотренный! метод аминодегидроксилирования в некото-
рых отношениях дополняет метод Шеррера и Беатти (см.4-Хлор-
2-фенилхиназолин, этот том). Последний метод требует высоких
температур и включает стадию щелочного гидролиза и, следо-
вательно, непригоден в случае термически неустойчивых или
чувствительных к щелочам соединений. Зато он применим к га-
логен- и нитрозамещенным фенолам, тогда как в условиях ре-
акции Росси и Беннета эти замещенные фенолы претерпевают
и другие превращения.
1. R о s s 1 R. А., В u n n е t t J. F., J. Org. Chem., 37, 3570 (1972).
ДИЭТИЛЦИАНМЕТИЛФОСФОНАТ (I, 437; V, 197—199-
VI, 120).
Одна из важных стадий в синтезе 3[3-ацетокси-5[3,14а-буфа-
диен-20,22-олида из дегидроэпиандростеиа заключается в кон-
денсации карбаниона, полученного из Д., с кетоиом (1) [1]:
1. Pettit G. R., Dias J. R., J. Org. Chem., 36, 3207 (1971).
221
диэтил цинк — ЙОДОФОРМ.
Расширение цикла в аренах [1], Иодкарбеноидиый реагент,
полученный из диэтилцинка и йодоформа, превращает бензол
в 7-этилциклогептатриеи-1,3,5 (1). Из алкилбензолов этим ме-
тодом получают смеси алкилзамещеиных 7-этилциклогептатрие-
нов-1,3,5 как показано в приведенном примере:
57% 22% 21%
1. М. i у а п о S., Hashimoto Н., J, С. S.
Chem. Comm., 1973, 216.
ДИЭТИЛ ЦИН К ~ МЕТИЛ ЕН ИОДИСТЫЙ (I, 437 V,
199—200).
Образование циклопропанов из олефинов (I, 437; V, 199—
200). Образование циклопропанов из олефинов при действии
Д.—м.и. заметно ускоряется кислородом; кроме того, повы-
шаются и выходы циклопропанов. Так, реакция циклогексена
с карбеноидом в присутствии воздуха завершается за 30 мин, и
норкаран получают с выходом 90%; в атмосфере азота выход
иоркараиа составляет лишь 53% [1].
1. М. i у а и о S., Hashimoto Н., Chem. Comm., 1971, 1418.
ДИЭТОКСИКАРБОНИЯ БОРФТОРИД, [HC(OCH2CH3)2]+BF;.
Мол. вес 189,96.
См. также Диалкоксикарбоиия борфториды, этот том.
Д. б. получают реакцией триэтилортоформиата с эфиратом
трифторида бора [1]:
HC(OCH3CH3)3 + BF3-C2H5OC3HS —> HC(OCH2CH3)4BF7
222
Алкилирование. Реагент использовали для получения N-этил-
бензиламина (2) из бензонитрила (1):
C = N HC(OCH2CH3)3+BF4‘ —> c s N C H2 CH3 | || BF4- CHjOH
(1) H. “[ GH2CH3 Г OCH3 bf4' ^r\,CH2NHCH2CHj NaBH3CN^ Г [T (2)
1. Borch R. F., Org. Syn., submitted 1972.
ж
ЖЕЛЕЗА(Ш) АЦЕТИЛАЦЕТОНАТ,
Fe(CH3COCHCOCH3)3. Мол. вес 353,17.
Дециаиирование [1]. Алкилнитрилы можно децианировать
(с восстановлением) до углеводородов действием Ж. а. и мел-
кораздробленного натрия в сухом бензоле при комнатной тем-
RCN —> RH
пературе в атмосфере аргона в течение 50—70 час. При дециа-
нировании насыщенных первичных, вторичных и третичных
алкилцианидов выходы углеводородов достигают 58—100%.
В случае аллил- и арилцианидов выходы несколько ниже.
1. VanTamelen Е. Е., R u d 1 е г Н., Bjorklund С., J. Am. Chem. Soc.,
93, 7113 (1971).
ЖЕЛЕЗА ДОДЕКАКАРБОНИЛ, Fe3(CO)i2. Мол. вес. 513,67.
Продажный реагент содержит около 10% по весу метанола
в качестве стабилизатора. Чтобы удалить метанол, реагент на-
до выдержать при 0,5 мм не менее 5 час.
Восстановление [1]. Растворы Ж. д. в метаноле избиратель-
но и с высоким выходом восстанавливают ароматические нитро-
соединения до первичных аминов; при этом другие функцио-
Бензол, кипячение
ArNO24-Fe3(CO)u4-CH3OH ----—--------> ArNH2
i о—УЬ7о
0,0Ш О,ОШ о,ш
нальные группы (С=С, С = О, COOR, NHAc) не затрагивают-
ся. В отсутствие метанола восстановление не идет. Судя по
спектральным данным, Ж-Д. образует с метанолом анион гид-
ридоундекакарбонилтрижелеза [HFe3(CO)n], который, по-види-
мому, и является собственно восстановителем.
Альпер [2] использовал эту же систему для восстановления
азот-углеродной двойной связи. Кипячение фталазина (1) с
Ж. д. в смеси метанол — бензол в течение 12—16 час приводит
к образованию 1,2-дигидрофталазина (2) с выходом 54 %-
СН3ОН, С6Н6
Fe3 (СО)12
54%
(?)
224
Рассмотренная система с успехом применялась также для вос-
становления целого ряда шиффовых оснований. Например,
N-бензилиденанилин восстанавливается до N-бензилаиилина с
88%-ным выходом.
i. Landesberg J. М,, К a tn L., Olsen С., J. Org. Chem., 37, 930 (1972).
2. Al per Н., J. Org. Chem., 37, 3972 (1972).
ЖЕЛЕЗА НОНАКАРБОНИЛ (II, 5—6; V, 202—203; VI,
123).
Циклогептеноны [1]. Дегалогенирование оца'-дибромкетонов
с помощью Ж. и. в присутствии 1,3-диенов позволяет в одну
стадию получить 7-членные циклические кетоны. Так, например,
в результате реакции 2,4-дибром-2,4-диметилпентанона-З (1) с
Ж. н. и 2,3-диметилбутадиеном (2) (молярное соотношение реа-
о
0)
гейтов 1,0: 1,2: 9,0) при 60° в течение 40 час в атмосфере азота
образуется 2,2,4,5,7,7-гексаметилциклогептен-4-он (3) с выходом
71%. Продукты реакции, полученные из соответствующих вто-
ричных бромидов и ациклических 1,3-диенов, бромированием и
последующим дегидробромированием можно превратить в тро-
поны. Например, обработка кетона (4) 4 же дибромида бром-
гидрата пирролидона-2 (VI, 211) в ТГФ и последующее дегид-
робромирование образовавшегося дибромида с помощью хло-
рида лития (11,204—205; V, 257—258) в ДМФА дает с 64%-ным
выходом 2,7-диметилтропон (5).
К)
64%>
Синтез циклопентенонов [2]. Реакция а,а'-дибромкетонов
с енаминами в присутствии Ж. н, приводит к производным цик-
лопентенона с выходом 50—100%. Так, при взаимодействии ке-
тона (1) с а-морфолиностиролом (2) в присутствии Ж-н. как
8 Зак. 590
225
восстановителя образуется циклопентенон
(1)
(3) с выходом 94%.
о
нзсч хсн3
нс сн +
1 I
Вг Вг ,
Fea(CO),
-C4H9NO^
90
HSC6
(3)
1. N о у о г I R., М a k i п о S., Т а к а у а Н., J. Am. Chem. Soc., 93, 1272 (1971);
N о у о г i R., Н а у а к a w a Y., F и п а к и г а М., Т а к а у а Н., Murai S.,
Kobayashi R., Tsutsumi S., J. Am. Chem. Soc., 94, 7202 (1972).
2. N о у о г i R., Y о к о у a m а К., M a к i n о S., H а у a к a w a Y., J. Am. Chem.
Soc., 94, 1772 (1972).
ЖЕЛЕЗА ПЕНТАКАРБОНИЛ (II, 6—7; V, 203; VI, 123—
124).
Селективное гидрирование а,р-ненасыщеиных карбонильных
соединений. Японские химики [1] осуществили селективное гид-
рирование а,р-ненасыщенных карбонильных соединений с по-
мощью винно-красного гидридокарбонильного комплекса желе-
за, полученного in situ из Ж-п. во влажных растворителях в
присутствии небольших количеств основания, Для этой цели
можно пользоваться тремя различными методиками: 1) обраба-
тывать Ж.п. небольшим количеством NaOH в 95%-ном мета-
ноле при 0—60° в атмосфере N2; 2) вместо 95%-ного метанола
брать смесь эфира с водой (4:1 по объему); 3) в качестве осно-
вания применять 1,4-диазабицикло-[2,2,2]-октан (ДАБЦО, V,
89—92; этот том) во влажном ДМФА или ГМТФК. Все эти ме-
тоды дают почти одинаково удовлетворительные результаты.
Для восстановления ос,p-ненасыщенных карбонильных соеди-
нений (кетоиы, альдегиды, эфиры или лактоны) их прибав-
ляют к растворам реагента, полученным, как описано выше, и
выдерживают реакционную смесь при комнатной температуре
в течение 12 час. Выходы соответствующих насыщенных карбо-
нильных соединений обычно составляют 90—98%. Восстанов-
ление кетонной или сложноэфирной группы при этом происхо-
дит в незначительной степени; восстановление до пинаконов не
наблюдается.
Примеры:
Бензальацетон —> бензилацетон (> 98%)
М е ти л в и в ил кетон —> Мет ил этил кетон (> 98 %)
LV —Холестенон —> Копростанон (32%)
Коричный альдегид —> 3-Фенилпропионовый альдегид (98%)
Диметиловый эфир малеиновой кислоты —> Диметиловый эфир янтарной
кислоты (96%)
б-Лактон 5-оксигексеи-2-овой кислоты —> б-Лактон 5-оксигексановой ки-
слоты (90%).
226
Различные реакции восстановления. Нельсон и сотр. [2]
с помощью ж. П, осуществили селективное восстановление еиол-
ацетатов винилхлоридов и а,0-иенасыщенных альдегидов до со-
ответствующих олефинов. Ж-п. также восстанавливает а-аце-
токсикетоны до соответствующих кетонов. /Монофункциональные
кетоны и сложные эфиры этим реагентом не восстанавливаются.
\ Ке(СО)5 \ ^С=О Fe(CO)s ^С=О
хс=<% ------I -------------------------> I
/ \ / \ у С—О Ас /С—Н
X = OAc, Cl, сно
Высокие выходы продуктов восстановления получают только в
дибутиловом эфире или кумоле; в ДМФА и толуоле образова-
ния олефинов не наблюдается. Выходы колеблются в интервале
30—75%. По обычной методике субстрат и Ж. п. [в молярном
отношении 1 : (5—10)] кипятят в дибутиловом эфире в атмосфе-
ре азота. Избыток реагента затем разрушают добавлением рас-
твора хлорной меди в ацетоне или хлорного железа в этаноле.
Синтез олефинов по Кори — Уинтеру. Синтез олефинов по
методу Кори — Уинтера (III, 389) осуществляют взаимодейст-
вием тиоикарбонатов с триметилфосфитом:
\=s Y + СОг + (Сн3о)зрд
о
Термически нестойкие олефины этим методом получаются с
плохими выходами. Дауб и сотр. [3], заменив триметилфосфит
на Ж- л., получили олефины с удовлетворительными выходами:
Fe(CQ)5
Тионкарбонат и Ж- п. (молярное соотношение примерно 1:1)
нагревали в атмосфере азота при температуре ~ 100° в течение
~2 час. Этим методом, например, был получен дибензобарре-
лен (1) с выходом 79,1%.
(1)
Дезоксигенирование N-окисей. Окиси аминов дезоксигени-
руют до соответствующих аминов кипячением с Ж- п. в ди-н-бу-
з* 227
тиловом эфире [4]:
R'\
R2—N->0
R3/
Fe(CO)6
— Fe(CO)4, —CO2
45-80%
R’\
R-—N
Ra/
Амины из N-ннтрозосоединений. Ароматические N-иит-розо-
соединения восстанавливают Ж. п. в кипящем ди-и-бутиловом
эфире до соответствующих аминов. Диалкилнитрозамины прн
такой обработке дают мочевины [4].
1. Noyori R., Umeda I., I s h i g a m i Т„ J. Org. Chem., 37, 1542 (1972).
2. Nelson S. J., Det re G., Tanabe M,, Tetrahedron Letters, 447 (1973).
3. Daub J., Trautz V., E г h a r d t U., Tetrahedron Letters, 4435 (1972).
4. A 1 p e r H., E d w a r d J. T„ Can. J, Chem., 48, 1543 (1970).
ЖЕЛЕЗА(П) СУЛЬФАТ (II, 7—8).
Карбоксилирование ароматических гетероциклических осно-
ваний [1]. Карбоксилирование гетероциклических ароматических
оснований можно проводить окислительно-восстановительным
разложением в их присутствии оксигидроперекисей эфиров а-ке-
токислот.
НО/ /ООН НО/ /О *
R— С— COOR + Fe3+ —> R—С—COOR + Fe3+ + ОН’
RCOOH + • COOR
Реакция протекает легко, перекисное соединение при этом мож-
но не выделять. Например, этиловый эфир пировиноградной
кислоты СН3СОСООС2Н5 обрабатывают при охлаждении до
—10° водным раствором перекиси водорода. Затем эту смесь и
водный раствор FeSO4-7H2O добавляют при перемешивании к
раствору гетероциклического основания в разб. H2SCU (0—5°).
Таким способом из бензотиазола
2-карбэтоксибензотиазол (2). Из
чают 58% 2-карбэтоксипиридина
(1) получали с выходом 82%
пиридина этим методом полу-
и 34% 4-карбэтоксипиридина.
1. Bernardi R., С а г о n n а Т., Galli R., М 1 п i s с i F.a Perchinun-
no М.., Tetrahedron Letters, 645 (1973),
228
ЖЕЛЕЗА(Ш) ХЛОРИД (II, 8—11; V, 203—204; VI, 124).
Алкеннлнрованне реактивов Гриньяра [1]. Реактивы Гринья-
ра легко реагируют с бромистым винилом в ТГФ в присутствии
Ж- х. в качестве катализатора. Например, реакция гексен-5-ил-
магиийбромида с бромистым винилом в ТГФ при 25°, катали-
зируемая Ж.Х., приводит к образованию октадиеиа-1,7 с выхо-
дом 64 °/о •
FeCi3
H2C=CH(CHa)4MgBr + BrCH=CH2 Н2С~СН(СН2)4СН=СН2 + MgBr2
Рассматриваемая реакция, по-видимому, стереоселективна,
поскольку взаимодействие метилмагнийбромида с цис- и транс-
1-бромпропенами-1 приводит исключительно к цис- и транс-бу-
тенам-2 соответственно:
HjCMgBr 4- вг CHj
HjC СН3 4- MgBr2
HjCMgBr 4-
СН,
сн,
+ MgBrj
I. Tamura M., Kochi J., Synthesis, 1971, 303.
ЖЕЛЕЗА(Ш) ХЛОРИД— ДИМЕТИЛФОРМАМИД.
Окислительная конденсация. При изучении окислительной
конденсации фенолов японские химики [1] получили обнадежи-
вающие результаты, применяя Ж> х. в ДМФА. В дальнейшем
они выделили новый кристаллический комплекс Ж-х. и ДМФА,
отвечающий формуле [Ре(ДМФА)3С12][ГеСи]; мол. вес 543,74,
т. ил. 220°. Комплекс получают с выходом 95,2%, прибавляя
раствор Ж.х. (1 моль) в сухом эфире к ДМФА (1,5 моля).
Этот комплекс оказался весьма эффективным реагентом как
внутримолекулярной, так и межмолекулярной окислительной
конденсации фенолов. Обычная методика заключается в при-
бавлении водного раствора комплекса (10 жмолей) к эфирному
раствору фенола (1лшоль); образующуюся смесь кипятят при
перемешивании в течение 1 час. Таким способом фенол (1) был
окислен до диенона (2) с выходом 67%.
229
Примером межмолекулярной окислительной конденсации
под действием нового комплекса может служить окисление
п-крезола (3) в кетон Пуммерера (4) с выходом 28%:
сн.
1. То b i в a g a S., К о t а п i Е., J. Am. Chem. Soc., 94, 309 (1972).
И30АМИЛНИТРИТ, (CH3)2CHCH2CH2ONO. Мол. вес.
117,15, т. кип. 99°.
Сложные эфнры а-замещеииых а-диазокарбоновых кислот.
Эти эфиры можно получить просто и с довольно высокими вы-
ходами кипячением эфиров а-амипокислот с И. в хлороформе
или бензоле в присутствии небольшого количества кислоты (на-
пример, уксусной) [1]:
R— CHCOOR' R CCOOR'
4O-ss% 1
Дифеиохиионы. 2,6-Дизамещенные феиолы окисляются И.
в хлористом метилене до 3,3',5,5'- тетразамещениых дифенохино-
нов [2]:
1. Takamura N., Mizoguchi Т., Koga К., Yamada S., Tetrahedron
Letters, 4495 (1971).
2. J е г и s s i R. A., J. Org. Chem., 35, 2105 (1970).
ИЗОВАЛИН (а-амино-а-метилмасляная кислота),
CH3
CH3—СН2—С—СООН. Мол. вес 117,15, т. пл. > 300°.
nh2
Бензиламины [1]. Бензальдегиды можно превратить в бен-
зиламины с выходом 60—75% кипячением в ДМФА в присутст-
вии (Д-изовалина с последующим кислотным гидролизом про-
межуточно образующегося имина. В диглиме реакция идет мед-
леннее.
СН3
АгСНО + СН3СН3ССООН
NLL
/СН3
CHN=C^
\сн2сн3
Н\
^CHNH2 +
Аг^
сн3<
;с=о
СН3СН/
н3о*
60—75 Уб
231
Если в качестве реагента использовать сс-метилаланин (а-ами-
ноизомасляную кислоту), то выходы снижаются.
1. Rizzi G. Р, J. Org. Chem., 36, 1710 (1971). ’
ИЗОПРОПЕНИЛАЦЕТАТ (II, 22 - 25; V, 206, 475; VI, 19).
Конденсация с янтарным ангидридом (II, 25) [1]. Опубли-
кована подробная методика.
1. М е г ё п у i F., Nilsson М., Org. Syn., 52, 1 (1972).
ИЗОПРОПИЛАТ АЛЮМИНИЯ (II, 25—27; VI, 125).
Восстановление по Меервейну — Понндорфу (II, 25—26).
В недавно опубликованной работе [1], посвященной восстанов-
лению по Меервейну — Понндорфу моно- и бициклических ке-
тонов, было показано, что в противоположность общепринятым
взглядам такие кетоны восстанавливаются с относительно вы-
сокой скоростью. Циклогексанон и 2-метилциклогексанон вос-
станавливаются с не поддающейся измерению скоростью. Даже
(з)
ментон почти полностью восстанавливается за 2 час. Хак [1]
изучал стереохимию восстановления 3-изотуйона (1) и 3-туйона
(4). При восстановлении кетона (1) преимущественно образует-
ся спирт цмс-строения (2). В случае 3-туйона (4) реакция го-
раздо менее стереоселективна, однако и здесь образуется боль-
ше спирта ^построения (5). Понижая концентрацию кетона и
алкоголята, можно еще больше повысить содержание в продук-
тах реакции спиртов 1{поконфигурации.
1. Н а с h V., J. Org. Chem., 38, 293 (1973).
232
(—)- И (4-)“2,3-0-ИЗОПРОПИЛ ИДЕН-2,З-ДИОКСИ-1,4-
бис-(ДИФЕНИЛФОСФИНО)-БУТАН (1). Мол. вес. 498,52,
т. пл. 91—92°.
н
Н3С ОЩ^снгр(сбн5)3
Н
Н3С О^СН^С^
Н3С^о-КснгР(С*Нв)2
И
(т)-(О, -12,3’
(+)-(!), »D +12,3’
Хиральные дифосфиновые лиганды получают в несколько
стадий из ь- ( + )-винион и d-(—)-винной кислот соответствен-
но [1].
Синтез d- и L-аминокислот [1]. Гидрирование (З-замещенных
а-ацетиламиноакриловых кислот (1) над оптически активным
комплексом родия (I) дает оптически активные N-ацетилами-
нокислоты (2) с оптическими выходами 70—80%.
R1. ZNHCOCH3 .NHCOCH3
'С=С Н2, катализатор ^СН—СН
R2// ^СООН * R2/ \соон
(1) (2)
Катализатор получают следующей реакцией:
[RhCl (циклооктен)2]2 + 2И. + 2СеНе —> 2RhС![И.]С6Нв + 4 циклоокген
Вместо [КйС1(циклооктен)2Ь можно исходить из [Кй(эти-
лен)2-С1]2 [2]. Если в качестве лиганда брать (%-)-И., то обра-
зуются производные ь-аминокислот; если же взять (—)-И., по-
лучаются производные D-аминокислот.
Наличие карбоксильной группы в субстрате не обязательно.
Так, енамид (3) восстанавливается до соединения (4) с опти-
ческим выходом 78% над комплексом родия(I), содержащим
в качестве лиганда (—)-И.
/NHCOCH3 f.j2! каталИаатор I
СН3СН=С; ------------CHsCONH—с—н
А н
1- Kagan 11. В, Dang Т. Р„ J. .Am. Chem Soc., 94, 6429 (1972).
ООН
ИМИНОНАДБЕНЗОЙНАЯ кислота, С6Н5—C=NH (in,
74—76).
233
Эпоксидирование. Карлсон и сотр. [1] сравнили скорости
эпоксидирования с помощью И. к. и надуксусной кислоты и от-
метили почти полное отсутствие между ними корреляции. Авто-
ры пришли к выводу, что рассматриваемая надкислота обла-
дает сравнительно низкой избирательностью и поэтому мало
пригодна для селективного эпоксидирования полиненасыщен-
ных субстратов.
1. Carlson R. G., В е h n N, S., Cowles С., J. Org, Chem., 36, 3832 (1971).
«ИНДАНОЦИКЛОН» (1). Мол. вес 286,31, т, пл. 205—206°.
И. получают из нингидрина и дибензилкетона в присутствии
этанольного раствора гидроокиси калия [1]:
Синтез по Дильсу — Альдеру с алкенами при высоких темпе-
ратурах и под давлением [2]:
1. Ried W., Freitag D., Chem. Ber., 99, 2675 (1966).
2. Ried W., Wagner R„ Ann., 716, 186 (1968).
Иод (II, 32—39; V, 209—211; VI, 125—127).
Окисление (II, 35—36; V, 209—210). Иод в метанольном рас-
творе гидроокиси натрия окисляет 1,3-дибензоилпропан до транс-
1,2-дибензоилциклопропана, вероятно, по следующему меха-
234
низму [1]:
Na+
С аСОССНДСОС^ - НаО1-> С4Н5СОСНгСНгСНСОС4Нв-Ь—> C6H5COCH,CH2CHCOCiH5
& СНЭОН I
NaOH^ CiHjCOCHCHiGHCOOiHj
CHjOH [1
н СОС^Щ
ZSj
С6Н3СО н
+ f
Окислением d-(—)-аскорбиновой кислоты под действием И.
в смеси растворителей уксусная кислота — метаиол получают
с выходом 25% чистую кристаллическую дегидро-о-(—)-аскор-
биновую кислоту (2) в виде призм с т. пл, 191 —193° (разл.) [2]:
но он
но-с
н но н
:г (СН3ОН;НОАс)
%
(2)
о
Ароматическое иодирование (V, 209). Для получения иодду-
рола [3] кипящую смесь 13,4 г (0,1 моля) дурола, 4,56 г (0,02
моля) дигидрата иодной кислоты и 10,2 г (0,04 моля) иода при
перемешивании (магнитная мешалка) обрабатывают раствором
3 мл концентрированной серной кислоты и 20 мл воды в 100 мл
уксусной кислоты. Образующийся пурпурный раствор переме-
шивают еще около 1 час при 65—70° до тех пор, пока не исчез-
нет окраска иода. Затем реакционную смесь разбавляют 250 мл
воды, выпавший желтовато-белый осадок отделяют и промывают
тремя порциями воды по 100 мл (кристаллы иоддурола, обра-
зовавшиеся во время нагревания, могут иметь пурпурный цвет,
обусловленный окклюдированным иодом, который удаляется при
235
перекристаллизации). Продукт растворяют в минимальном ко-
личестве кипящего ацетона (около 25 мл) и раствору дают по-
стоять при 0°. Выход иоддурола в виде бесцветных тонких игл
с т. пл. 78—80° составляет 20,8—22,6 г (80—87%).
Дегидратация [4]. Смесь di- и жезо-форм 3,4-диметилгексан-
диола-3,4 (1) (в отношении приблизительно 1 : 1) при нагрева-
нии с иодом в пропионовом ангидриде дегидратируется с обра-
зованием в основном ^ис,1{йс-3,4-диметилгексадиена-2,4 (2) и
цис,транс-3,4-диметилгексадиена-2,4 (3) по Зайцеву. Примене-
сн3 снэ
СНзСНгС —ССНгСН}
ОН он
Н3С\ CHj
с=с/ н +
Н Z /С=с/
HjCZ сн3
н,с сн,
X /
с=с сн3
н 'чс=с^
Н3С / ЧН
(1)
(2) 35% (3) 38%
ние в качестве реагента фенилизоцианата СбН5МСО ведет к об-
разованию преимущественно tpc-2-этил-З-метил пентадиена-1,3
(4) по Гофману.
н снгсн3
с—с н
нх ЧС=яС^
н3с 4 сн3
(4)
аез4-Дииодалканы [5]. При взаимодействии гидразонов (1)
с иодом в эфире в присутствии триэтиламина образуются гем-
дииодалканы (2) с выходом 40—60%.
R\
V=nnh2
h: (C2h6)3n
----------->
(1) (2)
Если R1 или R2 в a-положении содержат атом водорода, то в ка-
честве побочных продуктов могут получиться ВИНИЛИОДИДЫ.
Алкилмагнийфториды (VI, 126). Опубликована заключитель-
ная работа по получению алкилмагиийфторидов [6].
1. Colon I., Grilfin G. W., O’Connell E. J., Jr., Org. Syn., 52, 33
(1972).
2. Weiss W., Staudinger H., Ann., 754, 152 (1971).
3. Suzuki H., Org. Syn., 51, 94 (1971).
4. Ree ve W., Re i c h e D. M„ J. Org. Chem., 37, 68 (1972).
5. P г о s s A., S t e r n h e 11 S., Australian. J. Chem., 23, 989 (1970).
6. Y u S. W„ A s h b у E. C., J, Org. Chem., 36, 2123 (1971).
236
ИОДАЗИД (II, 39—40; V, 211—212; VI, 127—128).
Реакция с циклопентен-2-оном и циклогексен-2-оном [1].
Единственным идентифицированным продуктом реакции И. с
циклопентен-2-оном оказался 2-иодциклопентен-2-он (2) (вы-
ход (30%), Реакция И. с циклогексен-2-оном (3) также приво-
дит к 2-иодциклогексен-2-ону (4), однако в этом случае с выхо-
дом около 20% образуются и ожидаемые продукты присоедине-
ния (5) и (6).
а-Азидовинилкетоны [2]. При обработке И. раствора а,p-не-
насыщенного кетона (1) транс-конфигурации в ацетоиитрнле
получается эрцтро-форма иодазида (2) с выходом, близким к
количественному. При взаимодействии с азидом натрия в ДМФА
н ,с6н5
Зс=сх
с6н5с чн
и
о
(1)
IN3 a CH3CN^
г-^100% >
Чг,-Н NaN3
G6HSC -
N3. ,С(
C4HSC< H
II
О
это соединение превращается в азидов ин ил кетон (4) с выходом
72 %- По-видимому, первоначально ион азида атакует по
(Э^-механизму) атом углерода в соединении (2), связанный
с иодом, с образованием бас-азида (3), от молекулы которого
затем отщепляется азотистоводородная кислота.
1, McIntosh J. М, Can. J. Chem., 49, 3045 (1971).
2. L’abbfe G, Н a ssner A, J. Org. Chem, 36, 258 (1971).
237
ИОДА трис-(ТРИФТОРАЦЕТАТ), I(OCOCF3)3, Мол. вес
465,9, т. пл. 120° (разл.).
Получение [1]. И. т. получают взаимодействием иода с ды-
мящей азотной кислотой в трифторуксусиом ангидриде при—30°:
I2 + 6HNO3 + 6(CF3CO)2O 2I(OCOCF3)3 + 6CF3COOH + 6NO2
Стереоселективиое окисление алкеиов [2]. При комнатной
температуре реагент окисляет алкены до сложных эфиров вици-
нальных диолов с выходом 50—70%. Например, изобутилен(I)
превращается в 1,1 -диметилэтилен-бис-(трифторацетат) (2).
с=снг + l(OCOCF3)j ——И—> ?с—= снг
н5сх сн3 [ |
о о
(О со со
+ [l(OCOCF3)J
(2)
Окисление происходит с высокой степенью стереоселектив-
ности. Так, цис-бутен-2 дает главным образом мезоформу сое-
динения (3), а транс-бутен-2 в основном превращается в раце-
мат (4). Основным продуктом окисления циклогексена является
соединение цис-конфигурации (5).
4.OCOCF3
H3G-C
1
н3с-сч
й OCOCFj
(3)
¥ ncOCFj
HsC-6
Н-С.
’ OCOCF3
GH3
(4)
aOCOCFj
^^^OCOCFj
(5)
Предложен двухстадийный механизм этой реакции:
1(ОСОСГ3Ц
1. Schmelsser M., Dahmen К., Satori P., Chem. Ber., 100, 1633 (1967).
2. Buddrus J., Angew. Chem., Internat. Ed., 12, 163 (1973).
ИОДБЕНЗОЛА ДИХЛОРИД (II, 43; V, 213—214; VI, 128).
Избирательное свободнорадикальное хлорирование [1]. Об- -
лучение стероида (1) в присутствии И, д. в бензоле в течение
10 мин светом лампы мощностью 275 вт с последующей обра-
боткой реакционной смеси раствором перхлората серебра ввод-
ном ацетоне приводит к образованию с 75%-ным выходом смеси
олефииов (2) и (3) в отношении приблизительно 1 : 1. Анало-
гично, Зр-холестанилацетат превращается в смесь Д9(и). и Д4-хо-
238
лестенолацетатов в отношении 1 : 1. В реакции промежуточно
образуются радикалы CeHsICl- (вместо И. д. в качестве источ-
ника радикалов можно взять ВгСС1з); следует отметить, что
атака избирательно направляется лишь по третичным аксиаль-
ным атомам водорода.
Джерасси и сотр. [2] использовали этот метод введения
Д9(* О-двойной связи в синтезе диацетата (5) природного генина
морской звезды — Д9(11)-прегнен-5сс-диол-3(3,6а-она-20 [3] из со-
единения (4).
1. В г eslow R., D а 1 е J. A., Kalicky Р., Liu S. Y., Washburn W. N.,
J. Am. Chem. Soc., 94, 3276 (1972).
2. G u r s t J. E. Sheikh Y. M., Djerassi C., J. Am. Chem. Soc., 05, 628
(1973).
3. Sheikh Y. M., T u г s c h В, M., Djerassi C., J, Am. Chem. Soc., 94, 3278
(1972).
940
ИОД —КАЛИЯ ИОД ИД.
Изоксазолы [1]. Оксимы некоторых а,(3-неиасыщенных кето-
нов окисляются до изоксазолов раствором И. — к. и. в водном
ТГФ в присутствии бикарбоната натрия. Например, при обра-
ботке оксима (Е)- или (2)-|3-ионона (1) этой комбинацией реа-
гентов с выходом 91% образуется изоксазол (2), причем доро-
гостоящий иодид калия расходуется лишь в каталитических ко-
личествах. Изоксазол (2) восстанавливается натрием в жидком
аммиаке в присутствии Зэке трет-бутанола до енолята, который
после обработки реакционной смеси водой переходит в р-амино-
кетон (3). При кипячении последнего в толуоле в присутствии
следов n-толуолсульфокислоты образуется (З-дамаскон (4) с вы-
ходом 84%, считая на изоксазол (2).
Рассмотренный метод, таким образом, служит для изомери-
зации сс,(3-ненасыщенных кетонов, а также для изомеризации ке-
тонов в альдегиды, однако альдоксимы в эту реакцию ввести
не удается из-за чрезвычайной неустойчивости образующегося
изоксазола. Таким методом этилизопропенилкетон (5) был пре-
вращен в 2-метилпентен-2-аль (9).
1. В й с h i G., V е d е г a s J. С., J. Am. Chem. Soc,, 94, 9128 (19/2).
ИОДНАЯ КИСЛОТА (II, 48—53; V, 217—219; VI, 131—132).
Окислительное расщепление азинов [1]. Ароматические ази-
ны, например бензальазин (1), под действием И. к, в ледяной
уксусной кислоте или Ь1,Ь1-диметилформамиде при комнатной
температуре (мольное соотношение субстрата н окислителя 1: 2)
подвергаются окислительному расщеплению до соответствующе-
го альдегида или кетона с высоким выходом.
ЩЮв
CBH6CH=N—N=CHCgH6 —2CeH5CHO + N2
96%
(В
Метиловый и этиловый эфиры глиоксиловой кислоты [2]. Рас-
щеплением метиловых и этиловых эфиров d-винной кислоты в
сухом эфире под действием И. к, можно получить соответствую-
щие эфиры глиоксиловой кислоты с выходом 80—85%:
Н
I О
НО—С—COOR н51ов. эфир ||
| ---------5- 2 с
НО—С—COOR н/ \COOR
Н
R=CH3 или СаН5
1. F a t i a d i A. J., Chem. Ind., 1971, 64.
2. К e 1 I у T. R., S c h m i d t T. E., Ha g gerf у J. G., Synthesis, 1972, 544.
ИОДОЗОБЕНЗОЛА ДИАЦЕТАТ (II, 57—59; VI, 132—133).
о-Хниоиы. Пирокатехииы (1) реагируют при комнатной тем-
пературе с И.д. с образованием о-хинонов (3) и иодбеизола.
В качестве промежуточных продуктов образуются 2-фенилбен-
зо-1,3,2-диоксаиодолы (2) [1].
1. В а 1 a b а п А. Т„ Rev. Roumaine Chim., 14, 1281 (1969).
ЙОДОФОРМ (VI, 133).
Фотохимическая реакция с 2,3-днметилбутеном-2 приводит к
образованию 2,2,3,3-тетраметилиодциклопропана [1]:
1. М а г о 1 е v. s k i Т. A., Yang N. С,, Org. Syn., 52, 132 (1972).
1
241
ИОД —СЕРЕБРА АЦЕТАТ.
Присоединение к двойной связи в 22 (23)-положении эргосте-
рина [1]. Смесь И. — с. а. во влажной уксусной кислоте, явля-
ющаяся типичным электрофильным агентом, с За,5а-циклоэрго-
стадиен-7,22-оном-6 (1) образует единственный продукт — иод-
ацетат, имеющий, как было показано, строение (2). Таким об-
разом, данное присоединение происходит стереоселективно и
региоспецифично [2].
1. Barton D. Н. R,, Poyser J. Р., Sanimes Р. G,, Hursthouse М, В,,
N е i d 1 е S., Chem, Comm,, 1971, 715.
2. Н a s s и е г A., J. Org. Chem., 33, 2684 (1968).
ИОДЭТИНИЛТРИМЕТИЛСИЛАН, IC = CSi(CH3)3. Мол.
вес. 132,67, т. кип. 55720 мм, 1,5110.
Получение [1]. И. получают с выходом 95% реакцией хло-
ристого иода с бис-(триметилсилил)-ацетиленом в хлористом
метилене при комнатной температуре. бис-Триметилсилилацети-
лен в свою очередь получают с 86%-ным выходом из ацетиле-
нида лития и триметилхлорсилана в смеси эфир — ТГФ [2].
(CH3)3SiC=CSi(CH3)3+IC1 (CH3)3SiCsCI-HCH3)3SiCl
Арилацетилены [3]. При взаимодействии арилмедного соеди-
нения с И. при 0° (3 час), а затем при 20° (6 час) легко и с до-
вольно хорошими выходами (30—80%) образуются арилацети-
лены. Защитная триметилсилильная группа количественно уда-
ляется под действием щелочи [4].
“он
ArCu + IC==CSi(CH3)3 —> ArC=CSi(CH3)3 ----> ArC=CH
Арилмедные соединения лучше всего получать прибавлением
эфирного или тетрагидрофуранового раствора соответствующего
реактива Гриньяра или ариллитиевого соединения (1 моль) к
суспензии свежеприготовленной однобромистой меди (1,1 моля)
в эфире при интенсивном перемешивании. К образующемуся
при этом раствору арнлмедного соединения приливают затем
раствор И. (0,8 моля) в ТГФ и ведут реакцию, как описано выше.
242
1. Walton D. R. M„ Webb M. J., J. Organometallic Chem., 37, 41 (1972).
2. Walton D. R. M., Waugh F., J. Organometallic Chem., 37, 45 (1972).
3. OllverR., WaltonD. R. M., Tetrahedron Letters, 5209 (1972).
4, Eaborn C., Walton D. R. M., J. Organometallic Chem., 4, 217 (1966).
ИОНООБМЕННЫЕ СМОЛЫ (II, 62—71; V, 221—222).
Циклодегидратация [1]. Было показано, что смола амбер-
лит-15, содержащая сульфогруппы, является более эффектив-
ным катализатором циклодегидратации 2-метил-4-фенилбутано-
ла-2 (1), приводящей к образованию 1,1-диметил индан а (2),
чем реагент Брэдшера (V, 45—46), серная или муравьиная кис-
лота [1]:
Этерификация [2]. Кислоты легко этерифицируются в при-
сутствии кислой И. с. [рексин 101(H) R-231 и рексин 101(H)
R-204] в сочетании с осушителем, например сульфатом кальция.
Перед употреблением смолу высушивают в сушильном шкафу
при 100°, а сульфат кальция — при 180°. Этерификация проте-
кает при комнатной температуре, как правило, с хорошим выхо-
дом. Обработка реакционной смеси не представляет трудностей,
поскольку оба компонента этого катализатора этерификации
нерастворимы в реакционной среде.
Синтез ацеталей и кеталей. Кислая И, с. в сочетании с осу-
шителем оказалась также эффективной в синтезе ацеталей и
кеталей [3].
Никелевые катализаторы Ренея (III, 157). Обычный метод
приготовления никелевых катализаторов Ренея W-2 [4] и W-6
[5] включает утомительную операцию отмывания их дистил-
лированной водой до полного удаления следов соли. Уинберг
с сотр. [6] нашли, что деминерализацию легко осуществить с по-
мощью И. с. (амберлит IR 120 в протонированной форме). При-
готовленный таким образом катализатор несколько более акти-
вен, чем полученный согласно методике, приведенной в Organic
Syntheses.
1. Harms W. M., Eisenbraun E. J., Org. Prep. Proc. Int,, 3, 239 (1971).
2. Vesley G. F., Stenberg V. I., J. Org. Chem., 36, 2548 (1971).
3. Stenberg V. I., Vesley G. F., Rubik D., J. Org. Chem., 36, 2550
(1971).
4. Mozingo R., Org. Syn., Coll. Vol., 3, 181 (1955).
5. В И 1 i с a H. R, Adkins H„ Org. Syn., Coll. Vol., 3, 176 (1955).
6. Van Drlel H„ van Reljendam J. W., Buter J., Wynberg H.,
Syn. Comm., 1, 25 (1971).
243
ИОНООБМЕННЫЕ СМОЛЫ, ОСНОВНЫЕ (II, 65-70).
Синтез циклопропанов в результате аномального элиминиро-
вания по Гофману. Предлагаемая методика [1] позволяет су-
щественно улучшить выходы этилового эфира 1-бензилцнкло-
пропанкарбоновой кислоты. Метод является общим и применим
для синтеза эфиров циклопропанкарбоновых кислот, замещен-
ных в положение 1.
ClCH2CH2N(CHa)2 сн3г
СвН6СН2СН(СО2С2Н6), --------------> CeH6CHaC(CO2C2HB)s —>
CH2CICN(CH3)2
ОН"
—> СаН6СН2С(СО2С2НБ)2 —------->
I И. с.. о,,
I + FtOH
CH2CH2N(CH3)3r
CO2C2HS
Нагревание I
СвН5СН2С(СО2С2Н5)г ---------->• с3нБсн2 с—сн2
CH2CH2N(CH3)3, (OC2Hs)
В литровую трехгорлую круглодонную колбу, предваритель-
но прогретую открытым пламенем горелки для удаления следов
влаги и снабженную механической мешалкой, капельной ворон-
кой и обратным холодильником с хлоркальциевой трубкой, по-
мещают 20,5 г (0,5 моля) 58,5%-ной дисперсии гидрида натрия
в минеральном масле и 200 жл диметилсульфоксида. Смесь
перемешивают прн 20—25°, время от времени охлаждая ее в
случае необходимости на ледяной бане, и медленно добавляют
по каплям раствор 125,1 а (0,5 моля) диэтнлового эфира бен-
зилмалоновой кислоты. Перемешивают при 20—25° еще около
30 мин (до прекращения выделения водорода), а затем с такой
же скоростью прибавляют по каплям раствор 53,8 г (0,5 моля)
fj-диметиламнноэтилхлорида, который выделяют из соответст-
вующего хлоргндрата непосредственно перед употреблением.
Для этого 86,4 г (0,6 моля) хлоргидрата растворяют в 100 мл
воды и медленно прибавляют избыток твердого карбоната ка-
лия. Основание экстрагируют из густой реакционной смеси эфи-
ром, эфирный раствор высушивают (MgSO4) и отгоняют эфир
в вакууме при 25°. Остаток представляет собой бесцветную
жидкость. При длительном хранении основания даже прн 0° на-
блюдается его кватернизация. По окончании прибавления ами-
нохлорнда реакционную смесь постепенно нагревают до 100°,
не прекращая перемешнвання. Через 30 мин смесь охлаждают,
выливают в 1 л ледяной воды и экстрагируют тремя порциями
эфира по 150 мл, Объединенные эфирные вытяжки высушивают
сульфатом магния, переносят в литровую трехгорлую кругло-
донную колбу, снабженную механической мешалкой, капельной
воронкой и обратным холодильником, и прибавляют по каплям
244
78,1 г (0,6 моля) йодистого метила. Смесь перемешивают при
комнатной температуре в течение 30 мин, затем с сиропообраз-
ного иодметилата декантируют жидкость, а иодметилат промы-
вают декантацией 100 мл эфира. К сиропообразному иодмети-
лату (в той же колбе) добавляют 100 мл этанола и затем при-
близительно двукратный избыток (400 а) анионообменной смо-
лы в ОН_-форме. Для этого одинаково пригодны как рексин
201 Фишера, так н амберлит IRA 400 Маллинкродта. Исполь-
зованную анионообменную смолу переводят в ОН_-форму, про-
пуская через колонку с регенерируемой И. с. 10%-ный водный
раствор гидроокиси натрия до отрицательной пробы (AgNOs—
HNO3) на галогенид-ион, а затем воду до нейтральной реакции.
Отфильтровывают И. с. от реакционной смеси (см. выше) и оса-
док иа фильтре промывают тремя порциями этанола по 100 мл.
Объединенные фильтраты переносят в литровую одногорлую
круглодонную колбу, снабженную колонкой Вигрэ высотой
15,24 см, и почти бесцветный раствор концентрируют при пони-
женном давлении. (Внимание'. Вспенивается.) Оставшуюся
жидкость постепенно нагревают до 150—200° в вакууме водо-
струйного насоса (10—25 мин). По окончании термического
разложения (прекращение выделения газа) остаток растворяют
в 150 мл эфира, раствор высушивают сульфатом магния, фильт-
руют и концентрируют фильтрат. Перегонка остатка дает 66,5—
75,5 г (общий выход 65—75%) этилового эфира 1-бензилцнкло-
пропанкарбоновой кислоты в виде бесцветной жидкости с т.
кип. 109—12072 мм.
I, Kaiser С,, Weinstock J., Org. Syn,, submitted 1971,
КАДМИЯ КАРБОНАТ, CdCO3. Мол. вес 172,42.
Синтез арилглюкуронидов по Кенигсу — Кнорру. Стероидные
глюкурониды эстрона, 17(3-эстрадиола, эстриола и эквиленнна
можно получить с прекрасным выходом реакцией фенольных
стероидов с бромпроизводным сахара (1) в толуоле, катализи-
руемой К-к. [1]. Применение карбоната серебра — обычного ка-
(2)
тализатора реакции Кёнигса — Кнорра [2] — дает только при-
мерно 7°/о глюкуронида (2). Новый катализатор позволяет так-
же получить с хорошими выходами и некоторые стероидные
алициклические глюкурониды.
1. Conrow R. В., Bernstein S., J. Org. Chem., 36, 863 (1971).
2. Koenigs W., Knorr E., Ber., 34, 957 (1901); see Pigman W., The
Carbohydrates, Academic Press, New York, 150, 191 (1957).
КАЛИЙ В ГРАФИТЕ. Калий очень легко внедрить в графит
нагреванием смеси этих двух простых веществ в отсутствие
воздуха при 70°. Меняя соотношения реагентов, можно получить
соединения, отвечающие формулам С8К, С24К, СзеК и С4вК.
Восстановление кетонов [1]. Реагент восстанавливает насы-
щенные н сопряженные кетоны до спиртов; механизм реакции,
вероятно, аналогичен механизму электрохимического восстанов-
ления (как это показано, например, для ацетофенона на схеме I).
В некоторых случаях происходит также образование пинаконов.
Так, ацетофенон дает при восстановлении смесь спирта (выход
45%) н пинакона (выход 45%). Однако преобладающим про-
дуктом реакции обычно является спирт. Например, в результате
восстановления бензофенона с выходом 98% образуется бенз-
246
гидрол. а,р*Ненасыщениые кетоны восстанавливаются до насы-.
щенных спиртов. При восстановлении камфоры преимуществен-
но образуется спирт экзо-строения. Следует отметить, что вос-
становление натрием в спирте или калием в присутствии гра-
фита (но не внедренным) дает в основном спирт эндо-строеиия.
Схема I:
с6н5сосн3
о" "I
L
с6н5с
сн3
2 С6Ю
он
I
с6н5с- сн3
।
н
о' о"
I I
едс-сс.щ
HsC СНЭ
2 С8К+
НО он
I 1
C6H5C-CC6HS
I I
Н3С СНЭ
1. Lalancette J.-AL, Rollin G., Dumas P., Can. J. Chem., 50, 3058
(1972).
КАЛИЯ БОРГИДРИД, KBH4. Мол. вес 53,95.
Удаление тиазолидинового цикла пенициллина [1]. В резуль-
тате обработки производного пенициллина, сульфона (1) избыт-
ок
(1) R - о -ОгМС6Н4
D,io г
квн*
ком К-б. в водном пропаноле-2 при охлаждении образуется азе-
тидинон-2 (2). К такому же расщеплению приводит действие
избытка трис- (трз7’-бутокси)-алюмогндрида лития (II, 187—193;
V, 250—251; VI, 149) в ТГФ.
1. Sheehan J. С., Pane tta С. A., J. Org. Chem., 38, 940 (1973).
КАЛИЯ ГЕКСАМЕТИЛДИСИЛАЗАН [калия бш>(триме^
тилсилил)-амид], KN[Si (СН3)з]г. Мол. вес 199,49.
Получение [1]. К- г. получают кипячением амида калня (I,
54—57; V, 24; VI, 13—14) с гексаметилдиснлазаном в бензоле
247
(Na). Ср. методику [2] получения соответствующего натриевого
производного.
Циклизация галогеикеталей [1]. Кеталь нитрила (1) под дей-
ствием 1—3 экв К-г. в бензоле при комнатной температуре (8—
20 час) или при кипячении (1—4 час) циклизуется в производ-
ное циклогексанона (2), Под действием реагента можно цикли-
зовать даже вторичные а-галогенкетали [(3) -+ (4)].
Циклизация галогеикеталей оказалась особенно ценной в
случае синтеза полициклических систем. Так, соединение (5)
превращается в цмс-9-циандекалон-2 (6), а (7)—в (8). Гид-
риндан (10) был получен из бромкеталя (9).
(10)
248
Циклизация может привести и к одновременному образова-
нию двух циклов. Например, соединение (11) превращается в
смесь изомеров (12) и (13) в соотношении 95:5.
(И) (12) -(13)
Если при циклизации соединения (11) К-г. заменить гекса-
метилдисил азаном лития, то в продуктах реакции преобладает
соединение (13), образующееся с выходом 95% [3]. Авторы при-
водят возможное объяснение этому поразительному влиянию
природы катиона на рассматриваемую циклизацию.
1. Stork G., Gardner J. О., Boeck man R. К., Jr., Parker K. A.,
J. Am. Chem. Soc., 95, 2014 (1973).
2, Wannagat V., Niederspr.um H., Chem. Ber., 94, 1540 (1961).
3. Stork G., Boeckman R. R., Jr.. J. Am. Chem. Soc., 95, 2016 (1973).
КАЛИЯ ГЕКСАХЛОРОВОЛЬФРАМАТЦУ), K2WC16. Мол.
вес 474,86, красная кристаллическая соль.
К. г. приготавливают нагреванием прн 130° гексахлорида
вольфрама с нодндом калня в запаянной трубке Кариуса в те-
чение 3 диен [1].
Синтез олефинов. Шарплес и Флуд [2] описали новый метод
получения олефинов, состоящий в восстановительном дезокси-
генировании вицинальных цис- и транс-диолов при кипячении
с К. г. в ТГФ. Согласно типовой методике, диол, растворенный
в ТГФ, действием метиллития (2 мол. же) в диэтнловом эфире
в атмосфере превращают в дилитиевую соль. Затем добав-
ляют K.2WC16 (2 мол. же) н кипятят образовавшуюся гетеро-
генную смесь от 4 час до 4 дней в зависимости от строения ис-
ходного диола. Тетразамещенные диолы восстанавливаются
быстро с удовлетворительным выходом; моно-, дн- и тризаме-
щенные днолы восстанавливаются медленнее, но тоже с хоро-
шим выходом. Олефнны образуются в основном в результате
цис-эл ими ниров ания.
1. RennedyC. D.. Peacock R. D., J. Chem. Soc., 1963, 3392.
2. Sharpless R. В., Flood T, C., Chem. Comm,, 1972, 370.
КАЛИЯ ГИДРИД (II, 76; V, 224).
ВниманиеХ К. г. в масле медленно реагирует с влагой воз-
духа; лучше всего хранить его в атмосфере азота или аргона.
К. г. энергично реагирует с водой. С остатками К. г. следует об-
ращаться так же, как с остатками натрия.
249
Металлирование аминов и слабых органических кислот [1].
К. г. при комнатной температуре быстро взаимодействует с ами-
нами с образованием моно- и диалкилаыидов калия, которые
являются исключительно сильными основаниями. Так, N-калий-
этилендиамин (КЭДА) при 25° за 5 мин превращает лимонен
в «-цимол, а та же реакция с литийэтиленднамииом требует
двухчасового нагревания прн 90°.
нэдд.
—----
+ н2
100%
К-г. реагирует при 25° с ДМСО, образуя димснлкалий; водо-
род выделяется полностью за 8 мин.
Пространственно затрудненные третичные спирты также
энергично реагируют с К. г. с количественным выделением во-
дорода.
I. Б г о w п С. A., J. Am. Chem. Soc., 95, 982 (1973).
КАЛИЯ ГИДРООКИСЬ (II, 77—-80; V, 224; VI, 135).
Синтез мезо-иафтодиаитрона [1]. Нагревание эмодина (1) с
0,6 н. КОН в запаянной трубке (гидрохинон, Ь12, 3 недели при
110°) приводит к образованию нзогиперицина (2) с выходом
35%. Полагают, что в реакции происходит присоединение по
Михаэлю эмодин-аниона к самому эмодину, а затем еще целый
ряд таутомерных превращений и реакций конденсации и элими-
нирования.
Дигидрогалогенирование — гидролиз. Хиткок и Хасснер ис-
пользовали К-г. в этаноле на завершающей стадии синтеза ази-
ридина (4) из 1,2-дигидронафталнна (1) [2] через метил-(транс-
250
2-И0Д-1-тетралин) -карбамат (3) [3].
AgNCO + Iz --> Agl + INCO
Д2-Пиразолиноны-5 [4]. Прн обработке 5-окси-3,5-дифенил-Д2-
пиразолннона-4 (1) метанольным раствором К. г. образуется
4-окси-3,4-дифенил-Д2-пнразолинон-5 (2) с выходом 76—80%.
Реакция, вероятно, включает пннаколиновую перегруппировку.
Соединение (2) можно также получить, правда с несколько мень-
шим выходом, кипячением (4) в метанольном растворе карбо-
ната натрия в присутствии воздуха.
Алкилирование индола и пиррола [5], Индол и пиррол можно
с высоким выходом алкилировать по азоту, превратив каждый
из них сначала в калиевую соль действием КОН в ДМСО и об-
работав затем полученную соль галогеналкилом. Этим методом
кон/дмсо
С6Н5СНгВг
92-97%
из индола (1) получают 1 -бензилиндол с выходом 92—97%.
251
В случае вторичных галогеналкилов выходы ниже, третичные
галогеналкилы вообще не вступают в реакцию.
I. Steglich W., Arnold R., Angew. Chem., Internal. Ed., 12, 79 (1973),
2. Heathcock С. H., Hassner A., Org. Syn., 51, 53 (1971),
3. H e a t h с о с k С. H., H a s s n e r A,, Org. Syn., 51, 112 (1971),
4, Nye M. J., Tang W. P„ Can. J. Chem., 51, 338 (1973).
5. Heaney H„ Org. Syn., submitted 1973.
КАЛИЯ (НАТРИЯ) ИМИНОКСИЛДИСУЛЬФОНАТ (соль
Фреми) (II, 81—83; V, 225—226; VI, 135—136).
Получение [1].
NaNO2 + 2SO2 + NaHCO3 —HON(SO3Na)2 + СО2
HON(SO3Na)2 + ОН' —--------------> : 6—N(SO3Na)2 + Н2О
Анод из нержавеющей стали •* v 1
Реагент можно использовать для получения 4,5-диметил-1,2-
бензохиноиа [2]:
HjCx^X^OH
L J
+ 2ib-R(SOjK)a
HjO, (флр
25°
49-50%
+ HON(SOjK)2 + HN(SO3K)
Опубликован подробный обзор реакций окисления с помощью
К- И. [3J.
1, W е h г I i Р. A., Pigott F,, Org. Syn., 52, 83 (1972).
2. Т е u b е г H.-J„ Org. Syn., 52, 88 (1972).
3. Zimmer H., V a n k i n D, C., Horgan S. W., Chem. Rev., 71, 229 (1971).
КАЛИЯ ИОДИД —ЦИНК-МЕДНАЯ ПАРА.
Восстановительное отщепление вицинальных метан сульфо-
нилоксигрупп в 2,3-О-димезил-а-о-глюкопиранозидах первона-
чально осуществляли с помощью нодида натрия н цинковой пы-
ли [1]. Фразер-Рейд и Боктор [2] сообщают, что для улучшения
выходов этой реакции, а также ее воспроизводимости следует
с6н5соосн.
О Ms
KI, Zn-Cu
ДМФА ,кипячение
29-32%
С4Н5СООСНг
применять такую же цинк-медную пару, как и в реакции Сим-
монса— Смита [3]. Реакцию проводят в кипящем ДМФА,
1. Tipson R. S,, Cohen А„ Carbohydrate Res., 1, 338 (1965).
2. Fraser-Reid В., Boctor Б., Can. J. Chem., 47, 393 (1969),
3. Смит P. Д., Симмонс X. Э., «Синтезы органических препаратов», ИЛ,
М., 1964, сб. 12, стр. НО.
252
КАЛИЯ ОСМАТ, K2OsO4. Мол. вес 332,40. Реагент следует
хранить в сухом месте.
Гидроксилирование олефинов. Ллойд и сотр. [1] описали два
активных катализатора в растворе для гидроксилирования оле-
финов с помощью К-о- В состав одного из них (катализатор А)
входит перекись водорода в качестве окислителя и уксусная
кислота для нейтрализации раствора К. о. В другом (катализа-
тор Б) окислителем служит хлорат натрия (II, 424—425), Хотя
катализатор Б получить легче, чем катализатор А, его не сле-
дует применять в тех случаях, когда диол отгоняют непосред-
ственно из реакционной смеси, так как при нагревании оставше-
гося хлората натрия с органическими соединениями может про-
изойти взрыв. Гидроксилирование аллилового спирта с помощью
катализатора А приводит к образованию глицерина с выходом
67%. В результате гидроксилирования циклогексена с участием
катализатора Б был получен ^«с-цнклогександнол-1,2 с выхо-
дом 76%.
KaOsO-i, Н2О, НОАс, Н2О2
—А—он
—С—ОН
K2OSO4, НгО, НОАс, NaClOg
I. L 1 о у d W. D., Navarette В. J., Shaw М. F., Synthesis, 1972 , 610.,
КАЛИЯ ПЕРМАНГАНАТ-УКСУСНЫЙ АНГИДРИД.
а-Дикетоны из олефинов [1]. Олефины в результате реакции
с 3—4 мол. же перманганата калия в уксусном ангидриде при
температуре ниже 10° окисляются непосредственно в а-дике-
тоны. Например, циклододецен (1) окисляется в днон (2) и при
этом побочно образуется кетоацетат (3) и дикарбоновая кисло-
та (4). Поскольку кетоацетат (3) гидролизуется до а-оксике-
тоиа, который затем ацетатом медн(П) можно окислить до
днона (2), общий выход диона (2) удается повысить до 63%.
Реакцию можно проводить с большими загрузками [50 г цикло-
олефина (I)].
9.R.4
Другим примером может служить окисление децена-1 (5):
О
СН3(СН2)7СН=СН2 —> СН3(СН,)7(2СН2ОАс+ СНэ(СН2)7СООН
(5) (6)36% (7)50%
Если субстрат при низких температурах нерастворим в уксус-
ном ангидриде, то раствор можно разбавить диметоксиэтаиом.
1. Sharpless К, В., Lauer R. F., Repic О., Teranlshi A. Y., Wil-
li a m s D. R., J. Am. Chem. Soc., 93, 3303 (1971).
КАЛИЯ ПЕРХРОМАТ, КзСгО8. Мол. вес 297,31.
Синглетный кислород [1]. С целью получения К- и. водный
раствор хромата калия (KsCrCU), содержащий гидроокись ка-
лия, прибавляют к водно-метанольному раствору 30%-ной пе-
рекиси водорода. Эта реакция служит удобным источником син-
глетного кислорода. Например, генерирование К- п. в прнсутст-
Н3Сч /СН3 ц3сго8 С3Нч >^СН2
Cz > ХС___с
н3с/_________________________\сн3 35%-с3н/|_\сы2
хД Л
вии 2,3-диметилбутена-2 приводит к образованию с выходом
35% гидроперекиси 2,3-диметилбутен-1-ила-3. Последняя обра-
зуется также в результате сенсибилизированного фотоокисле-
ния. К- и. является обычным источником синглетного кислорода,
выделяющегося с контролируемой скоростью в зависимости от
содержания воды в растворителе и от температуры.
1. Peters J. W., Pitts J. Н., Jr., Rosenthal E, Fuhr H., J. Am. Chem.
Soc., 94, 4348 (1972).
КАЛИЯ ТЕТРАХЛОРОПЛАТИHAT(II) (V, 362).
Декарбонилирование. При изучении гомогенного дейтериро-
вания а-оксикнслот в присутствии К. т. Кальф и Гарнатт [1] об-
/ОН
(С9Н5)2С( —(СеН5)2С=О
хсоон
наружили, что бензиловая кислота при этом легко декарбонили-
руется, образуя с высоким выходом бензофенон. Бензгидрол
в этих условиях в бензофенон не превращается.
1. Calf G. Е., Garnatt J. L., Tetrahedron Letters, 511 (1973).
КАЛИЯ ФЕРРАТ(У1), KsFeO^. Мол. вес 198,05, кристаллы
пурпурного цвета. Соль следует защищать от действия влаги.
254
Получение. Томпсон и сотр, [1] рекомендуют получать К. ф-
окислеиием нитрата железа(III) гипохлоритом натрия. Для по-
лучения продукта 92—96%-ной чистоты с выходом 44—76% не-
обходима довольно утомительная процедура очистки.
Окисление [2]. К- ф. служит реагентом для селективного
окисления первичных спиртов или аминов до альдегидов и вто-
ричных спиртов до кетонов. Двойные связи, альдегидные груп-
пы, третичные спирты н третичные амины не окисляются этим
реагентом. Реакцию осуществляют при комнатной температуре
в воде или в водных растворителях при начальном pH 11,5 и
конечном pH 13,5; вода для окисления необходима. Согласно
обычной методике, к раствору спирта или амина (0,003 моля)
в 10 мл воды прибавляют КаРеО4 (0,002 моля) и образующуюся
смесь энергично встряхивают до исчезновения пурпурной окрас-
ки феррата (VI) (1—90 мин) и выпадения бурого осадка
Fe(OH)3. Продукт реакции выделяют обычным путем — экс-
тракцией эфиром или бензолом.
Примеры:
«-Гептанол —> н-Гептаналь (30%)
Бензиловый спирт —Бензальдегид (80%)
Циклогексанол —> Циклогексанон (20—30%)
Коричный спирт -—> Коричный альдегид (75%)
Бензиламин —> Бензальдегид (70%)
1-Метилбензиламип -—> Ацетофенон (70%)
Окснметильные группы углеводов селективно окисляются
К. ф. до альдегидных; при этом вторичные оксигруппы не за-
трагиваются [3].
1. Thompson G. W„ Ockerman L. Т., Schreyer J. M., J. Am. Chem.
Soc., 73, 1379 (1951); Wood R. H„ J. Am. Chem. Soc., 80, 2038 (1958).
2. Au det te R. J., Quail J. W,, Smith P. J., Tetrahedron Letters, 279
(1971).
3, В e M i 11 e г J. N., Kumar! V. G., Darling S. D., Tetrahedron Letters,
4143 (1972).
КАЛИЯ ФЕРРИЦИАНИД (II, 102—107; V, 227—228),
Окисление 2“амиио-3(6-ди-трет-бутилфенола К. ф. при pH 7,2
приводит к получению 3,6-ди-трвт-бутилбензохинона-1,2 в виде
темно-синнх кристаллов с т.пл. 204°, охарактеризованных УФ- н
ПК-спектрами [1].
K3Fe(CN)6
------------------>
СН3ОН, рНЦбуфер1)
Окислительная циклизация фенолов. Окисление вторичного
амина (1). К. ф. в присутствии бикарбоната натрия в хлороформе
255
приводит к норэритринадиенону (2) [2]
2-Карбэтоксшюрпротозиноменин (3) окисляется К-ф. в при-
сутствии ацетата аммония и аммиака (pH 9,2) до проэрптрнн-
адиенона (4) с выходом около 2%. Последний обладает угле-
родным скелетом ключевого промежуточного соединения в пред-
полагаемом биогенезе алкалоидов Erythrlna [3].
1. В г о с k m а п n Н., S е е 1 a F., Chem. Вег., 104, 2751 (1971).
2. К a m е t а п i Т., Takahashi К., Shibuya S., F и к u m э t о К., J. Chem.
Soc. (C), 1971, 1800.
3. К ametani T., Charubala R., lhara M., Koizumi M., Fuku tno-
to K, Chem. Comm., 1971, 289.
КАЛЬЦИЯ КАРБОНАТ (11,113—114; V, 230—231).
Пинаколиновая перегруппировка. 7,7-Диметнлбицнкло-[4,1,1]-
октанон-3 (2) удобно получать [1] нз тозилата (1) по методике
пииаколиновой перегруппировки Кори [2]. Тознлат (4,0 жмоля)
СаСО3, ТГФ
LiClO,
50^
обрабатывают К- к. (4,0 жмоля) в ТГФ в присутствии каталити-
ческих количеств перхлората лнтня (72 час, 60°).
1. Tubiani W., Waegell В., Angew. Chem., Internal. Ed., 11, 640 (1972).
2. Corey E. J., J. Am. Chem. Soc., 83, 1251 (1961); 86, 478 (1964).
256
(МО-КАМФОРСУЛЬФОКИСЛОТА (II, 115; V, 231-232).
Получение хиральных триарилфосфииов [1]. Последователь-
ная обработка фенилдихлорфосфина (1) металлоорганическими
соединениями, обладающими различной реакционной способ-
ностью, позволяет с высоким выходом получать хиральные фос-
фины (2). При взаимодействии фосфина (2) с параформом и
СбН6РС1г + 71 -(CHj)3CCf,H4ZnCl — > п-(СН3)эСС6Н4РС6Н5
(1)
----> (AHjn-fCHjJsCC^PCiH^Ar1)
(2) C6H4-C4Hs-n(Ar’)
л-С6Н5- С6Н41Л
0,5 экв К- образуется кристаллический ( + )-10-камфорсульфо-
нат триарилоксиметилфосфония. Из маточного раствора можно
выделить его антипод. Соли (3) расщепляются до оптически
активных три арил фосфинов (2) под действием оснований
(NaOH, КОН, NH3).
1. Wittig G., Braun Н., С г i s t a u H.-J., Ann., 751, 17 (1971).
п-КАРБМЕТОКСИНАДБЕНЗОЙНАЯ КИСЛОТА (п-мето-
ксикарбонилнадбеизойная кислота) (2). Мол. вес 196,15, начи-
нает разлагаться при 125°.
Эту надкислоту получают [1] с выходом 80—95% фотоокис-
лением метилового эфира п-формилбензойной кислоты (1) в че-
тыреххлористом углероде. Эфир (1) в свою очередь получают
окислением метилового эфира n-толунловой кислоты по методу
Либермана н Коннора [2].
СНО СОООН
СООСНз СООСНз
(1) &
9 Зак. 506
257
По устойчивости К. К. аналогична ж-хлорнадбензойной кис-
лоте: при хранении в течение одного года при 10° она разла-
гается менее чем на 5%. Надкислота довольно хорошо раство-
ряется в диоксане, этаноле, ацетоне и ацетонитриле и хуже —
в хлороформе, бензоле и эфире.
К. к. использовали для эпоксидирования и окисления по
Байеру — Виллигеру; в первом случае выходы колеблются в ин-
тервале от 60 до 90%, во втором —от 64 до 85%. Таким обра-
зом, по активности кислота соответствует надбензойной и над-
фталевой кислотам.
1. Kawabe N., Okada К., Ohno М., J. Org. Chem., 37, 4210 (1972).
2. Lieberman S. V., Connor R., Org. Syn., Coll. Vol. 11, 441 (1955).
бис-(КАРБЭТОКСИД ИАЗОМЕТИЛ )-РТУТЬ,
n2 -
_C2H5O2C—C—_2Hg (1). Мол. вес 426,80, т. пл. 103—105° (разл.).
Получение [1]. К. получают из диазоуксусного эфира и окнси
ртути; светло-желтое вещество кристаллизуют нз эфира.
Синтез этиловых эфиров галогеидиазоуксусиых кислот [2].
w <2SOi. неС1а Lc.HsChC—с—J2Hg
(2), Hal = I
(3), Hal = Br
(4), Hal = CI
1. В u c h n e r E., Ber., 28, 215 (1895).
2. Schollkopf U., Gerhart F., Reetz M., Frasnelli H., Schuma-
cher H., Ann., 716, 204 (1968).
2-КАРБЭТОКСИ-1.3-ДИТИАН (1). Мол. вес 192,30, т. кип.
75—77°/0,2 мм.
Получение [1]. Реагент (1) получают с выходом 70% реак-
цией этилового эфира диэтоксиуксусиой кислоты [2] с пропан-
днтнолом-1,3 в присутствии эфирата трифторнда бора.
ВГ3-(СгН5)2О
(CzHbOjjCHCOOCiHs + HS(CHZ)3SH
Эфиры а-кетокислот [1]. При взаимодействии с гидридом
натрия в смеси ДМФА и бензола К- (1) превращается в нат-
риевое производное, которое реагирует с первичными или вто-
ричными алкнлгалогенидамн с образованием 2-алкил-2-карбэт-
окси-1,3-днтиана (2) с выходом 75—95%. Десульфуризация эфи-
ра (2) никелем Ренея дает эфиры алкилированной уксусной кис-
лоты (3). Однако гораздо важнее то, что при обработке эфира
(2) N-бромсукцииимидом (см. 1,3-Днтиан, этот том) с выходом
60—85% образуются сложные эфиры а-кетокислот (4). Этот но-
вый метод синтеза эфиров а-кетокислот обладает тем преимуще-
ством, что неходкое соединение (1) легкодоступно, и, кроме того,
можно обойтись без применения алкиллитневых реагентов.
(i)
1) NaH, ДМФА—С4Н4
2) Ц’Вг
----------------->
(4)
1. Е 1 i е I Е. L., Н а г t m a n п A. A., J. Org. Chem., 37, 505 (1972).
2. Moffett R. В., Org. Syn., Coll. Vol., 4, 427 (1963).
N-КАРБЭТОКСИИЗОТИОЦИАНАТ, C2II5OOCN = C = S.
Мол. вес 131,15, т. кип. 44—46°/Ю мм.
Получение [1]. К. получают реакцией роданида калия с эти-
ловым эфиром хлоругольной кислоты в ацетонитриле (паровая
баня).
Синтез гетероциклов. Реагент взаимодействует с третичными
енаминами, такими, как, например, (1), с образованием аддук-
тов (2). При обработке первичными аминами (или аммиаком)
эти аддукты переаминируются и циклизуются, образуя 4-тио-
урацилы (3) [1].
+ c2h5oocn=c=s---------
65-9 0%
9й
259
к. взаимодействует с пирролом (4) при температуре ниже
40°, давая с выходом 93% N'-карбэтоксиамид пиррол-2-тиокар-
боновой кислоты (5) [2]. Амид (5) при нагревании в хинолине
превращается в имид пиррол-2-тио-1,2-днкарбоновой кислоты
(6). Взаимодействие пирролкалня (7), полученного реакцией
пиррола с калием в ТГФ, с реагентом приводит с выходом 45%
к N'-карбэтокси амиду пиррол-1-тиокарбоновой кислоты (8).
Амид (8) при непродолжительном кипячении с хинолином дает
с выходом 58% имид пиррол-1-тио-1,2-дикарбоновой кислоты (9).
(7)
CzH5OOCN=C=S
45%
C,HrN; 150-160”
-СгН5ОН
58%
4nhcooczh5
(8)
I. La mon R. W., J, Heterocyclic Chem., 5, 837 (1968).
2. Papadopoulos E. P., J. Org. Chem., 38, 667 (1973).
КАРБЭТОКСИМЕТИЛ МЕДЬ, CuCH2COOC2H5. Мол. вес
150,64.
Получение [1]. Это медьорганическое соединение получают
добавлением эквимолекулярного количества диизопропиламнда
лития к этилацетату в присутствии нодида меди(1) при —110°
в атмосфере N2 и затем смеси дают постепенно нагреться до
—30°. При этом образуется светло-коричневый гомогенный рас-
твор. Глубокое охлаждение необходимо; поскольку если прово-
дить реакцию даже при —78°, то выход реагента будет гораздо
ниже.
СН3СООС2Н6
ЫЩСНЮНзШ ст
73 %
CuCH2COOC2HB
Сложные эфиры у,&-неиасыщениых кислот [1]. С аллилгало-
генндами в ТГФ К- дает сложные эфиры у,6-ненасыщенных кис-
лот.
260
Примеры:
СН2 = CBrCH2Br
СН3ООССН=СНСН2Вг
С0Н5СН2Вг
----> СН2=СВгСН2СНэСООС2Н5
83
СН2СООС2Н5
69% \____/
----> СН3ООССН=СНСН2СН2СООС2Н5
89%
—> C6H5CH2CH2COOC2HS
62%
I, Ku w a j i m a L., Doi Y., Tetrahedron Letters, 1163 (1972).
4-КАРБЭТ0КСИ-5-МЕТИЛ-3-ЭТИЛИ30КСА30Л (cp. 1,361;
V, 148—149).
Синтез [1]
Реакция аннелирования изоксазола [2].
1. McMurry J. E., Org. Syn., submitted 1972.
2. M с M u г г у J. E,, Org. Syn., submitted 1972.
261
N - КАРБЭТОКСИ - 2 - ЭТОКСИ - 1,2 - ДИГИДРОХИНОЛ ИН
(КЭД) (V, 236—237; VI, 136—137).
Синтез пептидов по Меррифилду [1]. В качестве конденси-
рующего агента в синтезе пептидов из БОК-аминокислот и пеп-
тидных смол на твердом носителе по Меррифилду КЭД имеет
преимущество перед дициклогексилкарбодиимидом.
Твердофазный конденсирующий агент в пептидном синтезе [2].
КЭД был введен в нерастворимый полимер, полученный из сти-
рола и дивинилбензола. В пептидном синтезе полимерная фор-
ма по своей активности несколько уступает мономерной, однако
она не менее эффективна, чем широко применяемое сочетание
дициклогексил карбодиимид — N-оксисукцинимид.
Стероидные амиды. Герц и Мантекон [3], используя КЭД,
получили с удовлетворительным выходом амиды 3-формиллнто-
холевой кислоты.
1. S i р о s F., G a s t о n D. W., Synthesis, 1971, 321.
2. Brown J., Williams R. E., Can. J. Chem., 49, 3765 (1971).
3. Herz J. E., Mantecdn R. E., Org. Prep. Proc. Int., 4, 129 (1972).
КЕТЕН (I, 125—129; V, 237—238).
Ацетилирование СН-кислотиых соединений [1]. СН-Кислот-
ные соединения можно ацетилировать действием К- в присут-
ствии катализатора (пиперидин, метилат натрия, хлорацетат
натрия).
Примеры:
/СООС2Н5
/ Катализатор
2 н2с=с=о 4- СН2 ---------> (СН3СО)3ССООС2Н5
' \ 85—95% v 7
\СООСН3
2Н2С—С=О ф CH2(COOC2HS)2 KaTQ7ZaJ"> (СНэСО)2С(СООСгНЕ)2
о 5— У5 то
Катализатор
Н2С=С=О + НС(СООС2НЁ)3 -----——+ СНзСО—С(СООС2Нв)з
55%
1. Eek Н., Prigge N_, Ann., 731, 12 (1970).
КИСЛОРОД, о2.
Окисление триалкилбораиов [1]. Триалкилбораны реагируют
с К. в ТГФ, образуя спирты с практически количественными вы-
ходами:
зн2о
R3B+ 1</гО2 —> 1(RO)3B] 3ROH + В(ОН)3
Для завершения быстрой вначале реакции может потребо-
ваться до 75 мин.
К. генерируют разложением раствора перекиси водорода, ка-
тализируемым основной двуокисью марганца, в автоматическом
автоклаве, специально приспособленном для этой цели.
1. Brown Н. С., Midland М. М., Rabalka G. W., J. Am. Chem. Soc.,
93, 1024 (1971).
262
КИСЛОРОД СИНГЛЕТНЫЙ,1 О2.
1,4-Циилоприсоединеиие [1]. При фотоокислении 1,1-дифенил-
2-метоксиэтилена (1) в бензоле в присутствии сенсибилизатора
(динафталинтиофена) в качестве главного продукта образуется
у-лактои (5). Первичным продуктом реакции при —78°, по-ви-
димому, является энбо-перекись (2), которую можно выделить
и охарактеризовать методами ЯМР и УФ-спектроскопии. Кроме
того, удалось выделить и охарактеризовать метиловый эфир (3)
и перекись (4). Оба этих вещества при непродолжительном на-
гревании или при обработке кислотой превращаются в лактон
(5), Реакция, таким образом, представляет собой пример взаи-
модействия К,, с. по Дильсу — Альдеру.
Фотоокисление индена (6) при —78° в присутствии фотосеи-
сибилизатора (бенгальская роза) дает в качестве основного
продукта соединение (9), структура которого была установлена
на основании данных ИК и ЯМР спектров. По-видимому, сна-
чала К. с. присоединяется к индену (6) по реакции Днльса —
Альдера, а образующаяся перекись (7) перегруппировывается
затем в диэпоксид (8). Присоединение второй молекулы К. с. при-
водит к соединению (9). При нагревании последнее перегруппи-
ровывается с образованием двух продуктов, основным из кото-
рых является соединение (10). На основании спектральных
данных ему приписана структура тетраэпоксида (Ю).
263
Реакции со стероидными моноолефинами [2]. В результате
фотоокисления Д2-3-метилхолестена-5а (1) в присутствии гема-
топорфирина в качестве сенсибилизатора и последующего вос-
становления образующихся гидроперекисей раствором натрия
в метаноле получаются два аллильных спирта (2) и (3). Окис-
ление изомерного Д2-2-метилхолестена-5а в аналогичных уело-
н
(7) 30%
виях дает три аллильных спирта (5) — (7). Следует отметить,
что предпочтительно образуются а-перекиси. Полученные ре-
зультаты лучше всего объясняются, если принять, что переход-
ное состояние реакции ближе к исходному олефину, чем к ал-
лильной гидроперекиси.
1. Foote С. S.t Mazur S., Burns Р. A., Lerdal D., J. Am. Chem. Soc.,
95, 586 (1973).
2. Nickon A., DiGiorgio J. B., Daniels P. J. L,, J. Org. Chem., 38,
533 (1973).
КОБАЛЬТА (II) АЦЕТАТ, Co(OAc)2 4H2O. Мол. вес 249,08.
Окисление алкилтолуолов [I]. Группа химиков компании
«Шелл Ойл» исследовала окисление алкилтолуолов кислородом
с участием в качестве катализатора К- а- В реакции происходит
постоянное превращение ионов Со(П) в ионы Со(Ш), промо-
тируемое метилэтилкетоном. Преимущественно окисляется ме-
тильная группа. Так, например, в результате окисления п-ци-
мола образуется 90% /г-изопропилбензойной и ~Ю% гс-ацетил-
бензойной кислоты. По относительной скорости окисления ал-
кильных групп их можно расположить в ряд: метильная >
264
> этильная > изопропильная > srop-бутильная. Предложен
механизм реакции, включающий одиоэлектрониый перенос с об-
разованием катион-радикалов.
Окисление н-бутана [2]. В присутствии кислорода Со(П)
переходит в Со(III), который и является истинным катализато-
ром окисления алканов кислородом. Например, в результате
окисления н-бутана, катализируемого ионами Со(III), при 100°
и давлении 17—24 атм образуется уксусная кислота (83,5%) и
следы м-масляной и пропионовой кислот и метилэтилкетона.
Окисление w-пентана в тех же условиях дает уксусную (48%)
и пропионовую (27%) кислоты. Изобутан относительно мало
активен, В основе механизма реакции лежит одноэлектронный
перенос, причем в качестве переносчиков цепи выступают ионы
кобальта.
1. Onopchenko A., Schulz J. G. D., Seekircher R., J. Org. Chem.,
37, 1414 (1972).
2. Onopchenko A., Schulz J. G. D., J. Org. Chem., 38, 909 (1973).
бис-( КОБАЛЬТТЕТРАКАРБОНИЛ) ЦИНК (V, 240—241;
VI, 137).
Диамантаи. Шлейер и сотр, [1] описали применение К. в ка-
честве катализатора димеризации, осуществляемой на первой
стадии синтеза диамантана из норборнадиена. Гидрирование
образующегося на этой стадии димера последнего — биснора-S
дает тетрагидрид (3), изомеризующийся в диамантан (4) под
действием бромистого алюминия в присутствии трнфторида бо-
ра как сокатализатора.
Норборнадиен (1)
Zn[Co(CO)4]; ,
'— -----: ?
80-85%
Биспор- S (Я)
Нг, РЮг/АсОН-НСу
90-97%
-----> синго
Тетрагидробиснор-S (3)
Диамантан (4)
1 G и п d Т. М., Thielecke
1971.
W., Schley er Р. v. R„ Org. Syn., submitted
265
КОРИ ВОССТАНОВИТЕЛЬ (1), н3с.
(1)
Мол. вес. 239, 15.
Получение [1]. Реагент получают из (+)-лимонена и тексил-
борана (V, 115—117; этот том); образующийся триалкилборан
действием трет-бутиллития в ТГФ превращают в анион боргид-
Стереоселективное восстановление кетонов [2]. Ключевым
промежуточным соединением в синтезе простагландинов по Ко-
ри является енон (I). Одна из главных трудностей этого син-
теза состоит в стереоселективном восстановлении карбонильной
группы этого соединения до искомого 155-спирта (Па). Восста-
новление различными боргидридами или триалкилборгидридами
(RiR2R3BH“Li+) дает приблизительно равные количества (Па)
и (Пб). Кори и сотр. [2] с помощью реагента (1) в ТГФ удалось
осуществить восстановление енона (I) (R == «-СбН5С6Н4МНСО)
в высшей степени стереоселективно и получить спирты (Па) и
(Пб) в отношении 92:8, причем их суммарный выход был ко-
личественным. Реакцию проводили сначала в течение 4 час при
—130°, а затем в течение 2 час при —115°. Почти такой же се-
лективностью обладает трис-етор-бутилборгидрид (этот том),
(I) (На) , (15 S) R1 = ОН; R2 = Н
(116), (15 R) Rt = Н; R2 = ОН
который восстанавливает уретан (I) (R = h-C6H5C6H4NHCO)
доспиртов (Па) и (Пб) в отношении 89; 11.
Для осуществления стереонаправленного восстановления кар-
бонильной группы енона(I) наряду с правильным выбором вос-
266
становителя важно также подобрать и подходящий заместитель
R. Одной из причин, затрудняющих селективное восстановление
енона(I), является то обстоятельство, что молекула енона мо-
жет принимать как s-цис, так н s-транс-конформацию. Для осу-
ществления атаки гидрида в желательном направлении с тыль-
ной стороны [по оси а соединения(III)] необходимо, чтобы
енон(1) принял s-фщ-конформацию, как показано на рисунке.
••н
О—к
(Ш)
Рассмотрение молекулярных моделей показало, что конформа-
ция енона сильно зависит от природы заместителя R. Эта груп-
па должна быть достаточно объемистой, чтобы затруднить под-
ход реагента в нежелательном направлении (по оси б). Как
показало испытание большого числа группировок R, наибо-
лее эффективный стереохимический контроль обеспечивает
n-бифенилилкарбамоильная группировка. Енон (I) (R = п-
С0НзСбН4МНСО) получают из спирта (I) (R — Н) н п-бифени-
лилизоцианата (n-CeHsCgERN — С — О, т. пл. 58—58,5° [3]) в
ТГФ в присутствии 1,2 же триэтиламина при 25° в течение
3 час (выход >90%). Защитную группу удаляют с выходом
>90% гидролизом уретана 1 А4 водным раствором гидроокиси
лития при 120° в течение 72 час. Полученную щелочную реак-
ционную смесь экстрагируют при 0° смесью эфира с этилацета-
том для удаления нейтральных и основных компонентов, а за-
тем проводят релактонизацию, добавляя 2 же этилового эфира
хлоруголыюй кислоты к предварительно нейтрализованному
двуокисью углерода водному слою.
1. Corey Е. J., Albonico S. М., Koelliker U., Schaaf Т. К., Var-
ma R. К., J. Am. Chem. Soc., 93, 1491 (1971).
2. Corey Е. J., Becker К- Varma R. К., J. Am. Chem. Soc., 94, 8616
(1972).
3. v a n Gelderen M. J., Rec. trav., 52, 969 (1933).
ЛИНДЛАРА КАТАЛИЗАТОР (II, 136—i37; VI, 138).
Восстановление. Гидрирование р-дамасценона (1) над ката-
лизатором Линдлара дает с почти количественным выходом
(3-дамаскон (2). Гидрирование над палладием на угле приво-
дит к образованию смеси продуктов [1].
1. Buehl G., Wuest Н., Helv. Chim. Acta, 54, 1767 (1971).
ЛИТИЙ (11,137—141; VI, 138—139).
Л. для реакции готовят следующим образом. Отвешивают
необходимое количество металла, промывают его петролейным
эфиром (т. кип. 40—60°) и нарезают литиевую проволоку нож-
ницами непосредственно над горлом реакционного сосуда. Каж-
дые 2,5 см проволоки разрезают на 5 частей весом в среднем по
0,3 г (поскольку для этих реакций необходим избыток Л., то
в точном взвешивании нет необходимости).
Одностадийный синтез 2-метил-3-бутилгептен-1-ола-3, эквива-
лентный синтезу Гриньяра [1].
О C4Hg-fi
I! Li j
2н-С4НаВг + СН2—С—С—ОСНз -------> СН2=С—С—OLi + CH3OLi
I I I
CH3 CH3 C«H9-H
^разб. HC1
С4На-н
I
CH2-C—C—OH + LiCl
I 1
CH3 C4H9-H
В четырехгорлую колбу объемом 2 л, снабженную мешал-
кой с затвором, термометром, трубкой для ввода газа и капель-
ной воронкой с обводной трубкой, в атмосфере азота помещают
9КЯ
1200 мл сухого тетрагидрофурана и 50 г (7,1 моля) нарезан-
ного, как описано выше, металлического Л. Смесь при переме-
шивании охлаждают до —20° н медленно прикапывают в тече-
ние 3—4 час смесь 100 г {1,0 моля) метилметакрилата и 411 г
(3,0 моля) н-бутилбромида. Постоянную температуру, необхо-
димую для управления этой чрезвычайно экзотермической ре-
акцией, поддерживают с помощью специального устройства. Оно
состоит из охлаждающей бани, соединенной с термостатом, уп-
равляемым контактным термометром, который погружен в реак-
ционную колбу и поддерживает температуру реакционной смеси
при —20°. По окончании прикапывания всей смеси реакцион-
ную массу продолжают перемешивать при той же температуре
еще 30 мин, в течение которых блестящая серебристая поверх-
ность оставшегося металлического Л, становится пятнисто-туск-
лой. Содержимое колбы затем фильтруют для удаления избыт-
ка Л., растворитель удаляют на роторном испарителе при пони-
женном давлении. Оставшийся алкоголят литня гидролизуют
разб. соляной кислотой, образовавшийся спирт экстрагируют
эфиром и высушивают сульфатом магния. Остаток после фильт-
рования и удаления эфира на роторном испарителе перегоняют
с коротким дефлегматором; при этом получают 147 г (80%)
2-метил-3-бутилгептен-1-ола-3, т. кип. 80°/1 мм. Чистота полу-
ченного спирта по данным ГЖХ анализа составляет >>99%.
Этот метод нашел широкое применение. Карбонильное сое-
динение может быть альдегидом, кетоном или сложным эфиром,
а галогеналкил — хлоридом, бромидом или иодидом, хотя в слу-
чае иодидов выходы обычно ниже. Галогеналкилы и галоген-
арилы реагируют с одинаковой скоростью. Можно использовать
как первичные, так и вторичные или третичные галогеналкилы.
Ниже приведены выходы спиртов, определенные методом
ГЖХ, для реакций с участием 20%-ного избытка галогенал-
кила. Преимущества рассмотренного метода перед стандартной
реакцией Гриньяра сводятся к следующему: 1) это одностадий-
ный процесс; 2) обычно он дает более высокие выходы; 3) обра-
ботка реакционной смеси чище и удобнее.
Карбонильное соединение Г алогеналкил Продукт Выход, %
Пропионовый альдегид Бромистый ЭТИЛ Пентанол-3 90
Бензальдегид Хлорбензол Бензгидрол 100
Ди-н-бутилкетон Бромистый н-бу- тил Три-н-бутил-кар- бинол 91
Этилформиат Бромистый /^-бу- тил Нонанол-5 91
Акролеин Бромистый этил Пентен-1-ол-З 90
Масляный альдегид Бромистый втор- бутил 3-Метилгепта нол-4 89
269
^uc-Стильбен [2]. Дифенилацетилсн восстанавливается с ко-
личественным выходом до рнс-стнльбеиа под действием Л. в
ТГФ при —78° (1—2 час) с последующим протонированием ме-
танолом при этой же температуре. Метод можно использовать
для получения СбН5СО = СОСбН5.
Восстановление натрием в тех же условиях приводит к обра-
зованию смеси продуктов, включая трянс-стильбен, но не дает
рпс-изомера.
1. Р е а г с е Р. J., Richards D. Н., Scilly N. F., Chem. Comm., 1970,
1160; англ. пат. заявка No. 61956 (1969), J. С. S. Perkin I, 1972, 1655;
Pearce Р. J., Richards D. Н., Scilly N. F., Org. Syn., 52, 19 (1972).
2. Levin G., Jagur-Grodzinski J., Szwarc M., J. Org. Chem., 35,
1702 (1970).
ЛИТИЙ — АЛКИЛАМИНЫ (II, 141 — 150; IV, 141; V, 242—
243; VI, 139).
Восстановление карбоновых кислот (VI, 139). Беденбаух и
сотр, предложили для Org. Syn. [1] подробную методику восста-
новления декановой кислоты (1) до деканаля (3) и N-метилде-
циламина (4):
Li/cH3NH2
СН3(СН2)8СООН --------->
(1)
н3о+
—> [CH3(CH2)8CH=NCH3] „ > СНз(СН2)8СНО
(2) (3)
68% | H2, Pd/C
CH3(CH2)8CH2NHCH3
(41
Зр-Аминостероиды [2]. Самым лучшим методом получения
Зр-аминостероидов служит восстановление оксимов 3-кетосте-
роидов литием в этиламине.
Ьтрет-Бутилциклогексен [3]. Наиболее удобным методом по-
лучения 1-трет-бутилциклогексена является восстановление
трет-бутилбензола литием, растворенным в смеси этилендиами-
на и морфолина; при этом образуется смесь, содержащая 84%
желаемого продукта и 16% трет-бутил циклогексана. Компонен-
ты смеси легко разделяются колоночной хроматографией на си-
ликагеле. Присутствие морфолина обеспечивает более селектив-
ное восстановление, страхуя от образования 3- и 4-трет-бутил-
циклогексенов.
?Нз <гнз
Н 3 С — С С / у Н3 С — С — сн3
1) Li/H3NCH2CH2NH2, HN р
2) Н2О [
270
1. Bedenbaugh A, О., Bergin W. A., Be den ba ugh J. H., A d-
kins J. D., Org. Syn., submitted 1972.
2. Khuong-Huu F„ Tassel M., Bull. soc. chim. France, 1971, 4072.
3 Benkeser R. A., Agnihotri R. K., Hoogeboom T. J., Synthesis,
1971, 147.
ЛИТИЙ-АММИАК (11, 151 —153; V, 243; VI, 139—143).
Восстановительное раскрытие циклопропановых колец. Вос-
становительное расщепление циклопропанов можно осущест-
вить раствором щелочного металла в жидком аммиаке в том
случае, если циклопропановое кольцо сопряжено с карбониль-
ной (пример I) [1], карбалкоксильной (пример II) [2] или фе-
нильной (пример III) [3] группами. Приведенные примеры не
дают однозначного ответа на вопрос, какими факторами, сте-
реоэлектронными или стерическими, определяется направление
раскрытия циклопропанового кольца.
н соосн,
и)
Li - NH3
н
I
CH2CHZOH
Г лавный
продукт
СН2СНгОН
Побочн ый
продукт
III)
Na-NHj
9 0$
С^Нг. /СН,
СНСНгСНгСНд 4- снсн
Главный Побочны и
продукт продукт
271
96*
Вальборский и сотр. [Зв], исследовавшие целый ряд восста-
новительных систем, нашли, что самый высокий процент пре-
вращения наблюдается в случае Л. — а. (—78°) и в случае си-
стемы натрий — нафталин (II, 357—359; V; 316; этот том) в мо-
ноглиме (1,2-диметоксиэтан) прн 25°. Электролиз в ацетонит-
риле (25°) оказался также очень эффективным.
Пакьетт и Фур [4] использовали эту реакцию расщепления
для синтеза напряженных трициклических систем. Обработка
эпоксида (2), полученного окислением трициклогептена (I)
^-хлорнадбензойной кислотой, 2 же лития в жидком аммиаке
приводит к образованию спирта (3) с выходом 90%. Аналогич-
ным путем эпоксид (4) превращают в спирт (5) с выходом 95%.
В обоих случаях, как в соединении (2), так и в (4), преимуще-
ственно разрывается циклопропановая связь, несущая два фе-
нильных заместителя; и при этом генерируется радикал-анион,
атакующий затем ближайшую С—О-связь с образованием но-
вого циклопропанового кольца. Циклопропановые кольца в со-
единениях (3) и (5), по-видимому, защищены от дальнейшего
восстановления электростатическими факторами.
Восстановление ароматических кетонов [5]. До сих пор счи-
талось, что восстановление ароматических кетонов металлами
272
в жидком аммиаке идет до спиртов, по аналогии с тем, что бен-
зофенон (I) натрием в жидком аммиаке восстанавливается до
дифенилкарбинола (2) (бензгидрола). Однако Холл и сотруд-
ники показали, что действием Л. — а. с последующим разложе-
нием хлористым аммонием ароматические кетоны почти количе-
ственно восстанавливаются до углеводородов. Восстановление
катализируется следами таких металлов, как кобальт и алюми-
ний. В этих условиях а-тетралон (3) восстанавливается до те-
тралина (4) с выходом 95%.
Другие примеры (выходы даны на выделенные соединения):
1. Dau ben W. G„ Deviny E. 1., J. Org. Chem., 31, 3794 (1966); Dau-
ben W. G., Wolf R. E., J. Org. Chem., 35, 374 (1970).
2. Hou se H. О., В 1 a nkl ey C. J., J. Org. Chem., 33, 47 (1968).
3. a) Walborsky H. M., Pierce J. B., J. Org. Chem., 33, 4102 (1968);
6) Staley S. W., Rocchio J. J., J. Am. Chem. Soc., 91, 1565 (1969);
в) Walborsky H. M., A r n о И M. S., Sc h u 1 ma n M. F., J. Org. Chem.
36, 1036 (1971).
4. Paquette L. A., Fuhr К. H., J. Am. Chem. Soc., 94, 9221 (1972),
5- H a 1 1 S. S., Lipsky S. D., McEnroe F. J., Bartels .A. P„ J. Org
Chem., 36, 2588 (1971).
273
ЛИТИЙ — трег-БУТА НОЛ — ТЕТРАГИДРОФУРАН
(И, 153—155).
Восстановительное дехлорирование [1]. При нагревании 0,084
моля соединения (1) с 0,87 г-атом лития и 20 мл трет-бут анол а
в 140 мл ТГФ с выходом 72% образуется эндо-2-фенил-7,7-ди-
фторнорборнен-5 (2). В случае 7,7-дифторнорборнена-2 выход
невысок, вероятно, из-за трудности выделения этого очень лету-
чего соединения. Метод неприменим для получения 7,7-дифтор-
норборнадиена, так как при этом восстанавливается также и
одна из двойных связей.
1. Jef ford С. W., Broeckx W., Helv. Chim Acta, 54, 1479 (1971).
ЛИТИЙДИМЕТИЛ КАРБАМОИЛ НИКЕЛЬТРИ КАРБОНИЛ
(1). Мол, вес 221,75, чувствителен к воздуху.
Реагент получают следующим образом. Эфирный раствор
диметиламина обрабатывают при 0° н-бутиллитием в «-гексане.
Затем к суспензии образовавшегося диметиламида лития в эфи-
ре при температуре ниже 10° прибавляют эфирный раствор кар-
бонила никеля и смесь перемешивают 1 час.
ы°.
L>iN(CHj)2 + С-Ы1(СО)з
(CH3);N<'
(1)
Реагент взаимодействует с органическими галогенидами илн
хлорангидрндами кислот, образуя обычно с хорошим выходом
соответствующие карбоновые или а-кетокарбоновые кислоты[1].
Алкилиодиды и трет-бутилбромид не вступают в эту реакцию.
Ari и алкенилгалогениды (RCH = CHX), однако, реагируют
легко.
RX(RCOX) + (1) —> RCON(CH3)2
Бензофенон при взаимодействии с Л. (1) в ТГФ дает после
гидролиза М,М-днметиламид ct-фенилминдальной кислоты;
1) ТГФ, 67° ОН
2) НзО+ |
C6H5CCeHs + (l) ----> C6HsCCON(CH3)2
6 сан5
274
Бензальдегид образует с Л. (1) М,М-диметилбензамид (вы-
ход 84,2%).
1. Fukuoka S„ R у а п g М., Tsutsumi S., J. Org. Chem., 36, 2721 (1971).
ЛИТИЙМЕТИЛИЗОНИТРИЛ, LiCH2N=C. Мол. вес 46,98.
Реагент получают реакцией метилизонитрила (0,1 моля) с
«-бутиллитием (0,1 моля) в ТГФ при—60° в атмосфере азота[1].
Гомологизация циклических кетонов [1].При взаимодействии
с циклогексаноном (1) Л. дает с выходом 77% 1-(нзонитрилмс-
тил) -1-циклогексанол (2), который под действием метанольного
о
| LiCHzN=C; Н*
77%
(1)
ОН
^J^CH2N=C
СН,ОН, НС1
—i>
>63%
он
^Цснгнн2‘ НС1
(2) (3)
HNOj
-N,, -Н
--------->
71; 5%
(4)
раствора хлористого водорода превращается в хлоргидрат ами-
на (3). Соединение (3) вступает в реакцию расширения цикла
по Тиффено — Демьянову [2], образуя с хорошим выходом ци-
клогептанон (4).
1. Schollkopf U., Bohme Р., Angew. Chem., Internal. Ed., 10, 490
(1971).
2. Смит П, А., Боер Д. Р., Органические реакции, «Мир», М., 1965, сб. 11,
стр. 167.
ЛИТИЙ — НАФТАЛИН (V, 244—245; VI, 143).
Восстановительное диалкилирование. При действии 2 же
Л.—н. иа соединение (1) при —78° в ТГФ образуется димети-
ловый эфир циклооктатриен-1,4,6-дикарбоиовой-1,2 кислоты (2)
с выходом 60%[1]:
MsOCHj
Д. COOCHj
(1
X^/^XCOOCHj
MsOCHj
[C!0H3jX Li+
----->
60%
CL)
a COOCHj
COOCHj
(г)
275
Дианион диизопрена [2]. При добавлении изопрена к раство-
ру Л. — н. в ТГФ получается дианион диизопрена (1). Добав-
ление в качестве сорастворителя ГМТФК, а затем ацетона при-
водит к образованию смеси одноатомных спиртов (2) и (3). Ис-
Н, СН]
2 Li + - V 1 I CHjCOCH,
2 Н,С=С-СИ = СН2 —---> Li+[H2C-C=CHCHJ-CHI-C ~ C-CHilLi ---------- —
(О
нс I 1 I 1 1 X I 1
’ х'С=СН-СНг-СН2 — С=СН-СНгС-ОН + C-CH —СН1-СН1-СН = С-СН1-С-ОН
,. _ > I u.r X J...
пользование ГМТФК имеет существенное значение для успеш-
ного проведения реакции.
Реакция «.^-ненасыщенных кислот с кетонами [3]. Л.—н, в
присутствии диэтиламина реагирует с «^-ненасыщенными кис-
лотами, например с кротоновой кислотой (1), образуя «-анион
кротоната лития. Этот анион взаимодействует с кетонами, на-
пример циклогексаноном (2), давая 6-оксикислоты, например
6-(1'-оксициклогекснл-1')-кротоновую кислоту (3).
[ СюНв] Li
О + СНЭСН=СНСООН ...
ТГФ , -20°
СНгСН = СНСООН
Реакция с тетрагидрофураном. Л.— н. (1) реагирует при 65°
с тетрагидрофураиом, образуя спирты (2) и (3) [4].
снгснгснгснаон
(2) 92#
,снгснгснгснгон
(3) 8$
Реакция с алкил- и арилгалогеиидами [5]. Алкил- и арилга-
логениды превращают в литнйорганические реагенты взаимо-
действием с Л, — н.:
RX + 2C10H~Li+
RLi + LiX + 2С10Н3
276
Как было показано методом карбонизации, выходы колеблются
в пределах от 19 до 90%. Нафталин отделяют от продуктов ре-
акции возгонкой или перегонкой с паром.
1, Whitlock Н. W., Jr., Schatz Р. F., J. Am. Chem. Soc., 93, 3837 (1971).
2, Suga К., Watanabe S., Synthesis, 1971, 91.
i Watanabe S., Suga K., Fujita T., F u j i у о s h i K-, Chem, Ind., 1972,
80; Suga K., Watanabe S., Fujita T., Australian J. Chem., 25, 2393
(1972).
4, F u j i t a T, Suga K., Watanabe S., Synthesis, 1972 , 630.
5. S с г e 11 a s C. G., Chem. Comm., 1972, 752.
2-ЛИТИЙПРОПИЛИДЕНЧЧ-трет’БУТИЛИМИН,
CH3
LiCHCH=NC(CH3)3. Мол. вес 107,12.
Получают из пропилиден-трет-бутилимина [1].
Направленная альдольная конденсация (V, 252). Бюхи и
Бюст (2) использовали метод альдольной конденсации по Вит-
титу для осуществления чрезвычайно стереоселективного син-
теза рацемического сесквитерпенового альдегида иуцифераля
(3) реакцией альдегида (1) с Л. (2). Желаемый продукт был
получен с выходом 83%.
Реагент использовали также [3] для синтеза из альдегида (4)
0-синенсаля (5), одного из компонентов, ответственных за за-
пах китайского апельсина:
1. TioIIais R., Bull. soc. chim. France, 14, 708 (1947).
2. Buchi G., Wuest H„ J. Org. Chem., 34, 1122 (1969).
3. Buchi G., Wile st H., Helv, Chim. Acta, 50, 2440 (1967).
277
2-Л ИТИЙ-2-ТРИМЕТИЛСИЛ ИЛ-1,3-ДИТИАН (V, 160).
s s Мол. вес. 198,35.
Получение [1]. Реагент получают с почти количественным
выходом взаимодействием 2-литий-1,3-дитиана (V, 160—161) со
свежеперегнанным триметилхлорсиланом в ТГФ прн —55°.
Смесь выдерживают при этой температуре 20 мин, а затем 5 час
при 20°. При обработке водой Л. превращается в 2-триметилси-
лил-1,3-днтиан.
Тиоацетали кетенов. В двух лабораториях [2, 3] одновре-
менно был разработан удобный общий метод синтеза тиоацета-
лей кетенов (2) реакцией Л. (1) с альдегидами или кетонами
>Ri
(I) + О-С
(2)
в ТГФ при низких температурах. Выходы тиоацеталей в этой
реакции обычно высокие (50—90 %) Тиоацетали кетенов —пер-
спективные полупродукты для синтеза. Так, при гидролизе они
гидролиз
(2) ---------->
R1\
^снсоон
к/
(3)
дают карбоновые кислоты (3) [4]. Протонирование их с после-
дующим восстановлением гидридами приводит к образованию
тиоацеталей (4), которые при гидролизе с участием NBC в вод-
ном ацетонитриле дают альдегиды (5) [3]:
(2) RjRiCH—А \ RjRzCH-X \ R^CHCHO
СНгО1а \__/ S---/
<41 (5)
Тиоацетали винилкетенов [5]. С а,р-ненасыщенными альде-
гидами н кетонами Л. (1) реагируют исключительно по карбо-
нильной группе с образованием тиоацеталей винилкетенов (2).
Последние вступают в реакцию циклоприсоединения по Диль-
су — Альдеру; эту реакцию Кари и Курт использовали для син-
278
теза циклогексенонов. Так, тиоацеталь винил кетен а (3) при ки-
пячении с малеиновым ангидридом в ксилоле (3 час) образует
(I) + в.
+ (CH3)3SiOLl
аддукт (4) с выходом 60%. Гидролиз тиоацетальной группы
проводят кипячением в 90%-ном водном метаноле в присутст-
вии хлорной ртути. Первоначально образующаяся р-кетокисло-
та (5) в этих условиях декарбоксилируется, давая (6) с примесью
метилового эфира (7). При этерификации полученного сырого
продукта (6) получается эфир циклогексенонкарбоновой кисло-
ты (7) в виде смеси эпимеров; суммарный выход (7) из (4) со-
ставляет 58%.
К сожалению, реакция Днльса — Альдера с тиоацеталями
винилкетенов лимитируется использованием лишь реакционно-
способных диенофилов: например, тиоацеталь (3) не реагирует
ни с диэтиловым эфиром малеиновой кислоты, ни с дифеннлаце-
тиленом. Было показано, что тиоацетали некоторых винилкете-
нов не реагируют и с малеиновым ангидридом.
Тиоацетали винилкетенов при температурах от —80 до —20°
в ТГФ реагируют с алкиллитием по типу присоединения по Ми-
хаэлю. Например последовательная обработка соединения (8)
н-бутиллнтием и нодистым метилом дает с почти количествен-
ным выходом тиоацеталь а,р-венасыщениого метилкетона (9).
279
Гидролиз тиоацеталя в метаноле в присутствии хлорной ртути
и окиси ртути
1) «-BuLi
г) снэ1
—---------->
приводит к образованию свободного карбонильного соединения,
например (10)-*( 11). Йодистый метил можно заменить другими
электрофилами [6].
(10)
1) H-BuLl
2) СН31
3)гидролиз
------------>
(П)
1. Seebach D., Synthesis, 1969, 17.
2. Seebach D., Grobel B.-T., Beck A. K., Braun M., Geiss K.-H.,
Angew. Chem., Internal. Ed., 11, 443 (1972).
3. С a г e у F. A., С 0 u r t A. S., J. Org. Chem., 37, 1926 (1972).
4. Marshall J. A., Bell etire J. L., Tetrahedron Letters, 871 (1971).
5. С а г e у F, А., С 0 u г t A. S., J. Org. Chem., 37, 4474 (1972).
6. Seebach D., Kolb M., Grobel B.-T., Angew. Chem., Internat. Ed., 12,
69 (1973).
ЛИТИЙ — ЭТИЛЕНДИАМИН (II, 158—159; V, 245—246;
VI, 144-145).
Восстановительное расщепление оксетанов [1]. Оксетаны,
подобно эпоксидам (VI, 144—145), легко восстанавливаются
литием в этилендиамине (ЭДА). При восстановлении оксетана
(1) образуется смесь двух спиртов (2) и (3) в соотношении
3:1. Применимость этого метода восстановления ограничена
из-за отсутствия селективности в направлении расщепления не-
симметричных оксетанов.
1. Sauers R. R., Schinski W., Mason M. M., O’Hara Е., Byrne В.
J. Org. Chem., 38, 642 (1973).
ЛИТИЯ АЛЮМОГИДРИД (II, 163—179; V, 246—247- VI,
145—146).
Внутримолекулярная циклизация [1]. Восстановление солей
2-(3-аминопропил)-изохинолиния (1) (п = 2) Л. а. в эфире при-
280
водит к образованию нового гетероцикла (3) (а — 2) с выходом
53%. Аналогичная восстановительная циклизация соли (1) (п —
= I) дает гетероцикл (3) (п — 1) с выходом 83%. Реакция
протекает через 1,2-дигидроизохинолины (2) (п — 1 или 2).
При восстановлении бромида М-(3-бромпропил)-изохиноли-
ния (4) трис- (трет-бутокси)-алюмогидридом лития (II, 187—
193; V, 250—251; VI, 149) образуется 1,2-дигидроизохинолин
(5), который прн обработке этанольным раствором метиламина
дает соединение (6), являющееся N-метильным аналогом сое-
динения (3) (п = 2).
(6)
Восстановительное дегалогенирование галогеналкилов [2].
Л. а. обычно используют для восстановительного дегалогениро-
вания только реакционноспособных субстратов. Для восстанов-
ления инертных галогенидов используют оловоорганические гид-
риды, например три-н-бутилстаннан (III, 372—373; V, 431—432;
VI, 263—264). Недавно Джеффорд и сотрудники сообщили, что
считавшиеся до сих пор инертными галогеналкилы можно вос-
становить Л. а. Так, винилгалогеиид (1) при кипячении с Л. а.
в эфире (24 час) восстанавливается до зндо-2-фенилбицикло-
[3,2,1]-октена-3 (2). 3-Бромбицикло-[3,2,1]-октен-2 при нагрева-
нии с Л. а. в диметоксиэтане (ДМЭ) в течение 100 час восста-
навливается до соответствующего углеводорода, бицикло-[3,2,1]-
281
октепа-2, с выходом 80% [3].
С6Н5
(1)
LiAlH*
С6Н5
(2)
Аналогичная обработка 2-бром-2-метилстирола приводит к об-
разованию 2-метнлстирола с выходом 81 %.
Галогениды с атомом галогена в голове моста также могут
быть восстановлены LiA!H4. Например, 1-бром адамантан при
кипячении с LiA!H4 в эфире в течение 18 час восстанавливается
до адамантана с выходом 80%. Даже 1-бромтриптицен при дей-
ствии LiA!H4 в ДМЭ в течение 48 час восстанавливается до со-
ответствующего углеводорода.
С помощью LiAlH4 можно восстановить циклопропилгалоге-
ниды [3]. Например, 7,7-дибромбицикло-[4,1,0]-гептан (3) при
действии LiAlH4 в эфире в течение 18 час дает наряду с соот-
ветствующим углеводородом (6) анти- н сдн-монобромпроизвод-
ные (4) и (5) в отношении 6,8 : 23,2 : 70,0.
И действительно, было показано, что геминальные атомы
галогена восстанавливаются даже легче, чем атомы галогена,
расположенные в голове моста.
Стереоселективное восстановление. Л. а. в эфире восстанав-
ливает кетон бицикло-[4,3,1]-декатриен-2,4,8-он-7 (1) до прост-
ранственно менее затрудненного спирта эндо-7-оксибицикло-
[4,3,1]-декатриена-2,4,8 (2) [4].
(1)
(2)
3,3-Дизамещенные ЗН-индолы [5]. 3,3-Дизамещенные ЗН-ин-
долы (3) можно легко получить восстановлением 3,3-дизаме-
282
щенных оксиндолов (1) Л. а. или бис-(2-метоксиэтокси)-алтомо-
гидридом натрия (VI, 189—190; этот том) с последующим окис-
лением индолина (2) активированной двуокисью марганца или
перманганатом калия в ацетоне.
1. F i л с h N., G е m е n d е п С. W., J. Org. Chem., 38, 437 (1973).
2. Jefford С. W., Sweeney A., Delay F., Helv. Chim. Acta, 55, 2214
(1972).
3. J e f f о г d C. W., Kirkpatrick D., Delay F., J. Am. Chem. Soc., 94,
8905 (1972).
4. Schroder G., Prange U., Putze B., Thio T., Oth J. F. M., Chem.
Ber., 104, 3406 (1971).
5. Takayama K-, I so be M,, Har ano K-, Tag cichi T., Tetrahedron
Letters, 365 (1973).
ЛИТИЯ АЛЮМОГИДРИД — АЛЮМИНИЯ ХЛОРИД
(алан, AIH3) (II, 179—183; V, 249—250; VI, 146).
Восстановление оксетанов [1]. Оксетаны, подобно эпоксидам
(И, 182), восстанавливаются Л. а. — а.х. (соотношение 3:1) в
кипящем эфире. Например, оксетан (1) восстанавливается с об-
разованием смеси спиртов (2) и (3) в отношении 4:1. Спирт
(2), по-видимому, образуется через альдегид (а).
(3)
Циклобутеиы из циклопропенов [2]. Этиловый эфир 1,2-ди-
пропилциклопропен-1-карбоновой-З кислоты (1), полученный
действием диазоуксусного эфира в эфире на октин-4, при обра-
ботке Л. а.—а.х, (1:1) в эфире при —80° дает 1,2-дипропил-
283
циклобутен (2) с выходом 80%:
соосгн5
сн
с3н7с=сс3н.
Ъ1А1Н4/а1С13 ?
80%
(1)
нгс~сн2
с3н7с=сс3н7
(г)
Восстановление 6£-метокси-3а,5-цикло-5а-стероидов [3]. Вос-
становление 6[3-метокси-3а,5-циклохолестана (1) смешанным
гидридом приводит к образованию главным образом Зсс,5-ци-
кло-5а-холестана (2).
I Sauers R. A, Schinski W, Mason М. М, O’Hara Е, Byrne В.,
J. Org. Chem, 38, 642 (1973).
2. G е n s 1 ег W. J, L a n go n e J. J, Floyd M. B, J. Am. Chem. Soc, 93,
3828 (1971).
3. Romeo A, Paradis! M. P, J. Org. Chem, 37, 46 (1972).
ЛИТИЯ АЛЮМОГИДРИД —НАТРИЯ МЕТИЛАТ.
Восстановление пропаргиловых спиртов до аллиловых. Кори
и сотр. [1] сообщили в 1967 г, что восстановление пропаргило-
вого спирта (1) 0,1 М Л. а.— н.м. (молярное соотношение 1:2)
при кипячении (3 час} в ТГФ с последующей обработкой иодом
приводит к образованию только у-иодзамещенного спирта (2).
Реакция спирта (2) с диметилмедьлитием приводит к образова-
нию транс,транс-фарнезола.
Предложенный Кори метод был использован им же в новом
стереоспецифическом синтезе З-этил-7-метил-транс, ^цс-нонадиен-
2,6-ола-1 (IV), представляющего собой важное промежуточное
соединение в синтезе Ci8-ювенильного гормона Cecropia [2].
Так, восстановление октадиин-2,6-ола-1 (1) большим избытком
Л. а. — н. м. при кипячении в диметоксиэтане приводит к про-
межуточному образованию алюминийорганического соедине-
ния (II), которое при обработке иодом дает 3,7-дииод-транс,транс-
октадиен-2,6-ол-1 (Ш). Взаимодействие последнего с 8 эка ди-
284
этилмедьлнтия приводит к желаемому спирту(1У),
(С2Н5 )гСи1Л
60%
(IV)
1. Corey Е. J„ Katzenellenbogen J. A., Posner G. H., J. Am.
Chem. Soc., 89, 4245 (1967).
2. С о г e у E. J., Katzenellenbogen J. A., Roman S. A., G11-
m a n N. W., Tetrahedron Letters, 1821 (1971).
ЛИТИЯ АЛЮМОГИДРИД-ПИРИДИН [л ития тетракис-
N-дигидропиридил) алюминат] (II, 183—185).
З-Замещенные пиридины [1]. При нагревании Л. а. — п. с ал-
кнлгалогенидами, бензилхлоридом, бромом или другими элек-
трофильными агентами образуются З-замещенные пиридины с
выходом 40—90%• Реакция, вероятно, заключается в алкили-
ровании 1,2-дигидропиридиловых группировок в Л. а. — п. с об-
разованием 2,5-дигидропиридина, который окисляется затем до
конечного продукта. Эта реакция представляет значительный
интерес в синтетическом плане, поскольку прямое взаимодейст-
вие пиридина с нуклеофильными агентами приводит к 2- и 4-за-
мещенным пиридинам.
1. G 1 a m С. S., A b b о 11 S. D., J. Am. Chem. Soc., 93, 1294 (1971).
ЛИТИЯ АМАЛЬГАМА (VI, 147—148).
Дегалогенирование [1]. Дегалогенирование 1,4-дибром-Д2-8-
^«огексалина (1) до цис-\,4,9,10-тетрагидронафталина (2) удоб-
но проводить действием 4 мол. же 0,5%-ной амальгамы лития
Вт
Вг
(0 (2)
285
в эфире. Этот метод лучше метода
кипящем метаноле.
с использованием цинка в
1. van Т е m е 1 е n Е. Е., Pappas В. С.
(1971).
Т„ J. Am, Chem. Soc., 93, 6111
ЛИТИЯ БОРГИДРИД (II, 186).
5а-Буфалии [1]. Восстановление
пиридине приводит к образованию
Фалина (2):
сцилларенона (1) Л. б. в
ранее неизвестного 5а-бу-
1. Stache U., Radscheit
R u s c h i g H., Ann., 750, 149
К., Fritsch W,
(1971).
(2)
Haede W, К о 111 Н„
ЛИТИЯ БРОМИД (II, 186, 187; V, 250; VI, 53, 119, 295).
Превращение тетратозилатов (1а) и (2а) в 1,1,6,6-тетрабром-
метилспиро-[3,3]-гептан (16) и 1,1,10,10-тетрабромметилтриспи-
ро-[3,1,1,3,1,1]-тридекан (26) осуществляют нагреванием с без-
водным Л. б. в ацетоне [1]:
Перегруппировка эпоксигруппы в карбонильную [2]. Л. б.
вызывает быструю перегруппировку эпоксидов в бензольном
растворе в альдегиды или кетоны. Соль нерастворима в бензоле,
но при добавлении 1 моля ГМТФК или окиси три-м-бутилфос-
фина на каждый моль Л. б. происходит образование раствори-
мого комплекса, который и вызывает перегруппировку эпокси-
дов. Предполагают, что механизм реакции включает промежу-
точное образование алкоголята бромгидрина.
286
Примеры:
70%
сосн3
30%
1. Buchtа Е, Merk W., Ann., 716, 106 (1968).
2 Rickborn B., Gerkin R. M., J. Am. Chem. Soc., 90, 4193 (1968); 93,
’ 1693 (1971).
ЛИТИЯ-трет-БУТИЛАТ (VI, 149).
трет-Бутиловые эфиры кислот. Недавно опубликован метод
получения трет-бутилового эфира п-толуиловой кислоты [I].
1. Growther G. Р., Kaiser Е. М., Woodruff R. A., Hauser С. R.,
Org. Syn., 51, 96 (1971).
ЛИТИЯ трис-(втор-БУТИЛ)-БОРГИДРИД, 1л-втор-Ви3ВН.
Мол. вес. 190,10.
Этот восстановитель получают с количественным выходом ре-
акцией триметоксналюмогидрида лития (II, 203—204; V, 257)
с пространственно сильно затрудненным три-втор-бутилбораном
в ТГФ при 25°. Прн этом образуется также метнлат алюминия,
но вследствие его инертности можно использовать непосредст-
венно реакционную смесь [1].
ТГФ
ЫА1Н(ОМе)3 + втор-Ви3В ->L!-erop-BusBH + А1(ОМе)3
Этот новый реагент, являющийся активным восстановителем,
восстанавливает циклические и бициклические кетоны в выс-
шей степени стереоселективно [1]. Так, восстановление 2-метил-
циклогексанона (1) дает цис-2-метилциклогексанол (2) 99,3%-
ной чистоты. Следует отметить, что прн восстановлении кетона
(1) одним триметоксиалюмогидридом лития спирт (2) образует-
ся с выходом 69%. Таким образом, увеличение объема алкиль-
287
ных заместителей у атома бора приводит к увеличению стерео-
селективности действия аннона боргидрида. Даже простые про-
странственно незатрудненные циклогексаноны с алкильными за-
местителями в положении 3 или 4 восстанавливаются преимуще-
ственно с образованием новой экваториальной связи. Например,
4-трет-бутилциклогексанон (3) постанавливается при —78° до
(1) (2) 99,33
zjwc-4-тр^т-бутилциклогексанола (4) 96,5%-ной чистоты. При вос-
становлении соединения (3) пергидро-9Ь-борафеналил гидридом
лития (VI, 152—153) образуется всего 54% спирта (4). Таким
образом, Л. б. является, по-вндимому, самым стереоселективным
нз всех известных агентов восстановления циклических и бицик-
лических кетонов. Он имеет также то преимущество, что может
быть получен in situ и непосредственно использован для восста-
новления кетонов.
1. Brown Н. С., К г I s h и a m u с t h у S., J. Am. Chem. Soc., 94, 7159 (1972),
ЛИТИЯ трис-(трет-БУТОКСИ)-АЛ ЮМОГИДРИД (II, 187—
193; V, 250—251; VI, 149).
Восстановление циклических кетонов [1]. Сравнительное
восстановление циклических кетонов различных типов с по-
мощью Л. б. показало, что несопряженные еноны менее реакци-
онноспособны, чем циклические насыщенные кетоны, но более
активны по сравнению с сопряженными енонами. Стерические
эффекты, очевидно, не играют важной роли, так как 3-кетоцик-
логексен (1) оказался менее реакционноспособным, чем р-изо-
(1) (2)
288
форон — 3,5,5-триметилциклогексен-3-оп-1 (2). Главным факто-
ром, влияющим на реакционную способность, является, по-ви-
димому, сопряжение. По сравнению с Л. б. алюмогидрид лития
менее селективен при восстановлении «^-ненасыщенных и на-
сыщенных шестичленных циклических кетонов [2].
Восстановительное раскрытие тетрагидрофуранового кольца
[3]. Л. б. в сочетании с триэтилбораном вызывает быстрое и
почти количественное раскрытие тетрагидрофуранового кольца:
+ LiAlH(O-mpem-Bu)3
(С2Н5)3В
—X
1. Haubenstock Н., Quezada Р., J. Org. Chem., 37, 4067 (1972).
2. Haubenstock H., J. Org. Chem., 37, 656 (1972).
3. Brown H. С., К r i s h n a m u r t h у S., Coleman R. A., J. Am. Chem.
Soc., 94, 1750 (1972).
ЛИТИЯ ГИДРИД (V, 252).
Д5 д5,7_сТероиды. Даубен и Фуллертон [1] выбрали следую-
щий путь превращения диацетата Д5-андростендиола-Зр, 17£ (1)
в диацетат Д5’7-андростадиендиола-Зр,17р (4). Исходное со-
единение превращают в 7-кетон (2) окислением комплексом хро-
мового ангидрида с пиридином в хлористом метилене (V, 540).
Кетон (2) затем переводят в тозилгидразон (3), восстановление
последнего Л. г. в кипящем толуоле дает диен (4). Общий его
выход (40%) сравним с выходом, полученным при использова-
нии NEC. Преимущество предложенного метода восстановления
в том, что он не приводит к образованию побочного Д4-6-днена.
10 Зчк.
289
1. Dau ben W. G„ Fullerton D. S„ J. Org. Chem., 36, 3277 (1971).
2. C a g 1 i о t i L., C a s s e 11 i P,, Chim. Ind. (Milan), 1963, 559.
ЛИТИЯ ДИИ30ПР0ПИЛАМИД (V, 252—253; VI, 149—
151).
а-Оксиметилнрование V“ и 6-лактонов [1]. Обработка рас-
твора транс-бнциклического лактона (1) в ТГФ небольшим из-
бытком Л.д. в том же растворителе при —78° приводит к обра-
зованию енолята лактона. Реакционной смеси дают нагреться
до —20° и пропускают через нее газообразный формальдегид
(Ns). Реакцию прекращают добавлением 10% НС1. Соединение
(2) выделяют с выходом >-95%. Его можно превратить в транс-
а-метилен-у-бутиролактон (3) мезилированнем с последующим
кипячением в пиридине. Суммарный выход лактона (3), считая
на исходный лактон (1), составляет около 80%.
По этой же методике из б-валеролактона (4) был получен
а-оксиметил-6-валеролактон (5) с выходом >95%:
(4) (5)
Литиевые еноляты несимметричных кетонов. Хауз и сотр.
[2] нашли, что менее замещенный из двух литиевых енолятов
несимметричного кетона лучше всего получать при кинетически
контролируемом депротонировании кетона под действием прост-
ранственно затрудненного основания (Л.д.). Например, при
взаимодействии декалона-1 с 1,03 же основания в 1,2-диметок-
сиэтане в течение 10 мин образуется преимущественно литие-
вый енолят (2), а алкилирование реакционной смеси большим
избытком йодистого метила дает главным образом моноалкилн-
рованный кетон (4). Таким методом, следовательно, несиммет-
ричные кетоны можно алкилировать преимущественно в а-поло-
290
жение, которое пространственно менее экранировано.
(4) 66$ (5) з$
а-Метил-у-бутиролактои [3]. у-Бутиролактон (1) действием
Л.д. или изопропилциклогексиламида литня (этот том) в ТГФ
при —78° превращают в енолят. Затем при той же температуре
прибавляют избыток нодистого метила, дают реакционной сме-
си нагреться до —30° и выдерживают ее при этой температуре
в течение 2—3 час. Если взять 1 же основания, то а-метил-у-
бутиролактон получается с выходом 36%, а при использовании
1) LiN(Pr - изо )г
2) СН31
(2)
1,2 же основания выход его возрастает до 56% • Однако при
введении в реакцию более 1 же основания параллельно проис-
ходит а,«-диметилирование. Это — первый пример прямого пре-
вращения у-бутиролактона в а-метил-у-бутиролактон.
Моноалкилироваиие а,р-ненасыщеииых кетонов. Сторк и
Бенэйм [4] разработали метод моноалкилирования «^-ненасы-
щенных кетонов. Например, 10-метил-Д]<9)-окталон-2 (1) сна-
чала превращают в N-циклогексилимин (2) конденсацией с ци-
291
клогексиламнном (катализатор TsOH, азеотропная перегонка с
толуолом). Затем имин 8 час кипятят с Л. д. (это основание дает
лучшие результаты, чем н-бутиллитий, гидрид натрия или литие-
вая соль гексаметилдисилазана) в ТГФ в атмосфере N2, после
этого вводят избыток йодистого метила и кипятят реакционную
смесь в течение 12 час. После гидролиза выделяют с выходом
около 90% 1,10-диметил-А1<9)-окталон-2(3) и возвращают около
10% исходного енона (1).
Синтез кетонов. Сторк и Мальдонадо [5] нашли новый метод
синтеза кетонов из альдегидов: RCHO->RCOR'. Альдегид сна-
чала превращают в циангидрин и защищают его гидроксиль-
ную группу реакцией с этилвиниловым эфиром. При этом обра-
зуется соединение (1), которое превращают в анион реакцией
с Л.д. в тщательно контролируемых условиях: взаимодействием
бутиллития с диизопропиламином в ТГФ получают основание,
а затем к нему при перемешивании по каплям добавляют рас-
твор соединения (1) в гексаметаполе. После этого по каплям
ОС2Н5 ОС2Н5
1 1
однсн3 о LiN(M3O.c3H7)2 ОСНСНз ин* о
I 2) R'X 1 2) ОН- II
RCHO —> RCH—CN ---------------------> RCR' ---------:> RCR'
I
CN
(1) (21 (3)
вводят галогеналкил R'X. Полученный продукт (2) можно очи-
стить перегонкой или ввести в следующую стадию прямо без
очистки. Кетон (3) получают гидролизом соединения (2), про-
водимым сначала в присутствии кислоты, а затем основания.
Такая последовательность реакций применима и к а.^-ненасы-
щенным альдегидам для получения а,|3-ненасыщенных кетонов.
С-Алкилироваиие циклических а-дикетонов [6]. В обычных
условиях алкилирование цикл огександиона-1,2 (1), который,
как показано ниже, существует преимущественно в енольной
форме, приводит к образованию только продуктов О-алкилиро-
вання. Кенде и Эйлерман обнаружили, что если дион (1) обра-
ботать 2 экв Л.д. в ТГФ при —78° в атмосфере N2 и выждать,
пока не израсходуется все исходное вещество, а затем при этой
же температуре добавить йодистый метил, то соединение (2)
можно получить с выходом 70%. Если же дион (1) обработать
основанием при —78°, а затем добавить иодистый метил при
комнатной температуре, то образуются продукты (2), (3) и (4)
1) L1N[CH(CH3)2]2
I) ch3i
—78°
70%
(2)
292
о
о
(3) (4)
в соотношении 6:3:1. Рассмотренный метод с успехом исполь-
зовали для алкилирования циклопентандиона-1,2.
р-Оксикислоты [7]. Литиевые соли а-литированных карбоно-
вых кислот можно получить реакцией карбоновых кислот сЛ.д.
(VI, 149). Эти соединения реагируют с альдегидами или кето-
нами, образуя с умеренным выходом р-оксикислоты (метод яв-
ляется вариантом классической реакции Реформатского). В ре-
акции можно использовать моно- и дизамещенные уксусные
С=О + Li—С—COOLi
Y
R\ *
НО—С—С—соон
кислоты. Однако реакция чувствительна к пространственным
затруднениям в молекулах реагентов, и поэтому некоторые кис-
лоты (например, дифеннлуксусная кислота) и кетоны (напри-
мер, камфора) не вступают в эту реакцию.
а-Алкилгндракриловые н а-алкилакриловые кислоты [8].
При взаимодействии карбоновых кислот типа RCH2COOH (1)
с 2 экв Л.д. в ТГФ—ГМТФК образуется дианион (2) [9], кото-
рый реагирует с формальдегидом (полученным из парафор-
мальдегида, IV, 65). Последующий кислотный гидролиз разб.
НС1 дает а-алкилгидракриловые кислоты (3) с высоким вы-
RCH2COOH
2 LiN[CH(CH31E]2
ТГФ—ГМТФК
RCHCOOLi
I
Li
1) нсно
2) Н3О*
80—93%
(I) (2)
ню*
—> НОСНЕСНСООН -------------> СН2=ССООН + Н2О
2 I 90—94% | 1 2
R R
(3) (4)
ходом. Полученные кислоты дегидратируются под действием
кислотных катализаторов (фосфорная кислота) при повышен-
ной температуре, образуя с высоким выходом а-алкилакрило-
вые кислоты (4).
1. Grieco Р. А., Н t г о i К- J. С. S. Chem. Comm., 1972, 1317.
2. Н о u s е Н. О., G а 11 М., О I in s t е a d Н. D., J. Org. Chem , 36, 2361
(1971).
293
3. Р о s n e r G. Н., Loomis G. L,, Chem. Comm., 1972, 892.
4. S fork G., В e nai m J., J. Am. Chem. Soc., 93, 5938 (1971).
5. Stork G., Maldonado L., J. Am. Chem. Soc., 93, 5286 (1971).
6. Kende A. S., Eilerman R. G., Tetrahedron Leiters, 697 (1973).
7. Moersch G. W., Burkett A. R, J. Org. Chem., 36, 1149 (1971).
8. Pfeffer P. E., Kinsel E., Sil her t L. S., J. Org. Chem., 37, 1256
(1972).
9. Pfeffer P. E„ Silber t L. S., J. Org. Chem., 35, 262 (1970); Pfef-
fer P. E., S i 1 b e r t L. S., C h I r i n к о J. M., J. Org. Chem., 37, 451 (1972).
ЛИТИЯ ДИФЕНИЛФОСФИД (ЛДФ), ЫР(С6Н5)2. Мол.
вес. 192,12.
Получение [1]. Хлордифенилфосфин прибавляют по каплям
к 4 г-атом литиевой проволоки в сухом ТГФ при перемешива-
нии и охлаждении. Реакция завершается через 2 час прн ком-
натной температуре. Растворы можно хранить в течение не-
скольких дней под аргоном.
Инверсия олефинов [2]. ЛДФ в ТГФ стереоспецифически
раскрывает эпоксидные циклы. Кватернизация сырого продукта
иодистым метилом дает бетаины, которые в мягких условиях
(25°) фрагментируются с образованием олефинов и окиси ме-
тилдифенилфосфина. Конечный олефин образуется с обращен-
ной относительно исходного эпоксида конфигурацией, что объ-
ясняется раскрытием оксиранового цикла по 5я2-механизму с
последующим fpc-элиминированием окиси фосфина. Так, напри-
мер, окись транс-стильбена (1) превращается в бетаин (2), ко-
торый при 25° дает ^нс-стнльбен (3) с общим выходом 95%.
