/









































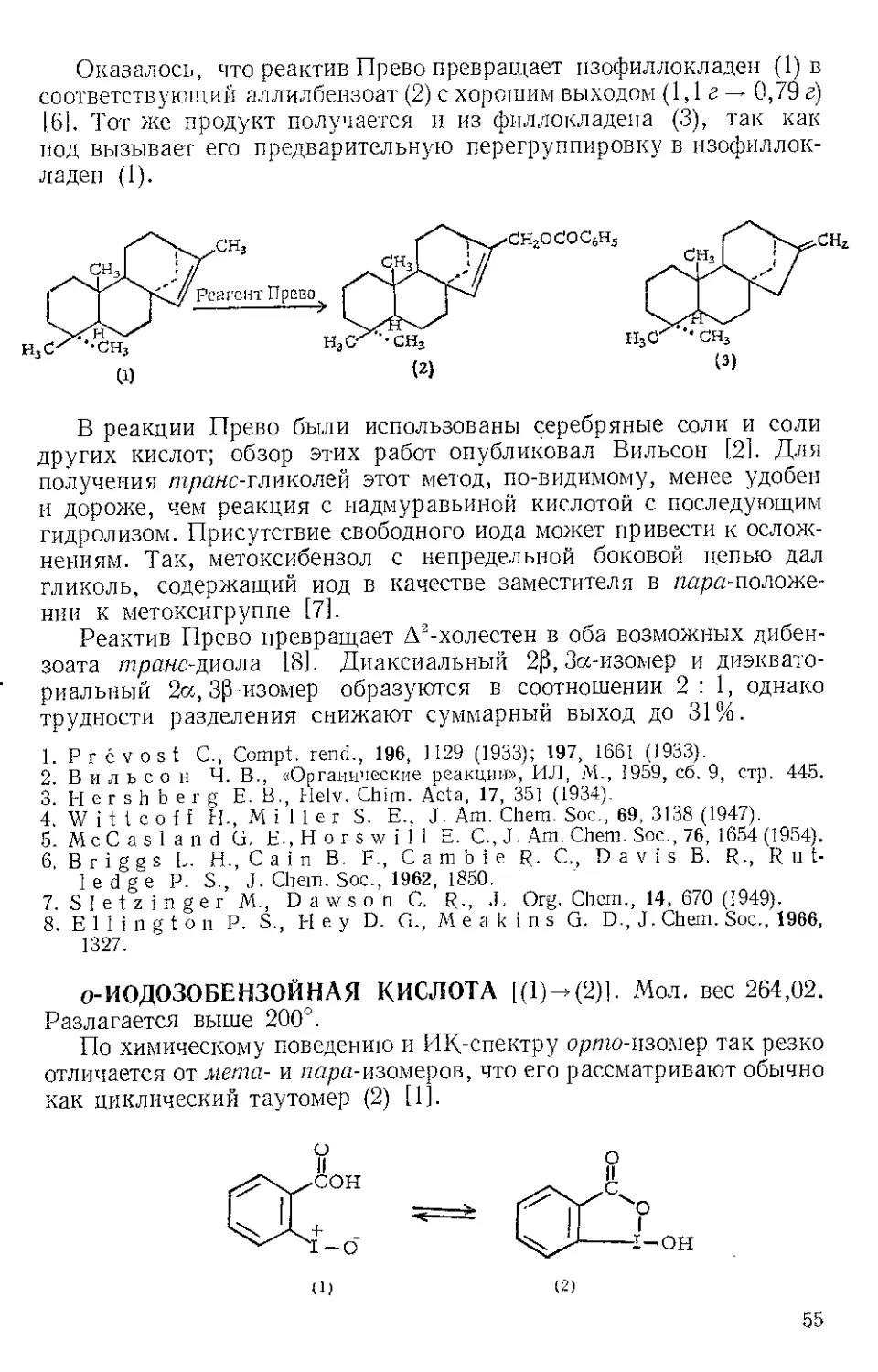













































































































































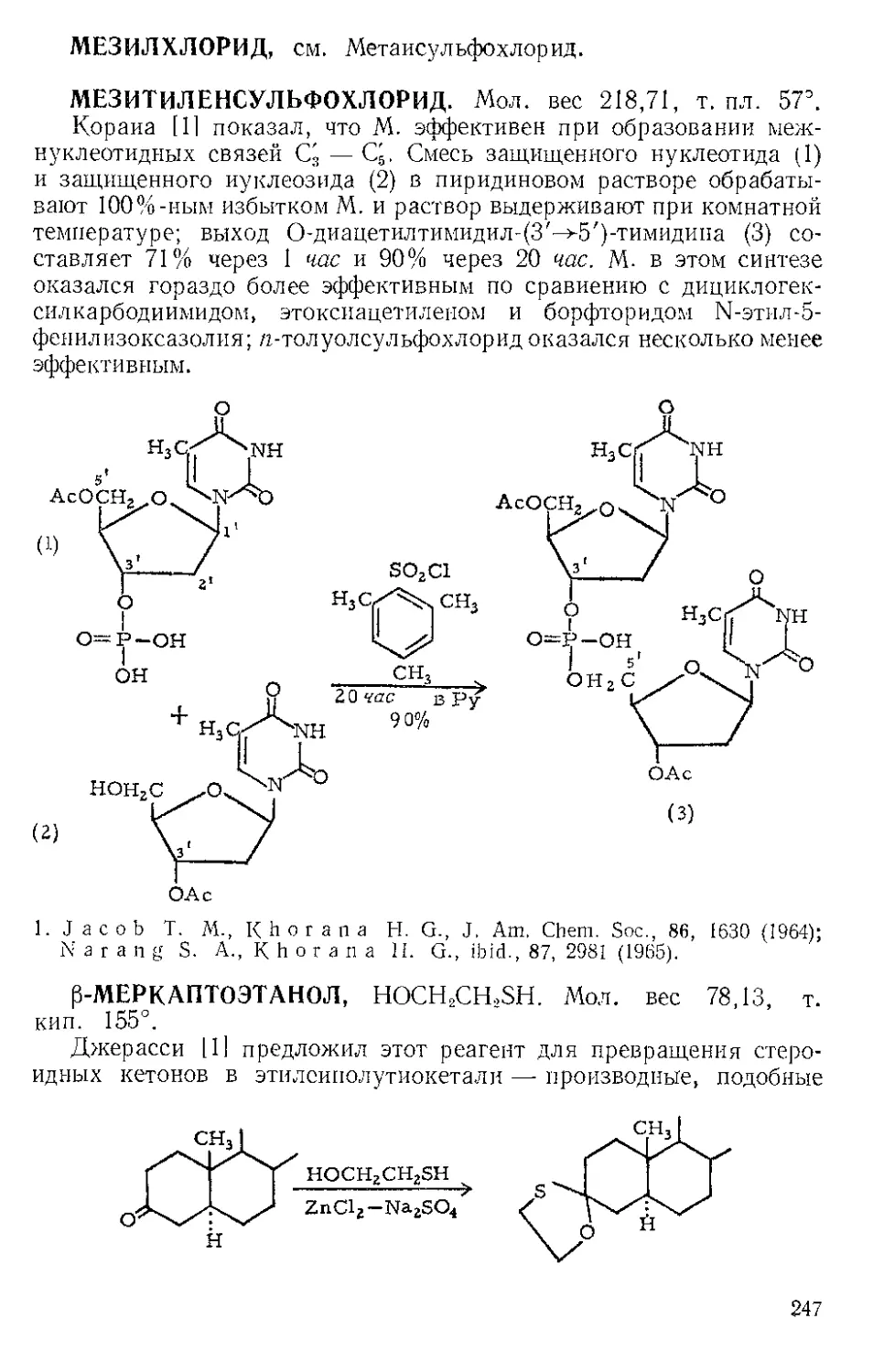
















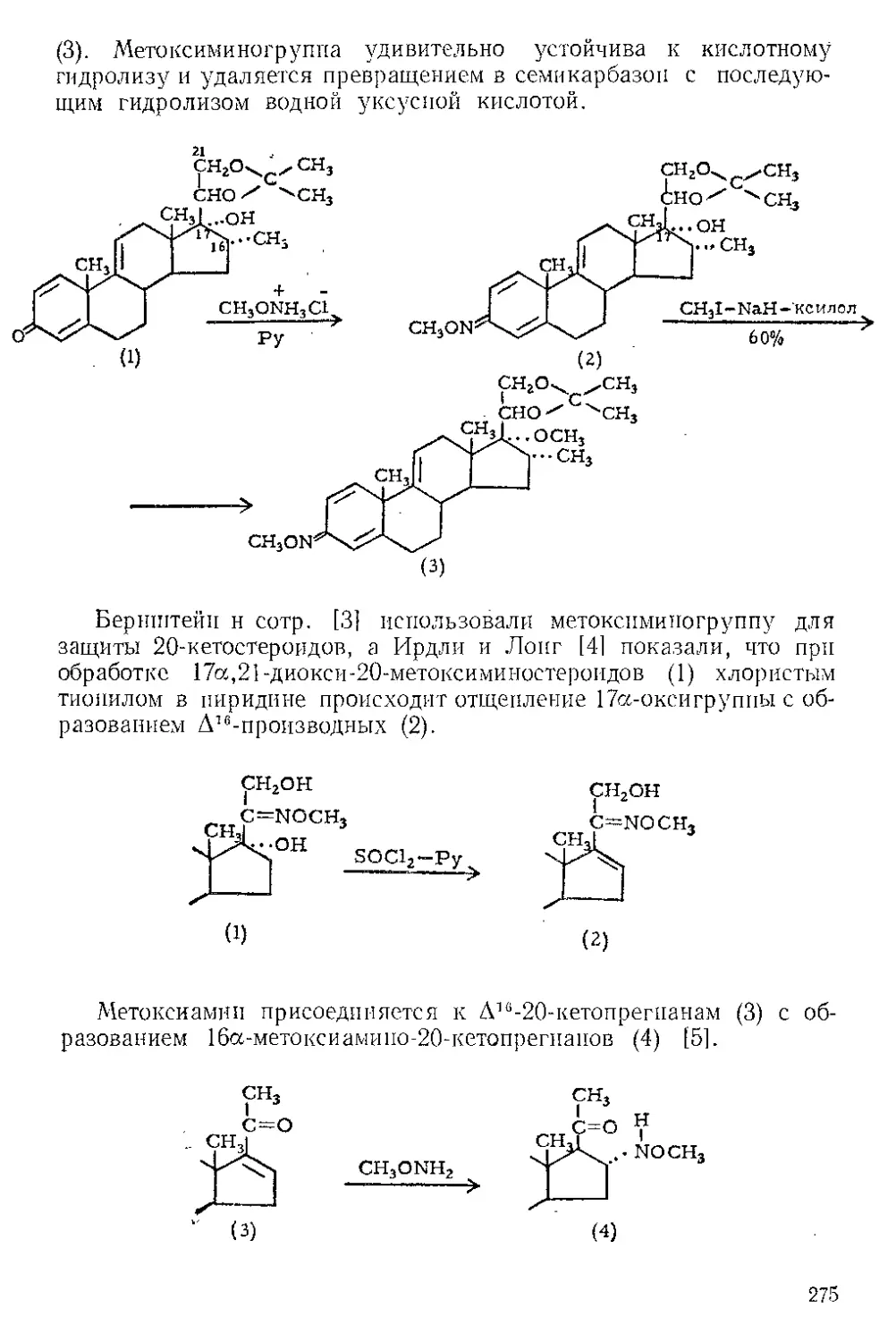
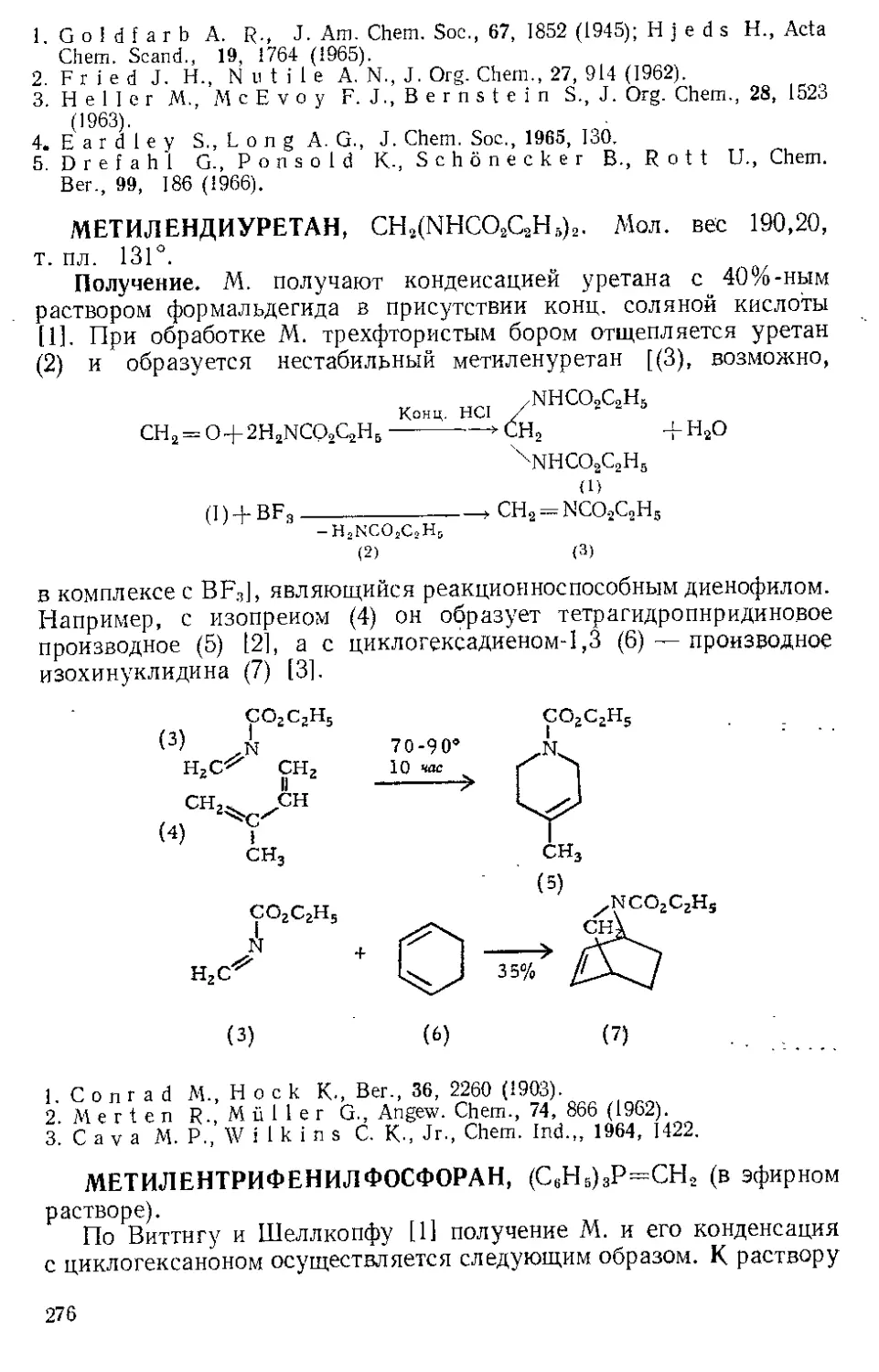









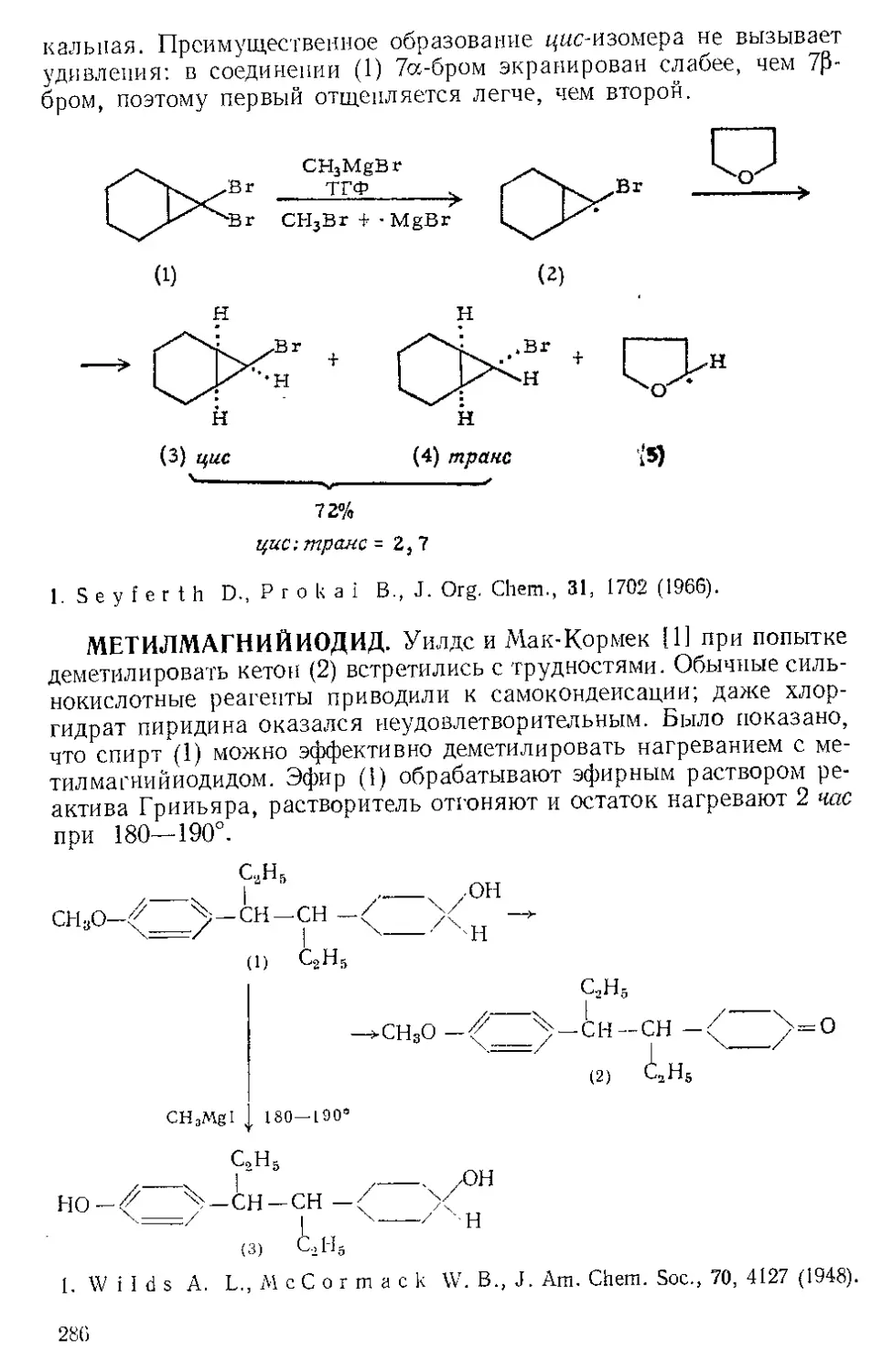




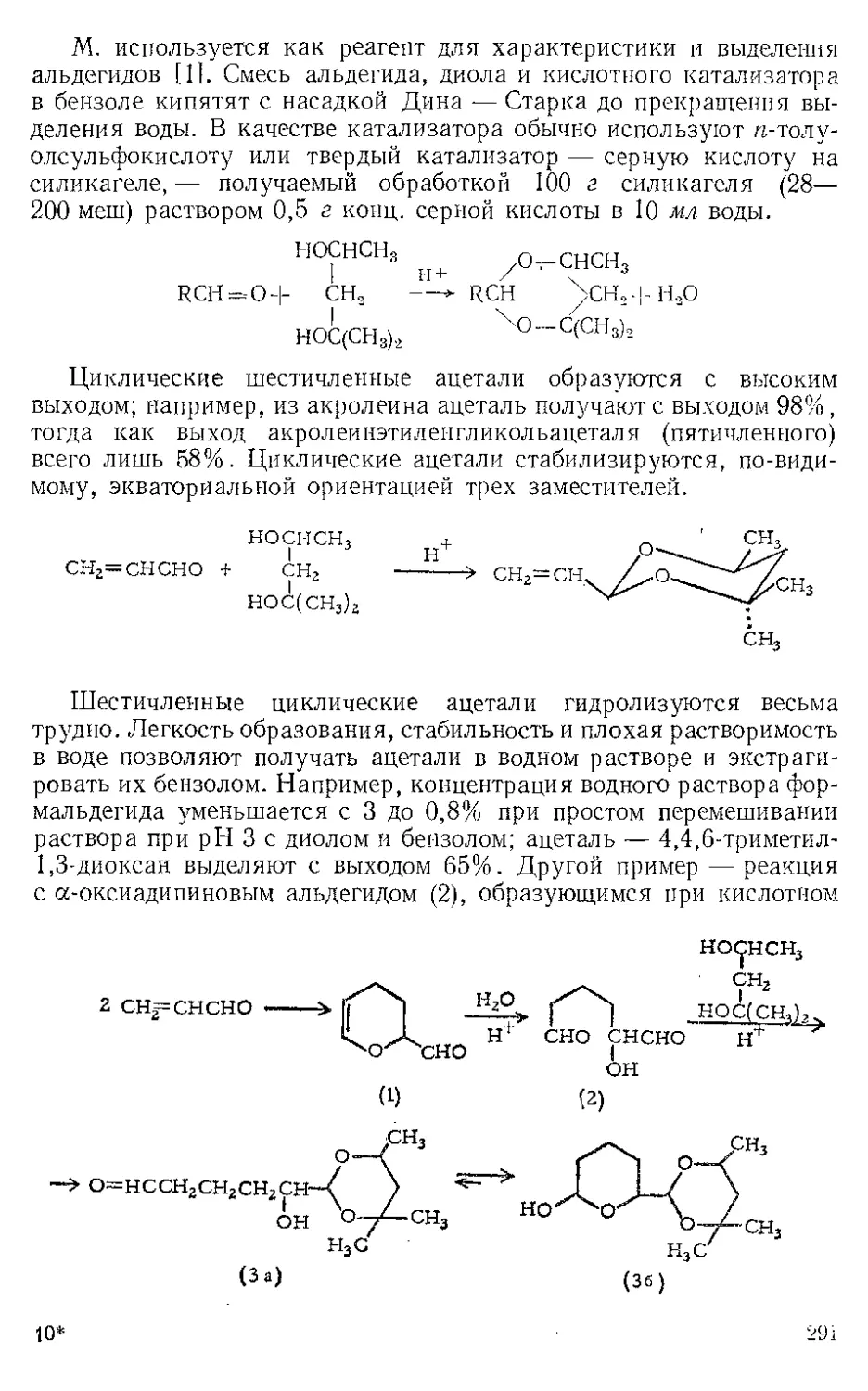




























































































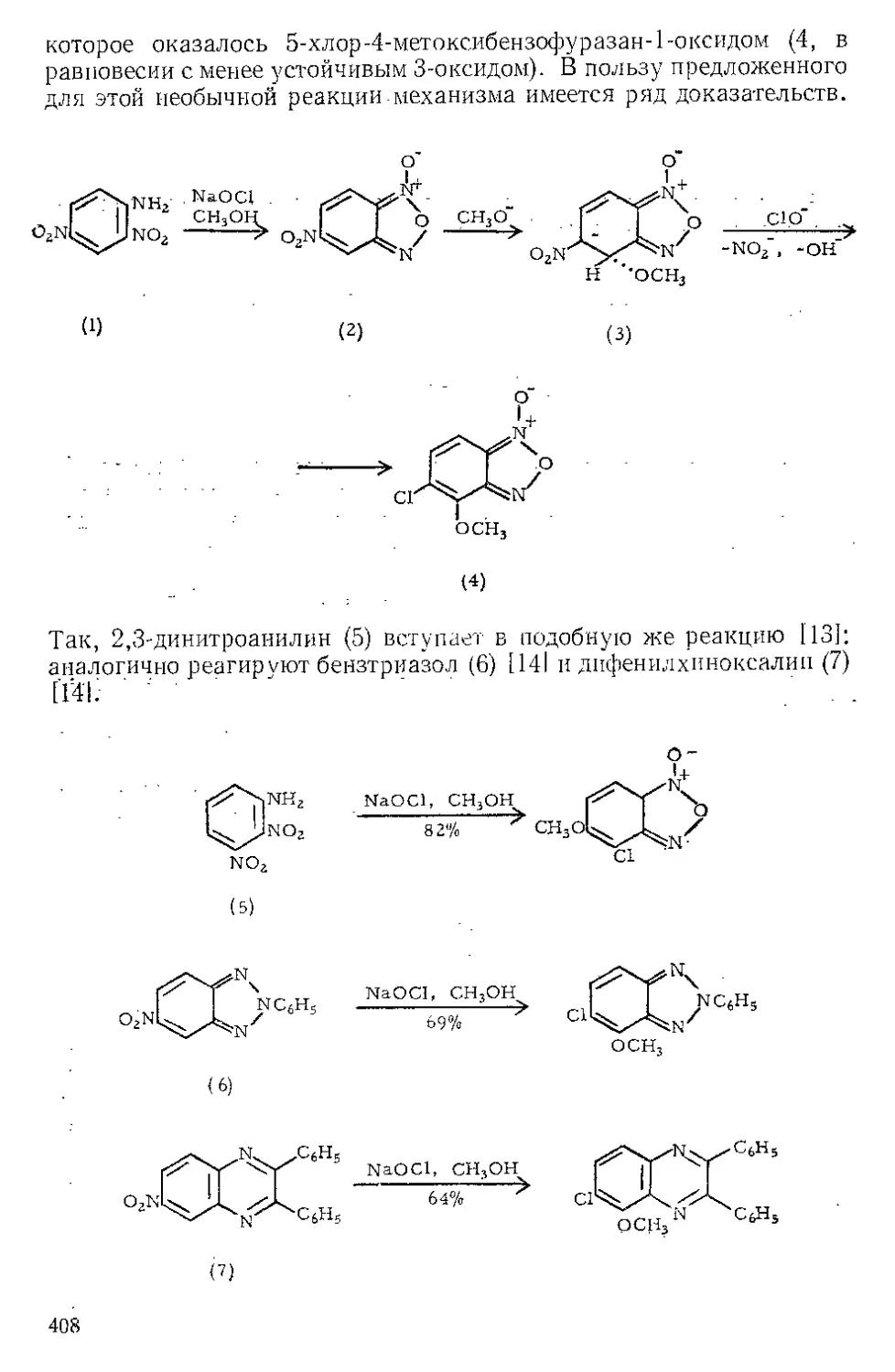



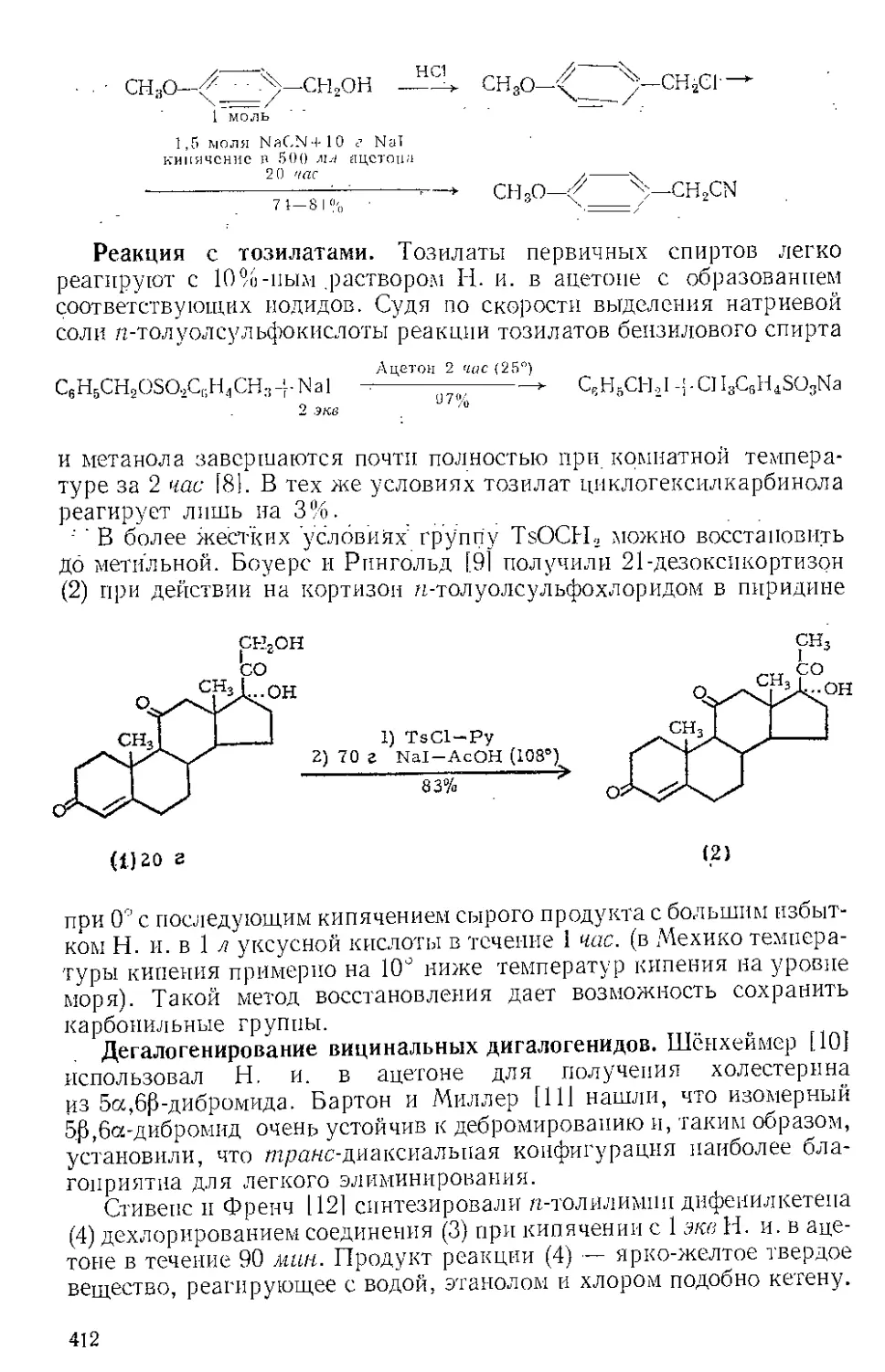



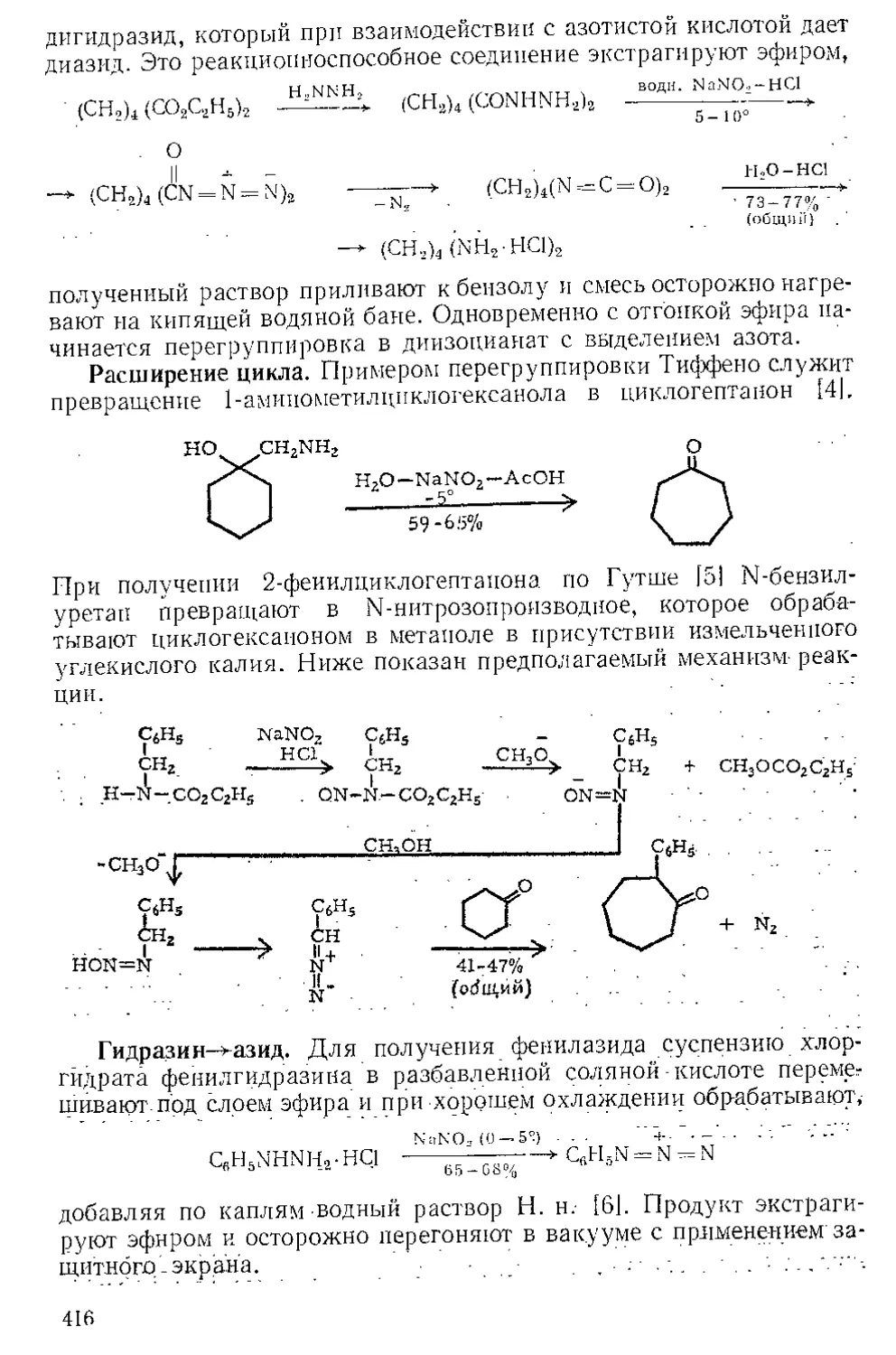

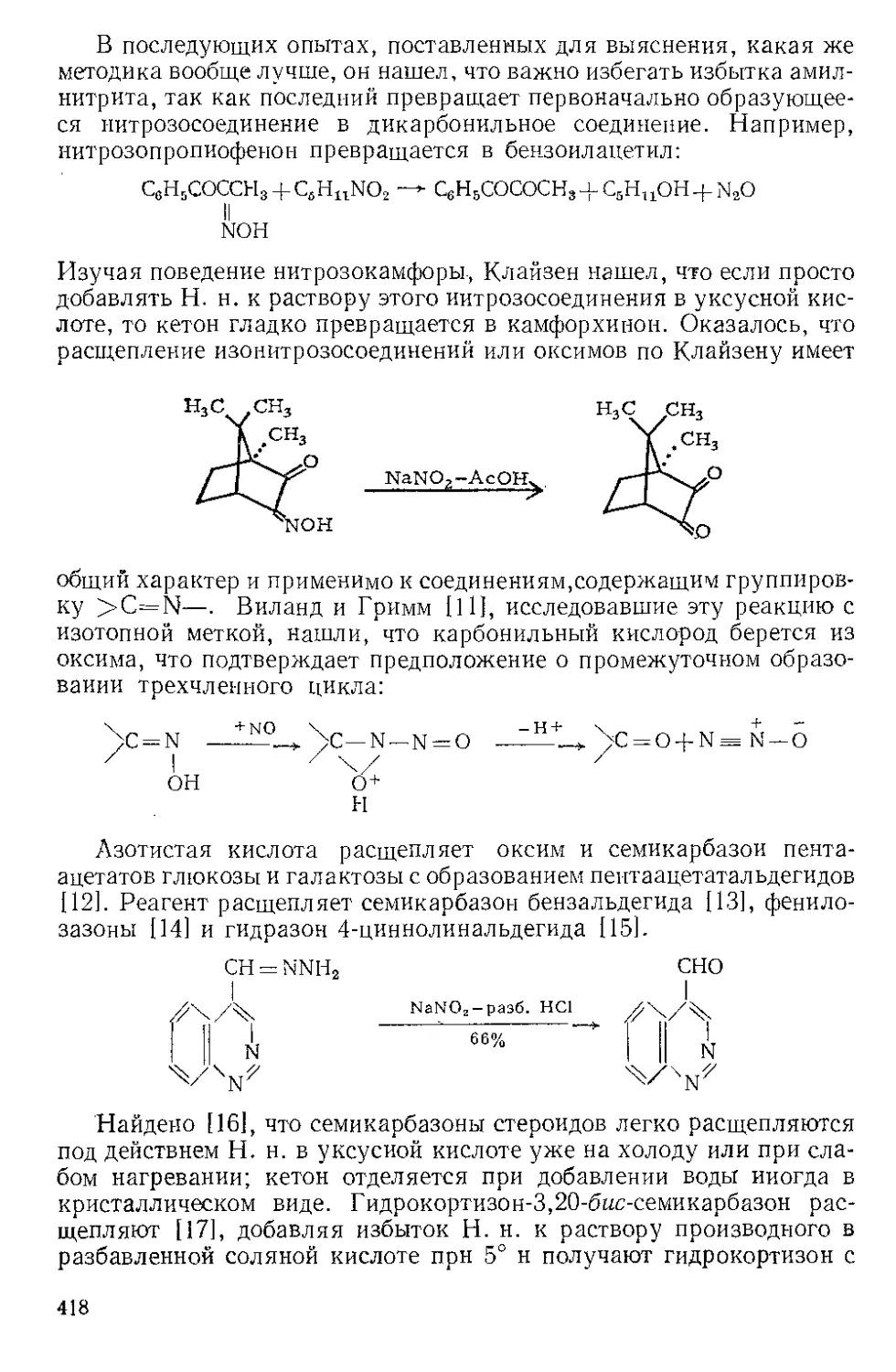
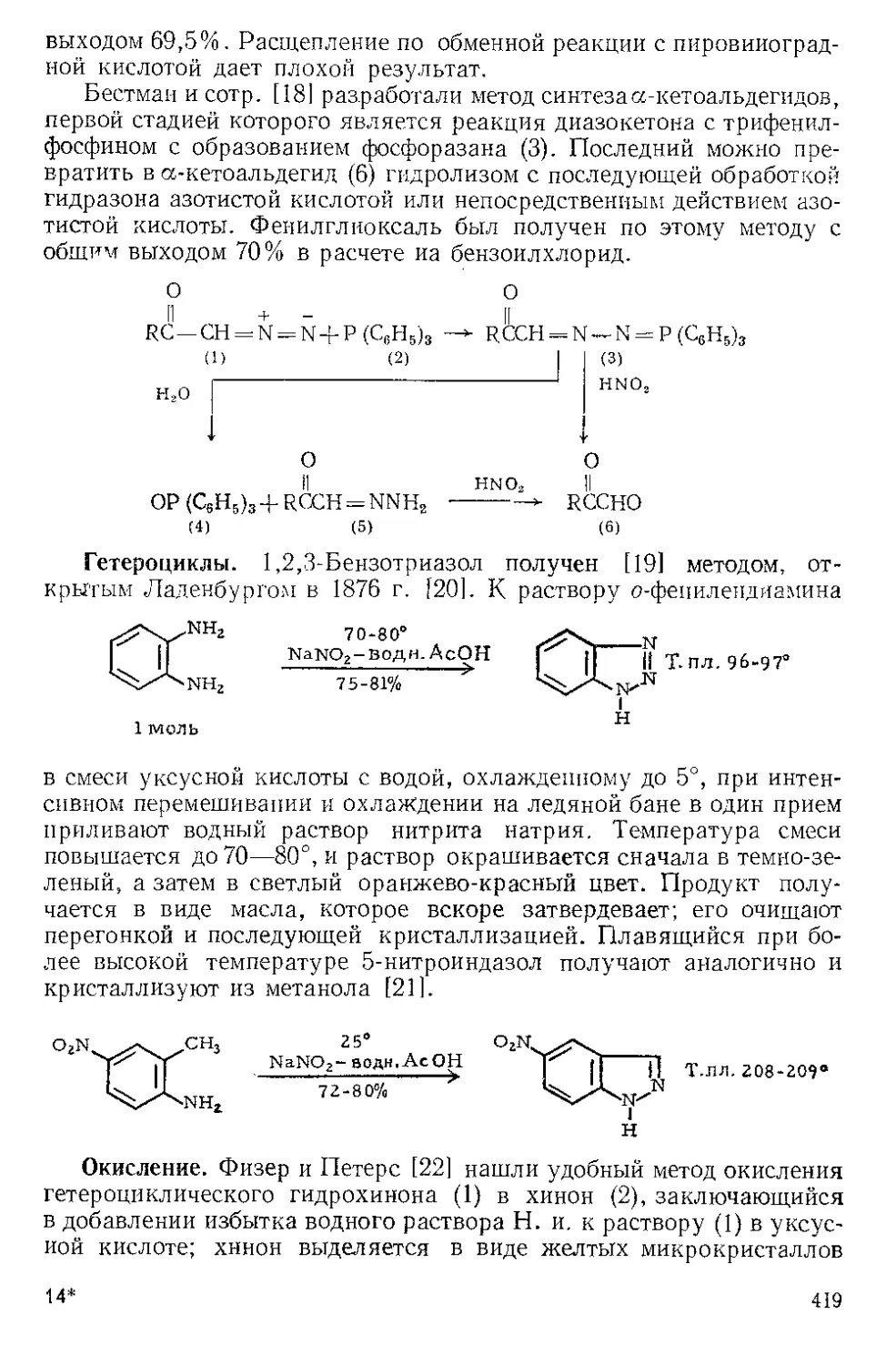
















































Текст
ИЗДАТЕЛЬСТВО'
«МИР»
Reagents for
Organic Synthesis
LOUIS F. F I E S E R
Sheldon Emery Professor ol Organic Chemistry
Harvard University
MARY FIESER
Research Fellow in Chemistry
Harvard University
JOHN WILEY AND SONS, INC.
NEW YORK . LONDON . SYDNEY
196S
Л. ФИЗЕР, М. ФИЗЕР
Реагенты
для органического
синтеза
томи
(Ж — Н)
ПЕРЕВОД С АНГЛИЙСКОГО
доктора хим. наук Н. С. ЗЕФИРОВА,
канд. хим. наук В. С. ПЕТРОСЯНА,
доктора хим. наук А. Ф. ПЛАТЭ
и канд. хим. наук С. С. ЧУРАНОВА
ПОД ‘РЕДАКЦИЕЙ
академика И. Л. КНУНЯНЦА
4 доктора хим. наук Р. Г. КОСТЯНОВСКОГО
МОСКВА 1970
ИЗДАТЕЛЬСТВО «МИР»
1*
УДК 661.7/54-41
Редакция литературы fib химии
Инд. 2-5-3
70
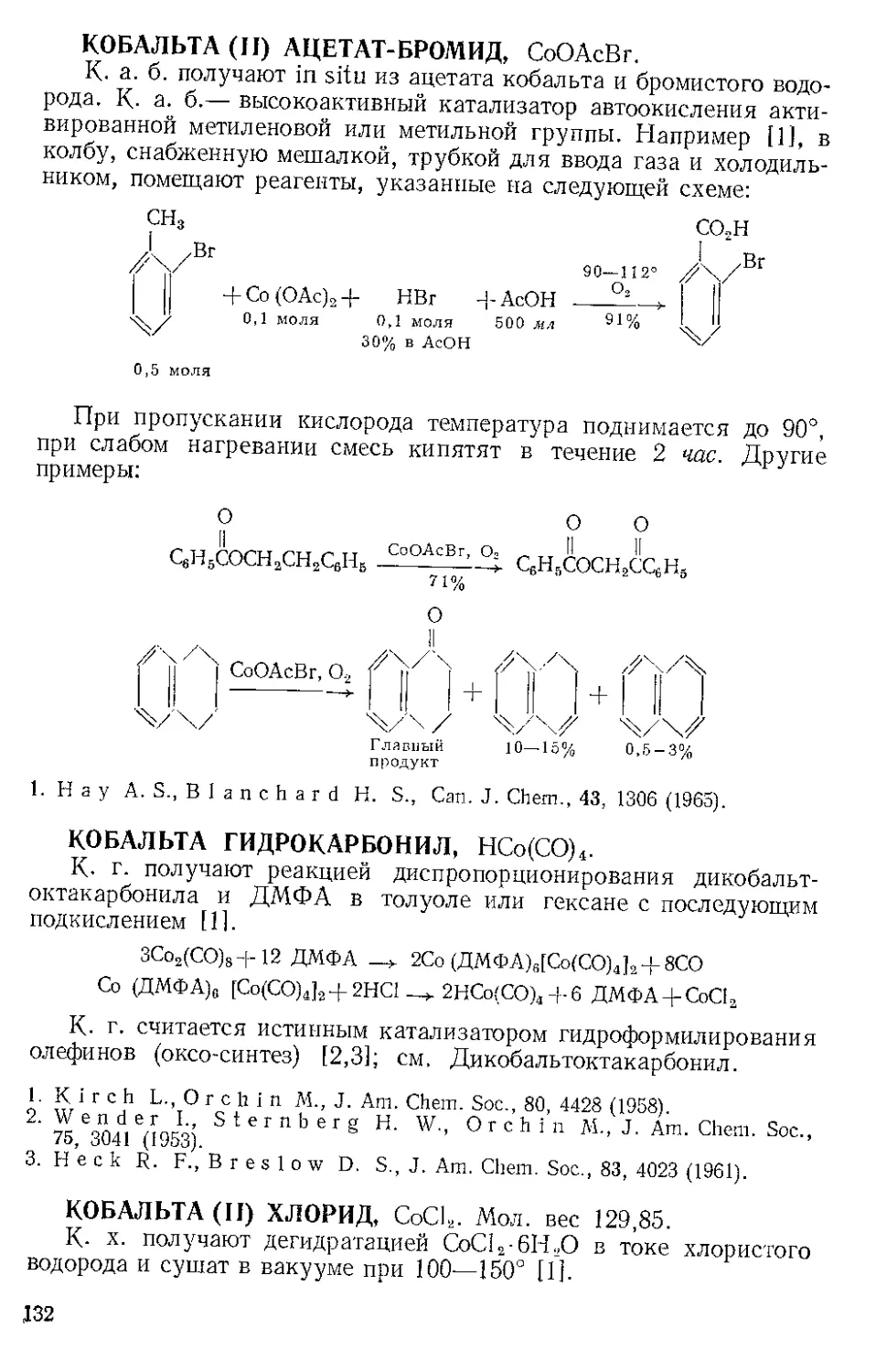
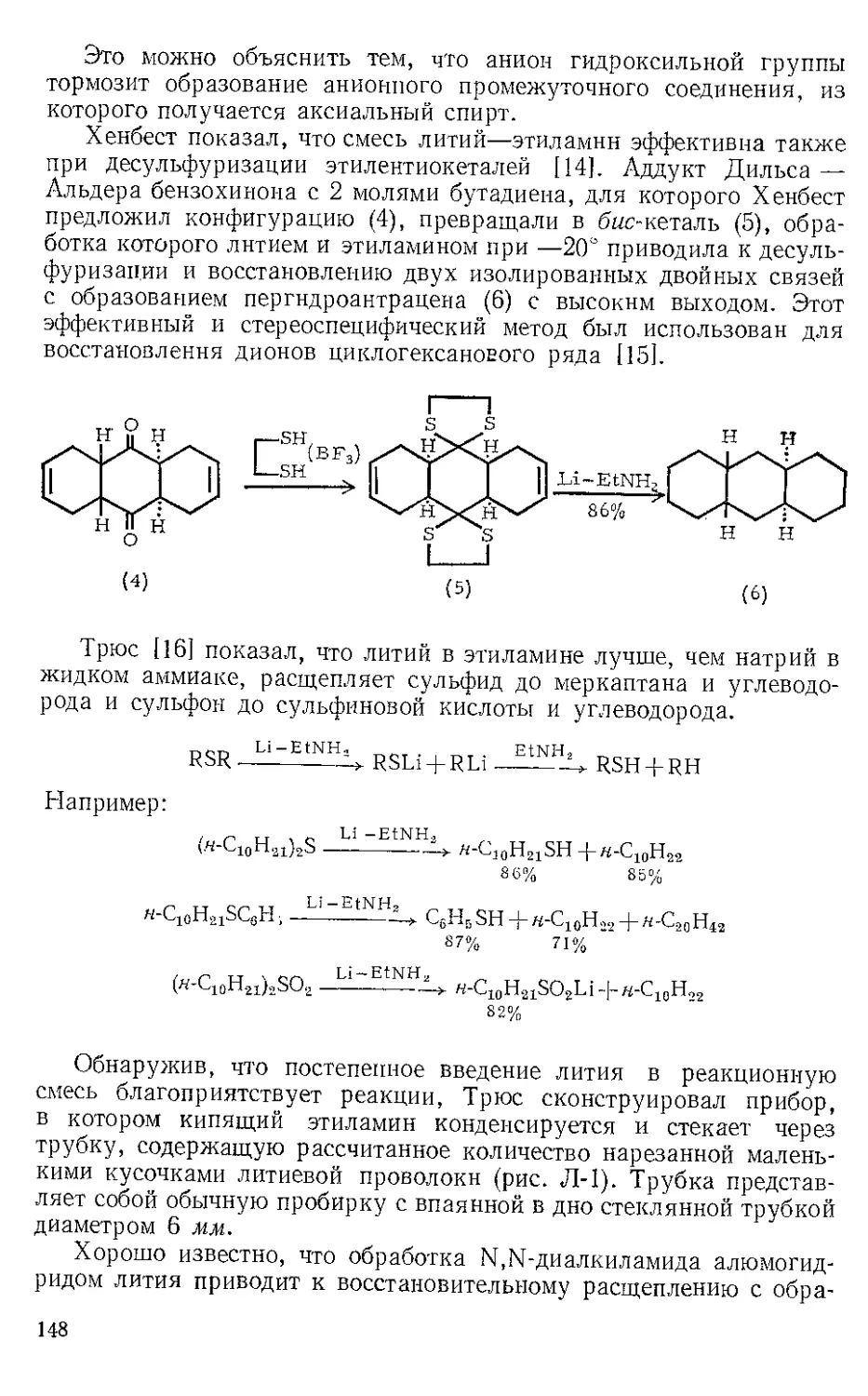
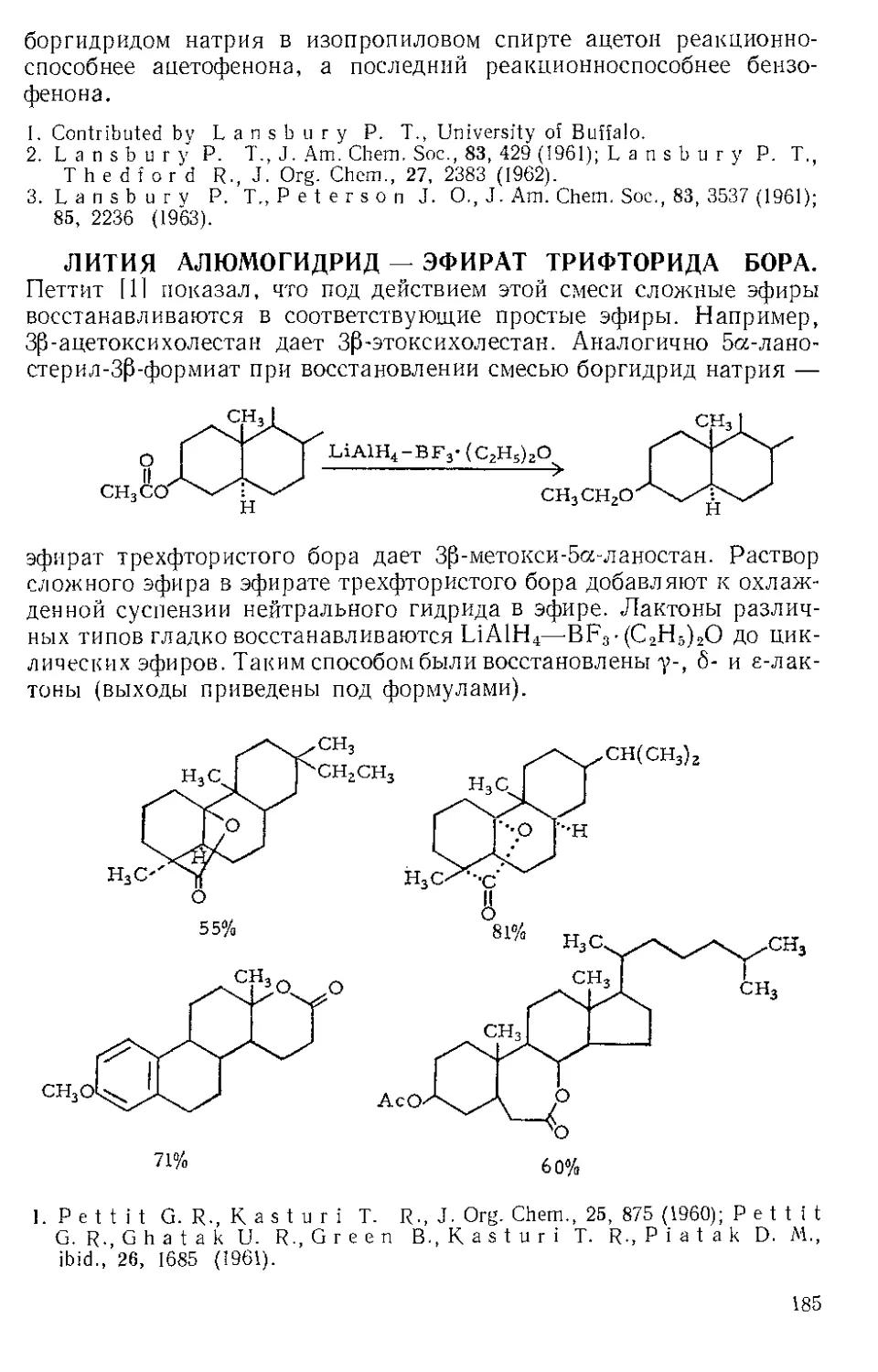
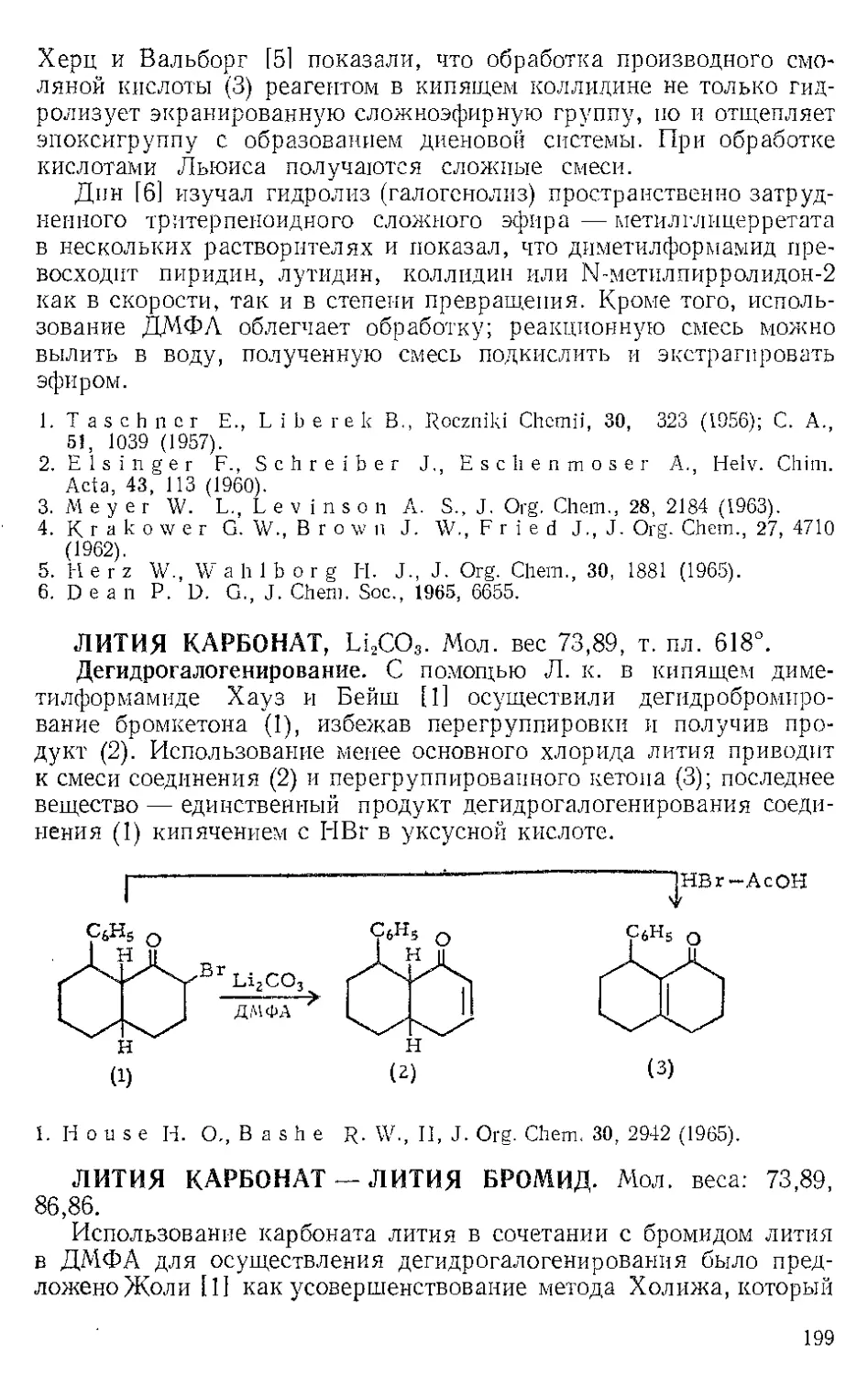
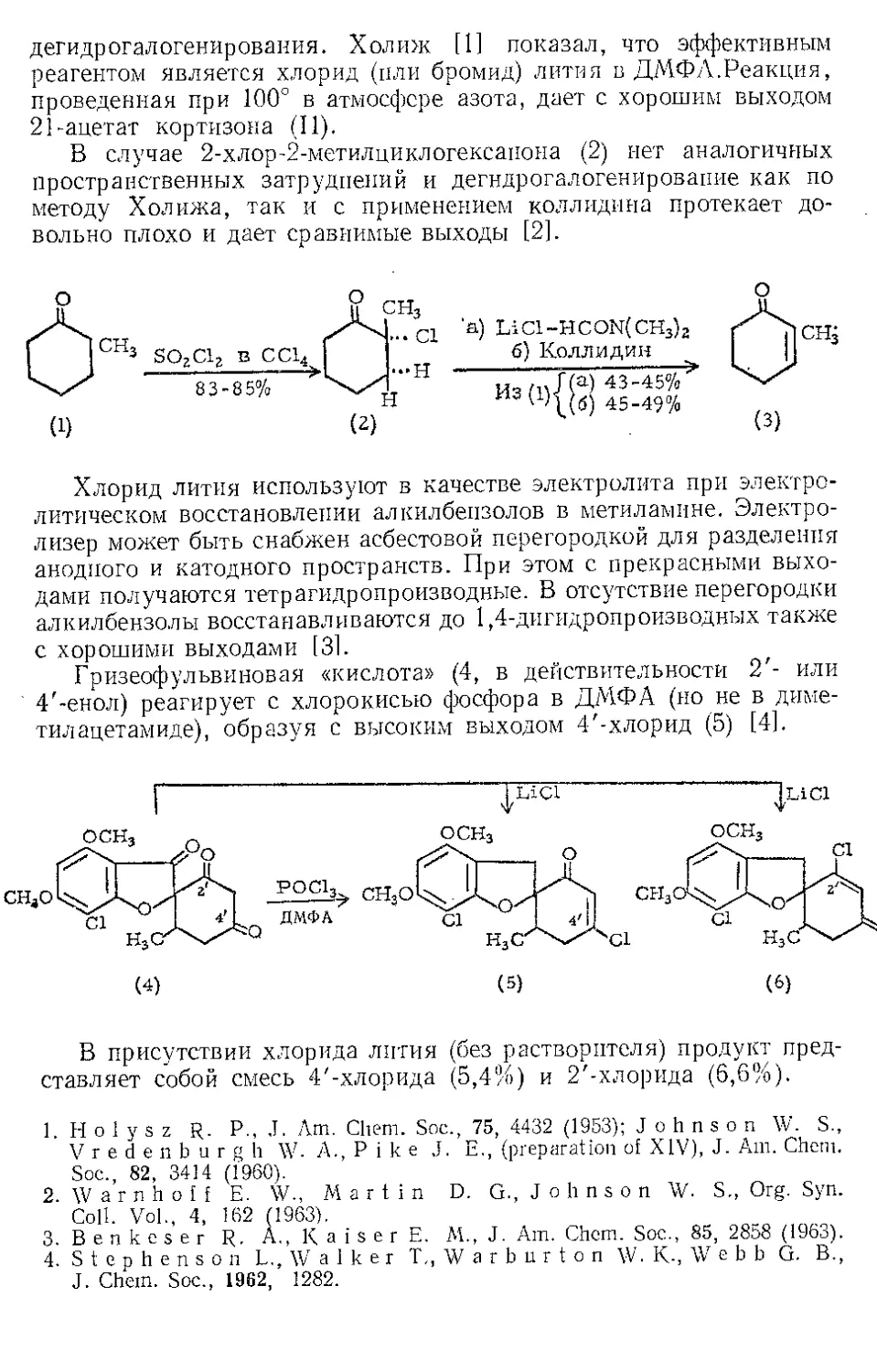
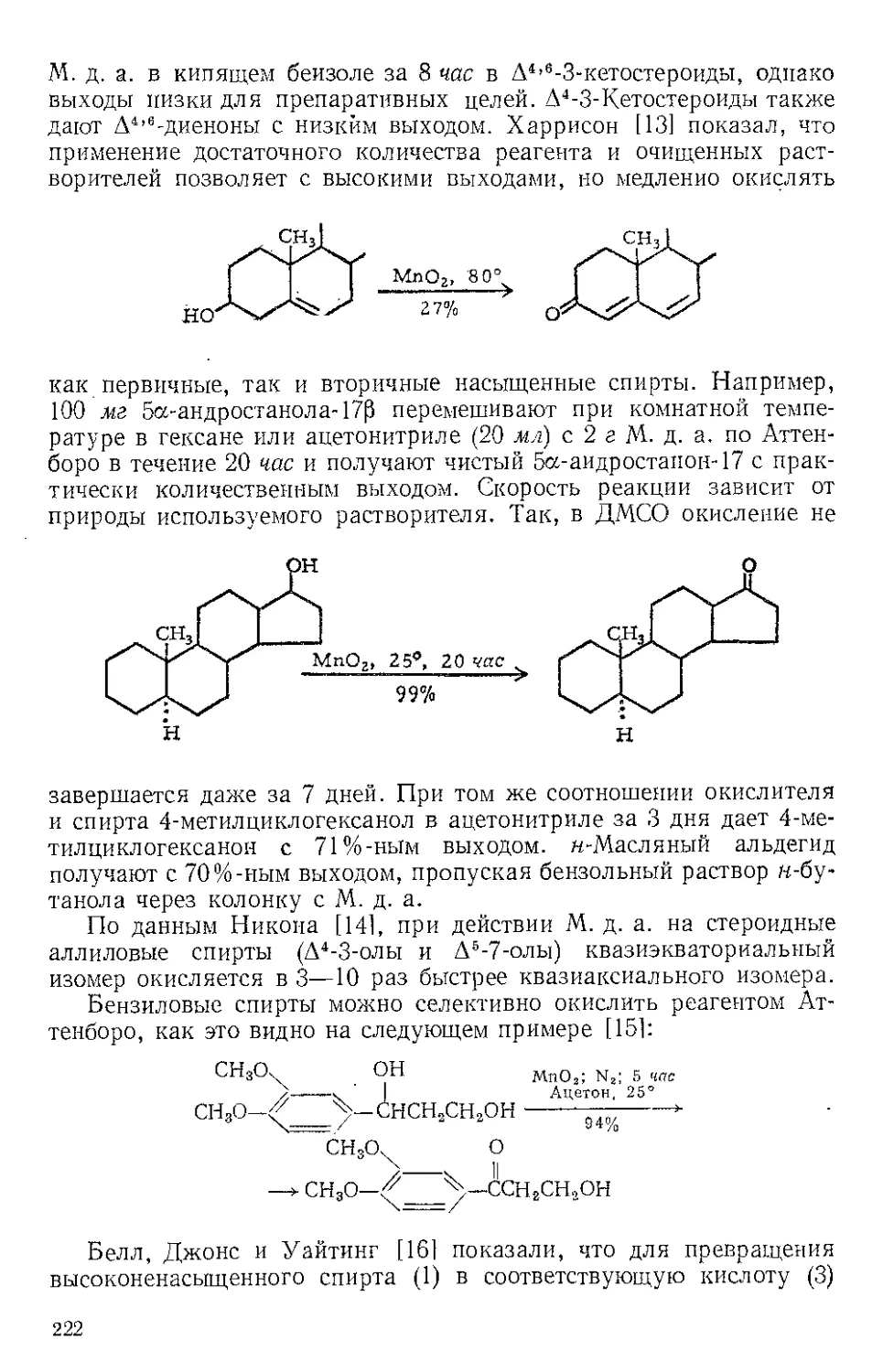
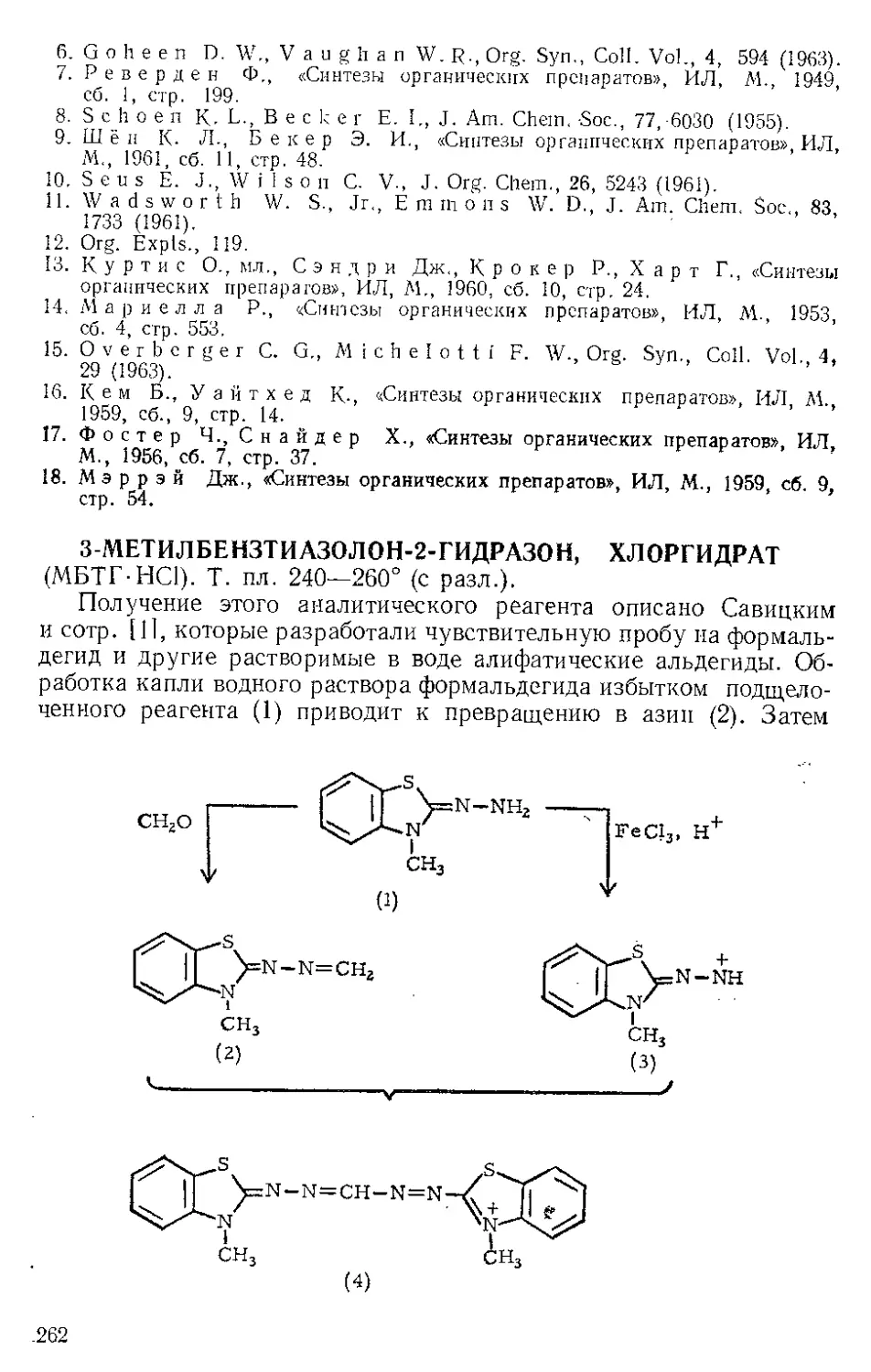
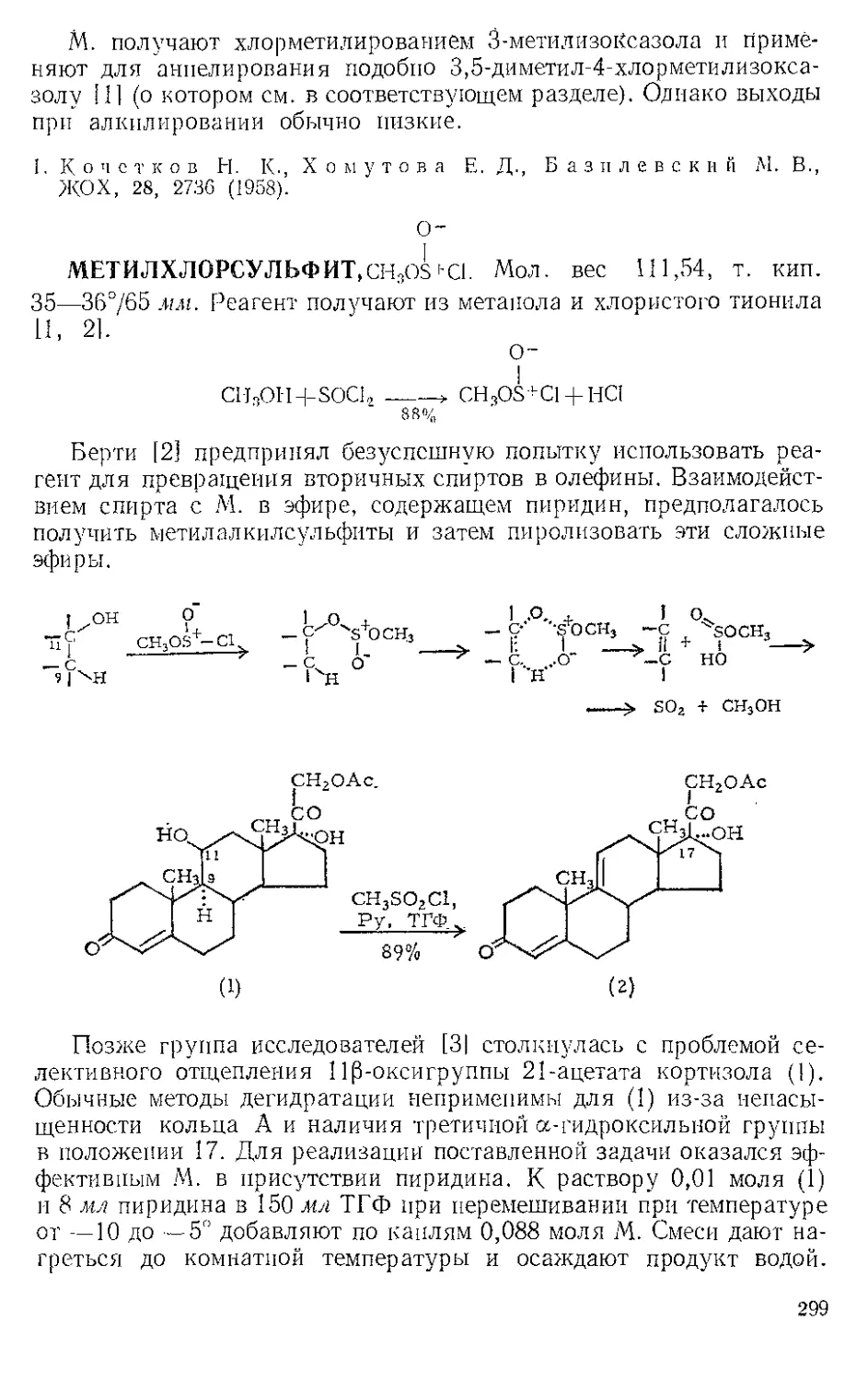
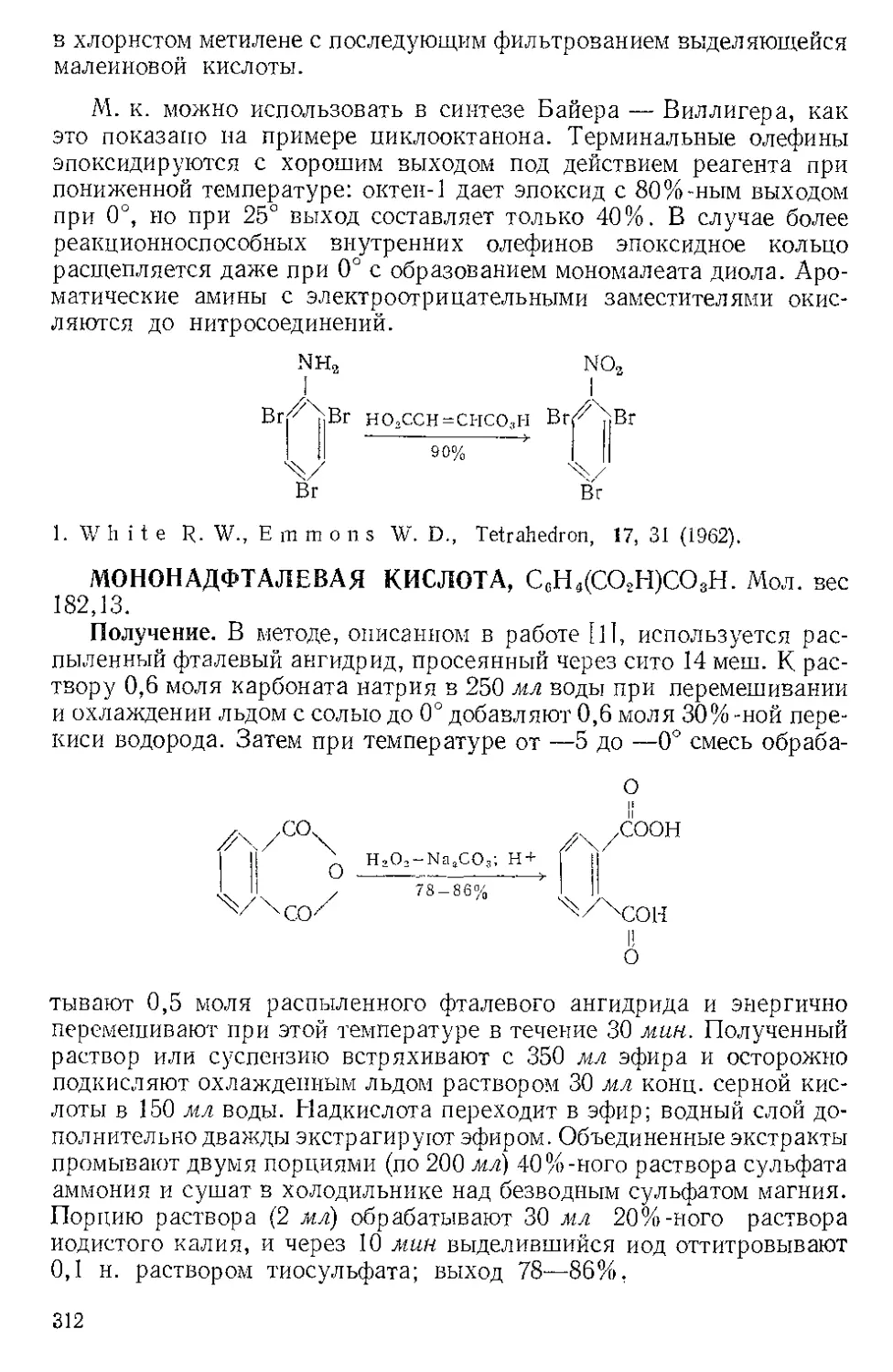
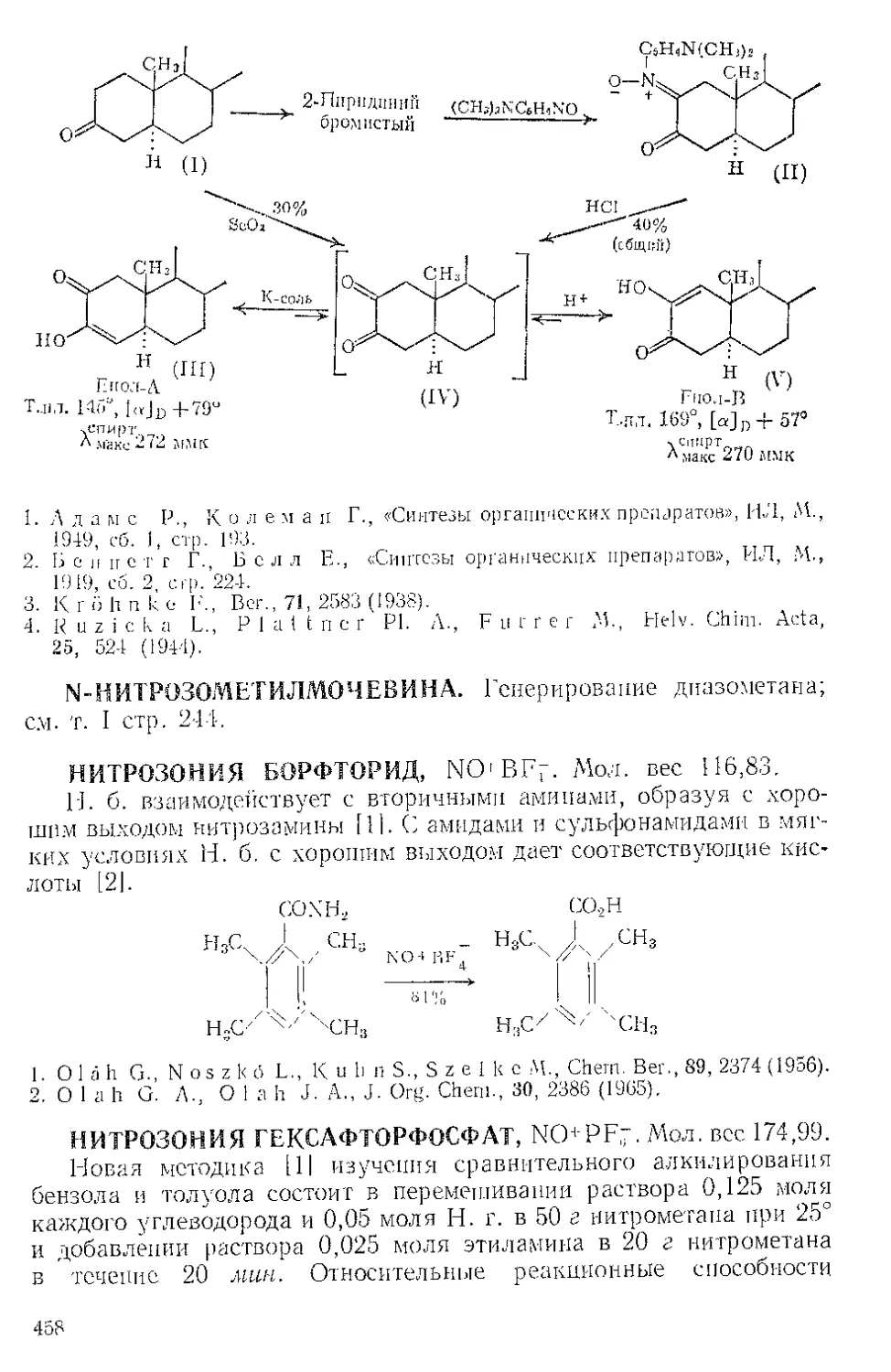
ЖЕЛЕЗА НОНАКАРБОНИЛ, Fe2(CO)9 (1). Мол. вес 273,74. По-
лучение [1].
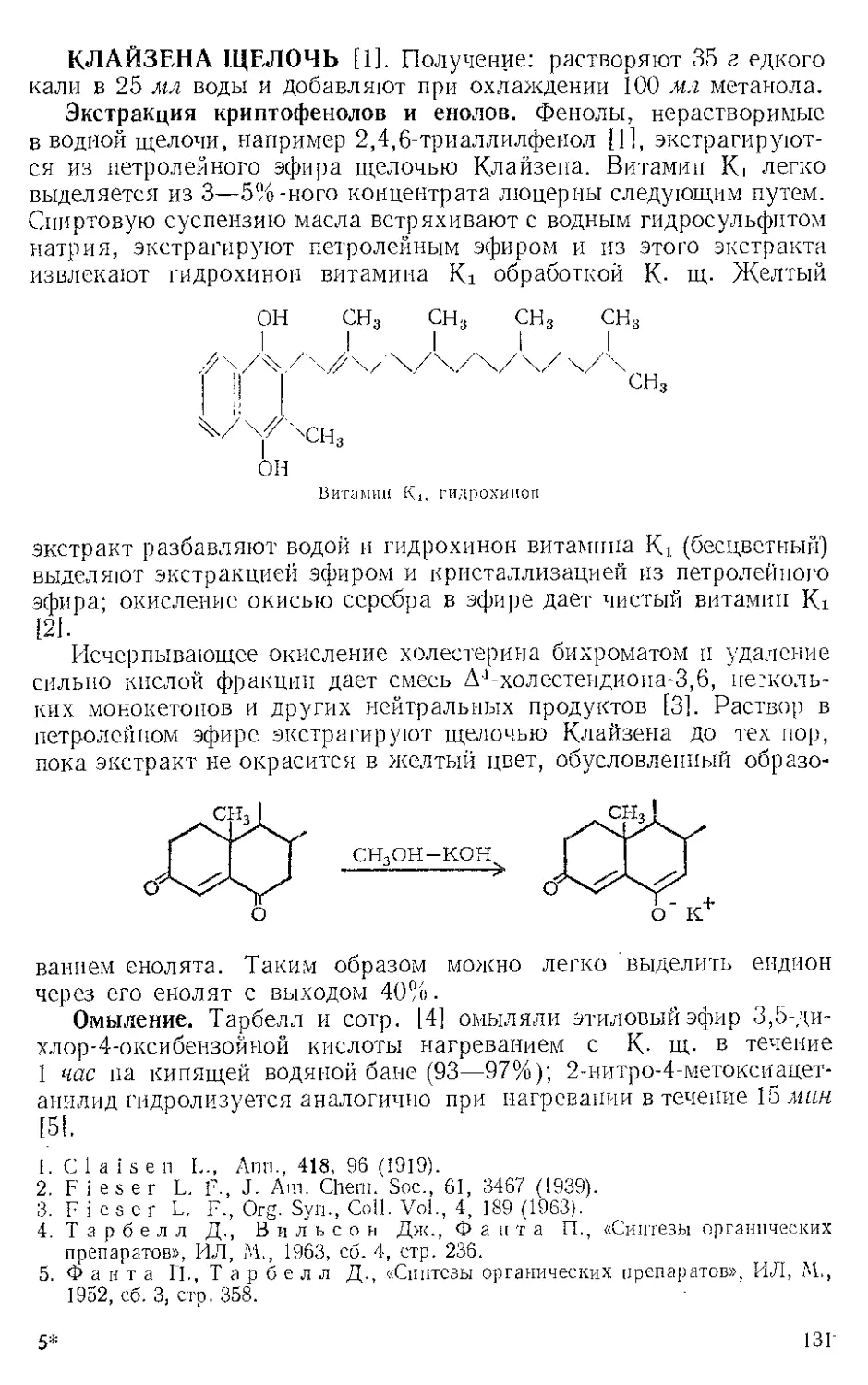
(1)
(С6Н5)ЭР
(2)
о=с'^
o'sC-Fe"
(CeHshP*'
О
(3) (4)
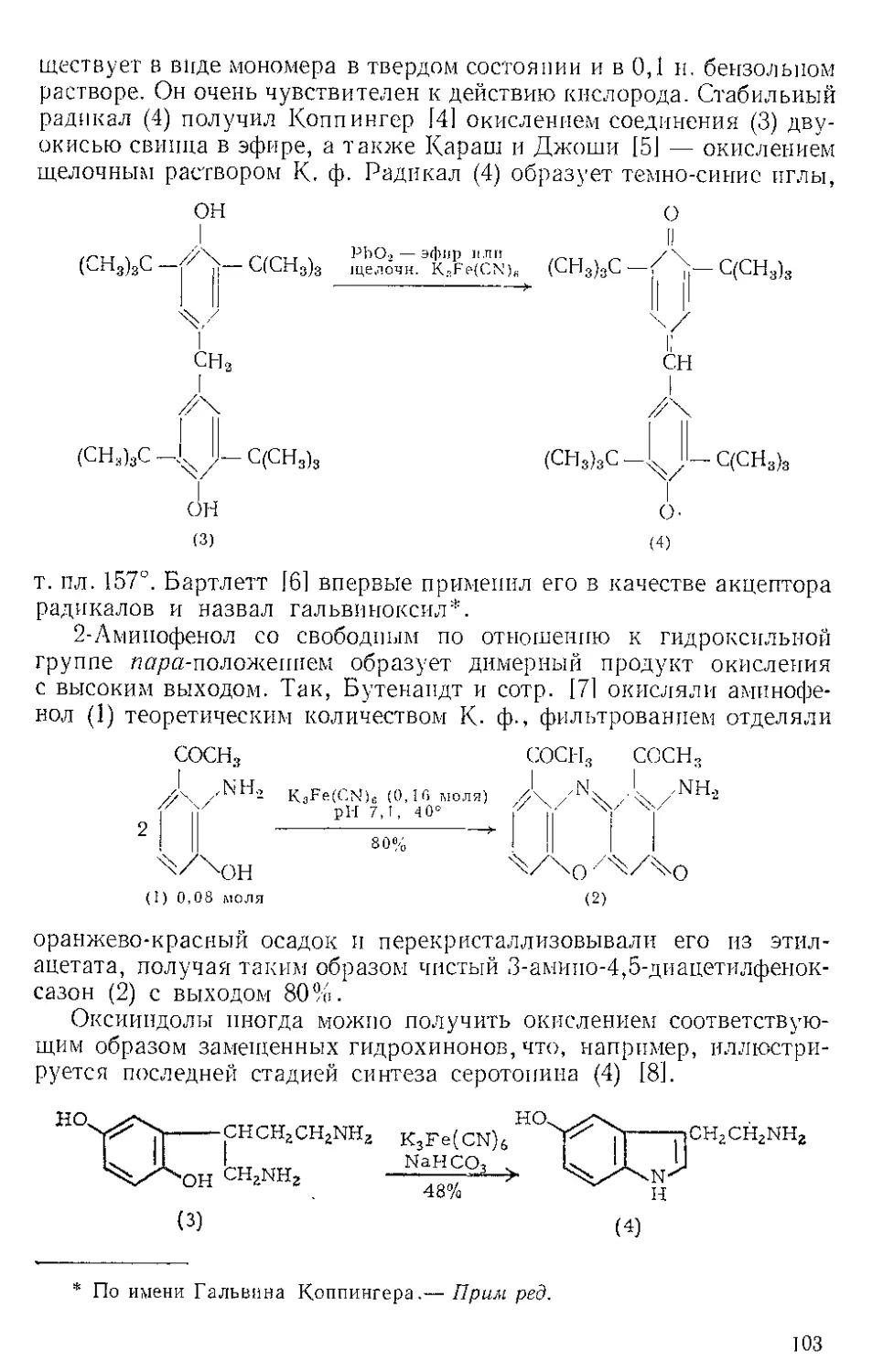
Этот комплекс содержит шесть концевых и три мостиковых кар-
бонильных групп, связанных с обоими атомами железа. Ацетилен
при 20—25° и 20—24 атм замещает мостиковые карбонильные груп-
пы Ж- н. с образованием тропонжелезотрикарбонила (2), представ-
ляющего собой оранжевое твердое вещество с двумя температурами
плавления 63,5—64° и 83—84°. Из карбонилов Fe(CO)5 и Fe(CO)2
получаются только следы этого соединения. Обработка комплекса
(2) трифенилфосфином дает тропой (3) с выходом 69% и новый комп-
лекс (4), тропоижелезодикарбонилтрифенилфосфин. Тропой также
можно получить окислением комплекса (2) хлорным железом, но
выход в этой реакции очень мал. г
C7H6OFe(CO)3+2FeCl3 Тропом+ 3FeCJ3 + 3CO
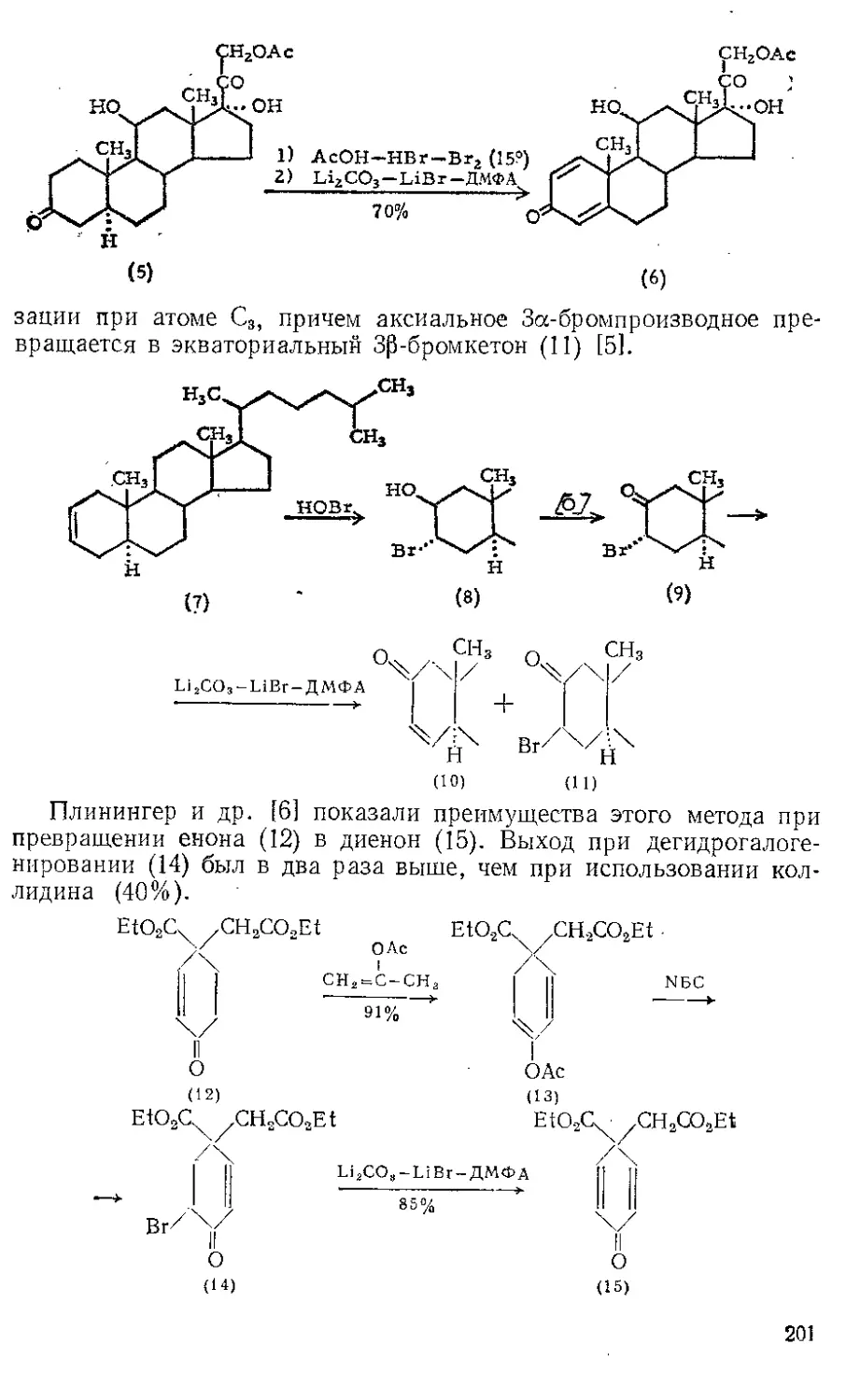
Действием Ж. и. на норборнадиен в темноте Куксон и сотр. (21
получили пять кетонов и четыре димерных углеводорода. Наиболее
высоко плав кий димер (т. пл. 163—164°) легко выделяется из смеси
с выходом 4%, поскольку ои первым элюируется с колонки при
хроматографировании на силикагеле. Лемаль и Шим [3] выделили
тот же димер из сложной смеси, полученной при облучении смеси
нор бор надиена и пентакарбонила железа. Для этого димера были
предложены две альтернативные структуры (1) и (2). При парал-
лельной ориентации двух молекул норбор надиена получится димер
5
(2), при ориентации под прямым угло.м — димер (1). В работе [2]
более вероятной считают структуру (2),
Гег(СО)^
1. We iss Е., Н й b е 1 W., Chem. Вег., 95, 1179 (1962).
2. Bird С. W-, С о 1 i и е s е D. L.,Cookson R.C., Hudec J., Wil-
liams R. О., Tetrahedron Letters, 373 (1961).
3. Lcmal D. M-, Shim K. S., Tetrahedron Letters, 368 (1961).
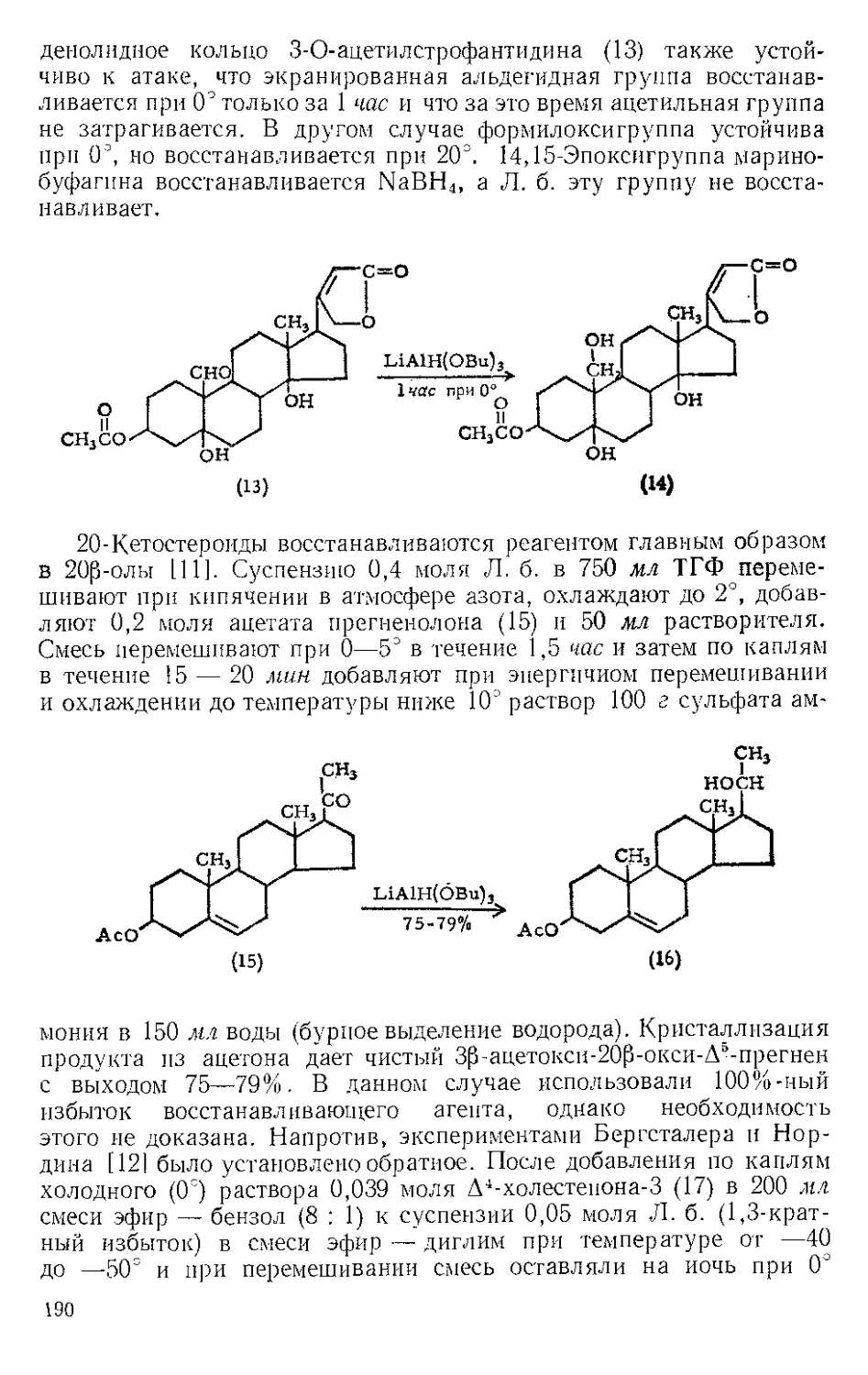
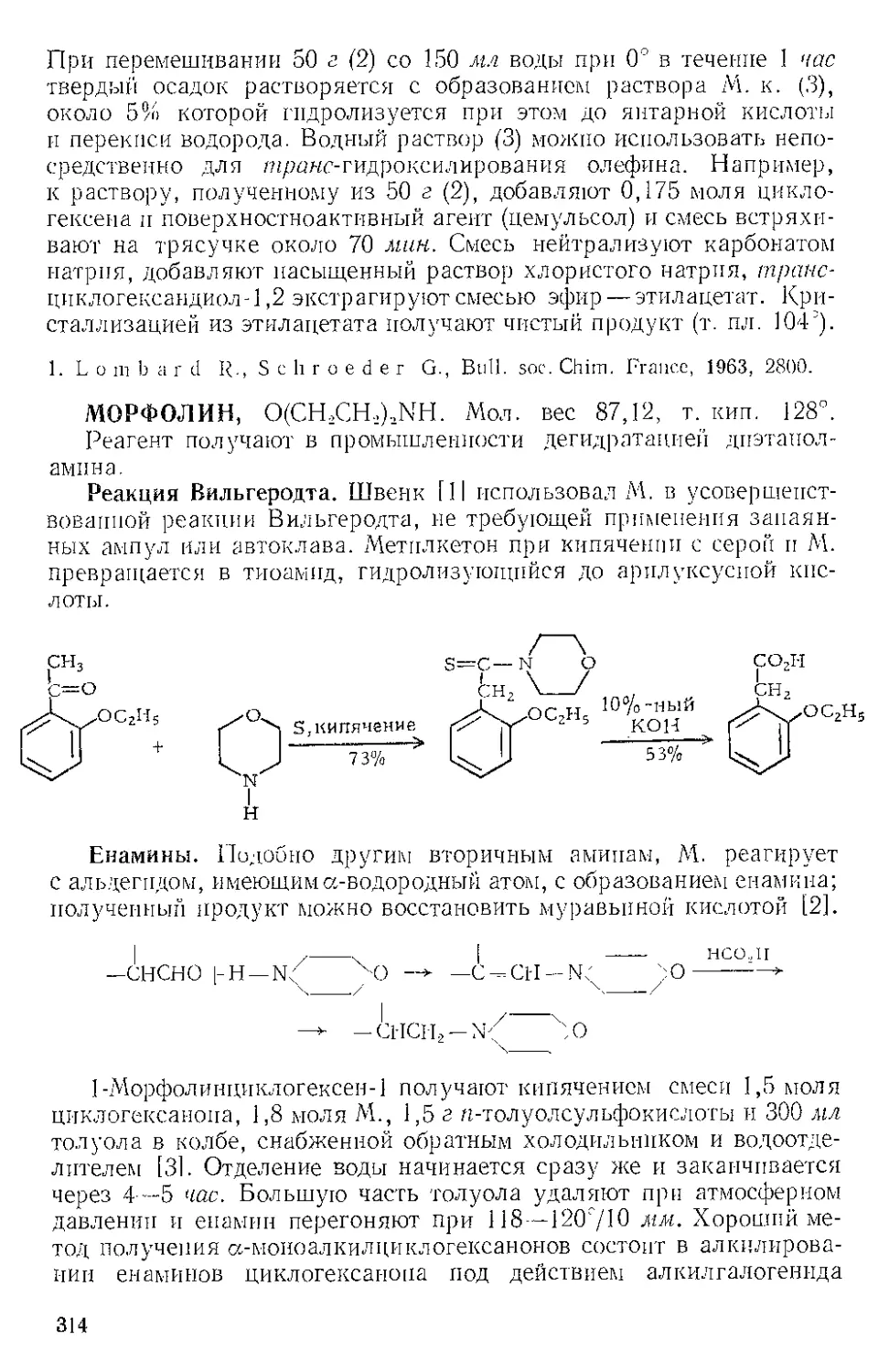
ЖЕЛЕЗА ПЕНТАКАРБОНИЛ, Fe(CO)3. Мол. вес 195,90, жел-
тый, т. пл. —20°, т. кип. 103°.
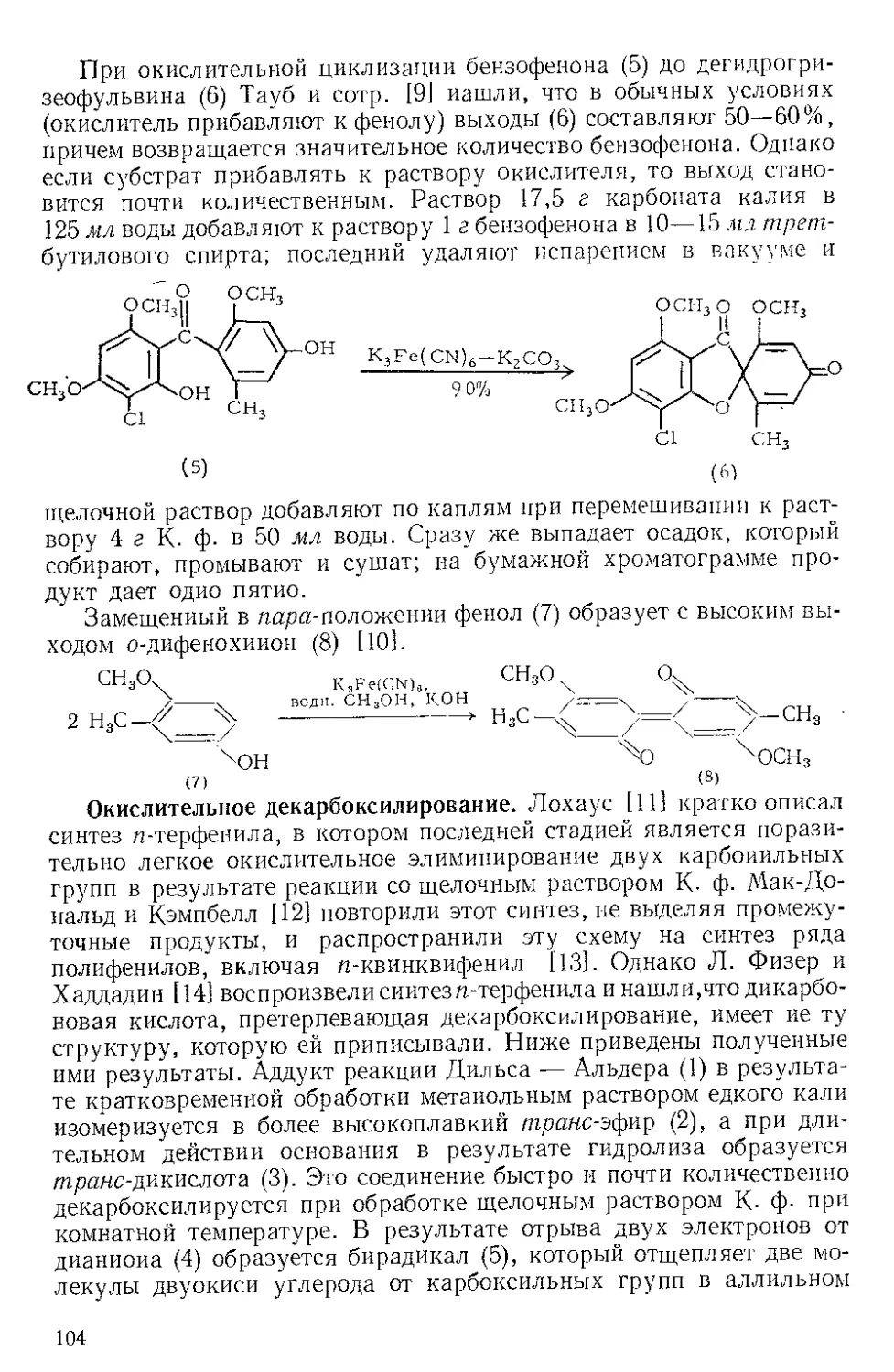
Реакция с ацетиленами. Реппе и Веттер [1] нашли, что ацетилено-
вые углеводороды реагируют с Ж.и. во влажном этаноле при 50—80° и
давлении 40 атм с образованием гидрохинонов. При более высоких
температурах главный продукт — этиловый эфир акриловой кислоты
(см. Никеля карбонил). Некоторое представление о ходе этой реакции
дает наблюдение Стернберга и corp. [2], которые отметили, что при
выдерживании на солнечном свету смеси диметил ацетилена и Fe(CO)5
выделяются оранжевые кристаллы состава Ре(СО)5(СН3С^ССН3).>.Это
вещество представляют в виде л-комплекса двух молекул алкина
с двумя карбонильными группами, так как при выдерживании
на воздухе комплекс разлагается с выделением дурохинона, а при
обработке его кислотой количественно образуются дурогидрохинон
и окись углерода.
СН3
2 111 + Fe(CO)#
?
сн3
НзСС^ ^ССНг
|| —Н----Fe(CO)3
Н3СС< /ССН3
S
о
Комплекс
2HCI
'-FeCh/
СО
он
он
Из реакционной смеси тетракарбонила железа с дифенилацети-
леном Шройцер [31 выделил в виде л-комплекса продукт внедрения
лишь одной карбонильной группы — тетрафенилциклопентадиенои»
6
3
Тримеризация бензонитрила. Кеттль и Оргел 14] показали, что
Ж. п. с хорошим выходом превращает бензонитрил в тример
2,4,6-трифенил-1,3,5-триазин. При охлаждении продукт кристалли-
зуется из реакционной смеси.
с0н5
I
n2.
кипячение песколЬ' N N
ко часов L и
C6H5C^N + Fe(CC%---------------------- [
Сочетание гелг-дигалогеиидов. Ж- п. осуществляет дегалогени-
рующее сочетание дихлор- и дибромдифенилметанов с образова-
нием с хорошим выходом тетрафенилэтилена [5]. Реакция возможна
Кипячение в СД-Ц
2Fe (СО)Э -Р 2 (C6HS), CCL---------------(С6НВ)2 С=- С (С6Н5). 2FeCl2 Д- ЮСО
к /□ 1 k - 95% (Сырой) Ч Ь к Ь э/- . 2
лишь для аелг-дигалогенидов и только в тех случаях, когда атомы
галогена активированы группами типа фенильной, циан- или кар-
бал кокси ль ной.
1. R е р р е W-, Vetter И., Ann., 582, 133 (1953).
2. S I е г п b е г g Н. W., М а г k Ъ у R., W ender I., J. Am. Chem. Soc.,
80, 1009 (1958).
3. Schr auzer G. N., Chem. Ind., 1958, 1403.
4. К e t t 1 e S. F. A., Orgel L. E., Proc. Chem. Soc., 1959, 307.
5. С о f f e у С. E., J. Am. Chem. Soc., 83, 1623 (1961).
ЖЕЛЕЗА(П) СУЛЬФАТ, FeSO, -7H,O. Мол. вес 278,03.
Восстановление нитрогруппы ароматических соединений, имею-
щих в орто-положении карбонильную функцию, действием Ж. с.
в щелочной среде было предложено Клайзеном [1] и развито Бам-
бергером [2]. На этой реакции основана современная методика
7
восстановления о-иитробензальдегида в о-аминобензальдегид [3].
/N0* /nh2
I || FcSO4, nh4oh / >['
I II 69—75%~I- II
"^/XCHO XCHO
До сих пор неясна роль Ж. с. как регулятора, смягчающего
обычно очень бурную реакцию анилина, глицерина, нитробензола
и конц. серной кислоты в синтезе хинолина по Скраупу [4].
СН2ОН
II -р I CeH6NO2. H2SO„ FeSOj f II I
И сноп -----------------пгщ-------н II I
NH"- CH.OH ' N
I. Claisen L.,Shadwell J., Ber., 12, 353 (1879); Claisen L., Thom-
pson С. M., Ber., 12, 1947 (1879).
2. Bamberger E., Dem ut h Ed., Ber., 34, 1330 (1901).
3. Смит Л., О и и Дж., «Синтезы органических препаратов», ИЛ, М., 1953,
сб. 4, стр. 27.
4. Кларк, Дэвис, «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 460.
ЖЕЛЕЗА(Ш) ХЛОРИД, FeCl3-6H2O. Мол. вес 270,32.
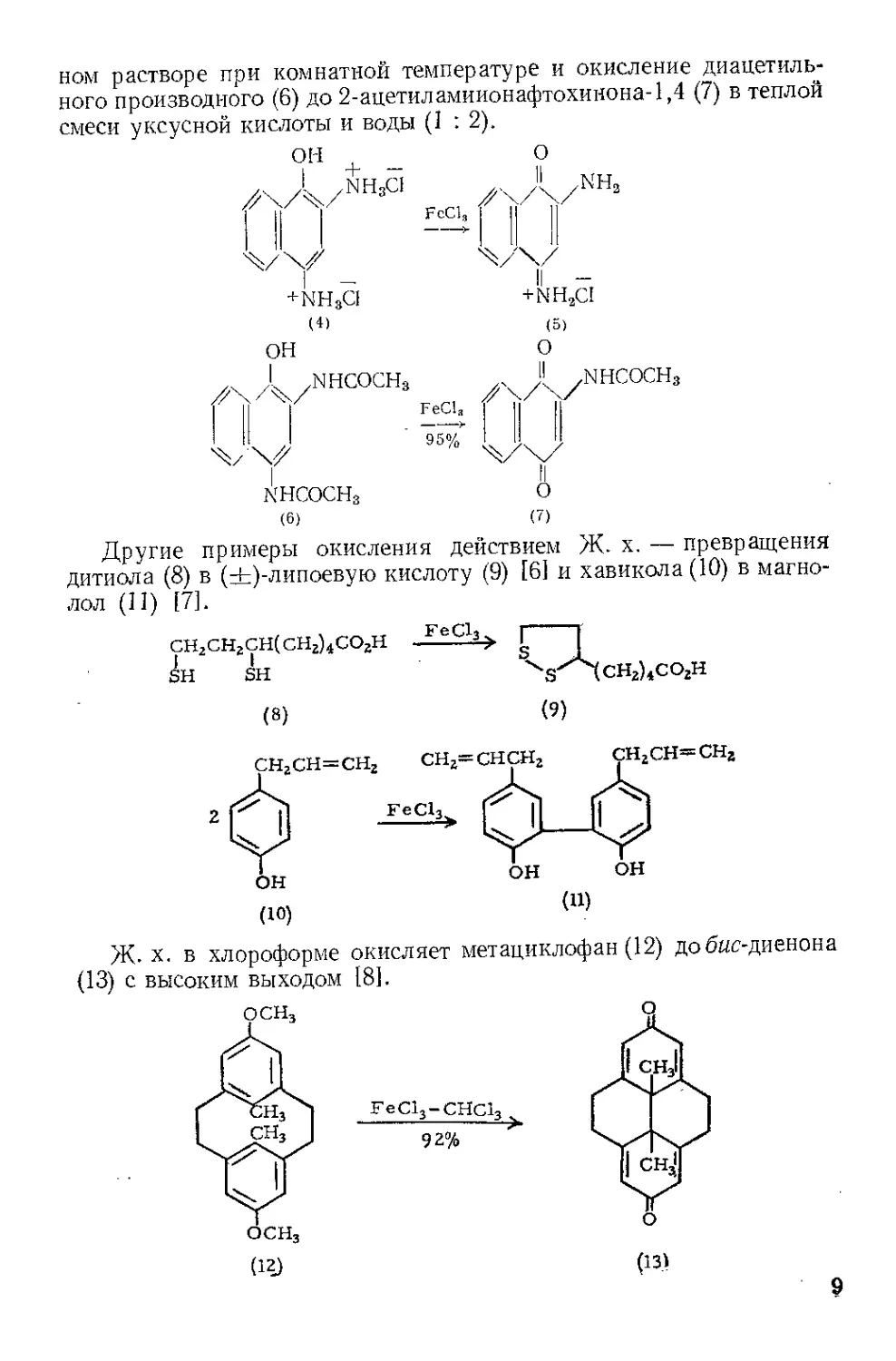
Окисление. При окислении хлор гидрата 1-амияо-2-нафтола (1)
под действием Ж. х. в водном растворе при 25—35° получается
сразу чистый нафтохинон-1,2 (2) [11. При перемешивании 10 г этого
желто-оранжевого хинона (2) с водным раствором 40 г FeCl3 •
• 6Н2О в 400 мл воды около 1 час при 65—70° наблюдается сначала
частичное растворение хинона, затем раствор внезапно светлеет и
начинается выпадение желтого осадка продукта окисления (3),
2-окси-3,3'-дииафтилдихинона-1,4,Г, 2' [2,3]. Получение 6-метокси-
и 5,6-диметоксинафтохинонов-1,2 по вышеуказанному методу опи-
сал Гэйтс [4]. С высоким выходом осуществляется [5] окисление
диамина (4) до хлоргидрата 2-аминонафтохинон-4-имина (5) в вод-
8
ном растворе при комнатной температуре и окисление диацетиль-
ного производного (6) до 2-ацетиламиионафтохинона-1,4 (7) в теплой
смеси уксусной кислоты и воды (1 : 2).
+NH3CI +NHaCl
NHCOCH3 О
(6) (7)
Другие примеры окисления действием Ж. х. — превращения
дитиола (8) в (±)-липоевую кислоту (9) [61 и хавикола (10) в магно-
лол (11) [7].
сн2снгсн(сн2)4со2н CU» Г 7
SH ен ^гДснЛсОгН
(8) (9)
(10) С11)
Ж. х. в хлороформе окисляет метациклофан (12) добис-диенона
(13) с высоким выходом [81.
(12) (13)
9
При получении нитрилов взаимодействием цианида меди (I)
с арилгалогенидами промежуточно образуется комплекс нитрила
с галогенидом одновалентной меди. Фридман и Шехтер [9] нашли,
что нитрилы при этом можно легко выделить из комплекса добавле-
нием водного раствора Ж. х., окисляющего ион меди до двухвалент-
ного состояния, в котором он не образует комплекса с нитрилами.
Вг CN
Ф Кипячение 3 ««с; затем FeCls
+ CuCN ДМФА ---------------—-------------
у 0,30 моля 150 мл
\ I
СОСНз СОСНз
0,25 моля
Наиболее удобным методом синтеза циклогександиона-1,2 яв-
ляется окисление 2-оксициклогексанона Ж, х. в кислом растворе
19а].
FeCL, — 2 н. НС1
Катализатор для восстановительного расщепления. При вос-
становительном расщеплении нитропропена (1) до о-метоксифенил-
ацетона (2) действием порошкообразного железа и конц. соляной
кислоты прибавляют в качестве катализатора небольшое количество
Ж. х. [10].
^Ч/СНО сн3
I II !
4/\q(2H3 СН2ХО2
СН3
сн=с\ю,
± Fe + НС1 +
/ч 200 г 360 мл
' ОСН3
(1)
Н„0тС6Н,СН3
+ FeCL 6ELO —„-------->
J ~ 63~71'“
СН3
/х/сн2с=о
хосн3
Ы)
Цветная проба. Некоторые фенолы дают характерную окраску
при добавлении к их разбавленному водному или спиртовому раст-
вору капли раствора Ж. х. Эта проба отрицательна для нитрофено-
лов и /г- и щ-оксибензойных кислот. [З-Кетоэфир (1) [11] и р-дике-
10
тон (2) [12] дают с Ж- х. положительную пробу, указывающую на
присутствие енольных форм.
О
li
СНз (СН2)4 ССНСО2СН (СН3)2
сн3
(1)
Х'''ХСССНаСОС(,Н5
(2)
1. Физер Л., «Синтезы органических препаратов», ИЛ, 14., 1949, сб. 2,
стр. 353.
2. W i с h е 1 h а и s Н., Вег., 30, 2199 (1897).
3. Hooker S. С., Ficser L. F., J. Am. Chem. Soc., 58, 1216 (1936).
The original structure assignment has been revised by Fieser L. F. and
unpublished work.
Am. Chem. Soc., 72, 228 (1950).
Fieser M., J. Am. Chem. Soc., 56, 1565 (1934).
Raphael R. A., J. Chem. Soc., 1962, 4263.
Runeberg J., Acta Chem. Scand., 11, 1060 (1957).
Sachs D. H. ,
4. Gates M., J.
5. Fieser L. F.,
6. L e w i s B. A.,
7. E r d t m a n H.,
1545
8. Bockelhei de V., Phillips J. B., J. Am. Chem. Soc., 85,
(1963).
9. Friedman L., S h e c h t e r H., J. Org. Chem., 26, 2522 (1961).
9a. De В о г g e r L.,Anteunis M., L a m m e n s H., Verzele M., Bull.
Soc. chiin. Belg., 73, 73 (1964).
10. Heinzelman R. V., Org. Syn., Coll. Vol., 4, 573 (1963).
11. Rinehart K. L., Org. Syn., Coil. Vol., 4, 120 (1963).
12. Уилер T., «Синтезы органических препаратов», ИЛ, М.,1953, сб. 4, стр. 516.
ЖЕЛЕЗА(Ш) ХЛОРИД БЕЗВОДНЫЙ, FeCl3. Мол. вес 162,22,
т. пл. 282°, т. кип. 315°.
Цветную пробу на фенолы и енолы проводят, добавляя 1 каплю
1%-ного раствора воз огн а иного Ж- х. б. в метаноле к раствору
1—2 капель вещества в 2—3 мл метанола [11.
Ж- х. б. восстанавливается порошкообразным железом в кипя-
щем ТГФ в атмосфере азота до хлористого железа, которое приме-
няют для получения ферроцена [2]. При получении раствора амида
натрия в жидком аммиаке к последнему добавляют каталитическое
количество Ж. х. б. и затем натрий в количестве, достаточном для
превращения соли железа в каталитически активное железо [3,4].
1. Henecka Н., Chem. Вег., 81, 188 (1948).
2. Уилкинсон Дж., «Синтезы органических препаратов», ИЛ, М., 1958,
сб. 8, стр. 64.
3. Khan N. A., Deatherage F. Е., Brown J. В., Org. Syn., Coll.
Vol., 4, 851 (1963),
4, Кхан H., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4, стр. 494.
ЖЕЛЕЗО, Fe. Ат. вес 55,85.
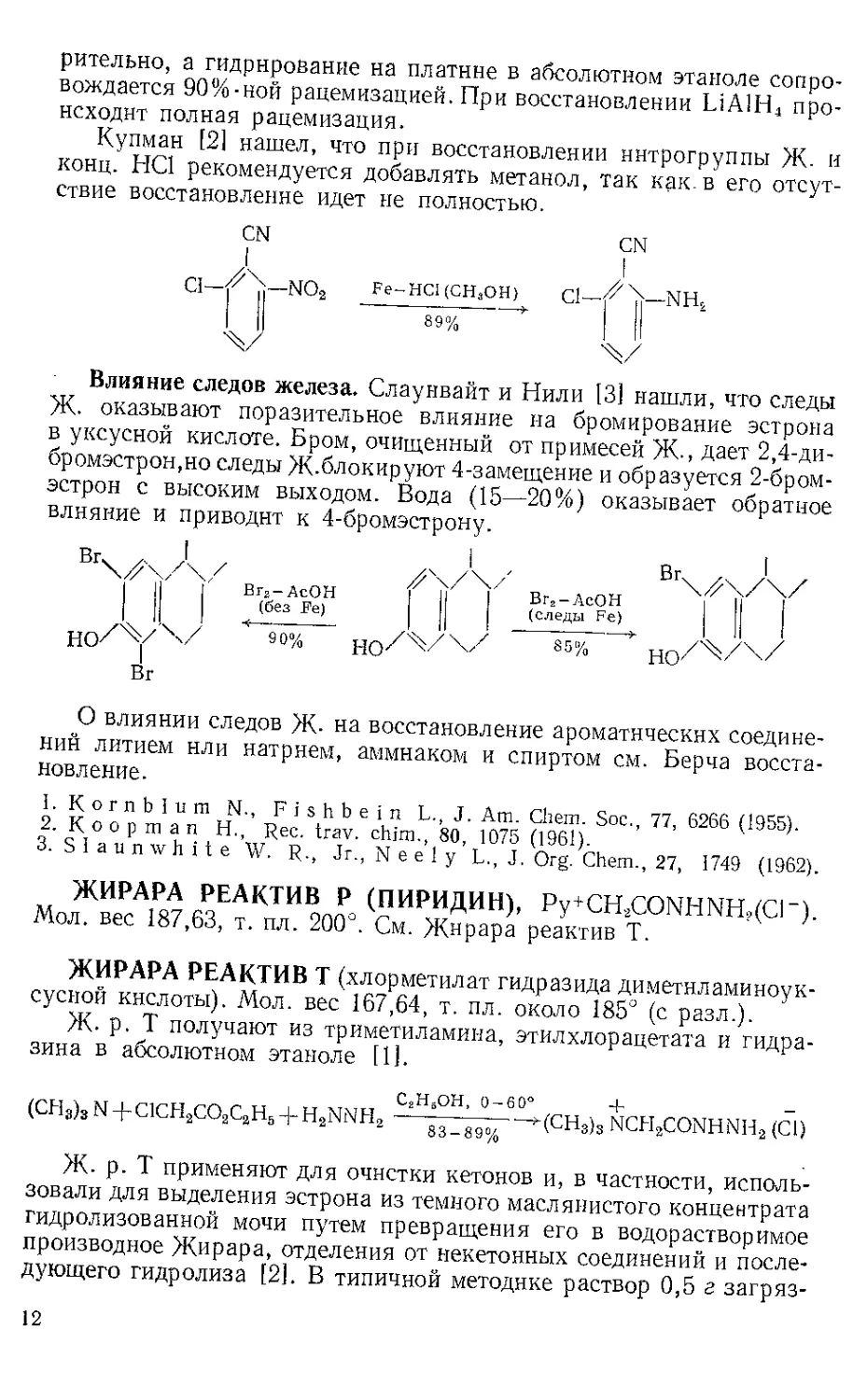
Восстановление. Корнблюм [11 показал, что восстановление
(—)-2-нитрооктана до амина Ж. и уксусной кислотой протекает
по крайней мере с 82%-ным сохранением оптической чистоты. Гидри-
рование на платине в уксусной кислоте несколько менее удовлетво-
рительно, а гидрирование на платине в абсолютном этаноле сопро-
вождается 90%-ной рацемизацией. При восстановлении LiА1Н4 про-
исходит полная рацемизация.
Купман [2] нашел, что при восстановлении ннтрогруппы Ж. и
конц. НС1 рекомендуется добавлять метанол, так как. в его отсут-
ствие восстановление идет не полностью.
CN
I
NO2
Ж/
Fe-HCl (СНаОН)
CN
Cl—NFL
Влияние следов железа. Слаунвайт и Нили [31 нашли, что следы
Ж. оказывают поразительное влияние на бромирование эстрона
в уксусной кислоте. Бром, очищенный от примесей Ж., дает 2,4-ди-
бромэстрон,но следы Ж-блокируют 4-замещение и образуется 2-бром-
эстрон с высоким выходом. Вода (15—20%) оказывает обратное
влияние и приводит к 4-бромэстрону.
Вг
9 0%
Вг2-АсОН
(без Fe)
Br3-AcOH
(следы Fe)
85%
Вг\у/у
О влиянии следов Ж- на восстановление ароматических соедине-
ний литием нли натрием, аммиаком и спиртом см. Берча восста-
новление.
I. Kornblum N., F i s h b e i n L., J. Am. Chem. Soc., 77, 6266 (1955).
2. Koopnian H., Rec. trav. chirm, 80, 1075 (1961).
3. Slaunwhite W. R., Jr., Neely L., J. Org. Chem., 27, 1749 (1962).
ЖИРАРА РЕАКТИВ P (ПИРИДИН), Py+CH2CONHNH?(Cr).
Мол. вес 187,63, т. пл. 200°. См. Жирара реактив Т.
ЖИРАРА РЕАКТИВ Т (хлорметилат гидразида ди метилами ноук-
сусной кислоты). Мол. вес 167,64, т. пл. около 185° (с разл.).
Ж- р. Т получают из триметиламина, этил хлор ацетата и гидра-
зина в абсолютном этаноле [1].
СгН6ОН, 0-60° +
(CH3)3N + ClCH2CO2CaH5 + H2NNH2 — --^(СН3)3 NCH3CONHNH2 (Cl)
О 3 - О У Уо
Ж- р. Т применяют для очистки кетонов и, в частности, исполь-
зовали для выделения эстрона из темного маслянистого концентрата
гидролизованной мочи путем превращения его в водорастворимое
производное Жирара, отделения от некетонных соединений и после-
дующего гидролиза [2]. В типичной методике раствор 0,5 г загряз-
12
о
ненного флуоренона (мол, вес 180), 0,5 г Ж- р. Т и 0,5 мл уксусной
кислоты в 5 мл 95%-ного этанола кипятят 30 мин для получения
производного. Раствор охлаждают и переносят в делительную
воронку. После добавления эфира, воды и хлористого натрия
(чтобы избежать образования эмульсии) слои разделяют и водный
слой обрабатывают 1 мл конц. соляной кислоты. Смесь нагревают
на кипящей водяной бане для гидролиза производного Жирара й
удаления растворенного эфира. При этом выделяется флуоренон
в виде желтого масла, кристаллизующегося при охлаждении. 1
Насыщенные кетоны,, например холестано.н-3, образуют ироиз-'
водные Жирара, которые гидролизуются очень разбавленной кис-
лотой; А4-3-кетоны взаимодействуют с Ж- Р- Т легче, чем насыщен-
ные кетоны, но для их гидролиза требуется значительно более
высокая концентрация кислоты [3]. (Примеры разделения насы-
щенных и сх,р-непредельных кетонов см. в работах [За].) Бензо-
фенон реагирует с Ж- р. Т, но только очень медленно, тогда как
флуоренон — быстро. Производные Жирара альдегидов также мож-
но расщепить с высоким выходом [4].
Производное Жирара выделяется следующим образом: суспен-
зию Ж- р. Т и кетона в уксусной кислоте нагревают на кипящей
водяной бане до растворения, раствор упаривают досуха в вакууме
и остаток кристаллизуют из смеси метанол — ацетон [5]. 17-Кето-
стероиды можно определять полярографически в виде производных
Жирара в водном буферном растворе; эти производные восстанавли-
ваются на ртутном капельном электроде при потенциале полувол-
ны— 1,4 в [5,6]. Производные Жирара 3-кетостероидов не восста-
навливаются в условиях такого определения, тогда как А4-3-кетосте-
роидные производные Жирара восстанавливаются при потенциале
— 1,1 в и могут быть определены в присутствии вышеуказанных
соединений. Андростенолон можно определить микроаналити-
ческим окислением по Оппенауэру в сопряженный кетон и поляро-
графией производного Жирара. Описано большое число модифика-
ций и усовершенствований первоначальной методики анали-
за 17].
13
Дегидратация. Эренштейн и сотр. 18] нашли, что 3-кето-5£-ок-
систероиды можно дегидратировать до Л4 -3-кетостероидов кипяче-
нием с Ж. р. Т в уксуснокислом растворе.
См, также обзор [9].
1. Жир ар А., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. ЮО.
2. G i г а г d A., S a n d а 1 es с о G., Helv. Chim. Acta, 19, 1095 (1936).
3. Reichstein T., Helv. Chim. Acta, 19, 1107 (1936).
3a. Zaffaroni A., Burton R.B., Keutmann E. H., J. Biol. Chem.,
177, 109 (1949); Kuehne M. E., .1. Am. Chem. Soc., 83, 1492 (1961).
4. Le derer E., N achm ias G., Bull. soc. chem. France, 16, 400 (1949).
5. W о 1 f e J. K., Hershberg E. B., Fieser L. F., J. Biol. Chem.,
136, 653 (1940).
6. Hersh berg E, B., W о 1 f e J. K-, Fieser L. F., J. Biol. Chem.,
140, 215 (1941).
7. Kab asak al ian P., McGlot ten J., J. Electrochem. Soc., 105, 261
(1958),
8. Ehrenstein AC, Dunnenbergen M., J. Org. Chem., 21, 774
(1956); Ehrenstein M., Otto K., ibid., 24, 2506 (1959).
9. Wheeler О. H., Chem. Rev,, 62, 205 (1962).
3
ЗОЛОТОХЛОРИСТОВОДОРОДНАЯ КИСЛОТА, HAuC14-3H2O. Мол.
sec 394,08.
3. к. — специфический реагент для окисления а-токоферола в
а-токоферилхинон [1].
НзСч /(СН2)3 СН (СН2)з СН (СН,)3 СНСН3
1 ! i
СН3 СН3 СН3
2AuCl»+3H.O
* *
он
Н3СЧ А /(СН2)2С (СН2)3 СН (СН»)3 СН (СН2)3 СНСН3
|/ I 1 ' I } +6НС1
—*з сн3 сн3 сн3 сн3
h3c/x/'Vh3
о
При добавлении водного раствора 3. к. к раствору а-токоферола
в этаноле происходит быстрое окисление с выделением тонкодисперс-
ного золота. Выпариванием профильтрованного раствора получают
аналитически чистый а-токоферилхинон в виде золотисто-желтого
масла. Окисление хлорным железом или нитратом серебра дает
очень загрязненный продукт.
I. К arrer Р. et al., HeJv. Chim. Acta, 21, 951 (1938); 23, 455 (1940).
15
ИЗО АМИЛ НИТРАТ («а мн л нитрат») ,пзо“С‘ Мол. вес 133,15;
уд. вес 0,996.
- . Фонер [11 показал, что при нитровании циклических кетонов
действием И. лучшим основанием является трет-бутилат калия,,
а лучшим растворителем — ТГФ. В этих условиях с хорошим
выходом получена ди калиевая соль 2,5-динитроциклопентанона.
о
KO2N^ U ^NO2K
+- (СН3)3СОК + c5huono2 —21°->
Л 72%
п---------------------------0, Нмоля
0,05 моля 1 5моля в 35 л/л.ТГФ
"в 70 л/л ТГФ
Реагент . используют для превращения А4-3-кетостероидов
в 2а-нитро-А4-3-кетоны, 3-кето-5а-стероидов в 2а-нитро-3-кетоны
и 3-кето-5р-стерондов в 4₽-нитро-3-кетоны [2]. 17-Кетоны'дают
16-нитропроизводные [2,3].
1 . Feuer Н., Shepherd J. AV., S a v i d е s C., J. Am. Chem. Soc., 78,
4364 (1956); F e u e г H., Anderson R. S., ibid., 83, 2960 (1961).
2 . Schaub R. -E.., Fulmor W., Weiss M. J., Tetrahedron, 20, 373
'(1964).
3 .- Hassner A., Larkin J. M., J. Am. Chem., Soc., 85, 2181 (1963).
ИЗО АМИЛ НИТРИТ, U30<eHuONO. Мол. вес 117,15, т. кип. 99°,
уд. .вес 0,872.
Получение [1].
CsHhONO
нс1 У
16:
ос-Оксимииокетоны. В присутствии mpem-бутилата калия [21
или соляной кислоты [31 И. превращает кетоны, имеющие соседнюю
метиленовую группу, в а-оксиминопроизводные.
Арилирование ароматических соединений. При разложении
диазониевой соли в водном растворе при pH 8 в присутствии арома-
тического соединения получаются неудовлетворительные резуль-
таты вследствие гетерогенности смеси и из-за побочных реакций.
Процесс значительно улучшается при диазотировании без кислоты
под действием И. в присутствии субстрата [41.
•С1-^~7^ын2+свн6+.с5НйОМО
ч / ТУ /9 \ — / : -/
7 г 300 лгл У е 5,43г ”
Получение дегидробензола, см. Бензолдиазонийкарбоксилат-2.
Реакция с виииламииами. При попытке получить стабильную
диазониевую группу, связанную с не а ром этическим атомом угле-
рода, Кертин и сотр. [5] обрабатывали дифенил винил амин (1) И.
и неожиданно получили дифен ил ацетилен (2). Э-(Аминометилен)-
флуорен (3) дает при этом с небольшим выходом дивиниламин (4).
Механизм этих процессов неизвестен.
с6н5^ ^nh2
с4н5 х н
Z/30-C5HnONO
85% :
к.Нойс В., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 132.
2. L i t v a n F., R obinson R., J. Chem. Soc., 1938, 1997.
3. Са unt D.,Cr о w W. D-.r Haworth R. D. , Vocloz С. А., У. Chem.
Soc., 1950, 163b- '
4. Cadogan J. I. G., J. Chem. Soc., 1962, 4257.
5. C u rt in D. Y.,Ka mp nieier J. A., O’C о n ii о r B. R., J. Am. Chem.
Sdc., 87, 863 (1965).
ИЗОАМИЛНИТРИТ - ТРИФШРУКСУСНАЯ КИСЛОТА. Для
диазотирования слабоосновного аминофенола (I), нестабильного
в ^присутствии, следов воды, к перемешиваемому раствору амино-
фенЬла в этилайетате, содержащем трифторуксусную кислоту, до-
бавляют при 0° раствор изоамилнитрита в этилацетате [I]. После
17
концентрирования раствора и промывания эфиром получают желтое
диазосоединение (2).
СО2С2Н5 СО2С2Н5
1 zNH2 J N
//у/ (CH3),CHCH,CH»ONO, //\/^^^
| || " CFaCO"3H | ||
НО 8° /о 4x0"
(1) (2)
1. Papanastassiou Z. В., McMillan A., Czebot аг V- J.,
Bardos Т. J., J. Am. Chem. Soc., 81, 6056 (1959).
ИЗОБЕНЗОФУРАН, (Villa, не выделен).
И. обычно получают термическим разложением аддукта Ша
1,4-эндоксо-1,4-дигидронафталина (1а) и тетрафенилциклопента-
диенона (II) (см. схему на стр. 19). Прн нагревании до 165° этот
аддукт отщепляет окнсь углерода; выделен и идентифицирован при
этом только один продукт— 1,2,3,4-тетрафе.нилбензол (IX) [1].
Промежуточное образование И. было установлено путем его
захвата 1,4-эмдо-окисыо (1а) и выделением аддуктов X и XI. И.
получают также пиролизом аддукта Va, полученного из 1,4-эндо-
окиси (1а) и а-пирона (IV), и идентифицируют превращением в X
и XI.
I. F i е s е г L. F., И a d d a d i п М. J., Can. J. Chem., 43, 1599 (1965).
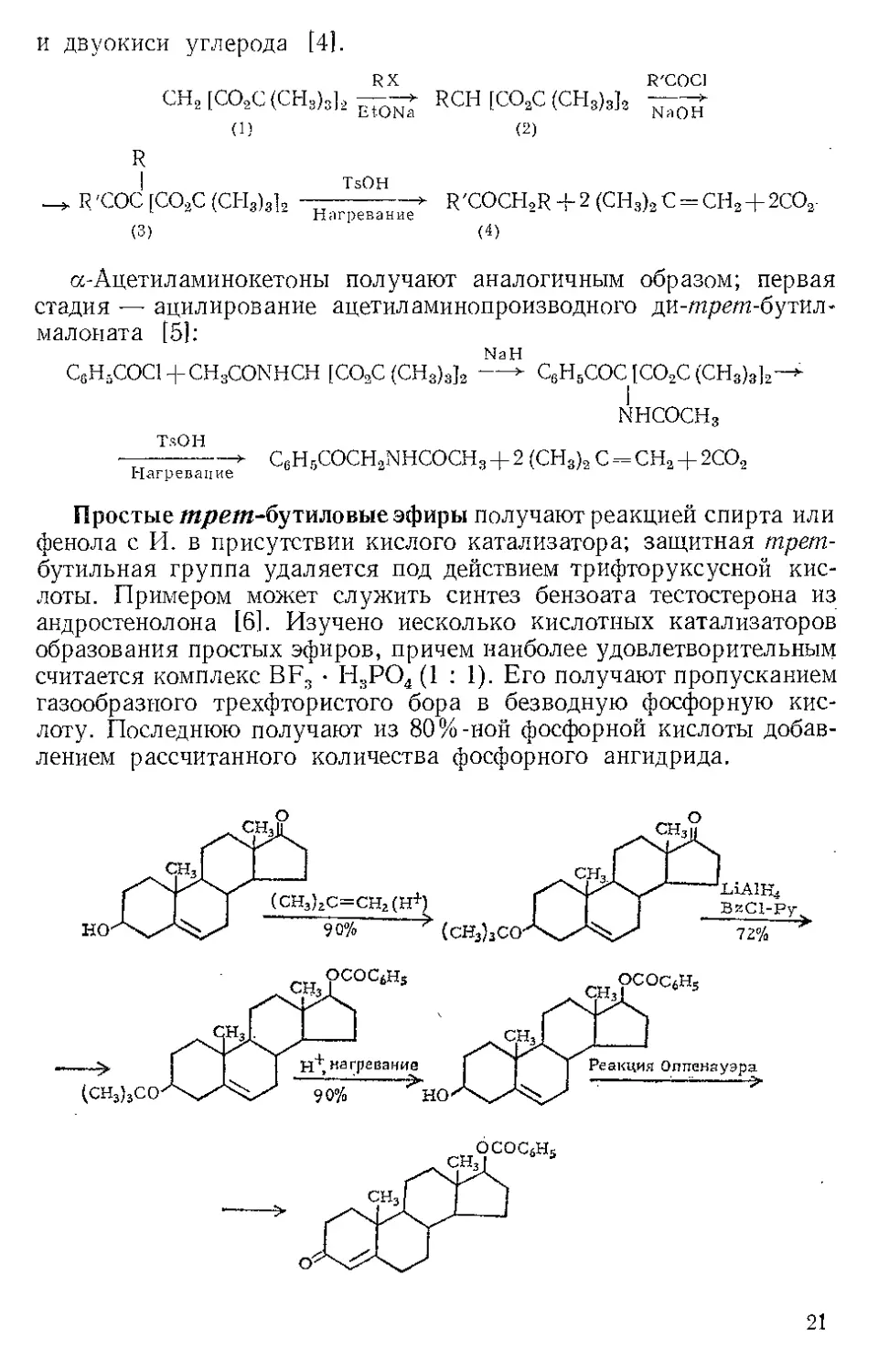
ИЗОБОРНИЛДИХЛОРАЛЮМИНАТ (1).
Получение. И. получают обработкой эфирного раствора изо-
борнеола (0,1 моля) хлористым алюминием (0,09 моля) и LiAlH4
(0,025 моля); добавляют небольшое количество трет-бутанола и ос-
тавляют до прекращения выделения водорода [1].
Восстановление [11. И.—восстанавливающий реагент, ана-
логичный смешанному гидриду (HA1CL); он проявляет исключи-
(5)
18
тельную стереоспецифичность при восстановлении производных
циклогексанона, давая главным образом аксиальные спирты.
Например, И. реагирует с холестаноном (2), образуя камфору
(3) и (после гидролиза и кристаллизации) холестанол-За (5) с почти
70%-ным выходом. Хроматография сырого спирта указывает на
образование смеси 86% а- н 14% (3-изомеров.
1. Е 1 i е 1 Е. L., N a s i р u г i D., J. Org. Chem., 30, 3809 (1965).
ИЗОБУТИЛЕН, (СН3)2С=СЫ2. Мол. вес 56,10, т. кип. —6,9°,
уд. вес 0,59.
трет-Бутиловые сложные эфиры. Получение трет-бутиловых
эфиров карбоновых кислот путем кислотно-катализируемого при-
соединения И. 'иллюстрируется методикой получения дн-трет-бу-
тилмалоната [1]. И. из баллона конденсируют при охлаждении
смесью ацетон — сухой лед и добавляют к охлажденной . смеси
малоновой кислоты, эфира и конц. серной кислоты в толстостенной
склянке для реакций под давлением; склянку закрывают и встряхи-
вают при комнатной температуре до растворения кислоты.
’ .у112 . /адиснз)^.
СН2 (СО2Н)24-С (СН3)а +Эфир-J-конц. H2SO4 —> СН2 ’ ;
0,48 моля 1,5 моля 100 мл 5 мл 58—60% \(2О-С(СН )
mpem-Бутиловые эфиры кислот имеют большое значение в син-
тезе, так как эта пространственно затрудненная эфирная группа
устойчива к гидролизу, ., но при необходимости легко удаляется
отщеплением И. в условиях кислотного катализа. Например, общий
метод синтеза этиловых эфиров (3-кетокислот [2] заключается в аци-
лировании этил-mpem-бутилмалойата через этокснмагниевое произ-
водное с последующим отщеплением И. и двуокиси углерода.. Необ-
ходимый для этого синтеза этил-трет-бутнлмалонат. получают из
диэтилмалоната и 1 же едкого кали в этаноле с образованием моно-
этилмалоната н обработкой последнего И. и серной кислотой [3].
,СО2С3Н5 /СО2С,Н5
5 Мо--ЬС2НГ1ОН / 325 RCOC1
СН2-------------------> C2H5OMgCH . ---->
^CCRC (СН3)3 ^ССЦС (СН3)3
/СОаСаН5 .
/.. TsOH
RCOCH ' ' НаГреван^ RCOCH2CO2C2H5 + (CHa)aC = CH2 + CO2
^СО2С(СН3)з агревание .
Синтез кетонов типа R'COCH2R включает алкилирование ди-
mpem-бутилмалоната (в виде Na-производного) с образованием (2),
ацилирование с образованием (3) и расщепление с выделением И.
20
и двуокиси углерода [4].
RX R'COC]
СН2 [СО2С (СН3)3]2 RCH [СО2С (СН3)3]2
(1) С (2)
R
I TsOH
—R'СОС [СО.,С (СН3)зЬ--------------> R'COCH,R + 2(CH3)2C = CH2 + 2CO3
Нагревание
(3) (П
а-Ацетил аминокетоны получают аналогичным образом; первая
стадия — ацилирование ацетил аминопроизводного ди-трет-б ути л-
маловата [5]:
NaH
C6H5COC14-CH3CONHCH [СО3С (СН3)3]2-> С6Н5СОС1СО2С (СН3)з]2—
I
NHCOCH3
TsOH
---------> C6H5COCH2NHCOCH3 + 2 (СН3)2 С = СН2 + 2СО2
Нагревание
Простые m/mm-бутиловые эфиры получают реакцией спирта или
фенола с И. в присутствии кислого катализатора; защитная трет-
бутильная группа удаляется под действием трифтор уксусной кис-
лоты. Примером может служить синтез бензоата тестостерона из
андростенолона [6]. Изучено несколько кислотных катализаторов
образования простых эфиров, причем наиболее удовлетворительным
считается комплекс BF3 • Н,РО4 (1 : 1). Его получают пропусканием
газообразного трехфтористого бора в безводную фосфорную кис-
лоту. Последнюю получают из 80 %-нон фосфорной кислоты добав-
лением рассчитанного количества фосфорного ангидрида.
21
Синтез пептидов. N-Защищенную аминокислоту (1) можно
легко превратить в тргт-бутиловый эфир (2) реакцией с избытком
И. в присутствии серной кислоты или ц-толуолсульфокислоты [7].
Удаление N-защитной группы гидрогенолизом дает трет-бутило-
вый эфир аминокислоты, более стабильный, чем метиловый или
СН,=С(СНЩ Н.-Pci
CbNHCHCO,H ---У-----> CbNHCHCO3C (CHS)S--> H2NCHCO2C (СН3)3
I HiS°4 I ' I
R R R
(1) (2) (3)
этиловый эфир, так как объемная /преш-бутильная группа создает
пространственные затруднения для межмолекулярной конденсации,
трет-Бутильную группу при необходимости можно удалить отщеп-
лением И. при кипячении в бензоле с кислым катализатором (H2SO4,
TsOH) или с НВг—АсОН. Свободную аминокислоту можно также
этерифицировать непосредственно под действием И. в смеси диок-
сан— серная кислота (10:1) [8].
mpem-Бутильную группу применяют также для защиты гидро-
ксильной группы серина или тирозина [9]. Эфиры расщепляются
смесью НВг—АсОН или НС1—СНС13. Для защиты сульфгидриль-
ных групп mpem-бутильная группа неудобна, так как для расщеп-
ления эфиров необходимы жесткие условия.
1. McCloskey A. L.,Fonken G. S., К. 1 u i b е г R. W., Johnson
W. S., Org. Syn., Coll. Vol., 4, 261 (1963).
2. Breslow D. S., В aumgarten E., HauscrC. R., J. Am. Chem.
Soc., 66, 1286 (1944).
3. Strube R. E., Org. Syn., Coll. Vol., 4, 417 (1963).
4. Fonken G. S. , J о h n s о n W. S., J. Am. Chem. Soc., 74, 831 (1952);
Puterbaugh W. H., Swamer F. W., Hauser C. R., ibid., 74,
3438 (1952).
5. Schrecker A. W., Trail M. M., J. Am. Chem. Soc., 80, 6077 (1958).
6. Beyerman H. C., Heiszwolf G. J., J. Chem. Soc., 1963, 755; Rec.
trav., 84, 203 (1965).
7. Anderson G. W., Callahan F. M., J. Am. Chem. Soc,, 82, 3359 (1960).
8. R о e s k e R., J. Org. Chem., 28, 1251 (1963).
9, В e у e r m a n FI. C., Bontekoe J. S., Rec. trav., 81, 691 (1962); Cal-
lahan F. M., Anderson G. \V., P а и 1 R., Zimmerman J. E.,
J. Am. Chem. Soc., 85, 201 (1963); W ii n s c h E., J e n t s c h J., Chem. Ber.,
97, 2490 (1964); Po d iisk а К.., T i t о v M. 1., Coll. Czech., 30, 1611 (1965).
OCOCH-,
И30ПР0ПЕНИЛАЦЕТАТ, j . Мол. вес 100,11,
сн3с=сн2
т. кип. 967750 мм, уд. вес 0,93.
Ацетилирование енолов. Промышленное получение И. основано
на катализируемой серной кислотой реакции ацетона с кетеном [11:
О ОН ососн3
II I СН^С=О I
сн3—с-сн3^±сн3с=сн2 —•сн3с=сн2
22
И, представляет собой енолацетат ацетона и в присутствии
каталитического количества серной или n-толуолсульфокислоты
вступает в обменную реакцию с высшими кетонами с образованием
высших енолацетатов и ацетона, который удаляют отгонкой для
смещения равновесия. Обычно реакцию проводят в бензоле илн
в избытке реагента.
