/





































































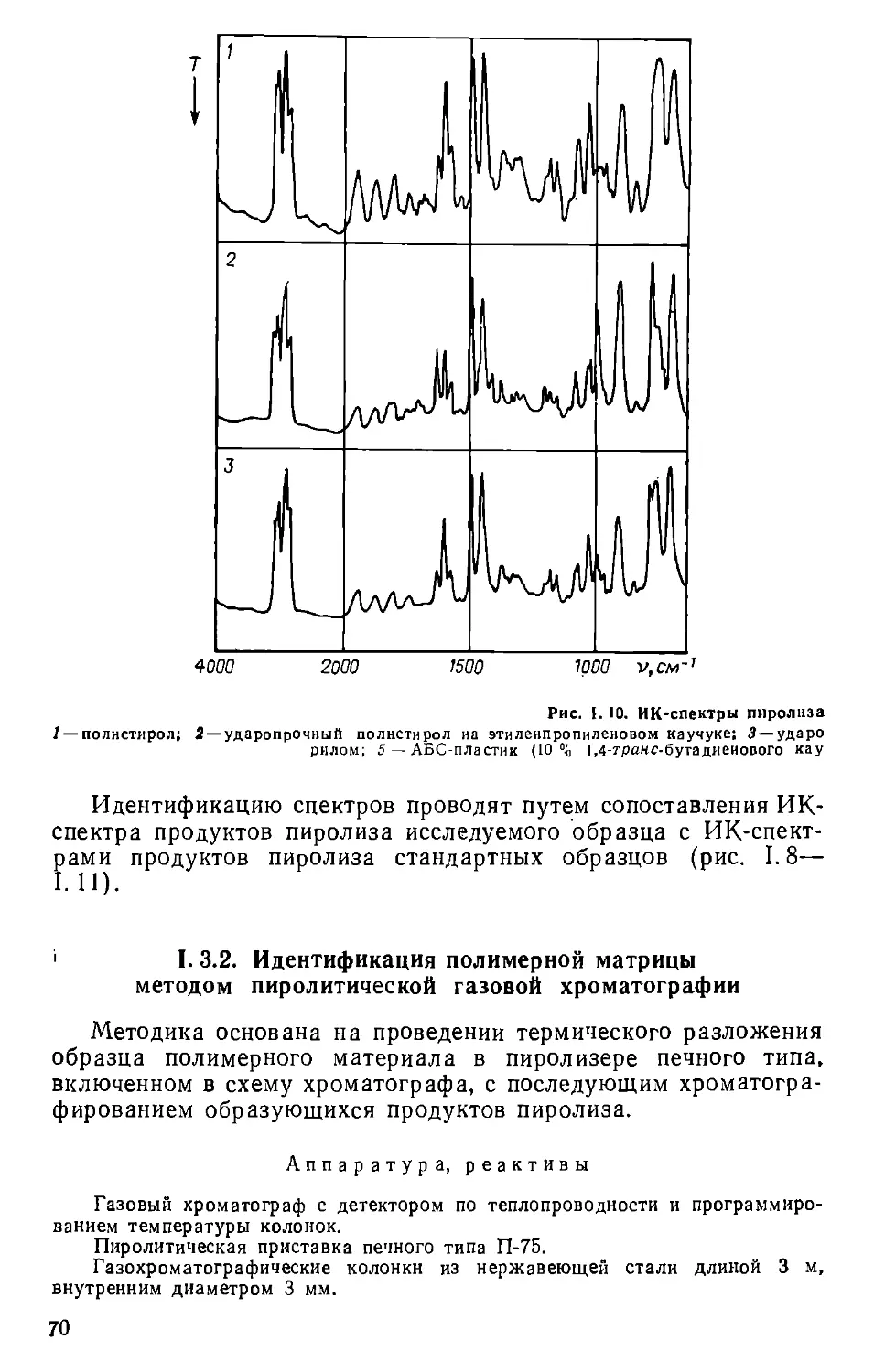


































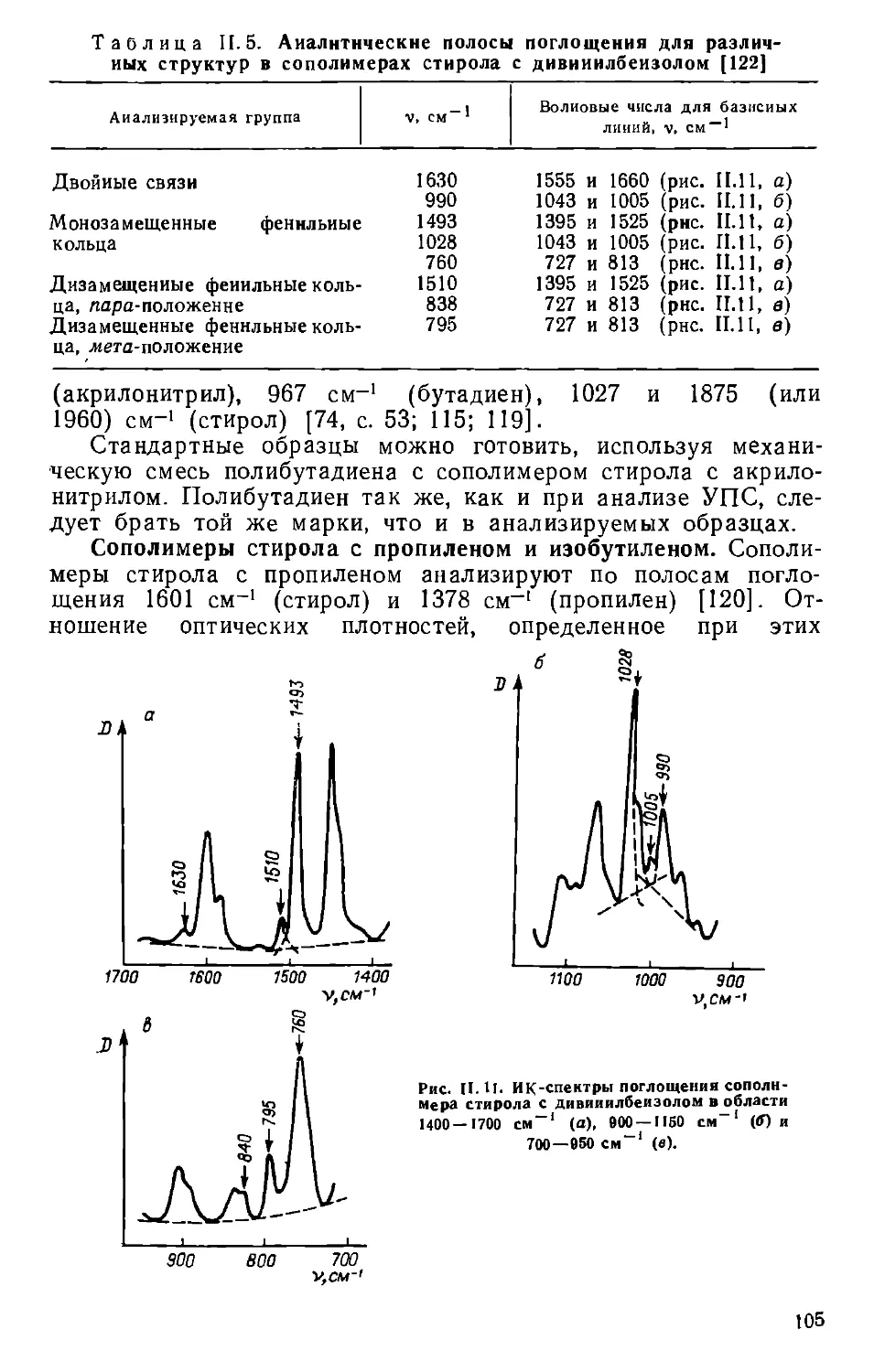












































































































































































































Текст
АНАЛИЗ
ПОЛИМЕРИЗАЦИОННЫХ
ПЛАСТМАСС
#
ЛЕНИНГРАД • «ХИМИЯ»
ЛЕНИНГРАДСКОЕ ОТДЕЛЕНИЕ
1988
1H>K G43
A64
УДК 678.01 : 678.7
Авторы: Г. С. Попова, В. П. Будтов, В. М. Рябикова,
Г. В. Худобина
Рецензент: канд. хим. наук А. И. Малышев
Анализ полимеризационных пластмасс/Г. С. По-
А64 пова, В. П. Будтов, В. М. Рябикова, Г. В.
Худобина.—Л.: Химия, 1988.— 304 с: ил.
ISBN 5—7245—0050—7
Рассмотрено применение различных химических, физико-химических
и физических методов для анализа крупнотоннажных полимерных
материалов - идентификации, определения состава, структуры, молекулярно-мас-
сового распределения, содержания примесей и добавок. Особое внимание
обращено на возможности и ограничения методов анализа.
Для хпмиков-аналптиков заводских и научно-исследовательских
лабораторий, занимающихся контролем состава и свойств полимерных
материалов, а также преподавателям и студентам химико-техиологическнх вузов.
. .804000000-008 ^ ББК 543
050(01)—88
ISBN 5-7245-0050-7
© Издательство «Химия», 1988 г.
ПРЕДИСЛОВИЕ
В связи с интенсивным развитием производства пластмасс
и их широким использованием в народном хозяйстве проблема
анализа полимерных материалов приобрела в аналитической
химии большое значение. Основными направлениями
экономического и социального развития СССР на 1986—1990 годы и на
период до 2000 года предусматривается довести в 1990 году
выпуск синтетических смол и пластических масс до 6,8—7,1 млн. т,
обеспечить ускоренное развитие производства современных
конструкционных пластических масс и других полимеров.
Решение этих задач невозможно без надежного аналитического
обеспечения.
Анализ полимерных материалов имеет специфические
особенности и существенно отличается от анализа других
органических веществ. Эта специфика обусловлена тем, что полимерные
материалы, находящиеся в обычных условиях в твердом
состоянии, растворяются в небольшом числе растворителей, образуя
даже при очень низких концентрациях высоковязкие растворы.
Вследствие этого некоторые традиционные для анализа
органических соединений методы оказываются вообще
неприменимыми к полимерам, а другие требуют модификации с учетом
особенностей полимерных матералов.
Наиболее полным и широко известным руководством по
анализу полимеров является трехтомная монография под ред.
Г. Клайна «Аналитическая химия полимеров», переведенная
па русский язык в 1963—1966 гг. В этой книге, написанной
коллективом авторов, каждый из которых является крупнейшим
специалистом в своей области, рассмотрено применение
различных методов для изучения строения полимеров, их
количественного и качественного анализа. Однако за время, прошедшее
с момента написания этой книги, очень многое изменилось как
в химии полимеров, так и в аналитической химии. Большинство
книг по анализу полимеров, вышедших за последние годы,
посвящено применению какого-нибудь одного метода для
аналитических целей. Такие книги, рассматривающие теоретические
основы метода и их практическое применение для решения
различных аналитических задач, безусловно необходимы, не
не могут полностью удовлетворить аналитика-практика.
В повседневной жизни перед химиком-аналитиком обычно
стоит конкретная аналитическая задача, и нужны руководства,
которые позволяли бы быстро находить пути ее решения.
Подобные книги по анализу резин и поликонденсационных пластмасс
пышли в последние годы: Малышев А. И., Помогайбо А. С.
Анализ резин. М.: Химия, 1977. 232 с; Исакова Н. А., Белова Г.А.,
Фихтенгольц В. С. Контроль производства синтетических кау-
чуков. Л.: Химия, 1987. 184 с; Анализ конденсационных
полиморов / Л. С. Калинина, М. А. Моторина, Н. И. Никитина, Н. А. Ха-
чапуридзе. М.: Химия. 1984. 296 с.
1*
3
По анализу полимеризационных пластмасс подобная книга
вышла в 1968 г. и, естественно, не может полностью отвечать
современным требованиям. В настоящей книге сделана попытка
заполнить этот пробел.
При отборе материала для книги авторы исходили из того,
что в первую очередь должны рассматриваться те вопросы,
которые необходимы широкому кругу химиков-аналитиков и
химиков-технологов. Поэтому в книгу не включены такие вопросы,
как определение характера чередования звеньев и
композиционной неоднородности сополимеров, исследование
поверхностных слоев полимерных образцов. Эти вопросы, как правило,
являются предметом специальных научных исследований и пока
не стали объектами серийных анализов.
Число методов и их различных модификаций, которые можно
использовать для решения различных аналитических задач,
в настоящее время достигает нескольких десятков (Рабек #.
Экспериментальные методы в химии полимеров. М.: Мир, 1983.
874 с). Однако в повседневной работе аналитических
лабораторий используются лишь отдельные методы. Особенно это
относится к методам, включенным в различные нормативные
документы. В книге сделан упор на те из них, которые являются
сейчас наиболее массовыми и стандартными.
В книге не рассматриваются теоретические основы методов,
так как они подробно изложены в посвященных этим вопросам
монографиях, но имеются небольшие вводные части, в которых
отмечена специфика применения метода к данной задаче.
Несмотря на то, что книга посвящена в основном
проблемам анализа полимеризационных пластмасс, она может быть
полезна всем, занимающимся анализом полимерных
материалов, так как многие вопросы являются общими для полимеров
разного химического строения.
Глава I написана В. М. Рябиковой, глава II — Г.С.Поповой,
глава III — В. П. Будтовым, глава IV — Г. В. Худобиной.
Авторы выражают благодарность сотрудникам методико-ана-
литической лаборатории ОНПО «Пластполимер» за оказание
помощи при подготовке книги; сотрудникам отдела
исследования структуры и свойств полимеризационных пластмасс
Л. Н. Пирожной и В. Л. Максимову за обсуждение I и II глав
книги, Н. Г. Подосеновой, Е. Л. Пономаревой, И. Б.
Цветковскому и В. В. Нестерову за обсуждение главы III; E. И.
Новиковой, В. А. Зинченко, Л. С. Мальцевой и Н. И. Малкиной за
предоставление отдельных методик по анализу полимеров.
Авторы будут признательны читателям за советы,
критические замечания и пожелания по содержанию книги и учтут
их при последующих изданиях.
ГЛАВА 1
ИДЕНТИФИКАЦИЯ ПОЛИМЕРНЫХ КОМПОЗИЦИЙ
Необходимость идентификации полимеров возникла
практически одновременно с появлением промышленной полимерной
химии. В связи с широким применением пластмасс в различных
областях народного хозяйства систематически приходится
идентифицировать полимерные продукты в зависимости от их
целевого назначения.
Для идентификации полимеров и полимерной основы
композиций используются различные методы: простые, основанные на
физико-химических и физико-механических свойствах
полимеров, химические, инструментальные. Наибольшее
распространение из инструментальных методов получили ИК-спектроскопия,
пиролитическая газовая хроматография, ЯМР-спектроскопия.
Применяются газовая, тонкослойная, гель-проникающая
хроматография, хромато-масс-спектроскопия, пиролитическая масс-
спектроскопия, термический анализ, а также разнообразные
комбинации этих и других методов. Инструментальные методы
позволяют значительно сократить время анализа и снизить
предел обнаружения ряда анализируемых компонентов [1—6].
На современном этапе развития аналитической химии
большое внимание уделяется разработке новых типов сенсорных
систем, основного компонента аналитического мониторинга.
Быстродействие инструментальных методов существенно возросло
с оснащением приборов компьютерной техникой и созданием
автоматизированных систем и комплексов по измерению
свойств, математической обработке результатов и их
интерпретации. Создаются библиотеки данных для идентификации
полимеров с помощью машинного поиска их масс- и ИК-спектров,
спектроаналитическпе комплексы для идентификации
органических соединений в смесях [1, 7]. Хотя анализ становится все
более эффективным, но при этом и все более дорогим. Все
чаще при выборе метода для идентификации полимеров наряду
с качеством аналитической информации приходится учитывать
стоимость аппаратуры и ее эксплуатации, уровень и стоимость
подготовки специалистов и их труда при работе на сложных
приборах и оборудовании.
Вместе с тем применение любых из имеющихся в арсенале
исследователя методов для индентификации
многокомпонентного полимерного материала в его изначальном виде, как
правило, не может дать ответа на целевые вопросы по составу
образца. Поэтому перед проведением идентификации необходимо
выбрать оптимальный путь ее осуществления с использованием
тех или иных методов и их комбинации в зависимости от
конечной цели.
5
Одним из первых и необходимых этапов является
предварительная идентификация с использованием простых методов и
химических тестов [8—11; 12, гл. 2; 13]. Установление с их
помощью класса полимерной основы образца в значительной
степени облегчает задачу последующего исследования
аппаратурными методами (ИК-спектроскопия, пиролитическая газовая
хроматография и др.). После предварительной идентификации
рассматривается уже более узкий круг возможных полимеров,
конкретизируется работа с атласами и справочниками, а также
в определенной степени уменьшается опасность ошибок,
которые могут возникнуть при наличии в ИК-спектрах полос,
относящихся к примесям, или перекрывании спектров основного
полимера спектрами других полимеров. В связи с тем, что на
идентификацию поступают образцы неизвестных полимеров,
природу которых предстоит установить, в настоящей главе
приводится ряд методов, позволяющих различить полимеризацион-
ные и поликонденсационные пластмассы.
Следует отметить, что в большинстве предлагаемые простые
методы идентификации распространяются на полимеры и
композиции на их основе с небольшим количеством
неорганических добавок (до 5 %) • Для многокомпонентных композиций,
состоящих из различных по химической природе полимеров, и
композиций с высоким содержанием органических и
неорганических добавок и наполнителей простые методы (в том числе
и химические тесты) могут дать ненадежные результаты. В
таких случаях следует предварительно выделять добавки и
разделять полимеры.
1.1. ИДЕНТИФИКАЦИЯ ПОЛИМЕРНОЙ ОСНОВЫ
1.1.1. Внешний вид и физические свойства
При первоначальном осмотре устанавливается визуально
однородность или же отмечается число полимерных
(неполимерных) слоев, составляющих образец. Если образец многослойный
или неоднородный, то составляющие компоненты отделяют с
учетом их свойств и целей идентификации и затем каждый из
них идентифицируют отдельно.
Физические свойства полимеров и их внешний вид во
многом определяются структурой макромолекулярных цепей,
которые могут быть гибкими или жесткими с линейной,
разветвленной или сетчатой структурой. По физическому состоянию и
внешнему виду образца (твердый, полужесткий, эластичный,
воскообразный, дисперсный, прозрачный, мутный, непрозрачный
и т. п.) можно сделать некоторые предположения о природе
полимерной основы.
Полимерной основой эластичных образцов, как правило,
являются полимеры-эластомеры. Наиболее типичные
представители эластомеров — полибутадиеновые, полиизопреновые, полиси-
локсановые, полиуретановые каучуки и резины на их основе,
к
Из термопластов, которые при повышении температуры
размягчаются, а при охлаждении возвращаются в исходное состояние,
идентификации чаще всего подвергаются полиэтилен,
полипропилен, полиамиды. Подобные изменения при нагревании не
характерны для термореактопластов, которые с изменением
температуры практически не изменяют агрегатного состояния, а
при высоких температурах подвергаются пиролизу с
выделением газообразных продуктов разложения. Характерные
свойства термореактопластов — высокая твердость, жесткость,
хрупкость, неплавкость, незначительная растворимость в
органических растворителях. Их излом имеет характерную зернистую
структуру." Типичные термореактопласты — фенопласты,
эпоксидные смолы.
Полимерной основой прозрачных твердых, полужестких
образцов и пленок, как правило, являются термопласты.
Воскообразные образцы обычно относятся к полиолефинам. В
дисперсиях дисперсной фазой могут быть полиолефины, поливинил-
ацетатные пластики, фторполимеры, а дисперсионной средой —
органические растворители, вода или их смеси.
Газонаполненные полимеры, которые имеют промышленное значение, могут
быть представлены эластичными и жесткими
пенополиуретанами на основе простых и сложных полиэфиров, пенополиоле-
финами, вспененными полистиролами, пенополивинилхлоридом,
вспененными мочевино-формальдегидными смолами, пенополи-
эпоксидами, пенофенопластами.
Цвет образца не является достоверной характеристикой
принадлежности образца полимера к тому или иному классу,
поскольку красители, пигменты и добавки могут изменить
природную окраску полимера. Однако для фенопластов коричневый
II черный цвета являются естественными, что отличает их от
других полимерных композиций с красителями, техническим
углеродом и графитом.
Образцы термопластов в отличие от термореактопластов
можно проткнуть насквозь нагретой металлической спицей.
После этих испытаний твердый образец, отнесенный к
термопластам, можно подвергнуть испытанию на изгиб, характер
которого свойственен определенным видам полимеров. Для
этого один конец образца размером 10 X 100 X 2 мм (при
сравнительных испытаниях желательно иметь образцы одинаковых
размеров) зажимают в тиски или лабораторный зажим и
изгибают под прямым углом. Образцы из полиэтилена, АБС-пла-
стика (акрилонитрилбутадиенстирольного пластика) изгибают-'
ся и сохраняют изгиб; образцы из полистирола изгибаются с
растрескиванием в месте изгиба и сохраняют изгиб; образцы
из полиметилметакрилата и сополимера стирола с метилмет-
мкрилатом растрескиваются при изгибе; образцы из жесткого
ноливинилхлорида, сополимеров этилена с пропиленом,
композиции из смеси АБС-пластика и поливинилхорида легко
изгибаются и выпрямляются; образцы из политетрафторэтилена и
7
полиамида изгибаются и пружинят при сгибании; образцы из
полипропилена, пентапласта практически не изгибаются.
Твердые полимерные образцы (без наполнителя) можно
различить по разнице в плотности следующими дополнительными
испытаниями [10]. В один из подготовленных двух стаканов
вместимостью 100 мл наливают 70 мл дистиллированной воды,
в другой — раствор тиосульфата натрия (70 г тиосульфата
натрия в 60 мл воды). Образец размером не более 2 х 10 X 10 мм
помещают вначале в стакан с водой. В зависимости от
плотности образец либо погрузится на дно стакана, либо будет
плавать на поверхности воды. Образцы, погрузившиеся на дно,
извлекают из стакана с водой и помещают в стакан с
раствором тиосульфата натрия. На этот раз на дно опускаются
образцы, плотность которых больше 1,1, остальные плавают на
поверхности раствора (табл. 1.1).
Для пленочных материалов проводится проба на раздир.
Кусочек пленки размером 15 X 50 мм слегка надрезают с
любой стороны ножницами и тянут в противоположные стороны
за концы надреза, пытаясь разорвать пленку. Это же
испытание проводят при надрезе пленки с другой стороны,
перпендикулярно первому. Легко и прямо разрываются пленки,
изготовленные из ацетата целлюлозы, целлофана. Также без особых
Таблица 1.1. Температура плавления и плотность полимеров
Полимер
Температура
плавления, °С
[281
Плотность,
г/смЗ
[28]
Идентификация по
плотности [10]
вода
раствор
тиосульфата
натрия
190—210
160-170
105-108
130-135
240-250
230—240
210—230
210—240
210—215
210—220
190-200
170-180
200—210
173—180
190—205
170—180
210—215
327
0,83
0,90
0,92
0,95
1,07
1,07
1,10
1,05
1,13
1,20
1,20
1,33
1,40
1,40
1,40
1,70—1,80
2,08—2,09
2,20—2,30
Образец
плавает
То же
»
»
Образец
тонет
То же
»
»
»
»
»
»
»
»
»
»
»
»
—
—
—
—
Образец
плавает
То же
»
»
»
Образец
тонет
То же
»
»
»
»
»
»
»
8
Поли-4-метилпентен-1
Полипропилен
Полиэтилен высокого давления
Полиэтилен низкого давления
Полистирол
Ударопрочный полистирол
Сополимер стирола с акрило-
нитрилом
АБС-пластик
Полиамид
Поликарбонат
Полиметилметакрилат
Пентапласт
Поливинилхлорид
Полиформальдегид
Поливинилфторид
Поливин илиден фтор ид
Политрифторхлорэтилен
Политетрафторэтилен
усилий рвутся пленки из полистирола с образованием неровных
краев. Пленки из полиэтилена и полипропилена при
разрывании становятся вязкими, растягиваются перед разрывом
(сильнее— полипропиленовые пленки), образуют при обрыве
волнистые края; пленки из поливинилхлорида растягиваются и
разрываются с образованием рваных краев; пленки из
поливинилового спирта растягиваются, затем под влиянием усилия
разрыв сдвигается в сторону; пленки из политетрафторэтилена
и других фторполимеров разрываются с треском. Для
высокопрочных ориентированных пленочных материалов это
испытание не подходит.
Более точно может характеризовать образец температура
плавления. Плавление полимеров, в отличие от
низкомолекулярных веществ, происходит в некотором интервале температур
[14, с. 134]. Для термопластов в табл. 1.1 приводится
температурный интервал, в котором полимер переходит в
расплавленное состояние. Определение можно проводить в приборе для
установления температуры плавления низкомолекулярных
веществ. Образец полимера массой 2—3 мг тщательно измельчают
и вносят в капилляр диаметром 2—3 мм, помещают в прибор
и наблюдают за его размягчением и плавлением.
1.1.2. Воздействие пламени и высокой температуры
Поведение полимерного образца в пламени изучают
следующим образом [9]. На конце предварительно очищенной медной
проволоки диаметром 0,5 мм делают небольшую петлю; на
другой конец проволоки надевают корковую или резиновую
пробку, за которую держат проволоку в процессе ипытания.
Проволочную петлю прокаливают в пламени спиртовки. На
охлажденную петлю помещают небольшой (1—2 мм) образец
полимера и вносят в среднюю часть бесцветного пламени. Если
образец воспламеняется и горит, выводят проволочную петлю
из пламени на 5—6 см и продолжают наблюдение. Отмечают
характер горения (горит, не горит, самозатухает), цвет
пламени, его форму, наличие дыма, копоти, зольного остатка. Свои
наблюдения сопоставляют с данными табл. 1.2 и делают
соответствующие выводы. Следует учесть, что присутствие галоген-
содержащих антипиренов в самозатухающих полимерных
материалах может привести к ошибочным выводам, поскольку они
дают такую же окраску пламени при сгорании полимерной
композиции, как и галогенсодержащие полимеры.
При термическом разложении полимерных образцов
образуются разнообразные низкомолекулярные продукты. Органо-
лептическая характеристика по запаху продуктов термического
разложения является довольно субъективной оценкой и требует
практического опыта и сравнения с известными образцами. Для
проведения этого испытания * небольшой образец полимера
* Испытания по п. I. 1.2 и I. 1.3 проводятся в вытяжном шкафу.
9
Таблица 1.2. Идентификация полимеров по воздействию пламени и высокой температуры [9]
Полимер
Поведение в
пламени
Характеристика
пламени
Характер горения
Поведение при пиролизе
Запах продуктов
термического разложения
Полиэтилен
Полипропилен
Горит и после Голубое с жел-
удаления из пла- той верхушкой,
мени светящееся
То же
Сополимер этилена с »
винилацетатом
Поливинилацетат »
Поливинилбутираль
Поливииилформаль
Поливин илацеталь,
поливин илэтилаль
Сополимер винилаце-
тата с бутилакрила-
том
Полиакрилаты
Полиметилметакри-
лат
То же
Желтое
Желтое с
искрами
Синеватое
светящееся
Голубая кайма
у основания
пламени
То же
Желтое
Голубое,
светящееся
Синее
Небольшое
количество копоти,
расплав капает
То же
Небольшое
количество копоти
Небольшое
количество копоти,
расплав капает
Нет дыма
То же
Небольшое
количество копоти
То же
Небольшое
количество копоти,
горит с
потрескиванием
Плавится, на стенках
пробирки
конденсируется жидкость,
застывающая при
охлаждении
То же
Разлагается с
образованием жидких
продуктов
Разлагается с
образованием жидких
продуктов
Разлагается
практически без жидких
продуктов
То же
Разлагается с
образованием жидких
продуктов
Плавится, разлагается
Плавится, пузырится
при разложении
Горящего парафина
Горящего парафина,
но более
ароматический
Уксусной кислоты и
горящего парафина
Уксусной кислоты
Прогорклого масла
или сыра
Слабый
формальдегида
Сладковатый
фруктовый
Уксусной кислоты и
раздражающий —
акрилатов
Острый, резко
ароматический
Сильный цветочно-
плодовый
Полистирол,
ударопрочный полистирол
Акрилонитрилбута-
диенстирольный
пластик
Триацетат целлюлозы
Целлюлоза, целлофан
Нитрат целлюлозы
Полиэтилентерефта-
лат
Полиуретан
Полиформальдегид
Эпоксидные смолы
(неотвержденные) *
Поликарбонат
Полимеры и
сополимеры на основе
винил- и винилиденхло-
рида, хлорированный
полиэтилен, полихло-
ропрен, пентапласт
Горит вне
пламени горелки,
постепенно
самозатухает
То же
Горит, но при
удалении из
пламени горелки
гаснет
Оранжево-желтое, светящееся
Оранжево-желтое
Желтое
Яркое
светящееся
Желтое,
светящееся
Много копоти,
при поджигании
вспыхивает,
горит легко
То же
Легкий дым,
горит легко
То же
Вспыхивает,
горит быстро, с
разбрызгиванием
искр, без дыма и
копоти
Немного копоти
Синеватое с жел- Легкий дым
тыми краями
Синеватое
Желтое,
светящееся
Желтое,
светящееся,
неспокойное
Зеленое с
голубой верхушкой
Без копоти,
расплав капает
Немного копоти
То же
Немного
дыма
белого
Плавится, на стенках
пробирки
конденсируется жидкость
Цветочный (стирола)
Плавится, разлагается Горящей резины и
цветочный (стирола)
Плавится, испускает
белый дым, кипит и
обугливается
Разлагается
Разлагается с
выделением бурых оксидов
азота
Уксусной кислоты
Жженой бумаги
Оксидов азота
Плавится, мутнеет, раз- Сладковатый
лагается, на стенках
пробирки образуется
белый налет
Разлагается Острый
Плавится
Чернеет, плавится,
разлагается
Очень резкий
альдегида
Жженой кости
форм-
Плавится, пузырится, Фенола
разлагается
Размягчается,
разлагается с выделением
белого дыма
Очень резкий
роводорода)
(хло-
Продолжение
Полимер
Поведение в
пламени
Характеристика
пламени
Характер горения
Поведение при пиролизе
Запах продуктов
термического разложения
Политрифторхлор-
этнлен
Поливинилфторид
Поливинилиден
фторид
Политетрафторэтилен
Кремнийорганический
эластомер
Горит, но при Зеленое с голу-
удалении из пла- бой верхушкой
мени горелки
гаснет
То же Желтое
Желтый
Фенолоформальде-
гидные смолы
Горит с трудом Желтое со светло-
только в пламе- голубыми кромка-
нн горелки, по- мн
степенно гаснет
Меламиноформальде- Не горит —
гндная смола
Немного белого
дыма
С копотью
Без копоти
Без копоти
При горении
трескается
Разлагается
Очень резкий (хло-
ро- и фтороводоро-
да)
Резкий (фтороводо-
рода)
То же
Разлагается с
образованием слабого белого
налета на стенках
пробирки и конденсата
коричневого цвета; на
дне в месте
нахождения образца — белый
налет
Разлагается с
образованием плотного
белого налета на стенках
пробирки; на дне в
месте нахождения
образца — белый налет
Разлагается, остается Слабый, горящего во-
большой зольный оста- ска
ток
Не плавится, разлага- Смешанный фенола
ется с выделением па- и формальдегида
ров
То же
Смешанный аммиака
и формальдегида
* Запах продуктов разложения отвержденной смолы в основном определяется введенными отвердителями.