О (CjHsJjPLi н Н СцНе Г.Н
ед,, °\ н 2) снд у х
У—\ с----C---C6HS . с=с ,
Н СНэР+(С4Н;)г о- g Z \
(1) (2) (3)
о
1!
(ед)грсн3
Превращение протекает стереоспецифично (>98%). Аналогич-
ным путем цис-стилъбен можно превратить в транс-стильбен.
Взаимопревращение цис- и транс-октенов-2 происходит с выхо-
дом 75% и в высшей степени стереоспецифично (>99,5%).
Рассмотренный метод рекомендуется для инверсии конфигу-
рации ациклических ди- и тризамещенных алкенов. Сильно
экранированные эпоксиды очень медленно реагируют с ЛДФ,
и поэтому алкены из них не удается получить с хорошими вы-
ходами. Наличие кетогруппы мешает реакции, поскольку тогда
реагент расходуется (2 экв) на образование енолята. Эпокси-
эфиры не удается дезоксигенировать с практически приемлемы-
ми выходами.
Новый метод оказался очень удобным для получения чистого
транс-циклооктена. Окись ^ас-циклооктена (4) превращают та-
ким путем в транс-циклооктен (7) >99,5 %-ной чистоты с вы-
294
ходом >90% [3]. Для обеспечения стереоселективности реакции
необходимо тщательно следить за соблюдением апротонности
среды. Аналогичным образом из соответствующих эпоксидов
были получены транс- 1-метилциклооктен, цнс.транс-циклоокта-
диен-1,4 и ^«с.трш/с-циклооктадненЛД
—---->
>90%
Этот метод, однако, не дал положительных результатов при
попытке превращения окиси цнклогептена (8) в транс-цикло-
гептен. В этом случае реакция дает борфторнд (тр ос-2’окси-
циклогептил) -метилдифеннлфосфония (9).
+
ЛДФ, н ,
CH3I, NaBF4
65%
СНЭ
(?)
(8)
1. V е d е j s Е., F u с h s Р. L., J. Am. Chem. Soc., 94, 822 (1972).
2. Vedejs E„ Fuchs P. L., J. Am. Chem. Soc., 93, 4070 (1971).
3. Vedejs E., S n о b 1 e K. A. J., Fuchs P. L., J. Org. Chem., 38, 1178
(1973).
ЛИТИЯ ДИЭТИЛАМИД (II, 193—195; V, 254).
Реакция с эпоксидами (II, 193—195; V, 254). Крэндолл н
Кроули [1] разработали подробную методику превращения оки-
си а-пинена (1) в пинокарвеол (2) под действием Л.д. в эфире:
1. Crandall J, К., Crawley L. С., Org. Syn., submitted 1972,
295
ЛИТИЯ N-ИЗОПРОП ИЛ ЦИКЛОГЕКСИЛ АМИД.
LiN
ХСН(СН3)2
Мол, вес 147,18.
Реагент получают из «-бутиллития и N-изопропилциклогек-
силамина.
Литиевые еноляты сложных эфиров [1]. Л. и. взаимодейст-
вует с разнообразными сложными эфирами в ТГФ при низких
температурах, давая соответствующие литиевые еноляты:
HCCOOR + LiN^
I СН(СН3)г
----> LiCCOOR + HNx
-78° I "сЩСН^
Применение различных других оснований не привело к успе-
ху. Уникальные свойства Л. и. можно отчасти отнести за счет
прекрасной растворимости этого соединения в ТГФ при низких
температурах.
Литиевые еноляты сложных эфиров при 20° в присутствии
ДМСО реагируют с большим числом органических галогенидов,
образуя с хорошим выходом эфиры а-алкилкарбоновых кислот:
LiCCOOR + R'X
дмсо-ТГФ
20°
50—95%
Rz—CCOOR + LiX
С бромом или иодом литиевые еноляты эфиров уже при—78°
образуют эфиры а-галогенкарбоновых кислот с высоким выхо-
дом. Этот метод лучше обычной методики кипячения сложного
| ТГФ, —78° J
LICCOOR + Вг2 (12) ——----> Br(I)CCOOR+ LiBr(I)
I 8g—95 % |
эфира с галогеном и треххлористым фосфором [2].
Еиоляты эфиров «^-ненасыщенных кислот [3]. Добавление
этилового эфира кротоновой кислоты к 0,50 М раствору Л. и. в
ТГФ, содержащему 20% (по объему) ГМТФК, при —-78° и по-
следующая обработка реакционной смеси разбавленной соляной
кислотой приводят к образованию эфира 0,у-ненасыщенной кис-
лоты (этиловый эфир бутен-3-овой кислоты) с выходом 87%,
1) Л. и.
2) Н3О+
снйсн=снсоос~н5 --------> СН. = CHCH,COOC2HS
в: %
29Н
причем 13% исходного эфира кротоновой кислоты остается не-
израсходованным. В отсутствие ГМТФК эфир |3,у-ненасыщенной
кислоты получается с гораздо меньшим выходом (23%). Приве-
денная методика дает в руки исследователя простой общий ме-
тод превращения сложных эфиров «^-ненасыщенных кислот в
соответствующие эфиры |3,у-ненасыщенных карбоновых кислот.
Эта реакция позволяет также легко и гладко получить эфир
алленкарбоновой кислоты — метиловый эфир бутадиен-2,3-овой
кислоты, исходя из метилового эфира тетроловой (метилпропио-
ловой) кислоты.
Л. и.
Н3О*
СН3С=ССООСН3 СН2=С=СНСООСН3
Алкилирование литиевого енолята этилового эфира кротоно-
вой кислоты в растворе приводит к образованию алкилирован-
ных несопряженных сложных эфиров:
СН3СН = СНСООС2Н5
0°, СН31
———> СН2==СНСНСООС2Н5
87% |
СН3
62%
———СН2=СНСНСООС2Н5
СдНзСНяВг I
0° I
сн2с6н5
Добавление ацетона к раствору литиевого енолята этилкро-
тоната дает с хорошим выходом эфир соответствующей р,у-не-
насыщенной £-оксикарбоновой кислоты, например этиловый
эфир З-окси-2-винилизовалериановой кислоты:
I) л. и.
2) СН3СОСН3
СН3СН=СНСООС2Н5 --------——>
-—78°
- СН2=СН—СНСООС2Н5-
н3с/С\сн3
L OLi -I
Н3О*
78%
СН2==СН—СНСООС2Н3
н3с/1 \сн3
он
а-Метил-у-бутиролактои. Познер и Лумис [4] осуществили
с хорошим выходом «-метилирование у-бутиролактона, превра-
тив его в енолят лактона обработкой избытком Л. и. или диизо-
пропиламида лития в ТГФ при —78°. К полученному раствору
§97
енолята при —78° добавляли избыток йодистого метила и дово-
дили температуру реакционной смеси до —30°
?.П-л-и.,тгф,-78°
2) СН31, -78°, затем -30’
Клянзеновская перегруппировка аллиловых сложных эфиров.
Айрланд н Мюллер [5] сообщили, что литиевые еноляты алли-
ловых эфиров при комнатной или немного более высокой тем-
пературе быстро перегруппировываются в соответствующие
у,6-ненасыщениые кислоты. Так, например, аллиловый эфир (1)
под действием Л, н. в ТГФ при —78° превращается в литиевый
енолят (2). Затем температуру раствора енолята (2) в течение
10 мин доводят до 25°. у,6-Ненасыщенную кислоту (3) получают
25°
----->
80%
CHj
сн3
сн,
(3)
с выходом 80%. В тех случаях, когда перегруппировка проте-
кает медленно, перед нагреванием реакционной смеси до ком-
натной температуры полезно обработать литиевый енолят при
—78° триметилхлорсиланом. Такая операция была проделана
с ацетатом (4). Промежуточный ацеталь кетена перегруппнро-
(CH,),sici
----------->
67®, 9 0 Ш
(4) (5)
вывается при 67° в соединение (5) с выходом 70%.
Метилирование а,р-ненасыщенных кетонов [6]. Алкилирова-
ние «,^-ненасыщенных кетонов обычно протекает почти исклю-
чительно в «-положение. Метилирование анионов «^-ненасы-
щенных кетоиов, образующихся под действием Л. и., напротив,
дает преимущественно а/-метил производные.
298
Примеры:
237»
При использовании в описанных реакциях в качестве осно-
вания трифеннлметиллнтия (III, 396; V, 443) в основном обра-
зуются а-метнлпроизводные. При действии этого основания на
5,5-диметнлциклогексен-2-он-1 (1) был получен аномальный про-
1) (CtH5)3CLi, ТГФ , О’
2) СН31
дукт (2) [7]. Трнфенилметиллитнй в ТГФ при —23° дает также
продукт сопряженного присоединения (4) к этилкротонату (3).
ТГФ
-23°
СН3СН=СНСООС2Н5 + (C6H5)3CLi —-> (С6Н6)3ССНСН2СООС2Н3
> оэ А> I
СНз
(3) (4)
1, Rathke М. W., Lin der t A., J. Am. Chem. Soc., 93, 2318 (1971).
2. Кларк X. Т„ Тейлор Е. Р., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 1, стр. 126.
3. R a t h k е М. W., Sullivan D., Tetrahedron Letters, 4249 (1972).
4. P о s n e r G. H., Loomis G. L,, Chem, Comm., 1972, 892.
5, Ireland R. E., Mueller R. H., J. Am. Chem. Soc., 94, 5897 (1972).
299
6. Lee R. A.. McAndrews C., Patel К. M., Reusch W., Tetrahedron
Letters, 965 (1973).
7. Lee R. A., Reusch W., Tetrahedron Letters, 969 (1973).
ЛИТИЯ ИОДИД (II, 196—199).
Циклобутанон. Салаюн н Комья [1] недавно опубликовали
удобный метод синтеза циклобутанона, ключевой стадией кото-
рого является изомеризация эпоксида под действием Л. и., реа-
гирующего как кислота Лыоиса. Исходное соединение — метал-
лилхлорид(1)—дегидрохлорируют амидом натрия в присутствии
трет-бутилата натрия до метиленциклопропана (2) с выходом
60%. Эпоксидирование последнего п-нитроиадбеизойной кисло-
той в хлористом метилене при —10° дает с почти количествен-
ным выходом оксаспиропентан (3). При нагревании этого сое-
динения в отсутствие катализатора образуется 40% циклобута-
нона (4) и другие продукты, а добавление к нему каталитиче-
ских количеств Л. и. (Lil• l,5HsO) вызывает экзотермическую
n-N02C6H4CO3H
-10°
количеств»
сн,
1
С1СНгС—СНг
(0
NaNH2
(СН3)3СО№щ
60%
перегруппировку его в кетон (4), который можно выделить с вы-
ходом >95%.
Расщепление алкилариловых эфиров [2]. ArOR -> АгОН.
Алкнлариловые эфиры расщепляются с высоким выходом под
действием Л. и. в сухом 2,4,6-коллидине. Реакционная среда в
процессе расщепления становится умеренно щелочной, однако
этого можно избежать, добавляя для создания буфера такую
кислоту, как бензойная. Реакцию можно также проводить в от-
сутствие растворителя при 180—200°, используя LiI-3H2O.
1. Salatin J. R., Cont a J. M., Chem. Comm., 1971, 1579; Salatin J. R.,
Champion J., Conla J. M., Org. Syn., submitted 1972.
2. Harrison 1. T., Chem. Comm., 1969, 616.
ЛИТИЯ ИОДИД—БОРА ТРИФТОРИД.
Восстановление п-галогенкетонов [1]. Взаимодействие а-
бромкетонов с Л. и. — б.т. в эфире или ТГФ при комнатной тем-
пературе приводит к образованию с высоким выходом соответ-
ствующего незамещенного кетона. Водород, встающий на место
атома галогена, как было в конце концов установлено, берется,
по-вндимому, из воды, содержащейся в «безводном» Л. и. Един-
ственное исключение представляет а-бромкамфора, которая в
300
этих условиях не восстанавливается. Приведенная методика
подходит и для восстановления некоторых а-хлоркетонов (фе-
нацилхлорида, 2-хлорпентанона), исключая пространственно за-
I1'. (2)
1. Townsend J. М., Spencer Т. A., Tetrahedron Letters, 137 (1971).
ЛИТИЯ КАРБОНАТ-ЛИТИЯ ГАЛОГЕНИД (II, 199—202;
V, 256).
Дегидрохлорирование 2-хлор-5-трет-бутилциклогексаиоиа [1].
Дегидрохлорирование 2-хлор-5-трет-бутилциклогексанона (1)
(хлор аксиальный или экваториальный) под действием смеси
карбоната и хлорида лития (метод Жоли и Варнана; II, 199—
202) приводит к образованию только 5-трет-бутилциклогексен-2-
она-1 (2). Использование для этой цели хлорида лития в ДМФА
(метод Холижа; II, 204—205) нлн коллидина дает кроме (2)
еще два продукта— (3) и (4).
а) ЫС^/дМФА
(3) 12-24% (4) 6_9 %
1. Aioreau Р., Casadevall Е., Compt. rend., 272, 801 (1971).
ЛИТИЯ ПЕРГИДРО-9Ь-БОРАФЕН АЛ ИЛ ГИДРИД (VI,
152-153).
Специфическое восстановление простагландинов Е до проста-
гландинов Fa. Одной из важных реакций в химии простаглан-
динов является взаимопревращение первичных [1] простаглан-
динов, например восстановление простагландина Ег (1) до про-
стагландина Fsa (2). Под действием боргидрида натрия эта ре-
акция протекает неизбирательно с образованием смеси (2) и
его Cg-эпимера Fsp, обладающего гораздо меньшей биологиче-
301
ской активностью, чем ?2а- Кори и Варма [2] недавно сообщили
что с помощью объемистого боргидридного реагента Л. п. в
ТГФ при —78° простагландин Е2 (1) можно восстановить сте-
рео специфически (выход 98,7%).
1. Bergstrom S., Science, 157, 382 (1967).
2. Corey E. J., Varma R. K., J. Am. Chem. Soc,, 93, 7319 (1971)
ЛИТИЯ 2,2,6,6-ТЕТРАМЕТИЛ П ИП ЕРИДИД,
Мол. вес 147,18.
О)
Это основание получают обработкой очищенного 2,2,6,6-тет-
раметилпиперидина метиллитием илн н-бутиллитием.
Стабилизированные бором карбанионы. Ратке и Коу [1]
обнаружили, что в отлнчие от обычных оснований, которые при
взаимодействии с борорганическими соединениями координи-
руются по атому бора, пространственно сильно затрудненные
литий-амиды, такие, как Л. т. или литий трет-бутилнеопентил-
амид [2], могут отщеплять а-протон борорганического соедине-
ния, генерируя карбанион. Так, например, взаимодействие бор-
органического соединения В-метил-9-борабициклононана (1) [3]
в бензоле с Л. т. в течение 12 час при комнатной температуре
с последующей обработкой реакционной смеси D2O приводит к
образованию дейтеропроизводного (3) с выходом 50%.
Ратке и Коу сообщили также об одном синтетическом при-
менении открытой ими реакции. Так, с циклогексаноном соеди-
нение (2) образует метиленциклогексан (4) с выходом 55—65%,
302
считая на (1).
(2) +
о -
в-он +
снг
(4)
Синтез арилциклопропанов [4]. Прн действии Л. т. на хло-
ристый бензил в эфире генерируется фенилкарбен или карбено-
идные частицы, реагирующие с олефином (большой избыток)
АгСН2С1
—-
с образованием арилциклопропана с выходом 50—80%. Цикло-
пропаны образуются в результате стереоспецифического ^не-
присоединения, причем менее устойчивый сан-изомер преобла-
дает над анти-изомером. Так, например, этим методом можно
получить с выходом 54% 7-фенилноркар ан, соотношение
син^&нти в котором составляет 2,2. Олофсон и Даурти [4] срав-
нили выходы 7-фенилноркарана, полученные при использова-
нии других оснований общей формулы LiNR2. Лишь несколько
оснований оказались столь же эффективными, как Л.т. Это —
дициклогексиламид лития [LiN(Су)2], LiN(трет-amyl)-трет-Bu и
литиевое производное N-(1-этилциклогексил)-1,1,3,3-тетраметил-
бутиламина (НЭТА). В двух последних реагентах, как и в Л.т.,
атом азота соединен с двумя третичными атомами углерода.
Если вместо алкенов в реакции участвуют нетерминальные
ацетилены, то образуются 3-арилциклопропены. Так, например,
при добавлении Л.-т. к эфирному раствору п-СН3ОСбН4СН2С1
и гексина-3 получается 1,2-диэтил-З-п-анизилциклопропен (1) с
выходом 60%.
.CZHS
Л-СН3ОС6Н4
:2HS
(1)
Бензины [5]. Л. т. является также эффективным основанием
для генерирования бензинов из АгС1. Так, например, при обра-
ботке о-хлораннзола Л. т. и ЫС = ССбН5 образуется //-метокси-
толан (1) (толан —это дифенилацетилен) с выходом 80%. Из
С6НбС1, CeHsSLi и Л. т. с выходом 93% был получен дифенил-
сульфид.
осн3
J^-C1
Г + LiC=CC6H5 Ад С6Н£С=СС6Н4ОСНз-л(
Д > 80%
(О
303
Сложноэфирные конденсации, Олофсон и Даурти [5], ис-
пользовав в качестве основания Л.т., осуществили синтез р-ке-
тоэфиров (1) с выходом 89%.
СНз\ п Г _ 1 c9hscoci
ЧСНСООС2Н5 ' -> хсоос2н5 -------------------->
СН3/ LcH3/ J
сн3
—> CeH5COCCOOC2H5
сн3
(И
1. Rathke М. W., Row R., J. Am. Chem. Soc., 94, 6854 (1972).
2. Hennion G. F., Hanzel R. S., J. Am. Chem. Soc., 82, 4908 (I960).
3. Brown H. C., Rogic iM. M., J. Am. Chem. Soc., 91, 4304 (1969).
4. Olofson R. A., Dougherty С. M., J. Am. Chem. Soc., 95, 581 (1973).
5. Olofson R. A., Dougherty С. M., J. Am. Chem. Soc., 95, 582 (1973).
ЛИТИЯ ТЕТРАХЛОРКУПРАТ, Li2CuCl4. Мол. вес 219,25.
Получение [1]. Л. т получают взаимодействием хлорида
лития (0,2 моля) и хлорида меди (II) (0,1 моля) в ТГФ.
Конденсация реактивов Гриньяра с алкилбромидами [1].
При взаимодействии реактивов Гриньяра с алкилбромидами в
ТГФ при 0° или ниже, катализируемом медью(I), выход гомо-
димеров R1—R1 и R2—R2 незначителен. Реакция представляет
собой нуклеофильное замещение атома галогена в R2X радика-
лом реактива Гриньяра. Наиболее высокие выходы получены
в случае
Ы2СиСЦ
Ri-MgX + R!X ----->• R’-Ra4-MgX2
первичных алкилгалогенидов (иодиды > бромиды хлориды).
Строение реактива Гриньяра не так важно.
Примеры:
Ь12СиСЦ
0°, 3 час
K-CgHijMgBr -f- w-CiHgBr ~~~ н-СюДз
73 %
Ы 2 С и С14
0°, 3 час
H2C=CH(CH2)4MgBr + С2Н5Вг —н2с=снс6н!3
Этот новый метод конденсации был использован недавно в
стереоселективном синтезе полового аттрактанта яблонной пло-
дожорки — транс,тра«с-додекадиен-8,10-ола-1 (4) [2]. Конденса-
ция бромида (1) и реактива Гриньяра (2), катализируемая Л.т,,
дает соединение (3) с выходом 85%. Полученное вещество со-
стоит на 78% из желаемого транс,транс-изомера. Гидролиз за-
щитной группы (n-TsOH в водном метаноле) приводит к обра-
304
зованию свободного спирта (4)’, содержащего около 80% нуж-
ного транс,т ране-дуитола, выделяемого из спирта (4) кристал-
лизацией последнего из пентана при —5°.
Активным катализатором конденсации реактива Гриньяра
и алкилгалогенидов является также серебро, но его применяют
только в том случае, когда алкильные группы обоих компонен-
тов реакции одинаковы:
Ag, ТГФ
25°, 5 час
H-C4H9MgBr + w-C4H9Br ——н-С8Н18
/У /0
Если эти алкильные группы различны, образуется смесь трех
продуктов конденсации. Растворимый серебряный катализатор
получают реакцией нитрата серебра (5,0 жмоля) с 1,0 М рас-
твором (30 мл} этилмагнийбромида при 0° в течение 3 час. По-
лученный раствор с концентрацией 7,5 М по растворимому се-
ребру устойчив в течение некоторого времени при хранении в
холодильнике [1].
1. Tamura М., Kochi J., Synthesis, 1971, 303.
2. Descoins С., Henrick С. A., Tetrahedron Letters, 2999 (1972).
ЛИТИЯ ТРИЭТИЛБОРГИДРИД, Ы(С2Н5)3ВН. Мол. вес
105,95.
Л.т. получают реакцией гидрида лития (V, 252) с триэтил-
бораном в ТГФ. Он устойчив в течение неопределенно долгого
времени.
ТГФ
ЫН + В(С2Н5)з ---> Li(C2H5)3BH
Восстановление алкилгалогенидов [1]. Этот реагент при 25°
почти количественно восстанавливает (5ь?2-замещенне) хлори-
стые и бромистые аллилы и бензилы. Незамещенные первичные
галогениды полностью восстанавливаются за 2 мин. Даже нео-
305
пентилбромид при 3-часовом кипячении с Л. т. дает неопентан
(выход 96%). Вторичные циклоалкилбромиды восстанавливают-
ся при 25° в течение 24 час, н даже экзо-2-бромнорборнан (1)
может быть восстановлен количественно. Восстановление тре-
тичных алкилбромидов протекает медленно и приводит преиму-
щественно к продуктам элиминирования, а с арилгалогенндами
Л.т. совсем не реагирует.
Скорость восстановления Л.т. намного выше, чем тиофено-
лятом натрия, алюмогидридом лнтия или боргидридом лития.
Алкилгалогениды можно восстанавливать также гидридом
лития в присутствии каталитических количеств триэтилборана.
1. Brown Н. С., К г i s h п a m u г t h у S., J. Am. Chem. Soc., 95, 1669 (1973).
ЛИТИЯ ТРИЭТИЛМЕТИЛАТ, LiOC(C2H5)3. Мол. вес.
122,13.
Это основание получают из н-бутиллития и триэтилкарби-
нола.
Триалкилкарбинолы из триалкилборанов. Триалкилкарбино-
лы были получены реакцией триалкилборанов с окисью углеро-
да в диглиме с последующим окислением перекисью водорода
[уравнение (1)]. См. V, 471. Этот метод имеет, правда, тот недо-
статок, что при температуре, при которой проводится реакция,
может в заметной степени протекать изомеризация бороргани-
ческих соединений.
Диглим Н2О2
1)«-Ви3В + СО .— [ 1 ----> н-Ви3СОН
90%
Браун и сотр. [1] недавно нашли, что под действием Л.т. ре-
акция три-н-бутилборана с хлордифторметаном (или другим
тризамещенным метаном) [уравнение (2)] с почти количествен-
ным выходом дает три-н-бутилкарбинол. Эта реакция быстро
протекает при 65°, а при такой температуре изомеризация бор-
органических соединений незначительна. Из всех испытанных
ТГФ. ЫОС(С2Н51з н2о2
2) W-BU3B + HCCIF2 --м0 ; ----> [ ] ----> w-Bu3COH
98%
алкоголятов наилучшие результаты были получены при исполь-
зовании пространственно затрудненных соединений. К послед-
ним относится и Л.т.» который даже превосходит трет-бутилат
306
калия и триэтилметилат калия. При замене хлордифторметана
хлороформом выходы несколько снижаются (85%), но зато ме-
тодика становится проще и удобнее.
1. Brown Н. С., Carlson В. A., Prager R. Н., J. Am. Chem., Soc., 93,
2070 (1971).
ЛИТИЯ ФОСФАТ, Ы3РО4. Мол. вес 115,80.
Получение. Раствор Na3PO4‘12H2O (570 г) в воде приливают
при перемешивании к водному раствору гидроокиси лития
(126 г). Образующийся осадок отфильтровывают, промывают
водой, затем диспергируют в воде (1,5—2 л) при 60° в течение
20 мин. Твердое вещество снова отфильтровывают и промывают.
Эту операцию выщелачивания повторяют еще четыре раза. За-
тем отфильтрованное вещество сушат в печи при 200° в течение
16 час.
Изомеризация окиси циклооктена. Изомеризация окиси цик-
лооктена (1) под действием диэтиламида лития дает бицикли-
ческий спирт (2) и 3-оксициклооктен (3) в отношении прибли-
зительно 4:1 (V, 254). Аномальный продукт (2) получается
только при изомеризации а-окисей средних циклов, макроцикли-
ческие окиси дают исключительно аллиловые спирты. По сооб-
щению Шенга [1], окись циклооктена можно изомеризовать ис-
ключительно в 3-оксициклооктен (3), если в качестве основа-
ния использовать трет-бутилат калия в ДМСО (выход 46%).
З-Оксициклооктен (3) можно получить даже с более высоким
выходом (70%) при пропускании раствора окиси циклооктена
в бензоле через трубку, заполненную гранулами Л. ф., при 180°.
1. Sheng М. N., Synthesis, 1972, 194.
МАГНИЙ (I!, 207—210; V, 259; VI, 155).
Активированный магний. Рике и Худналл [I] разработали
способ приготовления высокоактивной формы магния в мелко-
раздробленном состоянии, пригодного для получения труднооб-
разующихся реактивов Гриньяра. По этому методу безводный
хлорид магния восстанавливают щелочным металлом в инерт-
ном. растворителе типа эфира в атмосфере инертного газа. Для
этой цели особенно подходит натрий в диглиме или калий в
ТГФ. При этом нет необходимости удалять образующиеся соли
натрия и калия, а реактив Гриньяра можно получать прибавле-
нием алкил- или арилгалогенида непосредственно к суспензии
порошкообразного металлического магния. Например, фенил-
магнийбромид был получен из бромбензола и активированного
магния в ТГФ при —78° в течение 30 мин с выходом более 60%.
Таким путем синтезирован даже неизвестный до настоящего
времени фенилмагнийфторид, правда, с выходом лишь около
5%.
Димеризация остатков алкилгалогеиидов. Японские химики
[2] описали синтез 8,8'-дигептафульвенила (4) из бромида (1).
При взаимодействии бромида (1) с магнием в эфире с выходом
85% образуется 1,2-ди (циклогептатриенил-3)-этан (2). Отщеп-
ление гидрид-иона под действием трифенилметилборфторида
(III, 399—401; V, 442—443; этот том) в хлористом метилене
приводит к соли (3). При обработке этой соли триэтиламином
в хлористом метилене происходит отщепление борфтористоводо-
родной кислоты с образованием соединения (4) с выходом 46%,
308
Кетали циклопропанонов и аллены [13]. С а,а'-дибромкета-
лями (1) магний в ТГФ (или цинк в ГМТФК) образует аллены
(2) и кетали циклопропанонов (3), Если вместо магния исполь-
зуют цинк, в качестве растворителя необходимо брать ГМТФК-
в
R Rl R2 R3
X/ \/
гС—— С-—СВг
о М
Mg R-^ x-R2
тгф х ^с--с=с
~ В I 3
г< /К1
,о-сн
о-сн
Rz"xr3
(1)
(2) 20-40%
(3) 40-9 0%
2
2
1. Rieke R. D., Hudnall P. M.. J. Am. Chem. Soc., 94, 7178 (1972).
2. Kuroda S., Oda M.., Kit ah ar a Y.T Angew. Chem., Internal. Ed., 12, 76
(1973).
3. Giusti G., Morales C., Bull. soc. chim. France, 1973, 382
!Ч,1Ч-бас-МАГНИЙБРОМАНИЛИН, C6H5N(MgBr)2.
Получение [1]. К эфирному раствору этилмагнийбромида по
каплям при перемешивании прибавляют анилин и кипятят смесь
в течение 30 мин:
2 C2H5MgBr + CgH5NH2 —> C6H5N(MgBr)2 + 2C2HS
Реакция с 2-ацетиламино-3-карбэтокси-5-этилтиофеиом [1].
.COjCjHj
СН3СНг----II + (ВгМ§)гМС6Н5
'NHCOCH3
5
>СН3СН2
1. R i е d W., К a h г Е., Ann., 716, 219 (1968).
МАГНИЯ АЛЮМОГИДРИД, Mg(AlH4)2- Мол. вес 86,34.
М. а. лучше всего получать реакцией иодида магния с алю-
могидридом натрия в ТГФ. В этом растворителе (а также в
эфире) М.а. нерастворим и выделяется с выходом 58% в виде
сольвата Mg(AlH4)2’T^ [1].
Восстановление. В первом сообщении группы чехословацких
химиков [2] указывалось, что органические соединения с трудом
и с ничтожными выходами восстанавливаются М. а. Однако
Джеймс [3] недавно сообщил, что М. а., суспендированный в
эфире, с высоким выходом и довольно быстро (4—12 час) вос-
станавливает альдегиды, кетоны, кислоты и оксимы. Таким об-
разом, реагент Ай. а. по своему действию, но не по удобству
использования может конкурировать с алюмогидридом лития.
I. Ashby Е. С., Schwartz R. D., James В. D., Inorg. Chem., 9, 325
(1970).
2. Plesek J., Hefmanek S.. Coll. Czech., 31, 3060 (1960).
3. J a m e s B. D., Chem. Ind., 1971, 227,
309
МАЛЕИНОВЫЙ АНГИДРИД. Мол. вес 98,06, т. пл. 52—54°.
Присоединение тиоуксусной кислоты: I, 82
Присоединение к аллену: I, 34
Синтез нафтазарина: II, 363—364
Реакция с фталоил хлоридом: IV, 207
Присоединение к диенам по Дильсу — Альдеру: I, 293; III,
51, 310—311; IV, 200—201.
Присоединение по Дильсу — Альдеру в присутствии хлор-
анила [1].
В отсутствие дегидрирующего
можно выделить промежуточный
агента в некоторых случаях
аддукт Дильса—Альдера.
1. Zander М„ Ann., 723, 27 (1969).
МАЛОНОНИТРИЛ, CH2(CN)2. Мол. вес 66,07, г. пл. 32—34°,
т. кип. 220°.
Получение [1].
1) CNCH2CONH2 + PCls —> CH2(CN)2-!-POCl3 + 2HCl
2) 2CNCH2CONH2 + POC13 —> 2CH2(CN)2 + HPO3 + 3HC!
Реакция с тетрацианэтиленом (III, 113, 317).
Бромирование (III, 314).
грет-Бутилирование [2] действием;
(CH3)CBr - A1CI3
(CH3)3CC1 - BF3
(CH3)3CBr- AgC104
(CH3)2C=CH2-HC1O4
Выход, %
60
43-56
68
59
1. Корсон Б., Скотт Р., Бозе К., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 355.
2. Boldt Р., Mllitzer Н., Thielecke W., Schulz L., Ann., 718, 101
(1968).
МАРГАНЦА(Ш) АЦЕТАТ (V, 261—262).
Получение [1]. М. а. можно получить окислением тетрагид-
рата ацетата марганца(II) перманганатом.
Окисление толуолов [2]. М. а. окисляет толуолы до бензил-
ацетатов. Реакция заметно ускоряется галогенидами металлов,
такими, как бромид калия. По-видимому, реакция протекает че-
310
рез образование промежуточного соединения радикального ха-
рактера:
Мп(ОАс)з, КВт
ХС6Н4СН3 ----------> ХС6Н4СН2. XCsH4CH2Br —> XCsH4CH2OAc
НОАс
Аллильное окисление [1]. Циклогексен при 70° в присутствии
бромида калия в качестве катализатора примерно за 8 час
окисляется реагентом до циклогексен-2-и л ацетата с выходом
75%; без катализатора реакция протекает медленно. Норбор-
нен не вступает в эту реакцию, а бицикло-[3,2,1]-октен-2 быстро
окисляется с образованием приблизительно эквимолекулярной
смеси эндо- и экзо-бицикло-[3,2,1]-октен-3-ил-2-ацетатов. Ацикли-
ческие олефины, а также циклические олефины, склонные к ал-
лильной перегруппировке, дают сложную смесь аллилацетатов.
1. G i I m о г е J. R., М е 1 1 о г J. М., J. Chem. Soc. (С), 1971, 2355.
2. G i 1 m о г е J. R., М е 11 о г J. iH., Chem. Comm., 1970, 507.
МАРГАНЦА ДВУОКИСЬ АКТИВНАЯ (II, 218—227; V,
263—270; VI, 156—158).
Окисление 4,4-диметил-1,7-дифенилгептадиин- 1,6-диола-З,5
(1) с помощью М. д. а. (фирмы «Мерк») [1]:
ОН
Н3С. ,СНО Н3С. ZCHC=CC6HS Мн02
;с; + 2c6H3C£sCMgBr —-> ;с —*
Н3с/ \сно 77% H3CZ \снс^ссен5 79%
I
он
(1)
о
Н3С^ ZCC=CC6H6
Н3с/ ^сс—СС8Н3
(2)
Амиды карбоновых кислот. Гилман [2] применил метод окис-
ления аллиловых спиртов до сложных эфиров карбоновых кис-
лот, разработанный Кори и сотр. (V, 267—268), для получения
амидов карбоновых кислот. Так, например, в результате окис-
ления ароматических или ссф-ненасыщенных альдегидов МпО2
в присутствии цианистого натрия и амина с высокими выходами
образуются соответствующие амиды карбоновых кислот. В от-
сутствие цианистого натрия из альдегидов, аммиака и М. д. а.
С высокими выходами образуются нитрилы.
81!
Окислительная димеризация альдегидов [3]. Алифатические
альдегиды, имеющие хотя бы один а-водородный атом, под дей-
ствием М.д. а. претерпевают окислительную димеризацию с об-
разованием С—С и С—О-димеров, Например, изомасляный аль-
дегид (1) превращается с выходом около 80% в смесь 2,2,3,3-тет-
раметилянтарного альдегида (2) и 2-метил-2-(-2/-метил-1-про-
пенокси)-пропионового альдегида (3) в соотношении 43:57.
сн3
нзсх МпОг нзс-ссно
снсно -. -*-> |
н3сх н3с-ссно
сн3
(О (2)
сн3н
НзС-С-^^О
I хсн3
О
СН,
(3)
Дегидрирование. На последней стадии полного синтеза алка-
лоида спорыньи сИ-изосетоклавина (2) осуществляют дегидри-
рование соединения (1) с выходом 36% действием М.д.а. в
хлороформе при 25° [4].
1. Miiller Е., S е g п i t z A., Chem. Вег., 106, 35 (1973),
2. Gilman N. W., Chem. Comm., 1971, 733.
3. L e f f i n g w e 11 J. C., Chem. Comm., 1970, 357.
4. Ko r nf e 1 d E. С., В a c h N. J., Chem. Ind., 1971, 1233.
R-( — )-МЕВАЛОНОЛАКТОН (III, 85—86).
Выделение. Чеше и сотр. [1] нашли, что мевалозид пред-
ставляет собой глюкозид М. и может быть выделен с выходом
1,75%. Лактон легко получить ферментативным гидролизом из
листьев мушмулы германской с выходом 0,63%.
1. Tschesche R., Struckmeyer К., Wulff G., Chem. Вег. Ю4, 3567
(1971).
МЕДИ(П) АЦЕТАТА МОНОГИДРАТ (II, 228—230; V, 271).
Окислительная тримеризация гексадиина-1,5 в циклооктаде-
кагексаин-1,3,7,9,13,15 [1]. В трехгорлую круглодонную колбу
объемом 6 л, снабженную мешалкой с двумя 7,5-сантиметровы-
ми лопастями, а также обратным холодильником с хлоркальцие-
вой трубкой и капельной воронкой иа 500 ли (с трубкой для
выравнивания давления), помещают 600 а М. а. м. и 3,8 л пири-
312
дина. Колбу погружают в водяную баню, предварительно нагре-
тую до 55°, и смесь при этой температуре медленно перемеши-
Си(е>СОСН3)г-Н2О
Ру ’
вают в течение 1 час. Затем к суспензии зеленого цвета при 55°
в течение 30 мин при энергичном перемешивании добавляют
раствор 50 г гексадиина-1,5 (т. кип. 84—85°) в 400 мл сухого
(КОН) пиридина и интенсивно перемешивают смесь при 55° еще
2 час. После обработки получают около 35 г сырого циклоокта-
декагексаина (2) в виде темно-коричневого вещества.
1. Stockel К-, Sondheimer F., Org. Syn., submitted 1971.
МЕДИ(1) АЦЕТИЛАЦЕТОНАТ, CUC5H7O2. Розово-красный.
Мол. вес. 162,65.
Получение [1]. М. а. получают реакцией ацетилацетона с ам-
миачным раствором гидроокиси меди(1).
1,4-Дикетоны [2]. М. а. является весьма эффективным мед-
ным катализатором присоединения а-кетокарбенов по двойной
С—С-связи. Так, например, кетон (3) можно получить с выхо-
дом 55% реакций 1-диазобутанона-2 (1) с изопропенилацета-
том (2). При кипячении кетона (3) с 4%-ным раствором гидро-
окиси натрия в метаноле образуется 2,3-диметилциклопентен-2-
ои-1 (5) с выходом 85%.
313
Этот новый метод получения 1,4-дикетонов был использован
в синтезе qwe-жасмона (6).
о
(6)
1. Emmert В., Gsottschneider W., Stanger Н., Ber., 69В, 1319
(1936).
2. М с М и г г у J. Е., Glass Т. Е., Tetrahedron Letters, 2575 (1971).
МЕДИ(1) трет-БУТИЛАТ, (СН3)3СОСи. Мол. вес 136,65 воз-
гоняется около 170° (1 мм).
Получение [1]. М. б. получают реакцией безводного C112CI2
с грет-BuOLi в ТГФ в атмосфере N2 и очищают его возгонкой.
Металлирование [1]. М. б. является удобным металлирую-
щим агентом, поскольку алкоксильная группа, обладающая
основным характером, отщепляет активный водород с образова-
нием спирта. Таким образом, при этом нет необходимости про-
водить реакцию в основном или буферном растворе. В резуль-
тате реакции М. б. с большим избытком фенилацетилена с
97%-ным выходом образуется светло-желтый ацетиленид ме-
ди (I).
С6Н5С=СН Д- (СН3)3СОСи —> СеН5С=ССи+(СНз)зСОН
Избыток 97% 97
1. Tsuda Т., Hashimoto Т., Saegusa Т., J. Am. Chem. Soc., 94, 658
(1972).
МЕДИ ЗАКИСЬ —трет-БУТИЛ ИЗОНИТРИЛ,
Cu2O—(CH3)3CNC.
трет-Бутнлизонитрил можно получить по методике Уги и
Мейра [1].
Димеризация а,р-ненасыщеиных кетонов, эфиров и нитрилов
а,р-ненасыщенных карбоновых кислот [2]. Предлагаемое соче-
тание реагентов вызывает димеризацию а,р-ненасыщенных ке-
тонов, а также сложных эфиров и нитрилов сс,р-ненасыщенных
карбоновых кислот:
Cu2O-(CH3)3CNC
RR'CHCH=CHX ---------------> RR'CHCH=CX
rr'chc!hch2x
X=CN, COOR", COR*
Синтез циклопропанов [3]. Замещенные циклопропаны мож-
но с прекрасным выходом получить катализируемой М. з.—б.
314
реакцией олефина с а-хлорзамещенными кетонами или эфира-
ми и нитрилами а-хлоркарбоновых кислот:
ClCHjY ^C^-C<CU‘O-(CH»>»CN<:: >
Y - COOR, COR, CN
1. U gi I, М еу г R., Chem. Вег., 93, 239 (1960).
2. S а е g и s а Т, Ito У, Т о m i t a S., К 1 л о s h 11 a H, J. Org. Chem., 35,
670 (1970).
3. S ae gи s a T, Ito Y, Yoneza wa К., I n и b и s h 1 Y„ Tom i t a S., J. Am.
Chem. Soc, 93, 4049 (1971).
МЕДИ КАРБОНАТ ОСНОВНОЙ, CuCO3Cu(OH)2 (11,231—
232). Мол. вес 221,11.
Окислительное декарбоксилирование [1]. При нагревании
флуорен-9-карбоновой кислоты (1) с 2,0 мол. же М. к. о. обра-
зуется флуоренон (2) с выходом 56%. Эта реакция является
первым примером прямого окислительного декарбоксилирова-
ния, катализируемого солями меди.
О) (2) (3)
1. Trost В. М, Kinson Р. L, J. Org. Chem, 37, 1273 (1972).
МЕДИ(П) МЕТИЛАТ, Си(ОСН3)2. Мол. вес 125,61. М. м.
получают из CuCI2 и L1OCH3 [1],
Реакция с окисью углерода [2]. М. м. реагирует с окисью
углерода в пиридине при 35—70° с образованием диметилового
эфира угольной кислоты с выходами, достигающими 84%:
Си(ОСН3)2 + СО —> (СН3О)2СО
Предполагается, что реакция протекает через неустойчивое
карбметоксимедное промежуточное соединение и Си (II) при
этом восстанавливается до Cu(I).
Если реакцию проводить в присутствии вторичного амина,
с хорошим выходом образуются карбаматы:
со
Си(ОСН3)2 + ВДН ----► R2NCOCH3
А
315
Карбонилировать можно и другие алкоксильные производ-
ные меди (II), например Си (ОСН3) С1, Си(ОС3Н5)2 и
Си(асас) (ОСН3).
1. Brubaker С Н., J г., W 1 с h о 1 a s М., J. Inorg. Nucl. Chem., 27, 59 (1965).
2. Saegusa T., Tsuda T., Isayama K„ J- Org. Chem., 35, 2976 (1970).
МЕДИ(П) НИТРАТ— ПИРИДИН, Cu(NO3)2 • (C5H5N)4,
(II, 232).
Окисление [1]. При окислении метоксициклопропанола (1)
нитратом меди(II) промежуточно образуется радикал (2), ко-
торый превращается далее в метилакрилат (3) и диметиловын
эфир адипиновой кислоты (4). Для той же цели можно исполь-
зовать также нитрат железа Fe(NO3)3-
COOCHj
ОН Си2* . !
---—ССНгСНгСООСНэ] >H2C = CHCOOCHj + (СН2)4
OCHj |
(2) (3) COOCHj
(4)
Реакция метоксициклопропанола (1) с метилвинилкетоном
(5) в присутствии М. н.—п. приводит к образованию с выходом
45% метилового эфира 6-кетоэиантовой кислоты (6). [При ис-
пользовании нитрата железа (Ш) эфир (6) получается с выхо-
дом 30%.]
CH3 CH;
CH3 c=o 1 C=O -1
. OH 4- 1 c=o _ 1 Cu > 'CH 1 CH2 Cu+ CH 1 CH, нго CHjCOfCH^COOCHj (6)
/'"'осн3 U) CH 11 CH2 [ СЩ 1 1 СЩ 45%
(S) CH2 CHZ
COOCH; COOCHj
Присоединение радикала (2) к бутадиену (в СН3ОН) приво-
дит к возникновению аллильного радикала (7), который далее
димеризуется с образованием смеси диэфиров ненасыщенных
Си-кислот.
(2) + СН2=СНСН=СН2 —>
[СН2СН=СНСН2 СН2СНСН=СН2 1
I "tc* f Сн-диэфиры
СН2СН2СООС1-1з <7) СН2СН2СООСН3 J 52^
1. Schaaf sma S. Е., Jorrltsma R., Steinberg IL, de Boer Th. J.,
Tetrahedron Letters, 827 (1973).
316
МЕДИ ОКИСЬ, CuO.
Газофазное окисление спиртов. Газофазное окисление спир-
тов на поверхности целого ряда металлов и их окислов исполь-
зуется в основном в промышленности. Шейк и Идон [1] недавно
предложили простой лабораторный метод окисления первичных
и вторичных спиртов до соответствующих альдегидов и кетонов.
Пары спирта и инертный газ-поситель (обычно гелий) пропу-
скают через неплотно заполненную окисью меди в проволоке
2-метровую колонку препаративного газового хроматографа, ко-
торый и обеспечивает контроль температуры и скорости потока.
Выходы, как правило, высокие; исключение составляют лишь
аллиловые и гомоаллиловые спирты, дающие обычно смеси
продуктов.
1. Sheikh М. Y., Еа don G., Tetrahedron Letters, 257 (1972).
МЕДИ(1) ФЕНИЛАЦЕТИ Л ЕНИД, С6Н5С = С — Си. Мол.
вес 164,66.
Получение [1]. Раствор медного купороса в конц. водном
аммиаке перемешивают в атмосфере азота, охлаждают и вос-
станавливают хлоргидратом гидрокснламина. Затем к образо-
вавшемуся бледно-голубому раствору добавляют раствор фе-
нилацетилена в этаноле. М. ф. выпадает в виде обильного жел-
того осадка, который отделяют и высушивают в роторном
испарителе.
HONHgC!-
Cun(NH3)^ -----Cu’(NH3)J
Cu\NH3g + C6H5feCH —C0HSC=C—Cu + NHt + NH3
Синтез 2-фенилфуро-[3,2-Ь] -пиридина [1].
Си
I
С
III
с
ед
Ру
кипячение
С6Н5 + Cui
В реакционную колбу помещают навеску М. ф., систему про-
дувают азотом, добавляют пиридин и затем 2-иодпиридинол-З.
Смесь, цвет которой изменяется от желтого до темно-зеленого,
нагревают на масляной бане при ПО—120° в течение 9 час при
перемешивании в атмосфере азота. Обычная обработка смеси
и очистка продукта реакции дают 75—82% 2-фенилфуро-[3,2-Ь]-
пиридина (2).
I. Owsley D. С., С a s t г о С. Е., Org. Syn., 52, 128 (1972).
317
МЕДНАЯ БРОНЗА (11,235).
Продажную М. б. активируют по методу Фогеля [1].
Реакция Ульмана. Реакцию Ульмана можно проводить в за-
паянной ампуле при 90—115° (12 час) без растворителя. Таким
способом был получен 2,2'-дипиримидил с выходом около
45% [2].
1. Vogel A. L., A Textbook of Practical Organic Chemistry, 3rd Ed., Wiley,
New York, 1957, 192.
2. Musgrave T. R., Westcott P. A., Org, Syn., submitted 1972.
МЕДНЫЙ ПОРОШОК (II, 236—237; V, 276—278;
VI, 163—165).
Реакция Ульмана. Коэи и Поэт [1] показали, что в реакции
Ульмана [2] промежуточно образуются, по-видимому, медьорга-
нические соединения, а не радикалы. Так, например, конденса-
ция эфира иодфумаровой кислоты (1) при 100° в присутствии
активированного [3] М. п. приводит к чистому транс,трансЛ,2,3,4-
тетракарбэтоксибутадиену-1,3 (2) с выходом 96%. В результате
конденсации эфира иодмалеиновой кислоты (3) образуется в
основном ццс,цис-1,2,3,4-тетракарбэтоксибутадиен-1,3 (4) и не-
большое количество соединения (2).
С2Н5ООС^ /I
с =с^
соосгн5
0)
Си
100°
96%
сгн5оос^ /Н
СгН5ООСх zC =с
zc=c \;ooc2hs
н СООС2Н5
(2)
С2Н5ООСх х,СООС2Н5
с=сх
1% I
Си
100°
8?%
С2Н5ООС
СгН5ООС
н
чс=с
.СООС2Н5
+ (2) 13%
СООСДД
Н
(з) (4) 87%
Если нагревать с медью при 75° 2 части эфира (3) и 1 часть
эфира (1), то продукт реакции представляет собой чистый
транс,транс-эфир (2). При нагревании эфиров (1) или (3) по
отдельности с медью и бензойной кислотой главным продуктом
реакции является диэтиловый эфир фумаровой или малеиновой
кислоты соответственно. Эти эфиры образуются с полным сохра-
нением конфигурации, вероятно, в результате протолиза проме-
жуточного медьорганического соединения.
318
а-Диазокетоны (V, 276—278; VI, 163—165). Ключевой ста-
дией полного синтеза сесквитерпена (±)-р-кубебена (4) являет-
ся внутримолекулярная циклизация и-диазокетона (1) [4]. Эту
реакцию осуществили кипячением диазокетона (1) в циклогек-
сане в присутствии сульфата меди (II) в течение 1,5 час; при
99% (с6н5)3р=сн2
(4)
этом образовывались два изомерных кетона (2) и (3) прибли-
зительно в отношении 3 :5. Полученный в меньшем количестве
кетон — норкетон (±)-|3-кубебена— превращали затем по реак-
ции Виттига в природный сесквитерпен (4).
1. С о h е n Т., Р о е t h Т., J. Am. Chem. Soc., 94, 4363 (1972).
2. Fanta P. E„ Chem. Rev., 64, 613 (1964).
3. Lewin A. H, Zovko M. J., Rosewater W. H., Cohen T., Chem.
Comm., 1967, 80.
4. Piers E., Britton R. W., de Waal W., Canad. J. Chem., 49, 12 (1971).
МЕДЬ БРОМИСТАЯ (II, 237—239; V, 278—280; VI, 165).
Реакции сопряженного присоединения. В присутствии М. б.
было осуществлено сопряженное присоединение фениллития к
З-метилциклопентен-2-ону (1). По-видимому, на самом деле
реагентом здесь является дифенилмедьлитий.
1. Wolff S-, Agosta W. С., J. С. Sf Chem. Comm., 1972, 226.
319
МЕДЬ(1) ТЕТРАПИРИДИНПЕРХЛОРАТ, Cu+ (Ру)4СЮ:.
Мол. вес 479,39.
Получение [1].
Разложение солей арилдиазония. Закись меди (II, 230—
231) —лучший катализатор гомолитического разложения солей
арилдиазония [2]. Однако ее недостаток в том, что она эффек-
тивна только в кислой среде. Левин и Михль [3] исследовали
эффективность различных комплексов перхлората меди(1) с ге-
тероциклическими аминами. Из всех солей меди(1) М. обладает
наивысшей эффективностью в нейтральной среде. В этой среде
эффективен также медь(1)-тр«с-(2-пиколин)перхлорат [4].
При разложении борфторидов бензофеион-2-диазония (1) в
нейтральной среде в присутствии этих комплексов Cu(I) в каче-
стве главных продуктов образуются 2,2/-дибензонлдифенил (2)
с выходом ~70% и 9*флуоренон (3) с выходом ~30%. Раз-
ложение соли диазония (1) в присутствии закиси меди при pH 1
дает в качестве главных продуктов бензофенон, 2-оксибензофе-
нон и 9-флуоренон.
1. Chen К. L., Twansato R. Т., Inorg. Nucl. Chem. Letters, 4, 499 (1968).
2. Lewin A. H., Dinwoodie A. H., Cohen T., Tetrahedron, 22, 1527
(1966)* Lewin A. H., Cohen T. J. Org. Chem., 32, 3844 (1967).
3. L e w i n A. H., Michl R. J., J. Org. Chem., 38, 1126 (1973)
4. L e w i n A. H., Michl R. J., Ganis P„ Lepore U., J. C, S. Chem. Comm.,
1972, 661.
МЕДЬ ХЛОРИСТАЯ (II, 241—246; V, 280—281;
VI, 165—167).
транс-Циклооктен [1]. £{«с-Циклооктен (1) при облучении
в присутствии М. х. частично изомеризуется в тралс-циклооктен
(2). При разделении этих изомеров действием водного нитрата
(О
hv, СигС1;
(2)
320
серебра [2] изомер (2) можно получить с выходом 19% с чи-
стотой >99%- Изомеризация протекает через комплекс Cu(I)-
олефин. Подобным же образом ^ис,цис-циклооктадиен-1,5 с вы-
ходом 30—40% можно превратить в цыс.транс-циклоокта-
диен-1,5. Бромистая медь гораздо менее эффективна, чем М. х.
Дегидроконденсация терминальных ацетиленов (II, 244—245)
[3]. Ямамото и Зондхеймер сообщили о синтезе из соединения
(1) тетраалкилированного тетрадегидро-[18]-аниулендиона (5),
представляющего интерес в качестве потенциального хинона.
Ацетилен (1) под действием ацетата меди(П) в пиридине (II,
228) вступает в реакцию дегидроконденсации, образуя «димер»
(2) [4]. Обработка этого димера этинилмагнийбромидом (IV,
268) приводит с высоким выходом к диолу (3), который окис-
ляют активной двуокисью марганца до дикетона (4). Полученный
дикетон нельзя ввести в реакцию дегидроконденсации с уча-
стием ацетата медн(П) в пиридине, так как в пиридине он раз-
лагается. Желаемую реакцию все же удалось осуществить, дей-
ствуя на дикетон (4) кислородом в присутствии Ай. х. и хлори-
стого аммония в водном этаноле и бензоле. Соединение (5)
было получено с выходом 25%; при этом вернулось 50% непро-
реагировавшего дикетона (4).
1. Deyrup J. A., Betkouski М., J. Org. Chem., 37, 3561 (1972).
2. С о р е А. С., Bach R. D., Org. Syn., 49, 39 (1969).
1 I Зак, 596
321
3. Yamamoto K-, Sondheimer F., Angew. Chem., Internal. Ed., 12, 68
(1973).
4. M c G i г к R. H., Sondheimer F., Angew. Chem., Internal, Ed., 11, 834
(1972).
МЕДЬ ХЛОРНАЯ (II, 246; V, 281—284; VI, 167).
Арилиодиды [1]. Арилиодиды можно получать с умеренными
и даже хорошими выходами, действуя на ароматические угле-
водороды иодом в присутствии соли меди. Реакционноспособ-
ность различных солей меди изменяется в следующем порядке:
CuCl2 > CuFs > CuCl > Си(ООССНз)2. Реакция, по-видимому,
включает две последовательные стадии:
C11CI2
АгН 4- F---Ari 4- HI
ArH 4-HI 4-2CuCI2 —> ArI4-2HCI4-2CuCI
Общая стехиометрия реакции представлена уравнением (1).
Иногда (например, в случае бензола, алкилбензолов) в качест-
ве источника иода используют более активные в этой реакции,
чем сам иод, иодиды некоторых металлов, такие, как иодид
алюминия, иодид железа (П) или иодид меди(1).
1) 2АгН 4-12 4-2CuCI2 —> 2ArI 4-2HCI 4-2CuCl
Полихлорированные циклогександионы [2]. Циклогексанон
и его метильные, гомологи при кипячении с большим избытком
СиС12-2Н2О в 50%-ной уксусной кислоте или 50%-ном диоксане
(2 час) образуют дихлор- или трихлорпроизводные циклогексан-
днона-1,2 с выходом 60—70%,
Реакция окислительного сужения кольца [3]. 2-Окси-3,6-ди-
трег-бутил-1,4-бензохинон (1) при обработке СиС12-6Н2О в го-
рячей уксусной кислоте дает 2-хлор-2,4-ди-тре7,-бутилциклопен-
тендион-1,3 (2):
(1)
СиС1г>
5 5%
(2)
р-Хлорвинилсульфоиы. Соли меди(1) или меди(И) катализи-
руют присоединение сульфохлоридов к ацетиленам с образова-
нием р-хлорвинилсульфонов [4]. Без катализатора реакция не
322
идет. В качестве растворителя используют ацетонитрил или хло-
ристый метилен. Для повышения растворимости медных солей
добавляют небольшое количество четвертичной аммониевой со-
ли (например, хлорида тетраэтиламмония). Реакцию проводят
при кипячении с обратным холодильником или лучше в запаян-
ной ампуле. Присоединение идет стереоселективно: например,
реакция бензолсульфохлорида (2) с фенилацетиленом (1) при-
водит к смеси двух изомерных 2-бензолсульфонил-1 -хлорстиро-
лов (3) и (4). В больших количествах образуется продукт
традс-пр и соединен ия (3), а в меньших — продукт г^ас-присоеди-
нения (4). Последний может принимать копланарную конфор-
мацию, в то время как для продукта транс-присоединения ко-
планарность недостижима из-за наличия в молекуле простран-
ственных затруднений. Присоединение протекает по механизму
свободнорадикальной цепной реакции, в которой медный ката-
лизатор выступает в роли переносчика хлора:
RSO2C1 —> RSO2.
RSO. • + HCssCR' —> RSO2CH=CR'
RSO3CH=CR' + CuCl2 —> RSO2CH=CC1R' + CuCI
CuCl + RSO2CI —CuC12+RSO2.
Соотношение ^нс,транс-изомеров можно изменить, варьируя
условия эксперимента. Избыток хлорид-иона или сильно поляр-
ные растворители способствуют транс-присоединению; раствори-
тели же низкой полярности (например, сероуглерод) благопри-
ятствуют б{ас-присоединению.
1. В a ir d W. С., J г., S и г г i d g е J. Н, J. Org. Chem., 35, 3436 (1970).
2. S a t о h J. Y, Nisbiza wa K„ J. C. S. Chem. Comm., 1973, 83.
3. M oo r e H. W., W ikho1m R. J., J. C. S. Chem. Comm., 1971, 1073.
4. Amiel Y., J. Org. Chem., 36, 3691 (1971).
5. Amiel Y., J. Org. Chem, 36, 3697 (1971).
МЕТАНСУЛЬФОКИСЛОТА (11,248—249; V, 286).
Расщепление эпоксида. На первой стадии синтеза формил-
циклобутана (4) (1] к раствору 48,0 г (0,50 моля) М. (т. кип.
14070,2 мм) в 500 мл безводного эфира при перемешивании ме-
ханической мешалкой и при охлаждении на ледяной бане в те-
чение 3—4 час добавляют раствор 37 а окиси транс-бутена-2
II*
323
в 500 мл безводного эфира. Через 6 час охлаждение прекра-
щают, а смесь продолжают перемешивать еще в течение 12 час.
Эфир упаривают на роторном испарителе с водоструйным на-
сосом при 25°; остаток—мономезилат эратро-бутандиола-2,3 (1)
(83—84 г, 99—100%) представляет собой прозрачную, бесцвет-
ную немного вязкую жидкость.
1. Jоh п sо n М. R., R i с k b о г п В., Org. Syn., 61, Il (1971).
МЕТАНСУЛЬФОХЛОРИД (мезилхлорид) (II, 250—253;
V, 286—287).
Эфиры метансульфокислоты (мезилаты) [1]. Мезилаты мож-
но получить с выходом 85—95%, прибавляя при 0—10° в тече-
ние 5—10 мин взятый с 10 %'Ним избытком М. к раствору спирта
в хлористом метилене (либо в циклогексане или пентане), со-
держащему 50%-ный мольный избыток триэтиламина. Реакция
протекает, вероятно, через стадию сульфена СН2 —SO2, обра-
зующегося в результате дегидрохлорирования М. Таким спосо-
бом можно получать мезилаты третичных и неопентиловых
спиртов. Если проводить реакцию, а также обработку реакцион-
ной смеси при температурах не выше 0°, то можно получить да-
же реакционноспособные мезилаты.
Мезилаты сольволизуются труднее соответствующих тозила-
тов. Они удобнее тозилатов (II, 387—388) и при восстановлении
алюмогидридом лития, поскольку мезилатный фрагмент восста-
навливается до метилмеркаптана, который легко удалить.
Элиминирование виц-1- и -ОН-групп [2]. Обработка иодлакто-
на (1) 1,3же М. в сухом пиридине при —20° в течение 2 час и
затем при 0° в течение 1,5 час приводит с почти количественным
выходом к ненасыщенному лактону (2). Эта реакция является
ключевой стадией синтеза простагландинов А, которые ранее
получали из первичных простагландинов Е дегидратацией фраг-
мента {3-кетола молекулы простагландина.
324
Новая реакция элиминирования, по-видимому, родственна
методу превращения эпоксидов в олефины по Корнфорзу (III,
29—30; IV, 89—90, 220).
снгоснгс6н5
Краббе и Гузман осуществили аналогичную реакцию элими-
нирования с помощью хлорокиси фосфора в пиридине при ком-
натной температуре (см. Фосфора хлорокись, этот том).
Аллилхлориды [3]. Аллиловые спирты (0,10 моля) легко
превратить в аллилхлориды, действуя на них М. (0,11 моля) и
смесью хлорида лития (0,10 моля), сухого ДМФА и симм-кол-
лндина (0,11 моля) при 0°. Если в молекуле есть гидроксильные
группы не в аллильном положении, то они превращаются в ме-
зилаты.
си2 /
сн,
сн/ СН2
С = СНСН2ОН
CH3SO2C1
ЫСЦДМФД
Ноллидин
“ 83^ ’
сиЧсн^снг
/CHW
сн/ СНг
С = СНСН2С1
1. Crossland R. К., Servis К. L., J. Org. Chem., 35, 3195 (1970).
2. С о г е у Е. J., G г 1 е с о Р. A., Tetrahedron Letters, 107 (1972).
3. CoHlngton Е. W., Myers A. I., J. Org. Chem., 36, 3044 (1971)
МЕТИЛАТ НАТРИЯ (II, 256—262; V, 289).
Перегруппировка Фаворского (2)->(3) [1].
Br2
бензол, эфир'
( СНВг
> (СНг), 6 = 0
\ СНВг
NaOCH3
ад
(О (2)
210 г (1 моль) циклододеканона превращают в а,а'-дибро-
мид (2) в смеси бензола с эфиром при 20—25°, бромистый во-
дород и эфир отсасывают с помощью водоструйного насоса, а
оставшийся бензольный раствор 2,12-дибром циклододеканона
(2) обрабатывают при перемешивании в течение 30—40 мин
125 г (2,3 моля) порошкообразного М. н. Последний вносят пор-
325
циями через центральное горло колбы, поддерживая при этом
температуру реакционной смеси в пределах 25—30° добавле-
нием льда в водяную баню. Реакционную смесь перемешивают
еще 20 мин и затем экстрагируют последовательно 500-милли-
литровыми порциями воды, 5%-ной соляной кислоты и конц.
раствора хлористого натрия. Водные фазы экстрагируют 400 мл
диэтилового эфира и отбрасывают их. Органические фазы филь-
труют через безводный сульфат натрия и из объединенного
фильтрата отгоняют растворители при пониженном давлении.
Остаток перегоняют в вакууме с короткой колонкой Вигре, со-
бирая фракцию, кипящую ниже 104°/0,4 мм. При этом получают
191 —196 г (91—93%) метилового эфира циклоундецен-1-карбо-
новой кислоты в виде бледно-желтого масла, большая часть ко-
торого перегоняется при 83—8770,4 мм.
Синтез алкиларилсульфидов [2]. Для получения «-бутил-1-
нафтилсульфида в литровую трехгорлую круглодонную колбу,
снабженную термометром, обратным холодильником, капельной
воронкой и магнитной мешалкой, помещают 586,7 г (7,5 моля)
диметилсульфоксида и 225,5 г (2,4 моля) бутилмеркаптана. Рас-
твор при перемешивании нагревают с помощью колбонагрева-
теля до 70° и медленно прибавляют 108 г (2 моля) М. н. После
этого реакционную смесь нагревают до температуры кипения
(115°). Через капельную воронку быстро вливают 81,3 г 1-хлор-
нафталина (0,5 моля) и кипятят желтую реакционную смесь
при перемешивании в течение 45 час. Приблизительно через
8 час смесь приобретает коричневый цвет, на внутренних стен-
ках колбы оседает коричневое твердое вещество, а температура
кипения падает до 98°.
Затем реакционную смесь выливают в 500 мл холодной во-
ды, насыщенной хлористым натрием. Продукт реакции экстра-
гируют эфиром и перегоняют, получая 75—86 г (70—80%) н-бу-
тил-1-нафтилсульфида с т. кип. 115—130°/1 мм и немного побоч-
ного ди-«-бутилдисульфида.
1,2-Окись нафталина. Яги и Джерина [3] предложили новый
метод синтеза окисей аренов. Исходным веществом для синте-
за 1,2-окиси нафталина (5) является 1-окси-2-бромтетралин (1),
который ацилируют ангидридом трифторуксусной кислоты в
хлороформе, получая ацильное производное (2) с выходом 84%.
Последнее превращают в дибромид (3), действуя NEC (следует
отметить, что 1,2-эпокись тетралина неустойчива к бромирова-
нию NEC). На следующей стадии защитную группу удаляют,
326
обрабатывая дибромид (3) водным диэтиламином в ацетонит-
риле. При взаимодействии бромгидрина (4) с твердым М. н. в
ТГФ в одну стадию происходит замыкание эпоксидного кольца
и дегидробромирование до 1,2-окиси нафталина (5). Общий вы-
ход последней, считая на исходный тетралин (1), составляет
65%. Ранее соединение (5) синтезировали из тетралина (1)
иным путем с общим выходом 14% [4].
Аналогичную последовательность реакций использовали для
получения лабильной 3,4-окиси фенантрена, за тем лишь исклю-
чением, что в этом случае вместо М. н. применяли 1,5-диазаби-
цикло-[4,3,0]-нонен-5 (этот том).
1. Wohllebe J., Garbisch Е. W., Jr., Org. Syn., submitted 1971.
2. Bradshaw J. S., Chen E. Y., procedure submitted to Org. Syn., 1972.
3. Y a gi H, Jerina D. M., J. Am. Chem. Soc., 95, 243 (1973).
4. Vogel E., ftlarner F. G„ Angew. Chem., Internat. Ed., 7, 374 (1968).
МЕТИЛ-трет-БУТИЛОВЫЙ ЭФИР, CH3OC(CH3)3. Мол. вес.
88,15, т. кип. 53—56°.
Реакция этерификации. Карбоновые кислоты реагируют с
алкил-трет-бутиловыми эфирами в присутствии каталитических
количеств серной или я-толуолсульфокислоты с образованием
сложных эфиров [1]. Выходы, как правило, достаточно высокие.
Реакцию проводят кипячением реагентов с обратным холодиль-
ником или нагреванием их на кипящей водяной бане в течение
нескольких минут до прекращения выделения изобутилена.
н*
ROC(CH3)3 + R'COOH ---> R'COOR + СН2=С (СН3)г + Н2О
Этот метод был использован также для получения алкилгли-
козидов [2].
I Derevitskaya V. A., Klimov Е. М., Kochetkov N. К., Tetrahedron
Letters, 4269 (1970).
2. Кочетков Н. К., Климов Е, М., Деревицкая В, А., ДАН СССР,
192, 336 (1970).
327
бис- (3-МЕТИЛ-5-трет-БУТИЛ-4-ОКСИ)-ДИФЕН ИЛ СУЛЬ-
ФИД («этил» антиоксидант 736). Мол. вес 358,5 т. пл. 127°,
т. кип. 312°/40 мм.
Растворимость при 20°, вес. %
Этанол............. 39 Вода.............нерастворим
Толуол........... 8,8
Изопентан .... 0,2 10%-ный NaOH . <0,002
Н3С
НО—
(Н3С)3С
СНз
—он
С(СН3)
Ингибитор окисления натурального и синтетических каучу-
ков, полиолефиновых пластмасс, смол, клеев, нефтетоплива и
пластиков.
МЕТИЛГИДРАЗИН, H2NNHCH3. Мол. вес 46,07, т. кип.
87°, 1,4325.
А2-Пиразолиноны-5 Г1]. Взаимодействие 2-бром-1,3-дифенил-
пропандиона-1,3 (1) с М. в этаноле при комнатной температуре
приводит с выходом 56% к 1-метил-3,4-дифенил-Д2-пиразолино-
ну-5 (2). Последний образуется, вероятно, в результате пере-
группировки галогенгидрина типа пинаколиновой, как показано
на приведенной схеме;
(б) (2)
1. N у е М. J., Tang W. Р., Can. J. Chem., 51, 338 (1973).
328
О-МЕТИЛДИБЕНЗОФУРАНИЯ БОРФТОРИД (2). Мол.
вес. 270,04.
М. б. получают нагреванием соли диазония (1) в безводном
бензоле, Он представляет собой неустойчивое твердое вещество
чистого белого цвета. Предшественник М. б. (1) получают вос-
становлением 2-метокси-2/-ннтродифенила гидразингидратом в
присутствии палладия до 2-амино-2/-метоксидифенила, который
затем диазотируют нитритом натрия в присутствии водной бор-
фтористоводородной кислоты до соли диазония (1). Реагент
можно также генерировать in situ нагреванием (1) в хлори-
стом метилене [1].
Метилирование [1]. Эта новая соль борфтористоводородной
кислоты является эффективным метилирующим агентом, срав-
нимым по силе с метиловым эфиром фторсульфоновой кислоты
(VI, 321; этот том).
1. Сор son A. J., Heaney Н,, Logon Л. A., Sharma R. Р., Chem.
Comm., 1972, 315.
МЕТИЛЕН ХЛОРИСТЫЙ (И, 277; V, 293).
Синтез p-тетралонов. Буркхальтер и Кэмпбелл [1] получали
p-тетралоны, барботируя этилен в течение нескольких часов при
0° через раствор хлорангидридов фенилуксусных кислот в серо-
углероде в присутствии хлористого алюминия. Образующиеся
p-хлорэтилкетоны не выделяя превращали обработкой соляной
кислотой в р-тетралоны.
Симс н сотр. [2] сообщают, что этот метод можно существен-
но улучшить, применяя вместо сероуглерода М. х., выходы в
этом случае значительно возрастают, а для завершения реакции
требуются минуты, а не несколько часов. Улучшение результа-
тов при замене растворителя объясняется, вероятно, тем, что
329
М, х. растворяет комплекс хлористый ацил — хлористый алюми-
ний, и, таким образом, реакция протекает в гомогенной среде.
I. Bure k halter J. Н., Campbell J. R., J, Org. Chem., 26, 4232 (1961).
2. S i m s J. J., Cadogan M.., Selman L. H., Tetrahedron Letters, 951
(1971).
МЕТИЛЕН ХЛОРИСТЫЙ-w-БУТИЛЛИТИЙ.
Синтез углеводородов с помощью карбеноидов. Кац и сотр.
[I, 2] предложили простой метод синтеза из циклопентадиена
(1) ранее труднодоступного валентного изомера бензола беиз-
валена (3) (это вещество ранее получали в очень малых коли-
чествах фотолизом бензола [3]). Сначала диен превращают в
циклопентадиенил-анион (2) взаимодействием с метиллитием
в диметиловом эфире, затем при —45° прибавляют раствор хло-
ристого метилена и метиллития в диэтиловом эфире. Бензвален
(3) был получен с выходом 24% совместно с небольшим коли-
чеством (6,4%) бензола.
Внимание^. Бензвален легко взрывается и обладает отврати-
тельным запахом.
Подобным же образом получен соответствующий валентный
изомер нафталина, бензобензвален или нафтвален, (4) с при-
месью нафталина (5), однако в этом случае изомеры (4) и (5)
разделить не удалось.
СН2СК
«-C4H9Li
(4) ю%
(5) 18%
Недавно из двух лабораторий [4, 5] поступили сообщения об
аналогичном синтезе изобуллвалена, трицикло-[5,3,0,02>!0]-дека-
гриена-3,5,8, (7) из циклононатетраенида лития (6) и М. х. — б.
при низкой температуре. Наряду с основным продуктом реакции
изобуллваленом (7) побочно образуются также [10]-аннулен (8)
и цис-9,10-дигидронафталин (9). Аннулен (8) при температуре
ззо
около —20° изомеризуется в траяс-9,10-дигидронафталин (10).
(ю)
Две лаборатории [5, 6] одновременно сообщили о синтезе ва-
лентного изомера плейадиена (14) нафто-[1,8]-трицикло-
[4,1,0Д2,1]’Гептена (13) реакцией литиевого производного фена-
лена (И) с М.х.—б. при низких температурах. В результате
реакции получается смесь соединений (13) и (14) в отношении
4:1. Новый углеводород (13), т, пл. 76—78°, необыкновенно
устойчив, но при нагревании до 150° в циклогексане он превра-
щается в уже известный нафтоциклобутен (15). Под действием
иона Ag(I) или при облучении (2537 А) углеводород (13) изо-
меризуется в плейадиен (14).
I. Katz Т. J., Wang Е. J., Acton N., J. Am. Chem. Soc., 93, 3782 (1971).
2. Katz T. J., Cheung J. J., Acton N„ J. Am. Chem. Soc., 92, 6643 (1970).
331
3. W i 1 z b a c h K- E., R i t s c h e r J, S., Kaplan L., J. Am. Chem. Soc., 89,
1031 (1967); Kaplan L., W i 1 z b a c h K- E„ J. Am. Chem. Soc.., 90, 3291
(1968); Ward H. R., Wishnok J. S., J. Am. Chem. Soc., 90, 1085 (1968).
4. H о j о Km Sei drier R. T., Masamune S., J. Am. Chem. Soc., 92, 6641
(1970).
5. Mura t a I., N akasu ji K-, Tetrahedron Letters, 47 (1973).
6. P a g n i R. M„ Watson C. R., Tetrahedron Letters, 59 (1973).
МЕТИЛ ИЗОЦИАНАТ, CH3N = C = О. Мол. вес 57,05,
т. пл. 37—39°.
Превращение альдоксимов в нитрилы [1]. В результате де-
гидратации альдоксимов под действием М. в ДМФА в присут-
ствии трнэтнламина образуется О-(метилкарбамоил)-альдоксим.
Этот карбамат разлагается при 110—120°, давая нитрил, дву-
окись углерода и метиламин. Выходы нитрилов составляют 65—
99%.
ДМФА
(C2H5)3N
RCH=NOH + CH3N=C=O —1-------►
О
RCH=N—OCNHCH
110—120°
-------> RCN + СО2 + CH3NH2
1. Albright J. A., Alexander M. L., Org. Prep. Proc. Int„ 4, 215 (1972).
МЕТИЛ ИОДИСТЫЙ (11, 278—281; V, 293—294; VI, 169).
Тиоацетали->карбонильные соединения. Гидролиз тиоацета-
лей до исходных карбонильных соединений нередко сопряжен
с определенными трудностями. Фетизон и Джурион [1] описали
два новых метода для осуществления этого превращения. Со-
гласно одному из них, раствор тиоацеталя во влажном ацетоне
кипятят в течение нескольких часов с избытком М.и. В случае
соединений, чувствительных к кислотам, целесообразно добав-
лять в реакционную смесь порошкообразный карбонат натрия,
кальция или бария для нейтрализации образующейся HI.
Согласно второму методу, раствор тиоацеталя в жидком SOa
обрабатывают избытком метилового эфира фтор сульфоновой
кислоты (VI, 321; этот том).
1. F е 11 z о n М., J и г i о n М., Chem. Comm., 1972, 382.
МЕТИЛМЕДЬ, СНзСи. Мол. вес 102,59.
Получение [ 1]. М. можно получить из иодида меди(I)
(4,4 л/моля) и метиллития (4 лгмоля) в ТГФ (40 мл) при —20°.
332
Реакция с хлорангидридами пространственно затрудненных
карбоновых кислот. Израильские химики [1] нашли, что хлор-
ангидрид пространственно затрудненной З-О-ацетилглициррето-
вой кислоты (1) легко реагирует с М., образуя с выходом 87%
метилкетон (2). Реакция хлорангидрида (1) с метилмагнийбро-
мидом при 0° дает кетон (2) лишь с выходом 25%, а выходы
кетона в реакции с диметилкадмием еще ниже. В результате
реакции с диметилмедьлитием получается около 30—40% ке-
тона (2). М. гладко реагирует также с хлорангидридом 1-метил-
циклогексанкарбоновой кислоты (3) с образованием соответ-
ствующего метилкетона (4).
(з)
(4)
I. Rozen S., Shahak I., Bergmann E. D., Synthesis, 1971, 646.
МЕТИЛ-(МЕТИЛТИОМЕТИЛ)-СУЛЬФОКСИД,
CH3SOCH2SCH3. Мол. вес 125, 23, т. кип. 92—93°/2,5 мм.
Получение. М. получают с выходом 78% окислением диметил-
меркапталя формальдегида 30%-ной перекисью водорода в ук-
сусной кислоте [1].
Синтез альдегидов [2]. Генерированный из М. (1) карбанион
при взаимодействии с галогеналкилом образует S-окись диме-
тилмеркапталя альдегида (2). Это соединение легко гидроли-
зуется в присутствии каталитического количества серной кис-
лоты до соответствующего альдегида (3) и днметилдисульфида.
Выходы альдегида составляют 80—90%. Чувствительные к кис-
ZSOCH3 +
NaH, ТГФ / i Н+
CH3SOCH2SCH3 + RX ------> RCH —> RCHO + CH3SSCH3
^SCHg
(1) 12) (3)
333
лоте альдегиды можно выделить сразу в виде ацеталей, обраба-
тывая соединение (2) серной кислотой в этаноле в присутствии
трнэтилового эфира ортомуравьиной кислоты.
Производные фенилуксусной кислоты [3]. При конденсации
М. с бензальдегидом по реакции типа Кневенагеля (тритон Б)
образуется 1-метилсульфииил-1-метилтио-2-фенилэтилен (2). В
результате обработки соединения (2) соляной кислотой в эта-
ноле при комнатной температуре с выходом 78% получается
этиловый эфир фенилуксусной кислоты (3).
С6Н5СНО + CH3SOCH2SCH3
ТГФ
Тритон в
-------->
C6HSy ZSCH3
\¥ ^SOCH3
н+, с2н5он
78%
U)
(2)
—> С6Н5СН2СООС2Н5
(3)
«-Оксиальдегиды [4]. Литиевое производное М. гладко реа-
гирует с кетоном (1), образуя S-окись диметилмеркапталя
а-оксиальдегида (2), которая при обработке концентрирован-
ной соляной кислотой в ТГФ при комнатной температуре дает
а-оксиальдегид (3). В случае бензофенона (R = R' = CgHg)
D /SOCH3 V=o + СН2 ^sch. 1) H-BuLi 9H уЗОСНз + ?Н 2) Н2О 1 / 3 Н+ I > R—С—СН > R— С—СНО 4 Чен, 4
(1) (2) (3)
выход дифенилгликолевого альдегида (3) составляет 59%.
Если в соединении (2) R'= Н, то последняя операция при-
водит к изомеризации. Так, например, S-окись диметилмеркап-
таля фенилгликолевого альдегида (2а) превращается в ы-окси-
ацетофенон (4). В этом случае производное желаемого а-окси-
альдегида можно получить или защитив гидроксильную груп-
пу в (2а) превращением ее в простой эфир, или защищая вновь
°Н ySOCH, н.
с6н5с—сн —> С6Н5СОСН2ОН
i \sch.
11
(2а1
(4)
образующуюся формильную группу в виде диалкилацеталя.
334
1. Ogura K„ Tsuchihashi G.-L, Bull. Chem. Soc. Japan, 45, 2203 (1972).
2. Ogura K., Tsuchihashi G.-L, Tetrahedron Letters, 3151 (1971).
3. Ogura K., Tsuchihashi G.-L, Tetrahedron Letters. 1383 (1972).
4. Ogura !<, Tsuchihashi G.-L, Tetrahedron Letters, 2681 (1972).
2-МЕТИЛ-3-ТИА30ЛИН.
Мол. вес 101,17,
пъ 1,5200.
Синтез альдегидов. Мейерс и сотр. [1] описали метод синтеза
альдегидов, аналогичный методу с применением 2,4-диметилти-
азола (этот том). Первый метод иллюстрируется на примере
синтеза 2-(1-оксициклогексил)-уксусного альдегида (5). Тиазо-
лин (1) обрабатывают в ТГФ в атмосфере азота при —78° не-
большим избытком н-бутиллития. При взаимодействии обра-
зовавшегося литиевого производного тиазолина (2) с циклогек-
саноном сначала при —78°, а затем при комнатной температуре
получается замещенный тиазолин (3), который восстанавли-
вают амальгамой алюминия до тиазолидина (4). Сырой про-
дукт затем расщепляют до р-оксиальдегида (5), действуя хлор-
ной ртутью в водном ацетонитриле при комнатной температуре.
Следует отметить, что промежуточные тиазолины (3) можно
обработать еще и другим электрофильным агентом. Основное
преимущество рассмотренного метода заключается в том, что
все операции проводятся в нейтральной среде.
I. Meyers A. L, Munavu Durandetta J., Tetrahedron Letters, 3929
(1972).
1,3-бис-(МЕТИЛТИО)-АЛЛ ИЛЛ ИТИЙ, сщ5^Ч^зсн3ы +
Мол. вес. 142,21.
Получение [1]. М. получают из эпихлоргидрина глицерина
в ТГФ, как показано на приведенной схеме. Темно-пурпурный
раствор устойчив в течение 12 час при 0°.
335
OH I) NaH
CH2-CHCHjCI cHsS^A^SCH; _2LCbV >
\/ S4$ 90$
ОСИ, 2UN(m-C3H,)2
--^CHjS^/k^/SCHj------- > CH^S^O^SCHjL?
(1)
аф-Неиасыщенные альдегиды [1]. M. (1) выступает в каче-
стве нуклеофильного эквивалента группировки —CH —СНСНО.
Так, например, при взаимодействии соединения (1) с 1-бром-
пентаном при —75° в течение 2 час образуется с выходом 90%
1,3-бас-(метилтио)-октен-1 (2). Этот бас-тиоэфир в водном аце-
тонитриле в присутствии хлорной ртути гидролизуется до транс-
октеналя (3) с выходом 84%.
HgC!2 i
н-СэНнВг + (1) я-С5НиСНСН=!СН5СН3 /^С5НнСН=СНСНО
SCH3
(2)
(3)
В результате реакции соединения (1) с альдегидами или ке-
тонами с последующим гидролизом образуются у-окси-а,р-нена-
сыщенные альдегиды. Например, при добавлении соединения (1)
к пропионовому альдегиду образуется 1,3-бас-(метилтио)-гек-
сен-1-ол-4 (4) с выходом 89%, гидролиз которого, катализируе-
мый хлорной ртутью, позволяет получить с 48%-ным выходом
транс-4-оксигексен-2-аль (5).
HgC!2
СНзСН.СНО + (1) --> СН3СН2СН—CHCH=CHSCH3
89% । । 48%
ОН S СНз
(4)
—> СН3СН2СНСН=СНСНО
%
(5)
Реакция соединения (1) с окисью циклогексена приводит к
транс-замещенному циклогексанолу (6), При ацетилировании
последнего с последующим гидролизом, промотируемым ионом
ртути (II), образуется ацетоксиальдегид (7) с общим выходом
82%, считая на окнсь циклогексена.
sen
11 Ае2О, Ру
2) HgCl2j GaCOj
82$ (общий)
(И
(6)
336
М. (I) использовали также в ключевой стадии полного син-
теза простагландина [2]. А именно, обработка эпоксида аце-
таля (8) реагентом (1) в атмосфере аргона при —78° приводила
к образованию смеси требуемого продукта конденсации (9) и
его изомера (10). Смесь изомеров не разделяя гидролизовали
водным карбонатом кальция в присутствии хлорной ртути при
50° под аргоном. Два образовавшихся ненасыщенных альдегида
(11) и (12) разделяли методом препаративной тонкослойной
хроматографии. Требуемый оксиальдегид (И) был получен с
выходом 30%, выход изомерного альдегида составил 40%.Окси-
альдегид (11) превращали в несколько стадий в (Д-простаглан-
дин F2(Z (13).
Насколько стадий
HQ
НО' ОН
Простагландин
Такой путь синтеза простагландина (13) привлекает про-
стотой и изяществом; к сожалению, его ценность умаляется тем
337
обстоятельством, что при реакции окиси (8) с М. (1) раскрытие
ее цикла происходит в обоих возможных направлениях.
1. Corey Е. J., Erickson В. W., Noyori R., J. Am, Chem. Soc., 93, 1724
(1971).
2. Corey E. J., Noyori R., Tetrahedron Letters, 311 (1970).
N-МЕТИЛ-М-ТОЗИЛПИРРОЛИДИНИЯ ПЕРХЛОРАТ,
C^'n^^cio/ Мол. вес 326,79, т. пл. 148 — 150°.
XTs
М. п. получают с выходом 67,7% реакцией N-метилпирроли-
дина с тозилхлоридом в присутствии перхлората серебра в тем-
ноте [1].
Избирательное тозилирование [1]. Реагент селективно тозили-
рует аминогруппу в присутствии гидроксильной группы:
сю4
>NH
СНгС12
> NTs +
1. Oishi Т., К a in at а К., Rosuda S., Ban Y., Chem. Comm., 1972, 1148.
бис-(тра«с-2-МЕТИЛЦИКЛОГЕКСИЛ-1)-БОРАН,
J Мол. вес. 204,16.
'М. получают взаимодействием 1-метилциклогексена с дибо-
раном в ТГФ [1].
Гидроборирование — иодирование алкинов [2]. Методом гид-
роборирования — иодирования ’ алкинов можно стереоселективно
ввести в циклические системы боковую цепь с двойной связью.
Например, при обработке винилборана (2), полученного из гек-
сина-1 и М. (1), сначала 6 н. раствором гидроокиси натрия, а
затем раствором иода в ТГФ [3] с выходом 85% образуется
транс-1 -метил-2- (ty/zc-1-гексенил)-циклогексан (3). Таким обра-
(з)
зом, перенос 2-метилциклогексильной группы происходит с со-
хранением конфигурации этого фрагмента.
338
В эту реакцию могут вступать любые замещенные цикличе-
ские олефины при условии, что гидроборирование можно пре-
рвать на стадии диалкилборана.
L Brown Н. С,, Zweifel G., J. Am. Chem. Soc., 83, 2544 (1961).
2. Zweifel G., Fisher R. P., Snow J. T., Whitney С. C., J. Am. Chem.
Soc., 93, 6309 (1971).
3. Zweifel G., Arzoumanian H., Whitney С, C., J. Am. Chem. Soc.,
89, 3652 (1967).
OCH3
2-МЕТОКСИАЛЛИЛ БРОМИСТЫЙ, ,c Мол.
HjC CHjBr.
вес 135,04, разлагается прн комнатной температуре.
Получение [1]. М. б. синтезируют двумя различными путями.
Его можно получить с выходом около 50% реакцией 2-метокси-
пропена [2] с NEC в CCI4 (метод А) или пиролизом 1-бром-2,2-
диметоксипропана (который в свою очередь получают из бром-
ацетона). Реагент (в чистом виде) разлагается при комнатной
температуре и его лучше всего хранить в виде 50%-ного раство-
ра в СС14 в темноте. В лабораторной практике предпочтителен
- осн,
1
,.с
Н2С^ ^"CHj
СС14
55-65'
54%
осщ
,с.
^СНгЕг
ОСЩ ОСН3
I >150° ।
Н3 С О — С—С Н3----> хс
> 22% H,CZ CH,Br
метод А, метод Б применим для синтеза реагента в больших
масштабах.
Реакции циклоприсоединения [3]. Обработка М. б. трифтор-
ацетатом серебра (III, 434—435) в тщательно контролируемых
условиях позволяет генерировать 2-метоксиаллил-катион (а),
Щу— осн3
(а)
который вступает в реакцию циклоприсоединения с олефинами.
Например, раствор бромида в бензоле смешивают с раствором
фурана (1) в изопентане в присутствии суспензии твердого
NagCOs (буфер). Затем маленькими порциями при комнатной
339
температуре в темноте в течение 10 час добавляют трифтораце-
тат серебра и На2СОз, перемешивают образующуюся суспензию
с помощью эффективной вибромешалки (частота 100 гц), после
этого осторожно добавляют азотную кислоту и отфильтровы-
вают темный осадок, содержащий соединения серебра. Выход
8-оксабицикло-[3,2,1]-октен-6-она-3 (2) составляет 15%. Цикло-
пентадиен в аналогичных условиях дает с выходом 17% бицик-
ло-[3,2,1]-октен-6-он-3.
Н,С CHj
(3)
осн,
.С
ХСН2Вг
Реакция с 6,6-диметилфульвеном (3) приводит к образова-
нию 8-изопропилиденбицикло-[3,2,1]-октен-6-она-3 (4) с выхо-
дом 1%.
I. Greenwood G., Hoffman Н. М. R., J. Org. Chem., 37, 611 (1972).
2. S а не у G„М а гb еt R., Helv, Chim. Acta, 50, 1158 (1967).
3. Hill A. E., Greenwood G., Hoffmann H. M. R., J. Am. Chem. Soc.,
95, 1338 (1973).
а-МЕТОКСИБЕНЗИЛИДЕНВОЛЬФРАМ(О) ПЕНТАКАРБО-
НИЛ (1). Мол. вес 441,1, т. пл. 59°.
Получение [1].
I) CflHfjU
2) (CH3)4NOH
W(CO)6 -------—> [N(CH3)4]+ [W(CO)6COC6H6]-
/Сен5
—> (OC)6W=c(
^ОСНз
(1)
H+; CH2N2
зш
Виниловые эфиры [2]. Этот устойчивый металл-карбеновый
комплекс (1) взаимодействует при комнатной температуре с ре-
активами Виттига, образуя с высокими выходами виниловые
340
эфиры (2) и трифенилфосфинвольфрам (0) пентакарбонил (3).
С6Н5
еды и Из со 1 .W(CO)5
С—W(CO)5 + С=Р(С6Н5)3 >
HjCCT R|X 1
И [ Р(с6н5)з
(1) И'
НзССк
;с=с( + (c6h6)3pw(co)5
с0н/
(2) (3)
I. Fischer Е. О,, Maasbol A., Angew. Chem., Internal. Ed., 3, 580 (1964);
Chem. Ber., 100, 2445 (1967),
2. Casey С. P., Burkhardt T. J., J. Am. Chem. Soc,, 94, 6543 (1972).
CH3
3-МЕТОКСИ ИЗОПРЕН, нгс^%^осн3 . (VI, 202—203),
11
СНг
Мол. вес 146,18, т. кип. 93—95°.
Получение [1]. М. (1) получают присоединением метанола
к 2-метилбутен-1-ину-3 в присутствии катализатора, приготов-
ленного из красной окиси ртути, трихлоруксусной кислоты, эфи-
рата трифторида бора и метанола:
сн
нгс^
Катализатор
+ CHjOH --------------»
НгС'
СН3 СН5
xOCHj + НзСО^ ^.оси
н,с '
ьгосн
6н3ОСН'
12%
45%
77%
РУ
СИ,
хосн3
II
снг
траноТризамещеиные олефины (VI, 202—203). Опубликована
заключительная работа по использованию М. в синтезе поли-
изопреноидов, в которых все двойные связи имеют траяс-кон-
фнгурацию [1]. Исходным соединением для синтеза сквалена
341
(7) служил бисаллиловый спирт (2), полученный, как показано
на схеме, из янтарного альдегида и изопропенилмагнийбромида.
Спирт (2) обрабатывали 5 мол. же М. (1) в толуоле в присут-
ствии малых количеств щавелевой кислоты и гидрохинона и
нагревали при 140° в течение 24 час; при этом образовывался
(7)
Сзо-тетраеидион (3), который сразу же восстанавливали (NaBH-J
до С2о-тетраеидиола (4). При повторении этого двухстадийного
синтеза из (4) получали Сзо-Диол (5). Надо отметить, что с воз-
растанием длины изопреноидной цепи выход продукта перегруп-
пировки Кляйзена уменьшается. Сз0-Диол (5) далее подвергали
кляйзеновской перегруппировке в сквален (7) под действием
тионилхлорида (реакция SnK) с последующим восстановлением
алюмогидридом лития. Практически все двойные связи (>99%)
полученного сквалена имели транс-конфигурацию.
Фолкнер и Петерсон [1] применили перегруппировку Кляйзе-
на в синтезе оптически активных форм ювенильного гормона
Cecropia (8). В этом случае стереоселективность оказалась не
такой высокой, как в синтезе сквалена.
(8)
1. Faulkner D. J., Petersen М. R., J. Am, Chem. Soc., 95, 553 (1973).
342
МЕТОКСИКАРБЕНИЯ ГЕКСАФТОРАНТИМОН AT,
H3C~~O^CH2SbF6. Мол. вес 134,01, т. пл. 115-118°.
Эту устойчивую алкоксикарбениевую соль можно получить
[1] с выходом около 90% реакцией хлорангидрида метоксиук-
сусной кислоты или хлор метил метилового эфира с безводной
гексафторсурьмяной кислотой в жидкой двуокиси серы или в
хлористом метилене:
о +
HjCOCIbC^ Н3С—О—’CH^SbFb + со + НС1
'"С! 91%
Н3СОСНгС1 - HSbF^ ->Н3С — O-CHjSbFf, + НС1
89%
Ион метоксикарбения является амбидентным электрофилом,
способным реагировать как по атому С, так и по атому О. В ре-
зультате нуклеофильной атаки по атому С происходит метокси-
метилирование. Вытеснение формальдегида из иона метоксикар-
бения приводит к метилированию нуклеофила. В случае арома-
тических соединений преобладает последняя реакция. Напри-
мер, в результате взаимодействия М, г. с бензолом с высоким
выходом образуется толуол.
СН3
+ Н3С—О—СН, ---->- I [ + нсно + н+
1. Olah G. A., Svoboda J. J., Synthesis, 1973, 52.
6 - МЕТОКСИ - 7 - ОКСИ - 3, 4-ДИГИДРОИЗОХИНОЛ ИНИЯ
ИОДМЕТИЛАТ (1). Мол. вес 319,14, т. пл. 216—218°.
Получение [1].
(О
Синтез апорфина по Пшорру. Кутппан и сотр. [2] предложили
улучшенный метод синтеза апорфинов, основанный на реакции
конденсации реагента (1) с о-нитротолуолами (2) в присутствии
трет-бутилата калия в диметилацетамиде с образованием
нитробензил) - 2 - метил - 6-метокси-7-окси-1,2,3,4-тетрагидроизохи-
нолинов (3) с выходом 88—95%. Последние гидрируют до соот-
343
ветствующих аминофенолов (4), которые превращают
затем в 1-оксиапорфины (5) классическим методом Пшорра—
диазотированием в смеси 20 %-ной H2SO4 —НОАс (1:1) и цик-
лизацией под действием медного порошка.
1. Brossi A., O’Brien J., Те It el S., Org. Prep. Proc., 2, 281 (1970).
2. К u p c h a n S. M., Kames wa r a n V., F1 n d 1 a у J. M. A., J. Org. Chem.,
38, 405 (1973).
МОЛЕКУЛЯРНЫЕ СИТА (II, 309—311; V, 306—307;
VI, 177—178).
Кетимины и енамины. Кетимины и енамины удобно получать
взаимодействием кетонов с аминами в присутствии М. с. (были
использованы сита Линде 5А). Выходы составляют. 40—88% [1].
Ацетали из альдегидов или кетонов и вторичных спиртов.
Как правило,’ синтез ди-втор-алкилацеталей кетонов протекает
с низким выходом. Релофсен и ван Беккум [2] нашли, что такие
ацетали можно получить с выходом 70—90%, используя М, с.
типа 5А для избирательной адсорбции образующейся воды.
В качестве кислотного катализатора используют л-толуолсуль-
фокислоту.
уОС3Н7-изо
/С=О + 2«зо-С3Н7ОН + Н2О
' ' чЭС3Нгпзо
I. Taguchi К., Westhelmer F. Н., Л. Org. Chem,, 36, 1570 (1971).
2. R о е 1 о I s е n D. Р„ v а п В е к к u m 1-1., Synthesis, 1972, 419.
344
МОЛИБДЕНА ГЕКСАКАРБОНИЛ (V, 307; VI, 178—179).
Реакция простых эфиров с хлорангидридами кислот [1]. При
кипячении без растворителя, а также в гексановом или изоок-
тановом растворе ациклических простых эфиров и хлорангид-
ридов карбоновых кислот в присутствии М. г. образуются слож-
ные эфиры, органические хлорпроизводные и в некоторых слу-
чаях алкены.
Мо(СО)е
ROR + R'COCl ---->• RC' + R'COOR
Пример:
м-С4Н9ОС4Н9^ + СеНйСОС1 —► С6Н5СООС4Нй-« +н-С4Н9С1
73 %
Циклические простые эфиры в тех же условиях дают галогенсо-
держащие сложные эфиры или продукты элиминирования:
I J + CHjCOCl —CHjCOOCHjCHjCHiCHjCL
О 78%
I J + CHjCOCl—сНзСО0СНгСН=СНСНгС1
C 61%
Карбонилы металлов VI группы по своей каталитической
активности располагаются в следующий ряд: Мо(СО)б2>
>W(CO6) >Сг(СО)6.
В реакции 3{3-этокси-Д5-холестена с хлористым ацетилом на-
блюдается полное сохранение конфигурации: 3[3-хлор-А5-холе-
стен образуется с высоким выходом и лишь с небольшой при-
месью Д3-5-холестадиена.
Эпоксидирование олефинов (V, 307). Опубликована методи-
ка эпоксидирования олефинов гидроперекисью трет-бутила, ка-
тализируемого М. г. [2]. Для этой реакции получены кинетиче-
ские данные [3]. Предполагают, что механизм реакции включает
1) обратимое образование комплекса катализатора с гидропе-
рекисью, 2) обратимое ингибирование этой реакции одновремен-
но образующимся спиртом и 3) реакцию комплекса гидропере-
кись— М. г. с олефином с образованием эпоксида и в качестве
побочного продукта реакции спирта.
1. Alper Н., Huang C.-С., J, Org. Chem., 38, 64 (1973).
2. S h е и g М. N., Z a j а с е k J. G., J. Org. Chem., 35, 1839 (1970).
3. Baker T. N,, Ill, Mains G. J., Sheng M. N., Z a j a c e k J. C., J. Org.
Chem., 38, 1145 (1973).
МОЛИБДЕНА ПЯТИОКИСЬ —ГЕКСАМЕТАПОЛ (гидрат
комплекса), МоО5-ГМТФК- Мол. вес 371,14.
Получение. Мимун и сотр, [1] предложили получать реагент
растворением трехокиси молибдена (МоОз, 50 г) при 40° в 30 % -
345
ной Н2О2 (250 м.i). Образовавшийся желтый раствор охлаж-
дают до 10° и при энергичном перемешивании прибавляют к нему
ГМТФК (62,3 г). При этом сразу выпадает желтый осадок, ко-
торый собирают на фильтре и несколько раз промывают эфиром.
После сушки продукта и двукратной кристаллизации из мета-
нола комплекс получается в виде желтых кристаллов с выходом
84%. Французские химики показали, что он имеет структуру
гмтфн цго
(Г
Реагент следует хранить при низких температурах, при его
хранении при комнатной температуре в течение месяца был за-
фиксирован взрыв [2].
Не содержащий воды комплекс Мо05-ГМТФК можно полу-
чить дегидратацией реагента (1) над пятиокисью фосфора в
вакууме [3].
Эпоксидирование. По сообщению Мимуна и сотр. [3],М. п,—
г. реагирует с олефинами, образуя с высоким выходом эпок-
сиды. Алкильные заместители при двойной связи повышают
скорость эпоксидирования. Апротонные растворители также
ускоряют реакцию; с наибольшей скоростью эпоксидирование
протекает в хлористом метилене. В ДМФА и ТГФ реакция идет
очень медленно. Эпоксидирование проходит стереоспецифично,
с сохранением конфигурации олефина. Так, например, грс-бу-
тен-2 превращается в грс-2,3-эпоксибутан, а транс-бутен-2 — в
трш/с-2,3-эпоксибутан. Французские химики предложили сле-
дующий механизм реакции:
Шарплесс и сотр. [4] частично подтвердили этот механизм.
Методом меченых атомов они показали, что эпоксидный атом
кислорода образуется исключительно из пероксо-лигандов ком-
плекса, а не за счет оксо-кислорода. По своей реакционной спо-
собности по отношению к олефинам, однако, молибденовый
346
комплекс очень напоминал надкислоты, в случае которых пред-
почтительно реализуется трехчленное циклическое переходное
состояние [5].
Гидроксамовые кислоты. [6]. Окисление N-триметилсилил-
амидов (1), полученных реакцией соответствующих N-алкил-
амидов с гексаметилдисилазаном, комплексом MoOg-ГМТФК, в
хлористом метилене при комнатной температуре (продолжитель-
ность реакции колеблется от нескольких часов до нескольких
дней) приводит к образованию с умеренным выходом (15—40%)
диоксомолибденового комплекса (2). Обрабатывая комплекс
(2) этилендиаминтетрауксусной кислотой (ЭДТК), выделяют
свободные гидроксамовые кислоты (3).
r’con
вцснд
О)
(2)
ЭД Т.Н , / '
~> r’con
'он
(3)
[ Mimoun Н., de Roch I. S., Sajus L., Bull, soc, chim. France, 1969,
1481,
2. F u c h s P. L., personal communication.
3. Mimoun H., de Roch I, S., Sajus L., Tetrahedron, 26, 37 (1970).
4, Sharpless К. B., Townsend J. M., Williams D, R., J. Am, Chem.
Soc,, 94, 295 (1972).
5. В i ngh a m K. D., M eaki ns G, D,, W h i t h a m G. H., Chem. Comm.,
1966, 445.
6. Mail in S. A., Sammes P, G., J, C. S. Chem. Comm,, 1972, 1222.
МОНОХЛОРБОРАНА ДИЭТИЛЭФИРАТ, BH2C1-O(C2H5)2.
Получение [1], Реагент получают взаимодействием боргидри-
да лития с трихлоридом бора в эфирном растворе.
LiBH4 + ВС13 + 2 (С2Н5)2О 2ВН2С1-О(С2Н5)2 + LiCl
Гидроборирование. Монохлорборан в диэтиловом эфире гид-
роборирует самые разнообразные олефины с образованием с вы-
соким выходом соответствующих диалкилхлорборанов:
г
(сгн5)го
+ внга —0 ’ А Л—
BCL
В противоположность этому монохлорборан в ТГФ дает смесь
продуктов R3B, К2ВС1 и КВС12 [2]. Гидроборирование — окисле-
ние алкенов с помощью М. д., протекающее как бы против пра-
вила Марковникова, — дает соответствующие спирты более
347
99,5%-ной чистоты, содержащие менее 0,5% другого изомера.
Так, например, под действием этого реагента норборнен превра-
щается в экзо-норборнанол-2 с выходом >99,8%), а стирол — в
2-фенилэтанол (96%) и 1-фенилэтанол (4%), Таким образом,
реагент более селективен, чем диборан, и в этом отношении
почти сравним с бис- (1,2-диметилпропил-1)-бораном (I, 313—
316; V, 123; VI, 84—86).
1, Brown Н. С., Ravindran N., J. Org. Chem., 38, 182 (1973).
2. Brown H. C., Ravindran N., J. Am. Chem. Soc., 94, 2112 (1972).
МУРАВЬИНАЯ КИСЛОТА (II, 327—331; V, 308—310),
Карбоксилирование (II, 329—330). гре-Бицикло-[3,3,0]-ок-
тан-1-карбоновая кислота (2) легко получается [1] с выходом
около 25% взаимодействием ^ис-бицикло-[3,3,0]-октена-2 (1) с
98—100%-ной М. к, в присутствии серной кислоты при комнат-
(1)
ной температуре. Реакция включает гидридный перенос с обра-
зованием более устойчивого третичного катиона. Бициклооктен-2
(1) получен в свою очередь с выходом 55 % изомеризацией ци-
клооктадиена-1,3 под действием калия [2],
Бициклические простые эфиры [3]. При обработке 3-арил- или
З-алкил-З-оксигептадиенов-1,6 (1) смесью М. к. и 70%-ной сер-
ной кислоты прн комнатной температуре с выходом 60—70%)
образуются 1-арил- или 1-алкил-8-оксабицикло-[3,2,1]-октаны
(2):
нс=снг нсоон,
R - с - снг—снг - сн=сн, 7О?3~ная н?5а
он
(2)
Ароматизация кольца С в стероидах [4], В результате 15-ча-
сового кипячения 17а-метил-Д6’9(10-тестостерона (1) с 90%-пой
М.к. с выходом 45% образуется продукт дегидратации с пере-
348
группировкой — 17,17-диметиландростатетраеп-4,8,11,13-он-3 (2):
Перегруппировка
М. к. дает формиат
(2) [5]:
арилпротоадамантанола (1) в кипящей
соответствующего арилокси адамантана
1. McKervey М. A., Quinn Н. A., Rooney J. J., J. Chem, Soc. (С),
1971, 2430.
2, Stapp Р. R., Kleinschmidt R. F., J. Org. Chem., 30, 3006 (1965).
3. Watanabe S., Suga K, Fujita T., Takahashi Y., Synthesis, 1972,
422.
4. T u r n e г A. B., Chem. Ind., 1972, 932.
5. Lenoir D., Chem. Ber., 106, 78 (1973); Lenoir D., Glaser R., Mi-
son P., von R. Schley er P., J. Org. Chem., 36, 1821 (1971).
МУРАВЬИНОЙ КИСЛОТЫ ЭТИЛОВЫЙ ЭФИР (II, 332—
335; V, 310).
Оксиметилирование кетонов [1]. Кетоны можно превратить
в ct-оксиметильные производные в две стадии: ацилированием
с помощью М.к.э.э. и последующим восстановлением гидридом
алюминия. Эта последовательность реакций иллюстрируется на
примере превращения 4-трет-бутилциклогексанона (1) в 2-окси-
метил-4-трет-бутилциклогекса нон (3):
I. Corey Е. J., Cane D. Е., J. Org. Chem., 36, 3070 (1971).
н
НАДБЕНЗОЙНОЙ КИСЛОТЫ трет-БУТИЛОВЫЙ ЭФИР
(II, 342—346; V, 311).
Реакция ацилоксилирования [1]. Опубликован подробный об-
зор по реакции ацилоксилирования (реакция Хараша— Соснов-
ского).
1. Rawlinson D. J-, S о s п о v s ky G., Synthesis. 1972, 1,
НАДМУРАВЬИНАЯ КИСЛОТА (П, 346)
Циклические карбонаты. Окисление метилового эфира ат-
трактилигенина (1) муравьиной кислотой (99%) и перекисью
водорода (36%) неожиданно привело к образованию трех цик-
лических карбонатов (2)—(4):
1. Piozzi F., Savona G._. Marino M. L., Tetrahedron, 29, 621 (1973),
НАДУКСУСНАЯ КИСЛОТА (II, 351—353; V, 312—314;
VI, 182—183).
Окисление витамина А. Окисление витамина А (1) Н. к.
в ТГФ при комнатной температуре дает 11,12-эпоксиальдегид
витамина А (2) с выходом 45%. Атаке по двойным связям в по-
ложениях 5,6 и 7,8, вероятно, препятствуют стерические фак-
торы [1].
350
(1)
1. Ogata Y,, Tomizawa K., Takagi K-, Tetrahedron, 29, 47 (1973).
(2)
НАДУКСУСНАЯ КИСЛОТА — БОРА ТРИФТОРИДА
ЭФИРАТ.
Окисление макроциклических кетонов по Байеру-Виллигеру
(III, 65). Мукхержи и сотр. [1] нашли, что макроциклические
кетоны легко и с хорошим выходом превращаются в соответ-
ствующие лактоны под действием 40%-ной надуксусной кисло-
ты в присутствии эфирата трифторида бора.
СН3СОООН
ВГ3-О(СгН5)г
{2)
Взаимодействие циклододеканона (1) в хлороформе с эфи-
ратом трифторида бора (1 моль) и 40 %-ной надуксусной кисло-
той (3 моля) при 25° в темноте в течение двух недель приводит
к образованию циклододеканолида (2) с выходом 77,2%. Если
эфират трифторида бора заменить серной кислотой (II, 347),
выход целевого продукта уменьшается.
I, Mookherjee В. D., Trenkle R. W., Patel R. R-, J. Org. Chem., 37,
3846 (1972).
НАДУКСУСНАЯ КИСЛОТА —СЕРНАЯ КИСЛОТА.
Окисление по азоту ароматических азотистых гетероциклов
с пониженной основностью. Окисление по азоту полигалогенпи-
ридинов трудно осуществимо вследствие низкой основности ато-
ма азота. Например, при обработке пентахлорпиридина 90%-ной
перекисью водорода в трифторуксусной кислоте даже в опти-
мальных условиях N-окись образуется только с 20%-ным выхо-
дом. Чиверс и Сушицкий [1] недавно нашли, что это окисление
можно осуществить с выходом 95%, действуя 90%-ной пере-
кисью водорода в уксусной кислоте в присутствии конц. серной
кислоты. Вместо серной можно применить также и более доро-
гую, трифторуксусную кислоту, однако это нисколько не улуч-
шает выхода целевого продукта. При использовании 75%-ного
водного раствора серной кислоты окисление не идет. Функция
351
серной кислоты заключается, вероятно, в том, что она протони-
рует надкислоту и тем самым облегчает перенос ОНЛ
I. Chivers G, Е., Suschitzky Н,, J. Chem. Soc. (С), 1971, 2867.
НАТРИЙ (II, 354—355).
Расщепление циклических p-хлорэфиров. Тетрагидрофурфу-
рилхлорид (1) расщепляется до пентен-4-ола-1 (2) [1] действием
мелкодисперсного натрия в эфире, который готовят, измельчая
натрий в горячем ксилоле с помощью мешалки Гершберга, за-
тем ксилол декантируют и заменяют эфиром:
1) 2 Na
2) Нг°--> НОСНгСНгСНгСН = СНг
снгс1 76-83%
(2)
Опубликована подробная методика расщепления З-хлор-2-
метилтетрагидропирана (3) до тра«с-гексен-4-ола-1 (4) [2]:
1) 2 Na
2) HZO
89-94%
(4)
Внутримолекулярное дегалогенирование по Вюрцу: бицикло-
[ 1,1,0]-бутан [3] (т. пл. 8°). Реакцию проводят в атмосфере азо-
та в кипящем диоксане при перемешивании. Диоксан предвари-
тельно кипятят с кетилом натрия, полученным из 10 г бензофе-
нона и 1 г натрия (и 2 л растворителя), до образования темно-
синего раствора. Не содержащий после этого перекисей диоксан
отгоняют из той же колбы и сразу же используют, В работе
подробно описана применяемая аппаратура и правила обраще-
ния с ней. Выход бициклобутана составляет 78—94%.
78-94%
+ NaCl + NaBr
1. Brooks L, A., Synder H. R., Org. Syn., Coll. Vol., 3, 698 (1955).
2. Paul R., R i о b e O., Maumy M, Org. Syn., submitted 1973.
3. Lampman G. M., Aumiller J. C„ Org. Syn., 51, 55 (1971),
НАТРИЙ — АММИАК (II, 355; V, 314—315; VI, 183),
Восстановление аллеиов (V, 314—315). Моорти и Девапраб-
хакара [1] опубликовали изящный метод синтеза цне.грс-цикло-
352
ундекадиена-1,6 (4) из цис,чис-циклодекадиена-1,6 (1):
I. Moor thy S. N., Devaprabhakara D., Synthesis, 1972, 612.
НАТРИЙ — НАФТАЛИН (нафталенид натрия) (II, 357—
359; V, 316).
Реакция Вюрца [1]. Реакцией 3-бромметилциклобутилбро-
мида (1) с Н.— н. в моноглиме в атмосфере гелия можно полу-
чить бицикло-[1,1,1]-пентан (2) с выходом 6,5 % [2]:
ВгНгС
[ClqHa]~Na+
(9 (2>
Олефины из димезилатов в«ц-диолов [3]. Димезилаты виц*
диолов при обработке Н.— н. или натрий — антраценом в ТГФ
или ДМЭ быстро и с высоким выходом превращаются в соот-
ветствующие алкены. К предварительно обезгаженному раство-
ру димезилата диола при перемешивании медленно приливают
0,3 М раствор анион-радикала до появления устойчивой интен-
сивной окраски анион-радикала, Избыток реагента разлагают
воздухом или водой, а алкен выделяют обычным способом. Эта
реакция нестереоспецифична, так как цис- и транс-алкены обра-
зуются в отношении, близком к равновесному. В реакции можно
применять также тримезитилборан — натрий (VI, 265—266), но
выход алкена по крайней мере в одном случае был меньше, чем
при использовании ароматического анион-радикала. При замене
мезилатов тозилатами или брозилатами образуются лишь дио-
лы и некоторое количество соответствующих эпоксидов.
Алкилирование аминов сопряженными олефинами [4]. При
прибавлении изопрена и диэтиламина к раствору Н.— н. в ТГФ
образуется диэтилизопентениламин (1) с выходом 70%:
СН3.
;с-сн=сн2 + nh(c2h5)2
h2cZ
[CioHg] Na+
ТГФ
-------------
70 %
(C2H5)2NCH2CH=C(CH3)3
(1)
12 Зак. 596
353
При использовании нафталенида лития выход (1) ниже [4].
Другие примеры:
сйн5сн=сн2 + nh(c2h5)2---------> с6н5сн2сн2м(с2н5)г
95%
СН2
1Г
( СН2 +
/сх
Н3С сн3
HN(C2H5)2
с
Н3с/ СНз
Реакция с фенилацетонитрилом [5], Н. — н. реагирует с фе-
нилацетонитрилом, образуя продукты реакции с переносом
электрона или протона в зависимости от природы растворителя.
Реакциям с переносом электрона особенно благоприятствует
использование смеси растворителей ТГФ — тетраглим.
1. К о s о 1 а р о f f G, М., Org. React., 6, 326 (1951).
2. W i b e.r g K. B.„ W i 111 a m s V. Z., J r., J. Org. Chem., 35, 369 (1970).
3. Carnahan J. C., Jr., C lesson W. D., Tetrahedron Letters, 3447 (1972).
4. Fujita T., Suga K, Wat anabe S., Chem. Ind., 1973, 231.
5. Bank S., Thomas S. P., Tetrahedron Letters, 305 (1973).
НАТРИЙ —ФЕНАНТРЕН (фенантренид натрия), [C14H10]~Na+
Мол. вес 153,18.
Реагент получают прибавлением 0,014 г-атом натрия к рас-
твору 0,015 моля фенантрена в 30 мл сухого ТГФ. Образующий-
ся темно-зеленый раствор перемешивают в течение 18 час при
комнатной температуре.
Дехлорирование [1]. Обработка смеси цис- и транс,экзо,
экзо-3,4-дихлортетрацикло-[4,4,2,02’5,07’10]-додекадиенов-8,11 (1) и
(2) Н.— ф. до появления устойчивой зеленой окраски раствора
приводит к образованию экзо,экзо-тетрацикло [4,4,2,02>5,07>10]-до-
354
декатриена (3) с выходом 69%:
1, Paquette L. A., Stowell J. С., J. Am. Chem. Soc.,
93, 5735 (1971).
НАТРИЯ АЗИД (II, 359—362; V, 316—317; VI, 183),
Реакция Шмидта [1,2].
NaNj НгО
H2SO4, CHCl* ' * (снг)10 с-0
(4)
Внимание} Азотистоводородная кислота очень токсична и,
как сообщалось, может взрываться без видимых причин.
Реакцию проводят в вытяжном шкафу с хорошо действую-
щей вентиляцией в трехгорлой колбе емкостью 3 л, снабженной
магнитной мешалкой с яйцеобразным 5-сантиметровым рото-
ром, покрытым тефлоном, Колбу помещают на водяную баню,
в одно горло колбы вставляют термометр, в другое — обратный
холодильник, защищенный хлоркальциевой трубкой, а третье
горло закрывают резиновой пробкой. В колбе охлаждают до 5°
600 мл концентрированной серной кислоты.
К медленно перемешиваемой кислоте с помощью капельной
воронки с длинным носиком прибавляют постепенно 191 —196 г
метилового эфира циклоундецен-1-карбоновой кислоты (3).
Затем скорость перемешивания увеличивают, чтобы добиться
образования гомогенного раствора, прибавляют 500 мл хлоро-
форма и доводят температуру реакционной смеси до 35°, напол-
няя водяную баню теплой водой. К интенсивно перемешиваемой
реакционной смеси прибавляют (защитный экран!) в течение
50 мин 78 г (1,2 моля) азида натрия (комки предварительно
раздробить, избегая попадания на кожу), поддерживая темпе-
ратуру в интервале 35—40°, После этого реакционную смесь
охлаждают до 5°, выливают на 1 кг измельченного льда, раз-
бавляют 1,5—2 л воды и переносят в 5-литровую трехгорлую
колбу для перегонки с паром. Сначала отгоняют хлороформ, а
затем перегоняют с паром циклоундеканон, собирая 3,5—4 л
дистиллята. Дистиллят экстрагируют отогнанным хлороформом,
вытяжки сушат фильтрованием через безводный сульфат нат-
рия. Водный слой еще раз экстрагируют 500 мл эфира, экстракт
12* 355
промывают насыщенным раствором хлористого натрия и фильт-
руют через безводный сульфат натрия. Органические вытяжки
объединяют, растворитель удаляют при пониженном давлении.
Перегонка в вакууме маслообразного остатка дает 139—143 г
(83—85%) циклоундеканона (4) в виде бесцветного или светло-
желтого масла, т. кип. 84—85°/2 мм, т. пл. 16,2—16,6°.
Модифицированная реакция Курциуса. Кайзер и Вейншток
[3] разработали подробную методику синтеза 1-фенилциклопен-
тиламина из 1-фенилциклопентанкарбоновой кислоты через сме-
шанный ангидрид карбоновой кислоты и кислого эфира уголь-
ной кислоты:
с6н
соон
СбН5
С1СО2СгН5
-ОС2Н5
ЬГаОН
Нагревание
<-----------С6Н6
NH2
G6HS
1,2,3-Триазолы [4]. трацс-Арилнитроэтилены [например, (1)]
при взаимодействии с Н. а. в ДМСО или ДМФА дают 1,2,3-три-
c6h5ch=chno2 6 5 Я \\
+ (С6н5)3-с6н3
(3)
азолы (2) и неожиданно сюиж-три ар и л бен золы (3). В реакцию
могут вступать также аф-ненасыщенные нитрилы.
NaN3
C6H5CH=C-CN ДМСО >
(4)
С6н^—,CN
N Д?
N
Н
(5)
356
1. Garbisch E. W., Jr., Wohllebe J., J. Org. Chem., 33, 2157 (1968).
2. Garbisch E. W., Jr., Wohllebe J., Org. Syn., submitted 1971.
3. К a i s e r C., W e i n s t о с к J., Org. Syn., 51, 48 (1971),
4, 3 e ф и p о в H. С., Ч а п о в с к а я H. K-, Апсалой У. П., К о л e с н и-
к о в В. В., ЖОрХ, 8, 1335 (1972).
НАТРИЯ АЛЮМОХЛОРИД (II, 363—365; V, 318).
1-Амино-4-оксиантрахинон [ 1 ]. Конденсация 4-аминофенола
и его производных (1) с фталевым ангидридом, осуществляемая
сплавлением компонентов с хлористым алюминием и хлоридом
натрия (170—210°) с последующим гидролизом (2н. НО), при-
водит к образованию с довольно низким выходом 1-амино-4-ок-
сиантрахннона (2). Наибольший выход этого соединения был
получен при использовании триацетильного производного (1).
N(COCH3k
ососн.
1) AlCl3“NaCl
2) H3Q+______
4 5%
(О
1, Aggarwala V. Р., Gopal R., Garg S. Р„ J. Org. Chem., 38, 1247
(1973).
НАТРИЯ АМАЛЬГАМА (II, 366—370; V, 318; VI, 184).
1-Этил-4-карбметокснпиридинил (2). Этот устойчивый ради-
кал удобно получать восстановлением иодида 1-этил-4-карбме-
токсипиридиния (1) 3%-ной амальгамой натрия в ацетонитриле,
а не восстановлением цинком или магнием [2]:
сн2сн3
Na(Hg), CH3CN^
' 10-30%
(1)
снгсн3
(2)
1. Ко sower Е. М., Waits Н. Р., Org, Prep, Proc. Int., 3, 261 (1971).
2, Schwarz W. M., К о s о w e r E. M,, S h a i п I., J. Am. Chem. Soc., 83,
3164 (1961).
НАТРИЯ БОРГИДРИД (II, 381—388; V, 319—320;
VI, 185—187).
Восстановление ароматических иитросоединений. Продолжая
исследования по восстановлению боргидридом натрия в поляр-
ных апротонных растворителях (VI, 185), Хатчинс и сотр. [1]
357'
установили, что ароматические нитросоединения восстанавли-
ваются этим реагентом в ДМСО или сульфолане сначала до азо-
ксисоединений, которые затем восстанавливаются до азосоеди-
нений и аминов. Электроноакцепторные группы в ароматиче-
ском ядре нитросоединений облегчают и первую, и последующие
стадии восстановления. Электронодонорные группы, напротив,
замедляют восстановление азоксисоединений до такой степени,
что эти продукты можно получить лишь с умеренным выходом.
Нитроалканы [2]. Нитроалканы легко получить с выходом
60—95% восстановлением 1-нитро-2-алкилнитратов (приготов-
лены из 1-алкенов, окислов азота и кислорода [3]) с помощью
Н. б. в 95%-ном этаноле:
I КаВН4 I I I
R—с—ch2no2 —> Lr -c=chno2J —> R—CHCH2NO3
o2no
Восстановление альдегидов и кетоиов в очень мягких усло-
виях [4] (V, 319—320).
Смесь 1 г 5а-андростанол-17р-она-3, 0,90 г тозилгидразина и
70 мл метанола кипятят в течение 3 час, а затем охлаждают до
комнатной температуры. Маленькими порциями в течение 1 час
добавляют 2,5 г Н. б. и кипятят смесь еще 8 час. Растворитель
удаляют при пониженном давлении. Остаток растворяют в эфи-
ре и промывают водой, водным раствором карбоната натрия,
2 М раствором соляной кислоты и водой. Хроматографирование
дает 73—76% чистого 5а-андростанола-17{3 [4].
Восстановление третичных галогенидов [5]. Третичные гало-
гениды восстанавливаются Н. б. в тетраметиленсульфоне (суль-
фолане, III, 305—306; V, 409—411) до углеводородов. Реакция
протекает через стадии элиминирования, гидроборирования и
358
протолиза. Галогениды, нё содержащие а-водородного атома,
X
I NaBHi R\ ВН3 II н+, Д R\
R—С—CH2R" -----> X=CHR" --------> /С—CHR" ------> Yhch2r"
I R'/ R'/ R'/
также восстанавливаются H. б. В этом случае реакция, по-види-
мому, протекает через стадию ионизации, сопровождающуюся
захватом образующимся карбкатионом гидрид-иона. Выходы
углеводородов колеблются в пределах 45—90%.
Сульфурированный боргидрид натрия (VI, 186—187). Лалан-
цетте н сотр. [6] рассмотрели восстановление функциональных
групп сульфурированным Н. б. Реагент, в частности, пригоден
для селективного восстановления. При его использовании мож-
но осуществить селективное восстановление нитрооксимной н
нитрильной групп в присутствии сложноэфирной группировки.
В стероидных кетонах наиболее реакционноспособным местом
молекулы является карбонильная группа уС3-атома.С помощью
сульфурированного Н. б. удается селективно восстановить эту
карбонильную группу в присутствии карбонильных групп у ато-
мов Си, С]2, С17 или С20. Полученный спирт представляет собой
экваториальный изомер. При условии подбора подходящего мо-
лярного соотношения ЫаВНгЗз/субстрат можно селективно вос-
становить альдегиды в присутствии других карбонильных групп.
Реагент восстанавливает также ароматические нитросоедине-
ния до соответствующих аминов с выходом около 80% [7]. При
этом не затрагиваются присутствующие в молекуле сложно-
эфирные, нитрильные, алкоксигруппы, галогены и двойные свя-
зи. Первичные алифатические нитросоединения восстанавли-
ваются этим реагентом с высоким выходом до соответствующих
нитрилов. При восстановлении амидов соответствующие амины
образуются лишь с умеренным выходом [8].
С эпоксидами реагент образует симметричные бис- (2-окси-
этил)-дисульфиды (1), которые восстанавливаются с помощью
L1AIH4 до 1,2-меркаптолов (2) [8].
2 ХС-----С/ NaBHzS3 g LiAlH^ 2 \С —
Z Х Нго но
. 1
/С — Сх
(1) (2)
1. Hutchins R. О., Lamson D. W., Rua L., Mi lewski C., Marya-
n 0 f f B., J. Org. Chem., 36, 803 (1971).
2. L a r k i n J. M., Kreuz K. L,, J. Org. Chem., 36, 2574 (1971).
3. Lachowicz D. R., Kreuz K. L., пат. США 3282983 (Nov. 1, 1966);
C. A., 66, 10577s (1967).
4. C a g 11 0 t i L., Org. Syn., 52, 122 (1972).
359
5. Hutchins R. 0., Bertsch R. J., Hoke D,, J. Org. Chem,, 36, 1568
(1971).
6. Lalancette J. M., Freche A,, Brindle J. R,, Laliberte M., Syn-
thesis 1972, 526.
7. Lalancette J. M., Brindle J. R., Can. J. Chem., 49, 2990 (1971).
8. Lalancette J. M., Freche A., Can. J. Chem., 49, 4047 (1971).
НАТРИЯ ГИДРИД (II, 392—401; V, 320—321).
О-Алкилирование гидрохинонов [1]. Гидрохинон (1) можно
с хорошим выходом алкилировать по кислороду, обрабатывая
его сначала Н.г. в ДМСО, а затем галогеналкилом. Тем же спо-
собом были проалкилированы и некоторые другие фенолы [2].
но
N(C2H5)2
Ы(С2Н5)г
НаН, ДМСО
СН31
70%
Эта методика пригодна и для N-алкилирования ароматиче-
ских аминов [3]. Например, из 1,4-диамино-2,5-дибромбензола
(3) был получен М,Ь1,М',Н'-тетраэтил-1,4-диамино-2,5-дибром-
бензол (4) с выходом 49%.
49%
И(СгН5)з
N(C2H5)a
(4)
Диметиловые эфиры циклоалкен-1 -дикарбоновых-1,2 кислот
[4]. Диметиловые эфиры сса'-дибромалкандикарбоновых кислот
(1) (п = 2-i-5) под действием 2 же Н.г. в ДМФА вступают в
реакции циклизации — элиминирования, в результате которых
образуются диметиловые эфиры циклоалкен-1-дикарбоновых-1,2
СНВгСООСНз
(СНг)л
СНВгСООСНз
NaH
ДМФА
^_ССООСН3
(СН2)Я ||
""" ССООСНз
(1) п = 2-5
(2)
360
кислот (2). Диметиловый эфир а,а'-дибромглутаровой кислоты
(1) (лг= 1) дает в этих условиях диметиловые эфиры цис- и
трш/с-1-бромциклопропан-1,2-дикарбоновых кислот (3) в соот-
ношении 1 : 3.
СНВгСООСН3
снг
с!:нвгсоосн3
(1) п = 1
сн3
NaH
---->
ДМФА
соосн3
(3)
Сужение цикла в бицикло-[2,2,1]-гептанах. Паукстелис и
Махариа [5] разработали удобный метод сужения цикла в 1-за-
мещенных бицикло-[2,2,1]-гептанолах-2, приводящий к получе-
нию 1-замещенных бицпкло-[2,1,1]-гексанов. Предложенный ме-
тод иллюстрируется синтезом, в котором в качестве исходного
соединения используется d-камфора (1). Под действием смеси
треххлористого и пятихлористого фосфора (кипячение 6 час)
d-камфора превращается с выходом 70% в 1-хлоркамфеп (2)
(по номенклатуре ИЮПАД 1-хлор-2-метилен-3,3-диметилбицик-
ло-[2,2,1]-гептан). Озонолиз последнего, а затем восстановление
озонида боргидридом натрия приводят к образованию эпимер-
ных спиртов (3) и (4) в соотношении 5:1. При непродолжи-
5 7%, ечи’“ N- (1)
тельной обработке смеси спиртов (3) и (4) Н.г. (50% в масле)
в ДМФА получается альдегид (5), а при длительном его дей-
ствии образуется смесь кислоты (6) (5,5-диметилбицикл о-[2,1,1]-
гексан-1 -карбоновой кислоты) и спирта (7) (5,5-диметилбицик-
361
ло-[2,1,1]-гексил-1-карбинола). Полагают, что сужение кольца
является результатом транс-коплаиарной перегруппировки типа
пинаколиновой.
Восстановление. Бензофенон и флуоренон восстанавливаются
Н. г, соответственно до бензгидрола и флуоренола. Наилучшие
результаты (92—97%) получены в таких растворителях, как
ГМТФК, ДМФА или пиридин. Неенолизующиеся кетоны, а так-
же кетоны, которые енолизуются с трудом, восстанавливаются
Н. г. до соответствующих спиртов с очень хорошими выходами.
Восстановление альдегидов и сложных эфиров может ослож-
ниться побочными реакциями. В реакции с окисями стирола и
циклогексена (7) реагент ведет себя как основание.
NaH/ ГМТФН^
4 0%
NaH/ГМТФН^
35-40%
С6Н5СОСН3
Метилирование фенолов и спиртов [8]. Фенолы и спирты при
действии Н. г. и йодистого метила в ТГФ при комнатной темпе-
ратуре превращаются в метиловые эфиры. В реакцию вступают
даже пространственно-затрудненные фенолы (2,6-ди-трет-бутил-
ц-крезол, выход 86%) и спирты (трифенилкарбинол, выход
85%). Фенолы повышенной кислотности (п-нитрофенол), одна-
ко, ие метилируются даже при 80°. Этим рассматриваемый реа-
гент отличается от диазометана, который легко метилирует фе-
нолы кислого характера.
1. D о о г п b о s Т., Strating J., Syn. Comm., 1, 175 (1971).
2, Doornbos T., personal communication.
3. Doornbos T., Strating J., Org. Prep. Proc., 1, 287 (1969).
4. McDonald R. N., Reitz R. R., Chem. Comm., 1971, 90.
5. P auks tel is J. V., Ma ch aria B. W., J. Org. Chem., 38, 646 (1973),
6. C a u b ё r e P., Ad о r e a u J., Bull. soc. chim. France, 1971, 3270.
7. Caubere P., Moreau J., Bull. soc. chim. France, 1971, 3276.
8. Stoochnoff В. A., В eno it on N. L., Tetrahedron Letters 21 (1973).
НАТРИЯ ГИДРИД —трег-БУТИЛ ГИПОХЛОРИТ.
1,1-Диоксотиадиазиридины. Тимберлейк и Ходжес [1] полу-
чили 1,1-диоксо-2,3-ди-трет-бутилтиадиазиридин (2) с выходом
46%, действуя на соединение (1) Н. г.— б. в пентане. Получен-
ная трехчленная гетероциклическая система удивительно устой-
чива. Но при окислении трет-бутилгипохлоритом она превра-
щается в азоалкаи (3). Последний был получен ранее окисле-
нием соединения (1) гипохлоритом натрия [2]. Пиролиз соеди-
362
нения (2) в кипящем бензоле также приводит к образованию
(CH3)3CNHSO2NHC(CH3)3
(1)
NaOCl
NaQH
о,
NaH S
(СН313СОС1 / \
---------> (CH3)3CN-NC(CH3)3
&)
(CHahCOCl
* (CH3)SCN=NC(CH3)3
(3)
азоалкана (3) (транс-2,2/-диметил-2,2/-азопропан) с почти коли-
чественным выходом. С увеличением объема алкильных групп
у атома азота в 1,1-диоксотиадиазиридине возрастает его тер-
мическая стабильность, однако устойчивость соответствующего
азоалкана при этом понижается.
1, Timberlake J. W„ Hodges ДА. L., J, Am, Chem, Soc., 95, 634 (1973).
2, О h m e R,, P r e u s c h h о f H., Ann., 713, 74 (1968).