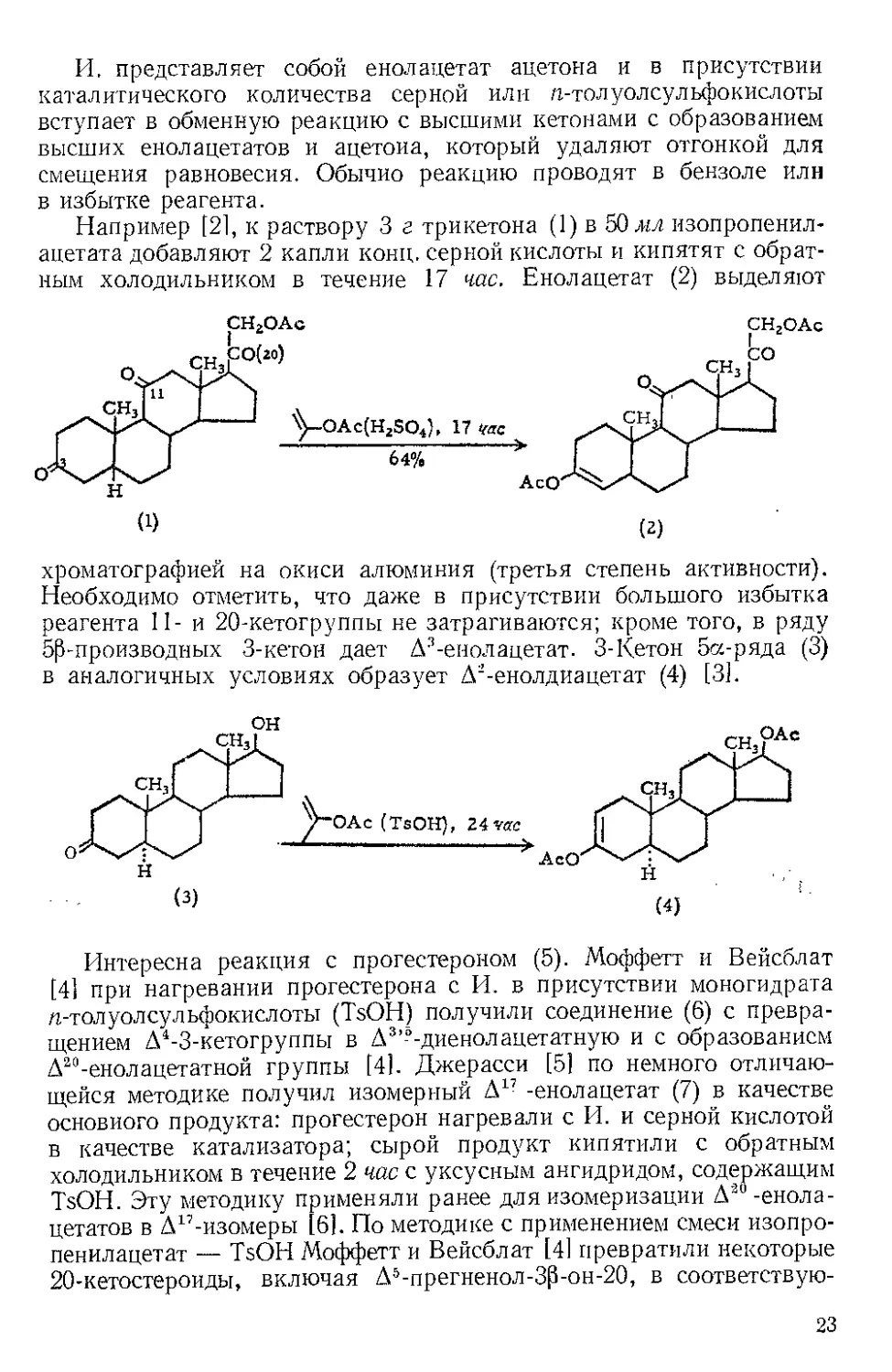
Например [21, к раствору 3 г трикетона (1) в 50 мл изопропенил-
ацетата добавляют 2 капли конц. серной кислоты и кипятят с обрат-
ным холодильником в течение 17 час. Енолацетат (2) выделяют
хроматографией на окиси алюминия (третья степень активности).
Необходимо отметить, что даже в присутствии большого избытка
реагента 11- и 20-кетогруппы не затрагиваются; кроме того, в ряду
5р-производных 3-кетон дает Д3-енол ацет ат. 3-Кетон 5а-ряда (3)
в аналогичных условиях образует Д -енолдиацетат (4) [31.
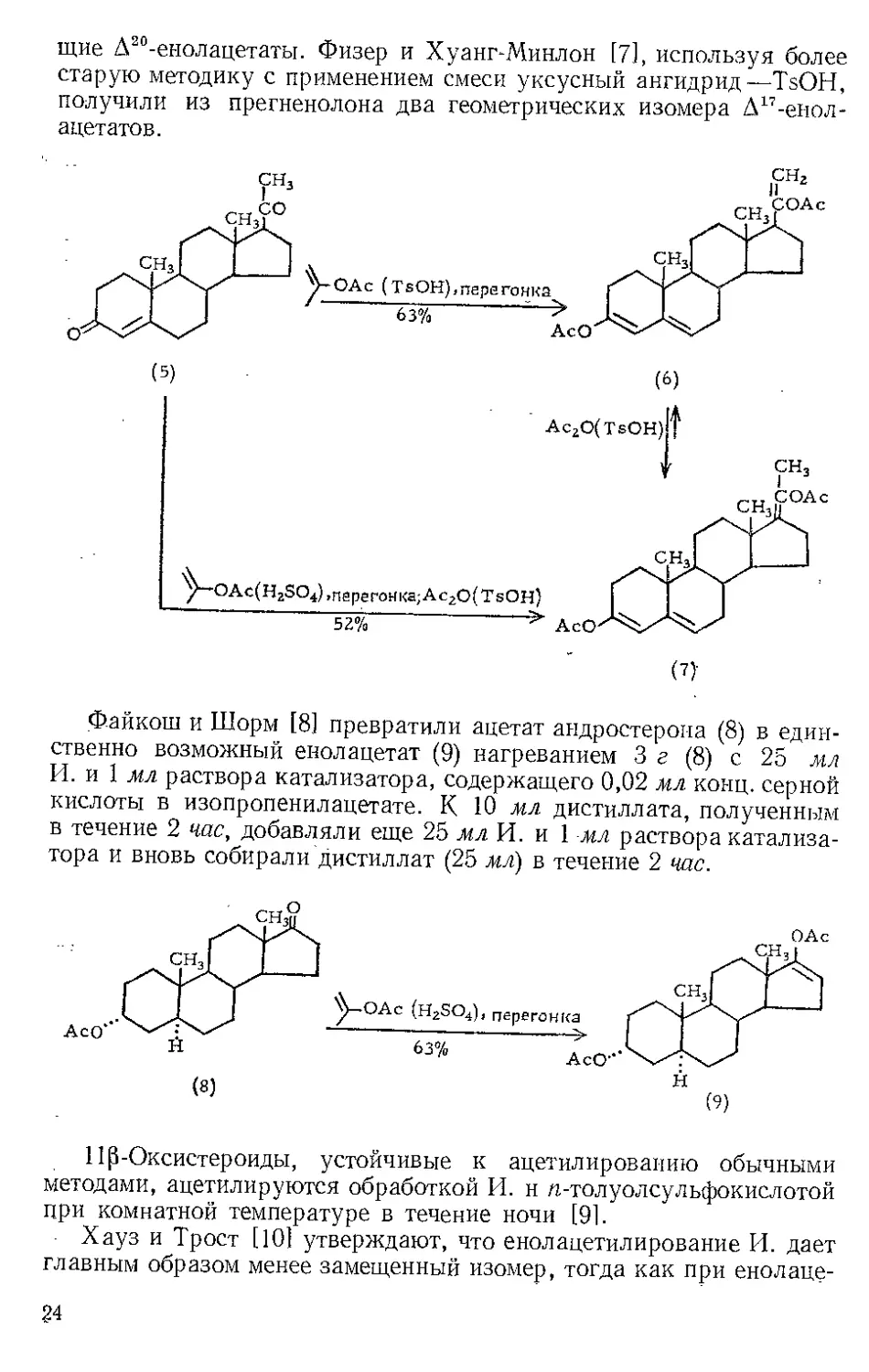
Интересна реакция с прогестероном (5). Моффетт и Вейсблат
[41 при нагревании прогестерона с И. в присутствии моногидрата
ц-толуолсульфокислоты (TsOH) получили соединение (6) с превра-
щением Д4-3-кетогруппы в Д3,?-диенолацетатную и с образованием
Д20-енол ацетатной группы [41. Джерасси [5] по немного отличаю-
щейся методике получил изомерный Д17 -енолацетат (7) в качестве
основного продукта: прогестерон нагревали с И. и серной кислотой
в качестве катализатора; сырой продукт кипятили с обратным
холодильником в течение 2 час с уксусным ангидридом, содержащим
TsOH. Эту методику применяли ранее для изомеризации Д30 -енола-
цетатов в Д17-изомеры [6]. По методике с применением смеси изопро-
пенилацетат — TsOH Моффетт и Вейсблат [4] превратили некоторые
20-кетостероиды, включая Д5-прегненол-Зр-он-20, в соответствую-
23
щие Д20-енолацетаты. Физер и Хуанг-Минлон [7], используя более
старую методику с применением смеси уксусный ангидрид—TsOH,
получили из прегненолона два геометрических изомера Д17-енол-
ацетатов.
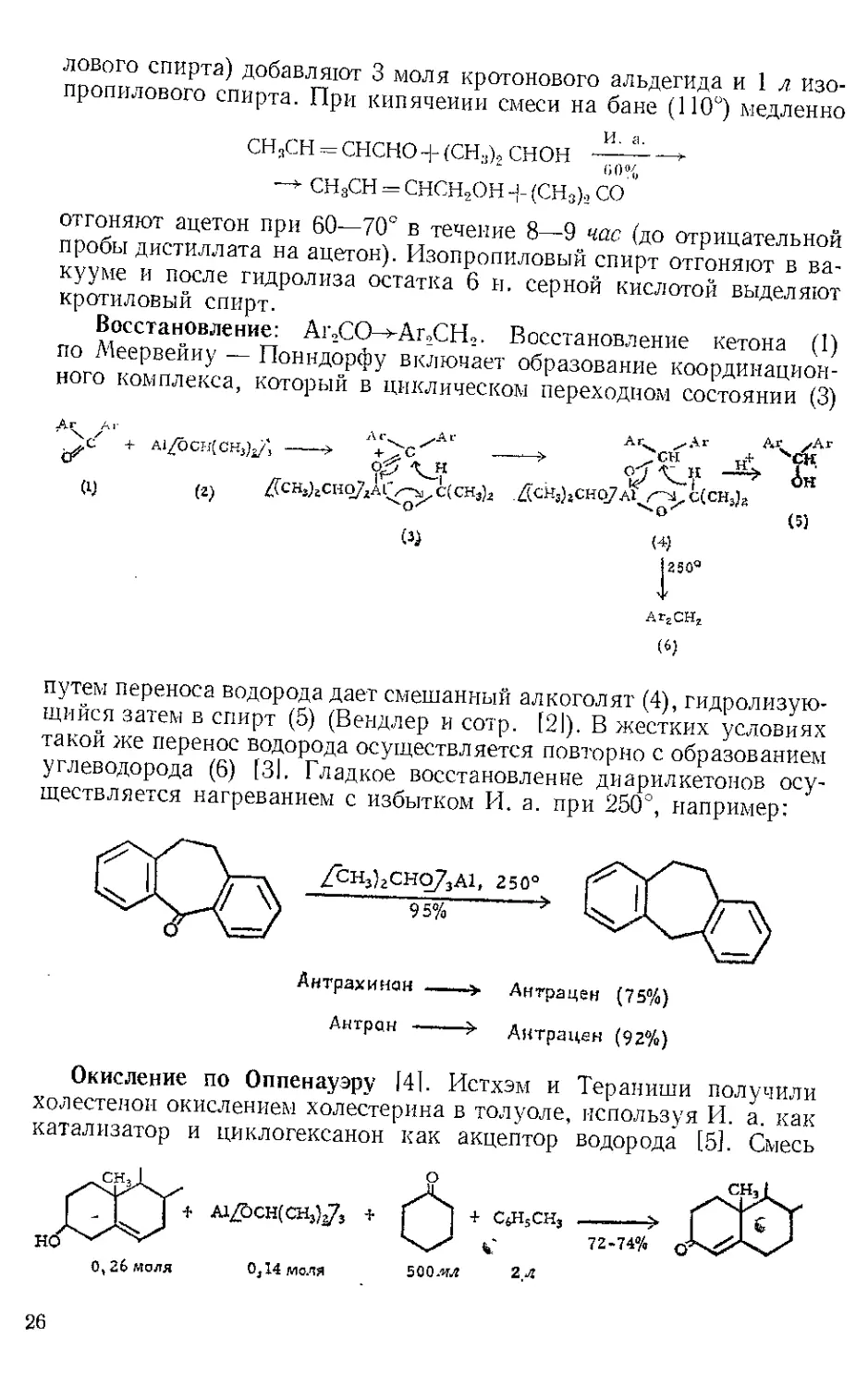
Файкош и Шорм [8] превратили ацетат андростерона (8) в един-
ственно возможный енол ацетат (9) нагреванием 3 г (8) с 25 мл
И. и 1 мл раствора катализатора, содержащего 0,02 мл конц. серной
кислоты в изопропенилацетате. К 10 мл дистиллата, полученным
в течение 2 час, добавляли еще 25 мл И. и 1 -мл раствора катализа-
тора и вновь собирали’дистилл ат (25 мл) в течение 2 час.
llp-Оксистероиды, устойчивые к ацетилированию обычными
методами, ацетилируются обработкой И. н п.-толуолсульфокислотой
при комнатной температуре в течение ночи [9].
Хауз и Трост [101 утверждают, что енолацетилирование И. дает
главным образом менее замещенный изомер, тогда как при енолаие-
24
тилировании уксусным ангидридом наблюдается обратная картина,
однако экспериментально эти выводы не подтверждены.
Конденсация с янтарным ангидридом [111. И. реагирует с ян-
тарным ангидридом (или хлор ангидридом) в дихлорэтане в присут-
ствии хлористого алюминия, давая 2-ацетилциклопентандион-1,3
с умеренным выходом.
ососн3
сн2=ссн3
А1С13, С1СН2СН2С1
55%
Более низкие выходы получаются при конденсации с глутаро-
вым (40%) и малеиновым ангидридами (16%).
Г. Hagemeyer Н. J., Jr., Hull D. С., Ind. Eng. Chem., 41, 2920 (1949),
2. Deghenghl R:, Engel C. R-, J. Am, Chem. Soc., 82, 3201 (1960).
3; V i I 1 о 11 I R., R I n g о 1 d H. J., D j erassi C., J. Am. Chem. Soc.,
82, 5693 (1960); the experimental data are cited by D j e r a s s i C., «Steroid
Reactions», p. 41, Holden-Day, Inc. (1963).
4. Moffett R. B,, W e i s b 1 a 11 D. I., J. Am. Chem. Soc., 74, 2183 (1952).
5. Dj erassi C., Grossman J., Thomas G. H., J. Am. Chem. Soc.,
77, 3826 (1955).
6. V a n d e r h a e g h e H., Ratzenell enbogen E. R., D о b г i-
n e r K.., Gallagher T. F., J. Am. Chem. Soc,, 74, 28Ю (1952).
7. F i e s e r L. F;, H uang-Minlon, J. Am. Chem. Soc., 71, 1840 (1949).
8. Faj kos J., Sorm F-, Coll. Czech., 24, 766 (1959).
9. О 1 i v e t о E. P. et al., J. Am. Chem. Soc., 75, 5486 (1953); see also FI a 1-
s a 1 1 T. G., T h e о b a 1 d D. W., Wal-shaw К.. B., J. Chem. Soc.f
1964, 1029.
10. House H. O., Trost В. M., J. Org. Chem., 30, 2502 (1965).
11. Mereny 1 F., Nilsson M., Chem.. Scand., 17, 1801 (1963); 18, 1368
(1964); Nilsson M., ibid., 18, 441 (1964).
И30ПР0ПИЛАТ АЛЮМИНИЯ, Al[OCH(CH3)al3.Мол. вес204,24,
т. пл. 118°, т. кип. 130—14077 мм. Получение и применение [1].
Восстановление по Меервейну — Поиндорфу. И. а. применяют
для восстановления карбонильных соединений и, в частности, для
избирательного восстановления карбонильной группы непредель-
ных альдегидов и кетонов. При восстановлении кротонового альде-
гида до кротилового спирта fl] смесь 27 г очищенной алюминиевой
фольги, 300 мл изопропилового спирта и 0,5 г сулемы нагревают до
кипения, добавляют 2 мл четыреххлористого углерода в качестве
катализатора и продолжают нагревание. Наблюдается бурное выде-
ление водорода и потемнение смеси. Кипячение продолжают почти
до прекращения выделения газа (6—12 час). И. а. можно использо-
вать в растворе (темном из-за присутствия суспендированных твер-
дых примесей) или после отгонки растворителя и перегонки в ваку-
уме выделить в виде бесцветной жидкости. К полученному таким
образом раствору И. а. (из 1,74 моля алюминия в 500 мл изопропи-
25
лового спирта) добавляют 3 моля кротонового альдегида и 1 л изо-
пропилового спирта. При кипячении смеси на бане (110°) медленно
И. а.
СН3СН -= СНСНО + (СН3), СНОН —----->
СН3СН = СНСН2ОН -J- (СН3)3 со ’
отгоняют ацетон при 60—70° в течение 8—9 час (до отрицательной
пробы дистиллята на ацетон). Изопропиловый спирт отгоняют в ва-
кууме и после гидролиза остатка 6 и. серной кислотой выделяют
кротиловый спирт.
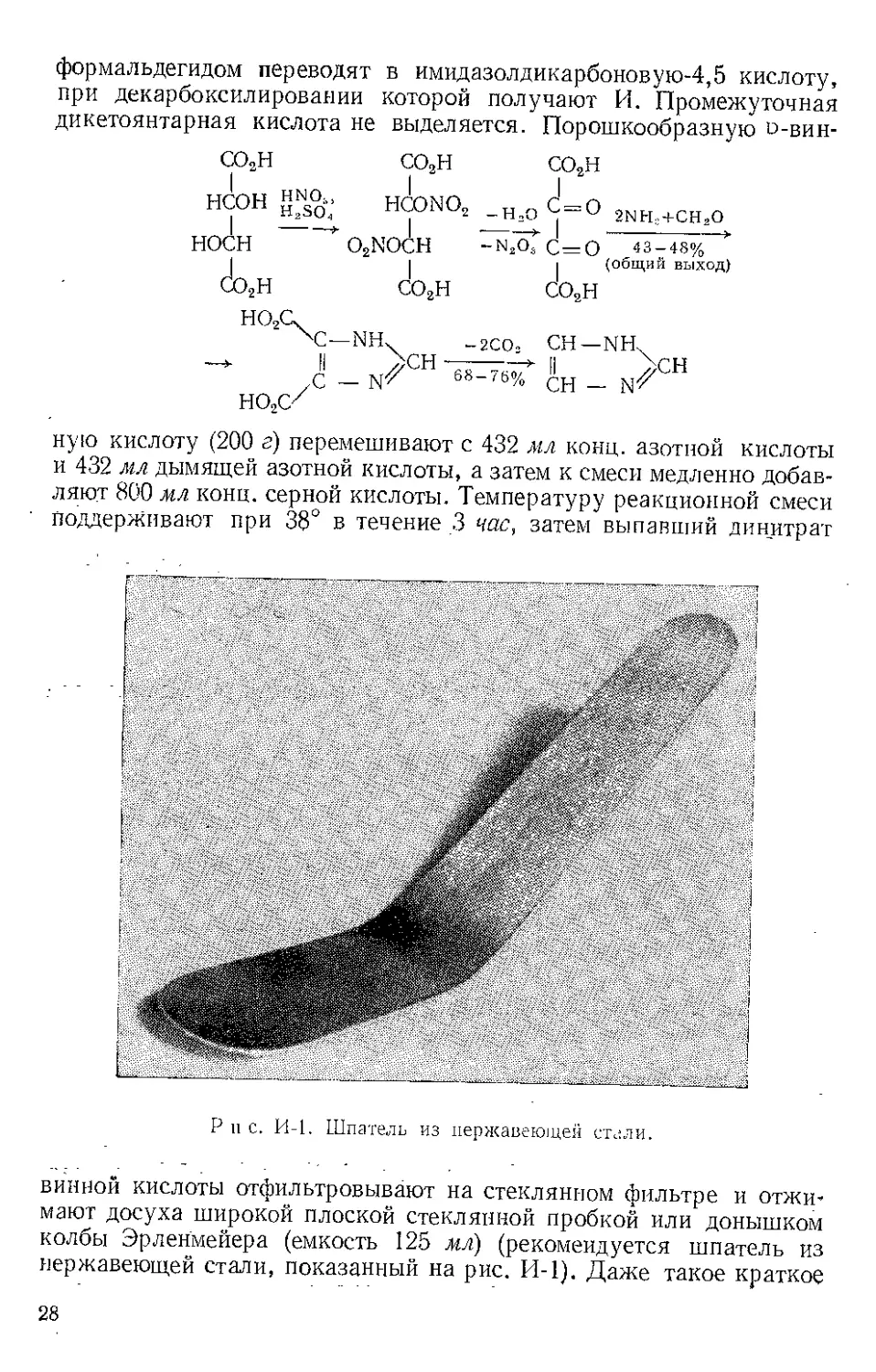
Восстановление: Аг2СО-^Аг2СН2. Восстановление кетона (1)
по Меервейиу — Понндорфу включает образование координацион-
ного комплекса, который в циклическом переходном состоянии (3)
'СН
А1/ОСИ(СНЖЛ ------>
ор VJ1
f2' /Гсн»)6сно74А1>-^.с(сн3)2 _/(СИ5)гСНО7А1 r~^,G(CHja
(3)
АГ.СН,
(6)
(5)
путем переноса водорода дает смешанный алкоголят (4), гидролизую-
щийся затем в спирт (5) (Вендлер и сотр. [21). В жестких условиях
такой же перенос водорода осуществляется повторно с образованием
углеводорода (6) [31. Гладкое восстановление диарилкетонов осу-
ществляется нагреванием с избытком И. а. при 250°, например:
Антрахинон ----> Антрацен (75%)
Антрон ----> Антрацен (92%)
Окисление по Оппенауэру [41. Истхэм и Тераниши получили
холестенон окислением холестерина в толуоле, используя И. а. как
катализатор и циклогексанон как акцептор водорода [5]. Смесь
А1/ЬСН(СНэ)з7э т
5 00.-ил 24
0,26 моля 0,14 моля
26
нагревают при перемешивании с отгонкой 900 мл толуола, затем
обрабатывают водным раствором K-Na-тартрата (чтобы перевести
ионы алюминия в раствор) и перегоняют с паром до получения 6 л
дистиллата. Продукт экстрагируют хлороформом и после двукрат-
ной перекристаллизации получают достаточно чистый холестенои.
Авторы считают, что это —менее трудоемкая модификация метода
Оппенауэра, применявшего mpem-бутилат алюминия. Однако этот
вывод сомнителен, так как не требующее внимания кипячение в те-
чение 8 час (по Оппенауэру), по-видимому, менее трудоемкая опе-
рация, чем длительные отгонки и перегонка с паром, описанные
выше.
Найдено [6], что формиаты в отличие от ацетатов легко окис-
ляются И. а. и дают те же продукты, что и незамещенные спирты.
Соединение (1) окисляли в (2) смесью И. а. и циклогексанона в кси-
лоле (т. кип. 1407760 мм, работа проводилась в Мехико при
570 мм) или толуоле (т. кип. 1117760 мм).
1, Уайлдс А. Л., «Органические реакции», ИЛ, М., 1950, сб. 2, стр. 194.
2. Woodward R. В., Wendler N. L., Brulschy F. J.,J. Am. Chem.
Soc., 67, 1425 (1945).
3. H of Isomni er R. D., Ta u b D., W e n d 1 e r N. L., Chem. Ind., 1964,
482.
4. Д ж e p а с с и К.-, «Органические реакции», ИЛ, М., 1953, сб. 6, стр. 235.
5. И с т х э м Дж., Тер ан иши Р., «Синтезы органических препаратов»,
ИЛ, М., 1956, сб. 7, стр. 75.
6. R Ingold Н. J., Loken В., Rosenkranz G., S о n d h е i m с г F.,
J. Am. Chem. Soc., 78, 816 (1956).
ИЗОЦИАНОВАЯ КИСЛОТА, см. Циановая кислота.
ИМИДАЗОЛ,
н
Мол. вес 68,08, т. пл. 90°, т. кип. 257°, рКЬ — 7,0; пикрат,
т. пл. 212°.
Получение. И. можно получить [11 по интересному методу,
предложенному Макуинном [21: D-винную кислоту превращают
в динитрат и этот эфир взаимодействием с гидроокисью аммония и
27
формальдегидом переводят в имидазолдикарбоновую-4,5 кислоту,
при декарбоксилировании которой получают И. Промежуточная
дикетоянтарная кислота не выделяется. Порошкообразную о-вин-
СО2Н СО2Н СО2Н
НСОН HCONO2 _н=о С = О 2NH;;+CH2O
HOCH ~*O2NOCH ^Хс=о“ 43 — 48%
j । । (общий выход)
CO2H СО2Н С02Н
но,о.
V—NH4 — 2С0» CH—NHX
н II >СН
ZC — Ж 68-/ь% сн _
HO2CZ
ную кислоту (200 а) перемешивают с 432 мл конц. азотной кислоты
и 432 мл дымящей азотной кислоты, а затем к смеси медленно добав-
ляют 800 мл конц. серной кислоты. Температуру реакционной смеси
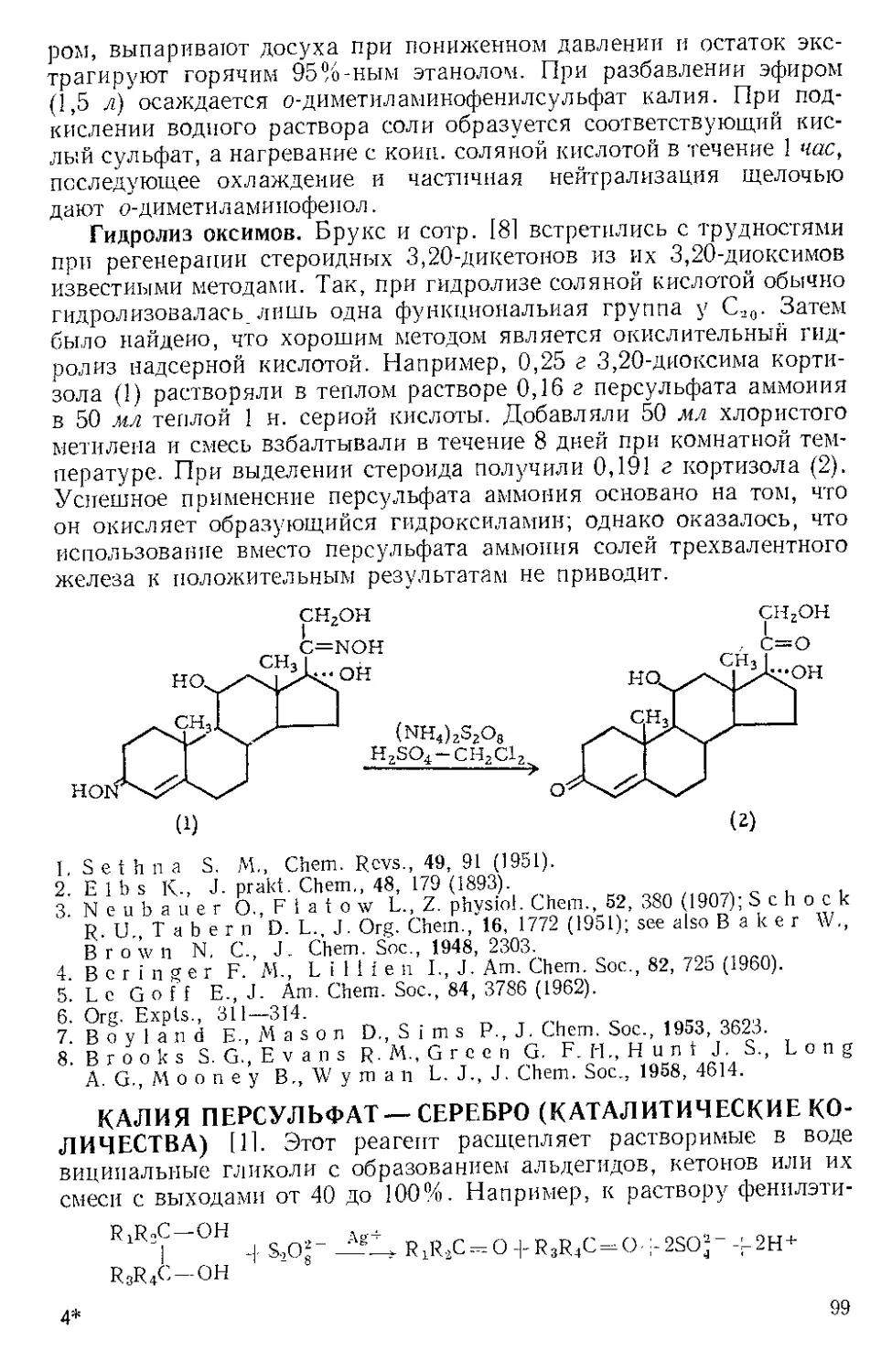
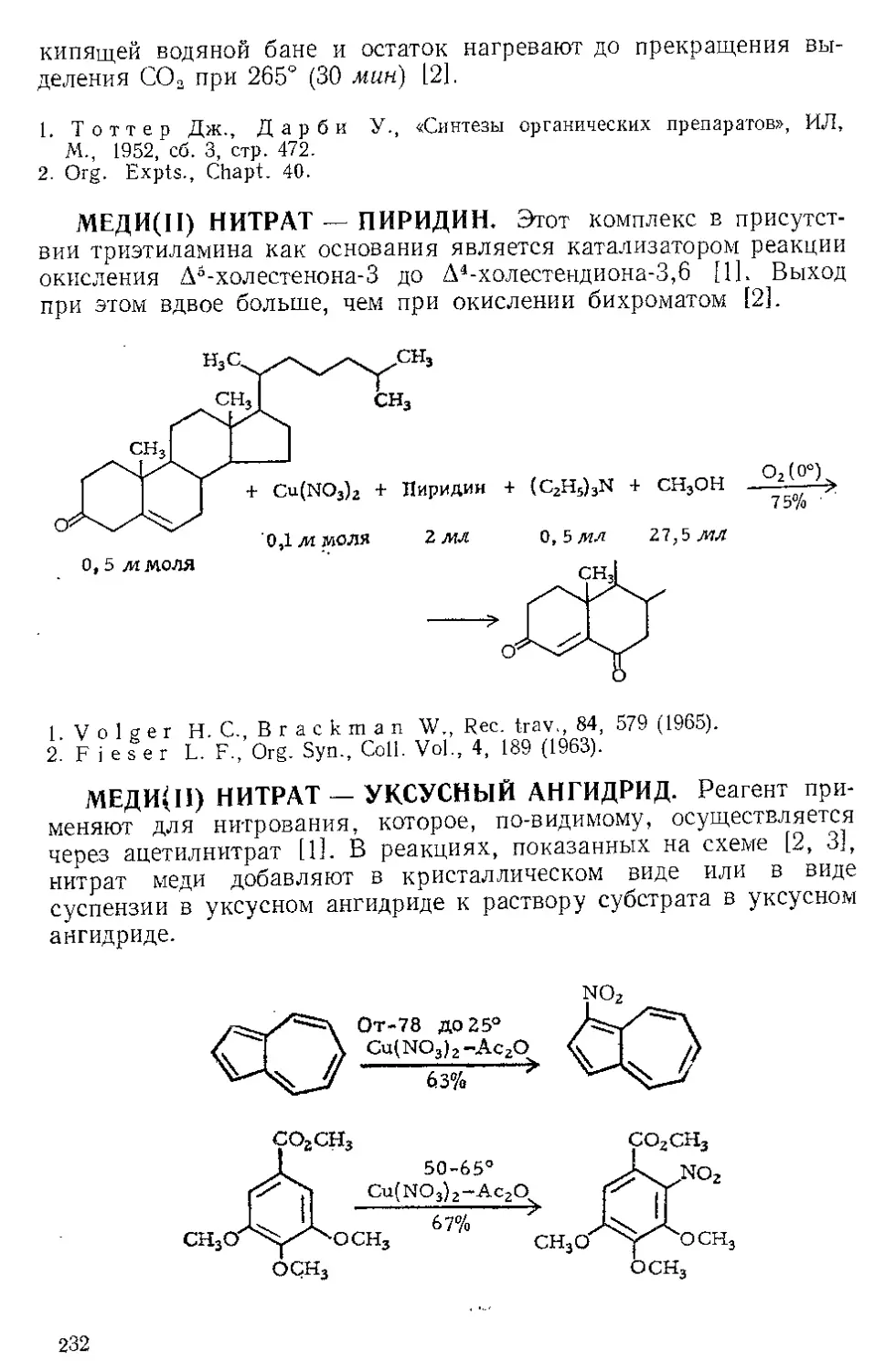
поддерживают при 38° в течение 3 час, затем выпавший динитрат
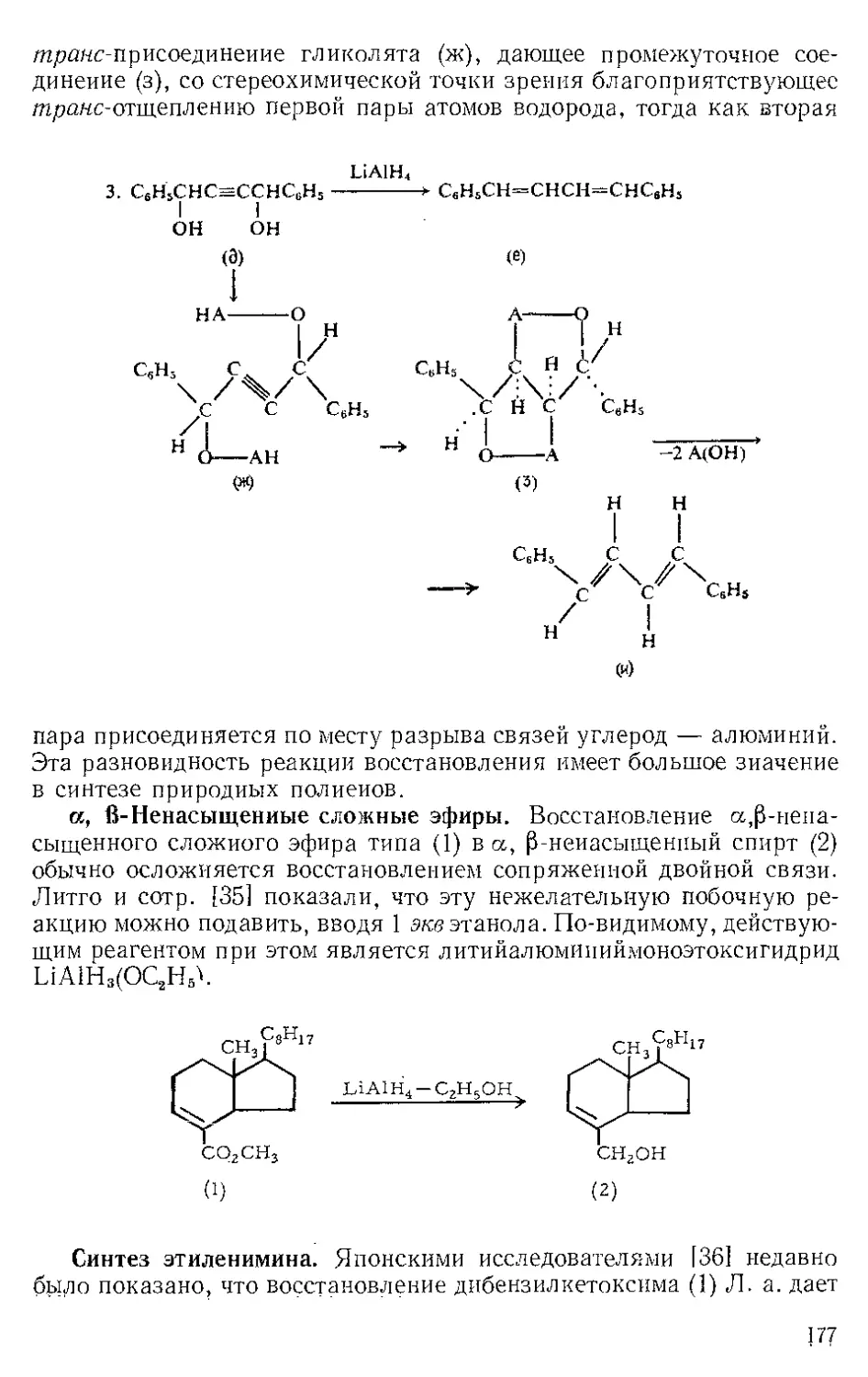
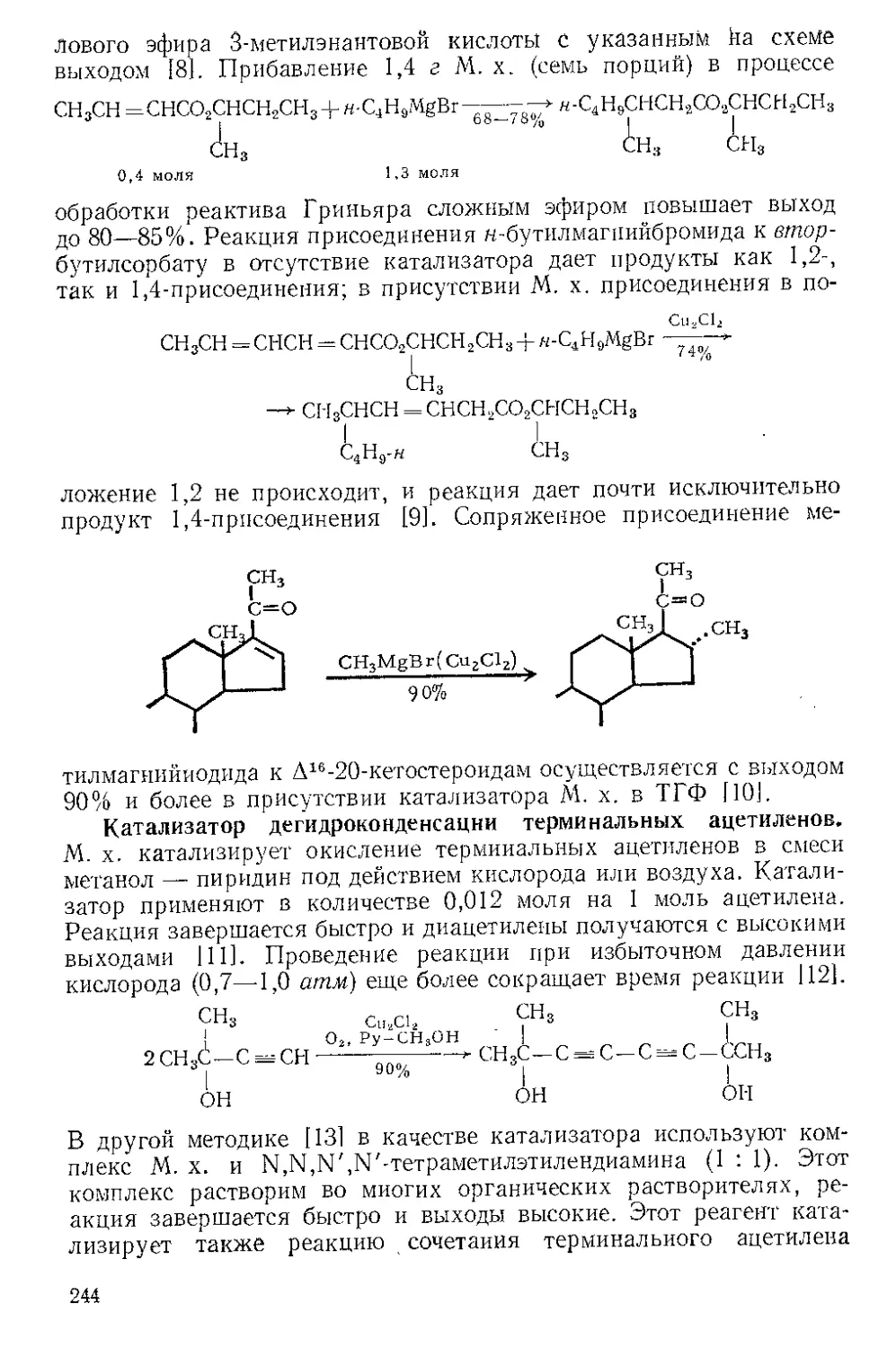
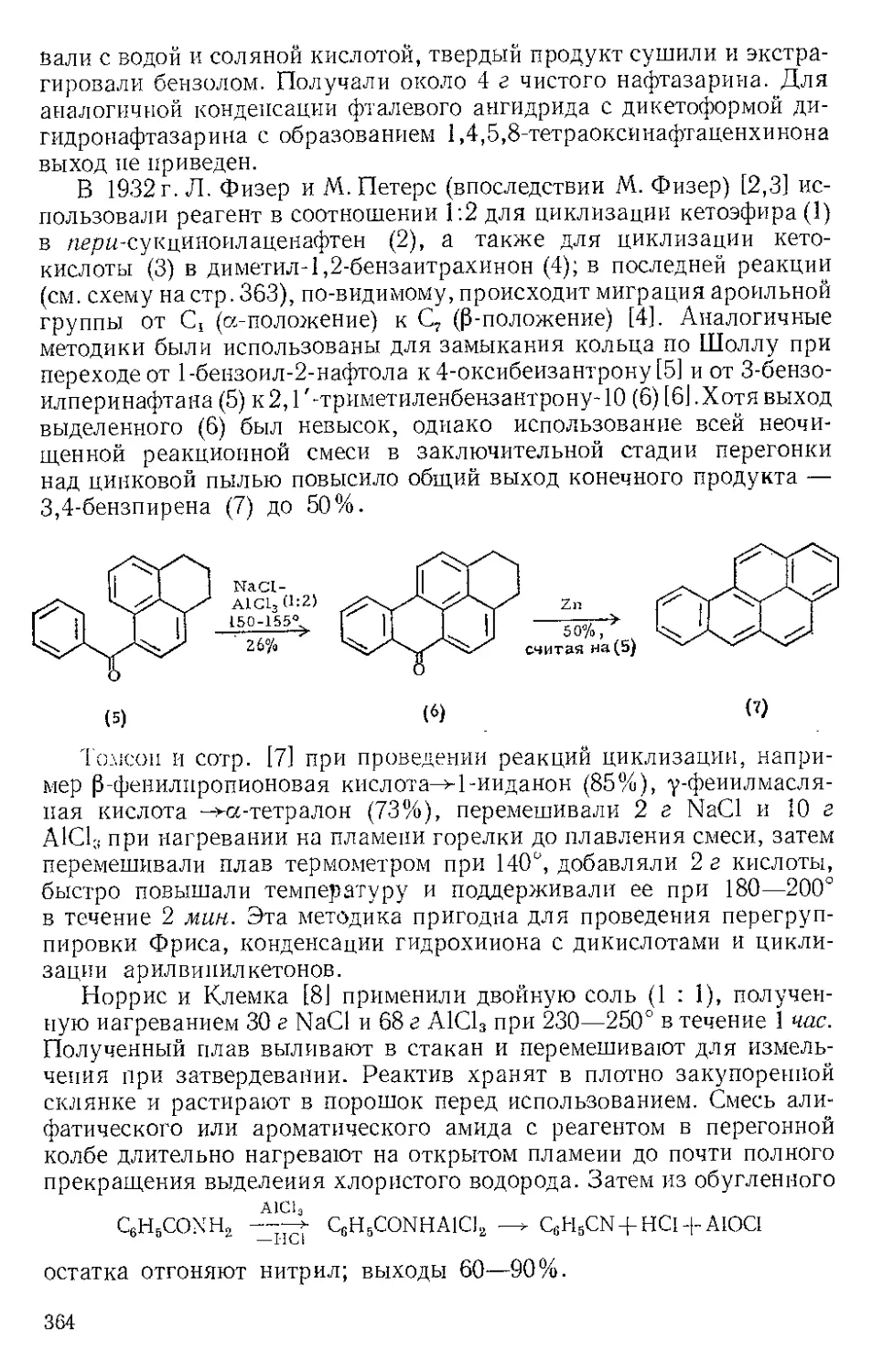
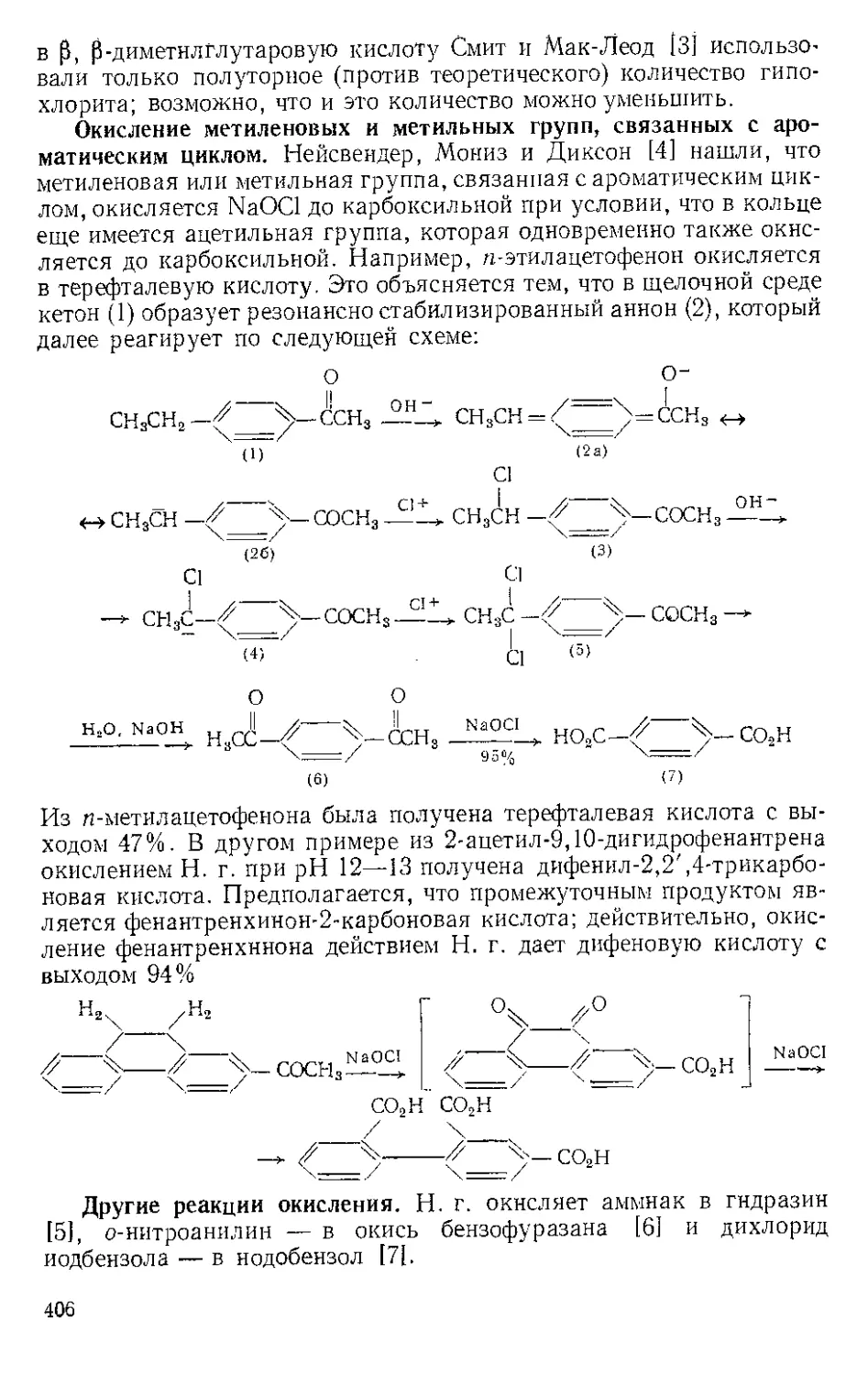
Р п с. И-1. Шпатель из нержавеющей стели.
винной кислоты отфильтровывают на стеклянном фильтре и отжи-
мают досуха широкой плоской стеклянной пробкой или донышком
колбы Эрленмейера (емкость 125 мл) (рекомендуется шпатель из
нержавеющей стали, показанный на рис. И-1). Даже такое краткое
28
описание показывает простоту и-изящество этого метода. В других
методах используют глиоксаль, аммиак и формальдегид 13].
В новом простом методе, описанном Бредереком и сотр. 14],
исходят из диэтил ацеталя бром ацетальдегид а (4), который легко
получается по Бедокяну [5] бромированием винилацетата и обра-
боткой бромировэнного продукта этанолом. Немецкие исследователи
сн2 - сно со сн3. ВГа> В Г СН2 СНО СО СН3 - В г СН2СН=О tOH>
-ЕЮАс НВ г
(1) (2) (3)
x*OEt
—>Br2CH2CH +
^ОЕГ
?Н*0Н 4- КОНЦ. НС1
СН2ОН
Кипячение
5 vac
94%
/ОСН2
->ВгСН,СН I
"S''OCH2
(4) 338 г 124 г 2 'МЛ (5)
^О—сн2
БгСНгСН |
^О-СН2
+ 2 HCONH2
6 уас'при’ 175°
50%
(5) 334 z
500 МЛ
показали, что сначала лучше превратить диэтилацеталь (4) в этилен-
кеталь (5) путем катализируемого кислотами обмена с этиленглико-
лем. Затем они нагревали циклический ацеталь (5) с избытком фор-
мамида, одновременно пропуская в раствор аммиак для предотвра-
щения разложения чувствительного к кислотам И., который был
получен с выходом 50%.
Синтез карбонильных соединений. Штааб разработал интересный
метод синтеза кетонов [6] и альдегидов f71 с использованием
И. Алифатическую или ароматическую кислоту переводят через хлор-
ангидрид в имидазолид, карбонильная группа которого чувстви-
тельна к нуклеофильной атаке. Реакцией имидазолида с реактивом
СбН5С—ci
C6H5MgBr; НгО
77,5% LiAlH4
С6Н5С—И.
II
с^гц— С—
Гриньяра можно получать кетоны, часто с хорошими выходами.
Имидазолид гладко восстанавливается алюмогидридом лития до
соответствующего альдегида. Избирательность реакции дает воз-
можность синтезировать альдегидоэфиры из моноэфиров дикарбо-
29
новых кислот и ациламиноальдегнды из ациламииокарбоиовых
кислот.
0 0 0 0
л-С2Н5ОС—СвН4 —С—имидазол /г-С9Н50С—С6Н4 — С— Н
78.5%
См. также NNr-Карбонил диимидазол, N,N'-Thohh л диимидазол,
КГ,М'-Тиокарбонилди имидазол.
Каталитическая активность. Имеются данные о том, что имида-
зольное кольцо гистидинового остатка некоторых гидролитических
ферментов ответственно за их протеолитическую активность. В связи
с этим было найдено [8,9], что И, обладает каталитической актив-
ностью при гидролизе фенилацетата и замещенных феии л ацетатов.
Эффективный катализ осуществляется лишь непротонированными
молекулами И. Заметно ускоряется И. также реакция ацилирования
эфиров аминокислот ц-нитрофениловым или другим активирован-
ным эфиром; возможно, что при этом в качестве нромежуточного
соединения образуется ацильное производное И. [10, 11].
Следует, однако, отметить, что, по данным Бейермана, Вейганда
и сотр. [12|, И, вызывает рацемизацию в большинстве пептидных
синтезов и менее эффективен, чем 1,2,4-триазол.
1, S п у d е г Н. R,,Handrick R. G., Brooks L. A., Org. Syn., Coll.
Vol., 3, 471 (1955).
2. Ma quenne M,, Ann. Chfm., 16], 24, 525 (1891).
3. R adziszewski B., Chem. Ber., 15, 1493 (1882); Be h rend R.,
Schmitz J., Ann., 277, 338 (1893).
4. В r e d e r e c k H., G о m p p e r R., Banger! R.,Herlinger H.,
Chem. Ber., 97, 827 (1964).
5. Bedouk ian P. Z., J. Am. Chem. Soc., 66, 651 (1944).
6. S t a a b H. A., Jost E., Ann., 655, 90 (1962); S t a a b H. A., Angew,
Chem., Internat. Ed., 1, 351 (1962).
7. S t a a b H. A., В r a u n 1 i n g H., Ann., 654, 119 (1962).
8. Ben der M. L., Tu m q nest B. W., J. Am. Chem. Soc., 79, 1652 (1957).
9. Bru ice T. C., S c h m i r G. L., J. Am. Chem. Soc., 79, 1663 (1957).
10. Mazur R. H., J. Org. Chem., 28, 2498 (1963).
11. W i e I an d T., V о ge 1 e r K., Angew. Chem., Internat, Ed, 2, 42 (1963);
Ann. 680, 125 (1964).
12. Beyerman H. C., Weygand F., et al., Rec. trav., 84, 213 (1965).
N-ИМИДАЗОЛКАРБОНОВОЙ КИСЛОТЫ шрвш-БУТИЛОВЫЙ
О
ЭФИР, ы.....у Мол. вес 168,20, т. пл. 46—47°.
| >N-COC (СН3)3
И. к. б. э. получают реакцией карбоиилдиимидазола с трет-
бутанолом и используют для /нретг-бутоксикарбоиилирования ами-
нокислот [1].
I. Klee W., Brenner М., Helv. Chim. Acta, 44, 2151 (1961).
ИМИНОДИКАРБОНОВОЙ КИСЛОТЫ ДИ-л«рел»-БУТИЛОВЫЙ
ЭФИР, НМ[СО2С(СН3)зк. Мол. вес 217,26, две формы, т. пл. 90 и 121°.
30
И.к. д. э. получают из этил-трет-бутилоксалата по следующей
схеме:
О
I!
СО2С2Н5 H2NNHa CONHNH2 hno, CN = N+ = N" (сн3)эсон
CO3C(CH3)3 CO2C(CH:i), CO2C(CH3)3
/CO.2C(CH3)3
HN< +\’o
ч:о2с(сн3)3
На последней стадии перегруппировка Курииуса осуществляется
при нагревании азнда с /црс/тг-бутанолом [11.
И.к. д. э. применяют для синтеза первичных аминов по реакции
Габриэля нли родственным реакциям. Например, суспензию гид-
рида натрия в растворе этого реагента в ДМФА перемешивают
6 час при 60° и к образовавшемуся натриевому производному добав-
ляют а, а'-дибром-о-ксилол, продолжая нагревание до завершения
алкилирования. Смесь разбавляют водой, тетраэфир экстрагируют
/СО2С(СН3)з
,СНаВг ,CH,N<
/||' H2NaN/CO'3C(CH3b 60’4№| || " СО,С(СН3)3
(Il Vo2C(CH3)3 4(1 /СО2С(СН3)3
хСН2Вг CHnN<
X2O3C(CHS)3
ZCH,NH,-HC1
57%* | Il
^/4'CH2NH2.HC1
хлористым метиленом и нагреванием с конц. соляной кислотой уда-
ляют сложноэфириые группы (в виде изобутилена и СОг) с образова-
нием дихлоргидрата о-ксилилемдиамина.
1. С а г р i п о L. A,, J. Org. Chem., 29, 2820 (1964).
ИНДИКАТОРЫ pH. Велыоз [1] опубликовал таблицу, содер-
жащую более 100 индикаторов, с примечаниями и ссылками на лите-
ратуру.
1. V е 1 1 u z L., Substances Naturelies de Synthese, 7, Part 3 (1953).
ИНДИКАТОРЫ pH, КИСЛОТНОСТЬ ПО ГАММЕТУ.
С помощью набора из 17 основных индикаторов можно опреде-
лить в любом растворителе любую кислотность в пределах от раз-
бавленной водной минеральной кислоты до чистой серной кислоты
[1]. Пределы индикаторов: от +2,8 (4-аминоазобензол) до—9,3
(2,4,6-тринитроаиилии).
1. Hammett L. Р., Deyrup A. J., J. Am. Chem. Soc., 54, 2721 (1932);
Hammett L. Р., Paul М. A., ibid., 56, 827 (1934).
31
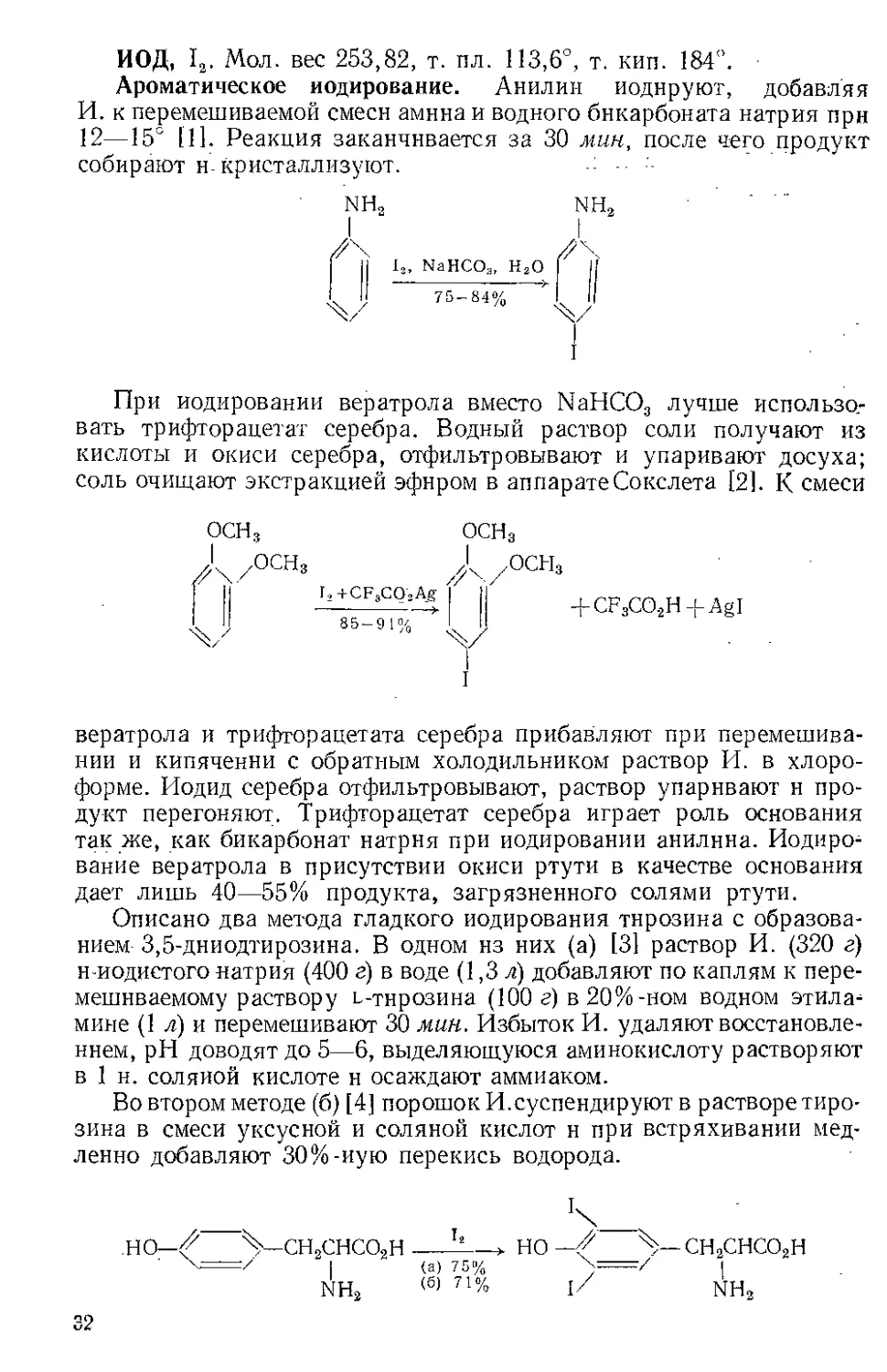
ИОД, 12. Мол. вес 253,82, т. пл. 113,6°, т. кип. 184”.
Ароматическое иодирование. Анилин иодируют, добавляя
И. к перемешиваемой смеси амина и водного бикарбоната натрия прн
12—15° 111. Реакция заканчивается за 30 мин, после чего продукт
собирают н-кристаллизуют.
NH, NH,
I р
\ Н I3, NaHCO:j, Н2О \ II
'Ч/ 75~84% V
I
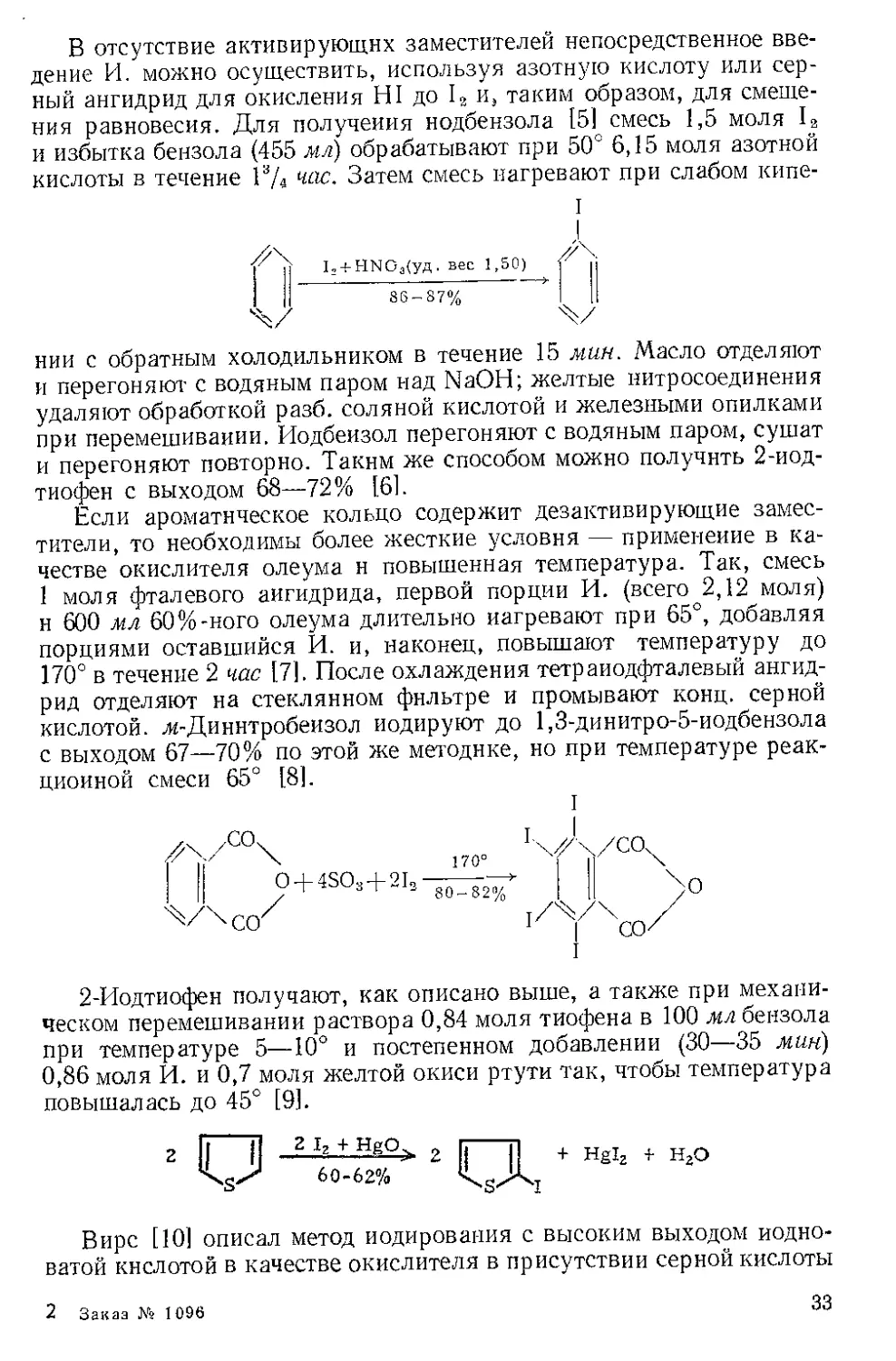
При иодировании вератрола вместо NaHCO3 лучше использо-
вать трифторацетат серебра. Водный раствор соли получают из
кислоты и окиси серебра, отфильтровывают и упаривают досуха;
соль очищают экстракцией эфиром в аппарате Сокслета [2]. К смеси
ОСН3
/ОСНз
'Ч/
ОСН3
/ОСНз
r2+CFsCQ’3Ag I 11
~85-91%“*
I
+ CF3CO.2H-f-AgI
вератрола и трифторацетата серебра прибавляют при перемешива-
нии и кипячении с обратным холодильником раствор И. в хлоро-
форме. Иод ид серебра отфильтровывают, раствор упаривают н про-
дукт перегоняют. Трифторацетат серебра играет роль основания
так же, как бикарбонат натрия при иодировании анилина. Иодиро-
вание вератрола в присутствии окиси ртути в качестве основания
дает лишь 40—55% продукта, загрязненного солями ртути.
Описано два метода гладкого иодирования тирозина с образова-
нием 3,5-дниодти розин а. В одном из них (а) [31 раствор И. (320 г)
н-иодистого натрия (400 г) в воде (1,3 л) добавляют по каплям к пере-
мешиваемому раствору ь-тнрозина (100 г) в 20%-ном водном этила-
мине (1 л) и перемешивают 30 мин. Избыток И. удаляют восстановле-
нием, pH доводят до 5—6, выделяющуюся аминокислоту растворяют
в 1 н. соляной кислоте н осаждают аммиаком.
Во втором методе (б) [4] порошок И.суспендируют в растворе тиро-
зина в смеси уксусной и соляной кислот и при встряхивании мед-
ленно добавляют 30%-иую перекись водорода.
НО—Ч—СН3СНСО3Н___- >НО—Ч Ч—СН3СНСО3Н
| (а) 75% \=7/ |
NH2 (б) 71% l/ NH3
32
В отсутствие активирующих заместителей непосредственное вве-
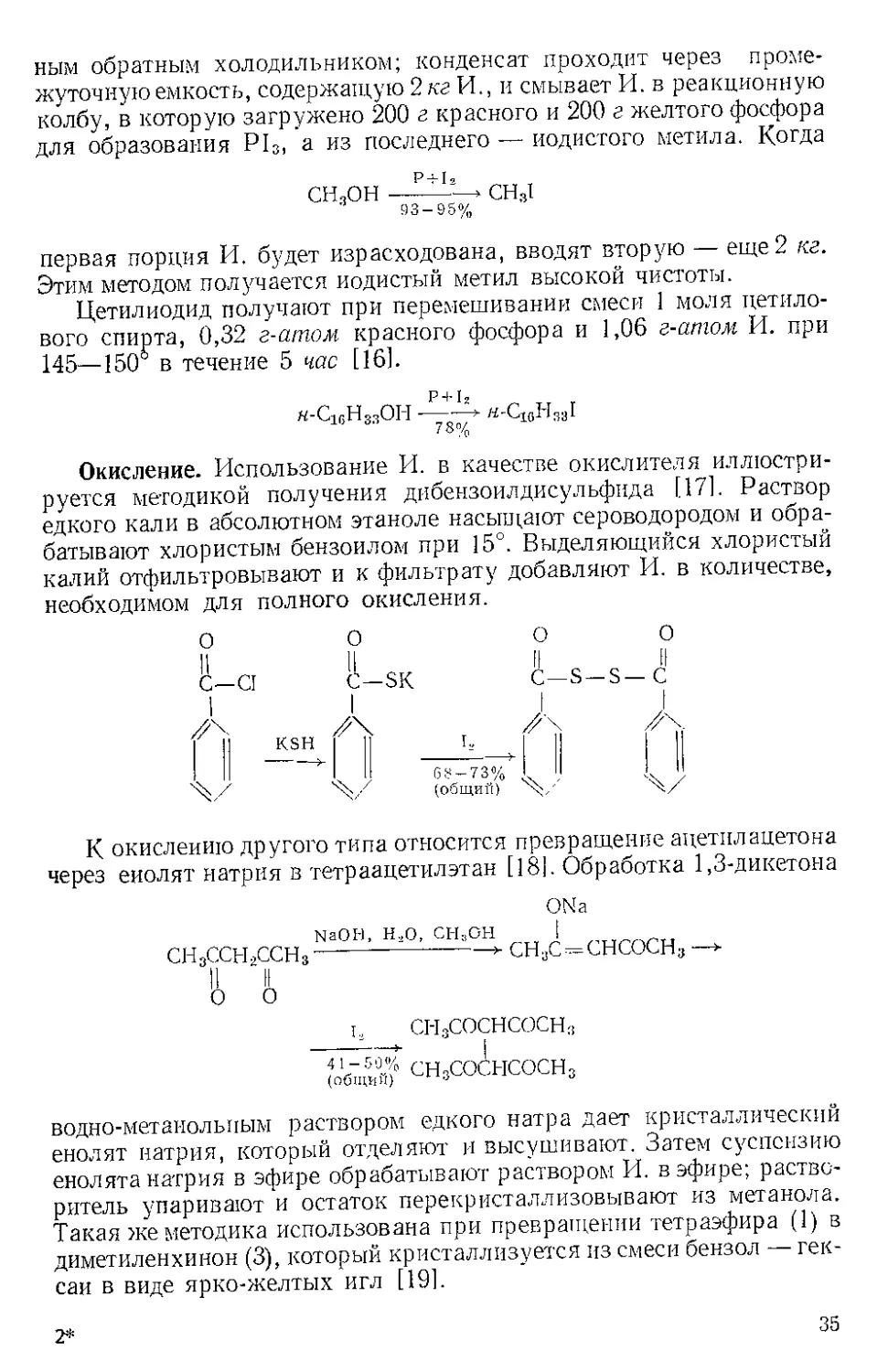
дение И. можно осуществить, используя азотную кислоту или сер-
ный ангидрид для окисления HI до 13 и, таким образом, для смеще-
ния равновесия. Для получения нодбензола [51 смесь 1,5 моля 1а
и избытка бензола (455 мл) обрабатывают при 50е 6,15 моля азотной
кислоты в течение 1ИД час. Затем смесь нагревают при слабом кипе-
I
J
Ц 4-HN О Дуд. вес 1,50)
I JI 86-8 7% Ml
нии с обратным холодильником в течение 15 мин. Масло отделяют
и перегоняют с водяным паром над NaOH; желтые нитросоединения
удаляют обработкой разб. соляной кислотой и железными опилками
при перемешивании. Иодбеизол перегоняют с водяным паром, сушат
и перегоняют повторно. Таким же способом можно получить 2-иод-
тиофен с выходом 68—72% [61.
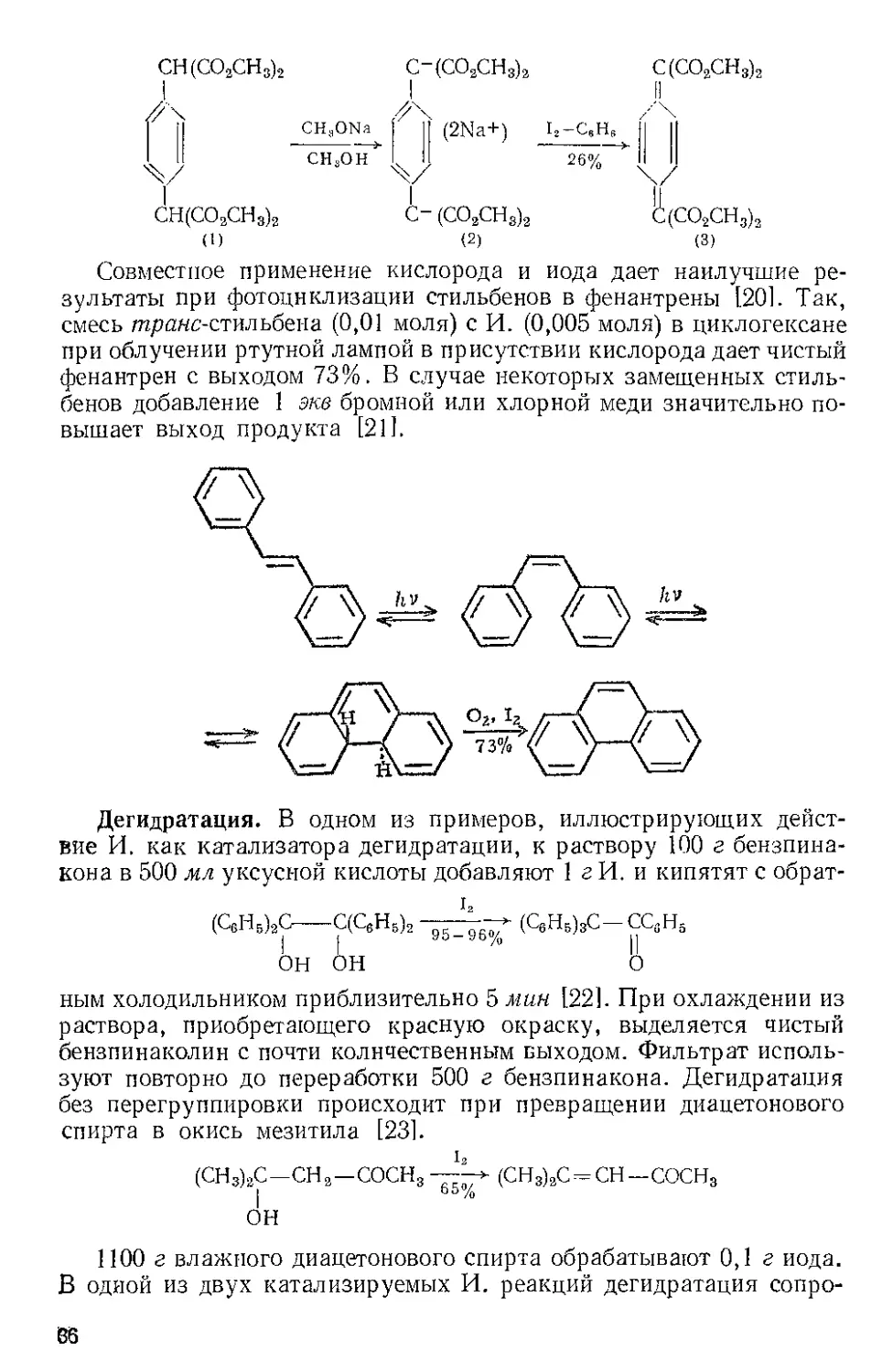
Если ароматическое кольцо содержит дезактивирующие замес-
тители, то необходимы более жесткие условия — применение в ка-
честве окислителя олеума н повышенная температура. Так, смесь
1 моля фталевого ангидрида, первой порции И. (всего 2,12 моля)
и 600 мл 60%-кого олеума длительно нагревают при 65°, добавляя
порциями оставшийся И. и, наконец, повышают температуру до
170° в течение 2 час [7]. После охлаждения тетраиодфталевый ангид-
рид отделяют на стеклянном фильтре и промывают конц. серной
кислотой, м- Ди нитробензол иодируют до 1,3-динитро-5-иодбензола
с выходом 67—70% по этой же методике, но при температуре реак-
ционной смеси 65° [8].
I
I
2-Иодтиофен получают, как описано выше, а также при механи-
ческом перемешивании раствора 0,84 моля тиофена в 100 мл бензола
при температуре 5—10° и постепенном добавлении (30—35 мин)
0,86 моля И. и 0,7 моля желтой окиси ртути так, чтобы температура
повышалась до 45° [9].
2 U 2 UL,+ Нб1г + НгО
Вире [10] описал метод иодирования с высоким выходом йодно-
ватой кислотой в качестве окислителя в присутствии серной кислоты
2 Заказ № 1096 33
как катализатора. Например, n-ксилол растворяют в уксусной кис-
5АгН + 21, + НЮз —5Лг1+ЗН,О
лоте, добавляют воду, серную кислоту, И. и йодноватую кислоту.
Смесь энергично перемешивают при 80° до исчезновения окраски И.
Выходы для ароматических углеводородов и эфиров составляют
75—90%.
сн3 сн3
к // - /
и % 4 час при К0я р н'
-j-L —HIО3-ф конц. HoS04----;—:----
‘С/ з°б'% V
СН3 СНз
Избыток
Огата 111] описал иодирование ароматических соединений с хо-
рошим выходом И. и надуксусной кислотой в уксусной кислоте;
эффективным реагентом служит, по-видимому, ацетилгипоиодит
СН3СОО1 .С циклогексеном реагент дает 1 -иод-2-ацетоксициклогексан,
но с плохим выходом 112]. Синтез по Прево идет значительно лучше.
Непрямое иодирование. Введение И. в ароматическое кольцо
замещением группы HgCl иллюстрируется двумя примерами. В од-
ном из них [131 ацетат ртути растворяют в горячем феноле с образо-
ванием о-ацетоксиртутного производного; полученный раствор вы-
ливают в горячую воду и для осаждения о-хлормеркурата добавляют
горячий раствор NaCl. Это вещество при реакции с И. в хлоро-
форме дает о-иодфенол. В другом примере 114] хлормеркурат полу-
чают реакцией замещения из сульфоната. Реакцию с И. проводят
в кипящем этаноле.
Замещение гидроксильной группы. В сб. «Синтезы органических
препаратов» описано [15] получение йодистого метила в больших
количествах (4150—4250 г) (эта методика проверена Физером).
Метанол вместе с образующимся иодистым метилом кипятят с длин-
34
ним обратным холодильником; конденсат проходит через проме-
жуточную емкость, содержащую 2кг И., и смывает И. в реакционную
колбу, в которую загружено 200 а красного и 200 г желтого фосфора
для образования Р13, а из последнего — йодистого метила. Когда
СН3ОН —СН31
93 — 95%
первая порция И. будет израсходована, вводят вторую — еще 2 кг.
Этим методом получается иодистый метил высокой чистоты.
Цетилиодид получают при перемешивании смеси 1 моля цетило-
вого спирта, 0,32 г-атом красного фосфора и 1,06 г-атом И. при
145—150° в течение 5 час [16].
h-CkjH33OII —I
Окисление. Использование И. в качестве окислителя иллюстри-
руется методикой получения дибензоилдисульфида [17]. Раствор
едкого кали в абсолютном этаноле насыщают сероводородом и обра-
батывают хлористым бензоилом при 15°. Выделяющийся хлористый
калий отфильтровывают и к фильтрату добавляют И. в количестве,
необходимом для полного окисления.
К окислению другого типа относится превращение ацетил ацетона
через енолят натрия в тетраацетил этан [18]. Обработка 1,3-дикетона
ONa
сн3ссн2ссн3
№ОН, Нзо, С1-ЦОН
СНЩ^СНСОСН
ц С1-13СОСНСОСНз
сн3со5нсосн,
водно-метанольпым раствором едкого натра дает кристаллический
енолят натрия, который отделяют и высушивают. Затем суспензию
енолята натрия в эфире обрабатывают раствором И. в эфире; раство-
ритель упаривают и остаток перекристаллизовывают из метанола.
Такая же методика использована при превращении тетраэфира (1) в
диметилен хи нон (3), который кристаллизуется из смеси бензол — гек-
сан в виде ярко-желтых игл [19].
2*
35
СН(СО3СН3)2 С-(СО3СН3)3 С(СО3СН3)2
I I н
/\
II CH4ONa 1| (2Na+) Ь—с8нв || ||
Y CHSO>T H) 26%~>O
I I Y
CH(CO3CH3)2 C-(CO3CHS)3 C(CO2CH3)3
(1) (2) (3)
Совместное применение кислорода и иода дает наилучшие ре-
зультаты при фотоцнклизации стильбенов в фенантрены [20]. Так,
смесь транс-стильбена (0,01 моля) с И. (0,005 моля) в циклогексане
при облучении ртутной лампой в присутствии кислорода дает чистый
фенантрен с выходом 73%. В случае некоторых замещенных стиль-
бенов добавление 1 же бромной или хлорной меди значительно по-
вышает выход продукта [21].
Дегидратация. В одном из примеров, иллюстрирующих дейст-
вие И. как катализатора дегидратации, к раствору 100 г бензпина-
кона в 500 мл уксусной кислоты добавляют 1 г И. и кипятят с обрат-
(СеН5)2С-С(СеН5)2 *• (С6Н5)3С—ссан5
j | УЪ-УЬ% ||
ОН он о
ным холодильником приблизительно 5 мин [22]. При охлаждении из
раствора, приобретающего красную окраску, выделяется чистый
бензпинаколин с почти количественным выходом. Фильтрат исполь-
зуют повторно до переработки 500 г бензпинакона. Дегидратация
без перегруппировки происходит при превращении диацетонового
спирта в окись мезитила [23].
(CH3)3C-CH3-COCH3--Y (СН3)3С = СН-СОСН3
।
ОН
1100 г влажного диацетонового спирта обрабатывают 0,1 г иода.
В одной из двух катализируемых И. реакций дегидратация сопро-
BS
вождается перегруппировкой, поэтому можно предположить, что
обе реакции осуществляются через стадию образования карбоние-
вых ионов, например:
6+ 6-
н ОН И НО • • • I • -. I
II II
—С—С—р J3 —С—С— —>
II II
н
—С-С—4-HOI4-I-
—-С— С—- -рН2О -р 12
Обычная лабораторная методика получения р-мирцена (2) включает
нагревание линалоола (1) со следами И. в вакууме при 150—160°
[241. По данным последних исследований [251, наряду с р-мирце-
ном образуется /пранс-Р-оцимен (3), а также следы ^иоизомера.
Главные продукты легко отделяются перегонкой и получаются
с указанными на схеме выходами.
Катализ. При получении 4-бром-о-ксилола [26] бром в течение
3 час добавляют при перемешивании к смеси 500 г о-ксилола и 12 а
железных опилок, содержащей кристаллик И. Следует отметить,
что в отдельности эти катализаторы не эффективны.
^\/с"3 ^/сн’
] I! Вг,(катал.) I II
L К 94 —97%*" 1 Н
ХСН3 Вг/ хсн3
Использование И. в качестве инициатора реакций Гриньяра уже
отмечалось. В работе Цехмайстера [27] показано действие И. как
катализатора ^нс-транс-изомеризации каротиноидных пигментов.
И. в каталитических количествах применяют при ацилировании
по Фриделю — Крафтсу фурана и тиофена [28], а также более актив-
ных производных бензола, таких, как анизол и ацетанилид [29].
Однако И. не эффективен при бензоилировании антрацена [30].
37
И. используют для активации цинка в реакции Реформатского
[311; описан пример, когда реакцию Реформатского удалось осу-
ществить лишь после активации металла иодом [32].
Получение HI. Бензиловую кислоту восстанавливают с хорошим
выходом до дифенилуксусной кислоты по следующей методике [33].
Смесь 250 мл уксусной кислоты, 15 г красного фосфора и 5 г И. вы-
держивают 15—20 мин, пока весь иод не превратится в иодистый
водород; затем добавляют 5 мл воды, 0,44 моля бензиловой кислоты и
смесь кипятят с обратным холодильником в течение 2,5 час.
(С6Н5)3С-СО,Н (С6Н5)3СНСО2Н
| J “ J /о
он
Иодирование кетонов. При 21-ацетоксилировании 20-кетопре-
гнанов кетон обрабатывают И. в ТГФ в присутствии окиси калышя,
затем иод замещают под действием ацетата калия в ацетоне [34]:
Халперн и Джерасси [351, использовавшие эту методику, устано-
вили, что ТГФ должен содержать некоторое количество перекисей.
Уолл и сотр. [36], получившие несколько неожиданные результаты,
предположили, что эта реакция — радикальная, и показали, что
добавление азодиизобутиронитрила в качестве инициатора дает
хорошие результаты.
1. Brewster R. Q., Org. Syn., Coll. Vol., 2, 347 (1943).
2. J anssen D. E., Wilson С. V., Org. Syn., Coll. Vol., 4, 547 (1963).
3. В a r n e s J. Ii., В о г г о w s E.T., Elks J.,Hems B. A.,LungA.G.,
J. Chem. Soc., 1950, 2824.
4. Jurd L., J. Am. Chem. Soc., 77, 5747 (1955).
5. D a i n s F. B., Brewster R. Q., Org. Syn., Coll. Vol., 1, 323 (1941).
6, Lew H. Y., Noller C. R., Org. Syn., Coil. Vol., 4, 545 (1963).
7. A I 1 e n C. F. H., Cressman H. W. J., Org. Syn., Coll. Vol., 3, 796
’ (1955).
8, Fletcher T. L., Nam k u n g M. J., Wetzel W. H., Pan II.-L.,
J. Org. Chem., 25, 1342 (1960).
9. i n n i s W., Org. Syn., Coll. Vol., 2, 357 (1943); Miller К. E., Lex
C. G. procedure submitted to Org. Syn.
10. W i r t h H. О., К 6 n i g s t e i n О., К e r n W., Ann., 634, 84 (1960).
11. Ogata Y., N aka j i m a K., Tetrahedron, 20, 43 (1964).
12. О g a t a Y., Aok i K., Fur uy a Y-, Chem. Ind., 1965, 304.
38
13. Whitmore F. C,, Hanson E. R., Org. Syn,, Coll. Vol., 1, 326 (1941).
14. Whitmore F. C. et al., Org. Syn., Coll. Vol., 1, 159, 325, 519 (1941).
15. К i n g H. S., Org. Syn., Coll. Vol., 2, 399 (19-43).
16. Il a rima n W. W., Byers J. R., Dicke у J. B., Org. Syn., Coll.
Vol. 2, 322 (1943).
17. Frank R. L.,BIegcn J. R., Org. Syn., Coll. Vol., 3, 116 (1955).
18. Charles R. G., Org, Syn., Coll. Vol., 4, 869 (1963).
19. Acker D. S., Hcrllcr W. R., J, Am. Chem. Soc., 84, 3370 (1962).
20. Mallory F. B., Wood C. S., Gordon J. T., J. Am. Chem., Soc.,
86, 3094 (1964); Wood C. S., Mallor у F. B., J. Org. Chem., 29, 3373
(1964).
21. С о 1 1 1 n s D. J., Hobbs J. J., Chem. Ind., 1965, 1725.
22. Bachman n W. E., Org. Syn., Coll. Vol., 2, 73 (1943).
23. С о n a n t J. B., Tuttle N., Org. Syn., Coll. Vol., 1, 345 (1941).
24. Арбузов Б. Л., Абрамов В. С. , Chem. Вег., 67, 1942 (1934).
25. Naves Y--R., В о п d a v а 1 1 i F. Helv., Chim. Acta, 48, 563 (1965).
26. W i s a n s к у W. A., Ansbach er S., Org. Syn., Coll,, Vol,, 3, 138
(1955).
27. Zech meis t er L., Progress in the Chemistry of Organic Products, 18,
223 (1960).
28. H a r t о u g h H. D., К о s а к Л. I., J, Am. Chem. Soc., 68, 2639 (1946).
29. C h о d г о f f S., Klein H. C., J. Am. Chem. Soc., 70, 1647 (1948); Kaye
I. A., Klein H. С., В nr I ant W. J., ibid., 75, 745 (1953).
30. G о r e P. H., Hoskins J. A., J. Chem. Soc., 1965, 5744,
31. Bachmann W, E., Cole W., Wilds A. L., J. Am. Chem, Soc., 62,
824 (1940).
32. Hardstone J. D., Schofield K., J. Chem. Soc., 1965, 5194,
33. Marvel C. S,, H a g e r F. D., Caudle E. C., Org. Syn., CoII.Vol., 1,
224 (1941).
34. R i n g о 1 d H. J., S t о г к G., J. Am. Chem. Soc., 80, 250 (1958).
35. H a 1 p e r n O., Djcrassi C., J. Am. Chem. Soc., 81, 439 (1959).
36. R о t h m a n E. S., P e r 1 s t e i n T., Wall M. E,, J. Org. Chem. 25,
1966 (1960).
ИОДАЗИД, I—N=N=N (не выделен).
И. получают реакцией водной суспензии азида серебра с эфир-
ным раствором иода. Ганч |1] синтезировал И. в виде неустойчивого
твердого соединения. Хасснер и Леви [2] получили этот псевдогало-
ген более удобным способом из монохлорида иода и азида натрия
в ДМФ или ацетонитриле и обнаружили, что он стереоспецифично
присоединяется к олефинам. Так, А3-холестен превращается в транс-
диаксиальный 2[3-азидо-3а-иодхолестаи. Выходы аддуктов с цикло-
гексеном, стиролом, цис- и транс-стильбенами равны соответст-
венно 80, 70, 63 и 80%.
Аддукт (1) И. и стирола можно превратить либо в этиленимин
(3), либо в азнрин (5) [3J:
39
CeH5CH— СНП
I +
N = N = N
(i)
в,н,
HCl
50%
CeH5CH —CHJ
'I
NH.rHCl
(2)
90%
C6H5CH —CH3
N
H
(3)
70% | OH-
C6H5C = CH2
I -h -
N = N = N
hv
80%
C^C-CHa + Ns
N
(4) (5)
1. Hantzsch A., Ber., 33, 524 (1900).
2. Hassner A., Levy L. A., J. Air. Chem. Soc., 87, 4203 (1965).
3. Hassner A., Private communication, Univ. Colorado.
ИОДА МОНОБРОМИД. Мол. вес 206,83, т. пл. 41°, т. кип. 116°.
В отличие от монохлорида иода, который служит иодирующим
агентом, И. м. является мягким бромирующим агентом, способным
превратить фенол и анилин в соответствующие и-бромпроизводные
[1]. Реагент получают нагреванием смеси иода и брома в уксусной
кислоте до образования гомогенного раствора. При добавлении суб-
страта выделяются НВг и иод. Выходы: 1-бромнафталин 55%,
4-бром-1 -нафтол (сырой) 63%.
1. Militzer W., J. Am. Chem. Soc., 60, 256 (1938).
ИОДА МОНОХЛОРИД. Мол. вес 162,38, т. пл. 14°, 27°; т. кип. 97°.
Раствор И. м. в уксусной кислоте (раствор Вийса) используют
для определения иодного числа жиров и масел.
И. м. получают пропусканием хлора в 254 г иода при встряхива-
нии до увеличения веса реакционной смеси на 71 г [1,2]. Реагент
используют сразу же [1] или перегоняют; выход фракции, кипящей
при 97—105°, составляет 87%. И. м. используют при иодировании
п-нитроанилина, антраниловой и салициловой кислот в уксусной
кислоте. Полученные продукты и выход указаны на следующей
схеме:
67-7 4% [3]
40
Феноловые кислоты типов (1) и (2) с И. м. в уксусной кислоте
дают трииодп рои вводные [41:
ОН ОН
I I
й Й
^/^СНХН — (СН,)Й- СН3 ^/\сн2—СН—СО2Н
1 “ I
СО3Н R
(1) (2)
1. S а и d i и R. В,, D г a k е W. V., L е g е г F., Org. Syn., Coll. Vol., 2,
196 (1943).
2. W о о 1 1 e t t G. H., Johnson W. W., Org. Syn., Coll. Vol., 2, 343 (1943).
3. Wallingford V- H., Krueger P. A., Org. Syn., Coll. Vol., 2, 349
(1943).
4. Papa D., Ginsberg H. F., Lederman I., DeCamp V., J. Am.
Chem. Soc., 75, 1107 (1953).
ИОДА ПЕНТАФТОРИД, IFS. Мол. вес 221,91, т. пл. —8°,
т. кип. 97° (с разл.), уд. вес 3,29.
Первичные амины, имеющие ct-метиленовую группу, окисляются
И. п. в растворе хлористого метилена до нитрила и альдегида, но
с очень низким выходом [1]. И. п. окисляет гидразобензол до азо-
бензола и вызывает бекмановскую перегруппировку оксимов. Наи-
большее значение И. и. имеет как окислитель трет-бутиламина
Перемешивание
QH5CH2NH3 4- IF5 при.-15° 2_^с’ЛеЛ C6H5CN + C6H5CHO
0,040 моля 0,043 моля 20% 9%
в азоизобутан, поскольку реакция идет с хорошими выходами, и
продукт более доступен, чем при получении другими методами.
СН3 СН3 СН3
I IFr, । I
2СН3—С—NH2-------» СН3—С—N = N—С—СН3
| 48% J |
сн3 сн3 сн3
Алкил- и ар ил изотиоцианаты превращаются при действии И. п.
в пиридине в тио-бис-М-(трифторметил)-амины [2].
CF3 CF3
IF- I I
2RN = C = S --RN —S—NR
25—!)()%
1. S t e v e n s T. E., J. Org. Chem., 26, 2531 (1961).
2. S t e v e n s T. E., J. Org. Chem., 26, 3451 (1961).
о-ИОДБЕНЗОЙНАЯ КИСЛОТА, o-ICfiH4COOH. Мол. вес 216,02,
т. пл. 162°.
И. к. получают с почти количественным выходом диазотирова-
нием антраниловой кислоты в разб. серной кислоте и прибавлением
41
раствора йодистого калия в разб. серной кислоте [1]. Продукт можно
перекристаллизовать из горячей воды.
По новой методике [2] антраниловую кислоту диазотируют в
разб. серной кислоте и отфильтрованный раствор прибавляют
к раствору йодистого калия в разб. серной кислоте. Сырую кислоту
(92%), окрашенную в коричневый цвет, превращают в этиловый
эфир с т. кип. 150—15Г/13 мм (71%), который при омылении едким
кали в этаноле дает чистую кислоту (т. пл. 163°) с почти теоретиче-
ским выходом.
Об использовании И. к. см. Дифенилиодоний-2-карбоксилат;
о-Иодозобензойная кислота.
1. Wachter W., Вег., 26, 1744 (1893).
2. Baker G. Р. Mann F. G., Sheppard N.,Tetlow A. J., J. Chem.
Soc., 1965, 3721.
И0ДБЕН30Л, CBH5I. Мол. вес 204,01, т. пл. —29°, т. кип.
188,5° (77—78°/30 лш), уд. вес 1,824.
И., содержащий незначительные примеси нитр осоед и нений, по-
лучают с выходом 86—87% реакцией бензола с иодом и азотной кис-
лотой [1]. Более чистый продукт образуется с выходом 74—76% по
реакции Зандмейера [2]. В более поздней методике [31 рекомендуют
перемешивать при 70° смесь 0,079 моля иода, 10 г конц. серной кис-
лоты и 100 г бензола и обрабатывать ее в течение 1,5 час 122 мл 1,5 М
раствора надуксусной кислоты. Последнюю получают перемешива-
нием 40 г 30 %-ной перекиси водорода и 1 г конц. серной кислоты на
бане при 30° и добавлением к этой смеси по каплям в течение 3,5 час
180 г уксусного ангидрида (раствор устойчив на холоду и в тем-
ноте не менее недели). После окончания реакции, когда коричневый
раствор становится почти бесцветным, смесь разбавляют водой,
отделяют органический слой, промывают его, высушивают и пере-
гоняют.
70°
2СяНв +1г-рСН3СОзН -----> 2CeHsI -ДСН3СО2Н -|-НаО
67—77%
Трифениламин можно получать реакцией дифениламина с И.
в кипящем нитробензоле в присутствии 1 экв поташа и следов медной
пудры [4]. Непрореагировавший дифениламин осаждают в виде хлор-
CeII,NO.(Cu)
2(C6H5)2NH + CbH5I-|-K,CO3 ——2(C6H5)3N4-2K1+CO,
о 2 —— о 5 /q
гидрата пропусканием хлористого водорода в бензольный раствор
сырого продукта.
1. Dains F. В., Brewster R. Q., Org. Syn., Coll. Vol., 1, 323 (1941).
2. Lucas H. J.} Kennedy E. R., Org. Syn., Coll. Vol., 2, 351 (1943).
3. Ogata Y., Nakaj ima K., Tetrahedron, 20, 43 (1964); procedure submitted
to Org. Syn.
4. Hager F. D.f Org. Syn., Coll. Vol., 1, 544 (1941).
42
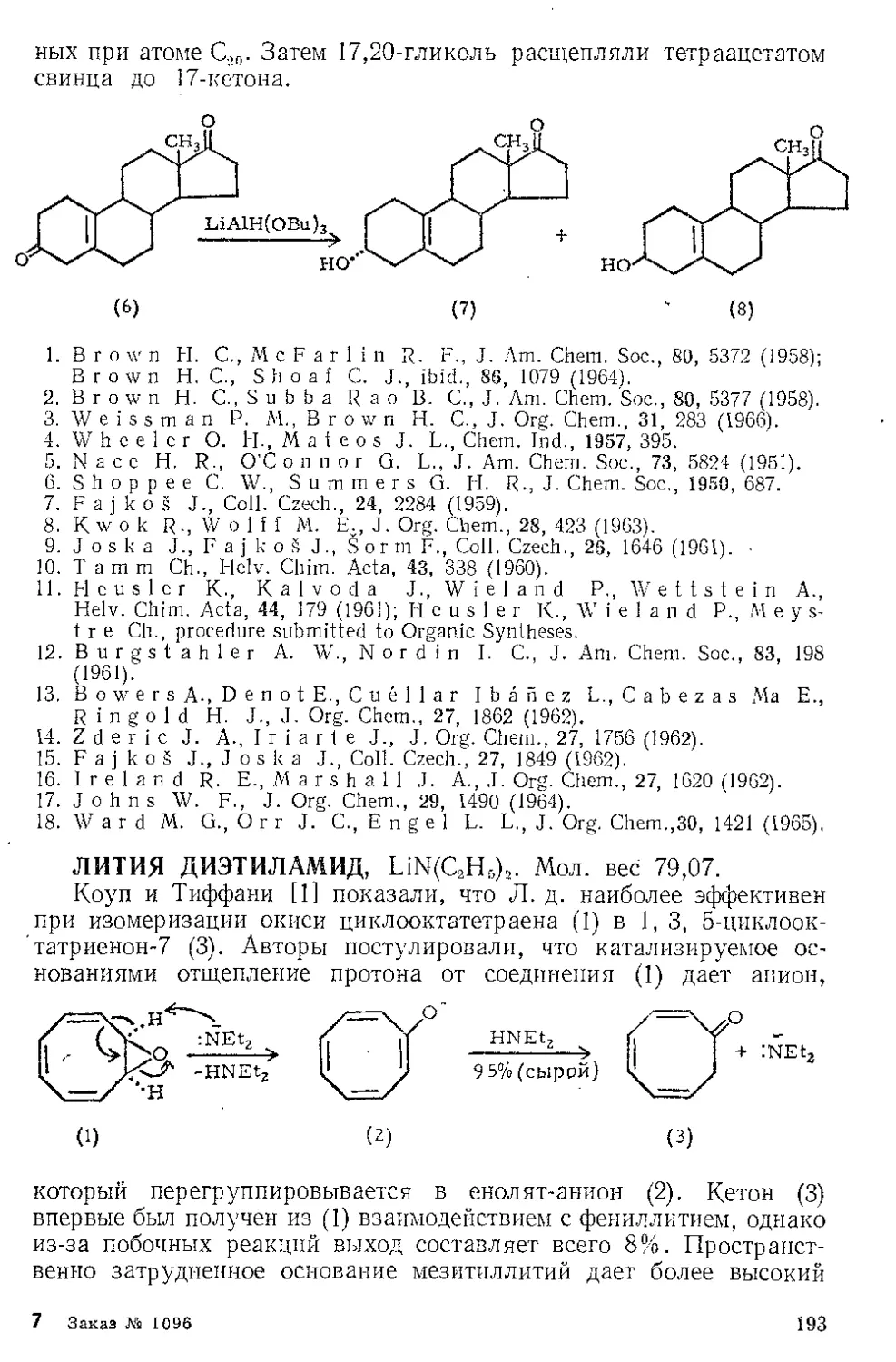
И0ДБЕН30ЛА ДИХЛОРИД (фенилиодидхлорид), СЙН5+1С1(С1“).
Мол. вес 274,92, т. пл. 115—120° (с разл.), желтый, разлагается при
хранении.
И. д. получают пропусканием сухого хлора в раствор иодбен-
зола в хлороформе при охлаждении льдом и солью 111. Продукт
выделяется в виде желтых кристаллов с выходом 87—94%.
Применение (см. также Иодозобензол, Иодобензол). Интересно
использование И. д. для превращения холестерина в 5а,6а-дихло-
рид 12,31. При хлорировании молекулярным хлором по ионному
механизму получается продукт /7г/?дас-лрисоединения — 5а,6р-ди-
снл
1 В СНС1з
Но g НО М :
6 с‘с1
хлорид. В присутствии воды И. д. дает частично продукт транс-
присоединения, при полном отсутствии влаги единственным про-
дуктом является цнс-дихлорид. Предполагается, что реакция осу-
ществляется через циклическое переходное состояние.
^>с==с<1 ^>с с<^ + с6н5г
ci + сГ —> ci .ci ’—> ci ci
ч1-’
с6н5 с6н3
При ультрафиолетовом облучении И. д. реагирует с насыщен-
ными углеводородами с образованием хлорированных углеводоро-
дов, иодбензола и хлористого водорода |41.
| | -|-СсН5! +С1(СГ) | j 4 С0Н51+НС1
Н/<ХС1
1. W i I 1 g е г о d t С., J. prakt. Chem., (2), 33, 155 (1886); Lucas H. J.,
Kennedy E. R., Org. Syn., Coll. Vol. 3, 482 (1955).
2. В e r g C. J., Wallis E. S., J. Biol. Chem., 162, 683 (1946).
3. В a r t 0 n D. H. R., M i 1 1 e r E., J. Am. Chem. Soc., 72, 370 (1950).
4. Banks D. F., Huy ser E. S., К 1 e i n b e г g J., J. Org. Chem., 29,
3692 (1964).
5-ИОД-2,4-ДИНИТРОФЕНИЛГИДРАЗИН (2). Мол. вес 328,47,
т. пл. 248°. Cl NHNH2
Получение [11: I I
'/ N'O2 H,\'Nl-b ['
I! ——j_i li
1-м/ М/
NO3 NO,
(1) (O
43
Применение [1]. И. взаимодействует с кетостероиДамй с обра-
зованием кристаллических высокоплавящихся производных.
1. К а г 1 s о п Р., Hoffmelster Н., Ann., 662, 1 (1963).
И0ДИ30ЦИАНАТ, IN=C = O. Мол. вес 168,93.
И. получают из цианата серебра и иода [11. И. присоеди-
няется к олефинам в транс-положение, давая иоднзоцнанаты [21,
интересные тем, что их можно превратить в этиленнмнны [31.
Свежеприготовленный цианат серебра добавляют к раствору оле-
фина в абсолютном эфире; смесь перемешивают, охлаждают и к ней
добавляют твердый иод. И. присоединяется к Д2-холестену, давая
транс-№аксиальный За-иод-2р-холестанилизоцианат [4].
1. Birckenbach L., L 1 n d h а г d М., Вег., 64, 961 (1931).
2. D г е f a h 1 G., Р о n s о 1 d R., Chem. Вег., 93, 519 (1960).
3. Н assner А., 14 е a t h с о с к С. С., Tetrahedron, 20, 1037 (1964).
4. Hassner А., Н е a t h с о с к С. С., Tetrahedron Letters, 1125 (1964); J.
Org. Chem., 30, 1748 (1965).
ИОДИСТОВОДОРОДНАЯ КИСЛОТА. Постоянно кипящая иоди-
стоводородная кислота содержит 57% HI, т. кип. 127°, уд. вес 1,7.
Восстановление. Азлактои а-бензоиламинокоричиой кислоты
восстанавливают до фенилаланина кипячением с И. к., красным
фосфором и уксусным ангидридом [11. Красный фосфор предназначен
для связывания образующегося иода.
..СО—О ,,
С6Н5СН = С< 1 +57%Н1+Р (красный)-J- Ас.,0 _ ПЯЧе".Х
1Ч = ССЙН5 64—67%
0,1 моля 0,76 моля 0,64 г-атом 125 мл
с6н&сн2снсо2н
nh2
44
Аналогичную смесь (исключая добавление уксусного ангидрида)
применяют для восстановительного дегалогенирования 3-хлорин-
дола [21. При восстановлении лмштробензолсульфохлорида до ди-
ж-нитрофенилдисульфида красный фосфор не применяют, но в конце
+ 57%-ная НИКрасный Р
0,1 моля
Кипячение 4 гас
------------
82-86%
100лм 0,15 моля
реакции прибавляют бисульфит натрия для связывания выделяю-
щегося иода [3]. Этот пример показывает, что под действием И. к.
нитрогруппа не восстанавливается.
SO.,C1 S ...-- S
I II
„ 1 I] , — Кипячение 3 нас, затем KaHSO;{ I II I 11
2 що5 —58%Ы1 --------------------->- |
। II—NO., 7 5 моля 74—79% I H-NOA jl-NO2
А/ "
1,5 моля
В кипящей уксусной кислоте И. к. восстанавливает сс-дикетоны
и а - кетолы главным образом до насыщенных кетонов [4]. Примеры
приведены на следующей схеме:
И. к. — наиболее подходящий реагент для превращения диазо-
кетонов в метилкетоны [5]. Например, 5 мл 57%-ной И. к. добав-
ляют к раствору 22 г диазоацетофенона в хлороформе. По окончании
С6Н5СОСН = N = N С6Н0СОСН3 ЩN2 +I,
96%
45
выделения азота прибавляют воду, отделяют хлороформный слой
и встряхивают его с раствором тиосульфата для удаления иода.
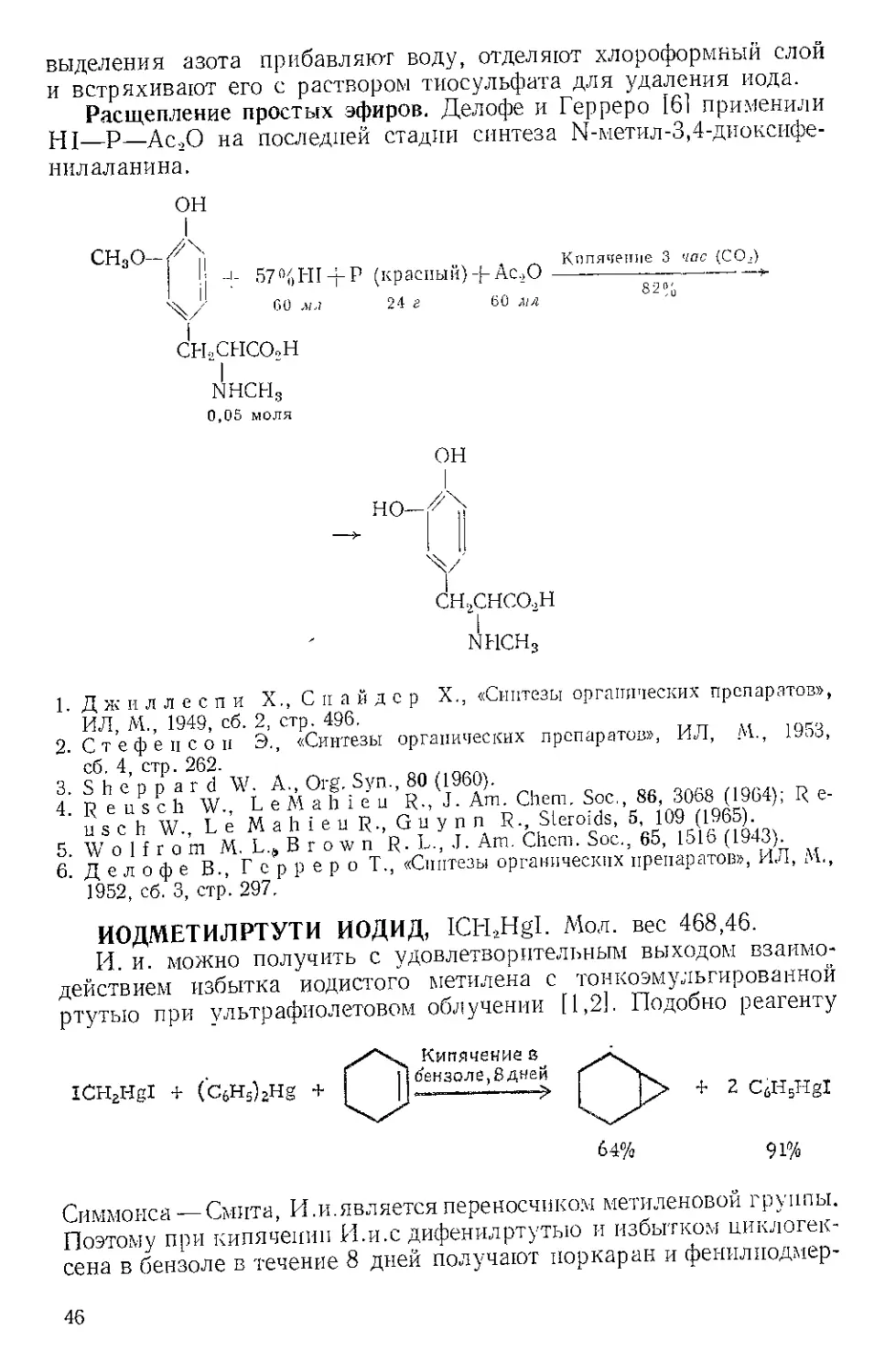
Расщепление простых эфиров. Делофе и Герреро 161 применили
HI—Р—Ас.,0 на последней стадии синтеза П-метил-3,4-диоксифе-
нил ал анина.
ОН
СН3О ( и . Кипячение 3 час (СО.)
!; -I- 57%Н1-рР (красный)-рАс.,0----л.%
60 лм 24 г 60 мл
I
СНХНСОоН
“I
NHCHg
0,05 моля
ОН
-> но-Н!
'У
снгснсо.,н
NHCH3
1. Джиллеспи X., Си а й д с р X., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 496.
2. С т е ф е и с о п Э., «Синтезы органических препаратов», ИЛ, М., 1953,
сб. 4, стр. 262.
3. Sheppard W. A., Org. Syn., 80 (1960).
4. Reusch W., LeMahieu R., J. Am. Chem. Soc,, 86, 3068 (1964); R e-
u s c h W., L e Mahieu R., Guynn R., Steroids, 5, 109 (1965).
5. W о 1 f г о m M. L.j, Brown R. L., J. Am. Chem. Soc., 65, 1516 (1943).
6. Делофе В., Герреро T., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 297.
ИОДМЕТИЛРТУТИ ИОДИД, ICH2HgI. Мол. вес 468,46.
И. и. можно получить с удовлетворительным выходом взаимо-
действием избытка йодистого метилена с тонкоэмульгированной
ртутью при ультрафиолетовом облучении fl,2]. Подобно реагенту
lCH2HgI + (C6H5)2Hg +
О Кипячение s
бензоле, Вдней
—---------------
+ 2 C6H5HgI
64% 91%
Си ммонса — Смита, И.и. является переносчиком мети лен ов ой г р у 11 п ы.
Поэтому при кипячении И.и.с дифенил ртутью и избытком циклогек-
сена в бензоле в течение 8 дней получают пор кар ан и фенил подмер-
46
кур ат с указанными на схеме выходами [2]. Хотя реакция требует
длительного времени, этот метод в большинстве случаев можно пред-
почесть методам с использованием диазометана. Для соединений,
чувствительных к кислоте Льюиса Znl2, этот метод лучше метода
Симмонса —-Смита.
1. Simmons Н. Е., S m I t h R. D., J. Am. Chem. Soc., SI, 4256 (1959).
2. Scyferth D., E isert M. A., J. Am. Chem. Soc., 86, 121 (1964).
ИОДМОРФОЛИНОВЫЙ КОМПЛЕКС, (1), (2). Мол. вес 213,03.
Об образовании кристаллического оранжевого комплекса иода
с морфолином (1 : 1) сообщалось в патентной литературе [1]. Сутвик
и Кирхнер [21, рассматривавшие это соединение как комплекс с пе-
реносом заряда (2), нашли, что при избытке морфолина он является
эффективным агентом, иодирующим терминальные ацетилены. Так,
его успешно применяют при иодировании фен ил ацетилен а и 1-эти-
ннлциклогексанола.
0, И моля
Иодирование бензофурана (1) было осуществлено только при
использовании комплекса морфолина с иодом [3].
13~ .Морфолин,. СН3ОН
30-35°
53%
(2)
(О
Шабрие и сотр. [4] нашли, что фенолы и арил амины иодируются
с хорошим выходом иодом и морфолином при соотношении реаген-
тов 1:1:3 в безводных растворителях (этанол, эфир, бензол).
Например, фенол дает 2,4,6-трииодфенол с выходом 90%.
47
1. Rice R. V., Beal G. D., пат. США 2290710 [С. A., 37, 502 (1943)],
2. Southwick P. L., К i r c h и e r J. R., J. Org. Chem., 27, 3305 (1962).
3. G i z a C. A., Hinm an R. L., J. Org. Chem., 29, 1453 (1964).
4. Chabrier P., Sey den-Penne J., Fouace A.-M., Compt. rend.,
245, 174 (1957).
ИОДНАЯ КИСЛОТА, H5IOe. Мол. вес 227,95, т. пл. 122",
т. разл. 130—140°; растворяется в воде; pKat 1,6, рКа3 5,7.
Расщепление вйц-гликолей. Применение И. к. для расщепления
виц-гликолей до карбонильных соединений (1) было предложено
Малапрейдом в 1928 г. [1]. Соединения, имеющие оксн- и амино-
1) RCHCR2
I 1
ОН он
RCH = O-|-O = CR3
2) RCH —CHR
С)Н г1н3
НГ1Ю,
RCH = O-]-O = CHR
группы при соседних атомах углерода, также подвергаются расщеп-
лению (2) (см. обзор Джэксона [2]). В отличие от тетраацетата свинца
И. к. не окисляет щавелевую кислоту, но медленно окисляет а-окси-
кислоты. Реакцию проводят в водном растворе или в растворителях,
содержащих воду, обычно при комнатной температуре. Исследова-
ния кинетики показывают [3, 41, что реагирующей частицей является
ион II и что реакция протекает с образованием циклического про-
межуточного продукта — гидратированного иона III или дегидра-
тированного нона IV.
пги НО, °,Н/ОН RCH - О ,О.Н/ОН RCH—О. °
KGH — Ог1 \ | / I \ I / । \| |
I -р 1 I г—о--|-н2о
R’CH-OH НО'Д^О- R'CH-o/(J\)- R'CH-O^
I п ш iv
RCH—О
+
R'CH = O
о
II
I —o-
Примером применения реакции в аналитических целях служит
окисление глюкозы, маннозы и галактозы, протекающее с образова-
нием 1 моля формальдегида и 5 молей муравьиной кислоты 12].Этот
метод применяется при исследовании сахаров. Например, а-метил-
о-глюкопиранозид (1) окисляют, растворяя 12,5 г (1) в водном раст-
воре, содержащем 2,1 же водной И. к., и выдерживая раствор
24 час при 20—25°. Окисление дает ди альдегид (2) с ликвидацией
асимметрических центров прн С3, С3 н С4. Тот же днальдегид
получается из семи других а-метил-о-альдопиранозидов [5].
Диальдегнд (2) можно окислить бромом в растворе, нейтраль-
ность которого обеспечивается добавлением карбоната. При этом
48
выделяют кальциевую соль ди кислоты (3), которая легко гидроли-
зуется. Описанный процесс — наилучший метод синтеза оптически
чистой о-глицериновой кислоты.
нсосн3 НСОСНз нсосн3 СНО
неон ОНО О—С = О 1 СО2Н
HOCH 9 НДОс с ' ил Са С ПЛЗ . он- со2н
1 — у ——> 1 ——* 1
неон сно О—0 = 0 неон
1 1
НС ПС 11С
СН20Н СН2ОН сн2он
(1) (2) О)
Расщепление диолов, нерастворимых в воде, показано на при-
мере окисления эритро^, 10-диоксистеариновой кислоты [(4), т. пл.
132°] [6]. Раствор 6 г йодата калия (КЮ4) в 300 мл 1 н. серной кис-
Н Н
СН3(СНг)7С—С(СН2)7СО2Н СН3(СН2)7СНО+ОНС(СН2)7СО.,Н
I I (5) (6)
он он '
(4)
лоты при 20° быстро добавляют к раствору 8 а соединения (4) в 400 мл
95%-ного этанола при 40°. Через 10 мин бесцветный раствор охлаж-
дают до 15°, добавляют воду для растворения осадка сульфата ка-
лия, экстрагируют эфиром. Перегонкой с паром выделяют 3,3 г
(76%) пеларгонового альдегида. Водный раствор фильтруют, охлаж-
дают льдом и выделяют сырой азелаиновый полуальдсгид (6) (3,3 а,
76%). Экстракцией петролейным эфиром выделяют тример (0,5 а),
а очистка через семикарбазон дает 1,5 а чистой альдегиде кислоты
(т. пл. 38°).
Препаративное применение реакции к частично защищенным
производным сахаров показано на примере расщепления о-галак-
тоза-4,5-моноацетониддиметнлацеталя (7) до о-треоза-2,3-моноаце-
тонид а (8) и глноксальдиметилацеталя (9) [7].
сн(осн3)2
сно
(9)
сно
I
осн
Ч—;с(сн3)2 (8)
СО"^
I
сн2он
49
Рейхштейн широко применял расщепление гликолей И. к, при
исследовании боковых цепей адренокортикоидных стероидов. Сте-
роид с глицериновой боковой цепью (1) сначала дает и-оксиальдегпд
(2), а затем 17-кетои (3) [8|. Боковая цепь диоксиацетона (4) расщеп-
ляется до а-оксикислоты (5), окисляющейся хромовой кислотой до
(4) (5)
CHjMgBr
---------->
17-кетона [91. Окисление И. к. применялось также в интересном
процессе отщепления 21-оксигруппы от (4): присоединение реактива
Гриньяра (6) и расщепление гликоля [101.
Расщепление эфирным раствором И. к. [111. В случае соеди-
нений, нерастворимых в воде или нестойких в водной кислоте, рас-
щепление йгщ-гликоля или эпоксида можно проводить раствором
И. к. в эфире или ТГФ. Раствор, содержащий около 16 мг И. к. на
1 мл, получают перемешиванием избытка порошкообразной И. к.
с сухим эфиром или ТГФ в течение 1 час. Смесь выдерживают до
ОН ОН
Н51О6
Эфир
или ТГФ
2 У^О-Н1О3 1,
или
образования осадка и затем раствор декантируют. Необходимое
количество титрованного раствора добавляют к раствору диола
50
в эфире пли ТГФ. Йодноватая кислота сразу же отделяется; она па-
столько плохо растворима, что обработку реакционной смеси можно
осуществлять простым фильтрованием и выпариванием. Этот метол
использован для превращения изол им ар адиена (1) в семикарбазон
альдегида (3) [12]. При осмилировании образуется диол (2) —
масло, которое без очистки расщепляли И. к. в растворе ТГФ.
Альдегид выделяли в виде семикарбазона (3), с общим выходом
51% в расчете на (1).
И. к. является гидратом (ЩОв), поэтому ее можно применять
при гидролитическом расщеплении эпоксидов. Этим методом из
эпоксида (4) был получен у-броммасляный альдегид (5), чрезвычайно
чувствительный к кислоте [13].
ВгСН.СНХН,
сн—сн., вгсн?сн.,сн2сно
’ 80% -5)
(4)
Гидролиз 1,3-диоксоланов и эпоксидов [14]. Карбонильную
группу часто защищают превращением в 1,3-диоксолан (этилен-
ацеталь или кеталь) реакцией с этиленгликолем.Если количественное
удаление блокирующей группы затруднено из-за неблагоприятного
равновесия, полный гидролиз можно провести в водном диоксане
добавлением эквивалентного количества И. к. Таким образом,
образовавшийся этиленгликоль разрушается, и равновесие сме-
51
щастся до полного завершения реакции. Линолевую кислоту
О —СН2
с5н]1с.^ссн2с=с(сн.,)7сн |
\| ню
О —СН2 3 5
QH, (С ССН,С -1= С(СН2)7СНО 4- 2СНаО
синтезируют следующим путем [15]. Смесь 1,6 а (0,0070 моля) йодата
калия*, 2 мл 6 н. серной кислоты и 100 мл воды быстро подогревают
для растворения, охлаждают и обрабатывают раствором 2 г
(0,0066 моля) этнленацеталя Аэ,12-октадекадииналя (1) в 240 мл
диоксана. Образовавшуюся мутную смесь быстро нагревают до
кипения, пока раствор не станет прозрачным. Колбу плотно за-
крывают и оставляют на 3 час. Добавляют 300 мл воды и бикар-
бонат натрия до слабоосновной реакции. Продукт экстрагируют
петролейным эфиром. Выход сырого альдегида количественный.
Альдегид не выделяют, а сразу же окисляют в Д0,12-октадекадиино-
вую кислоту.
Гидролиз эпоксида серной или соляной кислотой проходит
с плохим выходом из-за образования сульфата или хлорида, тогда
как И. к. не способна к образованию эфира. Отдается предпочтение
водной хлорной кислоте в ТГФ, однако применение И. к. хотя
и дороже, но так же эффективно. Раствор I г а-окиси холестерина
в 30 мл горячего ацетона обрабатывают раствором 0,625 г диги-
драта И. к. в 10 ляд воды [16]. Тонкие пластинки холестантриола-ЗР,
5<х,6р начинают выпадать до растворения осадка окиси. Смесь
кипятят 30 мин, охлаждают и отделяют продукт; выход 0,83 г
(81%), т. пл. 231—232°. Вторая порция продукта плавится при
225—226° (0,14 г).
l.Malaprade L., Bull. soc. chim. France, (4), 43, 683 (1928); Compt,
rend., 186, 382 (1928).
2. J a c k s о n E. L., Org. Reactions, 2, 341 (1944).
3. P r i с e С. C. et al., J. Am. Chem. Soc., 60, 2726 (1938); 64, 552 (1942).
4. В u i s t G. J„ Bunton C. A., J. Chem. Soc. 1954, 1406.
5. J a c k s о n E. L., Hudson C. S., J. Am. Chem. Soc., 59, 994 (1937).
6. King G., J. Chem. Soc., 1938, 1826.
7. P a c s u E., T г i s t e r S. M., Green J. W., J. Am. Chem. Soc., 61,
2444 (1939).
8. Pr ins D. A., R e i c h s t e i n T., Helv. Chim. Acta, 24, 396, 945 (1941).
* Рекомендуется вместо йодата и серной кислоты применять предварительно
приготовленную иодную кислоту, так как в этом случае для получения гомогенно-
го раствора необходимы меньшие количества воды и диоксана.
52
9. R e i c h s t e i n T., Meys tr e Ch., v on E uw J., Helv, Chim. Acta, 22,
1107 (1939).
10. von E n w J., Reichstein T,, Helv. Chim. Acta, 24, 408 (194I);F u c h s
H. G., R e ichst e i n T., Helv. Chim. Acta, 24, 804 (1941),
11. Contributed by Ireland R. E., University of Michigan.
12. Ireland R. E,, Newboul d J., J. Org. Chem,, 28, 23 (1963).
13. I r e 1 a ii d R. E., unpublished resulls.
14. Contributed by Walborsky H. AU, Florida State University.
15. Walborsky И. M., Davis R. H., H о w t о n D. R., J. Am. Chem.
Soc., 73, 2590 (1951).
16. F i e s e г L. F., R a j a g о p a 1 a n S., J. Am. Chem. Soc., 71, 3938 (1949).
ИОДОБЕНЗОЛ, С(;Н6Ю2> Мол. вес 236,02, т. пл. 230° (взрывает),
желтый. Растворимость в I л Н2О: 2,8 г при 12\ 12 г при 100°.
О~
I
С6Н5Н=О
Получение. Одна из стандартных методик получения И. [1]
основана на диспропорционировании иодозобепзола. Тонкую пасту
110 г иодозобепзола в воде перегоняют' с водяным паром в колбе
емкостью 5 л до полного удаления иодбензола; выход чистого гало-
генида составляет около 90%. Горячую смесь охлаждают и белый
осадок отделяют, сушат и измельчают в ступке. Йодометрически
показано, что чистота продукта составляет 99%.
„ °’
Перегонка с ।
2CgHs н- - О- СаН51 + - О + С6Н61
92 — 95%
Другой метод заключается в окислении гипохлоритом свеже-
приготовленного порошкообразного дихлорида иодбензола |2|.
Смесь 0,4 моля дихлорида иодбензола, 1 моля продажного раствора
гипохлорита натрия (1,15 л «хлорокса») и 2 мл уксусной кислоты
энергично перемешивают мешалкой Гершберга при 65—75° до вспе-
нивания и изменения цвета осадка с желтого на белый. Обработка
продукта аналогична вышеописанной, но высушенный продукт для
удаления иодбензола промывают хлороформом.
о-
I
С0Н5! + СЦС1 -) + NaOCl + Н2О-> CsHnl - -ОД-NaCl +2HCI
87- 92%
Третий метод [3] представляет собой простой одностадийный
процесс и дает более высокие выходы. Иодбензол (0,1 моля) переме-
шивают при 35°, добавляя в течение 30 мин 0,5 моля (65 мл) 40%-ной
надуксусной кислоты. При этом выделяется диацетат иодозобензола.
После добавления 80 мл воды температуру медленно повышают до
100° и смесь выдерживают при этой температуре в течение 45 мин
для гидролиза диацетата в иодозобензол и дальнейшего окисления
в И., который собирают после охлаждения, высушивают и
53
экстрагируют хлороформом (чистота 99,0—99,9%).
о-
2СН.,С0,Н |
СЙН,1--:---> СЙН51 + —О
72 — 80%
I. Lucas И. J., Kennedy Е. R., Org. Syn., Coll. Vol., 3, 485 (1955).
2. Formo М. W-, Johnson J, R., Org. Syn., Coll. Vol., 3, 486 (1955).
3. Sh aref kin J, G., Saltzman FL, Org. Syn., 43, G5 (1963).
ИОДОДИБЕНЗОАТ СЕРЕБРА (реактив Прево LU). Обзор [2].
Получение. Реактив Прево получают кипячением смеси бензоата
серебра п иода в бензоле [3]. Выпадает кристаллический комплекс,
который можно очистить экстракцией бензолом в аппарате Сокслета.
2C0H5CO,Ag +
2X228,99 = 457.98
ь-
253,82
Ag(C6H5CO2)2I-hAgI
477,10
Превращение олефинов в дибензоаты вицинальных гликолей.
Эту характерную реакцию проводят в бензоле; она протекает сле-
дующим образом: О
П
О-ССоНд
Ag(C6H0COa)2I---^С-t/ -pAgl
СеН5СО
й
Осадок йодистого серебра отфильтровывают и выпариванием филь-
трата получают ди бензоат. Реакцию можно проводить как с заранее
приготовленным реагентом, так и с полученным in situ. В качестве
одной из стадий синтеза альдегидов Гершберг [3] кипятил смесь
^0,1 моля аллилбензола, 45,8 г бензоата серебра, 25,4 г иода и 300 мл
бензола в течение 15 час, защищая смесь от действия влаги. Йодистое
Г м wtp.CH2 = CHCH^B'V u Гп Ги СП 2Cf,H5CO3Ag+h
Cf>HsMgBr--------—- C6Hr,CH2CH = CH.-> ———-----—>
82% " 85%
C(iH-CH.,CHCH,OCOCr,Hs —C6Hr,CH.,CHCH2OH -b(0A—> CeH5CH.,CHO
| “ 8 4% ' 3 72%
OCOC6H6 OH
серебро отфильтровали и после обработки выделили 3-фенилпро-
пандиол-1,2, омылением которого и расщеплением гликоля полу-
чили фенилацетальдегид.
Специальные исследования показали, что при реакции Прево
происходит транс-присоединение. Так, гидролиз метилового эфира
олеиновой кислоты дает низко кипящую /прео-9,10-диоксистеарино-
вую кислоту с выходом 75%, а гидролиз метилового эфира элаиди-
новой кислоты — эритро-ррюл (69%) [41. При реакции Д1,4-цикло-
гексадиена образуется или дибензоат, или тетрабензоат — в зависи-
мости от количества взятого реагента [5].
54
Оказалось, что реактив Прево превращает пзофиллокладен (1) в
соответствующий аллилбензоат (2) с хорошим выходом (1,1 а 0,79 г)
161. Тот же продукт получается и из филлокладена (3), так как
иод вызывает его предварительную перегруппировку в изофиллок-
ладен (1).
В реакции Прево были использованы серебряные соли и соли
других кислот; обзор этих работ опубликовал Вильсон [2]. Для
получения m/мнс-гликолей этот метод, по-видимому, менее удобен
и дороже, чем реакция с надмуравьиной кислотой с последующим
гидролизом. Присутствие свободного иода может привести к ослож-
нениям. Так, метоксибензол с непредельной боковой цепью дал
гликоль, содержащий иод в качестве заместителя в /гйрй-положе-
нии к метоксигруппе [7].
Реактив Прево превращает Д2-холестен в оба возможных дибен-
зоата /77/шнс-диола [8]. Диаксиальный 20, За-изомер и диэквато-
риальный 2а, 30-изомер образуются в соотношении 2:1, однако
трудности разделения снижают суммарный выход до 31%.
1. Prevost С., Compt. rend., 196, 1129 (1933); 197, 1661 (1933).
2. В и л ь с о в Ч. В., «Органические реакции», ИЛ, М., 1959, сб. 9, стр, 445.
3. Hershberg Е. В., Helv. Chiin. Acta, 17, 351 (1934).
4. W i t t с о f f H-, Miller S. E., J. Am. Chem. Soc., 69, 3138 (1947).
5. McCas 1 an d G, E.,Hors w i 1 1 E. C., J. Am. Chem. Soc., 76, 1654 (1954).
6, В г i g g s L. H., C a i n B. F., Cambie R. C., Da v i s B. R., R u t-
ledge P. S., J. Chem. Soc., 1962, 1850.
7. S I e t z inger M., Dawson C, R., J, Org, Chem., 14, 670 (1949).
8. E 1 I i n g t о n P. S., Hey D. G., M ea k ins G. D., J. Chem. Soc., 1966,
1327.
о-ИОДОЗОБЕНЗОЙНАЯ КИСЛОТА [(1)^(2)]. Мол. вес 264,02.
Разлагается выше 200°.
По химическому поведению и И К-спектр у орп го-изомер так резко
отличается от мета- и пара-изомеров, что его рассматривают обычно
как циклический таутомер (2) [1].
55
Получение. И. к. получают окислением о-иодбензойной кислоты
дымящей азотной кислотой при 50° [21.
Определение SH-групп. Приблизительно 0,02 н. раствор стан-
дартизуют иодометрически и используют в слабощелочном растворе
для определения цистеина, глутатиона и сульфгидрильных групп
в белках [3].
+NH3
А|—СО2" sch3chco3 со_
2HSCH3CHCO2 + SCH3CHCO- I II J
X "° i v1
+NH3
1. В aker G. P., Mann F. G.,Sheppard N.,Tetlow A. J., J. Chem.
Soc., 1965, 3721.
2. Meyer V., Wachter W., Ber., 25, 2632 (1892); As ken as у P„
Meyer V., Ber., 26, 1354 (1893).
3. Hellerman L., Ch inard F. P., Ramsd ell P. A., J. Am. Chem.
Soc., 63, 2551 (1941).
И0Д030БЕН30Л, CcH5I‘-Cr. Мол. вес 220,01; разлагается
(co взрывом) при температуре около 210°, желтый.
Получение. И. получают перемешиванием диацетата И. с 3 н.
едким натром; твердый продукт собирают, высушивают и промывают
хлороформом для удаления небольших количеств иодбензола [1].
Na ОН
С6Н6Н-ОСОСН3(СН3СО") —С6НГ,И-О"
ОV Ju уд
Этот метод предпочитают использовать вместо более старого метода
[21, основанного на щелочном гидролизе дихлорида иодбензола
так как диацетат иодозобензола устойчивее и доступнее его дихло-
рида, а выходы продукта выше (75% против 54%). В некоторых
реакциях можно использовать неочищенный влажный И. Для ана-
лиза [21 образец И. добавляют к смеси разб. серной кислоты, йоди-
стого калия и хлороформа; встряхивают в течение 15 мин и титруют
0,1 н. тиосульфатом натрия. Аналогичным образом можно анализи-
ровать иодобензол; эти два реагента можно отличить друг от
друга, основываясь на том, что И. восстанавливает иодид-ион в на-
сыщенном растворе бората натрия, тогда как иодобензол не
восстанавливает его.
Диарилиодониевые соли. Иодозоароматические соединения при-
меняют для получения диарилиодониевых солей. Описан метод
конденсации типа Фриделя — Крафтса с ароматическим соедине-
нием в присутствии кислот [3]. Например, холодный раствор 5 а
C8HS1+-O-+CSH„ НДДВг^ СвН5-1+-СОН6(ВГ-)
И., 12,5 мл бензола, 65 мл уксусной кислоты и 12,5 мл уксусного
ангидрида перемешивают, добавляя по каплям 5 мл конц. серной
56
кислоты. Через 24 час прибавляют воду, раствор дважды экстраги-
руют эфиром, обесцвечивают активированным углем и фильтруют.
При добавлении бромида натрия осаждается дифенилиодонийбромид.
Дифенилиодонийиодид можно получить перемешиванием 0,1 моля
И. и 0,1 моля иодобензола с 1 н. едким натром в течение 24 час [4].
Образующуюся коричневую массу перемешивают с водой и всплы-
вающий раствор йодата дифенилиодония декантируют через фильтр
и обрабатывают иодистым калием для осаждения иодида дифенил-
О
1| OH- KI
с6н51+-о- + с6н5и-о- (СвН^ВцЮ^)^^ (С6Н&)г1 + (1_)
иодония.
Окисление. Предложено использовать И. в качестве реагента
для окисления сульфидов в сульфоксиды[51. Так, раствор 12атиоди-
гликоля (1) в 50 мл воды нагревают на кипящей водяной бане
с 1,15 же Й. в течение 15 мин, охлаждают и фильтруют. После отде-
ления слоя иодбензола раствор упаривают досуха и образующийся
2,2'-диоксидиэтилсульфоксид (2) очищают перекристаллизацией.
HOCH2CH2SCH,CH2OH 4- С6Н51 + ~ о- —
81%
(I)
HOCH3CH3S+CH2CH2OH + C6H5I
о-
(2)
1. Saltzman Н., Share! k in J. G., Org, Syn., 43, 60 (1963).
2. Lucas H. J,, Kennedy E. R., Formo At. W., Org. Syn., Coll.
Vol., 3, 483 (1955).
3. В eringer F. M., et. al., J. Am. Chem. Soc., 75, 2705 (1953); 81, 342 (1959).
4. Lucas H. J., Kennedy E. R. Org., Syn., Coll. Vol., 3, 355 (1955).
5. Ford-Moore A. H., J. Chem. Soc., 1949, 2126.
ИОДОЗОБЕНЗОЛА ДИАЦЕТАТ (фенилиодозодиацетат),
CeH5I+OCOCH3(CH3COO~). Мол. вес 322,10, т. пл. 158°.
Получение. Й. д. получают, перемешивая 0,1 моля иодбензола
при 30° и прибавляя по каплям 0,24 моля (0,48 же) продажной
40%-ной надуксусной кислоты [1]. Кристаллизующийся продукт
отделяют после охлаждения и промывают водой. Чистоту продукта
определяют обработкой образца разб. серной кислотой, иодистым
калием и хлороформом с последующим титрованием тиосульфатом
натрия 121.
30°
С6Н51+СН3СО3Н -|- СН3СОгН —-—> С6Н51 +ООЭСН3(СН3СО") + Н2О
У / Jo yQ
И. д. служит главным источником получения иодозобензола,
однако при взаимодействии иодозобензола и уксусной кислоты на-
блюдается обратная реакция [3].
Сравнение с тетраацетатом свинца. И. д. расщепляет вици-
нальные гликоли в уксусной кислоте при 50—80°, но константа
57
скорости этой реакции почти в сто раз меньше константы скорости
аналогичной реакции с тетраацетатом свинца [4,5]. Аналогия с тетра-
ацетатом свинца заключается также в том, что И. д. способен пре-
вращать олефины в ди ацетаты гликолей [6], а при разложении в ки-
пящей уксусной кислоте, содержащей 2,4,6-тринитротолуол, может
метилировать последний до тринитро-ш-ксилола с выходом около
20% [3].
Окисление. И. д. окисляет первичные ароматические амины
в бензольном растворе при комнатной температуре в азосоединения
(1—2 дня), однако с различными выходами: толуидины—42, 56 и
6%; л-нитроанилин — 53%, а-нафтиламин — 3% [6]. Изучено
СД-Ц 1 + ОСОСН,(СИ3СОО-)
2CbH5NH2 ---------------------> C6H5N = NC6H5
также окисление с помощью И. д. в уксуснокислом растворе [7].
Некоторые N-ар ил ацетамиды ацетилируются этим реагентом в коль-
цо [8]. В бензольном растворе о-нитроанилпн окисляется И. д.
с высоким выходом в окись бензофуразана [6].
{/ЩНД + ОСОСНДСНзСО,-) // у/ X
----------------------—— /О
51,/’ Uw
Шмавт [9] показал, что И. д. окисляет диаминосульфон (1)
в циклическое азосоединение (2). Раствор реагирующих веществ
выдерживали при комнатной температуре три дня. Сульфон, ана-
логичный (1), но не содержащий галогенов, превращается в соответ-
ствующее циклоазосоединение с выходом 95% [101.
При использовании каталитических количеств четырехокиси
осмия в смеси трет-бутанол— пиридин И. д. вызывает окислитель-
ное гидроксилирование двойных связей [111.
СЩОАс
I
с=о
ЛсОСН2. ,Н
V/
CflHnl + OCOCH;t(CH.tCO7)-OsO4
65%
.он
58
И, д. был использован в качестве реагента, вызывающего соче-
тание, при получении диарилиодониевых солей [12].
Аг1+ОСОСН3(СН3СОг) + Ar'H+H,SO,
АгГАг'(НО5О;г) + 2СН3СООН
1. S h а г с f 1< i n J. G., Saltzman Н., Org. Syn., 43, 62 (1963).
2. Lucas Н. J., Kennedy E. R., Formo M. W-, Org. Syn., Coll.
Vol., 3, 483 (1955).
3. S and in R. B., McCormack W. B., J. Am. Chem. Soc,, 67, 2051
(1945).
4. Cr iegce R., Bcucker H., Ann., 541, 218 (1939).
5. Angyal S. J., Young R. J., J. Am. Chem., Soc., 81, 5251 (1959).
6, Pausacker К. H., J. Chem. Soc., 1953, 1989.
7. В а г I i n G. B., Pausacker К- H., Riggs N- V., J. Chem. Soc.,
1954, 3122.
8. В a r I i n G. B., R i g g s N. V-, J. Chem. Soc., 1954 , 3125.
9. Szmant H. H., Infante R,, J. Org., 26, 4173 (1961).
10. Szmant H. H., Lap inski R. L., J. Am. Chem. Soc., 78, 458 (1956).
11. Hogg J. A. ct al., .1. Am. Chem. Soc., 77, 4436, 4438, 6401 (1955).
12. В eringer F. M. ct al., J. Am. Chem, Soc., 81, 342 (1959).
ИОД — ПИРИДИН.
Метил кетоны типа АгСОСН3 и R2CHCOCH3 реагируют с иодом
в растворе пиридина, давая иодиды пиридиния с выходами до 95%
[1,2]. Соли гидролизуются щелочами до соответствующих кислот.
2-Метилхромон, являющийся винилогом метил кетон а, замещается
таким же образом [31.
1. Krohn k е F., Вег,, 66, 1386 (1933),
2. Kin g L. С., J. Am. Chem. Soc., 66, 894, 1612 (1944).
3. Sc h m u 1 z J., 11 i r t R., L a u en er 11., Helv. Chim. Acta, 35, 1168 (1952).
ИОД — СЕРЕБРЯНЫЕ СОЛИ (CF3COOAg или AgF).
Хасцельдин [1] получил трифторацетат серебра смешиванием
водных растворов трифторацетата натрия и нитрата серебра, филь-
трованием раствора от примеси хлорида серебра и непрерывной
экстракцией эфиром. Эта серебряная соль взаимодействует с иодом
в нитробензоле, давая раствор комплекса, в котором активным
компонентом является гипоиодит CF3COOI [21. Этот комплекс иоди-
59
рует бензол и галогенбензолы с выходом 60—80%. Однако 2-нафтол
дает 1-иодпроизводное с выходом только 20%, а 1-нафтол образует
продукт неопределенного состава [3]. Комплекс окисляет первичные
спирты до альдегидов с различными выходами (25—80%), а кетоны—
до ct-дикетонов с низкими выходами (33—50%) [31. 13 в сочетании
с AgF иодирует простые ароматические соединения с низкими выхо-
дами (10—50%) [31.
1. Haszeldine R. N., J. Chem. Soc., 1951, 584.
2. Has zel dine R. N., Sharpe A. G., J. Chem. Soc., 1952, 993.
3. Bergmann E. D., Shaliak I., J. Chem. Soc., 1959, 1418.
N-ИОДСУКЦИНИМИД, (CH2CO)2NI. Мол. вес 224,99, т. пл. 201°
(с разл.).
Получение [1, 2]. Сукцинимид серебра получают добавлением
в темноте свежеосажденной влажной окиси серебра к кипящему
раствору сукцинимида в воде. Образующуюся суспензию фильтруют
с отсасыванием, и фильтрат выдерживают до выпадения серебряной
соли. Соль сушат на воздухе, измельчают и высушивают в вакууме
при 110°; выход 47%. Соль добавляют в течение часа к смеси иода
и диоксана в темном сосуде, иногда встряхивая его, и затем нагре-
вают на бане при 50° . Иодид серебра удаляют фильтрованием,
фильтрат разбавляют четырех хлор истым углеродом и охлаждают
для кристаллизации И. Выход продукта ст. пл. 193—199° составляет
81—85%.
Иодирование енолацетатов. Джерасси и Ленк [1] показали, что
обработка гептанона-2 уксусным ангидридом или изопропенил аце-
татом приводит к енолацетату (2), реагирующему с И, без раствори-
теля или в диоксане с образованием чистого а-иодкетона (3) с выхо-
дом 82% и N-ацетилсукцинимида (4) с выходом 90%. Образование
(4) и тот факт, что И. не проявляет склонности к образованию ради-
калов, как это наблюдается в случае N-бромсукцинимида, говорит
о ионном механизме реакции, возможно протекающей через про-
межуточный иодониевый комплекс.
И. реагирует селективно с енолацетатной группой стероидов, не
затрагивая 5,6-двойную связь. Так, енолацетат (6), легко получае-
С' ососн3
^-одсн.сснз —^одсн^ссНз
(1)
о
I!
Н О-ССН,
1 3
гс4н,с<—ссн3
< сн2 со
I X
I >
^сн2со
(2)
О
'С^СНССНз (3>
I
< СН2СО
: I | /ЫСОСНз (4)
СН2СО
ч *
60
мый из прегненолона и изопропенилацетата, взаимодействует с И.
в диоксане при 85°, образуя 21-иод-20-он (7), который превращается
в 3,21-диацетат (8). Енолацетаты А4-3-кетостероидов более реакци-
онноспособны и взаимодействуют с И. при комнатной температуре,
образуя соответствующие 6- и од-А4 -3-кетоны [3].
1. D j е г a s s i С., Lenk С. Т., J. Am. Chem. Soc., 75, 3494 (1953).
2. В enson W. R., M с В e e Е. Т., R and L., Org. Syn., 42, 73 (1962).
3. D jerassi C., Gr ossm an J., Thomas G. H., J. Am. Chem. Soc.,
77, 3826 (1955).
ИОДУКСУСНОЙ КИСЛОТЫ АМИД, ICH,CONH2. Мол. вес
184,97, т. пл. 95°.
Виткоп [1] осуществил селективное расщепление метионилпеп-
тидов методом, основанным на разложении сульфониевых солей
метионина (2) с выделением сер у содержащего фрагмента н образо-
ванием лактона гомосерина (3). Наибольшие выходы получены с ис-
пользованием И. в качестве алкилирующего агента.
CH3S CH3s+R I-
I I
Н2С ОН KI нгс он Нагревание НаС---------------------О
I I------------I I ----------------। I
Н,СЧ ;С=О Н,С ,с = о “CH°SR н.,Сч .С = О
ХУ С7 ' /С7
Hz JxNH3 и/ XNH2 Hz ^NH.,
(1) (2) (3)
Применение реакции для расщепления
пептида показано на следующей схеме:
метионинсодержащего
CH3S
СН2
I
О сн2
R — CHN н'с---CHNHCOR'
I (4)
С02Н
ICH3C0NHa
Г СН2 п
I" /\
9 5’ но СН2
ch2conh2 С—CHNHCOR'
с со2н
I-CH3SCHaCONH2
СН3
I
О сн,
II I
RCHNHC-------CHNHCOR' —>
I (5)
С02Н
(6)
61
сн.
Т Нснг
I I
RCHN1T-H14-0 —С———-CHNHCOR'
СО.Н
(7} {8)
I. Lawson W. В., G г о s s Е., F о 11 z С, М., W i t к о р В., J. Ат.
Chem. Soc., 84, 1715(1962).
ИОНООБМЕННЫЕ СМОЛЫ. Большинство продажных И. с.
имеют полистирольиую матрицу, сшитую 3—5% дивинил бензол а.
Катионные И. с. обычно содержат сульфогруппы, введенные сульфи-
рованием, а анионные И. с. — четвертичные аминогруппы, введен-
ные хлорметилированием с последующим аминированием.
Преимущество И. с. как катализаторов состоит в том, что они
нерастворимы в воде и органических растворителях и их можно
отделить от реакционной смеси фильтрованием, причем полученный
раствор не будет содержать отделяемых ионов. Использованные
катионообменники можно регенерировать промыванием минераль-
ной кислотой, а использованные анионообменники — щелочью.
В следующих примерах И. с. типа амберлит (Rohm and Haas
Со.) обозначены IR.
Кислые смолы. Гидролиз. В качестве примеров приведем
гидролиз енамина (1) [1] и амида (2) 12]. Так, IR-120 (Н+) эффек-
IR-120 (Н+)
Н2о .
тивиее серной кислоты при гидролитическом декарбоксилировании
эфиров ацетоуксусной кислоты [31. IR-100 (Н‘!‘) эффективнее соля-
ной кислоты при гидролизе обычных сложных эфиров, причем ее
преимущества тем явственнее, чем выше молекулярный вес эфира [4].
Та же И. с. гидролизует П-(2-О-метил-d-глюкозил)-пиперидии (3)
с 80%-ным выходом, тогда как 1 и. серная кислота (90мин, кипящая
водяная баня) дает только 61%-ный выход [5].
IR-120 (н+)
87°, 1 7ДС
сн,он
но
он
OCHJ'H
(4)
62
Образование ацетонида. Для превращения метилового эфира
оротидипа (1) в ацетонид (2) лучше всего использовать кислую И.с.
161. Небольшое количество 2,2-диметоксипропана добавляли для
удаления воды к суспензии эфира и И. с. в ацетоне и смесь встряхи-
вали при комнатной температуре в течение 3 час.
(2)
Метилглнкозиды. И. с. дауэкс-50 (Н+) использовали для полу-
чения метил гликозидов 1.71.
Образование амида. Кислые И. с. оказались прекрасными катали-
заторами конденсации амина (1) с кислотой (2) с образованием амица
(3) 18].
Реакционную смесь кипятили с обратным холодильником в кси-
лоле в присутствии И. с. в концентрации 3 вес. 9'6 по отношению
к (2) до тех пор, пока в водоотделителе не собирался 1 моль воды.
Выход почти количественный.
Этерификация [9]. Метиловые сложные эфиры аминокислот
обычно получают, используя в качестве катализатора эффективные
катионные И. с.—IR-120 (Н + ) или зео-карб 222 (Н + ). И. с. обраба-
тывают 2—4 объемами 2 н. соляной кислоты, промывают до нейт-
ральной реакции п высушивают. Смесь И. с., аминокислоты и мета-
63
нола кипятят с обратным холодильником при перемешивании в тече-
ние 3 час и удаляют И. с. фильтрованием.
Перегруппировка. Ньюмен [10] использовал дауэкс-50 для пере-
группировки этинилкарбинолов в а, р-ненасыщенные кетоны. На-
пример, смесь 1-этинилциклогексанола, уксусной кислоты, воды
и И. с. кипятят с обратным холодильником в течение 45 мин,
НОЧ/С^СН сосн3
/К Дауэкс-50(Н + ). АсОН, Н2О /К
| I 84-87% |
Конденсация кетонов. Лоретти [11] использовал ту же И. с. для
конденсации кетонов в а, р-непасыщенные кетоны. Реакции проте-
кают очень медленно, но с хорошими выходами.
Дауэкс-50(Н + )
(СН3)2СО + СН3СОСН, —-----------> (СН3)^С = СНСОСН3
Конденсация фенолов. Катионная И. с. является очень хорошим
катализатором алкилирования фенолов изобутиленом [12]. Смесь
фенола и И. с. перемешивают при 80° и пропускают в нее в течение
3 час изобутилен. Затем в течение 4 час повышают температуру до
120° и отфильтровывают смолу. Выход н-т^ет-бутилфенола 88%
(57,5% конверсии). Бензол и ксилол не алкилируются.
ОН ОН
А 80° А
I || + IR-120(H + )4-(CHa)aC = CH2— | ||
1 моль 10 г 0,92 моля Н3С—С — СН3
сн3
Кислотную И. с. можно использовать вместо серной кислоты
при синтезе кумарина по Пехману [13]. Для реакций типа конден-
сации резорцина с этилацетоацетатом пригодны обе вышеуказанные
И. с. (зео-карб — английская полистиролсульфокислота) [14]. Ис-
пользование смолы упрощает очистку продукта.
ОС2Н5
но, .он I УУ 6о + i СЫа 1R-1 20(Н +) или зео-карб 1 || | 80% А/ КА
со 1 сн3
:н3
64
Вышеупомянутые И. с. использовали также для С-ацилирования
фенолов ангидридами, по-видимому, через О-ацилирование и пере-
группировку Фриса [15], Однако выходы оказались низкими даже
с 1,3-диокснбензолами.
СОСН3
нох ЮН НОХ । юн
1/ [00° I я
I II +(СН3СО)Ю + Ш-120(Н+) | ||
'Ч/ '° ЧЧ/
[ I
СНз СНд
Гидратация ацетиленов. Дауэкс-50 не является эффективным
катализатором гидратации ацетиленов, но Ньюмен [101 приготовил
прекрасный катализатор для этой реакции, пропитав И. с. 1%-ным
сульфатом ртути (обработка окисью ртути в разбавленной серной
кислоте). Этот метод использовали на одной из стадий синтеза
19-норпрогестерона 1161. Биллимория и Маклагаи [171 осуществили
гидратацию 1-этин ил циклогексанол а при помощи зео-карба 225,
пропитанного 1%-ным сульфатом ртути. В некоторых случаях
(например, с ]-этииил-2-метилциклогексанолом) необходима пред-
варительная обработка 20%-ным сульфатом ртути.