(л;0,02 г) помещают на дно пробирки из термостойкого стекла
со шлифом и нагревают в средней части пламени спиртовки в
течение 5 мин. Во время этой процедуры полимер начинает
разлагаться, и образующиеся летучие продукты заполняют
объем пробирки. Перед определением запаха продуктов
разложения отверстие пробирки закрывают стеклянной пробкой и
переворачивают пробирку вверх дном. Это облегчает
определение запаха в случае образования небольшого количества паров.
Через 1—2 мин пробку открывают и осторожно вдыхают
запах продуктов разложения. Свои впечатления сопоставляют с
данными табл. 1.2.
Одновременно наблюдают за поведением образца при
нагревании (размягчается, растекается, пузырится и т. п.), а
также за видом продуктов, конденсирующихся на стенках
пробирки. Полученные результаты сравнивают с данными табл. I. 2.
Во время пиролиза образца в пробирке проверяют реакцию
образующихся летучих продуктов с помощью увлажненной
индикаторной лакмусовой бумаги. Слабокислые пары
образуются при разложении углеводных полимеров, например,
нитрата и ацетата целлюлозы. Сильнокислые пары выделяются
при разложении поливинилхлорида и его сополимеров, поливи-
нилиденфторида, политрифторхлорэтилена и их сополимеров,
хлорированного полиэтилена. Щелочная реакция продуктов
разложения характерна для азотсодержащих полимеров:
полиамидов, полиуретанов, аминоформальдегидных смол. Нейтральные
продукты разложения выделяются при пиролизе полиолефи-
нов, изопреновых и бутадиеновых каучуков, силоксанов,
полиэфиров.
1.1.3. Растворимость
После предварительных заключений о принадлежности
полимера к тому или иному классу по п. I. 1.1 и I. 1.2 проводят
определение способности полимера растворяться в органических
растворителях (табл. 1.3), что дополняет сведения о его
свойствах [9]. Для этой цели подготавливают несколько пробирок,
помещают в каждую из них около 0,02 г тонкоизмельченного
или нарезанного мелкими кусочками полимера и добавляют по
5 мл растворителя, время от времени встряхивая пробирки.
Наблюдают за поведением полимера в течение 2 3 ч. На
первой стадии растворения полимер, как правило, набухает, а
затем набухшая твердая фаза переходит в раствор. Если
образец не растворяется при комнатной температуре, то
содержимое пробирки доводят до кипения при непрерывном
взбалтывании. Если образец не растворяется и после этого, удваивают
количество растворителя и повторяют определение. Следует
учитывать, что в общем случае увеличение молекулярной
массы полимера приводит к росту сил межмолекулярного
взаимодействия и ухудшению растворимости. Кроме того, резко ухуд-
13
Таблица 1.3. Растворители, применяемые в анализе полимеров
Растворитель
Химическая
формула
Гкип' °с
115, с 3571
е [15,
с. 357J
б".ю-з.
Дж'/г-м-3'2 [161
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
Ацетон
Бензол
Вода
Гексан
•/V.JV-Диметилацетамид
Диметилсульфоксид
МДГ-Диметилформамид
Диоксан
Дихлорэтан
Диэтиловый эфир
Изопропиловый спирт
Ксилол (смесь орто-,
мета и пара-изомеров)
Метиленхлорид
Метиловый спирт
Метил этилкетон
Муравьиная кислота
Петролейный эфир
Пиридин
Тетрагидрофуран
Тетрахлорметан
Тетрахлорэтан
Толуол
Уксусная кислота
Хлорбензол
Хлороформ
Циклогексан
Циклогексанол
Циклогексанон
Этилацетат
Этиловый спирт
* Диэлектрическая проница
** Параметр растворимости
СН3СОСНз
с6н6
Н20
СН3(СН2)4СН3
CH3CON(CH3)2
(CH3)2SO
(CH3)2NCHO
С4НвОг
С2Н4С12
С2Н5ОС2Нб
СНэСН(ОН)СН3
С6Н4(СН3)2
СН2С12
СН3ОН
снэсос2н5
нсоон
Смесь легких
бензиновых
фракций
C5H5N
С4Н80
СС14
С2Н4С14
С6Н5СН3
СН3СООН
С6Н5С1
СНС13
С6Н12
С6Н„ОН
С6Н10О
СН3СООС2Н5
С2Н5ОН
емость.
56,24
80,10
100,0
68,74
165,50
189,0
152,50
101,32
83,48
34,48
82,40
138-144,0
40,1
64,51
79,50
100,7
40—100,0
115,58
65,4
76,75
142,0
110,62
117,7
131,69
61,15
80,74
161,10
155,65
77,2
78,32
20,70
2,297
78,35
1,883
38,50
46,60
36,71
2,21
10,16
4,22
18,30
2,374
—
32,63
15,9
56,1
2,0
12,3
7,39
2,23
—
2,379
6,19
5,62
4,72
2,02
15,0
18,3
—
24,30
19,85
19,03
47,06
15,60
23,12
27,21
24,14
22,10
19,23
14,73
—
17,80
—
30,28
19,13
—
14,32—15,96
20,66
20,66
15,96
21,07
18,21
26,19
19,44
17,80
16,16
22,40
19,23
18,41
26,60
шается растворимость сшитых полимеров и полимеров,
прошедших стадию термообработки.
Результаты, полученные в опыте, сравнивают с данными
табл. 1.4. Кроме обычного сравнения с табличными данными
предлагаются различные схемы для идентификации полимеров
по растворимости. Одна из них, основанная на исследовании
растворимости образца полимера в шести растворителях (то-
14
+ Метиловый
спирт
Образец
+ толуол
нр
нр
Метиловый спирт
+ Этил ацетат
нр
+
Тетрахлорметан
- Этилацетат
+ Тетрахлорметан
нр
нр
+ Тетрахлор-
метан
9
10
- Толуол
+ Этилацетат
нр
11
12
13
нр
■ Этилацетат
+ Вода
14
- Тетрахлорметан
+ Циклогехсанон
16
17
18
нр
-Вода
+ Тетрахлорметан
"P
15
нр
19
20
21
Рмс. I. 1. Схема идентификации полимеров по растворимости (р — растворим; нр — нерастворим; + растворитель приливают; — растворитель
сливают):
/—этилцсллюлоза; 2— полистирол; 3— поли-а-метнлстпрол; 4— иолнметплметакрплат; 5 — поллмстплакрплат; б — полиизобутплсн; 7 — полиэтилен;
8— полипропилен; 9— полипппилацетат; 10— полшшпилбутпраль; // — нитрат целлюлозы; 12—сополимер впнилхлорпда с винплацетатом; 13 — полп-
вииилформаль; 14— поливиниловый спирт; 15—поликарбонат; 16—полмвппплхлорнд; 17—сополимер стирола с акрилоиитрнлом; 18—сополимер
впнилхлорпда с акрилоиитрнлом; 19—полиамид; 20 —полиформальдегид; 21 — политетрафторэтилен.
Таблица 1.4. Растворимость полимеров [12, 14, 17, 28]
Полимер
Номер растворителя
по табл. 1.3
Дополнительные сведения
о растворимости
По л и в и ни л ацетатные пластики
Поливинилацетат
Поливиниловый спирт
Сополимер винилацета-
та с бутилакрилатом
Поливинилбутираль
Поливинилбутиральфур-
фураль
Поливинилформаль
1, 2, 8, 9, 14, 18, 20,
23, 25, 28, 29, 30
3, 7, 16, 23
1, 2, 7, 12, 29, 30
1, 8, 9, 18, 19, 25,
28, 29, 30
30
8, 9, 18, 23, 25
Поливинилформаль
этилаль
Поливинилэтилаль
9, 14, 25, 30
Поливинилхлоридные пластики
Растворим в формамиде
Растворим в смеси
этилового спирта с бензолом
Растворим в смеси ацетона
и этилового спирта
Растворим в бензиловом
спирте, крезоле, фурфуроле
Растворим в бензиловом
спирте
Поливинилхлорид
Поливинилхлорид
хлорированный
(перхлорвинил)
Поливинилиденхлорид
Сополимер винилхлори-
да с винилацетатом т
Сополимер винилхлори-
да с метилметакрилатом
8, 9, 19, 21, 28
1, 2, 7, 8, 12, 15, 19,
21, 25, 29
8, 19
1, 8, 9, 19, 23, 25,
28
1, 8, 9, 18, 19, 25,
28
Полиолефины
В пиридине растворим при
нагревании
В пиридине растворим при
нагревании
Полиэтилен 12. 22 ^
Полипропилен
Поли-4-метилпентен-1
Сополимеры этилена с
пропиленом
Сополимеры этилена с
высшими а-олефииами
Сополимеры этилена с
винилацетатом
Полиэтилен хлорсульфи-
рованиый
Полистирольные пластики
12, 22
12, 22
12, 22
12, 22
12, 22
12, 22
2, 9, 13, 20, 22, 25
Полностью растворимы при
температуре кипения
растворителя, при охлаждении
раствора осаждаются вновь;
так же растворяются в аце-
тофеноне
Полистирол
Сополимер стирола с
а-метилстиролом
Сополимер стирола с
акрилонитрилом
Сополимер стирола с
метилметакрилатом
2, 7, 8, 9, 13, 15, 19,
20, 22, 25, 26
2, 9, 13, 19, 20, 23,
25, 28, 29
1, 7, 8, 9, 15, 19, 23,
25, 27, 28, 29
1, 2, 8, 9, 13, 19, 20,
23, 25, 28, 29
16
Продолжение
Полимер
Номер растворителя
по табл. 1.3
Дополнительные сведения
о растворимости
Сополимер стирола с ме-
тилметакрилатом и ак-
рилонитрилом
АБС-пластик
Поливинилфторид
Полининилиденфторид
Политрифторхлорэтилен
Политетрафторэтилен
1, 2, 8, 9, 13, 20, 23,
25, 28
22
Фторполимеры
5, 6, 7, 8, 28
1, 5, 6, 7, 12, 15, 20,
22, 25
2, 12, 20, 22
В метиловом спирте
растворим при нагревании до
50 °С; в тетрахлорметане и
этилацетате набухает
Растворим при 100—110°С,
при охлаждении раствора
полимер выпадает в виде
геля
Растворим под давлением и
выше температуры кипения
растворителей
Полимеры на основе акриловой и метакриловой
кислот и их производных
Полиакриламид
Полиакрилаты
Полиакрилонитрил
Полибутилметакрилат
Полиметилметакрилат
1, 2, 13, 22, 25, 28,
29
5, 6, 7
1, 2, 8, 9, 19, 20, 22,
25, 29
1, 2, 8, 9, 13, 18, 19,
22, 23, 25, 28
Растворим в формамиде,
диэтилсульфоксиде, этилен-
гликоле
Растворим в водных
растворах ZaCb, HN03>
NHiSCN
Поливинилкарбазол
Поли-Л/-винилпирроли
дон
Другие полимеризационные пластмассы
2, 9, 19, 20, 25 —
3, 8, 14, 18, 25, 30 Растворим в этиленгликоле
Полиэфиры простые
Полиформальдегид
Полиэтиленоксид
(высокомолекулярный)
Полиэтиленгликоль
(низкомолекулярный)
Поли-2,6-диметил-я-фе-
ниленоксид(норил)
Пентапласт [поли-3,3-
бис (хлорметил) оксацик-
лобутан]
Растворим
практически во всех
растворителях
То же
2, 6, 7, 8, 13, 19, 20,
25
8, 9, 19, 25, 28, 30
Растворим в ароматических
аминах и фенолах при
температуре выше 100°С
17
Продолжение
Полимер
Номер растворителя
по табл. 1.3
Дополнительные сведения
о растворимости
Полиэфиры сложные
Поликарбонаты
Полиэтилентерефталат
Алкидные смолы
Поли-е-капроамид
(полиамид П-6)
Полигексаметиленади-
пинамид (полиамид
П-6,6)
Полигексаметиленсе-
бацинамид (полиамид
П-6,10)
Полидодеканамид
(полиамид П-12)
Поли-со-ундеканамид
(полиамид П-11)
7, 13, 25
7
12, 22
Полиамиды
5, 6, 7, 16
5, 6, 7, 16, 23
5, 6, 7, 16, 23
5, 6, 7
5, 6, 7
Другие
Полиимиды
ароматические
алифатические
Полиуретаны
поликонденег
5, 7, 8
25
6, 7, 15, 19
Полифосфазены
Мочевиноформальдегид-
ные (карбамидные)
смолы неотверждениые
Меламиноформальдегид-
ные смолы,
неотверждениые
Фенолоформальдегидные
и фенолофурфурольные
смолы, неотверждениые
Фурановые смолы,
неотверждениые
Эпоксидные смолы (ди-
ановые),
неотверждениые
4, 12, 18, 19, 22, 24
3, 23
3, 23
1, 20, 25, 30
1, 2, 10, 12, 13, 15,
20, 22, 25
Растворим в ЛГ.Л/'-диметил-
формамиде, о-дихлорбензо-
ле и нитробензоле при
нагревании
Растворимы в уайт-спирите,
сольвент-нафте
Растворим в
концентрированных серной и соляной
кислотах
Растворим в
концентрированных серной и соляной
кислотах, крезоле, феноле
То же
Растворим в
концентрированных серной и соляной
кислотах
Растворим в
концентрированных серной и соляной
кислотах, крезоле,
фторированных спиртах
Растворимы в
концентрированной серной кислоте,
растворение сопровождается
деструкцией
Растворимы в 60 %-ной
серной кислоте,
концентрированных растворах
щелочей, в смеси фенол : вода
(90: 10)
Растворимы только
линейные полимеры
Растворимы в 1-гептаноле
Растворимы в 1-гептаноле.
Растворимы в ледяной
уксусной кислоте
18
Продолжение
Полимер
Номер растворителя
по табл. 1.3
Дополнительные сведения
о растворимости
Эпоксидированные фе- 1, 2, 15, 22
нолоальдегидные смолы,
неотвержденные
Кремнийорганические 2, 7, 10, 20, 25, 30
полимеры
Растворимы
спиртах
в высших
Производные
Ацетаты целлюлозы 8, 9, 11, 13,
23, 25, 28
Ацетобутират целлюлозы 1, 2, 8, 9, 13,
25, 29
Нитраты целлюлозы
Целлофан
Этилцеллюлоза
1, 8, 15, 29
11
1, 2, 6, 8, 11,
Растворимость зависит от
вида кремнийорганического
полимера
целлюлозы
18, 20, Ацетаты целлюлозы с
содержанием 60—61,5 %
СНзСООН растворимы в 7,
с содержанием 52—56 %
СНзСООН-в 1
18, 23, Растворим в метилацетате,
трихлорэтилене, этиленгли-
коле
Растворимы в бутилацета-
те
18, 29 Растворима в смеси
этилового спирта с бензолом
луоле, метиловом спирте, этилацетате, тетрахлорметане, цикло-
гексаноне и воде), приводится на рис. 1.1 [11].
Данные по растворимости необходимы в дальнейшем при
операциях выделения добавок, разделения на фракции,
подготовки образцов для снятия ИК- и ЯМР-спектров.
1.1.4. Качественное обнаружение элементов
Проведение качественных реакций на CI, Br, I, F, S, N,
Р, В, Si и некоторые металлы позволяет сделать заключение
о принадлежности анализируемого образца к той или иной
группе полимеров, содержащих гетероатомы (табл. 1.5).
Качественное определение элементов проводится после
минерализации полимера. Для этой цели предложены различные
сгособы: сжигание в колбе, наполненной кислородом [12, гл.2;
13; 17, с. 20], пиролиз в атмосфере водорода и гелия (20:80)
[18], минерализация с использованием низкотемпературной
плазмы [19], сплавление с бинарным сплавом металлического
натрия со свинцом (1:9) [20], восстановительное разложение
вещества с металлическим натрием или калием [21, с. 12].
Обзор методов сплавления с различными реагентами для
обработки трудноразлагаемых органических образцов, в том числе
и полимерных, приводится в работе [22]. Способ
восстановительного разложения с металлическим натрием (калием)
прошел многолетнюю проверку в органическом анализе и в
анализе полимеров [10, 12, 13, 17, 20, 21, 22] и, несмотря на не-
19
Таблица 1.5 Классификация полимеров по наличию
гетероатомов
Гетероатомы
Полимеры
N
S
N, S
С1
CI, S
Вг
F
F, N
F, C1
F, S
Р
Р, N
Р, S, N
Si
В
Си, Zn, Fe,
Со, Mo, Ni,
С г, Мп
Полиамиды, полиимиды, полиакрилоиитрил, полиакриламид,
поливииилпирролидон, поливинилкарбазол, полиуретаны,
нитрат целлюлозы, бутадиениитрильные каучуки, сополимеры
стирола с акрилонитрилом, сополимеры стирола с метилмет-
акрилатом и акрилонитрилом, АБС-пластики, меламиноформ-
альдегидные смолы, мочевиноформальдегидные смолы, ани-
линоформальдегидные смолы, мочевлнофурфурольные смолы
Полиариленсульфоны, полиариленсульфиды, полисульфиды,
каучуки бутадиеновые и изопреиовые(вулканизоваиные серой)
Тиомочевиноформальдегидные смолы, сульфонамидформаль-
дегидпые смолы
Поливинилхлорид, поливинилиденхлорид, пентапласт,
перхлорвинил, полидихлорстирол, полиэтилен хлорированный,
сополимеры в.инилхлорида с винилиденхлоридом, сополимеры
впнилхлорида с винилацетатом
Поливинилсульфохлорид, полиэтилен хлорсульфированный,
каучук хлорированный (вулканизованный серой)
Бромбутилкаучук
Политетрафторэтилен, поливинилфторид, поливииилиденфто-
рид, сополимеры тетрафторэтилена с этиленом, сополимеры
винилиденфторида с этиленом, сополимеры тетрафторэтилена
с гексафторпропиленом, сополимеры винилиденфторида с пер-
фторалкилвиниловыми эфирами, каучук фторированный
Фторнитрозокаучук
Политрифторхлорэтилен, сополимеры трифторхлорэтилена с
трифторэтиленом, сополимеры трифторхлорэтилена с винили-
денфторидом
Поливинилсульфофторид, полифторсульфон
Полимеры виниловых и аллиловых эфиров фосфорной
кислоты
Полифосфазены
Белковые полимеры
Кремнийорганические полимеры
Борорганические полимеры
Полихелатные полимеры
достатки, связанные с использованием металлического натрия
(калия) является довольно надежным, дающим возможность в
одной пробе открыть галогены, азот, серу, фосфор и кремний.
Для обнаружения бора можно применять минерализацию
полимерного образца в низкотемпературной плазме [19] или
сжигание в колбе с кислородом в присутствии платины.
Для обнаружения фтора, хлора, брома, иода, азота, серы,
фосфора и кремния кроме разнообразных химических методов
в последние годы находит все более широкое применение
ионная хроматография, позволяющая разделять и определять их
в продуктах разложения полимеров и в добавках органической
природы [27] с чувствительностью 1-10~4% [23,24].
20
Для обнаружения гетероатомов в полимерах и органических
добавках применяется также пиролитическая газовая
хроматография. Идентификация продуктов пиролиза, содержащих гете-
роатомы, осуществляется как на основе избирательной
способности хроматографических фаз к удерживанию различных
по природе химических соединений с использованием обычных
детекторов (ДИП, катарометр), так и с применением
селективных детекторов [18,25].
Обнаружение металлов в полимерах после разложения
можно осуществлять флуоресцентным, рентгенофлуоресцент-
ным, эмиссионным и атомно-адсорбционным методами [26,
гл. 19; 20].
1.1.4.1. Разложение образцов
При проведении разложения необходимо соблюдать правила
безопасной работы с металлическим натрием (калием),
работать в вытяжном шкафу, пользуясь защитной маской или
очками.
Небольшое количество металлического натрия (~0,5 г) и
не более 0,1 г полимера (или 10—20 мг органического
вещества— добавки) вносят в пробирку из тугоплавкого стекла и
нагревают на пламени спиртовки до появления паров натрия в
средней части пробирки. Затем в ту же пробирку вносят такое
же количество полимера таким образом, чтобы частицы
образца не попали на стенки пробирки, и продолжают нагревание
дна пробирки до красного каления и прекращения выделения
паров. После этого пробирку опускают, не охлаждая, в стакан
с 10 мл дистиллированной воды. Если пробирка не лопается, ее
необходимо разбить изнутри стеклянной палочкой. Полученный
раствор фильтруют от остатков стекла и фильтрат используют
для качественного обнаружения элементов.
1.1.4.2. Обнаружение хлора, брома, иода
Для полимеров из данных элементов наиболее характерно
наличие хлора (см. табл. 1.5). Хлор и бром содержатся в ан-
типиренах, наиболее распространенные из которых: хлорсодер-
жащие — хлорпарафины (содержание хлора до 70%); бромсо-
держащие — гексабромбензол, декабромдифенилоксид, бис(пен-
табромфениловый) эфир.гексабромциклодекан (содержание
брома до 83%) и другие; хлорбромсодержащие — синтекс ВС-26
(29% Вг и 40% С1), хлорфосфорсодержащие — три(1,3-ди-
хлорпропил)фосфат; бромфосфорсодержащие — три(2,3-дибром-
пропил) фосфат и другие [27, 28Jj иод содержащие антипире-
ны — тетраиодфталаты, триодбензоаты и другие — практически
не встречаются в промышленных образцах. Кроме антипиренов
галогены могут содержаться в термо- и светостабилиза-
торах — хлорпроизводных бензтриазола, пластификаторах —
21
хлорированных нафталинах, дифенилах, парафинах,
инициаторах полимеризации (2,4-дихлорбензоилпероксид), антистатиках
(алкилбензилпиридинийхлорид).
Перед проведением реакций на хлор, бром и иод фильтрат
(см. п. I. 1.4.1) подкисляют азотной кислотой и 2—3 мин
кипятят для удаления циано- и сероводорода, если в
анализируемом продукте есть азот и сера, затем охлаждают.
Проба Бейльштейна. В пламени спиртовки прокаливают
петлю из медной проволоки, которая при этом покрывается
оксидом меди. После охлаждения на петлю помещают небольшое
количество анализируемого продукта и вносят в бесцветную
зону пламени. Летучие галогениды меди окрашивают пламя в
чисто-зеленый цвет при наличии иода, в голубовато-зеленый —
при наличии хлора или брома. Следует учитывать, что
подобное окрашивание пламени летучими соединениями меди могут
дать мочевина, муравьиная кислота, нитрилы, борорганические
соединения и некоторые производные пиридина, не содержащие
галогена, но имеющие в своем составе азот и серу.
Реакция с AgN03. К 2 мл фильтрата добавляют 1 мл 2
о/оного раствора AgNOs- В присутствии галогенов выпадают
творожистые осадки серебряных солей: белый —хлорида, бледно-
желтый — бромида, желтый — иодида. В отличие от бромида и
иодида серебра хлорид серебра растворим в 12%-ном растворе
(ЫН4)гСОз и в растворе аммиака и вновь выпадает в осадок
при подкислении раствора. AgBr растворим в растворе аммиака
частично, Agl — практически нерастворим.
Реакция с KN02. К 1—2 мл фильтрата добавляют 5—
10 капель 10 %-ного раствора KN02 и 0,5—1 мл хлороформа.
Пробирку несколько раз встряхивают и дают отстояться.
Фиолетовая окраска хлороформенного слоя указывает на выделение
свободного иода. Ионы С1~ и Вг~ в отличие от I- не
окисляются нитрит-ионом.
I. 1.4.3. Обнаружение фтора
Наличие этого элемента является отличительным признаком
большого класса фторполимеров (см. табл. 1.5). Кроме того,
фтор содержится во фторированных поверхностно-активных
веществах (ПАВ) фтортензидах — производных перфторалифати-
ческих кислот, перфторалкилсульфонатах, используемых для
интенсификации процессов эмульсионной и суспензионной
полимеризации.
Реакция с цирконийализариновым реактивом. Метод основан
на изменении в присутствии фторид-иона ярко-красной окраски
комплекса цирконийхлорида с ализарином на желтую
вследствие образования анионов ZrFe" и выделения свободного
ализарина.
Реактив готовят смешением 10 мл 1 %-ного спиртового
раствора ализарина с 10 мл 2 %-ного раствора нитрата или
хлорида циркония в 5%-ной соляной кислоте и последующим раз-
:22
бавлением до 30 мл водой. Для проведения реакции можно
пользоваться раствором реактива или подготовить полоски
фильтровальной бумаги, пропитанные реактивом и высушенные.
К 1 мл фильтрата (п.1.1.4.1) добавляют 5—б капель
концентрированной соляной кислоты, нагревают до кипения,
охлаждают, прибавляют 5—6 капель реактива. В присутствии
фтора красно-фиолетовая окраска реактива изменяется на
желтую.
Каплю раствора (после кипячения с соляной кислотой)
можно нанести на полоску реактивной бумаги, на которую перед
этим наносят каплю 0,1 н. соляной кислоты. Наличие фтора
подтверждается образованием желтого пятна на красной
бумаге.
I. 1.4.4. Обнаружение азота
Азот содержится в ряде полимеров разных классов (см.
табл. 1.5). Этот элемент встречается во многих добавках,
вводимых в полимерную композицию: в стабилизаторах —
производных алкил- и ариламинов, мочевины и тиомочевины, бенз-
триазолов; во вспенивающих агентах—азобисизобутиронитри-
ле, азоформамиде, диазоаминобензоле и других; в антистатиках
и ПАВ — высших нитрилах, аминах и их солях, четвертичных
аммониевых солях, оксиэтилированных жирных аминах, алк-
аноламидах и продуктах их оксиэтилирования и др.
Реакция с солями Fe(II) и Fe(III). Разложение
азотсодержащего соединения сопровождается образованием цианида натрия
(калия), который открывают реакцией взаимодействия цианида
с солями Fe{II) и Fe(III), ведущей к образованию осадка
берлинской лазури Fe3[Fe(CN)6h.
К 1—2 мл фильтрата (п.1. 1.4.1) прибавляют несколько
капель 10%-ного раствора NaOH (до рН 13,0), 1—2 мл
свежеприготовленного насыщенного раствора FeS04 и 1—2 капли
раствора FeCb. [Вместо растворов Fe(II) и Fe(III) можно
применять раствор соли (NH4)2Fe(S04)2-6H20, которая всегда
содержит следы Fe(III).] Раствор кипятят в течение 30 с
и после охлаждения подкисляют, добавляя по каплям 30%-ную
серную или концентрированную соляную кислоту до
растворения осадка оксида Fe(III). Образование синего осадка
указывает на присутствие в образце азота. При малых содержаниях
азота раствор окрашивается в зеленовато-голубой цвет. Желтая
окраска раствора свидетельствует об отсутствии азота.
Реакция с я-нитробензальдегидом для соединений,
содержащих азот и серу [30]. К 1—2 мл фильтрата (п. I. 1.4.1)
добавляют по каплям 1 %-ный раствор n-нитробензальдегида в ди-
метилсульфоксиде. При наличии азота появляется пурпурное
окрашивание, сера дает зеленый цвет. В присутствии обоих
элементов окраска пурпурная, поэтому серу следует определить
по ее характерным реакциям (см. п. I. 1.4.5) в отдельной
порции фильтрата.
23
1.1.4.5. Обнаружение серы
Сера содержится в полимерах нередко в сочетании с
другими гетероатомами (см. табл. 1.5), на что следует обращать
внимание при идентификации. Органические серусодержащие
соединения применяются в производстве пластмасс в качестве
стабилизаторов — производные тиоэфиров, тиомочевины, ксан-
тогенатов; вспенивающих агентов — бензолдисульфоногидразид
и др.; антистатиков и ПАВ — алкилсульфаты, алкансульфона-
ты, нафталинсульфонаты, некоторые амфолитные и катионные
ПАВ.
Серу открывают в фильтрате (п.1.1.4.1), где она находится
в виде сульфида натрия (калия).
Реакция с ацетатом свинца. 1—2 капли фильтрата,
подкисленного уксусной кислотой, наносят на полоску
фильтровальной бумаги, предварительно смоченной 2—3 каплями
10%-ного раствора ацетата свинца. Можно к 1 мл этого же
фильтрата прибавить 0,5 мл 10 %-ного раствора РЬ(СНзСОО)г.
Мгновенное появление коричнево-черного или черного
окрашивания свидетельствует о наличии в пробе серы (образование
сульфида свинца).
Реакция с нитропруссидом натрия Na2 [Fe(CN)sNO]. К 1 мл
фильтрата добавляют несколько капель раствора
Na2[Fe(CN)5NO]. В присутствии серы появляется
красно-фиолетовая окраска вследствие образования нестойкого комплекса
Na4[Fe(CN)5NOS]. При подкислении раствора окраска
исчезает.
Реакция с я-нитробензальдегидом (см. п. I. 1.4.4).
1.1.4.6. Обнаружение кремния
Кремний является элементом, образующим, как правило,
основную полимерную цепь кремнийорганических полимеров,
иногда входит в боковые группы (см. табл. 1.5). Из химических
добавок к полимерам применяются кремнийсодержащие
стабилизаторы — диметилбис [4- (2-нафтиламино) фенокси] силан, ди-
метилбис(4-фениламинофенокси)силан; ПАВ — полиметилсил-
оксановые жидкости (смесь полимеров линейной структуры),
полиэтилсилоксан.
Реакция образования восстановленной молибденовой гете-
рополикислоты H4[SiMoi2O40] [20]. 1—2 мл фильтрата
(п.1. 1.4.1) разбавляют до 10 мл водой, подкисляют 5 мл
раствора серной кислоты (30 мл H2S04 и 0,4 мл HNO3
разбавляют водой до 100 мл), добавляют 0,2 г персульфата калия и
кипятят 10 мин. После охлаждения добавляют 1 каплю
фенолфталеина и водный раствор аммиака до щелочной реакции, затем
вновь добавляют раствор серной кислоты до исчезновения
розового окрашивания и еще 1 каплю. Затем постепенно вводят
8—10 капель раствора молибдата аммония (2,5 г молибдата
24
аммония растворяют в 20 мл воды и добавляют к
предварительно охлажденному раствору 30 мл концентрированной
серной кислоты в 40 мл воды; после охлаждения раствор
разбавляют водой до 100 мл) и 2 капли 2,5%-ного раствора SnCb
в глицерине. Раствор перемешивают и оставляют на 10 мин.
При этом появляется темно-синее окрашивание,
свидетельствующее о присутствии кремния. Такую же реакцию дает фосфор.
Кремнийорганические полимеры, в отличие от
фосфорсодержащих, после прокаливания в муфельной печи в тигле при
700 °С образуют большой зольный остаток диоксида кремния,
который нацело растворим в плавиковой кислоте.
1.1.4.7. Обнаружение фосфора
Фосфорсодержащие полимеры сравнительно редко
встречаются в аналитической практике. Чаще аналитику приходится
сталкиваться с фосфорсодержащими органическими добавками
в полимерных композициях: стабилизаторами — эфиры
фосфористой, пирокатехинфосфористой и салицилфосфористой кислот,
элементорганические — глицерофосфаты; пластификаторами —
трибутилфосфат, три-о-крезилфосфат и др.; антипиренами —
три (1,3-дихлорпропил) фосфат, три (2,3-дибромпропил) фосфат;
антистатиками—ди(2-этилгексил) фосфат натрия, бис(диэта-
ноламид) додецилфосфиновой кислоты; анионными ПАВ —
производные алкилфосфорных и алканфосфорных кислот.
Реакция образования молибденованадофосфорной гетеропо-
ликислоты H4[PVMon04o]. 1—2 мл фильтрата (п. 1.1.4.1)
доводят водой до 10 мл, добавляют 1 мл разбавленной азотной
кислоты (1:2), 5 мл раствора NH4N03 (0,25 г NH4N03
растворяют в 50 мл горячей воды, охлаждают, прибавляют 2 мл
концентрированной H2SO4 и разбавляют водой до 100 мл), 5 мл
молибденового реагента (2,5 г молибдата аммония растворяют
в 20 мл воды и добавляют к предварительно охлажденному
раствору 30 мл концентрированной H2S04 в 40 мл воды; после
охлаждения раствор разбавляют водой до 100 мл),
перемешивают и добавляют 20 мл воды. Через 5—10 мин раствор
приобретает устойчивое желтое окрашивание, свидетельствующее
об образовании молибденованадофосфорной кислоты.
1.1.4.8. Обнаружение бора
Борорганические полимеры (поликарбораны) в основном
отличаются высокой термостойкостью. Применяются карборансо-
держащие фенолоформальдегидные и эпоксидные смолы в
качестве основы термостойких клеев и связующих
композиционных материалов.