НАТРИЯ ГИДРИД-ДИМЕТИЛСУЛЬФОКСИД (11,392—
401; V, 320—321).
Амиды [1], Реакция эквимолярных количеств сложного эфщ
ра, амина и гидрида натрия (дисперсия в масле) в ДМСО (или
ГМТФК, ЬТИ-диметилацетамиде) представляет собой простой и
удобный метод синтеза амидов. Выходы колеблются в пределах
68—94%.
1, Singh В., Tetrahedron Letters, 321 (1971),
НАТРИЯ ГИПОХЛОРИТ (11,405—409; V, 515; VI,219,322),
Окисление ГфМ'-диалкилгидразинов. Реагент применяют для
получения азоэтана окислением ГфМ'-диэтилгидразина [1],
Ру, Петр, эфир NaOCl, NaOH
2 C2HsNH-> + SO2C12 ----------> (C2HsNH)2SO2 -----
н2о
"NaOaS—N—C2H5 HN—C2HS1 NaOCl, NaOH N—C2HS
L NH-C3H5 HN—C2HS] h2o, 25“.^ ^_С2Нб
1, Ohme R., Preuschhof H., Heyne H.U., Org. Syn,, 52, 11 (1972),
НАТРИЯ ДИГИДРОФОСФАТА МОНОГИДРАТ, NaH2PO4-
•H2O. Мол. вес 137,99.
Гидролиз [1]. Неочищенный 1,5-дихлорпентанон-З, получен-
ный в виде темно-коричневого масла (555—585 а) в результате
катализируемого хлористым алюминием присоединения 3-хлор-
пропионилхлорида к этилену в хлористом метилене, гидроли-
зуют, разделив на две равные части. В круглодонную трехгор-
363
лую колбу на 3 л, снабженную термометром, обратным холо-
дильником и капельной воронкой на 250 мл, помещают 600 мл
воды, 600 мл диоксана, 510 г (3,70 моля) Н. д. м. и два или три
о
и
НгСГ^СНг
НгС снг
С1 С1
+ Н2О + NaHjPO,
кипятильника № 12 из карборунда и нагревают ее иа песчаной
бане, температура которой поддерживается на уровне 140—-
160°. Когда температура внутри колбы поднимется выше 90° и
ее содержимое начнет энергично кипеть, в течение часа прибав-
ляют по каплям одну порцию неочищенного 1,5-дихлорпеит-
анона-3. Затем колбу нагревают еще в течение 5 час, так чтобы
ее содержимое энергично кипело при температуре 90—92°.
Реакционной смеси дают охладиться до комнатной темпера-
туры а затем колбу помещают в ледяную баню. При перемеши-
ваний медленно приливают к ней раствор 160—180 г (4—
4,5 моля) гидроокиси натрия в 500 мл воды до pH 5. Затем до-
бавляют 400 мл эфира, переносят реакционную смесь в дели-
тельную воронку на 5 л и энергично встряхивают. Темноокра-
шениый верхний органический слой отделяют, а водный экстра-
гируют двумя порциями эфира по 500 мл. Объединенные эфир-
ные вытяжки высушивают над безводным сульфатом магния и
концентрируют до объема около 800 мл на роторном испарителе
при пониженном давлении. Концентрированный экстракт пере-
носят в круглодонную колбу на 1 л и фракционируют иа 25-сан-
тиметровой колонке Вигре при пониженном давлении. Сначала
собирают предгон диоксана, затем перегоняется тетрагидро-4Н-
пиранон-4 в виде бесцветной жидкости- с т. кип. 59—60713 мм.
Выход его из двух порций 1,5-дихлорпеитанона-З составляет
102—112 г (50—55%, считая на введенный в реакцию 3-хлор-
пропиоиилхлорид).
1. О w е п G. R., R е е s е С. В„ Org. Syn., submitted 1972.
НАТРИЯ ДИЭТИЛАЛЮМОДИГИДРИД, NaAl(C2H5)2H2.
Мол. вес 110,12.
Восстановитель. [1]. Этот относительно новый восстановитель
получают в виде 25%-иого раствора в толуоле, содержащем 3—-
4% ТГФ для повышения растворимости. По своей способности
восстанавливать разнообразные функциональные группы Н. д.
сходен с LiAlH4. В отличие от LiAlH4 он растворим в аромати-
ческих углеводородах, кроме того, его стоимость несколько ни-
же, если считать на реально используемый в реакции активный
водород.
1. S а п d е г s Н, J., Chem, Eng. News, June 19, 1972, p. 29.
364
НАТРИЯ ЖЕЛЕЗА(П) ТЕТРАКАРБОНИЛ (VI, 188)
Синтез алифатических кетонов. Коллман и сотр. [1] описали
четыре близких по существу, но отличающихся по используе-
мым реагентам метода синтеза несимметричных кетонов с уча-
стием Na2Fe(CO)4, как показано на схемах реакций (а)— (г}1
О
R'X ||
a) RX-J-Na2Fe(CO)4 —> RFe"(CO)4 -------> RCR'
О О
j R'X 11
б) RX + Na2Fe(CO)4 + P(C6Hs)3 —> RCFe"(CO)3[P(CsHs)3I -----> RCR'
О О
1! R'X II
в) RX 4-Na2Fe(CO)4 + CO —> RCFe’(CO)4 -------> RCR7
О О
II R'X ||
г) RCOC1 -J- Na2Fe(CO)4 —> RCFe’(CO)4 -------> RCR'
По методу (а) при взаимодействии галогеналкила (или то-
зилата) с ионом [Fe(CO)4]2* в М-метилпирролидоне-2 образуют-
ся алкилжелезокарбонильиые анионы (1), причем некоторые из
них были выделены и охарактеризованы. В результате дальней-
шей реакции с другим галогеналкилом образуется комплекс
предположительной структуры (2), который может разлагаться
до кетона через комплекс (3), образующийся в результате вне-
дрения по связи Fe—R одной из карбонильных групп, под дей-
ствием растворителя. Продуктов конденсации галогеналкилов в
реакции не обнаружено.
к
"R X I ’ СО
a) Na2Fe(CO)4 > ОС —Fe'**
to со
растворитель
---------->
с—о
OC.t | .R’
ос*^ | ^со
растворитель
О
И ,
RCR + Fe(CO)3*'Растворитель
В методе (б) также образуется комплекс (1)'; присоедине-
ние трифенилфосфинового лиганда приводит к внедрению одной
из СО-групп по связи Fe—R с образованием анионного комп-
365
лекса ацилжелеза (4). Реакция с другим галогеналкилом дает
комплекс (5), соответствующий комплексу (3).
б) NazFe(CO)4 -----> (I)
(СбН;)^
R
?=?со
ОС —Fej"
I ▼СО
Р(С6Н5)з
(4)
R
I
с=о
ОС. I . R1
*
ocv | ▼со
Р(с6н^
(5)
RCR1 + Fe(CO)3 •
Метод (в) отличается от метода (б) только тем, что трифе-
нилфосфиновый лиганд заменен карбонильным. В одном из при-
веденных примеров в качестве растворителя использовали си-
стему ТГФ—ГМТФК (2:1).
Согласно методу (г), из Na2Fe(CO)4 и RCOX образуется не-
посредственно комплекс со связью ацил—железо (6). Дальней-
шая реакция приводит к образованию комплекса (3), который
при разложении дает кетон. Этот новый метод синтеза был
описан на восьми примерах; выходы выделенных кетонов со-
ставляют 30—85%.
г) NasFe(CO)4
I _
ОС —Fe '
| ▼СО
Растворитель
(6)
О
И
-> R.CR1 + Ге(СО)} * Растворитель
Синтез алифатических карбоновых кислот, сложных эфиров
и амидов [2]. С алифатическими галогеналкилами и тозилатами
Н.ж.т. (1) образует анионные комплексы алкилтетракарбонил-
железа(О) (2). В присутствии окиси углерода в комплексе (2)
происходит миграция алкильного радикала с образованием ани-
онного комплекса со связью ацил—железо (3). Окисление кис-
366
лородом или гипохлоритом натрия комплексов (2) или (3) в
ТГФ или М-метилпирролидоне-2 (МП) с последующим гидроли-
R
I СО
ЫагГе(СО)4 + FX —> OC-FeT’
I ^СО
со
I ,со
OC-Fe“ ‘
j ^*60
СО
(1)
(2)
(3)
зом дает карбоновые кислоты. Кислоты образуются также в ре-
зультате обработки комплексов (2) или (3) иодом и последую-
щего гидролиза [уравнения (I) и (II)]. Реакция комплексов (2)
или (3) с 12 и спиртом в ТГФ, ТГФ—ГМТФК или МП дает
сложные эфиры [уравнение (HI)]. При взаимодействии (2) или
(3) с h и аминами в тех же растворителях образуются амиды
[уравнение (IV)].
Оз или NaOCl Н2О
I) (2) или (3) --------> RCOOH
1а, Н20
II) (2) или (3) ------RCOOH
i2, R'OH
III) (2) или (3) -----> RCOOR'
I2, R'R"NH
IV) (2) или (3) --------> RCONR'R"
В случае первичных алифатических галоген алкилов и то-
зилатов выходы (выделенных соединений) составляют около
80%. В случае вторичных субстратов выходы менее удовлетво-
рительны, что объясняется образованием олефинов в результате
элиминирования, которое можно, однако, свести до минимума,
используя в качестве растворителя ТГФ. Первичные бромиды
реагируют с Н.ж.т. (1) в ТГФ гораздо быстрее соответствую-
щих хлоридов. Так, например, 1-бром-6-хлоргексан можно пре-
С1СН2(СН2\СН2Вг -—>• С1СН2(СН2)4СНаСООН
вратить в 7-хлорэнантовую кислоту с выходом 84%. Сложио-
эфирная и карбонильная группы в этих условиях не затраги-
ваются: так, из бромкетостероида (4) метиловый эфир (5)
получали с выходом 71 %,
367
Альдегиды из галогенангидридов кислот [3—5]. Галогенам-
гидриды кислот при действии Н.ж.т. в присутствии трифенил-
фосфина и кислоты дают альдегиды, Бензоилхлорид, например,
[О Л Р(СвН5)з
II I НОАс
Na+C6HSCFe-(CO)J
—> С6Н5СНО -J- Fe(CO)4P(C6H5)3
превращается в бензальдегид. При работе с малыми количест-
вами веществ, судя по данным ГЖХ или ТСХ, альдегиды обра-
зуются с хорошими выходами, одиако при проведении реакции
в большом масштабе с выделением альдегидов перегонкой по-
следние получаются с довольно низкими выходами.
Сейгел и Колман [5] показали, что промежуточными соедине-
ниями в этих реакциях действительно являются ацилтетракарбо-
нилферрат(О)-анионы. Некоторые из них они выделили в виде
солей тремя методами, наиболее удобный из которых иллюстри-
рует приведенная ниже схема. Выходы солей колеблются от 20
до 72%, Аналогичные соли алкилтетракарбонилферрат(О)-анио-
нов выделены и в реакции бромалкилов с Н.ж, т, с последующей
их * 'со d
Na3Fe(CO^ ----> OC~Fe' Na
ioc°
со
Г°со
ОС—Fe‘*
1 \;o
co
Na+ E (CeffsbFJaNC1 > j- R=o)Fe (CO )Д (C(. н5p^N
обработкой реакционной смеси хлоридом бмс-(трифенилфосфин)-
имииия. Показано, что эти соли являются промежуточными со-
единениями в превращении алифатических галогеналкилов (то-
зил атов) в кетоны.
Под действием реагента фталоилдихлорид (1) превращается
в дифталидилиден (2) с выходом 23% [6]:
(О
NatFe(COK
гз%
368
1. Collman J. P., Winter S, R.t Clark D. R,, J. Am. Chem. Soc., 94,
1788 (1972).
2. Collman J. R., Winter S. R„ Ko mo to R, G., J. Am. Chem. Soc., 95,
249 (1973).
3. W a t a n a b e Y., M i t s u d о T., Tanaka M., Yamamoto К., О к a j i-
ma T., Take garni Y., Bull, Chem. Soc, Japan, 44, 2569 (1971).
4. W a t a n a b e Y., M 11 s u d о T., T a к e g a m i Y„ Org, Syn., submitted 1973,
5. Seigel W. O., Collman J. P,, J. Am. Chem. Soc., 94, 2516 (1972).
6. M11 s u d о T.t Watanabe Y., Tanaka M., Y am a mo to K.> T a к e-
gami Y., Bull. Chem. Soc. Japan, 45, 305 (1972).
НАТРИЯ ИОДИД (II, 410—414; V, 323; VI, 188—189).
Реакция с тозилатами (ср. II, 412). Превращение метил-
2,3, 6-трибензоил-4-тозил-а-п-глюкозида (1) в иодид (2) [1]:
I
НС-ОСН3
I
HC-OBz
BzOCH
I
НС — OTs
I
нс-------
СНгОВ2
(1)
Nal-ДМФА
------->
ГД/Х
75/0
I-----
НС-ОСН3
I
HC-OBz
BzOCH
I
CHI
CHjOBz
(2)
о
Превр ащение метил-2,3-дибензоил-4,6-дитозил-а-в-глюкозида
(3) в метил-2,3-дибензоил-4-тозил-6-дезокси-6-иод-а-в-глюкозид
.(4) [2]:
I------
НС-ОСН3
HC-OBz
I
BzOCH
i
HC-OTs
I
НС------
CH2OTs
(3)
Nal—CHCI3—CH3OH
50%
I-----
HC-OCH3
HC-OBz
BzOCH
HC-OTs
HC------
CH3I
(4)
Спиро-[4,4]-нонатриен-1,3,7 (5) [3]. Это интересное спирано-
вое соединение было недавно синтезировано по следующей
369
схеме:
1. Siewert G., Westphal 0., Ann., 720, 161 (1968),
2, S i e w e r t G., W e s t p h a 1 O,, Ann,, 720, 171 (1968),
3, Semmelh ack M. F., Foos J. S., Katz S„ J. Am, Chem. Soc,, 94, 8637
(1972).
НАТРИЯ б«с-(2-МЕТОКСИЭТОКСИ)-АЛЮМОГИДРИД
(VI, 189—190).
Алкилирование [1], При непродолжительном действии реа-
гента на бензофенон (1) в w-пропилбензоле при 162° образуются
бензгидрол (2) (50%), а также дифенилметан с выходом 38,5%.
С увеличением продолжительности реакции выход этих двух ве-
ществ уменьшается и основным продуктом реакции становится
1,1-дифенилэтаи (4). Наконец, при увеличении времени контак-
та бензофенона и Н. м. до 3,5 час из реакции были выделены
2,2-дифенилпропаи (5), выход 71%, и 1,1-дифенилциклопропан
(6), выход 11%.
СН3
(С6Н5)2С=О (C6HS)2CHOH (СеН5)2СН2 C6H5iHC6Hs
(I) (2) (3) (4)
СНз
CgHgiceHs
СНз
(5)
CSH5-C-CSHE
Н2С—~сн2
(6)
Примеры алкилирования комплексными гидридами металлов
до сих пор в литературе не встречались. Механизм реакции, по-
видимому, свободнорадикальный с участием метильных ради-
калов, генерируемых в процессе фрагментации 2-метоксиэтокси-
групп. Следует отметить, что замещаемые радикалами атомы
водорода активированы в рассмотренных случаях фенильными
группами. Эту реакцию удалось распространить на флуоренон
и антрахинон, а также на другие диарилметаны.
37Q
Восстановление 2-ацетилнафталина Н. м. в кипящем бензоле
дает ожидаемый продукт 1-(2-нафтил)-этанол (выход 97%).
Одиако при восстановлении 2-ацетилнафталииа этим гидридом
в ^-ксилоле при 140° получается 27% бис- (2-нафтил)-бутана
(1) и 16% 2-этилнафталина (2). Образование продукта (1) яв-
ляется, по-видимому, первым примером превращения алкил-
арилкетоиа в диарилалкан под действием гидрида металла.
сн, сн
Сульфонамиды под действием реагента вступают в реакцию
восстановительного расщепления, регенерируя амины. Следует
отметить, что L1AIH4 для этой цели непригоден [3],
Обзор. Малек и Черни [4] опубликовали обзор способов по-
лучения и реакций алкоксиалюмогидридов.
1. Cerny М., Malek J., Tetrahedron Letters, 691 (1972),
2. Cerny M., M й 1 е k J., Synthesis, 1973, 53.
3. G о 1 d Е. H„ В a b a d Е„ J. Org. Chem., 37, 2208 (1972).
4. М а 1 е k J., С е г п у М., Synthesis, 1972, 217.
НАТРИЯ НИТРИТ (II, 415—421; V, 323—324).
а-Хлор-сС-изонитрозоацетои (альдоксим хлорметилглиоксаля)
(3). Тейлор и Портной [1] разработали удобный метод синтеза
альдоксима хлорметилглиоксаля (3), включающий хлорирова-
ние дикетена (1) и последующую обработку образующегося
хлорангидрида у-хлорацетоуксусной кислоты (2) в эфире вод-
ным раствором Н. н. Последняя реакция протекает через стадии
гидролиза, нитрозирования и декарбоксилирования. Конденса-
ция оксима (3) с динитрилом аминомалоновой кислоты приводите
о
и
ClCH;CCHjCOOH
-> CICHjCCHCOOH
N = O
-COz
---> Cl СНг C CH=NOH >
бо^считая на(1)
HjN V
0‘
371
образованию 1-окиси 2-амино-3-циап-5-хлорметилпиразина (4)—
перспективного предшественника 6-з вмещенных птеридинов.
Альдоксимы галогенметилглиоксаля — сильные лакриматоры.
При температурах выше —20° они неустойчивы.
З-Нитроизоксазолы [2]. Реакция бромметилацетиленов (1)
с Н.и. дает с выходом 20—60% 3-нитроизоксазолы (5), обра-
зующиеся через указанные на схеме промежуточные продукты:
Na NO, ДМФА
2 ДС=ССНгВг г> ----------> В.С^ССНгМОг + RC=CCH3ONO
О) (2) (3)
1. Taylor Е. C.t Portnoy R. С., J. Org. Chem,, 38, 806 (1973),
2, R о s s i S„ D u r a n 11 E,, Tetrahedron Letters, 485 (1973),
НАТРИЯ СУЛЬФИД, Na2S-9H2O (11,422—423).
Синтез олефинов. Корнблюм и сотр. [1] описали новый метод
синтеза тетразамещенных олефинов из вицинальных динитро-
соединений. Существует несколько способов превращения али-
фатических и алициклических нитросоедииений в вицинальные
дииитросоединения; наиболее употребительным из них является
метод Сейгла и Хасса [2]. Нитропарафии сначала превращают
в соль аци-формы (1), действуя иа него метилатом лития; обра-
ботка этой соли бромом дает затем бромиитросоединение (2),
r
J^C-NOa
R’ Z
Вгг
R. /NQa
R1
(1)
(2)
реагирующее co вторым эквивалентом соли нитропарафина (1)
с образованием симметричного вицинального дииитросоедине-
ния (3) с общим выходом 88—93%. Несимметричные вициналь-
ные динитросоединения (4) получают из а,а-динитросоединений
по следующей схеме:
R. /NOa R"
+ ^C-NOa
R,Z XNO2 R"’Z
%C—cz
R' NOa NO*"'
(4)
372
Обработка динитросоедииений типа (3) или (4) И. с. вДМФА
при облучении (флуоресцентная лампа на 20 вт) приводит к об-
разованию с высоким выходом олефинов. Столь же эффективен
в этой реакции и тиофенолят натрия в ГМТФК.
R >R
—e
R’ NOa КоД'
(3)
R R”
—C
ВТ I I XR'"
NO; ИОД
(4)
Na,S
---
R >R
R,Z R‘
RX ZR"
j;c=cz
R,Z 4R"‘«
NazS^
Примеры:
Новый метод элиминирования заслуживает особого внима-
ния, поскольку он позволяет получать олефины с высокими вы-
ходами и не дает изомеров. Реакция представляет собой, оче-
видно, цепной процесс, так как ее ингибирует ди-г^ет-бутилимин-
оксил.
I. Kornblum N., Boyd S. D., Pinnick H. W-, Smith p, G., J. Am.
Chem. Soc., 93, 4316 (1971).
2, S e i g 1 e L. W., H a s s H, B„ J. Org. Chem., 5, 100 (1940).
НАТРИЯ ТЕТРАБОРАТ, Na2B4O7. Мол. вес 201,27.
N-Ацетилнейраминовая кислота [1]. При добавлении Н. т.
возрастает стереоизбирательиость альдольной конденсации
2-ацетамидо-2-дезокси-о-маниозы (1) со щавелевоуксусиой кис-
лотой (2), проводимой в водном растворе при pH 10, а также
улучшается выход продукта конденсации — N-ацетилнейрамино-
вой кислоты (3). Так, например, в отсутствие борат-ионов вы-
ход соединения (3) составляет 11,5%, в то время как прибавлен
ние Н.т. повышает его до 21,6%. Борат-ион ингибирует катали-
зируемую щелочью эпимеризацию различных 2-ациламиио-2-
373
Дезокси альдоз.
нгсон
(з)
NaOH., НгО, Na2B4O-
21, 6%
I. How М. J., Halford М. D. A,, Stacey М., Vickers Е., Carbohydrate
Res., 11, 313 (1969).
НАТРИЯ ТИОСУЛЬФАТ (П, 423).
Дебромирование вш$-дибромидов [1]. в«ц-Дибромиды под
действием избытка Н. т. в ДМСО (60°, 8 час) дебромируются
обычно с хорошим выходом, например эритро-стильбендибро-
мид превращается в тра^с-стильбен с выходом 99%. Реакция,
по-видимому, является стереоспецифическим гранс-эламиниро-
ванием.
1. Ibne-Rasa R. М., Tahir A. R., Rahman A., Chem. Ind,, 1973, 232.
НАТРИЯ ХЛОРИД— ДИМЕТИЛСУЛЬФОКСИД.
Декарбалкоксилнрование [1], Геминальные сложные эфиры
декарбалкоксилируют с выходом 90—95%, нагревая их прн
140—180° в течение нескольких часов с небольшим избытком
хлористого натрия (можно взять поваренную соль) в ДМСО,
содержащем 2 моля воды иа моль субстрата.
NaCI-ДМСО
140-180°
RRzC(COOC2H5)2 —RR'CHCOOC2H5 + СО2 + СН3СН2ОН
Этим методом с прекрасным выходом декарбалкоксилируют-
,ся также p-кетоэфиры и а-цианэфиры.
Примеры:
о о
Js.coor NaC1_ дмсо JL
\__/ —-------------> \__/
85-95%
374
|| 153- 165° II
СН3(СНа)3СНССН3 ———>- СН3(СН2)ДСН3
СООС2Н5
NaCl-ДМСО
135-165=
ncch2cooc2h5 ---------CH3CN
NaCl-ДМСО
152-168°
CH3(CH2)3CHCN -----------CH3(CH2)4CN
COOC2H5
Вместо ДМСО (т. кип. 189°) можно использовать диметил-
формамид (т. кип. 153°), однако в последнем случае необходимо
увеличить продолжительность реакций.
1. Krapcho А, Р„ Lovey A. J., Tetrahedron Letters, 957 (1973).
НАТРИЯ ЦИАНБОРГИДРИД, NaBH3CN. Мол. вес 62,84,
т. пл. 240—242° (разл.).
Для большинства целей пригоден продажный препарат, ко-
торый при необходимости можно очистить по методу Вейда и
сотр. [1].
Восстановление алкилгалогенидов и тозилатов [2,3]. Восста-
новлением с помощью Н. ц. в ГМТФК удается быстро, селек-
тивно и с высоким выходом удалить иод-, бром- и тозилокси-
группы. Так, например, 1-иоддекан восстанавливается этим ме-
тодом до «-декана с выходом 93 %, а 1 -додецилтозилат — до
«-додекана с выходом 61—80%. Для получения удовлетвори-
NaBH3CN, ГМТФК
СН3(СН2)8СН21 ----—-------> СН3(СН2)8СН3
NaBH3CN. ГМТФК , ru
CH3(CH3)10CH3OTs ---—— -------> СНз(СН2)10СН3
О 1 О J 7U
тельных выходов при восстановлении тозилатов требуется боль-
шой избыток Н. ц.
Поскольку Н. ц. представляет собой очень мягкий восстано-
витель, рассматриваемый метод особенно перспективен в случае
соединений, содержащих группировки, чувствительные к дейст-
вию более сильных восстановителей, такие, как СООН, COOR,
CN, NO2 и С —О и, следовательно, он более селективен, чем
восстановление боргидридом натрия в ДМФА (VI, 185).
Первичные спирты можно восстановить до углеводородов в
две стадии: сначала превратить их в иодиды, действуя иодме-
тилатом трифенилфосфита (III, 416—417; V, 456; относительно
очистки этого реагента см. [4]), а затем полученные иодиды вос-
становить с помощью Н. ц. в ГМТФК.
375
Восстановительное аминирование. Согласно методике Борха
[5], к раствору 21 г (0,25 моля) хлоргидрата диметиламина в
150 мл метанола, находящемуся в круглодонной колбе на 500,«л,
добавляют при перемешивании магнитной мешалкой сразу 4 г
гидроокиси калия (в виде гранул). Тотчас же выделяется оса-
док хлористого калия, однако это не мешает дальнейшей реак-
ции, Когда гранулы КОН полностью растворятся, прибавляют
в один прием 20 мл (0,20 моля) циклогексанона. Образовав-
шуюся суспензию перемешивают при комнатной температуре в
течение 15 мин и затем по каплям в течение 30 мин при переме-
J?
Г I + (СН3)гЫН'НС1 + NaBH3CN ------> Г ]
шивании приливают раствор 4,75 г (0,075 моля) Н.ц. в 50 мл
метанола. По окончании прибавления Н.ц, суспензию переме-
шивают в течение 30 мин. Затем добавляют гидроокись калия
(15 а) и суспензию продолжают перемешивать до полного рас-
творения гранул КОН, Реакционную смесь фильтруют с отсасы-
ванием, фильтрат упаривают на роторном испарителе при тем-
пературе бани ниже 45° приблизительно до объема 50 мл.
К остатку приливают 10 мл воды и 25 мл насыщенного раствора
хлористого натрия и разделяют слои. Водный слой экстраги-
руют эфиром, эфирные вытяжки объединяют с органическим
слоем и извлекают 6 М НС1. Кислотные вытяжки насыщают
хлористым натрием и экстрагируют четырьмя порциями эфира
(по данным газохроматографического анализа, эфир экстраги-
рует циклогексанол), К водному раствору при перемешивании
и охлаждении в бане со льдом осторожно добавляют гранулы
КОН до pH > 12. Образовавшиеся слои разделяют и водный
слой экстрагируют двумя порциями эфира по 40 мл. Органиче-
ские вытяжки объединяют, промывают 10 мл насыщенного рас-
твора хлористого натрия, высушивают над поташом и удаляют
эфир иа роторном испарителе. Неочищенный продукт фракцио-
нируют с 15-сантиметровой колонкой Вигре. После удаления
остатков растворителя собирают 1—3 г предгона с т. кип. 144—
155°, по данным ГЖХ содержащего 80—85% 1Ч,М-диметилцик-
логексиламина и 15—20% циклогексанола, Бесцветный N,N-hh-
метилциклогексиламин с т. кип,- 156—159°, п? 1,4521 (чистота
99,2%) собирают в количестве 15,7—17,5 а (62—69%).
Приведенная методика применима для восстановительного
аминирования циклогептанопа, циклооктанона, ацетофенона,
норборнанона и бензальдегида, В качестве амина в реакции
можно использовать аммиак, метиламин, диметиламип, морфо-
лин, анилин и гидроксиламин.
376
Селективное восстановление алифатических кетонов и альде-
гидов до углеводородов [6], Алифатические кетоны и альдегиды
можно с высоким выходом селективно восстановить до углево-
дородов, действуя Н.ц. и n-толуолсульфонилгидразином в смеси
ДМ.ФА — сульфолан в присутствии n-толуолсульфокислоты при
100—105°. При этом нет необходимости в предварительном по-
лучении тозилгидразонов, так как свободные СО-группы Н. ц.
восстанавливает медленно. Максимальные выходы углеводоро-
дов получены при использовании четырехкратного молярного
избытка NaBH3CN. Выходы колеблются в пределах 62—98%.
Сложиоэфирные группы при этом не затрагиваются, аромати-
ческие кетоны не восстанавливаются.
Примеры:
CH3CO(CH3)3COO(CH2)0CN
Холестанон-3 --->
88%
4-трет-Бутилциклогексанон ---
СН3(СН2)9СНО
CH3CH2(CH2)3COO(CH2)6CN
Холе стан
тр ет- Бут ил циклогексан
СН3(СН2)9СН3
Выходы углеводородов, получаемых этим методом, выше, чем
при восстановлении тозилгидразонов с помощью NaBH4 (V,
319—320), кроме того, Н.ц. действует гораздо селективнее, чем
NaBH4.
Селективное восстановление. Борх и сотр. [7] недавно опуб-
ликовали результаты исследования восстановления различных
функциональных групп под действием Н.ц. Карбонильные
группы в нейтральной среде восстанавливаются в незначитель-
ной степени, одиако при pH 3—4 восстановление происходит
быстро. Кетоксимы при pH ~4 гладко восстанавливаются до
соответствующих алкил гидрокси л аминов. При восстановлении
альдоксимов образуются преимущественно диалкилгидроксил-
амины.
NOH
СН3ССН2СН3
C3H6CH=NOH
NaBH3CN, рН~4
73%
NaBH3CN, рН~4
60%
NHOH
сн3Ансн2сн3
(C6H6CH2)2NOH
Енамины не восстанавливаются в нейтральных средах, в кис-
лых же превращаются в соли имииия, которые восстанавли-
ваются очень легко:
ЫаВНдСЫ
377
Выходы продуктов восстановления колеблются в пределах 50—
85%. Такое легко протекающее восстановление солей иминия
позволяет осуществить с помощью Н. ц. восстановительное ами-
нирование альдегидов и кетонов при pH ^6:
С—O + HNR2
+ /К NaBHgCN \
C=bK ---------► HCNR2
XR /
Так, например, взаимодействие циклогексанона, н-пропиламина
и Н. ц. в метаноле при 25° и pH 6—8 в течение 24 час приводит
к образованию н-пропилциклогексиламина с выходом 85%. Эта
реакция восстановительного аминирования является общей для
аммиака, первичных и вторичных аминов, ароматические амины
реагируют вяло. В реакцию вступают все альдегиды и относи-
тельно пространственно незатрудненные кетоны. Выходы ами-
нов можно повысить использованием молекулярных сит марки
ЗА для связывания выделяющейся в реакции воды. Необходимо
отметить, что восстановительное аминирование аммиаком заме-
щенных пировиноградных кислот приводит к «-аминокислотам.
Так, например, из пировиноградной кислоты можно получить
аланин с выходом 50%. Оптимальным для синтеза «.-аминокис-
лот является pH 7.
Амиды и нитрилы не восстанавливаются Н. ц. даже при pH 2.
Сложные эфиры, кислоты, лактоны также инертны по отноше-
нию к реагенту. Хлораигидриды кислот восстанавливаются в
ТГФ до соответствующих спиртов.
Метилирование аминов [8]. Алифатические и ароматические
амины, основность которых соответствует значениям рКа от
10,66 до 2,47, под действием водного раствора формальдегида
и Н. ц. в ацетонитриле вступают в реакцию восстановительного
метилирования с выходами 45—90%. Рассматриваемая методи-
ка лучше метода Кларка — Эшвейлера [9], приводящего к обра-
зованию сложных смесей продуктов [10].
1. Wade R, С., Sullivan Е. А., Berschied J. R., Jr., Purcell К. F.,
Inorg. Chem., 9, 2146 (1970).
2, Hutchins R. O„ M a г у a и о f f В. E., M 11 e w s k 1 C. A„ Chem. Comm.,
1971, 1097.
3, Hutchins R. O., Maryanoff В, E., MHewski C. A., Org, Syn.,
submitted 1972.
4. V e r h e у d e n J., M о f t a 11 J. G., J. Org. Chem,, 35, 2319' (1970).
5, Borch R. F>, Org. Syn,, 52, 124 (1972),
6. Hutchins R. O., Maryanoff В. E., M11 e w sk I C. A,, J, Am. Chem,,
Soc,, 93, 1793 (1971).
7. Borch R. F., Bernstein M. D., Durst H. D„ J. Am, Chem, Soc., 93,
2897 (1971).
8, В о г c h R, F., H a s s i d A, 1., J, Org. Chem,, 37, 1673 (1972).
9. Moore M. L,, Org, React., 5, 301 (1949),
10. Pine S, H., Sanchez B. L., J. Org. Chem., 36, 829 (1971).
378
НАТРИЯ ЦИАНИД, NaCN. Мол. вес 49,02.
Цианборирование. Метод цианборирования объединяет реак-
ции, в которых в результате миграции алкильных остатков от
бора к углероду промежуточно образуются соли циаибориой
кислоты. При гидроборировании дибораном 1-метилциклопен-
теиа (1) образуется трис- (транс-2-метил циклопентил) -боран
(2), окисление которого приводит к т’ра/-/с-2-метилциклопеитано-
лу (3) [1]. Если к раствору борана (2) добавить Н. ц., а затем
обработать реакционную смесь ангидридом трифторуксусной
кислоты, то последующее окисление образовавшегося промежу-
точного соединения щелочным раствором перекиси водорода
приводит к кетону (4) с выходом 82%.
В этой реакции наблюдается миграция двух алкильных
остатков от атома бора к атому углерода.
В среде пиридина или ДМФА возможна миграция и третье-
го алкила от бора к углероду. При взаимодействии соединения
(2) с Н. ц. в пиридине или ДМФА с последующим окислением
в щелочной среде образуется карбинол (5) [3], т. е. происходит
миграция третьей объемистой группы от бора к углероду. Лег-
кость миграции алкильных групп уменьшается в ряду: первич-
NaCN
(2)
Ру или ДМФА
щец, ОН- '
I) Н
2) О3
3) Zn/HOAc
(5)
(6)
ные > вторичные > третичные для миграции каждой группы.
Необходимо отметить, что конфигурация мигрирующих групп
в реакциях цианборирования сохраняется.
379
Присоединение альдегидов к активированным двойным свя-
зям [4]. Альдегиды (1) реагируют с эфирами а,р-ненасыщенных
карбоновых кислот (2а), кетонами (26) или нитрилами в при-
сутствии каталитических количеств Н. ц., образуя эфиры у-ке-
токарбоновых кислот (За), у-дикетоны (36) или у-кетонитрилы
с выходом 30—90%. Реакцию проводят в полярных растворите-
лях, таких, как ДМФА или ДМСО.
I.
RC
I
CN
J
ОН R1 О
I 1 I ,
R-С—СН-СН=С-X
I
CN
+ RICH=CHC-X -------->
(2)
а) X = OR*
б) X = R*. Аг
OR1 О
I! I II
RC —СН —СНгС—X + CN
(3)
а) X » OR*
б) X « иг, Аг
1. Brown Н. С., R о g 1 с М. М.., Rathke М. W., К а Ь а 1 k a G. W., J. Ат.
Chem. Soc., 91, 2150 (1969).
2. Pel ter A., Hutchings M. G-, Smith K-, Chem. Comm., 1970, 1529;
1971, 1048.
3. Pel ter A., Hutchings M. G., Smith K„ J. C. S. Chem. Comm., 1973,
186.
4. Stet ter H., Schreckenberg M., Angew. Chem., Internat, Ed., 12, 61
(1973).
НАТРИЯ ЦИАНИД —ДИМЕТИЛСУЛЬФОКСИД (V, 326—
327).
Декарбалкоксилироваиие (V, 326—327). Заключительная ра-
бота [1].
Усовершенствованную методику см. Натрия хлорид—диме-
тилсульфоксид, этот том. Ван Тамелен и Андерсон [2] осущест-
вили декарбметоксилирование соединения (1) с выходом 81%,
действуя на него раствором цианида натрия в ДМСО при 130°.
Они отметили, что для получения удовлетворительных выходов
снгсоосн3
(2)
38Q
требуются условия более мягкие, чем те, в которых реакция про-
водилась первоначально (V, 326—327, ссылка 1).
1. Krapcho А. Р., Mundy В. Р„ Tetrahedron, 26, 5437 (1970).
2 van Tamelen Е. Е., Anderson R. Т., J. Am. Chem. Soc., 94, 8225
(1972).
НАТРИЯ ЭТИЛМЕРКАПТИД, NaSC2H5. Мол. вес 84,13.
Реагент получают in situ при взаимодействии гидрида нат-
рия (60 %-ной дисперсии в масле) с этилмеркаптаном в ДМФА.
Расщепление арилметиловых эфиров [1]. Реагент расщепляет
арилметиловые эфиры с высоким выходом. Реакцию следует
проводить в вытяжном шкафу, так как происходит выделение
метилэтилсульфида, обладающего отвратительным запахом.
NaSCaHs
ДМФА
АгОСНз ------> Аг ОН 4- CH3SC2H5
Метод представляет особую ценность в случае эфиров, чувстви-
тельных к кислотам. Другой важной особенностью рассматри-
ваемой реакции является то обстоятельство, что с помощью
этого реагента можно осуществить избирательное монодемети-
лирование диметиловых эфиров, например превратить димети-
ловый эфир орсина (1) в монометиловый эфир орсина (2) с вы-
ходом 88—96%.
I. Feu trill G. I., Mir rington R. N., Tetrahedron Letters, 1327 (1970);
Org. Syn., submitted 1972.
НИКЕЛЬАЛЮМИНИЕВЫЙ СПЛАВ (1:1, сплав Ренея)
(II, 434—435; V, 327—328).
Восстановление ароматических нитрилов до альдегидов по
Стефену [1]. В 2-литровую двугорлую круглодонную колбу,
снабженную механической мешалкой и обратным холодильни-
ком, помещают 40 г п-цианбеизолсульфамида, 600 мл 75 %-ной
(по объему) муравьиной кислоты и 40 а никеля Ренея. Смесь
нагревают при перемешивании в течение 1 час, затем фильт-
руют с отсасыванием на воронке Бюхнера, а остаток промы-
вают двумя порциями 95%-ного этанола по 160 мл каждая.
сно
CN
ni—ai/hco2h
— ------>
SOaNH2
SO2NH2
381
Объединенные фильтраты упаривают досуха на роторном испа-
рителе при пониженном давлении. Твердый остаток растворяют
в 400 мл кипящей воды, небольшое количество нерастворимых
частиц удаляют фильтрованием через тампон из стекловаты,
помещенный в коническую воронку. Фильтрат охлаждают в ба-
не со льдом, выпавший осадок отсасывают, промывают и высу-
шивают в вакууме.
Сырой продукт весит около 32 а, т. пл. 112—114°. Перекри-
сталлизация из этанола с активированным углем дает 25,6—
28,0 г (62,9—68,8%) n-формилбензолсульфамида, т. пл. 117—
118°.
1. van EsT,, StaskunB., Org. Syn., 51, 20 (1971).
НИКЕЛЯ БОРИД, Ni2B (II, 435—436; V, 328; VI, 190—192).
Катализатор гидрирования. Расселл и Хой [1] применили
Н.б. в качестве катализатора селективного восстановления
С=С-связей, не сопровождающегося ни гидрогенолизом гидро-
ксильных заместителей, ни гидрированием карбонильных или
эпоксигрупп. Катализатор в виде коллоидного раствора черного
цвета получают восстановлением ацетата никеля 1,0 М раство-
ром боргидрида натрия в этаноле.
Катализатор рекомендуется также для гидрирования
С —С-связей в субстратах, имеющих в своем составе азотсо-
держащие функции. Аминные и амидные группы не затраги-
ваются, а нитрильные восстанавливаются до первичных ами-
нов [2].
1. Russell Т. W., Hoy R. С., J. Org. Chem., 36, 2018 (1971).
2. Russell Т. W., Hoy R. C., Cornelius J. E., J. Org. Chem., 37, 3552
(1972).
НИКЕЛЯ БРОМИД, NiBr2. Ata. вес 218,52.
Реакция 1,3-диеиов с соединениями, содержащими активный
водород. Н. б. в сочетании с диалкоксифенилфосфином катали-
зирует конденсацию 1,3-диенов с соединениями, содержащими
активный водород, с образованием бутенильных и октадиениль-
ных аддуктов. Так, например, при взаимодействии 0,05 моля
морфолина (1) с 0,15 моля бутадиена в присутствии 1 ж моля
Н.б. и 1,1 .чмоля диизопропоксифенилфосфина при 100° в тече-
ние 30 мин исходные вещества на 79% превращаются в аддук-
ты (2), (3), (4) и (5).
Ni кат ал изатор
(2) 32%
(3) 33%
382
(4) 33%
(5) 22^
Подобного рода конденсация уже была осуществлена ранее,
правда с более низкими выходами, под действием сложного ка-
тализатора, состоящего из ацетил ацето ната никеля (II),
Ni(C5H7O2)2, диизопропоксифенилфосфина и боргидрида натрия
[2]. Наличие в составе катализатора боргидрида весьма суще-
ственно для достижения высоких степеней превращения. Соли
никеля по каталитической способности в системе бутадиен —-
амин — диалкоксифенилфосфин можно расположить в ряд
NiBr2 NiCl2 > Ni(OAc)2 №(лаурат)2 ^>Ni(acac)2.
Порядок реакционной способности диалкоксифенилфосфинов
следующий:
С6Н5Р[ОСН(СН3)2]2 > CSH5P(OC3H5)2 > CQH5P(OCH3)2
Н. б. оказался эффективнее ацетилацетоната никеля (II) так-
же и в конденсации метилбензилкетона с бутадиеном-1,3. Реак-
ция этого соединения, содержащего активную метиленовую
группу, с бутадиеном-1,3 в присутствии NiBr2 и фенолята нат-
рия в качестве сокатализатора при 75° в течение 16 час приво-
дит к образованию аддуктов (6), (7), (8) и (9) со степенью
превращения 82%.
Ni COCH,
с.и.си,тер катализатор _ I -
/снс$н5 +
- Т
сн
С(н,х '"'GOCH,
(М 13^ (7) 8^
CSHS
(с) 745; (9) 55j
В случае соединений, содержащих активную метиленовую
группу, соли никеля по реакционной способности составляют
ряд
NiBr2 ~ NiCl2 > Ni(acac)2 > Ni(OAc)2 ~ М1(лаурат)2
В катализируемых никелем реакциях с аминами боргидрид
натрия, по-видимому, предотвращает образование комплексов
383
между Ni(acac)2 и амином. Даже в тех случаях, где боргидрид
натрия не требуется, его добавление вызывает некоторое увели-
чение скорости реакции. Роль фосфина заключается в восста-
новлении Ni (II) до Ni (0).
1. Baker R., С о о k А. Н., Smith Т. N., Tetrahedron Letters, 503 (1973).
2. Shields Т. С., Walker W. Е., Chem. Comm., 1971, 193; Baker R.,
Halliday D. E., Smith T. N„ Chem. Comm., 1971, 1583; Baker R.,
Halliday D. E., Smith T. N., J. Organometallic Chem., 35, C61 (1972),
НИКЕЛЯ КАРБОНИЛ (II, 436—439; V, 328—331; VI, 192—
194).
Внимание^. H. к. чрезвычайно токсичен и обращаться с ним
следует крайне осторожно [1].
Обзор. В опубликованном Земмельхаком [2] обзоре рассмот-
рено образование углерод-углеродных связей через л-аллилни-
келевые соединения.
Реакция арилгалогенидов с галогенидами л-аллилникеля.
Получение металлилбензола [3]. Выход металлилбензола состав-
ляет 67—72%.
сн3
2 CUz СНгВг + 2 Ni(CO)4
—> СН3
50-70°
^,СН2^В^ СНг
-С- Ni Ni-—-С — СН3
СНг В г СНг
C6H5Br
(CH3)ZNCHO + 2
+ 2 NiBra
Диарил- или диалкилкетоны [4]. Ртутьорганические галоге-
ниды реагируют с Н.к. в ДМФА или ТГФ при 60—70° в течение
20 час, образуя с высоким выходом кетоны. Например, фенил-
меркурхлорид или фенилмеркурбромид превращаются в бензо-
фенон с выходом 92—95%:
ДМФА
2 C6H5HgCl(Br) + Ni(CO)< —> С6Н5СС6Н5 + 2 Hg + NiCI2(Br2) + ЗСО
92—95 ||
о
Несимметричные диарилкетоны получают из иодбензола и Н.к.
в присутствии ртутьорганического галогенида:
-СО ArHgCI
C6H5I -J- Ni(CO)4 rC6H5CNi(CO)2"| ► CgHsCAr
III II
L oi J о
Реакция с циклооктииом [5], При взаимодействии Н. к. с
циклооктином в эфире образуется никельсодержащий комплекс
384
красного цвета (1), который разлагается при 300°, давая жел-
того цвета кетон (2):
При окислении комплекса Н.к, с циклооктином (3) гекса-
нитратоцерратом (IV) аммония (IV, 184—185; V, 541—543; VI,
342—343; этот том) с последующей обработкой тетрафенилцик-
лопентадиеноном (III, 310—311) соединение (4) образуется
лишь в качестве побочного продукта, а главным продуктом яв-
ляется тример циклооктина (5).
Реакция л-аллилникельбромидных комплексов с хинонами.
Аллилбромиды реагируют с Н.к., образуя л-аллилникельбромид-
ные комплексы, структура которых приведена ниже для случая
самого аллилбромида (1) (V, 329; VI, 192). Хегедас и сотр. [6]
изучали взаимодействие этих комплексов с хинонами. ц-Бензо-
хиион реагирует с л-аллилникельбромидным комплексом, обра-
Нг
Hc'f Ni
Х'С/ ' Вг/
Н2
нг
^.С
; СН
с'х
(1)
зуя аллилгидрохинон (2) с выходом 58%. Другие хиноны, одна-
ко, дают главным образом аллилзамещенные хиноны. Например,
13 Зак. 506 385
---->
58^
(2)
л-комплекс, полученный из 2-металлилбромида и Н. к., дает с
2-метилбензохиноном соединение (3) с выходом 45%.
сн,
I
СН2=ССНгВг + Ni(CO)| , > Ц-Комплекс
(3)
Этот новый метод алкилирования был использован в изящ-
ном синтезе пластохинона-1 (4), исходя из 2,3-диметилбензо-
хинона и З-метилбутен-2-илбромида; выход 61 %. Реакцией
2,3-диметокси-5-метилбензохинона с л-комплексом
(GHjjC — СНСЩВг •+ Ni(CO)4 -- >П-Номплене
(4)
З-метилбутен-2-илбромида и Н. к, был получен с выходом 40%,
и кофермент Qi (5).
(CHJ)jC=CHCHiBr
Основным побочным продуктом в этом синтезе является со-
ответствующий исходному хинону гидрохинон, который можно
выделить из реакционной смеси и окислить. Хиноны с несколь-
кими незанятыми положениями алкилируются неспецифично, но
с образованием лишь моноалкилироваиных продуктов.
386
Синтез макролактоиов [7]. Недавно было показано, что ме-
тодом внутримолекулярной аллильной конденсации (II, 438—
439; V, 328—331; VI, 193) можно синтезировать макроцикличе-
ские лактоны. При добавлении дибромэфира (1) к 6 же Н. к.
в N-метилпирролидоне происходит циклизация с образованием
в качестве главного продукта макролактона (2).
1. «Nickel Carbonyl is Dangerous», Technical Bulletin, Mathesoe Co.
2. SemmelhackM, F., Org. React., 19, 115 (1972).
3. Semmelhack M. F., Helquist P. M., Org. Syn., 52, 115 (1972).
4. H i г о t a Y., Ryang M„ Tsutsumi S., Tetrahedron Letters, 1531 (1971).
5. Wittig G., Fritz e P, Ann., 712, 79 (1968).
6. H e g e d u s L. S. Waterman E. L., Catlin J., J. Am. Chem. Soc., 94,
7155 (1972).
7, С о г e у E. J., К i r s t H. A., J. Am. Chem. Soc., 94, 667 (1972).
НИКЕЛЯ ЦИАНИД, Ni(CN)2. Мол. вес 110,73.
Тримеризация циклооктина [1]. Ацетиленовый углеводород
(540 мг) кипятят в ТГФ (40 мл) с катализатором (550 мг) в
атмосфере азота в течение 20 час. Хроматографированием
выделяют 392 мг чистого тримера октадекагидротрициклоокта-
бензола.
I. W i 11 i g G., F r i t z e P., Ann., 712, 79 (1968).
НИНГИДРИН (индантрион-1,2,3). Мол. вес 178,14, т. разл.
125 .
Конденсация с эфирами фосфористой кислоты [1]. Трион (1)
реагирует в безводном хлористом метилеие с триметил-, три-
этил- и тринзопропилфосфитами, образуя бесцветные аддукты
13*
387
состава 2:1, которым приписывают структуру циклических на-
сыщенных пентаоксифосфоранов (2).
(RQ)3p
1, Mustafa A., S i d к у M. М., Z а у е d S. М. A. D., М a h г a n М. R., Ann,,
712, 116 (1968).
2-НИТРО-1-ДИМЕТИЛАМИНОЭТИЛЕН,
(CH3)2NCH = CHNO2 (2).
Нитровииилирование альдегидов и кетонов [1]. Альдегиды и
кетоны, содержащие метиленовую группу в a-положении, обра-
зуют с 2-нитро-1-диметилам иноэтиленом (2) 2-а^ц-нитроэтил-
иденпроизводные (3). Кетоны с двумя активными метиленовы-
ми группами могут реагировать с 1 или 2 же иитросоединения
(2). При взаимодействии производных 4-а^ц-нитрокротонового
альдегида с нитроуксусным альдегидом образуются соответст-
вующие производные 1,3-динитробензола.
Х\
/СН2 + (CH3)2NCH=CHNO2
у/
(1) (2)
с2н5ок
---------
-(CH3)2NH
Х\
^c=chch=no;k’
(3)
1. Severin R., Adhikary P., Dehmel E., Eberhard I., Chem. Ben,
104, 2856 (1971).
НИТРОЗОНИЯ БОРФТОРИД, NOBF4. Мол. вес 116, 83.
Реакция с олефинами [1]. Н. б. взаимодействует в ацето-
нитриле при температурах от —15° до 0° с моно- или симмет-
ричными дизамещенными олефинами (исключая тетразамещен-
ные), образуя соли 2-метил-Ы-оксиимидазолия (1), восстанав-
ливающиеся под действием бис- (2-метоксиэтокси)-алюмогидри-
да натрия (VI, 189—190; этот том) до 2,4,5-тризамещенных
имидазолов (3) с общим выходом 50—80%. Ацетонитрил можно
заменить бензонитрилом.
RCH=CHR
nobf4
CH3CN
0)
(2)
1. Scheinbaum Л1. L„ Dines M. B., Tetrahedron Letters, 2205 (1971).
388
НИТРОМЕТАН (11,459—461).
Спиро-[4,4]-нонатетраен (5) [1]. Это спиросоединение, кото-
рое долгое время никак не удавалось получить, было наконец
синтезировано из дихлорангидрида диаллилмалоновой кисло-
ты (1). Раствор вещества (1) в хлористом метилене, содержа-
щий несколько молярных эквивалентов нитрометана, обрабаты-
вают хлористым алюминием при 25° и после гидролиза водным
раствором хлористого аммония получают спиро-[4,4]-нонадиен-
2,6-дион-1,5 (2) с выходом 51%. Последний восстанавливают
гидридом алюминия (I, 38—39; V, 19; VI, 9—10) до диола (3),
снг=снснг СОС1
СНг=СНСНг COC1
CH3NO2
AICIj
СНгС1г
51%
А1НЭ
-----
30%
еос1г
Ру
С1 н
ко- mpem-Bu
15%,считая на(3)
обработка которого хлористым тионилом при 0° в ТГФ в при-
сутствии пиридина дает смесь дихлоридов (4). К раствору ди-
хлоридов (4) в диметиловом эфире тетраэтиленгликоля добав-
ляют затем избыток трет-бутилата калия. Раствор упаривают
при 0,1 мм с ловушкой, охлаждаемой до —196°. По истечении
1 час в ловушке собирается спиран (5), который выделяют в
индивидуальном состоянии методом препаративной ГЖХ. Прн
65° спиросоединение перегруппировывается в инден (выход
67%) и частично превращается в высокомолекулярные продук-
(6)
ты. Спиран быстро взаимодействует с тетрациаиэтиленом и ди-
метиловым эфиром азодикарбоновой кислоты, образуя соответ-
ственно моноаддукты (6) и (7).
389
1. Semmelhack M. F„ Foos J. S,, Katz S., J. Am. Chem. Soc,, 94, 8637
(1972).
НИТРОНИЯ БОРФТОРИД (II, 463—464; V, 336—337).
Реакция с олефинами [1]. Олефины (моно-, ди- и триз а ме-
шенные) реагируют с Н. б. в безводном ацетонитриле при —15°
с образованием продуктов, которые при гидролизе дают
N-(р-нитроалкил)-ацетамиды (производные ви^-нитроацетами-
да) с выходом 15—50%. Эта реакция представляет собой более
R'/ I
NHCOCH3
общий случай реакции Риттера [2], служащей для получения
аминов и амидов взаимодействием олефинов в кислой среде с
цианистым водородом или нитрилами.
1. S ch е inb aum М. L., Dines М., J. Org. Chem., 36, 3641 (1971).
2. R i 11 е г J. J., M1 n i e г 1 P. P„ J. Am. Chem. Soc., 70, 4045 (1948).
OOH
2-НИТРОПРОПАНА 2-ГИДРОПЕРЕКИСЬ, (CH3)2C
xno2
Мол. вес 121,10.
Получение. H. г, получают in situ окислением 2-нитропропана
кислородом воздуха, катализируемым хлористой медью.
N-Нитрозамины. Н. г. (2) и третичные амины (1) в пиридине
при 50° образуют диалкилнитрозамины (3) и альдегиды (4) [1]:
С2Н5ч /ООН C2H5s
XNCH2CH3 + (СН3)2с/ —> 41—NO + ОНССНз
CjHg^ -NO2 C2H5
(3) 50 %
1. Franck В., Conard J., Misbach P., Angew. Chem, Internal. Ed., 9,
892 (1970).
4-(4'-НИТРОФЕНИЛАЗО)-БЕНЗОИЛХЛОРИД,
°2N"C^N~N""ObCOC1' Мол- вес 289’67> т* пл- 163-165°.
890
Получение.
Идентификация спиртов [1]. Эфиры 4-(4'-нитрофенилазо)-
бензойной кислоты обычно представляют собой твердые веще-
ства; яркая красно-оранжевая окраска облегчает их хромато-
графическую очистку. Молекулярный вес этих эфиров легко
определить по поглощению хромофора в ультрафиолетовой об-
ласти спектра.
1. Nutting W. Н., Jewell R. A., Rapoport Н., J. Org. Chem., 35, 505
(1970).
о-НИТРОФЕНИЛСУЛЬФЕНХЛОРИД (II, 467—468).
и-Циаиаминокислоты. Чаймиак и Пастужак [1], предложив-
шие новый метод синтеза ш-цианаминокислот, нашли, что для
защиты аминогруппы в этой реакции наиболее пригодна о-нит-
рофенилсульфенильная (НФС) группировка. Так, НФС-произ-
водные ь-аспарагина (1) (п=1) и ь-глутамина (1) (п — 2)
удалось дегидратировать с помощью ДЦК до соединений фор-
мулы (2). Защитную группу удаляли действием тиоацета-
мида [2].
дцк, ch3csnh2
o-NO2C6H4S—NHCHCOOH ----> o-NO2C6H4S—NHCHCOOH ------->
I I
(CH3)rt (CH2)n
AoNH2 CN
(I, « = 1.2) (2)
—* H2N—CHCOOH
(<!:н2)ге
iN
(3)
N-Карбоксиангидриды N-сульфеииламинокислот [3]. Реагент
взаимодействует с N-карбоксиаигидридами аминокислот (NKA),
образуя с высоким выходом о-нитрофенилсульфенильные
391
(НФС) производные. Например, ь-фенилаланин-МКА (1) в этил-
ацетате или ТГФ дает с реагентом в присутствии триэтиламина
или 4-метилморфолина НФС-ь-фенилаланин-МКА (2) с выхо-
дом 93%. НФС-МКА-Аминокислоты перспективны для исполь-
HN-CHCH2C6H? 0-NO2C6H4SCl o-NOaCbH^-S-N-CHCHzCbHj
оХо-^о 93% > О^О^О
зования в пептидном синтезе, поскольку онн являются актив-
ными эфирами и реагируют не рацемизуясь. Например, при вза-
имодействии соединения (2) с триметилсилиловым эфиром
N-(три метил си л ил)-глицина в диоксане в присутствии сульфата
аммония (выделяется СОа) образуется с выходом 94% НФС-ь-
фенилаланилглицин.
1. С h i m i a k A., Pastuszak J. J., Chem. Ind., 1971, 427.
2. Kessler W., Iselin B., Helv. Chim. Acta, 49, 1330 (1966).
3. Kricheldorf H. R., Angew. Chem., Internat Ed., j2, 73 (1973).
НИТРОЭТАН. CH3CH2NO2. Мол. вес 75,07, т. кип. 112—114°.
Ацилирование по Фриделю — Крафтсу [1]. Наиболее суще-
ственное усовершенствование методики ацилирования «-ксилола
ангидридом метилянтарной кислоты по Фриделю — Крафтсу со-
стоит в замене нитробензола более летучим нитроэтаном;
3-(2,5-диметилбензоил)-2-метилпропионовую кислоту (3) можно
получить таким путем с выходом 82%. Этот растворитель спо-
соон
с ООН
собствует также преимущественному образованию кислоты (3),
а не (4).
1. Eisenbraun Е. J., Hinman С. W., Springer J. М., Burnham J. W.,
Chou Т. S., J. Org. Chem,, 36, 2480 (1971).
ОЗОН (in, 5—10).
Озонирование 2,6-б«с-метиленспиро-13,3]-гептана (2а) и 2,10-
бис-метилентриспиро-[3,1,1,3,1,1]-тридекана (4а) приводит к об-
разованию 2,6-диоксоспиро-[3,3]-гептана (26) и 2,10-диоксотри-
спиро-[3,1,1,3,1,1]-тридекана (46) соответственно [1]:
нгс
%
СНг --> О:
(4а)
(2а)
(26)
СН; -----* О.
(46)
Альдегиды -> сложные эфиры. Оз практически количественно
реагирует с ацеталями, образуя соответствующие сложные эфи-
ры. По-видимому, реакция начинается с внедрения Оз по
С—Н-связи ацеталя [2].
Примеры:
ОСН:
С6Н13С'^
осн3
I осн3
О-О-О-Н
» С6Н13СОСН
о
п
с5н13сн
4 о
о
II
С6Н13СОСНгСНгОН
1. Buchta Е., KronigerA., Ann,, 716, 112 (1968).
2. D е s 1 о n gc h a m p s P., Moreau C., Can. J. Chem., 49, 2465 (1971).
ОКИСЬ ЭТИЛЕНА (HI, 11 — 12; V, 339—340; VI, 197).
Генерирование дигалогенкарбеиов (V, 339—340). Будрус [1]
опубликовал обзор по применению О. э. для генерирования ди-
393
галогенкарбеиов согласно приведенному ниже уравнению:
(C2H5)4NBr
СН2Х
1
снгон
2,3,За,8а-Тетрагидрофуро-[3,3-Ь]-индолы. Эту циклическую си-
стему содержат некоторые алкалоиды калабарских бобов. Не-
давно один из них, физовенин (3), был синтезирован [2]. Ключе-
вой стадией синтеза этого алкалоида является реакция йодисто-
го 5-метоксискатолилмагния (1), полученного из 5-метоксиска-
тола и йодистого метилмагния, с безводной О. э. в эфире. Выход
продукта (2) составляет 13%.
1. В u d d г u s J., Angew. Chem., Internal. Ed., 11, 1041 (1972).
2. Onaka T., Tetrahedron Letters, 4391 (1971).
ОКСАЛИЛХЛОРИД (in, 13—19; V, 340; VI, 197—198).
Изоцианатхиноны. Первый представитель этого класса соеди-
нений 2,5-дихлор-3,6-диизоцианат-1,4-бензохииои (2) был полу-
чен реакцией 2,5-диамино-3,6-дихлор-1,4-бензохинона (1) и О.
в смеси бутил ацетата и хлорбензола [1].
(1) 1002 .
(2) 50г
1. von Gizycki U., Angew. Chem., Internat. Ed., 10, 403 (1971).
4-ОКСИМЕТИЛ-2,6-ДИ-трет-БУТИЛФЕНОЛ («этил» анти-
оксидант 754). Мол. вес 218,33, т. пл. 141°, т. кип. 16272,6 мм.
Растворимость при 20°, вес. %
Метанол...............39,2
Ацетон................37,1
Этанол................23,4
Изопропиловый спирт . . 18,7
Бензол................ 7,2
Изопентан............. 0,5
Вода.................... < 0,0035
394
он
(СН3)ЭС
С(СН3)
СН2ОН
Эффективный антиоксидант для пластмасс, резин и восков.
Его применяют для стабилизации жиров и масел в пищевой
промышленности, особенно в тех случаях, когда желателен ма-
лолетучий антиоксидант, не обладающий вкусом и запахом.
ОРТОМУРАВЬИ НОЙ КИСЛОТЫ ТРИМЕТИЛОВЫЙ
ЭФИР (VI, 200—201).
Об упрощенном методе синтеза см. Ортомуравьиной кисло-
ты триэтиловый эфир (этот том).
ОРТОМУРАВЬИНОЙ КИСЛОТЫ ТРИЭТИЛОВЫЙ ЭФИР
(III, 33—40; V, 342—343).
Описан более простой метод синтеза О. к. т. э. взаимодейст-
вием хлористого бензоила или этилового эфира хлоругольиой
кислоты с этанолом и формамидом [1]:
OCHNH2 + ЗС2Н5ОН + С3Н5СОС1 НС(ОС2Н5)3 + с6н5со2н + NH4C1
OCHNH2 + 3C2HeOH+C1CO2C2H5 > HC(OC2H5)3 + CO2 + NH4C1
1. О h гл е R., Schmitz Е., Ann., 716, 207 (1968).
ОРТОУКСУСНОЙ КИСЛОТЫ ТРИЭТИЛОВЫЙ ЭФИР
(VI, 202—204).
грамс-Тризамещенные олефины [1]. Труст и Айрланд [1]
предложили метод получения этилового эфира транс-4-метил-
нонадиен-4,8-овой кислоты клайзеновской перегруппировкой
аллилвииилового эфира, являющегося производным 2-метилгеп-
тадиен-1,6-ола-3 (1):
ОН СНз
СН3С(ОС2НЕ)3
СНзСНоСООН
- ~ 83-88%
(П
Н< .CHjCHjCOOCsHs
—>
СН2=СНСН2СН2'/ \сн3
(2)
1. Trust R. I., Ireland R. Е., Org. Syn., submitted 1972.
ОСМИЯ ЧЕТЫРЕХОКИСЬ—-КАЛ ИЯ ХЛОРАТ, (см. III,
46—47; V, 343—344).
395
Окисление. На одной из стадий синтеза фуранеола (3), ос-
новного компонента запаха ананасов и земляники, Бюхи и
сотр. [1] попытались осуществить гидроксилирование 2,5-диме-
тил-2,5-диметокси-2,5-дигидрофурана (1) путем окисления его
(15,8 ммоля) хлоратом калия (22,8 ммоля) и четырехокисыо
осмия (0,3 ммоля) в водном ТГФ в присутствии бикарбоната
натрия при 30° в течение 63 час [2], однако выход ожидаемого
диола составил лишь 10%. Бюхи предположил, что причиной
(1) (г) ' (з)
низкого выхода был гидролиз. Тогда окисление провели в среде
с большим содержанием воды, но в отсутствие бикарбоната
натрия. В этих условиях диоксикетон (2) эритро-конфигурации
был получен с выходом, близким к количественному. Дегидра-
тация кетона (2) под действием бикарбоната натрия или кис-
лого динатрийфосфата (Na2HPO4) привела к фуранеолу (3) с
выходом более 50%.
1, Buehl G., Demole Е., Thomas A. F„ J, Org. Chem., 38, 123 (1973).
2. М u х f e 1 d t H., Har dim ann G., Ann., 669, 113 (1963).
ПАЛЛАДИЕВАЯ ЧЕРНЬ (111, 48—53; V, 345).
3^-Окси-5а,8а-эпндиоксиаидростеи-6-он-17 (1) -* андроста-
триен-4,6,8(14)-днон-3,17 (3). В 1966 г. на собрании Американ-
ского химического общества Додсон и сотр. [1] сообщили, что
эпидиокись эргостерина при перемешивании с палладиевым ка-
тализатором в этаноле с высоким выходом превращается в эрго-
статриен-4,6,8(14) -он-3. Открытая Додсоном реакция недавно
была подробно описана в ряду андростана [2]. Например, при
обработке 2,14 г 3|3-окси-5а,8а-эпидиоксиандростен-6-она-17 (1)
10 г П. ч. (по Энгельгарду) в этаноле в течение 10 дней обра-
зуется андростатриен-4,6,8 (14)-дион-3,17 (3) с выходом 60%
(УФ-анализ). Триендион, вероятно, образуется через триол (2).
В самом деле, обработкой эпидиокиси (1) П. ч. (по Фишеру)
в этаноле при комнатной температуре в течение 18 час можно
получить триол (2) с выходом 70%. Затем триол (2) превра-
щается в триендион (3), по-видимому, путем каталитического
переноса водорода на растворитель, что приводит к окислению
ОН-группы в положении 3, сопровождающемуся отщеплением
воды.
1. Dodson R. М., Valiaveedan G. D,, Ogasawara Н., Т s и-
chiya Н. М., 151st National Meeting of the A. C. S., March, 1966, p. 1—33
2. Johns W. F., J. Org. Chem., 36, 2391 (1971).
ПАЛЛАДИЕВЫЕ КАТАЛИЗАТОРЫ (III, 48—53; V, 345).
, Восстановление по Розенмунду (IV, 120). Петерс и Ван Бек-
кум [1] описали удобный вариант метода восстановления хлор-
ангидридов кислот до альдегидов по Розенмунду [2], Этот спо-
соб основан на методике японских химиков [3], на которую
в свое время не обратили внимания. В качестве катализатора
используют палладий на угле, в качестве акцептора хлористого
397
водорода — этилдиизопропиламин, а в качестве растворителя —
ацетон (хлоргидрат амина растворим в ацетоне). Восстановле-
ние проводят при комнатной температуре и атмосферном дав-
лении.
Синтез аминокислот [4]. Фенилгидразоны а-кетокислот (1),
получаемые сочетанием солей фенилдиазония с активными ме-
тиленовыми соединениями по так называемой реакции Яппа —
Клингемана [5], легко гидрируются над 5% Pd/C (а также над
платиной) до аминокислоты (2). Фенилгидразон (1) можно
восстановить до аминокислоты (2) также цинковой пылью в
С6Н5
(1)
> Г о “1 1! _ RCCO |у+ NNC6H5 Нг/нго -H2NC6H5 ' О II RCC-OH - II NH -> RCHCOOH nh2
А
(2)
75%-ном спирте в присутствии сулемы или хлористого кальция.
Изомеризация сочленения колец А и В в дитерпенах
[6]. Стероидный тип сочленения колец А и В дитерпенов можно
изомеризовать в антиподный, обрабатывая дитерпен катализа-
тором 10% Pd/C [7] в кипящем триглиме. Например, метиловый
эфир 5а, 10а-подокарпатриен-8,11,13-овой-15 кислоты (1) можно
изомеризовать с выходом 83% в метиловый эфир 5|3,10а-подо-
карпатриен-8,11,13-овой-15 кислоты (2). Реакцию использовали
в синтезе (—)-подокарповоЙ кислоты из (4-)-дегидроабиетиио-
вой кислоты.
Такая же обработка эстрона (3) с умеренным выходом
(48%) дает (-%)-изоэквиленин (4):
398
1. Peters J. A., Van Bekkum H., Rec. trav., 90, 1323 (1971).
2. Мозеттиг Э., Мозинго P., «Органические реакции». ИЛ, М., 1951,
сб. 4, стр. 337.
3. S a k u г a i Y., Т a n a b е Y., J. Pharm. Soc. Japan, 64, 25 (1944).
4. Khan N. H., К 1 d w a 1 A. R., J. Org. Chem., 38, 822 (1973).
5. Japp R. F., Klingeman/i F., Ber., 20, 2942, 3284, 3398 (1887).
6. Pelletier S. W„ 1 c h i л о h e Y„ Herald D. L., Jr., Tetrahedron Letters,
4179 (1971).
7. Ross A., Smith P. A. S„ Dreiding A. S„ J. Org. Chem., 20, 905
(1955); Mathew C. T„ Gupta G. S., Dutta P. C., Proc. Chem. Soc.,
1964, 336.
ПАЛЛАДИЙ (10%) НА КАРБОНАТЕ КАЛЬЦИЯ — КАТА-
ЛИЗАТОР ГИДРИРОВАНИЯ (HI, 48—53; V, 345; VI, 206).
Гидрирование тридегидро-[18]-аннулена (3) до [18]'аннулена
(4) проводят в бензоле в присутствии суспензии катализатора
в аппарате для гидрирования. Общий выход [18]-аниулена из
гексадиина-1,5 составляет 0,63% [1].
(з) (4)
1. S t б с k е 1 К.., So и dheimer F., Org. Syn., submitted 1971.
ПАЛЛАДИЙ НА УГЛЕ (III, 172; V, 345; VI, 228).
Дегидрирование гидроароматических соединений было ис-
пользовано на последней стадии синтеза 3,4-бензпирена из
1-оксо-2-формил-1,2,3,4,5,6,7,8-октагидроантрацена [1]:
399
Катализатор гидрирования в реакции Розенмунда [2]. При
высушивании в вакууме при 115° в течение 48 час П.и.у. ста-
новится очень пирофорным. В автоклав емкостью 1,2 л загру-
жают 600 мл сухого толуола, 25 г (0,3 моля) высушенного в
вакуумной печи при 115° в течение 48 час ацетата натрия (слу-
жит внутренним акцептором хлористого водорода), 3 г сухого
10% Pd/C, 23 г (0,1 моля) 3,4,5-тр и метоксибензоил хлорида и
1 мл хинолина S [Org, Syn., Coll. Vol., 3, 629 (1955)]. Автоклав
продувают азотом, закрывают и после непродолжительного ва-
куумирования заполняют водородом до давления 3,5 атм. Смесь
встряхивают в течение 1 час при комнатной температуре и дав-
лении 3,5 атм, при необходимости подкачивают водород и на-
гревают при 35—40° в течение 2 час. Встряхивание продолжают
в течение 12 час, при этом смесь охлаждается до комнатной
температуры. Избыточное давление снимают и открывают авто-
клав. Смесь фильтруют через 10 г целита, а нерастворимый
остаток промывают 25 мл толуола. Объединенные фильтраты
промывают сначала 25 мл 5%-иого раствора карбоната натрия,
а затем 25 мл воды. Толуольный раствор высушивают над без-
водным сульфатом натрия, фильтруют и концентрируют в ва-
кууме водоструйного иасоса. Перегонка остатка с 10-саитимет-
ровой колонкой Вигре (для предотвращения кристаллизации
дистиллята через холодильник пропускают теплую воду) дает
12,8—16,2 а (64—83%) 3,4,5-триметоксибензальдегида, т. кип.
158—16177—8 мм, т. пл. 74—75°.
1. В и с h t а Е., Z о 1 1 л е г R., Ann., 716, 102 (1968).
2. Rachlin A. I., Gurien Н., Wagner D. Р., Org. Syn., 51, 8 (1971).
ПАЛЛАДИЙ ХЛОРИСТЫЙ (III, 53; V, 345—346).
it-Аллилпалладиевые комплексы (V, 346) [1]. Комплексы
л-аллилпалладия можно получить с выходом 80—100%, дейст-
вуя иа олефин либо П. х. в хлористом метилене в присутствии
карбоната натрия, либо П.х. в присутствии хлористого натрия
в уксусной кислоте, содержащей ацетат натрия. Так, например,
2-метилоктен-1 (1) количественно превращается в два л-аллил-
палладиевых комплекса (2) и (3) в отношении 1 : 1,6. Эти ком-
плексы можно алкилировать карбанионами. Например, смесь
комплексов (2) и (3) вступает в реакцию с карбанионом мало-
го
сн.
100%'
нового эфира в присутствии не менее 4 экв грифенилфосфина
[и отсутствие трифенилфосфииа реакция не идет; в качестве ли-
ганда можно использовать также бис-(дифенил)-фосфиноэтан]
В ТГФ или ДМФА при комнатной температуре за несколько ми-
нут с образованием смеси алкилированных олефинов (4) и (5)
с выходом 63%.
МаСН(СООСгН5)г
(г1 + (з) >
6 35S
(4)
। j- н
(5)
Гомологизация метильной группы в этильную была осуще-
ствлена следующим путем:
Конденсация фенолов [2]. П. х. использовали для конденса-
ции фенола (1) в соединение (2). Последнее при нагревании
в течение 2 час при 38° превращается в карпанон — лигиан, по-
лучаемый из коры карпаиового дерева. Следует отметить, что
401
в карпапоне (3) имеется пять смежных асимметрических цент-
ров.
1. Trost В. М., Fullerton Т. J., J. Am. Chem. Soc., 95, 292 (1973).
2. Ch а р та п О. L., Engel И. R., Springer J. Р., С 1 а г d у J. С., J. Ат,
Chem. Soc., 93, 6696 (1971).
ПАЛЛАДИЯ(П) АЦЕТАТ (111,53—54; V, 347).
л-Аллнлпалладневые комплексы [1]. Енамин 1-пиперидин-
циклогексен (1) при кипячении с аллилфениловым эфиром, П. а.
и трифенилфосфииом в бензоле дает после гидролиза 2-аллил-
циклогексаион (2) и 2,6-диаллилциклогексанон (3), образую-
щиеся через л-аллилпалладиевые комплексы:
Реакция енамина с 1,4-днацетоксибутеном-2 в присутствии
этого палладиевого катализатора приводит к бициклическому
кетоиу. Например, 1-пнперидинциклопентен (4) дает бицикло-
[4,2,1]-нонен-3-он-9 (5) с выходом 28%. В этой реакции проме-
жуточно образуется аллилзамещениый енамин, который в свою
очередь может образовывать л-аллилпалладиевый комплекс.
АсОСНгСН=СНСН>ОАс
1) Pd(OAc)2, Р(С6Н5)3
2) НгО
28%
1. Onoue Н., Moritani I., Murahashi S.-L, Tetrahedron Letters, 121
(1973).
402
1,2,2,6,6-ПЕНТАМЕТИЛ ПИПЕРИДИН, н3сХк><сн3
।
сн3
Мол. вес 155,28, бесцветная жидкость, р/<а 11,25
Получение [1]. Это осиоваиие получают метилированием
2,2,6,6-тетраметнлпиперидина иодистым метилом в метаноле при
комнатной температуре в течение 12 час. Образующийся с вы-
ходом 71 % иодгидрат 1,2,2,6,6-пентаметилпиперидина обраба-
тывают 5%-ным раствором NaOH, выделяя свободный амин.
Исчерпывающее алкилирование аминов. Соммер и сотр. [1,2]
разработали новый метод исчерпывающего алкилирования пер-
вичных и вторичных аминов сразу до четвертичных солей, про-
текающего при комнатной температуре. По обычной методике
кватернизацию аминов проводят при высоких температурах, а
образующуюся кислоту связывают сильным неорганическим
основанием (гидроокисью натрия, карбонатом натрия). В но-
вом методе используют сильное органическое основание, а в ка-
честве растворителя — ДМФА или ацетонитрил; реакция закан-
чивается за несколько часов. Особенно строгие требования
предъявляются к органическому основанию. Чтобы реакция бы-
ла гомогенной, оно должно обладать такой же растворимостью,
как и исходный амии. Основание должно быть более основным,
чем исходный амии, но алкилироваться оно должно медленнее.
В первой публикации об этом методе сообщалось, что для ква-
тернизации ароматических аминов с р/<а 3,86—5,34 подходит
2,6-лутиднн. В последней работе было показано, что для исчер-
пывающего алкилирования самых разнообразных аминов при-
годен 1,2,2,6,6-пеитаметилпиперидин. Это одно из самых силь-
ных из известных органических оснований, и, кроме того, ме-
тильные группы создают сильные пространственные затрудне-
ния для его алкилирования. Согласно общей методике, к рас-
твору амина или гидрата амииа и стехиометрического количе-
ства П. в подходящем растворителе (ДМФА или ацетонитрил)
прибавляют избыток йодистого метила и реакционную смесь
оставляют на 12 час прн комнатной температуре. Четвертичное
аммониевое соединение обычно выпадает в осадок; если этого
не происходит, то соль можно высадить добавлением менее по-
лярного растворителя (ацетона, этилацетата или эфира). Вы-
ходы иодидов четвертичных аммониевых катионов колеблются
в интервале 50—95%.
1. Sommer Н. Z., Lipp Н. I., Jackson L. L., J. Org. Chem, 36, 824
(1971).
2. Sommer H. Z., Jackson L. L., J. Org. Chem,, 35, 1558 (1970),
1,1,1,3,3-ПЕНТАХЛ ОРАЗАПРОПЕН (трихлорметил изонит-
рила дихлорид), C13CN=CC12. Мол. вес 215,31.
403
Реагент получают высокотемпературным хлорированием ди-
хлорида метилизонитрила [1],
Обзор [2]. N-Бензоилантраниловая кислота под действием П.
о
циклизуется в 4-кето-2-фенилбензо-[с/]-1,3-оксазин (1). Амиды
с помощью П. дегидратируются до нитрилов обычно с хороши-
ми выходами:
2R—С" 4-С13С—N=CCh —► R— CN + 4НС1 4-C1CN 4-СО;
\nh.
Карбоновые и сульфоновые кислоты с удовлетворительными вы-
ходами превращаются в соответствующие хлорангидриды:
2RCOOH+С13С—N=CC12 —> 2RCOC1 + СО2 + CICN + 2НС1
Реагент используют в синтезе гетероциклов, например
1,3,5-трназинов, пиримидинов, хиназолинов, 1,2,4-триазолов и
1,2,4-оксадиазолов:
С1
R-C + С13С-Н=СС1г — RCH2CsN + СЦС — М —СС1г + С13С—N—СС1г НС1 —> j | 1 3 5-Триазин Cl СГ-^n -^"CI Пиримидин CI - оЛ xX4:;' 'ci 2y4 “ Дихлорхииазолин
404
RNHNHa + C13C —N —CCi2 >
1,2,4-ТриаЗол
H;NOH + HC1 + C13C —N —CC12 g
3 5-Дихлор- 1, 2 ,A~ Онсадиазол
LHoltschmidt H., Degener E., Schmelzer I-I.-G., Tar now H.,
Zecher W., Angew. Chem., Internal. Ed., 7, 856 (1968).
2. Find ei sen K., Wagner K-, Holfschmidt H„ Synthesis, 1972,
599.
траяс-ПЕНТЕН-З-ОН-2 (V, 349--350; VIt 206—207).
Получение. Метод получения П., предложенный Одомом н
Пиндером (VI, 207), недавно опубликован [1],
Аинелнрование по Робинсону. Аннелирование по Робинсону
неактивироваиных циклогексанонов с П. в обычных условиях
если и протекает, то с малыми выходами. Однако Скаиио и
Старретт нашли, что натриевый енолят 2-метилциклогексанона
(1), полученный кипячением эквимолярной смеси NaH и (1) в
диоксане в течение 3 час, реагирует с 1 экв. П. [2] с обра-
зованием цис-4,10-диметил-Д1(9Аокталона-2 (3) с выходом 65%.
Если вместо диоксана взять ДМСО, то с выходом 72% обра-
зуется изомерный траке-окталон (4). Сканио и Старретт пола-
гают, что такой значительный эффект растворителя в случае
очень полярного ДМСО может быть связан с изменением обыч-
ного порядка стадий в реакции нивелирования, а именно
405
сначала имеет место альдольная конденсация, а затем — внут-
римолекулярная реакция Михаэля.
1. Odom Н. С., Pinder A. R., Org. Syn., 51, 115 (1971).
2. Scan io C. J. V., Starrett R. M., J. Am. Chem. Soc., 93, 1539 (1971),
ПЕРЕКИСЬ ВОДОРОДА, H2O2(III, 56).
Концентрированная П. в. (около 90%) — труднодоступный
реагент. Харт и сотр. [1] нашли способ повышения окислитель-
ной способности 30%-ной П. в. (обычно легкодоступной). На-
пример, 30%-ную П. в. добавляют при охлаждении к избытку
уксусного ангидрида в присутствии коиц. серной кислоты. Ре-
акция сопровождается большим выделением тепла, но легко
контролируется. Получающийся раствор легко окисляет гекса-
метилбензол (1) до гексаметилциклогексадиеи-2,4-она (2) с вы-
ходом (86%), сравнимым с тем, который получают при исполь-
зовании 90%-ной П.в. или смеси трифторнадуксусной кислоты
С BF3.
яиц-Гидропероксиспирты [2]. Взаимодействие эпоксидов с
90%-ной П.в. в эфире при комнатной температуре (в течение
приблизительно 2 недель) приводит к образованию яиц-гидро-
пероксиспиртов. Так, например, окись изобутилена (1) превра-
щается с выходом 70% в соединение (2), из которого действием
безводного сульфата меди (II) в ацетоне можно получить
нгог,
(с2н5)го
70% *
CuSO4
(СН3)гСЮ
50%
(3)
3,3,6,6-тетраметил-1,2,4-триоксан (3).
1. Blum J., Pickholtz Y., Hart H., Synthesis, 1972, 195.
2. Adam \V„ Rios A., Chem. Comm., 1971, 822.
ПЕРЕКИСЬ ВОДОРОДА —АЛЮМИНИЯ ХЛОРИД.
Гидроксилирование ароматических соединений [1]. Прибав-
ление 90 %-ной перекиси водорода (0,05 моля) при 0—5° к пе-
406
ремешнваемому раствору или взвеси хлористого алюминия
(0,075—0,10 моля) в избытке ароматического соединения при-
водит к образованию фенолов, выделяемых после разложения
АгН4~Н2О2 +А1С13 — > АгОН + НС1 + А1С12ОН
реакционной смеси льдом. Если взять А1С13 и Н2О2 в молярном
отношении . 2 : 1, можно использовать также более доступную
30%-ную перекись водорода. Конкурирующей реакцией являет-
ся хлорирование в ядро, протекающее, по-видимому, под дей-
ствием хлорноватистой кислоты. Выход фенола нз бензола со-
ставляет всего 10%; нитробензол не вступает в реакцию.
I. Kurz М. Е„ Johnson G. J., J. Org. Chem., 36, 3184 (1971).
ПЕРЕКИСЬ ВОДОРОДА В КИСЛОЙ СРЕДЕ (111,57—68;
VI, 207-208).
Изохроманон-3. Окислением инданона-2 (1) по Байеру-Вил-
лигеру смесью 90%-ной перекиси водорода с уксусным ангидри-
дом в присутствии серной кислоты [1,2] можно получить изо-
хромаион-3 (2) с выходом 68% (считая на чистый продукт).
Пиролиз соединения (2) при 565° (N2) дает с высоким выходом
565°
81%
(О
(И (3)
бензоциклобутен (3). Этот двухстадийный синтез является на-
иболее удобным методом получения соединения (3). На-
пример, в недавно описанном Маркграфом и сотр. [3] синтезе
407
бензоциклобутена (3), осуществленном, как показано ниже, об-
щий выход (3) составил всего около 20%.
1) «ЗО-AmONO
2) СНг—СНОАс
nh2
Li, NH3
трет- ВиОНДГФ
ОАс
42%
> (3)
Окисление циклобутаноиов в у-лактоны (VI, 208). Другие
примеры см. в работе Corey E.J., Pavindranathan Т., Tet-
rahedron Letters, 4753 (1971).
I. Markgrai J. H., Basta S, J„ Syn. Commun., 2, 139 (1972).
2. Spangler R. J., Kim J. H., Synthesis, 1973, 107,
3. Markgrai J. H., Basta S. J., Wege P. M., J. Org. Chem., 37, 2361
(1972).
ПЕРЕКИСЬ ВОДОРОДА В ЩЕЛОЧНОЙ СРЕДЕ (III,
70—77; V, 351; VI, 208).
у-Бутиролактоны [1]. Циклобутаноиы, ставшие легкодоступ-
ными благодаря открытию реакции карбонильных соединений,
с дифенилсульфонийциклопропилидом (этот том), окисляются
по Байеру — Виллигеру при обработке 30%-ной перекисью во-
дорода в присутствии гидроокиси натрия в среде метанол — во-
да, давая у-бутиролактоиы с высокими выходами (80—100%).
Реакцию можно осуществить так же, действуя гипобромнтом
натрия, полученным in situ из гидроокиси натрия и брома.
Легкость миграции алкильных групп уменьшается в ряду
третичные > вторичные > первичные, причем у мигрирующего
атома углерода сохраняется конфигурация.
Гидратация нитрилов алленкарбоновых кислот [2]. Нитрилы
4.4-диалкилзамещенных алленкарбоновых кислот (1) под дей-
ствием перекиси водорода в спиртовом растворе гидроокиси
натрия превращаются с выходом 60—70% в соответствующие
амиды (2):
R\
V=C=CHCN
Rz/
н2о2, он*
60 -70 И
R\
V=C==CHCONH2
Rz/
(i)
(2)
Гидратацию можно осуществить также в условиях реакции
Риттера [3] действием трет-бутанола в смеси серная кислота —
уксусная кислота.
408
] Bogdanowicz AL, Ambelang T., Trost В. M., Tetrahedron Letters,
' 923 (1973).
2 Greaves P. M., L a n d о r P. D., L a n d о r S. R., О d у с к О., Tetrahedron
Letters, 209 (1973).
3, Ritter J. J., Kalish J., J. Am. Chem. Soc., 70, 4045, 4048 (1948).
ПЕРЙОДАТЫ (III, 84—92; V, 351—353).
Спнроэпоксициклогексадиеи-2,4-оны. Окисление перйодатом
натрия о-оксиметилфенолов, имеющих хотя бы один объемистый
заместитель, дает с хорошим выходом спироэпоксициклогекса-
диен-2,4-оны [1]. Так, например, при окислении раствора 2,4-ди-
трет-бутил-б-окси метил фенола в метаноле небольшим избытком
перйодата натрия в водном растворе образуется соединение (2)
с выходом 95%.
В случае фенолов, не содержащих объемистых заместителей,
образующийся вначале эпоксикетон (2) тотчас самопроизвольно
димеризуется по типу реакции Дильса — Альдера, образуя де-
гидродимер оксиметилфенола [2]. Диеион (2) при облучении
(ртутная лампа высокого давления фирмы «Филлипс» иа
125 вт) при комнатной температуре ароматизуется, давая с вы-
соким выходом замещенный салициловый альдегид (3). Изоме-
ризацию можно также осуществить, правда с низким выходом,
нагреванием при 120°.
Окисление карбинола (4) с помощью перйодата привело к
образованию не ожидаемого спироэпоксисоединения, а 3,5-ди-
трег-бутилпирокатехинацеталя бензальдегида (5). Такого рода
окислительная перегруппировка ограничивается, по-видимому,
о-оксизамещенными днарил- и триарилкарбинолами [3].
409
Окислительная перегруппировка эфиров и амидов а-кетокнс-
лот [4]. Эфиры и амиды а-кетокислот при обработке перйодатом
натрия при pH 7—9 подвергаются окислительной перегруппи-
ровке в производные малоновой кислоты. Так, например, 1-ме-
МаЮ4_
80%
(1) (2)
тилпиперидинднон-2,3 (1) превращается в З-карбокси-1-метил-
пирролидон-2 (2) с выходом 80%. Другим примером является
окисление Ы,Ы-диметиламида 2-кето масляной кислоты (3), при-
водящее к образованию Ь1,Ы-диметилмоноамида метилмалоно-
вой кислоты (4) с выходом 69% • Относительно предполагае-
мого механизма реакции см. оригинальную статью.
О О
II II
СН3СН2С—CN(CH3)2
(3}
О СНз О
NaiO-f II | 1|
—-> НОС—СН—С]
69 м
N(CH3)2
(4)
I. Becker H.-D., Bremholt Т., Adler Е., Tetrahedron Letters, 4205
(1972).
2. Adler E., Brasen S., Miyake H., Acta Chem. Scand., 25, 2055 (1971).
3. Becker H.-D.. Bremholt T., Tetrahedron Letters, 197 (1973).
4. Rueppel M. L„ Rapoport H., J. Am. Chem. Soc., 94, 3877 (1972).
ПИКРИЛХЛОРИД (III, 100—101).
Конденсация П. с бромидом N-фенацнлхинолиния и после-
дующее дезацилироваиие приводят к 8,10-динитроизоиидоло-
[2,1-а]-хинол и ну [1]:
410
Циклизация, включающая элиминирование HNO2 [2]. 2-Ме-
тилциклоиммониевая соль, содержащая у атома азота актив-
ную метиленовую группу (при условии, что эта метиленовая
группа обладает лишь умеренной активностью), конденсируется
с П. по своей метильной группе в положении 2, образуя заме-
щенный в этом положении 2,4,6-тринитробеизилидеиовой груп-
пировкой 1,2-дигидрогетероцикл, который в присутствии такого
основания, как пиперидин, циклизуется с отщеплением HNO2
в новый шестичлеииый цикл.
8 ,10-Дмнитро-12 -карбэтокси-
"12 Н-изокинолино-f 2,3-а]-хинолин
1. Reuschling D.-B., Krohnke Е., Chem. Вег., 104, 2103 (1971).
2. Reuschling D.-B., Krohnke E., Chem. Ber., 104, 2ПО (1971).
ПИКРИНОВАЯ КИСЛОТА (III, 101 — 102).
Расщепление пикратов углеводородов. Пикраты диметилнаф-
талинов обычно расщепляют абсорбцией на колонке с основной
окисью алюминия и элюируют углеводород бензолом. Эйзен-
брауи и сотр. [1] нашли, что петролейный эфир растворяет
только арен, почти ие растворяя пикриновую кислоту. Предло-
женный ими метод значительно сокращает расход растворителя
и окиси алюминия и, кроме того, позволяет регенерировать пи-
криновую кислоту. Однако в случае труднорасщепляемых пи-
кратов более сложных аренов (пнкраты флуорантена и пирена)
этот метод себя не оправдывает.
I. Eisenbraun Е. J., W е b b Т. Е., Burnham J. W., Harris L. Е., Org.
Prep. Proc., 3, 249 (1971),
H
ПИПЕРАЗИН, [ J Мол. вес 86,14, т. пл. 108—110°.
ев
411
Лактонизация метилового эфира о-формилбензойной кислоты.
П,— необычайно эффективный реагент в получении лактона (2)
из метилового эфира о-формилбензойной кислоты (1) в воде [1]
или диоксане [2]. Он намного эффективнее пиперидина (III,
102—107; V, 355) или морфолина (II, 314—316).
1. Dahlgren G., Schell D. М., J. Org. Chem., 32, 3200 (1967).
2. Henderson G. H„ Dahlgren G., J. Org. Chem., 38, 754 (1973).
ПИПЕРИДИН (III, 102 -107; V, 355).
Поправка (V, 355). Ссылка должна быть: Baldvin D. E.,
L о e b 11 c h V. M., Lawrence R. V., J. Org. Chem., 23, 25
(1958).
ПИРЕН. Мол. вес 202,24, т. пл. 151°.
Синтез 3,4-бензпнрена (7) [I]. Усовершенствования, внесен-
ные в методику синтеза этого потенциального карцииогеиа с
использованием янтарного ангидрида, уже повысили его выход
от первоначальных 3% (Кук и Хьюэтт, 1933) примерно до 40%.
Дальнейшие усовершенствования метода на стадиях (2а)-+(3)
и (4)—>(7) не привели к увеличению выхода, но значительно
упростили синтез (7), позволив работать с 50-граммовыми
загрузками. Особенно важным оказалось использование «-бута-
нола в качестве растворителя на стадии восстановления кето-
эфира (2а) по Вольфу — Кижнеру — Хуангу-Миилону. Соеди-
нение (6) дегидрировали до (7) с помощью броманила в кси-
лоле.
1. S с h 1 u d е Н,, Chem. Вег., 104, 3995 (1971).
412
ПИРИДИН (III, 107—114; V, 355—357).
Дезацетокснлнрование. На одной из стадий синтеза 11-дезо-
кси-10а-оксипростаглаидинов Краббе и сотр. [1] провели окисле-
ние спирта (1) реактивом Коллинза (комплекс СгОз — пиридин,
этот том), получив диацетоксиальдегид (2). В результате
обработки последнего пиридином в течение нескольких минут
образуется а,₽-ненасыщенный альдегид (3) с общим выходом
50%jC4HTaH на (1)
Ру
количеств.
И)
50%, считая на спирт (1). Альдегид (3) количественно гидри-
руется до соединения (4).
1. Crabbfc Р., Cervantes A., Meana М. С., J. С. S, Chem. Comm., 1973.
119.
ПИРИДИНА ХЛОРГИДРАТ (III, 115—118; V, 358—360:
VI, 210).
Ксантоны [1]. 2,2/-Диметоксибензофеионы (1) при нагрева-
нии в течение 24—48 час с избытком П. х. превращаются в
о
413
ксантоны (2), которые можно получить с выходом до 80—90%.
Метод пригоден также для получения фураиоксантонов
(3)-(4).
Кумарины [2]. Как цис-, так и гра«с-2-метоксикоричные кис-
лоты (1) при нагревании с П. х. превращаются в кумарины (2):
1. Royer R., Lechartier J.-P., Demerseman P., Bull. soc. chim. Fran-
ce, 1971, 1707.
2. Royer R., Bodo B,, Demerseman P., Clavel J.-M., Bull, soc. chim.
France, 1971, 2929.
бис- (ПИРИДИН) -ДИМЕТИЛФОРМАМИДДИХЛОРОРО-
ДИЯ БОРГИДРИД, (Ру)2(ДМФА) (RhCl2)BH4. Мол. вес. 419,96,
темно-красные кристаллы.
Катализатор получают из (PyJsRhCU и боргидрида натрия
в ДМФА.
Гомогенное гидрирование. Этот комплекс — чрезвычайно ак-
тивный катализатор гомогенного гидрирования [1].
1. Abley Р., Jardine I., Me Quillin F. J., J. Chem. Soc. (С), 1971, 840.
ПИРОКАТЕХИНА дихлорметиленовый эфир
(III, 124).
Карбоксилирование. Реагент использовали на одной из ста-
дий синтеза диметилового эфира тиофен-2,4-днкарбоновой кис-
414
лоты (3) из метилового эфира тиофен-3-карбоновой кислоты (1)
[1]:
(О
(г)
(з)
1. J a n d а М.,
295 (1971).
Srogl J., Nemec М„ Stibo г I., Org. Prep. Proc. Int., 3,
4-(М-ПИРРОЛИдИЛ) ПИРИДИН.
Мол. вес
148,21, т. пл. 57°.
Получение [1].
Катализатор ацилирования [2]. П. (1), подобно М,М-диметил-
4-пиридиламииу (VI, 83—84), является очень активным катали-
затором ацилирования пространственно затрудненных спиртов.
Согласно типовой методике, спирт обрабатывают при комнатной
температуре ангидридом карбоновой кислоты и эквимоляр-
ным количеством дналкиламинопиридина. При работе с боль-
шими загрузками для связывания образующейся при ацилиро-
вании кислоты добавляют еще 1 экв триэтиламина. Например,
метиловый эфир холевой кислоты с П. в качестве катализатора
415
в течение 2 час при комнатной температуре превращается в три-
ацетат. Ацетилирование его с помощью Ас2О в пиридине прн
комнатной температуре дает только 3,7-диацетат (12а-окси-
группа находится в аксиальном положении) [3]. 4-Диалкилами-
нопнрндины представляют особую ценность как катализаторы
ацилирования чувствительных к кислотам третичных спиртов,
таких, как, например, линалоол (2), который по этой новой ме-
тодике можно ацетилировать с выходом 80%.
сн3
н Зс
Н3С ^он
W
I. Verbruggen Н, Angew. Chem., Internal Ed., 11, 305 (1972).
2. HofleG., Steglich W., Synthesis, 1972, 619.
3. Fieser L. F., Rajagopalan S., J. Am. Chem. Soc., 72, 5530 (1950).
ПЛАТИНА НА ОКИСИ АЛЮМИНИЯ (КАТАЛИЗАТОР).
Синтез адамантана. Ирландские химики [1] предложили но-
вый катализатор изомеризации напряженных трициклических
углеводородов в адамантаны, гораздо более эффективный, чем
описанный ранее так называемый «шлам» А1Вг3 (V, 17). Новый
катализатор готовят осаждением платинохлористоводородной
кислоты иа окиси алюминия, а затем последовательно обраба-
тывают ее Н2, НС1 и SOC12. Катализатор можно регенериро-
вать, обработав его сначала О2 при 500°, а затем повторив об-
работку Н2 — НС1 — SOC12.
I. Johnston D. Ei, McKervey M. A., Rooney J. J., J. Am. Chem., Soc.,
93, 2798 (1971).
2 Gianetti J. P., Sebulsky R. T., Ind. Eng. Chem., Prod. Res. Develop.,
8, 356 (1969).
ПЛАТИНОХЛОРИСТОВОДОРОДНАЯ КИСЛОТА — ОЛО-
ВО ХЛОРИСТОЕ.
Карбонилирование терминальных олефинов [1]. Терминаль-
ные олефииы в присутствии метанола с высоким выходом кар-
боиилируются до эфиров соответствующих карбоновых кислот
при участии в качестве катализатора П. к. — о. х. при 90° и дав-
лении СО 210 атм в ацетоне, метилизобутилкетоне или 1,2-ди-
метоксиэтане. Если вместо метанола взять воду, получаются
кислоты.
RCH=CH2-1-CO-1-CH3OH ^tC^~'SnCI^ > RCH2CH3COOCH3+RCH(CH3)COOCH3
I. Kehoe L. J., Schell R. A., J. Org. Chem., 35, 2846 (1970).
ПОЛ ИМЕТИЛ ГИДРОСИЛОКСАН, (CH3)3SiO(CH3HSiO)„.
Si(CH3)3i где n ~ 35. П. — удобная в обращении химически
инертная жидкость. См. VI, 264.
416
Восстановление [1]. При кипячении с П. в присутствии оло-
воорганического катализатора сшил-диацетокситетрабутилди-
станноксана [бис-(дибутилацетоксиолово)-оксида (ДБАОО)] в
протонсодержащем растворителе (обычно этаноле) альдегиды и
кетоны с высоким выходом специфически восстанавливаются до
спиртов (ДБАОО удобнее всего получать реакцией окиси дибу-
тилолова с уксусной кислотой, т. пл. 54—-57°, выход 80%). На
каждый моль субстрата требуется 1 экв гидросилоксана и моле-
кула растворителя, отдающая протон:
t ДБАОО i
ХС=О + - [CH3HSiO]„ + ROH ------> А2НОН + — [CH3Si(OR)O]«
R2/ " R’/
Активным восстановителем в этой системе на самом деле
является, вероятно, гидрид дибутилацетокснолова.
Примеры:
(СеН5)2С=О —> (С6НБ)2СНОН (80%)
С6Н5СНО —> CSHECH2OH (100%)
СН2—СНСОСНз —> СН2=СНСНОНСН3 (65%)+С2Н5СОСН3 (35%)
п-Бензохинон —> Гидрохинон (81%)
Сложные эфиры, карбоновые кислоты, лактоны, амиды, ни-
трилы, алкилгалогеииды и нитросоедииения в этих условиях ие
восстанавливаются. Некоторые из этих групп могут, одиако,
восстанавливаться в кипящем 2-этилгексаноле (130°).
П. в присутствии каталитических количеств 5% Pd/C можно
использовать также для гидрирования терминальных и нетерми-
нальных цнс-олефииов, ароматических нитросоединеиий и арома-
тических альдегидов. Растворителем в этом случае служит эта-
нол (95%), содержащий 1 каплю конц. НС1; реакцию проводят
при 40—60°. (Внимание} Гидрирование иногда протекает экзо-
термично.) Настоящий метод представляет собой удобный спо-
соб гидрирования при низком давлении.
Примеры:
Пг, Pd/C
с2н5он
CeHsNO2 — > C6HsNH2 (89%)
с6н5сно — э- с5н3сн3 (84%)
Октен-1 э- н- Октан (88%)
сн2=снсосн3 — > С2Н5СОСН3 (100%)
1. L i р о wi t z J., Bowman S. A., J. Org. Chem., 38, 162 (1973).
ПОЛ ИФОСФОРНАЯ КИСЛОТА (ПФК) (HI, 132—146;
V, 363—365; VI, 212).
Циклизация у-арилмасляных кислот. Эйзенбраун и сотр. [1]
считают, что циклизацию 4-(2,5-диметплфенил)-2-метилмасляной
кислоты (1) в 2,5,8-триметилтетралон-1 (2) лучше проводить
14 Зак. 596
417
под действием ПФК, чем по методике с применением хлоран-
гидрида кислоты в присутствии ALCI3.
Ароматические альдазниы и кетазины [2]. ПФК является пре-
восходным катализатором и одновременно растворителем при
получении азинов из ароматических альдегидов и кетонов и
гидразина, а также различных его производных, например его
солей: хлоргидрата семикарбазида, «.-толуол сульфонил гидра-
зина и гидразидов карбоновых кислот. При 100° реакция обыч-
но заканчивается в течение 15 мин. В случае алифатических
карбонильных соединений реакция не нашла применения.
Внутримолекулярное ацилирование. Обычно для внутримоле-
кулярного ацилирования используют большой избыток ПФК*
Метц [3], детально изучивший реакцию иа примере циклизации
2-фен окси бензойной кислоты (1) в ксантои (2), иашел, что
большой избыток ПФК не нужен и даже вреден, так как при-
водит к понижению выходов. Метц рекомендует использовать
350 г 84%-ной ПФК иа каждый моль карбоновой кислоты при
продолжительности реакции, протекающей в интервале темпе-
ратур 80—120°, 1—2 час. Соблюдение этих условий (70°) позво-
лило ему [4] получить с максимальным выходом инданои-1 (4)
из 3-феиилпропиоиовой кислоты (3).
(3)
2-Аминохннолины [5]. Замещенные амидииы хлоругольной
кислоты (1) реагируют с алкинилмагнийгалогенидами, образуя
неизвестные до настоящего времени амидины пропиоловой кис-
418
лоты (2), которые можно циклизовать
действуя ПФК при температурах около
в 2-аминохинолины (3j,
150°.
R
1) 2 BrMgC^CR2
I Ri 2> H*° , >
>Cx / 55-9 5%
Mi1
(I)
Внутримолекулярная конденсация Клайзена. Синтез р-дике-
тонов конденсацией Клайзена [6] обычно удается лишь в том
случае, если продукт реакции может стабилизироваться в виде
енолята. Герлах и Мюллер [7] нашли, что если раствор 3-кето-
1-циклогексилуксусиой кислоты (1) в ледяной уксусной кислоте
обработать ПФК, то можно получить с выходом 75% и неено-
лизующийся р-дикетон бицикло-[2,2,2]-октаидион-2,6 (2). В от-
сутствие уксусной кислоты выход понижается, причем уксусным
ангидридом ее заменить нельзя. Бартлетту и Вудсу [8] не уда-
лось получить дикетон (2) катализируемой основанием конден-
сацией метилового эфира З-кето-1-циклогексилуксусной кислоты;
этот дикетон онн получили лишь с низким выходом, пропуская
З-кето-1-циклогексилуксусную кислоту иад нагретой до 340е
окисью марганца(II).
В тех же условиях из соединения (3) с выходом 81% полу-
чен спиро-[4,4]-нонандион-1,6 (4).
14*
419
1 E i s e nb r a un E. J., H in m a n C. W., S p r in g e г J. Bur nham J.W.,
ChouT.S., J. Org. Chem., 36, 2480 (1971).
2 . Mobb s D. B., Suschitzky H., J. Chem. Soc. (C), 1971, 175.
,3. Metz G., Synthesis, 1972, 612.
4 Metz G., Synthesis, 1972, 614.
5. Ried W., Wei dema nn P., Chem. Ber., 104, 3329 (1971).
6 Хаузер Ч. P., С в э м e p Ф. В., Адамс Дж. Т„ «Органические реакции»,
ИЛ, М„ 1956, сб. 8, стр. 90.
7. Gerlach Н., Milller W., Angew. Chem, Internal, Ed., 11, 1030 (1972).
8. В art let t P. D., W о о d s G. F., J. Am. Chem. Soc., 62, 2935 (1940).
ПОЛИФОСФОРНОЙ КИСЛОТЫ ЭФИРЫ (ПФЭ)
(III, 146—148; V, 365—366; VI, 212—214).
Реакция Бишлера — Напиральского [1]. Нитроамид (1) не
циклизуется под действием обычно употребляемых конденси-
рующих агентов [2], однако эту реакцию удалось осуществить с
умеренным выходом с помощью ПФЭ, полученных по методике
Кава (VI, 212). Реакцию использовали на одной из стадий пол-
ного синтеза алкалоида василистника адиантифолина.
Производные 1,3-тиазииа. Цианамид (1) [3] конденсируется
с ароматическими кислотами в присутствии ПФЭ с образова-
нием 5-карбамоил-6-метилтио-1,3-тиазиноиов-4 (2) с выходом
27—90%. Реакция с алифатическими кислотами дает с низкими
выходами 5-циан-6-метилтио-1-кето-1,3-тиазиноиы-4(3) [4].
о
ПФЭ
RCOOH
П ФЭ
АгСООН
о
(3)
1. Doskotch R. W., Phil Ир son J. D., Ray A. B., Beal J. L., J. Org.
Chem., 36, 2409 (1971).
2. Уэли В., Говиндачари T,, «Органические реакции», ИЛ, М., 1953,
сб. 6, стр. 98.
420
3 TakeshimaT., Yokoyama M„ Fukada N., Aka no M., J. Org.
' Chem., 35, 2438 (1970).
4. Yokoyama M., Sawachi Y., Isso T., J. Org. Chem., 38, 802 (1973),
ПРОПАНДИТИОЛ-1.3, HS(CH2)3SH (III, 148—149). T. кип.
169—170° (приведенная ранее цифра неверна).
Гетеролитическая фрагментация 1,3-дитианилтознлатов. Мар-
шалл и Беллетайр [1] показали, что эта реакция идет, по-види-
мому, с участием аниона 1,3-дитиана [2]. Так, например, в ре-
зультате конденсации альдегида (1) с П. образуется 1,3-дити-
ан (2), который при действии фениллития дает с выходом 52%
CHz(CH2SHh
Б 1 Е tgO
----з---г-->
тиоацеталь кетена (3) 75 %-нон чистоты. Последний превра-
щают в кислоту (4): сначала кислотным гидролизом в присут-
ствии n-толуолсульфокислоты в водном метаноле получают
эфир этой кислоты и тиола, гидролиз которого в присутствии
основания дает затем кислоту (4).
1. Marshall J. А,, В el let! re J. L., Tetrahedron Letters, 871 (1971).
2. Grob C. A., S ch less P. W., Angew. Chem., Internat. Ed., 6, 1 (1967).
ПРОПАРГИЛОВЫЙ АЛЬДЕГИД (называемый также про-
пиоловым альдегидом), СН = ССНО. Мол. вес 54,05, т. кип
54—57°.
Получение. П. а. получают окислением пропаргилового спир-
Реакцня с о-фениленднамнном приводит к образованию ди-
гидродибензотетраза-[14]-аннулена [2]:
1. Sauer J. С., Org. Syn. Coll. Vol., 4, 813 (1962).
2. Hiller H., D i m г о t h P„ P f 11 z n e r H., Ann., 717, 137 (1968).
РОДАНИОДИД, ICNS. Мол. вес 185,00.
Получение [1]. Эфирные растворы Р. получают в основном
по методу Рэби [2]. К суспензии роданида свинца (0,025 моля)
прибавляют в один прием бром (0,025 моля). Смесь перемеши-
вают при комнатной температуре до исчезновения окраски
брома несколько минут. Затем к полученному раствору родана
прибавляют раствор иода (0,025 моля) в сухом эфире и пере-
мешивают смесь в течение 15 мин при комнатной температуре
в темноте.
Эписульфиды [3]. Хиншау высказал предположение, что по
аналогии с реакцией присоединения иодизоцианата (II, 44; V,
215—216; VI, 129—131) к алкенам прибавление алкена к эфир-
ным растворам Р. должно приводить к образованию [3-иодтио-
цианатов. И, действительно, при взаимодействии циклогексена
О) (2)
с Р. образуется [3-иодтиоцианат (2), который при обработке рас-
твором гидроокиси калия в метаноле превращается в эписуль-
фид циклогексена (3) с выходом 57%. Промежуточные иодтио-
циацаты при этом можно не выделять.
Для реакции с Д2-холестеном Р. получали в хлористом мети-
лене; выход 2|3,3|3-эпитиохолестана составил 46%.
К сожалению, реакцией Р. с ациклическими олефинами
нельзя получить эписульфиды со сколько-нибудь существен-
ными выходами.
1. Hinshaw J. С„ personal communication.
2. Raby С., Ann. chim., 6, 481 (1961).
3. Hinshaw J. C., Tetrahedron Letters, 3567 (1972).
РОДИИ (5%) НА ОКИСИ АЛЮМИНИЯ (III, 168—172).
Гидрирование ароматических ядер: цис, ^«с-декалол-1 [1].
422
В сосуд Парра для гидрирований емкостью 500 мл, запол-
ненный азотом, помещают 20,0 г Р.н.о. а. Катализатор осто-
рожно смачивают 25 мл 95% -ного этанола, после чего прили-
вают раствор 40,0 г (0,28 моля) сс-нафтола (очищенного пере-
гонкой при атмосферном давлении) в 125 мл 95%-ного эта-
нола и 3 мл уксусной кислоты. Смесь встряхивают в приборе
Парра при начальном давлении водорода 3,8—4,2 атм. Теорети-
ческое количество водорода поглощается приблизительно через
12 час. Затем катализатор отфильтровывают (с отсасыванием)
и дважды промывают 50-миллилитровыми порциями этанола.
Объединенные фильтраты упаривают на роторном испарителе и
вязкий остаток растворяют в 150 мл бензола. Раствор промы-
вают 75 мл 10%-ного раствора гидроокиси натрия, затем 75 мл
воды, сушат над сульфатом магния в течение 3 час и упаривают
досуха на роторном испарителе, получая 39—41 г (94—97%)
смеси геометрических изомеров декалола-1. ^«с,^«с-Декалол-1
можно выделить в виде кристаллического вещества, добавив
к смеси 15—20 мл гептана и охладив ее. После перекристалли-
зации из минимального количества н-гептана получают 13—14 г
(30—33%) днс,днс-декалола-1, т. пл. 92—93°.
Тем же методом, взяв 0,33 г катализатора на каждый грамм
субстрата, гидрируют гидрохинон и резорцин, получая соответ-
ственно 90% циклогександиола-1,4 и 85% циклогександиола-1,3;
[3-нафтол в тех же условиях дает смесь геометрических изоме-
ров декалола-2 с выходом 88%.
1. Meyers А. 1., Beverung W. М., Gault R., Org. Syn,, 51, 103 (1971).
РОДИЙ (5%) НА УГЛЕ (норит) (III, 172-173).
Димеризация и тримеризация норборнадиена [1]. Кругло-
донную колбу на 1 л с магнитной мешалкой снабжают обрат-
ным холодильником, соединенным с ртутным маностатом, по-
зволяющим откачивать из системы воздух, заменять его азотом
и сравнивать давление в колбе с атмосферным. В колбу поме-
щают норборнадиен (463 г, 5,03 моля), перегнанный над гидри-
О5*к1,/С.
кипячение^ L /У
дом кальция, Р.н. у. (2,5—4,5 в, 0,001—0,002 г-атом Rh) и кипя-
тят смесь при перемешивании в течение 1 дня, после чего Р.н.у.
отфильтровывают. После перегонки фильтрата при атмосферном
давлении часть норборнадиена возвращается, а в остатке содер-
жится продукт реакции, Повторяют всю операцию с возвращен-
ным норборнадиеном до тех пор, пока он не прореагирует весь.
При первичной обработке из норборнадиена, по-видимому,
удаляются вещества, отравляющие катализатор. Так, если после
423
первого или второго кипячения возвращается около 95% нор-
борнадиена, то при пятом кипячении с 2,5 г Р.н.у. в течение
10 час полностью олигомеризуется 120 г норборнадиена. Нор-
борнаднен, предварительно прокипяченный с норитом, дает оли-
гомеры с большим выходом, чем необработанный, однако про-
стейший способ проведения олигомеризации заключается в мно-
гократном кипячении норборнадиена, каждый раз со свежей
порцией Р. н.у., пока не будет достигнут высокий выход продук-
та реакции. Для этого может потребоваться пятикратное повто-
рение описанных операций с использованием в сумме 20 г
Р.н.у.
Остатки после перегонки объединяют и еще раз перегоняют
(т. кип, 70—7370,5 мм), получая 326 г (выход 70%) димера
норборнадиена. Остаток в колбе по охлаждении до комнатной
температуры застывает. Перекристаллизация его из 95%-кого
этанола дает 24,5 г (5,3%) тримера норборнадиена с т. пл.
176—178°.
Димер норборнадиена представляет собой бесцветную
жидкость; это — смесь изомеров: 84% (1), 12% (2) и 4% (3).
Их можно разделить разгонкой на роторной ректификационной
колонке с вращающейся лентой из тефлона. Например, фрак-
ционирование 73 г димера дает 50 г изомера (1) ст. кип.
136725 мм 98 %-ной чистоты и 5 а изомера (2) с т. кип.
(2)
140722 мм 97%-ной чистоты.
Тетрагидрофуран-1(«с-2,5-днкарбоновая кислота [2]. Р.н.у.—
лучший катализатор гидрирования фуран-2,5-дикарбоновой кис-
лоты (1) до тетрагидрофуран-ц«с-2,5-дикарбоновой кислоты (2):
1. Mrowca J. J., Roth R. J., Acton N., Katz T. J., Org. Syn., submitted
1972.
2. M о о r e J. A., К e 11 у J. E., Org. Prep. Proc,, 4, 289 (1972).
РОДИЯ ОКИСЬ, Rh2O3. Мол. вес 253,82.
Аминометнлироваиие алкенов. В первоначальной методике
амннометилироваиия алкенов Реппе [1] использовал в качестве
424
катализатора пеитакарбонил железа Fe(CO)s (II, 6 7; V, 203;
VI, 123—124). Согласно последним данным Айкбола [2], гораздо
более эффективным катализатором является Р. о., а при исполь-
+ ЗСО + Н2О + HN^
Катализатор
----------->
НС—С—CH2N'
+ 2СО2
зованин в качестве сокатализатора небольших количеств пента-
карбонила железа можно получить еще более высокие выходы
(90%).
Имидазолы [3], а-Олефины реагируют с окисью углерода и
конп. водным раствором аммиака в автоклаве (150°) с образо-
ванием 2,4,5-триалкилимидазолов с выходом 50—60%. Напри-
мер, этилен с выходом 52% дает 2,4,5-триэтилимидазол:
нь2о3 ?н*СНз
сн2=сн2 + со + nh3 ch3oh-h2o> Cch3ch2cchnhcch2ch3]
о о
NH3
52%
снэсн2
СН3СН2
СН2СН3
Углеродная часть имидазольного цикла строится, пО-видимому,
с участием окиси углерода. Р. о. служит катализатором восста-
новления и, кроме того, возможно, превращается в HRh(CO)3l
который выступает в качестве карбонилирующего агента.
Гидроформилирование. Швейцарские химики [4] рекомен-
дуют Р.о. в качестве катализатора гидроформилирования оле-
финов. Так, из циклогексена (1) они получили формилциклогек-
сан (2) с выходом 95%. Катализаторы на основе кобальта при-
водят к более низким выходам.
+ со + н2
Rh2O3
*
(2)
1. Reppe W., Experimentia, 5, 93 (1949).
2. Iqbal A. F. М., Helv. Chim. Acta, 54, 1440 (1971).
3. I w a s h i t a Y., S a k u г a b a M., J. Org. Chem., 36, 3927 (1971).
4. Pino P., В о 11 e g h i C., Org. Syn., submitted 1972.
РТУТИ АЗИД (VI, 216).
Внимание] Хиткок и Бах [1] сообщили, что при генерирова-
нии азида in situ, когда Hg(N3)2 осаждали из раствора при
Комнатной температуре, произошел сильный взрыв. Причиной
425
детонации, по-видимому, явилось перемешивание осадка рото-
ром магнитной мешалки. Авторы призывают соблюдать исклю-
чительную осторожность при проведении реакции азидомерку-
рирования олефинов. Галле и Хасснер [2] установили, что важ-
но добавлять ацетат ртути к раствору азида натрия в смеси
ТГФ — вода, а не наоборот.
Цнклопропилазнды [2]. Р. а. реагирует с циклопропенами
(1) по механизму с«н-присоединения, образуя £{Цс-2-азидоцик-
лопропилмеркуразиды (2) с прекрасным выходом. Восстанови-
тельное демер курирование под действием NaBH4 приводит с
умеренным выходом к циклопропилазидам (3).
(1) R) (з)
1. Н е a t h с о с k С. Н,, В а с h R. D., personal communication.
2. G а 11 е J. Е., Н a s s n er A., J. Am. Chem. Soc., 94, 3930 (1972).
РТУТИ(П) АЦЕТАТ (III, 173—184; ¥,368—372; ¥1,216—218).
Оксимеркурнрованне (¥, 368; ¥1, 217). Браун и Джеоджеган
[1] исследовали относительную реакционноспособность ряда оле-
финов в реакции оксимеркурирования в смеси вода — ТГФ
(20:80, по объему). По своей реакционной способности изучен-
ные олефины составляют следующий ряд: терминальные диза-
мещенные > терминальные монозамещенные Д> нетерминальные
дизамещенные Д> нетерминальные тризамещенные Д> нетерми-
нальные тетр аз а мешенные. Таким образом, главную роль в ре-
акционной способности олефинов играют пространственные фак-
торы. Увеличение числа заместителей при двойной связи, а так-
же возрастание пространственных препятствий с той стороны,
куда направляется атака гидроксила или ртутьсодержащего
заместителя, приводят к уменьшению скорости реакции. В слу-
чае олефинов общей формулы RCH = CHRZ -^«с-изомеры более
реакционноспособны, чем соответствующие олефины транс-
строения. Циклические олефины реагируют заметно скорее, при-
чем скорость реакции несколько варьирует при переходе от
одной циклической системы к другой: циклогексен Д> цикло-
пентен циклооктен; норбориен бицнкло-[2,2,2]-октен-2.
Полученные результаты позволяли надеяться, что возможно
избирательное монооксимеркурирование диенов, и действитель-
но эта возможность была реализована [2]. В случае симметрич-
ных диенов, таких, как пентадиен-1,4, выход енола, полученного
в результате оксимеркурирования — восстановления, оказался
ниже того, который соответствовал бы чисто статистическому
результату реакции (50% енола). С увеличением длины цепи
4Й$
диена выход приближался к среднестатистическому. Выходы
удалось повысить, используя трифторацетат ртути (VI, 217).
В случае несимметричных диенов реакцию гидратации осуще-
ствляли избирательно. Например, лимонен (1) превращали в
енол (2) с выходом 70%.
СИз сн3
2 Ф
НзС СНЙ Н3С'Щ4'4 он
сн3
(1) (2)
Оксимер курирование бицикло-[3,2,1]-октадиена-2,6 (3) в ук-
сусной кислоте при 50° в течение 7 час приводит к образованию
экзо, экзо-6-ацетоксимеркур-7-ацетоксибицикло-[3, 2, 1]-октена-2
Na ВН4
(4) (5)
(4) с выходом 86%, из чего следует, что напряженная двойная
связь диена (3) реагирует легче. Это справедливо также и для
реакции диена (3) с ацетатом палладия (II) (III, 52—54; V, 347)
в уксусной кислоте при 25°, в результате которой образуется
соединение (6) [3].
-HPdOAc
-------->
(6)
Олефины можно превратить в кетоны, если продукт оксимер-
курирования переметаллировать хлористым палладием. В отли-
чие от оксимеркурпроизводных аналогичные соединения палла*
дня в растворе разлагаются до кетонов и металлического пал-
ладия [4].
Н2о PdCl2
СН3(СН2)3СН=СН2 + Hg(OAc)2 --> CH3(CH2)3CHCH2HgOAc ---->
ОН
CH3(CH2)3CHCH2PdCi; СН3(СН2)3СОСН3 4- Pd° 4- НС1 + сг
Китчинг [5] опубликовал обзор. реакции оксимеркурирования.
427
Окисление стероидных алкенов [6], При взаимодействии Р. а.
(2 экв) с Д14-стероидами (1) образуются в основном Д8<14)-оны-15
(2), а не ожидаемые аллилацетаты 16£-ацетоксиены-14. Тетра-
замещенпая двойная связь Д8(14)-холестена в этих условиях не
вступает в реакцию. Д2-Холестен (дизамещенный) лишь медлен-
но реагирует с образованием Д^холестенона-З с выходом 20%,
причем возвращается 70% исходного соединения. Эта неожидан-
ная реакция стероидных тризамещенных двойных связей, по-
видимому, протекает через стадию аллильного ацетоксилирова-
ния с последующим окислением образовавшегося соединения.
Синтез третичных аминов [7]. Р. а. реагирует с енамииом (1)
в ДМФА по [3-углеродному атому с образованием иминиевой
соли (2). Восстановление этой соли боргидридом натрия дает
соответствующий третичный амин (3) с выходом 50—90%.
r2NCH=C
Hg(OAc)2 + / ~ К'аВГЦ
-------► R2N=CHC7 OAc ---------> r2nch2ch:
HgOAc
(2)
(3)
Окислительное расщепление циклопропенов [8]. 1,3,3-Триме-
тилциклопропен (1) окисляется Р. а. в хлористом метилене при
комнатной температуре в 1,1-диацетокси-2,3-диметилбутен-2 (2).
Реакция циклопропеиа (1) с триацетатом таллия или тетрааце-
татом свинца помимо бутена (2) дает 3,3-диацетокси-2-метилпро-
Hg(OAc)2
СНгС1г
65%
(АсО)гСНч ZCH3
С
II
с
нэс чснэ
(2)
пен (3) и 2-ацетокси-4-метнлпентадиен-1,3 (4). Окисление, по-
видимому, происходит путем присоединения ацетата металла с
428
последующим расщеплением цикла, приводящим к промежутоЧ'
С /ОАс
Н3С"' СН
хОАс
(3)
Н. /Н
с
II
н. .с
с ОАс
А
н3с сд
Н. /Н
с
ному образованию комплекса винилкарбен — ацетат металла.
Окислительная циклизация. Последней стадией синтеза ра-
цемического аймалицина (2) является окислительная циклиза-
ция соединения (1) [9], проводимая в 2,5%-ном растворе уксус-
ной кислоты под действием избытка смеси Р. а. с динатриевой
солыо этилендиаминтетрауксусиой кислоты 1 : 1 (ЭДТК) (IV,
259—261), и последующее восстановление иминиевых промежу-
точных соединений боргидридом натрия:
Сторк и Гутиконда [10] в тех же условиях осуществили цик-
лизацию секоспирта (3) до рацемического иохимбина (4) с вы-
ходом 32%. Сторк отмечает также, что для этой реакции тре-
буется лишь 2 же Р.а., так как в присутствии ЭДТК образуется
ртуть, а не ацетат ртути (I).
Аллильное окисление олефинов [II]. В результате окисления
аллилбензола (1} при кипячении с Р. а. в НОАс в течение 50 час
429
образуется 70% металлической ртути и 72% органических про*
дуктов:
Hg(OAc)2
С6Н5СН,СН=СН2 —С6Н5СН=СНСН2ОАс + C6HSCHCH=CH2 +
ОАс
(1) (2) 95% (3) 0,5%
t
4- CsH5CH2CH(OAc)CH2OAc + С6Н6СИ=СНСНз
(4) 2% (5) 0,1%
Основным продуктом реакции является циннамилацетат(2),
а-фенилаллилацетат (3), 1,2-диацетокси-З-фенилпропан (4) и
транс-пропенилбензол (5) образуются в следовых количествах.
Диалогичный набор аллилацетатов получается при сольволизе
цнниамилмеркурацетата CsH5CH = CHCH2HgOAc, который, по-
видимому, промежуточно образуется при окислении аллилбен-
зола.
Бутен-1 окисляется Р.а. при 25—50° в единственный продукт
а-метилаллнлацетат (6), являющийся также основным продук-
том окисления цас- и транс-бутенов-2 при умеренных темпера-
турах.
Hg(OAc)2
СН3СН2СН=СН2 -------> СНзСН(ОАс)СН=СН3
(Ф
Превращение олефинов в этиленкеталн. Хант и Роудхивер [12]
описали метод превращения олефинов в этилен кетали в три
стадии: 1) сольвомеркурированием, 2) переметаллированием и
3) разложением промежуточного палладийоргаиического соеди-
нения:
и
1) H.g(OAc)3 + RCH=CHj + НОСНгСНгОН——->RCHCH2HgOAc + НОАс
OCH2CH2OH
2) RCHCHzHgOAc + PdCI2—> RCHCH2PdCL + HgOAcCl
dcHjCH2OH OCH2CH2OH
3) RCHCH2PdCl ---> °\/0 + Pd + HC1
OCH2CI^OH R^CH3
Например, из гексена-1 этим методом получен 2-бутил-2-ме-
тил-1,3-диоксолан с выходом 77%.
1 . Brown Н. С., Geoghegan Р. J,, Jr., J. Org. Chem., 37, 1937 (1972).
2 . Brown H. C., Geoghegan Р. J., Jr., Lynch G. J., Kurek J. T.,
J Org. Chem., 37, 1941 (1972).
3 . SakaiM., Tetrahedron Letters, 347 (1973).
430
4 Rodeh eaver G. T., Hunt D. F„ Chem. Comm., 1971, 818.
5 Kit ching W., Organometallic Chem. Rev., 3, 61 (1968).
6 Blossev E C, Kucinski P., J. C. S., Chem. Comm., 1973, 56.
7 Bach RD.,'Mitra D. K-, Chem. Comm., 1971, 1433.
8* S h i r a f u j i T., N о z a к i H., Tetrahedron, 29, 77 (1973).
9* G u t z w i 11 e r J., P i z z о 1 a t о G., U s к о к о v i 6 M., J. Am. Chem. Soc.,
10 SuTk7G^Guthikonda R. N., J. Am. Chem. Soc., 94, 5109 (1972).
IL Rappoport Z., W instein S., Young W. G., J. Am. Chem. Soc., 94,
2320 (1972).
12. Hunt D. F„ Rodeheaver G. T., Tetrahedron Letters, 3595 (1972).
РТУТИ ОКИСЬ (III, 184—187; V, 372—373).
Реакция Хунсдикера — Кристола (III, 186—187). Опублико-
вана [1] подробная методика получения 1-бром-З-хлорциклобу-
тана модифицированной реакцией Хунсдикера. В 1-литровую
трехгорлую круглодонную колбу, закрытую от света алюминие-
вой фольгой и снабженную механической мешалкой, обратным
холодильником и капельной воронкой, помещают 37 г (0,17 мо-
ля) красной окиси ртути и 330 мл четыреххлористого углерода.
В колбу прибавляют 30,0 г (0,22 моля) 3-хлорциклобутанкарбо-
z C1-Z >-СО2Н + HgO + 2Brz
2 CI-Z V-Br + НгО + 2 СО2 + HgBr2
новой кислоты и при перемешивании нагревают смесь до кипе-
ния. Поддерживая температуру бани около 120°, как можно
быстрее (47 мин) приливают раствор 40 г (0,25 моля) брома
в 180 мл четыреххлористого углерода, не допуская потерь брома
через обратный холодильник. После короткого индукционного
периода начинает выделяться двуокись углерода со скоростью
150—200 пузырьков в минуту. За скоростью можно следить, бар-
ботируя с помощью резиновой трубки выходящий через обрат-
ный холодильник газ через небольшое количество воды (при
этом происходит небольшая потеря брома). Раствор кипятят
до тех пор, пока скорость выделения двуокиси углерода не
уменьшится до 5 пузырьков в минуту (25—30 мин). Затем смесь
охлаждают в ледяной бане и отфильтровывают осадок. Раство-
ритель отгоняют, используя модифицированную колбу Кляй-
зена с колонкой Вигре. Перегонкой в вакууме оставшегося ма-
сла получают 13—17 г (35—46%) 1-бром-З-хлорциклобутана
(т. кип. 67—72°/45 мм, п§ 1,5065).
Деструкция карбоновых кислот в галогеналкилы с помощью
Р.о. и галогенов включает первоначальное образование ртутной
соли кислоты, вступающей затем в нормальную реакцию Хунс-
дикера с галогеном. Выделяющаяся вода не мешает реакции,
431
что объясняется растворимостью ртутных солей в применяемом
растворителе (CCU). Реакция имеет два ограничения: во-пер-
вых, кислоты, несущие карбоксильную группу у третичного ато-
ма углерода, не подвергаются деструкции и, во-вторых, если в
качестве галогена выступает иод, реакция часто приводит в
основном к сложным эфирам RCOOR. Выходы в модифициро-
ванной реакции, как правило, ниже выходов, достигаемых в ме-
тоде с использованием серебряных солей [2].
Кейзон и Уэлба [3] получили наибольшие выходы продукта
реакции по модифицированному методу, применяя избыток кис-
лоты и проводя реакцию в кипящем ССЦ с удалением воды
азеотропной перегонкой. Им удалось получить тридецилбромид
из миристиновой кислоты с выходом 90%.
Окисление бисгидразона циклооктандиона-1,2 (1) с помощью
Р.о. приводит в основном к соединению (2), а также к неболь-
шим количествам циклооктнна, триазолов и их ртутных пронз^
водных (4):
1. Lampman G. М., Aumiller J. С., Org. Syn., 51, 106 (1971).
2. Bunce N.J., J. Org. Chem., 37, 664 (1972).
3. Cason J., Walba D. M., J. Org. Chem., 37, 669 (1972).
4. M ii 11 e r E., Meier H., Ann., 716, 11 (1968).
РТУТИ ОКИСЬ —БРОМ, HgO — Вг2.
Бромирование алканов [1]. Бром в сочетании с окисью рту-
ти— весьма активный реагент для свободнорадикального бро*
мирования алканов:
2RH + 2Вг2 + HgO —> 2RBr + Н2О + HgBr2
Этот комбинированный реагент значительно более реакцион-
носпособен, чем элементарный бром или NBC, и, следовательно,
он должен быть полезен в тех случаях, где использование Вг2
и NBC не приводит к успеху. Реакция, по-видимому, протекает
через образование окиси брома, выступающей в качестве актив-
ного бронирующего агента. Новая реакция применима для за*
432
мещения водорода в первичных или вторичных С—Н-связях;
выходы большей частью удовлетворительны.
