/






























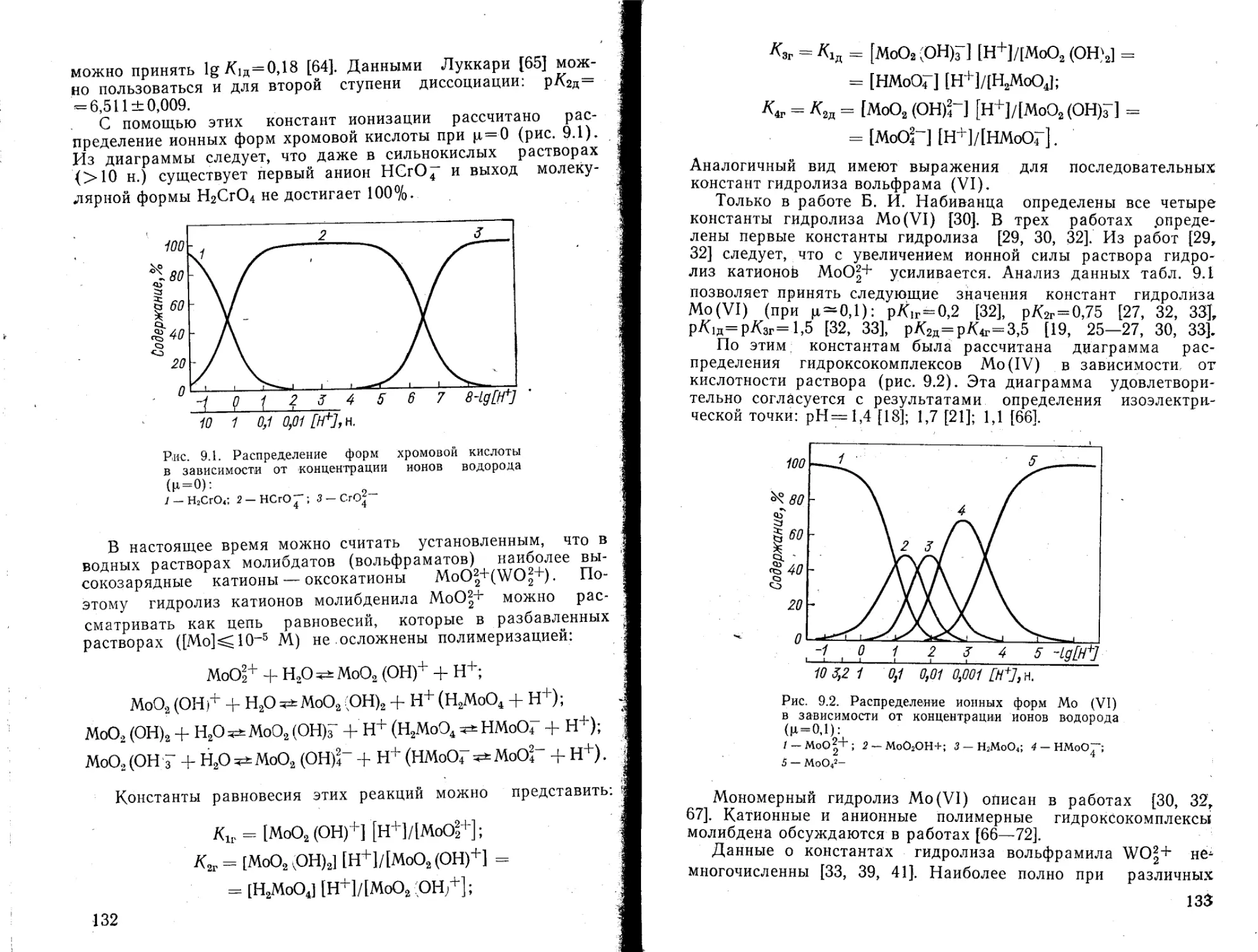



























Текст
В. А. НАЗАРЕНКО
В.П. АНТОНОВИЧ Е. М. НЕВСКАЯ
ГИДРОЛИЗ ионов металлов в разбавленных растворах
МОСКВА АТОМИЗДАТ 1979
УДК 66.093:669
Назаренко В. А., Антонович В. П., Невская Е. М. Гидролиз ионов металлов в разбавленных растворах. М.: Атомиздат, 1979. — 192 с.
В книге рассмотрены наиболее распространенные методы определения констант гидролиза ионов металлов, систематизированы константы гидролиза мономерных ионов элементов по группам, дана критическая их оценка и, где это было возможно, рекомендованы достоверные значения констант.
Книга может быть полезна работникам исследовательских институтов, изучающим состояние ионов металлов в природных водах, радиохимикам, биохимикам, геохимикам, технологам-гидрометаллур-гам, аналитикам и неорганикам, работающим в области координационной химии и теории ионных равновесий, а также аспирантам и студентам университетов и химико-технологических институтов.
Рис. 42. Табл. 20. Список литературы 953 наименования.
Nazarenko V. A., Antonovich V. Р., Nevskaja Е. М.
Metal Ions Hydrolysis in Dilute Solutions. M.: Atomizdat, 1979.
The work is a reference book on the ions monomer hydrolysis constants of the elements of all the groups of the Periodic Chart.
The main methods in studying hydrolitic reactions in dilute solutions has been investigated.
Hydrolysis constants of the monomer metal ions were systematized and tabulated.
Sometimes the most reliable constant data are recommended.
The results obtained by the authors of the work within the period 1968—1976 are given parallel to literary data.
The work may by helpful for investigators studying ionic equilibrium in dilute water solutions: analysts, inorganists, hydrometallurgists as well as for radiochemists, hydrochemists who deals with the problem of metal ions behavior in natural waters, and geochemists.
The present work can also be recommended as a reference book for students and post-graduates of the chemical departments of the Institutes.
20503—068 H ------------ 68—79 • 1802000000
034(01)—79
© Атомиздат, 1979
ПРЕДИСЛОВИЕ
Процессы гидролиза ионов металлов весьма распространены в природе и знание их необходимо химикам любой специальности, работающим с водными растворами солей металлов.
Гидролиз ионов металлов в их концентрированных и разбавленных растворах существенно различается — в первом случае образуются полимерные, во втором — мономерные гидроксо-комплексы. Исследователям в разных областях требуются количественные характеристики процессов образования именна мономерных гидроксокомплексов.
В отечественной литературе, оригинальной и переводной,, нет работ, многосторонне трактующих вопросы гидролиза ионов металлов. Авторы предлагаемой книги попытались заполнить существующий пробел, хотя бы в области процессов мономерного гидролиза.
Книга будет полезна химикам, изучающим равновесие с участием продуктов мономерного гидролиза ионов металлов^ Авторы заранее благодарны читателям за все замечания, которые будут сделаны по этой книге.
СПИСОК СОКРАЩЕНИЙ
Ам. эл. — амальгамный электрод
Калор. —калориметрический метод
Кат. — каталитический метод
Кинет. — кинетический метод
Кол. — колориметрия
Магн. воспр. —метод магнитной восприимчивости
Н-эл. — водородный электрод
Обмен. — метод ионного обмена
ПМР — протонный магнитный резонанс
Поляр. — полярографический метод
Распр. — метод распределения (жидкостная экстракция)
Раств. — метод растворимости
Редокс. пот. —метод окислительно-восстановительного потенциала (оксред-метрия)
Сорбц. — сорбционные методы (на бумаге, стекле и т. п.)
Спектр. — спектрофотометрический метод
Ст. эл. — стеклянный электрод
Тенз. — тензиметрический метод
Т.-д. —термодинамический анализ
Турб. — метод турбидиметрии (светорассеяния)
Ультрацентр. — метод ультрацентрифугирования
F-эл. — мембранный фторидный электрод
Хинг. эл. — хингидронный электрод
ЭДС —метод электродвижущих сил, потенциометрия с различными
электродами
Эл. диализ — электродиализ
Электромигр. — электромиграционный метод
Эл. пр. — метод электропроводности (кондуктометрия)
ЯСЭ —метод ядерного спинового эха
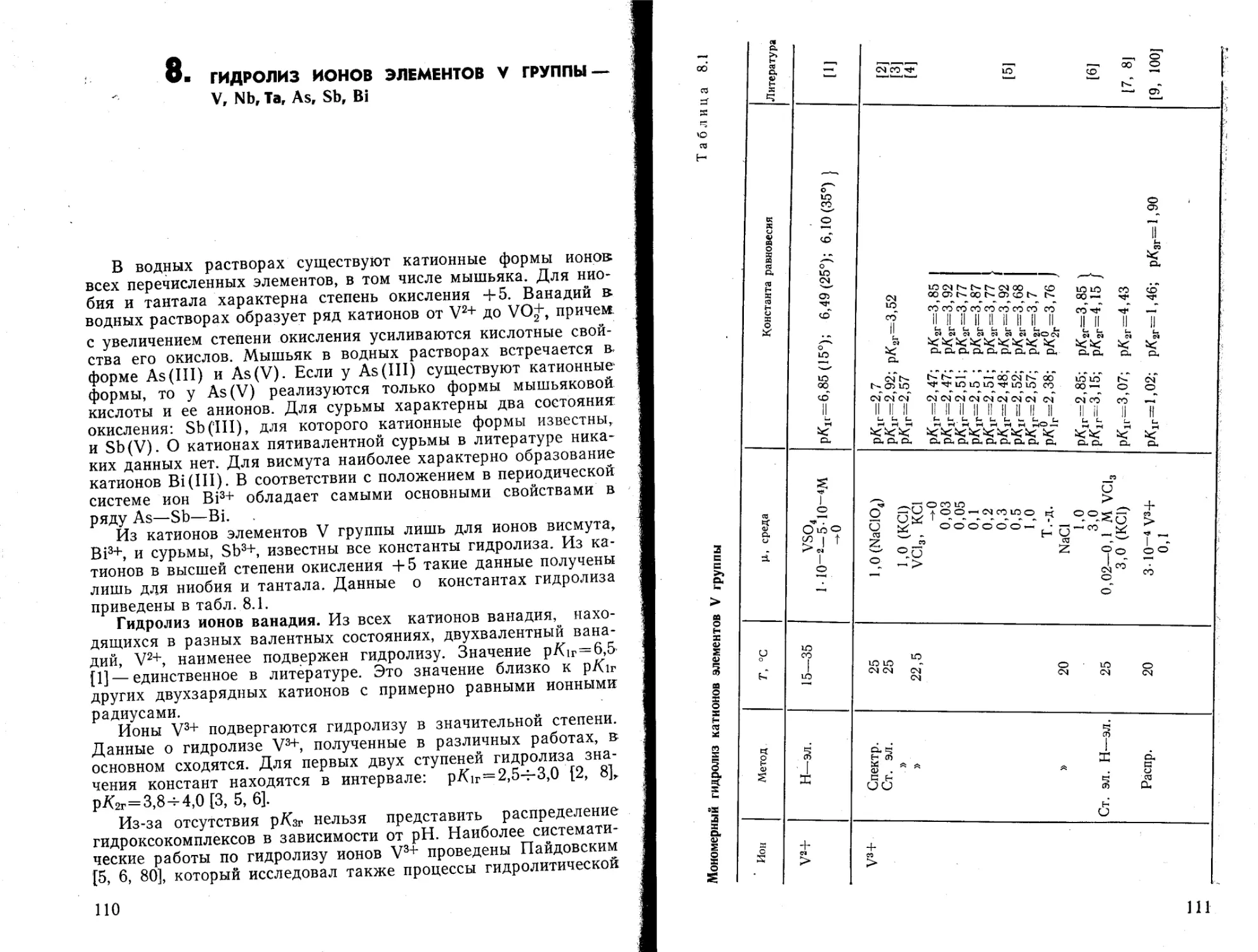
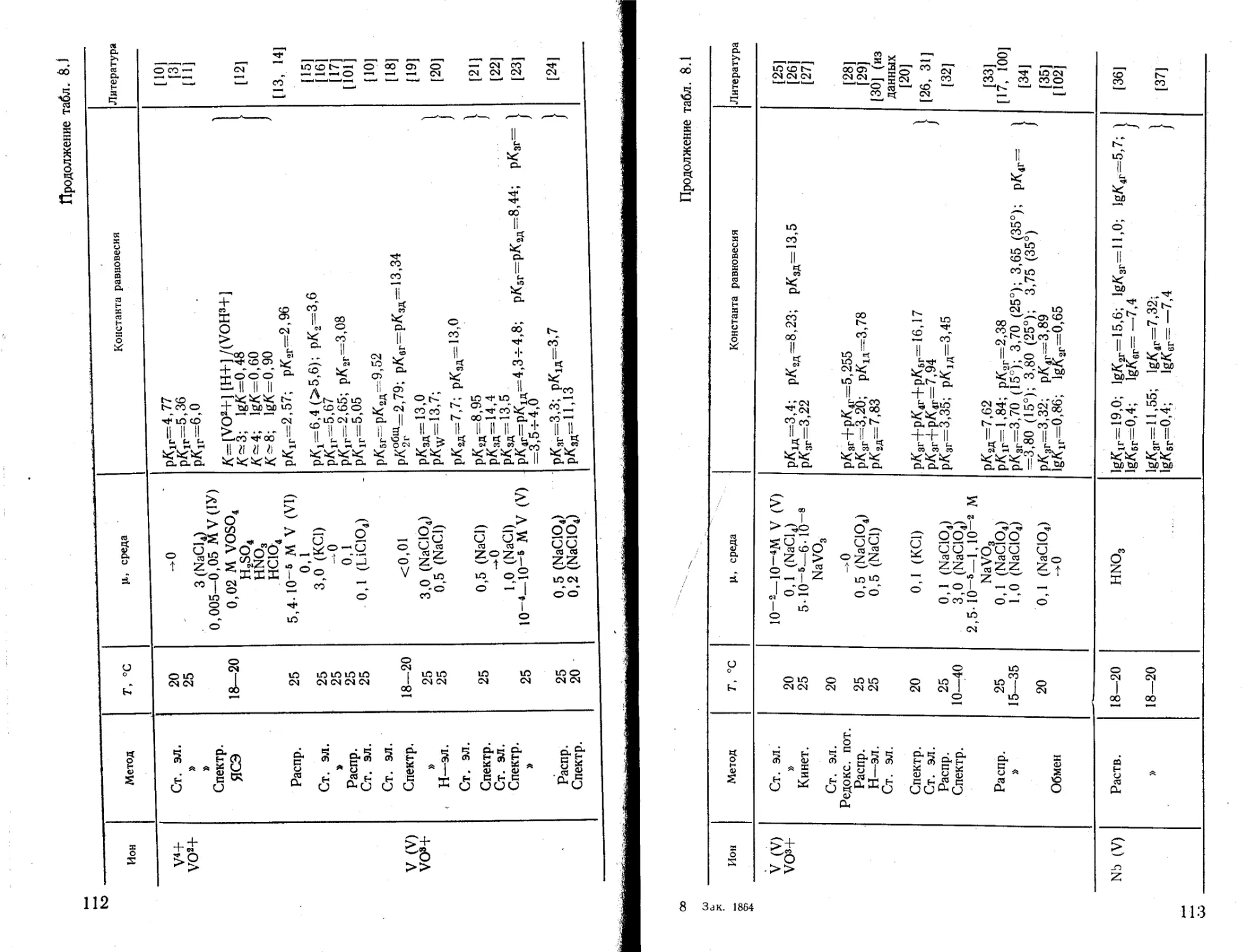
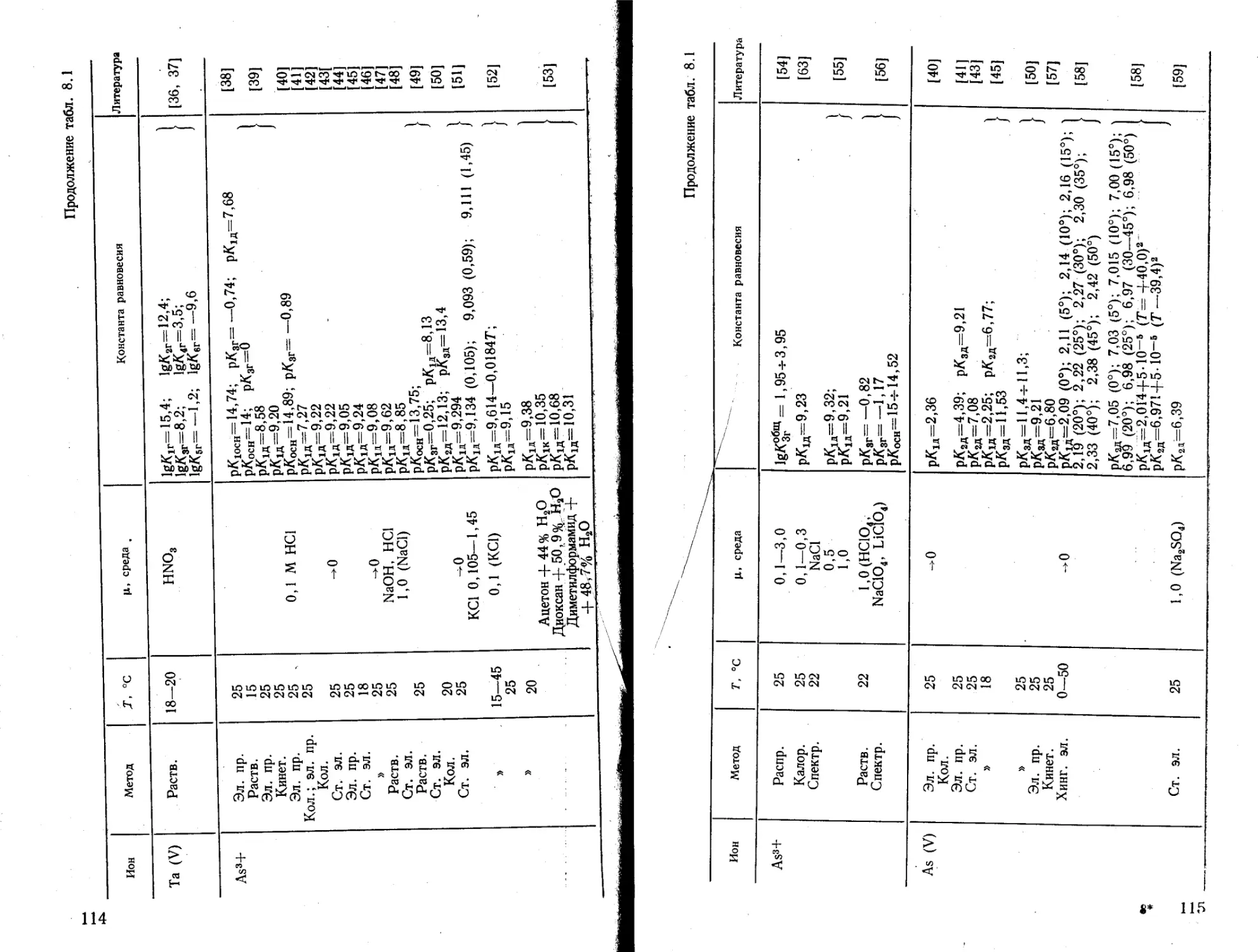
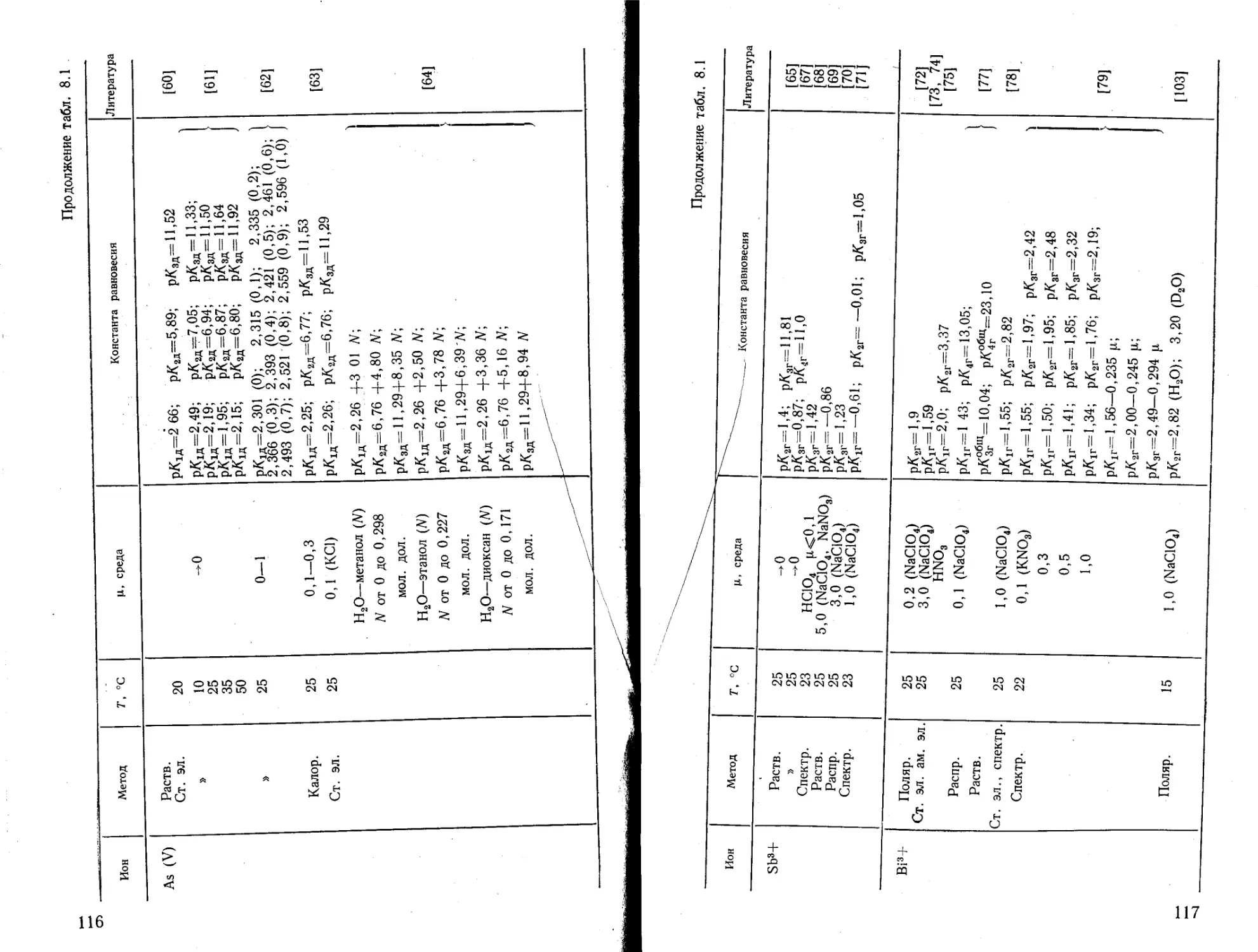
ВВЕДЕНИЕ
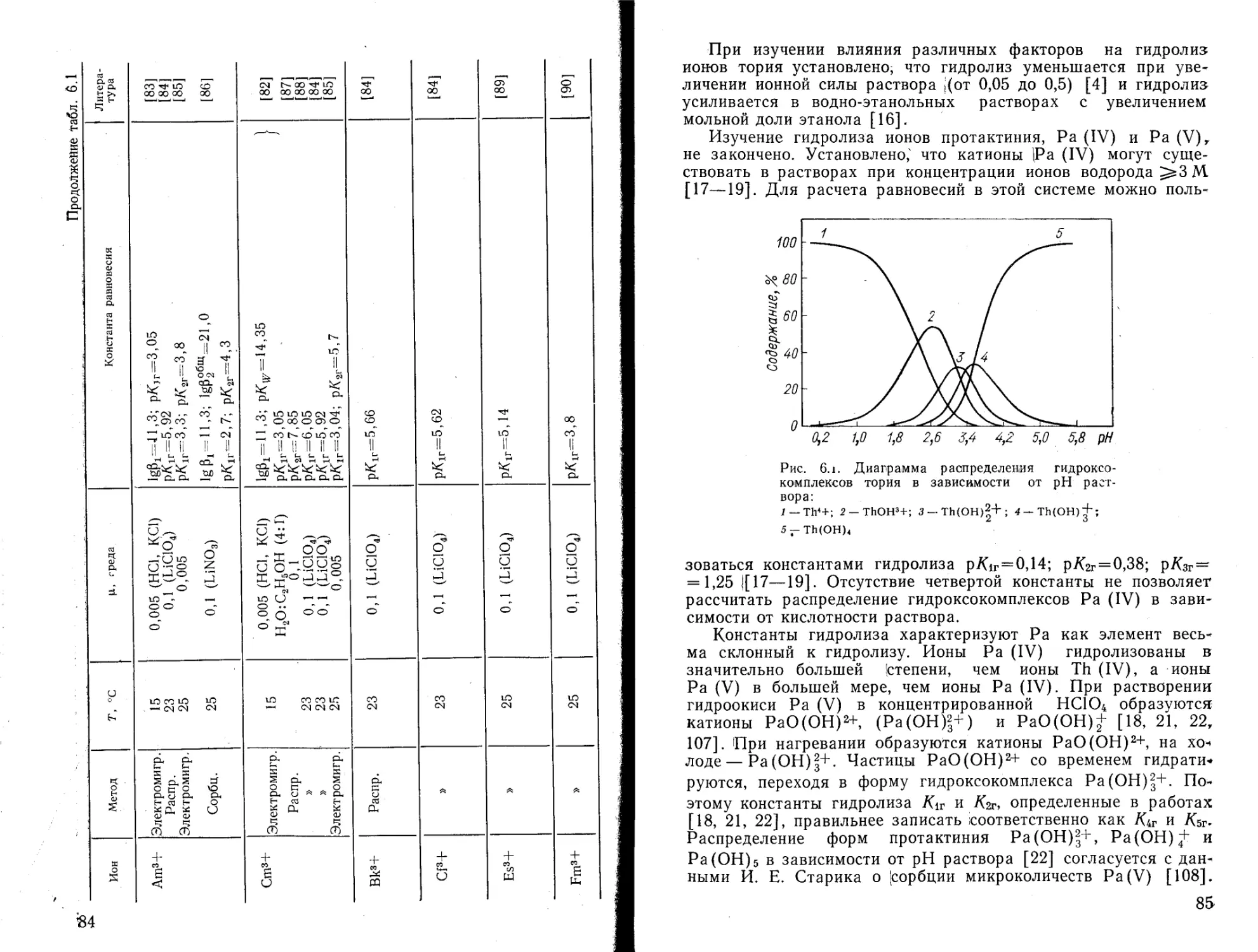
Химическое взаимодействие воды с различными соединениями, в частности с ионами металлов, приводит к образованию гидратов, гидроксокомплексов или гидролитических полимеров. Термодинамическое описание реакций и продуктов гидролиза позволяет выяснить направление и характер различных химических превращений. Для расчетов химических равновесий в водных растворах требуются значения констант равновесия реакций гидролиза (констант гидролиза). Гидролиз ионов металлов представляет собой реакцию между водой и катионами металла, сопровождающуюся выделением ионов водорода. По этой причине при определении констант гидролиза ионов металлов в первую очередь различными методами определяют концентрацию ионов водорода в равновесной системе.
При изучении гидролиза солей металлов в достаточно концентрированных растворах (0,01—0,5 моль/л) установлено образование гидролитических полимеров. Явление полимеризации характерно для большинства гидроксокомплексов металлов, но каждый из них имеет свои особенности, обусловленные концентрационными интервалами для ионов металла и ионов водорода, природой иона металла и аниона среды. Количественные характеристики полимерных гидроксокомплексов определить трудно, а применить для расчетов равновесных концентраций можно только в таких же условиях, в которых эти константы были рассчитаны. Достоверность значений и схем реакций полимеризации, которые эти константы характеризуют, трудно доказуемы прямым экспериментом.
Механизм реакций гидролиза ионов металлов в разбавленных растворах (10“4—10“6 г-ион/л) в основном характеризуется образованием мономерных гидроксокомплексов. В настоящее время накоплен большой материал по методам исследования гидролиза ионов металлов и константам гидролиза мономерных катионов металлов. Необходимо констатировать, что для многих ионов металлов константы равновесия, определенные Для одного и того же процесса разными авторами, значительно расходятся и в некоторых случаях трудно указать при
5
чину расхождения. К сожалению, в литературе отсутствуют руководства, в которых был бы систематически проанализирован материал по константам гидролиза ионов металлов.
В известной книге L. G. Sillen, А. Е. Martell «Stability Constants», Special Publication № 17, The Chemical Society, Burlington House, London (1964); Supplement, Special Publication N 25 (1968) приводятся константы образования гидро-ксокомплексов металлов (следовательно, и константы гидролиза их ионов), но без какой-либо их критической оценки. Обзор данных о термодинамических характеристиках реакций гидролиза и образования моно- и полиядерных гидроксоком-плексов приведен в работе К. А. Буркова, Е. А. Бусько, Л. С. Лилича «Химия и термодинамика растворов», 1977, № 4, с. 15—43. Нам известна лишь одна работа, где систематизирован материал по гидролизу ионов металлов и выбор рекомендованных значений констант термодинамически обоснован — Baes С. F., Mesmer R. Е. «The Hydrolysis of Cations», N. Y., John Wiley and Sons, 1976.
В данной работе авторы ограничились мономерным гидролизом, так как константы этого процесса более достоверны, чем константы процесса полимерного гидролиза. Отсутствие термодинамической строгости при выборе рекомендованных констант мы попытались возместить полнотой обзора и расчетами распределения гидроксокомплексов металлов в зависимости от pH среды там, где это было возможно. Мы попытались также установить зависимость склонности к гидролизу ионов элемента от положения его в Периодической системе Д. И. Менделеева.
Авторы сочли также целесообразным рассмотреть методы определения констант равновесия гидролиза, полагая, что это также может быть полезным при оценке достоверности констант.
I
ОСНОВНЫЕ ПРИНЦИПЫ РАСЧЕТА МОНОМЕРНОГО ГИДРОЛИЗА ИОНОВ МЕТАЛЛОВ
Гидролиз многовалентных катионов протекает ступенчато с последовательным образованием гидролитических продуктов, вплоть до труднорастворимой гидроокиси, и возможной их полимеризацией на любой ступени реакции. Суммарное уравнение гидролиза запишется в общем виде как
<?Мг+ + i Н2О =₽* М, (ОН)(Л‘И + i Н+ (1.1)
с термодинамической константой
г = [м|?(ОН)^--')+] [H+]< g.„-gH+ (1 2
Г [АИ+И ’
Задача по определению термодинамических констант гидролиза, сформулированная в виде такого уравнения, неразрешима, так как не известна активность ионов. Экспериментальное определение коэффициентов активности возможно лишь в некоторых простых системах в условиях малых концентраций и низкой ионной силы. При изучении гидролиза имеют дело с большим набором гидролитических форм, идентификация которых представляет сложную задачу и обычно проводится параллельно с расчетом констант или иногда даже с помощью уже рассчитанных констант. При этом в слишком разбавленных растворах сложно определить равновесные концентрации частиц.
Возможен различный подход к принципиальному решению задачи: 1) определение констант для различных концентраций катионов и последующая экстраполяция на бесконечное разбавление; 2) определение констант при каком-либо одном постоянном значении ионной силы и расчет термодинамических констант по уравнениям Дебая—Хюккеля или Дэвиса; 3) определение констант при нескольких постоянных значениях ц с последующей экстраполяцией к нулевому ее значению.
Первый способ мало распространен и применялся в основном в ранних работах. Изменение концентраций катиона в широких пределах может влиять на картину гидролиза и, следовательно, нарушать схему расчета, ее соответствие экспериментальным данным.
7
Исследователи гидролиза пользуются в основном двумя последними способами, отдавая предпочтение экстраполяции к нулевому значению ионной силы в большей степени, чем теоретическому расчету.
Постоянство ионной силы обеспечивает постоянство коэффициентов активности и независимость константы от концентрации. Константы, определенные при постоянной ионной силе^ включают поэтому коэффициенты активности форм, постоянные для данной ионной силы и зависят только от нее. Выражение для концентрационной константы в таком случае имеет вид
Кг = 1мг (OH)(/?-°+J [Н+] 7[Mz+]9. (1.3)
Теоретический расчет термодинамической константы из концентрационных при постоянной ионной силе ограничен тем, что теория Дебая—Хюккеля применима только к простым ионам при небольшой ионной силе. Предельный закон Дебая—Хюккеля можно использовать только для растворов с ц^10~3 М, а при использовании расширенной формы уравнения ц не должна превышать 0,1 М. Подробный сравнительный анализ расчетного и экстраполяционного методов получения термодинамических констант равновесия сделан в монографии Россотти [1]. Там же приведены различные приемы экстраполяции.
Значения констант, полученные экстраполяцией к нулевому значению ионной силы, более надежны, чем рассчитанные из измерений при одном постоянном значении ц, поскольку они не зависят от выбора параметров в уравнениях для коэффициентов активности. Однако при изучении гидролиза часто приходится использовать высокие концентрации ионов водорода для поддержания негидролизованных форм и значения р, при которых проводят эксперимент, довольно высоки, поэтому при экстраполяции к нулевому значению р возможны неточности.
Получение наиболее точных термодинамических констант — конечная цель изучения гидролитических равновесий. Большинство исследователей, однако, из-за сложности этой задачи ог* раничиваются определением констант гидролиза, выраженных* через концентрации. Такие результаты тоже очень важны, они характеризуют свойства металлов и при достаточном наборе значений ионной силы дают ценную информацию.
Постоянную ионную силу раствора поддерживают с помощью постоянных, высоких (по отношению к компонентам реакции гидролиза) концентраций фонового электролита. Основные теоретические предпосылки, которыми следует руководствоваться при выборе фонового электролита, изложены в работе [1].
В качестве фонового электролита обычно используют соли щелочных металлов с однозарядными анионами сильных кис-8
лот, чаще всего с перхлорат-ионом. В литературе почти нет указаний на образование сколько-нибудь прочных комплексов металлов с перхлорат-ионом, за исключением нескольких дискуссионных статей по гидролизу ионов железа в перхлоратной среде [2—8].
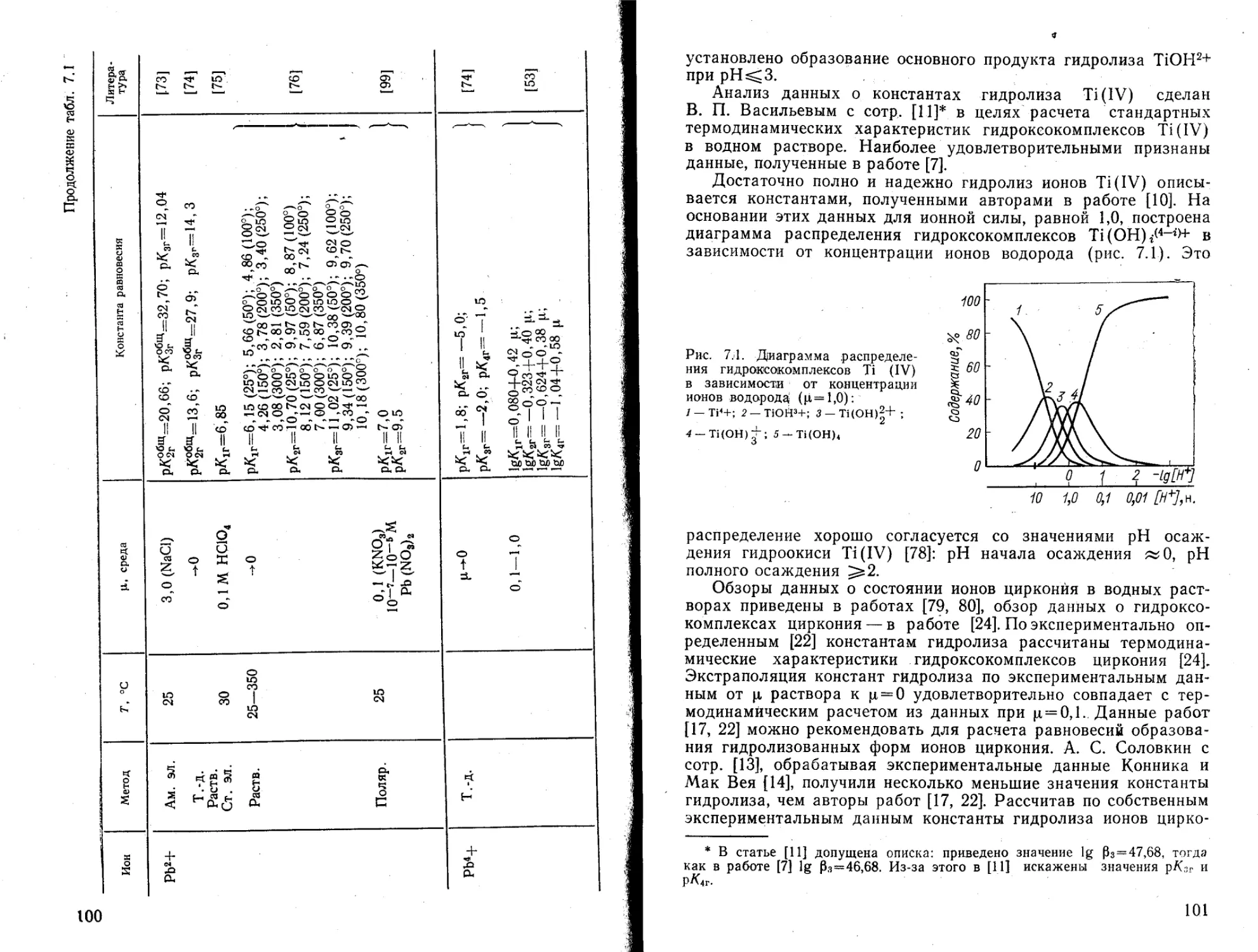
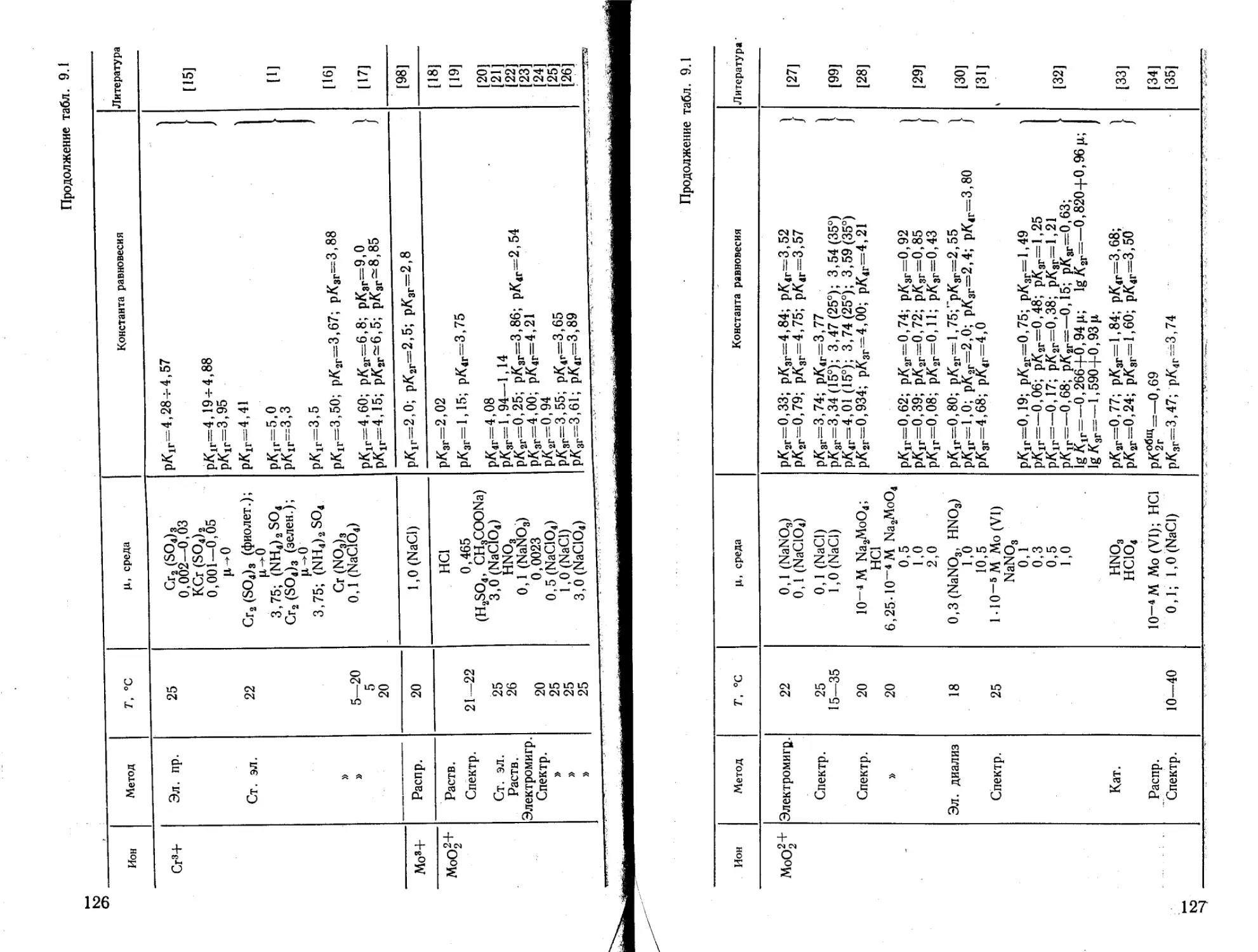
В литературе приведены данные о влиянии состава ионной среды на Гидролиз ионов металлов. Так, при изучении гидролиза ионов U (VI) [9—12] и Th (IV) [13, 14] (табл. 1.1 и 1.2) обнаружены различные гидроксокомплексы в разных по составу ионных средах. При изучении гидролиза ионов никеля (II) было найдено, что состав продуктов гидролиза в перхлоратной и хлоридной средах одинаков, но различаются константы устойчивости гидроксокомплексов [15]. Влияние среды на гидро-
Таблица 1.1
Константы устойчивости Igp^ гидроксокомплексов U (VI) (UO2)^ в различных средах [16]
Гидроксокомплекс урана ЗМ (Na)Cl [9] ЗМ (Mg) С1О4 [10, 11] ЗМ (Са) СЮ4 [10, 11]
(UO2)2 (ОН)3+ — —3,81 —3,96
(UO2)2 (ОН)2+ —6,64 —6,25 —6,20
(UO2)3 (ОН)2+ —12,54 —13,3 —13,4
(UO2)3(OH)+ — 18,57 —17,18 —16,91
(UO2)4(OH)2+ — 19,96 —20,2 —
(UO2) (ОН)+ — — —
(UO2)4(OH)+ —24,9 — —•
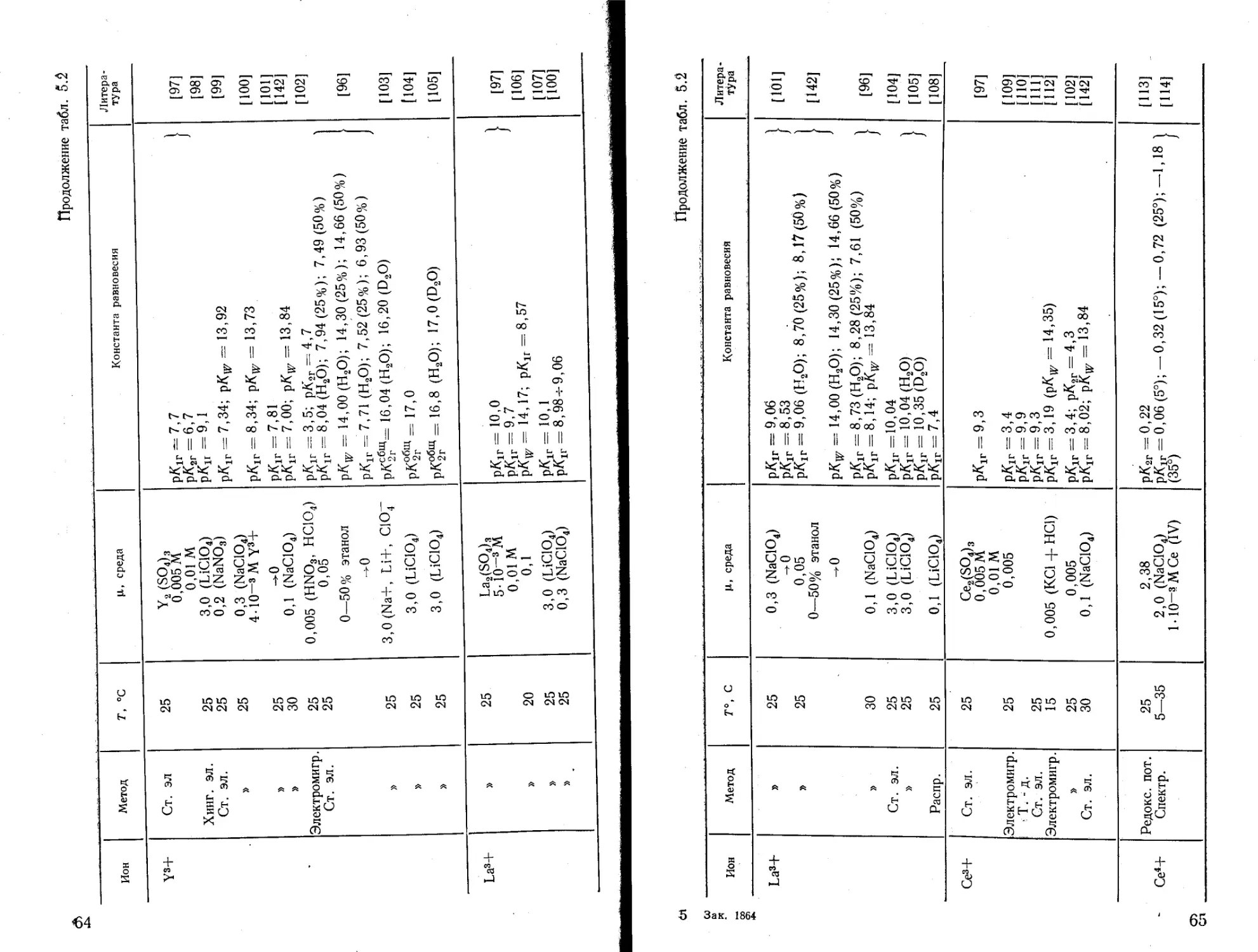
Продолжение табл. 1.1
Гидроксокомплекс урана ЗМ (Na) СЮ4 [10, 11] ЗМ (К) NOS [10,11] ЗМ (Mg) NO3 [12] 5М (Mg) NO3 [12]
(UO2)2 (ОН)3+ — —4,1 .— —
(UO2)2 (ОН)2+ —6,04 —5,96 —6,34 —6,52
(UO2)3 (ОН)2+ — 13,2 —12,8 — —
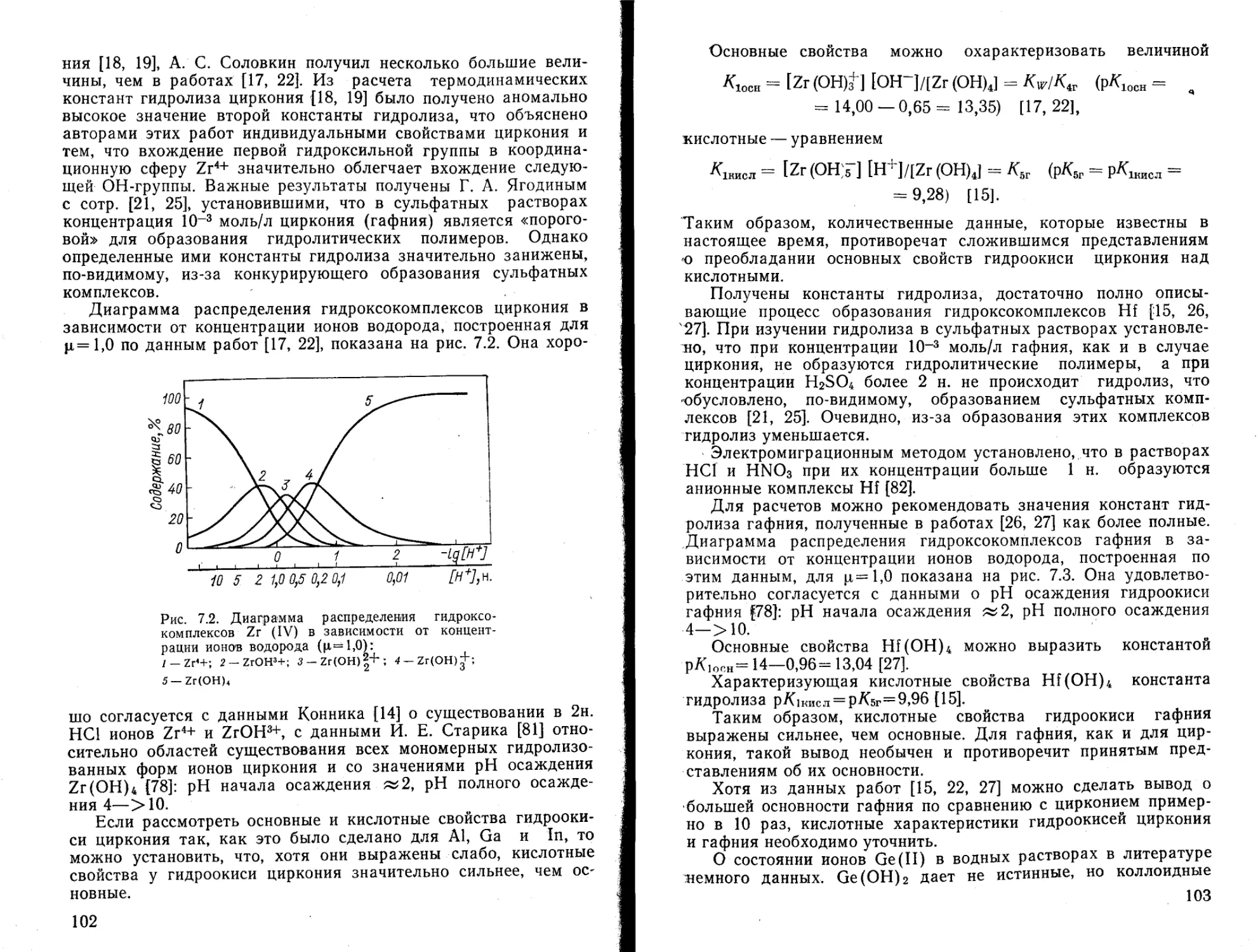
(UO2)3(OH)+ —16,53 —16,21 — 17,37 — 17,76
(UO2)4(OH)2+ — — —19,7 —
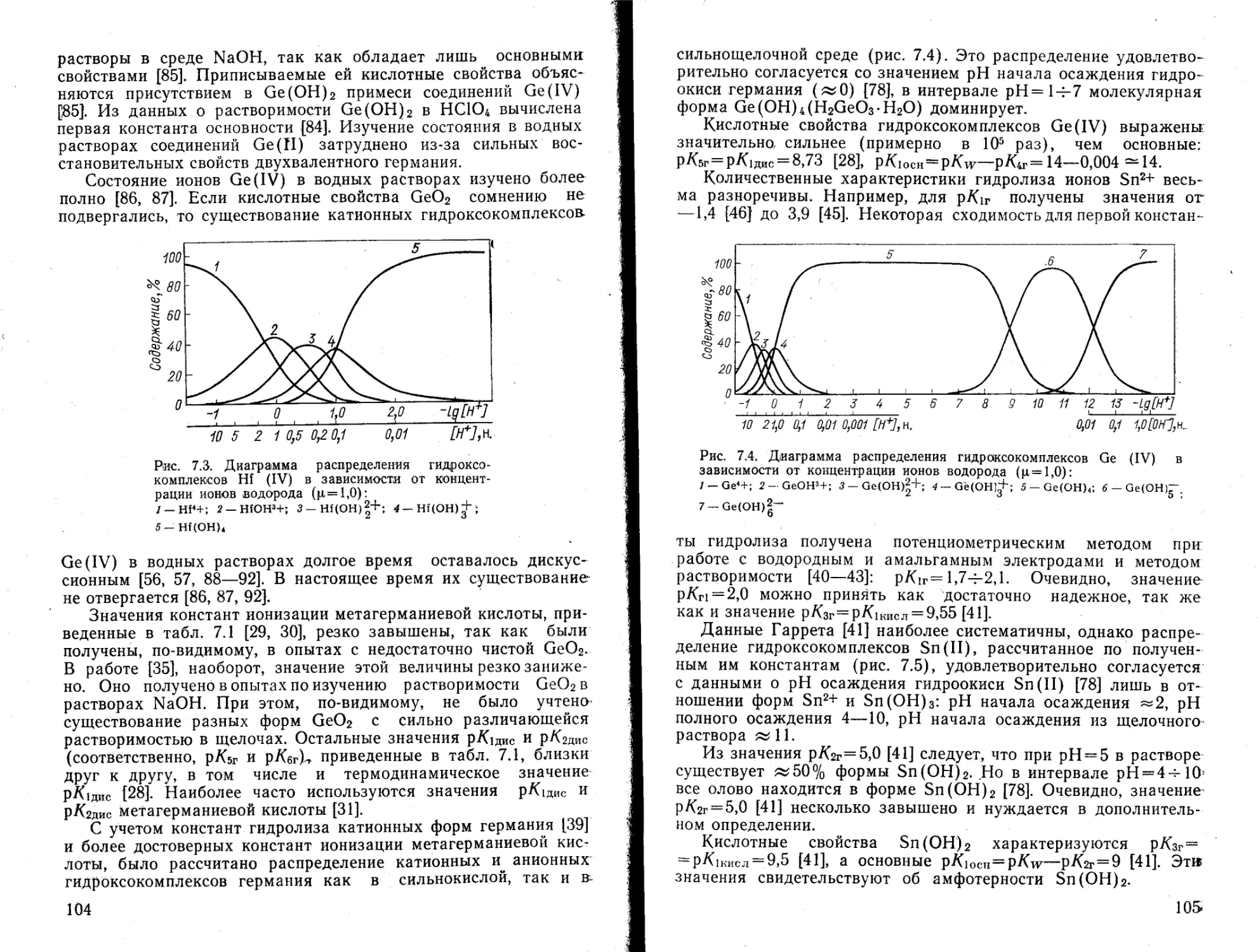
(UO2) (ОН)+ —6,1 — —5,4 —5,53
(UO2)4 (ОН)+ — — — •—•
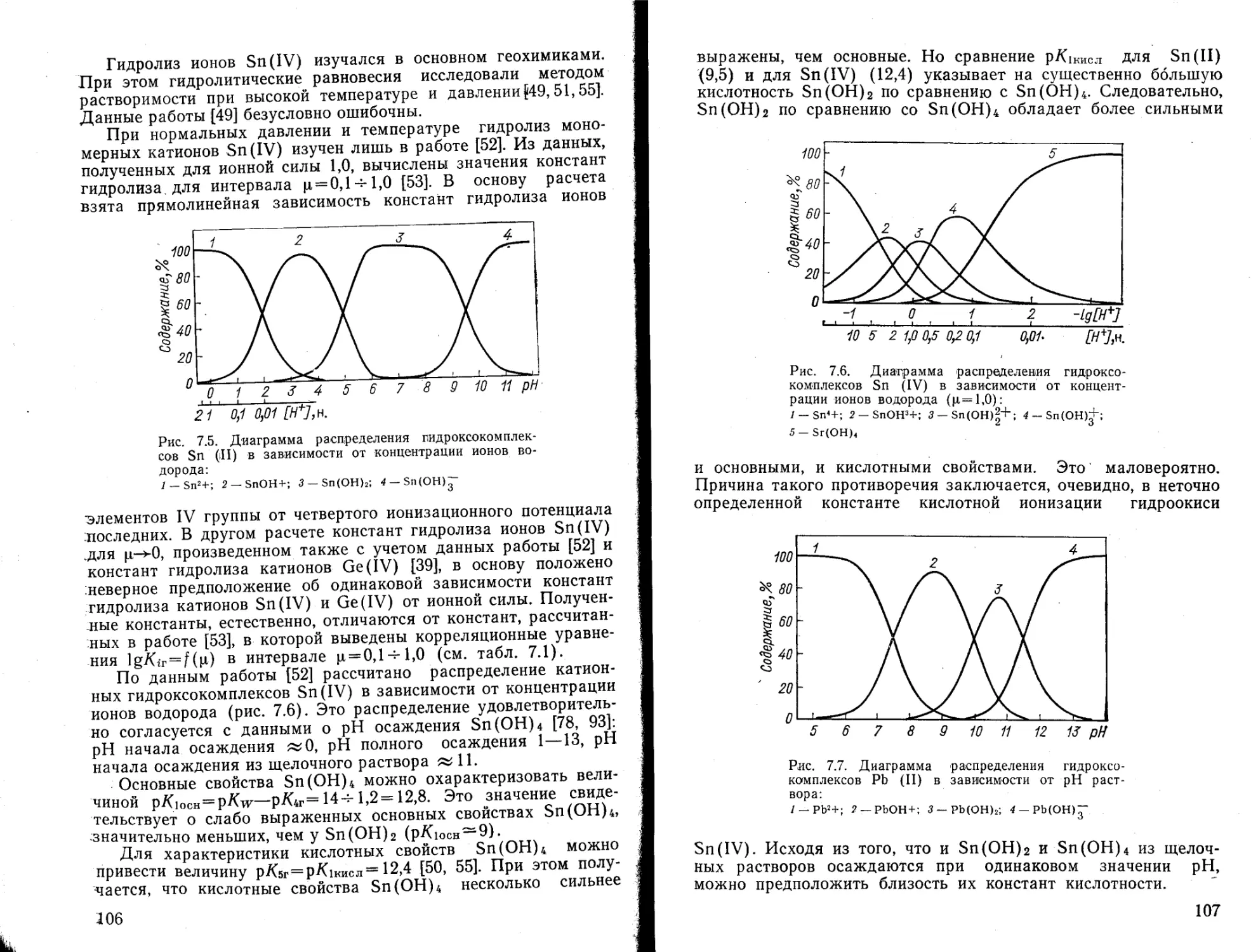
&
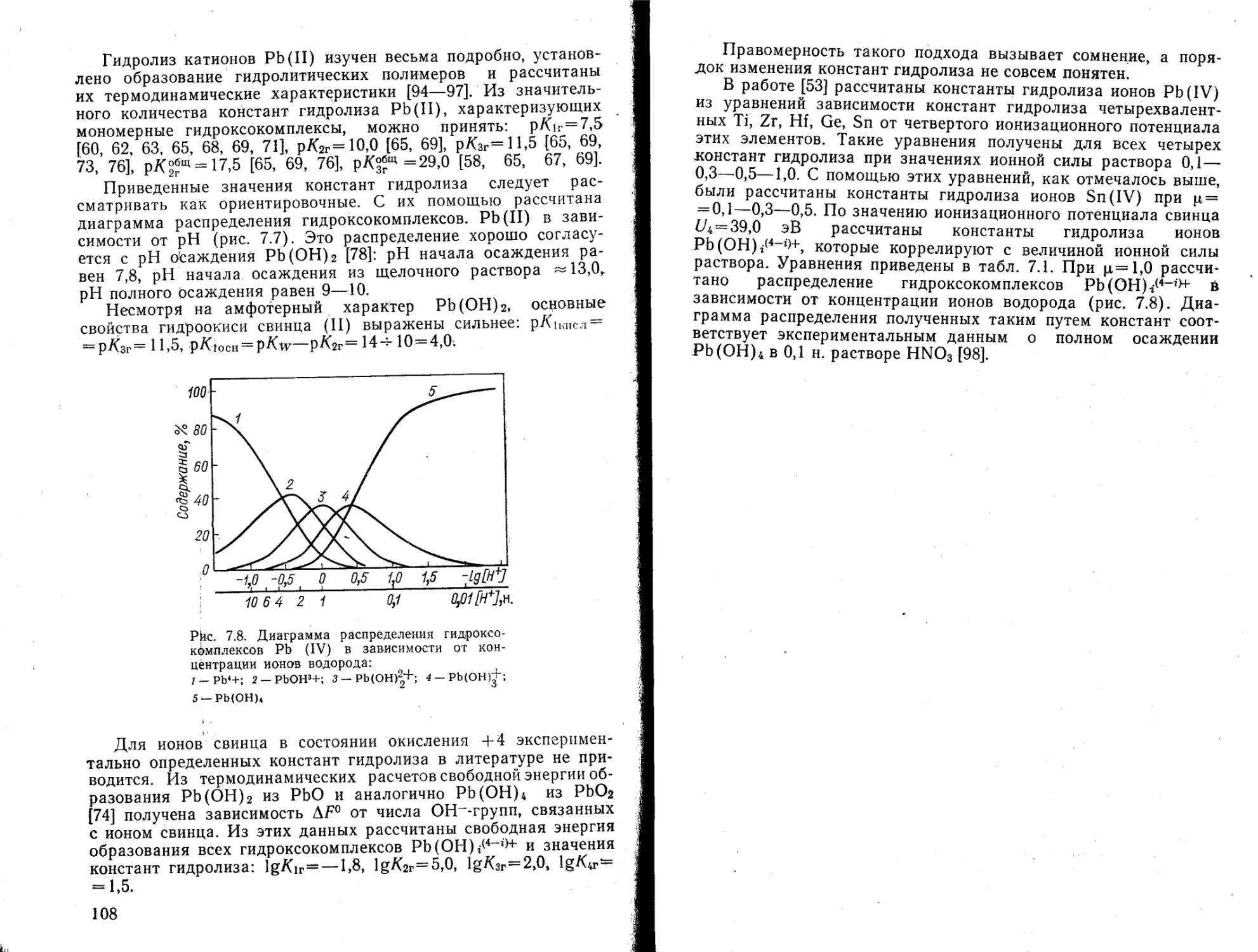
Таблица 1.2
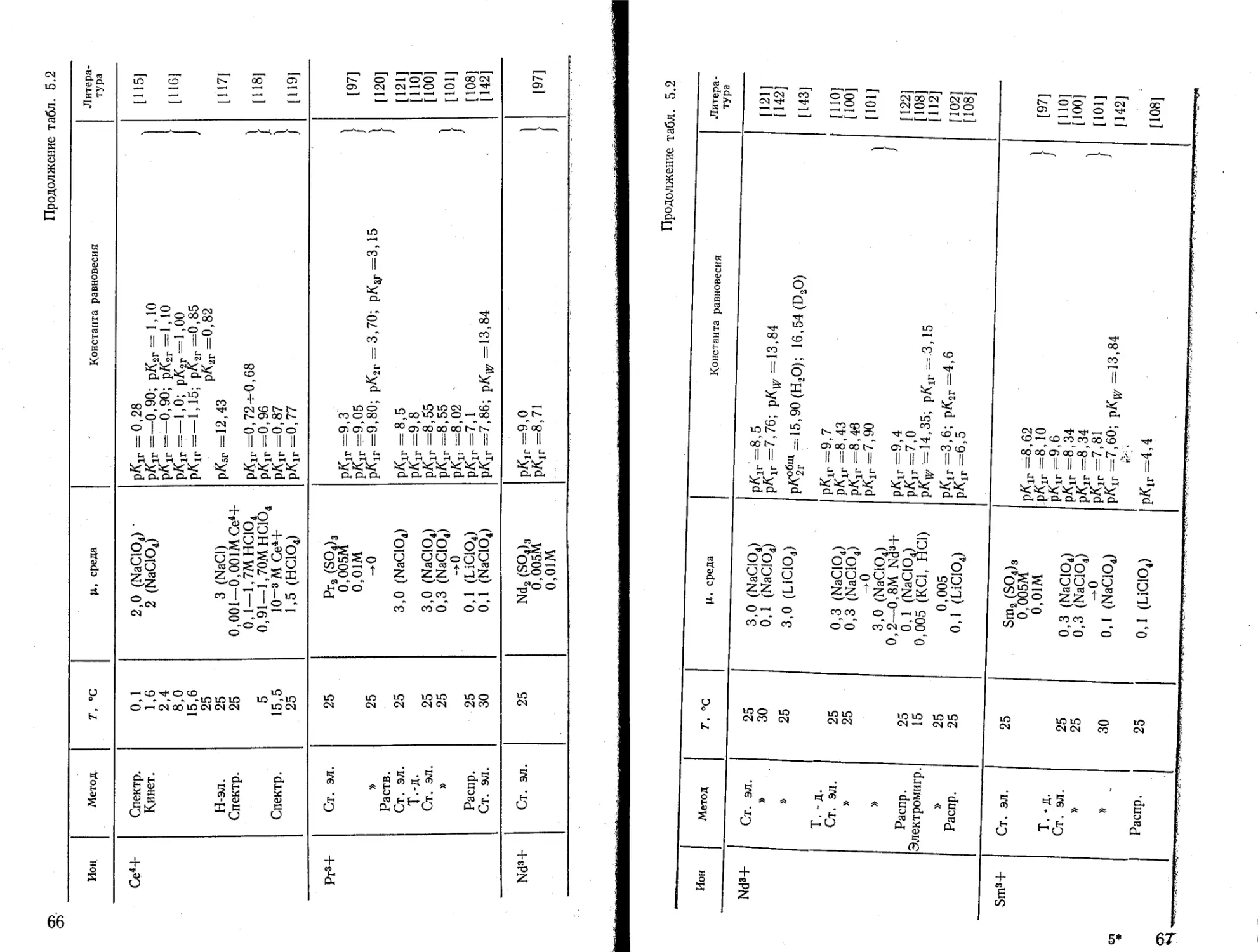
Константы устойчивости гидроксокомплексов тория Th^ (OH)J4flr“O+ в различных средах [16]
Гидроксо комплекс тория 3M (Li) NO3 [14] 3M (K)NO3 [H] 3M (Mg) NO3 [14] 4M (Na) NO3 [13] 4M (Na) CIO* [13]
Th (ОН) |+ — —9,67 — — —
Th/(OH)’+ — — — — —2,72
Th2(OH)|+ —5,26 —5,10 —5,17 —5,5
Th3(OH)53+ —8,98 '— — — •—-
Tha(OH)*+ — — — — —10,49
Th3(OH)t+ —8,0 — — — •—
Th3(OH)£+ —14,20 — —14,29 — —12,42
Th3(OH)|+ — — — —17,92 —
Th4(OH)|+ — ’— — — • —19,2
Th4(OH)t+ — — — —37,2 —
The (OH)}0+ — —- — — —36,2
The(OH)|+ —• —40,95 —43,20 — —
лиз ионов тория (IV) при одинаковых прочих условиях исследовали авторы работы [16]. Эксперимент проводился в различных нитратных средах. Установлено, что степень гидролиза ионов металла уменьшается с увеличением энергии гидратации, концентрации ионов среды и заряда аниона, но увеличивается с увеличением заряда катиона фоновой соли.