НО... /С^СН НОХ /СОСНз
( 5 сн,он-н2о
Дауэкс-5 0 - HgSO4
\/ 84% Х X/
Основные смолы. Гидролиз. Раствор 5 г 1-метил-3-карбметоксн-
пирндпнийподнда в 50 мл воды пропускают через колонку 2х 15 см
с силыюосновной И. с. в ОН-форме; элюирование 50 мл воды, упа-
ривание и обработка этанолом приводят к чистому бетаину с высо-
ким выходом [18].
Даузкс -1 (ОН~)^
87,4%
Свободное основание из хлоргидрата. Раствор хлоргидрата алкил-
гу анид ина в абсолютном этаноле пропускают через колонку с основ-
ной И. с., предварительно промытой абсолютным этанолом. Элюи-
рование этанолом позволяет получить алкилгуапидин, не содержа-
щий неорганических примесей; выход количественный [19].
h2n.
C=NH-HC1
RNZ
Н
1 RAM00 (ОН")
С2НЬОН
н2м^
C=NH
RNZ
Н
3 Заказ № 1 096
65
Дегидрогалогенирование. Хинман и Ланг [20] показали, что обыч-
ные реагенты INH3, (C.2H5)3Nl плохо дегидрохлорируют 3-хлор-
N,N-диметил индол инийхлорид (]), так как при этом за счет демети-
лирования образуются значительные количества 1-метилиндола. Ав-
торы показали, что дегидрохлорирование можно легко осуществить
(О
IRA-400 (Otf~X
с основной И. с, и выделить продукт в виде перхлората (3) с общим
выходом 83%.
IRA-400(OH_) служит отличным реагентом для циклизации
(3) и (5) в лактамы [211. Субстрат растворяют в абсолютном этаноле,
добавляют И. с. и перемешивают при комнатной температуре в те-
чение 1 час.
R' R'
RN-CHr-'RA-40°(O-HZ RN-L-R'
I I I
о = C—CHaCl О - G—CH,
(3) (4)
R'
RN —CHR"
O = C CH^Br
XCH2
(5)
R'
IRA-400 (OH’) RN —C —R"
----—------I I
О - C CH.
CH.
(6)
Гидратация. Галат [22] показал, что основная И. с. может быть
использована для гидратации никотинонитрила в никотинамид.
При использовании щелочи в качестве побочного продукта образует-
ся кислота. В другом случае амид был получен с выходом 83% [23].
1RA-40O (ОН“)
86—90%
х/СО\На
Конденсации. Альдольную конденсацию альдегида с нитроалка-
ном можно осуществить с помощью основной И. с, IRA-400 [24].
CHaCH2CHO4-CHsNO,+ lRA-400(OH") -5° ‘Д npPT CH3CH,CHCH„NOi
68% “ )
он
66
Бергман [25] показал, что И. с. IR-400 (ОН ) и IR-410 (ОН~)—
удовлетворительные катализаторы реакции Михаэля.
(CH3),CHNO2 + CHa = CHCN -IR'"100(0tL^ (CH3)oCCHaCH3CN
90%
no2
Сильноосновную И. с. дауэкс 1-Х 10 (четвертичноаммониево-
гидроокисная И. с.) используют при конденсации карбонильных
соединений с циклопентадиеном с образованием фульвенов [26],
Система имеет то преимущество, что время реакции можно менять,
регулируя время контакта катализатора с реагентами.
(сн3)гсо +
Дауэкс 1-Х10
47°/Г *
Эстль и сотр. [27] использовали слабоосновную И. с. IR-4B
или дауэкс-3 в качестве катализатора конденсации альдегидов
RCHO + СНХО2Н IR~4B или дауэк14 RCH - ССОоН
I I
CN CN
с циануксусиой кислотой по Кневенагелю. Еще более эффективным
катализатором является соль, полученная обработкой основной
И. с. избытком уксусной млн бензойной кислоты. Так, ацетатная
форма дауэкс-3 — эффективный катализатор конденсации прост-
ранственно незатрудненных кетонов с активными метиленовыми сое-
динениями, содержащими цианогруппу. Дауэкс-3, полистиролполи-
амндная смола, содержащая первичную, вторичную и третичную
аминогруппы, образует ацетатную форму, которая так же эффекти-
вна, как ацетат пиперидина.
Замещения. Для замещения ArCH2X^-ArCH2CN эффективную
анионную И. с. (амберлит IRA-400 или дауэкс-2П<) сначала перево-
дят в цианидную форму промыванием 20%-ным водным цианидом
натрия [28]. Затем перемешивают в течение 3 час раствор бензил-
галогенида в этаноле с 1,2 же И. с.
CsH6CH2Br+IRA-400(CN-) СгЩ0Н’ 3 час (6!Х CcH5CH2CN
69%
Аналогичный метод был использован для получения бензиловых
эфиров фенолов, причем сильноосновную И. с. промывали раство-
ром фенола в воде и в разбавленной щелочи, а затем спиртом [29].
Модифицированную смолу перемешивают с раствором бензнлгало-
генида до окончания реакции.
Вейншток и Бокелхайд [30] применили амберлит IRA-400 (ОН-)
для превращения алкилтриметиламмониниодидов в соответствую-
3*
67
щие четвертичные аммониевые основания. Эта методика проще, чем
обычно применяемый метод с использованием окиси серебра, и поз-
воляет исключить нежелательные побочные реакции. В случае про-
изводного ^-эритроидина этот метод повышает выход в расщеплении
по Гофману с 40 до 78%.
Образование эпоксидов. Австралийские исследователи [31] ис-
пользовали сильноосновную И. с. деацидит FF для превращения
1-О-гс-толурл сульфон и л инозита (1) в 1,2-ангидро-(±)-инозит (2). Ис-
пользование водной щелочи ведет к образованию 1,2-ангидромиоино-
зита (3) за счет «эпоксидной миграции».
Синтез пептидов на твердой фазе. Новый принцип пептидного
синтеза, описанный Меррифилдом [32], включает ковалентное при-
соединение первой аминокислоты к цепи твердого полимера, после-
довательное присоединение последующих единиц до образования
необходимой структуры и отделение пептида от твердой основы.
Когда растущая пептидная цепь присоединена к нерастворимой
твердой основе, ее можно отфильтровать и отмыть от реагентов и
побочных продуктов. И. с., сополимер стирола и дивин ил бензол а,
была частично хлорметилнрована, чтобы обеспечить возможность
присоединения N-защищенной аминокислоты при реакции с ее
триэтил аммониевой солью (I). Для повышения стойкости образую-
щейся бензильной сложноэфирной группы к кислотному расщепле-
нию хлорметилированный полимер нитровали (2). После взаимо-
действия (1) и (2) с образованием (3) N-защитную группу можно уда-
лить бромистым водородом в уксусной кислоте, чему сопутствует
незначительное сложноэфирное расщепление. Нейтрализация из-
бытком трнэтиламина дает продукт (4), содержащий свободную
N-концевую аминокислоту, способную к связыванию со второй
N-защищенной аминокислотой под действием N,N'-дициклогек-
сил карбоди имида в ДМФА с образованием (5). Описанные проце-
дуры завершают первый цикл. Дальнейшие циклы можно осущест-
вить последовательным снятием защиты и связыванием с соответ-
ствующей карбобепзоксиаминокислотой. Наконец, полностью защи-
щенный пептид типа (5) декарбобензоксилпруют бромистым водоро-
дом и омылением отделяют от полимера свободный пептид (6),
68
н
CbNHCHC07N+ HEt3-]-Cl — С —C6H4(NO2)—полистирол —>
R i <i> H (2)
H
—r CbNHCHCO—C —CeH4(NO„) — полистирол ,^Br. АсОВф
I li I
Rj О H (3)
H
—> H,NCHC—О—С — СеН4(ХОг)— полистирол £bN_c нсомЦ
I !| | RN=C=NR
Rv О H (4)
H
I HBr; NaOH
—CbNHCHCNHCHCO—C—CgHJNO.,)— полистирол---------------—>
S II I If I
R, О Rt о II
(5)
h„nchcnhchco2h
I II I
R2 О R4
(6)
Возможность такого превращения была доказана синтезом
ь-лейцил-ь-аланилглицил-ь-валина, который оказался идентичным
с образцом, полученным конденсацией через активированные п-
яитрофениловые эфиры.
Летсингер 1331 сообщил об исследовании синтеза пептидов па
твердой фазе с использованием полимера стирол — дивин ил бензол
с карбоксильными группами, введенными реакцией с дифенилкарба-
миноилхлоридом и хлористым алюминием с последующим гидроли-
зом. Восстановление литий алюмогид ридом в оксиметил ыщй полимер,
реакция с фосгеном с образованием хлорформильного производного,
конденсация с этиловым эфиром i-лейцина и щелочной гидролиз
привели к продукту полимер-лейцин. Это промежуточное соедине-
ние при последовательной обработке изобутилхлоркарбонатом и
триэтил амином в толуоле, а также глицином, бензиловым эфиром
n-толуолсульфопилглицина п триэтил амином в ДМФА дало лейцил -
глицинбензиловый эфир-полимер. Обработка 15%-ной бромисто-
водородной кислотой в уксусной кислоте привела к отщеплению
бензильной группы и отделению пептида от полимера, а добавле-
ние эфира к отфильтрованному раствору вызывает осаждение б ром-
гидра та дипептида.
В более поздней работе Летсингер [341 получил функциональный
полимер непосредственно сополимеризацией стирола и дивинил-
бензола с п-винилбензиловым спиртом или н-винилбензойной кисло-
той. Автор рекомендует вводить в полимер не более 0,2—0,5% дн-
виннлбензола, так как малое число сшивок облегчает диффузию.
69
Ионообменная хроматография. Мор и Штейн [35] разработали ме-
тод хроматографии пептидов и аминокислот на И. с., который стал
мощным средством анализа белковых гидролизатов. Однако этот
метод не всегда пригоден для белков и полинуклеотидов в связи
с тем, что большие полимерные молекулы построены не по прин-
ципу плотнейшей упаковки, а И. с. имеют малую емкость. В таких
случаях рекомендуют применять сорбенты на основе целлю-
лозы (см. следующий раздел).
Анионообменные целлюлозы. Обычные И. с. имеют ограниченное
применение в хроматографии белков, поэтому Петерсон и Собер
[36] изучили сорбенты на основе целлюлозы, из которых наибо-
лее широко применяется ДЭАЭ-целлюлоза. Ее получают обработкой
очень чистой древесной целлюлозы хлоргидратом 2-хлорэтилди-
этиламина, приводящей к диэтиламиноэтильному (ДЭАЭ) производ-
ному. Это вещество с успехом использовали для разделения сыво-
роточных белков [37] и олигонуклеотидов [381.
R —ОН C1CH3CHsN(C2H5)3 ~~+ ROCH2CH2N + (С3Н&)2(CI-)
(Целлюлоза) |
н
1. 1-1 a s е k R. IL, et al., J. Org. Chem., 28, 2496 (1963).
2. Collins R. F., Chem. Ind., 1957, 736.
3. A s t I e M. J.,Oscar J. A., J. Org. Chem., 26, 1713 (1961).
4. Davies C. W., Thomas G. G., J. Chem. Soc., 1952, 1607.
5. Hodge J, E., Rist C. E.,J. Am. Chem. Soc., 74, 1498 (1952).
6. Moffatt J. G., .1. Am. Chem. Soc., 85, 1118 (1963); see also Erne K.j
Chem. Scand., 9, 893 (1955).
7. Mowery D. F., Jr., J. Org. Chem., 26, 3484 (1961).
8. Walter M., Besendor f H., Schuider O., Helv. Chim. Acta,
44, 1546 (1961).
9. M i I 1 P. J., Crimmin W. R- C., Biochim, Biophys. Acta, 23, 432 (1 £ 5 7).
10, N e w m a n M. S., J, Am. Chem. Soc., 75, 4740 (1953).
H.Lorctte N. B., J. Org. Chem., 22, 346 (1957).
12, Loe v B., Massengale J. T., J. Org. Chem., 22, 988 (1957).
13. von Pech m апп H., D u i s b e r g C., Ber., 16, 2119 (1883).
14. J о h n E. V. О., I s r a e I s t a m S. S-, J. Org. Chem., 26, 240 (1961).
15. P r i c e P., 1 s r a e 1 s t a in S. S., J. Org. Chem., 29, 2800 (1964).
16. В n court R., Tessier J., N о m i n ё G., Bull. soc. chim. France,
1963, 1923.
17. В i I 1 i m о r i a J. D., M a c 1 a g a n N. F., J. Chem. Soc,, 1954, 3257.
18. К о s о w e г E. M., Patton J. W., J. Org. Chem., 26,1318 (1961).
19. Greenhalgh R., Bannard R. A. B., Can. J. Chem., 39, 1017
(1961); M e у e r s G. Y., Miller L. E., Org. Syn., Coll. Vol., 4, 39 (1963):
описано выделение аминокислоты из хлоргидрата аминокапроновой кис-
лоты на колонке со смолой амберлит IR-4B,
20. Hinman R. L., Lang J., J. Org. Chem., 29, 1449 (1964).
21. Chatterjee B. G., Rao V. V-, Mazumdar B. N. G., J. Org.
Chem., 30, 4101 (1965).
22. G a 1 a t A., J. Am, Chem., Soc., 70, 3945 (1948); see also Bobbitt J. M.,
S с о 1 a D. A. J. Org. Chem., 25, 560 (1960).
23. В о b b i t t J. M., Doolittle R. E., J. Org. Chem., 29, 2298 (1964).
24. A s t 1 e M. J., Abbott F. P., J. Org. Chem., 21, 1228 (1956).
25. В e r g m a n в E. D,, Corett R., J. Org. Chem., 21, 107 (1956); 23,
1507 (1958).
70
26. М с C a i n G. Н., J. Org. Chem., 23, 632 (1958).
27. A s t 1 е М. J., G е г g е 1 W. С., J. Org. Chem., 21,493 (1956); Hein R. W.,
A s t 1 e Л1. J., Shelton J. R., J. Org. Chem., 26, 4874 (1961).
28. Gordon M., DePamphi I is M. L., G r j f f i n С. E., J. Org. Chem.,
28, 698 (1953).
29. Rowe E. J., Kaufman K. L., P i an t a d os i C., J. Org. Chem.,
23, 1622 (1958).
30. Weinstock J., Boekelheide V., J. Am. Chem. Soc., 75, 2546 (1953).
31. A n g у a 1 S. J., Bender V., Curtin J. H., J. Chem. Soc., 1966,
798.
32. M e r r i f i e 1 d R. B., J. Am. Chem. Soc., 85, 2149 (1963). For references to
later papers, see Merrifield R. B,, Stewart J. M., Nature, 207,
522 (1965); S t e w a г t J. M., W о о 1 e у D. W., ibid., 206, 619 (1965).
33. Letsinger R. L., Kornet M. J., J. Am. Ch m. Soc., 85, 3045 (1963).
34. Letsinger R. L-, Mahadevan V., J. Am.Chem. Soc., 87, 3526 (1965).
35. Moore S., S t e i n W. H., Advances in Protein Chemistry, 11, 191 (1956).
36. P e t e r s о п E. A., Sober H. A., J. Am. Chem. Soc., 78, 751 (1956).
37. S о b e г H. A. et al., J. Am. Chem. Soc., 78, 756 (1956).
38. Coutsogeorgopoulos C., Khorana H. G., J. Am. Chem. Soc.,
86, 2926 (1964), and earlier papers by Khorana H. G.
К - ......... ------------------,
КАДМИЯ АЦЕТАТ —ЦИНКА АЦЕТАТ. Катализатор, получен-
ный нагреванием смеси 2,5 г дигидрата ацетата кадмия и 2,5 а ди-
гидрата ацетата цинка до прекращения выделения кристаллизаци-
онной воды [11, ускоряет присоединение первичных алифатических
аминов к ацетилену с образованием этилиденаминов. Например,
при нагревании этиламина с ацетиленом в автоклаве при 120—140°
в течение 29 час в присутствии этого катализатора получается
C,H&NНа + сн = сн _ КатаД^ СН 3С Н — N — СаН6
R г. и/
/у
(1)
CHS
(снзрхн н- сн3с г сн сн3с - сн„ ™ сьцс- с ссн3
| ‘ 71% ']
N(CH;!)., N(CH3)3
(2)
N-этилиденэтиламин (1) [11. При взаимодействии метил ацетилена
с диметиламином (в соотношении 2 : 1) в присутствии этого катали-
затора образуется 2-диметиламиио-2-метилпентин-3 (2) [2].
1. Kruse С. W., Kleinschmidt R. F., J. Am. Chem. Soc., 83, 213
(1961).
2. Kruse C. W., Kleinschmidt R. F., J. Ain. Chem. Soc., 83, 216
(1961).
КАЛИЙ, Ат. вес 39,10, т. пл. 62,3°.
При получении mpm-алкоголятов калия необходимо сначала
удалить с поверхности металла (кусочки весом до 20 а) окисленный
слой, затем взять навеску чистого металла и перенести его в атмосфе-
ре азота в колбу со спиртом. Ниже описаны две методики получения
алкоголята.
Методика Джонсона [ 1, 2]. (Ниже приведено лишь краткое
изложение; подробные даниые и сведения о мерах предосторожности
см. в оригинальной работе.) Окисленный слой с поверхности металла
срезают скальпелем под ксилолом в ступке и каждый кусок немед-
ленно переносят пинцетом в другую ступку с ксилолом. Чистый
кусок металла вынимают пинцетом, быстро удаляют остатки кси-
лола фильтровальной бумагой и помещают металл во взвешенный
стаканчик с ксилолом. Для получения mpem-бутилата калия взве-
шенный К. вносят в колбу с /ирет-бутанолом, из которой воздух
вытеснен азотом. Все обрезки и оставшийся металл немедленно
разлагают в ступке под тягой mpem-бутанолом, добавляя его пипет-
72
кой с такой скоростью, чтобы реакция была не слишком бурной.
В случае воспламенения ступку накрывают заранее приготовленным
куском асбеста.
Методика Пирсона [3, 4]. Приведенная ниже методика получения
шреш-амилата калия более экономна в отношении расхода К.,
кроме того, она проще и менее опасна, чем описанная выше.
Для абсолютирования к 2 л трет,-амилового спирта добавляют
10 г натрия, кипятят с обратным холодильником несколько часов
и перегоняют; т. кип. 101 —103,5°, п2^’6 1,4018.
Реакцию проводят в трехгорлой круглодонной колбе (емкость
2 л), снабженной обратным холодильником с осушительной труб-
кой. Центральное горло — широкий шлиф, через который можно
вводить сравнительно большие куски очищенного К. (см. ниже).
Нет необходимости создавать атмосферу азота (выделяющийся
водород и пары растворителя защищают К-), но при желании это
можно сделать путем последовательного откачивания и введения
азота в реакционный сосуд (см. Джонсон и Шнейдер [2]). В колбу
загружают 1 л сухого трет-амилового спирта.
Для очистки К- необходим стакан емкостью 400 мл, часовое
стекло для накрывания стакана и баня с ледяной водой такой формы,
чтобы стакан в ней не опрокинулся при погружении для охлажде-
ния. 150 мл сухого треш-амилового спирта* нагревают в стакане
до 70° на электрической плитке закрытого типа. Кусок продажного
К. (обычно весом не более 20 г) добавляют к нагретому спирту и ста-
кан сразу же накрывают часовым стеклом (так как мелкие кусочки
К. могут загореться). Немедленно начинается выделение водорода,
и вскоре расплавленный К- вытекает из окисной оболочки и собира-
ется на дне стакана. В этот момент накрытый стакан помещают
в баню с ледяной водой и выдерживают до затвердевания К. в виде
яйца. Выделение водорода почти прекращается. Накрытый стакан
взвешивают и кусок К. (светится с голубой флуоресценцией) с по-
мощью длинного пинцета вносят через центральное горло в реакци-
онную колбу; вес К. определяют по разности. Хотя, ио-видимому,
кратковременное действие воздуха ие оказывает вредного влияния,
К. можно переносить в колбу с помощью воронки с пористым стек-
лянным фильтром, через которую продувают сухой азот. Описан-
ную выше процедуру повторяют с новыми порциями К. и с тем же
трет-амиловым спиртом, который был взят вначале для очистки,
до тех пор пока не будет добавлено 1,5 г-атом металла к 1 л трет-
амплового спирта. В «яичной скорлупе» остается очень мало К.;
его разлагают после декантации растворителя обработкой остатков
отрт-бутанолом.
Смесь очищенного К. и трет-амилового спирта кипятят в тече-
ние 3—4 час и получают раствор шрет-амилата калия: 0,19 Л1 —
* Лучше взять смесь 120 мл сухого толуола и 30 мл /пре/п-амшювого спирта.
73
вычисленное значение и 0,16 Л1 найдено титрованием стандарт-
ной кислотой. Вид раствора, по-видимому, зависит от качества
применяемого К. Металл, который образует яркий и блестящий
расплав, дает прозрачный слегка окрашенный раствор mpem-ами-
лата калия. Если же при плавлении К. крошится и образует полу-
твердую массу, то раствор алкоголята будет окрашен в более темный
цвет.
mpem-Бутанол нельзя использовать для очистки К-, так как он
реагирует с металлом слишком бурно. Однако очищенный по указан-
ной методике с помощью трет-амилового спирта, К- содержит так
мало этого спирта, что может быть использован для получення
трет-бутил ат а калия.
Де Пюи [51 описал сходный метод очистки К., согласно которому
металл расплавляют под слоем гептана в атмосфере азота.
1. Джонсо и У. С., Д о б Г. X. т «Органические реакции*, ИЛ, М., 1953,
си. 6, стр. 7.
2. Д ж о н с о н У., Ш ней д е р У., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 279.
3. Contributed by Pearson D. E., Vanderbilt University.
4, Pearson D, E., Keaton O. D., J., Org. Chem., 28, 1557 (1963).
5. D e P u у С. 11., Morris G. I7., Smith J. S., Smat J,, J. Am.
Chem. Soc., 87, 2421 (1965).
КАЛИЯ БИСУЛЬФАТ, KHSOd. Мол. вес 136,17, т. пл. 197°.
Перед применением соль рекомендуется расплавить при 400° и из-
мельчить в ступке.
Дегидратация. Берцелиус в 1835 г. получил пировиноградную
кислоту деструктивной перегонкой виноградной или глицериновой
кислоты; однако этот процесс можно значительно улучшить, если
в качестве дегидратирующего катализатора использовать К- б. [11.
Для этого смесь виноградной кислоты (2,7 моля) и К- б. (4,5 моля)
измельчают в ступке, помещают в колбу (емкость 3 л) с коротким
ОН О О
1 I' II
сионсо.2н и п ссогн ссо2н гп ССО.2Н
। и — । 1
снонсо.н снсо2н снгсо2н сн3
дефлегматором и нагревают на масляной бане при 200—220° до
прекращения отгонки жидкости. Фракционированием получают пи-
ровиноградную кислоту с выходом 50—55%.
Селективную дегидратацию диола (1) с образованием соединения
(2) осуществляют следующим образом: смешивают 12 г исходного
соединения с За измельченного К- б., перегоняют при 245—260°,
промывают эфирный раствор продукта реакции раствором бикарбо-
СН, СН3
«-С4Н9СН,СС == ССН.,СНоОН н-С4нэСН = С — С = ССН2СН2ОН
" I ~ 92%
ОН (Г) (2)
74
ната натрия, высушивают и перегоняют [2]. Пропаргильный третич-
ный гидроксил при этом элиминируется без изменения первичной
спиртовой функции.
Инхоффен и сотр. [31 применяли К- б. в качестве дегидратирую-
щего агента при синтезе каротиноидов. Монометиловый эфир не-
предельного триола (3) (3 г) нагревают с 3 г К- б. при температуре
бани 150° в течение 10 мин и получают 2,6 г непредельного альде-
гида с двумя двойными связями (4).
Для получения jn-хлорстирола (6) в колбу, снабженную капель-
ной воронкой и коротким дефлегматором, помещают 12,5 г плавле-
ного, хорошо измельченного К- б. и 0,05 г /г-трет-бутилфенола
(в качестве антиоксиданта) и при нагревании до 220—230° при
давлении 125 мм к смеси добавляют по каплям 145 г карбинола (5)
с такой скоростью (5,5 — 5,8 час), чтобы температура паров в верх-
ОН
/>\-СНСН3
80—:
С!
KHSO<, 220—230°
—сн^сн.
(5) (6)
ней части дефлегматора была ПО—120° (Оверберджер и Саундерс
[4]). Продукт дегидратации отгоняется вместе с водой; органичес-
кий слой отделяют и после перегонки при 20 мм получают,w-хлор
стирол.
Хаузер и сотр. [51 циклодегидратацией диола (7) получили прос-
той эфир «дифенан» (8). Диол (7) тщательно перемешивают с К. б.
108 г khso4
160“ 40дан
89%
(7) 37,6 2
(8)
75
и нагревают. После охлаждения смесь обрабатывают водой и экстра-
гируют бензолом; для отделения окрашенных примесей бензольный
раствор пропускают через короткую колонку с окисью алюминия,
продукт реакции выделяют и перекристаллизовывают из лигроина.
«1,8-Нафталан» получен этим способом с выходом 81%.
Дегидратационная конденсация. Хиршман, Миллер и Вендлер
[6] разработали улучшенный метод синтеза витамина Ki, заклю-
чающийся в конденсации фитола с 1-моноацетатом 2-метил-1,4-наф-
тогидрохинона в диоксане при 76° в присутствии 1\. б. Этот кислый
ОСОСНз ососн3
/v /СН3 СН3 z. L ZCH3
3 I 3 3
I |l I +HOCH2CH = CCieH33——|| | CH3
Y 4CH2CH = CCieH33
OH OH
катализатор значительно более эффективен, чем щавелевая кислота.
Витамин получают при последующем гидролизе и окислении.
I. Хауард, Фрезер, «Синтезы органических препаратов», ИЛ, М,, 1949, сб. 1,
стр. 345.
2. L е е s е С. L., Raphael R. Л., .1. Chem. Soc,, 1950, 2725.
3. I п h о f f е п Н. Н , S i е m е г Н., М б h 1 с K.-D., Ann., 585, 126 (1954).
4. О в е р б е р д ж е р К., С а у ндерс Д ж., «Синтезы органических препа-
ратов», ИЛ, М., 1953, сб. 4, стр. 540-
5. W е i п h е i m е г А. .1., К antor S. W-, Hauser С. R., J, Org. Chem.,
18, 801 (1953).
6. Hirschman n R., Miller R., Wendler N. L., J. Am. Chem.
Soc., 76, 4592 (1954).
КАЛИЯ ГИДРИД В МИНЕРАЛЬНОМ МАСЛЕ, КН. Мол. вес
40,11.
Продажный препарат представляет собой 40—50%-иую суспен-
зию микрокристаллического КН в промышленном бесцветном ми-
неральном масле (байоль 85), которое легко растворяется в гексане,
гептане, диэтиловом и дибутиловом эфирах, толуоле. Поскольку
гидрид калия нерастворим в этих растворителях (а также в жидком
аммиаке и аминах), суспензию можно разбавить одним из этих
инертных растворителей, не оказывающих влияния на активность
реагента.
Гидрид калия как более основный и более активный реагент,
чем гидрид натрия, можно использовать в качестве конденсирую-
щего агента в конденсациях с ацетоуксусным эфиром, конденсациях
Клайзена, Штоббе и подобных реакциях, протекающих с трудом.
Техника эксперимента такая же, как и при работе с гидридом
натрия в минеральном масле.
76
КАЛИЯ ГИДРООКИСЬ, КОН. Мол. вес 56,10.
Калия гидроокись, спиртовый раствор [1]. Приготовление
спиртового раствора К. г. кипячением гранулированной К. г. с 95%-
ным этанолом — трудоемкая операция. Применение эффективного
водоструйного насоса позволяет значительно ускорить этот процесс.
Гранулированную К- г. и растворитель загружают в толстостенную
склянку для отсасывания, объем которой в 3—5 раз превышает
объем взятого спирта, склянку закрывают пробкой, отмечают вос-
ковым карандашом объем жидкости и склянку присоединяют к во-
доструйному насосу. Сразу же начинается бурное кипение и быстрое
растворение большей части тцелочи, причем иногда необходимо
вращать склянку рукой, чтобы предотвратить потери раствора
щелочи. Если кипение затихнет и смесь начнет охлаждаться до
того, как все твердое вещество растворится, колбу следует отсое-
динить от водоструйного насоса и нагреть до полного растворения
К. г. Часть спирта испарится, поэтому объем смеси доводят до
метки, добавляя этанол. Эффективно также механическое переме-
шивание. Трехгорлую колбу на 1 л укрепляют над баней со льдом,
снабжают ее мешалкой из тефлона с изогнутой лопастью и термо-
метром. В колбу помещают 75 г гранулированной К- г. и 300 мл
95%-кого этанола. При энергичном перемешивании растворение за-
капчивается через 2,5 мин, причем температура смеси повышается
от 26 до 56“. Если же погрузить колбу в баню со льдом, то через
2,5 мин температура достигает 26" 121.
30%-ный раствор К< г. в метаноле получают растворением
680 г гранулированной К. г. в 2 л метанола |3].
Расщепление олефинов и окисление альдегидов. В 1840 г. Вар-
рентрап [4] отделил олеиновую кислоту от предельных кислот,
экстрагировав эфиром ее свинцовую соль, и получил, по-видимому,
очень чистую кислоту. Он провел многочисленные анализы как
самой кислоты, так и ее солей, но не смог установить ее структуру
только на основании данных анализа. В поисках химических дока-
зательств он обработал олеиновую кислоту азотной кислотой и от-
крыл элаидиновую кислоту. В другом опыте Варрентрап переме-
шивал олеиновую кислоту, К- г. и несколько капель воды и посте-
пенно повышал температуру до плавления К. г. При этом началась
бурная реакция с выделением газа, который оказался водородом.
Горячую желтоватую смесь выливали в ограниченное количество
воды, и калиевая соль выделялась в виде твердого вещества, кото-
рое удалось отфильтровать. При подкислении была получена твер-
дая кислота, которая после двукратной перекристаллизации имела
постоянную т. пл. 62". Было доказано, что это пальмитиновая кис-
лота. Элаидиновая кислота дала тот же самый продукт расщепления.
Вторым продуктом расщепления, который в то время не был иден-
тифицирован, является уксусная кислота.
77
н н
I I
СН3(СН2)7С = С(СН2)7СОгН
н
I
СН3(СНфС = С(СНЧ)7СО2Н
)
н
Сплавление с КОН
СН3(СН2)14СОаН + СН3СО2Н + н2
Спустя более чем 100 лет Корнфорз, Хантер и Попжак 151 ис-
пользовали эту реакцию для определения схемы изотопного распре-
деления в холестерине, полученном биосинтезом из меченой уксус-
ной кислоты. При его расщеплении высвобождается кольцо А
в виде 2-метилциклогексанона (I), который по реакции Шмидта
превращали в лактам (2), а последний путем гидролиза и метили-
рования— в бетаин-(3). При нагревании этого бетаина с К- г.
сн3
сн2сн2сн2
сн=снсо2н
(5)
сн3
I I * 3
^h2ch2chnh
сн2сн2—со
сн3
ch2ch2Ahn(ch3)3
I
СН2СН2СОО
сн2
кон^ Сн2сн2сн
СН2СН2СО2Н
МОН'
’— >
350°
СИ3 сн3
СНгСН2СН2 ___ СН2СН2СН2
снси2со2н . з сн~о
+ СН3СО2Н
(8)
I сн3
L,A.KOH> сн2сн2сн2
СО2К
+ н2
(9)
гладко отщеплялся триметиламин, однако при этом не получилось
кислоты (4) с терминальной двойной связью. Вместо этого двойная
связь перемещалась по цепи в результате ряда отщеплений аллиль-
ных протонов и образовалась а, fi-непредельная кислота (5); однако
на этой стадии прошло расщепление, по-видимому, в результате
присоединения воды с образованием (6) и последующего процесса,
обратного альдолизации, который дал альдегид (7) и уксусную
кислоту (8). И, наконец, при повышенной температуре (350°) аль-
дегид реагировал с К- г. с образованием калиевой соли карбоновой
кислоты (9) и водорода.
При расщеплении кольца D и боковой цепи полученного биосин-
тезом холестерина Корнфорз, Гор и Попжак [61 показали, что
сплавление с К- г. можно использовать для окисления альдегида
78
в кислоту. Альдегид превращают в оксим, а последний сплавляют
с К- г.; возможно, что промежуточно образуется нитрил:
RCHO RCH = NOH 2522L [RC = N] —* RCO2K
[50°
Примером реакции альдегида с расплавленным К- г. с образо-
ванием соли кислоты и водорода [(7)->(9), см, выше] может служить
получение ванилиновой кислоты [7]. В стакан из нержавеющей
стали, снабженный мешалкой из нихрома или монель-металла,
помещают гранулированные КОН и NaOH и немного воды. Нагре-
сно
2,7 моля КОН, 4 3 моля NaOH, 50 H2O
180—195°; НС1
89—95%
осн3
1 моль
+ Н2
осн3
СО,н
вают на электрической плитке и при температуре плава 160° добав-
ляют небольшое количество ванилина. Начинается бурная реакция
и температура смеси повышается до 180—195°. Поддерживая эту
температуру, продолжают добавлять ванилин, общее количество
которого должно составлять 1 моль. Затем раствор реакционной
смеси в воде обрабатывают сернистым ангидридом, чтобы предот-
вратить появление коричневой окраски.
Восстановление. Более 30 лет назад Физер Л. и Физер М. [8]
наблюдали, что при сплавлении лактондикарбоновой кислоты (1)
с К- г. образуется с небольшим выходом дифенилметантрикарбоно-
вая кислота (2), однако механизм этого превращения до сих пор
неясен. Возможно, что (1) претерпевает диспропорционирование
в (2) и соответствующую беизофенонтр и карбоновую кислоту.
кон^
180°
со2н
(2) 3,5-4, 2 г
Другие реакции. См. [91.
1. Contributed by S п у d е г С. Н. University of Miami.
2, Experiment by F i e s e г L. F.
3. House H. O., Org. Syn., Coll., Vol., 4, 367 (1963).
4. Varrentrapp F., Ann., 35, 196 (1840).
5. Cornfort h J. W., Hunter G. D., PopjakG., Biochem. J., 54,
590, 597 (1953).
6. Cornf or t h J. W,, Gore I. Y., Popjak G., Biochem. J., 65, 94
(1957).
79
7. Пир л И., «Синтезы органических препаратов», ИЛ, ЛА,, 1953, сб. 4, стр. 120.
8. F i е s е г L. F,, F i е s е г М., J. Am. Chem. Soc., 55, ЗОЮ (1933).
9. See review of alkali fusion, Weedon В. C. L., Techn. Org. Chem., 11, 655
(1963).
КАЛИЯ ГИДРООКИСЬ — АЦЕТОН. Рёдиг и сотр. Ш получили
перхлоргексадиен-1,5 (4) известными методами, а именно присоеди-
нением хлороформа к перхлорэтилену (1), дегидрохлорированием
до перхлорпропилена (3) п конденсацией двух молекул последнего
в присутствии медной бронзы. Пер хлоргексадиен-1,5 (4) неспособен
к дегидрогалогенированию и, -подобно другим полнгалогенсоедине-
ниям этого особого типа, легко дехлорируется спиртовым едким
кали в ацетоне, образуя с высоким выходом перхлоргексатрпен-
1,3,5 (5).
Ci Cl Ci Cl CI С1
СС1а = СС12 + СНС13—^S'c^CIC-C-CCi ~r-.pL -°Д С1С — С — CCI —>
(1) 1
Н Ci Ci Ci
(2) (3)
Cl Cl Cl Cl Cl Cl
I I Illi Спирт. l\OH —
STlETX cic-c~c~c~c=cci cci., = ccicci = ccicci=cci3
CH3OH [ I 88-90% J (5]
ci ci
(4)
1. R о e d i g A., VossG,, К u c h i n k e E., Ann., 580, 24 (1953).
КАЛИЯ ГИДРОСУЛЬФИД, KSH. Циннер 111 подробно описал
получение раствора К. г. в этаноле и его использование в реак-
циях с алкилбромидамп, приводящих к алкилмеркаптанам, на-
пример CH3CH,SH (89%), (CH3),CHSH (79%), C6H5CH2SH (85%),
HSCH2CH2SH (69%).
1. Zinner H., Chem. Вег., 86, 825 (1953),
КАЛИЯ ГИПОХЛОРИТ, КОС1. к. Г. можно получить из ги-
похлорита кальция и углекислого калия HI.
При получении 5-формилфснаитрен-4-карбоповой кислоты озо-
нированием пирена в ДД1ФА Десси и Ньюмен 121 перемешивали
раствор озонида с 1%-ной уксусной кислотой, отфильтровали вы-
Oj в HCON(CH3h;
KOBr; NaOH; НС1у
32-33%
СОгН СНО О—С^о_/СНОН
павший осадок и многократно экстрагировали его горячим водным
раствором едкого кали. Темно-бурый фильтрат обрабатывали
60
раствором К- г., выдерживали в течение ночи, а затем нагревали
на кипящей водяной бане 4 час; при этом окраска становилась оран-
жевой. После добавления концентрированного раствора едкого
натра продукт реакции выпадал в осадок в виде натриевой соли.
Раствор, полученный добавлением углекислого калия к раство-
ру гипохлорита кальция до pH 9—11, окисляет бензиловые спирты
до соответствующих альдегидов (Мейерс 131). Например, встряхи-
ванием смеси водного раствора КОС1 и раствора бензилового спирта
в метаноле в течение ночи при комнатной температуре и последую-
щей экстракцией бензолом получен бензальдегид с выходом 77%.
1. Н ыоме п М., Холмс X., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 351.
2. Д е с с и Р., Н ыомен М., «Синтезы органических препаратов», ИЛ, М.,
1960, сб. 10, стр. 70.
3. Meyers С. Y., J., Огу. Chem., 26, 1046 (1961).
КАЛИЯ ИМИНОКСИЛДИСУЛЬФОНАТ (соль Фреми),
• О—N(SO3K), Мол. вес 268,33, стабильный растворимый в воде
радикал красного цвета.
К. и. получают [1] окислением гидроксилам и ндисульфоната
натрия перманганатом; МпО2 отфильтровывают и К- и. высаливают
хлористым калием. Тейбер и сотр. [21 изучали окисление замещен-
ных фенолов и производных анилина в хиноны. Лучшие результаты
были получены для соединений, в которых имеется по меиыпей
мере два алкильных или алкокенльиых заместителя и свободное
НО—N(SO3Na), N(SO3K)2
(бесцветный) (фиолетовый раствор)
орто- или пара-положение по отношению к гидроксильной или ами-
ногруппе [например, соединение (1)1.
nh2 о
I ,сн3 Д /Сн3
fY 2-c_N(SO3K)2 if 7
6 II ------------------'ll II +2HON(SO^)2
^'^ОСНз 7ХоСН»
О
О) (2)
Данн и Целлер [31 окислили фенол (3) в келлинхинон (4). 1 г
исходного вещества растворяют’ в 75 щ л. ДМФА в крутлодонной колбе
о
2 -O-N(SO3K)2
ДМФА-водн. КНгРО4
СЫз 75%
ОН
(3)
(4)
81
со стеклянной пробкой, в один прием приливают раствор 3,1 г соли
Фреми и 1,2 г КН.2РО4 в 230 мл воды, закрывают колбу пробкой,
энергично взбалтывают содержимое в течение нескольких минут до
перехода фиолетовой окраски в красно-бурую. Примерно через
10 мин начинают выпадать желтые иглы. Выход 0,8 г.
Тейбер и Штайгер [4] показали, что К. и. не только вызывает
дегидрирование 2,3-ди гидроиндол а, но и гидроксилирование, в ре-
зультате чего получается 5-оксииндол. Эта реакция использована
для дегидрирования — гидроксилирования амидов 2,3-дигидроли-
зергиновой кислоты (5) в амиды 12-оксилизергиновой кислоты (6)151.
С целью получения моделей для изучения продуктов фотохи-
мического растепления антибиотиков группы митомицина изучено
окисление различных производных аминонафтола солью Фреми;
при этом получены неоднозначные результаты [6]. Раствор 0,045
моля 7-ацети лам ин о-2-нафтола в 200 мл метанола прибавляют к ох-
лажденному льдом раствору 0,1 моля соли Фреми в 2 л воды и 400 .ил
ОН он 0
,0
ОН СН3С—N-
• о-кчзо.жь
88%
СН3С—N,
0,167 /И КН2РО4. Сразу же выпадает кирпично-красный 7-ацетил-
амино-1,2-нафтохинон. Из 8-ацетиламино-2-нафтола по той же мето-
дике был получен о-хинон с выходом 73%, но с 5-ацетиламино-2-
иафтолом реакция не пошла. В случае 5-амино-1-нафтола легко
происходит избирательное окисление фенольного кольца с образова-
нием 5-амино-1,4-нафтохинона с высоким выходом, однако 8-амино-
ОН О
О—N(SO,K)Z
nh2
2-нафтол не реагирует.
NH2O
82
Тейбер 17] окислял экви лен ин К. и. в водном ацетоне и выделил
ярко-красный о-хинон с высоким выходом.
Пирогаллол окисляется К. и. в водном буфере до пурпурогал-
лина [8], однако выход ниже (39%), чем при окислении йодатом
ОН
Пурпурогаллин
натрия. Трополон в реакцию не вступает и, следовательно, скорее
ведет себя как карбоновая кислота, а не как фенол.
1. Teuber II.-J., J е 1 1 i n е k G., Chem. Вег., 85, 95 (1952).
2. Principal papers: Teuber H.-J. ct a]., Chem. Ber., 86, 1036 (1953); 87, 1841
(1954); 92, 674 (1959).
3. Dann O-, Z e 1 1 e r H.-G., Chem. Ber., 93, 2829 (1960).
4. Teuber II.-J., Staiger G., Chem. Ber., 87, 1251 (1954); 89 489 (1956).
5. Stadler P. A., Erev A. J,, TroxlerF., Hofmann A., Helv.
Chim. Acta, 47, 756 (1964).
6. R e m er s W. A., J a m e s P. N., W eiss M. J., J. Org. Chem., 28, 1169
(1963).
7. Teuber H.-J., Chem. Ber., 86, 1495 (1953).
8. Teuber H.-J., G 1 о s a u e r O., Chem., Ber., 98, 2643 (1965).
КАЛИЯ МАНГАНАТ, КаМпО4. Мол. вес 197,13. При нагре-
вании водного раствора перманганата калия с едким кали происхо-
дит диспропорционирование, образуется К- м. и выделяется кисло-
род:
4КМпО4 + 4КОН 4[<.2МпО4 + 2Н2О + 0.2
Ригби [1] получил К. м. следующим образом: раствор 1 кг 85%-
ного едкого кали в 500 мл воды нагревают при 120—1403 и прибав-
ляют порциями по 40—50 г (всего 700 г) КМпО4; следующую порцию
добавляют лишь после прекращения выделения газа (О2). Получен-
ную суспензию кристаллического К- м. нагревают при температуре
кипения в течение 1 час при перемешивании стальным шпателем,
добавляя при этом воду, чтобы компенсировать потери за счет испа-
рения. Затем смесь охлаждают, осадок отфильтровывают и хорошо
83
отжимают. Он представляет собой почти сухую массу блестящих
кристаллов зеленого цвета, содержащих около 88% К,МпО4; выход
около 1 кг. Твердый К- М- стабилен, однако даже в умеренно щелоч-
ных растворах он медленно диспропорционпрует па МпО2 и КМпО4
уже при комнатной температуре.
Ригби исследовал окисление нескольких непредельных кислот
(коричной, олеиновой, аддукта циклопентадиена и малеиновой
кислоты) К. м. в водной щелочи при О ' и выделил соответствующие
ф/е-диолы с выходом сырых продуктов, равным 20—40%.
1. Rigby W., J. Chem, Soc., 195(i, 2452.
КАЛИЯ МОНОН АДСУЛ ЬФ AT КИСЛЫЙ,
о-
I
КО— S+— OOH(KHSO5).
I
о
Мол. вес 152,17. Получение II].
Смесь 1 моля К. м. к. (стабильная соль мононадсерной кисло-
ты), 0,5 моля бисульфата калия и 0,5 моля сульфата калия является
сильным окислителем, похожим на кислоту Каро, но более устой-
чивым [2]. Эта смесь нерастворима в органических растворителях,
однако, ио данным Кеннеди и Стока [21, ее можно использовать в
смесях этиловый спирт — вода, уксусная кислота — вода и этиловый
спирт — уксусная кислота — вода. Эти авторы не приводят подроб-
ного описания методики и выходов, однако указывают, что смесь
KHSO-,— KHSO4— K.,SO4 может иметь широкое применение. Так,
ее можно использовать в следующих реакциях: циклогексен —>
/прйнс-циклогександиол-1,2; толуол бензойная кислота; цик-
лические кетоны -> лактоны (умеренные выходы); меркаптаны
сульфокислоты (количественные выходы); первичные ариламины
и цнклоал кил амины иитрозосоедииения.
I. Stcplianon S. Е., пат. США 2802722 (1957).
2. Kennedy R. J., S t о с k А. М., J. Org. Chem., 25, 1901 (I960).
КАЛИЯ ПЕРМАНГАНАТ, КМпО4. Мол. вес 158,05; раствори-
мость в 100 ч. воды: 2,83 г при 0", 32,5 г при 75°.
Стехиометрия. Обычно считают, что кислородный эквивалент
К- п. равен 1,5, как это видно, например, из следующего уравнения
окисления вторичного спирта:
3R2CHOH-|-2KMnO4 3RoCO-T2MnO2-|-2KOH + 2H.2O
Однако данные, приведенные ниже, ставят под сомнение правиль-
ность общепринятой стехиометрии. Согласно одной методике, К-п.,
взятый с избытком 103% от теоретического количества, расходовался
полностью, и выход продукта реакции составлял 78—84%. В другом
84
случае К. п. был взят с 90%-ным избытком и при его Полном расходе
выход продукта реакции составил 82%; когда К- п. был взят с избыт-
ком только 50%, выход снизился до 73%. Аналогичные результаты
получены и при окислении неорганического вещества — сульфата
марганца (II). Болл, Гудвин и Мортон [11 получали активную дву-
окись марганца для окисления витамина А в ретинен путем смешения
водных растворов эквивалентных количеств К- п. и сульфата мар-
ганца (II). Значительно более активная двуокись марганца была
получена Аттенборо и сотр. [2] реакцией сульфата марганца (II)
в щелочном растворе с К-п., взятым с 85%-ным избытком от теоре-
тического количества:
2KMnO4-j-3MnS04 + 4NaOH 5МпО2-|-K2SO4+ 2Na2SO4
Как и в случаях с органическими исходными соединениями, большой
избыток К- п. оказался полностью израсходованным. Коричневый
осадок, судя по весу, представлял собой МпО2, но анализ его не
проводился.
Приведенные выше наблюдения можно в какой-то мере объяснить
на основе фактов, известных из аналитической химии: в определен-
ных условиях К. п. разлагается до двуокиси марганца с выделением
кислорода [3]. 0,04 п. раствор К- п. в серной кислоте разлагается
примерно в 20 раз быстрее, чем в нейтральном водном растворе. С
другой стороны, разложение ускоряется щелочью, а также двуо-
кисью марганца. Разложение реагента вовремя окисления представ-
ляет собой автокаталитический процесс. Эта реакция, по-видимому,
обусловливает избыточный расход К- п. в опыте Аттенборо.
Эксперимент, специально проведенный одним из авторов этой
книги, наглядно показал, что при типичном окислении К- п,
органического соединения выделяется кислород. Раствор К- п.,
взятого с 50%-ным избытком (от теорет.), в 500 мл воды перемеши-
О
II
11 5С,°
| I +КМпО4 НОоС(СН2)4СО,Н 4-МпО2 4-О3
20 лы 91,5 с (0,58 моля) 16.5 г (58%) 64,8 г (75%-пая 660 лл
(0,19 моля) 1,5хтеорет. (0,11 моля) степень чистоты) (0,03 моля)
количество (0,56 моля)
вали с помощью механической мешалки и нагревали до 52°, затем
охлаждали ледяной водой до 493, добавляли 20 мл циклогексанона
и колбу соединяли с системой для сбора выделяющегося кислорода.
Щелочи в качестве катализатора не требовалось; температуру под-
держивали точно при 50° путем энергичного охлаждения. Выделе-
ние кислорода начиналось сразу и происходило весьма энергично.
По истечении 47 мин необходимо время от времени нагревать смесь,
поддерживая температуру при 50° до обесцвечивания верхнего слоя
жидкости. Коричневый осадок отфильтровывали, тщательно про-
мывали, перемешивали с 500 мл воды при 70°, вновь собирали
85
и промывали. Осадок, высушенный до постоянного веса, содержал
75% чистой МпОй. Вторые промывные воды упаривали досуха
и остаток (8,9 г) растворяли в концентрате (130 мл) первогофильт-
рата. После подкисления и охлаждения получали белый осадок
(16,8 г), из которого экстракцией эфиром в аппарате Сокслета полу-
чали 16,5 г адипиновой кислоты (т. пл. 152—154°). Количество вы-
делившегося кислорода составляло 660 мл (н. д. т.), или 0,03 моля,
тогда как адипиновой кислоты образовалось 0,11 моля. Подвести
баланс для этого опыта невозможно, поскольку отсутствуют данные
о количестве К- п., израсходованного на дальнейшее окисление
адипиновой кислоты. Из этого эксперимента следует, что стандарт-
ное определение количества выделяющегося кислорода может мно-
гое объяснить и что количественное изучение реакций окисления
К- п., осуществляющихся с высоким выходом, может служить более
прочной основой для толкования механизмов реакций [4]. Такая
информация могла бы также способствовать повышению выходов.
Методы выделения. Избыток К- п. подвергается автокаталити-
ческому распаду, поэтому конец окисления в водной среде обычно
определяют с помощью следующей пробы. Каплю бурой суспензии
наносят на фильтровальную бумагу, и в присутствии следов К- п.
влажное кольцо вокруг бурого пятна окрашивается в розовый цвет.
Согласно многим методикам, объемистый бурый осадок отфиль-
тровывают, тщательно промывают водой, как в описанном выше
опыте, и даже экстрагируют в аппарате Сокслета для полного из-
влечения продукта реакции, который часто очень прочно адсорби-
руется осадком. Энергичное кипячение приводит к коагуляции
осадка и ускоряет фильтрование, однако эта операция требует
времени. Во всех случаях получают большой объем водного раство-
ра, из которого после подкисления извлекают продукт реакции
путем испарения и экстрагирования растворителями. Там, где это
применимо, более простой методикой является подкисление реак-
ционной смеси и пропускание сернистого газа (или добавление
NaHSO3+HCl) для восстановления МпО2 до растворимого суль-
фата. Таким путем удается избежать длительного процесса фильтро-
вания и промывки и уменьшить объем водного раствора, так как
растворенное неорганическое вещество снижает растворимость ор-
ганического соединения. Вероятно, многие методики, в которых
рекомендуется отделять МпО2 фильтрованием, можно улучшить,
если ввести восстановление сернистым газом.
Альдегид-^ кислот а. н-Гепта новую кислоту получают [5] сле-
дующим образом. Раствор 350 мл конц. серной кислоты в 2,7 л воды
перемешивают при охлаждении в бане со льдом; когда температура
понизится до 15°, добавляют 3 моля н-гсптальдегида, а затем
Зн-СвН13СНО-]- 2KMnO4 + H2SO4__________> Зн-СйН13СО,Н +
76-78%
3 моля 2,15 моля (теория:2 моля)
4 KaSO4 + 2MnO.,-|-H„O
86
К. п., взятый с небольшим избытком, с такой скоростью, чтобы
температура не превышала 20°. Затем в суспензию двуокиси мар-
ганца пропускают сернистый газ до полного растворения. Масля-
нистый слой отделяют, промывают водой, сушат и перегоняют.
Ниже приведена методика, которую Шрайнер и Клейдерер [6]
предложили для окисления пипероналя до пиперониловой кислоты.
Смесь пипероналя и 1,5 л воды нагревают на кипящей водяной бане
при энергичном перемешивании для образования эмульсии, после
0, 4 Моля
КМпО4 —>
О, 56 моля 78-84%
(теория'. 0,27 моля)
чего приливают раствор К. и. (большой избыток) в 1,8 л воды в те-
чение 45 мин. Смесь нагревают еще 1 час, раствор подщелачивают,
горячую смесь фильтруют и двуокись марганца промывают три
раза горячей водой порциями по 200 мл. При подкислении фильтра-
та выпадает осадок, который кристаллизуют из 95%-ного этанола,
и получают чистую пиперониловую кислоту.
Следует отметить, что окисление альдегидов в кислой среде
соответствует общепринятой стехиометрии, тогда как в щелочной
среде не соответствует.
АгСН3~^АгСОгН. Обе приведенные ниже методики предусмат-
ривают отделение МпОг фильтрованием. Согласно первой [71, ко-
личество взятого К. п. меньше теоретического и непрореагировав-
шее исходное вещество выделяют перегонкой с водяным паром.
По второй методике [8] а-пиколин нагревают с небольшим избытком
К. п. в 3 л воды на кипящей водяной бане; кислый продукт реакции
+ КМпСц
3,8 моля
(теориям,8 моля)
Кипячение с НЮ
(считая на вошедший
в реакцию)
выделяют в виде хлоргидрата.
Л
^NZ\CH3
0,54 моля
1 Л 4 моля
Избыток 5,6%
х:огн
Расщепление ароматического кольца. По мнению авторов, мето-
дика окисления нафталина до фталоновой кислоты (2) и декарбок-
87
силирования последней (без выделения) через бисульфит ное соеди-
нение до о-карбоксибензальдегида, приведенная в сб. «Синтезы
органических препаратов» [91, менее удовлетворительна, чем двух-
стадийная методика Гребе и Трюмпи [101, на которой, видимо, ос-
нована первая методика. По методике, приведенной в сб. «Синтезы
органических препаратов», исходят из 1/3 реагентов по сравнению
с оригинальной методикой; добавляют 1,5 л кипящего раствора
К- п. небольшими порциями в течение 1,5 час, что является трудо-
емкой операцией. По Гребе и Трюмпи, смесь 100 г нафталина, 625 г
К. п. (96% от теорет.) и 6,25 л воды энергично кипятят в течение
HOx/S03Na
+ кмпо4 МДк ЧСОгН Л^/¥ХсОаН
53-63% ф I Н
' со2н ^х^соан
625 г (2) (3)
(96% от теорет.)
НО SO3\'a
СНО
Н HCI
I II 63-65%* I 11
хсо.н 113 (2) хсоан
(4) (5)
3—4 час. Кипячение прекращают, когда качественная проба пока-
жет, что верхний слой жидкости стал бесцветным. Механическое
перемешивание ускоряет реакцию, обеспечивает спокойное кипение
и позволяет быстро охлаждать смесь. При охлаждении непрореаги-
ровавший нафталин затвердевает и его отфильтровывают вместе с
МпО2. В данном случае фильтрование необходимо для отделения
исходного вещества и его легко осуществить, поскольку двуокись
марганца в результате энергичного кипячения образует твердые
гранулы. Раствор упаривают, нейтрализуют 220—240 г конц. сер-
ной кислоты и выпаривают досуха. Органическое вещество отделя-
ют от сульфата калия экстракцией эфиром. Кристаллизацией из
небольшого количества воды отделяют 8—10 г (6—7%) фталевой
кислоты от значительно более растворимой фталоновой кислоты
(80—95 а). Для превращения в фталевый моиоальдегид раствор
10 г фталоновой кислоты и 5,5 г Na.,CO3 в воде упаривают досуха и
полученную соль прибавляют к 45 мл 40%-ного раствора бисуль-
фита натрия при 60°. Раствор упаривают досуха, остаток переме-
шивают с конц. соляной кислотой для разложения бисульфитного
соединения (4) и вновь упаривают досуха. Экстрагируют эфиром
и получают чистый фталевый моноальдегид; т. пл. 96°, выход 63—
65%.
Для окисления хиноксалина в пи разин-2,6-дикарбоновую кис-
лоту Джонс и Мак-Лафлин [11] брали 20%-ный избыток К. п.
88
(от теорет.); по-видимому, весь К. п. был израсходован и непрореа-
гировавшего хиноксалина не оставалось. Смесь хиноксалина и 4 л
воды при 90° перемешивали механической мешалкой с обратным
Д '. /X 90е ZN\/C°2H
I II р™ U
^N/x-CO2H
1,12 моля 6,6 моля
(20%-пы|1
избыток)
холодильником в колбе на 12 л и добавляли при 90—100° раствор
1050 ? К- п. примерно в 4 л воды из капельной воронки тонкой
струей с такой скоростью, чтобы смесь слабо кипела (1,5 час). Смесь
немного охлаждали, фильтровали и отделенную МпО.2 перемешива-
ли с 1 л воды до получения однородной массы, которую вновь филь-
тровали и промывали еще раз. (По-видимому, при слабом кипячении
не образуется компактная, легко отделяющаяся при фильтровании
МпО2.) Количество дикарбоновой кислоты, которое можно выделить
из 10 л фильтрата, зависит от многих условий, и вопрос этот не пред-
ставляет особого интереса. Выход указан для препарата, перекрис-
таллизованного из воды.
Вторичные спиргы^кетоны. Корнфорз [121 получил этиловый
эфир пировиноградной кислоты путем перемешивания этилового
эфира молочной кислоты, 130 мл насыщенного раствора сульфата
магния, 0,13 моля дигидрата мононатриевой соли фосфорной кисло-
ты и 500 мл петролейного эфира, не содержащего олефинов, на
водяной бане при 15°. После этого добавляли 55 г измельченного
15е
СН3СН0НСО,СяН54-КМпО4 сн3сосойсгн5
" “ 51-54%
0,42 моля 0,35 моля
(25%-ный
избыток)
К- п. в течение 25—30 мин. Перемешивание продолжали до тех пор,
пока проба на фильтровальной бумаге не показывала, что весь К- п.
израсходован; раствор в петро л ей ном эфире отделяли декантацией,
а остаток промывали тремя порциями растворителя. Вытяжки сое-
диняли и растворитель отгоняли на маленькой колонке. Масля-
нистый остаток взбалтывали с двумя порциями насыщенного водного
раствора хлористого кальция, чтобы отделить этиловый эфир молоч-
ной кислоты; этиловый эфир пировиноградной кислоты перегоняли
при 56—57°/20 мм. Дальнейшая очистка возможна путем превраще-
ния препарата в бисульфитное производное.
Окисление диола (1) в о-ди ацетил бензол осуществляют в раство-
ре, к которому в качестве буфера добавлен азотнокислый магний,
по-видимому, для осаждения гидроксил-иоиа и, следовательно,
89
для предотвращения альдолизации продукта реакции (Гольдшмидт
[13]). Смесь диола и буфера в 250 мл воды перемешивали при 68-
ОН
z ,СНСН3
/х / 3
| )| +КМпО4
СНСН3
I
ОН
(18-75%
Mg (NO;,). (0, 12 моля)
Gb%
ч /СОСН3
11'
хсосн3
(1)0,059 моля 0,09 моля
(избыток 14%)
75° и порциями в течение 3 час добавляли тонкоизмельченный К. п.
Двуокись марганца отделяли фильтрованием, промывали эфиром
и экстрагировали эфиром в аппарате Сокслета; фильтрат экстраги-
ровали эфиром в течение 12 час в перколяторе. Полученные экстрак-
ты соединяли, отгоняли растворитель и перегонкой (НО—116°/
0,1 мм) получали о-диацетилбеизол с выходом 66%. Маслянистое
вещество закристаллизовывалось при потирании стеклянной палоч-
кой о стенки стакана при —20°; после кристаллизации из смеси эфи-
ра и иетролейиого эфира получали длинные иглы, т. пл. 39—40°.
Эту методику можно, по-видимому, упростить, если использо-
вать восстановление с помощью SO2.
AiCILCH - АгСОСНз. Холстен и Питтс [141 получили и-диаце-
тилбензол путем добавления по каплям n-эти л ацетофенона к смеси
К. п., взятого с 90%-ным избытком, азотной кислоты и окиси магния
в 1034 мл воды при перемешивании и охлаждении (температура 60~).
СОСН3 СОСНз
I . I
// Ч GOT 4,5 час //\
I I! „ 2,1 моля IINO4- 1 июль MgO I [I
+ КМпО4 ---------------—------—
82% Ц II
f I
СН,СН3 СОСНз
0,4 мулл 1 моль
(избыток 9 0%)
Затем смесь перемешивали при 60° еще 4,5 час. После охлаждения
смесь МпО2 и продукта реакции отфильтровывали и продукт реак-
ции экстрагировали бензолом; выход 82%. Если К. п. взять только
с 50%-ным избытком (от теорет.), выход падает до 70—76%.
Олефии - кислота. Хилл и Мак-Ювеп [151 получали азелаиновую
кислоту по следующей методике. 1<. п. перемешивают с водой при
35° в колбе на 12 л до полного растворения, затем в один прием
добавляют щелочной раствор неочищенной рицинолевой кисло-
ты, полученной омылением касторового масла. Температура повы-
90
шается до 75° и примерно через 30 мин качественная проба па К. п.
показывает его отсутствие. Для обработки смесь делят па двечасти.
ОН
СН3(СН,);,СНСН2СН=СН(СН-J7CO,JI-I- КОН4- KMnOj —
0,8 моля (съ’ра:|) 1 моль 3 ,5 моля 3 2 — 86%
------------------------.----.--—-------' 7 5 л НЮ
1,6 л 1ЦО
—> 1-Ю,С(СН.р7СО.,Н
Каждую часть осторожно подкисляют серной кислотой (выделяющая-
ся двуокись углерода вызывает вспенивание), затем нагревают
на водяной бане, чтобы вызвать частичную коагуляцию МпО2; пос-
леднюю отделяют фильтрованием, помещают в стакан и кипятят с
2 л воды для растворения адсорбированной азелаиновой кислоты.
Соединенные фильтраты упаривают до объема приблизительно 4 л,
охлаждают льдом и получают азелаиновую кислоту, которая после
кристаллизации из воды оказалась препаратом удовлетворительного
качества (т. пл. 104—106 е).
В этом случае восстановление с помощью SO., может уменьшить
время, необходимое на фильтрование, промывание и упаривание.
Весьма возможно, что выход улучшится, если взять большее коли-
чество К- п. Наличие СО., в реакционной смеси указывает на частич-
ное расщепление по следующему уравнению:
ОН
СН3(СН2)Г1СНС1-12СН = СН(СН.,)7СО.ф1 11 экв кислоро^
—СН3(СН.2)6СО.2Н + СОа-1-С0.,-!-НО2С(СН2)7СО.2Н
Для такого процесса необходимо 5,85 моля К. п., тогда как взято
только 3,5 моля. Однако, по-видимому, рицинолевая кислота в
данном случае менее подходящее исходное соединение, чем олеино-
вая кислота.
—С=С——>—СОСО—. Методика Кана и Ньюмена [161 для
окисления стеароловой кислоты до 9,10-дикетостеариновой кислоты
интересна высоким выходом, получаемым в результате применения
в качестве буфера двуокиси углерода; кроме того, в реакции расхо-
дуется не весь К- п. Кислоту растворяют при перемешивании в разб.
щелочи, в раствор пропускают ток углекислого газа до pH 7,5,
затем в один прием приливают раствор К. п., взятого в избытке,
в 300 мл воды, поддерживая температуру 25° и pH раствора 7—7,5.
Через 1 час добавляют бисульфит натрия и соляную кислоту, чтобы
СН3(СН,)уС = С(СН2)7СОаН+ КМпО4 .СОд
92 — 96%
0,02 моля 0,4 моля
(0,0 23 моля КОН, (теория:
3 -г ЬЦО) 0,026 моля)
’ О О
il il
СН3(СН2)7С—С(СН.,)7СОаН
91
разрушить избыток К- и., восстановить МпО2 и осадить дикетокис-
лоту. После кристаллизации из абсолютного этанола получают пре-
парат (две порции) с т. пл. 84,5—85:;.
Брусон п сотр. [171 окисляли пространственно затрудненный
тетраметилтетралон (1) большим избытком К. п. в растворе 0,5 е
едкого натра в 255 мл воды, добавляли серную кислоту и бисульфит
натрия для растворения МпО., и охарактеризовали бесцветный пре-
парат, выпавший в осадок, как а-оксикислоту (3). По-видимому,
при окислении в а-дикетон (2) произошло сужение цикла, а в силь-
нощелочной среде прошла перегруппировка типа перегруппировки
бензиловой кислоты. При окислении в присутствии сульфата маг-
ния в качестве буфера (для осаждения гидроксил-иона) реакция
протекала значительно дольше, причем образовалась дикарбоновая
кислота (4). К. п. был взят с 72%-ным избытком и, по-видимому,
он весь прореагировал, поскольку при переработке «для удаления
МпО2 добавляли бисульфит натрия, раствор подкисляли серной
кислотой н многократно экстрагировали эфиром».
(1) 0, 016 моля (J)
О, 02гмоля4-KMnO.fO, Имдля) + MgSO4 (0, 33 моля)
Кипячение 40 час, 83%
(4)
Олсфин^виципальный диол в водной среде. Непредельные
жирные кислоты можно превратить окислением К. п. в щелочной
среде в вицинальные диолы с высоким выходом. Дж, М. Робинсон
и Р. Робинсон 1181 кратко описали окисление олеиновой кислоты
в 300 ч. воды и Д. ч. едкого кали при 0й путем постепенного добавле-
ния 0,5 н. раствора К. п. После восстановления МпО2 двуокисью
Н Н Н Н
сн3(сн3)7с=с(снг)7со2н сн3(сн2)7с-с(снг)7со3н
он он
серы и кристаллизации продукта реакции из смеси спирта и бен-
зола получили эрнтро-9,10-диоксистеаринову1о кислоту (т, пл. 132°)
с почти теоретическим выходом. Лэпуорс и Моттрам [191 нашли
условия для почти полного превращения в диол и разработали ана-
литическую методику, согласно которой достаточно 5 г непредель-
ной жирной кислоты. 1Четодика эта не имеет большого препаратнв-
92
кого значения, поскольку на 1 г олеиновой кислоты требуется 1 л
раствора. Трейнар [201 получил эритро-щюл с выходом 70—75%,
израсходовав только 40 мл раствора на 1 г непредельной кислоты.
Группа сотрудников Свериа [21] показала, что окисление олеиновой
кислоты К. п. зависит от pH. При pH, равном в конце реакции 11,8,
продуктом реакции оказалась смесь диола (60%) и 9,10- и 10,9-ок-
снкетостеарииовых кислот; при pH 9—9,5 продуктами реакции были
диол (4%) и а-кетолы (60?о). Окисление элаидиновой кислоты мало
зависит от pH.
В условиях, аналогичных тем, при которых из циклогексена
образуется цис-циклогексаидиол-1,2 с выходом 30—33% [22—23!,
Эванс и сотр. [241 получили диэтил ацеталь d,l -глицеринового аль-
дегида из диэтилацеталя акролеина с выходом 67%. Суспензию
ацеталя в воде перемешивали при 5° и прибавляли к ней водный
СН,-СНСН(ОС2Н5)г -р КМпО4-------
A Е - > - -т 12 А Л А С > г 67
0,5 моля, 600 мл 0,5 моля в 67%
Н2О 600 л.?
СН2СНСН(ОС2Н5)3
Н2О (теория:
0,33 моля)
ОН ОН
раствор К- п., взятого с 52%-ным избытком, со скоростью около
25 jmImuh. Вскоре после завершения добавления смесь преврати-
лась в гель. Смесь выдерживали в течение 2 час, нагревали 2 час
на водяной бане, МпОа отфильтровывали и
промывали водой (по-видимому, весь К- п.
был израсходован). Фильтрат обрабатывали
1,2 кг свежевысушенного поташа. Водный снг _^сн
слой отделяли, экстрагировали эфиром, пре-
парат выделяли и перегоняли. Относительно
высокий выход, полученный при применении
избытка К- л., можно объяснить стабилизацией диола за счет обра-
зования хелатного соединения.
Олефин * диол в безводном ацетоне. Тишлер и сотр. [25] раз-
работали эффективную методику для введения 17а-гидроксильной
СНэОАс
। г
с=о
СИЛ ..ОН
сн3
+ KMnCU
2 л ацетона н-
108 лгл'пиперидина
0, 545 моля 98,4%
(теория: 1,5 моля)
(1) (0, 226 моля)
В 2л ацетона
группы в 20-кетостероиды, используя для гидроксилирования К- п.
вместо четырехокиси осмия. 20-Кетой превращают через 20-циан-
гидрнн в 17,20-непредельный 20-нитрил (1). Раствор 90 а (1) в сухом
93
ацетоне перемешивают при —5°, обрабатывают пиперидином и из-
бытком К. п., перемешивают 30 мин и обрабатывают при темпера-
туре ниже 2° 18 мл уксусной кислоты в 225 мл ацетона. Реакционную
смесь выдерживают 4 час при 0—2°, приливают 1,8 л хлороформа,
а затем 270 мл коиц. соляной кислоты в 1350 мл воды и 126 г бисуль-
фита натрия в 900 мл воды. После экстрагирования хлороформом
и кристаллизации получают чистый 21-ацетат прегнаидиол-17а,
21-триона-3,11,20.
Окисление водным К. п. в смеси с метилциклогексаиом. В раннем
исследовании, целью которого было выяснение строения эргосте-
рина, Рейндел и сотр. [26] окисляли эргостерин по несколько нео-
бычной методике и получили продукт окисления, который удалось
идентифицировать спустя четверть века в исследовании М. Физер,
б)
(г)
А. Квилико и сотр. [271. Продуктом реакции оказалась смесь, со-
стоящая примерно из 7 ч. О3-соединения и 3 ч. Отсоединения, при-
чем главный продукт реакции был охарактеризован как трижды
ненасыщенный триол (2). Возможно, что он образуется в результате
({мс-гидроксилирования 5,6-двойной связи, аллильного гидрок-
силирования у С14 и отщепления воды. Квилико разработал воспро-
изводимую методику получения продукта окисления, не содержа-
щего эргостерина, с выходом 80% (по весу), используя 3 кислород-
ных эквивалента К- п. К горячему раствору 4 г эргостерина в 100 мл
метил циклогекса на (который является значительно лучшим раство-
рителем для стеринов, чем w-алканы) добавляют 75—80 мл 4%-ного
водного раствора К- п., предварительно нагретого до 90—100°,
колбу закрывают пробкой и смесь энергично взбалтывают, часто
вынимая пробку для выравнивания давления. Вскоре начинает
выделяться двуокись марганца,и густая вначале масса превращается
в жидкую суспензию. Все количество К- п. расходуется за 8—
10 мин. В смесь пропускают ток сернистого газа, после чего образо-
вавшуюся суспензию переносят в делительную вороику и промыва-
ют несколькими порциями воды для экстрагирования неорганиче-
ских солей. Углеводородный растворитель отгоняют с водяным па-
ром, раствор упаривают и после нагревания с метанолом получают
кристаллический продукт.
94
он
— СН (СН3)г —> — С(СН3)г. Ранние наблюдения Бючера [28],
а также полученные еще раньше данные о положении метильной
и изопропильной групп в ретене доказали гидроксилирование изо-
пропильной группы. Окисление ретепхнпона (1) в приведенных
СН (СН3)3
Щ КМпО4 4- Пиридин
Кипячение с 100 л/л НЮ
СН3
(1) 10 г
ОН
С(СН3)3
I
П
V чсо2н
I 7СО2Н
)!
(2) 2,2 г
на схеме условиях привело к образованию продукта (2).
Сравнительно недавно Истмен и Квини [291 нашли, что третич-
ный спирт (3), имеющий третичный атом водорода, окисляется К- и.
в соединение (4) со значительным выходом. К суспензии спирта в
воде в течение 2 дней прибавляют измельченный К- п. (немного
КМпО4-ОН
-----d--—>
40%
более 1 моля). Реакция протекает с сохранением конфигурации. По-
видимому, для реакции существенно наличие третичной гидроксиль-
ной группы в исходном соединении (1).
RCH2N02-^RCHO. Шехтер и Уильямс [30] показали, что пер-
вичный питроалкан окисляется в виде нитроната калия по урав-
нению 2:
1) rch2no2 25°Д rch = мо2к
2) 3RCH = NO2K + 2КМпО4Щ Н,0 —> 3RCHO %- 2MnO,+ 3RNO2 Щ2КОН
Для поддержания нейтральной среды добавляют сульфат магния,
осаждающий гидроксильный ион в виде гидроокиси магния. Хотя
образующийся альдегид окисляется медленнее, чем нитронат, к
концу реакции окисление до карбоновой кислоты становится кон-
курирующей реакцией, если количество взятого К. п. не составляет
95
70—90% от теоретического. Раствор нитросоединения в едком кали
обрабатывают раствором сульфата магния, разбавленного до 500 ли,
и по каплям при перемешивании и температуре 0—5° добавляют
СН3СНХН,СН.,т,-Н<МпО4л-2М MgSO4 —X СНХНХНоСНО
89%
Молих Ю8 = 6,88 6.22 20 л/л (в виде ДМФ)
раствор К- п. Альдегид перегоняют с водяным паром, собирая
дистиллат в приемник с водным раствором соляной кислоты, насы-
щенным 2,4 - ди питрофепи л гидр азином. Эта методика особенно удоб-
на для некоторых вторичных ш [тросоед и иен им, например нитро цик-
лобутана.
NOZ
< X KMnO4 X \
94%
RNFL->RNO>. Корнблгом и Джонс 1311 перемешивали смесь
гп pern-октил амина в 500 мл ацетона и раствора сернокислого магния
в 125 мл воды и добавляли большой избыток измельченного К- п.
в течение примерно 30 лшн, поддерживая температуру 25—30°.
(СН3)3ССН.,С(СН-Д,4-2К1МпО4-|-Водн. MgSO4 ..... '
| ‘ 69-8 2%
NH.,
0,2 0 моля 1,20 моля 0,14 моля
(теория: 0.40 моля)
(СН3), ССЫ,С (СН-Щ -у Mg (ОН), 4- K,SO4 -Ц2МпО„
* I
NO,
Перемешивание при 25—30° продолжали еще в течение 48 час. Для
удаления ацетона смесь перемешивали при 30° и создавали в колбе
вакуум с помощью водоструйного иасоса. Полученную вязкую мас-
су перегоняли с водяным паром и получали светло-голубой органи-
ческий слой, из которого после высушивания и перегонки при 53—
54°/3 мм получали бесцветный 4-иптро-2,2,4-триметилпентан, т. пл.
23,5—23,7°. По-видимому, роль сернокислого магния заключается
в осаждении гидроксильных ионов и, следовательно, предотвраще-
нии образования изонитрозоната калия % При температурах значи-
тельно выше 30° К- п. быстро расходуется «по-видимому, за счет
реакции с ацетоном». Этот метод был с успехом использован для
синтеза семи других три ал к ил нитрометанов.
АгСОСН ^ АгСНО. Ар илметилкетон, полученный по реакции
Фриделя — Крафтса или Фриса, окисляют щелочным К- п. в пи-
ридине при 10—15° в арилглиоксиловую кислоту, которую декар-
боксилируют обработкой П,ЭД-диметил-/г-толуидином при 140—
160° [321.
Агсосн3 22Д24 Агсосо,н Агсно з-со2
140-160°
* Объяснение не верно, так как тре/п-алкилнитрозосоединение не может
дать изонитрозонаты.— Прим. ред.
96
1, Ball S.,Goodwin T. W., Morton R. A,, Biochem. J., 42, 516
(1948).
2. Attenb arrow J., Cameron A. F. B., Chapman J. H.,
Evans R. AC, Hems B. A., J ansen A. B, A., Walker T., J.
Chem. Soc., 1952, 1094.
3. К о 1 t h о f f 1. M., Belcher R., Volumetric Analysis, 3, 37—38 (1957).
4. Ladbury J. W., C u 1 1 i s C. F., Chem, Revs., 58, 403 (1958).
5. Рухоф Дж., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 571.
6. Ш р а й н е р Р., К л е и д с р е р Е., «Синтезы органических препаратов»,
ИЛ, М,, 1949, сб, 2, стр. 417.
7. Кларк X., Тэйлор Е., «Синтезы органических препаратов», ИЛ, АТ,
1949, сб. 2, стр. 556.
8, С и и г е р А., Ма к-Эл ьвец С., «Синтезы органических препаратов»,
ИЛ, М„ 1952, сб, 3, стр, 145.
9. Гарднер Дж., Нэйлор К-, мл., «Синтезы органических препаратов»,
ИЛ,, М., 1949, сб. 2, стр. 27.
10, G г а е b е С,, Т г й m р у F., Вег,, 31, 369 (1898).
11, Д ж о н е с Р., М а к - Л а ф л и н, К,, «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 426.
12. Корнфорз Дж., «Синтезы органических препаратов», ИЛ, М,, 1953,
сб. 4, стр, 589.
13, Goldschmidt S,, Zoebelein A., Chem. Вег,, 94, 169 (1961).
14. Н о 1 s t е n J. R., Р i t t s E. Fl., Jr., J. Org. Chem., 26, 4151 (1961).
15. Хилл Дж., M а к - IO в e н В,, «Синтезы органических препаратов», ИЛ,
М-, 1949, сб. 2, стр. 9,
16. К h а п N, A., Newman М. S., J. Org. Chem., 17, 1063 (1952).
17. В г п s о n И. A., G г a n t F. W., Bobko Е., J. Am. Chem. Soc,, 80, 3633
(1958).
18. R ob inson G. M., Robinson R., J. Chem, Soc., 127, 175 (1925).
19, Lapworth A., Mottram E, N., J. Chem. Soc., 127, 1628 (1925).
20, Traynard J.-C., Bull. soc. chim. France, 19, 323 (1952).
21. Coleman J. E,, Ricciutf C., Swern D., J. Am. Chem. Soc., 78,
5342 (1956).
22. Физер Л., Ф изер M., «Органическая химия», изд-во «Химия», М., 1966.
23. С 1 а г k е М. F., Owen L, N., J. Chem. Soc., 1949, 315; окисление в смеси
этанол — вода с MgSO4 в качестве буфера при —15° с образованием диола,
выход 33%
24. Вицеман Е., Э в а и с У,, Хасс Г., Шредер Е,, «Синтезы органи-
ческих препаратов», ИЛ, М., 1949, сб. 2, стр. 65.
25. Т u 1 I R,, J о n е s R. Е., R о b i n s о n S. А., Т i s h 1 е г М., J. Ат.
Chem. Soc., 77, 196 (1955).
26, R е i n d е 1 F., et al,, Ann., 452, 34 (1927); 460, 212 (1928).
27. F i e s e г M,, Q u i 1 1 с о A., N ick on A., R о s c n W. E., Tarlton
E. J., Fieser L. F., J. Am. Chem. Soc., 75, 4066 (1953).
28. Bucher J. E., .J. Am. Chem. Soc., 32, 374 (1910).
29, E astm an R. H., Quinn R. A., J. Am. Chem. Soc., 82, 4249 (1960).
30. Shechter H., Williams F. T., Jr., J. Org. Chem., 27, 3699 (1962).
31. Kornblum N..Jones W. J., Org. Syn,, 43, 87 (1963).
32. Cy merman - Craig J,, L о der J. W., Moore B., Australian J.
Chem., 9, 222 (1956); see also Ferrari J. L., Hunsberger 1. M,
Gutowsky H. S,,J. Am. Chem. Soc., 87, 1247 (1965).
КАЛИЯ ПЕРСУЛЬФАТ И АММОНИЯ ПЕРСУЛЬФАТ, K2S3O8,
(NH4)2S2O8. Мол. вес 270,33; 228,21. См. также Каро кислота.
Фенолы двухатомные фенолы [11 (реакция Элбса [2]).
Примером может служить окисление салицилового альдегида
4 Заказ № 1096
97
в гентнзииовый альдегид [31:
он он он
1 сно OSOjOK ено д сно
\\\ -ЩИ’' +HOSO.OK
U кон V V
OSO,OK он
Ar 1соль иодония. Примером может служить получение ди-
феиилиодоний-2-карбоксилата 141 (IV) — предшественника дегид-
робензола 15]. По упрощенной методике [6] к смеси 2 г о-иодбензой-
ной кислоты и 2,6 г персульфата калия прибавляют 8 мл охлажден-
ной льдом серной кислоты, Смесь взбалтывают, вращая колбу от
(OSO2OK)
Нгзо4
ОЗОгОК
О5ОгОК
1-сщ5
со2н
IV
руки в течение 4—5 мин при О’, и выдерживают 20 мин при 25°.
Полученный раствор соединения II охлаждают до 0°, прибавляют
2 мл бензола и вновь взбалтывают таким же образом, после чего
дают нагреться до комнатной температуры, чтобы произошло обра-
зование соединения III. Хорошо охлаждаемую смесь разбавляют,
а затем нейтрализуют аммиаком в присутствии хлористого метилена,
экстрагирующего IV, который кристаллизуется из воды в виде круп-
ных призм моногидрата; выход 2,1 г.
Ариламин —> о-сульфат 17]. В щелочной среде ариламины окис-
ляются этим реагентом до солей о-сульфата. Так, раствор 5 г N,N-
диметиланилина в 250 мл воды, 400 мл ацетона и 30 мл 2 н. едкого
кали перемешивают при комнатной температуре в течение 8 час и за
это время добавляют 11,2 а персульфата калия в виде насыщенного
водного раствора. Раствор упаривают до 250 мл, промывают эфи-
Х(СН3)2 OSO.2OK N(CH-t)3 А/он Конц. HCI (100°) | |[
//л OSO,OK 1 OSO,OK
V 40% и 83»/О ' 1 II X/
98
ром, выпаривают досуха при пониженном давлении и остаток экс-
трагируют горячим 95%-ным этанолом. При разбавлении эфиром
(1,5 л) осаждается о-диметиламинофенилсульфат калия. При под-
кислении водного раствора соли образуется соответствующий кис-
лый сульфат, а нагревание с коин. соляной кислотой в течение 1 час,
последующее охлаждение и частичная нейтрализация щелочью
дают о-диметиламинофеиол.
Гидролиз оксимов. Брукс и сотр. 18] встретились с трудностями
при регенерации стероидных 3,20-дикетоиов из их 3,20-диоксимов
известными методами. Так, при гидролизе соляной кислотой обычно
гидролизовалась лишь одна функциональная группа у С,о. Затем
было найдено, что хорошим методом является окислительный гид-
ролиз надсерной кислотой. Например, 0,25 а 3,20-диоксима корти-
зола (1) растворяли в теплом растворе 0,16 г персульфата аммония
в 50 мл теплой 1 н. сериой кислоты. Добавляли 50 мл хлористого
метилена и смесь взбалтывали в течение 8 дней при комнатной тем-
пературе. При выделении стероида получили 0,191 г кортизола (2).
Успешное применение персульфата аммония основано на том, что
он окисляет образующийся гидроксиламин; однако оказалось, что
использование вместо персульфата аммония солей трехвалентного
железа к положительным результатам не приводит.
I, S е t h п a S. М., Chem. Revs., 49, 91 (1951).
2. Е 1 b s К., J. prakt. Chem., 48, 179 (1893).
3. Neubauer O., Flatow L.,Z. physiol. Chem., 52, 380 (1907); Schock
R. U., Tabern D. L., J. Org. Chem./16, 1772 (1951); see also Baker W.,
Brown N, C., J. Chem. Soc., 1948, 2303.
4. Bcringer F. M., Lilllen I., J. Am. Chem. Soc., 82, 725 (1960).
5. Le Goff E., J. Am. Chem. Soc., 84, 3786 (1962).
6. Org. Expts., 311—314.
7. Boyland Б-, Mason D., Sims P., J. Chem. Soc., 1953, 3623.
8. Brooks S.G., Evans R.M.,Grcc n G. F. f-E, Hunt J. S., Long
A. G., Mooney B., Wyman L. J.,J. Chem. Soc., 1958, 4614.
КАЛИЯ ПЕРСУЛЬФАТ — СЕРЕБРО (КАТАЛИТИЧЕСКИЕ КО-
ЛИЧЕСТВА) [1]. Этот реагент расщепляет растворимые в воде
вицинальные гликоли с образованием альдегидов, кетонов или их
смеси с выходами от 40 до 100%. Например, к раствору фенплэти-
R.RX-OH
I 4 S,O82- — RjR.^C —ОR3R4C = O--2SO2--L2H+
R3R4C-OH
4*
99
ленгликоля в 300 мл воды при перемешивании и температуре 30°
добавляют персульфат калия, а затем 1,3 мл 10?4-ного раствора
нитрата серебра. После перемешивания в течение 1 час температура
Д(г+ Т4 П 30°
CgH5CHCH2OH4OSO,OK — ' С6Н5СНО
' I I 61%
ОН OSO3OK
О, I моля 0,11 моля
повышается до 40° и бензальдегид выделяется в виде маслянистого
слоя. Этот реагент ие является специфичным только для вициналь-
ных гликолей; одноатомные спирты и альдегиды также окисля-
ются персульфатом при каталитическом действии серебра. Кине-
тическое исследование расщепления цис- и т/?а«с-циклогександио-
лов-1,2 неожиданно показало, что скорость реакции для обоих изо-
меров совершенно одинакова.
1. Greensp an F. Р., Woo d burn Н. М., J. Am. Chem, Soc., 76, 6345
(1954).
КАЛИЯ ТИОЦИАНАТ, KSCN. Мол вес 97,18.
К. т. реагирует с алкилгалогенидами, диалкилсульфатами 11]
и тозилатами 12] с образованием тиоцианопроизводных, которые
можно подвергать десульфуризации под действием никеля Ренея
[3]. К. т. в водном растворе легко реагирует с З-метокси-1,2-эпокси-
RX(OTs)4" KSCN RSCN _^ДД; RH
пропаном (1), давая 2-метоксиметилтииран (2) [41. Десульфуриза-
ция (2) гладко проходит в присутствии триэтил фосфит а с образова-
нием (3).
/°. ' Sx
сн3осн,-с —хсн, —сн3осн2—с~сн, .(с^н.фДД
] “92% ( “ 94%
н н
(I) (2)
СН3ОСН3СН = СН2-R (C2Hr,O)3PS
(3) (4)
1. Walden Р., Вег,, 40, 3214 (1907).
2. И a n n R. М., Richtmyer N. К-, Diehl Н. W.f Hudson C. S.,
J. Am. Chem. Soc., 72, 561 (1950).
3. Snyder H. R,, Stewart J. M., Ziegler J. B., J. Am. Chem. Soc.,
69, 2672 (1947).
4. S c h u e t z R. D,, Jacobs R. L., J. Org. Chem., 26, 3467 (1961).
О
I!
КАЛИЯ ТРИАЦЕТИЛОСМАТ, (CH3CO3)3Os—OK, и ДИКАЛИЯ
ТЕТРАМЕТИЛОСМАТ, (CH3O)dOs(OK)2.
Криге [1] эти реагенты применил для идентификации вициналь-
ных диолов. Дикалийтетраметилосмат (1), кристаллическое зеленое
100
вещество, получают' из четырехокиси осмия и раствора едкого кали
в метаноле. В растворе в метаноле он реагирует с цис-днолами, а
также с достаточно гибкими транс-диолами. транс-Циклогексан-
диол-1,2, например, растворяется в растворе этого реагента в мета-
ноле (КОН), причем происходит изменение окраски и умеренно
растворимую соль (2) можно выделить с хорошим выходом. Вещество
это весьма чувствительно к воде, но при подкислении суспензии
соли в хлористом метилене диэфир (3) переходит в органическую
фазу и его можно выделить в кристаллическом виде. Для получения
триацетил осмата калия (6) тетраметилосмат (1) растворяют в уксус-
ной кислоте; при соответствующей концентрации из синего раствора
(СНзСЩОв' ----------> (CHsOhOs' ----------> (СНзСОЩОз' --------->
\к хок ок
(1) (4) w (5)
(7) + 2 АсОН + АсОК + НгО
выпадают кристаллы (6). Это вещество в уксусной кислоте необра-
тимо реагирует с цис-диолами с исчезновением синей окраски и об-
разованием диэфира (7). Оно реагирует также со многими транс-
диолами, но реакция может протекать и в обратном направлении:
прибавление ацетата калия к раствору диэфира транс-диол а сопро-
вождается появлением синей окраски соли (6); в тех же условиях
диэфиры цис-диолов в реакцию не вступают.
l.Criegce R. cl al., Ann., 550, 99 (1942); 599, 81 (195 t };Chem. Ber., 90, 417
(1957).
101
КАЛИЯ ТРИФЕНИЛМЕТИД, см. Трифенилметилкалий.
КАЛИЯ ФЕРРИЦИАНИД I калия гексацианоферрат (III)],
K3Fe(CN)fi. Мол. вес 329,26.
Окисление фенолов. К- ф- в щелочном растворе действует как
комплексный ион, отщепляющий электрон, и многие реакции окис-
ления фенолов, описанные в литературе III, по-видимому, включают
окисление фенолят-аииопа в О-радикал, изомеризацию его в С-ра-
дикал и димеризацию углеродного радикала или соединение его
с кислородным радикалом. Интересным примером является двух-
стадийный синтез (±)-усниновой кислоты по Бартону [2]. Метил-
фл о р ацетофено и (5 а) растворяют в освобожденном от воздуха ра-
створе 12,5 г карбоната натрия в воде, раствор охлаждают до 0° и
при перемешивании в атмосфере азота добавляют I же К. ф. в воде.
По-видимому, в результате сдваивания С-радикалов образуется про-
межуточное соединение (иа схеме заключенное в скобки) и фенольный
гидроксил присоединяется к епоновой системе с образованием
димера, выделенного с помощью препаративной хроматографии
с выходом 15%. При дегидратации серной кислотой при 0° получена
усниновая кислота с выходом около 2%.
СОСНз
Уснииовая кислота
То, что первой стадией окисления фенола является образование
О-радикала, убедительно доказано выделением стабильных кисло-
родных радикалов. Мюллер и сотр. [31 получили несколько радика-
лов путем взбалтывания бензольного раствора 2,4,6-тризамещенного
фенола со щелочным раствором К. ф. Ароксильный радикал (2)
окрашен в темио-синий цвет, является сильным окислителем и су-
ОН О-
/QCHjjjg (СН 3 - С(СН3)3
J || K3Fe(CN)6, ОН- | ||
C(CH3)3 i(CH3)3
(1) (2)
102
шествует в виде .мономера в твердом состоянии и в 0,1 и, бензольном
растворе. Он очень чувствителен к действию кислорода. Стабильный
радикал (4) получил Коппингер 14] окислением соединения (3) дву-
окисью свинца в эфире, а также Караш и Джоши [5] — окислением
щелочным раствором К. ф. Радикал (4) образует темно-синие иглы,
ОН О
,1 з [I
(ТН3)3С——С(СН3)3 щелочи. KsFc‘(CN)fi (СН3)3С——С(СН3)3
I ||-------------------------* II ||
X/ \/
I II
сн3 сн
I I
XX /X
(сн8)3с ;L С(Сн3)3 (сн3)3с - J- с(сна)3
I , I
он о-
(3) (4)
т. пл. 157°. Бартлетт 16] впервые применил его в качестве акцептора
радикалов и назвал гальвиноксил*.
2-Амипофенол со свободным по отношению к гидроксильной
группе пара-пол ожени ем образует димерный продукт окисления
с высоким выходом. Так, Бутенапдт и сотр. [7] окисляли аминофе-
нол (1) теоретическим количеством К- ф., фильтрованием отделяли
СОСНд СОСНз СССН,
(!) 0,08 моля (2)
оранжево-красный осадок и перекристаллизовывали его из этил-
ацетата, получая таким образом чистый З-амипо-4,5-ди ацетилфенок-
сазон (2) с выходом 80%.
Оксииндолы иногда можно получить окислением соответствую-
щим образом замещенных гидрохинонов, что, например, иллюстри-
руется последней стадией синтеза серотонина (4) [8].
chch2ch2nh2
ch2nh2
K3Fe(CN)
NaHCO3
48%
CH2CH2NH2
(4)
* По имени Гальвана Коппингера.— Прим ред.
103
При окислительной циклизации бензофенона (5) до дегидрогри-
зеофульвина (6) Тауб и сотр. [9] нашли, что в обычных условиях
(окислитель прибавляют к фенолу) выходы (6) составляют 50—60%,
причем возвращается значительное количество бензофенона. Однако
если субстрат прибавлять к раствору окислителя, то выход стано-
вится почти количественным. Раствор 17,5 г карбоната калия в
125 мл воды добавляют к раствору 1 г бензофенона в 10—15 мл трет-
бутилового спирта; последний удаляют испарением в вакууме и
щелочной раствор добавляют по каплям при перемешивании к раст-
вору 4 г К. ф. в 50 мл воды. Сразу же выпадает осадок, который
собирают, промывают и сушат; на бумажной хроматограмме про-
дукт дает одно пятио.
Замещенный в пара-положении фенол (7) образует с высоким вы-
ходом о-дифепохииои (8) [101.
СН3СХ КяВеЩкщ. СН3О Ох
>---X води. СНзОН, КОН у---X
2Н3С-^_____-------------------> Н3С-^ 7-СН3
ЧОН ^ОСНз
(7) (8)
Окислительное декарбоксилирование. Лохаус 111] кратко описал
синтез n-терфенила, в котором последней стадией является порази-
тельно легкое окислительное элиминирование двух карбонильных
групп в результате реакции со щелочным раствором К. ф. Мак-До-
нальд и Кэмпбелл 112] повторили этот синтез, не выделяя промежу-
точные продукты, и распространили эту схему на синтез ряда
полифенилов, включая н-квинквифенил 113]. Однако Л. Физер и
Хаддадин [14] воспроизвели синтезn-терфенила и нашли,что дикарбо-
новая кислота, претерпевающая декарбоксилирование, имеет не ту
структуру, которую ей приписывали. Ниже приведены полученные
ими результаты. Аддукт реакции Дильса — Альдера (1) в результа-
те кратковременной обработки метанольным раствором едкого кали
изомеризуется в более высокоплавкий транс-эфир (2), а при дли-
тельном действии основания в результате гидролиза образуется
транс-дикислота (3). Это соединение быстро и почти количественно
декарбоксилируется при обработке щелочным раствором К- ф- при
комнатной температуре. В результате отрыва двух электронов от
дианиоиа (4) образуется бирадикал (5), который отщепляет две мо-
лекулы двуокиси углерода от карбоксильных групп в аллильном
104
положении с образованием п-терфенила (6). В той же работе приве-
дены и другие примеры окислительного декарбоксилирования, опи-
санные в литературе.
Окисление метильной группы ароматических соединений.
Нойс 1151 показал, что метильную группу в о- и л-иитрото-
луоле и в о-, м- и /г-толуолсульфамиде можно окислить до карбо-
ксильной группы кипячением исходного соединения с большим из-
бытком водного К- ф- Хотя в других случаях выходы очень иизки,
реакцию иногда можно использовать для выяснения структуры.
Так, Вейссгербер и Крубер 116] применили ее для идентификации
выделенного ими из каменноугольной смолы 1,6-ди мет ил нафтали на:
4 г 1,6-диметилнафталина перемешивали с 330 г К. ф. и 57 г едкого
кали (водный раствор) в течение 24 час, перегонкой с паром удаляли
K:iFe(CN)s
кон
СО2Н
не вступивший в реакцию углеводород, а подкислением и экстраги-
рованием эфиром выделяли 0,5 г неочищенной нафталин-1,6-дикар-
боновой кислоты. Аналогичные опыты приводит в своей работе
Ружичка [171.
Получение 1-метилпиридона-2 (2). Согласно методике, опи-
санной Приллом и Мак-Эльвеном [18], пиридин обрабатывают
эквивалентным количеством диметилсульфата для получения метил-
сульфата 1-метилпиридиния (1), раствор этой соли в воде окисляют
105
при 0°, одновременно добавляя растворы К. ф. и щелочи.
1) 83 моля
K3Fe(CN)6(3, 65 моля)
NaOH (7, 5моля)?10°
CH3SO4~ 65-7 0%
(оЗщий)
Деметилирование третичных аминов. Перрин [19] при попытке
окислить тропин (1) щелочным раствором К. ф. вместо троп ином а
получил портропин (2):
(О
K3Fe(CN)6^
87% (сырой)
(2)
Аналогичным образом из З-диметилампноцнклогексанола (3)
был получен 3-монометиламиноциклогексанол (4). При реакциях с
N-метил морфолином, кодеином, никотином и диметил анилином
также не удалось выделить вторичный амии, хотя в каждом из этих
случаев К. ф. восстанавливался.
Н /ОН
I / н
N-CH3
сн3
КзГе(СК)й
93% (сырой)
(3)
Нх / ОН
f н
X/N — Н
I
СН3
(4)
l.Thyagarajan В. S., Chem. Revs., 58, 439 (1958). For recent e.xemples
in the terpene scries, see Falshaw С. P., Johnson A. W., King
T. J., J. Chem. Soc., 1963, 2422; Day A. C., ibid., 1964, 3001.
2. В artoil D. H. [?., Oefloriu A. M., Edwards О. E., J. Chem.
Soc., 1956, 530,
3. Miillcr E., Sc hick A., Mayer R,, Scheffler K,, Che.m.Ber., 93,
2649 (1960). Paper XIV on oxygen radicals,
4, Coppinger G. M., J. Am. Chem. Soc., 79, 501 (1957).
5. Kharasch M. S., Joshi B. S., J. Org, Chem,, 22, 1435 (1957),
6. В a г t 1 e t t P. D. ct al., J. Am, Chem. Soc., 82, 1756 (I960); 84, 2596 (1962).
7. Butenandl A., S c h i e d t U., Biekert E., Ann., 588, 106 (1954).
8, Harlcy-Maso n J., Jackson A. H., J. Chem. Soc,, 1954, 3651.
9, Taub D., Kuo C. IE, Slates H. L., Wendler N. L., Tetrahed-
ron, 19, I (1963); see also Taub D,, К и о С. Н., Wendler N. L., J. Org.
Chem., 28, 2752 (1963).
10. Schu 1 t e-FrohHn de D., E г h a r d t F., Ann., 671, 92 (1964).
11. Lohans H,, Ann., 516, 295 (1935).
12, McDonal d R. N., Campbell T, W., J. Org. Chem., 24, 1969 (1959).
13, Campbell T. W., McDonald R. N., Org. Syn. 40, 85 (I960).
14. F i e s с г L. F., H a d d a d i n M. J., J, Am. Chem. Soc., 86, 2392 (1964).
106
15. Noves W. A, et al., Am. Chem, J,, 5, 97 (1883); 7, 145, 149 (1885); 8, 167,
176/185 (1886).
16. Weissgerber R., Kr uber O., Ber., 52, 352 (1919).
17. R uzick a L. ct al., Helv. Chim. Acta, 9, 976 (1926); 14, 238 (1931).
18. Г1 p и л л E., M ак-Эль вен С., «Синтезы органических препаратов»,
ИЛ; М., 1949, сб. 2, стр. 333.
19. Perrine Т. D>, J. Org. Chem., 16, 1303 (1951).
КАЛИЯ ФТОРИД, KF. Мол. вес 58,10, т. пл. 860°, раствори-
мость в 100 г Н2О: 92,3 г при 18°, 150 г при 80°.
Катализатор декарбоксилирования. Эту соль можно применять
как щелочной катализатор* при циклизации адипиновой кислоты
в циклопентанои [1]. Лучший выход (81%) получен при высоком
молярном отношении кислоты к катализатору (20 : 1); в этом слу-
СН2СН2СО2Н кр 250—280° СН2— СН2.
| . ---------->- | )с=о + ссп-н3о
СН2СН2СО2Н 81% сн, — сн/
чае выход вполне сравним с выходом 75—80% при использовании
классической методики [2] с применением гидроокиси бария, взя-
той в аналогичном соотношении (15 : 1).
Катализатор конденсации. Рэнд 11, 31 и ЛеГофф [4] цитируют
более ранние японские работы о способности К. ф. катализировать
конденсацию по Киевенагелю. Рэнд и сотр. 13] обрабатывали экви-
молярные количества кетона и соединения с активной метиленовой
группой 0,5 моля К. ф. в этаноле или ДМФА. Они пришли к выводу,
что фториды цезия и рубидия более активны, чем К- ф., тогда как
фториды натрия и лития значительно менее активны.
ЛеГофф [4] обрабатывал смесь 1,2,3-трифенилциклопептадиепа
и 1,2,3-трифенилпропенона К. ф. в ДМСО и таким образом осущест-
CsHs
со
I
СС6Н?
СНСеН5
--------_у.
вил дегидратациоиную конденсацию по Михаэлю — Киевенагелю
и получил ярко-желтый дигидрогексафенилпентален (выход 53%;
под действием N-бромсукцинимида он был превращен в гексафенил-
пенталеи с выходом 77%). ЛеГофф предположил, что способность
F- действовать в качестве основания Льюиса объясняется тем, что
HFp образует очень прочную водородную связь:
^>CH3 + 2F~ —>^>CH-j-FHF"
* Впервые основные свойства К. ф, обнаружены Кнунянцем в 1947 г. па
примерах получения окиси этилена из этиленхлоргидрина и камфена из бор-
нилхлорида,.—Прим, ред.
107
Рэнд и Долииски [5] нашли, что KF действует как основание
в реакции Гофмана при применении ее к N-хлорбеизамиду (1). Обра-
зуется фенил изоцианат, который далее реагирует с исходным сое-
динением, как показано на схеме, и дает (3).
ОН 0 0
II I KF (1) II ।' H20
CeH5C—NCI (C(!H5N = C-01 CGHr,C—N—C—NCcH-,------------
। । 95% (оощнй)
Cl 1'1
(1) (2) (3)
О о
ii i;
С6ЫЬС— N —C— ncbh5
i I
H H
(0
Алкилфториды. Для получения н- гексил фтори да [6] в кругло-
донную колбу, снабженную мешалкой, капельной воронкой и не-
большим дефлегматором, помещают 2 моля безводного тонкоиз-
мельчениого К- ф. и 200 г этиленгликоля. Смесь нагревают на мас-
ляной баие до 160—170° и по каплям прибавляют в течение 5 час
1 моль н-гексилбромида. Отгоняющийся продукт реакции освобож-
дают от гексеиа-1 действием брома и перегоняют при 91—92°.
носи,,СП,он
CHsfCH^CH^r+KF-------—-------* СПДСН,ДСН2Р
4 U —
Фторирование ароматических соединений [7|. Финджер и Крузе
[8] показали, что атом хлора или брома в ароматическом ядре, ак-
тивированный о- или п-нитрогруппой, может подвергаться нуклео-
фильному замещению на ион фтора при взаимодействии с К- Ф- в та-
ких растворителях, как динитрил янтарной кислоты, ДМФА или
ДМСО. В неопубликованных работах фирмы «Олин Мэтьесои ке-
микал корпорейши» [8, 9] указано, что лучшие результаты можно
получить при использовании ДМСО в качестве растворителя и К- ф.,
предварительно хорошо измельченного (<..100 меси) и высушенного
в вакуумном сушильном шкафу при 100° минимум за 4 час до
применения. Обычно [91 смесь n-нитрохлорбензола, К- ф. и ДМСО
перемешивают при нагревании до 180° в течение 8—9 час, после ох-
лаждения смеси отфильтровывают неорганические соли и промы-
вают их свежим ДМСО (степень протекания реакции можно опреде-
CJ
I
I II +KF
1,1 моля
I
N03
1 моль
о-
!
CH;!S + CH3 (4 моля), 18 0°, 8—9 час
108
лить по количеству Ионов хлора в смеси солей). Продукт реакции
выделяют перегонкой с водяным паром. Аналогично получают о-
фторнитробензол с выходом 58% [10].
Наиболее высокие выходы получаются при молярном отношении
ДМСО к органическому галогениду, равном 4 : 1. По мере умень-
шения этого отношения как конверсия, так и выход падают. Кине-
тические исследования при оптимальных условиях показали, что
максимальные конверсия и выход достигаются через 8—9 час. Если
реакцию вести дольше указанного времени, арилгалогеиид реаги-
рует с растворителем и образуются побочные продукты. Оптималь-
ная температура для системы с ДМСО 175—180°. При температуре
кипения (189°) растворитель медленно разлагается и образуются
фрагменты, которые реагируют с арилгалогенидом; например, в
указанном выше примере фторирование при 189° дает н-нитрофенил-
метилсульфид.
1. Rand L., W a g п е г W.,War пег Р. О., К о v а с L. R., J, Org. Chem.,
27, 1034 (1962).
2. Т о р п е, Кон, «Синтезы органических препаратов», ИЛ, М., 1949, сб. I,
стр. 518.
3. R a n d L., S w i s h e г J. V., Cronin C. J., J. Org. Chem., 27, 3505
(1962).
4, LeGoIf E., J. Am. Chem. Soc., 84, 3975 (1962).
5. Rand L., Doi insk i Org. Chem., 30, 48 (1965).
6. Вогель А., Л ейсесте p Дж., Мейси У., «Синтезы органических
препаратов», ИЛ, М., 1958, сб. 8, стр. 68.
7. Contributed by S h i р к о w s k i Е. R., Olin Mathieson Chemical Corporation.
8. F i n g e г G.C., Kruse C. W-, J. Am. Chem. Soc., 78, 6034 (1956).
9. Boudakian M. M., Polak R.J., unpublished work at Olin Mathieson
Chemical Corporation.
10. Lapkin M., Ford M., Shipkowski E. R., unpublished work at Olin
Mathieson Chemical Corporation.
КАЛИЯ ХЛОРАТ, KC1O3. Мол. вес 122,56.
Бромирование. Применение К. х. для удаления НВг при бро-
мировании простых кетонов в водном растворе было использовано
в Германии в промышленном масштабе [1]. Джонс и сотр. [2] пока-
зали, что этот метод пригоден для получения чистого бромацетона
бСН3СОСН3-1-ЗВгг + КС1О3 6СН3СОСН2ВгЧ- КС1+ ЗН2О
из ацетона и метил-1-бромэтилкетона из метилэтилкетона. Анало-
гичным образом метил-«-пропил кетон дает-оба а-броыпроизводных.
Вода, hv
СН3СОСН2СН2СН3 + Вгг+КС1О3 -у—
J I Z А I 4 I <5 40_450//
СН3СОСНСН.2СН3 + ВгСН,СОСН2СН3СН3
I
Вг
1. N о г г i s J. Г., J. Ind. Eng. Chem., 11, 828 (19[9).
2. С a t с h J. R., Elliot D.F.,Hcy D. H., Jones E. R. H., J. Chem.
Soc,, 1948, 272; C a t c h J. R., Hey D. H., J о n e s E. R. H., W i 1-
son \V., ibid., 1948, 276.
109
КАЛЬЦИЙГЕКСАМИН, Ca(NH3)«. Мол. вес 136,23.
Этот твердый комплекс получают пропусканием аммиака в сус-
пензию металлического кальция в эфире [11. К- восстанавливает
двойные связи ароматических углеводородов:
Ca(NH3)64- \С —
Ca(NH2)2-|~ 4NHn4- )С~ С
Н Н
Полициклические ароматические углеводороды восстанавлива-
ются до производных с одним изолированным бензольным циклом,
например:
Превращение фенантрена в симм- и йсижж-октагидрофеиантрены
осуществляется, по-видимому, через промежуточные 1,2,3,4-тетра-
гидро- и 9,10-дигидропроизводные. Бензол восстанавливается до
циклогексадиена-1,4 (выход 30%).
К. восстанавливает ал кил бензил сульфиды до меркаптана и то-
луола [2].
1. Boer Н., D u i n k е г Р. М., Rec. trav., 77, 346 (1958).
2. Van Schooten J., Knotnerus J., В о e г H., Du inker P, M.,
Rec. traw., 77, 935 (1958).
КАЛЬЦИЙ — ЖИДКИЙ АММИАК- Восстановление 12а- или
120-ацетокси-11-кетостероидов до 11-кетонов можно осуществить,
используя кальций в жидком аммиаке [1]. Ни натрий, ни калий
АсО
при этом не дают удовлетворительных результатов и только с ба-
рием получаются результаты, сравнимые с кальцием. Свободный
кетол, соответствующий (1), образует главным образом 11а, 120-
110
диол. 170-Ацетокси-17а-ацетостероид (3) превращается при действии
кальция в жидком аммиаке в нормальный (170) 20-кетостероид
(5) 12].
(3) (4) (5)
I. С h а р ш а п J. Н., Е J k s J>} Р h i 1 1 i р р s G. Н., W у ш а п L. J., J.
Chem. Soc., 1956, 4344.
2. М j 1 I s J. S., R i n g о 1 d H. J., Djerass i C., J. Am. Chem. Soc.,
80, 6118 (1958).
КАЛЬЦИЯ ГИДРИД, CaH.,. Мол. вес 42,10.
Осушитель, К. г. необратимо реагирует с водой по следующему
уравнению:
СаН.;-|-2Н2О Са(ОН), + 2Нг
Так, 1 г К. г. количественно взаимодействует с 0,85 г воды с об-
разованием I л водорода. Эту реакцию можно использовать для
получения чистого, ио влажного водорода.
К- г-— прекрасный осушитель для углеводородов, простых
эфиров (включая диоксан и ТГФ), аминов (включая пиридин),
сложных эфиров и бутиловых спиртов (но не метанола и этанола,
которые образуют алкоголяты). К. г.— эффективный катализатор
конденсаций, поэтому его нельзя использовать для высушивания
альдегидов, реакционноспособных кетонов, кислот и хлорангидри-
дов. Хлориды и бромиды можно сушить К. г. только при умеренных
температурах.
По одному из способов сушки растворитель в течение несколь-
ких часов выдерживают над К- г., причем для защиты от влаги
воздуха и отвода водорода применяют диффузионную трубку (рис.
К-1), представляющую собой спиральный капилляр диаметром 1 —
2 мм 11]. Этот способ предложен Гроссе [21 для проведения реакций
под давлением с малыми количествами вещества. Реакционную
смесь загружают в прибор Гершберга [31 (рис. К-2), который затем
помещают в автоклав. После этого начинают очень медленно (во
избежание поломки капилляра) подавать в автоклав азот. Давление
азота должно значительно превышать давление, ожидаемое при
реакции. Газ под давлением диффундирует через капилляр настоль-
ко медленно, что вероятность проникновения летучего реагента
или растворителя в камеру автоклава очень мала.
Два метода ускоренного высушивания описаны в сб. «Синтезы
органических препаратов» [4]. Айрланд и Чайковский [41 сушили
111
mpem-бутанол кипячением над К. г. в течение ночи и затем перего-
няли. Однако кипячение mpem-бутанола с обратным холодильником
опасно, так как холодильник может забиваться затвердевающим
спиртом (т. пл. 24—25°). Тишлер и Замбито сушили mpem-бутанол,
перемешивая его с К. г. в течение нескольких часов, а'затем перего-
няли.
Рис. К-1. Диффузионная трубка. Рис. К-2. Прибор Гершберга.
Катализатор конденсации. p-Пропионилфенилгидразин цикли-
зуется в 3-метилоксиндол при нагревании со свежеизмельченным
К. г. [5].
+ СаН2
0, 2 моля
0,33 моля
170-215°
41-44%
1. Contributed by Newman iM. S. Ohio State University.
2. Grosse A. V., J. Am. Chem. Soc., 60, 212 (1938).
3. F i e s e г L. F.} «Experiments in Organic Chemistry», 3rd Ed., D. C. Heath,
1957, 249.
4. Айрланд P. Э., Чайковский M., «Синтезы органических препара-
тов», изд-во «Мир», М., 1964, сб. 12, стр. 14.
5. Е п d 1 е г A. S., Becker Е. J., Org. Syn., Coll. Vol., 4, 657 (1963).
112
КАЛЬЦИЯ КАРБОНАТ, СаСО3.
Дегидрогалогенирование. Тонкоизмельченный К. к. применяют
для дегидрогалогенирования стероидных сс-бромкетонов II, 2\
В качестве растворителя используют ДМФА [31.
Перегруппировка тозилатов. На основе этой реакции Мазур [4]
предложил изящную схему синтеза пергидроазулеиов и впервые
использовал эту реакцию для изучения стереохимии стероидов.
6-Тозилат холестандиола-5а,6а (1) количественно превращается
в А-гомо-В-нор-5-кетостероид (2) кипячением с тонкоизмельченным
И)
К- к. в ДМФА в течение 6—8 час или нагреванием с /лрет-бутилатом
калия в mpem-бутаноле при 100°. 4-Тозилат холестандиола-4<х,5а
(3) дает А-нор-В-гомо-5-кетохолестан (4). Конфигурация и сочле-
нение циклов были установлены при изучении дисперсии оптичес-
кого вращения: эффект Коттона положителен для соединения (2)
8
ИЗ
и отрицателен для соединения (4). Такеда и сотр. 15] применили
метод Мазура для синтеза 5-оксипергидроазуленона-8 (6).
+ СаСО;
4,1 г
(5) 15 г в 140 мл ДМФА
1. G г е с п G. F. Н., L о п g A. G., J. Chem. Soc., 1961, 2532.
2. D j e r a s s i C., Williams D. H., Berkoz B., J. Org. Chem., 27,
2205 (1962); Vorbriiggen H, Pakrashi S. C., Djerassi C.,
Ami., 688, 57 (1963).
3. Zder ic J. A., Carpio H., Bowers A., Djerassi C., Steroids,
1, 233 (1963).
4. M az u r Y., N n s s i m M., J. Am. Chem. Soc., 83, 3911 (1961).
5. Takeda K- et al., J. Chem. Soc., 1964, 3577.
КАЛЬЦИЯ СУЛЬФАТ БЕЗВОДНЫЙ (драйерит), CaSO4.
Этот осушитель, называемый растворимым ангидритом в отли-
чие от обладающего иными свойствами нерастворимого минерала
того же состава, получают по Хэммонду и Визроу 11] нагреванием
полугидрата или дигидрата при 230—250“ в течение 2—3 час. Ис-
пользованный реагент можно регенерировать тем же путем. При
сушке спиртов и других органических растворителей растворимый
ангидрит превращается в полугидрат (CaSOj ЛСЕСО) и поглощает
10—12 вес. % воды (считая на вес соли), причем не наблюдается
усадки гранул и их каналов.
К. с. б.— стабильное нейтральное инертное вещество, нераст-
воримое в органических жидкостях. При добавлении его к 95%-ному
спирту происходит заметное повышение температуры. Как осуши-
тель воздуха драйерит занимает промежуточное положение между
пятнокисью фосфора и конц. серной кислотой и уступает только
первой. Сравнение с другими осушителями жидкостей показы-
вает, что сульфаты магния и натрия действуют медленно и менее
эффективны, чем К- с. б.
Применяют также «индикаторные» формы драйерита, изменяю-
щие окраску с синей на красную, когда осушающее действие пол-
ностью исчерпано. Это достигается нанесением на поверхность
гранул сульфата кальция 3,5—5 % безводного хлорида кобальта.
Синяя соль кобальта индифферентна к апротонным растворителям,
нерастворимым в воде (бензол) или частично растворимым в воде
(диэтиловый эфир), но вымывается растворителями, смешивающи-
мися с водой, как протонными (этанол, уксусная кислота), так п
апротонными (ДМФА, ДМСО, триглим).
Одни из авторов этой книги [2] ранее предложил сушить эфир-
ные экстракты встряхиванием с насыщенным раствором хлористого
натрия и последующим фильтрованием через воронку, наполненную
114
безводным сульфатом натрия (см.); однако сейчас считают, что
К. с. б. имеет несомненное преимущество. В одном поучительном
эксперименте эфир насыщали водой при 25° и 100 мл эфира, содер-
жащего около 1,2 г воды, встряхивали в колбе с притертой стеклян-
ной пробкой с 10 а индикаторного драйерита (способен поглотить
1 — 1,2 г воды). Уже спустя 70 сек. синяя окраска осушителя почти
нацело изменилась на красную. Другую порцию (100 мл) насыщен-
ного водой эфира добавляли по каплям из делительной воронки
и фильтровали через слой (Юг) этого осушителя. При этом также
происходит практически полное изменение окраски (вначале все
же следует промывать раствором хлористого патрия).
1. Hammond W. Л., Withrow J. R., Ind. Eng. Chem., 25, 650,
1112 (1933).
2. Org. Expts., 53.
КАЛЬЦИЯ ХЛОРИД, CaCl,. Мол. вес 110,99, т. пл. 772°.
Бланшар 111 применил К. х. (12 мет) вместо обычно используе-
мых поташа и окиси кальция в качестве катализатора и дегидрати-
рующего агента при конденсации кетонов со вторичными аминами.
В этом случае енамины образуются, как правило, с более высоким
выходом. Этим способом легко получаются N.N-диметил- и N.N-ди-
этиленамины, синтез которых старым методом представлял большие
трудности.
О ВД,)8
83%
(превращение о2%)
2 моля 3,4 моля 400 мл 150 г
I. В 1 а п с h а г (1 Е. Р., J. Org. Chem., 28, 1397 (1963).
гМО-КАМФОРСУЛЬФОКИСЛОТА (I). Мол. вес 232,30, т. пл.
194° (с разл.), ап+24°.
Получение. Природную камфору сульфируют смешанным ан-
гидридом серной и уксусной кислот [1].
НЭС ?,СН3
WCHj CH2SO3H
+ Конц. HZSO4 + Дс2О ------------------------>
33(м(6молей) 117.WZ(12молей) 38-42Л
912 2 (6.молей)
К. применяют для расщепления рацематов оснований.
1. Bartlett Р. D., Knox L. Н., Org. Syn., 45, 12 (1965).
115
10-КАМФОРСУЛЬФОХЛОРИД. Мол. вес 250,74, т. пл. 67—68°
(d' ), т. пл. 83—84' (d, 1-).
Получение [1]. Смесь камфорсульфокислоты (2 моля) и РС15
(2 моля) охлаждают и выдерживают до разжижения, после чего пере-
мешивают сначала при охлаждении, а затем при комнатной темпера-
туре. Смесь обрабатывают льдом, промывают водой и получают К.
удовлетворительной чистоты с почти количественным выходом.
Применение. Оптически активный К. применяют для расщеп-
ления рацематов спиртов и аминов. Окислением неочищенного ра-
цемического К. (т. пл. 81—83°) получают (±)-кетопиновую кислоту
[2], которая интересна как пример устойчивой к декарбоксилиро-
ванию р-кетокислоты.
КМпО4
100г в 600дм Н.О
4-
100 г в 9 00МЛН2О
NazSO3; Н+
----£--1---->
38-43%
1. Bartlett Р. D., Knox L. Н., Org. Syn., 45, 14 (i960).
2- В a г t 1 e t t P. D., Knox L. H., Org. Syn„ 45, 55 (1965).
КАРБМЕТОКСИМЕТИЛЕНТРИФЕНИЛФОСФОРАН, (C6H5);JP =
=CHCOoCH3 (мол. вес 333,34, т. пл. 163°) и КАРБЭТОКСИМЕТИ-
ЛЕНТРЙФЕНИЛ ФОСФОРАН, (С6Н5)3Р=СНСОгС2Н5 (мол. вес
348,37, т. пл. 117°).
Эти реа’генты’ Виттига получают взаимодействием этил- или ме-
тилбром ацетата с трифенилфосфином и обработкой аддукта основа-
нием:
(СеН5)-,Р + он-
RO2CCH2Br----—> RO?CC[-l2P(C6HRMBr-)--> RO2CCH-P (С6Н5)3
Айслер [1] применил к арбэтокс и производное для синтеза диэтил-
норбиксина:
СНз СН3
Диэтилнорйиксин
116
Бестмапн [2] показал, что илид (1) — достаточно сильное осно-
вание, чтобы алкилироваться реакционноспособными галогенидами
(например, аллил бромидом, мети л бромацетатом, бензил бромидом)
с образованием продукта (2), который при омылении дает соответ-
ствующую карбоновую кислоту (3).
СН., = СНСНоВг у- СНСОХН-. — СН3 = СНСНо — СНСОХН., —>
li 1
Р(С6Н5)з + Р (С6Н5)3 (Вг~)
(1) (2)
он- +
----> СНг-СНСН2СН2СО2Н +0 —Р(СЙН,)3
(3)
Реакция илида (1) с а-бромкетоном приводит к а, 0-непредельно-
му у-кетоэфиру [31:
RCOCH .,Br-|-С11С0.,СН3 ----► RCOCH = CHCO.,CH3
|| GO—8 и0/,
p (Ctil -I5)3
В достаточно жестких условиях карбэтоксиметилентрифенил-
фосфоран реагирует с эпоксидами с образованием карбэтоксицик-
лопропанов [4|. Предпринимались безуспешные попытки проведе-
ния этой реакции с другими фосфоранами.
+ (сйн5)3р=снсо2с2н5 200> Н
46% со2с2н5
и
(С6Н5)3Р=СНСО2С2Н5 8 Р.и_?00'
63%
Эйтер [5] использовал реагент (метиловый эфир) в наиболее
удобном синтезе «вещества царицы», выделенного из маточного
молочка пчел.
(1)
НС CCH,Br; Zn
О = СН(СН2)3СН = СНСО2СН3........... --------
(2)
НС=- ССНХН(СН.>)3СН = СНСО2СН3 2H,s0<(Hg2>>
I “ би %
он
(3)
117
8 7 Нг, Pd
_> СН3СОСН=СН(СН2)3СН = СНСО2СН3 ———>
(•1)
Na.. С 03
— 7,8-Дигндропроизволное 40% [и ~
(S)
Н
СЫ3СО(СН.?)г,С=ССО3Н
н
«Вещество царицы»
Петрагнани и Шилл [61 применил реагент (этиловый эфир) для
синтеза непредельных жирных кислот, например:
1) С,ЩМкВг
2) RCHO
1) НС — ССН(ОС,НГ>), ------->
Н-Ь
RCHC--ССН (ОС,Н5)2 -* RCHCe=CCHO—►
ОН ОН
(С«НЬ)ЭР - снсо.с.н5
———---------— RCHC = ссн=СНСО2С2Н5
36-3 8%
ОН
0Н“; Н +
-....RCHC — ссн - снсо.н
он
о
C1C(CH,)8CO,C,HS
2) (СвН5)3Р = СНСО2С.2Н5----:----►
—► (CGH5)yP = ССО3С2Н5 Вакуум, 230°
О = С(СН2)8СОаС2Н5 -(С„Н5)3РО
_> С2Н5О2СС С(СН3)8СО3С2Н5
68% (общий)
1. 1 s 1 е г O.,Gulmann H.,Montavon М., R u е g g R., R у s е г G.,
Z e i 1 e г P-, Helv. Chim. Acta, 40, 1242 (1957).
2. Bestman n H. J., S chu Iz H., Chem. Ber95, 2921 (1962).
3. Bestmann H. J., Seng F., Schulz H., Chem. Ber., 96, 465 (1963).
4. D e п п у D. В., V i 1 1 J. J., Boskin M. J., J, Am. Chem. Soc., 84, 3944
(1962).
5. E i t e r K., Ann., 658, 91 (1962).
6, P e t г a g n a n i N., S c h i 1 I G., Chem. Ber., 97, 3293 (1964).
КАРБОБЕНЗОКСИХЛОРИД, см. Хлоругольной кислоты бен-
зиловый эфир.
я-КАРБОКСИФЕНИЛСЕМИКАРБАЗИД. Мол. вес 195,17, т. пл.
218—220°.
118
к. получают конденсацией п-аминобензойной кислоты с нптро-
мочевиной и кипячением образующейся и- карбоксифенил мочевины
с гидразином в метаноле [II.
О О
II I'
NH, HN—CNH-, HNCNHNHa
I " о I ’ |
II /X
I [1 HzNCNHNO., I II H;NNH, Г и
I II 8i—8и%~* I || 34—51"%* | II
'Ч/
I I I
CO2H CO2H CO.2H
К. применяют для расщепления асимметрических карбонильных
соединений: конденсация К. с карбонильным соединением дает
семикарбазон, за счет карбоксильной группы которого образуются
диастереомерные соли с оптически активными основаниями. К.,
например, использовали для расщепления d,l-3-мети л цикл о гекса-
нона.
1. Shillington J. К.,
Hill Т., Jr., R ams ay
Denning G. S., Greenou gh W. B., HI,
О. B., J. Am. Chem. Soc., 80, 6551 (1958).
N, N -КАРБОНИЛДИИМИДАЗОЛ, n=\ 9 /=rN
Мол. вес 162,15, т. пл. 116—118°. I n~c—n I
К. получают из фосгена и имидазола (в со-
отношении I : 4) в ТГФ 11]. Опубликован обзор методов приме-
нения К. 12].
К. реагирует с карбоновыми кислотами с образованием имида-
золидов (1) [31, восстановлением которых получают альдегиды (2) [4].
В условиях катализа основаниями К- применяют для синтеза слож-
ных эфиров (3) [5]; с первичными аминами К- дает производные (4),
которые разлагаются при комнатной температуре на имидазол и
изоцианат (5) 16].
119
Поль и Андерсон 1.7] применили К- в пептидном синтезе (см. схе-
му) и указали, что при этом наблюдается слабая рацемизация, од-
нако по данным Вейганда [81 происходит интенсивная рацемизация.
Поль [9] показал, что в синтезе валилтирозина метод смешанных
ангидридов дает лучшие выходы и более чистые продукты.
R
i
CbNHCHCO2H
R'
I
H2NCHCO2C2HS
ROHR’
I 11 I 1
CbNHCHC - N - CH CO2 C2H5
К. использован также для превращения карбоновых кислот
в mpem-бутилоБые эфиры соответствующих надкислот [10] с выхо-
дом от 64 до 80%. N,N'-Тио ни л ди имид азол реагирует аналогично,
но с меньшим выходом.
Штааб и Датта [11] нашли, что даже очень неустойчивые хлор-
ангидриды можно получить с хорошим выходом, пропуская хло-
ристый водород в холодный раствор имидазолида (6) в хлороформе
или хлористом метилене. Эта реакция per se обратима, так как хлор-
ангидриды с имидазолом дают имидазолиды; однако в данном слу-
чае имидазол связывается хлористым водородом и образующийся
120
хлор гидр ат имидазола выпадает в осадок, что способствует завер-
шению реакции.
(6)
НС1
т
(7)
о
п
Reel
НС1
(8)
Неизвестный ранее хлорангидрид муравьиной кислоты НСОС1
получен впервые обработкой хлороформного раствора 1-формил-
имидазола газообразным НО при —60°. После добавления к смеси
метанола выделяют метилформиат с выходом 65-—70%. Если мета-
нол добавлять при —40°, то выход метилформиата снижается до 9%.
1. Staab Н. A., Wen del К., Chem. Вег., 93, 2910 (I960).
2. S t a a b Н. A., Angew. Chem., Internal. Ed., 1, 351 (1962).
3. Staab 1-1. A., Liik jng M., D й г г F. H, Chem. Ber., 95, 1275 (1962).
4. S t a a b H. А., В r a un 1 i ng H., Ann., 654, 119 (1962).
5. Staab H. A., Mannschreck A., Chem. Ber., 95, 1284 (1962).
6. S t a a b H. A., Benz W., Ann., 648, 72 (1961).
7. P a u 1 R., Anderson G. W., J. Am. Chem. Soc., 82, 4596 (I960);
J. Org. Chem., 27, 2094 (1962).
8. Weygand F. W., Prox A., Schmidhammer L., К о n 1 g W.,
Angew. Chem., Internal. Ed., 2, 183 (1963).
9. Paul R., J. Org. Chem., 28, 236 (1963).
10. H e c h t R., R u c h a r d t C., Chem. Ber., 96, 1281 (1963).
11. S t a a b H. A., D a t t a A. P., Angew. Chem., Internal. Ed., 3, 132 (1964).
N,N -КАРБОНИЛДИ-СЯММ-ТРИАЗОЛ, __________________ о ______
(структура точно не известна), i q | |
К. получают реакцией сш-ш-триазола с
фосгеном в ТГФ в присутствии пиридина, свя-
зывающего НО, с выходом 80% в виде белого порошка (степень
чистоты 95%) [1J.
Пептидные синтезы (ср. N.N’-Карбонилдиимидазол). Раце-
мизация если и наблюдается, то очень незначительная. В качестве
растворителя можно использовать ДМФА — хороший растворитель
пептидов с различным размером молекулы.
1. Beyer m an Н. С., Van Den Brink W. M., Rec. traw., 80, 1372
(1961).
КАРБОН ИЛФТОРИД, COF2. Мол. вес 60,01, токсичный газ,
т. кип. —81°.
121
К. получают из окиси углерода и фторида серебра(П) [11 или
при добавлении жидкого фосгена к суспензии фторида натрия в
ацетонитриле [2].
Реакция с кетонами и амидами. В присутствии следов ДМФА
или пиридина К. присоединяется к карбонильной группе цикло-
гексанона с образованием 1-фторциклогексилфторформиата (2) [21,
О
О F O-C-FF F
II \/ \/
А СОГ,<ДМФЛ) / \ EF,; f > +са
I 04% \ 5 4% | [
(1) (2) (3)
который можно перегнать. Это соединение при действии эфирата
трехфтористого бора расщепляется па 1,1-днфторциклогексан (3)
и углекислый газ. При взаимодействии К. с ДА1ФА (4) при 25° про-
межуточный аддукт выделить не удается и продуктом реакции яв-
ляется а, а-дифтор три мети л амин (5).
О
II COF,
(CHa):,NC----(CI-LHNCF., АССХ
| 82% |
Н И
(4) 5)
Присоединение к перфторолефинам и азаолефииам [2, 3]. С пер-
фторолефинами К. дает простые продукты присоединения, например
(6) (7):
COF
cf,cf=cf2 cf3cf-cf3
'(б) 80% (7)
Присоединение проводят с избытком К. в ацетонитриле при 100—
150° в присутствии каталитических количеств фторида цезия.
COF
CF3N-CF., — -A CF.NCFs
(8) 5С% (9)
Реакция с фенолами и спиртами. К. реагирует с фенолами и спир-
тами (автоклав, 100—200°) с образованием фторформиатов (10).
Под действием тетрафторида серы (150—200е) фторформиат дает
арил(алкил)трифторметиловый эфир [4]:
О
COF. II
Ar(R)OH -----» Ar(R)OCF —Д Ar(R>OCF3
HF (M)J
1. Farlow M. W., Man E. H., Tullock C. \V., Inorg. Syn., 6, 155
(I960).
122
2. Fawcett F. S., Tul lock C. W., Coffman D. D., J. Am. Chem.
Soc., 84, 4275 (1962).
3. S in i t h R. D., Fawcett F. S., Coffman D. D., J. Am, Chem.
Soc., 84, 4285 (1962).
4. Sheppard W. A., J. Org. Chem., 29, 1 (1964); Aldrich P, E., Shep-
pard W. A., J. Org. Chem., 29, Il (1964).
КАРБЭТОКСИМЕТИЛЕНТРИФЕНИЛФОСФОРАН, (CeH3)8P=
=CHCO2C2H5. Мол. вес 348,37, т. пл. 117°,
См. Карбметокси метил ентр и фен и л фосфор ан.
КАРБЭТОКСИМЕТИЛТРИФЕНИЛФОСФОНИЙБРОМИД,
C2H5O2CCHaP(C(iH5)3Br. Мол. вес 429,29.
К — предшественник соответствующего реагента Виттига.
N-КАРБЭТОКСИФТАЛИМИД,
Мол. вес 219,19, т. пл. 80°.
К. получают реакцией этилового эфира
хлоруголыюй кислоты с фталимидом калия
или с фталимидом и триэтил амином в ДМФА
Ц]. К- взаимодействует с аминокислотами в водном растворе карбо-
ната натрия с образованием оптически чистых фталоильных про-
изводных (4) с выходом 85—96%.
(1)
R
I
H.NCHCOtH
(2)
.NHCOjEt
N—CHRCOaH
N—СНСОзН + HjNCOjEt
(3)
(4)
I. Nefkens G. H. L., Nature, 185, 309 (1960); N e f k e n s G. H. L.,
T e s s с г G. I., N i v a r d R. J. G., Rec. trav., 79, 688 (1960); В о i s-
sonnas R. A., Adv. Org. Chem,, 3, 181 (1963).
КАРО КИСЛОТА (мопонадсерная кислота) I II,
О
!i
~O~S + —ООН
он
123
К- к. получают по мере необходимости перемешиванием 10 г
тонкоизмельченкого персульфата калия (мол. вес 270,33) и 7 мл
конц. серной кислоты, охлажденной льдом; в образовавшуюся го-
могенную смесь добавляют 40—50 г льда [21.
Применение, а) Окисление ариламинов до нитрозосоединений
[1, 3]. б) Окисление кетонов по Байеру — Виллигеру до лактонов
(1) 12, 4] или сложных эфиров (3) [5]. Если присутствует аромати-
ческое кольцо, расщепление происходит по связи с этим кольцом
(2) [61:
Окислительное расщепление кетонов осуществляют в лигроине
при 50—65° [4] кислотой Каро, полученной, как указано выше,
либо в растворе, содержащем больше серной кислоты, меньше воды
и не содержащем органического растворителя [21, либо кипячением
кетона со смесью персульфата калия (4 г) и конц. серной кислоты
(1 мл) в 150 мл 90%-ной уксусной кислоты [71, либо, наконец, с су-
хим реагентом Байера — Виллигера [2|, полученным тщательным
измельчением в ступке 10 г персульфата калия с 6 мл конц. серной
кислоты и 30 г сульфата калия до превращения смеси в сухой поро-
шок. Порошок прибавляют к холодному раствору кетона в уксус-
ной кислоте и смесь выдерживают при комнатной температуре в
течение.7—10 дней.
Аналогичный реагент для окислительного расщепления кетонов
можно получить при осторожном смешивании уксусного ангидрида
(65 а), конц. серной кислоты (30 г) и пергидроля (25 г) [51. Если
бензофенон (20 г) выдерживать с этой смесью в течение нескольких
дней при 0°, то он почти количественно превращается в фенилбен-
зоат. .
Окислительное расщепление А^-З-кетостероидов [8, 9] иллюстри-
руется реакцией пропионата тестостерона (1). 9 г персульфата калия
и Юг конц. серной кислоты смешивали в ступке, разбавляли 150 мл
уксусной кислоты, полученную смесь добавляли к раствору 9 г
пропионата тестостерона в 150 jo уксусной кислоты и выдерживали
при комнатной температуре в течение 7 дней при периодическом
встряхивании (Петтит и Кастури [9]). Обработка смеси, включаю-
124
шдя омыление и повторное подкисление, дает 2 г лактона 3-оксо-
170-окси-4-окса-5а-андростана (4). Возможная последовательность
реакций показана на следующей схеме:
1. Саго Н., Angew. Chem., 11, 845 (1893).
2. В а с у е г А., V i 1 1 i g е г V., Вег., 32, 3625 (1899); 33, 858 (1900); Л а н г-
л и В., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3, стр. 211.
(См. Белов В. И, Хейфиц Л. А., Вирезуб С. И., РЕМИОС,
Госхимиздат, М., 1961, том 10, стр. 7.— Прим. перев.)
3. В a m b е г g е г Е., Hiibner R., Вег., 36, 3803 (1903); Borel Е.,
Deuel Л., Helv. Chim. Acta, 36, 805 (1953) (З-нитро-4-аминотолуол —З-цит-
ро-4-нитрозотолуол, выход 92%).
4. Ruzicka L., S t о 1 1 M., Helv. Chim. Acta, 11, 1159 (1928).
5. Marker R. E. el al., J. Am. Chem. Soc., 62, 525, 650, 2543, 2621 (1940).
6. Schroef er G., герм. пат. 562827 (1928) [C. Z., 104, 127 (1933)].
7. R о 1 1 e t t A., Bfatke K., Monatsh., 43, 685 (1922).
8. Sal amon A., Z. physiol, chem., 272, 61 (1942).
9. Pell it G. 1%, Kasturi T. R., J. Org. Chem., 26, 4557 (1961).
КЕТЕН, CH2=C=O. Мол. вес 42,04; т. кип. —48". К. высо-
котоксичен.
Получение. Старый метод получения К. сводился к перегонке
ацетона через трубку для сожжения, наполненную битым пористым
фарфором, и нагреванию при 695—705° газом или в электрической
печи для сожжения; выход К. 25—29% [1].
СН3
сн,со сн8=с-0+сн,
Позднее Уильямс и Херд [2] описали усовершенствованный
генератор («кетеновая лампа»), который позволяет увеличить вы-
ход до 80—90% и получить 0,45 моля К- в час. На рис. К-3 показан
125
генератор Уильямса — Херда, усовершенствованный Гершбергом.
Пары ацетона, контактируя с раскаленной проволочной сеткой,
разлагаются на К. и метан; неразложившийся ацетон конден-
сируется и стекает обратно в испаритель, а газообразный К- вместе
с метаном пропускают непосредственно в жидкий реагент или его
Лента
накаливания
Вольфрамовые
Р и с. К-3. Кетеновая лампа.
раствор либо конденсируют в ловушке, охлаждаемой сухим льдом.
В генераторе другого типа проволочная спираль для пиролиза по-
гружена непосредственно в ацетон в колбе на 1 л, охлаждаемой па
водяной бане [31. Другой метод получения К. состоит в пиролизе
уксусного ангидрида при 500—510° [4| в приборе, в котором обеспе-
ОСОСН3
CH;i—С = О 500~~'-1Х СН, = С=0 + СН3С0011
чивается быстрое отделение К. от побочно образующейся уксусной
126
кислоты и, таким образом, предупреждается рекомбинация. Пре-
имущество этого метода — отсутствие газообразных побочных про-
дуктов. Опубликованы обзоры промышленных методов получения
К. 151.
Самый лучший и надежный метод получения К. в лабораторных
масштабах — пиролиз в кетеновой лампе дикетена 161, который
в настоящее время является доступным промышленным продуктом.
СН, —С = О
I ' I ----------> 2СН,=С--О
СНг=--С — О ‘16-з,'%
Реакции. К. довольно устойчив при температуре —80°; при 0°
он быстро димеризуется. Реакции с К. обычно проводят, пропуская
газообразный К- в жидкий реагент или в раствор реагента. Напри-
мер, при получении «-капронового ангидрида |7| в охлаждаемую
льдом газопромывную склянку помещают 1 моль капроновой кис-
лоты и пропускают 0,5—0,55 моля газообразного К., получаемого
из ацетона.
СнДСН.рС-О
СП3 (ClP).t СО. 41 -Р С.Н, - С = О->- '>0 -I- сн3 соон
80~S7% СНДСН,).1С = О
Образующуюся смесь медленно фракционируют для полного
превращения первоначально образующегося смешанного ангидри-
да; низкокипящие фракции содержат ацетон, кетен, уксусную кис-
лоту и уксусный ангидрид.
К. легко и с высоким выходом ацилирует первичные амины.
н О
I II
C0H5NH, 4 СН, С = о —> CfiH-N -С-СЩ
К. медленно реагирует с водой и поэтому плохо ею поглощается.
Реакция катализируется кислотами, и разбавленная уксусная кис-
лота легко поглощает К. Гораздо легче ацетилируется перекись
водорода с образованием перекиси ацетила 181.
О О
II II
2СНг = С—О~|- IIOOH —> СН3С—ОО—ССН3
С mpem-бутанолом К- взаимодействует лишь в присутствии
сильной кислоты [5]. Смесь 74 г тдт-бутанола и 0,5 мл конц. сер-
ной кислоты обрабатывают К. при 60° в течение 1,5 час.
H..SO,
(СН3)зСОИ4 СНо = С-0 — (СН.ЩСОСОСШ
60%
Благодаря медленному взаимодействию К- с водой в нейтральной
или щелочной среде Бергманну и Штерну |91 удалось осуществить
N-ацетилирование аминокислот действием К- на щелочной раствор
аминокислоты при комнатной температуре. Джексон и Кэхилл
127
[10] изучили ацетилирование ь-триптофана и показали, что если
при обработке К. поддерживать щелочную (по фенолфталеину)
реакцию раствора, то рацемизации не происходит. Если же среда
такова, что в растворе возможно образование уксусной кислоты,
то рацемизация осуществляется в пределах часа.
сн2=с=о
н2о '
СН2^НСО2Н
NHCOCH3
Цистеин легко реагирует с К. в слабощелочной среде с образо-
ванием N,S- диацетил производного [11]:
СН ,—СН — СО2Н СН 2—снсо2 н
| I сьц=с=о I I
SH NH.--------------> CH3COS NHCOCH3
Кюпг [12] обнаружил, что при соблюдении определенных усло-
вий К- гладко присоединяет формальдегид с образованием 0-про-
пиолактона. Получение этого ценного для многих синтезов высоко
реакционноспособного продукта можно осуществить в промышлен-
ном масштабе [13]. Заугг [14] обобщил данные по образованию 0-
лактонов при взаимодействии К. с кетонами в присутствии катали-
заторов.
СН., = С=О 7пГ1 СН,—С—-О
'+ -ЦСД j - I
СН, = О СН,— о
6-Оксикумаранон-З с уксусным ангидридом дает смесь О-аце-
тата (2) и енолацетата (3); использование же К- позволяет осущест-
вить избирательное О-ацетилирование [15].
(з)
Рид и Мепглер [16] обнаружили, что при взаимодействии К- с
дназокетонами при комнатной температуре (2 дня в толуоле) обра-
зуются 0,у -буте нол иды (1), гидролизующиеся до у-кето кислот (2),
которые можно восстановить (по Вольфу — Кижнеру) в соответ-
ствующие кислоты (3).
RC — CTI = N = N
й +
O=C=CHZ
°C ?снг
й
(1)
rcchzch,cozh
(1 г г г
О
(2)
------> вснгсн2снгсогн
(3J
128
Такая последовательность реакций позволяет удлинить боко-
вую цепь кислоты на три атома углерода, однако существенным ог-
раничением этого метода являются низкие выходы на первой стадии
(30—50%).
Раствор чистого циклопропанона можно получить добавлением
холодного (—78°) раствора диазометана в хлористом метилене к
раствору избытка К. в хлористом метилене и отгонкой избытка К.
в вакууме при —78° [17].
CH2=C=O + CH2N2
СН2С12, -78°
50-60%
^>=0
I. Hurd С. D., Org. Syn., Coll. Vol., 1, 330 (1941).
2. Williams J. W. , H u г d C. D., J. Org. Chem., 5, 122 (1940).
3. Hamalainen C., Reid J. D., Ind. Eng. Chem., 41, 1018 (1949).
4. Fischer G. J., MacLean A. F., Schnizer A. W., J. Org.
Chem., 18, 1055 (1953).
5. Lacey R. N-, Adv. Org. Chem., 2, 213 (i960); Q u a b e c k G., Newer
Methods of Preparative Org. Chem., 2, 133 (1963).
6. В 0 e s e A. B., Jr., пат. США 2218066 [С. A., 35, 1072 (1941)]; Andrea-
des S., Carlson H. D., Org. Syn. Chem., 45, 50 (1965).
7. Will iams J. W., Krynitsky J. A., Org. Syn., Coll. Vol., 3,
164 (1955).
8. d’Ans J., Frey W-, Ber., 45, 1845 (1912).
9. Bergmann M., Stern F., Ber., 63, 437 (1930).
10. J a c k s о n R. W., C a h i 11 W. M., J., Biol. Chem. 126, 37 (1938).
II. Neuberger A., Biochem. J., 32, 1452 (1938).
12, Kiing F. E., пат. США 2356459 [С. A., 39, 88 (1945)].
13. Gresham T. L., J ansen J. E. et al., J. Am. Chem. Soc., 70, 998, 999,
1001, 1003, 1004 (1948).
14. Zaugge H. E., Org. Reactions, 8, 305 (1954).
15. Logemann W., Cavagna G., Tosolini G., Chem. Ber., 96,
1680 (1963).
16. Ried W., Men g ler H-, Ann., 678, 113 (1964).
17. T u г г 0 N. J., Hammond W. B., J. Am. Chem. Soc., 88, 3673 (1966).
COCH2CO2H
a-KETO ГЛУТАРОВАЯ КИСЛОТА, (
СН2СН3СО3Н.
Мол. вес 148,11, т. пл. ПО—111°.
К. к. получают сложноэфирной конденсацией диэтилсукцината
и диэтилоксалата в присутствии этилата калия с последующим гид-
ролизом [1].
Реагент используют для обменной реакции расщепления 2,4-
динитрофенилгидразонов кетонов, перегоняющихся с водяным па-
ром [2].
I. Friedman L., Kosower Е., Org. Syn., Coll. Vol,, 3, 510 (1955).
2. FI a г r i s о n H. R., E isenbr aun E. J., J. Org. Chem., 31, 1294 (I960).
КИЛИАНИ РЕАГЕНТ, см. Хромовая кислота.
КИПЯТИЛЬНЫЕ КАМЕШКИ («кипятильники»).
5 Заказ № 1096 129
В качестве кипятильников лучше всего применять легко доступ-
ные карборундовые камешки, поскольку они не содержат пыли, од-
нородны и легко заметны среди продуктов реакции. Для обеспече-
ния равномерного кипения достаточно одного камешка величиной
с зерно. Для упаривания фильтрованного раствора до кристалли-
зации лучше применять блестящие сине-черные алундовые камеш-
ки, так как они хорошо заметны среди обычных кристаллических
веществ (белых, желтых и др,); белые кипятильники для этой цели
неудобны.
КИСЛОРОДА ДИФТОРИД, OR. Мол. вес 54,00, т. пл. —223,8°,
т. кип. —144,8°.
К- д. получают пропусканием фтора в 2%-пый раствор едкого
натра Ц]. В выделяющемся газе содержатся приблизительно равные
количества OF« и О2. Для улавливания OFa из тока фтора Меррит
и Руфф [2] конденсировали газ при температуре жидкого кислорода,
отгоняли кислород и фтор и получали продукт (степень чистоты
приблизительно 99%), который хранится при комнатной темпера-
туре в больших стеклянных баллонах. Меррнт и Руфф показали,
что К- Д. гладко окисляет первичные алифатические амины до нит-
розопроизводных при —78° в фреоне II в качестве растворителя.
3RNH2-ROF2—> RN = 0 + 2RNH3F
Эти же исследователи сообщили о присоединении К- д. к ацетиленам
и к некоторым олефинам при —78° в фреоне 11 [3]. Например, тет-
раметилэтилен с умеренным выходом дает фторгидрии (2); необхо-
димый при этом дополнительный протон, по-видимому, отщепля-
ется от молекулы растворителя. При кратковременном воздействии
влажного воздуха фторгидрии перегруппировывается в пинаколин
(3) с выделением фтористого водорода.
(СН3)2С^С(СН3)3 (СН3)ВС-С(СН3),_=
(1) ^-70% | j
ОН F
(2)
(CH3)3CCCH3-PHF
il
О
(3)
1,1- Дифепилэтилен дает с К. Д. <о-фтор ацетофенон (4). Ацетиле-
ны взаимодействуют с К- Д. с образованием а, ct-ди фтор кетонов (5).
О
of, !1
(CgHalaC^CH,___CcH5CCH2F (4)
RC CR RCOCF.R (5)
1. Йо ст Д. М., «Неорганические синтезы», ИЛ, М., 1951, сб. 1, стр. 107.
2. Мег г i 1 t R. F., R u f f J. K., J. Am. Chem. Soc., 86, 1392 (1964).
3. Merrill R. F., Ruff J. K., J. Org. Chem., 30, 328 (19G5).
130
КЛАЙЗЕНА ЩЕЛОЧЬ [1]. Получение: растворяют 35 а едкого
кали в 25 мл воды и добавляют при охлаждении 100 мл метанола.
Экстракция криптофенолов и енолов. Фенолы, нерастворимые
в водной щелочи, например 2,4,6-триал л илфеиол [11, экстрагируют-
ся из петролейного эфира щелочью Клайзена. Витамин Ki легко
выделяется из 3—5%-ного концентрата люцерны следующим путем.
Спиртовую суспензию масла встряхивают с водным гидросульфитом
натрия, экстрагируют петролейным эфиром и из этого экстракта
извлекают гидрохинон витамина Кд обработкой К. щ. Желтый
ОН СН3 Сн3 сн3 сн3
Витамин Щ, гидрохинон
ОН
экстракт разбавляют водой и гидрохинон витамина Ki (бесцветный)
выделяют экстракцией эфиром и кристаллизацией из петролейного
эфира; окисление окисью серебра в эфире дает чистый витамин Ki
[21.
Исчерпывающее окисление холестерина би хром атом п удаление
сильно кислой фракции дает смесь Д^-холестендиона-ЗД ие?коль-
ких монокетонов и других нейтральных продуктов [3]. Раствор в
петролейцом эфире экстрагируют щелочью Клайзена до тех пор,
пока экстракт не окрасится в желтый цвет, обусловленный образо-
ванием енолята. Таким образом можно легко выделить ендион
через его енолят с выходом 40%.
Омыление. Тарбелл и сотр. 14] омыляли этиловый эфир 3,5-ди-
хлор-4-оксибензойной кислоты нагреванием с К- щ. в течение
1 час на кипящей водяной бане (93—97%); 2-нитро-4-метоксиацет-
анилид гидролизуется аналогично при нагревании в течение 15 мин
[51.
I. Claisen L., Ann., 418, 96 (1919).
2. F i е s е г L. I7., J. Am. Chem. Soc., 61, 3467 (1939).
3. F 1 C s c 1- L, F., Org. Syn., Coll. Vol., 4, 189 (1963).
4. Тарбелл Д., Вильсон Дж., Файта П., «Синтезы органических
препаратов», ИЛ, М., 1963, сб. 4, стр. 236.
5. Фанта I],, Тарбелл Д., «Синтезы органических препаратов», ИЛ,
1952, сб. 3, стр. 358.