2Вг2 + HgO —> HgBr2 + Br2O
Br2o + 2RH — > 2RBr + H2O
Следует отметить, что из использованного ртутного остатка
можно регенерировать окись ртути [2].
1. Bunce N. J., Can. J. Chem., 1972, 3109.
2. С a d у G. Н., Inorg. Syn., 5, 156 (1957).
РТУТИ(П) ТРИФТОРАЦЕТАТ (VI, 217—218).
Циклизация изопреноидов [1]. Ациклические изопреноиды
циклизуются с выходом 30—60% под действием 1,2—2,0 же Р. т.
в нитрометане или ацетонитриле в интервале температур от —20
до 0°. Через 5—30 мин реакционную смесь обрабатывают 2 же
NaBH4 в ЗА! растворе NaOH при 0°. В качестве примера приве-
дена циклизация транс-геранилацетона (1) в 2,5,5,9-тетраметил-
гексагидрохромен (2).
1. К u г b а п о v М., Semenov sky А. V., Smit W. А„ Shmelev L. V.,
Kucherov V. К., Tetrahedron Letters, 2175 (1972).
РУТЕНИЯ ТРИХЛОРИДА ГИДРАТ (V, 368; VI, 215—216).
Селективное гидрирование цнклододекатриеиа-1,5,9 до цнкло-
додецена. Смесь транс, транс, транс-циклододекатриена-1,5,9 (1)
и транс, тра«с,цас-циклододекатриена-1,5,9 (2) в отношении 3:2
можно гидрировать до циклододецена (3) (цис и транс} с выхо-
дами до 98% методом гомогенного гидрирования с использова-
нием в качестве катализатора [(С6Н5)зР]2(СО)2КиС12 [1]. Ката-
(2)
лизатор удобнее готовить in situ. Смесь Р. т. г., трифенилфос-
фина, СО (0,28 атм}, триена (1+2) и этанола перемешивают
при 140° в атмосфере водорода. Циклододецен и в этих условиях
433
получается с высоким выходом лишь с небольшой примесью
циклододекана (2%) [2].
1. Stepheson Т. A., Wilkinson G., J. Inorg. Nucl. Chem., 28, 945 (1966).
2. Fahey D. R., J. Org. Chem., 38, 80 (1973).
РУТЕНИЯ ЧЕТЫРЕХОКИСЬ (III, 197—201; V, 373—376;
VI, 219—220).
Окисление вторичных спиртов в двухфазной системе
(VI, 219—220). Опубликованы подробные указания к окислению
оксилактоиов до кетолактонов и лактонов до кетокислот [1].
Окисление алкниов [2]. Окисление алкинов Р. ч. приводит к
соответствующим дикетонам, причем терминальные алкины
дают лишь соответствующие кислоты.
Примеры:
RuO2, NaOCl
С6Н5С=СС0Н5 ргт "->' СеН5СО-СОС6Н5+С6Н5СООН
83^ 7,5#
С3Н7С^СС3Н7 —> С3Н7СО—СОС3Н7 + С3Н7СООН
58,5% 40%
сен5с=сн —> С6Н5СООН
66%
1. Go pal Н,, Adams Т., Moriarty R. M., Tetrahedron, 28, 4259 (1972).
2. G о p a 1 H., Gordon A. J„ Tetrahedron Letters, 2941 (1971).
с -—:-----------=---------------------------------
CBHHUA(IV) АЦЕТАТА АЗИДЫ, Pb(OAc)4_n(N3)n. Разла-
гается даже при —20°.
Обзор [I]. Реагент получают, смешивая тетраацетат свинца
с триметилсилилазидом (III, 383; VI, 271) в апротонном раство-
рителе при температурах от —40 до —20°.
С простыми олефинами, такими, как транс-стильбен (1),
С. а. а. образуют исключительно sw^-диазндо- (2) и ацетокси-
азидосоединения (3). Мостиковые олефины подвергаются ске-
’ S'^C=C^’ РЬ(ОАс)4.л(Щ)^ CtH5CH-CHCtHs
« СЛ А, А,
N, ОАс
C6HSCN
tn
{27.43%
:dl, 70%
30$
(3).42$
эритро 75$
трео 25$
(C6HS сно
летным перегруппировкам. Например, норборнен (4) при взаи-
модействии с С. а. а. дает соединение (5) с выходом 75%.
Реагент взаимодействует также с соединениями, содержа-
щими тройную связь, при этом состав продуктов реакции зави-
сит от температуры. Главным продуктом реакции дифенилаце-
тилена (6) с С. а, а. при 20° является бензонитрил, образующий-
ся при фрагментации диазидостильбена. При —20° эта реакция
дает следующие продукты:
C6H5C=CCeHs —>
(6)
N3
“—> СвН6С—CCgHg 4~ CgHgC—С—CSH5 + CsHeCN3 2CgH5CN
ОО О N3 А
(35%) (30%) (15%) (15%)
435
Д5-Стероиды (7) расщепляются реагентом при —15° до 5-ке-
то-6-циаи-5,6-секостероидов (8) с выходом 50—70%, вероятно,
через промежуточные соединения, показанные на приводимой
схеме:
Дизамещенные стероидные олефины под действием реагента
превращаются в а-азидокетоиы. Например, Дй-холестен (9) при
взаимодействии с реагентом в хлористом метилене при —20°
дает с выходом 50—60% За-азидохолестанои-2 (10). Ниже при-
веден возможный механизм реакции:
сн3
РЬ(ОАс)ч_П(И3)Л
----------------?
-AcN3 .
50-60$
5 0?£
Силикагель
ZO час
(10) (11)
Аксиальный азидокетон (10) при продолжительном действии
силикагеля (кислого) перегруппировывается в термодинамиче-
ски более устойчивый Зр-азидохолестанон-2 (11).
При взаимодействии Д5-стероидов (7) с реагентом при ком-
натной температуре реакция идет в ином направлении, чем при
—15°, с образованием преимущественно 7а-азидо-Д5-стероидов
(12) с выходом 35—55%. В этом случае реакция может носить
радикальный характер. Д6-Стеронды также превращаются в 7сс-
436
азидо-Д5-стероидЫ,
Аналогично С. а. а. с олефинами взаимодействует комплекс-
ный реагент иодозобензола диацетат — триметилсилилазид
[C6H5I(OAc)2]—(CH3)3SiN3.
1. Z b i г а 1 Е., Synthesis, 1972, 285.
СВИНЦА ДИАЦЕТАТ, Pb(OCOCH3)2 (III, 206—207; V, 379).
Мол. вес. 325,30.
Циклические дисульфиды [1]. При обработке дитиола (1)
водным раствором С.д. с почти количественным выходом обра-
зуется дитиолат свинца (2), образующий с серой в бензоле при
комнатной температуре с высоким выходом циклический ди-
сульфид (3):
HS(CH2)nSH Pb(°Ac)z > Pb[S(CH3)^S] -
(1),Л = 3, 4, 5, Ь (г) (3)
Серу можно заменить селеном; использование иода или хло-
ра приводит к уменьшению выходов,
1. Cragg R. Н., Weston A. F., Tetrahedron Letters, 655 (1973),
СВИНЦА ТЕТРААЦЕТАТ (III, 208—242; V, 379—383;
VI, 222—225).
Галогендекарбоксилирование. Цочи [1] нашел, что соли гало-
геноводородных кислот в присутствии С. т. превращают карбо-
новые кислоты с хорошим выходом в алкилгалогениды. Рассма-
триваемый метод особенно удобен для получения алкилхлори-
дов. В отличие от классической реакции Хунсдикера новый
метод применим к карбоновым кислотам, карбоксильная группа
которых находится не только у первичного, но также н у вто-
ричного и третичного атомов углерода. Стехиометрия реакции
представлена уравнением
RCOOH + Pb(OAc)4 + MX —> RX + СО2 + РЬ(ОАс)2 + НОАс + МОАс
Реакцию проводят в бензольном растворе; при этом для облег-
чения декарбоксилирования желательно исключить доступ кис-
лорода. Предложен свободнорадикальный механизм реакции.
437
Предложенный метод неоднократно использовался, напри*
мер Рейх и Крам [2] осуществили превращение (1)—>(2) с вы-
ходом 15%:
Японские химики воспользовались этой реакцией при изуче-
нии стереохимии зизановой кислоты [3], превращая карбоновую
кислоту (3) путем хлордекарбоксилирования в хлоркетон (4);
Австралийские химики [6] описали пример успешного хлор-
декарбоксилирования в случае, когда реакция Хунсдикера (мо-
дификация Кристола — Фирта) не идет. Так, при обработке
кислоты (5) С, т. в бензоле в присутствии хлористого лития
были получены желаемый хлорид (6), соответствующий третич-
ный ацетат (7) и смесь олефинов:
Столоу и Джайнтс [5] исследовали стереохимию реакции
Кочи. Как цис-, так и транс-4-трет-бутилциклогексанкарбоновые
кислоты (8) и (9) дают в этой реакции одинаковую по составу
смесь изомеров: 67% цис- и 33% транс-4-трет-бутилциклогек--
438
силхлоридов (10) и (11). Точно такая же смесь изомеров обра-
зуется и при разложении в четыреххлористом углероде диметил-
(цис- или транс-4-грет-бутил циклогексил) метилгипохлоритов
(12) и (13), которое, как известно, включает образование 4-
трет-бути лциклогексильного радикала [6].
Реакцию Кочи использовали также для того, чтобы просле-
дить влияние удаленных от реакционного центра полярных за-
местителей на стереоселективность свободнорадикального пере-
носа атома хлора [7].
Окислительное декарбоксилирование кислот (III, 230—234;
V, 379—382; VI, 222—223). Шелдон и Кочи [8] опубликовали
обзор, посвященный окислительному декарбоксилированию кис-
лот под действием С.т.
Окислительное декарбоксилирование ангидрида бицикло-
[2,2,0]-гексен-5-дикарбоновой-2,3 кислоты (1) до бицикло-[2,2,0]-
гексадиена-2,5 (2) (бензол Дьюара) требует строгого контроля
условий: температуры реакционной смеси до 43—45°, бани до
5- г со2
439
47—48°, продолжительности реакции 20 мин и пониженного дав-
ления [9].
Окисление амидов. Амиды кислот окисляются С. т. в ДМФА
или бензоле при 50—60° до изоцианатов по реакции типа гоф-
мановского расщепления [10]:
РЬ(ОАс)4
RCONH2 ------> RN=C==O
При обработке амидов р-оксикислот С. т. в пиридине при
комнатной температуре происходит очень быстрое образование
2-оксазолидинонов (1) с выходом около 95%. [11]. Поскольку
2-оксазолидиноиы гидролизуются основаниями до р-окси аминов,
R' ,-R2
"’CH — CH
НО^ %С=О
H2N’X
РЬ(ОАс)4
Ру
ОН
--->
R1 /R2
^СН— CHNHj
нох
(1) (?J
эту реакцию можно использовать для превращения незамещен-
ных амидов кислот в амииы (2). Реакция протекает с сохране-
нием конфигурации. Так, например, амид (3) превращается в
р-оксиамин (5) с общим выходом 72%.
1,2,3-Бензотриазины. Окисление гидразонов о-аминофенилал-
кил- или -арилкетонов представляет простой путь к 4-алкил- или
4-арил-1,2,3-бензотриазинам. Так, например, окисление соедине-
ния (1) дает продукты (2) и (3) [12].
(1) R = СН3 или с6н5
1,2,3-Бензотриазииы быстро и почти количественно обра-
зуются также при окислении 1- и 2-амино-З-замещенных инда-
440
золов С.т. в хлористом метилене [13]:
R
Реакция с альдо- и кетоннтронами [14]. С ароматическими
альдонитронами (1) С.т. дает N-ароил-О-ацетилгидроксилами-
ны (3);
ArCH—NR Pb(OAc)4 ГагС^Ыи!---------- АгС—NR
* v ГТ I
о ДсО О о ОАс
9) (2) (3)
В случае кетонитронов имеет место другое направление оки-
сления, приводящее к расщеплению С — N-связи. Так, например,
С,С,М-трифенилнитрон (4) превращается в бензгидрилиденди-
ацетат (5) с выходом около 90%:
(C6HS)ZC=NQHS . > (С6Н5)2С(ОАс)г
О
(4) &
Окисление 4,6-диамино-5-нитрозопиримидина [15]. На первой
стадии нового общего синтеза 2-, 8- и 9-замещенных аденинов
4,6-диамино-5-нитрозопиримидин (1) окисляют небольшим из-
бытком С.т. в 7-аминофуразано-[3,4-йГ]-пиримидин (2):
РЪ(ОАс)4
(2)
1. Kochi J. К, J. Am. Chem. Soc., 87, 2500 (1965); J. Org. Chem., 30, 3265
(1965). .
2. Reich H J., Cram D. J., J, Am. Chem. Soc., 91, 3517 (1969).
3. Ki do F., Uda H., Yoshikoshi A., Tetrahedron Letters, 1247 (1968).
4. Ritchie E., Senior R. G., T а у 1 о г W. C., Australian J. Chem., 22, 2371
(1969).
5. Stolow R. D., G i a n t s T. W., Tetrahedron Letters, 695 (1971).
6. Greene F. D., Chu C.-С., W a 1 i a J„ J. Org. Chem., 29, 1285 (1964).
7. Stolow R. D., Giants T. W., J. Am. Chem. Soc., 93, 3536 (1971).
8. Sheldon R. A., Kochi J. K., Org. React., 19, 279 (1972).
441
9. V a n T a m e I en E. E., Pappas S, R., К i г к К. L., J. Am, Chem. Sod.,
93, 6092 (1971).
10. Aylward J. B., Quart. Rev. Chem. Soc., 25, 407 (1971).
11. Simons S. S., Jr., J. Org. Chem., 38, 414 (1973).
12. Bradbury S., Keating M., Rees C. W., Storr R. C., Chem. Comm.,
1971, 827.
13. Adams D. J. C., Bradbury S., Horwell D. C., Keating M.,
Rees C. W., Storr R. C., Chem. Comm., 1971, 828.
14. Neiman L. A., Zhukova S. V., Tetrahedron Letters, 499 (1973).
15. Taylor E, C,, Beardsley G. P., Maki Y., J. Org. Chem., 36, 3211
(1971).
СВИНЦА ТЕТРА-(ТРИФТОРАЦЕТАТ) (V, 384).
Трнфторацетоксилнрование производных бензола. Австралий-
ские химики [1], изучавшие взаимодействие С.т. в трифторуксус-
ной кислоте с целым рядом производных бензола, показали, что
реакция протекает по типичному механизму электрофильного
замещения (3£). Так, например, алкилбеизолы реагируют бы-
стрее галогеибензолов, а нитробензол вообще ие вступает в ре-
акцию в умеренно жестких условиях. Авторы получили также
доказательство (ЯМР) того, что по крайней мере в случае гало-
генбензолов (1) в реакции промежуточно образуются т/шс-три-
фторацетаты п-галогенфенилсвинца (2). Фторзамещеиное соеди-
нение (2) (X=F) было даже выделено. Таким образом, атака
реагента приводит к замещению протона на свинец(1У). Те же
авторы, изучая возможность замещения других групп с по-
мощью С.т., обнаружили, что особенно гладко обмен металл —
металл протекает в случае /z-замещеиных фенилтриметилсила-
нов (4) (получение см. [2]) и выходы арилтрифторацетатов (6)
оказались практически количественными. Обсуждаемая реакция,
таким образом, представляет собой перспективный метод син-
теза галогенфенолов из арилгалогеиидов.
Pb(OCOCF3)4
CF3C00H
(4)
Х = СН3, Р, CI, Вг
X X
442
Нет необходимости заранее получать очень гигроскопичный
С.т., так как его можно генерировать in situ из тетраацетата
свинца и трифторуксусной кислоты.
1 Campbell J. R., Kalman J. R., P i n h e у J. T., S t e г n h e 11 S., Tetra-
hedron Letters, 1763 (1972); Kalman J. R., Pinhey J. T., Stern-
hell S., Tetrahedron Letters, 5369 (1972).
2 . G i 1 m a n H., Marshall F. J., J. Am. Chem. Soc., 71, 2066 (1949); Chva-
lovsky V., В а г a n t V., Coll. Czech., 16, 580 (1951).
СЕЛЕН (IH, 243—246).
Синтез мочевин [1]. Мочевины можно синтезировать из али-
фатических аминов, окиси углерода и кислорода в присутствии
С. в качестве катализатора. Например, н-бутиламии (0,1 моля)
растворяют в ТГФ (100 мл), добавляют аморфный селен
(0,005 г-атом), пропускают через раствор окись углерода, а за-
тем кислород (или воздух). 1,3-Ди-н-бутилмочевина получается
при этом со стехиометрическим выходом. Реакция протекает
в две стадии:
О
. II Ог
2RNH2 4- Se + СО —> (RNH3)+(RNH-C—Se)- —> (RNH)2CO + H2O + Se
Окисление 3,5-диалкилпиридинов [2]. Трехокись серы или
олеум в присутствии каталитических количеств (1—4%) Se оки-
сляет 3,5-диалкилпиридины до пиридин-3,5-дикарбоновых кис-
лот с очень хорошими выходами:
1. Sono da N., Yas uh ага T., Kondo K-, Ikeda T., Tsutsumi S.,
J. Am. Chem. Soc., 93, 6344 (1971).
2. Dieterich D., Synthesis, 1972, 631.
СЕЛЕНА ДВУОКИСЬ (III, 246—256; V, 385—386;
VI, 226—227).
Аллильное окисление. Мейнвальд и сотр. [1] использовали
эту реакцию иа первой стадии синтеза, исходя из гераиилаце-
тата (5), гиринидаля (9) [по номенклатуре ИЮПАК: (£',£',£)-
3,7-диметил-8,11 -дикето-2,6,9-додекатриена^ь] — норсесквитерпе-
ноида, являющегося главной составной частью секрета брюшных
443
желез водяных жуков вертячек (Gyrinidae).
(6)
Окисление геранилацетата (5) свежевозогнанной С.д. в ки-
пящем 97%-ном этаноле дает альдегид (6) с выходом 54%. При
добавлении последнего к 10 мол. же дилитиевого производного
бутин-З-ола-2 в ТГФ образуется ацетиленовый триол (7), кото-
рый восстанавливают до триеиа (8) избытком натрия в смеси
жидкий аммиак — ТГФ в присутствии следов этанола. На по-
следней стадии синтеза соединение (8) окисляют активирован-
ной двуокисью марганца при 0° в хлороформе. Главной особен-
ностью этого синтеза является то, что чрезвычайно реакционно-
способная 1,4-диен-3,6-дионовая система образуется лишь на
последней его стадии.
Практически идентичный синтез гиринидаля (9) одновре-
менно был описан Катценелленбогеном и сотр. [2]. В качестве
исходного соединения эти авторы вместо геранилацетата ис-
пользовали геранилмезитоат. Защитная группа удалялась при
восстановлении тройной связи под действием L1AIH4 и метилата
натрия. Этим химикам удалось провести стадию окисления
(8)-+(9) двуокисью марганца с более высоким выходом (40%).
,снгон(сно)
Й)
Окисление г^лг-диметилтризамещеиных олефинов С. д. в вод-
ном этаноле протекает стереоспецифичио с образованием транс-
спиртов и траме-альдегидов. Так, например, при окислении 2-
444
метилгептена-2 (10) образуется траш>2-метилгептеи-2-ол-1 (11)
с выходом > 98% [3].
сн3 <рн3
( -
Н3С'"^''СН3 НЭС сн2он
(IO) (и)
Реакцию применяли и для окисления 2,7-диметилоктадиена-
2,6 (12) в 2,7-дим етил-трамс,трамс-октадиен-2,6-диаль-1,8 (13).
Это бифункциональное соединение тра«с,тра«с-конфигурации,
в котором два изопреноидных остатка соединены друг с другом
по типу «хвост к хвосту», было использовано в качестве одного
из фрагментов в изящном синтезе сквалена с траме-конфигура-
цией всех двойных связей [4].
сн3 сн3
Hi С. Sepj ОНС. a. а.
48%’*
CH3 CH3
(12) (13)
Селективное окисление. Проводя синтез dZ-сиренина (3), Ра-
попорт и сотр. [5] заметили, что окисление ненасыщенного эфира
(1) С.д. в этаноле происходит в высшей степени селективно и
приводит к образованию тра«с-а,р-ненасыщениого альдегида
(2) с выходом 55 % • Последний восстанавливали смешанным
гидридом до dZ-сиренина с выходом 86%.
(0 (2) (3)
Флавоны. 2'-Оксихалконы при кипячении в амиловом спирте
или ксилоле в присутствии С.д. с хорошим выходом окисляются
до флавонов [6]. Это первый пример использования реагента
для окислительной циклизации и один из двух лучших методов
синтеза флавонов [7].
445
1. M e i n w a 1 d J., Opheim K., Eisner T., Tetrahedron Letters, 281 (1973).
2. Miller С. H., Katzenellenbogen J. A., Bowlus S. B., Tetrahedron
Letters, 285 (1973).
3. В h a 1 e г a о U. T., Rapoport H., J. Am. Chem. Soc., 93, 4835 (1971).
4. Bhalerao U. T., Rapoport H., J. Am. Chem. Soc., 93, 5311 (1971).
5. Plattner J. J., Bhalerao U. T., Rapoport H., J. Am. Chem. Soc.,
91, 4933 (1969).
6. Mahal H. S., Rai H. S., Venkatara man K-. J- Chem. Soc., 1935, 866.
7. В a r g e 11 i n i Cf. С., M a r i n i - В e 11 о 1 о G. В., in Recent Progress in
the Chemistry of Natural and Synthetic Colouring Matters, Core T. S. et al.,
Eds,, Academic Press, New York, 1962, 261.
СЕРА ДВУХЛОРИСТАЯ (III, 261; V, 387).
С. д. присоединяется к норборнадиену (1), образуя с высоким
выходом 2,8-дихлор-4-тиатрицикло-[3,2,1,03’6]-октан (2) [1]. Пола-
гают, что промежуточно образуется соль эписульфоння (а) [2].
Оба атома хлора в соединении (2) можно количественно заме-
стить бромом, обрабатывая его трибромидом бора (I, ПО—111;
V, 37—38; VI, 30—31).
1. L a u t е n s с h 1 а е g е г F., J. Org. Chem., 31, 1669 (1966); 34, 3998 (1969).
2. Ziman S. D., T г о s t В. M., J. Org. Chem., 38, 649 (1973).
СЕРЕБРА БОРФТОРИД (III, 264—266; V, 388—389-
VI, 229—231).
Реакции циклоприсоединения дегидробензола с циклическими
олефинами. Дегидробензол реагирует с циклогексадиеном, да-
вая в качестве главных продуктов углеводороды (1) —(4) [1].
Углеводород (1) образуется в результате [2-j-4]-циклоприсоеди-
иения, соединения (2) и (3) — путем енового циклоприсоедине-
ния, а продукт (4) — [2 + 2]-циклоприсоединением. На результат
реакции заметно влияет добавление каталитических коли-
честв С. б.; в этом случае почти единственным продуктом реак-
ции становится углеводород (1). Ион серебра, однако, не ока-
зывает практически никакого влияния иа реакцию дегидробен-
зола с ациклическими диенами [2]. Постулируемый механизм
(1) 48# (2) 18# (з) 21# (4) 13#
443
этой реакции включает образование комплекса Ag+—олефин,
разлагающегося с образованием нового комплекса: дегидробен-
зол—олефин—Ag+.
Перегруппировки трицикло-[3,2,0,024]-гептанов [3]. Обра-
ботка углеводорода (1) небольшим количеством С. б. вызывает
быструю перегруппировку его в изомерный углеводород (2).
Аналогично (3) превращается в (4).
Это — первое сообщение о регио- и стереоселективной мигра-
ции метильных групп, промотированной Ag+.
AgBF4
(3) (4)
1. Crews Р., В е а г d J., J. Org. Chem., 38, 522 (1973) и ссылки, цитируемые
в работе.
2. С г е w s Р„ В е а г d J., J. Org. Chem,, 38, 529 (1973).
3. Paquette L. A., Lei ch ter L. M„ J. Am. Chem. Soc., 94, 3653 (1972).
СЕРЕБРА КАРБОНАТ (111,267; V, 389—390).
Эфиры а-ациламиноакрнловых кислот. Кипячение метилового
эфира N-ацетилцистеина (1) с небольшим избытком С. к. в ме-
таноле в течение 1 час дает метиловый эфир а-ацетиламиноакри-
ловой кислоты (2) с выходом 78%. Описанный метод приложим
к разнообразным производным цистеина и пеницилламина [1].
HS-.
Сн2 Ag;CO3
НС^ 78% '
AcN СООСН,
Н 3
Н. ZH
if
С
AcN СООСН,
Н
(О (2)
Окисление гидрохинонов. Для окисления гидрохинонов в хи-
ноны Шефер и сотр. [2] использовали С. к. в сочетании с сульфа-
том магния в абсолютном бензоле. Приведенные авторами вы-
ходы лежат в пределах 60—100%.
1. Gravel D,, Gauthier R., В е г s е С., J. С. S. Chem, Comm., 1972, 1322.
2. Schafer W., Leute R., Schlude H., Chem. Ber., 104, 3211 (1971).
447
СЕРЕБРА КАРБОНАТ - ЦЕЛИТ (V, 389—390; VI,231—233).
Механизм. Фетизон и сотр. [1] предложили согласованный
механизм окисления карбонатом серебра на целите, первая ста-
дия которого заключается в обратимой абсорбции спирта на
оу хн
£. + +
Ag Ag
О О'
чсх
II
о
-----> )с=он
2 Ag, С0/“, Н+
целите. Это облегчает координацию между катионом и
спиртовым атомом кислорода, который в результате этого ста-
новится положительно заряженным. На второй стадии окисле-
ния происходит согласованный перенос электронов, включаю-
щий и другой катион серебра.
Окисление р-аминоспиртов [2]. Окисление спирта (1)
С. к.—‘ ц. в бензоле привело к образованию вместо ожидаемого
насыщенного кетона смеси соединений (2) и (3) приблизитель-
но в отношении 2:1. Этот одностадийный процесс может пред-
ставить интерес для получения а-амино-а,|3-неиасыщенных ке-
тонов. 2-Ацетил-1,4,5,6-тетрагидропир идин (2) является одним из
соединений, создающих аромат свежеиспеченного хлеба.
Окисление углеводов. Фетизон и Моро [3] опубликовали
краткое сообщение об окислении некоторых гексоз. Окисление
глюкозы и маннозы является сложным процессом. В отличие
от упомянутых сахаров окисление галактозы (1) протекает чи-
сто. При проведении реакции в воде при 80° образуется ц-га-
лактоиолактон (2). Если окисление проводить в кипящем эта-
ноле, главным продуктом становится D-ликсоза (3), выделенная
с выходом 36%,
448
Окисление «-гликолей. ct-Гликоли типа соединения (1) селек-
тивно окисляются реагентом Фетизона до а-кетолов (2) с выхо-
дом примерно 65—80% [4]. сс-Кетолы можно окислить окисью
висмута дальше до сс-дикетонов (3) с выходом около 80%.
А^аСОз
СНзСН=СН—СН—СН—СН=СНСН; ———>
| | 80%
ОН он
(и
В12О3
—> СН3СН=СН— СО— СН— СН=СН— СНз ----->
I
он
(Л
—> сн3сн=снсососн=снсн3
(3)
Окислительная конденсация фенольных соединений [5].
Окисление ретикулина (1) под действием реагента в присут-
ствии бикарбоната натрия в хлороформе дает изоболдин (2) с
выходом 3% и паллидин (3) с выходом 1%, тогда как при окис-
лении ретикулина окситрихлоридом ванадия (VI, 42—43) изо-
болдин (2) образуется с выходом 1%, а паллидин (3)—с выхо-
дом 0,3%:
С-Нитрозосоединения [6]. С-Нитрозосоединения можно полу-
чить с выходом 55—95% окислением гидроксиламинов С. к. — ц.
Реакцию проводят при комнатной или более низкой темпера-
туре в СН2С12 или CFC13,
RNHOH —RNO
В практическом отношении эта реакция имеет то преимущество,
что вследствие ее большой скорости непрореагировавший гидро-
ксиламин не успевает конденсироваться с нитрозосоединением
в азоксипроизводное; в условиях реакции подавляется также
изомеризация первичных и вторичных нитрозосоединений в ок-
симы.
15 Зак. 596
449
Реакции фрагментации [7]. С. к.—ц. (реагент Фетизона) ко-
личественно расщепляет стероидные 17-этинилзамещенные
спирты. Так, например, при кипячении моиометилового эфира
17сс-этинилэстрадиола (1) с С. к.— ц. в толуоле количественно
образуется метиловый эфир эстрона (2). Аналогичным образом
из соединения (3) был получен дегидроэпиандростерон (4).
Изучена также фрагментация стероидных циангидринов. Ки-
пячение циангидрина прегнеиолонацетата (5) с реагентом в то-
луоле приводит с количественным выходом к прегненолонаце-
тату (6).
1. Fetizon М., Go 11 i er М., Mourgues Р., Tetrahedron Letters 4445
(1972).
2. Bile hi G, Wiles t H., J. Org. Chem., 36, 609 (1971).
3. Fetizon M., Moreau N„ Compt. Rend., 275 (C), 62! (1972).
4. Thu an S., Wlemann J., Compt. Rend., 272 (C), 233 (1971).
5. Kametani T., Kozuka A., Fukumoto I\., J. Chem. Soc. (C), 1971,
1021.
6, Alaassen J. A,, de Boer Th. J., Rec. trav., 90, 373 (1971).
7. Lenz G, R., Chem. Comm., 1972, 468,
450
СЕРЕБРА НИТРАТ (111,267—271; V, 390—393; VI, 233—234).
Окисление 1,1-бис-диалкнламнноэтиленов [1]. 1,1-бис-Диме-
тиламиноэтилен (1) окисляется С. и. в ацетонитриле до нитрата
1,1,4,4-тетракис-(диметиламино)-бутен-2-диилия-1,4, вероятно, че-
рез промежуточные продукты, приведенные на схеме в квадрат-
ных скобках:
2 AgNOj
2 CHf=C[N(CH3)2;2 CHjCN
(1)
> [(СН3)гН1гССНгСН2сЕК(СН3)Д
2 NO3"
—> I (CHj)jN)jC=CHCH=C[N(CHj)2}2
+ 2 СН3С[К(СНэ)г]гКОз"
2 AgN°3—> [ (СН3)гЮгССН=СНС( N(CH3)^ 2 NOf
(2)
В случае диентетраминов при такой обработке имеет место
преимущественно циклизация, а не димеризация. Так, например,
при окислении N,N,N',N\N" Г4",Ь1///,Ь1//,-октаметилпентадиен-1,4-
тетрамина- 1,1,5,5 (3) с высоким выходом образуется смесь цис-
и транс-циклопропилен-бис- (диметиламинометилий) -нитратов
(4):
AgNO3
г , , 1 CHiCN
СнЛсН С (NMez )г 1г------->
85%
(3)
+
xC(NMeA
4-
C(NMe2)j
2 NOj"
Этим методом можно синтезировать даже бициклические си-
стемы. Так, при окислении \,3-бис[бис- (диметиламино)-метилен]-
циклогексана (5) образуется бицикло-[3,1,0]-гексанилен-1,5-бис-
(диметиламииометилий)-нитрат (6):
(0
Превращение элемола в эйдесмолы [2]. Термолиз элемола
(1) при 210—220° приводит к частичной изомеризации его в ге-
дикариол (2), а при термолизе (1) в присутствии следов С.н.
наряду с продуктом (2) образуется примерно по 0,8% а-эйде-
смола (3) и р-эйдесмола (4). Эта реакция является, по-види-
мому, первым примером катализируемых ионами серебра (I)
15*
451
превращений, затрагивающих л-связи.
р-Лактамы. Взаимодействие N-хлорпроизводного карбииол-
амина (1) с избытком С. и. в ацетонитриле (3 час) приводит
к образованию р-лактама (2) [3]:
(n
AgNO3
CH3C=N
-AgCl '
(2)
Диалкнлы и диарилы [4]. Триалкил- или триарилбораны при
обработке С. и. в присутствии щелочи дают диалкилы и диари-
лы (выход 50—90 %):
AgNO3, он-
ИзВ ------------> R—R
!. Weingarten Н., Wager J. S-, J. Org. Chem., 35, 1750 (1970).
2. Jain Т. C., McCloskey J. E., Tetrahedron Letters, 5139 (1972).
3. Wasserman H. H., Adickes H. W., Espejo de Ochoa 0., J. Am.
Chem. Soc., 93, 5586 (1971).
4. Breuer S. W., В roster F. A., Tetrahedron Letters, 2193 (1972).
СЕРЕБРА ОКИСЬ (III, 272—276; V, 393; VI, 234—235).
Переалкилнрование. Классический реагент переалкилирова-
ния простых эфиров — алкоголят натрия в соответствующем
спирте. Браун и Сугимото [1] нашли, что превосходным катали-
затором переалкилирования некоторых алкокси-1,2,4,6,8-пента-
азанафталинов (1), алкоксинитропиримидинов (2) и родствен-
452
ных гетероциклических простых эфиров кипящими спиртами
является С. о. Ацетат серебра менее активен.
Окислительное метилирование. При попытке синтезировать
метиловый эфир 5-ванилилиденбарбитуровой кислоты (1) с по-
мощью С. о. и йодистого метила в ДМФА методом Куна (III,
278) Этье и Невилль [2] неожиданно получили соответствующий
бензальдегид (2) и тетраметилбарбитуровую кислоту (3). Реак-
ция промотируется, вероятно, ионом серебра, взаимодействую-
щим с олефиновой двойной связью.
Алкилирование 2-арил-1,3,4-оксадиазолов [3]. При алкили-
ровании 2-арил-1,3,4-оксадиазолов (1) галогеналкилами в ацето-
нитриле в присутствии С. о. с хорошим выходом образуются О-
и N-алкилпроизводные (2) и (3), легко разделяющиеся хрома-
тографически.
(I)
RX, AgzO
CH3CN
70-90$
(2) (3)
. Гидролиз тнокеталей [4]. При гидролизе тиокеталей десяти-
кратным избытком С.о. в кипящем водном метаноле (1:10)
продолжительностью от 16 час до 4 дней образуются исходные
кетоны (выход 75—80%).
1. Brown D. J., S u g i m о t о T., J. Chem. Soc. (C), 1970, 2661.
2. E t h i e r J. C„ N e v i 11 e G. A., Tetrahedron Letters, 5297 (1972).
3. G о 1 f i e г M., M i 1 с e n t R., Bull. soc. chim. France, 1973, 254.
4.. Gravel D., Vazlri C., Rahal S., J. C. S. Chem. Comm., 1972, 1323
СЕРЕБРА(П) ОКИСЬ (V, 393—394).
Окислительное деметилирование. Рапопорт и сотр. [1], пы-
таясь окислить с помощью С. о. метиленовую группу диметило-
вого эфира менахинола-1 (1), являющуюся одновременно ал-
463
лильной и бензильной, неожиданно выделили лишь меиахинон-1.
Благодаря этой реакции стало возможным защищать хиноиы,
превращая их в метиловые эфиры гидрохинонов.
При действии пиколината серебра (II) (VI, 209) на соедине-
ние (1) в ДМСО происходит окисление метильной группы в по-
ложении 2 с образованием соответствующего бензилового спир-
та (3) с выходом 25%.
Окислительное расщепление простых эфиров гидрохино-
на [2]. Реагент вызывает окислительное расщепление диметило-
вых эфиров «-гидрохинонов до /z-хинонов; выходы хорошие. Для
обеспечения высокой степени превращения небходимо добав-
лять минеральную кислоту (наиболее эффективные азотиая и
хлорная кислоты) и использовать избыток окислителя. о-Хиио-
ны можно получить лишь с умеренными выходами.
Различные реакции окисления [3]. Некоторые ароматические
амины (например, анилин, «-толуидин, «-хлоранилин) при окис-
лении С. о. дают с умеренными выходами соответствующие азо-
производные. Гидрохинон быстро и количественно окисляется
AgO до «-бензохинона. Окисление дигидразона бензила AgO
приводит к образованию дифенилацетилена с выходом 95%.
1. Snyder С. D., Bondinell W. Е., Rapoport Н., J. Org. Chem., 36,
3951 (1971).
2. Snyder С. D., Rapoport H., J. Am. Chem. Soc., 94, 227 (1972).
3. Oritz B., Villanueva p., Walls F., J. Org. Chem., 37, 2748 (1972).
СЕРЕБРА ПЕРХЛОРАТ (V, 394—396).
Метанолиз галогенкарбеновых аддуктов. С. п. катализирует
метанолиз э/сзо-8-бром- и 8,8-дибромбицикло-[5,1,0]-октанов (1а)
и (16) соответственно до 3-метокси- и 2-бром-З-метокси-транс-
454
циклооктенов (2а) и (26) с высокими выходами [1]. В тех же
условиях соответствующие производные бицикло-[5,1,0]-октепа-3
(1)
а)Х = Н
.5) X = В г
СН3ОН
AgC104
(2) X
а) Х = Н
б) Х= Вг
(За) и (36) дают в качестве главных продуктов 6-метокси- и
5-бром-6-метокси-грс,граж>циклооктадиены-1,4 (4а) и (46) [2].
CHjOH
AgC104^
э/сзо-8-Бромбицикло-[5,1,0]-октен-2 (5а) в тех же условиях
превращается в смесь (примерно 1:1) двух изомерных метокси-
циклооктадиенов — 3-метокси-г{йс,транс-циклооктадиена-1,4 (6а)
и 5-метокси-цис,транс-циклооктадиена-1,3 (7а) [3]. Аналогичный
результат был получен в случае 8,8-дибромбицикло-[5,1,0]-окте-
на-2 (56), образующего диены (66) и (76). Эти продукты полу-
н
(5)
а) X = Н
б ) X а ВГ
чаются, по-видимому, в результате атаки метанола по обоим
концам промежуточно образующегося тргшс.транс-аллильного
катиона.
Декарбонилирование [4]. Действие С. п. на трицикло-
[2,1,0,02’5]-пентаноны-3 в бензоле при комнатной температуре вы-
зывает их декарбонилирование. Обработка соединения (1) Ag+
в метаноле приводит к образованию циклобутена (2): анало-
455
гично кетон (3) превращается в соединения (4) и (5). Предпо-
о
(2)
лагают, что в этих реакциях промежуточно образуется катион
(6).
Бициклобутаны-> бутадиены [5]. Бициклобутан (1) под дей-
ствием С.п. при комнатной температуре перегруппировывается
в бутадиен (2) с выходом около 90%:
AgClO4
(О
(2)
жзо,экзо-2,4-Диметилбициклобутан (3) и эн5о,экзо-2,4-диметил-
бициклобутан (4) в тех же условиях превращаются в транс-
транс- и г{Цс,7’ранг-гексадиены-2,4 (5) и (6) соответственно. Ре-
акция, таким образом, в большинстве случаев стереоспецифич-
на, причем стереохимия ее противоположна наблюдаемой при
термолизе.
AgClO4
------
456
Перегруппировки (V, 395). Если эндо-2-метокситрицикло-
[4,1,0,03’7]-гептан (1) обработать 1—3 мол.% С. п. в безводном
бензоле, он быстро и количественно перегруппировывается в
антн-7-метоксинорборнен (2). Изомеризация вызывается перво-
начальной ионизацией метоксигруппы, а не каким-либо взаимо-
действием Ag+ с напряженной связью. Незамещенный трицик-
ло-[4,1,0,03’7]-гептан устойчив к действию С. п. [6].
Перегруппировки с участием напряженных а-связей (см. Се-
ребра борфторид, VI, 229—231). Пакьетт и сотр. [7] недавно ис-
следовали катализируемую Ag+ перегруппировку производных
менее напряженных, чем кубаны, секокубанов, эндо,эндо-Д,и-
эфир (1) при обработке раствором безводного С. п. в абсолют-
ном бензоле количественно и стереоспецифически превращается
в производное эис?о,энс?о-тетрацикло-[3,3,0,0,2’8,04’6]-октаиа (2), а
эндо,экзо-изомер (3) дает в этих условиях изомер (4) диэфи-
ра (2). При перегруппировке соединения (5) образуется един-
ственный продукт — экзо,эюзо-диэфир (6). Скорости перегруп-
пировки трех диэфиров, производных секокубана, сравнимы со
скоростью перегруппировки более напряженных кубановых си-
стем. Отсюда ясно, что ослабление напряженности не является
фактором, определяющим скорость перегруппировки.
457
Пакьетт [8] опубликовал обзор реакций изомеризации напря-
женных о-связей под действием иоиов Ag(I).
1. R е е s е С. В., S h a w A., J. Am, Chem. Soc., 92, 2566 (1970).
2. Baird М. S., R е е s е С. В., Chem. Сотт., 1970, 1644.
3. Baird М. S., Reese С, В., Tetrahedron Letters. 4637 (1971).
4. О n а Н., Sakai М., Suda М., Masamune S.. J. С. S., Chem. Сотт,,
1973, 45.
5. Sakai М., Yamaguchi Н., Westberg Н Н., Masamune S., J. Ат,
Chem. Soc., 93, 1043 (1971).
б. Р a q u е 11 е L. A., Z о n G., J. Am. Chem. Soc., 94, 5096 (1972).
7. Paquette L. A., Beckley R. S,, McCreadie T., Tetrahedron Letters,
775 (1971).
8. Paquette L. A., Accts. Chem. Res. 4, 280 (1971).
СЕРЕБРА СУЛЬФАТ (III, 276—277; VI, 235—236).
Гидролиз 1,3,5,7-тетрабромадамантана [1].
1. S tetter H., Kr a use ,M„ Ann., 717, 60 (1968),
СЕРНАЯ КИСЛОТА, H2SO4.
2-Оксаадамантан [1]. Под действием 95 %-ной С. к. бицикло-
[3,3,1]-нонандиол-2,6 (1) при комнатной температуре образует
с выходом 35—40% 2-оксаадамаитан (2). Механизм реакции
включает образование карбоииевых ионов и 1,2-гидридиые
сдвиги. Использование как более концентрированной кислоты
но
Ю (2)
(8% олеума), так и 75%-ной кислоты заметно уменьшает выход
продукта (2).
Сужение цикла 2-алкилнденциклобутанолов с образованием
циклопропилкарбонильных соединений [2]. 2-Метиленциклобу-
танол (1) при нагревании с 5%-ной H2SO4 при 100° в течение
30 мин количественно превращается в 1-метил-1-формилцикло-
пропан (2); такую же перегруппировку можно стимулировать
458
нагреванием при 245° в запаянной ампуле в течение 4 час. В
H;SO4
количеств.
СНО
СН3
(2)
этой реакции сужения цикла требуются более жесткие условия,
чем в реакции расширения цикла 1-винилциклопропанола, в
результате которой образуется 2-метилциклобутанол (см. Бро-
мистый водород, этот том).
Образование полуацеталя [3]. Обработка 2-(2'-метил-3'-
хлораллил)-циклогексанона (1) конц. H2SO4 приводит к обра-
зованию полуацеталя (2) с выходом 85%. Постулировано, что
промежуточным продуктом в реакции является ион хлоро-
ния (а).
Циклизация цитраля в а-циклоцитраль. Цитраль (1) (смесь
цис- и траис-изомеров) под действием пирролидина в присут-
ствии молекулярных сит марки 4А превращается в енамин (2).
Обработка последнего смесью конц. С. к. и воды (10-; 1 по
объему) дает а-циклоцитраль (3) с выходом 41% [4].
Использование оптически активного пирролидина (4), полу-
ченного из ь-пролина, позволило синтезировать этим методом
(S)-(+)-а-циклоцитраль с оптическим выходом 12%. Оптиче-
459
ский выход (27%) удалось повысить, применив замещенный
пирролидин (5) [5].
(4)
(5)
Циклизация (Е)- и (7)-4,8-диметилнонадиен-3,7-оиов-2. Ци-
клизация полиенов, катализируемая кислотами, в настоящее
время изучена достаточно хорошо (VI, 294) [6]. Например, цик-
лизация 6,10-диметилундекатриен-3,5,10-она-2 (1) под дейст-
вием конц, С. к. дает р-нонон (2) с выходом 85% [7],
Циклизация (Е)- н (Z)-4,8-диметилнонадиен-3,7-онов-2 (3)
под действием 75%-ной H2SO4 дает в качестве главных продук-
тов 1,3,5-триметил-2-оксабицикло-[3,3,1]-нонен-3 (4) и третичный
спирт (5), а ожидаемый продукт 1,1,3-триметил-2-ацетилцикло-
гексен-3 (6) образуется лишь в небольших количествах:
о
(4) 1,5 г
(5) 1,04 г
(6) О, 35 г
По мнению Бюхн [8], главный продукт реакции
ся из диенона (7);
(4) образует-
Детиоацетализация. Действие холодной конц. С.к, (10л/м«)
вызывает гидролиз дитноацеталей [9]. Этот метод был исполь-
460
зован в синтезе дигидрожасмона (4). При обработке (2) конц.
С, к. с одновременным гидролизом хлорвинильной группировки
к - BuLi
(реакция Вихтерле) образуется продукт (3) [10]. В жасмон (4)
дикетон (3) был превращен уже ранее действием гидроокиси
натрия в этаноле [11].
Выход соединения (3) довольно низок, но детноацеталнза-
ция простых кетонов обычно протекает с высокими выходами
(90-98%).
1. Аверина Н. В., Зефиров И. С., Кадзяускас П. П., Садо-
вая Н. К., ЖОрХ, II, 77 (1975).
2. Barnier J. Р„ Denis J. М„ Salatin J, R., С onia J. М., J. С. S.
Chem. Comm., 1973, 103.
3. Woo Е. Р., Mak К. Т., Wong Н. N. С., Tetrahedron Letters, 585 (1973).
4. Yamad a S,, S h i b a s a ki M., Ter a shima S., Tetrahedron Letters,
377 (1973).
5. Yamada S., Shibasaki M., Ter a shima S., Tetrahedron Letters,
381 (1973).
6. Johnsonn W. S-, Accts. Chem. Res., I, 1 (1968).
7. Hoffmann W., Pasedach H„ Pommer H., Reif W., Ann., 747,
60 (1971).
8. В ii c h i G., Pickenh agen W., J. Org. Chem., 38, 894 (1973).
9. И о T.-L., H о H, C., Wong С. M., Can. J. Chem., 51, 153 (1973).
10. Marshall J. A., Schaeffer D. J„ J. Org. Chem., 30, 3642 (1965).
11. HoH. C„ Ho T.-L., Wong С. M., Can. J. Chem., 50, 2718 (1972).
СЕРНИСТЫЙ АНГИДРИД (HI, 278—279; V, 398).
1,1-Д иокись тиепина (4) [1]. При пропускании С. а. в хо-
лодный эфирный раствор винилдиазометана (1) выделяется
азот н образуется сульфон, 1,1-диокись 4,5-дигидротиепнна
(3), с выходом 29%. В этой реакции, вероятно, промежуточно
461
образуется ддс-дивинилэписульфон (2), вступающий в быст-
рую перегруппировку Коупа. 1,1-Диокись тиепнна (4) получают
z ch2=chchn2 —5Ос >
- 2N;
(2) (3) [4)
аллильным бромированием сульфона (3) с последующим де-
гидробромированием.
1. Paquette L. A., Ma ioran a S., Chem. Comm., 1971, 313.
СЕРНЫЙ АНГИДРИД (111, 279).
Селективное окисление [1]. При кипячении 2,3,4,5,6-пента-
хлортолуола (1) с избытком С. а. (стабилизированного) в тече-
ние 2—3 час образуется вещество состава СуНзСК-ОуЭг (2),
гидролиз которого дает 2,3,4,5,6-пентахлорбензнловый спирт (3)
с выходом 91%. Если увеличить продолжительность реакции до
24 час, то с почти количественным выходом образуется пента-
хлор бензальдегид.
1. М а г k V., Zengierski L., Pattison V. A., Walker L. Е.. J. Am.
Chem. Soc., 93, 3538 (1971).
СЕРНЫЙ АНГИДРИД — ПИРИДИН (Ш, 282—283; V,
399—400; IV, 236—237).
Восстановление (VI, 236—237). На последней стадии синтеза
природного терпена, рацемического а-транс-бергамотена (2),
авторы применили разработанный ими ранее метод двухстадий-
ного восстановления спиртов: (1) обработкой С. а. — п. сначала
превращают в алкилсульфат пиридиния, который восстанавли-
462
вают затем с помощью L1A1H4 [1].
Этим методом восстанавливали также кеталь диенола (3)
до кеталя диена (4) с выходом 70%. Последний интересен тем,
что он является одним из промежуточных продуктов в полном
синтезе сесквитерпена (±)-оццнденталола (5).
(з)
I) so,- С,НЩ
2) ЫА1Щ
7О?6
(4)
Сульфирование индолов [3]. Индол, 1-метил-, 2-метил- и 1,2-
диметилиндолы сульфируются реагентом в кипящем пиридине
по С3*атому. З-Метил- и 1,3-диметилиндолы сульфируются по
Сз-атому. 2,3-Диметилиндол в этих условиях не вступает в ре-
акцию.
1. Corey Е. J., Cane D. Е., Libit L., J. Am. Chem. Soc., 93, 7016 (1971).
2. Watt D. S., Corey E. J., Tetrahedron Letters, 4651 (1972).
3. Smith G. F., Taylor Г). A., Tetrahedron, 29, 669 (1973).
СЕРОВОДОРОД, H2S. Мол. вес 34,08.
Нуклеофильное замещение в хлорцианбензолах [1].
Cl SH
1. В е с k G., D е g е п е г Е., Н е i t zет И., Ann., 716, 47 (1968).
СИММОНСА - СМИТА РЕАГЕНТ (III, 286—290; V, 400—
402; VI, 237—241).
Усовершенствованная методика. Конья и сотр. [1] описали
две усовершенствованные реакции Симмонса — Смита, приводя-
щие к заметному улучшению выхода. Согласно одной из них,
463
вместо цинк-медной пары применяют цинк-серебряную. Ее по-
лучают прибавлением гранулированного цинка к перемешивае-
мому горячему раствору ацетата серебра в уксусной кислоте и
перемешивают смесь еще 30 сек,. Образовавшуюся цинк-сереб-
ряную пару отделяют декантацией, промывают уксусной кис-
лотой и эфиром и затем стабилизируют добавлением небольшо-
го количества тонкой серебряной проволоки. Другое усовершен-
ствование состоит в том, что реакционную смесь не подвергают
кислотному гидролизу, а вместо этого добавляют амин, напри-
мер пиридин. Последний образует нерастворимые комплексы
ZnLrCsHgN и ICH2ZnI (CgHgNJs; циклопропановые продукты
реакции выделяют затем из фильтрата.
Используя эти усовершенствования, Денис и Конья [2] пре-
вратили бис- (триметилсилокси) бутадиен-1,3 (1) в дициклопро-
пан (2) с выходом 78%. бис-Силиловый эфир гидролизовали
НгС^О51(СН3)3
(СЩ)35Ю'хЛ^СНг
ш
1) ICH,Znl/Ag A^OSi(CH3h
W J-7
(CH3)3siO''V
СНдОН
100%
(2) (3)
до свободного диола (3) с количественным выходом кипячением
в метаноле в течение 3 час. Соединение (2) можно гидролизо-
вать также кипячением в ацетоне, содержащем 2 же воды.
Синтез метилового эфира стеркуловой кислоты (VI, 237).
Уильямс и Сгоутас [3] сократили число стадий в синтезе мети-
лового эфира стеркуловой кислоты (3) по Генслеру, осуществив
прямое декарбэтоксилирование одного из промежуточных 'сое-
динений (1) под действием фторсульфоновой кислоты в хлори-
стом метилене при комнатной температуре:
сн3(снг)тс = с(снг)7соосн3
Хсн
соосгн3
(1)
FSOjH или
. . С150?У > СНЭ(СН:)7С == С(СНг)7СООСН3 ->
+ сн
NaBH* > СН5(СНг)7С = С(СНг)7СООСН3
бЭ’/^считая наЦ)
(3)
Циклопропанилирование аллилового спирта [4]. Аллиловый
спирт (1) в присутствии реагента Симмонса — Смита чрезвы-
чайно легко дегидратируется до диена (3). При тщательном кон-
троле температуры (31—38°) соединение (2) было получено с
максимальным выходом 36%, выход диена в этих условиях со-
464
ставляет 11%.
1. D е n i s J, M„ Girard C., Conia J, Synthesis, 1972, 549.
2. D e n i s J. M., Conia J. M., Tetrahedron Letters, 4593 (1972),
3, Williams J. L,, Sgout as D. S., J. Org. Chem., 36, 3064 (1971).
4. DaubenW. G., Fullerton D. S., J. Org. Chem., 36, 3277 (1971).
5. Wittig G., Hutchison J. J., Ann., 741, 79 (1970).
СОЛЯНАЯ КИСЛОТА.
Селективное О-деметилированне. Селективное О-деметили-
рование хлоргидрата (±)-О-метилангалонида (1) до хлоргнд-
рата (+)-7,8-диокси-6-метокси-1-метил-1,2,3,4-тетрагидроизохи-
нолииа (2) было осуществлено кипячением в 20%-ной С. к. Эта
реакция является ключевой стадией в синтезе двух тетрагидро-
изохинолиновых алкалоидов кактуса ангалонина и лофофори-
на [1].
1. В г о s s i A., Blount J. F„ О’В г i е n J., Т е i t е 1 S., J. Am. Chem. Soc.,
93, 6248 (1971).
( + )-(К)-траяс-р-СТИРИЛ-п-ТОЛ ИЛ СУЛЬФОКСИД.
Мол. вес 242,33, т. пл. 81,5—82°, aD + 164,3°.
до
n-CH3C6H4S^ хн
с=сх
Нх с6н5
(П
465
Получение [1]. Реагент получают из (R)-(+)-метил-п-толил-
сульфоксида, как показано ниже:
о О ОН NaH
II I К II । СН31
П-СНэСбН45СН3 + С6Н5СНО л-СНэС6Н45СНгСНС6Н5 —
n-CHjCiHiS^ хн
-----> С=сч
Нх с(н5
(1)
Асимметрический синтез по реакции Михаэля [1]. Японские
химики установили, что а,р-ненасыщеиные сульфоксиды легко
вступают в реакцию присоединения по Михаэлю. Так, напри-
мер, я-толилвинилсульфоксид (2) реагирует с малоновым и аце-
тоуксусным эфирами в присутствии эквимолярного количества
этилата натрия в этаноле, давая аддукты (3) и (4).
о
СН2(СООСгН5)2 II
_ ,---------------> и-СН3СвН45СН2СИ2СН(СООСгН5)2
° I 6|% И
«-CH3C6HtSCH=CH2 ~
(2) СН3СОСН2СООС2Н5 ? /СООСгЙБ
w ।----------------> n-CH3CeH4SCH2CH2CH
71 % \юсн3
(4)
Те же авторы изучили реакцию Михаэля оптически актив-
ного а,р-ненасыщенного сульфоксида (1) с малоновым эфиром
в присутствии эквимолярного количества этилата натрия при
80° в аргоне. При этом был получен диэтиловый эфир 2-(/г-то-
лилсульфинил) -1-фенилэтилмалоновой кислоты (5) с выходом
О
C2H5ONa II
(1) + СН2(СООС2Н5)2 ——-> n-CH3C6H4SCH2CHCH(COOC2H5)2 —>
82% ।
с6н5
(5а). (56)
N1 Ренея
С2Н5ОН
— (+)-(R)S“(5a) qoci С6Н6СНСН(СООС2Н5)2
У2 /о |
сн3
ад+93.10 (-)’(б)
82% в виде смеси диастереомеров (5а) и (56) в отношении 4: 1.
Изомер, образующийся в большем количестве, (5а) удалось
выделить дробной кристаллизацией с общим выходом 51%, счи-
тая на (1). Десульфуризация его под действием никеля Ренея
дает (—)-диэтиловый эфир 1-феиилэтилмалоновой кислоты (6),
который путем гидролиза и декарбоксилирования превращали
в известную оптически активную (—) -3-фенилмасляную кислоту
466
СбН5СН(СНз)СН2СООН. Изомер (5а) был получен, следователь-
но, с оптической чистотой не менее 95% и обладай (R)s — (К)с-
конфигурацией (а).
П-СНзС^ СНг С6Н5
с
о
j н
сн(соосгн5)2
1. Tsiichihashi G,, М i t a m u r a S., Inoue S., Ogura К., Tetrahedron
Letters, 323 (1973),
СУЛЬФОЛЕН-3 (V, 402—403).
Озонирование и превращение в 1,1-диокись 4Н-1,4-тиазнна [1]:
1) о3, СгН5ОН
СНгС1г, -78°
2) 5Ог
1. Nol and W. Е„ DeM aster R. D., Org, Syn., 52, 135 (1972),
о-СУЛЬФОНАДБЕНЗОЙНАЯ КИСЛОТА,
Мол. вес 218,18.
Надкислоту получают с выходом 65—75% реакцией ангид-
рида о-сульфобензойной кислоты с 30 % -ной перекисью водо-
рода в ацетоновом растворе в интервале температур от —4 до
0°. Полученный раствор используют непосредственно для эпок-
сидирования олефинов. Так как реагент содержит сильнокнслую
группировку, то если не добавить буфер, например твердый
карбонат иатрия, первоначально образующийся эпоксид под-
вергается кислотному расщеплению до транс-диола. Надкисло-
ту можно также использовать для окисления по Байеру — Вил-
лигеру и для окисления третичных гетероциклических аминов
до N-окисей [1].
I. Bachhawat J, М„ Math ur N, К., Tetrahedron Letters, 691 (1971).
СУЛЬФОУКСУСНАЯ КИСЛОТА (III, 291—292; V, 404).
См. Ацетилсерная кислота, этот том,
СУЛЬФУРИЛ ХЛОРИСТЫЙ (Ш, 292—296; V, 404—405;
VI, 245).
467
Свободнорадикальное хлорирование циклобутан-1,1-дикарбо-
новой кислоты [1],
В 2-литровую трехгорлую круглодонную колбу, снабженную
мешалкой Трубора с лопастями, загружают 172,8 г (1,2 моля)
циклобутан-1,1-дикарбоновой кислоты и 1500 мл бензола. Смесь
нагревают до кипения при перемешивании и отгоняют для уда-
ления влаги 200 мл бензола (или азеотропной смеси бензола
с водой). Затем колбу снабжают капельной воронкой и обрат-
ным холодильником с хлоркальциевой трубкой. Продолжая пе-
ремешивание и нагревание, в течение 40 мин вносят с помощью
капельной воронки 170 г (102 мл, 1,26 моля) С. х. (предвари-
тельно перегнанного) и одновременно через верх холодильника
маленькими порциями добавляют 4,0 г перекиси бензоила. Пос-
ле короткого индукционного периода начинают выделяться хло-
ристый водород и сернистый газ. По окончании прибавления
реагентов смесь кипятят еще 22 час. Твердый осадок раство-
ряется через 1 час, образуя светло-коричневый раствор. По
окончании кипячения бензол отгоняют, а остаток нагревают
при 190—210° в течение 45 мин, чтобы прошло декарбоксилиро-
вание. Черный остаток переносят в небольшую колбу и перего-
няют в вакууме с 6-сантиметровой колонкой Вигре. Сначала
собирают 25—30 г предгона с т. кип. 100—130°/15 мм (смесь
циклобутанкарбоновой и 3-хлорциклобутанкарбоновой кислот),
а затем 65—79 а (40—49%) смеси цис- н транс-изомеров 3-хлор-
циклобутанкарбоновой кислоты в виде светло-желтой жидко-
сти; т. кип. 131 —137°/15 мм, 1,4790.
а-Хлорироваиие сульфоксидов (VI, 245). Тсучихаши
и сотр. [2] рекомендуют использовать С. х. для «-хлорирования
пространственно затрудненных сульфоксидов. Онн проводили
реакцию в хлористом метилене в присутствии пиридина.
При хлорировании бензгидрилбензилсульфоксида (1) хло-
ристым тионилом в хлористом метилене в присутствии либо
окнси кальция, либо пиридина он расщепляется на бензгидрил-
468
хлорид (3) и а-толуолсульфиновую кислоту (4). Предполагают,
что предшественником кислоты (4) является фенил сульфин
(5) [3].
О
II SOCI2
(C6H3)2CHSCH2C6H5 ------>
(П
CaO
---> (CSH5)2CHCI+ c6h5ch2so2h
(3) 98% (4) 80%
Py
---* (CGH5)2CHC] + неидентифицированные
продукты
O=S=CHC6H5
(5)
Хлорирование сульфонов [4]. Сульфоны в отличие от алкил-
сульфидов и алкил сульфоксидов хлорируются С. х. почти ис-
ключительно в p-положение. Так, например, тетраметиленсуль-
(1)
SOZC13) 60'
~ 100%
(2)
фон (сульфолан) (1) превращается в р-хлорпроизводное (2) с
почти количественным выходом,
N-Монозамещенные амидосульфохлориды [5]. N-Моноалкил-
амидосульфохлориды получают с высоким выходом при кипяче-
нии хлоргидрата алкиламина с С. х. в ацетонитриле в присут-
ствии кислоты Льюиса, такой, как пятихлористая сурьма.
SbCl5 в CHgCN
RNH2 • НС1 + SO2C12 —RNH~S°2Cl
Ионное хлорирование алканов [6], Алканы легко хлори-
руются С. х. в сульфолане (Ш, 305—306; V, 409—411). Так, на-
пример, адамантан (1) превращается почти исключительно в
1-хлорадамантан (2), а норборнан— исключительно в 2-эдзо-
хлорнорборнан. «-Гексан дает смесь 20% 1-хлоргексана, 56%
2-хлоргексана и 24% 3-хлоргексаиа. Хлорирование протекает
по ионному механизму.
469
L L a m p m a n G. M., A u m i 11 e r J. C., Org. Syn., 51, 73 (1971).
2. Tsuchihashi G., Ogura K-, I r i u c h i j i m a S., Tomisa w a S., Syn-
thesis, 1971, 89.
3. Meyers C. Y., McCollum G. J., Tetrahedron Letters, 289 (1973).
4. T a b u s h i I., Tamaru Y,, Yoshida Z., Tetrahedron Letters, 3893 (1971),
5. Weiss G., Schulze G., Ann., 729, 40 (1969).
6. T a b u s h i L, Yoshida Z., Tamaru Y., Tetrahedron, 29, 81 (1973).
СУРЬМА ПЯТИХЛОРИСТАЯ (111,296).
Селективное цис-хлорирование олефинов [1]. С. п. гладко
реагирует с олефинами в СС14, образуя преимущественно цис,-
виц-дихлоралканы. Так, например, циклогексен дает три приве-
денных ниже соединения и никаких других органических про-
дуктов:
SbcL
----->
i. U е m ura S., Sasaki О., О к а п о М., Chem. Comm., 1971, 1064,
ТАЛЛИЯ(1П)НИТРАТ, T1(NO3)3. Мол. вес 390,38.
Получение [1]. Т. н. получают растворением 50 г окиси тал-
лия (III) в 150 мд теплой конц. азотной кислоты. Светло-жел-
тый раствор охлаждают до 0°, тригидрат Т.н. отделяют и вы-
сушивают в вакууме иад Р2О5. Соль устойчива при хранении
в плотно закупоренной склянке.
Окисление олефинов [2]. Т. н. легко и быстро окисляет оле-
фины в метаноле при комнатной температуре в альдегиды и ке-
тоны. Если олефин содержит хотя бы один заместитель, обла-
дающий повышенной способностью к миграции, то окисление
сопровождается перегруппировкой, которая может стать глав-
ным и даже единственным направлением реакции.
Примеры:
TKNOsh
------>.
R1
V—со—i—R3
1V
С6Н5С(СН3)=СНС6Н5 (C6H5)ZCHCOCH3
Циклогексен окисляется в формилциклопентан с выходом
85%, т. е. индуцируемая Т.н. реакция сужения цикла является,
в сущности, наилучшим методом получения этого альдегида.
Кори и Равиндранатан [3] использовали эту реакцию на одной
из стадий синтеза 11-дезоксипростагландинов для превращения
соединения (1) в (2). Однако при проведении реакции в перво-
начально предложенных условиях образовывалась сложная
смесь продуктов, содержащая (2) лишь в небольшом количе-
стве. Удовлетворительные результаты удалось получить при
471
действии на (1) 1,6 экв Т.н. в водном 0,5 М растворе хлорной
T1(NO3)3
" —------
(2)
кислоты в присутствии 4,0 экв перхлората натрия при 25—28°
в течение 0,5 час.
Хал коныбензилы [2]. Окисление халконов под действием
Т.н. в среде водный раствор кислоты — моноглим приводит к
образованию бензилов с выходом около 50%. Эта реакция вклю-
ArCH=CHCOArz —> АгСОСОАг'
чает несколько стадий, и на трех из них происходит окисление:
это окислительная перегруппировка с образованием кетоальде-
гида (2), расщепляющегося затем до дезоксибензоина (3),
окисление последнего под действием Т.н. до бензоина (4) н,
наконец, окисление (4) до бензила (5).
СНО
I н+
АгСН=СНСОАг' —> АгСНСОАг' —► АгСН2СОАг' —►
(D (2) (3)
ОН
I
—► АгСНСОАг' —> АгСО—СОАг'
(4) (5)
Эфиры алкин-2-овых кислот [4]. Пиразолоны-5, получаемые
с количественным выходом реакцией гидразина с р-кетоэфира-
ми [5], под действием 2 экв Т. н. превращаются в эфиры алкин-
2-овых кислот. Выходы в этой реакции высокие, а условия чре-
звычайно мягкие.
‘.^(МО3)Эч
CH3OH J
-TIONOz RC^CCOOCI-l,
472
Окисление ацетиленов [6]. Диарилацетилены при действии
Т.п. в диметоксиэтане, содержащем водный раствор кислоты
(хлорная кислота), или в метаноле превращаются в бензилы
с выходом 45—95%:
н3о+
ArC^CAr'+ 2T1(NO3)3 --> Аг С— CAr'
А А
Диалкилацетилены в водной среде дают ацилоины, а в метано-
ле — а-метоксикетоны:
RC==CR' + Tl(NOa)3
ОН О О ОН
H2O I j , II 1 ,
--> RCH—CR + RC—CHR
ОСН3О О ОСН3
СНзОн II! j I
RC=CR' +TI(NO3)3 ----> RCH—CR'+ RC—CHR
Терминальные ацетилены подвергаются деструкции в карбоно-
вые кислоты, содержащие на один атом углерода меньше;
Н3О+
RC^CH + 2T1(NO3)3 - > RCOOH
□D-Л)
Алкиларилацетилены в метаноле претерпевают окислительную
перегруппировку с образованием метиловых эфиров а-алкил-
арилуксусных кислот с выходом 80—98%;
R
снэон |
ArC=CR + T1(NO3)3 яп йя1/> АгСНСООСНз
60—Уо
Окисление циклогексанона [7]. Циклогексанон быстро окис-
ляется Т.н. в уксусной кислоте при комнатной температуре в
адипоин (5) с выходом 84%. Если проводить окисление сначала
при комнатной температуре, затем Т.н. отфильтровать, а фильт-
рат нагревать в течение нескольких минут при ~40°, то полу-
чается циклопентанкарбоновая кислота (4) с выходом 84%.
Мак-Кщллоп и сотр. [1] полагают, что оба продукта имеют об-
473
щего предшественника — эпоксиенол (3).
Карбаматы [8]. Т. н. реагирует с изонитрилами в метаноль-
ном растворе при комнатной температуре в течение нескольких
минут с высоким выходом (35—97%), образуя карбаматы;
1) сн3он
2) н2о
RNC + TI(NO3)3 ----> RNHCOOCH3 + T1NO3 + 2HNO3
Можно использовать нитрат ртути(II), ио в этом случае вы-
ходы карбаматов много ниже, Одиако реакцию с нитратом рту-
ти в отлнчие от Т. и. можно проводить и в других спиртах, а не
только в метаноле. Т.н., по-видимому, стабилен только в мета-
нольном растворе.
Дезоксимирование [9]. При действии Т. н. в метаноле при
комнатной температуре оксимы в течение нескольких минут
превращаются в альдегиды и кетоны с выходом 70—95%. Оса-
док нитрата таллия (I) удаляют, фильтрат встряхивают с разб.
серной кислотой, а образовавшийся альдегид или кетон экстра-
гируют эфиром или хлороформом. Схема реакции приведена
474
ниже:
R1\
C=NOH
tl(noj5>
R[ HpCH3
XC=N CoNO,
R/ О — T1
XONO2
-tiono2
-HONO?
Rt ^OCHj
4c
r/ n=o
Rt
*4'C=O
RaZ
В случае ароматических альдегидов или кетонов, содержащих
в орто- или на^а-положениях свободные окси- или аминогруп-
пы, этот метод неприменим из-за параллельно происходящего
окисления ароматического ядра до хииоиа. Однако, если ОН-
или №Н2-группы защитить ацетилированием, реакция проходит
успешно.
Семикарбазоны и фенилгидразоны карбонильных соединений
реагируют аналогично, ио в этом случае реакция требует для
своего завершения от 1 до 5 мин.
Ацетофеноны —> метиловые эфиры арилуксусных кислот,
АгСОСНз —► АгСН2СООСН3 [10]. Обработка ацетофенонов Т.н.
в метаноле, содержащем следы хлорной кислоты, при комнат-
ной температуре в течение 2—18 час приводит к образованию
метиловых эфиров арилуксусных кислот с выходом 60—95%.
Механизм, предложенный для этого превращения, показан на
схеме. Реакция равноценна реакции Вильгеродта — Канцле-
ра [11].
ArCOCHj
он
I
Аг-С=СНг
CH3OH
Н-О-С — СНг —T1-ONO
I
оснэ
----> О=С~СНгАг + TlONOj + HONOZ
ОСН3
Эфиры аллен карбоновых кислот [12]. а-Алкил-р-кетоэфи-
ры (1) (недавно опубликован метод их получения [13]) можно
в две стадии превратить в эфиры алленкарбоновых кислот (6).
Сначала, действуя на соединение (1) 1 же гидразина, полу-
чают 3,4-дизамещенный пиразолон-5 (2), который, не выделяя,
обрабатывают затем раствором 2 же Т.н. в метаноле. Общий
выход эфиров алленкарбоновых кислот колеблется в интервале
50—70%. Следует отметить, что R3 должен быть алкилом (а не
Н), в противном случае образуются эфиры а,р-ацетиленкарбо-
новых кислот (см. выше). Превращение пиразолонов-5 (2) в
эфиры алленкарбоновых кислот (6) мо?кно представить как
475
электрофильное присоединение таллия
к енаминной форме (2а)
о О
\ IL, 11
^CHCCHCOR
HZNNHZ
Tl(NOa)3
CH3OH>
Ri
C = C=C
'''COOCHj
(6)
пиразолона (2) с последующим отщеплением протона и образо-
ванием соединения (4). Окисление последнего в продукт (5) и
его метаиолиз приводят к эфиру алленкарбоновой кислоты (6).
O2NO. ЮЬ’О2
Ri б
: R.J _
СН3ОН
TI(NO3)j
------->
(4)
(6) 4- TIONOi
Окислительное расщепление гликолей. Мак-Киллоп и
сотр, [14] исследовали окислительное расщепление гликолей под
действием триацетата таллия (HI, 297—298; V, 405—407; VI,
247), трифторацетата таллия (III), (VI, 247-—250) и Т.н. Наибо-
лее эффективным реагентом оказался Т. и. в уксусной кислоте,
ОГ1 ОП жт ,
। Т1^°зЬ/НОАс
С — С. ------------>
С6Н5 \6Н; 61%
н
2 Jjc=O
с6н5
(1)
(2)
„ он
С6Н5 I
yz — c
ОН
-----> 2
91%
(3)
С6Щ
С6Н5
(4)
однако он расщепляет только гликоли, содержащие вициналь-
ные ароматические заместителя, такие, как (1) и тетраарилза-
476
мешенные гликоли (3). Последние, например (3), гладко рас-
щепляются также этилатом таллия (I) (V, 558—564) в этаноле.
1 М с К i I о р A., Hunt J. D., Taylor Е. С., Kienzie F., Tetrahedron
Letters, 5275 (1970).
2. McKiliop A., Swann В. P., Taylor E. C., Tetrahedron Letters, 5281
(1970).
3. Corey E. J., Ravindrathan T., Tetrahedron Letters, 4753 (1971).
4 Taylor E. C., Robey R, L., McKillop A., Angew. Chem., Internat.
Ed., 11, 48 (1972).
5. W i 1 e у R. H., w i 1 e у P., in «The Chemistry of Heterocyclic Compounds»,
Weissberger A., Ed., Interscience, New York, Vol. 20 (1964).
6. M с К i 11 о p А., О 1 d e n z i e 1 О. H., Swann В. P., Taylor E. C., R o-
bey R. L., J. Am. Chem., Soc., 95, 1296 (1973).
7. McKillop A., Hunt J. D., Taylor E. C., J. Org. Chem., 37, 3381
(1972).
8. Kienzie F., Tetrahedron Letters, 1771 (1972).
9. M с К i 11 о p A., Hunt J. D., Naylor R. D., Taylor E. C., J. Am.
Chem. Soc., 93, 4918 (1971).
10. McKillop A., Swann В. P., Taylor E. C., J. Am. Chem. Soc., 93,
4919 (1971).
il. Кармак M., Шпильман M., «Органические реакции», ИЛ, M., 1951,
сб. 3, стр. 88; AsingerF., Schafer W., Halcour К.., Angew. Chem.,
Internat. Ed., 3, 19 (1964); Wegler R., I< u h 1 e E., Schaler W., Newer
Methods Prep. Org. Chem., 3, I (1964).
12. Taylor E. C., Robey R. L., McKillop A., J. Org. Chem., 37, 2797
(1972). .
13. Taylor E. C., Hawks G. H„ ill, McKillop A., J. Am. Chem. Soc., 90,
2421 (1968).
14. McKillop A., Raphael R. A., Taylor E. C., J. Org. Chem., 37, 4204
(1972).
ТАЛЛИЯ(Ш) СУЛЬФАТ, T12(SO4)3. Мол. вес 696,94.
Окисление циклогексенов [1]. При окислении 3-трет-бутил-
циклогексена (1) под действием Т. с. образуются 73% транс-
диаксиального диола (2), 10% тра«с-диэкваториального диола
(3) и 18% неидентифицированного продукта, а при окислении
4-трет-бутилциклогексена — исключительно т/шнс-диаксиальный
1,2-диол. Тот факт, что при этом окислении образуются только
транс-диолы, исключает возможность замещения таллия по ме-
ханизму Sn2 и наводит на мысль об участии а-оксигруппы в
разрыве связи С—Т1 промежуточного таллийорганического сое-
динения.
1. F г е р р е 1 С., F a v i е г R., Richer J.-C., Z a d о г М., Can. J. Chem., 49,
2586 (1971).
477
ТАЛЛИЯ ТРИАЦЕТАТ (111, 297—298; V, 406—407; VI, 247).
Ароматические бромиды (VI, 247). Опубликована заключи-
тельная статья по электрофильному бромированию ароматиче-
ских соединений действием брома в присутствии Т. т. [1]. Наи-
более характерные особенности этой реакции сводятся к тому,
что, во-первых, почти во всех случаях наблюдается моноброми-
рование, а во-вторых, почти для всех монозамещенных бензо-
лов наблюдается замещение исключительно в пара-положение,
хотя электроноакцепторные группировки затрудняют бромиро-
вание. Из этого можно заключить, что бром в сочетании с Т. т.
(1) Т1(ООССНз)з + ЗАгН + ЗВг2 —> ЗАгВг + ЗСН3СООН + Т1Вг3
является слабым электрофилом, но с довольно высокими сте-
рическими требованиями. Стехиометрия реакции представлена
уравнением 1. Судя по экспериментальным результатам, в этих
условиях имеет место бромирование молекулярным бромом, ка-
тализируемое бромидом таллия (III) или Т. т.
а-Ацилоксикарбоновые кислоты [2]. Реакция Т. т. с алифа-
тическими карбоновыми кислотами приводит к образованию
а-ацилоксикарбоновых кислот, которые легко гидролизуются до
а-оксикислот:
Т1(ОСОСНЭ)3 + 3R’R2CHCOOH .. п -> Т1(ОСОСВД^2)3 —>
“ЗиНзСииМ
к-^ссоо-тГ
OCOCHiW
1. М с К i 11 о р A., Bromley D., Taylor Е. С., J. Org. Chem., 37, 88
(1972).
2. Taylor Е. С., Al Hand Н. W„ McGillivray G., McKillop А.,
Tetrahedron Letters, 5285 (1970).
ТАЛЛИЯ(Ш) ТРИФТОРАЦЕТАТ (ТТФА) (VI, 247—250).
Ароматические иодиды (VI, 247—248). Опубликована заклю-
чительная статья по синтезу ароматических иодидов реакцией
бпс-трифторацетатов арилталлия с иодистым калием [1]. Раз-
работаны четыре методики этого синтеза. 1) Таллирование осу-
ществляют как обычно, а затем к полученному таллийоргани-
ческому соединению, не выделяя его, приливают водный рас-
твор йодистого калия. 2) Выделяют промежуточный бас-три-
фторацетат арилталлия и затем обрабатывают его иодистым
калием. 3) Субстраты, чувствительные к кислотам, таллируют
твердым ТТФА в ацетонитриле. 4') Перечисленные методы, од-
нако, непригодны для реакционноспособных соединений, таких,
как нафталин и дифеинл. В этих случаях используют элемен-
тарный иод в качестве электрофильного агента, а ТТФА служит
478
окислителем образующегося в ходе реакции йодистого водо-
рода.
Установлен ряд относительных скоростей иодирования моно-
замещенных производных бензола [2]. В условиях термодинами-
ческого контроля (повышенная температура) наблюдается ме-
та-замещение. В условиях кинетического контроля (комнатная
температура) для соединений с орго-пара-ориентантами наблю-
дается значительное преобладание продуктов лара-замещения.
орто-Замещение преобладает в тех случаях, когда координация
молекулы ТТФА с заместителем приводит к внутримолекуляр-
ному переносу электрофила. Например, метилбензоат дает почти
исключительно орто-таллированмое производное (95%).
Окислительная конденсация фенолов. Шварц и сотр. [3]
нашли, что ТТФА является эффективным реагентом для окис-
лительной конденсации фенолов. Так, например, окисление про-
изводного диарилпропана (1) 1 мол. же ТТФА в хлористом
метилене приводит к образованию диенона (2) с выходом 87%.
Этот метод, правда с существенно меньшим выходом, позд-
нее был использован для построения циклической системы
алкалоидов растений семейства амариллисовых. Так, например,
окисление производного норбелладина (3) с помощью ТТФА
дает дненон (4) только с выходом 19%. При гидролизе диено-
на (4) NasCO3 в водном метаноле образуется (±)-оксокринин
479
(выход 95%).
Новую методику с успехом применяли и в синтезе предшест-
венника колхицина (±)-О-метиландроцимбина (8) из фенетил-
нзохинолина (6). Последний превращали в аминоборан (7), об-
рабатывая дибораном в ТГФ — СНС13 [4]. (Окисление свобод-
ного амина приводит к образованию множества продуктов.)
При окислении соединения (7) избытком ТТФА с последующим
удалением защитной группы образуется продукт (8) с общим
выходом 20%.
480
1,6-Диокса-6а-тиапентален (4) [4]. При сливании раствора
ТТФА в ацетонитриле с раствором 4Н-пирантиона-4 (1) тоже
в ацетонитриле при комнатной температуре образуется лабиль-
ная соль пирилия (2), которая при добавлении воды превра-
щается в результате замыкания двух новых связей сера — кис-
лород в 1,6-диокса-6а-тиапенталеи (4). Полагают, что промежу-
точно образуется соединение (3). Движущей силой реакции яв-
ляется энергетически выгодное превращение Т1 (Ш)->Т1(1).
U)
Получение n-хинонов (VI, 249—250). Мак-Киллоп и сотр.
(VI, 250, [5]) постулировали, что при окислении 2,6-днзамещен-
ных 4-трет-бутил фенолов в 2,6-дизамещенные д-беизохиноны
промежуточно образуются 2,6-дизамещенные 4-трет-бутил-4-
трифторацетоксициклогексадиен-2,5-оны-1. Недавно устойчивый
трифторацетат хинола такого типа (2) был выделен из реакции
эстрона (1) с 2 же ТТФА [5].
I. McKillop A., Hunt J. D., Zelesko М. J., Fowler J. S., Tay-
lor E. C„ McGillivray G., Rienzi e F.f J. Am. Chem. Soc., 93, 4841
(1971).
2. Taylor E. С., К i e n z 1 e F., Robey R. L„ McKillop A, Hunt J. D.,
J. Am. Chem. Soc., 93, 4845 (1971).
3. Schwartz M. A., Rose B. F., V i s h n u v a j j a 1 a B., J. Am. Chem. Soc.,
95, 612 (1973).
4. R e i d D. H., Webster R. G., J. C. S. Chem. Comm., 1972, 1283.
5. Coombs M. M., Jones M. B., Chem. Ind,, 1972, 169.
]6 Зак, 590 481
ТЕЛЛУР ЧЕТЫРЕХХЛОРИСТЫЙ, ТеС14. Мол. вес 269,44
Синтез б«с-2,2х-дифенилентеллура (1) [1]:
1. HellwinkelD., Fahrbach G., Ann., 712, 1 (1968),
ТЕТРА-(БРОММЕТИЛ)-МЕТАН, С(СН2Вг)4. Мол. вес
433,80, т. пл. 158—160°.
Получение [1]. Продажный препарат следует перекристал-*
лизовать из хлороформа.
Электролитическое восстановление [2].
С(СН2Вг)4
Электролитическое восстановление
47—58%
СН- >СН2Вг
I + 2Вг"
СН/ \СН2Вг
1. Herzog Н. L., Org. Syn. Coll. Vol., 4, 763 (1963).
2. Rif i M. R., Org. Syn., 52, 22 (1972).
О
2,4,4,6-ТЕТРАБРОМЦИКЛОГЕКСАДИЕН-2,5-ОН,
Мол. вес 409,44, т. пл. 125—30° (с разл.). Бг Бг
Т. получают [1,2] с выходом 80—90% бромированием три-
бромфенола в уксусной кислоте в присутствии ацетата натрия.
Реагент используют для селективного монобромирования
фенолов [3] и ароматических аминов [2] преимущественно в па-
482
pa-положение. В качестве растворителей применяют хлористый
метилен и хлороформ. Выход обычно выше 90%.
Методики получения Т. и превращения с его помощью
NjN-диметнл-З-трифтор метил анилина в 4-бром-Ь1,Ь1-диметил-3-
трифторметиланилин (выход 82—90%) [4] рекомендованы для
сборника Organic Syntheses.
Т. легко взаимодействует с имидазолом (1), образуя смесь
изомерных бромимидазолов с преобладанием 4-бромимидазола
Й
(2) [5]. N-Метилимидазол бронируется до 5-бром-1-метилимида-
зола с выходом 66%. Бромирование индола приводит к 3-бро-
миндолу с выходом 88%.
1. Benedikt R, Ann., 199, 127 (1879).
2. Calo V-, Ciminale F, Lopez L., To de sc о P. E., J. Chem. Soc. (C),
1971, 3652.
3. Calo V, Ciminale F., Lopez L., Pesce G., Todesco P. E., Chim.
Ind, 53, 467 (1971).
4. Fox G. J, Hall as G, Hep worth J. D, Pa skins K- N, procedure
submitted to Org. Syn, 1972.
5. С a 1 6 V, Ciminale F, Lopez L, Naso F, Todesco P. E, J. C. S.
Perkin I, 1972, 2567.
ТЕТРА-н-БУТИЛАММОНИЯ БРОМИД,
[СНз(СН2)з]4И+Вг“. Мол. вес 322,38, т. пл. 100—102°.
Дегидробромирование. Ллойд и Паркер [1] рекомендуют
проводить дегидробромирование с помощью Т. б. в ацетоне в
присутствии 2,6-лутидина. В этих условиях образуется в основ-
ном олефин с ориентацией отщепления по Зайцеву, содержащий
транс-олефина больше, чем соответствует термодинамическим
соотношениям.
В описанных условиях была осуществлена последняя ста-
дия синтеза 3,4-бензоциклобута-[1,2-Ь]-циклогептатриена (2) [2].
Выход, однако, составил всего 20%.
i) N6°-
2) Вг*,
20^
1. Lloyd D. J, Parker A. J, Tetrahedron Letters, 637 (1971).
2. L о m b а г d о L, W е g е D, Tetrahedron Letters, 4859 (1972).
483
о
ТЕТРА-н-БУТИЛАММОНИЯ ФОРМИАТ, (C4Hg)4N(6CH).
Мол. вес 119,16.
Реагент получают нейтрализацией водного раствора гидро-
окиси тетра-к-бутиламмония муравьиной кислотой и выделяют
упариванием полученного раствора досуха.
Эпимеризация гидроксильных групп. Эпимеризацию гидро-
ксильных групп часто осуществляют реакцией 5г<2-замещения в
тозилатах спиртов под действием ацетата тетраэтиламмония (III,
318—319; V, 416) или ацетата тетра-«-бутиламмония (VI, 250).
В связи с изучением зависимости между конфигурацией и био-
логической активностью простагландинов Кори и Терашима[1]
исследовали реакцию тозилата модельного (±)-оксилактона (1)
с 5,0 экв ацетата тетр а-к-бутил аммония в ацетоне при 25° в те-
чение 2 час. Продукт реакции, полученный с выходом 95%,
^У^СНгОСНгС6Н3
TsO
a) R » СН3СО
6} R = НСО
СНгОСН2С6Н5
представлял собой смесь ожидаемого соединения (2а) и обра-
зовавшегося в результате элиминирования циклопентена (3) в
отношении 1,2:1. В отличие от этого реакция тозилата (1) с
Т. ф. (см. также Тетраэтиламмония формиат, III, 319—320) в
тех же условиях дает формиат эпимерного спирта (26) и цик-
лопентен (3) в отношении 3:1. Формиат-ион по сравнению с
ацетат-ионом проявляет большую склонность к замещению у
углеродного атома, чем к элиминированию, что можно объяс-
нить меньшим размером, а также меньшей основностью пер-
вого.
При взаимодействии соединения (1) с оксалатом тетра-н-
бутиламмония (средняя соль) образуется единственный про-
дукт— ненасыщенный лактон (3), выделенный с выходом 82%.
Таким образом оксалат-ион оказался превосходным нуклеофи-
лом для проведения элиминирования в очень мягких условиях.
Кори и Терашима [2] использовали реакцию Б^-замещения
с помощью Т. ф. на одной из стадий синтеза е/гМ 1,15-эпи-РОЕз
(6). Так, например, при взаимодействии соединения (4) с
6,7 же Т. ф. при 25° в ацетоне в течение 16 час образуется ин-
вертированный формиат (5), который превращали в соедине-
484
ние (6) последовательностью реакций, использовавшейся ранее.
В ряду простагландинов в отличие от большинства других
природных соединений оптические антиподы природных форм
по своей биологической активности близки к соединениям,
встречающимся в природе.
1. Core у Е. J., Тега shim a S., Tetrahedron Letters, 111 (1972).
2. Corey E. J., T e r a s h i m a S.t J. Org. Chem., 37, 3043 (1972).
ТЕТРА-я-БУТИЛАММОНИЯ ФТОРИД, (CH3CH2CH2CH2)4NF‘.
Мол. вес 261,46.
Получение [1]. Реагент можно получить нейтрализацией
10%-ного водного раствора гидроокиси тетра-и-бутиламмония
48 %-ной фтористоводородной кислотой. Полученный раствор
концентрируют при пониженном давлении, остатки воды уда-
ляют азеотропной отгонкой со смесью бензол — ацетонитрил
(1:1) при пониженном давлении, и, наконец, препарат высу-
шивают при 30° и 0,5 мм в течение 20 час. Относительно друго-
го способа получения см, [2].
Расщепление силиловых эфиров [1]. Т. ф. является мощным
агентом расщепления силиловых эфиров [3], в частности он при-
годен для расщепления недавно синтезированных ди мети л-трат-
бутилсилиловых эфиров (см. Диметил-трет-бутилхл орсил ан,
этот том). При обработке 2—3 экв Т. ф. в ТГФ при 25° эти эфи-
ры быстро расщепляются до спиртов. Однако фторид-ион в ТГФ
является довольно сильным основанием, поэтому Т. ф. нельзя
использовать в случае субстратов, содержащих достаточно чув-
ствительные к основаниям группы.
1. Corey Е. J., V е n k a t е s w а г 1 u A.. J. Am. Chein. Soc., 94, 6190 (1972).
2. Fowler D. L„ L о e b e n s t e i n W. V., Pall D. B., Kraus C. A., J. Am.
Chem. Soc., 62, 1140 (1940).
3. Corey E. J., Snider В. B., J. Am. Chem. Soc., 94, 2549 (1972).
485
ТЕТРАМЕТИЛ ГУАНИДИН (111,304).
Присоединение нитрометана по Михаэлю. Итальянским хи-
микам удалось осуществить присоединение по Михаэлю нитро-
метана к эфирам <х,|3-ненасыщенных карбоновых кислот, ката-
лизируемое Т. Если R1 или R2 —алкильные или арильные груп-
пы, выходы аддуктов состава 1:1 (1) колеблются в интервале
от 50 до 85%. Если R1 и R2 — Н, то в значительных количе-
ствах образуются также аддукты состава 1:2 (2).
к1 R3
^с=сг +
R2^ ЧСООСН3(СгН5)
R1 R’
----> R2—C-CHCOOCHjfC^Hj +
CH2NO2
H3CNOZ
HN=C
XN(CH3)Z
Rs
I
СНСООСН3(СгН5)
GHNO,
I
R2— C - C H C OOC H3 (C2 H5)
(1)
<2>
Присоединение нитрометана к кетону (3) по Айихаэлю, ка-
тализируемое Т., было использовано для получения нитрокето-
на (4), выход 84% [2]. Эта реакция является первой стадией в
синтезе (±)-11-дезоксипростагландинов Ец FI(Z и Fip.
о
(снг)6соосн3 HaCNO^
84<6 *
(3)
О
JL (снг)6соосн3
CHZNOZ
(4)
1. Р о 11 i л i G. Р., В а г с о A., D е G u i I i G., Synthesis, 1972, 44.
2. Alvarez F. S., Wren D., Tetrahedron Letters, 569 (1973).
^^Ж-ТЕТРАМЕТИЛДИАМИДОХЛОРФОСФАТ,
[(CH3)2N]2POC1. Мол. вес 170,60, т. кип. 98°/15 мм.
Получение [1]. Реагент получают взаимодействием диметил-
амина с хлорокисью фосфора.
Защита или восстановительное дезоксигенирование спиртов
и кетонов. Айрланд и сотр. [2] установили, что N,N,N',N'-TeTpa-
метилдиамидоалкнлфосфаты или -алкенилфосфаты (ТМДАФ-
производные спиртов или енолов) с высоким выходом восста-
навливаются литием в этиламине до углеводородов. ТМДАФ-
Производные легко получить фосфорилированием алкоголят-
или енолят-анионов. Алкоголят-анионы получают простой обра-
боткой спирта небольшим молярным избытком к-бутиллития, а
енолят-анноны насыщенных кетоиов — обработкой соответст-
вующих кетонов диизопропиламидом лития. Для получения тех
486
же енолят-анионов из «.^-ненасыщенных кетонов применяют
восстановление литием в аммиаке или сопряженное присоеди-
нение металлоорганического соединения. Фосфорилируют анио-
ны пятикратным избытком Т. в среде (4: 1) диметоксиэтана
(или ТГФ) и П.П.ЬГ.ЬГ-тетраметилэтилендиамина (1—2 час пе-
ремешивание, 25°); эфиры образуются с высокими выходами.
Для этой цели также можно использовать диэтилхлорфосфат
(I, 435—436; VI, 119—120), который в случае третичных спир-
тов даже более предпочтителен.
Восстановление проводят, приливая раствор ТМДАФ-произ-
водного в ТГФ и 2—4 экв трет-бутанола при охлаждении льдом
в атмосфере аргона к раствору 10 экв лития в сухом этил амине.
Очистка веществ на промежуточных стадиях необязательна.
Примеры:
I) я-BuLi, дмэ
2) [(CH3)2nJ2PQC1
92$
Li, C2HSNH2
ТГФ(трет-ВиОн
97$
I) LiAlH,,. C2H5OC2HS
2) н-BuLi, ДМЭ , N(C2Hs)3,
clpoLn(ch3)2]2
3) Li, C2HsNH2; H3O+
ТМДАФ-Группировка применяется и для защиты гидро-
ксильной группы; соответствующие производные спиртов устой-
чивы к действию CH3Li, LiAlH4, Ш КОН—С2Н5ОН и 0,2А4 вод-
ной НС1 — ацетон. Спиртовую группу можно регенерировать
обработкой 5 экв н-бутиллития в Ы,М,М/,М/-тетраметилэтилен-
диамине в течение 30 мин. при 25°.
1. Crunden Е. W., Hudson R. F., J. Chem. Soc., 1962, 3591; Cook H. G.,
I let t J. D., Saunders В. C., Stacey G. J., Watson H. G., W i 1 d-
Ing I. G. E., Woodcock S. J., 3. Chem. Soc., 1949, 2921.
2. I r e 1 a n d R. E., Muchmore D. C., Hen gar tn er U,, J. Am, Chem.
Soc., 94, 5098 (1972).
487
2,4,4,6-ТЕТРАМЕТИЛ-5,6-ДИГИДРО-1,3-(4Н)-ОКСАЗИН
(VI, 251—256).
Синтез альдегидов (VI, 252—254). Недавно подробно описано
получение альдегидов с помощью Т. [1 ]. Опубликован метод
синтеза 1-фенилциклопентанкарбоксальдегида (4) из 2-бензил-
4,4,6-триметил-5,6-дигидро-1,3- (4Н) -оксазина (2).
1) к-С4Н,Ы
2) Вт(СНЩВг
NaBH.
(2) (3) (4)
В новом варианте метода двухуглеродной гомологизации
алкилгалогенидов, приводящей к образованию альдегидов [3],
Т. сначала превращают в устойчивый кристаллический иодме-
тилат (5) действием 4,0 экв йодистого метила в темноте в тече-
ние 20 час. Соль (5) обрабатывают гидридом натрия в сухом
ДМФА при комнатной температуре. Затем добавляют алкилга-
логенид и перемешивают реакционную смесь при 50—55° до
прекращения выделения водорода. Затем смесь охлаждают и
(5)
восстанавливают боргидридом натрия в абсолютном этаноле.
На последней стадии проводят гидролиз водным раствором
щавелевой кислоты. Выходы альдегидов (6) колеблются в ин-
тервале 50—60%.
488
Синтез кетонов. Дигидро-1,3-оксазиновая система сама по
себе инертна к действию реактивов Гриньяра. Мейерс и Смит
[4] недавно сообщили, что при обработке оксазинов (1) избыт-
ком йодистого метила образуются устойчивые четвертичные
N-иодметилаты (2). Последние при комнатной температуре
взаимодействуют с реактивами Гриньяра (2—2,5 экв), образуя
аддукты (3), дающие при гидролизе водным раствором щаве-
левой кислоты кетоны (4) и аминоспирты (5). На самом деле
(2) (з)
нет необходимости получать иодметилаты отдельной операцией,
их можно получить в той же реакционной колбе, в которую при-
бавляют затем реактив Гриньяра. Выходы кетонов по этой ме-
тодике колеблются в зависимости от природы R и используе-
мого реактива Гриньяра от низких (12—20%) до хороших (50—
85%).
Только при R = СН3 выходы очень низкие, так как реактив
Гриньяра отрывает протон от метильной группы с образова-
нием неустойчивого Г4,О-ацеталя кетена (6). Этот ацеталь луч-
ше получать in situ, обрабатывая оксазин (1) 2 экв гидрида
(6)
натрия в ДМФА. Его используют в синтезе альдегидов (см.
выше).
489
Синтез метилового эфира жасминовой кислоты (8). Мейерс
и Назаренко [5] описали синтез метилового эфира жасмино-
вой кислоты, одного из компонентов, обусловливающих харак-
терный запах жасмина, исходя из оксазина (1). Оксазин (1)
металлируют, как обычно, и затем обрабатывают 1,0 экв 2-иод-
метил- 1,3-диоксолана, получая алкилированный оксазин (2) с
выходом 83°/о- Последний превращают в иодметилат (который
можно не выделять) и обрабатывают его реактивом Гриньяра,
полученным из ^мс-1-бромгексена-З в ТГФ, При нагревании про-
дукта (3) с водным раствором щавелевой кислоты удаляются
обе защитные группы и с хорошим выходом образуется кето-
альдегид (4), нагревание которого с 1%-ным раствором гидро-
окиси натрия в течение 30 мин, приводит к образованию цикло-
пентенона (5). Последний затем обрабатывают N.O-ацеталем
кетена (6) и разлагают реакционную смесь водой. Переэтери-
фикация образовавшегося кетоэфира (7) метанолом (катализа-
тор — п-толуолсульфокислота) дает метиловый эфир (±)-жас-
миновой кислоты.
Этот синтез интересен с двух точек зрения. Он дает в руки
синтетикам, во-первых, метод синтеза 2-алкилциклопентенонов,
а во-вторых, метод введения фрагмента —СН^СООСНз в моле-
490
кулу электрофильного олефина. Так, например, реакция цикЛо-
гексен-2-она (9) с Ы,О-ацеталем кетена (6) и последующая об-
работка по описанной выше методике приводят к кетоэфиру
(10) с выходом 55%.
1. Meyers A, I., Nabeya A., Adi ekes Н. W., Politzer I. R., Malo-
ne G. R,, Kovelesky A. C., Nolen R. L., Portnoy R., J, Org. Chem.,
38, 36 (1973).
2. Politzer I. R., M e у e г з A. I., Org. Syn., 51, 24 (1971).
3. Meyers A. I., Nazarenko N., J. Am. Chem. Soc., 94, 3243 (1972).
4. M e у e r s A. I., Smith E. M., J. Org. Chem., 37, 4289 (1972).
5. Meyers A. L, Nazarenko N., J. Org. Chem., 38, 175 (1973).
МЛ^М'-ТЕТРАМЕТИЛЭТИЛЕНДИАМИН (ТМЭДА) (V,
411; VI, 256).
Дегидрирование. В синтезе полициклических ароматических
углеводородов последней стадией часто является дегидрирова-
ние. Его можно с успехом осуществить действием комплекса
«-бутиллития с ТМЭДА [1]. Так, например, при кипячении цис-
9-этил- 10-метил-9,10-дигидроантрацена (1) с этим комплексом
в циклогексане генерируется дианион (а), обладающий интен-
сивной пурпурной окраской. При добавлении хлорида кад-
мия (II) окраска исчезает и образуется-9-этил-10-метилантрацен
(2) с выходом 99%.
9,10-Дигидрофенантрен (3) дегидрируется без добавления
соли металла. В этом случае предполагается образование про-
491
межуточного комплекса (б):
Активирование переметаллирования (V, 411). Металлирова-
ние некоторых вторичных бензиламинов проходит гораздо пол-
нее при использовании н-бутиллития, активированного ТМЭДА,
чем при действии одного н-бутиллития. Так, например, N-метил-
бензиламин под действием комплекса н-бутиллитий — ТМЭДА
металлируется в два положения: преимущественно по азоту, а
также в о-положение бензольного ядра [2].
Металлирование олефинов [3]. 1,1-Диметил-2,2-дифенилэти-
лен (1) металлируется комплексом /z-BuLi— ТМЭДА с образо-
ванием аниона (2), при обработке которого триметилхлорсила-
ном с хорошим выходом образуется соединение (3). Реакция
олефина (1) с н-BuLi в смеси гексан —ТГФ идет намного мед-
леннее.
НзС \-_г/СбН5 H- BuLi-ТМЭДА НзСХ /СбНг; (CHjjSiCl А'бнз
с—с, —--------------С — С Щ ±3 С= СД
НзС ед Н2С-'‘ - С6Н5 9 0^ / ед
• Li+ CHs5i(CH3)3
(Г (2) (3)
Реакция сс-циклопропилстирола (4) с комплексом н-ВпЫ —
ТМЭДА приводит к раскрытию циклопропанового кольца с об-
разованием литиевых, производных (5) и (6).
с=снг
ед
Н СН2~Н-Ви
+ - J?C=C’X’ +
Li CHjCHj ^"Сед
Li CHjCH2 Сн2—H- Bu
с=с^
Нх С6Н5
(4)
Н Снг-Я-Ви
с=с:Г +
сн,сн2^ хсед
СИ 3СНг СН2 - к- Ви
С=С^
сед
1, 0:1,I
17) (8)
492
Дифеиилметиленциклобутан (9) одинаково хорошо металли-
руется как н-BuLi в среде гексан — ТГФ, так и комплексом
и-BuLi — ТМЭДА. В этом случае удалось выделить кристалли-
ческое литиевое производное (10).
Н- Bu Li
------->
с(с4н5)г
(11)
(9) (Ю)
Металлирование лимонена. Крауфорд и сотр. [4] показали,
что лимонен (1) под действием комплекса к-бутиллития с
ТМЭДА состава 1 : 1 избирательно металлируется в положение
Сю с образованием 2-замещенного аллиллитиевого производно-
го, изображенного формулой (2). Металлирование проводят,
выдерживая смесь 2 экв лимонена с 1 экв комплекса (прибли-
зительно 1,5М раствор в н-гексане) в течение ^12 час при
комнатной температуре. Растворы соединения (2) при хранении
в атмосфере азота или аргона устойчивы в течение нескольких
дней.
Для металлирования можно использовать как ( + )-(R)-ли-
монен (3), так и (—)-(5)-лимонен (4).
Эту новую реакцию избирательного металлирования приме-
няли для получения 10-замещениых производных лимонена. Из-
493
быток лимонена легко отделяется от продукта реакции фрак-
ционированной перегонкой. Например, металлирование (.ф-)-ли-
монена (3) с последующей карбонизацией, а затем этерифика-
цией приводит к эфиру р,у-ненасыщенной кислоты (5) с выхо-
дом 19%. Под действием метилата натрия в метаноле эфир (5)
изомеризуется в эфир а,р-ненасыщенной кис-лоты (6).
1) Я-ВиЫ-ТМЭД'А
2) СО,
3) Н3О+
( , 4) СН3ОН, HZSO4
— ... 2
(5) (6)
Избирательное металлирование было использовано в синтезе
некоторых сесквитерпенов группы бисаболана. Так, сесквитер-
пен (—)-р-бисаболен (7) можно синтезировать в одну стадию
из металлированного (—)-лимонена реакцией с 1-бром-З-метил-
бутеном-2. При этом образуются два продукта (7) и (8) в от-
(8)
ношении 4: 1, которые можно разделить перегонкой на колонке
с вращающейся лентой или препаративной ГЖХ.
Крауфорд [5] описал синтез моноциклического дитерпена
аг-артемизена (15). В этом синтезе селективное металлирова-
ние используется на двух из четырех стадий. Так, (4-)-лимонен
(3) превращают в металлированное производное (9), из кото-
рого реакцией с параформом получают затем спирт (10). Спирт
кипятят с N-литийэтилендиамином в этилендиамине (П, 159—
163), превращая в ароматический спирт (И), из которого в
свою очередь обработкой трехбромистым фосфором получают
бромид (12). Параллельно гераниолен (13) избирательно ме-
таллируют комплексом н-бутиллитий— ТМЭДА. Прибавлением
бромида (12) к полученному раствору металлированного произ-
494
водного гераниолена (14) получают (±)-аг-артемизен (15).
(15)
Методику Крауфорда и сотр. использовали для проведения
аллильного алкилирования в синтезе сесквитерпена (—)-крип-
томериона (19) из (—)-карвона [6]. Так, действием комплекса
H-BuLi —ТМЭДА с последующим добавлением 1-хлор-З-метил-
бутена-2 было осуществлено аллильное алкилирование ацеталя
(4-)-дигидрокарвона (16). Дигидрокрцптомерион (17) был по-
495
лучен с общим выходом 70%. Бромирование этого соединения
пербромидом фенилтриметиламмония (IV, 56—57; V, 500) с по-
следующим дегидробромированием приводит к образованию
j) H-Bulji, ТМ'ЭДА
2) Н3С-С=СНСНгС1
СН,
3) н3о+___________
70%
(18) (19)
с хорошим выходом (—)-криптомериона (19).
1. Н а г v е у R. G,, Naz аг ело L., С h о Н„ J. Am. Chem. Soc., 95, 2376
(1973).
2. L u d t R. Е., Н a u s е г С. R., J. Org. Chem., 36, 1607 (1971).
3. DunkelbiurnE., Brenner S., Tetrahedron Letters, 669 (1973).
4. Crawford R. J., Erman W. F., Broaddus C. D., J. Am. Chem. Soc.,
94, 4298 (1972).
5. Crawford R. J., J. Org. Chem., 37, 3543 (1972).
6. Hodgson G. L., M ac S weeney D. F., Money T., J. C. S. Chem. Comm.,
1973, 236.
ТЕТРАФЕНИЛ ЦИКЛОПЕНТАДИЕНОН (тетрациклон) (I,
291—299, 390—391; П, 18—19, 98, 308; III, 310—311; VI, 99,
105—106, 146, 224). Мол. вес 384,45, т. пл. 219°.
Получение [1].
С6Н5С=О
I
С6Н3С =О
е-йН’5
снгх
С=О
chZ
ад
+
С6Н5СНгМ(СНз)зОН
-2 НгО
Конденсация с 1,2-дегидроциклооктатетраеном (5) [2].
496
Конденсация с дифенилацетиленом [3].
C6HS
с
III
с
I
ОД
С^СООД^
' 84^ '
Г ен сафенил бензол
+ СО
т.пл 454-456*
Использование для улавливания дегидротиофена [4].
1. F i е s е г L, F., Org. Syn., 46, 44 (1966).
2. К г е b s А., В у г d D., Ann., 707, 66 (1967).
3. Fieser L. F., Org. Exps., 2nd Ed., Heath D. C. and Co., Boston, 1968,
299; Org. Syn., 46, 44 (1966).
4. Wittig G., Rings M., Ann., 719, 127 (1968).
ТЕТРАХЛОРСИЛАН, (VI, 256—257), SiCl4. Мол. вес 169,92.
Амиды [ 1]. T. — эффективный конденсирующий агент для
получения амидов из карбоновых кислот и аминов:
О О
II R'NH2 ||
SiCI4 4- RCO2H —> Si(OCR)4 ----R'NHCR
Пептидный синтез [2]. Взаимодействием защищенной по
азоту аминокислоты с SiCl4 в пиридине и последующим добав-
лением к образовавшемуся хлорангидриду эфира аминокислоты
со свободной аминогруппой можно получить дипептиды с уме-
ренными выходами. Однако реакция сопровождается значитель-
но?
ной рацемизацией, а защитная карбобензоксигруппа в этих
условиях неустойчива.
1. С h а n Т. Н., Wo n g L. Т. L., J. Org. Chem., 34, 2766 (1969).
2. Chan Т. Н.. Wong L. Т. L„ J. Org. Chem., 36, 850 (1971).
ТЕТРАХЛОРЭТАН — НИТРОБЕНЗОЛ.
Ацилирование янтарным ангидридом [1]. Ацилирование ани-
зола янтарным ангидридом с целью получения 3- (я-метоксибен-
зоил) -пропионовой кислоты удобнее всего проводить в смеси
тетрахлорэтана с нитробензолом (4 : 1):
А1С13^
82%
1. Rao Y. S., Кг etch тег R. A., Org. Prep. Proc. Int., 3, 177 (1971).
s
I
ТИОАЦЕТАМИД, CH3C—NH2. Мол. вес 75,13, т. пл.
111-114°.
Тэйлор и Золтевич [1] разработали общий метод синтеза тио-
амидов кислот из соответствующих нитрилов с использованием
в качестве источника сероводорода Т. в кислой среде. Этот ме-
тод обеспечивает хорошие выходы тиоамидов высокой степени
чистоты. В реакцию вступают нитрилы алифатических кислот
и ароматических, содержащих в ядре электронодонорные и
электроноакцепторные заместители, а также группировки, по-
тенциально способные к восстановлению. Нагревают на паровой
бане в течение 15—30 мин 1 экв нитрила с 2 же Т. в ДМФА,
S S
I! нс| |j
RCN+CH3C—NH2 ';> RC—NH2 4- CH3CN
насыщенном сухим хлористым водородом. Реакционную смесь
упаривают в вакууме водоструйного насоса до четверти перво-
начального объема и избыток кислоты нейтрализуют водным
раствором бикарбоната натрия. По охлаждении профильтро-
ванного горячего раствора выпадает тиоамид. Выходы сведены
в таблицу:
Нитрил Тиоамид Выход, % T. пл., °C
ft-O2NC6H4CN n-O2NCGH4CSNH2 83 158,5— 159,5
ft-C[-I3OCGH<CN n-CH3OC6H4CSNH2 87 148,5—149,5
CH2(CN)2 CH2(CSNH2)2 63 211-212
NC(CH2)4CN H2NCS(CH2)4CSNH2 78 178.5-179,5
49?
Полагают, что равновесие реакции (1) при удалении низко-
кипящего ацетонитрила необратимо сдвигается вправо (2):
RCN 4- CH3CSNH, RC—S—ССН3
1,1 I II т
NH NH
—> RCSH-|-CH3CN
NH
RC=S
NH2
1. Taylor E. C., Zoltewicz J. A., J. Am. Chem. Soc., 82, 2656 (1960).
нА
ТИОМОЧЕВИНЫ ДИОКСИД, ^C—SOI. Мол. вес 108,13,
H2n/
t. пл. 144°.
Реагент, известный также под названием формамидинсуль-
финовая кислота, получают окислением тиомочевины перекисью
водорода [1].
Восстановление кетонов [2]. Т. д. восстанавливает кетоны до
вторичных спиртов с выходом 75—100%:
ч ч h2nx
V=O + 'С—sol 4- 2NaOH —> %НОН 4- 4- Na,SO3
/ H2NZ z h2nz
В типичном эксперименте флуоренон (0,01 моля) растворяют
в этаноле, приливают к нему при перемешивании водный рас-
твор гидроокиси натрия (0,02 моля) и Т. д. (0,01 моля) и на-
гревают смесь в течение 2 час при 90°. Флуоренол получают
с выходом 95,6%
1. В arnett Е. В., J. Chem. Soc., 97, 63 (1910).
2, Nakagawa К., Mina mi К, Tetrahedron Letters, 343 (1972).
ТИОНИЛ ХЛОРИСТЫЙ (HI, 329—335; V, 418—419;
VI, 258).
Синтез изонитрилов. Первую стадию синтеза rf-2-метилбута-
наля-1 по методу Вальборского и сотр. [1] проводят в трехгор-
лой круглодонной колбе емкостью 3 л, снабженной мешалкой
Гершберга, трубкой для ввода азота и капельной воронкой на
500 мл с соединительной трубкой для выравнивания давления.
Колбу высушивают над открытым пламенем, дают ей остыть и
499
вместо трубки для ввода азота вставляют низкотемпературный
термометр, а ввод азота подключают к капельной воронке,
В колбу помещают 118 а (0,75 моля) М-(1,1,3,3-тетраметил-
бутил) формамида и 1500 мл диметилформамида (т. кип. 76—
77°/1 мм). Затем смешивают 89 а (55 мл, 0,75 моля). Т. х. с 250 мл
сн3 сн3 сн3 сн3
I I <9 soci, । । ..
CH3-C-CH,-C-NH-C^ „ тт/Д» CH3-C-CH2-C-N = C
f t \ II”
сн3 сн3 н сн3 сн3
сн3 сн3
СН3 — С —СН, —С—N —С + сн3снсн2сн3.
СН3 СН3 | Li
СН3 СН3 Li 1} D2O, „
( l( 2 ) Н О+
CHj-C-CHz-C-N = C - - 1 х C-CtI-CHjCIf3
- СН3 СН3 СНСНгСН3 /
СН3
диметилформамида (температура поднимается почти до 30°) и
помещают смесь в капельную воронку. Колбу погружают в ба-
ню со смесью сухого льда с ацетоном и начинают перемешивать
содержимое с умеренной скоростью. Когда температура в колбе
достигнет —50°, начинают прикапывать раствор из капельной
воронки с такой скоростью, чтобы температура реакционной
смеси держалась в интервале между —55 и —50° (около
10 мин). По окончании прикапывания баню на короткое время
убирают, чтобы температура реакционной смеси поднялась до
“35°. Затем баню со льдом подставляют снова и в течение
Юлшн через воронку для внесения твердых веществ в атмо-
сфере азота прибавляют 159 г (1,5 моля) сухого карбоната на-
трия (высушенного в вакууме при 130°). Перемешивают смесь
в течение ~ 12 час и выливают в 6-литровую колбу Эрленмейе-
ра, содержащую 3 л воды со льдом (тяга)! Ополаскивают реак-
ционную колбу 300 мл пентана и необходимым для растворения
неорганических солей количеством воды и переносят промывные
растворы в ту же колбу Эрленмейера. Смесь энергично переме-
шивают в течение 5 мин и разделяют слои. Верхний слой про-
мывают двумя порциями воды по 100 мл, высушивают над
сульфатом натрия, фильтруют и отгоняют пентан. Сырой тет-
раметилбутилизонитрил перегоняют с 1,5 X 15-сантиметровой
колонкой Вигре. Выход продукта (т, кип. 55,5—56,5711 мм,
1,4178, d25 0,7944) составляет 86 — 90 г (82 — 87%).
4-Оксибеизолсульфохлорид. 4-Оксибензолсульфонат натрия
(1) при взаимодействии с Т. х. в присутствии каталитических
количеств ДМФА превращается в 4-оксибензолсульфохлорнд
500
(2) с выходом 80—90%- [2]. Другие катализаторы (например,
ОН ОН
(D
(2)
трифенилфосфин, ГМТФК) менее эффективны.
В этой реакции Т. х. нельзя заменить пятихлористым фосфо-
ром, однако для получения 3,5-дизамещенных 4-оксибензолсуль-
фохлоридов применять последний можно [3].
Аценафто-[1,2-с]-фуразан (2) [4]. Этот фуразан (2) может
быть получен с выходом 55% дегидратацией диоксима аценаф-
техинона (1) с помощью Т. х. в хлористом метилене:
1. Niznik G. Е„ Morrison W. Н., Ill, Walborsky Н, М., Org. Syn.,
51, 31 (1971).
2. С a m р b е 11 R. W., Н 111 Н. W„ J r„ J. Org. Chem., 38, 1047 (1973).
3. На 11 W. L., J. Org. Chem., 31, 2672 (1966).
4. Boulton A. J., Mathur S. S., J. Org. Chem., 38, 1054 (1973).
ТИОНХЛОРУГОЛЬНОЙ КИСЛОТЫ л-ТОЛИЛОВЫЙ
ЭФИР, n-CH3C6H4OC(S)Cl. Мол. вес 176,66, т. кип.
52—5370,1 мм.
Реагент (1) получают обработкой раствора тиофосгена в
хлороформе и-крезолом в водном растворе гидроокиси нат-
рия fl]:
NaOH
n-CH3CsH4OH+ CSC12 ---->- n-CH3CsH4OC(S)Cl
(О
Синтез олефинов из пространственно-затрудненных спиртов.
Герлах н сотр. [I] предложили удобную модификацию реакции
Чугаева, с помощью которой им удалось получить термически
нестойкие олефины из пространственно затрудненных спиртов,
трале, транс-С пи ро-[4,4ф но на и диол-1,6 (2) при взаимодействии с
раствором Т. к. т. э. в пиридине при комнатной температуре в
течение 4 час превращается в средний смешанный бас-О-эфир
501
тионугольной кислоты (3) с выходом 80%. Полученное произ-
водное при температуре ^135° гладко разлагается с образова-
нием спиро-[4,4]-нонадиена-1,6 (4). Пиролиз соответствующего ди-
ацетата или ди-Э-метилксантогената в этом случае не дает диен.
(D
80%
9С(3)ОС6ЩСНЭ
OC(S)OC6H4CHj
U)
+ 2 CHjCjHjOH + i COS
I. Gerlach H., Huong T. T., Muller W„ J. C. S. Chem. Comm., 1972,
1215.
ТИОФЕНОЛ — АЗОДИИЗОБУТИРОНИТРИЛ.
цис,транс-Изомеризация олефинов. В 1969 г. Сгоутас и Кам-
мероу [1] сообщили, что эфиры ненасыщенных жирных кислот
цас-конфигурации при нагревании с тиолами или дифенилфос-
фином в присутствии азодиизобутиронитрила при 65° в запаян-
ной ампуле изомеризуются в тряяс-изомеры. Равновесная смесь
содержит 75—80% 7 ран с-изомер а. Миграции двойной связи не
наблюдалось. По-видимому, реакция включает обратимое при-
соединение тиильных или фосфинильных радикалов к двойной
связи.
Недавно этот метод был использован Балерао и Рапопор-
том [2]. Так, смесь цис- и тряяс-изомеров 2,3-диметилоктена-3
(а) и (б) в отношении 85: 15 при нагревании с Т.— а. в запаян-
ной ампуле при 65° в течение 8 час превращается в смесь цис,-
трянс-изомеров в отношении 25:75. Как показано методами
сн3 снэ
(сн3)2сн-'''^сн3 н3с ^сн(снр2
а 6
ГЖХ и ЯМР, использование в этом случае селена (220°, 1 —
2 час) [3] приводит к сложной смеси продуктов.
1. S g о u t a s D. S., Kummerow F. A., Lipids, 4, 283 (1969).
2. В h а 1 е г а о U. Т., Rapoport Н., J. Am. Chem. Soc., 93, 4835 (1971) .
3. Stowell J. С., J. Org. Chem., 35, 244 (1970).
ТИТАН ТРЕХХЛОРИСТЫЙ, TiCl3 (V, 419—420). Мол. вес
154,27.
в«;ц-Диолы. япц-Диолы получают взаимодействием карбоно-
вых кислот и алкиллитиевых соединений в 1,2-диметоксиэтане
502
в присутствии Т. т. с последующей обработкой водой [1]. Напри-
мер, из реакции бензойной кислоты с метиллитием в этих усло-
виях выделены 2,3-дифенилбутандиол-2,3 (33%), ацетофенон
(27%) и сс-фенилпропиофенон (11%).
1) Ticis 9Нз 9Нз
2) Гидролиз J ]
C6HSCOOH + CH3Li ------------> C6I-I5C—СС0Н3 + С6Н5СОСН3 4-
он он
33%
СН3
+ С6НЙСНСОС6Н5
11%
Диол образуется в виде смеси (±)- и мезо-изомеров в соот-
ношении 2,4: 1.
Восстановление. Недавно Тиммс и Уайлдсмит [2] иашли,
что оксимы быстро восстанавливаются 15%-ным водным рас-
твором TiCl3, содержащим ZnCl2 и НС1, до иминов, которые
легко гидролизуются до карбонильных соединений. Поскольку
при восстановлении нитросоединений можно ожидать промежу-
точного образования оксимов, Мак-Мери и Мелтон [3] изучили
СН3СН2СНСН2СН2СОСН3 —> СН3СН2СОСН2СИ2СОСН3
j 85 %
no2
(I) (2)
восстановление 5-иитрогептанона-2 (1) водным раствором Т. т.
(TiCIg следует брать из полной банки и работать с иим в атмо-
сфере инертного газа) и получили гептандион-2,5 (2) с выходом
85%. Хлориды хрома и ванадия низшей валентности также вы-
зывают это превращение, но со значительно меньшими выхода-
ми (~25%). Описанная выше реакция была использована в
изящном синтезе ^«с-жасмона (6)
503
Рассмотренный метод с успехом использовали для превра-
щения соединения (7) в альдегид (9) [4]. Предшественник (8)
альдегида (9) получали присоединением нитрометана по Ми-
хаэлю к стероидному 3-кетотриену-1,4,6 в трет-бутаноле в при-
сутствии трет-бутилата калия.
1. Axelrod Е. N., Chem. Comm., 1970, 451.
2. Т i m m s G. H., W i 1 d s m i t h E., Tetrahedron Letters, 195 (1971).
3. McMurry J. E., Melton J., J. Am. Chem. Soc., 93, 5309 (1971).
4. Кос or M., Gumulka M., Cynkowski T., Tetrahedron Letters, 4625
(1972).
ТИТАН ЧЕТЫРЕХХЛОРИСТЫЙ (III, 342—344; V, 420; VI,
259—260).
Конденсация по Киёвеиагелю [1]. Раствор Т. ч. в ТГФ в
присутствии пиридина превосходно катализирует конденсацию
по Кневенагелю [2]. Например, ацетоуксусный эфир и этиловый
эфир нитроуксусной кислоты в этих условиях легко конденси-
руются с алифатическими, ароматическими и гетероциклически-
ми альдегидами.
>СООС2Н5
RCHO + Н.2С^
х:осн3
>СООС2Н5
RCHO + Н2С^
^NO2
50—90%
>СООС2Н5
RCH=C(
ХзосНз
>СООС2Н5
КСН=С^
\NO2
Малоновый эфир конденсируется с алифатическими и аро-
матическими кетонами, образуя с удовлетворительными выхо-
дами а,(3-ненасыщениые соединения [3]:
TiCI4
Ру, ТГФ
R\
}C=C(COOC2HS)2
Хлорирование ароматических соединений [4]. Т. ч. в присут-
ствии такого окислителя, как трифторнадуксусная кислота, хло-
рирует различные ароматические субстраты. Активированные
ароматические соединения, такие, как фенол, хлорируются с вы-
504
сокими выходами, бензойная кислота и нитробензол не всту-
пают в реакцию.
Избирательное орто-замещеиие в о-оксикарбоиильных соеди-
нениях [5]. Бромирование соединений типа (1) дает исключи-
тельно 5-бромпроизводные. Однако в присутствии Т. ч. обра-
зуются также значительные количества 3-бромпроизводных. По-
лагают, что ответственным за селективность о/?то-замещения
соосн,
(1)
(2)
является комплекс типа (2). Аналогичные результаты были по-
лучены при формилировании соединений типа (1) дихлорметил-
метиловым эфиром.
Оксимы-> нитрилы [6]. Альдоксимы под действием раствора
Т. ч. в ТГФ или диоксане в присутствии пиридина дегидрати-
руются до нитрилов. С алифатическими альдоксимами реакция
протекает при комнатной температуре, а в случае ароматиче-
ских требуется нагревание до 80°. Выходы высокие (80—97%).
Амиды нитрилы [7]. При обработке раствором Т. ч. в ТГФ
в присутствии основания (триэтиламин, N-метилморфолин) при
0° амиды карбоновых кислот дегидратируются до нитрилов с
выходами в пределах 65—95%.
1. Johnson J. R., Org. React, 1, 226, 233 (1942).
2. Lehnert \V., Tetrahedron Letters, 4723 (1970); Tetrahedron, 28, 663 (1972).
3. Lehnert W., Tetrahedron, 29, 635 (1973).
4. Chip G. K, Grosser! J. S., Can. J. Chem., 50, 1233 (1972).
5. Cresp T. M., Sargent M. V., S 11 x J. A., Chem. Comm., 1972, 214.
6. Lehnert W., Tetrahedron Letters, 559 (1971).
7. Lehnert W., Tetrahedron Letters, 1501 (1971).
ТОЗИЛМЕТИЛИЗОНИТРИЛ, Z2-CH3C6H4SO2CH2N=C (I),
Мол. вес 195,24, т. пл. 116—117°.
Получение. Реагент (1) получают с 66%-ным выходом из
метилизонитрила, как показано на схеме:
H-BuLi
H3CNC — > LiCH2NC + n-CH3C6H4SO2F —> rt-CH3C6H4SO3CH2NC
11Ф, —
Голландские химики [1] разработали две новые методики
получения Т. (1) [уравнения (I) и (II)]. По методу 11 он полу-
505
чается с несколько большим выходом, с другой стороны, ме-
тод I позволяет избежать работы с дурно пахнущими летучими
изонитрилами.
Н2О2/Ас2О
I) n-CH3C6H4SCH2NHCHO -------> n-CH3C6H4SO2CH,NHCHO —>
РОС1з
N(C2h5)3
----—n-CH3CeH4SO2CH2N=C
глим, 0—10°
80%
(1)
II) n-CH3C6H4SO2F + CH2N=C —> (1)
I 87%
Li
Превращение карбонильного соединения в ближайшую выс-
шую карбоновую кислоту [2]. Т. (1) при действии трет-бутилата
калия в ТГФ при 7—10° превращается в а-металлированное
производное, реагирующее с альдегидом или кетоиом с образо-
ванием аддукта (2). Кипячение последнего с 2 н. НС1 в течение
10 час дает кислоту (3).
(1)
1) КОС(СНз)з
2)
Y=o
R2/
3) НО А с
>
40-80%
Rk /NHCHO
/”С\
\SO2C6H4CH3-rt
Н3О*
55-65%
;снсоон
R2Z
Синтез гетероциклов. Реакцией Т. с карбонильными соеди-
нениями можно с высоким выходом получить оксазолы [3]. На-
пример, 5-фенилоксазол (2) синтезируют с выходом 91% кипя-
чением бензальдегида с Т. и К2СО3 в метаноле 2 час. Если ре-
акцию проводить при 20°, то элиминирования толуолсульфино-
вой кислоты не происходит.
TsCH3N=C
К,СО3
СНзОН^
TsCH-N=C + C^HgCHO-----> TsCH-N=C----->
K+ CjHj-CHO* K+
ТвСН-Щ
—=> I . >c
C6HS-CH-OZ
-TsH
S06
Вместо альдегидов в реакцию можно вводить хлораигидриды
или ангидриды кислот. В этом случае образующиеся оксазолы
(3) несут тозильную группу в положении 4.
TsCH—N-C + RCx
TscH-N=C
I
RC=O
TsC—N=C
' II
RCOH
55-80%
(3)
При взаимодействии T. (1) с имидхлоридами (4) [5] обра-
зуются тозилзамещенные имидазолы (5) [4]. Смесь Т. и имид-
хлорида (4) 1 : 1 в глиме или ТГФ прибавляют к перемешивае-
мой суспензии NaH в ДМСО (20°, Ns).
TSCH-N=C + RC=NR' —----->
w + Cl
Na
(4)
TSCH-N-C
RC=NR'
Tsc-N=C
II
RC-NHR1
—------>
60-887»
(5)
n-Толилтиометилизонитрилы. Родственный реагент n-толил*
тиометилизонитрил n-CHsCgFUSCHsN — C (I) с мол. вес. 163,24,
т. кип. 6070,001 мм был получен [6, 7] дегидратацией формами-
да (2) с помощью РОС13 в глиме или триэтиламиие:
О
n-CH3C6H,iSCH2NHCH
—* az-CH3C6H4SCH2N=C
53%
(2)
(1)
а-Литий-п-толилтиометилизоиитрил (3), полученный из (1)’
и я-BuLi в ТГФ при —70°, взаимодействует с уксусным ангид-
ридом в ТГФ при постепенном повышении температуры от —70
до 20° (2 час), образуя 5-метил-4-п-толилтиооксазол (4) с вы-
507
ходом около 30 %:
re-CH3CtH4SCHN=C +
(3)
.о
СН3<
о
CH3CZ
ч
г
K-CH3CtH4S-CH-N=C
сн3—с=о
(4)
С кетонами, например ацетоном, изонитрил (3) реагирует
аналогичным образом, давая с выходом 74% оксазолин (5), а с
сн3
(3) + /С=О --->
сн3
(5)
метилформпатом он образует с выходом 52% 4-п-толилтиоокса-
зол (6).
(з)
+ НСООСН3 -->
(6)
1. v a n L е u s е n А. AL, Boerma G. J. AL, Н е 1 m h о 11 R. В., S i d е-
r i и s H., Strafing J„ Tetrahedron Letters, 2367 (1972).
2. Schollkopf U„ Schroder R., Angew. Chem., internat. Ed., 11, 31 i
(1972).
3. van Leusen A. M., Hoogenboom В. E., Siderius H., Tetrahedron
Letters, 2369 (1972).
4. van Leusen A. AL, О 1 d e n z i e 1 О. H., Tetrahedron Letters, 2373 (1972).
5. Cramer E, В aer U., Chem. Ber., 93, 1231 (i960).
6. van Leu sen A. AL, van Gen ne p H. E., Tetrahedron Letters, 627
(1973).
7. Schollkopf U., Blume E., Tetrahedron Letters, 629 (1973).
n-ТОЛ ИЛСУЛЬФИНИЛМЕТИЛИДЛИТИЙ (2). Мол. вес
144,16.
Получение. Т. (2) получают с количественным выходом из
метил-я-тол ил сульфоксида (1) и диэтиламида лития (II, 193—
195; V, 254—256) в ТГФ; н-бутиллитий для этой цели меиее
пригоден.
О О
|| LiH(C2H5l2 " II
CH3SC6H4CH3-rt ----->• Li+CH2SCeH4CH3-,7
(I) (g)
508
Синтез оптически активных спиртов [1]. Для асимметриче-
ского синтеза спиртов Т. (2) генерируют, исходя из оптически
активного (R) -мети л-n-тол ил сульфоксида (1) [2]. Оптически
активный карбанион реагирует с бензальдегидом (3) с образо-
ванием смеси диастереомеров 2-окси-2-фенилэтил-и-толилсуль-
фоксидов (4а) и (46) в отношении 1:1с выходом 84%- Смесь
можно разделить хроматографией на силикагеле и дробной кри-
сталлизацией на диастереомеры (4а) (ат> + 91,7°, выход 17%)
и (46) (ccd + 202,8°, выход 15,5 %). Десульфуризация (4а) и
(46) действием никеля Ренея дает (S)-(—)-1-фенилэтанол (5а)
(ccd—42,6°) и (R)-(4~)-1-фенилэтанол (56) (ccd +42.Г) соот-
ветственно с выходом около 60%. Поскольку удельное враще-
ние оптически чистого спирта (56) составляет czd +43,5°, этим
путем, следовательно, можно получить спирты высокой оптиче-
ской чистоты.
Реакция оптически активного карбаниона (2) с сс-тетрало-
ном (6) приводит к образованию с количественным выходом
смеси (1,8:1) диастереомеров 1-окси-1-(/г-толилсульфинилме-
тил)-1,2,3,4-тетрагидроиафталинов (7). Главный компонент сме-
си с ccd +77,6° был выделен хроматографически с выходом
45,5%. Десульфуризация с помощью никеля Ренея дала с вы-
ходом 68% (—)-1-окси-1-метил-1,2,3,4-тетрагидронафталин (8)
(aD —31,0°).
Реакция оптически активного (2) с 1,2-эпоксициклогексаном
(9) в глиме (кипячение) приводит к смеси (1,5 2: 1) соедине-
509
ний (10а) и (106) с выходом 63,5%. В результате их разделе-
ния методом хроматографии с последующей дробной кристал-
лизацией были выделены продукты (10а) (ссо +118,2°, выход
13,5%) и (106) (ас +225,3°, выход 13%)- Десульфуризация
приводит к образованию (R,R)-(—)-трсшс-2-метилциклогекса-
нола (11а) с выходом 62% и (S,S) (~\~)-транс-2-метилциклогек-
санола (Иб) (ан +40,7°, выход 77%). Удельное вращение czD
чистого спирта (Иб), как сообщалось, составляет +42,9° [2],
т. е. с помощью карбаниона (2), как уже было сказано выше,
можно получать спирты высокой оптической чистоты.