Реакция окрашивания пламени. 10—20 мг борсодержащего
полимера сжигают в колбе с кислородом в пакетике из
фильтровальной бумаги, помещенном в платиновую сетку (см.
25
рис. 11.32). В колбу налит поглотительный раствор из 2,5 мл
Ь,5 н. раствора NaOH и 2,5 мл воды. Спустя 20 мин содержимое
колбы переносят в фарфоровую чашку и упаривают на водяной
бане досуха. Сухой остаток после охлаждения обрабатывают
1—2 каплями концентрированной серной кислоты и 5—6
каплями этилового спирта, хорошо перемешивают и поджигают.
Пламя по краям окрашивается в зеленый цвет в результате
образования летучего борноэтилового эфира:
НэВОз + ЗС2Н5ОН = B(OC2H5)3f + ЗН20.
Обнаружение бора можно проводить этой же реакцией, не
■сжигая образец в колбе, а подвергая его непосредственно
обработке теми же реактивами, как сухой остаток. При
минерализации образца в процессе сгорания образуется борноэтиловый
эфир.
1.1.4.9. Обнаружение Си, Zn, Fe, Co, Ni, Mo, Сг, Мп, Na,
Са, Mg, Al, Sb, Pb и других элементов
Некоторые из этих элементов входят в структуру полихелат-
ных полимеров (см. табл. 1.5), другие содержатся во вводимых
в полимерные композиции добавках: в стабилизаторах —
цинковая соль 2-меркаптобензимидазола, сгеарат, рицинолеат и лак-
тат кальция, лактат и ацетат натрия, стеарат и глицерофосфат
магния, стеарат цинка; в антистатиках и анионных ПАВ —
натриевые соли алкилсерных кислот, натрийсульфонаты.
Особенно широк ассортимент неорганических добавок к полимерам:
минеральные дисперсные наполнители (аэросил, тальк,
каолин); металлические порошки (латуни, бронзы, алюминий,
железо, цинк, свинец); керамические порошковые материалы
(ферриты); соли и гидраты оксидов металлов [MgCl2-6H20,
А1С13-бН20, CaS04-2H20, Co(NH3)6Cl3, (Li)3P04-0,5H2O,
(NH4)2S04-2SbF3, Al(OH)3]; одноосноориентированные
нитевидные монокристаллы волокон из оксидов металлов (АЬОз,
Fe203, ВеО, Мо03, NiO, Сг203).
Полимеры перед определением в них металлов отделяют от
наполнителей, добавки экстрагируют из полимерной
композиции, затем озоляют по ГОСТ 15973—82 и анализируют зольный
остаток спектрально-эмиссионным или атомно-адсорбционным
методом [26, гл. 20]. Обнаружение металлов в неорганических
наполнителях проводят теми же методами после отделения
полимерной основы композиционного материала и органических
добавок.
1.1.5. Цветные реакции полимеров
Идентификацию ряда полимеров можно осуществить также
по окрашенным соединениям, которые образуются при
взаимодействии некоторых функциональных групп или гетероатомов с
определенными реактивами [10; 12, гл. 2; 13, 17]. Реакции
проводят с полимерами или продуктами их пиролиза.
26
1.1.5.1. Поливинилацетат, сополимеры винилацетата с этиленом,
сополимеры винилацетата с винилхлоридом
Реакция с иодом. 0,05—0,1 г полимера помещают в
пробирку и добавляют 1 мл раствора йода (0,1 г 12 и 1 г KI
растворяют в нескольких каплях воды, разбавляют при
перемешивании 20 мл смеси воды с этиловым спиртом 1 : 1 и доливают
2 н. раствором НС1 до 100 мл). В присутствии поливинилаце-
тата содержимое пробирки приобретает темно-красное
окрашивание.
Реакция с иодом и нитратом лантана. 0,2 г полимера
помещают в пробирку и нагревают на пламени спиртовки до
разложения. В другой пробирке несколько капель пиролизата
смешивают с 1—2 мл дистиллированной воды, добавляют
0,5 мл 5%-ного раствора нитрата лантана и 0,5 мл 0,01 н.
раствора иода, нагревают до кипения. После охлаждения до
40—50 °С осторожно по стенке пробирки вводят пипеткой по
каплям 1 н. раствор аммиака. В присутствии уксусной кислоты,
выделяющейся при пиролизе полимера, на границе
соприкосновения растворов образуется синее кольцо, расширяющееся
книзу. Сульфат, фториды и другие анионы, образующие с лантаном
нерастворимые соли, мешают реакции. Такую же реакцию дает
пропйоновая кислота.
1.1.5.2. Поливиниловый спирт
Реакция с иодом и бурой. 0,05 г полимера растворяют в-
5 мл дистиллированной воды, добавляют 2 капли раствора иода
(п.1. 1.5.1). При появлении синей, зеленой или желто-зеленой
окраски раствор разбавляют водой таким образом, чтобы
окрашивание стало едва заметным. К 5 мл полученного раствора
добавляют 10—20 мг буры, энергично перемешивают и вносят
5 капель концентрированной соляной кислоты, в присутствии
которой появляется зеленая окраска, особенно заметная на
кристаллах буры.
При идентификации поливинилового спирта с большим
содержанием ацетатных групп после прибавления иода раствор
окрашивается в винно-красный цвет, который после добавления
буры и соляной кислоты изменяется на сине-зеленый.
I. 1.5.3. Поливинилацетали
Все поливинилацетали при кипячении с 25 %-ным раствором
серной кислоты отщепляют альдегиды, обладающие
характерным запахом. Альдегиды дают положительную реакцию с фук-
синсернистой кислотой.
Реакция с фуксинсернистой кислотой. 0,5 мл раствора поли-
винилацеталя в серной кислоте переносят в пробирку,
добавляют 2 мл фуксинсернистой кислоты — реактива Шиффа (0,2 г
27
фуксина растворяют в 120 мл горячей дистиллированной воды,
раствор фильтруют и после охлаждения прибавляют 2 г
безводного бисульфита натрия и 2 мл концентрированной соляной
кислоты. Раствор переносят в мерную колбу вместимостью
200 мл, доводят до метки дистиллированной водой. Раствор
применим в течение года.) В присутствии альдегидов раствор
принимает окраску от красной до сине-фиолетовой вследствие
образования красителя хиноидной структуры.
Реакция с анилинацетатом. 0,5 мл раствора поливинил-
ацеталя в серной кислоте переносят в пробирку, добавляют 1 мл
50 %-ного раствора анилина в ледяной уксусной кислоте.
В присутствии фурфурола появляется винно-красная окраска.
Эту реакцию дает только поливинилбутиральфурфураль.
Реакция с хромотроповой кислотой. 0,1 г образца помещают
на дно пробирки, добавляют 2 мл 75 %-ной серной кислоты и
несколько кристаллов хромотроповой кислоты (1,8-диоксинаф-
талин-3,6-дисульфокислоты); смесь нагревают на водяной бане
при 60—70 °С в течение 10 мин. При наличии формальдегида
(полнвинилформаль, поливинилформальэтилаль) появляется
фиолетовое окрашивание. Подобную реакцию дает акролеин.
Красное окрашивание в этой реакции образуется в присутствии
поливинилацетата, поливинилбутираля, нитрата целлюлозы.
Реакция с иодом. Небольшой кусочек образца помещают
на дно фарфоровой чашки, наносят на него 1—2 капли
реактива (10 г 50 %-ной уксусной кислоты смешивают с 7 г
раствора, состоящего из 1 г KI, 0,9 г lj, 40 мл воды и 2 г
глицерина). Окраска образца зависит от природы альдегида в составе
поливинилацеталя и содержания свободных гидроксильных
групп. Поливинилбутираль с высоким содержанием бутираль-
ных групп окрашивается в желтый цвет; при содержании
гидроксильных групп не более 15 % желтая окраска сохраняется,
до 18% —светло-зеленая, от 18 до 27 % зеленая, а при
содержании гидроксильных групп 28 % — синяя. Соответственно
окраска поливинилформаля изменяется от синего к черному,
поливинилэтилаля — от зеленого к темно-зеленому.
I. 1.5.4. Поливинилхлоридные пластики
Реакция с хлоруксусными кислотами. В пробирке из
термостойкого стекла расплавляют несколько граммов моно- или ди-
хлоруксусной кислоты, добавляют около 0,1 г измельченного
полимера. Нагревают на пламени спиртовки до кипения при
постоянном перемешивании. Через 2 мин в присутствии поли-
винилхлорида расплав монохлоруксусной кислоты приобретает
синий цвет, дихлоруксусной—красно-фиолетовый; сополимеров
винилхлорида с винилацетатом — вишнево-красный и
сине-фиолетовый, соответственно. В присутствии сополимеров
винилхлорида с метакрилатами расплав монохлоруксусной кислоты
окрашивается в бледно-красный, переходящий в сине-фиолетовый
28
цвет. Сополимеры дают положительную реакцию при наличии
в составе 2/з винилового компонента.
Реакции мешают поливиниловый спирт, канифоль,
модифицированные алкидные смолы.
Реакция с пиридином. 0,01 г образца помещают в пробирку
и растворяют в 1 мл пиридина, нагревая пробирку на водяной
бане. К горячему раствору добавляют 0,5 мл 2 %-ного раствора
гидроксида натрия в метиловом спирте. В присутствии поливи-
нилхлорида образуется темно-коричневый осадок. Поливинили-
денхлорид, в отличие от поливинилхлорида, дает эту реакцию
без подогревания.
I. 1.5.5. Полистирол
Реакция с метиленхлоридом и азотной кислотой. 0,05 г
полимера помещают на дно фарфоровой чашки и наносят на него
3—5 капель реактива (смесь 55 % метиленхлорида и 45 %
концентрированной азотной кислоты). Красно-коричневая окраска
свидетельствует о том, что полимер является полистиролом.
Реакция образования я-нитробензойной кислоты. 0,1 г
полимера в круглодонной колбе с обратным холодильником
нагревают до кипения с 5 мл концентрированной азотной кислоты и
кипятят около 1 ч до получения прозрачного раствора. Затем
раствор выливают в 20 мл дистиллированной воды. При
наличии стирола выпадает желтый осадок n-нитробензойной
кислоты, которая может быть восстановлена, диазотирована и
соединена с р-нафтолом (см. п.1. 1.5.6).
I. 1.5.6. Сополимеры стирола
Реакция с р-нафтолом. Около 1 г полимера нагревают до
кипения с 20 мл концентрированной азотной кислоты в колбе
с обратным холодильником и кипятят в течение 1 ч. Затем к
смеси в колбе приливают 100 мл воды, переносят в
делительную воронку и экстрагируют диэтиловым эфиром (2 X 25 мл).
Экстракты объединяют, промывают водой (2X15 мл), затем
5 %-ным раствором гидроксида натрия (3 X 20 мл) и вновь
водой (1 X 20 мл). Промывные воды и щелочные экстракты
объединяют, раствор подкисляют концентрированной соляной
кислотой и прибавляют избыток кислоты (20 мл). К раствору
добавляют около 5 г гранулированного цинка для
восстановления n-нитробензойной кислоты и нагревают на водяной бане
20 мин.
После растворения цинка содержимое колбы нейтрализуют
20 %-ным раствором гидроксида натрия, прибавляют его
избыток для растворения гидроксида цинка и экстрагируют
диэтиловым эфиром. Эфирный слой отбрасывают. Щелочной раствор
подкисляют соляной кислотой, охлаждают до комнатной
температуры и добавляют 2 мл 0,5 н. свежеприготовленного раствора
29
нитрита натрия. Полученный раствор вливают в 10 мл
насыщенного р-нафтола в 5 %-ном растворе гидроксида натрия.
При наличии стирола появляется ярко-красное окрашивание,
усиливающееся при добавлении 20 %-ного раствора гидроксида
натрия.
I. 1.5.7. Полиакрилаты и полиметакрилаты
Реакция с азотной кислотой и нитритом натрия. 0,1—0,2 г
полимера помещают на дно термостойкой пробирки, нагревают
до образования продуктов пиролиза. Переносят 3 капли пиро-
лизата в пробирку, добавляют 1 мл концентрированной азотной
кислоты и нагревают до кипения (раствор иногда
окрашивается в желтый цвет). После охлаждения добавляют 2 мл
дистиллированной воды и 10—20 мг кристаллического нитрита натрия.
После перемешивания в присутствии метакрилатов развивается
синее окрашивание, которое постепенно бледнеет. Если к
раствору добавить хлороформ, то окраска переходит в хлорофор-
менный слой. Сложные эфиры акриловой кислоты дают желто-
коричневую окраску.
Реакция с азотной кислотой и цинковой пылью. К другой
части пиролизата (0,5 мл) добавляют 1 мл концентрированной
азотной кислоты и слегка нагревают. После охлаждения
приливают 0,7 мл воды и добавляют немного цинковой пыли. В
присутствии метакрилатов появляется синее окрашивание, которое
переходит в хлороформенный слой после взбалтывания
раствора с 1—2 мл хлороформа.
I. 1.5.8. Полиакрилонитрил
Работу проводить только в вытяжном шкафу, пары не
вдыхать!
Реакция с серой и солями Fe(III). 0,01 г полимера
нагревают в пробирке из термостойкого стекла с 0,01 г элементарной
серы. После появления паров разложения полимера к
отверстию пробирки подносят фильтровальную бумагу, пропитанную
раствором соли железа(III). Бумага окрашивается в красный
цвет (образование роданида железа), что является
доказательством наличия нитрильной группы.
Реакция с солями Fe(II) и (III). 0,05 г полимера помещают
па дно пробирки и нагревают до образования продуктов
пиролиза. Отбирают около 0,1 мл пиролизата и переносят в
небольшую пробирку. К пиролнзату добавляют 2—3 капли
свежеприготовленного насыщенного на холоду раствора FeSCu [в нем
всегда есть следы Fe (III) ]. Смесь нагревают до кипения,
вводят по каплям концентрированную соляную кислоту до
появления голубого осадка берлинской лазури Fe4[Fe(CN)6]3,
образование которого указывает на присутствие нитрильной
группы.
30
Реакция с ацетатом меди и ацетатом бензидина. 0,05 г
полимера вносят на дно пробирки из термостойкого стекла,
добавляют 0,06 г смеси СаО + СаСОз (1:1) и нагревают. При
появлении паров разложения полимера отверстие пробирки
закрывают полоской фильтровальной бумаги, смоченной 2—3
каплями раствора ацетата меди и ацетата бензидина. В
присутствии нитрильной группы бумага окрашивается в синий цвет.
1.1.5.9. Поливинилкарбазол
Реакция с хлоруксусными кислотами (см. п. I. I.5.4).
Поливинилкарбазол дает светло-зеленое окрашивание с монохлорук-
сусной кислотой и синее с дихлоруксусной кислотой.
1.1.5.10. Поли-Ы-винилпирролидон
Реакция с хлоруксусными кислотами (см. п. I. 1.5.4). Поли-
N-винилпирролидон дает с монохлоруксусной кислотой розово-
фиолетовое, переходящее при умеренном нагревании в сине-
зеленое окрашивание. С дихлоруксусной кислотой поли-N-bh-
ннлпирролидон не образует окрашивания.
1.1.5.11. Полиформальдегид
Реакция с хромотроповой кислотой (см. п. I. 1.5.3).
1.1.5.12. Поли-е-капроамид (полиамид П-6),
полигексаметиленадипинамид (полиамид П-6,6),
полигексаметиленсебацинамид (полиамид П-6,10),
поли-со-ундеканамид (полиамид П-11),
полидодеканамид (полиамид П-12), сополимеры полиамидов
Реакция с п-диметиламинобензальдегидом. 0,05 г полимера
помещают на дно пробирки из термостойкого стекла и
нагревают на пламени спиртовки. В отверстие пробирки вводят
полоску фильтровальной бумаги, смоченной 5 %-ным раствором п-ди-
метиламинобензальдегида. В результате пиролиза полиамидов
образуется пиррол или его производные, которые реагируют с
я-диметиламинобензальдегидом, вызывая красное окрашивание
полоски фильтровальной бумаги. Подобную реакцию могут
давать продукты пиролиза некоторых видов эпоксидных смол и
волокна животного происхождения.
I. 1.5.13. Полигексаметиленадипинамид
Реакция с о-нитробензальдегидом. 0,05 г полимера
нагревают в пробирке из термостойкого стекла, в отверстие которой
помещают полоску фильтровальной бумаги, смоченной
насыщенным раствором о-нитробензальдегида в 2 н. растворе гидр-
оксида натрия. Бумага окрашивается от розово-сиреневатого
31
до черного с лиловым оттенком цвета при наличии в продуктах
пиролиза адипокетона, образующегося при распаде полигекса-
метиленадипинамида. Окрашивание исчезает при смачивании
бумажной полоски разбавленной серной кислотой.
1.1.5.14. Поли-е-капроамид
Реакция с иодовисмутатом калия. 0,2 г полимера помещают
в колбу с 20 мл дистиллированной воды и кипятят 10—15 мин.
После охлаждения добавляют 2—3 капли концентрированной
серной кислоты и 2 мл реактива иодовисмутата калия (5 г
основного нитрата висмута и 2,5 г KI растворяют в 10 мл 2 %-ной
H2S04). Оранжево-красный осадок подтверждает присутствие
поли-е-капроамида.
I. 1.5.15. Полиуретаны
Реакция с л-диметиламинобензальдегидом. 0,5 г полимера
растворяют в 10 мл ледяной уксусной кислоты. Если продукт
плохо растворяется, его следует подогреть до растворения и
после охлаждения добавить 0,1 г /г-диметиламинобензальдегида.
Через 20—30 мин при наличии полиуретана раствор желтеет.
Реакция с гидроксиламинийхлоридом. 0,05 г полимера
помещают в пробирку, добавляют по 3—5 капли 1 н. спиртовых
растворов гидроксида калия и гидроксиламинийхлорида и
нагревают около 1 мин при температуре не более 50 °С,
подкисляют 1 н. соляной кислотой и добавляют 1—2 капли 2 %-ного
водного раствора хлорида железа(III). Сложноэфирные
полиуретаны, в отличие от простых эфирных, реагируют с гидроксил-
амином с образованием гидроксамовой кислоты, дающей с
Fe(III) фиолетовую окраску. Реакции мешают другие сложные
эфиры.
1.1.5.16. Поликарбонат
Реакция с я-диметиламинобензальдегидом. 0,1 г полимера
пиролизуют в пробирке из термостойкого стекла, охлаждают и
добавляют 2 мл 1 %-ного раствора п-диметиламинобензальдеги-
да в метаноле и 1 каплю 5 н. соляной кислоты. В присутствии
продуктов пиролиза поликарбоната раствор окрашивается в
голубой цвет.
I. 1.5.17. Полиэтилентерефталат
Реакция с о-нитробензальдегидом (см. п. 1.1.6.13). При
разложении полиэтилентерефталата образуется циклопентанон,
дающий с о-нитробензальдегидом сине-зеленое окрашивание,
которое после смачивания полоски фильтровальной бумаги
раствором разбавленной серной кислоты приобретает оттенок
индиго (в отличие от полиамидов на основе адипиновой кислоты).
32
1.1.5.18. Алкидные смолы
Реакция с фенолом. 0,05 г полимера вносят в пробирку,
содержащую 0,5 мл дистиллированной воды и 2 мл
концентрированной серной кислоты. Смесь кипятят в течение 1 мин,
охлаждают, отбирают пипеткой 0,5 мл раствора и переносят в
другую пробирку. К раствору добавляют 0,1 г фенола,
нагревают до кипения и фильтруют. К фильтрату добавляют по
каплям 5 н. раствор гидроксида натрия. При наличии фталатов
появляется фиолетово-розовое окрашивание вследстие
образования фенолфталеина.
Реакция с резорцином. 0,05 г полимера сплавляют с 0,1 г
резорцина и 2—3 каплями концентрированной серной кислоты в
пробирке из термостойкого стекла на пламени спиртовки. Сплав
охлаждают, обрабатывают 2—3 мл горячей воды, фильтруют.
К фильтрату добавляют 0,5 н. раствор гидроксида натрия до
щелочной реакции. В присутствии себациновой кислоты
наблюдается интенсивное оранжево-красное окрашивание с зеленой
флуоресценцией вследствие образования флуоресцеина.
Если алкидная смола получена на основе адипиновой
кислоты, то по этой реакции после подщелачивания образуется
вишнево-красное окрашивание. Если алкидная смола получена на
основе янтарной кислоты, то после обработки фильтрата
5 %-ным раствором гидроксида аммония образуется
темно-красное окрашивание с зеленой флуоресценцией.
1.1.5.19. Фенолоформальдегидные смолы
Реакция с солями Fe(III). 0,1 г полимера подвергают
пиролизу в пробирке из термостойкого стекла, пиролизат
охлаждают, добавляют 1—2 мл дистиллированной воды,
перемешивают и вводят 2—3 капли 10 %-ного раствора хлорида
железа (III). В присутствии фенола раствор окрашивается в
фиолетовый цвет.
Реакция с фталевым ангидридом. 0,5 г полимера нагревают
в пробирке из термостойкого стекла с 0,5 г фталевого
ангидрида и 3 каплями концентрированной серной кислоты до
получения расплава темно-коричневого цвета. Расплав охлаждают в
воде и добавляют по каплям 10 %-ный раствор гидроксида
натрия до появления фиолетово-розового окрашивания вследстие
образования фенолфталеина.
Реакция с хромотроповой кислотой (см. п. I. 1.5.3).
1.1.5.20. Фурановые и фуриловые смолы
Реакция с анилином. 0,5 г полимера пиролизуют в
пробирке из термостойкого стекла. 1 мл пиролизата переносят в
другую пробирку и добавляют 1 мл 5 %-ного раствора анилина в
ледяной уксусной кислоте. При наличии фурфурола, образую-
2 Зак. 632
33
щегося в продуктах пиролиза фурановых смол, развивается
красное окрашивание.
Реакция с бензидином. 0,5 г полимера пиролизуют в
пробирке из термостойкого стекла. 1 мл пиролизата переносят в
другую пробирку, добавлют 1 мл уксусной кислоты и 5 мл
5 %-ного раствора бензидина в уксусной кислоте. В
присутствии фурфурола образуется красно-фиолетовое окрашивание.
Реакция с соляной кислотой. 0,5 г полимера пиролизуют в
пробирке из термостойкого стекла. В отверстие пробирки
вводят сосновую лучинку, смоченную соляной кислотой. В
присутствии фурилового спирта, образующегося при пиролизе фури-
ловых смол, лучинка окрашивается в зеленый цвет.
Реакция с солями Fe(III) и с хромотроповой кислотой (см.
п. I. 1.6.19 и п. I. 1.5.3).
I. I.5.21. Мочевино- и меламиноформальдегидные смолы
Реакция с я-диметиламинобензальдегидом. 0,05 г неотверж-
денной смолы (0,1 г отвержденного полимера) помещают на
дно круглодонной колбы, добавляют 10 мл смеси уксусной
кислоты и уксусного ангидрида (3: 10), вносят 0,015 г п-диметил-
аминобензальдегида и присоединяют к колбе воздушный
холодильник. Смесь нагревают на плитке в течение 30—50 мин.
В присутствии мочевиноформальдегидной смолы образуется
синее окрашивание. Меламиноформальдегидные смолы этой
реакции не дают.
Реакция с пикриновой кислотой. 0,5 г полимера вносят в
круглодонную колбу, добавляют 25 мл 80%-ной уксусной
кислоты, соединяют с воздушным холодильником и нагревают
30 мин. Раствор охлаждают, фильтруют, фильтрат выпаривают
на водяной бане досуха. Добавляют к сухому остатку 2 мл
дистиллированной воды, охлаждают и вновь фильтруют. К
фильтрату прибавляют 1 каплю насыщенного водного раствора
пикриновой кислоты. Образуется желтый осадок,
свидетельствующий о наличии меламина. Мочевиноформальдегидные смолы не
мешают определению.
Реакция с хромотроповой кислотой (см. п. I. 1.5.3).
1.1.5.22. Эпоксидные смолы
Реакция с NaOH. 0,02 г тонкоизмельченного полимера
вносят в пробирку и осторожно растворяют в 1 мл
концентрированной серной кислоты. Раствор охлаждают и добавляют 1 мл
концентрированной азотной кислоты. Полученную смесь
выливают в 100 мл 1 н. раствора гидроксида натрия. В присутствии
эпоксидной смолы сразу же появляется яркое
оранжево-красное окрашивание.
Реакция с я-фенилендиамином. В пробирке растворяют
30 мг n-фенилендиамина в 8 мл дистиллированной воды. В по-
34
лученный раствор вносят 0,5—1 г тонкоизмельченного
полимера. При наличии эпоксидной смолы мгновенно появляется
розовое окрашивание, которое становится еще более интенсивным
через 12 ч.
Реакция с формальдегидом. Растворяют 0,01 г
тонкоизмельченного полимера в 1 мл концентрированной серной кислоты,
добавляют 3 капли 40%-ного раствора формальдегида. При
положительной реакции образуется красно-коричневое
окрашивание, которое при разбавлении водой меняется на зеленое.
Реакция с солями Fe(III). 0,01—0,02 г тонкоизмельченного
полимера растворяют в 1—2 мл концентрированной серной
кислоты и осторожно приливают 5 мл 10 %-ного
свежеприготовленного раствора Fe2(S04)3. В присутствии эпоксидного полимера
появляется темно-фиолетовое окрашивание.
I. 1.5.23. Ацетаты и ацетобутират целлюлозы, целлофан, этилцеллюлоэа,
нитрат целлюлозы
Реакция с ос-нафтолом. 0,05 г полимера растворяют в 3—
5 мл 50 %-ной (по объему) серной кислоты. К 2—3 каплям
полученного раствора добавляют 1 мл дистиллированной воды и
2 капли свежеприготовленного 10 %-ного раствора а-нафтола
в хлороформе. Затем осторожно по стенке пробирки приливают
пипеткой 3 мл концентрированной серной кислоты. Фиолетовое
кольцо на границе соприкосновения двух жидкостей указывает
на присутствие производных целлюлозы. При наличии нитрата
целлюлозы кольцо окрашивается в желтый цвет.
1.1.5.24. Нитрат целлюлозы
Реакция с дифениламином. На дно фарфоровой
выпарительной чашки помещают кусочек анализируемого полимера и
наносят на него 2—3 капли свежеприготовленного реактива
(0,02 г -дифениламина растворяют в 1 мл концентрированной
H2S04). В присутствии нитрогруппы мгновенно появляется
интенсивное голубое окрашивание.
Реакция с резорцином. 0,01 г полимера растворяют в 1 мл
концентрированной серной кислоты и добавляют кристаллик
резорцина. В присутствии нитрата целлюлозы появляется
пурпурно-голубое окрашивание.
1.1.6. Инфракрасная спектроскопия
Для качественного анализа полимерной матрицы из
инструментальных методов наиболее, часто используют методы ИК-
спектроскопии и пиролитической газовой хроматографии. Все
инструментальные методы отличаются высокой степенью
автоматизации и возможностью использовать ЭВМ для обработки
информации, поступающей непосредственно с прибора. Это во
2*
35
много раз повышает скорость получения результатов анализа
по сравнению с химическими методами. Высокая
производительность труда компенсирует затраты на дорогостоящее
оборудование и содержание специального персонала для его
обслуживания, но только при условии достаточно эффективной
его загрузки. Основным недостатком всех инструментальных
методов является то, что они применимы в принципе только к
индивидуальным соединениям. Поэтому для проведения
качественного анализа необходимо предварительно из полимерной
матрицы удалить примеси и добавки (см. 1.2).
Метод ИК-спектроскопии является одним из самых
распространенных методов структурного анализа и идентификации
органических соединений. Теоретические основы метода и вопросы
техники эксперимента изложены в [32, 33].
ИК-спектроскопия полимеров имеет свои специфические
особенности по сравнению со спектроскопией органических
соединений и представляет ее самостоятельный раздел. Имеются
книги, специально посвященные вопросам теории спектров
полимеров [34, 35] и их практическому применению для
качественного анализа [7, 17, 36—39].
Для образца полимера, не содержащего примесей, методом
ИК-спектроскопии можно установить с высокой степенью
надежности не только основную структуру полимерной цепи, но даже
незначительные ее детали, обусловленные способом получения
или переработки. Метод применим для анализа разнообразных
объектов, т. е. практически любой образец может быть
препарирован в виде, пригодном для получения его ИК-спектра, и
при этом не требуется перестройки аппаратуры в зависимости
от физической или химической природы образца. Если в
образце содержатся другие соединения в количестве 5—10 %, не
обладающие очень большой поглощающей способностью и слабо
рассеивающие излучение, то они существенно не будут
искажать основные полосы поглощения спектра полимера. В этом
случае удается провести его идентификацию, хотя надежность
и информативность получаемых результатов снижается.
Наиболее характерной задачей качественного анализа
полимеров по ИК-спектрам является определение хотя бы основных
компонентов, когда их количество и концентрация в смеси
неизвестны. Использование ЭВМ открывает новые возможности
для решения этой задачи и позволяет идентифицировать смеси,
содержащие до 4 компонентов. Имеется ряд программ, с
помощью которых возможно разделение ИК-спектра смеси на
спектры исходных веществ. Особенно успешно эта задача
решается [7, с. 78], если иметь не один спектр смеси, а
несколько спектров смесей, одинаковых по качественному составу, но
различающихся соотношением компонентов. Обычно для снятия
таких спектров используют образцы, полученные в результате
ступенчатой экстракции или экстракции различными
растворителями. Необходимое условие применимости метода—наличие
36
взаимно неперекрывающихся участков в спектрах веществ,
образующих смесь. На основании спектров смесей составляют
систему линейных уравнений, в результате решения которой можно
получить спектр каждого компонента.
I. 1.6.1. Подготовка образцов
Качество ИК-спектра и его последующая интерпретация во
многом зависят от правильной подготовки образца: В
зависимости от свойств образцов применяют различные способы их
подготовки. Обычно образцы исследуют в виде пленок. Такой
образец может быть использован многократно, храниться дли-
.тельное время, в спектре пленок (в отличие от растворов и
суспензий) отсутствуют посторонние полосы поглощения,
упрощается подготовка образца к съемке. Рекомендуемые толщины
пленок приведены в табл. I. 6. Пленки сополимеров готовят
такой же толщины, что и пленки соответствующих гомополимеров.
В некоторых случаях, когда требуется определить наличие
модифицирующих добавок или уточнить марку полимера,
необходимо готовить образцы нескольких толщин. Так, например, что-
■бы отличить полиэтилен высокого давления (ПЭВД) от
полиэтилена низкого давления (ПЭНД), необходимо получить
спектры образцов толщиной 0,5 мм.
Пленки готовят методом прессования, отливают из раствора
или, значительно реже, срезают резцом от заготовки. Прессуют
пленки на прессах, способных развивать усилия до 9,8 кН.
Прессование проводят в небольших пресс-формах. Толщину
образца регулируют с помощью вкладышей-ограничителей,
которые готовят из фольги или металлических пластинок.
Наружный размер вкладыша совпадает с площадью пуансона, а
внутреннее отверстие имеет форму, необходимую для образца.
Для предотвращения прилипания образцов к поверхности
пуансона применяют прокладки из триацетата целлюлозы. Условия
прессования выбирают в соответствии с природой прессуемого
материала (табл. 1.7).
Таблица 1/6. Подготовка образцов полимеров для
ИК-спектроскопии
Класс полимера
Способ получения пленки
Толщина пленки, мм
Полиолефииы
Полистирол и
сополимеры стирола
Фторполимеры
Поливииилацетатиые
пластики
Поливииилхлорид и
сополимеры вииилхлорида
Прессование
Из раствора
Прессование
Из раствора
0,02-0,04; 0,2—0,5
0,02—0,04
0,005—0,02
0,003-0,01
0,02—0,04
37
Таблица I. 7_ Рекомендуемые температуры н давления для
прессования пленок полимеров
Полимер
Полиэтилен высокого давления
Полиэтилен низкого давления
Полипропилен
Сополимер этилена с пропиленом
Сополимер этилена с бутеном-1
Сополимер этилена с винилацетатом
(нерастворимый)
Сополнмер этилена с гексеном-1
Политетрафторэтилен
Сополимер тетрафторэтилена с гексафторпропи-
леном
Сополимер этилена с тетрафторэтиленом
Поливинилиден фторид
Сополимеры винилиденфторида
Поли винил фторид
Политрифторхлорэтилен
Сополимер трифторхлорэтилена с этиленом
Температура
прессования, °С
150
175
215
175
175
130
175
Холодное
таблетирование
300—330
300
190
180—200
220
250
230
Давление,
МПа
3,5
7—10
10
7-10
7—10
3,5
7-10
30
30
20
30
30
20
30
25
Пленки растворимых полимеров готовят из 1—3 %-ных
растворов методом полива. В качестве растворителей можно
использовать любой из указанных для данного полимера в
табл. I. 4. Однако предпочтение отдается более летучим и
химически нейтральным растворителям. Наиболее часто применяют:
тетрахлорметан, хлороформ, ацетон, метиловый спирт,
дихлорэтан и гексан. Пленку отливают на стеклянных или
металлических пластинах с хорошо отполированной поверхностью.