131-
КОБАЛЬТА (II) АЦЕТАТ-БРОМИД, СоОАсВг.
К. а. б. получают in situ из ацетата кобальта и бромистого водо-
рода. К. а. б.— высокоактивный катализатор автоокисления акти-
вированной метиленовой или метильной группы. Например [1], в
колбу, снабженную мешалкой, трубкой для ввода газа и холодиль-
ником, помещают реагенты, указанные на следующей схеме:
СН3
Л/Вг
I II q-Со (ОАс)2+ НВг Н-АсОН
л У 0,1 моля 0,1 моля 500 мл
30% в АсОН
91%
90—112°
О3
СО2Н
0,5 моля
При пропускании кислорода температура поднимается до 90°,
при слабом нагревании смесь кипятят в течение 2 час. Другие
примеры:
О ОО
С6Н5СОСНаСН2С6Н& С6НаСОСН2ССбН5
71%
Главный 10—15% 0,5-3%
продукт
1. Н а у A. S., В I a n с h а г d Н. S., Сап. J. Chem., 43, 1306 (1965).
КОБАЛЬТА ГИДРОКАРБОНИЛ, НСо(СО)4.
К. г. получают реакцией диспропорционирования дикобальт-
октакарбонила и ДМФА в толуоле или гексане с последующим
подкислением [1].
ЗСо2(СО)8Ч-12 ДМФА —> 2Со(ДМФА)е[Со(СО)4]2 + 8СО
Со (ДМФА)е [Со(СОД]гД2НС1_^2НСо(СО)4-|-6 ДМФАЧ-СоС12
К. г. считается истинным катализатором гидроформилирования
олефинов (оксо-синтез) [2,3]; см, Дикобальтоктакарбонил.
I. Kirch L., О г с h i и М., J. Am. Chem. Soc., 80, 4428 (1958).
2. Wen der I., Sternberg H. W., О r c h i n M., J. Am. Chem. Soc.,
75, 3041 (1953).
3. Heck R. F., Br eslow D. S., J. Am. Chem. Soc., 83, 4023 (1961).
КОБАЛЬТА (II) ХЛОРИД, СоС1й. Мол. вес 129,85.
К. х. получают дегидратацией СоС12-6Н.2О в токе хлористого
водорода и сушат в вакууме при 100—150° [1].
132
Каталитическое количество К- х. оказывает влияние на направ-
ление реакции Гриньяра, так как промежуточно образующееся
RCoCl активирует конденсацию с органическими галогенидами
(Караш [1, 2]):
СоС1г
1 [l])C6H5MgBr + C6H5Br—-> С6Н5-С0Н5
СоС12
2 [3]) C6H5MgBr Д-С6Н5С = СВг———► C6H5feCC6H5
75%
1. К h а г a s с h М. S., Fields Е. К., J. Am. Chem. Soc., 63, 2316 (1941),
and others papers.
2. Reviewed by Hey D. H., Ann. Reports, Chem. Soc., 41, 195 (1944); 45, 160
(1948).
3. В 1 a с k H. K-, HornD. H. S., W e e d о n В. C. L., J. Chem. Soc.,
1954, 1704.
cumm-КОЛЛИДИН (2,4,6-триметилпиридин). Мол. вес 121,18,
т. кип. 171°. Перегоняют перед использованием.
Для дегидрогалогенирования вместо пиридина лучше применять
это основание. Рекомендуется добавлять 4% 3,5-лутидина (синтезы
каротиноидных полиенов реакцией при 140° в атмосфере азота) [1].
1. Inhoffen Н. Н., Krause Н. -J., В о г k S., Ann., 585, 132 (1954).
КРЕМНИЯ ТЕТРАИЗОЦИАНАТ, Si(N=C=O)4. Мол. вес 196,16,
т. пл. 26°, т. кип. 185,6°.
К- т. получают взаимодействием цианата серебра с четыреххло-
ристым кремнием с выходом 85% [1].
К. т. в бензольном растворе экзотермически реагирует с первич-
ными и вторичными аминами с образованием соответствующих
производных мочевины с выходом 97—100% [1].
О
К
4RR'NH + S1 (NCO)4 -+ 4RR'NCNH2 + SiO.,4- 2H.2O
Аналогичным образом тетраизотиоцианат кремния дает произ-
водные тиомочевины. Этот способ значительно лучше старой мето-
дики с использованием водных растворов циановой или тиоциано-
вой кислоты.
1. N е v i 1 1 е R. G., McGee J. J., Can. J. Chem., 41, 2123 (1963).
ЛАУРИ Л ПОЛ ИЭТИЛЕНГЛИКОЛЬ. Поверхностноактивпос веще-
ство используется для озонирования циклоолефинов до двухоснов-
ных кислот в водной щелочной перекиси водорода [1].
1. Fremery М. I., F i е 1 d s Е. К-, J. Org. Chem., 28, 2537 (1963).
ЛЕВУЛИНОВАЯ КИСЛОТА, СН3СОСН,СН2СО,Н. Мол. вес
116,11, т. пл. 33—35°.
Получение. Толленс [Г1 обнаружил, что Л. к. можно получить
действием разбавленных минеральных кислот на углеводы: саха-
розу [1], крахмал [2], глюкозу [3], фурфурол [4] и 4-оксиметил-
фурфурол 14]. По методике, приведенной в работе [5], раствор 500 а
сахарозы в 1 л воды обрабатывают 250 мл конц. соляной кислоты и
нагревают на кипящей водяной бане в течение 24 час. Образующий-
ся в результате обугливания черный осадок отфильтровывают и
фильтрат упаривают досуха. Экстракцией эфиром (6—7 час) и пе-
регонкой получают вещество, почти полностью затвердевающее
при 30°. Выход составляет 72-—76 г или 21—22% в расчете на ис-
пользование обоих компонентов дисахарида. Промежуточные про-
изводные фурана типа (3) и (4), очевидно, отщепляют формальдегид,
под действием которого восстанавливаются. Согласно статье, поя-
вившейся после опубликования приведенной методики, глюкоза
как исходное вещество лучше сахарозы, поскольку дает меньше
продуктов обугливания и обеспечивает несколько более высокий
выход [3].
Сахароза —>
( НОСН — СНОН СН-СН
II К Р
НОСН..СН СНСНО ,сч . с
" [ [ НОН.,с/ \z х сно
он он ' и
(1) (3)
НОСН —СНОН СН — СН
i i [[ и
НОСН,СН CCFLOH—/С /Сч
| [1 НОН2С/ Мснаон
он о и
(2) (4)
СН—СН
(5)
СНа —СН..
I I
НО.,С СОСН34-НСО.,Н
(6>
Г34
Расщепление 2,4-динитрофенилгидразонов 161 и оксимов [7].
Кини [6] разработал полумикроколориметрический метод определе-
ния 2,4-динитрофенилгидразонов, в котором Л. к. служит акцеп-
тором 2,4-динитрофенил гидразина и регенерирует кетон при об-
менной реакции с его производным. Он нашел, что гидразоны иесо-
пряженпых кетонов расщепляются гораздо легче, чем гидразоны
сопряженных или ароматических кетонов .
Де Пюи 171 встретился с трудностями при гидролизе ди оксима
(I) (димер оксима циклопе нтадиенон а) кислотой, а также при гид-
ролизе кислотой в сочетании с формальдегидом, так как соединения
(1), (2) и (3) исключительно чувствительны к действию сильных
кислот. Используя в качестве акцептора Л. к., он нашел, что если
перемешивать при комнатной температуре соединение (1) с Л. к.,
ft) (*) (3)
к которой добавлено 10 об. % 1 н. соляной кислоты, то диоксим
растворится в течение 3 час, и из раствора почти с количественным
выходом выделяется кетоксим (2). Кетоксим (2) гидролизуется в
ди кетон (3) нагреванием с той же смесью левулиновой и соляной
кислот в течение 3 час на кипящей водяной бане. Те же условия в
общем пригодны и для гидролиза насыщенных оксимов (25°) и со-
пряженных оксимов (90°). Мейнвальди сотр. [81 превращали нитро-
зохлориды ряда норборнана в хлоркетоны (выход 79—85%) по
методике Де Пюи — Пондера.
По усовершенствованной Мэттоксом и Кендаллом [91 методике
выделения ацетата кортизона из С3-2,4-динитрофенил гидразон а
используется смесь пировиноградной кислоты, уксусной кислоты,
хлороформа и бромистого водорода; необходимое соединение полу-
чается с выходом 80% . Для сравнения Де Пюи [7] приготовил смесь
1 г Д4-холестен-3-он-2,4-динитрофенилгидразоиа со 100 мл хлоро-
форма и 100 мл обычной смеси левулиновой и соляной кислот и ки-
пятил ее в течение 3 час. Хроматография дала с высоким выходом
А4-холестеион-3.
135
1. Grote A., von, Tollens B., Ann., 175, 181 (1875); 206, 226 (1880).
2. Rischbieth P., Ber., 20, 1773 (1887).
3. Sah P. P. T., Ma S.-Y., J. Am. Chem. Soc., 52, 4881 (1930).
4. T e u n i s s e n H. P., Rec. trav., 50, 1 (1931).
5. McKenzie B. F., Org. Syn., Coll. Vol., 1, 335 (1941).
6. Keeney M., Anal. Chem., 29, 1489 (1957).
7. D e P u у C. FI., Ponder B. W., J. Am. Chem. Soc., 81, 4629 (1959).
8. ’Meinwal d J., Meinwald Y. С., В а к e r T. N., Ill, J. Am. Chem.
Soc., 86, 4074 (1964).
9. Mattox V. R., Kendall E. C., J. Am. Chem. Soc., 70, 882 (1948);
J. Biol. Chem., 188, 287 (1951).
ЛЕМЬЕ — ДЖОНСОНА ОКИСЛЕНИЕ, см. Перйодаты, окис-
ление смесью перйодат — тетраокись осмия.
ЛИНДЛАРА КАТАЛИЗАТОР, Pd — СаСО3 — РЬО. Линдлар
и Дюбуи [1] детально описали методику Линдлара [2], немного
усовершенствовав ее. Раствор хлорида палладия в разбавленной
соляной кислоте нейтрализуют до pH 4—4,5 и добавляют к суспен-
зии осажденного карбоната кальция. Суспензию перемешивают и
нагревают на кипящей водяной бане до осаждения красно-корич-
невого окисла. К перемешиваемой теплой суспензии добавляют
НС1 Н2О НСО2Н
PdCl3 H2PdCl2------► PdO —> Pd
н2о
РЬ(ОАс)2--РЬО
раствор формиата натрия; через 40 мин восстановление заканчива-
ется и выпадает черный осадок, который собирают, промывают на
воронке Бюхнера и частично дезактивируют шламмом 7,7%-ного
водного ацетата свинца (80°, перемешивание в течение 45 мин).
Отравленный катализатор промывают и высушивают при 60—70°.
Выход катализатора в расчете на 1,48 г хлорида палладия составля-
ет 19,5 г.
Л. к. чрезвычайно эффективен для селективного гидрирования
тройных связей До ^цс-двойных связей. Линдлар и Дюбуи привели
пример гидрирования фенил ацетилен а в стирол. В колбу для гид-
рирования вводят 2,04 г (0,02 моля) фенил ацетилена, 0,10 г Л. к.,
1,0 мл хинолина и 150 мл гексана. Воздух из системы удаляют
трехкратным откачиванием и промыванием водородом. Гидрируют
при небольшом избыточном давлении с перемешиванием на трясуч-
ке или магнитной мешалкой. Из более поздних работ приведем
лишь некоторые [3]. В присутствии Л. к. кумулены частично гид-
рируются в цис-полиены [4].
CgHs. zCeH5 н pd-Pb
>С=С = С = С = С=С< ’ Л
с6н/ Чн5
Нх
СаН5 /С=С\ /С6Н5
>с = с< /с=с<
СеН/ \н Н 'С6Н6
136
Крам и Аллинджер [5] использовали вместо Л. к. палладий на
сульфате бария, отравленный синтетическим хинолином (хинолин,
полученный из каменноугольной смолы, непригоден). Например,
при гидрировании диметилового эфира 5-деци нди к ар боновой кис-
лоты (1) в метаноле слабо экзотермическая реакция завершается
через 20 мин с поглощением точно 1 моля водорода; при этом
образуется 97% диметилового эфира ^ttc-5-децендикарбоновой кис-
лоты (2). Шнейдер [6] считает такую замену Л. к. удобной, однако
СН3ОЙС(СН2)3С С (СН2)3 СО2СН3 + 5%Pd-BaSO4ф Н2 —
(1) 19,2 г в 100 мл СН3ОН 0,4 г
СН3ОаС (СН2К /(СНа)3СО2С1-13
)с=с<
н/ \н
(2)
предлагает еще более эффективный катализатор —5% палладия
на сульфате бария и пиридин в качестве растворителя. Такой ката-
лизатор восстанавливает ацетилены в цис-олефины и резко обры-
вает реакцию на стадии образования олефина.
1. Lindlar Н., Dub uis R., procedure submitted to Organic Syntheses.
2. Lindlar H., Helv. Chim. Acta, 35, 446 (1952).
3. Arnol d R. T., Smol insky G., J. Am. Chem. Soc., 82, 4918 (1960);
Riiegg R., G 1 о о r U-, Langemann A., ler M., von Plan-
t a C., R у s e г G., Isler O., Helv. Chim. Acta, 43, 1745 (1960); Surma-
tis J. D., Ofner A., J. Org. Chem., 26, 1171 (1961); Os b on d J. M.,
Philpott P. G., Wickens J. C., J. Chem. Soc., 1961, 2779.
4. Kuhn R., Fischer H., Chim. Ber., 93, 2285 (1960).
5. С r a m D. J., A 1 1 i n g e r N. L., J. Am. Chem. Soc., 78, 2518 (1956).
6. S c h n e i d e r W. P., private communication.
ЛИТИЙ. Ат. вес 6,94, т. пл. 180,5°, т. кип. 1332°, уд. вес 0,53420°;
растворимость: 10,9 г/100 г NH3 при —33,2°; 36,5 а/л CH3NH2 при
—22,8°.
Техника эксперимента. Большинство методик, приведенных
в литературе, было разработано в то время, когда Л. выпускался
лишь в виде больших брусков или слитков, покрытых слоем пара-
фина. Такой кусок металла расплющивают молотком в тонкую
фольгу, а затем отмывают ее эфиром от парафина и нарезают нож-
ницами на тонкие полоски непосредственно над горлом реакционной
колбы с эфиром [1, 2]. Гершберг [3] вытягивал литиевую проволоку
с помощью пресса для натрия, снабженного фильерой с трехмил-
лиметровым отверстием. Бартлетт и Леффертс [4] получали мелкую
дробь плавлением Л. в минеральном масле, добавляли несколько
капель олеиновой кислоты для предотвращения слипания и смесь
энергично встряхивали в течение нескольких секунд. Смесь охлаж-
дали, фильтровали через стеклянную вату, промывали эфиром и
серебристо-серые частицы диаметром около 1 мм переносили в реак-
ционную колбу. Бергсталер [5] рекомендует осторожно добавлять
лигроин к еще горячей суспензии в минеральном масле, прежде
137
чем она успеет охладиться и образовать гель (что обусловлено при-
сутствием олеиновой кислоты). Дробь в чистом виде поднимается
на поверхность к горлу колбы и легко извлекается шпателем, име-
ющим форму ложки.
Выпускаемую в настоящее время литиевую ленту очищают от
защитного слоя масла и помещают в петролейный эфир 16!. Нож-
ницами нарезают ее на кусочки и после отдувания растворителя
сухим аргоном помещают в реакционную колбу.
В настоящее время Л., содержащий обычно около 0,02% натрия,
выпускается* в нескольких видах. Проволоку, как это рекомендо-
вано Гершбергом [31, выпускают диаметром около 3 мм в катушках
весом 100, 400 и 800 г. Гершберг рекомендует обращаться с такой
проволокой следующим образом. Длину проволоки следует брать
из расчета, что 1 см сухой проволоки весит 0,0423 г (12 см весят
0,508 г). Покрытую маслом проволоку разрезают на соответствую-
щее число отрезков длиной 12 см и помещают их в трубку, снаб-
женную внизу краном и вводом для аргона через резиновую пробку
сверху. Литиевую проволоку отмывают от вазелина последователь-
но порциями бензола и эфира в атмосфере аргона и промывную
жидкость выдавливают аргоном через кран. Перед внесением Л.
реакционную колбу продувают аргоном, выводя газ через самое ши-
рокое горло колбы. Трубку с Л. также продувают аргоном все
время, пока она открыта, укрепляя ее под прямым углом к горлу
колбы. Проволоку вытягивают пинцетом и разрезают ножницами
на небольшие кусочки, которые надают в колбу. Таким образом
обеспечивается защита свежеобнаженной серебристой поверхности.
Площадь поверхности 3 мм проволоки составляет 23 см2 fa.
Литиевая дробь имеет размер частиц от 4 до 16 меш. Частицы Л.,
диспергированные в минеральном масле, вазелине или парафине,
имеют диаметр 15 мк и площадь поверхности 7,3-103 см2 fa. Ниже
приведены составы смесей (вес. %). Дисперсию в парафине (т. пл.
около 52°) выпускают в виде стержней диаметром 2,5 см и длиной
Li Минеральное масло Вазелин Олеиновая кислота Парафин
30 69 1
30 61 8 1 —
30 — 70 —
30 '— — 2 68
20 смл в таком стержне содержится 22,7 г металлического Л. Стерж-
ни запаяны в полиэтиленовые трубки и упакованы по 20 стержней
в банке. Дисперсия в парафине исключительно устойчива при хра-
нении, с ней легко и надежно работать на воздухе. Необходимое
* Американская литиевая корпорация (Lithium Corporation of America).
138
количество металла легко получить, отрезав и взвесив рассчитан-
ное количество стержня. Из него получают сухой литиевый порошок,
вымывая парафиновую матрицу пентаном или гексаном в закрытом
сосуде в атмосфере сухого аргона с последующим удалением раст-
ворителя и высушиванием в токе аргона. Сухой литиевый порошок
чрезвычайно чувствителен к действию воздуха, влаги и азота.
Дисперсия в минеральном масле разделяется при хранении, но
ее однородность можно восстановить перемешиванием (частицы не
слипаются). Дисперсию извлекают из контейнера ложкой и взве-
шивают.
Противопожарные средства. Загоревшийся Л. засыпают без-
водным хлористым литием.
Алкил- и ариллитиевые производные, см. также и-Бутиллитий;
Литий-натриевый сплав; Метиллитий; Фениллитий.
Алкил- и ариллитиевые производные получают с высокими
выходами обработкой галогенида 2 же Л. в сухом эфире в основном
по методике, используемой при получении реактива Гриньяра [7, 8].
Л. реагирует легче и с большей скоростью, чем магний.
RBr4-2Li RLi-pLiBr
В отличие от магния Л, всплывает на поверхность эфира и его
нельзя нагревать в токе азота, так как при этом образуется нитрид.
Алкиллитиевые производные получают не только в эфире, но и в
низкокипящем петролейном эфире [9]; углеводородный раствори-
тель позволяет получать литийорганические соединения (например,
изопропиллитий), трудно образующиеся в других условиях.
Литиевые производные несколько более реакционноспособны,
чем соответствующие реагенты Гриньяра [10], и в реакции с а, р-
ненасыщенными кетонами в отличие от магнийорганических гало-
генидов дают более высокое содержание продукта 1,2-присоеди-
нения [11]. Через литиевые производные удается вовлечь в синтез
хлор ароматические соединения, что не выполнимо методом Гринь-
яра. Алкил- и ариллитиевые производные легко реагируют с окисью
этилена без нагревания, которое необходимо при взаимодействии
с реактивами Гриньяра. Для предотвращения образования продук-
тов типа ROCH 2СН 2(ОСН 2СН 2)Л.ОН на 1 моль R (Ar) Li следует
расходовать точно 1 моль окиси этилена.
Не все органические галогениды удовлетворительно реагируют
с Л.; часто необходимое соединение Л. можно получить косвенно
обменной реакцией с легко доступным соединением Л., например
с w-бутиллитием:
я-СДГЛ + RX —RLi-pH-C4HBX
Реакция обмена, открытая в 1938 г. Гилманом [12] и Виттитом [13],
нашла широкое применение [14]. Получение н-бутиллития (а также
фениллития, хотя он и менее реакционноспособен), реакцию обмена
и реакцию с карбонильным соединением можно с успехом провести
139
в одной и той же колбе, причем для образования промежуточного
соединения RLi требуется всего 30—60 мин. Атом Л. можно замес-
тить на остаток MgBr, что иногда является наиболее удобным ме-
тодом получения реактива Гриньяра [15].
Выход алкил- и ариллития можно определить методом кислот-
ного титрования, используемого для анализа реактивов Гриньяра,
или добавлением бензофенона к аликвотной порции и взвешиванием
образующегося третичного спирта [16]. Длительное время приме-
нялся метод двойного титрования Гилмана и Хаубейна [17]. Для
определения алкиллития в углеводородном растворе простым и точ-
ным является оке иди метр и чес кий метод с использованием V2O5 [18].
Однако ни один из двух последних методов нельзя применить для
определения ариллития.
При разработке метода синтеза плазмалогенов типа (1) Крэйг
и сотр. [19] нашли, что дегидробромирование а-бромацеталя гли-
церина (2) с образованием алкениловых эфиров (3) и (4) лучше про-
водить под действием Л. в 1,2-диметоксиэтане, чем под действием
натрия или магния.
н н 1 1 снаоч | >CHCHR ы СН„ОСН = CHR 1 сн2он
ch2oc=cr СНО/ 1 снон 4- CHOCH=CHR
1 1 Вг [ 1
CHOCOR' снаон сн2он СН2ОН
1 /ОН СНЁОР< |ПОСН2СНаКНг (2) (3) (4)
О
(1)
Циклизация. Для осуществления синтеза интересной трицикло-
13,2, 1,03,е1-октановой системы (1) Соуерс и др. [20] использовали
в качестве исходного вещества аддукт Дильс а-Альдер а (2), легко
получаемый из аллилового спирта и циклопентадиена. Ненасыщен-
ный спирт превращали в 6-хлорметил-3-оксатрицикло-[3,2,1,02’4]-
октан (3). Для циклизации (3) в трициклический спирт (4) Л. в ТГФ
более эффективен, чем натрий в толуоле. Спирт (4) имеет относитель-
но напряженное циклобутановое кольцо, но окислением СгО3 в пи-
ридине и восстановлением по Вольфу — Кижнеру его легко прев-
ратить в желаемый углеводород.
140
CH;
CH
I.
CHjOH
CHZOH
(2)
(3)
1. Woodward R. B., Kornf eld E. C., Org. Syn., Coll, Vol., 3, 413
(1955),
2. Heaney H., Millar I. T., Org. Syn., 40, 105 (1960).
3. Fieser L. F., Hershberg E. B., J. Am. Chem. Soc., 59, 396 (1937).
4. Bartlett P. D., Lefferts E. B., J. Am. Chem. Soc., 77, 2804 (1955).
5. Burgstahler A. W., private communication.
6. Gutsche C. D., Peter H. H., Org. Syn., Coll. Vol., 4, 887 (1963).
7. Ziegler K-, С о 1 о n i u s H., Ann., 479, 135 (1930).
8. G i 1 m a n H. et al., J. Am. Chem. Soc., 54, 1957 (1932).
9. Gilman H., Langham W., Moore F. W., J. Am. Chem. Soc., 62,
2327 (1940).
10. Gilman H., Kirby R. H., J. Am. Chem. Soc., 55, 1265 (1933).
11. Luttringhaus A., Ber., 67, 1602 (1934).
12. G i 1 m a n H., J а с о b у A. L., J. Org., 3, 108 (1938).
13. W i t t i g G., P о с к e 1 s U., D r 6 g e H., Ber., 71, 1903 (1938).
14. J о n e s R. G., G i 1 m a n H., Org. Reactions, 6, 339 (1951).
15. Gilman H., Arntzen С. E., J. Am. Chem. Soc., 72, 3823 (1950).
16. G i 1 m a n H, Zoellner E. A., S e 1 b у W. M., J. Am. Chem. Soc.,
54, 1957 (1932).
17. Gilm an H., H aube in A. H., J. Am. Chem. Soc., 66, 1515 (1944).
18. С о 1 1 i n s P. F., Kamienski C. W-, Esmay D. L., Ellestad
R. B., Anal. Chem., 33, 468 (1961).
19. C r a i g J. C., Hamon D. P. G., Brewer H. W., H a r 1 e H., J.
Org., 30, 907 (1965).
20. S a u e r s R. R., Parent R. A., D anile S. B., J. Am. Chem. Soc., 88,
2257 (1966).
ЛИТИЙ-АЛКИЛАМИННОЕ ВОССТАНОВЛЕНИЕ.
Li -f-
f Метиламин
I Диметиламин
J Этиламин
'l Изопропиламин
н-Пропиламин
k Морфолин
кип., °C Уд. вес
—6,5 0,699
7,4 0,680
16,6 0,689
34 0,694
48,7 0,719
128 -—
Бенкесер [1] показал, что комбинация лития с низкомолекулярным
алкиламином является энергичным восстановителем для аромати-
ческих углеводородов и некоторых других соединений. Наиболее
часто используется этил амин. Например, при восстановлении на-
фталина литием и этиламином с высоким выходом образуется смесь
А9’10- и А1,9-окталинов. Поданным Бергсталера [2], на другом при-
мере реакция тормозится при использовании неперегнанного этил-
амина из баллона, возможно, из-за того, что следы соединений
141
железа, попадающие из баллона, катализируют образование амида
за счет восстановления. Обычно используют промытую эфиром и
нарезанную мелкими кусочками литиевую проволоку, хотя Бергста-
лер рекомендует мелкую литиевую дробь, полученную по Бартлетту
(см. Литий).
Восстанавливаемое вещество растворяют в достаточно большом
избытке перегнанного безводного этиламина в колбе, снабженной
конденсатором дьюаровского типа, охлаждаемым сухим льдом.
Добавляют рассчитанное количество лития и смесь перемешивают
с помощью магнитной мешалки. Появляющееся сначала ярко-синее
окрашивание исчезает по окончании восстановления. В отсутствие
субстрата литий медленно растворяется в кипящем этиламине без
выделения водорода с образованием ярко-синего раствора, который,
очевидно, содержит Li+, связанный в комплекс с отрицательно за-
ряженным растворителем. Эту реакцию, хотя она и необратима,
обычно проводят в присутствии субстрата, который реагирует с
сипим комплексом почти со скоростью его образования, возможно,
путем 1,4-присоединения лития. Восстановление нафталина иллю-
стрируется следующей гипотетической схемой:
1,4-Дилиткйаддукт (2) реагирует с этиламином с образованием
Д3-диалина (3), который частично претерпевает второе 1,4-присое-
динение к бензольному кольцу с образованием после обмена металла
с растворителем изотетралина (4). Возможность того, что этот угле-
водород является промежуточным соединением, подтверждается
тем, что Гроб [3] при восстановлении нафталина натрием в смеси
142
жидкий аммиак — эфир — абсолютный этанол при —70° получил
(4) с выходом 82%. При более высокой температуре в реакции с
Li—EtNH2 вероятна катализируемая основанием изомеризация
изотетралина в сопряженный триен (5). Последний путем 1,6-при-
соединения (6) и последующего 1,4-присоединения превращается
в А9’10-окталин (7)—основной конечный продукт. Побочный
окталин (11) образуется, по-видимому, в результате катализируе-
мой основанием изомеризации А2-ди ал и на (3) в сопряженный AL-
диалин (8), последующего 1,8-присоединения (9), 1,6-присоедине-
ния (10) и 1,4-присоедипепия (11).
Обработка толуола в метиламине 4 же лития дает смесь метил-
циклогексеиов, содержащую, по данным газовой хроматографии,
59% 1-метилциклогсксена (V) и 41 % 3- и 4-метил циклогексено в 14].
I
II их
В этом случае также предполагается 1,4-присоедииепие, изо-
меризация несопряженных диенов в сопряженные и последующее
1,4-присоединение. Бенкесер и сотр. 15] нашли, что реакции более
селективны при замене значительной части метиламина или этила-
мина на диметиламин, н- или изопропиламин либо морфолин. До-
бавление этих аминов способствует образованию термодинамически
более устойчивого продукта восстановления. Например, при вос-
становлении толуола 4 же лития в смеси 10 об. % метиламина и 90%
диметиламина образуется смесь 82% 1-метилциклогексена (V) и
18% 3- и 4-метил цикло гексенов. Добавление более высококипящего
амина позволяет также проводить реакцию в менее летучей системе
растворителей с высокой растворяющей способностью. Интересно,
однако, что в чистом диметиламине или морфолине восстановление
не происходит, для этого необходимо небольшое количество метил-
нли этиламина. Восстановление можно провести и в чистом «-про-
пиламиие, но при кипячении (49°); в этом растворителе наряду с
присоединением металла наблюдается конкурентная реакция ме-
талла с растворителем с образованием водорода и амида.
В улучшенной методике синтеза А9’10-окт ал и на [61 предусмат-
ривается использование второго амина. Смесь 0,2 моля нафталина
и по 250 мл этиламина и диметиламина помещают в колбу, снаб-
женную ловушкой с сухим льдом. 1,65 г-атом литиевой проволоки
нарезают на кусочки длиной 0,5 слг, добавляют их к смеси и переме-
шивают ее 14 час магнитной мешалкой. Смесь растворителей отго-
няют, а серовато-белый остаток (содержащий избыток лития) осто-
рожно разлагают 100 мл воды (при охлаждении). Выпавший осадок
143
отделяют и фильтрат экстрагируют эфиром; перегонка дает 19—20 г
углеводорода, содержащего, поданным газовой хроматографии, 80%
А9,10-окталина и 20% А1,9-окталина. Основной продукт выделяют
в чистом виде с помощью бас-(1,2-диметилпропил-1)-борана, который
избирательно присоединяется к менее пространственно затруднен-
ному Д1,3-изомеру. Окисление продукта перекисью водорода для
превращения аддукта в легко отделяемый спирт и последующая
перегонка дает АВ 9’10-окталин 99%-ной чистоты с выходом 50—54%
(в расчете на нафталин).
Еще до того как стал доступен метод газовой хроматографии,
Бенкесер [1] наблюдал, что при температуре кипения этилами на
(16,6°) этилбензол восстанавливается избытком лития до этилцик-
логексена и этилциклогексана, тогда как при —78° единственным
продуктом является 1-этилциклогексен. В более поздней работе
14], уже цитировавшейся выше, показано, что восстановление то-
луола в метиламине 4 экв лития дает 59% 1-метилциклогексена (V)
и 37% смеси 3-метилциклогексена (VI) и 4-метилциклогексена
(VII). Возможные пути образования этих изомеров показаны ниже.
VII
В присутствии соответствующего количества лития эти изомеры
чрезвычайно легко восстанавливаются до метилциклогексана, оче-
видно, за счет 1,2-присоединения лития и переметаллирования на
амин. В тех же условиях 1-метилциклогексен (V) восстанавливается
гораздо медленнее [например, лишь на 4% за 3 час (Li-J-CH3NH3)],
чем 4-метилциклогексен (59%). Индуктивный эффект метильной
группы, очевидно, затрудняет захват электронов двойной связью.
Ацетиленовые связи восстанавливаются легче, причем только
путем транс-присоединения [7]. Например, 5-децин (0,1 моля)
в этиламине при — 78° с прекрасным выходом восстанавливается
литием (0,25 моля) в транс-5-децен, а 3-октин дает транс-3-ок-
тен (52%). Однако реакция при температуре кипения этиламина
(16,6°) дает значительные количества алканов, особенно в при-
144
сутствии избытка лития. Эта реакция отличается от восстановле-
ния алкинов натрием в жидком аммиаке, которое останавливается
на стадии образования олефинов, и сходна с поведением производ-
н
Li —СгН5ЫН2 |
СН3(СН3)3С С(СН2)3СН3“——------СН3(СН2)3С = С(СН2)3СН3
/о |
н
ных циклогексена, в которых неконцевая дизамещенпая двойная
связь очень легко восстанавливается системой Li—RNHa.
По данным Беикесера и сотр. [8], восстановление ароматических
нитросоединений реагентом полностью останавливается на стадии
образования ароматических аминов, хотя ариламины могут быть
восстановлены избытком лития в этил амине до производных цикло-
гексена. Этот неожиданный результат объясняется следующим
образом. При восстановлении нитрогрупп образуются алкиламнн-
ные ионы, и по мере увеличения их концентрации между ариламин-
ными и алкиламинными ионами устанавливается равновесие:
ArNOo L1~EtNI^ ArNH3 + EtNH(Li+)
4-------------------—
ArNH(Li+)+ EtNH2
Так как ал кил а минный ион — гораздо более сильное основание,
чем ариламинный, равновесие смещается в сторону образования
последнего. Отрицательно заряженные ар ил аминные ионы не склон-
ны принимать электроны, поэтому восстановление заканчивается
на данной стадии. С другой стороны, если исходным веществом яв-
ляется ариламин, то вначале, когда в растворе практически нет
н.
(а) (б)
ар ил аминных ионов, быстро восстанавливается кольцо и с достаточ-
но хорошим выходом образуется продукт восстановления. Напри-
мер, n-толуидин восстанавливается литием и этил а ми ном до транс-
1-метнл-4-аминоциклогексана с 50 %-ным выходом за 6 час. Реакция
не останавливается на стадии 1-аминоциклогексена (а), как в слу-
чае алкилбензола, поскольку соединение (а) является енамином и
145
находится в равновесии с имином (б), который легко восстанавли-
вается. Данные по восстановлению N-метиланилина и N.N-диме-
тиланилина подтверждают этот механизм [4]. Следует отметить,
что при восстановлении /г-толуидина образуется продукт с эквато-
риальным положением обоих заместителей. Восстановление как
о-, так и лг-толуидинов также дает наиболее стабильные производ-
ные циклогексана с обоими заместителями в экваториальном поло-
жении. Таким образом, эту реакцию можно использовать для сте-
реоспецифического синтеза производных циклогексана.
Для синтеза абиетиновой кислоты (3) из дегидроабиетиновой
кислоты (1) Бергсталер н Уорден [2] вначале безуспешно пытались
восстановить ароматическое кольцо натрием или литием и этано-
лом в жидком аммиаке в обычных условиях по Берчу (и даже в при-
сутствии эфира в качестве второго растворителя). Затем они обра-
тились к более сильной восстанавливающей системе, разработанной
Бенкесером. Под действием лития и этиламина восстанавливается
не только ароматическое кольцо, но и карбоксильная группа до
альдегидной (наблюдалось восстановление и других кислот в аль-
дегиды, но в общем с низкими выходами [9]). Однако в присутствии
очень слабой протонной кислоты типа mpem-амилового спирта или
трет-бутанола осуществляется селективное восстановление дсгпд-
роабиетиновой кислоты в А8,1--абиетадиеновую кислоту (2) с выхо-
дом 90%. Из образующейся в результате кислотной изомеризации
смеси абиетиновую кислоту (3) выделяют в виде соли диизоамила-
мина. Спирты, более кислые, чем два вышеуказанных, например
этанол или изопропанол, почти полностью тормозили восстановле-
ние. 300 мг (1л1моль) дегидроабиетиновой кислоты и 4,3 ли (40щмо-
лей) mpem-амилового спирта помещали в колбу, снабженную ло-
вушкой, охлаждаемой сухим льдом, и высокоскоростной мешалкой
(медленное перемешивание приводит к неполному восстановлению).
Смесь обрабатывали безводным этиламином (30 ли), 240 мг (35 Me-
tt том) тон ко из мельчен ной литиевой дроби (Бартлетт, см. Литий).
Через 15 мин литий растворяется и раствор приобретает бледно-
голубую окраску, затем добавляют вторую порцию литня весом
240 мг. При появлении характерной ярко-синей окраски раствор
обесцвечивают трет-амиловым спиртом (~5 мл) и обрабатывают.
146
Хенбест [10] показал, что смесь литий — этил амин является
эффективным восстановителем простых и сложных аллиловых эфи-
ров в олефины. Например, раствор 0,4 а бензоата Д4-холестенола-Зр
в 20 мл этиламина при реакции с 0,1 а лития дает 0,224 а чистого
Д4-холестена. Для восстановления полученного из Зр-ацетокси-Д7-
ланостена 7а,8а-эпоксида до 7а-ола Фрид и сотр. [11] добавляли в
качестве источника протонов mpe/n-бутанол. Одним из преимуществ
этого реагента перед системой Li—NH3 является большая раство-
римость многих органических соединений в этиламине по сравне-
нию с жидким аммиаком. Для восстановления эпоксидов стероидов
в аксиальные спирты такой реагент более эффективен и более спе-
цифичен, чем алюмогидрид лития [12]. Так, 7а,8а- и 9а,11а-эпок-
сиды не восстанавливаются гидридом металла, но реагируют с ли-
тий— этилампном с образованием 8а- и 9а-спиртов:
Если молекула содержит гидроксильную группу, как в случае
а-окиси холестерина (1), то восстановление литием — этиламином
дает как аксиальный спирт (2), так и непредельный холестерин (3)
[ 13]. При реакции с (3-эпоксидом получают только исходное вещест-
во и олефин (3).
147
Это можно объяснить тем, что анион гидроксильной группы
тормозит образование анионного промежуточного соединения, из
которого получается аксиальный спирт.
Хенбест показал, что смесь литий—этиламнн эффективна также
при десульфуризации этилентиокеталей [14]. Аддукт Дильса —
Альдера бензохинона с 2 молями бутадиена, для которого Хенбест
предложил конфигурацию (4), превращали в бис-кеталь (5), обра-
ботка которого литием и этиламином при —20" приводила к десуль-
фуризации и восстановлению двух изолированных двойных связей
с образованием пергндроантрацена (6) с высоким выходом. Этот
эффективный и стереоспецифический метод был использован для
восстановления дионов циклогексанового ряда [15].
(4)
Трюс [16] показал, что литий в этиламине лучше, чем натрий в
жидком аммиаке, расщепляет сульфид до меркаптана и углеводо-
рода и сульфон до сульфиновой кислоты и углеводорода.
RSR .. LL~EtNHi> RSLi + RLi RSH + RH
Например:
(.4-C10H31)2S —-—-—-—> /i-C]0H21SH-[-/z-C10H22
86% 85%
h-C10H21SCsH ; -L1 C6H5 SH 4- w-C10H22 + «-C20H42
87% 71%
(*-C10H21).2SO3 H-C10H21SO2Li~|-«-C10H22
82%
Обнаружив, что постепенное введение лития в реакционную
смесь благоприятствует реакции, Трюс сконструировал прибор,
в котором кипящий этиламин конденсируется и стекает через
трубку, содержащую рассчитанное количество нарезанной малень-
кими кусочками литиевой проволоки (рис. Л-1). Трубка представ-
ляет собой обычную пробирку с впаянной в дно стеклянной трубкой
диаметром 6 мм.
Хорошо известно, что обработка N.N-ди алии л амида алюмогид-
ридом лития приводит к восстановительному расщеплению с обра-
148
Рис. Л-1. Прибор для медленного ввода
лития.
/ — конденсатор с сухим льдом; 2 —- стеклянная
трубка; Й — ввод CH3NH2; 4 — литиевая прово-
лока; 5 — стеклянная вата; 6 — магнит; 7 — маг-
нитная мешалка.
зованием альдегида и вторичного амина 117]. По данным Патчор-
ника [18], пептидная связь N-пролина при восстановлении литием
О о
!1 ПАШ 11
RC-NR2_ RCH + HNR;
и метиламином расщепляется аналогичным образом. Если пептид
содержит С-копцевой пролин, то при восстановлении выделяется
свободный пролин, который может быть определен колориметри-
чески:
149
Например, ь-алаиил- ь-пролин ацетилируют, N-ацетильное произ-
водное растворяют в метиламине и обрабатывают избытком лития
при —70°. Через час добавляют хлористый аммоний до исчезнове-
ния синей окраски, а растворитель отгоняют. Остаток растворяю!'
в воде и колориметрически определяют пролин. Пептид, содержащий
пролин в середине цепи, дает при восстановлении новый пептид с
N-концевым пролином, из которого пролин можно выделить фер-
ментативным гидролизом ь-пролиниминопептидазой.
1. Benkeser R. A., Robinson R. Е,, Sauve D. М,, Thomas
О. Н., J. Am. Chem. Soc., 77, 3230 (1955).
2. Burgstahler A, W., Worden L. R., J. Am. Chem. Soc., 86, 96
(1964).
3, Grob C. A., S c h i e s s P. W., Helv. Chim. Acta, 43, 1546 (1960).
4. Benkeser R. A., H az dr a J. J., L ambert R. F., R у a n P. W.,
J. Org. Chem., 24, 854 (1959).
5. Benkeser R. A., Agnihotr i R. К., В urrous M. L., Tet-
rahedron Letters, 1, (1960); Benkeser R. A., A g n i h о t r i R. K.,
В u r ro u s M. L., К a i s er E. M., Malian J. M., R у a n P. W.,
J. Org. Chem., 29, 1313 (1964).
6. Benkeser R. A., К a i s с г E. M., J. Org. Chem., 29, 955 (1964).
7. Benkeser R. A., S c h г о 1 I G., Sauve D. M., J. Am, Chem. Soc.,
77, 3378 (1955),
8. Benkeser R. A., Lambert R. F.,R yan P. W., S t о f f e у D, G.,
J. Am. Chem. Soc., 80, 6573 (1958).
9. Burgstahler A. W., Worden L. R., Lewis T. B., J. Org.
Chem., 28, 2918 (1963).
10. H a 1 1 s w о г t h A. S., 14 e n b e s t H. B., W r i g 1 с у T. I., J. Chem.
Soc., 1957, 1969.
11. Fried J., Brown J. W., Borkenhagen L., Tetrahedron Letters,
2499 (1965).
12. H allsworth A. S., H enbesl H. B., J. Chem. Soc., 1957, 4604.
13. H a 1 1 s w о r t h A. S., Henbest 14. B., J. Chem. Soc., I960, 3571.
14. С г о s s 1 e у N. S., 14 e n b e s t H. B., J. Chem. Soc., 1960, 4413.
15. S t о 1 о w R. D.,B ona vent ur a M. M. Tetrahedron Letters, 95 (1964).
16. Truce W. E., T a t e D. P., В ur dge D. N., J. Am. Chem. Soc., 82,
2872 (1960).
17. В i r k о f e r L., Frankus E., Chem. Ber., 94, 216 (1961).
18. Patchornik A., W i 1 c h e k M., S ar i d S., J. Am. Chem. Soc., 86,
1457 (1964).
150
ЛИТИЙ - АММИАК, см. также Берча восстановление.
Сторк [1] предсказал, что октален (1) при взаимодействии с ли-
тием в жидком аммиаке должен превращаться в менее устойчивый
из двух возможных енолятов; опыт подтвердил это предска-
зание. Восстановление ненасыщенного кетона (1) дало в качестве
первоначального продукта лишь /и/7й«о-2-декалон (3). Реакцию рас-
сматривают как присоединение электрона от лития к кетону (1)
с образованием гибридного промежуточного соединения (2), р-уг-
леродный атом которого имеет карбанионный характер: промежуточ-
ное соединение отрывает от аммиака протон с образованием про-
дукта (3). Эти данные способствовали появлению нового метода
алкилирования [2]. Алкилирование трцнс-2-декалона (4) в присут-
ствии основания протекает через более устойчивый енолят и дает
соединение (5). Однако менее устойчивый енолят можно получить
восстановлением ненасыщенного кетона (1) Л.— а. и алкилировать
с образованием изомерного метилированного продукта(6)
Шауб и Вейсс [31 алкилировали тестостерон (7) восстановлением
его литием в жидком аммиаке н добавлением йодистого метила в
эфире. Согласно данным ЯМР, этот продукт представляет собой
(7) (8) (9)
151
4а-метилкетон (8), что было подтверждено также образованием иден-
тичного продукта при восстановлении 4-метилтестостерона (9).
Шауб и Вейсс показали также, что Д*>6-3-диеноны (10) гладко
восстанавливаются Л.— а. до Д5-3-кетостероидов (12). Раствор
де -тестостерона (10) в ТГФ (12) добавляют к литию в жидком амми-
аке до исчезновения синей окраски и обрабатывают реакционную
смесь хлористым аммонием. Получающийся с хорошим выходом
несопряженный кетон (12) в присутствии КОН легко изомеризуется
в тестостерон (7), однако аммиак недостаточно основен, чтобы ката-
лизировать изомеризацию. Венкерт [41 использовал последнюю
реакцию в работе, посвященной синтезу ряда смоляных кислот.
Раствор 100 мг диенона (13) и 100 мг лития в 5 мл тетрагидрофурана
и 80 мл жидкого аммиака перемешивают в течение 3 мин, добавляют
хлористый аммоний и дают смеси испариться в атмосфере азота.
СН3| )|
/\ /-у
Li-NH3
0АА/ ' *
(13)
(15)
Экстракция хлороформом, промывание и выпаривание дают 90 мг
кетона (14) в виде масла; его кипятят 30 мин с 10 % -ным раствором
едкого натра в метаноле и после перекристаллизации получают
71 мг сопряженного кетона (15).
По мнению Дира и Паттисона [5], Л.— а. более предпочтителен,
чем Na—NH3 для стереоспецифического транс-восстановления ал-
кинов. Реакцию проводят в ТГФ в автоклаве под давлением при
комнатной температуре.
(СН3)(СН3)7С С(СН3)7СО3Н
Стеароловая кислота
СНз(СН2)7 н
Li-NH3 в ТГФ /
Н7 (СН2)7СО2Н
Элаидиновая кислота
Метиловый эфир дезоксиподокарповой кислоты (1), имеющий
сильно экранированную сложиоэфирную группу, реагирует с Л.—
а., образуя соответствующую кислоту (выход 75%), а предполагае-
152
мый спирт является побочным продуктом [6]. Метилдегидроабиетат
(3), в котором сложноэфирная группа экваториальна, восстанавли-
вается главным образом до спирта (4). Сходный пример опублико-
ван Мейером и Левинсоном 17].
1. S t о г k G., D а г 1 i n g S. D., J. Am. Chem. Soc., 82, 1512 (1960); StorkG.,
Tsuj i J., J. Am. Chem. Soc., 83, 2783 (1961).
2. S t о г к G., R osen P., Gol dman N. L., J. Am. Chem. Soc., 83, 2965
(1961).
3. Sch aub R. E., Weiss M. J., Chem. Ind., 1961, 2003.
4. Weaker t E., Afonso A., Bredenberg J. В-son, К a n e к о С.,
T a h а г a A., J. Am. Chem. Soc., 86, 2038 (1964).
5. D ear R. E. A., Pall Ison F. L. Al., J. Am. Chem. Soc., 85, 622 (1963).
6. W e п к e г t E., J ackson B. G., J. Am. Chem. Soc., 80, 217 (1958).
7. Meyer W. L. , Lev inson A. S., J. Org. Chem., 28, 2184 (1963).
ЛИТИЙ — АММИАК—ROH, см. Берча восстановление.
ЛИТИЙ — трет-БУТА НОЛ — ТЕТРАГИДРОФУРАН. Инсек-
тициды альдрин и изодрин с открытием Бруком, Томпсоном и Вин-
штейном [1] простого метода дехлорирования этих соединений, при-
водящего к замещению всех атомов хлора на атомы водорода, стали
привлекательными исходными веществами для синтеза интересных
циклических систем. Полигалоидное соединение обрабатывают ли-
тиевой стружкой (3 г-атом на галоген) и mpem-бутанолом (2 же
на галоген) при кипячении в ТГФ в инертной атмосфере. В этом
153
ряду выходы углеводородов составили около 60%. Следует отме-
тить, что при этом замещаются винильные, аллильные и геминаль-
ные галогены и даже галогены в голове моста бициклических сис-
тем. Замещение ароматического галогена эффективно осуществля-
ется при превращении З'Д'Д'Д'-тетрахлорбепзонорбориена (1)
в бензопорборнен (2) [2].
Дехлорированию ГЗ] подвергался также аддукт (5), который лег-
ко получается по приведенной ниже реакции. Дехлорирование ли-
тием и треш-бутанол ом в ТГФ сопровождалось частичным во с ста-
новлением; продукт реакции — смесь ненасыщенного кеталя (7)
и насыщенного кеталя (8) в отношении 65 : 35. Ненасыщенный ке-
таль (7) выделяли из смеси повторной экстракцией раствора в пен-
тане 20%-ным водным раствором нитрата серебра и обработкой
экстракта гидроокисью аммония. Однако при замене натрия на
литий продуктом реакции дехлорирования был чистый ненасыщен-
ный кеталь (7), превращающийся в результате кислотного гидролиза
2 з 13 20 мл ТГФ
+ Li + (СНЭ)3СОН—>Н
2 2 30 МА Н
154
в бицикло-12, 2, 1]-А2-гептепон-7. Метод эффективен при восстанов-
лении цнс-бнс-аддукта дихлоркарбепа в цис, цис- 1,5-циклооктади-
ен 14]. Раствор галогенида в ТГФ перемешивали магнитной мешал-
кой на ледяной бане и добавляли расплющенную молотком литие-
вую проволоку и шрет-бутанол в количестве, достаточном для раст-
ворения металла. Аддукт (2), полученный из эргостерилтетрагидро-
пираиилового эфира (1) и дибромкарбепа, успешно восстанавливали
аналогичным методом [5, 6].
1, В ruck Р., Т h ompson D., W i и s t с i n S., Chem. Ind., I960, 405.
2. Bruck P., Tetrahedron Letters, 449 (1962).
3. G a s s m a n P. G., Pape P. G., J. Org. Chem., 29, 160 (1964).
4. F i c s e r L. F., S achs D. H., J. Org. Chem., 29, 1113 (1964).
5. N a z e r M. Z,, J. Org. Chem., 30, 1737 (1965).
6, For further examples, see Schuster D. L, Lee F.-T., Tetr ah edr on Let-
ters, 4119 (1965).
ЛИТИЙ — ДИФЕНИЛ, Мол. вес 166,06.
Образуемый литием с дифенилом в ТГФ ^{7- \=/
сине-зеленый аддукт состава 2 : 1 более эффе- \—~/\—/
ктивен в некоторых реакциях расщепления,
чем литий [1]. Одно из преимуществ его использования — проте-
кание реакции в гомогенной среде [1]. Например, трифенил амин
дает с комплексом 58% дифениламина, а с литием только 8% дифе-
ниламина. Анизол и дифениловый эфир расщепляются с выходами
80 и 96%.
(CeHr,)3N (C6HJ,NHH-CgH5NH2
10 час, 66° 5go.,o 1,6%
Эстрон — основной промежуточный продукт при синтезе при-
меняемых в медицине 19-порстероидов—можно получать с высоким
выходом при температуре 35° реакцией 17-этилеиксталя Д1,4-андро-
стадиендиопа-3,17 (1) с Л.— д. в ТГФ в присутствии подходящего
кислого углеводорода типа дифенил метана. Последнее необходимо
для связывания побочного продукта — метиллития— и предотвра-
щения возможности его присоединения к потенциальной 17-кар-
бонильной группе [2].
155
(1) (2)
I. E i s c h J. J., J. Org. Chem., 28, 707 (1963).
2. Dryden H. L., Jr., Webber G. M., Wieczorek J. J., J. Am.
Chem. Soc., 86, 742 (1964).
ЛИТИЙ — N-МЕТИЛАНИЛИН. Этот реагент в эфире осу-
ществляет 1,4-восстановление сопряженных диенов в олефины [11:
RCH -= СНСН =CHR' + 2Li Щ 2CeH5NHCH3 ~►
RCH3CH=CHCH.,R' + 2C(iH5N-CH3
I
Li
Примеры: бутадиен -s- бутен-2; нафталин 1,4-дигидронафта-
лим.
I. Ziegler К. et al., Ann., 511, 64 (1934); 528, 101 (1937); 567, I (1950).
ЛИТИЙ-НАТРИЕВЫЙ СПЛАВ ДЛЯ ПОЛУЧЕНИЯ ЛИТИЙ-
АЛ КИЛОВ [1]. Бил и др. 121 получили «-бутиллитий с выходом
80—95% из «-бутилбромида, диэтилового эфира и металлического
лития, содержащего 0,05% натрия [3]. Если литий содержит лишь
0,005% натрия, максимальный выход «-бут ил лития составляет
только 48% (выходы определяли методом обратного титрования
[41). Позже группой Била было показано, что выходы можно повы-
сить, если расплавить литий высокой чистоты под слоем масла
и сплавить его не менее чем с 0,5% натрия. Такая концентрация
обеспечивает быстрое протекание реакции и высокие выходы алкил-
литиевых соединений. Для получения каталитического эффекта
недостаточно добавить к реакционной смеси нужное количество
натрия: необходимо сплавить натрий хотя бы с частью лития. Со-
держание натрия можно увеличить до 5%, не вызвав при этом
побочных реакций.
2RCI3-2Na —RR+2NaCI
Эти наблюдения были подтверждены и дополнены Мак-А рту ром
и др. [5.1 при получении больших количеств различных алкиллитие-
вых соединений. Для получения алкильных производных лития,
содержащих пять и меньше атомов углерода, достаточно быстрое
протекание реакции обеспечивается содержанием в сплаве 0,5%
натрия. Для высших гомологов (С10—С24) оптимальное содержание
натрия несколько выше; например, с децилхлоридом и октадецил-
хлоридом оно составляет приблизительно 1,33%. Размер частиц
лития не имеет значения; если содержание натрия оптимально,
156
то одинаково хорошо реагируют куски металла, дробь, стружки,
лента, фольга и порошок.
Например, 1,18 моля лития, содержащего 1,33% натрия, сус-
пендируют в 200 мл эфира при 7—10° в трехгорл ой круглодонной
колбе, предварительно продутой сухим азотом, и в течение часа
добавляют 0,5 моля октадецилхлорида в 60 мл эфира. Перемешивают
смесь еще 1,5 час при 7—10°, фильтруют раствор через неплотный
слой стеклянной ваты в атмосфере азота. Выход октадециллития
72—88%. Дециллитий был получен аналогичным образом с выхо-
дом 67—92%.
Было показано, что наилучший растворитель для реакции —
диэтиловый эфир; качество растворителя значительно улучшалось
после кипячения с литием в течение 1 час перед добавлением алкил-
галогенида. После образования ал кил лития диэтил овый эфир мож-
но заменить другим растворителем, поддерживая при этом темпера-
туру ниже 10°.
При получении больших количеств алкиллитиевых соединений
(38—3800 л) желательно увеличить количество натрия до ~25%
и активировать диэтиловый эфир продолжительным кипячением
с литием, чтобы избежать длительного индукционного периода (15—
24 час) и бурной реакции.
1. Contributed by McArthur R. E., Olin Mathieson Chemical Corporation,
New Haven, Connecticut.
2. В e e 1 J. A., Koch W. G., Tomasi G. E., Her mansen D. E.,
F 1 e e t w о о d P., J. Org. Chem., 24, 2036 (1959).
3. G i 1 m a n H., Beel J. A., Brannen C. G., BuHock M. W.,
DunnG. E., M i I I e г Ь. S., J. Am. Chem. Soc., 71, 1499 (1949).
4. Gilman H-, H aubein A. H., J. Am. Chem. Soc., 66, 1515 (1944).
5. Chiras S. J., F r u I 1 a F. F., Al i n k 1 e i A. O., Kaufman C. W.,
Hugos H., Motcock G. T., McArthur R. E,, unpublished work
at Olin Mathieson Chemical Corporation under Supply Contracts to U. S. Air
Force; AF (33-600) 36839 (1957); AF (33-601) 57-10186 (1957); AF (33-601) 57-573
(1957).
ЛИТИЙ — я-ПРОПИЛАМИН. Леонард и др. [1] использовали
этот восстановитель для получения енаминов, которые устойчивы
к гидролизу благодаря тому, что функциональная группа включена
в би- или трициклическую систему. Например:
157
j, Leona rd N. J., S t e inh ar d t С. K., Lee C., J. Org. Chem., 27, 4027
(1962).
ЛИТИЙ — ЭТИЛЕНДИ АМИН (т. кип. 117°), см. также N-Литий-
эти лен ди амин. При восстановлении системами металл—амии ис-
пользуют аммиак, метил- и этиламины; высшие же алифатические
первичные амины мало пригодны (для этой цели), вероятно, потому,
что растворимость щелочных металлов в них уменьшается с увели-
чением отношения углерода к азоту. Реггель и сотр. [1], отметив-
шие, что этилендиамин содержит одну аминогруппу на каждый
атом углерода и что его относительно высокая точка кипения (117°)
должна давать определенные преимущества, действительно, пока-
зали, что система литий — этилендиамин при 90—100° является
эффективным восстанавливающим агентом. Она восстанавливает
тетралин (1) до А9(1и)-окталина (2), а последний до транс-декалина
(3). Колбу, снабженную мешалкой, холодильником и термометром,
продувают азотом и загружают в нее этилендиамии (высушенный
и перегнанный над натрием) и восстанавливаемое вещество и добав-
ляют через холодильник литий (4—8 порций, 0,5—1 а) при 90—100°
в течение 1,5—3 час. Скорость его загрузки контролируется по ко-
личеству выделяющегося водорода; свежую порцию металла добав-
ляют перед исчезновением синей окраски от предыдущего добавле-
ния. Обработка тетралина в диамине литием, взятым лишь в 10 % -
158
ном избытке от теории, дает соединение (2) с выходом 32%; 46%
тетралина выделяются обратно. При 100%-ном избытке лития выход
соединения (2) увеличивается до 68%, и извлекают 31 % исходного
вещества. Беглое изучение других реакций указывает в большин-
стве случаев па низкие выходы, неполное превращение и образова-
ние смесей.
Кори и Кэнтролл [2] показали, что реагент эффективен при вос-
становлении метиленовых групп микроколичеств 3-метиленхоле-
стана (4) и тритерпенойдного производного (6). В обоих случаях
с хорошими выходами были получены продукты, в которых метиль-
ная группа занимает экваториальное положение. Отметим, что три-
замещепная 12,13-двойная связь а-амирина (7) остается незатро-
нутой.
1. R с g g е 1 L., F г i е d е 1 R.A., Wender I., J. Org. Chem., 22, 891 (1957).
2. Corey Е. J., С a n t г а 1 1 Е. W., J. Am. Chem. Soc., 81, 1745 (1959).
N-ЛИТИЙЭТИЛЕНДИАМИН, H,.NCH2CH2NH2 (мол. вес 60,10)%
%-Li (ат. вес 6,94) H2NCH3CH.,NHLi-rH2.
Реггель, Фридман и Вендер 111 открыли, что Л. обладает заме-
чательной способностью изомеризовать олефины и дегидрировать
дигидробензоидные системы. Безводный этилендиамин получают
либо нагреванием продажного продукта с натрием в течение 1—2
дней и перегонкой [1], либо азеотропной перегонкой с очищенным
от тиофена бензолом и последующей перегонкой над гранулирован-
ным едким кали, а затем над натрием [21. Для получения Л. к очи-
щенному этилендиамину при перемешивании в атмосфере азота
при 90—110° добавляют порциями литий; первоначальная темно-
синяя окраска в конце добавления исчезает и раствор становится
бледно-желтым 11].
Реггель и сотр. [11 наблюдали, что Л. в избытке этплеидиамииа
быстро изомеризует терминальные алкены во внутренние алкены.
Например, октен-1 при кипячении с Л. в течение 2 час дает 90%
внутренних алкенов, главным образом октена-2.
Еще более замечательно то, что дегидрирование осуществляется
в очень мягких условиях. При добавлении 4-винилциклогексепа
(1) к раствору Л. при комнатной температуре выделяется водород
и в качестве единственного органического продукта получается
этилбензол (3). Первая стадия, по-видимому, заключается в миг ра-
СН3-СН3
I
сы., NHL Г
I
сн.мн.
<3>
ции терминальной двойной связи, возможно, с образованием сопря-
женного диена (2). Удивительно, что следующая стадия, дегидриро-
159
вание (2) в (3), осуществляется при комнатной температуре. В дру-
гом примере к раствору Л., полученному из 2 молей лития и 375 мл
этиленди амина, добавляют 2 моля d-лимонена и перемешивают при
100—115° в течение 1,5 час (бурное выделение газа) и смесь кипятят
0,5 час. В результате реакции образуется исключительно «.-цимол
с т. кип. 170—173°. В контрольном эксперименте Л., полученный
й-Лимонен
н,с/ хсн
2
з
ге-Цимол
из 0,25 моля лития и избытка этилендиамина, обрабатывают I молем
d-лимонена и смесь нагревают в течение 1 час. Водород при этом
не выделяется и продукт реакции состоит исключительно из d-лимо-
нена с т. кип. 169—172°, возвращенного с 92%-ным выходом. Нам
кажется, это наблюдение исключает предложенный Реггелем согла-
сованный механизм, включающий девятичленное переходное состо-
яние и предполагающий, что Л. играет каталитическую роль; другой
аргумент против этого механизма—то, что литий-триэтилендиамин
unch2ch2nh2
-h2nch2ch2nhz
h2nch2ch2nh2
(4) (з)
+ i!nch2ch2nh2 + н2
н
также обладает активной дегидрирующей способностью. Наш со-
трудник Хаддадин предложил следующий механизм. Сильно основ-
ное производное лития атакует углеводород (2) с замещением одного
из кислых водородов на литий (4). На второй стадии (4) реагирует
с этилендиамином, образуя (3) и гидрид лития, причем последний,
в свою очередь, реагирует с этилендиамином, давая водород и Л.
Для реакции необходима основная среда; она не осуществляется
в присутствии значительного количества кислого углеводорода (2)
и лишь после полной нейтрализации углеводорода возможна реали-
зация заключительной стадии. Для этого требуется эквимолярное
(или чуть большее) количество Л. Хотя Л. регенерируется и не рас-
ходуется, он не является катализатором, поскольку, реагируя
160
в одной реакции, он регенерируется в другой независимо от первой.
Л. дегидрирует а-фел л андрен (5) в и-цимол с выходом 74% (Рег-
гель и сотр.). Группа индийских химиков [2] получила и-цимол с
высоким выходом аналогичными реакциями терпинолена, перил-
лового спирта (6) и карвеола (7). Они нашли, что циклопропановое
кольцо А3-карена (8) расщепляется в обоих направлениях с образо-
ванием смеси, состоящей примерно наполовину из п-цимола (9) и
.м-цимола (10):
Сескв итер пен-у х-кадинен (11) превращается в каламепел (12), веро-
ятно, за счет миграции экзоциклической двойной связи к двузаме-
щенному кольцу и ароматизации. При повторной обработке второе
(ID
н
I
NHoCH,CH4N-Li
<12)
кольцо не ароматизируется. Такая частичная ароматизация откры-
вает некоторые возможности, но устраняет другие. Например, один
из авторов книги получил отрицательный результат при использо-
вании реагента для ароматизации гвайена в гвайазулен. Аромати-
зация гидроароматического кольца, очевидно, требует наличия
двух двойных связей или двойной связи и циклопропанового кольца
[21. Сопряженный кетон (//-пи пер Итон (13) при обработке Л. не из-
меняется; таки.м образом, двойная связь, образующаяся при эиоли-
зации, не способна выполнять функции одной из двух необходимых
6 Заказ № 1096
161
кратных связей. Карвон (14), содержащий две двойные связи, дает
карвакрол (15) с количественным выходом. Гераниол (16) изомери-
зуется Л. в сопряженный диен (17). Хотя используется избыток
СН3 н
/Г-TJ гпги г-и Д /1Г гтт ли НЛСНЛНЛ-Н
(CH;HnC = CHCHnCH2C-=CII — СНоОН -----— -----эн
(1бГ СНз
—> (СН3).,С^ смен = снсн -~сн„ -СН2ОН
(17)
реагепта и кипячение продолжается 8 час, последующего дегидри-
рования в триен не происходит.
Согласно краткому сообщению [3], Л. дегидрирует циклогекса-
нол и 2-мет ил циклогекса нол в циклогексанон и 2-мети л циклогек-
санон с выходами 54 н 38% соответственно. Насыщенные первичные
спирты дают альдегиды с выходами 25—66% и бензиловый спирт
дает бензальдегид с выходом 80%.
Хотя Л. был получен и интенсивно использовался Реггелем
и др., выделение соединения было впервые описано Гомелем и Хар-
рисом 141.
Ниже приведены другие примеры изомеризации и частичного
дегидрирования [5].
ГУН
I-/\(СН2)12СОгН
162
I. Reggel L., Friedman S., Wen der I,, J. Org. Chem., 23, 1136 (1958).
2. Ту agi B. S., G h a t g e B. B.,Bhatt ach aryy aS, C., J. Org. Chem.,
27, 1430 (1962).
3. Smith S., Thompson R., Woolfol к E. O., J. Org. Chem., 27,
2662 (1962).
4. В e u m e 1 O. F., Jr., Harris R. F-, J. Org. Chem., 28, 2775 (1963), 30,
814 (1965).
5. Tyagi B. S., Gha tgc В. B,, Bh a I 1 a ch а г у v a S. C., Tetra-
hedron, 19, 1189 (1963).
ЛИТИЯ АЛЮМОГИДРИД, LiAlH<. Мол. вес 37,95, Раство-
римость, г/100 г при 252: в диэтиловом эфире 35—40 (для достижения
такой растворимости необходимо сконцентрировать более разбав-
ленный раствор); в ТГФ 13; в дн-н-бутиловом эфире 2; в диоксане
0,1. Обзоры [1а].
Определение. Методика и прибор для анализа по выделению
водорода описаны в Metal Hydrides Technical Bulletin № 401.
История. Л. а. был открыт Финхольтом, Бойдом и Шлезинге-
ром [1] в 1947 г. Его получали постепенным добавлением хлористого
алюминия к суспензии гидрида лития в эфире и отфильтровыванием
выпавшего хлорида лития и избытка гидрида лития. В это же время
Нанстром и Браун 121 сообщили о множестве примеров восстанов-
ления Л. а. органических соединений с высокими выходами.
4LiH-i- А1С13 LiAlH4 i-3LlGl
Л. а. мгновенно реагирует с избытком воды или этанола с выде-
лением 4 молей водорода. Хохштейн [3] количественным изучением
восстановления Л. а. показал, что соединения, образующие метан
при взаимодействии с мети л магии йиоди дом в пробе Церевитинова,
выделяют водород и из Л. а., хотя имеются некоторые количествен-
ные различия и различия в поведении снолизпрующихся соеднне-
1лА1Н4 4 4НОН-^ LiOH-|-Al(OH),H-4H,
LiAlH4 + 4C,H5OH LiOC2H5--Al(OC.Jls)34-4H3
ний. Например/под действием первичного амина из реактива Гринь-
яра выделяется 1 моль метана, а из Л. а. 2 моля водорода. Хохш-
тейн [31 разработал аналитический метод определения активного
водорода и общего расхода Л. а.
LiAlH* + <
4RCO..H
4С6Н/ОН
4R=, NH —>
4RSH
4RC = CH
RCO.,Li4AI(OCOR)3
CGH5bLi--Al(OCcI-I5}3
LiNR.,5-Al(\'R.,)3
LiSR -i-Al(SR)3
LiC = CR-l-Al(C--CR)3
4Н2 Щ
Формулу реагента обычно пишут в виде LiAlH.i, но интерпре-
тируют как LH [А!Н4]~или LHAlHy. Так, в эфирном растворе он
существует в виде сольватированных ионов лития и алюмогидрид-
ных ионов. Первая стадия реакции с кетоном состоит в переносе
гидрид-иона и образовании комплекса (1), который, обладая еще
6*
163
тремя реакционноспособными атомами водорода, может реагировать
еще с тремя молями кетона с образованием алкоголята (2), разлага-
ющегося водой на вторичный спирт и гидроокиси алюминия и лития.
Ввиду низкого молекулярного веса и способности 1 молем восста-
R2C = O+H — AlH3(Li + )—>R2C-OAIH3(Li + ) 2^.52^
н
(1)
(R2C-O)4AlLi+ 4R2C-OH~:~AI(OH)3+LiOH
I I
Н Н
(2)
навливать 4 моля альдегида или кетона, Л. а. обладает высокой
удельной восстановительной активностью.
Техника эксперимента. Реагент можно хранить в склянке со
стеклянной пробкой, склянку предварительно продувают азотом,
а открыв, вновь тщательно закрывают пробкой н заливают пара-
фином.
Главная опасность при работе с Л. а. заключается в его чрезвы-
чайной чувствительности к действию влаги н к растиранию. Контакт
с влагой (например, недостаточно высушенный прибор, влажные
руки нли перчатки и т. поможет вызвать воспламенение Л. а. Из-
мельчать реагент лучше всего эбонитовым молотком в противне с
покрытием из алюминиевой фольги. Измельчение Л. а. пестиком в
ступке следует проводить в сухой камере в атмосфере азота или
аргона. Энергичное растирание Л. а. на воздухе может вызвать вос-
пламенение. Кроме того, порошок Л. а. очень едкий н вызывает раз-
дражение слизистых оболочек. Однако, если измельчать небольшое
количество Л. а. и принять соответствующие меры предосторожности,
то с помощью ступки и пестика можно разбить один-два куска
и легко растереть их и не в инертной атмосфере. Следует отметить,
что в приведенной ниже методике восстановления амида Коуп 1251
использовал кускн Л. а. без предварительного растирания и показал,
что при перемешивании суспензии в эфире магнитной мешалкой
осуществляется достаточное измельчение.
В целях противопожарной безопасности при работе с Л. а. реко-
мендуется иметь ведро измельченного известняка и лопату с длинной
ручкой. Можно использовать только специальные огнетушители,
но нн в коем случае нельзя пользоваться водой, углекислотными
или другими химическими огнетушителями.
Раствор Л. а. в диэтиловом эфире лучше всего получать слабым
кипячением и перемешиванием в течение нескольких часов в атмос-
фере азота в колбе с осушительной трубкой. При обработке, по-ви-
димому, вследствие контакта с атмосферой обычно образуется не-
растворимый осадок. Этот осадок кажется очень объемным, по он
.легко удаляется фильтрованием, и, как было показано, составляет
164
менее I % от количества Л. а., последний в растворе медленно реа-
гирует с атмосферным кислородом, выделяя водород. Растворы Л. а.
неустойчивы, как и твердое вещество, и нх следует хранить в атмо-
сфере азота, поскольку они могут самопроизвольно воспламениться.
Методика восстановления. Ранее для восстановления исполь-
зовались прозрачные растворы Л. а. в эфире, получение которых
требовало продолжительного кипячения, трудоемкого фильтрова-
ния, переноса раствора без доступа воздуха и удаления загрязнений.
В настоящее время растворитель (эфир или ТГФ) помещают в колбу,
снабженную мешалкой, холодильником, капельной воронкой, до-
бавляют Л. а. (обычно измельченный, а иногда в виде кусков) н вво-
дят раствор восстанавливаемого вещества со скоростью, обеспечи-
вающей слабое кипение,
Разложение избытка реагента. Л. а. часто берут в большом
избытке от теории (2—4-кратный избыток), хотя необходимость
этого часто не подтверждается контрольными опытами. При разло-
жении большого количества Л. а. даже медленное добавление воды
и перемешивание необходимо проводить очень осторожно, так как
выделяющийся водород вызывает бурное вспенивание. Использова-
ние 10%-иого раствора едкого натра или хлористого аммония более
удобно, поскольку выделяющаяся при этом гранулированная окись
алюминия легко отфильтровывается. Часто предпочитают использо-
вать этилацетат, так как при этом не выделяется водород и продукт
восстановления (этанол) не мешает выделению конечного продукта.
4СН3СО2С2Нй + LiAIH4 -> LiOC2H5 + А1(ОС2Н5)3-Г4С2Н5ОН
Выделению продукта восстановления часто мешает объемный
желатинообразный осадок гидроокисей, обработка которого избыт-
ком кислоты или щелочи приводит к увеличению объемов и образо-
ванию эмульсий. Штейнхардт 14] установил, что этого можно избе-
жать последовательным добавлением к восстановленной смеси
из п граммов Л. а. по каплям п мл воды, п мл 15%-ного раствора ед-
кого натра и Зп мл воды (при перемешивании). Такая методика [5]
приводит к образованию сухого гранулированного осадка, который
легко отфильтровать и отмыть.
Восстановление альдегидов и кетонов [6]. Для приведенных
ниже типичных реакций восстановления, проводившихся со стан-
дартными растворами Л. а., требуется 0,25 моля реагента на моль
карбонильного соединения. Современная методика иллюстрируется
восстановлением [7] карбонильной группы З-этоксициклогексен-2-
СНз(СН.>)5СНО . °’25L1A1^ СН3(СНЯ)5СН.ОН
86%
СНзСНоСОСН, L1A1^2 СН3СН,СН-СНз
80%
он
сн3сн-снсно °-25 L1A1.^ СН3СН-СНСН2ОН
70%
165
она (I) — промежуточного соединения при синтезе циклогексен-
она-2. В колбу, снабженную обратным холодильником, механичес-
Эфир
L i Л1Н.,
(1 1 0 % - иь; ii избыток)
кой мешалкой и капелы-юй воронкой и защищенную от атмосферной
влаги осушительными трубками, помещают 0,16 моля Л. а. и 200 мл
абсолютного эфира, причем количество используемого Л. а. в 2,1
раза превышает теоретическое. Раствор 0,31 моля енольного эфира
(1) в 50 мл эфира добавляют со скоростью, обеспечивающей слабое
кипение раствора (1,5 час). Кипячение продолжают в течение 30 мин,
после охлаждения к смеси добавляют по каплям 15 мл воды (вспе-
нивание). Затем смесь обрабатывают и енольный эфир (2) гидроли-
зуют кислотой до циклогексенона-2 (3).
Карбоновые кислоты и производные. Особенно поразительным
свойством Л. а. является его способность восстанавливать карбоно-
вые кислоты в первичные спирты. На замещение кислотного водо-
рода расходуется 3/4 экс реагента, а на восстановление еще т/2 же.
Хорошие выходы в этой реакции были получены прн использовании
0,75 моля Л. а. на 1 моль кислоты [81. Трифен ил уксус на я кислота
не восстанавливается при обычных условиях (эфир), однако ее
можно превратить в спирт с хорошим выходом либо в более высоко-
кипящем ТГФ, либо превращением в ацилхлорид, который легко
восстанавливается в эфире [91. Аксиальная 4р-карбокснльная груп-
па подокарповой кислоты сильно пространственно экранирована
1,5Ь1АП-Ц
НО.,С(СНг)8СО.2Н ———> НОСНДСН,)ЙС1'ГОН
9 (%
сн3
СН3-С-СО.,Н
Ан3
LiAlH,
ТГФ
сн.,
СН3 — A—CITOH
t ।
СНз
сн3
I I
СН3—С —СОС1
сн3
LiAiH4 (Эфир)
аксиальной р-метильной группой при атоме С10. Активные водороды
карбоксильной и фенольной гидроксильных групп также потреб-
ляют реагент с образованием производных с меньшей раствори-
мостью. Поэтому восстановление свободной кислоты в течение
4 дней при комнатной температуре дает только 56% иодокарпинола;
1G6
метиловый эфир О-метилподокарповой кислоты восстанавливается
более удовлетворительно:
При этом расходуется 0,5 моля реагента; восстановление осу-
ществляется, по-видимому, через промежуточное образование аль-
дегида:
О
О -
li
С6Н5СН
0,25ЫАПЖ
--------> С,ЩСН.,ОН
90%
В подробной методике восстановления эти л-а-(1 -пиррол иди л )-
пропионата (1) рекомендуется [101 кипятить при перемешивании
0,56 моля распыленного Л. а. с 300 мл эфира до тех пор, пока боль-
шая часть Л. а. не растворится, н добавить по каплям раствор 0,92
моля сложного эфира в 200 мл эфира со скоростью, обеспечивающей
слабое кипение растворителя. После перемешивания и кипячения
в течение 30 мин медленно добавляют 50 мл этил ацетата и затем
600 мл 4 и. соляной кислоты.
___ 1ЛД1Н4- эфир —.
N —СНСО2С2Н5 ('2°%"НЫИ избыток) N—СНСНгОН
/1 80-90% / I
--f бы3 ---< бн3
(1) (2)
При получении 2,2-дихлорэтанола из ди хлора цетил хлорида
(Сруг и Вудберн [1 И) реакция,по-видимому,протекает более плавно,
чем со свободной кислотой или этиловым эфиром. Эта методика
по существу совпадает с методикой получения сложного эфира (1).
ЫА1Н4
(теорет.)
CHC1XOCI-------> СНС1,СН.,ОН
Й4-Ь5% - -
13) (4)
К суспензии 0,3 моля Л, а. в эфире добавляют 0,6 моля ацилхло-
рида в эфире.
Так как ацетильное производное спирта легко расщепляется
Л. а., реагент часто применяют для деацетилирования соединений,
О
CH3COR 2-L-lA2^ CH3CH2OH+HOR
167
чувствительных к действию кислот и оснований. Например, А8(14)-
х ол естен д иол-Зр,7а-ди ацетат (1) легко превращается под дейст-
вием кислоты в А8,14-диен (2), а при щелочном гидролизе образует
очень мало продукта; однако кратковременная обработка его Л. а.
в эфире дает почти чистый диол (3) с количественным выходом [12].
(1)
Бухта показал [13], что Л. а. восстанавливает одну из двух кар-
бонильных групп ангидрида (4), не затрагивая другой. Очевидно,
препятствия двух орто-метильных групп позволят лишь одной
карбонильной группе образовывать комплекс с металлом. Восста-
новление фталида в диол (6) требует образования лишь одного ком-
плекса и в тех же экспериментальных условиях.
Эпоксиды. Для химии стероидов имеет большое значение син-
тетическая последовательность, заключающаяся в превращении
олефина в эпоксид и его восстановительном расщеплении на один
из двух возможных спиртов. А1-Холестен (1)дает 1а, 2а-эпоксид (2),
который восстанавливается Л. а. в холестанол-1а (3) [141. Анало-
гичная картина наблюдается для А11-хол естен а (4) [15]. В обоих
случаях образующийся спирт аксиален, тогда как раскрытие окис-
ного кольца в другом направлении должно приводить к образова-
нию экваториального изомера. Были изучены и многие другие слу-
168
чан (в частности, Платнером и corp. [16]), показавшие, что окись
почти всегда расщепляется с образованием аксиального спирта.
До применения Л. а. эпоксиды восстанавливали каталитическим
гидрированием. Теперь предпочитают использовать гидриды метал-
лов, которые дают хорошие выходы там, где гидрирование либо во-
обще не удавалось, либо давало плохие результаты.
Галогениды и тозилаты. Тревой и Браун [171 показали, что Л. а.
восстанавливает бромистый и иодистый бензил в эфире или ТГФ
при 35° с высокими выходами. В ТГФ при 65° бензилхлорид и
1-бромдекан восстанавливаются в толуол ин-декан с выходами72%.
Джонсон, Близзард н Кархарт [181 показали, что скорость восста-
новления Л. а. в этом случае ниже, чем в других реакциях. Ими
представлены экспериментальные доказательства в пользу того,
что не все четыре атома водорода одинаково реакционноспособны
по отношению к алкилгалогенидам. По-видимому, реакция проте-
кает по крайней мере в две стадии, одна из которых более быстрая,
чем другие:
ЫА1Н4-|-RX -^ RH + LiX+AIH3
A1H3 + 3RX AlX3-]-3RH
1-Бромоктан при кипячении в ТГФ в течение 1 час с 1,4-кратным
избытком Л. а. (от теории) дает н-октан с выходом 64%, а при кипя-
чении с 3,2-кратным избытком выход н-октана увеличивается до
96%. Впоследствии была разработана улучшенная методика, по
которой гидрирование осуществлялось гидридом лития и небольшим
количеством Л. а. в качестве переносчика водорода. Например,
I-бромдекан при кипячении в ТГФ в течение 0,5 час с 0,13 моля
Л. а. и 1,5 моля А1На на моль RX дает н-декан с выходом 96%.-
Бухта 1131 использовал гидрирование Л. а. в синтезе пренитола,
а Бреслоу [191—для восстановления снмм-трифенилциклопропе-
169
сн3 сн3
,1 ,CH2Br ) ZCH4
\ I LiAiH, V I.'
| Кипячение | ||
'Y' ХСН2Вг Г1 Ф "V 4 СНз
СН3 СН3
нилбромида в трифенилциклопропен. Гейлорду F201 не удалось
восстановить 1-оксиметилбензотрназол (1) в 1-метилбензотриазол
н5с6 Вг
(3) гидрированием в присутствии никеля Ренея или палладия на
угле. 1-Метилбензотриазол был падучей с высоким выходом вос-
становлением хлорида (2) в ТГФ 10-к ратным избытком Л. а.
Тозила! ы, в некотором отношении сходные с галогенидами,
иногда восстанавливают следующим образом:
R — OSO2CeH4CH3-rt —R НHO3SC(iH4CH3-n
Шмид и Каррер [21] показали, что Z-ментилтозилат восстанав-
ливается, в основном, в /-ментан и чтоб-тозилат диацетонид-о-галак-
топиранозида (1) дает диацетонид-в-фукопиранозид (2). Однако два
нс-оч
I >С(СН3)а
НС —СИ
.О—СН
0 LiAIH
О-СН
(СН3)2С
НС —о,.
I >С(СН3)3
НС—О-
О —^н
О —сн
О
НС
CH,OTs
НС
СНз
других сахаридных тозилата, один из которых является первичным
тозил атом, расщепляются следующим образом:
ROSO2C6H4CH8-n —ROH-!-HOaSC6H4CH3-n
170
Зорбах 1221 использовал эту реакцию при синтезе о-рамнозы, хотя
выход на стадии восстановления был низок.
СНО
— CHjSH.
— и с л
2 52%—
СН2ОН
D-Мавноза
а-цэсНзу, ch(Sch3)2
TsCI _ LiAlH4
“ 66% — 23%
СН2ОН CH2OTs
О) (2)
CH(SCHs)3 сно
HgO-HgCl.
60%
СН3
(3)
СН3
D-Рамноза
Амиды. Восстановление амида Л. а. с хорошим выходом дает
соответствующий амин. (Порядок реакционной способности:
RCONR2>RCONHR>RCONH.2.) По методике Моффетта [23] 5,5-
о
RC\rH> 0,5LiAJlL
rch,\h.
диметилпирролидон-2 (1) восстанавливают кипячением I моля
распыленного Л. а. в ТГФ при перемешивании с постепенным до-
бавлением 0,8 моля (1) в ТГФ. Для восстановления N-метиламида
(I)
_ЦА1Щ(150%-ный избыток)—
ТГФ
н
(2)
67-79%
лауриновой кислоты (3), ограниченно растворимого в эфире, Вильсон
и Стенберг I24I поместили в колбу, снабженную мешалкой и экст-
рактором Сокслета, 1 моль тонкоизмельченного Л. а., а в патрон
экстрактора 0,75 моля амида. Экстракцию заканчивают через 3 час\
LiAIH4 (110%-пыii избыток) — эфир
СН3(СН9_)1(1СО\ГНСН3 ----------——---------------> СНя(СН2)1аСНЛ’НСН3
о 1 “ У О Л
(3) (4)
реакцию продолжают еще 2 час. По методике Коупа и Циганека
[25] N,N-диметил а мид циклогексан карбоновой кислоты (5) восста-
навливают обычным добавлением эфирного раствора этого амида
(0,86 моля) к суспензии в эфире большого избытка Л. а. (0,85 моля).
Эта методика, однако, имеет одну интересную особенность: Л. а.
171
используется не в распыленном состоянии, а в виде кусков. Суспен-
зию перемешивают магнитной мешалкой, добавляют амид в эфире
в течение часа и кипятят смесь при перемешивании в течение 15 час.
О
/Х/С\Г(СН3)2
I Гхн
LiAlH4 (100%-Hbiii избыток) — s-фир
88%
/X/CH2N(CH3)2
I |хн
(fl/
Затем разлагают избыток Л. а. добавлением по каплям 70 мл воды,
подщелачивают и отгоняют продукт на кипящей водяной бане. Срав-
нение шести приведенных ниже методик по количеству использо-
ванного Л. а. обнаруживает следующее важное обстоятельство.
Тип восстановлен- ного соединения Избыток LiAlHx Средний выход, %
Кетон 2,1 63,5
Сложный эфир 1,2 85
RCOC1 1,0 64,5
RCONHR' 2-5 73
RCONHR' 2,7 88
RCONR'.2 2,0 88
На практике в четырех из шести случаев результаты свидетель-
ствуют не в пользу большого избытка реагента. Это не только спо-
собствует сбережению дорогого реагента, но и сокращает время,
необходимое для разложения избытка Л. а., особенно при исполь-
зовании воды. В работе Хох штейн а [3] было показано, что ненасы-
щенные соединения восстанавливаются Л. а. в разбавленных раст-
ворах количественно. При использовании 1,2-кратного избытка
учитывается возможность слабого разложения реагента кислородом
воздуха. Простая методика Коупа с использованием реагента в виде
кусков и перемешивания магнитной мешалкой [25] представляется
наиболее привлекательной, что подтвердилось экспериментально.
Если куски Л. а. перемешивать с эфиром магнитной мешалкой
1 час при комнатной температуре, то они превращаются в порошок.
Синтез альдегидов. N-Монозамещенный амид (1) восстанавли-
вается, по-видимому, за счет присоединения Л. а. к карбонильной
группе и образования геминального аминоспирта (2), который при
расщеплении дает имин (3), восстанавливающийся в конечный про-
дукт (4). Для NjN-дизамещенного амида промежуточное соединение
типа (3) будет солью иммония. При соответствующем контроле за
172
количеством Л. а. гидролиз (2) или (3) должен давать альдегид (5).
Однако решающее значение, по-видимому, имеет строение амида;
удовлетворительные результаты были получены лишь с амидами
трех типов. Вейганд [26] получил ароматические альдегиды реак-
цией N-метил анилина с фосгеном, приводящей к метил фенил кар -
баминоилхлориду (1), реагирующему с ароматическими углеводоро-
О OAIH3(Li+)
RC-NCH3 R(2—N—СН3 -____________ RC-NCH3RCH,NHCH3
| | | — HOAlHs(Li + ) |
H H H \ / H
(1) (2) \ H2O^/Z (3) (4)
RCH-О
(5;
дамп по Фриделю — Крафтсу с образованием N-метиланилида
карбоновой кислоты; это соединение восстанавливается контроли-
руемым количеством Л. а. в альдегид (3). Избыток Л. а. превращает
СН3 СН3
//—ч^ос. С^СНАА1С|^
'''Л — / " / го о/
—-- ^0/0
U)
сн3
— сн3 —НС—1—сн3
il \=/ 66% II \=/
о о
(2) (3)
альдегид в соответствующий первичный спирт. Браун [27] описал
сходный синтез на примере реакции хлорангидрида циклопропан-
карбоновой кислоты (4) с этиленимином (5) в эфире, содержащем
О
Н,СХ ,СН2 Et N Н2С. || /СН, LiA1H
| Ichcoci + hnI I рснс-н/l ~ _________L1A Hj__,
H,CZ %CH2 -Bt.NHCI H2C/ 4CH, 67%, считая на (4)
(4) (5) (6)
о
н2с II ,сн2
I фснсн-рнм/ I
НоС/ хсн2
(7)
1 же триэтиламина, при 0°. Хлоргидрат триэтиламина выпадает
в осадок; отфильтрованный раствор 1-ацилазиридина (6) при восста-
новлении Л. а. дает после гидролиза альдегид кислоты (7). Из
н-бутироил-, н-капроил- и н-пивалоилхлоридов этим методом были
получены соответствующие альдегиды с выходом 75, 81 и 79%; при-
менение в двух опытах 100%-ного избытка ацилхлорида не приво-
дило к уменьшению выхода.
173
Разработанный Шт а абом [281 метод синтеза альдегидов вклю-
чает конденсацию ацил хлорида с имидазолом с образованием ими-
дазолида (1) и восстановительное расщепление Л. а. на альдегид
(2) и имидазол. В одностадийном процессе обработка карбоновой
С6Н5СОС1
°
LiAlHj (20°)^ СбН5СН + H-N I
77,5%
кислоты в ТГФ или эфире N.N'-карбонил диимидазол ом дает имида-
золид; двуокись углерода удаляют вакуумированием (так как она
восстанавливается), после чего добавляют Л. а. в количестве, до-
статочном для взаимодействия с имидазолом, активным по Це рев и-
тинову, и для восстановления. Выходы реакции составляют
30—83%.
jLiAlHi
А г СНО
В синтезе метионина-а-С14 Клаус и Моргентау 12у1 исследовали
возможность превращения p-метилмеркаптопропионитрила в [3-
метилмеркаптопропионовый альдегид, однако восстановление по
Стефену и прямое восстановление Л. а. дали неудовлетворительные
ci-i3scii2ch,cn —+ CH3SCH2CH.2CIIO
результаты. Ими разработан общий метод превращения нитрилов
в альдегиды, приведенный ниже:
+ NH.,
„„ _ EtOII SI , 2E(OH 1ЛА1Н4
RC=N —__> RC — OEt(Cl-) ——RC(OEt)3 ——>
i-ici
О) (2) (3)
—> RCH(OEt)2 Ь? RCHO
(Ц
Обработка нитрила (1) этанолом и хлористым водородом дает хлор-
гидрат иминоэфира (2), реагирующий с этанолом с образованием
ортоэфира (3). Ортоэфир (3) восстанавливается в диэтилацеталь (4)
добавлением 0,25 моля 1 А4 раствора Л. а. в эфире к кипящему раст-
вору 1 моля ортоэфира в бензоле и кипячением в течение 4 час. Раз-
174
ложение комплекса 30%-ным раствором смешанной соли виннокис-
лого калия и натрия (сегнетова соль) дает диэтилацеталь fJ-метилмер-
каптопропионового альдегида с выходом 73%; выход диметилацета-
ля 97%). При кислотном гидролизе образуется р-метил меркапто-
пропионовый альдегид.
Восстановление аллиловых и пропаргиловых спиртов. Хотя
Л. а. не восстанавливает этиленовых и ацетиленовых углеводородов,
он восстанавливает аллиловые спирты и их ацетиленовые аналогу!
[30]. Реакция была открыта Найстромом и Брауном при восстанов-
лении коричной кислоты в ди гидрокор ич и ый спирт. Исследование
показало, что карбоновая кислота восстанавливается по обычной
схеме, а из коричного спирта получается насыщенный спирт. Ре-
LiAUI,
СДКИСЯ — СНССцН-----► C0H5CH.,CH.,CII.,OH
85%
зультаты эксперимента исключали другие возможные пути и указы-
вали, что необычная реакция протекает, по-видимому, следующим
образом. Сначала Л. а. быстро реагирует со спиртом с выделением
водорода н образованием литпйалюмннийалкоголята, который
затем восстанавливается с умеренной скоростью в бициклическое
комплексное соединение. При этом атом водорода связывается с
одним из ненасыщенных атомов углерода, а атом алюминия — со
вторым. Второй атом водорода, необходимый для насыщения двой-
ной связи, поставляется на последней стадии гидролиза комплекса
при расщеплении водой связи углерод — алюминий. В действитель-
ности, комплекс выделяется в виде нерастворимого осадка и явля-
4СеН5СН СНСН2ОНLiAHi.! (CeII5CH = CHCH2O)4AILi д- 4Н2
Коричный спирт
СН2-СН2
С6Н5СН - СНСН3ОЧ . ОСН,СН = СНС6Н5 СсН5С11 о
А11Л + 2 Al~Li +
С8Н5СН-СНСНоО7 \хн3сн = снс6н5 (/Хне Н-
____________________________4НЮ_______ I
1 I
4С6Н5СН2СН2СНпОН 4 А 1(0Н)3 Д- L1OH СНг-СН2
Дигндрокоричпый спирт
ется, по-видимому, полимерным аналогом приведенного здесь для
простоты циклического мономера. Хохштейн и Браун 1311 показа-
ли, что коричный альдегид восстанавливается в обычный продукт —
коричный спирт— с 90%-ным выходом обратной реакцией — при-
бавлением по каплям раствора Л. а. (10%)-ный избыток) к эфирному
раствору коричного альдегида при —10°.
Иоргенсеи и сотр. 132] показали, что аллиловый спирт можно
восстановить Л. а., минуя стадию образования насыщенного спир-
175
та, и получить циклопропан. Например, коричная кислота, слож-
ный эфир, альдегид или кетон при восстановлении 100%-ным избыт-
ком Л. а, в кипящем ТГФ или диметоксиэтане дает 45—80% феиил-
циклопропана. Реакция является хорошим дополнением к синтезу
циклопропанов по Симмонсу — Смиту.
N2, кипячение
3 час
CC-CHCOoH + LiAIHi + ТГФ с
С Н / 57-61%
0,09 моля 0,135 моля 200 ,чл
Группой авторов [33] было показано, что реакция применима
к пропаргиловым спиртам; они использовали ее при синтезе вита-
мина А; другие примеры были приведены Джонсом и сотр. [341.
Фенилпропиоловая кислота (1) сначала восстанавливается в фенил-
пропаргиловый спирт, который дает комплекс, разлагающийся во-
дой с образованием коричного спирта. В примере 2 диацетиленовый
гликоль (а) восстанавливается в диэтиленовый гликоль (б), содер-
LiA!H,t LiAIH4
1. с6н5с = ссогн —С6Н5С^ССН2ОН ——> С6Н5СН = СНСН2ОН
Фенилпропиоловая Фенилпропаргиловый Коричный спирт
кислота спирт
ылп-ц
2. (СвН5)оСС^СС^СС(С6Н5)г-----> (С6Н5)2ССН = СНСН-СНС(С6Н5)2
он он он он
(а) (б)
НА--------О
/^\\\ С (С,
(С6Н5)гС с с /
I \\\г/
О-АН Г
(в) А = — Al ^OR)3 Li
жащий две двойные транс-связи. Возможный промежуточный гли-
колят (в) циклизуется транс-присоединением к тройным связям
с образованием связей углерод — алюминий (г), при расщеплении
которых вторая пара водородов вводится в транс-положение отно-
сительно водородов, введенных на первой стадии. Моноацетилено-
вый гликоль (д) восстанавливается по-другому, с отщеплением обеих
гидроксильных групп и образованием транс, транс-дрена (и). Пред-
полагаемый (уравнение 3) механизм включает внутримолекулярное
176
трднс-присоединеиие гликолята (ж), дающее промежуточное сое-
динение (з), со стереохимической точки зрения благоприятствующее
транс-отщеплению первой пары атомов водорода, тогда как вторая
LiAlH4
3. С6НД НС.ЩСНС,,Н5-------► СвН5СН=СНСН=СНСвНз
I 1
он он
пара присоединяется по месту разрыва связей углерод — алюминий.
Эта разновидность реакции восстановления имеет большое значение
в синтезе природных полиенов.
a, fl-Ненасыщенные сложные эфиры. Восстановление os,^-нена-
сыщенного сложного эфира типа (1) bos, ^-ненасыщенный спирт (2)
обычно осложняется восстановлением сопряженной двойной связи.
Литго и сотр. [35] показали, что эту нежелательную побочную ре-
акцию можно подавить, вводя 1 же этанола. По-видимому, действую-
щим реагентом при этом является лнтийалюминиймоноэтоксигидрид
1лА1Н3(ОСгНД
СО2СН3
1яА1Н4-СгН5ОН
О)
СНгОН
Синтез этиленимина. Японскими исследователями [36] недавно
было показано, что восстановление дибензилкетоксима (1) Л. а. дает
177
quc-2-бензил-З-фенилэтиленИхМИН (2) с хорошим выходом и лишь
небольшое количество продукта полного восстановления (3).
CtiH-,CH, С8н5
V -— с/
C(3HsCH.,CCI-I.,C6H5-pLiAlH4 н-ТГФ —> Н' N7 'Н|-
'! моля 80 ,ir.i [
кон н
(1) 0,05 моля
(2) 75%
+ СеН5СНХНСНоС6Н5
|
КН2
(3) 7Т6%
1. F i п 11 о 1 t Л. Е, Bond А. С,, Jr., Schlesinger И. 1,, J. Am.
Chem. Soc., 69, 1199 (1947).
la. Alctal Hydrides Technical Bulletin No. 401, October, 1963; Solms U., C h i-
in i a , 5, 25 (1951); Bro w n \V. G., Org. Reactions, 6, 649 (1951); Gay-
lord N. G., «Reduction with Complex Metal Hydrides», Intersciencc, N. Y.,
1956; Gaylord N. G., J. Chem. Ed., 34, 367 (1957); H о r in a u n H-,
«Newer Methods in Preparative Organic Chemistry», 2, 213 (1963).
2. N у s t г о m R. 1?., Brown W. G., J. Am. Chem. Soc., 69, 1197 (1947).
3. H о c li s t e i n F. A., J. Am. Chem. Soc., 71, 305 (1949).
4. S t e i n h a r d I С. K., private communication.
5. M i if о vic V. M., Mihail о у i с M. L. J., J. Org. Chem., 18, 1190
(1953).
6. Г e й лор д 14., «Восстановление комплексными гидридами металлов»,
ИЛ, М., 1959.
7. Ganno n W. F.T House Н. О., Org. Syn., 40, 14 (I960).
8. N у s t г о rn R. F. Bro w n W., G., J. Am. Chem. Soc., 69, 2548 (1947).
9. Unpublished observation cited in Brown’s review in Organic Reactions, VI,
469 (1951).
10. Moffett R. B., Org. Svn., Coll. Vol., 4, 834 (|963).
11. Sr oo gC. E., Woodburn H. M., Org. Syn., Coll. Vol., 4, 271 (1963).
12. Fieser L. F., Ourisson G., ,1. Am. Chem. Soc., 75, 4404 (1953).
13. Bucht a E., toe w G., Ann., 597, 123 (1955).
14. Hen best H. B., W i 1 s о n R. A. L. J. Chem. Soc. 1956, 3289.
15. Furst A., Platlner Pl. A., Helv. Chim. Acla, 32, 275 (1949).
16. Platt n с г PI. A., Hcusscr H;, К и 1 k а г n i А. В., Helv. Chim.
Acta, 31, 1822, 1885 (1948); 32, 265, 1070 (1949); P 1 a t t n e r Pl. A., H e и fi-
sc r H;, Fcurer M., Helv. Chim. Acla, 32, 587 (1949).
17. T r c v о у L. W., Bro w n AV. G., J. Am. Chem. Soc., 71, 1675 (1949).
18. Johnson J. E., Blizzard R, H., C a r h a r t H. W., J. Am. Chem.
Soc., 70, 3664 (1948).
19. Brcslo w R., D о w d D., J. Am. Chem. Soc., 85, 272) (1963).
20. Gaylor d N. G., J. Am. Chem. Soc., 76, 285 (1954).
21. S c h m i d 11., Karrer P., Helv. Chim. Acta, 32, 1371 (1949).
22. Zorbacli W. W., T i о С, O., J. Org. Chem., 26, 3513 (1961),
23. Moffett R. B., Org. Syn., Coll. Vol., 4, 355 (1963).
24. W ilson С. V., Stenberg J. F., Org. Syn., Coll. Vol., 4, 664 (1963).
25, Cope A. C., Ciganek E,, Org. Syn., Coll. Vol., 4, 339 (1963).
26. W e у g a n d F., M i t g a и R., (Paper VI), Chem. Ber., 88, 301 (1935).
27. Bro w n H. C., Ts и k aniot о A., J. Am. Chem. Soc., 83, 4549 (1961).
28. S t a a b H. A., В ra uni ing H., Ann., 654, 119 (1962).
29. Claus C. J., Al urgent h a и .1. L., Jr., J. Am, Chem. Soc., 73, 5005
(1951); пат. США 2786872 (1957); С. A., 51, 12130 (1957).
178
30. N v s t г о tn R. F., Bro w n W. G.T J. Am. Chem. Soc., 69, 2548 (1947);
70,’3738 (1948); G i 1 s d о г f R. T,, Nord F. F., J. Org. Chem., 15, 807
(1950).
31. 14 о c 11 s t c i n F. A., Bro w n W-, G., .1. Am. Chem. Soc,, 70, 3484 (1948).
32, J о r g e n s e n M. .1.. Friend
Jorgensen M. J., T h a c h e
33. Attenb urrow J., C a m e r
Evans R. M., Hems B. A.,
Chem. Soc., 1952, 1094.
34. В a t c s E, B., J ones E. R.
1954, 1854,
35. Davidson R. S., Giinther
1 h e r S. M., Ly Ihgoc B., J.
36, К о t e г а К , К i t a 11 о п о 1< i
also Kitahonoki I\. cl al.,
A, \V., J. Am, Chem. Soc,, 87, 1815 (1965);
r A. F,, procedure submitted to Org. Syn.
о n A. F. B., Chapman J. Id.,
J a ii s c n A. B. A., W a I ker T., J.
H., \V h i t i n g M, C., J. Chem. Soc,,
W. H, FI,, Waddington - F e a-
Chetn. Soc., 1964, 4907.
K-, procedure submitted to Org- Syn.; see
Tetrahedron Letters, 1059 (1965).
ЛИТИЯ АЛЮМОГИДРИД — АЛЮМИНИЯ ХЛОРИД, см. так-
же Алюминия гидрид. Мол. вес: LiА1Н4 37,95; A1CL 133,35. Обзоры
[1а, 161.
Природа смешанных гидридов. В зависимости от молярных
соотношений двух компонентов возможно образование различных
соединений:
LiAIH,-i-AICI3 LiCl-]-2A1FLC1 (1:1)
LiAlH4-у 3AlCI;i LiCl --4A1HCL (1:3)
3IJA1H4A1CI3 — 3LiCl -|-4A1H3 (3:1)
Реакции проводят в эфире, так как ТГФ расщепляется смешанными
гидридами до «-бутанола 111; смеси состава 1 : 1, 1 : 3 и 3 : 1 дают
растворы восстанавливающего агента или агентов. Хотя хлорид
лития нерастворим в эфире, образующийся в приведенных выше
уравнениях хлорид не выпадает в осадок и, по-видимому, присут-
ствует в виде комплекса. Генерируемый в результате третьей реак-
ции гидрид алюминия, по-видимому, также стабилизирован комп-
лексообразованием. Измерения электропроводности указывают на
присутствие ионных частиц типа ALCl't, А1Н4“ и /\1,Н,СЦА1Нр.
Хотя использовались различные отношения гидрида к галогениду,
что, вероятно, приводило к изменению состава и концентрации ак-
тивных частиц, все смешанные гидриды оказались несколько менее
активными восстановителями, чем сам LiAlH4; однако они обладали
большей тенденцией к восстановительному расщеплению, анало-
гичному гидрированию водородом над катализатором.
Восстановительное расщепление,
гидрида впервые описано Брауном
Использование смешанного
[21, исследовавшим действие
179
смеси 1 : 1,5 на енолацетат холестенона (1). Им была получена смесь
продуктов, из которой представлял интерес лишь один продукт—
Д4-холестен (2), образующийся в результате восстановительного
отщепления и гидрирования. Восстановление холестенона (3) сме-
шанным гидридом (1 : 2) даете хорошим выходом Д4-холестеи (2) [3].
В противоположность этому, при восстановлении енолацетата хо-
лестенона (1) LiAlH4 получают смесь пяти продуктов, в которых
при атоме С3 сохраняются кислородсодержащие группы [41.
Смешанный гидрид используют преимущественно для восста-
новления диар ил кетонов, ал кил арил кетонов и некоторых аромати-
ческих спиртов в соответствующие углеводороды [5]. Самые высокие
выходы получают при добавлении к смеси ЫА1Н4—А1С13
(1 : 1) эквимолярной смеси кетона и хлористого алюминия в
эфире.
Метод Маркера раскрытия F-кольца стероида сапогенина с об-
разованием дигидросапогенина гидрированием в сильно кислой
среде неприменим к диосгенину, так как двойная связь в положении
5,6 является первым объектом атаки. Доукас и Фонтэн [6] показа-
ли, что насыщенные и ненасыщенные сапогенины можно превратить
в дигидропроизводные добавлением твердого LiAlH4 в эфирный
раствор стероида, насыщенный хлористым водородом. Другие кис-
лоты оказались неэффективными. Последующее изучение реакций
восстановления смешанными гидридами позволило предположить,
что активным реагентом при превращении кеталя (1) в эфир (2) яв-
ляется смешанный гидрид; это стимулировало дальнейшие исследо-
вания. Петтит и Боуэр 17] установили, что сочетание LiAlH4—А1С13
действительно эффективно. Например, 6,4 г хлорида алюминия в
50 мл эфира добавляют к охлаждаемой льдом смеси 0,45 г LiAlH4
и раствора 0,5 г тигогенина в 75 мл эфира.
Илиел и сотр. [8], изучавшие некоторые ацетали н кетали, по-
казали, что восстановление смешанным гидридом в эфире является
общей реакцией. Наилучшие выходы получены при отношении
1лЛ1Н4 к А1С13, равном 1:4, и при 100%-ном избытке реагента.
180
Типичные примеры приведены ниже:
Li А 1Н„ - А1СЦ „ ,, , ,
С6Н5СН (ОСН3)2———> с6н/н2осн3
88%
СД LiД1Н4-А)С1, С6Н5ч
>С (ОС,НГТ,--— — /СН — 0СЛ-Т5
СН/ 81 % сн/
Разработана подробная методика восстановительного расщепления
этиленкеталя циклогексанона в циклогексил оксиэтанол [9]. Смесь
1,76 моля хлорида алюминия и 500 мл эфира перемешивают при
охлаждении на бане со льдом и солью до образования светло-серого
раствора. В сухой камере распыляют 0,44 моля LiAlH4, помещают
в колбу Эрленмейера, охлаждают на бане со льдом и заливают 100 мл
эфира. Раствор перемешивают до насыщения его ЫА1Н4 и через
капельную воронку переносят в реакционную колбу. Перемеши-
вают еще в течение часа. Раствор 0,88 моля этиленкеталя цикло-
гексанона в 200 мл эфира добавляют со скоростью, обеспечивающей
слабое кипение. Убирают баню со льдом и кипятят смесь 3 час. Из-
быток LiAlH4 разлагают, добавляя по каплям 12 мл воды, затем
вводят разбавленную серную кислоту, смесь обрабатывают и про-
дукт перегоняют.
LiAlH/О, 5)-А1С13(2)
94%
аО-СН2СНгОН
Н
Енамины расщепляют до алкенов кипячением в эфире в течение
5—24 час с эквимолярной смесью LiА1Н4 и А1С13, как это показано
для енамина, полученного из пирролидина и циклопентанона [101:
L1A1H4-A1C13%
83%
Восстановление кетонов. Уилер и Матеос [11] показали, что при
восстановлении 3-холестанона смешанным гидридом в качестве ос-
новного продукта получается чистый холестенол-Зр с экваториаль-
ной гидроксильной группой, тогда как при восстановлении одним
LiAlH4 образуется смесь Зр- и За-эпимеров в соотношении прибли-
зительно 9:1. Илиел и сотр. [12, 13] при попытках получить чистый
трдас-4-щрет-бутилциклогексанол (4) показали, что восстановле-
ние 4-трет-бутилциклогексанона (1) смешанным гидридом при
обычных условиях кинетического контроля дает транс- и ^ис-спирты
181
в отношении 80 : 20, Однако они отметили, что промежуточные
,4’0.,,-производные (2) и (3) обладают двумя объемными группами
и потому идеальны для аксиально —> экваториальной изомериза-
ции, По стандартизованной методике [131 смешанный гидрид по-
лучают из 0,5 моля АЮз и 0,145 моля LiAlHj в эфире и затем до-
бавляют 0,5 моля кетона (11 в эфире. После окончания реакции вво-
pasg-Hzso4 он
—(снз)зг^4—
(4)
дят 10 мл mpcm-бутанола, который взаимодействует с избытком
LiAlH4.Затем добавляют 3 г кетона (1) в качестве катализатора
равновесия и кипятят смесь 4 час. Последующая обработка дает
вещество, содержащее поданным газовой хроматографии 96% транс-
спирта, 0,8% цпс-спирта и 3,2% кетона. При кристаллизации об-
разуется чистый транс-спирт с т. пл. 75—78° и выходом 78%.
Восстановление эпоксидов. Илиел и сотр. [14] показали, что
восстановительное расщепление окиси трифенилэтилена (1) порази-
тельно изменяется при использовании смешанных гидридов. В при-
сутствии только LiAlH.j атака идет по наименее замещенному атому
углерода с образованием карбинола (2), Смешанный гидрид с вы-
соким содержанием хлорида алюминия (1 : 4) дает трифенил метил -
карбинол (3), образующийся в результате перегруппировки соеди-
нения (1) в трифенилацетальдегид под действием А1С% или BF:i.
Смешанный гидрид с низким содержанием А1С13 (1 : 0,33) дает фе-
нилбеизгидрил карбинол (4),
LiAJH,
-----------------(С«Н5), СНСН,СЙНЭ
ОН
(2)
I.iД1Н,:AJCL (1:4)
----------:----> (СеН5)3 ССНО —> (Сен,)з ссн2он
(3)
I.i Л1Н ASCI., (1:0 331
---------------—(С3Н5).г снснсйн5
он
(4}
/°\
(С6Н5)2 с—СНС6НД.
(1)
182
Другие реакции восстановления. Смешанный гидрид состава
1 : 1 превосходит алюмогидрид лития как агент для восстановле-
ния нитрилов в амины [15]. В отличие от LiAlH4 смешанный реа-
гент не восстанавливает нитрогруппы; например, л-нитробензаль-
дегид восстанавливается смешанным гидридом в п-нитробензиловый
спирт с выходом 75%. Под действием LiAlHj отщепляются атомы
галогенов, в присутствии же хлорида алюминия восстановление
замедляется или вовсе прекращается. Например, р-бромпропио-
нилхлорид восстанавливается смешанным гидридом (1 : 1) в 3-бром-
иропанол-I с выходом 77%; в отсутствие А1С1Ч выход понижается
до 44% [15, 16].
При восстановлении оксима LiА1Н4 наряду с первичным амином
часто образуется и вторичный амин. При использовании смешан-
ного гидрида количество образующегося вторичного амина увели-
чивается [17].
I. В a i 1 е у W. J., М а г k t s с h с f f е 1 Г., J. Org. Chem., 25, 1797 (I960),
la. Remick M. N., «The Mixed Hydrides», Metal Hydrides, Inc. (1959).
16. E 1 i e I E. L., Record of Chemical Progress, 22, 129 (1961).
2. Brown B. R., J. Chem. Soc., 1952, 2756; For other examples of hydrogeno-
lysis sec Birch A. J., S 1 aytor M., Chem. Ind., 1 956, 1524.
3. В г о о m e J., В r own B. R., R о b er 1 s A., W h i t e A. M. S., J.
Chem. Soc., 1960, 1406.
4. D a u b on W. G., Eastham J. F., J. Am. Chem. Soc., 73, 3260 (1951).
5. N v s t г о m R. F., Berger C. R. A., J. Am. Chem. Soc., 80, 2896
(1958); Brown B. R., White A. M. S-, J- Chem. Soc., 1957, 3755;
Blackwell J., II i с к i n b о t t о m W. J., ibid., 1961, 1405.
6. D о u к a s H. M., Fontaine T. D., J. Am. Chem. Soc., 73, 5917 (1951).
7. P e t t i t G. R., Bowyer W. J-, J. Org. Chem., 25, 84 (I960).
8. E 1 i e 1 E. L., В a d d i n g V. G., R e r i с к M. N., J. Am. Chem.
Soc., 84, 2371 (1962).
9. Daignault R. A., E 1 i e 1 E. L., procedure submitted io Organic
Syntheses.
10. Lewis J. W., Lynch P. P., Proc. Chem. Soc., 1963, 19.
II. Wheeler О. H., M a t cos J. L., Can. J. Chem., 36, 1431 (1958).
12. E 1 1 e 1 E. L., R c r i с к M. N., J. Am. Chem. Soc., 82, 1367 (i960).
13. E 1 i e 1 E. L, M a r t i n R. J. L., N as i p и r i D., procedure submitted
to Organic Syntheses.
14. E 1 i e 1 E. L., Delmonte D. W., J. Am. Chem. Soc., 80, 1744 (1958);
E 1 i e I E. L., R e r i с к M. N., ibid., 82, 1362 (I960); R e r i с к M. N.,
FI i e 1 E. L., ibid., 84, 2356 (1962).
15. N у stro m R. к'., J. Am. Chem. Soc., 77, 2544 (1955).
16. N у s t г о rn R. I?., J. Am. Chem. Soc., 81, 610 (1959).
17. R e r i с к AL NL ct al., Tetrahedron Letters, 629 (1963).
ЛИТИЯ АЛЮМОГИДРИД — ПИРИДИН [11. для восстанов-
ления карбонильных соединений LiAIHL Лэнсбури |2| использовал
в качестве растворителя пиридин, который обладает большей соль-
ватирующей способностью, чем ТГФ. Небольшие количества ве-
щества восстанавливают добавлением тоикопзмельченного гидрида
к раствору субстрата в пиридине, перемешиванием раствора без
охлаждения в течение нескольких минут до окончания слабой эк-
зотермической реакции, добавлением метанола для разложения
183
избытка реагента, выливанием раствора в 5%-ную соляную кислоту
и экстракцией продукта реакции эфиром. Использование пиридина
в качестве растворителя иногда повышает реакционную способность
гидрида металла. Так, бензпинаколин восстанавливается LiAlH4
в эфире в соответствующий спирт, а в пиридиновом растворе он рас-
щепляется до трифенилметиллития и бензилового спирта. Присут-
ствие трифенил метильных карбаниснов в растворе доказано карбо-
низацией и реакцией с беизилхлор идом. Обработка кислотой дает
О
II LiAlH4 - +
(СвН,)3 СССаН5 —-(С6НЭ)3 CLi -РСвЩСНЮН
^У ж
_________________1и -
I со3 н+ I т , „
V /' Т-Т г[ т L1AIH4 —Ру
(С6Н5)3ССО2Н j<-.ntCH2ci
(С6Н5)3ССНаС6Н5 (Сбн5)3сн
трифеиилметан; под действием LiAlH4 в пиридине эта реакция об-
ращается. Еще более кислый углеводород — флуорен легко метал-
лируется гидридом в пиридине. Этим простым методом с последую-
щей карбонизацией из углеводородов в течение 1 час можно полу-
чить 0,5 г трифенилуксусной или 9-флуоренкарбоновой кислоты.
Используя пиридин в качестве растворителя, Лэисбури [3] по-
казал, что при растворении порошкообразного Li Л1Н4 (0,5 г) в пи-
ридине (50 мл) и выдерживании оранжевого раствора в закрытом
сосуде в течение не менее 24 час получают раствор нового более мяг-
кого восстанавливающего агента. По данным ЯМР и ИКС получен-
ное вещество не содержит связей А1—Н и имеет связанные с алюми-
нием 1,2- и 1,4-дигидропиридиновые группы. Это соединение рас-
L1AIH4 + 4
сматривается как тещракис-(И-дигидропиридил)-алюминат. Под
действием такого длительно стоявшего раствора эффективно восста-
навливаются кетоны, однако карбоновая кислота или сложный эфир
при этом не восстанавливаются. Этот слабый восстановитель можно
использовать для селективного восстановления кетокислоты в со-
ответствующую оксикислоту. Показано, что диарилкетоны более
реакционноспособны по отношению к щетра/шс-(И-дигидропири-
дил)-алюминату, чем диалкил- или арилалкилкетоны. Этот вывод
противоречит данным Брауна, показавшего, что при восстановлении
184
боргидридом натрия в изопропиловом спирте ацетон реакционно-
способнее ацетофенона, а последний реакционноспособнее бензо-
фенона.
I. Contributed by Lansbury Р. Т., University of Buffalo.
2. Lansbury P. T., J. Am. Chem. Soc., 83, 429 (1961); Lansbury P. T.,
Thedford R., J. Org. Chem., 27, 2383 (1962).
3. L a n s b u г v P. T„Peterson J. О., J. Am. Chem. Soc., 83, 3537 (1961);
85, 2236 (1963).
ЛИТИЯ АЛЮМОГИДРИД — ЭФИРАТ ТРИФТОРИДА БОРА.
Петтит [1] показал, что под действием этой смеси сложные эфиры
восстанавливаются в соответствующие простые эфиры. Например,
Зр-ацетоксихолестан дает ЗР-этоксихолестан. Аналогично 5а-лано-
стерил-ЗР-формиат при восстановлении смесью боргидрид натрия —
эфир ат трехфтористого бора дает Зр-метокси-5а-л аностан. Раствор
сложного эфира в эфирате трехфтористого бора добавляют к охлаж-
денной суспензии нейтрального гидрида в эфире. Лактоны различ-
ных типов гладко восстанавливаются LiAlH4—BF3- (С2Н5)2О до цик-
лических эфиров. Таким способом были восстановлены у-, б- и е-лак-
тоны (выходы приведены под формулами).
1. Р е t t i t G. R., К a s t u r i T. R., J. Org. Chem., 25, 875 (1960); Pettit
G. R., G h a t a k U. R.,Green В., К a s t u r i T. R.,Piatak D. M.,
ibid., 26, 1685 (1961).
185
ЛИТИЯ БОРГИДРИД, LiBH4. Мол. вес 21,79, т. пл. 284° (с разд.)
исключительно гигроскопичен, растворим в низших первичных
аминах и эфирах (диэтиловый эфир, 4 г/100 а; тетрагидрофуран,
21 г/100 г); в низших спиртах (при этом происходит реакции), в воде
с медленным разложением:
LiBH4+2HsO—* 1лВ0г-j-4Нг +АН
Л. б. можно получить в растворе реакцией боргидрнда натрия
с хлоридом лития в абсолютном этаноле ! 1 ], изопропилампне [21 или
диглиме |31. Л. б. более активный восстановитель, чем боргид-
NaBH4+LiCl —► LiBH4 + NaCl
рид натрия, и менее активный, чем алюмогидрид лития. Например,
он восстанавливает альдегиды, кетоны, ацил хлор иды, окиси, слож-
ные эфиры и лактоны, по не восстанавливает карбоновые кислоты,
нитрилы или нитросоединения 141. Главным преимуществом Л. б.
по сравнению с боргидридом натрия является его высокая раство-
римость в эфирах.
1. К о 1 1 о n i t s с h J,, Fuchs О., Gabor V., Nature, 173, 125 (1954).
{’.Schlesinger H. L.Brow n H. C., Hvde E. K.., J. Am. Chem. Soc.,
75, 209 (1953).
3. Bro w n bl. C., Mead E. J., Subba Rao В. C., J. Am. Chem. Soc.,
77, 6209 (1955).
4. Brown H. C., «Hydroboration», Benjamin, 1962, p. 245.
ЛИТИЯ БРОМИД, LiBr. Мол. вес 86,86.
Под действием Л. б. в эфире при 0° из диазоалкена (1) выде-
ляется азот и образуются эквивалентные количества (по 2,5%)
метилтрициклогексана (2) и 1-метил-Д1,3-циклогексадиена (3) [1].
По-видимому, (2) образуется через промежуточный карбен. Большой
избыток бромистого лития в кипящем ацетонитриле медленно
(12 час) превращает перхлорат хннолиния (4) в основание (5) [2].
сн(сн3)2
(5)
186
1. С 1 о s s G. L., Larrabee R, B,, Tetrahedron Letters, 287 (1965).
2. Good w i n S., Fl orning E. C., J. Am. Chem. Soc., 81, 1908 (1959);
Huffman J. W,, Browder L. E., J. Org. Chem., 29, 2598 (1964).
ЛИТИЯ БРОМИД — ЭФИРАТ ТРИФТОРИДА БОРА. Алифа-
тические простые эфиры можно расщепить под действием реагента
в уксусном ангидриде при комнатной температуре в течение 30 час
[1]. Например, мстоксициклогексан дает смесь (7:1) ацетокси-
циклогексана и циклогексена. Насыщенные стероидные эфиры
расщепляются в этих условиях с образованием смеси ацетатов и не-
предельных производных; холестерилмстиловый эфир дает при-
близительно равные количества холестерил ацетата и холестерил-
бромнда. Однако, по данным Нараянана [2|, присутствие гало-
генида лития не обязательно и часто даже вредно. Например, об-
работка холестерилметплового эфира эфиратом трифторида бора
и уксусным ангидридом в эфире при 0° (14 час) дает холестерил-
ацетат с выходом 93%.
1. Y о и s s е f у е h R. D., М azur Y., Tetrahedron Letters, 1287 (1962).
2. N а г а у а п а п С. R., Iyer К. N., Tetrahedron Letters, 759 (I9G4).
ЛИТИЯ ш/п<с-(////2Ц77ыБУК)КСИ)-АЛЮМО ГИДРИД,
LiAlH 10C(CH:t);t];t. Мол. вес 254,30, возгоняется в вакууме при 280°,
растворимость при 25“ (в а/100 г): диглим 41, ТГФ 36, эфир 2,
1,2-диметоксиэтан 4, mpem-бутанол 0. Содержание активного
водорода на грамм реагента составляет лишь 3,7% по сравнению
с LiAlH4.
Браун и сотр. 11] показали, что L1А1Н4 в эфире реагирует с 4 мо-
лями метанола, этанола или изопропанола и лишь с 3 молями трет-
бутанола. Добавление по каплям 1 моля //грт-бутанола при ком-
натной температуре и перемешивании к раствору 0,31 моля LiAlH4
в эфире приводит к образованию белого осадка Л. б. с почти ко-
личественным выходом. Показано, что новый реагент является
более мягким восстановителем, чем LiAlH4. Л. б. восстанавливает
альдегиды, кетоны и хлорангидриды в диэтиловом эфире или диг-
лиме при 0:;, однако не реагирует со сложными эфирами и нит-
рилами.
Браун и Рао [2] показали, что добавление Л. б. в диглиме
к раствору хлор ан гидр ида при —78“ представляет удобный метод
синтеза альдегидов. Хлорангидриды ароматических кислот с мета-
и /ш/ш-заместителями образуют альдегиды с выходами 60—90%.
При этом не затрагиваются такие заместители, как нитро-, цпан-
и карбэтоксигруппы; opmo-заместители снижают выходы; при —40°
выходы на 5—10% ниже, чем при —78е1.
Вейссмаи и Браун [3] показали, что в противоположность ал-
киловым сложным эфирам, которые инертны по отношению к реа-
генту, фениловые эфиры алифатических и алициклических кислот
реагируют с образованием альдегидов. Однако, так как фениловые
187
эфиры обычно получают из хлорангидридов, лучше восстанавливать
хлор ангидрид непосредственно.
Уилер и Матеос [41 в предварительном сообщении указали, что
восстановление холестанона-Зр Л. б. или смесью LiAlH4 — А1С13
дает 99% экваториального холестанола-3, тогда как восстановле-
ние алюмогидридом лития дает лишь 88—91% Зр-ола [5, 61.
Файкош [7] изучил восстановление некоторых стероидных
кетонов Л. б., однако не проверил результаты Уилера и Матеоса.
Восстановление Зр-формилокси-Д5-аидростенона-17 (1) в 17р-ол (2)
с высоким выходом и высокой степенью чистоты указывает на
исключительную стереоспецифичность реакции, а также на то, что
активная сложноэфирная группа в реакции не затрагивается.
Восстановление 2а-бромхолестанона-3 (3) боргидридом натрия
дает смесь, содержащую значительные количества обоих эпимерных
спиртов; восстановление Л. б. дает лишь Зр-ол (4); это наблюдение
позже было подтверждено другими работами [8]. Файкош сравнил
скорости восстановления кетонов 5а- и Ду-рядов при комнатной
температуре и установил, что они изменяются в ряду: 3-она>
>17-она>>Д4-3-она.
188
Он также показал возможность селективного восстановления
более реакционноспособной группы диона (5). Для восстановления
менее реакционноспособной из двух кетогрупп диона (7) чешские
исследователи [9] защищали 3-кетогруппу превращением в пирро-
лидиновый енамин (8), восстанавливали 17-кетогруппу и удаляли
защитную группировку. Следует отметить, что енамины восстанав-
ливаются LiAlHa — А1С13 в эфире.
Тамм [10] использовал реагент для восстановления карбониль-
ных групп в ряду буфадиенолидов. Например, восстановление
кетона (11) боргидридом натрия неудовлетворительно из-за ча-
стичной атаки ненасыщенного лактонного кольца; выход соеди-
нения (12) составляем 40—50%. Восстановление Л. б. в ТГФ
при 0° осуществляется быстро (15 мин) без затрагивания лактонного
кольца с удвоенным выходом соединения (12) (детали методики
не опубликованы).
Контрольные опыты по восстановлению микроколичеств и анализ
продуктов методом бумажной хроматографии показали, что кар-
189
денолидное кольцо З-О-ацетилстрофантидина (13) также устой-
чиво к атаке, что экранированная альдегидная группа восстанав-
ливается при 0° только за 1 час и что за это время ацетильная группа
не затрагивается. В другом случае формилоксигруппа устойчива
при О’, но восстанавливается при 20°. 14,15-Эпоксигруппа марино-
буфагпна восстанавливается NaBHd, а Л. б. эту группу не восста-
навливает.
20-Кетостероиды восстанавливаются реагентом главным образом
в 20[3-олы [11]. Суспензию 0,4 моля Л. б. в 750 мл ТГФ переме-
шивают при кипячении в атмосфере азота, охлаждают до 2°, добав-
ляют 0,2 моля ацетата прегненолона (15) и 50 мл растворителя.
Смесь перемешивают при 0—5 е в течение 1,5 час и затем по каплям
в течение 15 — 20 мин добавляют при энергичном перемешивании
и охлаждении до температуры ниже 10е раствор 100 г сульфата ам-
мония в 150 мл воды (бурноевыделение водорода). Кристаллизация
продукта из ацетона дает чистый 3(3-ацетокси-20(3-окси-А-прегнен
с выходом 75—79%. В данном случае использовали 100%-ный
избыток восстанавливающего агента, однако необходимость
этого не доказана. Напротив, экспериментами Бергсталера и Нор-
дина [ 121 было установлено обратное. После добавления по каплям
холодного (0е) раствора 0,039 моля А4-холестенона-3 (17) в 200 мл
смеси эфир — бензол (8 : 1) к суспензии 0,05 моля Л. б. (1,3-к рат-
ный избыток) в смеси эфир — диглим при температуре от —40
до —50е и при перемешивании смесь оставляли на ночь при О’
190
и затем гидролизовали обработкой льдом, 5 н. NaOH и сегнетовой
солью. Отгонка промытого и высушенного эфирного экстракта
и кристаллизация из этилацетата дает 13 г (87%) Д^холестенола-Зр
с т. пл. 126—129’. После перекристаллизации был получен чистый
продукт с т. пл. 131 —132°.
сн3
(17)
(18)
При стереоспецифическом восстановлении 1131 ацетата 6-кето-
тигогенина (19) в 6[3-ол (20) к перемешиваемому раствору 10 г
соединения (19) в 400 мл ТГФ при комнатной температуре добав-
ляют 32 г Л. б. (6-кратный избыток). Через 60 час смесь выливают
в 3 л 5%-ной уксусной кислоты для гидролиза комплекса и раз-
ложения избытка реагента.
1лД1Н(ОВи)3
Здерик н Ириарт [14] изучали восстановление бмс-метилендиок-
сипроизводного преднизона (или преднизона-БМД), предполагая,
что этот объемный реагент будет селективно восстанавливать кар-
бонильную группу при атоме С3. Однако вопреки ожиданиям
избытком Л. б. при комнатной температуре восстанавливается
только экранированная 11-кетогруппа и с хорошим выходом об-
разуется преднизолон-БМД.
Преднизон-БЛ/1Д
Преднизолон-БМД
191
Файкош и Йоска [15] показали, что восстановление 16а-бром-17-
кетостероида (21) гидридами металлов в полярных растворителях
сопровождается значительной инверсией при атоме СХе, а восста-
новление Л. б. в неполярных растворителях дает 17-эпимерные
16а-бромсппрты (22) и (23).
Айрланд и Маршалл [161 нашли способ превращения аддукта
(1) бутадиена и 1,4-бензохинона в Д0-окталон-1 (5). Восстановление
ендионовой системы аддукта (1) цинком в уксусной кислоте до сое-
динения (2) было осуществлено Альдером и Штерном в 1933 г.;
следующая задача состояла в селективном восстановлении одной
из двух эквивалентных карбонильных групп. Это достигается при
(3) (4) (5)
восстановлении рассчитанным количеством LiAlHd, но с очень низ-
ким выходом. Восстановление теоретическим количеством Л. б.
в разбавленном растворе в ТГФ протекает удовлетворительно и дает
(3), а восстановление по Вольфу — Кижнеру с последующим окис-
лением хромовой кислотой дает Д6-тра«с-окталон-1 (5) с общим
выходом в расчете на бензохинон 52%.
В противоположность обычному восстановлению 3-кетостероидов
в 3[3-оксистероиды, Д5а0)-эстрепдион-3,17 (6) восстанавливается
Л. б. в За- и Зр-окси производные с соотношением 15 : 1 [17]. Энгел
и др. [18] использовали реагент для восстановления Па-окси-Д4-
прегнендиона-3,20 в смесь Д4-прегнен-Зр, 17а, 20-триолов, эпимер-
192
ных при атоме С.,о. Затем 17,20-гликоль расщепляли тетраацетатом
свинца до 17-кетона.
1. Brown Ы. С., М с F а г 1 i n R. F., J. Am. Chem. Soc., 80, 5372 (1958);
Brown Н, С., Shoaf С. J., ibid., 86, 1079 (1964).
2. Brown Н. С., S и b b a Rao В. С., J. Ат. Chem. Soc., 80, 5377 (1958).
3. Weissman P. M., Brow п H. C., J. Org. Chem., 31, 283 (1966).
4. Wheeler O. hl., Mateos J. L., Chem. Ind., 1957, 395.
5. N а с с H. R., O’C о n п о r G. L., J. Am. Chem. Soc., 73, 5824 (1951).
6. Shoppee C. W., Summers G. FI. R., J. Chem. Soc., 1950, 687.
7. Fajkos J., Coll. Czech., 24, 2284 (1959).
8. Kwok R., W о 1 f I M. E., J. Org. Chem., 28, 423 (19G3).
9. J о s k a J., FajkoS J., Sorm F., Coll. Czech., 26, 1646 (19GI).
10. Tamm Ch., Helv. Chim. Acta, 43, 338 (1960).
11. Heusler К., К a 1 v о cl a J., Wieland P., W e 11 s t e i n A.,
Helv. Chim. Acta, 44, 179 (1961); Heusler !<., W ieland P., M e у s-
t r e Ch., procedure submitted to Organic Syntheses.
12. Burgstahler A. W., Nor d i n I. C., J. Am. Chem. Soc., 83, 198
(1961).
13. Bowers A., DenotE., Cuellar Ibanez L., Cabezas Ma E.,
R i n g о 1 d H. J., J. Org. Chem., 27, 1862 (1962).
14. Z d e r i c J. A., Iriarte J., J. Org. Chern., 27, 1756 (1962).
15. F a j k о § J., Joska J., Coll. Czech., 27, 1849 (1962).
16. Ireland R. E., Marshall ,J. A., J. Org. Chem., 27, 1G20 (19G2).
17. J о h n s W. F., J. Org. Chem., 29, 1490 (1964).
18. W a r d M. G., О r r J. С., E n g e 1 L. L., J. Org. Chem.,30, 1421 (1965),
ЛИТИЯ ДИЭТИЛАМИД, LiN(C2H&)2. Мол. вес 79,07.
Коуп и Тиффани [1] показали, что Л. д. наиболее эффективен
при изомеризации окиси циклооктатетраена (1) в 1, 3, 5-циклоок-
татриенон-7 (3). Авторы постулировали, что катализируемое ос-
нованиями отщепление протона от соединения (1) дает анион,
(1)
HNEt,
----,-$--
9 5% (сырой)
(2)
/~\Л’
[I Т + :NEt2
(3)
который перегруппировывается в енолят-анион (2). Кетон (3)
впервые был получен из (1) взаимодействием с фениллитием, однако
из-за побочных реакций выход составляет всего 8%. Пространст-
венно затрудненное основание мезитиллитий дает более высокий
7 Заказ № 1096
193
выход (39%) кетона (3), однако при использовании более слабого
основания Л. д. был получен лучший результат; общий выход
кристаллического семикарбазона кетона (3) из (1) составил 71%.
Раствор Л. д. получают добавлением раствора 0,60 моля диэти-
ламина в эфире в течение 15 мин к раствору фепиллития, получен-
ному в эфире в атмосфере азота из 0,66 моля бромбензола. Завер-
шение реакции определяется отрицательной пробой на фениллитий
кетоном Михл ер а [2]. Добавление 0,3 моля окиси циклооктатет-
раена в эфире при температуре от —8 до —12° вызывает появление
ярко-оранжевой окраски; через 5 мин смесь подкисляют и обра-
батывают.
Впоследствии [31 было показано, что под действием Л. д.,
полученного из н-бутиллития, осуществляются следующие пре-
вращения:
Окись с-стильбе и а -----> Дезокси бензо ин (70%)
Окись /прайс-стиль бен а ——Дифенил ацетальдегид (66%)
- Окись трифенилэтилена ----------у Бензгидрилфенилкетоп (80%)
Окись 1,1-дифенил-2-п-то- ——Бензгидрил-/г-толилкетои (41 %)
лплэтнлена
Окись тетрафепилэтилена — Не реагирует
Окись 1,1-дифенилэтилена — Не реагирует
Бергсталер [4] получил Л. д. перемешиванием литиевой дроби
в этиламине до полного исчезновения синей окраски и использовал
его для изомеризации несовряженного диена (1) в соединение (2).
Под действием минеральных кислот эта изомеризация не осуществ-
С (СНз)3
JX/C(CH,),
С (СН3)3
(СН3)3
LiN (СгН5),,| Г
V
(2)
ляется. Л. д. изомеризует кислоту (3) в палюстриновую кислоту (4),
однако в этом случае более удовлетворительные результаты дает
кипячение с едким кали в этиленгликоле [5].
194
Бенкесер и др. [6] показали, что под действием Л. д. 2,5-дигидро-
кумол быстро изомеризуется с образованием, по-видимому, смеси
всех возможных сопряженных диенов. Циглер [7] использовал это
основание для алкилирования алифатических нитрилов. Кейзон [8]
показал, что это — лучший метод получения 2,2-диметилоктаде-
кановой кислоты.
Li R’
LiN (С2Н6)2 [ R'X [
R2CHCN---------► RgCCN R,C—CN
Под действием сильноосновного Л. д. окись нор бор йена (1)
изомеризуется исключительно в нор три цикл анол (2) с умеренным
выходом [9]. Кислотные катализаторы действуют на (1) с образо-
ванием смеси продуктов.
1. С о р е А. С., Т i f f а п у В. D., J. Ага. Chem. Soc., 73, 4158 (1951).
2. Gilman Н., Schulze F., J. Am. Chem. Soc., 47, 2002 (1925).
3. С о p e A. C., Trumbull P. A., Trumbull E. R., J. Am. Chem.
Soc., 80, 2844 (1958).
4. Burgstahler A. W., Chien P.-L., A b de I - R ah m a n M. O., J.
Am. Chem. Soc., 86, 5281 (1964).
5. Burgstahler A. W., W о г d e n L, R., J. Am. Chem. Soc., 86, 96 (1964).
6. Benkeser R. А., В u г г о u s M. L.,Hazdra J. J., К a i s e г E. М.»
J. Org. Chem., 28, 1094 (1963).
7. Ziegler К., О h 1 i n g e r H., Ann., 495, 84 (1932).
8. C a s о n J. et al., J. Org. Chem., 15, 850 (1950).
9. Crandall J. K-, J. Org. Chem., 29, 2830 (1964).
ЛИТИЯ ДИ ЭТО КСИ АЛЮМО ГИДР ИД, LiAlH2(OC2H5)2.
Реагент получают в растворе прибавлением 2 молей этанола
или 1 моля этилацетата к 1 молю LiAlHa в эфире при 0° [1]. Он
восстанавливает N, N-диметиламиды до альдегидов с выходом 70—
90% [1]. По данным Тарбелла и сотр. [2] 3,4,5-триметоксибензаль-
CH3CH2CH2CON (СН3)2 СН3СН3СН3СН0
90%
CON (СН3)2 СНО
I I
//\
I I L1A1H, (ОС2НБ)3 I !|
с о % I II
СНзО/^/4 ОСН3 СН3О'^/\осн3
осн3 осн3
дегид гораздо легче получается этим методом, чем восстановлением
хлор ангидридов по Розенмунду. Суспензию 0,26 моля амида в эфире
7
195
обрабатывают при 0° раствором Л. д., полученным из 0т17 моля
LiAlH4 (в 1,3 раза больше рассчитанного), который .добавляют
в течение 2 час при 0°. Обработка и кристаллизация дают чистый
альдегид с выходом 60%.
1. Brown Н. С., Tsukamoto A., J. Ага. Chem. Soc., 81, 502 (1959).
2. Perun Т. J., Z е f t е 1 L., N e I b R. G.,T arb el 1 D. S-, J. Org. Chem.,
28, 2937 (1963).
ЛИТИЯ ИОДИДА ДИГИДРАТ, Lil-2HaO. Мол. вес 169,88.
Безводный иодид лития в кипящем основании с соответствую-
щей температурой кипения расщепляет сложные эфиры на кислоты,
со2,сн3
сн2с6н5
LiP 2 Н2О +
0,17 7 моля
Кипячение 19ад<?
-----&----->
снз 72-76%
140лм
Н
снгс6н5
0,129 моля
+ СО2 + ЫОН + СНЭ1 + Н2О
а Л. и. д. осуществляет и расщепление, и декарбоксилирование,
как это показано для 2-бензил-2-кар бметокси циклопентанона. Эл-
сингер 1П кипячением смеси этого кетоэфира, Л. и. д. и 2,4,6-
колл иди на в атмосфере азота получил с хорошим выходом 2-бен-
зилциклопентанон. Он отметил, что метиловые сложные эфиры
Рис. Л-2. Ловушка Дина—Старка [3].
реагируют быстрее соответствующих этиловых эфиров, которые,
в свою очередь, реагируют быстрее сложных эфиров вторичных
спиртов. трет-Бутиловые сложные эфиры очень легко расщеп- .
196
ляются каталитическим количеством Л. и. д. Последний получали
азеотропной перегонкой тригидрата с коллидином до отделения
1 моля воды при помощи ловушки Дина — Старка [2] (рис. Л-2).
1. Elsinger F., Org. Syn., 45, 7 (1965).
2. D е a n Е. W., Stark D. D., Ind. Eng. Chem., 12, 486 (1920).
3. Ace Glass 7725.
ЛИТИЯ ИОДИД БЕЗВОДНЫЙ, Lil. Мол. вес 133,85, т. пл. 446°.
Ташнер и Либерек [1] показали, что сложноэфирную и N-карб-
метоксильную группы можно эффективно расщепить действием
Л, и. б. в кипящем пиридине. N-Ацильные и пептидные связи в эту
RCO2CH3-|-Lil —RCO2Li + CH3I
реакцию не вступают. Встретившись с трудностями при гидролизе
трициклического сложного эфира I вследствие чувствительности
этого продукта к действию кислот и оснований, Эшенмозер [2]
испытал новый метод и нашел, что соединение I при кипячении
СО2СН3
в пиридине в течение 2 час с большим избытком Л. и. б. расщеп-
ляется с высоким выходом до соответствующей кислоты. В случае
стероидного ацетоксильного сложного эфира II температура ки-
пения пиридина оказалась недостаточно высокой (115°) и вместо
этого растворителя использовали 2,6-лутидин (т. кип. 143°). После
кипячения соединения II в течение 8 час с 6,5 же Л. и. б. в 20 мл
растворителя на 1 лгмоль сложного эфира выход ацетокси к и слоты
составил 68—74%; при этом было выделено 3—6% исходного
вещества, а оксикислота образовывалась с выходом 5—7%, Для
этой реакции характерно высокоселективное отщепление метильной
группы и очень низкая степень атаки на ацетоксигруппу, что резко
отличает ее от реакции гидролиза, при которой экваториальная
Зр-ацетоксигруппа гидролизуется с большей легкостью. В случае
метилового эфира О-ацетилолеаноловой кислоты (III) 17-карбме-
токсигруппа пространственно сильно экранирована, а 3-ацето-
ксигруппа экранирована до некоторой степенищ поэтому применение
приведенной выше методики с использованием лутидина дает аце-
токсикислоту IV с выходом 45—56%, а 28—35% соединения III воз-
197.
вращается. Однако если в качестве растворителя использовать
2,4,6-коллидин (т. кип. 172°, 6,5 же Л. и. б., кипячение 8 час),
то ацетоксикислота (IV) образуется с выходом 90%.
Некоторые сложные эфиры, исключительно устойчивые к ос-
новному гидролизу, были успешно гидролизованы кипячением
с Л. и. б. в коллидине. Реакция часто идет с низкой скоростью,
например, сложный эфир (1) гидролизуется лишь при кипячении
в течение 72 час в атмосфере азота, однако выход соответствующей
кислоты 94% [3]. Щелочной гидролиз в кипящем диэтиленгликоле
протекает лишь с умеренным выходом. Другим примером является
пространственно затрудненное, устойчивое к щелочи производное
эфира эбурикоевой кислоты (2), дающее 95% соответствующей
кислоты лишь по методу с использованием Л. и. б,— коллидина [4].
198
Херц и Вальборг [51 показали, что обработка производного смо-
ляной кислоты (3) реагентом в кипящем коллидине не только гид-
ролизует экранированную сложноэфирную группу, по и отщепляет
эпоксигруппу с образованием диеновой системы. При обработке
кислотами Льюиса получаются сложные смеси.
Дин [6] изучал гидролиз (галогснолиз) пространственно затруд-
ненного тритерпеиоидного сложного эфира — метил гл ицерретата
в нескольких растворителях и показал, что диметилформамид пре-
восходит пиридин, лутидин, коллидин или N-метилпирролидон-2
как в скорости, так и в степени превращения. Кроме того, исполь-
зование ДМФА облегчает обработку; реакционную смесь можно
вылить в воду, полученную смесь подкислить и экстрагировать
эфиром.
1. Taschncr Е., L I b е г е к В., Roczniki Chcmii, 30, 323 (1956); С. А.,
51, 1039 (1957).
2. Elsinger F., Schreiber J,, Eschenmoser A., Helv. Chim.
Acta, 43, 113 (1960).
3. Meyer W. L., Lev inson A. S., J. Org. Chem., 28, 2184 (1963).
4. Krakower G. W., Brow n J. W., Fried J., J. Org. Chem., 27, 4710
(1962).
5. Herz W., Wall lb org FI. J., J. Org. Chem., 30, 1881 (1965).
6. Dean P. D. G., J. Chem. Soc., 1965, 6655.
ЛИТИЯ КАРБОНАТ, Li2CO3. Мол. вес 73,89, т. пл. 618°.
Дегидрогалогенирование. С помощью Л. к. в кипящем диме-
тилформамиде Хауз и Бейш [1] осуществили дегидробромиро-
вание бромкетона (1), избежав перегруппировки и получив про-
дукт (2). Использование менее основного хлорида лития приводит
к смеси соединения (2) и перегруппированного кетона (3); последнее
вещество — единственный продукт дегидрогалогенирования соеди-
нения (1) кипячением с НВт в уксусной кислоте.
1. House И. О., Bashe R. W., II, J. Org. Chem. 30, 2942 (1965).
ЛИТИЯ КАРБОНАТ —ЛИТИЯ БРОМИД. Мол. веса: 73,89,
86,86.
Использование карбоната лития в сочетании с бромидом лития
в ДМФА для осуществления дегидрогалогенирования было пред-
ложено Жоли [1] как усовершенствование метода Холижа, который
199
применял хлорид лития в ДМФА. В применении к 2р, 4£-дибром-17р-
ацетоксиэтиохолану (1) методика Холижа дает смесь 1,4-дненона
(2) и 4,6-диенона (3). Французские исследователи нагревали 4,9 а
дибромида (1) с 4,5 г карбоната лития и 5,2 г бромида лития в 150 мл
ДМФА в атмосфере азота при температуре кипения (около 150и)
в течение 1 час и выделили чистый 1,4-диенон (2) с выходом 92%.
(з)
В тех же условиях, но с применением одного карбоната лития выход
соединения (2) составил лишь 42%. Дегидрогалогенирование трех
других 2р,4р-дибром-3-кетонов 5(1-ряда Li3CO3 — LiBr — ДМФА
при температуре кипения дало 1,4-диеноны с выходами 71, 90
и 93%. В более мягких условиях можно выделить промежуточный
4р-бром-А1-ен-3-он (4) [2]. Так, кетон (1), обработанный, как и ранее,
при 100° в течение 30 мин дает с умеренным выходом соедине-
ние (4).
Хотя опубликованные результаты противоречивы, примененная
к 2ос,4а-дибром-3-кето нам 5а-ряда реакция гораздо менее удовле-
творительна. Пельк и др. [3] обрабатывали 17р-ацетокси-2а,4а-
дибром-3-кетоандростан смесью Li2CO3 — LiBr — ДМФА и полу-
чили смесь 1,4- и 4,6-диенонов с преобладанием последнего. Однако
Виланд, Хейслер и Веттштейн [4] получили 1,4-диенон (6) с удов-
летворительным выходом из (5) следующим образом. Раствор (5)
в уксусной кислоте, содержащей следы бромистого водорода, обра-
батывали бромом при 15—16°; выпавший при разбавлении осадок
собирали, а фильтрат экстрагировали эфиром. При нагревании
всего продукта с Li3CO3— LiBr — ДМФА образуется 21-ацетат
преднизолона (6) с выходом 70%.
Применение этой реакции к За-бромхолестанону-2 (9) дает
продукт дегидрогалогенирования З-енон-2 (10) и продукт эпимери-
200
Плинингер и др. [6] показали преимущества этого метода при
превращении енона (12) в диенон (15). Выход при дегидрогалоге-
нировании (14) был в два раза выше, чем при использовании кол-
лидина (40%).
(14)
(15)
201
Кори и Хортман [7] синтезировали ди гидро посту иол ид (6) из
сантонина (1) приведенным ниже способом. Для дегидробромнро-
вания (2) в (3) метод Жоли (выход 80—90%) намного превосходит
метод с использованием коллидина (выход 30%). Смесь 7,26 г
соединения (2), 3 а бромида лития, 4 г карбоната лития и 50 мл
ДМФА нагревали в атмосфере азота при 120—125° в течение
75 мин.
1. J о 1 у R., Warn а п t J., Nomine G., Berlin D., Bull. soc. chim.
France, 366, (1958).
2. Joly R., Warn ant J., Bull. soc. chim. France, 1958, 367.
3. P e 1 с B., Her nianck S., Holubek J., Coll. Czech., 26, 1852, (196-1).
4. Wieland P., Heusler K-, Wettstein A., Helv. Chim. Acta, 43,
523 (1960).
5. Nakano T., Hasegawa iM., D j e г a s s i C., Chem. Pharm. Bull., 11,
465 (1963).
6. Plieninger H., Ege G. E., G r a s s h о f f H. J., К e i 1 i c h G.,
Hoffmann W., Chem. Ber., 94, 2115 (1961).
7. Corey E. J., Hortmann A. G., J. Am. Chem. Soc., 87, 5736 (1965).
ЛИТИЯ МОНОЦИАНБОРГИДРИД, LiB(CN)H3 -Диоксин. Мол.
вес 134,90.
Л. м. получают реакцией боргидрида лития с жидким циани-
стым водородом в эфире и выделяют в виде комплекса с диокса-
ном [1]. Реагент исключительно устойчив к действию воды и кислот,
LiBH4-|-HCN —> H3BCN (Li+) + H3
не разлагается при длительном кипячении в водном диоксане. Л. м.
является слабым восстановителем; он восстанавливает алифати-
ческие и ароматические альдегиды, ct-оксикетоны, но не восста-
навливает кетоны, ароматические нитросоединения, карбоновые
кислоты и их сложные эфиры, азоксисоединения [2]. Пирен-З-аль-
дегид (2 щмоля) при нагревании на водяной бане в течение 15 час
с 2 л/молями диоксанового комплекса Л. м. дает соответствующий
спирт с выходом 85%.
1. W i 11 i g G., R a f f P., Ann,, 573, 202, 209 (1951).
2. Drefahl G.} К e i 1 E., J. prakt. Chem., 6, 80 (1958).
ЛИТИЯ НИТРИД, Li3N. Мол. вес 34,83. Получение [1].
Л. н. практически нерастворим в большинстве инертных раст-
ворителей и считался нереакционноспособным. Однако, по данным
Кенига и др. 12], диоксан, диглим и другие растворители силы-ю
повышают реакционную способность Л. н., возможно, за счет ча-
стичного его растворения. Флуорен не реагирует с Л. н. в эфире
или в отсутствие растворителя при температуре плавления угле-
водорода. Но при кипячении флуорена с Л. н. в диглиме в течение
ночи с последующей карбонизацией раствора получают флуорен-9-
карбоиовую кислоту с 31%-ным выходом.
1. Masdupuy Е., G а 11 a i s F., Inorg. Syn., 4, 1 (1953).
2. К о е n 1 g Р. Е., М о г г i s J. М., Blanchard Е. J., М ason Р. S,,
J. Org. Chem., 26, 4777 (1961).
ЛИТИЯ ТРИМЕТОКСИАЛЮМОГИДРИД, LiAlH(OCH3)3.
Получение стандартного раствора. Браун и Вейссман [1] пока-
зали, что растворы этого реагента нестабильны; например, кон-
центрация раствора 0,67 М по гидриду через неделю уменьшается
до 0,57 Л4. Поэтому авторы предпочли использовать свежеприго-
товленные растворы, полученные добавлением метанола к стандарт-
ному раствору Ь1Л1Н4 в ТГФ. LiAlHd перемешивают в ТГФ в те-
чение 2 час в атмосфере азота, фильтруют раствор, передавливая
его азотом через слой целита на пористом стеклянном фильтре;
прозрачный раствор хранят в колбе емкостью 1 л, плотно закрытой
резиновой пробкой для ввода шприца. Для переноса раствора реа-
гента используется шприц с длинной иглой. Раствор при хранении
устойчив.