Влияние состава ионной среды на гидролиз окончательно не выяснено, но его необходимо принимать во внимание при сравнении констант гидролиза, полученных в различных средах.
В настоящее время разработаны методы, которые позволяют учесть максимально возможное число продуктов гидролиза, рассчитать с помощью ЭВМ константы их образования, составить несколько схем гидролиза с участием различных форм и при согласовании с экспериментальными данными выбрать нужную схему [17, 18]. Наряду с этим применяются различные графические методы, основанные на определении путем экстраполяции. Принципы и приемы этих методов подробно изложены в работах [1, 19].
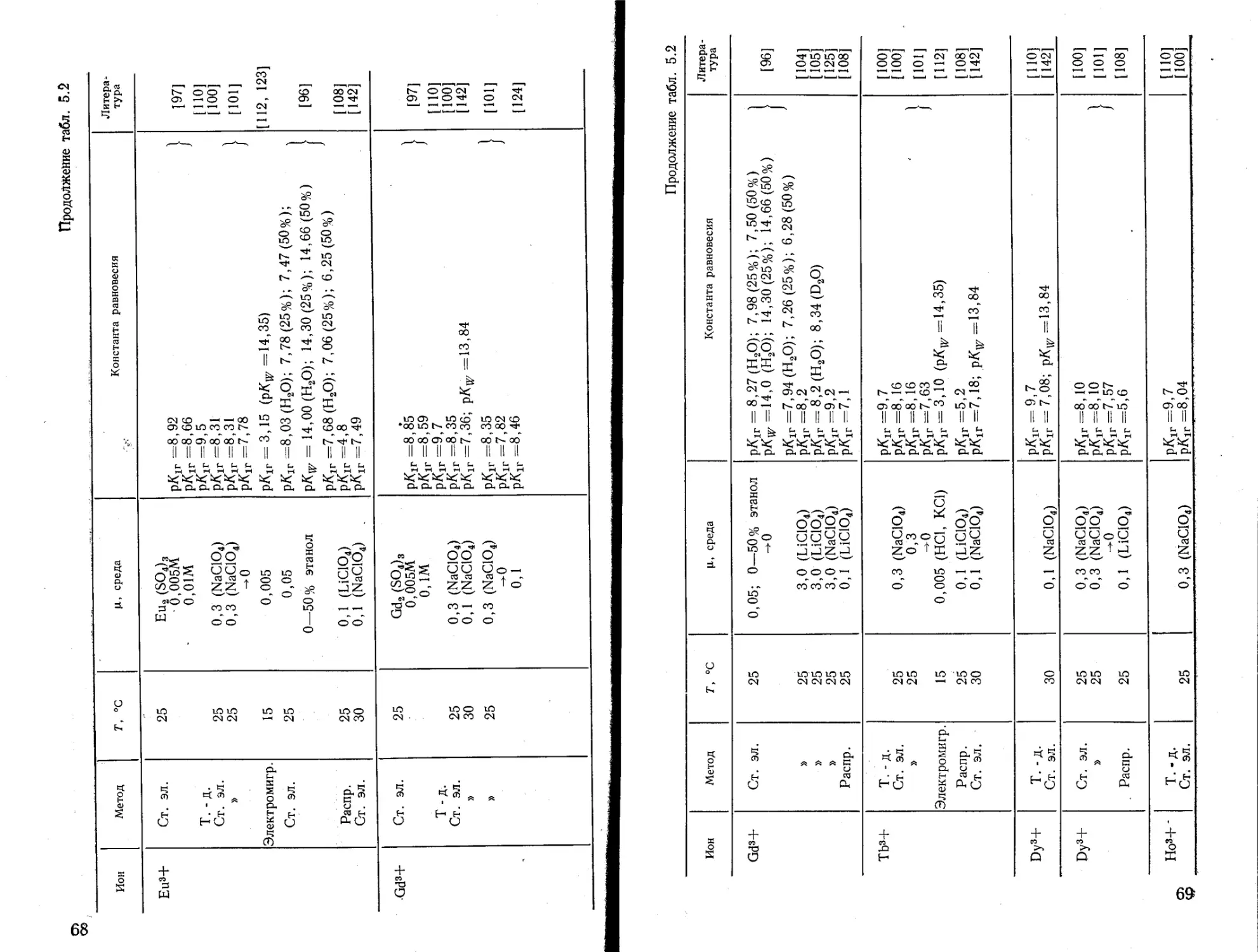
Использование сильно разбавленных растворов обычно исключает образование полимерных форм. Принято считать порогом полимеризации концентрацию металла, равную Ю-3 М 10
{20—22]. Концентрация ниже 10~4—10-5 М гарантирует присутствие в растворе только мономерных форм.
Из работ по изучению коллоидных свойств полония, протактиния, циркония, ниобия и других элементов следует, что в принципе возможно образование истинных коллоидов (точнее, коллоидных частичек гидроокисей) при микроконцентрациях элементов в растворе [23], хотя для этого, как правило, требуются высокие значения pH. Показано, например, что Pu (IV) при концентрации в растворе 10”7 М и рН>7,5 образует истинные коллоиды, a Eu (III) при такой же концентрации образует их в растворах с рН> 11,0 [24]. Граница значений pH, с которой начинается образование коллоидных частиц, очевидно, обусловлена (так же как и pH начала осаждения) гидролизом катионов металла. Количественной связи не установлено, но можно сказать, что при равных концентрациях образование полимерных коллоидных частиц начнется в более кислых растворах металла, константа гидролиза ионов которого больше.
В настоящее время невозможно экспериментально определить границу концентраций процесса полимеризации, так как невозможно полностью исключить влияние коллоидных загрязнений на состояние в растворах элементов при их микроконцентрациях. Поэтому вывод о переходе соединения элемента из ионного состояния в коллоидное (полимерное) часто делается на основании нарушения закономерностей, установленных для ионных форм (например, отклонения от закона Бугера—Ламберта—Бера при спектрофотометрических исследованиях). При строгом подходе к вопросам гидролиза присутствие моно- или полиядерных видов определяется предварительным изучением данных о зависимости функции образования от концентрации металла [19]. Если этой зависимости нет, то в системе присутствуют только моноядерные виды.
Моноядерный гидролиз представляет собой комплексообразование ионов металла с ионами гидроксила и к нему применимы методы расчета комплексообразования в растворах. Количественно моноядерный гидролиз характеризуется константами гидролиза, образования и основности гидроксокомплексов.
Константами гидролиза называют константы равновесия
реакций гидролиза
Мг+ + Н2О^МОН(г-1)+ + Н+; (1.4)
М (ОН)^~г)+ + Н2О М (ОНЯ+Л1)+ + Н+; (1.5)
общее уравнение
Мг+ + i Н2О М (ОН)*2-1)+ + i Н+. (1.6)
Константы образования характеризуют процессы
Мг+ + ОН-^МОН(г-,)+; (1.7)
11
М (0Н)^“‘)+ + ОН- М (ОН)<+Л")+; (1.8)
общее уравнение
Mz+ + i ОН- =₽ь М (ОН)|г“0+. (1.9}
Константы основности представляют собой величины, обратные константам образования.
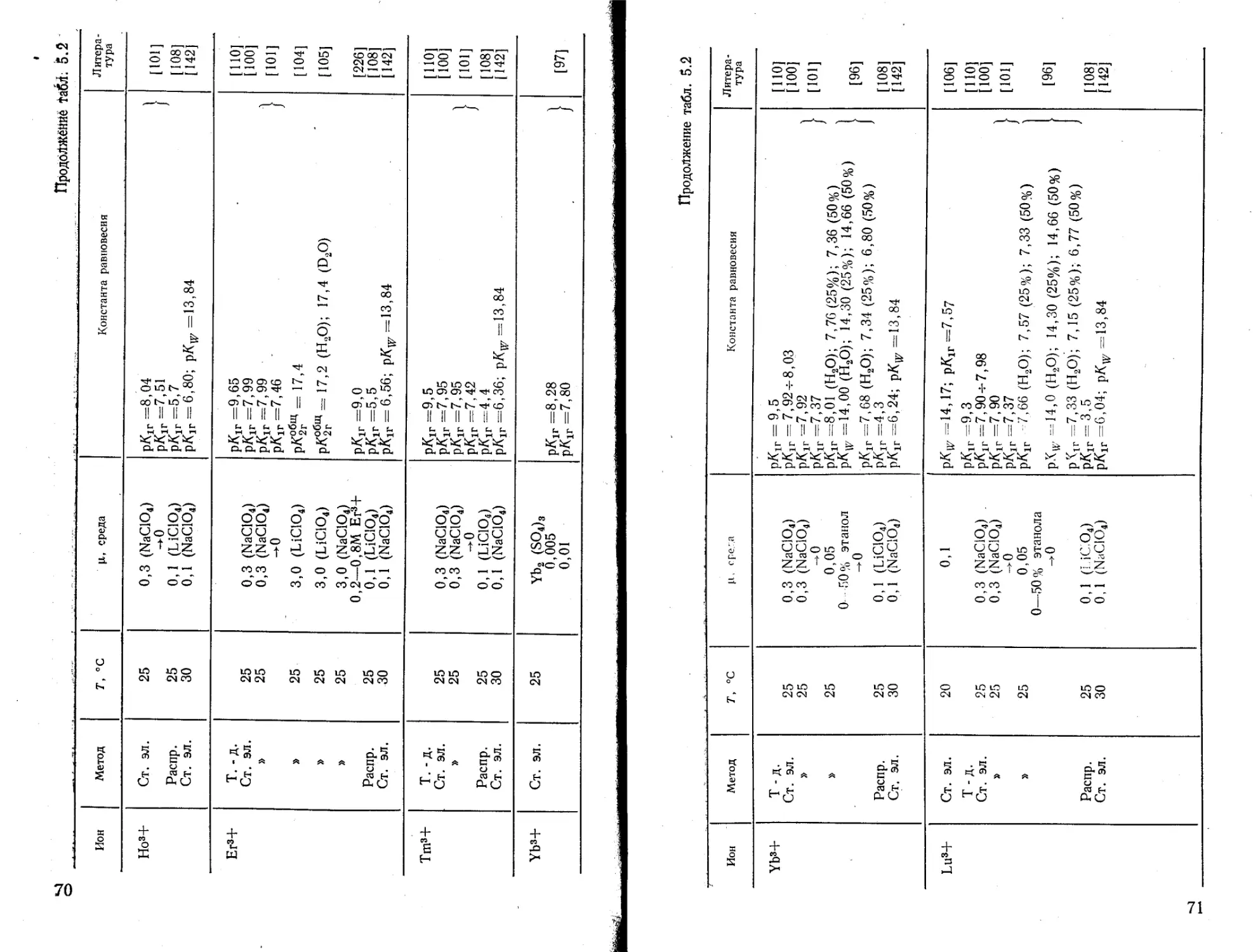
При постоянной ионной силе и, следовательно, постоянных коэффициентах активности ионов соответствующие константы:
Klr = [М (ОН)(г-1)+1 [н+]/[Мг+]; Kiv = [М (0Н),2“‘)+] х
X [H+]/[M(OH)fcz-n+]; (1.10)
^Мм(онр>ш+] (1.11}
0, = [М(ОН)(г-*)+]/[М*+] [ОН-]; (1.12)
= [М (ОН)Н)+]/[М (0Н)£Л1)+1 [ОН-]; (1.13)
р”64 = [М (0Н)(Г1)+]/[Мг+1 [ОН-]' = РА . . . pf; (1.14) в=[Мг+][ОН-]'/[М(ОН)]<г-°+ = КоснЛоон2 . . .tf0CHi. (1.15) Эти константы связаны между собой через ионное произведение воды Kw-
— Kw Pi! = ^Pz/PiJ — Kw Pi/Pf—i; (1.16)
^1Лосн1 = Kw; KiTK0&ii = Kw- (1.17)
Теоретические и экспериментальные данные по диссоциации многоосновных кислот свидетельствуют о следующем порядке величин ступенчатых констант ионизации: Л1ион>^2ион> 3>Хзион> ... >Кгион*
Реакции гидролиза ионов металлов — реакции протолиза^ и поэтому в ряду ступенчатых констант гидролиза должен соблюдаться такой же порядок. О соотношении между ступенчатыми константами гидролиза можно судить по изменению порядка ступенчатых констант образования гидроксокомплексов. За исключением случаев, когда в системе образуются гидролитические полимеры или в результате присоединения последующей гидроксильной группы резко изменяется геометрия комплекса, отношение между последовательными константами образования р определяется статистическим эффектом [25, 26]
Pn __ n 4~ 1 г — гг -|- 1 « * — п *
где i — максимальное координационное число; п — число уже присоединенных гидроксильных групп. Из этой формулы следует, ЧТО р1>р2> ... >рг.
При изучении гидролиза обычно определяют константы образования гидроксокомплексов рг-, из которых вычисляют част-12
ные и общие константы гидролиза. Методика определения констант образования (или устойчивости) отдельных комплексов описана в работах [1, 27, 28].
Константы устойчивости рассчитывают с помощью функций, легко вычисляемых из опытных данных и связанных с константами довольно простыми зависимостями. Наиболее широко пользуются функцией образования п (лигандное число), предложенной Бьеррумом [29]. Применительно к гидроксокомплек-сам она запишется в общем виде как
П = (Сон- - [ОН~])/СМ, (1.18)
где Сон" и См — общие концентрации ионов гидроксила и ме-талла-комплексообразователя.
С константами устойчивости функция образования связана соотношением
- = рх [он-] + 2рг [он-р + • . • + nprt [он-]» (1 19)
1+PJOH-1+PJOH-P + . . .+рл[ОН—]”
или
jfipzpH-f
п = . (1.20)
i + $f РИОН-Г t=l
Леден [30] и Фронеус (31] ввели функцию Ф, представляющую собой отношение общей концентрации металла к его равновесной концентрации:
Ф = См/[М]. (1.21)
К. Б. Яцимирский (32] назвал эту функцию закомплексованностью, так как она характеризует глубину протекания комплексообразования в системе.
Закомплексованность выражается через
Ф = 1 + рх [ОН-] + р2 [ОН-]2 + . . . +Р„[ОН-]П (1.22) или
п
ф = 1 + ^Рнон-ь (1.23)
Иногда находят долю данного комплекса а,:
az = [М (ОН)(]/СМ. (1.24)
Значение щ меняется от 0 до 1 и связано с константами устойчивости уравнением [32]:
/п
У РНОН-Г*. (1.25)
т^о
13
После нахождения функций остается еще задача определения с их помощью констант устойчивости. Для этого применяют различные графические методы [32, 33]. В работе [1] подробно рассмотрены варианты графического и экстраполяционного решений систем с различным количеством комплексных частиц.
С развитием электронно-вычислительной техники стало возможным совместное решение набора уравнений, включающих все экспериментальные данные, методом наименьших квадратов с применением статистических приемов.
Подробно принцип выбора схемы гидролиза с помощью» ЭВМ излагается в работе [19].
2
МЕТОДЫ ОПРЕДЕЛЕНИЯ КОНСТАНТ ГИДРОЛИЗА
Для изучения гидролиза применяют различные экспериментальные методы как кинетические, так и равновесные. Среди равновесных используют методы, которые включают определение концентрационных переменных (потенциометрия, метод распределения), а также методы, с помощью которых определяется изменение тех или иных свойств системы в зависимости от накопления продуктов гидролиза (спектрофотометрия, рассеяние света, магнитная восприимчивость). Задача любого метода в конечном итоге сводится к получению сведений о равновесных концентрациях ионов металла или водорода и расчету из этих данных функций, непосредственно связанных с константами образования гидроксокомплексов. В идеале желательно иметь метод, который позволил бы Фиксировать каждую гидролизованную частицу. К сожалению, такого метода пока нет. Прямые физические методы (ядерный магнитный резонанс, спектроскопия комбинационного рассеяния, ультрацентрифугирование) позволяют идентифицировать ту или иную частицу в наиболее простых системах, но они применимы пока лишь к растворам с высокими концентрациями и не относятся к точным и чувствительным. Бейес и Месмер [1] пишут: «...Ни один из этих методов,, взятых поодиночке или вместе, не может конку рировать с pH-метрией, более чувствительной и позволяющее выделять среди различных альтернатив механизм гидролиза... все эти методы используются чаще вместе с потенциометрическим методом, чем вместо него».
2.1. Потенциометрический метод
Существуют два способа потенциометрического исследования гидролиза: определение концентрации свободных ионов металла и определение pH.
Н. П. Комарь [2] показал, что для исследования ступенчатого моноядерного гидролиза достаточно знать исходные концентрации ионов металла и водорода и определить равновесную концентрацию водородных ионов, т. е. pH.
Потенциометрическое определение pH проводят с помощью
15
водородного, стеклянного или хингидронного электродов, которые обратимы к ионам водорода. Особенности и конструкция этих электродов достаточно подробно рассмотрены в работе [3].
Полное выражение для п — функция образования при использовании потенциометрического измерения pH для кислых растворов запишется как
й= ([Н+] —[ОН“] — Сн)/См, (2.1)
а для щелочных, где — Сн = Сон'-
п = (Сон - [ОН-] + (Н+])/См. {2.2)
Для растворов, где концентрации гидроксильных и водородных ионов сравнимы, необходимо пользоваться полными выражениями. Если одна концентрация преобладает, эти выражения упрощаются:
й=([Н+]-Сн)/См; (2.3)
й=(Сон-[ОН-])/См. (2.4)
При изучении гидролиза обычно титруют кислые растворы, начиная с тех значений кислотности, при которых гидролиз практически не протекает, и поэтому чаще используют выражение (2.3). Например, при изучении моноядерного гидролиза Н. П. Комарь [2] вычисляет функцию образования следующим образом.
Если обозначить начальные концентрации ионов металла и водорода См и Сн, а равновесные [М]=Ь, [Н+]=й и ,[M(OH)d=zb то
= (2.5)
См = & + Дг(.; (2.6)
h = Сн + Д izt. (2.7)
В тех случаях, когда разность h—Сн значительно больше погрешности измерения Л, система уравнений (2.5) — (2.7) сводится к уравнению
_ п _
П/ = Х (/-п,)₽ЛГ, (2.8)
где
n^lhj-C^IC^ (2.9)
(nj — функция образования в /-м опыте).
16
Константы pf могут быть вычислены методом наименьших квадратов. Н. П. Комарь [2] вычислил константы гидролиза ртути таким методом из данных работы [4] и получил значения констант, близкие к значениям Хиетанена и Силлена [4], которые использовали кроме измерений pH измерения концентрации ионов ртути.
Такой же метод был использован для изучения первой ступени гидролиза редкоземельных металлов и иттрия [5], а также ионов неодима [6]. В этой же работе полученные потенциометрическим методом результаты были уточнены расчетами по методу «самосреды» [7].
2.2. Спектрофотометрический метод
Спектрофотометрический метод основан на определении при разных pH светопоглощения растворов, содержащих гидролизованные формы ионов металла. В отличие от pH-метрии он не позволяет определить равновесные концентрации участвующих в равновесии форм, так как при образовании нескольких комплексов сложно определить их коэффициенты молярного погашения. В результате серийных измерений оптической плотности при разных pH можно найти средний (наблюдаемый) молярный коэффициент погашения, который определяется одновременным присутствием всех поглощающих форм:
e=A/CMZ, (2.10)
где А — оптическая плотность раствора; См —исходная концентрация металла; I — толщина поглощающего слоя.
С учетом всех поглощающих форм оптическая плотность раствора по закону Бугера—Ламберта—Бера выражается как
АЦ = е0 [М] + 61 [М (ОН)!] + е2 [М (ОН)2] + . . . + 8г [М (ОН),]. (2.11)
Из материального баланса следует, что
См = [М] + [М(ОН)1] + [М(ОН)2]+ . . . + [М(ОН)(]. (2.12)
Из уравнений (2.10) — (2.12) получаем
е= e° + eiMOH-] + s2MOH~]2+. . . +е£МОН-]г (2 13)
1+₽1[ОН-] + ₽г[ОН-р+ . . .+р, (он-г ' ’ }
Если из обеих частей уравнения вычесть значение ео, то получим
д- = AeiPi 1°н~1 + Де8Рг ЮН-]2 + . + ДегР/ [ОН-]‘ „
1 + ₽! [ОН-] + р2 [ОН-]2 + . . .+₽г[ОН-Г 1 ’
Из серии измерений можно получить большое число значений Де и, следовательно, большое число уравнений. Необходимо
2 Зак. 1864 1 7
решить эти уравнения относительно Аег- и Способ решения таких уравнений дан К. Б. Ядимирским [8, 9].
Такой вариант спектрофотометрического метода используется для изучения гидролиза в том случае, если в растворах наблюдается одновременно не более двух поглощающих свет частиц [10—13]. При определении констант гидролиза ионов Fe (III) установлено, что в области 260—360 нм и концентрации Fe, равной 4,7-10~4 г-ион/л, в растворе существуют два поглощающих вида — негидролизованные ионы Fe3+ и первый продукт гидролиза Fe(OH)2+ [10]. Для кюветы с /=1 см наблюдаемый молярный коэффициент погашения
е = [Fe3+] + [FeOH2+] е2, (2.15}
а исходная концентрация CM=[Fe3+]+[FeOH2+]. На основании этого уравнения и выражения для константы образования FeOH2+ получаем
fFe3+] = [Н+] ——; (2.16)
ЦН+] + Д1Г)
[FeOH2+] = J (2.17)
([H-rj 4- Kir)
е(Я1г + [Н+]) - С№ [Н+] 81 + К1гСм82. (2.18)
Последнее уравнение используют для расчета. Измерения при постоянном значении См и трех значениях pH дают систему из трех уравнений с неизвестными 8ь 82 и /Gr. Из этой системы уравнений находят значение Ллг.
Спектрофотометрический метод в таком варианте применяется довольно широко. Он используется для металлов, обладающих собственным хромофорным действием и в тех случаях,, когда продукт гидролиза имеет достаточно высокий молярный коэффициент погашения и вносит значительный вклад в наблюдаемую оптическую плотность.
В. А. Назаренко с сотр. [15—32] разработан метод спектрофотометрического определения констант моноядерного гидролиза с использованием конкурирующего лиганда. Для этой цели используют окрашенные органические реактивы, образующие комплексы с изучаемым металлом.
В растворе соли многовалентный металл находится в виде гидроксокомплексов М(ОН) Окрашенный органический реактив, вступая в реакцию комплексообразования с металлом,, действует как конкурирующий лиганд по отношению к гидроксильным ионам. Если известен химизм комплексообразования металла с конкурирующим лигандом и точно определена концентрация образующегося комплекса, то можно определить константы образования гидроксокомплексов металла. Непременное условие заключается в отсутствии полимеризации в системе металл—гидроксил—ион—окрашенный органический ре-18
актив. Это достигается использованием низких концентраций металла (около 10~5 М) и поэтому при выборе конкурирующего лиганда необходимо учитывать его чувствительность по отношению к изучаемому металлу. Реактив, который играет роль, конкурирующего лиганда, должен образовывать одно комплексное соединение с изучаемым металлом, и комплексообразование должно происходить в той области концентраций ионов водорода, где существуют гидролизованные ионы металла. При соблюдении этих условий уравнение реакции взаимодействия металла с окрашенным органическим реактивом можно записать так:
М (ОН)<-г~‘>+ + [М (ОН), (Hm_„ R)9]<z-‘-^)+ + qntt+ .(2.19) Образующийся комплекс диссоциирует по схеме
[М (ОН), (Hm_„ R)9](z~z~9")+ М (ОН)*г-‘)+ + qHm_n R"~ (2.20) и характеризуется константой нестойкости
Кн = [М (ОН),] [Hm_n Rp/[M (ОН), (Hm_n R)J. (2.21)
Реактив в растворе диссоциирует. Последовательные константы диссоциации реактива равны
. [HOT_1R] [Н+]
1 [HmR]
ь [Hm-nR] 1Н+] ,
* * П г т т Г) 1 > * * * 2 /Ц
[Нщ—п+х R]
IR] [Н+] [HR]
(2.22)
Если обозначить концентрацию комплекса Ск, общую концентрацию реактива Cr, то концентрация реактива, не связанного в комплекс, будет
CR - qCK = [HmR] + [Hm_, R]+ . . . + [Hm_„R] + . .. + [R]. (2.23)
Из выражений (2.22) и (2.23) можно определить равновесную концентрацию иона реактива [Hm_„R]:
[Hm_„ R] =----—--------(Cr~-Ck)------------ . (2.24)
fi. . __[H±r__________\
km + • • • + k^. . .kn. . .kmJ
Концентрация не связанного в комплекс металла равна сумме концентраций гидроксоформ:
z
Co-CK = V М(ОН)Г°+. (2.25)
1 = 0
Выразив равновесную концентрацию негидролизованного иона металла через общие константы устойчивости гидроксокомплексов с учетом (2.25), находят
2*
19
[Mz+] = (Co - CK) / 1 + V ₽z [OH-]'. (2.26)
Общая константа устойчивости гидроксокомплекса равна pt. = [М (OH)^-,)+]/[Mz+] [ОН-]1 (2.27)
и с учетом (2.26)
₽f [ОН-]' (Со — CJ = [М (ОН)Г°+1 (1 + Д ₽' t°H~]z) • (2-28>
Делением равенства (2.28) на (2.21) получают:
________Pf [ОН-]' (Со - Ск) (CR - дСку_=
( [н+] [Н+]'" V
^iZ-k ( 1 + ъ + • • • + bfc k ь J
\ кт • • • Иц . . . кщ /
= 1+Д₽ДОН~]'. (2.29)
Если обозначить
S = [ОН-]'(С0 —CK)(CR — уСк)*7
( [Н+] [Н+Г у’ '
Ск ( 1 + k + • • - + Т"£ £ £ )
то уравнение (2.29) примет вид
S₽A=1 + Дрдон-]'. (2.31)
Дальнейшие преобразования проводят по методу В. В. Фомина и Е. П. Майоровой [14]. Продифференцировав выражение (2.31), получим:
м$/адон-] - Д [ОН-]'-1. :
Разделив выражение (2.32) на (2.31) и записав функцию обра- • зования Бьеррума
« = 5} Ф, [ОН-] / 1 + V р( [ОН-]', (2.33)
i=I / i=l
получим результат в виде
dlnS/d[OH“] = n/[OH-J. (2.34)
Величина гё/[ОН~] представляет собой наклон касательной в данной точке кривой зависимости In 5 от [ОН-]; S находят из спектрофотометрических и потенциометрических измерений. 20
Дифференцирование проводят графически. Значение 0,- находят из уравнения
limG, = lim —-— = В,;
[он—]+о [oh-j-,-0 [ОН~]
lim G2 = lim = 202 — 0? ;
[он~]->о [он“]^о [OH—]
G2“(2P2~" Pl) no QRR lR3.
—— = 3₽з — 3₽1P2 + Pl ;
.. n -r G3 —' (ЗРз — 3pxp2 + Pi)
limG4 = lim -------------------------- =
[OH“]+0 [OH^]->0 [OH-]
= 404 - 40X03 + 40i 02 - 202 — 01.
(2.35)
(2.36)
limGg = lim [ОН~]-»0 [ОН-]-^0
' (2.37)
(2.38)
Этот метод применим также и в том случае, когда металл образует с окрашенным органическим лигандом нерастворимое соединение, которое остается в растворе в коллоидном состоя' нии, может быть стабилизировано защитным коллоидом и если эти коллоидные растворы следуют закону Бугера — Ламберта — Бера. Тогда вместо выражения для константы не-стойкости (2.21) следует пользоваться выражением для произведения растворимости комплекса:
ПР = [М (OH)J [Н^ RK (2.39)
Все подстановки и преобразования выполняют аналогично преобразованиям, приведенным выше, и получают те же конечные выражения, где
S = [ОН-Н(С0- Ск)(Ск-4СкГ . (2 40}
[ [н+] ______[н+]т у *
\ km "b , kn . . . km )
Этим методом были определены константы моноядерного гидролиза ионов Ga [15, 161, Al [17, 18], Sc [19], In [20], T1 (III) [21], Ti [22], Zr [23], Hf [24], Ge [25], Sn [26], Mo [27], W [28, 29], Bi [30], Sb [31], Te( IV) [32].
2.3. Метод распределения (экстракция]
Этот метод основан на способности органических растворителей экстрагировать катионы металлов в виде их солей или комплексных соединений. Распределение вещества между двумя растворителями подчиняется закону Нернста. Если вещество. растворимо в обеих ограниченно смешивающихся (в идеальном случае несмешивающихся) фазах, то отношение концентраций его в обеих фазах постоянно при данной температуре: £ = Сорг/СВОдн, где D — коэффициент распределения.
21
Это соотношение справедливо для тех случаев, когда растворенное вещество находится в обеих фазах в одном и том же .молекулярном состоянии.
Если предположить, что комплексы или соли металла присутствуют только в форме простых моноядерных частиц и в органической фазе присутствует только один незаряженный комплекс, то простейший механизм экстракции, пригодный для изучения гидролитических равновесий, можно записать в следующем виде:
Мг+ + zHR(opr) MR, (opr) + ZH+ . (2.41)
Если коэффициенты активности в каждой фазе сохраняются постоянными при всех концентрациях в опыте, что достигается применением фонового электролита, то константу равновесия реакции (2.41) будет иметь вид:
Q = [MRz]opr [H+J7[HR]*pr [Мг+]. (2.42)
Наблюдаемый коэффициент распределения ионов металла равен
£>М ~ См (орг)/См (водн) = [MRjopf/lMz+] (1 + Д Г°И ]‘) • (2ЛЗ)
Из (2.42) и (2.43) следует, что
2
1 + 2 0. [ОН-]' = Q- [HR]opr/(DM- [Н+И)- (2.44)
1—1
Если обозначить
[HRjopr/^м [Н+р = S, (2.45)
то уравнение (2.44) примет вид
QS = 1+ Д 0ДОН-Г. (2.46)
Продифференцировав и разделив полученный результат на (2.46), получим уравнения
2 фдон-р-1
—------= ; (2.47)
Sd[OH-] *
i+Дмон-р
d In S/d [ОН"] = n/[OH~]. (2.48)
Уравнение (2.48) тождественно уравнению (2.34), которое используют при вычислении констант образования гидроксокомплексов металлов спектрофотометрическим методом конкурирующего лиганда. Дальнейшее вычисление констант Рг произ-22
водят также с помощью вспомогательных функций G2-, которые определяются уравнениями (2.35) — (2.38).
Этот метод расчета предложен В. В. Фоминым и Е.П. Майоровой [14] и использован многими исследователями при изучении мономерного гидролиза [33—37].
По методу Фронеуса [38, 39], если есть функция
X([OH-])=l+^z[OH-]\ ' (2.49)
то
[ОН-]
1пх([ОН-])= ( d[OH“], (2.50)
[ОН ]
о
Значения х([ОН~]) находят из зависимости пДОН”] от [ОН~].
При использовании экстракции для изучения гидролиза необходимо четкое представление о механизме распределения, который, по возможности, должен быть наиболее простым. Особенно важно знать, что экстрагент и экстрагируемый комплекс не изменяются в водной и органической фазах на протяжении всей изучаемой области pH, что растворитель нерастворим в водной фазе и не влияет на поведение катиона, а продукты экстракции и формы реактива не ассоциированы.
Методом распределения с применением расчетов В. В. Фомина и Е. П. Майоровой и параллельно по Фронеусу изучен гидролиз ионов Ge (IV) [33].
С помощью распределения ионов Zr4+ между водными растворами HC104+LiC104 (ц = 1,0) и растворами теноилтри-фторацетона [37] в бензоле изучен гидролиз ионов циркония. Термодинамические константы устойчивости гидроксокомплексов циркония
рг(ОН)«~'>+]й7<}+
Pi — : (2.51)
[Zr4+] gZr4+£0H-
определены из данных по распределению и значений коэффициентов активности ионов, найденных по эмпирическим уравнениям [40—42].
2.4. Метод растворимости
По мере увеличения pH ионы металлов осаждаются в виде гидроокисей или окислов. В насыщенном растворе твердой гидроокиси устанавливается равновесие
М (ОН)2 (тв) + (г — 0 Н+ М (ОН)Г~о+ + (z — I) Н2О. (2.52)
23
Константу равновесия такого процесса можно записать в виде xs = [м (ОН)(/-°+]/[Н+]<г~° . (2.53)
Концентрация металла в насыщенном растворе, т. е. растворимость, будет равна
Л 2V
См (насыщ) “ 2 [М (ОН)‘г“,)+] = 2 Ks [Н+]<2-° . (2.54)
о и
Таким образом, с помощью растворимости соединения в насыщенном растворе при различных pH можно характеризовать гидролиз ионов металла. При этом справедливость равенства (2.54) сохраняется при следующих условиях: 1) коэффициенты активности поддерживаются постоянными; 2) в растворе нет полиядерных комплексов; 3) состав твердой фазы не меняется.
Последнее условие наиболее трудно выполнимо, и так как на состав осадка влияет много различных факторов (температура, время, скорость осаждения, концентрации в маточном растворе, общий ионный состав раствора), то всегда остаются сомнения относительно постоянства состава твердой фазы.
Существует много методов для установления состава твердого осадка [43, 44], однако в большинстве работ по изучению гидролиза идентификации твердой фазы не уделяется должного внимания. Все расчеты в методе растворимости основаны на предположении об отсутствии полиядерного комплексообразования, но концентрация растворимых форм часто остается довольно высокой (~10~2 М), что не исключает полимеризации.
Этот метод использовали, например, для изучения гидролиза ионов Sn (II) [45], Hg(II) [46], Nd [47], Np [48] в различных степенях окисления и др.
В работе [48] концентрацию металла в растворе, находящемся в равновесии с твердой фазой, определяли радиометрически. Одновременно проводили потенциометрические измерения. Твердые осадки гидроокисей нептуния различных степеней окисления приготовляли осаждением их из соответствующих растворов. Были приняты меры для стабилизации той или иной формы. Идентификацию ионных форм в растворе проводили спектрофотометрически. Состав осадка дополнительно не исследовали. Растворимость принимали равной концентрации Np4+, NpO^ или NpO|+ соответственно изучаемой твердой фазе. Метод растворимости был применен для определения констант гидролиза ионов Th (IV) [49]. В основу положено уравнение равновесия гидроксокомплексов тория с твердой фазой
Th (ОН)4 Th (ОН)4° =₽± Th (ОН)ТХ + т ОН~ (2.55) твердая фаза
24
Константа диссоциации гидроокиси тория согласно этой реакции будет иметь вид
Кт = [Th (OHtf+j (OH-r/[Th (ОН)4°], (2.56)
причем, естественно, [Th (ОН) J]=const=[Th]0. Далее принималось, что концентрация ионных форм тория, находящихся в равновесии с твердой фазой, равна разности между общей концентрацией (Сть) и концентрацией молекулярных форм. Это положение можно представить равенством
[Th (ОЩХ] = CTh - [Th]0. (2.57)
Однако это равенство ошибочно, так как в действительности
£ [Th(OH)?+„] =CTh-[Th]0, (2.58)
т. е. необходимо учитывать образование всех ионных форм или быть уверенным, что только данная ионная форма участвует в равновесии. Фактическое пренебрежение всех ионных форм, кроме одной, видимо, и привело в данной работе к получению констант, отличающихся от других констант, известных в литературе.
2.5. Метод рксредметрии
Первоначально измерение окислительного потенциала применительно к гидролизу использовали для нахождения равновесной концентрации окисленной формы металла при условии» что гидролизом восстановленной формы можно пренебречь и ее равновесную концентрацию считать равной общей концентрации. Измеряли ЭДС цепи, в которую входила данная ионная пара, параллельное измерение pH проводили стеклянным электродом. Так были определены, например, константы гидролиза Hg (II) [4], Т1 (Ш)Д50], Fe (III) [51].
Рассмотрение совокупности всех равновесных реакций в окислительно-восстановительной системе проведено Б. П. Никольским [52]. Окислительный потенциал связан функциональной зависимостью с составом раствора. С помощью этой зависимости можно определить вид и число частиц, входящих в комплексы (в том числе и в гидроксокомплексы), и определить их константы устойчивости. Если окислительно-восстановительная реакция
Мт+ + пе^М{т~п}+ (2.59)
протекает в условиях постоянства ионной силы, то окислительный потенциал равен
Ф = Ф<> + &[М]/п[7И], (2.60)
25
где <р° — кажущийся стандартный окислительный потенциал, учитывающий коэффициенты активности аквакомплексов металла в обеих степенях окисления; '6 = 2,3 RT/F; F — число Фарадея. Подробные выводы уравнения <p=f(pH, с°, сг), методика определения состава комплексов и констант устойчивости приведены в работе [52].
Методом оксредметрии была изучена система Fe2+/Fe3+ и определены первая константа гидролиза иона Fe3+ и константа димеризации [53—55].
2.6. Ионообменный метод
Если допустить, что негидролизованные ионы металла, например М.4+, а также гидроксокатионы МОН3+, М(0Н)2+ могут сорбироваться на катионите в Na-форме (RNa), то уравнения реакций сорбции можно записать
nRNa + Mn+=PtR„M+nNa+; . (2.61)
п — 1) RNa + M (ОН )+ ч* R(„_i > М (ОН) + (п — 1) Na+; (2.62) (п — 2) RNa + М(ОН)^-2)+ R(„_2) МЮН)2 + <п — 2. Na+. (2.63)
Изучение гидролиза М4+ методом катионного обмена, где возможна сорбция М(ОН)^- , чрезвычайно трудно. Это обусловлено тем, что высокозарядные ионы металлов, как правило, быстро гидролизуются. Ион М4-*- может существовать лишь в сильно разбавленных (см®1СН—10~5 М) и сильно кислых растворах, где обязательно наряду с процессами сорбции М4+ будут проходить процессы перехода катионита в Н-форму.
Приведем уравнения изотерм реакции сорбции:
(R„M)/lMn+] = Ко (NaR)"/[Na+]n; (2.64)
(R„_i М (ОН))/[М (ОН)(П-’)+] = (NaR)"-1/[Na+]n~1; (2.65)
(R„_2M (ОН)2)/[М (ОН)<21-2)+] = К2 (NaR)n-2/[Na+]"-2. (2.66) В круглых скобках указаны концентрации ионов в фазе катионита, в квадратных — активности ионов в растворе. Концентрация фонового электролита обязательно должна быть достаточно высокой, чтобы соблюдалось требование постоянства ионной силы в водной фазе и в фазе катионита. Кроме того, необходимо соблюдение условия C^V^tnS0, где См — общая концентрация металла; V — объем раствора; т — масса катионита; 5° — полная обменная емкость катионита. В этом случае концентрацию смолы и активность ионов натрия, т. е. (NaR) и [Na+], можно считать постоянными величинами и пользоваться законом действующих масс для ионообменного равновесия.
26
Коэффициент распределения металла между раствором и катионитом Kd можно записать
(2.67)
где Cmr — общая концентрация металла в смоле; См — общая концентрация металла в растворе.
При отсутствии в растворе других лигандов, кроме ионов гидроксила, в фазе катионита могут быть сорбированы различные гидроксокомплексы и негидролизованный ион металла, которые в растворе находятся в равновесии с катионитом.
В отсутствие гидролиза коэффициент распределения
' . (2.68)
Парциальные коэффициенты распределения отдельных гидроксокомплексов при мольной доде, равной единице, можно записать
Kd = (Rn—i MOH)/[MOH(n~1)+], Kd = (R„_2 M (OH)2)/[M (OH)ST2)+1, тогда коэффициент распределения металла
CMR [M"+]K° + IMOH]KJ + [M(OH)S]K2 . -----------------------------------,
С1Л [Mn+] + [МОН] + . . . + [М (ОН)„1
Если выразить концентрации гидроксокомплексов константы образования, то выражение для Kd примет
~17п+фм°н~Г).
Из этого уравнения следует, что при [ОН“]~>0, .
и определив Kd при различных концентрациях [ОН~], методом Фронеуса рассчитывают значения рг*. Расчет по методу Фронеуса применим при изучении комплексообразования в системах, когда к центральному иону присоединяется не более двух лигандов и где существуют только мономерные комплексы. Более общий подход представлен в работе [56]. На примере гидролиза ионов Fe(III) авторам удалось установить образование полиядерных гидроксокомплексов и рассчитать их константы наряду с константами образования всех трех мономерных гидроксокомплексов.
Следует отметить, что работ по использованию ионного обмена для расчета констант гидролиза ионов металла немного. Это обусловлено многими причинами, важнейшие из которых: сложность эксперимента и расчета, в который необходимо вводить промежуточные функции и операции, невысокая точность метода, неоднозначность в толковании механизма сорбции.
(2.69)
(2.70)
(2-71)
через
вид
(2.72)
Зная
27
2.7. Электромиграционный метод
Электромиграционный метод для изучения гидролиза имеет важное значение, так как подобно спектрофотометрическому методу он позволяет работать при низких (10~5—10-6г-ион/л) концентрациях металла, способствующих образованию только моноядерных гидроксокомплексов. Кроме того, электромиграционный метод экспериментально устанавливает, при какой концентрации начинается полимеризация, и таким образом выбор общей концентрации металла при изучении моноядер-ного гидролиза становится обоснованным.
В электромиграционном методе используют наблюдения за подвижностью гидратированных ионов металла в электрическом поле, которая зависит как от характеристик состава и свойств ионов (радиуса, заряда, прочности комплексных частиц и ассоциатов), так и от состава и свойств раствора. Для лабильных систем, в которых скорость всех процессов значительно выше скорости электромиграционного переноса ионов; и при наложении электрического поля динамическое равновесие не смещается, средняя скорость миграции элемента Uc связана с равновесной концентрацией лиганда (при гидролизе— это ионы гидроксила) выражением
(п \ / / п \
UK + У и[ОН-]') / 1 + У Pi [ОН-]'), (2.73}
где UK— скорость миграции негидролизованного катиона металла; п — количество видов комплексных ‘частиц.
Для одноядерных комплексов при постоянной концентрации лиганда средняя скорость миграции не зависит от общей концентрации металла в растворе. Это очень важное обстоятельство позволяет работать в любых разбавленных растворах,, причем пока это условие соблюдается, образованием поли-ядерных форм можно пренебречь. Нахождение неизвестных констант устойчивости мономерных гидроксоформ принципиально возможно решением п уравнений такого типа, записанных для любых п точек электромиграционной кривой.
Решение возможно несколькими приближенными способами, которые применительно к методу электромиграции изложены в работе [57]. Там же описаны экспериментальные установки для определения подвижности и разделения ионов.
В практике изучения гидролиза наиболее часто используется графический способ расчета констант. Он применим в тех случаях, когда ступенчатые константы гидролиза значительно различаются: /Cr(i-i)3>Kri. Тогда на кривой электромиграции наблюдают горизонтальные участки, соответствующие областям доминирования отдельных видов гидроксокомплексов. Каждый из этих участков можно описать уравнением электромиграцищ
28
которое в логарифмической форме представляет собой уравнение прямой с угловым коэффициентом, равным 1:
1g [°н-] + (2-74)
с/ с — U i
при этом lg Ki = —lg[OH“].
Электромиграционный метод был применен для изучения гидролиза лантанидов и актинидов [58—61].
Константы, полученные в различных вариантах электро-миграционного метода, согласуются между собой, но значения их больше, чем значения, полученные другими методами. Авторы работ [58, 59] объясняют это различие влиянием полимеров в тех случаях, когда метод требует работы с концентрациями большими, чем 10”5 г-ион/л.
2.8. Метод протонного магнитного резонанса
Метод применяется главным образом для измерения магнитных моментов и измерения относительных изменений времени протонной релаксации 7\. Эти измерения дают возможность получить информацию о состоянии парамагнитных ионов в растворе. Эффективный магнитный момент иона зависит от ряда факторов, в том числе от расстояния предельного сближения иона и протонов растворителя, времени жизни протона в гидратной оболочке парамагнитного иона, состава среды. В ряде случаев зависимость от состава выражена настолько отчетливо, что позволяет использовать метод ПМР для выяснения состава и прочности комплексов. Было высказано предположение, что гидролиз парамагнитных ионов будет вызывать эффект, аналогичный комплексообразованию [62].
В работе [63] исследована зависимость времени спин-ре-шеточной релаксации протонов водных растворов U(IV) (0,03 М) от pH. Эффективный магнитный момент цдг возрастает с увеличением pH (т. е. с увеличением гидролиза). Из этой зависимости могут быть. рассчитаны константы образования гидроксокомплексов. В случае U(IV) первая константа' гидролиза определена равной p/Gr= 1,7, что хорошо соответствует данным, полученным другими методами. .Способы расчета констант образования, возможные области применения метода списаны в работах [64, 65].
3. ДИССОЦИАЦИЯ ВОДЫ. ГИДРОЛИЗ ионов ЭЛЕМЕНТОВ I ГРУППЫ
3.1. Диссоциация воды
Реакция ионизации воды — одна из важнейших реакций в; водных растворах:
Н2О^Н+ + ОН~
К = [Н+] [ОН-]/[Н2О],
К-[Н2О] = [н+] [OH-]=tfuz
Произведение молярных концентраций ионов гидроксила и ионов Водорода называется ионным произведением воды. Ионное произведение воды прямо пропорционально зависит от температуры, а также от природы и величины ионной среды из-за влияния ее на активность воды.
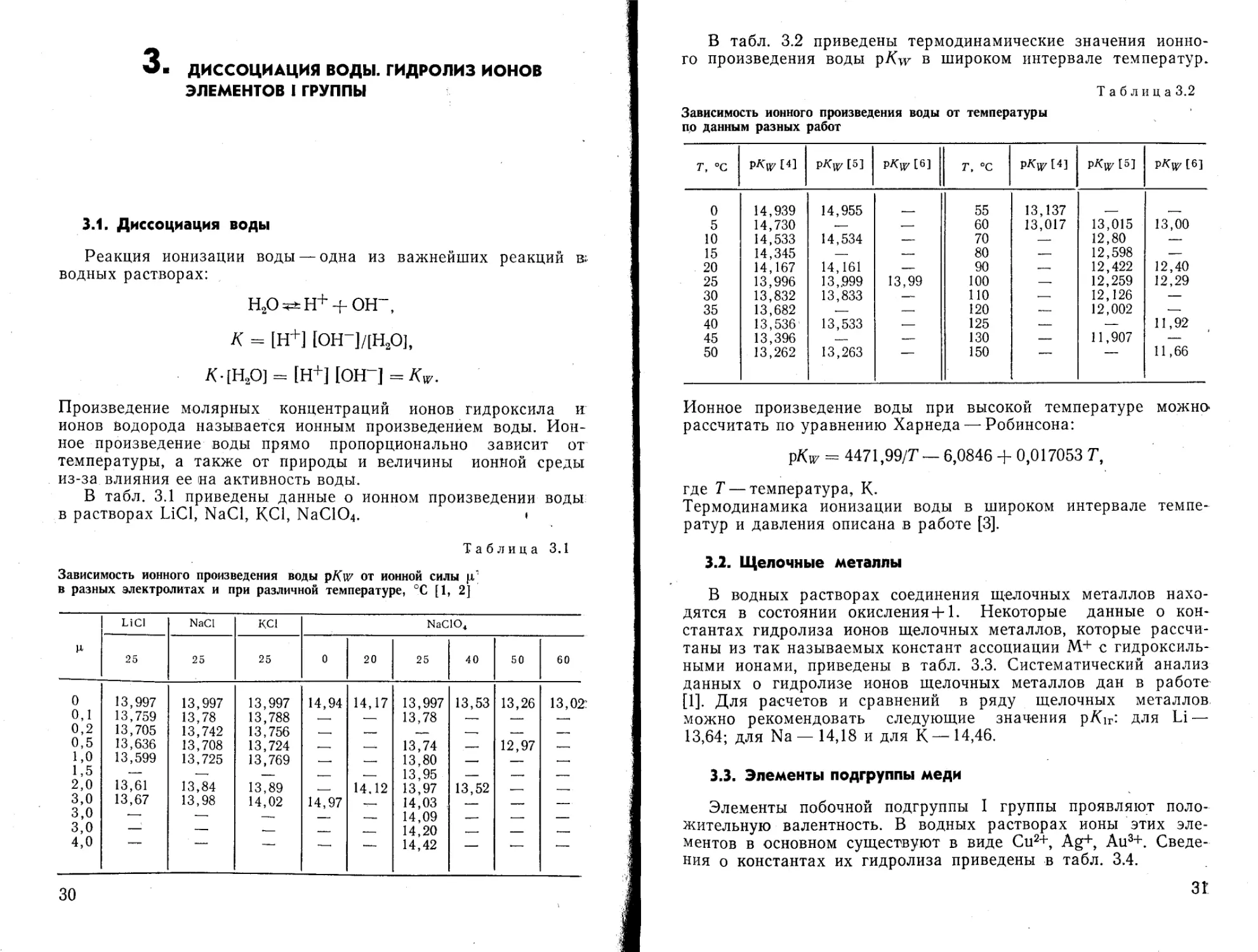
В табл. 3.1 приведены данные о ионном произведении воды в растворах LiCl, NaCl, КС1, NaC104. »
Таблица 3.1
Зависимость ионного произведения воды pKw от ионной силы ц' в разных электролитах и при различной температуре, °C [1, 2]
И LiCl NaCI КС1 NaClO4
25 25 25 0 20 25 40 50 60
0 13,997 13,997 13,997 14,94 14,17 13,997 13,53 13,26 13,02:
0,1 13,759 13,78 13,788 — 13,78 .— — —,
0,2 13,705 13,742 13,756 —. — — —. — —
0,5 13,636 13,708 13,724 .— —. 13,74 — 12,97 .—
1,0 13,599 13,725 13,769 —. —, 13,80 —, — .—.
1,5 — — — .— — 13,95 — — —
2,0 13,61 13,84 13,89 14.12 13,97 13,52 —
3,0 13,67 13,98 14,02 14,97 14,03 — — —
3,0 • — —. — — 14,09 — —. —
3,0 — — — — — 14,20 — —
4,0 — — —. '—• — 14,42 — — —
30
Таблица 3.2
В табл. 3.2 приведены термодинамические значения ионного произведения воды pAw в широком интервале температур.
Зависимость ионного произведения воды от температуры по данным разных работ
Т, °C [4] И P^nz 16] т, °C рХ"^ [4] P^U7 [5] pA'uz [6]
0 14,939 14,955 55 13,137
5 14,730 .— — 60 13,017 13,015 13,00
10 14,533 14,534 — 70 — 12,80 —
15 14,345 — — 80 — 12,598 —
20 14,167 14,161 — 90 — 12,422 12,40
25 13,996 13,.999 13,99 100 — 12,259 12,29
30 13,832 13,833 110 .— 12,126 —
35 13,682 .— —. 120 — 12,002 —
40 13,536 13,533 .— 125 — — 11,92
45 13,396 — — 130 — 11,907 —
50 13,262 13,263 — 150 — — 11,66
Ионное произведение воды при высокой температуре можно* рассчитать по уравнению Харнеда — Робинсона:
pKw = 4471,99/7 — 6,0846 + 0,017053 7,
где 7 — температура, К.
Термодинамика ионизации воды в широком интервале температур и давления описана в работе [3].
3.2. Щелочные металлы
В водных растворах соединения щелочных металлов находятся в состоянии окисления + 1. Некоторые данные о константах гидролиза ионов щелочных металлов, которые рассчитаны из так называемых констант ассоциации М+ с гидроксильными ионами, приведены в табл. 3.3. Систематический анализ данных о гидролизе ионов щелочных металлов дан в работе [1]. Для расчетов и сравнений в ряду щелочных металлов можно рекомендовать следующие значения p/Gr: для Li — 13,64; для Na — 14,18 и для К—14,46.
3.3. Элементы подгруппы меди
Элементы побочной подгруппы I группы проявляют положительную валентность. В водных растворах ионы этих элементов в основном существуют в виде Cu2+, Ag+, Au3+. Сведения о константах их гидролиза приведены в табл. 3.4.
31
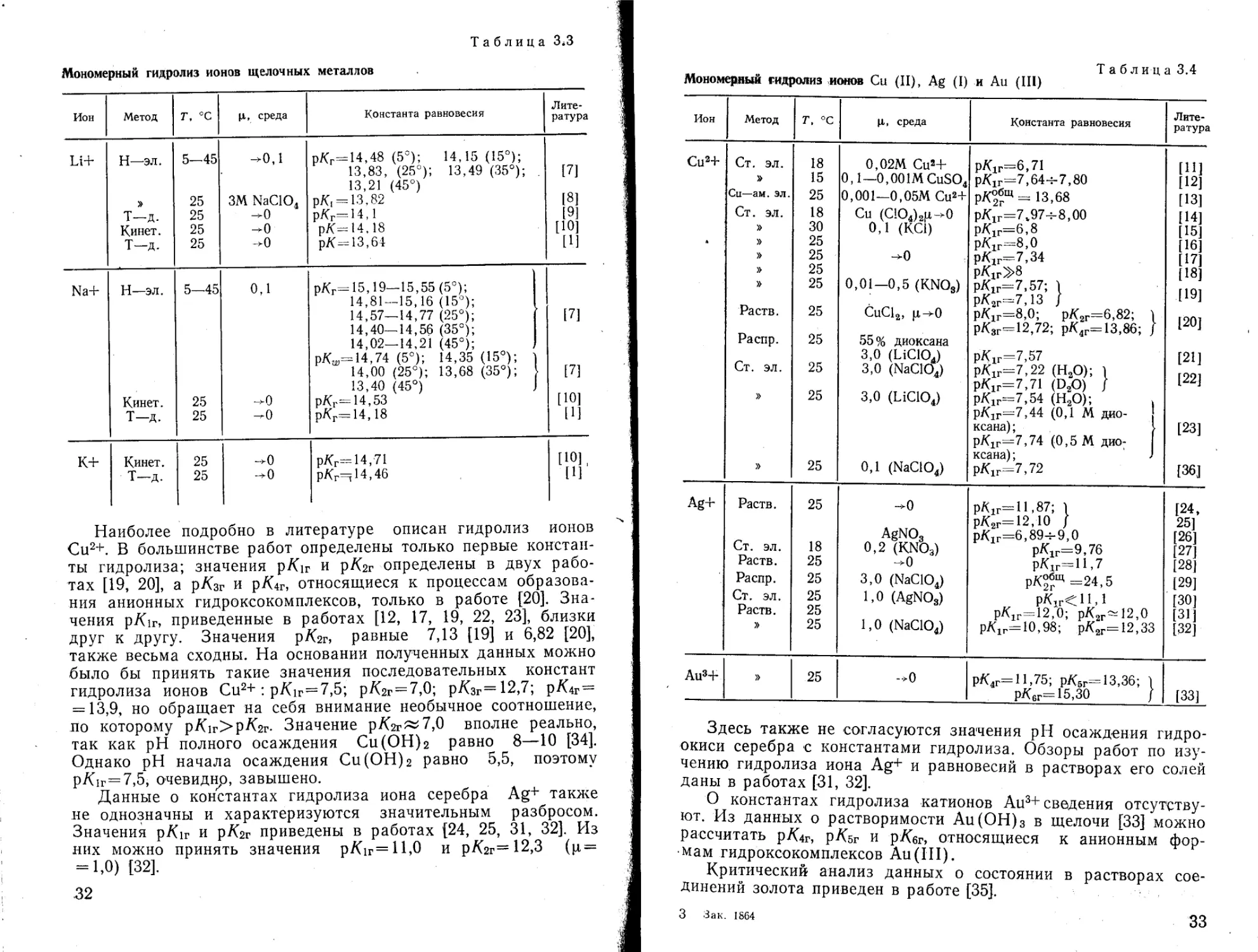
Таблица 3*3
Мономерный гидролиз ионов щелочных металлов
Ион Метод Т, °C И, среда Константа равновесия Литература
ы+ Н—ЭЛ. 5-45 ^0,1 рКг=44,48 (5°); 14,15 (15°); 13,83, (25°); 13,49 (35°); 13,21 (45°) [7]
» 25 ЗМ NaClO. рк^гзлг [8]
Т-д. 25 -0 рКг-14,1 [9]
Кинет. 25 -0 рК-14,18 [Ю]
Т-д. 25 -0 рК—13,64 [1]
Na+ И—эл. 5—45 0,1 рКг=15,19—15,55 (5°); 14,81 — 15,16 (15°); 14,57—14,77 (25°); 14,40—14,56 (35°); 14,02—14,21 (45°); pKw^14,74 (5°); 14,35 (15°); ) 14,00 (25°); 13,68 (35°); 13,40 (45°) J [7] [7]
Кинет. 25 ^0 рКг-14,53 [Ю]
Т-д. 25 -0 рКг=14,18 [1]
К+ Кинет. 25 ->0 рКг= 14,71 [Ю],
Т-д. 25 -^0 pKr=jl4,46 [1]
Наиболее подробно в литературе описан гидролиз ионов Си2+. В большинстве работ определены только первые константы гидролиза; значения рАлг и рЛгг определены в двух работах [19, 20], а рЛзг и p/Gr, относящиеся к процессам образования анионных гидроксокомплексов, только в работе [20]. Значения рЛщ приведенные в работах [12, 17, 19, 22, 23], близки друг к другу. Значения рА2г, равные 7,13 [19] и 6,82 [20], также весьма сходны. На основании полученных данных можно было бы принять такие значения последовательных констант гидролиза ионов Си2+: рЛлг=7,5; рКгг = 7,0; р/С3г=12,7; p^4г= = 13,9, но обращает на себя внимание необычное соотношение, по которому рЛ\г>рЛ2г. Значение p/Gr~7,0 вполне реально, так как pH полного осаждения Си (ОН) 2 равно 8—10 [34]. Однако pH начала осаждения Си(ОН)2 равно 5,5, поэтому р/С1г=7,5, очевидно, завышено.
Данные о константах гидролиза иона серебра Ag+ также не однозначны и характеризуются значительным разбросом. Значения р/Сг и p^2г приведены в работах [24, 25, 31, 32]. Из них можно принять значения рА1Г=Н,0 и рА2г=12,3 (ц = -1,0) [32].
32
Таблица 3.4
Мономерный гидролиз ионов Си (II), Ag (I) и Au (III)
Ион Метод Т, °C Ц, среда Константа равновесия Литература
Си2+ Ст. эл. 18 0,02М Си2+ рК1Г=6,71 [11]
» 15 0,1—0,001MCuS04 р/(1Г=7,64->7,80 [12]
Си—ам. эл. 25 0,001—0,05МСи2+ р/^бщ = 1368 [13]
Ст. эл. 18 Си (СЮ4)2|х-*0 рК1Г=7.97ч-8,00 [14]
» 30 0,1 (КС1) рК1Г=6,8 [15]
* < » 25 рК1Г=8,0 [16]
» 25 ->0 рК1Г=7,34 [17]
» 25 pKirS’S [18]
» 25 0,01—0,5 (KNO3) р.К1Г—7,57; 1 рК2Г=7,13 } [19]
Раств. 25 СиС12, р^0 рК1Г—-8,0; рК2Г=6,82; 1 Г 901
рК3г-=12,72; рК4Г= 13,86; J
Распр. 25 55% диоксана
3,0 (LiClOJ рК1Г=7,57 [21]
Ст. эл. 25 3,0 (NaClOJ рК1Г=7,22 (Н3О); I Г 991
рК1Г=7,71 (DaO) f
» 25 3,0 (LiClOJ РК1г-7,54 (Н2О);
рК1Г—7,44 (0,1 М дио-
ксана) ; [23]
р/(1Г=7,74 (0,5 М дио- 17 001101 *
» 25 0,1 (NaClO4) KLdHd , * рК1Г=7,72 [36]
Ag+ Раств. 25 -0 рЛ1Г=11,87; 1 [24,
рК2Г=12,10 f 25]
AgNO3 рК1Г=6,89-^9,0 [26]
Ст. эл. 18 0,2 (KNO3) рК1г=9,76 [27]
Раств. 25 -0 рК1г=П,7 [28]
Распр. 25 3,0 (NaClOJ рК^Щ=24,5 [29]
Ст. эл. 25 1,0 (AgNO3) [30]
Раств. 25 рК1Г=12,0; рК2ГМ2,0 [31]
» 25 1,0 (NaClOj рК1г=10,98; рК2Г=12,33 [32]
Au34- » 25 ~>0 рК4Г= 11,75; рКвг= 13,36; 1
рК6Г= 15,30 / [33]
Здесь также не согласуются значения pH осаждения гидроокиси серебра с константами гидролиза. Обзоры работ по изучению гидролиза иона Ag+ и равновесий в растворах его солей даны в работах [31, 32].
О константах гидролиза катионов Аи3+сведения отсутствуют. Из данных о растворимости Аи(ОН)3 в щелочи [33] можно рассчитать р/<4г, pKsr и р/Сбг, относящиеся к анионным формам гидроксокомплексов Au (III).
Критический анализ данных о состоянии в растворах соединений золота приведен в работе [35].
3 Зак. 1864
33
4. ГИДРОЛИЗ ИОНОВ ЭЛЕМЕНТОВ II ГРУППЫ
4.1. Элементы главной подгруппы — Be, Mg, Са, Sr, Ba
Щелочноземельные металлы проявляют положительную валентность 2+. По своему химическому поведению бериллий существенно отличается от других элементов этой подгруппы. Ион Ве2+ более «кислый», чем ионы Mg2+, Са2+, Sr2+, Ba2*, которые проявляют только основные свойства. В водных растворах солей этих элементов сильно гидролизованы лишь ионы Ве2+.
Константы гидролиза ионов элементов главной подгруппы II группы приведены в табл. 4.1.
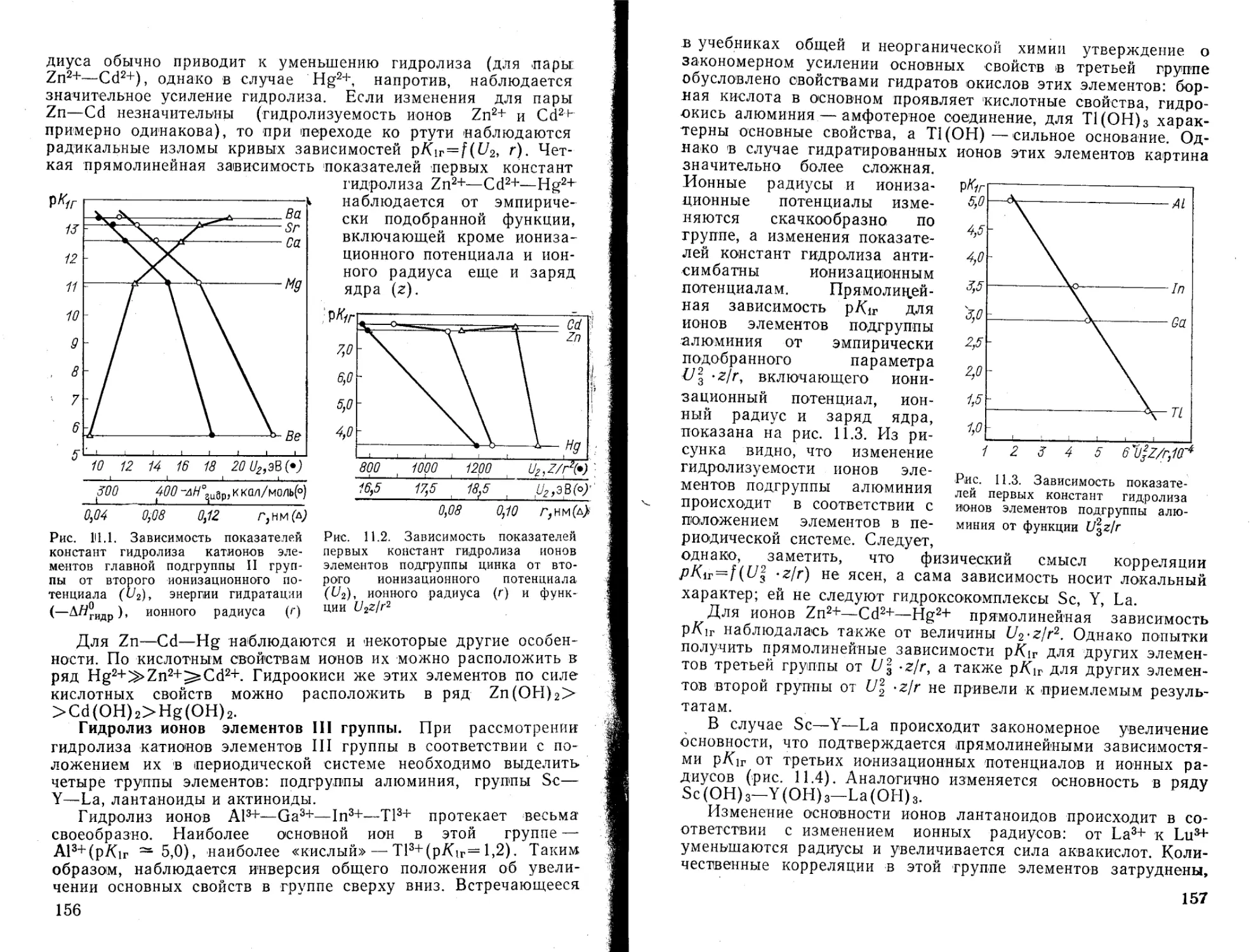
Для большинства химических элементов различными авторами определены константы гидролиза, расхождения между которыми часто весьма значительны. Обычно предпочтение отдают результатам таких работ, в которых использован наиболее адекватный системе метод. В случае гидроксокомплексов наиболее надежные результаты получены потенциометрическим (с измерением концентрации Н+ водородным и стеклянным электродами) и спектрофотометрическим методами в разбавленных растворах при постоянной ионной силе раствора, создаваемой индифферентным к ионам металла электролитом. Как правило, наиболее ценны те работы, где проведено систематическое изучение гидролиза иона металла при различных значениях ионной силы раствора в широком интервале температур, в которых определены константы равновесия нескольких ступеней гидролиза. При отсутствии таких обстоятельных исследований ориентируются на совокупность работ, в которых получены близкие значения константы гидролиза одной и той же ступени.