63%
(106)
Ni Ренея
62%
Ni Ренея
77%
(Пб)
1. Tsuchihashi G., Iriuchijima S., Ishibashi M., Tetrahedron Let-
ters, 4605 (1972).
2. Andersen К. K., Tetrahedron Letters, 93 (1962).
П-ТОЛУОЛСУЛЬФОКИСЛОТА (III, 347—354).
Еиолацетилирование (III, 349—350). Первую стадию синтеза
З-н-бутилпентандиона-2,4 [1] осуществляют следующим образом.
В закрытой круглодонной колбе на 500 мл с помощью магнит-
ной мешалки перемешивают 30 мин при комнатной температуре
смесь 28,6 г (0,25 моля) гептанона-2, 51,0 г (0,50 моля) уксус-
ного ангидрида и 1,9 а (0,01 моля) моногидрата Т. Затем при-
бавляют 55 г (0,43 моля) комплекса трифторида бора с уксус-
ной кислотой 1:1 [I, 112—113, Reagents, 1, 69 (1967)], что со-
провождается выделением некоторого количества тепла. Обра-
зовавшийся янтарно-желтый раствор перемешивают 16—2^ час
ОС ОСН,
н-С^эСНгСОСНз + (СН3СО)»О «-С4Н9СН=С-СН3 -*
сн3
ВГ3-СН3СООН „ тт ^С-О\т, CH3CO,Na
----5> «-С4н9-С .BF -------------------*--*--* к-С4Н9СН(СОСН3Ь
(СН3СО)3О С==О’’ Н2О,нагреа.
in3
в закрытой колбе при комнатной температуре, а затем к нему
добавляют раствор 136 г (1,00 моля) тригидрата ацетата нат-
510
рия в 250 мл воды. Колбу снабжают обратным холодильником
и кипятят реакционную смесь в течение 3 час, а затем охлаж-
дают и извлекают продукт реакции тремя порциями петролей-
ного эфира (т. кип. 30—60°) по 100 мл. Объединенные вытяжки
промывают 5%-яым раствором бикарбоната натрия, затем на-
сыщенным раствором хлористого натрия и высушивают безвод-
ным сульфатом кальция. Растворитель удаляют на роторном
испарителе и маслообразный остаток перегоняют в вакууме.
З-я-Бутилпентандион-2,4 представляет собой бесцветную жид-
кость с т. кип. 84—86°/6мм. Выход 25—30 а (64—77%).
Перегруппировка Вагнера — Меервейна. Кипячение соеди-
нения (1) с ТзОН в бензоле дает с количественным выходом
продукт (2) [3], образующийся в результате двух последова-
(D
тельных перегруппировок Вагнера — Меервейна [2]. Соедине-
ние (3), представляющее собой смесь изомеров, аналогичным
образом перегруппировывается в 3,4-диметилтрицикло-[3,3,3,0]-
ундеканон-2 (4).
Гидролиз—декарбоксилирование. Согласно методике, описан-
ной в Organic Syntheses [4], диэтиловый эфир 2,5-дикетоцикло-
гексан-1,4-дикарбоновой кислоты (1) превращают в циклогек-
сандион-1,4 (2), обрабатывая (I) водой в стальном автоклаве
прн 185—195° в течение 10—15 мин (выход 72—80%). Для
упрощения методики и улучшения выхода предложено [5] ки-
пятить эфир (1) в водном этиленгликоле в присутствии неболь-
шого количества Т.
О Jl>COOC2H5 C2H5OOC'xksjzJ (1) О TsOH. НОСН2СН2ОН 85-92^ L \ О (2) 511
1. М а о C.-L„ Hauser С. R., Org. Syn., 51, 90 (1971).
2. Р о р р F. D., McEwen W. Е„ Chem. Rev., 58, 375 (1958).
3. Peet N. P., Cargill R. L, Bushey D. F„ J. Org. Chem., 38,1218 (1973).
4. N i e 1 s e п A. T., С a r p e n t e г W. R., Org. Syn., 45, 25 (1965).
5. Patwardhan S. A., Dev S., Synthesis, 1971, 427.
n-ТОЛУОЛСУЛЬФОНИЛАЗИД (тозилазид) (III, 355—357;
V, 421—423; VI, 260—261).
Введение диазогруппы. Региц [1] опубликовал обзор мето-
дов введения диазогруппы с помощью Т. в соединения с актив-
ной метиленовой группой.
Реакция с тетраметокснэтиленом. Тетраметоксиэтилен (1) и
Т. (2) образуют имидокарбонат (3). Реакция имеет первый по-
рядок по каждому реагенту. Эти данные, а также проведение
контрольных экспериментов с соединением (1), содержащим
изотопную метку, исключают его диссоциацию в условиях реак-
ции на две молекулы диметоксикарбена [2].
сн3оч ,ОСН3
4- 2TsN3 —>
CH3CZ \)СН3
(1) (2)
СН3О. СН3СХ
—► ^C=NTs + N3 + \y-=NN=NTs
СН3СИ СНзСИ
(3) (41
а-Диазокетоиы (VI, 260—261). Опубликована методика по-
лучения 2-диазоциклогексанона [3]:
1. Regitz М, Synthesis, 1972, 351.
2. Hoffman R. \V., В r e s s e 1 U., G e h 1 h a u s J., Hauser H., M u h 1 G.,
Chem. Ber., 104, 2611 (1971).
3. Regitz M., Rtiter J., Liedhegener A., Org. Syn., 51, 86 (1971).
n-ТОЛУОЛСУЛЬФОНИЛГИДРАЗИН (тозилгидразин) (III,
357—360; V, 423—429; VI, 261—262).
Новый метод синтеза олефинов (V, 424—426). Шапиро и
Дункан [1] опубликовали подробную методику получения Т., а
также привели пример его использования для синтеза борнена-2.
Реакция с циклопентен-2-оном-1 и циклогексен-2-оиом-1 [2].
< TsNHNH; . кД ° CHjOH/Fjr 7 TsNHHN (1) 1 .TsNHNH3 1 СН3ОН-НС1 Na ОСН) + 7=NNHTs ; > L p—OCHj + /L. / ДИГЛИМ Ts (3) 16°S (4) = П=1,2 | NNHTs (2) X-\ 0+ U (5) (6) X - CHj, (CHgJi, ch-ch2
512
Полагают, что в реакции соединения (3) с метилатом натрия
при 160° промежуточно образуются карбены.
Пиролиз солей тозилгидразонов лактонов, Агоста и сотр. [3]
разработали процесс, формально обратный окислению по Байе-
ру— Виллигеру, т. е, метод превращения лактонов в цикличе-
ские кетоны. Примером такой реакции является синтез спиро-
[3,4]-октаноиа-1 (5) из лактона (1), который действием сначала
борфторида триэтилоксония (III, 459—461; V, 467—468; VI,
ТбЫНЫНг
301—302), а затем этанола в присутствии этилата натрия пре-
вращают в лактон соответствующей орто-кислоты (2). При обра-
ботке лактона (2) Т. образуется тозилгидразон лактона (3), пре-
вращающийся под действием метилата натрия в метаноле или
гидрида натрия в диэтиловом эфире диэтилеигликоля в натрие-
вое производное (4), в результате пиролиза которого при 310°
образуется циклобутанои (5) с выходом 42%. Это превращение,
по-видимому, включает термическое элиминирование азота и
ге-толуолсульфинат-аниона с образованием алкоксикарбена, ко-
торый затем перегруппировывается в спирокетон (5).
Следует отметить, что (5) можно также получить с выходом
35% облучением циклопентенил-1-изобутилкетона (6) в течение
125 мин в кварцевой иммерсионной ячейке ртутной лампой На-
novia L (N679A-36) с фильтром из уранового стекла марки Кор-
нинг.
о
(6)
17 Зак. 596
513
Фостер и Агоста [4] недавно обсудили возможный механизм
превращения алкоксикарбенов в кетоны с сужением цикла.
Кетоны -> нитрилы [51. Кетоны можно превратить в нитрилы
через тозилгидразоны. Так, тозилгидразон циклогексанона (1),
обработанный сначала цианистым калием в смеси метанол—
уксусная кислота при комнатной температуре, а затем 2 н. рас-
твором гидроокиси калия, превращается с выходом 85% в соеди-
NNHTs
(U
HCN
----->
85*4
(2)
180°
---->
60&
+ Nj,. + TsH
(3)
нение (2), разлагающееся при 180° в течение 2 час на нитрил
(3) с выходом около 60%, N2 и TsH.
1. Shapiro R. Н., D u п с а п J. Н., Org. Syn., 51, 66 (1971).
2. Kirmse W., Ruetz L., Ann., 728, 30 (1969); Kirmse W., Ruetz L,
Ann., 726, 36 (1968); Kirmse W., Miincher G., Ann., 726, 42 (1969).
3. Smith A. B., Ill, Roster A. №., Ago st a W. C., J. Am. Chem. Soc.,
94, 5100 (1972); Foster A. №., Agosta W. C„ J. Am. Chem. Soc., 94,
5777 (1972).
4. Foster A. №., Agosta W. C., J. Am. Chem. Soc., 95, 608 (1973).
5. Cacchi S., Caglioti L, Paolucci G., Chem. Ind., 1972, 213.
п-ТОЛУОЛСУЛЬФОХЛОРИД (III, 361—367; VI, 262).
Алкилиитроиы N-алкиламиды [1]. Метилнитроны (1), по-
лученные из Д4-3-кетостероидов, при обработке Т. в пиридине
перегруппировываются в А-аза-А-гомостероиды (2). Реакция,
вероятно, начинается с ц-толуолсульфонилирования нитрона,
г.-СН3С6Н4ЗОгС1
(П (2)
сопровождающегося захватом нуклеофила Сз-атомом. Образую-
щийся О-тозилат гидроксиламина перегруппировывается в со-
единение (2). Эту перегруппировку можно использовать вместо
перегруппировки Бекмана.
1. Barton D. Н. R., Day М. J., Hesse R. Н., Р echet №.. №., Chem.
Comm., 1971, 945.
514
ТРЕХХЛОРИСТЫЙ АЗОТ (III, 367—368; V, 429; VI, 263).
Вицинальные дихлориды. Опубликована заключительная ра-
бота по хлорированию алкенов с помощью Т. а. [1].
1. Fie! d К. W., Kovacic Р.( J. Org. Chem., 36, 3566 (1971).
ТРИ-н-БУТИЛ-н-БУТЕН~3-ИЛ-ОЛОВО,
(н-С4Н9)з5пСН2СН2СН=СН2. Мол. вес 345,13, т. кип. 91 —
9370,35 мм.
Получение [I]. Реагент получают с выходом 81% из три-
н-бутилхлорстаннана и СН2—CHCH2CH2MgBr.
Циклопропилметиленовые производные [2]. Электрофильные
агенты (Cl2, SO3, RSC1, HgCl2) присоединяются к Т., образуя
циклопропилметиленовые производные с выходом 72—86%:
(«-C^JjSnCHjCHjjCH^CH;, + Е +--------->
------> £>СНгЕ + («-C4H,)3Sn+
1. Seyferth D., Weiner M. A., J. Am. Chem. Soc., 84, 36! (1962).
2. Peterson D. J., Robbins M. D„ Tetrahedron Letters, 2135 (1972).
ТРИ-н-БУТИЛСТАННАН (III, 372—373; V, 431—432; VI,
263—264).
Восстановление галогенидов по радикальному механизму.
Полагают, что восстановление галогенидов с помощью Т., про-
текающее по радикальному механизму, представляет собой цеп-
ной процесс [1]:
Инициатор + BujSnH —-> Bu3Sn •
Bu3Sn • RX —> R • -)- BtijSnX
R • 4- Bu3SnH —> RH 4- Bu3Sn •
R» + Bu3Sn* -—> Обрыв цепи
Если в ^-положении промежуточного радикала R- имеется атом
водорода, часто наблюдается отщепление НХ. Так, например,
обработка 2,3-дибромбутана 2 экв BusSnH дает 99% бутена-2 и
только 1 % бутана [2].
Лёффлер [3], проводя восстановление аддукта (2) буллвале-
на (1) с бромистым водородом с помощью Т. в бензоле (80°),
инициируемое азодиизобутироиитрилом (I, 11), выделил пять
17*
515
продуктов [(3) —(7)]:
Продукты (5), (6) и (7) образуются, по-видимому, в результате
перегруппировки промежуточных радикалов (а), (б) и (в).
Реакция дибромида буллвалена (8) с BusSnH также дает
сложную смесь продуктов. Однако при повышении температуры
и уменьшении концентрации Т. в этом случае с высоким выхо-
дом (82%) образуется продукт элиминирования (9). Следует
заметить, что дибромировапие буллвалена (1) протекает сте-
(8) (9)
реоизбирательно, давая менее устойчивый т/шнс-ди бромид, об-
разующийся в результате транс-1,4-присоединения.
С другой стороны, дибромид (11) семибуллвалена (10) вза-
имодействует с Т. сравнительно просто, давая в качестве осиов-
616
пых продуктов два диена (12) и (13) в отношении 3: 1 [4]. Сле-
дует отметить, что бромирование семибуллвалена (10) проте-
кает стереоизбирательно путем экзо-1,4-присоединения.
(Ю)
Циклопропеион (1). Бреслоу и Ода [5] восстановлением
тетрахлорциклопропена под действием Т. в вазелиновом масле
в атмосфере аргона удалось получить и выделить чистый цик-
лопропенон (I) ст. пл. после перекристаллизации от —29 до
'О
—28°. При восстановлении тетрахлорида образуется смесь три-
хлор-, дихлор- и 3-хлорциклопропенов, из которой обработкой
NaHCOs, а затем NasSO^ можно получить циклопропенои с об-
щим выходом 41—46%.
Синтез простых эфиров нз олефинов и спиртов. Грэйди и
Чокский [6] описали двухстадийиый синтез простых эфиров. При
обработке олефина (1) безводным спиртом в присутствии NBC
с выходом 50—60% образуется бромзамещеиный простой эфир
(2) [7], который затем восстанавливают Т. до простого эфира
(3) с выходом 70—90%. Восстановитель получали по методике
(О
R'OM,
OR’
R-C-C-R
R R
(2)
R—С—С—R
1 I
R R
(3)
в
R R
Грэйди и Кювилы (VI, 264, [3]). Новый метод синтеза простых
эфиров позволяет избежать применения сильных оснований, не-
обходимых в классическом синтезе Вильямсона.
Внутримолекулярная циклизация хлорангидридов ненасы-
щенных кислот. Т. восстанавливает ацилхлориды до альдегидов
по цепному свободнорадикальному механизму, включающему
образование ацильных радикалов (III, 373). Жекович [8] не-
давно нашел, что при действии Т. (инициатор— азодиизобути-
ронитрил) на хлор ангидриды алифатических кислот, содержа-
517
щих двойную связь в положении 5 или 6, образуются производ-
ные циклогексанона, например хлорангидрид гексен-5-овой кис-
лоты (1) превращается в циклогексанон (2), а хлорангидрид
цитронелловой кислоты (3) —в ментон (4).
1. Kuivila Н. G., Accts. Chem. Res., 1, 299 (1968).
2. Strunk R. J., Di Gia сото P. M., As о К., Kuivila H. G., J. Am.
Chem. Soc., 92, 2849 (1970).
3. Loffler H.-Р., Chem. Ber., 104, 1981 (1971).
4. Paquette L. A., Birnberg G. H., Clardy J., Parkinson B.,
J.C. S. Chem. Comm., 1973, 129.
5. Breslo w R., Oda M., J. Am. Chem. Soc., 94, 4787 (1972).
6. Grady G. L., Chokski S. K-, Synthesis, 1972, 483.
7. Erickson K' L., К i m K., J. Org. Chem., 36, 2915 (1971).
8. Cekovic Z„ Tetrahedron Letters, 749 (1972).
ТРИ-н-БУТИЛФОСФИНА — МЕДИ(1) ИОДИДА КОМП-
ЛЕКС, (h-Bu)3P-CuI. Мол. вес 221,20, т. пл. 75°.
Получение [1]. Реагент получают с выходом 96,7% взаимодей-
ствием иодида меди(1) (избыток) со свежеперегнанным три-я-
бутилфосфином.
Построение скелета простаиовой кислоты [2]. Ключевая ста-
дия полного синтеза простагландина Ej (PGEi) (4) включает
конденсацию соединения (1) с 2 мол. экв соединения (2) в при-
сутствии 1 мол. экв Т. — м. и. к:
518
Шауб и Вейс [3] осуществили с помощью этого комплекса
сопряженное присоединение З-трет-бутокси-1-октил магнийбро-
мида (6) к циклопентенону (5), получив с выходом 35% про-
дукт (7), который после снятия защитной группы трифторуксус-
иой кислотой и омыления дает кетон (8). Восстановление пос-
леднего пергидро-9Ь-борафеналилгидридом лнтия (этот том)
приводит к (±)-11-дезокси-13-дигидропростагландину Еь
1. К a u f m a n G. В., Т е t е r L. А„ Inorg. Syn., 7, 9 (1963).
2. Sih С. J., Price P., Sood R., Salomon R. Peruzzotti G.. Ca-
sey M„ J. Am. Chem. Soc., 94, 3643 (1972).
3. Schaub R. E., Weiss M. J., Tetrahedron Letters, 129 (1973).
ТРИИЗОБУТИЛАЛЮМИНИЙ, A1[CH2CH(CH3)2]3 (Ш,
375—376). Мол. вес 198,32.
Восстановление а-кетолов. Катценелленбоген и Боулас [1]
исследовали стереоселективность восстановления алифатических
он
к1. с^о
т/г Т
СН3
Е>\ .он
R2
Н СНа
Н
сн3
трео
эритро
сс-кетолов различными алюмогидридами. Согласно циклической
модели Крама [2], при этом преимущественно должен образо-
вываться эрщгро-диол. Наиболее селективным восстановителем
оказался Т. Меньшую избирательность действия алюмогидридов
приписывают их способности образовывать ассоциаты, например
519
существующий в виде тримера диизобутилалюминийгидрид (I,
275—278) проявляет слабую селективность.
1. К a t z е n е 11 е n b о g е п J, A,, Bowlus S. В., J. Org. Chem., 38, 627
(1973).
2. С г a m D. J., W i 1 s о n D. R., J. Am. Chem. Soc., 85, 1245 (1963).
2,4,6-ТРИИЗОПРОПИЛБЕНЗОЛСУЛЬФОКИСЛОТЫ ГИД-
so,nhnh2
РАЗИД, (сн3)гнсх^Д^.сн(сн3)г Мол. вес 310,46, т. пл. 121 — 122'
II | (с разл.).
сн(сн3)2
Реагент можно получить [1] с выходом около 80% при взаи-
модействии 2,4,6-триизопропилбензолсульфохлорида [2] с гидра-
зингидратом в ТГФ.
Диимид [1]. Т. г. в интервале температур 35—65° разлагается
с образованием диимида. Поэтому с его помощью можно с хо-
рошим выходом гидрировать олефины. В качестве предшествен-
ников диимида использовались также гидразиды бензолсульфо-
кислоты [3] и п-толуолсульфокислоты [4], однако оба этих гидра-
зида генерируют диимид при более высоких температурах.
1. Cusack N. J., Reese С. В., Roozpeikar В., Chem. Comm., [972, 1132.
2. Lohrmann R., fthorana H. G., J. Am. Chem. Soc., 88, 829 (1966).
3. H u n 1 g S., M u 11 e r H.-R., T h i e r W., Tetrahedron Letters, 353 (1961).
4. van Tamelen E. E., Dewey R. S., J. Am. Chem. Soc., 83, 3729 (1961).
1,3)5-ТРИМЕТИЛ-2,4,6-трпс-(3,5-ДИ-трет-БУТИЛ-4-ОКСИ-
БЕНЗИЛ)-БЕНЗОЛ. Мол. вес 775,2, т. пл. 244°.
Растворимость при 18°, вес. %
Хлористый метилен .
Бензол.............
Метил циклогекса и
31,9
20,0
1,7
Метанол...........
Изопропиловый спирт
Вода..............
0,20
0,10
Нераство-
рим
Пространственно затрудненный, термически устойчивый, фе-
нольный антиоксидант. Это — чрезвычайно эффективный, лишен-
520
ный цвета и запаха стабилизатор для пластмасс, смол, резин и
восков. Обладает исключительно низкой летучестью и особенно
ценен в процессах, требующих высоких температур.
ТРИМЕТИЛ ЕНДИТИОТОЗИЛАТ, TsS(CH2)8STs. Мол. вес
416,59, т. пл. 67,5°.
Получение. Сначала реакцией гидросульфида калия с тозил-
хлоридом получают тиотозилат калия, который затем обраба-
тывают триметилендибромидом, получая Т.
2KHS + TsCl —> TsSK + H2S+KCl
2TsSK + Br(CH2)3Br —> TsS(CH2)3STs + 2KBr
Реакция с активированными метиленовыми группами [1].
С соединениями, содержащими активированные метиленовые
группы, Т. образует 2,2-дизамещенные 1,3-дитианы (1) с выде-
лением 2 экв п-толуолсульфиновой кислоты. Дитиокетальиая
группа соединения (1), в отличие от ацетальной группы анало-
гичных кислородсодержащих соединений, удивительно устой-
чива к действию кислот, одиако дитианы легко превратить об-
ратно в метиленовые соединения восстановлением никелем
Ренея или гидразином. Дитиокетальную группу можно также
превратить в карбонильную гидролизом, катализируемым иона-
ми Hg(II). Таким образом, Т. удобен для защиты активных
Cs ,х
"X;
S Y
(1)
метиленовых групп. Карбонильные соединения, которые не вза-
имодействуют с Т., перед реакцией можно активировать, превра-
тив их в енамины или оксиметиленовые производные. Так, цик-
логексанон превращают в 2,2-триметилендитиоциклогексанон
через енамин пирролидина. Реагент впервые был использован
521
в изящном синтезе ланостерина из холестерина для защиты
Са-метиленовой группы Д4-холестеноиа-3 [2].
Аналогичным образом можно использовать и этилендитио-
тозилат, TsS(CHaJsSTs.
1. Woodward R. В., P a c h t e г 1. J., Scheinbaum M. L., J. Org. Chem.,
36, 1137 (1971); Org. Syn., submitted 1972.
2. Woodward R. B„ Patchett A. A., Barton D. H. R., Ives D. A. J.,
Kelley R. B., J. Chem. Soc., 1957, 1131.
1Ч,4,4-ТРИМЕТИЛ-2-ОКСАЗОЛИНИЯ ИОДИД,
Мол. вес 114,17.
CH3
Получение реагента из 2-амино-2-метилпропанола-1 и му-
равьиной кислоты описано в примечании к методике синтеза
о-анисового альдегида [1]:
(О (2)
В круглодонную трехгорлую колбу на 1 л, снабженную ка-
пельной воронкой на 500 мл с обводной трубкой для выравни-
вания давления, механической мешалкой и трубкой для ввода
азота, помещают 80 г (0,33 моля) Т. и. Систему заполняют азо-
том, вливают в колбу 150 мл тетрагидрофураиа, перегнанного
над алюмогидридом лития, и полученную суспензию при пере-
мешивании охлаждают в бане со льдом. Тем временем к охлаж-
денному раствору о-метоксифенилмагнийбромида (0,414 моля),
свежеприготовленного из о-броманизола (77,5 г, 0,414 моля)
и магниевых стружек (11 а, 0,458 г-атом), прибавляют 146 мл
(0,828 моля) сухого ГМТФК) (высушенного перегонкой над гид-
ридом кальция). Полученный раствор переносят в атмосфере
аргона в капельную воронку на 500 мл. Раствор медленно при-
капывают к охлажденной суспензии, и при этом иодметилат
постепенно растворяется. По окончании прибавления раствора
реакционную смесь перемешивают при комнатной температуре
в течение ~ 12 час.
Образовавшуюся суспензию охлаждают до —5°, выливают
в 600 мл ледяной воды и быстро подкисляют (pH 2—3) охлаж-
денной 3 н. соляной кислотой. Кислый раствор тотчас экстраги-
руют 300 мл холодного гексана и экстракт отбрасывают. Вод-
ный раствор затем подщелачивают 20%-ным раствором гидро-
окиси натрия (добавляя лед, если необходимо). Образовавшуюся
522
суспензию экстрагируют тремя порциями эфира по 1 л каждая,
объединенные вытяжки высушивают над поташом, фильтруют
и концентрируют, получая при этом 75—85 г светло-желтой
смеси оксазолидина (1) и ГМТФК. Последний можно удалить
пропусканием эфирного раствора через силикагель.
1. Brinkmeyer R. S., Collington Е. W., Meyers A. I., Org. Syn.,
submitted 1971.
ТРИМЕТИЛОКСОНИЯ БОРФТОРИД (III, 382, V, 433; VI,
269—270).
Метилирование 1,3-диазабицнкло-[3,1,0]-гексенов-3 [1]. Т. б.
метилирует 1,3-диазабицикло-[3,1,0]-гексен-3 (1) селективно по
ненасыщенному атому N с образованием борфторида 2,2,3-три-
метил-4-феиил-6-п-нитрофенил-1-аза-3-азоииабицикло-[3, 1, 0]-ге-
ксена-3 (2) с выходом 93%. При взаимодействии (1) с л/-хлор-
(СН3)3О bf4‘
СН,С1,
(3)
надбензойной кислотой (IV, 138—143; V, 517—519; VI, 324—
325) атака реагента направляется в то же положение и обра-
зуется N-окись 2,2-диметил-4-фенил-6-п-нитрофенил-1,3-диазаби-
цикло-[3,1,0]-гексена-3 (3).
1. Н е i n е Н. W., Newton Т. А., В 1 о s i с k G. J., Irving К. С., М е у е г С.,
Corcoran G. В., Ill, J. Org. Chem., 38, 651 (1973).
2.4,4-ТРИМЕТИЛ ПЕНТИЛ-2-ИЗОНИТРИ Л (VI, 270—271).
Получение. Методика получения этого реагента недавно
опубликована [1].
Альдегиды из алкилмагннйбромндов [2]. Алифатические ре-
активы Гриньяра взаимодействуют с 1 же Т. в ТГФ, образуя
альдимины (1), гидролиз которых (щавелевая кислота) дает
523
альдегид (2) с выходом 48—67%. Карбонизация соединения
сн3
сн3 ,n-c-ch2-c(ch3)3 + о
RMgBr + C=N-C-CH2-C(CH3)3---->НС СН3 RCM
СН3 \lgBr
U (2)
(1) приводит к соответствующей а-кетокислоте.
1. Walborsky Ы. М., Niznik G. Е., J. Org. Chem, 37, 187 (1972).
2. Walborsky Н. М., Morrison W. Н., Ill, Niznik G. E., J. Am.
Chem. Soc., 92, 6675 (1970).
ТРИМЕТИЛСИЛ ИЛДИАЗОМЕТАН, (CH3)3SiCHN2. Мол.
вес. 114,23, т. кип. 96°/775 мм.
Получение [1]. Этот необычайно устойчивый диазоалкан
можно получить по следующей схеме:
NH3 Мочевина, НС1
(CH3)3SiCH2Cl ----> (CH3)3SICH2NH2 ------------->
hno2
—> (CH3)3SiCH2NHCONH2 ---------->
T. пл. 113-114°
20%-ная КОН
СдНе
—> (CH3)3SiCH2N(NO)CONH2 ---------—--->• (CH3)3SiCHN2
(1)
1,3- Диполярное присоединение [1]. Т. (1) в декалиновом
растворе вступает в реакцию 1,3-диполярного присоединения с
акрилонитрилом, в результате которой образуется с выходом
73% 2-пиразолин (2):
NC ___
(1) + СН2=СНСМ ---> //\
73$ X Z^SifCHM
N >
H
(2)
Триметил си лил карбен [1]. При добавлении CU2CI2 к рас-
твору Т. (1) в бензоле в присутствии циклогексена образуется
в качестве главного продукта лнт’й-7-триметил сил ил норкар ан
524
(2) с выходом 65%:
Синтез ацетиленов из карбонильных соединений [2]. Т. (1)
(или диметилфосфонодиазометан (2) [3]) обрабатывают н-бу-
тиллитием в ТГФ при —78°, а затем карбонильным соединением,
например бензофеноном, и получают в результате этого ацети-
лен, в приведенном примере дифенилацетилен:
(CH3)3SiCHN2
о)
1) н-ВиЫ
3) 1йо 5hG=0 (CH3)3SiOH
(CH3O)2PCHN2---------> C6H5C^CCBI-15 + N2 + или
li 80 А (СН3О)2
(2)
РОН
А
В случае карбонильных соединений, содержащих а-водород-
ные атомы, наблюдается конкурирующая реакция енолизации.
Так, например, из Т. и ацетофенона 1-фенилпропин образуется
лишь с выходом 16%, а 50% кетона возвращается обратно.
Альдегиды превращаются в терминальные ацетилены, например
фенилацетальдегид превращается в 3-фенилпропин с выхо-
дом 30%.
Эта реакция, вероятно, включает перегруппировку Вольфа
[4].
1. Seyferth D., Dow A. W., Menzel Н„ F 1 о о d Т. С„ J. Am. Chem, Soc.,
90, 1080 (1968).
2. Colvin Е. W, Hamill В. J., J. C. S. Chem. Comm., 1973, 15].
3. Seyferth D., Marmor R. S., Hilbert P., J. Org. Chem., 36, 1384
(1971).
4. Bacnman W. E., Struve W. S., Org. React., 1, 38 (1942).
ТРИМЕТИЛСИЛИЛДИЭТИЛАМИН (VI, 272).
Превращение простагландинов F в E. Янки и Банди [1]
установили, что метиловый эфир 15-метил-РОР2а (1) под дей-
ствием Т. силилируется преимущественно в положение 11. Окис-
ление образовавшегося 11-триметилсилильного производного (2)
реактивом Коллинза с последующим гидролитическим отщепле-
525
нием ТМС-группы в водном метаноле в присутствии следов ук-
сусной кислоты дает метиловый эфир 15-метил-РйЕ2 (3) с выхо-
дом 45%, считая на 15-метил-РОЕ2а (1)-
Этот метод, как было недавно показано, является общим для
превращения F- в Е-простагландины (2). Так, Т. силилирует
метиловый эфир PGF2a (4) дважды по ОН-группам в положе-
(6)
киях 11 и 15, оставляя Cg-оксигруппу свободной для окисления.
Метиловый эфир PGE2 (6) был получен этим способом с общим
выходом 35—50%-
1. Yankee Е. W., Bundy G. L., J. Am. Chem. Soc., 94, 3651 (1972).
2. Y a n k e e E. W., L i n C. H„ F г i e d J., Chem. Comm., 1972, 1120.
бис-(ТРИМЕТИЛСИЛИЛ)-Л ИТИЙАМИД, [ (CH3) 3S i] 2NLi.
Мол. вес 167,33.
Получение (VI, 144).
Синтез глицидных эфиров. Реакцию Дарзана [1] не удается
осуществить с такими соединениями, как ацетальдегид, который
526
в условиях реакции дает только продукты самокоиденсации.
Борх [2] описал новую методику, применимую к любым карбо-
нильным соединениям. При взаимодействии а-бромэфира (1) с
Т. при —78° в ТГФ гладко генерируется анион (2). Добавление
к этому раствору альдегида или кетона при —78° дает после
обычной обработки реакционной смеси с высоким выходом соот-
ВСНСООС2Н5 ЦСНзБ51!^!'1 > вссоосгн5
Li
(1} (2)
55:45
(За)
(36)
ветствующий глицидный эфир (3), причем образуется преиму-
щественно транс-изомер эфира (За).
I. Newman М. S-, М a g е г 1 е i п В. J., Org. React,, 5, 413 (1949).
2. Borch R. F., Tetrahedron Letters, 3761 (1972).
бмс-(ТРИМЕТИЛСИЛИЛ)-НАТРИЙАМИД (III, 385—386;
VI, 272—273).
Моиобромциклопропаиы. Мартел и Хириарт [1] обнаружили,
что реакция бромистого метилена с сильным основанием Т. дает
возможность с прекрасным выходом генерировать бромкарбен.
Эта реакция лежит в основе нового удобного метода ситеза мо-
нобромциклопропанов:
Г(СНД51]2МЫа * СНгВгг + >С = С< >>С-С< + [(CH^SHsNh + NaBr
А
Н вг
До этого самым лучшим способом получения монобромцикло-
пропанов было восстановление гем-дибромциклопропанов три-
бутилстанианом. По новой методике можно получать также и
монохлорциклопропаны, если вместо бромистого метилена взять
хлористый метилен.
1. Martel В., Н i г i а г t J. М,, Synthesis, 1972, 201.
ТРИМЕТИЛСИЛИЛЦИАНИД, (CH3)3SiCN. Мол. вес 81,15
т, кип. 117°, 1,3883.
527
Получение [1]. Реагент можно получать несколькими спосо-
бами:
(CH3)3SiCl 4- AgCN ——* (Cria)aSiCN + AgCl
оо >6
2(CH3)3SiCl + K2Hg(CN)4 2(CH3)3SiCN + 2KCI + Hg(CN)2
(CH3)3SiNHSi(CH3)3 + 3HCN ~~2(CH3)3SiCN + NH<CN
45,o %
Циансилнлироваиие карбонильных соединений [2]. Т. легко
реагирует с альдегидами [(1), R2 = Н], давая с хорошим выхо-
дом продукты циансилилироваиия [(2) К2 = Н]. Кетоны в тех
же условиях реагируют медленно, однако при катализе иоди-
R1\
)С==0 +(CH3)3SICN
OSi(CH3)3
R1—C—R2
I
CN
стым цинком они даже при комнатной температуре образуют
соответствующие производные (2) с превосходным выходом. Из
а.р-ненасыщенных альдегидов и кетонов получаются только
OSi(CH3)3
OSi(CH3)3
продукты 1,2-присоединения. Бензохинон дает аддукты (3) или
(4), образующиеся в результате присоединения 1 или 2 же Т.,
1ДА1Й4 восстанавливает эти аддукты до р-аминоспиртов.
Аддукты можно использовать также как средство защиты
карбонильных групп. Они устойчивы в апротонной среде, одна-
ко в разбавленной кислоте или щелочи быстро регенерируют
карбонильную группу.
1. Bither Т. A., Knoth W. Н., Lindsey R. V., Jr., Sharkey W. H.,
J. Am. Chem, Soc., 80, 4151 (1958).
2. Evans D. A., Truesdale L. K., Car roll G. L., J. C. S. Chem. Comm.,
1973, 55.
ТРИМЕТИЛФОСФИТ (III, 388—390; V, 436—438; VI, 275—
276).
Синтез олефинов по Кори — Уинтеру (III, 389). Чонг и Уайз-
мэн [1] показали, что в одном из синтезов олефинов по Кори —
528
Уинтеру промежуточно образуется бицикло-[3,2,1]-октеи-1 (2) —
алкен с двойной связью в голове моста — с нарушением правила
Бредта. Так, кипячение (165°) тионкарбоната (1) с триэтилфос-
фитом в течение 24 час в присутствии в качестве акцептора оле-
фина 1,3-дифеиилизобензофурана (I, 388—390; V, 164) приводит
к двум аддуктам Дильса — Альдера (3) и (4), которые могли
образоваться только из соединения (2).
Этерификация. 1 -Метилиндол-2-карбоновую кислоту (1) ки-
пячением в Т. можно превратить с выходом 94% в метиловый
эфир (2) [2];
(1)
I. С h о п g J. A., W i s е m a n J. R., J. Am. Chem. Soc., 94, 8627 (1972).
2. SzmuszkoviczJ., Org. Prep. Proc. Ini., 4, 51 (1972).
ТРИМЕТИЛХЛОРСИЛАН (III, 390—391; V, 439—442; VI,
276—278).
Ацилоиновая конденсация (VI, 277—278). Было показано
[1], что разработанный на примере замещенных бис- (триметил-
силокси)-циклобутенов способ окислительного расщепления бро-
мом в апротонной среде с образованием соответствующих 1,2
529
дикетоциклобутанов является прекрасным методом получения
нееиолизующихся а-дикетоиов [1]:
R-C-—OSi(CH3)3 Br2/cch R—С=О
[| ------> [ 4-2BrSi(CH3)3
Р—С—OS i (СН3)3 R—С=О
Для этой же цели пригодны хлор и иод, однако обычно пред-
почитают использовать бром. Например, при взаимодействии
соединения (1) с бромом в четыреххлористом углероде обра-
зуется дипивалоил (2) с выходом 81%:
(СН3)зС^ zOSi(CH3)3
С ВГ2/СС14
(CH3)3SiOZ ^С(СН3)3
(1)
(СН3)3СЧ .0
0^ ^С(СН3)3
Реакцию можно использовать также для получения цикличе-
ских нееиолизующихся а-дикетонов. Например, соединение (3)
превращается в 1,2-дикето-3,3,7,7-тетраметилциклогептан (2) с
выходом 98%:
Вгг/ССЦ
9й% *
Для получения енолизующихся сс-дикетонов реакция оказа-
лась мало подходящей [2], поскольку оиа осложняется тем, что
выделяющийся триметилсилилбромид взаимодействует с вновь
образовавшимся енольным гидроксилом, приводя к появлению
новой двойной связи, к которой может присоединиться бром.
Таким образом, в этой реакции образуется несколько продуктов
бромирования а-дикетонов наряду с продуктами разложения.
C^OSi(CH3)3
^OSi(CH3)3
Br2 ;
-2 (CHjJjSiBr
(CH3)3SiBr
-HBr *
О О
OSi(CH3)3
Кюльманн [3] опубликовал обзор реакций эфиров карбоно-
вых кислот с натрием в присутствии Т.
4Na 4- (CII3)3SiCI R—С—-OSi(CH3)3
2RC00R' -------------> [|
R—C—OSi(CH3)3
530
fJ-Аминоспирты. Пархам и Рузвельт [4] показали, что три-
метилсилиловые эфиры енолов, полученные как по методу Стор-
ка и Худрлика (V, 440), так и по методу Хауза и сотр. (VI,
276), могут быть превращены в p-аминоспирты. Например, три-
метилсилиловый эфир енольной формы циклогексанона (1) при
взаимодействии с безводным цианистым водородом, содержащим
каплю серной кислоты, дает с выходом 49% а-цианциклогексил-
триметилсилиловый эфир (2). Восстановление циангруппы (алю-
могидрид лития) с последующим гидролизом эфира приводит к
образованию с выходом 74,5% слегка загрязненного (3-амино-
спирта. Последний был очищен и охарактеризован в виде хлор-
гидрата.
С^ОЗЦСН^з
у
НС?\
49%
1) LiAIH4
нго ,
74,5% '
GCH2NH3
ОН
(3)
Пархам и Рузвельт отмечают различия в двух методах полу-
чения силиловых эфиров енолов. Для пространственно-затруд-
ненных кетонов более пригоден, по-видимому, метод Сторка,
в то время как метод Хауза лучше применять в случае простран-
ственно менее затрудненных, легко конденсирующихся кетонов.
Декарбоксилирование эфиров дизамещенных малоновых кис-
лот. При попытке осуществить ацилоиновую конденсацию эфи-
ров дизамещенных малоновых кислот Эйнсворз, Блумфилд и
сотр. [5] вместо ожидаемых продуктов получили с высоким вы-
ходом алкилсилилацетали кетена (1); при этом образуется
эквивалентное количество окиси углерода. Поскольку ацетали
RWlCOOCHsh + NaHHCH^SiCl —> WC=C(OCH3)OSi(CH3)3+
(i)
+ CH3OSi(CH3)3 + CO
(1) + СНзОН —> R1R2CHCOOCH3 + CH3OSi(CH3)3
(2)
(1) легко гидролизуются до эфиров (2), этот двухстадийный
синтез представляет собой удобный метод декарбоксилирования
эфиров дизамещенных малоновых кислот.
Кетокислоты. В новом методе синтеза р-кетокислот диа-
нионы карбоновых кислот взаимодействуют со сложными эфи-
рами [6]. Промежуточные продукты реакции улавливают с по-
мощью Т. и выделяют в виде триметилсилиловых эфиров. На-
пример, изомасляную кислоту обработкой 2 экв диизопропил-
амида лития в ТГФ при 0° превращают в дианион (1). При до-
бавлении к нему 1 экв метилового эфира пивалиновой кислоты
(2) и избытка Т. с выходом 70% образуется триметилсилило-
531
вый эфир fj-кетокарбоновой кислоты (3). Метанолизом эфира (3)
(СН3)2ССо6 + (СНз)зССООСНз
(1) (2)
СН3ОН
------>- (СН3)3ССОС(СН3)2
количеств. ।
соон
(CH3)3SiCl
——(СН3)3ССОС(СН3)2
да |
COOSi(CH3)3
(3)
Нагревание
----------> (СНз)3ССОСН(СН3)г
ИЛЛННРПТЙ
(4)
(5)
с количественным выходом получают р-кетокислоту (4), пиролиз
которой дает с количественным выходом трет-бутилизопропил-
кетон (5).
Новый метод синтеза гептаметилдисилазана (2) [7]. Реакция
метиламина с Т. (1) в н-пеитане (растворитель) приводит к об*
разованию (2) с выходом 85%:
2(CH3)3SiCl + 3CH3NH2 —> (CH3)3Si—N—Si(CH3)3 + 2CH3NH3C1
CH3
(i) ' (2)
1. Str a ting J., Reiffers S., Wynberg H., Synthesis; 1971, 209.
2. S t r a t i n g J., R e i f f e r s S., Wynberg H., Synthesis, 1971, 211.
3. Kuhlmann R., Synthesis, 1971, 236.
4. Parham W. E., Roosevelt C. S., Tetrahedron Letters, 923 (1971).
5. Kuo Y.-N., Chen F., Ainsworth C., Bloomfield J. J., Chem. Comm.,
1971, 136.
6. Ruo Y.'N., Yahner Y. A., Ainsworth C., J. Am. Chem. Soc., 93, 6321
(1971).
7. Birkofer L., S c h ni i d t b e r g G., Chem. Ber., 104, 3831 (1971).
1,2,3-ТРИОКСО-2,3-ДИГИДРОФЕНАЛЕН (1). Мол. вес
210,1, кристаллы красного цвета, разл. при 255—260°.
Получают из ангидрида нафталин-1,8-дикарбоиовой кислоты
в три стадии с общим выходом 60—65%:
532
Конденсация с дибензилкетоном.
1. Ried W, Meh rot га М. L, Ann., 718, 120 (1968).
сош-ТРИТИАН (VI 279).
Синтез альдегидов. Описан синтез я-пентадеканаля из Т. и
1-бромтетрадекана (VI, 279) [1]. Т. очищают экстракцией 30 г
продажного препарата 300 мл толуола.
I. Seeb ach D, В еск А. К, Org. Syn, 51, 39 (1971).
ТРИФЕНИЛАРСИНА ОКИСЬ, (C6H5)3As = O. Мол. вес
332,21.
Безводную Т. о. получают из продажного реактива (содер-
жащего воду) кипячением в бензоле с насадкой Дина — Старка
примерно 5 час. Полученный твердый продукт затем сушат при
135°/0,1 мм.
Обратная реакция Виттига [1]. Дициаиацетилен реагирует
с окисью трифенилфосфииа в бензоле при 160°, давая с выходом
78% дииитрил а-трифенилфосфоранилиден-а'-кетоянтарной кис-
лоты (1). Образующиеся в реакции промежуточные соединения
показаны на схеме:
NC-C=C-CN + (СЩ5)3Р-О
NC-C=C-CN
+ I
(СЩ5ЦР-0
(СДЦЦР-О
XCN
(СьН})зР=С
COCN
(И
533
Реакцию с окисью трифенилфосфина не удается распростра-
нить на другие ацетилены с электроноакцепторными заместите-
лями. Однако с Т. о. реакция протекает гораздо легче. Этот реа-
гент взаимодействует с метиловым эфиром пропиоловой кисло-
ты, диметиловым эфиром ацетилендикарбоновой кислоты, эти-
ловым эфиром фенилпропиоловой кислоты и гексафторбути-
ном-2, давая с хорошими выходами (55—90%) продукты типа
(2).
Ri-C=CR2 + (C$H6)3As=C
(C^HjJjAs— О
R'C—C—R2
^(C6H5)3As-О
R1C=C-R2
zCOR!
> (C6Hp3As = C
XR'
(2)
Окись трифенилстибина (СбНе)з5Ь=О реагирует с дициана-
цетиленом при комнатной температуре, но чистых продуктов при
этом выделить не удалось. Реакция с метиловым эфиром про-
пиоловой кислоты при 115° дает метиловый эфир феиилпропио-
ловой кислоты с выходом 40%.
1. С i g a n е k Е., J. Org. Chem., 35, 1725 (1970).
ТРИФЕНИЛМЕТИЛБОРФТОРИД (III, 399—401; V, 442—
443).
Усовершенствованный метод получения [1]. Т. можно легко
и просто получить реакцией трифеиилхлорметана [2] с комплек-
сом безводной борфтористоводородной кислоты с диметиловым
эфиром в сухом бензоле:
сеНв +
(С6Н5)3СС1 + HBF4 • О(СН3)2 (CsH5)3CBF4' + НС1 + (СН3)2О
73%
Из безводной фторсурьмяной кислоты и комплекса эфира с без-
водной гексафторофосфорной кислотой можно аналогичным об-
разом получить трифеиилметилгексафторантимонат (C6H5)3CSbF6
и трифенилметилгексафторфосфат (С6Н5)3СРРб соответственно.
Перекись эргостеринацетата [3]. При облучении (~10 мин)
эргостеринацетата (1) вольфрамовой лампой (500 вт) на воз-
духе или в чистом кислороде при —78° в присутствии каталити-
ческих количеств Т. образуется перекись эргостеринацетата (2)
с количественным выходом. Фотоокисление при более высоких
534
температурах не дает индивидуальных продуктов.
В тех же условиях из а-терпинена с высоким выходом обра-
зуется .аскаридол.
Относительно возможного механизма этого катализируемого
фотоокисления см. оригинальную статью.
Окисление ацеталей кетонов и простых эфиров [4]. Из эти-
ленацеталей обработкой Т. в сухом хлористом метилене (N2)
при комнатной температуре можно регенерировать кетоны. Так,
например, реакция Т. с этиленацеталем циклогексанона приво-
дит к образованию циклогексанона (80%) и трифенилметана.
Реакция, таким образом, включает гидридный перенос на ионы
трифенилкарбония. Для этой цели можно использовать также
борфторид триэтилоксония, однако последний несколько менее
эффективен, чем Т.
Реакция с ацеталем (1) приводит к образованию циклогек-
санона и бензоина (выход 64%). Судя по полученным резуль-
татам, эту реакцию можно использовать для окисления диолов
в виде их ацетоновых производных или других ацеталей. И дей-
с6н3 с6н3
(П
(с6н5)3свк4“
------------>
с6н5с-с—ед
о он
ствительно, ацетонид 2р,3р-диоксихолестана (2) при действии Т.
дает Зр-оксихолестанон-2 (3) с выходом 79%.
535
Целый ряд экспериментальных данных свидетельствует в
пользу следующей схемы окисления:
Реакция оказалась пригодной для получения кстосахаров.
Так, например, при взаимодействии Т. с ацетоновым производ-
ным D-маннита (4) образуется с выходом 65% соответствующий
кетосахар (5).
CH2OBz СНгОВх
BzO----- EzO-----
CHjOBz СНгОВи
(4) (5)
Окислительное расщепление бензиловых эфиров с помощью
Т. может быть использовано как для получения ароматических
альдегидов, так и для расщепления бензиловых эфиров спиртов.
Примеры:
С6НБСН2ОСН3 --> СВН5СНО
7Ь%
С6Н5СН2О Холестерин —> Холестерин + А4-Холестеной
60% го %
1. Olah G. A., Svoboda J. J., Olah J. A., Synthesis, 1972, 544.
2. Bachman W. Е., Org. Syn., Coll. Vol., 3, 842 (1955).
3. Barton D. H, R., Leclerc G., Magnus P. D., Menzies I. D.t Org.
Syn., 1972, 447.
4. В a r t о n D. H. R., Magnus P. D., Smith G., S t r ecker t G., Z u r r D..
J. C. S. Perkin I, 1972, 542.
ТРИФЕНИЛМЕТИЛГЕКСАФТОРФОСФАТ, (С6Н5)3С*РРб.
Мол. вес 388,28.
Синтез альдегидов и кетонов [1]. Трифенилметиловые эфиры
[их легко получить с высоким выходом из трифенилхлорметана
(HI, 417—419) и спиртов] диспропорционируют на трифенилме-
тан и альдегиды пли кетоны в присутствии небольших количеств
солей трифенил мети ла: Т., трифен ил метил гексафтор антимона-
536
та (С6Н5)3С SbF6-
(C6H5)3C+AsF6-.
или три фенил метилгекса фтор арсената
zR1
(С6н6)3сосеГ
\R2
R\
—> (С6Н5)3СН+ чс=о
R2/
Согласно обычной методике, трифенилметилбензиловый эфир
(5,0 жмоля) добавляют при перемешивании к раствору соли
трифенилметила (0,50 жмоля) в ацетонитриле. Через 4 час при-
ливают воду и выделяют продукты реакции. Бензальдегид и
трифенилметан получаются с количественными выходами. Таким
образом, эта реакция может служить прекрасным методом окис-
ления спиртов до альдегидов и кетонов.
Реакция, вероятно, представляет собой катионный цепной
процесс, включающий гидридный перенос. И действительно,
было найдено, что при окислении трифенилметилбензилового
эфира стадией, лимитирующей скорость реакции, является ста-
дия продолжения цепи:
(cshs)3coch2csh6 + (с6н5)3с+ —> (с6н5)3с+ + едено + (еднен
1. Doyle М. Р., DeBruyn D. J., Scholten D. J., J. Org. Chem., 38, 625
(1973).
ТРИФЕНИЛМЕТИЛЛИТИЙ (III, 396; V, 443).
Еноляты металлов в качестве защитных групп кетонов. Бар-
тон и сотр. [1] осуществили селективное восстановление 11-кето-
группы преднизона BMD (1), превратив с помощью Т. 3-кето-
группу в енолят металла (2). Восстановление 11-кетогруппы in
situ алюмогидридом лития дает с хорошим выходом преднизо-
лон BMD. Можно использовать также бцс-триметилсилиламид
натрия или лития [2], однако выходы в этих случаях ниже. Про-
толиз Т. сопровождается заметным изменением окраски, что яв-
ляется дополнительным преимуществом данного реагента. Этот
537
метод селективной защиты представляет интерес в случае неко-
торых кетостероидов. Реакции обычно проводят при —78° в ат-
мосфере аргона в ТГФ, используя 4—10 экв активного водорода
алюмогидрида лития на моль субстрата.
1. Barton D. Н. R., Hesse R. Н., Р е с h е t М. М., Wiltshire С., Chem.
Comm., 1972, 1017.
2. Wannagat U., Niederpriim H., Chem. Ber., 94, 1540 (1961).
ТРИФЕНИЛСТАННАН (HI, 401—402; V, 444—445; VI, 280).
Восстановление (VI, 280). При кипячении кетона (1) с Т. в
толуоле (4 час) можно избирательно восстановить в нем двой-
ную связь, находящуюся в а,(3-положеиии, получив при этом
соответствующий кетон (2) с выходом 81 % [1]:
1. Yamasaki М., J. С. S. Chem. Comm., 1972, 606.
ТРИФЕНИЛФОСФИН (III, 402—413; V, 445—447; VI, 280—
283).
Восстановление гидроперекиси аллоцимена (1) до оцимено-
вого спирта (2) [1].
Синтез полициклических углеводородов внутримолекулярным
С-алкилированием фосфиналкиленов [2]. Ароматическое соеди-
нение типа (1) реакцией с 1 молем Т. можно превратить в мо-
нофосфониевую соль (2), которая при действии основания дает
соответствующий фосфоран (5) :
538
>СНгВг
Ar
\(СНг)дСНгВг
Р( CfeH5)^ Аг СН2₽(С6нз)з основан^
'''pCHj^CHj Вг
(1) W
Н
С~Р(С6Н5)3
Аг
*** (СНг)л—СН2Вг
хсн
Ar-C X СНг
(снг)„
(3)
(4)
JWj
Ar^ СН2
(CHa)/j
(5)
J, RCH°
HCR
II
,С
Аг ^СИ,
(6)
Реакция с р-хлорвииилкетоиами [3],
(С6Н5)3Р + С1СН=СНСОСНз —+ 1(СвН8)зР—СН=СНСОСН3]СГ
Изонитрилы [4]. Наиболее часто применявшийся до сих пор
метод получения изонитрилов заключается в дегидратации фор-
мамида под действием хлорокиси фосфора в присутствии пири-
дина или трет-бутилата калия (I, 163—164). Недавно была опу-
RNHCHO+(CeH5)3P + CC14 + (C2H6)3N —> R—N^C: +
+ (CSH5)3PO + НСС13 + (C2Hs)3N • НС1
бликована методика, в которой для этой цели применяют трой-
ную систему: трифенилфосфин — четыреххлористый углерод —
триэтиламин. При использовании 20%-ного избытка трифенил-
фосфина изонитрилы образуются с выходом приблизительно
90% Растворителями служат 1,2-дихлорэтан, хлористый мети-
лен или хлороформ.
Дибромкетеи [5]. При взаимодействии триметилсилилтрибром-
ацетата (1) с Т. в присутствии избытка циклопентадиена с вы-
ходом 89% образуется продукт циклоприсоединения 7,7-дибром-
бицикло-[3,2,0]-гептен-2-он-6 (2). Эфир (1) получают с выходом
539
75% реакцией гексаметилдисилазана с трибромуксусной кисло-
той.
(CH3)3SiBr + (C6HS)3PO +
82% 87%
(21 89%
Олефинирование по Виттигу [6]. Бициклические амиды с
атомом азота в голове моста (1) и (2) взаимодействуют с (карб-
этоксиметилен) -трифенилфосфораном или (цианметилен) -три-
фенилфосфораном с образованием моио- и диолефинов. Реакция
направляется преимущественно по карбонильной группе в пяти-
членном кольце.
R = СОгСгН5
R-CN
Синтез олефинов методом «двойного элиминирования» (VI,
282—283). Опубликована первая статья из серии работ, посвя-
щенных этому новому методу синтеза олефинов [7].
1. von Gustorf Е. К., Greve)s F.-W., Schenck G. О., Ann., 719, 1
(1968).
2. Bestmann H. J., Hartl R„ Haberlein H., Ann., 718, 33 (1968).
3, Z b i r a 1 E., Werner E., Ann., 707, 130 (1967).
4. A p p e 1 R„ Kleinstiick R., Ziehn K.-D., Angew. Chem., Internal. Ed.,
10, 132 (1971).
5. Okada T., Okawara R., Tetrahedron Letters, 2801 (1971).
6. Flitsch W., Milter B., Chem. Ber., 104, 2852 (1971).
7. В a г to n D. H. R., Will is B. J„ J. C. S. Perkin I, 1972, 305.
540
ТРИФЕНИЛФОСФИН - АЗОДИКАРБОНОВОЙ КИСЛО-
ТЫ ДИЭТИЛОВЫЙ ЭФИР (I, 13—15).
Избирательное фосфорилирование 5'-гидроксильных групп
нуклеозидов [1]. Японские химики осуществили избирательное
фосфорилирование б'-гидроксильной группы нуклеозидов тими-
дина и уридина действием кислого дибензилфосфата в присут-
он он
Т ими дим
У ридин
ствии диэтилового эфира азодикарбоновой кислоты и трифенил-
фосфииа. Авторы полагают, что реакция включает стадию обра-
зования четвертичных фосфониевых солей. Селективность фос-
форилирования, направляющегося лишь по первичной гидрок-
сильном группе, авторы связывают с большим объемом проме-
жуточной соли (а).
о о о
11 II , - 11
C2H5OG-N=N-COC,HS * (C6HS)3P ----> CzHsOC=N-N-COC2H^
+
^Р(С6Н5)Э
г о
]|
C3H5OC=N-NHCOC2H3
—-----> + -°
ЧР(СЙ)3 ОР(ОСН2СгН5)г
(едсВДгРОН
о о о
" “ II , , II н н II
(С6Н5)зРОВ. О-Р(ОСНгС6Н3)2 + СгН5ОС-Ы-Ы-СОС3Н3
| о о
(С6Н5)гР~О + НОР(ОСН2С6Н3). —> ДОР(ОН)3
Тимидин-б'-фосфат был получен по этой методике с выходом
47%, уридин-б'-фосфат—с выходом 63%.
Превращение спиртов в амииы [2]. Реакция фталимида (1)
с различными спиртами в присутствии Т.— а.к. д. э. приводит к
образованию N-алкилфталимидов (2) с выходом 60—90%. Так
как последние под действием гидразиигидрата (I, 207—208) пре-
541
вращаются в амины, эта реакция может служить методом пре-
вращения спиртов в амины.
(1)
CjHjOOGN Н — MHCOOC£HS +
(CtHs)3P=O +
H;NNH
RNHj
(2)
Использование в данной реакции (S)-(+)-октанола-2 высо-
кой оптической активности приводит к образованию (R)
октиламина-2, также обладающего высокой оптической актив-
ностью. Таким образом, реакция протекает с почти полным об-
ращением конфигурации.
Н
/
НО-С^СНз
* с6н13
н
> CH3e^C-NH2
СбН15
(S)-(+) (Ц)-(-)
Изоцианнды. Уги и сотр. [3] пытались разработать метод
синтеза изонитрилов (2) путем дегидратации N-монозамещенных
формамидов (1) действием Т.— а. к. д. э. В шести из исследован-
ных случаев авторам не удалось получить изонитрилы. В осталь-
ных пяти случаях выходы изонитрилов колебались в пределах
RNHCHO —-> RN=C:
— Н20
(1) (2)
от 25 до 58%. Метод, однако, представляет некоторую ценность
для получения таких изоиитрилов, которые рацемизуются или
разлагаются в щелочной среде.
Алкилирование соединений, содержащих активный водород.
Японские химики [4] описали несколько примеров конденсации
542
спиртов с соединениями, содержащими активный водород, про-
текающей с отщеплением воды под действием Т, — а. к.д. э. Вы-
дон + сн^ + сгн5ос-м— n-coc2h5 + (С6Н5)зР----->
XY
X = Y = CN
X = CN; Y = СООСгН5
X - CH3CO; Y = СООСгН5
О н н О
►-------> r-сн + сгн5ос-А - А- сосгн5 + (Сьн5)3р=о
XY
ходы умеренные. Реакцию применяли и для алкилирования
амидов.
1. Mitsunobu О., Kato К., Kimura J., J. Am. Chem. Soc., 91, 6510
(1969).
2. Mitsunobu О., Wada M., S a n о T., J. Am. Chem. Soc., 94, 679 (1972).
3. Beijer B., von H i n r i c h s E„ U g i I., Angew. Chem., 11, 929 (1972).
4. Wada M., Mitsunobu O., Tetrahedron Letters, 1279 (1972).
ТРИФЕНИЛФОСФИН - N-ГАЛОГЕНСУКЦИНИМИД.
OH —>X [1]. Оксигруппы углеводов можно с высокими вы-
ходами заместить на галоген (бром, хлор, иод) под действием
Т. — г. в ДМФА. Для замещения одного гидроксила требуется
по 2 же N-галогенсукцинимнда и трифенилфосфина, которые
превращаются при этом в сукцинимид и окись трифенилфос-
фина. При этом первичные гидроксильные группы можно изби-
рательно заместить галогеном в присутствии вторичных.
I. Hanessian S., Ponpipom М. М., Lavalle Р., Carbohydrate Res.,
24, 45 (1972).
ТРИФЕНИЛ ФОСФИНДИБРОМИД (III, 413—415; V, 447;
VI, 283—285).
Карбодиимнды [1]. При взаимодействии Т. с Ы,Кт-дизамещен-
ными мочевинами в присутствии триэтиламина образуются кар-
бодиимиды:
НОН
+ I II I
[(CGH5)3PBr]Br~ + RN—С—N—R'
-(СвН5)3Р-Вг
О
—► r—n=c=N—R' 4- (С6Н5)3Р=О
543
Бензоины -> бензилы [2]. Бензоины при обработке Т. в аце-
тонитриле окисляются с высоким выходом (75—98%) в бен-
зилы:
(СвН5)зРВг21 ch3cn
АгСН—САг -------------------* АгС—САг
1. Bestmann Н. J., Lienert J., Mott L„ Ann., 718, 24 (1968).
2. Ho T.-L., Synthesis, 1972, 697.
б«с-(ТРИФЕНИЛФОСФИН)-НИКЕЛЬ(П)ДИЦИАНИД,
[(C6H5)3P]2Ni(CN)2 (1). Мол. вес 635,29, т. пл. 212° (разл.).
Получение [1]. Комплекс получают с выходом 62% кипяче-
нием цианида никеля с трифеиилфосфииом в этаноле в течение
48 час. Этот комплекс подобен бис-(трифенилфосфин)-никельди-
карбоиилу (III, 415), однако более устойчив на воздухе и более
безопасен в обращении.
Катализатор Дильса — Альдера [1]. Т. катализирует гомо-
конденсацию Дильса—Альдера винильных соединений с нор-
бориадиеном. Так, например, при взаимодействии акрилони-
трила с норборнадиеном в присутствии этого комплекса с вы-
ходом 93% образуется в виде смеси эпимеров 8-циантетрацикло-
[4,3,0,02’403’7]-нонан (2). (Для циклического углеводорода, произ-
водным которого является цианид (2), предложено название
дельтациклан [2].) Если в качестве катализатора использовать
бис-(трифенилфосфин)-никельдикарбонил, выход (2) составляет
86%.
120% 15 час
93^
(2)
Т. катализирует также присоединение алкинов к норборна-
диену, но эта реакция протекает с несколько более низкими вы-
ходами, чем присоединение винильных соединений.
+ снгоосс=ссоосн3
(1)
120”
32%
НзСООС СООСН;
(3)
544
1. Schrauzer G. N., Glockner P., Chem. Ber., 97, 2451 (1964).
2. Freeman P. K., Balls D. M., В 1 a z e v i c h J. N., J. Am. Chem. Soc.,
92, 2051 (1970).
б«с-(ТРИФЕНИЛФОСФИН)-КОБАЛЬТ(П)ДИБРОМИД,
CoBr2-2P(C6H5)3 (cm. VI, 137).
Димеризация норборнадиена в биснор-S (V, 240—241; VI,
137) [1]. Димеризацию норборнадиена в бисиор-S, катализируе-
мую Т. в сочетании с эфиратом трифторида бора, можно осу-
ществить с выходом 8О°/о, если тщательно контролировать тем-
пературу в начале реакции (реакция экзотермическая).
I, Courtney Т., Johnston D. Е., McKervey М. A., Rooney J. J.,
J. С. S. Perkin 1, 1972, 2691.
б«с-(ТРИФЕНИЛФОСФИН)-НИКЕЛЬ(0), [(C6H5)3P]2Ni(0).
Мол. вес 295,00, красно-бурые кристаллы, чувствительные к дей-
ствию воздуха.
Реагент получают восстановлением бис- (трифенилфосфип) -
никельдибромида амальгамой натрия в ацетонитриле или бен-
золе.
Циклоолигомеризация аллена [1] (VI, 288—289). Т. катали-
зирует превращение аллена в смесь изомерных тримеров, тетра-
мера, пентамера, изомерных гексамеров и полимеров. В реакции
первоначально образуется [(СбН5)3Р]2М1*СзН4, который можно
выделить.
Алкенкарбонаты [2]. При нагревании окиси этилена с дву-
окисью углерода в бензольном растворе при 100° в присутствии
Т, образуется этиленкарбоиат (I). В тех же условиях 2-метил-
1,2-эпоксипропан превращается в 1,1-диметилэтиленкарбоиат, а
2,3-эпоксибутан — в 1,2-диметилэтиленкарбоиат. Эффективными
катализаторами этой реакции являются и некоторые другие
комплексы типа L2Ni(0).
о
СН2ч Катализатор г \ л
) о + сог -------> I )=о
сн/ о
(1)
1. D е Р a s q u а 1 е R. J., J. Organometallic Chem., 32, 381 (1971).
2. D е Р a s q u а 1 е R. J., J. С. S. Chem. Comm., 1973, 157.
трцсДТРИФЕНИЛ ФОСФИН)-РОДИЙ (1)КАРБОНИЛ ГИД-
РИД, HRh(CO)[(C6H5)3P]3 (1). Мол. вес 918,77.
Тризамещенные олефины [1]. Т. стереоспецифически присо-
единяется к диметиловому эфиру ацетилендикарбоновой кис-
лоты (ДАДК) с образованием цмс-винилродиевого (1) соедине-
18 Зак. 59S
645
ния (2), которое при взаимодействии с Иодистым метилом без
растворителя при 25° (30 мин.) дает диметиловый эфир [иодкар-
бонил-бис- (трифенилфосфии) -метилродий (Ш)]-малеиновой кис-
лоты (3). Для того чтобы сохранить конфигурацию у двойной
связи при образовании новой С—С-связи, соединение (3) поме-
(1) 4- ДАДН
ь
СН3ООСЧ J
C-RHCO
CHjOOC-c/ L —
хн
СН31
88%
(2)Д« [(С6Н5)3р]
,сх
СН3ООС н
Н3СООС
Н3СОО<Д /СН3
с
> II
85Z H3COOCZ хн
Й) >98%
Н3СООС /СН3
f ZL
+ с + /Е<
HZ >оосн3 L с
(5) <2% (6)
щают в сосуд, вакуумированный до остаточного давления
8-10~2лш и заранее нагретый до 115°. При этом образуются ди-
метиловые эфиры цитраконовой (4) и мезаконовой (5) кислот
с выходами, указанными выше на схеме. Если соединение (3),
находящееся в вакуумированном сосуде, нагревать медленно до
115°, то эфир (5) получается с выходом >75%, а эфир (4) —
с выходом <25%. Переходный металл регенерируется в виде
соединения IRh (СО)[(С6Н5)зР]2.
Металлоорганическое соединение (1) реагирует по приведен-
ной выше схеме и с этиловым эфиром тетроловой кислоты, об-
разуя этиловый эфир р,р-диметилакриловой кислоты (7).
2) СНз1(С0’4 СН. /СООС2Нд
СН3С=С—СООС2Н5 --------->- /С- СС
сн/ Хн
(7)
1. Schwartz J., Hart D. W-, Holden J. L., J. Am. Chem. Soc., 94, 9269
(1972).
трис-(ТРИФЕНИЛФОСФИН)-РОДИЙХЛОРИД (HI, 415; V,
448—454; VI, 286—291).
Декарбоннлирование (V, 452; VI, 289—290). Недавно опуб-
ликована заключительная статья Вальборского и Аллена [1] по
стереохимии этой реакции. Авторы пришли к заключению, что
546
в реакции образуется радикальная пара:
/Р(С6Н;)3
------------>
CZ ХР(С6Н5)3
О
р(ад3
Р(С6Н5)3
Радикальная пара
> RH
(c6hs)3pzZ \р(с6н5)3
Десульфоиилирование [2]. Т. катализирует десул ьфонилиро-
вание аренсульфохлоридов и ареисульфобромидов, осуществляе-
мое с выходом 70—90% путем перегонки (в атмосфере азота
или на воздухе) либо при нагревании в гексахлорбензоле:
ArSO2Cl(Br) —> ArCl(Br) + SO2
Для той же цели был опробован целый ряд комплексов ири-
дия, рутения, платины и палладия, однако десульфонилирова-
ние происходит с наиболее высоким выходом при катализе ро-
диевым комплексом. Полагают, что оно протекает через образо-
вание металлсульфинатного комплекса, который затем теряет
SO2.
Циклизация. Обработка у,6-ненасыщенного альдегида, содер-
жащего в молекуле систему 4-ен-1-аль(1), родиевым комплексом
(1 экв) в хлороформе, бензоле или ацетонитриле при комнатной
спо
.(1)
“ " .........
температуре приводит к 2,3-диал кил циклопентанону (2) и про-
изводному циклопропана (3), образующемуся в результате де-
карбонилирования. Оба этих продукта получаются с практиче-
ски одинаковыми выходами (20—35%). Обработка альдегида (1)
SnCl4 в нитрометане приводит исключительно к циклопентанону
(2) с выходом 20-~55 % [3].
Иного типа циклизация под действием родиевого комплекса
наблюдается в случае соединений, содержащих систему 6-ен-1-
аль [4]. Так, например, реакция (+)-цитронеллаля (4) с 1 экв Т.
в свежеперегнапном хлороформе при комнатной температуре
приводит к образованию (-р)-неоизопулегола (5) и (—)-изопу-
18*
547
легола (6) в отношении 3: 1.
сн-
[(ОДЬрЗзИЪС!
55%
Н3СХ ^снг
3:1
(4) (5) (6)
Флуоренон из бензойного ангидрида [5]. Т. катализирует
превращение бензойного ангидрида в флуоренон. Реакция про-
текает при температуре около 240°.
г
f 2 СИССОН + СО
Гомогенное каталитическое гидрирование пространственно-
незатрудненных двойных связей [6]. В двугорлую колбу на
500 мл, в которой находится ротор магнитной мешалки и кото-
рая присоединена к прибору для гидрирования при атмосфер-
ном давлении, снабженному градуированной бюреткой для из-
мерения объема поглощенного водорода, помещают 0,9 г свеже»
н^с^ снэ
Нг, С6НЙ
[(С6Н5)3Р13Ш1С1
приготовленного Т. и 160 мл бензола, перегнанного иад гидри-
дом кальция. Одно горло колбы закрывают резиновым колпач-
ком и смесь энергично перемешивают до образования гомоген-
ного раствора. Затем систему заполняют водородом. В колбу
через резиновый колпачок с помощью шприца вводят 10 г
(0,066 моля) свежеперегнанного карвона (т. кип. 105—
106°/14 мм). Шприц ополаскивают двумя порциями бензола по
10 мл, вводя каждую из них в колбу, и возобновляют перемеши-
вание. При использовании свежеприготовленного катализатора
сразу начинается поглощение водорода, заканчивающееся через
3,5 час, когда поглотится теоретическое количество газа. Раствор
фильтруют через сухую колонку (диаметр 4 см) с флорисилом
(10/100 меш) (IV, 62—63). Колонку промывают 300 мл эфира
548
и объединенный элюат концентрируют при пониженном давле-
нии. Желтый остаток перегоняют с колонкой Вигре (II см) в
вакууме, получая 9,1 г (90%) дигидрокарвона с т. кип. 100—
102714 мм.
Избирательное гидрирование простагландинов [7]. Гидриро-
вание PGE2 (1) в присутствии Т. в смеси бензола н ацетона дает
главным образом PGEi (2) с выходом 50% - Наблюдаемая изби-
рательность восстановления 5,6-двойной связи по сравнению с
13,14-двойной связью, которая почти не затрагивается, никак не
связана с тем, что одна из них имеет цис-, а другая транс-кон-
фигурацию, поскольку соединение (3) гидрируется также с об-
разованием в основном продукта (2).. Такое же избирательное
гидрирование 5,6-двойной связи наблюдается и в других про-
стагландинах.
Гидрирование алленов [8]. Т. катализирует гидрирование
алленов до алкенов с умеренным выходом, причем преимуще-
ственно восстанавливается менее замещенная двойная связь.
Реакция стереоселективна: там, где возможна изомеризация, об-
разуются цис-алкены.
Восстановление алкенов и алкинов [9]. Алкены и алкины
восстанавливаются до алканов смесью муравьиной кислоты и
формиата лития в присутствии Т. при 40—60°. В качестве ката-
лизаторов эффективны также трифенилфосфиновые комплексы
рутения и иридия.
Восстановление борфторидов диазония [10]. Т.[(С6Н5)3Р]3-RhCl,
а также [(C6Hs)3P]2RhCl(CO) катализируют восстановление бор-
фторидов арилдиазония с помощью ДМФА (комнатная темпера-
тура в течение 2 дней или 1 день при 80°):
ДМФА, Rh-комплекс
ArN+BF; -------------------* АгН
549
Алкоголиз диарилсилаиов [11]. Т. и трис- (трифенилфосфин) -
рутенийдихлорид являются эффективными катализаторами алко-
голиза диарилсилаиов:
/н
R‘SiH3+R2OH —> R’s/ +Н2
^OR2
Они также эффективно катализируют гидросилилирование
карбонильных соединений:
R\ yR2
R‘siH2 + )с=о —-> R'siOCH;
RSZ 2| \R.
ri
1. Walborsky H. M., Alien L. E., J. Am. Chem. Soc., 93, 5465 (1971).
2. Blum J., Tetrahedron Letters, 35, 3041 (1966); Blum J., Scharf G.,
J. Org. Chem., 35, 1895 (1970).
3. Sakai K., Ide J., Oda 0., Nakamura N., Tetrahedron Letters, 1287
(1972).
4. Sakai R., Oda 0., Tetrahedron Letters, 4375 (1972).
5. В 1 u m J., L i p s h e s Z., J. Org. Chem., 34, 3076 (1969).
6. I r e I a n d R. E., В e у P., Org. Syn., submitted 1972.
7. Lincoln F. H., Schneider W. P., Pike J. E., J. Org. Chem., 38,
951 (1973).
8. Bhagwat M. M., Dev a prabhakar a D., Tetrahedron Letters, 1391
(1972).
9. а) К о л о м н и к о в И. С., К у к о л е в В. П., Чернышев В. О., В о л ь-
п и н М. Е., Изв. АН СССР, Сер. хим., 1972, 693; б) Ко л о м н и ков И. С.,
Корешков Ю. Д., Куколев В. П., Мосин В. А., Вольпин М. Е.,
Изв. АН СССР, Сер. хим., 1973, 175.
10. Marx G. S., J. Org. Chem., 36, 1725 (1971).
11. Corriu R. J. P., Moreau J. J. E., J. C. S. Chem. Comm., 1973, 38.
трмс-(ТРИФЕНИЛФОСФИН)-РОДИЙХЛОРИД—ТРИ-
ЭТИЛСИЛАН.
Избирательное восстаиовлеиие а,р-ненасыщеиных карбо-
нильных соединений. Японские химики [1] осуществили изби-
рательное восстановление «.^-ненасыщенных карбонильных со-
единений в ряду терпенов с помощью триэтнлсилана, катализи-
руемое комплексом родия(1). Например, обработка а-ионона
|НЗ К2С0э-(СНз)2С=О
X^"osi( ед), снэон—н2о ;
CHj 965?, считая на (1)
СН,
(2)
CHS
-^о
550
(1) небольшим избытком триэтилсилана в присутствии катали-
тического количества трас- (трифенилфосфин) -родийхлорида в
атмосфере азота при 50° в течение 2 час приводит к силило-
вому эфиру енола (2), который гидролизуется смесью К2СО3—
ацетон — метанол — вода до дигидро-а-ионона (3) с общим вы-
ходом 96%. Аналогичным образом цитраль (4) восстанавли-
вается через силиловый эфир енола (5) до цитронеллаля (6) с
выходом 97%. Следует отметить, что в этих условиях комплекс
родия (I) не вызывает декарбонилирования (V, 452; VI, 289—
290).
(4) (5} (6)
При восстановлении триэтилсиланом в тех же условиях со-
пряженного диенона р-иоиона (7) образуются дигидро-р-ионон
(8) и спирт (9) в отношении 44 : 56. Было найдено, что на состав
продуктов существенное влияние оказывает природа R3S1H. При
использовании фенилдиметилсилана соединения (8) и (9) полу-
чаются в отношении 91:9. С другой стороны, восстановление
(7) диэтилсиланом или дифенилсиланом дает в качестве един-
ственного продукта спирт (9).
IjHSiR,.
[(C^P^RhCl
2) Гидролиз
(7) (8) (9)
1. Ojima I., Kogura Т., Nagai Y., Tetrahedron Leiters, 5085 (1972).
трмс-(ТРИФЕНИЛФОСФИН)-РУТЕНИЙДИХЛОРИД,
[(СбН5)3Р]3 RuCh- Мол. вес 959, 44.
Гомогенное каталитическое гидрирование с переносом во-
дорода. Сассон и Блюм [1] впервые применили этот металлоор-
ганический катализатор для переноса водорода от первичных
спиртов к «.^-ненасыщенным карбонильным соединениям. Так,
при нагревании бензилового спирта в атмосфере азота при 200°
в течение 2 час в присутствии бензальацетона и катализатора
551
образуются бензальдегид с выходом 90% и 4-фенилбутанон-2
[(C6I-I5)3P]3I?UC12
C6HSCH2OH + СсН6СН=СНСОС1-13 ---------------+
С6НВСНО + С6Н5СН2СН2СОСН3
с выходом 92%. Реакцию можно проводить в растворителе (ки-
пячение в толуоле, ксилоле или мезитилене), но ее скорость при
этом уменьшается. В качестве доноров водорода вместо спиртов
можно использовать гидроароматические соединения, однако в
этом случае требуется большая продолжительность реакции и
более высокие температуры. Реакция применима как для окис-
ления спиртов, так и для восстановления сс,р-ненасыщенных ке-
тонов. С альдегидами она протекает не так гладко.
Реген и Уайтсайдз [2] использовали этот метод для прямого
окисления вицинальных диолов в сс-дикетоны, проведение кото-
рого ранее было связано со значительными трудностями (см.,
например, [3]). Так, грс-циклододекандиол-1,2 (1) окисляется до
дикетона (2) с выходом 53%. Если реакцию прервать, не до-
(2)
ведя до конца, то в реакционной смеси можно обнаружить а-ок-
сициклододеканон. Следовательно, окисление протекает, веро-
ятно, в две стадии.
I. S a s s о n Y., В 1 u m J., Tetrahedron Letters, 2167 (1971).
2. Regen S. L., Whitesides G. M., J. Org. Chem., 37, 1832 (1972).
3. Newman M. S., Da vis С. C., J. Org. Chem., 32, 66 (1967).
ТРИФЕНИЛФОСФИНСЕЛЕНИД, (C6H5)3P = Se. Мол. вес
341,24, т. пл. 183—184°,
Получение [1]. Т. получают нагреванием трифенилфосфина
(3 моля) с селеном (1 моль).
Дезоксигенирование эпоксидов [2]. Реагент в сочетании с
трифторуксусной кислотой при комнатной температуре быстро
и стереоспецифично превращает эпоксиды в олефины с сохране-
Е52
нием стереохимии у С—С-связи эпоксида:
(C6H5)3P=Se
CF3COOH
нч /Н
с=с.
R1' R2
Примеры:
1,2-Эпоксиоктан —» Октен-1 (71%)
Окись ^ыс-стильбена —> цис-Стильбен (около 71%)
.Окись циклогексена —> Циклогексен (53%)
1. Michaelis A., v. Soden Н., Ann., 229, 295 (1885).
2. С 1 i v е D. L. J., D е п у е г С. V., J. С. S. Chem. Comm., 1973, 253.
ТРИФЕНИЛФОСФИН — УГЛЕРОД ЧЕТЫРЕХБРОМИС-
ТЫЙ (см. также Трифенилфосфин — углерод четыреххлористый).
RCHO ^RC=CH. Кори и Фукс [1] осуществили превраще-
ние RCHO -► RC = СН или RC = CRZ в две стадии:
RCHO —> RCH=CBr2 —> RC^CH или RC=CR'
(1) <2) (3) (4)
Для проведения первой стадии имеются две методики. По од-
ной из них альдегид (1 экв) прибавляют к смеси трифенилфос-
фина (4 экв) и четырехбромистого углерода (2 экв) в хлори-
стом метилене при 0°; продолжительность реакции 5 мин. Со-
гласно другой, более совершенной методике, сначала готовят
реагент взаимодействием цинковой пыли (2 экв), трифенилфос-
фина (2 экв) и четырехбромистого углерода (2 экв) в хлори-
стом метилене при комнатной температуре в течение 24—30 час.
К полученному реагенту прибавляют альдегид (1 экв) и остав-
ляют реакционную смесь на 1—2 час. Выходы дибромалкенов
по второй методике составляют 80—90%.
Полученные таким путем дибромолефины превращают в
ацетилениды лития действием н-бутиллития или тонко измель-
ченной 1,5%-ной амальгамы лития в ТГФ. Гидролиз ацетиле-
нида дает терминальный ацетилен (3), карбонизация ацетнле-
2BuLi H2O
RCH=CBr2 RCsCLi ------> RQsCH
(2) (3)
|со2
]К.-с со он
(4)
нида приводит к пропаргиловой кислоте (4). Реакция промежу-
точных ацетиленидов лития с другими электрофилами (алкил-
553-
галогенидами, альдегидами, кетонами) позволяет получить са-
мые разнообразные ацетилены.
I. Corey Е. J., Fuchs Р. L., Tetrahedron Letters, 3769 (1972).
ТРИФЕНИЛФОСФИН — УГЛЕРОД ЧЕТЫРЕХХЛОРИС-
ТЫЙ (III, 415—416; V, 455—456; VI, 291).
Реакция с еиолизующимися кетонами [1]. Трифенилфосфин
взаимодействует с циклогексаноном при кипячении в четырех-
хлористом углероде, образуя продукты (1) и (2) в отношении
93:7. Аналогичные продукты получены и в случае циклопента-
нона, только здесь в большем количестве образуется продукт
(4), соответствующий продукту (2). Дихлорметилеиовые аддук-
ты (2) и (4), вероятно, образуются через промежуточное сое-
динение (5), которое было идентифицировано как продукт ре-
акции трифенилфосфина с четыреххлористым углеродом [2].
2(СеН5)3Р 4-СС14 —> (С6Н5)3Р=СС124-(С6Не)3РС12.
(5) (6)
Реакция с эпоксидами [3]. При кипячении эпоксидов с Т.—у.
ч. в течение 1—2 час в атмосфере N2 образуются соответствую-
щие 1,2-дихлоралканы и окись трифенилфосфина. Так, напри-
мер, окись циклогексена превращается в ^ис-1,2-дихлорцнкло-
гексан с выходом 80%. При этом образуются лишь следы транс-
1,2-дихлорциклогексана.
Аллилхлориды. Аллиловые спирты с помощью Т. — у. ч.
можно превратить в аллилхлориды без (или почти без) пере-
группировки [4]:
СН3СН=СНСН2ОН СН3СН=СНСН>С1
СН3СНОНСН-=СН2 —СН3СНС1СН=СН2 + СН3СН=СНСН2С1
89% 11 %
Этот реагент рекомендуется для осуществления превраще-
ния аллиловых спиртов в аллилхлориды без перегруппировок,
$54
Так, например, из гераниола (1) можно получить геранилхло-
рид (2) с выходом 75—81 % [5].
+ (С6Н£)3РО + НСС13
Четыреххлористый углерод служит как растворителем, так
и источником галогена, трифенилфосфин желательно использо-
вать в небольшом избытке. По мнению Хуза, эта реакция про-
текает по 5м2-механизму
+ ROH + -
(СвН5)зР + CCU —> (С6Н5)3РС1СС13- ——(C6H5)3PC1OR —>
— <-<13
--> (CgHsJgPORCl ---> RC1 + (СбНд)зРО
Синтез карбодиимида [6]. При взаимодействии 1,3-дизаме-
щенных мочевин или тиомочевии с трифенилфосфином и четы-
реххлористым углеродом в присутствии триэтиламина в хлори-
стом метилене при 40° образуются карбодиимид с выходом
85—92%.
X
II ,
RNHCNHR + (С0Н5)3Р + ССЦ + (C2H5)3N —>
X=O, s
—> R—n=C=N—R' + (С6Н5)3Р=Х + НСС13 + (C2H5)3N • НС1
Пептидный синтез. Виланд и Зеелигер [7] использовали
Т.— у. ч. в сочетании с триэтиламином для получения пептидов
конденсацией БОК-аминокислот с эфирами аминокислот, но при
этом наблюдается очень сильная рацемизация.
Реакция с ацилглицеринами [8]. При кипячении 1,3-дисте-
ароилглицерииа (1) с Р(СбН5)3—СОЦ образуется 2-хлордезок-
си-1,3-дистеароилглицерин (2) с выходом 95%:
^HjOCOCxyHjj CH2OCOC17H3S
ноДн p(qhJ,-cc.4 > с I н
ЧНгОСОСцН,, СН2ОСОС,,Н„
W (2)
Нуклеофильное замещение у Q в ацилглицеринах (глицери-
дах) протекает, как правило, с миграцией ацилоксигруппы от
Ci или С3 к С2. Рассматриваемая реакция происходит без обра-
щения конфигурации у Cs-атома.
555
1. Isaacs N. S., Kirkpatrick D., Chem. Comm., 1972, 443.
2. Ramirez F., Desai N. B., McKelvie N., J. Am. Chem. Soc., 84, 1745
(1962).
3. Isaacs N, S., Kirkpatrick D.. Tetrahedron Letters, 3869 (1972).
4. Collington E. W., Meyers A. 1. M., J. Org. Chem., 36, 3044 (1971);
Snyder E. 1., J. Org. Chem., 37, 1466 (1972).
5. Calzada J. G., H о о z J., Org. Syn., submitted 1973.
6. Appel R., Kleinsttick R., Ziehn R.-D., Chem. Ber,, 104, 1335 (1971).
7. Wieland T., S e e 1 i g e г A,, Chem. Ber., 104, 3992 (1971).
8. Aneja R., Davies A. P., Rnaggs J. A., J. C. S. Chem. Comm., 1973,
110.
ТРИФЕНИЛФОСФИТ (III, 416; V, 456; VI, 291).
Методика иодирования A [1]. Настоящая методика, предло-
женная Райдоном, наиболее проста в экспериментальном отно-
шении и особенно удобна для получения иодидов из простран-
ственно затрудненных спиртов, например неопентилового спир-
та. Согласно этой методике, в двугорлую круглодонную колбу
на 500 мл, снабженную обратным холодильником с хлоркаль-
циевой трубкой, помещают 136 г (0,44 моля) Т., 35,2 г (0,4 мо-
ля) неопентилового спирта и 85 г (37 мл, 0,60 моля) йодистого
СН3
А) СН3ССН2ОН + СН31 + (С6Н5О)3Р —>
СНз
сн3
—> СНзССНг! + (CeHsO)2POCH3 + С6Н5ОН
Ан
СНз
метила. В другое горло колбы вставляют термометр так, чтобы
его шарик был погружен в жидкость. Смесь доводят до слабого
кипения, нагревая колбу электрическим колбонагревателем, и
кипятят ее до тех пор, пока температура жидкости в колбе не
поднимется от первоначального значения 75—80° приблизитель-
но до 130°, а смесь не потемнеет и не начнет дымить (около
24 час). Нагрев колбы необходимо регулировать, поскольку с
течением времени интенсивность кипения постепенно умень-
шается. За ходом реакции удобно следить с помощью ИК-спект-
роскопии. По мере протекания реакции широкая интенсивная
полоса поглощения при 865 см~[ с плечом при 880 см~1 исче-
зает и появляется другая широкая интенсивная полоса при
945 см~1 и узкая полоса средней интенсивности при 1310 см~1.
Перегонкой при пониженном давлении, промыванием водой и
затем холодным 1 н. раствором гидроокиси натрия для удале-
ния фенола, снова водой, высушиванием и повторной перегон-
кой получают 51—60 г (64—75%) неопентилиодида с т. кип.
54-55755 мм.
556
Методика иодирования Б. Эта методика рекомендуется в
случае чувствительных к кислотам спиртов, легко дегидра-
тирующихся с образованием олефинов. Она иллюстрируется на
примере получения циклогексилиодида. В двугорлую кругло-
донную колбу на 500 мл, снабженную обратным холодильником
с хлоркальциевой трубкой, помещают 124 г (107 мл, 0,4 моля)
Т. и 85 г (37 лы, 0,6 моля) йодистого метила, в другое горло
колбы вставляют термометр так, чтобы его шарик был погру-
жен в жидкость. Смесь доводят до слабого кипения с помощью
колбонагревателя и кипятят ее до тех пор, пока температура
внутри колбы не поднимется приблизительно до 120°, а смесь
Б) (С6Н5О)3Р + сн31 —> Е(с6н5о)3рсн3]1
h i
[(C6H5O)3PCH33l + [ 1 ----> (С6Н5О)гРОСН3 + Г | + С6Н5ОН
не станет темной и вязкой. Колбу охлаждают и к маслообраз-
ному иодметилату Т. добавляют 40 г (0,4 моля) циклогекса-
нола. Смесь осторожно встряхивают до образования однород-
ной массы и оставляют иа ~ 12 час при комнатной температу-
ре. За ходом реакции можно следить с помощью ИК-спектро-
скопии по исчезновению интенсивной широкой полосы поглоще-
ния при 1040 см~1 и появлению такой же полосы при 945 слг-1;
реакция завершается примерно через 6 час. Смесь перегоняют
с 13-сантиметровой колонкой Вигре, получая 62,5—63 г (74—
75%) циклогексилиодида с т. кип. 66—68°/12 мм, 1,5475.
Стероидные амиды. Херц и Мантекой [2] с удовлетворитель-
ным выходом получили с помощью Т. по методу Митина и Глин-
ской амиды 3-формокси-5р-холановой кислоты (VI, 291).
1. R у d о n Н. N., Org. Syn., 51, 44 (1971).
2. Herz J. E., Ma nt econ R. E., Org. Prep. Proc. Int., 4, 123 (1972).
ТРИФЕНИЛФОСФИТА ИОДМЕТИЛАТ (111,416—417).
Продажный препарат можно очистить промыванием этил-
ацетатом до получения янтарной окраски [1].
Получение. Ферхейден и Моффат [1] внесли некоторые усо-
вершенствования в первоначальную методику Ландауера и Рай-
дона (III, 416—417, [1]). Колбу со смесью трифенилфосфита
(0,2 моля) и йодистого метила (0,26 моля) помещают в нагре-
тую до 90° масляную баню и в течение 8 час повышают мед-
ленно ее температуру до 125°. Температура внутри колбы мед-
ленно поднимается от 70 до 85°, а затем быстро подскакивает
до 115°. Эту температуру поддерживают в течение 12—14 час.
557
После охлаждения и внесения затравки смесь закристаллизо-
вывается в твердую массу бурого цвета. Добавляют сухой эфир
и получившееся вещество несколько раз промывают деканта-
цией свежими порциями сухого этилацетата. Реагент получают
в виде янтарно-желтых кристаллов с выходом 90%. Работать
с реагентом следует в сухой камере в атмосфере азота.
ОН—» 1. В качестве растворителя при получении иодидов из
спиртов действием Т. и. Ферхейден и Моффат [1] рекомендуют
не обычно применяемый бензол, а ДМФА. В этом растворителе
реакция протекает при комнатной температуре и, как правило,
быстро. Холестанол под действием реагента в безводном ДМФА
в течение 2 час при 25° превращается в За-иодхолестан с вы-
ходом 57%. Взаимодействие реагента со спиртами, по-видимо-
(C6HsO)3P+CH3l" + ДОН > (C6HsO)2P^OR I -------> SI + (C6H5O)ZPCH3
сн3
му, начинается с нуклеофильной атаки по атому фосфора с от-
щеплением фенола и образованием соли алкоксифосфония, ко-
торая затем распадается на иодистый алкил и дифенилметил-
фосфонат. Этот предполагаемый механизм находится в согласии
с наблюдаемым в реакции обращением конфигурации.
Ферхейден н Моффат [1] применили раствор реагента в
ДМФА для замещения на иод первичных гидроксильных групп
нуклеозидов. Например, реакция 2',3'-0-изопропилиденуриднна
(1а) с 2 экв Т. и. в ДМФА в течение 15 мин приводит к б'-дезок-
си-5'-иод-2',3'-0-изопропилиденуридииу (16) с выходом 96,5%’.
С помощью раствора Т. и. в ДМФА удается также осуществить
избирательное замещение на иод только первичной гидроксиль-
ной группы. Так, непродолжительная обработка тимидина (2а)
«) R = I
он он
(2)
a) R = ОН
б) К=1
1,1 экв реагента дает с выходом 63% 5'-дезокси-5'-иодтими-
дин (26).
558
Дегидратация вторичных спиртов. Хатчинс и сотр. [2] пред-
приняли попытку превратить транс-4-трет-бутилциклогексанол в
соответствующий ^иоиодид, действуя на него раствором Т. и. в
ГМТФК при комнатной температуре. Вместо этого они получи-
ли с выходом 88% 4-трет-бутилциклогексен. Впоследствии ав-
торы нашли, что вторичные спирты под действием двукратного
избытка Т. и. в ГМТФК при 25—75°, как правило, дегидрати-
руются в течение 0,25—25 час, первичные спирты в этих усло-
виях с превосходными выходами превращаются в соответствую-
щие иодиды, а третичные спирты практически не вступают в ре-
акцию. Дегидратация в этом случае, по-видимому, осуществля-
ется путем первоначального образования соответствующего
иодида с обращенной конфигурацией и последующего его де-
гидрогалогенирования, индуцируемого иодид-ионом и ГМТФК
(см. V, 79—80). В большинстве случаев образование более
устойчивого алкена по Зайцеву значительно преобладает над
образованием алкена по Гофману.
1. Verheyden J. Р. Н., Moffatt J. G., J. Org. Chem., 35, 2319 (1970).
2. Hutchins R. О., Hutchins M. G., Milewski C. A., J. Org. Chem.,
37, 4190 (1972).
ТРИФЕНИЛФОСФИТА ОЗОНИД (VI, 291—293).
Окисление дисульфидов [1]. Простые диалкилдисульфиды
окисляются Т. б. при —78° вначале с образованием смеси тиол-
сульфинатов и тиолсульфонатов в отношении приблизительно
10 : 1. После 5—6-дневного стояния при комнатной температуре
отношение продуктов становится равным примерно 1 : 10 с пре-
обладанием тиолсульфоната. Окисление может быть вызвано
о о
II II
RSSR + (C6H5O)3PO3 —> RSSR + RSSR
II
О
действием синглетного кислорода, выделяющегося из озонида,
хотя при —78° озонид не разлагается.
1. Murray R. W., Smetana R. D„ Block E., Tetrahedron Letters, 299
(1971).
ТРЙФЕНИЛХЛОРМЕТАН (III, 417—419; V, 456—457).
Ацетаты хлоргидринов. Ньюмен и Чен [1] предложили удоб-
ный двухстадийный метод превращения 1,2- или 1,3-гликолей
в ацетаты хлоргидринов. Сначала гликоль действием триметн-
лового эфира ортоуксусной кислоты в присутствии кислых ка-
тализаторов (бензойной или хлоруксусной кислот) переводят
в циклический ортоэфир с выходом 80—90%. Циклический ор-
559
RCHcHGHOHR
СН3С(ОСН3)з
н
-2 СН30н"^"
н3с осн3
с
о хо
) I
RCH—CHR
(С6НЁ)3СС1
С1 о сосн3
•-----RCH-CHR + (С6Н5)ЭСОСН3
тоэфир кипятят затем с Т. (1 же) в хлористом метилене в те-
чение 1—2 час. Ацетаты хлоргидринов получают с выходом
около 85—95%* Согласно высказанному Ньюменом предполо-
жению, трифенилметильный катион атакует метоксигруппу орто-
эфира, давая амбидентный катион (а), который затем реагирует
с Т. или ионом хлора с образованием конечного продукта.
В случае 1,4-диолов этот метод дает низкие выходы.
R R
НС-0 ,СН3 НС-0
I V > |+ -Jc-CH3 + (С6Н5)3СОСНЛ
НС-О ОСН3 HC-OZ
R R
(а)
(СЬН5)3СС1
С1 ОСОСН3
11 +
RCH-—CHR + (С6Н53С
I. Newman М. S., Chen С. Н., J. Am. Chem. Soc., 95, 278 (1973).
ТРИФТОРМЕТАНСУЛ ЬФОКИСЛ ОТА, CF3SO3H. Мол. вес
150,09.
Т. является самой сильной из известных кислот, на воздухе
дымит, С этой кислотой следует обращаться с величайшей осто-
рожностью.
Вииилтрифторметансульфонаты (трифлаты) [1]. Существуют
два метода получения винилтрифлатов. Один из них заключает-
ся во взаимодействии ацетилена с кислотой при низких темпе-
ратурах:
CH3C^CCH3 + CF3SO3H --> CH3CH=C(CH3)OS02CF3
55%
цис и транс
Согласно другому, кетон обрабатывают ангидридом три-
фторметансульфокислоты, полученным перегонкой кислоты над
большим избытком Р2О5 (т. кип. 83—85°):
(СНз)2СНСОСНз + (CF3SO2)2O —(CH3)2C=C(CH3)0SO2CF3,
Oq 70
560
Трифлаты широко применяются в изучении сольволиза,
1. Stang Р. J., J. Am, Chem. Soc,, 91, 4600 (1969); Org, Syn., submitted
1972; Du eb er T. E. et al., Angew. Chem., Internal. Ed., 9, 521 (1970);
Stang P. J., D ueber T. E., Org, Syn., submitted 1972.
ТРИФТОРМЕТАИСУЛЬФОКИСЛОТЫ АНГИДРИД,
(CF3SO2)2O. Мол. вес 266,15, т. кип. 84°.
Получение [1]. К 32,1 г трифторметаисульфокислоты, охлаж-
денной до 0°, тремя порциями прибавляют 25 а Р2О5 и отгоняют
Т. а., постепенно повышая температуру реакционной смеси до
110° (температура бани) в течение 1 час. Фракцию, кипящую в
интервале 80—100° (760 жж), для удаления следов кислоты пе-
регоняют над ~8 г P2Og. Перегонку повторяют до тех пор, пока
(1) + (cf3so2)2o
hc=cch2ch2oso2Cf3
1) CF3CO2H + CF3CO2Na
2) Н2О
стеклянная палочка, смоченная дистиллятом, не перестанет ды-
мить на воздухе. Выход ангидрида 25 г (83%), т. кип. 84°.
Синтез циклобутаноиа [2]. Синтез циклобутанона осуще-
ствляют в три стадии, как показано на вышеприведенной схеме.
На последней стадии требуется нагревание при 60—65° и пере-
мешивание в течение 7—8 дней.
1. Burdon J., Far azmand 1., Stacey M., T a 11 о w J. C., J. Chem. Soc.,
1957, 2574.
2. H a n a c k M., Dehesch T., Hummel K., N i e г t h A., Org. Syn., sub-
mitted 1971.
ТРИФТОРМЕТАНСУЛ ЬФОНОВОЙ И КАРБОНОВЫХ
КИСЛОТ СМЕШАННЫЕ АНГИДРИДЫ, CF3SO2—О—
Получение [1]. Реагенты получают с выходами 80—100% при
взаимодействии серебряной соли трифторметаисульфокислоты
с хлорангидридами кислот:
561
Ацилирование [1]. Эти ангидриды — сильные ацилирующие
агенты. Так, неактивированные арены, такие, как, например,
бензол, легко ацилируются без добавления катализаторов реак-
ции Фриделя — Крафтса:
Z0
с6н6+ CF3SO2—ОС^
хсен5
60°, 5 час
90%
CcH5COCsH5
Трифторметансульфокислоту можно при этом почти количест-
венно регенерировать в виде бариевой соли.
Смешанные ангидриды могут образоваться также in situ в
реакции хлорангидридов кислот с аренами, катализируемой
трифторметансульфокислотой [2]:
RC^ +
xci
CF3SO3H
------->
1. Effenberger F., Epple G., Angew. Chem., Internal. Ed., 11, 299 (1972).
2. Effenberger F., Epple G., Angew. Chem., Internal. Ed., 11, 300 (1972),
ТРИФТОРМЕТИЛ ГИПОФТОРИТ (V, 457—458; VI, 293—
294).
Фторированные стероиды. Действие T. на еполацетаты 3-ке-
то-Д4- и 3-кето-Д4-6-стероидов приводит к получению производ-
ных, фторированных в положения 6 н 2 соответственно [1]:
1. Chavis С., Mousseron-Canet М., Bull. Soc. Chim. France, 1971, 632.
ТРИФТОРНАДУКСУСНАЯ КИСЛОТА (III, 422—431; V,
458—459; VI, 294).
Т. к. — бора трифторид (III, 429—430; V, 459). Харт [1]
опубликовал обзор реакций окисления этой смесью реагентов.
1. Hart Н., Accts. Chem, Res., 4, 337 (1971).
662
ТРИФТОРУКСУСНАЯ КИСЛОТА (ТФК) (III, 432—434; V,
459—460; VI, 294—297).
Дегидрирование. Андерсен и сотр, [11] обнаружили, что об-
работка сесквитерпенов ТФК приводит к их дегидрированию с
частичной ароматизацией. Реакция, в частности, удобна для по-
лучения тетралинов, Например, при обработке углеводородов
группы кадинена (1) — (4) в я-декане избытком ТФК при ком-
натной температуре в течение нескольких минут образуется
каламенен (5) с хорошим выходом. Для превращения лимонена
(6) в цимол (7) необходимо затратить несколько часов.
(6) (7)
Циклизация олефинов (VI, 294—296). В продолжение изуче-
ния биогенетически подобной циклизации олефииов Джонсон
и сотр. [2] нашли, что в ней может принимать участие опре-
деленным образом расположенная тройная связь с образова-
нием транс-сочлененного пятичленного кольца. Например, обра-
ботка триенинола (1) хлористым метиленом, содержащим
553
2 вес.°/о ТФК, при —70° в течение 5 мин дает трициклический
триен (2) с выходом около 70%.
он
(1) (2)
Эту циклизацию очень соблазнительно использовать в полном
синтезе стероидов, и действительно, Джонсон и сотр. [3] приме-
нили ее в синтезе ^/-прогестерона. На ключевой стадии синтеза
происходит циклизация соединения (3) с образованием (4), Эту
реакцию проводят под действием ТФК, как описано выше, толь-
ко в реакционную смесь добавляют еще этиленкарбонат для за-
хвата винильного катиона. По окончании циклизации к реак-
ционной смеси добавляют раствор поташа, чтобы гидролизовать
полученный комплекс енола. Соединение (3) превращали, та-
ким образом, в (4) с выходом 71 %- Тетрациклический кетон
(4) озонированием с последующей внутримолекулярной аль-
дольной конденсацией превращают в прогестерон (6). Следует
отметить, что соединение (4) представляет собой смесь (5:1)
17₽- и 17а-эпимерных кетонов. Ее превращают в эпимеры про-
гестерона (6), которые затем разделяют дробной кристаллиза-
цией.
сн,
564
Перегруппировка Кляйзена в орто-положение [4]. Фенилал-
лиловые эфиры при растворении в ТФК при комнатной темпе-
ратуре подвергаются перегруппировке Кляйзена. Константы
скорости реакции для некоторых пара-замещенных соединений
составляют около 105, т. е. они того же порядка, что и наблю-
даемые в обычной термической перегруппировке. Перегруппи-
ровку применяют и в целенаправленном синтезе. Например, по-
сле обработки диаллилового эфира гидрохинона ТФК в течение
1 час при 50° можно выделить примерно с выходом 70% 2,5-ди-
алл ил гидро хи нон.
1. Andersen N. Н„ Sy г dal D. D., Graham С., Tetrahedron Letters, 903
(1972).
2. J о h n s о n W. S., Grave stock M. B., Parry R. J., Myers R. F.,
Bryson T. A., Miles D. H., J. Am. Chem. Soc., 93, 4330 (1971).
3. Johnson W. S., Gravestock ЛА. В., McGarry В. E., J. Am. Chem.
Soc., 93, 4330 (1971).
4. Svanholm U., Parker V. D.. Chem. Comm., 1972, 645.
ТРИХЛОРАЦЕТИЛКЛОРИД, CC13COC1. Мол. вес 181,85,
т. кип. 114—116°, n'p 1,4689.
Ацетилирование по Фриделю — Крафтсу [1]. Первая стадия
синтеза этилового эфира пиррол-2-карбоновой кислоты легко
протекает в эфирном растворе без катализатора. Так, при до-
бавлении к раствору 225 г Т. в 200 мл абсолютного эфира рас-
твора 77 г пиррола в 640 мл абсолютного эфира при перемеши-
вании в течение 3 час выделяется тепло, достаточное для того,
чтобы смесь кипела в течение всего времени прибавления. Смесь
о
----, и ----------, о.
П и . седеет п п к
и н •> || И ССС1
N
н н
О-^с>.+ СН’СНг0Н ОАоснгсН1 + снсь
Н Н
перемешивают 1 час, после чего медленно добавляют к ней рас-
твор 100 г (0,72 моля) поташа в 300 мл воды. Органический
слой высушивают над сульфатом магния, обрабатывают 6 г
активированного угля (норит) и фильтруют. Растворитель от-
гоняют на кипящей водяной бане, а остаток растворяют в 225 ли
гексана. При охлаждении темного раствора льдом продукт вы-
деляется в виде кристаллов. Рыжевато-коричневые кристаллы
отфильтровывают и промывают 100 мл охлажденного гексана,
получая 189—196 г (77—80%) 2-трихлорацетилпиррола с т. пл.
73—75°.
1. Bailey D. М., Johnson R. Е,, Albertson N. F., Org. Syn., 51, 100
(1971).
565
ТРИХЛОРЙЗОЦИАНУРОВАЯ КИСЛОТА (цианурхлорид)
(V, 461; VI, 298). Мол. вес 184,41, т. пл. 145^148°.
Хлористые алкилы [1]. При нагревании в отсутствие влаги
с избытком реагента при температуре несколько ниже темпера-
туры кипения первичные, вторичные и третичные спирты пре-
вращаются в хлористые алкилы:
3ROH + W зо-9'w? 3RCI +
он
Йодистые алкилы [2]. Йодистые алкилы можно получить с
выходом около 40—75% нагреванием реагента с избытком
спирта и иодида натрия:
3 ROH + j |Г + з Nal ----------------> з RI + Н j
N^N -3 NaCl
C1 OH.
Нитрилы [3]. Альдоксимы под действием Т. к. в пиридине
при комнатной температуре с хорошим выходом дегидрати-
руются в нитрилы.
1. S а л d 1 er S. R., J. Org. Chem., 35, 3967 (1970).
2. Sandler S. R., Chem. Ind., 1971, 1416.
3. Chakraharti J. K-, Hot ten T. M., J. C. S. Chem. Comm., 1972, 1226.
ТРИХЛОРМЕТИЛЛИТИЙ (I, 416—417; V, 184—185).
Реакция с циклооктином [1].
1, \V i 11 i g G., Hutchison J. J., Ann., 741, 79 (1970).
566
ТРИХЛОРСИЛАН (VI, 298—299).
Для очистки реагент обрабатывают хинолином для связыва-
ния хлористого водорода и отгоняют Т, из полученного раство-
ра при атмосферном давлении. Перегонкой остатка в вакууме
регенерируют хинолин.
Бенкесер [1] опубликовал обзор химии комплексного реаген-
та Т. — третичный амин.
Образование метильных групп в результате восстановления
ароматических карбоновых кислот Т. — три-н-пропиламииом [2].
Синтез следует проводить в хорошо действующем вытяжном
шкафу для удаления образующегося в ходе реакции газообраз-
ного хлористого водорода и захватываемого им Т. Прибор со-
стоит из трехгорлой круглодонной колбы на 300 мл, снабжен-
ной магнитной мешалкой, термометром, стеклянным краном
HS1C1, /^\ XX кон ХХ—ХХ
у V \—/ «-Рг3/ X Y \—/ СН3ОН \=У
ХОгН ^CH2SiCl3 хсн3
для ввода азота и эффективным обратным холодильником, сое-
диненным со счетчиком пузырьков. Всю стеклянную посуду
тщательно высушивают открытым пламенем горелки или в су-
шильном шкафу, после чего систему заполняют сухим азотом.
В колбу помещают 19,8 г (0,1 моля) дифенил-2-карбоновой
кислоты, 60 мл (0,6 моля) Т. и 80 мл ацетонитрила (высушен-
ного с помощью молекулярных сит 4А). Сухую инертную атмо-
сферу в системе поддерживают медленным током азота. Смесь
кипятят (40—45°) при перемешивании в течение 1 час или до
прекращения выделения газа и полного растворения карбоно-
вой кислоты. Затем раствор охлаждают в бане с сухим льдом по
крайней мере до 0°. Стеклянный кран заменяют капельной во-
ронкой на 100 мл с соединительной трубкой для выравнивания
давления, содержащей 37,8 а (0,264 моля) три-/г-пропиламина,
который быстро приливают при перемешивании к реакционной
смесн. Баню с сухим льдом удаляют и перемешивают содержи-
мое колбы до прекращения экзотермической реакции. Затем
реакционную смесь кипятят на колбонагревателе 16 час; за это
время температура поднимается до 70—75°.
Раствор быстро переливают в 1-литровую колбу Эрленмейе-
ра и разбавляют абсолютным эфиром до объема около 850 мл,
при этом большая часть хлоргидрата три-н-пропиламина выпа-
дает в осадок. После охлаждения смеси в течение 1 час в плот-
но закрытой колбе Эрленмейера осадок отфильтровывают на
150-миллилитровой воронке Бюхнера и промывают тремя пор-
циями абсолютного эфира по 50 мл. Прозрачный желтый фильт-
рат помещают в капельную воронку на 1 л с соединительной
трубкой и прибавляют по каплям с постоянной скоростью через
567
10-сантиметровую колонку Вигре с головкой для перегонки в
300-миллилнтровую круглодониую колбу, снабженную магнит-
ной .мешалкой, Колбу нагревают при перемешивании, пока не
отгонится почти весь эфир. Оставшийся темный раствор затем
нагревают при 40° (80 мм) для удаления остатков легколетучих
веществ. Колонку Вигре, 1-литровую капельную воронку и го-
ловку для перегонки заменяют капельной воронкой на 100 мл
тоже с соединительной трубкой, содержащей 100 мл метанола,
который медленно прибавляют к маслообразному содержимому
колбы, оставив горло воронки открытым для выпуска образую-
щегося хлористого водорода. После прекращения самопроиз-
вольного кипения раствор кипятят на плитке в течение 1 час, за-
тем охлаждают в ледяной бане и медленно обрабатывают рас-
твором 56 г (1 моль) гидроокиси калия в 25 мл воды и 50 мл
метанола. Получившуюся смесь кипятят около 19 час, затем
растворяют в 600 мл воды и экстрагируют тремя порциями эфи-
ра по 100 мл. Объединенные эфирные вытяжки промывают в
один прием 50 ли 5 н. раствора соляной кислоты и высушивают
над безводным сульфатом магния. Эфир удаляют перегонкой,
как описано выше, а перегонка в вакууме оставшейся жидко-
сти дает 12,5—13,4 г (74—80%) 2-метилдифенила, т. кип. 76—
7870,5 мм, 1,5920 (лит. данные 1,5914).
(П (2)
Следует отметить, что Рамакришнам и Бикарт [3] при вос-
становительном силилировании фенантрен-3,4-дикарбоновой
кислоты (1) выделили аномальный продукт (2).
Диалкиловые эфиры [4]. При у-облучении Т. может восста-
навливать алкиловые эфиры алифатических карбоновых кислот
до диалкиловых эфиров в некоторых случаях с количественным
выходом. Предполагают, что реакция протекает по свободнора-
дикальному цепному механизму [5].
v-лучи
ClgSiH
RCOOR' ----> RCH,OR'
1. Benkeser R. A., Accts. Chem. Res., 4, 94 (1971).
2. L i G. S., Ehler D, F., Benkeser R. A., Org. Syn., submitted 1972;
Benkeser R. A., Foley К. M., Gaul J. M„ L i G. S., J. Am. Chem. Soc.,
92, 3232 (1970).
3. Ramakrishnan K-t Bickart P., J. C. S. Chem. Comm., 1972, 1338,
568
4. T s и г и g i J., Макао R., Fukumoto T„ J. Am. Chem. Soc., 9l, 4587
(1969).
5. Nagata Y., Dohmaru T., Tsurugi J., J. Org. Chem., 38, 795 (1973).
ТРИХЛОРУКСУСНАЯ КИСЛОТА (III, 446; V, 461—462).
Гидролиз ацеталей циклопропанкарбоксальдегида [1]. На
последней стадии описанного недавно синтеза циклопропанкар-
боксальдегида (2) осуществляют гидролиз ацеталей (1) аль-
дегида. Применение для этой цели неорганических кислот не
привело к успеху. Самые лучшие результаты были получены
ch(or)2 сно
6) (И
при использовании 1 н. раствора Т, к, в таком количестве, чтобы
при перемешивании образовался гомогенный раствор.
1. Stewart J. М., Carlisle С., Кет К., Lee G., J. Org. Chem., 35, 2040
(1970).
ТРИХЛОРУКСУСНОЙ кислоты ЭТИЛОВЫЙ ЭФИР
(III, 448—449; VI, 300).
Дихлоркарбен (VI, 300). Т. к. э. э. используют на первой
стадии трехстадийного синтеза бицикло-[3,2,1]-октанона-3 из нор-
борнена [1] (выход первичного аддукта составляет 74—88%).
С13ССО2С2Н5
NaOCH3
«---£—
H2SO.
В 1-литровой четырехгорлой круглодоиной колбе при охлажде-
нии льдом с солью перемешивают механической мешалкой рас-
твор 52,5 г (0,56 моля) норборнена в 400 мл петролейного эфи-
ра и 112 г NaOCH3. В капельную воронку помещают 112 г
(2,06 моля) Т. к. э. э. и при перемешивании медленно прикапы-
вают его к реакционной смеси с такой скоростью, чтобы темпе-
ратура не поднималась выше 0°. Прибавление Т. к. э.э. зани-
мает около 4 час, в течение которых вначале белая смесь при-
569
обретает все более интенсивную желтую окраску. Смесь остав-
ляют при комнатной температуре в течение ночи, после чего
выливают на толченый лед с водой. Органический слой отде-
ляют, а водный экстрагируют четырьмя порциями эфира, ней-
трализуют соляной кислотой и продолжают экстракцию эфиром.
Затем эфирные вытяжки объединяют, промывают насыщенным
раствором хлористого натрия, высушивают над сульфатом маг-
ния, растворитель упаривают, а остаток перегоняют. Получают
72,5—87,0 г (74—88%) э/сзо-3,4-дихлорбицикло [3,2,1]-октена-2,
т. кип. 72—73°C (0,9 мм).
Следующая стадия синтеза — избирательное замещение хло-
ра на водород у насыщенного Статома без затрагивания хлора
у ненасыщенного Сз-атома — гладко протекает под действием
алюмогидрида лития в эфире и приводит к 3-хлорбицикло-
[3,2,1]-октену-2 (выход 74—75%). Последняя стадия, осуществ-
ляемая перемешиванием полученного на второй стадии хлорбн-
циклооктена с конц. серной кислотой и последующим разложе-
нием реакционной смеси льдом, без сомнения, протекает через
образование енола:
I I I
RCH=C—Cl -> RCH—С—ОН —> RCH2C=O
Реактив Реформатского [2]. Т. к. э. э. реагирует с цинком в
ТГФ при —15° с образованием устойчивого хлорцинк-енолята
(1). Этот реагент может вступать в реакцию конденсации с це-
лым рядом электрофильных агентов, таких, как карбонильные
zn. тгф zOZnCl
cue—> C12C=CZ
Х)С2Н5 \эс2н5
(1)
соединения, ангидриды карбоновых кислот или галогеналкилы.
(1) + С1СН2ОС2Н5 С2Н5ОСН2СС12СООС2Н5
(!) + СН3СН3СНО СН3СН2СНОНСС12СООС2Н5
(1) -I- (СН3СО)2О CH3COCC12COOC2HS
1. Jef ford С. W., Gunsher J., Hill D. T., Brun P., Le Gras J.,
Waegell B., Org. Syn., 51, 60 (1971).
2. Castro B., Villieras J., Ferracutti N., Bull. Soc. Chim. France,
1969, 3521.
2,2,2-ТРИКЛОРЭТАНОЛ (VI, 300).
Защита альдегидов и кетоиов [1]. При кипячении диметил-
или диэтилацеталей с 1,5 экв Т. в бензоле в присутствии кисло-
го катализатора (л-TsOH) образуются смешанные ацетали.
570
Увеличение количества Т, до 4 экв приводит к получению ди-
2,2,2-трихлорэтилацеталя.
ZOCH3(C2H5) н+
С' + СС1ЭСН2ОН >
RZ ОСН3(С2Н5) 1, 5 эм
В1 2 ОСНэ(С2Н5)
X
К ОСНгСС13
сн3он
(СгН5ОН)
рЭСН3(С2Н5)
ц/ хосн3(с2н5)
+ СС13СН2ОН
4»
В> ОСН2СС1Э
Щ ОСН2СС13
2 СН3ОН
* (2 С2Н5ОН)
н
Карбонильное соединение из последнего можно регенерировать
обработкой активированной цинковой пылью в этилацетате или
ТГФ в отсутствие кислот.
Защита карбоксильной группы в пептидном синтезе [2],
Взаимодействием соответствующих хлористых ацнлов с Т. мож-
но получить 2,2,2-трихлорэтиловые эфиры N-карбобензоксиами-
нокислот. Карбобензоксигруппу селективно отщепляют дейст-
вием НВг—НОАс. Образующиеся эфиры затем конденсируют
с N-карбобензоксиаминокислотамн или пептидами в присутст-
вии дициклогексилкарбодиимида в ацетонитриле, получая трн-
хлорэтиловые эфиры N-карбобензоксипептидов. Защитную груп-
пу избирательно удаляют восстановлением цинком в уксусной
кислоте. Выходы довольно хорошие (63—83%). Факт полного
отсутствия изомеризации еще нуждается в проверке, однако
оптическая активность полученных таким путем N-карбобензок-
сипептидов согласуется с литературными данными.
1. I s i d о г J. L., Carlson R. М., J. Org. Chem., 38, 554 (1973).
2. Marinier В., Kim Y. C., Navarre J.-M., Can. J. Chem., 51, 208 (1973).
ТРИЭТИЛАЛЮМИНИЙ (in, 450—451; V, 463—464; VI,
301).
Присоединение HCN (V, 463—464). Из кетона (1) по методу
Нагаты был получен 7-циан-Д1’9-окталиион-2 (2) с выходом 48 %
Г11, который не удавалось синтезировать классическими мето-
дами:
(1) (2)
Опубликовано подробное описание [2] реакции 30-ацетокси-
Д5-холестенона-7 (V, 463—464).
1. Finnegan R. A., Bachman Р. L., J. Org. Chem., 36, 3196 (1971).
2. Nagata W., Y о s h i о k a M., Org. Syn., 52, 100 (1972).
671
ТРИЭТИЛАМИН (111,451—458; V, 464—466).
Дегидрогалогенирование (III, 455—456; V, 464—465), До-
бавлением Т. по каплям при перемешивании н охлаждении
льдом к эфирному раствору дифенилацетилхлорнда в атмосфе-
ре азота и последующей перегонкой при 1 мм получают дифе-
нилкетен в виде оранжевого масла с выходом 53—57% [1]:
(C2H3)3N
(СеН5)2СНСОС1 ——> (С0Н5)2С=С=О + (C2H5)3N • НС1
1, Taylor Е. С., McKillop A., Hawks G. Н., Org. Syn., 52, 36 (1972).
N, N, М-ТРИЭТИЛ-М'-КАРБМЕТОКСИСУЛЬФАМИДА ВНУ-
ТРЕННЯЯ СОЛЬ— ТРИЭТИЛАМИН (комплекс),
CH3O2CNSO2N(C2H5)3-(C2H5)3NHC1 (1). Мол. вес 173,59, т. пл.
70 — 78° (разл.).
Получение [1, 2]. Реагент получают взаимодействием хлор-
сульфонилизоцианата с метанолом в бензольном растворе. К об-
разовавшемуся N-карбметоксисульфа мои л хлориду прибавляют
триэтиламнн.
С3Н5 2N(C2Hs)3
ciso2nco + ch3oh •-----> CH3O2CNHSO2CI —-Я-7И-» (1)
Уретаны [1, 2]. Реагент взаимодействует с первичными
спиртами, например гексанолом-1 (2), с образованием соли (3),
которая после нагревания в течение 1 час при 95° и последую-
щей обработки водой превращается с выходом 55% в метнл-N-
гексилкарбамат (4). Аналогичным образом бензиловый спирт
превращают в метил-ЬТбензилкарбамат с выходом 80%.
Нагревание
+ Н20
н-С6Н13ОН + (!) —> C6HI3OSO2NCO2CH3-HN(C2H5)3 —----->
(2) (3)
—> я CgHJ3NHCOOCH3
(4)
Дегидратация [1]. Третичные и вторичные спирты под дей-
ствием Т. (1) дегидратируются до олефинов. Например, 1,2-ди-
фенилэтанол (2) реагирует с (1) в бензоле или ДМФА, обра-
С6Н5СН2СНСаН5 + (I) —>
in
(2)
“° С6Н5 Н
—> с6н5сн2снс6н5
so2ncooch3 н с6н5
HN+(C3H5)3
(3) w
572
зуя триэтиламмонневую соль 1,2-дифенилэтил-ЬКкарбметоксн-
сульфамата (3). Эту соль затем нагревают 30 мин при 50° и
обрабатывают водой. В результате получают тушнс-стильбен с
выходом 96°/о- С помощью дейтерированных производных сое-
динения (2) было показано, что дегидратация имеет механизм
стереоизбирательиого ццс-элиминирования.
Другие примеры:
/СН3
Н3с
снг-с-он
сн3
ч Нх Л'-Щ
-----> С=С 4-
9 5% И, СХ
3 3 24 ;1
лн2-с
ЩС- СНз
сн3
Н3С^ \ = сн2
н—с—с—сн3
H3GZ С(СН3)3
н3с^ с(снэ)3
н—с—с—он
Н3с \(СНЭ)3
----->
70%
1. Burgess Е. М., Р еп t о п Н. R., Jr., Taylor Е. A., Williams W. М„
J. Org. Chem., 38, 26 (1973).
2. Burgess Е. М., Р е n t о и Н. R., Jr,, Taylor Е. A., Williams W. М.,
Org. Syn., submitted 1973.
N,N,\'-TP ИЭТИ Л-Х'-КАРБЭТО КСИСУЛ ЬФАМИДА ВНУТ-
РЕННЯЯ СОЛЬ, C2H502CN-SO2N+(C2H5)3. Мол. вес 252,34,
т. пл. 66—69°.
Получение [1]. Реагент получают с выходом 81 % взаимо-
действием карбэтоксисульфамоилхлорида с 2 экв триэтиламина
в бензольном растворе при 30°:
2(C2H5)3N - +
C2H5O2CNHSO,C1 -------► C2H5O2CNSO2N(C2H5)3 + (C2H5)3N.HC1
Дегидратация. Бургесс и сотр. [2] нашли, что Т. в. с. при-
менима для дегидратации простых спиртов. Реакция является
примером стереоизбирательиого цис-элиминирования и подчиня-
ется правилу Зайцева. Краббе и Леон [3] использовали этот
метод для дегидратации целого ряда вторичных и третичных
стероидных спиртов. Они пришли к заключению, что основными
факторами, определяющими направление дегидратации, являют-
ся природа спиртовой группы, ее конфигурация и окружение.
Реакцию проводят при комнатной температуре в безводном бен-
573
золе в течение 2 час, а промежуточно образующийся сульфонат
разлагают в вакууме при 90°, Выходы обычно удовлетворитель-
ные. Таким путем За-оксиандростен-5а-он-17 (1) превращается
в Д2-андростен-5а-ои-17 (2) с выходом 75%.
1. Atkins G. М, Jr., Burgess Е. М., J. Am. Chem. Soc., 90, 4744 (1968),
2. Burgess Е. М., Taylor Е. А., Р е n t о n Н. Р., Jr., 159th National Meet-
ing, A. C. S. Houston, Texas, Feb, 1970, Abstracts OpGN-105.
3. Ora bbe P„ Leon C„ J. Org. Chem., 35, 2594 (1970).
ТРИЭТИЛОКСОНИЯ БОРФТОРИД (II!, 459—461; V, 467—
468; VI, 301—302).
Ацетали амидов [1]. Амиды карбоновых кислот (1) с высо-
ким выходом (95—100%) алкилируются Т.б.в соответствующие
борфториды карбония (2). Взаимодействие борфторидов (2) с
этилатом натрия при 0°, как правило, приводит к получению
ацеталей амидов (3) с выходом 50—70% В результате реакции
RC^° Btv NaOC,H„ 0°
XH(CH3)2 9 5-100% X'N(CH3)2 -NaBF4 " XN(CH3)2
(О (M (3)
апеталей (3) с C—Н-кислотными соединениями при 80° проис-
ходит отщепление этанола и образуется амннометиленовое про-
изводное. Например, с нитрометаном ацеталь амида (4) обра-
зует соединение (5) с выходом 60%.
(4)
^,ОСгН5 „
(с2н5о)2снс—ос,н5 ----1-Ц>(С,Н30)2СНС=СННО, 4- 2 СгН5ОН
^м(сн,)г 60% 1,(снд
(5)
Азоксиалкаиы [2].. Реакция алкилдиазотатов (вероятно,
спн-формы) с суспензией Т. б. в 'СН2С12 дает азоксиалканы
574
о выходом 50—60%:
Азоксналканы можно также получить взаимодействием алкил-
диазотатов с галогеналкилами (желательно иодидами) в гекса-
метаполе:
R ок+ ГМТФК R о
х \ / + R4 ------> \ /7 f KI
XN=NZ + N=N
Этерификация [3]. T. б, в хлористом метилене при комнат-
ной температуре легко этерифицирует пространственно затруд-
ненные карбоновые кислоты (равно, как и незатрудненные),
причем карбоксилат-анион генерируют добавлением объемисто-
го органического основания, например диизопропилэтиламина
[4], Выходы в этой реакции составляют 85—95%-
° ы <-сл
R_C-0 + СН3СН2-СИ
RC-OC2H5
Расщепление тиокеталей [5]. Защиту кетонных и альдегид-
ных групп превращением их в тиокетали применяют редко, по-
тому что тиокетали устойчивы к кислотному и щелочному гид-
ролизу. Наиболее удобным методом их расщепления до сих пор
считался гидролиз в присутствии солей ртути (III, 193; V, 158;
VI, 101 —102). Японские химики недавно с успехом осуществили
расщепление тиокеталей, применив предварительное алкилиро-
вание их под действием Т.б. Так, алкилирование реагентом эти-
лентиокеталя циклогексанона (1) дает соль (2). Щелочной гид-
ролиз соли (2) приводит к получению циклогексанона с выходом
лишь 36%. Однако если соль (2) встряхивать с 3%-ным раство-
ром CuSO.i в хлористом метилене, то выход циклогексанона по-
вышается до 81 %.
О)
(СгЩ)3О+ВГ4"
(2)
575
По другой методике дважды алкилируют тиокеталь (1) с
образованием соли (3), гидролизом которой уже 10%-ным рас-
твором NaOH получают циклогексанон с выходом 81%. Исполь-
зуя эту методику, японские химики осуществили расщепление
|— йсгН5
1— SCaH5
(3)
8Ц
тнокеталей целого ряда альдегидов и кетонов с удовлетвори-
тельными выходами.
I. Bredereck Н., Kantlehner W., Schweizer D., Chem. Ber., 104,
3475 (1971).
2. Moss R. A., Landon M. J., Luchter К. M.., Mamantov A., J. Am.
Chem. Soc., 94, 4392 (1972).
3. Raber D. J., Gariano P., Tetrahedron Letters, 4741 (1971).
4. H u n i g S., К i e s s e I M., Chem. Ber., 91, 380 (1958).
5. Oishl T., Kamemoto K-, Ban Y., Tetrahedron Letters, 1085 (1972).
ТРИЭТИЛСИЛАН (III, 461—462; V, 468; VI, 302).
Восстановление альдегидов и кетонов до простых эфиров [1].
Т. в спирте в присутствии кислоты восстанавливает карбониль-
ные соединения до простых эфиров:
R2C=O+(C2H5)3SiH + R'OH > R2CHOR' + (C2H5)3SiOH
<4-0 ' Уи ZQ
В качестве активных катализаторов могут с равным успехом
выступать серная, трифторуксусная или трихлоруксусная кис-
лоты. Авторы предлагают следующий механизм реакции:
R2C=O + н+
r2C=OH
.он
ОН;
ReC=OH + R'OH R2cf\ =ё=± R2q R3C=OR' + Н2О
\OR' W
н
R2C=OR' + (C2H5)3SiH 4- Н2О —> R2CHOR' + (C2H5)3SiOH
1. Doyle M. P., DeBruyn D. J., Koo i str a D. A., J. Am. Chem. Soc., 94,
3659 (1972),
576
ТРИЭТИЛФОСФИТ (III, 463—469; V, 468—470; VI, 303).
Дехлорирование [1]. Т. — наиболее подходящий реагент для
дехлорирования соединения (1) до гексахлорфульвена (2).
(1) (2)
В этом превращении эффективны также иодид натрия (выход
98,2%) и хлористое олово (выход 63%).
Восстановление азотистых соединений (III, 467—468; V, 470).
Брук и сотр. [2] доказали, что циклизация соединения (1) в
карбазол (2) под действием Т. протекает не через нитрены, как
считалось ранее, а вероятно, через промежуточную биполярную
структуру (а):
. - +
(1) a) X=N-OP(OC2H5)3
1. McBee Е. J.( Wessel er Е. Р„ Crain D. L., Hurnaus R., Hod-
gins T., J. Org. Chem., 37, 683 (1972).
2. Brooke P. K-, Herbert R. B., Holliman E. G., Tetrahedron Letters,
761 (1973).
ТРОПИЛИЯ СОЛИ (2).
Получение путем замещения водорода [1]:
+ (С6НДС+¥'
y“ + (С6ндсн
Реакционная способность катионов хлортропилия по отно-
шению к нуклеофилам [2]. Взаимодействием солей хлортропи-
лия со спиртами, тиолами н М-алкил-ЬБариламинами получают
соли алкокси-, алкилтио- и N-алкил-М-ариламинотропилия со-
ответственно. В присутствии диметиламина или бензолсульфа-
мида соли хлортропилия перегруппировываются в бис-(диме-
тиламино) -фенилметан или N-бензилиденсульфамид. Полагают,
что образование хлорциклогептатриенов (1а)— (1в) происходит
в условиях кинетического контроля, тогда как замещенные
19 Зак. 593
577
ионы тропилия (3) образуются в условиях термодинамического
контроля.
1. Dauben Н. J,, Jr., Ga deck! F. A,, Harmon К. М., Pearson D. L.,
J. Am, Chem. Soc., 79, 4557 (1957).
2. F о h 1 i s c h В., H a u g E., Chem, Ber., 104, 2324 (1971),
УГЛЕРОДА ОКИСЬ (V, 471; VI, 305—307).
Три ал кил карбинолы [I]. Смешанные триалкилбораны, на-
пример боран (1), реагируют с У. о. в присутствии этиленгли-
коля при 150° и давлении 70 атм с образованием 2-трет-алкил-
1,3,2-диоксабороланов (2), которые не выделяя окисляют пере-
кисью водорода в водном растворе гидроокиси натрия до трн-
алкилкарбинолов (3). Выходы выделенных продуктов состав-
ляют 60—90 %,
«-ОД^ \с
«-c4Hj--b + со + £
о
я-с4н,—с~в
(2)
H^Oj, NaOH, H2O
62%
«-ОД
К-ОД —СОН
Я -CsHif^
(3)
I. Brown Н. C.t N е g i s h I E., Gupta S. !<> J. Am. Chem. Soc., 92, 6648
(1970).