Восстановление. В противоположность LiAlH(OBu)3 тримето-
ксипроизводное по восстанавливающим свойствам приблизительно
так же активно, как LiAlHd [1]. Однако эпоксиды (особенно внут-
ренние эпоксиды) оно восстанавливает медленнее, чем ЫА1Н4.
Коричный альдегид восстанавливается в гидрокоричный альдегид.
Прн восстановлении бициклических кетонов Л. т. более стереоспе-
цифичен, чем LiA!Hd [2]. Например, камфора восстанавливается
в изобориеол (экзо) Л, т. с выходом 98%, a LiAlHd с выходом 91%.
Реагент также более селективен при восстановлении несимметричных
203
окисей [3]. Так, окись стирола (1) восстанавливается LiА1Н4
с образованием 96% вторичного спирта (2) и 4% первичного спирта
(3); в присутствии Л. т. количество (3) уменьшается до 1%. В общем
случае восстановление эпоксидов, оксимов, азобензола и циклогек-
силтозилата Л. т. протекает медленнее, чем LiAlH4.
/°Х Г
С6Н5СН—СНа + ЫА1Н4—* С6Н5СНСН2ОН-|-СвН5СНгСН3ОН
(1) (2) (3)
1. Brown Н. С,, Weissman Р. М., J. Am, Chem, Soc., 87, 5614 (1965).
2. Brown Н. С., Deck Н. R,, J. Am. Chem. Soc,, 87, 5620 (1965).
3. Brown Н. С., Weissman Р. М., Yoon Н. М., J. Am. Chem. Soc.,
88, 1458 (1966).
ЛИТИЯ ТРИЭТОКСИАЛЮМОГИДРИД, LiAlH(OC3H5)3.
Реагент получают in situ добавлением либо 3 молей этанола,
либо 1,5 моля этилацетата к 1 молю LiAlH4 в 1,3 /И эфирном раст-
воре при 0° [11. В эфирном растворе при 0° Л. т. восстанавливает
как алифатические (70—80%), так и ароматические нитрилы (80—
90%). Л. т восстанавливает также с высоким выходом диметил а-
Н
LiAlH (OEt), j - Н-,0
RC^N ----------RC = NAJ(OEt)3(Li+) —RCHO
миды карбоновых кислот в альдегиды [2].
N(LH3)2
LiAlH(OEt)s | - Н2О
RCON(CHS)3 ——-----RC----------OAl(OEt)3(Lj+)-----> RCHO
H
1. Brown H. C., Garg С. P., J. Am. Chem, Soc., 86, 1085 (1964).
2, В г о w п H. С., Т s и k a m о t о A., J. Am. Chem. Soc.', 86, 1089 (1964).
ЛИТИЯ ХЛОРИД, LiCl. Мол. вес 42,40, т. пл. 613°.
Тот факт, что на последней стадии оригинального синтеза кор-
тизона должно происходить ^ис-отщепление бромистого водорода
от 21-ацетата 4|3-бромдигидр©кортизона (I), что создает опре-
деленные трудности, стимулировал поиски лучших методов
204
дегидрогалогенирования. Холиж fl] показал, что эффективным
реагентом является хлорид (пли бромид) лития в ДМФА.Реакция,
проведенная при 100° в атмосфере азота, дает с хорошим выходом
21-ацетат кортизона (II).
В случае 2-хлор-2-метилциклогексапона (2) нет аналогичных
пространственных затруднений и дегидрогалогенирование как по
методу Холижа, так и с применением коллидина протекает до-
вольно плохо и дает сравнимые выходы [2].
a) LiCl-HCON(CH3)2
б) Коллидин
Из 43-45%'
ОД(б) 45-49%
Хлорид лития используют в качестве электролита при электро-
литическом восстановлении алкилбепзолов в метиламине. Электро-
лизер может быть снабжен асбестовой перегородкой для разделения
анодного и катодного пространств. При этом с прекрасными выхо-
дами получаются тетрагидропроизводные. В отсутствие перегородки
алкилбензолы восстанавливаются до 1,4-дигидропроизводных также
с хорошими выходами [31.
Гризеофульвиновая «кислота» (4, в действительности 2'- или
4'-енол) реагирует с хлорокисью фосфора в ДМФА (но не в диме-
тилацетамиде), образуя с высоким выходом 4'’-хлорид (5) [4].
В присутствии хлорида лития (без растворителя) продукт пред-
ставляет собой смесь 4'-хлорида (5,4%) и 2'-хлорида (6,6%).
1. Н о 1 у s z R. Р., J. Am. Chem. Soc., 75, 4432 (1953); Johnson W. S.,
Vredenburgh W- A., Pike J. E., (preparation of XIV), J. Ain. Chem.
Soc., 82, 3414 (1960).
2. Warnhoff E. W., Martin D. G., Johnson W. S,, Org. Syn.
Coll. Vol., 4, 162 (1963).
3. Benkeser R, A., Kaiser E. M., J. Am. Chem. Soc., 85, 2858 (1963).
4. Stephenson L., W alker T,, Warburton \V. K-, W e b b G. B.,
J. Chem. Soc., 1962, 1282.
2,6-ЛУТИДИН. Л4ол. вес 107,15; уд. вес 0,923, т. кип. 142—143°.
Н3(У N ХСН3
Мазурек и Перлин [1] показали, что этот гомолог пиридина
превосходит карбонат серебра — обычный акцептор кислот — при
получении ортоэфиров (3) из 2,3,4,6-тетра-О-ацетил-а-о-маннопира-
нозилбромида (1). Значительный интерес представляет тот факт,
что при этом почти исключительно образуется экзо-ОР-изомер.
Вг
О) (2) (3)
Реакцию лучше проводить в хлороформе, а не в избытке осно-
вания.
1. Mazurek М., Р е г 1 i п A. S., Can. J, Chem., 43, 1918 (1965).
2,6-ЛУТИДИН-З,5-ДИКАРБОНОВОЙ КИСЛОТЫ ГИДРАЗИД.
Мол. вес 223,23.
H.2NHNOC-X\— CONHXH.2
н3с~! Lch3
Вельюз и Руссо [1.1 описали получение реагента и его исполь-
зование для осаждения прогестерона в виде производного 1 : 1
с более чем 95%-ным выходом. Прогестерон выделяют обменной
реакцией с бензальдегидом.
1, V е 1 1 u z L., Rousseau G., Bull. Soc. chim. France, 13, 288 (1946).
WM JWIIURUM
МАГНИЙ, ат. вес 24,32,т. пл. 650°; см. также Гриньяра реактивы.
Абсолютные метанол и этанол в 1931 г. получали кипячением
над магниевыми стружками в течение 4 час (1 г/100 мл) [1, 2].
Восстановление. Амальгама магния является восстанавливаю-
щим агентом в классической реакции восстановления ацетона в пи-
након [3]. В колбу, содержащую М. в бензоле, постепенно при
кипячении добавляют раствор сулемы в ацетоне. Выход пинакон-
гидрата в расчете на М, составляет 43—50%.
Эффективным методом восстановления алкил- и арилгалогеиидов
является реакция с 1М. и изопропанолом [4]. 1-Бромнафталии
восстанавливается при температуре кипения изопропанола (82J).
С менее реакционноспособным галогенидом, например хлорбен-
золом, реакцию проводят следующим образом. Смесь 0,25 г-атом
Вг
ххА
1 II I + (СНОСНОЙ +Mg —| || | + (CH3)2CHOMgBr
Х/ХХ '° Х/ХХ
порошка М. я 50 мл декалина (т. кип. ~190°) кипятят без переме-
шивания, добавляют небольшой кристаллик иода и из капельной
воронки примерно одну пятую смеси 0,1 моля хлорбензола и 0,15 мо-
ля изопроианола [4]. Реакция начинается сразу в том месте, где
находится иод, и становится более энергичной при перемешивании
и уменьшении подогрева. После этого в течение 30 мин добавляют
остаток раствора галогенида, затем еще 25 мл декалина для лучшего
перемешивания и кипятят смесь еще 1 час. К охлажденной смеси
по каплям прибавляют разбавленную соляную кислоту до раство-
рения твердого вещества, органический слой тщательно промывают
водой, сушат и перегоняют; выход чистого бензола 89%.
В случае еще менее реакционноспособного галогенида или гало-
генида, склонного к отщеплению галогенводорода, следует для
ускорения реакции добавлять реакционноспособный галогенид
типа 1-бромнафталина или н-бутилбромида. Например, при взаи-
модействии 0,05 моля цикло гексилхлорида и 0,05 моля 1-бромнаф-
талина с 0,33 г-атом Ж. и изопропанолом (0,3 моля) в 50+20 мл
декалина получают смесь 83% циклогексана и 10% циклогексена
(удаляемого серной кислотой). По этой методике циклогексилфторид
дает циклогексан (33%), а бензотрифторид — толуол (10%). Фтор-
бензол в реакцию не вступает. В этой работе использован порошок
207
М., приготовленный за 6 месяцев до употребления. Использование
более старого или более грубо измельченного магния может удли-
нять индукционный период.
Синтезы через этоксимагниймалоиовый эфир. Четыре методики,
приведенные в сб. «Синтезы органических препаратов», начинаются
с реакции абсолютного этанола, малонового эфира и М. в присутст-
вии каталитического количества СС14 с образованием этоксимаг-
ссц
С2Н5ОН СН2(СО2С2Н5)г Mg —-* C,H5OMgCH(CO2C2H5)2 -j- н2
ни ймалонового эфира. М. (1 моль) и катализатор (1 мл) обрабатывают
небольшой порцией раствора малонового эфира (1 моль) в этаноле;
как только начнется реакция, добавляют остальной раствор так,
чтобы продолжалось кипение. Когда будет выпадать кристалли-
ческий продукт реакции, смесь охлаждают, добавляют эфир -для
растворения этоксимагниипроизводного и продолжают кипячение
до полного расходования металла. Следующую стадию — ацили-
рование обычно проводят с эфирным раствором сразу после его
приготовления. Этоксимагниевое производное образуется, по-ви-
димому, количественно. Так, в синтезе трикарбэтоксиметана, опи-
санном Лундом и Фойгтом [51, при добавлении раствора этилхлор-
формиата к эфирному раствору этоксимагниипроизводного общий
выход из малонового эфира составляет 88—93%. Превосходство
С1СО,СЛЦ
C2H50MgCH(CO2C2H5)2——CIMgC(CO2C2H5)3
м иц гЦ
АсОН
СН(СО2С2Н5)з
88-93% (из малонового эфира)
этоксимагниипроизводного как реагента видно из сравнения с ме-
тодикой получения трикарбметоксиметана через натрийдиметилма-
лоновый эфир, в котором общий выход составляет 40—42% [6].
В другом синтезе продукт ацилирования этоксимагниймал©но-
вого эфира о-нитро бензо и л хлоридом превращают кислотным гид-
ролизом и декарбоксилированием в о-нитроацетофенон [7]. Ацили-
COCI
X NO;
C2Hr,OMgCH(CO2C,H.0,
СОСН(СОгС2Н5)3
1 ,NOa
СОСНд
A /NO.
82—83.% (общий)
208
рование этоксимагиийпроизводного фенилацетилхлоридом дает про-
дукт, превращаемый в 1,3-диоксинафталин последующей схеме [81:
С6Н5СН,СОС1
CsH,OMgCH(CO2C:Hsb
H2SO4
50—59% (из малонового эфира}
UU\0H
Ва(ОН)г: H.SO,
90%
н.о: юо°
54—56%
Для синтеза диэтил бензоил мал он ат а Прайс и Тарбелл [91
получали эт оксимагниймалоновый эфир, как обычно, из малонового
эфира (0,2 моля), М. (0,2 моля), СС14 (0,2 мл) й небольшого избытка
этанола (0,275 моля) в эфире; затем отгоняли растворитель при
пониженном давлении. Частично кристаллический остаток раство-
ряли в бензоле и растворитель снова отгоняли; этот прием позволяет
удалить следы этанола, который может мешать на следующей
стадии — в синтезе смешанного ангидрида бензойной кислоты
и этилового эфира угольной кислоты. Бензойную кислоту и триэтил-
амии растворяют в толуоле с образованием соли амина, раствор
охлаждают ниже 0° и обрабатывают этилхлорформиатом. Выпа-
дающий хлоргидрат не мешает последующему ацилированию, про-
водимому добавлением по каплям раствора этоксимагниймалонового
эфира в сухом эфире.
О ОО
-+ II Толуол, 0° II II
C6H6COONHEt3 + ClCOEt-—CsH5C-O-COEt
— l..jN “г НС1 “*
СгН-.ОМяСНЩОрСЩв).
— 68-75% (06^)^ C6H5CCH(C02Et)3 + CO2 + Mg(OEt)3
о
Ацилирование трет-спиртов и ацетоуксусного эфира, трет-
Бутилацетат можно получить с умеренным выходом реакцией трет-
бутанола с ацетил хлоридом и М. в эфире [10]. Ацетилирование
ацетоуксусного эфира ацетил хлоридом в присутствии М. в кипящем
О О
II Эфир 1|
2СН3СС1-[-2HOC(CHj3Ц-Mg —2CH3COC(CH3)3 + MgCl3-pH3
209
бензоле (2 час) и очистка продукта в виде голубого медного произ-
водного дает с умеренным выходом чистый этилди ацето ацет ат [11],
Однако Висконтини и Мерклинг [12] подобной реакцией в абсолют-
ном этаноле при комнатной температуре получили этилдиацето-
ацетат (диацетоуксусный эфир) с 73%-пым выходом. По этой ме-
СОСН3
CII,COCI+’/3Mff |
CH3COCH3CO3Et 46_52<i/----> CH3COCHC03Et
тодике швейцарские исследователи получили 11 С-ацильных про-
изводных ацетоуксусного эфира со средним выходом 73% и расще-
пили некоторые из них в p-кетоэфиры с хорошим выходом либо
аммиаком в этаноле, либо метилатом натрия.
СОСН3 О сосн;з
| Mg-ЕЮН I ] EiOH-НН, пли CH3ONa
RCOC1CH2CO3Et ——7^ RC—CHCO3Et...................o;-----
•" / 3 % — 61%
RCOCHJ2OsEt
Реакция Реформатского. Эту реакцию можно провести лучше
и с более высоким выходом с М. вместо менее реакционноспособ-
ного цинка при условии, что используется шде/п-бутилгалогенэфир
для замедления самокондснсации а-галогенэфира [131.
Сн3 СН3
] Mg; эфир; Н,о |
С6Н5С=04-ВгСН3С02С(СН3)3————* С6Н5С-СН3СО3С(СН3)3
й J % I
он
1. Lund H.,Bjerruin J,, Вег., 64, 210 (1931).
2. Баумгартен Г.,Петер сен Д., «Синтезы органических препаратов»,
изд-во «Мир», М., 1964, сб. 12, стр. 171.
3. Адамс Р., Адамс Е., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 342.
4. Bryce-Smith D., Wakefield В. J,, В 1 u е s Е. Т., Proc. Chem,
Soc., 1963, 219; procedure submitted to Org. Syn.
5, Л у ид X., Фойгт А., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 2, етр. 472.
6. К о р с о и Б., С э й р Дм;., «Синтезы органических препаратов», ИЛ, At,,
1949, сб. 2, стр. 471.
7. Reynolds G. A., Hauser С. R., Org. Syn., Coll. Vol., 4, 708 (1963).
8. M e у e г K-, Bloch H. S., Org. Syn., Coll. Vol., 3, 637 (1955).
9. Price J. A.,TarbelI D. S., Org. Syn., Coll. Vol., 4, 285 (1963).
10. Sp assow A., Org. Syn., Coll. Vol., 3, 144 (1955).
11. Sp assow A., Org. Syn., Coll, Vol., 3, 390 (1955).
12. V iscon t in i At., Merckl ing N., Helv. chim. Acta, 35, 2280 (1952).
13. M oriwake T., J., Org. Chem., 31, 983 (1966).
МАГНИЙ — ИОД — ЭФИР [II. Суспензия металлического маг-
ния в эфирном растворе иодида магния является удобным реагентом
для отщепления вицинальных галогенов с образованием двойной
связи [2]. Для дегалогенирования 2 молей 2,3-дихлордиоксана
к 3,4 моля магниевых стружек в 1,2 л сухого эфира при перемеши-
вании и кипячении небольшими порциями добавляют 0,4 моля иода.
210
После обесцвечивания к смеси приливают раствор 2 молей дихлор-
диоксана в 200 мл эфира с такой скоростью, чтобы цвет раствора не
становился темнее светло-коричневого (9 час). Уменьшение ско-
рости реакции по мере ее завершения обусловлено, по-видимому,
соосаждением иодид-иона с образующимся нерастворимым хлори-
дом магния. Этой реакцией захвата можно объяснить также и то,
что требуется более чем каталитическое количество иода [3]. Ре-
акционную смесь выливают в воду со льдом, органический слой
отделяют, сушат и перегоняют; выход диоксена с т. кип 93—95°
Mg С
/О /С1 - О-
I |Z Эфир i Ф
j | +M8I, -- | H+MgCL + L
х0/
составляет 84 г (49%). Для дегалогенирования менее реакционно-
способных соединений в качестве растворителя нужно исполь-
зовать более высококипящий ди-я-бутиловый эфир [4]. Реагент
применяется также для получения продуктов, чувствительных
к действию водного раствора иода, например виниловых эфиров [5].
1. Contributed by S u m m е г b е 1 1 R. К-, Northwestern Univ.
2. Summerbell R. K-, Umhoefer R. R., J. Am. Chem. Soc,, 61, 3016
(1939).
3. R о c h e L. K-, Ph, D. Thesis, Northwestern University (1940).
4. Snmmerbel 1 R, !(., Umhoefer R. R., J. Am. Chem. Soc., 61, 3020
(1939).
5. Snmmerbel 1 R. K„ Hyde D. K. A., J. Org. Chem., 25, 1809 (1960).
МАГНИЙ — НАФТАЛИН. Смесь 60 щ.молей магниевой пыли
и 60 лшолей нафталина перемешивают в 150 мл. жидкого аммиака,
добавляют 50 мл эфира и продолжают перемешивание до почти
полного растворения магния с образованием ярко-зеленого раствора
(1,5 час) [I]. Зеленый комплекс, по-видимому, имеет состав С1()Н8М^.
Добавление 120 нгмолей абсолютного этанола приводит к исчезно-
вению окраски, и обработка дает 1,4-дигидронафталин с т. пл. 24—
25°. Комплекс металлирует бензилцианид в эфире с образованием
желтого осадка; замена аммиака на эфир и кипячение с этилиодидом
дает сс-фенилбутиронитрил с выходом 34%. Попытка металлировать
C,0HdMg С2Н.,1
C6H6CHaCN -------> Осадок --> CeH&CHCN
СН2СН3
этилфенилацетат оказалась безуспешной, а вместо этого прошла
сложноэфирная конденсация в этил-а,у-дифенилацетоацетат.
С6Н5 С6Н5
I c,oi-isMg 1
СйН5СН>2СО2С2Н5-|- сн2со2сан6 —--> сен5сн8соснсо3с3н5
1. I v а п о f f Chr., Markov P., Naturwiss., 50, 688 (1963).
211
МАГНИЯ БРОМИДА ЭФИРАТ.
Получение. Смесь 0,3 г магниевого порошка, 60 мл абсолютного
эфира и 30 мл бензола обрабатывают 2,16 г бромной ртути, кипя-
тят 2 час, раствор фильтруют и сразу используют 111. Известно
12], что 9(11)-моноэпоксиэргостерил-D-ацетат (1) изомеризуется
эфиратом трехфтористого бора в сопряженный кетон (3). Бахман[1]
показал, что изомеризацию в несопряженный кетон (2) можно осу-
ществить кипячением в течение 2 час смеси 75 мл свежеприготовлен-
ного эфирного раствора бромида магния и раствора 1,23 г (1) в 140 мл
эфира. Изомеризацию в (3) проводят или под действием следов
соляной кислоты в этаноле или пропусканием раствора в смеси
бензол — петролейный эфир через колонку с активированной
окисью алюминия.
Хауз 13] показал, что как цис-, так и транс-2,3-эпоксибутаны
изомеризуются бромидом магния в эфире в бутанон-2. цис-Эпокись
в присутствии трехфтористого бора также дает бутанон-2, а транс-
эпокись наряду с кетоном образует изомасляный альдегид. При
низкотемпературной изомеризации окиси циклогексена был вы-
делен также бромгидрин [4].
1. Bachmann W. Е., Horwitz J. Р., Warzy П ski R. J.,J. Am.
Chem. Soc., 75, 3268 (1953).
2. H e u s s e r H. et al., Helv. chim. Acta, 34, 2106 (1951).
3. House H. O., J. Am. Chem. Soc., 77, 5083 (1955).
4. Naq vi S. M., Horwitz J. P., Filler R., J. Am. Chem. Soc., 79,
6283 (1957).
МАГНИЯ ОКИСЬ, MgO. Мол. вес 40,32, т. пл. 2800°.
Сообщения о получении пиримидина (2) восстановительным
дехлорированием 2,4-дихлорпиримидина (1) и 2,5-дихлорпирими-
дина (3) были опубликованы почти одновременно Уиттекером [1]
и Литго и Райнером [2]. В обоих случаях наилучшим акцептором
хлористого водорода была признана М. о.; сырой продукт осаж-
дали в виде пиримидинмеркурхлорида, который расщепляли кипя-
чением с сульфидом натрия. В улучшенной методике 13] к суспензии
84 г 2,4-дихлорпиримидина в смеси 420 мл этанола и 840 мл воды
212
добавляли 50 г М. о., 21 г 3%-ного палладия на угле и при переме-
шивании гидрировали под давлением водорода 2,1—2,8 атм до
завершения поглощения водорода (80 мин). Выход очищенного
вышеуказанным способом пиримидина составляет 78%.
Реагент был использован в качестве акцептора хлористого
водорода при защите аминогруппы аминокислот реакцией с карбо-
бензоксихлоридом [4].
(1) (2) (3)
1. Whittaker N., J. Chem. Soc., 1951, 1565.
2. L у t h g о е В., R ay ner L. S., J. Chem. Soc., 1951, 2323.
3. Whittaker N., J. Chem. Soc., 1953, 1646.
4. du V i g n e a u d V., Miller G. L., Biochem. Preps., 2, 79 (1952).
МАГНИЯ СУЛЬФАТ, MgSO4; MgSO4-7H3O.
Следует иметь в виду, что безводный М. с., применяющийся
иногда в качестве осушителя, может оказаться неподходящим,
так как обладает кислотным характером. Робертсон [11 разработал
эффективный метод присоединения третичного этинил карбинола
к дигидропирану с образованием защищенного производного: смесь
реагентов с небольшим избытком М. с. и следами н-толуолсульфо-
кислоты нагревают в течение 1 час на кипящей водяной бане (вы-
ходы 60—84%). Отмечено, что если при обработке проводить эк-
СН3 /ч сн3 /ч
I TsOH |
нс^с-ё-онI —нс^с-с-о-1 +Н2о
CH3 f/ сн3
стракцию эфиром, то осушитель должен быть нейтральным или
основным; сульфат магния обладает достаточной кислотностью
и способен вызвать обращение реакции за несколько часов.
213
Это свойство М. с. использовано для осуществления селектив-
ного гидролиза А5-3,20-бпс-этиленкеталя (I) [2]. Раствор (I) в бен-
золе, насыщенном водой, встряхивают с безводным М. с. в течение
1 час; при этом количественно удаляется 3-этиленкетальная группа
и образуется 20-моноэтиленкеталь (II) (см. схему на стр. 213).
1. Robertson D. N., J. Org. Chem., 25, 931 (I960).
2. Bro w n J. J., Lenhard R. II., Bernstein S., J. Am. Chem.
Soc., 86, 2183 (1964).
H —C—CHO
МАЛЕИНОВЫЙ ДИАЛЬДЕГИД, H-cLcho Мол. вес
86,09, желтое масло, т. кип. 56—5979,5 мм; растворяется в уксусной
кислоте, ацетоне или хлороформе; ограниченно растворим в бен-
золе, эфире, четыреххлористом углероде; быстро полимеризуется;
желтые растворы в воде и метаноле при стоянии обесцвечиваются;
дисемикарбазон кристаллизуется из воды в виде игл, т. пл. 246—
247° (с разл.).
Несмотря на указанные неблагоприятные свойства, Волю и сотр.
[1] удалось выделить высокочувствительный диальдегид в доста-
точно чистом виде и охарактеризовать в виде кристаллических
производных типа дисемикарбазона. Синтез М. д. включает ре-
акцию ацетилендимагнийбромида (1) и ортомуравьиного эфира
с образованием бнс-диэтил ацеталя ацетилендиальдегида (2), се-
лективное гидрирование последнего в присутствии катализатора
Линдлара в соединение (3) и последующий тщательно проводимый
низкотемпературный гидролиз. Строение и конфигурация продукта
подтверждаются окислением бнс-диэтилацеталя (3) перманганатом
и последующим гидролизом до соединения, идентифицированного
как мезовинный диальдегид (5).
2HC(OEt)3 Н2 —Pd(Pb)
BrMgC ss CMgBr "" 0 (EtO)XHC = CCH(OEt).,----------->
(1) (2)
H-C—CH(OEt)2 н+ H—C—CHO
II ----► II
H—C-CI1(OEI)2 H^C— CHO
(3) (4)
j H +
CHO
нАэн
HtioH
^ho
(5)
Синтез Воля, по-видимому, второй по значению метод полу-
чения стабильного исходного вещества, гидролизующегося до М. д.
Третьим считается описанный Маркисом 12] процесс, в котором
фуран нитруют в уксусном ангидриде при низкой температуре
с образованием некристаллизующегося нестабильного продукта,
214
дающего при обработке пиридином 2-ннтрофурап. Джонсон [3]
приводит доказательства в пользу того, что это лабильное промежу-
точное соединение является продуктом 1,^присоединения ацетил-
ннтрата (7), и таким образом объясняет1 возможность превращения
его в ЛА. д.
(6) (7) (8)
Лучшим исходным веществом для синтеза М. д. в настоящее
время является, по-видимому, 2,5-диметокси-2,5-дигидрофур ан (9),
предложенный Олдричем и Истманом и получаемый по методике,
разработанной шведскими химиками [4]; в дальнейшем она была
усовершенствована Вернессом [5]. Охлажденный (—25°) раствор
брома в метаноле добавляют к холодному (—40е) раствору фурана
в смеси метанол — эфир и поддерживают температуру ниже —25 л
Затем реакционную смесь обрабатывают газообразным аммиаком
при температуре ниже —15° до появления щелочной реакции по
индикаторной бумажке. После перемешивания в течение 1,5 час
раствор оставляют* для нагревания до —5°; при этом он должен
обесцветиться. Бромид аммония удаляют фильтрованием, фильтрат
концентрируют, повторно фильтруют и продукт выделяют пере-
гонкой; т. кип. 50—51 °/12 мм.
Кислотным гидролизом 2,5-диметокси-2,5-дигидрофурана по-
лучают М. д. наряду с небольшим количеством фумарового диаль-
дегида [6, 71.
Вгг-СНэОН-(С2Н5)2О
От -40 ДО -25°
72-76%
Н ’Н
I 1
с~с
I I
осн сно
(9)
При исследовании реакции фурана с бромом и метанолом ин-
тересные результаты получены советскими химиками [81. В усло-
виях, очень сходных с методикой Вернесса, но при более высокой
Вг2-СН3ОН
3 -10° 3 . У,
Метанол-фуран
12:1
СН3ОН(Н+)^
83%
(9)
•> (СН3О)2СНСН=СНСН(ОСН3)г
(10)
215
температуре и без эфира они после нейтрализации смеси аммиаком
получили 1,1,4,4-тетраметокси-2-бутен (10) с хорошим выходом
и подробно изучили влияние условий реакции. Показано, что при
проведении реакции в смеси метанол — фуран (21 : 1), начиная
с —10° и с дальнейшим понижением температуры наблюдается
увеличение соотношения диметоксиди гидрофурана (9) к тетраме-
токсибутену (10):
Температура, °C (9). % (Ю), %
0 2 19
-10 18,4 53
—20 36,6 19,6
—30 56,5 3
Другим определяющим фактором реакции является концент-
рация бромистого водорода, образующегося на стадии бромиро-
вания. Полученное в реакции при —10° изменение соотношения
метанол : фуран от 21 : 1 до 12 : 1 приводит к повышению выхода
тетраметоксибутена от 53 до 83%. Таким образом, повышение кон-
центрации бромистого водорода способствует превращению диме-
токсидигидрофурана (9) в тетраметоксибутен (10). Высокий выход
соединения (9) в методике Вернесса объясняется главным образом
низкой температурой реакции.
1. W о h I А., М у 1 о В., Вег., 45, 322, 1746 (1912); Wohl A., Bernreu-
t h er E., Ann., 481, 11, 29 (1930).
2. Marquis R,, Ann. chim., (8), 4, 216 (1905).
3. Freure В. T., J о h n s о n J. R., J. Am. Chem, Soc., 53, 1142 (1931).
4. Cl a u s о n -K a a s N., Limborg F., Fakstorp J., Chem. Scand.,
2, 109 (1948)' Fakstorp J., Raleigh D., S c h n i e p p L. E., J. Am.
Chem. Soc., 72, 869 (1950).
5. Бернесе Д., «Синтезы органических препаратов», изд-во «Мир», М„
1964, сб. 12, стр. 45.
6. Hufford D. L., Т а г b е 1 1 D. S., К о s z а 1 k а Т. R., J. Am. Chem.
Soc., 74, 3014 (1952).
7. Alder К., Betzing Н., Heimbach К., Ann., 638, 187 (1960).
8. М а к и н С. М., Те л егин а Н. И., ЖОХ, 32, 1104 (1962).
МАЛОНОВОЙ КИСЛОТЫ ДИБЕНЗИЛОВЫЙ ЭФИР (дибензил-
малонат), СН3(СО2СНаС6Н5)2. Мол. вес 284,30, т. кип. 188°/0,2 мм.
М. к. д. э. получают с выходом 95% кипячением с водоотдели-
телем смеси малоновой кислоты, бензилового спирта и следов сер-
ной кислоты в толуоле (метод А [1]). Этот эфир разлагается при
нагревании, поэтому перегонку следует проводить быстро н без
дефлегматора [2].
М. к. д. э. применяют в общем методе синтеза метилкетонов [3],
включающем превращение хлорангидрида кислоты в ацилмалоновый
эфир с последующим гидрогенолизом и декарбоксилированием.
216
Указанный метод использован на одной из стадий полного синтеза
альдостерона [2].
NaH RCOC1
сн2(со2сн2сви5)2 Ха+с“н(со2снгс6н5)2 ——-
Н5( Pd Нагревание
RCOCFI (CO2CH2Cr,H5)2--RCOCH(CO2H)2 -—— —> RCOCH3
1. Baker B. R., Schaub R. E.,Querry M. V., Williams J. A.,
J. Org. Chem., 17, 77, see also p. 88 (1952).
2. Johnson W. S., et al., J. Am. Chem. Soc., 85, 1409, see p. 1418 (1963).
3. В 0 w man R. E-, J. Chem. Soc., 1950, 325.
МАЛОНОВОЙ КИСЛОТЫ ДИ-/црсда-БУТИЛОВЫЙ ЭФИР,
СНо[СО2С(СН3)3]2. Мол. вес 216,27, т. пл. —6°, т. кип. 112—115°/
31 мм.
Реагент получают из мало новой кислоты, изобутилена и ката-
литических количеств конц. серной кислоты в эфире под давлением
2,8 атм II, 2]. Соединение получают также через хлорангидрид
малоновой кислоты [3]; первая, стадия требует нагревания при
H2SO4
СН2(СО2Н)2ф2(СН3)2С=СН2 "77~7-го- [(СН3)3СО2С]2СН2
| О о—О (J уо
SOC12
72—85%
83-84%
2HOC(CH3)3
— — C6H3N(CH3),
45—50° в течение 3 дней. Этот реагент применяют для синтеза ке-
тонов, поскольку трет-бутильные группы ацильных производных
СНг[СО2С(СНвЫг н +
RCOC1-----———у RCOCH[CO3C(CH3)312 —*
RCOCH3 + 2(CH3)2C=CHa^2CO2
легко элиминировать пиролизом; реакцию проводят в кипящем
толуоле или уксусной кислоте в присутствии n-толуолсульфокис-
лоты как катализатора [11.
1. Fonken G. S., JohnsonW. S., J. Am. Chem, Soc., 74, 831 (1952).
2. McCloskey A. L., Fonken G. S.,Kluiber R. W., Johnson
W. S., Org. Syn., Coll. Vol., 4, 261 (1963).
3. P a x а, «Синтезы органических препаратов», ИЛ, М., 1954, сб. 5, стр. 19.
МАЛОНОВОЙ КИСЛОТЫ ИЗОПРОПИЛИДЕНОВЫЙ ЭФИР,
Мол. вес 144,12, т. пл. 97°, рКа 5,2.
М. к. и. э. получают с выходом почти" 50% реакцией малоновой
кислоты и ацетона с уксусным ангидридом [1, 2] или с изопропенил-
217
ацетатом, а обоих случаях в присутствии каталитических коли-
честв серной кислоты. Этот ацетонид является сильной кислотой
и гидролизуется в очень мягких условиях.
о
4 сод-1
ососщ
сн,=с.сщ
(Щзод
I!
С—О. ZCH3
Н2С.( )С/ .
хс—о/ хсн3
Высокая реакционная способность метиленовой группы делает
это соединение интересным с точки зрения замены малонового эфира
в синтезах. Метилирование (.31, которое лучше всего проводить
обработкой изопропнлпдепмалоната окисью серебра и иодистым
метилом в ацетонитриле [2], обычно протекает до стадии диалки-
лирования. Алкилирование бензилхлоридом в метаноле, этаноле
или ДМФА дает только изопропилидендибепзилмалонат [4].
(1)
°А-°уСН3 Ру
СНз 70%
(3)
асн3 CO2CZH5
>-сн==с
СН3 \ю2Н
(4)
Кори [5] нашел, что М. к. и. э. можно применять для синтеза
полуэфиров арилиденмалоновой кислоты. М. к. и. э. легко кон-
денсируется с карбонильными соединениями, даже с сильно про-
странственно затрудненным мезнтальдегидом (1). Так, конден-
сация соединений (1) и (2) проведена в пиридиновом растворе,
и продукт (3) при кипячении с абсолютным этанолом и следами
хлористого водорода дает моиоэтилмезитилиденмалонат (4).
1. М eldrum A. N., J. Chem. Soc., 93, 598 (1908).
2. D a v i d s о л D., Bernhard S. A., J. Am. Chem. Soc., 70, 3426 (1948).
3. О t t E., Ann., 401, 159 (1913).
4. Hedge J. A., Kruse C. W., Snyder H. R., J. Org. Chem., 26,
992 (1961).
5. С о г e у E. J., J. Am. Chem. Soc., 74, 5897 (1952).
МАРГАНЦА ДВУОКИСЬ АКТИВНАЯ. Мол. вес 86,93. Техни-
ческий продукт получается по методу, отличающемуся от приве-
денных ниже; по существу, он представляет собой МпО2, содер-
жащую 5% гидратационной воды.
218
Получение. Мортон и сотр. [1] получили активированную форму
двуокиси марганца смешением водных растворов эквивалентных
количеств сульфата марганца и перманганата калин, промыванием
и высушиванием осадка. Они показали, что М. д. а. является
эффективным реагентом для окисления витамина Ах в ретинен;
обычная МпО2 в этой реакции не эффективна. Для окисления смесь
Витамин Aj
Ретинен (альдягид А;)
раствора 0,2 г витамина Аг в 50 мл петролейного эфира и 10 а М. д. а.
в закрытой колбе выдерживали 6—10 дней при периодическом
встряхивании. По Мортону успешно окисляются другие первичные
аллиловые спирты, включая 2Р-нонилидсиэтанол 12] и витамин АДЗ!.
При разработке синтеза витамина Ах Аттенборо и сотр. 141
исследовали окисление пяти аллиловых спиртов и разработали
метод получения МпСУ, более активной, чем у Мортона. Растворы
сульфата марганца и едкого натра добавляют одновременно к пе-
ремешиваемому раствору перманганата калия, осадок отделяют
центрифугированием и промывают до тех пор, пока промывная
жидкость не станет бесцветной. Твердый продукт высушивают при
100—120° и растирают в мелкий порошок. Эго активное вещество
представляет собой гидратированную окись, содержащую 3—4%
прочно связанной воды. В типичном окислении раствор 1,0 г вто-
ричного спирта (1) в 50 мл петролейного эфира встряхивают с 1 г
М. д. а. в течение 1 час, после выделения перегонкой получают
0,8 г чистого кетона (2). Для сравнения можно указать, что при
окислении (1) по Оппенауэру и очисткой через семикарбазон кетон
(2) получается с выходом лишь 10%. М. д. а., полученная по методу
Аттенборо, оказалась удовлетворительной в пяти исследованных
реакциях, а также в опытах с аллиловым и коричным спиртами
и с А3-октинолом-2 и З-дегидро-fJ-ионол ом. Выходы так же высоки,
как и при использовании М. д. а., полученной по Мортону, а окис-
ление обычно заканчивается за 0,5—1 час при комнатной темпера-
туре, тогда как с М. д, а. по Мортону для его завершения требуется
219
6—10 дней. В работе Аттенборо и сотр. [4] показано, что этот метод
окисления является общим для аллиловых спиртов, как первичных,
так и вторичных, а также для спиртов типа RC=CCH(OH)R'.
Опубликованы также другие методы получения М. д. а, [5, 6].
Гриттер и Уоллес [7] сравнили образцы М. д. а., полученные по
всем вышеупомянутым, а также по усовершенствованным мето-
дикам, и не нашли существенных различий. Большинство исследо-
вателей используют окись Аттенборо и находят ее вполне удовлет-
ворительной.
Окисление спиртов. Приведенные примеры иллюстрируют окис-
ление аллиловых и пропаргиловых первичных и вторичных спиртов
до альдегидов и кетонов. Интересным использованием реакции
является синтез 4,5,6,7-тетрагидроинданона-1 (5), описанный Брауде
и Колсом [81. Кромби и Кроссли [91 показали, что циклопропил-
карбинолы удовлетворительно окисляются суспензией М. д. а.
£>—снгон
МпОг>
61%
он
МпОг ,
51%
в я-пентане. С помощью нейтрализованной окиси в хлористом ме-
тилене Сторку и Томашу [10] удалось окислить очень неустойчивый
карбинол (3) в кетон (4), первый представитель винилэтинилке-
тонов.
220
н
С2Н5ОС CLi + СН3С= tcHO
I
н
(I) (2)
ОН н он
i i МлОз II |
—> с,н 5ос = ссн —с=ссн3 ——с2нбос с—с - с=ссн3
। CH^Cla ।
н н
О) (4)
Пратт и Ван-де-Кастль [11] изучили окисление М. д. а. различ-
ных фенил карбинолов. После кипячения реагента с бензолом до
тех пор, пока вода ие будет больше собираться в ловушке Дина —
Старка, добавляют карбинол и продолжают кипячение до прекра-
щения выделения воды. Результаты можно суммировать следую-
щим образом:
АгСН2ОН АгСНО (75—85%)
Аг2СНОН^Аг.,СО (88 — 96%)
ArCHOHR ArCOR (72—95%)
Мансера, Розенкранц и Зондхеймер [5] осуществили селективное
окисление аллильной вторичной спиртовой группы без затраги-
вания обычных вторичных спиртовых групп. Адреностерон (1)
при восстановлении LiAlHs дает смесь За, 110,170- и 30,110,170-
триолов (II), при обработке которой М. дла. в хлороформе прн
комнатной температуре происходит окисление только аллильной
спиртовой группы и образуется 110-окситестостерон (III).
Зондхеймер, Амендолла и Розеикраиц [12] показали, что хо-
лестерин и другие гомоалл ильные А5-3-оке и стер о иды окисляются
221
М. д. а. в кипящем бензоле за 8 час в А4,6-3-кетостероиды, однако
выходы низки для препаративных целей. А4-3-Кетостероиды также
дают А4,6-диеноны с низким выходом. Харрисон [13] показал, что
применение достаточного количества реагента и очищенных раст-
ворителей позволяет с высокими выходами, но медленно окислять
как первичные, так и вторичные насыщенные спирты. Например,
100 мг 5а-андростанола-17Р перемешивают при комнатной темпе-
ратуре в гексане или ацетонитриле (20 мл) с 2 г М. д. а. по Аттен-
боро в течение 20 час и получают чистый 5а-аидростапон-17 с прак-
тически количественным выходом. Скорость реакции зависит от
природы используемого растворителя. Так, в ДМСО окисление не
завершается даже за 7 дней. При том же соотношении окислителя
и спирта 4-метилциклогексанол в ацетонитриле за 3 дня дает 4-ме-
тилциклогексанон с 71%-ным выходом. w-Масляный альдегид
получают с 70%-ным выходом, пропуская бензольный раствор «-бу-
танола через колонку с М. д. а.
По данным Никона [14], при действии М. д. а. на стероидные
аллиловые спирты (А4-3-олы и А5-7-олы) квазиэкваториальный
изомер окисляется в 3—10 раз быстрее квазиаксиального изомера.
Бензиловые спирты можно селективно окислить реагентом Ат-
тенборо, как это видно на следующем примере [15]:
СН3Оч ОН МпО3; Щ; 5 час
л-*—— ч Ацетон, 25°
CH3O-<f у—GHCHSCH3OH-----г----*
сн3о^ о
сн3о—сснгснаон
Белл, Джонс и Уайтинг [16] показали, что для превращения
высоконенасыщенного спирта (1) в соответствующую кислоту (3)
222
удобно окислять (1) М. д. а. до альдегида (2) с последующим окис-
лением смесью хромовой и серной кислот.
с Мп О и
снчснгсн.,с сс — ссн - снсн.он ------->
J Z - - 9 0%
(1)
—> CI 13СН.2СН2С = сс = ссн снсно
(2)
сн3сн2сн.-с сс = ссн = снсо,н
(3)
Бекер использовал окись Аттенборо для превращения про-
странственно затрудненных фенолов в хинол иды. Например, 2,4,6-
три-mpem-бутилфенол окисляют в бензоле с образованием голубого
раствора радикала (2), который реагирует с фенолом (1) с образо-
ванием хшюлидного эфира (3).
он ° в
(сн3)3с с(сн3)3 ; МпОг^СНз]зСГфАЙС^СПз)з АгОН_/СН^зСу%С^С11^3, + Ц)
с(снэ)3 с4н6 С(СНз>3 АгО'С(СН3)3
(1) (2) (3)
Азотистые соединения. Показано, что М. д. а. в эфире является
эффективным окислителем бнс-гидразопа (1) в темно-красное бис-
диазосоединение (2). Желтая окись ртути, обычно используемая
для окислений такого типа, дает низкие выходы 1181. Пратт и Мак-
Говерн [19] показали, что N-бензиланилипы окисляются с выходом
75—90% в соответствующие бепзальапилины. В этом случае окись
высушивают кипячением в бензоле в течение 5 час с водоотдели-
телем и степень окисления контролируют по количеству выделив-
шейся воды. Гидразобензолы эффективно окисляются этим методом
ArCH2NHArf + МпОа —> АгСН — \тАг' -ф Мп О -| - Н./Э
в азобензолы. Анилин окисляется в азобензол, но в этом случае
223
реагент не должен быть сухим. Сообщалось 119, 20] об окислении
с хорошими выходами некоторых замещенных анилинов в азобен-
золы; нитроанилины и амин обе изо иные кислоты не окисляются.
Ейтс [21] нашел, что М. д. а. предпочтительнее окиси ртути для
окисления 1-мезитилглиоксаль-2-гидразона в а-диазокетон; с оки-
сью ртути выход составляет лишь 75%. Аллинджер [22] исполь-
зовал эту методику в синтезе 4-карбокси-]8]-парациклофана (3)
/СНз /СН3 О
H3C—f COCH = NNHa — Н3С—<С --------с—CH = N
ХСН3 ХСН3
из 4,5-дикето-[91-парациклофанмоиогидразона (1).
Келли [23] показал, что М. д. а. легко окисляет фенилгидра-
зиды в водной уксусной кислоте при комнатной температуре с об-
разованием соответствующих кислот с хорошими выходами; аро-
матическими продуктами реакции являются бензол (30%), фенол
(27%) и фенилацетат (41 %). Реакция была применена к модельным
дипептидам на примере у-фенилгидразида N-карбобензокси-а-ь-
О Н Н
II I I МпО2
RC—N - NC6Hfi—RCO2H + N2-PC6H6
с U У U уо
глутамил-ь-метиогшнметилового эфира (4); при этом с хорошим
выходом была получена карбоновая кислота (5) без затрагивания
защитных карбобензокси- и сложноэфириой групп. Полученные
результаты говорят о возможности использования фенилгидра-
зидной группы для защиты карбоксильных групп в синтезе пептидов.
НОН НОН
I II I МпО2 | || |
CbN—СНС—- NCHCOaCH3 —CbN—СНС- МСНСО2СН3
I I 73/e I I
СНа СНа СНа СН2
с:на ^н2зсн3 iH2 ch2sch3
nc6h5 toaH
II (5)
Н H
(4)
224
Хенбест Г24] показал, что под действием большого избытка
М. д. а. N-алкил- и М,]\Т-диалкиланилины окисляются по трем
направлениям:
\хсн3 >NCHO
\nch2r -^NH+O-CHR
^NCH2CHaR-^- CHr] У NCHO + O — CHR
Co многими аминами осуществляется преимущественно одни
тип реакции; например, диметиланилин дает N-метилфор манил ид
с выходом 80%. Три-н-ал кил амины окисляются в N-диалкилфор-
мамиды [25]:
МпО^
(CH3CH2CH2CH2)3N —(CH3CH2CH2CH2)2NCHO
4 и %
Х,5Г-Дифеиилпиразолидин (2) получают окислением М. д. а.
1,3-дианилинопропана. Виттиг и сотр. [26], используя окись Ат-
тенборо, получили (2) с выходом 31%; Даниэльс и Мартин [27],
использовав МпО,, полученную по Хенбесту, Джонсу и Оуэну
[28], увеличили выход до 45%.
C6H5NH HNQHs
МпО2
45% >
СбН5Ы----NC6H3
(Ч
(2)
Джансен и сотр. [29] показали, что окись Аттенборо — лучший
реагент для дегидрирования индолина и родственных соединений
типа (1) до индолов (2). С перекисью никеля, следующим по эф-
фективности реагентом, выход составлял только 33%. Реймлингер
МпО2-СН2С12
24 час при 20%
6 4%
(2)
[30] считает М. д. а. и окись серебра лучшими окислителями диарил -
гидразонов в диарилдиазометаны; другие окислители дают в ка-
честве побочных продуктов азины.
Агч Агч
С = N NH2 с = N N
Аг/ Аг/
8 Заказ № 1096
225
Диалкилсульфиды. Ди-и-бутилсульфид и дибензилсульфид окис-
ляют в сульфоксиды продолжительным встряхиванием их раст-
воров в петролейном эфире с М. д. а. 1311. Диаллилсульфоксид
был получен этим методом с выходом 13?6.
МлО.>, 7'2 чае
(Сан5сн.,д5 ———-- (сддсн.рз — о-
7 4 %
Другие примеры окисления. Реагент, полученный в работе [51,
использовали для окисления пирндилкарбинолов в соответствую-
щие альдегиды, меркаптанов — в дисульфиды, алифатических
а-кетолов — в а-дикетоны, N-фенилгидроксиламина — в нитро-
зобензол. Суспензию М. д. а. в эфирном растворе субстрата энер-
гично перемешивают в течение 5—6 час, окись удаляют и промы-
вают эфиром. Фильтрат и промывные воды концентрируют при
пониженном давлении и продукт выделяют перегонкой или кри-
сталлизацией.
I. В а 1 I S., Goodwin Т. W., М orton R. A., J. Biochem., 42, 516
(1948).
2. W е л d 1 е г N. L., S 1 a t е s Н. L., Т i s h 1 е г М., J. Am. Chem. Soc.,
71, 3267 (1949).
3. Farrar К. R., Hamlet J. C., Henbest FL B., Jones E. R. H,
Chem. Ind., 1951, 49.
4, Attenburrow J., Camero n A. F. В., C h a p m a n J, H.,
Evans R. AL, H e m s B. A., J aiiscn A. B. A., Walker T.,
J. Chem. Soc., 1952, 1094.
5. Mancera O., Rosenkranz G., Sondhe imer F., J. Chem. Soc.,
1953, 2189; Sondheimer F., Mancera O., Urqulza AL, R o-
senkranz G., J. Am. Chem. Soc., 77, 4145 (1955).
6. Harfcn is! M., Bavley A., Lazier W. A., J. Org. Chem., 19,
1608 (1954).
7. G г i t t e r R. L., Wallace T. J., J. Org. Chem,, 24, 1051 (1959).
8. Braud e E. A., Coles J. A., J. Chem. Soc., 1950, 2014; 1952, 1430.
9. Crombie L., Crossley J., J. Chem. Soc., 1963, 4983.
10. S t о r k G., Tomasz M.T J. Am. Chem. Soc., 86, 471 (1964).
11. Pratt E. F., Van de C a s t 1 e J. F., J. Org. Chem., 26, 2973 (1961).
12. Sondh ei mer F., Amen dol laC., R ose n kr anz, G., J. Am. Chem,
Soc., 75, 5932 (1953).
13. Harrison I. T., Proc. Chem. Soc., 1964, 110.
14. Nickon Д., В a g 1 i J. F., J. Am. Chem, Soc., 83, 1498 (1961); N ickon
A., Schwartz N., Di-Giorgio J. B., Widdowson D, A.,
J. Org. Chem., 30, 1711 (1965).
15. Adler E., Becker H.- D., Chem. Scand., 15, 849 (1961); see also P a-
p adopou los E. P., J а г r a r A., I s s i d о r i d e s С. H., J. Org.
Chem., 31, 615 (1966).
16. В e 1 1 I., J о ii e s E. R. FL, Whiting M. C., J. Chem. Soc., 1958, 1313.
17. Becker H.-D., J. Org. Chem., 29, 3068 (1964).
18. M urray R. W-, T г о z z о 1 о A. AL, J. Org. Chem., 26, 3109 (1961).
19. Pratt E, F-, McGovern T. P., J. Org. Chem., 29, 1540 (1964).
20. Wheeler О. H., Gonzalez D., Tetrahedron, 20. 189 (1964).
21. Morrison II., D a ii is h e is k у S., Y at es P., J. Org. Chem., 26, 2617
(1961).
22. A I 1 inger N. L,, F re iberg L. A., Hermann R. B., Al filer
Al. A., J. Am. Chem. Soc., 85, 1171 (1963).
226
23. К е 1 1 e v R. В., J, Org. Chem., 28, 453 (1963); Kelley R. В., Um fe-
re it G. R., Liggett W. R., ibid., 29, 1273 (1964).
24. Henbest H. B., Thomas A., J. Chem. Soc., 1957, 3032.
25. Henbest H. B„ Stratford M. J. W., Chem. Ind., 1961, 1170.
26. W i t t i g G, Joos R., R at hfcl dcr P., Ann., 610, 180 (1957).
27. Daniels R., Mart in B. D., J. Org. Chem., 27, 178 (1962).
28. Henbest H. B., J ones E. R. 11., Owen T. C., J. Chem, Soc.,
1957, 4909.
29. Jansen A. B. A., Johnson J. M., Surtees J. R., J. Chem, Soc.,
1964, 5573.
30. R e 1 in 1 i ii g e г H., Chem. Ber., 97, 3493 (1964).
31. Edwards D., S tenl a к e J. B., J. Chem. Soc., 1954, 3272.
МАРГАНЦА ДВУОКИСЬ ОБЫЧНАЯ. Мол. вес 86,93.
Ранняя методика [II, являющаяся быстрым, хотя и неэффек-
тивным способом получения толухипона, заключается в перегонке
с водяным паром смеси о-толуидина, М. д. о. и серной кислоты.
Реагент используют для окисления пиридоксина (1) до пиридок-
саля (2) [2] и пиридоксаминфосфата (3) до пиридоксальфосфата
(4) [3].
сн,он сно
(1) (2)
chsnh2 сно
НО.Х У /CH.OPOsH,, НО ,.СН.ОРО3Иг
|| Х| 1! Т
НзС V У
(3)
(-1)
Фурфурол (5) под действием М. д, о. и концентрированной
соляной кислоты дает с 75°6-ным выходом соединение, известное
как мукохлористая кислота (7) [4]. Промежуточный продукт иден-
тифицирован как Р’Хлор-у-оксилактоп (6) [5].
(5) (6) (7)
1. С 1 а г k Т. Н., J. Am. Chem, Soc., 14, 565 (1892).
2. Н с у 1 D., J. Am. Chem. Soc., 70, 3434 (1948).
3. Wilson A. N., Harris S. A., J. Am. Chem. Soc., 73, 4693 (1951).
4. Yanagita M., J. Pharm. Chem. Soc. Japan., 72, 1383 (1952).
5, H acb ih ama Y., Shon о T„ I к e d a S., J, Org. Chem,, 29, 1371 (1964).
8*
227
МЕДИ(П) АЦЕТАТ БЕЗВОДНЫЙ, Си(ОСОСНя)о. Мол. вес
181,63.
Безводную соль получают кипячением ее гидрата с уксусным
ангидридом; нерастворимый продукт промывают сухим эфиром ГН.
М. а. б. применяют в смеси пиридин — метанол для окислительной
дегйдроконденсацип терминальных ацетиленов в диины 12—4].
Механизм см. 121. Обзор см. [5|.
2RC СН Си10АрД RС = СС = CR
Внутримолекулярная окислительная дегидроконденсация
со,со-диацетиленового эфира (1) в диинолид (2) является ключевой ста-
дией в новом синтезе макроциклического лактона экзальтолида [6].
О сЦОАсЦ-Ру
НС=С(СН,)эСО(СНг)3С=СН ~СбНб >
(СН2)е (СН2)2 pt> (снг)14 Р
^_JL. У Ю
1. S р a t Ii Е., Si'tzungsber. Akad. Wiss. Wien., 120, 117 (1911).
2. С 1 i f f о r d A. A., W atcrs W. A., J. Chem. Soc., 1963, 3056.
3. E g 1 i n t о n G., G a 1 b r a i t h A. 1-?., J. Chem, Soc., 1959, 889.
4. Sondheinier F., Wolovsky R., J. Am. Chem. Soc., 81, 1771 (1959);
Sondheimer F. el al., J, Am. Chem. Soc., 81, 1771 (1959),
5. Эл и iitoii Г., ЛА а крае В., «Успехи органической химии», изд-во
«Мир», М., 1966, том 4, стр. 239.
6. McCrae W., Tetrahedron, 20, 1773 (1964).
МЕДИ (I I) АЦЕТАТ, МОНОГИДРАТ, Сн(ОСОСН3)г-Н.,0. Мол.
вес. 199,64.
а) М. а. м.— один из наиболее употребительных реагентов для
выделения 1,3-дикетонов в виде медных хелатов: ацетилацетоп Ш,
диизовалеро идметан [2], диацетоуксуспый эфир 13], Дикетон или
его метанольный раствор обрабатывают водным раствором М. а. м.
П, 31,
б) Этет реагент является эффективным катализатором моноци-
анэтилирования ароматических аминов [4]:
/,---Ы СщОАсЦ /—— - х
" Ы— Nl-Io + CHa-CHCN-------' Д Д- NHCH2CH2CN
/ 53 —э6% .- /
'-Ci ХС1
в) М. а. м, окисляет а-кетолы в а-дикетоиы; например, себа-
циноип (1) окисляется в себапил' (2); реакцию проводят в смеси
228
метанол — водная уксусная кислота; при 75° окраска реакционной
смеси изменяется от синей до красной Г51. Стероидные в-гомокетолы
(3) дают диосфенолы (4) [6], а стероидный кетодиол (5) дает диен-1,4-
Си(ОАс)2
88 -89%
он-З-ол-4 (6) [7]. а-Кетол боковой цепи стероида (7), например кор-
тизона, под действием М. а. м. в метаноле легко окисляется (30 мин)
до глиоксаля (8), который затем медленно перегруппировывается
Са(ОАс^
------->
под действием окислителя и в результате реакции с метанолом
дает эпимерные метилгликоляты (9) и (10) [8]. Глиоксаль (8) можно
получить с выходом около 90% при окислении кетола (7) воздухом
в присутствии М. а, м. как катализатора [91; при соотношении
М. а. м. : стероид, равном 1 : 8, окисление завершается менее
чем за 1 час.
сн2он
Си(ОАс)
СНЭОН
соосн3
носы
(8) (9)
Кэмпбелл и Эглинтон [10] осуществили окислительную диме-
ризацию фен ил ацетилена в дифенилдиацетилен по Глазеру под
действием М. а. м. в смеси пиридин — метанол. Темно-синяя сус-
пензия приобретает зеленую окраску при кипячении в- течение.
229
1- час. Охлажденную смесь подкисляют прн перемешивании и ох-
лаждении и продукт экстрагируют эфиром.
70-УСЦ,
,—Кипячение; H..SO.
< С = СН + CufOAch • Н<,0 4- Ру СН3ОН
2 г 5,5 г 10 мл 10 мл
—> ±_С^С-С^С±
Берч и Смит [111 нашли, что М, а. м. лучше обычно использу-
емой бромистой меди в качестве катализатора 1,4-прпсоединенпя
реактива Гриньяра к ос, 0-непредельным кетонам. Так, 19-норте-
стостероп (1) в присутствии бромистой меди дает с высоким выходом
продукт 1,4-присоединен и я метилмагпийоидида:
(Ч (2)
1. Де лун К., «Синтезы органических препаратов», ИЛ, М., 1952, сб. 3,
стр. 92.
2. X а у 3 е Р Ч-, Адамс Дж., Левин Р., «Синтезы органических препа-
ратов», ИЛ, М., 1953, стр. 160.
3. Спасов А., «Синтезы органических препаратов», ИЛ, АТ, 1952, сб. 3,
стр. 513.
4. Heininger S. A., J. Org. Chem., 22, 1213 (1957); Org. Svn., Coll. Vol.,
4, 146 (1693).
5. Б л о м к в и с т А., Г о л ь д ш теин А., «Синтезы органических препа-
ратов», ИЛ, М., 1958, стр. 84.
6. Wendler N. L., Т a li b D.,Gr aber R. Р., Tetrahedron, 7, 173 (1959).
7. Sasaki К., Chern. Pharm. Bull., 9, 684 (1961).
8. Lewbarl M. L., M a t t о x V. R.T J. Org. Chem., 28, 1773, 1779, 2001
(1963).
9. Lewbarl M. L-, Mattox V. R., J. Org. Chem., 28, 2001 (1963).
10. Campbell 1. D., Egl 1 л to n G., Org. Syn., 45, 39 (1965).
11. В 1 r c h A. ,1., Smit li M.. Proc. Chem. Soc., 1962, 356; see also M a г s-
11 a 1 1 J. A., pant a \V. I., Roebk e H., J. Org. Chem., 31, 1016 (1966).
МЕДИ ЗАКИСЬ, Cu,O, Мол. вес 143,08.
Видон и сотр. 1.11 получили 1,4-дизамещенвые винилацетилены
с выходом 30—60% путем добавления терминального ацетилена
к раствору каталитического количества М. з, в горячей уксусной
кислоте. Реакция, по-видимому, включает первоначальное образо-
СнП А,с
2RC СИ ±1—2% R СН = СНС CR
ванне ацетиленида меди.
230
Бэкон п Хилл [2] применили М. з. в апротонном растворителе
(обычно ДМФА, а также ДМСО) в качестве катализатора реакции
нуклеофильного замещения галогена в арилгалогенидах. Эта
реакция имеет особое значение при получении ароматических
простых эфиров и тиоэфиров;
SCfiH5 Вг OCriHs
М, з. имеет преимущество перед другими производными меди
как реагент и катализатор при получении диариловых эфиров мо-
дифицированной конденсацией Ульмана [ЗБ
1. Garwood R. I7., О s к а у Е., Weedon В. С. L., Chem. Ind., 1962,
1684,
2. Bacon R. G. R., H i I 1 H. A. O., J. Chem. Soc., 1964, 1108.
3. Вас о n R. G. R., S 1 e w art O. J., .1. Chem. Soc., 1965, 4953.
МЕДИ(1) ИОДИД, Cui. Мол. вес 190,45.
Перегруппировка Вольфа (реакция Арндта — Эйстерта). Для
проведения перегруппировки в гомогенных условиях этот реагент
используют в смеси ацетонитрил — метанол, например в случае
О
!' + - Си!
С6Н5ССН -N N + СН3ОН—СН3СХ СеН-СН2СО2СН3 + N3
75 — 85%
а-диазоацетофенона [1|. Смесь выдерживают в течение нескольких
часов при комнатной температуре и получают метиловый эфир фе-
нилуксусной кислоты с хорошим выходом.
1. Y а t cs Р., Bugger J., Chem. Ind., 1957, 1511.
МЕДИ(П) КАРБОНАТ ОСНОВНОЙ, СиССБ -Сн(ОН)п. Мол. вес
221,11.
Реагент получают добавлением эквивалентного количества раст-
вора карбоната натрия к раствору сульфата меди и промыванием
осадка до освобождения от сульфат-иопа. Влажный М. к. о. при-
меняют без высушивания и взвешивания. В присутствии гидроокиси
аммония реагент является эффективным окислителем [1|:
снон
2 !
с- о
I _
СиСОИ-'ЫОН), С-° . „
------------2 I 2Cu СО2 ЗгЫО
С-0
Декарбоксилирование. Смесь 15 г бензоилбензойной кислоты
и 0,5 г М. к. о, осторожно нагревают до расплавления кислоты,
выделяющуюся воду удаляют в вакууме при нагревании на
231
кипящей водяной бане и остаток нагревают до прекращения вы-
деления СО2 при 265° (30 мин) 12].
1, Т оттер Дж., Дарби У., «Синтезы органических препаратов», ИЛ,
М., 1952, сб. 3, стр. 472.
2. Org. Expts., Chapt. 40.
МЕДИ(П) НИТРАТ — ПИРИДИН. Этот комплекс в присутст-
вии триэтиламина как основания является катализатором реакции
окисления Д5-холестенона-3 до Д4-холестендиона-3,6 ]1]. Выход
при этом вдвое больше, чем при окислении бихроматом [2].
l.Volger Н. С., Brackman W., Rec. trav., 84, 579 (1965).
2. F i e s e r L. F., Org. Syn., Coll. Vol., 4, 189 (1963).
МЕДИ(П) НИТРАТ — УКСУСНЫЙ АНГИДРИД. Реагент при-
меняют для нитрования, которое, по-видимому, осуществляется
через ацетилнитрат [1]. В реакциях, показанных на схеме [2, 3],
нитрат меди добавляют в кристаллическом виде или в виде
суспензии в уксусном ангидриде к раствору субстрата в уксусном
ангидриде.
232
1. B a c h a r a c h G., J. Am. Chem. Soc., 49, 1522 (1927).
2. Anderson A. G., Jr., Nelson J. A., Tazuma J. J., J. Am. Chem.
Soc., 75, 4980 (1953).
3. WilliamsK. 1. H., Cremer S. E., KentF. W., Sehm E. J., Tar-
bell D. S., J. Am. Chem. Soc., 82, 3982 (1960).
МЕДИ(П) СУЛЬФАТ, CuSO4-5H2O. Мол. вес 249,69.
Окисление. М. с. в смеси пиридин — вода окисляет а-кетолы до
ос-дикетонов; отработанный раствор можно регенерировать окис-
лением воздухом [1].
CttSO+-Py(90°)
СЙН5СН—ССеН5-------—--------> СвН3С-ССйН5
I II 86 % II II
ОН О 0 0
Инданондиоксикарбоновая кислота (2) выделена в качестве
промежуточного соединения при окислении по Хукеру 2-окси-З-ал-
килнафтохинона-1,4 (1) в его низший гомолог (3). Кислота (2)
получается с высоким выходом при действии на (1) перекиси во-
дорода в присутствии карбоната натрия и далее окисляется в (3)
с очень хорошим выходом действием М. с. в щелочном растворе 121.
Катализатор гидролиза. М. с. катализирует гидролиз арил йоди-
дов в фенолы; бромиды и хлориды гидролизуются медленнее [31.
В случае 5-иодванилина реакционную смесь кипятят 4,5 час при
перемешивании в атмосфере азота.
СНО
у й Кипячение 4,5 час
+ CuSO4.5H2O + 4H.NaOH — -—------->
I | О О /о
ОН
2,8 г
СНО
СН3О I он
он
233
1. Кларк, Дреджер, «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 83.
2. F i с s е г L. F., F i с s е г М., J. Am. Chem. Soc., 70, 3215 (1948).
3. Banerjee S. К., M а л о 1 о p о u 1 о М., Pepper J. М., Can. J. Chem.,
40, 2175 (1962),
МЕДИ(П) ХРОМИТ (ката лизатор Лэзира). Для получения
катализатора [II к водному раствору нитрата бария и тригидрата
цитрата меди добавляют при перемешивании раствор хромата ам-
мония, полученный из бихромата аммония и водного аммиака.
Красновато-коричневый осадок хроматов меди, бария и аммония
промывают, сушат н разлагают прокаливанием в муфельной печи
при 350—450°. Остаток измельчают, промывают 10%-ной уксусной
кислотой, сушат и растирают в ступке в тонкий черный порошок.
Применение М. х. в качестве катализатора гидрирования ил-
люстрируется гидрированием себациноина с образованием легко
разделимой смеси цис- и /пщшс-цнклодеканднолов-1,2 [21. цис-Диол
(т. пл. 138°) кристаллизуется из смеси бензол — этанол (1 : 1),
т/^нс-диол (т. пл. 54е) — из пентана.
Синтез р-метил-6-вал еролактона из 3-метил пентандиол а-1,5 [3]
свидетельствует о том, что М. х. является эффективным катали-
затором дегидрирования. Промежуточные продукты этой реакции,
по-видимому,— оксиальдегид и полуацеталь. Реакция сопровож-
сн3 сн3 сн3 сн3
Г III
СН, хромит меди zChL /\ /''•
/ 200° 4 ii [ j
СН. СН., ----------^сн„ СИ. -L Ln -L I +2Н.>
I j “ 3 час । ’ j ! у II
СН2 СН2ОН СН.2 СНО 'L ОН С'О
ОН 01-1 90—95%
дается почти количественным выделением водорода и образованием
лактоиа с высоким выходом.
М. х. и окисный медно-хромовый катализатор эффективны для
декарбоксилирования кислот при нагревании в хинолине. На-
пример, цис-а -фен ил коричная кислота в кипящем хинолине дает
цис-стильбен; оба вышеупомянутых катализатора одинаково эф-
фективны в этой реакции [4|. Сходные катализаторы применялись
СВН5-С--CO.J1 с»-сг се115—С—И
il Хинолпп , |. — СО.
С6115— С — Н T. кип. 237° (10 мин) С[;Н5 — С — Н
234
для реакций декарбоксилирования в хинолине [5, 6]. М. х. приме-
няли без растворителя в качестве катализатора декарбоксилиро-
вания имидазол-4,5-дикарбоновой кислоты 171 и изодегидрацетовой
кислоты [81. См. Медный порошок, Медные соли.
1. Л эз ир В., Арнольд X., «Синтезы органических препаратов», ИЛ, М.,
1949,сб.2,стр.301.
2. Б л о м к в и с т А., Г о л ь д ш т е й и А., «Синтезы органических препа-
ратов», ИЛ, М., 1958, сб. 8, стр. 83.
3. L о п g 1 е у R. 1., Emerson W. S., Org. Syn., Coll. Vol., 4, 677 (1963).
4. Org. Expts., Chapt. 42.9
5. Kinney C. R., Langlois D. P., J. Arn. Chem. Soc., 53, 2189 (1931).
6. Reichstein T., Gruss пег A., Helv. Chim. Acta, 15, 1067 (1932).
7. С H a ii д с p X., X а п д p и к P., Брукс Л., «Синтезы органических пре-
паратов», ИЛ, М., 1952, сб. 3, стр. 254.
8. Б э к л с Р., Уилер Н., «Синтезы органических препаратов», ИЛ, М.,
1954, сб. 5, стр. 64.
МЕДНАЯ БРОНЗА. Обычная медная бронза не дает таких
удовлетворительных результатов в реакции Ульмана, как бронза,
активированная 2%-ным раствором иода в ацетоне [1].
NO. NO.Z NO2
1. Ф ьюзон Р., Кл и вл э кд Э., «Синтезы органических препаратов»,ИЛ, М.,
1952, сб. 3, стр. 216.
МЕДНАЯ ПУДРА — БЕНЗОЙНАЯ КИСЛОТА (т. кип. 249 ).
Смит [1] показал, что в ароматических нитрох^юрсоединениях Про-
исходит восстановительное отщепление хлора йод действием медной
пудры в расплавленной бензойной кислоте при 150—200°. Источ-
ником водорода является, по-вндимому, карбоксильная группа
кислоты. 2,4-Ди нитро-1-хлор нафталин дает в этих условиях 1,3-ди-
нитроиафталин с выходом 74%. Ньюмен |2|, применивший этот
метод для превращения 4-бром- и 4-иод-2,5,7-тринитрофлуоренонов
в 2,5,7-тринптрофлуоренон с выходом 78 и 22%, указывает на не-
обходимость строгого температурного контроля реакции в пределах
170—180° (в реакционной смеси).
1. Smit h W. Т., Jr., J. Am. Chem. Soc., 71, 2855 (1949).
2. N e w m a n M- S., В 1 u m J., J. Am. Chem. Soc., 86, 5600 (1964).
МЕДНЫЕ СОЛИ. и-Бензоилбензойная кислота декарбоксили-
руется с высоким выходом при нагревании до 260—270° с небольшим
количеством ее медной соли до окончания выделения СО2 [1]. По
более простой методике к кислоте (15 г) в перегонной колбе добав-
ляют небольшое количество основного карбоната меди (0,5 г),
смесь расплавляют и после удаления воды в вакууме нагревают
235
при 260—270° [2]. По другой методике кислоту нагревают в хино-
лине с каталитическим количеством ее медной соли [3].
о
и
Соль меди
82-84%
Т. Dougherty G., J. Am. Chem. Soc., 50, 571 (1928).
2. Org. Expts., Chapt. 40.1.
3. P i e r s E., Brown R. K-, Can. J. Chem., 40, 559 (1962).
МЕДНЫЙ ПОРОШОК- Продажный M. п. применяют как ката-
лизатор декарбоксилирования при нагревании в хинолине, например
для декарбоксилирования 3-метилпирослизевой [1] и лг-нитроко-
ричной [21 кислот.
При нагревании р-оксикислоты или ее mpcm-бутилового эфира
в хинолине со следами М. п. происходит декарбоксилирование
и дегидратация с образованием экзо-циклического олефина [3].
Си-хинолин,
85% *
Си-хинолин
СН2 <--------
Как катализатор декарбоксилирования замещенных коричных
кислот в хинолине М. п. лучше, чем сульфат или ацетат меди [4].
М. п. применяют как катализатор реакции натрия с анилином
~ 4-
с образованием натриевого производного C6H5NHNa [5]. Активный
М. п.— лучший катализатор в синтезах и-нитродифенилового [6]
и о-метоксидифенилового [7] эфиров. Активный катализатор полу-
чают [6] восстановлением сульфата меди цинковой пылью в водном
растворе; образующийся осадок промывают 5%-ной соляной кис-
лотой для удаления избытка цинка. Активный катализатор, полу-
Си (150—160°)
n-NOaC6H,C! 4- КОСеН5-- n-NO2C6H4OC6H5
О U'— О уф
Си (200°)
С6НеВг 4- КОС6Н4ОСН3-о —— ~+ СеН5ОСеН4ОСН3-о
О т’ о 1 /о
ченный аналогичным способом [8], является наиболее эффективным
в синтезе 2,2'-динитродифенила по Ульману.
236
1. Бернесе Д., «Синтезы органических препаратов», ИЛ, М., 1961, сб. 11,
стр. 50.
2. У а й л и Р., Смит Н., «Синтезы органических препаратов», ИЛ, М., 1954,
сб. 5, сгр. 51.
3. V i 1 k a s М., Abraham N. A., Bull. soc. chim. France, I960, 1196.
4. Wa 11 ing C., W о 1 f s t i r n К- B., J. Am. Chem. Soc., 69, 852 (1947).
5. В о г а и В., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4, стр. 194.
6. Брюстер Р„ Грениг Т., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 370.
7. Унгнаде X,, Орволл Э., «Синтезы органических препаратов», ИЛ,
М., 1953, сб. 4, стр. 334.
8. G о г е Р. Н., Hughes G. К.-> J- Chem. Soc., 1959, 1615.
МЕДЬ(или Cu2 + ) — АСКОРБИНОВАЯ КИСЛОТА. Анбар и сотр.
Ill нашли, что о-иодбензойиая кислота легко восстанавливается до
бензойной кислоты ионами меди(П) и аскорбиновой кислотой
в нейтральном водном растворе; n-иодбензойная кислота в тех же
условиях восстанавливается в 1000 раз медленнее. Действием
металлической меди в щелочном растворе аскорбиновой кислоты
Сейф и Мойр [2] осуществили селективное восстановление атома
иода в соединении (1) без затрагивания нитрогруппы. Для восста-
новления соответствующих бромидов метод непригоден.
NO3
СН3О I 7С0 о он он н он
Y)/ \ II 1 ! I
О-|-С*—С = С — С—С—СН3ОН + Си-бронза ф-
Д К / 1_________О—___J 0,02 г
СН3О у СН2 0,1 г
I
сн3о J, .со
Кипячение 2 час । \
4-4%-ное води. КОН —. ..> О
г - ., , 1 J /и III /
СН3о/^/чСН.;>
1. АлЬаг М., Guttmann S„ Friedman С., Proc. Chem. Soc., 1963, 10.
2, S a f e S., M о i r R. Y., Can. J. Chem., 43, 337 (1965).
МЕДЬ БРОМИСТАЯ, Си2Вг,. Мол. вес 286,91, т. пл. 504°.
Получение и применение для реакции Зандмейера [1].
Гомологизация. Этот реагент катализирует реакцию ацеталей
кетенов с диазометаном, приводящую к образованию ацеталя
циклопропанона [2].
0О>Н5 Си,вг, .ОС,Нг>
СН2 = С< “ -|-CH,Na———>сн2-с(
'ОС3Н5 \ / xOC,H54-N2
снг
В улучшенном синтезе тропилидена (5) [3] раствор диазометана
в бензоле медленно добавляют к кипящему бензолу, содержащему
М. б. в качестве катализатора. Бензол применяется в большом
237
избытке, и продукт легче всего выделяется фильтрованием (отде-
ление катализатора) с последующим добавлением фильтрата к раст-
вору РС1Г, в СС14. Хлористый тропилий отделяют, растворяют
в воде и обработкой хлорной кислотой получают перхлорат тро-
пилия с выходом 85%. Эффективность этого метода объясняется
образованием промежуточного соединения (3) с электрофильным
атомом углерода, стабилизированного в комплексе с металлом.
Троп ил идеи выделяют из перхлората восстановлением боргидридом
натрия и экстракцией эфиром. Как и ожидалось, моноалкилбен-
золы образуют смесь трех продуктов расширения цикла прибли-
ch2n2
----г__2^
Си2В г 2
(3)
зительно с одинаковым выходом [4]. При температуре 120—190°
нафталин дает смесь углеводородов (1) — (3); углеводород (2)
перегруппировывается в (1) при нагревании до 260° 15]. Фенантрен
образует два продукта.
Катализатор 1,4-присоедииения реактива Гриньяра. В присутст-
вии М. б. в качестве катализатора метилмагнийбромид реагирует
с /А1(9)-окталоиом-2 только по схеме 1,4-присоединения с образо-
ванием 9-метил-цис-декалона-2 [61.
I. Вис k J. S., Ide W. S., Org. Syn., Coll. Vol., 2, 132 (1943).
2. D и 1 1 M- F., Abend P, G., J. Am. Chem. Soc., 81, 2588 (1959).
3. M ii 1 1 e r E, Fricke H., Ann., 661, 38 (1963).
4. M й 1 1 e r E., Fricke H., Kessler H., Tetrahedron Letters, 1501
(1963).
238
5. M u 1 1 е г Е. et al., Tetrahedron Letters, 1525 (1964).
6. Birch Л. J. , Robinson R., J .Chem. Soc., 1943, 501; see also Church
R. F., Ireland R. E.,Shridhar D. R., J. Org. Chem., 27, 707 (1962).
МЕДЬ БРОМНАЯ, CuBr2. Мол. вес 223,37, т. пл. 498°, черное
твердое вещество, хорошо растворимое в воде (зеленый раствор).
Бромирование карбонильных соединений. Коши [1] показал,
что хлорная медь реагирует с ацетоном по следующему уравнению:
CH3COCH3 + 2CuC12 С1СН2СОСН3 + 2СиС1 -pHCl
В последующих работах была использована также более реакцион-
носпособная М. б. Изофорон реагирует с 14. б. в метаноле при
комнатной температуре с образованием а-бромпроизводного (1),
которое неустойчиво в присутствии НВг и метанола и перегруппи-
ровывается в 3,4,5-триметиланизол (2) [21. Последующее броми-
рование (2) дает (3) с хорошим выходом.
М. б. реагирует с а, р-непредельным и альдегидами в спирте,
как показано на примере акролеина [31:
CHa = CHCHO4-3EtOH + 2CuBr.,— EtOCH2CHCH(OEt)2 + 2CuBr-R
6 7 % |
Br
-i-HBr + H.2O
При взаимодействии М. б. с Л4-3-кетостероидом в метаноле об-
разуется 6-бромпроизводное, которое за счет реакции с раствори-
телем дает бр-метоксипроизводное [4]. Этот реагент был исполь-
зован для бромирования 17-кетостероидов, прямое бромирование
которых приводит к плохим результатам [5]. Одно из преимуществ
239
состоит в том, что при бромировании не затрагивается 5,6-двойная
связь [6].
Дойфоуд и Мар аз ей [7], а также Кинг и Острум [81 нашли, что
при взаимодействии М. б. с фенолом, содержащим ацетильную
группу, в отличие от реакции с бромом или с NEC наблюдается
преимущественная атака ацетильной группы, а не ароматического
СН3ОЧ /ОН
Кипячение 3 час 'у и
45% I ||
^/\С0СНгВг
СН3Оу /х^/ОН
4-СиВг3-Р Диоксан
Ух 'к 4,5 г 50 ял
ХСОСН3(0.02 моля)
1,75г
(0,01 моля)
цикла. Однако экспериментальные условия у этих авторов сильно
различаются. В первой работе [7[ реакцию проводили в гомогенной
среде в диоксане с выходами на двух примерах 45 и 54%. Другие
авторы применяли гомогенный раствор в воде, спирте или ДМФА.
Однако в работе [8] было найдено, что лучше всего применять
0,03 моля в
25 мл CHCI,,
-f- CuBr.2 Этилацетат
0,05 25 мл
мол я
Перемеш ивание
и кипячение
около 100%
/. /ОН
III
^/ЧСОСН2ВГ
гетерогенную суспензию тонкоизмельченнон М. б. (80 меш) в смеси
СНС13 — этилацетат. Суспензию М. б. в этилацетате кипятят при
перемешивании магнитной мешалкой и обрабатывают хлороформ-
ным раствором кетона, взятым во избежание дибромирования в не-
большом избытке. Бурная реакция сопровождается выделением
бромистого водорода, плохо растворимого в этой системе раство-
рителей, и превращением черной М. б. в белую бромистую медь.
Завершение реакции определяется по прекращению выделения
бромистого водорода, полному исчезновению черного осадка и из-
менению зеленой окраски раствора до янтарной. Бромистая медь
практически нерастворима и легко удаляется фильтрованием,
со-Б ром кетон образуется с почти количественным выходом. Полу-
чающийся раствор можно использовать без выделения этого лак-
римогенного продукта. Так, например, получен чистый бромистый
2'-оксибензоил метил пиридиний с выходом 85—93%.
Бромирование ароматических соединений. Углеводороды бро-
нируют при кипячении с этим реагентом в СС14: антрацен дает 9-бром-
антрацен с выходом 99%, пирен— 1-бромпирен с выходом 94%
[9]. Бромирование антрацена в нитробензоле при 210° дает 9,10-
дибромантрацен с низким выходом [10].
В абсолютно безводных условиях М. б. реагирует с толуолом
с образованием о-бромтолуола (63%) [11]. В присутствии небольших
количеств воды в основном образуется смесь фенилтолилметанов,
СсН5СНйС6Н4СН3.
240
Реакция с этиленовыми и ацетиленовыми соединениями. Под
действием М. б, осуществляется галогенирование изолированной
двойной связи [12]. Ацетилены с тройной связью внутри цепи
претерпевают транс-галогенирование, а терминальные ацетилены
превращаются в трибромолефины:
Кипячение в СН3ОН
СН2 - СНСН2ОН 2СиВга-------ВгСН,СНСНгОН + 2СиВг
9 9 % ।
Вг
Кипячение в СН3ОН ОН г... zBr
24 час ь \ /
С6Н5С ’= СС0Н5 + 2СиВг2— ~~ >• С = С -р 2СиВг
О 1 /о / \
Вг/ \н5
Кипячение в СГЦОН
[ 5 час
С6 н 5С Eze СН -Т 4Сц В г2 ~ ’
о ' /о
С6Н5С = СВга + 4CuBr + НВг
Вг
1. К о с h i J. К,, J. Am. Chem. Soc., 77, 5274 (1955).
2. Fort A. W-, J- Org. Chem., 26, 765 (1961).
3. Castro С. E., J. Org. Chem., 26, 4183 (1961).
4. Soilman P. B., D о d s о n R. M., J. Org. Chem., 26, 4180 (1961).
5. Glazier E. R., J. Org. Chem., 27, 2937 (1962).
6. G 1 a z i e г E. R., J. Org. Chem., 27, 4397 (1962).
7. Doifode К. B., Marathey M. G., J, Org. Chem., 29, 2025 (1964).
8. King L. C., Ostrum G. K., J. Org. Chem., 29, 3459 (1964); procedure
submitted to Org. Syn.
9. Nonhebel D, C., Proc. Chem. Soc., 1961, 307.
10. Ware J. C., Borchert E. E., J. Org. Chem. 26, 2263 (1961).
11. Kovacic P., Davis К,- E., J. Am. Chem. Soc., 86, 427 (1964).
12. C a s t г о С. E., Gaughan E. J., J. Org. Chem., 30, 587 (1965).
МЕДЬ ХЛОРИСТАЯ, Cu3CI3. Мол. вес 197,99, т. пл. 422°, очень
слабо растворима в воде.
Получение и применение М. х. в реакции Зандмейера [1].
Реакция Зандмейера. Эта реакция в случае 2,6-динитроанилина
представляет особый интерес, так как является специфическим
методом диазотирования этого слабого основания [2]. Твердый
нитрит натрия прибавляют при перемешивании в течение 10—
15 мин к конц. серной кислоте и смесь нагревают при 70° до раст-
NH3 N==NC1
o2n4 I /no2 o2nx /Jx/no2
II H-NaNO2H-KoHU. H2SO4—> I II —ь
0,22 моля 160 .it.?
0,2 моля
в 400 мл АсОН
С1
0,44 моля Си2С1, °-N\ /NO3
в 400 .v.j конц. HCI
71-74%
24!
ворения твердого осадка. К раствору при охлаждении до 25—30°
и перемешивании добавляют раствор 2,6-динитроанилина в теплой
уксусной кислоте с такой скоростью, чтобы температура не пре-
вышала критическую (40°). Раствор соли диазония приливают в те-
чение 5 мин в охлажденный льдом раствор М. х. в конц. соляной
кислоте. Затем температуру повышают до 20° для разложения
комплекса и после прибавления равного объема воды раствор
оставляют для кристаллизации желтого 2,6-дииитрохлорбензола
(т. пл. 86—88°).
Реакция Барта. Для получения п-нитрофенил мышьяковой кис-
лоты L3I смесь борфторида /г-нитрофенилдиазония и воды добавляют
NssNBFp AsO3H2
NaOHAt
+ NaAsO. -ф NaOH Щ Cu2Cl,
0,4 моля 1,4 моля 6 г
NO., N03
0,25 моля
300 мл воды
к суспензии М. х. в растворе метаарсенита натрия и едкого натра
в 600 мл воды. Смесь нагревают, прибавляют дополнительное
количество едкого натра и, наконец, соляную кислоту для выделе-
ния мышьяковой кислоты из ее соли.
Синтезы Гаттермана — Коха. Методика синтеза п толуилового
альдегида [4] состоит в перемешивании суспензии М. х. и хлори-
2,17 моля СсНоСНа
30 г Cu.CI. 2 моля А1С13 /~..х
СО + НС1-----НСОС! — Н3ССНО
стого алюминия в толуоле, пропускании тока СО и НС1 с после-
дующим гидролизом получающегося комплекса льдом.
Замещение ароматического галогена. 4-Бром-о-ксилол превра-
щают в соответствующий амин, применяя смесь М. х. п металли-
Вг
4-28% NH3-H,O4-Cu3C12 4-Си (проволока)
G10 мл 12s 14г
14 нас при 195°
79% (сырoil)
3
СН3
1,08 моля
NH3
С1!3
242
ческой меди в качестве катализатора 15]. Реагенты нагревают до
195° в качающемся автоклаве в течение 14 час.
Полупродукт в синтезе важного растворимого кубового кра-
сителя — 2-ацетиламино-З-хлораитрахинон (2) получается только
многостадийным синтезом. Однако соответствующее 3-б ром произ-
вол мое (1) можно легко получить с высоким выходом бромированием
2-а миноа игра хи попа и последующим ацетилированием. Харди
и Фортепбо 161 нашли, что бромид (1) под действием М. х. в «-пико-
лине при 130° дает хлорид (2) с почти количественным выходом.
а-Пыколни, 130°
Катализатор реакции присоединения реактива Гриньяра. Караш
17] обнаружил, что в реакциях реактива Гриньяра с а, 0-ненасы-
щенными кетонами, катализируемых М. х., происходит преиму-
щественное 1,4-присоединение по сравнению с 1,2-присоединением.
В отсутствие катализатора изофорон реагирует с метилмагнийбро-
мидом независимо от способа прибавления реагентов почти исклю-
чительно с образованием продукта 1,2-присоединения. Если же
Изофорон
82,5% ( + 7% диена)
кетон добавлять к раствору метилмагнийбромида, содержащему
1 мол. % М. х., присоединение осуществляется почти исключительно
в 1,4-положение.
В обычных условиях реакции Гриньяра н-бутилмагиийбромид
присоединяется к smop-бутилкротоиату с образованием втор-бутл-
243
левого эфира 3-метилэнантовой кислоты с указанным ha схеме
выходом [8]. Прибавление 1,4 г М. х. (семь порций) в процессе
СН3СН =СНСО2СНСН2СН3 + H-C4H9MgBr--—н-С4Н9СНСНгСОаСНСН,СН3
i 68—(8% I |
СНз СНз СН3
0,4 моля 1,3 моля
обработки реактива Гриньяра сложным эфиром повышает выход
до 80—85%. Реакция присоединения н-бутилмагиийбромида к втор-
бутилсорбату в отсутствие катализатора дает продукты как 1,2-,
так и 1,4-присоединения; в присутствии М. х, присоединения в по-
Си.СЦ
СН3СН = СНСН = СНСО2СНСН2СН3 + w-C4H9MgBr
I * %
СНз
СН3СНСН = СНСН2СО2СНСНХН3
I I
С4Нэ-н СН3
ложение 1,2 не происходит, и реакция дает почти исключительно
продукт 1,4-прпсоединения [9]. Сопряженное присоединение ме-
CH3MgBr(Cu2Clz)
90%
сн3
с=о
тилмагнийиодида к Д16-20-кетостероидам осуществляется с выходом
90% и более в присутствии катализатора М. х. в ТГФ 110].
Катализатор дегидроконденсацни терминальных ацетиленов.
М. х. катализирует окисление терминальных ацетиленов в смеси
метанол — пиридин под действием кислорода или воздуха. Катали-
затор применяют в количестве 0,012 моля на 1 моль ацетилена.
Реакция завершается быстро и диацетилены получаются с высокими
выходами 111]. Проведение реакции при избыточном давлении
кислорода (0,7—1,0 атм) еще более сокращает время реакции 112].
СНз Сийсц
I О2, Ру —сн3он 1
2 СН 3С—С = СН------ -- СН 3С—С = С - С С - ССНз
ОН ОН ОН
В другой методике 113] в качестве катализатора используют ком-
плекс М. х. и П,П,КГ,КГ-тетраметилэтилендиамина (1 : 1). Этот
комплекс растворим во многих органических растворителях, ре-
акция завершается быстро и выходы высокие. Этот реагент ката-
лизирует также реакцию сочетания терминального ацетилена
244
с б ром ацет иленом [141; выходы в пределах 70—95%. Оба эти
Способа синтеза диацетиленов имеют большое значение в оргаии-
Си +
Водн. C2HsNHs
СвН5С СВг+ НС СС(С6Н5)2--------87%-----» С6Н5С СС = СС(С6Н5)2
он он
Т. пл. 86°
ческом синтезе (обзор [15]). М. х. катализирует конденсацию тер-
минального ацетилена с хлористым аллилом, приводящую к об-
разованию енина-1,4 [16]. В смеси с основными аминами М. х.
Си -1-
RC = СН ш С1СНаСН = СН2----RCССНХН СН.,
катализирует сочетание терминального ацетилена, с алленгалоге-
н идами [17].
Си2С12-СЛЦНН2
ДМФА
(СН3)гС=С = СНВг+НС=ССН.2ОН---------------->
(СН3)2С = с = снс^ ССН2ОН
Фотохимическая перегруппировка. Облучение л-комплекса цис-
цис- циклооктадиена-1,5 с М. х. дает трицикло-[3,3,0,02,6]-октан —
исключительно стабильный углеводород [18]:
Cu2Cl2, ДУ
30%
1. Марвел л, МакЭл ьвсн, «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 491.
2. Гэнстон Ф., Т е к к е р С., «Синтезы органических препаратов», ИЛ, М.,
1953, сб. 4, стр. 532.
3, Рэдди А., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 378.
4. Колеман Дж., Крэг Д., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 464.
5. Виз анский В., Ансбахер С., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 170,
6. II а г d у W. В., For tenb augh R. В., J. Am. Chem. Soc., 80, 17[6
(1958).
7, Rharasch M. S,, Tawney P. O., J. Am. Chem. Soc., 63, 2308 (194!).
8. Munch-Petersen J., Org. Syn., 41, 63 (1961).
9. Munch - Petersen J., В re t t i ng C., J orgenscnP. M., R ef n
S., Andersen V. K-, J art A., Chem. Scand., 15, 277 (1961).
10. Heusler K- et al., Helv. Chim. Acta, 42, 2043 (1959).
11. Stansbury H. A., Jr., P г о о p s W. R., J. Org. Chem., 27, 320 (1962).
12. Org. Expts., Chapt. 14.
13. Hay A. S., J. Org. Chem., 27, 3320 (1962).
14. Chodkiewicz W., Ann. chim., [13], 2, 819 (1957).
245
15. Эгл питон Г., МакРае В., «Успехи органической химии», изд-во
«Мир», М., 1966, стр. 228.
16. Kurtz Р., Anti., 658, 6 (1962),
17, Baker С. S. L., Land or Р. D., Landor S. R., Proc. Chem. Soc.,
1963. 340.
18. Srinivasan R., J. Am. Chem. Soc., 85, 3048 (1963).
МЕДЬ ХЛОРИСТАЯ, КОМПЛЕКС С ХЛОРИСТЫМ АММОНИЕМ.
Эта смесь в слабо кислом водном растворе катализирует окисли-
тельную (воздухом) дегидрокоиденсацию терминальных ацетиленов
в диацетилены П, 2]. Группы NH2, ОН, СО2Н и CO2R не мешают
реакции. Указанную на схеме конденсацию проводят в смеси спирта
с 0,08 н. соляной кислотой, содержащей катализатор.
о.,
С1ф -СН(СН.,)4С-2 СН I- ИС С(СНг)7СО,Н
СН2 СН (СН,.)4С СС С(СН2)7СО2Н
Эритрогеиовпя кислота
1, В о w d е п К. et al., J. Chem. Soc., 1947, 1579; Rose J. D.( Weedon
В. C. L., J. Chem. Soc., 1949, 782; Riley J. P., J. Chem. Soc., 1953, 2193.
2. Black H. K., Weedon В. C. L., J. Chem. Soc., 1953, 1785.
МЕДЬ ХЛОРНАЯ БЕЗВОДНАЯ, CuCl2. Мол. вес 134,45. М. х. б.
получают нагреванием дигидрата хлорной меди (мол. вес 170,49)
при 110° в течение ночи [11.
Хлорирование ароматических соединений. Хлорированием ант-
рацена по приведенной ниже схеме Ноихебель 12] получил 9-хлор-
антрацен с очень высоким выходом. Реакцию контролируют по
превращению коричневой хлорной меди в белую хлористую медь.
По окончании реакции хлористую медь отфильтровывают и раствор
С1
011 моля 0,202 500 мл
моля
в ССЦ хроматографируют на окиси алюминия; продукт получают
в виде лимонно-желтого кристаллического вещества. По анало-
гичной методике пиреи превращают в 1-хлорпиреи с выходом 90%;
фенантрен не хлорируется.
Хлорирование диэтилфосфита дает диэтилхлорфосфат 13];
О
II
(СгН-,О)2РОНф2СиС12 (С2Н5О).2Р-Cl -|-2CuCl-|- НС1
{ %
1. W а г е J. С., Borchert Е. Е., J. Org. Chem., 26, 2263 (1961).
2. N onhebel D. C., Org. Syn,, 43, 15 (1963).
3. Smith T. D., J. Chem. Soc., 1962, 1122.
246
МЕЗИЛХЛОРИД, см. Метаисульфохлорид.
МЕЗИТИЛЕНСУЛЬФОХЛОРИД. Мол. вес 218,71, т. пл. 57°.
Кораиа [1] показал, что М. эффективен при образовании меж-
нуклеотидных связей Сз — G,. Смесь защищенного нуклеотида (1)
и защищенного нуклеозида (2) в пиридиновом растворе обрабаты-
вают 100%-ным избытком М. и раствор выдерживают при комнатной
температуре; выход ОщиацетилтимидилДЗ'^бС-тиммдина (3) со-
ставляет 71% через 1 час и 90% через 20 час. М. в этом синтезе
оказался гораздо более эффективным по сравнению с дициклогек-
сил карбодиимидом, этоксиацетиленом и борфторидом М-этил-5-
фенил изоксазолин; /2-толуол сульфохлор ид оказался несколько менее
эффективным.
1. J а с о b Т. М., Khor ana Н. G., J. Am. Chem. Soc., 86,
Nara и g S. A., Khorana II. G., ibid., 87, 2981 (1965).
1630 (1964);
Р-МЕРКАПТОЭТАНОЛ, HOCH2CH.,SH. Мол. вес 78,13, т.
кип. 155°.
Джерасси Ц] предложил этот реагент для превращения стеро-
идных кетонов в этилсиполутиокетали — производные, подобные
247
этилентиокеталям, но более легко подвергающиеся обратному
превращению в кетоны (никель Ренея) [2]. В первоначальной ме-
тодике для насыщенных кетонов раствор кетона в диоксане обра-
батывают свежеплавленым хлористым цинком и безводным суль-
фатом натрия и оставляют на 20 час при комнатной температуре.
В случае Л4-3-кетонов используют более активный катализатор —
п-толуолсульфокислоту, но выходы при этом низкие.
В более простой методике в качестве катализатора используют
эфират трехфтористого бора или его раствор в уксусной кислоте
13]. Так, из раствора 300 мг андростенолоиацетата, 0,3 мл 0-мер-
каптоэтанола и 0,3 мл эфирата трехфтористого бора в уксусной
кислоте при 25° очень быстро выпадают кристаллы. Добавляют
немного воды, кристаллы собирают и промывают метанолом; полу-
чают 310 мг продукта с т. пл. 166—170°. Одна кристаллизация из
95%-ного этанола дает чистый продукт с т. пл. 183—184°.
1. R о m о J., Rosenkranz G,, Dj erassi С., J. Am. Chem. Soc., 73,
4961 (1951).
2. Review: Dj erassi C,, «Steroid Reactions», Holden-Day, 1963, 21—34.
3. F i e s e r L. F., J. Am. Chem. Soc., 76, 1945 (1954).
МЕТАНСУЛЬФОКИСЛОТА, CH3SO3H. Мол. вес 96,11.
Надкислоты. Сверн и сотр. [I] показали, что эта кислота лучше,
чем серная, в качестве растворителя и катализатора для превра-
щения карбоновых кислот в надкислоты. Реагент можно исполь-
зовать даже в случае кислот, содержащих группы, неустойчивые
к действию кислот. Карбоновые кислоты хорошо растворимы в М.,
однако надкислоты из нее обычно осаждаются по мере образования.
Надкислоты образуются с выходами 75—98% из и-трет-бутил-,
о- и п-нитро- и /г-цианбензойиых кислот, терефталевой, лауриновой,
пальмитиновой, стеариновой, 12-оксистеариновой и а-бромстеарино-
вой кислот. Метод неприменим к м-и и-метоксибензойиым кислотам.
Надбензойную кислоту получают [2] добавлением по каплям
22 г (0,45 моля) 70%-ной перекиси водорода к перемешиваемой
смеси 0,3 моля бензойной кислоты (растворяется ие полностью)
и 86,5 г М. в высоком стакане на 500 мл при охлаждении до 25—30°.
Добавление проводят в течение 30 мин; при этом вся бензойная
кислота растворяется. Еще через 2 час раствор охлаждают до 15°,
обрабатывают льдом и холодным насыщенным раствором сульфата
248
аммония (для уменьшения растворимости) и экстрагируют бен-
золом. Экстракт промывают раствором сульфата аммония, сушат
и фильтруют. Раствор используют непосредственно для эпоксиди-
рования или других реакций. Указанный выход определен по
данным йодометрического титрования.
СОгН СОООН
1
(А 70% Н2О; CH3SO3H
I || 85-90% | ||
ZZ
«Гидролиз» сложных эфиров 13]. Метод расщепления сложного
эфира в нещелочных условиях состоит в кипячении 0,1 моля слож-
ного эфира и 0,1 моля М. в 100 мл 90%-ной муравьиной кислоты
в течение 5 час. Выход кислоты 64—97%; спиртовый остаток слож-
ного эфира превращается в формиат. М. является наилучшим
реагентом для этой цели; серная кислота дает гораздо более низкие
выходы, и-толуолсульфокислота — еще ниже, а трифторуксусная
и фосфорная кислоты не реагируют вовсе.
1. S 1 1 b е г t L. S., S I е g е 1 E.,Swern D., J. Org. Chem., 27, 1336 (1962).
2. S i 1 b е г t L. S., Siegel E, Swern D., Org. Syn., 43, 93 (1963).
3. Loev B., Chem. Ind., 1964, 193.
МЕТАНСУЛЬФОКИСЛОТЫ АЛКИЛОВЫЕ ЭФИРЫ, CH3SO3R.
получают реакцией метансульфохлорида со спиртами в пиридине [1],
1. S е ker а V. С., М ar v е I С. S., J. Am. Chem. Soc,, 55, 345 (1933).
МЕТАНСУЛЬФОКИСЛОТЫ АНГИДРИД, (CH3SO2)2O. Мол. вес
174,20, т. пл. 70°, т. кип. 138°/10 мм. М. а. получают нагреванием
метансульфокислоты с хлористым тионилом (выход 80%) [1] или
с пятиокнсью фосфора (выход 55%) [21.
М. а. взаимодействует с бензолом в присутствии хлористого
алюминия быстрее, чем мезилхлорид, и дает с 77%-ным выходом
метилфеиилсульфон [2]; ацилирует без катализатора анизол и не-
которые алкилбензолы [3]; образует метансульфоизоцианат с лучшим
+ (CH3SO2)2O4-CI3CHCHC13
0,05 2 моля 50 мл
Кипячение
16 час
70%
0СН3
SO2CH3
выходом (38%), чем мезилхлорид (5%), и используется вместо
ангидрида трифторуксусной кислоты при этерификации уксусной
249
и бензойной кислот [2]. Линстед [4] показал, что при обработке
дилактона манносахарной кислоты (1) мезил хлоридом, тозилхло-
ридом или тионилхлоридом в различных условиях образуется
смола, тогда как М. а. с почти количественным выходом дает кри-
сталлический 2,5-димезилат (2).
НОСИ о
(СНа50А-0
о j
ИСОН
СО
CH3SOBOCH о
I
Г—сн
нс —
0 HCOSO.,CH3
со
(2)
1, Owen L. N., Whitelaw S. Р., J. Chem, Soc,, 1953, 3723,
2. F f e I d L., Set t 1 age P. H., J, Am. Chem. Soc,, 76, 1222 (1954).
3. Gilbert E, E., J. Org, Chem,, 28, 1945 (1963).
4. Linst ea d R. P., Owen L. N., W ebb R, F., J, Chem. Soc,, 1953,
1255.
МЕТАНСУЛЬФОХЛОРИД (мезил хлор ид), CH3SO2C1. Мол. вес
114,56, т. кип. 70°/20 мм, уд. вес 1,47.
М. получают из технической метансульфокислоты и хлористого
тиопила (71—83%) [1].
Первичные и вторичные спирты реагируют с М. в сухом пиридине
при комнатной температуре с образованием мезилатов, обычно
с хорошими выходами. М. несколько более реакционноспособен,
чем тозилхлорид, и мезилаты иногда лучше кристаллизуются, чем
тозилаты. Мезилокси- и тозилоксигруппы являются эффективно
/X
ROH + CH3SOaCl+ | ||-*ROSO2CH3+ I ||
"V на
_1_
отщепляющимися группами. Например, мезилирование 3-ацетата
холестаптриола-Зр, 5а, 6|3 (1) сопровождается реакцией 6-мезилата
(2) с едким кали в метаноле с образованием 5а, 6а-эпоксида (3) [21.
Эта последовательность реакций была использована для пре-
вращения холестап-2а, За-эпоксида (4) в 2|3, Зр-изомер (7) [3]. Аце-
толиз соединения (4) дает диаксиальный 2р-ацетоксиол-3а (5),
превращающийся в ацетат-мезил ат (6). Тот факт, что взаимодействие
(6) с основанием сопровождается инверсией у атома С:! с образо-
ванием р-эпоксида (7), показывает, что эффективно отщепляющейся
250
является мезилокси-, а не ацетоксигруппа. Аналогия мезилокси-
группы и атома брома демонстрируется поведением двух из четырех
и зв ест пых 5а-с и и р ост аион-12-диол -2,3-ди мез ил атов. Соед и н е ш i я (8)
и (10) при взаимодействии с иодистым натрием в ацетоне при 100°
дают А2-олефин (9) [41. В тех же условиях димезилаты 2р, Зр- и 2р,
За-диолов не изменяются.
Мезилат, неспособный к превращению в олефин путем транс-
отщепления, может реагировать с основанием со скелетной пере-
группировкой. Один 15] из двух случаев 15, 61 в ряду сапогенина
представляет собой превращение (11) в альдегид (12) с сужением
цикла.
251
В ряду углеводов мезилаты имеют некоторые преимущества
перед тозилатами ввиду меньшего размера группы. Первичные
спиртовые группы реагируют с М. быстрее вторичных, однако
никаких затруднений не встречается при этерификации всех гид-
роксильных групп. Так, метил-а,о-глюкопиранозид (1) при взаи-
модействии с 1 молем АТ в пиридине в течение 10 час при —20°
и затем в течение 14 час при 0° с последующей обработкой уксусным
1 нсосн3 Н^ОСНз 1 НСОСНз
неон НСОАс HCOMs
10 10 I
носн АсОСН MsOCH о
неон НСОАс HCOMs
1-4С
11С llCr
сн,он CH2OMs CH2OMs
(1) (2) (3)
ангидридом в течение 20 час при 25° дает кристаллический 6-О-мезил-
2,3,4-три-О-ацет ил-а-метил-D-глюкопиранозид (2) с хорошим вы-
ходом [7]. Реакцией (1) с М. (немного более 4 молей) в пиридине
(4) (5)
в течение 24 час при комнатной температуре получен 2,3,4,6-тетра-
О-мезил-а-метил-D-глюкопиранозид (3) с т. пл. 146° с выходом
63%. р-Метил-D-целлобиозид реагирует с образованием 6,6'-диме-
зилата, выделенного с хорошим выходом в виде кристаллического
пентаацетильного производного (4) [8]. Все гидроксильные группы
трегалозы мезилируются; кристаллическая окта-О-мезилтрегалоза
(5) образуется с выходом 90%. Обработанная 1,1 моля мезилхло-
рида в очень мягких условиях и затем подвергнутая ацетилиро-
ванию, трегалоза дает 6-О-мезилгепта-О-ацетилтрегалозу.
Гельферих [9] показал, что фенолы можно мезилировать либо
в пиридине, либо по Шоттеп — Бауманну, и иашел, что мезилаты
исключительно устойчивы к сильным кислотам, но легко гидроли-
зуются щелочами. Так, ди мезил ат гидрохинона (7) можно превра-
252
тить в мономезилат (8) при выдерживании с ацетоном и водной
щелочью при комнатной температуре. Гельферих 110] показал также
1 моль CHaSOXl-Py (40 час)
|’ ’ ’ ~ 50-60%
ОН OSO3CH3 ОН
Д I I
' й CHsSO»CI; Ру Ф NaOH-ацетон \|
ОН OSOaCH3 0SO2CH3
(6) (7), т. пл. 167° (8), т. пл. 76°
возможность получения N-мезиламиноэфиров и их щелочного гид-
ролиза в N-мез ил аминокислоты.
н
Эфир |
CH3SO2CI + 2HaNCH2CO2C2H5——CH3SOaNCH2CO2C2H5 +
+ HCI.HoNCH3CO2C3H5
1. Н е а г s t Р. JN о I 1 е г С. R., Org. Syn., Coll. Vol., 4, 571 (1963).
2. F u r s t А., К о 1 1 e r F., Helv. chim. Acta, 30, 1454 (1947).
3. F ii r s t A., P 1 a 11 n e r Pl. A., Helv. chim. Acta, 32, 275 (1949).
4. SI ates H. L., W endler N. L., J. Am. Chem. Soc., 78, 3749 (1956).
5. Wendler N. L., HirschmannR. F., Slates H. L,, Walker
R. W., J. Am. Chem. Soc., 77, 1632 (1955).
6. H irschmann R., S n о d d v C. S., Jr., Hiskey C. F., Wendler
N. L., J. Am. Chem. Soc., 76, 4013 (1954).
7. He If erich B., Gniichtel A., Ber., 71, 712 (1938).
8. H e И e r i c h B., von S t г у k F., Ber., 74, 1794 (1941).
9. 11 e 1 f e r i c h В., P a p a 1 a m b г о u P., Ann., 551, 235 (1942).
10. H e I f e r i c h В., M i 11 a g R., Ber., 71, 1480 (1938).
МЕТАНСУЛЬФОХЛОРИД — ДВУОКИСЬ СЕРЫ. Хазен и Ро-
зенбург 11] обнаружили, что под действием перегнанного в вакууме
метансульфо хлорида не происходит дегидратации 11|3-оксистероида
в Дй(11)-ен в ДМФА, содержащем коллидин. С метансульфохлоридом,
перегнанным при атмосферном давлении, эта реакция осуществ-
ляется при 25—30° в течение нескольких минут. Было установлено,
(1)
(2)
253
что при перегонке при атмосферном давлении образуется двуокись
серы. Действительно, метансульфохлорид, не вступающий в ре-
акцию, становится эффективным при добавлении небольшого
количества двуокиси серы.
Методика дегидратации состоит в следующем. Раствор 0,04 моля
(1) в смеси 33 мл природного коллидина и 100 мл ДМФА охлаждают
при перемешивании до 10°, затем охлаждающую баню убирают
и в течение 1—2 мин прозрачный раствор обрабатывают 10 мл
(0,128 моля) метансульфохлорида, содержащего 5% безводной
двуокиси серы. Смесь охлаждают, поддерживая температуру в ин-
тервале 25—35°. За 5 мин выделяется окрашенный осадок, а рас-
твор становится красноватым; затем охлаждение прекращают и мед-
ленно добавляют 17 мл воды для разложения избытка метансульфо-
хлорида. Последующим осаждением водой получают 15,3 г (96 ° 6)
соединения (2) с т. ил. 203—211°. Кристаллизация из этанола дает
чистый продукт с т. пл. 216,5—-218°.
Хазен и Розеибург 11] предполагают, что метансульфохлорид
и двуокись серы реагируют обратимо с образованием смешанного
ангидрида — хлорангидрида, который содержит менее простран-
ственно затрудненную хлорангидридную группу, чем исходный
метансульфохлорид, и поэтому способен к атаке экранированной
0-гидроксильной группы. Распад образующегося сложного эфира
О” О О" ст
I !| I I
СН3—S+— Cl+ O = S —> СН3—S1'— О—S+—С!
можно представить через циклическое переходное состояние.
О SO2CH
—С._ тт .O-SO,CH,
I
-с
II
-с
1
LHazcn G. G., Rosenburg D. W., J. Org. Chem., 29, 1930 (1964).
МЕТАФОСФОРНАЯ КИСЛОТА. М. к. получают нагреванием
85 К)-ной ортофосфор ной кислоты до образования прозрачной жид-
кости Ц|. ДА. к. используется в синтезе пиридоксаль-5'-фосфата (3)
(по указанной схеме), который выделяют в виде плохо растворимой
о
it
3 НО-Р-ОН
он
------>
-з Н2О
(НРО3)3
254
желтой кальциевой соли 121. При кислотном гидролизе соединения
(2) для отщепления NjN-диметнл глицил гидразина используют
азотную кислоту.
Пиридоксаль
HZNNHCOCHZN(CH3)Z
CH=NNHCOCH,N(CH3),
CH=NNHCOCH2NH( СИЛ г
I он ст ст
>сн,ор-о-р~о-рон Il II II
H3ct^ у О о о
н
НС1 >
HNOj
(2)
СНО О'
О|
СН2ОР-ОН
II
о
N
f3)
1. V i s с о n t i и 1 М., В о п е t t i G., К агг er Р., Helv. Chim. Acta, 32,
1482 (1949).
2. V iscont in i M., Ebnot h er C., Karrer P., Helv. Chim. Acta, 34,
1834, 2198 (1951).
МЕТИЛАЛЬ, CH-2(OCH3).2. Мол. вес 76,09, т. пл. 42°, уд. вес
0,57. М. получают из формалина и конц. соляной кислоты [1].
Влияние растворителя. Рунге 12] превратил хлорметилмети-
ловый эфир в соответствующий реактив Гриньяра в М. или ТГФ.
М. предпочтителен вследствие большей стабильности реактива
Гриньяра в этом растворителе. Галогенид и магний не взаимодейст-
вуют в диэтиловом эфире, ди-изо-амиловом эфире, анизоле, диоксане,
бензоле или петролейном эфире. Рунге 12] показал, что метокси-
метилмагнийхлорид реагирует с циклогексеном с образованием
следов норкарана; однако выход по данным газожидкостной хро-
Mg
СН3ОСН2С1 ,п CH3OCH2MgCI
f-и Гт I/ ( U 1 1 у } 9
матографии только около 1%.
Шеллкопф и Кюпперс [31 получили метокси метил литий в М.
при температуре от —25 до —30°. Реагент стабилен в течение дня
при —70°, но разлагается за несколько часов при 0J. Реакции с про-
2 Li
СН,ОСН2С1---------CHsOCHsLi-4-LiCI
3 2 CH,(OCHS)„ 3 2 1
255
стыми альдегидами и кетонами дали ожидаемые продукты с выхо-
дами 54—75%. Хлорметилметиловый эфир не реагирует с литием
в диэтиловом эфире или ТГФ.
Хлорметилирование. Хлорметилирование /г-нитрофенол а осу-
ществляют следующим путем. В смесь 0,36 моля /г-нитрофенола,
650 мл конц. соляной кислоты, 5 мл конц. серной кислоты и 1 моля
М. пропускают хлористый водород при 70±2° и перемешивают
в течение 4—5 час. Хлорметильное производное начинает кристал-
лизоваться в первый час. Реакционную смесь охлаждают льдом
и кислотный слой декантируют. Кристаллизация из бензола дает
чистое хлорметильное производное с т. пл. 129—130°; выход 69%.
ОН ОН
А А/СНаС1
" 4 сн2(осна)г+нс1 V
I II 69% | ||
I I
NO2 NOa
1. Buehler С. A., Kirchner F. К., D е е b е 1 G. F., Org. Syn,, Coll.
Vol., 3, 468 (1955),
2. Runge F., Taeger E., Fiedler С., К a h 1 e r t E., J. prakt. Chem.,
19, 37 (1963).
3. Schollkopf U., Kiippers FI., Tetrahedron Letters, 1503 (1964).
МЕТИЛАТ НАТРИЯ, CH3ONa. Мол. вес 54,03, растворяется
в пентане.
Раствор в метаноле. Раствор 1 моля М. н. в 300 мл метанола
можно получить по методике синтеза этилата натрия по Тишлеру,
но при обратном порядке прибавления реагентов. Обратная мето-
дика, сходная с получением этилата натрия по Цауггу, отличается
лишь тем, что натрий не расплавляют, а нарезают мелкими кусоч-
ками [ 1 ].
Применение в качестве основания. Для превращения перекиси
бензоила в натриевую соль надбензойной кислоты лучше использо-
вать не этилат натрия, а М. н., так как последний лучше растворим
в метаноле и не осаждается при охлаждении не ниже —5° [2]. М. н.
используют в качестве основания в реакции электролитической
HCCL 0
(C6H6CO2C)3 + 2CH3ONa —•—*' -
0,21 моля 0,22 моля
в 100 мл
СН3ОН
2С6Н5СО3Ма +С6Н5СО2СН3
H2S04
------> С6НЙСО3Н (в СНС13)
82,05-86%
димеризации по Кольбе в метаноле; при этом достаточно каталити-
ческого количества М. н., так как он регенерируется на катоде [3].
CH3O2C(CHa)8CO2H + CH3ONa ЭлектролХ СН3О.2С(СНа)16СО2СН3
1 моль 0,05 моля 68- 74%
в 500 мл
CHSOH
256
Продажный М. н. обычно используют в качестве основания для
получения дихлоркарбена из этилового эфира трихлоруксусной кис-
лоты, например в реакции с дигидропираном [4]. В колбу после
продувания азотом помещают порошок М. и., дигидропиран и 600 мл
пентана. Светло-желтый раствор перемешивают в течение 15 мин,
0,3 моля
СС13СОгС2Н5 + CHjONa . ________:>
л 68-75%
0, 86 моля о, 9.2 миля
охлаждая в бане с водой и льдом, добавляют сложный эфир в течение
3—4 мин, после чего смесь перемешивают 6 час при 0° и еще в тече-
ние ночи при 25°.
Дегидрогалогенирование. Первой стадией получения дибензо-
ил метана из бензальацетофенондибромида является отщепление
одного атома брома в виде НВг и замещение второго атома брома на
метоксильную группу [51. Конечная стадия — кислотный гидролиз
эфира енола.
H..O-HC1
C6H5CHCHCOCeH5~f-2CH3ONa С6Н6С = СНСОСвН5 —--------
| [ 1 МОЛЬ В I 8С-0
Вт Вг 230 ллСНаОН ОСН3 (сырой)
0,5 моля
—► С6Н5СОСН2СОСвН5
Полагают, что промежуточным соединением в перегруппировке
Фаворского является продукт дегидрохлорирования. Перегруппи-
ровку 2-хлорциклогексанона Гохин и Воган [6] осуществили следу-
1 моль
ющим путем. Суспензию продажного М. и. в 330 мл эфира переме-
шивали, прибавляли в течение 40 мин хлоркетон и вновь переме-
шивали при кипячении в течение 2 час. М. н. был взят с небольшим
избытком, поскольку применение точно 1 экв снижало выход.
Баумгартен и Петерсен [11 разработали общий метод синтеза
а-аминокетонов, который можно иллюстрировать превращением
а-фенилэтиламина в аминоацетофенон. Чтобы превратить N.-N-ди-
257
9 Заказ № 1096
хлорамин (1) в циклическое промежуточное соединение (2), ра-
створ М. н. в метаноле добавляли к раствору (1) в бензоле.
0,6 моля CH3ONa
I 2(СНЩС-ОС1 | в 140 .и.1 CH.OH
сйн5снсн3- — ---------> с6щсн-сн,---------------
0 6 С9Н0, 5—10° h л
0,2 моля (1)
Н
,N4
/ \ НС1-Н,О
—► СВН5С—СН2 СЙН5СОСНЛ1 + Н3СГ
ОСН (общин) (3)
(2)
Нуклеофильное замещение. Одна нитрогруппа в 1,3,5-трииитро-
беизоле замещается на метоксильную группу при кратковременном
кипячении с М. н. в метаноле [71.
NO., | ОСН3
Кипячение 20 мин LCH3ONa >
O,N \ NO, (1,23 моля в 350 лиг СН3ОН ' 0,22 моля 63~77% 1 II Вс.щонЛ O2N^/\NO2
Метилирование флуорена. Шён и Бекер [8] обнаружили, что
9-я-ал кил флуорены можно получать нагреванием флуорена с нор-
мальным спиртом и соответствующим алкоголятом натрия при
210—220° в автоклаве. В качестве примера приведена схема полу-
чения 9-метилфлуорена [9].
+ CH3ONa-
I моль в
220°
78-86%
0, 68 моля 450 мл СН3ОН
Для этой реакции был предложен следующий механизм:
RCHO RCHsO.\’a ,\а
АггСН, —> Аг2С = CHR -------АгХ< -% RCHO
xch2r
RCH.OH
,н
Аг„С/ Щ RCbLONa
X2H,R
Катализатор реакций конденсации. Осуществляя синтез транс-
стильбена с помощью фосфонатной модификации реакции Виттита,
258
Зейс и Вильсон ПО] провели последнюю стадию конденсации с помо-
щью М. н. в ДМФА. Реакцию можно проводить и в 1,2-диметокси-
ОС, Нг,
С6Н6СЩВгН-Р(ОС2Н5)3 СнН5СН2Р + (ОС2Н5).,Вг -
(О
о
-*С8НбСНгР-НОС.,Н5).,
(6)
О’
| NaOCHs
сен,,снар+(ОСгн5)г-р о - снс8н5 —
О ° %
н
I
СбН5С = ССеН5 + (C2H6O)2POONa + СН3ОН
н
этане с гидридом натрия, однако выход в этом случае ниже [11].
Подробно разработан синтез транс,транс-1,4-днфенилбутаднена-1,3
по методике Зейса и Вильсона [121.
Харт и сотр. [13] разработали синтез ди циклопропил кетоиа;
первая стадия заключается в самоконденсации у-бутиролактона
(1) в присутствии М. н. в качестве катализатора с образованием ди-
мера (2). Метанол, используемый в качестве растворителя, отгоняют
и окончательно удаляют в вакууме. При нагревании с конц.
соляной кислотой образуется дихлоркетокислота (3), декарбоксили-
рованием которой получают (4), а это соединение циклизуется под
действием водной щелочи в ди циклопропил кетон (5). Промежуточные
соединения (2) и (4) удалось выделить.
СН,СС
2 | До
сн2сн2
сн2с==сс^
+ CHjONa--->| )о I
сн2сн2 сн/5н2
2,17 моля в
4 моля
(1)
520 мл СН3ОН
(2)
О
СН2С—СНСО2Н
I I
СН2СН2 СН2СН2С1
С1
О
____> С Н 2 с— сн2 Na0H .
"CO2 | [ 55% (общий)
СН2СН2 СН2СН2С1
С1
!? ^СН2
I .СН-С-СН I
снХ ^сн2
(з) (4)
Другие реакции конденсации, осуществленные под действием
М. и., рассмотрены в следующем разделе.
Гетероциклы. При добавлении по каплям смеси ацетона и этил-
формиата (по 0,8 моля каждого) к суспензии 0,86 моля М. н. в эфире
9* 259
при 0° происходит конденсация и образуется натриевое производное
формилацетона (1) [14]. Эфир отгоняют и остаток — енолят нат-
рия — обрабатывают 0,3 моля цианацетамида в 400 мл воды и ра-
створом 8 мл уксусной кислоты в 20 мл воды, который имеет слабо-
СН3
СН3СО4-НСОйС2Н5 СНз°^
0,8 моля 0,8 моля
,CHONa
//
СН
сн3([:о
со
CH.CN
1
H,N-C = O
55-62%
н
(2)
основную реакцию в результате добавления пиперидина; смесь ки-
пятят в течение 2 час, подкисляют уксусной кислотой и получают
3-циан-6-метилпиридон-2 (2) в виде желтого осадка.
Для синтеза 2-амино-4-анилино-6-хлорметил-снжм-триазина из
хлоргидрата фенилдигуанида (0,3 моля) и этилового эфира мопо-
хлоруксусной кислоты (0,3 моля) хлоргидрат добавляют к раствору
0,3 моля М. н. в метаноле, раствор отфильтровывают от хлористого
натрия, добавляют этиловый эфир монохлоруксусной кислоты
и смесь перемешивают при комнатной температуре в течение 14 час
115]. Продукт выпадает в осадок и его кристаллизуют из диоксана.
СНгС1
C2H5O(t = O
NH
1[
CflI-hN/
н и
CH3ONa—СН3ОН
44-47%
СН„С1
|ч
n/^n
CeHsN/^N^4 NH2
’Н
З-нТептил-5-цианцитозин (3) синтезируют [16] из н-гептил амина,
который сначала превращают в н-гептил моче вину (1). Последнюю
кипятят с малононитрилом и триэтиловым эфиром ортомуравьиной
кислоты, причем образуется кристаллический 3-н-гептилуреидоме-
тиленмалононитрил (2). Циклизацию в (3) осуществляют, постепенно
добавляя М. н. (0,16 моля) к раствору 0,145 моля (2) в 70 мл мета-
нола, после чего смесь выдерживают при 25° в течение 3 дней. Вы-
павшую соль растворяют в воде, продукт осаждают уксусной кис-
лотой и перекристаллизовывают из спирта.
h-C7H1sNH2 2J-aCNO-i-H21 h-C7H15NHCONH2
86-88%
(1)
260
I
h2n
С2Н5ОС(ОС2Н6)Й
I
н
CN
нА
"CN
C7H15-h
I
C% /NH
'Cz CN
- 3CZHSOH I |
80—83% , J, J,
CH3ONa-CHaOH;
CH3C02H
88—92%
H1N4 zC.
'C/Z "CN
H
(3)
C?Hi5-«
HN
x^\:n
(3)
2-Тио-6-метилурацил получают [17] нагреванием на кипящей
водяной бане смесн 1 моля ацетоуксусного эфира, 1 моля тиомочеви-
ны и 3,3 моля продажного М. н., затем смесь выпаривают досуха.
Водный раствор полученной соли подкисляют уксусной кислотой.
z СН. Z/o
СН36Х \С'/
! I
он осгн5
H2N /NHa
X'CZ
CHaONa;
CH3COOH
69—84%
H3Cw\/0H
I II
N N
Парабановая кислота получена аналогичной конденсацией моче-
вины с диэтил оксалатом [18]. В этой реакции можно использовать
и этилат натрия в этаноле, но выход будет ниже. О конденсации
фенилазида с фенилацетонитрилом, катализируемой М. н., см.
Фенил азид, раздел Гетероциклы,
о о
и [I
С2Н5ОС —сос2н5
Н2КХ ✓NH,
с
II
о
CH3ONa-CH3OH; НС1^
" 71, 5-76%
1. Баумгартен Г. Э., Петерсен Д. М., «Синтезы органических пре-
паратов», изд-во «Мир», М., 1964, сб. 12, стр. 171.
2. Браун Г., «Синтезыорганических препаратов», ИЛ., М., 1949, сб. 1,стр.337.
3. Сванн Ш,, мл., Г а р р и с о н У. Э., мл., «Синтезы органических препа-
ратов», изд-во «Мир», М., 1964, сб. 12, стр. 42.
4. Пар х ам У. Э., Швейцер Э.Э., Миерцва 3. А., мл., «Синтезы
органических препаратов», изд-во «Мир», М., 1964, сб. 12, стр. 113.
5. А л л е н Ч. Ф. X., А бе л л Р. Д., Норм и н гтон, «Синтезы органи-
ческих препаратов», ИЛ, М., 1949, сб. стр, 186.
261
6. Goheen D. W., Vaughan W.R., Org. Syn., Coll. Vol., 4, 594 (1963).
7. Ревер дев Ф., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 199.
8. Schoen К.. L., В е с к е г Е. I., J. Am. Chem, Soc., 'll, 6030 (1955).
9. Шён К. Л., Бекер Э. И., «Синтезы органических препаратов», ИЛ,
М., 1961, сб. 11, стр. 48.
10. S е u s Е. J., W i 1 s о и С. V-, J. Org. Chem., 26, 5243 (1961).
11. W a d s w о г t h W. S., Jr., E m in ons W. D., J. Am. Chem. Soc., 83,
1733 (1961),
12. Org. Expts., 119.
13. К у p т и с О., мл,, Сэндри Дж., Крокер P., X a p т Г., «Синтезы
органических препаратов», ИЛ, М,, 1960, сб. 10, стр, 24.
14. 'Мариелла Р., «Синтезы органических препаратов», ИЛ, М., 1953,
сб. 4, стр. 553,
15. О v е г b с г g е г С. G,, Micheletti F. W., Org. Syn., Coll. Vol., 4t
29 (1963).
16. К e м Б., У айтхед K-, «Синтезы органических препаратов», ИЛ, Л1.,
1959, сб., 9, стр. 14.
17. Фостер Ч., Снайдер X., «Синтезы органических препаратов», ИЛ,
М., 1956, сб. 7, стр. 37.
18. Мэ р р э й Дж., «Синтезы органических препаратов», ИЛ, М., 1959, сб. 9,
стр. 54.
ЗМЕТИЛБЕНЗТИАЗОЛОН-2-ГИДРАЗОН, ХЛОРГИДРАТ
(МБТГ-НС1). Т. пл. 240—260° (с разл.).
Получение этого аналитического реагента описано Савицким
и сотр. [11, которые разработали чувствительную пробу на формаль-
дегид и другие растворимые в воде алифатические альдегиды. Об-
работка капли водного раствора формальдегида избытком подщело-
ченного реагента (1) приводит к превращению в азип (2). Затем
(4)
.262
хлорным железом окисляют (1) в (3), который конденсируется с ази-
ном с образованием ярко-синего катиона [(4), одна из резонансных
структур]. Описаны методики обнаружения и определения альдеги-
дов капельным анализом, а также хроматографией па бумаге,
силикагеле и на колонках; эти методы используют, в частности,
для определения формальдегида в выхлопных газах автомобилей
и в загрязненном воздухе.
Реагент взаимодействует с большинством производных анилина
с образованием, после окисления хлорным железом, красителей,
обладающих характеристическим поглощением в области 545—-
675 ммк [2]. Реагент можно использовать также для идентификации
и определения карбазолов [3].
1. Sawicki Е., Н a u s е г Т. R. .Stanley Т. W., Е 1 b е г t W-, Anal.
Chem., 33, 93 (1961).
2. S a w i с k i E., Stanley T. W., Hauser T. R., Elbert W., Noe
J. L., Anal. Chem., 33, 722 (1961).
3. S a w i c k i E., Hauser T. R. Stanley T. W., Elbert W., Fox
F. T., Anal. Chem., 33, 1574 (1961).
МЕТИЛ БРОМИСТЫЙ, СН3Вг. Мол. вес 94,95; т. кип. 4,6Э,
уд. вес 1,732.
М. б. получают из метанола, бромида натрия и серной кислоты
[1] и хранят в запаянных ампулах.
Для метилирования малонового эфира [2] к раствору 2 г-атом
натрия в 1 л абсолютного этанола добавляют 2 моля диэтилмало-
пата. Затем в смесь при перемешивании приблизительно в течение
4 час пропускают 2,1 моля М. б. Наблюдается спокойное взаимо-
действие с осаждением бромида натрия и выделением тепла, доста-
точного для поддержания слабого кипения. Оранжевую слабоще-
СНа(СОпСгН5), NaOc^ NaCH(CO2C2H5)n-^Ji-^ СН3СН(СОоС.,Н5)г
79-83%
л очную суспензию нейтрализуют уксусной кислотой, бромид натрия
отделяют фильтрованием и сохраняют. Фильтрат переносят в реак-
ционную колбу и при перемешивании (для предотвращения толчков
при кипении) отгоняют большую часть этанола. К остатку добавляют
водный раствор бромида натрия для уменьшения растворимости
сложного эфира и повышения плотности водного слоя, чтобы слой,
содержащий сложный эфир, находился сверху. Водный слой отде-
263
ляют и экстрагируют эфиром. Органический слой и эфирные вытяж-
ки объединяют, сушат кратковременным встряхиванием с хлори-
стым кальцием и отгоняют эфир. Непрореагировавший малоновый,
эфир удаляют по Михаэлю 13]: смесь эфиров встряхивают точно
1 мин с холодным раствором 10 г едкого натра в 30 мл воды (алки-
лированный малоновый эфир подвергается воздействию лишь не-
значительно). Продукт вновь высушивают и перегоняют.
1. W е i n е г N., Org. Syn., Coll. Vol., 2, 280 (1943).
2. Вейнер H., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 589.
3. М i с h а е 1 A., J. prakt. Chem., (2), 72, 537 (1905).
2-МЕТИЛБУТИЛАТ-2 КАЛИЯ (mpem-амнлат калия),
СН3
сн3сн,сок
V
Получение см. Калий, методика Пирсона.
«Летаргическое» оксимирование. Кадеш [1] описал ряд не-
удачных попыток превратить ди-орто-замещеиные ацетофеноны
в оксимы. Однако Грир и Пирсон [2] получили оксим 2,4,6-три-
метилацетофенона с выходом 40% по весьма простой методике —
выдерживанием раствора кетона и хлоргндрата гидроксиламина
в пиперидине при комнатной температуре в течение 1 месяца. Про-
странственно затрудненный оксим претерпевает перегруппировку
Бекмана с высокой скоростью; в конц. серной кислоте при 0° реакция
проходит почти нацело (на 94%) за 75 мин. Такую высокую реакци-
онную способность объясняют тем, что а-оксиминоэтильная группа
расположена перпендикулярно к плоскости кольца, в результате
чего невозможно резонансное взаимодействие между боковой цепью
и кольцом. Позднее Пирсон и Китон [3] значительно улучшили
эту методику путем применения сильного щелочного агента —
М. к. в трет-амиловом спирте. Реагенты в указанных ниже количе-
ствах прибавляли к 125 мл раствора алкоголята, колбу закрывали
пробкой и выдерживали при комнатной температуре 32 дня.
СН3 СН3
( I
UD C=NOH
H8Cx Jx/CH3-PHONH3C1 + CH3CH2C(CH3)2OK —- н3сх^|х/сн3
| ||' 0,03 моля 0,2 моля j Н
"Ч/ V
сн3 сн,
0,063 моля
Никаких изменений, кроме выпадения хлористого калия, не отме-
чено. После соответствующей обработки был получен неочищенный
264
оксим (т. пл. 98—101°) с выходом 98%. Возгонка дает чистый пре-
парат, т. пл. 101,5—102,5°. При кипячении реакционной смеси
выход оксима не превышает 50% , по-видимому, вследствие разложе-
ния гидроксилами на на аммиак, воду и другие продукты. В таблице
приведены результаты оксимирования других замещенных ацето-
фенонов, а также бензоил мезитил ена.
Ацетофенон (или кетон) Время, сутки Выход неочи- щенного ок- сима. %
2,4,6-Триметил- 32 98
10 53
2,3,4,6-Тетраметил- 180 90
10 30
Пентаметил- 420 81
2,6-Диметил-4-трет-бу- 180 95
тил-
Бензоилмезитилен 450 16
Пирсон предложил термин «летаргическая реакция» для обозна-
чения медленно протекающего процесса, который нельзя ускорить
вследствие побочных реакций разложения, начинающихся при повы-
шении температуры. Этот термин подчеркивает потенциальные воз-
можности применения данной методики к другим реакциям.
Считают, что М. к. более эффективен, чем mpem-бутилат калия,
для алкилировани я 1, 10-диметил-Д1(9’-окталона-2 141.
1. Kadesch R. G., J. Am. Chem. Soc., 66, 1207 (1944).
2. G r e e r F., Pearson D. E., J. Am. Chem. Soc., 77, 6649 (1955).
3. Pearson D. E., Keaton O. D., J. Org. Chem., 28, 1557 (1963).
4. Mo kher jee S. L., Dutt a P. C., J. Chem. Soc., 1960, 67.
2-МЕТИ Л БУТИЛ AT-2 НАТРИЯ (трети-амил ат натрия),
СН3
CH3CH2CONa
СН3
М. н. растворим в ароматических углеводородах, поэтому для
получения его раствора (1—3 Л4) кипятят раствор mpem-амилового
спирта в бензоле, толуоле или ксилоле в атмосфере азота с избытком
натрия (в виде проволоки или ленты) до прекращения выделения
водорода [1]. По охлаждении в атмосфере азота раствор остается
265
прозрачным и пробу его можно оттитровать стандартным раствором
кислоты, применяя в качестве индикатора фенолфталеин. Алкилиро-
вание можно осуществить и в гомогенном растворе. Этот алкоголят
является эффективным основанием при алкилировании кетонов,
особенно склонных к самоконденсации [2]. Например, алкилирова-
нием изофорона (1) 1,3-дихлорбутеном-2 в бензоле в присутствии
М. н. получают кетон (2) с выходом 50% [3]. При алкилировании
в присутствии трет-бутилата натрия выход составляет только 20%
121. Окталон (3) при метилировании в бензоле с тлре/п-амилатом
СН3
О С1
!l I
/\/СН2СН = ССН3
НзС\1 II
:х/чсн3
сн3
(2)
калия в качестве основания дает кетон (4) с выходом 93% [41, а при
использовании mpem-бутилата калия в трет-бут ловом спирте вы-
ход составляет только 24%. Другие исследования см. 16],
1. Coni a J.-M,, Bull, soc, chim. France, 1950, 533.
2. Review: Co n i a J.-M., Rcc. Chem. Progr., 24, 43 (1963).
3. J u I i a S., Bull, soc, chim. France, 1954, 780.
4. M ukherjee S. L., D u t t a P. C., J. Chem, Soc., 1900, 67.
5. Y anagit a M., Hir akur a M-, S e k i F., J. Org. Chem., 23, 841 (1958).
6. Con i a J,-M. and co-workers, Bull. soc. chim. France, 1954, 690, 943; 1956
1040,1392; 1957 1064; 1960, 1929; 1961, 836; Tetrahedron, 16, 45 (1961); Tetrahedron
Letters, 505 (1962).
OH
3-МЕТИЛБУТИН1-ОЛ-3, (CH3).,C—С —CH. Мол. вес 84,11,.
т. пл. 2—3°, т. кип. 104°.
Применение. См. Медь хлористая, Диацетилен.
МЕТИЛ ВИНИЛ КЕТОН, СН3СОСН=СН„. Мол. вес. 70,09,
т. кип. 81°, 35—36°/140 мм, п2^ Л,4086.
М. получают в промышленности гидратацией винилацетилена.
Продажный продукт представляет собой азеотропную смесь 85%
метилвинилкетона и 15% воды. Этот продукт сушат избытком без-
водного карбоната калия, органический слой отделяют, охлаждают
266
льдом, сушат сначала хлористым кальцием, затем поташом и перего-
няют; т. кип. 30—32°/120 мм или 35—36'7140 мм [1].
Синтез стероидов. При разработке метода циклизации, который
теперь имеет громадное значение в полном синтезе стероидов, Ро-
бинсон [21, изучая вначале конденсацию кетонов с М., встретился
с трудностями, обусловленными склонностью М. к полимеризации.
Затем он обратился к исследованию доступных предшественников
М. и получил неплохие результаты с метил-|3-хлор ацетил кетоном,
xCHjNEt2
Дц "сН3ОГ1
°" С1Д
СУ '
CH;JCOCHaCH2Cl. Однако лучшим реагентом оказался иодмети-
лат (2) основания Мании ха (1), 1-диэтиламинобутанона-З. При об-
работке сильным основанием типа амида натрия в присутствии 2-
метнл циклогексанона подметил ат расщепляется до метил винил ке-
тона (3), который реагирует с кетоном с одновременным присоеди-
нением по Михаэлю и альдолизацией с образованием бицикличе-
ского кетона (5). Эта реакция позже стала ключевой стадией в син-
тезе эпиандростерона по Корнфорзу — Робинсону: превращение
бициклического кетона (7) в трициклический кетон (8).
Джонсон [1] в своем гидрохризеновом синтезе стероидов исполь-
зовал ту же схему конструирования кольца А: (9)—>-(10). 1-Диэтил-
амипобутанон-3 превращают в и од мети л ат де ист в и ем поди сто го метила
в эфире в присутствии небольшого количества бензилового спирта
267
в качестве катализатора четвертизации. Эфир отгоняют,кристалли-
ческую соль и кетон (9) растворяют в метаноле и раствор обрабаты-
вают метилатом натрия в метаноле. В другом синтезе Джонсон
использовал непосредственно М. для осуществления циклизации по
Робинсону. Синтез кортизона поСаретту [4] включает одностадийное
построение кольца А на основе (11) с образованием (12). В описан-
ной методике используется 85%-ный водный азеотроп метилвннид-
кетона и тритон Б в качестве катализатора, однако указывается,
что применение подметил ата основания Мании ха (с добавлением
эквивалента основания) также приводит к хорошим результатам.
Рамачандран и Ньюмен [5] в синтезе метилокталиндиона (III)
осуществили циклизацию Робинсона в две стадии. Присоединение
к (II) по Михаэлю проводят кипячением в течение 3 час смеси 0,5 моля
(I), 0,75 моля М., 0,25 г едкого кали и 250 мл этанола. Промежуточ-
ный аддукт (II) выделяют в виде жидкости, которую без очистки на-
гревают в бензоле с 3 мл пирролидина с насадкой Днна — Старка
для удаления воды. Соответствующей обработкой и кристаллиза-
цией из эфира получают бициклический ди кетон (Ш) с т. пл. 47—
50°.
Метод получения 1-диэтиламинобутаноиа-З, впервые опублико-
ванный Уилдсом и Шунком [6] в 1943 г., подробно описан в сб.
СН3СОСНд СН2 = О (C2H5)2NH2C1 -
г!
4- — КОН
—> CH3COCH2CH2NH(C2H5)2(C1) CH3COCH2CH2N(C2H5)g
(общий)
«Синтезы органических препаратов» [7]. Смесь 1,6 моля хлоргидрата
диэтиламнна, 2,26 моля параформальдегида, 600 мл (8,2 моля) аце-
тона, 80 мл метанола и 0,2 мл конц. соляной кислоты кипятят 12 час;
светло-желтый раствор охлаждают и нейтрализуют раствором 65 г
268
едкого натра в 300 мл воды, экстрагируют эфиром основание Ман-
ниха и перегоняют его. Хагемайер 181, по-видимому, не знавший
о работе Уилдса, описал методику, в которой смесь 6 молей хлор-
гидрата диэтнламина (30%-иый водный раствор), 12 молей ацетона,
8 молей параформальдегида и 150 мл изопропанола кипятят 6 час
н затем концентрируют при пониженном давлении на водяной бане.
Раствор нейтрализуют 50%-ным водным раствором едкого натра,
отделяют третичный амин, сушат и перегоняют. Выход основания
Манниха 80,5%.
Хагемайер показал, что 1-диэтиламинобутанон-З можно превра-
тить в М. суспендированием третичного амина в даутерме (инертная
NEL
I
,СН2
снй
Х'СН3
НС1.
нагре ванне
zCH2
Ж
^Г1
ХСЧ
хсн3
теплообменная жидкость), пропусканием хлористого водорода для
образования хлоргидрата амина и нагреванием при атмосферном
давлении; перегонка дает чистый М. с количественным выходом.
Ньюмен 191, столкнувшись с некоторыми трудностями при получе-
нии основания Манниха по методике Уилдса, указал на обратимость
реакции. Смесь 2,2 моля диэтиламина и 4,5 мл уксусной кислоты
добавляли по каплям при охлаждении льдом к 2 молям М. Смесь
выдерживали 2 час при 0°, затем давали нагреться до 27°; жид-
кость промывали 50%-ным водным раствором едкого кали и водой,
сушили и перегоняли при 69—73°/16 мм.; выход 68%.
В вышеприведенной работе Рамачандрана и Ньюмена 15] пока-
зано, что 2-метнлциклогександион-1,3 (1) гладко реагирует с М. пу-
тем одновременного присоединения по Михаэлю и альдолизации
с образованием с хорошим выходом метилокталиндиона (III). В про-
тивоположность этому конденсация 2-мети л цикло гекса нона (2)
с-М. (1) в сравнимых условиях протекает очень плохо. Маршалл
и Фанта [10] после нескольких безуспешных попыток увеличить
выход показали, что медленное прибавление М. к эквивалентному
количеству 2-метилциклогексанона при —10° в присутствии катали-
тического количества метнлата натрия приводит к циклизации в цис-
кетол (3) с удовлетворительным выходом. Этот продукт был де-
гидратирован в (4) перегонкой с водяным паром над щавелевой кис-
лотой или едким кали.
269
При осуществлении циклизации по Робинсону часто бывает по-
лезно повысить реакционную способность соседнего с карбонильной
группой положения путем введения заместителя, который можно
впоследствии удалить. Так, Тернер и сотр. [11] показали, что непо-
средственная конденсация 7-метокситетралона-1 (1) с М. или соот-
ветствующим основанием Манниха оставляет «мало надежд». Они
превратили (1) в 2-оксиметилеповое производное (2), легко конденси-
рующееся с М. с образованием соединения (3), которое без очистки
было превращено в (4) обработкой метанольным раствором едкого
кали.
Кори и Нозое [12] использовали этот прием в синтезе гельмин-
тоспор ал я (6) из (—)-карвомеитопа (1). Конденсацией 3-формильного
производного (2) с ЛА. получили соединение (3), дсформилировапие
которого 2%-ным раствором поташа в этаноле не привело к продукту
альдольной конденсации, а циклизация в (5) была успешно осуществ-
лена с трехфтористым бором. Последующие пять стадий привели
к гельминтоспоралю (6) и подтвердили строение и стереохимию,
предложенные для этого ядохимиката, и позволили установить аб-
солютную конфигурацию.
Синтез витамина А. Присоединение ацетилена к М.— главная
стадия в промышленном синтезе витамина А по Айслеру [131.
Образующийся спирт (2) при кислотном катализе претерпевает
аллильную перегруппировку в первичный спирт (3), который пре-
вращают в ди-Mg В г- производное (4) с последующим присоедине-
нием к альдегиду (5). Селективное гидрирование тройной связи
и ацетилирование в (7) с последующей катализируемой иодом де-
гидратацией завершают синтез.
Реакции Дильса — Альдера. М.— реакционноспособный диено-
фил и реагирует с циклопентадиеном в эфирно.м растворе с выделе-
нием тепла и образованием с почти количественным выходом смеси
эндо- и экзо-аддуктов в соотношениях, указанных на схеме [14].
270
(1) (2)
(3) (4)
Na, NH3
(1) (2)
CH3 CH3
--> HOCH2CH=C-C=CH 1 2 3 EtMgB^ BzMgOCH2CH-C-CSCMgBr
(3) (4)
CHCOCH3
COCH3
эндо - (62%)
^КЗО - (38%)
271
Циммерман и Пауфлер [15] нагревали tz-пирон с избытком М.,
осуществив двойной диеновый синтез с декарбоксилированием
и образованием диацетилбицикло- [2,2,2]-октена-2. Полученный
твердый ди кетон обычными реакциями был превращен в би цикл о-
[2,2,2]-октатриен-2,5,7 с т. пл. 16°.
‘(Синтез 4-метилхинолинов. Кемпбелл и Шефнер [16] разрабо-
тали эффективный метод синтеза лепидина из хлоргидрата анилина
ОСН.,
CH,OH(Hg= + ) ] н +
СН2 = СН — С == C1T ---СНнОСН3СН2С—СН3-------->
ОСН3
Н +
СН3ОСН3СН2СОСН3 —СН3 = СНСОСНз
и М. или одного из двух болёе стабильных его предшественников —
1 ДЗ-триметоксибутаиа или 4-метоксибутаиоиа-21
Лепидин
В колбу загружают 0,625 моля хлоргидрата анилина, 1 моль гек-
сагидрата хлорного железа, 10 г безводного хлорида цинка и 450 мл
95%-кого этанола. Смесь перемешивают при 60—65° и в течение
1,5—2 час добавляют по каплям 0,5 моля М., 1 ДЗ-триметоксибутана
или 4-метокси бута но па-2. После кипячения в течение 2 час удаляют
большую часть спирта, смесь подщелачивают и перегоняют с паром.
Ацетоэтилирование карбонильных соединений. Простые кетоны
присоединяются по Михаэлю к М. в присутствии амида лития, три-
тона Б или этанольного раствора КОН [17]. Как это показано для
ацетофенона, сначала образуется [3-ацетоэтильное производное ке-
тона (1) или производное циклогексанона (2), являющееся продук-
том альдольной циклизации (1). Некоторые сложные эфиры, 0-
кетоэфиры, малоновые эфиры и амины ацето этилируются, часто
272
с хорошими выходами, реакцией с М. в присутствии щелочного ка-
тализатора [18|.
О
О О 0 0 J1 11
I: I; LiNH3 :l II ( >
СвН5ССН3 + СН3 = СНССН3-----^С6Н5ССН,СН3СН2ССН3^ HO.J |
с6н3/х/
(I) (2)
Синтез пирролов [191. Главной стадией нового синтеза пирролов
является катализируемая основанием конденсация М. со сложным
эфиром N-тозиламинокислоты, например этиловым эфиром N-тозил-
глицииа (1). Дегидратация пирролидина (2) с пятиокисыо фосфора
в кипящем бензоле дает А3-пирролин (3), обработка которого гидри-
дом натрия в кипящем ТГФ приводит к отщеплению натриевой соли
п-толуолсульфокислоты и образованию 2-карбэтокси-З-метилпир-
рола (5).
о
СН— ССП3 КОС(СН3)3
|| s Эфир,отреот-ВиОИ
СНг/ CH2CO2C2HS ’
HN
I
Ts
ОН
сн—ссн3 _
I I РгО5>
СН2 СНСО2С2Н5 1Т
сбН6
I
Ts
(2)
сн=ссн3
-~»СИ2 СНСО2С2Н5
Ts
NaH-ТГФ
-------->
-NaTs
СН=ССП,
I I
СНг ССО2С2Н5
сн—С СИ,
II II
СН ССО2С2Н5
(3) (4) (5)
I. Johnson W. S., Szmuszkovicz J., R о g i е г E. R., Hadler
H. I., Wynberg H., J. Am. Chem. Soc., 78, 6285 (1956).
2. Du Fe u E. C., Mc.Q u i 1 I i fi F. J., R ob i ns on R., J. Chem. Soc.,
1937, 53.
3. Corn forth J. W., R о b i n s о n R., J. Chem. Soc., 1949, 1855.
4. P о о s G. I., Ar t h G. E., В e у I er R. E., S a r e t t L. H., J. Am.
Chem. Soc., 75, 422 (1953).
5. Ramachandran S., Newman M. S., Org. Syn., 41, 38 (1961).
6. W 1 1 d s A. L.,Shunk С. H., J. Am. Chem. Soc., 65, 469 (1943)
7. W i 1 d s A. L., N о w a k R. M., McCalebR. E., Org. Syn., Coll. Vol
4, 281 (1963).
8. Hagerncyer II. J., Jr., J. Am. Chem. Soc., 71, 1119 (1949).
9. Swaminathan S., Newman M. S., Tetrahedron, 2, 88 (1958).
10. Marshall J. A., Fanta W- I., J. Org. Chem., 29, 2501 (1964).
11. Turner R. B., Net tieton D. E.,Jr.,Ferebee R..J, Am. Chem.
Soc., 78, 5923 (1956).
12. Corey E. J., Nozoe S., J. Am. Chem. Soc., 87, 5728 (1965).
13. I s 1 e г О., H uber W., Ronco А., К о f 1 e r AT, Helv. chim. Acta
30, 1911 (1947).
273
14. Dinwiddle J. G., Jr., McManus S. P., J. Org. Chem., 30, 766 (1965).
15. Z i mm erman H. E,, P a u f 1 er R. AL, J. Am. Chem. Soc., 82, 1514
(1960).
16. C a in p b e 1 1 K- N., Schaffner I. J., J. Am. Chem. Soc., 67, 86 (1945).
17. Ross N. C., Levine R., J. Org. Chem., 29, 2341 (1964).
18. Ross N. C., Levine R., J. Org. Chem., 29, 2346 (1964).
19. j а с к s о n A. H., Kenner G. AV., Terry W. G., Tetrahedron Lei-
ters, 921 (1962).
МЕТИЛВИНИЛОВЫЙ ЭФИР, СН3ОСН = СНг. Мол. вес 58,08,
т. кип. 13°.