Таким образом, при оценке констант мономерного гидролиза ионов металлов наиболее надежными можно считать константы, полученные при исследовании в разбавленных растворах (исключается полимеризация гидроксокомплексов); полученные потенциометрическим методом с измерением концентрации Н+ стеклянным или водородным электродом; спектрофотометрическим методом, а также уменьшающиеся в последовательности /С1г>К2Т>Азг.
34
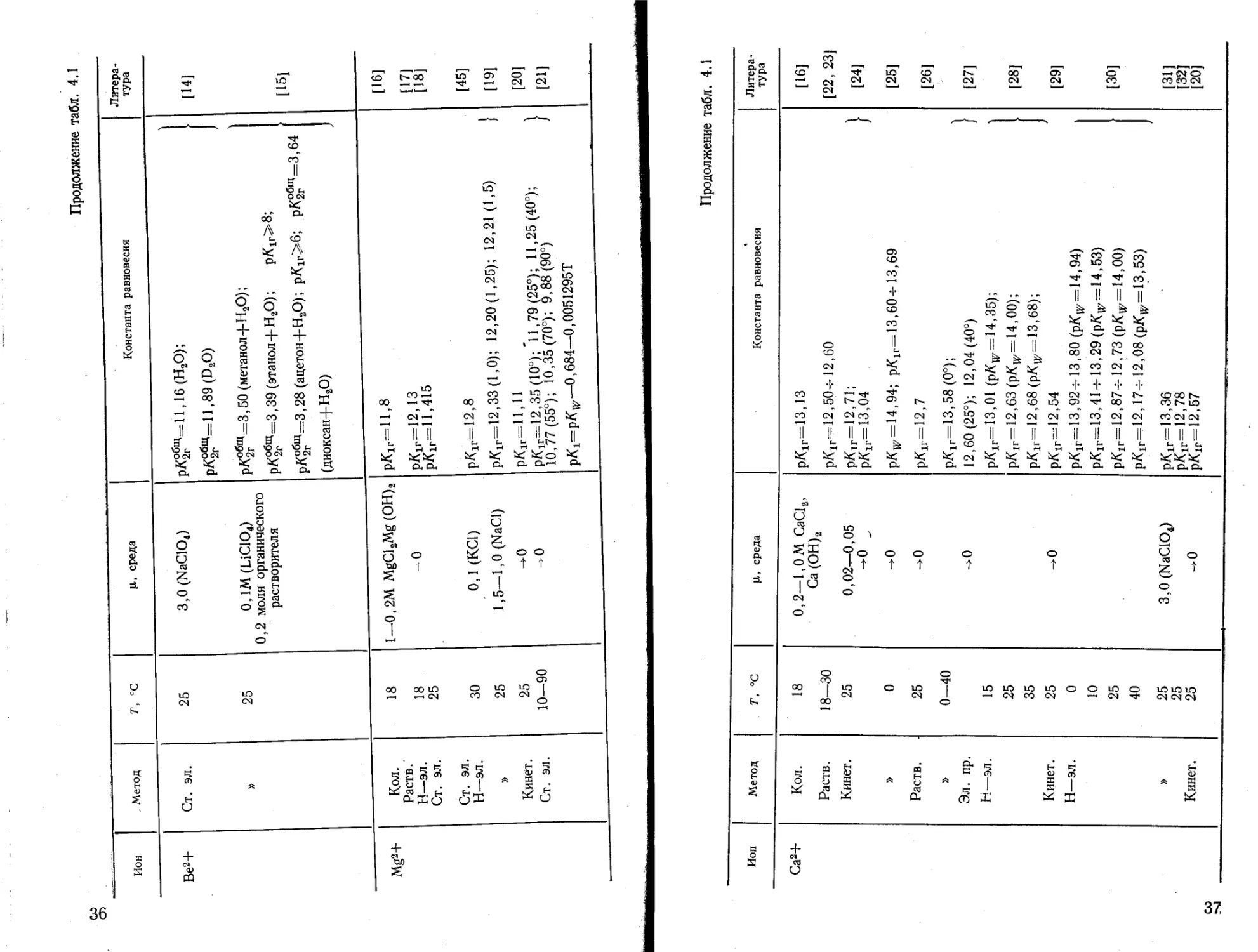
Мономерный гидролиз ионов Ве2+, Mg2+, Са3+, Sr,4- Ва24-
3*
35
Продолжение табл. 4.1
Ион , Метод т, °C ц, среда Константа равновесия Литература
Ве2+ Ст. ЭЛ. 25 3,0(NaC104) рК^щ=П,16(Н2О); pK°*4=ll,89(D2O) [14]
» 25 0,lM(LiC104) 0,2 моля органического растворителя рЯ^щ=3,50 (метанол-|-Н2О); рЯ°бщ=3,39 (этанол-]- Н2О); рй\г>8; рК^щ=3,28(ацетон+Н2О); рК1г>6; рК^щ=3,64 (диоксан-|-Н2О) [15]
Mg2+ Кол. Раств.' Н—эл. Ст. эл. 18 18 25 1—0,2М MgCl2Mg(OH)2 0 11 1 — to — 4*- — ОО СП [16] [17] [18]
Ст. эл. Н—эл. » Кинет. Ст. эл. 30 25 25 10—90 0,1 (КС1) 1,5—1,0 (NaCl) -+0 ^0 рК1Г=12,8 рК1г=12,33 (1,0); 12,20(1,25); 12,21(1,5) } рК1г=11,11 рК1Г==12,35 (10°); 11,79(25°); 11,25 (40°); 1 10,77(55°); 10,35(70°); 9,88(90°) / рК1= pKw -0,684—0,0051295Т [45] [19] [20] [21]
Продолжение табл. 4.1
Ион Метод г, °с Ц, среда 1 Константа равновесия Литература
Са2+ Кол. Раств. Кинет. » Раств. » Эл. пр. Н—эл. Кинет. Н—эл. » Кинет. 18 18—30 25 0 25 0—40 15 25 35 25 0 10 25 40 25 25 25 0,2-1,ОМ СаС12, Са (ОН)2 0,02—0,05 ^0 ^0 ->0 ->0 ->0 3,0 (NaClOJ ~>0 .. рК1г=13,13 рК1Г=12,50~ 12,60 рК1Г=12,71; ч рК1Г=13,04 } PKW = 14,94; рК1Г=13,60ч-13,69 рК1г=12,7 рК1Г=13,58 (0°); 12,60(25°); 12,04(40°) } рК1Г=13,01 (рЛг=14,35); p/Cjr—12,63 (рХу/— 14,00); pKlr=12,68 (p^=13,68); p/fir^12,54 рК1Г=13,924-13,80 (рЛИ7=14,94) рК1Г=13,414-13,29 (pKltz=14,53) pKir=12,874-12,73 (рХ^=14,00) рК1Г= 12,174-12,08 (pKBZ=13,53) рК1г=13,36 pXir=12,78 рК1Г=12,57 [16] [22, 23] [24] [25] 126] [27] [28] [29] [30] [31] [321 [20]
Продолжение табл. 4.1
38
Образование осадков гидроокисей металлов наблюдается в достаточно концентрированных растворах и относится к явлениям полимерного гидролиза. Тем не менее наблюдают соответствие значений констант мономерного гидролиза области pH начала или полного осаждения гидроокисей. Если металл образует анионные гидроксокомплексы (Be, Zn, Al и др.), значения констант, соответствующих переходу М(ОН)г в M(OH)rj_t согласуются со значениями pH осаждения гидроокисей из щелочного раствора. Все это в известной степени может служить ориентировочным критерием правильности констант гидролиза.
Авторы книги в некоторых случаях рекомендуют значения констант гидролиза. Эти рекомендации, не имеющие, разумеется, абсолютного характера, бывают двух типов.
В первом случае рекомендуются значения констант, полученные в одной работе при постоянной ионной силе и фиксированной температуре, либо термодинамически строгие величины. Такие константы можно использовать для расчетов ионных равновесий и термодинамических параметров реакций гидролиза.
Во втором случае рекомендуются «оценочные» значения констант гидролиза. Часто при этом в рекомендуемом наборе констант разные ступени гидролиза охарактеризованы константами разных авторов, которые, работая в различных условиях (Т, ц, солевой состав), тем не менее получили близкие между собой значения. Такие константы пригодны для ориентировочных оценок областей существования отдельных гидроксокомплексов. Для расчетов же ионных равновесий в определенных условиях следует пользоваться результатами работы, в которой получены константы близкие к оценочной. Так как разброс данных весьма значителен, близкими значениями считаются значения, различающиеся на 0,5 единицы рА.
В концентрированных растворах солей бериллия гидролиз протекает с образованием многрядерных гидроксокомплексов. Определен состав комплексов Ben(OH)m и рассчитаны константы равновесия реакций их образования [37—43]. Критический анализ работ о полимерном гидролизе ионов» бериллия дан в работе [44].
Из данных о мономерных константах гидролиза бериллия можно выбрать значения, полученные для разбавленных растворов с ц = 04-0,Г. рА1г=5,7 [6, 13]; рА^=13,2 [13]; рЛ^щ = 24,11 [8]; рА^щ =37,56 [4], тогда последовательные константы гидролиза ионов бериллия равны: p/Cir=5,7; р/С2г= = 7,5; рЯ3г= 10,91 ; рЯ4г= 13,45.
Эти значения хорошо согласуются с данными о pH начала осаждения гидроокиси бериллия (pH ^6), о pH полного осаждения Be (ОН) 2 (pH ^74-10), о pH осаждения гидроокиси бе
39
риллия из щелочного раствора (рН~ 12). На основании этих констант можно рассчитать распределение мономерных гидроксокомплексов бериллия в зависимости от pH раствора (рис. 4.1).
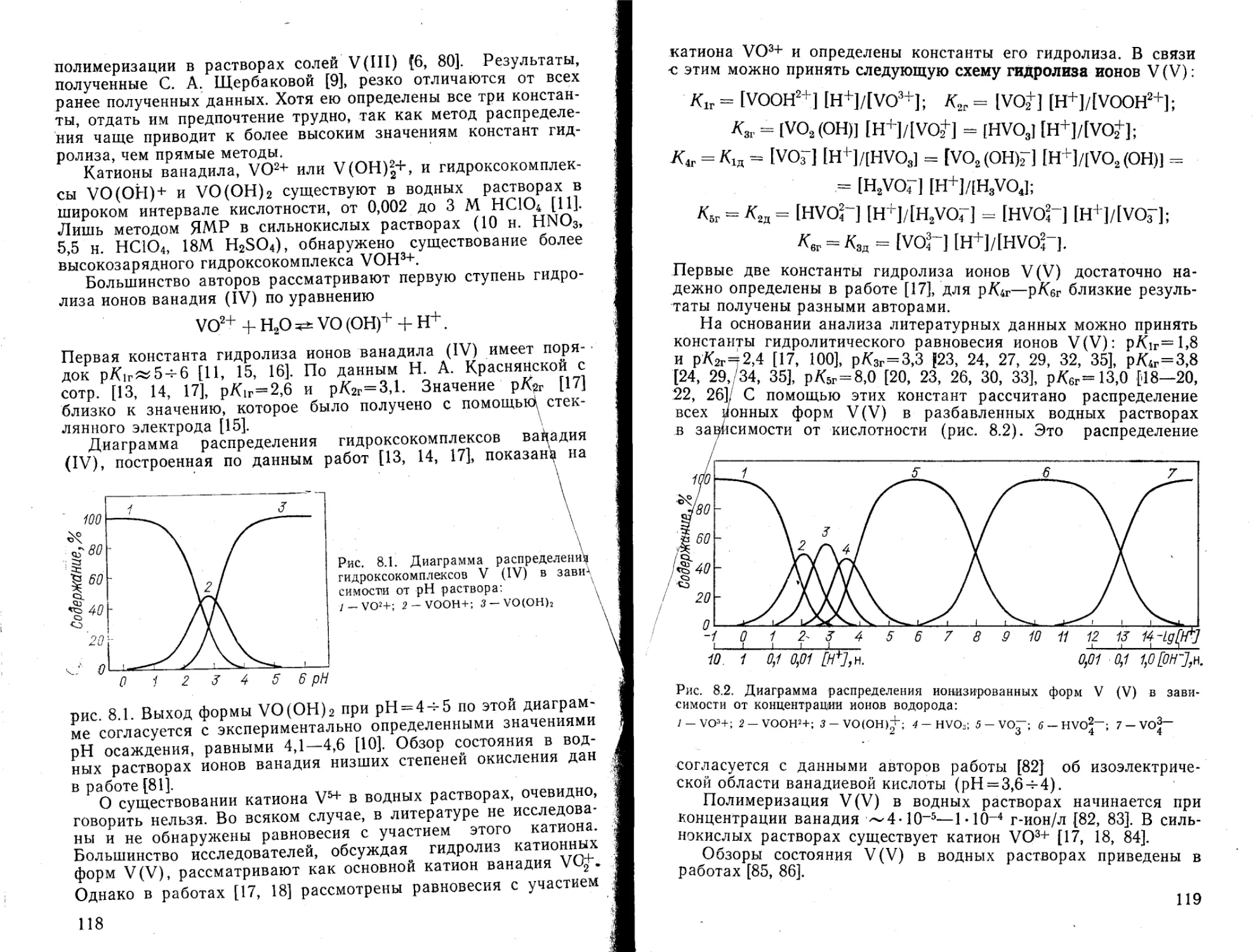
Из немногих значений константы гидролиза иона магния [16—21, 45] можно выбрать значения рАлг= 11,1-^11,4, полу-
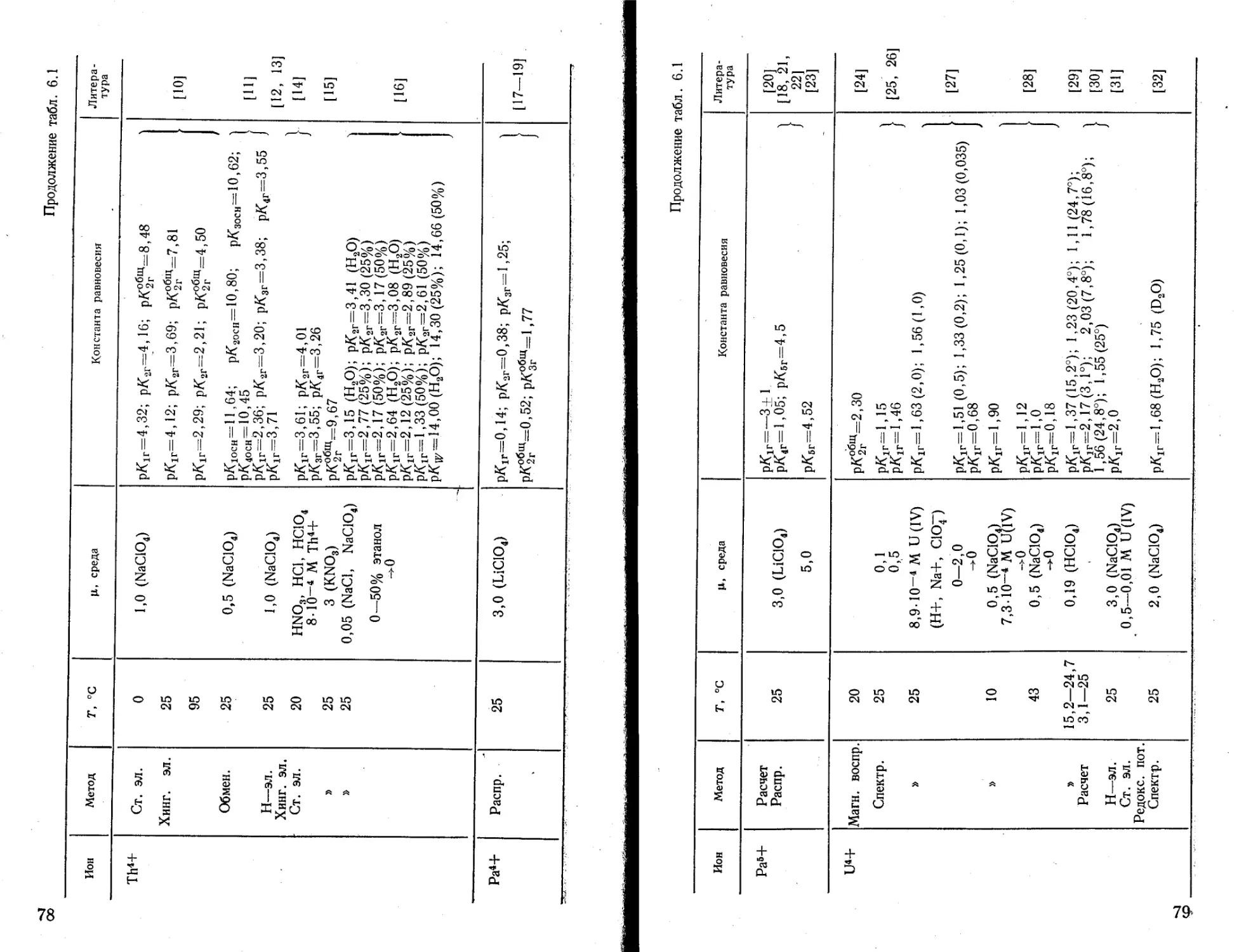
Рис. 4.1. Диаграмма распределения гидроксокомплексов бериллия в зависимости от pH раствора:
J — Ве2+; 2 — ВеОН+; 3 — Ве(ОН)2; 4 — Ве(ОН)3'; 5-Ве(ОН)2“
ченные для разбавленных растворов [18, 20]. Данных о второй константе гидролиза ионов магния в литературе нет.
Значение р/С1Г для иона кальция на основании многих согласующихся между собой результатов работ можно принять равным 12,5—-12,7 [20, 22—24, 26—30, 32]. Для иона стронция можно принять значение p/Cir=13,2 [28], полученное для разбавленных растворов (ц = 0,1; 25° С). К нему близки данные работ [16, 33]. Для иона бария р/С1Г= 13,4 [16, 28, 34, 35]. Вторые константы гидролиза ионов Са2+, Sr2+ и Ва2+ не определены.
4.2. Элементы подгруппы цинка — Zn, Cd, Hg
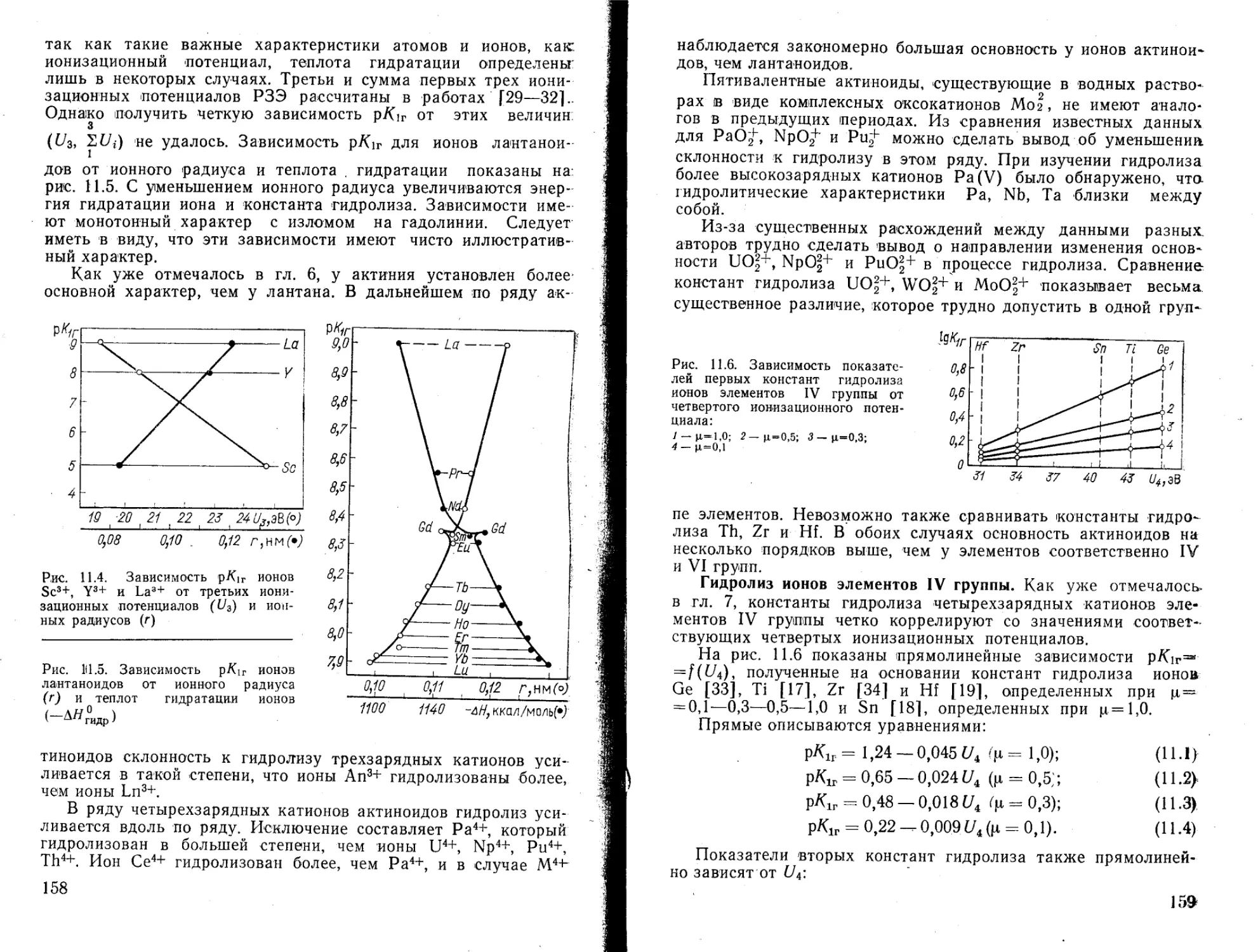
Элементы Zn, Cd и Hg в своих соединениях проявляют положительную валентность 2 + . Ртуть образует также соединения, в которых она одновалентна, хотя и здесь формально валентность ртути равна двум.
Гидроокись цинка обладает амфотерными свойствами. Гидроокись кадмия проявляет в большей степени основные свойства, хотя амфотерность свойственна и ей. Окись ртути HgO растворяется в кислотах и лишь в незначительной степени— в концентрированных щелочах.
Данные о мономерном гидролизе ионов элементов погруппы цинка приведены в табл. 4.2.
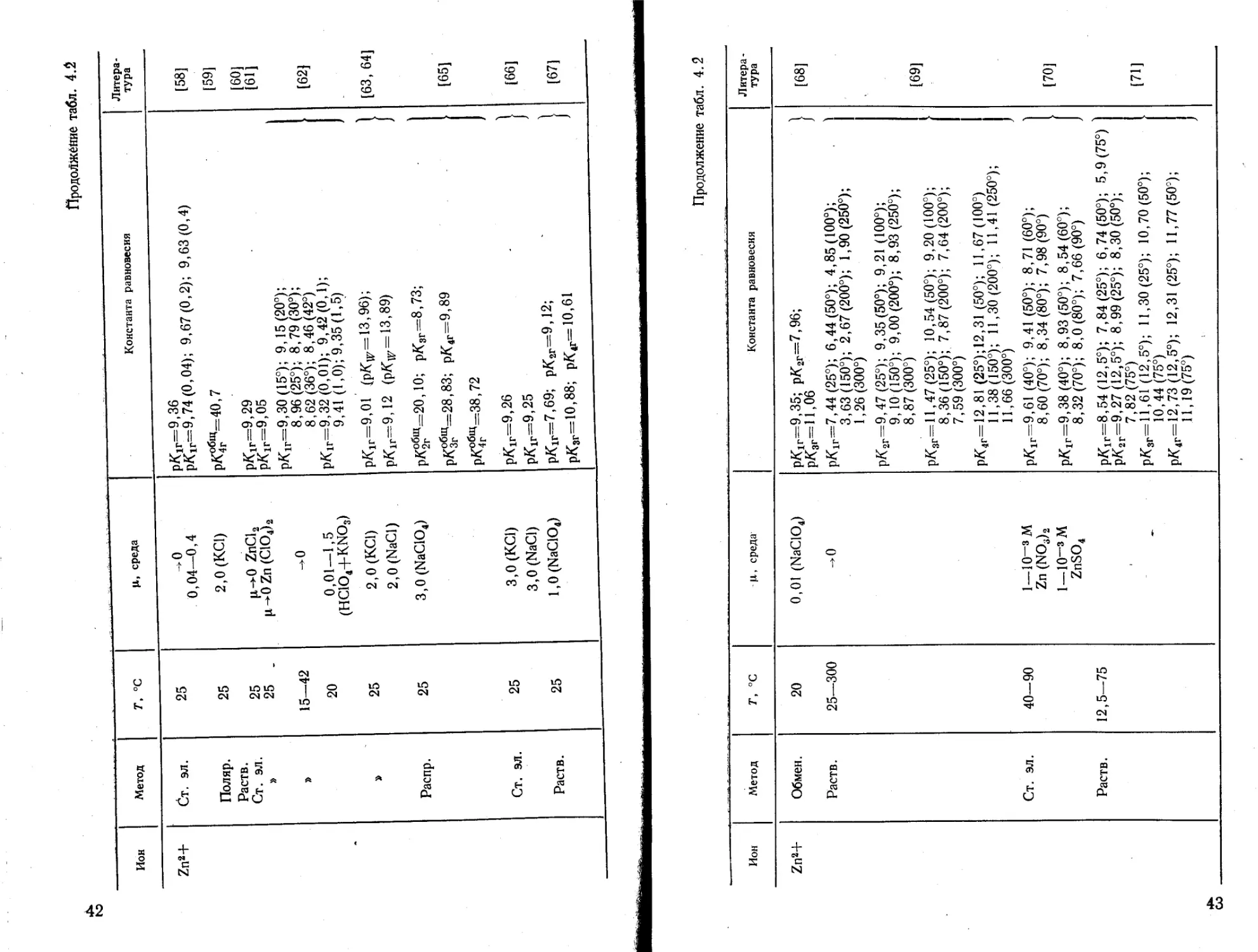
Гидролиз ионов цинка изучен весьма подробно в растворах различных солей, в широком интервале концентраций и с использованием разнообразных методов [45—71]. Обзоры о состоянии ионов цинка в кислых и щелочных растворах даны в работах [60, 67]. Значения pH осаждения для Zn(OH)z 40
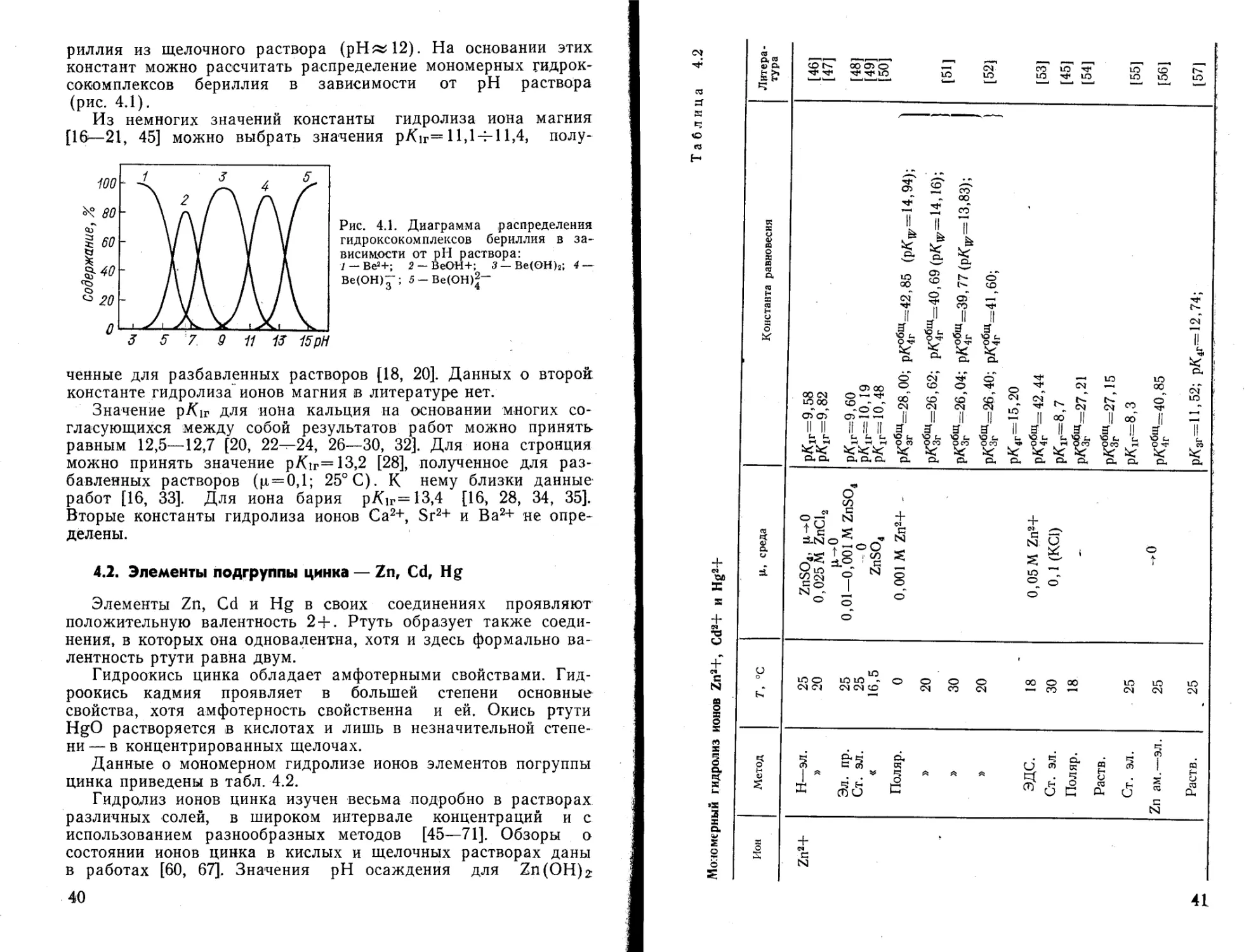
Мономерный гидролиз ионов Zn2+, Cd2+ и Hg2+
41
Продолжение табл. 4.2
Ион Метод 7\ °C |1, среда Константа равновесия Литература
Zn2+ Ст. ЭЛ. Поляр. Раств. Ст. эл. » » Распр. Ст. эл. Раств. 25 25 25 25 _ 15—42 20 25 25 25 25 -.0 0,04—0,4 2,0 (КС!) ц ->0 ZnCl2 И— 0Zn(ClO4)a ->0 0,01—1,5 (HC1O4+KNO3) 2,0 (КС1) 2,0 (NaCl) 3,0(NaC104) 3,0 (КС1) 3,0 (NaCl) l,0(NaC104) рКг=9,36 р^г=9,74(0,04); 9,67(0,2); 9,63(0,4) рК^и’=40,7 ptfir=9,29 рК1Г=9,05 рК1Г=9,30 (15°); 9,15(20°); 8,96(25°); 8,79(30°); 8,62(36°); 8,46(42°) р/<1г=9,32 (0,01); 9,42(0,1); 9,41 (1,0); 9,35(1,5) РК1Г=9,О1 (рКг= 13,96); 1 pKlr=9,12 (pKw= 13,89) j рК^Щ=20,10; рКзг=8,73; рКзгЩ=28,83; РК4Г=9,89 р/(общ=з8,72 рК1г=9,26 . ) рК1Г=9,25 ) рК1г=7,69; рКзг=9,12; ' 1 рКзг=10,88; рК4Г= 10,61 J [58] [59] [60] [61] [62] [63, 64] [65] [66] [67]
Продолжение табл. 4.2
Ион Метод т, °с Ц, среда Константа равновесия Литера -тура
Zn2+ Обмен. 20 0,01 (NaClOJ pKir=9,35; р/С,г=7,96; РК8Г=11,О6 [68]
Раств. 25—300 рК1г=7,44 (25°); 6,44(50°); 4,85(100°); 3,63(150°); 2,67(200°); 1,90 (250°); 1,26(300°) рКзг=9,47 (25°); 9,35(50°); 9,21(100°); 9,10(150°); 9,00(200°); 8,93(250°); 8,87 (300°) 1691
р/Сзг=11,47 (25°); 10,54(50°); 9,20 (100°); 8,36(150°); 7,87(200°); 7,64(200°); 7,59(300°) рК4Г=12,81 (25°);12,31 (50°); 11,67(100°) 11,38 (150°); 11,30 (200°); 11,41(250°); 11,66(300°)
Ст. эл. 40-90 1_Ю-з м Zn (NO3)2 р/<1Г=9,61 (40°); 9,41 (50°); 8,71 (60°); 1 8,60(70°); 8,34(80°); 7,98(90°) [70]
1—10“3 М ZnSO4 рК1Г=9,38 (40°); 8,93 (50°); 8,54(60°); 8,32(70°); 8,0(80°); 7,66(90°)
Раств. 12,5—75 — рК1г=8,54 (12,5°); 7,84(25°); 6,74(50°); 5,9(75°) ' р/Сзг=9,27 (12,5°); 8,99(25°); 8,30(50°); 7,82 (75°) рКзг=11,61 (12,5°); 11,30 (25°); 10,70 (50°); 10,44 (75°) р/Сг=12,73 (12,5°); 12,31(25°); 11,77(50°); 11,19(75°) [71]
Продолжение табл. 4.2
Ион Метод г, °C Ц, среда Константа равновесия Литература
Zn2+ Обмен 18 0,05 (NaClOJ рК1Г=7,52; рК2Г=10,33 [90]
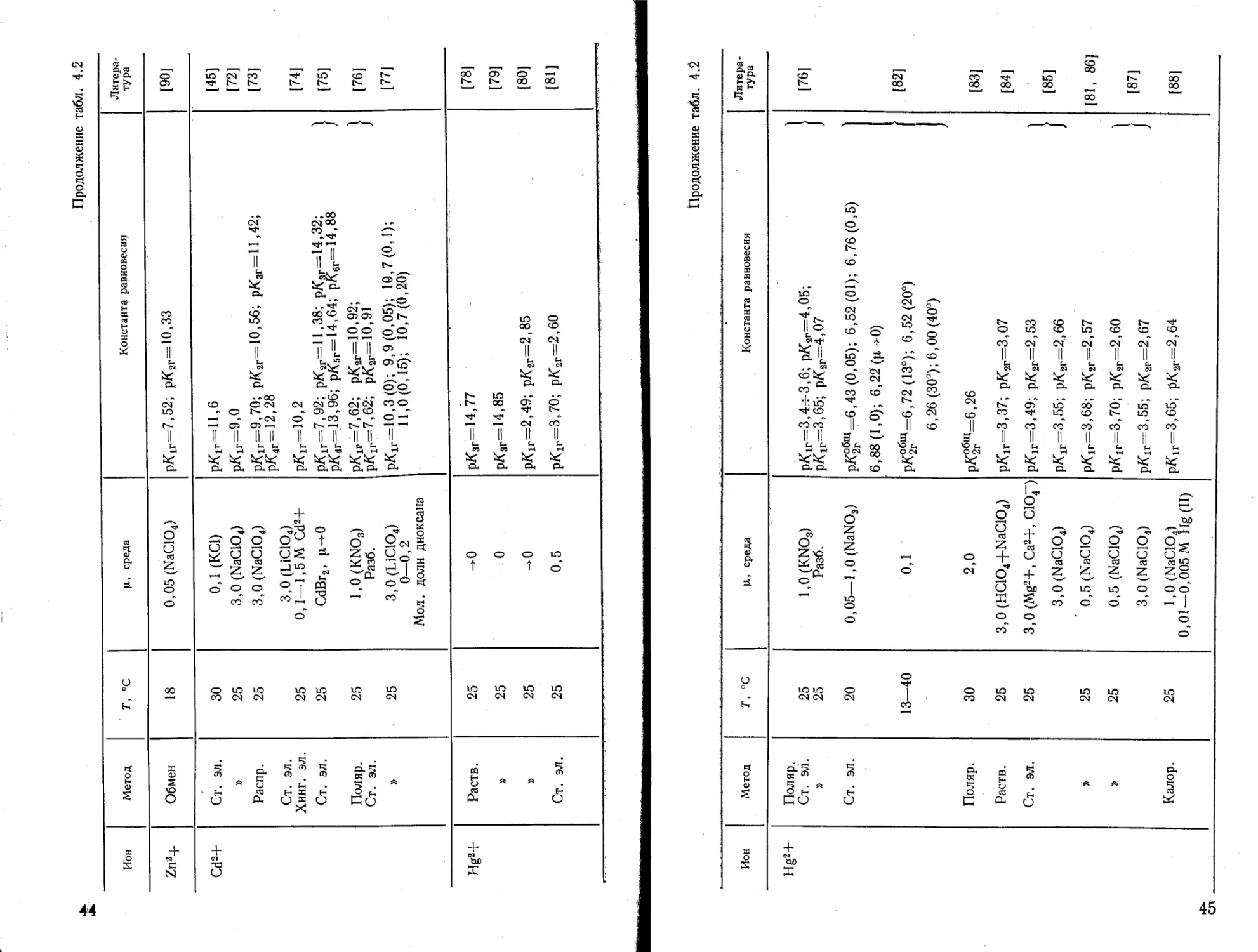
Cd2+ Ст. эл. 30 0,1 (КС1) рК1Г=11,6 [45]
» 25 3,0(NaC104) рК1г=9,0 [72]
Распр. Ст. эл. Хинг. эл. 25 25 3,0(NaC104) 3,0 (LiC104) 0,1—1,5M Cd2+ р/С1г=9.70; рК2Г=10,56; рКзг=11,42; рК4Г=12,28 рК,г=Ю [73] [74]
Ст. эл. 25 CdBr2> |л-*0 рК 1Г=7,92; ркаг=11.38; рКзг=14,32; 1 рК4г=13,96; рК6г=14,64; рКвг=14,88 / [75]
Поляр. Ст. эл. 25 l,0(KNO3) Разб. рК1Р=7)62; рК2Г=10,92; 1 рК1Г=7,62; рК2Г=10,91 f [76]
» , 25 3,0(ЫСЮ4) 0—0,2 Мол. доли диоксана рК1г=10,3(0); 9,9(0,05); 10,7(0,1); 11,0(0,15); 10,7(0,20) [77]
Hg2+ Раств. 25 ^0 рКзг=14,77 [78]
» 25 О рКзг=14,85 [79]
25 ->0 РК1Г=2,49; рК2Г=2,85 [80]
Ст. эл. 25 0,5 рК1Г=3,70; рК2Г=2,60 [81]
сл
Продолжение табл. 4.2
Ион Метод г, °C Ц, среда Константа равновесия Литература
Hg2+ Поляр. Ст. эл. » Ст. эл. Поляр. Раств. Ст. эл. » Калор. 25 25 20 13—40 30 25 25 25 25 25 l,0(KNO3) Разб. 0,05—1,0 (NaNO3) 0,1 2,0 3,0 (HC104+NaC104) 3,0 (Mg2+, Са2+, СЮр 3,0(NaC104) ' 0,5(NaC104) 0,5 (NaClO4) 3,0(NaC104) l,0(NaC104) 0,01—0,005 M Hg(II) "E ci та тата F F F Д д go II II II II II II II В В ~ £ Illi OO w CO CO GO GO OO || Cl || || OO GO Cl СЛ Cl СИ Ф» GO ° lo O1 S a “o V сл СИ О 00 сл о -q ° ч м' 1^ СП ^7 *тз. та пс х? та 05 л^-^^та^ ts re го Ц to ro to GO * • 1 II ll ll ll ll ll > ~ 8 Il’S, ю ьэ ю ю JO 00 о i Cl Cl Cl СП ci СП о ° 5 ^^o^oico-q Й S ll to СЛ to to ,—ч о 3 2 5? Cl Cl о СП [76] [82] [83] [84] [85] 1[81, 86] [87] [88]
можно привести следующие [89]: pH начала осаждения гидроокиси ~6; pH полного осаждения гидроокиси «8; pH осаждения гидроокиси из щелочного раствора ~ 12.
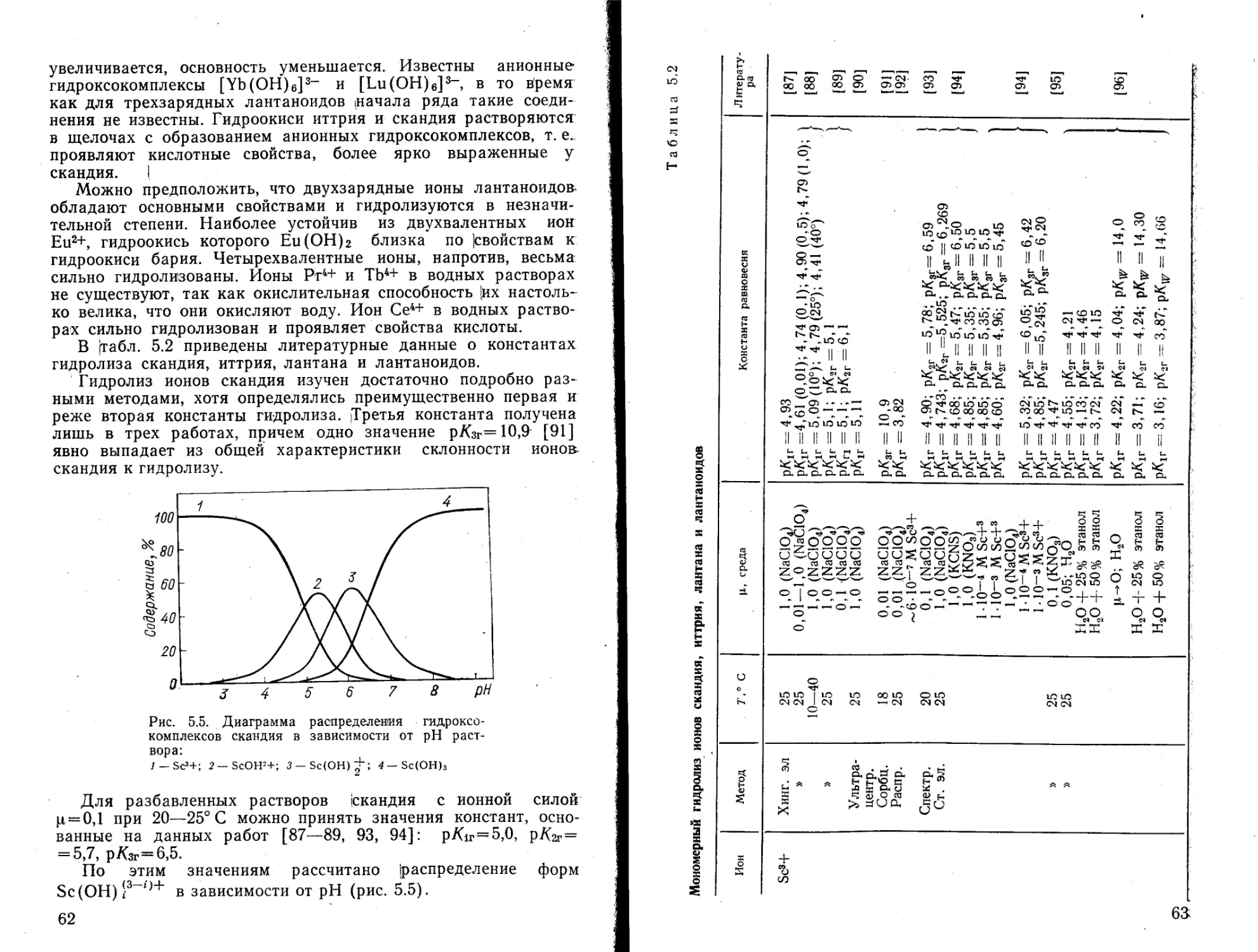
Руководствуясь критериями выбора (стр. 00), данными pH осаждения гирдоокиси и данными табл. 4.2, можно принять следующие значения констант гидролиза ионов цинка: p/Cir= =7,7 [67]; р/<2г=9,1 [67, 71]; ptf3r= 11,5 [57, 69, 71]; рК4г=12,7 £57]; рК*“< =16,8 [67, 68]; рК*“* =28,3 [65, 67, 68]; рЛ°*« = =41 [52, 56, 59].
С помощью этих констант рассчитано распределение гидроксокомплексов цинка в зависимости от pH раствора (рис. 4.2). Приведенные диаграммы соответствуют значениям
Содержание, % Содержание^
Рис. 4.2. Диаграмма распределения гидроксокомплексов цинка в зависимости от pH раствора:
/-Zn4; 2 — ZnOH+; <3 — Zn(OH)2; 4 — Zn(OH)“; 5-Zn(OH)2~
Рис. 4.3. Диаграмма распределения, гидроксокомплексов кадмия в зависимости от pH раствора:
1 - Cd2+; 2 - CdOH + ; 3 - Cd(OH)2; 4 -Cd(OH)~
Рис. 4.4. Диаграмма распределения, гидроксокомплексов Hg (II) в зависимости от pH раствора:
1 - Hg2+; 2 - HgOH+; 3-Hg(OH)2; 4 — Hg(OH)-
pH осаждения гидроокиси цинка Zn(OH)2. Данные термодинамического анализа [69] совпадают с этими рекомендациями.
Данные о константах гидролиза ионов кадмия немногочисленны. Принимая во внимание то, что Cd(OH)2 начинает осаждаться при pH л?8, а растворяется только в достаточноконцентрированных растворах щелочей, можно принять следующие константы [73, 75, 76]: p/Gr—7,9, рКгг^ЮД р/Сзг*^ = 14,3.
Диаграмма распределения гидроксокомплексов кадмия в. зависимости от pH раствора изображена на рис. 4.3.
46
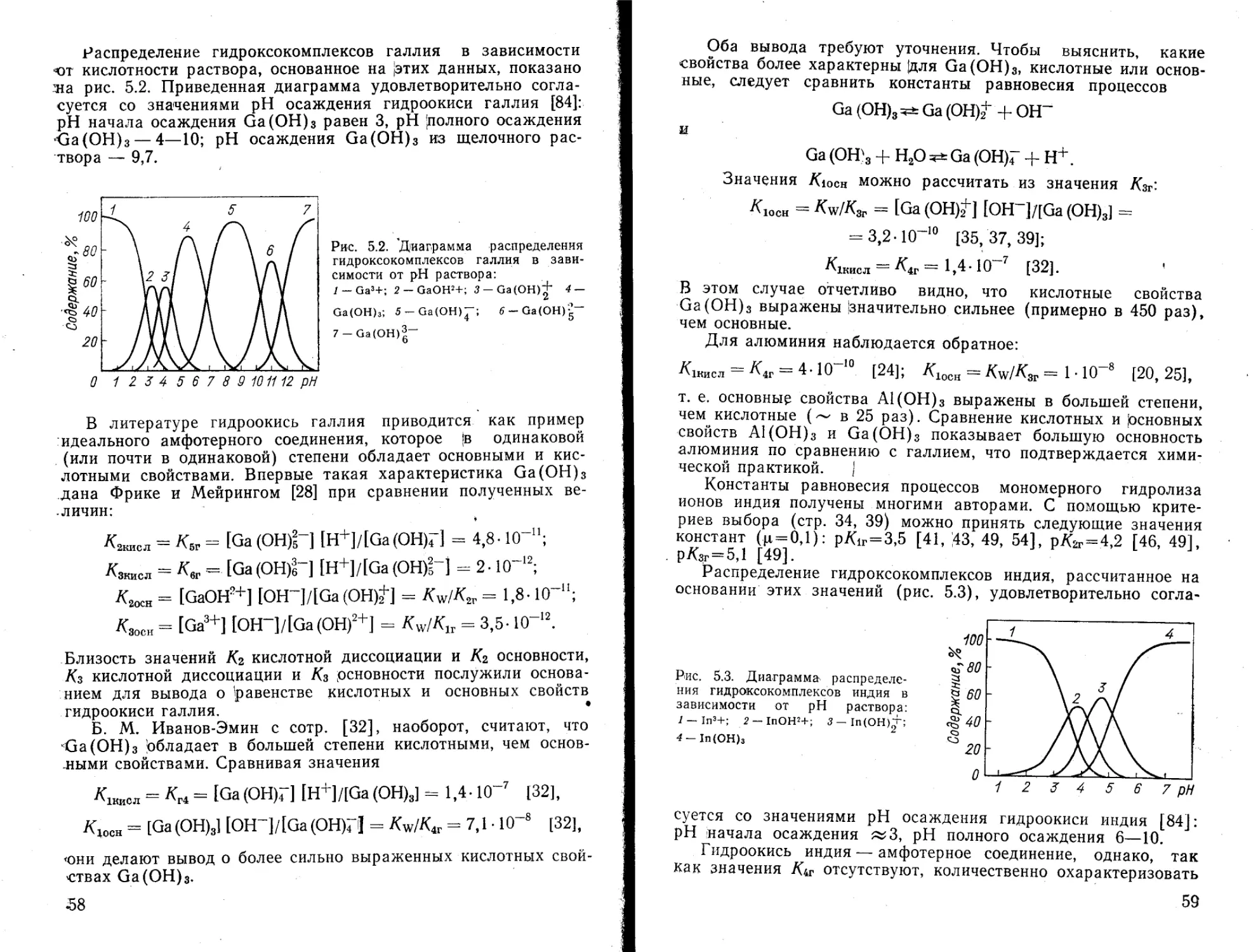
Гидроокись ртути начинает осаждаться при рН^2; полное осаждение происходит при pH ^54-12 [89]. Растворяется гидроокись ртути в концентрированных щелочах. С учетом этого можно принять следующие значения констант гидролиза иона Hg2+: рЛ1г=3,5 [76, 81, 84—87], рЛ2г-4,0 [76], рЛ3г=14,8 [78, 79]. С их помощью рассчитано распределение гидроксокомплексов Hg2+ в зависимости от pH (рис. 4.4). Диаграмма распределения хорошо согласуется со значениями pH осаждения гидроокиси ртути (II). Методом окислительно-восстановительного потенциала при ц~3,0 (NaCICU) и 25° определена р/Сг=4,48 для иона Hg2+ [91].
5 ГИДРОЛИЗ ИОНОВ ЭЛЕМЕНТОВ III ГРУППЫ
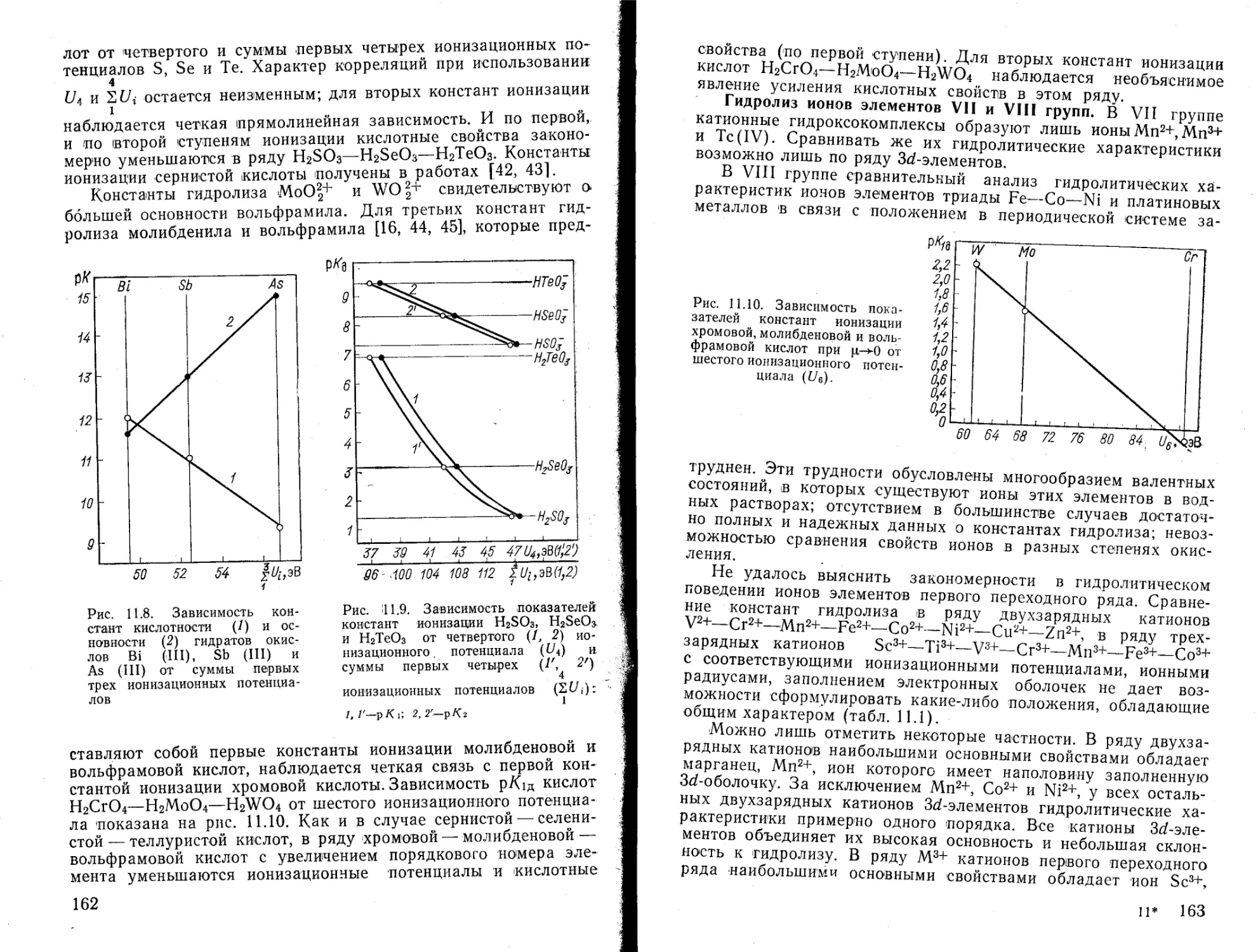
5.1. Элементы подгруппы В, Al, Ga, In, Ti
Элементы главной подгруппы III группы в своих соединениях проявляют высшую валентность +3. Бор и алюминий могут быть переведены [в низшие валентные состояния с большим трудом. Галлий и индий восстанавливаются до низших валентных состояний легче. В водных растворах соединения этих элементов в низшей валентности неустойчивы. Таллий, напротив, чаще встречается |в виде иона Т1+, чем Т134-.
Принято считать, что в этой группе элементов сверху вниз происходит закономерное усиление основности: бор образует борные кислоты и в катионной форме В3+ не обнаружен; Алюминий и галлий — металлы, но их гидроокиси обладают амфотерными свойствами; для Т1(ОН)3 и 1п(0Н)3 амфотерность не характерна, это основания, хотя и слабые. Аналогичным образом характеризуют кислотные и основные свойства окислов МгО3 этих элементов. Иная картина возникает при рассмотрении основности ионов элементов в водных растворах. Константу гидролиза можно (расценивать как константу кислотности катиона. Основность катионов элементов главной подгруппы III группы изменяется сверху вниз не симбатно положению в периодической системе, о чем можно судить по значениям констант ^гидролиза (табл. 5.1).
В этой таблице не рассмотрен бор, хотя в литературе известны константы, характеризующие ионизацию борной кислоты по катионному типу:
HsBO3 + H3O+^H4BOt + Н2О (рК = 0,13) [1].
Константы гидролиза катионов бора:
В3+ + Н2О^ВОН2+ + Н+ (рК1г = 6,82) [2,3]
ВОН2+ + Н2О^В(ОН)^ + Н+ (рК2г = 7,12) [2,3];
В(ОН)2+ + Н2О^В(ОН)3 + Н+ (рКзг = 7,62) [2,3]
Существование катионов бора в слабокислых водных растворах представляется маловероятным.
48
ю
со
Д’
S
\0
н
Мономерный гидролиз ионов АР4-, Ga3+, In3-h, Т1+, Tl34-
4 Зак. 1864
49
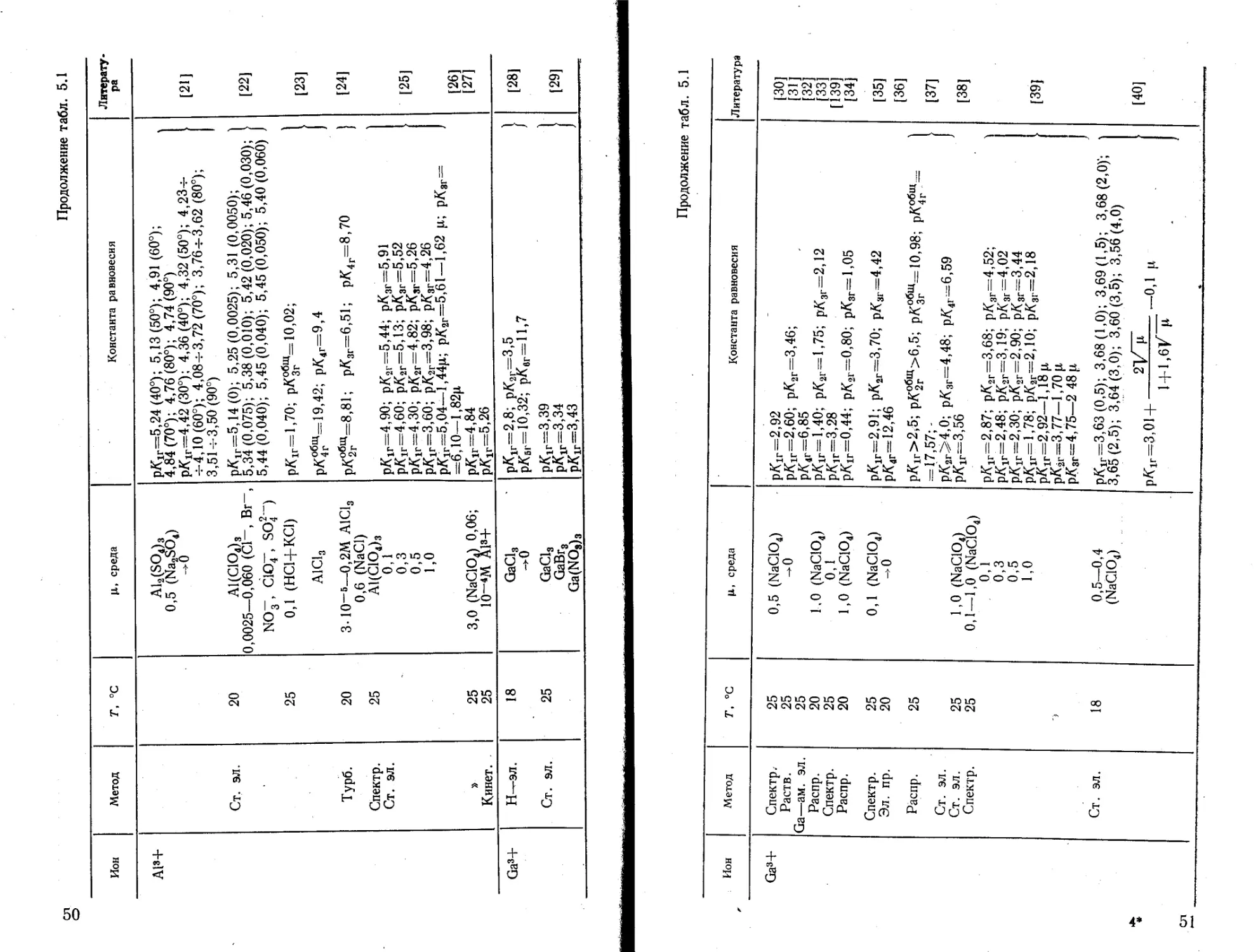
Продолжение табл. 5.1
сл о
Ион Метод г, °C ц, среда Константа равновесия Литература
А1»+ Ст. эл. 20 25 ai2(so4)3 0,5 (Na2SO4) Л) А1(СЮ4)з 0,0025—0,060 (Cl-, Br~, NO” СЮ7, SO^“) 0,1 (HC1+KC1) AICI3 pKlr=5,24 (40°); 5,13(50°); 4,91(60°); 4,84(70°); 4,76(80°); 4,74 (90°) pf( =4,42(30°); 4,36 (40°); 4,32(50°); 4,23= =4,10(60°); 4,08=3,72(70°); 3,764-3,62 (80°); 3,51=3,50(90°) pAlr=5,14(0); 5,25(0,0025); 5,31 (0,0050); 1 5,34(0,075); 5,38(0,010); 5,42 (0,020); 5,46 (0,030); 5,44 (0,040); 5,45(0,040); 5,45 (0,050); 5,40 (0,060) J рК1Г=1,70; рК°®щ=10,02; ] рА^щ= 19,42; рА4Г=9,4 1 [21] [22] [23]
Турб. Спектр. Ст. эл. » Кинет. . 20 25 25 25 3.Ю-6—0,2M Aici3 0,6 (NaCl) A1(C1O4)3 0,1 0,3 0,5 1,0 3,0 (NaClOA 0,06; 10~4M Al3+ рК°2бгщ=8,81; PA3r=6,51; pK4r=8,70 ' pKir=4,90; pA2r=5,44; pK3r=5,91 pAlr=4,60; p/<2r=5,13; pK3r—5,52 p/Cir=4,30; pA2r=4,82; pK3r=5,26 pKir=3,60; pK2r=3,98; pK3r=4,26 ptflr=5,04—l,44p; pK2r=5,61—1,62 p; pK3r= =6,10—1,82р. pKir=4,84 pKir=5,26 • [24] [25] [26] [27]
Ga3+ Н—эл. Ст. эл. 18 25 GaCl3 ->0 GaCl3 GaBr3 Ga(NOs)3 Ю CO II II J? к 1 . -CO оз ’f co QO CO^CO^ CM co" co co ii_ii_ 'L'L'L C2u Qh CX* J [28] [29]
Продолжение табл. 5.1
Ион Метод г, °C Ц, среда Константа равновесия Литература
Ga3+ Спектр Раств. Ga—ам. эл. Распр. Спектр. Распр. Спектр. Эл. пр. Распр. Ст. эл. Ст. эл. Спектр. Ст. эл. 25 25 25 20 25 20 25 20 25 25 25 18 0,5 (NaClOA -0 1,0 (NaClOA 0,1 1,0 (NaClO4) 0,1 (NaClOA ->0 1,0 (NaClOA 0,1—1,0 (NaClOA 0,1 0,3 0,5 1,0 0,5—0,4 (NaClO4) II toll II II 'll 'll ’ll llll llVbtV 'll'll 11'll'll ll li "ii 5° cjv?° ** 7*“ 7° j43 jo 5° 7° ’““5° 2 cn 1 + S°o II ъ xj ° >: jx “ >- >; -U OO О ООт ’ll Ц 13 II II II ll 1 v И ll il II O —'= 3°6ОСлЗС*э цх СЛ GO О = ОЭ О 8 32 -°? Ь S s a £ 1 °o тз n "o TEL тз —n < i -cr С Л ' ° il II II II / II ll ll -• (£3 ЬОСО^.4^ - II 4^ — кэ _ * Сл м 1 COO — tU О СИ CO о 4^ о 00 4^ to Ю 5-^ ЬО СП to - £ О Ф. О 00 т СК - £ ъ II [30] [31] [32] [33] [139] [34] [35] [36] [37] [38] [39] [40]
Продолжение табл* 5Л
Ион Метод Т, °C среда Константа равновесия Литература
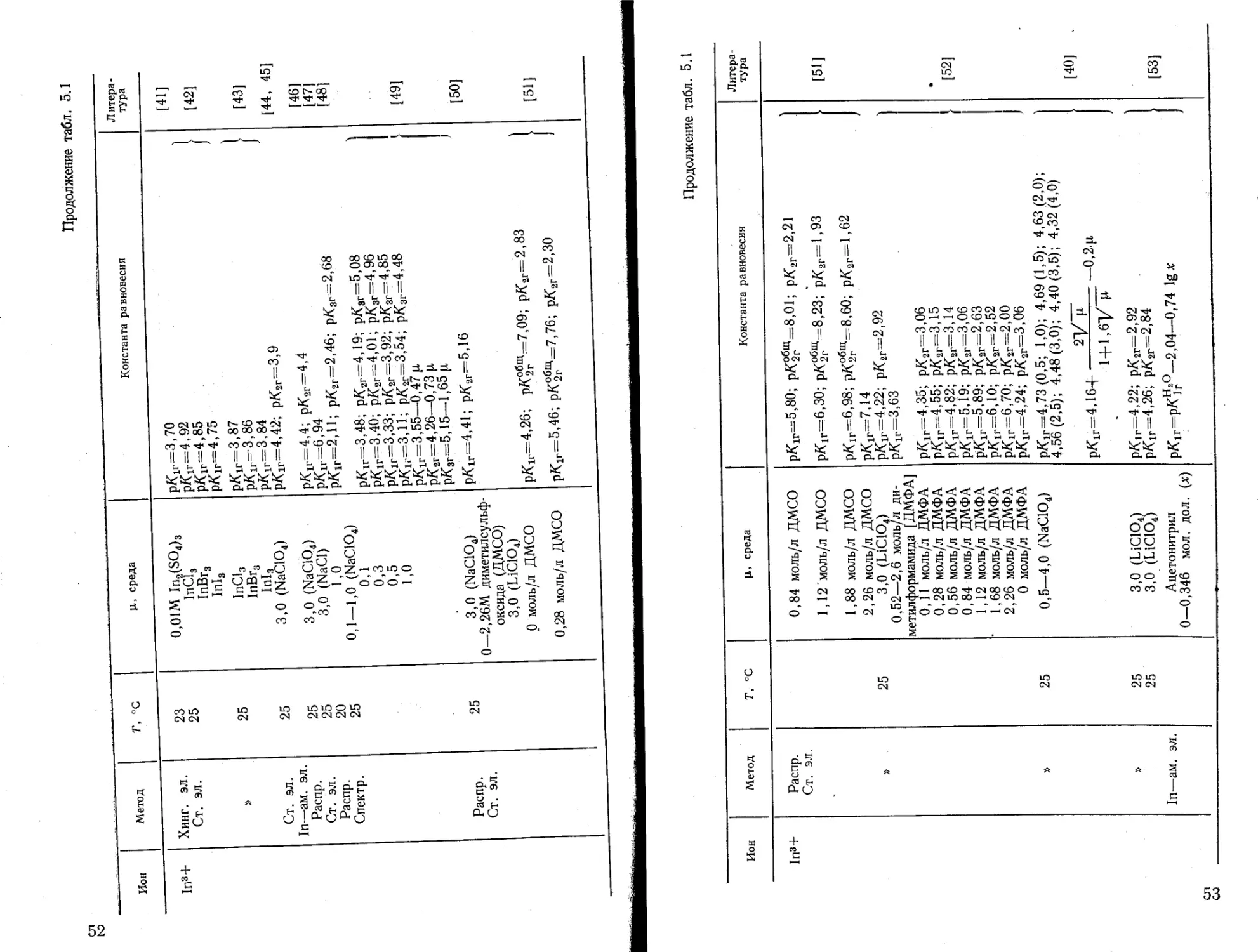
1п3+ Хинг. ЭЛ. Ст. эл. » Ст. эл. In—ам. эл. Распр. Ст. эл. Распр. Спектр. Распр. Ст. эл. 23 25 25 25 25 25 20 25 25 0,01М In2(SO4)3 1пС13 InBr3 Inl3‘ 1пС13 1пВг3 Inl3 3,0 (NaClO4) 3,0 (NaC104) 3,0 (NaCl) 1,0 0,1—1,0 (NaC104) 0,1 0,3 0,5 1,0 3,0 (NaC104) 0—2,26M диметил сульфоксида (ДМСО) 3,0 (LiClOJ 0 моль/л ДМСО 0,28 моль/л ДМСО СО со о OO OO in ОО 04°^ о ОФ ОО и 04 СЧ Ю L Н и iiiiiiii к? от от от, от от & “С (X СХ СХ СХ сх о о о о сл 1 04 о? СЛ rt* —< О ’-'7 II г- 00 04 'Ф со 00 ш д' II || * || || || || || 1X4 ||_ |u * % * сх сх (X сх .. °- Ill-- •- -н QOOCO—‘Юсоио*— о С- О 00 о* 00 00 00 -ф rf Ф -- 'ef со' LO о^~* сч СО Д ТГ оо со СО TF CD of со" СО со СО СО Ю 'Ф М4 К? 11 ч ч ч ч ч ч ч ч ч ч ч ч ч ч ч ч ч ч i СХ СХ СХ СХ сх СХ СХ СХ СХ СХ СХ сх сх сх сх сх сх сх сх сх сх [41] [42] [43] [44, 45] [46] [47] [48] [49] [50] [51]
Продолжение табл. 5Л
Ион Метод Т, °C Ц> среда Константа равновесия Литература
1пЗ+ Распр. Ст. эл. » » » In—ам. эл. 25 25 25 25 0,84 моль/л ДМСО 1,12 моль/л ДМСО 1,88 моль/л ДМСО 2,26 моль/л ДМСО 3,0 (ЫС1О4) 0,52—2,6 моль/л диметил формамид а [ДМФА] 0,11 моль/л ДМФА 0,28 моль/л ДМФА 0,56 моль/л ДМФА 0,84 моль/л ДМФА 1,12 моль/л ДМФА 1,68 моль/л ДМФА 2,26 моль/л ДМФА 0 моль/л ДМФА 0,5—4,0 (NaC104) 3,0 (LiClOA 3,0 (LiClO4) Ацетонитрил 0—0,346 мол. дол. (х) *5, *5^ ХХХХ.ХХХХ XXX X X X 4 11 • 1 *>li li 1 li li li li li li li li li li li li -a ^ФФФСЛХХХ W X 4 0 CT> СЛ “Ж о? КП 00 b-<e> Сл CO фр-ф co OO -iw ф5\Ь? COJxoOCOOfrOCnCH CO he X. OO о о о 1 *O *« *15 XJ *O *O T5 "И. "P - *1 w n ts t; ta to ьа Ц 2 to О О || и г *3 IlMi MNIMI ll £ > ] О "—'45 5^ to Ю to 00 CO CO CO 5° 1! ii || -° Sf§ ’’x. xl ООФфЬ'--О CO 00 QO \^|44ДФОЮФФХФФ ЬЭ о to о 4^ X 5? 5? гг 0Q [ х X ' *О * Д JX сл СП п « — II Н II 4* JX р- J— to Ctf сг> Ъ CD to to 00 to со — ^"to [51] [52] [40] [53]
Продолжение табл. 5.1
Ион Метод Г, °C Ц, среда Константа равновесия Литература
1п3+ In—ам. эл. 25 3,0 (LiC104) Н2О-диоксан х=0,009 мол. дол. диоксана р/С1Г=4,18; рКгг=2,85; х=0,063; рК1Г=4,05; р/С2Г=2,85 х=0,180; рК1г=3,89; рК2Г=2,84 [54]
х=0,260; рК1г=3,78; р/<2Г=2,75 р/С1Г=3,54 (Н2О)
ц^О х=0,0615 мол. дол. диоксана; рК1Г=3,42; рд2Г=
Н3О—диоксан =2,63; х=0,172; рК1Г=3,22; рК2Г=2,60; х=0,237; рК1г=3,12; рК2Г=2,42 J
Ст. эл. 25 3,0 (ЫС1О4) рК1г=4,26 (Н2О) 0,046 мол. дол. ацетона
Н2О—ацетон рК1г=4,22; рК2Г=3,19; х=0,096; рК1Г=4,05; рК2Г=3,31; [60]
х=0,201; рК1Г=3,84; рК2Г=2,97;
х=0,36; рК1г=3,64; рК2Г=2,84
Т1+ Кинет. 25 0,02—0,04 р/С1г = 13,78; рК2Г= 13,58 . 1 [55]
ц-0
Раств. Эл. пр. 0—40 -+0 рК1Г=14,14(0°); 13,18(25°); 12,69(40°) 1 [56]
Кинет. 25 ->0 рК1Г= 13,58 J [57]
25 ->0 рК1г=13,15
Спектр. 25 2-10-3 М Т1ОН РК1Г=13,3 [58]
ц>0 рК1г=13,70 (0,5); 13,75(1,0); 13,91(3,0); 1
» 25 0,5—5,0
(NaClO4, LiC104) 14,08(5,0) [59]
|W“>0 рК1г= 13,31
Продолжение табл. 5.1
сл сл Ион Метод T, °C ц, среда Константа равновесия Литература
Т13+ Ст. эл. Спектр. Кинет. Редокс. пот. Кинет. Редокс. пот. Спектр. Редокс. пот. Ст. эл. Ст. эл. Редокс. пот. Ст. эл. » » Спектр. » 18 25—41,8 25-45 25 50 25 25—40 25 25 25 25 25 25 25 25 Т1Вг3 р->-0 6,0 (NaC104) 3,0 (NaClO4) 3,0 (NaClO.) 6,0 3,68 1,5 (NaClO4) 3,0 (NaClO4) H2O, H2o+D2o 3,0 (NaClO4) 3,0 (LiC104) 4MPy*HNO3 10 (NH4NO3) 2M En**HNO3 2M Mg(NO3)2 3,0 (LiCICL) 3,0 (LiC104) (NaClOJ 0,1 0,3 0,5 1,0 'P’PTL’P P P "° та та та та n та та та % ъ "1 Illi li mi li lili "ini 1i iiiiii IniIiImi 1i 1i 1i . ~ ~ III | ~~ 1 1 - 1 1 ° oo о to о о — w — 2- Л- 1. — — - о A J ’ L 1 ^СЛЧОООДОО -ч OO - - ,, ~ OO4^ О О ЬЭ | 1 [ Х^ОО-Л 00 bl о оузта/р та та та • та та ^та ~ ° “З.ч’ч’ ? g g' . I11 '-^ii si! —li - ' — СЛ СЛ о О 1 й- 4^ 4^ О •— IL 4х | | ^boL о1*03 Soo » « « « ^0 GO и и JL и 3 сл оо г '77' 4*. 00 СЛЮ -= А> ° 00 00 [61] [62] [63] [45, 64] [65] [66] [67] [68] [69] [70] [71] [72] [73] • [74]
* Ру—'Пиридин.