Пластину устанавливают горизонтально с помощью уровня, на нее
помещают и плотно прижимают ограничитель в виде,
например, кольца высотой 1—3 см с хорошо отполированными
торцами; внутрь кольца наливают раствор полимера. Количество1
раствора полимера, необходимого для получения пленки
заданной толщины, можно приближенно рассчитать по формуле:
V=dS-\Q0/C,
где V—объем раствора полимера, см3; d — толщина пленки, см; С —
массовая концентрация раствора полимера, %; S — площадь образца, см2.
Растворитель медленно испаряют при комнатной
температуре. Образовавшуюся пленку снимают с подложки и помещают
в держатель. Следует иметь в виду, что некоторые
растворители, такие, например, как диметилсульфоксид и вода, довольно
трудно удаляются из пленок. В этом случае необходимо
досушивать пленки в вакууме и контролировать наличие
растворителя по их наиболее интенсивным полосам поглощения. Для
диметилсульфоксида такой полосой является 1050 см-1, а для
воды — 1650 см-1. Если растворитель не является агрессивным
по отношению к материалу, из которого изготовлены пластины,
38
то пленку можно отливать непосредственно на них и затем
получать спектр, не снимая пленки с пластины. Такой метод
обычно применяется в том случае, когда исследуются образцы
толщиной менее 0,01 мм.
Если образец нерастворим и из него не удается приготовить
пленку, то необходимо измельчить образец в тонкий порошок.
В случае хрупких материалов это легко достигается с помощью
перетирания в ступке, но для большинства полимеров такой
метод не пригоден. Образец полимера приходится измельчать
в замороженном виде или применять ультразвук, бормашину,
соскабливать лезвием бритвы. Измельченные образцы готовят для
съемки в виде суспензий или таблеток, отпрессованных с КВг.
Суспензию готовят, смешивая порошок полимера с
вазелиновым маслом, и для получения спектра помещают между
двумя пластинками из NaCl. Вазелиновое масло поглощает в
области 2600—3000 см-1 и 1350—1470 см-1, поэтому для
анализа в этих областях спектра можно применять фторированное
масло. Измельченные образцы смешивают с сухим порошком
КВг (1—3% от массы КВг) и прессуют таблетки под
давлением 500—700 МПа. КВг гигроскопичен и в процессе
приготовления образца может поглощать некоторое количество паров
воды из атмосферы, что приводит к появлению в спектре полос
поглощения при 1640 см-1 и 3400 см-1. При разделении
полимеров методом колоночной или тонкослойной хроматографии с
использованием негигроскопичного растворителя в качестве
элюента полученные фракции, не выделяя из раствора, также
можно смешивать с порошком КВг; затем растворитель
удаляют испарением и прессуют таблетки. Растворы полимеров при
качественном анализе практически не используются.
В последнее время часто приходится иметь дело с
образцами, высоконаполненными или модифицированными различными
способами, благодаря чему они становятся нерастворимыми и
неплавкими. Анализ таких образцов методом ИК-спектроскопии
представляет большие экспериментальные трудности. Если
образец прозрачен, то иногда удается приготовить микротомом
достаточно тонкий срез, пригодный для получения ИК-спектров.
Высоконаполненные образцы вообще не могут быть
исследованы методом ИК-спектроскопии. Следует иметь в виду, что при
среднем наполнении образцы сильно рассеивают свет в
видимой области спектра, но остаются достаточно прозрачными в
ИК-области, что позволяет получать спектры
удовлетворительного качества, компенсируя при необходимости рассеяние света
образцом с помощью диафрагмы или сеток, помещаемых в
канал сравнения.
Для качественного анализа высоконаполненных образцов
применяется метод, основанный на изучении ИК-спектров их
пиролизатов. Часто этот метод применяют и для качественного
анализа сшитых образцов, так как их препарирование с
применением техники КВг или микротома очень трудоемко и
39
требует определенных навыков. Этот метод нашел широкое
применение в анализе резин [40, с. 16]. Метод используется и при
идентификации полимеризационных пластиков в
композиционных материалах [41].
1.1.6.2. Получение ИК-спектров полимеров
Для качественного анализа ИК-спектр образца снимают
обычно в интервале 700—4000 см-1, хотя современные
спектрометры позволяют это делать в более широком интервале
волновых чисел. Это. объясняется тем, что информация,
содержащаяся в области спектра 700—4000 см-1, вполне достаточна
для качественного анализа и в большинстве атласов
ИК-спектров приводятся спектры только в этом интервале.
В настоящее время создан ряд так называемых
информационно-поисковых систем (ИПС), которые позволяют наряду с
другими задачами решать задачу поиска заложенного в память
ИПС спектра, тождественного по ряду закодированных
признаков спектру, предъявленному машине исследователем. Когда
речь идет об универсальных ИПС, т. е. системах, стремящихся
охватить весь фактический спектральный материал,
накопленный к настоящему времени ИК-спектроскопией, то требуется
ЭВМ с большой памятью и предварительная кодировка
спектров. Однако большее распространение, вероятно, будут иметь
системы, банк данных которых формируется с учетом интересов,
их постоянных пользователей [42]. Это позволяет резко
сократить количество вводимой в память информации и использовать
для поисковой системы микро-ЭВМ, которой снабжаются
современные ИК-спектрометры.
Разрабатываются и другие поисковые системы, основанные
на структурно-групповом анализе, в основе которого лежат
представления о связи между определенными —
характеристическими — полосами поглощения в спектре и структурными
фрагментами молекулы. Эти системы позволяют
идентифицировать даже те соединения,-спектры которых отсутствуют в
атласах. Подробно эти вопросы рассмотрены в книге [7].
Как показывает практика, внедрение таких сложных систем
требует много времени и сил, поэтому далеко не всегда
доступно и оправдано для небольших аналитических и химических
лабораторий. Занимаясь сравнительно узким кругом вопросов,
такие лаборатории решают подобные задачи более простыми и
дешевыми методами. Вначале определяют класс полимера,
используя информацию, получаемую с помощью простейших
методов анализа, а затем на основании ИК-спектров. Примером-
использования ИК-спектров для первичной идентификации
полимеров может служить схема, приведенная на рис. 1.2, которая
позволяет резко сократить круг поиска. Дальнейшее уточнение
и окончательную идентификацию можно осуществлять путем-
прямого сравнения спектров, имеющихся в различных книгах
и атласах [17, 34, 36, 38, 39]. -
40
1.1.6.3. Идентификация полимеров по ИК-спектрам их пиролизатов
В тех случаях, когда образцы для ИК-спектроскопии не
могут быть получены или когда подготовка их для съемки
является слишком трудоемкой, можно использовать ИК-спектры не
самих образцов, а их пиролизатов.
В процессе термического разложения полимеров, как
правило, наряду с газообразными продуктами, в основном
состоящими из мономеров и исследуемыми методом газо-жидкостной
хроматографии (ГЖХ), образуются также жидкие или смоло-
образные продукты, представляющие собой чаще всего
фрагменты исходной полимерной молекулы со сравнительно низкой
молекулярной массой. Эти продукты удобны для изучения
методом ИК-спектроскопии и могут быть легко получены, так как
при разложении конденсируются на холодных частях
установки, в то время как неорганические добавки остаются на дне, а
мономеры улетают.
Исследования показали, что для полиолефинов, полистиро-
лов, полиамидов, поликарбонатов информативность ИК-спект-
ров пиролизатов, полученных на воздухе, такая же, что и для
пиролизатов, полученных в инертной среде или вакууме [41].
Пиролизаты полимеров этих классов получали в атмосфере
воздуха, так как это легче осуществляется и сокращаются
затраты времени на подготовку образцов. Для получения
пиролизатов фторполимеров необходима инертная среда.
Пиролизаты, в принципе, могут быть приготовлены в
обычной пробирке из термостойкого стекла, нагреваемой на
спиртовке. Однако практика показывает, что в этом случае очень
низка воспроизводимость состава продуктов пиролиза. С целью
повышения воспроизводимости была разработана методика,
которая позволила стабилизировать условия пиролиза (см. 1.31).
С применением этой методики был создан атлас ИК-спектров
пиролизатов основных промышленных полимеров, которым
успешно пользуются при анализе высоконаполненных образцов
полимеров.
Образцы из пиролизатов готовят, помещая их между
пластинками КВг и раздавливая до нужной толщины. Если
продукты пиролиза имеют жидкую консистенцию, то толщину
регулируют, помещая между пластинками кольцо из фольги или
из полимерной пленки необходимой толщины. Если продукты
смолообразные, то их нагревают после нанесения на пластинку
для того, чтобы раздавить до нужной толщины.
ИК-спектры пиролизатов снимают в интервале 700—
4000 см-1 и сравнивают с ИК-спектрами пиролизатов
известных образцов.
1.1.7. Пиролитическая газовая хроматография
При термическом воздействии полимерные молекулы
претерпевают определенные изменения, характер которых
зависит от температуры и длительности нагревания. До 350 °С эти
41
ИК-спектр
+
695, 758 см"1
3484-3333 см"1
1605, 1590,1490 см"1
(ароматическое кольцо)
Эпоксидная
смола
Фвнопоформальдегидная
смола
1000-1100 см"1
Силиконовый
каучук
1540 см"1
Полистирол, стирол-
бутадиеновый каучук
1670 см"'
Полиамиды, амино-
формальдегидные смолы
2220 см"1
Сополимеры
акрилонитрила
3333 см"1
Нитрат целлюлозы,
целлофан
143Uсм -
Метилэтилцеллюлоэа,
меламиновая смола,
этиленпропилеиовый
каучук,
поливиниловый
спирт
950-925 см"1
Бутадиеновый
каучук, бутилкаучук
850 см'
Изопреновый
каучук
125 0-1000 см"1
Тиокол
Поливинилхлорид
Рис. 1.2. Схема идентификации поли
42
Образца
1740 см"' (С=0)
1605, 1590, 1490 см1
830 см"'
(ароматическое копьцо)
1430 см"'
Модифицированная
эпоксидная смола
1430 см"
Ппастифициров анный
ШЛИвинилхпорид
1330-1212 см"1
Алкидные смолы,
сложные полиэфиры
ароматического ряда
+
690 см"1
1050 см"
Поливиниловый
спирт,
поливинил формапь
Сополимеры
винилхлорида
Простые эфиры
целлюлозы
Сложные
эфиры
целлюлозы
Полиакрилаты, полиэфиры
алифатического ряда
1250 см"'
Силиконовый
каучук
760,833 с
Хлоропреновый каучук
м"'
Полиэтилен
меров методом ИК-спектросиопии [5].
43
изменения идут довольно медленно и связаны в основном с
реакциями окисления и дегидрирования; при этом образуются в
небольшом количестве летучие низкомолекулярные продукты,
снижается молекулярная масса полимера, образуются сшивки.
При нагревании выше 350°С начинается интенсивный распад
полимерной молекулы с выделением исходных мономеров и
других фрагментов основной цепи.
Первичной реакцией термораспада полимерной молекулы
является разрыв связей с наиболее низкой энергией активации.
Это можно продемонстрировать на примере молекулы
полистирола (у связей указаны энергии активации в кДж/моль) [43]:
н5 ;
>4^1Сн2Гсн-снг-
Образующиеся при разрыве полимерной цепи
макрорадикалы могут распадаться либо по реакции деполимеризации
с отщеплением мономерных звеньев, либо с образованием
фрагментов полимерной цепи различной длины. Типичными
представителями полимеров, распадающихся по первому механизму,,
являются полиметилметакрилат, поли-а-метилстирол и
политетрафторэтилен, а по второму — полиэтилен и другие поли-
олефины:
СН3 СНз СН3 СНз
-СН2-
-СН2—C-j-CHg—С—j—CH2-
СН3
СН-.
СНз
СНЭ
->-~СН2—С- + СН2=С + СН2=С +-СН2—С~ и т.д.
п.
П ? 7тт,ттТ Т7
с—с— с-с-с-с+с-с-с-с-с ~
7 7 Г 7 7 7 7 7 7 7 7
■ с—с—с-с-с-с- + -с-с-с-с-с-
Н Н (HlH Н Н
44
~c— с—с— с—с—с-н + н^:—с~с4-с_с~ .
U ' й A i i U'i '
f T т г" V У v т f ? ¥
~c- + c=c—с—с—с—н + h-c—с—о + c=c-
II ill I I I
и и н н л н м и й
И Т.Д
В первом случае образовавшийся радикал в силу стериче-
ских препятствий не может участвовать в реакциях передачи
цепи, а во втором случае реакция передачи цепи возможна
через любой атом водорода. При пиролизе политетрафторэтилена
наблюдается высокий выход мономера. Это обусловлено тем,
что атом фтора, в отличие от атома водорода, не может
принимать участия в передаче цепи из-за более высокой прочности
связи С—F по сравнению со связью С—Н [44, с. 355].
В большинстве случаев эти два механизма конкурируют
между собой и вклад того или иного в процесс пиролиза
зависит от условий его протекания.
Иногда наряду с разрывом связей основной цепи
энергетически выгодным оказывается и отрыв заместителей. Это
характерно, например, для поливинилацетата и поливинилхлорида.
Состав продуктов пиролиза является характерным только для
данного полимера, поэтому хроматограммы продуктов
пиролиза — пирограммы — могут быть использованы для качественного
анализа. Пирограмму анализируемого образца сопоставляют по
характерным признакам (временам удерживания и
интенсивности пиков) с пирограммами стандартных образцов. Все
пирограммы должны быть получены при одинаковых, специально
подобранных условиях пиролиза и хроматографического
разделения.
Преимуществами метода пиролитической газовой
хроматографии (ПГХ) является возможность работать с высоконапол-
ненными и сшитыми образцами без всякой предварительной их
подготовки, сравнительная простота и доступность аппаратуры,
минимальное количество образца, необходимое для анализа.
Основным недостатком ПГХ, препятствующим еще более
широкому внедрению этого метода в практику работы
аналитических лабораторий, является его плохая воспроизводимость,
особенно при межлабораторных испытаниях. Состав продуктов
пиролиза зависит от многих факторов: температуры пиролиза,
типа пиролизера, продолжительности пиролиза, навески и
условий газохроматографического разделения продуктов
пиролиза. В течение последних 10—15 лет была проведена большая
работа по совершенствованию пиролизеров и методических
45
приемов, направленная на повышение воспроизводимости
метода ПГХ. В настоящее время, благодаря появлению серийной
аппаратуры, выпускаемой промышленностью, а также
уточнению условий эксперимента на базе многочисленных
исследований уже достигнут значительный прогресс по внутрилаборатор-
ной воспроизводимости. Опыт показывает, что для успешного
проведения не только количественного, но и качественного
анализа необходима строгая стандартизация всех условий
пиролиза. Основной проблемой при разработке методик качественного
анализа является выбор таких условий, которые подходили бы
для получения характерных пирограмм для широкого круга
полимерных материалов.
I. 1.7.1. Пиролиэеры
В 60-е годы, в период становления ПГХ как
аналитического метода, различными исследователями были предложены
разнообразные конструкции пиролизеров, основанные на
различных принципах. В настоящее время практическое значение
имеют три типа пиролизеров, получивших наибольшее
распространение и выпускаемых промышленностью,— пиролизер по
точке Кюри, пиролизер фнламентного типа и пиролизер
печного типа.
В 70-е годы наибольшее распространение получил пиролизер
фнламентного типа, так как он прост по конструкции и его
можно легко изготовить в лабораторных условиях. В последние
годы исследователи отдают предпочтение пиролизеру по точке
Кюри, выпускаемому промышленностью. Были попытки
использовать для разложения полимеров излучение лазера, однако,
при этом трудно регулировать и контролировать температуру
образца, метод применим только для темных образцов,
сравнительно дорог [47].
При работе на пиролизерах различных типов [45, 46, 48]
необходимо учитывать следующие рекомендации:
1) нельзя переносить условия пиролиза, являющиеся
оптимальными для одного ппролизера, на другой пиролизер, даже
работающий по тому же принципу, но отличающийся
конструктивным оформлением;
2) для пиролизера любого типа почти всегда удается
подобрать условия анализа, обеспечивающие внутрилабораторную
воспроизводимость пирограмм с точностью, необходимой для
качественного анализа.
Пиролизер по точке Кюри. Пиролизером по точке Кюри
сокращенно называют пиролизеры, в которых пиролиз образца
осуществляется за счет нагревания держателя образца из
ферромагнитного сплава токами высокой частоты до определенной
температуры — точки Кюри [45, с. 23; 46, с. 20]. Эти
пиролизеры позволяют проводить исследование только при строго
определенных температурах, зависящих от состава
ферромагнитного сплава. Например, температуры 358, 480, 510, 600,770
46
и 980 °С достигаются при использовании 6 различных по
составу сплавов. Держатель образца может быть выполнен в
форме трубки, лодочки, спирали, ленты. Образец рекомендуют
вводить из раствора, но иногда пользуются и образцами в
твердом виде. Достоинством этих пиролизеров является быстрое
достижение заданной температуры (за сотые доли секунды), ее
стабильность и воспроизводимость.
Пиролизер филаментного типа. Пиролизеры филаментного
типа представляют собой металлическую спираль (чаще всего
из нихромовой или вольфрамовой проволоки), которая
нагревается электрическим током. Температура пиролиза может быть
установлена любая, но требуется предварительная градуировка
с помощью веществ с известной температурой плавления с
целью построения графика зависимости температуры спирали —
напряжение, подаваемое на спираль. Образцы наносят на
спираль из раствора, однако в некоторых работах образец
помещали в спираль в твердом виде и при этом были получены
хорошие количественные результаты [46, с. 15]. Скорость
подъема температуры до заданной составляет приблизительно 5 с
для обычного филамента и сотые доли секунды для филамента
с подводкой дополнительной мощности в момент включения в
виде импульса.
Пиролизер печного типа. Пиролизер представляет собой
печь цилиндрической формы с заключенной в нее кварцевой
трубкой, в которую образец вводится в лодочке (кварцевой
или металлической) специальным держателем или другими
способами, после чего печь герметизируется [46, с. 11]. В отличие
от двух выше описанных пиролизеров, где образцы помещаются
в пиролизер до его нагревания, в печном пиролизере образец
вводят в печь, уже нагретую до заданной температуры.
Поэтому скорость нагревания образца в печном пиролизере никак не
может быть меньше, чем в других пиролизерах, как
утверждается иногда в литературе. Главным недостатком печного пиро-
лизера является возможность протекания вторичных реакций,
так как продукты пиролиза более длительное время находятся
в зоне нагрева. Температура пиролиза может быть установлена
любая, но предварительно требуется произвести градуировку с
помощью термопары и получить зависимость температура
пиролиза — напряжение, подаваемое на печь. Следует учитывать,
что температура сильно отличается в разных точках трубки и
ее надо замерять точно в месте нахождения образца. Образец
может быть использован любой формы и вида, его масса
может варьироваться в широких пределах—от 0,5 до 500 мг.
I. 1.7.2. Условия пиролиза
Температура существенно влияет на скорость и механизм
реакций и для ПГХ является решающим фактором воздействия
на состав и количество образующихся при пиролизе летучих
47
S/m
'мс. 1.3. зависимость выхода мономера стирола из полисти-
ола от температуры пиролиза (5 —площадь пика, мм2: т —
навеска полистирола, мг).
800 t°C
продуктов. На рис. 1.3 и 1.4 это показано
на примере полистирола и АБС-пластика.
Температура поверхности образца ts
совпадает с температурой окружающей среды
ta только до определенной температуры
ta, индивидуальной для каждого
полимера [49]. Повышение tx выше этого
значения приводит лишь к увеличению
скорости распада образца, но почти не
изменяет его температуры ts (рис. 1.5, табл.
1.8). Очевидно, что целесообразнее вести
пиролиз при температурах выше ta, так как при этом
колебание t оо мало влияет на качественный состав продуктов
пиролиза. Однако не рекомендуется проводить пиролиз при
/оо ^> ta, поскольку в этом случае возрастает роль вторичных
реакций, и вследствие этого увеличивается выход
малоспецифических легких продуктов (см. рис. 1.4). Поэтому наиболее
предпочтительным для пиролиза большинства полимеров с целью
их идентификации является интервал температур 600—700 °С.
В последние годы получил распространение ступенчатый
пиролиз, когда один и тот же образец нагревается
последовательно при различных температурах в интервале от 100 до 800 °С.
Этот вид пиролиза осуществим только при использовании пи-
ролизеров филаментного типа. При температурах 100—250 °С
выходят летучие компоненты полимерной композиции
(растворители, остаточные мономеры), при температурах 250—
350 °С — стабилизаторы и пластификаторы, а затем по
продуктам пиролиза, выделяющимся при температурах 500—
600 °С, можно идентифицировать полимерную основу.
Продолжительность пиролиза во многом зависит от
конструкции пиролизера и массы образца. В пиролизерах
филаментного типа образцы растворимых в органических
растворителях полимеров наносятся из растворов в виде тонкой пленки;
Таблица 1.8. Температура поверхности образцов полимеров ts
в зависимости от температуры окружающей среды tx при пиролизе
в потоке азота [49]
Полимер
<-. °С, пр„ *
500 °С 600 "С 700 "С 800 °С 900 "С 1000 °С
Полиметилметакрилат
Полистирол
Полиэтилен
Полипропилен
410
465
485
480
440
500
535
525
460
525
570
560
470
545
595
585
480
555
610
600
485
565
620
610
48
300° С
Рис. I. 4. Вид пирограммы АБС-
пластика в зависимости от
температуры пиролиза.
после испарения
растворителя они
подвергаются пиролизу за
доли секунды. Большая
часть полимерных
образцов представляет
собой
малорастворимые продукты. Перед
пиролизом некоторые
из них, поддающиеся
измельчению,
превращают в порошок и
наносят на ленту или
нить пиролизера;
иногда кусочек образца или
порошок помещают в
фольгу, изготовленную
из ферромагнитного
сплава, а затем
подвергают пиролизу при
температуре,
соответствующей точке Кюри
для данного сплава.
Кусочек
нерастворимого образца можно
помещать на витки
спирали и специально
сделанные на филаменте
крючки. В пиролизерах
печного типа образец
вносят в лодочку из
кварца или платины.
В любом случае для
пиролиза
нерастворимых образцов
необходимо более длительное 25 20 15 10 s
время, чем для пиролиза тонких пленок. Это связано с
затратами времени на прогрев образца, диффузию и испарение
летучих продуктов с поверхности образца (см. рис. 1.5). При
использовании пиролизеров филаментного типа и по точке Кюри
продолжительность пиролиза составляет 1—15 с, а для
пиролизеров печного типа— 10—30 с.
Вид и масса образца. Для идентификации представляются
различные образцы: пленки, дисперсии, клеи, лаки, гранулы и
порошки, высоконаполненные композиции, товарные изделия.
Перед проведением идентификации методом ПГХ необходимо
49
Рис. I. 5. Изменение массы (/) и температуры (2)
ТП MS образца в процессе разложения полиметил-
,.. ' метакрилата:
I — прогревание образца; // — удаление
продуктов разложения; /// — регистрация температуры
окружающей среды (азот).
подготовить образец — удалить
растворители отгонкой под
вакуумом, после чего высушить об-
2 4 6 разец в вакуум-сушильном шка-
Время,с фу. Процедура вакуумной
сушки необходима также после
нанесения образца из раствора в органическом растворителе на
филамент или ленту пиролизера во избежание влияния
остаточного растворителя на вид пирограммы, особенно при
использовании чувствительной газохроматографической аппаратуры
(капиллярные колонки, пламенно-ионизационный детектор).
Как показано в работе [48], пиролиз нескольких не
соприкасающихся друг с другом кусочков полимера протекает
независимо, что приводит к увеличению вероятности протекания
вторичных реакций. Поэтому для пиролиза желательно брать
один кусочек полимера. При использовании пиролизера печного
типа порошкообразные образцы нередко выносятся из лодочки
током газа-носителя, поэтому порошкообразные
мелкодисперсные образцы лучше таблетировать механическим прессом и для
пиролиза брать кусочек таблетки.
Наиболее воспроизводимые пирограммы получают при
пиролизе образцов массой 5—20 мкг, поскольку низкая
концентрация летучих продуктов в пиролизере уменьшает вероятность
протекания вторичных реакций. В большинстве работ по
идентификации масса образца для растворимых полимеров
колеблется от 10 до 100 мкг, а нерастворимых—от 1 до 3 мг и
более в зависимости от типа применяемого пиролизера и газохро-
матографического детектора.
I. 1.7.3. Условия газохроматографического разделения летучих
продуктов пиролиза
Газохроматографические условия разделения продуктов
пиролиза весьма важный фактор в ПГХ и требуют не меньше
внимания, чем выбор пиролизера, температуры пиролиза и
подготовка образца. Продукты пиролиза полимеров часто
представляют собой многокомпонентную смесь соединений различной
полярности, с различными функциональными группами и
температурами кипения. Поэтому колонки с неподвижными
фазами, способными с высокой эффективностью разделять
специфические продукты, мало пригодны в ПГХ. В работах по
идентификации методом ПГХ используются различные набивные
колонки. Длина колонок меняется от 1 до 15 м, диаметр — от 3
до 6 мм, чаше применяются стальные колонки, чем стеклян-
50
ные; неподвижные жидкие фазы весьма разнообразны и
наносятся на твердый носитель в различных количествах [40, с. 27].
При их выборе рекомендуется [45, 46, с. 70] учитывать природу
образующихся при пиролизе продуктов и использовать 2—3
колонки с различными по полярности фазами; для разделения
углеводородов целесообразно использовать неполярные фазы, а
если предполагается исследовать полимер, содержащий гетеро-
атомы,— слабополярные или полярные фазы. Несомненно ряд
задач при идентификации полимеров — определение стереорегу-
лярности, степени разветвления, чередования звеньев —
наиболее целесообразно решать с применением капиллярных колонок
[48, 50]. Ответы на многие вопросы по газохроматографическо-
му разделению продуктов пиролиза полимеров и выбору фаз
можно найти в монографиях [51, 52].
Для идентификации полимеров разных классов методом ПГХ
наиболее широкое применение нашли неспецифические
силиконовые фазы низкой полярности— полисилоксановые эластомеры
SE-30, Е-301 (аналог — ПМС-100), полиметилсилоксановые
эластомеры OV-1, OV-101 (аналог—SP-2100), а также
углеводородная фаза апиезон L (аналоги — сквалан, вазелиновое
масло). Из пористых сорбентов чаще других используется по-
рапак Q (этилвинилбензол, сшитый дивинилбензолом;
аналог—полисорб). Поскольку продукты пиролиза представлены
весьма широким набором соединений с различными
температурами кипения, наиболее информативными результаты ПГХ
могут быть только при работе колонок в режиме
программирования и в случае необходимости при температурах ниже 0°С.
При этом необходимо учитывать, что использование
неподвижных жидких фаз в режиме программирования ограничивается
интервалом их рабочих температур:
SE-30, Е-301 50-300 "С
OV-1, OV-101 0-350 °С
•Апиезои L 50—250 °С
При выборе твердых носителей следует обращать
внимание на удельную поверхность (2—5 м2/г) носителей и на
возможное каталитическое действие необработанной поверхности
некоторых носителей (сферохром-3, динохром-Н) на летучие
продукты пиролиза при повышенной температуре колонки.
Поэтому перед применением активные центры на поверхности этих
носителей следует дезактивировать [46, с. 75].
При пиролизе полимеров образуются не только летучие
продукты малой молекулярной массы, но и тяжелые фракции,
которые задерживаются на входе в колонку и загрязняют ее.
Для предотвращения этого между пиролизером и колонкой
ставят форколонку, заполненную стекловатой, стеклобисером
или инертным твердым носителем, не влияющим на хроматогра-
фическое разделение продуктов пиролиза, или тем же
сорбентом, что и колонка. Наполнение форколонки следует постоянно
51
обновлять через 20—30 определений, форколонку перед
заполнением новой порцией сорбента необходимо промыть
растворителями и высушить.
Кроме нелетучих фрагментов при пиролизе фтор- и хлорсо-
держащих полимеров могут выделяться агрессивные газы — фто-
роводород и хлороводород соответственно. С целью
предотвращения их корродирующего воздействия на аппаратуру
форколонку обычно заполняют носителем, покрытым NaOH или
гранулированным ЫагСОз [40, с. 30]. Пиролизер должен иметь
не только минимальный объем, но и соединяться кратчайшим
путем с колонкой для максимально быстрой эвакуации летучих
продуктов в колонку. Сокращение «мертвого объема» и
предотвращение конденсации летучих продуктов на коммуникациях
газохроматографической системы является залогом получения
высоковоспроизводимых пирограмм.
На распределение продуктов пиролиза может оказывать
влияние природа и скорость газа-носителя. При исследовании
продуктов пиролиза сополимера стирола с изопреном в пиролизе-
рах филаментного типа и по точке Кюри с использованием
двух газов-носителей — гелия и азота — было показано, что в
случае азота пирограмма дает завышенное количество
низкомолекулярных продуктов [53]. Скорость газа-носителя меньше
влияет на состав продуктов пиролиза в пиролизерах названных
типов, поскольку количество образующихся продуктов невелико
и они разбавляются газом-носителем настолько, что
уменьшается протекание вторичных реакций. Более заметно скорость
газа-носителя сказывается на пиролизе в пиролизерах печного
типа. При невысокой скорости газа-носителя растет время
пребывания продуктов пиролиза в пиролизере, что способствует
вероятности протекания вторичных реакций.
Чувствительность метода ПГХ зависит не только от системы
газохроматографического разделения, но и от вида
используемого детектора. Одним из наиболее широко применяемых
детекторов является пламенно-ионизационный. Как показывает
опыт, его применение оправданно, если масса анализируемого
образца меньше милиграмма. При большей массе образца
(1 мг и выше) следует применять катарометр. При
идентификации полимеров, содержащих гетероатомы, рекомендуется
использовать селективные детекторы. Комбинация детектора
поверхностной ионизации, пламенно-ионизационного и двух
пламенно-фотометрических детекторов с различными
светофильтрами позволила достоверно определить N, F, CI, Br, S, Р в
нелетучих органических добавках и полимерах. Идентификация
проводилась по низкомолекулярным продуктам — НС1, НВг,
H2S, РНз и другим характерным соединениям, образующимся
при 2—3-минутном пиролизе при 650—800 °С в среде
различных газов [25].
Несомненно, одним из самых информативных детекторов
является масс-спектрометр, который позволяет проводить иденти-
52
фикацию разделяемых на колонке продуктов по их
масс-спектрам [54]. Для идентификации продуктов пиролиза успешно
используются ИК-спектрометры с Фурье-преобразованием. Они
позволяют идентифицировать компоненты при их массовой доле
в смеси не менее 1 %, имеющие не очень высокую температуру
кипения [55].
Следует помнить также, что для получения
воспроизводимых пирограмм необходимо постоянно контролировать скорость
движения бумаги в самописце, поскольку трудно сравнивать
пирограммы, записанные при разных скоростных режимах.
I. 1.7.4. Обработка результатов пиролитической газовой хроматографии
Для идентификации полимерной основы неизвестного
образца по пирограммам предложены различные способы. Наиболее
распространено сравнение пирограммы неизвестного образца с
пирограммой эталонного полимера, полученной в аналогичных
условиях, по методу «отпечатка пальца». Для этого метода не
нужно выделять и идентифицировать отдельные продукты
пиролиза. Экспериментатор лишь должен иметь атлас эталонных
пирограмм, полученных при строго стандартизованных условиях
пиролиза. К сожалению, вследствие разнообразия конструкций
пиролизеров, газохроматографических условий разделения
летучих продуктов пиролиза, а также вида и массы образцов
межлабораторная воспроизводимость метода недостаточно высока.
Поэтому каждая лаборатория должна иметь свой набор
пирограмм.