В синтезе 3-мети л пентанди ола-1,5 (5) смесь 4,08 моля кротоно-
вого альдегида, 5,06 моля М. э. и 1,1 а гидрохинона (в качестве ин-
гибитора полимеризации) нагревают в автоклаве при 200° в течение
12 час [1]. Образующийся метилметоксидигидропиран (3) гидроли-
зуют в p-метилглутаровый альдегид (4) перемешиванием при 50°
с разб. соляной кислотой, добавляют твердый бикарбонат натрия
до нейтральной реакции раствора (индикаторная бумага) и полу-
ченную смесь гидрируют в качающемся автоклаве при 125° в тече-
ние 4 час [2]. По этой методике р-метилглутаровый альдегид полу-
чается с выходом 90%.
СН3
СН3
СНОСНз
200®
HCI
нго
СН3
сн3
ОСНСН2^НСН2СНО Ti _“&з% ИЗ (зГ носн2сн2снсн2сн2он
Ы2, NI Ренея
(5)
1. L о и g 1 е у R. 1., Jr., Emerson W- S., В 1 a r d i n e 1 1 i A. J., Org.
Syn., Coll. Vol., 4, 311 (1963).
2. Longley R. I., Jr., Emerson W. S., Org. Syn., Coll. Vol., 4, 660 (1963).
О-МЕТИЛГИДРОКСИЛАМИН (метокснамин), CH3ONH3. Мол.
вес 47,06, т. кип. 49—50°. Хлоргидрат, мол. вес 83,53, т. пл. 149°.
П олу че н и е [ 1 ]. Г и др о кс ил а м и ид и с ул ьфон ат натрия мет и л н р уют
ди мети л с у л ьфатом.
Защита кетогрупп. Фрид и Нутиле [2] использовали метокс-
иминопроизводное (2) для защиты А‘^-диенон-З-овой системы сте-
роида (1), который превращали в (2) реакцией с хлоргидратом М.
в пиридине. Метоксиминогруппа производного (2) устойчива в же-
стких условиях, необходимых для метилирования сильно простран-
ственно затрудненной 17а-гидроксильной группы с образованием
274
(3 ). Метоксиминогруппа удивительно устойчива к кислотному
гидролизу и удаляется превращением в семикарбазоп с последую-
щим гидролизом водной уксусной кислотой.
Бернштейн н сотр, [3] использовали метокспми по группу для
защиты 20-кетостероидов, а Ирдли и Лонг [4] показали, что при
обработке 17а,21 -диокси-20-метоксиминостероидов (1) хлористым
тио пил ом в пиридине происходит отщепление 17а-оке и группы с об-
разованием Д16-производных (2).
СН2ОН
I
„ C=NCCH3
РЩ.он „
[ ЬОС12~-Ру
Метоксиамнп присоединяется к Д1й-20-кетопрегианам (3) с об-
разованием 16а-метоксиамипо-20-кетопрегнапов (4) [5].
CH3ONH2
(4)
275
1. Goldfarb A. R., J. Am. Chem. Soc., 67, 1852 (1945); H j e d s H., Acta
Chem. Scand., 19, 1764 (1965).
2. Fr led J. H., Nut ile A. N., J. Org. Chem., 27, 914 (1962).
3. Heller M., McEvoy F. J., В e r n s t e i n S., J. Org. Chem., 28, 1523
(1963).
4. Ear dley S., L о n g A. G., J. Chem. Soc., 1965, 130,
5. Dref ahi G., Ponso Id K-, Schonecker B., Rott U., Chem.
Ber., 99, 186 (1966).
МЕТИЛЕНДИУРЕТАН, CH2(NHCO2C2H5)2. Мол. вес 190,20,
т. пл. 131°.
Получение. М. получают конденсацией уретана с 40%-ным
раствором формальдегида в присутствии конц. соляной кислоты
[1]. При обработке М. трехфтористым бором отщепляется уретан
(2) и образуется нестабильный метиленуретан [(3), возможно,
ZNHCO2C2H5
Конц. HCI / 2 2 5
CH2=O+2H2NCQ,C2H&——-------->СН2 4-НаО
XNHCO2C2H5
(1}
(I) + BF3-------------> СНа - NCO,C,HS
-H2NCOsC2H5
(2) (3)
в комплексе с BF3], являющийся реакционноспособным диенофилом.
Например, с изопреном (4) он образует тетрагидропнридиновое
производное (5) 12], а с циклогексадиеном-1,3 (6) — производное
изохинуклидина (7) [3].
согсгн5
Н2С^ сн,
II
СНг ^СН
(4) |
СН3
согсгн5
N
Н2С^
70-90’
10 час
------->
(7)
(3) (6)
1. Conrad М., Hock К., Вег., 36 , 2260 (1903).
2. М е r t е п R., М ii 1 1 е г G., Angew. Chem., 74 , 866 (1962).
3. С a v а М. Р., W з 1 k i n s С. К., Jr., Chem. Ind.,, 1964, 1422.
МЕТИЛЕНТРИФЕНИЛФОСФОРАН, (С6Н5)3Р=СН2 (в эфирном
растворе).
По Виттиту и Шеллкопфу [1] получение М. и его конденсация
с циклогексаноном осуществляется следующим образом. К раствору
276
0,1 моля н-бутиллития в—100 мл эфира добавляют еще 200 мл
эфира, перемешивают в атмосфере азота и затем в течение 5 мин
постепенно добавляют 0,1 моля кристаллического метилтрифенил-
фосфонийбромида (выделение бутана вызывает вспенивание). Пере-
мешивание в течение 4 час при комнатной температуре дает оранже-
вый раствор, содержащий небольшое количество осадка реактива
Виттига. После добавления по каплям 0,11 моля циклогексанона
наблюдается обесцвечивание раствора и образование белого осадка
окиси трифенилфосфима. Смесь кипятят в течение ночи, охлаждают,
осадок отфильтровывают на воронке Бюхнера и промывают эфиром.
Эфирный раствор промывают водой, высушивают, эфир отгоняют
на 80-сантиметровой колонке со стеклянной насадкой и перегонкой
на колонке получают метиленциклогексан 99%-ной чистоты.
+ С4Н»Ы \__
(СеН5)3Р—СН3(Вг_) —-~~—-—> (С6Н5)оР = СН2-—*
1 е 3 ' -C4Hl0, -LiBr b S S s 35—40%
—/-—CH a (C6H6)3P —O“
1, W i t t i g G., Schoellkopf U., Org, Syn., 40, 66 (1960).
МЕТИЛЕН ХЛОРИСТЫЙ, CH2CI2, T. кип. 40,8°, уд. вес 1,34.
M. х. широко применяется в качестве растворителя для ацили-
рования по Фриделю — Крафтсу и в одном случае меняет направ-
ление замещения. Ацетилирование хризена в нитробензоле или серо-
углероде дает 2-, 4- и 5-ацетилхризены, тогда как в М. х. единствен-
ным продуктом является 2-ацетилхризен [1].
fAП—С0СНз
CHjCOCl
z^-.А/W3 —AI(A /С\А/А
I II I 4 750/0 I II I
6
В М, х. хорошо растворяются эфираты магнийгалогенидов
и даже те производные терминальных ацетиленов, которые плохо
растворимы в эфире, М. х. используют для проведения реакции
Гриньяра вместо более дорогого ТГФ [2]. Реактив Гриньяра, как
обычно, получают в эфире, но перед реакцией растворитель заме-
няют на М, х.
Кертин [3] использовал смесь М. х.— эфир для восстановления
алюмогидрндом лития сложных эфиров, нерастворимых в эфире.
I. Carruthers W., J. Chem. Soc., 1953, 3486.
2. V 1 е h е H. G., Reinstein M., Chem. Ber., 95, 2557 (1962).
3. C u r t i n D. Y., К ampm eier J. A., Farmer M. L., J. Am. Chem,
Soc., 87, 874 (1965).
277
МЕТИЛ ИОДИСТЫЙ, СНД. Мол. вес 141,94, т. кип. 42,3%
уд. вес 2,279.
Реагент получают (а) вымыванием иода конденсатом
СНзОН — СН31 в камеру, содержащую смесь красного и желтого
фосфора (выход 93—95%) [11; (б) добавлением диметилсульфата
к суспензии карбоната кальция в водном растворе йодистого
калия при перемешивании; М. и. отгоняют по мере образования
(выход 90—94%) [2].
О-Метилирование. Для этерификации карбоновой кислоты раст-
воряют 0,1 моля кислоты в метаноле, добавляют 15,5 а М. и. и 14 а
карбоната калия и перемешивают смесь в течение ночи [3]. 2,5-Ди-
оксиацетофенон превращают в монометиловый эфир охлаждением
ацетонового раствора до комнатной температуры, добавлением кар-
боната калия и М. и. и кипячением в течение 6 час 14]. Ацетон отго-
няют и продукт выделяют перегонкой с паром.
KI + (CH3),SO;J —> CH3I-'-CH3SO4K
СОСНз ССОД
/I ОН 1 /01
Д\ / Ацетон (300 мл), K2CO;t(0,2 моля) //\/
I || СН31 (0,295 моля) | |1
|]| я r_(ijo/ 1 II
0,197 моля
/[ля исчерпывающего метилирования углеводов было предложено
использовать М. и. в сочетании с окисью серебра [51. В улучшенной
методике [6] раствор углевода в метаноле встряхивали при 25°
с М. и. и окисью серебра и через каждые 12 час прибавляли следую-1
щис четыре порции, периодически отфильтровывая серебряные
соли.
Кун [7| показал, что исчерпывающее одностадийное метилирова-
ние углеводов происходит под действием окиси серебра и М. и.,
если в качестве растворителя использовать ДМФА. Углевод рас-
творяют в ДМФА, взятом в количестве, в 10—25 раз превышающем
вес углевода, и обрабатывают при комнатной температуре М. и.
(3 же па каждую OII-группу) и окисью серебра (2 же па каждую
ОН-гругшу). Можно использовать также окись бария [8]. 1,2-Дике-
топы по этой методике превращают в монокетали. Например, бен-
зил превращается в (1), фенантренхинои — в (2), нингидрин —
в (3) [9].
с ссн3
п 1
с6н5с~с-с6н5
ссн3
О)
278
Уолкер и др. [10] показали, что использование и. и окиси
серебра в ДМФА эффективно для превращения углеводов, обладаю-
щих восстановительной активностью, в полностью метилированные
гликозиды без защиты активной группы. Реакция гладко протекает
с различными альдозами, кетозами и уроновыми кислотами; за
16 час можно получить полностью метилированные продукты с вы-
ходом 80—90%.
Кун и Тришманн [111 показали, что метилирование в ДЛ4ФА
лучше проводить не М. и., а диметилсульфатом. Вещества, плохо
растворимые в ДМФА, можно метилировать в Д^МСО или в смеси
ДМСО — ДМФА. Инозитол был превращен таким методом в гекса-
метиловый эфир (т. пл. 18°).
Сривастава [12] показал, что полисахариды типа кукурузного
крахмала можно перметилнровать растворением их в ДМСО и пере-
мешиванием при 25° в течение 48 час с М. и. и окисью бария.
Однако в более поздней работе [13] утверждается, что более высокая
степень метилирования крахмала достигается при использовании
ди метиле ул ьфата и едкого натра в ДМСО.
Сильно хелатированную пери-гидр оке ильную группу юглона
можно прометилировать М. и. и окисью серебра в хлороформе, а не
в ДМСО [14|.
CH3I-AgzO>
СНС1Э
ОМетилирование. Для метилирования 1-зтилпропилиденпиан-
уксусного эфира (1, получен из диэтилкетона и этилцианоацетата)
в 1-этилпропемилметилциануксусный эфир (3) [15] к раствору
0,4 моля этилата натрия в этаноле добавляют при —5° по каплям
0,4 моля (I); к образовавшемуся еноляту (2) прибавляют 0,44 моля
М. и. со скоростью, обеспечивающей медленное повышение темпе-
ратуры. Смесь кипятят до получения нейтральной реакции на лак-
мус. Затем доливают воду, органический слой отделяют, высуши-
вают и перегоняют.
СН3СН2 СН3СН,
I NaOC..I-Ц [ СНа1
СН8СН3С^ССО3С2Н, — > CH3Cl-I^C -C"CO2C„H-,(Na+)--—ц
[ I “ 31—8/%
CN CN
(О (2)
СН3СП3СН3
СНаСН — (!— ССОгС2Н5
(.3) CN
279
C-Метилирование ди гидрорезорцина (2) проводится без' выделе-
ния продукта, полученного гидрированием резорцина (2 моля)
в щелочном растворе [16]. Раствор отфильтровывают от катализа-
тора, обрабатывают диоксаном и 2,64 моля М. и. и кипятят смесь
12—14 час. При охлаждении льдом в почти чистом виде выделяется
2-метилциклогександион-1,3. Кристаллизация из 95%-ного эта-
нола дает с небольшими потерями бесцветные кристаллы, плавя-
щиеся при 208—210° с разложением.
ОН
(1)
58—60%
(2)
Na ОН — Диоксан
СНа1
Синтез 1-метилизохинолина из изохинолина осуществляют ме-
тилированием соединения Рейссерта (2) [17]. Это твердое вещество
(0,32 моля) высушивают и растворяют в смеси диоксан — эфир при
перемешивании в атмосфере азота, раствор охлаждают до —10°
и добавляют по каплям 0,35 моля фениллитйя в эфире; раствор при-
обретает ярко-красную окраску и из него выпадает красный осадок.
Затем добавляют М. и. (0,4 моля), смесь перемешивают на холоду
в течение 2 час и оставляют на ночь. При обработке смеси получают
(3) в виде кристаллов кремового цвета, дающих при щелочном
гидролизе 1-метилизохииолин (4).
С,Н5СОС1.
KCN
58—77%
1) CnH5Li; эфир — диоксан
2)СНг1
Присоединение к трех валентным азоту и фосфору. При получении
иодида бензилтриметиламмоиия (2) к раствору 1 моля третичного
амина (1) в 200 мл абсолютного этанола при перемешивании добав-
ляют 1,34 моля М. и. со скоростью, обеспечивающей слабое кипение
(30 мин) [18]. Значительное количество (2) выделяется в процессе
добавления М.; и., остальное осаждается при добавлении 1 л аб-
солютного эфира.
C6H5CH3N(CH3)3 + CH3I — C6HSCH2N(CH3)3(I~)
у ч — у у /0
(I) (2)
280
Диизопропилметилфосфонат (4) получают реакцией 2 молей трн-
изо пропил фосфита с 2 молями М. и. [19]. В колбу, снабженную хо-
лодильником, помещают М. и. и к нему приливают 50 мл триизопро-
пилфосфита, нагревают до начала реакции, затем нагревание пре-
кращают и остаток реагента прибавляют со скоростью, обеспечиваю-
щей слабое кипение. После кипячения в течение 1 час смесь разго-
няют. Количество выделенного изопропилиодида составляет 91%.
о-
[(СН3)2СНО]3Р+ СН31 — [(СН3)2СНО]3Р^-СН3-Н(СН3)2СН1
V 3 У О ; О
(3) ‘ (4)
Фосфонат перегоняется в виде бесцветной жидкости при 51°/1 мм.
Метилирование каротиноидных спиртов. Работы в этой области
развиты и освещены Каррером [20].
1. Кинг Г., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 281.
2. Н а г t m a n W. W-, Org. Syn., Coll. Vol., 2, 404 (1943).
3. S a r e t t L. H., private communication.
4. V у a s G. N-, Shan N- jM., Org. Syn., Coll. Vol,, 4, 836 (1963).
5. Purdie T., Irvine J. C., J. Am. Chem. Soc., 83, 1021 (1903).
6. A n d e r s о n A. S., Barker G. R., Gulland J. M., Lock AL V.,
J. Chem. Soc., 1952, 369.
7. Kuhn R., Trischmann LL, Low I., Angew. Chem., 67, 32 (1955);
Kuhn R., В a e r H., Chem. Ber., 88, 1537 (1955); 89, 504 (1956).
8. Kuhn R., Baer H., S e e 1 i g e r A., Ann., 611, 236 (1958).
9. Kuhn R., Trischmann FL, Chem. Ber., 94, 2258 (1961).
10. W a Iker H. G., Jr., G e e M., McCr eady R. M., J. Org. Chem.,
27, 2100 (1962).
11. Kahn R., Trischmann H., Chem. Ber., 96, 284 (1963). Review:
Wallenfels К., В ech t 1 e r G., Kuhn R., Trischmann H.,
Egge H., Angew. Chem., Internat. Ed., 2, 515 (1963).
12. S r i v a s t a v a H. С. H a r s h e S. N., S i n g h P. P., Tetrahedron Let-
ters, 1869 (1963).
13. Sr i vast a v a H. C., Singh P. P., Harshe S. N., V. irk K-,
Tetrahedron Letters, 493 (1964).
14. G a r d e n J. F., Thomson R. H., J. Chem. Soc., 1957, 2483.
15. H a n с о c k E. M., Cope A. C., Org. Syn., Coll. Vol., 3, 397 (1955).
16. Меклер А., Рамашандран С., Сваминатхан С., H ь ю-
м e н AL, «Синтезы органических препаратов», изд-во «Мир», М., 1964, сб. 12,
стр. 93.
17. Weinstock J., Boekelheide V., Org. Syn., Coll. Vol., 4, 641
(1963).
18. Brasen W. R., Hauser C. R., Org. Svn., Coll. Vol., 4, 585 (1963).
19. Ford-Moore A. H., P e г г у B. J., Org. Syn., Coll. Vol., 4, 325 (1963).
20. Muller H., Rarrer P., Helv. chim. Acta, 48, 291 (1965).
МЕТИЛКЕТЕНА ДИЭТИЛАЦЕТАЛЬ, CH3CH=C(OC2H5)2. Мол.
вес 130,18, т. кип. 133—134°.
М. д. получают [1], прибавляя по каплям триэтил-а-бромортопро-
пиопат при перемешивании к суспензии натрия в кипящем бензоле.
СН3СНВгС(ОС2Н5)3 + 2Ма СН3СН ^С(ОС2Н5)3 +NaBr NaOC,H5
281
Дегидратация альдоксимов [2]. М. д. образует с син-альдоксимом
аддукт (1), разлагающийся с хорошим выходом каталитическим ко-
личеством трехфторисюго бора и окиси ртути в изонитрил (2); ад-
дукт (3) антц-оксима разлагается с образованием нитрила (4).
HON=CHC6H4CH3 сн,с,н = С(ОС2Ищ СН3СН2С(ОС,Н5),
Оч /Н
XN=CZ
чс5н4сн3
(п
—> CH;iCH2CO2C2H5 + C2H5OH-pCH3CfjH4N^C
(2)
СН3СНПС(ОС3Н5)., СН;!СНаСО,С.Н5 У- С2НГ1ОНч- СН3СЙН4С = N
I (Ъ
о. свн4сн3
XN-C<
(3) ' н
1. Wallers Р. М., М cElvain S. ДР, J. Am. Chem. Soc., 62, 1482 (1940).
2. М uka iy ата Т., Tonooka К., Inoue КД, J. Org. Chem., 26, 2202
(1961).
МЕТИЛЛИТИЙ, СНДл.
Алленовый синтез. Деринг [1] открыл новый метод синтеза
алленов, состоящий в присоединении дибромкарбена к олефииу
и дегалогенировании магнием с раскрытием трехчленного цикла
[2]. На второй стадии были получены очень чистые продукты, но
с низкими выходами (16—35%). Позднее Моор [3] и Скаттебол [4]
Вг.^ Вг
/ х
— СН = СН- СН —ХСН- — CH = C = CH-4-MgBra
показали, что дегалогенирование с перегруппировкой гораздо более
эффективно в присутствии алкиллития, а не магния. Например,
Вгч Вг
+CH3Li — СН=С = СН—ДСН3Вг<ЫВг
-СН-СН —
в синтезе циклоноиадиева-1,2 [3, 4], подробно описанном в мето-
дике [5], исходным веществом является ^ис-цикл ооктеи (2), получен-
ный частичным гидрированием гщс,гщс-циклооктадиена-1,5 [6].
В колбу, снабженную мешалкой и холодильником, продувают азот
и загружают 1,87 г-атом калия и 2 л безводного mpem-бутанола,
смесь перемешивают при кипячении под небольшим избыточным
давлением азота до растворения металла (водород выводится через
ртутную ловушку). При атмосферном давлении в среде азота отго-
няют в высушенную колбу около 1,5 л mpem-бутанола, который мож-
но использовать повторно. Оставшийся растворитель удаляют при
пониженном давлении и образующийся белый mpem-бутилат калия
282
нагревают при 150° и давлении 1—0,1 мм в течение 2 час. Затем
в систему снова вводят азот, загружают 1,62 моля цис-циклооктсна
и 200 мл пентана (разбавитель, облегчающий перемешивание),
снабжают колбу капельной воронкой с 1,66 моля бромоформа
и охлаждают реакционную смесь в бане со льдом и солью. К смеси
в течение 6—7 час при перемешивании добавляют по каплям бромо-
форм; при этом цвет реакционной смеси постепенно изменяется от
светло-желтого до коричневого. Затем температуру смеси доводят
до комнатной и перемешивают в течение ночи. Добавление воды,
нейтрализация, экстракция пентаном и перегонка дают чистый 9,9-
дибромби цикло-16,1,0]-нонан (3) с т. кип. 6270,4 мм. Следующую
стадию проводят в колбе, снабженной мешалкой, капельной ворон-
кой с противодавлением и трубкой для ввода азота, соединенной
с предохранительным клапаном (см. Гриньяра реактивы, рис. Г-3).
СН3СН2СН2СН2СНгСНг
В колбу загружают 0,66 моля дибромида (3) и при охлаждении сме-
сью ацетон — сухой лед до температуры от —30 до —40° и переме-
шивании в течение одного часа добавляют по каплям раствор 0,85 мо-
ля М. в 450 мл эфира. После перемешивания в течение еще 30 мин
избыток М. разлагают, добавляя по каплям 100 мл воды. Затем
приливают большее количество воды, продукт экстрагируют эфи-
ром и выделяют разгонкой. Полученный с выходом 90—93% цикло-
октадиен-1,2 имеет, согласно данным газовой хроматографии, 99%-
ную чистоту. Гарднер [6! под действием магния получил из (3)
углеводород с выходом 59% и восстановил его натрием в жидком
аммиаке до цис-циклононена (5).
Для синтеза алленов преимущественно используют М., но он
менее реакционноспособен, чем /-t-бутиллитий. Моор [3] показал,
что 1,1-ди хлор-2-гексил циклопропан выделяется неизмененным по-
сле обработки М. в кипящем эфире, тогда как под действием «-бу-
тиллития в эфире при —10° он дает нонадиен-1,2 с выходом 31%
(конверсия 52%):
1 сн3снгснгснгснгсн2сн—с=снг
1
283
Хотя реакция М. с se.w-дибромциклопропанами обычно дает алле-
ны, взаимодействие с напряженным 8,8-дибромцикло-[5,1,0]-окта-
ном приводит к большому числу указанных продуктов [6а];
CH3L1
EtjO
8% 17% 33%
10%
32%
Унч и др. [7] описали превращение олефина в аллен по следующей
схеме:
\с = С^+СВг4 + СНзЕ1_>^С = С^+:СВг2 —►
Четырехкратный избыток олефина обрабатывают 1 экв четырех-
бромистого углерода и 2 экв №. в эфире при —65ъ. Выхбд продукта
вдвое выше, чем при реакции с «-бутиллитием.
Скаттебол [8] использовал эту реакцию для синтеза кумуленов
из алленов. Однако на первой стадии реакции выход желаемого
бис-аддукта составляет лишь 6%, тогда как главный продукт реак-
ции — моноаддукт.
СВг2
(СН3)гС = С=С(СН3)а (СН3),_С— С-С(СН3)2
^СВп,
СН31Л
—"Г (СН3)3С=С = С = С = С(СН3)3
-7 8%
CH3Li вместо CH3MgBr. В превращении кетолов типа (1) в
eutf-гликоли (2) с применением реактива Гриньяра выходы не пре-
вышают 20%, тогда как с М. они составляют ~58% [9].
R R СН3
R-C-CCH3 R_C-(icH3
in II
он о он он
(1) (2)
284
Метилкетоны из кислот. Карбоновые кислоты или их литиевые
солн реагируют с алкиллитием с образованием кетона [10]. Пример
такой реакции приведен в следующем разделе.
О
II
RCO2H-|-2R'Li RCR' + Li,O+ R'H
Расщепление ацетатов. Де Пюи [111 использовал М. в первой
и последней стадиях общего метода синтеза циклопропанолов из
циклопропанкарбоновых кислот, например из трднс-2-фенилцикло-
пропанкарбоиовой кислоты (1). Реакция с 2 же М. дает метилкетон
(2), который по Байеру — Виллигеру превращают в ацетат (3).
Расщепление в циклопропанол (4) легко осуществляется под дейст-
вием М., более пригодного для этих целей, чем сильноосновный
гидрид лития, к действию которого циклопропанолы очень чувстви-
тельны.
со2н
с6н5 н
(1)
2 CH3Li- эфир; Д CF3CO3H д
NH4CI—НгО Нч/ \/ССН3 СН2С1г Ну\/ОССНз
* сД'н 77’A сД'н
(2) (3)
CH3Li — эфир; вода.Н3Р03
79%
(4)
1. Von Doering W., Hoffmann A. К., J. Am. Chem. Soc., 76, 6t61
(1954).
2. Von Doering W., LaFlammc P. M., Tetrahedron, 2, 75 (1958).
3. Moore W. R-, Ward H. R., J. Org. Chem., 27, 4179 (1962).
4. Skatteb# 1 L., S 0 I 0 m 0 n S., Chem, Scand,, 17, 1683 (1963).
5. Skattebjgrl L., Solomon S., procedure submitted to Org. Syn.
6. G a r d n e r P. D., Narayana M., J. Org. Chem., 26 3518 (1961).
6a. Marquis E. T„ Gardner P. D., Tetrahedron Letters, 2793 (1966).
7. U n t c h K., G., Martin D. J., Castell ucci N. T., J. Org. Chem.,
30, 3572 (1965).
8. S k a 11 e b 0 1 L., Tetrahedron Letters, 2175 (1965).
9. -H i c k i n b о t t о m W. J., Hyatt A. A., Sparke M. B., J. Chem.
Soc., 1954, 2533.
10. Gilman H., van Ess P. R., J. Am. Chem. Soc., 55, 1258 (1933); G i I-
m a n H., Langham W., Moore F. W., J. Am. Chem. Soc., 62, 2327
(1940); Tegner C., Chem. Scand., 6, 782 (1952); Arens J. F., van Dorp
D. A., Rec. trav. chim., 65, 338 (1946); 66, 759 (1947).
11. DePuyC. H., Dappen G. M., Eilers K. L., KleinR. A.,
J. Org. Chem., 29, 2813 (1964).
МЕТИЛМАГНИЙБРОМИД. Реагент в ТГФ при комнатной тем-
пературе с хорошим выходом восстанавливает гелг-дибромциклопро-
пан в монобромциклопропан [1]. Эта реакция, по-видимому, ради-
285
кальпая. Преимущественное образование цпе-изомера не вызывает
удивления: в соединении (1) 7а-бром экранирован слабее, чем 7f3~
бром, поэтому первый отщепляется легче, чем второй.
72%
цис; транс = 2} 7
1. Seyferth D., Р г о k а 1 В., J. Org. Chem., 31, 1702 (1966).
МЕТИЛМАГНИЙИОДИД. У и лдс и Ма к Ко р ме к [ 1 ] при п оп ыт ке
деметилировать кетон (2) встретились с трудностями. Обычные силь-
нокислотные реагенты приводили к самокондеисации; даже хлор-
гидрат пиридина оказался неудовлетворительным. Было показано,
что спирт (1) можно эффективно деметилировать нагреванием с ме-
тилмагнийиодидом. Эфир (I) обрабатывают эфирным раствором ре-
актива Гриньяра, растворитель отгоняют и остаток нагревают 2 час
при 180—190°.
СаН5
/—\ I ✓--х /ОН
сн3о-/ сн—сн —< х —>
I \—/хн
(1) сгн5
-+сн3о
С2Н,5
^-сн-сн -
н
(2) С2Н6
CH3MgI , 180—190°
I. Wilds A. L., McCormack W. В., J. Am. Chem. Soc., 70, 4127 (1948).
286
N-МЕТИЛМОРФОЛИН, I J . Мол. вес 101,15, т. кип.
113—115° V
СН3
N-МЕТИЛМОРФОЛИНА ОКИСЬ — ПЕРЕКИСЬ
ВОДОРОДА [I]. ЛАол. вес 151,16, т. пл. 73—75°.
Реагент получают [21. путем медленного добавления 34 а
(0,5 моля) 50%-ной перекиси водорода к 26 г (0,25 моля) N-метил-
морфолина в 100 мл трет-бутанола при охлаждении реакционной
смеси па водяной бане до температуры 30—35°. Смесь разбавляют
170 мл трет-бутанола и оставляют на 48 час для полного окисления
N-метилморфолина. Раствор после определения содержания пере-
киси титрованием * можно непосредственно использовать для окис-
ления олефинов или высушить сульфатом магния и отогнать в ва-
кууме летучие вещества и получить кристаллический комплекс
окиси N-мети л морфолина с перекисью водорода, который расти-
рают с ацетоном и отделяют.
Реагент используют с каталитическим количеством четырех-
окиси осмия для превращения А1?(20)-прегнепа типа (I) в 17а-окси-
20-кетопрегнан (2) [31 **. Раствор 3,24 г (1) в ПО мл трет-бутанола,
30 мл хлористого метилена, 4,1 мл пиридина, 10,9 мл раствора 2,1 экв
комплекса в трет-бута ноле и раствор 5 мг четырехокиси осмия
в 1,7 мл трет-бутанола перемешивают при комнатной температуре
в течение ночи. Реакционную смесь обрабатывают 30 мл 0,5%-ного
гидросульфита натрия и 2 г порошка для фильтрования, переме-
* Порпшо 2 мл добавляют к смеси 20 мл уксусной кислоты и ~ 1 г бикарбо-
ната натрия (хранить без доступа воздуха) и 2 мл насыщенного раствора нодида ка-
лия. Закрывают колбу непритертой пробкой, выдерживают в темноте в течение
5 мин и образующийся иод титруют 0,1 н. тиосульфатом натрия.
Для осуществления этого превращения-использовали перекись водорода
и четырехокись осмия.
237
шивают 30 мин и фильтруют. Фильтрат и промывные воды отгоняют
досуха и продукт хроматографируют.
1. Contributed by Schneider W. P., Upjohn Co.
2. Schneider W. P., H anze A. R., пат. США 2769823.
3. Nathan A. H., Magerlein B, J., Hogg J. A., J. Org. Chem., 24,
1517 (1959); M i e s c h e K., Schmi dl in J., Helv. chim. Acta, 33, 1840
(1950).
МЕТИЛНИТРАТ, CH3ONCK. Мол. вес 74,02, т. кип. 65°, уд. вес
1,20.
Получение [1]. Конц, азотную кислоту (300 мл) охлаждают
льдом и обрабатывают при охлаждении 300 мл конц. серной кислоты.
Охлаждают льдом 150 мл метанола и при температуре ниже 10°
осторожно добавляют 50 мл конц. сериой кислоты. В три колбы
Эрленмейера емкостью по 500 мл разливают поровну холодную смесь
азотной и сериой кислот, добавляют к каждой порции соответствую-
щую равную долю смеси метанола с серной кислотой и перемешивают
в течение 2—3 мин. М. отделяется в виде почти бесцветного масло-
образного верхнего слоя. После выдерживания в течение 15 мин
нижний слой отработанной кислоты отделяют и смешивают с боль-
шим количеством воды для предотвращения бурного разложения.
Объединенные порции М. промывают 25 мл охлажденного льдом
22%-ного раствора хлористого натрия и этот процесс повторяют,
добавляя щелочь до слабощелочной реакции. М. промывают для
удаления щелочи охлажденным льдом раствором соли и затем
дважды порциями по 15 мл ледяной воды, сушат хлористым кальцием
и отделяют декантацией; выход 190—230 г (66—80%). Перегонять
продукт ие рекомендуется; в неочищенном виде он вполне пригоден
для синтетических целей.
Получение фенилнитрометана 121. К раствору этилата натрия,
полученному из 2 г-атом натрия и 400 мл абсолютного этанола, до-
бавляют 100 мл абсолютного этанола и раствор охлаждают до 0°;
при этом этилат натрия выделяется в виде твердого слоя. Затем
добавляют еще 100 мл этанола и охлажденную смесь 2 молей бензил-
цианида и 2,8 моля М. (перемешивание при 5—15°). Спокойная ре-
акция сопровождается выделением д^и-нитронатриевой соли (1).
О“
CsH5CH.2CN + CH3pNO2+ N.aOC.2Hs 75-82%" СвНбС= N ONa+—►
о- ’ о-
-ЕЛЛЕЦ С6Н5С = N+—ONa + ЛЕЦ CaH5CH = N+—ОН
“N-H3 j -СО2 (3)
CO2Na
(2)
CeH6GH3NO2.
(4)
288
Соль отделяют (300—320 г), помещают в стакан емкостью 4 л и об-
рабатывают раствором 300 г едкого натра в 1,5 л воды. Для гидро-
лиза иитрильной группы смесь кипятят до прекращения выделения
аммиака (3 час), затем при перемешивании охлаждают льдом с солью
до 30°. Добавляют 500 г льда и 850 мл конц. соляной кислоты и пере-
мешивают при 0—10°. Фенил нитрометан экстрагируют эфиром и пе-
регоняют; т. кип. 92—94°/4 мм, выход 56—60% (в расчете на бен-
зилцианид).
I. Блэк А., Б е б е р с Ф., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 2, стр. 329.
2. Блэк А., Б ебс р с Ф., «Синтезы органических препаратов», ИЛ, М-,
1949, сб. 2, стр. 513.
МЕТИЛ НИТРИТ, CH3ONO. Мол. вес 61,04, т. кип. —12°, уд. вес
0,99.
Методика [1] превращения пропиофенона в а-окснминопроиз-
водное — изонитрозопропиофенон — включает обработку раствора
О О NOH
II НС1 II II
С6Н5ССН2СН3 + СН3ОИО —СеНг,С— ссн3
65-68%
пропиофенона в эфире хлористым водородом и М., который получают
добавлением по каплям смеси серной кислоты и воды (1 : 2 по объ-
ему) в смесь нитрита натрия, метанола и воды. Через 4,5 час корич-
нево-красный раствор становится желтым и продукт выделяют
повторной экстракцией 10%-ной водной щелочью; подкислением
объединенных экстрактов получают с выходом 65—68% продукт,
плавящийся при 111—113°. Кристаллизация из толуола дает бело-
снежный изонитрозопропиофенон (т. пл. 112—-113°).
Феррис 12] разработал интересный метод получения о,-амино-
кислот, рассмотренный ниже на примере синтеза гщ-фенилаланина
из 1-фепилбутанона-З (1). Раствор 1,8 моля кетона в 800 мл эфира
CH3ONO (CHs)2SO4
с6нвсн2сн2сосн3 —-СвН5СН2ССОСН3
(1) НС1 (2) NaOH
NOH
NaOCi; Н+ Н2, Pd/C
СеН5СНгССОСН3 —%------> С6Н5СНХСО2Н--------* CgH5CH2CHCOaH
” Г| Диоксап “ц J । “
NOCHs NOC2H5 (5) NHa
(3) (4)
обрабатывают M., получаемым из 2,58 моля метанола, 2,28 моля
95%-ного нитрита натрия в 100 мл воды и 2,86 моля конц. серной
кислоты, разбавленной 145 мл воды. Под действием диметилсуль-
фата, и щелочи оксиминокетон (2) превращается в О-метиловый эфир
(3), гипохлоритное расщепление которого в водном ди оксане дает
кислоту (4). Синтез D.L-фенилаланина (5) завершается гидрирова-
нием с общим выходом 50% [в расчете на (1)].
10 Заказ № 1096
289
Феррис и сотр. [3] разработали синтез лизина, в котором цикло-
гексанон нитрозируют действием М. и соляной кислоты в эфире,
а натриевую соль 2,6-диокси.миноциклогексанона обрабатывают ук-
сусным ангидридом в этаноле для осуществления бекмановскон
перегруппировки в этиловый эфир 2-оксимино-5-циаивалериаиовой
кислоты. Гидрирование над никелем Ренея и щелочным сокатализа-
тором и последующий гидролиз дают моно хлор гидр ат о,вблизи на
с общим выходом 63% (в расчете на циклогексанон). В менее под-
О
J' /NOH
С Н О КГ а Н Ni
NC(CH,)3CCO.,C2Hr, СЩСН.фзСНСОф!
I I Лс2о II НС1 | I
\/ NOH NH2-HC1 NH.2
роб по изученном варианте этой реакции с цикло пентаноном полу-
чают D.L-ОрПИТИН с общим выходом 21%.
1. Хартунг В., Кросслей Ф., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 264.
2. F е г г i s А. Г., J. Org. Chem., 24, 1726 (1959).
3. Ferris A. F., J. Org. Chem.,25, 12 (1960); Ferris A. F.,Joh nsonG, S.,
Gould F. E., Lal ourct t e H. K., ibid,, 25, 492 (I960); Ferris A. F.,
.1 olinso n G. S., Gould F. E., ibid., 25, 496 (1960); Ferris A. F.,
Johnson G. S., Gould F. E., Stange. H., ibid., 25, 1302 (I960);
26, 2602 (1961).
МЕТИЛОВЫЕ ЭФИРЫ КИСЛОТ [4]. Удобным методом превра-
щения кислот в их метиловые эфиры является кипячение кислоты
(1 моль) с метанолом (3 моля), дихлорэтаном (300 мл, можно
использовать также метиленхлорид) и серной кислотой (3 мл для
алифатических и 15 мл для ароматических кислот) в течение 6—
15 час (например, в течение ночи). Реакционную смесь разбавляют
водой, органический слой промывают раствором карбоната натрия,
высушивают и отгоняют растворитель. Большинство алифатических
и ароматических кислот дает метиловые эфиры с 87—98%-ным вы-
ходом [1].
Этот метод представляет собой улучшенный вариант метода
Вайсбергера и Килбера [2], в котором требуется сложное оборудова-
ние, и метода Бейкера [31, который удовлетворителен для малых,
но непригоден для больших количеств.
1. С 1 i п t о п R. О., Laskowski S. С., J. Am. Chem. Soc., 70, 3135 (1948).
2. W е iss b er ge г A., I< i b 1 с r C. J., Org. Syn., Coll. Vol., 3, 610 (1955).
3. Baker B. R,, J. Am. Chem. Soc., 65, 1577 (1943).
4. Contributed byNewma n M. S., Ohio State University.
CH3
2-МЕТИЛПЕНТАНДИОЛ-2,4, CH;1CHCHsicH3. Мол. вес 118,17,
ОН ОН
т. кип. 135—136740 мм; растворим в воде.
290
М. используется как реагент для характеристики и выделения
альдегидов [11. Смесь альдегида, диола и кислотного катализатора
в бензоле кипятят с насадкой Дина — Старка до прекращения вы-
деления воды. В качестве катализатора обычно используют ^-толу-
ол сульфо кислоту или твердый катализатор — серную кислоту на
силикагеле,— получаемый обработкой 100 а силикагеля (28—
200 меш) раствором 0,5 г конц. серной кислоты в 10 мл воды.
НОСНСНз
I I'
RCH—-О-|- СН.,
I
НОС(СН3)2
zO-.-CHCH3
RCH \сН2-|- Н3О
ХО-С(СН3)2
Циклические шестичлениые ацетали образуются с высоким
выходом; например, из акролеина ацеталь получают с выходом 98%,
тогда как выход акрол ей иэтил ей гликольацеталя (пятичленного)
всего лишь 58%. Циклические ацетали стабилизируются, по-види-
мому, экваториальной ориентацией трех заместителей.
НОСНСНз
I
снг=снсно + сн,
I
НОС(СН3)г
Шестичленные циклические ацетали гидролизуются весьма
трудно. Легкость образования, стабильность и плохая растворимость
в воде позволяют получать ацетали в водном растворе и экстраги-
ровать их бензолом. Например, концентрация водного раствора фор-
мальдегида уменьшается с 3 до 0,8% при простом перемешивании
раствора при pH 3 с диолом и бензолом; ацеталь — 4,4,6-триметил-
1,3-диоксан выделяют с выходом 65%. Другой пример — реакция
с а-окси адипиновым альдегидом (2), образующимся при кислотном
но
<fHCH3
10*
гидролизе продукта термической димеризации акролеина (1). Окси-
диальдегид устойчив только в водном растворе кислоты, в котором
он образуется. При попытках его перегонки или отделения от воды
каким-либо другим способом наблюдается конденсация. Однако при
добавлении к водному раствору М. и бензола получают полуаце-
таль (3) с выходом 90—95%. Полуацеталь перегоняется и хранится
без разложения, не реагирует далее с диолом и существует, по-ви-
димому, в циклической форме (36).
1. F i s с h е г R. F., Smith С. W.( J. Org. Chem., 25, 319 (1960); 28, 594
(1963).
1Ч-МЕТИЛПИРРОЛИДОН-2, Г } Мол. вес 99,13, т. кип.
202° (78—79710 мм).
сн3
Специальный растворитель. Ньюмен [1, 2] рекомендует М.
в качестве растворителя при получении нитрилов реакцией арил-
галогенида с цианидом меди(1). Вместо первоначально использо-
вавшегося пиридина (т. кип. 115°) был предложен [31 более подхо-
дящий ДМФА (т. кип. 153°), ноЛЕ еще более удобен благодаря более
высокой температуре кипения. Например, при синтезе 1-цианнаф-
талина из 1-хлорнафталина 121 для выделения нитрила охлажденную
Кипячение
о, 3 моля
0,4 моля I
СН3
.N^O 88-90%
150 МЛ
реакционную смесь, окрашенную в темный цвет, выливают в рас-
твор 60 г этилендиамина в 800 мл воды, затем выдерживают и экст-
рагируют смесью эфир — бензол; экстракт охлаждают, высушивают
и продукт перегоняют. При получении 1-бромнафталина реакцион-
ную смесь достаточно кипятить 3 час.
Хенбест [4] показал, что М. лучше других испытанных апро-
тонных растворителей для нуклеофильного замещения тозилатной
группы. При превращении тозилата холестанола-30 (i) в За-циан-
холестан (2) наилучший выход был получен в случае М., содержа-
сь
сн3
NaCN
TsO'
80%
NC’’’
U)
292
щего 5% трет- бутанол а при 90°, хотя в отсутствие спирта выход
лишь немного ниже.
1. Newman М. S., В oden Н., J. Org. Chem., 26, 2525 (1961).
2. Newman М. S., Ramachandran S., procedure submitted to Org.
Syn.
3. Frie dm an L., Shechter H., J. Org. Chem., 26, 2522 (1961).
4. Henbest H. B., Jackson W. R., J. Chem. Soc., 1962, 954.
1-МЕТИ Л-З-я-ТОЛИЛ ТРИ A3EH, n-CH3CeHdN=NNHCH3. Мол.
вес 149,19, т. пл. 78—81°.
Получение [1]. Диазотируют хлор гидрат /г-толуидина и с по-
мощью холодного конц. раствора карбоната натрия доводят (при
0°) pH раствора хлорида диазония до 6,8—7,2. Образующийся ярко-
красный раствор медленно добавляют при энергичном перемешива-
нии к смеси карбоната натрия, водного метиламина и льда. Экстрак-
ция эфиром дает сырой триазен, возгонкой которого при 50°/0,1 мм
л-СН3СсИ4Йн3С1 /2-CH3C6H4N N(C1)
CHsNH2
—n-CH3CsHdN = N-NHCH3
получают желтый продукт с т. пл. 77—80°. Кристаллизацией из
гексана или смеси эфир — гексан выделяют чистое бесцветное ве-
щество.
Этерификация кислот. Использование реагента иллюстрируется
метилированием 3,5-ди нитро бенз ой ной кислоты. Эфирный раствор
кислоты медленно добавляют к эфирному раствору реагента; при
этом выделяется азот и раствор приобретает красную окраску. По
завершении реакции (около 1 час) эфирный раствор промывают со-
ляной кислотой для удаления n-толуидина, затем раствором соды;
высушивают и упаривают.
СО2Н СО2СН3
I I
I |] +tt-CH3CeHdN = NNHCH3 -_______> || +
/К / 7,1 моля в 10 мл 85—90% I
O2N -/ xNO2 И>ира О2Ж 'NO.
7,1 лмоля в 25 мл
эфира
Щ /2-CH3C6HdNH2 Щ
1. W h i t е Е. Н., В a и ш А. А., Е i t е 1 D. Е., procedure submitted to
Org. Syn.
МЕТИЛТРИФЕНИЛФОСФОНИЙБРОМИД, (CGH5)3PCH3(Br-).
Мол. вес 257,23, т. пл. 232—233°.
Получение [1]. В толстостенную склянку для реакций под
давлением помещают раствор 0,21 моля трифенилфосфина в 45 мл
бензола, охлаждают льдом с солью, добавляют 0,29 моля предвари-
тельно перегнанного бромистого метила, закрывают и оставляют
при комнатной температуре на 2 дня. Затем склянку открывают,
293
белый осадок переносят на воронку, используя 500 мл горячего
бензола, фильтруют с отсасыванием и высушивают в вакууме при
100° над пяти окисью фосфора; выход 74 г (9996).
(С6П5)3Р-|-СН3Вг (C(iH5)3PCH3(Bi-“)
1. W i t t i g G., Schoellkopf U., Org. Syn., 40, 66 (I960).
CH3
1-МЕТИЛФЕНИЛГИДРАЗИН, C6H6NNH2 Мол. вес 122, 17,
т. ки п. 117—118721 мм.
Получение. М. получают восстановлением М-нитрозо-Ы-метил-
анилина цинковой пылью 111.
СН3 сн3
| Zn—АсОН I
CGHr,N — N = ОC8H5NNH2
Ь 2 — 0 о уб
Алказоны. Тот факт, что окисление сахаров фенил гидразином
завершается на стадии образования фенилозазона, авторы объясняли
образованием хелатного кольца [2]; это предположение было под-
тверждено экспериментально [31. Чепмен [4] высказал мысль о том,
что если такая интерпретация верна, то реакция с М. должна осу-
ществляться, минуя стадию образования озазона, и, таким образом,
открыл новый класс соединений, названных алказонами. Например,
1,3-диокси ацетон, эритроза и арабиноза при обработке избытком
реагента в смеси вода — этанол — уксусная кислота реагируют
с 3, 4 и 5 молями М. с образованием алказоиов [плавятся при 127,
151 и 159° (полиморфные вещества) и 152°1 с выходами 94, 55 и 30%
соответственно. Аналогичные продукты получены из ксилозы (39/6)
СН3
СН2ОН сНз ch = nnc6h5
со C = NN(CH3)CcHb
I [
СН.ОН СН —NNCeH5
I
СН3
и арабинозы. С фруктозой образуется с низким выходом загрязнен-
ный алказон.
Арабиноза')
V C6H5NNH3
Ксилоза ------------i
СНЭ
с6н5
294
1. X артман В., Ролл Л,, «Синтезы органических препаратов», ИЛ, №..
1949, сб. 2, стр. 337.
2. Ф и з е р Л., Физер М., «Органическая химия», ИЛ, М,, 1949, стр. 314.
3. Mester L., J. Am. Chem. Soc., 77, 4301 (1955); Angew. Chem., Internal’. Ed.,
4, 574 (19G5); Mester L.,Moczar E., Parel Io J., .1. Am. Chem. Soc.,
87, 596 (1965); Henseke G., Koh ler H., Ann., 614, 105 (1958).
4. Chapman O. L., Welstead W. J., Jr., Murphy T. J., King
R. W., J. Am. Chem. Soc., 86, 732, 4968 (1964).
^метил-^фенилкарбаминоилхлорид,
СНз
c6h5ncoci
Мол. sec 169,61, т. пл. 85—86°.
Вейганд Lil получил M. насыщением 500 мл этилацетата фосге-
ном и одновременным добавлением в течение 1,5—2 час раствора
150 г N-метиланилина в 1,5 л этилацетата. Отгонка растворителя и
кристаллизация из этанола дают чистый продукт с выходом 75—85% .
М. взаимодействует по Фриделю — Крафтсу с ароматическими
соединениями с образованием амидов, восстанавливающихся алю-
могидридом лития в альдегиды,
СН3 СН3 СН;9
J\ /сНз снз А/СНя /\/СНз
I II СДТ-NCOCI, АЮЩ100—130°) | || LIA1H., l" ||
А ~ kJ'
С = О СИ = о
Н3С-г!-СД15
1. Weygand F., М i t g a u R., Chem. Вег., 88, 301 (1955).
N-МЕТИЛФОРМАНИЛИД, CcH5N(CH3)CHO. Мол. вес 135,16,
т. пл. 14°, т. кип. 131°/22 мм.
М. получают азеотропной перегонкой смеси N-метиланилина,
муравьиной кислоты и толуола для удаления образующейся при
конденсации воды с последующей вакуумной перегонкой продукта;
выход составляет 93—97% [Ц.
Вильсмайер и Хаак [21 показали, что реагент образует комплекс
с хлорокисью фосфора (1 : 1), который атакует ароматическое соеди-
нение соответствующей реакционной способности с образованием
продукта, гидролизуемого водой в ароматический альдегид. Авторы
получили п-диметиламинобеизальдегид, но не описали деталей син-
теза:
СН3
Г ТД \run I ПАГ1 тл CtHjN (СНЩ
CGHSMCH(J“|- РОС13 —> Комплекс .——---— > Промежуточное соединение
-2^ C6H5NHCH3+ (CH3)2NC6H4CHO-n
295
Первая подробная методика опубликована для синтеза пирен-
3-альдегида [3]. М. (135 а) смешивают с 100 мл о-дихлорбензола
(для растворения углеводорода) и к полученной смеси при 20—25°
и перемешивании добавляют в течение 2 час 135 г хлорокиси фос-
фора. Затем добавляют пирен (100 а) и смесь перемешивают в тече-
ние 2 час при 90—95°. Из образующегося ярко-красного раствора
при охлаждении выделяется комплекс, который собирают, промы-
вают бензолом и разлагают водой. Кристаллизация из этанола дает
ярко-желтые длинные иглы пирен-3-альдегида.
сн3
С6Н5ЫСНО + РОС13^
53%
Методика получения 9-антральдегида аналогична вышеописан-
ной, но реагенты и растворитель смешивают сразу и смесь
перемешивают механически на кипящей бане в течение 20лшн [4].
К красному комплексу для разложения продукта конденсации М.
с хлорокисью фосфора добавляют водный раствор ацетата натрия.
Перегонка с паром и кристаллизация из уксусной кислоты дают ярко-
СНО
I I
Z\ Z\ /К CeHBNCH0 (0,26 моля), РОС13 (0,23 моля) Zx Z\ /Ы
77 77 '7 о-Дихлорбензол (20 мл) 77 77
желтые кристаллы альдегида. По аналогичной методике 3,4-бензпи-
рен дает 5-альдегид с выходом 90% [51. С менее реакционноспособ-
ным 1,2-бензантраценом выделяют около половины непрореагиро-
вавшего углеводорода, а выход продукта составляет 63% (в расчете
на вступившее в реакцию вещество) [61- Высокая реакционная спо-
собность тиофена позволяет проводить синтез при более низких
температурах [7]. Для образования комплекса смешивают 1 моль
реагента с 1 молем хлорокиси фосфора и оставляют на 30 мин.
В течение этого времени температура немного повышается, а цвет
296
изменяется от желтого до красного. Затем к смеси при перемешива-
нии в течение 2 часпри25—30°добавляют 1,1 моля тиофена. 2-Тио-
фенальдегид образуется с выходом 71—74%. При нагревании
смеси компонентов на кипящей водяной бане в течение 6 час 2-это-
ксинафталин превращается в 1-альдегид с 74—84%-ным выходом [4].
1. Fieser L. F,, Jones J. Е., Org. Syn., Coll. Vol. 3, 590 (1955).
2. Vilsmeier A., Haack A., Ber., 60, 119 (1927).
3. V о I I ni a n и H., Becker H., С о г e I I M., Streeck H., Ann., 531,
1 (1937).
4. Fieser L. F., Hartwell J. L., J ones J. E., Org. Syn., Coll. Vol., 3,
98 (1955).
5. Fieser L. F., Hershberg E. B., J. Am. Chem. Soc. 60, 2542 (1938).
6. Fieser L. F., H artwell J. L., J. Am. Chem. Soc., 60, 2555 (1938).
7. W e s t о n A. W., Michaels R. J., Jr., Org. Syn., Coll. Vol., 4, 915 (1963).
МЕТИЛФОСФОНОВОЙ КИСЛОТЫ ДИХЛОРАН ГИДР ид,
СН3 Cl
¥
O^XCI
Мол. вес 132,92, т. пл. 32°.
Гофман [1] получил М. к. д. хлорированием мети л фосфоновой
кислоты действием 2 же пяти хлор исто го фосфора. Киннер и Перрен
[2] получили это соединение реакцией хлористого метила с трех-
хлористым фосфором в присутствии хлористого алюминия и осто-
рожным разложением комплекса водой.
Мак-Кей [3] показал, что М. к. д. взаимодействует по крайней
мерее некоторыми 1,2-, 1,3-и 1,4-гликолями с образованием цикли-
ческих сложных эфиров, которые расщепляются кислотами.
RCHOH RCH — О О
I I \ //
(СН2)„ —> (СНа)„ Р
RCHOH RCH — О7 СН3
и=-0, 1, 2
1. Hofmann A, W., Вег., 6, 303 (1873).
2. К i п и е а г А. М., Р е г г е n Е. A., J, Chem. Soc., 1952, 3437.
3. McKay A. F. et al., J. Am. Chem. Soc., 74, 5540 (1952); 76, 3546 (1954.)
2-МЕТИЛФУРАН. Мол. вес 82,10, т. кип. 65°, уд. вес 0,916.
Хо/Х'СН3
Илиел и Фиск [1], описавшие конденсацию Манниха с М., при-
водящую к 5-метилфурфурилдиметиламину, установили, что про-
дажный продукт содержит стабилизатор, который можно удалить
выдерживанием над едким кали в течение 24 час, декантацией и по-
вторным выдерживанием над свежим едким кали.
297
Бухта [2] разработал интересный синтез ундекановой кислоты
(5), исходя из конденсации 2-метилфурана (1) с винил-(р-карбметок-
сиэтил)-кетоном (2).
+
(1)
о
СНг=СНС(СНг)2СО2СН3 А1С1?
н3с'
(2)
о
'сы2сн2с:(снг)гсогсн;
(3)
о
Н CJ II
сн3сснг|
о у
,СНгС( СН2)гС( сн2)2со2сн3
Восстановление,
гидролиз
СН3(СНг)9СОгН
(4)
(5)
I .Eliel Е. L,, Fisk М. Т., Org. Syn., Coll.Vol., 4, 626 (1963).
2 . Buehl a E., В ay er W., Naturwiss., 46, 14 (1959),
МЕТИЛ ХЛОРИСТЫЙ, CH3C1. Мол. вес 50,49, т. кин. —23,7°,
уд. вес 0,920.
Прибор для синтеза М. х. (NaCl, СНаОН, H2SOJ описан [1| в ме-
тодике получения дурола из ксилола, выделенного из каменноуголь-
ной смолы. Реакцию проводят в колбе емкостью 5 л, снабженной
трубкой для ввода газа и обратным холодильником с трубкой для
отвода газа в пробулыснватель, содержащий слой ртути высотой
10 см и соединенный с ловушкой для НС1. В колбу загружают
3,7 л. ксилола и 1 кг AlCl;i и в реакционную смесь пропускают М. х.
в течение 100 час. Дурол выделяют из тетраметилбеизольной фракции
2CHaCJ j А1С13
H3c/^Z ' СН3 НзС/^^СНз II3C/4V 'сн3
СПз
вымораживанием и после кристаллизации получают чи т яй про-
дукт с выходом 10—11%. В качестве побочных продутой выделяют
пента- и гексаметилбензолы.
1. Smit h L. 1., Org. Syn., Coll. Vol., 2, 248 (1943'.
З-МЕТИЛ-4-ХЛОРМЕТИЛИЗОКСАЗОЛ,
Мол. вес 119,56, т. кип. 62°/1 мм.
Н3 СЦ 4^СНгС1
О'
1
298
М. получают хлор метилированием 3-метилизоКсазола и приме-
няют для анпелирования подобно 3,5-ди мети л-4-хлор мети ли зокса-
золу II] (о котором см. в соответствующем разделе). Однако выходы
при алкилировании обычно низкие.
!, Кочетков Н. К., Хомутова Е. Д., Базилевский М. В.,
ЖОХ, 28, 27.3G (1958).
О~
МЕТИЛХЛ0РСУЛЬФИТ,СН3ОЗ Ю Мол. вес 111,54, т. кип.
35—36о/65 мм. Реагент получают из метанола и хлористого тионила
11, 2].
О~
CIJ3OI-I4-SOCL, СН3О5+С1ЩНС1
Берти [2] предпринял безуспешную попытку использовать реа-
гент для превращения вторичных спиртов в олефины. Взаимодейст-
вием спирта с М. в эфире, содержащем пиридин, предполагалось
получить метилалкилсульфиты и затем пиролизовать эти сложные
эфиры.
I он о 1 о j. 1 .,о*. + 1 °*.
-У сн,оТ-а -г7оа1’_^ “7 “сн’-
1 с * - _ с , о- ^„с н0
91 I хн I 'Н* I
--> £Ог + СН3ОН
Позже группа исследователей [3| столкнулась с проблемой се-
лективного отщепления 110-оксигруппы 21-ацетата кортизола (1).
Обычные методы дегидратации неприменимы для (1) из-за ненасы-
щенности кольца А и наличия третичной а-гидроксильной группы
в положении 17. Для реализации поставленной задачи оказался эф-
фективным М. в присутствии пиридина. К раствору 0,01 моля (1)
и 8 мл пиридина в 150 мл ТГФ при перемешивании при температуре
от —10 до —5° добавляют по каплям 0,088 моля М. Смеси дают на-
греться до комнатной температуры и осаждают продукт водой.
299
Выход сырого продукта (2) (т. пл. 226—230°) составляет 89%; чистое
соединение плавится при 232,5—236,5°. При использовании в каче-
стве растворителя диметил ацетамида выход составляет 65%. Отме-
тим, что дегидратация при низкой температуре сопровождается
транс-отщепленисм и, по-видимому, не включает циклическое пере-
ходное состояние для пиролитического ^нс-отщеплепия.
1. Саггё Р., L iberm апп D., Bull. soc. chim. France, [4], 53, 1050 (1933).
2. В e г t i G., J. Am. Chem. Soc., 76, 1213 (1954).
3. Chamberlin E. M., Tristram E. W., UtneT., Chemerd a
J. M., J. Org. Chem., 25, 295 (1960).
МЕТИЛ ЦИКЛОПРОПИЛ КЕТОН, О CH2
Мол. вес 84,11, т. кип. 110—112°. r
Исходное соединение для синтеза М.— а-ацетил-у-бутиро лактон
(1). Ниже указан один из запатентованных методов получения (1).
При расщеплении (1) соляной кислотой выделяется двуокись угле-
рода и образуется хлоркетон (2), который под действием основания
циклизуется в М. (3) [1].
О
COaEt п О С
I / \ ОН- II / \ HCI
СН3СОСН2 + СН2 — СН. —СН3С—СН о —
60% I | -со3
сн2 —сн2
(1)
СН3ССН2СН2СНгС1-------СН3ССН — сн2
80%
(2) (3)
М.— ключевой реагент в общем методе синтеза терпеноидов
[2]. Взаимодействием (3) с метилмагнийбромидом получают карби-
нол (4), претерпевающий при обработке бромистым водородом
гомоаллильную перегруппировку с образованием С6-бромида (5).
Превращение в реактив Гриньяра (6) и повторение предыдущей
(8)
(9) Неролидол
300
серии реакций дают Сц-бромид (8). Синтез можно закончить на
этой стадии превращением (8) в реактив Гриньяра и взаимодействием
с метилвинилкетоном получить неролидол. Можно также продол-
жить синтез для получения высших терпеноидов.
1. Cannon G. W., Е 1 1 I з R. С., L е а 1 J. R., Org. Syn., Coll. Vol., 4, 597
(1963).
2. Julia M., Julia S., Guegan R., Bull. soc. chim. France, I960, 1072.
CH2O zCHaCH3
2-МЕТИ Л-2-ЭТИЛ ДИОКСОЛ AH-1,3, I
CH2O -CH3
Мол. вес. 116,16, т. кип. 117°.
М. получают и используют при обменном образовании кеталей
(«диоксол а низания») [1]. Выходы в ряде случаев выше и селектив-
ность несколько больше, чем при прямой реакции с этиленгликолем.
В ряду стероидов порядок реакционной способности следующий:
насыщенные 3- и 20-кетоны>а,р-непасыщенные 3- и 20-кетоныу>2-
или 4-бром-3-кетоны^>17-кетоны; 2,4-дибром-З-кетоны (не реаги-
руют) [2].
I. Dauben Н. J., L 6 k е п В., R i n g о 1 d Н. J., J. Am. Chem. Soc., 76,
1359 (1954); Rosenkranz G., Velasco M., Sondheimer F.,
J. Am. Chem. Soc., 76, 5024 (1954).
2. D j e r a s s i C., «Steroid Reactions», Holden-Day, 14-16 (1963).
МЕТИЛЭТИЛКЕТОН, CH3COCH2CH3. T. кип. 79,6°.
Кноф [1] показал, что этот растворитель лучше уксусной кис-
лоты для окисления хромовой кислотой смеси а- и р-эпоксидов,
полученных из Зр-ацетокси-А5-андростенона-17. Раствор 18,38 г
Суспензия в метилэтилкетоне
смеси в 180 мл горячего М. охлаждают до комнатной температуры
и к образующейся суспензии мелких кристаллов добавляют 9 мл
75%-ного раствора СгО3 в воде. Через 10 мин смесь разбавляют 1 л
воды, продукт отделяют, промывают, высушивают и получают 17,13 а
(89%) вещества, плавящегося при 198—199°, а после перекристал-
лизации — при 200—201°.
I. Knot L., Ann., 657, 171 (1962).
МЕТОКСИАЦЕТИЛЕН, СН3ОС=СН. Мол. вес 56,06, т. кип. 23°.
Методика получения эфира из диметилхлорацеталя и амида нат-
рия аналогична методике получения этокси ацетилен а; улучшенная
301
методика получения больших количеств М. опубликована Вассер-
маном 1.1].
Аренс [2] показал, что кислоты, например бензойную, можно пре-
вратить в ангидрид в очень мягких условиях реакцией с 0,5 же М.
Вассерман [1! нашел, что при взаимодействии бензойной кислоты
с 1 же М. в определенных условиях можно выделить 1-метоксиви-
нилбензоат (1); реакцию проводят при низкой температуре в при-
сутствии следов бензоата ртути, катализирующих селективное при-
соединение по тройной связи. Джонс [31 предположил, что дальней-
шая реакция осуществляется через нестабильный ортоэфир (2),
разлагающийся внутримолекулярно через переходное состояние
(3) в ангидрид (4) и метилацетат. Вассерман [4] подтвердил это
экспериментально: реакция моноаддукта (1) с бензойной кислотои-
О,а привела к равномерному распределению изотопа между ангид-
ридом и метилацетатом.
ш 0
соси» II II
СаНЕСОгН —ОДС-ОСОСН3
сацссън
еда—о
.с:
СДЦС—о
о
(1)
(2)
ОДС “О СНз
/• ‘.Л
о с
с6н;с---о’^^оснз
О
(3)
О
од<д
----о
08% Z /
с6ньс
о
(4) тпл., 45°
СНз
О-Д
ОСНз
1. Wasserman Н. Н., W li а г t о n Р. S., J. Am. Chem. Soc., 82, 6G1 (1960).
2. Arens J. F., Door n h о s T., Rec. trav., 74, 79 (1955), and earlier papers
cited.
3. Egl inion G., J ones E. R. H., Shaw B. L., Whiting M. C.,
J. Chem., Soc., 1954, 1860.
4. Wasser m a n H. IL, W harlon P. S., J. Am. Chem. Soc., 82, 1411
(1960).
n-МЕТОКСИБЕНЗИЛ ХЛОРИСТЫЙ, n-CH3OC(iH4CH2Cl. Мол.
вес 157,62; т. кип. 101—10378—10 мм. Получение [Г].
Пептидный синтез. Для защиты сульфгидрильной группы п-
метоксибензильная группа считается более подходящей, чем бен-
зильная. Она легко отщепляется от конечного пептида либо под
действием натрия в жидком аммиаке, либо кипячением с трифтор-
уксусной кислотой [2] или с безводным фтористым водородом [3].
302
1. S h r i п е г I?. L., Hull C. J., J. Org. Chem., 10, 228 (1945).
2. Ak abor i S., S а к a к i b a r a S.,Sh i mon isb i Y.,Nob uhara Y.,
Bull. Chem. Soc. Japan, 37, 433 (1964).
3. Sa к akib a r a S., Nob u bar a Y., S h i m о n i s h 1 Y,, Kiy oi R.,
Bull. Chem. Soc. Japan, 38, 120 (1905).
1-МЕТОКСИБУТЕН-1-ИН-3, HC=CCH=CHOCH3. Мол. вес
82,10, т. кип. 52°/28 мм.
1-Метокси- и 1 -этоксибутен-1 hiн-З используют для осуществле-
ния превращения RCHCM>R (СН=СН).2СНО. По Инхоффену [П
этоксисоединение превращают в литиевое производное, добавляют
его к альдегиду с образованием (1), селективно гидрируют тройную
связь и гидролизуют енольный эфир (2) с образованием полиенового
альдегида (3).
RCHC)-[-LiC = CC[-[ = CHOQH5 —> RCHC = ССН _-С11ОС2Н, —
он (1)
rchch =--chch-=choc2h5 -2L'\ rch-chch-chcho
он (2) (3)
По Маршаллу и Уайтингу [21 М. превращают в магнийбромпро-
изводное, добавляют его к бензальдегиду с образованием пропарги-
лового спирта (4), который восстанавливают алюмогидридом лития
в диен (5). Кислотным гидролизом получают с выходом 75% диено-
вый альдегид (6) (т. пл. 431').
С0Н5СНО | BrMgC = CCH=.CHOCH3 —> СеН5СН — С = ССН = СНОСЫ3 —-А;
I
он
Ы)
СВН5СНСН^СНСН^СНОСН, СаН-СН-СНСН^СНСНО
I
он
(5) (6)
1. I п h о f I е п Г1. Н., Bohlmann F., Rummert G., Ann,, 569, 226
(1950).
2. Marshall D., Whiting M. C., J. Chem. Soc., 1956, 4082.
МЕТОКСИМАГНИЙМЕТИЛКАРБОНАТ, CH3OMgOCO,CH3-F
+xco2.
Жарвазы [1] пропускал двуокись углерода в суспензию метилата
магния в метаноле до растворения твердого вещества, отгонял
растворитель и получал белое твердое вещество, по результатам
анализа и свойствам которого можно предположить следующую
реакцию:
О О
'i II
Mg(OCH3), + 2CO2 CH3OCOMgOCOCH3
303
Стилз и Финкбайнер [21 предложили интересное применение
реагента в синтезах, которые свидетельствуют об ошибочности
первоначального вывода о реакции образования реагента. На
самом деле название «метоксимагнийметилкарбонат» относится к ра-
створу, полученному насыщением метилата магния двуокисью угле-
рода, активный реагент которого вернее всего описывается формулой
CH3OMgOCO2CH3+XCO2, где X широко варьирует в зависимости
от растворителя н температуры. Детальные и почти идентичные ме-
тодики получения больших количеств таких растворов приведены
Финкбайнером и Стилзом [3] и Финкбайнером и Вагнером [4].По
этим методикам магниевую стружку прибавляют в течение несколь-
ких часов к сухому метанолу при перемешивании и кипячении до
полного превращения металла в метилат магния. Метанол частично
отгоняют при 50° и пониженном давлении, добавляют ДМФА и в рас-
твор пропускают двуокись углерода с такой скоростью, чтобы она
успевала поглощаться. Затем остаток метанола отгоняют и получают
светло-желтый раствор М. в ДМФА.
Хотя хелатная магниевая соль (1) £Щ«-формы нитроуксусной кис-
лоты и не идентифицирована, ее образование в растворе предпола-
гается на основании поразительного влияния магниевых ионов на
скорость декарбоксилирования нитроуксусной кислоты. Стилз
О- о-
CH3N+=0 CHa = N+— ОН НС.----СОО —ЕЦ. СН2СО,Н
II I I
+ N Mg NO2
-о/\о/
(1)
и Финкбайнер [2] показали, что при обработке нитрометана 4 мол.
же 2 М раствора М. в ДМФА при 50° в течение 4—5 час происходит
количественное превращение его в нитроацетат магния (1), что было
доказано спектрофотометрически. Гидролиз смеси льдом и соляной
кислотой дает чистую нитроуксусную кислоту с выходом 63%. Это
указывает на обратимость реакции декарбоксилирования нитроук-
сусной кислоты, равновесие которой полностью смещается хелати-
зацией. Следовательно, хелатизация является движущей силой в но-
вом способе получения а-нитрокислот и, следовательно, а-аминокис-
лот путем последующего каталитического гидрирования (Pd — С).
Нитроэтан, 1-нитропропаи и 1-нитробутап превращают этим спо-
собом (выходы указаны в скобках) в н,ъ-аланин (46%), э.ь-а-ами-
номасляиую кислоту (34%) и н.ь-норвалин (42%). 2-Нитропропан,
не образующий хелата типа (1), в реакцию не вступает. а-Карбо-
ксилированием М. восьми других нитроалкаиов и последующей эте-
рификацией получены эфиры а-нитрокислот со средним выходом
45% [41.
Стилз [51 использовал реагент для а-карбоксилирования кето-
нов с образованием |3-кетокислот (3). Промежуточно образующуюся
304
магниевую соль (2) можно проалкилировать in situ и затем декарбо-
ксилировать в (4), осуществляя в результате а-ал кил ирование. Аце-
тофенон при нагревании с М. (3—4 моля) при 110—120° в течение
1 час дает бензоил уксусную кислоту с выходом 68%.
О , Mg о
II м О О Tj_f. [1
RCCH2R' —-1^ I I----> RCCHCCbH
R —.CO |
Acz R'
I
R'
(2) (3)
О
R" X • H+ II
- RCCHR"
-CO2 [
R'
Пельтье [6] применил новую реакцию к 5-метокситетралону-2
(5) и установил, что единственным образующимся при этом продук-
том является 3-карбоновая кислота (6). По-видимому, пространствен-
ное строение хелатной магниевой соли благоприятствует смещению
равновесия в положение 3 по сравнению с более экранированным по-
ложением 1.
305
Финкбайнер [7] распространил «-карбоксилирование и после-
дующее алкилирование — декарбоксилирование на 3-фенилгидан-
топн (7). Реакция с М. в последующая этерификация (СН3ОН —
НС1) дает 3-фенил-5-карбметоксигидантоин (8) с выходом 72%. Это
о
HNi ?N‘
н2с5—4-е.
M.
HC1-CH3OH
72%
HN N
RX
О
А Д%Н5
HN XN
(7)
—С*О
СО2СН3
(8)
-со2
(?)
н?_с<о
R
RCHCO2H
nh2
c6h5nh2 +
со2
(10)
соединение алкилируют бензилхлоридом в присутствии избытка ме-
тилата натрия в метаноле и после гидролиза водной соляной кисло-
той и декарбоксилирования получают 3-фенил-5-бензилгидантоин.
В синтезе рацематов природных аминокислот по Финкбайнеру 3-фе-
пилгидантоин (7) обрабатываютМ. для получения хелатной магние-
вой соли, добавляют соответствующий алкилирующий агент, смесь
выливают в лед с соляной кислотой и получают З-фенил-5-алкил-
гидантоин. Гидролиз до свободной аминокислоты осуществляют по
Годри [8] нагреванием с гидроокисью бария под давлением при
160° в течение 15—30 мин‘, ионы бария осаждают карбонатом ам-
мония, смесь упаривают досуха и остаток нагревают на кипящей
водяной бане для удаления избытка карбоната аммония. В синтезах
D.L-валина, лейцина, триптофана, пролина и лизина средние выходы
З-фенил-5-а л кил гидантоинов 53%, а при гидролизе 45%. В син-
тезе о,L-фенилаланина выходы на этих стадиях составляли 98
и 97%. Еще один пример «-карбоксилирования этим методом
опубликован Гриффином и др. [9].
l.Szarvasy Е., Вег., 30, 1836 (1897).
2. Stiles М., F i п k b е i n е г II. L., J. Am. Chem. Soc., 81, 505 (1959).
3. F i п к b е i ii с г H, L., Stiles M., J. Org. Chem., 85, 616 (1963).
4. Fin к bei'ner H. L., Wagner G. W., J. Am. Chem. Soc., 28, 215 (1963).
5. S t f 1 e s M., J. Am. Chem. Soc., 81, 2598 (1959).
6. P e 1 1 e t i e r S. W., Parthasarathy P. C., Tetrahedron Letters, 103
(1964); Pelletier S. W., C h a p pe 1 1 IE L., P a r t h a s a r a 111 у P. C.,
Lewi и N., J. Org. Chem., 31, 1747 (1966).
7. Fin kbeiner H. L., J. Am. Chem. Soc., 86, 961 (1964).
306
8. G a u d г у R., Can. J. Res., 26B, 387 (1948). M.
9. G r i f f 1 n R. W., Jr., Gass J, D,, В cr w i с к h.
J. Org. Chem., 29, 2109 (1964).
МЕТОКСИМЕТИЛЕНТРИФЕНИЛФОСФОРАН, (C(iH5)3P=
“СНОСНз (эфирный раствор).
Этот реактив Виттита получают [П взаимодействием трифенил-
фосфина с хлорметилметиловым эфиром с образованием метоксиме-
тилтрифенилфосфоиийхлорида (1) и перемешиванием суспензии
тонкоизмельченной соли в эфире в атмосфере азота с постепенным
добавлением 1 же фенил лития в эфире fl L Левин [1] использовал об-
разующийся ярко-красный раствор М. (2) в двукратном избытке для
реакции с тигогеноном (3) и получил метоксиметилен производное
(4), гидролизом которого при кратковременной обработке эфиром,
насыщенным 72%-ной НС1О4, с почти количественным выходом об-
разуется смесь 3-эпимерных альдегидов (5). Реакцией 100%-ного
избытка М. с циклогексаноном и ацетофеноном ожидаемые альдегиды
получаются лишь с 40%-ными выходами.
Виттиг [2] позже сообщил о реакции этого нлида с некоторыми
альдегидами, кетонами и циклическими кетонами. Он показал, что
виниловые эфиры образуются с высокими выходами при использо-
вании реагента (8), получаемого, подобно М., из п-толнлхлорметило-
в )го эфира (6).
(С1-)
(СГ1Н-)3Р -[-С1СН2О _Ч-СН3_..> (CfiH5)3P—
(Ы (7)
-СН.О (C(iH5)3Р CI-1O СНз
(8)
307
1. L e v i n e S. G., J. Am. Chem. Soc., 80, 6150 (1958).
2. Wittig G., Schlosser M., Chem. Ber., 94, 1373 (1961); Wittig G.,
Boll W., Kruck К.-H., Chem. Ber., 95, 2514 (1962).
2,5 - бис- (п- МЕТОКСИФЕНИЛ)-3,4-ДИФЕНИЛЦИКЛОПЕН-
ТАДИЕНОН. Мол. вес 390,46, т. пл. 196°.
79%
М. получают по Дильтею [1]. М, эффективнее тетр афенил цикл о-
пентадиенона [2] для связывания дегидробензола при распаде
внутренней соли дифенилиодонийкарбоновой-2 кислоты. В опытах
с эквимолярными количествами реагентов выходы тетраарилнафта-
линов, выделенных хроматографически, составляют 79 и 57% соот-
ветственно.
1. Coan S. В., Т г и с k е г D. Е., В е с k е г Е., J. Am. Chem. Soc., 77, 60
(1955).
2. Beringer F. M., Huang S. J., J. Org. Chem., 29, 445 (1964).
а-МЕТОКСИЭТИЛИДЕНТРИФЕНИЛФОСФОРАН [1]. Этот реа-
гент Виттита (4) получают реакцией трифенил фосфина (1) са-хлор-
этилметиловым эфиром (2) в бензоле, суспендированием белой твер-
дой соли (3) в диметоксиэтане в атмосфере азота при —40° и пере-
мешиванием с mpe/n-б утил атом калия в течение 5 мин. При этом
получают красный раствор нестабильного высокореакционноспо-
собного илида (4), который применяют для синтеза метилкетонов
из альдегидов и кетонов. Например, при добавлении бензальдегида
к илиду (4) образуется эфир енола (5), превращаемый кислотным
гидролизом в метилкетон (6).
СН3
(СеН5)3Р Д-Cl СНОСИ 3
(1) (2)
СН3
—> (С6Н5)3Р=СОСН3
(4)
I d KOC(CH3)3
ЛЩ (с,н,),р-сносн,(С1-)
88% -40°;
(3)
СН3 СН3
—£н±2 с6н5сн=сосн3 —сен5сн2с=о
88%
(5) (6)
1. Coulson D. R,, Tetrahedron Letters, 3323 (1964).
308
МОЛЕКУЛЯРНЫЕ СИТА [11. Известно, что природные цеолиты
(алюмосиликаты) имеют трехмерную систему полостей, соединенных
каналами, в которых обычно содержится гидратная вода. Уникаль-
ным свойством кристалла цеолита является его способность терять
гидратную воду при нагревании без усадки кристалла, который ос-
тается неизмененным и прочным с сетью свободных полостей или
пор. Обезвоженный природный цеолит обладал бы способностью
адсорбировать газы илн жидкости, если бы имел одинаковые размеры
Рис. М-1.
пор. В 1954 г. были получены синтетические цеолиты с исключитель-
но регулярной кристаллической структурой и одинаковыми раз-
мерами пор, что имеет большое значение для процессов адсорбции
[2]. Синтетический цеолит определенного типа сорбирует лишь те
молекулы, объем которых строго соответствует размерам пор, и по-
этому может быть использован для высокоселективного разделения;
поглошая малые молекулы и не вмещая молекулы большие, чем его
поры, он действует как М. с. На рис. М-1 показана модель кристалла
цеолита н цилиндрические гранулы диаметром 0,15 см. Кристалл
цеолита прочен и не изменяет своей структуры прн нагревании до
320° прн удалении адсорбированного вещества и регенерации М. с.
Свойства. Наиболее широко используют М. с. марок ЗА, 4А, 5А
и 13Х в виде порошков, гранул диаметром 0,15 и 0,30 см, а также
в виде шариков трех размеров (сита марки 4А).
Марка
Формула
Диа-
метр
пор, А
ЗА KgNag j (AlOgJjn (SiO2)12l-27 H2O 3
4А Na13[(A102h3(Si02)la].2/ HSO 4
5А Ca4i5Na3[(A103h2] * 30 H2O 5
13Х NagylfAlOsJgfi (SiO2)10g].xH2O 10
309
М. с. ЗА адсорбируют воду, аммиак и не поглощают этан; они
высушивают полярные жидкости и газы типа метанола, этанола,
пропилена. М. с. 4А адсорбируют этанол, сероводород, СО2, SO2,
СД14, C2Hfi, C3HG и не поглощают пропан и высшие углеводороды;
они С максимальной производительностью высушивают неполярные
жидкости и газы до минимального содержания воды. М. с. 5А ад-
сорбируют w-бутанол, //-бутан и не поглощают изосоединепия и цик-
лы с четырьмя и большим числом атомов углерода. М. с. 13Х, по-
добные природному цеолиту фоязиту, поглощают ди-н-бутиламип
п не адсорбируют три-//-бутиламин. Их используют для осушки
гексаметилтриамидофосфата [(СНа).,N]3PO.
Применение. М. с. марок 4А и 5А, широко применяющиеся для
осушки газов (воздух, водород) и химических реагентов в потоке,
более эффективны, чем любые другие сорбенты. В органической
химии их применяют главным образом для эффективной осушки рас-
творителей. Осушка растворителей простым выдерживанием над по-
рошком пли гранулами требует довольно длительного времени и по-
следующей отгонки или фильтрования; вероятно, более целесооб-
разно использовать перемешивание или аппарат Сокслета, как в опи-
санной ниже методике этерификации. Для повышения эффективно-
сти можно также медленно пропускать растворитель через 50-сан-
тиметровую колонку, заполненную М. с. в виде гранул диаметром
0,15 см. Шрисхейм п др. 13, 41 проводили осушку при отношении
объемов растворителя п М. с. 10 : 1.
Для осушки различных растворителей применяются следующие
М. с.:
ЗА Метанол, см. ниже Этерификация
4Л Ацетон J5], ДМСО |5, 6], пиридин [6], гпгтрометан [5J;
каприловая кислота, ДМФА [7, 8]
5Л ТГФ [9], диоксап [10]
13Х Гексаметилтриамидофосфат [4]
Этерификация 111!. Для получения метилового эфира пеларго-
новой кислоты в колбу, снабженную термометром, экстрактором Сок-
слета, в который помещено 160 г М. с. ЗА в виде гранул диаметром
0,15 см, и мощным холодильником, помещают смесь пеларгоновой
кислоты, метанола и конц. серной кислоты. Экстрактор заполняют
метанолом, пускают охлаждающую воду и кипятят содержимое
колбы на электрической плитке при перемешивании магнитной ме-
шалкой в течение 15 час при температуре 65—67°. После охлаждения
Кипячение 15 час
сщсн.усо.н -ьсНйОН-i- h„so4 _ сн3(снд7со.,сн.;
96%
1 мол;. 4 моля 0,5 моля
добавляют 5,3 г безводного карбоната натрия для нейтрализации
серной кислоты, удаляют метанол перегонкой при атмосферном дав-
310
лении и перегоняют продукт при 45°/0,2 мм. Нагреванием при 320°
в течение 3 час получают свежие М. с. Перед повторным использо-
ванием М, с. их следует тщательно высушить на воздухе для предот-
вращения возможного загорания или взрыва и нагревать при 320°
в течение 12 час в хорошо вентилируемой печи.
Енамнны. Шмушкович [12] установил, что енамины, с трудом
получаемые обычными методами, можно получить с использованием
М. с. 4А в виде гранул диаметром 0,15 см.
1. Bulletin: «Union Carbide Molecular Sieves for Selective Absorption». The
British Drug Houses Ltd.; В reck D. W., «Crystalline Molecular Sieves»,
J. Chem. Ed., 41, 678 (1964).
2. Breck D. W. ct al., J. Am. Chem. Soc., 78, 5963, 5972 (1956).
3. Hofmann J. E., W a 1 1 a c e T. J., Schr i eshei m A., J. Am. Chem.
Soc., 86, 1561 (1964).
4. Wallace T. J., P о b i n с r 14., Scliriesheim A., J. Org. Chem.,
30, 3768 (1965).
5. S in i t h S. G., Fa i nb erg A. H., Winstein S., J. Am. Chem. Soc.,
83, 618 (1961).
6. Khorana 11. G- cl al,, J. Am. Chem. Soc., 87, 350 (1965).
7. Thomas A. B., Roclio w E. G., J. Am. Chem. Soc., 79, 1843 (1957).
8. P c t t i t G. R., Ka 1 vins M. V., L i u T. M., T h о m a s E. G-, P a-
rent K-, J. Org. Chem., 26, 2563 (1961).
9. Cain M. E., .1. Chem. Soc., 1964, 3532.
10. Burst ein S. 14., R ingol d 14. ,1., J. Am. Chem. Soc., 86, 4952 (1964).
II. Harrison H. R., Haynes W. M., Arthur P., E is en br a u n
E. J. (Oklahoma State Univ,), procedure submitted to Org. Syn.
12. Szrauszkovicz J., Adv. Org. Chem., 4, 11 (1963).
H H
МОНОНАДМАЛЕИ НОВАЯ КИСЛОТА, H02C(LcC03H.
Мол. вес 132,07.
Для приготовления раствора реагента 39,2 г (0,4 моля) свежеиз-
мельчеиного малеинового ангидрида добавляют при охлаждении
льдом к 11,6 г 90 %-ной перекиси водорода в 150 мл хлористого ме-
тилена и смесь перемешивают до полного растворения, осадка [11.
Реакционная способность ЛА. к. несколько ниже реакционной спо-
собности трифтор надуксусной кислоты и намного выше реакцион-
ной способности обычных надкислот. При комнатной температуре
титр раствора падает приблизительно на 5% в течение 6 час. Реакцию
проводят добавлением окисляемого вещества к раствору реагента
но2ссн=сн.со3н (0°)^
8.0%
СН3(СН2)5СН=СН2 НО2ССН-СНСО3Н(0°)* Сн3(сНг)э-СН“СН2
80%
311
в хлористом метилене с последующим фильтрованием выделяющейся
малеиновой кислоты.
М. к. можно использовать в синтезе Байера — Виллигера, как
это показано на примере циклооктанона. Терминальные олефины
эпоксидируются с хорошим выходом под действием реагента при
пониженной температуре: октен-1 дает эпоксид с 80%-ным выходом
при 0°, но при 25° выход составляет только 40%. В случае более
реакционноспособных внутренних олефинов эпоксидное кольцо
расщепляется даже при 0° с образованием мономалеата диола. Аро-
матические амины с электроотрицательными заместителями окис-
ляются до нитросоединений.
NHa
Br^%Br
90%
НО,ССН = СЫСО3Н
1. W h i t е R. W., Emmons W. D., Tetrahedron, 17, 31 (1962).
МОНОНАДФТАЛЕВАЯ КИСЛОТА, СсНв(СО2Н)СО3Н. Мол. вес
182,13.
Получение. В методе, описанном в работе [1], используется рас-
пыленный фталевый ангидрид, просеянный через сито 14 меш. К рас-
твору 0,6 моля карбоната натрия в 250 мл воды при перемешивании
и охлаждении льдом с солью до 0° добавляют 0,6 моля 30% -ной пере-
киси водорода. Затем при температуре от —5 до —0° смесь обраба-
О
А/С°°Н
НгОз-Иа/Юд Н+ | |1
78-86% ' * 1 Я
I!
О
тывают 0,5 моля распыленного фталевого ангидрида и энергично
перемешивают при этой температуре в течение 30 мин. Полученный
раствор или суспензию встряхивают с 350 мл эфира и осторожно
подкисляют охлажденным льдом раствором 30 мл конц. серной кис-
лоты в 150 мл воды. Надкислота переходит в эфир; водный слой до-
полнительно дважды экстрагируют эфиром. Объединенные экстракты
промывают двумя порциями (по 200 мл) 40 %-кого раствора сульфата
аммония и сушат в холодильнике над безводным сульфатом магния.
Порцию раствора (2 мл) обрабатывают 30 мл 20%-ного раствора
йодистого калия, и через 10 мин выделившийся иод оттитровывают
0,1 н. раствором тиосульфата; выход 78—86%,
312
Поскольку распыление фталевого ангидрида усложняет мето-
дику, авторы книги рекомендуют кристаллизацию, которая позво-
ляет подготовить этот продукт к реакции и удалить из него фталевую
кислоту, обычно образующуюся при открывании бутыли. Ангид-
рид кипятят с произвольным количеством бензола, горячий раствор
отфильтровывают от нерастворившейся фталевой кислоты и разбав-
ляют лигроином. Усовершенствование ранее опубликованного ме-
тода [2] состоит в следующем. Смесь 66 г перекристаллизованного
ангидрида, 92 г пербората натрия и 280 мл воды перемешивают в те-
чение 2 час при 0°, встряхивают с эфиром и подкисляют 120 мл
30 %-ной серной кислоты, охлажденной ледяной водой. Экстракцию
проводят, как описано выше.
Применение. Реагент находит то же применение, что и надбен-
зойная кислота,— в основном для превращения олефинов в эпок-
сиды. Однако по сравнению с надбензойной кислотой в хлороформе
М. к. в эфире удобнее, так как реакцию можно контролировать по
количеству осадка образующейся фталевой кислоты или путем филь-
трования раствора и наблюдения, не осаждается ли дополнительное
количество фталевой кислоты. Следует также заметить, что при стоя-
нии в холодильнике в закрытой колбе реагент разлагается.
Метиловый эфир р-нафтола окисляется с расщеплением кольца
под действием надбензойной кислоты в бензоле и надуксусной кис-
лоты в уксусной кислоте при комнатной температуре, тогда как под
действием М. к. в эфире он изменяется незначительно [31.
1. Payne G. В., Org. Syn., 42, 77 (1962).
2. S t ahm ann M. A., Bergm ann M., J. Org. Chem., 11, 589 (1946),
3. Fernholz H., Chem. Ber., 84, 110 (1951).
МОНОНАДЯНТАРНАЯ КИСЛОТА (3). Мол. вес 134,09, т. пл.
107°.
При перемешивании 50 г тонкоизмельченного янтарного ангид-
рида с 85 мл 30 %-ной перекиси водорода при охлаждении до 25°
получают водонерастворимую ди ацил перекись (2), которую отде-
ляют и промывают водой [1]. Продукт, плавящийся при 132—133°
с разложением, при комнатной температуре устойчив более 6 мес.
СН3СО СН2СО2Н СН2СО2Н
2 о снг- о о-ссщ 2 |НгС0,Н
/ 85% II II СН2СО3Н
сн3со
(1)
Циклогексен
85%
!—н
он
И)
313
При перемешивании 50 г (2) со 150 мл воды при 0° в течение I час
твердый осадок растворяется с образованием раствора М. к. (3),
около 5% которой гидролизуется при этом до янтарной кислоты
и перекиси водорода. Водный раствор (3) можно использовать непо-
средственно для /?ф«нс-гидроксилирования олефина. Например,
к раствору, полученному из 50 а (2), добавляют 0,175 моля цикло-
гексена и поверхностноактивный агент (цемульсол) и смесь встряхи-
вают на трясучке около 70 мин. Смесь нейтрализуют карбонатом
натрия, добавляют насыщенный раствор хлористого натрия, транс-
циклогександиол-1,2 экстрагируют смесью эфир — этил ацетат. Кри-
сталлизацией из этилацетата получают чистый продукт (т. пл. 104=).
1. L о in b а г d R-, Schroeder G., Bull. soc. Chim. Prance, 1963, 2800.
МОРФОЛИН, O(CH2CH,).2NH. Мол. вес 87,12, т. кип. 128°.
Реагент получают в промышленности дегидратацией диэтанол-
амина.
Реакция Вильгеродта. Швейк [II использовал М. в усовершенст-
вованной реакции Вильгеродта, не требующей применения запаян-
ных ампул или автоклава. гМетнлкетон при кипячении с серой и М.
превращается в тиоамид, гидролизующийся до арплуксусиой кис-
лоты.
S.кипячение
----------->
73%
СОД-1
Енамины. Подобно другим вторичным аминам, М. реагирует
с альдегидом, имеющим а-водородный атом, с образованием енамина;
полученный продукт можно восстановить муравьиной кислотой [2].
- СНСНО [-Н —Х'О -> —С^СН--Мф 70
-СИСЩ-Уц7 7 О
\--.
I-Морфолинциклогексен-1 получают кипячением смеси 1,5 моля
циклогексанона, 1,8 моля М., 1,5 а ц-толуолсульфокислоты и 300 мл
толуола в колбе, снабженной обратным холодильником и водоотде-
лителем [31. Отделение воды начинается сразу же и заканчивается
через 4—5 час. Большую часть толуола удаляют при атмосферном
давлении и енамин перегоняют при 118 — 120710 мм. Хороший ме-
тод получения а-моно ал кил циклогексанонов состоит в алкилирова-
нии енаминов циклогексанона под действием ал кил галогенида
314
[4—6] или электрофильного олефина [4, 5] с последующим гидроли-
зом. Ацилирование енаминов циклопентанона и циклогексанона яв-
ляется первой стадиен общего метода удлинения цепи карбоновых
О
72—80%
Кипячение,
толуол (TsOH)
кислот на 5—6 атомов углерода и дикарбоновых кислот на 10 —
12 атомов углерода 1.7].
N-Нитроморфолин [81. Нитрат ацетонциангидрина (2) получают
перемешиванием уксусного ангидрида при охлаждении на ледяной
бане до 3—5° и добавлением по каплям дымящей азотной кислоты
с последующим добавлением по каплям в течение 45 мин при 5—Ж
ацетонциангидрина. Смесь охлаждают льдом и экстракцией хлори-
стым метиленом выделяют нитрат ацетонциангидрина (т. кип. 62—
657Ю мм\ умеренно взрывчат). При медленном нагревании смеси
0,4 моля М. и 0,2 моля нитрата ацетонциангидрина до 110° обра-
зуется N-нитроморфолин (3), ацетон и цианистый водород. Эти реа-
генты при взаимодействии со вторым молем морфолина образуют
a-N-морфолиноизобутиронитрил (4).
(СН ;,С0)Ю-HNO3
57—61
ONO,
(CM3)3CCN
i\r — NO.,
(4)
ОН
(CH3).2CCN
(1)
(3)
Этот побочный продукт основен и остается в кислотном слое при
охлаждении и выливании смеси в 10%-ную соляную кислоту и экст-
ракции ее хлористым метиленом. N-Ннтроморфолин выделяют из
абсолютного этанола в виде белых кристаллов с т. пл. 57—64°.
Синтез иллюстрирует общий метод получения первичных и вторич-
ных нитраминов.
Альдегиды из гел£-дигалогенидов [9]. М. используют в двух-
стадийном гидролизе аещ-дигалогенидов до альдегидов. Дигалогенид
И.О
RCHX2 + 4HN(CHaCH2)2O RCH[N(CH.2CH2)3O]2 RCHO
315
нагревают в течение 2—3 час на кипящей водяной баие с избытком
М. до полного прекращения осаждения хлоргидрата морфолина.
Смесь добавляют при энергичном перемешивании к соляной кислоте
со льдом и водой. Выходы 60—90%.
гелг-Дитиолы получают с хорошим выходом, пропуская серо-
водород в раствор кетона и М. в этаноле, ДМФА или ДМСО при
комнатной температуре [10]. При этом, несомненно, вначале обра-
зуется соответствующий енамин, так как морфолиновые енамины
реагируют с сероводородом в ДМФА в мягких условиях с образова-
нием асж-дитиолов [11].
)c=o+H2s
RZ
м.
R ^SH
r/ ^sh
1. Schwenk E., В 1 о c h E., J. Am. Chem. Soc., 64, 3051 (1942).
2. DeBenneville P. L., Macartney J. FL, Am. Chem. Soc., 72,
3073 (1950).
3. X ю н и г С., Л ю к к e Э., Бреннингер У., «Синтезы органических
препаратов», изд-во «Мир», М., 1964, сб. 12, стр. 96.
4. S t о г k G., Т е г г е 1 1 R., Szmuszkovicz J., J. Am. Chem. Soc.,
76, 2029 (1954).
5. Stork G., В r i zzo 1 ar a A., Landes man H., Szmuszkovicz
J., Terrell R., J. Am. Chem. Soc., 85, 207 (1963).
6. Locke D. M., Pelletier S. W., J. Am. Chem. Soc., 80, 2588 (1958).
7. H tin i g S. et al., Chem. Ber., 91, 129 (1958); 92, 652 (1959); 93 909, 913
(1960).
8. Freeman J. P., Shepard I. G., Org. Syn., 43, 83 (1963).
9. Kerf an to M., Compt. rend., 252, 3457 (1961); 254, 493 (1962); Angew. Chem.,
Intcrnat. Ed., 1, 459 (1962).
10. Jentzsch J., Fabian J., Mayer R., Chem. Ber., 95, 1764, (1962);
Mayer R., Hiller G., N i tzsch ke M., Jentzsch J., Angew.
Chem., Intcrnat. Ed., 2, 370 (1963).
11. Djerassi C., Tursch B., J. Org. Chem., 27, 1041 (1962).
МОЧЕВИНА, H2NCONH2. Мол. вес 60,06, т. пл. 133°, 1 г М.
растворим в 1.щгводы, 10 мл 95%-ного этанола, 1 мл кипящего 95%-
ного этанола, 20 мл абсолютного этанола, 6 мл метанола; почти не-
растворима в эфире или хлороформе, растворима в конц. НС1.
Соединения включения мочевины. Нормальные алканы, имеющие
7 или более атомов углерода, образуют соединения включения, где
молекулы М., связанные водородными связями, образуют спи-
ральную решетку, полость которой заполняется неразветвленными
молекулами углеводорода. Молекулы гостя не связаны химическими
связями с молекулами хозяина, а просто заполняют каналы. Откры-
тые в 1940 г. Бенгеном [1] подобные комплексы углеводородов с М.
изучали Шленк [2], Шисслер, Флиттер [3] и Смит (рентгеноструктур-
ный анализ) [4]. При этом были получены следующие основные
данные.
С удлинением цепи молекул углеводорода необходимо больше
молекул М. для построения каналов, но величины, приведенные
в табл. 1, показывают, что соотношение молекул хозяина и гостя
316
Таблица 1
Число молекул мочевины на молекулу я-алкана
н-Алкан Соотношение «-Алкан Соотношение
Се Комплекс не обра- ед 8,7
зуется
С7 6,1 С12 9,7
С8 7,0 Сц5 12,3
Q 7,3 с 24 18,0
Qo 8,3 ^28 21,2
не стехиометрично. Для получения комплекса жидкий углеводород
добавляют к насыщенному раствору М. при комнатной температуре;
образующийся аддукт выделяется в виде объемистого кристалличе-
ского осадка, обычно сразу достаточно чистого. Для выделения угле-
водорода комплекс обрабатывают водой, чтобы растворить М., и уг-
леводород экстрагируют эфиром. Каналы, в которых умещаются
нормальные алканы, не соответствуют по размерам более объеми-
стым молекулам углеводородов с разветвленной цепью, например
2,2,4-триметилпентану (изооктану), который не образует комплекса.
Введение метильной группы у центрального атома нормального
углеводорода Ci3 также препятствует образованию комплекса.
Строение комплекса w-декана с М. можно продемонстрировать
с помощью молекулярной модели н-дек ан а [5] и листа прозрачного
целлофана (рис. М-2). Целлофановый цилиндр плотно охватывает
модель и определяет объем комплекса, однако фактически вандер-
ваальсовы силы препятствуют контакту и удерживают атомы на не-
котором удалении друг от друга. Диаметр цилиндра соответствует
14,3x0,2=2,86 А, но рентгеноструктурные измерения свидетель-
ствуют, что истинный диаметр равен 5,30 А. Пол у разность, равная
1,22 А, соответствует истинному расстоянию до стенок канала. Эта
оценка применима только к комплексам нормальных алканов и, ве-
роятно, является верхним пределом. Так, нонин-3 образует комплекс
с М., хотя модель не входит в цилиндр диаметром 14,3 см.
СН3СН2С=ССН2СНаСНгСН.2СН3 СН3С=еС—С=С—С=ССН3
Нонин-3 Октатриип-2, 4, G
Она входит в цилиндр диаметром 16,3 см, что соответствует диаметру
канала 3,24 А; в этом случае расстояние до стенок составляет
только 1,03 А. Таким образом, «-декан занимает большее простран-
ство в канале, чем это ему действительно необходимо. С другой
стороны, молекула гостя должна достаточно плотно заполнять вну-
треннюю полость каналов, чтобы образовать устойчивый комплекс.
317
Например, модель октатриина-2,4,6 оказывается очень тонкой вслед-
ствие наличия трех линейных ацетиленовых групп, п углеводород
не образует комплекса с М. Модель 7-метилтридекана ие входит
Р и с. М-2. Модель комплекса /-(-декана с мочевиной (диаметр цилиндра Ь'1,.3 см,
длина его окружности 45 см).
даже в цилиндр диаметром 16,2 см; это соответствует данным о том,
что 7-метнлтридекан ие образует комплекса с М.
СЬ13СН.СН2СН2С1-1оСН2СНСНоСН2СН,СН2СН2СН3
I
сн3
I 7 13
Сверн и сотр. [6] изучили комплексы высших жирных кислот
с М. и показали, что каждый комплекс имеет характерную темпера-
туру диссоциации, при которой прозрачные кристаллы комплекса
становятся непрозрачными. Из данных, приведенных в табл. 2,
ясно, что эти константы можно использовать для идентификации.
В данном ряду температура диссоциации повышается с увеличе-
нием молекулярного веса и приближается к температуре плавления
М. В табл. 2 также приведено соотношение мочевины и жирной кис-
лоты; этот параметр изменяется почти так же, как и в случае н-ал-
канов. Наиболее часто применяют следующий метод получения:
растворяют 1 ч. жирной кислоты и 5 ч. М. в 20 ч. метанола, при не-
обходимости нагревают, оставляют для самопроизвольной кристал-
лизации и получают чистый комплекс. Твердую кислоту можно
растворить в изопропаноле и раствор разбавить метанолом.
318
Таблица 2
Комплексы жирных кислот с М.
Ч nc.no атомои С Кислота От ношеf[ но Темпера- '1УР11 ДД(С- соцасции, °C
мочеин н молярное с 'кислоту
весовое
8 Каприловая 6,7 2,8 73
9 Пеларгоновая 7,4 2,8 80,5
10 Каприновая 8,0 2,8 85
12 Лауриновая 9,7 2,9 92,5
13 Тридекановая 11,8 3,3 96
14 Миристиновая 10,6 2,8 103
16 Пальмитиновая 12,0 2,8 114
18 Стеариновая 14,7 3,1 126
18 Олеиновая (цис) 13,6 2,9 110
18 Элаидиновая (транс) 13,6 2,9 116
18 трео-9,10-Диоксистеариповяя 14,7 2,8 107
Св ер п [7] показал, что низкоплавкий изомер образует комплекс
с М. (табл. 2), а высокоплавкая кислота не образует его, подтверж-
дая тем самым наличие эритро- и трео-конфигураций у 9,10-диокси-
стеариновых кислот, плавящихся соответственно при 132 и 95’.
Кислоте с температурой плавления 132“', образующейся при цис-
гидроксилировании олеиновой кислоты перманганатом, условно
(СНг)7СН3 (СНг)7СН3
Й-С КМ пОх или Н — С — ОН
11 тт 1
н-с ОзОл н-с-он * 1
(СН2)7СОгН (СН2)7СОгН
(1) (2) Эритро
ОН
СОгН
ОН
(2а)
приписывают формулу (2), но если цепь развернуть, как это изобра-
жено формулой (2а), то становится ясно, что гидроксильные группы
расположены по разные стороны цепи. Анализ молекулярных
моделей показывает, что эти выступающие группы препятствуют
внедрению цепи в цилиндр М. диаметром 16,2 см. В шрсо-диоле гид-
роксильные группы находятся в скошенной конформации, и моле-
кула может включаться в капал кристаллов мочевины.
319
Шленк и Холман [81 выделили метилолеат 97—98%-ной чистоты
из метиловых эфиров кислот оливкового масла с помощью комплекса
с М. Оливковое масло (10 а) омыляют (КОН в триэтиленгликоле,
5 мин, 160°) и гидролизат (9,5 г) кристаллизуют из ацетона (70 мл)
при—15° для удаления насыщенных кислот. Упаривание фильтрата
дает 5—7 г смеси олеиновой и линолевой кислот, которые растворяют
в 50 мл горячего метанола в присутствии 11 г М. При охлаждении
комплекс М. с олеиновой кислотой выпадает в виде длинных тяже-
лых кристаллов (10—12 г). Ранее [91 разделение объясняли тем,
что олеиновая кислота имеет две неразветвленные углеводородные
цепи, достаточно длинные для комплексообразования, тогда как
линолевая кислота имеет только одну цепь. Данные Сверна опровер-
гают этот аргумент, так как эрширо-9,10-диоксистеариновая кислота,
которая имеет такую же потенциальную возможность для образо-
вания двухканального комплекса, как и олеиновая кислота, не дает
его. Более того, линолевая кислота все же образует комплекс, правда
с большим трудом. Шленк и Холман [8] с помощью методики (1ч.
кислоты, 5 ч. М., 30 ч. метанола), по существу аналогичной методике
Сверна, получили для некоторых кислот, приведенных в табл. 2,
комплексы, сходные по составу. Обработанная таким же образом
линолевая кислота не дает кристаллических продуктов. Однако
Н>С = С<Н
СН3(СН2)/ >СНа)7СО2Н
Олеиновая кислота
Н. 12 ННЧ 9 ZH
>С = С< )С = С<
СН3(СНа)/ Чсн/ Х(СН2)7СО2Н
Линолевая кислота
комплекс, содержащий 13,1 моля М. на 1 моль кислоты, был полу-
чен в более жестких условиях при нагревании смеси на кипящей
водяной бане (соотношение 1:3: 3,2) и перемешивании. Хотя
нельзя исключать возможность изомеризации цис, цнс-Л9,12-кислоты
в этом процессе, тем не менее рентгеноструктурные данные Нико-
лаидеса и Лэйвса [10] показывают, что укорочение молекулы, обус-
ловленное присутствием двух двойных цнс-связей в комплексе ли-
нолевой кислоты с М., очень близко удвоенному укорочению в ком-
плексе олеиновой кислоты с М. Пересмотр размеров моделей и ци-
линдров в свете ранее неизвестных данных приводит нас к следую-
щим заключениям. Олеиновая кислота не входит в цилиндр диамет-
ром 14,3 см, имеющийся у я-ал капов, но помещается в цилиндр диа-
метром 16,2 см, требующийся для нонина-3. Линолевая кислота не
может войти и в этот больший цилиндр;требуется некоторое расши-
рение канала или сжатие молекулы, или и то и другое в соответствии
с более жесткими условиями получения комплекса.
Линстед и Уэлли [11] показали, что комплексы М. могут исполь-
зоваться для эффективного разделения эфиров карбоновых кислот
320
С разветвленной и перазветвлеппой цепями. Группа Сверна [ 12| с ус-
пехом использовала этот метод при исследовании автоокисления
метилолеата для разделения гидроперекисей с длинной цепью. Эти
перекиси ведут себя подобно соединениям с разветвленной цепью
ввиду объемистости перекисной группы и остаются в растворе, а не-
перекисные компоненты осаждаются в виде комплексов с М.
Переаминирование. При нагревании с М. кислота превращается
в соответствующий амид [13, 14].
Нагрсвги-i не
RCO2H-tH2NCONH? -------------> RCONH2-yCOa-!-NH3
Например, при пол учении нитрила себациновой кислоты и w-циан-
п ел ар тоновой кислоты [151 смесь себациновой кислоты и М. нагре-
вают при 160° при перемешивании в течение 4 час, после чего про-
СООН NHg 4 час, 160'
I I -.....-
(СН2)В+ со
СООН NHo
2,9 МОЛЯ а моля
conh2
I
(СНЛ)3
I
conh2
220—340° CN CN
Перегонка* (5|,)S + (Lh„)8
CN СООН
46—49% 32-34%
дукт реакции состоит почти исключительно из амида себациновой
кислоты, который можно выделить. Затем колбу вынимают из ма-
сляной бани, вытирают, изолируют асбестовым шнуром и асбесто-
вой бумагой и содержимое перемешивают в течение непродолжи-
тельного времени при 220% Мешалку затем заменяют насадкой с
холодильником для перегонки и, по мере того как отгоняются вода,
динитрил, нитрил кислоты и себациновая кислота, постепенно
повышают температуру до 340°. После экстракции эфиром кислот-
ную фракцию экстрагируют 5%-ным карбонатом аммония. Нитрил
себациновой кислоты перегоняют при 12 мм\ со-цианпеларгоновую
кислоту получают в виде твердого вещества (т. пл. 48—49°). Коли-
чество М., необходимое для синтеза, определяется эксперимен-
тально. Если на 1 моль себациновой кислоты расходуется 2 моля
М., то выход нитрила только 27%.
Получение т/>е/и-бутиламииа [16]. mpam-Бутилмочевину, полу-
ченную из mpm-бутанола, М. и конц. серной кислоты, нагре-
вают с фталевым ангидридом до образования трет-бутил фтал имида,
который при кипячении с водноспиртовым раствором гидразина
расщепляется на фтал ил гидр азид и mpem-бутиламин. После охлаж-
дения смесь подкисляют (НС1) и гетероциклический продукт отфиль-
тровывают. Соответствующей обработкой из фильтрата получают
чистый хлоргидрат ш/лта-бутиламина.
(СН3)3СОН+ HqNCONH.j-P конц. H.SO4
2 моля , 1 моль 1,98 моля
20—26°
(CH3)3CNHCONH.
11 Заказ № 1096
321
фталевый ангидрид(200-240°\
72-76%
1) h2nnh2
2) НС1
72-88%
Катализатор. Тетрацианэтилен можно перекристаллизовать из
метилового или этилового спирта. Однако в присутствии М. как
катализатора в результате гладкой реакции образуется ацеталь
дицианкетена [ 17]. Другим примером является получение этилен-
ацеталя дицианкетена [18]. Суспензию тонкоизмельченного тетра-
цианэтилена в растворе М. в этиленгликоле перемешивают при
NC., .CN
NCr ^CN
ИОСИ, '
) 4- H2NCONH2
НОСН2
77-85%
Ч 2HCN
25,6 г
50Д7Л
70—75° до полного растворения (15 мин). При охлаждении из
коричневатого раствора кристаллизуется достаточно чистый про-
дукт.
Действие М. в данном случае не объяснено. Добавление
М. к раствору тетрацианэтилена в спирте вызывает появление
ярко-фиолетовой окраски; по мере выделения цианистого водорода
окраска исчезает и раствор почти обесцвечивается. Пиридин и аце-
тат цинка также катализируют эту реакцию, но М. — наилучший
из известных катализаторов.
Образование хлоргидринов [19]. М. хлорируется в аппарате,
предназначенном для быстрого поглощения газа. На рис. М-3 и
М-4 приведены принципиальные схемы двух приборов, которые
можно использовать для этой цели. В первом приборе (рис. М-3),
описанном Расселом и Вандерверфом [20], стеклянная трубка ме-
шалки аа входит во внутреннюю трубку вращающегося ртутного
затвора в. Нижний конец газоподводящей трубки б погружен
в ртуть на 8 см. При правильной установке шариковых подшипни-
ков а, д, д подвижная часть свободно н плавно вращается с помощью
мотора ж и шкива е. В качестве направляющего подшипника можно
использовать подшипник от старого автомобильного генератора.
Для предотвращения засорения отверстия мешалки она должна
вращаться в направлении, противоположном потоку хлора.
322
На рис. М-4 дана схема прибора с мешалкой, имеющей жидкост-
ной затвор и г аз о подводящую трубку 2 с входящим в нее стеклянным
стержнем; этим стержнем можно прочистить трубку, если она ока-
жется забитой [2]. Муфта мешалки 1 опущена довольно глубоко, так
что сама жидкость служит как бы затвором. Можно также исполь-
зовать простую резиновую муфту. В этом случае муфта мешалки,
подобная /, должна быть опущена
в колбу неглубоко, к ней прикреп-
ляется отрезок резиновой трубки
(2 см), плотно облегающий стер-
жень мешалки. Место контакта
резиновой трубки с осью смазы-
вается глицерином.
Для получения хлоргидрина
смесь М., карбоната кальция и
воды перемешивают при 0—15° и
пропускают в нее с большой ско-
ростью хлор до тех пор, пока привес не составит 95 г (30——40 мин).
К суспензии добавляют воду, смесь фильтруют и фильтрат, содер-
жащий моиохлормочевину, выдерживают на холоду. Осадок на
фильтре экстрагируют водой, фильтраты объединяют, добавляют
лед и для получения хлорноватистой кислоты уксусную кислоту
и затем обрабатывают избытком циклопентена. Содержимое колбы
11*
323
перемешивают и выдерживают в бане со льдом до тех пор, пока не
исчезнет верхний слой и на дне колбы не осядет тяжелое масло
(12—15 час). Водный слой насыщают хлористым натрием и транс-
2-хлорциклопентанол выделяют перегонкой с паром и последующей
перегонкой при 81—82°/15 мм.
H2NCONH2 + СаСО3 + Н2О Clz > H2NCONHC1
0-15°
2, 5 моля 1,25моля 150 мл
Н2О, А с ОН,
НОС1
(2 Моля)
52-56%
Применение при диазотировании. В обычном методе диазоти-
рования аминов нитрит натрия добавляют до тех пор, пока проба
с иодкрахмальной бумажкой не покажет избыток последнего. Перед
использованием раствора в следующей стадии избыток реагента
можно разложить добавлением М. [22, 23].
Стабилизатор. Вильсон [241 рекомендует стабилизировать све-
жеперегнаыный 2-фурфуриловый спирт 0,5—1,0% М.
Гетероциклы. Катализируемая щелочами конденсация М. с
диэтил о кс ал атом дает парабановую кислоту (Мюррей [25]); конден-
сация с диэтилмалонатом дает барбитуровую кислоту (Дикки и Грей
[261).
ос2н3
о-с
1
о=с
ОС2Н5
nh2
с=о
nh2
+ 2 CH3ONa
1 МОЛЬ
20-25“ Г* НС1 „
> I ___кт -------->
кг rSW 71.5-76%
NaO *
0, 5 моля 0, 5 моля
л?\
О ЫН2
" I
СОС2Н5 + £.=о +
I
COC,HS NHz
II 2 5
О 0,5 моля
0,5 МОЛЯ
C2H5ONa
0, 5 моля
Кипячение 7 час
------------>
72-78%
В первой реакции используют раствор натрия в метаноле, во
второй — раствор натрия в абсолютном этаноле. В отличие от Мюр-
рея, бравшего 2 моля ал к оголят а на 1 моль эфира, Дикки и Грей
применяли 1 моль основания. Михаэль впервые осуществил обе
реакции и описал их в одной статье [27]. Михаэль использовал в каж-
324
дом случае Г же алкоголята и получил удовлетворительные резуль-
таты. Так как первая реакция протекает быстрее и при более низкой
температуре, чем вторая, возможно, что избыток алкоголята будет
ускорять обе реакции, но для решения этого вопроса необходимы
дальнейшие эксперименты.
Родственной реакцией является катализируемая алкоголятом
конденсация М. с этиловым эфиром ппануксусной кислоты — пер-
вая стадия синтеза хлоргидрата 5,6-диаминоурацила (Тэйлор [28]).
Поскольку общий выход высокий, выход в решающей стадии кон-
денсации должен быть почти количественным. И опять количество
примененного этилата натрия, по-видимому, вдвое превышало
необходимое, но нет данных, указывающих на нецелесообразность
этого избытка.
zNH.2 CsN
0-0 + I
I сн2
NH3 /
0,86 моля C2H5OC
Я
о
0,86 моля
Т CaH&ONa
1,72 моля
в I л EtOH
Кип, 2
Н(\^zNH.
ОН
НО, X znh2
x/ч./
II I Na^SjO*: НС1
J. 68-81%
\]Я0 (общий)
ОН
он
6-Метилурацил получают из ацетоуксусного эфира и М. в
две стадии 129]. Тонкоизмельченную М. перемешивают со смесью
небольшого количества абсолютного этанола и соляной кислоты
в кристаллизаторе, который помещают в герметичный эксикатор
над конц. серной кислотой для поглощения воды, образующейся
СН3СОСН2СО,С2Н5+ H,NCONH,
1,23 моля 1,33 моля
1 0 капель НС1
25 СД-ЦОН
-НЮ
води. NaOH
,СНСО,Н
CH3-Cf
xnhconh2
HCl
71-77%*
(общий)
/СНСО-^Н, —►
СНз-Сф
XNHCONH3
HN
Н
при конденсации до |3-ураминокротонового эфира. Эксикатор в те-
чение продолжительного времени откачивают водоструйным насо-
сом; ежедневно в него заливают свежую серную кислоту. Когда сы-
рой продукт полностью высохнет (5—7 дней), его перемешивают
с разбавленным раствором едкого натра при 95°. Прозрачный раст-
вор охлаждают до 65° и при перемешивании медленно добавляют
325
кислоту. Пиримидин почти сразу же осаждается в виде бесцветного
порошка высокой чистоты.
Конденсацию бензоина с М. до 4,5-дифенилглиоксалона проводят
кипячением реагентов в уксусной кислоте [301.
Замещение ароматического галогена. Эштои [31] описал улуч-
шенный метод превращения 2,4-динитрохлорбензо л а в 2,4-динитро-
анилин кипячением с водным аммиаком в этаноле в течение 3 час.
Ферри [32] установил, что метод с кратковременным кипячением
с М. в этаноле «гораздо быстрее и дает очень хорошие выходы».
Однако выходы составляли лишь 53 и 57%, и в каждом случае
закристаллизованный продукт плавился примерно на 10° ниже при-
нятой для него температуры плавления.
1. В еп gen М. F., герм. пат. март 18 (1940).
2. Schlenk W., Jr., Ann., 565, 204 (1949); 573, 142 (1951); Fortschr.
chem. Fortsch., 2, 92 (1951).
3. Sell i easier R. W. F I i tier D., J. Am. Chem. Soc., 74, 1720 (1952).
4. Smit h A. E,, J. Chem. Phys., 18, 150 (1950).
5. Fieser L. F., J. Chem. Ed.,'40, 457 (1963); 42, 408 (1965).
6. Knight H. B., W iln auer L.P.,Co leman J. E., N о b 1 e W- R.,
Jr., Swern D., Anal. Chem., 24, 1331 (1952).
7. Swern D., W i t n a u e r L. P., Knight H, B., J. Am. Chem. Soc.,
74, 1655 (1952).
8. S c h 1 e n k H., II о 1 ni a n R. T., J. Am. Chem. Soc., 72, 5001 (1950).
9. Org. Expts., 162.
10. Nicolaides N., Laves F., J. Am. Chem. Soc., 76, 2596 (1954); 80,
5752 (1958).
H. Linstead R. P., Whalley iM,, J. Chem. Soc., 1950, 2987.
12. Golem an J. E., К n i g h t 11. B., Swern D., J. Am. Chem. Soc., 74,
4886 (1952).
13. В i g g s B. S., В i s 11 о p W. S., J. Am. Chem. Soc., 63, 944 (1941).
14. C h e г b u 1 i e z E., L a n d о 1 t F., Helv. Chim. Acta, 29, 1438 (1946).
15. Биггс Б,, Бишоп В., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 208.
16. С м и т Л., Эмерсон О,, «Синтезы органических препаратов», ИЛ, М.»
1953, сб. 4, стр. 105.
17. Middleton W. J., Е п g е 1 h а г d t V. A., J. Am. Chem. Soc., 80,
2788 (1958).
18. Д ii к и п с о п Ч. Л., М е л ь б и Л. Р., «Синтезы органических препара-
тов», ИЛ, М., 1961, сб. 11, стр. 72.
19. Д о и а х о X., Вандервсрф К., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр., 550.
20. R и s s е 1 1 R. R., Vanderwerf С. A., Ind. Eng. Chem., Anal. Ed.,
17, 269 (1945).
21. Fieser L. F., «Experiments in Organic Chemistry», 2nd Ed., 310, Heath
F. C. (1941).
22. Ungnade H, E., О r w о I 1 E. F-, Org. Syn., Coll. Vol., 3, 130 (1955).
23. Кэзлоу К', Сэм мер с Р., «Синтезы органических препаратов», ИЛ,
М., 1956, сб. 5, стр. 46.
24. Вильсон В. К., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 351.
25. Мюррэй Дж., «Синтезы органических препаратов», ИЛ, М., 1959, сб. 9,
стр. 54.
26. Дикки Дж., Грей А., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 79.
27. М i ch ae’l A,’, J. Prakt. Chem., [2], 35, 456, 458 (1887).
326
28. Шерман У., Тэйлор Э., мл., «Синтезы органических препаратов»,
ИЛ, М., 1959, сб. 9, стр. 74.
29. Донливи Дж., К а й з М., «Синтезы органических препаратов», ИЛ, 2М.,
1949, сб. 2, стр, 335.
30. К о р с о н Б,, Фриборн Э., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 232.
31. A s h t о п A. A., J. Chem. Ed., 40, 545 (1963).
32. Ferry N., J. Chem. Ed., 41, 404 (1964).
МУРАВЬИНАЯ КИСЛОТА, HCOOH. Мол. вес 46,03, т. пл. 8,4°,
т. кип. 100,5°, уд. вес. 1,220, рКа 3,77.
Формилироваиие (см. также Уксусномуравьиный ангидрид).
М. к. — активный реагент для ацилирования спиртов и аминов
и в ряде случаев не требует применения катализатора или дополни-
тельного растворителя. При нагревании холевой кислоты с 87%-ной
М. к. (уд. вес 1,2; соотношение 2 мл на 1 г ) при 50—55° в течение
5 час образуется триформильное производное [11. 11а-Гидроксиль-
ную группу 11-эпикортизола формилируют в пиридине при 0°,
добавляя по каплям смесь 1 мл 99%-ной М. к. и 0,4 мл уксусного
ангидрида [2]. d.l-Цистин формилируют прибавлением при пере-
мешивании к смеси 40 г аминокислоты и 600 мл 87%-ной М. к.
50 мл уксусного ангидрида с такой скоростью, чтобы поддерживать
температуру реакционной смеси ~60° [31.
На первой стадии получения о-толилизонитрила [4] раствор
о-толуидина и 98%-ной М. к. в толуоле кипятят с водоотделителем
около 3 час. После отгонки толуола и избытка М. к, остающийся
СН3
нсоон
75—80%
N—С = О
Н Н
РОС1,
63-7 3%
КОС(СНз)
N-о-толилформамид кристаллизуется. При добавлении этого амида
к горячей суспензии mpcm-бутилата калия в трет-бут аноле при
327
перемешивании алкоголят быстро растворяется. После этого раст-
вор охлаждают до 10—20° и обрабатывают хлорокисью фосфора
в течение 30—40 мин. По окончании реакции смесь прибавляют
к холодному раствору бикарбоната натрия, маслообразный о-толил-
изонитрил экстрагируют петролейным эфиром и перегоняют.
Метод получения аллилового спирта, впервые предложенный
Толлеисом (1870 г.) и в дальнейшем улучшенный [51, состоит в на-
гревании глицерина до 260° при последовательном добавлении трех
носнгснонсн2он
|нсо2н
^г°^°
нонгссн i >
А
сн, с
I: :
нон2ссн ,н -
‘••О**
I
н
СН2 Щ
II
> НОСНгСН ХН
о
порций М. к. (полтора дня) и выделении аллилового спирта из ди-
стиллата (выход 46%). Образующийся при этом моноформиат гли-
церина дает при пиролизе непредельный спирт, углекислоту и воду.
Ацидолиз. Один из способов получения безводной акриловой
кислоты заключается в нагревании метилакрилата с 98%-ной М. к.
в присутствии каталитических количеств серной кислоты и гидро-
хинона как ингибитора полимеризации [6].
н+
СН2 -- СНСО2СН3 + НСООН СН2 - СНСО2Н 4- НСООСНз
Восстановление. Прн нагревании трифенилкарбинола с 5 ч.
М. к. некоторое количество вещества растворяется с образованием
желтого раствора галохромной соли. При этом наблюдается мед-
(СвН5)3СОН + НСООН —> (С6Н5)3СН + СО2 + Н2О
ленное выделение углекислоты, и по мере завершения реакции
восстановления до трифенил метана окраска раствора постепенно
исчезает [7].
М. к. применяется как восстановитель при алкилировании ами-
нов формальдегидом (см. Формальдегид, метилирование).
Синтез гетероциклов. Бензимидазол обычно получают нагре-
ванием смеси о-фенилендиамина (0,5 моля) и 90%-ной М. к. (0,75
моля) при 100° в течение 2 час с последующим охлаждением и нейт-
рализацией щелочью [8]. Продукт реакции отфильтровывают и пе-
рекристаллизовывают из воды (т. пл. 170—172°).
н
328
В синтезе 3-амино-1,2,4-триазола смесь бикарбоната аминогуа-
нидина и 98%-ной М. к. осторожно нагревают (вспенивание) до
прекращения выделения углекислоты [9]. Раствор, содержащий
н NH
и ктхт Дкттт и НСО2Н
H2MN cnh2- н2со3 ----------—
(1)
120° НМ------СМН2 120°
Тн2(? ОСН
н
(3)
Н NH
I II
H2NM СМН2‘НСО2Н —>
(2)
N____ МН2
о
N''"
I
Н
(4)
формиат аминогуанидина (2), нагревают 5 час при 120°; при этом
формиат (2) теряет воду и превращается в N-формильное производ-
ное (3), которое подвергается циклодегидратации с образованием
амипотриазола (4). Выход кристаллического продукта с т. пл.
152—153° составляет 68—70%.
Синтез самого 1,2,4-триазола (9) [9] начинают с получения
формильного производного (6) из тиосемикарбазида (5) при кратко-
временном нагревании его с 90%-ной М. к. Соединение (6) цикли-
зуется в 3-меркапто-1,2,4-триазол (8) под действием щелочи, по-
видимому, через таутомерную форму (7). Как это ни удивительно,
Yi-siy?
HN
НОСН ^C-SH
II I
N---МН
NaOH >
72-81%
н2м—мн
(5)
при окислении соединения (8) азотной кислотой в присутствии азо-
тистой кислоты в качестве катализатора происходит элиминирова-
ние меркаптогруппы с образованием 1,2,4-триазола.
Карбоксилирование. Адамантан превращается в 1-адамантан-
карбоновую кислоту при реакции с большим избытком М. к. в че-
тыреххлористом углероде в присутствии mpm-бутанола и конц.
серной кислоты [10]. Побочная реакция — восстановление трет-
бутанола до пзобутана, который частично карбоксилируется в три-
метил уксусную кислоту. Обработка смеси включает кристалл иза-
329
цию аммонийной соли адамантанкарбоновой кислоты, в то время
как соль побочно образующейся кислоты остается в маточном
растворе.
+ нсо2н + (сн3)3сон
0,1 МОЛЯ
H2SO4
2 55 МЛ
17-25°
СС14 ----->
5б-61%(чистый}
100ЛМ
1,2 МОЛЯ 0,4 МОЛЯ
+ (СН3)3СН + (СН3)3ССО2Н
+
Эта методика иллюстрирует общий метод карбоксилирования
насыщенных углеводородов с водородом при третичном углеродном
атоме, например изопентана, 2,2-диметилбутана и метилциклогек-
сана.
Другим примером является синтез 1-метилциклогексанкарбоно-
вой кислоты из метилциклогексанола-2 [10а].
/си® /снз
। । h.so4 । V
+ нсоон — хСОаН +Н,0
х/\эн
Перегруппировка Бекмана [111. Кетоксимы гладко перегруппи-
ровываются при кипячении с М. к. в течение I—6 час [II].
NOH
—-х ]| z-----\ Кипячение 6 час
Н3С-^ С >-СНз 4- нсоон -------------------
х~ — / / У О /о
1г 15 мл
О н
Н3с^ С - N У~СН3
l. Cortese F., Baum an L., J, Am. Chem. Soc., 57, 1393 (1935).
2. R eb er F., L ar don A., R e i c hst e i n T., Helv. Chim. Acta, 37, 45
(1954).
3. du Vignea ud V., Dorfman R., Lor ing H. S., J. Biol. Chem.,
98, 577 (1932).
4. Ugi I., Me yr R., Org, Syn., 41, 101 (1961).
5. К а м м О., Марвел С., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 25.
б. Реберг Ч., «Синтезы органических препаратов», ИЛ, М., 1949, сб. I,
стр. 25.
7. KauHmann Н., Pannwitz Р., Вег., 45,766 (1912); К о v а с h е А.,
Ann. chim., [9], 10, 184 (1918).
330
8. Вагнер Е., Миллетт В., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 85.
9. Ainsworth С., Org. Syn., 40, 99 (1960).
10. К о с h Н., Н a a f W., Org. Syn., 44, 1 (1964).
10а. Н a af W., Org. Syn., 46, 72 (1966).
И. van Е s Т., J. Chem. Soc., 1965, 3881.
МУРАВЬИНАЯ КИСЛОТА — ФОРМАМИД. При кипячении
в течение 1 час сМ. к. — Ф. 3,4-дигидроизохииолины подвергаются
восстановительному формилированию с образованием N-формил-
1, 2, 3, 4-тетрагидроизохинолииов [1] с хорошим выходом. Наличие
гидроксильных групп препятствует реакции; их необходимо блоки-
ровать путем превращения в простые эфирные группы.
1. G а г d е n t J., Bull. Soc. chim, France, 1960, 118; В a x t e г I., A 1 1 a n
L. I.,Swan G. A., J. Chem. Soc., 1965, 3645; Baxter I., S w a n G, A.,
J. Chem. Soc., 1965, 4015.
МУРАВЬИНОЙ КИСЛОТЫ л-НИТРОФЕНИЛОВЫЙ ЭФИР,
n-NO2C6H4OCHO. Мол. вес 167,12, т. пл. 72—74°.
Получение. Реагент получают ацилированием л-нитрофенола
муравьиной кислотой в присутствии дициклогексил карбодиимида
(ДГК) В ТГФ [11.
дгк-тгф
n-NOaCeH4OH + НСООН ---zi-NO2CfiH4OCHO
63%
Формилироваиие. Реагент селективно формилирует концевые
аминогруппы орнитина и лизина [1], тогда как по Шихану [2] под
действием муравьиной кислоты и уксусного ангидрида формили-
руются обе аминогруппы.
О
II ТГФ, 25°. б час
Н2М (СН2)ПСНСО.2Н + HCOCeH4NO.rn------"
nh2
о и
II I я = з или 4
НСО— N(CH2)Z1 СНСО2Н + HOC6H4NO2
NH3
1. О k a w а К-, Hase S., Bull. Chem. Soc. Japan, 36, 754 (1963).
2. Sheehan J.C., Yang D. D., J. Am. Chem. Soc., 80, 1154 (1958).
МУРАВЬИНОЙ КИСЛОТЫ ТРИМЕТИЛАММОНИЕВАЯ СОЛЬ,
5НСО2Н-2N(CH3)3. Мол. вес 348,35, т. кип. 91—-93718 мм. Эту
слабокислую жидкую соль с высокой и постоянной температурой
331
кипения получают, пропуская газообразный триметиламин в охлаж-
денную льдом 80%-ную муравьиную кислоту до щелочной реакции
раствора [1].
Восстановительное действие. М, к. т. с. используют для восста-
новительного расщепления N-ацпламино- и N-сульфамн домети ль-
ных соединений [1]. Можно использовать также и муравьиную
кислоту, но выходы при этом ниже.
65% .
1. Sekiya М., 1 t о К., Chem. Pharm. Bull. (Japan), 12, 677 (1964).
МУРАВЬИНОЙ кислоты ЭТИЛОВЫЙ ЭФИР, НСООС2Н5.
Мол. вес 74,08, т. кип. 54°, уд. вес 0,92.
Технический препарат очищают добавлением 15 г карбоната ка-
лия на 100 г жидкости, после чего оставляют смесь на 1 час, перио-
дически взбалтывая, затем декантируют в сухую перегонную колбу,
прибавляют 5 г пятнокиси фосфора и фракционируют без доступа
влаги.
Синтез Гриньяра. Применение этого реагента для синтеза вто-
ричных спиртов при взаимодействии с реактивами Гриньяра иллю-
стрируется следующей схемой [11:
нсоос..н6 цао
H-C4HyMgBr---------> (я-С4Н9)гСНОМёВг ---7 (н-С4Н9)аСНОН
Формилирование кетонов. Конденсация циклогексанона с
с М. к. э. э. в присутствии этилата или гидрида натрия дает соеди-
нение, которое можно представить как 2-оксиметнленциклогскса-
нои (1) или как его таутомер — 2-формилциклогексанон (2) [2];
для простоты этот процесс называют формилированием. В описан-
ной выше методике после формилирования проводят реакцию с гид-
разином, дающую тетрагидроиндазол (3), дегидрированием которого
получают индазол (4). Формилирование циклических кетонов часто
применяют для блокирования одного из а-положеннй, после чего
можно алкилировать по (//-положению. Блокирующая группировка
удаляется затем щелочным гидролизом. Джонсон [3] усовершенство-
332
вал этот метод, применив О-алкилированне оксиметиленового про*
изводного (6) изопропилиодидом, метилирование производного (7)
с образованием соединения (8) и элиминированиеО-алкильной груп-
пы кислым, а формильной группы щелочным гидролизом до соеди-
нения (10).
Интересным методом синтеза 1,3,5-триацетил бензол а является
формилированне ацетона [4]. К раствору этилата натрия в абсолют-
ном этаноле после разбавления эфиром при кипячении и перемеши-
вании добавляют по каплям в течение 2 час смесь ацетона и М. к.э.э.
В этих условиях формильное производное конденсируется в арома-
тический трнкетон.
СН3СОСН3 + НСООС2Н5 + NaOC,H6 СН3СОС112СНО
СОСН3
I
СН2 СОСНз
! I
О СНО СНО ~зн2О /ч,
СН3С-СН2 СНгС0СН3 30-38% || |
ОСН СН3СО^ХсОСНз
Т.пл. 163°
333
Методика синтеза 3-циан-6-метилпиридона-2 (3) [5] включает
выделение натриевого производного формилацетона (1). Для этого
к суспензии метилата иатрня в эфире при перемешивании и охлаж-
дении льдом добавляют по каплям смесь ацетона и М. к. э. э.,
после чего удаляют эфир при температуре ниже 70°. Твердый оста-
ток обрабатывают водным раствором цианацетамида и ацетата
СН3СОСН3-|-НСООС2НГ1-|-СН3ОУа —> CH3COCH^CHONa+
(1)
(I) ,CHONa+ ZWCN
04 CH3CN cn3io to 1 H,N (2) Ацетат пиперидиния, AcOH |
55 — 62% (общий) /\/Х1 H3C N U H (3)
пиперидиния и раствор кипятят 2 час. После разбавления водой и
подкисления уксусной кислотой образуется объемистый желтый
осадок соединения (3); т. пл. 294° (с разл.).
В синтезе [6] 6,7-диметокси-3,4-ди гидронафтой ной-2 кислоты
(IV) по методу Кроули и Робинсона [7] этиловый эфир у-вератрил-
масляной кислоты (1) форматируют М.к.э.э., добавляя по каплям
раствор М.к.э.э. и (I) в эфире к суспензии этилата натрия в эфире
при —10°. После подкисления формильное производное (II) выде-
ляют экстракцией эфиром и циклизуют в III под действием смеси
90%-ной фосфорной и конц. серной кислот при 0—10°. Омыление
дает кислоту (IV); т. пл. 193°.
СН3ОХ СН2СООС,Н5
I И ।
I II
I
сно
НСООСЛЦ
NaOC2Ha
НаРО4
I II /сн“
СН3о/^/ХСЩ
h2so4
40—13%
(общий)
он-, н +
СНзО^^/Х/
Неочищенный формилгидразин, полученный осторожным при-
бавлением гидразиигидрата к раствору М.к.э.э. в этаноле и кипяче-
334
иием смеси в течение 18 час, конденсируется в 4-амино-1,2,4-триазол
при нагревании до 200 при атмосферном давлении в течение 3 час 18].
О
H,NNH, il
НСООС2Н5 - . Д. HCNHNH.
СН-- О -2Н,о ,.CH^N
H,NNH HOCH=N C5~7I% " XCH = N
N-Цпклогекснлформамид, полученный медленным добавлением
М.к.э.э. к циклогексиламину при перемешивании и охлаждении,
является промежуточным продуктом при превращении этого амина
в циклогексил изонитрил [91.
НСООС, ГЦ РОСЦ — п приди п
C6HhNH., ----—- C6HnNHCHO'-------——-----> C6HnN = C
О / -- t Л /q
Первичные и некоторые вторичные амины можно формилировать
с хорошим выходом при нагревании амина с М.к.э.э. при 100—110°
в автоклаве в течение 1 час 1101. В тех же условиях этилацетат не
реагирует.
1. Колем air Дж., Крэг Д., «Синтезы органических препаратов», ИЛ,
194 9, сб. 2, стр. 193.
2. Эйпсворз Ч., «Синтезы органических препаратов», ИЛ, ЛК, 1961,сб.П,
стр. 27. For application in the steroid series, see also C i i n t о n R. O- et al.,
J. Am. Chem. Soc., 83, 1478 (1961).
3. Johnson W. S,, P о s v i с H., J. Am. Chem. Soc., 69, 1361 (1947).
4. С 1 a i s e n L., S t у 1 о s N., Ber., 21, 1145 (1888); Франк P., Бор-
ланд P., «Синтезы органических препаратов», ИЛ, 1953, сб. 4, стр. 488.
5. Марвелла Р., «Синтезы органических препаратов», ИЛ, М., 1953, сб. 4,
стр. 553.
6. X о л м с X., Т р е в о й Л., «Синтезы органических препаратов», ИЛ, М,,
1953, сб. 4, стр. 197.
7. Crowley G. Р., Robinson R., J. Chem. Soc., 1938, 2001.
8. Аллен Ч., Белл А., «Синтезы органических препаратов», ИЛ, М., 1952,
сб. 3, стр. 56.
9. И g i I., М е у г R,, L i р i n s k i М., Bodesheim F., Rosen d h al
E, Org. Syn., 41, 13 (1961).
10. M о f f a t J., N ewton Al. V., Papenm aier G. J., J. Org. Chem.j
27, 4058 (1962).
НАДБЕНЗОЙНАЯ КИСЛОТА, C(JH5CO3H. Мол. вес 138,12.
Получение. В ранее предложенном методе [II охлажденный
до —5° раствор метилата натрия в метаноле обрабатывают холодным
раствором перекиси бензоила в хлороформе, надбензоат натрия экст-
рагируют ледяной водой, экстракт промывают холодным хлорофор-
CHaONa HjSO,
((ДЩСОЛ, •----------> C6ILCO3Na----CEiH-,CO3H
k - а -С,ИЬСО2СНЯ 1 ° J 82,5—86% ь 13
мом для удаления метилбензоата и выделяющуюся при подкислении
Н. к. экстрагируют хлороформом. Продажную перекись бензоила
необходимо предварительно перекристаллизовать, растворяя в го-
рячем хлороформе и добавляя метанол до начала кристаллизации.
О совершенствовании метода получения Н. к. из 70%-ной пере-
киси водорода и бензойной кислоты в метансульфо кислоте и выделе-
нии экстракцией бензолом (Сверн) см. Перекись водорода. Мойер
и Менли [21, очевидно не зная о методе Сверна, опубликовали
статью «Усовершенствованный синтез надбензойной кислоты».В их
методе, основанном иа работе Вилкаса [3], 8 г перекиси натрия
растворяют при 20° в 135 мл воды, фильтруют и к раствору при пе-
ремешивании магнитной мешалкой добавляют 175 мл денатурата и
раствор 0,5 г MgSO4 7 Н2О в 15 мл воды. Эта соль ингибирует ката-
литическое действие следов ионов металлов и создает возможность
проведения реакции при комнатной температуре с таким же выхо-
дом, как и при охлаждении до — 5°. Температуру раствора доводят
до комнатной и при перемешивании в течение 10—12 мин добавляют
11,6 мл хлористого бензоила. Перекись бензоила отделяют фильтро-
ванием, подкисляют 20%-ной серной кислотой и продукт экстраги-
руют шестью порциями четыреххлористого углерода, хлороформа
или бензола по 75 мл каждая; выход около 75%. При экстракции
в органическую фазу переходит около 25 мл этанола.
Реакция с олефинами. Реакция Н. к. с олефином обычно осу-
ществляется гладко при охлаждении до 0—25° с высоким выходом
эпоксида. Олефин растворяют при комнатной температуре в растворе
Н. к. в хлороформе или бензоле (при необходимости добавляют
другой растворитель) и раствор оставляют при комнатной или пони-
женной температуре па несколько часов. Реакцию можно контроли-
ровать йодометрическим титрованием. Используя избыток реагента,
можно определять его расход и, таким образом, число имеющихся
двойных связей.
336
В классических исследованиях эргостерина и витамина D Вин-
даус описал все известные в настоящее время типы реакций Н. к.
с ненасыщенными соединениями. Наблюдение Виндауса и Лют-
трингхауса, заключающееся в том, что в присутствии избытка Н. к.
эргостерин поглощает точно 3 моля надкислоты, было первым пря-
мым доказательством наличия в нем трех двойных связей. Реакция
стерина с 1 молем Н. к. дает продукт, идентифицированный
впоследствии как 3|3,5«,6а-триол-6-беизоат. транс-22,23-Двойпая
связь значительно менее реакционноспособна. Цисоидная 5,8-дпено-
вая система эргостерина обладает высокой реакцион неспособностью
по отношению к диенофилам типа малеинового ангидрида, а при
фотосенспбилизации присоединяет кислород в положения 1,4 с об-
разованием 5а,8а-трансаннулярной перекиси. Преимущественное
присоединение к 5,6-двойиой связи показывает, что Н. к. не склонна
к 1,4-присоединению. Очевидно, в этой реакции первоначально
образующийся аллильный «-эпоксид легко расщепляется до 5,6-
диол-6-бензоата. Если насыщенные эпоксиды расщепляются до
/пранопроизводных, то аллильный эпоксид может расщепляться
или с обращением, или с сохранением конфигурации в зависимости
от структуры и условий реакции. Сохранение конфигурации 6а
можно, по-видимому, объяснить экваториальной ориентацией
ба-бензоатной группы; наличие объемной группы в 6|3-аксиалъном
положении привело бы к очень сильному взаимодействию с аксиаль-
ной 10|3-метильной группой. Однако поведение лумистернна проти-
воречит этой интерпретации [51. Лумистерин отличается от эрго-
стерина конфигурацией при Ся и С10, а также тем, что его «-область
экранирована ангулярной метильной группой.
Таким образом, Н. к. атакует эргостерин из «-области, тогда как
лумистерин атакуется из ^-области, и первичным продуктом реак-
ции с Н. к. в бензоле является р-окись (2). Эта окись гидролизуется
при непродолжительном нагревании суспензии в воде. Подобным же
образом, не затрагивая ацетильной группы, осуществляется гидро-
лиз 3-ацетата-5,6-[3-эпоксида. Реакция сопровождается обычной
инверсией при С6 даже в том случае, когда аксиальная 6а-оксн-
группа экранирована аксиальной 10а-метильной группой. При
обработке лумистерина Н. к. в хлороформе по обычной методике
337
или при выдерживании бензольного раствора в течение 3 дней обра-
зуется 3|3,5|3,6|3-триол-6-бензоат (4), омыляющийся в триол (5).
6-Эпимеры (3) и (5) окислением сведены к дикетону (6). Аномальная
в этом ряду реакция раскрытия р-окиси (2) бензойной кислотой
с сохранением конфигурации при С6 обусловлена аллильной при-
родой окиси и кислотным характером реагента. 7,8-Двойная связь
способствует появлению частичного положительного заряда, доста-
точного для подхода нуклеофила с последующим цис-раскрытием
эпоксидного цикла.
В ряду стероидов гидрирование четырехзамещенных двойных
связей затруднено или не осуществляется совсем, тем не менее
такие соединения реагируют с Н. к. Однако в этом случае продук-
том реакции оказывается не эпоксид, а аллиловый спирт или соп-
ряженный диен. Первый продукт получен Виндаусом и Люттринг-
хаусом [41, но лучшим примером является описание Виидауса, Лнн-
зерта и Экхардта [6]. Реакцией Ав-холестерилацетата (1) с Н. к.
в хлороформе получают ненасыщенный диол-3-ацетат, описанный
как третичный аллиловый спирт (3), поскольку при обработке
уксусным ангидридом из него образуется диен (4). Поскольку Хен-
бест и Райли [7] показали, что А8-эргостерилацетат с Н. к. в бензоле
дает с хорошим выходом 8а, 9<х-эпоксид, а также с точки зрения до-
казательств, приведенных ниже, образование аллилового спирта
(3) можно рассматривать через промежуточный эпоксид (2), расщеп-
ляющийся до (3) под действием следов кислоты, содержащейся
338
в хлороформе. При уточнении результатов Виитерштейнера и Моора
19] Физером и Гото 18] были получены следующие доказательства.
А7-Холестерилацетат (I) под действием мононадфталевой кислоты
в эфире дает 7а, 8а-эгюксид (II). Последний не изменяется при по-
следующей обработке мононадфталевой кислотой в эфире, но в хло-
роформе с 1 молем Н. к. образует эпоксиспирт IV. Окись II устой-
чива в сухом или влажном хлороформе, содержащем бензойную
кислоту, таким образом, расщепление и дальнейшая реакция не
in
IV
339
могут быть обусловлены действием этой кислоты. В методе получе-
ния Н. к., применявшемся в то вре.мя, раствор натриевой соли под-
кисляли серной кислотой и экстрагировали хлороформом. Встря-
хиванием хлороформа с 10%-ной серной кислотой с последующей
сушкой получали кислый раствор, пригодный для изомеризации
окиси II в аллиловый спирт III, выделяемый в виде диацетата. При
титровании оказалось, что хлороформ содержит 0,2 мг серной кис-
лоты на 100 мл.
Окисление а-дикетонов, о-хинонов и производных фенола (в не-
которых реакциях, приведенных в этом разделе, применяли над-
уксусную и монон адфт ал ев у ю кислоты). Каррер [101, имевший бо-
гатый опыт превращения каротиноидов в эпоксиды, часть которых
встречается в природе, исследовал реакцию ненасыщенного а-дике-
топа (1) с мононадфталевой кислотой в эфире. Кристаллический про-
дукт реакции по составу отвечал моноэпокснду, но пе соответствовал
/ЮзН
О о Пр
СеНГ((СН = СН)2С—С(СН-СН)8С6Н5 ------
(1)
() о
И !|
СвН5(СН - СН)2С- О - С(СН = СН)2С0Н5
(2)
ему по свойствам и оказался ангидридом (2). Таким образом, под
действием надкислоты происходит расщепление связи между карбо-
нильными группами с внедрением атома кислорода. Бозекен [111
охарактеризовал продукт окисления [Кнафтохинона надуксусной
кислотой как о-карбоксиаллокоричную кислоту (5). Каррер и Шней-
дер 112] обрабатывали раствор 3,5 г р-нафтохинона в 70 мл хлоро-
форма раствором 3,4 г (1,1 эки) Н. к. в 50 мл хлороформа при ком-
натной температуре. Раствор вскоре принимал темно-красную ок-
раску, но через 5 дней становился ярко-желтым. Кислоты отделяли
экстракцией раствором бикарбоната и упариванием высушенного
хлороформного раствора с последующей многократной кристалли-
зацией остатка получили 0,85 г чистого ангидрида (4) с т. пл. 115°.
Другие примеры реакции [13—151 приведены на схеме:
340
с6н5с=о
СО2С2Н5
C6HgC=O
°
СО2СгН5
СН3С = О
СН3С = О
0^СО3Н
I
со,н
------->
сн3с=о
/°
СН3С=О
Фернхольц 116] изучил окисление метилового эфира Р-нафтола
под действием Н. к. и представил убедительное доказательство нап-
равления этой интересной реакции превращения в монометиловый
эфир о-карбоксиаллокоричной кислоты (б) с 35%-ным выходом. 6 а
эфира (1) добавляют при охлаждении к 692 мл 2,5%-ного раствора
Н. к. в бензоле и раствор, вскоре окрашивающийся в красный цвет,
оставляют на две недели при 10°. Экстракцией бикарбонатом отде-
ляют кислотную фракцию, из которой выделяют кислый эфир (б), а
из оставшейся небольшой нейтральной фракции удается выделить
2-метокси-1,4-нафтохинон (4) с выходом 2,5%. Предполагаемая
последовательность стадий основной реакции состоит в окислении
до р-нафтохинона (2) с отщеплением метанола, расщеплении до ангид-
рида Каррера (3) и реакции последнего с метанолом, выделяющимся
на первой стадии, с образованием кислого эфира (б). При окислении
раствором надуксусной кислоты, полученной из 300 мл уксусной
кислоты и 30 мл 30%-ной перекиси водорода (14 дней при 25°),
свободная о-карбоксиаллокоричная кислота (соответствующая (6)1
341
получена с 65%-ным выходом. Очевидно, в этом случае с ангидридом
реагирует преимущественно вода, имеющаяся в большом избытке
по отношению к образующемуся метанолу. Окисление 3 а метило-
вого эфира (1) Н. к. в бензоле с добавлением 30 г абсолютного эта-
нола дает 0,9 г кислого этилового эфира дикислоты. Окисление
а-нафтола под действием Н. к. в смеси бензола с метанолом также
дает кислый метиловый эфир (6), который, по-видимому, образуется
через хинои (2) и ангидрид (3).
Интересно, что в условиях указанных реакций окисления под
действием Н. к. и надуксусной кислоты мононадфталевая кислота
реагирует лишь в незначительной степени.
Окисление диарнламинов. Токумару и сотр. [17] обнаружили,
что при добавлении Н. к. в эфире к эфирному раствору 4,4'-димето-
ксидифен ил амина (1) образуется кристаллический осадок стабиль-
ного радикала 4,4'-диметоксидифенилиминоксила (2). Соединения
такого типа ранее рассматривались Вейландом как содержащие
четырехвалептный азот [18].
(rt-CH3OCcH4)2NH+C0Ht,CO3H— (п-СН3ОС6Н4)2\-О
7 0 %
(1) 0,6 г в эфире 0,(56% и эфире (2) Т. пл. 150—151%
1. Брони Г., «Синтезы органических препаратов», ИЛ, 1М., 1949, сб. 1,
стр. 337.
2. Moyer J. R., Manley N. С., J. Org. Chem., 29, 2099 (1964).
3. V i 1 k as M., Bull. soc. Chiin. France, 1959, 1501.
4. W i n dans A., L ii t i r i n g h a u s A,, Ann., 481, 119 (1930).
5. Физер JL, Ф и в e p M., Стероиды, изд-во «Мир», M., 1964.