** Еп—этилендиамин.
56
Гидролиз ионов алюминия изучен многими исследователями. Методом ЯМР на протонах, 17О и 27А1 получены данные о числах гидратации иона А13* в водных (растворах, о механизме обмена свободных и координированных молекул воды, о механизме гидролиза с образованием мономеров и димеров [78— 83]. Можно считать доказанным механизм [образования моно-гидроксокомплекса алюминия по реакции А1 (Н2О)|+ + Н2О^ ^[А1(Н2О)5ОН]2++Н3О+ [80, 81].
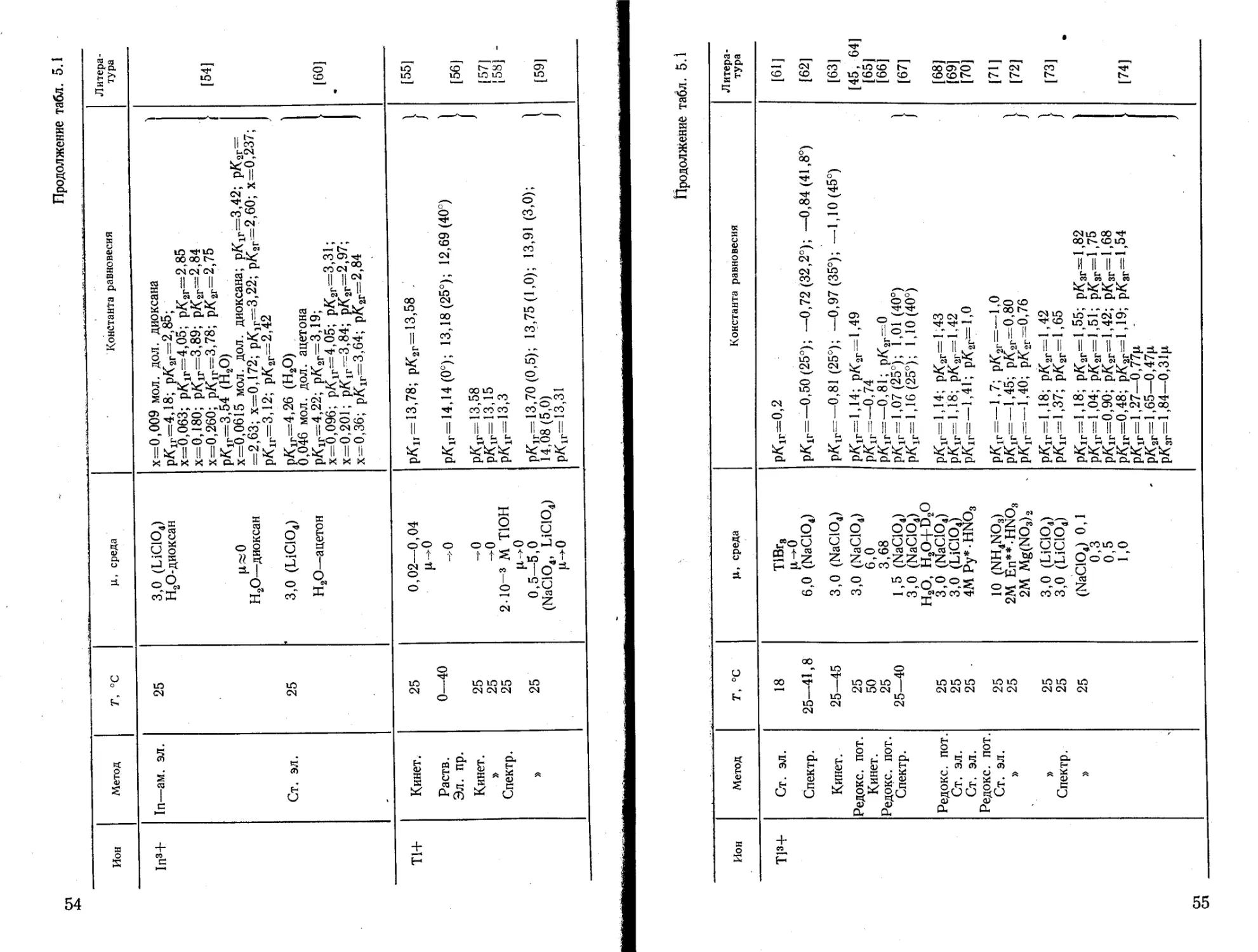
Из анализа данных о константах мономерного гидролиза ионов алюминия можно рекомендовать значения: рЛ\г=5,0 [4—6, 9—11, 13—15, 20—22, 25—27], рК2г = 5,5 [20, 25], рК3г= = 6,0 [20, 25], рК4г=9,4 [23].
На основании этих констант гидролиза построено распределение гидролизованных форм алюминия (рис. 5.1), которое
Рис. 5.1. Диаграмма распределения гидроксокомплексов алюминия в зависимости от pH раствора:
/ —А13+; 2-АЮН2+; 3-А1(ОН)+; 4-А1(ОН)3;
5 —А1(ОН)“
хорошо согласуется со значениями pH осаждения гидроокиси алюминия [84]: pH начала осаждения А1(ОН)3 равен 4; pH полного осаждения А1(ОН)3 составляет 5—7,5; pH осаждения А1(ОН)3 из щелочного раствора примерно равен 11. Рассчитаны константы гидролиза иона алюминия от 25 до 350° С [141].
С учетом принятых критериев (стр. 00) можно рекомендовать значения констант, которые получены в результате наиболее полного исследования последовательного гидролиза ионов галлия. Это работы, в которых определены константы равновесия нескольких ступеней гидролиза [130, 137, 139, 142].. Такими рекомендуемыми значениями могут быть: p/Gr = 2,9 [28, 30, 31, 35, 37, 39, 40], !рК2г = 3,7 [31, 35, 37, 39], рЛ3г = 4,5 Г35, 37, 39], рК4г=6,8 [32, 37], рК5г= 10,3 [28], рА6г= 11,7 [28].
57'
Распределение гидроксокомплексов галлия в зависимости ют кислотности раствора, основанное на |этих данных, показано ’на рис. 5.2. Приведенная диаграмма удовлетворительно согласуется со значениями pH осаждения гидроокиси галлия [84]: pH начала осаждения Са(ОН)з равен 3, pH 'долного осаждения Ga(OH)3— 4—10; pH осаждения Ga(OH)3 из щелочного раствора — 9,7.
Рис. 5.2. ’Диаграмма распределения гидроксокомплексов галлия в зависимости от pH раствора:
У — Ga3+; 2 — GaOH24-; 3-Ga(OH) + 4 —
Ga(OH)3; 5-Ga(OH)“; £-Ga(OH)-H 7-Ga(OH)|“
В литературе гидроокись галлия приводится как пример идеального амфотерного соединения, которое в одинаковой (или почти в одинаковой) степени обладает основными и кислотными свойствами. Впервые такая характеристика Ga(OH)3 дана Фрике и Мейрингом [28] при сравнении полученных ве-.личин:
Я2КИСЛ = = [Ga (OH)i-] [H+]/[Ga (ОН)?] = 4,8 • 10-11;
Язкисл = = [Ga(OH)H [H+]/[Ga (ОН)М = 2- 10~12;
/С20сн = [GaOrf+j [OH~]/[Ga (ОН)^ = Kw/K^ = 1,8- КГ11;
tf3oCH = [Ga3+] [ОН-]/[Ga(OH)2+] = KW/K1T = 3,5- КГ12.
Близость значений Кг кислотной диссоциации и Кг основности, Кз кислотной диссоциации и Кз основности послужили основанием для вывода о 'равенстве кислотных и основных свойств гидроокиси галлия. *
Б. М. Иванов-Эмин с сотр. [32], наоборот, считают, что Ga(OH)3 'обладает в большей степени кислотными, чем основ-лыми свойствами. Сравнивая значения
*1кисл = = iGa (ОН)?] [H+]/[Ga (ОН)3] = 1,4- 1СГ7 [32],
К10СН = [Ga(ОН)3] [OH-]/[Ga (ОН)?] = K^lK^ = 7,1 • 1(Г8 [32], «они делают вывод о более сильно выраженных кислотных свойствах Ga (ОН) з.
58
Оба вывода требуют уточнения. Чтобы выяснить, какие свойства более характерны |для Ga(OH)3, кислотные или основные, следует сравнить константы равновесия процессов
Ga (ОН)3 Ga (ОН)2+ + 0Н~
И
Ga (ОН'Я + Н2О =₽* Ga (ОН); + Н+.
Значения /Сюсн можно рассчитать из значения К3т:
Кюсн = *w/K3r = [Ga (ОН)2+] [OH-]/[Ga (OH)3] =
= 3,2 •КГ’10 [35,37,39];
К1кИсЛ = ^4г=1,4-10-7 [32].
В этом случае отчетливо видно, что кислотные свойства Ga(OH)3 выражены |значительно сильнее (примерно в 450 раз), чем основные.
Для алюминия наблюдается обратное:
Кишел = 7<4г = 4.1О-10 [24]; tf10CH = = 1 • 10~8 [20,25],
т. е. основные свойства А1(ОН)з выражены в большей степени, чем кислотные (~ в 25 раз). Сравнение кислотных и [основных свойств А1(ОН)3 и Ga(OH)3 показывает большую основность алюминия по сравнению с галлием, что подтверждается химической практикой. )
Константы равновесия процессов мономерного гидролиза ионов индия получены многими авторами. С помощью критериев выбора (стр. 34, 39) можно принять следующие значения констант (ц = 0,1): рЛ1г=3,5 [41, [43, 49, 54], рА2г-4,2 [46, 49], рАзг = 5,1 [49].
Распределение гидроксокомплексов индия, рассчитанное на основании этих значений (рис. 5.3), удовлетворительно согла-
Рис. 5.3. Диаграмма распределения гидроксокомплексов индия в зависимости от pH раствора: 1 — 1п3+; 2 —1пОН2+; 3-1п(ОН) + ;
4~ 1п(ОН)з
суется со значениями pH осаждения гидроокиси индия [84]: pH начала осаждения ^3, pH полного осаждения 6—10.
Гидроокись индия — амфотерное соединение, однако, так как значения /С4г отсутствуют, количественно охарактеризовать
59
меру основности и кислотности 1п(ОН)3 не представляется возможным. Установлено, что 1п(ОН)3 в большей степени обладает ^основными свойствами [85]. Изучена растворимость 1п(ОН)3 в растворах NaOH [85, 86]. Определены концентрационные константы образования анионных гидроксокомплексов:
pt- = [1п(ОН)з+,]/[ОН“].
Для 1-й ступени константа равна Pi = (1,0— 1,1) • 10~3. По фор-^ муле Ki = nPin(OH)3/Pi рассчитаны константы неустойчивости гидроксокомплексов [86], |из которых можно получить общие константы гидролиза: рЛ^щ = 26,89, р/С^щ =42,52, р/С^щ=58,3 и частные константы рКбг= 15,63 и рЛ6г= 15,78. По данным работы [49], рЛ^щ = 12,8, и тогда рЛ4г“14,1. Последняя величина, несмотря на условность (ее расчета и относительный характер точности, характеризует гидроокись индия как соединение^ обладающее лишь в незначительной степени кислотными свойствами: Л1кисл=Л4г=8-10“15, Kioch^Kw/K3~ 10~9.
О гидролизе ионов Т1(1) в литературе нет (разногласий. Гидроокись Т1ОН представляет собой сильное основание. Эта видно при сравнении констант гидролиза Tl (I) и ионов щелочных металлов. Данные Белла /[55—57] и Ф. Я. Кульбы с сотр. [58, 59] согласуются между собой. Если данные работы [59], полученные для растворов с разными значениями ионной силы, экстраполировать к р=0 методом наименьших квадратов, то получим рЛ1г-= 13,67. С учетом этого по данным работ [55, 56, 59] можно принять pKir=13,6.
Гидролиз ионов Т1(Ш) изучен как в водных, [гак и в водноорганических растворах, причем в большинстве случаев при высокой ионной силе. Можно рекомендовать константы (для ц-^0 и 25°С): рК1г=1,27, рК2г=1,65, )рК3г=1,84 [74]. К ним близки значения рЛ\г и рЛгг, полученные в работах [64, 68, 69,73,77].
Рассчитанное с помощью констант распределение гидроксокомплексов таллия [в зависимости от pH раствора показано на рис. 5.4. Эта диаграмма удовлетворительно согласуется со значением pH начала осаждения гидроокиси таллия, равном -1,0.
Методом растворимости, аналогично тому, как это было сделано для индия, рассчитаны константы нестойкости анионных гидроксокомплексов таллия [86]: Pi=l,3-10~5; р2 = 8,8Х Х10~6; Рз = 6,1-10“6. С помощью, значения ПРТ1(он)3 и Kw можно рассчитать константы гидролиза: рК°®щ= 17,62, = 31,79, р^щ =46,61.
С учетом первых констант гидролиза можно вычислить: р/<4г= 13,02, рК5г= 14,17, рК6г= 14,82 [74], а также Л^кисл^ = ^Ы0^, 7(10CH==Kw//<3r=6,3*10-13.
60
Основные свойства у гидроокиси таллия выражены сильнее, чем кислотные. (По склонности к гидролизу элементы можно расположить в ряд Tl(III)>Ga>In>AL Основность М(ОН)3 изменяется в обратном порядке. Ряд кислотности несколько иной: Ga>Al>Tl>In. Такой порядок, возможно, обусловлен тем, что p/Ctr для индия и таллия вычислены с использованием значений ПРм(он)8 и констант образования гидроксокомплексов. При более достоверных значениях /<4г(Л1кисл) могут быть получены иные значения констант.
Рис. 5.4. Диаграмма распределения гидроксокомплексов Т1 (III) в зависимости от концентрации ионов водорода:
/-ТР+; 2 —Т1ОН2+; 3 — Т1(ОН)+;
4-Т1(ОН)3
Из рассмотрения приведенных :в табл. 5.1 констант следует, что наиболее основной и наименее ^склонный к гидролизу элемент в подгруппе — алюминий, а наименее основной — таллий. Обращает также внимание большая склонность к гидролизу ионов (галлия, чем индия, Все это не согласуется с положением элементов подгруппы в периодической системе: обычно основность элементов увеличивается сверху вниз в группе. Связь этого явления с ионными радиусами и ионизационными потенциалами атомов элементов этой подгруппы будет обсуждаться на с. 156—157.
5.2. Элементы подгруппы скандия — Sc, Y и лантаноиды
Скандий, иттрий и лантаноиды образуют ионы М3+, которые в этом состоянии весьма устойчивы. В степени окисления 2+ 'существуют ионы европия (электронная конфигурация /7), иттербия (jf14), малоустойчивые ионы самария (f6), тулия (f13), неодима (f4). Известны доказательства существования в кристаллах ионов Рг2+ и Се24-. В степени окисления 4+ существуют ионы (церия (f°), тербия (f7), малоустойчивые ионы празеодима (f1), неодима (f2), диспрозия (/8).
В ряду скандий — иттрий — лантан основность ионов М34-увеличивается. Аналогично увеличивается основность гидроокисей М(ОН)3. Скандий наиболее подвержен гидролизу, ион уз+ гидролизован |в меньшей степени, La3+ гидролизован незначительно. В ряду от La3+ к Lu3+ способность к гидролизу
61
увеличивается, основность уменьшается. Известны анионные гидроксокомплексы [Yb(OH)6]3" и [Lu(OH)6]3-, в то время как для трехзарядных лантаноидов (Начала ряда такие соединения не известны. Гидроокиси иттрия и скандия растворяются в щелочах с образованием анионных гидроксокомплексов, т. е.. проявляют кислотные свойства, более ярко выраженные у скандия. I
Можно предположить, что двухзарядные ионы лантаноидов^ обладают основными свойствами и гидролизуются в незначительной степени. Наиболее устойчив из двухвалентных ион Еи2+, гидроокись которого Еи(ОН)г близка по Двойствам к гидроокиси бария. Четырехвалентные ионы, напротив, весьма сильно гидролизованы. Ионы Рг4+ и ТЬ4+ в водных растворах не существуют, так как окислительная способность [их настолько велика, что они окисляют воду. Ион Се4+ в водных растворах сильно гидролизован и проявляет свойства кислоты.
В ггабл. 5.2 приведены литературные данные о константах гидролиза скандия, иттрия, лантана и лантаноидов.
Гидролиз ионов скандия изучен достаточно подробно разными методами, хотя определялись преимущественно первая и реже вторая константы гидролиза. (Третья константа получена лишь в трех работах, причем одно значение рК3г=10,9 [91] явно выпадает из общей характеристики склонности ионов-скандия к гидролизу.
Рис. 5.5. Диаграмма распределения гидроксокомплексов скандия в зависимости от pH раствора:
1 - Sc3+; 2~ ScOH2+; 3-Sc(OH)+; 4— Sc(OH)3
Для разбавленных растворов [скандия с ионной силой Р“0,1 при 20—25° С можно принять значения констант, основанные на данных работ [87—89, 93, 94]: рЛ1г=5,0, p/Gr = = 5,7, рК3г=6,5.
По этим значениям рассчитано [распределение форм Sc (ОН) в зависимости от pH (рис. 5.5).
62
<М 16
ГЗ
Я
S
ю
СЧ
Мономерный гидролиз ионов скандия, иттрия, лантана и лантаноидов
Литература гТ 00 © ос о° ро. ©. — CM' co* ф1 CT> 03 CD [95] [96]
Константа равновесия ;о,01); 4,74 (0,1); 4,90 (0,5); 4,79 (1,0); (10°); 4,79(25°); 4,41 (40°) Р^2Г ~= 6j 1 рКгг = 6,1 03 CO 03 ^4.0 Ю Ю o Ю1О IQ Ф co" || co LQ IQ LQ II fellIIIIII ex. „ cl cl cl ex 00 £3 r-T 16 LQ co Г^^Ф CO co 03 Ю to 16 l6 iQ Ф и ; ii ii и и u, N u |_ , к , w. e’. M CL CX CL CL CL ; рК2Г 3=6,05; рХзг —6,42 , рК2Г —5,245; рКзг = 6,20 > — со in (МФ — Т1и N N N CL CL CL ; рК2Г = 4,04; рК^ = 14,0 о со 7 < сч т С1 СО II сх 00 со ii ) ci f а.
со . 03 - - . Ф ^10 Ш IQ Ю II I! II II II II CL CL СХ CX CL CL 2 coll II co w CU Cu o co Ю Ю о Оз r- CO 00 OO CO Tf ’Ф Tt* II II II II II II CL CL CL CX CL CL <М 1О ь- ш со см со 00 ш — ь- Щ "Ф м* Тр 'ф со II IE II II II II CL СХ СХ CL CL СХ а и сх СО II 5< СХ CD co II CL
ц,, среда 1,0 (NaC104) 0,01—1,0 (NaClOJ 1,0 (NaClOJ 1,0 (NaClO4) 0,1 (NaC104) 1,0 (NaClO4) 0,01 (NaClO4) 0,01 (NaClOJ i ~6-10~7MSc3+ ч 5 —л. 1 +' 8 S w Z? ? .-inc> i i * ю см ю - О О о о О о - О , . o'-~- ТТ- • ' ° О ТГ ^6, оо о X о t =1 Н20 + 25 % этанол H20 + 50% этанол
и о о Ю Ю I Ш LQ CM CM 1 M CM о 00 ю — CM о ю см <м LQ Ш (М (М
Метод § S.(LS<i u л £ Си о К <p о TO rS Спектр. Ст. эл. Л А
Ион + ба
Продолжение табл. 5.2
Ион Метод Т, °C Ц, среда Константа равновесия Литература
Y3+ Ст. эл 25 Y2 (so4)3
0,005М pKir = 7,7 | [97]
0,01 М pKar = 6,7 J
Хинг. эл. 25 3,0 (LiC104) рК1Г =9,1 [98]
Ст. эл. 25 0,2 (NaNO3) pKir = 7,34; pKuz = 13,92 [99]
» 25 0,3 (NaClO4)
4-10“SM Y3+ pKlr = 8,34; pKvi7= 13,73 [100]
» 25 ->0 pKir = 7,81 [101]
» 30 0,1 (NaClO4) pKlr = 7,00; pKu7 = 13,84 [142]
• Электромигр. 25 0,005 (HNO3, HC1O4) p/Cir = 3,5; pA'2r=4,7 [Ю2]
Ст. эл. 25 0,05 pKir = 8,04 (H2O); 7,94(25%); 7,49 (50%)
0—50% этанол pKw = 14,00 (H2O); 14,30(25%); 14,66 (50%) [96]
>0 pKir = 7,71 (H2O); 7,52(25%); 6,93(50%)
» 25 3,0 (Na+, Li+, C1O7 рК^щ= 16 04 (Ha0). 16|20 (D2O) [ЮЗ]
» 25 3,0 (LiClOJ рКобщ= pc 1104]
» 25 3,0 (LiClOJ рК°бщ= 1б)8 (н2О); 17,0 (D2O) [Ю5]
La3+ » 25 La2(SO4)3
5-10- 3M pKlr=10 0 I [97]
0,01 M pKlr = 9,7 I
» 20 0,1 pKw= 14,17; pKlr =8,57 [Ю6]
» 25 3,0 (LiClOJ pK = 10,1 [Ю7]
» 25 0,3 (NaClO4) pKir =8,98+9,06 [100]
Зак.
Продолжение табл. 5.2
Ион Метод Г>, С ц, среда Константа равновесия Литература
La3~h » » » Ст. эл. » Распр. 25 25 30 25 25 25 0,3 (NaClO4) >0 0,05 0—50% этанол =0 0,1 (NaClOJ 3,0 (LiClOJ 3,0 (LiClO4) 0,1 (LiClOJ P^Cir = 9,06 ] pKir=8,53 / pKir = 9,06 (H2O); 8,70 (25%); 8,17(50%) j pKw = 14,00 (H2O); 14,30(25%); 14,66(50%) ’ pKir = 8,73 (H2O); 8,28(25%); 7,61 (50%) i p/(ir = 8,14; pKw = 13,84 | pKlr =10,04 pKir= 10,04 (H2O) ) PKir= 10,35 (D2O) / P^ir = 7,4 [101] [142] [96] [Ю4] [Ю5] [108]
Сез+ Ст. эл. Электромигр. т.-д. Ст. эл. Электромигр. » Ст. эл. 25 25 25 15 25 30 Ce2(SO4)3 0,005M 0,01 M 0,005 0,005 (KC1 + HC1) 0,005 0,1 (NaClOJ ll II 'll 'll 'll 'll ’ll 00 co CO CD co co CO >;s II 11 * JL O° CaD фь ^СЛ [97] [109] ПО] [111] [112] [Ю2] [142]
Ce4+ Редокс. пот. Спектр. 25 5—35 2,38 2,0 (NaClO4) b!0-зМСе (IV) рК2г = 0,22 р/^г = 0,06 (5°);— 0,32 (15°);— 0,72 (25°);—1,18 } [ИЗ] [И4]
Продолжение табл. 5.2
Константа равновесия Литера-
Ион, Метод. Г, °C Ц, среда тура
СеЧ- Спектр. 0,1 2,0 (NaC104) ' p/Gr = 0,28 [115]
Кинет. 1,6 2 (NaClOJ pK r ==—0,90; рК2Г= 1,10
2,4 рК1Г=—0,90; рК2г =1,10 [Пб]
8,0 рК1Г=—-1,0; рК2г=1,00
15,6 рК1г=—1,15; рК2г=0,85
25 рК2Г=0,82 [117]
Н-эл. 25 3 (NaCl) рК5г =12,43
Спектр. 25 0,001-0,001МСе4+
5 0,1—1,7М НСЮ4 0,91—1,70М НС1О4 рЯ1Г =0,72 = 0,68 1 р/С1Г=0,96 1 [118]
Спектр. 15,5 IO'3 м Се4+ рК1г=0,87 1 [119]
25 1,5 (НС1О4) рК1Г=0,77 /
Рг3+ Ст. эл. 25 Рг2 (SO4)3 рК1Г —9,3 1
0,005М [97]
0,01М рК =9,05 /
» 25 ->0 рК1Г=9,80; рК2г = 3,70; pK^=3,15 j [120]
Раств.
Ст. эл. 25 3,0 (NaC104) р/С^г — 8,5 [121]
Т.-Д. рК1Г=9,8 [НО]
Ст. эл. 25 3,0 (NaClO4) pKir =8,55 [100]
» 25 0,3 (NaC104) ->0 рК1г=8,55 1 рК1Г=8,02 / [101]
Распр. 25 0,1 (LiC104) рК1Г=7,1 [108]
Ст. эл. 30 0,1 (NaC104) рК1Г=7,86; pK^ =13,84 [142]
Nd34- Ст. эл. 25 Nd2 (SO4)3 рХ1Г=9,0 1
0,005M 0,01M рК1Г=8,71 [97]
Продолжение табл. 5.2
СЛ * О Ион Метод г, °C Ц, среда Константа равновесия Литература
Nd34- Ст. эл. » Т.-д. Ст. эл. » » Распр. Электромигр. » Распр. 25 30 25 25 25 25 15 25 25 3,0 (NaCIO.) 0,1 (NaC104) 3,0 (LiC104) 0,3 (NaCIO.) 0,3 (NaCIO.) ->0 3,0 (NaCIO.) 0,2—0.8M Nd3+ 0,1 (NaCIO.) 0,005 (KC1, HC1) 0,005 0,1 (LiClO4) <—'—. § Q QO Ч, 52 ~ cO 11 S 1 T 1 а. о io ~ г? ю „ co.- г _ ° 'Т о D ю OO r- || . Ст>оо*'о6'г^? coo . II 11 3 H II II II II II .11 II II cu Ou GL, d. Q. CL, CX< Cl. £X ct [121] [142] [143] [HO] [100] [101] [122] [Ю8] [H2] [Ю2] [Ю8]
Sm3+ Ст. эл. Т.-д. Ст. эл. » Распр. j 25 25 25 30 25 । So,ao©)3 0.01M 0,3 (NaCIO.) 0,3 (NaClOJ =0 0,1 (NaClOJ 0,1 (LiC104) £ u ii и imil *£- . у --4 -J OO OO О 00 00 cr> OO CO CO •— cr> о i— 4* 4». о to 1 OO OO [97] [ИО] [100] [101] [142] [Ю8]
Продолжение табл. 5.2
Ион Метод Т, °C Ц, среда Константа равновесия Литература
Еи3+ Ст. ЭЛ. Т.-д. Ст. эл. » 25 25 25 Eu2 (SO4)3 0,005М 0,01М 0,3 (NaC104) 0,3 (NaC104) -0 CM CD < —< OO CT> CD ID OO OO O- OO OO OO oo II II II II II II CL ex Du CL ex cl 197] [HO] [100] [101]
Электромигр. 15 0,005 pXlr = 3,15 (pX^ =14,35) [112, 123]
Ст. эл. 25 0,05 pXlr =8,03 (H2O); 7,78(25%); 7,47(50%); ,
0—50% этанол pXn7 = 14,00 (H2O); 14,30(25%); 14,66(50%) ) [96]
Распр. Ст. эл. 25 30 0,1 (LiC104) 0,1 (NaC104) pX,r =7,68 (H2O); 7,06(25%); 6,25(50%) J pXlr=4,8 pXir=7,49 [108] [142]
Gd3+ Ст. эл. Т-д. Ст. эл. » 25 25 30 Gd2 (SO4)3 0,005M 0,lM 0,3 (NaC104) 0,1 (NaC104) pKir=8,85 ) pXlr=8,59 J pXlr=9,7 pXir =8,35 pXir =7,36; pX^ =13,84 [97] [HO] [100! [142 1
» 25 0,3 (NaClO4) -+0 0,1 .—-—. Ю (M CD OO OO QO Г- ОО 11 1! 1! сх q- ex [101] [124]
Продолжение табл. 5.2
Ион Метод Т, °C Щ среда Константа равновесия Литература
Gd3+ Ст. эл. 25 0,05; 0—50% этанол рХ]г = 8,27 (H2O); 7,98 (25%); 7,50 (50%) ] рХ^ =14,0 (Н2О); 14,30 (25%); 14,66(50%)
->0 [96]
25 3,0 (LiC104) рХ1Г =7,94 (Н2О); 7,26(25%); 6,28(50%) '
» рХ1Г =8,2 [Ю41
» 25 3,0 (LiC104) рХ1Г = 8,2 (Н2О); 8,34 (D2O) 105]
» 25 3,0 (NaClOJ рХ1Г =9,2 125]
Распр. 25 0,1 (LiClOJ pKir =7,1 108]
Tb3+ Т.-д. рк1г =9,7 [100]
Ст. эл. 25 0,3 (NaClO4) РХ1Г =8,16 [100]
» 25 0,3 рК1Г =8,16 ' 1
,0 рХ1Г=7,63 f Liui]
Электромигр. 15 0,005 (HC1, KC1) рК1Г = 3,10 (рК^ =14,35) 1 :н2]
Распр. 25 0,1 (LiC104) pKir —5,2 1 [1081
Ст. эл. 30 0,1 (NaClOJ рК1Г =7,18; рК^ =13,84 [142]
Dy3+ Т.-д. рК1Г = 9,7 [ИО]
Ст. эл. 30 0,1 (NaClO4) рК1Г = 7,08; рК^ —13,84 [142]
Py3+ Ст. эл. 25 0,3 (NaClO4) рХ1Г =8,10 [ЮО]
» 25 0,3 (NaC104) ^0 рК1Г=8,10 1 РХ1Г=7,57 / [Ю1]
Распр. 25 0,1 (LiC104) рХ1Г =5,6 [Ю8]
Ho3+- Т.-д. 0,3 (NaClO4) рХ1Г =9,7 [ПО]
Ст. эл. 25 рК1г =8,04 [100]
’Ч ©
ПродоЛЖёййе ТабЛ. 5.2
Ион Метод г, °C Ц, среда Константа равновесия Литература
Но3+ Ст. эл. 25 0,3 (NaC104) -.0 рК1Г=8,04 1 рЛ1г=7,51 / [101]
Распр. 25 0,1 (LiClOJ рК1Г =5,7 [108]
Ст. эл. 30 0,1 (NaClO4) pKlr = 6,80; pKw =13,84 [142]
Ег3+ Т.-д. рК1Г=9,65 [ИО]
Ст. эл. 25 0,3 (NaClOJ рК1Г =7,99 [ЮО]
» 25 0,3 (NaClOJ ->0 рК1Г ~7,99 1 РК1Г=7,46 * / [Ю1]
» 25 3,0 (LiClOJ р/(^щ = 17,4 [Ю4]
» 25 3,0 (LiClO4) рК^щ=17,2 (Н2О); 17,4 (D2O) [Ю5]
» Распр. 25 25 3,0 (NaClO4) 0,2—0,8M Er»+ рК1Г =9,0 [226]
0,1 (LiClO4) рК1Г —5,5 [108]
Ст. эл. 30 0,1 (NaClO4) рХ1Г = 6,56; рК^ =13,84 [142]
Тт3+ Т.-д. 25 рК1г =9,5 [ИО]
Ст. эл. 0,3 (NaClO4) рЛ1Г =7,95 [ЮО]
» 25 0,3 (NaClOJ ->0 рК1г=7,95 1 рК1Г=7,42 / [Ю1]
Распр. 25 0,1 (LiClOJ pKir —4,4 [Ю8]
Ст. эл. 30 0,1 (NaClO4) рК1г=6,36; рК^, =13,84 [142]
Yb3+ Ст. эл. 25 Yb2 (SO4)3
0,005 рК1Г=8,28 1
0,01 рК1Г=7,80 . f I97]
Продолжение табл. 5.2
7— Ион Метод Т, °C |.i, с ре; а Константа равновесия Литература
Yb3+ Т-д. Ст. эл. » » Распр. Ст. эл. 25 25 25 25 30 0,3 (NaCiO4) 0,3 (NaClO4) ->0 0,05 0- - 50% этанол ^0 0,1 (LiClOJ 0,1 (NaClOJ р/(1Г = 9,5 рК1Г = 7,92-8,03 рК1Г=7,92 ) рКгг=7,37 ) рК1г=8,01 (Н2О); 7,76 (25%); 7,36 (50%) . рКг =14,00 (Н2О); 14,30 (25%); 14,66 (50%) | р/<1Г =7,68 (Н2О); 7,34 (25%); 6,80 (50%) J pKjp =4,3 рК1Г =6,24; рК^ =13,84 [НО] [100] [101] [96] [108] [142]
Lu3+ Ст. эл. Т-д. Ст. эл. » Распр. Ст. эл. 20 25 25 25 25 30 0,1 0,3 (NaClO4) . 0,3 (NaClO4) ->0 0,05 0—50% этанола 4) 0,1 (I iCO4) 0,1 (NaClOJ рКц, =14,17; рК1Г =7,57 рК1Г =9,3 рК1Г =7,90 = 7,98 р/(1Г=7,90 1 рК1Г=7,37 рК1Г -7,66 (Н2О); 7,57 (25%); 7,33 (50%) p.Kw, =14,0 (Н2О); 14,30 (25%); 14,66 (50%) рК1Г =7,33 (Н2О); 7,15 (25%); 6,77 (50%) р^,г 8,5 рК1г =6,04; р/С^, =13,84 [Ю6] [НО] [ЮО] [Ю1] [96] [Ю8] [142]
Диаграмма распределения удовлетворительно согласуется с экспериментальными данными о pH начала и конца образования гидроокиси скандия: pH начала осаждения равен 4,9 [127], 4,5—5,2 [128], 5,50—4,75 [129], 5,9 [84]; pH полного осаждения равен 7—9 |[130].
Эти данные весьма близки к аналогичным характеристикам иона алюминия. Ионы Sc3+ и А13* обладают приблизительно равными кислотными свойствами. Однако гидроокись скандия в большей степени, чем гидроокись алюминия, обладает основными свойствами. Это можно оценить по данным о |раствори-мости Sc(OH)3 в щелочах [86, 131, 132].
Для скандия рассчитаны [86] приближенные значения констант образования анионных гидроксокомплексов рК^щ^зззд* рК^щ =48,08, ipK°^ = 63,28.
Частные константы, вычисленные из этих общих констант, изменяются непоследовательно: рК4г=16,7, рК5г= 14,19, рК6г= = 15,20.
При сравнении этих значений с данными для элементов подгруппы алюминия можно заключить, что Sc(OH)3 обладает более основными свойствами, чем АГ(ОН)3, Ga(OH)3, 1п(ОН)3 и Т1(ОН)3. В ряду Sc—Y—La ионы лантаноидов как ион Sc3+, так и гидроокись Sc(OH)3 обладает кислотными свойствами в самой большой степени.
Количественных данных о гидролизе иттрия недостаточно для построения диаграммы распределения его гидроксокомплексов. Для первых двух констант гидролиза можно принять следующие значения: рК1Г = 8,0, рК°^щ=16,8, р/<2г=8,8 [96, 97, 100, 101, 105], хотя они не очень согласуются с pH начала осаждения гидроокиси иттрия, равным 6,8 [84].
Как и в случае скандия, иттрий более гидролизован в вод-но-этанольных смесях, чем ;в воде. С увеличением мольной доли этанола гидролиз усиливается. Аналогичное явление установлено для лантаноидов [96].
Резко отличаются от всех других константы гидролиза иттрия, полученные электромиграционным методом. Из данных работы [102] [следует, что катион Y3+ сильно гидролизуется уже в кислой области. Аналогичные результаты получены в работе [133]: при сорбции микроколичества иттрия на катионите КУ-2 гидролиз Y3+ наблюдался уже при рН^1. Эти результаты противоречат большинству других данных, которые характеризуют иттрий как высокоосновный элемент.
При изучении растворимости гидроокиси иттрия в щелочах выяснилось, что Y(OH)3 обладает в большей степени основными, чем кислотными свойствами. По данным работы [86] приближенно оценены константы гидролиза иттрия с образованием анионных форм: рК^щ =40,15, рК^щ =55,73, рКбгЩ ~ = 71,20.
72
Для лантана определена только первая константа гидролиза p/Cir=9,0 [96, 100, 101]. Полимерные гидроксокомплексы лантана (описаны в работе [107].
Данных о гидролизе Се3+ в литературе недостаточно. Меллером было проведено потенциометрическое определение констант гидролиза Се3+ в сульфатных растворах, где гидролиз мог быть осложнен образованием сульфатных (комплексов [97]<
Данные Меллера согласуются со значением рЛдг=9,3, полученным при измерении pH растворов солей церия стеклянным электродом [111]. С учетом данных о константах гидролиза Y, La и лантаноидов (см. табл. 5.2) можно предположить для Се34- значение рК1Г^9. Подтверждением того, что рК1г для [Се3+ должна быть несколько меньше, чем p/Cr для La34-, служат результаты расчетов по корреляционному уравнению [ПО]. Расчеты основывались назначении р/Сг=10,1 для лантана [107], а в результате их бЬло получено значение рЛ1г = 9,9 для Се3+ [НО].
Установлено, что гидролиз Се3+ начинается при pH >6,3 [134], а гидроокись Се(ОН)3 начинает осаждаться при pH —7,6 [84]. Самостоятельная фаза Се(ОН)3 существует при pH 12г9,3 [135], а по данным работы [84] в интервале pH = 84-10. Электромиграционным методом и методом электрофореза на бумаге получены данные, которые характеризуют Се (III) как весьма сильно гидролизующийся элемент: р/(1г=3,2-?3,4 [102, 109, 112]. -В такой же степени гидролизован и иттрий, (для которого p/Cir = 3,5 [102]. Методом переменной «концентрации» массы сорбента обнаружено, что гидролиз церия начинается при pH = 2,8, а при рН = = 3,15 преобладает катион СеОН2+ [135]. Несомненное химическое (сходство Се(III) со всеми трехвалентными лантаноидами позволяет сделать вывод, что гидролиз Се34- должен происходить аналогично гидролизу Ln3+ и для Се3+ p/Gr=C9,0. Полимерный гидролиз ионов Се(III) описан в работе [134].
Из приведенных в табл. 5.2 данных следует, что ион Се4+ гидролизован в значительно большей степени, чем ион Се3+. Хороший обзор по определению констант гидролиза церия (IV) дан в работе [119]. На основании сведений из работ [118, 119] можно принять значение первой константы гидролиза рК1г —0,7, что согласуется с pH начала осаждения гидроокиси церия (IV), равным 1—1,2 [84]. Полное осаждение происходит при рН = Зч-10, [что соответствует р/Сг= 12,43. Полимерные гидроксокомплексы Се(IV) рассмотрены в работе [137]. Гидролиз Се4+ аналогичен гидролизу U4+ [138].
Для всех остальных лантаноидов в литературе известны только данные о константах гидролиза [трехзарядных ионов. В работах [96, 100, 101] потенциометрическим методом исследовано наибольшее число ионов лантаноидов, поэтому значения, полученные в этих работах, можно рекомендовать .как
73
более надежные. Для прометия данные потенциометрического исследования отсутствуют. Как и для Y и Се (III), методами
Таблица 5.3 Константы гидролиза анионных форм ионов Sc3+, Y3+, Er3+, Yb3+t Lu3-г электромиграции и электрофореза на бумаге получены константы гидролиза иона Рт3+ на несколько порядков большие, чем для других лан-
Элемент р<щ рк°5бгщ Р<Щ таноидов. Нет достаточных данных считать, что основность Pm3+, Y3+, Се3+ значительно меньше, чем основность других лантаноидов. Поэтому для прометия в настоящее время нельзя рекомендовать какую-либо коне-
Er Yb Lu Y Sc 38,59 37,89 37,51 40,15 33,89 53,58 52,52 52,18 55,73 48,08 68,42 . 67,11 66,91 71,20 63,28
танту гидролиза.
Изучение растворимости гидроокисей эрбия, иттербия и лютеция в ^щелочи позволило ориентировочно оценить константы образования анионных гидроксокомплексов [86], из которых рассчитаны общие константы гидролиза, суммирован-, ные в табл. 5.3 вместе с данными для скандия и иттрия.
Из табл. 5.3 следует, что в ряду Ег(ОН)3—Yb(OH)3— Lu(OH)3 основные свойства убывают.
6
ГИДРОЛИЗ ИОНОВ ЭЛЕМЕНТОВ СЕМЕЙСТВА АКТИНОИДОВ
К актиноидам относятся элементы с порядковыми номерами от 89 (актиний) до 103 (лоуренсий). В атомах актиноидов происходит заполнение 5/-оболочек, как в ’атомах лантаноидов 4/-оболочек. В химическом поведении актиноидов и лантаноидов существует много аналогий, но по химическим свойствам актиноиды, особенно в начале ряда, больше различаются 'между собой, чем лантаноиды.
В водных растворах все актиноиды находятся в виде катионов различного строения и заряда. [Основные виды катионов М3+, М4+, МО^ и МО|+ соответствуют валентным состояниям 3, 4, 5 и 6. Оксокатионы МО^~ и МО^+ весьма устойчивы и характеризуются прочной связью М — О. Доказано, что такие катионы находятся в твердых структурах. При различных химических реакциях в растворах эти* катионы остаются неизменными. Данные изотопного обмена с использованием 18О подтвердили существование в растворах катионов UOg4" и РиО|+. Окислительные потенциалы в системах U(VI)/U(V), Pu(VI)/Pu(V) не зависят от pH, что может служить доказательством присутствия в растворах катионов UO^ и РиО^. Это справедливо также для нептуния и америция. При изучении кислородного обмена между уранилом и водой [1] высказано предположение, что в [водных растворах солей уранила происходит взаимное превращение структур
[UO2(H2O)2]2+^[U(OH)24+],
Возможно, что аналогичные гидроксокомплексы наряду с оксокатионами существуют для нептуния, плутония и америция.
Все актиноидные элементы характеризуются сильными основными 'свойствами. Стандартные электродные потенциалы .перехода М3+/М характеризуют актиноиды как сильные электроположительные элементы, аналогичные лантаноидам. Для актиния известно, что это более основной элемент, чем лантан. Соли актиния в разбавленных водных растворах гидролизо-ваны в меньшей степени, чем соли лантана. Вообще же для
75
валентного состояния 3+ характерна несколько большая склонность к гидролизу у актиноидов по сравнению с лантаноидами.
Разнообразие форм валентных состояний актиноидов приводит к тому, что анализ основности ионов этих элементов и склонности их к гидролизу необходимо проводить в рядах однотипных ионов. Такой анализ возможен только на основе количественных характеристик гидролиза, которые приведены в табл. 6.1.
Гидролиз ионов тория изучали многие исследователи различными методами. (В табл. 6.1 приведены данные, относящиеся только к мономерным гидроксокомплексам. Большинство исследователей, изучавших поведение ионов тория при гидролизе, отмечают большую склонность его гидроксокомплексов к полимеризации. При концентрации ^10“2 моль/л в [растворах преобладают гидролитические полимеры и, возможно, поэтому результаты расчета констант мономерного гидролиза ионов тория в системах, где наряду с моноядерными установлено образование полиядерных гидроксокомплексов, существенно различаются [3, 5, 7, 8, 10, 12—15]. Обзоры исследований о гидролизе ионов тория даны в работах [91—98]; гидролитические полимеры описаны в работах [99—103].
На основании данных о растворимости гидроокиси тория в едком натре и хлорной кислоте [Ю4] сделан вывод, что гидроокись тория ведет себя как основание с образованием в растворе катионных форм ThO2+ ^и ThO(OH)+. Эти формы следует рассматривать как Th(OH)2+ и Th(OH) + e учетом данных работы [105], в которой при изучении кристаллической структуры солей тория установлено присутствие Th(OH)^+.
Сведения об условиях образования и устойчивости мономерных гидроксокомплексов тория весьма разноречивы. Гидролиз солей тория начинается, в зависимости от концентрации металла, при различных значениях pH раствора: рН = 3,0± ±0,2 при 10-2—10“4 моль/л Th4+ [7, 106] и pH 0,5 при 0,5— 0,7 моль/л Th44- [99]. Все четыре последовательные константы гидролиза ионов тория определены методом растворимости [9], ионообменным (методом [11] и потенциометрическим со стеклянным электродом [14]. Из них предпочтение следует отдать значениям, определенным ионообменным методом [11]: рЛ1г = 2,36, рЛ2г=3,30, рК3г=3,38, р/<4г=3,55, хотя для первой и второй констант они отличаются ^на 1—1,5 порядка от значений, определенных потенциометрическим методом другими авторами [3, 4, 10, 16]. Диаграмма распределения гидроксокомплексов тория в зависимости от pH (рис. 6.1), Построенная по этим данным, не противоречит значениям pH начала осаждения (pH ж 3,5) и полного осаждения (pH—4±10) гидроокиси тория. Константы, определенные методом растворимости [9]» вероятно, занижены.
76
СЗ
S ч ло
Мономерный гидролиз ионов актиноидоб
ч g
у—*.
1 CQ
^9 О О г? ° т z - о н и t °. «г
£ J Н о о о о
1 — 7
о 1
—< о
*
!Л
сч
к
77
оо
Продолжение табл. 6.1
Ион Метод г, °C Ц, среда Константа равновесия Литература
Th4+ Ст. ЭЛ. Хинг. ЭЛ. Обмен. Н—эл. Хинг. эл. Ст. эл. » » 0 25 95 25 25 20 25 25 1,0 (NaC104) 0,5 (NaClOj 1,0 (NaC104) HNO3, HC1, HC1O, 8-10-4 M Th4+ 3 (KNO3) 0,05 (NaCl, NaC104) 0—50% этанол ->0 pKlr=4,32; pK2r=4,16; pK°^=8,48 рК1Г=4,12; рКзг=3,69; p^=7,81 р^г=2,29; рК2Г=2,21; pK°^=4,50 рКхоси=Н,64; p/C2OCH=10,80; pK3OCH=10,62; 1 Р^4осн=Ю> 45 рК1Г=2,36; р/<2Г=3,20; рКзг=3,38; р/С,г=3,55 j р/С1Г=3,71 pKir=3,61; р/С2Г=4,01 j рКзг=3,55; рК4Г=3,26 рК°бЩ=9>67 рК1г=3,15 (Н2О); рК2Г=3,41 (Н2О) 1 рК1Г=2,77 (25%); рК2Г=3,30 (25%) рК1г=2,17 (50%); рК2Г=3,17 (50%) рК1г=2,64 (Н2О); рК2Г=3,08 (Н2О) рК1г=2,12 (25%); рЛГ2Г=2,89 (25%) рК1г=1,33(50%); рК2Г=2,61 (50%) p/Cltz==14,00 (Н2О); 14,30(25%); 14,66(50%) [10] [11] [12, 13] [14] [15] [16]
Ра4+ Распр. 25 3,0 (LiC104) р/С1Г=0,14; р/Сзг=0,38; р/Сзг=1,25; 1 р^щ=0,52; р/^^1,77 J [17—19]
Продолжение табл. 6.1
Ион Метод т, °C ц, среда Константа равновесия Литература
Раб+ Расчет Распр. 25 3,0 (LiClO,) 5,0 рК,г=—3+1 [20] [18, 21, 22] [23]
рК4Г=1,05; рК5г=4,5 |
рА 5г—4,02
U4+ Магн. воспр. Спектр. » » Расчет Н—эл. Ст. эл. Редокс. пот. Спектр. 20 25 25 10 43 15,2—24,7 3,1—25 25 25 0,1 0,5 8,9-10—4 М U (IV) (Н+, Na+, СЮр 0—2,0 ->0 0,5 (NaClO,) 7,3-10“4М U(IV) ->0 0,5 (NaClO4) ->0 0,19 (НС1О4) 3,0 (NaClOJ 0,5—0,01 М U (IV) 2,0 (NaClO4) pK°6m=2,30 рК1Г=1,15 ) рК1г=1,46 J рК1Г= 1,63 (2,0); 1,56(1,0) рК1Г=1,51 (0,5); 1,33(0,2); 1,25(0,1); 1,03(0,035) рК1Г=0,68 рК1г=1,90 рК1Г=1,12 рК1Г=1.0 рК1Г=0,18 рК =1,37(15,2°); 1,23(20,4°); 1,11(24,7°); рК1г=2,17 (3,1°); 2,03(7,8°); 1,78(16,8°); 1 1,56(24,8°); 1,55(25°) рК1Г=2,0 рК1Г=1,68(НаО); 1,75 (DaO) [24] [25, 26] [27] [28] [29] [30] [31] [32]
Продолжение табл. 6.1
00 О
Ион Метод Т, °C Ц, среда Константа равновесия Литература
и4+ Н. ЭЛ. 25 1,6-10-* М UF. рК1г=2,48; рК2Г=3,735; рКзг=4,165; рК4Г=6,00 [33]
0,0001—0,008 [34]
» 25 UC14, 1,5% этанол рК1Г=— 3,05; рК2Г=1,95; рКзг=3,14
Эл. пр. ПМР 25 0,03 М и (IV) рК1Г=1,52 [35]
Спектр. 20 1,0 (LiClOJ рК1г=1,57 [36]
Расчет 25 ->0 pKir=l, и [37]по[27]
Спектр. 20 10—2—10—3 М и (С1О4)4 рК1Г=1,28 [38]
0,4
» 25 0,5 (НС1, NH4C1) р/С,г=1,56; рК2Г=2,55 1 [39]
0,5 (НС1О4, NH4C1O4) рК1г=1,75; рК2Г=2,42 J
ио?+ рК1г=4,22; рК2Г=3,34 [40]
Хинг. эл. 25 ^0 рК1г=4,3 [41]
Кол. рК1Г=4,5 [42]
Поляр. 25 ->0 10-2—10-3 м UO2C12 рК1Г=4,09 [43]
Ст. эл. Хинг. эл. 20 1,0 (NaClOj РК,г=4,7 рК1Г=4,2; рК2Г=5,2; pKuz=13,8 [44]
Распр. 25 0,1 (NaClO4) [45]
Хинг. эл. 4-Ю-з М и (VI) рК1г=4,19 [46]
Раств. 25 -0 рК =4,14 [47]
Ст. эл. 25—40 10-2—Ю-з М рК,г=5,82 (25°; 0,035); рК1Г=5,40 (25°; 0,35) 1 [48]
U02(ClO4)2 р/(1Г=5,10 (40°; 0,035) 1
Ва(С1О4)2 0,35—0,035 [49]
» 25 0,1 (KNO3) pKir=6,! „ „ г п
Распр. 20 0,1 (NaClO4) рК,г—5,0; рК2Г—6,2; рКзг-а,9 [50]
Ст. эл. 20 рК1г=8,74; рК2Г=8,04; рКИ7=14,07 [51]
Зак. 1864
Продолжение табл. 6.1
Ион Метод Т, °C Ц, среда Константа равновесий Литература
UOf+ Ст. эл. Электромигр. Ст. эл. Хинг. эл. Эл. пр. Кинет-. Ст. эл. » » Раств. Спектр. 25 94,4 16 27 25 25—200 25 25 50—150 50—150 25—350 25 50 100 150 200 300 20 HNO3, KNO3 0,5 1,510-2—Ю-з М ио|+ 3,0 (NaClOJ 2-10—5 м UO2(NO3)2 <4-10—4 1,5 М Mg(NO3)2 2,5 М Mg(NO3)2 UO2(C1O4)2, g .0 10-2— Ю~5 М UO2(NO3)2 10-2—10-3 М UO2(NO3)2 . ->0 10-5—10-е м UO2(NO3)2 1,0 (NaNO3) 1 рК1г=5,7 1 рЯ1Г=4,19 J рК1г=5,05 1 рК1Г=4,59 1 рК1Г=6,10 (>5,86); рК1г=5,2 (25°); 4,7(50°); 4,0(100°); 3,5(150°); ) 3,4(200°) J рК1Г=5,98 рК1г=5,38(5,14) 1 рК1Г=5,53 } рК1Г=4,21 |g/(ir=3,668—2633/7° ] lgtf2r=13,2—7000/7 / lg/(lr=3,668—2633/Т lgKir-3,39 (100°); 2,95 (125°); 2,55 (150°) ) lg/(2r=4,148—3268/7’ / lg/Cir=2795,3/T—4,295 lgK2r= 1249,0/74-3,014 pKlr=5,08; p/<2r=7,20 р7Г1г=4,36; рК2Г=6,88 рК1Г=3,20; pK2r=6,36 pKlr=2,31; pK2r=5,97 рК,г=1>61; pK2r=5,65 pKir=0,58; pK2r=5,19 P^ir=3,43 [52] [53] [54] [55] [56] [57] [ИО] [58] [59] [60] [61]
Продолжение табл. 6.1
Ион Метод Т, °C среда Константа равновесия Литература
Np3+ Ст. эл. 25 0,3 рК1Г=7,43 [62]
Np4+ Ст. эл. спектр. 25 2,0 (NaClO,) - pKlr=2,3 (H2O); 2,5 (D2O) [32, 63]
NpO+ Ст. эл. Раств. 25 25 0,1 (НС1) -0 pKir=8,9 pKir=10,l [25] [64]
NpO|+ » Ст. эл. 25 25 ~0 1,0 (NaClO4) рК1Г=3,37; pK2r=5,44; pK3r=9,7 } pKlr=5,17 [64] [65]
Pu’+ » Распр. 25 23 0,069 (НС1О4) 0,024 (НС1) 0,2 (LiClO4) T5 T3 X) 11 11 ll ' GO oo co io ч to [66] [67]
Ст. эл. 0,1 М HI+0,9 М NaClO4 pKir=5,67 [Hl]
Pu4+ Спектр. » » Редокс. пот. Кинет. 25 25 25 25 25 1,1 (NaC104 NaNO3) 0,5 (NaClO4) 0,5 (NaClO4) 1,0 (NaC104) 2,0 (LiC104) X X X3 x x 11111 tOQlOm W ч^-осл [68] [25] [27] [69] [70]
Продолжение табл. 6.1
Ион J Метод т, гс И. среда Константа равновесия 1 , и . 1 Литература
Pu4+ Редокс. пот. » » Спектр. » Распр. 12,5 0 14,9—34,3 25 15,4 25 20 2,0 (LiClO4) 1,0 (NaClO4) 2,0 (NaC104) HNO3 (1—5) IO-6 M Pu (IV) 1,0 (LiC104) 10-4—10-8 M pu pKlr==l,51; , рЛ1Г=1,77 } '.«><34.3., j рК1Г=1,93(Н2О); 2,42 (D2O); , pKir = l ,72 (H2O); 1,93 (D2O) } pKir=l,54; p7C2r=l,70 P^ir=0,45; p/(2r=0,75 , рКзг=3,3; рЛ4Г=6,3 [70] [71] [72] [73] [74] [75]
PuO^ Ст. эл. 25 0,0026 (HC1) 0,003 (HC1O4) p/C]r=9,6 , pKir=9,7 ) [76]
Pup2+ » » » Ст. эл. Раств. Ст. эл, 25 20 1,0 (NaClO4, NaNCU 10-2—10-3 M Pu (IV) HNO3 1,0 (N3C1O4) p7^ -—5,3 pKlr=5,71; pK2r=5,7I; pK3r=9,7 pKxr=3,33; рК2Г=4,05 . pKjr=3,39; pK2r=5,25; pX3r=9,52 pKir^5,97 [77] [78] [79] [80] [81]
Am3 4- : Электромигр. 15 0,005 (HC1, KC1) H2O:C2H5OH (4:1) lgP”filll^22,60; p^=14,35; pK°®uL=6,10 [82]
Fm3+ » 25 0,1 (LiC104) 1рК1Г-3,8
84
При изучении влияния различных факторов на гидролиз: ионов тория установлено, что гидролиз уменьшается при увеличении ионной силы раствора [(от 0,05 до 0,5) [4] и гидролиз усиливается в водно-этанольных растворах с увеличением мольной доли этанола [16].
Изучение гидролиза ионов протактиния, Ра (IV) и Ра (V), не закончено. Установлено,4 что катионы ]Ра (IV) могут существовать в растворах при концентрации ионов водорода ^ЗМ [17—19]. Для расчета равновесий в этой системе можно поль-
Рис. 6.1. Диаграмма распределения гидроксо-комплексов тория в зависимости от pH раствора:
-Th4+; 2 —ThOH3+; 3 — Th(OH)2+; 4 —Th(OH) + ;
5 — Th(OH)4
зоваться константами гидролиза p/Cir = 0,14; р7<2г = 0,38; рКзг — = 1,25 1[17—19]. Отсутствие четвертой константы не позволяет рассчитать распределение гидроксокомплексов Ра (IV) в зависимости от кислотности раствора.
Константы гидролиза характеризуют Ра как элемент весьма склонный к гидролизу. Ионы Ра (IV) гидролизованы в значительно большей [степени, чем ионы Th (IV), а ионы Ра (V) в большей мере, чем ионы Ра (IV). При растворении гидроокиси Ра (V) в концентрированной НСЮ4 образуются катионы РаО(ОН)2+, (Ра(ОН)2+) и РаО(ОН)+ [18, 21, 22, 107]. При нагревании образуются катионы РаО(ОН)2+, на хо-* лоде — Ра(ОН)2+. Частицы РаО(ОН)2+ со временем гидрати* руются, переходя в форму гидроксокомплекса Ра(ОН)|+. Поэтому константы гидролиза Kir и определенные в работах [18, 21, 22], правильнее записать соответственно как К4г и К5г-Распределение форм протактиния Ра (ОН)2+, Ра (ОН) + и Ра (ОН) 5 в зависимости от pH раствора [22] согласуется с данными И. Е. Старика о [сорбции микроколичеств Pa(V) [108].
85
Тидролиз Pa(V) существенно отличается от гидролиза U(V), 'Np(V), Pu(V). Сравнение рК5г(Ра)^4,5 с pKir(Np, Pu) 9— 10, т. е. сравнение констант равновесия перехода от однозарядных катионов к нейтральным молекулам свидетельствует о большей склонности к гидролизу Pa(V), которая сопоставима с гидролизуемостью ионов ниобия и’тантала.