УГЛЕРОД ЧЕТЫРЕХХЛОРИСТЫЙ (VI, 307).
Теломеризация монохлортрифторэтилена [1], У. ч. реагирует
с монохлортрифторэтиленом в ацетонитриле в присутствии хлор-
ного железа, хлоргидрата триэтиламина и бензоина (восстано-
витель) с образованием теломеров, отвечающих формуле (1):
CCU + ftCF2=CFCl —>
CCl3(CF2CFCf)„Cl
(1), n== 1 —15
1. Boutevin В., Pietrasanta Y., Tetrahedron Letters, 887 (1973).
УГОЛЬНОЙ КИСЛОТЫ трет-БУТИЛОВОГО ЭФИРА
АЗИД (трет-бутилазидоформнат) (IV, 6; V, 471—473; VI, 307).
Получение. Сакай и Ансельм [1] рекомендуют получать ре-
агент взаимодействием трет-бутилового эфира хлоругольной
1.9*
579
кислоты с азидом тетраметилгуанидиния (V, 409):
СОС12 + НОС(СНз)з > С1СООС(СН3)3
С1СООС(СН3)3 + [(CH3)2N]2C=NH2N3
—> N3COOC(CH3)3 + [(CH3)2N]2C=NH2C1
Внимание! Все операции следует проводить в хорошем вы-
тяжном шкафу и за защитным экраном.
N-Защита аминокислот (VI, 307). трет-Бутоксикарбоиилами-
нокислоты (2) можно получить с выходом 80—100% взаимо-
действием 1,1,3,3-тетраметилгуанидиииевых солей аминокислот
(1) с реагентом в ДМФА [2]:
(CH3)3COCON3
(CH3)2N— С—N(CH3)3RCHCOO ----------> RCHCOOH
+NHo nh2 NH
I
COOC(CH3)3
(1) (2)
1. Sakai K., Anselme J.-P., J. Org. Chem., 36, 2387 (1971); Org. Syn.,
submitted 1972.
2. A Ji A., Fahrenholz F., Weinstein B., Angew. Chem., Internal. Ed.,
II, 289 (1972).
УГОЛЬНОЙ КИСЛОТЫ трет-БУТИЛ ОВОГО ЭФИРА
ФТОРАНГИДРИД (V, 473).
Получение при —25°.
сн3 сн3
I I
F—С—CI + НОС—СНз ~~> F—С—О—С—СН3
II 1 “II
о сн, о сн,
Т кип. 4°/15 мм
Применяется для синтеза БОК-аминокислот [1].
I. Schnabel Е., Herzog Н., Hoffman Р., Klauke Е., Ugi I., Ann.,
716, 175 (1968).
УГОЛЬНОЙ КИСЛОТЫ МЕТИЛОВОГО ЭФИРА ГИДРА-
ЗИД, NH2NHCOOCH3. Мол. вес 90,08, т. пл. 70—72,5°.
Кетоиы -> нитрилы [1]. Карбонильные соединения реагируют
с У.к.м.э.г, (1), образуя с высоким выходом карбметоксигид-
разоны (2) [2], которые при последовательной обработке циани-
стым водородом и бромом превращаются с почти количествен-
ным выходом в метиловые эфиры замещенной метилазокарбо-
новой кислоты (4). При обработке последних раствором мети-
лата натрия в метаноле (7) выделяется азот и с высоким вы-
ходом образуются нитрилы (5). Если диазеиы (4) обрабаты-
вать метилатом лития в ДМЭ в присутствии диметилкарбона-
580
та, то образуются эфиры замещенной циануксусной кислоты (6)
RjR2C=O + NH2NHCOOCH3 -- >
(1)
HCN Вгй, водн. NaHCO3
СНзОН CH2CI2
—> R1R2C=NNHCOOCH3 ---------> RiR2C(CN)NHNHCOOCH3 -------------->
(2) (3)
CHgONa—CH3OH
—> R1R2C(CN)N=NCOOCH3 ------------------> RtR2CHCN
(4) (5)
CH3OL!, ДМЭ
(CH3O)2CO
СН3ОЫ, CH3I
ДМЭ
RjR2C(COOCH3)CN
(6)
1ШССН3СМ + (6)
(7)
и лишь следовые количества нитрилов (5). Третьим вариантом
превращения диазенов (4) является метилирование при дейст-
вии йодистого метила и метилата лития в ДМЭ с образованием
а-метилнитрилов (7). Для превращений (4)-* (5) и (4)-* (6)
достаточно каталитических количеств основания, тогда как ре-
акция (4)-* (7) требует уже стехиометрического количества
основания.
1, Z i е g 1 е г F. Е., W е n d е г Р. A., J. Am. Chem. Soc., 93, 4318 (1971).
2. С h а с о М. С., R a b j о h п N., J. Org. Chem., 27, 2765 (1962).
УГОЛЬНОЙ КИСЛОТЫ ЭТИЛОВОГО ЭФИРА АЗИД
(IV, 9—11; V, 475; VI, 308).
Реакция с этоксиацетиленом fl]. Реакция циклоприсоедине-
ния У. к. э. э, а. с этоксиацетиленом (комнатная температура,
35 дней) не региоспецифична и приводит к образованию при-
мерно равных количеств 1-карбэтокси-5-этокси-1,2,3-триазола
(1) и 1-карбэтокси-4-этокси-1,2,3-триазола (2). Кроме того, в
результате изомеризации (вероятно, термической) триазолов
(1) и (2) образуются следы 2-карбэтокси-4-этокси-1,2,3-триазола
нс^сосгН5 + c2hsocn3
НС = СОС,Н= НС==СОС,Нс
I I z 5 + I I 3
c2h5oocn n
'Зн5
+
N;
(1)
нс-----сос,н=
II II 3
N XN
N
I
соосгн5
(3)
581
(3). Триазолы (1) и (2) легко изомеризуются в (3) под дей-
ствием 1,4-диазабицикло-[2,2,2]-октана (ДАБЦО) (V, 89—90).
Реакция ииамина, 1-(М,№диэтиламино)-пропина-1, с избыт-
ком У. к.э.э.а,, напротив, региоспецифичиа. Если ее проводить
в СС14 при комнатной температуре в течение 20 мин, то методом
ЯМР можно обнаружить лишь триазол (4). Если реакцию про-
о
СНэСвС-ШСаНД +JC:HsOCNj-
* H3C-<j = CN(C2H5)2
n^n^nC°OC2H5
(41
Н3с-с-----CN(C2H3)a
-N
COOC3HS
водить с эквимолекулярными количествами обоих реагентов в
хлористом метилене при комнатной температуре, то получается
смесь триазолов (4) и (5) в отношении 95: 5. Добавление ииа-
мина к этой смеси вызывает дальнейшее превращение триазола
(4) в (5). Таким образом, инамииы, подобно ДАБЦО, катали-
зируют изомеризацию 1-карбэтокси-1,2,3-триазолов в 2-карбэт-
оксиизомеры.
1. Ykman Р., L’a bb ё G., S m еt s G., Chem. Ind., 1972, 886.
УКСУСНОГО АЛЬДЕГИДА ЭТИ Л-З-БРОМП РОЛИ ЛА ДЕ-
ТАЛЬ, Вгсн2сн2сн2оснсн3. Мол. вес 212,12, т. кип. 49—5Г/1 мм.
ОСН2СНЙ
Реагент (1) получают с выходом 92% присоединением
3-бромпропанола к этилвиниловому эфиру, катализируемым ди-
хлоруксусной кислотой [1]:
Вг(СН2)2СН2ОН + СНг=СНОСН2СНз ВгСН3СН2СН2ОСНСН3
осн2сн3
Оксипропилироваиие [1]. Реакция У. а. э. (1) с литиевой
проволокой (1% натрия) в эфире при 0° приводит к образова-
нию металлоорганического реагента (2), с помощью которого
удобно вводить оксипропильную группу. Так, соединение (2) при
взаимодействии с циклогексаноном в эфире дает аддукт (3) с
582
выходом 90%, Гидролиз (3) соляной кислотой в водном этаноле
(1) -Ы» ыснаснгснгоснсн3
ОСН2СН
(2)
оснасн3
дает с выходом 96% 1-(3-оксипропил)-циклогексанол (4). Об-
работка аддукта (3) ц-толуолсульфокислотой при пониженном
давлении при 150—200° приводит к образованию 1-оксаспиро-
[4,5]-декана (5).
Взаимодействие литиевого производного (2) с 0,5 экв иоди-
да меди(1) при —60° приводит к образованию литийорганиче-
ского купрата (6). При сопряженном присоединении последнего
к циклопентенону образуется аддукт (7), гидролиз которого ди-
хлоруксусной кислотой дает 3- (3-оксипропил) -циклопента-
нон (8).
(2)
Ь1(СНгСНгСНгОСНСНз)гСи
оснгсн3
(6)
,снгснгснгоснсн3
/~\ оснгсн3
о
(7) (8)
583
1. Eaton P. E., Cooper G. F., Johnson R, C,, Mueller R. H,, J. Org,
Chem,, 37, 1947 (1972),
УКСУСНОЙ КИСЛОТЫ ХЛОРАНГИДРИД (IV, 21; V, 321).
Ацетилирование этилового эфира у-(иафтил-2)-масляной
кислоты в нитробензоле в присутствии А1С1з см [1].
Восстановление сульфоксидов [2]. При взаимодействии ал-
кил-о-карбоксифенилсульфоксида (1) с У, к. х. в хлористом ме-
тилене при комнатной температуре происходит экзотермическая
реакция с выделением хлора и почти количественным образо-
ванием сульфида (2). Следует отметить, что действие на суль-
фоксид (1) уксусного ангидрида приводит к перегруппировке
Пуммерера с образованием 3,1-бензоксатианона-4 (3) с хоро-
шим выходом [3].
Восстановление сульфоксидов действием У, к. х. представ-
ляет собой общую реакцию обычно с высокими выходами целе-
вых продуктов. Однако при восстановлении ди-н-бутилсульфо-
ксида ди-н-бутилсульфид образуется только с 70%-ным выхо-
дом.
Предложен следующий механизм восстановления:
г ci л
АсС! + । 1
R—S—R' —-> PR—S—R'lCC —> LR—S—R'JAcO*
А I +
О L OAc J
|дсс1
R—S—R' + C12
Иминосульфураны (4) также восстанавливаются У. к. х. до
сульфидов (5) почти с количественным выходом:
С6Н5—S—R > CsH5S—R
NTs
(4) (5)
R = СН3, СНгСвНэ, CgHs
584
1. R a h m a n A. U., P e r 1 C„ Ann., 718, 127 (1968).
2. Numata T., Oae S„ Chem. Ind., 1973, 277.
3. Numata T., Oae S., Chem. Ind., 1972, 726.
УКСУСНОМУРАВЬИНЫЙ АНГИДРИД (IV, 21—22; V,
480—482; VI, 308).
Диазоацетальдегид [1]. Диазоацетальдегид удобно получать
реакцией У. а. с диазометаиом (выход 46%). Побочно образую-
щийся метилацетат легко удаляется при упаривании на ротор-
ном испарителе. Диазо ацетальдегид является предшественни-
о о
0 ]! эфир
СН3С—О—С—Н + CH2N2 —NoCHCHO + СНзСООСНз + n2
46%
ком формилкарбена (V, 91—92) и используется для удлинения
цепи олефинов на два атома углерода (VI, 52).
1. Hooz J., Morrison G. F., Org. Prep. Proc. Int, 8, 227 (1971).
УКСУСНЫЙ АНГИДРИД-ПИРИДИНА ХЛОРГИДРАТ.
Расщепление простых эфиров [1]. При кипячении кетоэфира
9-оксатрицикло-[4,3,3,0]-додеканона-3 (1) с У. а. — п. х. в течение
5,5 час образуется диацетат 4-ацетокси-1-(2-ацетоксиэтил)-би-
цикло-[4,3,0]-ионадиен-4,6 (2) с выходом 93%:
CSHSN' НС1
93% $
1. Peet N. Р., С а г g i 11 R. L., J. Org. Chem., 38, 1215 (1973).
УКСУСНЫЙ АНГИДРИД—ЦИНКА ХЛОРИД.
Ч«с-Гидринданол-1 [1]. Реакция дибромида (1) [2] с У. а.—
ц. х. в хлористом метилене приводит к образованию смеси ке-
тона (2) с выходом 30% и дибромида (3) с выходом 21%. Ук-
сусный ангидрид играет существенную роль в реакции трансан-
нулярной циклизации. При действии одного хлористого цинка
в хлористом метилене дибромид (1) лишь изомеризуется в
транс-изомер бромида (3). Кетон (2) окисляют по Байеру —
Виллигеру в ацетат (4), который гидролизуют до спирта, и
окисляют последний реагентом Джойса. Восстановление кето-
на (5) литием в аммиаке дает только спирт (6), а гидрирова-
ние этого спирта приводит к ф/с-гидринданолу-1 (7).
585
1. Boyle L. W., Sutherland J. K., Tetrahedron Letters, 839 (1973).
2. Skattebol L., Acta Chem. Scand., 17, 1683 (1963).
УРУШИБАРЫ ГИДРИРОВАНИЯ КАТАЛИЗАТОРЫ.
Обзоры [1—3]. Эти катализаторы были разработаны глав-
ным образом Урушибарой, Их получают осаждением металла-
катализатора (никеля, кобальта или железа) из водного рас-
твора его соли (обычно хлорида) цинковой пылью или грану-
лированным алюминием. Осажденный металл затем обрабаты-
вают щелочью или кислотой (обычно гидроокисью натрия или
уксусной кислотой). У. г. к. сравнимы с катализаторами Ренея.
Их можно использовать для гидрирования алкинов и алкеиов
до алканов, карбонильных соединений до спиртов, ароматиче-
ских нитросоедииений до аминов, а также в качестве катализа-
торов дегидрогенизации. Так, например, стигмастерии дегид-
рируется до соответствующего Д4-3-кетона, причем акцептором
водорода служит циклогексанон. Кроме того, У. г. х. применя-
лись для осуществления восстановительной десульфуризации.
1. Urushibara Catalysts, University of Tokyo Press, Tokyo, Japan (1971).
2. New Hydrogenating Catalysts, Halsted Press, Wiley, New York (1972).
3. Hata K„ Motoyama I., Sakai K, Org. Prep. Proc. Int,, 4, 180 (1972).
ФЕНИЛАЗИД (IV, 25—29; V, 473; VI, 24).
Реакция присоединения [1]. Эфиры гетероциклических р-еи-
аминокарбоновых кислот дигидрофуранового ряда (1а), (16)
взаимодействуют с Ф., образуя триазолы (4а), (46). В реакции
происходит раскрытие дигидрофуранового цикла и миграция
сложиоэфирной группы.
2- А мина - 3-нарбэтонси -
4, 5-ДИ гн дрофу раны
ajR-HJ б) К = СН3
I. W a m h о f f Н., S о h а г Р., Chem. Вег., 104, 3510 (1971).
№
N Нагревание
I!
N 17 0 час
1
ОД
л-ФЕНИЛБЕНЗОИЛ ХЛОРИСТЫЙ,
Мол. вес 216,67, т. пл. 110—112°.
Кори и сотр. [1] в ходе синтеза простагландинов с помощью
Ф. х. получали эфиры л-фенилбензойной кислоты —удобные в
обращении кристаллические вещества, легко идентифицируемые
и легко поддающиеся хроматографической очистке.
1. Core у Е. J., Albonico S. М., Koelliker U,, Schaaf Т. К,, V а г-
m a R. К., J. Am. Chem. Soc., 93, 1491 (1971),
587
ФЕНИЛ-(1-БРОМ-1-ХЛОР-2,2,2-ТРИФТОРЭТИЛ )-РТУТЬ,
C6H5HgCClBrCF3. Мол. вес 474,14, т. пл. 140—144°.
Получение [1],
ТГФ, от —10 до 0°
CaH5HgCi + (СН3)3СОК+CF3CBrClH -—-----* C0H5HgCClBrCF3
Перенос CF3CCl-rpynnw [1]. Реагент можно использовать
в реакциях переноса CF3CCI при 130—140°.
+ C6H5HgCClBrCF3
138°
1. Seyferth D., Mueller D. C., J. Am, Chem. Soc., 93, 3714 (1971).
ФЕНИЛДИАЗОНИЯ ХЛОРИД.
Конденсация с фенилгидразоном альдегида [ 1 ]. Конденса-
ция реагента с фенилгидразоном триметилацетальдегида в сре-
де пиридин — ДМФА приводит к образованию бцс-азосоедине-
ния (1), которое под действием сильного основания перегруп-
пировывается в форм азан (2), ие переходящий в открытую
форму, обладающую желтой окраской, даже при облучении.
+
(CH3)3C-CH-N-N-C6H5 + С6Н;№КСГ
А
ХС6Н5
С(СН3)з N-N
1 ОН , \
<—> c6h5n-n-c-n=nc6h5 -- > (СН3)3С-С н
н
'c6Hs
(I) {2)
Азотная кислота окисляет формазаи (2) в нитрат б-трет-бу-
тил-2,3-дифенилтетразолия (3), превращающийся при облуче-
нии в нитрат 5-трет-бутил-2,3-(2,2/-дифенилен)-тетразолия (4).
Гидросульфит натрия восстанавливает последний до радикала
588
(5), обладающего темно-сиией окраской.
(5)
CjHjBCl; +
CbHjNBCIj
-Nj
С6НИ
(1)
1. Neugebauer F. A., Trlschmann Н., Ann., 706, 107 (1967).
ФЕНИЛДИХЛОРБОРАН, С6Н5ВС12. Мол., вес 158,83.
Стереоспецифический синтез вторичных аминов [1]. Ф. (1)
реагирует с азидом, например циклогексилазидом (2) (полу-
чают из циклогексилбромида и избытка азида натрия в ДМФА
[2]), в бензоле сначала при 20°, а затем при 80°, образуя после
подщелачивания с выходом 96% N-циклогексиланилин (3):
n3
NaOH
—> C6H5NHC6Hn
96%
(з)
Реакция является общей для арилдихлорборанов.
С равным успехом можно использовать и алкилдихлорбора-
ны [3]. Так, например, при взаимодействии циклогексилдихлор-
борана с н-бутилазидом образуется N-н-бутилциклогексиламин
с выходом 95%.
Реакция стереоспецифична, например т’ранс-2-метилцикло-
пентилдихлорбораи (4) реагирует с циклогексилазидом (2), да-
вая N- (гранс-2-метилциклопентил) -циклогексиламин (5).
(4) (5)
689
1. Brown H, C., Midland М. М., Levy А. В., J. Am. Chem., Soc. 95,
2394 (1973).
2. Р ar ker A. J., J. Chem. Soc., 1961, 1328.
3. Brown H. C„ Levy A, B., J. Organometallic Chem., 2l4, 233 (1972);
Brown H. C., Ravindran N., J, Am. Chem. Soc., 95, 2396 (1973).
ФЕНИЛИЗОЦИАНАТ (IV, 45—46; V, 492).
Дезоксигенирование фенолов [1]. Фенолы превращают в уре-
таны обработкой Ф., используя бензол в качестве растворителя,
а также для перекристаллизации. Гидрогенолиз уретанов над
Pd/C в уксусной кислоте приводит к аренам с выходом 20—
80%. Разброс в выходах говорит о том, что здесь важную роль,
возможно, играют стерические факторы.
н2
АгОН + СДМ=С=Ю — ArOCONHC6H5 —> АгН
1. Weaver J. D., Eisenbrann Е. J., Harris L. Е., Chem. Ind., 1973, 187.
N-ФЕНИЛИМИД АЗОДИКАРБОНОВОЙ КИСЛОТЫ (IV,
46—47; V, 492—494; VI, 311—312).
Получение. Опубликован [1] метод получения Ф, а. к., пред-
ложенный Куксоном и сотр. (VI, 312, [1]).
Недавно появилась [2] заключительная статья, посвященная
использованию реагента для защиты диеновой системы кольца
В стероидов (VI, 311—312).
Реакции циклоприсоединения. Метиленциклопропаны (1)
очень медленно вступают в реакцию[2 -ф 2]-циклоприсоединения
по своей экзоциклической двойной связи с Ф.а.к., образуя ад-
дукты состава 1:1 (2) [3]:
С другой стороны, алкенилиденциклопропаны (3) очень бы-
стро взаимодействуют с Ф. а. к., давая аддукты (4), являющиеся
результатом [4 + 2]-циклоприсоединения [4]
(3) (4)
Обсуждается различие в поведении (1) и (3) с привлече-
нием метода молекулярных орбиталей.
690
Реакции циклоприсоединения с вииилциклопропаиами [5].
сс-Циклопропи л стирол (1) не реагирует с малеиновым ангидри-
дом, но с более реакционноспособным диенофилом Ф. а. к. он
дает аддукт состава 2: 1 (4). Этот аддукт, вероятно, образуется
путем циклоприсоединения Ф.а.к. по псевдодиеновой системе
соединения (1). Образующийся при этом продукт (3) вступает
затем в реакцию енового синтеза со второй молекулой Ф.а.к.,
регенерируя ароматическую систему.
транс-2-Фенил-1-изопропенилциклопропан (5) быстро реаги-
рует с соединением (2) (15 лшн, 25°, СН2С12), давая только про-
дукт енового присоединения (6) с выходом 91 %:
•с=снг
снл
(5)
(2)
91$
С£н5
ц HN'
'’jC-CHj-N.
сн2
.О
NC6HS
(6)
О
Винилциклопропан (7) медленно вступает в реакцию с сое-
динением (2), образуя с выходом 87% продукт [2-J-2]-цикло-
присоединения (8):
£>-сн=снг + (2)
(7)
---
87$
(3)
591
Винилциклопропаиы (1) и (5) по-разиому реагируют с хлор-
сульфоизоциаиатом (ХСИ) (IV, 155—156; V, 521—522; VI, 326—
329; этот том). Так, например, при взаимодействии (1) с ХСИ
(CH2CI2, 0°) образуется продукт (9). В этом случае циклопро-
паиовое кольцо сохраняется, а в реакции соединения (5) цик-
лопропановый цикл раскрывается. Предложено возможное объ-
яснение различного поведения соединений (1) и (5):
°
хси-> С—CHCNHSOZC1 ОИ->
88$ С6Н<
(Ю)
ХСИ^
(5)
(12)
Реакция с циклооктатетраеиом [6]. С большинством диено-
филов циклооктатетраеи (1) взаимодействует в форме своего
валентного таутомера бицикло-[4,2,0]-октатриеиа (2) с квазипла-
нарной диеновой системой. Например, тетрацианэтилен, реаги-
руя таким путем, дает (3). Однако более реакционноспособный
диенофил Ф.а.к, взаимодействует не только с валентным тауто-
мером (2), давая (4), ио также вступает в реакцию 1,4-цикло-
присоединения с соединением (1) с образованием (5).
(5)
К92
1. Cookson R. C., Gupte S. S„ Stevens 1. D. R., Watts С. T„ Org.
Syn., 51, 121 (1971).
2. Barton D. H. R-, S h i о r I T., W i d d о w s о n D. A., J. Chem. Soc, (C),
1971, 1968.
3. P a s t о D. J., C h e n A. F.-T., Tetrahedron Letters, 2995 (1972).
4 Paste D. J., Chen A. F.-T„ Binsch G., J. Am. Chem. Soc., 95, 1553
(1973).
5. P a s t о D. J., C h e n A. F.-T., Tetrahedron Letters, 713 (1973).
б' Huis gen R., Konz W. E., Schnegg LI., Angew. Chem., Internal. Ed.,
’ 11, 715 (1972).
N.N-ФЕНИЛМЕТИЛНАТРИЙАМИД (IV, 49—50).
Циклизация Торпа — Циглера [1]. Дурнбос и Стратинг при-
менили этот реагент для циклизации соединения (1) в (2). Про-
вести ацилоиновую конденсацию аналогичных эфиров в этом
случае не удалось.
(1) 8,56 г
1, Doornbos Т., Strating J., Syn. Comm., 1, 193 (1971).
9-ФЕ Н ИЛ-9-0 КС ИАНТРО Н (тритилоиовый спирт). Мол. вес
286,31, т. пл, 213—214°.
Реагент получают с выходом 73% окислением 9-фенилан-
трацеиа.
Ф. быстро реагирует со спиртами, образуя тритилоновые
эфиры. При этом можно осуществить селективную реакцию
первичных спиртов в присутствии вторичных. Эти новые эфиры
устойчивее к действию кислот, чем тритиловые. Их можно рас-
щепить восстановлением по Вольфу — Кижнеру [1].
1. Barnett W. Е., Needham L. L., Powell R. W., Tetrahedron, 28, 419
(1972).
593
ФЕНИЛ ПАЛЛАДИЯ АЦЕТАТ, генерированный in situ из
фенилмеркурацетата (мол, вес. 336,74) и ацетата палладия
(мол. вес 224,49).
Синтез 2-метил-З-феиилпропионового альдегида [1]. Суспен-
зию 33,6 г (0,10 моля) фенилмеркурацетата в 200 мл ацетонит-
рила и 14,4 а (17 мл, 0,2 моля) металлилового спирта переме-
шивают механической мешалкой в трехгорлой колбе на 500 мл,
снабженной обратным холодильником и термометром, охлаж-
дают при перемешивании в бане со льдом и в течение 1 мин в
CsH5HgOCOCH3 + Pd(OCOCH3)2 —> [C6H5PdOCOCH3] + Hg(OCOCH3)2
СНз - СНз
[C6H5PdOCOCH3] + сн2=ссн2он —> сан5сн2<!сн2он —►
1
L PdOCOCH3-l
CH3
—> CSH5CH2CHCHO+CH3COOH+Pd
колбу прибавляют 22,4 г (0,1 моля) порошкообразного ацетата
палладия. Смесь перемешивают при охлаждении 1 час, а затем
при комнатной температуре в течение ~ 12 час.
Реакционную смесь черного цвета разбавляют 100 мл эфира
и'переносят в хроматографическую колонку 45X2,5 см, запол-
ненную смоченной эфиром окисью алюминия (Woelm, 1-й сте-
пени активности). Продукт элюируют с колонки приблизитель-
но 1 л эфира. Коричневый элюат концентрируют отгонкой
эфира при атмосферном давлении на водяной бане с колонкой
Вигре (45 см}. Затем в небольшом вакууме удаляют большую
часть ацетонитрила. Когда в колбе останется около 50 мл рас-
твора, его фильтруют в перегонную колбу на 100 мл, чтобы
удалить небольшой осадок металлического палладия. Перегон-
кой фильтрата с 10-сантиметровой колонкой Вигре получают
9,4 г (69%) 2-метил-З-фенилпропиоиового альдегида ие менее
95%-ной чистоты с т. кип, 77—80°/3 мм.
1. Heck R. F., J. Am. Chem. Soc., 90, 5526 (1968); Org. Syn., 51, 17 (1971).
ФЕНИЛ-®-СТИРИЛ-(ДИМЕТИЛАМИНО)-СУЛЬФОКСОНИЯ
C6HS н
БОРФТОРИД, hzC = C^s^°
с6н/ X'N(CH3)2
0)
ПЛ, 130-131,5°.
Получение [1]. При взаимодействии
нимидметилида лития с бензальдегидом
Мол, вес 345,19, т.
N-метил бензол сульфо-
образуется соединение
594
(2) [2], которое превращают в реагент (1) дегидратацией
(TsOH) и последующим метилированием борфторидом триме-
тилоксония:
он о
CBHeCHCH2SC6H5
(1)
(2)
Циклоприсоединение нуклеофилов по активированной этиле-
новой связи. Реагент взаимодействует с первичными аминами,
енаминами и соединениями с активной метиленовой группой:
mpem-BuNH* , > г и / \ „„„„ о.
8 ^6^5 N - ^Р € П? - В\1
сн3со сьн5
СН3СОСН2СОСН3 —Д \
85’/» Н3С<0>
1. Johnson С. R., Lockard J. Р., Tetrahedron Letters, 4589 (1971),
2. Y a n k е е Е. W., Cram D. J., J. Am. Chem. Soc., 92, 6329 (1970).
ФЕНИЛТИОМЕТИЛЛИТИЙ (V, 496—497).
Метилеиироваиие кетоиов [1]. Реагент образует с кетонами
аддукты (2), которые можно выделить с прекрасным выходом,
разложив реакционную смесь водой [2], или не выделяя превра-
тить в сложные эфиры (3) обработкой м-бутиллитием, а затем
ацилирующим агентом (хлорангидридом или ангидридом кис-
лоты). Восстановление эфира (3) литием в жидком аммиаке
дает метиленовое соединение типа (4) обычно с высокими вы-
ходами. Выделять промежуточный карбинол, как правило, нет
С6Н5БСНгЫ
Т ГФ
R1. ОН
CH2SC6H6
Н-ВиЫ-ТГФ
<СНэСО)-гО
(2)
R* .ОСОСНз
Rs СНгЗСДЦ
(>)
Li/NH3
R1
> = СНа
RaZ
(4)
необходимости. Рассмотренная реакция применима даже в слу-
чае сильно пространственно затрудненного трициклического ке-
695
тона (±)-норзизанона (5).
Этот метод был использован для превращения Д5-холестеио-
на-3 в 3-метилен-Д5-холестен (выход ~40%). Реакция по Вит-
титу Д5-холестенона-3 с метилентрифенилфосфораном дает за-
грязненный примесями 3-метилен-Д5-холестен с выходом лишь
<15%.
С помощью Ф. можно осуществить два новых синтеза. Мети-
ловый эфир декановой кислоты (8) при —25° реагирует с двумя
молями реагента с образованием бис-фенилтиометилкарбинола
(9) с выходом 73%. Последний бензоилируют, а затем восста-
навливают литием в аммиаке. При этом в результате отщепле-
ния обеих СбН5-групп и вытеснения группы OR образуется 2-ме-
тилундецен-1 (10).
OR
I ZH2
СН3(СНо)вСООСН3 CH3(CH2)8C(CH2SC6Hs)2 —> СНз(СН2)8С
”* 'СИ,
(8) (9) (10)
a) R = H, 73%
б) R = COC6H5, 82%
Взаимодействие сс-н-бутилтиометилеициклогексаноиа (11) с
Ф. и последующая обработка продукта реакции 10 %-ной
НС1 и сулемой дает альдегид (12). Восстановление, а затем
метилирование последнего приводят к образованию экзоциклн-
ческого р,у-ненасыщеиного альдегида (13).
1) Li —NH3
2) CH3I
50%
(13)
1. Sow er by R. L., Coates R. M., J. Am. Chem. Soc., 94, 4758 (1972),
2. С о г eу E. J., Seebach D., J. Org. Chem., 31, 4097 (1966).
ФЕНИЛ-(ТРИГАЛОГЕНМЕТИЛ)-РТУТЬ (IV, 52—56- V,
497—500; VI, 312).
Обзор. Сейферт [1] опубликовал обзор химии фенил-(три-
галогеиметил) -ртутиых соединений.
596
Присоединение дихлоркарбена к эфирам азодикарбоновой
кислоты [2]. Фенил- (бромдихлорметил) -ртуть взаимодействует
с диэтиловым эфиром азодикарбоновой кислоты, образуя фе-
нилмеркурбромид (выход 98%) и продукт (87%), который
представляет собой ие ожидаемый диазиридин (1), а соедине-
ние структуры (2). Идентичный продукт получен при декарбок-
силировании CCl3COONa в присутствии диэтилового эфира азо-
дикарбоновой кислоты в кипящем 1,2-диметоксиэтаие (выход
69%).
СгН5ООСх /СООСгНа
XN---N
/С
G1 GL
(CjH5OOC)2NN=CG12
U)
1, Seyferth D., Accts. Chem, Res., 5, 65 (1972).
2, S e у f e r t h D., S h i h H., J. Am. Chem. Soc., 94, 2508 (1972).
ФЕНИЛТРИМЕТИЛАММОНИЯ ПЕРБРОМИД (IV, 56—
57; V, 500).
Получение [1].
+ Br, HBr
C6H5N(CH3)2 + (CH3)2SO4 —> C5H5N(CH3)3 • CH3SO7 -*
CcHsN(CH3)3 Br3 + CH3SO4H
В широкогорлую колбу емкостью 250 мл, снабженную тер-
мометром, наливают раствор 26 мл (0,2 моля) свежеперегиан-
иого диметилаиилина (т. кип. 78713 мм) в 100 мл толуола. -То-
луол предпочитают бензолу вследствие его меньшей токсично-
сти. Смесь перемешивают магнитной мешалкой и нагревают
приблизительно до 40°, затем в течение 20 мин вносят из ка-
пельной воронки 19 мл (0,2 моля) перегнанного диметилсуль-
фата. Все операции необходимо проводить в хорошо действую-
щем вытяжном шкафу. Диметилсульфат берут в количестве, не-
много меньшем стехиометрического, чтобы полностью израсхо-
довать этот токсичный реактив. Через несколько минут начинает
выкристаллизовываться бесцветный метилсульфат аммония. Во
время прибавления диметилсульфата температура реакционной
смеси изменяется незначительно, а потом в течение 1 час мед-
ленно поднимается примерно до 50°. Через 1,5 час после окон-
чания прибавления диметилсульфата смесь нагревают на паро-
вой баие в течение 1 час. По охлаждении выделившийся метил-
сульфат фенилтриметиламмоиия отфильтровывают, промывают
20 мл толуола и высушивают в вакууме; выход продукта 47,5 а
(96%). Этот продукт слегка гигроскопичен, но специальных
мер предосторожности при обращении с ним не требуется.
697
В колбу емкостью 125 мл с широким горлом помещают рас-
твор 10 г (0,04 моля) полученного метилсульфата аммония в
10 мл продажной 48%-ной бромистоводородной кислоты, раз-
бавленной 10 мл воды. Смесь перемешивают магнитной мешал-
кой и в течение примерно 20 мин прикапывают к ней из капель-
ной воронки 2,5 мл брома. Моментально образуется оранжево-
желтый осадок. Перемешивают при комнатной температуре
еще в течение 5—6 час. Продукт собирают на фильтре, промы-
вают водой (около 10 мл) и высушивают и а воздухе в хорошо
действующем вытяжном шкафу. При перекристаллизации 14,9 а
сырого продукта из 25 мл уксусной кислоты получают 14,2 а
(93%) воздушно-сухого кристаллического вещества оранжевого
цвета с т. пл. 115—116°.
1. Jacques J., Marque t A., procedure submitted to Org. Syn., 1971.
ФЕНИЛТРИФТОРМЕТИЛ КЕТЕН (1). Мол. вес 220,15.
Получение [1]. Реагент получают из а-трифторметилмин-
дальней кислоты следующим образом:
F3C< /СООН рС15 FsC. ZCOC1 РС15
;CZ ------------> -----Л
СеН5/ \он 20 25° сен/ \он 160 170
F3C. ZCOC1 Zn F3CX
—> —> V=c=o
C6HS/ \ci CeH5/
(1)
Стереоселективиое ацилирование рацемических вторичных
спиртов [1]. Реагент ацилирует хиральный вторичный спирт (2)
в (Э)-эфир (За) и (К)-эфир (36). Отношение продуктов (За) и
(36) было определено методом ЯМР-19Е. (S)-Диастереоизомеры
преобладают.
C6HS
(1)
(36),R
1. Anders E., Ruch E., Ugi I., Angew. Chem., Internal. Ed., 12, 26 (1973),
598
ФЕНИЛ-(ТРИФТОРМЕТИЛ)-РТУТЬ (VI, 312—313).
Опубликована [1] заключительная статья об использовании
Ф. в качестве переносчика дифторкарбена. Ниже приведен наи-
более употребительный в настоящее время метод получения Ф.
[1,2]:
HgO + 2CF3COOH — > HgfOsCCFgls -j- Н2О
300°
Hg(O2CCF3)2 CF3HgO2CCFs
CF3HgO2CCF3 + (C6H5)2Hg CsH5HgCF3 + C6HsHgO3CCF3
. _ H2°
C6H5HgO3CCF3 + NHtCl --> C6H5HgCl+ NHtO2CCF3’
Сейферт нашел, что Ф. — превосходный предшественник ди-
фторкарбена. Согласно общей методике, используют 1 мол. же
C6H5HgCF3, 2,5—3,0 мол.экв хорошо высушенного йодистого
натрия и 3,0 мол.эка высушенного олефина (Ns). Бензол, абсо-
лютированный в присутствии натрий-кетила из бензофенона, пе-
регоняют прямо в реакционную колбу. Реакционную смесь ки-
пятят при перемешивании в атмосфере Ns в течение 12—18 час.
Феиилмеркуриодид, Nal и NaF удаляют фильтрованием, гем-
дифторциклопропаны выделяют перегонкой или газохромато-
графическими методами обычно с хорошим выходом.
Примеры:
с2н3 сгн5
^с—с
нz '''и
83%
93^
F. F
Некоторые соединения, достаточно хорошо реагирующие с
дихлоркарбеном или хлорфгоркарбеиом, не взаимодействуют с
CeHsHgCFg/NaL К ним относятся кумол, сшил-ди хлор тетр а-
фторацетон и диэтиловый эфир азодикарбоновой кислоты.
699
1. Seyferth D„ Hopper S. P„ J. Org, Chem., 37, 4070 (1972).
2. Seyferth D., Hopper S. P., Murphy G. J., J. Organometallic Chem.,
46,201 (1972).
ЬФЕНИЛ-5-ХЛОРТЕТРАЗОЛ (V, 500—501).
Замещение фенольной ОН-группы на Н. Опубликована [1]
подробная методика превращения и-феинлфенола в дифенил.
1 , М u s 1 i n е г W. J., Gates J. W., J г., Org. Syn., 51, 82 (1971).
ФОРМАЛЬДЕГИД (IV, 65—72; V, 503—504).
Метилирование аминов. Классическим методом метилирова-
ния первичных или вторичных аминов является реакция Эшвей-
лера [1]—Кларка [2]. Реакция заключается во взаимодействии
амииа с Ф. и муравьиной кислотой:
R\ R\
J)NH 4- Н.С=О + НСООН —> \jCH3 + СО2 + Н2О
R2/ R2/
В некоторых случаях получаются сложные смеси продуктов [3].
Недавно были предложены два новых метода метилирования
аминов. В одном из них [4] муравьиную кислоту заменили бор-
гидридом натрия. Например, при обработке стероидного амина
типа (1) большим избытком Ф. в метаноле с последующим вос-
становлением боргидридом натрия получают метилированный
амин (2) с выходом около 85%.
По другому методу [5] амин обрабатывают водным Ф. в аце-
тонитриле, а образовавшийся имин восстанавливают цианбор-
гидридом натрия (этот том). Выходы большей частью высокие
(80—90%). Даже весьма слабое основание n-нитроаиилин (рКо
1,00) превращается в смесь моно- и диметилированного продук-
тов. В реакцию вступают и пространственно-затрудненные ами-
ны (например, N-изопропилциклогексиламин метилируется с
выходом 87%).
Хлорметилирование. Реагент можно использовать для полу-
чения бензилхлорметилового эфира [6]. При этом вместо
600
37%-ного водного раствора Ф. можно, не меняя методики, взять
симм-триоксан (0,493 моля).
10°
С6Н5СН2ОН + НС1 + НСНО — СвН5СН2ОСН3С1
88 —У7 %
j Е s с h w е i 1 е г W., Вег., 38, 880 (1905).
2 С 1 а г k е Н. Т„ Gillespie Н. В., Weisshaus S. Z., J. Am. Chem. Soc.,
’ 55, 4571 (1933).
3 . PineS. H., Sanchez B. L., J. Org. Chem., 36, 829 (1971).
4 Sondegam B. L., Hemo J. H., Charles G., Tetrahedron Letters,
261 (1973).
5 Borch R. F., Hassid A. I., J. Org. Chem., 37, 1673 (1972).
6 Connor D. S., Klein G. W., Taylor G. N., Org. Syn., 52, 16 (1972).
ФОСФОРА ХЛОРОКИСЬ (IV, 84—90; V, 507—508; VI, 315).
Синтез хииазолина [1]. При нагревании З-нитрозо-2-фени*
линдола (1) с 3 молями Ф.х. (или тозилхлорида) в тетраме-
тиленсульфоне (III, 305—306; V, 409—411) при 200° в течение
1 час образуется 2-фенилхиназолин-4(ЗН)-он (2) с выходом
(1) (2)
90%. Реакция, по-видимому, включает перегруппировку Бек-
мана [2].
Элиминирование вицЛ- и -ОН-групп [3]. При взаимодействии
иодгидрина (1) со свежепер егнанным Ф.х. в пиридине сначала
при 0°, а затем при комнатной температуре (30 мин) с высоким
выходом образуется бициклический олефин (2). Реакцию обыч-
но применяют для синтеза олефинов.
Кори и Гренко провели это элиминирование с помощью ме-
зилхлорида (см. Метансульфохлорид, этот том).
Холестерилдихлорфосфат. Холестерилдихлорфосфат (2) мож-
но получить с выходом 87% при обработке холестерина (1)
Ф.х. в присутствии эквимолярного количества триэтиламина в
эфире [4]. Реакция холестерина с Ф. х. в присутствии пиридина
601
в ацетоне дает загрязненный продукт [5].
I. Yoneda F.} Higuchi М„ Nonaka R., Tetrahedron Letters, 359 (1973).
2, Р о р р F. D., М с Е w е п W. Е., Chem. Rev., 58, 370 (1958).
3. Crabbe Р., Guzman A., Tetrahedron Letters, 115 (1972).
4. C r e m 1 у n R. J. W., Dewhurst В. B., W a k e f о r d D. H., Synthesis,
1971, 648.
5. Venn er H., J. prakt. Chem. [4], 12, 59 (1960).
ФОСФОРНАЯ КИСЛОТА (IV, 95).
Перегруппировка ^/-ц-окси-сс- (метиламинометил)-беизилового
спирта (1) в n-оксифеиил ацетальдегид [1]. Колбу, снабженную
термометром и содержащую 4,0 а спирта (1) и 60 мл 85%-ной
Ф. к., погружают в нагретую до 170—180° масляную баню и осто-
рожно вращают до тех пор, пока образовавшийся раствор не
нагреется до 118° [2]. Затем колбу вынимают из бани ровно на
15 сек и за это время с ее поверхности удаляют избыток мине-
рального масла. Сразу же после этого горячий раствор выли-
вают в 720 мл воды при комнатной температуре. Согласно мето-
дике Роббинса [2], через 90лшн раствор насыщают хлористым
(1)
СН2С-ОН
JxSO3Na
ф
он
натрием и смесь встряхивают последовательно со 120-, 70- и
50-миллилитровыми порциями эфира ие менее чем по 15 мин
с каждой. Полученные эфирные вытяжки объединяют со вто-
рым эфирным экстрактом, полученным аналогичной обработкой
еще 4,0 а спирта (1). Объединенный экстракт в течение 10 мин
встряхивают с 40 мл фосфатного буфера (0,1 М, pH 7,6), полу-
ченного добавлением 100 мл воды к 13 мл 0,2 М раствора
NaH2PO4 и 87 мл 0,2 М раствора Na2HPO4-7H2O. Эфирный
слой декантируют, эфир упаривают в токе сухого азота до по-
лучения желтой жидкости, представляющей собой сырой аль-
602
дегнд. В методике даны указания, как получить из соединения
(3) п-оксифенилацетальдегид.
1 Fellman J. Н., Nature, 182, 311 (1958); Arch. Blochem. Blophys., 85, 345
’ (1959).
2 . Robbins! H., Org. Syn., submitted 1972.
ФОСФОРНОЙ КИСЛОТЫ ДИЭТИЛОВОГО ЭФИРА СЕРЕ-
о
f
БРЯНАЯ СОЛЬ, (C2HsO)2POAg. Мол. вес 260,98.
Реагент получают взаимодействием диэтилфосфата (0,12 мо-
ля) с карбонатом серебра (0,05 моля) в эфире при 25°; вы-
ход 82 % •
Надкислоты. Конен и Зильберт [1] разработали общин ме-
тод синтеза надкислот в мягких условиях, заключающийся в
том, что сначала проводят ацилирование реагента в эфире, а
затем полученный смешанный ангидрид обрабатывают пере-
кисью водорода (98 °/о -ной концентрации). Обе стадии проте-
кают с высоким выходом. В качестве кислотного катализатора
О 0 0
f f н2о2, н+
RCOC1 + (C2H50)2P0Ag > (C2H50)2P0CR ------
“AgUI
о
—> RCO3H + (C2HSO)2 JoH + H+
обычно используют метансульфокислоту.
1. KonenD. A., Silbert L. S., J. Org. Chem., 36, 2162 (1971).
ФОСФОР ПЯТИСЕРНИСТЫЙ, P4Si0 (IV, 103; VI, 318—320).
Дегидрирование — дезоксигенирование [1]:
с6н,
1. Cava М. Р., Scheel F. М„ J. Org. Chem., 32, 1304 (1967).
ФОСФОР ПЯТИХЛОРИСТЫЙ (IV, 104—109).
Сложные эфиры галогенгидринов [1]. Взаимодействие 2-кар-
бокси-1,3-диоксоланов (1) с Ф. п. в хлористом метилене при
комнатной температуре приводит к получению сложных эфиров
603
1,2- хлоргидринов (2) с выходом около 80%. Аналогичная обра-
ботка 2-карбокси-1,3-диоксаиа (3) позволяет получить слож-
ный эфир 1,3-хлоргидрииа (4). Реакция 2-карбокси-2-метил-1,3-
диоксапана (5) дает сложный эфир 1,4-хлоргидрииа (6). Для
О С ООН
O^R3
РС15
СН2С12
------> R1 CHC1CHRZOCOR3 + СО + НС1
(2)
zCH3d /С оон fH3
(СН3)2С j;c ---------- ’ > С1СНгССН2ОСОСН3 + СО + НС1 •
СНгО СН3 I
^^С1СНгСН2СН2СН2ОСОСН3 + СО + НС1
(6)
той же цели можно использовать и хлористый тионил, но в этом
случае выходы ниже.
С достаточной убедительностью показано, что при —60° из
диоксолана (7) образуется непосредственно соединение (8),
при 0° оно превращается в сложный эфир (9):
н2с-о^ 7соон
{ X
Н2С-О сн3
РС15
-60\
------
о
II
НгС-О^ С—О
I
Н2С —О I "ci
-РОС13
-со
Н2С-О С1
। X
Н2С-0 сн3
(8)
» С1СН2СНгОСОСН3
(9)
Реакция протекает стереоспецифически с обращением кон-
фигурации у углерод-кислородной связи, превращающейся в
связь углерод — хлор. Например, обработка о-(—)-2-карбокси-
2,4,5-триметил-1,3-диоксолаиа (10) приводит к получению
ь-(+)-э/штро-3-хлор-2-бутилового эфира уксусной кислоты (11).
Взаимодействие эфира (11) с КОН в этиленгликоле дает
604
d-(+)-2,3-эпоксибутан (12), а относительно этой реакции изве-
стно, что она протекает с обращением у связи углерод — гало-
геи.
(ю)
РС15
9 Я
------С — С
81%---\ / ХСНа
о э
(12)
О-Алкилбензогидроксимоилхлориды. О-Алкилбензогидрокси-
моилхлориды (2) можно получить с хорошим выходом взаимо-
действием алкиловых эфиров беизогидроксамовой кислоты (1)
с Ф. п. [2]:
о
и
c4h5-c-nor
н
(I)
(2)
« Cl
>=NZ
--W
(3)
pci С1^
Ч > с=ы,
В недавно проведенном исследовании [3] было показано, что
соединение (2) получается в виде Е-изомера. Под действием
УФ-облучения оно частично изомеризуется в Z-изомер (3).
1-Хлоралкеиы [4]. Взаимодействие енолов (1), где R1 и
R2—электроотрицательные заместители (CN, СООС2Н5, СОСН3,
R\ /R3
zG==Cx
R2^ ОН
PC15
65-85%
СОС6Н5), с Ф.п. (кипячение) приводит к получению 1-хлорал-
кенов (2). Если енол (1) брать в виде енолята щелочного ме-
талла, то лучше пользоваться хлорокисью фосфора (IV, 84—
90; V, 507—508; VI, 315); выход 45—75%.
1. Newman И. S., Chen С. Н., J. Am. Chem. Soc., 94, 2149 (1972); J. Org.
Chem., 38, 1173 (1973).
2. Johnson J. E., Springfield J. R., Hwang J. S., Hayes L. J.,
Cunningham W. C., McClaugherty D. C., J. Org. Chem., 36, 284
(1971).
3. Johnson J. E., Nalley E. A., W e i d i g C., J. Am. Chem. Soc., 95, 2051
(1973).
4. Friedrich K., Thieme H. K., Synthesis, 1973, 111.
ФТОРИСТЫЙ ВОДОРОД БЕЗВОДНЫЙ (IV, 116—118; V,
510—511).
605
Оксазолы. N-Ароил-а-аминокетоны (1) можно с высоким
выходом циклизовать действием Ф. в. б. в замещенные оксазсн
лы [1]:
о r о
и i п
С6Н5С —N - СН- СС6Н5
HF
(1)
1. Daub G. Н., Ackerman М, Е., Hayes F. N,, J. Org. Chem,, 38, 828
(1973),
ФТОРИСТЫЙ ВОДОРОД-БОРА ТРИФТОРИД.
Изомеризация ацетиленов и алленов [1]. HF и BF3> по-ви-
димому, образуют кислоту HBF4, a HF и PF6 дают HPFe. В та-
блице приведено примерное время, необходимое для изомериза-
ции на 20% пяти С6-изомеров с прямой цепью: гексина-1, гек-
сина-2, гексина-3, гексадиеиа-1,2 и гексадиена-2,3 — при обра-
ботке их каждым из четырех катализаторов в сухом сульф-
олане.
Таблица
Примерное время, необходимое для изомеризации иа 20% при 25°
Катализатор Г ексин-1 Гексадиен-1,2 Гексин-2 Гекса- диен-2,3 Гекеин-3
0,1 М hbf4 30 мин 25 сек 40 мин 35 сек 25 мин
0,1 М HPFe 25 мин < 5 сек 2 час 6 час 30 мин
0,96 М H2SO4 24 час 2 час > 100 час 20 мин 40 час
2,07 М H2SO4 — — — — 2 час
Эти результаты подтверждают последовательность реакций,
показанную на следующей схеме:
Г енсин-3
Г енсин-1
Г енсин-2
Генсаднен -1, 2 Генсадиен -2, 3
1. Barry В. J,, Beale W. J., Carr М. D., Hei S.-К., Reid I., J. C S
Chem. Comm., 1973, 177.
бис-(ФТОРОКСИ)-ДИФТОРМЕТАН, CF2(OF)2. Мол. вес
101,01, при —184° бесцветная жидкость.
Реагент получают фторированием СО2 в присутствии CsF{l].
606
Электрофильное фторирование. Бартон и сотр. [2], исследо-
вав целый ряд фтороксисоединеиий в качестве агентов электро-
фильного фторирования, пришли к заключению, что Ф. можно
использовать там же, где и трифторметилгипофторит (V, 457—
458; VI, 293—294), однако в перспективе Ф. может оказаться
дешевле.
1. Hohorst F. A., Shreeve J. М., J. Am. Chem. Soc., 89, 1809 (1967);
Thompson Р. G., J. Am. Chem. Soc., 89, 1811 (1967); Cauble R. L.,
CadyG. H„ J. Am. Chem. Soc., 89, 1962 (1967).
2. Barton D. H. R., Hesse R. H., P e c h e t M. M., T a г z i a G., To h H. T.,
Westcott N. D., Chem. Comniun., 1972, 122.
ФТОРСУЛЬФОНОВОЙ КИСЛОТЫ МЕТИЛОВЫЙ ЭФИР
(VI, 321).
Алкилирование [1]. Кетон (1) под действием реагента в при-
сутствии основного катализатора дает продукт О-алкилироваиия
7-метоксибицикло-[4,2,2]-декатетраеи-2,4,7,9 (2) с выходом 93—
95%. Так, например, при обработке кетона (1) сначала трет-бу-
тилатом калия (3 экв, 4 мин), а затем Ф.к. м.э. (3 экв, 3 мин)
в ГМТФК при 5° образуется соединение (2) с высоким выхо-
дом. В меиее полярных средах наблюдается значительное С-ал-
килирование.
(I) (2)
Удаление тиоацетальной группы [2]. Дитиоацетали (1) под
действием Ф.к. м.э. в безводном бензоле легко метилируются
с образованием солей сульфония (2). Последние гидролитиче-
ски расщепляются гидроокисью натрия до соответствующих ке-
тонов (3). Общие выходы составляют 60—90%.
сн3
(СНг)« 2 CHyOSQiF, '^с/' ~7снг)г 2 FSO3" J5aQH
CHj
(1) П = 2 илиЗ (2)
607
Метилсульфоиаты [3]. Фенолы, а также эфиры фенолов
при нагревании с Ф. к. м. э. при 100° в течение 12 час превра-
щаются с умеренными выходами в метилсульфонаты:
H3COSOZF
' 40*
OCHj
зогосн3
1. Press J. В., Shechter Н„ Tetrahedron Letters, 2677 (1972).
2. Н о T.-L., Wong С. М,, Synthesis, 1972, 561.
3. Kametani Т., Takahashi К., Ogasawara К., Synthesis, 1972, 473.
ХИНУКЛИДИН (IV, 122).
Деметилирование 2-метокситропона [1]. 2-Метокситропои (1)
деметилируется при кипячении в бензоле с хииуклидином, взя-
тым в четырехкратном избытке, давая с выходом 60% соедине-
ние (2). С ДАБЦО (V, 89—90; этот том) реакция идет медлен-
нее и дает более низкий выход целевого продукта. Деметилиро-
вание можно осуществить также при действии меркаптидов в
гексаметаполе.
1. Biggi G., Ci ma F. D., Pietra F., Tetrahedron Letters, 183 (1973).
«-ХЛОРАКРИЛОВОЙ КИСЛОТЫ ХЛОРАНГИДРИД,
CH2=CC1COC1. Мол. вес 124,96, т. кип. 45—48778—80 мм.
Получение [1]. Реагент получают с выходом 37% дегидро-
хлорированием а,р*дихлорпропионилхлорида с помощью ди-
этйланилина.
Реакция Дильса — Альдера [2]. Реагент является диенофи-
лом с высокой реакционной способностью, сравнимой с реакци-
оиноспособностью малеинового ангидрида. X. к. х. взаимодейст-
вует с 5-замещенными циклопентадиенами с образованием
аддуктов, в которых заместитель у седьмого углеродного атома
находится исключительно в дмти-положеиии по отношению к
мостику, несущему хлор и хлорформильную группу. Так, напри-
мер, реакция X. к. х. с 5-бензилоксиметилциклопеитадиеном (1)
дает с выходом около 99% аддукт (2), представляющий собой
смесь (2:1) экзо- и эндо-хлорангидридов кислот. Обработка
аддукта (2) азидом натрия приводит к соответствующему аци-
лазиду, который при нагревании подвергается перегруппировке
Курциуса в изоцианат, дающий после гидролиза бициклический
20 Зак, 596
609
кетон (3) с общим выходом около 90%. Рассмотренная после-
Не сколько
стадйй
90%”
С6Н6СН2ОСНг
(-3)
довательиость реакций, таким образом, служит методом 1,4-при-
соединения фрагмента —СН2СО— к 1,3-диеиам.
1 Marvel С. S., Dec J., Cooke Н. G,, Jr., Cowan J. C., J. Am. Chem.
Soc., 62, 3495 (1940).
2. Corey E. J., Ravindranathan T., Terashima S., J. Am. Chem,
Soc., 93, 4326 (1971).
2-ХЛОРАКРИЛОНИТРИЛ, CH2=CC1CN. Мол. вес 87,51.
п™ 1,4323.
Реакция Дильса — Альдера [1]. X. (VI, 160), а также 2-аце-
токсиакрилоиитрил (I, 83; V, 30) используют иногда как экви-
валенты кетена в реакциях Дильса—> Альдера. Эванс н сотр.[1]
недавно сравнили поведение этих реагентов в реакции с дигид-
роанизолом (1), в результате которой образуются аддукты (2)
и (3). При этом X. оказался не только более реакционноспособ-
ным, ио также и более региоспецифичиым агентом. Так, он реа-
гирует с дигидроанизолом (1) с образованием почти исключи-
тельно аддукта (2), а не (3). 2-Ацетоксиакрилоиитрил в этой
реакции дает в основном продукт, аналогичный аддукту (2), и,
кроме того, некоторое количество изомера, соответствующего
аддукту (3).
ch2=ccicn
80$
Те же химики улучшили метод превращения полученных из
X. аддуктов в соответствующие кетоны. Согласно их рекомен-
дациям, это превращение можно реализовать с высоким выхо-
610
дом (>80%) с помощью сульфида натрия в кипящем этаноле.
NaaS- 9 Н2О
о
1 Evans D. A., Scott W. L., Truesdale L. К., Tetrahedron Letters, 121
' (1972).
ХЛОРАМИН (IV, 123—126; V, 513—514; VI, 322).
Окисление. Анилин окисляется хлорамином (комнатная тем-
пература, 12 час) до тра«с-азобензола (50%), содержащего
следы цис-азобензола, и 4-аиилиноазобензола (12%). В некото-
рых случаях этот метод приводит и к цис-азобензолам, которые
2CeH5NH2 + 2NH2C1 —> C6H6N==NC6H6 + 2NH4C1
иначе можно получить лишь с низким выходом облучением со-
ответствующих транс-изомеров.
Спирты окисляются до карбонильных соединений с высоки-
ми выходами, сравнимыми с выходами, достигаемыми при дей-
ствии 1-хлорбеизотриазола (V, 515; VI, 323—324).
Бензиловый спирт окисляется до бензальдегида (выход
74%), а дифеиилкарбинол дает бензофенон (выход 87%) [1].
1,2,3-Триазолы [2]. При аминировании 1,2,4-триазин- (2Н)-
онов-3 (которые легко получить реакцией бензилов с семикар-
базидом) под действием эфириого раствора X. неожиданно
произошло сужение цикла с образованием 1,2,3-триазолов
(формально с отщеплением окиси углерода). 5,6-Дифеиил-1,2,4-
NH;C1
94%
Т х + nh4ci+ со
it
триазии- (2Н) -он-3 (1), например, превращается в 4,5-дифенил-
триазол (2) с выходом 94%- X.,. следовательно, в этой реакции
проявляет себя как окислитель, вероятно первоначально хлори-
рующий соединение (1) по азоту.
1. J a f f а г i G. A., N u n n A. J., J. Chem. Soc. (С), 1971, 823.
2. Rees С. W., S а 1 е A, A., Chem. Comm., 1971, 532.
3. Elderfield R. C., Heterocyclic Compounds, Wiley, New York, 7, 759
(1961).
ХЛОРАМИН-Т (N-хлор-н-толуолсульфамид-натрий),
H-CH3C6H4SO2NClNa.3H2O. Мол. вес. 281,68.
Удаление этилеитиокетальиой. защитной группы (1). Боль-
шое число разнообразных 1,3-дитиоланов (этилеитиокеталей)
гладко реагирует с X. в водном метаноле или этаноле с образо-
ванием соответствующих карбонильных соединений с хороши-
20*
611
ми} а часто даже и с превосходными выходами. Например, при
обработке соединения (1) X. в водном метаноле или 85%-ном
водном этаноле образуется флуоренон (2) с выходом 86%.Для
получения карбонильных соединений с оптимальным выходом
достаточно 2 экв реагента, однако для более быстрого заверше-
rt-CH3C6H4SO;iNClNa- ЗНгО
СН3ОН, С2Н5ОН, НгО
(2)
ния реакции рекомендуется брать 4 моля X.
Реагент можно использовать также для регенерации кето-
нов и альдегидов из 1,3-оксатиоланов [2]. Например, 1,4-окса-
тиаспиро-[4,4]-нонан (1) при обработке X. в водном 85%-ном
СН3ОН при 25° в течение 2 мин дает с выходом 91% циклопен-
танон (2).
rt-CH3C6H4SO2NClNa- 3 Н2О
(П (2)
1. Huurdeman W. F. J., Wynberg FL, Emerson D. W., Tetrahedron
Letters, 3449 (1971).
2. Emerson D. W., Wynberg H., Tetrahedron Letters., 3445 (1971).
ХЛОРАНИЛ (IV, 127—130; V, 514—515; VI, 323).
Дегидрирование дикетона (3) до углеводорода октацетрепа
(4) синего цвета [1]:
612
1. Erii niii R. К., Ann., 721, 43 (1969).
о
о-ХЛОРЛНИЛ (IV, 130—132).
Окисление 4-метилпирокатехина в эфире до 4-метил-о-беизо-
хинона [1]:
I. Horner L., В it г g е г Т., Ann., 70S, 105 (1967).
л-ХЛОРБЕНЗОИЛ НИТРИТ, ft-ClC6H4CO«ONO. Мол. вес
185,57, т. кип. 50°/0,2 мм.
Реагент синтезируют из л-хлорбеизоата серебра и хлористо-
го иитрознла и хранят в виде бензольного раствора [1]:
n-ClCgHiCOOAg + NOC1 —► п-С1С6Н4СО • ONO + AgCl
Дегидробеизол. Реакции ацетанилида с X. в сухом бензоле
в присутствии тетрафенилциклопеитадиенона, антрацена, 9,10-
диметоксиантрацена и метилметакрилата, служащих для улав-
ливания дегидробензола, приводят к образованию соответст-
613
вующих аддуктов дегидробеизола—1,2,3,4-тетрафенилнафталн-
CeH8NHAc
CbHeN(NO)Ac + п-С1С6Н4СООН
C6H5NUcO’
на (выход 70%), триптицеиа (выход 16%), 9,10-диметокситри-
птицеиа (выход 14%) и метилового эфира 2-метилеи-З-феиил-
пропионовой кислоты (выход 31%) соответственно.
Последняя реакция [2] представляет собой пример енового
синтеза (обзор реакций енового синтеза см. [3]):
СНз
СН2
—> С8НбСН2А-СООСН3
о! л
При взаимодействии X. со смесью анилина и уксусного ангид-
рида выходы аддуктов дегидробензола снижаются. Можно
использовать также и замещенные анилины, причем из мета-
замещенных анилинов аддукты получаются с гораздо более вы-
сокими выходами, чем из соответствующих пара-изомеров, a
многие орто-изомеры вообще не дают ариновых аддуктов.
Авторами [1] было показано также, что N-нитрозо-и-хлор-
беизоилаиилины дают арииы с гораздо более высокими выхо-
дами по сравнению с соответствующими ацетильными производ-
ными.
1. Baigrie В,, Cadogan J. 1. G., Mitchell J. R., Robertson A. R.,
S h a r p J. T., J. C. S. Perkin I, 1972, 2563.
2. T a b u s h i Okazaki K., Oda R., Tetrahedron, 25, 4401 (1969).
3. Hoffman H. M. R., Angew. Chem., Internal. Ed., 8, 556 (1969).
1-ХЛОРБЕНЗОТРИАЗОЛ (V, 515; VI, 323—324).
Производившая этот реагент американская фирма сообщи-
ла, что во время упаковки одна из партий реагента загорелась,
после чего фирма перестала его выпускать [1].
Хлорирование алкалоидов ряда иидола [2]. Алкалоиды типа
(1) при взаимодействии с X., взятом в 5—10%-ном молярном
избытке, в хлористом метилене или бензоле с высоким выходом
превращаются в хлориидоленины (2). Ранее для такого рода
614
превращений использовали трет-бутилгипохлорит, но он дает
целевой продукт не с такими высокими выходами и, кроме то-
го, более взрывоопасен по сравнению с X. Рассмотренную реак-
цию с успехом использовали для хлорирования иохимбииа без
побочного окисления вторичной гидроксильной группы.
Хлорирование карбазола [3]. При обработке 1 мол. экв
реагента в хлористом метилене при комнатной температуре кар-
базол (1) превращается в 3-хлоркарбазол (2) с выходом 79%.
3,6-Днхлоркарбазол получают с выходом 64%, используя 2 мол.
экв. реагента, при действии 4 мол. экв реагента получают с вы-
ходом 61 % 1,3,6,8-тетрахлоркарбазол. Этим методом можно
(2)
(О
хлорировать и N-алкилированиые карбазолы.
Окисление сульфидов [4]. Сульфиды окисляются реагентом
с промежуточным образованием аддуктов (1), обработка кото-
рых спиртом, а затем борфторидом серебра дает борфториды
алкоксисульфониев (2) с выходом 30—40%:
ri_s-r3 +
(О
R3OH
AgBF4^
OR’
R1- S—'R3
+
bf4'
При взаимодействии аддуктов (1) с первичными или вто-
ричными аминами получаются соли аминосульфоиия с выходом
615
40—85%:
R3R4NH R3—N—R4
AgBF4 I
-----> R!—S—R2
+
BF?
Тиоацетали стероидов-> кетоны [5]. Хотя этилен- и триме-
тилентиоацетали кетонов представляют собой устойчивые в кис-
лой среде защитные группы карбонильной функции в ряду сте-
роидов (1), однако до сих пор регенерация из иих карбонилы
ного соединения была трудно осуществима. Теперь в распоря-
жении химиков имеется простой метод регенерации. Тиоацеталь
окисляют с помощью X. до дисульфоксида (2), который не вы*
деляя разлагают избытком гидроокиси натрия до кетона (3).
1. Н о р р s Н. В., Chem. Eng. News, July 26, 1971, p. 3.
2. Lichman R. V., J. Chem Soc. (C), 1971, 2539.
3. Bowyer P. M., Iles D. H., L e d w i t h A., J. Chem. Soc., 1971, 2775.
4. J о h n s о n C. R., Bacon С. C., Kingsbury W. D., Tetrahedron Letters,
501 (1972).
5. Heaton P. R., Midgley J. M., Whalley W. B., Chem. Comm., 1971,
750.
ХЛОРДИМЕТИЛСУЛЬФИД, C1CH2SCH3. Мол. вес 96,58,
т. пл. 105°.
Метилтиометиловые эфиры карбоновых кислот [1]. Превра-
щение карбоновых кислот в метилтиометиловые эфиры исполь-
зуется в целях защиты карбоксильных групп. Эти эфиры полу-
чают кипячением кислоты с реагентом в ацетонитриле в присущ
ствии триэтиламииа в течение 24 час [2]. Полученные эфиры
CH3CN; Et3N
CICH2SCH3 + RCOOH —-----> RCOOCH2SCH3
вполне устойчивы к водным растворам щелочей и к действию
мягких восстановителей (NaBEU, Zn—СН3ОН). Их можно гид-
ролизовать в кислых или нейтральных растворах. В одной из
методик рекомендуется гидролизовать эфир, обрабатывая его
CF3COOH (15 мин, при комнатной температуре, выход 80—
98%). Согласно другой методике, эфир кипятят с иодистым ме-
616
тилом в водном ацетоне в течение 17 час (выход 62—90%). На
первой стадии в этом случае происходит S-метилирование:
СН31 +
RCOOCH2SCH3 -----> RCOOCH3S(CH3)ar
1. Н о T.-L., Wong С. М., J. С. S. Chem. Comm., 1973, 224.
2. Mills R. H., F а г г a r M. W., Weinkauff 0. J.. Chem, Ind., 1962, 2144;
Wagenkneckt J. H., Balzer M. M., Chruma J. L., Syn. Comm., 2,
215 (1972).
ХЛОРИРИДИЕВАЯ КИСЛОТА (IV, 133—134; V, 516; VI,
324).
Хенбеста восстановление. Восстановление кетона (1) с по-
мощью X. к. в присутствии триметилфосфита приводит к обра-
зованию с выходом 73% аксиального 3[3-спирта (2). Восстанов-
ление кетоиа (1) с помощью боргидрида натрия дает в каче-
стве главного продукта 74% экваториального За-спирта и толь-
ко 17% искомого спирта (2) [1].
1. Р е 11 i t G. R„ D i a s J. R„ J. Org. Chem., 36, 3207 (1971).
ХЛОРИСТЫЙ ВОДОРОД (V, 516).
Ароматизация стероида [1]. Если соединение (1), получен-
ное из холевой кислоты, растворить в метаноле и в течение
4 час пропускать в раствор X. в., то с высоким выходом полу-
чается «ароматический стероид» (2):
(1), 12 а и 12 р - изомерм
1,935s
(2) 1,85 г
617
1. Meney J., Kim Y.-H., Stevenson R., Margulis T. N., Tetrahedron,
29, 21 (1973).
ХДОРМЕТИЛМЕТИЛОВЫЙ ЭФИР (IV, 134—137).
Вниманий Реагент канцерогенен. бис-Хлорметиловый эфир —
еще более сильный канцероген [1].
Хлорметилирование иитрофенолов [2]. Описано хлорметили-
рование с помощью реагента о-нитрофеиолов и па нескольких
примерах n-нитрофеиолов. С динитрофенолами реакция не идет.
Для реакции требуется катализатор (хлористый цинк).
Бисхлорметилирование. Тетразамещеыные ароматические со-
единения, такие, как нитромезитилеи (1), при обработке Х.э.
в присутствии 60% -ного олеума хлорметилируются дважды [3].
Метоксиметилироваиие эфиров р-кетокислот [4]. Алкилирова-
ние натриевых производных эфиров р-кетокислот с помощью
Х.э. в гексаметаполе дает почти исключительно продукты О-ал-
килирования (96—100%). В менее полярных апротонных рас-
творителях выходы О-алкилироваииых продуктов ниже. Эти
простые эфиры енолов при действии лития в жидком аммиаке
восстанавливаются до эфиров соответствующих насыщенных
кислот (выход 23—61 % ) ..Рассмотренный метод особенно удобен
для восстановления пространственно затрудненных кетогрупп
618
в эфирах р-кетокнслот.
/СООС2Н5
с:
1) NaH. гексаметапол
2) СН3ОСНаС1
СН3ОСН2О
СООС2Н
LI, NH3
---------
1. Van D u i s е n B. L., S i v a k A., Goldschmidt В. Katz С., M el-
ch i о n n e S., J. Nat. Cancer Inst., 43, 481 (1969); Drew R. T., С a p p I e 1-
lo V., Larkin S., Kuschner M., 1970 Conference American Industrial
Hygiene Association, Abstract 164.
2. Bohmer V., Deveaux J., Org. Prep. Proc. Int., 4, 283 (1972).
3. Suzuki H„ Bull. Chem. Soc. Japan, 43, 3299 (1970).
4. Coates R. M., Shaw J. E., J. Org. Chem., 35, 2597, 2601 (1970).
CH2C1
у-ХЛОРМЕТИЛПИРИДИНА ХЛОРГИДРАТ, .HC1
Мол. вес 164,04, т. пл. 160—163°.
Пептидный синтез с помощью пиколиловых эфиров амино-
кислот. Янг и сотр. [1] разработали метод синтеза пептидов, в
котором концевые карбоксильные группы защищают, превра-
щая их в у-пиколиловые эфиры (1). Эфиры (1) получают взаи-
модействием X. х. с тетраметилгуанидиииевой солью бензилок-
сикарбониламинокислоты в ДМФА при 90—100°. Из этих про-
изводных N-защитную группу можно удалить действием броми-
стого водорода в уксусной кислоте. у-Пиколиловые эфиры
можно расщепить обработкой холодной щелочью, каталитиче-
ским гидрированием, электролитическим восстановлением или
R
I
Ь1Н2СНСООСН
О)
действием натрия в жидком аммиаке. Новый метод равноценен
методу синтеза пептидов иа твердом носителе, в котором конце-
вая карбоксильная группа этерифицируется оксиметилполисти-
рольной смолой. Но его преимущество в том, что пептидный
синтез в данном случае протекает в гомогенном растворе. Сла-
боосновиой характер остатка, которым защищен пептид, дает
возможность отделить последний от избытка ацилирующего
619
агента и побочных продуктов. Выделение пептида осуществля-
ется с помощью катионообменных смол или экстракцией раз-
бавленной кислотой. Эту новую методику впервые использовали
в синтезе тетрапептида ь-лейцил-ь-аланилглицил-ь-валина [2],
а позже —в синтезе Уа15-ангиотенсина-П (общий выход 38%)
[2] и брадикинина (общий выход 42%) [3].
1. С amble R., Garner R., Young G. T„ J. Chem. Soc. (C), 1969, 1911.
2. Garner R., Young G. T., J. Chem. Soc. (C), 1971, 50.
3. Schafer D. J., Young G. T., Elliott D. F., Wade R., J. Chem, Soc.
(C), 1971, 46.
лг-ХЛОРНАДБЕНЗОЙНАЯ КИСЛОТА (IV, 138—143; V,
517—519; VI, 324—325).
Окисление углеводов по Байеру — Виллигеру. Хейнс и сотр.
[1] окисляли углеводы по Байеру — Виллигеру с помощью X. к.
в хлороформе, не содержащем этанола, при комнатной темпера-
туре.
Пример:
Эпоксидирование при повышенных температурах. На одной
нз стадий синтеза тетродотоксина японским химикам [2,3] пона-
добилось эпоксидировать двойную связь олефииа (1). Однако
этот олефин оказался инертным к действию X. к., надуксусной
(1)
и надмуравьиной кислот в обычных условиях. Тогда авторы по-
пытались провести эпоксидирование при повышенной темпера-
туре (90°) и нашли, что в этом случае необходимо присутствие
ингибитора радикальных реакций. Наилучшим ингибитором ока-
зался продажный препарат 4,4'-тио-бис- (6-трет-бутил-З-метил-
фенол) . Олефин (1) при взаимодействии с X. к. в дихлорэтане
620
в присутствии каталитических количеств ингибитора в течение
2 час при 90° превращается в искомый эпоксид с выходом бо-
лее 95%.
Новый метод был с успехом применен для эпоксидирования
октена-1, додецена-1 и метилметакрилата, которые ранее уда-
валось эпоксидировать только под действием такого сильного
эпоксидирующего агента, как трифторнадуксусная кислота.
1. Кб 11 Р., Dilrrfeld R., Wolfmeier U., Неу ns К-, Tetrahedron Let-
ters, 5081 (1972).
2. К i s h i Y., Aratani M.., Tanino H., Fukuyama T., Goto T,,
InoueS., S u g i u r a S., К a k о i H., Chem, Comm., 1972, 64.
3. Kishi Y., Aratani M., Fukuyama T., Nakatsubo F., Goto T.,
Inoue S., Tanino H, Sugiura S., Kakoi H., J. Am. Chem. Soc., 94,
9217 (1972).
N-ХЛОРПОЛ ИМАЛЕИНИМИД,
Полималеинимид получают свободнорадикальной полимери-
зацией малеииимида в присутствии дивинилбензола (5%) или
Г^М'-метилен-б/щ-акриламида (5—10%), используемых в каче-
стве сшивающих агентов. X. получают, обрабатывая суспензию
полимера в водном растворе гидроокиси натрия раствором хло-
ра в четыреххлористом углероде.
Хлорирование алкилбензолов [1]. Хлорирование алкилбен-
золов с помощью X. при 100—140° в течение 16—20 час дает
исключительно продукты ароматического замещения в отличие
от хлорирования с помощью N-хлорсукцинимида (NXC) в тех
же условиях. В последнем случае образуется смесь продуктов
хлорирования как бензольного кольца, так и боковых алкиль-
ных групп. Различие в поведении X. и NXC приписывают влия-
нию полимерного скелета первого из них. В самом деле, хлори-
рование алкилбензолов с помощью NXC в присутствии сукцини-
мида направляется главным образом в бензольное кольцо. Ср.
N-Бромполималеинимид, этот том.
1. Yaroslavsky С., Katchalski Е., Tetrahedron Letters, 5173 (1972).
ХЛОРСУЛЬФОИЗОЦИАНАТ (IV, 155—156; V, 521—522;
VI, 326-329).
[2 + 2]-Циклоприсоединение с олефинами (VI, 326—327).
При взаимодействии реагента с олефинами, содержащими внут-
реннюю пространственно-незатрудненную двойную связь, в ка-
621
честве основных продуктов образуются N-сульфохлориды а,р-ди-
замещенных р-лактамов (1) и (2) [1]:
R’CHjCHCHC^R2
R1CH2CH=CHCH2RZ
O = C=NSOzC1
SO2C1
(1)
^CHaCHCHCHaR5
O=C-N
SO2C1
(2)
R1CH=CHCHCH2R2 +
I
O=C-NHSO2C1
(3)
RnCH2CHCH=CHR2
O = C — NHSOjCl
(4)
+
Малпасс и Тведл [2] в результате реакции X. с камфеном
(5) в CH2CI2 при —60° выделили производное р-лактама (6).
При более высоких температурах оно перегруппировывается в
термодинамически более стабильные изомеры (7) и (8).
Присоединение к ненасыщенным бициклическим монотерпе-
иам [3]. X. реагирует с сс-пиненом (1) в эфире при —73° регио-
специфично, давая нестабильный аддукт состава 1 : 1 3-хлорсуль-
фонил-2,8,8-триметил-3-азатрицикло-[5,1,1,02>5]-нонанон-4 (2) с
выходом 65%. При нагревании, а также при хроматографирова-
нии на силикагеле аддукт (2) подвергается перегруппировке
типа перегруппировки Вагнера — Меервейна, превращаясь с вы-
ходом 40% в N-сульфохлорид лактама (3). Сульфит натрия вос-
622
станавливает аддукт (2) до р-лактама 2,8,8-триметил-З-азатри-
цикло-[5,1,1,02’5]-нонанона-4 (4) с выходом 61%.
р-Пинен (5) реагирует с X. при —73° с образованием край-
не нестабильного аддукта состава 1:1 (6), строение которого
установлено восстановлением до р-лактама 2-азетидинон-4-спи-
ро-2/(6/,6/-диметилбицикло-[3,1,1]-гептана) (9). При хроматогра-
фировании на колонке с силикагелем аддукт (6) перегруппиро-
вывается, образуя смесь соединений (7) и (8).
Камфен (10) реагирует с X. в эфире при температурах от
—3 до 0°, давая аддукт 4-хлорсульфонил-10,10-диметил-4-азатри-
623
цикло-[5,2,1.01’5]-деканон-3 (12) с выходом 77%':
В результате присоединения X. к Д3-карену (13) в кипящем
эфире (23 час} с выходом 72% образуется аддукт состава 1 : 1
4 - хлор сульфонил - 3,9,9 -трнметил-4-азатрицикл о-[6,1,0,03-6]-нона-
нон-5 (14):
о
(13) (141
Циклоприсоединение к напряженным бициклическим углево-
дородам [4]. С простыми алкенами X. вступает в реакцию [2 -ф 2]-
О)
(3)
I) X.
2) NaOH, аодн.ацетон
43%
D X.
2) C6H5SH, Ру
51%
СН3
(6)
624
циклоприсоединения. Однако по мере усложнения участвующих
в реакции циклических систем возрастает возможность реализа-
ции [14-4]-, [1+5]- и [1 + 6]-циклоприсоединения. Так, напри-
мер, X. реагирует с трицикло-[4,1,0,02’7]-гептаном (1), давая после
гидролиза в качестве основного продукта лактам (2). В тех же
условиях из 1,2,2-триметилбицикло-[1,1,0]-бутана (3) в качестве
основного продукта образуется лактам (4). 1,2,2,3-Тетраметил-
бицикло-[1,1,0]-бутан (5) образует лактам (6). В аналогичных
условиях 1,3-дим ети л би цикл о -[1,1,0]-бутан (7) дает бицикличе-
ский лактам (8) [4, 5].
1)Х.
2) C6H5SH, Ру
15,5%
1. Bestian Н,, Biener Н., Clauss К-, Heyn Н., Ann., 718, 94 (1968).
2. Malpass J. R., T w e d d 1 e N. J., J. C. S. Chem. Comm., 1972, 1244.
3. Sasaki T., Eguchi S., Yamada H,, J. Org. Chem., 38, 679 (1973);
Malpass J. R., Tetrahedron Letters, 4951 (1972).
4. Paquette L. A., Allen G. R., Jr., Broadhurst M. J., J. Am. Chem.
Soc., 93, 4503 (1971).
5. P a q u e 11 e L. A., private communication.
1-ХЛОР-Х,Х,2-ТРИМЕТИЛПРОПЕНИЛАМИН,
ZC1
(CH3)2C=C(
XN(CH3)2
Мол. вес 133,63, т. кип. 40°/25 мм.
Получение. Этот а-хлоренамин (1) получают с выходом
около 80% при взаимодействии фосгена с Ь1,Ь1-диметиламидом
изомасляной кислоты с последующим элиминированием хлори-
стого водорода с помощью триэтиламина [1]
11 COU2, р, pi-
ll (CZH5)2O (C£H5)aN
(CH3)2CHCN(CH3)2 ---------> (СН3)2СНС' ------->
МЧ(СН3)2
ZC1 +
—> (СН3)2С=С' (CH3)2C=C=N(CH3)2Cr
М4(СНз)2
(I> (la)
Аминоалкенилироваиие [2]. Реагент в полярной форме (1а)
легко реагирует с обладающими повышенной электронной плот-
ностью ароматическими системами, давая продукты аминоалке-
нилирования:
А 0
(CSH5)3N /Аг н3о+ II
АгН+(1а) --------(СН3)2С=»+ ----> (СН3)2СНСАг
Ж(СН3)2
625
X., например, легко реагирует с фураном, пирролом и NjN-диме-
тиланилином, образуя продукты замещения; выход 85—95%.
Алкилированные циклобутаноны [3]. Хлоренамин X. при вза-
имодействии с борфторидом серебра в хлористом метилене при
—60° превращается в борфто'рид тетраметилкетениммония (2).
Эта соль легко вступает в реакцию [2 2]-циклоприсоединения
с олефинами, давая иминиевые соли (3), которые легко гидро-
/С1 д +
(СН3)2С=С( (CH3)2C==C=N(CH3)2BF-
\n(CH,)2 -АгС1
(I) (2)
(2) t >С=С<
ОН
--->
лизуются до алкилированных циклобутанонов (4). Выходы цик-
лоаддуктов высокие (80—95%).
I. Ghosez L., Have aux В., Viehe Н, G., Angew. Chem., Internal Ed„
6, 454 (1969).
2 M arc ha nd - В ry п ae г t J., Ghosez L., J. Am. Chem. Soc., 94, 2869
(1972).
3. Marchand-Brynaert J., Ghosez L., J. Am. Chem. Soc., 94, 2871
(1972).
ХЛОРТРИФТОРЭТИЛЕН (2) (IV, 246).
Присоединение хлортрифторэтилена (2) к n-нитрофенилаце-
тилену или к n-метоксифенилацетилену приводит к образованию
циклобутенов (За) и (36), обработка которых серной кислотой
дает соответствующие циклобутендноны (4а) и (46) [1]:
626
Аналогично был получен дион (5), однако авторы не выяс-
нили, обладает ли он хиноноподобными свойствами.
I. Ried W, Schmidt А. Н., Kuhn W, Chem. Ber, 104, 2622 (1971).
ХЛОРУГОЛЬНОЙ КИСЛОТЫ р,р,р-ТРИХЛОРЭТИЛОВЫЙ
ЭФИР (V, 527—528).
Смесь эндо- и экзо-бицикло-[3,3,1]-нонанол-2-онов-9 (1) и (2)
удалось разделить кристаллизацией и хроматографией их три-
хлорэтоксикарбонильных производных [1]. Альдоли можно реге-
нерировать без эпимеризации при обработке трихлорэтоксикар-
бонильных производных цинковой пылью в уксусной кислоте.
I. Gravel D, Rahal S, Regnault A, Can. J. Chem, 50, 3846 (1972).
ХЛОРУГОЛЬНОЙ КИСЛОТЫ 9-ФЛУОРЕНИЛМЕТИЛО-
ВЫЙ ЭФИР (VI, 330—331).
Опубликована заключительная работа, посвященная 9-флуо-
ренилметилоксикарбонильной защите аминогруппы [1].
1. С а г р i по L. А, Н a n G. Y„ J. Org. Chem, 37, 3404 (1972).
ХЛОРУГОЛЬНОЙ КИСЛОТЫ ЭТИЛОВЫЙ ЭФИР,
CICO2C2H5 (IV, 162—165; V, 529).
Реагент применяют на первой стадии синтеза 1-фенилцикло-
пентиламина для получения смешанного ангидрида карбоновой
и угольной кислот.
Алкилирование имидов [2]. X. к. э. э. в ДМФА реагирует с
фталимидом лития при температурах 60—110° с образованием
N-этилфталимида (2) с выходом 80%:
1) ын
2) С1СООСгН5
8 0%
Ш (2)
1. Kaiser С, Weinstock J, Org. Syn, 51, 48 (1972),
2. V i d a J. A, Tetrahedron Letters, 3921 (1972),
627
4-ХЛОР-2-ФЕНИЛХИНАЗОЛИН,
Мол. вес 240,69, т. пл. 124—124,5°.
Получение [1]. Реагент можно получить из амида N-беизо-
илантраниловой кислоты.
Фенолы-> анилины [2]. Шеррер и Битти [2] разработали
новый общий метод превращения фенолов в анилины. Фенолят
натрия (лучше всего полученный с помощью гидрида натрия)
конденсируют с X. при температуре около 100°. Реакция закан-
чивается в течение 10 мин, приводя к образованию 4-арилокси-
2-фенилхиназолина (2) с выходом 70—85%. Ключевой стадией
синтеза является термическая перегруппировка соединения (2)
(,4)
в 3-арил-2-фенил-4(ЗН) -хиназолинон (3). 1,3-Миграцию арила
от О к N впервые наблюдали Чичибабин и Елецкий [3]. Пере-
группировка происходит с достаточной скоростью в интервале
температур 275—325°, причем в тяжелом минеральном масле
образуются более чистые продукты. На последней стадии син-
теза продукт перегруппировки гидролизуют до анилина и 2-фе-
нил-4Н-3,1-бензоксазинона-4 (4). Образующийся при щелочном
гидролизе промежуточный амидин при подкислении дает анилин
и соединение (4). Соединение (3) можно гидролизовать раство-
ром КОН в этиленгликоле при нагревании.
Общий выход колеблется в пределах 40—70%.
1. Endicott М., Wick Е., Mercury М. L., Sherrill М. L„ J. Ат.
Chem. Soc., 68, 1299 (1946).
2. Scherrer R, A., Beatty H. R., J. Org. Chem., 37, 1681 (1972).
3. Tschitschibabin A. E., J e 1 e t z k у N. P., Ber., 57, 1158 J1924).
628
ХЛ ОРФТОРМЕТИЛ ЕНТРИФЕНИЛ ФОСФОРАН,
(C6H5)3P=CFC1. Мол. вес. 328,75.
Реагент получают in situ при действии трифенилфосфииа на
дихлорфторацетат натрия, который в свою очередь получают
нейтрализацией дихлорфторуксусной кислоты в эфире раство-
ром соды. Эфир и воду удаляют при пониженном давлении,
остаток высушивают нагреванием при 50° (4 мм) в течение
—- 12 час. Таким же образом можно приготовить дихлорфтор-
ацетат лития.
Зр-Замещенные 1-хлорперфторолефииы [1]. При взаимодей-
ствии с полифторированными кетонами X. дает смеси цис-
v. транс-р-замещенных 1-хлорперфторолефинов с выходами
34—70%. При взаимодействии с нефторированными альдегида-
ми и кетонами реагент образует соответствующие хлорфтороле-
фины с гораздо более низкими выходами (9—49%).
Триглим
90°
Ar(R)CORF + (C6H5)3P=CFC1 ----> Ar(R)—C=CFC1 + (C6H5)3P=O
Rf
(Rf=CF3, C2F5, CF2C1 и t. д.)
Наиболее подходящим растворителем для этой реакции Виттига
является триглим.
1. Burton D. J., Krutzsch Н. С., J. Org. Chem., 35, 2125 (1970).
ХЛОРЦИАН (IV, 166—167).
Алифатические изоцианаты [1]. При алкилировании X. алкил-
хлоридом в присутствии безводного хлорного железа образуется
комплекс дихлорида алкилизоцианида с FeCh. Например, с хло-
ристым изопропилом в присутствии хлорного железа X. обра-
зует с высоким выходом гигроскопичный комплекс (1). Этот
комплекс реагирует с этанолом или с бензилатом натрия с об-
разованием карбаматов (3) и (4) соответственно.
3C1CN + 3(СН3)2СНС1 + 2FeCl3 —> [(СН3)2СН—N=CCl2]3(FeCl3)2
(D
н2о
(1) ---> (СН3)2СН—NH,
(2)
С2Н5ОН
(I) >• (СНз)2С1-1ИНСООС2Н5
(3)
СаН5СН2ОМа
(1) > (CH3)2CHNHCOOCHaC6H5
(4)
zno
(1) ---> 3(CH,)2CHN=C=O + 3ZnCl2 + 2FeCl3
(5) 85%
629
С окнсыо цинка (IV, 204—205) он дает нзопропилизоцианат
(5), который образуется с высоким выходом, если не выделять
чрезвычайно чувствительный к влаге комплекс (1). Реакция
комплекса (1) с окисью серебра протекает со взрывом. С ацета-
том натрия этот комплекс образует изоцианат (5) с выходом
24%.
1. F u k s R„ Н а г t е m i п к М., Tetrahedron, 29, 297 (1973).
а- ХЛОР - N - ЦИКЛОГЕКСИЛ ПРОПАНАЛЬДОНИТРОН.
Мол. вес 189,69, т. пл. 73—75° (разл.).
Получение [1]. а-Хлориитрон получают из а-хлорпропаналя
(получен согласно методике [2]) н N-циклогексилгидроксил-
амина в сухом эфире при 0°. По истечении второго часа в реак-
ционную смесь добавляют еще некоторое количество эфира и
хлористый метилен. Полученный раствор выдерживают над
MgSO4 при 0° в течение 15 час. После этого осушитель отфиль-
тровывают, растворитель удаляют, а остаток кристаллизуют из
смеси эфир — пентан. Выход продукта 79%. Реагент устойчив
Эфир, О’
, MgSO4
79%
при температуре —20°, но желтеет при хранении при комнатной
температуре. Так же получают а-хлор-Ы-циклогексилацетальдо-
нитрон. Обычно применяют именно циклогексилнитроны, по-
скольку циклогексильная группа повышает нх стабильность.
1,4-Циклоприсоединение [1]. Обычно олефнны с неактивнро*
ванными двойными связями не вступают ни в реакции Диль-
са— Альдера, ни в реакции 1,3-циклоприсоединения. Замеча-
тельная особенность а-хлорнитронов заключается в том, что в
присутствии ионов серебра они способны вступать в реакции
циклоприсоединения с неактивированными двойными связями
олефннов. Следовательно, их можно отнести к классу енофилов.
Истинным реагентом является, по-видимому, М-алкил-М-виннл-
нитрозониевый ион (а). Например, а-хлорнитрон (1) в. присут-
630
ствии борфторида серебра реагирует с циклогексеном в 1,2-ди-
хлорэтане (растворитель для AgBF4) в темноте при интенсив-
ном перемешивании. Осадок AgCl отделяют, а продукт цикло-
присоединения выделяют в виде цианпроизводного (2) (два
эпимера) с выходом 85%. Децианированием последнего с по-
мощью AgBF4 можно получить иминиевый ион (3), который при
действии тетрафеннлбората натрия осаждается в виде соответ-
ствующего тетрафенилбората с выходом 94%.
1) AgBF4,
СН2С1СНгС1
2) КСЬ1/НгО
KCb/' 1) AgBF4,
НгО СНгС1СНгС1
I 2) Na(C6H5)4B
Эшенмозер и сотр. [3] описали два примера синтетического
использования только что рассмотренных продуктов присоеди-
нения. При обработке смеси эпимерных нитрилов (2) 1,1 мол.
же трет-бутилата калия в трет-бутаноле при 80° в атмосфере
азота образуется с выходом 85% иминолактон (4). Последний в
результате кислотного гидролиза превращается в у-лактон (5)
с выходом 82%. Два изомера лактона (5а, 5р) можно разде-
631
лить методом газовой хроматографии.
Другим интересным примером синтетического применения
реакции циклоприсоединения является фрагментация двойной
связи исходного олефина, называемая непрямым «карбоксолити-
ческим» расщеплением [4]. Сначала аддукт олефина с X. обра-
боткой поташом в хлористом метилене переводят в енамин (6),
который при последующем нагревании до сравнительно невысо-
кой температуры гладко рециклизуется с образованием альд-
имина (7). Последнюю стадию гидролиза осуществляют, пропу-
ская альдимин (7) через колонку с влажным силикагелем, что
приводит к образованию а,р-ненасыщенного альдегида (8).
Селективный гидролиз амида. Аналогичный реагент а-хлор-
N-циклогексилацетальдонитрон был использован в полном син-
тезе витамина В!2 [5] для осуществления селективного гидролиза
амидной функции в присутствии шести карбметоксигрупп. Гене-
рируемый из а-хлорнитрона под действием борфторида серебра
электрофильный катион (1) реагирует с амидом (2), давая 2-ок-
соэтиловый эфир (3), который при взаимодействии с диметил-
амином в изопропиловом спирте превращается в лабильный
эфир (4). Гидролиз (4) приводит к кислоте (5).
632
I. Kempe U. M., DasGupta T. K-, Blatt K., Gygax P., Felix D.,
E s c h e n m о s e r A., Helv. Chim. Acta, 55, 2187 (1972).
2. Dick C. R„ J. Org. Chem., 27, 272 (1962).
3 DasGupta T. K-, Felix D., K^pe U. M., Eschenmoser A., Helv.
' Chim. Acta, 55, 2198 (1972).
4 Gygax P., DasGupta T. K., Eschenmoser A., Helv. Chim. Acta,
’ 55, 2205 (1972).
5. Woodward R. B., Pure Appl. Chem., 33, 145 (1973).
XPOMA(II) — АМИНОВ КОМПЛЕКСЫ (VI, 332—334):
Гидролиз связей углерод — галоген [1].
Вт
+ г СгП(еп)л_ + Нго
-
(CH3)ZNCHO
+ 2 СгШ(еп)д_+ Вг' + нб-
Смесь диметилформамида и этилендиамииа перемешивают
магнитной мешалкой в атмосфере азота и под поверхность
жидкости с помощью шприца вводят водный раствор перхло-
рата хрома(II) до образования пурпурного раствора комплекса.
Затем к полученному комплексу добавляют раствор 1,66 г
1-бромнафталина в несодержащем кислорода диметил формами-
де н перемешивают смесь до тех пор, пока ее цвет не изменится
от пурпурного до темно-красного, а затем выливают ее в рас-
твор соляной кислоты. Продукт реакции экстрагируют эфиром
и после обычной обработки получают 0,96—1,00 г нафталина.
I. W a d е R. S., С a s t г о С. Е., Org. Syn., 52, 62 (1972).
ХРОМА(П) АЦЕТАТ (IV, 169—170; V, 530; VI, 334—335).
Восстановление а,^-эпоксикетонов. В поисках пути синтеза
А/В-бициклической системы, входящей в состав молекулы кар-
диотонических стероидов (периплогенин, строфантидин), Робин-
сон и Хендерсон [1] изучили восстановление 4р,5р-эпоксихоле-
станона-3 (1) с помощью X. а. Наилучшие результаты были
получены при восстановлении эпоксида (1) большим избытком
свежеприготовленного X. а. в водном ацетоне. В этом случае
целевой продукт (2) может быть получен с выходом около 50%.
Побочный продукт (3) можно снова превратить в (1), следова-
тельно, при размыкании окисного цикла оксикетон (2) удается
получить с удовлетворительным выходом.
633
Восстановление 3-кетогруппы оксикетона (2) с целью полу-
чения целевого Зр-аксиального спирта лучше всего осуществ-
лять с помощью никеля Ренея W-2 в кипящем этаноле.
1. Robinson С. Н., Henderson R., J. Org. Chem., 37, 565 (1972).
XPOMA(VI) ОКИСИ — ПИРИДИНА КОМПЛЕКС (реак-
тив Коллинза), CrO3(C5H5N)2 (V, 540; VI, 339—340). Мол. вес
258,21, кристаллы красного цвета.
Получение [1]. Коллинз и Гесс опубликовали подробную ме-
тодику получения реагента нз окиси хрома(VI) (высушенной
над Р2О5) и пиридина. Внимание! Реакция чрезвычайно экзо-
термична. Окись хрома(VI) следует прибавлять к пиридину с
такой скоростью, чтобы температура не поднималась выше 20°,
и следить за тем, чтобы окись быстро смешивалась с пиридином.
Сразу после начала прибавления окиси хрома начинает выпа-
дать желтый хлопьевидный осадок. По окончании прибавления
окиси хрома (VI) смеси дают медленно нагреться до комнатной
температуры при перемешивании. Выпавшее вначале желтое
вещество в течение часа превращается в кристаллы темно-крас-
ного цвета. Пиридин с кристаллов декантируют, а кристаллы
промывают декантацией сухим петролейиым эфиром. Продукт
отжимают на фильтре и еще раз промывают тем же растворите-
лем. Комплекс высушивают при 10 мм (выход 85—91%). Он
чрезвычайно гигроскопичен и его следует хранить при 0° в
склянке темного стекла.
Окисление спиртов [1]. Окисление спиртов реактивом Кол-
линза проводят в хлористом метилене обычно при 25°, исполь-
зуя шестикратный молярный избыток реагента. Реакция завер-
шается в течение 5—15 мин. Реагент, в частности, рекомен-
дуется для окисления первичных спиртов до альдегидов. Приме-
нение в этих случаях реактива Саретта (IV, 180—181; V, 540;
VI, 339—340) обычно дает более низкие выходы [2]. Так, напри-
мер, гептанол-1 можно окислить реактивом Коллинза в хлори-
стом метилене до гептаналя-1 с выходом 70—84%.
Окисление мзо-7-оксибицикло [4,3,1]-декатриена-2,4,8 (1) ре-
активом Коллинза до бицикло-[4,3,1]-декатриен-2,4,8-она-7 (2)
было осуществлено с выходом 64% [3]:
(1) (2)
1. С о 111 n s J. С., Н е s s W. W., Org. Syn., 52, 5 (1972).
2. Holum J. R., J. Org. Chem., 26, 4814 (1961).
3. Schroder G., Prange U., Putze B., Thio J., Oth J. F. M., Chem.
Ber,, 104, 3406 (1971).
634
ХРОМИЛА ХЛОРИД (IV, 174—175; ¥,534—535; ¥1,337—
338).
Окисление терминальных олефинов до альдегидов. Согласно
подробной прописи [1], смесь 1,0 моля 2,4,4-триметнлпентена-1 и
1 л хлористого метилена перемешивают механической мешалкой
в 5-литровой трехгорлой колбе, снабженной термометром и ка-
пельной воронкой с хлоркальциевой трубкой и погруженной в
баню со льдом и солью. Раствор охлаждают при перемешивании
1) СгОеС1е СНз
| 2) 2п, Н2О [
СН3—С—СН2—С=СН2 —-—СН3—С—СНо—СН—СНО
[ | 70-78% j [
сна сн3 сн3 сн3
до 0—5° и, продолжая перемешивать, при той же температуре
прикапывают к нему из капельной воронки раствор 158 з
(1,02 моля) свежеперегнанного X. х. в 200 мл хлористого мети-
лена (в течение примерно 60 мин). Реакционную смесь переме-
шивают еще 15 мин, после чего добавляют 184 г 90—95 %-ной
технической цинковой пыли. Этот приблизительно пятикратный
избыток Zn необходим, чтобы восстановить соли хрома высшей
валентности и тем самым предотвратить дальнейшее окисление
и расщепление двойной связи. Смесь перемешивают в течение
5 мин, затем как можно быстрее прибавляют 1 л ледяной воды
и 400 г льда (температура при этом поднимается до 8—10°) и
перемешивают смесь еще в течение 15 мин. Охлаждающую
баню заменяют колбонагревателем, а колбу присоединяют к
прибору для перегонки с паром и отгоняют хлористый метилен.
Остаток перегоняют с паром, пока дестиллат не будет давать
отрицательную пробу с 2,4-динитрофенилгидразином. Объединен-
ный органический слой не высушивая перегоняют для удаления
растворителя на 56-сантиметровой колонке Вигре с вакуумной
рубашкой. Остаток переносят в круглодонную колбу на 250 мл,
отгоняют остатки хлористого метилена при атмосферном давле-
нии, а основную массу вещества перегоняют в вакууме, собирая
фракцию, кипящую в пределах 45—52°/15 мм. Выход 2,4,4-три-
метилпентаналя составляет 90—100 г (70—78%).
а-Хлоркетоны [2]. Ди- и тризамещенные олефины реагируют
с Х.х. в ацетоновом растворе при низких температурах, давая
с хорошим выходом (40—90%) а-хлоркетоны:
О С1
н\ /RI cro2ci2 II I zRl
> R—С—C
rZ \rs
Успех этого метода определяется использованием в качестве
растворителя ацетона. При добавлении цинковой пыли к необ-
работанной реакционной смеси хлоркетон восстанавливается до
635
соответствующего кетона. Наибольшие выходы хлоркетонов до-
стигаются при —70°, ио даже при температурах от —5 до 3°
выходы еще приемлемые. Например, при окислении циклододе-
цена с помощью X. х. при —75° а-хлорциклододеканон обра-
зуется с выходом 79°/о, а при —5° выход хлоркетона составляет
69%.
Окисление циклогексена до окиси. При разложении комп-
лекса X. х. с циклогексеном, суспендированного в CH2CI2, хо-
лодным 5%-ным водным раствором бикарбоната натрия окись
циклогексена удалось выделить с выходом лишь 1,5% [3].
1. Freeman F., DuBois R. Н., McLaughlin Т. G., Org. Syn., 51, 4
(1971).
2. Sharpless К. B., Teranishi A. Y., J. Org. Chem., 38, 185 (1973).
3. Bachelor F. W., Cher iy an U. O., J. C. S., Chem. Comm., 1973, 195.
ХРОМОВАЯ КИСЛОТА (IV, 175—179; V, 535—536; VI, 338).
Реактив Джонса в ацетоне (IV, 176—177; V, 535; VI, 338).
С помощью реактива Джонса в ацетоне бензоины окисляются
в бензилы с выходом около 90—95% [1]. Для этой же цели мо-
Н2СгО4 в
СвН5СН(ОН)СОС6Н6 ацет°не > С6Н5СОСОС0НЙ
жно применить и метод двухфазного окисления, разработанный
Брауном и Гартом (IV, 178; V, 535—536), однако выходы в этом
случае несколько ниже (75—90%).
Двухфазное окисление (IV, 178; V, 535—536). Браун и сотр.
[2] опубликовали завершающую работу по двухфазному окисле-
нию вторичных спиртов в диэтиловом эфире X. к. в воде. Ав-
торы исследовали целый ряд не смешивающихся с водой раство-
рителей, однако диэтиловый эфир оказался наиболее подходя-
щим. Были разработаны две стандартные методики. Согласно
одной из них, водный раствор взятых в стехиометрических коли-
чествах бихромата натрия и серной кислоты добавляют при
25—30° к эфирному раствору спирта и оставляют реакционную
смесь на 2 час.
3R2CHOH+Na2Cr2O7 + 4H2S04 —> 3R2CO + Na2SO4 + Cr2(SO4)3-J-7Н2О
Выходы кетонов, как правило, составляют 85—97%, но в случае
бициклических спиртов с жесткой структурой они значительно
ниже. Для таких случаев рекомендована модифицированная
методика, согласно которой реакцию проводят при 0° в течение
15 мин со 100%-ным избытком окислителя.
1. Но T.-L., Chem. Ind., 1972, 807.
2. Brown Н. С., Garg С. Р., Liu K-Т., J- Org. Chem., 36, 387 (1971)
ХРОМОВЫЙ АНГИДРИД (IV, 179-183; V, 536—540;
VI, 338—341).
636
Комплекс СгО3 —пиридин (IV, 180—181; V, 540; VI, 339—
340). Ратклиф [1] (VI, 341, [4]) подробно описал методику полу-
чения комплекса СгОз — пиридин in situ и его использование
для окисления деканола-1 (1) в деканаль-1 (2). Раствор пири-
СгОз-2Ру
СН2С12
СНЭ(СН2)8СН2ОН ——СН3(СН2)8СНО
ad %
(1) (2)
дина (1,2 моля) в хлористом метилене охлаждают до 5° и добав-
ляют к нему X. а. (0,6 моля). Темно-красного винного цвета рас-
твор перемешивают при 5° в течение 5 мин или несколько доль-
ше и затем дают ему медленно нагреться до 20°. После этого
быстро прибавляют раствор спирта (1) (0,1 моля) в хлористом
метилене. После обычной обработки получают альдегид (2) с
выходом 83%. В действительности выход еще выше, но дека-
наль очень летуч.
Реагент сравним по характеру действия с реактивом Кол-
линза комплексом пиридина с СгО3 (этот том), однако чистый
реактив Коллинза крайне гигроскопичен.
При окислении алкинов комплексом СгО3 — пиридин можно
получить с выходом около 40% кетоны с сопряженными свя-
зями С = С и С —О:
СгОз-РУ2
RC=CCH2R ---------> RC=CCR
Например, при окислении октина-4 (0,015 моля) с помощью
СгО3-Ру2 (0,225 моля) в хлористом метилене при комнатной
температуре в течение 24 час (N2) образуется октин-4-он-З с вы-
ходом 42%. Для этой же цели можно использовать также без-
водный хромат натрия (Na2CrC>4), но он дает более низкие вы-
ходы. При этом значительные количества исходного алкина воз-
вращаются в неизмененном виде [2].
Хромовый ангидрид в графите [3]. Крофт [4] нашел, что
можно внедрить хромовую кислоту в кристаллическую решетку
графита путем диффузии паров СгО3 в графите при пониженном
давлении и высокой температуре. Предложенный Крофтом ме-
тод был усовершенствован Лалансеттом и сотр. [3], получивши-
ми реагент, содержащий 55—60 вес.% СгОз; материал по вне-
шнему виду напоминает графит.
Новый реагент — окислитель очень специфичного действия
для превращения первичных спиртов в соответствующие альде-
гиды. Спирт обрабатывают реагентом в кипящем толуоле и че-
рез 24 час отгоняют растворитель. Альдегид получается с высо-
ким выходом (70—100%). Участвовавшие в реакции соединения
хрома остаются в кристаллической решетке графита. Вторичные
и третичные спирты этим методом не окисляются. 1,2-Диолы
637
окисляются с разрывом углеродной цепи и образованием альде-
гидов с меньшим числом углеродных атомов.
Бензиловый спирт —> Бензальдегид (98%)
Коричный спирт —> Коричный альдегид (100%)
Фенилэтиленгликоль —> Бензальдегид (80%)
Циклогексанол —> Циклогексанон (2%)
I. Ratcliffe R. W„ Org. Syn., submitted 1973.
2. Shaw J. E., Sherry J. J., Tetrahedron Letters, 4379 (1971).
3. Lalancette J.-M., Rollin G., Cumas P., Can. J. Chem,, 50, 3058
(1972).
4. Croft R. C., Australian J. Chem., 9, 201 (1956).
ЦЕРИЙ, АММИАКАТ НИТРАТА.
См. Гексанитратоцеррат(1У) аммония, этот том.
ЦЕРИЯ ТЕТРААЦЕТАТ, Се(ОАс)4. Мол. вес 376,31.
Получение. Ц. т. получают озонолизом ацетата церия(III) в
присутствии нитрат-иона [1]. Ц. т. — стабильное твердое веще-
ство желтого цвета.
Реакция с олефинами [2]. Если Ц. т. в течение 20 час нагре-
вать со стиролом при 110° в ледяной уксусной кислоте, содержа-
щей 10% ацетата калия, то при этом в качестве основного про-
дукта образуется лактон (1) с выходом 70%. Для препаратив-
ного получения лактонов вместо Ц. т. можно использовать более
доступный гексанитратоцеррат(1У) аммония.
с6н5бн=снг
Се(ОАс)4
СНдСООН
70%
С6Н5СН— CHi
о сна
с
й
(1)
В отличие от этого фотохимическая реакция Ц. т. с транс-$~
метилстиролом приводит к образованию главным образом слож-
ного эфира (2) н только небольших количеств лактона (3).
с6н5сн=снснэ
Се(ОАс)4
95%
с6н5сн-сн-сн3
ОАс СН,
C6HS-CH-^H-CH3
,СНг
44 С
•(2)
Г лавный
продукт
(3)
Побочный
продукт
Реакцию Ц. т. с ароматическими углеводородами можно ис-
пользовать для синтеза арилуксусных кислот. Так, реакция Ц. т.
с толуолом в смеси уксусного ангидрида с уксусной кислотой
дает в качестве главных продуктов бензилацетат и толилуксус-
ную кислоту (выход 29%).
I. Hay N., Kochi J. К-, J. Inorg. Nucl. Chem., 30, 884 (1988).
2. Heiba E. I., Dessau R. M., J. Am. Chem, Soc., 93, 995 (1971).
ЦИАНИСТЫЙ ВОДОРОД (IV, 187—188).
Асимметрический синтез аминокислот [1]. Путем присоеди-
нения Ц. в. к шиффовым основаниям, полученным из алифати-
639
ческих альдегидов и оптически активных бензиламинов, можно
синтезировать оптически активные аминокислоты:
нск нэо+
RCH=NR] ----> RCHNHR1 -----> RCHNHR1 ----ь
CN соон
(1) (2) н2 Pd(OH)2/C > RCHCOOH NH, И) (3)
Аминокислоты были получены с выходами 19—58%, оптическая
чистота полученных таким путем природных аминокислот со-
ставляет 22—58%, Ее можно повысить путем дробной кристал-
лизации.
I. Н а г a d а К., О k a w а г а Т., J. Org. Chem., 38, 707 (1973),
ЦИАНМЕТИЛИДЕН-^с-(ТРИФЕНИЛФОСФОНИЯ) ДИ-
БРОМИД, (С6Н5)3РСНР(С6Н5)32Вг-. Мол. вес 723,42, т. пл.
CN
268 — 269°.
Реагент (1) получают [1] с выходом 93,4% взаимодействием
трифенилфосфина с дибромацетонитрилом [2]:
CgHe + +
Br2CHCN + Р(С6Н5)3 —> (C6H5)3P-CH-P(C6H5)32Br“
CN
(1)
Реакция Внттига [1]. Альдегиды при кипячении с бнс-фосфо-
ниевой солью (1) в водно-бензольной среде в присутствии гидро-
окиси натрия превращаются в нитрилы а,|3-ненасыщенных кис-
он~, с6на
н2о
Ar(R)CHO + (1) - Ar(R)CH=CHCN + 2(С6Н5)3Р=0
4U — оО то
(2)
лот (2). Кетоны не реагируют с солью (1). В случае алифатиче-
ских альдегидов продукт реакции частично, а иногда и пол-
ностью изомеризуется в нитрил (фу-ненасыщенной кислоты.
I. Wilt J. W., Но A. J„ J, Org. Chem., 36, "026 (1971).
2, Уилт Дж„ Д а й б о л д Дж,, «Синтезы органических препаратов», ИЛ,
. М„ I960, сб, 10, стр. 19.
640
ЦИАНМЕТИЛМЕДЬ, CuCH2CN. Мол. вес 103,59.
Реагент получают [1] взаимодействием цианметиллития (по-
лучен из ацетонитрила и «-бугиллития при —78° [2]) с 1 же
иодида меди(1) в ТГФ при —25° в течение 10 мин в атмосфере
аргона или азота.
у,д-Неиасыщенные нитрилы [1]. Ц. реагирует с аллилгало-
генидами, образуя с высоким выходом нитрилы у,6-ненасыщен-
ных карбоновых кислот:
С=С—СВг + CuCH3CN
^С=С—С—CH2CN + CuBr
/ I I
Например, Ц. при взаимодействии с трацс-геранилбромидом
(1) дает гранс-гомогеранилцианид (2) с выходом 92%• Охлаж-
денный раствор н-бутиллития (2,0 мл, 1,3 Af) в пентане медлен-
но прибавляют к раствору ацетонитрила (3,34 ммоля) в 5 мл
ТГФ при —78° в атмосфере аргона. Смесь выдерживают 40 мин
при температуре —78°, а затем дают ей нагреться до —25° и
вносят иодид меди(1) (3,37 лшоля). После 15-минутного переме-
шивания при —25° раствор Ц. кирпично-красного цвета обра-
батывают транс-геранил бромидом в ТГФ (3 мл), оставляют
смесь на 1 час при —25° и добавляют к ней водный раствор
хлористого аммония. Продукт реакции экстрагируют эфиром, а
затем выделяют хроматографически или перегонкой при пони-
женном давлении.
Реагент не взаимодействует с бромистым бензилом н неак-
тивированнымн бромистыми алкилами. Реакция Ц. с ненасы-
щенными бромэфирами типа ВгСН2СН=СНСООСН3 настолько
сложна, что ее нецелесообразно использовать в синтетических
целях.
1. С о г е у Е. J., К u w a j i m a I., Tetrahedron Letters, 487 (1972).
2. Kaiser E. M„ Hauser C. R., J. Org. Chem., 33, 3402 (1968).
ЦИКЛОГЕКСИЛАТ АЛЮМИНИЯ (IV, 197—198).
Восстановление. Хиншоу [1] осуществил восстановление тет-
раметилантрахинона (1) до 2,3,6,7-тетраметнлантрацена (2) с
21 Зак. ЗОВ
641
выходом 85% с помощью Ц, а. в кипящем циклогексаноле.
1. Hinshaw J, С., Org. Prep. Proc. Int., 4, 211 (1972).
б«с-(ЦИКЛООКТАДИЕН-1,5)-НИКЕЛЬ(0),
Ni (ЦОД)2.
Мол. вес 230,27, желтые кристаллы, в растворе
чрезвычайно чувствителен к кислороду.
Получение. Реагент лучше всего получать по видоизмененной
[1] методике Вильке [2] восстановлением ацетилацетоната никеля
триэтил алюминием в присутствии циклооктадиена-1,5. Вслед-
ствие чувствительности Ы1(ЦОД)2 к кислороду на всех опера-
циях необходима атмосфера инертного газа. Выход 81%.
Ni( ОгС5Н7)2
2 Al(CzH-,)3
81#
Ni(UO4)j
л-Аллнлннкельгалогеннды (V, 329). Вальтер и Вильке [3] со-
общили, что Ы1(ЦОД)2 реагирует с аллилгадогенидами с обра-
зованием л-аллилникельгалогенидов активнее, чем карбонил
никеля. Реакция при температурах ниже 0° приводит к количе-
ственным выходам. Однако, несмотря на это, данный метод не
получил широкого распространения, поскольку карбонил никеля
в отличие от Ы1(ЦОД)2— продажный реактив. Тем не менее
Земмельхак рекомендует пользоваться ЙП(ЦОД)2 для получе-
ния термически нестабильных л-аллилникельгалогенидов, таких,
как комплекс л-(2-карбэтоксиаллил)-никельбромид(4), получен-
ный из этил-2-бромметилакрилата с помощью этого никелевого
реагента с выходом 76%.
Конденсация арилгалогенидов [5]. С целым рядом арилгало-
генидов при температурах 25—40° в ДМФА Ы1(ЦОД)2 дает ди-
арилы, дигалогениды никеля (II) и циклооктадиен:
ДМФА
Ж(ЦОД)2 + 2АгХ Ar—Ar + NiX2 + 2ЦОД
Выходы диарилов, как правило, высокие (80—90%), но наличие
орто-заместителей замедляет реакцию, а с некоторыми орто-
642
замещенными арнлгалогенидами реакция вообще не идет. Арил-
галогениды, содержащие кислотные функциональные группы
(ОН, СООН), не вступают в реакцию конденсации, а восста-
навливаются по связи углерод — галоген. Единственным подхо-
дящим растворителем для этой реакции является, по-видимому,
ДМФА; в ТГФ или толуоле результат отрицателен. ДМСО и
гексаметапол не подходят по той причине, что в этих раствори-
телях никелевые реагенты быстро разлагаются.
Конденсация алкенилгалогенидов [6]. Алкенилгалогениды ре-
агируют с ЫДЦОДД в ДМФА при 25° с образованием 1,3-ди-
енов:
2RCH=CHX + Ni (ЦОД)3 —> RCH=CHCH=CHR + NiX2 + 2ЦОД
Если в качестве растворителя применять эфир, то приходится
добавлять электронодонорный лиганд (трифенилфосфин или
три-н-бутилфосфин). Алкенилгалогениды с электроноакцептор-
ными группировками димеризуются с высокими выходами. Так,
транс-2-бромстирол превращается в транс,транс-1,4-дифенилбу-
тадиен-1,3 с выходом 70%.
с6щх Н
В случае простых алкенилгалогенидов продукты конденсации
получаются лишь с умеренными выходами (34—60%), причем
образуются смеси геометрических изомеров, в которых несколь-
ко преобладает более стабильный изомер. Причиной низких вы-
ходов в данном случае, по-видимому, является олигомеризация
первоначально образующихся 1,3-диенов под действием
М1(ЦОД)2.
Конденсация 2-галоген- и 3- галогеиакрилатов протекает с
высокими выходами и с полным сохранением конфигурации, как
это показано на примере реакций (а) и (б):
а) ВгСН=СНСООСН3 СН3ОСОСН=СНСН=СНСООСН3
б) ВгСН^СНСООСНз д-^> СН3ОСОСН=СНСН=СНСООСН3
Земмельхак [1] привел один пример внутримолекулярной кон-
денсации под действием ЫДЦОДД в ДМФА, Так, цис,цис-1,6-
дииодгексадиеи-1,5 (1) превращается в циклогексаднен-1,3 (2)
с выходом 67% (считая на выделенный продукт):
IHC
IHC.
•67’/
21*
643
[2 + 2]-Кросс-присоедннение олефинов [7]. Реакция норбор-
надиена (1) (4,0 Л4моля) с метиленциклопропаном (2) (4,0 ммо-
ля), катализируемая М1(ЦОД)2 (0,2 лшоля) в присутствии три-
фенилфосфина (0,22 лшоля), в бензоле при 20° в течение 24 час
приводит к образованию циклоаддукта 1 : 1 (3) с выходом 86%:
Реакция между (1) и (2) в присутствии Ь11(ЦОД)2 и (—)-
бензилметилфенилфосфина [7] приводит к оптически активной
форме аддука (3), aD' —0,8° [8].
I. Semmelhack М. F., Org. React., 19, 115 (1972).
2. Bogdanovic B., Kroner M., Wilke G., Ann., 699, I (1966).
3. Walter D., Wilke G., Angew. Chem., Internat. Ed., 5, 897 (1966).
4. Semmelhack M. F., Org. React., 19, 179 (1972),
5. Semmelhack M. F., H e 1 q u i s t P. M., Jones L. D., J. Am. Chem. Soc..
93, 5908 (1971).
6. Semmelhack M. F., Helquist P. M., Gorzynski J. D., J. Am.
Chem. Soc., 94, 9234 (1972).
7, N о у о г i R., I s h i g a m i T., Hayashi N., T a k а у a H., J. Am. Chem.
Soc., 95, 1674 (1973).
8. N a li m a n n K., Zon G., Mislow K-> J- Am. Chem. Soc., 91, 7012 (1969).
ЦИКЛОПЕНТАДИЕНИЛТРИБЕНЗИЛТИТАН,
C5H5T1 (СН2СбН5)3. Мод. вес 386,37. Получают из CsH5TiCl3 и
C6H5CH2MgCl.
Циклосодимеризация этилена и бутадиен а-1,3 [1]. Этот го-
могенный титановый катализатор промотирует циклосодимери-
зацию этилена и бутадиена-1,3, приводящую к образованию ви-
нилциклобутана. Обычным продуктом содимеризации (в отсут-
ствие катализатора) является гексадиен-1,4.
Ti
СН=СН2
снг=сн2
снг=снсн=снг
катализатор,
54$
4*
При использовании этого катализатора норборнен (1) н бу-
тадиен содимеризуются в соединение (2) с выходом около 90%.
Менее напряженные олефины дают замещенные винилциклобу-
644
таны с более низкими выходами.
и
+ СНг=СНСН==СНг натали3ат°Р.
1. Cannell L. G., J, Am. Chem. Soc., 94, 6867 (1972).
ЦИКЛОПРОПЕН, Мол. вес 40,06, нестабилен.
Получение (I, 65),
Спектры [1].
Реакции Дильса — Альдера. О реакции Дильса — Альдера
между циклопропеном и циклопентадиеном уже упоминалось
(1, 65). Ц., в сущности, один из самых реакционноспособных
диенофилов. Он реагирует с тропоном (1) в хлористом метилене
при 0° с образованием продукта 1,4-присоединения (2). Анало-
гично реакция с трополоном (3) приводит к образованию аддук-
та (4). Эта реакция является первым примером реакции Диль-
са— Альдера с трополоном. Избирательное образование аддук-
тов зн(?о-строения является, по-видимому, общим свойством,
присущим циклопропену [2].
1. Wiberg К. В., Nist В. J, J. Am. Chem. Soc., 83, 1226 (1961).
2. Uyehara Т., S а к о N., К i t a h а г a Y., Chem. Ind., 1973, 41.
ЦИНК (IV, 210—227; V, 550—555; VI, 344—347).
Децианирование. Швейцарские химики [1] осуществили де-
пианирование цианамида (1), полученного нз лизергиновой кис-
лоты, цинковой пылью в 80%-ной уксусной кислоте (3 час,
100°). Это превращение можно осуществить также действием
645
водорода в присутствии никеля Ренея. Алюмогидрид лития для
этой цели непригоден, поскольку в молекуле (1) имеется легко
восстанавливающаяся группировка 0,у-ненасьгщеного сложного
эфира.
Сторк [2] использовал Ц. в уксусной кислоте для децианиро-
вания цианамида (3) в соединение (4), которое участвовало да-
лее в качестве одного из компонентов в полном синтезе (±)-
иохнмбина (5):
Восстановительное расщепление аллиловых спиртов, их прос-
тых эфиров или ацетатов с помощью амальгамированного Ц. и
НС1 [3, 4]. Амальгамированный Ц. получают обработкой цинко-
вой пыли водным раствором HgCU. Получившуюся амальгаму
промывают дистиллированной водой, спиртом и, наконец, сухим
эфиром.
Аллиловые спирты, их простые эфиры или ацетаты можно
восстановить до олефинов, обрабатывая большим избытком
амальгамированного Ц. и хлористым водородом в эфире. Новый
метод отличается от обычной методики (например, действие
натрия в жидком аммиаке) тем, что в условиях реакции обра-
зуется преимущественно, а в ряде случаев исключительно менее
стабильный изомер. Так, метилциклогексен-1-ол-З (1) превра-
щается в З-метилциклогексен-1 (2) с выходом 73%• Изомерные
аллиловые спирты (3) и (4) дают одну и ту же смесь олефинов.
646
с6нБсн=сн2—СН2ОН
(3)
С6Н5СНОНСН=СН2
(4)
с8нБсн2сн=сн2 + C6HSCH=CHCH
80% 17%
Ацилоины [5]. а-Дикетоны при кипячении с цинковой пылью
в водном ДМФА (N2) с высоким выходом восстанавливаются
Zti.aoAH. ДМФД^
в ацилоины. Этот способ служит препаративным методом полу-
чения олефинов из неенолизующихся а-дикетонов, как показано
в приводимом примере:
(О
Синтез прегнана н боковых цепей кортикоидов [6]. Ацетат
17а-этинилкарбинола (1) восстанавливают цинковой пылью в
кипящем диглиме, что сопровождается перегруппировкой и эли-
минированием с образованием алленилстероида (2) с выходом
86%. Реакцией с четырехокисью осмия в присутствии пиридина
в бензоле с последующим расщеплением осмата сульфитом нат-
рия и бикарбонатом калия боковую цепь стероида (2) можно
647
превратить в боковую цепь искомого кортикоида (3)1
О s СЬ,
53%
снгон
(3)
При окислении надкислотами соединение (2) превращается
в 17а-оксн-20-кетопрегнан, Возможным промежуточным продук-
том в этом окислении является 17,20-окись аллена.
у,д-Неиасыщенные кислоты. Болдуин и Уолкер [7] разрабо-
тали новый путь синтеза у,6-ненасыщенных кислот, включающий
перегруппировку Клайзена [8]. Эфиры а-бромкарбоновых кис-
лот (1), полученные этерификацией аллиловых спиртов бром-
ангидридами а-бромкарбоновых кислот [9], кипячением с цин-
ковой пылью в ароматическом углеводороде превращают по ре-
акции типа реакции Реформатского в цинк-еноляты (2). Послед-
ние в результате перегруппировки Клайзена дают бромцинковые
соли (3) у,д-ненасыщенных кислот. При наличии в соединении
(1) а-алкильных заместителей выходы понижаются, например
соль кислоты (3) получается с выходом 100%, если R1 — R2 =
= СН3) a R3 = R4 = Н. Если R1 = СН3, R2 = R4 = Н, a R3 —
= СзНб, то выход соли (3) составляет лишь 16% •
Рассматриваемая реакция в высокой степени стереоселектив-
на. Так, например, эфир а-бромпропионовой кислоты (1) (R1 =
= R4 = СНз; R2 = R3 = Н) дает 2-метилгексен-4-овую кислоту
648
с соотношением изомеров транс: цис, равным 93:7. Реакция
о н,с
н3с \ ^соон
Вг —С '"о HjC-c
„ L J > нс
Н3С X*
нс^ ^сн3
(5) Сб)
применима также к ацетиленовым эфирам. Эфир (5), например,
превращается этим методом в алленовую кислоту (6).
1. Fehr Т., Stadler Р. A., Hofmann A., Helv, Chim. Acta, 53, 2197
(1970).
2. Stork G., Nath Guthikonda R., J. Am. Chem. Soc., 94, 5109 (1972),
3. Elphimoff-Felkin I., Sards P., Tetrahedron Letters, 725 (1972).
4. E 1 p h i m о f f - F e 1 k i n L, Sard a P., Org. Syn., submitted 1972.
5. К г e i s e r W., Ann., 745, 164 (1971).
6. В i о 11 a z M., H a e f 1 i g e r W-, V e r I a d e E., Crabbe P., Fried J. H,
Chem. Comm., 1971, 1322.
7. Baldwin J. E., W a I k e r J. A., J. C. S. Chem. Comm., 1973, 117.
8. T a r b e 11 D. S., Org. React., 2, I (1944); Jefferson A., Schein-
in a nh F., Quart. Rev., 22, 391 (1968).
9. Smith C. W., Norton D. G., Org. Syn., Coll. Vol., 4, 348 (1963).
10. Shriner R. L., Org. React, 1, I (1942); Diaper D. G. M., Kuksis A.,
Chem. Rev., 59, 89 (1959).
ЦИНКА КАРБОНАТ, ZnCO3. Мол. вес 125, 39.
Получение, Ц. к. осаждают добавлением 0,1 М раствора кар-
боната натрия к 0,1 М раствору сульфата цинка и высушивают
в вакууме.
Синтез кумаринов. Синтез кумаринов по Пехману осущест-
вляется реакцией фенолов с эфирами р-кетокислот в присут-
ствии конденсирующего агента (обычно серной кислоты). Пы-
таясь синтезировать природный кумарин — метаболит плесени
афлатоксин Mi (3), Бюхи и Вейнреб [2] обнаружили, что для
этой цели обычные условия синтеза кумаринов непригодны из-за
чрезвычайно высокой чувствительности фенола (1) к кислотным
агентам. Однако авторам удалось осуществить желаемую кон-
649
денсацию фенола (1) и эфира (2) под действием Ц. к. или кар-
боната магния в хлористом метилене. С другой стороны, карбо-
наты натрия и калия оказались неэффективными в этой реак-
ции. Новый метод пригоден для синтеза и других родственных
афлатоксинов.
I. Сетна С., П х а д к е Р., «Органические реакции», ИЛ, М., 1956, сб. 7, стр,7.
2. В й с h i G., Weinreb S. M., J. Am. Chem. Soc., 93, 746 (1971).
ЦИНК —АНГИДРИД КИСЛОТЫ — КАТАЛИЗАТОР.
Восстановительное ацетилирование. Тиле [1] установил, что
при восстановлении бензила цинковой пылью в уксусном ангид-
риде в присутствии серной кислоты происходит 1,4-присоедине-
ние водорода к а-дикетонной группировке и ацетилирование об-
разовавшегося ендиола, приводящее в смеси геометрических изо-
меров (1) и (2). Тиле и более поздним исследователям удавалось
выделить лишь не вполне чистый более растворимый диацетат
ct,р-диокси-^ностильбена (2) с более низкой т. пл. (около 110°).
Разделение смеси с помощью хроматографии оказалось невоз-
можным, поскольку оба изомера обладают одинаковой способ-
ностью адсорбироваться на окиси алюминия. Оба изомера уда-
лось выделить в чистом состоянии путем фракционированной
кристаллизации, описанной в руководстве для студентов [2].
Г он он
—> с6н5 - с = с - с6н5
1,4--.присоед; L
2 АсгО, Н
о о
и п
с6н5-с - С—с6н5
(1), транс
Т.пл.155°
''ЮН 271 у = 23 400)
(2), час
Т. пл. 11$°
ОН
2 65 и и (с = 12 800)
Серная кислота может восстановиться до серы и даже до серо-
водорода, и поэтому в методе, рекомендуемом для приготовле-
ния смеси изомеров, оиа заменена соляной. Если в этой реакции
вместо смеси соляной кислоты с уксусным ангидридом исполь-
зовать хлористый ацетил (2ло), то единственным продуктом бу-
дет ^нг-изомер (2).
Разработана удобная методика получения диацетильного про-
изводного легко окисляющегося на воздухе продукта восстанов-
ления хинона (3). Хинон (3) суспендируют в уксусном ангид-
риде (5—6жл/г), добавляют столько же по весу цинковой пыли,
650
0,2 части свежесплавленного порошкообразного ацетата натрия
н осторожно нагревают смесь до исчезновения окрашенного ве-
щества, пока жидкость над осадком не станет бесцветной или
светло-желтой: в зависимости от чистоты исходных реагентов.
После непродолжительного кипячения для завершения реакции
продукт и часть выделившегося ацетата цинка растворяют в ук-
сусной кислоте, кипящий раствор фильтруют, промывают оста-
ток горячим растворителем, а объединенный фильтрат кипятят
с обратным холодильником, осторожно добавляя к нему воду
в количестве, достаточном для гидролиза избытка уксусного ан-
гидрида и получения насыщенного раствора. Из бесцветного
раствора по охлаждении выделяется 0,6 г чистого диацетата
2-метил-1,4-нафтогидрохинона с т. пл. 112—113°. Вместо ацетата
натрня можно применять бромид тетра метил аммония (0,2 ча-
сти), который является даже более эффективным катализато-
ром [3].
В более ранних методиках [4] рекомендовалось использовать
в качестве катализатора пиридин, однако это оказалось неудоб-
ным, так как реакционная смесь в результате побочной реакции
между уксусным ангидридом, пиридином и цинком может окра-
шиваться в желтый цвет. Этого удается полностью избежать,
применяя третичные алифатические амины, например триэтил-
амин, как это делают в методике получения диацетата 2-метил-
1,4-нафтогидрохинона (см. III, 456).