Кроме сравнения пирограмм с эталонными путем наложения
применяется также идентификация только по временам
удерживания, поскольку эта величина в меньшей степени
подвержена изменениям, чем количественный выход летучих
продуктов пиролиза. Подобный метод наиболее удобен для контроля
определенных продуктов, химическая природа и состав которых
известны.
С целью снижения возможных погрешностей и повышения
надежности результатов идентификации времена удерживания и
площади пиков продуктов пиролиза представляют в
относительных величинах. Расчет относительного времени удерживания и
площадей пиков проводят, либо выбирая один из пиков на пи-
рограмме в качестве стандартного, либо по пику внешнего или
внутреннего стандарта. В качестве внешнего стандарта
применяют н-нонан, что позволяет рассчитывать эти величины для
продуктов пиролиза любых полимеров по отношению к одному
и тому же стандартному веществу. На этом может быть
основано качественное и количественное сравнение при создании
атласа пирограмм [56]. Для внутреннего стандарта
используют, как правило, полимер, разлагающийся при пиролизе с
преимущественным выделением мономера (поли-сх-метилсти-
рол, полиметилметакрилат), и пик мономера принимают за
S3
стандартный, по отношению к которому проводят расчеты [57].
Кроме мономера используют также в качестве внутреннего
стандарта бензол, образующийся при пиролизе многих
полимеров [40, с. 30]. Для нормализации времен удерживания пиков
на пирограмме предложено [58] проводить линейную
интерполяцию в интервале между двумя пиками стандарта, в пределы
которого попадает неидентифицированный пик. Стандартные
пики получают при пиролизе образца полиэтилена, основные
продукты термораспада которого составляют гомологический
ряд а-ненасыщенных алкенов. В начале рабочего дня
записывают пирограмму полиэтилена и относительно нее нормализуют
время удерживания летучих продуктов пиролиза всех
последующих полимеров. Результаты обрабатывают с применением
ЭВМ.
Довольно удобно проводить сравнение эталонных пирограмм
с пирограммами неизвестных образцов в виде штрихдиаграмм
в координатах: высота пика — время удерживания, отношение
площади пика к сумме площадей всех пиков (в
%)—относительное время удерживания, относительная площадь пика —
относительное время удерживания. Сравнение можно проводить
в логарифмических координатах. Кроме штрихдиаграмм можно
пользоваться табулированными значениями относительных
времен удерживания и площадей пиков (в том числе и в
логарифмической форме) [46, с. 122; 48, 57, 58]. Для ускорения
идентификации пирограммы неизвестных полимеров обрабатывают
на ЭВМ и полученные результаты сравнивают с данными,
имеющимися в памяти ЭВМ [59].
Флуктуации времен удерживания, высот и площадей пиков
во времени, влияние замены колонки или пиролизера и другие
факторы осложняют идентификацию образца методом
сравнения с эталонными пирограммами. Метод «отпечатка пальца»
практически неприменим для качественного обнаружения
небольших количеств полимеров в смеси. Также весьма сложно,
а иногда практически невозможно идентифицировать
структурные различия в однотипных полимерах с неодинаковыми
свойствами (например, атактический и изотактический
полипропилен).
Более надежной считается идентификация по пирограммам,
на которых известны пики летучих фрагментов, характерные
только для термораспада данного полимера в стандартных
условиях метода ПГХ. Для идентификации марки полимера
предлагается рассматривать не только качественно наличие или
отсутствие определенных пиков, но также использовать данные о
соотношении пиков характеристических продуктов, учитывать
выход этих продуктов на единицу массы полимера, поскольку
соотношение пиков определяется структурой полимера [59].
Идентификация пиков проводится по определению времен
удерживания в сравнении с временем удерживания эталонных
веществ, по индексам удерживания, а также химическими мето-
54
дами и методами ИК- и масс-спектрометрии выделенных пиков
[45, с. 72; 46, с. 122; 48].
С учетом имеющихся в литературе данных авторами работ
[41, 60] была разработана единая методика (см. 1.3.2),
позволяющая идентифицировать методом ПГХ полиолефины, поли-
стирольные и поливинилацетатные пластики, полиакрилаты,
фторопласты и другие полимеры. Неорганические соединения —
тальк, оксид кремния, оксиды титана и алюминия,
стекловолокно, асбест и другие инертные наполнители — практически не
влияют на вид пирограмм. Добавки органического
происхождения не оказывают существенного влияния на характер
пирограмм при содержании 1—5 % при данных условиях пиролиза
и газохроматографического разделения летучих продуктов
пиролиза.
Методика нашла широкое применение для
экспресс-идентификации полимеров различных классов; особенно необходима
она в тех случаях, когда метод ИК-спектроскопии имеет
ограничения, например, при идентификации полимерной основы
нерастворимых и неплавких композиционных материалов или
каучуковой части ударопрочных полистирольных пластиков.
В последнем случае используется сочетание методов ПГХ,
ИК-спектроскопии и химического [61] (см. 1.3.3), что
значительно сокращает трудоемкость проведения идентификации.
Практически во всех случаях идентификации полимерной
основы композиционных материалов сочетание ПГХ с ИК-спектро-
скопией дает хорошие результаты.
1.1.8. Другие инструментальные методы
Для идентификации полимеров помимо методов,
рассмотренных выше, можно использовать также ЯМР, термический
анализ, УФ-спектроскопию.
Метод ЯМР по своей информативности в целом не уступает
методу ИК-спектроскопии [2]. Однако по ряду причин он
сравнительно редко используется для целей идентификации
полимеров. Во-первых, ЯМР неприменим к твердым нерастворимым
образцам, вследствие чего многие полимерные образцы не
могут быть исследованы этим методом. Например, нельзя
получить ЯМР-спектры большинства фторполимеров, сшитых
полимеров, полиолефины можно изучать только при нагревании.
В настоящее время существует метод получения ЯМР-спектров
на 13С для твердых образцов. Однако этот метод пока
технически сложен, требует дорогой аппаратуры. Во-вторых,
органические и неорганические наполнители в большинстве случаев
мешают анализу методом ЯМР. Кроме того, спектр ЯМР
сильно зависит от приборных факторов: от частоты, на которой
был получен, от того, на резонанс с каким атомом настроен
прибор, а также от природы растворителя. Все это делает
вопрос о создании атласов спектров ЯМР более неопределенным,
55.
чем атласов ИК-спектров. Таким образом, если есть выбор, то
исследователь всегда предпочтет использовать для
качественного анализа полимеров более универсальные и менее
трудоемкие методы — ПГХ и Ш<-спектроскопию.
Метод ЯМР обычно применяется для идентификации
растворимых образцов или для образцов, способных давать сильно
набухшие гели, когда нельзя применить другие методы.
Например, когда необходимо отличить статистический сополимер от
блочного; когда требуется определить длину боковых цепей в
сополимерах этилена с а-олефинами; когда приходится
идентифицировать образец, аналога которому нет в атласах
ИК-спектров и ПГХ; когда нужно идентифицировать отдельные
структурные единицы в молекуле.
Возможности УФ-спектроскопии более ограниченны в
сравнении с другими методами. Во-первых, потому, что большинство
полимеров не имеют характерных полос поглощения в УФ- и
видимой областях спектра, а во-вторых, из-за высокой
чувствительности метода к некоторым случайным примесям и добавкам,
которые могут существенно изменять поглощение образца. Этот
метод целесообразно использовать для обнаружения
стабилизирующих добавок в полиолефинах.
Термический анализ и рентгеноскопия могут применяться на
последних стадиях идентификации для определения степени
кристалличности, что бывает необходимо для уточнения марки
образца.
1.2. ВЫДЕЛЕНИЕ И ИДЕНТИФИКАЦИЯ
ДОБАВОК И НАПОЛНИТЕЛЕЙ
Несмотря на широкое внедрение инструментальных методов
в практику идентификации полимеров, приходится признать,
что не существует таких методов, которые можно
гарантированно применять для анализа полимерных композиций,
содержащих иногда по массе больше наполнителей и добавок, чем
полимерной основы, без предварительного их отделения.
Препарирование исследуемых образцов, отделение добавок и
наполнителей методами экстракции, центрифугирования,
препаративной и жидкостной хроматографии должно предшествовать как
предварительной идентификации полимерной основы, так и
последующей ее детальной идентификации инструментальными
методами, если полимерная композиция является высоконапол-
ненной. Проблема идентификации полимерных композиций
весьма сложна, поскольку постоянно не только разрабатываются
принципиально новые полимеры, но различными способами
изменяются физико-химические свойства существующих
полимеров, расширяется их марочный ассортимент. Поэтому методы,
предложенные для отделения определенного типа полимера от
добавок и наполнителей, могут оказаться неприемлемыми для
его модификаций. Методов препарирования и идентификации
56
композиционных материалов, которые были бы применимы ff
неизменном виде для различных образцов, пока не существует.
Однако общая схема проведения идентификации сохраняется
для любых анализируемых полимерных объектов.
1.2.1. Характеристика добавок и наполнителей в полимерных
композициях
Введение химических добавок [27—29] в полимерные
композиции преследует разнообразные технологические цели и
задачи регулирования свойств полимерных материалов, что в
свою очередь предопределяет многообразие свойств и
структуры применяемых веществ. Ассортимент этих соединений в связи
с требованиями промышленности пластмасс постоянно
расширяется и обновляется, а рецептуры композиций усложняются.
Наряду с добавками, влияющими на потребительские и
технологические свойства пластмасс (стабилизаторы,
пластификаторы, поверхностно-активные вещества, антипирены, биоцидные
добавки, оптические добавки и т. п.), массовая доля которых в
композиции составляет от сотых долей до десятков процентов,
в полимерной матрице, как правило, в сравнительно небольших
количествах (от тысячных до десятых долей процента)
содержатся продукты, непосредственно участвующие в синтезе
самого полимера или образующиеся из него (остатки мономеров,
каталитических и инициирующих систем, ускорителей,
прерывателей цепи, регуляторов молекулярной массы, эмульгаторов
и т. п.).
Химическая структура и свойства добавок весьма
разнообразны, поэтому идентификация их и количественное
определение является сложнейшей аналитической задачей.
Стабилизаторы, (антиоксиданты, светостабилизаторы, анти-
озонанты, антирады), вводимые в композиции в количествах от
0,01 до 10 % от массы полимера, представлены в большинстве
своем производными алкил- и ариламинов, фенолов, бензтри-
азолов, бензофенонов, нафталина и антрацена, эфирами
бензойной, салициловой, фосфористой и салицилфосфористой
кислот, тиоэфирами, тиофенолами, элементорганическими
соединениями, техническим углеродом (сажей) различных марок и др.
Для придания пластичности при переработке и эксплуатации
в полимерные композиции вводят пластификаторы—от 1 до
100 % от массы полимера: эфиры фталевой, себациновой, ади-
пиновой и ортофосфорной кислот, бутилстеараты, галогенсодер-
жащие углеводороды (хлорированные парафины, нафталины,
дифенилы и др.) [28, т. 1, с. 337].
Поверхностно-активные вещества (ПАВ) используются в
технологических процессах, а также в качестве
пластификаторов, антистатиков и модификаторов поверхности наполнителей.
Среди них чаще других применяются анионные ПАВ — соли
алкилсерных и алкилфосфорных кислот, алкиларилсульфонаты,
57
'перфторалкилсульфонаты, карбоксиметилцеллюлоза и др.
Неионные (неионогенные) ПАВ представены полиоксиэтиленовы-
ми эфирами алифатических спиртов и кислот, оксиэтилирован-
ными жирными аминами, алканоламинами, алкилфенолами,
высокомолекулярными соединениями (блоксополимер этилена и
пропилена, поливиниловый спирт, полиэтиленгликоль и др.).
К катионным ПАВ относятся высшие нитрилы, алкиламины и
их соли, четвертичные аммониевые соли, поли-1Ч-винилпирроли-
дон и др.; к амфолитным ПАВ — циклимид, сульфононат и др.
Все шире применяются кремнийорганические ПАВ [29, с. 290,
317].
Широкое распространение в последние годы получили
полимерные композиции с пониженной горючестью. Это свойство
композиций обеспечивается во многих случаях введением от
0,1 до 25 % (от массы полимера) и более антипиренов
органического и неорганического происхождения. Из неорганических
-антипиренов применяются оксиды металлов (Sb203, MgO,
А1203, Fe203, BeO, M0O3, NiO, Сг203), их смеси, гидроксид
алюминия, хлориды, фториды и фосфаты магния, алюминия,
аммония. Органические антипирены, как правило, отличаются
высоким содержанием брома (до 80 %) и хлора. К ним
относятся: декабромдифенилоксид, гексабромбензол, гексабромцик-
лодекан, хлорпарафины с различным содержанием хлора, хлор-
фосфорсодержащие соединения [например, три(1,3-дихлорпро-
пил)фосфат] и др. Органические фосфаты (трикрезилфосфат,
триксиленилфосфат, изодецилдифенилфосфат и др.)
одновременно являются пластификаторами и антипиренами [27, с. 226;
28, т. 1, с. 337].
Для улучшения эксплуатационных свойств и снижения
стоимости в полимерные материалы часто вводят наполнители —
твердые, жидкие и газообразные вещества, которые достаточно
равномерно распределяются в объеме полимерной композиции
и имеют четко выраженную границу раздела с непрерывной
полимерной фазой [31]. Наибольшее распространение в
производстве пластмасс получили твердые наполнители. Это, как
правило, высокодисперсные порошки, волокна, гранулы, листы и т. п.
При этом некоторые наполнители (графит, стекло, металлы)
могут применяться в различном виде. В зависимости от
характера взаимодействия с полимером наполнители условно делят
на инертные (не изменяющие свойств полимера) и активные
(упрочняющие, армирующие). Из органических
порошкообразных наполнителей применяются целлюлоза, газовый канальный
технический углерод, графит, политетрафторэтилен, поливинил-
хлорид и др. Группа неорганических наполнителей включает:
мел, каолин, тальк, слюду, кварц, оксиды металлов, гидроксид
алюминия, фториды и сульфаты кальция, стронция и бария,
порошки металлов и их сплавов (железа, меди, свинца, цинка,
алюминия, бронзы, латуни), керамические магнитные
порошковые материалы (ферриты).
58
Содержание порошкообразных наполнителей обычно
находится в пределах 1—50 ч. (масс), но в случае высоконапол-
ненных композиций возрастает до 200—300 ч.(масс) на
100 ч. (масс.) полимера. Для лучшей диспергации в массе
полимера наполнители часто обрабатывают растворами или
эмульсиями ПАВ, кремнийорганическими жидкостями, а
поверхность технического углерода подвергают химической
обработке — галогенированию, алкилированию, аминированию,
восстановлению и модификации капролактамом, полиэтилен-
имином.
Доля волокнистых наполнителей в термопластах составляет
15—40%. в реактопластах — 30—80 %• Из волокон
органической природы используются целлюлозные, полиакрилонитриль-
ные, на основе ароматических полиамидов (фенилон, кевлар),
ароматических полиимидов (аримид-ПМ); из неорганических
волокон — стеклянные, асбестовые, керамические, нитевидные
монокристаллические. Наполнители в виде зерен (гранул)
представлены полыми сферами из стекла и полимеров, углеродными
микросферами. Листовые наполнители (бумага, ткани, шпон,
сетки, холсты), как правило, служат основой для получения
слоистых пластиков из термореактопластов.
В качестве жидких наполнителей применяют воду,
минеральные масла; из газообразных наполнителей — диоксид углерода,
азот, аммиак, изопентан, изооктан (для получения вспененных
пластмасс).
1.2.2. Органические добавки и наполнители
Первоначальной операцией в ходе идентификации добавок
является выделение их из полимерной композиции. Для этой
цели чаще всего применяют экстракцию, подбирая экстрагент
таким образом, чтобы полимер в нем набухал, а не
растворялся полностью. Возможен и другой путь выделения добавок —
растворение полимера с последующим его высаждением осади-
телем и отделением от оставшихся в растворе добавок (см.
табл. 1.4).
На практике для оценки способности растворителя
растворять или вызывать набухание полимера обычно пользуются
эмпирическим правилом — подобное растворяется в подобном:
неполярные полимеры растворяются в неполярных растворителях,
а полярные — в полярных. Полярность растворителя
оценивается значением дипольного момента, а также диэлектрической
проницаемости е (см. табл. 1.3). По нашему мнению, выбирать
растворитель по значению дипольного момента нерационально.
К выбору растворителя для полимера можно подойти с
позиций концепции параметра растворителя б. В нем, согласно
теории регулярных растворов, находит отражение связь между
растворимостью полимера и природой растворителя [63]:
б = -у/ьЕ/Умол.
59
где Д£ — энергия межмолекулярного взаимодействия растворителя (энергия
когезии); Умол — молярный объем растворителя.
Для ряда растворителей значение параметра растворимости
■5 приведены в табл. 1.3, а для полимеров—ниже [62, 63]:
в-Ю-3, Дж'/'.м-'/!
в-Ю-3. Дж'/'-м-''1
15,75—16,98
15,96—16,57
16,16—16,98
16,57—17,60
18,82—19,23
17,39—19,03
18,62—19,44
25,58-31,51
18,21-18,62
18,00
16,98—18,00
20,05—21,28
18,82-19,23
18,41
Полибутилакрилат
Поливииилацетат
Поливинилхлорид
Полихлоропрен
Политетрафторэтилен
Полиэтилеитерефталат
Полигексаметилеиади-
пинамид
Полидиметилсилоксаи
Амииоальдегидные
смолы
Эпоксидные смолы
Фенолоформальдегид-
ные смолы
17,80—18,41
19,23—19,64
19,23—20,66
16,78-18,82
12,68
21,89
27,82
14,94—15,55
. 19,64—22,30
19,85-22,30
23,53
ч Полиэтилен
Полииэобутилен
Полиизопрен
Полибутадиен
Полипропилен
Полистирол
Полиметилметакрилат
Полиакрилонитрил
Полиэтилметакрилат
Полипропилметакрилат
Полибутилметакрилат
Полиметилакрилат
Полиэтил акрилат
Полипропилакрилат
По значениям параметра растворимости полимера и
растворителей можно сразу ограничить круг растворителей, среди
которых находятся способные растворить данный полимер. Полимер
не будет растворяться в жидкостях, параметр растворимости
которых намного меньше или больше параметра
растворимости полимера. Например, полистирол, у которого б = 19,03 X
X Ю3 Дж'л-м_3/'2 не будет растворяться в растворителях с 14X
X Ю3 > б > 23-103 Дж'^-м-1'1, но это не является гарантией
растворимости полистирола во всех растворителях с б = 14 X
ХЮ3-Ь23-103 Дж'А-мЛ
Параметр растворимости, к сожалению, не имеет
предсказательной силы в отношении растворимости полимеров, а может
лишь указать, что весьма немаловажно, в каких растворителях
полимер безусловно нерастворим. Это связано с тем, что
параметр растворимости — интегральная характеристика
межмолекулярного взаимодействия, а растворимость полимеров имеет
сложный характер и зависит от присутствия в молекулах
компонентов раствора различных функциональных групп,
вступающих или не вступающих во взаимодействие друг с другом.
Для растворения и экстракции желательно выбирать
инертные растворители, которые не вступали бы в химическое
взаимодействие с экстрагируемыми и растворяемыми
соединениями. При выборе растворителя следует обращать внимание на
возможность его удаления испарением, стабильность, удобство
в работе, воспламеняемость, стоимость, токсичность и другие
свойства. Тип растворителя, степень его очистки определяются
во многом аналитическими задачами.
Для идентификации выделенных веществ используют
различные физические методы: ИК- и ЯМР-спектроскопию, масс-
спектрометрию, газожидкостную и высокоэффективную
жидкостную хроматографию и др. Если вещество по каким-либо при-
60
чинам не выделяется из раствора и исследуется в растворителе,
при выборе растворителя для выделения и переосаждения
добавок необходимо учитывать требование несовпадения
характеристических сигналов, принадлежащих растворителю и
анализируемому продукту. Растворители для выделения добавок
должны быть весьма чистыми, поскольку физические методы
отличаются высокой чувствительностью и наличие примесей может
оказывать влияние на результаты идентификации. Желательно
перед проведением идентификации добавки тем или иным
методом предварительно получить спектр или хроматограмму
растворителя для сравнения.
Обычно проводят последовательное выделение добавок
различными растворителями, например, диэтиловым и петролейным
эфиром, ацетоном и др. Перед экстракцией (или растворением)
образцы полимера обязательно измельчают, что значительно
облегчает протекание этих процессов: гранулированные и
твердые образцы подвергают дроблению в замороженном состоянии
или получают срезы (бритвой, микротомом), пленочные
образцы нарезают ножницами на квадраты со стороной около 1 мм.
Многослойные композиции разделяют на составляющие части,
каждую из которых подвергают экстракции.
Наиболее распространен метод выделения добавок путем
экстрагирования навески образца массой 4—5 г, взвешенной с
точностью до 0,01 г, в аппарате Сокслета. Образец помещают в
пакет из фильтровальной бумаги, вставляют в насадку
экстрактора, соединяют с колбой, в которую налит растворитель-экст-
рагент. Как показывает практика, при нагревании на водяной
бане необходимо, чтобы экстракционное пространство
заполнялось экстрагентом 20—30 раз в 1 ч. При этом полная
экстракция компонентов из пленочных материалов достигается за 20 ч,
а из твердых полимеров в зависимости от вида — за 40—50 ч.
По окончании экстракции растворитель отгоняют, остаток из
колбы количественно переносят в предварительно взвешенный
бюкс и высушивают до постоянной массы в вакуум-сушильном
шкафу. Содержание добавок рассчитывают в процентах к
взятой навеске.
Возможен и другой путь выделения добавок—методом
растворения и переосаждения полимерного образца, который
описан в разделе 1.2.3. После отделения полимера и наполнителя
из маточного раствора, в котором растворены добавки в рас-
творителе-осадителе, отгонкой удаляют большую часть
растворителя, остаток количественно переносят в предварительно
взвешенный бюкс и высушивают в вакуум-сушильном шкафу до
постоянной массы. Содержание добавки рассчитывают в
процентах к взятой навеске.
В настоящее время для селективного выделения добавок
широко используется препаративная жидкостная хроматография.
Подробнее применение этого метода в сочетании с
количественным определением будет изложено в гл. IV.
61
Идентификацию выделенных добавок проводят в
соответствии с руководствами по органическому и функциональному
анализу [21, 64, 65]. Добавки идентифицируют по внешнему
виду, плотности, показателю преломления. Гетероэлементы
открывают реакциями, описанными в разделе I. 14 с
последующим обязательным количественным определением элементного
состава.
Для пластификаторов характерно число омыления — число
мг КОН, необходимое для нейтрализации свободных и
связанных кислот и для омыления сложных эфиров, содержащихся в
1 г вещества. Омыление образца проводят 0,5 н. или 1 н.
спиртовым раствором КОН при нагревании в колбе с обратным
холодильником. Избыток щелочи оттитровывают в присутствии
фенолфталеина соляной кислотой или определяют потенциомет-
рически [12, с. 368].
Наиболее целесообразно проводить идентификацию
выделенных добавок по их УФ-, ИК- и масс-спектрам, а также с
использованием пиролитической газовой хроматографии [66—68].
Эффективность физических методов возрастает при их
сочетании: например, стабилизаторы можно идентифицировать
методом высокоэффективной жидкостной хроматографии с
последовательным применением ультрафиолетового (280 нм) и
масс-спектрометрического (с химической ионизацией)
детектирования при содержании этих соединений в полимерной матрице-
порядка Ю-9 г [66].
Наиболее затрудняющим работу по идентификации
полимерной основы наполнителем является технический углерод,
который применяется не только в качестве наполнителя
композиционных материалов, но и как термо- и светостабилизатор и
электропроводящая добавка. Основным элементом технического
углерода (до 90%) является углерод. Помимо углорода
технический углерод содержит кислород, водород, серу, следы азота,,
минеральные соединения, количество которых зависит от
способов получения технического углерода. Поверхность
технического углерода неоднородна, имеются дефектные, энергетически:
неравноценные и шероховатые участки. Молекулы
высокомолекулярных соединений, как правило, не проникают в углубления
рельефа, но низкомолекулярные компоненты диффундируют в
них достаточно легко и адсорбируются поверхностью
технического углерода. На это следует обращать внимание при
выделении различных добавок (стабилизаторов, пластификаторов:
и др.) из образцов полимеров, наполненных техническим
углеродом.
Для установления типа технического углерода и
исследования его трехмерной структуры в наполненных полимерах
применялась просвечивающая электронная микроскопия. Путем
изготовления серий электронных стереомикрофотографий срезов
образцов с последующей реконструкцией объемной структуры
детально изучено распределение частиц технического углерода:
62
и их агрегатов в объеме полимерной матрицы [69].
Идентифицировать технический углерод методом рентгеноструктурного
анализа не представляется возможным, так как не удается
разделить рассеяние, вносимое техническим углеродом, от
рассеяния аморфной части полиэтилена или другого полимера. По
этой же причине невозможно этим методом идентифицировать
органические красители и отличить их от технического
углерода, если они черного цвета.
Графит на рентгенограммах имеет четко выраженный
рефлекс при углах рассеяния 26 = 26°30', что позволяет
определять его в полимерных образцах при содержании выше 1 % и
отличить от технического углерода и черных красителей.
Выделить технический углерод из полимеризационных
пластмасс путем подбора растворителей и осадителей для
полимерной части или другими аналитическими приемами практически
невозможно. Разработаны методы выделения и количественного
определения технического углерода только из полиолефинов,
основанные на пиролизе образцов в кварцевой трубке в токе
азота с постепенным подъемом температуры от 50 до 350 °С
и затем до 600—750°С [70] или на нагревании образцов
только при 500 °С в течение 30 мин также в атмосфере азота [71].
При содержании в композиционных материалах менее 1 %
технический углерод не мешает идентификации полимерной
матрицы спектральными методами ИК и ЯМР. При более высоких
содержаниях создаются большие трудности, поэтому для
идентификации полимера в подобных случаях применяется метод
пиролитической газовой хроматографии или ИК-спектроскопия
пиролизатов (см. разделы 1.1.6 и I. 1.7).
Другие добавки органического и неорганического
происхождения при наличии их в полимерной композиции до 5 % не
оказывают существенного влияния на идентификацию
полимерной матрицы методами ИК- и ЯМР-спектроскопии, но их
возможное влияние на спектры необходимо учитывать.
1.2.3. Неорганические наполнители и добавки
При идентификации неорганических наполнителей, как и
других вводимых в полимеры добавок, очень важно выделить
их в удобной для анализа форме. Неорганические наполнители
и добавки выявляют путем определения зольности полимерного
материала (ГОСТ 15973—82). При температурах 650—750°С
многие из неорганических наполнителей не претерпевают
существенных изменений, а полимеры при этих температурах
обугливаются и сгорают. Следует учитывать, что при сжигании
могут улетучиваться некоторые металлы (As, Cd, Hg); в
присутствии хлорированных полимеров и высокогалогенированных
добавок образуются летучие галогениды Sb, Sn, Zn; при
определении золы во фторполимерах, как правило, весьма ощутимы
потери в виде летучих фторидов металлов и SiF4, если в ка-
63
честве наполнителя присутствует SiC>2 или тальк. При сложном
составе наполнителя, представляющего собой смесь
минеральных веществ, гравиметрическое определение зольного остатка
дает лишь суммарное массовое содержание соединений,
причем большинство из них находится в виде оксидов. Зола
является удобной аналитической формой для определения
различными методами отдельных металлов (Fe, Ti, A1) и других
элементов, входящих в состав наполнителя, а также остатков
каталитических систем. Информацию о качественном и
количественном составе зольного остатка можно получить, анализируя
его атомно-адсорбционным, рентгено-флуоресцентным,
эмиссионным и другими спектральными методами [26, гл. 19, 20].
Кроме того, отдельные элементы можно определять спектрофо-
тометрически, а также с использованием электрохимических
методов [72, 73].
Однако при определении наполнителей по зольности
безвозвратно теряется полимерная матрица. Поэтому установив
наличие минеральных соединений, следует растворить полимер,
отделить наполнители фильтрованием или центрифугированием и
далее высадить чистый полимер из раствора. Перед
проведением операции отделения неорганических наполнителей
необходимо убедиться в отсутствии органических добавок
(стабилизаторов, пластификаторов и т. п.), проведя экстракцию
полимерного образца органическими растворителями (п. 1.2.2). Затем
навеску измельченного образца (1—2 г), взвешенную с
погрешностью не более 0,0002 г, растворяют в 50—100 мл выбранного
растворителя (см. табл. 1.4) при комнатной температуре или,
если необходимо, при нагревании на водяной бане. Раствор с
осадком наполнителя количественно переносят во взвешенный
стакан для центрифугирования и отделяют наполнитель на
центрифуге при частоте вращения 6000 об/мин. После
центрифугирования раствор полимера сливают с осадка. Наполнитель
2—3 раза промывают растворителем, каждый раз проводя
центрифугирование. Промывной раствор соединяют с основным
раствором. Наполнитель высушивают до постоянной массы в
вакуум-сушильном шкафу при 80 °С. Его содержание
рассчитывают в процентах к навеске образца полимера.
Идентификацию наполнителя проводят эмиссионным,
атомно-адсорбционным, рентгено-структурным и другими методами.
Для выделения полимера к полученному раствору по каплям
добавляют осадитель, в котором данный полимер нерастворим
(см. табл. 1.4). После промывки осадителем осадок полимера
отделяют, высушивают в вакуум-сушильном шкафу при 50—
60°С и взвешивают. Содержание полимера рассчитывают в
процентах к навеске. Выделенный полимер может быть
представителем одного класса, но иногда в композицию вводится
механическая смесь полимеров, которую следует разделять с
использованием растворителей, специфических для каждого
полимера. При необходимости изучения фракционного состава по-
64
лимер подвергают фракционированию путем разделения на
фракции избирательным растворением [74].
Следует обратить внимание на то, что при отделении
наполнителей вместе с ними может выделиться в осадок
нерастворимая сшитая фракция полимеров, каучуков и других продуктов.
Если их не удается отделить с применением других
растворителей, то осадок следует высушить и озолить.
Для выделения наполнителей из дисперсий, эмульсий,
лаков, клеев и других продуктов предварительно удаляют
растворители отгонкой под вакуумом, а оставшийся полимер с
добавками и наполнителями разделяют по описанной схеме.
Полиолефиновые композиционные материалы растворимы
только в горячих ароматических углеводородах, а при
охлаждении раствора до комнатной температуры выпадают в осадок,
захватывая частицы минеральных наполнителей, что делает
невозможным применение ИК-спектроскопии для идентификации
высоконаполненных образцов. Предложен способ выделения
минеральных наполнителей [75], основанный на смешении навески
композиционного материала с ксилолом и нагревании ее в
присутствии соляной кислоты при температуре кипения ксилола
(140°С) в течение 30—40 мин с последующим разделением
водного и ксилольного слоев. Из ксилольного слоя после
удаления ксилола выделяли полиолефпн в виде прозрачной пленки,
из которой приготовляли образец, пригодный для исследования
методом ИК-спектроскопии. Наполнители после выпаривания
раствора (если они растворялись в растворе соляной кислоты)
и нерастворимые наполнители после отделения фильтрованием
и высушивания до постоянной массы (см. п. 1.3.4)
анализировали эмиссионным методом. Предложен также способ [76]
отделения полиолефина (полипропилена) путем кипячения
навески образца композиционного материала в течение 2,5—3 ч
в ацетофеноне с последующим охлаждением раствора до
формирования на поверхности полимерной пленки, которую
механически отделяли. Выпавшую в осадок неорганическую часть
(тальк) и выкристаллизовавшийся из раствора хлорпарафин
разделяли фильтрованием, после чего взвешивали.
Прямым деструктивным методом определения наполнителей
может служить термогравиметрия. Этот метод применим для
идентификации минеральных наполнителей, разложение
которых сопровождается характерными экзо- или эндотермическими
эффектами, заметными на фоне термораспада полимеров. Для
остальных наполнителей это обычное гравиметрическое
определение зольности.
Более привлекательны прямые недеструктивные методы
анализа наполненных полимеров с использованием электронного
микрозонда [77], позволяющего проводить измерение
содержания элементов с порядковым номером более 10, входящих в
состав наполнителей, как на поверхности, так и в поперечном
сечении образца.