Гидролиз ионов U(III) количественно не охарактеризован. В работе [91] была сделана попытка теоретически оценить константу гидролиза иона U(III) методом Каспера (pKir= = 9,2). Растворы 'солей U(III) крайне неустойчивы, поэтому получить достоверные экспериментальные данные очень трудно. При измерении pH 0,02 М растворов солей U(III), U(IV) и U(VI) получены значения равные соответственно 2,4; 1,3 и 2,9 [91], из чего можно заключить лишь, что ионы U(III) тидролизованы в меньшей степени, чем ионы U(IV). Вывод о большей гидролизуемости ионов U(III) по сравнению с ионами UO*+, нельзя признать обоснованным. Диспропорционирование U(III) в водных [растворах приводит к образованию U(IV), гидролиз ионов которого может приводить к искаженным представлениям. Существует мнение [109], что гидролитические характеристики U(III) должны быть близки к характеристикам Pu (III) и лантаноидов. Тогда можно считать, что для ионов U3+ p/(ir~7.
Обзор исследований гидролиза U(IV) дан в работе [39]. Все четыре константы гидролиза были получены при изучении гидролиза UF4 [33] и не [могут быть рекомендованы для расчетов равновесия реакций образования гидроксокомплексов. Комплексообразование с фтор-ионами затрудняет гидролиз, из-за чего pKir гораздо больше соответствующих констант, полученных другими авторами. Можно принять следующие значения констант для 25° С: рК1г = 1,5 [25—27, 30, 32, 35, 36, 39], р7<2г=2,5 [34, 36],рКЗГ = 3,1 [34].
Отсутствие четвертой константы гидролиза не позволяет рассчитать распределение гидроксокомплексов U(IV) в зависимости от pH.
Образование коллоидных форм гидроксокомплексов U(IV) происходит при рН>3 даже при концентрации урана 10~6— 10“7 моль/л [112].
Степень полимеризации прямо пропорционально зависит от pH раствора и концентрации урана [39]. С повышением температуры гидролиз соединений U(IV) усиливается [28—30], константы гидролиза увеличиваются с разбавлением и уменьшением ионной силы [раствора [25—28]. Тяжелая вода в незначительной степени затрудняет гидролиз U(IV) [32].
Количественных данных о гидролизе ионов U(V) в литературе нет. Установлено, что ионы U5+ в свободном состоянии не существуют, a U(V) находится в водных растворах в виде оксокатионов UO+ которые устойчивы при pH = 2—4 [25]. 86
в такой же форме существуют ионы нептуния и плутония, соответственно NpO^ и РиО^~, схема гидролиза которых
МО^ + Н2О МО2 (ОН) + н+
и константы гидролиза [соответственно р/Сг=8,9 и рКг=9,7.
По аналогии для UO+ р/Сг должен иметь порядок 8 [25]. Таким образом, в этом небольшом ряду актиноидов гидролиз уменьшается с увеличением порядкового номера элемента. !
Гидролиз ионов уранила, UO|+ из соединений всех актиноидов изучен наиболее подробно. Образование мономерных гидроксокомплексов обсуждается в работе [59, 61, 92—94, 109]; полимерный гидролиз описан в работах [113—115].
Для ионов UO^ можно принять следующие значения констант гидролиза: при 20—25° С p/Gr —5,0 [44, 50, 53, 55, 61], р^-бД [50].
Построенная по этим константам диаграмма распределения гидроксокомплексов уранила в зависимости от pH (рис. 6.2)
100
^80
Рис. 6.2. Диаграмма распределения g гидроксокомплексов U (VI) в зави- g 60 оимости от pH раствора:
1~ UO|+; 2 —UO2OH+; 3 — UO2(OH)2 ^0
20
2,0 ^0 4,0 5,0 60 7,0 8,0 О,0рН
хорошо согласуется со значениями pH осаждения гидроокиси уранила: pH начала осаждения ^5, pH полного осаждения ^8.
Трехокись урана обладает амфотерными свойствами. Из данных о растворимости трехокиси урана по кислотному и основному типу следует, что UO3 обладает в большей степени основными свойствами [47]. Гидролиз солей уранила протекает медленно, и равновесие устанавливается в течение многих часов [51].
. Анализ гидролиза ионов урана в различных валентных состояниях позволяет сделать вывод, что наиболее склонны к гидролизу ионы U (IV). С учетом ориентировочных оценок гидролитической способности ионов U(III) и UO^ можно расположить ионы урана по склонности к гидролизу в ряд U(IV)> >U(VI)>U(III)>U(V).
В водных растворах существуют ионы нептуния в различных валентных состояниях: Np(III), Np(IV), Np(V), Np(VI) и Np(VII). Количественные данные о константах гидролиза
87
ионов нептуния ограничены. Можно | лишь заключить, что склонность к гидролизу усиливается в ряду Np(IV) >Np(VI) > >Np(III) >Np(V), который аналогичен такому же ряду для: урана.
В тяжелой воде гидролиз ионов Np(IV), |как и ионов других металлов, протекает труднее, чем в обычной [32, 63]. Данные А. И. Москвина [64] о константах гидролиза NpO^ существенно (отличаются от других результатов [65] и не соответствуют гидролитическому поведению уранила и плуто-нила.
Для трехзарядного катиона Ри3+ определены константы первой ступени гидролиза p/Gr = 7,2—7,4 [66] и рК1г=3,8 [67]. Предпочтительны данные Крауса [66], (по которым ион Pu34* ведет себя как слабогидролизующийся катион, аналог Np34- и других трехзарядных актиноидов и лантаноидов.
В большинстве работ, авторы которых исследовали гидролиз Pu(IV), констатируется значительная гидролизуемость ето ионов. Определена первая константа гидролиза Pu(IV) pKir — 1,3“ 1,7 [25, 27, 68—74]. Данные Ю. П. Давыдова
Рис. 6.3. Диаграмма распределения гидроксокомплексов Pu (IV) в зависимости от концентрации ионов водорода:
/ - Ри4+; 2 —PuOH3+; 3-Pu(OH)2+; 4-Ри(ОН)+;
5 —Ри(ОН)4
pKir= 1,54 и рК2г=1,70 [74] указывают на близость количественных (характеристик гидролиза U(IV) и Pu(IV). Наиболее полно определены все четыре константы гидролиза Pu(IV) в работе [75]: pKir=0,45, pK2r = 0,75, рК3г = 3,3, рК4г = 6,3. По этим значениям рассчитано распределение гидроксокомплексов Pu(IV) в зависимости | от концентрации ионов водорода (рис. 6.3).
Гидролиз ионов РиО+ изучен Краусом [76]: pKir = 9,64-9,7. Количественные характеристики гидролиза плутонила у
88
разных авторов различны. Для первой константы гидролиза эти различия существенны: рАлг = 3,34-3,4 [79, 80] и 5,3—5,7—5,97 [77, 78, 81]. Различия для второй константы гидролиза менее значительны: рА2г=4,05 [79], 5,71 J78] и 5,25 [80]. Для третьей константы гидролиза ионов плутонила данные Крауса и А. И. Москвина согласуются лучше: рКзг = 9,7 [78] и рК3г = 9,52 [80].
Можно принять для расчетов гидролитических равновесий ионов плутонила значения констант [79, 80]: p/Cir = 3,4, рК2г= — 5,2, рКзг-9,5.
По этим значениям рассчитано распределение гидроксокомплексов плутонила в зависимости от pH (рис. 6.4).
Полимерные гидроксокомплексы Pu(VI) рассмотрены в работах [81, 116]. Гидролиз ионов плутония в различных валентных состояниях усиливается в ряду
Pu (IV) > Pu (VI) > Pu (III) > Pu (V).
Аналогичный ряд установлен для нептуния и урана.
Рис. 6.4. Диаграмма распределения гидроксокомплексов плутонила в зависимости от pH раствора:
1 — PuO^+j 2 —PuO2OH + ; 3— PuO2(OH)2; 4-PuO2(OH)~
Америций в водных растворах существует преимущественно в трехвалентном состоянии. Различными исследователями получены противоречивые данные о гидролизе ионов Аш3+, о pH начала гидролиза, и о значениях констант гидролиза. По данным И. Е. Старика и Н. Н. Гинзбурга, которые получены методами ионообменной сорбции, электромиграции и сорбции на стекле, при концентрации америция в растворах 10“8—10-9 г-ион/л катион Ат3+ существует в сильнокислых и слабокислых растворах от 1 н. HNO3, до рН = 4,0-44,5. Гидролиз начинается при рН>4, а при pH = 64-7 образуются коллоидные формы [117]. Другие авторы полагают, что при pH = 64-7 гид
89
ролиз ионов Am3+ только начинается [118]. При изучении сорбции Am (III) на поверхности полированной платины в азотнокислых растворах установлено, что максимальная сорбция наблюдается при рН = 2-?3. Полагают, что в этом интервале и начинается гидролиз Am (III) [119]. Систематические исследования гидролиза трансурановых элементов, в частности Am (III), разнообразными методами проведены Ю. С. Корот-киным [86, 120], который установил, что гидролиз Am (III) начинается при низких значениях pH (~1,0). Константы гидролиза иона Ат3+, определенные в работах [83, 85, 86], согласуются удовлетворительно, можно принять рК1г^З,0. В работах [85, 86] принимают р/Сгг — 4,0. В работе французских исследователей [84] рК1Г = 5,92. Эти величины согласуются с константами гидролиза трехзарядных ионов актиноидов, определенных для кюрия, берклия, калифорния и эйнштейния этой же группой авторов [84, 88—90].
Для констант гидролиза иона кюрия Ст3+, как и для америция, получены несогласующиеся данные. Для констант гидролиза берклия, калифорния, эйнштейния и фермия известны лишь единичные определения.
Систематический анализ гидролиза ионов актиноидов провести в настоящее время трудно. Разнообразие валентных состояний и окислительно-восстановительных свойств ионов элементов этой группы, радиоактивность, необходимость исследования при низких концентрациях элементов, образование радиоколлоидов, сорбция элементов на стенках сосудов — все это усложняет изучение реакций гидролиза и приводит к невоспроизводимым результатам при определении констант, к расхождениям в результатах, полученных различными методами и различными авторами.
Изучение гидролиза протактиния, америция и других актиноидов проводилось при концентрациях элемента 10~8— 10 -12 г-ион/л. При этом процессы сорбции ионов радиоактивных элементов, с одной стороны, служили исследователям индикатором гидролиза, а с другой — затрудняли и искажали изучение равновесий в растворах. Очевидно, поэтому при исследовании гидролитических равновесий в растворах с концентрацией радиоактивных элементов 10-10 г-ион/л методами сорбции и электромиграции исследователи обычно получали значительно большие константы гидролиза и обнаруживали гидроксокомплексы в более кислых растворах, чем при использовании потенциометрии или экстракции в диапазоне концентраций 10~2— 10~5 г-ион/л. В настоящее время нет достаточного числа экспериментальных данных, чтобы можно было получить точные значения констант гидролиза некоторых актиноидов.
7
ГИДРОЛИЗ ИОНОВ ЭЛЕМЕНТОВ IV ГРУППЫ —
Ge, Sn, Pb, Ti, Zr, Hf
В четвертой группе наблюдается сглаживание различий между Элементами главной и побочной подгрупп. Поэтому гидролиз катионов подгруппы титана и подгруппы германия можно рассматривать совместно.
Из элементов IV группы лишь для свинца характерно двухвалентное состояние с преобладанием основных свойств. Хотя катион РЬ4+ известен и свойства его описаны, в водных растворах он неустойчив из-за высокой гидролизуемости и окислительной способности. Известны также катионы Ge2+, обладающие основными свойствами и сильной восстановительной способностью. Трехзарядный катион Ti3+ существует в водных растворах, проявляя основные и восстановительные свойства. Олово в водных растворах существует в виде катионов Sn2+ и Sn4+, причем для Sn(II) более характерны основные свойства, для Sn(IV) — амфотерные. Преобладающее валентное состояние для элементов четвертой группы 4+
У Ge (IV) и Ti (IV) кислотные свойства преобладают над основными, у Zr (IV) и Hf (IV) —наоборот.
Основные свойства у Ge(IV) выражены незначительно, так что долгое время ставилась под сомнение возможность существования в кислых и слабокислых водных растворах его катионов. Однако в последние годы прямые эксперименты по электромиграции и комплексообразованию в водных растворах подтвердили существование катионных форм Ge (IV). Следует отметить, что появление катионов Ti4+, Ge4+, Sn4+ возможно при высокой кислотности раствора (>2—5 н. Н+) и в реальных условиях трудно достигнуть области кислотности, где бы эти катионы преобладали.
Константы гидролиза элементов IV группы приведены в табл. 7.1.
Гидролиз ионов Ti(III) изучен недостаточно. Очевидно, из-за восстановительных свойств Ti3+ наряду со склонностью к образованию гидроксополимеров нельзя было рассчитать все три константы гидролиза. На основании данных работ [1, 2] можно принять, что для иона Ti3+ p/Cir=2,04-2,5. Это значение согласуется с результатами работы [77], в которой методом ЭПР
91
со Таблица 7.1
to
Мономерный гидролиз катионов элементов IV группы
Ион Метод г, °C ц, среда Константа равновесия Литература
TiH- Ст. ЭЛ. 15 0,0008—0,014М Ti3+ р/СИ7=14,34; рК1Г=1,40
0,25—1,50 (KBr, KI) (ц->0)
25 -»0 pKw,= 14,00; рХ1г=1,29
25 ->0 =13,68; рК1г=1,42 [1]
0,25 рК1г=2,27 (15°); 2,18(25°); 2,30(35°)
0,50 рК1Г=2,29 (15°); 2,20(25°); 2,42 (25°)
0,75 р/<1г=2,32 (15°); 2,25 (25°); 2,23 (35°)
1,50 рК1г=2,11 (15°); 2,00(25°); 2,13(35°)
Эл. пр. 25 3,0 (КВг) p/(ir=2,55 [2]
Ti4-F Тенз. 0 Переменная рК^щ=1,57 [3]
Обмен. 1,5(НС1О4) рК3г—о > з (4]
» 25 рКзГ=1 >3 [5]
Раств. р/Сзг=1,5
Распр. 25 0,1 (НС1О4, NaClO4) р/С2Г=1,8; рКзг=2,4 1 [6]
1.10-4-5.10-з М Ti (IV) рК4Г=2,1 J
Обмен. 18 ->0 рКй/=14,24 - I
Продолжение табл. 7.1
Ион Метод т, °с • щ среда Константа равновесия Литература
TiM- Обмен. 18 -0 РК1Г=-3,72; р/(2Г=—2,97 рКзг=1,73; рК4Г=3,24 [7]
Турб. 5-IO"3 М Ti (IV) рК1г=1,22; рК2Г=0,89 [8]
Распр. (1-5)-IO”4 М Ti (IV) 1,0(НСЮ4, NaClO4) рК1г=0,16; рК2Г=0,58 рКзг=0,92; р/<4Г=1,05 1 [9]
Стектр. 25 Ы0~5М Ti (IV) 10% этанол, HNO3, KNO3
0,1 0,3 0,5 1,0 рК1Г=-0,10; рК2Г=0,31; рКзг—0,64; p/Gr=0,96 р/<1Г=— 0,15; рХ2Г=0,027; р/Сзг=0,62; р/(4Г=0,96 рК1г=-0,29; рК2Г=0,11; рКзг=0,42; рК4Г=0,67 рК1г=— 0,40; рК2Г=—0,025; рКзг=0,24; рК4Г=О,55 р/<1г=—0,70; рК2Г=—0,32; р/<зг= —0,05; р/С4Г=О, 26; lg/fir=O,098+0,6065 р; 1g^r=—0,314+0,647 р; lg/C3r=—0,643+0,706 р; lgK4r=-0,954+0,726 р [Ю]
Раств. Обмен. Поляр. 25 25 0 рК1г=— 3,96; рК2Г=— 3,21; ) рКзг=1,49; J рК4Г=3,00 рК1Г= -0,03; рК2г=2,77; р/Сзг=6,77 [Н] [12]
о 4^
Продолжение табл. 7.1
Ион Метод т, °C Ц, среда Константа равновесия Литера -тура
ZH+ Распр. 25 2,0(НСЮ4) pKlr=0,22; рК2Г=0,62; рКзг=1,05; рК4Г=1,17 [13, 14]
Раств. 25 1 —Юн. NaOH рК«сн=4,72; рК6г=9,28 [15]
» 2,0(NaCl) рК2Г—- —0,36 [16]
Распр. 25 1,0 (NaClO4) рк1г=—0,32; рК2Г=0,06; р/Сзг=0,35; рК4Г=0,64 [17]
25 1,0 (LiClOJ pKir=—0,56; р/Сг =—0,20;
рКзг=0,07; рК4Г=0,17
-+0 рК1г=—0,58; рК2Г= — 0,80; [18, 19]
рКзг=-0,34; р/(4Г=-0,13
Турб. 20 HCiO4 рК°?щ=3,31; рКзг=156; p/cir=3i63 [20]
Сорбц. 25 H2SO4, р/<!£щ=3,50 [21]
Спектр. 25 ~>0 рК1Г=— 0,05; рК2Г=0,28; рКзг=0,43; р/(4Г=0,80
0,1 Рк1г=—0,08; рК2Г=0,25; рКзг=0,62; рК4Г-0,84
0,3 р/С1г=— 0,12; рК2Г=0,22; рКзг=0,40; рК4Г=0,76 [22]
0,5 рК1Г=-0,15; рК2Г=0,20; рКзг=0,38; рК4Г=0,72
1,0 рК1Г=—0,30; рК2Г=0,07; рКзг=0,32; р^г=0,66
lg/Cir=O,0489+0,246 р;
lg/C2r=—0,2807+0,200 р;
lgK3r= —0,4327+0,110 р; [22]
lgK4r= —0,8035+0,143 р
Продолжение табл. 7.1
Ион Метод т, °C Ц, среда Константа равновесия и Литература
Zr4+ F - эл. Распр. 25 20 НС1О4, NaClO4 То же рК1г=0,55 p/flr=—0,3 [23]
Т.-д. [22] ->0 pXir= -0,76; рК2Г=—0,19; | рКзг=0,21; рК4Г=0,81 [24]
Сорбц. 25 ->0,H2S04,10~10M Zr4+ рК1Г=1,98; рК2Г=2,51 ]25]
НН+ Раств. 25 1— 10 W NaOH рК°сн=4,04 рК5г=9,96 , [15]
Распр. 25 l,0(NaC104) РХ1Г=—0,13; рК2Г=0,23 ] рКзг=0,43; рК4Г=0,52 J [26]
Сорбц. 25 h2so4 рК^щ=2,396 [21]
Спектр. 25 0,1 (KNO3) 0,3 0,5 рК1Г=— 0,05; рК2Г=0,39; рКзг=0,80; р/<4г=1,50 ' рК1Г=— 0,07; рК2Г=0,42;. рКзг=0,91; рК4Г=1,19 рК1Г=— 0,10; рК2Г=0,43; рКзг=1,06; рК1Г=1,44 [27]
1,0 р/(1Г——0,15; р/<2Г=0,32; рКзг=0,77; рК4Г=0,96 1 lg/Cir=O, 043+0,181 р J [27]
F-эл. Распр. 25 20 HC1O4, NaClO4 To же рК1Г=1,10 ) рК1Г=0,155 ) [23]
О
Продолжение табл. 7.1
Ион Метод т, °C Ц, среда Константа равновесия Литература
Н11+ Сорбц. 10-4—10-ь м НН+ H2SO4, р+0 рК°^=3,57 [25]
Ge4+ Хинг. ЭЛ., Ст. эл. 25 КС1 р >0 рК5Г=8,73 [28]
Эл. пр. 18 Нас. р-р GeO2 рК6г=6,92 [29]
» 20 Нас. р-р То же рК-г=7,30 [30]
Н-эл. 20 0,025—0,1 М Na2GeO3 рК5г=8,585; 1 рЛвг=12,72 / [31]
Эл. пр. 25 Нас. р-р GeO2 рК6Г=8,5^9,0 [32]
эдс 12 рК6г=9,1; рК61=12,7 [33]
» >0,04 М GeO2 рК5г=9,07 [34]
Раств. 25 р/<5г=11,56 [35]
Н-эл. 25 3 (NaCl) рК1Г = 14,07; рК6г=12,43 [36]
Ст. эл. 25 0,5 (NaCl) pKw,=13,70; рК5Г=9,02 1 рК6г=12,24 1 [37]
Распр. 25 8,0 (LiCl) рК1г= -0,88; рК2Г= -0,51; 1 рКзг=-0,26; рК4Г=0,40 J [38]
Зак. 1864
Продолжение табл. 7.1
Ион Метод Г, °C М-, среда Константа равновесия Литература
Ge«+ Спектр. 25 HNO3, KNO3 4% этанол ^0 0,1 0,3 0,5 1,0 рК1Г=—0,11; рД-2Г=0,26; рКзг=0,52; рК4Г=0,72 рК1г=—0,18; рК2Г=0,19; рКзг=0,47; рК4Г=0,70 рК1г=—0,33; рК2Г=0,07; рКзг=0,37; рК4Г=0,68 р/С1г=—0,45; рК2Г=—0,05; рД’зг=0,27; рК4Г=0,60 рК1г=—0,82; рК2Г=-0,45; рКзг= —0,20; рК4Г=0,004 [39]“
Sn2+ Н-эл. 25 -+0,НСЮ* ₽К1г=1,7 [40]
Раств. 25 -0, НС1, NaOH рК1Г=2,07; рК2Г=5,00; рКзг=9,55 } [41]
эдс, ам. эл. 0—45 3,0 (NaCIO*) рК1Г=1,81 (0°); 1,70(25°); 1,63(35°); 1,60(45°) } [42, 43]
Ст. эл. 25 2 (NaNO3) р#1Г=3,2 [44]
Ам. эл. Ст. эл. Спектр. '25 3 (NaCIO*) 2.10-зм Sn2+ рК1г=3,92 рК1Г=—1,39 [45] [46]
Ст. эл. Ам. эл. эдс 25 3,0 (NaCIO*) рК1г=3,70 рК^*«==15,8 [47] [48]
25 3 (NaCIO*) рК°б*Ц=17,48; 17,2 [100, 101]
Продолжение табл. 7.1
Ион Метод Т, °C Ц, среда Константа равновесия Литература
Sn4+ Раств. 100 КОН (NaOH) рН=7ч-11 рК1Г=7,125; рК2Г=7,155; рКзг=7,54; 1 рК4Г=9,06; рК6г=9,24 f [49]
» 25 ^0 рК6г=12,4 [50]
300; 90 атм 0,5(Н3ВО3) рК4Г=15,26 [51]
Спектр. 25 l,0(KNO3) 8% этанола рЛ1г=-0,57; рК2Г=-0,11; 1 рКзг=0,33; рК4Г=1,22 / [52]
Из данных работы [52] 25 0,1—1,0 Д) lgKlr=0,090+0,480 и,; 1 lgK2r=—0,31+0,488 р.; lgKsr=—0,55+0,235 и; lgK4r=—1,01+0,654 р. [53]
То же ~>0 рК1Г=—0,49; рК2Г=0,19; 1 рКзг=0,88; рК4Г=2,03 J [54]
Раств. 25; 50 -.0 рК6г=12,4(25°); 11,6(50°) [55]
рК°бЩ=22 [48]
Pb3+ Поляр. рК§Сц’=29,85 [58]
Раств. 25 NaOH Р^2г+р^зг=22,7 [59]
Ст. эл. 15 Pb (NO3)2 рК1Г=7,7 [60]
Раств. Ст. эл. 25 18 -+0 Pb (NO3)3 рК1Г=6,18; рКзг=10,92 (красная РЬО); ] рКзг= 11,13 (желтая РЬО) > рК1Г—7,78 (р, - 0) J [61]
Продолжение табл. 7.1
Ион Метод Т, °C И. среда Константа равновесия Литера -тура
РЬ3+ ** со со Кол. Ст. эл. Поляр. » » Ам. эл. Ст. эл. Ам- эл. Ст/ эл. Поляр. Ст. эл. » 20 25 25 25 25 .25 25 25 Ba (NO3)2 Pb (NO3)3 Pb (NO3)2 2-10“4M 0,06—0,6 1,0 (KNO3) -Л) 2,0(NaC104) 0,3(NaC104) 3,0(NaC104) 0,3(NaC104) 3,0(NaC104) ->0 2,0(NaC104) 0,1—0,0001 M Pb2+ 2,0(NaNO3) 0,01—0,2M Pb2+ pKlr=7,778+0,996l/jT—0,617 p+0,432 p2 1 pKir=7,664-8,44 pKlr=8,37 (0,06); 8,66(0,6) ptfir=7,1; рК2Г=10,1; рКзг=11,5; р^щ=28,7 PK°^=28,05 рЛ°бЩ=29,38 pKir=7,8 1 рК1г=7,9 J рК1г=7,8; рК2Г=9,4; рКзг=10,8; рКг=13,76 р/С1Г=7,9; рК2Г=9,6; рХзг=11,5; pXuz = 14,22 рК°бЩ=;28,1 РК1Г=7,93; рК^.==13,97 рК1Г=8,84 [62] [63] [64] [65] [66] [67] “ [68] [69] [70] [71] [72]
Продолжение табл. 7.1
Литература 173] [74] [75] [76] [99] [74] [53]
- / ч
О сч Т[ и К СХ о о? со It =г сх СО СО 11 =г >О X осч СХ СО . -о O-.LO -—'О ° ° ° сч £ ° о ю Т\О)
Константа равновесия и fe ex о сч 11 Is ex со со It Is £ <o II ^4 1 рК1Г=6,15 (25°); 5,66(50°); 4,86 (1( 4,26(150°); 3,78(200°); 3,40( 3,08(300°); 2,81 (350°) р/( =10,70 (25°); 9,97(50°); 8,87 f 8,12(150°); 7,59(200°); 7,24 7,00(300°); 6,87(350°) рКзг=П,02(25°); 10,38(50°); 9,62 9,34(150°); 9,39(200°); 9,70 10,18(300°); 10,80(350°) рК1Г=:7,0 рК2Г—9,5 О' ю 1 ||_ сч а OD Т[ & 1Л |[ сх о 7 i[ 5< £Х lgKir=0,0804-0,42 ц; lgK2r=—0,3234-0,40 ц; lg/( =—0,6244-0,38 ц; lg^r=-1,044-0,58 И .
Ц, среда 3,0 (NaCl) © t 0,1 м нсю4 О t 0,1 (KNO3) 10-7— 10-5М Pb (NOs)2 О t =L 0,1—1,0 i -
О о 25 30 25—350 1 25
Метод S S < 1 Т.-д. « з о . Раств. Поляр. Нс Н
Ион Си РЬ*+
too
установлено образование основного продукта гидролиза ТЮН2+ при рН^З.
Анализ данных о константах гидролиза Ti(IV) сделан В. П. Васильевым с сотр. [11]* в целях расчета стандартных термодинамических характеристик гидроксокомплексов Ti (IV) в водном растворе. Наиболее удовлетворительными признаны данные, полученные в работе [7].
Достаточно полно и надежно гидролиз ионов Ti(IV) описывается константами, полученными авторами в работе [10]. На основании этих данных для ионной силы, равной 1,0, построена диаграмма распределения гидроксокомплексов Т1(ОН)/4^>+ в зависимости от концентрации ионов водорода (рис. 7.1). Это
Рис. 74. Диаграмма распределения гидроксокомплексов Ti (IV) в зависимости от концентрации ионов водорода (ц=1,0):
7 — ТН+; 2 — TiOH3+; 3 — Ti(OH)2+ ;
4~ Ti(OH) + ; 5-Ti(OH)4
распределение хорошо согласуется со значениями pH осаждения гидроокиси Ti(IV) [78]: pH начала осаждения ^0, pH полного осаждения ^2.
Обзоры данных о состоянии ионов циркония в водных растворах приведены в работах [79, 80], обзор данных о гидроксо-комплексах циркония — в работе [24]. По экспериментально определенным [22] константам гидролиза рассчитаны термодинамические характеристики гидроксокомплексов циркония [24]. Экстраполяция констант гидролиза по экспериментальным данным от ц раствора к ц = 0 удовлетворительно совпадает с термодинамическим расчетом из данных при ц = 0,1. Данные работ [17, 22] можно рекомендовать для расчета равновесий образования гидролизованных форм ионов циркония. А. С. Соловкин с сотр. [13], обрабатывая экспериментальные данные Конника и Мак Вея [14], получили несколько меньшие значения константы гидролиза, чем авторы работ [17, 22]. Рассчитав по собственным экспериментальным данным константы гидролиза ионов цирко
* В статье [11] допущена описка: приведено значение 1g р3=47,68, тогда как в работе [7] 1g р3“46,68. Из-за этого в [11] искажены значения рК3г и Р*4г-
101
ния [18, 19], А. С. Соловкин получил несколько большие величины, чем в работах [17, 22]. Из расчета термодинамических констант гидролиза циркония [18, 19] было получено аномально высокое значение второй константы гидролиза, что объяснено авторами этих работ индивидуальными свойствами циркония и тем, что вхождение первой гидроксильной группы в координационную сферу Zr4+ значительно облегчает вхождение следующей ОН-группы. Важные результаты получены Г. А. Ягодиным с сотр. [21, 25], установившими, что в сульфатных растворах концентрация 10~3 моль/л циркония (гафния) является «пороговой» для образования гидролитических полимеров. Однако определенные ими константы гидролиза значительно занижены, по-видимому, из-за конкурирующего образования сульфатных комплексов.
Диаграмма распределения гидроксокомплексов циркония в зависимости от концентрации ионов водорода, построенная для |а=1,0 по данным работ [17, 22], показана на рис. 7.2. Она хоро-
Рис. 7.2. Диаграмма распределения гидроксокомплексов Zr (IV) в зависимости от концентрации ионов водорода (ц= 1,0):
I - Zr*+; 2 —ZrOH3+; 3-Zr(OH)|+; 4-Zr(OH)+;
5 —Zr(OH)4
шо согласуется с данными Конника [14] о существовании в 2н. НС1 ионов Zr4+ и ZrOH3+, с данными И. Е. Старика [81] относительно областей существования всех мономерных гидролизованных форм ионов циркония и со значениями pH осаждения Zr(OH)4 [78]: pH начала осаждения «2, pH полного осаждения 4—>10.
Если рассмотреть основные и кислотные свойства гидроокиси циркония так, как это было сделано для Al, Ga и In, то можно установить, что, хотя они выражены слабо, кислотные свойства у гидроокиси циркония значительно сильнее, чем основные.
102
Основные свойства можно охарактеризовать величиной
*юСн = [Zr (ОН) Л [OH-]/[Zr (ОН)4] = KwlKK (рЛ\0СН = ч = 14,00 — 0,65 = 13,35) [17, 22],
кислотные — уравнением
^Хкисл ~ [zr (он;л [н+j/izr (OH)j = к5г (Рк5г = Рк
1КИСЛ
= 9,28) [15].
Таким образом, количественные данные, которые известны в настоящее время, противоречат сложившимся представлениям о преобладании основных свойств гидроокиси циркония над кислотными.
Получены константы гидролиза, достаточно полно описывающие процесс образования гидроксокомплексов Hf [15, 26, '27]. При изучении гидролиза в сульфатных растворах установлено, что при концентрации Ю-3 моль/л гафния, как и в случае циркония, не образуются гидролитические полимеры, а при концентрации H2SO4 более 2 н. не происходит гидролиз, что -обусловлено, по-видимому, образованием сульфатных комплексов [21, 25]. Очевидно, из-за образования этих комплексов гидролиз уменьшается.
Электромиграционным методом установлено, что в растворах HCI и HNO3 при их концентрации больше 1 н. образуются анионные комплексы Hf [82].
Для расчетов можно рекомендовать значения констант гидролиза гафния, полученные в работах [26, 27] как более полные. Диаграмма распределения гидроксокомплексов гафния в зависимости от концентрации ионов водорода, построенная по этим данным, для р,= 1,0 показана на рис. 7.3. Она удовлетворительно согласуется с данными о pH осаждения гидроокиси гафния £78J: pH начала осаждения «2, pH полного осаждения 4—>10.
Основные свойства Hf(OH)4 можно выразить константой рК1осн= 14—0,96— 13,04 [27].
Характеризующая кислотные свойства Hf(OH)4 константа гидролиза р/С1кисл = Р^5г=9,96 [15].
Таким образом, кислотные свойства гидроокиси гафния выражены сильнее, чем основные. Для гафния, как и для циркония, такой вывод необычен и противоречит принятым представлениям об их основности.
Хотя из данных работ [15, 22, 27] можно сделать вывод о •большей основности гафния по сравнению с цирконием примерно в 10 раз, кислотные характеристики гидроокисей циркония и гафния необходимо уточнить.
О состоянии ионов Ge(II) в водных растворах в литературе немного данных. Ge (ОН) 2 дает не истинные, но коллоидные 103
растворы в среде NaOH, так как обладает лишь основными свойствами [85]. Приписываемые ей кислотные свойства объясняются присутствием в Ge(OH)2 примеси соединений Ge(IV) [85]. Из данных о растворимости Ge(OH)2 в НС1О4 вычислена первая константа основности [84]. Изучение состояния в водных растворах соединений Ge (Г!) затруднено из-за сильных восстановительных свойств двухвалентного германия.
Состояние ионов Ge(IV) в водных растворах изучено более полно [86, 87]. Если кислотные свойства GeO2 сомнению не подвергались, то существование катионных гидроксокомплексов*
Вис. 7.3. Диаграмма распределения гидроксокомплексов Hf (IV) в зависимости от концентрации ионов водорода (р,= 1,0):
1 — Hf‘+; 2 — HfOH3+; 3 - Hf(OH)|+; 4—Hf(OH) + ;
5- Hf(OH).
Ge(IV) в водных растворах долгое время оставалось дискуссионным [56, 57, 88—92]. В настоящее время их существование-не отвергается [86, 87, 92].
Значения констант ионизации метагерманиевой кислоты, приведенные в табл. 7.1 [29, 30], резко завышены, так как были получены, по-видимому, в опытах с недостаточно чистой GeO2. В работе [35], наоборот, значение этой величины резко занижено. Оно получено в опытах по изучению растворимости GeO2B растворах NaOH. При этом, по-видимому, не было учтено-существование разных форм GeO2 с сильно различающейся растворимостью в щелочах. Остальные значения р/Сдис и р^дис (соответственно, рК5г и рЛбгХт приведенные в табл. 7.1, близки друг к другу, в том числе и термодинамическое значение рХ1дис [28]. Наиболее часто используются значения рК1ДИс и рКгдис метагерманиевой кислоты [31].
С учетом констант гидролиза катионных форм германия [39] и более достоверных констант ионизации метагерманиевой кислоты, было рассчитано распределение катионных и. анионных гидроксокомплексов германия как в сильнокислой, так и в-104
сильнощелочной среде (рис. 7.4). Это распределение удовлетворительно согласуется со значением pH начала осаждения гидроокиси германия (^0) [78], в интервале рН=14-7 молекулярная форма Ge(OH)4(H2GeO3-H2O) доминирует.
Кислотные свойства гидроксокомплексов Ge(IV) выражены: значительно, сильнее (примерно в 105 раз), чем основные: рКбг=рК1дис~ 8,73 [28], р/С1осн~рКиг—рЛ4г~14—0,004 —14.
Количественные характеристики гидролиза ионов Sn2+ весьма разноречивы. Например, для pKir получены значения ог —1,4 [46] до 3,9 [45]. Некоторая сходимость для первой констан-
Рис. 7.4. Диаграмма распределения гидроксокомплексов Ge (IV) в зависимости от концентрации ионов водорода (pi=l,0):
7 — Ge4+; 2 - GeOH3+; 3-Ge(OH)2+; 4-Ge(OH) + ; 5 - Ge(OH)4; £-Ge(OH)“ Z о о ;
7 —Ge(OH)|~
ты гидролиза получена потенциометрическим методом при работе с водородным и амальгамным электродами и методом растворимости [40—43]: pKir= 1,74-2,1. Очевидно, значение pXri=2,0 можно принять как достаточно надежное, так же как и значение рКзг=рК1кисл = 9,55 [41].
Данные Гаррета [41] наиболее систематичны, однако распределение гидроксокомплексов Sn(II), рассчитанное по полученным им константам (рис. 7.5), удовлетворительно согласуется с данными о pH осаждения гидроокиси Sn(II) [78] лишь в отношении форм Sn2+ и 5п(ОН)з: pH начала осаждения ~2, pH полного осаждения 4—10, pH начала осаждения из щелочного раствора «11.
Из значения p/Qr=5,0 [41] следует, что при pH = 5 в растворе существует «50% формы 8п(ОН)г. Но в интервале рН=4-4-105 все олово находится в форме Sn(OH)2 [78]. Очевидно, значение-р/С2г = 5,0 [41] несколько завышено и нуждается в дополнительном определении.
Кислотные свойства Sn(OH)2 характеризуются рКзг= = рК1кисл = 9,5 [41], а основные pKiocu=pKw—рК2г=9 [41]. Эти значения свидетельствуют об амфотерности 8п(ОН)г.
105
Гидролиз ионов Sn(IV) изучался в основном геохимиками. При этом гидролитические равновесия исследовали методом растворимости при высокой температуре и давлении {49,51,55]. Данные работы [49] безусловно ошибочны.
При нормальных давлении и температуре гидролиз мономерных катионов Sn(IV) изучен лишь в работе [52]. Из данных, полученных для ионной силы 1,0, вычислены значения констант гидролиза, для интервала ц=0,1^-1,0 [53]. В основу расчета взята прямолинейная зависимость констант гидролиза ионов
Рис. 7.5. Диаграмма распределения гздроксокомплек-сов Sn (II) в зависимости от концентрации ионов водорода:
1 — Sn2+; 2 — SnOH+; 3 — Sn(OH)2; 4 —Sn(OH)~
элементов IV группы от четвертого ионизационного потенциала последних. В другом расчете констант гидролиза ионов Sn(IV) .для ц-*0, произведенном также с учетом данных работы [52] и констант гидролиза катионов Ge (IV) [39], в основу положено :неверное предположение об одинаковой зависимости констант гидролиза катионов Sn(IV) и Ge(IV) от ионной силы. Получен--ные константы, естественно, отличаются от констант, рассчитанных в работе [53], в которой выведены корреляционные уравнения Ig/Cir^Hp) в интервале р.=0,1-Н,0 (см. табл. 7.1).
По данным работы [52] рассчитано распределение катионных гидроксокомплексов Sn(IV) в зависимости от концентрации ионов водорода (рис. 7.6). Это распределение удовлетворительно согласуется с данными о pH осаждения Sn(OH)4 [78, 93]: pH начала осаждения «0, pH полного осаждения 1—13, pH начала осаждения из щелочного раствора а; 11.
Основные свойства Sn(OH)4 можно охарактеризовать величиной pA\ocH=pKw—р/<4г=14-4-1,2=12,8. Это значение свидетельствует о слабо выраженных основных свойствах Sn(OH)4, значительно меньших, чем у Sn(OH)2 (р/Сюсн — 9).
Для характеристики кислотных свойств Sn(OH)4 можно привести величину р-К5г=рКн<исл= 12,4 [50, 55]. При этом получается, что кислотные свойства Sn(OH)4 несколько сильнее 206
выражены, чем основные. Но сравнение рКшисл для Sn(II) (9,5) и для Sn(IV) (12,4) указывает на существенно большую кислотность Sn(OH)2 по сравнению с Sn(OH)4. Следовательно, Sn(OH)2 по сравнению со Sn(OH)4 обладает более сильными
Рис. 7.6. Диаграмма распределения гидроксо-комплексов Sn (IV) в зависимости от концентрации ионов водорода (ц=1,0):
1 - Sn4+; 2 - SnOH3+; 3 - Sn(OH)2+; 4~Sn(OH)+;
5 — Sr(OH)4
и основными, и кислотными свойствами. Это маловероятно. Причина такого противоречия заключается, очевидно, в неточно определенной константе кислотной ионизации гидроокиси
Рис. 7.7. Диаграмма распределения гидроксокомплексов Pb (II) в зависимости от pH раствора:
/-РЬ2+; 2 —РЬОН+; 3 — РЬ(ОН)2; 4- РЬ(ОН)"
Sn(IV). Исходя из того, что и Sn(OH)2 и Sn(OH)4 из щелочных растворов осаждаются при одинаковом значении pH, можно предположить близость их констант кислотности.
107
Гидролиз катионов РЬ(П) изучен весьма подробно, установлено образование гидролитических полимеров и рассчитаны их термодинамические характеристики [94—97]. Из значительного количества констант гидролиза РЬ(П), характеризующих мономерные гидроксокомплексы, можно принять: р/<\г=7,5 [60, 62, 63, 65, 68, 69, 71], рХ2г=10,0 [65, 69], рК3г=П,5 [65, 69, 73, 76], рХ°бщ = 17)5 [65, 69, 76], рК^=29,0 [58, 65, 67, 69].
Приведенные значения констант гидролиза следует рассматривать как ориентировочные. С их помощью рассчитана диаграмма распределения гидроксокомплексов. РЬ(П) в зависимости от pH (рис. 7.7). Это распределение хорошо согласуется с pH осаждения РЬ(ОН)2 [78]: pH начала осаждения равен 7,8, pH начала осаждения из щелочного раствора — 13,0, pH полного осаждения равен 9—10.
Несмотря на амфотерный характер РЬ(ОН)2, основные свойства гидроокиси свинца (II) выражены сильнее: рК1КПсл = = рХзг=П,5, pXtocH = pKiv—р/<2г= 14ч-10=4,0.
Рйс. 7.8. Диаграмма распределения гидроксо-кбмплексов Pb (IV) в зависимости от концентрации ионов водорода:
/_рь++; 2 — РЬОН3+; 5— РЬ(ОН)2+; 4-РЬ(ОН)+;
5 — РЬ(ОН)4
Для ионов свинца в состоянии окисления +4 экспериментально определенных констант гидролиза в литературе не приводится. Из термодинамических расчетов свободной энергии образования РЬ(ОН)2 из РЬО и аналогично РЬ(ОН)4 из РЬО2 [74] получена зависимость Д/70 от числа ОН~-групп, связанных с ионом свинца. Из этих данных рассчитаны свободная энергия образования всех гидроксокомплексов Pb(OH)/4“i)+ и значения констант гидролиза: lgKir==—1,8, lgK2r=5,0, lg/<3r=2,04 IgA^r— = 1,5.
108
Правомерность такого подхода вызывает сомнение, а порядок изменения констант гидролиза не совсем понятен.
В работе [53] рассчитаны константы гидролиза ионов Pb(IV) из уравнений зависимости констант гидролиза четырехвалентных Ti, Zr, Hf, Ge, Sn от четвертого ионизационного потенциала этих элементов. Такие уравнения получены для всех четырех констант гидролиза при значениях ионной силы раствора 0,1— 0,3—0,5—1,0. С помощью этих уравнений, как отмечалось выше, были рассчитаны константы гидролиза ионов Sn(IV) при р.= = 0,1—0,3—0,5. По значению ионизационного потенциала свинца £/4=39,0 эВ рассчитаны константы гидролиза ионов Pb(OH)j(4-i)+, которые коррелируют с величиной ионной силы раствора. Уравнения приведены в табл. 7.1. При р,= 1,0 рассчитано распределение гидроксокомплексов РЬ(ОН)^4-,)+ в зависимости от концентрации ионов водорода (рис. 7.8). Диаграмма распределения полученных таким путем констант соответствует экспериментальным данным о полном осаждении РЬ(ОН)4 в 0,1 н. растворе HNO3 [98].
8.
ГИДРОЛИЗ ИОНОВ ЭЛЕМЕНТОВ V ГРУППЫ —
V, Nb, Та, As, Sb, Bi
В водных растворах существуют катионные формы ионов всех перечисленных элементов, в том числе мышьяка. Для ниобия и тантала характерна степень окисления +5. Ванадий в водных растворах образует ряд катионов от V2+ до VO+, причемг с увеличением степени окисления усиливаются кислотные свойства его окислов. Мышьяк в водных растворах встречается в форме As(III) и As(V). Если у As(III) существуют катионные формы, то у As(V) реализуются только формы мышьяковой кислоты и ее анионов. Для сурьмы характерны два состояния: окисления: Sbflll), для которого катионные формы известны, и Sb(V). О катионах пятивалентной сурьмы в литературе никаких данных нет. Для висмута наиболее характерно образование катионов Bi (III). В соответствии с положением в периодической системе ион Bi3+ обладает самыми основными свойствами в ряду As—Sb—Bi.
Из катионов элементов V группы лишь для ионов висмута, Bi3+, и сурьмы, Sb3+, известны все константы гидролиза. Из катионов в высшей степени окисления +5 такие данные получены лишь для ниобия и тантала. Данные о константах гидролиза приведены в табл. 8.1.
Гидролиз ионов ванадия. Из всех катионов ванадия, находящихся в разных валентных состояниях, двухвалентный ванадий, V2+, наименее подвержен гидролизу. Значение pKir=6,5 (1] — единственное в литературе. Это значение близко к pKir других двухзарядных катионов с примерно равными ионными радиусами.
Ионы V3+ подвергаются гидролизу в значительной степени. Данные о гидролизе V3*, полученные в различных работах, в основном сходятся. Для первых двух ступеней гидролиза значения констант находятся в интервале: pKir=2,54-3,0 [2, 8], рК2г=3,8 ч-4,0 [3, 5, 6].
Из-за отсутствия рКзг нельзя представить распределение гидроксокомплексов в зависимости от pH. Наиболее систематические работы по гидролизу ионов V3+ проведены Пайдовским [5, 6, 80], который исследовал также процессы гидролитической
ПО
111
Продолжение тайл. 8.1
1—* .... .. . .
to —>— — Ион Метод Т, °C И» среда Константа равновесия Литература
V4+ vo2+ V (V) vo»+ Ст. ЭЛ. » » Спектр, ясэ Распр. Ст. эл. » Распр. Ст. эл. Ст. эл. Спектр. » < Н—эл. Ст. эл. Спектр. Ст. эл. Спектр. » Распр. Спектр. 20 25 18—20 25 25 25 25 25 18-20 25 25 25 25 25 20 -+0 3 (NaCl4) 0,005—0,05 MV(IY) 0,02 М VOSO4 H2SO4 HNO. HC1O4 5,4-10-s м V (VI) 0,1 3,0 (КС1) -0 0,1 .0,1 (LiClOJ <0,01 3,0 (NaCIOJ 0,5 (NaCl) 0,5 (NaCl) -0 1,0 (NaCl) 10-4-10-» MV (V) . 0,5 (NaClO4) 0,2 (NaClO4) рК1Г=4,77 pKir=5,36 рК1Г=6,0 K=[VO2+][H+]/(VOH3+] №3; lgK=0,48 K-4-, lgK=0,60 /W; lgK=0,90 рК1Г=2,57; рК2Г=2,96 pK1=6,4(>5,6); pK2=3,6 pKir=5,67 рК1Г=2,65; рК2Г=3,08 pKir=5,05 рКы-=рК2д—9,52 рК^щ=2,79; рКвг=рКвд=13,34 рКзд=13,0 pKw = 13,7; j рКзд=7,7; рКзд=13,0 > рКэд=8,95 ) Р*зд=[М . 1 fe=pKiA=4,3+4,8; рК8Г=рК2д=8,44; рКзг= 1 =3,5+4,0 ’ рК3г=3,3; рК1д=3,7 | РКзЯ=П’13 [10] 13] [11] [12] [13, 14] [15] [16] [17] [Ю1] [Ю] [18] [19] [20] [21] [22] . [23] [24]
и
Зак. 1864
Продолжение табл. 8.1
Ион Метод Г, °C Ц, среда Константа равновесия Литература
V (V) VO3+ Ст. эл. » Кинет. Ст. эл. Редокс. пот. Распр. Н—эл. Ст. эл. Спектр. Ст. эл. Распр. Спектр. Распр. » Обмен 20 25 20 25 25 20 25 10—40 25 15—35 20 10-2— 10~4М V (V) 0,1 (NaCL) 5-10“5—6*10-8 NaVO3 ->0 0,5 (NaClOJ 0,5 (NaCl) 0,1 (КС1) 0,1 (NaClOJ 3,0 (NaClOJ 2,5* IO-5—1,10-2 M NaVO3 0,1 (NaClO4) 1,0 (NaClO4) 0,1 (NaClO4) ->0 P*w=3’04’ Р^2Д=8,23; ркзд=13,5 p/\ др — О, ЛЛ рКзг+рК4г=5,255 рКзг=3,20; рК1Д=3,78 рК2Д=7,83 рК3г+рК4г+рКзг= 16,17 ) рК3г~гРК4г=7,94 J рКзг=3,35; рК1д=3,45 рК3д=7,62 рК1Г=1,84; рК2Г=2,38 р/Сзг=3,70 (15°); 3,70 (25°); 3,65 (35°); рК4Г= 1 =3,80 (15°); 3,80 (25°); 3,75 (35°) | рКзг=3,32; рК4Г=3,89 lgKir=0,86; lgK2r=0,65 [25] [26] [27] [28] [29] [30] (из данных [20] [26, 31] [32] [33] [17, 100] [34] [35] [102]
№ ( V) Раств. » 18—20 18—20 HNO3 lgKir=19,0; lgK2r=15,6; lgK3r=ll,0; lgjf4r=5,7; | IgKer—0,4; lgKer= —7,4 J lgK3r= H.55; lgK4r=7,32; ] lgK6r=0,4; lgK6r= —7,4 f [36] [37]
Продолжение табл. 8.1
114 fl «* 115
Ион Метод Т, °C (1> среда > Константа равновесия Литература
Та (V) Раств. 18-20 HNO3 lgKir=15,4; lgKar=12,4; 1 lg^sr=®>2’> lgK4r—3,5; } lgK6r= —1,2; lgKer=-9,6 [36, 37]
As3+ Эл. пр. Раств. Эл. пр. Кинет. Эл. пр. Кол.; эл. пр. Кол. Ст. эл. Эл. пр. Ст. эл. » Раств. Ст. эл. Раств. Ст. эл. Кол. Ст. эл. » 25 15 25 25 25 . 25 25 25 18 25 25 25 20 25 15—45 25 20 0,1 М НС1 -^0 ^0 NaOH, НС1 1,0 (NaCl) ^0 КС1 0,105—1,45 0,1 <КС1) Ацетон + 44 % Н2О Диоксан + 50,9 % НаО Диметилформамид + + 48+7% Н2О рК1осн=14,74; pKsr= —0,74; рК1д=7,68 рКосн=14; pK3r 0 | рК1Д=8,58 pK„=9,20 n on J рКосн= 14,89; pKsr=-0,89 рКхд—7,27 рК1д=9,22 рК1д=9,22 рК1Д=9,05 рК1д=9,24 рК1д=9,08 pKH=9,62 pKH=8,85 рКосн= 13,75; 1 рКзг=0,25; рК1д=8,13 f рК2Д=12,13; рКзд=!3,4 оК =9 294 1 рК“=9’,134 (0,105); 9,093 (0,59); 9,111 (1,45) / рК1д=9,614—0,01847; ] рК1д=9,15 1 рХ1д=9,38 рК1к= 10,35 рК1Д= 10,68 Р*и= Ю,31 [38] [39] [40] [41] 42] [43[ 44] 45] (46] [47] [48] [49] [50] [51] [52] [53]
Продолжение табл. 8.1
Ион Метод т, °C Ц, среда Константа равновесия Литература
As3+ Распр. Калор. Спектр. Раств. Спектр. 25 25 22 22 0,1—3,0 0,1—0,3 NaCl 0,5 1,0 1,0 (НС1О4 , NaClO4, LiC104) lgK°6m= 1,95+3,95 рК1д=9,23 рХ1д=9,32; I рК1д=9,21 } рК8Г=—0,82 ) рКзг=-1,17 рКосн=15+14,52 f [54] [63] [55] [56]
As (V) Эл. пр. Кол. Эл. пр. Ст. эл. » » Эл. пр. Кинет. Хинг. эл. Ст. эл. 25 25 25 18 25 25 25 0-50 25 ->0 ->0 1,0 (Na2SO4) рК1д=2,36 рК2Д=4,39; рКзд=9,21 p^2Д=7,08 рК1д=2,25; рКзд=6,77; ) рКзд=П,53 } рКзд=11,4+11,3; ) рКзд=9,21 } рК2Д==6,80 рК1д=2,09 (0°); 2,11 (5°); 2,14 (10°); 2,16 (15е); 1 2,19 (20°); 2,22 (25°); 2,27 (30°); 2,30 (35°) 1 2,33 (40°); 2,38 (45°); 2,42 (50°) J рК2д=7,05 (0°); 7,03 (5°); 7,015 (10°); 7,00 (15°)• 6,99 (20°); 6,98 (25°); 6,97 (30—45°); 6,98 (50°) рК1д=2,01'4+5-IO-3 (Т= +40.0)2 ' | рАзд=6,971+5-IO-® (Г_39)4)2 рЛ2Д=6,39 [40] [41] [43] [45] [50] [57] [58] [58] [59]
Продолжение табл. 8.1
Ион Метод Т, °C Ц, среда Константа равновесия Литература
As (V) Раств. Ст. эл. 20 рК1д=2 66; рК2Д=5,89; рКзд=И>52 [60]
10 ->0 рК=2,49; рКзд^.Об; р£зд=!Чп
» 25 рК*д=2,19; рК2д=6,94; рКзд= ,50 [61]
35 рК^=1,95; рК2д=6,87; рКзд= ,64
50 рХ1д=2,15; рК2д=6,80; рКзд—11,92
» 25 0—1 оК,„=2,301 (0); 2,315 (0,1); 2,335 (0,2); 1 2 3§6 (0,3); 2,393 (0,4); 2,421 (0,5); 2 461 (0 6); [62]
2,493 (0,7); 2,521 (0,8); 2,559 (0,9); 2,596 (1,0) )
Калор. 25 0,1—0,3 рК1д=2,25; рК2д=6,77; рАзд=11,53 [63]
Ст. эл. 25 0,1 (КС1) РК1Д=2,26; рКзд=6,76; РАЗД= 11,29
Н2О—метанол (ЛГ) рК1д=2,26 +3 01 N-,
/V от 0 до 0,298 рКзд=6,76 +4,80 АГ;
мол. дол. рКзд=11,29+8,35 N-,
Н2О—этанол (W) РК1Д=2,26 +2,50 N-, [64]
N от 0 до 0,227 рК2Д=6,76 +3,78 А;
мол. дол. рКзд=11,29+6,39 А;
Н2О—диоксан (AQ рК1д=2,26 +3,36 N-,
N от 0 до 0,171 РКЗД=6,76 +5,16 N-,
мол. дол. рКзд=11,29+8,94 N
Продолжение табл. 8.1
Ион Метод Т, °C Ц, среда — Константа равновесия Литература
Sb3+ Раств. » Спектр. Раств. Распр. Спектр. 25 25 23 25 25 23 ->0 НС1О4 ц<0,1 5,0 (NaClO4, NaNOJ 3,0 (NaClO,) 1,0 (NaClO4) рА2Г=1,4; рКзг=Ц,81 рАзг=0,87; рА.г=11,0 рЛзг=1,42 рА2Г= —0,86 рА3г= 1,23 рК1г=— 0,61; рК^ —0,01; рКзг=1,05 [65] [67 [68 [69 [70 [7Г
ВР+ Поляр. Ст. эл. ам. эл. Распр. Раств. Ст. эл., спектр. Спектр. Поляр. 25 25 25 25 22 15 0,2 (NaClO.) 3,0 (NaClO.) HNOg 0,1 (NaClO,) 1,0 (NaClO,) 0,1 (KNO3) 0,3 0,5 1,0 1,0 (NaClO,) 11111 1 11 Д i V11 22 £ 2 Ь 23 сл сл •— ослф “ О СП СИ ° С J • СО -II ’• о X ° о о зт "о <0 (О ►+ OJ U ts ю « N *1 ” т “ и и li li li Л li 1 СО -Г г Г Г Г Г >. § <? 5? 5? Й * о -• to СЛ Z—ч СО U -о X о W W W СО ' ll II II ll КЗ го ЬО ю X со X X. О to ОО (О [72] [73, 74] [75] [77] [78] [79] [ЮЗ]
полимеризации в растворах солей V(III) (6, 80]. Результаты, полученные С. А. Щербаковой [9], резко отличаются от всех ранее полученных данных. Хотя ею определены все три константы, отдать им предпочтение трудно, так как метод распределения чаще приводит к более высоким значениям констант гидролиза, чем прямые методы.
Катионы ванадила, VO2+ или V(OH)|+, и гидроксокомплек-сы VO(OH)+ и VO(OH)2 существуют в водных растворах в широком интервале кислотности, от 0,002 до 3 М НС1О4 [11]. Лишь методом ЯМР в сильнокислых растворах (10 н. HNO3, 5,5 н. НС1О4, 18М H2SO4), обнаружено существование более высокозарядного гидроксокомплекса VOH3+.
Большинство авторов рассматривают первую ступень гидролиза ионов ванадия (IV) по уравнению
VO2+ + Н2О VO (ОН)+ + н+.
было получено с помощыб стек-
Первая константа гидролиза док рА1г^5ч-6 [11, 15, 16].
сотр. [13, 14, 17], рК1Г=2,б и рК2г=3,1. Значение р/^г [17] близко к значению, которое лянного электрода [15].
Диаграмма распределения гидроксокомплексов вайадия (IV), построенная по данным работ [13, 14, 17], показана на
ионов ванадила (IV) имеет поря- -По данным Н. А. Краснянской с
Рис. 8.1. Диаграмма распределений гидроксокомплексов V (IV) в зави\ си мости от pH раствора:
;_уо-+; 2 ~ VOOH + ; 3 — VO(OH)2
рис. 8.1. Выход формы VO(OH)2 при рН = 4-4-5 по этой диаграмме согласуется с экспериментально определенными значениями pH осаждения, равными 4,1—4,6 [10]. Обзор состояния в водных растворах ионов ванадия низших степеней окисления дан , в работе [81]. "
О существовании катиона V54- в водных растворах, очевидно, говорить нельзя. Во всяком случае, в литературе не исследованы и не обнаружены равновесия с участием этого катиона. Большинство исследователей, обсуждая гидролиз катионных форм V(V), рассматривают как основной катион ванадия VO+* J Однако в работах [17, 18] рассмотрены равновесия с участием 1
118
Кбг К2д
катиона VO3+ и определены константы его гидролиза. В связи -с этим можно принять следующую схему гидролиза ионов V (V):
К1Г = [VOOH2+] [H+]/[VO3+]; К2г = [VO2+ ] [H+]/[VOOH2+];
K3r = [VO2(OH)J [H+]/[VO2+] = [HVO3] [H+]/[VO2+];
K4r = к1д = Ivor] [H+]/[HVO3] = [VO2 (ОВД [H+]/[VO2 (OH)] =
- [H2vor] [H+]/[H3VO4];
= [HVorj [H+]/[H2vor] = [Hvori [h+]/[vo3-];
Ker = кзд = [VO43-] [h+]/[hvo|-j.
Первые две константы гидролиза ионов V(V) достаточно надежно определены в работе [17], для р/С4г—рКбг близкие результаты получены разными авторами.
На основании анализа литературных данных можно принять константы гидролитического равновесия ионов V(V): p/<ir= 1,8 и рХ2г=42,4 [17, 100], р/'Сзг=3,3 |23, 24, 27, 29, 32, 35], рК4г=3,8 [24, 29,/34, 35], р/<5г=8,0 [20, 23, 26, 30, 33], р/С6г= 13,0 [18—20, 22, 26]/ С помощью этих констант рассчитано распределение всех онных форм V(V) в разбавленных водных растворах в за симости от кислотности (рис. 8.2). Это распределение
80
60-
20-tfL.
-/ 9^10 12 13 14Чд^Г]
10. 1 0,1 0,01 [Н*],н. 0fl1 0,1 1,0 [ОН-],».
Рис. 8.2. Диаграмма распределения ионизированных форм V (V) в зависимости от концентрации ионов водорода:
J —VO3+; 2 —VOOH2+; 3-VO(OH)+; 4 - HVO3; 5 - VO~; 6 — HVO^; 7 - VO|~
согласуется с данными авторов работы [82] об изоэлектрической области ванадиевой кислоты (pH = 3,6-^4).
Полимеризация V(V) в водных растворах начинается при концентрации ванадия ~4-10-5—1 -10—4 г-ион/л [82, 83]. В сильнокислых растворах существует катион VO3+ [17, 18, 84].
Обзоры состояния V (V) в водных растворах приведены в работах [85, 86].
119
По склонности к гидролизу катионы ванадия в разных степенях окисления можно расположить в ряд
vo3+ > vo2+> v3+ > vot > v2+.
В случае V(V) сравнение кислотных и основных свойств свидетельствует о том, что HVO3 обладает в большей степени кислотными свойствами.
Соединения ниобия и тантала относятся к наиболее гидролизующимся. Равновесие в водных растворах соединений этих элементов осложнено гидролизом, гидролитической полимеризацией, образованием двойных и смешанных комплексов.
В литературе приведены все пять констант гидролиза катионных форм ниобия и тантала, константы кислотной ионизации гидроокисей этих элементов. Однако экспериментально были определены лишь константы основности и кислотной ионизации [36, 37]. Все остальные константы рассчитаны на основании теоретического соотношения между ступенчатыми константами нестойкости комплексных соединений.
Из экспериментальных данных [36, 37] получены константы:
^оси = [М (ОН)4+] [OH-J/IM (ОН)6] \
(рК^н= 14,6, рСан= 13,0); \
Квг = = [МОГ] [н+]/[М (ОН)6] \
(рК™ = 7,4, p/Cja = 9,6), \
с помощью которых сделан вывод о преобладании кислотных свойств над основными у Nb(OH)5 и Та(ОН)5. Основные свойства у тантала выражены сильнее, чем у ниобия (в 40 раз), К кислотные свойства — сильнее у ниобия, чем у тантала (в\ 160 раз). Это согласуется с положением элементов в периодической системе. Обзор литературных данных о гидролитическом поведении и состоянии в растворах ионов ниобия и тантала дан в работе [37].
Закономерное усиление основности и уменьшение кислотных свойств в ряду V—Nb—Та четко подтверждается при сравнении констант кислотной ионизации ванадиевой, ниобиевой и танталовой кислот:
P^(V) (3,8) < р№ь (7,4) < р/СГ (9,6).
Сравнение склонности к гидролизу однотипных ионов МО+ не приводит к таким же четким результатам:
р/Г<4 (3,3) > рЛТаО^ (1,2) > pKNbo^ (—0,4). г г г
В принципе этот ряд должен совпадать с рядом кислотности. Причина инверсии ванадия не ясна.
120
Существованием катионов As(III) в водных растворах, обусловлены равновесия:
As3+ + Н2О AsOH2+ + Н+, К1Г;
AsOH2+ + Н2О^ As (ОН)^ + Н+, К2г;
As (ОН)2+ + Н2О As (ОН)3 + Н+, Лзг;
As (ОН)3 H2AsO3- + Н+, А4Г = Л1Д;
H2AsOF HAsO23- 4- Н+, К№ = К2Д;
HAsOi- AsO33~ + Н+, Квг = А3д.
Первые две ступени гидролиза не охарактеризованы константами равновесия. Значение Азг может быть рассчитано из констант основности мышьяковистой кислоты
/Сосн= [AsO+] [OH-]/[H3AsO3] = [As(OH)2+] [OH-]/[As(OH)3],
которые определены в некоторых работах [38—40, 49, 56]. Интервал значений константы основности мышьяковистой кислоты можно принять равным (14-3) -10~15, как это установлено А. А. Ивакиным с сотр. [56], что согласуется с данными [ 38, 40]. Первая константа ионизации мышьяковистой кислоты (Л1Д) определена достаточно надежно в работах многих исследователей. Ее значение находится в интервале рК\д= = 9,1ч-9,3 [39, 42—46, 51, 52, 55]. Вторая и третья константы ионизации определены лишь в работе [50]: рКгд—12,1 и рКзд=13,4. Кислотные свойства мышьяковистой кислоты выражены значительно сильнее основных.
Ионизация мышьяковой кислоты может быть представлена реакциями:
H3AsO4 H2AsOr + Н+ (К1Д); H2AsO? HAsO2- + Н+ (/<2д); HAsO2~^AsO^ + H+ (К3д).
Для мышьяковой кислоты основные свойства не установлены, а для констант кислотной ионизации можно рекомендовать значения: рК1Д = 2,2, рЛ^д^бД; рК3д=Н,5 [61]. Близкие результаты в аналогичных условиях получены Бриттоном [45], а по отдельным ступеням ионизации в работах [57, 58, 60, 62, 63]. Кислотные свойства мышьяковой кислоты выражены значительно сильнее, чем у мышьяковистой.
Гидролиз ионов Sb3+ исследован недостаточно. В работе [65], очевидно, определены значения не рЛзг и рДзг, а рК2г= = 1,4 и рЛ4г= 11,81. В таком случае для рА3г у разных авторов получены близкие значения [65, 68, 70, 71]. По константам
121
гидролиза, определенным в работе [71], рассчитано распределение гидроксокомплексов Sb(III) в зависимости от кислотности раствора (рис. 8.3). Это распределение удовлетворительно согласуется с pH начала осаждения 5Ь(ОН)з, равном Ю,5—0,75, и pH конца осаждения гидроокиси, равном 2,2 [87], а также с областями существования ионов SbOH24- и Sb(OH)^ [69, 70]. В справочнике [88] приведены значения: pH начала осаждения ж 0, pH осаждения из щелочного раствора ~ 11. Этому соответствует рЛлг=р^1кисл~11,0[67]. Таким образом, кислотные свойства гидроокиси сурьмы характеризуются величиной рК4г=рК1кисл~ И, а основные рЛосн = Р#гг—рКзг~ ж 13. Преобладание кислотных свойств над основными выражено в меньшей степени, чем в случае мышьяка, что согла-
Рис. 8.4. Диаграмма распределения гидроксокомплексов висмута в зависимости от концентрации ионов водорода (ц—1,0):
1 - ВР+; 2 - BiOH2+; 3 - Bi(OH)+;
4 —Bi(OH)3
Рис. 8.3. Диаграмма распределения гидроксокомплексов Sb (III) :в зависимости от концентрации .ионов водорода (р—1,0):
J — Sb3+; 2 —SbOH2+; 3-Sb(OH)+;
4 — Sb(OH)3
«суется с положением этих элементов в периодической системе.