6. Wind aus A.,L insert O.,Eckhardt H. J., Ann., 534, 22(1938).
7. Henbest II. B., Wrigley T. I.. J. Chem. Soc., 1957, 4596.
8. F i e s e г L, F., Got о T., В h a t t a ch a г у у а В. К., .1. Am. Chem.
Soc,, 82, 1693 (1960).
9. Winters! e iner О., M core M,, J. Am, Chem. Soc., 65, 1507 (1943).
10. Karrcr P,, С о c h a n d Ch., Neuss N,, Helv. Chim. Acta, 29, 1836
(1946).
11. Boeseken J., S 1 о о f I G,, Rec. trav. Chim., 49, 100 (1930).
12, К a r r e r P,, S c h neidcr L., Helv, Chim. Acta, 30, 859 (1947),
13. Karrcr P., S c h w yzer R., Neu w i r t ii H., Helv. Chim. Acta, 31,
1210 (1948),
14. К а г г e r P., H a a b F., Helv. Chim. Acta, 32, 950 (1949).
15, Karrcr P., Hohl Th., Helv. Chim, Acta, 32, 1932 (1949).
16. Fernholz H., Chem. Ber.,84, 110 (1951).
17. T о k и ш а г u K-, Sakur ago H., Simani ига O., Tetrahedron
Leiters 3945 (1964),
18. W 1 с 1 a n d IL, О 11 e n b a c k e r M., Ber,, 43, 2111 (1914).
НАДБЕНЗОЙНОЙ КИСЛОТЫ mpem-БУТИЛОВЫЙ ЭФИР,
о
l)
СВНГ)СООС(СН3)3.
Мол. вес 194,22, т. кип. 75—7670,2 мм\ стабилен при комнатной
температуре; период полураспада при 100° 18 час [1].
342
Синтез /ирет-бутнловых эфиров. Взаимодействие этого реагента
с реактивами Гриньяра является удобным методом синтеза аромати-
ческих и алифатических /ире/п-бутиловых эфиров [2]. Поскольку
трет-бутиловый эфир фонола легко гидролизуется кислотами, эту
Mg
Чн3
С6И5С о
ХО/'ХС(СН3)3
ЧС(СН3)3
реакцию можно рассматривать как способ превращения арилгало-
генидов в фенолы [3,41 (2-бромтиофен —2-оксптиофен 1.31).
Реакция с эфирами. При разложении Н. к. б. э. под действием
хлористой меди в присутствии эфиров (RCFE)2O и спиртов R'OH
образуются ацетали RCH(OR'). [51:
н-С3Н7СН,ОС4Ня-н-рС6Н5С —О«С(СН3)3 —.
II
| /ОСдНд-Л
н-С3Н.С/
X>COCSH5
н-С3Н 7С(ОС6 Н j з - н) 9
С алифатическими и циклическими эфирами (например, ТГФ)
Н. к. б. э. реагирует в присутствии галогенидов меди(1) при 67—
90° с образованием нестабильного а-бензоилоксипроизводного (1)
[51, которое при реакции с ТГФ (4—6 час) выделяют и осторожным
о
С6Н5СОр(СН3)3 (Си\
-(СНЭ)3СОН
Q!CC6H5 -СДЦСОДГ
|] (СН3)3СОН
н+
кос(сн3)3
нагреванием превращают в 2,3-дигидрофураи (выход 66%), Если
исходную реакционную смесь сразу нагревать 14—48 час, то мед-
ленно образующийся ненасыщенный эфир (2) при каталитическом
действии бензойной кислоты присоединяет трет-бутакол и превра-
щается в ацеталь (3) — 2-трет-бутокситетрагидрофуран.
343
Реакция с ненасыщенными соединениями (обзор [6]). В при-
сутствии каталитических количеств соли меди или кобальта
Н.к.б.э. реагирует с олефинами с образованием замещенных аллил-
бензоатов (Караш, Сосновский, Янг [7]), При этом не наблюдается
аллильной перегруппировки; единственный продукт реакции с тер-
минальными олефинами — аллиловый эфир с концевой двойной
связью. Это подтверждается также реакцией с 1 -метилен-4-трет-
бутилциклогексаном [8]. Однако позднее с помощью ГЖХ было
нсн2сн=сн2
о
11
С6Н5СООС(СН3)Э^ RCHCH=CH2 + (СН3)3СОН
CuBr ОСОС6Н5
ОСОС6Н3
найдено, что эта реакция все-таки сопровождается аллильной пере-
группировкой. Так, бутен-1 и бутен-2 дают практически одинако-
вую смесь З-бензоилоксибутепа-1 и кротилбензоата. Было показано,
СН8СН2СН = СН3
о
II
СвН3С03С(СН9)_,
CuBr
СН3СН = СНСН3
р OCOC6Hs
СН3СНСН = СН2 90%
СН3СН-СНСН,
! ' ю°/о
V ОСОС6Н5
что эти олефины и эфиры в условиях реакции не изменяются. Кроме
того, оказалось [10], что реакция с оптически активным бпцикло-
(СНЩСО- >
-(сн3)3сон
СДЦСО^
(2) (3>
{3,2,1]-октеном-2 (1) дает рацемический жзо-бицикло-[3,2,1 [-Д3-
октенилбензоат-2 (4). Считают, что отщепление аллильного водорода
при С4 приводит к образованию симметричного радикала (2<->3)
с полной эквивалентностью углеродных атомов 2 и 4 и, таким об’
разом, к превращению в рацемическую форму жзо-бепзоата,
344
Другие примеры таких перегруппировок привел Денни [11].
ОСОС6Н5
СНВС^СНСН3 сн3=сснси3
I I
СН3 СН3
ОСОС6Н6
СН3С-ССН3 сн3с —ссн3
II' II I
СН3 С113 сн3 сн3
Эта реакция использована для получения оптически активных
аллиловых спиртов [ 12L Циклические олефины, например циклопен-
тен, обрабатывают трет-бутилгидроперекисью в присутствии мед-
ной соли оптически активной кислоты [обычно (-г)-«-этиловый эфир
камфорной кислоты! и получают оптически активные аллиловые
[^j| + НООС(СН3)з + (RCOjCu ----------->
(1)
(2)
эфиры типа (1), которые расщепляются LiAlH4 до спирта (2). Эффект
асимметрической индукции невелик; однако вследствие высокой
удельной активности оптически чистого вещества наблюдается зна-
чительное вращение. Реакция бензоата холестерина с mpem-бутил-
гидроперекисью в присутствии бромистой меди как катализатора
в бензоле дает равные количества Д5-холестендибензоатов-Зр, 7а и
-Зр, 7р [13]. В этом случае аллильная атака не сопровождается пере-
группировкой.
Очень интересная и препаративно ценная реакция Н. к. б. э.
с иорборнадиеном дает 7-трет-бутоксинорбор надиен. Примеча-
тельно, что при этом образуется именно эфир, а не бензоат. Как и
с6н5соос(сн3)3
СиВг
------:------->
20-25%
в вышеуказанных реакциях, аллильная атака в данном случае
невозможна, а отщепление водорода от С7 обусловлено, по-видимому,
анхимерным эффектом двойных связей. Под действием хлорной и
уксусной кислот 7тгрст-бутоксинорборнадиен превращается в
7-норборнадиеннлацетат (73%), восстановление которого алюмо-
гидридом лития дает норборнадиенол-7 с выходом 95%.
345
1. Lawcss on S.-О., Sosnovs к у G., Svensk. Kem. Tids., 75, 568
(1963); Angew. Chem., Infernal. Ed,, 3, 269 (1964).
2, Lawcss on S.-O., YangN. C., J. Am. Chem. Soc,, 81, 4230 (1959);
E г i s c 1 1 C., L awesso n S.-О., Org. Syn., 41, 91, (1961).
3. F г i s e 1 1 C., Lawesson S.-О., Org. Syn., 43, 55 (1963).
4. Lawesson S.-О., F r i s c 1 1 C., Arkiv. KemL, 17, 393 (1961).
5. Lawesson S.-О., Berglund C., Angew. Chem., 73, 65 (1961),
6. Sos no vs к у G., Tetrahedron, 13, 241 (1961); Lawesson S.-O,,
Berglund C,, G r 6 n w a 1 1 S., Chem. Scand., 14, 944 (1960).
7. Kh arasch M. S., Sosnovsky G., Yang N. C., J, Am- Chem.
Soc., 81, 5819 (1959); Sosnovsky G,, Yang N. C,, J. Org. Chem., 25,
899 (1960).
8. Cross B., W hit h a m G. H., J. Chem. Soc., 1961, 1650.
9. Kochi J. K-, J. Am. Chem. Soc., 84, 774 (1962),
10. Goering H. L., Al а у e г IL, J. Am. Chem, Soc., 86, 3753 (1964).
II. D e n n e у I). Z., Appelbaum A., Denney D, B., J. Am. Chem.
Soc., 84, 4969 (1962).
12. Denney D, B. Napier R., C a m m а г a t a A., J. Org. Chem., 30,
3151 (1965),
13. Beckwith A. L J,, Evans G. W., Proc. Chem. Soc., 1962, 63.
14. S t о г у P. R., J. Org. Chem., 26, 287 (1961); Story P. R., Fahren-
h о 1 t z S. R,, Org. Syn., 44, 12 (1964).
НАДМУРАВЬИНАЯ КИСЛОТА, см. Перекись водорода.
НАДУКСУСНАЯ КИСЛОТА, см. Перекись водорода — уксус-
ная кислота. Хотя этот раствор и содержит Н. к. и обычно назы-
вается Н. к,, по составу и свойствам он в значительной степени
отличается от Н. к. безводной и Н. к. продажной 40%-ной.
НАДУКСУСНАЯ КИСЛОТА БЕЗВОДНАЯ.
Филлипс, Фростик и Старчер П1 описывают эффективный двух-
стадийный синтез раствора Н. к. б. в этил ацетате или ацетоне. На
первой стадии ацетальдегид самоокисляется до моноперацетата
ацетальдегида (МПА) под действием ультрафиолетового облучения
при О3 в присутствии 0,01 % ацетата кобальта(1) или под действием
, н
2 сн3сно сНзС7 V сн3со3н + СН3СНО
Y___о' 'сн3 Т, пл. 0° Т.кип. 21’
МПА т.кип. 110°
озона. МПА образует взрывчатые кристаллы (т. пл. 20—22°), кото-
рые безопасны в растворе. Ацетоновый раствор, содержащий 48,5%
МПА, 7,7% уксусной кислоты и 0,1% стабилизатора, вводят через
подогреваемый паром змеевиковый испаритель в среднюю часть
колонки для фракционирования, снабженной вводом для ацетоно-
вых паров на расстоянии 2,5 см от дна. Таким образом, МПА пиро-
лизуется при ~70° до Н. к. б. и ацетальдегида, и более летучий
компонент отгоняется. Жидкость в нижней части колонки представ-
ляет собой ацетоновый раствор, содержащий 27,9% Н. к. б. и 9,1%
уксусной кислоты.
346
Этот раствор используют для эпоксидирования «^ненасы-
щенных эфиров до глицидиловых эфиров [2]. Например, реакцией
24 молей этилкротоната с 4130 г (12 молей) 22,2%-ного раствора
Н. к. б. в этил ацетате при 85° в течение 5,5 час и последующим фрак-
ционированием получают 1167 г этил-2,,3-эпоксибутирата. Избыток
этилкротоната способствует отделению уксусной кислоты от эпо-
ксида.
сн;!со3н
СН3СН==СНСО2С.,Н3 ——СН3СН-СНСО2СЩЦ
J л 74,8% г\ / г ~
По методу Байера — Виллигера лучшие результаты при окис-
лении циклических кетонов в лактоны получаются при использо-
вании безводного реагента [31. Например, из циклопентаноиа в ука-
занных условиях с высоким выходом получают 6-валеролактон, а
84%
циклогексанон дает 85% е-капролактона (продолжительность реак-
ции 6,25 час). В случае 2-замещенных циклогексанонов расщепле-
ние кольца происходит только с одной стороны карбонильной
группы, как было показано для 2-циклогексилциклогексрнона,
0, 5 моля СН3СО3Н
10 Vac при 50°
82%
Циклогептанои и циклооктанон менее реакционноспособны; при
80° циклогептанон дает приблизительно равные количества лактона
н дикислоты, а из циклооктанона в основном образуется дикислота.
/---X ° СОг14
* 1 10-30° ' \ 1
(СНг)п С=О + АсОН + КоНЦ-Н25О4+29,2%-НяЯ СН3СО3Н у (СЩЩ С = О + (СНг)10
2 00 МЛ
300 мл
(I) 30г
(2) 50%
Т "
согн
(3) 2 57,
35$ МЛ
При окислении циклододеканона (1) получают лактон (2) с вы-
ходом 50% при реакции в присутствии конц. серной кислоты в боль-
шом избытке реагента [41. Декандикарбоновая кислота при этом
получается в значительном количестве. При 20° в отсутствие серной
кислоты реакция заканчивается лишь через 36 дней. Заметный ката-
литический эффект серной кислоты в реакции Байера — Виллигера
отмечался ранее, в частности Дерингом [5].
347
Хорнер и Юргенс F61 получали Н. к, б. в растворе медленным
добавлением 60 мл продажной 40%-ной Н. к. б. к охлажденной
льдом суспензии 45 г Р2О5 в 200 мл бензола. Через 10—15 мин, про-
зрачный раствор отфильтровывают, титруют и хранят на холоду
в темноте. Безводный реагент применяют для эпоксидирования
шиффовых оснований (азометинов) до оксазиранов (изонитронов).
Например, реакцией циклогексилиденциклогексиламина (1) с бен-
зольным раствором надуксусной кислоты (2%-ный избыток) при
комнатной температуре в течение 5 час получают пентаметилен-N-
циклогексилоксазиран (2). За несколько месяцев до опубликования
статьи Хорнера Эммонс и сотр. [71 описали окисление нескольких
сн3со3н^
89%
(1)
(2)
азометинов в оксазираны, «полученные с хорошими выходами реак-
цией с Н. к. б. в хлористом метилене», не упомянув о методе приго-
товления безводного реагента. Позднее [81 он описал метод получе-
ния Н. к. б. in situ для окисления аминов, например п-толуидина.
К хлороформу (60 мл) при энергичном перемешивании и охлаждении
льдом добавляют 0,5 моля 90%-ной перекиси водорода и 2 капли
серной кислоты в качестве катализатора. Затем в течение 30 мин
приливают по каплям уксусный ангидрид (0,6 моля), который реа-
гирует с водой, содержащейся в перекиси; при этом часть перекиси
расходуется на образование перекиси ацетила. Через 15 мин про-
зрачный раствор разбавляют при комнатной температуре 40 мл
хлороформа и нагревают до кипения. К кипящему раствору добав-
ляют 0,1 моля «-толуидина в 25 мл хлороформа в течение 20 мин,
желтый раствор кипятят 1 час и выливают в воду. Обработкой и
Н3С-^
___nh2
ЗСП3СО3Н
72%
И3С ______S- no2
кристаллизацией из лигроина получают 72% чистого «-нитротолу-
ола. Некоторые другие нитробензолы получены с выходами 62—
83%. Этим методом при окислении втор-бутил амин а получают
2-пнтробутан с выходом 65%.
Представляет интерес применение Н. к. б. для получения гидро-
ксильных радикалов. До сообщения о методе Хейвуда, Филлипса
и Стенсбери [9] Шлейер и Николас [101 описали эксперимент,
в котором смесь 150 г адамантана (1,1 моля) в 1870 г хлорис-
того метилена и 1,35 моля раствора Н. к. б. в этнлацетате энер-
гично перемешивают при облучении ультрафиолетовым светом
(лампа 100 вт погруженного типа). Уже с самого начала реакции
начинается выделение газа. Через 21 час при 40—45° расходуется
348
94,8% надкислоты. Смесь концентрируют почти досуха в вакууме
водоструйного насоса и после добавления и упаривания двух пор-
ций толуола но 100 мл получают 155 г белого твердого вещества.
Одну его часть хроматографируют, другую окисляют в более легко
разделяемую смесь, содержащую адамаитанол-1 н адамантанол-2.
Выходы непрореагировавшпх углеводородов — адамантанола-1 и
адамаитапола-2 указаны под формулами. Полигидроксильный про-
дукт и неидеитифицированпое карбонильное соединение получены
с выходами соответственно 1,8 и 4,5%.
В других опытах по окислению с применением Н. к. б. в этила-
цетате Хейвуд и Филлипс [И] показали, что а,р-ненасыщенные
циклические ацетали, например (1), гладко окисляются в эпоксиды
О>снз
с^5сн=сн--сн
\О-
СН3СО3Н
------2—>
90%
(2)
сн3
(1)
(2) . Удивительно, что нециклический ненасыщенный ацеталь (3)
реагирует только при повышенной температуре и дает не эпоксид,
а эфир (4). Эта реакция катализируется серной кислотой, в присут-
ствии которой ненасыщенный циклический ацеталь (I) превращается
сн;со3н
СН3СН = СНСН(ОС1Нр)2 -—— СН3СН = СНСО2С4НЭЧ-
I V %
(3) (4)
+ С4Н9ОН+СН3СООН
CH,COsH(H,SO4)
н-С3Н7СН(ОС2Н5)2------—------«-СзНтСОХгНз + СзНзОН + СНзСООН
Ь9
(5) (6)
в нендентифицированное вещество и не образует эпоксида. Насы-
щенные ацетали, например (5), реагируют с Н. к. б. в присутствии
349
серной кислоты с образованием эфиров (6). Реакция осуществляется,
по-видимому, через полуацетальперацетат (8).
R уН
ЕЮ7 XOEt
стцсощ
-Е1ОН
/Н оч
/С<' ^ССН3
ЕЮ7 ХО—О7
(?)
(8)
1\ Н-О.
фссн3
EtOz Z'O - О'7
ЕЮ
но
;ссн.
R
(9)
Кросби [12] разработал интересный синтез скополетина (V).
Продажный изованилин (I, 1,28 моля) в этилацетате окисляют при
40° 1,40 моля Н. к. б. в том же растворителе. Продукт — 3-окси-
4-мстоксифенилформиат (II) перегоняют при 111 —11571 мм; через
2 недели он затвердевает при комнатной температуре (т. пл, 57—58°).
Гидролизом II спиртовым раствором едкого кали и последующей
конденсацией фенола III с щавелевоуксусным эфиром в при-
сутствии 85%-ной фосфорной кислоты получают эфир IV, который
омылением и декарбоксилированием (медь, хинолин) превращают
в скополетии (т. пл. 198—200“).
СН3О. /ч
] II СН3СОЯН
I II 7-1%
НО" ^/’7СНО
I
Т н кон
I 11„
НО7'7-' 7ОСНО
п
ссш
1аС\.^А
I II I D он
| || | 2) Нагревание
НО77:/ ~ ~~6О%
IV
V
(.Phillips В., Fr о s t i с k F. С., Jr., Starcher Р. S,, J. Am.
Chem. Soc., 79, 5982 (1957); Phillips B., Starcher P. S., Ash
B. D., J. Org. Chem., 23, 1823 (1958).
2. At a c Peek D. L., Starcher P. S., P h i I 1 i p s B., J. Am. Chem.
Soc., 81, 680 (1959).
3. Starcher P. S., Phillips B., J. Am. Chem. Soc., 80, 4079 (1958).
4. К о s s w i g K.., Stumpf AV., Kirch h of AV., Ann., 681, 28 (1965).
5. Von Doering AV., Speers L., Am. Chem. Soc., 72, 5515 (1950).
6. H о г n c r L., Jurgens E., Chem. Ber., 90, 2184 (1957).
350
7. E m m о n s W. D., J. Arn. Chem. Soc., 78, 6208 (1956).
8. Emmons AV. D., J. Am. Chem. Soc., 79, 5528 (1957).
9. Heywood D. L., Phillips B., Stansbury H. A., Jr., unpub-
lished.
10. Von Schleyer P., Nicholas R. D., J. Am. Chem. Soc., 83, 182
(1961).
11. П ey wood D. L., Phillip s B., J. Org. Chem., 25, 1699 (1960).
12. Crosby D. G., J.Org. Chem., 26, 1215 (1961).
НАДУКСУСНАЯ КИСЛОТА, ПРОДАЖНАЯ 40%-НАЯ. Со-
держит 40% Н. к., 5% перекиси водорода, 39% уксусной кислоты,
1% сериой кислоты и 15% воды. Плотность 1,15г/то. В свежеприго-
товленном образце содержится 0,77 моля Н. к. на 100 мл. Так как
при хранении содержание Н. к. уменьшается, необходимо произво-
дить анализ полученного ранее образца обработкой аликвотной
части иодистым калием и последующим титрованием выделившегося
иода стандартным раствором тиосульфата натрия. В некоторых
случаях рекомендуется нейтрализация имеющейся серной кислоты
ацетатом натрия.
Внимание! Реагент, попадая на кожу, вызывает сильные ожоги.
Эпоксидирование олефинов. Рейф и Хауз 111 разработали сле-
дующую методику синтеза окиси транс-стильбена. Титрованием
продажного 40%-ного препарата показано, что он содержит 0,497 г
Н. к. на 1 мл. Для реакции с 0,3 моля mpawc-стильбена берут 65 мл
(0,425 моля) реагента, к которому добавляют 5 г NaOAc-З Н2Одля
нейтрализации имеющейся серной кислоты. Раствор углеводорода
в 400 мл хлористого метилена охлаждают до 20°, затем охлаждаю-
щую баню убирают и добавляют при перемешивании Н. к. в течение
15 мин. Смесь перемешивают еще 15 час, нагревают до 32—35°
н/С С'ЧС6Н5 70-7 5%” н/' °\с3Н5
в течение 2 час и затем постепенно охлаждают. За это время погло-
щение смеси при 295 ммк снижается до значения, составляющего
менее 3% от первоначального. Если количество непрореагировав-
шего транс-стильбена превышает это значение, его нельзя удалить
многократной кристаллизацией, поэтому следует добавить Н. к.
По окончании реакции после соответствующей обработки и кристал-
лизации из метанола н затем гексана получают чистую окись с вы-
ходом 70—75%.
По Галахеру, введение 17а-оксигруппы в 20-кетостероид осуще-
ствляют через 17-енол ацетат (2), который эпоксидируют в (3) дей-
ствием надбензойной кислоты с последующим щелочным гидроли-
зом. В другой работе 121 эпоксидирование проводили 40%-ной
Н. к., нейтрализованной ацетатом натрия, и показали, что этот
дешевый реагент, как и надбензойная кислота, дает эпоксид (3).
Оливето и Гершберг 131 пытались использовать продажную Н. к.
без предварительной нейтрализации серной кислоты и получили
351
Непосредственно 17а-окси-20-кетон (4). Исключение стадии щелоч-
ного гидролиза (3) дает возможность сохранить другие ацетильные
группы в молекуле.
снэ
(9
(2)
Окисление. Ван Дуурен и сотр. [41 окисляли 1,2,5,6-дибензан-
трацен до 3,4-секо-дикислоты добавлением 10 г 40 %-ной Н. к.,
насыщенной ацетатом натрия, к суспензии 2 г углеводорода в 75 мл
хлороформа при 4° и перемешиванием при 4“ до образования про-
зрачного раствора (8 час). При последующем перемешивании при
4° начиналось выделение продукта, и по окончании реакции (8 дней)
продукт отделяли.
Пейдж и Тарбелл [51 окисляли ^-нафтол до о-карбоксикоричной
кислоты действием 1,1-кратного количества (от теоретического)
40%-ной Н. к. без добавления ацетата натрия. При перемешивании
к надкислоте (0,14 моля) при 25—30° добавляли в течение 4 час
раствор 0,14 моля [J-нафтола в 100 мл уксусной кислоты (при темпе-
ратуре не выше 40°). После добавления приблизительно одной трети
^\/С0*И
ЗСН3СОаН i I)
67—7 0% !
ЧСН-СНСО2Н
всего количества реагента из оранжевого раствора начиналось
выделение твердого вещества. Смесь оставляли при 25—30° на
6—8 час и при комнатной температуре на 4 дня. Удовлетворительная
чистота кислоты достигалась ее осаждением из отфильтрованного
бикарбоиатного раствора.
Для превращения пиридина в N-окись (Мошер 161) 1,5 моля
40%-ной Н. к. добавляют к 1,39 моля пиридина при перемешивании
с такой скоростью, чтобы поддерживать температуру 80°
(50—60 мин). Смесь перемешивают до тех пор, пока температура не
352
снизится до 40°. Растворитель отгоняют на кипящей водяной бане
с помощью водоструйного насоса и остаток — ацетат N-окиси пири-
дина помещают в колбу для перегонки легкокристаллизующихся
веществ (том III, рис. П-4). Перед вакуумным насосом ставят
охлаждаемую сухим льдом ловушку для сбора уксусной кислоты,
N
СН3СО3Н
—-—
78-83%
образующейся при диссоциации ацетата, и окись перегоняют при
95—98°/0,5лш. Гигроскопичное основание представляет собой бес-
цветное твердое вещество с т. пл. 65—66° (запаянный капилляр).
Иодбензол (0,1 моля) окисляют в иодозобензол д и ацетат дейст-
вием 0,24 моля 40%-ной Н. к. при перемешивании при 30° в течение
30—40 мин (Шарефкин и Зальцман 17]). При перемешивании еще
сн3соан
СеН51-------> С6Н61+ОСОСН3(СН3СОр
в течение 20 мин при 30° раствор желтеет и выпадают кристаллы
иодозобензолдиацетата. После охлаждения продукт собирают и
промывают холодной водой; т. пл. 158—159° (разл.), чистота
97—98%.
Н. к. окисляет 1,1-диалкилгидразины до тетразинов [реакция
(1)] с выходом 70—80% и расщепляет бензоилгидразон ацетофенона
[реакция (2)] [8]. В этих реакциях лучше применять Н. к., чем жел-
I) 2 (C3H5)aNNH., —(CaH5)2NN-NN(C2H6)2
/СН3 CH,COSH C6H5CONH ,CH3
2) 2(CcH5)CONHN-C( --------:---| -f-20 = C<
Xh5 “n- CgH5CONH XCeH5
59% 93%
тую окись ртути, так как кислота растворима в органических раст-
ворителях (хлороформе, водной уксусной кислоте).
I. Reif D. J., House Н. О., Org. Syn., Coll. Vol., 4, 860 (1963).
2. Anderson H. V., Garrett E. R., Lincoln F. H., Jr., Nat-
han A. H., Hogg J. A., J. Am. Chem. Soc., 76, 743 (1954).
3. О 1 i v e t о E. P,, Hershberg E. B., J. Am. Chem. Soc., 76, 5167
(1954).
4. Van Duuren B, L., Bekersky 1., Lefar M., J. Org. Chem., 29,
686 (1964).
5. Page G. A., Tar bell D. S., Org. Syn., Coll. Vol., 4, 136 (1963).
6. Mosher H. S,, Turner L., Carlsmith A., Org. Syn., Coll. Vol.,
4, 828 (1963).
7, Sharefkin J. G., Saltzman H., Org. Syn., 43, 62 (1963).
8. Horner L., Fernekess H., Chem. Ber., 94, 712 (1961).
12 Заказ Яс 1096
353
НАТРИЙ, Na. Атомный вес 23,00, т. пл. 97,5°. О мерах предо-
сторожности и технике безопасности см. Калий.
Получение чистого натрия [1]. Очистка Н. путем срезания с его
поверхности под ксилолом окисленных слоев металла трудоемка и
связана с потерями. Приведенная ниже методика облегчает эту
операцию, экономит металл, сводя к минимуму количество обрезков.
Количество Н., значительно большее требуемого, помещают в ши-
рокогорлую колбу ЭрленмеЙера или в высокий стакан и наливают
сухой ксилол. Колбу нагревают до тех пор, пока И. не распла-
вится, но все же останется в окисной пленке. Нагревание прекра-
щают и колбу осторожно вращают так, чтобы Н. вытек из своих
оболочек и образовал несколько глобул. Тогда колбу охлаждают,
осторожно перемешивая ее содержимое, чтобы глобулы не соедини-
лись. В качестве меры предосторожности можно во время охлажде-
ния пропускать через раствор сухой азот. Когда металл затвердеет,
светлые глобулы вынимают с помощью острой железной палочки или
другого приспособления, быстро сушат фильтровальной бумагой
или сухим полотенцем и помещают в заранее взвешенный сосуд,
в котором и отвешивают нужное количество Н. В окисных оболоч-
ках остается очень мало И.; растворитель отделяют декантацией,
а обработка трет-бут левым спиртом разрушает остатки активного
металла.
Суспензия натрия (натрий в виде дроби или песка). Для полу-
чения суспензии Н. Гершберг [2] расплавлял металл в ксилоле,
взбалтывал смесь, пользуясь тем же приемом, что и при получении
суспензии калия (только в другом растворителе); см. Калия этилат.
Описана также безопасная модификация этой методики [3]. Марвел
и Кинг [41 помещали Н. и ксилол в колбу, снабженную механиче-
ской мешалкой и обратным холодильником, нагревали Н. пока он
не расплавится, энергично перемешивали, чтобы раздробить металл
на мелкие частицы, а затем, прекратив нагревание, продолжали
перемешивание до затвердевания. Хафнер и Кайзер [51 полу-
чали суспензию Н, (23 а) в толуоле (150 мл) в колбе, снабженной
обратным холодильником, пробкой на шлифе и вибромешалкой.
Толуол нагревали до кипения, расплавленный металл дисперги-
ровали вибро мешал кой и смесь быстро охлаждали. Толуол отделяли
декантацией в атмосфере азота и заменяли его на ТГФ. Аналогич-
ной техникой пользовался и Элсингер [6].
Дисперсии натрия. Дисперсии, содержащие частицы диаметром
порядка 10 мк, величина поверхности которых в 100 раз больше
величины поверхности натриевого песка, лучше всего получать
непосредственно перед применением, однако технология и аппара-
тура детально не разработаны. Дисперсионной средой могут служить
углеводороды и простые эфиры, главным образом с температурой
кипения в пределах 100—200°. Часто применяют диспергирующий
агент, например олеиновую кислоту или лауринат алюминия, в ко-
личестве порядка 0,2—1,0%. Применение дисперсии часто приводит
354
к более высоким выходам, большим скоростям реакции, к пониже-
нию температуры процесса и более легкому контролю за реакцией.
Натрий иа окиси алюминия. Натриевый катализатор, имеющий
большую поверхность, получают энергичным перемешиванием при
150° в атмосфере азота сухой окиси алюминия и Н. [71. Он активен
при изомеризации бутенов и пентена-]. В отличие от изомеризации
при кислотном катализе скелетной изомеризации при этом не про-
исходит. Результаты свидетельствуют о некоторой стереоспеци-
фичности. Например, бутен-1 первоначально изомеризуется с обра-
зованием примерно равных количеств цис- и транс-бутенов-2, в даль-
нейшем же образуется термодинамически равновесная смесь, бога-
тая транс-изомером [8]. Этот катализатор был использован также
для почти количественного превращения метиленциклобутана
в 1-метилциклобутен [91.
1. Contributed by Newman Al. S., Ohio State University.
2. Гершберг E., Флзер Л., «Синтезы органических препаратов», ИЛ,
/М,, 1949, сб. 2, стр. 53.
3. Org. Expts., 142—143.
4. Марвел, Кинг В. Б., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. I, стр. 548.
5. Hafner К., Kaiser Н., Org. Syn., 44, 94 (1964).
6. Е 1 s i п g е г F., Org. Syn., 45, 7 (1965).
7. Pines H., Haag W. О., J. Org. Chem., 23, 328 (1958).
8. Haag W. O., Pines H., J. Am. Chem. Soc., 82, 387 (1960),
9. Shabtai J., Gil-Av E-, J. Org. Chem., 28, 2893 (1963).
НАТРИЙ — АММИАК. Для восстановления фенола в аромати-
ческий углеводород на раствор этого фенола и кислого диэтилфос-
фита в четыреххлористом углероде действуют триэтиламином, после
чего смесь оставляют на 24 час до полного выделения хлоргидрата
триэтиламина [реакция (1)]. Диэтилфени л фосфат отделяют, раство-
ряют в ТГФ и восстанавливают Н.—а. [11. Для реакции можно ис-
1) АгОН НО—Р{ОС2Н5)., 4- (С,Н5)3К -1- СС1й
о-
ArOP+(OCTIJ.,H-CHCl34-(C2Hs)E)NHCl
О~ о-
2) ArO —Р+(ОС2115)2-;-2Na-|-NH3 -•> ArH + KaNH., 4-NaOP +(ОС2НД
пользовать навески в 50—200 мг; выходы составляют 18—52% [2].
Другие примеры см. в работе [3].
I. К е л л е г G. W., Williams N. R., J. Chem. Soc., 1955, 522.
2. Р с 1 1 е t i е г S. W., L о с k е D. М., J. Org. Chem., 23, 131 (1958).
3. Fishma n J., Tomasz M., J. Org. Chem., 27, 365 (1962); C a s p i
E., Cullen E., Grover P. K-, J- Chem. Soc., 1963, 212.
12*
355
НАТРИЙ - АММИАК — ЭТАНОЛ. Сочетание натрия с жидким
аммиаком и этанолом обычно применяют для восстановления по
Берчу простых фениловых эфиров (см. Берча восстановление).
Пакетт и Нельсон [1| нашли, что такая комбинация дает лучшие
результаты, чем метод Буво — Блана при избирательном вос-
становлении сложноэфирной функции в соединении (1). Натрий
(1)
(2)
добавляют небольшими порциями при перемешивании к раствору
(1) в смеси аммиака и этилового спирта; раствор перемешивают до
тех пор, пока большая часть аммиака не испарится, после чего до-
бавляют воду и соляную кислоту и продукт реакции экстрагируют
эфиром.
1. Paquette L. A., Nelson N- A., J. Org. Chem., 27, 2272 (1962).
НАТРИЙ — mpcm-БУТАНОЛ — ТЕТРАГИДРОФУРАН, см. Ли-
тий — mpem-бутанол — тетрагидрофуран.
НАТРИЙ-КАЛИЕВЫЙ СПЛАВ (1 : 5, жидкий).
Сплав получают нагреванием 1 ч. натрия и 5 ч. калия в ксилоле
до плавления и осторожным перемешиванием расплава металлов
палочкой, сохраняя сплав в виде одной большой капли. После ох-
лаждения сплав остается жидким и его можно отбирать пипеткой
[1]. Реагент вступает в реакцию с образованием калиевых, но не
натриевых производных, например:
СН3 СН3
C0H5COCH3-j-NaK C6H5CK + NaOCH3
сн3 с!н3
(С6Н5),С=С(С6Н5)2-р2МаК (СеН5)3С-С(С6Н5)2
К К
(C6I-I5)3CLi + NaK (СЙН5)3СК
Металлирование бензола осуществляют реакцией с алкиллитие-
выми производными и Н. с. в эфире [21:
NaK С„НЛ
RLi ..... ---> RH + С^ЩК
356
Этот сплав был использован Циглером [3] для расщепления ме-
тиловых эфиров, а также Мюллером и Бунге [4] для расщепления
дифенилового эфира:
со,. н +
CsH50qjH5 + 2NaK С6Н5ОК-[-СеН&К —
С6Н5ОН + С6Н5СО2Н
Маркевич и Даусон [5] выделили олефиновые компоненты дейст-
вующего начала ядовитого плюща, «урушиол», в активной феноль-
ной форме бензилированием, хроматографией и дебензилированием
действием диспергированного Н. с. в лигроине. При дебензилиро-
вании этим реагентом положение или геометрическая конфигурация
олефиновых двойных связей, разделенных одной метиленовой
группой, не изменяется (под действием натрия такие соединения
изомеризуются в сопряженные системы, которые затем восстанав-
ливаются металлом).
Натрий в отсутствие калия не дает удовлетворительных резуль-
татов при ацилоиновой конденсации в ряду тиофена, однако Н. с.
может быть использован для этой цели. Так, действием этого реа-
гента из (1) легко получают (2) [6]:
(CHZ)5
Vo2CH3
СО2СН3
(сн2
Na-K^
70%
(1) &
1. Gilman ii., Young R. V., J. Org. Chem., 1, 315 (1936).
2. В г у c e-S m i t h D., Turner E. E., J. Clieni. Soc., 1953, 861.
3. Ziegler K-, Schnell B., A n n., 437, 227 (1924).
4. M ii 1 1 e г E., Bunge W., Chem. Ber., 69, 2164 (1936).
5. Markiewitz К. 14., Dawson C. R., J. Org. Chem., 30, 1610 (1965).
6. G о I’d f a r b Ya. L., Taits S. Z., Be le n’k i i L. I., Tetrahedron,
19, 1851 (1963).
НАТРИЙ — НАФТАЛИН, [C10HjNa+.
Нафталин не реагирует с натрием в эфире, но Скотт [1] показал,
что при введении натрия в раствор нафталина в диметиловом эфире
или диметиловом эфире этиленгликоля металл растворяется с обра-
зованием ярко-зеленого раствора. Карбонизация зеленого раствора
дает смесь нафталина, А1-диалин-3,4- и А3-диалин-1,4-дикарбоновых
кислот, что позволило Скотту предположить образование в растворе
комплекса (C10H8Na,)C10H8. Однако проведенное Полем [2] коли-
чественное изучение реакции в ТГФ показало, что при растворении
металла происходит перенос электрона к нафталину с образованием
окрашенного в зеленый цвет ион-радикала, указанного на схеме.
Несмотря на то что нафталин реагирует только с одним атомом
357
натрия, карбонизация 1 моля Н.— и. при —70° сопровождается дис-
пропорционированием с образованием 0,5 моля нафталина и 0,5 мо-
ля смеси 1,4- и 3,4-дикарбоновых кислот. Лисси [31 выделил неко-
торые компоненты этой смеси и установил их строение.
Норман [41 получил Н.— н. в ТГФ и показал, что этот реагент
эффективнее, чем натрий в ТГФ для металлирования терминальных
ацетиленов и соединений, содержащих активные метиленовые
группы. Однако при этом выходы кислот, полученных карбониза-
цией, не очень хорошие:
н-С4Н9Сг-СН |
НС^СН
Инден
Нафталин — Na
Флуорен
c6h5ch3cn
«-С4Н9С =•=- ССО2Н (50%)
с0; | НС^ССО.Н (20%)
---> -j Инден-1-СО3И (62%)
Флуореп-Э-СОоН (44%)
ч CGH5CH(CN)CO.2H (40%)
Хорнер [5] исследовал сс-металлирование нитрилов с последующим
алкилированием; типичные результаты приведены ниже:
Нафталин—N.) циклогскснлподид
CeH5CH2CN —-------ОДСНСХ
О>ни
Нафталин —Na. С.Н.Вг
(CgH5)2CHCN ------ 7-~—(C6H5)2CCN
с2н5
Нафталин - Na, СЛЦВг
СН3СМ ---------—-------ch3ch,ch2cn
о J %
Особенно хорошо под действием Н. — н. осуществляется конден-
сация бензилгалогенидов по Вюрцу [61:
2 Нафталин — Na
2QHaCH2Cl —----—--------СбН5СНгСНгС6Н,
358
Анион-радикал в ТГФ описан как «почти идеальный» реагент
для восстановительного расщепления тозилата в спирт [7). Реагент
получают перемешиванием чистых кусочков натрия с небольшим
избытком нафталина в ТГФ в атмосфере азота или аргона при ком-
натной температуре.
I. S с о t t N. D., Walker J. F., H a n s 1 ey V7. L., J. Am. Chem.
Soc., 58, 2442 (1936); Walker J. F., S с о t t N. D., ibid., 60, 951 (1938).
2. Paul D. IL, L i p k 1 n D., W eissm an S. I., J. Am. Chem. Soc., 78,
116 (1956).
3. Lyssy Th. M., J. Org. Chem., 27, 5 (1962).
4. N о r m a n t H., Angelo B., Bull. soc. chim. France, 1960, 354.
5. Horner L., Giistcn II., Ann., 652, 99 (1962).
6. (3 ii s t e n H., Horner L., Angew. Chem. Internal. Ed., 1, 455 (1962).
7. Closson W. D., Wriede P., Bank S., J. Am. Chem. Soc., 88, 158
(1966).
НАТРИЙТЕТРАКАРБОНИЛ КОБАЛЬТ, Na+Co(CO)r.
Бесцветный порошок, нерастворимый в воде. Его получают из
дикобальтоктакарбонила и амальгамы натрия в эфирном растворе
в атмосфере азота [II. Чтобы заменить растворитель, эфир отгоняют
в вакууме и добавляют новый растворитель, не допуская соприкос-
новения смеси с воздухом.
Хек и Бреслоу 121 нашли, что ал кил галогениды, сульфаты и
сульфонаты претерпевают карбалкоксилирование в присутствии
окиси углерода, спирта, основания и каталитического количества
Н. Например — реакция 1-иодоктана в метаноле в присутствии
сильно основного пространственно затрудненного дициклогексил-
этиламина в качестве основания. При этом применение пространст-
венно незатрудненного амина приводит к образованию амида.
Na+Co(CO)y, толуол (50°)
н-С8Н 171 Д- СО-р V-.H3OH -р (СсН j । )2NCH2C1-I3 “ ►
3t%
—> /r-CgH^CO.jCHs-P (CeHtl)aNCH2CH3(l _)
H
1. Hieber W., В г a u n G., Beck W., Chem. Ber., 93, 901 (1960).
2. Heck R. F,, Breslow D. S., .1. Am. Chem. Soc., 85, 2779 (1963).
НАТРИЯ АЗИД, NaN=NA-N. Мол. вес 65,02, растворимость
в 100 г воды: 39 г при 0°, 55 г при 100° (см. также А зотист овод о род-
ная кислота).
Синтез изоцианатов 111. Согласно стандартной методике 12], смесь
хлорангидрида лауриновой кислоты и ацетона добавляют из капель-
ной воронки при охлаждении и перемешивании к раствору Н. а.
в воде с такой скоростью, чтобы температура не превышала 10—15°.
По завершении реакции (примерно 1 час) нижний водный слой
осторожно отделяют отсасыванием с помощью стеклянного капил-
ляра. Верхний слой сырого ацилазида медленно добавляют к 500 мл
бензола, нагретого до 60°. При термическом разложении выделяется
359
азот и образуется бирадикал, который перегруппировывается в изо-
цианат. Примерно через 1 час бензол отгоняют и ундецилизоцианат
О
я-СпН23СОС1 -J- NaNa
0,5 моля 0.7 моля
в 150 мл ацетона
СиНс. 60-70°
10-15°
Вода
«-СпНгзС — N = N = N
81—86% (общий)
«CUH2JN=C=O+N2
перегоняют в вакууме при 3 мм.
Реакция Курциуса. Суммарный процесс превращения кислоты
через азид и изоцианат в амин известен как реакция Курциуса [3].
При исследовании этой реакции на примере zpc-2-фенилциклопро-
панкарбоновой кислоты Вейншток [4] нашел, что способ получения
азида через хлорангидрид кислоты неудовлетворителен ввиду его
легкой изомеризации в хлорангидрид транс-кислоты. Эффективным
методом оказалось применение в качестве промежуточного соеди-
нения смешанного ангидрида типа используемых в синтезе пептидов.
Раствор кислоты в водном ацетоне обрабатывают триэтил амином
с образованием соли амина, которая при добавлении этилового
эфира хлормуравьиной кислоты дает смешанный ангидрид. Обра-
боткой смеси водным раствором Н. а. получают ацил азид, который
экстрагируют эфиром и разлагают в толуоле. Общий выход чистого
zjwc-2-фенилциклопропиламина составляет 77%.
Мэнч-Петерсен [5] получил азид ж-нитробензойной кислоты до-
бавлением по каплям при перемешивании ж-нитробензоилхлорида
COCI
I
+ NaNs
1,2 моля
в 500 мл воды
1 моль, в 300 мл ацетона
20—25°
98%~*
360
в ацетоне к раствору Н. а. в воде. Азид выделяется в виде белого
осадка, достаточно чистого для идентификации и количественного
анализа алифатических и ароматических оксисоединений. Реактив
взаимодействует со спиртом с выделением азота и перегруппиров-
кой в арилизоцианат, к которому присоединяется спирт с образо-
ванием арилуретана [6]:
О
II + - —N2 ROH
ArC—N = N = N ----> ArN-C = O ArNHCO3R
Эти соединения являются хорошо кристаллизующимися произ-
водными, в которых содержание нитрогрупп можно определить тит-
рованием хлористым титаном. С аминами реактив образует jw-пит-
рофени л мочевины [6]:
О О
II + - -N2 RNHS ||
ArC —N —N=N ArN^C^O ------>• ArNHCNHR
Арилазиды. В одном из синтезов [7] окиси бензофуразана (3)
водный раствор Н. а. добавляют к раствору диазотированного о-пит-
роанилина при 0—5°. Выделяется азот и о-нитрофенилазид выпадает
Cl
+ NaN3 - у
хггч rv э . 63-69%’
Ж)г 0, 2 Маля
В толуоле три 87'
77-85%
(1) 0, 2 моля
в виде светло-кремового или бесцветного твердого вещества. После
кристаллизации из 95%-ного этанола получают препарат (т. пл.
52—54°), вполне пригодный для проведения следующей стадии —
термического разложения в толуоле. Окись бензофуразана еще удоб-
нее получать окислением о-иитроанилина гипохлоритом [8].
Реакции с эпоксидами. Вандерверф н сотр. [9] добавляли насы-
щенный водный раствор Н. а. (взятого в небольшом избытке)
к кипящему раствору окиси пропилена в диоксане и установили, что
сн3сн---сн2
Na№N—N +
CH3CHCH3-N=N=N
ОН
н
N=N==N
Н
361
главным продуктом реакции является 1-азидопропанол-2. Окись
циклопентена реагирует с образованием транс-2-азидоциклопента-
нол а.
Капельная проба 110]. /Меркаптаны и тиокетоны заметно ката-
лизируют реакцию Н. а. с иодом, протекающую с выделением азота;
этот эффект используется для обнаружения серу содержащих со-
единений.
2NaN3-|-I, 2Nal -3N,
Синтез бензофуразана [11]. Бензофуразаны образуются при
самопроизвольном разложении (при 100ф о-нитрозофенилазидов.
Например, 1,3-дихлор-2-нитрозобензол (2) при взаимодействии
с Н. а. дает 4-хлорбензофуразан (3); т. пл. 843
1. S с h г о с t е г G., Вег., 42, 3355 (1909).
2. А ,л л е и Ч., Белл А., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 432
3. Curl i us Th., J. prakL Chem., [2], 50, 275 (1894).
4. W c i n s t о c k J., J. Org. Chem., 26, 3511 (1961).
5. Мэнч-Петерсе и Дж., «Синтезы оргаинческх препаратов», ИЛ, 51.,
1954, сб. 5, стр. 44.
6. See references in preceding paper.
7. С м и т П., Бойер Д ж., «Синтезы органических препаратов», ИЛ,
М., 1953, сб. 4, стр. 395.
8. 54 э л л о р и Ф., «Синтезы органических препаратов», ИЛ, 54., 1959, сб. 9,
стр. 51.
9. V an derwerf С. A., Heisler R. ДУ, 51 cEwen \¥. Е., J. Ат.
Chem. Soc., 76, 1231 (1954).
10. F е i g 1 F., «Spot Tests», 2, 164, Elsevier (1954).
11. Boulton A. J., Ghosh P. B., Kairitzky A. R., Tetrahedron
Letters, 2887 (1966).
НАТРИЯ АЛЮМОГИДРИД, NaAlH4. Мол. вес 54,01. Получение
[1].
Как восстановительный агент этот гидрид подобен алюмогид-
риду лития [2]. В ТГФ или в смеси ТГФ с пиридином при темпера-
туре от — 65 до — 45° Н. а. восстанавливает эфиры карбоновых
кислот до альдегидов с выходами 50—85%. Алифатические альде-
гиды получаются с более высокими выходами, чем ароматические.
В тех же условиях алюмогидрид лития дает более низкие выходы [3].
1. С 1 a s е n Н., Angew. Chem., 73, 322 (1961).
2. Fin holt A. E., Jacobson E. C.,Ogard A. E., T h о in p s о n P.,
J. Am. Chem. Soc., 77, 4163 (1955).
3. Захаркин Л. И., Гавриленко В. В., Маслин Д. EL, X о р-
л и и а И. 54., Tetrahedron Letters, 2087 (1963).
362
НАТРИЯ АЛЮМОХЛОРИД, NaAlCl4. Мол. вес 191,80 (NaCl
58,45; А1С1з 133,35).
Во всех упомянутых ниже исследованиях, за исключением
одного, реагент получали путем тщательного смешения 1 же NaCl
с 2 же А1С13 с последующим нагреванием смеси пламенем горелки
до образования прозрачного плава (180—200°). Если реакцию про-
водят в пробирке, которую осторожно вращают по мере понижения
температуры, плав начинает затвердевать при 87° и его можно под-
держивать в полужидком состоянии путем нагревания на кипящей
водяной бане. Если таким же образом обрабатывать смесь состава
1:1, то часть твердого вещества не растворится при 180—250°;
жидкая часть начинает кристаллизоваться при охлаждении до 150°
и затвердевает при 130°. Реагент, полученный в соотношении 1 : 2,
эффективен для реакций дегидратации, конденсации и циклизации
при температурах от 90 до 220°.
Цан и Охват [1] перемешивали плав 20 г NaCl и 100 г А1С13
при 180°, добавляли смесь 10 г малеинового ангидрида и 11 г гидро-
хинона и довольно быстро повышали температуру до 200—220°,
пока сине-красный плав не затвердевал. Охлажденный плав нагре-
(3) 10 г
363
вали с водой и соляной кислотой, твердый продукт сушили и экстра-
гировали бензолом. Получали около 4 г чистого нафтазарина. Для
аналогичной конденсации фталевого ангидрида с дикетоформой ди-
гидронафтазарина с образованием 1,4,5,8-тетраоксинафтаценхинона
выход не приведен.
В 1932г. Л. Физер и М. Петерс (впоследствии М. Физер) [2,3] ис-
пользовали реагент в соотношении 1:2 для циклизации кетоэфира (1)
в перн-сукциноилаценафтен (2), а также для циклизации кето-
кислоты (3) в диметил-1,2-бензаитрахинон (4); в последней реакции
(см. схему на стр. 363), по-видимому, происходит миграция ароильной
группы от (a-положение) к С7 (0-положение) [4]. Аналогичные
методики были использованы для замыкания кольца по Шоллу при
переходе от 1-бензоил-2-нафтол а к 4-оксибеизантрону [5] и от 3-бензо-
илперинафта на (5) к2,1'-триметиленбензантрону-10 (6) [61 .Хотя выход
выделенного (6) был невысок, однако использование всей неочи-
щенной реакционной смеси в заключительной стадии перегонки
над цинковой пылью повысило общий выход конечного продукта —
3,4-бензпирена (7) до 50%.
Томсон и сотр. [7] при проведении реакций циклизации, напри-
мер 0-фенилиропионовая кислота-Ч-ииданон (85%), у-феии л масля-
ная кислота -нкх-тетралон (73%), перемешивали 2 г NaCl и 10 г
А1С1;; при нагревании на пламени горелки до плавления смеси, затем
перемешивали плав термометром при 140°, добавляли 2 г кислоты,
быстро повышали температуру и поддерживали ее при 180—200°
в течение 2 мин. Эта методика пригодна для проведения перегруп-
пировки Фриса, конденсации гидрохинона с дикислотами и цикли-
зации а рил винил кетонов.
Норрис и Клемка [8] применили двойную соль (1 : 1), получен-
ную нагреванием 30 г NaCl и 68 г А1С13 при 230—250° в течение 1 час.
Полученный плав выливают в стакан и перемешивают для измель-
чения при затвердевании. Реактив хранят в плотно закупоренной
склянке и растирают в порошок перед использованием. Смесь али-
фатического или ароматического амида с реагентом в перегонной
колбе длительно нагревают на открытом пламени до почти полного
прекращения выделения хлористого водорода. Затем из обугленного
А1С13
С6НаСОХН2 C6H5CONHA1C12 C6H5CN + HCI-|-AIOCI
остатка отгоняют нитрил; выходы 60—90%.
364
бсосн
Под действием реагента Партридж и др. [91 осуществили дегидро-
циклизацию триазабензантрацена (1) в трициклохиназолии (2) и
получили значительно более высокие выходы (45%), чем в присут-
NaAlQli
320° -
45%
ствии палладированного угля (3%). При нагревании (1) с серой при
280° или в кипящем ДМФА выделения сероводорода не наблюдается.
1. Zahn К., О ch w at Р., Ann., 462, 72 (1928),
2. F i е s е г L, F., Peters M. A., J. Am. Chem. Soc., 54, 4347 (1932).
3. For wedding announcement see F i e s e r L. F., Fieser M., J. Am. Chem.
Soc., 55, 3012, footnote 8 (1933).
4. F i e s e r L. F., Peters M. A., J. Am. Chem. Soc., 54, 3742 (1932); F i e-
ser L. F., Fieser M., J. Am. Chem. Soc., 55, 3347 (1933).
5. Fieser L. F., J. Am. Chem. Soc., 53, 3546 (1931).
6. F i e s e r L. F., Hershberg E. B., J. Am. Chem. Soc., 60, 1658 (1938).
7. В r u c e D. B., Sorrie A. J. S., Thomson R, H., J. Chem. Soc.,
1953, 2403.
8. Norr is J. F., Klcmka A. J., J. Am, Chem. Soc., 62, 1432 (1940).
9. Partridge M. W., S I о г a c h S. A., Vipon d H. J., J. Chem. Soc.,
1964, 3670.
365
НАТРИЯ АМАЛЬГАМА РАЗБАВЛЕННАЯ. Полутвердая при
комнатной температуре 1,2%-ная Н. а. р. плавится при 50°;
Н. а. р. более высокой концентрации (за исключением 38—
41?/о-ной)— твердое вещество, которое можно измельчать.
Получение. Методики получения Н. а. р. [1—3], приведенные
в Сб. «Синтезы органических препаратов», по-видимому, менее
удобны и безопасны, чем методика получения 2%-ной Н. а. р. 141.
Натрий (6,9 г, 0,3 моля) помещают в круглодопную трехгорлую
колбу на 250 мл, в боковые горла которой вставлены трубки для
подачи и отвода азота, а в центральное горло — капельная воронка.
Колбу продувают азотом и в воронку помещают 340 г ртути. В колбу
наливают около 10 мл ртути и слегка нагревают пламенем горелки
до начала реакции. Начиная с этого момента, нагревания почти не
требуется, так как реакцию можно поддерживать постепенным добав-
лением ртути из воронки. По окончании реакции еще горячую рас-
плавленную Н. а. р. выливают на твердую поверхность. Пока сере-
бристая Н. а. р. еще теплая и мягкая, ее измельчают в ступке и
помещают в герметичную склянку. Перед использованием можно
определить содержание натрия в Н. а. р., оттитровав навеску в 10 г
0,1 н. серной или соляной кислотой.
Метод также пригоден для получения 3%-ной Н. а. р. В этом
случае смесь начинает затвердевать после прибавления половины
всей ртути; смесь сохраняют в расплавленном состоянии, нагревая
и периодически встряхивая.
Восстановление кетонов. Ксантон восстанавливают до ксант-
гидрола, встряхивая суспензию кетона в 95%-ном этаноле при
60—70° с жидкой Н. а. р., полученной из 9 а натрия и 55 ты
ртути [1].
Трифенилметилнатрий. К раствору трифенил хлор метана в 1,5 л
абсолютного эфира в склянке на 2 л с притертой пробкой добавляют
1%-ную Н. а. р.; на шлиф наносят специальную смазку и заку-
(С8Н5)3СС1 + 2NaHg Na+C-(C0H5)3-hNaCl + 2Hg
0,236 моля 0,5 моля Колич.
порениую склянку взбалтывают на качалке, охлаждая мокрыми
полотенцами [3]. Красное окрашивание за счет трифеи ил метил кар-
баниона становится заметным через несколько минут, а реакция
заканчивается через 3 час. Смесь охлаждают до комнатной темпера-
туры и дают осесть хлористому натрию. Эфирный раствор трифенил-
метилнатрия отделяют от хлористого натрия и ртути фильтрова-
нием под давлением азота.
366
Восстановление а, p-непредельных карбонильных соединений.
Следующий пример заимствован из классического «Руководства»
Э. Фишера L51 — первого лабораторного практикума для студентов
младших курсов.
Растертую в порошок коричную кислоту (10 а) растворяют
в 50 г воды в закрытой склянке на 200 мл и добавляют рассчитанное
количество едкого натра. Затем при взбалтывании постепенно при-
бавляют 2,5%-ную Н. а. р.; при хорошем качестве Н. а. р. доста-
точно 200—250 г. Щелочной раствор декантируют и подкисляют;
осадок кристаллизуют «из не слишком малого количества воды...
NaOI-I, NaH'f, НС!
сбн5сн = снсо3н —------ —C6II3CH3CHZCO2H
Выход очень хороший».
При синтезе Ы-метил-3,4-диоксифенилалаиипа (6) из ванилина
(1) и креатинина (2) одной из стадий является восстановление (3)->
->(4) действием Н. а. р., для чего к суспензии (3) в воде добавляют
прн перемешивании порциями 3%-ную Н. а. р. [61. Твердое веще-
ство растворяется; первоначальная оранжево-красная окраска
раствора медленно исчезает по мере восстановления. При интенсив-
ном перемешивании полное обесцвечивание наступает через 45—
60 мин.
NaHg
72-7
Ва(ОНЦ,
HZSO4
74%
Расщепление по Эмде [71. Расщепление по Эмде состоит в дейст-
вии Н. а. р. на четвертичную галоидную соль в спиртовом или вод-
ном растворе. Четвертичная соль аммония (1а) при расщеплении по
Эмде дает тот же продукт (2), который образуется при расщеплении
по Гофману четвертичного основания (16).
Дальнейшее расщепление (3) по методу Гофмана не приводит
к положительным результатам, в то время как расщепление по
Эмде дает о-метилстирол (4) [8]. Согласно методике Бразена и Хау-
зера 19], для получения гемимеллнтола (6) к суспензии четвертич-
367
(la) X = Cl"
(16) X = OH
(2)
.CH=CH;
CHZN(CH3)3
NaHg
+ (CH3)3N
(4)
иого иодида (5) при перемешивании и нагревании на кипящей водя-
ной бане добавляют большой избыток Н. а. р.
CH2N(CH3)3I-
—Nti Hg
6 г-атом
натрия
87°
85-90%
(5) 0,33 моля
Суспензия в 2 л Н2О
Восстановительное детозилирование. Гроб и Принс [101 к ме-
танольному раствору дитозилата (1) при перемешивании добавляли
Н. а, р. 10 порциями в течение 7 час. Раствор декантировали, выпа-
Н—С—осн3
TsO—С—Н
Н—с—осн3
Н—С—OTs
I
Н —С-----
с!н3
(1)
7,2 г в 200 мл
сн3он
О 4-2%-ная NaHg
400 г
Н —С—ОСН3
I
но—с—н
---> | O + CHaC6H4SO2Xa
91% Н —С—ОСН3
н—С—он
Н—С-------
ii3
(О
ривали и экстракцией остатка горячим ацетоном получали кристал-
лическую натриевую соль ц-толуолсульфокислоты с высоким выхо-
дом. Из ацетонового фильтрата получали чистый (2) в виде сиропо-
образной жидкости.
Альдоиолактоиы >альдозы. Восстановление альдонолактонов в
альдозы действием Н. а. р., открытое Фишером [11],— одна из наи-
более важных реакций в химии углеводов. При тщательном исследо-
вании условий реакции Спербер, Заугг и Сандстром [121 нашли,
368
что наиболее важным условием для получения высоких и вос-
производимых выходов является значение pH 3—3,5. Температура
реакции не должна превышать 15°, а оптимальное количество реа-
гента на 1 моль лактона составляет 2—3 г-атом натрия в виде
2,5%-ной амальгамы, от которой отделяли мелкие частицы, просеи-
вая через сита в 4 и 8 меш. Например, к раствору 11 г крпсталличе-
О
с—
но—i—н
н—с—он
H-ct-OH
О 2,5%-ная NaHg-K H2SO,
200 г
рНЗ-3,5
56%
сн2
СН-0
НО—t—н
н-i—он
н—с—он
сы2он
ского арабонолактона в 200 мл дистиллированной воды в реакцион-
ном сосуде, снабженном pH-метром, добавляют сразу 200 г Н. а. р.
Стандартный раствор серной кислоты приливают из бюретки в тече-
ние первых 2 мин реакции; значение pH определяют через интер-
валы в несколько секунд; восстановление заканчивается через
8 мин. После соответствующей обработки получают чистую кристал-
лическую арабинозу. При замене кристаллического арабонолактона
на сиропообразный арабонолактон или на метиларабонат никаких
изменений в выходе или в чистоте полученного продукта не обна-
ружено.
Оксим->амин. Хохштейн и Райт [13] восстанавливали 5-хлорфур-
фуральдоксим в этаноле добавлением пропорциональных количеств
Н. а. р. и уксусной кислоты при перемешивании в течение 1 час.
Раствор разбавляли и экстрагировали эфиром, после чего амин
+ 2,5%-HbmNaHg
CH=NOH
237 г
+ АсОН
15, 5 г
76%
С1
сн2гшг
7,252»
100 лгл 95%-ного EtOH
выделяли щелочью и экстрагировали эфиром.
1. Гол л ем ан А. Ф., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 234.
2. Б р а з е н У., Хаузер Ч., «Синтезы органических препаратов», ИЛ,
М., 1956, сб. 6, стр. 17.
3. Ренфроу В., мл., Хаузер Ч., «Синтезы органических препаратов»,
ИЛ, АС, 1949, сб. 2, стр. 481.
4. Contributed by Т 1 s h I е г М.
5. F i s с h е г Е., «Anleitung zur DarsteHung organischen Praparate», 8th
edition, p. 39, Braunschweig (1908).
369
6, D e u 1 о f e u V., Guerrero T. J., Org. Syn., Coll. Vol., 3, 586 (1955).
7. E m d e H. Ber., 42, 2590 (1909); Ann., 391, 88 (1912).
8. Emd e H., Kull H., Arch. Pharm., 274, 173 (1936).
9. Brasen W. R., Hauser C. R., Org. Syn,, Coll. Vol., 4, 508 (1963).
10. Grob C. A., Prins D. Л-, Helv. Chim. Acta, 28, 840 (1945).
11. Fischer E., Ber., 22, 2204 (1889); 23, 930 (1890).
12. Spc r b e г N., Zaugg H. E., Sandstro m W. M., J. Am. Chem.
Soc., 69, 915 (1947).
13. Hoclistoin F. A., Wright G. F., J. Am. Chem. Soc, 71, 2257
(1949).
НАТРИЯ АМАЛЬГАМА, 40% НАТРИЯ, ЖИДКАЯ.
Получение 111. В стакан па 600 мл помещают парафин (около
80 г), чтобы получить после плавления слой высотой 1,2 см. Парафин
взвешивают и нагревают па электрической плитке выше темпера-
туры плавления натрия (97,5°). Около 100 г (97 мл) натрия нарезают
на небольшие кусочки, очищают их кратковременным погружением
в спирт и быстро переносят в расплавленный парафин. Вес натрия
определяют путем взвешивания стакана в горячем состоянии. По-
гружают оттянутый конец (диаметр 1 мм) стеклянной трубки диа-
метром 4 мм в расплавленный натрий и закрепляют трубку (по-
скольку возможно разбрызгивание, трубку не рекомендуется дер-
жать рукой). Через трубку капельной пипеткой медленно вводят
ртуть (1,50 г на 1 г натрия), и после добавления каждой порции
смесь осторожно перемешивают, вращая стакан. Теплота, выде-
ляющаяся при амальгамировании, достаточна для того, чтобы Н. а.
оставалась расплавленной при добавлении всей ртути (30—50 мин).
В полутвердый парафин с противоположных сторон вставляют две
стеклянные палочки. После того как парафин охладится и застынет,
палочки вынимают. В результате образуются два отверстия: через
одно выливают Н. а., а второе служит для входа воздуха. При 20—
30° Н. а. переносят в склянку и хранят под слоем лигроина (т. кип.
60—90°). Отбирают Н. а. с помощью шприца.
Из фазовой диаграммы [21 следует, что амальгамы, содержащие
38—41% натрия, при температуре 20—30° находятся в жидком
состоянии. Такую Н. а. обычно используют — в зависимости от
условий — после небольшого нагревания или охлаждения.
Карбоксилирование ароматических углеводородов [1]. В узко-
горлую склянку на 225 мл помещают 16,6 г (0,1 моля) флуорена
(т. пл. 113—114°) и закрывают склянку медицинской резиновой
пробкой. В склянку через пробку вводят две иглы № 20 от меди-
цинского шприца длиной 50 мм и через одну из них в течение
15 мин пропускают ток очищенного азота, после чего ее вынимают.
370
1,2-Диметоксиэтаи очищают или непосредственно перед употреб-
лением перегонкой над кетилом бензофенона или же после пере-
гонки хранят над гидридом кальция; 32 мл этого чистого раство-
рителя вводят в склянку с помощью шприца. После этого иглы
вынимают и склянку вместе с содержимым взвешивают. Затем
снова вводят иглу для отвода вытесняемого азота и с помощью
шприца подают около 80 мл 40 %-ной Н. а. (10-кратный избыток).
Содержание натрия в Н. а. можно определить до и после реак-
ции, добавляя 1 каплю Н. а. к взятой в избытке стандартной
0,1 н. соляной кислоте, избыток которой оттитровывают 0,1 н.
едким натром. Оставшийся шарик ртути сушат и взвешивают.
Реакционную смесь взбалтывают, пока температура не пони-
зится до комнатной, но не более 1 час. Натрийорганическое соеди-
нение растворяется в эфирном слое с темно-бурой окраской.
Н. а. замораживают, помещая склянку в баню со смесью су-
хого льда и ацетона, после чего эфирный слой декантируют во
взвесь сухого льда в диметоксиэтане. Н. а. промывают, приливая
диметоксиэтан, расплавляя и снова замораживая.
Реакционной смеси дают нагреться до комнатной температуры,
приливают воду и двухфазную систему экстрагируют три раза бен-
золом, чтобы удалить нейтральные вещества. При подкислении вод-
ной фазы конц. соляной кислотой в осадок выпадает флуорен-
9-карбоновая кислота в виде белого вещества; т. пл. 224—225°
(15 а, 72 %). После кристаллизации из уксусной кислоты температура
плавления повышается до 230—231°.
1. Contributed by Blatchford J. K.f Shupack S. I., О r c h i n M.,
University of Cincinnati.
2. S i t t i g M., «Sodium, its Manufacture, Properties, and Uses», 54, Reinhold
Publishing Corp. (1956).
НАТРИЯ тре/и-АМИЛАТ, см. 2-Метилбутилат-2 натрия.
НАТРИЯ БИСУЛЬФИТ, NaHSO3. Мол. вес 104,07 (см. также
Натрия метабисульфит).
Альдегиды. Удобный метод очистки альдегидов состоит в пре-
вращении их под действием избытка Н. б. в твердые бисульфитные
соединения, которые для удаления примесей неальдегидного ха-
рактера промывают спиртом или эфиром. Альдегид обычно выделяют
разложением аддукта кислотой, основанием или содой. Примеры:
сиреневый альдегид [1], н-капроновый альдегид [21.
Чтобы превратить формальдегид в диэтиламиноацетонитрил
[3] или бензальдегид 141 в нитрил миндальной кислоты, рекомен-
дуется сначала получить бисульфитные соединения альдегидов.
C6H5CHSO3Na + KCN -+ СгН5СНСЧф KNaSO3
I I
ОН он
(C2H5)2NHH-HOCH2S08Na+KCN (C2H5)2NCH2CN Д- KNaSO3+ Н2О
371
Глиоксаль выделяют в виде бисульфитного соединения
O=CHCH=O-2NaHSO3-2H-,O [5].
Хотя некоторые метилкетоны и образуют аддукты с избытком
реагента, а-метоксифенилацетон достаточно инертен по отношению
к Н. б. и может быть очищен от исходного альдегида промыванием
его раствора в толуоле водным бисульфитом [6].
СН3
/ч /СНО сн-C — no2
II щщыо, I и
I II «-с.н.аЦ । 1|
Х/ ' ОСН3 -юсн3
Fe — HCI
63—71%
(общий)
Циклические кетоны. В результате расширения цикла под
действием диазометана из циклогексанона получают циклогептанон
с выходом до 63% и небольшие количества циклооктанона и окиси
6з%
метиленциклогексана [7]. Цикл о гепта нон легко выделяют из реак-
ционной смеси встряхиванием эфирного раствора с водным Н. б.,
который образует аддукт с циклогептаноном; циклооктанон бисуль-
фитного соединения не образует [8].
Эфиры а-кето карбо но вых кислот. Корнфорз [9] для очистки
этилового эфира пировиноградной кислоты превращал его неболь-
шими порциями в бисульфитное производное. К эфиру (2,2 мл)
в длинной пробирке приливают 3,6 мл насыщенного раствора би-
сульфита, который образует нижний слой. Пробирку встряхивают
при охлаждении в охладительной смеси. Происходит быстрая кри-
сталлизация и через 3 мин добавляют 10 мл этанола. Аддукт отфиль-
тровывают, промывают этанолом и эфиром; выход 3,0 г. Для выде-
ления эфира 16 а аддукта смешивают с 32 мл насыщенного
раствора сульфата магния и обрабатывают 5 мл 40%-ного форма-
лина. После взбалтывания образовавшийся маслянистый слой
экстрагируют эфиром, сушат и перегоняют; выход этилового эфира
пировиноградной кислоты 5,5 а.
Этиловый эфир бензоилмуравьиной кислоты очищают экстрак-
цией из бензольного раствора раствором бисульфита с последующей
регенерацией серной кислотой [10].
Замещеииые орто-хиноны. Для очистки сырого фенантренхи-
нона, полученного окислением технического (90%) фенантрена, от
372
антрахинона и других примесей его обрабатывают 4—6 порциями
горячего 40%-ного раствора бисульфита, затем раствор фильтруют,
охлаждают, отфильтровывают бисульфитное соединение и к суспен-
зии его в воде добавляют соляную кислоту [11].
Физер [12] нашел, что при алкилировании серебряной соли
2-окси-1,4-нафтохинона иодистым метилом в эфире или бензоле
образуется смесь примерно равных частей метиловых эфиров пара-
и орто-хинонов и что эту смесь легко разделить обработкой раство-
ром Н. б. /шра-Хинон растворяется лишь в ничтожных количест-
Нерастворим
в NaHSO3
О
ОСН3
Растворим
в NaHSO3
вах, тогда как орто-изомер хорошо растворим и его можно выделить
из фильтрата добавлением карбоната натрия.
Присоединение к хиноидным системам. Л. Физер и М. Физер
[13] описали методику получения калиевой соли 1,4-нафтохинон-
2-сульфокислоты путем присоединения Н. б. с образованием гидро-
О
[I
il || Н- NaHSO3
Д\/\/ 3S г в
х || 400 мл Н2О
О
32 г
ОН
КйСгг07-Н2304; КС1
79% (общий)
ОН
хинонсульфоната, последующим окислением и высаливанием хло-
ристым калием. Соль выпадает в виде ярко-желтых пластинок моно-
гидрата.
О 1,4-присоединении Н. б. к хиноидной форме 1-иитрозо-2-наф-
тола см. 1,2-Нафтохинон-4-сульфокислоты аммониевая и калиевая
соли.
Меиотти [14] и Палладии [15] нашли, что 2-метил-1,4-нафтохи-
нон под действием Н. б. при комнатной или несколько более высокой
373
температуре образует моноаддукт, из которого метилнафтохинон
можно выделить действием кислоты или основании. Если же реак-
цию проводить при 70—100°, нестабильный продукт присоединения
необратимо превращается в натриевую соль 2-метилиафтогидрохи-
иоп-3-сульфокислоты.
Присоединение к эфиру а, 0-непредельной кислоты. Коуп и Хэн-
кок 116] для очистки сложного эфира (1) перемешивали неочищен-
ный препарат с водным Н. б., образующийся мутный водный
раствор промывали бензолом и из осветленного раствора выделяли
(1) после нейтрализации едким натром.
(С,Н5)2С-ССО.,СоН5
CN
Л)
По-видимому, водорастворимый продукт представляет собой
1,4-аддукт, который в отличие от аддукта с хиноном неспособен
к ароматизации.
I. Аллен Ч., Л е й б и е р Г., «Синтезы органических препаратов», ИЛ,
М., 1953, сб. 4,, стр. 441.
2. Б а х м а и Д ж., «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 295.
3. А л л с и Ч., В ап Алл а и Дж., «Синтезы органических препаратов»,
ИЛ, М., 1953, сб. 4, стр. 249.
4. Org. Expts., 109.
5. Рои з и о А., Во Т., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 106.
6. Хейнцел ьман Р., «Синтезы органических препаратов», ИЛ, М., 1958,
сб. 7, стр. 44.
7. К о h 1 е г Е. Р., Tishler М., Potter Н., Thompson Н. Т.,
J. Am. Chem. Soc., 61, 1059 (1939).
8. М о s е t t i g E., Burger A., J. Am. Chem. Soc., 52, 3456 (1930).
9. Корнфорз Дж., «Синтезы органических препаратов», ИЛ, М., 1953,
сб. 4, стр. 589.
10. Корсон, Д о д ж, Г а р р и с, Хазе и, «Синтезы органических препа-
ратов», ИЛ, М., 1949, сб. I, стр. 537.
11. У э н д л э и д Р., Лалонд Дж., «Синтезы органических препаратов»,
ИЛ, М., 1956, сб. 6, стр. 73.
12. Fieser L. F., J. Am. Chem. Soc., 48, 2922 (1926).
13. Fieser L. I7., Fieser AT, J. Am. Chem. Soc., 57, 491 (1935).
14. M e n о t t i A. R., J. Am. Chem. Soc., 65, 1209 (1943).
15. Пал л а д и и А. В., ДАН СССР, 41, 258 (1943).
16. Коуп А., Хэнкок Э., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 520.
НАТРИЯ БИХРОМАТ, ДИГИДРАТ, Na2Cr2O7 -2Н,О. Мол. вес
298,05. Кислородный эквивалент на 1 моль 3.
Водная H2SO4. Серная кислота ускоряет окисление шестива-
лентным хромом, и в присутствии кислоты восстановленный реагент
находится в растворе в виде сернокислого хрома, как это видно из
уравнения реакции окисления а, у-дихлоргидрина. Согласно мето-
374
дикс Конанта и Квейла [11, к смеси спирта и Н. б. в 225 мл воды
при перемешивании добавляют охлажденную смесь 245 мл конц.
ОН
3C1CH2CHCH2C1 J-Na2Cr2O7-2H2O + 4H2SO4
2,3 моля 1,3 моля 4,3 моля 68-/5%
(30%-ный избыток)
О
[
-> ЗС1СН,ССН2С1 + №nSO4 •-[- Cr2(SO4)3 Щ 9Н,0
серной кислоты и 115 мл воды в течение 7—8 час при охлаждении;
таким путем легко поддерживать температуру 20—35°. В данном
случае берут Н. б. в умеренном избытке по сравнению с теоретиче-
ским количеством. При окислении ь-ментола Сэидборп [21 исполь-
зовал неоправданно большой избыток окислителя. В этом случае
СН3 СН3
1 J
l74! 55°
,Н -'r-Na,Cr,O7-2H.,O 4 HaSO4 ---------
L X “ 0.9 7 83-85’" ! J,
''ОН (130%-ubiii избыток) \/ До
СН (СН3)а (СН3)2
0,53 моля
постепенным добавлением спирта к раствору Н. б. и кислоты темпе-
ратуру реакции доводят до 55°. Для полуокисления и-бутанола и
превращения его в н-бутиловый эфир масляной кислоты в водной
среде Робертсон [3]. использовал теоретическое количество окисли-
теля; полученный низкий выход скорее связан со стадией этерифи-
н-CjHgOH + Na2Cr,O7 2Н,0 + H,SO4 20 ~3-> я-С3Н7СО2С4Нэ-и
3,24 моля 1,07 моля 4,3 моля 41 — 47%
(теорет.)
нации, чем со стадией окисления, Влие [4] получил хинон с выходом
76—81% окислением гидрохинона 1,6-кратным избытком Н. б. (от
теоретического) в разбавленной серной кислоте при 30°.
При исследовании действительно необходимых количеств реа-
гентов Хассей и Бэкер [51 нашли, что выходы кетонов значительно
повышаются при использовании лишь 20%-пого избытка Н. б.
(0,4 моля на 1 моль R..CHO11) и стехиометрического количества сер-
ной кислоты (1,33 моля). Добавлением этих количеств реагентов
к теплой взвеси спирта в воде получают следующие выходы: 4-этил-
ци кл огекс анол ->4-этил циклогекс апо и (90%); ментол -яиентон (94%).
Камм и Мэтьюс [61 окислили /2-нитротолуол в /г-иитробензойиую
кислоту, пользуясь несколько необычной методикой. К смеси нитро-
соединения и Н. б. с 1,5 л воды при перемешивании добавляли
1700 г серной кислоты в течение 30 мин. Затем смесь нагревали до
375
слабого кипения в течение 30 мин. Применение большого избытка
+ №гСг2Ог2Н2О H2SO4
2,3 моля 1 7,2 моля
(130%-ныЙ избыток)
юо
1,7 моля
СО2Н
NO3
кислоты привело к сокращению времени реакции с 40 час до 1 час.
Аналогично окисляли и тринитротолуол, но при медленном добав-
лении И. б., чтобы поддерживать температуру 45—55° [7].
Двухфазное окисление. Для получения холестанона Брюс 18]
к раствору И. б., серной и уксусной кислот в воде при перемешива-
нии и охлаждении приливал раствор холестанола в 500 мл бензола.
После перемешивания при 25—30° в течение 6 час слои разделяли
и водный слои экстрагировали бензолом.
+ МагСггО?‘ гнго > нг$о4
0,7.3 моля
Л400%-ль;й изо'ыток) 90лм
0,13 моля
+ АсОН ________>
83-87%'
50 мл
В 3 00 МЛ Н2О
Джонсон и др. 19] окисляли 2-метпл циклогексанол по аналогич-
ной методике, по без большого избытка окислителя. Водный раствор
указанных реагентов перемешивают и при температуре 10° прили-
вают бензольный раствор спирта в течение 2,5 час, перемешивание
2 моля в 1 л С0Н0
-LNa,Cr3O7-2H3O Щ H2SO4 + СН3СО3Н
0,8 моля 4,3 моля 100 .пл
(20%-ньгй избыток)
1 л Н20
продолжают еще в течение 3 час.
В воде при 250°. Иллюстрацией весьма эффективной методики
окисления, которая была описана в патентной литературе и в даль-
нейшем улучшена Фридманом [10], является окисление 2,3-диметил-
пафталина в нафталин-2,3-дикарбоновую кислоту [11]. В автоклав
загружают указанные реагенты и непрерывно взбалтывают при
250° в течение 18 час. По охлаждении смесь фильтруют, чтобы отде-
лить зеленую гидроокись хрома, и фильтрат подкисляют. Ньюмен
и Боден [12] окислили по этой методике четыре метилбензо-[с]-фе-
нантрена в соответствующие карбоновые кислоты с выходами
376
1Ф28 моля
9 S0°
+ 2Na2Cr.Or2H2O----------
3,14 моля 18 час
(23%-ный избыток)
в 1,8л воды
„ ,CO,Na
/уу
+ 2Cr2O3 + 2NaOH +
+2Н2О
^ZXY ZCO2\.
а
HCl 87-93%
85—95%. Рейтсемаи Аллфин[13] сообщили об окислении по этой же
методике этилбензола в фенилуксусную кислоту, однако их резуль-
таты не получили подтверждения.
В безводной уксусной кислоте. Н. б. растворим в уксусной
кислоте без добавления воды; 25%-ный раствор в горячей уксусной
кислоте можно охладить до 10° без осаждения Н. б. [14]. При полу-
чении циклогексанона [15] растворы спирта и Н. б. в уксусной
+ Na2Cr207-2H20
0,052 моля (теорет.)
в 25 мл СН3СО2Н
60-65%
85%""
0,15 моля
в 1 0 мл СНаСОгН
кислоте смешивают при 15°. Затем температуру повышают до
60°, а через 15 мин она самопроизвольно поднимается
до 65° (25—30 мин), после чего начинает снижаться, и раствор при-
обретает темно-зеленую окраску. При низкой температуре в начале
реакции возможно образование хромата спирта, а также высокий
выход кетона.
Кетон, образующийся при окислении холестерин-5а, 60-дибро-
мида, быстро темнеет под действием света или при нагревании до
70°, но тем не менее он был получен с высоким выходом по следующей
методике [16]. К суспензии дибромида в 2 л уксусной кислоты при
о, 37 мода
+ Ыа2Сг2О/ 2 Н2О
96% '
0,28моля (100%-ный
избыток)
перемешивании при комнатной температуре через воронку прили-
вают раствор IT. б. в 2 л уксусной кислоты, предварительно нагре-
тый до 90°. Вскоре температура реакционной смеси повышается до
377
55—58°; твердое вещество растворяется через 3—5 мин, а еще через
2 мин колбу с помощью подъемного столика помещают в баню со
льдом, прекращают перемешивание и препарату дают закристалли-
зоваться. Затем смесь охлаждают до 25° при перемешивании, при-
ливают 400 мл воды и доводят температуру до 15°; чистый кристал-
лический препарат отфильтровывают и тщательно промывают мета-
нолом. Рекомендуется теоретическое количество 14. б.
Методика ] 17] получения аценафтенхииопа основана па резуль-
татах работы [18], в которой в качестве растворителя применяли
безводную уксусную кислоту, а в качестве катализатора — анстат
0, 65 МОЛЯ
Т Na2Cr2O7-ZH2O Т
1,1 моля (30%-ныЙ
избыток)
Се(ОАс)3
5 г
800 мл АсОН
40°_____.
38-60%
церия. 14. б. прибавляли при перемешивании к смеси остальных
реагентов при 40°, после чего перемешивали при комнатной темпе-
ратуре еще 8 час. а-Дикетон очищали через бисульфитное производ-
ное. Большой разброс выходов показывает, что эта методика нуж-
дается в дальнейшем совершенствовании.
В смеси безводной уксусной кислоты и бензола. Смесь этих
растворителей, использованная одним из авторов книги при изуче-
нии окисления холестерина, обладает следующими преимущест-
вами: бензол лучше растворяет стерины, чем уксусная кислота;
О, 06 5 моля (20
В 225 МЛ с6П6 +
-г 225 МЛ АсОН
Na2Cr2O7- 2 Н2О
225 мл. АсОН (642)
------->
39-40%
меньше вероятности ацетилирования; окисление можно проводить
в гомогенном растворе при температурах от 0° 1191 до 121° [20].
По этой методике из холестерина (а не из сопутствующих веществ)
удалось выделить не менее шести нейтральных продуктов реакции
и, кроме того, две дикарбоновые кислоты. В работе 121] для
получения А4-холестеидиона-3,6 был предложен новый способ.
Смесь 14. б. и уксусной кислоты нагревают, вращая колбу, до пол-
ного растворения, после чего раствор охлаждают до 15°. Раствор
холестерина в бензоле охлаждают до 20”, приливают равный объем
уксусной кислоты, охлаждают раствор до 15э и выливают в раствор
14. б; холестерилхромат выделяется в виде, густой пасты оранжевого
цвета. Колбу погружают в баню со льдом и помещают в холодиль-
378
ник на 40 час. При этом температура быстро снижается до 0°, через
несколько часов растворяется эфир хромовой кислоты. Бурый
раствор выливают в делительную воронку и взбалтывают с 225 мл
петролейного эфира (т. кип. 30—60°). После непродолжительного
стояния смесь расслаивается на верхний красноватый углеводород-
ный слой и меньший по объему очень темный нижний слой, содержа-
щий соединения хрома и уксусную кислоту. Нижний слой сливают,
а верхний взбалтывают с 50 мл воды, после чего дают смеси отсто-
яться. Образовавшийся сравнительно объемный нижний слой сли-
вают и отбрасывают, а затем повторяют эту операцию. Полученный
слабоокрашенный углеводородный слой несколько раз экстраги-
руют щелочью Клайзена для избирательного удаления ендиопа
в виде желтого енолята. Каждую порцию желтого экстракта поме-
щают в делительную воронку, содержащую соляную кислоту, лед
и эфир для экстракции свободного ендиона, который кристалли-
зуется из смеси метанола и эфира в виде желтых пластинок.
Четыре других продукта можно легко получить окислением
холестерина по такой же методике, но при более высокой темпера-
туре. В результате окисления при 60°, выделения кислой фракции
СОД1
(3) при 121’, 3,9’/4
и экстракции из этой фракции кетонов получают кислоту Буте-
нандта (1). Нейтральный продукт реакции, оказавшийся комплек-
сом (1 : 1) эпихолестерина и А4-холестепол-бр-она-3, получен
с выходом 7% при выливании раствора 5,6 г Н. б. в уксусной кис-
лоте, предварительно нагретого до 90°, в раствор 20 а холестерина
в бензоле при 50° [19]. Любопытный продукт (2), «кетон 104», имеет
карбонильную группу, инертную по отношению к реактиву Гринь-
яра и очень устойчивую к окислению. Наилучший препаративный
метод его получения заключается в окислении в таких жестких
условиях, при которых все остальные нейтральные продукты рас-
щепляются [20]. Раствор 150 а Н. б. в уксусной кислоте нагре-
вают до 70° и выливают в раствор холестерина в бензоле, нагре-
тый до температуры кипения, после чего раствор кипятят в тече-
ние 1 час при температуре смеси 100°. Затем «кетон 104» легко выде-
ляют из небольшой по объему нейтральной фракции. Следующий
продукт окисления — дуаннеловую кислоту (3) получают по анало-
гичной методике. Когда первоначальная бурная реакция закончит-
ся, отгоняют растворитель до повышения температуры жидкости
379
до 121°, после чего смесь кипятят в течение 4 час. Из нейтральной
фракции выделяют 1,1% «кетона 104»,а в результате обработки кис-
лотной фракции реактивом Жирара отделяют кетонную фракцию,
из которой получают чистую дуаннеловую кислоту с выходом 3,9%.
В смеси уксусной кислоты с уксусным ангидридом. Применение
такой комбинации растворителей описано в патенте Ривешла и Рая
[22], а также в методике получения флуоренон-2-карбоновой кис-
лоты [23]. К раствору 2-ацетилфлуорепа в уксусной кислоте при
нагревании на кипящей водяной бане и перемешивании в течение
о
о, 24 моля 1,5 моля (370%-ный
’ избыток}
в 650 МА АсОН
45 мин добавляют порциями растертый в порошок Н. б. Затем раст-
вор доводят до слабого кипения на открытом пламени горелки,после
чего приливают в течение 1,5 час 200 мл уксусного ангидрида. Затем
смесь кипятят еще 8 час. Дальнейшая обработка продуктов реакции
состоит в превращении кетокислоты в ее калиевую соль; из неболь-
шой по объему нейтральной фракции получают 1—5 г чистого 2-аце-
ти л флуоренона.
I. Конант, Квейл, «Синтезы органических препаратов», ИЛ, М., 1949,
сб. 1, стр. 211.
2. С э н д б о р и, «Синтезы органических препаратов», ИЛ, М., 1949, сб. 1,
стр. 246.
3. Робертсон, «Синтезы органических препаратов», ИЛ. М., 1949, сб. I,
стр. 147.
4. В л н е, «Синтезы органических препаратов», ИЛ, М., 1949, сб. I, стр. 463.
5. Hussey A. S., Baker К. И., J. Org. Chem., 25, 1434 (I960).
6. Дамм О., Мэтьюс, «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 296.
7. Кларк, Хартма н, «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр. 416.
8. Брюс У., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2, стр. 563.
9. У о р п х о ф Э., Мартин Д., Джонсон У., «Синтезы органических
препаратов», ИЛ, М., 1959, сб. 9, стр. 77.
10. F г 1 е d m a n L., Fishel D. L., Shechter Н., J. Org. Chem., 30,
1453 (1965).
И. Friedman L., Org. Syn., 43, 80 (1963).
12. N e w m a n M. S., Boden H., J. Org. Chem., 26, 1759 (1961).
13. Reitsema R. H., A 1 1 p h i n N. L., J. Org. Chem., 27, 27 (1962).
14. F i e s e г L. F., J. Am. Chem. Soc., 75, 4377 (1953)-
15. Org. Expts., 106.
16. Физер JI., «Синтезы органических препаратов», ИЛ, М., 1956, сб. 7,
стр. 78.
17. Allen С. F. Н., van Allan J. A., Org. Syn., Coll. Vol., 3, I (1955).
18. Пат. ФРГ 73485, 1579.
19. Fieser L. F., J. Am. Chem. Soc., 75, 4377, 4386, 4395 (1953).
20. F i e s e r L. F-, Huang W. Y., Goto Т.» J. Am. Cnem. Soc., 82,
1688 (196QX.
380
21. Физер Л., «Синтезы органических препаратов», ИЛ, М., 1956, сб. 7,
стр. 72.
22. Rievcschl G., Jr., Ray F. E., пат. США 2377040; С. А., 39, 3305
(1945).
23. Ривешил Дж. мл., Рай Ф., «Синтезы органических препаратов»,
ИЛ, М-, 1953, сб. 4, стр. 520.
НАТРИЯ БОРГИДРИД, NaBH4. Мол. вес 37,85.
Историческая справка. Н. б. был открыт Шлезингером и Брау-
ном с сотр. в Чикагском университете в 1943 г. [11. Препаративный
метод состоит во взаимодействии триметилбората и гидрида натрия
при повышенных температурах:
4NaH + B(OCHs)3 NaBH^p3NaOCH3
В 1949 г. Чайкин и Браун [2] сообщили, что этот гидрид в водном
или спиртовом растворе является исключительно эффективным и из-
бирательным реагентом для восстановления альдегидов, кетонов и
хлорангидридов кислот, содержащих также и другие группы, спо-
собные к восстановлению. В 1953 г. Шлезингер, Браун и др. [3]
в серии работ описали детальную методику получения и химические
свойства гидридов щелочных металлов и диборана, а в другой
работе [4] сообщили о применении Н. б. в качестве восстановителя
и источника водорода. В 1950 г. был взят патент и начато промыш-
ленное производство боргидридов. В течение последующих несколь-
ких лет были усовершенствованы методы производства боргидри-
дов, и они нашли новые области применения, например в текстиль-
ной, целлюлозной и бумажной промышленности, в нефтехимии.
Продажный реагент. Н. б. выпускают в виде порошка, а также
в виде гранул, содержащих около 99% реагента. Единственными
активными примесями являются метилат натрия (0,3%) и едкий
натр (0,2%). Если продажный реагент имеет степень чистоты
79—94%, его очищают перекристаллизацией из диглима [51 или из
изопропилами на [6].
Выпускают также стабилизированный водный раствор, содержа-
щий (в среднем) 40% едкого натра и 12% Н. б. (уд. вес 1,4). Этот
реагент не меняет свои свойства при длительном хранении и может
быть использован в продажном виде или после разбавления водой,
метанолом или этиловым спиртом для восстановления карбонильных
соединений. Большинство альдегидов восстанавливаются настолько
быстро, что реакция конденсации под действием щелочи не мешает
восстановлению. Для восстановления а, 0-непредельных альдеги-
дов этот сильно основной реактив не пригоден и его следует разба-
вить до желаемой концентрации и нейтрализовать двуокисью угле-
рода. После нейтрализации реагент следует немедленно использо-
вать, поскольку 1-1. б. не стабилен и при 25° разлагается на 4,5% за
1 час.
Растворители. В противоположность алюмогидриду лития Н. б.
нерастворим в эфире и растворяется в метаноле и этаноле, причем
381
лучше в первом. Но поскольку с метанолом он реагирует с заметной
скоростью, а с этанолом очень медленно, в качестве растворителя
лучше применять этанол 18]. Н. б. еще более стабилен в изопропа-
нолс, и этот растворитель использовали для исследования кинетики
восстановления альдегидов и кетонов [9].
Растворимость Н. б, [71
Растворитель Темпера- тура, °C Раствори- мость, г/1 00 г растворите л 51 Растворитель Темпера- тура, °C Раствори - моеть, г/100s растворителя
Вода 0 25 Тетрагп дрофу - р а н 20 0,1
Вода 60 88,5 1,2-Днметокси- этан 20 0,8
Метанол (реа- гирует) 20 16,4 Диглим 25 5,5
Этанол (реаги- рует медлен- но) 20 4,0 Диметилформа- мид 20 18
Н. б. — восстановитель гораздо более мягкий, чем LiAlH4.
В гидроксилсодержащих растворителях при 25° он быстро восста-
навливает альдегиды и кетоны, оставаясь совершенно инертным по
отношению к другим функциональным группам: эпоксидным,
сложноэфирным, лактонным, карбоксильным и их солям, нитриль-
ным и нитрогруппам [8]. Хлорангидриды быстро восстанавливаются
в диглиме или диоксане.
ню
4R2C=O + Na+BH.r Na ^B (OCHR2)J- —> 4R,CHOII
Восстановление кетонов. Бернштейн с сотр. [10] нашли, что
восстановление 11-кетостероида (1) до 3,20-бис-эти лен кеталя (2)
в присутствии алюмогидрида лития приводит к неудовлетворитель-
ным выходам, небезопасно и т. п. Однако это восстановление можно
осуществить с высоким выходом действием чрезвычайно большого
382
избытка Н. б. в смеси ТГФ с водным едким натром и кипячением
смеси в течение 20 час.
При восстановлении 4 молей кетона 1 молем И. б. за медленной
первой стадией следуют три быстрых реакции. Джонс и Уайз [11]
предложили возможную интерпретацию этого процесса, которую
подтвердили экспериментально.
Сложные эфиры. В обычных условиях Н. б. не восстанавливает
эфиры карбоновых кислот. Например, эфиры 3-кетожелчных кислот
можно восстановить у С3, не затронув при этом сложноэфирнон
функции [12]. Метиловый эфир 4|3-бром-3-кетохолаповой кислоты
(1) восстанавливается Н. -б. в метаноле в смесь За- и 30-бромгидри-
нов (2) ]13]. В таких реакциях восстановления необходимо исполь-
зовать в качестве растворителя спирт, соответствующий сложно-
эфирпой группировке кетокислоты, поскольку Н. б. является эф-
фективным катализатором переэтерификации сложных эфиров.
Сложные эфиры недостаточно реакционноспособны, что позво-
ляет провести их селективное восстановление (типа, приведенного
выше), однако эфиры алифатических, ароматических и гетероцик-
лических кислот могут быть восстановлены в различной степени
действием большого избытка Н. б. в метаноле П4].
Галогениды. Н. б. восстанавливает вторичные и третичные
алкил галогениды, которые способны образовывать сравнительно
стабильные карбониевые ионы, например дифенил хлор метан [15].
Эта реакция ускоряется водой. Так, на 0,5 моля галогенида действо-
вали при 50°65об. % раствора в диглиме, содержащего 4 моля Н. б.
и 1 моль едкого натра, чтобы свести к минимуму гидролиз Н. б.
01-1- + NaBH4
(С6Н5)2СНС1 —- (С6Н5)2СН ------> (С6Ы5)2СН2
94%
Тозилгидразоны. Конденсация С3-, С7-, С17- или С2()- кетостероидов
с /г-толуол сульфонил гидр азином и восстановление образовавшегося
тозил гидразона избытком Н. б. в диоксане дает способ отщепления
карбонильного кислорода (выходы 60—80%) [16]. Этот метод с ус-
пехом используется в химии углеводов (применяется КВН4) [17].
383
Например, при восстановлении тозил гидразона о-глюкозы (5 а)
получают 1-дсзокси-d-сорбит.
Озониды. Н. б. восстанавливает озонид непосредственно в
спирт; при этом нет необходимости выделять озонид. Суза и Блюм
[18] озонировали циклогексен в хлороформе при 0° до исчезнове-
ния в ИК-спектре полосы при 6,2 мк. Затем добавляли этанол для
растворения смолообразного озонида и при энергичном перемеши-
вании приливали раствор Н. б. в 50%-ном водном этаноле. В этом
Оз >
СНС1Э .0°'
НО(СН2)6ОН
63%
случае можно также использовать алюмогидрид лития, но только
в соответствующем растворителе (эфире или углеводороде), и если
желаемый продукт не является оксикислотой. Диапер и Митчелл
[19] озонировали Д10-ундециленовую кислоту в метаноле при 0°,
добавляли раствор озонида по каплям при перемешивании к охлаж-
денному льдом до 0° раствору Н. б. в 50%-ном водном этаноле и
О----о
О„ I I NaBH*
СН2 = СН(СН2)8СО2Н —------| ——
снаон, Сн2 СН(СН2)8СО2Н 91%
—> НОСН2(СН2)8СО2Н
перемешивали в течение 10 час при комнатной температуре. После
отгонки растворителя в вакууме и подкисления получали чистую
твердую 10-оксикаприновую кислоту. 9-Оксипеларгоновая кислота
была получена из линолевой кислоты (выход 42%), олеиновой кис-
лоты (выход 50%), элаидиновой кислоты (выход 74%) [20].
Борфториды диазоиия. Борфторид диазония можно восстано-
вить добавлением твердого Н. б. порциями к охлажденному мета-
нольному раствору или суспензии этой соли [21]:
N^NBF;
I .осн,
СН3с/^/
NaBH4-CH30H
61%
384
По другой методике охлажденный раствор Н. б. в ДМФА добав-
ляют к охлажденному раствору соли диазония в том же раство-
рителе.
Соль Кенига. Соль Кенига (красная), непредельная система-
которой построена на основе пиридинового цикла, при 0° восстанав-
ливается н. б. в водном этаноле до енамина, дающего при кислотном
гидролизе пентадиеиаль (Греве 1221).
GCH3
।
hnc6h5
1N-CH3
СбН5 сГ
CHS сн3
NaEH/y c6hsnch=chch=chch2nc6hs о=снсн=снсн=сн2
9 5% 9 0%
Енамин
Соль' Кенига
Восстановление азидов в амины [23]. Серусодержащий азид
практически не восстанавливается в соответствующий амин обычным
гидрированием. В этом случае, если в молекуле не содержится дру-
гих легко восстанавливающихся групп, для восстановления можно
использовать И. б. в изопропаноле. Эту методику можно применять
и к соединениям, неустойчивым в кислой среде.
При восстановлении ароматических азидов [24] 0,1 моля азида
кипятят с 0,072 моля Н. б. в 100 мл изопропанола в течение 2 час.
В случае алифатических азидов, по-видимом у, следует брать боль-
ший избыток гидрида металла и увеличить продолжительность
реакции, Азид 1 (2,84 .имоля) и И. б. (6,61 лшоля) в 10 мл изопропа-
нола кипятят в течение 16 час. Остаток после отгонки растворителя
экстрагируют хлористым метиленом, промывают водой и высуши-
вают, растворитель отгоняют и получают амин II с выходом
более 90%.
. Эта методика менее опасна и значительно проще, чем использо-
вание алюмогидрида лития.
Гидрирование. Об использовании Н. б. для получения активных
катализаторов и водорода in situ см. Платиновые катализаторы (Ка-
тализатор Брауна и Брауна).
Применение в. химии целлюлозы и сахаров 125].
_Натрия боргидрид — хлористый алюминий [26]. Смесь свеже-
приготовленных растворов Н. б. и А1С1а в диглиме в отношении
13 заказ X 1096
385
3 : 1 образует прозрачный раствор, обладающий сильными восста-
новительными свойствами [27]. Порядок смешивания не имеет зна-
чения, по обычно раствор А1С13 приливают к раствору Н. б. и вос-
станавливаемого соединения. Смешанный гидрид, как и Н. б., вос-
станавливает альдегиды, кетоны и хлорангидриды до спиртов, а
кроме того — сложные эфиры, лактоны и кислоты (но не натриевые
соли кислот). Поскольку при восстановлении кислоты требуется
еще один эквивалент гидрида, который вступает в реакцию с водо-
родом кислоты, целесообразнее восстанавливать сложный эфир, а не
кислоту. Эпоксиды легко восстанавливаются при 25°, но ангидриды
при 25й реагируют медленно и для них требуется температура 75°.
Нитрилы восстанавливаются до первичных аминов. Незамещенные
амиды образуют натриевые соли и не восстанавливаются; дизаме-
щенные амиды восстанавливаются легко:
К’аВЩ-АЮЦ
CfiHBCON(C,H5), - ---C6H5CH2N(C,H5),
N-Окись пиридина гладко восстанавливается до пиридина при 25°,
но дальнейшее восстановление требует более высокой температуры.
Нитросоединения и ароматические галогениды не восстанавли-
ваются.
Н. б. не пригоден для восстановления соединений, имеющих
реакционноспособную двойную связь, так как они реагируют с об-
разованием алкилборанов [28]. Поэтому этот реагент можно исполь-
зовать для гидроборирования, однако он уступает диборану,
поскольку при этом используются только три из четырех атомов
водорода в Н. б.; четвертый атом идет на образование гидрида
алюминия.
9^C = c/+?,NaBH4+AiCl3--->3 (>СН-С/) B + 3NaCl-yAlH3
NaBH4 — BF3. Петтит и Пиатак [29] нашли, что эта комбинация
эффективно восстанавливает лактоны до циклических простых
эфиров.
При восстановлении у-лактопа дигидроабиетиновой кислоты (1)
в окись (2) наилучшие результаты были получены при использовании
30 молей эфирата трехфтористого бора и 2 молей Н. б. на 1 моль
лактона в смеси диглима с ТГФ. Так как LiAlH4 и LiBH4 давали
несколько менее удовлетворительные результаты, чем Н. б., комби-
нация И. б. — BF3 была использована для сравнения поведения
трех изомерных бутилхоленатов. При восстановлении по описанной
выше методике из mpem-бутилевого сложного (3) был получен
mpe/n-бутиловый простой эфир (4) с выходом 76%. Выход при реак-
ции с втор-бутиловым сложным эфиром составлял 41%, а с «-бути-
ловым сложным эфиром — 7% . Это восстановление сложного трет-
бутилового эфира является удобным путем получения простого
mpem-бутилового эфира, <
336
NaBH4 — Pd — С. Ароматические нитросоединения обычно не
восстанавливаются Н. б,, но их можно гладко восстановить в воде
или метаноле действием Н. б. и 10% -ным Pd на угле в качестве ката-
лизатора [30]. Азобензол восстанавливается по этой методике до
гидразобензола, а не до анилина.
ROH^RX. Н. б. в смеси с галогеном превращает спирт или
эфир в алкил галогенид [311.
3ROH + 2X,-!-NaBH4 —> 3RX + Ы3ВО3-|- NaX % 2Н,
3ROR + 2ХЭ +NaBH4 3R X + B(0R)3---ХаХ -|-2Н,
Восстановление нитроолефинов. Хасспер и Хиткок [321 нашли,
что 6-нитрохолестерилацетат можно восстановить Н. б. в этаноле до
предельного 6а-нитро-5а-стсроида.
Восстановление алкнлгалогенидов. ЫА1Н4 восстанавливает пер-
вичные и вторичные галогениды или тозилаты до углеводородов,
однако в случае третичного галогенида продуктом реакции является
преимущественно олефин. В 65%-ном водном диглиме алкилгало-
гениды и тозилаты, даже третичные, восстанавливаются Н. б. с хо-
рошими или высокими выходами [33]. Когда образуется сравни-
тельно устойчивый карбониевый ион, не вступающий в реакцию
отщепления, получают высокие выходы. Выходы остаются удовлет--
387
верительными даже в случае отщепления. В отсутствие воды реак-
ция протекает медленно. Гомогенный раствор для проведения кине-
тических исследований получают из 80 об. % водного диглима,
содержащего 1,80 моля реагента.
I. Schlesinger И. J., Brown Н. С,, пат. США 2461661, 2534533,
2683721.
2. Chaikin S. W., Brown W. G., J. Am. Chem. Soc., 71, 122 (1949).
3. S ch lesi nger H. I., Brown H. C., et al., J. Am. Chem. Soc., 75,
186, 191, 192, 199, 205 (1953).
4. Sch lesinger FI. I-, Brown H. C. et al., J. Am. Chem. Soc., 75,
215 (1953).
5. Brown IT. C. ct al., J. Am. Chem. Soc., 77, 6209 (1955); Tetrahedron, 1,
214 (1957).
6. Cocker J. D,, II a I s a 1 I T. G., J. Chem. Soc., 1957, 3441.
7. Technical Bulletin No. 550 (1958).
8. Reviews: Schenker E., Angew. Chem., 73, 81 (1961); Brown H. C.,
«Hydroboration», 242 (1963).
9. В г о w n H. C., Ichikawa K., J. Am. Chem. Soc., 84, 373 (1962).
10. A 1 1 e n W. S., Bernstein S., L i t t e 1 1 R., J. Am. Chem. Soc., 76,
6116 (1954).
II. Jones W, M., AV j s e H. E., Jr., J. Am. Chem. Soc., 84, 997 (1962).
12. Hey шапп H., Fieser L. F., J. Am. Chem. Soc., 73,5252 (1951).
13. Fieser L. F., E t t о r r e R., J. Am. Chem. Soc., 75, 1700 (1953),
14. Bro w и M, S-, R a p о port FE, J, Org. Chem., 28, 3261 (1963).
15. Brown FI. C., Bell H. M., J. Org. Chem., 27, 1928 (1962).
16. C a g I i о t о L., G r a s s e 1 1 i P., Chem. Ind., 1964, 153.
17. В e 1 d e r A. N. de, Weigel FI., Chem. Ind., 1964, 1689.
18. Sousa J. А., Bluh in A. L., J. Org. Chern., 25, 108 (1960).
19. D i a p e r D. G. M., Mitchell D. L., Can. J. Chem., 38, 1976 (1960).
20. В e n t о n F. L., К i ess A. A., J. Org. Chem., 25, 470 (1960).
21. Hendrickson J. B., J. Am. Chern. Soc., 83, 1251 (1961).
22. G r e w c R., Boni n W. von, Chem, Ber., 94, 234 (1961).
23. Contributed by Christen sen J. E., Stanford Research Inst.
24. S m i t h P. A. S., II a I I J. FI., Kan R. O., J. Am. Chem. Soc., 84,
485 (1962).
25. «The Borohydrides in Cellulose and Sugar Chemistry», Metal Hydrides Inc.,
1959,
26. Rerick M. N., «The .Metal Hydrides», Metal Hydrides Inc. (Ventron).
27. В г о w п H. C., Sub ba Rao В. C., J. Arm Chem. Soc., 78, 2582 (1956).
28. Brow u IT. C., S u b b a Rao В. C., J. Am. Chem. Soc., 78, 5694 (1956).
29. P c t t i t G. R., P i a t a k D. M., J. Org. Chem., 27, 2127 (1962).
30. Neilson T., Wood IT. C. S., W v I i e A. G., J. Chem. Soc., 1962,
371.
31. L о n g N. H., Frecguard G. F., Chem. Ind., 1965, 223.
32. H ass ner A., FI c a t h с о c k C., J. Org. Chem., 29, 1350 (1964).
33. Bell Fl. M., Brown H. C., J. Am. Chem. Soc., 88, 1473 (1966).
НАТРИЯ БРОМАТ (или ИОДАТ), NaBrO3 (NaIO3). Мол. вес
150,91 (197,91). Растворимость в 100 г воды NaBrO3 27,5 г при 0°,
90,9 г при 100°; NalO;i 2,5 г при 0°, 24 г при 100°.
Дэн [11 нашел, что Н. б. (или иодат) — прекрасный реагент для
проведения некоторых специфических окислительных реакций,
например, окисление бензоина в бензил в таких условиях, когда
дикетон перегруппировывается в бензиловую кислоту.
388
NaCN NaBrOs-NaOH-Вода
2CgHsCHO ~-----C0H5CHCOCSH5--------------------->
4 7 моля Этиловый I 84—90%, считая на
’ ' сиирг— кц бензальдегид
вода ''7П
(СбН&)2ССОчН
I
он
В методике [2] синтеза бензиловой кислоты в больших количест-
вах бензоин, полученный из бензальдегида [3J, промывают и, не
высушивая, прибавляют ио частям при перемешивании к раствору
0,76 моля NaBrO:} (8%-ный избыток) и 12,5 моля едкого натра
в 880 мл воды. Смесь нагревают на кипящей водяной бане при пере-
мешивании, добавляя еще воды (800 мл) по мере необходимости до
такого разведения, чтобы взятая проба полностью растворялась
в воде (5—6 тос). Смесь разбавляют водой, фильтруют для удаления
небольшой примеси бензгидрола и подкисляют. Выпадающая в оса-
док бензиловая кислота получается сразу чистой; т. пл, 149—150°.
Сравнивая различные реагенты для окисления пирогаллола
в пурпурогаллин, Перкин и Стивен [4.1 пришли к выводу, что лучшим
является нитрит натрия в уксусной кислоте, однако полученный
при этом выход составлял лишь 30—40%, Эвансу и Дэну 11] удалось
удвоить этот выход применением йодата натрия, К раствору 10 г
пирогаллола в небольшом количестве воды при перемешивании
постепенно добавляли холодный раствор 8 г йодата натрия в 100 мл
воды, В результате гладкого взаимодействия краситель был полу-
чен с хорошим выходом. Структура пурпурогаллина была неиз-
вестна до 1945 г. Позднее Хорнер [5], пользуясь специальными
71% (общий)*5
приемами улавливания, установил, что первичным продуктом
окисления является З-окси-1,2-бснзохинон и предложил обоснован-
ный механизм его превращения в пурпурогаллин [6, 7].
1. Е v а п s Т. W., D е h п W. М., J. Am, Chem. Soc., 52, 3649 (1930),
2. Баллард, Дэп, «Синтезы, органических препаратов», ИЛ, М., 1949,
сб. 1, стр_ 88.
3. A dams R-, М arvel С. S., Org. Syn., Coll. Vol., 1, 94 (1941).
4. Perkin A- G., Steven A. B., J. Chem. Soc., 83, 194 (1903).
5. Horner L., Gowecke S., Chem. Ber., 94, 1267 (1961).
6. H о r n e r L., W e b e г К. H., Durckheimer W., Chem. Ber.,
94, 2881 (1961).
7. For a formulation, see Fieser L. F., Fieser M., «Topics in Qrganic
Chemistry», Reinhold Publishing Corp., 1963, p. 567—568.
НАТРИЯ ВИСМУТАТ, NaBiO3. Мол. вес 280,000.
Ригби 111 нашел, что этот реагент по окислительному действию
до некоторой степени сходен с тетраацетатом свинца и иодной
кислотой, Н, в. применяют в смеси с фосфорной кислотой обычно
389
в водном спирте, водном диоксане или водной уксусной кислоте;
реагент восстанавливается до нерастворимого фосфата висмута.
Расход реагента определяется по исчезновению оранжево-желтой
окраски реакционной смеси. Под действием Н. в. вицинальные гли-
коли расщепляются до карбонильных соединений; образующийся
при этом альдегид далее реагентом не окисляется. При количест-
венном сравнении окисления цис- и транс-циклогександиолов-1,2
в адипиновый ди альдегид Ригби не обнаружил заметного различия
в скорости реакции. Были осуществлены также следующие реакции
окисления;
RCH(OH)CO2H RCHO
R2C(OH)CO2H R2C = O
RCOCH(OH)R RCO2H + O = CHR
HCO3H CO,
Штолль и сотр. [2] разработали методы получения глутарового
диальдегида из циклопентадиена и циклопентанона, причем в обоих
он н
NaBiO,
—— 3> о=снсн2сн2сн2сн=о
случаях одной из стадий является окисление транс-циклопентан-
ди ола-1,2 под действием Н. в. К раствору 3 г диола в 10 мл уксусной
кислоты и 8 мл воды при перемешивании добавляют 12 г NaBiO3 и
С1НОН
снон
(1)
+ СН2О
СН2ОН
(4)
СН2ОН
(2)
СО2Н
A-н
+ сн2о
СНз
СНОН
(5)
о
'хХ^.(без СН2О)
<jJH2OH
снон
(3)
- + СН2О
СН3
СО
ДпОН
(неизмененный)
(6)
390
14 мл 33%-ной фосфорной кислоты отдельными порциями так,
чтобы температура поддерживалась при 30°. Затем смесь фильтруют
для отделения фосфата висмута и непрореагировавшего реагента,
после чего диальдегид выделяют в виде бис-п-нитрофенил гидразона.
Брукс и Норимберски [3] использовали Н. в. для аналитического
определения боковых цепей типов (1)—(5) в адренокортикостероид-
ных гормонах. При этом не затрагивается только 21-дезоксикетол
(6). За ходом окисления (1)—(5) можно следить, определяя отщеп-
ляющийся формальдегид, если он образуется, и проводя количест-
венный анализ 17-кетостероидов. При анализе на суммарное содер-
жание 17а-оксипрегианов, например в моче, прежде чем окислять
смесь восстанавливают боргидридом натрия для превращения не-
реакционноспособных кеталей (6) в а-гликоли (5), которые далее
окисляются в 17-кетоны.
1. Rigby W., J. Chem. Soc., 1950,1907.
2. S t о И A., Lindenmann A., J u c k e r E., Helv. Chim. Acta, 36
268 (1953).
3. Brooks C. J. W., Norymberski J. K., Biochem. J., 55, 371 (1953).
НАТРИЯ ГИДРАЗИД, H2NNHNa [11. Мол. вес 54,04; желтое
кристаллическое вещество, взрывается при температуре выше 70°
или в присутствии кислорода. Реагент получают взаимодействием
амида натрия с безводным гидразином в атмосфере азота при 0°,
в большинстве случаев в виде суспензии в эфире, диизопропиловом
эфире, бензоле или в смеси этих растворителей [2].
NaNH2-pH2NNH2 —> H,NNHNa + NH3
Восстановление [3]. В присутствии гидразина этот реагент
восстанавливает многие непредельные и ароматические углеводо-
роды. Примеры:
транс-Стильбен > Дибевзил (выход 92%)
Д1,з,5,7_циклооктатетраен ->. ДЬз.Б.Циклооктатрпен (85%)
Аценафтилен > Аценафтен (81%)
Антрацен > 9,10-Дигпдроантрацен (90%)
Восстановительное расщепление олефинов [4]. В кипящем
эфире в атмосфере азота с помощью Н. г. осуществляют восстанови-
тельное расщепление олефинов типа (1). Как правило, выходы про-
дуктов реакции (3), не содержащих азот, выше 80%. Изолированная
двойная связь при этом не затрагивается.
СеН5х /R' 7Н С6НК Na yR'
>С = С< + H2NN< >СН2-% >NN = C<
RZ XR" xNa RZ Hz XR"
(1) (2) (3) (4)
Реакция с сопряженными диенами [5]. H. г. в присутствии
гидразина реагирует с изопреном с образованием азина (1) и пира-
зола (2).
391
сн3 , сн3 С113
„ ' 35° । I
2 СН2=С-СН=СНг + NANHNH2 > ch3c=chch=n-n=chch=cch5
(1) 62%
+
н3с
н3с
н
(2) 1, 6%
Нуклеофильная атака пиридинового цикла [61. Н. г. н гидразине
реагирует с пиридином, изохпполипом и 4-метил хи полином, обра-
зуя а-гидразинзамещенные производные с выходами 25—76%.
NaXHNH;
п
xnhnh2
N
Реакция с нитрилами [71. Этот реагент взаимодействует с нит-
рилом при 0° и диэтиловом или диизопропиловом эфире с образова-
нием аддукта, при гидролизе которого образуется иминогидразид —
соединение, трудно доступное при получении другими методами; \
NaNI-INH,, Н3О ZNH2 -NH
RC^N ........- ~ —> RC.< RC<f
^NNH2 xNIINH2
1..Review: К a u f f тп a n n T., Angew. Chem., Internal. Ed., 3, 342 (1964)...
2. Kaul imann T., Kosel C., W о 1 I D., Chem. Ber., 95, 1540 (1962).
3. Rauf [ma nn T., Kosel C., Schoeneck W-, Chem. Ber., 96, 999
(1963).
4. Kauflmann T., Hen hi.er H., Kosel C., Schulz J., Weber
R., Angew. Chem., Internat. Ed., 1, 456 (1962).
5. Kauflmann T., M u 11 e r H., Chem. Ber., 96, 2206 (1963).
6. Rauff ma n n T., Hanse n -J., Rosel C., Schoeneck W.,
Ann., 656, 103 (1962).
7. Kauifmann T., Sp aude S., Wolf D., Chem. .Ber., 97, 3436 (1964).
НАТРИЯ ГИДРИД, NaH. Мол. вес 24,01. Реактив поступает
в продажу в виде гранул и в виде серого порошка (средняя величина
частиц 25 мк), тонко диспергированного в белом масле [1]. Диспер-
сия содержит 50—55 вес. % Н. г.; точная концентрация указана на
этикетке для каждой партии.-Масло легко смешивается с углеводо-
родами и простыми эфирами и его можно разбавить гексаном, эфи-
ром, ТГФ, толуолом и др., причем активность реагента в резуль-
тате разбавления не снижается. Н. г. нерастворим во всех инертных
органических растворителях, а также в жидком аммиаке. Он бур по
реагирует с* водой. Реакция - Н. г.; диспергированного в масле,
с водой протекает бурно, но не представляет такой опасности, как
392*
реакция сухого препарата; самовоспламенение водорода происходит
редко.
Сложноэфнрная конденсация. При самоконденсации этилаце-
тата 1 элг; II. г. расходуется на образование натриевого производ-
ного этилацетата и еще 1 же на реакцию с отщепляющимся спиртом.
Толуол О Na+
I ’ШС 7fi° I
2СН3СО.,С,Н5 т..2ХаН ----> СН3С - СНСО,С.,Н5 4- С,Н5ОНа ЩН2
1 моль 1 моль 88%
В мягких условиях, создающихся при использовании дисперсии
в масле, как это указано в методике [11, образующийся этилат нат-
рия, по-видимому, ие принимает участия в сложноэфирной кон-
денсации. Выход высокий, и реакция протекает быстро, поскольку
при этом нет необходимости удалять этанол, как при использовании
в качестве конденсирующего агента этилата натрия. Более того,
реакцию можно контролировать по количеству выделяющегося
водорода.
При изучении синтеза нингидрина конденсацией диметилфта-
лата с этилацетатом один из авторов книги разработал безопасную
методику получения хороню измельченного натрия [21. Еще более
безопасный способ предложили Груэн и Норкросс [3], которые
вместо измельченного натрия применили дисперсию Н. г. При этом
они добились большего выхода (36—72%), чем при использовании
измельченного натрия. Основания способствую!' переэтерификации
с образованием енолята метилового эфира.
асо2сн3
СО2СН3
+ СН3СОгС2Н= Я №Н __________>
54-83%
Большой Избыток 0,17 моля
0,122М0ЛЯ
Робертсу и Мак-Эльвену [4] не удалось осуществить самокон-
денсацию этилового эфира изовалериановой кислоты в присутствии
этилата натрия даже при отгонке этанола, однако Суомер и
Хаузер [5], используя Н. г., получили этот пространственно зат-
С11з
\снсн2со3сан5+CH3CO3C3H5
сн I
3 СН(СН3)3
рудненный (3-кетоэфир.
' \сНСНаССНСОоС,Н5
ci£ I
13 сн<
ХСН3
393
Конденсации по Штоббе. Джонсон и Дауб [6] показали, что
mpem-бутилат калия лучше этилата натрия в качестве основания
при конденсации по Штоббе, но еще лучшим основанием является
Н. г. При перемешивании смеси бензофенона, диэтилового эфира
янтарной кислоты и Н. г. даже при 87° реакция не пойдет, пока не
будет добавлено небольшое количество этанола (10 капель); после
этого она легко протекает даже при комнатной температуре.
CH3CO2C3HS CH3CO2Na
| 25°, 8 час |
(С6Н5)2СО -|-CH2CO3C2H54-2NaH — —> (СйН5)2С = ССО3С3Н54-С2Н5ОХа
9 7%
0.05 моля 0,15 моля 0,1 моля
Через 5 час к смеси для облегчения перемешивания добавляют эфир;
выделение водорода прекращается примерно через 8 час. Хинклей
и др. [1], использовавшие Н. г., тонко диспергированный в гексане,
при 40°, нашли, что прибавлять этанол для инициирования реак-
ции не обязательно. Они получили продукт конденсации в чистом
виде с выходом 95,7% всего лишь за 1 час.
Конденсация Дикмана. Для циклизации диэтилового эфира
адипиновой кислоты в 2-к ар бэтокси циклопентанон использовали
натрий, амид и этилат натрия. Пинкни [7] проводил реакцию с нат-
рием при ПО—115° в течение 7 час и получил кетоэфир с выходом
74—81 % . Позднее Хинклей и др. [1] приливали сложный эфир к гу-
стой дисперсии Н. г. в толуоле при 45° и за 1,5 час получили препа-
рат с выходом 65—80%.
сн2со2с2н5 А согсгн5
4- 2 ман Голуд71- 45°> \ | + СгН5ОЫа + 2 Нг
сн2 65-80% 1----1
СН2СО2СгН5
Ацилирование кетоиов. При алкилировании нитрила малоновой
кислоты в этаноле или бензоле действием этилата натрия в ка-
честве основания выход достигает лишь 70%, причем образование
имидоэфиров является основной побочной реакцией. Блумфилд [81
использовал ненуклеофильное основание — Н. г. в ДМСО (сравни-
тельно ненуклеофильный растворитель, способный растворять про-
межуточно образующиеся соли). Используя это сочетание, ему уда-
лось получить диметилмалононитрил с выходом 60%. Затем он ис-
следовал ацилирование кетона (1) сложным эфиром (2) по той же
394
Методике, взяв исходные реагенты в указанных в уравнении соотно-
шениях. Ди кетон (4) был получен с высоким выходом. При исполь-
зовании только 1 моля Н. г. выход снижался. Если реакцию прово-
дить в толуоле, действуя метилатом натрия в качестве основания, то
выход (4) составит только 36 %.
При фор мили ровании циклогексанона Эйнсворз [9] к смеси дис-
персии Н. г., 2 л эфира и 20 мл этанола в течение 1 час добавлял при
перемешивании и охлаждении циклогексанон и этилформиат, пере-
мешивал еще 6 час и оставлял смесь на ночь. Выход оказался таким
-L НСОЙС2Н5-Д NaH
1 моль
Ч/Чнон
же, как и при использовании натрия (нарезанного в виде кубиков
объемом I см:') в качестве основания. При формилировании холесте-
иона Вейзенборн, Реми и Джекобс [10] добавляли твердый Н. г.
сн3
+ нсо2с2н5
20 мл
20 г
к раствору кетона и этилформиата в 250 мл бензола. Реакция начи-
нается сразу же и сопровождается выделением соли енолята. Через
2 суток непрореагировавший Н. г. разлагают 20 мл метанола и
100 мл эфира, и затем соль отфильтровывают. Для выделения сво-
бодного енола суспензию соли взбалтывают с соляной кислотой, а
затем кристаллизуют енол. Аналогично формилировали и тестосте-
рон (83%).
Алкилирование и нитрование малоновых эфиров. Заугг [11]
применил Н. г. для превращения н-бутилмалонового эфира в его
натриевое производное. При этом он нашел, что скорость алкили-
рования этого вещества ц-бутилбромидом в ДМФА в 1000 раз выше,
чем в бензоле. Эммонс и Фримен [12] разработали методику нитро-
вания соединений с активной метиленовой группой нитратом ацетон-
циангидрина путем предварительного превращения их в натриевые
производные действием Н. г. Продукт реакции (3) способствует от-
щеплению иона натрия от (2), но даже при трехкратном избытке
натриймалонового эфира выход достигнет только 45%.
СНДСОаС2Н5)2 —NaCH(CO2C2H5)2 O2NCH]CO2C2H5)24-
(2) 4 5 %
-E(CH3)BCO + NaCN
395
Конденсация по Дарзану. Хинклей [1J нашел, что конденсация
а-галогснзамещениых сложных эфиров с кетонами под действием
дисперсии Н. г. проходит гладко и не сопровождается обычными
побочными реакциями.
+ С1СН2С О2 С2Н5 + NaH •
Гексан
О,
---------
88%
СО2СгН5
СН
Внутримолекулярное алкилирование кетона. Ключевой стадией
в синтезе твистана по Уитлоку [13] является циклизация кетото-
зилата (1) в присутствии твердого Н. г. в ДМФА.
(CH2)2OMs
(1) d. или I
NaH—ДМФА (60°)
90%
(2) d- или I
Дегидрогалогенирование. Ньюмен и Меррилл Ц4[ разработали
методику дегидрогалогенирования этилового эфира дибромкорнчной
кислоты. К раствору сложного эфира в бензоле прибавляют Н. г.
и смесь кипятят, время от времени добавляя абсолютный этанол
порциями по 2 мл. Поскольку Н. г. не вступает в реакцию с дибро-
мидом в кипящем бензоле, это показывает, что активным реагентом
является этилат натрия, который образуется при взаимодействии
Кипячение 3 час, опыление,
двухкратная кристаллизация
C(iH5CHBrCHBrCOoC,H5-i-21\iaN -]-СвН5ОН —---— --------—
0.65 моля, 300 мл бензола 1,33 моля 51 %
с8н5с = ССОоН
этанола с Н. г. и регенерируется в результате реакции C3H5ONa+
+ НВг^СЛ-15ОН4-ХаВг. При этом следует отметить, что под дейст;
вием этилата натрия в этаноле этиловый эфир дибромкорнчной кис-
лоты превращается главным образом в этиловый эфир р-этоксико-
ричной кислоты. Методику Ньюмена используют и для дегидробро-
мирования (1), который оказался инертным по отношению к кипяче-
нию в 2,4,6-колл иди не [15]. Под действием Н. г. в кипящем бензоле
в атмосфере азота амиды а-галогенкарбоиовых кислот (3) превра-
щаются в азиридиноиы (4) [16].*
Синтез циклопропанов. А'Уак-Кой 117] нашел, что а-хлор-(или
бром-)замещенные сложные эфиры конденсируются с а, Р-пей редел ь-
* Эта работа опровергнута Шиханом [S he е h а п J. G., Frankenteld J. W.,
J. Am. Chem. Soc., 83, 4792 (1961)]. — Прим, ped-,
396
ними эфирами в бензоле или эфире под действием И. г., дисперги-
рованного в масле, с образованием эфиров циклопр опа иди кар боно-
вых кислот (в твердом состоянии Н. г. неактивен). Согласно общей
методике, смесь обоих компонентов (по 1 молю каждого) с 1 молем
(1)
О
[I
с
/ \
со \nhc6h5
R7
NaH-C6H3-
КаН-СгН5ОН
60%
О
В)
0)
дисперсии Н. г. перемешивают с бензолом или толуолом или про-
стым эфиром при 20—30°. В этих условиях всегда образуется глав-
ным образом /{ПС-изомер. При использовании растворителя с высо-
кой диэлектрической постоянной, например ДМФА или смеси гскса-
метилфосфамида с бензолом, преобладают более устойчивые транс-
изомер ы.
Н КаН
сн2=ссогсн_з + С1-С-СОгСН3_—
сн3 СН3 1
сн3
СО2СН3
93% цис, 7% тиране
Алкилирование аминов и амидов. Меджоо и Петерсон [18]
заместили два атома водорода в 2-амииопиридипс на две различные
группы, как это видно из уравнения. Амин (1) или (3) превращают
в натриевое производное, на которое действуют замещенным ал кил-
галогенидом.
Диоксан
СН;,
CHaONCH2CH2Cl
СНз
N N
4 Н
(4)
о-СН3ОСеН ,СЫ2Вг
(3)
397
А сн»
’ 11 /CHoCH,NrOCH3
N XN\
14 HH2CGH4OCH3-o
(5)
Фоне [19] нашел, что при алкилировании некоторых N-заме-
щепных амидов в качестве основания удобнее применять Н. г., чем
натрий, Марвел и Мойер.[20] использовали эту методику для алки-
Н v + СНз
| NoH i^a CH3I I
C6HaNCOCH3 —CcH5NCOCH3---------> CcH5NCOCH3
89%
лирования е-капролактама. К смеси лактама и Н, г. в толуоле после
кипячения в атмосфере азота в течение 10 час при перемешивании
добавляют раствор н-бутилбромида в ксилоле и кипятят еще 4 час.
CNH + NaH + я-С^Вг ------------->
' 7 0%
0,1 моля 0,1 моля 0,2 МОЛЯ
В 200 МЛ толуола В 50 МЛ КСИЛОЛЯ
С^О
Бензилтозилаты. К бензилтозплатам нельзя применять обычные
препаративные методы, поскольку эти сложные эфиры очень реак-
ционноспособны и нестойки. По методике, предложенной Кочи и
Хэммондом [211, сначала готовят суспензию бензилата натрия путем
Кипячение в эфире 0,097 моля TsCI
15 час —10а
С6Н5СН.,ОНШ МаН -----------------> CeH5CHaONa ------------—'
8 0%
0,04 7 моля 0,10 моля
—> CsHsCH2OTs
перемешивания эфирного раствора бензилового спирта с Н. г. без
доступа влаги и воздуха; оказалось, что удобно пользоваться бу-
сами и лопастной мешалкой для измельчения Н. г, in situ. Суспен-
зию охлаждают до — 10е, приливают эфирный раствор н-толуол-
сульфохлорида и смесь перемешивают при охлаждении до —10°
в течение 2 час, а затем еще 1 час при 25°. Раствор фильтруют и охла-
ждают в бане с сухим льдом; при этом тозил ат выпадает в виде мел-
ких белых игл.
В бензиловом спирте (1) имеется прочная водородная связь между
гидроксильной группой и нитрогруппой, и Тарбеллу и др, [22]
осуществить тознлирование в пиридине не удалось. Однако приме-
нение Н. г. в этом случае оказалось исключительно успешным. Ис-
398
СН3ОН
Д^/NOs
II I ""I"
СН3О'/Ч^\ОСН3
осн3
(1) 6 г в 140 МЛ СвНе
Перемешивание 1,3 час (25°)
1,2 г Nali-f-TsCi ------—-----—----------—>.
50%-ная 4 г 90—94%
дисперсия
CH3OTs
СН3О/ \^Х' осн3
осн3
(2)
пользуя тонко диспергированный в бензоле Н. г. с добавлением в на-
чале реакции n-толуолсульфохлорида, удалось провести превраще-
ние быстро и эффективно уже при комнатной температуре.
Бензилирование. Тейт и Бишоп [23] нашли, что Н. г, исклю-
чительно эффективен как основание при бензилировании углеводов
хлористым бензилом. Например, метил-cc-D-глюкопиранозид пре-
вращается в тетрабензиловый эфир с высоким выходом без образо-
вания побочных продуктов, тогда как по обычной методике с ис-
пользованием порошка КОН в качестве основания выход составляет
только 33%, и главным продуктом реакции является дибепзгловый
эфир.
. нс£ СН2ОН Перемешивание 3час, + сбН5СН2С1 + NaH 125~135° ОН : 125 мл 5 г
Восстановление. Кетоны, не содержащие а-водородиых атомов,
восстанавливаются Н. г. Например, бензофенон при нагревании с
небольшим избытком этого реагента в ксилоле при 145° в течение
6 час восстанавливается в бензгидрол; выход 83% [5]. В тех же усло-
виях метиловый эфир бензойной кислоты не восстанавливается.
Циклизация. Фолкерс и др. [24] использовали Н. г. для цик-
лизации хинонов группы кофермента Q в хроменоны (выходы
45—90%).
399
CH.O^ /СН3
JI II
CH3O 4'j-
° /
H3CZ ^CH3
OH
ONa
CH3O
CH3O
H3C/XCH3
CH3oX/JwCHs
h3c/Vii3
Феноляты натрия. Для получения натриевых солей фенолов
в виде суспензии в толуоле успешно используют суспензию Н. г.
в толуоле [25].
Ароилирование [3-дикетонов сложными эфирами. Н. г. в кипящем
1,2-диметоксиэтане более активен, чем амид калия в жидком аммиа-
ке для ароилирования концевой группы в [3-дикетонах под дейст-
вием сложных эфиров при получении трикетонов [26J. Наиболее
удобна следующая методика. Р-Дикетон (1 же) и сложный эфир
(1,2 же) прибавляют к реагенту (4 же). В приведенном здесь примере
выход при использовании KNH2 составляет 62%.
NaH
СЙН5СОСН2СОСН3 + С6Н5СО,С;Н5--> СЙНДОСНЧСОСН,СОС;Н5
Расщепление формиатов и формамидов [27]
ROCHO^NaH-^ R0Na + C0-pH2
R3NCHO-|-NaH —R2NNa |-CO + H3
Реакцию проводят в кипящем 1,2-диметоксиэтане до прекращения
выделения газа (20 мин). Выходы 30—80%.
1. Н i n с k 1 е у A. A., «Sodium Hydride Dispersions», Metal Hvdrides, Inc.,
July, 1964.
2. Org. Expts, 141.
3. Gruen H., Norcross В. E., J. Chem. Ed., 42, 268 (1965).
4. Roberts D, C., McElva in S. M., J. Am. Chem. Soc., 59, 2007
(1937).
5. S w amor F, W., Hauser C. R., J. Am. Chem, Soc,, 68, 2647 (1946).
6. Daub G. H., Johnson W. S., J. Am. Chem. Soc,, 70, 418 (1948);
72, 501 (1950).
7. 11 и н к н и II., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 297.
8, В 1 о о m f i е I d J. J., J. Org. Chem., 26, 4112 (1961); 27, 2742 (1962).
9, Ainsworth C., Org. Syn., Coll, Vol., 4, 536 (1963).
10. Weiscnborn F. L., Remy D. C., Jacobs T. L,, J, Am. Chem.
Soc., 76, 552 (1954).
11. Zaugg El, E, el. al., J. Am. Chem, Soc., 82, 2895, 2903 (1960); J. Org.
Chem., 26, 644 (1960).
12. E m m о n s W. D., Free m a n J. P., J. Am. Chem. Soc., 77, 4391 (1955).
13. W h i t 1 о c k H. W., Jr., J, Am. Chem. Soc., 84, 3412 (1962),
14. New m a n M. S., Merrill S. H., J. Am. Chem. Soc., 77, 5549 (1955).
15. C a r b о n J. H., Martin W. B., Swett L, R., J. Am. Chem. Soc.,
80, 1002 (1958).
400
16. -Sarel S., Leader H., J. Am. Chem. Soc., 82, 4752 (1960).
17. M cCoy L. L„ J. Am. Chem. Soc., 80, 6568 (1958); 84, 2246 (1962); J. Org.
Chem., 25, 20/8 (1960),
18- Major R. T., Peterson L. FL, J. Org. Chem., 22, 579 (1957).
19. Fones W. S., J. Org. Chem., 14, 1099 (1949).
20. M a r v e I C. S., M oyer W. W., Jr., J. Org. Chem., 22, 1065 (1957).
21. Kochi J. K-, FI ainmond G. S., J. Am. Chem. Soc., 75, 3443 (1953).
22. Williams К. I. H.., Cremer S. E., Kent F. W., Sehm E, J.,
T a r b e 1 1 D. S., J. Am, Chem. Soc., 82, 3982 (1960).
23. Tate M. E., Bishop С. T., Can, J. Chem., 41, 1801 (1963).
24; . Linn B. 0., Shurik С. H,, Wong E. L,, F о 1 к e r s K-, J, Am.
Chem. Soc., 85, 239 (1963); Wagner A. F., W i t t г e i c h P. E., Ari-
son B., Trenner N. R., Folkers K-, ibid., 85, 1178 (1965).
25. E 1 к о b a i s i F. M., Hick i n b ot tom W. J., J. Chem. Soc., 1958,
- 2431.
26. M i -1 e s M. L., Harris T. M., Hauser C. R., J, Org. Chem., 30,
1007 (1965).
27. Powers J. C., S e i d n e r R., Parsons T. G., Tetrahedron Letters,
' 1713 (1965).
НАТРИЯ ГИДРОСУЛЬФИТ, Na2S2O4 (см, также Физе pa раст-
вор). Мол.-вес 174,11, растворимость в 100 г воды 22 а при 20°.
Этот реагент — сильный восстановитель в щелочном растворе.'
При pH менее 7 гидросульфит разлагается с выделением серы.
Если его использовать в первоначально нейтральном водном раство-
ре без буфера, как, например, при восстановлении азосоединений,
раствор постепенно становится кислым в результате образования
бисульфита натрия.
ArN =- NAr + 2Na2S2O4 + 2Н2О —> 2ArNH3 4NaFISO3
Хиноны. H. г. восстанавливает большинство хинонов до соот-
ветствующих гидрохинонов. Хиноны бензольного или нафталинового
ряда можно даже идентифицировать по их обесцвечиванию под дей-
ствием гидросульфита и окрашиванию при окислении. Антрахиноны
можно отличить по образованию при восстановлении щелочным
гидросульфитом кроваво-красного кубового красителя, содержащего
анион антрагидрохииоиа; при встряхивании раствора на воздухе
красный кубовый краситель обесцвечивается. Кубовое окрашивание
можно иногда с успехом использовать для идентификации или раз-
деления. Нафтаценхиноп-и высшие линеарные бензологи представ-
ляют собой исключение в том отношении, что они не восстанавли-
ваются щелочным гидросульфитом [11. л ин-Нафтиндазол-4,9-хи нон
отличается тем, что, будучи бесцветным, он образует характерный
красный кубовый краситель под действием щелочного гидросуль-
фита [2].
401
2-Метил-1,4-нафтогидрохинон легко получают следующим обра-
зом [3]. Растворяют 2 г хинона в 20 мл эфира, раствор нагревают и
взбалтывают со свежеприготовленным раствором 4 г Н. г. в 30 мл
воды. На стадии образования хингидрона раствор приобретает ко-
ричневую окраску н через несколько минут обесцвечивается или ста-
новится светло-желтым. Водный слой отделяют, а эфирный взбалты-
вают с насыщенным раствором поваренной соли, фильтруют через
сульфат натрия и упаривают до небольшого объема. После добавле-
ния некоторого количества петролейного эфира гидрохинон отделя-
ется в виде белого или сероватого порошка (1,9 г).
Антрахинон—>антрои 14]. Смесь 5 г антрахинона, 6 г едкого
натра, 15 г Н. г. и 130 мл воды при нагревании и перемешивании
дает красный кубовый краситель; прн кипячении раствора в течение
45 мин красная окраска переходит в желтую, характерную для ани-
она антранола, а затем в результате нейтралнзации щелочи обра-
зующимся бисульфитом натрия антранол переходит в более устой-
чивый антрон, который отфильтровывают и промывают.
— N = N >— NH2 -|- H2N —. Удобный путь для перехода от
фенола к хинону заключается в сочетании его с диазотированной
сульфаниловой кислотой, восстановлении азосоединения водным
раствором Н. г., очистке через хлоргидрат аминофенола и окислении
хлорным железом, как и при получении 1,2-нафтохинона [5|.
—N02~>—NH2. Например, при восстановлении красителя жел-
того Марциуса (в виде аммониевой соли) в 2,4-диамино-1-нафтол [61.
С-нитрозосоедннения. Другой возможный путь, вместо диазо-
сочетания и восстановления, состоит в превращении фенола в о-
или п-нитрозопроизводное, которое затем восстанавливают; приме-
ром может служить синтез диаминоурацила по Шерману и Тэйлору
[7]. Розово-красное нитрозосоединение перемешивают с водой и
нагревают, а затем обрабатывают гидросульфитом до исчезновения
красной окраски и образования рыжевато-коричневой суспензии
бисульфитной соли диаминоурацила, которую затем превращают в
хлоргидрат.
402
N-Нитрозосоединения. Овербергер с сотр. 18] нашли, что вос-
становление N-нитрозодибензил амин а действием Н. г. сопровождает-
ся выделением азота и перегруппировкой в дибензил. Смешанные
бензиларнл- или диарил-N-нитрозо амины восстанавливаются в гид-
разины.
С8Н5СН2ХСН2С6Н5 С6Н5СН2СН2СеН-а + Х-Н,О
[ 77%
ко
C6H5CH2NC6H5 C6H5CH2NCbH5
I !
NO NH2
Дигидропиридины. H. г. широко применяют для восстановления
солей пиридиния в 1,4-дигидропиридины [9]. Важность этой реак-
ции заключается в том, что получаемые продукты моделируют вос-
становленную форму дигидрофосфопиридиннуклеотида (ДФПН-Н).
Na2S3O4
1. Fieser L. F., J. Am, Chem. Soc., 53, 2329 (1931).
2, Fieser L. F., Peters M. A., J. Am. Chem. Soc., 53, 4080 (1931).
3. Org. Expts., 250—251.
4, Org, Expts., 199.
5. Org. Expts., 249—250; Физер Л,, «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр, 353,
6. F i е s е г L. F., Fieser ЛЕ, J, Am. Chem. Soc., 56, 1565 (1934); Org,
Expts., 280.
7. Шерман У,, Тэйлор Э. мл., «Синтезы органических препаратов»,
ИЛ, ЛЕ, 1959, сб. 9., стр. 74,
8. Overberger С, G., Lombardino J. G,, Hiskey R, G,, J.
Am. Chem. Soc,, 80 , 3009 (1958).
9, К а г г e r P. et al., Helv. Chim. Acta, 19, 1028 (1936); 20, 55, 72, 418, 622 (1937);
21, 223, 1174 (1938); 29, 1152 (1946); 32, 960 (1949); MauzerallD., Westhei-
mer F, H., J, Am. Chem. Soc., 77, 2261 (1955); H u 1 t о n R. F., West-
h e i m e r F. FL, Tetrahedron, 3, 73 (1958).
НАТРИЯ ГИПОБРОМИТ, NaOBr.
Приготовление водного раствора: из брома и едкого натра [11;
прибавлением бромистого натрия к гипохлориту натрия [2],
Реакция Гофмана. При получении 3-аминопиридина из нико-
тинамида бром прибавляют к водному раствору едкого натра при
перемешивании и охлаждении в бане со льдом и солью [1]. По охлаж-
дении до 0° сразу добавляют все количество амида и после его раст-
ворения (15 мин) смесь нагревают до 70—75° в течение 45 мин.
Затем раствор охлаждают, насыщают хлористым натрием и экстра-
гируют эфиром в экстракторе непрерывного действия (15—20 час).
403
CONH.
NH<
+ NaOH Щ Bn,
0,49 моля
1,87 моля 0,6 моля
в 800 .и.; Н,0
После обработки получают темно-красный продукт, который обесцве-
чивают нагреванием в петролейном эфире с активированным углем
и гидросульфитом натрия. После перекристаллизации получают чис-
тый белый препарат (т. пл. 63—64°).
Окислительное расщепление кетонов. Левин и Стефенс 13]
показали, что гипогалогенитпое окисление кетонов в кислоты пе
ограничивается окислением метнлкетонов. Например Н. г. окисляет
пропиофенон до бензойной кислоты с выходом 96%, по-видимому,
по следующей схеме:
2NaOBr NaOH NaOBr + NaOH
С6Н5СОСН2СН:{-----С6Н5СОСВ1-2СН3--------> С6Н5СОСОСН3 ----—----*
CsHr,CO2Na + NaBr-[-H2O + CH3CO2Na
Н. г. окисляет циклогексанон до адипиновой кислоты (выход 82%)
и циклопентапои до глутаровой кислоты с удовлетворительным вы-
ходом [4].
Другие реакции. Кергомард [2], получавший реагент из гипо-
хлорита натрия, использовал его как источник НОВг для присоеди-
нения, например к циклогексену. К раствору НО г бромистого на-
трия в 250 мл воды прибавляют при 15° 500 мл продажного NaOCl
и эту смесь добавляют по каплям при. 10° и перемешивании к смеси
Ji 1
II + КОВГ + СН3СО2Н --
100 мл циклогексена, 100 мл уксусной кислоты и 750 мл воды. Ана-
логичным образом из фталимида получают N-бромфталимид с выхо-
дом 70%.
Н. г. реагирует с терминальными ацетиленами с образованием
1-бромалкинов [51. К раствору реагента, полученному из водного
раствора щелочи и брома, добавляют фенилацетилен (жидкость) и
смесь энергично взбалтывают на механической качалке в течение
60 час при комнатной температуре.
NaOBr
СЙН5С СН ----------> СеН5С = СВг
7 3—83%
1. А 1 1 е п С. F. Н., W о И С. N., Org. Syn., Coll. Vol., 4, 45 (1963).
2. Kergom ard A., Bull. soc. chim. France, 1961, 2360.
3. Lev ine R., Stephens J. R., J. Am. Chem. Soc., 72, 1642.(1950).
404
4. Farrar M. W , J. Org. Chem., 22, 1708 (1957).
5. M filer S, I., Ziegler G. R., Wlelesec к R., Org. Syn., 45, 86
(1965).
НАТРИЯ ГИПОФОСФИТ, NaHsPOo-HiO. Мол. вес 106,01,
растворимость 100 г в 100 а Н2О при 25°.
Никель Ренея выделяет водород из воды в присутствии Н. г.,
который при этом окисляется до фосфита натрия. Эта восстанови-
тельная система в водной уксусной кислоте или в смеси водной ук-
сусной кислоты и пиридина восстанавливает нитрилы в альдегиды
при комнатной температуре и под давлением; выход 50—90% [11.
1. В а с 1< е b с r’g О. G., St askun В., J. Chem. Soc., 1962, 3961.
НАТРИЯ ГИПОХЛОРИТА РАСТВОР, NaOCl. «Хлорокс» в США
продается как отбеливающее средство для бытовых целей; содержит
5,25% NaOCl..
Получение: из хлора и едкого натра [11 (см. также ссылки [5]
и [61); из гипохлорита кальция и углекислого натрия [11.
Галоформное окисление [2]. Ньюмен и Холмс [1] получали
раствор гипохлорита по методике Стерлинга, согласно которой не-
обходимое количество льда добавляют к холодному раствору щелочи
и без дополнительного охлаждения в раствор пропускают хлор до
моля
—- NaOH 4- Лед (1250
л,45 моля в 4,5
300 м.1 НЮ моля
(25°) ~
нужного привеса; к'этому времени температура'должна быть не
выше0°. Ньюмен и Холмс снабдили колбу термометром п эффектив-
ной мешалкой, нагревали раствор до 55’, добавляли метилф-наф-
ти л кетон и путем охлаждения поддерживали температуру при 60—
70°. Примерно через 30 мин по завершении экзотермической реакции
приливали раствор бисульфита для разложения избытка гипохло-
рита, смесь охлаждали и подкисляли. После кристаллизации из
95%:ного' этанола получали чистую р-нафтойную кислоту; т. пл.
184—185°. ' ' ..... .
Количество взятого в эту реакцию гипохлорита в три раза пре-
вышает теоретическое, однако доказательств в пользу необходимости
такого огромного избытка не представлено. Для окисления димедона
ОН -
0,5 моля в
води. КОН
NaOH
5,45 моля
в 3 00 мл
. воды.
35—4 0“ /СН3СО2Н
4-Лед (1250 г)-ДС1, —(СН3)„Сб +
2,27 81-9107 ЧСН,СО.,Н
+ СНС13
405
в р-диметнлглутаровую кислоту Смит и Мак-Лео д [3] использо-
вали только полуторное (против теоретического) количество гипо-
хлорита; возможно, что и это количество можно уменьшить.
Окисление метиленовых и метильных групп, связанных с аро-
матическим циклом. Нейсвендер, Мониз и Диксон [4] нашли, что
метиленовая или метильная группа, связанная с ароматическим цик-
лом, окисляется NaOCl до карбоксильной при условии, что в кольце
еще имеется ацетильная группа, которая одновременно также окис-
ляется до карбоксильной. Например, n-этилацетофенон окисляется
в терефталевую кислоту. Это объясняется тем, что в щелочной среде
кетон (1) образует резонансно стабилизированный аннон (2), который
далее реагирует по следующей схеме:
О О-
сн3сн2 — Z ссн3 сн3сн - ссн3
(I) (2а)
С1
сн3сн сосн3 сн3сн —/ сосн3 -ДД
(26) (3)
С1 С]
—9- сн3с—Д~ сосна _Д^Д сн3ссосн3
(4) q (М
о о
_наО^ЦаОД наСС—Z_CCH3 —ДДЦ НОоС—Z ^~со2н
95% \==/
(6) (7)
Из n-метилацетофенона была получена терефталевая кислота с вы-
ходом 47%. В другом примере из 2-ацетил-9,10-ди гидрофенантрен а
окислением Н. г. при pH 12—13 получена дифенил-2,2',4-трикарбо-
новая кислота. Предполагается, что промежуточным продуктом яв-
ляется фенантренхинон-2-карбоновая кислота; действительно, окис-
ление фенантренхннона действием Н. г. дает дифеновую кислоту с
выходом 94%
Другие реакции окисления. Н. г. окисляет аммиак в гидразин
[5], о-нитроанилин — в окись бензофуразана [6] и дихлорид
иодбензола — в нодобензол [7[.
406
+ NaOCl +
0)58 МОЛЯ
0,29 моля
КОН ________>
80-82%
0,29 МОЛЯ
В щелочной среде Н. г. окисляет сернокислый метилендиамин
до диазометана с выходом 26% [8].
,NH3
Н,С< + 1 2HSOT
' XNH,
он -
,NH,
Н2С<
xnh2
/NH2 он-
Н,С< —
XNHCI
/NH
^NH -
ОС1“
ОС1~
Эпоксидирование а, 0-не предельных соединений. Мармор [9]
нашел, что при добавлении «хлорокса» к бензальацетофенону в пи-
ридине желтый раствор почти мгновенно обесцвечивается, и при до-
бавлении воды с высоким выходом осаждается эпоксид.
С3Н,СН = СНСОС6Н3 - NaOcl X С6Н5СН-СНСОС6Н6
94% \ /
о
1,4-Нафтохинон при взаимодействии с Н. г. в диоксане дает
2,3-эпоксид с выходом 71,5%.