3 Зак. 632
65
Без выделения из композиционных полиолефнновых
материалов можно идентифицировать методом ИК-спектроскопии
такие наполнители, как ЭЮг, тальк, Ti02 (характеристические
полосы поглощения 1090 см-1, 1020 см-1, широкая полоса с
максимумами 650 и 515 см-1 соответственно), если их содержание
в образцах не превышает 5 % от массы полимера. В остальных
полимеризационных пластмассах не удается идентифицировать
минеральные наполнители методом ИК-спектроскопии без
отделения полимерной основы, так как характеристические полосы
поглощения полимеров перекрывают полосы поглощения
наполнителей. Газообразные наполнители идентифицируют хромато-
графическим методом [78, с. 140].
1.3. МЕТОДИКИ ВЫДЕЛЕНИЯ И ИДЕНТИФИКАЦИИ
ПОЛИМЕРНОЙ ОСНОВЫ
1.3.1. Идентификация полимерной основы
по продуктам пиролиза методом ИК-спектроскопии
Методика основана на термическом разложении образца
полимерного материала и улавливании образующихся летучих
продуктов для последующего анализа методом
ИК-спектроскопии.
Аппаратура, реактивы
Две электропечи трубчатые универсальные типа МА-0.2.20,
отрегулированные: первая на 120 ± 10 °С, вторая на 500—700 °С.
Термопара ХА.
Насос механический вакуумный с масляным уплотнением.
Манометр ртутный.
Секундомер.
Сосуд Дьюара вместимостью 0,35—0,5 л.
Пробирки со шлифом № 14 из термостойкого стекла диаметром 13—
14 мм, длиной 120—130 мм.
Пробирки из термостойкого стекла диаметром 13—14 мм, длиной 140—
150 мм с расширением посередине в виде шарика диаметром 18—20 мм
(рис. 1.6).
Ловушка стеклянная для газовых продуктов.
Кран соединительный трехходовой.
Стаканчики для взвешивания (бюксы) вместимостью 1—5 мл.
Азот жидкий.
Получение продуктов пиролиза на воздухе. 0,2 г
измельченного полимера, взвешенного с точностью до 0,001 г, помещают
в пиролизер, который вставляют на глубину около 6 см в
гнездо печи, укрепленной под углом 20—30° к горизонтали и
нагретой до 500—550 °С. Пиролиз образца проводят в течение
5 мин, контролируя время по секундомеру. Образующиеся
летучие продукты пиролиза конденсируются при комнатной
температуре на выступающей части пробирки в виде капель
маслянистой жидкости, которые стекают в «шарик».
Образовавшийся жидкий или мазеобразный конденсат переносят стеклянной
66
Рис. 1.6. Пиролизер для получения продуктов деструкции полимеров 7J
на воздухе. Н—(■<-
.гь,
<Г? s
пипеткой с изогнутым концом или шпателем в
стеклянный бюкс или колбочку с притертой пробкой.
Далее пиролизат анализируют методом ИК-спек-
троскопии.
Получение газообразных и твердых продуктов
пиролиза фторсодержащих полимеров. 0,2 г
измельченного полимера, взвешенного с точностью
до 0,001 г, помещают в стеклянную пробирку,
соединенную с вакуумной пиролитической системой
(рис. 1.7). В течение 1 ч всю систему вместе с
образцом вакуумируют при помощи вакуумного
насоса до остаточного давления 133,3 Па (1 мм рт. ст.).
Образец при этом прогревается электрической печью при
100—120°С для удаления влаги. После этого отодвигают
первую печь и быстро вводят пробирку во вторую электрическую
печь, нагретую до 700—750°С. Разложение проводят в течение
30—40 мин. Образующиеся летучие продукты конденсируются
в ловушке, охлаждаемой жидким азотом в сосуде Дьюара. При
проведении пиролиза следят за показанием ртутного манометра,
так как постоянство давления в системе гарантирует полноту
улавливания летучих продуктов. По окончании пиролиза всю
систему разгерметизируют с помощью трехходового крана 3.
На оба конца ловушки 2, отсоединенной от пиролизера с
образцом и крана, надевают резиновые заглушки. Лишь после этого
снимают охлаждение.
Для снятия ИК-спектров газообразных продуктов пиролиза
ловушку непосредственно соединяют с газовой кюветой и
отбирают в нее газовую пробу.
Твердые продукты пиролиза, нелетучие при комнатной
температуре, осаждаются в виде мазеобразного налета на холод-
S^7
Рис. 1. 7. Пиролизер для получения продуктов деструкции полимеров в инертной среде:
/ — пробирка; 2 — ловушка; 3—край; 4 — форвакуум; 5 — ртутный манометр; б — электропечь;
7—термопара.
3*
67
ных стенках пиролизера / вне зоны нагрева. Этот налет по
окончании пиролиза снимают со стенок шпателем. ИК-спектры
поглощения продуктов пиролиза получают на двухлучевом ИК-
спектрофотометре в интервале частот 4000—700 см-1. Образцы
готовят, раздавливая каплю между пластинками из КВг или
NaCl. Толщину слоя подбирают таким образом, чтобы
пропускание в максимумах наиболее интенсивных полос поглощения
в спектре составляло 10—15 %.
7 "1
4^J \
■А
'X
а
-А
,л
_^ь
_~-Ь
_J-
_А
^
^
IL.
(Lj
v_
й
а
й
а
и
4000 2000 1500 WOO v,cm
Рис. I. 8. ИК-спектры пиролнэатов полиэтилена и сополимеров этилена:
/ -пэллэтилен высокого давления; 2 —полиэтилен низкого давления; 3 — сополимер этилена
с пропиленом (12%); 4 —сополимер этилена с бутеном-1 (11%); 5—сополимер этилена
с винилацетатом (9,5 %).
68
J.
ж
л
л]
^v
JL
^x
X
__vJL
_jv
_A
._A
\ju
V
I\l
pJ
ч
.^
ih,
^
к
H
г
'iv
4000 2000 7500 7000 v^cw'
Phc. I. 9. ИК-спектры пнролнзатов полн-а-олефиков:
/ — изотактический полипропилен; 2 — атактический полипропилен; 3—блоксополимер
пропилена с этиленом (14%); 4 — полнбутен-1; 5 —полн-4-метилпентен-1; б — поли-З-метнлпентен-1.
69
2
1
3
^ч.
1
и
/Wwl
дл/Ъ-л^
/\л№^^
Ui
<wilUH
LyjiA
4000 2000 Ы0 WOO v,or'
Рис. 1. 10. ИК-спектры пиролиза
/ — полистирол; 2—ударопрочный полистирол иа этиленпропилеиовом каучуке; 3—ударо
рилом; 5 — АБС-пластик (10% 1,4-тро«с-бутадиенопого кау
Идентификацию спектров проводят путем сопоставления ИК-
спектра продуктов пиролиза исследуемого образца с ИК-спект-
рами продуктов пиролиза стандартных образцов (рис. 1.8—
1.11).
' 1.3.2. Идентификация полимерной матрицы
методом пиролитической газовой хроматографии
Методика основана на проведении термического разложения
образца полимерного материала в пиролизере печного типа,
включенном в схему хроматографа, с последующим хроматогра-
фированием образующихся продуктов пиролиза.
Аппаратура, реактивы
Газовый хроматограф с детектором по теплопроводности и
программированием температуры колонок.
Пиролитическая приставка печного типа П-75.
Газохроматографические колонки из нержавеющей стали длиной 3 м,
внутренним диаметром 3 мм.
70
4
1
J
)
u
ц
ц
/WlJ^
/\A/w
/V/vr
bill
кмЛ
Ч4
4000 2000 7500 WOO v,c/w-'
тов полнстнрольных пластиков:
лрочный полистирол на 1,4-цис-бутадиеновом каучуке; 4—сополимер стирола с акрнлонит-
чука); 6 — АБС-пластпк (25 % бутплакрплатного каучука).
Форколонка из нержавеющей стали длиной 100 мм, внутренним
диаметром 3 мм.
Секундомер.
Полумикровесы ВЛР-20.
Весы аналитические.
Ча шка фарфоровая вместимостью 450—850 мл.
Стекловолокно из стеклоткани.
Хлороформ.
Силиконовый эластомер Е-301.
Инертон AW (0,160—0,200 мм).
Силипор 600 (0,125—0,160 мм).
Ацетон.
Тетрахлорметан.
Гелий газообразный.
Воздух сжатый.
Азот газообразный.
Приготовление насадки для колонки хроматографа. 92 г
сухого твердого носителя инертона AW (0,16—0,20 мм),
взвешенного с точностью до 0,02 г, вносят в фарфоровую
выпарительную чашку. 8 г неподвижной фазы силиконового эластомера
Е-301, взвешенного с точностью до 0,02 г, растворяют в
хлороформе и заливают раствором носитель в чашке таким обра-
71
А^
2 » v
J
~^Л
уьДллА
дЪ
aJ
X
/ у
/vJ
Ul
V w
/f
4000 2000 7500 1000 ЦСМ'1
Рис. I. 11. ИК-спектры пиролиза
J—политетрафторэтилен; 2—сополимер тетрафторэтилена с гексафторпропиленом; 3 —сопо
лен; 5 — сополимер тетрафторэтилена с этиленом; 6 — полнвинил
зом, чтобы он оказался под слоем раствора. Полученную смесь
перемешивают, помещают на водяную баню (70—80 °С) и при
постоянном перемешивании удаляют хлороформ.
Приготовленную насадку сушат в сушильном шкафу при 120—130°С в
течение 2—3 ч.
Подготовка форколонки. Форколонка предназначена для
поглощения фтороводорода, образующегося при пиролизе фтор-
полимеров, с целью предохранения насадки и детектора от его
корродирующего действия. Поглотитель для форколонки готовят
путем смешения оксида алюминия с оксидом магния (0,16—
0,20 мм) в соотношении 1:1.
Подготовка колонок. Промытые тетрахлорметаном и
ацетоном колонки высушивают и заполняют сорбентом: колонку 1—
силипором 600 (0,125—0,160 мм), кондиционируют при
температуре 100 °С и скорости газа-носителя гелия 30 мл/мин в
течение 7—8 ч; колонку 2—приготовленной насадкой с неподвиж-
72
5
6
7
'
*——
_л
|Л
^
/Л
w
ll/i
V
к
4000 2000 1500 WOO v,cm-'
tOb фторполимеров:
лимер тетрафторэтилена с перфорированным виниловым эфиром; 4—политрифторхлорэти-
иденфторид; 7 —сополимер винилидеифторида с тетрафторэтилеиом.
ной фазой —силиконовым эластомером Е-301, кондиционируют
в течение 5 ч без газа-носителя при температуре 300°С,
затем продувают гелием при скорости 30 мл/мнн и той же
температуре в течение 5—7 ч.
Включение и подготовку хроматографа и пиролизера к
работе проводят по инструкциям, прилагаемым к приборам.
Выполнение анализа. Перед проведением пиролиза
определяют в полимерном образце наличие фтора (см. п. I. 1.4.3).
Фторсодержащие полимеры анализируют на колонке /,
остальные полимеры —на колонке 2 при условиях пиролиза:
Температура, °С 600±10
Продолжительность, с 60±1
Условия газохроматографического анализа продуктов
пиролиза для колонок 1 и 2 приведены на стр. 80.
73
20 15
Время, vu4
Рис. I. 12. Пнрограмма полиэтилена низкого давления.
«20
Время, мин
Рнс. 1. 13. Пнрограмма сополимера этилена с виннлацетатом (9,5 %).
74
х10
15
15
■t-J \^/\~+*а**~/
10 5 0 15 10 5
Время, мин Время, мин
Рнс. I. 14, Пнрограмма полнвиннлацетата.
Рис. 1.16. Пнрограмма ударопрочного полистирола (на бутадиеновом каучуке).
х20
_1_
1л>
10 5'
Время, мин
15 Ю 5 О
Время, мин
Рнс. I. 16. Пнрограмма АБС-пластнка.
Рнс. I. 17. Пнрограмма сополимера стирола с метнлметакрилатом и акрнлоинтрнлом.
75
75 10
Время, мин
Рис. I. IS. Пнрограмма полнвнннлхлорнда.
10
Время, мин
Рис Т. 1В. Пнрограмма пентапласта.
76
20
15 10
Время, мин
Рис. 1.20. Пнрограмма поликарбоната.
20
15 10
Время, мин
Рис. 1.21. Пнрограмма полифениленокснда.
77
25
20 15 Ю
Время, мин
Рис. I. 22. Ппрограмма полиамида П-в.
Ю 5
Время, мин
10 . 5
Время, мин
Рис. I. 23. Пирограмма политетрафторэтилена.
Рис. I. 24. Пирограмма поливииилидеифторида.
78
JO
JVaa^J
25 20 15 10 5 0 10 5
Время, мин Время, мин
Рнс. I. 25. Пнрограмма полнтрнфторхлорэтнлена.
Рнс. I. 26. Пнрограмма сополимера этнлеиа с тетрафторэтнлеиом.
х 10 *№
х?0
25 20 15 10 5
Время, мин
Рис. 1.27. Пнрограмма фторкаучука СКФ-26.
79
Температура колонок, °С
1 (изотермический режим) 80
2 (программирование со скоростью 11сС/мии через
2 мии 15 с от начала анализа) 50—250
Температура детектора для колонок / и 2, °С 100 и 300
Сила тока моста для колонок 1 и 2, мА 240 и 100
Скорость газа-носителя гелия (/ и 2), л/ч 2
Время выхода компонентов, мин, не более 30
Скорость движения диаграммной ленты, мм/ч 600
В предварительно прокаленную кварцевую лодочку
помещают примерно 2 мг образца, взвешенного с точностью до 0,1 мг.
Лодочку закрепляют с помощью держателя на штоке,
присоединяют к печи пиролиза и открывают кран. После
установления нулевой линии вручную отмечают начало анализа на пи-
рограмме, одновременно включают секундомер. Через 15 с
вводят направляющую до упора в печь пиролиза. По истечении
1 мин от начала пиролиза направляющую выдвигают из печи
до упора, кран закрывают.
Обработка результатов. Тип полимера в исследуемом
образце определяют путем сравнения полученной пирограммы с
эталонными (рис. 1.12—1.27). Вывод о типе идентифицируемого
полимера делают при условии идентичности пирограммы с
одной из эталонных пирограмм.
Пирограммы считают идентичными, если на пирограмме
анализируемого полимера, полученной на том же приборе, что и
эталонная, времена удерживания пиков газообразных продуктов
пиролиза с интенсивностью не менее 5 % от суммарной
интенсивности всех пиков совпадают с временами удерживания
подобных пиков на эталонной пирограмме в пределах
относительной погрешности ±3,5%. Интенсивность характеристических
пиков может различаться для сополимеров, что свидетельствует
об отличии их состава от состава эталонного образца.
1.3.3. Идентификация каучуков в ударопрочных полистирольных
пластиках
Предложена последовательность анализа для
идентификации 1,4-цис- и 1,4-гранс-бутадиенового, этилен-пропиленового и
бутилакрилатного каучуков в ударопрочном полистироле, АБС-
пластиках и в композициях на их основе сочетанием методов
ПГХ, ИКС и химического (определение двойных связей)
(рис. 1.28).
Аппаратура
Хроматограф с детектором по теплопроводности и программированием
температуры колонок.
Пиролитическая приставка печного типа.
Двухлучевой ИК-спектрофотометр.
Подготовка образца к анализу методом ИК-спектроскопии.
Для получения ИК-спектров ненаполценных полимеров
используют пленки толщиной 50 мкм, полученные поливом из 4 %-ных
80
растворов в тетрахлорметане или хлороформе. Для
наполненных полимеров отделение наполнителя и анализ пиролизата
проводят по методике 1.3.1.
Выполнение анализа. Исходный образец подвергается
пиролизу по методике 1.3.2, что позволяет определить тип поли-
стпрольного пластика: ударопрочный полистирол или
сополимер стирола с акрилонитрилом, привитый на разные каучуки.
Содержание каучука в этих сополимерах колеблется в пределах
от 2 до 40 %. На пирограммах сополимеров при таких
содержаниях каучука регистрируются лишь легкие продукты пиролиза
каучуковой части, времена удерживания которых совпадают
для 1,4-цис- и 1,4-гранс-бутадиеновых, этилен-пропиленового
и бутилакрилатного каучуков. Химический метод (см.
методику II. 5.11) позволяет определить непосредственно в
образце каучуки, имеющие в структуре двойные связи, т. е.
бутадиеновые каучуки (без различия изомеров).
Для определения транс-изомера используется ИК-спектро-
скопия. ИК-спектры полистирольных пластиков на
бутадиеновом каучуке, основу которого составляет 1,4-транс-изомер,
содержат полосу поглощения при 967 см-1, характерную для
колебаний СН-групп при двойной связи С=С в положении
Исходный образец
Пиролитическая газовая хроматография
Ударопрочный полистирол
АБС-пластики
Определение двойных связей
Бутадиеновый
каучук
Инфракрасная
спектроскопия
гранс-Изомер
цыс-Изомер
Этилеипропилеиовый и
бутилакрилатный каучуки
1
Инфракрасная
спектроскопия
■4>-^
Бутил -
акрил атный
каучук
\^_
Этиленпро-
пиленовый
каучук
Рис.
. 28. Схема идентификации каучука в ударопрочных полистирольных пластиках
(+ определяется; — не определяется).
81
1,4-транс. Полоса поглощения, характеризующая колебание СН-
групп при двойной связи С=С в положении 1,4-цис имеет
максимум при 730 см~', т. е. в области, где расположены
наиболее интенсивные полосы поглощения полистирола. Методом
ИК-спектроскопии определяется только 1,4-гранс-бутадиеновый
каучук. При отсутствии двойных связей каучук
идентифицируется как этилен-пропиленовый или бутилакрилатный. Методом
ИК-спектроскопии определяют буталакрилатный каучук по
наличию в ИК-спектре поглощения полос, характерных для
колебаний связей С=0 (1730 см^1) и С—О (1250 и 1170 см-')
сложноэфирной группировки бутилакрилатного каучука.
Этилен-пропиленовый каучук в полистирольных пластиках методом
ИК-спектроскопии не идентифицируется.
1.3.4. Выделение полиолефинов из композиционных материалов
на основе полиолефинов
Методика позволяет проводить выделение полиолефинов из
композиций, содержащих до 90 % неорганических
наполнителей.
Аппаратура и реактивы
Печь муфельная типа «Снол».
Эксикатор, заполненный хлоридом кальция.
Тигли платиновые с крышками.
Шкаф вакуум-сушильный.
Весы аналитические ВЛА-200.
Электроплитка с закрытой спиралью.
Термометр стеклянный ртутный, электроконтактный типа ТПК.
Баня масляная.
Колба круглодонная вместимостью 150 мл с притертым обратным
холодильником.
Воронка стеклянная делительная типа ВД.
Воронка стеклянная типа В.
Чашка фарфоровая выпарительная.
Ксилол.
Вода дистиллированная.
Соляная кислота, 2 н. раствор.
Бумага фильтровальная типа ФОМ.
Выполнение анализа. Вначале определяют содержание
зольного остатка. Навеску 3—4 г композиции, взвешенной с
точностью до 0,0002 г, помещают в платиновый тигель,
предварительно прокаленный до постоянной массы, охлажденный в
течение 1 ч в эксикаторе и взвешенный с точностью до 0,0002 г.
Навеску помещают в муфельную печь, предварительно
нагретую до 400±50°С и медленно сжигают, избегая
воспламенения. Постепенно поднимают температуру до 600±50°С.
После полного сгорания пробы температуру печи повышают до
850 ± 50 °С и прокаливают остаток 30 мин. Прокаливание
повторяют до постоянной массы. Массовую долю золы 3 (в %)
вычисляют по формуле:
3 = (т,/ш0). 100,
82
где /Й1 — масса золы после прокаливания в муфельной печи; /Ло — масса
пробы, г.
Состав зольного остатка идентифицируют методом
эмиссионной или атомно-адсорбционной спектроскопии.
0,5 г измельченного (2X2 мм) образца, взвешенного с
точностью до 0,0002 г, помещают в круглодонную колбу,
добавляют 50 мл ксилола, соединяют колбу с обратным холодильником
и нагревают на масляной бане при 140 °С (температура
кипения ксилола) до полного растворения полимера. Затем
добавляют 2 н. раствор соляной кислоты, объем которой зависит от
содержания минерального наполнителя в композиции:
Содержание минерального на- < 20 20—40 40—60 60—90
полнителя (определенное
вначале), %
Объем 2 и. раствора соляной 5 10 20 25
кислоты, мл
Смесь кипятят 30—40 мин при 140 °С и переносят в
обогреваемую делительную воронку. Отделяют ксилольный слой
от водного солянокислого слоя. Ксилольный слой промывают
горячей (85—95СС) дистиллированной водой до нейтральной
реакции промывных вод, отделяют от водного в обогреваемой
делительной воронке и фильтруют через обогреваемую
стеклянную воронку с бумажным фильтром в выпарительную чашку.
Ксилол удаляют выпариванием на водяной бане в вытяжном
шкафу. Полученную пленку сушат в вакуум-сушильном шкафу
при температуре 70—75 °С и давлении 66,5—133 Па.
Полученную пленку идентифицируют методом ИК-спектроскопии.
При необходимости анализа неорганического наполнителя
другими методами (кроме эмиссионной спектроскопии)
полученный водный солянокислый слой соединяют с промывными
водами, образующимися при промывке ксилольного слоя, а
также с порциями горячей дистиллированной воды (5—10 мл),
которыми смывают остатки частиц нерастворимого в
солянокислом растворе наполнителя и упаривают. Сухой остаток
анализируют.
ГЛАВА II
ОПРЕДЕЛЕНИЕ СОСТАВА СОПОЛИМЕРОВ
От состава сополимера зависят его основные физические,
механические и химические свойства, поэтому определение
состава является одной из основных задач аналитической химии
высокомолекулярных соединений. Знание состава необходимо
при научно-исследовательской работе для установления
основных закономерностей процесса полимеризации, при разработке
технологии и для контроля за технологическим процессом в
промышленных условиях.
83
Таблица П. 1 Применимость различных методов для анализа
состава сополимеров
+ + применим без ограничений; + применим с ограничениями; — применение
невозможно; ? теоретически применение возможно, но методика неизвестна
Сополимеры
ИКС
ПГХ
ЯМР
Анализ
элементный
Анализ
функциональный
Полиолефины'
Этилен — винилацетат
Этилен — акрилаты, метакрилаты
Ударопрочный полистирол на основе
полибутадиена
Ударопрочный полистирол на основе
СКЭПТ
АБС-пластики
Стирол — акрилонитрил
Стирол — метакрилаты и акрилаты
Тетрафторэтилен-гексафторпропилен
Тетрафторэтилеи — винилиденфторид
Тетрафторэтилеи — этилен
Тетрафторэтилеи — перфторвинило-
вые эфиры
Винилацетат — виниловый спирт
++
++
++
++
+
++
++
++
++
++
++
++
++
++
++
++
++
++
++
++
++
+
+
+
+
?
+
++
++
+
+
+
++
++
—
+
—
?
?
—
+
+
—
—
+
++
++
—
++
+
+
—
—
++
++
++
—
+
—
++
—
—
—
?
++
В настоящее время из большого числа различных методов,
которые в принципе могут быть применены для анализа
состава сополимеров можно выделить несколько основных. Из
спектральных методов это прежде всего ИК-спектроскопия; ЯМР-
и УФ-спектроскопия применяется значительно реже. Из хро-
матографических методов все большее значение приобретает
метод пиролитической газовой хроматографии (ПГХ). Кроме
того, сравнительно широко применяется функциональный и
элементный анализ. Из табл. II. 1 видно, что наибольшей
универсальностью обладают методы ИК-спектроскопии и ПГХ. Однако
следует иметь в виду, что эти методы являются зависимыми,
так как требуют при разработке методик наличия стандартных
образцов известного состава. Методы элементного и
функционального анализа, а также ЯМР являются абсолютными, их
часто применяют для оценки состава образцов, которые затем
используются в качестве стандартных при разработке методик
методами ИКС и ПГХ.
Табл. II. 1 отражает применимость методов только для
чистых сополимеров. При анализе большинства высоконаполнен-
ных и сшитых сополимеров возникают существенные
ограничения. Более подробно этот вопрос был рассмотрен в
разделах I. 1.6 и I. 1.7.
Наряду с анализом сополимеров целесообразно
рассматривать и вопросы анализа гомополимеров, так как данные,
полученные при анализе гомополимеров, очень часто служат
отправной точкой при разработке методики анализа сополимеров.
Строение реальных молекул гомополимеров отличается от тео-
84
ретического, поскольку они, как правило, обладают разнозвен-
ностью. Под разнозвенностью подразумевают наличие в
молекулах разветвленности, структур «голова к голове», стереорегу-
лярности и других отклонений от структуры, представляемой
обычной химической формулой. С аналитической точки зрения
оценка разнозвенности гомополимеров ничем не отличается от
анализа состава сополимеров.
Задача определения состава смесей гомополимеров
аналогична задаче определения состава блоксополимеров и является
частным случаем более общей задачи—определения состава
сополимеров.
II. 1. ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
В основе количественного анализа высокомолекулярных
соединений методом ИК-спектроскопии лежат те же принципы,
что в основе количественного анализа органических соединений
[33]. Однако имеется ряд вопросов, которые характерны
только для высокомолекулярных соединений. Полезная информация
по этим вопросам содержится в книгах [17, 34, 39, 79].
11.1.1. Специфика анализа состава сополимеров
методом ИК-спектроскопии
Количественный анализ состава сополимеров, как и любого
органического соединения, основывается на том, что каждый из
анализируемых компонентов имеет свое специфическое
химическое строение, а следовательно, и свои, характерные только
для него, полосы поглощения. Интенсивность этих полос
поглощения связана с концентрацией компонентов следующим
соотношением:
Dv = evrfC, (II. 1)
где Ov — оптическая плотность при волновом числе v (обычно в максимуме
поглощения аналитической полосы); ev — молярный показатель поглощения
при волновом числе v; d — толщина образца, см; С — концентрация вещества,
моль/л (для полимеров имеют в виду концентрацию определяемых
структурных единиц—мономерных звеньев).
°v = 1g(1/7'v) = ,g(/0v//v)- <"-2>
где /^ и / — интенсивность падающего и прошедшего через образец света;
7"v — пропускание.
Определив ev по стандартным образцам и измеряя Dv и d
для анализируемого образца, можно определить концентрацию
интересующего компонента. Из уравнения (II. 1) следует, что
коэффициент поглощения ev должен оставаться постоянным во
всем интервале определяемых концентраций. При анализе
смесей низкомолекулярных соединений эти условия в основном
выполняются, так как в этом случае значение е может
меняться только для некоторых полос, обусловленных колебаниями
85
групп, которые способны участвовать в образовании
водородных связей.
Для сополимеров все обстоит гораздо сложнее и на
значение е и положение полосы могут влиять следующие факторы.
1. В сополимере анализируемые компоненты—мономеры
связаны между собой химической связью и с изменением
состава меняется по существу строение молекулы. Естественно
ожидать, и это подтверждается экспериментом и расчетами, что
положение и интенсивность некоторых полос поглощения,
относящихся к определенной группе в мономере А, будут
чувствительны к тому, расположен ли мономер в окружении себе
подобных (БАААААААБ) или находится в окружении
мономеров Б (БББАБББ), а также от длины последовательностей
А и Б. Из теории сополимеризации известно, что и окружение,
и длина последовательностей зависят в значительной степени
от состава сополимеров.
2. Если сомономеры содержат группы, которые могут
образовывать водородные связи, то с изменением состава будет
меняться число групп, участвующих в образовании таких
связей. Зависимость числа таких связей от концентрации
мономеров имеет сложный характер. Помимо водородных связей
определенную роль в изменении спектральных характеристик полос
поглощения могут играть и диполь-дипольные взаимодействия.
3. Кристалличность сополимеров очень сильно зависит от
состава, а при переходе из аморфного состояния в
кристаллическое и наоборот существенным образом меняются и частоты,
и интенсивности полос поглощения.
Из всего сказанного следует, что правильный выбор
полосы поглощения имеет решающее значение для анализа состава
сополимеров.
Имеет свою специфику и получение стандартных образцов
для построения градуировочного графика. При анализе смесей
низкомолекулярных соединений получение стандартных
образцов не представляет трудностей, если имеются исходные
компоненты. Для сополимеров исходные компоненты в принципе
отсутствуют. Даже синтез низкомолекулярных аналогов,
процесс очень трудоемкий и сложный, не решает проблему
градуировки полностью. Для построения градуировочного графика
в случае анализа сополимеров применяется ряд приемов,
которые будут рассмотрены в дальнейшем.
II. 1.1.1. Выбор аналитических полос поглощения
К аналитическим полосам поглощения при определении
состава сополимеров предъявляются следующие требования.
1. Каждая из полос должна быть характерной только для
одного из сомономеров.
2. Коэффициент полосы поглощения должен оставаться
постоянным во всем интервале определяемых концентраций. Это
86
условие выполняется, если график зависимости между Dv и С
имеет линейный характер. Обычно достаточно хорошим
показателем того, что полоса может быть использована для
количественного анализа, служат такие простые признаки, как
неизменность частоты и контура при изменении состава сополимера.
Чаще всего таким требованиям отвечают полосы поглощения,
вызванные колебаниями атомов не в основной цепи, а в
боковых группах (например, колебания бензольного кольца в
сополимерах стирола или колебания, связанные с эфирной группой,
в акрилатах).
3. Полоса должна быть изолированной или как можно
меньше перекрываться с другими полосами. Это требование редко
удается выполнить, поэтому очень часто работают с
перекрывающимися полосами поглощения. Естественно, что при этом
стараются выбрать полосы так, чтобы мешающая полоса была
минимальной по интенсивности и нечувствительной к
чередованию звеньев.
4. Пропускание в измеряемом интервале концентраций
должно меняться в пределах от 10 до 80 % при толщине образца
желательно не менее 0,02 мм.
На практике довольно часто приходится идти на нарушение
этих условий, но в таких случаях всегда требуется
дополнительное исследование, чтобы правильно оценить последствия
допущенных отклонений и выработать метод их компенсации.
II. 1.1.2. Методические подходы к количественному анализу сополимеров
Как следует из уравнения (II. 1), для количественного
определения состава сополимеров в первую очередь надо
определить показатель поглощения ev. Значение ev постоянно для
данного вещества при данном волновом числе и должно
изменяться только в зависимости от физического состояния
вещества. Однако на практике при получении спектров одних и тех
же образцов на разных приборах количественные результаты
часто не совпадают. Это объясняется тем, что sv определяют
через экспериментальное значение Dv, на которое оказывают
влияние аппаратурные параметры, в частности оптическая
ширина щели и скорость сканирования.
На практике непостоянство ev при переходе с одного
прибора на другой приводит к тому, что использование литературных
данных для ev может приводить к значительным ошибкам и
при разработке методики эту величину приходится уточнять.
Имеются два следующих основных метода определения ev.
1. Если содержание анализируемых групп можно определить
другим независимым методом, то по этим стандартным
образцам строят график зависимости Dv от концентрации
определяемых групп. При пересчете Dv на толщину образца 1 см
тангенс угла наклона прямой этой зависимости будет равен ev.
87
В качестве независимых методов могут быть использованы
ЯМР, функциональный и элементный анализ.
Надежные стандартные образцы можно получать, синтезируя
сополимеры, в которых один из сомономеров содержит меченые
атомы. Определение состава таких сополимеров методом
радиометрии не представляет трудности. Если проводить
полимеризацию, определяя состав реакционной системы в начале и
конце процесса, то можно рассчитать состав получаемых при
этом сополимеров и использовать их в качестве стандартных
образцов. Такой способ трудоемок, но дает надежные
результаты.
2. Известно, что одинаковые группы в одинаковом
окружении имеют сходные спектральные характеристики — частоты и
интенсивности. Поэтому, когда имеется низкомолекулярное
соединение, которое содержит группу, аналогичную той, которую
надо определить в полимерной молекуле, и ближайшие
соседние группы также аналогичны, то определяют ev для этого
низкомолекулярного соединения и затем используют его для
анализа полимерного образца. Анализ образца
кристаллической структуры следует проводить при температуре выше
температуры плавления полимера. Если модельное соединение
имеет кристаллическую структуру, то это не означает, что она
аналогична кристаллической структуре полимера, поэтому спектры
должны быть получены для образцов в расплавленном
состоянии.
При определении состава сополимеров иногда в качестве
модельных соединений используют смеси гомополимеров.