Синтетическому веществу бицикло-[4,4,1]-ундекатетраен-3,5,-
8,10-диону-2,7 (5) можно приписать строение хинона на основа-
нии того факта, что при его обработке цииковой пылью н уксус-
ным ангидридом в присутствии пиридина происходит быстрое
восстановительное ацетилирование с образованием ароматиче-
ского 2,7-диацетокси-1,6-метаио-[10]-аинулена (6) [5]:
Восстановительное бензоилирование. К охлаждаемой льдом
смесн 0,5 г 2-метил-1,4-нафтохинона, 0,5 г цинковой пыли и 3 жл
651
пиридина добавляют по каплям 1 мл хлористого бензоила [46].
Реакция происходит сразу, и после 10-мннутного выдерживания
при комнатной температуре светло-желтую реакционную смесь
нагревают до кипения, охлаждают и экстрагируют продукт
смесью спирта и эфира. После промывания разб. кислотой и
сн3
осос6н5
Zn + Ру + 2 СДЦСОС!^
COCfcHs
раствором соды и упаривания высушенного раствора до неболь-
шого объема из него выделяется дибеизоат в виде бесцветных
игл с т. пл. 178—179,5°. Двукратная перекристаллизация из спир-
та (35 мл) повышает его температуру плавления до 180—180,5°.
1. Thiele J., Ann., 306, 142 (1899).
2. Fieser L. F., Org. Exp., 2nd Ed., Heath D. C. and Co., Boston, 1968, 221.
3. Fieser L. F., Experiments in Organic Chemistry, 2nd Ed., Heath D. C. and
Co., Boston, 1941, 399.
4. a) Fieser L, F., J. Am. Chem. Soc., 61, 3473 (1939); 6) Fieser L. F.,
Campbell W. P., Fry E. M., Gates M. D., Jr., J. Am. Chem. Soc., 61»
3216 (1929).
5. V о g e I E., L о h m а г E., Boll W. A., S 6 h n g e n B., Mullen K.,
Gtinther H., Angew. Chem,, Internat. Ed„ 10, 398 (1971).
ЭПИХЛОРГИДРИН (IV, 234—235).
Улавливающий агент для бромистого водорода. При окисле-
нии ациклических а-галогенкетонов с помощью ДМСО обра-
зуются а-дикетоны (I, 327). Однако циклические а-бромкетоны
окисляются необычным образом. 5-Бром-2-карбэтокси-2-метил-
циклопеитанон (1), например, при окислении ДМСО образует
3-бром-5-карбэтокси-2-окси-5-метилциклопентен-2-он-1 (2) с вы-
ходом 70% [1]. Эта необычная реакция объясняется, вероятно,
тем, что выделяющийся в реакции бромистый водород окис-
ляется ДМСО до элементарного брома. В самом деле, если ке-
сн3
соосгн5
(I) (2)
тон (1) окислять в присутствии Э., связывающего бромистый
водород, то получается нормальный продукт (3) с выходом
58,5% [2].
1. Sato К., Suzuki S., Kojinia Y., J. Org. Chem., 32, 339 (1967).
2. S a t о К., К о j i m a Y., Sato H., J. Org. Chem., 35, 2374 (1970).
ЭТАНОЛАМИH (IV, 235; V, 557—558).
Селективное галогенирование метилкетонов [1]. При взаимо-
действии прегненолона (1) сЭ. в толуоле в присутствии неболь-
шого количества ионообменной смолы дауэкс 20 (в Н+-форме)
образуется с выходом 94% кетимин (2), обработка которого N-
хлорсукцинимидом или NEC в эфире с последующим гидролизом
разб. соляной кислотой приводит к образованию 21-галогенпро-
изводного с высоким выходом (90—100%). Галогенирование
653
протекает, вероятно, через таутомерную енамииную форму со-
единения (2):
Следует особо отметить, что 5,6-двойную связь при этом не ну-
жно защищать.
Описанная методика была опробована также на пентаноне-2;
главным направлением реакции и в этом случае оказалось гало-
генирование соседней с карбонилом метильной группы.
I. Keana J. F. W., Schumaker R. R., Tetrahedron, 26, 5191 (1970).
ЭТИЛА ПЕРЕКИСЬ, С2Н5ООС2Н5. Мол. вес 90,12, т. кип.
62—63 °C, rtD 1,3724.
Получение [1]. Реагент получают с выходом 50% взаимо-
действием этилового эфира метансульфокислоты (2) с щелоч-
ным раствором 30 %-ной перекиси водорода; в качестве эмульга-
тора используют стеариновую кислоту.
Фрагментация фосфоранов [3]. З-М.етил-1 -фенилфосфолен-3
(1) образует с Э.п. фосфоран (2), который самопроизвольно
распадается при комнатной температуре на изопрен (3) и диэти-
ловый эфир фенилфосфонистой кислоты (4):
н3с
Ц P~C6HS
снг
+ С6Н5Р(ОСгЩ)2
снг
(4)
Эта же реакция фрагментации со смесью изомерных 1,2,5-
триметил фосфол енов-3 (5а) и (56) приводит к образованию
транс, трсшс-гексадиена-2,4 (6) с количественным выходом и ди-
этилового эфира метилфосфонистой кислоты (7). Поскольку
диен (6)—наиболее стабильный изомер, ничего нельзя сказать
о стереоспецифичности приведенной реакции.
654
+ CH3P(OC2Hsk
f6)
I. Pryor W. A, Huston D. M., J. Org. Chem., 29, 512 (1964).
2. Williams H. R., Mosher H. S., J. Am. Chem. Soc., 76, 2987 (1954).
3. H a 11 C. D„ В r a m b 1 e 11 J. D., L i n F. F, S., J. Am. Chem. Soc., 94, 9264
(1972).
ЭТИЛАТ НАТРИЯ (IV, 237—248).
Циклопропилкетоны. Замещенные циклопропилкетоны можно
получить путем катализируемой основаниями изомеризации у,д-
эпоксикетонов. Взаимодействие эпоксида (2) 4-нзопропилиден-
циклогексаноиа (1) сЭ.н. в этаноле в течение 15 мин приводит
к образованию 7-оксисабинкетона (3) — 1-(Г-окси-Г-метил)-
этилбицикло-[3,1,0]-гексанона-4 — с выходом более 90%. (Мож-
но также применять 2 н. NaOH в кипящем этаноле.) Кетон (3)
превращают далее в ряд производных туйана [1].
Эту изомеризацию использовали в синтезе некоторых произ-
водных иоркарана [2]. Реакция эпоксида (4) караханаенона [3]
с Э. н. в этаноле (кипячение 15 .инн) стереоспецифична и приво-
дит к образованию единственного бициклического оксикетона
655
5-эцдо-окси-3,3,6-триметилноркараноиа-2 (5) с выходом >-80%.
NaOG;H5
О
ОН
£5)
1. G а о n i Y., Tetrahedron, 28, 5525 (1972).
2. G а о n i Y., Tetrahedron, 28, 5533 (1972).
3. Demole E., E n g g i s t P,, Helv. Chim. Acta, 54, 456 (1971).
ЭТИЛАТ ТАЛЛИЯ(1) (V, 558—564).
Алкилирование азотистых гетероциклов [1]. Пиррол чисто
алкилируется по азоту, если предварительно превратить его в
пиррилталлий(I) реакцией с Э.т. Пиррилталлий представляет
собой относительно устойчивое твердое вещество, структура его
не установлена. Реакция этой соли с избытком алкилгалогенида
(или алкилтозилата) прн 20—60° в течение 1—32 час приводит
к N-алкилпирролам с хорошим выходом. Алкилирование пир-
рилмагнийбромида дает в качестве главных продуктов изомер-
ные 2- и 3-алкилпирролы.
Карбазол (1), фенотиазин (2) и (в меньшей степени) дибенз-
[&Д-азепин (3) таким путем можно проалкилировать по азоту в
мягких условиях с хорошими выходами [2]. Раствор гетероцикла
и небольшого избытка Э.т. в смеси ДМФА и эфира перемеши-
вают при комнатной температуре, затем обрабатывают избыт-
(1) (2) (3)
ком йодистого или бромистого алкила и, наконец, короткое вре-
мя нагревают на водяной бане.
2,3-ДигидрО’1,5-бензоксазепин-(5Н)-он-4 (4) метилируют по
азоту, действуя Э.т., а затем иодистым метилом [3], Однако,
К)
если к смеси соединения (4) и йодистого метила в диоксане не-
большими порциями прибавлять гидрид натрия, продукты алки-
656
лирования образуются с более высоким выходом. Было замече-
но, что (4) легко расщепляется основаниями.
Алкилирование циклических 1,3-дикетонов [4]. Реакция цик-
лических 1,3-дикетонов с Э.т., карбонатом таллия(I) или цикло-
пентадиенилталлием (I) приводит к образованию бесцветных
кристаллических солей одновалентного таллия (V, 559). Эти
соли алкилируются в неполярных растворителях (бензол, ДМЭ),
образуя с высоким выходом продукты О-алкилирования, что
можно проиллюстрировать на примере димедона (1), Наиболее
1) тюсгн5
2) CjHjI
3 0-100%
(2)
высокие выходы целевого продукта наблюдаются при алки-
лировании наименее активными первичными алкилгалогенида-
ми. В случае йодистого метила или хлористого бензила в не-
большом количестве получаются также С-диалкилпроизводные.
1. Candy С. F., Jones R. A., J. Org. Chem., 36, 3993 (1971).
2. К г I с k a L. J„ L е d w 11 h A., J. С. S. Perkin I, 1972, 2292.
3. Huckle D., Lockhart I. M., Wright M., J. C. S. Perkin I, 1972, 2425.
4. Pizzorno M. T, Albonico S. M., Chem. Ind., 1972, 425.
ЭТИЛВИНИЛОВЫЙ ЭФИР (IV, 251—253; V, 564).
у,ft-Ненасыщенные альдегиды [1]. Виниловые эфиры реаги-
руют с третичными винилкарбинолами, давая с хорошими выхо-
дами у,6-ненасыщенные альдегиды. В качестве катализатора
обычно используют фосфорную кислоту. Примером, иллюстри-
рующим эту реакцию, может служить получение 5-метилгексен-
4-аля (5) из 2-метилбутен-3-ола-2 (1) и этилвинилового эфира
(2). Вначале, по-видимому, образуется смешанный ацеталь (3),
который при нагреваний в присутствии кислотного катализатора
теряет молекулу этанола. Последний реагирует с избытком этил-
винилового эфира с образованием ацеталя (6). Образующийся
657
из ацеталя (3) аллилвиниловый эфир (4) претерпевает затем
перегруппировку Клайзена, превращаясь в продукт (5) с выхо-
дом 52—60%. Ср. синтез у,б-ненасыщенных кетонов из третич-
ных винилкарбинолов и изопропенилметилового эфира (V,
207—208).
Введение функциональнозамещенных аигулярных метильных
групп [2]. Реакция Э. с 10-метил-Ай9)-окталолом-2 (1), ката-
лизируемая ацетатом ртути(П), в запаянной трубке Кариуса
при 200° в течение 12 час дает Э-формилметил-Ю-метил-А^окта-
лин (2) с выходом 85% и смесь нескольких диенов (3) с выхо-
дом 15%. При катализе фосфорной кислотой образуются только
диены (3). Образование винилового эфира и последующую пе-
регруппировку Клайзена можно объединить в одну стадию, од-
(1) (2) 8 5% (3) 15%
нако это не удается с соединениями, содержащими циклическую
систему гидриндена, которые в таких условиях претерпевают
лишь элиминирование с образованием диенов. Чтобы преодолеть
указанные трудности, реакцию в данном случае проводят в две
стадии: сначала получают виниловый эфир, а затем осуществ-
ляют пиролиз.
I. Маг bet R., Saucy G., Helv. Chim. Acta, 50, 2095 (1967); Org. Syn.,
submitted 1971.
2. Dauben W. G., Dietsche T. J., J. Org. Chem., 37, 1212 (1972).
ЭТИЛДИИЗОПРОПИЛАМИН (IV, 253).
Пнрроло-[1,2-4?]-а«-триазнн [1]. Эта гетероароматическая си-
стема с 10 л-электронами (2) была синтезирована катализируе-
мой основаниями циклодегидратацией формилгидразона пир-
рол-2-альдегида (1) в кипящем ксилоле с выходом 56%:
1 (СНз^СН^СДЦ
5 6%
(2)
1. С г е s s J. Р., F о г к е у D. М, J. С. S. Chem. Comm., 1973, 35
ЭТИЛЕНДИАМИН (IV, 259).
Изомеризация алкинов [1]. Гексин-3 не изомеризуется при
действии сильных оснований (амид натрия, w-бутиллитий) при
658
комнатной температуре в течение 72 час, но в сочетании с Э. эти
основания быстро изомеризуют гексин-3. При молярном отно-
шении гексин-3 : амид натрия : Э. 18:1:18 образуются гексин-2
(80%), гексин-3 (12%), гексин-1 (4%) и гексадиен-2,3 (4%).
Действительным агентом изомеризации является ЫаЭ. Предло-
жен согласованный механизм этой изомеризации через девяти-
членное переходное состояние (а).
(а)
1. Wotiz J. Н,, В ar el ski Р. М,, Koster D. F., J. Org. Chem,, 38, 489
(1973).
ЯНТАРНОЙ КИСЛОТЫ ТЕТРА МЕТИЛДИАМИД,
CH2CON(CH3)2
CH2CON(CH3)2. Мол. вес 152,23, т. пл. 81—82°.
Реагент получают взаимодействием диэтилового эфира ян-
тарной кислоты с диметиламином (метилат натрия, бомба Пар-
ра, 100°, 24 час} с выходом 76%.
1,4-Дикетоны [1]. Я. к. т. реагирует при —78° с литийоргани-
ческими соединениями, образуя 1,4-дикетоны. Выходы дикетонов
зависят от природы литийорганического соединения. Из всех
изученных авторами литийорганических соединений наилучшие
результаты были получены при взаимодействии Я. к. т. с 6-бром-
2-пиридиллитием, синтезированным из 2,6-дибромпиридина и
я-бутиллития. При использовании фениллития выход соответ-
ствующего 1,4-дикетона составил только 4%.
СНгСОЬЦСНзЭг
снгсоы(сн3)г
(СгН5)2О
1,5-Дикетоны [1]. 1,5-Дикетоны получают аналогичным пу-
тем, но вместо Я. к. т. используют тетраметилдиамид глутаровой
кислоты. И здесь с самым большим выходом был получен 1,5-
дикетон из 6-бром-2-пиридиллития. С фениллитием, правда, в
этом случае соответствующий 1,5-дикетон образовался с умерен-
ным выходом (50%).
1. Owsley D. С., N е 1 k е J. М., Bloomfield J. J., J. Org. Chem., 38, 901
(1973).
ЯНТАРНЫЙ АНГИДРИД. Мол. вес 100,07, т. пл. 118—120°.
Двукратное последовательное ацилирование нндана Я. а. [1].
660
о
1. Rahman A. U., del Carmen Torre M„ Ann., 718, 136 (1968).
УКАЗАТЕЛЬ РЕАГЕНТОВ ПО ТИПАМ РЕАКЦИИ
Алкилирование. Бензилтриэтиламмопийхлорид. н-Бутилмеркаптан. Гексаме-
тилтриамид фосфорной кислоты. Диалкоксикарбония борфториды. Диме-
тилмедьлитий. Диметилсульфид. Диметилсульфонийметилид. Диметилфор-
мамида диметилацеталь. Диэтоксикарбония борфторид. Калия гидро-
окись. Лития диизопропиламид. Лития N-изопропилциклогексиламид.
О-Метилдибензофурания борфторид. Натрий — нафталин. Натрия гид-
рид. Натрия бнс-(2-метоксиэтокси) -алюмогпдрид. Палладий хлорис-
тый. 1,2,2,6,6-Пентаметилпиперидин. Серебра окись. Фторсульфоновой
кислоты метиловый эфир. Хлоругольной кислоты этиловый эфир, Этилат
таллия (f). Этилвиниловый эфир.
Алкилирование геминальное. Дпфенилсульфоцийциклопропилид.
Алкилирование хинонов. Никеля карбонил.
л-Аллилникеля галогениды, бис- (Циклооктадиен-1,5) -никель (0).
л-Аллилпалладия комплексы. Палладий хлористый. Палладия(П) ацетат.
Аллильное окисление. Марганца(Ш) ацетат. Ртути(П) ацетат. Селена дву-
окись.
Альдольная конденсация. трет-Бутилмагнийхлорид. 2-Литийпроиилиден-трег-
бутилампн. Натрия тетраборат.
Амидов гидролиз. а-Хлор-Ц-циклогексилпропанальдонитрон.
Аминоалкенилирование. 1-Хлор-Ц,!Ч,2-триметилпропениламин.
Аминодегидроксилирование. Амид калия. Диэтилхлорфосфат. 4-Хлор-2-фенил-
хиназолин.
Аминометилирование алкинов. Родия окись.
Аннелирование. Ацетилендикарбоновой кислоты диметиловый эфир. Дифенил-
сульфонийциклопропилид. траяс-Пептен-З-он-2.
Арилдиазония солей разложение. Медь(I)тетрапиридинперхлорат.
Ароматическое гидроксилирование. Перекись водорода — алюминия хлорид.
Асимметрический синтез. Диборан. Диизопинокамфилборап. (—)- и ( + )-
2,3,-О-Изопропилиден-2,3-диокси-1,4-бис (дифенилфосфино) -бутан. ( + )-
(R)-транс-р-Стирил-л-толилсульфоксид. н-Толилсульфинилметилидлитий.
Цианистый водород.
Ацеталей получение. Молекулярные сита.
Ацетали. Ионообменные смолы.
Ацетилирование. Висмута триацетат. Кетен.
Ацилирование. 4-(Ц-Пирролидил)пиридин. Трифторметансульфоиовой и кар-
боновых кислот смешанные ангидриды. Фенилтрифторметилкетен.
Ацилирование внутримолекулярное. Полифосфорная кислота.
Ацилоиновая конденсация. Триметилхлорсилан.
Байера — Виллигера окисление. и-Карбметоксинадбензойная кислота. Над-
уксусная кислота — бора трифторида эфпрат Перекись водорода в кис-
лой среде. о-Сульфонадбензойная кислота. ж-Хлорнадбензойная кислота.
Берча восстановление (см.).
662
Бишлера — Напиральского реакция. Полифосфорпой кислоты эфиры.
Бромирование. N-Бромполималеипимид. N-Бромсукцинимид. Ртути окись —
бром. 2,4,4,6-Тетрабромциклогексадиеп-2,5-ои. Фенилтриметиламмония пер-
бромид.
Вагнера — Меервейна перегруппировка. n-Т о лу о л сульф о кис л от а.
Винилирование. Дивинилмедьлитий. Диметилацетамид.
Виттита реагент. Хлорфторметилентрифенилфосфоран.
Виттига реакция (см.). Трифенилфосфин.
Виттига реакция обратная. Трифениларсипа окись.
Внутримолекулярная циклизация ненасыщенных ацилхлоридов. Три-я-бутил-
станнан.
Восстановительное аминирование. Натрия цианборгидрид.
Восстановительное ацетилирование. Цинковая пыль.
Восстановительное бензоилирование. Цинковая пыль.
Восстановительное дегалогенирование. Лития алюмогидрид.
Восстановительное дезоксигенирование спиртов и кетонов. N.N.N'.N'-Terpa-
метилдиамидохлорфосфат.
Восстановительное дехлорирование. Литий — трет-бутанол — тетрагидрофуран.
Восстановительное диалкилирование. Литий — нафталин.
Восстановительное расщепление. Калия боргидрид.
Восстановление, реагенты. Алюминий. Гексаметилтриамид фосфорной кис-
лоты. Диборан. Диизобутилалюминийгидрид. Диимид. 2,3-Диметилбутил-
2-боран. бис-(1,2-Диметилпропил-1)-боран. Ди мети л сульф ид — боран. Же-
леза пентакарбонил. Калий в графите. Калия иодид — цинк-медная пара.
Кори восстановитель. Литий. Литий — алкиламины. Литий — аммиак.
Литий — этилендиамин. Лития алюмогидрид. Лития алюмогидрид — алю-
миния хлорид. Лития алюмогидрид — натрия метилат. Лития амальгама.
Лития боргидрид. Лития трис-(ето/ьбутил)-боргидрид. Лития трис-{трет-
бутокси)-алюмогидрид. Лития гидрид. Лития иодид. Лития иодид — бора
трифторид. Лития пергидро-9Ь-борафеналилгидрид. Лития триэтилбор-
гидрид. Магния алюмогидрид. Натрия боргидрид. Натрия гидрид.
Натрия диэтилалюмогидрид. Натрия бис-(2-метоксиэтокси)-алюмогидрид.
Натрия цианборгидрид. Никеля борид. Полиметилгидросилоксан. Сер-
ный ангидрид—пиридин. Тиомочевины диоксид. Титан треххлористый.
Три-н-бутилстаннан. Триизобутилалюмнний. Трифепилстаннан. Трифе-
нилфосфин. трис- (Трифенилфосфин) -родийхлорид. трис- (Трифепилфос-
фин)-родийхлорид—триэтилсилан. Трихлорсилан. Триэтилсилан. Уксус-
ной кислоты хлорангидрид. 1-Фенил-5-хлортетразол. Хлориридиевая кис-
лота. Хрома (11) — аминов комплексы. Циклогексилат алюминия. Цинк.
Восстановления методы. Берча восстановление (см.).
Вюрца дегалогенирование. Натрий.
Вюрца реакция. Натрий — нафталин.
Галогендекарбоксилирование. Свинца тетраацетат.
Галогенирование. Гексаметилтриамид фосфорной кислоты. Этаиоламин.
Гидридного иона отщепление. Метилсульфинилметилиднатрий. Тропилия соли.
Гидрирование каталитическое гомогенное с переносом водорода, трис-(Три-
фенилфосфин) -рутенийдихлорид.
Гидрирования катализаторы. Железа додекакарбонил. Железа пентакарбопил.
Линдлара катализатор. Никеля борид. Палладий (10%) на карбонате
кальция, бис- (Пиридин) -диметилформамиддихлорородия боргидрид. Ро-
дий (5%) на окиси алюминия. Родий (5%) на угле. Рутения трихлорида
гидрат, трис- (Трифенилфосфин) родийхлорид. Урушибары катализаторы
гидрирования.
Гидроалюминирование. Диизобутилалюминийгидрид.
Гидроборирование. 1,3,2-Бензодиоксаборол. Диизопинокамфилборан бис- (3,6-
Диметил) борепан. 2,3-Диметилбутил-2-боран. Диметилсульфид — боран.
663
Дициклогексилборан. бис- (траис-2-Метилпиклогексил-1)-боран. Мопохлор-
борана диэтилэфират.
Гидроборирования реагенты. 1,3,2-Бензодиоксаборол. Диборап. Диизопино-
камфилборан. бис- (3,6-Диметил) -борепан. бис- (3,5-Диметил) -боринан.
2,3-Диметилбутил-2-боран. Дихлорборана диэтилэфират. Дициклогексил-
борап. бис- (траяс-2-Метилциклогексил-1) -боран. Монохлор борана диэтил-
эфират.
Гидроксилирование. Калия осмат. Перекись водорода — алюминия хлорид.
Гидролиз. Натрия дигидрофосфата моногидрат. Серебра сульфат.
Гидросилилирование, трдс-(Трифенилфосфин)-родийхлорид.
Гидроформилирование. Родия окись.
Гидроцианирование. Диэтилалюмипийцианид. Триэтилалюминий.
Гликолей расщепление. Таллия(III) нитрат.
Гомологизация. Бута ди ей а-1,3 1,2-окись. Диазоуксусный эфир. Литий метил-
изонитрил.
Гриньяровского типа реакция. Литий.
Даффа реакция. Гексаметилентетрамин.
Дегалогенирование. Железа нонакарбонил. Лития амальгама. Натрий — фе-
нантрен. Натрия тиосульфат. Триэтилфосфит.
Дегидратация. Алюминия окись. Бора трифторида ди-н-бутилэфират. Гекса-
метилтриамид фосфорной кислоты. Гексахлорциклотрифосфазатриен. Ди-
метилсульфоксид. Иод. Тионил хлористый. Трифенилфосфита иодметилат.
Н,Н,И-Триэтил-Ь1'-карбэтоксисульфамида внутренняя соль.
Дегидрирование. Азодикар боновой кислоты диэтиловый эфир. Антрахинон.
2)3-Дихлор-516-дициал-1,4-бензохинон. Калия гидрид. Марганца двуокись
активная. Палладиевые катализаторы. Палладий на угле. Тетраметил-
этилендиамин. Трифторуксусная кислота. Хлоранил.
Дегидробензола предшественники. Дифенилиодоний-2-карбоксилата моногид-
рат. п-Хлорбеизоилнитрит.
Дегидробензолов образование. Лития 2,2,6,6-тетраметилпиперидид.
Дегидрогалогенирование. Амид натрия — тдаг-бутилат натрия. Амидины би-
циклические. N-Бромсукцинимид. трет-Бутилат калия. Гексаметилтриамид
фосфорной кислоты. 1,5-Диазабицикло- [4,3,0] -нонен-5. Диметилсульфоксид.
Диэтил а мин. Лития бромид. Тетра-я-бутиламмония бромид. Триэтиламин.
Дегидрогалогенирование — гидролиз. Калия гидроокись.
Дегидротозилирование. 1,5-Диаза бицикло- [4,3,0] -нонен-5.
Дегидроцианирование. Аскарит.
Дезалкилирование. 1,4-Диазабицикло-[2,2,2]-октан.
Дезацетилирование. Бария гидроокись.
Дезацетоксилирование. Пиридин.
Дезоксигенирование. Альдегидов и кетонов: Вольфрама гексахлорид. N,N,N/,N/-
Тетраметилдиамидохлорфосфат. N-Окисей: Железа пентакарбонил. N-Со-
держащих функциональных групп: Триэтилфосфит. Сульфоксидов: Ди-
хлорборан. Фенолов: Фенилизоцианат. Эпоксидов: Вольфрама гексахло-
рид. Трифенил фосфинселенид.
Дезоксимирование. Таллия (III) нитрат. Титан треххлористый.
Дейтерирование. Галлия окись.
Декарбметоксилирование. 1,5-Диазабицикло-[4,3,0]-ноиен-5. Натрия хлорид —
диметилсульфоксид. Натрия цианид —ди метилсульфоксид.
Декарбоксилирование. n-Толуолсульфокпслота. Триметилхлорсилан.
Декарбонилирование. Симмонса — Смита реагент, трис-(Трифенилфосфин)-ро-
дийхлорид.
Деметилирование. Бора трихлорид. Бромистоводородная кислота. Диметил-
формамид. Хинуклидин.
Десульфуризация. н-Вутиллитип. Гексаэтилтриамид фосфористой кислоты.
трис- (Трифенил фосфин) -родийхлорид.
Детиоацетализация. Серная кислота. Фторсульфоновой кислоты метиловый
эфир.
664
Деформилирование. Калия тетрахлороплатинат(И).
Де цианирование. Гексаметилтриамнд фосфорной кислоты. Цинковая пыль.
Диазосоединения. Медный порошок. л-Толуолсульфонилазнд,
Диимида предшественник. 2,4.6-Триизопропилбепзолсульфокислоты гидразид.
Дильса-Альдера реакции. Дихлормалеиновый ангидрид. Дицианацетилен. «Ин-
даноциклон». Кислород синглетный, бис-(Трифенилфосфин)-никель (II)ди-
цианид. N-Фенилимид азодикарбоиовой кислоты. а-Хлоракриловой кис-
лоты хлорангидрид. 2-Хлоракрилоиитрил. Циклопропен.
Дильса — Альдера реакции катализаторы. Алюминия хлорид.
Димеризация, бис-(Трифенилфосфин)-кобальт(II)дибромид.
Енолацетилирование. «-Толуолсульф о кислота.
Еноляты. Лития диизопропиламид. Лития N-изопропилциклогексиламид.
Защита. Аминогруппы: Бора трифторида эфират. грет-Бутилоксикарбонил-
триазол. трет-Бутил-8-хинолилкарбонат. Ди-тдет-бутилпирокарбонат.
4-Диметиламино-1 -трет-бутилоксикарбонилпиридиния хлорид. Угольной
кислоты трет-бутилового эфира азид. Угольной кислоты трет-бутилового
эфира фторангидрид. Аминогруппы первичной: Бензоина енольной формы
циклический карбонат. Гидроксильной группы: Диметил-трет-бутилхлОр-
силан. Ц,Ц,Ц/,Ц/-Тетраметилднамидохлорфосфат. я-фенилбензоил хлори-
стый. 9-Фенил-9-оксиантрон. Хлоругольной кислоты РДР-трихлорэтило-
вый эфир, виц-цис- Диолов: Диметил ацетамид а диметилацеталь. Карбо-
нильной группы: Гексанитратоцеррат(1У) аммония. 1,2-Диокси-З-бромпро-
пан. Таллия (III) нитрат. Триметилсилилцианид. 2.2,2-Трихлорэтанол. Хлор-
амин-Т. Карбоксильной группы: Ы.Ы'-Диизопропилгидразин. Хлордиметил-
сульфид.
Идентификация спиртов. 4-(4/-Нитрофенилазо)-бензоилхлорид.
Изомеризация. трет-Бутилат калия. Ацетиленов и алленов: фтористый водо-
род—-бора трифторид. Диенов: Дихлормалеиновый ангидрид. Дитерпе-
нов: Палладиевые катализаторы. Оксаспиропентана: Лития иодид Оле-
финов: трет-Бутилат калия. Лития дифенилфосф ид. Лития фосфат. Медь
хлористая. Тиофенол — азодиизобутиронитрил.
Иодирование. Трифенилфосфит.
Карбенов образование. Бензилтриэтиламмонийхлорид, Дегидро-М,М/,Н"-три-
циклогексилгуанидиногексакарбонилдижелезо(О). Окись этилена. Триме-
тилсилилдиазометан. Трихлоруксусной кислоты этиловый эфир. Фенил-
(1 -бром-1 -хлор-2,2,2-трифторэтил) -ртуть. Фенил- (тригалогенметил) -ртуть.
Карбеноидов реакции. Метилен хлористый — н-бутиллитий.
Карбеноиды, Диэтилцинк — йодоформ.
Карбоксилирование. Железа(П)сульфат. Муравьиная кислота.
Карбонилирование. Платинохлористоводородная кислота — олово хлористое.
Кетали. Ионообменные смолы.
Кислород синглетный. Калия перхромат.
Кислоты неорганические. Азотная кислота. Борная кислота. Бромистоводород-
иая кислота. Бромистый водород. Иодная кислота. Ионообменные смолы.
Полифосфорная кислота. Серная кислота. Соляная кислота. Фосфорная
кислота. Фтористый водород. Фтористый водород — бора трифторид.
Хлориридиевая кислота. Хлористый водород.
Кислоты органические. Ацетилсерная кислота, d- 10-Камфорсульфокислота.
Ментансульфокислота. Муравьиная кислота. Пикриновая кислота.
n-Толуолсульфокислота. Трифторметансульфокислота. Трифторуксусная
кислота. Трихлоруксусная кислота.
665
Клайзена конденсация. Полифосфорная кислота.
Клайзена перегруппировка. N.N-Диметилацетамида диметилацеталь. Лития
N-изопропи л циклогексил амид. Трифторуксусная кислота. Цинк. Этилвини-
ловый эфир.
Кневенагеля конденсация. Титан четыреххлористый.
Конденсации катализатор. Лития тетрахлоркупрат.
Конденсации сложноэфирные. Лития 2,2,6,6-тетраметилпиперидид.
Конденсация арил- и алкенилгалогенидов. бис-(Циклооктадиен-1,5)-никель (0).
Курциуса реакция. Натрия азид.
Лактонизация. Пиперазин.
Лактоны. Церия тетраацетат.
Литиевые еноляты сложных эфиров. Лития N-изопропилциклогексиламид.
Манниха реагент. N,N-Диметил метилена ммония иодид.
Меервейна — Понндорфа восстановление. Изопропилат алюминия.
Мезилаты. Метансульфохлорид.
Металлирование. н-Бутиллитий. Калия гидрид. Меди (I) трет-бутилат.
N,N,NZ,N1 -Тетр а мети л мети лен ди а мин.
Металлоорганические реагенты. бис- (Бензонитрил)-палладий (II) дихлорид.
трег-Бутилмагнийхлорид. Винилмагнийхлорид. бис-(Дибутилацетоксиоло-
во)-оксид (см. Полиметилгидросилоксан). Ди-н-бутилмедьлитий. Диви-
пилмедьлитий. Диизобутилалюминийгидрид. Дикобальтоктакарбонил
2,2/-Дилитийдифенил. Диметилмедьлитий. Дихлорметиллитий. М,М-Ди-
хлоруретан. Диэтилалюминийхлорид. Диэтилцинк — йодоформ. Диэтил-
цинк—метилен иодистый. Железа(Ш) ацетилацетонат. Железа до-
декакарбонил. Железа нонакарбонил. Железа пентакарбонил. б«с-(Карбэто-
ксидиазометил) -ртуть. Карбэтоксиметилмедь. бис- (Кобальттетракарбо-
нил) -цинк. Литийдиметилкарбамоилникельтрикарбонил. Литийметили-
зонитрил. Литий — нафталин. 2-Литийпропилиден-трет-бутиламин. 2-Ли-
тий-2-триметилсилил-!,3-дитиан. Лития трет-бутилат. Лития трис- (трет-
бутокси)-алюмогидрид. Лития диизопропиламид. Лития дифенилфосфид.,
Лития диэтиламид. Лития N-изопропилциклогексиламид. Лития пер-
гидро-9Ь-борафеналилгидрид. Лития 2,2,6,6 - тетр а метил пип ер и ди д. Лития
триэтилметилат. М,М-б«с-(Магнийбром) -анилин. Медь(I) тетрапиридинпер-
хлорат. Метилмедь. 1,3-бис-(Метилтио)-аллиллитий. а-Метоксибензилиден-
вольфрам(О) пентакарбонил. Натрий — нафталин. Натрий — фенантрен.
Натрия диэтилалюмогидрид, Натрия железа(Н) тетракарбонил. Натрия
бис- (2-метоксиэтокс и)-алюмогидрид. Никеля карбонил. Палладий хлори-
стый. Палладия (II) ацетат, бис- (Пиридин)-диметилформамиддихлороро-
дия боргидрид. Ртути(Н) ацетат. Ртути(И) трифторацетат. Свинца(1У)
ацетата азиды. Свинца диацетат. Свинца тетраацетат. Свинца тетра-
(трифторацетат). Три-я-бутил-я-бутен-З-ил-олово. Три-н-бутилстаннан.
Три-я-бутилфосф ила — меди (I) иодида комплекс. Триизобутилалюминий.
бис- (Триметилсилил)-литийамид. Трифениларсина окись. Трифенилстан-
нан. бис-(Трифенилфосфин)-никель(II)дицианид. т’рис-(Трифенилфосфин)-
родий(])карбонилгидрид. трис- (Трифенилфосфин) -родийхлорид. трис~
(Трифенилфосфин)-родийхлорид — триэтилсилан. трис- (Трифенилфосфин) •
рутенийдихлорид. Триэтилалюминий. фенил-(1-бром-1-хлор-2,2,2-трифтор-
этил)-ртуть. Ц,Ц-Фенилметилнатрийамид. Фенилпалладия ацетат. Фенил-
тиометиллитий. Фенил- (тригалогенметнл) -ртуть. Фенил- (три фтор мети л) -
ртуть. Фосфорной кислоты диэтилового эфира серебряная соль. бис-(Цик-
лооктадиен-1,5)-никель (0).
Метанолиз. Серебра перхлорат.
Метиленирование кетонов. Фенилтиометиллитий.
Метилирование. Триметилоксония борфторид.
N-Метилирование. Натрия цианборгидрид. Формальдегид.
Метилсульфонилнрование. Фторсульфоновой кислоты метиловый эфир.
666
Метоксилирование. грет-Бутилгипохлорит.
Метоксиметилирование. м-Бутилмеркаптан Метоксикарбения гексафторанти-
монат. Хлорметилметиловый эфир.
Михаэля присоединение. Диметилмедьлитий. Диметилсульфоксопийметилид
(+) - (R)-транс-Р-Стирил-я-толилсульфоксид. Тетраметилгуанидин.
Нитрование. Азота четырехокись.
Нитровинилирование. 2-Нитро-1-диметиламиноэтилен.
Озонирование. Сульфолен-3.
Окисления катализатор. Селен.
Окисления реагенты. трет-Амила гидроперекись. N-Бромсукцииимид. Гексани-
тратоцеррат(1У) аммония. Диметил-Н-сукцинилсульфония хлорид. Диме-
тилсульфид — хлор. Диметилсульфоксид. Диметилсульфоксид — серы
трехокись. Диметилсульфоксид — уксусный ангидрид. Диметилсульфо-
ксид— хлор. 2,3-Дихлор-5,6-дициан-1,4-бензохинон. Ди цикло гексил- 18-кра-
ун-эфир-6— калия перманганат. Изоамилнитрит. Иод. Иод — калия иодид.
Иода трис- (трпфторацетат). Иодная кислота. Иодозобензола диацетат. Ка-
лия иминоксилдисульфонат. Калия феррат(У1). Калия феррицианид. Ко-
бальта(П) ацетат. Марганца двуокись активная. Меди(П) ацетата
моногидрат. Меди нитрат — пиридин. Натрия гипохлорит. Озон. Осмия
четырехокись — калия хлорат. Ртути (I I) ацетат. Ртути окись. Рутения
четырехокись. Свинца тетраацетат. Селена двуокись. Серебра карбонат
Серебра карбонат — целит. Серебра нитрат. Серебра окись. Серебра(П)
окись. Серный ангидрид. Таллия(Ш) нитрат. Таллия(III) сульфат. Тал-
лия (III) трифторацетат. Трифенил метил борфторид. Трифенилфосфинди-
бромид. Трифенилфосфита озонид. Трифторнадуксусная кислота. Хлор-
амин. о-Хлоранил. 1-Хлорбензотриазол. Хрома(VI) окиси — пиридина
комплекс. Хромила хлорид. Хромовая кислота. Хромовый ангидрид.
Окислительная конденсация. Железа(Ш) хлорид — диметилформамид. Калия
феррицианид. Марганца двуокись активная. Медь хлористая. Палладий
хлористый. Серебра карбонат — целит. Таллия(Ш) трифторацетат.
Окислительная перегруппировка. Медь хлорная. Перйодаты.
Окислительная циклизация. Ртути(П) ацетат.
Окислительное декарбоксилирование. Меди карбонат основной. Свинца тетра-
ацетат.
Окислительное метилирование. Серебра окись. Серебра (II) окись.
Окислительное расщепление циклопропенов. Ртути(П) ацетат.
Оксимеркурирование. Ртути(П) ацетат.
Оксиметилирование. Лития диизопропиламид. Муравьиной кислоты этиловый
эфир.
Оксипропилирование. Уксусного альдегида этил-3-бромпропилацеталь.
Олефинов диспропорционирование. Вольфрама гексахлорид.
Олефинов реакции присоединения. N-Бромацетамид. Нитрозония борфторид.
Свинца (IV) ацетата азиды. Сера двухлористая, бис-(Циклооктадиен-1,5)-
никель(О).
Основания. Аскарит. Бария гидроокись. «-Бутиламин. трет-Бутилат калия.
1,5-Диазабицикло-[4,3,0]-нонен-5 (ДБН). 1,4-Диазабицикло-[2,2.2]-октан
(ДАБЦО). 1,5- Диазабицикло-[5,4,0]-ундецен-5 (ДБУ). 1,8-бис- (Диметил-
амино)-нафталин. Диэтиламин. Калия гексаметилдисилазан. Калия гид-
рид. Калия гидроокись. 2-Литийпропилиден-т’рет-бутиламин. Лития трет-
бутилат. Лития диизопропиламид. Лития диэтиламид. Лития М-изопро-
пилциклогексиламид. Лития 2,2,6,6-тетраметилпиперидид. Лития триэтил-
метилат. Лития фосфат. Метилат натрия. Натрия этилмеркаптид. 1,2,2,6,6-
Пентаметилпиперидин. Пиперазин. Пиридин. 4-(N-Пирролидил)-пиридин.
Тетраметилгуанидин. бас-(Триметилсилил)-литийамид. Триэтиламин. Хи-
нуклидин. Этилат натрия. Этилат таллия (I). Этилендиамин.
Отщепление. Бензильных групп: Алюминия хлорид.
667
Пептидов синтез. Дифенилфосфорной кислоты азид. М-Карбэтокси-2-этокси-
1,2-дигидрохинолин. о-Нитрофенилсульфенхлорид. Тетрахлорсилан. Три-
фенилфосфин. Трифенилфосфин — углерод четыреххлористый. Трифенил-
фосфит. у-Хлорметилпиридина хлоргидрат.
Перегруппировки. Алюминия хлорид. N-Бромсукцинимид. Лития бромид. Му-
равьиная кислота. Серебра перхлорат. Фосфорная кислота. Хлористый
водород.
Перемещение метиленовой группы. трет-Бутилат калия.
Переэтерификация. Серебра окись.
Пикраты. Пикриновая кислота.
Померанца — Фрича реакция. Бора трифторид — трифторуксусный ангидрид.
Расщепление. Ацеталей: Трифенилметилборфторид. ви(|'Гликолей: Гексанитра-
тоцеррат(1У) аммония. Нуклеозидов: трет-Бути л ат калия. Тиоацеталей:
Метил иодистый. Серебра окись. Триэтилоксония борфторид. Этилентио-
кеталей: Хлорамин-Т. Эфиров простых: Лития иодид. Натрия этилмеркал-
тид. Эфиров простых силиловых: Тетра-н-бутиламмония фторид. Эфиров
простых циклических p-хлорзаметенных: Натрий. Эфиров сложных мети-
ловых: 1,5-Диазабицикло-[4,3,0]-нонен-5. 1,5-Диазабицикло-[5.4,0]-ундецен-5.
Ренея никелевый катализатор. Ионообменные смолы.
Реформатского реакция. Лития диизопропиламид. Трихлоруксусной кислоты
этиловый эфир.
Риттера реакция. Нитрония борфторид.
Розенмунда восстановление. Палладиевые катализаторы.
Силилирование. Триметилсилилдиэтиламин.
Симмонса—Смита реагент (см.).
Синтез. Адамантан-2-карбоновой кислоты: Диметилсульфоксонийметилид. Ази-
нов: Полифосфорная кислота. Азиридинов: Бензолсульфонилазид. Азо-
алканов: трет-Бутил гипохлорит. Азоксиалканов: Триэтилоксония борфто-
рид. а-Алкилакриловых кислот: Лития диизопропиламид. N-Алкил-
амидов: п-Толуолсульфохлорид N-Алкиланилинов: Амид натрия.
Алкиларилсульфидов: Метилат натрия. О-Алкилбензогидроксимоил-
хлоридов: Фосфор пятихлористый. Алкилбромидов: Диметилбром-
сульфония бромид. а-Алкилгидракриловых кислот: Лития диизопро-
пиламид. Алкилиодидов: Дициклогексилкарбодиимида иодметилат. Три-
хлоризоциануровая кислота Алкилнитратов: Азота четырехокись. Алкил-
силанов: Дихлорсилан. Алкилхлоридов: Молибдена гексакарбонил. Три-
хлоризоциануровая кислота. Алкинилдиэтилаланов: Диэтилалюминия хло-
рид. Алленов: Диизопинокамфилборан. Лития N-изопропилциклогексиламид.
Магний. Алленовых кислот: Диэтил- (1,1,2-трифтор-2-хлорэтил) -амин.
Аллиловых спиртов: Лития алюмогидрид — натрия метилат. транс-Алли-
ловых спиртов: Диизобутилалюминийгидрид. Аллилхлоридов: Диметил-N-
сукцинилсульфония хлорид. Метансульфохлорид. Трифенилфосфин — угле-
род четыреххлористый. Альдегидов: 2,4-Диметилтиазол. Метил-(метилтио-
метил) -сульфоксид. 2-Метил-З-тиазолин. 1,3-бис- (Метилтио) -аллиллитий.
Натрия железа (П) тетракарбонил. Никель-алюминиевый сплав. Тал-
лия (III) нитрат. 2,4,4,6-Тетр а метил-5,6-дигидро-1,3-(4Н) -оксазин. 2,4,4-
Триметилпентил-2-изонитрил. М,4,4-Триметил-2-оксазолиния иодид. симм-
Тритиан. Этилвиииловый эфир. Амидинов: Гексаметилтриамид фосфорной
кислоты. Амидов: Ь1-Карбэтокси-2-этокси-2-лигидрохинолин. Натрия гид-
рид — диметилсульфоксид. Тетрахлорсилан. Трифенилфосфит. Амидов аце-
талей: Триэтилоксония борфторид. Аминоантрахинонов: Диметилформ-
амид. Аминов: Трифенилфосфин — азодикарбоновой кислоты диэтиловый
эфир. Фенилдихлорборан. Аминокислот: (+)- и (—) -2,3-О-Изопропилиден-
2,3,-диокси-1,4Л«с-(дифенилфосфино)-бутан. Палладиевые катализаторы.
Цианистый водород. рАминоспиртов: Триметилхлорсилан. Анилинов:
4-Хлор-2-фенилхиназолин. Аннуленов: Пропаргиловый альдегид. Антро-
нов: Пиридина хлоргидрат. Апорфинов: 6-Метокси-7-окси-3,4-дигидроизо-
668
хинолииия иодметилат. Арилацетиленов: н-Бутиламин. Иодэтинилтриметил-
силаи. Арилбромидов: Таллия триацетат. Арилиодидов: Медь хлорная.
Таллия (III) трифторацетат. Арилуксусных кислот; Церия тетраацетат.
Арилциклопропанов и арилциклопропенов: Лития 2,2,6,6-тетраметилпипе-
ридид. Ацетилена: Триметилсилилдиазометан. Ацетиленов: Трифенилфос-
фин— углерод четыреххлористый. у,б-Ацетиленовых кетонов: Днэтилалю-
минийхлорид. Бензиламинов: Изовалин. Бензилгалогенидов: Диметил-N-
сукцинилсульфонця хлорид. Бензилов: Таллия(Ш) нитрат. Безоцикло-
пропена: грет-Бутилат калия. 3,4-Бензпирена: Пирен. «Биуреттрихлорида»
(1Ч,\1,\1',\Г-тетраметил-1,3-Дихлор-2-азатриметинцианина): Дихлормети-
лендиметиламмония хлорид. Бициклических эфиров: Муравьиная кислота.
Бромгидринов: N-Бромацетамид. а-Бромкислот и их эфиров: Бария гид-
роокись. у-Бутиролактонов: Перекись водорода в щелочной среде. Валент-
ных изомеров аренов: Метилен хлористый — я-бутиллитий. Винилизоци-
анидов: Диэтил изоциан мети л фосф он ат. Винилкетена тиоацеталей: 2-Ли-
тий-2-триметилсилил-1,3-дитиан. Виниловых эфиров простых: и-Метокси-
бензилиденвольфрам (0) пентакарбонил. Виниловых эфиров трифторметан-
сульфокислоты: Трифтор метансульфокислота. Винилтетрагидропиранило-
вых эфиров: Гексаметилтриамид фосфорной кислоты. Галогенгидринов
сложных эфиров: Фосфор пятихлористый. а-Галогенкетонов: Днхлорме-
тиллитий. Гетероциклов: Амид калия. Ацетилендикарбоновой кислоты
диметиловый эфир, бис-(Бензонитрил) -палладий (II)дихлорид. Диимино-
янтарной кислоты динитрил. Дитиоугольной кислоты N-цианимида дика-
лиевая соль. Дихлорметилиден диметилам иония хлорид. Диэтилизоцианме-
тилфосфонат. Калия гидроокись. N-Карбэтоксиизотиоцианат. Лития алю-
могидрид. Меди(1) фенилацетиленид. Метилгидразин. Окись этилена.
1,1,1,3,3-Пентахлоразапропен. Полифосфорная кислота. Полифосфорной
кислоты эфиры. Свинца тетраацетат. Тозилметилизонитрил. п-Толилтио-
метилизонитрил. Триметилсилилдиазометан. фосфора хлорокись. Фтори-
стый водород. Этилдиизопропиламин. Гидринданов: Уксусный ангидрид —
цинка хлорид. Гидроксамовых кислот; Молибдена пятиокись — гексаме-
тапол (гидрат комплекса), ем t|-Гидропер оксиспиртов: Перекись водорода.
1,2-Гликолей: Диметилсульфоксид. Глицидных кислот нитрилов: Бензил-
триэтиламмопийхлорид. Глицидных эфиров: бис- (Три мети леи лил)-литий-
амид. 0-Глюкопиранозидов: Бора трифторида эфират. 1,3-Дегидроада-
мантанов: я-Бутиллитий. 1,3,2,4-ДиазадифосфетиДинов: Г ексаметилтри-
амид фосфорной кислоты. Диазоацетофенона: Диазометаи. а-Диазокето-
нов: я-Толуолсульфонилазид. аелг-Диал кил алканов: Диметилмедьлитий.
Диалкилов и диарилов: Серебра нитрат. Диамантана: Алюминия хлорид.
Дибензо-1,4-диазепина: Гидроксиламин-О-сульфокислота. Дигидро-у-пиро-
нов: 2-(2,2-Диметоксиэтил)-1,3-дитиан. Дигидрофуранов-3: 2-(2,2-Диметок-
сиэтил)-1,3-дитиан. Диенов-1,3: Дивинилмедьлитий. Дициклогексил-
боран, а-Дикетонов: Калия перманганат — уксусный ангидрид. Рутения че-
тырехокись. 1,3-Дикетонов: Натрия цианид. 1,4-Дикетонов: Меди(1) аце-
тилацетопат. Янтарной кислоты тетра метилдиамид. 1,5-Дикетонов: Янтар-
ной кислоты Гетр а мети л диамид. 1,6-Дикетонов: Винилмагнийхлорид. 2-Ди-
метиламинобензо-1,3-диоксола: Диметилформамид — тионилхлорнд, 3,3-Ди-
метоксициклоиропена: Амид калия. 1,6-Диокса-6а-тиапенталена: Таллия(1П)
трифторацетат. еи^-Диолов: Титан треххлористый. 1,3-Диолов: Диборан.
транс- 1,2-Диолов: Диборан. Енаминов: Молекулярные сита. Енолов фосфа-
тов: Диметилмедьлитий. Изонитрилов: Бензилтриэтиламмонийхлорид. Диме-
тилформамид— тионилхлорнд. Тионил хлористый. Трифенилфосфин. Три-
фенилфосфин — азодикарбоиовой кислоты диэтиловый эфир. Изоциана-
тов: Бромциан. Изоцианатохинонов: Оксалилхлорид. Имидазолов: Родия
окись. Индолов: Этилендиамин. Йодидов: Трифенилфосфита иодметилат.
Карбаматов: Меди (И) метилат. Таллия (III) нитрат. Карбодиимидов:
Азодикарбоновой кислоты диэтиловый эфир. Трифенилфосфин — углерод
четыреххлористый. Карбонатов; бис-(Трифенилфосфин) -никель (0). Карбо-
новых кислот: Тозилметилизонитрил. Карбоновых кислот диметилами-
дов: Гекса метилтриамид фосфорной кислоты. Кетена тиоацетатов: 2-Ли-
669
тий-2-триметилснлил-1.3-дитиан. Кетиминов: Молекулярные сита. уКето-
карбоновых кислот эфиров; Натрия цианид. (J-Кетокислот: Триметилхлор-
силан. а-Кетокислот эфиров: 2-Карбэтокси-1,3-дитиац. Р-Кетокислот эфи-
ров: Диэтил-(1,1,2-трифтор-2-хлорэтил)-амин. у-Кето нитрил о в; Натрия
цианид. Кетонов: Лития диизопропиламид. Натрия железа(II) тетракар-
бонил. Никеля карбонил. Таллпя(Ш) нитрат. 2,4,4,6-Тетраметил-5,6-дпгид-
ро-1,3-(4Н) -оксазин. Трифенилметилгексафторфосфат. Кислот сложных
эфиров и амидов: Натрия железа(II) тетракдрбопил. Кумаринов: Пири-
дина хлоргидрат. Цинка карбонат. р-Лактонов: Серебра цитрат. у-Лак-
тонов; а-Хлор-Н-цикдогексидпропанальдопитроп. Макролактонов: Нике-
ля карбонил. Манниха оснований: N.N-Днметилмети ленам иония иодид.
Меркаптанов: N-Апетилтпомочевипа. Метансульфокислоты эфиров: Ме-
таисульфохлорид. а-Метил-у-бутиролактонов: Лития диизопропиламид.
Метиленацеталей: Диметилсульфоксид— N-бромсукцинимид. а-Метилен-
у-бутиролактонов: Бензоила перекись. 1,2-Дибромэтац. Метиленциклоалке-
нов: Дихлоркетен. Метилкетонов: Диметнлмедьлитий. Метилмедь. Метил-
тиометиловых эфиров сложных: Хлордиметилсульфид. Моиобромцикло-
пропанов: бис- (Триметилсилил)-натрийамид. Мочевин: Селен. Надкислот:
Фосфорной кислоты диэтилового эфира серебряная соль. Нафталина 1,2-
окиси: Метилат натрия. а,р-Ненасыщенных альдегидов: н-Бутиллитий.
1,3-бис- (Метилтио) -аллиллнтий р, у-Ненасыщенных альдегидов: !,3-Ди-
тиап. а,р-Ненасыщенных кетонов: Дициклогексилкарбодиимид. у,6-Нена-
сыщенных кетонов: Дивинилмедьлитий — три-н-бутилфосфип. ^'-Ненасы-
щенных кислот: Цинк, у,б-Ненасыщенных кислот эфиров: Карбэтоксиме-
тилмедь. у,б-Ненасыщенных нитрилов: Цианметилмедь. а,р-Ненасыщен-
ных сульфонов; н-Бутиллитий. Нитрилов: Гексаметилтриамнд фосфорной
кислоты. Гексахлорциклотрифосфазатриен. Глутаровой и янтарной кис-
лот динитрилы. Метилизоциаиат. Титан четыреххлористый. я-Толуолсуль-
фонилгидразин. Трихлоризоциануровая кислота. Угольной кислоты мети-
лового эфира гидразид. Нитроалканов: Азотная кислота. Натрия боргид-
рид. N-Нитрозаминов; 2-Нитропропана 2-гидроперекись. С-Нитрозосоеди-
нений: Серебра карбонат — целит. З-Нитроизоксазолов: Натрия нитрит.
N-Окисей: трет-Амила гидроперекись. 9-Оксабицикло-[3,3,1]-нонена-1:
Трет-Бутилат калия. Оксазиридинов: трет-Амила гидроперекись. Оксаспи-
ропентанов: Дифенилсульфонийциклопропилид. р-Оксидитиокоричных
кислот: трет -Бутилат калия. а-Оксикислот: Метил-(метилтиометил)-суль-
фоксид. Талия (III) ацетат. Р-Оксикислот: Лития диизопропиламид. р-Окси-
а,Р-ненасьиценных кислот: Литий — нафталин. Олефинов: Диметил-
формамида диметилацеталь. Железа пентакар бонпл. Калия гек-
сахлоровольфрамат. Метилсульфин и лметилиднатрий Натрий — нафталин.
Натрия сульфид. Тионхлоругольной кислоты л-толнловый эфир. л-Толуол-
су ль фон и лги др аз ин. Триметплфосфит. Трифенилфосфип. тркнс-Олефинов:
Бромциан. 2,3-Диметилбутил-2-боран (тексилборап) Пиридинов: 3,5-Ди-
метил-4-хлорметилизоксазол Лития алюмогидрид — пиридин. Полицикли-
ческих углеводородов: Трифенилфосфин Прегнана и боковых цепей корти-
коидов: Цинк. Пропаргиловой кислоты эфиров: Диазоуксусный эфир. Раз-
ветвленных сахаров: 1,3-Дитиан Спиробутанонов: Дифенилсульфоний-
циклопропилид. Спиро-[4,4]-нонатетраена. Нитрометан. Спиро-[4,4]-нона-
триена-1,3,7: Натрия иодид. Спиросоединений: N.N-Диметиламиноцикло-
пропилфенилсульфоксония борфторид. Спироэпоксициклогексадиен-2,4-
онов: Перйодаты. Спиртов: N-Бромсукцинимид. я-Бутиллитий. б«с-(3,6-Ди-
метил)-борепан. Кислород. д-Толилсульфинилметилидлитий. Р-Тетралонов:
Метилен хлористый. Тиадиазиридинов 1,1-диокисей: Натрия гидрид —
грет-бутилгипохлорит. Тиоамидов: Тиоацетамид. Триазолов: Натрия
азид. Угольной кислоты этилового эфира азид. Триалкилкарбинолов: Ли-
тия триэтилметилат. Углерода окись. Тризамеиценных олефинов: Броми-
стоводородиая кислота. З-Метоксиизопрен. Ортоуксуспой кислоты три-
этиловый эфир, трис- (Трифенилфосфин)-родий (I) карбон и л гидрид. Трифе-
нилметиловых эфиров простых: Трифенилметилгексафторфосфат. Урета-
нов: Дифенилфосфорной кислоты азид. N,N,N-TpH3Tiui-N -карбэтоксисуль-
Б70
фамида внутренняя соль. Арилуксусных кислоты метиловых эфиров; Тал-
лия (III) нитрат. 1-Формиламино-1-диэтилфосфонилалкенов; Диэтилизо-
цнанметилфосфопат. Фуранов; Диметилсулъфопийметилид. 2-Хлоралкенов:
Фосфор пятихлорпстый. Хлоргидринов: Трифенилхлорметан. ц-Хлоркето-
нов; Хромнла хлорид. а-Хлорсульфоксидов: Диазометан. 0-Хлорсульфо-
нов; Хлорная медь. Холестерилдихлорфосфата: Фосфора хлорокись.
Циангидринов: Диэтилалюминийцианид. (в-Цианоаминокислот: о-Нитро-
фенилсульфенхлорид. Циклических дисульфидов: Свинца диацетат.
Циклических карбонатов: Надмуравьиная кислота. Циклических кетонов:
n-Толуолсульфонилгидразин. Циклоалкенов окисей; грет-Бутилат калия.
Циклобутандиона-1,2: Бром. Циклобутандионов: Хлортрифторэтилен.
Циклобутанонов: Бромистый водород. Лития иодид. Трифтор метансуль-
фокислоты ангидрид. I -Хлор-М,Ы,2-триметилпропениламИн. Циклопента-
нонов: Дифенилсульфонийцнклопропилид. А3-Циклопентенонов: 1,3-Дитие-
ния борфторид. Циклопропанов: Диазопропионовой кислоты метиловый
эфир. Меди закись — трет-б утилизе и игр ил. Циклопропанонов кеталей:
Магний. Циклопропенона: Три-«-бутилстаннан. Циклопропилазидов: Рту-
ти азид. Циклопропилкетонов: трег-Бутилат калия. Серная кислота. Эти-
лат натрия. Циклопропилметиленовых производных: Три-н-бутил-я-бутен-
3-ил-олово. Эписульфидов: Роданиодид. Эпоксидов: Гексаметилтриамнд
фосфорной кислоты. Диметилсульфоксонийметилид. Эпоксиолефинов:
Гексаметилтриамид фосфорной кислоты. N-Этилбензиламина: Диэтоксп-
карбония борфторид. Этилеикеталей: Ртути(П) ацетат. Эфиров 2-алкинил-
карбоновых кислот: Таллия(Ш) нитрат. Эфиров простых: Алюминия хло-
рид. Диметил фосфит. Метилсульфииилметилиднатрий. Молибдена гекса-
карбонил. Натрия гидрид. Три-н-бутилстамнан. Трихлорсилан.
Соммле реакция. Гексаметилентетрамин.
Сопряженное присоединение. Дивинилмедьлитий. Диметнлмедьлитий Медь
бромистая. Три-н-бутилфосфина — меди(1) иодида комплекс.
Спироалкилирование. Дифенилсульфонийцнклопропилид.
Стефена восстановление. Никель-алюминиевый сплав.
Стивенса перегруппировка. Натрия гидрид.
Стивенса перегруппировка — элиминирование. Диалкоксикарбопия борфто-
риды.
Сукциноилирование. Янтарный ангидрид.
Сульфирование. Серный ангидрид — пиридин.
Теломеризация. Углерод четыреххлористый. бис-(Трифенилфосфин)-цикель(О).
Тозилирование. К’-Метил-М-тозилпирролидиния перхлорат.
Трифторацетоксилирование. Свинца тетра-(трифторацетат).
Ульмана реакция. Медная бронза. Медный порошок.
Уэдсворта — Эммонса реагент. Диэтил-(I-метилтио)-этилфосфонат.
Фаворского перегруппировка. Метилат натрия.
Фосфорилирование. Диацетила и триметилфосфита аддукт. Трифенилфос-
фин— азодикарбоновой кислоты диэтиловый эфир.
Фрагментация. Пропандитиол-1,3. Серебра карбонат—целит. Этила пере-
кись.
Фриделя — Крафтса ацилирование. Нитроэтан. Тетрахлорэтан — нитробензол.
Трихлорацетилхлорид.
Фторирование. Трифторметилгипофторит. бис- (Фторокси) -дифторметан.
Хлорирование. грег-Бутилгипохлорпт. Иодбензола дихлорид. Медь хлорная.
Сульфурил хлористый. Сурьма пятихлористая. Титан четыреххлористый.
Треххлористый азот. 1-Хлорбензотриазол. N-Хлорполималеинимид.
671
Хлорметилирование. Формальдегид. Хлорметилметиловый эфир.
бнс-Хлорметилирование. Хлорметилметиловый эфир.
Цианборирование. Натрия цианид.
Циансилилирование. Триметилсилилцпанид.
Циглера циклизация. 1Ч,Ы-фенилметилнатрийамид.
Циклизация. Бензоила перекись. Бора трифторида эфират. Диметилдихлор-
силаи. Диметилмедьлитий. Димстилформамида диметилацеталь. Лития
алюмогидрид. Метилсульфипилметилиднатрий. Натрия гидрид. Полифос-
форпая кислота. Ртути(II) трифторацетат. Серная кислота, трдс-(Три-
фенилфосфин) -родийхлорид.
Циклизация галогенкеталей. Калия гексаметилдисилазан.
Циклобутадиен. Гексацитратоцеррат(1У) аммония.
Циклодегидратация. Ионообменные смолы.
Циклодимеризация. Циклопентадиенилбензилтитан.
Циклоприсоединение. Ацетилендикарбоновой кислоты диметиловый эфир.
трет-Бутилцианкетен. 4,4-Диоксо-2-метокси-6-метиЛ’!-окса-4-тиа-3,5-Д!1-
азин. Дифенилкетен. Дихлоркетеи. Кислород синглетный. 2-Метоксиаллнл
бромистый. Серебра борфторид. Триметилсилилдиазометап. N-Фенилимид
азодикарбоновой кислоты. Хлорсульфоизоцианат. а-Хлор-Ы-циклогексил-
прогтанальдонитрон.
Циклопропанирование. Диэтилцинк — метилен иодистый. Симмонса — Смита
реагент.
Чугаева реакция. Метилсульфипилметилиднатрий.
Шмидта реакция. Натрия азид.
Элиминирование HOI. Метансульфохлорид. Фосфора хлорокись.
Эпимеризация. Тетра-«-бутиламмония формиат.
Эпоксидирование. N-Бромсукцинимид. Иминоиадбензойная кислота. п-Карб-
метоксинадбензойная кислота. Молибдена гексакарбонил. Молибдена пя-
тиокись—гексаметапол (гидрат комплекса). Надуксусная кислота.
о-Сульфонадбеизойная кислота. Л1-Хлорнадбензойная кислота.
Этерификация (сложные эфиры). Бора трифторида эфират. Борная кислота.
Гексаметилтриамид фосфорной кислоты. Ионообменные смолы Метил-
трет-бутиловый эфир. Триметилфосфит. Триэтилоксония борфторид.
Этиленового фрагмента введение. Фенил-и-стирилдиметиламиносульфоксония
борфторид.
Эфиров простых расщепление. Алюминия хлорид.
Эшвейлерй — Кларка реакция. Формальдегид.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
А-Аза-А-гомостероиды 514
Адамантан 37, 282, 416, 469
Адамантан-1-карбоновая кислота 53
Адамантан-2-карбоновая кислота 166
Адамантац-I-карбоновой кислоты ме-
тиловый эфир 52—53
Адамантанон 166
Адиантиф'олин 420
Адипиновой кислоты диметиловый
эфир 316
Адипоин 473, 474
2- Азетид инон-4 - спи по -2'- (6',6'-д.име~
тилбицикло-[3,1,!]-гептан) 623
Азетидиноны-2 247
а-Азидовинилиетоны 237
7а-Азидо-А5-стероиды 436, 437
Зсх-Азидохолестанон-2 436
ЗР-Азидохолестанон-2 436
цис-2- Азидоциклопропил мерку рази-
ды 426
Азиридин И
Азиридины 46, 47
Азоалканы 72, 362, 363
Азобензол 611
Азодиизобутиронитрил 515, 517
Азодикарбоновой кислоты диметило-
вый эфир 389
Азодикарбоновой кислоты диэтило-
вый эфир 11 —12, 597, 599
Азоксиалканы 574—575
Азоксисоединения 358
Азота четырехокись 12
Азотистоводородная кислота 355
Азотная кислота 12—13, 588
Азоэтан 363
Азулен-4,5-дикарбоновой кислоты ди-
метиловый эфир 32
Аймалицин 429
Акриловая кислота 26
Акрилонитрил 524, 544
Акролеин 269
Алан 283—284
Аланин 378
Алкалоиды ряда индола 614—615
Алкенил-1-аланы 126
2-Алкенил-1,3,2-бензодиокса боролы 43
Алкенилборные кислоты 43
Алкенилгалогениды 643
Алкенилмеркурацетаты 43
Алкенкарбонаты 545
а-Алкилакриловые кислоты 293
N-Алкиланилины 24
Алкиларилацетилены 473
сс-Алкиларилуксусиых кислот метило-
вые эфиры 473
О-Алкилбензогидроксимоилхлориды
605
2-Длкил-1,3,2-бензодиоксаборолы 42,
43
Алкилборные кислоты 42
Алкилборных кислот эфиры 198
4-Алкилбутен-2-олы-1 64
tx-Алкилгидр акриловые кислоты 293
Алкилдиазотаты 574, 575
2-трет- Алкил-1,3,2-диоксабороланы
579
Алкилдихлорбораны 198
Алкилиденсулъфоны 75
2-Алкилиденциклобутанолы 458—459
N-Алкилиндолы 94
Алкилирование кетонов 40, 78—80
2-Алкил-2-карбэтокси-1,3-дитианы 259
Алкилмагнийфториды 236
1-Алкил-2-метилгидразины 34
Алкилнитраты 12
N-Алкилпирролы 656
N-Алкилфталимиды 541, 542
2-Алкилциклопентеноны 490
Алкилы иодистые 210
Алкини л диэтил аланы 212—213
Алкин-2-овых кислот эфиры 472
Алкинил-1-уксусных кислот эфиры
112—113
Алкоксикарбонилов перекиси 96
Алкоксинитропиримидины 452, 453
Алкокси-1,2,4,6,8-пентаазанафталины
452, 453
Алкоксисульфониев борфториды 615
Аллен 310
Аллена циклоолигомеризация 545
22 Зак. 596
673
Алленкарбоновые кислоты 220—221
Алленкарбоновых кислот амиды 408
Алленкарбоновых кислот нитрилы 408
Алленкарбоновых кислот эфиры 475—
476
Аллены 128—129, 309, 352—353
Аллилбензол 117, 429, 430
Аллилбромид 385
Аллилгалогениды 154—155, 260, 325
642
Аллилгидрохинон 385, 386
Аллилмагиийхлорид 124
Аллилмедь 124
1-Аллил-2-нафтол 47
л-Аллилникельгалогениды 384, 385,
642
Аллиловые спирты 126, 127, 284, 285
Аллиловый спирт 253
л-Аллилпалладия комплексы 400—
401, 402
Аллилфениловый эфир 402
2-Аллилфенол 47
Аллилхлориды 554—555
2-Аллилциклогексанон 402
Аллильное окисление 61, 311, 429—
430, 443—445
Аллоцимена гидроперекись 538
Альдазины 418
Альдегидов фенилгидразоны 588—589
Альдольная конденсация 23, 78, 277
Альдонитроны 441
Алюминий 13—14, 586
Алюминия амальгама 335
Алюминия бромид 14, 49, 265
Алюминия бромид, in situ 14—15
Алюминия гидрид 349, 389
Алюминия(Ш) иодид 322
Алюминия окись 15, 161, 411
Алюминия хлорид 15 —20, 208, 329,
389, 392, 418, 584
Амберлит-15 243
Амберлит IR 243
Амидины бициклические 20—23
Амид калия 23—24, 221, 247
Амид натрия 24, 69, 300, 658
Амид натрия — трет-бутилат натрия
24—25
Амидов ацетали 574
Амиды 363, 497, 574
трет-Амила гидроперекись 25
трет-Амилгидросульфат 25
трег-Амил-8-хинолилкарбонат 25—26
Аминоалкенилироваиие 625—626
Аминоантрахиноны 171
н-Аминоацетофенон 26
4-Аминобензойная кислота 51
Аминов окиси 227—228
4-Амино-3-гидразино-5-меркапто-
1,2,4-триазол 26—27
Аминодегидроксилирование 23, 221
674
2-Амино-3,6-ди-трет-бутилфенол 255
а-Аминоизомасляная кислота 232
Аминокислоты 233, 398, 639—640
Аминомалоновой кислоты динитрил
371
Аминометилирование 424—425
а-Амино-а-метилмасляная кислота
231—232
2-Амиио-2-метилпропанол-1 522
2-Амино-2/-метоксидифенил 329
1-Амино-4-оксиантрахинон 357
Аминопиразолы 205
Аминопиримидины 205
2- (3-Аминопропил) -изохинолиния со-
ли 280
ЗР-Аминостероиды 270
2-Аминотиазол 27
3-Амино-],2,4-триазол 29
4-Аминофенол 357
7-Аминофуразано-[3,4-</]-пиримидин
441
2-Аминохинолины 418—419
2-Амино-1,4-хиноны 72
2-Амино-3-циан-5-хлорметилпирази-
на 1-окись 372
Ангалонин 465
Ангидропенициллин 73
УаР-Ангиотенсин-П 620
Д5-7-АндростадиенДиола-Зр,17Р диаце-
тат 289
5а-Андростанол-17(3 358
5а-Андростанол-17(3-он-3 358
Андростатриен-4,6,8( 14)-дион-3,17 397
Д5-Андростендиола-3£,17р диацетат
289
Д2-Андростен-5а-он-17 574
Анизол 498
Анилин 92, 157, 158, 309, 376, 454,611,
614
Анилиноазобензол 611
о-Анисовый альдегид 522
Аннелирование 29—30, 195, 261,405—
406
[10]-Аннулен 330
Г18]-Аннулен 70, 399
Антимонии см. Стибабензол
Антрахинон 27—28, 370
Антрацен 93, 613
Антрацена хингидрона калиевая соль
28
Аповербенон см , 6,6-Диметилнорпи-
нен-З-он-2
( + )-Арбускулин-В 44, 45
Аргон 89
Арилацетилены 64, 242
Арилгалогеииды 642—643
Арилглюкурониды 246
Арилдиазониев борфториды 549
Арилдиазоция соли 320
Арилди эти л фосф а ты 23
Арилизоциапаты 23
Арилиодиды 322
у-Арилмасляные кислоты 417—418
2-Арил-1,3,4-оксадиазолы 453
4-Арилокси-2-фенилхиназолины 628
Арилталлия бис-трифторацетаты 478
Арилуксусные кислоты 639
Арилуксусных кислот метиловые эфи-
ры 475
Арилуксусных кислот хлорангидриды
329
3-Арил-2-фенил-4 (ЗН) -хиназолиноны
628
Арилциклопропаны 303
N-Ароил-О-ацетилгидроксиламины
44!
Ароматизация 348—349
Ароматическое гидроксилирование
406—407
(±)-аг-Артемизен 494, 495
Асимметрическое гидроборирование
129
Аскаридол 535
Аскарит 28
D-(—)-Аскорбиновая кислота 235
Аспарагин 175
Аттрактилигенина метиловый эфир
350
Афлатоксины 649, 650
Аценафто-[1,2-с]-фуразан 501
Аценафтехинона диоксим 501
Ацетали 243, 344, 393
Ацетальдегид 526
Ацетиламиноакриловой кислоты ме-
тиловый эфир 447
2-Ацетамидо-2-дезокси-D-манноза
373, 374
З-Ацетаминоэстратриен-1,3,5 (10)-
ол-17р 199
Ацетанилид 613, 614
1-Ацетил-2-а лкил-2-тиопсевдомоче-
вины галогенгидраты 35
2- Ацетил амино-З-карбэтокси-5-этил-
тиофен 309
Ацетилацетон 313
u-Ацетилбензойная кислота 264
м-Ацетил-а-бромгидрокоричная кис-
лота 26
З-О-Ацетилглицирретовая кислота
333
I-Ацетил-2,2-диметилспиропентан 136
Ацетилендикарбоновой кислоты ди-
метиловый эфир 27, 28—34, 56
111 — 112, 168—173, 534, 545
Ацетилендикарбоновой кислоты мо-
нометиловый эфир 27
у.б-Ацетиленовые кетоны 214
Ацетилирование 86, 262
1-Ацетил-1-мети л гидр азин 34
Ацетилмочевина 35
2-Ацетилнафталин 37!
N-Ацетилнейраминовая кислота 373—
374
Ацетилсерная кислота 34, 467
2-Ацетил-1,4,5,6-тетрагидропиридин
448
N-Ацетилтиомочевина 34—35
Ацетил-п-толуолсульфонат 35
Ацетил флороглюцина диметиловый
эфир см. Флороацетофенона диме-
тиловый эфир 19
Ацетилфлороглюцина триметиловый
эфир см. Флороацетофенона триме-
тиловый эфир
Ацетил хлористый 345
N-Ацетилцистеина метиловый эфир
447
2-Ацетоксиакрилонитрил 610
4-Ацетокси-1 - (2-ацетоксиэтил) -би-
цикло-[4,3,0]-нонадиен-4,6 585
зр-Ацетокси-5[3,!4а-буфадиен-20,
22-олид 221
экзо,экзо-6-Ацетоксимеркур-7-ацет-
оксибицикло-[3,2,1]-октен-2 427
2-Ацетокси-4-метилпента диен-1,3 428,
429
З-Ацетоксипропилтрифенилфосфония
бромид 67, 68
3[3-Ацетокси-Д5-холестенон-7 571
Ацетоксициклооктатетраен 67
3(3-Ацетокси-Д5-этиеновой кислоты
метиловый эфир 106
Ацетона азин 129
1 - Ацетонил-1 -изппропенилциклопро-
пан 136
Ацетонитрил 12, 96—98, 132, 178, 237.
259, 323, 336, 357, 388, 403, 433, 451,
453, 469, 478, 481, 544, 571, 579, 600,
616, 641
Ацетоуксусный эфир 36, 102, 466,
504 '
Ацетофенон 35, 246, 247, 255, 376, 475,
503, 525
2-Ацил амино-2-дезоксиальдозы 373,
374
2-Ацил-1,3-дитианы 180
Ацилирование 415—416, 562, 598
Ацилирование внутримолекулярное
418
М-Ацилмочевины 206
Ацилиитраты 12, 13
Ацилоиновая конденсация 529—530,
593
Ацилоины 647
а-Ацилоксикарбоновые кислоты 478
Ацилоксилирование 350
З-Ацилоксипропилфосфония соли 67
Ацилтетракарбопилферраты 366—368
1 -Ацил-1,2,3-триазолы 582
2-Ацил-1,2,3-триазолы 582
675
Байера — Виллигера окисление 258.
351, 407, 408, 467, 585, 620
Бария гидроокись 36—37
Бекмана перегруппировка 514, 601
Бенгальская роза 263
Бензальазин 241
Бензальацетон 226, 551, 552
Бензальдегид 87 , 89, 168, 207, 255.
269, 275, 334, 368, 376, 509, 537, 552,
594, 611, 638
Бензанилид 185, 186
Бензантрацен 93
Бензвален 330
Бензгидрилбензилсульфоксид 468, 469
Бензгидрилидендиацетат 441
Бензгидрилхлорид 155, 468, 469
Бензгидрол 155, 247, 254, 269, 273,
362, 370, 61!
Бензил 61, 650
Бензила дигидразон 454
Бензиламин 255
Бензиламины 23!—232
N-Бензиланилип 225
Бензилат натрия 629
Бензилацетат см. Уксусной кислоты
бензиловый эфир 639
Бензилацетаты 310, 311
Бензилацетон 226
2-Бензилбензофуран 47
Бензилбромид 119, 641
Бензилгалогениды 154—155
N-Бензилидецанилин 225
N-Бензилиденсульфамиды 577
1-Бензилиндол 25!
Бензилмалоновой кислоты диэтило-
вый эфир 244
(—)-Бензилметилфенилфосфин 644
Бензиловая кислота 254
Бензиловые эфиры простые 536
Бензиловый спирт 38, 119, 255, 551,
552, 572, 611, 638
5-Бензилоксиметилциклопентадиен
609—610
Бензилтриметила ммония гидроокись
см. Тритон Б 81, 334
2-Бензил-4,4,6-триметил-5,6-дигидро-
1,3-(4Н)-оксазин 488
Бензилтриэтиламмонийхлорид 37—42
Бензилхлориды 303
Бензил хлористый 38, 171, 657
Бензилхлорметиловый эфир 600,
60!
1-Бензилциклопропанкарбоновой кис-
лоты этиловый эфир 244—245
Бензилы 472, 473, 544, 636
Бензины см. Дегидробензол 24, 188—
189, 303, 446—447, 613—614
Бензобензвален 330
Бензогидроксамовой кислоты алкило-
вые эфиры 605
Бензогомотропон 166
1,3,2-Бензодиоксаборол 42—43
N-Бензоилантраниловая кислота 404
М-Бецзоилантраниловой кислоты амид
628
Бензоила перекись 29, 30 44—45, 59,
90
Бепзоилтиомочевина 35
Бензоил хлористый 368, 395, 652
Бензоин 45, 535, 579
Бензоина енольной формы цикличе-
ский карбонат 45—46
Бензоины 97, 544, 636
Бензойная кислота 85, 212, 503, 505,
559
Бензойной кислоты фениловый эфир
54
Бензойный ангидрид 548
3,1-Бензоксатианон-4 584
Бензол 89, 222, 343
Бензолсульфамид 577
Бензолсульфокислоты гидразид 520
Бензолсульфоиилазид 46—47
2-Бензолсульфонил-1-хлорстиролы 323
М.Ы'-быс-бензолсульфо-гс-хинонди-
имнн 175
Бензолсульфохлорид 323
Бензонитрил 223, 388, 435
бис-(Бензонитрил) -палладий (II) ди-
хлорид 47
Бензотиазол 228
1,2,3-Бензотриазины 440
4,5-Бензотропон 166
Бензофенон 111, 163, 195, 246, 254,
273, 274, 320, 334, 362, 370, 384, 525,
611
Бензофенон-2-диазония борфторид
320
Бензофураны 47
Бензохинон 528
п-Бензохинон 96, 97, 385. 417, 454
3,4-Бензоциклобута-[1,2-6]-циклогеп-
татриен 483
Бензоциклобутен 407, 408
Бензо-[4,5фциклогепта-[с?е-1,2,3]-на-
фталин 199
Бензоциклопропен 68
3,4-Бензпирен 101, 399, 412
Бенз-[с]-тиофен 15
Бензтиофен 52
Бенкесера восстановление 48
а-трйис-Бергамотен 462, 463
Берча восстановление 48—49, 203
[З-Бисаболен 494 -
Биснор-S 265, 545
«Биуреттрихлорид» см. N,N,N',N'-Te-
траметил-1,3-дихлор-2-азатриме-
танциапин
Бициклические эфиры простые 348
Бицикло-[1,1,0]-бутан 352, 456
676
Бициклобутаны 456
Бицикло-[2,2,0]-гексадиен-2,5 439
Бицикло-[2,2,0]-гексен-5-дикарбоно-
вой-2,3 кислоты ангидрид 439
Бицикло-[2,2,1]-гегттадиен-2,5 35
Бицикло-[2,2,1]-гептанолы-2 36!
Бицикло-[4,! ,0]-гепт а и он-2 145
Бицикло-[4,3,!]-декатриен-2,4,8-он-7
282, 634
Бицикло [3,3,1
Бииикло-[3,3,Г
Бицикло-[4,2,Г
Бицикл о-[3,2,1
Бицикло-[2,2,2^
Бицикл о -[3,3,0;
кислота 348
Бицикло-[3,2,Г
Бицикло- 4,2,0.
Бицикло-[3,2,Г
Бицикло-[2,2,2;
Бицикло-[3,2,Г
-нонандиол-2,6 458
-нонанол-2-он-9 627
-нонен-З-он-9 402
-октадиеп-2,6 427
- окта нд ион-2,6 419
-октан-1 -карбоновая
Бицикло-[3,3,0]-октанон-2 195
-октанон-3 569
-октатриен 592
-октен-1 529
-октен-2 426
-октен-2 281, 311
цис-Бицикло-[3,3,0]-октен-2 348
Бицикло-[3,2,1]-октен-3-ил-2-ацетаты
31!
Бицикло-[3,2,1]-октен-6-он-3 340
Бицикло-[1,!,1]-пентан 353
Бицикло-[4,4,1]-ундекатетраен-3,5,8,
Ю-дион-2,7 651
Бишлера — Напиоальского реакция
420
9-Борабицикло-[3,3,1]-нонан 49
Бора трибромид 49, 53, 446
Бора трцс-(трифторацетат) 49—50
Бора трифторид 162, 222, 265, 300
Бора трифторида ди-н-бутилэфират
50
Бора трифторида эфират 50—52, 113,
114, 166, 258, 341, 545
Бора трифторид — трифторуксусный
ангидрид 52
Бора трифторид — уксусная кислота
510
Бора трихлорид 52—54, 197, 347
Борная кислота 54
Борпен-2 512
Борфтористоводородная кислота 329
Борфтористоводородная кислота —
диметиловый эфир, комплекс, 534
Брадикинин 620
Бредта правило 529
Бром 49, 54—55, 140, 372, 422, 431,
478, 529, 530, 580, 581, 598
1-Бромадамантан 119, 282
о-Броманизол 522
Броманил 412
Брома окись 432, 433
N-Бром ацетамид 55—56
N-Бромацетамид — фтористый водо-
род 56
Ы-Бромацетимино-2-бромалкиловые
эфиры 55
N-Б ром ацетимино-2-бромциклогекси-
ловый эфир 55
Бромацетон 339
бис- (а-Бромбензил) -фенилфосфино-
ксид 104, 105
Бромбензол 119, 308
э«зо-Бромбицикло-[5,1,0]-октан 454,
455
3-Бромбицикло~[3,2,1 -октен-2 28!
экзо-8-Бромбицикло-[5,1,0]-октен-2 455
экзо-8-Бромбицикло-[5,1,0]-октен-3 455
1-Бромбутан 119
цш?-1-Бромгексеп-3 490
1-Бромгексин-1 142
5/-Бром-5/-дезокснаденозин 93
4-Бром-Ь1,Ы-диметил-3-трифторме-
тиланилин 483
1-Бром-2,2-диметоксипропан 339
2-Бром-1,3-дифенилпропандиоп-!,3 328
4-Бромимидазол 483
З-Броминдол 483
Бромирование 59, 62
Бромистоводородная кислота 56—57
Бромистый водород 46, 57—58, 619
а-Бромкамфора 300
Бромкарбен 527
а-Бромкарбоновые кислоты 36—37
а-Бромкарбоновых кислот эфиоы
36—37
5-Бром-2-карбэтокси-2-метилцикло-
пентанон 653
3-Бром-5-карбэтокси-2-окси-5-метил-
циклопентеи-2-on-I 653
трднс-а-Бромкоричная кислота 150
Ь1-Броммагнезил-Ь1-метиланилин 16
1-Бром-3-метилбутен-2 494
5-Бром-1-метилимидазол 483
2-Бромметил-З-метилнафталин 90
(Е)-5-Бром-2-метилпентен-2-ол-1 56
(Е)-5-Бром-3-метилпентен-2-ол-1 57
3-Бром-4-метилпентен-3-он-2 15, 16
2-Бром-2-метилстирол 282
1-(Бромметил)-циклобутанол 59
З-Бромметилциклобутилбромид 353
Бромметилэтилеикетали 177
5-Бром-6-метокси-ц«с,тданс-циклоок-
тадиены-1,4 455
2-Бром-З-метокси-транс-циклооктен
454, 455
1-Бромнафтадин 633
Бромноватистая кислота 59—60
экзо-2-Бромнорборнан 306
Р-Бромнорпинон 160
1-Бромпентан 336
6-Бромпиридил-2-литий 660
N-Бромполималеипимид 58—59, 62!
З-Бромпропанол 582
1-Бромпропен 229
22 Зак. 595
677
N- (З-Бромпропил} -изохинолиния бро-
мид 281
транс-2-Бромстирол 643
N-Бромсукцинимид 59, 59—63, 90, 155,
176, 180, 259, 278, 289, 326, 327, 339,
432, 517, 653
I-Бромтетрадекая 533
1-Бромтриптицен 282
Бром фтористый 56
1-Бром-6-хлоргексан 367
1-Бром-3-хлор-2,2-диметоксипропан 24
1-Бром-З-хлорпропан 89
1-Бром-З-хлорциклобутан 43!
Бромциан 63—64
1-Бромциклогексен 25
Бромциклооктатетраен 67
1-Бромциклопропан-1,2-дикарбоно-
вой кислоты диметиловый эфир 361
Бромциклопропаны 527
4-Бр ом- 1,2-эпокси-1,2,3,4-тетрагидро -
нафталин 21
а-Бромэтилдиэтилборан 14
Брэдшера реагент 243
Буллвален 515, 516
Бутадиен-1,3 382, 383, 456, 644
Бутадиена-1,3 1,2-окись 64
Бутадиен-2,3-овой-1 кислоты метило-
вый эфир 297
Бутан 515
н-Бутан 265
еритро-Бутандиола-2,3 мономезилат
324
«-Бутанол 13, 119, 412
трет-Бутанол 148, 408
Бутанон-2 87
Бутатриены 69
Бутен-! 430
Бутен-2 238, 515
цис-Бутеп-2 111, 229, 346, 430
транс-Бутен-2 111, 229, 346, 430
гранс-Бутена-2 окись 323, 324
Бутен-З-овой-1 кислоты этиловый
эфир 296
трет-Бутила гидроперекись 345
н-Бутилазид 589
«-Бутиламин 17, 64, 443
о- (н-Бутил)-анилин 157, 158
трет-Бутила перекись 44
трет-Бутилат калия 22, 27, 64—72,
104, '164, 206, 218, 306, 307, 343 344,
389, 506, 607
трет-Бутилат натрия 300
Бутилацетат 394
трет-Бутилбензол 270
н-Бутилбромид 40, 269
трет-Бутилбромид 274
етор-Бутил бромистый 269
трет-Бутилгипохлорит 72—74, 157, 362,
363, 615
н-Бутилдивинилкарбинол 85
5-грст-Бутил-2,3- (2,2/-дифенилен)-те*
тразолия нитрат 588, 589
5-трет-Бутил-2,3-дифенилтетразолия
нитрат 588, 589
трет-Бутилизонитрил 314
трет-Бутилизопропилкетон 532
н-БутиллитИЙ 74—77, 87, 88, 94, 115,
117, 164, 171, 176, 178, 182, 207, 212,
213, 219, 274, 275, 279, 280, 292, 296,
302, 306, 335, 486, 487, 491, 492—
496, 507, 508, 525, 553, 595, 641, 658,
660
грет-Бутиллитий 65, 74, 142, 266
«-Бутиллития комплекс с N,N,N',
N'-тстраметилэтилеггдиамином 77—
78
трет-Бутилмагнийхлорид 78
н-Бутилмеркаптан 78—80, 326
трис-(н-Бутил)-метиламин 17
2-Бутил-2-метил-1,3-диоксолан 430
н-Бутилнафтил-1-сульфид 326
трет-Бутиловый спирт 93, 184
1 -трет-Бутилоксикарбонилтриазол-
1,2,4 80—81
З-н-Бутилпентадион-2,4 510, 511
2-н-Бутилтиометиленциклогексанон
80, 596
трет-Бутилфенилацетилен 83
трет-Бутилфенилкарбоиат 8!
трет-Бутил-8-хинолилкарбонат 81
трет-Бутилхлорформиат см. Хлор-
угольной кислоты трст-бутиловый
эфир
трет-Бутилхромат 81
трет-Бутилцианкетен 82—84, 216,
217
трет-Бутилциклогексан 270, 377
цис- и транс-Ь-трет- Бутилциклогек-
санкарбоновая кислота 438, 439
4-трет-Бутилциклогексанол 153, 185,
288, 559
4-трет-Бутилциклогексанон 153, 192,
193, 288, 349, 377
1-трет-Бути л цикл о гексен 270
З-трет-Бутилциклогексен 477
4-трет-Бутилциклогексен 185, 477,
559
5-трет'-Бутилциклогексен-2-он-1 301
N-н-Бутилциклогексиламин 589
цис- и гранс-4-трет-Бутилциклогек-
силхлорид 438, 439
етор-Бутилэтилбора бромид 14
Бутин-З-ол-2, дилитпевое производ-
ное 444
у-Бутиролактон 291, 297
у-Бутиролактоны 408
З-трет-Бутоксиоктил-1-магнийбро-
мид 519
Буфалин 55—56
5«-Буфалин 286
678
Вагнера—Меервейпа перегруппиров-
ка 51!. 622
н-Валериаиовая кислота 85
Валериановой кислоты az-бутиловый
эфир 13
Валероилпитрат 13
6-Валеролактон 290
Ванадия окептрихлорид 449
б-Вапилплиденбарбитуровая кислота
453
Вильгеродта—Киндлера реакция 475
Вильямса синтез 517
Випилаланы 126, 127
Винилалкилидепциклопропаяы 68—
69
Винил бромистый 229
Випилдиазометап 461,462
Впнилизоцианиды 217—218
Р-Випплировантгые кетоны 122
N-Випилкарбазол 136
Винилкетенов тиоацетали 278—279
Виниллитпй 122, 124. 125
Вппилмагпийбромпд 57
Вннилмагпинхлорид 85, 125
Виниловые эфиры 340—34!
3-Вицил-3,5.5-триметилциклогекса-
нон 125
Вииилтрнфторметапсульфонаты (три-
флаты) 560—561
Винилциклобутаны 644
1-Виннлпиклопропаиол 459
1-Винилциклоп ропаколы 57
Винили иклопропаны 182, 291—292
D-(-)-Винная кислота 233
L-(+)-Винная кислота 233
rf-Винной кислоты метиловый эфир
241
(/-Винной кислоты этиловый эфир
241
Висмута окись 85, 449
Висмута триацетат 85—86
Висмутилацетат 86
Витамин А 350, 351
Витамин Bia 632
Витамин А ацетат 21
Виттига олефииирование 540
Виттига реакция 86—87, 180, 540, 596,
629, 640
Вихтерле реакция 461
Вольфа—Кижнера восстановление
412, 593
Вольфа перегруппировка 525
Вольфрама гексакарбонил 345
Вольфрама гексахлорид 87—88
Восстановительное аминирование 376.
378
Восстановительное ацетилирование
650—652
Восстановительное бензоилирование
651—652
Восстановительное дезоксигенирова-
ние 486—487
Вюрца дегалогенирование 352
Вюрца реакция 353
Газы инертные 89
Галактоза 448
D-Галактонолактон 448
Галлия окись 89
Галогена обмен 14—15
Галогенгидринов эфиры сложные
603—605
Галогендекарбоксилирование 473—
438
Галогенирование 653—654
Галогеикеталей циклизация 248—219
а-Галогенкетоны 207
Галогенметилглиоксаля альдоксимы
372
3-Галоген-1,2,4-тиадиазолы 183
л-Галогенфенилсвинца трнс-трифто-
рацетаты 442
Гедикариол 451, 452
Гексабромциклопентадиеп 49
3,4,5,6,7,8-Гексагидрона фталин-1
(2Н)-он-7-карбоновая кислота 144
Гексадиен-1,2 606
Гексадиен-1,4 644
Гексадиен-2,3, 606, 659
транс, транс-Гекса диен-2,4, 654, 655
транс-Гексадиен-2,5-овой кислоты ме-
тиловый эфир 124
Гексадиены-2,4 456
Гексадиин-1,5 312, 313, 399
Гексаметилбензол 406
Гексаметилдисилазан 247, 347, 540
Гексаметилентетрамин 89—90
Гексаметилтриамнд фосфористой кис-
лоты 90
Гексаметилтриамид фосфорной кисло-
ты 75, 91—95, 100, 199, 219, 226,
276, 286, 292, 296, 297, 309, 346, 375,
501, 522, 559, 575, 607, 618, 643
Гексаметил циклогексадиен-2,4-он 406
2,2,4,5,7,7-Гексаметилциклогептен-4-он
225
н-Гексан 469
Гексанитратоцеррат(1У) аммония
95—99, 385, 639
Гексанитратоцеррат(IV) калия 96
Гексанол-1 372
1,2,4,5-Гексатетраен 32, 33
Гексафенилбензол 497
о-Гексафенилены 134
Гексафторбутин-2 534
Гексафтор-2-фепилпропанол-2 184
Гексафторсурьмяная кислота 343, 531
Гексафторфосфорная кислота 534
Гексахлорфульвён 577
22*
679
Гексахлорциклопентадиен 49
Гексахлорциклотрифосфазатриен 99—
100
Гексаэтилтриамид фосфористой кис-
лоты 100—101
гдДнс-Гексен-3 142
Гексен-1 430
Гексен-3 88
Гексен-3-диин-1,5 23
Гексен-5-илмагНийбромид 225
Гексен-5-овой кислоты хлорангидрид
518
транс-Гексен-4-ол-1 44, 352
Гексин-1 338, 606, 659
Гексин-2 606, 659
Гексин-3 126, 303, 606, 658, 659
Гематопорфирин 264
Геминальное алкилирование 193
Генкванин 19
Гептаметилдисилазан 532
Гептаналь-1 255, 634
Гептандиои-2,5 503
Гептановая кислота 41
Гептанол-1 255, 634
Гептанол-2 510
Гептен-3 88
Геранилацетат 443, 444
три с-Геранилацетон 433
Геранилбромид 155, 641
Геранилмезитоат см. 2,4,6-Триметил-
, бензойной кислоты гераниловый
эфир
Гераниол 155, 555
Гераниолен 494
Геранилхлорид 555
i/Z-Гибберелин Ais 215
Гидразин 45, 101, 418, 472, 475, 476,
521
Гидразингидрат 130, 329, 520, 541
Гидридоундекакарбонилтрижелеза
анион 224
цпс-Гидринданол-1 585—586
Гидрирование гомогенное 414
Гидрирования катализаторы 382,
548—549, 551—552, 586
Гидроалюминирование 125
Гидроборирование 43, 128—129, 139,
159, 209, 338, 347—348
Гидроксамовые кислоты 347
Гидроксиламин 376
Гидроксил амин-О-сульфокислота
101 — 102
а-Гидроперекиси 264
виц-Гидропероксиспирты 406
Гидросилилирование 550
Гидроформилирование 425
Гидрохинон 342, 417, 423, 454
Гидрохинона диаллиловый эфир 565
Гидрохинонов диметиловые эфиры 454
Гидрохиноны 97
680
Гидроцианнирование 215—216, 571
Гирннидаль 443, 444
1,2-Гликоли 41, 96, 449
Глиоксиловой кислоты метиловый
эфир 241
Глиоксиловой кислоты этиловый эфир
241
Глицерин 253
Глицеринборная кислота 89
Глицина гидразид Ill
Глицина этилового эфира хлоргидрат
122
Глицидные эфиры 526—527
Глицидных кислот нитрилы 40
Глутамин 175
Глутаровой и янтарной кислот ди-
нитрилы 102
Глутаровой кислоты тетраметилди-
амид 660
Глюкоза 448
|5-Глюкопиранозиды 50—51
транс-Гомогеранилциаипд 641
Гомологизация 275, 401
Г омосопряженное присоединение
122—123
Гриньяра реактивы 229, 304—305,308
Р-Дамаскон 240, 218
8-Дамаскон 16
[З-Дамасценон 15, 16, 268
Дарзана реакция 526
Дауномицинон 19—20
Дауиомицинона триметиловый эфир
19—20
Даффа реакция 89—90
Дегалогенирование 13, 202, 203, 281—
282, 285—286, 300—301, 354—355,
374, 577
Дегидратация 91—92, 99, 160—161,
184—185, 209, 236, 332, 541, 572—
574
Дегидрирование 27—28, 198—200,312,
491—492, 563, 612
Дегидрирование — дезоксигениро-
вание 603
Дегидроабиетиновая кислота 12, 398
Дегидроабиетиновой кислоты метило-
вый эфир 12
1,3-Дегидроадамантан 75—76
Дегидро-D -(—)-аскорбиновая кисло-
та 235
14-Дегидроацетилбуфалип 55
Дегидробензол 24, 188—189, 303,
446—447, 613—614
Дегидробензолы 24, 303
Дегидрогалогенирование 20—22, 24—
25, 30, 61, 65—67, 92, 103—104, 160,
216, 250—251, 483, 572, 609
3,6-Дегидрооксепин 106—107
Дегидротиофеп 497
Дегидро-М,1т,ЬГ''-трициклогексил-
гуанидипогексакарбонилдижеле-
30(0) 103
Дегидроцианирование 28
1,2- Дегидроциклооктатетраеп 496
Дегидроэпиандростен 221
Дегидроэпиандростерон 450
Дезалкилирование 107
Дезаминирование 12
Дезоксибензоипы 472
Дезоксигенирование 87—88, 197, 227—
228, 552, 577
(±)-11-Дезокси-13-дигидропростаг-
ландин Ei 519
5/-Дезокси-5/-иод-2/,3/-О-изопропц-
лидеиуридин 558
б'-Дезокси-б'-иодтимидин 558
Дезоксимпрование 474—475, 503
11-Дезоксипростагландины 471, 472,
486
Дейтерирование 89
Декалол-1 422—423
Декалон-1 147, 290
н-Декан 375
Деканаль-1 270, 637
1,10-Декандикарбоповая кислота 102
1,10-Декандикарбоновой кислоты ди-
нитрил 102
Декановая кислота 270
Декановой кислоты метиловый эфир
596
Деканол-1 637
Декарбалкоксилирование 375, 464
Декарбметоксилирование 105—108
Декарбоксилирование 48, 531—532
Декарбонилирование 194, 254, 455—
456, 546—547
Деметилирование 53, 57, 609
О-Деметилирование 465
4-О-Деметил-7-О-метилдаумицинон
19—20
Десмостерилацетат 39
Десульфонилирование 547
Десульфуризация 75, 100, 181, 259,
587
Децен-1 41, 254
Децианирование 92, 224, 645—646
Джонса реагент 69, 154, 166, 585,
636
1,3- Диазабицикло-[3,1,0] -гексены-3
523
1,5-Диазабицикло-[4,3,0]-нонен-5 20—
22, 103—107, 120, 160, 164, 327
1,4- Диазабицикло-[2,2,2]-октан 107,
226, 409, 582
1 5-Диазабицикло-[5,4,0]-ундецен-5
20—22, 107—108
1,3,2,4-Диаза дифосфетидины 92
2,5-Диази до-3,6-ди-трет-бути л-1,4-
бензохинон 82
Диазидостильбен 435
Диазоацетальдегид 108, 585
Диазоацетофенон 109—НО
1-Диазобутаион-2 313
Диазогруппы введение 512
а-Диазокетопы 28—29, 51, 319
Диазометан 108—110, 362, 585
Диазометил-н-пропил кетон 151
Диазопропионовой кислоты метило-
вый эфир НО—111
Диазоуксусной кислоты азид 111—
112
Диазоуксусный эфир 112—113, 283
Диазоциклогексанон 512
4-Диалкиламинопиридины 415, 416
1,1-бис-Диалкиламиноэтилены 451
Диалкилиитрозамины 228, 390
3,5-Диалкилпиридины 443
1Ч,1Ч'-Диалкилсульфамнды 72
Диалкилхлорбораны 113, 347
2,3,-Диалкилциклопентаноны 547
Диалкилы 452
Диалкоксикарбония борфториды
113 — 115
Диалленил см. 1,2,4,5-Гексатетраен
2,5-Диаллилгидрохинон 565
Диаллилмалоновой кислоты дихлор-
ангидрид 389
2,6-Диаллилциклогексаноп 402
Диамантан 18, 38, 365
1,4-Диамино-2,5-дибромбензрл 360
2,5-Диамино-3,6-дихлор-1,4-бензо-
хинон 394
Диаминомалеиновой кислоты дини-
трил 131
1,8-Диаминонафталин 135
4,6-Диамиио-5-нитрозопиримидины
441
Диарилацетилены 473
Диарилсиланы 550
Диарилсульфиды 97
Диарилсульфоксиды 97
Диарилы 452, 642
М.Ы'-Диацетилдииминосукцинонит-
рил 132
1,4-Диапетоксибутен 402
1,1 -Диацетокси-2-3-диметил бутен -2
428
2,7-Диацетокси-1,6-метано-[10]-анну-
лен 651
3,3-Диацетокси-2-метилпропец 428, 429
силм<-Диацетокситетрабутилдистай-
ноксан см. б/'с-(Дибутилацетокси-
олово)-оксид
1,2-Диацетокси-З-фенилпропан 430
Дибенз-[б, (]-азепин 656
5,5-Дибензилдитиогидантоин 17
Дибензилкетон 116, 234, 533
681
4,4-Дибензил-2,5-бис- (метилтио) -
4Н-имидазол 17
Дибеизплфосфат 54!
Дибензобарролеп 227
Дибензо-1,4-л пазепины 10!—102
о-Дибензоилбеизол 186
2,2/-Дибензоилдифеиил 320
1,3-Дибензоилпропан 234, 235
1,2-Дцбензо11ЛШ11<.лонропан 234, 235
Дибеизо-18-краун-эфир-6 210, 211
Ди-2-бензотпазолилдисульфид 100
Ди-2-бензотиазолилсульфид 100
Диборан 42, 116-119, 139, 140, 159,
338, 348, 379, 480
Диборан—диметплсульфид 116
Диборан—тетрагидрофуран 116
1,3-Дибромадамантан 75
ct, ctz- Диброма лкан дика рбоновых кис-
лот диметиловые эфиры 360
N.N-Дибромацетамид 55
Дибромацетонитрил 640
7,7-Дибромбицикло-[4,1,0] -гептан 282
7,7-Дибромбицикло-[3,2,0]-гептен-2-
он-6 539
здзо-8,8- Дибром цикло-[5,1,0]-октан
454, 455
8,8-Дибромбицикло45,1,0]-октеп-2 455
8,8-Днбромбицикло-[5,1,0]-октен-3 455
2,3-Дибромбутан 515
1,4-Дибром-Д2>6-цис-гексалин 285
цис-З,4- Дибромгексахлор-1,2-диме-
тиленциклобутан 13
сда'-Дибромглутаровой кислоты ди-
метиловып эфир 361
2,4,-Дибр ом-2,4-диметил пентанон 225
eutf-Дибромиды 374
Дибромкарбен 37
Дибромкетен 539—540
а.а'-Дибромкетенов кетали 309
2,6-Дибромпиридпн 660
9,9-Дибромфлуорен 134
2,12-Дибромцикло до деканон 325
транс-1,2- Дибром циклогексан 25
1,2-Дибром циклононадиен-1,5 585,
586
1,2-Дибромэтац (этилен бромистый)
119-120
Дибутилацетоксиолова гидрид 417
бас-(Дибутилацетоксиолово)-оксид
417
3,6-Ди-т/?ег-бутил-1,2-бензохинон 255
2,6-Ди-грег-бутил-а-диметиламино-
п-крезол 120—121
Ди-я-бутилдисульфид 326
1,3-Ди-грет-бутил-1,3-дициан аллен
83—84
Ди-грет-бутилиминоксил 121, 373
Ди-я-бути л кетон 17, 269
2,6-Ди-трет-бутил-га-крезол 362
Ди-я-бути л медь литий 121
1,3-Ди-я-бутилмочевина 443
Ди-я-бутиловый эфир 227, 228
2,4-Д11-грет-бутил-6-окспметплфсиол
409
Дибутилолова окись 417
Ди-трет-бутилпирокарбонат 121 — 122
З.б-Ди-грет-бутилпирокатехинаце-
таль бензальдегида 409
Ди-я-бутплсульфид 584
Ди-н-бутндсульфоксид 584
Ди-трет-бутплтрпкарбоиаг 121 '
Ди-трет-бутилхипопы 109
Дивпнилбепзол 621
Дивинилмедьлитий 122—124, 125
Дивинилмедьлитня комплекс с три-
я-бутилфосфином 124—125
Ы.М-Дигалогеиамины 17—18
Дигалогепкарбены 393—394
ц«с-7,8-Дигалогенциклооктатриены-
1,3,5 67
8,8/-Дигептафульвенил 308
4/,5/-Дигидроаденозин 60
Дигидроанизол 610
9,10-Дигидроаитрацен 93
2,3-Дигидро-1,5-бепзоксазепил-(5Н)-
он-4 656
1,3-Дегидробензо-[с]-тиофена 2-окись
15
Дигидродибензотетраза-[14]-апнулен
421
Дигидродиктамнин 199
Дигидрожасмон 461
Дигидро-а-ионон 551
Дигидро-р-иопон 551
Дигидрокарвон 549
(+)-Дигидрокарвона ацеталь 495, 496
Дигидрокриптомерион 495, 496
1,2-Дигидронафталин 250
цис-9,10-Дигидронафталпн 330, 331
транс-9,10-Дигидронафталин 331
!,3-Дигидронафто-[1,2-с]-тиофена
2-окись 15
4,5-Дигидро-7 (6Н) -оксопиридо-
[1,5-а)-пиразол-2,3-дикарбоновой
кислоты диметиловый эфир 28—29
2,3-Дигидропираи 51
Дигидро-у-цироны 176
4,5-Дигидротиепина 1,1-диокись 461,
462
9,10-Дигидрофенантрен 491, 492
1,2-Дигидрофталазин 224
Дигидрофураноны-3 176
1,4-Дигидро-1-хлорстиба бензол 107
Диены 123—126
Диизоамилборан см, бис-(1,2-Диме-
тилпропил-1) -боран
Диизобутилалюминийгидрид 125—
128
Диизопинокамфилборан 128—129
Диизопрена дианион 276
682
Диизопропиламид лития 486
М,1\г/-Диизопропилгидразип 129—130
Диизопропилсульфид 122
Диизопропилэтиламин 575
Диизопропоксифенилфосфин -382, 383
Диимид 130—131, 520
ДииМЦНОСуКЦИНОНИТрИЛ см. ДнИМИПО-
янтарной кислоты динитрил
Диимпнощавелевой кислоты димети-
ловый эфир 131, 132
Дииминоянтарной кислоты динитрил
131 — 133
гел/-Дииодалкапы 236
цис, цис-] ,6-Дииодгексадиен-] ,5 643
3,7-Дииод-трамс, грш/с-октадиен-
2,6-ол-1 284, 285
цис-3,6-Дикарбметокси-1,2-дитиан 100
транс-2,5- Дикарбметокситиол ан 100
Дикетен 371
а-Дикетоны 180, 253, 434, 449, 530,
552, 647, 653
Р-Дикетоны 419
1,4- Дикетоны 313—314, 380, 660
1,5- Дикетоны 660
1,6- Дикетоцы 85
1,2-Дикето-3,3,7,7-тетраметилцикло-
гептап 530
2,5-Дикетоциклогексан-1,4-дикар-
боновой кислоты диэтиловый эфир
511
Дикобальтоктакарбонил 134
Диктамш-ш 199
2,2/-Дилитийдифепил 134
Дильса—Альдера реакция 15—16,
203, 208—209, 234, 263, 278, 310,
529, 544, 609, 610, 613—614, 645
Димедон 657
Димезилаты виц-диолов 353
2.3-0-ДиМезил-а-В-Глюкопиранози-
ды 252
траис-2,2/-Диметил-2,2/-азопропан 363
Ы,Ы-Диметиламидины 92
Ы,Ы-Диметиламид 2-кетомасляной
кислоты 410
Р,Р-Диметилакриловой кислоты эти-
ловый эфир 546
1,3-Диметилаллен 128
N,N-Диметил амид из о масляной кис-
лоты 625
Диметиламин 204, 274, 376, 486, 577,
632, 660
Диметиламина хлоргидрат 376
1 -Диметиламиноантрахинон 171, 172
2-Диметиламиноантрахинон 171, 172
2-Диметиламинобензо-1,3-диоксол 174
1,1 ,^-тетракис- (Диметиламино) -
бутен-2-диилия-1,4-нитрат 451
4-Диметиламино-1-грет-бутилокси-
карбонилпиридиния хлорид 135
6- (2-Диметиламиновинил) -фульвен 32
2-Диметиламиио-гранс-4-5-Дпфеппл-
1,3-ди оксолан 172
Диметиламиномалеияовой кислоты
диметиловый эфир 32
З-Диметил аминометилгептаион-4 151
2-Диметиламинометилен-З-изопро-
пилидеиянтарной кислоты димети-
ловый эфир 30—31
1,3-бис-(бис (Диметиламино) -мети-
лен]-циклогексан 451
1,8-бис- (Диметнламипо) -нафталин
135
4-Диметиламинопиридии 135
бис-(Днметилиламино) -фенилметан
577
трис- (Диметиламино) -фосфин см.
Гексаметилтриамид фосфористой
кислоты
1-Диметиламиноциклогексен 93
Ы,Ы-Диметиламиноциклопропилфе-
нилсульфоксоиия борфторид 135—
136
1,1 -бис-Диметил аминоэтилен 451
Р-Диметиламиноэтилхлорид 244
17,17-Диметиландростатетраен-
4,8,11,!3-он-3 349
М,М-Диметиланилип 45, 597, 626
Диметилацетамид 136, 160, 172, 343,
363
Диметилацетамида диметилацеталь
137—139
Ы,Ы-Диметилбензамид 275
3- (2,5-Диметнлбензоил) -2-метил-
пропионовая кислота 392
2,3- Ди метил-1,4-бензохинон 386
4,5-Диметил-1,2-беизохинон 252
1,3-Диметилбицикло-[1,1,0]-бутан 625
2,4- Диметил бициклобутаны 456
5,5-Диметилбицикл о-[2,!,!]-гексан-
1 -карбоновая кислота 36!
5,5-Диметилбициклоф2,1,1]-гексил-
1-карбинол 361, 362
7,7-Диметилбицикло-[4,1,1]-октанон-3
256
бис- (3,6-Диметил) -борепан 139—140
бис- (3,5- Диметил) -боринаи 139
Диметилбромсульфония бромид 140
2,3-Диметилбутадиен-1,3 225
2,3-Диметил бутен-2- 241, 254
2,3-Диметилбутен-1-ила-3 гидропере-
кись 254
2,3-Диметилбутил-2-бораи (тексилбо-
раи) 140—142, 266
Диметил-грет-бути л си лиловые эфиры
485
Диметил-трет-бутилхлорсилан 142—
143, 485
Диметил- (цис- и траис-4-трет-бутил-
циклогексил) -метилгипохлориты 439
2,5- Диметилгексадиен-1,5 139
683
3,4-Диметилгексадиен-2,4 236
3,4-Диметилгександиол-3,4 236
16,16-Диметилдигидропиреп 114, 115
2,4-Диметил-3,5-дикарбэтоксипиррол
102
(Е ,Е ,Е) -3,7-Диметил-8-11-дикето-
2,6,9-додекатриеналь 443, 444
№^1-Диметил-№-(диметилкарба-
моил) -хлорформамидин 206
2,5-Диметил-2,5-диметокси-2,5-диги-
дрофурап 396
Диметилдисульфид 180, 333
4,4 • Диметил-1,7-дифенилгептадиин
1,6-диол-3,5 311
1,1-Диметил-2,2-дифепилэтилеи 492
Диметилдихлорсилан 142, 143—144
2,2- Диметил-4,5-дицианизоимида-
зол 133
1,1,-Диметилен-бис- (трифторацетат)
238
5},М-Диметилизобутени ламин 30, 31
1,1- Диметилиндан 243
1,2- Диметилиндол 463
1,3- Диметилиндол 463
2,3-Диметилиндол 463
Диметилкарбамоилировапие 274—275
5р-М,М-Диметилкарбоксамидометил-
холестеп-3 138
Диметилкарбонат 156, 315, 580
Диметилкетена метилтриметил сили-
лацеталь 48
2,10-Диметил-6-кетодекагпдроизохи-
полин 145
Диметилмалоновой кислоты димети-
ловый эфир 48, 49
Диметнлмедьлитий 144—150, 284, 333
4,12-Диметил-[2,2]-метациклофаи-
диен-1,9 114, 115
М,!\т-Диметилметиленаммония иодид
150—152
КЩ-Диметилметиленаммопия три-
фторацетат 152
N.N-Диметилмоноамид метилмало-
новой кислоты 410
2,3-Диметилнафталин 90
Диметилнафталинов пикраты 411
4,8-Диметилнонадиен-3,7-он-2 460
6,6-Диметилнорпииен-3-он-2 160
2,7-Диметилоктадиен-2,6 445
2,7-Диметил-тра«с7 гранс-октадиен-
2,6-диаль-1,8 445
3,5-Диметил-4-окси бензальдегид 90
1,Ю-Диметил-Д11е)-окталон-2 291, 292
цис- и тда^с-4,1О-Диметил-Д1(01-окта-
лон-2 405
2,3- Диметилоктен-3 502
2,4- Диметилпента диен-1,4 139
Т\1,Ь1-Диметил-4-пиридиламин 415
бис- (1,2-Д иметилпропил-1) -боран
(диизоамилборан) 140, 152, 209, 348
684
Диметил-М-сукцинилсульфония хло-
рид 152—155, 160
Диметилсульфат 135, 155—157, 597
Диметил сульф ат — калия карбонат
157
Диметилсульфид 140, 153, 157—159,
159
Диметилсульфид — боран 159
Диметилсульфид — хлор 159—160,
170
Диметилсульфоксид 61, 103, 104, 115,
151, 160—162, 163, 166, 167, 251,
307, 356, 405, 454, 358, 643, 653
Диметилсульф оксид — N-бром сук-
цинимид 167—168
Диметилсульфоксид — серы трех-
окись 168—169
Диметилсульфоксид — уксусный ан-
гидрид 168, 169—170
Диметилсульфоксид — хлор 170
Диметилсульфоксонийметилид 165—
167
Диметилсульфоиийметилид 57, 164—
165
2,4-Диметилтиазол 171, 335
N,N-Диметилтиокарбамоилхлорид 171
2,5- Диметилтиофен 208
3,3-Диметил-1,2-бис- (триметил-
силилокси)-циклопропан 49
N ,N -Диметил-З-трифторметил ани-
лин 483
3,4-Диметилтрицикло-[3,3,3,0]-унде-
канон-2 511
2,7-Диметилтропон 225
6,10-Диметилундекатриен-3,5,10-oii-2
460
4- (2,5-Дпметилфенил) -2-метилмасля-
ная кислота 417, 418
2,2-Диметил-4-фепил-6-/г-нитрофе-
нил-1,3-диазабицикло-[3,1,0]-гексе-
на-3 N-окись 523
Диметилформамид 44, 45, 58, 107,
160, 171-172, 185, 226, 229, 231,
259, 301, 325, 356, 375, 377, 379, 403,
500, 549, 558, 588, 643, 647
Диметилформамида диметилацеталь
172—173
Диметилформамида диэтилацеталь
172
Диметилформамид — тионилхлорид
173—174, 178
Диметилфосфит 174—175
Ди м етил ф ос ф он о ди азо мета и 525
3,6-Диметилфталевой кислоты ди-
нитрил 208
6,6-Диметилфульвен 340
3,5-Диметил-4-хлорметилизоксазол 175
4,4-Диметилхолестери л ацетат 61
Диметилцианамид 206
2,2-Диметилциклогексаыон 80
5,5-Диметилциклогексен-2-он-1 299
Ы,1\т-Диметилциклогексиламин 376
2,3-Диметилциклопентен-2-он-1 313
1,1-Диметилэтиленкарбонат 545
1,2-Диметилэтиленкарбонат 545
2,4-Диметил-6-этилпиридин 76
9,10-Диметоксиаитрацен 613
2,4-Диметокси бензальдоксим 175
2,4-Диметоксибензиламин 175
2,2/-Диметокси бензофеноны 413
4,5-Диметоксибензоцикло бутендион-
1,2 57
2,3-Диметокси-5-6-дициан-1,2,3,4-тет-
рагидропиразин 132
Диметоксикарбен 512
Диметоксикарбония борфторид 115
2,3-Диметокси-5-метилбепзохииои 386
2,2-Диметоксипропап 133
9,10-Диметокситриптицен 614
3,3-Диметоксициклопропен 23—24
1,2-Диметоксиэтан 44, 60, 118, 120,
195, 219, 254, 272, 281, 282, 290, 353,
416, 502, 580, 597, 657
2-(2,2-Диметоксиэтил)-1,3-дитиан
175—176
цис-1,2, -Диметоксиэтилен 132
Динатрийфосфат кислый 396
Динафталинтиофен 263
Дионеопентилкетон 65
2,6-Динитробензойная кислота 156
1,3-Динитробензолы 388
8,10-Динитроизоиндоло-[2,1 -а]-хнно-
лии 410
2,6-Дииитро-п-крезол 12
suq-Дииитросоединения 372, 373
2,4-Динитро фен и л гид раз ин 635
2,4-Динитрохлорбензол 162
Диоксан 61, 199, 405
1,6-Диокса-6а-тиапентален 481
1,2-Диокси-З-бромпропаи (а-бром-
гидрин) 177
4,4/-Диокси-2,6,2/,6/-гстракнс- (трет-
бутил)-дифенилметан 177
(±)-7,8-Диокси-6-метокси-1 -метил-
1,2,3,4-тетрагидроизохинолина
хлоргидрат 465
2,4-Ди- (2-окси-2-пропил) -циклогек-
сен-1 160
трео- и эритро-Q, 10-Диоксистеарино-
вые кислоты 161
а,|}-Диоксистильбена диацетат 650
2|1,3|}-Диоксихолестана ацетонид 535
3,4-Диоксициклобутен-3-дион-1,2 57,
177—178
1,3-Диоксо-2-диазо-1,2-дигидрофена-
лен 73
1,1 -Диоксо-2,3-ди-трет-бутилтиади-
азиридин 362, 363
1,1-Диоксо-2-карбметокси-3,3-дифе-
нил-1,2-тиазетидин 179
4,4-Диоксо-2-метокси-6-метпл-1 -окса-
4-тиа-3,5-диазин 178—180
Диоксомолибдена комплексы 347
2,6-Диоксоспиро-[3,3]-гептан 393
2,10-Диоксоспиро-[3,1,1,3,1,1]-триде-
кан 393
1,2-Диолы 118, 502—503
1,3-Диолы 117—118
Дипивалоил 530
2,2/-Дипиридил 25
2,2/-Дипиримидил 318
1,2-Дипропилциклобутен-1 284
1,2-Дипропилциклопропен-1-карбо-
новой-3 кислоты этиловый Эфир
283, 284
1,3-Дистеароилг лидер ин 555
2,11-Дитиа-[3,3]-метапарациклофан
114, 115
1,3-Дитиан 180—182, 182, 89
1,3-Дитианилтозилаты 421
1,3-Дитианы 98
1,3-Дитиения борфторид 182—183
1,3-Дитиоланы 98, 611—612
Дитиоуголыюй кислоты N-цианимида
дикалиевая соль 183
Дитиоциан 183, 422
Ди-2,2,2-трихлорэтилацетали 571
Дифенил 478, 600
1,3-Дифеиилаллен 128
Дифенилацетилен 61, 105, 270, 279,
303, 435, 454, 496, 524
Дифенилацетилхлорид 572
транс,транс-1,4-Дифенил бута диен-
1,3, 643
2,3-Дифенилбутандиол-2,3 503
Дифенилгликолевый альдегид 334
Дифенилдиазометан 184
Дифенилди- (1,1,1,3,3,3-гексафтор-
2-фенил-2-пропокси)-сульфуран
184—186
бис- (г^'-Дифенилеи) -теллур 482
Дифеиилизобензотиофен 187
1,3-Дифенилизобензофуран 186—187,
529
Дифенилиодопий-2-карбоксилата мо-
ногидрат 187—190
Н,Ы-Дифепилкарбамоилпяридиния
хлорид 191
Дифенилкарбинол см. Бензгидрол
Дифенилкарбонат 204
Дифенил-2-карбоновая кислота 567
Дифенилкетел 191, 572
Дифенилмедьлитий 319
Дифенилметан 370
Дифенилметиленциклобутан 493
Дифенилметилфосфонат 558
1,1 - Дифенил-2-метоксиэтилен 263
4,5-Дифенил-Д4-оксазол11ионы-2 45—46
Дифенилокса мидин 132
1,5-Дифенилпептацд11Ш1Оп 101
685
2,2-Дифенилпропан 370
1,3-Дифенилпропин-2-он-1 167
Дифенилсилап 551
Дифспилсульфид 184, 303
Дифенилсульфоксид 184
Дифенилсульфонийциклопропилид
191 — 198, 408
1,2-Дифенил-5,6,7,8-тетраметилнаф-
тялии-3,4-дикарбоновой кислоты
дпметиловый эфир 30
5,6-Дпфенил-1,2,4-триазин- (2Н) -
он-3 611
4,5-Дифенилтриазол 611
Дифепилфосфин 502
бис- (Дифенил)-фосфиноэтан 401
Дифенилфосфит 196
Дифенилуксусная кислота 293
Дифенилфосфорной кислоты азид
196—197
Дифенилхлорфосфат 196
1,ЬДифенилциклопропан 370
Дифенилциклопропепон 180
1,1-Дифенилэтан 370
1,2-Дифепилэтандиол-1,2 172
1,2-Дифенилэтапол 572
1,1-Дифенилэтилец 163, 179
Дифепилэтиленкарбопат см. Бензоина
енольной формы циклический кар-
бонат
1,2-Дифенилэтил-М-карбметокси-
сульфамата триэтиламмопиевая
соль 572, 573
тракс-3,4-Ди-(фенилэтинил)-1,2,3,4-
тетраметилциклобутен-1 30
Дифенохпноны 231
Дифталидилиден 368
Дифторкарбен 599
7,7-Дифторпорборнадиен 274
7,7-Дифторнорборнеп-2 274
гелг-Дифторциклопропаны 599
17а-Дйфторциклопропенил-5а-ан-
дростандиола-3|1,17(1 3-ацетат 220
Дихиноны 97
цис,аиц-Дихлоралканы 470
Й,М-Дихлораминоапокамфан 18
Дихлорацетилхлорид 202
7,7-Дихлорбицикло-[4,1,0]-гептен-3 68
экзо-3,4-Дихлорбицикло-[3,2,1]-
октен-2 569, 570
Дихлорборан 197
Дихлорборана диэтилэфират 197—
198
ИЖ-Дихлор-трис- (и-бутил) -метил-
амин 17
2,5-Дихлор-3,6-диизоцианат-1,4-
бензохинон 394
М.М'-Дихлордииминосукцинонит-
рил 132
7,7-Дихлор-2,5-дифенилбензоцик-
лопропен 65—66
Дихлордифеиилметан 134
1,1-Дихлор-2,7-дифенилпиклопропа-
[6]-нафталин 66
2,3-Дихлор-5,6-дициап-1,4-бензо-
хинон 198—201
4,5-Дихлор-4,5-дицианимидазоли-
дон 133
Дихлордиэтиловый эфир 210
Дихлориды вицинальные 515
3,6-Дихлоркарбазол 615
Дихлоркарбен 37—39. 68, 103, 198,
201—202, 569, 597, 599
Дихлоркетен 202—203
Дихлормалеиновый ангидрид 203—204
1,3-Дихлормалонилцианины 204, 205
1-Дихлорметиладамантан 37
1-Дихлорметилдиамантан 38
4-Дихлорметилдиамаптан 38
Дихлорметилендиметил аммония хло-
рид 204 —207
Дихлорметиленциклоалканы 202
Дихлорметиллитий 207
Дихлорметилметиловый эфир 505
2,3- Дихлор пор бор и а диен-2 66
Дихлорноркараи 37
7,7-Дихлорноркаран 103
3,5-Дихлор-1,2,4-оксадиазол 405
3,3-Дихлороксетаноны-2 202
1,5-Дихлорпентапон-З 16, 363
2,3-Дихлорпропен 24
а,|}-Дихлорпропионилхлорид 609
Дихлорсилан 207—208
силш-Дихлортетрафторацетоп 599
1,2-Дихлор-3,3,4,4-тетрафторцикло-
бутен 178
цис- и транс,зкзо,экзо-3,4-Днхлор-
тетрацикло-[4,4,2,02’5,07’10]-доде-
кадиен-8,11 354
2,8-Дихлор-4-тиатрицикло-
[3,2,1,03’е]-октан 446
Дихлоруксусная кислота 582, 583
Н,М-Дихлоруретан 208
Дихлорфторуксуспая кислота 629
2,4-Дихлорхииазолин 404
1,2-Дихлорциклогексан 554
3,6-Дихлорциклогександион-1,2 322
1,2-Дихлорциклооктан 170
Дихлорциклопропаны 37, 68
Дихлорциклопропен 517
1,2-Дихлорэтан 50, 631
1,1 -Дихлор-2-этил-З-метилцикло-
пропан 69
Дициан 131
Дицианацетилен 208—209, 533, 534
4,5-Дициан-4,5-диалкоксиимидазо-
лидоны-2 133
3,4-Дициан-1,2,5-тиадиазол 133
Дициклогексил бор ан 209
Дициклогексилкарбодиимид 103, 209*
210, 262, 391, 571
686
N,N-Дицикл огексилкарбодиимида
подметилат 210
Днцпклогекс1гл-18-крауи-эфир-6
210—212
Ди- (циклогексплокси) -метан 168
Дициклопропилкарбинол 18
бис-(Дициклопропилметнловый)
эфир 18
1 -Дгтэтпл алюминий алкины 2Г2
Диэтил а люмиипйх лори д 212—214
Дпэтилалюминийцианид 215—216
Днэтпламин 112, 216—217, 353
1-(N,N-Диэтил амино) -пропин-1 582
трис- (Диэтиламино) -фосфин 100—
101
1,2-Диэтил-З-гс-а низилциклоп ропен
303
Диэтиланилин 609
М,ЬГ-Диэтилгидразин 363
Диэтилизопентениламин 353
Диэтилизоцианметилфосфонаг 217—
218
Диэтилмедьлнтий 285
Диэтил- (метилтиометил) -фосфо-
нат 219
Диэтил-(1-метилтио) -этилфосфонат
218—219
4,5-Дцэтил-транс, транс-октадиеп-3,5
126
Диэтилсилаи 551
Диэтилсульфат 156
Диэтил-(1,1,2-трифтор-2-хлорэтил) -
амин 219—221
№,И-Диэтилформамид 172
Диэтилфосфат 603
4-Диэтилфосфонил-2-оксазолины 218
Диэтилхлорфосфат 23, 148, 221, 487
Диэтилцианмети.лфосфонат 221
Диэтилцинк — йодоформ 222
Диэтилцинк — метилен иодистый 222
1,2-Диэтинил оксираны 106
Диэтоксикарбоиия борфторид 222—
223
Диэтоксиуксусной кислоты этиловый
эфир 258
цис, ч^с-Додекадиеи-5,7 209
траяс,гранс-Додекадиен-8,10-ол-1
304, 305
Додек.адиин-5,7 209
н-Додекан 375
Додецен-1 621
цис- Додецеи-8-ола-1 ацетат 152
Додецил- 1-тозилат 375
Дурол 235
Дьюара бензол 439
Енамины 30—32, 344, 377
Еновый синтез 614
Енолацетилирование 510—511
Жасминовой кислоты метиловый
эфир 490—491
цис-Жасмон 98, 314, 503
Железа(Ш) а цетил ацето цат 244
Железа додекакарбонил 224—225
Железа(II) иодид 322
Железа (III) цитрат 316
Железа нонакарбонил 75, 225—226
Железа пентакарбонил 228—228, 425
Железа(П) сульфат 228
Железа (III) хлорид 57, 229, 579, 629
Железа (111) хлорид — диметилфор-
мамид 229—230
Зайцева правило 573
Изоамилнитрит 231
Изоболдин 449
Изобуллвалеи 330
Изобутап 265
Изобутилен 111, 185, 238, 327
Изобутилена окись 406
Изовалин (а-амино-а-метилмасля-
ная кислота) 231—232
Изогиперицин 250
Изоксазолы 240
Изомасляная кислота 141, 162, 531
Изомасляный альдегид 312
1- (Изонитрилметил) - циклогекса-
нон-1 275
Изонитрилы 38, 173—174, 474, 499—
500, 539, 542
Изопентан 339
Изопрен 353, 654
Изопропенилацетат 232, 313
Изопропенилмагнийбромид 342
Изопропилат алюминия 232
н-Изопропилбензойиая кислота 264
Изопропилдиметилсилилхлорид 154
8-Изопропилиденбицикло-[3,2,1]-
октен-6-он-З 340
(+)- и (—)-2,3,О-Изоггропилиден-
2,3-диокси-1,4-бис- (дифенилфос-
фино)-бутан 233
ЗфЗ'-О-Изопропилиденуридин 558
4-Изопропилиденциклогексанон 655
Изопропнлизоцианат 630
Изопропил хлористый 629
2-Изопропилциклогексанон 80
N-Изопропилциклогексиламин 296,
600
(—) -Изопулегол 547, 548
(Я-Изосетоклавиц 312
Изотетралин 198
З-Изотуйон 232
Изофорон 125
Изохинолины 52
Изохроманон-3- 407—408
687
Изоцианатхипоны 394
Изоцианаты 629—630
Изоциапиды см. Изонитрилы
(+) -Изоэквиленин 398
Изоэргостероп 200
Имидазол 142, 143, 483
Имидазолы 388
Имиды 627
Иминолактоны 631, 632
Имипонадбензойная кислота 233—
234
Иминосульфураны 185, 186, 584
Индазолы 440, 441
Индан 660, 661
Индалон-! 418
Инданон-2 407
Инданоциклон 234
Индантри о и-1,2,3 см. Нингидрин
Инден 263
Индол 251, 463, 483
Индолы 158, 165
ЗН-Индолы 282
Иод 234—236, 238, 284, 322, 338, 367,
422, 437, 478, 530
Иодазид 237
Иода трис-(трифторацетат) 238
о-Иодбензойиая кислота 187, 188
Иодбензол 384
Иодбензола дихлорид 238—239
Иодгидрины 324—325, 601
1-Иоддекан 375
Иоддурол 235, 236
Иодизоциапат 422
Иодирование 556, 557, 558
Иод — калия иодид 240
[Иодкарбонил-бис-(трифенилфос-
фин) -метилродий (Ш)]-малеино-
вой кислоты диметиловый эфир
546
Иодмалеиповой кислоты диэтиловый
эфир 318
2-Иод метил-1,3-диоксол ан 490
Иодная кислота 235, 241
Иодозобензола диацетат 241
Иодозобензола диацетат — триме-
тилсилилазид 437
Йодоформ 241
2-Иодпиридинол-З 317
Иод — серебра ацетат 242
р-Иодтиоцианаты 422
гегрйкыс-[Иод- (три-н-бутилфос-
фин)-медь(1)] 124, 125
Иодфумаровой кислоты диэтиловый
эфир 318
За-Иодхолестан 210, 558
За-Иод-Д5-холестен 210
Иодциан 63
2-Иодцкклогексец-2-он 237
Иодциклооктатетраен 67
2-Иодциклопентен-2-ои 237
688
Иодэтинилтриметилсилан 242—243
а-Ионон 550
р-Ионон 240, 460, 551
Ионообменные смолы 243, 653
Йохимбин 429, 615, 646
Кадинены 563
Кадмия карбонат 182, 246
Кадмия (11) хлорид 491
Каламенен 563
Калий 221, 260, 348
Калий в графите 246—247
Калий иодистый 478
N-Калийэтилендиамин 250
Калия боргидрид 247
Калия бромид 310, 311
Калия гексаметилдисилазан 247—
249
Калия гексахлоровольфрамат (IV)
Калия гидрид 249—250
Калия гидроокись 250—252
Калия иминоксилдисульфонат (соль
Фреми) 252
Калия иодид — цинк-медная пара
252
Калия карбонат 61
Калия осмат 253
Калия перманганат 41, 283
Калия перманганат — уксусный ан-
гидрид 253—254
Калия персульфат 187, 188
Калия перхромат 254
Калия роданид 259
Калия тетрахлороплатинат 254
Калия тиотозилат 521
Калия бис- (триметилсилил)-амид
247—249
Калия триэтилметилат 307
Калия феррат (VI) 254—255
Калия феррицианид 255—256
Калия цианид 514
Кальций хлористый 398
Кальция гидрид 522
Кальция гипохлорит 17
Кальция карбонат'6‘1, 256
Кальция сульфат 243
Камфен 622, 623
Камфора 247, 293, 361
d-10-Камфорсульфокислота 257
Караханаенон 61—62, 655, 656
Караша — Сосновского реакция 350
Карбазол 615, 656
Карбаматы 315, 474
5-Карбамоил-6-метилтио-1,3-тиази-
ноны-4 420
Карвон 495, 496, 548
Карен-3 81, 624
Карбметоксигидразоны 580
М-Карбметокси-р,Р-дифенилвинил-
сульфамид 179
1 -Карбметокси-1 -метилциклогексан
194
п-Карбметоксинад бензойная кислота
257—258
N-Карбметоксисульфамоилхлорид
178, 179, .542, 572
Карбодиимиды И—12, 543, 555
Карбоксилирование 228, 348, 414—
415
2-1<арбокси-2-метил-1,3-ДНОКсапан
604
2-1<арбокси-2-метил-1,3-диоксолан
604
3-Карбокси-1-метилпирролидипон-2
410
D (—) -2-Карбокси-2,4,5-триметил-
1,3-ди оксолан 604
Карбонилирование 315—316, 416
Карбонил борфториды 574
Карбоновые кислоты 129—130
Карбоновых кислот амиды 311
Карбоновых кислот гидразиды 97
Карбоновых кислот диметиламиды
91
Карбоновых кислот хлорангидриды
23
(—)-Карен-3-он-5 81
л- (2-Карбэтоксиаллил)-никельбро-
мид 642
N'-Карбэтоксиамид пиррол-1-тиокар-
боновой кислоты 260
N'-Карбэтоксиамид пиррол-2-тиокар-
боновой кислоты 260
2-Карбэтоксибензотиазол 228
б«с-1Ч,М'-Карбэтоксигидразин И
бис-(Карбэтоксидиазометил) -ртуть
258
2-Карбэтокси-1,3-дитиан 258—259
N-Карбэтоксиизотиоцианат 259—260
(Карбэтоксиметилен) -трифенилфос-
форан 540
Кароэтоксиметилмедь 260—261
4-Карбэтокси-5-метил-3-этилизокса-
зол 261
2-Карбэтоксинорпротозииоменин 256
2-Карбэтоксипиридин 228
4-Карбэтоксипиридин 228
Карбэтоксисульфамоилхлорид 573
N -Карбэтокси-2-этокси-1,2-дигидро-
хинолин 262
1 -Карбэтокси-4-этокси-1,2,3-триазол
581
2-Карбэтокси-4-этокси-1,2,3-триа-
зол 581, 582
1-Карбэтокси-5-этокси-1,2,3-триа-
зол 581
Карпанон 401
Катализ фазового переноса 41
Квадратная кислота см, 3,4-Диокси-
цикл об утен-3-дион-1,2
Квадрициклан 35
Кватернизация 403
Кепигса—Кнорра синтез 246
Кетазины 418
Кетали 243
Кетен 262, 610
Кетена тиоацетали 278, 421
Кетимины 344
а-Кетокарбены 313
|3-Кето кислоты 531—532
а-Кетокислот эфиры 259
у-Кетокислот эфиры 112, 113
Кетолактоны 434
ct-Кетолы 449
у-Кетонитрилы 380
Кетонитроны 441
4-Кето-2-фенилбензо-[<Д-1,3-оксазин
404
5-Кето-6-циан-5,6-секостероиды 436
3-Кетоциклогексен 288
З-Кетоциклогексил-1 -уксусная кисло-
та 419
З-Кетоциклогексил-l-уксусной кисло-
ты метиловый эфир 419
(2-Кетоциклопентил-1)-масляная
кислота 419
6-Кетоэнантовой кислоты метиловый
эфир 316
р-Кетоэфиры 304
Кислород 222, 262, 264, 321, 366, 367,
390, 443, 642
Кислород синглетный 254, 263—264
Клайзена конденсация 419
Клайзена перегруппировка 71, 298,
342, 395, 565, 648
Кларка — Эшвейлера алкилирование
378
Кневенагеля конденсация 334, 504
Кнорра синтез пирролов 102
Кобальта (II) ацетат 264—265
бис- (Кобальттетракарбонил) -цинк
265
Коллидин 61, 76, 105, 300, 301, 325
Коллинза реактив см. Хрома (VI) -
окиси — пиридина комплекс
(±)Р-Копаен 164
Копростанон 226
Кори восстановитель 266—267
Кори — Уинтера синтез олефинов
227, 528—529
Коричный альдегид 226, 255, 638
Коричный спирт 255, 638
Костанолид 5!