Значение первой константы гидролиза иона Sb34- характеризует этот катион как один из наиболее гидролизующихся. Ионы Sb(V) гидролизуются еще в большей степени. Изучены процессы гидролиза хлоридных комплексов Sb(V). Для реакции
SbCir + Н2о [Sb 1ОН) С1Б]“ + Н+ + Cl-
определена константа равновесия, равная 4,5 • 104 [89, 90]; .2,34 -104 [91]; 1,15 • 104 [92].
122
Для реакции
[Sb (ОН) С15]~ + Н2О [Sb (ОН)2 C1J ~ + Н+ + СГ
константа равновесия равна 2,7-103 (89], 4,44-103 [91], 3,05 X X Ю3 [92]. Третья константа гидролиза равна 1,67 • 103 [92].
Обзор работ о гидролизе ионов висмута дан в статье [93]. Полимерные гидроксокомплексы висмута изучались в работах [94—97]. В случае мономерных форм хорошая сходимость наблюдается для первой константы гидролиза рА\г = 1,3-е-1,6 [73, 74, 77—79] и для второй рК2г= 1,84-2,0 [72, 79]. Все три константы мономерного гидролиза висмута определены лишь в работе [79]. На основании этих констант рассчитано распределение гидроксокомплексов Bi(OH)(3~Z)+ (рис. 8.4). Это распределение хорошо согласуется со значениями pH осаждения гидроокиси висмута [98]: pH начала осаждения ~ 1,6—2,6, pH полного осаждения равен 4,7—4,8.
По данным работы [88], осаждение Bi(OH)3 начинается при pHж 2; полное осаждение происходит при pH = 74-8.
Кислотные свойства гидроокиси висмута можно охарактеризовать четвертой константой гидролиза [77, 99], равной рК4г=рК1кисл — 124-13. Константа основности рКОсн=рКтг— —рКзг= 11,6 [79]. Следовательно, в случае висмута основные свойства, хотя и незначительно, но преобладают над кислотными.
Если сравнить кислотные и основные свойства гидроокисей элементов сверху вниз по подгруппе, то от As к Sb и Bi видно закономерное увеличение основных рЛ^П1) ~ 144-14,3; Р^оснП1) * 13; рЛ^1П)=х 11,6 и уменьшение кислотных свойств ptfAsaii) =9-49,5; pKSbdii)^ji; pKBi(iii)=-124-13.
В случае H3AsO3[As(OH)3] кислотные свойства преобладают над основными в ~105 раз, в случае Sb(OH)3 — в ~102 раз, а в случае Bi(OH)3 основные свойства преобладают над кислотными. Такой порядок изменения кислотноосновных свойств в подгруппе мышьяка полностью соответствует периодическому закону.
9
ГИДРОЛИЗ ИОНОВ ЭЛЕМЕНТОВ VI И VII ГРУПП
9.1. Элементы VI группы — Сг, Mo, W, Те
В разбавленных водных растворах элементы подгруппы хрома могут находиться в различных степенях окисления. Для хрома наиболее устойчивы состояния Сг (3 + ) и Сг (6+). Катионные гидроксокомплексы образует только трехвалентный хром. Для молибдена и вольфрама более характерно состояние окисления +6. В кислых растворах существуют оксокатионы МоО|+, WO|+ при более высоких значениях pH — анионы молибденовой и вольфрамовой кислот. Для элементов подгруппы серы катионные гидроксокомплексы известны лишь для Te(IV) и Po(IV).
В подгруппе хрома от Cr(VI) к W(VI) кислотные свойства уменьшаются. Аналогичным образом уменьшаются кислотные свойства в ряду сернистой-селенистой-теллуристой кислот.
Данные о константах гидролиза элементов VI группы приведены в табл. 9.1.
Известна лишь одна работа, в которой была определена первая константа гидролиза сульфата Сг(П). Получены сходящиеся результаты при титровании растворов 0Д1М CrSO4 раствором КОН без постороннего электролита и в присутствии -2,5М (NH4)2SO4 [1L
Определены различными методами константы гидролиза различных солей Сг(Ш): хлорида, нитрата, перхлората, сульфата. Кроме данных о гидролизе зеленого сульфата Сг(Ш) [1] и зеленого хлорида Ст(III) [14], все результаты достаточно сходимы. Для первой ступени гидролиза иона Сг3+ можно принять рК1г=4,2±0,3 [2—6, 11 — 15, 17]. В случае зеленых сульфата и хлорида Сг(Ш) заметное различие значений рА1г вызвано, очевидно, взаимодействием между ионами хрома и Cl~, SO^“ с образованием гидроксополи-меров.
Удовлетворительная сходимость данных получена и для второй константы гидролиза: pK2r~5,5—5,6 [8, 13, 14]. Авторы работ [4, 17] получили рК2г~ 6,2-у6,8, работы [16] — рК2г^З,7. Лишь в двух работах [16, 17] приведены все три 124
Мономерный гидролиз катионов элементов VI группы
<л
а
125
Продолжение табл. 9.1
ьо
05 i— Ион Метод Т, °C ц, среда Константа равновесия Литература
Сг3+ Эл. пр. Ст. эл. » » 25 22 5—20 5 20 Cr2 (SO4)3 0,002—0,03 КСг (SO4)2 0,001—0,05 Cr2(SO4)3 (фиолет.); и-0 3,75; (NH4)2SO4 Cr2(SO4)3 (зелен.); ix->0 3,75; (NH4)2SO4 Cr (NO3)3 0,1 (NaClO4) рХ1Г=4,28-г4,57 рХ1Г=4,19ч-4,88 pKir=3,95 рХ1Г=4,41 рК1Г=5.0 p/Cjr—3,3 pK4r—3,5 pXir=3,50; pX2r=3,67; pXsr=3,88 рХ1Г=4,60; рК2Г=6,8; рХзг=9,0 1 pKir=4,15; pKjr—6,5; рХзг—8,85 J [15] [1] [16] [17]
Мо3+ Распр. 20 1,0 (NaCl) pKir—2,0; p/C2r==2,5; pKgr—2,8 [98]
МоО^+ Раств. Спектр. Ст. эл. Раств. Электромигр. Спектр. » » » 21—22 25 26 20 25 25 25 HC1 0,465 (H2SO4, CH3COONa) 3,0 (NaC104) HNO3 0,1 (NaNOg) 0,0023 0,5(NaC104) 1,0 (NaCl) 3,0(NaC104) pK3r=2,02 pX3r=l,15; pK4r=3,75 pKlr=4,08 pK-r= 1,94—1,14 pX’=0,25; pXar=3,86; pX4r=2,54 pK3r=4,00; pK4r=4,21 рХ2Г=0,94 pX r=3,55; pX4r=3,65 pX3r=3,61; pX«r=3»89 [18] [19] [20] (21 [22 [23 [24 [25 [26; ..J-.. . ".'LTTTCz
Продолжение табл. 9.1
Ион Метод т, °C Ц. среда Константа равновесия Литература
MoO|+ Электромигр. Спектр. Спектр. » Эл. диализ Спектр. Кат. Распр. Спектр. 22 25 15—35 20 20 18 25 10—40 0,1 (NaN03) 0,1 (NaClO4) 0,1 (NaCl) 1,0 (NaCl) 10-4 м Na2MoO4; HC1 6,25-10’4 до Na2Mo04 0,5 1,0 2,0 0,3(NaNO3, HNO3) 10,’5 l-I0~5M Mo (VI) NaNO3 0,1 0,3 0,5 1,0 HNOg HC1O4 10-4 м Mo (VI); HC1 0,1; l,0(NaCl) s 0 0 £ + I Sfc ^5 g?S23 5^4] «g 4е? ЗЙ II 000 COCO 1 1 «« 4J 4 4 1 tci “ II II °-°” й»8».. °-°-°- - - Г^оо 1 -hz* ХФ «I ГЧ lil^I 1MISSI1 Э : cSiJWsi < еГоГоо 6 .►do °? ЧЭСОООООО -"ООО 1 1 СЧ со есть o' o'd'o' 0"—o' I 1 1 II п o'o' 1! СГ< 14 4 4 4 4 11 11 11 11 11 I' и и и || И °® * ” СХ CL, ex, CL. О. Си Си Сх СХ Си СХ. сх Си СХ Си СХ^Р^Р *cL^cL V [27] [99] [28] [29] [30] [31] [32] [33] [34] [35]
127
продолжение табл. 9.1
Ион Метод Г, °C И, среда Константа равновесия Литература
МоО^+ Обмен. Спектр. 25 0,7 (NaCl); 1.5-10-4—410”бМ Mo (VI) NaC104 рК1Г=1,96; рК2Г=2,44; рКзг=5,82 рК2Г=1,06 (р=3,0); 0,98 (ц=2,3); 0,90 (р=1,5); 0,85 (И=0,25—ТО) [83] [100]
wo|+ Кинет. » Т.-д. Спектр. Кинет. Электро-мигр. Спектр. 25 25 50—350 25 20 1,0 (КС1) 0,003 0,1 10% этанол 2,5(NaNO3) HNO3 НСЮ4 NaNO3 10% этанол ' 0,1 0,3 0,5 0,7 1,0 рКзг=2,3; рК4Г=3,66 ’38 (50°)4[ 2,93 (100°); 3,62 (150°); 4 41 (200°); 5,27 (250°); 6,19 (300°); 7 15 (350°) 1 „ь- _3 96 (50°); 4,57 (100°); 5,28(150 ); 6,04(200°); 6,85(250°); 7,69(300°); 8,55(350°) рК1Г=0,82; рК2Г=1,28; рКзг=1,71 j рК = 0,72; рК2Г=1,14; Р^зг=1,50 рЛ,г=.0,02; рКзг=2,23; Р^«г=3,47 | рК2Г=1,02; рКзг=2,10; рК4г—3,42 ) рК4г=0,6 рК1Г—0,99; рК2Г=1,62; Р^г=2,19 рК1Г=0,96; рК2Г=1,52; рКзг—2,04 рК’г=0,89; РК2Г=1,46; рК3г= -94 =0,84; рК2Г=|,36; рКзг= ,84 } р/сг=0,76; рК2Г=1,28; рКзг=1,69 рк;г=1,022-0,254 |Х | рК2Г= 1,647-0,383 р ) рКзг=2,215-0,533ц [36] [37] [38] [39] [33] [40] [41]
CD
Продолжение табл. 9.1
Зак. 1864
Ион Метод Т, °C Ц, среда Константа равновесия Литература
Те4+ Кол. Эл. пр. Раств. эдс Раств. Распр. Раств. Ст. эл. » Эл. диализ Спектр. 25 18 25 18 20 20 28 0,1 (НС1О4) 3,0 (Н+, Na+, С1ОП 0,5 (NaClО4, NaCl) 1,5 NaClO4 1,0 0,5 1,0 1,5 0,1—1,0 0,1—1,0 2,5 1,0 0,5 KNO3 10% этанол 0,1 0,3 0,5 0,7 '-° А."." -ишщ 1 8=ч SSS8S8 38 4 SH'S 3 g 33333 1 ШЗЗЗ 3 ”5 8 = 11 и il il и ё “ii "ii ii .“-“«to 10 L01 1 й ° г, та та *2_та та II ’ll II ll ll КЭ to N3 N3 О CO cn 7^ o 5? op о та та та та fe tit *>> II ll ll ll ll N3 № tO CO СД 00 — CO jg [42] [43] [44] [45, 46] [47] [48] [49] [50] [51]
Продолжение табл. 9.1
I
130
константы гидролиза ионов Сг(Ш), однако вторая константа определена приближенно, а расчет p^зг основан на предположении, что р/(зг—рЛ2г = pK2r—pXir [17]. Нельзя признать надежными и данные, из которых следует, что все ступени гидролиза ионов Cr(III) протекают в интервале pH ^0,4 [16]. Попытка на основании констант гидролиза, определенных в работах [16, 17], построить диаграмму распределения гидроксокомплексов Сг(Ш) в зависимости от pH показывает существенное расхождение с табличными значениями pH осаждения Сг(ОН)з [56]: pH начала осаждения ~5; pH полного осаждения равен 6—11; pH осаждения из щелочного раствора — 11,5.
Сравнительные обзоры данных о гидролизе хрома (III) приведены в работах [13, 17]. Наиболее обстоятельна и подробна работа о гидролизе ионов хрома (III) при разных значениях ионной силы раствора и в виде, разных исходных солей (гексааквахром (III) сульфата и хромкалиевых квасцов) [15]. При экстраполяции к ц->0 получено значение рЛлг 4,0.
В разбавленных водных растворах присутствуют мономерные формы хрома (VI): Н2СгО4, НСгО^и CrC)2*~. Целесообразно рассмотреть более подробно данные о первой константе ионизации хромовой кислоты Ku=[HCrO“][H+]/ /[Н2СгО4], чтобы можно было провести сравнение кислотных свойств в ряду Н2СгО4—Н2МоО4—H2WO4 (табл. 9.2).
Первая константа ионизации хромовой кислоты
Таблица 9.2
Ион Метод Г, °C Ц, среда Константа равновесия Литератур»
Cr (VI) Ст. эл. 25 0,16 (КС1) РК1д = 0,75 .[571
Спектр. Распр. 25 1,0(ЫСЮ4) l,0(HNO3, НС1О4) рК1д = —0,08 рК1д = —0,10 (58J [59}
Спектр. 25 НСЮ, рК1Д = —0,98 [60Ц
» 25 —V -> 0 рК1д =-1,01 [61).
» 25 1—3 (NaC104) рК =-0,80(1,0); —0,72 (3,0) [62}
» 15—35 1,0 (1ЛС1О4) рКг =-0,83(15°); —0,61 (25°); —0,41 (35°) [63}
» 20 НС1О4 0 рК1д = —0,18 [64J i
Наиболее систематичный обзор литературных сведений а первой ступени ионизации хромовой кислоты дан ЛуккарИ; [64]. Для расчетов равновесий в растворах хромовой кислоты
9* 131
можно принять lgKm=0,18 [64]. Данными Луккари [65] можно пользоваться и для второй ступени диссоциации: рКгд= = 6,511 ±0,009.
С помощью этих констант ионизации рассчитано распределение ионных форм хромовой кислоты при ц.=0 (рис. 9.1). Из диаграммы следует, что даже в сильнокислых растворах (>10 н.) существует первый анион НСгО^~ и выход молекулярной формы НгСгО4 не достигает 100%-
Рис. 9.1. Распределение форм хромовой кислоты в зависимости от концентрации ионов водорода (ц = 0):
1 — НаСгО.; 2 —НСгОТ ; 3 - СгО?~ 4 4
В настоящее время можно считать установленным, что в водных растворах молибдатов (вольфраматов) наиболее высокозарядные катионы — оксокатионы MoO^WC^). Поэтому гидролиз катионов молибденила МоО^+ можно рассматривать как цепь равновесий, которые в разбавленных растворах ([Мо]^10~5 М) не .осложнены полимеризацией:
МоО2+ + Н2О =₽± МоОа (ОН)+ + Н+;
МоОа (ОН)+ + Н2Оч±МоО2 (ОН)2 + Н+ (Н2Мо04 + Н+);
Мо02 (ОН)2 + Н2О МоО2 (ОН)Г + н+ (Н2МоО4 НМоОГ + Н+); МоО2 (ОН Г + Н2О т МоО2 (ОН)24“ + Н+ (НМоОГ МоО2Г + Н+).
Константы равновесия этих реакций можно представить:
= [МоО2(ОН)+] [H+]/[MoOi+];
К2Г = [МоО2 ,ОН)2] [Н+]/[МоО2(ОН)+] =
= [H2MoOj[H+]/[Mo02:OH;+];
132
7<3г = л1д = [МоОг (ОН)Г] [Н+]/[МоО2 (0Н\] = = [НМоОГ] [H+]/[H2MoOj;
^4г = ^2д = 1МоО2 (ОН)2-] [Н+]/[МоО2 (ОВД = = [МоО2-] [Н+]/[НМоОГ].
Аналогичный вид имеют выражения для последовательных констант гидролиза вольфрама (VI).
Только в работе Б. И. Набиванца определены все четыре константы гидролиза Mo(VI) [30]. В трех работах .определены первые константы гидролиза [29, 30, 32]. Из работ [29, 32] следует, что с увеличением ионной силы раствора гидролиз катионов МоО^+ усиливается. Анализ данных табл. 9.1 позволяет принять следующие значения констант гидролиза Mo(VI) (при ц = 0,1): pXir-0,2 [32], р/<2г = 0,75 [27, 32, 33[, р/С1д=р/С3г= 1,5 [32, 33], р/С2д=рК4г=3,5 [19, 25—27, 30, 33].
По этим; константам была рассчитана диаграмма распределения гидроксокомплексов Mo(IV) в зависимости, от кислотности раствора (рис. 9.2). Эта диаграмма удовлетворительно согласуется с результатами определения изоэлектрической точки: рН“1,4 [18]; 1,7 [21]; 1,1 [66].
Рис. 9.2. Распределение ионных форм Mo (VI) в зависимости от концентрации ионов водорода (Ц-ОД):
/-МоО|+; 2 —Мо02ОН+; 3 - Н2МоО4; 4 - НМоО";
5 — МоО?-
Мономерный гидролиз Mo(VI) описан в работах [30, 32', 67]. Катионные и анионные полимерные гидроксокомплексы молибдена обсуждаются в работах [66—72].
Данные о константах гидролиза вольфрамила WOg+ не^ многочисленны [33, 39, 41]. Наиболее полно при различных
133
значениях ионной силы гидролиз вольфрама изучен в работе [41]. Получены уравнения связи констант гидролиза с ионной силой раствора, которыми можно пользоваться при определении констант для данной ионной силы при ц=0,14-1,0. Для второй константы диссоциации H2WO4 можно принять рК2д=рК4г-3,5 [33, 36, 37].
Диаграмма распределения гидроксокомплексов W(VI) в зависимости от pH, построенная по данным работы [41], показана на рис. 9.3.
Рис. 9.3. Распределение ионных форм W (VI) в зависимости от концентрации ионов водорода (ц=0,1):
/ —WC|+; 2 — WO2OH+; 3 —H2WO4; 4 —HWO“;
5 - Wq2~
По значениям первой и второй констат диссоциации проведен [36—38] термодинамический расчет этих констант при температуре 50°—350° С и получены уравнения, связывающие константы диссоциации с температурой: р/С1Д= = 1492,75/7—9,766 + 0,0233 7; рК2д=924,493/7—5,3286+ + 0,0199 Т, где 7 — температура, К.
Из этих данных следует, что с повышением температуры диссоциация вольфрамовой кислоты на обеих ступенях должна уменьшаться. Этот вывод был подвергнут сомнению [73] на том основании, что поведение вольфрамовой кислоты с повышением температуры должно напоминать поведение кремниевой кислоты, для которой экспериментально установлена прямая зависимость константы диссоциации от температуры до ~150°, затем константа уменьшается. Обзор состояния W(VI) в водных растворах приведен в работах [74, 75].
134
О существовании ионов Те (IV) в катионной форме в кислой области известно давно, однако лишь в отдельных работах были найдены следующие количественные характеристики кислотно-основных равновесий ионов Те(IV):
/С1Г = [ТеОН3+] [Н+]/[Те4+];
К2Г = [Те (ОН)22+] [Н+]/[ТеОН3+];
КЗГ = [Те (ОН'з+] [Н+]/[Те (ОН)!+];
= [Те(ОН)4] [Н+]/[Те (ОН Л;
=К1Д= [Те (ОВД [Н+]/[Те(ОН)4] - [НТеО3~] [Н+]/[Н2ТеО3];
Явг = ^2Д = [Те (ОН)!-] [Н+]/[Те (ОН)Г] = = [ТеОз“] [Н+]/[НТеОГ].
Константы образования катионных гидроксокомплексов определены в нескольких работах, результаты которых расходятся. Константы гидролиза всех четырех катионных форм Те(IV) можно взять из работы [51], в которой исследование гидролиза проведено при различной ионной силе раствора и получены соответствующие уравнения связи констант гидролиза с ц.
Для констант кислотной ионизации теллуристой кислоты предпочтительнее данные работы [49]. Эти константы при большой ионной силе (ц=1,5) хорошо согласуются с данными работы [48].
С помощью констант [49, 51] рассчитано распределение ионных форм Те (IV) в зависимости от кислотности среды в широком интервале значений pH раствора (рис. 9.4).
Обзор работ по определению констант ионизации теллуровой кислоты приведен в статьях [75—80, 101].
Рассмотрение и анализ работ, авторы которых исследовали гидролитические формы соединений S(II, IV, VI) и Se(II, IV, VI) и привели термодинамические расчеты констант ионизации ряда кислот в широком интервале температур, даны в исследованиях [75, 76]. Ионизация селенистой кислоты подробно изучена при различной ионной силе раствора [49] и при различных солевых составах [81]. Выведены уравнения [49], связывающие константы ионизации селенистой кислоты с ионной силой раствора: рК1Д=3,097—0,5703 р (р = 0,14-1,5); рК2д=8,304—1,070 |i (ц = 0,14-1,0), и установлено закономерное уменьшение кислотных свойств соединений в ряду H2SO3—H2SeO3—Н2ТеО3, что соответствует положению этих кислотообразующих элементов в периодической системе.
Методом потенциостатического титрования определены термодинамические значения первой константы ионизации селе
135
нистой кислоты в обычной и тяжелой воде: p/<i = 2,648± ±0,001 (Н2О); р^ = 3,015±0,013 (D2O). При ц = 0,1 (КС1) и 25° С эти величины равны: 2,433 (Н2О); 2,817 (D2O) [82].
Гидролиз ионов Po(IV) изучен в работах Н. И. Ампелого-вой [54, 55], которая подвергает критике работу [53], что» очевидно, справедливо, так как значения третьей и особенно четвертой константы гидролиза оценены приближенно, с точностью до порядка величины.
Рис. 9.4. Распределение ионных форм Те (IV) в зависимости от концентрации ионов водорода (]и=0,1):
/ — Те4 4-; j? — ТеОН3+; 3 — Те(ОН)|+; 4-Те(ОН)+; 5 — Те(ОН)4; 6 - НТеО“:
7 — ТеОд”
Если сравнивать первые константы гидролиза ионов Те4+ (р/С1г= 1,4 [51]) и Po4+(pAir = 0,48 [55]), то можно прийти к неверному выводу о большей основности теллура по сравнению с полонием. Если же сравнивать константы основности, которые можно рассчитать из четвертых констант гидролиза рАосн = рЛлг—pK4r, 'то получим следующие значения для Te(IV) рЛосн=П,9, для Po(IV) рАосн-10,6.
Эти результаты свидетельствуют о большей основности Ро(ОН)4 по сравнению с Те(ОН)4, что соответствует положению этих элементов в периодической системе.
9.2. Элементы VII группы — Мп, Тс
Из элементов VII группы могут существовать в водных растворах в виде катионных гидроксокомплексов лишь ионы Мп(II), Мп(III) и Тс(IV). Рений в водных растворах преимущественно существует в виде солей рениевой кислоты — перренатов. Технеций также предпочтительно существует в валентном состоянии +7. В разбавленных водных растворах 136
CO сп Литература иГ o' СО 00 00 Г- ОС? оо^оо. СП о СО СП СП 1—JI——1 1——1 СЧ СО1 СП СП 1—1 М*4 иэ1 о 2L о'г-' СП о
s
\o Константа равновесия рЯ1Г=10,6 рК1Г=10,93(15°); 10,76(20°); 10,59(25°); 10,38(30°); 10,19(36°); 10,10(42°) рК =10,81 (0,002); 10,88(0,015); 11,12(0,115); 10,17(0,215—0,315); 11,25(0,615); 11,04(1,015); 10,99(1,5); 11,05(1,515) J рК1Г= 10,22 рк1г=_ 0,7 рК1Г=—0,25 ) пХ1Г=—0,18 1 о Ю о о о сч СЭ ''"'о <0 LO о со С- 1 ° 1 см СО СП .<• -«Лет? oogj 1 II 11-11 < Ьи UC0 и СХО сх Ю 00 о о о" о" л о. о СЧ СЧ -ООО со" 1 1![ 1 а. о. а. со Tt< СЧ ||_ УГ СО О 17 СХ, СХ
HTOB VII группы ji, среда 0, 1 (КС1) MnCl2, KNO3 0,002—0,04 0,002—1,5 0 6,0 (НС1О.) 6,0 (HC1OJ 4,0 (НС1О4) 4,0 (HC1OJ 3,04(NaC104, НС1О4) й СО о LO д’ OZ У i 770 °.s о to 0,01—1,0 (KNO3) : 0,1 !
атионов элеме О сч О 1 о ю со 1 СМ СЧ LO СО со сч сч 25 1,2—34,5 12,4 23 со to СЧ СЧ 00
ли гидролиз к Метод г Ст. эл. » Спектр. . Спектр. Редокс. пот. Спектр. Кинет. Спектр. Спектр. Кинет. Н—эл. Поляр. Электромигр. Распр.
X 8-s О § s Ион 4* а 1 4* 'Ъ
137
галогены в любых степенях окисления существуют в анионных формах.
Катион Мп2+ подвержен гидролизу в незначительной степени, катион Мп3+ — весьма сильно. Гидроокись Мп(IV) проявляет амфотерные свойства.
Литературные данные о гидролизе катионов Мп (II), Мп(Ш) nTc(IV) приведены в табл. 9.3.
Данные о гидролизе ионов Мп2+ немногочисленны. Определена только первая константа гидролиза, которую, очевидно, можно принять равной 10,5—11,0 [84, 85]. Небольшой обзор данных о гидролизе Мп2+ приведен в работе [86]. Данные из таблицы pH осаждения Мп(ОН)2 не согласуются с pKir: pH начала осаждения равен 8,7, pH полного осаждения — 10—14.
Гидролиз ионов Мп(Ш), как и ионов Мп(П), изучен мало. Определена лишь первая константа гидролиза [89, 90, 92— ) *95]. Можно допустить, что Kir=0,9-ь 1,0. Следует отметить обстоятельные работы по изучению гидролиза ионов Мп(III) [94, 95], в которых дан критический анализ литературных данных.
Различие в склонности к гидролизу ионов Мп2+ и Мп3+ — одно из самых значительных среди ионов одного элемента с разными зарядами.
Гидролиз ионов Тс (IV) систематически не исследован. Данные, полученные разными авторами [96, 97], не согласуются между собой. Характеристики четвертой константы гидролиза Тс (IV) близки к аналогичным характеристикам для циркония и гафния. Каких-либо сравнений склонности к гидролизу ионов элементов VII группы провести нельзя, так как все исследованные системы относятся к ионам в различных степенях окисления.
ГИДРОЛИЗ ИОНОВ ЭЛЕМЕНТОВ VIII ГРУППЫ — Fe, Со, Ni, Ru, Rh, Pd, Os, Ir, Pt
Образование гидроксокомплексов характерно для ионов Fe (II), Fe (III), Co (II), Co (III), Ni (II), Rh (III), Pd (II), Pt (IV). Подробно и систематично изучены гидроксокомплексы ионов железа. Во всех остальных случаях проведены отдельные исследования. Разнообразие валентных состояний, склонность к образованию смешанолигандных и ацидогидроксо-комплексов, участие различных комплексов в кислотно-основных равновесиях затрудняют рассмотрение процессов гидролиза ионов металлов платиновой группы и делают невозможным сравнительный анализ склонности к гидролизу ионов этих металлов. Поэтому в случае ионов металлов платиновой группы приходится ограничиться лишь перечислением тех немногочисленных работ, где изучено образование гидроксокомплексов в разбавленных растворах. Более подробно эти вопросы рассмотрены в специальных монографиях и обзорах И, 2].
Литературные данные о мономерном гидролизе ионов металлов VIII группы приведены в табл. 10.1.
Три константы гидролиза ионов Fe2* определены лишь в одной работе [18]. Определенная здесь третья константа гидролиза (рЛзг= 10,62) должна свидетельствовать о легком образовании анионных гидроксокомплексов, что не соответствует известным свойствам Fe(OH)2. Значение общей третьей константы гидролиза (рК^щ = 25,77) резко отличается от значений этой константы, полученных другими авторами: 31, 78 [17] и 34, 15 [4].
Вторая константа гидролиза рассчитана лишь в двух работах: p^2г=5,0 [10] и 8,25 [18]. В основном исследователи рассчитывали лишь первую константу гидролиза, рЛ)г. Полученные значения этой константы можно объединить в три группы: рК1г=б4-7 [5, 8—11, 18], pKir= 8,04-8,3 [6, 8, 15], р/(1г=9,24-9,5 [7, 14, 16, 17]. Кинетическим методом получено значение р^1Г=3,3 [12, 13]. Из литературных данных рассчитаны значения термодинамических функций ионов Fe2*, FeOH+ и др. [17, 107].
139
Таблица ЮЛ
Мономерный гидролиз катионов VIII группы
Ион Метод Г, °C Ц. Среда Константа равновесия Литература
Fe2+ Раств. 25 18,83 [3]
Поляр. 10-4—10“5М Fe2+, pK°^ = 34,15 [4]
Н-эл. 20 Fe(ClO4)2 pKlr=5,92 j. [5]
Ст. эл. Раств. 25 -0 -0 pKlr = 8,3 [6]
Ст. эл. 25 l,0(NaC104) p/Cir = 9,5 17]
» 25 FeCla pKlr = 7,92 1 [8]
0,5 (KC1) pKlr = 7,17 f
» 12—30 Fe(C104)2 p/Clr = 8,50 (12°); 8,03(15°); 7,15(20°); 6,8(25°); 1 [9]
-1,0 5,95(30°) J [10] из [7]
Расчет 25 l,0(NaC104) pKlr=6,5 ) pK3r«5,0 f
Ст. эл. 20 l,0(NaC104) pKlr=6,93
25—40 0,5(NaC104) pKlr=6,74 (25°); 6,49(35°); 6,34(40°) [11]
Кинет. 25 1,0 (NaClOJ pKlr=3,32 [12]
Ст. эл. Кинет. 0—35 0,01 (NaC104) pKlr=4,64 (0°); 3,98(10°); 3,82(15°); 3,29(25°);]
Ст. эл. 2,82 (30°); 1,94 (35°) pKlr=3,24 [13]
25 l,0(NaC104)
Раств. 25 0 pKir=9,3 [14]
Спектр. 2M (NH4)2SO4 P^ir—8>3 | [15]
2M NaClO4 pKlr=8,18 )
Н-эл. 25 FeCl2; ->0 pKir=9,49 [16]
Т.-д. 25 ->0 pKlr=9,22; рК°бщ=31,78
50 pKlr=7,97; р/<°бгщ=29,06 [17]
150 pKlr=4,67; PK°6u’=22,60 ,
Продолжение табл. 10.1
Ион Метод Г, °C Ц, среда Константа равновесия Литература
Fe2+ Обмен 250 350 18 0,1 (NaC104) 2-10—«М Fe(ClO4)2 pKlr=2,79; рК°®щ=19,24 | pKlr=l,61; рК°бщ=17)42 j pKlr=6,90; рК2Г=8,25; рКзг=10,62 [17] [18]
Fe3+ Раств. Кинет. Редокс. пот. Ст. эл. Редокс. пот. эдс Спектр. Ст. эл. Спектр. Редокс. пот. Спектр. Ст. эл. » Спектр. Кинет. Спектр. 15 15 25 35 25 25 25 20 20 25 25 25 26,5 25 25—45,3 25 0—22 25 0,008—0,1 0,084 0,4—1,0 ^0 ^0 ->0 0,53 (KNO3) 2,0(NaC104)' -0 Fe(C104)3 0,04—0,09 (NaClO4) 0,5(NaC104) 0,00166 (NaClO4) -0 0,435 (KNO3) Fe2(SO4)3 -^0 0,55 0,5(NaC104) ptflr=2,63 (0,008); 2,59(0,01); 2,50(0,1); 2,2 (->0)} p/Cjr—2,4/ рК1Г=2,75 1 рК1Г=2,22 pKlr=l,92 J рК1г—2,46; рК2Г==4,70 рК1г=2,76 рК1Г=2,82 рК1Г=3,0 рК1Г=2,18 1 р/С1Г==1,96 f рК1г=2,53 (0,040); 2,55(0,046); 2,61 (0,09) рК1г=2,72 рД1г~2,33 ) рк ^2,19 / рК1г=2,68 (25°); 2,13 (30°); 2,28 (35,7°); 2,32 (40,3°);1 2,14(45,3°) f рК1г=£,90; рЛ2Г=:3,38 рК1г=К7 рК1г=3,59(0°); 3,33(7,3°); 3,07(14,7°); 2,85(21,6°) р/(1г=2,80 [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33
н- ' Продолжение табл. 10.1
to
Ион Метод Т, °C Ц, среда Константа равновесия Литература
Fe3+ Редокс. пот. Ст. эл. » Эл. пр. Редокс. пот. Спектр. Расчет Спектр. » » Ст. эл Магн. воспр. Ст. эл. Редокс. пот. Спектр. » 25 25 15 25 3—25 21 35 18—32 25 15—35 20 20 18—36 —60—20 3,0(NaC104) 0,5(NaC104) 0,0147—3,0 ->0 1,0 0,55 0,01—1,0 -+0 0—1 0,004—0,2 3,0(NaC104) l,0(NaC104) 0,025—0,15 (NaClO4) 0,15 80% метанола рКц.^3,05; рК2Г=3,27 рК1г=2,83; рК21—4,59 рК1г=2,93 рК1г=2,17 . } рК1г=3,51 (3,1°); 3,35 (7,8°); 3,07 (16,8°); 2,80 (24,8°) рК1Г=2,92 (D2O) рК1г=2,50 рК1г=2,38(18°); 2,19(25°); 2,02(32°) ' 1 рК1г=2,78(р=1,0); 2,17(ц=0) / рК1г=2,71 (15°, р=0,01); 2,41 (25°, р=0,004); ) 2,47 (25°, р=0,01); 2,50 (25°, р=0,02); 2,28 (35°3 и.=0,01) J рК1г=3,0; рК2Г=3,3 р/(1г=2,75; рК2Г=3,31 рК1г=2,74 (18,2°; и,=0,15); 2,70(18,7°; 0,1); ) 2,68(20°; 0,08); 2,60(20,1°; 0,06); 2,55 (20,3°; } 0,04); 2,52 (20,3°; 0,025); 2,32(35,5°; 0,15) J РК1Г=2,49(—59,2°); 2,45 (—49,7°); 1 2,09 (—41,8°); 2,04 (—25°); 1,49(0,0°); 1,46(20°) / [34] [35] [36] [37] [38] из [37] [39] [40] [41] [42] [43] [44] [45] [46]
143
Продолжение табл. 10.1
Ион Метод Т, °C Ц, среда Константа равновесия Литература
Fe3+ Спектр 20—22 0,1 (KNO3); метанол : во- pKlr=2,63 ]
да =66 : 34 рК1Г=1,77 - / [47]
» 25 0,9MFe(C104)3 pKlr=2,39 [48]
Редокс.
пот. 25 l,0(NaC104) pKlr=2,19 [49]
Спектр. 5—25 0,5 (D2O) pKlr=3,44 (5°); 3,20(15°); 2,95(25,2°) [50]
» (FeNO3)3 pKlr=l,98; ptf2r=2,31; ptf3r=2,49 [51]
CH3COONa
Ст. эл. 25—45 3,0 pKlr=3,00(25°); 2,73 (35°); 2,52 (45°) [52]
» 20 0,1 (NaClO4) pKlr=2,97; pK2r=4,01 [53]
Редокс.
пот.
Спектр. 25 0,25 (NaClO4) pKlr=2,66 [54]
» 0,15(NaC104) pKlr=2,84 )
0,15(NaCl) pXir—3,06 I [55]
0,15(CaCl2) рК1г=2,96 1
» 20 0,1 (NaClO4) pXlr=2,39 [56]
Кинет. 25 ->0 pKsr=5,12 [57]
Т.-д. 25 ->0 pKlr=2,43; pK2r=4,72
50 pKir=l,63; pK2r=3,81
150 pKlr=0,12; рК2Г=0,83 [17]
250 pKlr=—0,70; pK2r=—0,75
350 pKlr=—1,19; pK2r=—1,64
Спектр. 18 l,0(LiNO3) pKlr=2,58]
l,0(KNO3) pKlr=2,75 [58]
l,0(NaC104) pKlr=2,92J
Редокс.
пот. 25 3,0(NaC104) pKir=2,34 [59]
Продолжение табл. 10.1
Ион Метод Г, °C н. Среда Константа равновесия 'Литература
Fe3+ Обмен Спектр. Редокс. пот. Обмен. Ст. эл. Обмен. Спектр. Ст. эл. Спектр. » Редокс. пот. Спектр. \ 25 18 25 25 25 25 25 0—55 25 45 0,4 (NaNO3) 0,l(HNO3) 0,4 (NaNO3) 3,0 (NaClO4) 0,1 (NaClOJ l,0(NaC104) 0,4 (NaNO3) 0,5 (NaNO3) 3,0(NaC104) 0,01—1,0 (NaClO4, KNO3) 0,1 (NaClOJ 0—3,0 0—3,0 0—2,0 pKlr=2,57) pKlr=2,55) pKlr=2,43) pKlr=2,92 pKlr=2,58; pK2r=3,83; pK3r=5,61 pKir=2,51; рК2Г=2,78 pKlr=2,551 рК1Г=2,57/ dK, 3 1 pXir=2*43 (0,01); 2,67 (0,05); 2,84 (0,10); 2,94 (0,60);) 3,07(1,0) J pKlr=2,54 (I I2O); 2,85 (D3O) л /nro, , pKir=+a-0,145 p; a -2,96 (O’); 2,74 (10°); 2,49 (25°);) 2,35(35°); 2,24(45°); 2,14(55°) 1 pKlr=2,54 (0); 2,52(0,1); 2,45(0,5); 2,36(1,0); 2,18(2,0); 2,0(3,0) , p/(lr=2,22 (0); 2,20(0,1); 2,10(0,5); 2,06(1,0); 1,96(2,0) [60] [61] [62] [63] [64] [65] [66] [67] [И7] [H8]
Со2+ Раств. Ст. эл. » » » » » 25 25 30 25 15—40 25 25 28 25 CoCl2; p,—>0 CoCl2; p-*0 0,1 (KC1) Co(NO3)2; H-0 0,26 (NaClO4) 0,76 ->0 l,0(NaC104) 3,0 (Ba(C104)2) р7(°бщ=18,80; pK3I=12,7 pKlr=12,20 pKlr=8,9 рК4г=10’,00 (15°); 9,83 (25°); 9,58(35°); 9,50(40°) | pKir=9,80 i pKlr=8,9 > pKir=9,82 pKlr=9,75 [68] [69] [70] [71] [72, 73] [74] [75]
10 Зак. 1864
Продолжение табл. 10.1
Ион Метод г, °C Ц, среда Константа равновесия Литература
Со3+ Спектр. Кинет. » 12,5—28,2 25 7 l,0(NaC104) 3,0 (Cl—) 0,25 рК1Г=2,10 (12,5°); 1,98 (18,45°); 1,78 (23,6°); 1,71(28,2°) V h pKir=0,66 рк1г=1,3 [76] [77] [78]
Ni2+ Раств. Ст. эл. » » » » » » Поляр. 25 25 30 25 25 20 25 28 25—50 15—42 20 25 ->0 0,1 (КС1) Ni(NO3)2 NiCl2 Ni(ClO4)2 NiSO4 Ni(C104)2->0 Ni(NO3)2 Ni(NO3)2; -0 l,0(NaC104) l,0(NaC104) KNO3; -0 0,1 —1,5 0,2MK2SO4 pKir=10,82[ рК1г=Ю,64/ ptflr=9,4 p/flr=9,0 рК1г=9,23] 9,291 9,301 9,50' рК1г=8,94 9,10 рК1г=10,92; рК2Г^4 рК1г=10,01 рК1Г=9,76 (25°); 9,65(30°); 9,52 (40°); 9,3(50°) рК1г=10,22 (15°); 10,05 (20°); 9,86 (25°); 9,75(30°) ) 9,58 (36°); 9,43 (42°) ' ' рК1Г=10,35 (0,1); 10,28(0,6); 10,26(1,0); 10,17(1,5)/ рК1г=8,94 [69] \70] [79] [80] [81] [82, 83] [74] [84] [85] [86]
Ru(VIII) Распр. » 20 25 Р^кисл— П,17; рКосн—^4,24 Р^кисл—11,89 [87] [88]
Продолжение табл. ЮЛ
Ион Метод т°, с ц, среда Константа равновесия Литература
Rh (III) Ст. эл. Спектр. Кинет. Ст. эл. Раств, » Эл. диализ 20—25 25 64,4 25—85 16±2 18 + 2 0,0728 MRh(C104)3 l,0(NaC104) 12 2,5(NaC104) 0,1 (NaClO4) 0,1 (HC1O4) (1— 5). 10-«М Rh(lII) 0,1(НСЮ4) 0 рК1г=3,43 рКхг=2,92 рК,г—3,2 | рК1г=2,4 / рК.г=3,4 (25°); 3,2(45°); 3,08(60°); | 2,96 (75°); 2,92 (80°); 2,89 (85°) / рКзг=3,9 р/ф=14,25; РКЗГ=4,12; рК4г=6,85 рК,г=3,22; рК2Г=3,79; рКзг=4,20; рК4г=7,48; ] pKsr=9,48; pKer=10,55; / pKir=2,73; рК2Г=3,45; рКзг=3,96; рК4Г=7,48; ) рК6г=9,68; рК6г= 10,95 [89] [901 [91] [92] [93] [94] [95]
Pd(II) Ст. эл. Спектр. Раств. 25 25 17—18 : -Ю НС1О4, 0,05 0,10 0,18 *0 НС1О4 рЯ1г=1,0; рКаг=1,2 рК1г=1,4; рКгг=1,2 рК1г=1,6; рКгг=1,2 рК1Г=2,0; рК2Г=1,3 рК1г=1,6; рК2Г=—0,1 tn рК\р=14,25; рК1г=2,53; рК21 =2,40; рКзг=12,40; | рК4г=13,25 > [96] [97, 98]
Pd(IV) Раств. | 25 рК5Г=14,39 [99]
10* 147
Продолжение табл. 10.1
Ион Метод г, °с Ц, среда Константа равновесия Литература.
Os (VIII) Распр. Спектр. » эдс Распр. 25 25 20 24 0,006 l,0(NaC104) 8Л0“3_2 10-з MOs 1,0 1,0 рК, „ = 12,10 рКи=11,98 рК2д=14,87 z рК1д=2,20; рКзд=12,20; Р*зд={3,96; p4=U,7 } РЛ1д — [100] [101] [102, 103] [104]
Ir(IV) Сорбц. Раст. 20 18 2,06 (НС1О4, NaClO4) 0,1 IgKir=0,4 p/Clr=6,43; pKBr=9,63 [Ю5] [120]
Pt (IV) Раств. 17—18 °,! p^=14,25; р^г=1,55; 1 ptfar=2,40; р/Сзг=3,13; рК4г=3,85; p/C5r=4,59; ' рК,г=5,45 [Ю6]
Если воспользоваться рекомендациями работы [107], то по данным работы [18] p/(ir=6,90 и р/<2г=8,25 можно построить распределение гидроксокомплексов железа (II) в зависимости от pH (рис. 10.1), которое удовлетворительно согласуется с величинами pH осаждения гидроокиси Fe(OH)2: pH начала осаждения — 7,0, pH полного осаждения — 9—14.
Необходимо, однако, отметить, что с помощью спектрофотометрических измерений [15], выполненных в широком диапазоне концентраций железа в разбавленных растворах (10“3—10~5 М), рассчитана константа гидролиза p/Cir=8,24-
Рис. 10.1. Диаграмма распределения гидроксокомплексов Fe (II) в зависимости от pH раствора:
/ — Fe2+; 2 — FeOH+; 3 -- Fe(OH)2
4-8,3. Методом растворимости с учетом всех возможных схем гидролиза [14] и электрометрическим методом [16] получены еще меньшие значения константы (p/Gr=9,34-9,5). На основании этих данных рассчитана термодинамическая константа гидролиза иона Fe2+: pAir —9,22 (при 25° С) [17].
Гидролиз ионов Fe(II) изучали многие исследователи. Наиболее полные и систематические обзоры даны в работах [108, 109]. В обзоре [108] приведена наиболее полная сводка количественных данных по гидролизу ионов Fe(III). Автор обзора выделил отдельно электрометрические и спектрофотометрические методы, а также некоторые работы, использующие магнитный и кинетический методы. К сожалению, его таблица содержит много досадных неточностей и ошибок. В обзоре [109] рассмотрены работы по гидролизу ионов железа в двух состояниях: Fe(II) и Fe(III). Лишь в двух работах определены все три константы гидролиза иона Fe(III): рА1г= 1,98, р/С2г=2,31, рАзг-2,49 [51]; р/<1г-2,58, рК2г= = 3,83, р/<зг=5,61 [62]. Наиболее сходящиеся результаты получены для первой константы гидролиза: pAir=2,24-2,4 [17, 19—21, 25, 26, 28, 37, 41, 42, 45, 48, 49, 56, 59, 60, 62—64, 66, 67]. Термодинамическая константа pKJr=2,43 [17].
При высоких значениях ионной силы раствора в интервале температур 25—80° С определена первая константа гидролиза [119]. Для второй константы гидролиза наибольшая сходимость данных для значения р/С2г=3,34-3,4 [30, 34, 43, 44]. Значение термодинамической константы по данным ра-148
боты [17] существенно отличается от этого значения: = 4,72.
Несмотря на большое число литературных сведений о константах гидролиза иона трехвалентного железа, в настоящее время для приближенных расчетов гидролитических равновесий можно рекомендовать лишь первую константу.
Данные о гидролизе ионов Со(II) ограничены и разноречивы. Расхождения между крайними значениями показателей констант гидролиза достигают порядка 4,6. Данные последних лет более однородны [72—75]: р/<1г=9,74-9,8 (ц = 0,34-3,0, Т = = 254-28° С). Из результатов работ [72—75] следует, что с уменьшением ионной силы раствора гидролиз усиливается; термодинамическое значение рЛ°г = 8,9.
В водных растворах ионы трехвалентного Со (III) гидроли-зованы в значительно большей степени, чем Со(II).
Для приближенной оценки гидролиза ионов кобальта (III) при 25° С можно принять значение p/fir = 1,76 [75], которое получено из корреляционной зависимости рЛ1Г от 1/7. Величины, полученные кинетическим методом [77, 78], немного завышены.
Полимерные гидроксокомплексы Со(III) описаны в работе [ПО].
Гидролиз Ni(II), как и Со (II), изучен недостаточно. При 25° С для разбавленных растворов можно принять рЛ1г=9,0-49,5 [70, 79—81, 86].
Такое значение, как и pKirfCo(II)]=9,74-9,8, соответствует правилу Ирвинга — Вильямса о большей устойчивости комплексов никеля по сравнению с комплексами кобальта. В приложении к процессам гидролиза из этой закономерности следует, что соли никеля (II) должны быть в большей степени гидролизованы, чем соли Со(II).
Рутений в состоянии окисления 3+ в сульфатных растворах существует в виде катионов Ru3+ и Ru(OH)+ (при рН = = 1,5-44,5), а при рН>4,2 начинается образование Ru(OH)3 [111]. Нитрат четырехвалентного рутения в кислых растворах образует ряд гидроксокомплексов RuOH3+, Ru (ОН)2+, Ru(OH)+ [112]. В 1 М НСЮ4 доминирует гидроксокомплекс Ru(ОН)2+ [113, 114]. Обзор о состоянии в перхлоратных растворах Ru(IV) дан в работе [2].
Из немногочисленных данных о четырехокиси рутения можно заключить, что RuO4 обладает слабыми кислотными свойствами и в значительно меньшей степени — основными [87, 88].
Состояние Rh (III) в перхлоратных растворах рассмотрено в работе [2], гидролиз Rh(III) в перхлоратной и хлоридной средах, существовании полиядерных гидроксокомплексов описаны в работе [115].
149
Значения первой константы гидролиза в литературе согласуются хорошо: рК1г = 2,9-ьЗ,4 [89—92, 95]. Наиболее систематические и подробные исследования гидроксокомплексов Rh (III) приведены Б. И. Набиванием с сотр. [93—95]. По данным работы [95] при построено распределение гидроксокомплексов Rh(OH) в зависимости от pH (рис. 10.2), На основании данных [95] можно заключить, что кислотные свойства Rh(OH)3 выражены гораздо сильнее основных: Р Акисл = рА4г “ 7,5; р Аосн = рAw — рАзг ^10.
Состояние в растворах Pd(II) • обстоятельно рассмотрено в работе [2]. Константы гидролиза ионов двухвалентного палладия определены в работах [96—98] (см. табл. 10.1). Они
Рис. 10.2. Диаграмма распределения гидроксокомплексов Rh (III) в зависимости от pH раствора:
1 - Rh3+; 2 —RhOH2+; 3-Rh(OH)+; 4-Rh(OH)3; 5-Rh(OH)“; 6-Rh(OH)|~;
7 — Rh(OH)3”
сильно расходятся, но в обоих случаях вторая константа гидролиза больше первой (pAsr<CpAir). По тем же данным ион Pd2+ обладает достаточно высокой склонностью к гидролизу, хотя по размерам и заряду он близок к ионам Мп2+ и Zn2+, умеренно склонным к гидролизу.
В настоящее время нельзя рекомендовать какие-либо количественные характеристики гидролиза Pd(II) для расчета равновесий в водных растворах его солей.
Из количественных характеристик гидролиза ионов Pd (IV) известна лишь константа образования первого анионного гидроксокомплекса Pd(OH) [99].
Состояние в растворах ионов Os(IV, VI, VIII) подробно обсуждается в работе [116]. Приведены количественные характеристики ионизации осмиевой кислоты. Природа этого соединения, его основность все еще служат предметом дис-150
куссии. Многие авторы полагают, что четырехокись осмия представляет собой двухосновную кислоту H2OsOs [100] или H2nOsO4+n [Ю1]. По последним данным это трехосновная кислота Нз[ОзО4(ОН)2] [121]. Константы ионизации, хорошо согласующиеся на первой ступени диссоциации рК1Д^ 12,0 [100, 101], показывают, что эта кислота весьма слабая.
Французские исследователи полагают, что четырехокись осмия в водных растворах представляет собой четырехосновную кислоту OsO2(OH)4 либо H4OsO6 [102, 103] и первая константа ионизации (pKi = 2,2) характеризует ее как довольно сильную. Высказаны сомнения в правильности таких предположений [116].
Рис. 10.3. Диаграмма распределения гидро-ксокомплексов Pt (IV) в зависимости от pH раствора:
/ — Pt4+; 2-.-РЮНН; 3 —Pt(OH)^+; 4 — Pt(OH)+;
5-Pt(OH)4; 6 —Pt (OH)”; 7-Pt(OH)2_ o о
Кислотные свойства OsO4 представлены [104] как следствие гидролиза
OsO4 + H2O^OsO4(OH)-+H+ а константа гидролиза близка по значению к константе ионизации [100, 101].
Условиям образования гидроксокомплексов Ir (IV) и их термодинамическим характеристикам (К]Г = 2,5) посвящена лишь работа [105]. В статье [120] определены КоСН= 1,6-Ю-8 и Ккисл = 2,5« 10-5 гидроокиси Ir(IV).
Различными методами Б. И. Набиванец и Л. В. Калабина [106] рассчитали константы диссоциации всех шести гидроксокомплексов Pt(IV), из которых авторы данной книги пересчитали константы гидролиза, приведенные в табл. 10.1. По этим данным построена диаграмма распределения гидроксокомплек-сов Pt(IV) в зависимости от pH (рис. 10.3).
151
Гидроокись Pt (IV)—Pt (ОН) 4 — соединение амфотерное, может быть охарактеризовано константами основности и кислотности [106]: рЛкисл = Р^5г^4,6, рКосн^рД'W ~рК4г= 14,25“ 4-3,85=10,40. Из этих величин следует, что у Pt(OH)4 кислотные свойства существенно превосходят основные.
Так как в группе платиновых металлов не все ионы металлов изучены достаточно полно и многие металлы изучены в разных степенях окисления, сравнительный анализ в этой группе элементов сделать очень трудно. Сравнить можно лишь кислотные свойства четырехокисей рутения RuO4 и осмия OsO4. Из литературных данных следует, что RuO4 и OsO4 — слабые кислоты (для RuO4 рК1кисл = 11,17 [87] и для OsO4 рК1КИСЛ — ж 12—12,5). Увеличение основности от рутения (VIII) к осмию (VIII) объясняется положением элементов в периодической системе.
Сравнение свойств гидроксокомплексов элементов платиновой группы с гидроксокомплексами триады железа возможно лишь в отдельных случаях. В частности, можно отметить, что образование гидроокисей Fe(OH)3 и Ru(OH)s начинается при рН2>4; гидролиз ионов Со (III) (рК1Г=1,76 [76]) в соответствии с положением в периодической системе протекает в большей степени, чем гидролиз ионов Rh(III) (р/Сг=2,9Ч“3,4 [89—92,95]). Только ионы Pd(II) подвергаются гидролизу сильнее, чем ионы Ni(II), что еще не находит объяснения.
II
СВЯЗЬ МЕЖДУ ПОЛОЖЕНИЕМ ЭЛЕМЕНТА В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ Д. И, МЕНДЕЛЕЕВА И СПОСОБНОСТЬЮ ЕГО ИОНОВ К ГИДРОЛИЗУ
В предыдущих главах при обсуждении данных о констан ‘тах гидролиза ионов элементов было отмечено изменение способности ионов к гидролизу в соответствии с положением элементов в периодической системе. В этой главе рассматриваются кислотные свойства аквакомплексов ионов элементов, (склонность к гидролизу) по группам периодической системы. Д. И. Менделеева.
Гидролиз ионов металлов тесно связан с кислотно-основными свойствами их окислов и гидроокисей, ’комплексообразующей способностью элементов, величинами pH осаждения соответствующих гидроокисей, что имеет большое значение в химической технологии, радиохимии, аналитической химии и т. п. Попытки установить общие закономерности процессов гидролиза давно предпринимались различными авторами. Одной из первых работ было исследование Тепельманна [1], где четко сформулированы некоторые общие положения о кислотных свойствах гидроокисей элементов:
1) при максимальной валентности металлов кислотность соответствующих гидроокисей увеличивается в горизонтальных рядах слева направо;
2) в вертикальных рядах кислотность гидроокисей уменьшается с увеличением порядкового номера;
3) в различных гидроокисях одного и того же элемента с увеличением валентности соответствующего соединения усиливаются кислотные свойства.
Эти положения имеют ряд особенностей, когда рассматриваются кислотные свойства гидратированных катионов. Закономерности проявления кислотных свойств гидроокисей элементов-и гидратированных катионов подобны, но не тождественны. Гидролиз равнозарядных катионов одной подгруппы, как правило, уменьшается сверху вниз. Это четко видно на примере-катионов щелочных и щелочноземельных элементов Sc—Y—La, элементов IV группы в парах Nb—Та, Sb—Bi, Mo—W. Однако в подгруппах Zn—Cd—Hg, Al—Ga—In—TI изменение гидро-лизуемости катионов происходит иным образом. Сравнение констант гидролиза трехзарядных ионов З^-элементов Sc—Ti—
155
Cr—Мп—Fe—Co показывает расхождение с рядом Ирвинга — Уильямса [2].
Однозначный вывод можно сделать лишь в случае гидролиза разнозарядных ионов одного и того же элемента: Кислотные ’Свойства или склонность к гидролизу в большей степени выражены у более высокозарядных катионов. Это четко видно на примерах TH—Tl3+, Се3+—Се4+, Ti3+—Ti4+, Sn2+—Sn4+, Mn2+—Mn34-, Fe2+—Fe3+, Co2+—Co3+ В случае оксокатионов такая закономерность формально не соблюдается: константы гидролиза V3+ и VO2+ примерно одного порядка, кислотные свойства ионов Np3+ и Ри3+ выражены более сильно, чем ионов NpOt и PuOt, а ионы Np4+ и Ри4+ более гидролизованы, чем ионы нептунила и плутонила.
Установить взаимосвязь между положением элементов в периодической системе и склонностью ионов к комплексообразованию, в частности к образованию гидроксокомплексов, пытались многие авторы. Константы нестойкости (гидролиза) коррелировали с ионными радиусами, энергиями гидратации, ионными потенциалами (z/r, z/г2), стандартными окислительно-восстановительными потенциалами, ионизационными потенциалами (U), отношениями U/r, U/r2 [3—11]. Эти вопросы систематически рассмотрены в монографиях К. Б. Яцимирского и А. А. Гринберга [12, 13], обзорных статьях Л. С. Лилича и К. П. Мищенко [14, 15].
Выяснение периодических изменений в склонности к гидролизу ионов элементов сопряжено с трудностями, которые объясняются сложностью таких систем, как водные растворы [14]. Тем не менее авторам данной книги удалось обнаружить прямолинейную зависимость (между показателями констант гидролиза элементов VI группы Cr(VI), Mo(VI), W(VI) и 6-м ионизационным потенциалом [16], элементов четвертой группы и 4-м ионизационным потенциалом [17—19]. С помощью этой зависимости можно рассчитать не определенные экспериментально константы гидролиза ионов Sn(IV) при ц = 0,1—0,3—0,5 и константы гидролиза ионов Pb(IV) при р, = 0,14-1,0 [20]. Была показана связь между первой константой ионизации кислот H2SO3—H2SeO3—Н2ТеО3 и 4-м ионизационным потенциалом [21]. Связь констант гидролиза ионов подгруппы алюминия с положением элементов в периодической системе более сложна [22].
В дальнейшем изложении использованы различные параметры атомов и ионов из справочников [23—26] и монографий [27—30].
Гидролиз ионов элементов I группы. Сравнение кислотно-основных свойств ионов и соединений элементов I группы возможно только в ряду Li+—Na4"—К4-. Существование в водных растворах ионов меди, серебра и золота в разных степенях 154
окисления не позволяет проводить какие-либо корректные сравнения.
В ряду щелочных элементов наблюдается закономерное уси--ление основных свойств сверху вниз по группе (ослабление гидролиза, увеличение р/Сг).
Более резкое изменение основности при переходе от Na к Li, чем от К к Na обусловлено увеличением доли ковалентно-«сти связи у лития то сравнению ic ионными связями натрия и калия.
Гидролиз ионов элементов II группы. Гидролиз катионов элементов главной подгруппы II группы протекает в значительной степени лишь для бериллия.
Константы гидролиза закономерно изменяются в ряду Ве2+...Ва2+. В этой подгруппе лишь гидроокись бериллия наряду с основными свойствами проявляет и кислотные. Для Mg— Са—Sr—Ва характерны только основные свойства, которые усиливаются в группе сверху вниз, так же как в случае щелочных металлов.
На рис. 11.1 показаны зависимости рК\г катионов элементов главной подгруппы от 2-го ионизационного потенциала (1/2), энергий гидратации (—Л/7°гидр) и ионных радиусов (г). Из рисунка следует, что с увеличением ионизационного потенциала и энергии гидратации катионов усиливается их гидролитическая способность. В случае ионных радиусов зависимость, разумеется, обратная. Заметно монотонное изменение свойств в ряду Ba—Sr—Са—Mg и резкий излом при переходе к бериллию. Такой характер зависимостей полностью соответствует положению элементов в периодической системе и химическому поведению этих элементов. Бериллий занимает особое положение в этой подгруппе, это видно на графиках зависимости p/Gr=
Д^°гидр, И •
Такие четкие зависимости между показателями констант гидролиза ионов и физическими параметрами ионов (ионизационные потенциалы, энергии гидратации и т. п.) в соответствии с периодическим законом могут служить подтверждением правильности найденных констант. Если такое предположение справедливо, можно допустить, что значение р/Сг для бария должно быть больше, чем указано в табл. 4.1 (13,7—14,0).
Для элементов подгруппы цинка склонность ионов к гидролизу изменяется симбатно изменению ионизационных потенциалов и энергий гидратации, причем зависимости эти немонотонны, в отличие от зависимости для элементов главной подгруппы. Это объясняется явлением вторичной периодичности [6].
На рис. 11.2 показаны графики зависимости констант гидролиза Zn2+—Cd2+—Hg2+ от ионизационных потенциалов и ионных радиусов. С увеличением U2 (и суммы U\ и U2) увеличивается склонность иона к гидролизу. Увеличение ионного ра-
155
диуса обычно приводит к уменьшению гидролиза (для лары Zn2+—Cd2+), однако в случае Hg2+, напротив, наблюдается
значительное усиление гидролиза. Если изменения для пары Zn—Cd незначительны (гидролизуемость ионов Zn2+ и Cd2H
примерно одинакова), то при переходе ко ртути наблюдаются
радикальные изломы кривых зависимостей p/Cir=f(^2, г). Четкая прямолинейная зависимость показателей первых констант
0,04 0,08 0,12 г,нм (А?
Рис. И 1.1. Зависимость показателей констант гидролиза катионов элементов главной подгруппы И группы от второго ионизационного потенциала (U2), энергии гидратации (—ДЯ°идр), ионного радиуса (г)
гидролиза Zn2+—Cd2+—Hg2-h наблюдается от эмпирически подобранной функции, включающей кроме ионизационного потенциала и ионного радиуса еще и заряд ядра (г).
16,5 17,5 , 18?5 , Д,эВ(о/
0,08 0,10 /унм(д>
Рис. 11.2. Зависимость показателей первых констант гидролиза ионов элементов подгруппы цинка от второго ионизационного потенциала (й2), ионного радиуса (г) и функции Uzzjr2
Для Zn—Cd—Hg наблюдаются и некоторые другие особенности. По кислотным свойствам ионов их можно расположить в ряд Hg2+^>Zn2+^Cd2+. Гидроокиси же этих элементов по силе кислотных свойств можно расположить в ряд Zn(OH)2> >Cd(OH)2>Hg(OH)2.
Гидролиз ионов элементов III группы. При рассмотрении гидролиза катионов элементов III группы в соответствии с положением их в периодической системе необходимо выделить четыре труппы элементов: подгруппы алюминия, группы Sc— Y—La, лантаноиды и актиноиды.
Гидролиз ионов А13+— Ga3+— 1п3+—Т13+ протекает весьма своеобразно. Наиболее основной ион в этой группе — Al3+(pAir —5,0), наиболее «кислый» — Tl3+(p/Gr= 1,2). Таким образом, наблюдается инверсия общего положения об увеличении основных свойств в группе сверху вниз. Встречающееся 156
ионов этих элементов картина
Рис. 11.3. Зависимость показателей первых констант гидролиза ионов элементов подгруппы алюминия от функции Ulz/r
в учебниках общей и неорганической химии утверждение о закономерном усилении основных свойств в третьей группе обусловлено свойствами гидратов окислов этих элементов: борная кислота в основном проявляет кислотные свойства, гидроокись алюминия — амфотерное соединение, для Т1(ОН)3 характерны основные свойства, а Т1(ОН)—сильное основание. Однако в случае гидратированных значительно более сложная. Ионные радиусы и ионизационные потенциалы изменяются скачкообразно по труппе, а изменения показателей констант гидролиза анти-си м б а тн ы ион из ационн ы м
потенциалам. Прямолинейная зависимость рК1Г для ионов элементов подгруппы алюминия от эмпирически п о до бранного п а р а м етр а
U2 • г/г, включающего ионизационный потенциал, ионный радиус и заряд ядра, показана на рис. 11.3. Из рисунка видно, что изменение гидролизуемости ионов элементов подгруппы алюминия происходит в соответствии с положением элементов в периодической системе. Следует,
однако, заметить, что физический смысл корреляции -г/г) не ясен, а сама зависимость носит локальный характер; ей не следуют гидроксокомплексы Sc, Y, La.
Для ионов Zn2+—Cd2+—Hg2+ прямолинейная зависимость p/Cir наблюдалась также от величины Однако попытки
получить прямолинейные зависимости pKir для других элементов третьей группы от U% -z/r, а также рК1Г для других элементов второй группы от Uf -zjr не привели к приемлемым резуль
татам.
В случае Sc—Y—La происходит закономерное увеличение основности, что подтверждается прямолинейными зависимостями рЛлг от третьих ионизационных потенциалов и ионных радиусов (рис. 11.4). Аналогично изменяется основность в ряду Sc (ОН) з-Y (ОН) з—La (ОН) 3.
Изменение основности ионов лантаноидов происходит в соответствии с изменением ионных радиусов: от La3+ к Lu34* уменьшаются радиусы и увеличивается сила аквакислот. Количественные корреляции в этой группе элементов затруднены,
157
так как такие важные характеристики атомов и ионов, как: ионизационный потенциал, теплота гидратации определены: лишь в некоторых случаях. Третьи и сумма первых трех ионизационных потенциалов РЗЭ рассчитаны в работах [29—32].. Однако получить четкую зависимость p/Cir от этих величин.
3
({/3, 2^0 не удалось. Зависимость р/С1Г для ионов лантанои-
дов от ионного радиуса и теплота . гидратации показаны на: рис. 11.5. С уменьшением ионного радиуса увеличиваются энергия гидратации иона и константа гидролиза. Зависимости имеют монотонный характер с изломом на гадолинии. Следует иметь в виду, что эти зависимости имеют чисто иллюстративный характер.
Как уже отмечалось в гл. 6, у актиния установлен более> основной характер, чем у лантана. В дальнейшем по ряду ак-
тиноидов склонность к гидролизу трехзарядных катионов усиливается в такой степени, что ионы Ап3+ гидролизованы более, чем ионы Ln3+.