Однако, как будет показано в дальнейшем, этот прием может быть
применен далеко не для любой пары сомономеров и требует
специального рассмотрения.
Анализ по уравнению (П. 1) принято называть анализом по
абсолютной градуировке. Его применение требует точного
знания толщины образца, поэтому его чаще всего используют для
анализа сополимеров в виде растворов. Для растворов
воспроизводимость толщины кюветы и концентрации раствора может
быть надежно гарантирована. В этом случае обычно не
определяют истинное значение ev, а пользуются экспериментально
найденным коэффициентом для выбранной концентрации и
толщины кюветы.
Наибольшее распространение, как это следует из анализа
литературы, получил метод, при котором используют отношение
оптических плотностей двух полос — DVl и Dv,, принадлежащих
сомономерам А и Б. Исходя из уравнения (II. 1), можно
написать:
* eNCl e* С^к^. (п.з)
< *ldCB < св Съ
88
Имея стандартные образцы, строят график в координатах
DvjDv2 и Са/Сб- Если график имеет линейный характер, то его
можно выразить в виде уравнения:
В = а + КХ, (ПА)
где В = Dy /Ov ; К — коэффициент пропорциональности, равный тангенсу
угла наклона прямой; X — Сд/Св — отношение концентраций мономеров А и
В, искомая величина.
Следует иметь в виду, что К является просто
коэффициентом пропорциональности и, как правило, не равен ev,/ev,. Он
автоматически включает поправку за счет проведения базисной
линии и пересчетные коэффициенты при использовании
по-разному выраженных значений концентрации.
Аналогично можно анализировать не только двухкомпонент-
ные сополимеры, но и трех- и четырехкомпонентные. Для трех-
компонентной системы, например, состоящей из мономеров А,
Б и В можно написать 3 уравнения:
0£К. = * ,СА/СВ; Dl/Dl = К2СЪ/СВ;
СА + СБ+СВ= 100%.
По стандартным образцам определяют К\ и Kz и затем
решают систему трех уравнений относительно Сд, Съ и Св.
II. 1.1.3. Определение толщины образца
Толщину образца, если она больше 0,02 мм, обычно
измеряют с помощью микрометра, имеющего погрешность измерения
не более ±0,005 мм. Измерение проводят в нескольких точках
и затем берут среднее значение. Желательно, чтобы головка
микрометра имела плоскую площадку, а не форму шарика, так
как в последнем случае возможна деформация образца, что
приводит к большим ошибкам. Если образец имеет толщину
менее 0,02 мм, то определение с помощью микрометра дает
большую ошибку. В этом случае толщину образца можно
определять с помощью следующих приемов.
1. Гладкие плоскопараллельные пленки дают
интерференционную картину, которую удобно наблюдать в области 1900—
2700 см-1. Зная расстояние между интерференционными
полосами &v, можно рассчитать толщину пленки d, если известен
или может быть определен другим методом показатель
преломления п:
Av = 0,5nd.
2. Выбирают полосу поглощения, неперекрывающуюся и
имеющую среднюю интенсивность при толщине образца 0,02 мм,
и строят график зависимости ее интенсивности от толщины
(определенной с помощью микрометра) в широком интервале.
Такой прием позволяет определять толщины образцов до
89
0,003 мм с погрешностью не более 20%. Он особенно
незаменим для определения толщины образца, когда пленку отливают
прямо на пластинках КВг или NaCl.
Оба метода определения толщины пленок довольно
трудоемки, так как требуют проведения большой подготовительной
работы, поэтому при получении спектров тонких пленок стараются
избежать определения толщины и часто пользуются методом
внутреннего стандарта или отношения полос (см. II. 1.1.2).
II. 1.1.4. Определение оптической плотности
Инфракрасные спектрометры, не снабженные логарифмато-
ром или ЭВМ, записывают спектр пропускания Т. Значение
оптической плотности D вычисляется только для тех частот,
которые выбраны в качестве аналитических. D определяют
обычно по таблицам, рассчитанным по формуле (II. 2) или
вычисляют на ЭВМ. Современные приборы дают спектр, в
котором интенсивность поглощения выражается через оптическую
плотность D. Иногда определяют не оптическую плотность в
максимуме полосы поглощения, а площадь полосы или
произведение jDvAv (где &v — полуширина полосы поглощения).
Однако при решении аналитических задач этот прием
используется сравнительно редко.
В значение Dv обычно вносят поправку на рассеяние и
отражение света образцом, так как даже образцы,
приготовленные из одного и того же материала, несколько различаются
между собой по этим показателям. Особенно это относится к
образцам, анализируемым в виде пленок. Чтобы учесть потери
света, не связанные с поглощением, из DVl — оптической
плотности в максимуме полосы поглощения вычитают Dv,, где v2 —
частота, при которой данный образец не имеет полос поглощения.
Говоря о значении Z)v, обычно имеют в виду исправленное
значение.
Очень часто анализ приходится вести по перекрывающимся
полосам. Наличие прибора с ЭВМ позволяет проводить
надежное и быстрое разделение полос и сводить задачу к анализу по
изолированным полосам, что должно повышать точность
анализа. В литературе, как правило, кроме нескольких статей,
используют в случае перекрывания полос метод базисных линий, т. е.
тоже разделение полос, но условное. Результаты, полученные
этим методом, часто не уступают по точности анализу по
изолированным полосам. Дать общие рекомендации по проведению
базисных линий трудно, поэтому различные варианты их
использования будут рассмотрены на конкретных примерах.
II. 1.2. Определение состава сополимеров различных классов
В литературе часто для конкретных задач приводят
уравнения типа (II. 4), связывающие отношения оптических плотностей
с отношением концентраций мономеров. Однако приводимые в
90
статьях сведения иногда недостаточны для воспроизведения
результатов: отсутствуют данные по спектральной ширине щели
и толщине образца, не всегда ясно, в каких единицах
выражена концентрация, натуральный или десятичный логарифм от
1/Т использовали авторы. К сожалению, до настоящего
времени отсутствуют единые требования к изложению материала по
количественному анализу методом ИК-спектроскопии. Однако
нам кажется, что приводить уравнения типа (П. 4) в книге
целесообразно, так как они могут быть использованы тогда, когда
нет необходимости в высокой точности определения абсолютных
значений состава сополимеров, например, при идентификации
неизвестных образцов на последнем этапе расшифровки, когда
тип сополимера уже установлен. Применение этих уравнений
при отсутствии стандартных образцов позволяет быстро
получать приближенную оценку состава сополимеров, а когда такие
обрацы имеются, достигать большой точности даже при их
небольшом числе.
В разных работах для одних и тех же полос поглощения в
одинаковых соединениях различия в волновых числах иногда
достигают 10 см-1. Чтобы не вносить путаницы в текст, для
рассматриваемой полосы выбирали то значение, которое чаще
указывается в наиболее серьезных работах, и затем
использовали во всех случаях, независимо от того, какое значение было
указано в первоисточнике.
II. 1.2.1. Гомополнмеры и сополимеры олефинов
Полиэтилен. Полиэтилен при толщине образца менее 0,04 мм
имеет полосы поглощения только в областях спектра 2800—
3000, 1450—1475 и 720 см-1, относящиеся к колебаниям СН2-
групп [34, с. 190]. Однако реальная молекула полиэтилена
помимо СН2-групп содержит небольшое число СН3 и С=С-групп,
и при толщине образца более 0,1 мм их поглощение становится
заметным. Содержание этих групп зависит от технологии
получения полиэтилена, влияет на его надмолекулярную
структуру и, следовательно, на физико-механические свойства.
Определение СН3-групп. Содержание СН3-групп
характеризует число коротких боковых цепей в молекуле полиэтилена и
поэтому является важной характеристикой, так как сильно
влияет на кристалличность полимера. Степень разветвленности
обычно выражают числом СНз-групп на 100 атомов углерода.
Наиболее удобной и часто используемой для определения
содержания СНз-групп является полоса поглощения 1378 см-1,
относящаяся к симметричным деформационным колебаниям.
Однако на эту полосу накладываются полосы поглощения
СН2-групп 1368, 1352 и 1304 см-1, что затрудняет анализ по
этой полосе и снижает его точность (рис. II. 1).
В ранних исследованиях применяли графический способ
разделения полос. Можно вычислить поправку, если определить
91
п = лСНз -t- пСН2 ш ч\
^1378 ^1378 ~ ы1378> I»'-0J
JJ к Рис. II. I. ИК-слектр поглощения
полиэтилена в области 1320 —
1400 см~~ . Разделение спектра на
составляющие полосы метолом ЭВМ.
<1,21СНз/100 С).
отношение поглощения
полиметилена, т. е.
полили дао шГ— то" " меРа- практически не
содержащего СНз-групп,
при частотах 1368 и
1378 см-1 (а = £> 1368/^1378)- Зная поглощение анализируемого
образца при 1368 см-1, можно вычислить вклад этой полосы в
поглощение СН3-групп (aD136e) и затем вычесть из поглощения
образца при 1378 см-1; Ор3783 = Аз7в — aDi368 [80]. Более
правильно для учета перекрывания полос использовать систему двух
уравнений [81]
^1378 — "1378 "•" "1378
Д1368 = ^3683 + ^3682- ("-6)
Зная контур полос 1378 и 1368 см-1, можно записать:
^368Э=К3783- (И-8)
Подставляя (П. 7) и (II. 8) в (П. 5) и (П. 6) и решая
относительно £>|3783, получаем:
С = (Di378 - aOI36e)/(l - a?). (II. 9)
Определив а по спектру полиметилена, а р методом
графического деления, можно рассчитать поглощение СН3-групп по
уравнению (II. 9).
Классическим приемом учета перекрывания полос является
компенсационный метод [82, 83]. При этом методе в пучок
сравнения помещают образец в виде клина из линейного
полиэтилена, не содержащего СН3-групп, и, передвигая его в пучке,
подбирают эмпирически такую толщину в области 1368 см-1,
чтобы полностью компенсировать поглощение СНг-групп; тогда
полосу 1378 см-1 удается выделить в чистом виде (рис. II.2).
В последнее время появились работы, в которых для выделения
полосы 1378 см-1 применяется расчет на ЭВМ с
использованием соответствующей программы [83, 84]. Это позволяет
отказаться от компенсационного метода и по существу
вернуться к первоначальному графическому разделению, но на более
высоком уровне, и сделать его независимым от природы
полиэтилена, используемого для компенсирующего клина, и
экспрессным [85].
В ранних работах показатель поглощения ei37s
определяли на основании моделей, в качестве которых использовали
нормальные углеводороды. Молярный показатель поглощения е
92
Рис. II. 2. ИК-спектр поглощения j
полиэтилена в области 1320 —
поглощения 1378 см методом
компенсации.
для нормальных
углеводородов довольно
сильно зависит от
условий получения
спектров: его значение ко- v,«*-'
леблется в интервале
от 11 до 21 л/(см-моль).
Детальные исследования, проведенные в работе [86], показали
зависимость е для полосы 1378 см-1 от характера ответвления.
Значение е уменьшается с увеличением числа (СН2)л-групп,
находящихся между СНз-группой и основной цепью, и стремится
к пределу при п ^ 3.
Учитывая большую зависимость 8 от условий получения
спектров, многие авторы предпочитают рассматривать
относительный показатель поглощения Q = еша/еша. где ек относится
к концевым СНз-группам, а ер — к СН3 в разветвлениях. Ниже
представлены полученные разными авторами значения
относительного показателя поглощения Q для полосы 1378 см-1
углеводородов с различными ветвлениями [86]:
СНз
0,65
0,55
0,53
0,37
0,41
с2Н5
0,87
0,77
0,88
0,57
0,58
С3Н7
0,94
—
—
—
—
с4н9
0,82
0,83
0,95
0,74
0,79
CsH,,
1,00
1,0
1,00
1,00
1,00
с5н13
1,00
—
1,00
1,00
1,00
Результаты разных авторов довольно значительно
отличаются друг от друга, но общая тенденция в изменении 8]378
просматривается во всех результатах. Из этих данных следует, что
определение разветвленности с использованием одного общего
коэффициента не всегда справедливо. Определение
разветвленности для ПЭВД таким образом возможно, так как для него
характерны в основном бутильные и более длинные
разветвления; для ПЭНД, в котором возможны разветвления различного
типа, использование общего коэффициента может приводить к
ошибке ±20-^30 %.
Полиэтилен является частично кристаллическим веществом,
поэтому, чтобы можно было использовать показатели
поглощения, полученные на жидких образцах, для определения в нем
СНз-групп, необходимо ИК-спектр получать для полимера в
расплавленном состоянии. При плавлении образцов
полиэтилена интенсивность полосы 1378 см-1 уменьшается
приблизительно до 0,6 от исходного значения, но полуширина полосы не
изменяется и практически равна ее полуширине в жидких
углеводородах. Это позволяет не прибегать к плавлению анализи-
93
руемого образца каждый раз, а вводить поправочный
коэффициент, заранее определенный в тех же условиях, в которых
проводится анализ [81].
На практике показатель поглощения используют в основном
только в процессе разработки методики, а в аналитических
целях обычно применяют эмпирические уравнения, связывающие
число СН3-групп на 100 атомов углерода с интенсивностью
полосы 1378 см-1. Можно привести несколько примеров таких
уравнений:
СНз/ЮОС = (0,085 ±0,003) Кпп — (0,09 ± 0,04) [85, 87],
СН3/100С = ад,з7в, где В = 0,0735* [81],
В = 0,072; 0,068 [88],
В =0,105; 0,108 [85].
K = D/d (d в см).
Очевидно, для грубых расчетов можно использовать
уравнение:
СНз/ЮОС = 0, \D/d.
Значение d может меняться от 1,0 до 0,05 мм в
зависимости от содержания СН3-групп в образце.
В некоторых работах используют отношение полос
поглощения 1378 и 1368 см-1 [89]. Такой прием позволяет избегать
точного определения толщины образца. Для построения градуи-
ровочного графика использовали углеводороды известного
строения, а спектр полиэтилена снимали в расплаве. Толщина
образца составляла 0,05 мм. Полученная зависимость
описывается уравнением:
СНз/ЮОС = 7,2D,37b/D136b - 2,25.
Абсолютная погрешность определения числа метильных
групп по этому уравнению колеблется в пределах от 0,13 до
0,35СН3/Ю0С (ПЭВД содержит от 2 до 4, а ПЭНД от 0,5 до
1,5СНз/100С).
Можно отдельно определять число бутильных и этильных
ветвлений в полиэтилене. Бутильные ветвления поглощают при
745 см-1, а этильные — при 770 см~' и их число можно
рассчитать по уравнениям [87]:
С4н9/юос = o,8tf745; С2н5/юос = о,5#77„.
Определение ненасыщенности. Полиэтилен в зависимости от
способа получения может содержать ненасыщенные связи
различного типа (табл. II. 2, рис. II. 3). Обычно для анализа
используют образцы толщиной 0,5—1,0 мм. Определению цис-ви-
ниленовых групп мешает перекрывание характерных для него
полос поглощения с дублетом интенсивных полос 720—730 см-1,
а определению винилиденовых групп мешает полоса поглоще-
* Значение В из работы [81] пересчитано в единицах, принятых в
остальных работах.
94
Таблица II. 2. Характеристические полосы и показатели
поглощения неплоских колебаний СН-группы при двойной связи
в углеводородах
Тнп связи
Формула
Винильная
Винилиденовая
т/>а«с-Виниленовая
tfuc-Виниленовая
Н
R—С=СН2
R-C-R
сн2
NC=C/
н/ \r
ч /R
хс=с:
н/ \н
992, 910
888
965
720-790
45 и 125
150
ПО
Слабая
ния 894 см-1, которая обусловлена наличием бутильных
разветвлений. Чтобы повысить точность определения винилидено-
вых групп, можно использовать метод компенсации. Для этого
анализируемый образец бромируют и после этого используют
для компенсации полосы 894 см-1, помещая в пучок сравнения.
Бромирование проводят в запаянных ампулах в суспензии над
жидким бромом при температуре 20°С [90] или 5 %-ным
раствором брома в тетрахлориде метана [82]. Бромирование
длится не менее 2 ч и контролируется по ИК-спектру. После бро-
мирования образцы тщательно отмывают и сушат в вакууме.
Образцы, имеющие высокую степень кристалличности, требуют
большего времени для их полного бромирования.
Использование метода компенсации позволяет определять винилиденовые
группы с чувствительностью 0,005С=С/100С, а тра«с-виниле-
новые— 0,01С=С/100С.
В работе [89] предлагается проводить определение
С=С/1000С винилиденовых
связей по эмпирическому
уравнению:
С = С/1000С = 0,0266D888/d,
где d — толщина образца, мм.
Базисную линию проводят
между точками при 840 и
1000 см-1. Относительная
погрешность определения по этому
уравнению больше, чем при исполь-
Рис. П. 3. ИК-спектР гоглощеиин полиэтилена
в области 800 — 1100 см-1;
/ —полиметнлен; 2— ПЭНД; 3 — ПЭВД.
7700
1000
300 600
95
зовании бронированных образцов для компенсационного метода,
и составляет 25—32 %•
Кристалличность влияет на интенсивность полос
поглощения, поэтому при использовании аморфных модельных
соединений нужно получать спектры анализируемых образцов в
расплаве или вводить поправочный коэффициент. Общее
содержание С=С-связей можно определять по полосе 1640 см-1, но
различные типы С = С-связей имеют разные показатели
поглощения и эту полосу рекомендуется использовать для образцов,
содержащих преимущественно один тип связи.
Полипропилен. Полимер, получаемый в промышленности,
содержит изотактическую и атактическую фракции. Атактиче-
ский полипропилен обладает плохими механическими
свойствами и является нежелательной примесью. Поэтому было
выполнено довольно большое число работ, посвященных изучению и
количественному определению микротактичности
полипропилена [91, 92].
Если сравнивать ИК-спектры изотактического и атактиче-
ского полипропилена (рис. II.4), то можно выделить полосы,
которые сильно изменяются по интенсивности, и такие, которые
не меняются. К первым принадлежат полосы 1168, 995 и
842 см-1, ко вторым—1460 и 973 см-1. Первые обычно
используют для оценки микротактичности, а вторые в качестве
внутреннего стандарта. Например, используют отношение
/)995/-Оэ7з. Единый метод калибровки отсутствует, поэтому
определение микротактичности другими, не спектральными
методами не всегда согласуется с данными ИКС, полученными
на основании анализа сополимера по различным полосам по-
D k
в 4
1800 1000 1100 1200 1030 800 С00
Рис. II. 4. ИК-спектры изотактического (а) и атактического {б) полипропилена.
96
1600
Рис. II. 5. ИК-спектр поглощения сополимера эти- ** к 1462
лена с пропиленом в области 1200—1600 см- .
Аналитические полосы 1462 и I37S см~ (стрелками
отмечены точки, между которыми проводятся
базисные линии).
глощения. Например, результаты
определения содержания изотакти-
ческой фракции по температуре
плавления хорошо согласуются только
с данными, полученными по полосе
1168 см-1. Это объясняется тем, что
каждый метод, каждая полоса
связаны с наличием изотактической
спирали не менее какой-то
определенной длины. По данным работы
[93] оптическая плотность полосы 998 см-1 пропорциональна
концентрации изотактических спиралей, содержащих более
12 мономерных звеньев, а оптическая плотность полосы 973 см-1
характеризует регулярные последовательности, содержащие
более 4 мономерных звеньев.
Сополимер этилена с пропиленом. При анализе состава
сополимера этилена с пропиленом (СЭП) при малых
содержаниях последнего, очевидно, можно использовать те же полосы
поглощения и приемы, которые были описаны для определения
степени разветвленности полиэтилена. Задача даже упрощается,
так как длина боковой цепи не вызывает сомнения и поэтому
не возникает вопроса, какое модельное соединение надо
использовать при построении градуировочного графика.
Однако при увеличении в сополимере содержания
пропилена интенсивность полосы поглощения 1378 см-1 возрастает,
приходится работать с тонкими пленками, что приводит к
большим ошибкам в определении толщины образца, а
следовательно, и состава. Кроме того, имеются данные, что показатель
поглощения в этом случае зависит от характера стереорегуляр-
ности блоков пропилена [94].
Для определения состава СЭП можно использовать
различные пары полос поглощения: 720 и 1159 см-1, 1155 и
4310 см-1, 1378 и 1462 см-', 5917 и 5682 сиг' [94—96].
Однако наибольшее распространение получил метод анализа по
полосам поглощения 1462 и 1378 см-1 (рис. II. 5). Образцы
можно готовить в виде раствора в тетрахлорметане, если
полимер растворим, или в виде пленки. Полученные разными
авторами значения Z)i37e/Di462 при одинаковом содержании
пропилена в сополимере могут существенно различаться [97, с. 104]:
Доля пропилена в СЭП, %
^1378/Ol4S2
10
0,21
0,12
—
0,15
0,264
20
0,35
0,24
—
0,28
0,395
30
0,47
0,38
—
0,41
0,521
40
0,60
0,52
0,44
0,53
0,647
50
0,70
0,68
0,53
0,65
—
4 Зак. 632
97
Построение градуировочных кривых для растворимых
образцов обычно проводят по данным ЯМР, а для нерастворимых —
по стандартным образцам, специально синтезированным и
содержащим один из сомономеров с мечеными атомами, или по
анализу состава газовой смеси до и после полимеризации при
малой конверсии.
В тех случаях, когда сополимер был подвергнут
вулканизации, из него невозможно приготовить образец для получения
спектра, тогда можно подвергнуть образец пиролизу при 450—
500 °С. Отношение поглощения образующихся при пиролизе
винильных групп (909 см-1) к поглощению винилиденовых
групп может быть использовано для определения состава СЭП
[98].
СЭП так же, как и полиэтилен, содержит С=С-связи.
Определение их количества представляет интерес как с точки
зрения изучения механизма, так и для качественного анализа,
и проводится по тем же полосам, что и в случае полиэтилена.
Терполимеры на основе этилена и пропилена. В качестве
третьего компонента при полимеризации этилена с пропиленом
используют дициклопентадиен, гексен-1, бутен-1, 5-этилиден-2-
норборнен. Определение состава сополимера этилен —
пропилен— гексен-1 проводят по' полосам поглощения 730 и
1150 см-1 [99]. Для расчета используют уравнения:
Сп = О,15207зо - 0,064О'150 + 0,0335,
Сг = 0,031^150 + 0,050О730 + 0,094,
Сэ = 100 — (Сп + Сг),
где Сп, Сг и Сэ — содержание в сополимере пропилена, гексена-1 и этилена
соответственно; D' — оптическая плотность при 730 и 1150 см-1, деленная на
оптическую плотность при 4310 см-1, для введения поправки на толщину
образца.
Методика позволяет определять состав тройного сополимера
при содержании звеньев этилена 35—60%, пропилена 28—50%
и гексена-1,4— 18 %. Аналогично для определения состава эти-
ленпропиленбутадиенового сополимера можно использовать
следующие уравнения [100]:
Сэ=17.120732; Сб = 29,330765-1,730722, Сп = 100 - (Сэ + Сб),
где Сэ, Се и Сп — содержание этилена, бутадиена и пропилена
соответственно; D' = D/Duio.
Для определения состава сополимера этилен— пропилен — 5-
этилиден-2-нонборнен проводили пиролиз образца с
последующим ИКС-анализом пиролизата по полосе поглощения
1615 см-1. Методика позволяет определять от 2 до 8%
третьего сомономера [101].
Получение стандартных образцов известного состава для
терполимеров представляет значительные трудности, а исполь-
98
зование механических смесей гомополимеров может приводить
к существенным ошибкам. Поэтому представляет интерес опыт
использования в качестве стандартных образцов смеси двух
сополимеров [99, 100]. Например, в работе [99] использовали
механическую смесь сополимеров этилена с пропиленом и
пропилена с гексеном-1. Массовую долю акрилонитрила Са (в %),
привитого на сополимер этилена с пропиленом (СКЭП) или на
тройной сополимер этиленпропиленового каучука (СКЭПТ),
можно определять по уравнению:
С а = 7,20224б/^43ю-
Для проверки правильности коэффициента 7,2 можно
использовать в качестве стандартных образцов механическую
смесь исходного сополимера и полиакрилонитрила.
Сополимеры этилена с бутеном-1 и другими а-олефинами.
Для определения состава сополимеров этилена с бутеном-1
используют полосы поглощения 1378 и 770 см-1.
При анализе по полосе 1378 см-1 полосу поглощения
1368 см-1 вычитают из спектра образца, помещая в пучок
сравнения клин из полиэтилена, полученного в тех же условиях,
что и сополимер. При спектральной ширине щели 6 см-1
число С2Н5-групп на 100 атомов углерода может быть выражено
следующим уравнением [102]:
C2H5/100C = (D137e/rf)-41.102,
где d — толщина образца, мкм.
Результаты, полученные по этой формуле, дают хорошее
совпадение с результатами химического метода. Можно
использовать и отношение интенсивностей этой полосы и полосы 1368 см-1
^i37s/^i368 [81]. Можно также вести анализ по полосе 770 см-1
[87, 102], относящейся к маятниковым колебаниям СНг-группы
в этильном ответвлении. Показатель поглощения для этой
полосы равен (4,2 ± 0,2 см-1) при спектральной ширине щели
3,2 см-1. Полоса 770 см-1 имеет некоторые преимущества перед
полосой поглощения 1378 см-1: она менее перекрыта соседней
полосой и имеет полуширину 25 см-1, в то время как полуширина
полосы 1378 см-1 — 10 см-1. Вследствие этого контур полосы
770 см-1 менее чувствителен к искажениям, вносимым оптической
системой прибора, и поэтому значение оптической плотности
должно быть лучше воспроизводимо.
Массовую долю бутена-1 СБ (в %) или молярную долю
Cg (в %) можно определять также по уравнениям [103]:
С6 = (1,2950^/d) ± 1,94; D5910/D5680 = 0,018^ ± 0,01;
Д2959/Л2854 = °'023Сб±0'02-
4*
99
Таблица II. 3. Основные полосы поглощения в спектре СЭВ [105]
.-1
Отнесение полос
3460 С = 0 валентные колебания (1 обертон)
2678 СН2 валентные колебания
1740 С = 0 валентные колебания
1465 СН2 деформационные колебания
1250 С—О валентные колебания
1020 COOR ножничные деформационные колебания
720 — (СНг)п, я ^ 4 маятниковые колебания
610 COOR — веерные колебания
Молярную долю 4-метилпентена См (в %) в сополимере
этилена с 4-метилпентеном (при содержании последнего 0—
20 %) можно определить по уравнению [103]:
D92i/D770 = 0,038СМ.
Все сказанное относительно применения полосы поглощения
1378 см-1 или отношения Аз7в/Азбв можно распространить
и на другие сополимеры этилена с а-олефинами. В случае,
если содержание сомономера — сс-олефина мало, а
молекулярная масса сополимера невелика, на результаты анализа по
полосе 1378 см-1 могут оказывать влияние концевые СН3-группы,
приводя к завышению результатов. В работе [104] предложен
способ введения поправки для такого случая.
Сополимер этилена с винилацетатом. При анализе состава
сополимера этилена с винилацетатом (СЭВ) используются почти
все полосы поглощения, имеющиеся в спектре этого
сополимера (табл. II. 3, рис. II. 6). Чаще применяют отношение интен-
сивностей полос, причем выбор различных вариантов
достаточно большой. Каждый из авторов приводит аргументы в пользу
своего варианта и, так как обычно не хватает информации для
объективного сопоставления этих аргументов, сделать выбор
трудно (табл. И. 4).
' ' ■ L__l 1 1 L_
■ ■ ■ ■
3600 2800 2000
1600
1200
800
V,cm-<
Рис. II. 6. HK-спектр поглощения сополимера вннилацетата с этиленом (молярная доля
этилена 50 %) прн толщине образца 0,01 мм (/) и 0,1 мм (2).
100
Таблица II. 4. Аналитические полосы для определения
состава СЭВ
V, см , винил-
ацетат/этилен
Молярная
доля
винил-
ацетата.
%
Внд образца
Толщина
слоя, мм
Относительная
погрешность, %
графический
источник
1433/2925
1372/2925
1372/1465
1740/1170
1740/1465
720/610
3460/2678
610/2678
1020/2678
3460
5-60
30-85
80—95
0-10
0-20
0-15
2—25
—
—
4-25
Раствор в ССЦ
10 г/л
То же
Пленка
Таблетка КВг
Спектр МНПВО
—
Пленка
■»
>
»
0,25
0,25
0,01
—
—
—
0,05±0,02
0,05±0,02
0,05±0,02
0,4-0,5
10
10
10
—
—
15
—
—
—
10
[106
[106
[106
[107
[108
[109]
1105, ПО]
[105, ПО]
[105]
[111]
В литературе нет единого мнения в пользу того или другого
варианта.
Наиболее распространены и имеют промышленное
значение СЭП с молярной долей винилацетата от 1 до 20%,
поэтому большинство методик разработаны именно для этой
области.
При выборе полос в первую очередь следует
руководствоваться необходимым для анализа интервалом концентраций и
возможностью приготовления образцов. Однако нам кажется,
что полосы поглощения 1740 и 1250 см-1, на которые сильно
влияет чередование звеньев, надо использовать с большой
осторожностью. Стандартные образцы необходимо получать по той
же технологии, что и анализируемые. Состав стандартных
образцов можно определять методом омыления (см. раздел II. 4),
ЯМР или элементного анализа (см. раздел II. 3). Для СЭВ с
небольшим содержанием винилацетата, можно определять раз-
ветвленность этиленовой части. Для этого сополимер
предварительно омыляют, а затем анализируют по полосе поглощения
1378 см-'.
Другие сополимеры этилена. Сополимеры этилена с
другими мономерами, кроме рассмотренных выше, имеют меньшее
практическое значение, поэтому им посвящается меньше
исследований. Ниже приведены основные аналитические полосы для
этих сополимеров:
V, СМ_
Акрилонитрил 2234
Мономеры со сложноэфирной группировкой (малеи- 1725 и 1170
наты, акрилаты, метакрнлаты) [112, 113]
Акриловая кислота 1710
Виниловый спирт [114] 3340 и 2926
101
II. 1.2.2. Сополимеры стирола
Сополимер стирола с акрилонитрилом. Анализ сополимера
стирола с акрилонитрилом на первый взгляд не представляет
трудностей, так как у акрилонитрила имеется хорошо
изолированная и, как правило, никогда не прекрывающаяся полоса
2247 см-1, обусловленная валентными колебаниями C = N-rpyn-
пы, а спектр полистирольной части имеет большое число полос,
удобных для анализа и мало чувствительных к окружению.
Однако, как показывает эксперимент, частота колебаний
группы C = N меняется в зависимости от состава сополимера. Поли-
акрилонитрил имеет полосу поглощения 2240 см~', а
сополимер с небольшой массовой долей акрилонитрила (до 30%) —
2234 см-1; при большем содержании акрилонитрила полоса
поглощения занимает промежуточное положение. Следует
ожидать, что при этом не будет сохраняться прямо
пропорциональная зависимость между содержанием акрилонитрила и
интенсивностью полосы. В связи с этим для построения градуиро-
вочного графика нельзя использовать смеси гомополимеров, а
необходимо иметь стандартные образцы с составом, близким к;
определяемому. Обычно состав стандартных образцов
определяют методом Кьельдаля (см. раздел II. 3). Следует также иметь,
в виду, что полуширина полосы поглощения 2234 см-1 равна
14 см-1, поэтому спектральная ширина щели должна быть
меньше, так как в противном случае возможна значительна»
ошибка при использовании литературных данных.
Для определения состава сополимеров можно использовать»
отношения оптических плотностей при следующих волновых
числах: 2234 и 1875; 2234 и 1603; 3061 и 2234; 2234 и 540 см-'
[74, с. 48; 115, 116].
Для анализа по полосам поглощения 2234 и 1875 см-1
отливают пленку толщиной 0,03—0,04 мм и снимают спектр в
интервале частот 1800—2400 см~'. Базисные линии проводят,
как показано на рис. П. 7. Определяют отношение D22inlDmb:
п _ пмакс _ гчО . п _ гчмакс _ п0
"2247 — "2247 "2247' "1875 — "1875 "1875-
где D0 — оптическая плотность, соответствующая базисной линии при данном'
волновом числе.
Строят градуировочный график в координатах ^2234/^1875 —
массовая доля акрилонитрила в образце (в %). Состав
стандартных образцов определяют методом Кьельдаля.