Окислительное декарбоксилирование аминокислот. В работе
[10] приведена реакция, которую ранее применяли к природным
аминокислотам [11]. К раствору 4 кмолей (1) в 25 мл воды добавляли
в виде верхнего слоя 10 мл бензола; к смеси приливали по каплям
СН3 СН3
СН3О СН^СО СН3 °\/\/сн’с“0
I II I II
CH3OZ СНзО7^7"
(1) (2)
в течение 20 мин 14 мл продажного 5%-ного раствора Н. г. Капель-
ная проба на иодокрахмальпую бумажку после каждого прибавле-
ния (через 30 сек после нанесения капли) была отрицательной до
достижения конечной точки. Слои разделяли и водный слой, имев-
ший щелочную реакцию, экстрагировали смесью эфира и бензола.
Галооксизамещение. При повторном исследовании удивитель-
ного продукта, полученного Грином и Роу [12] при действии Н. г.
на 2,4-динитроанилин в щелочном метанольном растворе, Мэллори
н Варимбн [13] установили истинную структуру этого соединения,
407
которое оказалось 5-хлор-4-метокс,ибензофуразан-1 -оксидом (4, в
равновесии с менее устойчивым 3-оксидом). В пользу предложенного
для этой необычной реакции механизма имеется ряд доказательств.
Так, 2,3-динитроанилин (5) вступает в подобную же реакцию 113];
аналогично pearnpvioT бензтриазол (6) [141 идифенилхиноксалип (7)
[Г4Т
NaOCl, CHjOH
69%
осн3
(?)
NaOCl, СН3ОН
64%
408
1. Ньюмен М., Холмс X., «Синтезы органических препаратов», ИЛ,-
М., 1949, сб. 2, стр. 351.
2. Fuson R. С., В u 1 1 В. A., Chem. Revs., 15, 275 (1934).
3. Смит В., МакЛеод Дж\, «Синтезы органических препаратов», ИЛ,
М.,. 1953, сб. 4, стр-. 172.
4. Neis w ender D. D.., Jr., M oniz W. B., Dixon J. A., J. Am.
Chem. Soc., 82, 2879 (1960).
5. Адамс P., Браун Б. К., «Синтезы органических препаратов»,
ИЛ, 1М., 1949, сб. 1, стр. 158.
6. Мэллор и Ф., «Синтезы органических препаратов», ИЛ, М., 1959, сб. 9,
стр. 51.
7. Форме М., Д ж о н с о п Дж., «Синтезы органических препаратов»,
. ИЛ, М., 1952, сб. 3, стр. 262.
8. О 11 тп е R., Schmitz Е., Chem. Вег., 97, 297 (1964).
9. М а г ш о г S., J. Org. Chem., 28, 250 (1963).
10. Slates Н. L., Taub D., Кио С. H., Wendler N. L., J. Org.
. Chem., 29, 1424 (1964).
11. See preceding reference, note 4.
12. О г e e n A. G., Rowe F. M., J. Chem. Soc., 101, 2452 (1912).
13. M a 1 1 о г у F. В., V a r i m b i ' S. P., J. Org. Chem., 28; 1656
(1963).
14. M a 1 1 о г v Г. В., Wood С. S., Hurwitz В. M., J. Org. Chem., 29,
2605 (1964”).
НАТРИЯ ДИСУЛЬФИД (ИЛИ ПОЛИСУЛЬФИД), РАСТВОР,
Na2S2.. При получении тиосалициловой кислоты [11 этот реагент
приготовляют путем нагревания и перемешивания .смеси.1,.1 моля
Na.,S-9H.>0, 290 д/л воды и 1,1 моля порошкообразной серы до обра-
зования коричневато-красного' раствора. Затем приливают раствор
40 г едкого натра в 100 мл воды, раствор охлаждают в бане со льдом
и солью до 0—5° и при перемешивании добавляют раствор диазоти-
рованной антраниловой кислоты. В результате реакции типа пере?
группировки Зандмейера получают дисульфид, восстанавливающий-
ся в тиосалициловую кислоту.
При получении 2,4,5-триаминопитробензола исходя из ти-дихлор-
бензола [21 восстановление 0,114 моля диамииодинитробензола в
триа м и но и мт р об е нзбл (красные' игл ы) осуществ л я ют путем нагре-
вания смеси 0,125 моля Na2S-9HL-O, 0,20 моля серы и 125 мл воды;
образующийся прозрачный оранжевоысрасный раствор прибавляют
по каплям в течение 1,5 час при перемешивании к пастообразной сме-
си диаминодинитробензола в 150 мл воды при температуре кипения
смеси. . ( . ' ’. у
.409
Ск у уС1 NH3
KNOS \/Х/ (СНЮН).,
H2SOi I 1 140е
70—71,5% J II. 88-89,5°Г
Na2S2 + HjO i II
49,5-52%^ L If
h2\\ zx/nh2
1
ОМ2/^/ХМОг
При взаимодействии «-нитротолуола с щелочным полисульфидом
натрия происходит интересное диспропорционирование: метильная
группа окисляется в СНО-группу, а нитрогруппа восстанавливается
до NH2-rpynnbr [3.1, При кипячении в течение 3 час образуется темно-
красный раствор; этиловый спирт, н-толуидин и исходное вещество
удаляют перегонкой с водяным паром. При охлаждении раствора
кристаллизуется и-аминобензальдегид.
СН3 СНО
/\ Кипячение
Г II 3 час Г и
<к / 0,125 моля 0,47 г-атом /0 к )
X/ В 600 мл Н.О у
no2 nh2
0,36 моля в 300 мл
95%-ного С2НЬОН
1. Аллен К., Мак-Кей Д., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 455.
2. Б ой ер Дж., Бьюр и кс Р., «Синтезы органических препаратов», ИЛ,
М., 1964, сб. 12, стр. 149.
3. Кампень Э., Будде В., Шефер Дж., «Синтезы органических
препаратов», ИЛ, At, 1953, сб. 4, стр. 30.
НАТРИЯ ИОДИД, Nal. Мол. вес 149,91. Растворим в воде,
этаноле, ацетоне, уксусной кислоте, диметилформамиде, метил-
ат и л кетоне.
Замещение CI или Вг. Беннетт и Коннер [1] усовершенствовали
ранее предложенную методику получения 2,4-динитроиодбензола
из соответствующего хлор содержащего соединения, используя в ка-
честве растворителя ДМФА вместо этиленгликоля. Н. и. берут
в пятикратном молярном избытке, по-вндимому исходя из обрати-
мости этой реакции.
0,2 моля В
200 мл ДМФА
410
Однако Форд-Мур [2] замещал хлор на иод с гораздо более высоким
выходом путем нагревания в течение 24 час хлорида (1) с небольшим
избытком Н. и. в метилэтилкетоне.
QiHsCOXHXHXl + Nal
(1) 0,7 моля 1,1 моля
1,2 л мстилэтилкетона
24 час при 873
78—81%
С6Н5СО2СН2СНг1
(2)
Шуринк [3] превратил пентаэритриттетрабромид в
с очень высоким выходом реакцией в метилэтилкетоне
Н. н. вместо необходимого по теории 0,52.
тетраиодид
с 0,67 моля
C(CH2Br)4 + Nal
0,13 моля 0,67 моля
87°, 48 час
83-98%
С(СН31)4
В 240 м.г метилэтил кетон а
Замещение брома на иод является ключевой стадией в превра-
щении 3-кето-5а-стероида (1) в Д4-3-кетостероид [4]. Бромированием
получают 2а,4а-днбромкетон (2), который при кипячении с Н. и.
в ацетоне в течение 20 час дает А4-2а-иод-3-кетон (3). Последний
деиодир уют восстановлением хлористым хромом или цинковой
пылью и получают А4-3-кетостероид (4). Промежуточный иодкетон
(3) можно не выделять; суммарный выход достигает 50—60%. Пре-
вращение по этой методике 4,5«-дигидрокортизон-21 -ацетата в ацетат
кортизона [5] вначале удалось осуществить лишь с выходом 10%.
Поскольку из реакционной смеси был выделен кетой предельного
характера, это навело на мысль, что промежуточный иодкетон (3)
восстанавливается иодистым водородом. Действительно, выход уда-
лось улучшить при добавлении второго иодкетона (иодацетои), кон-
курентно реагирующего с иодистым водородом. Шрайбер 16] весьма
успешно использовал эту модификацию в синтезе пасленового алка-
лоида и путем увеличения относительного количества иодацетона
повысил выход до 73%.
Катализатор реакции замещения. В синтезе и-метоксибензил-
цианида из хлорида небольшое количество Н. и. служит, по-види-
мому, катализатором, хотя никаких доказательств в пользу такого
утверждения нет [71.
411
СН3О—CH2OH CH3O—сн2сг —
1 моль ' ' ' ' “
1,5 моля NaCN+10 г NaT
кипячение в 5(H) ,iu ацетона
2 0 час
7 1-8 1%
CH3O—____4—ch2cn
Реакция с тозилатами. Тозилаты первичных спиртов легко
реагируют с 10%-пым .раствором Н. и. в ацетоне с образованием
соответствующих иодидов. Судя по скорости выделения натриевой
соли n-тол у ол сульфокислоты реакции тозил атов бензилового спирта
Ацетон 2 час (25п)
C6H5CH2OSO2CJCCH:!-pNal --------—------С6НД],1 -!-CJI3C6H4SO3Na
2 же /l*
и метанола завершаются почти полностью при комнатной темпера-
туре за 2 чаи [81. В тех же условиях тозилат циклогексил карбинол а
реагирует лишь на 3%.
В более жестких условиях группу TsOCH2 можно восстановить
До метильной. Боуерс и Рингольд [91 получили 21-дезоксикортизон
(2) при действии на кортизон п-толуолсульфохлоридом в пиридине
(1)20 г
(2)
при 0° с последующим кипячением сырого продукта с большим избыт-
ком Н. и. в 1 л уксусной кислоты в течение 1 час. (в Мехико темпера-
туры кипения примерно на 10° ниже температур кипения на уровне
моря). Такой метод восстановления дает возможность сохранить
карбонильные группы.
Дегалогенирование вицинальных дигалогенидов. Шёнхеймер [10]
использовал Н. и. в ацетоне для получения холестерина
из 5а,60-дибромида. Бартон и Миллер [111 нашли, что изомерный
50,6а-дибромид очень устойчив к дебромированию и, таким образом,
установили, что т/щнс-днаксиальпая конфигурация наиболее бла-
гоприятна для легкого элиминирования.
Стивенс и Френч [12] синтезировали /г-толялимпп днфеиилкетепа
(4) дехлорированием соединения (3) при кипячении с 1 же Н. и. в аце-
тоне в течение 90 мин. Продукт реакции (4) — ярко-желтое твердое
вещество, реагирующее с водой, этанолом и хлором подобно кетену.
412
2H,NCcH,CH,^
(CfJH6)aCCOCl - — ——
Cl
(1)
(C6H5),C-C-NC6H4CH3-« —
I II I
Cl О 1-1
(2)
-* (C6H5)sC-~C = NC6H4CH3-rt
ci di
(3)
Nal—зцстон
(C6H5)3C = C = NC6H4CH3-rt
14)
Вицинальные ли1ози.чагы >о.чефины. Подобно вицинальным ди*
галогенидам вицинальные дитозилаты (или димезилаты) типа RCH
(ОТз) СН2ОТк при действии Н. и. в ацетоне превращаются в непре-
дельные соединения RCH--CIi2 [13.1. Однако отщепление осуществ-
ит
ляется медленно, если обе группы вторичные [14]. Слейте и Венд-
лер П5|, исследовавшие димезилаты четырех 2,3-диолов — произ-
водных 5а-спиростанона-12, нашли, что (1) и (2) реагируют с Н. и.
в ацетоне при 100° с образованием с хорошими выходами А’2-олефина,
тогда как в тех же условиях (3) и (4) практически не изменяются.
Такой результат весьма удивителен: по аналогии с дебромированием
5,6-дибромидов можно было бы ожидать, что трансу аксиальный
изомер (4) будет наиболее склонен к реакции отщепления под дей-
ствием иодид-иона. Возможно, что отсутствие реакции в случае
(3) и (4) связано с пространственным экранированием 2р-аксиальной
мезилатной группы аксиальной ангулярной метильной группой.
Восстановление. При получении 2,2'-дифенальдегида Бэйли
и Эриксон [16] озонировали фенантрен в метаноле и восстанавлива-
ли образующуюся перекись в диальдегид действием Н. и. (1,5-крат-
пое количество от теории) в уксусной кислоте.
413
Деалкилирование простых эфиров. Н. и. в уксусной кислоте —
мягкий реагент для эффективного деалкилирования алкоксипирнми-
динов [17]. Например, (1) превращается в (2) с выходом 82% за
4 час при 100°. Ту же реакцию можно осуществить и при действии
осн3 о
Л/Бг Л/Вг
N ]1 К'а1-СН3СО3Н (100°) HN II
CH3O/4N/ °
Н
(1) (2)
бромистого водорода в уксусной кислоте с почти количественным
выходом за 2 час при 70°.
J. Беннетт Дж. Ф., Коннер Р. М., «Синтезы органических препара-
тов», изд-во «Мир», М., 1964, сб. 12, стр. 47.
2. Форд-Мур А., «Синтезы органических препаратов», ИЛ, М_, 1953,
сб. 4, стр. 266.
3. Шуринк X., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 443.
4- Rosenkranz Q., Mancera О., Qatlca J., Djerassi С.,
J. A m. Chem. Soc., 72, 4077 (1950); Rosenkranz Q., Palaki J.,
Kaufmann St., Berlin J., Djerassi C., ibid., 72, 4081 (1950);
S on d h e i m er F., Rosenkranz G., Mancera O., Djerassi
C., ibid., 75, 2601 (1953).
5. Evans R. M., Hamlet J. C., Hunt J. C., Jones P. G.,
Long A. G., Oughton J. F-, Stephenson L., Walker T.,
Wilson Б. M., J. Chem. Soc., 1956 4356.
6. Schreiber K., Ronsch H., Ann., 681, 196 (1965).
7. Рориг K-, Джонстон Дж., Гамильтон Р., Телинский
Т., «Синтезы органических препаратов», ИЛ, М., 1958, сб. 8., стр. 32.
8. Т i р s о n R. S., Clapp М. А., Cretcher L. Н., J. Org. Chem.,
12, 133 (1947).
9. Bowers A., Ringold Н. J., J. Am. Chem. Soc., 80, 3091 (1958).
10. S c h о e n h e i m e г R., J. Biol. Chem., 110, 461 (1935).
11. В a r t о n D. H. R., Miller E., J. Am. Chem. Soc., 72, 1066 (1950).
12. S t e v e n s C. L., French J. C., J. Am. Chem. Soc., 75, 657 (1953).
13. В ladon P-, Owen L. N., J. Chem. Soc., 1950, 598; Foster A. B-,
Overend W. G., ibid., 1951, 3452.
14. L i n s t e a d R. P., Owen L. N., W ebb R. F., J. Chem. Soc., 1953,
1211.
15. S 1 a t e s H. L., Wendler N. L., J. Am. Chem. Soc., 78, 3749 (1956).
16. Б э й л и Ф. С., Эриксон P. Э., «Синтезы органических препаратов»,
изд-во «Мир», М., 1964, сб. 12, стр. 61.
17. U 1 b г i с h t Г. L. V., J. Chem. Soc., 1961, 3345.
414
НАТРИЯ МЕТАБИСУЛЬФИТ, Na2S203(+H2O->2NaHS03). Мол.
вес 190,11.
Очистка 3-кето-А'-стероидов [1, 21. Бромирование 3-кето-5а-
стероида и последующее дегидробромирование до 3-кето-Ах-произ-
водного (3) сопровождаются образованием побочных продуктов,
которые трудно удаляются хроматографией или кристаллизацией.
Полученная смесь состоит из исходного соединения (1), 2а-бромке-
тона (2) и, возможно, некоторого количества 3-кето-А1,4-стероида.
Удобный метод, который можно применять и в промышленном мас-
штабе для выделения чистого (3), состоит в следующем. Раствор не-
очищенного продукта реакции [0,05 моля, содержится примерно
50/6 (3)1 в 250 мл метанола смешивают с раствором 40 г Н. м. в 210 мл
воды в течение 2—3 мин и сразу же экстрагируют три раза хлори-
стым метиленом. При этом исходное вещество (1) остается в водной
фазе в виде бисульфитного соединения. Органический слой промы-
вают водой, сушат и упаривают, остаток растворяют в 150 мл ме-
танола и раствор кипятят 1—1,5 час с раствором 28 г Н. м. в 100мл
воды для превращения (3) в бисульфитное соединение. Затем добав-
ляют 100 мл воды, раствор охлаждают и экстрагируют три раза
хлористым метиленом. Для выделения (3) водный слой кипятят
в течение 15 мин с раствором 5 г едкого натра в 150 мл воды. После
охлаждения экстракцией хлористым метиленом выделяют чистый
З-кето-А1 -стероид; ^гаах 228,5 ммк\ log е 4,004.
1. Contributed by К 1 i m s t r a P. D., Searle G. D. and Co.
2. Klim st г a P. D., пат. США 3018296.
НАТРИЯ НИТРИТ, NaNO2. Мол. вес 69,01.
В число примеров по использованию этого реагента, приведен-
ных ниже, не включены диазотирование, нитрозирование фенолов
и третичных ароматических аминов и обыкновенное N-нитрозиро-
вание аминов.
Изоиитрозопроизводные. В методиках реакций ацетоуксус-
ного [1] и диэтилмалонового [2J эфиров с Н. н. и водной уксус-
ной кислотой для получения изонитрозосоедннений описывается
лишь первая стадия процесса.
О NOH
11 II
НОХ = с (СО2С2НЛ СН3С— ССО2С2Н5
Гидразид->азид. Для синтеза дихлоргндрата путресцина [31 дн-
этиловый эфир адипиновой кислоты превращают в соответствующий
415
дигидразид, который при взаимодействии с азотистой кислотой дает
диазид. Это реакционноспособное соединение экстрагируют эфиром,
' (СН,)ДСО2С2Н5)2 (СЫг)4 (CONHNHA ‘
О
(CH2)4(CN = N=N'% ------—> №)4(N=C = O)2 ——-°-.H5U
' ' 1 ’ 73 — 77%'
(общий)
(СН,)4 (NH2-HC1)2
полученный раствор приливают к бензолу и смесь осторожно нагре-
вают на кипящей водяной бане. Одновременно с отгонкой эфира на-
чинается перегруппировка в диизоцианат с выделением азота.
Расширение цикла. Примером перегруппировки Тиффено служит
превращение 1-амипометил циклогексанол а в циклогептанон [4],
Н2О-NaNO,~ АсОН
------>
59-65%
При получении 2-феиилциклогептапона по Гутше [51 N-бензил-
уретап превращают в N-нитрозопроязводное, которое обраба-
тывают циклогексаноном в метаноле в присутствии измельченного
углекислого калия. Ниже показан предполагаемый механизм-реак-
ции.
Гидразин—>-аз ид. Для получения фенил азида суспензию хлор-
гйдрата фенилгидразина в разбавленной соляной-кислоте перемет
шивают под слоем эфира и при хорошем охлаждении обрабатывают,
njiko, (о —... Д
CfiH5\’HNHa• НС1 -----:------> CflH,N = N = N
h ь -i. • 65- G3% °
добавляя по каплям водный раствор Н. н.- [61. Продукт экстраги-
руют эфиром и осторожно перегоняют’ в вакууме с применением за-
щитного экрана.
416
Диазоуксусный эфир получают действием Н. и. на хлоргидрат
этилового эфира глицина [7].
+ 0-2" - 4-
C1-H3NCH2CO2C<,H5-|-NaNO2 —— N =- N СНСО2С2Н5
Я’5 /о
Нитрометан по методу Кольбе (1872 г.). Согласно этому методу
атом хлора в хлорацетате натрия замещают на нитрогруппу, затем
раствор осторожно нагревают для осуществления декарбоксилиро-
вания [81.
NaNO, 80—35“
ClCH.,CO2Na ----NO2CH2CO,Na —CH3NO3 + NaHCO3
' “ Зо—38%
Реакция Зандмейера. При получении 1,4-динитронафталина
из 4-ннтро-1-нафтиламниа Ходгсон и сотр. 191 применили специаль-
ные приемы для диазотирования этого амина, обладающего очень
слабо выраженными основными свойствами. 10 г измельченного Н. н.
растворяют при охлаждении льдом в 50 мл конц. серной кислоты.
Затем раствор 10 г амина в 100 мл. уксусной кислоты охлаждают до
20° и полученную слегка мутную смесь медленно по каплям добав-
ляют к холодной иитрозилсерной кислоте при перемешивании меха-
нической мешалкой и при охлаждении. Затем перемешивают с эфи-
ром при 0° для осаждения кристаллического несколько вязкого суль-
фата диазопия, который отделяют и растворяют в 100 мл ледяной
воды. Реагент, содержащий медь и пригодный для осуществления
разложения по Заидмейеру, получают смешением водных растворов
сульфата меди и сульфита натрия (каждого по 50 г). Зеленовато-ко-
ричневый осадок отфильтровывают, перемешивают с водным раство-
ром 100 г И. н. и смесь медленно обрабатывают холодным раствором
N=NHSOf
NO,
+ NaNO2
Сульфиты меди
54-60%
диазон и йсульфата, добавляя эфир, чтобы сбить пену.
Расщепление \c=N------Клайзеп 110] в 1889 г. нашел,
что для некоторых кетонов превращение в соответствующие нитро-
зопроизводные (оксимы) наиболее эффективно достигается при вза-
имодействии с амил нитритом и этилатом натрия (а), тогда как другие
лучше реагируют при использовании в качестве конденсирующего
агента соляной кислоты (б).
(a) CeHr,COCH3 + C5HnNO2 + C2H5ONa CSH-COCH = NOH
(б) CH3COCH3 + CsHnNO2-!-HCl —» CH3COCH = NOH
14 Заказ № 4090
417
В последующих опытах, поставленных для выяснения, какая же
методика вообще лучше, он нашел, что важно избегать избытка амил-
нитрита, так как последний превращает первоначально образующее-
ся нитрозосоединение в дикарбонильное соединение. Например,
нитрозопропиофенон превращается в бензоил ацетил:
CeH5COCCH3 + C6HnNO2 C6H5COCOCH3 + C5HnOH + N2O
II
NOH
Изучая поведение нитрозокамфоры, Клайзен нашел, что если просто
добавлять Н. н. к раствору этого иитрозосоединения в уксусной кис-
лоте, то кетон гладко превращается в камфорхинон. Оказалось, что
расщепление изонитрозосоединений или оксимов по Клайзену имеет
NaNO2-АсОН,
общий характер и применимо к соединениям,содержащим группиров-
ку >C^N—. Виланд и Гримм [11], исследовавшие эту реакцию с
изотопной меткой, нашли, что карбонильный кислород берется из
оксима, что подтверждает предположение о промежуточном образо-
вании трехчленного цикла:
C-N
ОН
+ NO
-Н+ \ + -
;C = O + N= N —О
Азотистая кислота расщепляет оксим и семикарбазои пента-
ацетатов глюкозы и галактозы с образованием пентаацетатальдегидов
[12]. Реагент расщепляет семикарбазон бензальдегида [13], фенило-
зазоны [14] и гидразон 4-циннолинальдегида [15].
СН = NNH2
СНО
NaNO2-pas6. НС1
66%
^/XNZ/
Найдено [16], что семикарбазоны стероидов легко расщепляются
под действием Н. н. в уксусной кислоте уже на холоду или при сла-
бом нагревании; кетон отделяется при добавлении воды иногда в
кристаллическом виде. Гидрокортизон-3,20-бмс-семикарбазон рас-
щепляют [17], добавляя избыток Н. н. к раствору производного в
разбавленной соляной кислоте при 5° н получают гидрокортизон с
418
выходом 69,5%. Расщепление по обменной реакции с пировиноград-
ной кислотой дает плохой результат.
Бестман и сотр. [18] разработали метод синтезасс-кетоальдегидов,
первой стадией которого является реакция диазокетона с трифенил-
фосфином с образованием фосфоразана (3). Последний можно пре-
вратить в а-кетоальдегид (6) гидролизом с последующей обработкой
гидразона азотистой кислотой или непосредственным действием азо-
тистой кислоты. Фенилглиоксаль был получен по этому методу с
общим выходом 70% в расчете иа бензоилхлорид.
О О
11 + - II
RC—CH = N = N + P(C6H5)3 —RCCH = N — N = P(C6H5)3
(1) (2) I (3)
О о
II HNOa ||
OP(C6H5)3 + RCCH = NNHb--------> RCCHO
(4) (5) (6)
Гетероциклы. 1,2,3-Бензотриазол получен [191 методом, от-
крытым Ладенбургом в 1876 г. [20]. К раствору о-фепилендиамина
1 моль
70-80°
NaNO2-ВОДИ. АсОН
75-81%
в смеси уксусной кислоты с водой, охлажденному до 5°, при интен-
сивном перемешивании и охлаждении на ледяной бане в один прием
приливают водный раствор нитрита натрия. Температура смеси
повышается до 70—80°, и раствор окрашивается сначала в темно-зе-
леный, а затем в светлый оранжево-красный цвет. Продукт полу-
чается в виде масла, которое вскоре затвердевает; его очищают
перегонкой и последующей кристаллизацией. Плавящийся при бо-
лее высокой температуре 5-нитроиндазол получают аналогично и
кристаллизуют из метанола [21].
25°
NaNO2- води, Ас ОН
72-80%
1
Н
Окисление. Физер и Петерс [22] нашли удобный метод окисления
гетероциклического гидрохинона (1) в хинон (2), заключающийся
в добавлении избытка водного раствора Н. и. к раствору (1) в уксус-
ной кислоте; хинон выделяется в виде желтых микрокристаллов
14*
419
после разбавления водой. Один из авторов [23] позже нашел, что
по этому методу можно получить 1,4-нафтохинон диеновым син-
тезом из 1,4-бензохинона. Хотя 5,8-дигидро-1,4-нафтохинон (4) не
стабилен па свету и реакционноспособен, его можно получить с
высоким выходом в виде ярко-желтых игл окислением (3) азотистой
(1) (2)
кислотой при 100°. Если после окисления Н. н. температуру по-
низить до 60° и добавить водный раствор бихромата натрия и серной
кислоты, то с высоким выходом образуется нафтохинон высокой
(3) 0,1 моля
в 150 мл АсОН
91—97%
100°
16.2 г NaNO3
в 25 мл Н2О
88% в расчете
на (3)
60°
1 6 г Na2Cr,O7 2Н,0
1 мл HuSO4 в Г1ЕО
чистоты. Прямое окисление (3) в (5) би хроматом требует для полу-
чения удовлетворительных результатов более сложной методики.
Добавление Н. п. к водному раствору хлоргидрата 4-аминотимола
при 60° и перегонка с паром дает с высоким выходом тимохинон
[241.
I. Фишер Г., «Синтезы органических препаратов®, ИЛ, М., 1952, сб. 3,
стр. 269.
2. 3 амбито А. Дж., X о у Е. Э., «Синтезы органических препаратов»,
изд-во «Мир», М., 1964, сб. 12, стр. 65.
3. Смит П., «Синтезы органических препаратов», ИЛ, ЛЕ, 1958, сб. 8, стр. 12.
4, Д аубен X., мл., Ри и гол ьд Г., Вейд Р,, Пирсон Д., А н-
д е р с о и А. мл., «Синтезы органических препаратов», ИЛ, ЛЕ, 1956,
сб. 6, стр. 92.
5. Gulsche С. D., J. Лги. Chem. Soc., 71, 3513 (1949); G u t s c h e C. D.,
Johnson H. E., ibid., 77, 109 (1955); Org. Syn., Coll. Vol., 4, 780 (1963).
6. Линдсей P., Аллен Ч., «Синтезы органических препаратов», ИЛ,
М., 1952, сб. 3, стр. 433.
7. В о м а к Э., Нельсон А., «Синтезы органических препаратов», ИЛ,
ЛЕ, 1952, сб. 3, стр. 512.
8. У итмор Ф. К., Уитмор М. Г., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 1, стр. 303.
9. Ходгсон X., Махадеван А., У о р д Э., «Синтезы органических
препаратов», ИЛ, М-, 1953, сб. 4, стр. 206.
10. С I a i s е n L., М a n a s s е О., Вег., 22, 526, 530 (1889).
II. W i е I а п d Т., Grimm D., Chem. Вег., 96, 275 (1963).
420
12. W о 1 f г о m М. L., Georges L. W., Soltzb erg S., J. Am.
Chem. Soc., 56, 1794 (1934).
13. A u w e r s K. von, Ottens B-, Ber., 58, 2067 (1925).
14. О h 1 e H., Henseke G., Czyzewski A., Chern. Ber., 86, 316 (1953).
15. Castle R. N., О n d a M., J. Org. Chem., 26, 4465 (1961),
16. Goldschmidt St,, Veer W. L. C., Rec. trav,, 66, 238 (1947).
17. О 1 i v e t о E. P., Rausser R., Weber L., Shapiro E,,
Gould D., Hershberg E. B., J. Am. Chem. Soc., 78, 1736 (1956).
18. Bestmann H.-J., Klein O., G 6 t h I i c h L,, Buckschewski
H., Chem. Ber., 96, 2259 (1963).
19. Д а м ш p о д с p P., Петерсон В., «Синтезы органических препара-
тов», ИЛ, М., 1952, сб, 3, стр. 104.
20. Ladenburg К-, Вег,, 9, 219 (1876).
21. Портер X., Петерсон В., «Синтезы органических препаратов»,
ИЛ, М., 1952, сб. 3, стр. 357.
22. Fieser L. F., Peters М., J, Am. Chem. Soc, 53, 4080 (1931).
23. Fieser L. F., J. Am. Chem. Soc., 70, 3165 (1948).
24. Кремере, Уэкмаи, Хиксон, «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. I, стр. 378.
НАТРИЯ ПЕРБОРАТ, NaBO3-4H,O. Мол. вес 153,88.
В уксусной кислоте при температуре 35—60° Н. п. окисляет
n-замещенные производные анилина до азобензолов [11. Например,
[21:
500 мл АсОН
/.---ч 6 час при 50—60°
2AcHN _______у- NH2 + NaBO3-4H2O + H3BO3 ---- —
л ГТ 7 0,26 0,16 ° ’ /п
0,1.) моля моля моля
— AcHN — N - N NHAc
Прибавление борной кислоты увеличивает выход. Реагенты вначале
растворяются, и спустя 40 мин начинается выделение продукта реак-
ции.
О реакции IT. п. с фталевым ангидридом см. также Мононадфта-
левая кислота.
1. М е h t a S. М., V a k i 1 w а 1 а М. V., J. Am. Chem. Soc., 74, 563 (1952).
2. Сантурри П., Роббинс Ф., Стубб и иге Р., «Синтезы органи-
ческих препаратов», изд-во «Мир», М., 1964, сб, 12, стр. 28.
НАТРИЯ ПЕРСУЛЬФАТ, Na.,SaOs. Мол. вес 238,11.
Реакция окисления Н. и. в заметной степени катализируется
ионами серебра; предполагается, что истинным окислителем является
SaO|“Ag+. Толуол окисляется до бензальдегида (выход 50%) с об-
разованием побочных продуктов, а бензиловый спирт — до бен-
зальдегида (выход 75%) [1]. Фенолы окисляются до смолообразных
полифенолов [2].
I. В aeon R. G. R., D о g g а г t J. R., J. Chem. Soc,, 1960, 1332.
2. В а с о n R. G. R., Grime R., Munro D. J., J. Chem. Soc., 1954,
2275.
421
НАТРИЯ СУЛЬФАТ БЕЗВОДНЫЙ, Na2SO4. Мол. вес 142,05.
Реагент — эффективный осушитель, так как при температурах
ниже 33° он образует гидрат Na2SO4- ЮН2О. При высушивании эфир-
ного экстракта следует встряхивать экстракт с насыщенным раство-
ром соли для удаления большей части воды и затем фильтровать эфир-
ный раствор из делительной воронки через слой осушителя на филь-
тре. Фильтр легче наполнять гранулированным сульфатом натрия,
чем порошкообразным сульфатом магния.
НАТРИЯ СУЛЬФИД, Na2S-9H2O. Мол. вес 240,19, т. пл. 50°.
Поскольку реагент разлагается под действием воздуха, следует ис-
пользовать препарат, взятый из только что вскрытой склянки.
Реагент, взятый с 10%-ным избытком, довольно эффективен при
избирательном восстановлении одной иитрогруппы 2,4-динитрофе-
нола [1]. Примечательно, что при этом восстанавливается нитро-
группа в орто-положении по отношению к гидроксилу. В недавно
разработанном почти количественном избирательном восстановлении
-|- 3Na2S 4- 6NH4C14- 28 % NH3
5,4 I 1,6 100 мл
моля моля
ОН
NO3
2,5 л Н20
2,4-ДИ нитронафтол а хлористым оловом восстановленная группа на-
ходится в мара-пол ожении к гидроксилу (см. Олово хлористое).
В синтезе бнс-и-аминофенил дисульфид а [2] сульфид натрия сна-
чала замещает активный атом галогена, а затем восстанавливает
нитрогруппу. Конечной стадией является окислительное сочетание
O3N—'%— Ci
1,5 моля
4 моля Na2S
2,5 л Н,_О
°2N~ \ _SNa
Кипячение
в течение 20 час
HSO2
58—64%
H2N —S - S —
под действием перекиси водорода. Легко осуществляется замещение
хлора при получении р-тиодигликоля [31:
45 мин при 87°
НОСН2СН2С1 + NaaS -----р—-----► HOCH2CH2SCH2CH2OH
0,37 моля 2,05 79
моля
2 0%-ный водный
раствор
. В первой стадии синтеза диэтилмеркаптоацеталя [4] смесь Н. с.
и серы в этаноле кипятят до появления темно-красной окраски
422
раствора; нагревание прекращают и в течение 30 мин к смеси добав-
ляют диэтилбромацеталь. Затем добавляют бикарбонат натрия в ка-
честве буфера, большую часть этанола отгоняют, остаток разбавляют
водой и полисульфид экстрагируют эфиром.
уОСаН5
/ Кипячение 4 час
ВгСН2СН -|-Na2S-9H2O+ S + 95%-иый ЕЮН 83_9£O/-------->
Xqq Н 2,5 моля 4,5 г-ат ом. 2,7 л ,е
3 моля
С,Н5С\ /ОС2Н5 Na — жидк. NH3 /ОС2Н6
— >СНСН2 — (S)4 — сн2—сн hsch2Ch
С2Н5О/ хос2н5 HCI 'Ч'ОС2Н&
Состав полученной смеси в среднем соответствует тетрасульфиду;
исходя из этого рассчитывают выход. Если используют только Н. с.,
то образуется значительное количество моносульфида, который не
восстанавливается натрием в жидком аммиаке; полисульфиды же
восстанавливаются легко.
1, Хартмэн В., Силловей X., «Синтезы органических препаратов»,
ИЛ, М., 1952, сб. 3, стр. 54.
2, П р а й с Ч., Стэси Г,, «Синтезы органических препаратов», ИЛ, М.,
1953, сб. 4, стр. 150.
3. Фабер Е., Миллер Дж,, «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 2, стр. 453.
4. Р а г h a m W. Е., W у n b erg Н,, Org, Syn,, Coll. Vol,, 4, 295 (1963).
НАТРИЯ ТИОСУЛЬФАТ, Na2S2O3 • 5Н2О. Мол. вес 248,21.
Титрованием Н. т. определяют количество и чистоту иода,
монохлормочевины, N-бромацетамида [11 и т. д.
1. О ливе то Ю., Герольд К., «Синтезы органических препаратов»,
ИЛ, ДЕ, 1953, сб. 4, стр. 89.
НАТРИЯ ФТОРИД, NaF. Мол. вес 42,00.
ь-Триптофаи получают из казеина ферментативным гидролизом
под действием продажного панкреатина (две порции по 20 а) при 37°.
К суспензии 600 г казеина в 3,2 л воды добавляют раствор 60 г
Na2CO3 и 6 г NaF в 100 мл воды и прибавляют фермент в виде жидкой
пасты в воде [1]. К смеси в виде верхнего слоя добавляют толуол,
разбавляют смесь до объема 6 л, тщательно взбалтывают и оставляют
при 37° на 5дней; затем прибавляют вторую порцию фермента и смесь
оставляют при той же температуре еще на 12 дней. «По-видимому,
Н. ф. ингибирует действие оксидаз». После обработки получают
4,0—4,1 г чистого l-триптофана и 17,0—18,2 г чистого ь-тирозина;
последний легче получить в качестве основного продукта при гид-
ролизе шелка соляной кислотой с последующей нейтрализацией
кислоты едким натром и подкислением уксусной кислотой.
1. К о к с Д ж., Кинг Г., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 474.
423
НАТРИЯ ХЛОРАТ, NaClO3. Мол. вес 106,46.
Хлорирование. а-Хлорантрахинон получают с почти количест-
венным выходом по методике, первоначально разработанной Улль-
маном н Очснером [1] и усовершенствованной, как это описано в ра-
боте [2J. К раствору калиевой соли антрахинои-а-сульфокислоты и
соляной кислоты в 500 мл воды при перемешивании мешалкой Герш-
О S(.)3K
0,001 моля
NaCIO3-P ЗНС1
0,19 моля 1 моль
берга из танталовой проволоки и кипячении добавляют из капель-
ной воронки Гершберга (см. том III, рис. П-1) в течение 3 час
раствор Н. х. в 100 мл воды. Спустя 1 час ярко-желтый осадок
tz-хлорантрахипона отфильтровывают, промывают и сушат. Полу-
ченный препарат имеет высокую степень чистоты.
Окисление. Милас 131 разработал методику получения фума-
ровой кислоты окислением фурфурола под действием Н. х. в присут-
ствии пятиокиси ванадия в качестве катализатора. Смесь Н. х.,
NaClOj z >
50-58%
4, 2 моля
2, Об моля
НООС-С-Н
II
н-с-соон
1 л воды и 2г пятиокиси ванадия перемешивают при 70—75° и фур-
фурол добавляют со скоростью, обеспечивающей бурное взаимодей-
ствие в течение 70—80 мин. Затем смесь перемешивают при 70—75°
еще 10—II час и оставляют на ночь при комнатной температуре.
Первую порцию сырой фумаровой кислоты отделяют фильтрованием;
после удаления растворителя и обработки остатка соляной кислотой
выделяют вторую порцию; чистый препарат получают кристаллиза-
цией из разбавленной соляной кислоты.
В присутствии пятиокиси ванадия в качестве катализатора в кис-
лой среде Н. х. окисляет гидрохинон в хинон. Согласно методике
Ундервуда и Уолша [4], смесь реагирующих веществ в I л 2%-ной
серной кислоты перемешивают до превращения первоначально обра-
зующегося зеленого хингидрона в желтый хинон (около 3 час) с по-
вышением температуры до 40°. По охлаждении смеси продукт от-
424
он
о
II
фильтровывают; экстракция фильтрата и промывной жидкости бен-
золом дает еще 12—14 г препарата.
Для окисления антрацена в антрахинон смесь 90 г растертого
в порошок углеводорода, 0,5 г пятиокиси ванадия, 76 г Н. х., 1 л
уу&уъыул кислоты и 200 мл 2%-ной серной кислоты кипятят до на-
чала бурной реакции 14]. Кипячение продолжают еще некоторое
время и получают антрахинон с выходом 88—91%. Этот метод не-
пригоден для окисления углеводородов рядов нафталина и фенантре-
на в соответствующие хиноны, а также для окисления аценафтена
или флуорена.
В эффективном методе получения 2,5-бас-(2-карбоксианилино)-
1,4-бензохинона Браун и 1Чске 151 применяют в качестве окислителя
Н. х., а в качестве катализатора — ванадат аммония.
О
II
/\ 200 мл 60%-ного
if и водного СНдбН
+ +NH4VO3-|- NaClO3 — “7ДУ
0,1 г 15 г в 30 мл НД А
II
О
0.1 моля
F., О с h s п е г Р., Ann., 381, 1 (1911); герм. пат. 205195, 228875.
2. Скотт В., Аллен К-, «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 2, стр. 549.
3. М и л а с Н., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 541.
4. У п Д е р в у д X. мл., Уолш В., «Синтезы органических препаратов»,
ИЛ, М., 1949, сб. 2, стр. 545.
5. Braun W,, Mecke R., Chem. Вег., 99, 1991 (1966).
НАТРИЯ ХРОМАТ БЕЗВОДНЫЙ, Na..CrO4. Мол. вес 162,00.
Имеется патент [11 на применение этого реагента в смеси уксусной
кислоты и уксусного ангидрида для окисления А5-стероидов в
425
А5-7-кетостероиды. При окислении А5-стероидов по этой методике
[2] Н. х,, по-видимому, более предпочтителен [3], чем mpem-бутил-
хромат. Методика из последней работы в применении к ацетату пре-
гненолона приведена на схеме. Выход неочищенного препарата
(е 10 300) составляет 79%.
40 а
25—4 0е. в течение
ночи
90—94%
1. М е u 1 е у W. С., пат. США 2505646 (1950) [С. А,, 44, 6894 (1950)].
2. Marshall С. W., R a v R. Е., Laos I., Riegel В, J. Am. Qhem.
Soc., 79, 6308 (1957).
3. Atwater N. \V., J. Am. Chem. Soc,, 83, 3071 (1961).
НАТРИЯ ЦИАНАТ, NaOC^N. Мол. вес 65,02.
Лоев и Корменди [1] нашли, что ранее труднодоступные карба-
маты третичных спиртов можно получить простым перемешиванием
субстрата, Н. ц. и трифторуксуспой кислоты в инертном растворителе
при комнатной температуре. Для получения трет-бутилкарбамата
[2] к суспензии Н. ц. в смеси /лрт-бутанола с бензолом при мед-
ленном перемешивании (два оборота в секунду или еще медленнее)
С1-13
СН3 - С - ОН NaOC = N -4- CF3CO3H -4- CeHB
] 0,4. моля 0,42 моля 123 мл
сн3
0,2 моля
сн3 о
—* СН3С—OCNH,
I
сн3
быстро добавляют по каплям трифтор уксусную кислоту, Этот метод
применим к первичным и вторичным спиртам (включая стероидные
спирты), полиолам, фенолам, тиолам и оксимам. Наиболее удовлет-
ворительные растворители — бензол и хлористый метилен. При
замене Н. ц. на цианат калия выход падает до 5% или даже еще ниже.
Выход также снижается при использовании только 1 экв Н. ц.
и возрастает до максимального с применением 2 экв [31. Более низкий
выход при меньшем молярном отношении, по-видимому, обусловлен
разложением некоторого количества HOCNT. Этим же объясняется
низкий выход при быстром перемешивании.
1. L о е v В., Kormendy М. F., J. Org. Chem,, 28, 3421 (1963).
2. L о е v В., Kormendy М. F., Goodman М. М., procedure submit-
ted to Org. Syn.
3. L о e v B., private communication.
426
НАФТАЛИН-2,3-ДИКАРБОНОВАЯ КИСЛОТА. Мол. вес216,18.
т. пл. 239—241.
Получение [1]. В автоклав загружают 1,28 моля 2,3-диметил-
нафталина, 3,14 моля (23%-ный избыток) дигидрата бихромата
натрия и 1,8 л воды и закрывают его; смесь непрерывно встряхивают
в течение 18 час при 250°. После охлаждения (также при встряхива-
нии) автоклав открывают, выгружают содержимое и промывают.
Зеленую гидратированную окись хрома отделяют с помощью боль-
шой воронки Бюхнера; фильтрат подкисляют и после соответству-
ющей обработки осаждают кислоту.
I. Friedman L., Org. Syn., 43, 80 (1963).
НАФТАЛИН-2,6-ДИКАРБОНОВАЯ КИСЛОТА. Мол. вес 216,18,
т. пл. 310—313° (с разл,).
Получение [1]. Раствор 1,01 моля едкого кали в 300 мл воды
нагревают до 60—70° при перемешивании с 0,505 моля технического
нафтойного ангидрида. Доводят pH темно-коричневого раствора до 7
и дважды осветляют норитом. Фильтрат концентрируют до объема
180 мл, охлаждают, обрабатывают при перемешивании 800 мл мета-
нола и охлаждают до 0—5°. Выпавшую дикалиевую соль отделяют,
промывают метанолом, высушивают при 150°/150 мм, измельчают с
4%-пым безводным хлоридом кадмия в шаровой мельнице в течение
4 час и помещают в автоклав (роль хлорида кадмия не выяснена).
Автоклав вакуумируют для удаления кислорода, заполняют дву-
окисью углерода при^30 атм и при перемешивании нагревают смесь
в течение 1,5 час при средней температуре 400—430°. При этом карбо-
ксильные группы мигрируют в менее экранированные положения
и при подкислении охлажденного расплава выделяется Н. к.
I. Реке Б,, Ш и р п Г., «Синтезы органических препаратов», ИЛ, М., 1964,
сб. 12, стр. 97.
1,2-НАФТАЛИНДИКАРБОНОВОЙ кислоты ангидрид.
Мол. вес 198,17, т. пл. 166—167°.
Получение [1]. Сложноэфирная конденсация этил-у-фенилбу-
427
тирата с диэтило ксал атом дает сырой эти л-а-этоксалилу-фенил бу-
тират, который циклизуется под действием конц. серной кислоты
при 20—25° в 3, 4-дигидро-1,2-нафтойный ангидрид.
Для дегидрирования нагревают на предварительно нагретой до
230—235° бане [сплав Вуда или смесь KNO:i и NaNO2 (10:7,5)] смесь
0,1 моля полученного ангидрида и 0,1 моля серы (в течение 30 мин
температура повышается до 250°); перегоняют продукт при понижет
ном давлении и кристаллизуют из смеси бензола с лигроином.
I. Гсршбер г Е., Ф и з е р Л., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 53, 347.
1,8-НАФТАЛИНДИКАРБОНОВОЙ КИСЛОТЫ АНГИДРИД.
Мол. вес 198,17, т. пл. 274°.
Окисление И. к. а. перманганатом калия в щелочной среде дает
гемимеллитовую кислоту [11.
NaOH, КМпО,
1, Graebe С., Leonhardt М., Ann., 290, 217 (1896).
НАФТАЛИН-р-СУЛЬФОКИСЛОТА, p-C10H7SO3H -НгО. Мол. вес
226,24; т. пл. 120—122°.
Н. используется в качестве катализатора дегидратации. Напри-
мер, Колер и сотр. [1] обрабатывали циклогептанол Н. (1—2 г на
1 моль спирта) и по мере отгонки продукта медленно повышали
температуру бани от 180 до 250°:
/З-СюЩБОзН^
80% *
428
Брауде и Эванс [21, обнаружившие, что сложный эфир (1) чрезвы-
чайно трудно дегидратировать обычными методами (нагреванием с
КИ5О4или иодом, обработкой SOC12 в пиридине или Р2О5 и триэтил-
амином), осуществили дегидратацию с перегруппировкой в (3),
нагревая 1 г (1) с 20 мг Н. при 1307100 мм.
СН3 СН3 Г СН3 СН2
| | p-c1{,Hrso,H | li
сн3с=снссо2сн3-----------> [сн3с-снссогсн3] —
| ' /о
он
(I) (2)
сн3 сн3
I I
-> СН2 - ссн =. ССО2СН3
(3)
1. К О h 1 е г Е. Р-, 'fishier М., Potter Н., Thompson Н. Т.,
J. Aim Chem. Soc., 61, 1057 (1939).
2. В г а и d е Е. Л., Evans Е. A., J. Chem. Soc., 1955, 3324.
НАФТАЦЕН-9,10,11,12-ДИХИНОН (4). Мол. вес 288,24; т. пл.
330—333° (с разл.)
Получение. Исходное соединение 11,12-диокси-9,10-хинон (3)
429
было получено двумя методами, ни один из которых не описан до-
статочно подробно. По одному из них [1] фталевый ангидрид конден-
сируют с янтарной кислотой и ацетатом калия при 200—220° в те-
чение 2 час, образующийся а, 0-дифталиденэтан (1, «этиндифталид»)
при обработке метилатом натрия в метаноле с последующим подкис-
лением изомеризуется частично в диоксинафтацеихиион (3, «изоэтин-
дифталид») и частично в бас-дикетогидрипден (2); выход (3) очень
низок. Второй метод, представляющийся более перспективным, хотя
выход не указан, состоит в конденсации фталевого ангидрида с
1,4-пафтогидрохиионом в расплаве натрийалюминийхлорида [21
(можно проводить конденсацию с H2SO4 — Н3ВО3; см. Борная кис-
лота). Диоксихинон (3) (возгоняется в вакууме в виде темно-красных
игл с т. пл. 349°) при окислении дымящей азотной кислотой в уксус-
ной кислоте даст с хорошим выходом Н. (4).
Диенофил Дильса — Альдера. Дихинон имеет реакционноспо-
собную 15,16-двойную связь, о чем свидетельствует образование ди-
хлорида и эпоксида [3]. При 100° в уксусной кислоте он реагирует с
2,3-диметилбутадиеном с образованием бесцветного аддукта (5)
с т. пл. 255—256° [4]. Подобно другим неспособным к енолизации
1,3-дикетонам [5], аддукт (5) легко расщепляется 25%-ным раство-
ром едкого кали в метаноле; после изомеризации и окисления возду-
хом первоначально образовавшегося нейтрального продукта полу-
чаются 2,3-ди мети л антрахинон и фталевая кислота.
1. G a b г i е 1 S., L е и р о 1 d Е., Вег., 31, 1159, 1272 (1898).
2. R а и d п i t г Н., Вег., 62, 509 (1929).
3. Voswinckel Н., Вег., 38, 4015 (1905); 42, 458 (1909).
4. F i е s е г L. F., Dunn J. Т., Am. Chem. Soc., 58, 1054 (1936).
5. Beckham L. J., Adkins H., J. Am. Chem. Soc., 56, 2676 (1932).
430
а-НАФТИЛИЗОТИОЦИАНАТ, a-CL0H7N = С = S. Мол. вес 185,25,
т. пл. 56—57°. Получение (11.
Анализ концевых групп в белках. Реагент используется анало-
гично фенилизоцианату и обладает тем преимуществом для спектро-
фотометрического анализа, что нафтильные производные имеют очень
интенсивный максимум поглощения при 222 ммк [2].
1. Dyson G. М., Hunter R. F., Rec. trav. Chim., 45, 421 (1926).
2. Ka tsuki S., Scott J. E., Yamash in a 1., Biochem. J., 97, 25C
(1965).
а-НАФТИЛ ИЗОЦИАНАТ, a-C10H7N — C = О. Мол. вес 169,18;
т. пл. 3—5°.
Реагент используют для характеристики оксисоединений путем
превращения их в нафти л уретаны. Например, смесь 0,2 г фенола
и 0,2 г Н. с 1 каплей пиридина нагревают на небольшом пламени.
Через несколько минут смесь становится красной; ее охлаждают и
твердое вещество кристаллизуют из лигроина.
1,2-НАФТОХИНОН (5). Мол. вес 158,15, т. пл. 145—147°
(с разд.).
Получение [1]. К смеси 0,5 моля сульфаниловой кислоты и
0,25 моля карбоната натрия, растворенной в 500 мл воды и охлаж-
денной до 15°, добавляют раствор 0,54 моля нитрита натрия в 100 мл
воды и выливают в смесь 1,25 моля соляной кислоты и 600 г льда.
Раствор, из которого вскоре выпадает сульфонат га-фенилдиазония,
оставляют на 15—25 мин. За это время в теплом растворе 2,75 моля
едкого натра в 600 мл воды растворяют 0,5 моля р-нафтола. К раст-
вору, охлажденному до ~5° прибавлением 400 г льда, добавляют
суспензию диазотированной сульфаниловой кислоты и оставляют на
1 час. Азосоединение (2) выделяют в виде густой пасты. Твердое ве-
щество вносят в раствор, нагретый до 45—50°, и постепенно добав-
ляют примерно одну десятую часть от 230 г (1,1 моля) гидросульфита
натрия до оседания пены и затем загружают весь остаток. Чтобы
образующийся продукт легко фильтровался, смесь сильно нагревают
до вспенивания, после чего охлаждают до 25° и продукт (3) кремо-
вого цвета отделяют и промывают. Сырой аминонафтол промывают
в стакане раствором 0,63 моля конц. соляной кислоты и 2 а хлористо-
го олова (антиоксидант) в 1 л воды при 30°. После растворения амина
раствор осветляют 10 г норита, фильтруют с отсасыванием, нагре-
вают до кипения и, обработав 100 мл конц. соляной кислоты, охла-
N = NCeH4SO3Na NH2
(О (2) (3)
431
ждают. Хлоргидрат 1* амино-2-нафтол а (4) выделяется в виде абсо-
лютно бесцветных игл. Для окисления в хинон (5) раствор 0,89 моля
хлорного железа в смеси 90 мл конц. соляной кислоты и 200 мл
воды охлаждают, добавляя 250 г лъд,а, и фильтруют с отсасыванием.
Смесь 0,41 моля хлоргидрата (4), 5 мл конц. соляной кислоты и 3 л
воды, предварительно нагретой до 35°, перемешивают 1—-2 мин до
растворения осадка и фильтруют с отсасыванием. Быстро при пере-
мешивании добавляют окислитель; хинон, выделяющийся в виде
объемного микрокристаллического оранжево-желтого осадка, со-
бирают и тщательно промывают водой. При этом сразу же получает-
ся чистый неустойчивый продукт; перекристаллизация понижает его
температуру разложения.
I. Физер Л., «Синтезы органических препаратов», ИЛ, М., 1949,сб. 2, стр. 353.
1,4-НАФТОХИНОН. Мол. вес 158,15, т. пл. 124—125°.
Получение. Один из эффективных путей к этому соединению —
реакция диазосочетания, аналогичная описанной для 1,2-нафтохи-
нона [1]. Другим методом получения Н. является диеновый синтез
[2]. К суспензии 1 моля 1,4-бензохинона в 500 мл уксусной кислоты,
охлажденной до начала кристаллизации растворителя, добавляют
1,2 моля сконденсированного бутадиена. Колбу закрывают проб-
кой, которую обматывают проволокой, охлаждают проточной водой
и периодически встряхивают в течение нескольких часов до полного
растворения хинона. Спустя 40—48 час раствор фильтруют через
слой активированного угля и нагревают фильтрат и промывные
воды на кипящей водяной бане. Добавляют раствор 100 мл конц.
соляной кислоты и 15 а хлористого олова в 500 мл воды и в течение
15 мин нагревают раствор на кипящей водяной бане для изомериза-
ции в продукт (3), который вскоре начинает выпадать в виде тяже-
лых белых игл. Дигидронафтогидрохинон (3) растворяют в горячей
уксусной кислоте, дают раствору остыть до 100° и быстро при пере-
мешивании вливают в него раствор нитрита натрия. Под действием
432
азотистой кислоты осуществляется мягкое окисление в 5,8-Дигид-
ро-1,4-нафтохинон (4), который выделяется с 91—97%-ным выходом.
Температуру раствора доводят до 65° и добавляют к нему теплый
(65°) раствор бихромата натрия, содержащий немного серной кис-
лоты. Смесь выдерживают при 65—70° в течение 15 мин, а затем,
добавляя ледяную воду (45 мин), осаждают ярко-желтый нафтохи-
нон (т. пл. 124—125°).
1. Ф и з е р Л., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 1,
стр. 286; F i е s е г L. F., Org. Syn., Coll., Vol., 2, 39 (1943).
2. Fieser L. F., Am. Chem. Soc., 70, 3165 (1948).
3. Burton H., P r a i 1 1 P. F. G., J. Chem. Soc., 1952, 755.
1,2-НАФТОХИНОН-4-СУЛЬФОКИСЛОТЫ АММОНИЕВАЯ И
КАЛИЕВАЯ СОЛИ. Мол. веса 255,25 и 276,31.
Получение. К раствору 2,1 моля р-нафтола в щелочи, обрабо-
танному при 0° нитритом натрия, медленно приливают разбавлен-
ную серную кислоту [1]; при этом выделяется 1-нитрозо-2-нафтол.
Влажную массу переносят на широкую воронку Шотта и промывают
холодным раствором 5,8 моля бисульфита натрия и 100 мл 6 н. NaOH
в 2 л воды [2, 3]. Смесь разбавляют водой до 4—4,5 л и энергично
перемешивают лопастной мешалкой так, чтобы все растворимые
продукты растворились за 3—4 мин. Раствор фильтруют как можно
быстрее, и прозрачный золотисто-желтый фильтрат сразу же подкис-
ляют серной кислотой. Выделяющаяся 1-амино-2-нафтол-4-сульфо-
кислота * имеет светло-серый цвет; после промывания теплым эта-
нолом до обесцвечивания фильтрата и затем эфиром (в темноте)
получают 370—380 г (75—78% в расчете на 0-нафтол) чистого про-
дукта в виде белого сухого порошка [4]. Окисление проводят раз-
бавленной азотной кислотой при 25—30° и раствором хлористого
аммония высаливают аммониевую соль, выделяющуюся в виде ярко-
оранжевых микрокристаллов высокой чистоты, которые превраща-
ются в калиевую соль с высоким выходом.
N=O
NOH
* Продажную 1-амшю-2-иафтол-4-сульфокислоту можно использовать после
промывания теплым этанолом н затем эфиром.
433
I, Марвел K-, Портер П., «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 1, стр. 300.
2. Ф и з е р Л., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 2,
стр. 51.
3. Мартин Е., Физер Л., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 69.
4. Мартин Е., Физер Л., «Синтезы органических препаратов», ИЛ, М.,
1952, сб. 3, стр. 367.
НИКЕЛЬ-АЛЮМИНИЕВЫЙ СПЛАВ (1 : 1, сплав Ренея).
При действии на сплав водным едким натром алюминий «вымы-
вается» с выделением водорода, а никель остается в виде черной губ-
ки, которая очень эффективна в качестве катализатора гидрирова-
ния. В некоторых случаях химическое восстановление осуществ-
ляется при реакции сплава Ренея со щелочью. Папа, Швейк и др.
[1, 2] восстановили этим методом бензиловый спирт в толуол (70%),
NiAla-f-6NaOH —► Ni + 2\а3АЮ3 + ЗНа
ацетофенон в этилбензол (70%) и 2-метилциклогексанон в 2-метил-
циклогексанол (80%). Наилучшие результаты были получены в тех
случаях, когда восстанавливаемое вещество растворимо в щелочи,
как, например, дикислота (2), образующаяся при окислении |3-наф-
тола надуксусной кислотой 13]. К раствору дикислоты в 500 мл
0,14 моля (II
ЗСН3СО3Н
67-7 0%
со,н
со2н
I
54 г NI-A1
NaOH
0,094 моля (2)
j со.н
I II I
(3)
10%-него едкого натра, нагретому до ~87°, в течение 50 мин при
перемешивании добавляют 54 г сплава Ренея. После перемешивания
и нагревания на кипящей водяной бане еще в течение 1 час смесь
фильтруют с отсасыванием, следя за тем, чтобы металл оставался
влажным, так как, высыхая, он становится пирофорным. Затем
металл разрушают, осторожно обрабатывая разбавленной азотной
кислотой (бурная реакция). Если карбонильная группа связана не-
посредственно с ароматическим циклом, то степень восстановления
90?
8О/о
10-20°
80%
н
434
зависит от температуры: при 80—90° происходит восстановление по
Клемме нсену [1, 2], тогда как при 10—20° продуктом реакции яв-
ляется карбинол, как это показано для случая о-бензоилбензойной
кислоты [41. Салициловый альдегид при 10—20° дает салигенин
(77%), а при 90° о-крезол (76%).
Сплав Ренея в щелочной среде восстанавливает с хорошим выхо-
дом а рил галогениды в углеводороды 15, 61; его используют для вос-
становления хлорированной полиеновой кислоты (4) в 6-фенил-«-
валсриановую кислоту (5) [7].
Ni-AI, NaOH
С6Н5СС1 = СС1СС1 = СС1СО2Н ---—> СеН5 1СН2)4СОгН
86%
Н) (5)
Оксимы и нитрилы без нагревания восстанавливаются сплавом
Ренея со щелочью в амины, как правило, с удовлетворительными
выходами [81.
I. Р а р a D., Schwenk Е., Whitman В.. J. Org. Chem., 7, 587 (1942).
2. Schwenk Е., Papa D., Hankin H., Ginsberg H., Org.
Syn., Coll. Vol., 3, 742 (1955).
3. Page G. A., T a г b e 1 1 D. S., Org. Svn., Coll. Vol., 4, 136 (1953).
4. С о о k P. L., J. Org. Chem., 27, 3873 (1962).
5. Schwenk E., Pap a D., Whitman B., Ginsberg H., J. Org.
Chem., 9, 1 (1944).
6, Ma r k I G., Chem. Ber., 96, 1441 (1963).
7. Roed ig A., Mark! G., S c h a a 1 V-, Chem. Ber., 95, 2844 (1962).
8. S t a s k u п B., van E s T., J. Chem. Soc., 1966, 531.
НИКЕЛЯ БОРИД, NLB. Браун описал два активных катали-
затора гидрирования. Обозначенный через Р-1 Н. б. получают вос-
становлением ацетата никеля боргидридом натрия в водном раство-
ре 11]. Сравнительное гидрирование олефинов, начиная от очень
активного сафрола до нереакционноспособного циклогексена, по-
казало, что катализатор Р-1 значительно активнее никеля Ренея
и менее склонен изомеризовать олефины в процессе гидрирования.
Второй Н, б., обозначенный через Р-2, получают восстановлением
ацетата никеля боргидридом натрия в этаноле[2]. Этот катализатор
менее активен, чем Р-1, и чрезвычайно селективен при гидрирова-
нии олефинов различного типа, что следует из периодов полугидри-
рования (в минутах): сафрол 3,5 (с Р-1 6) октеп-1 7, З-метилбутен-1
15, 3,3-диметилбутен-1 56, циклогексен — 200, транс-пентен-2 —360,
2-метилбутен-1 —400. Катализатор Р-2 восстанавливает гексин-3
в гц/с-олефин 98—99%-ной чистоты и для такого рода восстановле-
ний превосходит по своим качествам катализатор Линдлара.
Трюс [3] получил аналогичный описанному выше никелевый
катализатор восстановлением гексагидрата хлорида никеля (II)
боргидридом натрия в этаноле и использовал его для десульфури-
зации. Например, дифенилмеркапталь бензила (1) десульфуризуется
в две стадии через промежуточное соединение (2), а с избытком
435
i Ni..B 60%
CsH5S О C6H5S о о
I Ii Ni-.B | ;l NizB -I |;
C6H-C-CCeHr,-4* CcH5CH—CC6H5 C6H5CH2-CC6H5
[ 7 0 %
C0H5S
(1) (2) (3)
реагента — в одну стадию, в продукт (3).
1. Brown С. A., Brown Н. С., J. Am. Chem. Soc., 85, 1003 (1963).
2. Brown Н. С., Brow п С. A., J. Am. Chem. Soc., 85, 1005 (1963),
3. Truce W. E., Roberts F. E., J. Org. Chem., 28, 961 (1963); Tru-
c e W. E., Per г у F. M., J. Org. Chem., 30, 1316 (1965).
НИКЕЛЯ КАРБОНИЛ, Ni(CO)4. Мол. вес 170,73, исключительно
ядовитая бесцветная жидкость, т. пл.—25°, т. кип. 43°, уд. вес 1,31.
Характер данного обзора. Взаимодействие Н. к. с ненасыщен-
ными органическими соединениями изучается немногим более 20 лет,
однако обзоры Берда [1] и Шройцера [21 охватывают обширную ли-
тературу, уже имеющуюся в этой узкой области металлоорганиче-
ской химии. Поэтому настоящий обзор ограничивается лишь крат-
ким рассмотрением тех типов реакций, которые если и не полностью
поняты, то уже достаточно ясно определены.
Гидрокарбоксилирование ацетиленов. В 1953 г. Реппе [3] опуб-
ликовал подробности реакции «карбонилирования», осуществленной
с использованием Н. к. и названной более правильно гидрокарбок-
силированием (присоединение муравьиной кислоты). Реакция, ко-
торую в настоящее время считают более сложной, первоначально
описывалась следующим уравнением:
4CH-= CH + 4H2O+Ni (CO)43-2HCI 4CH2-CHCO2H + NiCl2-LH2
Джонс и др. [4] показали, что эту реакцию можно проводить
при температурах 60—70° и атмосферном давлении с 40—50%-ными
выходами. Ниже приведено несколько примеров [1,2] реакции:
Ni (СО}4
С6Н5С=СН —-----> С6Н5С = СН2
ioaH
Ni (СОЦ
Cl(H5C CCI13 —----- С6ывс СНСНз + СД15СН = ССОоН
со3н сн3
NUCO1 АсОНйС СНаОАс
АсОН2СС.-=ССН2ОАс-------- с/С^С\н
rlOjv-/ г!
Гидрокарбоксилирование ацетиленов по Реппе сначала прово-
дилось в присутствии кислоты, однако о роли кислоты или о при-
роде агента — переносчика окиси углерода мало что было известно.
Стернберг и др. [51 показали, что дифенил ацетилен можно
436
гидрокарбоксилировать в щелочном растворе и что в этом случае
источником окиси углерода является анион И. к. При встряхивании
углеводорода с насыщенным раствором едкого натра в метаноле в
присутствии избытка Н. к. в атмосфере гелия реакционная смесь при-
обретала темно-красную окраску; спустя 80 час смесь подкисляли и
после соответствующей обработки выделяли а-фенил-т/хжс-корич-
ную кислоту и 1, 2, 3, 4-тетрафени л бутадиен с указанными на схеме
выходами. Отметим, что коричная кислота образуется в результате
цис-присоединения муравьиной кислоты.
СеН5
I
С6Н5 СН
С СЙН5\ /Н С — Св1-15
!|! CHsOH-NaOH '-С7 [
С + М(СО)4 —— I! 4-С-СвН&
7 80 час. Не q q
cel-l5 CGHr/ хсоан сн
CfjHr,
25% 67%
Образование алленкарбоновых кислот. Хлорзамещенные аце-
тилены типа (1) дают с Н. к. алленкарбоновые кислоты (2)
60°
RX-C-^CR' + Ni (СО)4 + НаО^ RaC-C^CR'
Cl CCbH
(1) (2)
16], выходы которых очень низки (5—15%), возможно, из-за неустой-
чивости продуктов. При обработке а-оксиацетиленов (3) Н. к.,
бутанолом и сухим хлористым водородом получаются алленкар-
боновые кислоты через промежуточное образование хлорида.
HCI-BnOH Ni (СО).,
(СН3)2С-С^СН-----(СН3)а С —С=зСН --------(СН3)аС=С = СНСО2Н
'I 6" I
ОН С1
(3) (4) (5)
Гидрокарбоксилирование олефинов. Если ацетилены обычно
претерпевают гидрокарбоксилирование при атмосферном давлении
и комнатной температуре, то олефины требуют более жестких усло-
вий. Например, для получения циклогексанкарбоновой кислоты
Реппе и Крепер [7] нагревали циклогексен с Н. к. при 270°/100 атм.
Берд и др. 18] высказали мнение, что напряженные олефины должны
реагировать более легко, и действительно показали, что норборнен
(6) взаимодействует с Н. к. в смеси этанол — вода — уксусная кис-
лота при атмосферном давлении и при 50° с образованием с хорошим
выходом смеси экзо-кислоты (7), соответствующего сложного эфира
437
(8) и следов кетона (9). Реакция состоите ц«с-присоединении муравьи-
ной кислоты из менее пространственно затрудненной области.
Реакция с диазоалканами [9]. Каталитические количества Н. к.
разлагают диазоалканы до продуктов, по-видимому, образующихся
из промежуточного кетена. Использование большого избытка
Н. к. в присутствии этанола приводит к образованию сложных
эфиров карбоновых кислот с 20—25%-ным выходом.
+ -СО - -Na +
RsC-N = N-[-Ni (СОц---* R2C—Ni (СО)3-->- R,C-Ni (СО)3——
j -N1 (СО)а
сан,он
— ру==С^О-------R2CHCOSC2H6
Взаимодействие с борфторидами диазония [10]. В присутствии
ацетона или уксусной кислоты Н. к. реагирует с борфторидом диа-
зония, образуя карбоновую кислоту с выходом порядка 5—70%.
ч- № (СО).
ArNs=N (BF7) —щ----+ АгСО2Н
1 4 1 АсОН-Н2О 2
Взаимодействие с арилиодидами [11]. При кипячении в мета-
ноле, этаноле или изоиропаполе Н. к. реагирует с арилиодидом с
образованием с хорошим выходом соответствующего сложного эфира.
О
И
Arl + Ni (СО)4+С3Н5ОН — АгСОС2Но
В тех же условиях арил бромиды, арилхлориды и а л кил галогениды
не вступают в реакцию. При использовании в качестве растворителя
ТГФ продуктом реакции является арил-а-ди кетон.
О О
ТГФ II ||
CeH5I -1- Ni (СО)4 ~~-у CGH5C ССвН5
о и %
Конденсация аллильных соединений. Болд [12] показал, что
аллил- и циннамилацетаты реагируют с И. к., образуя с умеренными
выходами диаллил и дициннамил.
45 — 6 5°, 2 час
2RCH ~CHCH2OAc-|-Ni (СО)4------>
—- RCH - СНСНаСН2СН = CHR ф Ni (ОАс)2 ф 4СО
R = H 50%-ный выход
R=CeH6 31%-ный выход
438
Кори и Хаманака [13] использовали внутримолекулярную ал-
лильную конденсацию в новом синтезе карбоциклических систем
средних размеров. Аллильный а,и-дигалогенид (1) при циклиза-
ции под действием Н. к. дает в качестве основного продукта транс,
mpawc(mpawc-диметил цикл ододекатриен (2), а также, по-видимому,
цис,транс, транс-изомер
?Нз транс. Ni(COb
ВгСНгСН=С(СНг)гСН=СН(СНг)гС=СНСНгВЕ—
1 63-68%
+ цис, трапе,
(2)
1. В i г d С. W., Chem. Revs,, 62, 283 (1962).
2. Schrauzer G. N., Advances in Organometallic Chemistry, 2, 1 (1964).
3. Rcppe W., Ann., 582, 1 (1953).
4. J ones E. R. H., Shen T. Y., Whiting M. C., J. Chem. Soc.,
1950, 230; 1951, 763, 766.
5. Sternberg H. W., Markby R., W c n d e г I., J. Am. Chem. Soc.,
82, 3633 (1960).
6. J о u e s E. R. H., W h i t h a m G. H., Whiting M. C., J. Chem.
Soc., 1957, 4628.
7. R e p p e W., К г 6 p e r H., Ann., 582, 38 (1953).
8. Bird C. W., Cookson R. С., H и d e c J., Williams R. O., J.
Chem. Soc., 1963, 410.
9. Riichardt C., Schrauzer G. N., Chem. Ber., 93, 1840 (1960).
10. С 1 a r k J. C., Cookson R. C., J. Chem. Soc., 1962, 686.
11. В a u 1 d N. L., Tetrahedron Letters, 1841 (1963).
12. В a u 1 d N. L., Tetrahedron Letters, 859 (1962).
13. Corey E. J., Hamanaka E., J. Am. Chem. Soc., 86, 1641 (1964).
НИКЕЛЯ ПЕРЕКИСЬ.
Реагент, представляющий собой черную водную смесь высших
окислов никеля, получают обработкой сульфата никеля в щелочном
растворе гипохлоритом натрия [11. В щелочном растворе насыщен-
ные алифатические первичные спирты, хорошо растворяющиеся
в воде, окисляются в карбоновые кислоты (3—5 час при 30°). Бензи-
ловый спирт окисляется щелочным реагентом в бензойную кислоту,
однако, если в качестве растворителя использовать бензол, при
небольшом избытке Н. п. окисление идет лишь до бензальдегида.
Этим методом можно также окислить аллиловые спирты до
альдегидов.
Суспензия реагента при 80° окисляет анилин в азобензол с выхо-
дом 80%, а бензиламин при 60° —• в бензонитрил с 78% -ным выходом
[2]. В апротонных растворителях (бензол, эфир) Н. п. окислительно
расщепляет сс-кетолы и а-кетокислоты. Этим свойством реагент на-
поминает тетраацетат свинца.
Н. п. окисляет основание Шиффа типа (1) в бензоксазол (2) в бен-
золе или эфире при комнатной температуре [4].
439
о-Феиилендиамины окисляются Н. п. в ^г/с,^«с4,4-дицианбутадие-
СНС6Н5
(1)
Окисление,15^
------------->
72%
ны-1,3, правда, с очень низкими выходами (9—26%) [5]. Реагент
был использован для окисления 4-цианпирокатехина в о-хинон [6!.
Окись серебра в этой реакции оказалась неэффективной.
В присутствии аммиака при —20° ароматические и а, Р-непре-
дельные альдегиды окисляются II. п. до амидов. При более высоких
температурах образуются нитрилы.
NHS
АгСНО----»-
г ОН л
I -Н..0
ArC— NH2-----> ArC— NH
_ Н Н
|[О] |[OJ
о
II
ArCNH2
ArC^N
1. Nakagawa К., Konaka R., Nakata T., J. Org. Chem., 27,
1597 (1962).
2. Nakagawa К., I s u j i T., Chem. Pharm. Bull., 11, 296 (1963).
3. Nakagawa K., Igano K., Sugita J., Chem. Pharm. Bull., 12, 403
(1964).
4. Nakagawa К., Олове H., Sugita J., Chem. Pharm Bull., 12
1135 (1964).
5. Nakagawa К., О n о u e M., Tetrahedron Letters, 1433 (1965).
6. A n s e 1 1 M. F., Gosden A. F., Chem. Comm., 1965, 520.
7. Nakagawa К., О n о u e H., Minam i K-, Chem. Comm., 1966, 17.
НИНГИДРИН (5). Мол. вес 178,14, т. пл. 253° (с разл.)
Шестистадийный синтез нингидрина исходя из двойной сложно-
эфирпой конденсации ди мети л фталата с этилацетатом описан в ка-
честве учебного опыта [1]. В новом синтезе [2] на первой стадии
диэтилфталат добавляют по каплям к суспензии метилата натрия в
ДМСО при перемешивании в токе азота. Первоначально образую-
щийся р-кетосульфоксид (2) претерпевает внутримолекулярную сло-
жпоэфирную конденсацию и образует 1,3-индандионовую систему
(3), которая в присутствии соляной кислоты перегруппировывается
440
по Пуммереру в 2-хлор-2-метилмеркаптоипдаидион-1,3 (4). Полу-
чепный кристаллический продукт с т, пл. 63° (80%-ный выход) гид-
ролизуется в кипящей воде с почти количественным выходом в нин-
гидрин (5).
1. Org. Expts., 138.
2. Becker Н. D., Russell G. A., J. Org. Chem., 28, 1896 (1963).
НИТРИЛ ИОДИСТЫЙ, INOa. Мол. вес 172,92.
Хасснер и сотр. [1] получили Н. и. in situ из 1 же нитрита сере-
бра и иода в атмосфере азота в эфире при комнатной температуре.
Авторы показали, что Н. и. присоединяется к некоторым олефинам,
например к А2-холестеиу:
Аддукты иногда нестабильны, но их можно дегидрогалогенировать
в ненасыщенные иитросоедипения:
ino2
с6н5сн=сн,—> ГС6Н3СН—CH3NO2'
L 1
--^c6h5ch^ci-ino3
1. Hassner A., Univ. Colorado, private communication.
НИТРИЛ ХЛОРИСТЫЙ, C1NO2. Мол. вес 81,47; т. кип. от —17
до —15° (бледно-желтая жидкость).
Получение из безводной азотной и хлорсульфоновой кислоты [11:
HNO3-рC1SO3H CINO, + H3SO4
441
Присоединение к олефинам. Н. х. присоединяется при 0° к
акриловой кислоте и акрилонитрилу:
CINOj
СН2 = СНСО2Н---► СН,—СНСОпН
2 2 71 % [ 2 | 2
NO2 Cl
CINOa
ch.2^chcn------► CH2 —CHCN
NO2 Cl
С несимметричными терминальными олефинами H. х. образует
1-нитро-2-хлоралканы:
RCI-I = СН2 —R СНСН2 NO3
Нитрование кетонов. Непосредственное нитрование протекает
плохо, и а-нитрокетоны были получены лишь с низким выходом
через енол ацетат [2].
СН3СН - ССН3 —СН3СН — ССН3
I 36% I Ii
OAc NO2 О
1. Shechter Н., Conrad F., Daulton A. L., Kaplan R. В.,
J. Am. Chem. Soc., 74, 3052 (1952).
2. Bachman G. B., Hokama T., J. Org. Chem., 25, 178 (1960).
4-НИТРОАЗОБЕНЗОЛ-4-КАРБОНОВОЙ КИСЛОТЫ ХЛОР-
АНГИДРИД (4). Мол. вес 289,67, т. пл. 163°.
Реагент получают по нижеприведенной реакции [1]. На первой
стадии суспензию 6,2 г ц-нитроанилина в 125 мл воды обрабатывают
16,2 г 51%-ной надуксусной кислоты, встряхивают в течение 72 час
и оставляют на 48 час. Фракционная перегонка с паром дает в пер-
вых двух фракциях п-нитронитрозобензол удовлетворительной чис-
тоты, а в последующих фракциях я-динитробензол и 4,4'-динитро-
азобензол.
o2n_^^_Nh2о,к-Г“У NO <А°0Н' Щ
33% \=/ 85-90%
(I) 12)
__ SOCR, ЩН.СН,
О2М— N = N—Ч-СО2Н -Ка2со
\=У \=/ 94%
_____(3) __
QgN—N = N —СОС1
(И
Сложные эфиры, полученные при взаимодействии Н. к. х. с пер-
вичным или вторичным спиртом в бензоле, содержащем пиридин,—
это интенсивно окрашенные (X 297—320; е ~30 000) высокоплавкие
442
кристаллические вещества: т. пл. метилового эфира 187°, этилового
эфира 163°, цетилового эфира 11°, циклогексилового эфира 165°
и кротилового эфира 136° [1]. Изучение модельных сложных эфиров
показало, что смеси легко разделяются методом противоточного рас-
пределения и хроматографией [2].
Бутенандт и сотр. [3] применяли окрашенные ацилхлориды для
исследования действующего начала полового аттрактанта, выделя-
емого самкой шелкопряда, — бомбикола, который они получили в
виде эфира 4'-нитроазобензол-4-карбоновой кислоты (2). Из
500000 пар пахучих желез было выделено 12 мг чистого сложного
эфира с т. пл. 95—96°. Окисление 1 мг этого сложного эфира пер-
манганатом калия в ацетоне, отделение окрашенного фрагмента и
этерификация диазометаном дают сложный эфир, идентичный, по
данным бумажной хроматографии, метиловому эфиру кислоты (3).
Перегонка с паром реакционной смеси дает дистиллат, содержащий
Н Н кмпо4
I I | ArCOCl в ацетоне
НО(СНа)9С = С—С = С(СНа)гСН3---> Эфир —
10 [ 12 (2)
н
(1)
о
—>Q.,N—М —N—Д—(5—О(СН2)эСО2НЧ-НОаССО2Н-Б
(3) (4)
+ НО3С(СН2)аСН3
(5)
масляную кислоту (5), тогда как в остаток переходит щавелевая
кислота (4); обе кислоты идентифицированы методом бумажной
хроматографии. Этим методом было установлено, что бомбикол
представляет собой Д10,13-гексадиенол-1.
Эмии [4] исследовал окрашенные производные, полученные при
взаимодействии Н. к. х. с первичными и вторичными аминами и
тиолами. Нейрат и Дерк [5] охарактеризовали 45 амидов, получен-
ных из Н. к. х. и первичных и вторичных аминов. Эти амиды
отлично кристаллизуются и поглощают при 336—337 ммк с высокими
коэффициентами экстинкции.
1. Hecker Е., Chem. Вег., 88, 1666 (1955).
2. A m 1 п Е. S., Hecker Е., Chem. Вег., 89, 695 (1956).
3. Butenandt A., Beckmann Т., Hecker Е., Z. physiol.
Chem., 324, 71 (1961); Butenandt A., Stamm D., Hecker E.,
Chem. Ber., 94, 1931 (1961).
4. Amin E. S., J. Chem. Soc., 1957, 3764; 1958, 4769; 1959, 1619.
5. Neurath G., D о e r k E., Chem. Ber., 97, 172 (1964).
я-НИТРОБЕНЗИЛ БРОМИСТЫЙ (ХЛОРИСТЫЙ),
n-NOaCeH4CHaBr(Cl). Мол. вес 216,04 (171,72), т. пл. 100° (71°).
443
Защитная группа. /г-Нитробензильная сложноэфирная группа
используется в пептидном синтезе для защиты карбоксильной груп-
пы [1]. Сложный эфир получают обработкой N-защищенной амино-
кислоты реагентом в присутствии триэтиламина. Группа очень устой-
чива к действию НВг в уксусной кислоте—реагента, обычно исполь-
зуемого для отщепления карбобеизоксигруппы. Она удаляется
гидрогенолизом.
1. Schwyzcr R., Sieber Р., Ileiv. Chim. Acta, 42, 972 (1959); Fos kcr
A. P., Law II. D., J. Chem. Soc., 1965, 4922.
я-НИТРОБЕНЗОИЛ ХЛОРИСТЫЙ, n-NO2CGH4COCl. Мол. вес
185,57, т. пл. 72—74°.
Кисс [1] сообщил, что, как правило, /г-бензоил аминобензоаты
сахаров очень легко кристаллизуются и подвергаются гидролизу
в условиях, в которых большинство сахаров устойчиво (0,1 н. NaOH
в НаО — СН3ОН). Получают эти эфиры ацилированием п-нитро-
беизоил хлоридом, восстановлением нитрогруппы над никелем
Ренея и бензоилированием.
О
' л-ЫОгС(Н4СОС1 Н H,Ni
ROH (сахар)----—----—ROCCfiH4NO2-n >
Осп она пне
о о
—> ROCC6H4NH.rn ROCC6H4NHCOC6H5
Основанне
1 .Kiss J., Chem. Ind., 1964, 32.
N-я-НИТРОБЕНЗОЛСУЛЬФОКСИУРЕТАН (3). Мол. вес 290,26,
т. пл. 116,5°.
Н. получают [1] из N-оксиуретана (1) и п-нитробензолсульфо-
х лорида в присутствии основания. Избыток триэтиламина отщепляет
п - нитробензолсульфокислоту, и образуется высокорсакционноспо-
соб иый карбэтоксииитрен (4), который в присутствии циклогексена
дает карбэтоксиэтиленимин (5) и З-циклогексенилуретаи (6).
он о н
с2н5ос&он + C1SO2C6H4NO2-7Z
444
I. L wowski W,, Mari ci ch T. J., Mattingly T. W-, Jr., J. Am.
Chem. Soc., 85, 1200 (1963).
1-НИТР0ГУАНИЛ-3,5-ДИМЕТИЛПИРА30Л (4). Мол. вес
171,16, т. пл. 125—126°.
Получение [1]. Конденсация нитрогуанидина (1) с гидразином
дает иитроаминогуапидин (2), который циклизуется с енольной фор-
мой ацетилацетона (3), образуя (4).
nh2 NH— NHZ Hjcl^ Й
C=NH OH (3)
1 NHNO2 -2H2O
(1) (2)
I
C=NH
I
NHNOz
(4)
Модификация белков [11. Н. взаимодействует со свободными
аминогруппами белка (5), при этом NH, замещается нитрогуанидиль-
иой группой; другими словами, он осуществляет нитрогуанидирова-
ние белка и модифицирует его замещением положительно заря-
женных аммонийных групп неосновными иитрогуанидилы-гыми
группами.Таким образом, реакция имеет значение для направленной
модификации ферментов. Остатки тирозина, триптофана и гисти-
дина действию IT. не подвергаются.
<5)
Белок—nh2
I
C=NH
NHNO2
(4)
-> Белок-NHC=NH
I
nhno2
(7)
(6)
1. Habceb A. F. S. A., Biochem biophysica Acta, 93, 533 (1964).
НИТРОЗИЛСЕРНАЯ КИСЛОТА, HOSO2NO2. Мол. вес 127,09.
Получение твердых сульфатов диазония по методу Ходгсона 111
включает образование нитроз плеер ной кислоты in situ. Холодный раст-
вор 1 г анилина в 10 мл уксусной кислоты перемешивают с раство-
ром 1 г нитрита натрия в 5 мл конц. серной кислоты при 0°. Через
10—30 мин при 0° добавляют при перемешивании 50 мл эфира для
осаждения твердого сульфата диазония, который собирают и промы-
вают смесью уксусная кислота — эфир и затем эфиром; выход 74%.
Получить этим методом соли из аминов, содержащих одну или две
нитрогруппы, не удалось. Образующиеся соли содержат примесь
бисульфата натрия.
445
Оба эти недостатка исключаются при использовании твердой Н. к.
[2], которую получают пропусканием сернистого ангидрида в азот-
ную кислоту с уд. весом 1,5 при охлаждении до затвердевания всей
реакционной смеси. Твердый продукт собирают на стеклянном филь-
тре и отмывают от азотной кислоты небольшим количеством уксус-
ной кислоты и затем четыреххлористым углеродом; выход 70%.
К раствору твердой Н. к. (избыток 10%) в 10 мл уксусной кислоты
добавляют раствор или суспензию 2 а амина в 10 мл уксусной кис-
лоты. Через 30 мин для осаждения сульфата диазония при переме-
шивании и охлаждении добавляют 100 мл эфира.
При обработке циклоалканкарбоновых кислот в хлороформе при
65—70° Н. к. в 15—30%-пой дымящей серной кислоте образуются
лактамы [3].
76, 5% (7Z= 10)
88, 5% ( 72 = 11)
hoso2no2-chci3
65-70°
СО2
е-Капролактам получают с 60%-ным выходом, обрабатывая
циклогексанкарбоновую кислоту жидкой двуокисью серы, азотной
и серной кислотами [4]. По-видимому, in situ получается нитрозил-
серная кислота.
При катализе сульфатом ртути происходит 1,4-присоединение
Н. к. к сопряженному диену с образованием С-нитрозосульфата
(1), который самопроизвольно циклизуется в бН-1,2-оксазин (3),
дающий при пиролизе пиррол (4) [51.
Н3С ^СН2
NO*
I
oso2oh
(HgsoJ
c
H3C CH2OSO3H
(1)
H-jC CH=NOH
c
II
c.
H3c CH2OSO3H
(2)
98%
сн2
н,с
(3)
О5ОгОН 16QO
NH + СН2О
(4)
Н3С^ ^CH2NO
1. Hodgson H. H., Mahadevan A. P., J. Chem. Soc., 1947, 325.
2. P i e г с e у M. R-, Ward E. R., J. Chem. Soc., 1962, 3841.
3. Ziegenbein W., Lang W., Angew. Chem., Internal. Ed., 2, 149 (1963).
4. Tokura N., Kawahara T„ Sato T., Bull. Chem. Soc., Japan,
38, 849 (1965).
5. К lama nn D., Fligge M., Weyer st ahi P., Kratzer J.,
Chem. Ber., 99, 556 (1966).
446
НИТРОЗИЛ ФТОРИСТЫЙ, FNO. Из нескольких методов полу-
чения этого высокореакционноспособного газа, приведенных в об-
зоре Эндридса [11, наиболее предпочтительным считается пиролиз
смеси борфторида нитрозония и фтористого натрия. Босвелл [2]
показал, что если ток получаемого таким методом газа пропускать
в раствор холестерил ацетата в хлористом метилене при 0°, то раствор
становится темно-синим и после обработки с высоким выходом полу-
чают 5а-фтор-6-нитримин (2). Этот продукт гидролизуется нейтраль-
ной окисью алюминия в 5а-фтор-6-кетон (3), который при восстанов-
лении цинком в уксусной кислоте дает дегалогенированный кетон
(4)-
1. Andreades S., J. Org. Chem., 27, 4157, 4163 (1962).
2. Boswell G. A., Jr., Chem. Ind., 1965, 1929.
НИТРОЗИЛ ХЛОРИСТЫЙ, CINO. Мол. вес 65,47, т. кип. —5,5°.
Для получения Н. х. газообразную двуокись серы пропускают
в холодную дымящую азотную кислоту; образующуюся при этом
нитрозилеерную кислоту нагревают с эквивалентным количеством
хлористого натрия и перегоняют И. х. в приемник, охлаждаемый
сухим льдом [1]. При другом методе [21 сухой хлористый водород
пропускают в нитрит натрия:
NaNO2-j-2HCl —> ClNO-j-NaCl+НаО
Н. х. можно также получить in situ из алкил нитритов (этил-
или амилнитрита) и соляной кислоты либо соляной кислоты в ме-
таноле, этаноле или ускусной кислоте [3].
N-Нитрозирование (NH—>NNO). Если вторичный амин доста-
точно хорошо растворим в разб. соляной кислоте, то простейший ме-
тод нитрозирования состоит в прибавлении к такому раствору нит-
рита натрия и отделении осаждающегося N-нитрозопроизводного.
447
В случае аминов или лактамов, нерастворимых в водных кислотах,
или в случае высокореакциониоспособиых нитрозосоединений сле-
дует использовать Н. х. в неводной среде.
Хейльброи и сотр. [4] растворяли 5 г ацетанилида в 35 мл уксус-
ной кислоты и 15мл уксусного ангидрида, добавляли 5 г расплавлен-
ного ацетата калия и 0,5 г пятиокиси фосфора, перемешивали смесь
при 8° и медленно добавляли 3 г 25%-кого раствора Н. х. в уксус-
ном ангидриде. Через 15 мин раствор выливали в воду со льдом,
после чего выделяли 4,7 г желтого кристаллического М-нитрозоаце-
танилида. Метод оказался пригодным для п-нитроацетанилида, ко-
CHgCONH CH3CONNO
А Л
I [| CINO. АсОП, Ac,O. КОЛс, P2O., f II
торый не удалось нитрозировать нитрозными газами. Хьюстон [5]
с успехом применил эту методику для получения очень реакционно-
способных N-нитрозолактамов типа I. Бэкер и сотр. [6] несколько
модифицировали методику Хейльбропа (не добавляли пятиокись
ni
фосфора) и получили с 90%-ным выходом N-нитрозосоеди пение II.
Ньюмен и Катнер [7] превратили ряд 2-оке а зол ид оно в в 3-нитроз о-
2-оксазолидопы (Ш) добавлением раствора Н. х. в уксусном ангид-
риде к раствору оксазолидона в пиридине при 15°. Чен и сотр. [8]
показали, что метод с применением пиридина пригоден для ряда диа-
риламинов, недостаточно растворимых в водной кислоте. Дифенила-
мин дает с 95%-ным выходом N-нитрозодифениламии.
Ранделу и Мюллеру 191 удалось получить N-нитрозоэтиленимин
в эфирном растворе реакцией этиленимииа, Н. х. и триэтиламина
[>-н
C1NO- Эфир
(Et)3N
-70°
ПНг + n2o
сн2
(все в эквимолярных количества/Х) при —70°; при комнатной темпе-
ратуре продукт разлагается на этилен и закись азота. Тем же мето-
дом Кларки Хелмкамп [10] получили М-нитрозо-траж>2,3-диметил-
эти лени мин в виде неустойчивого желтого масла.
448
Нитрозохлориды. Первый метод превращения терпеновых угле-
водородов в кристаллические производные был предложен в 1877 г.
Тильденом [111: присоединение Н. х. по двойной связи с образова-
нием нитрозохлорида. Раствор терпена в хлороформе охлаждали до
—10‘ и насыщали Н. х. При этом раствор становится ярко-зеленым,
и когда проба укажет на окончание реакции, раствор выливают в
2—3 объема метанола; нитрозохлорид выделяется в виде очень мел-
ких кристаллов. Все терпены, исследованные Тильденом, содержали
ди- и три замещенные двойные связи и давали бесцветные нитрозо-
хлориды. Более поздние исследования тетр аз вмещенных олефинов
показали, что они образуют темно-синие нптрозохлориды (1) [31:
R2C-CR2-|-C1NO —> R2C--CR-2
| | (синий)
CI N-0
(I)
Авторы книги считают, что цвет обусловлен вкладом ионной резо-
нансной структуры (а):
К тризамещенным несимметричным олефинам II. х. обязательно
присоединяется таким образом, что атом хлора идет к наиболее за-
мещенному атому углерода, как в (2). Однако в отличие от (1), про-
дукт бесцветен. В настоящее время принято считать, что менее
экранированное нитрозосоединение (2) димеризуется в более устой-
чивую бесцветную форму (3). Нагревание иногда приводит к частич-
R2C = CHR
CINO
R,C—CHR lyLtR2C-CHR
ii ci n+— o-
(2) -O--N+ Cl
L I
RCH- CR.
(3)
ной диссоциации в синий мономер (2). С d-ли моченом, имеющим трн-
замещениую эндоцикл и ческ у ю двойную связь и дизамещенную экзо-
циклическую двойную связь, И. х. реагирует преимущественно по
первой из функциональных групп [31 (см. схему реакции на стр 450).
Мейнвальд и сотр. 112] исследовали стереохимию димерных пит-
розохлоридов, полученных из норборнена [11 и нор бор и адиен а, и
пришли к выводу, что продукты имеют жзсн^ис-конфигурацию, как
15 Заказ JN5 1096
449
Нзс/'^СН2 И3С/^СН2
в (3). Отсутствие перегруппировки, цис-присоединение и отсутствие
взаимодействи я с ну к леофл л в ным р аствор ителем (С ЛI .->011, СН ;!СООН)
говорят против ионного механизма присоединения. Свободноради-
кальный механизм, инициируемый -NO, маловероятен, так как
(3, £&ИЛИ Z)
Димер
окись азота не реагирует с норборнадиепом. Meйнвальд предложил
четырехцентровой механизм с малым участием карбопиевого иона
в переходном состоянии (2).
В более поздней работе Мейпвальд [131 сделал вывод о /{«с-при-
соединеиии II. х. к напряженным двойным связям, таким, как в
нор бор пене, и о трап с- присоединении к ненапряженным олефинам
типа А9-окталипа. Сообщение Хасснера [14] о взаимодействии холе-
стерилацетата (4) согласуется с этой точкой зрения. Н. х. пропуска-
ли в хлористый метилен или четыреххлористый углерод до окраши-
вания раствора в темно-красный цвет, добавляли стероид и выдер-
живали смесь в темноте в течение 2—24 час при температуре от —16
до 0°. Продукт представляет собой хлор нитросоединение, очевидно
образующееся из нитрозохлорида путем окисления его И. х. Обра-
зовавшемуся продукту приписана 5а-хлор-6|3-нитроконфигурация,
возникающая при транс-присоединении, главным образом на осно-
вании того, что он взаимодействует с цинковой пылью и соляной
кислотой, отщепляя C1NO2 и регенерируя соединение (4). транс-
Диаксиальная ориентация групп способствует такому отщеплению.
450
С другой стороны, реакцию с пиридином при комнатной температуре
с образованием 6-нитрохолестерил ацетата (6) следует описать как
необычно легкое ^ис-отщепление.
Джонс и сотр. [15] отметили, что с тщательно очищенным Н. х.
холестерил ацетат реагирует очень медленно и объяснили это при-
сутствием в обычном реагенте двуокиси азота. При добавлении к чис-
тому реагенту двуокиси азота нитрохлорид образуется за 2 час с
74%-ным выходом.
Оно и сотр. 116] показали, что направление присоединения Н. х.
зависит от растворителя. Так, присоединение Н. х. к циклогексену
в двуокиси серы дает транс-аддукт, тогда как в хлористом метилене,
хлороформе или трихлорэтилене образуется цис-аддукт.
Регенерация олефина из аддукта с Н. х. является важной
стадией в синтезе чистого А’-окталина. Одним из двух эффективных
методов получения сырого углеводорода является гидрирование
р-пафтола до 2-декалола и дегидратация (H3PO,i или Н3ВО3); второй
метод — восстановление тетралина при добавлении лития к раствору
тетралина в этилендиамине [17]. Продукт, полученный первым ме-
тодом, нагревают с пятиокисью фосфора, чтобы сдвинуть равновесие
в сторону образования несколько большего количества А9-изомера.
Даубсн [17] сравнил эти два метода и показал, что в каждом из них
получается вещество, содержащее, согласно данным ЯМР, глав-
ным образом А9-окталин (I), а также 10—20% углеводорода с
ш
Синий,т.пл. 91° Белый,т.пл. 127°
ди- и тризамещенными двойными связями. Бенкссер [18] показал, что
главным побочным продуктом является Ак9’-окталин (II) и охарак-
теризовал оба изомера, проведя реакции с Н. х. и выделив синий
нитрозохлорид III и плавящееся при более высокой температуре
димерное производное IV (III и IV, по-видимому, транс-изомеры).
Так как А9-окталин преобладает в смеси, то синий нитрозохлорид
III легко получить в чистом виде кристаллизацией. Бепкесер реге-
нерировал чистый А9-окталин обработкой синего производного мети-
15* 451
л атом натрия. Дау бен [17] перемешивал раствор Ш в эфире с цин-
ковой пылью и добавлял по каплям соляную кислоту; выход Ан-окта-
лина равен 59%, что сравнимо с выходом, полученным с метилатом
натрия. Хассей [19] получил окталиновую смесь дегидратацией
2-декалола фосфорной кислотой и осуществил превращение в синий
нитрозохлорид III смешиванием 0,5 моля изоамил нитрита с 0,5 моля
конц. соляной кислоты при —10° и добавлением 0,5 моля окталино-
вой смеси. Через 1,5 час отделяли синий осадок и промывали его эта-
нолом, охлажденным льдом (выход сырого продукта 75%). Кристал-
лизация из смеси ацетон — эфир дала синие призмы чистого нитро-
зохлорида с т. пл. 91—92°; выход 60%. Для регенерации углеводо-
рода раствор 0,094 моля нитрозохлорида в 30 мл N, N-диметилани-
липа нагревают до 70°, причем начинается выделение газа (CINO).
Нагревание продолжают при 85° 2,5 час, затем смесь разбавляют во-
дой и экстрагируют пентаном чистый А9-декалин; выход 85—95%.
Хотя скорость реакции олефинов с Н. х. снижается с уменьше-
нием числа алкильных заместителей, кристаллические нитрозохло-
риды (димерные) были получены даже из этилена и пропилена [3].
Имеются доказательства, что И. х. с дигидропираном дает цис-
аддукт. Серфонтейп и сотр. [20] показали, что 3,4,6-три-О-ацетил-о-
глюкаль (1) дает кристаллический аддукт, представляющий собой,
поданным ЯМР, 1-хлор-2-нитрозо-1,2-дидезоксисахар (2). Аналогич-
ные результаты были получены Лемье и сотр. [211. Соединения типа
(2) представляют интерес благодаря своей способности превращаться
в 2-а ми по-2-дезокси глюкозиды.
Клосс [22] описал интересную реакцию восстановления нитро-
зохлоридов, полученных из тетразамещепных олефинов, в хлорами-
нопропзводные и их циклизацию основанием в этиленимины. Нит-
розохлориды менее замещенных олефинов в этих условиях разла-
гаются.
„„ CINO в CH3OH( —70°)
К 2^ — К 2 —------------—>
Кол ИЧ.
RSC—CR3
SnCla-HCl
NO Cl
2c - cr2 2^
nh3 i
r2c~cr3
N
H
452
Пуммерер [23] сообщил, что нитрозохлорид (2) при кипячении
с пиридином дает се, р-ненасыщенный оксим (3).
н3с /Сн2сн
СН3
C1NO
Кипячение в Ру
(2)
НзС^/СНзСНд
"Y ^NOH
CHg
(3)
Мейнвальд и сотр. [24] использовали Н. х. для удаления олефи-
новых побочных продуктов (3), образующихся при синтезе нортри-
циклилацетата (2) катализируемым BF3 присоединением уксусной
кислоты к норборнадиену (1). Реакционную смесь, содержащую
CINO СНС13(-10в)
(4)
10—15% непредельного ацетата (3), растворяли в хлороформе, про-
пускали при — 10° Н. х. до изменения цвета от ярко-зеленого до
коричневато-зелеиого. Димерный аддукт (4) выделяется в виде белого
осадка, и обработка отфильтрованного раствора даете 52—66%ным
выходом нортри цикли л ацетат (2) в виде светло-зеленой жидкости.
Дезаминирование амидов. Уолфром [25] показал, что пента-О-
ацетилманнонамид можно гладко дезаминировать в соответствую-
щий пентаацетат альдоновой кислоты взаимодействием с Н. х. в
хлороформе.
conh2 СО2н
НСОАс НСОАс
| C1NO в СНС13 |
НСОАс ------------> НСОАс
I % !
АсОСН АсОСН
I !
АсОСН АсОСН
СНаОАс (1н2ОАс
453
Алкилнитриты. Корнблюм и Оливето [26] описали эффек-
тивный метод получения 2-октнлнитрита. Раствор 0,6 моля октанола-
2 в 240 мл пиридина перемешивают при 0—10° и в течение 3 час
пропускают в раствор 0,95 моля Н. х. К смеси добавляют петролей-
иый эфир и воду, органический слой отделяют, промывают, высуши-
вают и перегоняют. Реакция спирта с нитритом натрия и серной
кислотой дает продукт лишь с 59%-ным выходом.
I CIN'O
СН.>СНгСН2СН„СИ,СНчСНСН3 —СН3С1-12СНаСН2СН<,СНХНСН3
'! * I
ОН ONO
Оксимированке углеводородов. Нэйлор и Андерсон 1271
облучением смеси циклогексана и Н. х. получили оксим циклогек-
санона с 71 %-ным выходом.Реакции приписан радикальный меха-
низм:
Присоединение по связи C~N. Н. х. взаимодействует (0°,
40 час) с оксимииоэфирами (I) в хлористом метилене или эфире, об-
разуя cz-хлор-а-иитрозоэфпры (3) [28|, которые представляют собой
мономерные синие жидкости, чувствительные к свету и нагреванию.
1 CINO " Cl Cl
rcco3c2h5 —- I] рссо2с,нй rcco3c2h5 1
Ii NOH NNO NO
(I) (2) (3)
Окисление кетонов. Маннинг и Стапсбери [291 обрабатывали
1 моль ацетофенона в этаноле при 23—30° 1,6 моля Н. х. и постепен-
но нагревали смесь до 60°. По окончании реакции побочно обра-
зующиеся сложные эфиры омыляли щелочью, и с 53%-ным выходом
был легко выделен диэтилацеталь фенилглиоксаля.
С6Н6СОСН3 <-.NO’ СгН.^ С6НдСОСН(ОС2Но),
53%
Гидразиды^Азиды. Пептидный синтез с применением азидов
кислот, например получение защищенного дипептида (3), имеет то
преимущество, что не сопровождается рацемизацией пептидных
компонентов [30]. Однако при превращении гидразида N-карбобен-
зокси-5-бснзил-н -цистеина (1) в азид реакцией с азотистой кислотой
в нормальных условиях в качестве побочного продукта образуется
454
значительное количество амида (RCONHMI INO->RCONH.,+N- -
-I-
= Nl -О). Образование амида можно подавить, проводя реакцию в
более кислой среде и при низкой температуре, одпако эти условии
благоприятствуют другой побочной реакции, приводящей к образо-
ванию сульфоксида. Хопцль и Рудпигер 1311 показали, что II, х.
превращает гидразид в азид при различных условиях без образо-
вания амида или сульфоксида. Так, суспензию 1 а соединения (1)
в ТГФ обрабатывают при --20° И. х. в ТГФ и диоксане и интенсивно
перемешивают 6 мин до растворения гидразида. Другой способ за-
О II О к
II I CINO ;| -г - ПЛ’СЧСО С II-
CbNHCHC— NNH., —- CbNHCHC—!_.—J........ ..Д
I I
СН„ CH.,
I I
SCH,CeH5 SCH,CfiI-L.
(I) (2)
О HR
II I I
CbNHCl-IC-N CHCO.2C,H5
CH,
SCII,COH5
(T)
ключается в обработке раствора 1 г соединения II) в ТГФ, насыщен-
ном хлористым водородом до 2,2 щ, при —20° н-бутил нитритом.
ArNH,-*ArOH. Н. х. оказался наилучшим агентом для гидро-
лиза IN-окиси аденозина в N-окись инозина [321.
1. Woilrom М. L., Konigsberg М., W е i s b 1 a t D. L., J. Ain.
Cbern. Soc., 61, 5Г4 (1939); К о л e м а и Г., Д ж и л л и с Г., К о э п Г,,
«Неорганические синтезы», ИЛ, М., 1951, сб. I, стр. 57.
2. М orton J. R., Wilcox Н, W.. Inorg. Syn., 4, 48, (1953),
3. See review by Beckham L. J., Fessler W. A., К i s e M. Д.,
Chem. Revs., 48, 324 (1951).
4. France H., H e i ] b г о п I. M., Iley D. II., J. Chem. Soc., 1940, 369.
5. H и i s g e n R., Ann., 574, 180 (1951).
6. Skinner W. A.,Gra m H. P., Baker B. R., J. Org. Chem., 25, 777
(1960),
455
7. Newman M. S., Kutner A,, J. Am. Chem. Soc., 73, 4199 (1951).
8. Chen M. M., D’Adamo A. F., Jr., Walter R. I., J. Org. Chem.,
26, 2721 (1961).
9. Rundel W-, M fi 1 1 e r E„ Chem. Ber., 96, 2528 (1963).
10. C 1 a г к R. D., Helmkamp G. K., J. Org. Chem., 29, 1316 (1964).
II. T I 1 d e n W. A., Sehnstone W. A., J. Chem. Soc., 31, 554 (1877).
12. Meinwald J., Meinwald Y. C., Baker T. N., Ill, J. Am.
Chem. Soc., 85, 2513 (1963).
13. Meinwald J., Meinwald Y. C., Baker T. N., Ill, J. Am.
Chem. Soc., 86, 4074 (1964).
14. Hassner A,, Hea t h cock C., J. Org. Chem., 29, 1350 (1964);
Tanabe !<., Hayashi R., Chem. Pharm. Brill. (Tokyo), 10, 1177
(1962).
15. Harrison W. A., Jones E. R. H., AT e a к i n s G. D., Wilkin-
son P. A., J. Chem. Soc., 1964, 3210,
16. О h n о M., Okamoto M., Nukada K., Tetrahedron Letters, 4047
(1965).
17. Dau ben W. G., Martin E. C., Fon ken G. J., J, Org. Chem,,
23 1205 (1958).
18. Benkeser R, A., Robinson R E., Sauve D. M., Thomas
О. H-, j. Am. Chem. Soc., 77, 3230 (1955).
19. H ussey A. S., S a u v a g e J. F., Baker R. H., J. Org. Chem.,26,
256 (1961).
20. S e r f о n t e i n W. J,, Jordaan J. H., White J., Tetrahedron
Letters, 1069 (1964).
21. Lamieux R. U., Nagabhushan T. L., O’N e i 1 1 I. K., Tetra-
hedron Letters, 1909 (1964).
22. С 1 о s s G. L., Brois S. J., J. Am. Chem. Soc., 82, 6068 (1960),
23. Pnmmerer R-, Graser F., Ann., 583, 207 (1953).
24. M e i n w a 1 d J., Crandall J., H v m a n s W. E., Org. Syn., 45,
74 (1965).
25. W о 1 I г о m M. L., Wood H. B., J. Am. Chem. Soc., 73, 730 (1951).
26. Kor n bl u m N., О 1 i v e t о E. P„ J. Am. Chem. Soc., 69, 465 (1947).
27. N а у I о r M. A., Anderson A. W., J. Org. Chem., 18, 115 (1953).
28. Kissinger L. W., U n g n a d e H. E., J. Org. Chem., 23, 1517 (1958).
29. Manning D. T., Stansbury H, A., Jr., J. Org. Chem., 26, 3755
(1961).
30. W i e 1 a n d T., Deter mann H., Angew. Chem., Internal. Ed., 2,
358 (1963).
31. Honzl J., R u dinger J., Coll. Czech., 26, 2333 (1961).
32. Sigel H., Brintzinger H,, Helv. Chim. Acta, 48, 433 (1965).
л-НИТРОЗО-N, N-ДИМЕТИЛАНИЛИН, (CH3)2NCeH4NO. Мол.
вес 150,18, т. пл. 86°.
Н. получают нитрозированием Н,Н-диметиланилина и выделяют
в виде хлоргидрата (мол. вес 185,66) [1,2].
В одном из методов синтеза альдегидов необходимый для альде-
гидной группы атом углерода вводится в виде формальдегида. Так,
для получения и-диметиламинобензальдегида [11 смесь М,М-диметил-
анилина, формалина и конц. соляной кислоты нагревают в течение
10 мин па кипящей водяной бане н быстро добавляют хлоргидрат Н.
Бурная реакция заканчивается примерно за 5 мин. Смесь разбав-
ляют водой и подщелачивают для осаждения желтого основания
Шиффа (1). Основание Шиффа расщепляют избытком формальдегида
в разб. уксусной кислоте.
456
N(CH3)2 N(CH3)2
J\ 1) СН2О + ОМС5Н4К(СИЩ+НС1
I [I 2) Na0H j jj
CH — NCeH4N(CH3)2
(1)
N(CH3)2
CH,O- AcOH
—+ CH2 = NCeH4N(CH3)2
56—59% (общин) | ||
ino
(2)
В других синтезах в альдегидную группу превращается ме-
тильная группа, активированная орто- нли пара-нитрозаместите-
лями, как это показано на примере получения 2,4-динитробен-
зальдегида [21.
СН3
no2
сн - NC6H4N(CH3)a
1 /NO,
О1\’С(1НЩ(СН,)а-НС! р jy H.O-HCl
СгНо0Н, Na2co7 I Ii 27-38%"
NO,
Реакция Крёнке [3L Галоидсодержащее соединение взаимодейст-
вует с пиридином в этаноле,образуя пиридиниевую соль (1), при об-
работке которой щнитрозо-И,К-диметиланилином и щелочью полу-
чают нитрон (2), дающий при кислотном гидролизе альдегид.
С6Н5СН3С14-1 || -+СеН3СН2---N оищнщщнщ
1 'I X - ,/ NaOH
о-
С6Н5СНN + - N(CH3)a CGH5CH = O
(2)
Ружичка, Платнер и Фуррер [4] использовали эту реакцию для
превращения холестанона в две енольные формы холестандиона-
2,3; бромирование 2-бромпроизводного, превращение в бромид 2-пи-
р иди ни я и конденсация его с Н. дают нитрон II, который при кис-
лотном гидролизе образует смесь двух енольных форм дионов.
457
1. Л д а м с Р., к о я емап Г., «Синтезы органических препаратов», ИЛ, М.,
1949, сб. 1, стр, 144
2. Б с п п с г г Г., Бе л л Е., «Синтезы органических препаратов», ИЛ, М.,
1919, сб. 2, стр. 224.
3, К г б h п к с- Р'., Вег., 71, 2583 (1938).
4. R ц z 1 с к a L., Platt п с г Pl. A., F и г г е г ?•!., Helv. Ch ini. Acta,
25, 524 (194'1).
N-НИТРОЗОМЕТИЛМОЧЕВИНА. Генер ировапие диазометана;
см. т. I стр. 244.
НИТРОЗОНИЯ БОРФТОРИД, NO'BFp. Мол. вес 116,83,
И. б. взаимодетгетвует с вторичными аминами, образуя с хоро-
ш 11 м выходом н и тр оз а ми нм 111. С а м идами и с у л ьфо н а ми д а м и в м яр-
ких условиях 14. б. с хорошим выходом дает соответствующие кис-
лоты [2].
сохи,
н’схА/ сн»
Н5С//'^'/Ч'СН;1
со,н
Н3сч ЛН3
т г
т:н3
1. О 1 a h G., N о s z к б L., К u b n S., Sz e 1 к с M., Chem. Вег., 89, 2374 (1956).
2. О 1 a h G. Д., О 1 a h J. A., J. Org. Chem., 30, 2386 (1965).
НИТРОЗОНИЯ ГЕКСАФТОРФОСФ AT, NOPFy. Мол. вес 174,99.
Новая методика HI изучения сравнительного алкилирования
бензола и толуола состоит в перемешивании раствора 0,125 моля
каждого углеводорода и 0,05 моля 14. г. в 50 г нитрометана при 25°
и добавлении раствора 0,025 моля этил а мина в 20 а нитрометана
в течение 20 мин. Относительные реакционные способности
458
толуола (Т) и бензола (В): 1,51. Данные ГЖХ указывают
на следующее соотношение изомеров (%): о-35,8; лг-48,5;
zz-15,7, С ацетонитрилом в качестве растворителя получаются ана-
логичные результаты. Благодаря большей растворимости гексафтор-
фосфатная соль предпочтительнее, чем борфторпд.
ArH + C2I-I5\H2 + NO + PFr~ ЛгС2115-|-Х,Д-11.,О-р HPF0
CHi(NO«
1. О I a h G. A., Overchuk N. A., Lapicrre J. C-, J. Am. Chem.
Soc., 87, 5785 (1965).
НИТРОМЕТАН, CH3NO2. Мол. вес 61,04, т. кип. 101°, уд.вес 1,31.
В сб. «Синтезы органических препаратов» [1] описана методика
получения нитрометана по Кольбе (1909 г.):
CICHaCO2Na NO2CH2CO2Na CH3NO2^-NaHCO3
35 — 38%
В настоящее время Н. получают нитрованием углеводородов нефти
в газовой фазе. Использование Н. в качестве растворителя при изуче-
нии кинетики описано Ола 12]. Спектроскопически чистый Н. трижды
промывают раствором, содержащим 25 a NaHCO3 и 25 a NaHSC);-
в 1 л, затем водой, 5%-пой серной кислотой, снова водой, водным
NaHCO3, высушивают в течение ночи драйеритом, пропускают через
колонку высотой 50 см, заполненную молекулярными ситами (Линде
4 А в виде стержней диаметром 0,15 см), и перегоняют при 587160 мм
над небольшим количеством молекулярных сит в виде порошка. При
этом получают растворитель, нейтральный по бромфенолу синему
в этаноле и сухой по отношению к реагенту Фишера (по 1,3790).
Влияние растворителя. Для Н. характерна высокая диэлектри-
ческая постоянная (39), поэтому его рекомендуют в качестве раство-
рителя для конденсации по Кёнигу — Кнорру гликозил бромидов с
этанолом в присутствии карбоната серебра, при которой образуются
р-глпкозиды [3]. В менее полярных растворителях главным продук-
том может быть а-гликозпд.
Конденсация с карбонильными соединениями. Н. конденси-
руется с бензальдегидом в смеси метанол — водный едкий натр
при 10—15° с образованием енолята натрия, который при под-
кислении даст с хорошим выходом |3-питр ости рол [4]. Катализирус-
о-
С6Н5СНО -у CI-I3NO0 5Д.1°Н ~ Na2S CeH5CHCI-I = N + ОХа~>
1
он
cch-ch^chno2
80—83%
мая щелочью конденсация с формальдегидом, приводящая к 2-нитро-
этанолу, осуществляется [5] при обработке смеси 4,2 моля парафор-
ма и 2,5 л (46,6 моля) Н. водным едким кали. После окончания
459
реакции смесь нейтрализуют кислотой и отгонкой отделяют не-
прореагировавший Н. (2,3 л). Оставшуюся золотисто-желтую жид-
1) кон
o2nch3+ch2o o2nch2ch2oh
46-49%
кость обрабатывают равным объемом дифенилового эфира (тепло-
отводящий агент) для уменьшения вероятности взрыва и отгоняют
2-нитроэтанол вместе с некоторым количеством дифенилового эфира.
Синтез циклогептаиона (5) начинается с катализируемого осно-
ванием присоединения Н. к циклогексанону [6], причем на 2,5 моля
кетона требуется 3,25 моля Н. Выделяющийся сиолят натрия (2)
собирают и подкисляют для освобождения нитроспирта (3), который
гидрируют до аминоспирта (4). Под действием азотистой кислоты
осуществляется дезаминирование с перегруппировкой, приводящей
к расширению кольца и образованию циклогептанона (5).
о
1 +
НО CH=N ONa
С некоторыми диальдегидами в щелочной среде Н. образует
циклические продукты конденсации, в которых метильные группы
двух молекул Н. включены в кольцо [7]. Так, с глиоксалем Н. об-
разует смесь изомерных производных инозита, одно из которых (1)
можно получить в чистом виде благодаря его нерастворимости в воде.
Винный альдегид при взаимодействии с Н. дает смесь стереоизомер-
ных нитро циклопентантетраолов-2, 3, 4, 5 (2). Глутаровый альде-
гид конденсируется с Н. в растворе карбоната натрия, образуя
смесь 2-нитроциклогександиолов-1,3, из которой один изомер можно
выделить с 51 %-ным выходом экстракцией эфиром. Этому соедине-
нию на основании данных ЯМР была приписана транс-конфигура-
ция (3). о-Фталевый альдегид с Н. в спиртовой щелочи дает при
подкислении желтый 2-иитро-З-оксиинден (4) с т. пл. 148°
[7, 8].
460
o2nch3
о о
II II
нс—СН
НС—СН
II II
CH3NOZ
но он
pH 10
он он
(1)
носнсно
носнсно
CH3NOZ
---1---
носн-снон
I \
CHNOj
носн-снон
Сено
сно
CH3NOZ, Nazco
51%
(4)
Исходя из инозина (5) конденсацией с Н. был синтезирован
2-ами но - 3-дезокси-p-D-глюко пираноз и л гипо кс анти и (8) [31.
(5) (6) (7) (8)
I. У ит м о р Ф., Уитмор АН, «Синтезы органических препаратов», ИЛ,
М., 1949, сб. 1, стр. 303.
2. Olah G. A., Kuhn S. J., Flood S. IL, Hardin В. A., J. Am.
Chem. Soc., 86, 1043 (1964).
3. L 1 о у d P. F., Roberts G. P., J. Chem. Soc., 1963, 2962; L i c h t ent-
haler F. W_, Albrecht H. P., Chem. Ber., 99, 575 (1966).
4. У op ролл Д., «Синтезы органических препаратов», ИЛ, М., 1949, сб. 1,
стр. 308.
5. Ноланд В., «Синтезы органических препаратов», ИЛ, М., 1964, сб. 12,
стр. 105.
6. D auben И. J., R i n g о 1 d Н. J., Wade R. Н., Pearson В. L.,
Anderson A. G., Jr., Org. Syn., Coll. Vol., 4, 221 (1963).
461
7. Review: I. i ch t с n 1 li a 1 er F. W., Angew. Chem., Internal. Ed., 3,
211 (1964).
8. В а с г H. H., Achmatowicz B., Angew. Chem., Internal. Ed.,
3, 224 (1964).
n-НИТРОН АД БЕНЗОЙНАЯ КИСЛОТА, n-NOAHKXKH. Мол.
вес 183,12, т. пл. 137°.
Н. к. получают постепенным добавлением /г-нитробензоилхло-
рида к перемешиваемой суспензии перекиси натрия в ТГФ при тем-
пературе от —20 до —5° в присутствии каталитического количества
льда [1]. Эта надкислота представляет собой твердое кристалличе-
ское вещество, исключительно устойчивое при комнатной темпера-
туре и в 7—20 раз более реакционноспособное, чем падбензойная
кислота, при эпоксидировании олефинов.
1. V i 1 k a s М., Bull. soc. chirn. France, 1959, 1401.
З-НИТРО-М-НИТРОЗОКАРБАЗОЛ (1). Мол. вес 241,20. Н.
получают из карбазола [1].
Н. можно использовать для нитрозирования вторичных аминов
[2]. Так, N-метил анилин при кипячении с Н. в бензоле в течение
20 мин дает с хорошим выходом N-иитрозометиланилин. Бумгард-
(0
(ArAr’NNO)
нер [2] пытался превратить этим методом этилепимпн в N-нитрозо-
производное, но выделил лишь этилен и закись азота. N-Нитрозо-
азетидин — стабильное соединение, кипящее при 195°,— был полу-
чен непосредственным нитрозированием [3].
Этилен имин
ArAr’NNO
НОИО,разб1 АсОН
сн2
---> ||_ + n2o
CHj
-N—NO
Аэетидин
Кларк и Хелмкамп [41 показали, что дезаминирование мезо- и
б/,/-2,3-диметилэтиленимина под действием Н. протекает очень сте-
реоспсцифичпо. .мезо-Изомер дает ^ис-бутен-2 99,9% -ной чисто-
ты, a d, /-изомер — трпне-бутен-2 99%-иой чистоты. Сравнимые
результаты получены [5] при использовании хлористого нитрозила
и триэтилами на.
462
H N Н
Н
ArAr’NNO, С6Н6
СН3 СН3
43-53%
С
1
Н
1. Lindemann Н., Вег., 57, 557 (1924); Shirley D. А., «Ргсрп. of
Organic Intermediates», Wiley, 1951, p. 218.
2, В u m g ar dner C. L., M cC a 11 u m K. S., Freeman J. P., J. Am.
Chem. Soc., 83, 4417 (1961).
3. Howard С. C., jMarckwald W-, Ber., 32, 2032 (1899).
4. С 1 а г к R. D., H e I in к a m p G. K., J. Org. Chem., 29, 1316 (1964).
5. В e r t i 1 e E. et. al., Angew. Chem., Internat. Ed., 3, 490 (note 13) (1964).
НИТРОНИЯ БОРФТОРИД, NO%BF-. Мол. вес 112,83, разла-
гается при 170°.
Получение [1, 2]. В охлажденный льдом раствор 1 моля без-
водного фтористого водорода и 1 моля 95%-ной дымящей азотной
кислоты (уд. вес 1,50) в 200 мл нитрометана при перемешивании
(температура от —20 до 0е) в кварцевом или полиэтиленовом сосуде
HNO3 % HF -- 2BF3 - NOJBFI тс H2OBF3
пропускают трехфтористый бор до насыщения. Выпадает белая
кристаллическая соль нитрония, которую отфильтровывают и триж-
ды промывают нитрометаном (порциями по 30 мл) и 30 мл 1,1,2-
трифтор-1,2,2-трихлорэтана или хлористого метилена. Влажную
соль переносят в круглодонную колбу и сушат в вакууме при 40—
50°. Выход бесцветного кристаллического Н. б. (чистота более 95%)
составляет 89—100 г (88—99%). Соль очень гигроскопична, но без
доступа влаги ее можно хранить при комнатной температуре.
Нитрование. В бензонитрилы можно ввести одну нитрогруппу
обычными методами, но ввести две иитрогрунпы не удавалось, так
как в необходимых для этого жестких условиях нитрильная группа
гидролизуется (или окисляется). С помощью I-1.6,можно осуществить
введение одной или двух нитрогрупп в неводной не содержащей
кислоты среде, в которой кислота появляется лишь в результате
отщепления протона при нитровании. В качестве растворителя ис-
пользуют растворимые в воде тетраметиленсульфоп или ацетонитрил,
достаточно основные для того, чтобы поддерживать концентрацию
кислоты ниже уровня, необходимого для осуществления заметного
гидролиза (или окисления).
Нитрование о-толу нитрила осуществляется в две стадии [1, 21.
Смесь 0,55 моля Н. б. и 300 а тетраметилепсульфона перемешивают
при 10—20° до образования гомогенной суспензии (растворяется
около одной трети общего количества соли) и добавляют по каплям
в течение 20—30 мин 0,5 моля о-толунитрила; по мере протека-
ния реакции соль растворяется и начинает выделяться продукт.
Охлаждающую баню убирают и перемешивание продолжают в те-
чение 15 мин при 30—35°. Затем смесь выливают в ледяную воду,
463
продукт отделяют, промывают, сушат и кристаллизуют из 100 мл
метанола. Выход 5-нитро-о-толунитрила с т. пл. 105—106° состав-
ляет 74—80%.
CN
А/сн*
O.2n/^/ANO3
На следующей стадии смесь 0,43 моля 5-нитро-о-толунитрила,
0,56 моля Н. б. и 350 г тетраметиленсульфо на перемешивают при
кипячении и нагревают до 100—115° (температура жидкости). Эту
температуру поддерживают в течение 1 час, затем смеси дают остыть
и выливают ее в ледяную воду. Кристаллизация сырого продукта из
80 мл метанола дает чистый 3,5-динитро-о-толунитрил (т. пл.
85,5—86,5°) с 74—77,5%-ным выходом.
1. Kuhn S. J_, Olah G. A., J. Am. Chem. Soc., 83, 4564 (1961).
2. О 1 a h G. A., Kuhn S. J-, procedure submitted to Org. Syn.
1-НИТРОПРОПАН, СНзСН3СН3КтО,. Мол. вес 89,09, т. кип. 130°.
Реакция и идол-3-альдегида с ди аммонийбифосфатом и 1-нитро-
пропаном, приводящая к образованию нитрила и ндол-3-кар боновой
кислоты, иллюстрирует общий метод превращения ароматических
альдегидов в соответствующие нитрилы [1, 21.
н о, 053 моля
О, 01 моля
СН3СНгСНгНО2 + Асон
0, 34 моля 10 МЛ
Кипячение 12,5 час
48-63% :
1. Blatter Н. М., Lukaszewsk i Н., de Stevens G, J. Am.
Chem. Soc., 83, 2203 (1961).
2. В 1 a t t e r H. M., Lukaszewski H., de Stevens G., Org.
Syn., 43, 58 (1963).
2-НИТРОПРОПАН, НАТРИЕВАЯ СОЛЬ аци-ФОРМЫ,
(CHS)2 C-N“(Cr)—O’Na+. Мол. вес 111,09.
Раствор реагента готовят добавлением 2-нитропропана к раствору
этилата натрия в абсолютном этаноле, причем при необходимости
добавляют этанол для растворения выпадающего в осадок нитрона-
та. Хасс и Бендер [11 использовали этот реагент для превращения
бензилгалогевидов в альдегиды. Компоненты перемешивают при
464
комнатной-температуре около 15 час, раствор фильтруют для отделе-
ния хлористого натрия, фильтрат разбавляют водой и экстрагируют
эфиром. Промыванием водным раствором едкого натра удаляют аце-
токсим и избыток 2-нитропропана. Метод непригоден для получения
о-
о-СН5С0Н4СНйС1 + (СН3)2С = N+ - O-Na+
68-73%
—> 0-СН3С6Н4СНО + (СН3)3С = NOH + NaCl
нитробензальдегидов и неприменим к некоторым полизамещенным
бензилгалогенидам.
Бломквист и сотр. [2] успешно использовали этот метод для полу-
чения [ 10]-парациклофан-12-альдегида (2). Если по реакции Соммле,
описанной в сб. «Синтезы органических препаратов», выход а-наф-
тальдегида равен 75—82%, то при получении альдегида (2) выход
в лучшем случае составляет 20% .
(1) 0,015 моля
Реакция реагента с 9, 10-б«с-(хлорметил)-антраценом позволяет
осуществить простейший синтез антрацен-9,10-ди альдегида [41. В ка-
честве растворителя ДМСО более предпочтителен, чем ДМФА.
СН2С1 СНО
I о- |
,гн , с Д /А\/АХ 'Ч
II । । (СНя)гС = N т — О Na+ J [[ I
I II I I II I
Х/Х./Х/ VW
СН2С1 СНО
1. Hass Н. В., Bender М. L., J. Am. Chem. Soc., 71, 1767 (1949).
2. В I о т q u 1 s t A. Т., Stahl R. E., Melnwald Y. C., Smi t h B.H.,
J. Org. Chem., 26, 1687 (1961).
3. Аижи ал С., Тстаз Дж., Вильсон Дж., «Синтезы органических
препаратов», ИЛ, М., 1953, сб. 4, стр. 346.
4. Klauderman В. Н., J. Org. Chem., 31, 2618 (1966).
1-(О-НИТРОФЕНИЛ)-БУТАДИЕН-1,3 (4). Мол. вес 175,18,
бледно-желтые иглы, т. пл. 67°.
Получение. Исходное вещество — 1-хлор-4-(о-иитрофенил)-бу-
тен-2 (3) получают по реакции Меервейна из диазотированного
р-нитроанилина (2) и бутадиена в ацетоне в присутствии хлорной
465
меди и ацетата натрия [1, 2]. Последующее дегидрогалогенирование
едким кали в метаноле дает диен (4) [2].
Реакция Дильса ™- Альдера. Н. более активен, чем фенилбу-
тадиен. Брауде и Фоусетт [2] использовали Н. для интересного
синтеза фенаптридина (9) (см. схему). Общий выход из о-нитроани-
лина составил около 15%.
1. S е 1 1 s t е d t J. И., Noland W. E., procedure submitted to Org. Syn.
2. В r a u de E. A., Fawcett J. S., J. Chem. Soc., 1951, 3113.
л-НИТРОФЕНИЛДИХЛОРФОСФАТ (3). Мол. вес 255,99, т. пл.
43,5—44,5°.
Получение [ 1 ]. Н. получают при взаимодействии безводного
n-нитрофенолята натрия (1) и хлорокиси фосфора (2).
О
II
n-O2NC6H4ONa-HPOCl3 n-O2NCeH4OPCl2-p NaCl
(t) (2) (3)
Фосфорилирование нуклеозидов (1). Кристаллический Н. луч-
ше незамещенного фенилового эфира; например, при реакции Ы.
466
с 5'-О-тритилтимпди11ом(4)с последующим удалением /г-нитрофенил ь-
ной группы (обработка щелочью) и тритильной группы (мягкое кис-
лотное расщепление) получают тимидин-З'-фосфат (7) с общим вы-
ходом 75%.
I. Turner A. F., Khorana И. G., J. Am, Chem. Soc., 81, 4651 (1959).
0-НИТРОФЕНИЛСУЛЬФЕНХЛОРИД, o-NO.>C6H4SC1. Мол. вес
189,63, т. пл. 74,5—75°.
Получение [1].
0-NO.,CcH4SSC0H4NO2-o^CL-!-CC14 —iL. 2o-X02eirH4SCl
Защита аминогруппы в пептидном синтезе. Зервас и
сотр. [2] использовали Н, для защиты аминогруппы в аминокислотах
и сложных эфирах. Последние взаимодействуют с II. в присутствии
триэтиламина; со свободными аминокислотами реакцию проводят в
присутствии диоксана и 2 н. едкого натра. Защитная группа легко от-
щепляется кислотами, даже уксусной кислотой, но лучше всего тео-
ретическим количеством хлористого водорода в этаноле. Метод широ-
ко применяется для синтеза пептидов, которые труднодоступны обыч-’
ными методами, например ь-фенилалаиил- ь-глутамиппл- ь-глута-
мил-ь-глутамин [3].
о-Нитрофенилсульфенильная защитная группа оказалась очень
удобной в крупномасштабном синтезе амида ь-лейцил-ь-метиоппна
[4]. Преимущество Н. состоит еще и в том, что его легко выделить
после отщепления защитной группы для повторного использования.
1. X у б а ч е р М„ «Синтезы органических препаратов», ИЛ, М,, 1949, сб. 2,
стр. 560.
2. Zervas L., Borovas D., Gazi's Е., J. Am. Chem. Soc., 85,
3660 (1963).
467
3. 2 е г v a s L., Hamaii d is C., J, Am. Chem. Soc., 87, 99 (1965).
4. Bentley P. H., Gregory H., Laird A. H., Morley J. S.,
J. Chem. Soc., 1964, 6130.
«-НИТРОФЕНОЛ, n-NCXCJWH. Мол. вес 139,11, т. пл. 114°,
рКа 7,16.
Пептидный синтез. Н. взаимодействует с карбобензоксиамино-
кислотой в этилацетате в присутствии дициклогексилкарбодиимида
с образованием реакционноспособного н-нитрофенилового сложного
эфира, который образует пептидную связь при реакции с эфиром
аминокислоты при комнатной температуре и без катализатора [1].
CbNHCHCO2H+ HOCeH4NO.-n -' М СД
I
R
О R'
li !
n.vurnrrv' и Н2ЫСНСОЮНз
—CbNHCHCOC6H4NO2-n •—————►
1
R
о НОН
и мм
—> CbNHCHC—NHCHCOaCHa-pCeHuN-С — NQjHn
R R'
Эта методика — одна из наиболее пригодных для пептидного синтеза.
1. Bodanszky М., du V i g n e a u d V., J. Am. Chem. Soc., 81, 5688
(1959); Bodanszky M., Meienlioter J., du VigneaudV.,
J. Am. Chem. Soc., 82, 3195 (1960); Bodanszky M., Denning G. S.,
Jr., du V igneaud V., Biochemical Preparations, 10, 122 (1963).
/1м/
НОРБОРНЕН, Мол. Bec 94J5, т. пл. 46°,
т. кип. 96°, уд. вес (30е) 0,8589.
Н. получают взаимодействием дициклопентадиена с 2 же этилена
при 200° [1]; по существу, для реакции необходимы эквимолярные
количества мономерного циклопентадиена и этилена;
сн,
II
СН;
Э/гзо-2-Норбориеол и 2-норборнанон. Кляйнфелтер н Шлейер
[2] осуществили гладкое присоединение муравьиной кислоты к нор-
борнену и получили зкзо-2-норборнилформиат (2) с прекрасным вы-
ходом. Авторы не ссылаются па более раннюю работу [31, в которой
в качестве катализатора использовали эфират трехфтористого бора,
468
но выход (2) составлял лишь 71 %. Формиат при омылении дает
чистый ахзо-2-норборнеол (4), который при окислении по Джонсу
образует 2-норборнанон (3).
Бицикло-[3,2,1]-октанон-3 [4|. Первая стадия этого синтеза —
присоединение дибромкарбена к норборнену — дает дибромцикло-
пропильное производное (1), перегруппировывающееся в олефино-
вый дибромид (2); при использовании ди хлор карбена был выделен
первоначально образующийся эхзо-аддукт. Восстановлением гид-
ридом металла удаляют аллильный 4р-атом брома в (2) и последую-
щим кислотным гидролизом получают 3-кетон (4).
экзо-2-Норборниламин. О получении этого амина из норборнена
гидроборированием и реакцией с C1NH3—NaOH см. Хлорамин
(Браун и сотр. [20]).
469
Норбориан-2-э/сзо-карбоновая кислота. О получении этой кис-
лоты гидрокарбоксилированием норборнена при 50° см. Никеля
карбонил.
Реакционная способность олефинов, см. Фенил азид.
1. Meinwal d J., Hudak N. J., Org. Syn., Coll. Vol., 4, 738 (1963).
2. К 1 e i n f e 1 t e r D. C., von SchleyerP. R., Org. Syn., 42, 79 (1962).
3. Schmerling L., Luvisi J. P., Welch R. W., J. Am. Chem.
Soc., 78 , 2819 (1956).
4. Jefford C. W., Gu usher J. A., (Temple Univ.), procedure submitted
to Org. Syn.
СОДЕРЖАНИЕ
Ж
Железа нонакарбонил ................................................ 5
Железа пента карбонил............................................... 6
Железа(Н) сульфат .................................................. 7
Железа(Ш) хлорид ................................................... 8
Железа(Ш) хлорид безводный ........................................ 11
Железо............................................................ 11
Жирара реактив Р (пиридин)......................................... 12
Жирара реактив Т . . . ............................................ 12
3
Золотохлористоводородная кислота .................................. 15
И
Изоаминилнитрат.................................................... 16
Изоамил нитрит..................................................... 16
Изоамил нитрит — трифторуксусная кислота........................... 17
Изобензофуран...................................................... 18
Изобор нилднхл орал том пяат....................................’ 18
Изобутилен......................................................... 20
Изопропепилацетат.................................................. 22
Изопропила? алюминия.............................................. 25
Изоциановая кислота ............................................... 27
Имидазол........................................................... 27
N-Имидазолкарбоновой кислоты трет-бутиловый эфир................... 30
Имиподикарбоновой кислоты ди-mpe/n-бутиловый эфир.................. 30
Индикаторы pH...................................................... 31
Индикаторы pH, кислотность но Гзммс-ту............................. 31
Иод................................................................ 32
Иодазид............................................................ 39
Иода монобромид.................................................... 40
Иода монохлорид.................................................... 40
Иода пентафторид................................................... 41
о-Иодбензойная кислота ............................................ 41
Иодбепзол ......................................................... 42
471
Иодбензола дихлорид.................................................. 43
5-Иод*2,4-динитрофеннлгидразин....................................... 43
Иодизоцианат......................................................... 44
Иодистоводородная кислота ........................................... 44
Иодметилртути иодид.................................................. 46
Иод морфолиновый комплекс.......................................‘ 47
Иодная кислота ...................................................... 48
Иодобензол........................................................... 53
Иододибензоат серебра ............................................... 54
о-Иодозобецзойная кислота............................................ 55
Иодозобензол ........................................................ 56
Иодозобеизола диацетат............................................... 57
Иод — пиридин...................................................... 59
Иод — серебряные соли................................................ 59
N-Иодсукцинимид...................................................... 60
Иодуксусной кислоты амид............................................. 61
Ионообменные смолы................................................... 62
К
Кадмия ацетат — цинка ацетат......................................... 72
Калий................................................................ 72
Калия бисульфат...................................................... 74
Калия гидрид в минеральном масле..........'...........................76
Калия гидроокись..................................................... 77
Калия гидроокись — ацетон............................................ 80
Калия гидросульфид................................................... 80
Калия гипохлорит .................................................... 80
Калия иминоксил дисульфонат.......................................... 81
Калия манганат....................................................... 83
Калия мононадсульфат кислый.......................................... 84
Калия перманганат.................................................... 84
Калия персульфат и аммония персульфат................................ 97
Калия персульфат — серебро (каталитические количества)............... 99
Калия тиоцианат..................................................... 100
Калия триацетил осмат и Дикалия тетраметил осмат.................... 100
Калия трнфенилметнд................................................. 102
Калия феррицианид................................................... 102
Калия фторид........................................................ 107
Калия хлорат........................................................ 109
Кальцийгексамин...................................................... НО
Кальций — жидкий аммиак........................................... НО
Кальция гидрид..................................................... И1
Кальция карбонат..................................................... ИЗ
Кальция сульфат безводный.......................................... И 4
Кальция хлорид .................................................... И5
10-Камфорсульфокислота......................................... . 115
47§
10-Камфорсульфохлорид............................................... 116
Карбметокси метилентрифен и л фосфор ан и К а рбэтоксн метилентрифенил фос-
форан .............................................................. 116
Карбобензоксихлорид................................................. 118
п-Карбоксифеплл семикарбазид........................................ 118
N, N'-Карбоиилднимидазол............................................ 119
N, \/-Карбонилдп‘С«лм1-триазол...................................... 121
Карбонилфторид..................................................... 121
Карбэтоксиметилеитрифенилфосфоран................................... 123
Карбэтокспметилтрнфенилфосфонийбромид............................... 123
N-Карбэтоксифталимид................................................ 123
Каро кислота ....................................................... 123
Кетен............................................................... 125
ct-Кетоглутаровая кислота........................................... 129
Кил на ни реагент................................................... 129
Кипятильные камешки................................................. 129
Кислорода дифторид ................................................. 130
Кланзена щелочь..................................................... 131
Кобальта(П) ацетат-бромид .......................................... 132
Кобальта гидрокарбоинл.............................................. 132
Кобальта(П) хлорид.................................................. 132
симм- Колл идин..................................................... 133
Кремния тетранзоцианат.............................................. 133
Л
Лаурил полиэтиленгликоль......................................... 134
Левулиновая кислота ............................................. 134
Лемье — Джонсона окисление....................................... 136
Линдлара катализатор............................................. 136
Литий............................................................ 137
Литий-алкиламииное восстановление ............................... 141
Литий — аммиак................................................... 151
Литий — аммиак — ROH............................................. 153
Литий — трет-бута нол — тетра гидрофуран......................... 153
Литий — дифенил.................................................. 155
Литий — N-метиланнлин............................................ 156
Литий-натриевый сплав для получения литийалкилов................. 156
Литий — н-пронпламня............................................. 157
Литий — этилендиамин............................................. 158
N-Литийэтилендиамин....................: ............. 159
Лития алюмогидрид................................................ 163
Лития алюмогидрид—алюминия хлорид................................ 179
Лития алюмогидрид — пиридин................................... 183
Лития алюмогидрид — эфират трифторида бора....................... 185
Лития боргидрид.................................................. 186
Лития бромид..................................................... 186
473
Лития бромид — эфират трифторпда бора.............................. 187
Лития глрпс-(/??рет-бутоксп)-ал1ОМогидрпд............................ 187
Лития диэтпламид..................................................... 193
Лития диэтоксиалюмогидрпд............................................ 195
Лития иодида дпгидрат................................................ 196
Лития лодлд безводный................................................ 197
Лития карбонат....................................................... 199
Лития карбонат — лития бромид........................................ 199
Лития моноциаиборгидрид ............................................. 202
Лития нитрид........................................................ 203
Лития трнметокслалгохмогидрид........................................ 203
Лития триэтоксиалюмогидрпд........................................... 204
Лития хлорид......................................................... 204
2,6-Л}Тидин........................................................ 206
2,6-Лутидин-3,5-дикарбоновой кислоты гидразид........................ 206
м
Магний............................................................... 207
Магнии — иод — эфир.................................................. 210
Магний — нафталин.................................................... 211
Магния бромида эфират.............................................. 212
Магния окись ........................................................ 212
Магния сульфат....................................................... 213
Малеиновый дпальдегид................................................ 214
Малоновой кислоты дибеизллоиыи эфир.................................. 216
Малоновой кислоты дп-/яреп2-бутпливып эфир........................... 217
Малоновой кислоты изопропилиденовый эфир............................. 217
Марганца двуокись активная......................................... 218
Марганца двуокись обычная............................................ 227
Меди(11) ацетат безводный............................................ 228
Меди(П) ацетат, моногидрат ..................................... 228
Меди закись.......................................................... 230
Меди (1) иодид ...................................................... 231
Меди(П) карбонат основной ........................................... 231
Меди(Л) нитрат — пиридин ..................................... 232
Меди(П) нитрат — уксусный ангидрид................................. 232
Меди(П) сульфат...................................................... 233
Медц(П) хромит....................................................... 234
Медная бронза ....................................................... 235
Медная пудра — бензойная кислота .................................... 235
Медные соли.......................................................... 235
Медный порошок....................................................... 236
Медь (или Си3’ J — аскорбиновая кислота.........................- • 237
Медь бромистая....................................................... 237
Медь бромная......................................................... 239
Медь хлористая...................................................... 241
474
Медь хлористая, комплекс с хлористым аммонием...................... 246
Медь хлорная безводная............................................. 246
Мезилхлорид........................................................ 247
Мезитил енсульфохлорид............................................. 247
р-Меркаптоэтанол .............................. 247
Метансульфокислота................................................. 248
Метансульфокислоты алкиловые эфиры................................. 249
Метансульфокислоты ангидрид . . . ,............................. 249
Метансульфохлорид.................................................. 250
Метансульфохлорид — двуокись серы.................................. 253
Метафосфорная кислота.............................................. 254
Метилаль........................................................... 255
Метилат натрия..................................................... 256
З-Метил бензтиазол о н-2-гпд разом, хлоргидрат..................... 262
Метил бромистый.................................................... 263
2-Метил бутилат-2 калия............................................ 264
2-Метилбутилат-2 натрия............................................ 265
З-Метилбутин-1-ол-З................................................ 266
Метил винил кетон.................................................. 266
Метил виниловый эфир............................................... 274
О-Метил гидроксил а мин............................................ 274
Метилендиуретан ................................................... 276
Метилентрифенил фосфор ан.......................................... 276
Метилен хлористый.................................................. 277
Метил йодистый.................................................... 278
Метилкетена диэтилацеталь ......................................... 281
Метиллитий......................................................... 282
Метилмагнийбромид.................................................. 285
Метилмагнийиодид................................................... 286
N-Метил морфолин................................................... 287
N-Метилморфолина окись — перекись водорода......................... 287
Метилнитрат........................................................ 288
Метил нитрит........................;............................. 289
Метиловые эфиры кислот............................................. 290
2-Метил пентандиол-2,4............................................. 290
N-Метил пир рол и дон-2 ........................................... 292
1-Метил-3-п-тол ил трназеп......................................... 293
Метилтрифенилфосфоиийбромид........................................ 293
1-Метилфеиилгидразнп............................................... 294
Ы-Метил-Ы-фенилкарбаминоилхлорнд................................... 295
N-Метилформалилид................................................ 295
Метилфосфо новой кислоты дпхлорангидрид............................ 297
2-Метилфуран....................................................... 297
Метил хлористый.................................................... 298
З-Метил-4-хлорметилпзоксазол ...................................... 298
Метилхлорсульфит................................................... 299
Метилциклопропилкетон.............................................. 300
475
2-Метпл-2-этилдиоксолан-1,3........................................ 301
Метил эти л кетон.................................................. 301
Метоксиацетилен .................................................. 301
/2-Метоксибензил хлористый......................................... 302
1-Метоксибутеи-1-ин-3.............................................. 303
Метоксимагнийметилкарбонат......................................... 303
Метоксиметилентрифенилфосфоран..................................... 307
2,5-бнс-(я-Метоксифенил)-3,4-Дифенилцнклопентадиенон............... 308
а-Метоксиэтилидевтрифенллфосфоран.................................. 308
Молекулярные сита.................................................. 309
Мононадмалеиновая кислота ......................................... 311
Моноиадфталевая кислота ........................................... 312
Моноиадянтарная кислота............................................ 313
Морфолин........................................................... 314
Мочевина........................................................... 316
Муравьиная кислота ................................................ 327
Муравьиная кислота — формамид...................................... 331
Муравьиной кислоты я-нитрофениловый эфир.......................... 331
Муравьиной кислоты триметиламмонневая соль......................... 331
Муравьиной кислоты этиловый эфир................................... 332
Н
Надбензойная кислота .............................................. 336
Надбензойной кислоты трет-бутиловый эфир.......................... 342
Надмуравьиная кислота ............................................. 346
Надуксусная кислота ............................................... 346
Надуксусная кислота безводная.................................... 346
Надуксусная кислота, продажная 40%-ная............................. 351
Натрий............................................................. 354
Натрий — аммиак.................................................... 355
Натрий — аммиак — этанол........................................... 356
Натрий — трет-бутанол — тетрагидрофуран............................ 356
Натрий-калиевый сплав.............................................. 356
Натрий — нафталин.................................................. 357
Натрийтетракарбонил кобальт........................................ 359
Натрия азид........................................................ 359
Натрия алюмогидрид................................................ 362
Натрия алюмохлорнд ................................................ 363
Натрия амальгама разбавленная ..................................... 366
Натрия амальгама, 40% натрия, жидкая............................... 370
Натрия трет-амилат................................................. 371
Натрия бисульфит................................................... 371
Натрия бихромат, дигидрат ......................................... 374
Натрия боргидрид................................................... 381
Натрия бромат (или иодат).......................................... 388
Натрия висмутат ................................................... 389
476
Натрия гидразид .................................................... 391
Натрия гидрид ....................................................... 392
Натрия гидросульфит................................................. 401
Натрия глпобромит ................................................... 403
Натрия гипофосфит.................................................... 405
Натрия гипохлорита раствор .......................................... 405
Натрия дисульфид (или полисульфид), раствор.......................... 409
Натрия иодид ........................................................ 410
Натрия метабисульфит................................................. 415
Натрия нитрит ....................................................... 415
Натрия перборат...................................................... 421
Натрия персульфат.................................................... 421
Натрия сульфат безводный............................................. 422
Натрия сульфид....................................................... 422
Натрия тиосульфат.................................................... 423
Натрия фторид........................................................ 423
Натрия хлорат ....................................................... 424
Натрия хромат безводный.............................................. 425
Натрия цианат ....................................................... 426
Нафталиц-2,3-дикарбоновая кислота.................................... 427
Нафтали и-2,6-дикарбоновая кислота................................... 427
1,2-Нафталиндикарбоновой кислоты ангидрид ......................... 427
1,8-Нафталиндикарбоцовой кислоты ангидрид ........................ 428
Нафталин-0-сульфокислота............................................. 428
Нафтацен-9,10,11,12-дихипон.......................................... 429
а-Нафтилизотиопианат................................................. 431
ос-Нафтил изоцианат.................................................. 431
1,2-Нафтохинон....................................................... 431
1,4-Нафтохинон....................................................... 432
1,2-Н афтох и но j 1-4-сульфокислоты аммониевая и калиевая соли..... 433
Никель-алюминиевый сплав............................................. 434
Никеля борид................................................... . . 435
Никеля карбонил...................................................... 436
Никеля перекись...................................................... 439
Нингидрин............................................................ 440
Нитрил иодистый...................................................... 441
Нитрил хлористый..................................................... 441
4'-11итроазобензол-4-карбоновой кислоты хлорангидрид................. 442
n-Нитробензил бромистый (хлористый).................................. 443
л-Нитробецзоил хлористый............................................. 444
N-n-Нитробензол сульфокси уретан..................................... 444
1-Нптрогуанил-3,5-диметилпиразол..................................... 445
Ннтрозилсерная кислота .............................................. 445
Нитрозил фтористый .................................................. 447
Нитрозил хлористый................................................... 447
п-Нитрозо-Г4,1\т-диметнл анилин...................................... 456
N-Нитрозометилмочевина , . , . ...................................... 458
477
Нитрозония борфтерид................................................. 458
Нитрозо ни я гексафторфосфат......................................... 458
Нитрометан........................................................... 459
п-Нитронадбензойная кислота.......................................... 462
З-Ннтро-N-нитрозокарбазол............................................ 462
Нитрония борфторид .................................................. 463
1-Питропропаи...................................................... 464
2-Нитропропан, натриевая соль лци-формы.............................. 464
1-((?-Нитрофе11Ил)-бутадиен-] ,3..................................... 465
/г-Нитрофеяилдихлорфосфат............................................ 466
о-Цитрофенилсульфенхлорид............................................ 467
«-Нитрофенол..............................-.......................... 468
Норборнен............................................................ 468