В ряду четырехзарядных катионов актиноидов гидролиз усиливается вдоль по ряду. Исключение составляет Ра4+, который гидролизован в большей степени, чем ионы U4+, Np4+, Pu4+, Th4+. Ион Ce4+ гидролизован более, чем Ра4+, и в случае М4^ 158
наблюдается закономерно большая основность у ионов актинон* дов, чем лантаноидов.
Пятивалентные актиноиды, существующие в водных растворах в виде комплексных оксокатионов М02, не имеют аналогов в предыдущих периодах. Из сравнения известных данных для РаО^-, NpOrf' и Ри+ можно сделать вывод об уменьшений склонности к гидролизу в этом ряду. При изучении гидролиза более высокозарядных катионов Pa(V) было обнаружено, что гидролитические характеристики Ра, Nb, Та близки между собой.
Из-за существенных расхождений между данными разных, авторов трудно сделать вывод о направлении изменения основ* ности UO^, NpO|+ и PuO|+ в процессе гидролиза. Сравнение констант гидролиза UO^, WO24" и МоО|+ показывает весьма существенное различие, которое трудно допустить в одной груп-
Рис. 11.6. Зависимость показателей первых констант гидролиза ионов элементов IV группы от четвертого ионизационного потенциала:
/ — ц=1,0; 2 — |Л = 0,5; 3 — |1=0,3;
4 — ц = 0,1
пе элементов. Невозможно также сравнивать константы гидролиза Th, Zr и Hf. В обоих случаях основность актиноидов на несколько порядков выше, чем у элементов соответственно IV и VI групп.
Гидролиз ионов элементов IV группы. Как уже отмечалось в гл. 7, константы гидролиза четырехзарядных катионов элементов IV группы четко коррелируют со значениями соответствующих четвертых ионизационных потенциалов.
На рис. 11.6 показаны прямолинейные зависимости pKir3* = f(t/4), полученные на основании констант гидролиза ионов Ge [33], Ti [17], Zr [34] и Hf [19], определенных при ц = = 0,1—0,3—0,5—1,0 и Sn [18], определенных при р=1,0.
Прямые описываются уравнениями:
рК1г.= 1,24— 0,045 t/4 'р = 1,0); (11.1)
рК1г = 0,65 —0,0241/4 (р = 0.5J; (11.2>
рЛ1Г = 0,48 — 0,018t/4 Гр = 0,3); (11.3).
рК1г = 0,22 0,009С/4 (р = 0,1). (11.4)
Показатели вторых констант гидролиза также прямолинейно зависят от U4:
15»
рК2г — 1,78— 0,048 £/4 (н- 1,0); (11.5)
р/С2г- 1,24 — 0,029 £74 (и = 0,5); (11.6)
рК2г- 1,00 — 0,021 <74 (И = 0,3); (11.7)
р/С2г = 0,64 —0,010(pt = 0,1). (11.8)
В работе [20] (получены уравнения связи pKir^HUi) Для 'третьих и четвертых констант гидролиза:
р7(зг - 2,22 — 0,051 £/4 (Ц =1,0); (11.9)
р^г- 1,75— 0,033 £74 (р-0,5); (11.10)
р/Сзг — 1,45 — 0,024 £/4 (р -0,3); (11.11)
рАзг- 1,00 —0,011 £/4 (р-0,1); (11.12)
р^-2,55 — 0,0541/4 (р- 1,0); (11.13)
рК4г - 2,20 — 0,036 £/4 (р - 0,5); (11.14)
рЛ4г= 1,87 —0,027£74 (р-0,3); (11.15)
р^4г- 1,45 — 0,012£/4 (р-0,1 . (11.16)
По уравнениям (11.1) — (11.16) можно рассчитать константы
гидролиза ионов Sn(IV) при р —ОД—0,3—0,5 и Pb(IV) при р = 0Д—0,3—0,5—1,0. Для расчетов использовали значения ионизационных потенциалов свинца £74 = 40,7 эВ и олова £/4= = 39,0 эВ. Несмотря на приближенный характер полученных значений рК2Г для ионов Pb(IV), распределение гидроксокомплексов РЬ (ОН) г<4”9+ и в частности область концентрации ионов водорода, в которой существует и доминирует РЬ(ОН)4, удовлетворительно согласуются с экспериментальными данными, что РЬ(ОН)4 полностью осаждается в ОД М HNO3 [35].
Полученные константы гидролиза и уравнения связи рКтг = —/(р) для Sn(IV) и Pb(IV) приведены в табл. 7.1, а на рис. 7.8 показано распределение РЬ(ОН)г(4-9-ь в зависимости ст концентрации ионов водорода. Таким образом, количественные корреляции констант гидролиза ионов элементов с ионизационными потенциалами могут носить не только иллюстративный характер, показывая связь гидролитической способности иона элемента с положением этого элемента в периодической системе, но и служить для оценки правильности найденных значений либо экспериментально неопределяемых величин.
Гидролиз ионов элементов V группы. Анализ взаимосвязи между положением элементов V группы в периодической системе и склонностью их ионов к гидролизу затруднен многими обстоятельствами. Для ниобия и тантала наиболее характерно состояние окисления +5 и сравнение свойств их ионов возможно лишь со свойствами ионов ванадия V(V). При этом следует иметь в виду, что экспериментально определены для Nb и Та лишь константы основности и кислотности. На рис. 11.7 пока-160
заны кривые зависимости р/С1Д HVO3, HNbO3, НТаО3 от 5-го и суммы первых пяти ионизационных потенциалов V, Nb, Та, которые четко иллюстрируют уменьшение кислотности в ряду ванадиевой-ниобиевой-танталовой кислот. Однако константы основности в этом ряду закономерно изменяются лишь для ниобия и тантала. Ванадиевая кислота по литературным данным о константе равновесия VO^+H2O^tHVO3 + H+ оказывается более основным соединением, чем ниобиевая и танталовая кислоты. Может быть, эта аномалия объясняется неточностями определения констант. Возможно, причина заключа-
рА/з
Рис. 11.7. Зависимость констант диссоциации ванадиевой, ниобиевой и танталовой кислот от пятого ионизационного потенциала (U5) и от суммы первых пяти ионизационных 5 потенциалов (2^)
45 50 55 60 65Ц5,эВ(<>)
125 135 145 155 'luh3B(*j
1
ется в том, что при ионизации по основному типу указанных кислот для ванадия механизм реакции иной, чем у ниобия и тантала.
Сравнение свойств элементов другой подгруппы V группы — As, Sb, Bi — также затруднено главным образом из-за отсутствия систематических данных о константах гидролиза As(III). Возможно сравнение лишь основных и кислотных свойств гидратов окислов As (III), Sb (III) и Bi (III).
Константы основности для Bi(III) и Sb(III) при 1,0 рассчитаны из третьих констант гидролиза их ионов М3+ [36, 37], для As (III)—из данных работы [38]. Константы кислотной диссоциации получены в работах: As—[39], Sb—[40], Bi—[41]. Зависимости констант кислотной и основной диссоциации гидратов окислов As (III), Sb (III) и Bi (III) от суммы первых трех ионизационных потенциалов представлены на рис. 11.8. Эти зависимости показывают, как в ряду As—Sb—Bi с уменьшением ионизационного потенциала уменьшаются кислотные и увеличиваются основные свойства гидратов окислов.
Гидролиз ионов элементов VI группы. При гидролизе ионов элементов VI группы возможны лишь некоторые локальные корреляции. В разд. 9 говорилось об установленной зависимости между первыми константами ионизации сернистой, селенистой и теллуристой кислот и четвертыми ионизационными потенциалами этих элементов [21]. На рис. 11.9 показаны зависимости первых и вторых констант ионизации указанных кис-11 Зак. 1864 1 61
лот от четвертого и суммы первых четырех ионизационных потенциалов S, Se и Те. Характер корреляций при использовании 4
(74 и S С7г- остается неизменным; для вторых констант ионизации 1
наблюдается четкая прямолинейная зависимость. И по первой, и по второй ступеням ионизации кислотные свойства закономерно уменьшаются в ряду H2SO3—Н25еОз—Н2ТеОз. Константы ионизации сернистой кислоты получены в работах [42, 43].
Константы гидролиза МоО|+ и WO^+ свидетельствуют о большей основности вольфрамила. Для третьих констант гидролиза молибденила и вольфрамила [16, 44, 45], которые пред-
Рис. 11.8. Зависимость констант кислотности (/) и основности (2) гидратов окис-лов Bi (III), Sb (III) и As (III) от суммы первых трех ионизационных потенциалов
Р*а
9
8
7
6
5
4
3
2
1
37 39 41 43 45 4? U4,35(1{2') 96-100 104 108 112 ^1,35(1,2)
Рис. 11.9. Зависимость показателей констант ионизации H2SO3, H2SeOa и Н2ТеОз от четвертого (/, 2) ионизационного, потенциала (U4) и суммы первых четырех (/', 2') 4 ионизационных потенциалов (StA):
Л г—рКг, 2, 2'-p/G
ставляют собой первые константы ионизации молибденовой и вольфрамовой кислот, наблюдается четкая связь с первой константой ионизации хромовой кислоты. Зависимость рКщ кислот Н2СгО4—Н2МоО4—H2WO4 от шестого ионизационного потенциала показана на рис. 11.10. Как и в случае сернистой — селенистой— теллуристой кислот, в ряду хромовой —молибденовой — вольфрамовой кислот с увеличением порядкового номера элемента уменьшаются ионизационные потенциалы и кислотные
162
свойства (по первой ступени). Для вторых констант ионизации кислот Н2СгО4—Н2МОО4—H2WO4 наблюдается необъяснимое явление усиления кислотных свойств в этом ряду.
Гидролиз ионов элементов VII и VIII групп. В VII группе катионные гидроксокомплексы образуют лишь ионы Мп2+, Мп3* и Тс(IV). Сравнивать же их гидролитические характеристики возможно лишь по ряду Зй-элементов.
В VIII группе сравнительный анализ гидролитических характеристик ионов элементов триады Fe—Со—Ni и платиновых металлов в связи с положением в периодической системе за-
Рис. 11.10. Зависимость показателей констант ионизации хромовой, молибденовой и вольфрамовой кислот при ц->0 от шестого ионизационного потенциала (t76).
трудней. Эти трудности обусловлены многообразием валентных состояний, в которых существуют ионы этих элементов в водных растворах; отсутствием в большинстве случаев достаточно полных и надежных данных о константах гидролиза; невозможностью сравнения свойств ионов в разных степенях окисления.
Не удалось выяснить закономерности в гидролитическом поведении ионов элементов первого переходного ряда. Сравнение констант гидролиза в ряду двухзарядных катионов V2+—Сг2+—Мп2+—Fe2+—Со2+—Ni2+—Cu2+—Zn2+, в ряду трехзарядных катионов Sc3+—Ti3+—V3+—Сг3+—Mn3+—Fe34"—Со3+ с соответствующими ионизационными потенциалами, ионными радиусами, заполнением электронных оболочек не дает возможности сформулировать какие-либо положения, обладающие общим характером (табл. 11.1).
Можно лишь отметить некоторые частности. В ряду двухзарядных катионов наибольшими основными свойствами обладает марганец, Мп2+, ион которого имеет наполовину заполненную Зй-оболочку. За исключением Мп2+, Со2+ и Ni2+, у всех остальных двухзарядных катионов Зй-элементов гидролитические характеристики примерно одного порядка. Все катионы Зс?-эле-ментов объединяет их высокая основность и небольшая склонность к гидролизу. В ряду М34" катионов первого переходного ряда наибольшими основными свойствами обладает ион Sc34-,
11* 163
Характеристика ионов З^-элементов
не имеющий Зй-электронов. В отличие от Мп2+, у которого на Зй-орбите 5 электронов, изоэлектронный ему ион Fe3+ не обладает никакими особенностями. Его гидролитические характеристики близки к характеристикам Ti3+, V3+, Со3+. Обращает на себя внимание марганец. Если в ряду М2+ катионов он обладал самыми основными свойствами, то в ряду М3+ катионов он обладает самыми кислыми свойствами. Возможно, такое различие объясняется высоким окислительно-восстановительным потенциалом £°Мпш/Мпп= + 1,49 В.
Известно, что стандартные электродные потенциалы металлов могут служить для сравнения устойчивости комплексов с одним типом лиганда [461. В случае Зй-элементов, которые в водных растворах могут существовать в виде двух- и трехзарядных ионов, отсутствуют четкие корреляции между Е° и рКг. Можно сформулировать лишь чисто качественное обобщение: чем больше стандартный окислительно-восстановительный потенциал системы, состоящий из двух разнозарядных ионов одного и того же элемента, тем больше различие между показателями констант гидролиза ионов, образующих редокс-систему.
В случае 4d- и Sd-элементов провести анализ изменения гидролитической способности ионов в соответствии с периодическим законом вдоль по ряду не представляется возможным.
В заключение можно отметить, что, несмотря на значительные трудности, удается во многих случаях получить достаточно точные корреляции между константами гидролиза ионов элементов и параметрами, характеризующими атомы и ионы этих элементов. Особенно успешным оказалось использование ионизационных потенциалов, соответствующих заряду гидролизующегося катиона. В ряде случаев установлено, что использование в качестве параметра сравнения i-ro ионизационного потенциала допустимо наряду с применением более корректной величины — суммы первых i-x ионизационных потенциалов. Корреляции с использованием ионных радиусов, кристаллографических по Гольдшмидту, Белову и Бокию, орбитальных по Уоберу и Крамеру [47] оказались успешными лишь для элементов I и II групп. Возможно, лучшие результаты могут быть получены при использовании радиусов гидратированных ионов, так как гидролиз представляет собой в конечном счете кислотную ионизацию гидратированного катиона. К сожалению, в настоящее время отсутствуют строго определенные значения таких радиусов. Недостаточно эффективной оказалась корреляция констант гидролиза ионов элементов с относительными ионизационными потенциалами ]n(Ui + Un)/2rn [48].
Нет сомнения, что гидролиз ионов элементов в группах периодической системы происходит в соответствии с периодическим законом. Особенности таких сложных систем, как разбавленные водные растворы, не всегда позволяют эту взаимосвязь увидеть в чистом виде. Поэтому в тех случаях, когда удается
165
обнаружить какую-либо закономерность в изменении гидролитических характеристик ионов, обусловленную положением элементов в периодической системе, установленные корреляции могут служить дополнительным критерием правильности определенных констант, для ориентировочных оценок констант гидролиза, которые по разным причинам либо не могли быть определены экспериментально, либо вызывают сомнение.
Следует также отметить, что, несмотря на огромное количество данных о константах гидролиза ионов различных элементов, в настоящее время еще не все элементы изучены достаточно полно. Во многих случаях, как это отмечалось в гл. 3—10, необходимы надежные данные о силе аквакислот, нужны полные наборы констант гидролиза ионов по всем ступеням. В настоящее время все еще существует необходимость в исследованиях по определению констант гидролиза отдельных ионов, так как без таких работ невозможны строгие обобщения.
СПИСОК ЛИТЕРАТУРЫ
К гл. 1.
1. Россотти Ф., Россотти X. Определение констант устойчивости, и других констант равновесия в растворах. Пер. с англ. Под ред. Д. И. Рябчи • кова. М., «Мир», 1965.
2. Weinland R. F., Ensgraber F.— «Z. anorgan. und allgem. Chem.», 1915, Bd 84, S. 340, 368.
3. Jander G., Jahr K. F.— «Kolloid Beihefte», 1936, Bd 43, S. 303, 305, 323.
4. Olson A. R., Simonson T. R.—«J. Chem. Phys.», 1949, v. 17, p. 1322.
5. Sykes K. W.—«J. Chem. Soc.», 1959, p. 2473.
6. Richards D. H., Sykes K. W.— «J. Chem. Soc.», 1960, p. 3626.
7. Sutton J.— «Nature», 1952, v. 169, p. 71.
8. Jones M. M., Jones E. A., Harmon D. F. e. a.— «J. Amer. Chem. Soc.», 1961, v. 83, p. 2038.
9. Dunsmore H. S., Sillen L. G.—«Acta chem. scand.», 1963, v. 17, p. 2657.
10. Dunsmore H. S., Hietanen S., Sillen L. G.— Ibid., p. 2644.
11. Hietanen S., Row B. R. L., Sillen L. G.— Ibid., p. 2735.
12. Scheidin U., Frydman M.— Ibid., 1968, v. 22, p. 115.
13. Danes! P. R., Magini M., Margherite S. e. a.— «Energia Nucl.», 1968, v. 15. p. 335.
14. Milic N. B.— «Acta chem. scand.», 1971, v. 25, p. 2487.
15. Бурков К. А., Л и лич Л. С,—«Вести. Ленингр. ун-та», 1965, № 10, с. 103.
16. Milic N. В.— «J. Chem. Soc., Dalton Trans». 1973, N 2, p. 229.
17. Contributions to Coordination Chemistry in Solution. Trans. Royal Inst. Technol. Stockholm, 1972.
18. Ahrlans S., Bovin J. O.— «Acta chem. scand. А», 1974, v. 28, p. 1089.
19. Baes C. F., Mesmer R. E. The Hydrolysis of Cations. N. Y., John Wiley and Sons, 1976.
20. Кульба Ф. Я., Яковлев Ю. Б., Зенченко Д. А.— «Журн. неорган. химии», 1975, т. 20, с. 1781.
21. Першин А. С.— «Радиохимия», 1972, т. 14, с. 159.
22. Biedermann G., Kilpatrik М., Pokras L. е. а.— «Acta chem. scand.», 1956, v. 10, p. 1327.
23. Старик И. E. Основы радиохимии. М.—Л., Изд:во АН СССР, 1959.
24. Давыдов Ю. П.— В кн.: Формы элементов и радионуклидов в морской воде. М., «Наука», 1974, с. 25.
25. Гринберг А. А. Введение в химию комплексных соединений. М.— Л., «Химия», 1966, с. 431.
26. Поляк Э. А.— «Журн. общ. химии», 1975, т. 45, с. 2357.
27. Яцимирский К. Б., Васильев В. П. Константы нестойкости комплексных соединений. М., Изд-во АН СССР, 1959.
28. Шлефер Г. Л. Комплексообразование в растворах. М.— Л., «Химия», 1964.
29. Бьеррум Я. Образование аминов металлов в водном растворе. М., Изд-во иностр, лит., 1961.
167
30. Leden I.— «Z. phys. Chem.», 1941, Bd A 188, S. 160.
31. Fronaeus S.— «Acta chem scand.», 1950, v. 4, p. 72.
32. Яцимирский К- Б.—«Журн. неорган. химии», 1956, № 1, с. 412.
33. Sillen L G.—«Acta chem. scand.», 1956, v. 10, p. 186.
К гл. 2
1. Baes C. F., Mesmer R. E. The Hydrolysis of Cations. N. Y., John Wiley and Sons, 1976.
2. Комарь H. П.— В кн.: Труды хим. фак-та и науч-исслед. ин-та химии Харьковск. ун-та», 1963, т. 19, с. 189.
3. Бейтс 3. Р. Определение pH. Теория и практика. Л., «Химия», 1968.
4. Hietanen S., Sillen L. G.— «Acta chem. scand.». 1952, v. 6, p. 747.
5. Фролова У. К., Кумок В. Н., Серебренников В. В.—«Изв. вузов. Химия и хим. технология», 1966, т. 9, с. 176.
6. Бурков К. А., Лилич Л. С., Нгуен Динь Нго и др.— «Жури. неорган. химии», 1973, т. 18, с. 1513.
7. Hietanen S., Sillen L. G.—«Acta chem. scand.», (1959, v. 13, p. 533.
8. Яцимирский К. Б.— «Журн. неорган. химии», 1956, т. 1, с. 2306.
9/ Яцимирский К. Б., Васильев В. П. Константы нестойкости комплексных соединений. М., Изд-во АН СССР, 1959, с. 47.
10. Olson А. К., Simonson Т. R.— «J. Chem. Phys.», 1949, v. 17, р. 1322.
11. Глебов В. А., Клыгин А. Е., Смирнова И. Д. и др.— «Журн. неорган. химии», 1972, т. 17, с. 3312.
12. Amjad Z., McAuleys A.—«J. Chem. Soc. Dalton Trans.», 1974, N 23,. p. 2521.
13. Offner H. G., Skoog D. A.— «Analyt. Chem.», 1966, v. 38, p. 1520.
14. Фомин В. В., Майорова Е. П.—«Журн. неорган. химии», 1956, т. 1Г с. 1703.
15. Назаренко В. А., Антонович В. П., Невская Е. М.—Там же, 1968, т. 13^ с. 1574.
16. Бирюк Е. А., Назаренко В. А.— Там же, 1973, т. 18, с. 2964.
17. Назаренко В. А., Невская Е. М.— Там же, 1969, т. 14, с. 3215.
18. Назаренко В. А., Бирюк Е. А.-— Там же, 1974, т. 19, с. 632.
19. Антонович В. П., Назаренко В. А.— Там же, 1968, т. 13, с. 1805.
20. Бирюк Е. А., Назаренко В. А., Равицкая Р. В.— Там же, 1969, т. 14^ с. 965.
21. Бирюк Е. А., Назаренко В. А., Лям Нгок Тху.— Там же, 1969, т. 14г с. 714.
22. Назаренко В. А., Антонович В. П., Невская Е. М.— Там же, 1971, т. 16г с. 997.
23. Назаренко В. А., Манджгаладзе О. В.—Там же, 1969, т. 14, с. 1219.
24. Назаренко В. А., Антонович В. П., Невская Е. М.— Там же, 1971, т. 16,. с. 2387.
25. Назаренко В. А., Флянтикова Г. В.— Там же, 1968, т. 13, с. 1855.
26. Назаренко В. А., Антонович В. П., Невская Е. М.— Там же, 1971, т. 16^ с. 1844.
27. Назаренко В. А., Шелихина Е. И.— Там же, 1971, т. 16, с. 166.
28. Назаренко В. А., Полуэктова Е. Н.— Там же, 1969, т. 14, с. 204.
29. Назаренко В. А., Полуэктова Е. Н., Шитарева Г. Г.—-Там же, 1977,. т. 22, с. 998.
30. Антонович В. П., Невская Е. М., Шелихина Е. И. и др.— Там же, 1975,. т. 20, с. 2968.
31. Антонович В. П., Невская Е. М., Суворова Е. Н.— Там же, 1977, т. 22,. с. 1278.
32. Назаренко В. А., Шитарева Г. Г., Полуэктова Е. Н.—Там же, 1977„ т. 22, с. 980.
33. Андрианов А. М., Назаренко В. А.~Там же, 1966, т. 11, с. 1527.
34. Соловкин А. С.—Там же, 1957, т. 2, с. 611.
168
35. Лобанов Ф. И., Савостина В. М., Серженко Л. В. и др.— Там же, 1969^ т., 14, с. 1077.
36. Пешкова В. М., Мельчакова Н. В., Пэн Ан. «Труды комиссии по аналит. химии», 1963, т. 14, с. 172.
37. Соловкин А. С., Иванцов А. И.—«Журн. неорган. химии», 1966, т. 11,.. с. 1897.
38. Fronaeus S.—«Acta chem. scand.», 1951, v. 5, p. 139.
39. Fronaeus S.— Ibid., 1950, v. 4, p. 72.
40. Соловкин A. C.— «Журн. физ. химии», 1963, т. 37, с. 447.
41. Соловкин А. С. Высаливание и количественное описание экстракционных, равновесий. М., Атомиздат, 1969.
42. Соловкин А. С. Высаливание и количественное описание экстракционных равновесий. «Неорганическая химия», т. 3 (Итоги науки и техники),, IM., ВИНИТИ, 1972.
43. Аксельруд Н. В., Спиваковский В. Б —«Журн. HteopraH. химии», 1957г т. 2, с. 2709.
44. Feitknecht W., Schindler Р.— «Риге Appl. Chem.», 1963, v. 6(2), р. 130.
45. Garrett А. В., Heiks R. Е —«J. Amer. Chem. Soc», 1941, v. 63, p. 562.
46. Garrett A. B., Howell W. W.—Ibid., 1939, v. 61, p. 1730.
47. Tobias R. S., Garrett A. B.— Ibid., 1958. v. 80, p. 3532.
48. Москвин А. И.— «Радиохимия», 1971, т. 13, с. 681.
49. Набиванец Б. И., Кудрицкая Л. Н.— «Укр. хим. журн.», 1964, т. 30?. с. 891.
50. Biedermann G —«Arkiv kemi», 1953, Bd 5, S. 441.
51. Brosset C.— «Svensk kem tidskr.», 1941, v. 53, p. 434.
52. Оксредметрия. Под ред. Б. П. Никольского. Л., «Химия», 1975.
53. Якубов X. М., Оффенгенден Е. Я., Пальчевский В. В. Комплексообразование в окислительно-восстановительных системах. Под ред. Б. П. Никольского. Душанбе, Изд-во Таджикск. гос. ун-та, 1972, с. 52—59.
54. Никольский Б. П., Пальчевский В. В., Пендин А. А. —«Докл. АН СССР»,. 1971, т. 196, № 3, с. 609.
55. Якубов X. М., Пальчевский В. В.—Докл. АН ТаджССР», 1964, т. 7, № 4, с. 16.
56. Колосов И. В., Инцкирвели Л, Н., Варшал Г. М.— «Журн. неорган. химии», 1975, т. 20, с. 2121.
57. Электромиграционный метод в физико-химических и радиохимических исследованиях. Под ред. В. П. Шведова. iM., Атомиздат, 1971.
58. Степанов А. В., Шведов В. П.—«Журн. неорган. химии», 1965, т. 10,, с. 1000.
59. Шалинец А. Б., Степанов А. В.— «Радиохимия», 1972, т. 14, с. 280.
60. Marin В., Kikindai Т., Gourisse D.— «Note СЕА», 1968, N 1044, р. 332.
61. Marin В., Kikindai Т.— «Compt. rend. Acad, sci.», 1969, v. C—-268, p. 1..
62. Козырев В. M., Ривкинд А. И.—«Журн. эксперим. и теор. физ.», 1954,. т. 27, с. 69.
63. Вдовенко В. М., Романов Г. А., Щербаков В. А.— «Радиохимия», 1963,. т. 5, с. 137.
64. Вдовенко В. М. Химия урана и трансурановых элементов. М.— Л.^ Изд-во АН СССР, 1960.
65. Попель А. А. Применение ядерной магнитной релаксации в анализе неорганических соединений. Казань, Изд-во Казанск. ун-та», 1975.
К гл. 3
1. Baes С. Е, Mesmer R. Е. The Hydrolysis of Cations. N. Y., John Wiley and Sons, 1976.
2. Бурков К А., Бусько E. А., Зиневич H. И.—«Вести. Ленингр. ун-та»,. 1975, вып. 1, с. 144.
3. Olofsson G., Hepler L. G.— «J. Solut. Chem.», 1975, v. 4, p. 127.
4. Harned H. S., Copson G. R.— «J. Amer. Chem. Soc.», 1933, v. 55, p. 2206.
169
3. Ackermann Т.—«Z. Elektrochem.», 1958, Bd 62, S. 411.
3. Перковец В. Д., Крюков П. А.—«Изв. СО АН СССР. Сер. хим.», 1969, № 7 (157), вып. 3, с. 9.
7. Gimblett F. G. R., Monk С. В.— «Trans. Faraday Soc.», 1950, v. 50, p. 965.
8. Ohtaki H.— «Acta chem. scand.», 1964, v. 18, p. 521.
9. Masterton W. L., Berka L. H.— «J. Phys. Chem.», 1966, v. 70, p. 1924.
10. Jones J. R.— «Trans Faraday Soc», 1968, v 64, p. 440.
11. Davies C. W.— «J. Chem. Soc.», 1935, p. 910.
12. Cranston J. A., Brown H. F.—«J. Roy. Techn. College», (Glasgow), 1937, v. 4, p. 54. Цит. no (17).
13. Quintin M.—«Compt. rend.», 1937, v. 204, p. 968.
"14. Pedersen K. J.—«Kgl. danske vid. seiscab. Mat.-fys. medd.», 1943, Bd 20, S. 7 (цит. no Berecki-Biedermann C. «Arkiv Kemi», 1956, v. 9, p. 175).
15. Chaberek S., Country R. C., Martell A., G.— «J. Amer. Chem. Soc.», 1952, v. 74, p. 5057.
16. Schwab G. M., Polydoropoulos K.— «Z. anorgan. und allgem. Chem.», 1953, Bd 274, S. 234.
17. Achenza F.~~ «Ann. Chimica», 1958, v. 48, p. 565; 1964. v. 54, p. 240.
18. Perrin D. D.— «J. Chem. Soc.», 1960, p. 3189.
19. Mahapatra S., Subrahmanya R. S — «Proc. Indian Acad. Sci.», 1967, v. A 65, N 5, p. 283.
20. Спиваковский В. Б., Маковская Г. В.—«Жури. неорган. химии», 1968, т. 13, с. 1555.
“21. Ohtaki Н.— «Inorg. Chem.», 1968, v. 7. р. 1205.
22. Kakihana Н., Amaya Т., Maeda М.—«Bull. Chem. Soc. Japan», 1970, v. 43, p. 3155.
23. Ohtaki H., Kawai T —Ibid., 1972, v. 45, p. 1735.
’24. Lorn E.—«Z. anorgan. und allgem. Chem.», 1927, Bd 165, S. 327.
25. Johnston H. L., Guta F., Garrett A. B.—«J. Amer. Chem. Soc.», 1933, v. 55, p. 2311.
26. Carriere E., Guiter H.— «Bull. soc. chim. France», 1947, p. 267.
*27. Faucherre J.— Ibid., 1954, p. 253.
28. Beck M. T.— «Acta chim. Acad, scient. hung.», 1954, v. 4, p. 227.
29. Antikainen P. J., Dyrssen D.— «Acta chem. scand.», 1960, v. 14, p. 86.
30. Biedermann G., Hietanen S.— Ibid., p. 711.
31. Biedermann G., Sillen L. G.— Ibid., p. 717.
32. Gubeli A. O., Ste-Marie J.— «Canad. J. Chem.», 1967, v. 45, p. 827.
33. Johnston H. L., Leland H. L.— «J. Amer. Chem. Soc.», 1938, v. 60, p. 1439.
34. Справочник химика. Изд. 2. T. 4. М.—Л., «Химия», с. 56—57.
35. Пещевицкий Б. И., Белеванцев В. И., Земсков С. В,— «Изв. СО АН СССР. Сер. хим.», 1976, вып. 2, № 4, с. 24.
36. Arena G., Cali R., Rizzarelli E. e. a.— «Thermochim. acta», 1976, v. 16, p. 315.
К г л . 4
1. Prytz M.— «Z. anorgan. und allgem. Chem.», 1929, Bd 180, S. 355.
2. Mattock G.— «J. Amer. Chem. Soc.», 1954, v. 76, p. 4835.
3. Kakihana H., Sillen L. G.— «Acta chem. scand.», 1956, v. 10, p. 985.
4. Gilbert R. A., Garrett A. B.— «J. Amer. Chem. Soc.», 1956, v. 78, p. 5501.
5. Адамович Л. П., Шупенко Г. С.—«Укр. хим. жури.», 1959, т. 25, с. 155.
6. Schwarzenbach G.— «Pure and Appl. Chem.», 1962, v. 2, p. 377.
7. Hietanen S., Sillen L. G.— «Acta chem. scand.», 1964, v. 18, p. 1015.
8. Green R. W., Alexander P. W.— «Austral. J. Chem.», 1965, v. 18, p. 651.
9. Bertin F., Thomas G., Merlin J. C.— «Compt. rend.», 1965, v. 260, p. 1670; «Bull. Soc. chim. France», 1967, p. 2393.
10. Ohtaki H.— «Inorg. Chem.», 1967, v. 6, p. 808.
11. Ohtaki H., Kato H.—Ibid., p. 1935.
170
12. Руденко Н. П., Севастьянов А. И.— «Журн. неорган. химии», 1969, т. 14, с. 848.
13. Schwarzenbach G., Wenger Н.— «Helv. chim. acta», 1969, v. 52, p. 644.
14. Kakihana H., Maeda M.— «Bull. Chem. Soc. Japan», 1970, v. 43, p. 109.
15. Tsukuda H., Kawai T., Maeda M. e. a.—Ibid., 1975, v. 48, p. 69L
16. Kolthoff I. M.— «Recueil trav. chim.», 1923, v. 42, p. 973.
17. Gjaldbaek J. K-—«Z. anorgan. und allgem. Chem.», 1925, Bd 144, S. 269.
18. Stock D. J., Davies C. W.— «Trans. Faraday Soc.», 1948, v. 44, p. 856.
19. Lewis D.—«Acta chem scand.», 1963, v. 17, p. 1891.
20. Jones J. R.—«Trans. Faraday Soc.», 1968, v. 64, p. 440.
.21. McGee K. A., Hostetler P. B.—«Amer. J. Sci.», 1975, v. 275, N 3, p. 304.
22. Kilde G.— «Z. anorgan. und allgem. Chem.», 1934, Bd 218, S. 118; 1936, Bd 229, S. 321.
23. Davies C. W.— «J. Chem. Soc.», 1938, p. 277.
24. Bell R. P., Prue J. Е,— Ibid., 1949, p. 362.
25. Bell R. P., Waind G. M.~ Ibid., 1950, p. 1979.
26. Davies C. W., Hoyle В. E.—Ibid., 1951, p. 233.
27. Bell R. P., George J. H. B.— «Trans. Faraday Soc.», 1953, v. 49, p. 619.
28. Gimblett F. G. R, Monk С. B.— Ibid., 1954, v. 50, p. 965.
29. Bell R. P., Panckhurst M. H.—«J. Chem. Soc.», 1956, p. 2836.
30. Bates R. W., Bower V. E., Canham R. G. e. a.— «Trans, Faraday Soc.», 1959, v. 55, p. 2062.
31. Carell B., Olin A,.—«Acta chem. scand.», 1961, v. 15, p. 727.
32. Hopkins H. P„ Wulff C. A.—«J. Phys. Chem.», 1965, v. 69, p. 6.
33. Colman-Porter C„ A., Monk С. B.— «J. Chem. Soc.», 1952, p. 1313.
34. Harned H. S., Mason С. M.— «J. Amer. Chem. Soc.», 1932, v. 54, p. 1439.
35. Davies C. W.— «J. Chem. Soc.», 1939, p. 349.
36. Kruger G., Thilo E.-—«Z. anorgan. und allgem. Chem.», 1961, Bd 308, S. 242.
37. Carell B., Olin A.— «Acta chem. scand.», 4961, v. 15, p. 1875.
38. Paris M. R., Gregoire Cl.— «Analyt. chim. acta», 1968, v. 42, p. 431.
39. Lanza E., Carpeni G.— «Electrochim. acta», 1968, v. 13, p. 519.
40. Lanza E.— «Rev. chim. miner.», 1969, v. 6, p. 653.
41. Kakihana H., Maeda M.— «Bull. Chem. Soc. Japan», 1969, v. 42, p. 1458.
42. Cooper M. K., Garman D. E. J., Yaniuk D. W.— «J. Chem. Soc. Dalton Trans.», 1974, p. 1282
43. Mesmer R. S.^ Baes C. F.— «Inorg. Chem.», 1967, v. 6, p. 1957.
44. Baes C. F., Mesmer R. E. The Hydrolysis of Cations. N. Y., John Wiley and Sons, 1976.
45. Chaberek S., Country R. C., Martell A., C —«J. Amer. Chem. Soc.», 1952, v. 74, p. 5057.
46. Kolthoff J. M., Kameda T — Ibid., 1929, v. 51, p. 2888; 1931, v. 53, p. 832.
47. Prytz M.— «Z. anorgan. und allgem. Chem.», 1931, Bd 200, S. 133.
48. Owen В. B., Gurry R. W.— «J. Amer. Chem. Soc.», 1938, v. 60, p. 3074.
49. Hagisawa H.— «Bull. Inst. Phys. Chem. Res. Tokyo», 1939, v. 18, p. 368. Цит. no [58].
50. Brown H. F., Cranston J. A.— «J. Chem. Soc.», 1940, p. 578.
51. Коршунов И. А., Хрулькова .E. Ф.^<Журн. общ. химии», 1-949, т. 19, с. 2045.
52. Bernheim Р., Quintin М.— «Compt. rend.», 1950, v. 230, р. 388.
53. Стабровский А. И.— «Жури. общ. химии», 1951, т. 21, с. 1223; «Журн. физ. химии», 1952, т. 26, с. 949.
54. Витченко Н. К., Тихонов А. С. Труды Воронежск. ун-та. Сб. работ хим. ф-та. Харьков, Изд-во Харьковск. ун-та, 1953, с. 129.
55 Schwab G. М., Polydoropoulos К.—«Z. anorgan. und allgem. Chem.», 1953, * Bd 274, S. 234.
56. Dirkse T. P.— «J. Electrochem. Soc.», 1954, v. 101, p. 328.
57. Fulton J. W., Swinehart D. F.—«J. Amer. Chem. Soc.», 1954, v. 76, p. 864.
58. Achenza F.— «Ann. Chimica», 1958, v. 48, p. 565; 1964, v. 54, p. 240.
171
59. Matsuda H., Ayabe Y.— «Z. Electrochem.», 1959, Bd 63, S. 1164.
60. Besson J., Eckert W.— «Bull. Soc. chim. France», 1959, p. 1676.
61. Dye J. L., Eqber M. P., Karl D. J.— «J. Amer. Chem. Soc.», 1960, v. 82r p. 314.
62. Perrin D. D.— «J. Chem. Soc.», 1962, p. 4500.
63. Schorsch G., Bye J.— «Compt. rend.», 1963, v. 257, p. 2833.
64. Schorsch G.— «Bull. Soc. chim. France», 1964, v. 1449.
65. Sekine T.— «Acta chem. scand.», 1965, v. 19, p. 1526.
66. Schorsch G.— «Bull. Soc. chim. France», 1965, p. 988.
67. Gubeli A. O., Ste-Marie J.— «Canad. J. Chem.», 1967, v. 45, p. 827.
68. Окунев M. C.— «Труды по химии и хим. технологии» (Горький), 1973г вып. 3 (34), с. 41.
69. Ходаковский И. Л., Елкин А. Е.—«Геохимия», 1975, № 10, с. 1490.
70. Николаева Н. М.—«Журн. неорган. химии», 1969, т. 14, с. 936.
71. Reichle R. A., McCurdy К. G., Hepler L. G.— «Canad. J. Chem.», 1975r v. 53, p. 3841.
72. Marcus J.— «Acta chem. scand.», 1957, v. 11, p. 690.
73. Dyrssen D., Lumme P. L.—Ibid., 1962, v. 16, p. 1785.
74. Biedermann G., Ciavatta L.— Ibid., p. 2221.
75. Спиваковский В. Б., Мойса Л. П.— «Журн. неорган. химии», 1964, т. 9, с. 2287.
76. Goward G. W. Thesis, Princeton, 1954. Univ. Microfilms, N 9414..
77. Matsui H., Ohtaki H.—«Bull. Chem. Soc. Japan», 1974, v. 47, p. 2603.
78. Fuseya G.—«J. Amer. Chem. Soc.», 1920, v. 42, p. 368.
79. Garrett A. B., Hirschler A. E.— Ibid., 1938, v. 60, p. 299.
80. Garrett A. B,, Howell W. W.— Ibid., 1939, v. 61, p. 1730.
81. Hietanen S., Sillen L. G.— «Acta chem. scand.», 1952, v. 6, p. 747; «Arkiv Kemi», 1956, Bd 10, S. 103.
82. Anderegg G., Schwarzenbach G., Padmoyo M. e. a.— «Helv. chim. acta»r 1958, v. 41, p. 988.
83. Newman L., Hume D. N.— «J. Amer. Chem. Soc.», 1959, v. 81, p. 5901.
84. Dyrssen D., Tyrrell V.— «Acta chem. scand.», 1961, v. 15, p. 393.
85. Ahlberg J.— Ibid., 1962, v. 16, p. 887.
86. Комарь H. П.— «Труды хим. фак-та и науч, исслед. ин-та химии Харь— ковск. ун-та», 1963, т. 19, с. 189.
87. Ahlberg JL, Leden J. Contributions to Coordination Chemistry in Solution.— «Trans. Roy. Inst. Technol. Stockholm», 1972, N 249, p. 16.
88. Ciavatta L., Grimaldi M., Palombari R.— «J. Inorg. and Nucl. Chem.»,. 1975, v. 37, p. 1685.
89. Справочник химика. Изд. 2. T. 4. М.— Л., «Химия», 1965, с. 56—57.
90. Колосов И. В.—«Координац. химия», 1978, т. 4, с. 531.
91. Hietanen S., Hogfeldt Е.— «Chem. scr.», 1976, v. 10, p. 41.
К гл. 5
1. Sadek H., Tadros Th. F.—«Z. phys. Chem.», 1959, Bd 19, S. 193.
2. Адамчук И. П., Пачаджанов Д. Н.— В кн.: Химия в Таджикистане. Душанбе, «Дониш», 1973, с. 156.
3. Адамчук И. П., Пачаджанов Д. Н., Валиев Ю. Я. Там же с. 157.
4. Bronsted J. N., Volqvartz К..— «Z. phys. Chem.», 1928, Bd 134, S. 97.
5. Hartford W. H.—«Ind. Engng Chem.», 1942, v. 34, p. 920.
6. Чернов В. А. О природе почвенной кислотности. М.— Л., Изд-во АН СССР, 1947, с. 42.
7. Lacroix S.— «Ann chimie», 1949, v. 4, p. 5.
8. Faucherre J —«Compt. rend.», 1948, v. 227, p. 1367; «Bull. soc. chim, France», 1954, p. 253.
9. Ko T., Yui N.—«Sci. Rep. Tohoku Univ. Ser. I», 1953, v. 37, p. 185; C. A. 1954, v. 48, p. 5613.
10. Schofield R. K., Taylor A. W.— «J. Chem. Soc.», 1954, p. 4445.
172
11. Kenttamaa J.—«Suomen kern.», 1955, v. 28B, p. 172.
12. Иванов A. H., Алешин С. Н,— «Докл. ВАСХНИЛ», 1956, т. 22, с. 386.
13. Kubota H.— Thesis Univ. Wisconsin, 1956. Univ. Microfilms, 16183; «Dissert. Abstrs», 1956, v. 16, p. 864. Цит. no [14].
14. Frink C. R., Peech M.—«Inorg. Chem.», 1963, v. 2, p. 473.
15. Nishida T., Tsuchiya R.— «Bull. Chem. Soc. Japan», 1965, v. 38, p. 1398.
16. Hem J. D., Roberson С. E.—«Geol. Surv. Water-Supply Paper», 1967, N 1827A. РЖХ, 1968, 16Б1247.
17. Holmes L. P., Cole D. L., Euring E. M.— «J. Phys. Chem.», 1968, v. 72, p. 30.
18. Srinivasan K., Rechititz G. A.— «Analyt. Chem.», 1968, v. 40, p. 1818.
19. Grunwald E., Fong Dodd-Wing. — «J. Phys. Chem.», t 1969, v. 73, p. 650.
20. Назаренко В. А., Невская E. M.—«Журн. неорган. химии», 1969, т. 14, с. 3215.
21. Николаева Н. М., Толпыгина Л. Н.— «Изв. СО АН СССР. Сер. хим.»,
1969, вып. 3, № 7, с. 49. ,
22. Волохов Ю. А., Павлов Л. Н., Еремин Н. И. и др.—«Журн. прикл. химии», 1971, т. 44, с. 246.
23. Dezelic N., Bilinski Н., Wolf R. Н.—«J. Inorg. and Nucl. Chem.», 1971, v. 33, p. 791.
24. Stefanowicz T., Kiciak S.— «Roczn. chem.», 1972, v. 46, p. 1209.
25. Назаренко В. А., Бирюк E. A.—«Журн. неорган. химии», 1974, т. 19, с. 632.
26. Turner R. С.—«Canad J. Chem.», 1975, v. 53, p. 2811.
27. Коненкова T. Я. ВИНИТИ № 164—75 Деп.
28. Fricke R., Meyring K.— «Z. anorgan. und allgem. Chem.», 1928, Bd 176, S 325
29. Moeller T., King G. L..—«J. Phys. Chem.», 1950, v. 54, p. 999.
30. Wilson A. S., Taube H.— «J. Amer. Chem. Soc.», 1952, v. 74, p. 3509.
31. Fetter N. R. Doctor Dissert., 1957 (University of Oregon).
32. Иванов-Эмин Б. H., Нисельсон Л. А., Ларионова Л. E.— «Журн. неорган. химии», 1962, т. 7, с. 522.
33. Алимарин И. П., Шериф Абдель Хамид, Пуздренкова И. В.—Там же, 1965, т. 10, с. 389.
34. Савостин А, П.— Там же, 1966, т. 11, с. 2817.
35. Назаренко В. А., Антонович В. П., Невская Е. М.—Там же, 1968, т. 13, с. 1574.
36. Яценко С. П.—'«Труды ин-та химии У ФАН СССР», 1970, вып. 20, с. 153.
37. Liljenzin J .О., Vadasdi К., Rydberg J. Contributions to Coordination Chemistry in Solution.— «Trans. Roy. Inst. Technol.», 1972, N 280, p. 407.
38. Гордиенко В. И.—«Журн. общ. химии», 1973, т. 43, с. 2117.
39. Бирюк Е. А., Назаренко В. А — «Журн. неорган. химии», 1973, т. 18, с. 2964.
40. Сидоренко В., Журавлев Е. Ф., Гордиенко В. И. Тезисы докладов II Всесоюзного совещания «Термодинамика и структура гидроксокомплексов в растворах». Л„ «Наука», 1975, с. 5.
41 Hattox Е. М., De Vries T,—«J. Amer. Chem. Soc.», 1936, v. 58, p. 2126.
42. Moeller T — Ibid., 1941, v. 63, p. 1206; 1942, v. 64, p. 953.
43. Hepler L. G., Hugus Z. Z — Ibid., 1952, v. 74, p. 6115.
44. Biedermann G.— «Arkiv kemi», 1956, Bd 9, S. 277.
45. Biedermann G.— «Recueil trav. chim.», 1956, v. 75, p. 716.
46. Rossotti F. J., Rossotti H.— «Acta chem. scand.», 1956, v. 10, p. 779.
47. Biedermann G., Li N. C., Yu J.—Ibid., 1961, v. 15, p. 555.
48. Шериф Абдель Хамид, Алимарин И. П., Пуздренкова И. В.— «Вести. Моск, ун-та. Сер. хим.», 1965, № 3, с. 71.
49. Ьирюк Е. А., Назаренко В. А., Равицкая Р. В—«Журн. неорган. химии», 1969, т. 14, с. 965.
50. Aziz A.. Lvle S. J.— «J. Inorg. and Nucl. Chem.», 1969, v. 31, p. 2431.
173
51. Кульба Ф. Ям Зенченко Д. А., Яковлев Ю. Б. В кн.: XXVI Герценов-ские чтения. Химия. Научные доклады. Вып. 1. Л., 1973, с. 3.
52. Кульба Ф. Я-, Яковлев Ю. Б., Зенченко Д. А.—«Журн. неорган. хи- , мии», 1974, т. 19, с. 923.
53. Кульба Ф. Ям Яковлев Ю. Б., Зенченко Д. А.— Там же, 1975, т. 20,. с. 1781.
54. Яковлев Ю. Б., Кульба Ф. Я., Зенченко Д. А.—Там же, 1976, т. 21, с. 61..
55. Bell R. Р., Prue J. Е.—«J. Chem. Soc.», 1949, р. 362.
56. Bell R. Р., George J. H. В.— «Trans. Faraday Soc.», 1953, v. 49, p. 6, 9.
57. Bell R. P., Panckhurst M. H.—«J. Chem. Soc.», 1956, p. 2836.
58. Кульба Ф. Ям Миронов В. Е. Химия таллия. М., Госхимиздат, 1963,. с. 82.
59. Кульба Ф. Ям Яковлев Ю. Б., Копылов Е. А.— «Журн. неорган. химии», 1970, т. 15, с. 2112.
60. Кульба Ф. Ям Зенченко Д. А., Яковлев Ю. Б.—Там же, 1975, т. 20,.. с. 2372.
61. Benoit R.— «Bull. soc. chim. France», 1949, p. 518.
62. Harbottle Gm Dodson R.—«J. Amer. Chem. Soc.», 1951, v. 73, p. 2442.
63. Johnson С. E.— Ibid., 1952, v. 74, p. 959.
64. Biedermann G.—«Arkiv kemi.», 1953, Bd 5, S. 441.
65. Dodson R. W.— «J. Amer. Chem. Soc.», 1953, v. 75, p. 1795.
66. Brubaker С. H., Mickel J. P.— «J. Inorg. and Nucl. Chem.», 1957, v. 4, p. 55.
67. Rogers T. E., Waind G. M.—«Trans. Faraday Soc.», 1961, v. 57, p. 1360.
68. Комарь H. П.—«Труды хим. фак-та и науч, исслед. ин-та химии Харь-ковск. ун-та», 1963, т. 19, с. 189.
69. Кульба Ф. Ям Яковлев Ю. Б., Миронов В. Е.—«Журн. неорган. химии»,. 1964, т. 9, с. 2573.
70. Лобов Б. И., Кульба Ф. Я., Миронов В. Е.—«Журн. физ. химии», 1966, т. 40, с. 2527.
71. Лобов Б. И., Кульба Ф. Ям Миронов В. Е.— «Журн. неорган. химии»,, т. 12, с. 334.
72. Лобов Б. И., Кульба Ф. Я., Миронов В. Е.— Там же, 1967, т. 12, с. 341.
73. Яковлев Ю. Б., Кульба Ф. Ям Миронов В. Е.— В кн.: Проблемы современной химии координационных соединений. Вып. 2. Л., Изд-во Ле-нингр, ун-та, 1968, с. 241.
74. Бирюк Е. А., Назаренко В. А., Лям Нгок Тху.~«Журн. неорган. химии», 1969, т. 14, с. 714.
75. Родина Т. Ф., Левин И. С., Ворсина И. А.— «Изв. СО АН СССР. Сер. хим. наук», 1971, вып. 3, с. 71.
76. Кульба Ф. Ям Копылов Е. А., Яковлев Ю. Б. и др.— «Журн. неорган. химии», 1972, т. 17, с. 2604.
77. Кульба Ф. Ям Зенченко Д. А., Яковлев Ю. Б.— Там же, 1975, т. 20,. с. 2645.
78. Fiat D., Connick R. Е.—«J. Amer. Chem. Soc.», 1968, v. 90, p. 608.
79. Malinowski E. R., Knapp P. S.—«J. Chem. Phys.», 1968, v. 48, p. 4989.
80. Takahashi A.— «J. Phys. Soc. Japan», 1968, v. 24, p. 657; Цит. no* РЖХим. 1969, 4Б379.
81. Akitt J. W., Greenwood N. N., Lester G. D.—«J. Chem. Soc., А», 1969, p. 803.
82. Akitt J. W., Greenwood N. N., Lester G. D.—«Chem. Comm.», 1969, N 17, p. 988.
83. Akitt J. W., Greenwood N. N, Kandelwahl B.—«Spectroscop. Lett.», 1971,. v. 4, N 5, p. 139.
84. Справочник химика. Изд. 2. T. 4. М.— Л., «Химия», 1965, с. 56—57.
85. Thompson L. С. A., Pacer R.— «J. Inorg. and Nucl. Chem.», 1963, v. 25,.
p. 1041.
86. Иванов-Эмин Б. H., Егоров А. Мч Романюк В. И. и др.—«Журн. неорган. химии», 1970, т. 15, с. 1224.
174
87. Kilpatrick M., Pokras L.—«J. Electrochem. Soc.», 1953, v. 100, N 2, p. 85_
88. Kilpatrick M., Pokras L.~ Ibid., 1954, v. 101, N 1, p. 39.
89. Biedermann G., Kilpatrik M., Pokras L. e. a.— «Acta chem. scand.», 1956, v. 10, p. 1327.
90. Aveston J.— «J. Chem. Soc.», 1966, p. 1599.
91. Заглядимова H. В. Исследование поведения микроколичеств скандия в растворах. Автореф. дис. на соиск. учен, степени канд. хим. наук. Горький, 1966 (Горьковск. ун-т).
92. Schweitzer G. К., Winkley D. С. Solvent Extraction Chemistry. Amsterdam, North-Holland, 1967, p. 39.
Швейцер Г., Уинкли Д.—6 кн.: Химия экстракции. М., Атомиздат, 1971, с. 34.
93. Антонович В. П., Назаренко В. А.— «Журн. неорган. химии», 1968, т. 13, с. 1805.
94. Комиссарова Л. Н., Пруткова Н. М., Пушкина Г. Я.— Там же, 1971,. т. 16, с. 1798.
95. Akalin S., Ozer U. Y.—«J. Inorg. and Nucl. Chem.», 1971, v. 33, p. 417L
96. Ушеренко Л. H., Скорик H. A.— «Журн. неорган. химии», 1972, т. 17, с. 2918.
97. Moeller Т.—«J. Phys. Chem.», 1946, v. 50, p. 242.
98. Biedermann G., Ciavatta L.— «Arkiv kemi», 1964, Bd 22, S. 253.
99. Терешин Г. С., Рубинштейн А. Р., Тананаев И. ,В.—«Журн. аналит. химии», 1965, т. 20, с. 1082.
100. Фролова У. К., Кумок В. H.f Серебренников В. В.— «Изв. вузов. Химия и хим. технология», 1966, т. 9, с. 176.
101. Кумок В. Н.— В кн.: Труды научной конференции Томского отделения-ВХО им. Д. И. Менделеева. Томск, Изд-во Томск, ун-та, 1969, с. 73.
102. Шалинец А. Б., Степанов А. В.— «Радиохимия», 1972, т. 14, с. 280.
103. Kakihana Н., Amaya Т., Maeda М.— In: Contributions to Coordination Chemistry in Solution.—«Trans. Roy. Inst. Technol.», 1972, N 251, p. 49.
104. Amaya T., Kakihana H., Maeda M.— «Bull. Chem. Soc. Japan», 1973,. v. 46, p. 1720.
105. Amaya T., Kakihana H., Maeda M.—- Ibid., p. 2889.
106. Wheelwright E. J., Speding F. H., Schwarzenbach G.—«J. Amer. Chem. Soc.» 1953, v. 75, p. 4196.
107. Biedermann G., Ciavatta L.— «Acta chem. scand.», 1961, v. 15, p. 1347.
108. Guillaumont R., Desire B., Galin M.— «Radiochem. Radioanalyt. Lett.»,. 1971, v. 8, p. 189.
109. Степанов А. В., Шведов В. П.— «Журн. неорган. химии», 1965, т. 10, с. 1000.
110. Кумок В. Н., Серебренников В. В.— Там же, с. 2011.
111. Sarma Р. L., Davis М. S.— «J. Inorg. and Nucl. Chem.», 1967, v. 29,. p. 2607.
112. Marin B., Kikindai T., Gourisse D.— «Note СЕА», 1968, v. 1044, p. 332,.
113. Sherrill M. S., King С. B., Spooner R. C.— «J. Amer. Chem. Soc.», 1943, v. 65, p. 170.
114. Hardwick T. J., Robertson E.—«Canad. J. Chem.», 1951, v. 29, p. 818.
115. Duke F. R., Parchen F. R.— «J. Amer. Soc.», 1956, v. 78, p. 1540.
116. Baker F. B., Newton T. W., Kahn M.— «J. Phys. Chem.», 1960, v. 64,. p. 109.
117. Ingri N., Schorsch G —«Acta ichem. scand.», 1963, v. 17, p. 590.
118. Offner H. G., Skoog D. A.— «Analyt. Chem.», 1966, v. 38, p. 1520.
119. Amjad Z., McAuleg A.— «J. Chem. Soc. Dalton Trans.», /1974, N 23,. p. 2521.
120. Rakowsky F. W. Thesis. Ohio State Univ., 1954. Univ. Microfilms,, p. 59—5443.
121. Tobias R. S., Garrett A. B.— «J. Amer. Chem. Soc.», 1958, v. 80, p. 3532.
122. Бурков К. А., Лилич Л. С., Нгуен Динь Нго и др.— «Журн. неорган. химии», 1973, т. 18, с. 1513.
123. Marin В., Kikindai Т,—«Compt. rend.», 1969. v. С268, р. 1.
175
124. Moutte A. These 3l cycle. Paris, 1968.
125. Нгуен Динь Нго, Бурков К. А.—«Журн. неорган. химии», 1974, т. 19, с. 1249.
126. Бурков К. А., Лилич Л. С., Нгуен Динь Нго.— «Изв. вузов. Химия и хим. технология», 1975, т. 18, с. 181.
127. Остроумов Э. А. Применение органических оснований в аналитической химии. М., Изд-во АН СССР, 1959, с. 75.
128. Самоделов А. П., Руденко О. А.— «Бюл. НТИ ЦНИИ. Олово» (Новосибирск), 1961, № 1, с. 39.
129. Коваленко Н. П., Багдасаров К. Н.— «Изв. вузов. Химия и хим. технология», 1963, т. 6, с. 546.
130. Fischer W., Werner Н. Е.— «Z. anorgan. und allgem. chem.», 1961, Bd ЗГ2, S. 221.
131. Иванов-Эмин Б. H., Нисельсон Л. А., Иволгина A. T.—«Журн. неорган. химии», 1960, т. 5, с. 2840.
132. Коренман И. М., Заглядимова Н. В.-~ «Труды по химии и хим. технологии» (Горький), 1965, вып. 3 (14), с. 191.
133. Бетенков Н. Д., Пузако В. Д., Егоров Ю. В.—«Радиохимия», 1971, . т. 13, с. 755.
134. Biedermann G.,, Newmann L.— «Arkiv kemi», 1964, Bd 22, S. 303.
M35. Спицын В. И., Берновская Р. Н., Попов Н. И.— «Докл. АН СССР», 1968, т. 182, с. 855.
136. Пузако В. Д., Бетенеков Н. Д., Егоров Ю. В. и др.— «Труды Уральск, политехи, ин-та» (Свердловск), 1971, сб. 193, с. 72.
137. Danesi Р. R.— «Acta chem, scand.», 1967, v. 21, p. 143.
138. Holzapfel H., Dittrich K — «Z. Chem.», 1965, Bd 5, S. 314.
339. Nishida H.—«Bunseki kagaku», 1976, v. 25, p. 866; РЖХим, 1977, 15Б200.
140. Кульба Ф. Я., Ушакова В. Г., Яковлев Ю. Б. и др.— В кн.: «Вопросы химии растворов электролитов», Барнаул, 1977, с. 52.
141. Соколова Н. Т., Ходаковский И. Л.— «Геохимия», 1977, с. 831.
142. Nayan R., Dey А. - К.—«Transit. Met. Chem.», 1977, v. 2, p. Hu.
143. Maeda M., Amaya T., Kakihana H.—«Z. Naturforsch.», 1977, Bd B32, S. 1493.
К гл. b
1. Пожарский Б. Г.— «Радиохимия», 1962, т. 4, с. 561.
2. Kasper J. Dissertation. The Johns Hopkins University. Baltimore, Mery-land, 1941. Цит. no [92].
3. Kraus K. A., Holmberg R. W.— «J. Phys. Chem.», 1954, v. 58, p. 325.
4. Pan K., Hseu T. M.— «Bull. Chem. Soc. Japan», 1955, v. 28, p. 162.
5. Lefebvre J.— «J. Chim. Phys.», 1958, v. 55, p. 227.
6. Янимирский К. Б., Жуков Ю. А.—«Радиохимия», 1961, т. 3, с. 466.
7. Bilinski Н., Furedi Н., Branica М. е. a.—«Croat. Chem. acta», 1963, v. 35, p. 19.
8. Hietanen S., Sillen L. G.— «Acta chem. scand.», 1964, v. 18, p. 1018.
9. Набиванец Б. И., Кудрицкая Л. Н.—«Укр. хим. журн.», 1964, т. 30, с. 891.
10. Baes С.. F., Meyer N. J., Roberts С. Е.— «Inorg. Chem.», 1965, v. 4, р. 518.
11. Beran M.— «Collect. Czechoslov. Chem. Commun.», 1967, v. 32, p. 1368.
12. Hietanen S., Sillen L. G.— «Acta chem. scand.», 1968, v. 22, p. 265.
13. Hietanen S.—Ibid., 1954, v. 8, p. 1626.
14. Kiciak S., Stefanowicz T.— «Roczn. chem.», 1971, v. 45, p. 1801.
15. Milic N.— «Acta chem. scand.», 1971, v. 25, p. 2487.
16. Ушеренко Л. H., Скорик H. A.—«Журн. неорган. химии», 1972, т. 17, с 2998.
176
17. Guillaumont R. M.— «Compt. rend.», 1965, v. 260, p. 1416.
18. Guillaumont R. M.— «Rev. chim. mineral.», 1966, v. 3, p. 339.
19. Guillaumont R. M.— «Bull. Soc. chim. France», 1968, p. 162.
20. Михайлов В. A.— «Журн. физ. химии», 1958, т. 32, с. 1421; «Радиохимия», 1959, т. 1, с. 395.
21. Guillaumont R. М,—«Bull. Soc. chim. France», 1965, N 1, p. 135.
22. Guillaumont R. M — Ibid., 1968, N 1, p. 168.
23. Liljenzin J. O., Rydberg J. Colloque sur la phys.-chim. du protactinium CNRS, N 154, Paris, 1966, p. 255. Цит. no [22].
24. Lawrence R. W.— «J. Amer. Chem. Soc.», 1934, v. 56, p. 776.
25. Kraus K. A., Nelson F., Johnson G.. L.— Ibid., 1949, v. 71, p. 2510.
26. Kraus K. A., Nelson F.— Ibid., 1949, v. 71, p. 2517.
27. Kraus K. A., Nelson F —Ibid., 1950, v. 72, p. 3901.
28. Kraus K. A., Nelson F.—Ibid., 1955, v. 77, p. 3721.
29. Betts R. H.— «Canad. J. Chem.», 1955, v. 33, p. 1775.
30. Betts R. H.—Ibid., p. 1780.
31. Hietanen S.—- «Acta chem. scand.», 1956, v. 10, p. 1531.
32. Sullivan J. C., Hindman J. C.— «J. Phys. Chem.», 1959, v. 63, p. 1332.
33. Николаев H. С., Лукьянычева Ю. A.— «Атомная энергия», 1961, т. 11, вып. 1, с. 67.
34. Roach D., Amis E. S.— «Z. phys. Chem.», 1962, Bd 35, S. 274.
35. Вдовенко В. M., Романов Г. А., Щербаков В. А.— «Радиохимия», 1963, т. 5, с. 137.
36. McKay Н. А. С., Woodhead J. L.— «J. Chem. Soc.», 1964, р. 717.
37. Соловкин А. С., Цветкова 3. Н., Иванцов А. И.— «Журн. неорган. химии», 1967, т. 12, с. 626.
38. Глебов В. А., Клыгин А. Е., Смирнова И. Д. и др.— Там же, 1972, т. 17, с. 3312.
39. Давыдов Ю. П., Ефременков В. М.— «Радиохимия», 1965, т. 17, с. 160.
40. Best R. J., Taub D., Longsworth L. G. US Atomic Energy Commission Report A-389, 1942, Nov., p. 24. Цит. no [80].
41. Heidt L. J.— «J. phys. Chem.», 1942, v. 46, p. 624.
42. Guiter H.— «Bull. Soc. chim. France», 1947, p. 64.
43. Harris W. E., Kolthoff I. M.— «J. Amer. Chem. Soc.», 1947, v. 69, p. 446.
44. Ahrland S.— «Acta chem. scand.», 1949, v. 3, p. 374.
45. Rydberg J.— «Arkiv kemi», 1955, Bd 8, N 2, S. 113.
46. Комарь H. IL, Третьяк 3. A.—«Журн. аналит. химии», 1955, т. 10, с. 296.
47. Gayer К. Н., Leider Н.— «J. Amer. Chem. Soc.», 1955, v. 77, p. 1448.
48. Hearne J. A., White A. G.— «J. Chem. Soc.», 1957, p. 2168.
49. Gustafson R. L., Richard C., Martell E.— «J. Amer. Chem. Soc.», 1960, v. 82, p. 1526.
50. Stary J.— «Collect. Czechoslov. Chem. Communs», 1960, v. 25, p. 890.
51. Брусиловский C. A.— «Труды ин-та геологии рудных месторождений, петрографии, минералогии и геохимии», 1960, вып. 42, с. 58.
52. Baes С. F., Meyer N. J.—«Inorg. Chem.», 1962, v. 1, р. 780.
53. Пожарский Б. Г., Стерлингова Т. Н., Петрова А. Е.— «Журн. неорган. химии», 1963, т. 8, с. 1594.
54. Dunsmore Н. S., Hietanen S., Sillen L. G.— «Acta chem. scand.», 1963, v. 17, p. 2644; Hietanen S., Rov B. R. L., Sillen L. G.— Ibid., p. 2735.
55. Рыженко Б. H., Наумов Г. Б., Гоглев В. С.— «Геохимия», 1967, с. 413.
56. Cole D. L., Eyring Е. М., Rampton D. Т. е. a.— «J. Phys. Chem.», 1967, v. 71, р. 2771.
57. Schedin U., Frydman M.—«Acta chem. scand.», 1968, v. 22, p. 115.
58. Николаева H. M., Антипина В. А., Пастухова E. Д.— ВИНИТИ, № 395-68 Деп.
59. Николаева Н. М.—«Изв. СО АН СССР. Сер. хим.», 1971, вып. 3, с. 61.
60. Никитин А. А., Сергеева Э. И., Ходаковский И. Л. и др.— «Геохимия», 1972, № 3, с. 297.
61. Давыдов Ю. П., Ефременков В. М.— «Изв. АН БССР. Серия физ.-энерг. наук», 1973, № 4, с. 21.
12 Зак. 1864 177
62. Мефодьев М. П., Крот Н. Н., Афанасьева Т. В. и др.— «Изв. АН CCCPl Сер. хим.», 1974, № 10, с. 2370.
63. Hindman J. С., Sullivan J. С., Cohen D.-— «J. Amer. Chem. Soc.», 1959y v. 81, p. 2316.
64. Москвин А. И.—-«Радиохимия», 1971, т. 13, с. 681.