Коупа перегруппировка 462
Кофермент Qi 386
Колхицина предшественник 480
689
ft-Крезол 12, 230
(—)-Криптомерион 495, 496
Кротоновая кислота 276
Кротоновой кислоты этиловый эфир
296, 297, 299
Ксантогенаты 163, 164
Ксантон 418
Ксантоны 413—414
2,6-Ксиленол 90
R-Ксилол 392
(±)-|3-Кубебен 319
(+)-[1-Кубсбена норкетон 319
Кумарины 414, 649—650
Кумол 59, 563, 599
Курциуса реакция 197, 356, 609
р-Лактаиы 452
Лактонизация 123, 124
Лактонов тозилгидрэзопов соли
513—514
Лактоны 639
у-Лактоны 408, 631, 632
Ланостерин 522
Левопимаровой кислоты метиловый
эфир 12
L-Лейцил-Е-алапилглицил-Е-валнн
620
Лизергиновая кислота 645
D-Ликсоза 448
Лимонен 250, 266, 427, 493, 563
Линалоол 61, 416
Линдлара катализатор 130, 268
Литий 268—270, 294, 582, 583
Литий — алкиламины 270—271
2- Литий — 2-алкил-1,3-дитианы 180
Литий — аммиак 79, 80, 27!—273,
585, 586, 595, 596, 618, 619
Литий-тре г-бут а но л — тетрагидро-
фураи 39, 274
Литий — трет-бутилнеопентилампд
302
Лптийдиметилкарбамоилникельтри-
карбонил 274—275
2-Литий-2- (2',2'-ди метоксиэтил) -
1,3-дитиан 176
2-Литий-1,3-Дитиан 181, 278
Литийметилизонитрил 275
Литий — нафталин 275—277, 354
1-Литий-тра«с-октен-1 125
2-Литийпропгглидеп-Ы-грет-бутил-
амин 277
а-Литий-я-толнлтиометилизонитрил
507, 508
2-Л итий-2-триметилсилил- 1,3-дитиан
278—280
Литий — этилендиамин 280
N-Литийэтилендиамин 250, 494
Лития алюмогидрид 127, 280—283,
289, 306, 324, 342, 359, 364, 371, 444
463, 487, 522, 528, 531, 537, 570,646
690
Лития алюмогидрид — алюминия хло-
рид 283—284
Лития алюмогидрид — натрия мети-
лат 284—285
Лития алюмогидрид — пиридин [ли-
тия тетро!/шс-(К-дигидропиридил)-
алюминат[ 285
Лития амальгама 285—286, 553
Лития ацетиленид 242
Лития боргидрид 197, 286, 306, 347
Лития бромид 160, 286—287
Лития трет-бутилат 287
Лития трис-(втор-бутил) -боргидрид
266, 287—288
Лития трис- (трет-бут окси) -алюмо-
гидрид 247, 281, 288—289
Лития гексаметилдисилазац 219, 292
Лития гидрид 289—290, 305, 306
Лития гидроокись 307
Лития тетракис- (N-дигидропиридил) -
алюминат см. Лития алюмогид-
рид — пиридин
Лития диизопропиламид 199, 260,
290—294, 297, 531
Лития диметиламид 274
Лития диметилкупрат см. Диметил-
медьлитий
Лития дифепилфосфид 294—295
Лития дихлор фторацетат 629
Лития дициклогексиламид 303
Лития диэтиламид 295, 307, 508
Лития N-изопропилциклогексиламид
291, 296—300
Лития иодид 106, 123, 300
Лития иодид — бора трифторид 300—
301
Лития карбонат 160
Лития карбонат — лития галогенид
30!
Лития метилат 73, 74, 315, 372, 580,
581
Лития нафталеиид см. Литий — на-
фталин
Лития пергидро-9Ь-борафеиалилгид-
рид 288, 301—302, 519
Лития перхлорат 193, 256
Лития м-пропилмеркаптид 106
Лития 2,2,6,6-тетраметилпиперидид
302—304
Лития тетр ах лорку прат 304—305
Лития триметоксиалюмогидрид 287
Лития триэтилборгидрид 305—306
Лития триэтилметилат 306—307
Лития фосфат 307
Лития фталимид 627
Лития хлорид 225, 301, 304, 325, 438
Логании 78, 79, 129
Лофофорин 465
2,6-Лутидин 403, 483
Льюиса кислоты 14
Магний 308—309
N,N-бис- (Магнийбром)’-анилин 309
Магния алюмогидрид 309
Магния амальгама 143
Магния иодид 309
Магния карбонат 650
Магния сульфат 447
Магния хлорид безводный 308
Макролиды см. Макроциклические
лактоны
Макроциклические лактоны 35], 387
Малеииимид 621
Малеиновой кислоты диметиловый
эфир 226
Малеиновой кислоты диэтиловый
эфир 279
Малеиновый ангидрид 279, 310, 591,
609
Малонового диальдегида бис-(диме-
тилацеталь) 175, 176
Малоновый эфир 400, 401, 466, 504
Малононитрил 310
Манноза 448
Марганца (III) ацетат 310—311
Марганца (II) ацетат тетрагидрат
310
Марганца двуокись 283, 311—312,
321, 444
Марганца(Ц) окись 419
Марковникова присоединение 56, 127
Масляный альдегид 269
Мевалозид 312
R-(— )-Мевалополактов 312
Меди(Ц) ацетат 112, 253, 321
Меди (11) ацетата моногидрат 312—
313
Меди(1) ацетилацетонат 313—314
Меди(1) трет-бутилат 314
Меди закись 320
Меди закись — трет-бутилизонитрил
314__gig
Меди(1) иодид 122, 124, 125, 260,
322, 332, 518, 583, 641
Меди карбонат основной 315
Меди(П) метилат 315—316
Меди(И) нитрат — пиридин 316
Меди окись 317
Меди (II) сульфат безводный 407
Меди(1) (Ьенилаиетиленид 314, 317
Медная бронза 318
Медный купорос 317
Медный порошок 318—319, 344
Медь бромистая 26, 242, 319, 321
Медь (1) — трис- (2-пиколин) -перхло-
рат 320
Медь(1)тетрапиридинперхлорат 320
Медь хлористая 126, 320—322, 390,
524
Медь хлорная 324, 322—323
Меервейиа арилирование 26
Меервейна — Понндорфа восстанов-
ление 232
Мезаконовой кислоты диметиловый,
эфир 546
Мезаконовой кислоты нитрил 27—28
Мезилаты см. Метансульфокислоты
эфиры
Мезилхлорид см. Метансульфохлорид
МезитоГиая кислота см, 2,4,6-Триме-
тилбеизойная кислота
Мезитойлой кислоты метиловый эфир
см. 2,4,6-Триметилбензойной кисло-
ты метиловый эфир
Менахинола-1 диметиловый эфир 453,
454
Менахипон-1 454
Ментон 232, 518
Меркаптаны 35
1,2-М.еркаптолы 359
2-Меркаптопиримидин 173
6-Меркапто-5-триазоло-[4,3-£]-£«ми-
тетразины 26
Меррифилда синтез пептидов 262
Металлилбензол 384
2-Металлилбромид 385, 386
Металлиловый спирт 594
Металлилхлорид 69, 300
Металлирование 250, 314
1,6-Метано-[10]-аннулен 198
Метанол 341
Метанолиз 454—455
Метансульфокислота 323—324, 603
Метансульфокислоты этиловый эфир
654
Метансульфокислоты эфиры 324
Метансульфохлорид (мезилхлорид)
324—325, 601
[2,2]-Метапарациклофандиен-1,9 114,
115
Метилазокарбоновой кислоты (заме-
щенной) метиловые эфиры 580, 581
N-Метилакридиния иодид 101, 102
Метилакрилат 111, 316
а-Метилаланин 232
ct-Метилаллилацетат 430
Метиламин 281, 376
1-Метиламиноантрахиион 171, 172
(±)-О-Метилангалонид 465
(±) -О-Метиландроцимбин 480
Метилацетат 585
1-Метилбезз-[Я-азепин-3,4-дикарбо-
новой кислоты диметиловый эфир
31
N-Метилбензиламин 255, 492
Метил-Ы-бензилкарбамат 572
Мети л бенз и жетон 40, 383
Метилбензоат 479
N-Метилбензолсульфонимидметилид
лития 594
2-Метилбензофуран 47
691
2-Метилбепзохиноп 386
4-Метил;0-бензохинон 58, 613
В-Метил-9-борабициклононан 302
d-2-Мети л бутаналь-1 499
2-МетИлбутена-З 1,2-окись 127
'З-Метилбутен-2-илбромид 386
2-Метилбутен-1-ип-3 341
2-Метилбутеп-3-ол-2 657
З-Метилбутен-З-он-2 151
бис-(З-Метилбутил-2)-боран см. бис-
(1,2-Диметилпропил-1)боран
2-Метил-3-бутилгеитеп-1-ол-З 268—
269
Метил-трет-бутиловый эфир 327
бис- (З-Метил-б-трет-бутилМ-окси) -
дифенилсульфид 328
а-Метил-у-бутиролактон 119, 291,
297—298
2-Метил-2-ВИНИЛ-5- (1-бром-1-метил-
этил)-тетрагидрофуран 61
Метилвинилкетон 226, 316
5-Метилгексен-4-аль-1 657
транс-1 -Метил-2- (цас-гексен-1 -ил) -
циклогексан 338
2-Метилгексен-4-овая кислота 648
З-Метил-транс-гексен-4-овой кислоты
М,М-диметиламид 138
цис-4-Метилгексеп-3-ол-1 130
Метил-Ы-гексилкарбамат 572
2-Метилгептадиен-1,6-ол-3 395
З-Метилгептанол-4 269
2-Метилгептен-2 444, 445
транс-2-Метилгептен-2-ол-1 445
Метилгидразин 34, 328
а-Метилглутаронитрил 102
N-Метилдециламин 270
1 -Метилдибепзо-[6, /]-1,4-диззепин
101, 102
Метил-2,3-дибензоил-4,6-дитозил-
a-D-глюкозид 369
Метил-2,3-дибеизоил-4-тозил-6-дез-
окси-6-иод-а-О-глюкозид 369
О-Метилдибензофурания борфторид
329
1 -Метил- 1,4-дигидрохинолин 32
1-Метил-3,4-дикарбметокси-1,6-ди-
гидр о бенз-[§]-азоцин 32
2-Метилдифенил 568
1-Метил-3,4-дифенил-Аа-пиразоли-
ноя-5 328
1-Метил-3,5-дифенилтиа бензола
1-окись 167
Метилдифенилфосфина окись 294
2-Метиленадамантана эпоксид 166
]Ч,М'-Метилен-б«с-акриламид 621
Метиленацетали 167—168
Метилен бромистый 126, 527
сс-Метилеи-у-бутиро лактоны 44—45,
119—120
Метилен иодистый 150
-692
Метиленирование 595—596
2,6-б«с-Метиленспиро-[3,3]-гептан 393
2,10-бис-Метилеитриспиро-
[3,1,1,3,1,1]-тридекан 393
Метилентрифенилфосфоран 147, 596
2-Метилен-З-фенилпропиоиовой кис-
лоты метиловый эфир 614
Метилен хлористый 16, 17, 18, 41, 49,
52, 53, 55, 77, 113, 114, 182,231,242,
289, 300, 308, 323, 329—330, 343,
346, 347, 363, 389, 400, 422,428,441,
464, 468, 483, 501, 527, 553, 555, 563,
584, 585, 614, 615, 630, 645
Метилен хлористый — и-бутиллитий
330—332
3-Метилеп-Д5-холестен 596
Д5-24,25-Метилеыхолестенол-3|1 39
Метиленциклоалканоны 209
Метиленциклобутан 59
2-Мети леи цикло бутанол 458, 459
Метилеициклогексаи 302, 303
Метиленциклопропан 69, 300, 644
Метиленциклопропацы 590
М-Метилизатин 112
Метилизобутилкетон 416
Метилизонитрил 275, 505
Метилизонитрила дихлорид 404
N-Метилизохинолиния иодид 172
Метилизоциапат 332
N-Метилимидазол 483
1-Метилиндол 31, 463
2-Метилипдол 463
З-Метилиндол 463
1-Метилиндол-2-карбоновая кислота
529
Метил иодистый 210, 219, 245, 279,
280, 290, 294, 298, 332, 362, 403, 453,
488, 489, 546, 556, 557, 581, 616,
617, 656, 657
Метил- (транс-2-исд-1 -тетралин) -
карбамат 250, 251
N-Метилирование 165, 378
S-Метилирование 617
О- (Метилкарбамоил) -альдоксимы 332
Метилкарбметоксикарбен 110—111
5-Метил-Д3-копростен 148
трдкс-а-Метилкоричная кислота 150
Метиллитий 139, 196, 219, 249, 302,
330, 332, 487, 503
Метилмагнийбромид 78, 333
Метилмагний иодистый 394
Метилмедь 332—333
Метилметакрилат 269, 613, 614, 621
2-Метил-2- (2/-метилпропен-1/-окси) -
пропионовый альдегид 312
Метил- (метил тио метил) -сульфоксид
333—335
N-Метилморфолин 392, 505
З-Метилпафталин-2-карбоновая кис-
лота 90
2-Метил-1,4-нафтогидрохинона ди-
ацетат 651
2-Метил-[2,1-6]-нафтофуран 47
2-Метил-1,4-нафтохинон 651, 652
ct-Метилнитрилы 581
транс А- Мети лнона диен-4,8-овой кис-
лоты этиловый эфир 395
2-Метил-М-оксимидазолия соли 388
4а-Метил-3,4-4а,5,6,7,8,8а-октагидро-
нафталин-1 (2Н)-он-7-карбоновые
кислоты 144
10-Метил-А1(9)-окталол-2 658
10-Метил-А1(91-окталон-2 291
2-Метилоктен-1 400, 401
З-Метил-транс-пепта диен-2,4-овой
кислоты метиловый эфир 124
З-Метилпентанол-З 62
2-Метилпеятен-2-аль 240
2-3-Метилпентен-2-диол-1,5 155
1-Метилпиперидиндион-2,3 410
N-Метилпиридиния иодид 172
4-Метилпирокатехин 613
N-Метилпирролидин 338
И-Метилпирролидон-2 365, 367, 382
О-Метилподокарповая кислота 52
О-Метилподокарповой кислоты мети-
ловый эфир 52, 53, 107, 108
Метил-[}-О-рибофуранозид 137
а-Метилстильбен 471
р-Метилстирол 282, 639
Метилсульфинилметилидкалий (дим-
силкалий) 250
Метилсульфинилметилиднатрий (дим-
силнатрий) 162—164
1- Метилсульфинил- 1-метилтио-2-фе-
нилэтилен 334
Метилсульф он аты 608
Метил-Г4-сульфонилуретан 178—179 -
17а-Метил-А5’9(10-тестостерон 348, 349
2-Метил-З-тиазолин 335
1,3- бис-(Метилтио)-аллиллитий 335—
338
Метилтиоацетальдегид 158
1,3-бис-(Метилтио) -гексен-1 -ол-4 336
Мети л тио метиловые эфиры 616—617
1,3-бис-(Метилтио)-октен-1 336
2-Метилтиопиримидин 173
Метилтиопропанон-2 158
М-Метил-М-тозилпирролидмния пер
хлорат 338
Метил-п-толилсулъфоксид 466, 508.
509
5-Метил-4-п-толилтиооксазол 507, 508
Метил-2,3,6-трибензоил-4-тозил-а-
D-глюкозид 369
2-Метилундецен-1 596
Метилфенацилстльфид 158
2-Метил-4-фенилбутанол-2 243
2-Метил-З-фенилпропионовый альде
гид 594
Метилфенилсульфид (тиоанизол) 154
З-Метил-1-фенилфосфолен-3 654
Метилфосфонистой кислоты диэтило-
вый эфир 654, 655
N-Метилформамид 172
Метилформиат 78, 79, 508
1 -Метил-1 -формилциклопропан 458,
459
N-Метилхинолиния иодид 172
2-(2'-Метил-З'-хлор аллил)-циклогек-
санон 459
А2-2-Метилхолестен-5а 264
Д2-3-Метилхолестен-5а 264
2-Метилциклобутанол 459
2-Метилциклобутанон 57
I -Метилциклогексанкарбоновой кис-
лоты хлорангидрид 333
^uc-2-Метилциклогексанол 287, 288
2-Метилциклогексанолы 510
2-Метилциклогексанон 118, 232, 287,
288, 405
4-Метилциклогексанон 118
1-Метилциклогексен 203, 338
З-Метилциклогексен-1 646
1-Метилциклогексен-1-ол-3 646
бис- (тронс-2-Метилциклогексил-1)
боран 338—339
5-Метилциклопентадиен 129
2-Метилциклопентанол 379
2-Метилциклопентанон 379
1-Метилциклопентен 203, 379
3-Мет и л цикло пентен-2-он-1 319
трис- (транс-2-Метилциклопентил) -
боран 379
транс-2-Метилциклопентилдихлор- '
боран 589
N- (транс-2-Метилциклопентил) -цик-
логексиламин 589
1-Метилциклопропен 69
Метилциклооктатетраен 67
Метилциклопропилкетон 57, 67, 68
транс-1-Метил цикл о октен 295
2-Метил-1,2-эпоксипропан 545
Д1.3,5(10)_4_Д^етилэсТратриены ду—gg
Метилэтилкетон 226, 264
Метилэтилсульфид 381
Метилянтарной кислоты ангидрид
392
2-Метоксиаллил бромистый 339—340
а-Метоксибензилиденвольфрам (0) -
пентакарбонил 340—341
3- (и-Метоксибензоил) -пропионовая
кислота 498
7-Метоксибицикло-[4,2,2]-декатет-
раеи-2,4,7,9 607
1 -Метокси-1 -диметиламиноэтилен 138
З-Метоксиизопрен 341—342
Метоксилирование 73—74
Метоксикарбения гексафторантимо-
нат 343
693
я-Метоксикарбонилнадбензойная кис-
лота 257—258
цис- и транс-2-Метоксикор ичные кис-
лоты 414
1 -Метокси-4,5; 10,11 -бис- (тетрамети-
лен)-6,8-бис-дегидро-[13]-аинуле-
нил-анион 163
Метоксиметилирование 618
2-Метокси-2-нитродифенил 329
анти-7-Метоксинорборнен 457
6-Метокси-7-окси-3,4-Дигидроизохи-
нолиния иодметилат 343—344
6-Метоксипенициллины 73, 74
2-Метоксипропен 339
5-Метоксискатол 394
5-Метоксискатолилмагний йодистый
394
л-Метоксистирол 471
6-Метокси-р-тетралон 16,17
8-Метокситетралин-2-карбоновая кис-
лота 147
.м-Метокситолан 303
энбо-2-Метокситрицикло-
[4,!,0,03'т]-гептан 457
2-Метокситропон 157, 609
Метоксиуксусной кислоты хлоран-
гидрид 343
п-Метоксифенилацетальдегид 47!
п-Метоксифенилацетилен 626
о-Метоксифенилмагнийбромид 522
Зр-Метоксихолестан 174
Зр-Метокси-А6-холестен 174
7-Метоксицефалоспорины 74
1 -Метоксициклогексадиены-1,3 203—
204
1 -Метоксициклогексадиены-1,4 203-
204
Метоксициклопропаиол 316
З-Метокси-цис,транс-цикл оокта-
диен-1,4 455
5-Метокси-цис,транс-циклоокта-
диен-1,3 455
7-Метокси-цис,транс-циклоокта-
диен-1,4 455
Метоксициклооктатетраен 67
З-Метокси-транс-циклооктен 454, 455
5|1-Метокси-За,5-цикло-5а-холестан
284
3-Метоксиэстратриен1,3,5(10)-он-17 58
Миристиновая кислота 432
Михаэля присоединение, см. Сопря-
женное присоединение
Молекулярные сита 344
Молекулярные сита Лииде 5А 344
Молибдена гексакарбонил 25, 345
Молибдена пятиокись — гексаметапол
(гидрат комплекса) 345—347
Молибдена трехокись 345
Монохлорборана диэтилэфират 347—
348
694
Морфолин 270, 376, 382, 412
а-Морфолиностирол 225, 226
Мочевины 443
Муравьиная кислота 348, 381, 522
Муравьиная кислота — лития фор-
миат 549
Муравьиной кислоты этиловый эфир
(этилформиат) 79, 80, 269, 349
rfZ-Мускон 103, 104
Надбензойная кислота 58, 258
Над бензойной кислоты трст-бутило-
вый эфир 350
Надмуравьиная кислота 350, 620
Надуксусная кислота 350—351, 620
Надуксусная кислота — бора трифто-
рида эфират 351
Надуксусная кислота — серная кис-
лота 351—352
Надфталевая кислота 258
Натрий 93, 224, 352, 530
Натрий — аммиак 46, 48—49, 202,
240, 352—353, 619, 646
Натрий — аммиак — тетрагидрофу-
ран 444
Натрий — антрацен 353
Натрий йодистый 599
Натрий — нафталин 272, 353—354
Натрий — фенантрен 354—355
Натрия азид 15, 196, 237, 355—357,
426, 589, 609
Натрия алюмогидрид 309
Натрия алюмохлорид 357
Натрия амальгама 148, 357, 545
Натрия ацетат 630, 651
Натрия бикарбонат 161, 256
Натрия бихромат 636
Натрия боргидрид 34, 144, 171, 186,
301, 342, 357—360, 361, 375, 377,382,
383, 414, 426, 428, 429, 433, 488, 600,
616, 617
Натрия боргидрид сульфированный
359
Натрия бромат 62
Натрия гидрид 36, 44, 75, 94, 115,
118, 135, 162, 178, 192, 244, 259,
292, 360-362, 381, 405, 488, 489,
507, 513, 628, 656
Натрия Гидрид — трст-бутилгипохло-
рит 362—363
Натрия гидрид — диметилсульфоксид
363
Натрия гидросульфит 588, 589
Натрия гипобромит 408
Натрия гипохлорит 255, 362, 363, 367
Натрия дигидрофосфата моногидрат
363—364
Натрия дитионит см. Натрия гидро-
сульфит
Натрия дихлорфторацетат 629
Натрия диэтилалюмогидрид 364
Натрия железа (II) тетракарбонил
365—369
Натрия иминоксилдисульфонат (соль
Фреми) 252
Натрия иодид 57, 369—370, 566, 577
Натрия карбонат 174
Натрия метилат 75, 141, 157, 162, 193,
194, 220, 262, 325—327, 444, 494,
513, 580
Натрия метилсульфинилметилид 162—
164
Натрия бис- (2-метоксиэтокси) -алю-
могидрид 175, 283, 370—371т
388
Натрия нитрит 371—372
Натрия 4-оксибензолсульфонат 500
Натрия перборат 73
Натрия сульфид 372—373, 611
Натрия сульфит 622, 623
Натрия тетраборат 373—374
Натрия тетрафенилборат 631
Натрия тиосульфат 374
Натрия тиофенолят 306, 373
Натрия тиоцианат 34, 183
Натрия фенолят 383
Натрия феноляты 628
Натрия фосфат додекагидрат 307
Натрия хлорацетат 262
Натрия хлорид — диметилсульфоксид
374—375
Натрия N-хлор-п-толуолсульфамид
см, Хлорамин-Т
Натрия хромат 637
Натрия цианборгидрид 223, 375—378,
600
Натрия цианид 311, 379—380
Натрия цианид — диметилсульфоксид
380—381
Натрия этилат 466, 574, 655—656
Натрия этилмеркаптид 381
Нафтазарин 310
Нафталин 198, 330, 478, 633
Нафталина 1,2-окись 21, 326—327
Нафталин-1,2,3,4-тетракарбоновой
кислоты тетраметиловый эфир 29,
30
Нафталинтиол-2 171
Нафтвален 330
2,3-бис-(Нафтил-2)-бутан 371
у-(Нафтил-2)-масляной кислоты эти-
ловый эфир 584
1-(Нафтил-2)-этанол 371
жезо-Нафтодиантрон 250
Нафталин-1,8-дикарбоновой кислоты
ангидрид 532
сс-Нафтол 21, 423.
р-Нафтол 171, 423
Нафто-[1,2-с]-тиофен 15
Нафто-fl,8]’трицикло-[4,1,0,0й’1]-
гептен 331
а, |}-Ненасыщенные альдегиды 77,
336—338, 632
[},у-Ненасыщенные альдегиды 180—
181
у,6-Ненасыщенные альдегиды 657—
658
у,б-Ненасыщенные кетоны 124—125
у,б-Ненасыщенные кислоты 648—649
у,б-Ненасыщенные нитрилы 641
а,|3-Ненасыщенные сульфоны 75
аф-Ненасыщенных кислот нитрилы
640
р,у-Ненасыщенных кислот нитрилы
640
у,д-Ненасыщенных кислот эфиры
260—261
(Н-)-Неоизопулегон 547, 548
Неопентан 306
Неопентилбромид 305—306
Неопентилиодид 556
Неопентиловый спирт 556
Неоэргостерин 199, 200
Никель-алюминиевый сплав 381—382
Никель Ренея 55, 56, 79, 157, 158,243,
259, 466, 509, 510, 521, 634, 646
Никеля(П) ацетилацетонат 383, 642
Никеля борид 382
Никеля бромид 382—384
Никеля карбонил 274, 384—387, 642
Никеля тетракарбонил см. Никеля
карбонил
Никеля цианид 387, 544
Нингидрин (индаитрион-1,2,3) 234,
387—388
Нитрамины 25
Нитрилы 91—92, 99, 102, 332,505,514,
566, 580—581
Нитроалканы 12—13
И-([1-Нитроалкил)-ацетамиды 390
1-Нитро-2-алкилнитраты 358
«-Нитроанилин 600
1 - (2/-Нитробензил) -2-метил-6-мет-
окси-7-окси-1,2,3,4-тетрагидроизо-
хинолины 343, 344
Нитробензол 407, 442, 505
4-Нитробензолсульфокислоты алки-
ловые эфиры 161—162
4-Нитробензолсульфохлорид 161
1-Нитробутан 13
Нитровинилирование 388
5-Нитрогептанон-2 503
2-Нитро-1-диметиламиноэтилен 388
Нитрозамины 25
Нитрозил хлористый 613
N-Нитрозоацетанилид 29
1-(Н-Нитрозоацетиламинометил)-
циклогексанол 40
Нитрозония борфторид 388
695
З-Нитрозооксазолидоны-2 64
N-Ннтрозосоединения 228
С-Нитрозосоединения 449—450
З-Нитрозо-2-фенилиндол 601
N-Нитрозо-п-хлорбензоиланилины 614
З-Нитроизоксазолы 372
4-ацм-Нитрокротоновый альдегид 388
Нитромезитилен 618
Нитрометан 182, 389—390, 433, 486,
504, 574
п-Нитронадбензойная кислота 300
Нитрония борфторид 390
2-Нитропропан 390
2-Нитропропана 2-гидрОперекись 390
Нитроуксусной кислоты этиловый
эфир 504
Нитроуксусный альдегид 388
4- (4-Нитрофенилазо) -бейзоилхлорид
390—391
п-Нитрофенилацетилен 626
о-Нитрофенилсульфенхлорид 391—392
д-Нитрооенол 362
о-Нитрооенолы 618
п-Нитрофенолы 618
Нитроэтан 392
2-яцц-Нитроэтилидены 388
Нонановая кислота 41
Нонанол-5 269
н-Нонанил-быс- (фенилтиометил) -
карбинол 596
Норбелладина производное 479, 480
Норборнадиен 46—47, 265, 423—424,
446, 544, 545, 644
Норборнадиена-димер 423, 424
Норборнадиена тример 423, 424
Норборнадиенол-7-железотрикарбо-
нил 168
Норборнадиенон-7-железотрикарбо-
нил 168
Норборнан 469
экзо-Норборнанол-2 348
Норборнанон 376
Норборнен 46, 47, 311, 348, 426, 435,
569, 644
(±)-Норзизанон 596
Норкарпан 222
19-Норэргостапентаен-5,7,9,14,22-
ол-Зр 199
Норэритринадиенон 256
Нуклеозиды 71
Нуцифераль 277
Озон 393
N-Окиси 25, 351
Окисление кеталей 535—536
Окисление перманганатом 41
Окислительная конденсация 229—230
312, 321
Окислительная циклизация 429
Окислительное • декарбоксилирование
315, 439—440
Окислительное деметилирование
453—454
Окислительное метилирование 453
Окислительное расщепление глико*
лей 476—477
Окись мезитила 16, 136
Окись этилена 393—394, 545
2-Оксаадамантаи 458
З-Оксабицикло- [3,2,0] -гептадиен-1,4
106, 107
9-Оксабицикло-[3,3,1]-нонен-1 69
8-Оксабицикло-[3,2,!]-октен-6-он-3
340
1,2,4-Оксадиазолы 404, 405
Оксазиридины 25
Оксазолидиноны-2 440
Д3-Оксазолинон 28
Оксазолины 508
Оксазолы 506, 507, 606
Оксалилхлорид 394
Оксалилцианид 131
1-Оксаспиро-[4,5]-декан 583
Оксаспиропентан 300
Оксаспиропентаны 194—195
1,4-Оксатиаспиро-[4,4]-нонан 612
1,3-Оксатиоланы 612
9-Оксатрицикло-[4,3,3,0]-додеканон-3
585
Оксетаны 280, 283
1-Оксиадамантан 38, 119
а-Оксиальдегиды 334
За-Оксиандростен-5а-он-17 574
1-Оксиапорфины 344
[З-Оксиацетилены 213
2-Оксиацетофенон 173
©-Оксиацетофенон 334
экзо-7-Оксибицикло-[4,3,1]-декат-
риен-2,4,8 634
эндо-7-Оксибицикло-[4,3,1]-декатри-
ен-2,4,8 282
4-Оксибензолсульфохлорид 500—501
2-Оксибензофенон 320
1-Окси-2-бромтетралин 326, 327
З-Окси-2-винилизовалериановой кис-
лоты этиловый эфир 297
5-Оксигексановой кислоты 6-лактон
226
цраж-4-Оксигексен-2-аль 336
5-Оксигексен-2-овой кислоты 6-лак-
тон 226
2-Окси-3,6-ди-т/?ет’-бутил-1,4-бензо-
хинон 322
р-Оксидитиокоричные кислоты 71
4-Окси-3,4-дифенил-Дг-пиразоли-
нон-5 251
5-Окси-3,5-дифенил-Д2-диразоли-
нон-4 251
(1-Оксикетоны 209
696
у-Оксикетоны 209
а-Оксикислоты 478
р-Оксикислоты 293
6-Оксикислоты 276
Оксилактоны 434
й/-п-Окси-а-(метил аминометил)-
бензиловый спирт 602
2-Оксиметил-4-трет-бутилцикло-
гексаион 349
ct-Оксиметил-б-валеролактон 290
3-Окси-17|3-метил-14|3-гонапентаен-
1,3,5 (10) ,6,8 58
4-Оксиметил-2,6-ди-т’/7гт-бутилфенол
394—395
Оксиметилирование 349
(—) -1 -Окси-1 -метил-1,2,3,4-тетра-
гидронафталин 509
2-Оксиметилциклобутанон 58
1 - (1 '-Окси-1 '-метил) -этилбицикло-
[3,1,0]-гекса нон-4 655
Оксимеркурирование 426—427
2-Оксимицо-1-фецилпропан 117
Оксиидолы 283
1-Окси-9-оксабицикло-[3,3,1]-нонан 69
Оксипропилирование 582—583
З-Оксииропилфосфония соли 67
1 - (3-Оксипропил) -циклогексанол 583
3- (З-Оксииропил) -циклопентанон 583
1-Окси-1 - (n-толилсульфинил метил)
1,2,3,4-тетрагидроиафталины 509
эритро-З-Окси-4-п-толуолсульфонил-
оксигексадиин-1,5 106
5-энОо-Окси-3,3,6-триметилноркара-
нон-2 6'56
п-Оксифенилацетальдегид 602
2-Окси-2-фенил-л-толилсульфоксид
509
2'-Оксихалконы 445
4-Оксихинолины 162
Зр-Оксихолестанон-2 535
а-Оксициклододеканон 552
6- (1'-Оксициклогексил-Г) -кротоно-
вая кислота 276
трц^с-2-Оксициклогексилмеркур-
хлорид 70
2- (1 -Оксициклогексил) -уксусный аль-
дегид 335
(трппс-2-Оксициклогептил) -метил-
дифенилфосфония борфторид 295
4а-Оксициклокостанолид 51
З-Оксицпклооктен 307
гранс-2-Оксициклопентилмеркур-
хлорид 70
3|}-Окси-5а,8а-эпидиоксиандростен-
б-он-17 397
бис-(2-Оксиэтил)-дисульфиды 359
(±) -Оксокринин 479, 480
5-Оксононен-1 85
2-Оксо-7Н-тиазо-[3,2-а]-пиримидин 27
5-Оксо-5-фенилпентеп-1 85
1-Оксо-2-формил-1,2,3,4,5,6,7,8-окта-
гидроантрацен 399
7-Оксохолестерилацетат 61
1,2,3,4,5,6,7,8-Октагидроакридин 93
Октадекагидро-т^ис-циклооктабен-
зол 387
Октадиен-1,7 229
Октадиин-2,6-ол-1 284
Октаметилнафталин 162
N,N,N',N',N"N'',N'",N'"-OKTaMe™-
пентадиен-1,4-тетрамин-1,1,5,5 451
Октаналь-1 154
Октанол-1 154
(S)-(-]-)-Октанол-2 542
(4-) -в тор- Октанол 140
Октацетрен 612, 613
Октен-1 41, 553, 621
Октен-4 88
Октен-2-аль-1 77, 336
цис- и г^анс-Октены-2 294
(R)-(—)-Октил-2-амин 542
(—)-srop-Октил бромид 140
Октин-4 283, 637
Октин-2-ол-1 77
Октан-4-он-З 637
Олеум 433
Олефинов взаимопревращения 88
Олефинов инверсия 294—295
Олово хлористое 577
Ортомуравьиной кислоты триметило-
вый эфир 395
Ортомуравьиной кислоты триэтило-
вый эфир 222, 334, 395
Ортоуксусной кислоты триметиловый
эфир 559, 560
Ортоуксусной кислоты триэтиловый
эфир 138, 395
Орсина диметиловый эфир 381
Орсина монометиловый эфир 38!
Осмия четырехокись 41, 647, 648
Осмия четырехокись — калия хлорат
395—396
(±)-Оциденталол 219, 463
Оцименовый спирт 538
Палладиевая чернь 397
Палладиевые катализаторы 397—399,
402
Палладий 397, 427, 590
Палладий (10%) на карбонате каль-
ция 399
Палладий на угле 399—400, 417
Палладий (II) хлористый 400—402,
427
Палладия (II) ацетат 402, 427, 594
Паллидин 449
Параформ 257, 293, 494
23 Зак. 596
697
Пеларгоновая кислота 95
и-Пептадеканаль 533
Пентадиен-1,3 15, 16
Пентадиен-1,4 426
1,2,2,6,6-Пентаметилпиперидин 403
1,2,2,6,6-Пентаметилпиперидина иод-
гидрат 403
к-Пентан 265
Пентанол-3 269
Пентаноп-2 654
Пентаоксифосфораны 388
1,1,1,3,3-Пентахлоразапропен 403—405
Пентахлорбензальдегид 462
2,3,4,5,6-Пеитахлорбензиловый спирт
462
Пентахлорпиридин 351
2,3,4,5,6-Пентахлортолуол 462
Пентен-1-ол-З 269
траяс-Пентеп-З-ол-2 138
Пентен-4-ол-1 352
транс-Пентен-З-он-2 405—406
Пептидный синтез 196, 197, 497—498
555, 571, 619-620
Пергидроазуленоны 195, 196
Переалкилирование 452—453
Перекись водорода 25, 117, 118, 119,
139, 140, 152, 228, 253, 254, 306, 333,
346, 406, 467, 499, 579, 603, 654
Перекись водорода — алюминия хло-
рид 406—407
Перекись водорода в кислой среде
407—408
Перекись водорода в щелочной среде
408—409
Переметаллироваиие 427, 430
Периллен 165
Перйодаты 409—410
Периплогенин 633
Пехмана кумаринов синтез 649
Пивалиновой кислоты метиловый
эфир, см. Три мет и л уксусной кисло-
ты метиловый эфир
у-Пиколиловые эфиры сложные 619
Пикраты 411
Пикрилхлорид 410—411
Пикриновая кислота 411
Пинаколииовая перегруппировка 251,
256
Пинаконы 246, 247
а-Пинеи 128, 212, 622, 623
р-Пинен 623
а-Пинена окись 295
Пинокарвеол 295
^мс-Пиноновая кислота 212
Пиперазин 411—412
Пиперидин 262, 412
1-Пиперидиноциклогексен 402
1-Пиперидиноциклопентен 402
Д2-Пиразолииопы-5 251, 328
Пиразолоны 472, 475, 476
Пиразол-3,4,5-трикарбоновой кислоты
диметилового эфира мопоамид 111,
112
4Н-Пирантион-4 481
Пирен 101, 412
Перена пикрат 411
Пиридин 123, 161, 196, 228, 285 324,
325, 379, 413, 464, 468, 504, 505 514,
601, 651
Пиридина хлоргидрат 413—414
Пиридин-3,5-дикарёоновая кислота
443
бис-(Пиридин)-диметилформамндди-
хлорородия боргидрид 414
Пиридиния бромид-пербромид 193
Пиридинсульфотрноксид 168
Пиридины 175
Пиримидины 404
Пировиноградная кислота 378
Пировиноградной кислоты этиловый
эфир 228
Пирокатехин 42, 174
Пирокатехина дихлорметилеповый
эфир 414—415
Пирокатехинборан см. 1,3,2-Бензоди-
оксаборол
Пирокатехины 241
Ппррилмагиийбромид 656
Пиррилталлий(1) 656
Пиррол 251, 260, 626, 656
Пиррол-2-альдегида формилгидразон
658
4-(N-Пирролидил) -пиридин 415—416
Пирролидин 451
Пирролидона-2 бромгпдрата дибро-
мид 225
Пирролкалий 260
Пиррол-2-карбоновой кислоты этило-
вый эфир 565
Пирроло-[1,2-dJ-flbS-триазин 658
Пиррол-1 -тио-1,2-дикарбоновой кис-
лоты имид 260
Пиррол-2-тио-1,2-дикарбоновой кис-
лоты имид 260
Пластохинон-1 386
Платина 398
Платина на окиси алюминия 416
Платинохлористоводородпзя кислота
207, 208, 416
Платинохлористоводородная кисло-
та — олово хлористое 416
Плейадиен 331
5|},10а-Подокарпатриен-8,11,13-овая-
15 кислота 398
5а,10а-Подокарпатриен-8,11,13-овой-
15 кислоты метиловый эфир 398
Подокарповая кислота 105, 398
ПодокарповоЙ кислоты метиловый
эфир 201
Полиизопреноиды 341—342
698
Полималеинимид 59, 621
Пол иметилгидросилоксан 416—417
Полипоровая кислота 170
Полистирол — алюминия хлорид 18—
19
Полифосфорная кислота 417—420
Полифосфорной КИСЛОТЫ Эфиры
420—421
Полуацеталь 459
Померанца—Фрича реакция 52
Порфирина производные 112
Дэ(11)-Прегнен-5а-диол-3[ф6а-он-20
239
Прегнаполон 653
Прегненолонаиетат 450
Прегненолонацетата циангидрин 450
Преднизолон BWD 537
Преднизон BMD 537
t/Z-Прогестерон 564
L-Пролин 459
Пропандитиол-1,3 175, 176, 258, 421
Пропанол-2 247
Пропаргиловые спирты 284
Пропаргиловый альдегид 421
Пропаргиловый спирт 421
грана-Пропен ил бензол 430
н-Пропиламии 378
н-Пропилбензол 370
Пропилиден-трет-бутилимин 277
5-Пропилнонанол-5 63
н-Пропилциклогексиламин 378
Пропиоловой кислоты амидины 418,
419
Пропиоловой кислоты метиловый
эфир 123, 534
Пропиоловой кислоты эфиры 27
Пропиоловый альдегид 421
Пропионовый альдегид 269, 336
Простагландины 125, 154, 213—214,
266, 267, 301—302, 324, 337, 413,
484, 485, 486, 518—519, 525—526,
549, 587
Проэритринадиенон 256
Птеридины 6-замещенные 372
Пульвиновой кислоты дилактон 170
Пуммерера кетон 230
Пуммерера перегруппировка 97, 584
Пшорра апорфина синтез 343—344
Расщепление простых эфиров 19—20,
585
Расщепление тиоацеталей 332, 460,
607
Резорцин 423
Ретикулин 449
Реформатского реактив 570
Реформатского реакция 293, 648
Рибонуклеозиды 92—93
Риттера реакция 390, 408
Родапиодпд 422
Родий (5%) на окиси алюминия
422____423
Родий (5°/о) па угле 423—424
Родия (III) окись 424—425
Розенмунда восстановление 397—
398, 400
Ртути азид 425—426
Ртути(П) ацетат 43, 426—431, 658
Ртути (11) нитрат 474
Ртути окись 180, 258, 341, 431—432
Ртути окись — бром 432—433
Ртути (II) трифтор ацетат 327, 433
Ртуть 429, 430
Ртуть хлорная (сулема) 171, 180,
182, 219, 279, 280, 335, 336, 337, 398,
596
Рутения трихлорида гидрат 433—434
Рутения четырехокись 434
а-Сантонин 44
Саретта реактив 634
Свинца (IV) ацетата азиды 435—437
Свинца диацетат 437
Свинца дитиолаты 437
Свинца роданид 422
Свинца тетра ацетат 96, 130, 428, 435,
437—442
Свинца тетра-(трифторацетат) 442—
443
Сейчеллен 144
Селен 437, 443, 502, 552
Селена двуокись 443—446
Селенокарбонильные соединения 162
Семибуллвален 516, 517
Семикарбазид 611
Семикарбазида хлоргидрат 418
Сера 437
Сера двухлористая 133, 446
Серебра ацетат 453, 464
Серебра борфторид 192, 446—447,615,
626, 631, 632
Серебра диэтилфосфат см. Фосфор-
ной кислоты диэтилевого эфира
серебряная соль
Серебра карбонат 246, 447, 603
Серебра карбонат — целит 448—450
Серебра нитрат 90, 194, 305, 320,
451—452
Серебра окись 452—453, 630
Серебра (II) окись 453—454
Серебра перхлорат 238, 239, 454—
458
Серебра (II) пиколинат 454
Серебра сульфат 458
Серебра тетрафтороборат см. Сереб-
ра борфторид
Серебра трифторацетат 339, 340
Серебра л-хлорбензоат 613
23*
699
Серебро 305
Серная кислота 52, 102, 162, 173, 185,
458—461, 576, 626, 636, 649, 650
Сернистый ангидрид 343, 461—462
Серный ангидрид 443, 462
Серный ангидрид — пиридин 462—
463
Сероводород 463
Сероуглерод 163, 183
Силиловые эфиры простые 485
Силикагель 622, 623
Симмонса—Смита реагент 463—465
Симмонса—Смита реакция 252
р-Синенсаль 277
f/1-Сиренин 445
Сквален 341, 342, 445
Сложноэфириые конденсации 304
Смешанные ангидриды 19!
Соляная кислота 465
Соммле реакция 90
Сопряженное присоединение 278—280,
316, 319, 466, 486
Спироалкилирование 192—193
Спиробутаны 192
Спиро-[4,4]-нонадиен-1,6 502
Спиро-[4.4]-нонадиен-2,6-дион-1,5 389
транс, транс-Спиро-[4,4]-нонан диол-
1,6 501, 502
Спиро-[4,4]-нонандион-1,6 419
Спиро-[4,4'-нонатетраен 389
Спиро-[4,4]-нонатриен-1,3,7 369—370
Спиро-[3,4]-октанон-1 513
Спироэпокси циклогекс а диен-2,4-оны
409
Стеариновая кислота 654
Стеркуловой кислоты метиловый
эфир 464
Стероидные амиды 262, 557
Стефена восстановление 381—382
Стибабензол 21, 107
Стивенса перегруппировка 114—115
Стигмастерилацетата диэпоксид 88
Стигмастерин 586
Стильбен 87, 88, 172, 212, 270, 294,
374, 435, 553, 573
qwc-Стильбена окись 553
транс-Стильбена окись 88, 294
э^нтро-Стильбендибромид 374
Стильбены 98
(+) - (R) -трацс-р-Стирил-п-толил-
сульфоксид 465—467
Стирол 348, 639
Стирола окись 362
L-Стрептоза 181
Строфантидин 633
Сукцинимид 543
Сульфамиды 371
Сульфиды 197, 615—616
Сульфинилхлориды 109
Сульфирование 463
о-Сульфобензойной кислоты ангидрид
467
Сульфоксидов дегидратация 15
Сульфоксиды 15, 197, 468—469, 584
Сульфолан 358, 469
Сульфолен-3 (2,5-дигидротиофенди-
оксид-1,1) 467
о-Сульфонадбензойная кислота 467
Сульфоны 469
Сульфоуксусная кислота 34,467
Сульфурил хлористый 467—470
Сурьма пятихлористая 470
Сцилларенон 286
Таллия (II) бромид 478
Таллия (I) карбонат 657
Таллия (I) нитрат 474
Таллия(III) нитрат 471—477
Таллия (III) окись 471
Таллия (III) сульфат 477
Таллия триацетат 428, 476, 478
Таллия (III) трифторацетат 476,478—
481
Таллия этилат 477, 656—657
Тексилборан см. 2,3-Диметилбутил-
2-боран
Теллур четыреххлористый 482
а-Терпинен 535
Тестостерон 5!
2,3,4,6-Тетра-0-ацетил-|}-0-глюко-
пираноза 50
1,3,5,7-Тетрабромадамантан 458
Тетра-(бромметил)-метан 482
1,1,6,6-Тетрабромметилспиро-[3,3]-
гептан 286
1,1,10,10-Тетрабромметилтриспиро-
[3,1,1,3,1,!]-тридекан 286
2,4,4,6-Тетрабромциклогексадиен-
2,5-он 482—483
Тетра-н-бутиламмония ацетат 484
Тетра-н-бутиламмония бисульфат 71
Тетра-н-бутилаымония бромид 483
Тетра-н-бутиламмония гидроокись
484, 485
Тетра-н-бутиламмония оксалат (сред-
няя соль) 484
Тетра-н-бутиламмония формиат
484—485
Тетра-н-бутиламмония фторид 142,
143, 485
Тетрадегидро-[18]-аннулендионы 321
Тетрагидробиснор-S 17, 265
цис-1,4,9,10-Тетрагидронафталин 285
О-Тетрагидропиран-2-иловые произ-
водные стероидов 51—52
Тетрагидро-4Н-пиранон-4 364
Тетрагидрофуран 36, 42, 63, 73—75,
77, 78, 87, 88, 93, 94, 99, 117—119,
122—124, 139, 166, 167, 171, 176,
180, 181, 192, 199, 229, 240, 249, 256,
700
260, 269, 270, 274—276, 287, 290,
291, 294, 296—299, 300, 305, 309,
335, 338, 350, 353, 366, 367, 387, 389,
396, 426, 443, 485, 506, 525, 527, 641
Тетрагидрофуран-цмс-2,5-дикарбоно-
вая кислота 424
2,3,За,8а-Тетрагидрофуро-[2,3-/>]-ин-
долы 394
н-Тетрадекандион-5,10 85
^ис,цис-1,2.3,4-Тетракарбэтокси-
бутадиен-1,3 318
трансуране-1,2,3,4-Тетракарбэток-
сибутадиен-1,3 318
Тетралин 273
Тетралина 1,2-эпокись 326
ct-Тетралон 273, 509
Р-Тетралоны 329
Тетраметиламмония бромид 651
2,3,6,7-Тетраметилантрахинон 641,642
2,3,6,7-Тетраметилантрацен 641, 642
Тетраметилбарбитуровая кислота 453
1,2,2,3-Тетраметилбицикло-[1,1,0]-
бутан 624, 625
1,1,3,3-Тетраметилбутилизонитрил см,
2,4,4-Триметилпентил-2-изонитрил
N- (1,1,3,3-Тетраметилбутил) -форм-
амид 500
2,5,5,9-Тетраметилгексагидрохромен
433
Тетраметилгуанидин 486
Тетраметилгуанидиния азид 580
Ы^М'.Ы'-Тетраметилдиамидохлор-
фосфат 486—487
2,4,4,6-Тетраметил-5,6-дигидро-1,3-
(4Н)-оксазин 171, 488—491
N,N,N',N'-TeTpa метил- 1,3-дихлор-
2-азатриметинцианин 206—207
Тетраметиленсульфид 157
Тетраметиленсульфон 601, 358; см.
также Сульфолан
2,2,3,3-Тетраметилиодциклопропан 241
Тетраметилкетениммония борфторид
626
Тетраметилмочевины дихлорид 204
2,2,7,7-Тетраметилоктадиин-3,5 209
2,2,6,6-Тетраметилпиперидин 302, 403
Тетраметилтиомочевина 204
2,3,4,5-Тетраметилтиофен 208
Тетраметилтиурамдисульфид 204
3,3,6,6-Тетраметил-1,2,4-триоксан 406
Тетраметилфталевой кислоты динит-
рил 209
Тетраметилэтилен 55
М^И'.М'-Тетраметилэтилендиамин
120, 487, 491—496
2,2,3,3-Тетраметилянтарный альдегид
312
Тетраметоксиэтилен 512
Тетра-З-пинанилдиборан см. Диизо-
пинокамфилборан
Тетрафенил бипикло-[3,1,0]-гексеноны
108
1.2,3.4-Тетрафенилнафталин 189—190
614
Тетрафенилциклопентадиенон 188,
189, 190, 385, 496—497, 613; см. так-
же Тетрациклон
1,6,7,7-Тетрахлор-2,5- Дифенил бицик-
ло-[4,1,0]-гептен-3 65, 66
1,3,6,8-Тетрахлоркарбазол 615
Тетрахлорсилап 497—498
Тетрахлорциклопропен 517
Тетрахлорэтан — нитробензол 498
Тетрацен 93
Тетрацианэтилен 310, 389, 592
зкзо,зкзо-Тетрацикло-[4,4,2,02’6,0т’10]-
додекатриен 354, 355
Тетрациклон см. Тетрафенилцикло-
пентадиенон
Тетрацикло-[3,3,0,02>8,04>е]-октанов
производные 457
Тетраэтиламмония ацетат 484
Тетраэтиламмония формиат 484
Тетраэтиламмония хлорид 323
М,Гч,Ы'^'-Тетраэтил-1,4-диамино-
2,5-дибромбензол 360
Тетродотоксин 620
Тетроловой кислоты метиловый эфир
124, 297
Тетроловой кислоты этиловый эфир
546
Тиабензола 1-окись 167
4Н-1,4-Тиазина 1,1-диокись 467
1,3-Тиазины 420
Тиазолидины 335
Тиазолины 335
Тиепина 1,1-диокись 461—462,
Тиле реакция 650
Тимидин 143, 541, 558
Тимидин-5'-фосфат 541
Тиоамиды 498, 499
Тиоанизол см. Метилфенилсульфид
Тиоацетали 332, 616
Тиоацетамид 391, 498—499
Гиобензальдегидоксид 469
4,4'-Тио-б«с- (6-трет-бутил-З-метил-
фенол) 620
Тиокарбонилилиды 191
Тиокарбонильные соединения 162
Тиокетали 453, 575
Тиометилиндол 158
Гиомочевина 499
Тиомочевины диоксид 499
Тионилбромид 92
Тионил хлористый 92, 342, 389, 468,
499—501, 604
Тионхлоругольной кислоты «-толило-
вый эфир 501—502
Тиоуксусная кислота 310
4-Тиофурацилы 259
701
Тиофен-2,4-дикарбоновой кислоты
диметиловый эфир 414, 415
Тиофен-З-карбоновой кислоты мети-
ловый эфир 414, 415
Тиофенол — азодиизобутиронитрил
502
Тиофенолы 171
Тиофены 208—209
Тиофосген 501
Титан треххлористый 502—504
Титан четыреххлористый 504—505
Тиффено — Демьянова реакция 275
Тозилазид см. п-Толуолсульфонил-
азид
Тозилгидразин см. «-Толуолсульфо-
нилгидразин
Тозилметилизонитрил 218, 505—508
Тозилхлорид 521
Толан 303
Толилалкилкарбонаты 96
п-Толилвинилсульфоксид 466
rt-Толилсульфинилметилидлитий
508—510
2- («-Толилсульфинил) -1 -фенил-
этилмалоновой кислоты диэтило-
вый эфир 466
Тозилтиометилизонитрнл 507—508
4-м-Толилтиооксазол 508
Толилуксусная кислота 639
«-Толуидин 454
п-Толуиловой кислоты грет-бутило-
вый эфир 287
«-Толуиловой кислоты метиловый
эфир 257
o-Толуиловый альдегид 90
«-Толуиловый альдегид 90
Толуол 90, 96, 212, 343, 639
а-Толуолсульфиновая кислота 469 .
«-Толуольсулъфиновая кислота 506,
521
«-Толуолсульфокислота 36, 50, 77,
131, 162, 174, 176, 177, 200, 240,
292, 304, 344, 377, 421, 490, 510—
512, 570, 583, 594
n-Толуолсульфонилазид (тозилазид)
512
п-Толуолсульфонилгидразин (тозил-
гидразин) 130, 358, 377, 418, 512—
514, 520
п-Толуольсульфохлорид 78, 79, 338,
514, 601
Торпе—Циглера циклизация 593
Треххлористый азот 515
1,2,4-Триазин-(2Н)-оны-3 611
1,3,5-Триазины 404
Триазолы 81, 207, 356, 404, 405, 432,
581, 582, 587, 611
Триалкилбораны 42—43, 62, 64, 262,
306, 579
Триалкилкарбиволы 306, 579
702
Триалкинилбораны 112
сшил/- Триарилбензолы 356
Триарилфосфины 257
1,3,5-Трибромадамантан 76
Трибромуксусная кислота 540
Трибромфенол 482
Три-м-бутилборан 306
Три-втор-бутилборан 287
Три-«-бутил-«-бутен-3-ил-олово 515
Три-«-бутилкарбинол 269, 306
Три-н-бутилстаннан 193, 194, 281,
515—518, 527
Три-н-бутилфосфин 518, 643
Три-н-бутилфосфина — меди I)’ иоди-
да комплекс 518—520
Три-к-бутилфосфина окись 286
Три-к-бутилхлорстаннан 515
Три-трет-бутилциклопропеяилия
борфторид 65
Триглим 629
Тридегидро-[18]-аннулен 399
Тридецилбромид 432
Триизобутилалюминий 519—520
2,4,6-Триизопропилбензолсульфо-
кислоты гидразид 520
2,4,6-Триизопропилбензолсульфохло-
рид 520
Триизопропилуксусная кислота 106
Триизопропилуксусной кислоты мети-
ловый эфир 105, 106
Триизопропилфосфит 387
Трикаприлилметиламмонийхлорид 39
Тримезитилборан — натрий 353
2,8,8-Триметил-З-азатрицикло-
[5,1,1,02’5]-нонанон-4 622, 623
Триметиламин 150
Триметилацетальдегида фенилгидра-
зон 588
1,1,3-Три мети л-2-а цетилцикло гек-
сен-3 460
2,4,6-Триметилбензойная кислота 53,
95, 105, 107, 108, 211
2,4,6-Триметилбензойной кислоты
гераниловый эфир 444
2,4,6-Триметилбензойной кислоты ме-
тиловый эфир 95, 103—107, 108
1,2,2-Триметилбицикло-[1,1,0]-бутан
624, 625
1,3,5-Триметил-2,4-6-тр«с-(3,5-ди-
тргт-бутил-4-оксибензил) -бензол
520
Триметилендибромид 521
Триметилендитиотозилат 521—522
2,2-Триметилендитиоциклогексанон
521
Триметилентиокетали 616
1,3,5-Триметил-2-оксабипикло-
[3,3,1]-нонен-3 460
Ц,4,4-Триметил-2-оксазолиния иодид
522—523
Триметилоксония борфторид 116, 171,
523, 595
2,4,4-Триметилпентаналь 635
2,4,4-Триметилпентен 635
2,4,4-Триметилпентил-2-изонитрил
500, 523—524
2,4,6-Триметилпиридин 76
Триметилсилилазид 435
N-Триметилсилиламиды 347
О,М-б«с-Триметилсилилацетамид 195
бпс-(Триметилсилил) 'ацетилен 242
Триметилсилил бромид 530
N-(Триметилсилил) -глицина триме-
тилсилиловый эфир 392
Триметилсилилдиазометан 524—525
2-Триметилсилил-1,3-дитиан 278
Триметилсилилдиэтиламин 525—526
Триметилсилилкарбен 524—525
бис- (Триметилсилил) -литийамид 130,
526—527, 537
бис- (Триметилсилил)-натрийамид 527
анти-7-Трпметилсилилноркаран 524,
525
Триметилсилиловые эфиры енолов 118,
531
1-Триметилсилйлтриазол-1,2,4 80
Триметилсилилтрибромацетат 539,
Триметилсилилцианид 527—528
бис- (Триметилсилокси) -бутадиен-1,3
464
4-Триметилсилоксинонин-2-овой кис-
лоты метиловый эфир 123, 124
1,2-бис- (Триметилсилокси) -циклобу-
теп-1 54
Триметилсульфония борфторид 164
Триметилсульфония иодид 164
2,5,8-Триметилтетралон-! 417, 418
Триметилуксусной кислоты метило-
вый эфир 531, 532
2,2,3-Триметил-4-фенил-6-п-нитро-
феиил-1-аза-З-азониабицикло-
[3,1,0]-гексена-3 борфторид 523
Триметилфосфит 227, 387, 528—529,
617
1,2,5-Триметилфосфолен-З 654, 655
Триметилхлорсилан 48, 143—144 , 195,
242, 278, 298, 492, 529—532
2,2,6-Триметилциклогексанон 80
3,5,5-Триметилциклогексен-3-он-1 289
1,3,3-Триметилциклопропен 428
3,4,5-Триметоксибензальдегид 101,
400
3,4,5-Триметоксибензоилхлорид 400
2,2,2-Три мето кси-4,5-ди мети л-1,3-
диоксафосфолен 115—116
(±) -2,3,4-Триметоксиэстратриен-
1,3,5(10)-ол-17р 48
2,4,6-Тринитробензолсульфокислота
161
сшог-Триоксан 601
1,2,3-Триоксо-2,3-дигидрофеналей
532—533
Три-н-пропиламин 567—568
Три-н-пропиламина хлоргидрат 567
Триптицен 282, 614
силш-Тритиан 533
Тритилбензиловый эфир см. Трифе-
нил метил бензиловый эфир
Тритилборфторид см. Трифенилметил
борфторид
Тритиллитий см. Трифенилметилли-
тий
Тритиловые эфиры см. Трифенилме-
тиловые эфиры
Тритилоновые эфиры простые см. 9-
Фенил-9-алкоксиантрон
Тритон Б 81, 334
Трифениларсина окись 533—534
Трифенилкарбинол 362
Трифенилметан 535, 536
Трифенилметилбензидовый эфир 537
Трифенилметилборфторид 182, 308,
534—536
Трифенилметилгексафторантимонат
534, 536, 537
Трифенилметилгексафторарсенат 537
Трифенилметилгексафторфосфат 534,
536—537
Трифенилметиллитий 44, 118—120,
299, 537—538
Трифенилметиловые эфиры простые
536
С,С,1Ч-Трифенилнитрон 441
Трифеиилстаннан 538
Трифенилстибина окись 534
Трифенил фосфин 11, 209, 365,366,368,
401, 402, 433, 501, 538—540, 541,
544, 552, 629, 640, 643, 644
Трифенил фосфина окись 533, 534,543,
554
Трифенилфосфин — азодикарбоно-
вой кислоты диэтиловый эфир 541 —
543
Трифенилфосфинвольфрам (0) пента-
карбонил 341
Трифенилфосфин — N-галогенсук-
цинимид 543
Трифенилфосфиндибромид 543—544
бис- (Трифенилф'осфин) -иминия хло-
рид 368
бис- (Т рифенилфосфин) -кобальт (II) -
дибромид 545
бис-(Трифенилфосфин)-никель (0) 545
бис- (Трифенилфосфин) -никельдибро-
мид 545
бис- (Трифенилфосфин) -никельди-
карбонил 544
бис- (Трифенилфосфин)-никель (II)-
дицианид 544—545
703
трис- (Трифенилфосфин) -родий (1)’-
карбонилгидрид 545—546
трис- (Трифеиилфосфин) -родийхло-
рид 194, 546—550
трис- (Трифенилфосфин) -родийхло-
рид — триэтилсилан 550—551
трис- (Трифенилфосф ин) -рутенийди-
хлорид 550, 551—552
Трифеиилфосфинселенид 552—553
Трифенил фосфин — углерод четырех-
бромистый 553—554; см. также
Трифенилфосфин — углерод четы-
реххлористый
Трифенилфосфин — углерод четырех-
хлористый 554—556
Трифенилфосфирена окись 104—105
Трифенилфосфит 556—557
Трифенилфосфита иодметилат 375,
557—559
Трифенилфосфита озонид 559
Трифенилфосфоранилиден-а'-кетоян-
тарной кислоты динитрил 533
Трифенилхлорметан 534, 536, 559—
560
Трифлаты см. Трифторметансульфо-
кислоты эфиры
Трифторацетоксилироваиие 442—443
Трифтор метансульфокислота 560—561
Трифторметаисульфокислоты серебря^
ная соль 561
Трифторметаисульфокислоты эфиры
(трифлаты) 560—561
Трифторметансульфоновой и карбо-
новых кислот смешанные ангидри-
ды 561—562
2,6-бйС-Трифторметилбензойная кис-
лота 156
Трифторметнлгипофторит 562, 607
а-Трифторметилминдальная кислота
598
Трифторнадуксусная кислота 504,
562, 621
Трифторнадуксусная кислота — бора
трифторид 406, 562
Трифторуксусная кислота 45, 46, 49,
50, 58, 89, 90, 162, 175, 519, 552,
563-565, 576, 616
Трифторуксусный ангидрид 52, 238,
326, 327, 379
2-Трихлорацетилпиррол 565
Трихлорацетилхлорид 202, 565
3,6,6-Трихлоргександион-1,2 322
Трихлоризоциануровая кислота (циа-
нурхлорид) 566
Трихлорметилизонитрила дихлорид
403—405
Трихлорметиллитий 566
Трихлорнорборнан 66
5Д6-Трихлорнорборнен-2 66
Трихлорсилан 567—569
Трихлоруксусная кислота 341, 569,
576
Трихлоруксусной кислоты натриевая
соль 597
Трихлоруксусной кислоты этиловый
эфир 569—570
Трихлорциклогександионы-1,2 322
Трихлорциклопропен 517
1,1,1-Трихлорэтан 137
2,2,2-Трихлорэтанол 570—571
Трихлорэтилен 66
Трицикло-[4,1,0,03’7]-гептан 457
Трицикло-[4,1,0,03'7]-гептан 624, 625
Трицикло-[3,2,0,02’4] -гептаны 447
эндо-Трицикло-[4,4,0,02’й]-декадиен-
3,8-дион-7,10 97
Трицикло-[5,3,0,0г>10]-декатриен-3,5,8
330, 331
Трицикло-[2,1,0,02'®]-пентапоны-3 455,
456
Трициклопропилкарбинол 185
Трицикло-[4,4,1,01>°}-ундека диен-3,8
198
Триэтилалюминий 571, 642
Триэтиламин 81, 83, 84, 99, 109, ПО,
153, 154, 165, 168, 170, 191, 202,236
267, 308, 324, 332, 392, 505, 539, 543,
'555, 572, 573, 601, 616, 625, 651
Триэтил амин а хлоргидрат 579
Триэтилборан 62, 151, 152, 289, 305,
306
Триэтиленгликоля диметиловый эфир
190
2,4,5-Триэтилимидазол 425
Трэтилкарбинол 306
Ы,М,М-Триэтил-М'-карбметоксисульф-
амида внутренняя соль — триэтил-
амин (комплекс) 178, 343—344,
572—573
М,М,М,-Триэтил-ЬГ-карбэтоксисульф-
амида внутренняя соль 573—574
Триэтилоксония борфторид 513, 535,
574—576
Триэтилсилан 576
Триэтилфосфит 387, 529, 577
Тропилиден 98
Тропилия азид 15
Тропилия соли 577—578
Трополон 157, 645
Тропой 15, 645
Тропоны 225
З-Туйон 232
Углерода двуокись 545, 606
Углерода окись 306, 315—316, 366,
367, 416, 425, 433, 443, 579
Углерод четыреххлористый 579
704
Угольной кислоты трет-бутил ового
эфира азид 80, 579—580
Угольной кислоты трет-бутилового
эфира фторангидрид 580
Угольной кислоты метилового эфира
гидразид 580—581
Угольной кислоты этилового эфира
азид 581—582
Ульмана реакция 318
Уксусная кислота 85, 142, 143, 417
Уксусного альдегида этил-3-бром-
пропилацеталь 582—584
Уксусной кислоты бензиловый эфир
639
Уксусной кислоты хлорангидрид (аце-
тил хлористый) 584—585, 650
Уксусномуравьиный ангидрид 585
Уксусный ангидрид 172, 186, 584, 614
Уксусный ангидрид — пиридина хлор-
гидрат 585
Уксусный ангидрид — цинка хлорид
585—586
Ундекандион-2,5 98
Уодсворта — Эммонса реакция 219
Уретаны 196—197, 572, 590
Уридин 541
Уридин-5'-фосфат 541
Урушибары гидрирования катализа-
торы 586
Фаворского перегруппировка 325—
326
фарнезилацетат 60
транс,трднс-Фарнезол 284
фенален 331
Фенантрен 39
фенантрена 3,4-окись 327
фенантрен-3,4-дикарбоновая кислота
568
Фенантридин 23
о-Фенантролин 25
N-Фенацилхинолиния бромид 410
Фенацилхлорид 207, 301
Фенилазид 587
9-Фенил-9-алкоксиаятрон 593
а-Фенилаллилацетат 430
Фенилаллиловые эфиры (простые) 565
6-Фенилантрацен 593
Фенилацетальдегид 525
Фенилацетилеи 83, 179, 314, 317, 323
феиилацетоцитрил 354
2-Фенилбензо-1,3,2-диоксаиодолы 241
n-Фенилбензоил хлористый 587
2-Фенил-4Н-3,1 -бензоксазинон-4 628
эндо-2-Фенилбицикло-[3,2 1]-октен-3
281, 282
фенил-(бромдихлорметил) -ртуть 597
Фенил-(1-бром-1-хлор-2,2,2-трифтор-
этил)-ртуть 588
4-Фенилбутанон-2 552
З-Фенилгептанон-2 40
Фенилгидразин 206, 207
Фенилгликолевого альдегида диме-
тилмеркапталя S-окись 334
Фенилдиазометан 98
Фенилдиазония хлорид 588—589
Фенилдиметилсилан 551
эндо-2-Фенил-7,7-дифторнорборнен-5
274
Фенилдихлорборан 589—590
Фенилдихлорфосфин 257
о-Фениленборат 43
о-Фенилендиамии 421
о-Фениленхлорфосфит 163
транс-2-Фенил-1-изопропенилцикло-
пропан 591
Фенилизоцианат 236, 590
N-Фенилимид азодикарбоновой кис-
лоты 590—593
2-Фенилиндолы 158
Фенилкарбен 303
Фениллитий 319, 421, 660
Фенилмагнийбромид 308
Фенилмагнийфторид 308
(—)-3-Фенилмасляная кислота 465
5-( + )-Фенилмасляной кислоты хлор-
ангидрид 117
Фенилмеркурацетат 594
Фенилмеркурбромид 384, 597
Фенилмеркуриодид 599
Фенилмеркурхлорид 384
М,М-Фенилметилнатрийамид 593
сс-Фенилминдальной кислоты М,М-ди-
метиламид 274
7-Фенилноркаран 303
5-Фенилоксазол 506
9-Фенил-9-оксиантрон 593
Фенилпалладия ацетат 594
1-Фенилпропандиол-1,3 117
1-Фенилпропин 525
З-Фенилпропин 525
Фенилпропиоловой кислоты этиловый
эфир 534
3-Фенилпропионовая кислота 418
З-Фенилпропиоиовый альдегид 171,
226
а-Феиилпропиофенон 503
Фенил-®-стирил-(диметиламино)’ -
сульфоксоиия борфторид 594
Фенилтиометиллитий 595—596
Фенил- (тригалогенметил) -ртуть 596—
597
Фенилтриметиламмония метилсуль-
фат 597
Фенилтриметиламмония пер бромид
496, 597—598
Фенилтрифторметилкетен 598
Фенил- (трифторметил) -ртуть 599—
600
705
Фенилуксусной кислоты этиловый
эфир 334
3(5) -Фенил-5 (3) -фенилэтинилпира-
зол 101
п-Фенилфенол 600
Фенилфосфонистой кислоты диэтило-
вый эфир 654
2-Фенилфуро-[3,2-&]-пиридин 317
2-Фенил-4 (ЗН) -хиназолинон 601
Фенил-З-хлорпропилсульфид 135
1-Фенил-5-хлортетразол 600
фенилциклобутендион 116
1-Фенилциклогексен 203
Фенилциклооктатетраен 67
1 -Фенилциклопентанкарбоксальде-
гид 488
1-Фенилциклопентанкарбоновая кис-
лота 356
1-Фенилциклопентил амин 356, 627
1-Фенилэтанол 348, 509
2-Фенилэтанол 156, 348
2 - (2-Фенилэтил) -3,4-диметилтиазо-
лидин 171
Фенилэтиленгликоль 638
(—) -1-Фенилэтилмалоновой кислоты
диэтиловый эфир 466
р-фенилэтилметилкарбонат 156
2- (2-Фенилэтил) -4-метилтиазол 171
2-Феноксибензойная кислота 418
Зр-Фенокси-Д5-холестен 174
Феноксициклооктатетраен 67
Фенол 119, 504
Фенолов конденсации 255—256, 401,
449, 479—480
Фенолы 118—119, 407, 442
Фенотиазин 656
Физовенин 394
Фишера синтез сложных эфиров
101
Флавоны 19, 445
Флорацетофенона диметиловый эфир
19
Флорацетофенона триметиловый эфир
19
Флуорантена пикрат 411
Флуорен-9-карбоновая кислота 315
Флуоренол 362, 499
Флуоренон 320, 362, 370, 499, 548
Формазаны 588
Формальдегид 290, 293, 378, 600—601
Формальдегида диметилмеркапталь
333
Формамид 395
Формамидинсульфиновая кислота см.
Тиомочевины диоксид
Формамиды 539
1 -Формил амино-1 -диэтил фосфонил-
алкены 218
о-Формилбензойной кислоты метило-
вый эфир 412
п-Формилбензойной кислоты метило-
вый эфир 257
п-Формилбензолсульфамид 382
Формилкарбен 585
З-Формиллитохолевой кислоты ами-
ды 262, 557
Э-Формилметил-Ю-метил-Д^окталин
658
Формилциклобутан 323, 324
Формилциклогексан 425
Формилциклопентан 471
Формилциклопропан см. Циклопро-
панкарбоксальдегид
3-Формокси-5Р-холановой кислоты
амиды см. З-Формиллитохолевой
кислоты амиды
Фосген 45, 133, 625
Фосфора илиды 23
Фосфора хлорокись 325, 486, 507, 539,
601—602, 605
Фосфорилирование 115—116, 541
Фосфорная кислота 102, 293, 602—
603, 657, 658
Фосфорной кислоты диэтилового эфи-
ра серебряная соль 603
Фосфорный ангидрид (пятиокись фо<>
фора) 185
Фосфор пятисернистый 603
Фосфор пятихлористый 67, 361, 501,
603—605
Фосфор трехбромистый 494
Фосфор треххлористый 296, 361
Фотоокисление 263
Фриделя — Крафтса ацилирование
392, 565
Фриделя — Крафтса циклизация 16—
17
(—)-Фрулланолид 119, 120
Фталазин 224
Фталевой кислоты дихлорангидрид
368, 310
Фталевый ангидрид 357
Фториодкарбен 40
1-Фтор-1-иодциклопропаны 40
Фторирование 607
Фтористый водород безводный 46, 50,
175, 605—606
Фтористый водород — бора трифто-
рид 606
бис- (Фторокси) -дифторметан 606—607
Фторсульфоновая кислота 464
Фторсульфоновой кислоты метило-
вый эфир 329, 332, 607—608
Фторсурьмяная кислота 534
Фуран 339, 340, 626
Фуран-2,5-дикарбоновая кислота 424
Фуранеол 396
фураноксантоны 414
фураны 164—165
706
Халконы 472
Хенбеста восстановление 617
Хииазолины 404
Хинолин 260, 567
Хинолин S 400
о-Хиионы 241
Хиноны 97
Хинуклидин 609
Хлор 437, 530, 621
1-Хлорадамантан 38, 469
tz-Хлоракриловой кислоты хлораягид-
рид 609—610
2-Хлоракрилонитрил 610—611
1-Хлоралкены 605
Хлорамин 611
Хлорамин-Т 98, 611—612
о-Хлоранизол 303
Хлоранил 199, 200, 310, 612—613
о-Хлоранил 613
д-Хлоранилин 454
1-Хлорантрахинон 171, 172
2-Хлорантрахинон 17f
Хлорацетонитрил 40
у-Хлорацетоуксусной кислоты хлор-
ангидрид 371
гс-Хлорбензоилнитрит 613—614
2-Хлорбензойной кислоты этиловый
эфир 162
Хлорбензол 202, 269, 394
1-Хлорбензотриазол 611, 614—616
L-( + ) -эритро-З-Хлор-2-ацетоксибу-
тан 604, 605
З-Хлорбицикло-[3,2,1]-октен-2 570
2-Хлор-5-трег-бутилциклогексанон 301
p-Хлорвинилкетоны 539
fJ-Х лор винил сульфоны 322—323
1-Хлоргексан 469
2-Хлоргексан 469
З-Хлоргексан 469
Хлоргидринов ацетаты 559—560
2-Х лор дезокси-1,3-дистеароилглице-
рин 555
б'-Хлор-б'-дезоксицитидин 92, 93
1-Хлордиамантан 18
4-Хлордиамантан 18
6-Хлордибензо-[а,с]-тропилийхлорид
39
2-Хлор-2,4-ди-трет-бутилциклопен-
теидион-1,3 322
Хлордиметилсульфид 153, 154 616—
617
Хлордифеиилфосфин 294
Хлордифторметан 306
а-Хлор-а'-изонитрозоацетон 371—372
м-Хлориодбензол 119
Хлориридиевая кислота 617
Хлорирование 72, 238—239, 470 614—
615, 621
Хлористый водород 45, 617—618
1-Хлоркамфен 361
З-Хлоркарбазол 615
а-Хлоркетоны 635—636
1-Хлор-3-метилбутен-2 495, 496
2-Хлор-2-метил-1,3-диоксол ан 604
1 -Хлор-2-метилен-3,3-диметилби-
цикло-[2,2,1]-гептан 361
Хлорметилирование 600, 618
бис-Хлорметилирование 618
Хлорметилметиловый эфир 343, 618—
619
бис-Хлорметиловый эфир 618
у-Хлорметилпиридина хлоргидрат
619—620
З-Хлор-2-метилтетрагидропиран 352
л(-Хлорнадбензойная кислота 46, 258,
272, 523, 620—621
1-Хлорнафталия 326
Хлорная кислота 472, 473
2-Хлор-5-нитробензойной кислоты
этиловый эфир 162
экзо-2-Хлорнорборнан 469
а-Хлорнорборпанон 301
Хлороформ 37, 38, 103, 307
2-Хлорпентанон 301
1-Хлорперфторолефины 629
N-Хлорполималеинимид 621
а-Хлорпропаналь 630
3-Хлорпропионовой кислоты хлоран-
гидрид 16, 363
N-Хлорсукцинимид 153, 157, 167, 180,
621, 653
Хлорсульфоизоцианат 572, 592, 621 —
625
а-Хлорсульфоксиды 109
4-Хлорсульфонил-10,10-диметил-4-
азатрииик-ло-[5,2,1,01>5]-деканон-3
624
3-Хлорсульфонил-2,8,8-триметил-3-
азатрицикло-[5,1,1,03’5|-нонаион-4
622
4-Хлорсульфонил-3,3,9,9-триметил-4-
азатрицикло-[6,1,0,03’6] -нонанон-5
624
1-Хлор-М,М,2-триметилпропениламин
625—626
Хлортрифторэтилен 579, 626—627
Хлортропилия соли 577
Хлоругольной кислоты амидины 418,
419
Хлоругольной кислоты трет-бутило-
вый эфир 81, 135, 579, 580
Хлоругольной кислоты рДр-трихлор-
этиловый эфир 627
Хлоругольной кислоты 9-флуорепил-
метиловый эфир 627
Хлоругольной кислоты этиловый
эфир 259, 267, 395, 627
Хлоруксусная кислота 559
N-Хлоруретан 208
707
4-Хлор-2-фенилхиназолин 221 628
л1-Хлорфенол 119
Хлорформамидиняя соли 206
Хлорфторкарбен 599
Хлор фтор метилентрифенилфосфоран
629
6-Хлорфульвен 201—202
Зр-Хлор-Д5-холестен 345
Хлорциан 629—630
З-Хлорциклобутанкарбоновая кислота
431, 468
2-Хлорциклогексанон 207
З-Хлорциклогексен 155
а-Хл о p-N-циклогексил ацетальдонит-
рон 630
а-Хлор-Х-циклогексилпропанальдо-
нитрон 630—633
Хлорциклогептатриены 577, 578
а-Хлорциклододеканон 636
Хлорциклооктатетраен 67
3-Хлорциклопропен 517
2-Хлорциклотридеканон 207
7-Хлорэнантовая кислота 367
р-Хлорэтилацетат 604
Холевая кислота 617
Холевой кислоты метиловый эфир 415
Д3.5-Холестадиен 345
Холестан 377
Зр-Холестанилацетат 238, 239
Холестанол 174, 558
ЗВ-Холестанол 210
Холестанон-3 377
Д3-Холестен 422, 428, 436
дв(14)-Холестен 428
Д4-Холестенол-ЗР 137
Д4-Холестенолацетат 238, 239
Д9(Н).Холестенол ацетат 238, 239
Д^Холестенон-З 428
Д4-Холестенон-3 522
Д4-Холестенон 226
Д5-Холестенон-3 596
Холестерилацетат 61
Холестерилдихлорфосфат 601—602
Холестерилметилфосфит 174
Холестерин 174, 210, 522, 601, 602
Д4-Холестон-3 148
Хорнера—Виттига реакция 75
Хрома (II) — аминов комплексы 633
Хрома (II) ацетат 633—634
Хрома гексакарбонил 345
Хрома(У1) окиси — пиридина ком-
плекс (реактив Коллинза) 168, 413,
525, 634, 637
Хрома(П) перхлорат 633
Хрома(Ш) хлорид 134
Хромила хлорид 635—636
Хромовая кислота 636
Хромовый ангидрид 636—638
Хромовый ангидрид в графите 637—
638
Хромовый ангидрид — пиридин 289,
636—637
Хромон 173
Хуанг—Минлона восстановление 101
Хунсдикера реакция 437, 438
Хунсдикера—Кристола реакция 431—
432
Цезия фторид 606
Церий аммиакат нитрата, см. Гекса-
нитратоцеррат(1У) аммония
Церия(III) ацетат 639
Церия гидроокись 96
Церия нитрат 96
Церия тетраацетат 639
Цетил три мети л а мм они их лор ид 39
Цефалоспорины 74
(о-Цианаминокислоты 391
Цианамид 183
п-Цианбензолсульфамид 381
Цианборирование 379
2-Циангексен-5-овой кислоты этило-
вый эфир 123
Циангидрины 216
^мс-9-Циандекалон-2 248
Цианистый водород 131, 390, 531,
580, 581, 639—640
Цианметилентрифенилфосфоран 540
Цианметилиден-бис-(трифенил фос-
фония) дибромид 640
Цианметиллитий 641
Цианметилмедь 641
5-Циан-6-метилтио-1 -кето-1,3-тиази-
ноны-4 420
1-Циан-2-метилциклогексанкар боно-
вой кислоты этиловый эфир 44
7-Циан-Д1<®-окталинон-2 571
транс-2-Цианоктен-б-овой кислоты
этиловый эфир 44
Циансилилирование 528
8-Циантетрацикло-[4,3,0,0г.4,03'7]-
нонан 544
Циануксусной кислоты этиловый
эфир 44
а-Цианциклогексилтриметилсилило-
вый эфир 531
а-Цианциклопропанкарбоновой кис-
лоты этиловый эфир 123
Циклизация 23, 24, 44, 143, 146, 164
183, 248—249, 280—281, 404-405,
411, 417—418, 433, 517—518, 547—
548, 563—564
Циклические карбонаты 350
Циклобутадиен 96—97
Циклобутадиенжелезотрикарбонил
96, 97
Циклобутаи-1,1-дикарбоновая кисло-
та 468
Циклобутан-1,2-дикарбоновой кисло-
ты динитрил 28
708
грдяс-Циклобутандиол-!,2 50
цис-Циклобутандиол-1,2 50
Циклобутандион-1,2 54—55
Циклобутанкарбоновая кислота 468
Циклобутанон 89, 300, 561
Циклобутаноны 57—58, 82, 83, 202,
203, 626
Циклобутендионы 626
Циклобутен-1 -карбоновой кислоты
нитрил 28
Циклобутены 283—284, 626
Циклогексадиен-1,3 446, 643
Циклогексадиен-1,4 68
Циклогексадиена-1,4 моноокись 149
Циклогексан диол-1,2 41, 118, 253
Цитслогександиол-1,3 423
Циклогександиол-1,4 423
Циклогександион-1,2 292
Циклогександион-1,4 511
Циклогексанол 119, 167, 168, 255, 376,
557, 638
Циклогексанон 79, 80, 93, 118, 136,232,
255, 275, 276, 302, 303, 322, 335,
376, 378, 473-474, 518, 521, 535,
554, 575, 576, 582—583, 586, 638
Циклогексанона оксим 99
Циклогексанона тозилгидразон 514
Циклогексанона этиленацеталь 535
Циклогексанона этилентиокеталь 575
Циклогексен 25, 37, 40, 55, 82, 222
238, 253, 311, 422, 425, 426, 470
471, 524—525, 553, 631, 636
Циклогексена окись 70—71, 336, 362,
509, 553, 554, 636
Циклогексена эписульфид 422
Циклогексен-2-илацетат 311
М-(Циклогексен-1-ил-1)-пирролидин
Циклогексен-2-ол-1 155
Циклогексен-2-он-1 237, 491, 512—513
Ци кл о гексен-1-ои-З 214
Циклогексилазид 589
Циклогексиламин 291—292
N-Циклогексиланилин 583
Циклогексилат алюминия 641—642
Циклогексилбромид 119, 589
N-Циклогексилгидроксиламин 630
Циклогексилдиметиламин 93
Циклогексилдихлор боран 589
Циклогексилиденкарбен 39
Циклогексилиодид 557
Циклогексилкарбинол 259
Циклогексилкарбоксальдегид 159
160
1-Циклогексилциклогексанол 63
Циклогептанон 71, 195, 196, 275, 376
1,2-бис-(Циклогептатриенил-3)-этан
308
трсяс-Циклогептен 295
Циклогептена окись 295
Циклогептеноны 225
Циклодегидратация 243, 658
цис, цяс-Цикл о дека диен -1,6 353
Циклододекан 434
Циклододекандиол-1,2 41, 552
Циклододекандион-1,2 552
Циклододеканолйд 351
Циклододеканон 207, 325, 351
Циклододекатриен-1,5,9 433—434
Циклододецен 41, 88, 253, 433—434,
636
Циклододецена окись 88
Циклононадиен-1,2 82, 83
Циклононатетраенид . лития 330, 331
Циклооктадекагексаин-1,3,7,9,13,15
70, 312, 313
Циклооктадиен-1,3 348
^ы^г^йяс-Циклооктадиен-1,4 295
цис.цис,-Циклооктадиен-1,5 321
цис,траяс-Циклооктадиен-1,5 295, 321
Циклооктадиен-1,5 69, 642
бис- (Циклооктадиен-1,5) -никель (0)
642—644
ц«с-Циклооктандиол-1,2 41
qwc-Циклооктандиол-1,5 69
Циклооктандиола-1,2 бяс-гидразон
432
Циклооктанон 376
Циклооктатетраен 592
Циклооктатриен-1,4,6-дикарбоиовой-
1,2 кислоты диметиловый эфир 275
({«с-Циклооктен 41, 82, 320
тряяс-Циклооктен 82, 294, 295, 320—
321
Циклооктен 170, 426
цяс-Циклооктена окись 294, 295
Циклооктена окись 306
Циклооктин 184, 384, 387, 432, 465,
566
Циклопентадиен 66, 201, 202, 330,
340, 539, 540, 645
Циклопентадиениллитий 330
Циклопентадиенилталлий (I) 657
Циклопентадиепилтрибензилтитан
625, 644—645
Циклопентандион 293
Циклопентанкарбоновая кислота 473,
474
Циклопентанон 195, 207, 374, 412,
554
Циклопентен 426
Циклопентена окись 70—71
Циклопентенил-1 -изобутилкетон 513
Циклопентенонов тиокетали 182
Циклопентен-2-он 237
Циклопентеноны 225—226, 512—513
583
Д3-Циклопентеноны 182—183
Циклопентин 187
709
Циклоприсоединение 82— 83, 132, 179,
180, 191, 202, 203, 263 339—340,
446—447, 539, 581, 582, 590—592,
621—625, 630—632
Циклопропанкарбоксальдегид 50, 569
Циклопропанонов кетали 309
Циклопропаны 244—245, 314—315
Циклопропен 645
Циклопропенон 517
Циклопропеноны 65
Циклопропены 37, 283—284, 426
Цнклопропилазиды 426
Циклопропилацетилен 67
Циклопропилдифенилсульфония бор-
фторид 194, 195
Циклопропилкетоны 67—68, 209,
655—656
Циклопропилметилкетон 195
а-Циклоп р опил стирол 492, 591
Циклопропилфенцлсульфид 135
цис,ц«с-Циклоундекадиен-1,6 352, 353
Циклоундеканон 355, 356
Ци клоун децен-1 -карбоновой кислоты
метиловый эфир 326, 355
[2,2]-«-Циклофаны 32—33
Зсх,5-Цикло-5я-холестан 284
а-Циклоцитраль 459—460
Зя,5а-Цшслоэргоста диен-7,22-он-6
242
Цинк 177, 202, 309, 398, 553, 570, 571,
586, 612, 616, 627, 635, 645—649
Цинка бромид 57
Цинка иодид 528
Цинка карбонат 649—650
Цинк — ангидрид кислоты — катали-
затор 650—652
Цинка окись 630
Цинк-медная пара 126, 463
Цинк-серебряная пара 463, 464
Цинк хлористый 618
Циннамилацетат 430
Циннамилмеркурацетат 430
2-Цимнамилфенол 47
tt-Цимол 250, 264, 563
Цитраконовой кислоты диметиловый
эфир 546
Цитраконовой кислоты нитрил 27—
28
Цитраль 459—460, 551
Цитронелл аль 166, 167, 551
(+)-Цитронеллаль 547, 548
Цитронелловой кислоты хлорангид-
рид 518
Чугаева реакция 163—164
Шиффа основания 25
Шмидта реакция 355—356
Щавелевая кислота 131, 342, 488, 489,
523
Щавелевоуксусная кислота 373, 374
а-Эйдесмол 451—452
р-Эйдесмол 144, 147, 451—452
Эквиленин 246
Экзальтон 103, 104
Элемол 451—452
Эмодин 250
Эпиаллогибберовая кислота 78
Эпибромгидрин 117
6-Эписантонин 119
Эписульфиды 75, 422
2р,Зр-Эпитио-5а-холестан 422
Эпихлоргидрин 335. 336, 653
2,3-Эпоксибутан 346, 545, 605
11,12-Эпоксивитамина А альдегид
350, 351
8,9-Эпокси-2,6-диметилнонсн-2 167
Эпоксидирование 60—61, 234, 258,
345, 346, 620
Эпоксиды (оксираны) 90 161, 186,
286—287, 294—295, 300, 552—553,
554
у,б-Эпоксикетоны 655
1,2-Эпоксиоктан 553
Эпоксиолефины 166—167
цис- и транс-9,10-эпоксистеариновые
кислоты 161
22,23-Эпоксистигмастерилацетат 88
10,11-Эпоксифарнезилацетат 60, 61
4р,5р-Эпоксихолестанон-3 633
Эпоксициклогексан, см. Циклогексена
окись
3,4-Эпоксициклогексен-1 149
д!,4,6,22 -Эргостатетраенон-3 200
Д4,в,8(14),22.ЭрГ0Статетраен0Н-3 200
Эргостатриен-4,6,8(14)-он-3 397
Эргостерилацетат 534
Эргостерилацетата перекись 534, 535
Эргостерина эпидиокись 397
Эргостерон 200
17|3-Эстрадно л 246
Эстриол 246
Эстрогены 199
Эстрон 246, 398, 481
Эстрона метиловый эфир 450
Этанол 395, 629
Этаноламин 653—654
Этерификация 51, 54, 94—95, 157,
174, 243, 327, 529, 575
Этила перекись 654—655
Этилат натрия 655—656
Этилат таллия (1) 656—657
N-Этилбензиламин 223
Этил бромистый 269
Этилвиниловый эфир 292, 582, 657-—.
658
710
траяС-4-Этилдецен-5 142
Этилдиизопропиламин 398, 658
1 -Этил-3- (З'-диметиламинопропил) -
карбодиимида хлоргидрат 46
Этилен 16, 329, 363, 425, 644
Этилен бромистый, см. 1,2-Дибром-
этан
Этиленгликоль 579
Этилендиамин 494, 633, 658—659
Этилендиаминтетрауксусная кислота
347
Этилендиаминтетрауксусной кислоты
динатриевая соль 429
Этилендитозилат 522
Этиленкарбонат 545, 564
Этиленкетали 430
Этилентиокетали 611—612, 616
Этилизопропенилкетон 240
1 -Этил-4-карбметоксипиридинил 357
1-Этил-4-карбметоксипиридиния
иодид 357
Этилмагиийбромид 305, 309
9-Этил-Ю-метилантраден 491
9-Этил-10-метил-9,10-дигидроантра-
цен 491
3-Этил-7-мети л-транс, цис-но на ди ен-
2,6-ол-1 284, 285
ц«с-2-Этил-3-метилпеитадиен-1,3 236
Этилмеркапган 381
2-Этилнафталин 371
N-Этилфталимид 627
7-Этилциклогептатриеи-1,3,5 222
Ы-(1-Этнлциклогексил)-1,1,3,3-тетра-
метилбутиламип и его литиевое
производное 303
Этинилыагиийбромид 321
Этинилмочевины 206
2-Этинилтиофен 64
17а-Этинилэстрадиола монометило-
вый эфир 450
Этоксиацетилен 581
2-Этоксибензазепин 32
2-Этокси-1-метилипдол 32
3|3-Этокси-Д5-холестен 345
Эфиры простые 18, 163
Эшвейлера—Кларка реакция 600
Cis-Ювенильный гормон Cecropia 61,
284, 342
Янтарной кислоты диметиловый эфир
226
Янтарной кислоты динитрил 102
Янтарной кислоты диэтиловый эфир
660
Янтарной кислоты тетраметилдиамид
660
Янтарный альдегид 342
Янтарный ангидрид 232, 412, 660—
661
Яппа—Клингемана реакция 398
СОДЕРЖАНИЕ
Предисловие авторов ................................................ 5
Введение ........................................................... 7
Основные сокращения...............................................* 7
А
Азиридин.......................................................
Азодикарбоновой кислоты диэтиловый эфир....................... 11
Азота четырехокись..................................................12
Азотная кислота . ..................................................12
Алюминий............................................................13
Алюминия бромид .................................................. 14
Алюминия бромид, in situ ...........................................14
Алюминия окись......................................................15
Алюминия хлорид.....................................................15
Амидины бициклические...............................................20
Амид калия..........................................................23
Амид натрия....................................................... 24
Амид натрия — трет-бутилат натрия................................. 24
трег-Амила гидроперекись............................................25
трет-Амил-8-хинолилкарбонат.........................................25
п-Аминоацетофенон ..................................................26
4-Амино-3-гидразино-5-меркапто-1,2,4-триазол...................... 26
2-Аминотиазол..................................................... 27
Антрахинон..........................................................27
Аскарит.............................................................28
Ацетилен дикар боновой кислоты диметиловый эфир.....................28
1-Ацетил-1-метилгидразин............................................34
Ацетилсерная кислота .............................................. 34
N-Ацетилтиомочевина.................................................34
Ацетил-га-толуолсульфонат...........................................35
Ацетофенон..........................................................35
Б
Бария гидроокись.................................................. 36
Бензилтриэтиламмонийхлорид............................................37
1.3,2-Бензодиоксаборол (пирокатехинборан).............................42
Бензоила перекись................................................. . 44
Бензоина енольной формы циклический карбонат (ди фе ни лэти л бикарбо-
нат) ..............................................................45
Бензолсульфонилазид...................................................46
бис-(Бензонитрил) палладий (II)дихлорид . . 47
Берча восстановление..................................................48
712
9-Борабицикло-[3,3,1]-нован '(9-ББН)'.......................... . 49
Бора трибромид...................................................49
Бора трис-(трифторацетат) .......................................49
Бора трифторида ди-н-бутилэфират.................................50
Бора трифторида эфират...........................................50
Бора трифторид — трифторуксусный ангидрид........................52
Бора трихлорид...................................................52
Борная кислота.................................................. 54
Бром............................................................ 54
N-Бромацетамид (NBA)..............................................55
N-Бромацетамид — фтористый водород................................56
Бромистоводородная кислота . ~....................................56
Бромистый водород . .................... 57
Бромистый водород безводный.......................................58
Бромистый водород — трифторуксусная кислота.......................58
N-Бромполималеинимид (ЫБПМИ)......................................58
N-Бромсукцинимид..................................................59
Бромциан..........................................................63
Бутадиена-1,3 1,2-окись ..........................................64
н-Бутиламин.......................................................64
трет-Бутилат калия................................................64
трет-Бутилгипохлорит..............................................72
к-Бутиллитий......................................................74
трет-Бутиллития — комплекс с Н,Н,Н',Н'-тетраметилэтилендиамином . . 77
трет-Бутилмагнийхлорид . 78
н-Бутилмеркаптан . 78
1-трат-Бутилоксикарбонилтриазол-1,2,4.............................80
трет-Бутил-8-хинолилкарбонат.................................... 81
трет-Бутилхромат..................................................81
трат-Бутилцианкетен...............................................82
В
Винилмагнийхлорид .............................................. 85
Висмута триацетат.................................................85
Виттига реакция...................................................86
Вольфрама гексахлорид .......................................... 87
Г
Газы инертные.....................................................89
Галлия окись ................................................. 89
Гексаметилентетрамин (уротропин)..................................89
Гексаметилтриамид фосфористой кислоты.............................90
Гексаметилтриамид фосфорной кислоты (гексаметапол, ГМТФК) ... 91
Гексанитратоцеррат(1У) аммония (ГПА)..............................95
Гексахлорциклотрифосфазатриен (фосфонитрилхлорид).................99
Гексаэтилтриамид фосфористой кислоты [трис-(диэтиламино)-фосфин] . .100
Гидразин.........................................................101
Гидроксиламин-О-сульфокислота....................................101
Глутаровой и янтарной кислот динитрилы....................... 102
Дегидро-N, ^№'-трициклогексилгуанидиногексакарбонилдижелезо(0) . .
1,5-Диазабицикло-‘4.3.01-нонен-5 (ДБН)................................
1,4-Диазабицикло-[2.2,21-октан (ДАВНО)................................
1,5-Диазабицикло- [5,4,0] -ундецен-5 (ДБУ)............................
Диазоапетальдегид . ..................................................
Диазометан ...........................................................
103
103
107
107
108
108
713
Диазопропионовой кислоты метиловый эфир...........................110
Диазоуксусной кислоты азид........................................111
Ди аз о уксусный эфир.............................................112
Диалкоксикарбония борфториды .....................................113
Диацетила и триметилфосфита аддукт (2Д,2-трнметокси-4,5-диметил-1,3-
диоксафосфолен) .................................................ИЗ
Дибензилкетон.....................................................116
Диборан ..........................................................116
1,2-Дибромэтан (этилен бромистый).................................119
2,6-Ди-трет-бутил-а-диметиламино-га-крезол (этилаитиоксидант 703) . . • 120
Ди-трет-бутилиминоксил............................................121
Ди-н-бутилмедьлитий .....................'........................121
Ди-трет-бутилпирокарбонат.........................................121
Дивинилмедьлитий..................................................122
Дивинилмедьлития комплекс с три-н-бутилфосфином...................124
Диизобутилалюминийгидрид..........................................125
Диизопинокамфилборан..............................................128
М,М'-Диизопропилгидразин..........................................129
Диимид........................................................... 130
Дииминоянтарной кислоты динитрил (дииминосукцинонитрил)...........131
Дикобальтоктакарбонил.............................................134
2,2'-Дилитийдифенил...............................................134
4-Диметиламино-1-трет-бутилоксикарбонилпири диния хлорид..........135
1,8-бис-(Диметиламино)-нафталин...................................135
ТфМ-Днметиламиноциклопропилфенилсульфоксония борфторид............135
Диметилацетамид.................................................. 136
Диметилацетамида дпметилацеталь...................................137
бис- (3,6-Диметил)-борепан........................................139
Диметилбромсульфония бромид.......................................140
2,3-Диметилбутил-2-боран (тексилборан)............................140
Диметил-трет-бутилхлорсилан.......................................142
Диметилдихлорсилан .............................................. 143
Диметнлмедьлитий ................................................ 144
ГДЫ-Диметилметиленаммония иодид...................................150
N,N-Диметил мети ленаммония три фтор ацетат.......................152
бис- (1,2-Диметилпропил-1) -боран («диизоамилборан»)..............152
Диметил-М-сукцинилсульфоиия хлорид................................152
Диметилсульфат....................................................155
Диметилсульфат— калия карбонат . . ........... . . 157
Диметилсульфид....................................................157
Диметилсульфид—боран..............................................159
Диметилсульфид — хлор........................................... 159
Диметилсульфоксид.................................................160
Диметилсульфоксида производные, а) Метилсульфинилметилиднатрий
(димсилнатрий). б) Диметилсульфонийметилид. в) Диметилсульфоксо-
нийметилид......................................................162
Диметилсульфоксид—N-бромсукцинимид (ДМСО—NEC).............167
Диметилсульфоксид — серы трехокись ...............................168
Диметилсульфоксид—уксусный ангидрид...............................169
Диметилсульфоксид — хлор..........................................170
2,4-Диметилтиазол.................................................171
Диметилтиокарбамоилхлорид.........................................171
Диметилформамид...................................................171
Диметилформамида диметилацеталь...................................172
Диметилформамид — тионилхлорид....................................173
Диметилфосфит.....................................................174
3,5-Диметил-4-хлорметилизоксазол..................................175
2,4-Диметоксибензиламип...........................................175
2-(2,2-Диметоксиэтил)-1,3-дитиан..................................175
1,2-Диокси-З-бромпропан (сс-бромгидрин) . . ..............177
714
4,4/-Диокси-2,6,2/,6/-7’е7’рак:«с-(трет-бутил)’-дифенилметан («этил антиокси-
дант 702»)..........................................................177
3,4-Диоксициклобутен-3-дион-1,2 (квадратная кислота)...............177
4(4-Диоксо-2-метокси-6-метил-1-окса-4-тиа-3,5-диазин...............178
1,3-Дитиан.........................................................18о
1,3-Дитиения борфторид........................................... 182
Дитиоугольной кислоты N-цианимида дикалиевая соль..................183
Дитиоциан (родан)..................................................183
Дифенилдиазометан................................................ 184
Дифенилди-(1,1,1,3,3,3-гексафтор-2-фенил-2-пропокси)-сульфуран . . .184
1,3-Дифенилизобеизофуран...........................................186
Дифенилиодоний-2-карбоксилата моногидрат...........................187
РфМ-Дифенилкарбамоилпиридиния хлорид...............................191
Дифенилкетен.......................................................191
Дифенилсульфонийциклопропилид .................................... 191
Дифенилфосфит..................................................... 196
Дифенилфосфорной кислоты азид..................................... 196
Дихлорборан........................................................197
Дихлорборана диэтилэфират ........................................ 197
2,3-Дихлор-5,6-дициан-1,4-бензохинон (ДДХ).........................198
Дихлоркарбен ..................................................... 20]
Дихлоркетен........................................................202
Дихлормалеиновый ангидрид ........................................ 20з
Дихлорметилендиметиламмония хлорид.................................204
Дихлорметиллитий.................................................. 207
Дихлорсилан........................................................207
N.N-Дихлоруретан . ................................................208
Дицианацетилен................................................... 208
Дициклогексилборан.................................................20g
Дициклогексилкарбодиимид (ДЦК)................................... 209
Дициклогексилкарбодиимида иодметилат...............................210
Дициклогексил-18-краун-эфир-6......................................21о
Диэтил алюминийхлорид..............................................212
Диэтилалюминийцианид............................................. 215
Диэтиламин.........................................................216
Диэтилизоцианметилфосфонат.........................................217
Диэтил-(1-метилтио)-этилфосфонат...................................218
Диэтил'(1,1,2-трифтор-2-хлорэтил)-амин........................... 219
Диэтилхлорфосфат...................................................221
Диэтилцианметилфосфонат........................................... 221
Диэтилцинк — йодоформ..............................................222
Диэтилцинк — метилен иодистый.................................... 222
Диэтоксикарбония борфторид.........................................222
Ж
Железа(Ш) ацетилацетонат...........................................224
Железа додекакарбонил .............................................224
Железа нонакарбонил.............................................. 225
Железа пентакарбонил...............................................226
Железа(И) сульфат..................................................228
Железа(Ш) хлорид...................................................229
Железа (111) хлорид — диметилформамид..............................229
И
Изоамилнитрит......................................................231
Изовалин (а-амино-сс-метилмасляная кислота) .......................231
Изопропенилацетат..................................................232
Изопропилат алюминия...............................................232
715
'(—J - и (+J -2,3-0-Изопропилиден-2,3-диокси-1,4-бш> (дифенилфосфиио)’ -
бутан..............................................................233
Иминонадбензойная кислота.........................................233
«Инданоциклон».....................................................234
Иод............................................................... 234
Иодазид............................................................237
Иода трис-(трифторацетат)..........................................238
Иодбензола дихлорид................................................238
Иод — калия иодид..................................................240
Иодная кислота ................................................... 241
Иодозобензола диацетат ............................................ 241
Йодоформ...........................................................241
Иод—серебра ацетат.................................................242
Иодэтинилтриметилсилан ........................................... 242
Ионообменные смолы.................................................243
Ионообменные смолы, основные.......................................244
К
Кадмия карбонат , . ...............................................246
Калий в графите..................................................246
Калия боргидрид..................................................247
Калия гексаметилдисилазан [калия бис- (триметилсилил) -амид] .... 247
Калия гексахлоровольфрамат(1У) ................................ 249
Калия гидрид................................................... 249
Калия гидроокись.................................................250
Калия (натрия) иминоксилдисульфонат (соль Фреми).................252
Калия иодид — цинк-медная пара...................................252
Калия осмат..................................................... 253
Калия перманганат — уксусный ангидрид..............................253
Калия перхромат..................................................254
Калия тетрахлороплатинат(II).....................................254
Калия феррат(у!)............................................... 254
Калия феррицианид................................................255
Кальция карбонат ................................................. 256
d-10-Камфорсульфокислота...........................................257
п-Карбметоксинадбензойная кислота ............................... 257
бнс-(Карбэтоксидиазометил)-ртуть...................................258
2-Карбэтокси-1,3-дитиан............................................258
N-Карбэтоксиизотиоцианат...........................................259
Карбэтоксиметилмедь................................................260
4-Карбэтокси-5-метил-3-этилизоксазол...............................261
М-Карбэтоксн-2-этокси-1,2-дигидрохинолин (КЭД).....................262
Кетен............................................................ 262
Кислород ..........................................................262
Кислород синглетный................................................263
Кобальта(II) ацетат .............................................. 264
бнс-(Кобальттетракарбонил)цинк.....................................265
Кори восстановитель................................................ . 266
Л
Линдлара катализаторы..............................................268
Литий..............................................................268
Литий — алкиламины.................................................270
Литий—аммиак.......................................................271
Литий—трет-бутанол—тетрагидрофуран.................................274
Литийдиметилкарбамоилникельтрикарбонил.............................2 74
Литийметилизонитрил ............................................... 275
Литий — нафталин...................................................275
2-Литийпропилиден-М-трет-бутилимин ................................277
716
2-Литий-2-триметилсилил-1,3 дитиан . ...............278
Литий — этилендиамин..................................................280
Лития алюмогидрид . .............................................280
Лития алюмогидрид — алюминия хлорид (алан).......................283
Лития алюмогидрид — натрия метилат...............................284
Лития алюмогидрид — пиридин [лития тетракис-(N-дигидропиридил) алю-
минат] ..........................................................285
Лития амальгама................................•..................285
Лития боргидрид....................................................286
Лития бромид.......................................................286
Лития трег-бутилат.................................................287
Лития трис-(втор-бутил)-боргидрид .................................287
Лития трис- (трет-бутокси) -алюмогидрид............................288
Лития гидрид ......................................................289
Лития диизопропиламид..............................................290
Лития дифенилфосфид (ЛДФ)..........................................294
Лития диэтиламид...................................................295
Лития N-изопропилциклогексиламид...................................296
Лития иодид........................................................300
Лития иодид—бора трифторид.........................................300
Лития карбонат—лития галогенид.....................................30]
Лития пергидро-ЭЬ-борафеналилгидрид................................301
Лития 2,2,6,6-тетраметилпиперидид..................................302
Лития тетрахлоркупрат..............................................304
Лития триэтилборгидрид............................................ 305
Лития триэтилметнлат...............................................ЗОб
Лития фосфат.......................................................307
М
Магний...................................................... 308
М,М-бис-Магнийбром анилин............................
Магния алюмогидрид . ................................
Малеиновый ангидрид..................................
Малононитрил...................................... . .
Марганца(Ш) ацетат...................................
Марганца двуокись активная ..........................
R- (—) -Мевалонолактон...............................
Меди(II) ацетата моногидрат..........................
Меди(1) ацетилацетонат...............................
Меди(1) трег-бутилат.................................
Меди закись — трет-бутилизонитрил....................
Меди карбонат основной ..............................
Меди (II) метилат....................................
Меди (II) нитрат—пиридин.............................
Меди окись . .....................................
Меди(1) фенилацетиленид..............................
Медная бронза .......................................
Медный порошок.......................................
Медь бромистая.......................................
Медь(1)тетрапиридинперхлорат.........................
Медь хлористая.......................................
Медь хлорная.........................................
Метансульфокислота...................................
Метансульфохлорид (мезилхлорид)......................
Метилат натрия ......................................
Метил-трет-Сгутиловый эфир ..........................
бис- (3-Метил-5-грвг-бутил-4-окси) -дифенилсульфид («этил»
736).................................................
Метилгидразин........................................
309
309
310
310
310
.............312
.............313
.............314
.............314
.............315
.............315
.............316
.............317
.............317
............31 8
.............318
.............319
.............320
............320
.............322
.............323
.............324
.............325
.............327
антиоксидант
............328
.............328
717
О-Метилдвбензофурания борфторид................... . ............329
Метилен хлористый ..............................................329
Метилен хлористый — н-бутиллитий..................................330
Метилизоцианат....................................................332
Метил иодистый....................................................332
Метилмедь '.......................................................332
Метил-(метилтиометил)-сульфоксид............................... . 333
2-Метил-З-тиазолин................................................335
1,3-бис-(Метилтио) -аллиллитий ...................................335
ЬФМетил-И-тозилпирроли диния перхлорат............................338
бУС-(трйяс-2-Метилциклогексцл-1)-боран............................338
2-Метоксиаллил бромистый....................................... 339
а-Метоксибснзилиденвольфрам (О)пентакарбоцил.................... 340
З-Метоксиизопрец . .............................................. 341
Метоксикарбения гексафторантимопат................................343
6-1Метокси-7-окси-3,4-Дигидроизохинолиния иодметилат..............343
Молекулярные сита................................................ 344
Молибдена гексакарбонил ..........................................345
Молибдена пятиокись — гексаметапол (гидрат комплекса) ............345
Монохлорборана диэтилэфират.......................................347
Муравьиная кислота................................................348
Муравьиной кислоты этиловый эфир..................................349
Н
Надбензойной кислоты трет-бутиловый эфир .........................350
Надмуравьиная кислота.............................................350
Надуксусная кислота............................................. 350
Надуксусная кислота — бора трифторида эфират......................351
Надуксусная кислота — серная кислота ............. 351
Натрий........................................................... 352
Натрий — аммиак...................................................352
Натрий — нафталин (нафталенид натрия).............................353
Натрий — фенантрен (фенантренид натрия) . . ......................354
Натрия азид.......................................................355
Натрия алюмохлорид.............................................. 357
Натрия амальгама .................................................357
Натрия боргидрид................................................ 357
Натрия гидрид ................................................... 360
Натрия гидрид — трет-бутилгипохлорит..............................362
Натрия гидрид — диметилсульфоксид.................................363
Натрия гипохлорит.................................................363
Натрия дигидрофосфата моногидрат..................................363
Натрия диэтилалюмодигидрид...................................... 364
Натрия железа (II) тетракарбонил..................................365
Натрия иодид......................................................369
Натрия бис-(2-метоксиэтокси)-алюмогидрид........................ 370
Натрия нитрит.....................................................371
Натрия сульфид....................................................372
Натрия тетраборат ............................................... 373
Натрия тиосульфат ................................................374
Натрия хлорид — диметилсульфоксид.................................374
Натрия цианборгидрид .............................................375
Натрия цианид................................................... 379
Натрия цианид — диметилсульфоксид.................................380
Натрия этилмеркаптид............................................. 381
Никель-алюминиевый сплав (1:1, сплав Ренея).......................381
Никеля борид .................................................... 382
Никеля бромид.....................................................382
Никеля карбонил .............................................. ... 384
718
Никеля цианид......................................................387
Нингидрин (ипдантрион-1,2,3).......................................387
2-Нитро-1 .........................................................388
Нитрозония борфторид...............................................388
Нитрометан ........................................................389
Нитрония борфторид.................................................390
2-Нитропропана 2-гидроперекись.................................... 390
4-(4'-Нитрофенилазо)-бензоилхлорид.................................399
о-Нитрофенилсульфенхлорид..........................................391
Нитроэтан..........................................................392
О
Озон...............................................................393
Окись этилена......................................................393
Оксалилхлорид......................................................394
4-Оксиметил-2,6-ди-трет-бутилфенол («этил» антиоксидант 754) .... 394
Ортомуравьиной кислоты трпметиловый эфир ......................... 39а
Ортомуравьиной кислоты триэтиловый эфир .......................... 395
Ортоуксусной кислоты триэтиловый эфир..............................395
Осмия четырехокись — калия хлорат..................................395
П
Палладиевая чернь.............................................. 397
Палладиевые катализаторы...........................................397
Палладий (10%) на карбонате кальция — катализатор гидрирования . . 399
Палладий на угле . ................................................399
Палладий хлористый ............................................... 400
Палладия (И) ацетат................................................402
1,2,2,6,6-Пенгаметилпиперидин......................................403
1,1,1,3,3-Пентахлоразапропен (трихлорметилизонитрила дихлорид) . . . 403
т’ранс-Пентен-З-он-2............................................. 405
Перекись водорода..................................................406
Перекись водорода — алюминия хлорид................................406
Перекись водорода в кислой среде...................................407
Перекись водорода в щелочной среде.................................408
Перйодаты......................................................... 409
Пикрилхлорид..................................................... 410
Пикриновая кислота .............................................. 411
Пиперазин........................................................ 411
Пиперидин......................................................... 412
Пирен..............................................................412
Пиридин . ................................................... ..... 413
Пиридина хлоргидрат .............................................. 413
б«с-(Пиридин)-диметилформамиддихлорородия боргидрид................414
Пирокатехина дихлорметиленовый эфир................................414
4-(N-Пирролидил)пиридин............................................415
Платина па окиси алюминия (катализатор)............................416
Платинохлористоводородная кислота — олово хлористое................416
Полиметилгидросилоксан........................................... 416
Полифосфорпая кислота (ПФК) .....................................417
Полифосфорпой кислоты эфиры (ПФЭ)..................................420
Пропандитиол-1,3...................................................421
Пропаргиловый альдегид.............................................421
Р
Роданиодид ........................................................422
Родий (5%) на окиси алюминия.......................................422
Родий (5%) па угле (норит).........................................423
Родия окись........................................................424
719
Ртути азид..........................................................425
Ртути(П) ацетат.....................................................426
Ртути окись.........................................................431
Ртути окись — бром ,................................................432
Ртути(II) трифторацетат.............................................433
Рутения трихлорида гидрат . ... ................................... 433
Рутения четыре.хокись...............................................434
С
Свинца (IV) ацетата азиды...........................................435
Свинца диацетат.....................................................437
Свинца тетраацетат ................................................ 437
Свинца тетра-(трифторацетат)...................................... 442
Селен ..............................................................443
Селена двуокись ................................................... 443
Сера двухлористая...................................................446
Серебра борфторид...................................................446
Серебра карбонат . .................................................447
Серебра карбонат — целит............................................448
Серебра нитрат . ...................................................451
Серебра окись ..................................................... 452
Серебра (II) окись ............................................... 453
Серебра перхлорат...................................................454
Серебра сульфат.....................................................458
Серная кислота .................................................... 458
Сернистый ангидрид..................................................461
Серный ангидрид ....................................................462
Серный ангидрид — пиридин . ........................................462
Сероводород ....................................................... 463
Симмонса — Смита реагент............................................463
Соляная кислота ....................................................465
(4-)-(К)-трйнс-р-Стирил-п-толилсульфоксид...........................465
Сульфолен-3........................................................ 467
о-Сульфонадбензойная кислота........................................467
Сульфоуксусная кислота..............................................467
Сульфурил хлористый ............................................... 467
Сурьма пятихлористая .............................................. 470
Т
Таллия (III)’ нитрат................................................471
Таллия (Ш) сульфат..................................................477
Таллия триацетат ...................................................478
Таллия (III) трифторацетат (ТТфА)'..................................478
Теллур четыреххлористый.............................................482
Тетра-(бромметил) -метан............................................482
2,4,4,6-Тетрабромциклогексадиен-2,5-он..............................482
Тетра-н-бутиламмония бромид ...................................... 483
Тетра-я-бутиламмония формиат........................................484
Тетра-я-бутиламмония фторид ........................................485
Тетраметилгуанидин..................................................486
ГфМ.М'.ГД-Тетраметилдиамидохлорфосфат...............................486
2,4,4,6-Тетраметил-5,6-дигидро-1,3-(4Н)-оксазин.....................488
М^КК.М'-Тетраметилэтилендиамин (ТМЭДА)..............................491
Тетрафенилциклопентадиеион (тетрациклон)............................496
Тетрахлорсилан.................................................. 497
Тетрахлорэтан — нитробензол....................................... 498
Тиоацетамид.........................................................498
Тиомочевины диоксид.................................................499
Тиоиил хлористый.....................................,..............499
720
Тионхлоругольной кислоты n-толиловый эфир ...... ...................
Тиофенол"— азодиизобутиронитрил.....................................
Титан треххлористый . ..............................................
Титан четыреххлористый .............................................
Тозилметилизонитрил ................................................
n-Т олилсульфинилметилидлитий.......................................
/i-Толуолсуль фоки слота............................................
n-Толуолсульфонилазид (тозилазид) . ................................
п-Толуолсульфонилгидразин(тозилгидразин) . .........................
я-Толуолсульфохлорид................................................
Треххлористый азот .................................................
Три-н-бутил^-бутен-З-ил-олово.......................................
Три-w-б у тил станнан...............................................
Три-н-бутилфосфина— меди(1) иодида комплекс.........................
Триизобутилалюминий.................................................
2,4,6-Триизопропилбензолсульфокислоты гидразид......................
1,3,5-Триметил-2,4,6-трис-(3,5-ди-трег-бутил-4-оксибензил)-бензол . .
Триметилендитиотозилат .............................................
ЬГ,4,4-Триметил-2-оксазолиния иодид.................................
Триметилоксония борфторид...........................................
2,4,4-Триметилпентил-2-изонитрил....................................
Триметилсилилдиазометан.............................................
Триметилсил ил диэтила мин .........................................
бис- (Триметилсилил) -литийамид.....................................
бис- (Триметилсилил)-натрийамид ....................................
Триметилсилилцианид ................................................
Триметилфосфит .....................................................
Триметилхлорсилан ..................................................
1,2,3-Триоксо-2,3-дигидрофенален....................................
силш-Тритиан .......................................................
Трифениларсина окись................................................
Три фенил мети л бор фторид.........................................
Трифенилметилгексафторфосфат........................................
Трифенилметиллитий..................................................
Трифенилстаннан ....................................................
Трифенилфосфин . ...................................................
Трифенилфосфин — азодикарбоновой кислоты диэтиловый эфир . . . .
Трифенилфосфин — N-галогенсукцинимид................................
Трифенилфосфиндибромид .............................................
бис-(Трифенилфосфин) -никель (II) дицианид..........................
бис- (Трифенилфосфин) кобальт (I I) дибромид........................
бис- (Т рифенилфосфин) -никель (0)..................................
трис- (Трифенилфосфин)-родий (1)карбонилгидрид......................
трис-(Трифенилфосфин) -родийхлорид..................................
трис-(Трифенилфосфин)-родийхлорид — триэтилсилан....................
трис- (Трифенилфосфин) -рутенийдихлорид.............................
Трифенилфосфинселенид ..............................................
Трифенилфосфин — углерод четырехбромистый ...................... .
Трифенилфосфин — углерод четыреххлористый...........................
Трифенилфосфит . ...................................................
Трифенилфосфита иодметилат..........................................
Трифенилфосфита озонид .............................................
Трифенилхлорметан...................................................
Трифторметансульфокислота ..........................................
Трифторметаисульфокислоты ангидрид..................................
Трифторметансульфоновой и карбоновых кислот смешанные ангидриды
Трифторметилгипофторит..............................................
Трифторнадуксусная кислота..........................................
Три фтор уксусная кислота (ТФК).....................................
Трихлорацетилхлорид ...............................................
501
502
502
504
505
508
510
512
512
514
515
515
515
518
519
520
520
521
522
523
523
524
525
526
527
527
528
529
532
533
533
534
536.
537
538
538
541
543
543
544
545
545
545
546
550
551
552
553
554
556
557
559
559
560
561
561
562
562
563
565
721
Трихлоризоциаиуровая кислота (цианурхлорид) .................. ..... 566
Трихлорметиллитий ..................................................566
Трихлорсилан ..................................................... 567
Трихлоруксусная кислота ........................................... 569
Трихлоруксусной кислоты этиловый эфир...............................569
2,2,2-Трнхлорэтапол.................................................570
Триэтилалюминий................................................... 571
Триэтилами .........................................................572
^1\р\-Триэтил-1\Ч<арбметоксисульфамида внутренняя соль — триэтнл-
амин (комплекс).....................................................572
К,1Ч,1\-Триэтил-1Ч'-карбэтоксисульфамида внутренняя соль............573
Триэтилоксония борфторид............................................574
Триэтилсилан . .....................................................576
Триэтилфосфит.......................................................577
Тропилия соли.......................................................577
У
Углерода окись ..................................................... 579
Углерод четыреххлористый.............................................579
Угольной кислоты грет-бутилового эфира азид (трет-бутил азидоформиат) 579
Угольной кислоты трет-бутилового эфира фторангидрид..................580
Угольной кислоты метилового эфира гидразид ......................... 580
Угольной кислоты этилового эфира азид ...... ....................... 581
Уксусного альдегида этил-3-бромп роли л ацеталь......................532
Уксусной кислоты хлорангидрид........................................584
Уксусномуравьиньш ангидрид...........................................585
Уксусный ангидрид — пиридина хлоргпдрат..............................585
Уксусный ангидрид — цинка хлорид.....................................585
Урушибары гидрирования катализаторы..................................536
Ф
Фенплазид.............................*............................587
n-феиилбензонл хлористый...........................................587
Фенил- (1 -бром-1 -хлор-2,2,2-трифторэтил) -ртуть.................;588
Фенилдиазония хлорид ............................................. 588
Фепилдихлорборан ................................................. 589
Фенилизоцианат ..................................................... 590
N-Фенилимид азодикарбоновой кислоты................................590
М,й1-Фенилметилнатрийамид........................................ 593
9-Фепил-9-оксиантрон (тритилоновый спирт)..........................593
Фенилпалладия ацетат ..............................................594
Фенил-ш-стирил-(диметиламиио)-сульфоксопия борфторид...............594
Фенилтиометиллитий ............................................... 595
Фенил-(тригалогенметил)-ртуть.................................... 596
Фепилтриметиламмония пербромид.....................................597
Фенилтрифторметилкетеи.............................................598
Фенил-(трифторметил)-ртуть.........................................599
1-Фенил-5-хлортетр азол............................................600
Формальдегид.......................................................600
Фосфора хлорокись..................................................601
Фосфорная кислота ................................................ 602
Фосфорной кислоты диэтилового эфира серебряная соль................603
Фосфор пятисернистый...............................................603
Фосфор пятихлористый ............................................ 603
Фтористый водород безводный........................................605
Фтористый водород —бора трифторид..................................606
бис-(Фторокси)-дифторметан.........................................606
Фтор сульф оно в ой кислоты метиловый эфир.........................607
722
X
Хинуклидин.........................................................609
а-Хлоракрвловой кислоты хлорангидрид ............................. 609
2-Хлоракрилонитрил............................................... 610
Хлорамин...........................................................611
Хлорамии-Т (N-хлор-п-толуолсульфамид-натрнй).......................611
Хлоранил...........................................................612
о-Хлоранил.........................................................613
«-Хлорбензоилнитрит .............................................. 613
1-Хлорбензотриазол ................................................614
Хлорди метилсульф ид...............................................616
Хлориридиевая кислота ............................................ 617
Хлористый водород..................................................617
Хлорметилметиловый эфир............................................618
у-Хлорметилпиридина хлоргидрат.....................................619
jw-Хлорнад бензоина я кислота......................................620
N-Хлорполималеинимид............................................. 621
Хлорсульфоизоциапат................................................621
1-Хлор-Ь1,Ы,2-триметилпропениламин.................................625
Хлортрифторэтилен..................................................626
Хлоругольной кислоты р,[3,|?-трихлорэтиловый эфир..................627
Хлоругольной кислоты 9-флуоренилметиловый эфир.....................627
Хлоруголыгой кислоты этиловый эфир.................................627
4-Хлор-2-фепилхиназолин........................................... 628
Хлорфторметилентрифенилфосфоран....................................629
Хлорниан...........................................................629
а-Хлор-Ц-циклогексилпропанальдонитрон..............................630
Хрома (11)—аминов комплексы . . ...................................633
Хрома(П) ацетат....................................................633
Хрома (VI) окиси — пиридина комплекс (реактив Коллинза) .... .634
Хромила хлорид ................................................... 635
Хромовая кислота ................................................. 636
Хромовый ангидрид..................................................636
Ц
Церий, аммиакат нитрата............................................639
Церия тетраацетат..................................................639
Цианистый водород..................................................639
Цианметилиден-бис-(трифенилфосфония) дибромид......................640
Цианметилмедь......................................................641
Циклогексилат алюминия ............................................641
б«с-(Циклооктадиен-1,5)-никель (0).................................642
Циклопептадиенилтрибензилтитан ................................... 644
Циклопропен........................................................645
Цинк ............................................................ 645
Цинка карбонат.....................................................649
Цинк — ангидрид кислоты — катализатор..............................650
Э
Эпихлоргидрин ................................................... 653
Этаноламин........................................................653
Этила перекись....................................................654
Этилат натрия ................................................... 655
Этилат таллия (1).................................................656
723
Этилвиниловый эфир..............................................657
Этилдиизопропиламип ........................................... 658
Этилевдиамин....................................................658
Я
Янтарной кислоты тетраметилдиамид ..............................660
Янтарный ангидрид ............................................. 660
Указатель реагентов по типам реакций.............................662
Предметный указатель........................................... 673
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании кни-
ги, ее оформлении, качестве перевода и
другие просим присылать по адресу:
129820, Москва, И-110, ГСП, 1-й Риж-
ский пер. д. 2.