65. Cassol A., Magon L., Tomat G. e. a.—«Inorg. Chem.», 1972, v. 11, p. 515.
66. Kraus K., Dam J. R.— «Transuranium Elements. Nat. Nucl. Energy Ser.»,, 1949, Div. IV, v. 14B, pt 4.14, p. 466.
67. Hubert S., Hussonnois M., Guillaumont R.— «J. Inorg. and Nucl. Chem.»*. 1975, v. 37, p. 1255.
68. Hidmann J. C.— «The Transuranium Elements. Nat. Nucl. Energy Ser.»r 1949, Div. IV, v. 14B, pt 4.4, p. 370.
69. Rabideau S. W., Lemons J. F.— «J. Amer. Chem. Soc.», 1951, v. 73„ p. 2895.
70. Keenan T; K.— «J. Phys. Chem.», 1957, v. 61, p. 1117.
71. Rabideau S. W.— «J. Amer. Chem. Soc.», 1957, v. 79, p. 3675.
72. Rabideau S. W., Kline R. J.— «J. Phys. Chem.», 1958, v. 62, p. 617.
73. Rabideau S. W., Kline R. J.~ Ibid., 1960, v. 64, p. 680.
74. Давыдов Ю. П — «Докл. АН БССР», 1972, т. 16, с. 524.
75. Metivier Н., Guillaumont R.— «Radiochem. Radioanalyt. Lett», 1972, v. I0r p. 27.
76. Kraus K. A., Dam J. R.— «The Transuranium Elements Nat. Nucl. Energy Ser.», 1949, Div. IV v. 14B, pt 4.15, p. 478.
77. O’Connor P. R. US Atomic Energy Commission Report. Sept. 1944, CN-2083.
78. Kraus K. A., Dam J. R.—US Atomic Energy Commission Report, 15 July,. 1945, CL-P-432; 15 Oct., 1945, CL-P-449; Oct. 1946, CN-2831.
79. Кревинский M. E., Никольский В. Д., Пожарский Б. Г. и др.— «Радиохимия», 1959, т. 1, с. 548.
80. Москвин А. И., Зайцева В. П.— Там же, 1962, т. 4, с. 73.
81. Cassol A., Magon L, Portanova R. е. a.—«Radiochim. acta», 1972, v. 17„ p. 28.
82. Marin R., Kikindai T., Gourisse D. «Note СЕА», 1968, N 1044, p. 332.
83. Marin R., Kikindai T.,— «Compt. rend.», 1969, v. C268, p. 1.
84. Desire B., Hussonnois M., Guillaumont R.—Ibid., 1969, v. C269, p. 448.
85. Шалинец А. Б., Степанов A. B.— «Радиохимия», 1972, т. 14, с. 280.
86. Короткий Ю. С.— «Радиохимия», 1973, т. 15, с. 766.
87. Moutte A. These З1 cycle. Paris, 1968. Цит. по {88].
88. Guillaumont R., Ferreira de Miranda C., Galin M.—«Compt. rend.», 1969,. v. C268, p. 140.
89. Hussonnois M., Hubert S., Brillard L. e. a.— «Radiochem. Radioanalyt. Lett.», 1973, v. 15, p. 47.
90. Hussonnois M., Hubert S., Aubin L. e. a —Ibid., 1972, v. 10, p. 231.
91. Актиниды. Пер. с англ. Под ред. Г. Сиборга и Дж. Каца. М., Изд-во-иностр, лит., 1955.
92. Сиборг Г. Т., Кац Дж. Дж. Химия актинидных элементов. Пер. с англ. Под ред. Г. Н. Яковлева. М., Атомиздат, 1960.
93. Вдовенко В. М. Химия урана и трансурановых элементов М.— Л.г Изд-во АН СССР, 1960.
94. Вдовенко В. М. Современная радиохимия. М., Атомиздат, 1969.
95. Рябчиков Д. И., Гольбрайх Е. К. Аналитическая химия тория. М.> Изд-во АН СССР, 1960.
96. Чарыков А. К. Вопросы аналитической химии минеральных веществ. Л., Изд-во Ленингр. ун-та, 1966, с. 129.
97. Оносов В. Н. «Труды Уральск, политехи, ин-та», 1971, сб. 193, с. 21.
98. Першин А. С.—«Радиохимия», 1972, т. 14, с. 88.
99. Hietanen S., Sillen L. G.— «Acta chem. scand.», 1959, v. 13, p. 533.
100. Matijevic E. e. a.—«J. Phys. Chem.», 1960, v. 64, p. 1157.
101. Hentz F. C., Tyree S. J.—«Inorg. Chem.», 1965, v. 4, p. 873.
178
102. Danesi P. R. e. a.—«Energia nucl.», 1968, v. 15, p. 335. -
103. Сергеев Г. M., Алмазова В. Д.—«Труды по химии и хим. технологии» (Горький), 1970, вып. 1, с. 31.
104. Gayer К. Н., Leider Н.— «J. Amer. Chem. Soc.», 1954, v. 76, p. 5938.
105. Lundgren G., Sillen L. G.—«Arkiv kemi», 1949, Bd 1, S. 277.
106. Bilinski H., Fiiredi H., Tezak B.— «Croat, chem. acta», 1963, v. 35, p. 31.
107. Guillaumont R.— «Bull. Soc. chim. France», 1965, N 7, p. 2106.
108. Старик И. E. и др.—«Радиохимия», 1959, т. 1, с. 168.
109. Москвин А. И. Координационная химия актиноидов. М., Атомиздат, 1975.
ПО. Волк В. И., Беликов А. Д.— «Радиохимия», 1977, т. 19, с. 811.
111. Nair G. М., Keshav С., Joshi Y. К. — Govt India Atom. Energy Commis. [Rept], 1976, N 900 p. 57. РЖХим. 1978, 10Б 1149.
112. Давыдов Ю. IL, Ефре^енков В. М.—«Радиохимия», 1975, т. 17, с. 155.
113. Sutton J.—«J. Chem. Soc. Suppl. Yssue», 1949, N 2, p. 275.
114. Давыдов Ю. IL, Ефременков В. M., Скрипцова А. В.— «Радиохимия», 1973, т. 15, с. 452.
115. Короткий Ю. С.—Там же, с. 671.
116. Schedin J.—«Acta Chem. Scand.», 197i, v. 25, p. 747.
117. Старик И. E., Гинзбург Ф. Л.—«Радиохимия», 1959, т. 1, с. 435; 1961, т. 3, с. 45, 685.
118. Delle Site А., В ay ba г z R. D.— «J Inorg. and Nucl. Chem.», 1969, v. 31, p. 2201.
119. Самарцева А. Г.—«Радиохимия», 1969, т. 11, с. 502.
120. Короткий Ю. С.—Там же, 1974, т. 16, с. 217, 221; 1975, т. 17, с. 547.
К г л. 7
1. Pecsok R. L., Fletcher A. N.— «Inorg. Chem.», 1962, v. 1, p. 155.
2. Paris M. R., Gregoire Cl.—«Analyt chim. acta», 1968, v. 42, p. 439.
3. Морозов И. С., Топтыгина Г. М.—«Жури, неорган. химии», 1957, т. 2, с. 1629.
4. Beukenkamp, Herrington К. D.— «J. Amer. Chem. Soc.», 1960, v. 82, p. 3025.
5. Набиванец Б. И.— «Жури, неорган. химии», 1962, т. 7, с. 417.
6. Liberti А. е. a.—«J. Inorg. and Nucl. Chem.», 1963, v. 25, p. 415.
7. Набиванец Б. И., Лукачина В. В.—«Укр. хим. журн.», 1964, т. 30, с. 1123.
8. Рязанцева И. М., Полуместная Э. И.— «Труды Воронежск. технол. ин-та», 1968, т. 17, № 1, с. 125.
9. Лобанов Ф. И., Савостина В. М., Серженко Л. В. и др.— «Журн. неорган. химии», 1969, т. 14, с. 1077.
10. Назаренко В. А.—Там же, 1971, т. 16, с. 997.
11. Васильев В. П.—Там же, 1974, т. 19, с. 2712.
12. Liegois С. These doct. Sci. Phys. Univ. Paris, 1972.
13. Соловкин А. С.—«Журн. неорган. химии», 1957, т. 2, с. 611.
14. Connick R., Е., McVey W.— «J. Amer. Chem. Soc.», 1949, v. 71, p. 3182.
15. Шека И. А., Певзнер Ц. В.—«Журн. неорган. химии», 1960, т. 5, с. 2311.
16. Пятницкий И. В., Пилипюк Е. С.—«Укр. хим. журн.», 1961, т. 27, с. 247.
17. Пешкова В. М., Мельчакова Н. В., Жемчужин С. Г.— «Журн. неорган. химии», 1961, т. 16, с. 1233.
18. Соловкин А. С., Иванцов А. И.—Там же, 1966, т. 11, с. 1897.
19. Соловкин А. С.—Там же, 1967, т. 12, с. 626.
20. Bilinski Н. е. а.—«Acta chem. scand.», 1966, v. 20, p. 853.
21. Чекмарев A. M.—«Докл. АН СССР», 1968, т. 183, с. 150.
22. Назаренко В. А., Манджгаладзе О. В.—«Журн. неорган. химии», 1969, т. 14, с. 1219.
23. Noren В.— «Acta chem. scand.», 1973, v. 27, p. 1369.
24. Васильев В. П., Кочергина Л. А., Лыткин А. И.—«Журн. неорган. химии», 1974, т. 19, с. 2998.
25. Чекмарев А. М.—«Радиохимия», 1975, т. 17, с. 165.
12* 179
26. Пешкова В. М., Пэн Ан.—«Журн. неорган. химии», 1962, т. 7, с. 2110.
27. Назаренко В. А., Антонович В. П., Невская Е. М.— Там же, 1971, т. 16, с. 2387.
28. Antikainen Р. J.— «Suomen kern.», 1957, v. ЗОВ, р. 123.
29. Roth W. A., Schwarz О.—«Вег.», 1926, Bd 59, S. 338.
30. Schwarz R., Huf E.— «Z. anorgan. und allgem. Chem.», 1931, Bd 203, S 188
31. Pugh W.— «J. Chem. Soc.», 1929, Sept., p. 1994.
32. Gulezian С. E., Muller J. H.— «J. Amer. Chem. Soc.», 1932, v. 54, p. 3142.
33. Carpeni G.— «Bull. Soc. chim. France», 1948, p. 629.
34. Lourijsen-Teyssedre M.— «Bull. Soc. chim. France», 1955, p. 1118.
35. Gayer К. H., Zajicek.— «J. Inorg. and Nucl. Chem.», 1964, v. 26, p. 951.
36. Ingri N., Schorsch G.—«Acta chem. scand.», 1963, v. 17, p. 590.
37. Ingri N.— Ibid., 1963, v. 17, p. 597.
38. Андрианов A. M., Назаренко В. A.— «Журн. неорган. химии», 1966, т. 11, с. 1527.
39. Назаренко В. А., Флянтикова Г. В.—-Там же, 1968, т. 13, с. 1855.
40. Gorman М.— «J. Amer. Chem. Soc.», 1939, v. 61, p. 3342.
41. Garrett A. B., Heiks R. E.— Ibid., 1941, v. 63, p. 562.
42. Vanderzee С. E., Rhodes D. E.— Ibid., 1952, v. 74, p. 3552.
43. Vanderzee С. E.— Ibid., p. 4806.
44. Davis J. A. Thesis Indiana, 1955. Univ. Microfilms, 14650.
45. Tobias R. E.— «Acta chem. scand.», 1958, v. 12, p. 198.
46. Gordon G., Brubaker C—«J. Amer. Chem. Soc.», 1960, v. 82, p. 4448.
47. Gobom S.— «Acta chem. scand.», 1974, v. A28, p. 1180; 1976, v. A30, p. 745.
48. Kragten J.—«Taianta», 1975, v. 22, p. 505.
49. Курильчикова Г. E., Барсуков В. A.— «Геохимия», 1970, № 1, с. 35.
50. Барсуков В. Л., Клинцова А. П.— «Геохимия», 1970, № 10, с. 1268.
51. Курильчикова Г. Е., Маров И. Н.— «Журн. неорган. химии», 1970, т. 15, с. 2978.
52. Назаренко В. А., Антонович В. П., Невская Е. М.— Там же, 1971, т. 16, с. 1844.
53. Антонович В. П., Невская Е. М., Шелихина Е. И.— Там же, 1977, т. 22, с. 363.
54. Клинцова А. П., Барсуков В. Л.— «Геохимия», 1973, № 5, с. 701.
55. Клинцова А. П. Экспериментальное исследование растворимости SnO2 в воде и водных растворах электролитов в интервале температур 25— 300° С. Автореф. дис. на соиск. учен, степени канд. хим. наук. М., 1974 (ГЕОХИ АН СССР).
56. Everest D. Е., Salmon J.— «J. Chem. Soc.», 1954, р. 2438.
57. Назаренко В. А., Флянтикова Г. В., Лебедева Н. В.— «Укр. хим. журн.», 1962, т. 28, с. 266.
58. Heyrovsky J.— «Trans. Faraday Soc.», 1923—1924, v. 19, p. 692.
59. Topelmann H.— «J. prakt. Chem.», 1929, Bd 121, N 3, S. 320.
60. Cranston J. A., Brown H„ F.— «J. Roy. Techn. Coll. (Glasgow)», 1937, v. 4, p. 54.
61. Garrett A. B., Velenga S., Fontana С. M.— «J. Amer. Chem. Soc.», 1939, v. 61, p. 367.
62. Pedersen К. X—«KgL danske vid. selskab. Mat.-fys. medd.», 1945, Bd 22, N 10, S. 1.
63. Guiter H.—«Bull. Soc. chim. France», 1947, p. 269.
64. Geloso M., Faucherre J.— «Compt. rend.», 1948, v. 227, p. 200, 430.
65. Goward G. W., Thesis Princeton, 1954. Univ. Microfilm, p. 9414.
66. Vicek A. A.—«Collect. Czechosl. Chem. Commun.», 1955, v. 20, p. 400.
67. Olver J. W., Hume D. N.— «Analyt. chim. acta», 1959, v. 20, p. 559.
68. Olin A.— «Acta chem. scand.», 1960, v. 14, p. 126.
69. Carell B., Olin A.—Ibid., p. 1999.
70. Nyman C. J., Roe D. K., Plane R. A.—«J. Amer. Chem. Soc», 1961, v. 83, p. 323.
180
71. Hugel R.—«Bull. Soc. chim. France», 1964, p. 1462.
72. Hugel R.—Ibid., 1965, p. 968.
73. Schorsch G, Ingri N.— «Acta chem. scand.», 1967, v. 21, p. 2727.
74. Gazikalovic M., Pavlovic Z., Markovic T.— «Arch, technol.», 1967, v. 5, N 3—4, p. 51.
75. Das D. C., Satpathy К. C., Pani S..— «J. Indian. Chem. Soc.», 1973, v. 50, N 12, p. 781.
76. Тугаринов И. А., Ганеев И. Г., Ходаковский И. Л.— «Геохимия», 1975, № 9, с. 1345.
77. Cookson D. J., Smith Т. D., Pilbrow J. R.— «Austral. J. Chem.», 1975, v. 28, p. 999.
78. Справочник химика. Изд. 2. T. 4. М.—Л., «Химия», 1965, с. 56—57.
79. Ларсен Э.— «Успехи химии», 1952, т. 21, с. 825.
80. Caletka R.— «Chem. Listy», 1964, v. 58, p. 349.
81. Старик И. E., Скульский И. А.— «Радиохимия», 1959, т. 1, с. 379.
82. Савенко Н. Ф., Щека И. А.— «Укр. хим. журн.», 1970, т. 36, с. 566.
S3. Pant К. М.—«J. Indian. Chem. Soc.», 1969, v. 46, p. 541.
84. Gayer К. H., Zajicek О. К—«J. Inorg. and Nucl. Chem.», 1964, v. 26, p. 2123.
85. Everest D. E., Terrey H.—«J. Chem. Soc.», 1950, p. 2282.
86. Тананаев И. В., Шпирт М. Я. Химия германия. М., «Химия», 1967, с. 258—263.
87. Назаренко В. А. Аналитическая химия германия. М., «Наука», 1973, с. 26 -29.
88. Драницкая Р. М., Гаврильченко А. И., Морозов. А. А.— «Укр. хим. журн.», 1962, т. 28, с. 866.
89. Фортунатов Н. С., Слобцов Л. Е.— Там же, 1964, т. 30, с. 1279.
90. Бабко А. К., Гридчина Г. И.— «Журн. неорган. химии», 1966, т. 11, с. 1973.
91. Вехов В. А., Давыдова В. В., Доронкина Р. Ф. В кн.: Химическая технология. Респ. Межведомств, науч.-техн. сб. Вып. II. Харьков, Изд-во Харьковск. ун-та, 1968, с. 101.
92. Алексеева И. И., Немзер И. И.— «Журн. неорган. химии», 1971, т. 16, с. 1857.
93. Коваленко П. Н.—«Укр. хим. журн.», 1958, т. 24, с. 656.
94. Olin А.— «Acta chem. scand.», 1960, v. 14, p. 711.
95. Pajdowski L., Olin A.—Ibid., 1962, v. 16, p. 983.
96. Carell B., Olin A.— Ibid., p. 2350.
97. Johanson G., Olin A.— Ibid,, 1968, v. 22. p. 3197.
98. Nagai T., Matsuda T., Ito S.— «Bunseki kagaku», 1975, v. 24, N 3, p. 203.
99. Bilinski H., Huston R., Stumm W.— «Analyt. chim. acta», 1976, v. 84, p. 157.
100. Mark W.— «Acta chem. scand.». 1977, v. A31, p. 157.
101. Смирнова В. А., Кравцов В. И., Иллювиева Г. В.— «Электрохимия», 1978, т. 14, с. 293.
К гл. 8
1. Podlaha J., Podlahova J.— «Collect. Czechoslov. Chem. Commun.», 1964, v. 29, p. 3164.
2. Furmann S. C., Garner C. S.— «J. Amer. Chem. Soc.», 1950, v. 72, p. 1785.
3. Meites LIbid., 1953, v. 75, p. 6059.
4. Jezowska-Trzebiatowska B., Pajdowski L.—«Roczn. chem.», 1960, v. 34, p. 787.
5. Pajdowski L.— «Roczn. chem.», 1963, v. 37, p. 1351.
6. Pajdowski L.— Ibid., p. 1363.
7. Brito F.— «An. Real. soc. esp. fis. у quim.», 1966, v. B62, N 2, p. 193.
8. Mateo D. S., Brito F.— Ibid, 1968, v. B64, N 2, p. 115.
9. Щербакова С. А. Комплексные соединения ванадия (III, IV. V) с ди
181
кетонами и их применение в аналитической химии. Автореф. дис. на соиск. учен, степени канд. хим. наук. М., 1976 (Моск. гос. ун-т им. М. В. Ломоносова).
10. Ducret L. Р.— «Ann. Chimie», 1951, v. 6, р. 705.
11. Rossotti F. J. C., Rossotti H. S.— «Acta chem. scand.», 1955, v. 9, p. 1177.
12. Ривкинд А. И — «Докл. АН СССР», 1962, т. 142, с. 137.
13. Мельчакова Н. В., Хаджидеметриу Д. Г., Краснянская Н. А. и др.— «Журн. неорган. химии», 1971, т. 16, с. 1981.
14. Краснянская Н. А., Эвентова И. И., Мельчакова Н. В. и др,—«Журн. аналит. химии», 1972, т. 27, с. 1842.
15. Mateo S., Brito F.—«Ап. Real. soc. esp. fis. у quim.», 1972, v. 68, N 1, p. 37.
16. Henry R. P., Mitchell P. С. H., Prue J. E.— «J. Chem. Soc. Dalton Trans.», 1973, N 11, p. 1156.
17. Краснянская H. А. Комплексообразование ванадия (IV, V) с дикетонами методом экстракции (распределения). Автореф. дис. на соиск. учен, степени канд. хим. наук. М., 1974 (Моск. гос. ун-т им. М. В. Ломоносова).
18. Толмачев В. Н., Серпухова Л. Н.— «Журн. физ. химии», 1956, т. 30, с 134.
19. Newman L., La Fleur W. J., Brousaides F. J. e. a.— «J. Amer. Chem. Soc.», 1958, v. 80, p. 4491.
20. Ingri N., Brito F.— «Acta chem. scand.», 1959, v. 13, p. 1971.
21. Bailey N., Carrington A., Lott К. A. K. e. a.— «J. Chem. Soc.», 1960, p. 290.
22. Chauveau F.—«Bull. Soc. chim. France», 1960, p. 819.
23. Schiller K-, Thilo E.—«Z. anorgan. und allgem. Chem.», 1961, Bd 310, S. 261.
24. Dyrssen D., Sekine T.— «Acta chem. scand.», 1961, v. 15, p. 1399.
25. Санников Ю. И., Золотавин В. Л., Безруков И. Я.— «Журн. неорган. химии», 1963, т. 8, с. 923.
26. Schwarzenbach G., Geier G.— «Helv. chim. acta», 1963, v. 46, p. 906.
27. Яцимирский К. Б., Калинина В. Е.— «Журн. неорган. химии», 1964, т. 9, с. 1117.
28. Джафаров Ф. 3., Горбачев С. В.— Там же, 1964, т. 9, с. 2399.
29. Dyrssen D., Sekine t.— «J. Inorg. and Nucl. Chem.», 1964, v. 26, p. 981.
30. Brito F., Ingri N., Sillen L. G.— «Acta chem. scand.», 1964. v. 18, p. 1557.
31. Przyborowski L., Schwarzenbach G.— «Helv. chim. acta», 1965, v. 48, p. 1556.
32. Tanaka M., Kojinia J.— «J. Inorg. and Nucl. Chem.», 1967, v. 29, p. 1769.
33. Rieger P. H.— «Austral. J. Chem.», 1973, v. 26, p. 1173.
34. Yamada S., Funahashi S., Tanaka M.—«J. Inorg. and Nucl. Chem.», 1975, v. 37, p. 835.
35. Kawamoto H., Akaiwa H.— «Nippon Kagaku Kaishi», 1973, p. 90.
36. Бабко A. K4 Лукачина В. В., Набиванец Б. И.— «Журн. неорган. химии», 1963, т. 8, с. 1839.
37. Бабко А. К., Набиванец Б. И. Проблемы современной химии координационных соединений. Вып. 2. Л., Изд-во Ленингр. ун-та, 1968, с. 111.
38. Zawidzky J.— «Вег.», 1903, v. 36, р. 1427.
39. Wood J. К —«J. Chem. Soc.», 1908, v. 93, р. 411.
40. Washburn Е. W., Strachan E. К.— «J. Amer. Chem. Soc.», 1913, v. 35, p. 681.
41. Blanc E.—«J. chim. phys.», 1920, v. 18, p. 28.
42. Kolthoff I. M.— «Pharmac. Weekbl.», 1920, v. 57, p. 514.
43. Hughes W. S.— «J. Chem. Soc.», 1928, p. 491.
44. Cernatescu R., Mayer A.— «Z. phys. Chem.», 1932, Bd 160A, S. 305.
45. Britton H. T. S., Jackson P.— «J. Chem. Soc.», 1934, p. 1048.
46. Ishikawa F., Aoki J.—«Bull. Inst. Phys. Chem. Res. Tokyo», 1940, v. 19, p. 136.
182
47. Garrett A. В., Holmes О., Laube A.—«J. Amer. Chem. Soc.» 1940, v. 62, p. 2024.
48. Carpeni G., Souchay P.— «J. chim. phys.», 1945, v. 42, p. 149.
49. Tourky A. R., Mousa A. A.— «J. Chem. Soc.», 1949, v. 1305.
50. Konopik N., Leberl O.— «Monatsh. Chem.», 1949, v. 80, p. 655.
51. Antikainen P. J., Rossi V. M. K-—«Suomen. kem.», 1959. v. 32B, p. 185.
52. Antikainen P. J., Tevanen K.—- Ibid., 1961, v. 34B, p. 3.
53. Hargreaves M. K., Stevinson E. A., Evans J.— «J. Chem. Soc.», 1965, p. 4582.
54. Левин И. С., Сергеева В. В., Колышев А. И.— «Изв. СО АН СССР. Сер. хим.», 1975, № 7, вып. 3, с. 113.
55. Ивакин А. А., Воробьева С. В., Гертман Е. М. и др.— «Журн. неорган. химии», 1976, т. 21, с. 442.
56. Ивакин А. А., Воробьева С. В., Горелов А. М. и др.— Там же, с. 2973.
57. Skrabal A., Zahorka A.— «Z. Elektrochem.», 1927, Bd 33, S. 42.
58. Агафонова А. Л., Агафонов И. Л.— «Журн. физ. химии», 1953, т. 27, с. J137.
59. Mader Р. М.—«J. Amer. Chem. Soc.», 1958, v. 80, p. 2634.
60. Чухланцев В. Г.— «Журн. физ. химии», 1959, т. 33, с. 3.
61. Флис И. Е., Мищенко К. П., Туманова Т. А.— «Журн. неорган. химии», 1959, т. 4, с. 277.
52. Salomaa Р., Schaleger L. L., Long F. A.—«J. Amer. Chem. Soc.», 1964, v. 86, p. 1.
53. Sellers P., Sunner S.t Wadso J.— «Acta chem. scand.», 1964, v. 18, p. 202.
64. Tossidis I.— «Inorg. and Nucl. Chem. Lett.», 1976, v. .12. p. 609.
65. Kasper J. Thesis Johns Hopkins Univ., 1941. Baltimore. Цит. no [66].
*66. Stability Constants of Metal-Ion Complexes. Spec. Publ. N 17, L., Burlington House, 1964.
67. Pitman A. L., Pourbaix M., Zoubov N.—«Cebelcor. Rapp. Tech.», 1957, N 55. Цит. no [66].
68. Mishra S. K*, Gupta J- К.—«Indian. J. Chem.», 1968, v. 6, p. 757.
69. Ahrland S.. Bovin J. O.— «Acta chem. scand.», 1974, v. 28, p. 1089.
70. Shoji H., Mabuchi H., Saito N.— «Bull. Chem, Soc. Japan.», 1974, v. 47, p. 2502.
71. Антонович В. IL, Невская E. M., Суворова E. H — «Журн. неорган. химии», 1977, т 22, с. 1278.
72. Goward G. W. Thesis Princeton, 1954. Univ. Microfilms 9414.
73. Graner F., Olin A., Sillen L. G.— «Acta chem. scand.», 1956, v. 10, p. 476.
74. Olin A.—Ibid., 1957, v. 11, p. 1445.
75. Van Muylder J., Pourraux M. Proc. 9th Meeting Intern. Comm. Electro-chem. Therma Kinetics, 47 London (1959). Цит. no [76].
76. Ишина В. А., Иванченко А. Ф., Зив Д. M.— «Радиохимия», 1960, т. 2, с. 691.
77. Bidleman Т. F.— «Analyt. chim. acta», 1971, v. 56, p. 221.
78. Dragulescu C., Nimera A., Julean J.—«Chim. analyt.», 1972, v. 17, p. 631.
79. Антонович В. IL, Невская E. M., Шелихина E. И. и др.—«Журн. неорган. химии», 1975, т. 20, с. 2968.
80. Pajdowskl L.— «J. Inorg. and Nucl. Chem.», 1966, v. 28, p. 433.
61. Григорьева M. Ф., Церковницкая И. A.—В кн.: Проблемы современной аналитической химии. Вып. 1. Л., Изд-во Ленингр. ун-та, 1976, с. 74.
52. Бабко А. К., Гридчина Г. И.— «Журн. неорган. химии», 1968, т. 13, с. 3029.
83. Морачевский Ю. В., Беляева Л. И.— «Журн. аналит. химии», 1956, т. 11, с. 672.
64. Золотавин В. Л., Безруков И. Л., Санников Ю. И.—«Журн. неорган. химии», 1961, т. 6, с. 581.
65. Ивакин А. А.— «Труды ин-та химии УФ АН СССР», 1968, вып. 18, с. 3.
66. Химия пятивалентного ванадия в водных растворах.—«Труды ин-та химии УНЦ АН СССР». 1971, вып. 24, (Свердловск).
67. Коваленко П. Н.— «Журн. прикл. химии», 1958, т. 31, с. 1488.
183
88. Справочник химика. Изд. 2. Т. 4. М.—Л., «Химия», 1965, с. 56—57.
89. Neumann Н. М.— «J. Amer. Chem. Soc.», 1954, v. 76, p. 2811.
90. Neumann H. M., Ramette R. W.— Ibid.. 1956, v. 78, p. 1848.
91. Bonner N. A., Goishi W.—Ibid., 1961, v. 83, p. 84.
92. Иофа Б. 3., Дакар Г. M.— «Радиохимия», 1963, т. 5, с. 490.
93. Moorhead Е. D., Lipcsey S.— «J. Electroanalyt. Chem. Interfac. Electro-chem.», 1970, v. 26, p. 27.
94. Olin A.— «Acta chem. scand.», 1959, v. 13, p. 1791.
95. Tobias R. S., Tyree S. C.— «J. Amer. Chem. Soc.», I960, v. 82, p. 3244.
96. Gattow G., Schott D.— «Z. anorgan. und allgem. chem.», 1963, Bd 324,.
S. 31.
97. Dragulescu C., Nimara A., Julean J.— «Rev. Roum. chim.», 1972, v. 17, p. 1181; 1974, v. 19, p. 1455.
98. Коваленко П. H.— «Хим. наука и пром-сть», 1957, т. 2, вып. 4, с. 531.
99. Коренман И. М., Воронцова Т. Е.—«Труды по химии и хим. технологии»’ (Горький), 1968, вып. 3 (21), с. 75.
100. Щербакова С. А., Краснянская Н. А., Мельчакова Н. В. и др.— «Журн. неорган. химии», 1978, т. 25, с. 170.
101, Komura A., Hayashi М., Imonaga Н.— «Bull. Chem. Soc. Japan», 1977, v. 50, p. 2927.
102. Лукачина В. В., Пилипенко А. Т., Карпова О. И.— «Журн. неорган. химии», 1977, т. 22, с. 1275.
103. Nimara A., Julean J.— «Rev. Roum. chim.», 1977, v. 22, p. 691.
К г л. 9
1. Кладницкая К. Б., Кублановский В. С., Зосимович Д. П.—«Укр. хим. журн.», 1972, т. 38, с. 104.
2. Bjerrum N.,— «Z. phys. Chem.», 1907, Bd 59, S. 336.
3. Denham H. G.— «J. Chem. Soc.», 1908, v. 93, p. 41.
4. Bjerrum N.—«Z. phys. Chem.», 1910, Bd 73, S. 724.
5. Lamb A. B., Fonda G. R.— «J. Amer. Chem. Soc.», 1921, v. 43, p. 1154.
6. Bronsted I. N., Volqvartz K.— «Z. phys. Chem.», 1928, Bd 134, S. 97.
7. Hartford W. N.— «Ind. Eng. Chem.», 1942, v. 34, p. 920.
8. Cranston J. D.. Brown H. F.— «J. Roy. Tech. College (Glasgow)», 1937,. v. 4, p. 54.
9. Postmus C., King E. L.— «J. Phys. Chem.», 1955, v. 59, p. 1208.
10. Charreton B.— «Compt. rend.», 1957, v. 244, p. 1208.
11. Bjerrum J., Jorgensen E.—«J. Inorg. and Nucl. Chem.», 1958, v. 8,. p. 313.
12. Jorgensen E., Bjerrum J.—«Acta chem. scand.», 1958, v. 12, p. 1047.
13. Emerson K., Graven W. M.— «J. Inorg. and Nucl, Chem.», 1959, v. 11,. p. 309.
14. Schwarzenbach G.—«Pure and Appl. Chem.», 1962, v. 5, p. 377.
15. Tsuchiya R., Umayahara A.— «Bull. Chem. Soc. Japan», 1963, v. 36,. p. 554.
16. Кондратов П. И., Кондратов T. С., Сурков К. К.— «Труды Воронежем технол. ин-та», 1968, т. 17, вып. 1, с. 102.
17. Meyenburg U., Siroky О, Schwarzenbach G.— «Helv. chim. acta», 1973,. v. 56, p. 1099.
18. Issa I. M., Khalifa H.— «J. Indian Chem. Soc.», 1954, v. 31. p. 91.
19. Яцимирский К. Б., Алексеева И. И.— «Изв. вузов СССР. Химия и хим. технология», 1958, № 1, с. 53.
20. Sasaki J., Lindqvist 1., Sillen L. G.— «J. Inorg. and Nucl. Chem.», 1959,. v. 9, p. 93.
21. Seshaish U. V., Banerji S. N.—«J. Indian Chem. Soc.», 1962, v. 39, p. 93.
22. Chojnacka J.— «Roczn. chem.», 1963, v. 37, p. 259.
23. Rohwer E. F. G. H., Cruywagen J. J.— «J, S. Afric. Chem. Inst.», 196-3», v. 16, p. 26.
24. Rohwer E. F. С. H., Cruywagen J. J.— Ibid., 1964, v. 17, p. 145.
184
25. Aveston J., Anacker E. W., Johnson J. S.— dnorg. Chem.», 1964, v. 3... p. 735.
26. Sasaki J., Sillen L. G.~ «Acta chem. scand.», 1964, v. 18, p. 1014.
27. Chojnacka J.— «Roczn. chem.», 1965, v. 39, p. 161.
28. Rohwer E. F. С. H., Cruywagen J. J.— «J. S. Afric. Chem. Inst.», 1966,. v. 19, p. 11-
29. Воробьев C. IL, Давыдов И. IL, Шилин И. В.— «Журн. неорган. химии», 1967, т. 12, с. 2142.
30. Набиванец Б. И.— «Журн. неорган. химии», 1969, т. 14, с. 653.
31. Jain D. V. S.— «Indian J. Chem.», 1970, v. 8, p. 945.
32. Назаренко В. А., Шелихина E. И.— «Журн. неорган. химии», 1971, т. 16,. с. 166.
33. Панталер Р. П. Изучение новых каталитических реакций и их применение в следовом анализе. Автореф. дис. на соиск. учен, степени канд. наук. Харьков, 1973. (Харьковск. гос. ун-т).
34. Титкова Т. Д., Левин В. И.— «Радиохимия», 1975, т. 17, с. 55.
35. Cruywagen J. J., Rohwer Е. F. С. Н.—«Inorg. Chem.», 1975, v. 14, р. 3136.
36. Яцимирский К. Б., Прик К. Е.— «Журн. неорган. химии», 1964, т. 9,. с. 1838.
37, Яцимирский К. Б., Романов В. Ф.—«Журн. неорган. химии», 1965, т. 10,. с. 1607.
38. Иванова Г. Ф., Ходаковский И. Л.—-«Геохимия», 1968, № 8, с. 931.
39. Назаренко В, А., Полуэктова Е. Н.— «Журн. неорган. химии», 1969,. т. 14, с. 204.
40. Chojnacka J.— «Roczn. Chem.», 1973, v. 47, p. 1359.
41. Назаренко В. А., Полуэктова E. H., Шитарева Г. Г.—«Журн. неорган. химии», 1977, т. 22, с. 998.
42. Blanc Е.— «J. Chim. Phys.», 1920, v. 18, р. 28.
43. Kasarnowsky Т.— «Z. phys. Chem.», 1924, Bd 109, S. 287.
44. Issa I. M., Awad S. A.—«J. Phys. Chem.», 1954, v. 58, p. 948.
45. Sekine T. Solvent Extraction Chemistry. Amsterdam, North-Holland, 1967f. p. 32.
46. Sekine T., Iwaki H., Sakairi M. e. a.— «Bull. Chem. Soc. Japan», 1968,. v. 41, p. 1.
47. Набиванец Б. И., Капанцян Э. Е.— «Журн. неорган. химии», 1968, т. 13, с. 1817.
48. Ганелина Е. Ш., Боргояков В. А.—Там же, 1971, т. 16, с. 596.
49. Назаренко В. А., Шитарева Г. Г., Полуэктова Е. Н.— Там же, 1973, т. 18, с. 1155.
50. Набиванец Б. И., Капанцян Э. Е., Оганесян Э. И.— Там же, 1974, т. 19, с. 729.
51. Назаренко В. А., Шитарева Г. Г., Полуэктова Е. Н.—Там же, 1977, т. 22, с. 980.
52. Masson М. R.—«J. Inorg. and Nucl. Chem.», 1976, v. 38, p. 545.
53. Koch H., Schmidt H.—«Z. Naturforsch.», 1963, Bd. 18b, S. 936.
54. Старик И. E., Ампелогова H. И.— «Радиохимия», 1964, т. 6, с. 519.
55. Ампелогова Н. И.— Там же, 1975, т. 17, с. 68.
56. Справочник химика, Изд. 2. Т. 4, М.—Л., «Химия», 1965, с. 56—57.
57. Neuss J. D., Rilman W.— «J. Amer. Chem. Soc.», 1934, v. 56, p. 2238.
58. Tong J., L, King E. L.—Ibid., 1953, v. 75, p. 6180.
59. Шевченко В. Б., Шилин И. В., Жданов Ю. Ф.— «Журн. неорган. химии», 1960, т. 5, с. 2832.
60. Bailey N. е. а.—«J. Amer. Chem. Soc.», 1960, v. 290.
61. Lee D. W., Steward R.— «J. Amer. Chem. Soc.», 1964, v. 86, p. 305L
62. Haight G. R., Richardson D. C., Colurn N. N.— «Inorg. Chem.», 1964», v. 3, p. 1777.
63. Tong J. V., Johnson R. L.— «Inorg. Chem.», 1966, v. 5, p. L902.
64. Lukkari O.— «Suomen kem.», 1970, v. 43B, p. 347.
65. Lukkari O., Lukkari H.— Ibid., 1971, v. 45B, p. 6.
18S
66. А* К., Набиванец Б. И.— «Журн. неорган. химии», 1957, т. 2,
67. Шарипова Н. С. Сборник работ по химии. -Вып. 3. Алма-Ата, Изд-во Казахск. ун-та, 1973, с. 217.
*68. Баб*0 А. К., Гридчина Г. И.—«Журн. неорган. химии», 1963, т. 8,
69. Воробьев С. П., Давыдов И. П., Шилин И. В.—Там же, 1967, т. 12, с. 2665.
70. Sasaki Y., Sillen L. G..— «Arkiv kemi», 1968, Bd 29, S. 253.
71. Jain D. V. S., Jain С. M.—«Indian J. Chem.», 1969, v. 7, p. 821.
72. Krumenacker L.— «Bull. Soc. chim. France», 1971, p. 2820, 2824.
73. Брызгалин О. В.—«Геохимия», 1972, № 1, с. 51.
74. Каримова Л. К., Шульц А. А. Химия и хим. технология редких и цветных металлов. Ташкент, «Фан», 1974, с. 59.
75. Дьячкова И. Б., Ходаковский И. Л.—«Геохимия», 1968, № 11, с. 1358.
76. Ходаковский И. Л., Рыженко Б. Н., Наумов Г. Б.— Там же, № 12, с. 1486.
77. Бирюков В. П., Ганелина Е. Ш.— «Журн. неорган. химии», 1971, т. 16, с. 600.
78. Ганелина Е. Ш., Боргояков В. А., Кузьмичева В. FL—«Изв. вузов. Химия и хим. технология», 1973, т. 16, с. 346.
79. Kaehler Н. С., Brito F.— «Ап. Real. soc. esp. fis. у quim.», 1971, v. 67, p. 1185.
80. Kaehler H. C., Mateo S., Brito F.—Ibid., 1974, v. 69, p. 1269, 1273.
81. Barcza L., Sillen L. G.— «Acta chem. scand.», 1971, v. 25, p. 1250.
82. Vesala A., Koskinen V.—«Finn. Chem. Lett.», 1975, N 5,
p. 145.
83. Шпак T. П., Колосов И. В., Сенявин М. М.— «Журн. неорган. химии», 1976, т. 21, с. 3309.
84. Chaberek S., Country R. C.t Martell A. C.—«J. Amer. Chem. Soc.», 1952, v. 74, p. 5057.
85. Perrin D. D.— «J. Chem. Soc.», 1962, p. 2197.
86. Заводнов С. С., Фесенко H. Г.— «Гидрохимические материалы», 1964, т. 36, с. 148.
87. Diebier Н., Sutin N.— «J. Phys. Chem.», 1964, v. 68, p. 174.
88. Facklel J. P., Chawla I. D.— «Inorg. Chem.», 1964, v. 3, p. 1130.
89. Wells C. F., Davies G.—«Nature», 1965, v. 205, p. 692.
90. Wells C. F., Davies G — «J. Chem. Soc. (A)», 1967, p. 1858.
91. Rosseinsky D. R., Nicol M. J.—Ibid., 1968, p. 1022.
92. Davies G., Kustin K.— «Inorg. Chem.», 1969, v. 8, p. 1196.
93. Гончарик В. П., Яцимирский К. Б., Тихонова Л. П.— В кн.: Тезисы докладов 7-й Украинской Республиканской конференции по неорганической химии. Одесса, 1972, с. 142.
94. Гончарик В. П., Тихонова Л. П., Яцимирский К. Б.— «Журн. неорган. химии», 1973, т. 18, с. 1248.
95. Rosseinsky D. R., Nicol М. J., Kite К. е. a —«J. Chem. Soc. Faraday Trans.», 1974, v. 1, N 12, p. 2232.
96. Gorski B., Koch H.— «J. Inorg. and Nucl. Chem.», 1969, v. 31, p. 5565.
97. Guennec J. Y. These doct. ing. Fac. Sci. Paris-Sud. 1973. Цит. по РЖХим., 1975, 21B2079.
98. Митькина Л. И., Мельчакова Н. В., Пешкова В. М.— «Журн. неорган. химии», 1978, т. 23, с. 1258.
99. Cruywagen J. J., Rohwer Е. F., С. Н.— «J. S. Afric. Chem. Inst.», 1976, v. 29, р. 30.
100. Cruywagen J. J., Heyns J. В. B., Rohwer E. F. С. H.— «J. Inorg. and Nucl. Chem.», .1976, v. 38, p. 2033.
101. Ганелина E. Ш., Боргояков В. А., Бубнова Л. А. и др —В кн.: Комп-лексообр. в растворах и на твердых поверхностях. Л., 1976,
с. 3.
186
К ГЛ. 10
1. Гинзбург С. И., Езерская Н. А., Прокофьева И. В. и др. Аналитическая химия платиновых металлов. М.» «Наука», 1972.
/2. Алимарин И. П., Шленская В. И., Бирюков А. А. и др.— «Журн. аналит. химии», 1970, т. 25, с. 1965.
3. Whitman W. G., Russell R. Р., Davis G. H. В.— «J. Amer. Chem. Soc.», 1925, v. 47, p. 70.
4. Schrager B.—«Chem. News», 1929, v. 138, N 7, p. 354.
3. Lindstrand F.— «Svensk kern, tidskr.», 1944, v. 56, N 8, p. 282.
6. Leussing L., Kolthoff J. M.— «J. Amer. Chem. Soc.», 1953, v. 75, p. 2476.
7. Hedstrom В. O. A.— «Arkiv kemi», 1953, v. 5, p. 457.
8. Gayer К. H., Wootner L.— «J. Amer. Chem. Soc.», 1956, v. 78, p. 3944.
9. Bolzan J. A.— «Rev. Fac. cienc. quim. Univ. nac. La Plata», 1961, v. 33, p. 67.
10. Комарь H. П,—««Труды хим. фак-та и науч, исслед. ин-та химии Харь-ковск. ун-та», 1963, т. 19, с. 189.
11. Bolzan J. A., Arvia A. J.— «Analyt. Asoc. Quim. Argentina», 1963, v. 51, p. 333; «Electrochim. acta», 1963, v. 8, p. 375.
12. Wells C. F., Salam M. A.—«Nature», 1965, v. 205, p. 690.
13. Wells C. F.t Salam M. A.— «J. Chem. Soc. А», 1968, v. 24.
14. Sweeton F. H., Baes C. F.— «J. Chem. Thermodyn.», 1970, N 2, p. 479.
15. Enrenfreund M., Leibenguth J. L.— «Bull. Soc. chim. France», 1970, N 7, p. 2494, 2498.
16. Messmer R. E.—«Inorg. Chem.», 1971, v. 10, p. 857.
17. Lewis D.— «АЕ-Repts», 1971, N 436.— In: Contributions to Coordination Chemistry in Solution. Trans. Royal Inst. Technol. Stockholm, 1972, N 252, p. 59.
18. Инцкирвели Л. H., Колосов И. В., Варшал Т. М.— «Журн. неорган. химии», 1975, т. 20, с. 2388.
19. Bronsted J. N., Volqvartz К.—«Z. phys. Chem.», 1928, Bd. 134, S. 97.
120. Bray W. C., Hershey A. V.—«J. Amer. Chem. Soc.», 1934, v. 56, p. 1889.
21. Lamb A. B., Jacques A. G.—Ibid., 1938, v. 60, p. 1215.
22. Brosset C.— «Svensk. kem. tidskr.», 1941, v. 53, N 8—12, p. 434.
23. Brosset C.— «Naturwissenschaften», 1941, Bd 29, S. 455.
24. Rabinovitch E., Brockmayer W. H.—«J. Amer. Chem. Soc.», 1942, v. 64, . p. 335.
225. Lindstrand F.— «Svensk. kem. tidskr.», 1944, v. 56, p. 251.
26. Olson A. K., Simonson T. R.— «J. Chem. Phys.», 1949, v. 17, p. 1322.
27. Dodgen H. W.f Rollefson G. K.— «J. Amer. Chem. Soc.», 1949, v. 71, p. 2600.
28. Siddall T. H., Vosburgh W. C.—Ibid., 1951, v. 73, p. 4270.
29. Barb W. G., Baxendale L H., George P. e. a.—«Trans. Faraday Soc.», 1951, v. 47, p. 591.
30. Arden T. V.—«J. Chem. Soc.», 1951, p. 350.
31. Sutton J.—«Nature», 1952, v. 169, p. 71.
32. Silverman J., Dodson R. W.— «J. Phys. Chem.», 1952, v. 56, p. 846.
33. Wilson A. S., Taube H„— «J. Amer. Chem. Soc.», 1952, v. 74, p. 3509.
34. Hedstrom В. O. A.—«Arkiv kemi», 1953, v. 6, N 1—2, p. 1.
35. Ito L, Yui N.— «Sci. Rept., Tohoku Univ.», 1953, v. 37, p. 19.
36. Connick R. E., Tsao M. S.—«J. Amer. Chem. Soc.». 1954, v. 76, p. 5311.
37. Milburn R. M., Vosburgh W. C.— Ibid., 1955, v. 77, p. 1352.
38. Betts R. H.—«Canad. J. Chem.», 1955, v. 33, p. 1780.
39. Hudis J., Dodson R. W.— «J. Amer. Chem. Soc.», 1956, v. 78, p. 911.
40. Connik R. E.. Hepler L. G., Hugus Z. Z. e. a.—Ibid., p. 1827.
41. Milburn R. M.— Ibid., 1957, v. 79, p. 537.
42. Turner R. C., Miles К. E.— «Canad. J. Chem.», 1957, v. 35, p. 1002.
43. Broersma S.—«J. Chem. Phys.», 1957, v. 26, p. 1505.
44. Perrin D. D.— «J. Chem. Soc.», 1959, N 5, p. 1710.
45. Richards D. H., Sykes K. W.—Ibid., 1960, p. 3626.
187
46. Bowers E. J., Weaver H. D.— «Proc. Indian Acad. Sci.», 1961, v. 71» p. 101.
47. Popa G., Luca C., Josif E.— «Z. phys. Chem.», 1963, Bd 222, N 1—2V S. 49.
48. Пальчевский В. В., Якубов X. М.—«Докл. АН ТаджССР», 1963, т. 6^ № 1, с. 17.
49. Якубов X. М., Пальчевский В. В.—Там же, 1964, т. 7, № 4, с. 16.
50. Fukishima S., Reynolds W. L.— «Taianta», 1964, v. 11, p. 283.
51. Кондратов П. И., Кондратова T. С., Сурков К. К-—«Труды Воронежем технол. ин-та», 1968, т. 17, № 1, с. 102.
52. Звягинцев О. Е., Ляхманов С. Б.— «Журн. неорган. химии», 1968. т. 13*. с. 1230.
53. Ropars С., Rougee М., Momenteau М. е. а.— «J. Chim. phys. et phys^ chim. biol.», 1968, v. 65, p. 816.
54. Behar B., Stein G.— «Isr. J. Chem.», 1969, v. 7. p. 827.
55. Fordham A. W.—«Austral. J. Chem.», 1969, v. 22, p. 1111.
56. Maslowska J.—- «Zesz. nauk. Politechn. lodzkiej», 1969, N 111, p. 7.
57. Hemmes P., Rich L. D„, Cole D. L. e. a.— «J. Phys. Chem.», 1971, v. 75,. p. 929.
58. Koren R., Perlmutter-Hayman B.— «Inorg. Chem.», 1972, v. 11, p. 3055..
59. Пальчевский В. В., Ткачук Е. X., Якубов X. М. В кн.: Комплексообразование в окислительно-восстановительных системах. Вып. 2. Душанбе,. Изд-во Тадж. ун-та, 1973, с. 49.
60. Давыдов Ю. П., Грачок М. А.— «Изв. АН БССР. Сер. физ.-инж. наук»,. 1973, № 4, с. 31.
61. Пальчевский В. В., Фан Ты Банг.—«Вести. Ленингр. ун-та», 1974, № 4, вып. 1, с.. 116.
62. Колосов И. В., Инцкирвели Л. Н., Варшал Г. М.— «Журн. неорган. химии», 1975, т. 20, с. 2121.
63. Гордиенко В. И., Журавлев Е. Ф., Сидоренко В. И. В кн.: Тезисы докладов II Всесоюзного совещания «Термодинамика и структура гидроксокомплексов в растворах». Л., «Наука», 1975, с. 21.
64. Давыдов Ю. П., Грачок М. А., Молчун Л. А. Там же, с. 10.
65. Ciavatta L., Grimaldi М.— «J. Inorg. and Nucl. Chem.», 1975, v. 37,.. p. 163.
66. Landry J. CL, Buffle J., Haerdi W. e. a.—«Chimia», 1975, v. 29, N 6, p. 253.
67. Knight R. J., Sylva R. N.— «J. Jnorg. and Nucl. Chem.», 1975, v. 37, p. 779.
68. Gayer К. H., Garrett A. B.— «J. Amer. Chem. Soc.», 1950, v. 72, p. 3921-
69. Gayer К. H., Woontner L— Ibid., 1952, v. 74, p. 1436.
70. Chaberek S., Country R. C., Martell A. C.— Ibid., p. 5057.
71. Achenza F.— «Rend. Fac. Scinz. Univ. Cagliari», 1959, v. 29, p. 52.
72. Bolzan J. A., Arvia A. J.— «Electrochim. acta», 1962, v. 7, p. 589.
73. Bolzan J.A., Podesta J. J., Arvia A. J.— «Analyt Asoc. Quim. Argentina», 1963, v. 51, p. 43.
74. Shankar J., De Souza В. C.— «Austral. J. Chem.», 1963, v. 16, p. 1119.
75. Collados M. P., Brito F., Diaz Calavieco R.—«An. Real. soc. esp. fis. yquim.», 1967, v. 63B, p. 843.
76. Sutclieffe L. H., Weber J. R.—«Trans. Faraday Soc.», 1956, v. 52, p. 1225.
77. Conocchioli T. J., Nancollas G. H., Sutin N.—«Inorg. Chem.», 1965, v. 5, p. 1.
78. Hill J., Me Auley A.—«J. Chem. Soc (A)», 1968, p. 2405.
79. Schwab G. M., Polydoropoulos K.— «Z. anorgan. und allgem. Chem.»r 1953, Bd 274, S. 234.
80. Ksandr Z., Hejtmanek M. Sb.— «J. Celost. Pracov. Konf. Analyt. Chem.»,. 1952, p. 42. C. A., 1956, v. 50, p. 3150h.
81. Guta F., Ksandr Z., Hejtmanek M.—«Collect. Czechoslov. Chem. Com-mun.», 1956, v. 21, p. 1388.
82. Achenza F.—«Ann. Chim.», 1959, v. 49, p. 624.
83. Achenza F.— Ibid., p. 848.
188
84. Bolzan J. A., Jauregui E. A., Arvia A. J.— «Electrochim. acta», 1963, v. 8, p. 841.
85. Perrin D. D.— «J. Chem. Soc.», 1964, p. 3644.
86. Matulis J., Slizys R.— «Electrochim. acta», 1964, v. 9, p. 1177.
87. Martin F. S.— «J. Chem. Soc.», 1954, p. 2564.
88. Silverman M. D., Levy H. A.,— «Amer. Chem. Soc.», 1950, 118-th Meeting, Chicago, Sept. Цит. no [87].
89. Forrester J. SM Ayres G. H.— «J. Phys. Chem.», 1959, v. 63, p. 1979.
$0. Cola M.— «Gazz. chim. Ital.», 1960, v. 90, p. 1037.
91. Plumb W., Harris G. M.— «Inorg. Chem.», 1964, v. 3, p. 542.
92. Swaminathan K., Harris G. M.— «J. Amer. Chem. Soc.», 1966, v. 88, p. 4411.
93. Маслей H. H., Набиванец Б. И., Cac Ф. Ф.— В кн.: Тезисы докладов 7-й Украинской республиканской конференции по неорганической химии. Одесса, 1972, с. 59.
94. Маслей Н. Н., Набиванец Б. И., Сас Ф. Ф.— «Укр. хим. журн.», 1974, т. 40, с. 1181.
95. Маслей Н. Н., Набиванец Б. И., Янцо Е. А.— Там же, 1976, т. 42, с. 247.
96. Izatt R. М., Eatough D., Christensen J. J.—«J. Chem. Soc. (A)», 1967, p. 1301.
97. Набиванец Б. И.., Калабина Л. В., Кудрицкая Л. Н.— «Изв. СО АН СССР. Сер. хим.», 1970, № 9, вып. 4, с. 51.
98. Набиванец Б. И., Калабина Л. В.—«Журн. неорган. химии», 1970, т. 15, с. 1595.
99. Иванов-Эмин Б. Н., Борзова Л. Д., Егоров А. М. и др.— Там же, 1974, т. 19, с. 1570.
100. Yost D. М., White R. J.— «J. Amer. Chem. Soc.», 1928, v. 50, p. 81.
101. Sauerbrunn R. D., Sandell E. B.— Ibid., 1953, v. 75, p. 4170.
102. Bavay J. C.,, Nowogrocki G.( Tridot G.—«Bull. Soc. chim. France», 1967, N 6, p. 2026.
103. Bavay J. C. These doct. sci. phys. Univ. Sci. et techn. Lille, 1972. Цит. РЖХ, 1975, 2В161Д.
104. Lee D. A.-—«J. Inorg. and Nucl. Chem.», 1972, v. 34, p. 375.
105. Тихонова Л. П., Заяц В. Я.— «Журн. неорган. химии», 1976, т. 21, с. 2154.
106. Набиванец Б. И., Калабина Л. В.— В кн.: Тезисы докладов 7-й Украинской республиканской конференции по неорганической химии. Одесса, 1972, с. 66.
107. Наумов Т. Б., Рыженко Б. Н., Ходаковский И. Л. Справочник термодинамических величин. М., Атомиздат, 1971.
108. Sylva R. N.—«Rev. Pure Appl. Chem.», 1972, v. 22, p. 115.
109. Якубов X. M. Применение оксредметрии к изучению комплексообразования. Душанбе, Изд-во Таджикск. ун-та, 1966, с. 32.
110. Davies G., Warnqvist В.— «Coord. Chem. Revs», 1970, v. 5, p. 349.
111. Bremard C.. Tridot G.— «Compt. rend.», 1974, v. C278, p. 1413.
112. Anderson J. S., McConnell J. D. M.— «J. Inorg. and Nucl. Chem.», 1951» v. 1, p. 371.
113. Wehner P., Hindman J.— «J. Amer. Chem. Soc.», 1950, v. 72, p. 3911.
114. Парамонова В. И., Латышев Е. Ф.—«Радиохимия», 1959, т. 1, с. 458.
115. Бурков К. А., Бусько Е. А., Ларионова С. В.— В кн.: Проблемы современной химии координационных соединений. Вып. 3. Л., Изд-во Ленингр. ун-та, 1970, с. 144.
116. Алимарин И. П., Хвостова В. П., Кадырова Г. И —«Журн. аналит. химии», 1975, т. 30, с. 2007.
117. Оффенгенден Е. Я*, Ибрагимов О. К.— В кн.: Комплексообразование в окислительно-восстановительных системах. Вып. 3. Душанбе, Изд-во Таджикск. ун-та, 1976, с. 14.
118. Шакирова Р. Г.— Там же, с. 37.
189
119. Sapleszko R. S., Patel R. C., Matijevic E.— «J. Phys. Chem.», 1977, v. 81,, p. 880.
120. Маслен H. H., Набиванец Б. И., Янцо E. A.— «Укр. хим. журн.», 1976,. т. 42, с. 1314.
121. Дакар Г. М., Иофа Б. 3., Несмеянов Ан. Н.— «Вести. Моск, ун-та. Химия», 1977, т. 18, с. 62.
К гл. 11.
1. Topelmann Н.~—«J. prakt. chem.», 1929, v. 121, р. 320.
2. Irving Н., Williams R. J. P.— «J. Chem. Soc.», 1953, p. 3192.
3. Davies C. W.—Ibid., 1951, v. 1256.
4. Van Panthaleon Van Eck C. L.— «Rec. Trav. Chim.». 1953, v. 72, p. 50.
5. Gimblett F. G. R., Monk С. B.—«Trans. Faraday Soc.», 1954, v. 50„ p. 965.
6. Лилич Л. С., Могилев M. E.— «Журн. общ. химии», 1956, т. 26, с. 312.
7. Лилич Л. С., Варшавский Ю. С.— Там же, с. 317.
8. Perrin D. D.— «J. Chem. Soc.», 1962, р. 2197.
9. Черкесов А. А.— «Изв. вузов. Химия и хим. технология», 1973, т. 16,. с. 351.
10. Rosseinsky D. R., Nicol М. J., Kite К. е. a.—«J. Chem. Soc. Faraday Trans.», 1974, v. 1, p. 2232.
11. Черкесов A. A.— «Изв. вузов. Химия и хим. технология», 1976, т. 19,. с. ИЗО.
12. Яцимирский К. Б., Васильев В. П. Константы нестойкости комплексных, соединений. М., Изд-во АН СССР, 1959.
13. Гринберг А. А. Введение в химию комплексных соединений. М.— Л.,. «Химия», 1966.
14. Лилич Л. С.— В кн.: Химия и термодинамика растворов. Л., Изд-во Ле-нингр. ун-та, 1964, с. 5.
15. Лилич Л. С., Мищенко К. П.— В кн.: Сто лет Периодического закона, химических элементов. М., «Наука», 1969, с. 302.
16. Назаренко В. А., Шелихина Е. И.—- «Журн. неорган. химии», 1971, т. 16, с. 166.
17. Назаренко В. А., Антонович В. П., Невская Е. М.— Там же, с. 997.
18. Назаренко В. А., Антонович В. П., Невская Е. М—Там же, с. 1844.
19. Назаренко В. А., Антонович В. П., Невская Е. М.— Там же, с. 2387.
20. Антонович В. П., Невская Е. М., Шелихина Е. И.— Там же, 1977, т. 22,. с. 363.
21. Назаренко В. А., Шитарева Г. Г., Полуэктова Е. Н.— Там же, 1973, т. 18, с. 1155.
22. Назаренко В. А., Бирюк Е. А.— Там же, 1974, т. 19, с. 632.
23. Справочник химика. Изд. 2. Т. 1. М.—Л., ГНТИХЛ, 1962, с. 325, 380—382.
24. Зайдель А. Н., Прокофьев В. К., Райский С. М. и др. Таблицы спектральных линий. М., Физматгиз, 1962, с. 604.
25. Bull М. С., Norbury А. Н. Physical Data for Inorganic Chemists. L., Longman, 1974.
26. Таблицы физических величин. Справочник. Под ред. И. К. Кикоина. М., Атомиздат, 1976.
27. Сиенко М., Плейн Р., Хестер Р. Структурная неорганическая химия. Пер., с англ. Под ред. М. Е. Дяткиной. М., «Мир», 1968.
28. Коттон Ф., Уилкинсон Дж. Современная неорганическая химия. Ч. 1—3.. Пер. с англ. Под ред. М. Е. Дяткиной. М., '«Мир», 1969.
29. Крестов Г. А. Термохимия соединений редкоземельных и актиноидных элементов. М., Атомиздат, 1972.
30. Крестов Г. А. Термодинамика ионных процессов в растворах Л., «Химия», 1973.
190
И;/Яцимирский К- Б., Костромина Н. А., Шека 3. А. и др. Химия комплексных соединений редкоземельных элементов. Киев, «Наукова думка>\
•4й. Martin W. С., Hagan L., Reader J. е. а.— «J. Phys. Chem. Ref. Data»* W’ 1974, v. 3, N 3, p. 771.
/§3. Назаренко В. А., Флянтикова Г. В.— «Журн. неорган. химии», 1968, т. 13, с. 1855.
2 34. Назаренко В. А., Манджгаладзе О. В.— Там же, 1969, т. 14, с. 1219.
35. Nagai Т., Matsuda Т., Ito S.—«Bunseki kagaku», 1975, v. 24, p. 203.
36. Антонович В. П., Невская Е. М., Шелихина Е. И. и др.— «Журн. неорган. химии», 1975, т. 20, с. 2968.
37. Антонович В. П., Невская Е. М., Суворова Е. Н.—Там же, 1977, т. 22, с. 1278.
38. Ивакин А. А., Воробьева С. В., Горелов А. М. и др.— Там же, 1976, т. 21, с. 2973.
39. Ивакин А. А., Воробьева С. В., Гертман Е. М. и др.—Там же, 1976, т. 21, с. 442.
40. Pitman A. L., Pourbaix М., Zoubov N.— «Cebelcor. Rapp. Tech.», 1957*. N 55.
41. Коренман И. M., Воронцова Т. Е.— «Труды по химии и хим. технологии» (Горький), 1968, вып. 3 (21), с. 75.
42. Архипова Г. IL, Флис И. Е., Мищенко К. П.— «Журн. прикл. химии», 1964, т. 37, с. 2306.
43. Флис И. Е., Архипова Г. П., Мищенко К. П.— Там же, 1965, т. 38, с. 1494.
44. Назаренко В. А., Полуэктова Е. Н., Шитарева Г. Г.— «Журн. неорган. химии», 1977, т. 22, с. 998.
45. lukkari О.— «Suomen kem.», 1970, v. 43В, р. 347.
46. Позин Л. М.— «Журн. прикл. химии», 1972, т. 45, с. 2328.
47. Waber J. Т., Cromer D. Т.— «J. Chem. Phys.», 1965, v. 42, р. 4116.
48. Воздвиженский В. М.-—«Журн. физ. химии», 1970, т. 44, с. 317.
ОГЛАВЛЕНИЕ
Предисловие.............................................................................3
Список сокращений....................................., , 4
Введение ........................................................................... 5
1. Основные принципы расчета мономерного гидролиза ионов металлов 7
2. Методы определения констант гидролиза..............................................15
2.1. Потенциометрический метод.....................................................15
. 2.2. Спектрометрический метод......................................................17
2.3. Метод распределения (экстракция).............................................21
2.4. Метод растворимости...........................................................23
2.5. Метод оксредметрии............................................................25
2.6. Ионообменный метод............................................................26
2.7. Электромиграционный метод.....................................................28
2.8. Метод протонного магнитного резонанса.........................................29
3. Диссоциация воды. Гидролиз ионов элементов I группы . 30
3.1. Диссоциация воды..............................................................30
3.2. Щелочные металлы..............................................................31
3.3. Элементы подгруппы меди.....................................................31
4. Гидролиз ионов элементов II группы.................................................34
4.1. Элементы главной подгруппы — Be, Mg, Са, Sr, В а . . . 34
4.2. Элементы подгруппы цинка—Zn, Cd, Hg ..........................................40
5. Гидролиз ионов элементов III группы................................................48
5.1. Элементы подгруппы В, Al, Ga, In, TI..........................................48
5.2. Элементы подгруппы скандия — Sc, Y и лантаноиды ... 61
6. Гидролиз ионов элементов семейства актиноидов .................................. 75
7. Гидролиз ионов элементов IV группы — Ge, Sn, Pb, Ti, Zr, Hf . 91
8. Гидролиз ионов элементов V группы — V, Nb, Та, As, Sb, Bi .HO
9. Гидролиз ионов элементов VI и VII групп....................124
9.1. Элементы VI группы — Сг, Mo, W, Те.......................124
9.2. Элементы VII группы — Мп, Тс............................................... 136
10. Гидролиз ионов элементов VIII группы — Fe, Со, Ni, Ru, Rh, Pd, Os, Ir, Pt................................................. . -139
11. Связь между положением элемента в периодической системе Д. И. Менделеева и способностью его ионов к гидролизу . .153
Список литературы.....................................................................167
ИБ № 1013
Василий Андреевич Назаренко
Валерий Павлович Антонович Елена Михайловна Невская
ГИДРОЛИЗ ИОНОВ МЕТАЛЛОВ В РАЗБАВЛЕННЫХ РАСТВОРАХ
Редактор Т. А. Молоканова
Художественный редактор А. Т. Кирьянов
Обложка художника Ю. С. Шлепера
Технические редакторы А. Л. Гулина, О. Н. Адаскина
Корректор Г. П. Домрачева
Сдано в набор 31.08.78. Подписано к печати 25.12.78. Т-19898 Формат 60 x 90’/i6.
Бумага тип. № 2. Гарнитура литературная. Печать высокая. Усл. печ. л. 12,0.
Уч.-изд. л. 13,19. Тираж 1560 экз. Зак. изд. 73224. Зак. тип. 1864. Цена 2 р.
Атомиздат, 103031. Москва, К-31, ул. Жданова, 5
Московская типография № 6 Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
.109088, Москва, Ж-88, Южнопортовая ул., 24.