Относительная погрешность 10%. При анализе по полосам поглощения
3061 и 2234 см-1 отношение D3oe\/D2234 связано с массовой долей:
акрилонитрила (АН) в сополимере СдН следующим уравнением:
САН = 0,362 (0,032 + Dml/Dom).
Можно использовать также область поглощения обертонов,,
беря отношение О597о/^52зо. Полоса поглощения 5970 см-1
является обертоном колебаний фенильного кольца, а полоса
5230 см-1 — комбинацией валентных колебаний C^N-связи с
102
т,%
20г 2234
60
so
1875
/vk/
2400 2200 2000
1900 180C
6000
5000
4000
v,CM~'
Рис. II. 7. ИК-спектр поглощении сополимера стирола с акрилонитрилом в области
1800 — 2400 см-1. Аналитические полосы 2234 и 1875 см-1.
Рис. U.S. ИК-спектр поглощения полиакрилонитрила (/), сополимера стирола с 26 % акри-
лоннтрила (2) и полистирола (3) в области 4000—6000 см . Аналитические полосы 5070
и 5230 см-1.
асимметричными валентными колебаниями СН2-группы. На
рис. П. 8 показан наилучший способ проведения базисной линии
для этих полос [117]. Массовая доля акрилонитрила в
сополимере Сдн (в %) связана с отношением D$97o/Ds2w следующим
уравнением:
0,0206 САН +0,0882Z)5970/D523o= 1.
В том случае, когда пленку приготовить трудно, можно
проводить анализ пиролизата [118]. Для этого образец подвергают
пиролизу при температуре 545—640 °С и снимают спектр
пиролизата, в зависимости от состава используя для анализа
частоты 1016, 1022, 3020 и 2234 см-'.
Сополимеры стирола с бутадиеном. Наиболее
распространенным сополимером стирола с бутадиеном является так
называемый ударопрочный полистирол (УПС) — блоксополимер,
получаемый прививкой стирола на полибутадиеновый каучук. Для
анализа состава этого сополимера используют отношение ин-
тенсивностей полос поглощения 967 см-1 (внеплоскостные
деформационные СН-колебания в группе гранс-СН = СН) и
1027 см-1 (СН-колебания бензольного кольца) [74, с. 51].
Пленки толщиной 0,02—0,04 мм отливают из 2 %-ного раствора
сополимера в бензоле
непосредственно на пластинку NaCl.
Снимают ИК-спектры в интервале
частот 800—1200 см-1, проводят
базисную линию, как это
показано на рис. П. 9, и определяют
отношение оптических
плотностей при 967 и 1027 см-1
Т,%
20
40
60
Рис. II. 9. ИК-спектр поглощения
ударопрочного полистирола в области 800 —1200 см- .
Аналитические полосы 067 и 1027 см-1.
80
1027
367
1200
1000
800
v,cm-
103
Aw/A 027-
'967"
пмакс nU .
^967 и9ЪТ
п _ пмакс _ п0
*Л027 — ^1027 ^1027'
Градуировочный график строят в координатах: D$67/DlQ27 —
отношение массовой доли каучука Ск (в %) к массовой доле
полистирола ССт (в %) (рис. 11.10). Относительная
погрешность определения 5—7 %.
Можно использовать для аналитических целей и отношение
других пар полос, например Дбзэ/^боь Dg67/D767. Для
последней пары полос было получено уравнение:
ССт = 94,63 - 29,485D957/D767.
Для УПС в качестве стандартных образцов для
градуировки можно использовать механические смеси полистирола и
полибутадиена. Однако надо иметь в виду, что полибутадиен при
этом должен быть той же марки, на которой был получен
анализируемый образец. Это вызвано тем, что все полосы
поглощения, используемые для анализа, связаны с группировкой,
содержащей двойную связь, а в полибутадиене, как известно,
эти связи могут быть трех типов и соотношение их от марки к
марке может сильно изменяться. По ИК-спектрам УПС можно
определить только содержание гра«с-СН=СН или СН=СН2-
групп, а содержание групп цис-СН=СН не определяется,
поэтому, не зная состава полибутадиена или не имея его в качестве
стандартного образца, нельзя проводить анализ. В этом случае
рекомендуется пользоваться химическим методом, который
позволяет определять сумму двойных связей (см. раздел П. 4).
Анализ состава статистических сополимеров стирола с
бутадиеном также рекомендуется проводить с использованием
стандартных образцов, состав которых определен химическим
методом.
АБС-пластики. АБС-пластики, получаемые прививкой
стирола и акрилонитрила на полибутадиен, можно анализировать
по тем же полосам поглощения, которые используются при
анализе сополимеров стирола с акрилонитрилом и УПС: 2234 см-*
Рис. П. 10. Градуировочиые графики для определения концентрации стирола и
бутадиенового каучука в ударопрочном полистироле для разных интервалов концентраций.
104
Таблица II. 5. Аналитические полосы поглощения для
различных структур в сополимерах стирола с дивинилбензолом [122]
Анализируемая группа
Двойные связи
Монозамещенные фенильные
кольца
Дизамещенные феннльные
кольца, яара-положение
Дизамещенные феннльные
кольца, л«ета-положение
V, СМ '
1630
990
1493
1028
760
1510
838
795
Волновые числа для базисных
ЛИНИЙ, V, СМ
1555 н 1660 (рис.
1043 и 1005 (рис.
1395 и 1525 (рнс.
1043 и 1005 (рис.
727 и 813 (рнс.
1395 и 1525 (рис.
727 н 813 (рнс.
727 и 813 (рис.
-1
11.11,
11.11,
11.11,
11.11,
11.11,
11.11,
11.11,
11.11,
а)
6)
а)
б)
в)
а)
в)
в)
(акрилонитрил), 967 см-1 (бутадиен), 1027 и 1875 (или
1960) см-1 (стирол) [74, с. 53; 115; 119].
Стандартные образцы можно готовить, используя
механическую смесь полибутадиена с сополимером стирола с акрило-
нитрилом. Полибутадиен так же, как и при анализе УПС,
следует брать той же марки, что и в анализируемых образцах.
Сополимеры стирола с пропиленом и изобутиленом.
Сополимеры стирола с пропиленом анализируют по полосам
поглощения 1601 см-1 (стирол) и 1378 см-1 (пропилен) [120].
Отношение оптических плотностей, определенное при этих
-DA
а
i
630
■
1 .
I
сэ
151
1
JL
. i
1700
J)'
TS00
1500 1400
V,CM->
1100
1000 900
v,cm-i
6
I
8
♦
°> 1 I
N I 1
-M
« t J
J A/ h
/l / \J ^*
J \_/ _—-"*"
i i
Рис. 11.11. ИК-спектры поглощения
сополимера стирола с дивинилбензолом в области
1400 — 1700 см-1 (а), 900 — 1150 см-1 {б) и
700 — 950 см-1 (в).
900
800 700
v,cm-'
105
волновых числах, связано с молярной долей пропилена тп
следующим уравнением:
D 137в/^1б01 = 0,235 + 1,80тп (1 - т„).
При расчете учитывается поглощение стирола при 1378 см-1.
Если сополимер содержит много пропилена, то для его
определения можно использовать полосу поглощения 973 см-1.
Тогда уравнение имеет вид:
Я.бо./А>7з = 3,18[1/(тп-1)].
Состав сополимера стирола с изобутиленом определяют по
отношению полос поглощения 1492 и 1350 см-1 [121].
Попытки разработать методику определения состава блоксополи-
мера, представляющего собой стирол привитой на сополимер
этилена с пропиленом (СКЭП), к успеху не привели.
Сополимер стирола с дивинилбензолом. В результате
полимеризации стирола с дивинилбензолом, образуются различные
структуры, количественная оценка которых позволяет лучше
понять механизм полимеризации и оценить степень сшивания,
что является важнейшей характеристикой продукта. Детальный
анализ возможностей метода ИКС для решения этого вопроса
проведен в работе [122]. Основные рекомендации авторов по
выбору полос поглощения для анализа структур разного типа
приведены в табл. П. 5. Проведение базисных линий указано
на рис. 11.11. Результаты, полученные по разным полосам,,
удовлетворительно совпадают между собой.
II. 1.2.3. Сополимеры винилацетата
Сополимер винилацетата с винилхлоридом. Наиболее
простым способом определения состава сополимеров винилацетата
с винилхлоридом является определение содержания
винилацетата по интенсивности полосы поглощения 1740 см-1, используя
метод абсолютной градуировки. Из-за большой интенсивности
полосы поглощения 1740 см-1 работать с пленками нельзя, так
как их толщина должна быть 3—5 мкм даже при небольших
содержаниях винилацетата. Поэтому приходится анализировать
образцы в виде растворов в хлороформе или тетрахлорметане.
Абсолютная погрешность анализа 0,5% [123]. Иногда
применяют метод внутреннего стандарта, в качестве которого
используют, например, полосу поглощения 1425 см-1. Для
определения содержания винилацетата от 2 до 12 % можно применять
отношение D]7Ao/D\A25, приготовляя образцы в виде тонких
пленок или таблеток КВг.
В ближней ИК-области имеется полоса поглощения
4700 см-1, которая также характерна для ацетатных групп.
Вести анализ по ней можно, используя пленки толщиной 20—
50 мкм, которая измеряется с достаточной точностью.
106
Если сополимер винилацетата подвергнуть частичному
омылению, то получается тройной сополимер, состоящий из звеньев
винилацетата, винилхлорида и винилового спирта. Анализ
винилацетата можно проводить по полосе поглощения 1740 см-1,
винилспиртовых групп — по полосе 3600 см-1. Образцы
исследовали в виде 0,5—3%-ных растворов в метиленхлориде,
толщина слоя 1 мм. При массовой доле звеньев винилового
спирта 0,5—7 % наблюдается линейная зависимость между этой
величиной и оптической плотностью полосы поглощения
.3600 см-1.
Сополимер винилацетата с виниловым спиртом. Состав
сополимера винилацетата (ВА) с виниловым спиртом (ВС),
получаемого при частичном омылении поливинилацетата,
определяют по полосам поглощения 1740 см-1 (ВА) и 850 см-1 (ВС)
(рис. 11.12) [124]. Толщина пленки 0,01—0,015 мм. Массовую
долю винилацетата СВА (в'%) рассчитывают по формуле:
СВА = -1,81 + 12,08£1740/Я850.
Состав стандартных образцов определяют методом
щелочного омыления (см. раздел II. 4). Анализируемый интервал
концентраций от 1 до 40%. Относительная погрешность
определения 2—2,5 %.
Надо иметь в виду, что полоса поглощения карбонильных
групп в сополимерах винилацетата с виниловым спиртом имеет
частоту 1740 см-1 только тогда, когда винилацетатные звенья
в сополимере не соседствуют с гидроксильными группами, а
расположены блоками. Если при омылении образуется
статистический сополимер, то с увеличением степени омыления в
спектре сополимера появляется полоса поглощения 1710 см-1,
1740
8оо
1200
1Б00
V,CM'
1200
1100
1000 900
Рис.
12. ИК-спектр поглощения поливинилового спирта, содержащего ацетатные группы.
Аналитические полосы 850 и 1740 см (толщина образца 0,01 мм).
Рис. II. 13. ИК-спектр поглощении сополимера винилацетата с бутилакрилатом
(молекулярное соотношение 1:1). 1,5 %-ный раствор в метиленхлориде, толщина слоя 0,26 мм.
Аналитические полосы 1025 и 1170 см- .
107
обусловленная наличием внутримолекулярной водородной связи
между группами С=0 и О—Н:
Н Н
I I
~ сн2—с—сн2—с—сн2~
I I
0 о
1 I
с=о. • • н
Сополимеры винилацетата с винилпирролидоном. Состав
сополимеров винилацетата с винилпирролидоном определяли [106]
по отношению интенсивностей полос поглощения 1740 см-1
(винилацетат) и 1672 см-1 (винилпирролидон). Для построения
градуировочного графика использовали механическую смесь
гомополимеров. Снимали спектры образцов в виде растворов
в дихлорэтане с концентрацией 5 г/л толщина слоя 0,1 мм.
Интервал анализируемых концентраций винилацетата 0—80%.
Можно использовать для анализа также отношение
интенсивностей полос поглощения 1250 и 1195 см-1.
Сополимеры винилацетата с акрилатами. Для определения
состава сополимеров винилацетата с любым акрилатом (Ак)
могут быть использованы одни и те же полосы поглощения:
1025 см-1 (ВА) и 1170 см-1 (Ак) (рис. 11.13). Градуировоч-
ные графики для молярного состава всех акрилатов совпадают
[106] (рис. II. 14). Если строить градуировочный график в
координатах Сдн/Сва — -Dak/Dba, to он будет иметь линейный
характер до молярной доли акрилата 50%. График, построенный
в координатах lg D\\7o/Dm5 — молярная доля акрилата (в %),.
будет линейным в интервале концентраций от 0 до 70 %.
Сополимеры винилацетата с малеинатами. Для малеинатов
имеется единственная удобная для анализа полоса поглощения
%2
0,8
0,4
0
0,4
0,8
/Л
■ х/\
уУ'
' S
11111
0 20 40 60 80 100 0 20 40 60 80 100
Молярная Золя второго мономера, %
\
Рис. II. 14. Градунровочные графики для определении состава сополимеров винилацетата-
(первый мономер) с этиленом (/—3), дибутилмалеииатом (4), винилпирролидоном (5)-,
бутил- и атилгексилакрилатами (6):
Кривая
2925 2925 1372 1672 1672 117J
1433 1372 1465 1020 1735
1020
108
1170 см-1. Для винилацетата используют полосы поглощения
1740, 1020 и 1240 см-1. Графики, построенные в координатах
Dza/Du7o — молярная доля винилацетата (в %), совпадают
для всех малеинатов [106].
II. 1.2.4. Гомополимеры и сополимеры фтормономеров
Политетрафторэтилен. Молекула политетрафторэтилена
(ПТФЭ) не содержит разветвлений, так как передача цепи при
полимеризации фторолефинов невозможна. Метод ИК-спектро-
скопии используется преимущественно для определения степени
кристалличности ПТФЭ по интенсивности полосы поглощения
аморфной структуры 780 см-1 [125]. Градуировочный график
строили с использованием данных рентгеновского рассеяния.
Для низкомолекулярных теломеров тетрафторэтилена
(ТФЭ) удается определять среднечисленную молекулярную
массу по полосам поглощения, характерным для концевых
групп, образовавшихся при взаимодействии растущей
макромолекулы с молекулой растворителя [126]. Содержание концевых
групп, образовавшихся при полимеризации ТФЭ в хлорсодер-
жащих растворителях и в диэтиловом эфире, определяют для
—CF2C1 по полосе 960 см-1, для групп —СН2—О—С2Н5 по
полосе 2970 см-1. В качестве внутреннего стандарта
используют полосу первого обертона валентных колебаний CF2 —
2350 см-1. Массовые доли водорода Сн (в %) и хлора СС\
(в °/о) определяют по формулам:
Сн = 0,5D2970/D2350; СС1 = 0,74D960/D2350.
Зная содержание Н и С1, можно определить число
концевых групп и, следовательно, среднечисленную молекулярную
массу.
Сополимеры ТФЭ с полностью фторированными
разветвленными мономерами — гексафторпропиленом, алкилвиниловыми
эфирами, перфторвинилсульфонилфторидом и перфторметил-
1,3-диоксаланом. В спектрах всех перечисленных сополимеров
имеются полосы, интенсивность которых изменяется прямо
пропорционально содержанию сомономера в полимерной цепи.
Состав сополимеров ТФЭ с гексафторпропиленом (ГФП)
определяют по полосе поглощения 985 см-1, характерной для звеньев
ГФП в этих сополимерах. В работе [127] рекомендуется
использовать отношение интенсивности этой полосы к
интенсивности полосы 2350 см-1 ПТФЭ (рис. II. 15):
СГФП = 4>5D98d/D2350-
Коэффициент пропорциональности был определен по
данным материального баланса при полимеризации. Эти
результаты получили дальнейшее подтверждение при сопоставлении
с данными ЯМР и пиролиза.
109
Состав сополимеров ТФЭ со следующими мономерами
CF2 = CF—О—CF3; CF2 = CF—S02F;
/0-CF2
CF2=CF( I
X)-CF2
определяют по полосам 890, 965 и 1005 см-1 соответственно,
характерным для этих эфиров. В некоторых случаях
интенсивность полос поглощения концевых групп оказывается
достаточной для определения среднечисленной молекулярной массы
перечисленных сополимеров. Типичные для полностью
фторированных полимеров концевые группы—-карбоксильная и фтор-
ангидридная, имеющие полосы поглощения 1780 см-1
(характерно для всех перфторкарбоновых кислот) и 1880 см-1
соответственно. Анализ проводят на образцах толщиной около
0,25 мм.
Поливинилиденфторид. Характерной особенностью поливи-
нилиденфторида (ПВДФ) является кристаллический
полиморфизм, связанный со способностью каждой конформации
макромолекулы кристаллизоваться в собственной ячейке со своими
параметрами. Специфические применения ПВДФ в качестве
пиро- и пьезоэлектриков, электретов потребовали развития
аналитических методов определения типа кристаллической фазы
(по данным рентгеновского рассеяния) и конформации
молекулы.
Основные кристаллические фазы полимера имеют
следующие конформации молекул:
а - (TGTG')a, Р - Та, У- (TTTG)n,
где Г, С и С обозначают транс- и гош-формы с углами поворота С—С-свя-
зей относительно предыдущих +180°, +120° и —120° соответственно.
Для количественной оценки соотношения долей фаз
а/(Р + 7) рекомендуется использовать отношение интенсивно-
I 1 1 i i 1
500 1000 1500 2000 3000
Рис. II. 16. ИК-спектр поглощения сополимера тетрафторатилена с генсафторпропиленом
в области 600 — 3000 см-1. Аналитические полосы 985 и 2360 см-1; (толщина образца 0,03 мм).
ПО
Рис. II. 16. Ик-спектр поглощения поливинилиденфто-
рида в области 400—650 см- . Аналитические полосы
дли количественной оценки соотношения фаз °/(p + v)
630 и 510 см- ; толщина образца 0,03 мм.
стей полос .D530/D510, определяемое
для пленок ПВДФ толщиной около
0,03 мм; р-фаза характеризуется
также полосой 410 см-1 (рис. II. 16).
Сополимеры этилена с ТФЭ и три-
фторхлорэтиленом. Для
рассмотренных выше сополимеров ТФЭ с
полностью фторированными мономерами
характерна линейная зависимость
интенсивности полос поглощения от
состава, за исключением полос,
чувствительных к кристалличности. В то же
время спектры сополимеров ТФЭ с во-
дородсодержащими фтормономерами сложным образом зависят
от состава [128]. Так, в спектрах сополимеров этилена с ТФЭ
(рис. II. 17) и трифторхлорэтиленом (ТФХЭ) интенсивность всех
полос поглощения нелинейно изменяется с составом (рис. II. 18).
Такая особенность спектров, с одной стороны, осложняет
определение состава фторводородсодержащих сополимеров, с
другой стороны, свидетельствует о большой чувствительности
спектра к распределению мономерных звеньев в цепи.
В работе [129] было предложено использовать отношение
оптических плотностей .D2950/-D2973 для характеристики «блоч-
ности» этиленовых звеньев в сополимерах этилена с ТФЭ и
ТФХЭ. В работе [130] показано, что графики изменения интен-
735 725
900
вОО 700
832см-1
\
770
845т-
40 50 ВО 70
Молярная доля этилена, %
Рис. II. 17. ИК-спектр поглощения сополимера этилена с тетрафторэтиленом в области
700 — 000 см"
с различным молярным содержанием звеньев этилена (толщина образцов
0,04 мм).
Рис. II. 18. Зависимость интенсивности полос поглощения в сополимере этилена с
тетрафторэтиленом от содержания этиленовых звеньев.
111
сивности полос в спектрах сополимеров этилена с ТФЭ близки
к кривым изменения содержания различных
последовательностей мономерных звеньев в сополимерах, рассчитанным по
формулам Медведева [131] для каждого состава сополимера с
использованием произведения констант сополимеризации. Это
позволило дать предварительное отнесение полос поглощения,
уточненное затем на основе теоретического анализа
колебательных спектров сополимеров [128]. Наиболее удобными полосами
для определения «блоков» этиленовых и тетрафторэтиленовых
звеньев в сополимере являются полосы 890 и 845 см-1
соответственно, а для определения содержания регулярно
чередующихся участков — полоса 770 см-1 [128, 130]. Состав
сополимера рекомендуется определять по отношению оптических
плотностей DB9o/DBi5, прокалибровав график по значениям состава
сополимера, определенным методом химического элементного
анализа.
Сополимеры ТФЭ с трифторэтиленом. Состав сополимера
определяют по интегральной интенсивности полосы 2987 см-1
С—Н-группы. При построении градуировочного графика
исходят из исходного состава газовых смесей мономеров для
полимеризации.
Сополимеры этилена с трифторпропиленом. В работе [132]
дано отнесение полос маятниковых колебаний СНг-групп в
ИК-спектрах сополимеров этилена с ТФП. Полосы 852, 755,
737 и 722 см-1 отнесены соответственно к последовательностям
(CH2)i, (CH2h, (СН2)з и (СН2)„, где п ^ 5. Следовательно,
полоса 852 см-1 относится к блокам ТФП, полоса 722 см-1 —
к блокам этилена, полоса 737 см-1 — к чередующейся
структуре, а слабая полоса 722 см-1 — к участкам, образующимся в
результате аномального присоединения трифторпропилена к
этилену. По этим полосам построены кривые зависимостей
содержания различных последовательностей мономерных звеньев
от состава сополимера.
II. 2. ПИРОЛИТИЧЕСКАЯ ГАЗОВАЯ ХРОМАТОГРАФИЯ
Применение метода ПГХ для определения состава
сополимеров рассматривается в ряде обзоров и монографий [46,
133—135], в которых основное внимание уделяется
методическим вопросам, чаще всего на примере каучуков и резин.
II. 2.1. Выбор условий количественного анализа
Определение состава сополимера методом ПГХ основано на
том, что выделяющиеся при его пиролизе продукты,
характерные для данного сомономера, образуются в количестве,
пропорциональном содержанию этого мономера в сополимере. Чтобы
определить состав сополимера, необходимо знать коэффициент
пропорциональности между его составом и сигналом детектора
112
на количество выделяющихся характеристических продуктов
пиролиза. Для этого необходимо иметь стандартные образцы,
состав которых определен другим независимым методом, и по
ним построить градуировочный график. Пользуясь таким
графиком, можно проводить анализ неизвестного образца.
Количественный состав продуктов пиролиза, к сожалению,
зависит не только от состава сополимера, но и от конструкции
пиролизера, температуры и продолжительности пиролиза,
навески и условий хроматографирования. Все эти факторы связаны
между собой, и от их удачного выбора зависит правильность и
воспроизводимость получаемых результатов анализа.
II. 2.1.1. Условия пиролиза
Основные- типы пиролизеров были рассмотрены в главе I.
Для количественного анализа обычно предпочтение отдают пи-
ролизерам по точке Кюри, но имеется большое число работ, в
которых применялись пиролизеры филаментного и печного
типов. Для всех трех типов пиролизеров, если условия подобраны
правильно, относительная погрешность определения колеблется
в пределах от 5 до 10 %•
Температуру пиролиза при проведении количественного
анализа следует выбирать таким образом, чтобы выход тех
продуктов, которые являются характеристическими, был
максимальным. Как правило, такую температуру определяют
экспериментальным путем, строя зависимость площади или высоты
соответствующих пиков от температуры пиролиза в интервале
400—900 °С. Различные продукты пиролиза одного и того же
сополимера могут иметь максимумы выделения при различных
10
■ ■ ■ ■■ ■ ■ I
300
■400
га Сополимеры Йимил-
I" ацетата
_ Сополимеры ЭфироЙ
I акриловой и мета-
крилоВой кислот
g Сополимеры оле<риноа
\\ Сополимеры стирала
а. Сополимеры сртор-
Н мономерой
500 В00 700
Температура пиролиза,"С
л. I . в i. a
800
Рис. 11.10. Распределение работ по количественному анализу различных сополимеров
в зависимости от используемой в них температуры пиролиза.
113
10 20 30 40
Время пиролиза, с
50
Рис. 11.20. Зависимость выхода стирола
из полистирола от продолжительности
пиролиза при 450 °С (пиролиэер
печного типа).
температурах. В этом
случае выбирают ту
температуру, которая соответствует
максимуму выделения
продукта, образующегося в-
наименьшем количестве.
Температура,
отвечающая максимуму выделения
того или иного продукта,
очень сильно зависит от других условий пиролиза, поэтому,
вероятно, в литературе приводятся очень противоречивые
данные по температурам пиролиза, используемым при
количественном анализе. Нами была построена диаграмма
распределения работ в зависимости от используемой в них температуры
пиролиза (рис. 11.19). Для построения диаграммы было
использовано 65 литературных источников, в которых
рассматривался количественный анализ сополимеров стирола,
этилена, эфиров акриловой и метакриловой кислот и фторсодержа-
щих мономеров. Наиболее часто при пиролизе используется
температура 500—600°С. Зависимость температуры пиролиза
от типа сополимера явно не прослеживается. Можно отметить
тенденцию к снижению температуры по сравнению со средней
для сополимеров акрилатов и метакрилатов и повышению для
фторсополимеров. Создается впечатление, что большое
разнообразие температурных режимов в работах по количественному
анализу состава сополимеров не всегда оправдано.
Продолжительность пиролиза в первую очередь
определяется температурой пиролиза и во вторую — массой и формой
образца. Чем выше температура, меньше масса и больше
поверхность образца, тем за меньшее время образец разлагается
полностью. При количественном анализе особенно существенно,
чтобы образец успевал при выбранных условиях пиролизовать-
ся полностью.
Рекомендуется строить зависимость выхода определяемого
продукта пиролиза (площади или высоты соответствующего
пика) от продолжительности пиролиза. На рис. 11.20 приведен
пример такой зависимости. Обычно, за продолжительность
пиролиза принимают время, соответствующее на графике точке А
или несколько большее, т. е. такое время, выше которого
выход продукта остается постоянным. Если работать при меньшей
продолжительности пиролиза, чем А, то наблюдается плохая
воспроизводимость результатов. Следует иметь в виду, что если
А > 30 с, то это означает, что температура пиролиза выбрана
неправильно и ее надо повысить. Как правило, стараются,
чтобы продолжительность пиролиза не превышала 30 с, так как
114
при работе с пиролизерами непрерывного действия более
длительный пиролиз может приводить к размыванию пиков, а в
случае пиролизеров с остановкой потока — к увеличению
вероятности протекания вторичных реакций.
Масса образца. Как указывалось в гл. I, чем меньше
образец, тем лучше качество получаемых хроматограмм и выше
воспроизводимость результатов. Однако работа с образцами,
масса которых меньше 0,5 мг, имеет и ряд неудобств.
Возрастает опасность влияния композиционной неоднородности
полимера и каталитического действия держателя на результаты
анализа; растет ошибка определения массы, что делает
неприменимым метод абсолютной градуировки. Работать с
нерастворимыми образцами массой менее 0,5 мг в принципе
невозможно. В большинстве аналитических работ используют образцы с
массой от 0,5 до 2 мг.
II. 2.1.2. Условия хроматографирования
Когда состав продуктов пиролиза известен, то условия
хроматографирования, в частности выбор насадки для колонки,
ничем не отличаются от проводимых в обычных хроматографи-
ческих методиках. Основные требования при этом — хорошее
разделение характеристических пиков. При работе с
образцами, состав продуктов пиролиза которых неизвестен, задача
несколько усложняется, но требования остаются теми же; в этом
случае условия хроматографирования чаще всего подбирают
часто эмпирически. Следует отметить, что сейчас редко
приходится встречаться с анализом полимеров, о продуктах пиролиза
которых отсутствовала бы информация.
Таким образом, на количественные результаты
хроматографирования влияет целый ряд факторов, тесно связанных
между собой. Прежде всего это температура и продолжительность
пиролиза, а также масса навески. Обычно эти условия
подбирают эмпирическим путем, но в последнее время все чаще
используют метод математического моделирования процесса
пиролиза с последующей оптимизацией по тем или иным
параметрам.
При определении состава сополимеров методом ПГХ часто
используют детектор по теплопроводности, так как для этой
задачи необходимо фиксировать только самые интенсивные
пики и чувствительности прибора бывает вполне достаточно при
навеске 0,5 мг и выше. Детектор по теплопроводности, в
отличие от ионизационно-пламенного детектора (ДИП), позволяет
определять некоторые неорганические продукты, например НС1
и HF. ДИП используют, когда работают с образцами массой
менее 0,1 мг или применяют капиллярные колонки. Этот
детектор применяют также тогда, когда изучают не основной состав
сополимера, а детали строения, т. е. когда существенными
становятся продукты пиролиза, выделяющиеся в незначительном
количестве.
115
II. 2.1.3. Выбор характеристических пиков
Под «характеристическими пиками» подразумевают те пики
на хроматограмме, которые относятся к продуктам пиролиза,
наиболее хорошо отражающим структуру сополимера и дающим
наименьшую ошибку при анализе. Это обычно достаточно
интенсивные пики, образование каждого из которых связано
только с одним из сомономеров. Иногда в качестве
характеристических используют пики, не идентифицируя их, но чаще, особенно
в последнее время, выбор пиков основывается на
предварительном изучении состава продуктов пиролиза и механизма
деструкции.
Характеристические пики, используемые различными
авторами для определения состава того или иного сополимера
приведены в обзорах [134, 135]. Наиболее часто пиролиз полимери-
зационных пластмасс протекает с выделением исходных
мономеров и тогда, естественно, в качестве характеристических
пиков используются пики, относящиеся к этим мономерам.
В других случаях выбирают характеристические пики исходя из
механизма пиролиза или эмпирическим путем. В работе [59]
предложен принцип выбора характеристических пиков,
обеспечивающих наибольшую чувствительность к исследуемой
характеристике полимера, с применением ЭВМ, на примере
определения состава смеси натурального и бутадиенстирольного ка-
учуков. На основании литературных данных калибровочные
кривые выражали в виде параболических функций второго
порядка: вида
у = А, + А2х + А3х\
где У—отношение площадей пиков на пирограмме; х — содержание одного
из компонентов в анализируемой системе; коэффициенты Лi, А2, А3 находили
из условий минимума суммарной квадратичной невязки.
Программа позволяет вычислить изменения х—линии
регрессии для всех сочетаний пиков и выбрать сочетания, для
которых квадратичная погрешность имеет наименьшее значение.
При переходе от двух- к многокомпонентным системам задача
значительно усложняется.
Наиболее простым методом выбора характеристических
пиков является сравнение пирограмм сополимеров с пирограмма-
ми соответствующих гомополимеров. В большинстве случаев
наиболее интенсивный пик в пирограмме гомополимера
присутствует и в пирограммах соответствующих сополимеров, и его
удобно использовать в качестве характеристического.
11.2.2. Методы расчета состава сополимеров
Для расчета состава сополимера обычно строят градуиро-
вочный график, связывающий интенсивность
характеристических пиков на пирограмме с составом сополимера. Для
построения градуировочного графика необходимо иметь образцы йз-
116
вестного состава — стандартные образцы. Интервал составов
стандартных образцов должен перекрывать весь интервал
составов анализируемых образцов. В случае линейной
зависимости можно ограничиться тремя стандартными образцами. Если
зависимость имеет нелинейный характер, то число стандартных
образцов должно быть больше. Исключение составляет случай,
когда необходимо контролировать соответствие состава образца
заданному значению. В этом случае достаточно иметь один
надежный образец заданного состава и сравнивать полученные-
результаты с ним.
Состав стандартных образцов определяют независимым
методом: ЯМР, элементным анализом, функциональным анализом
или на основании технологических данных — по количеству
вступивших в полимеризацию мономеров. Количество мономера
определяют методом газовой хроматографии в смесях до и
после полимеризации л вычисляют разницу между ними или
используют мономеры, меченные радиоактивными атомами.
Стандартные образцы должны быть получены по той же
технологии, что и анализируемые. Условия анализа и вид
образцов должны полностью совпадать с условиями анализа к
видом образцов, подвергающихся анализу. Для анализа
состава блоксополимеров можно, как правило, использовать смеси-
гомополимеров.
Содержание в полимере мономерных звеньев того или иного
типа или других структурных единиц С связано с количеством
выделяющихся из него продуктов пиролиза Q уравнением:
C = klQ. (II. 10)
Q пропорционально площади соответствующих пиков 5 на
хроматограмме:
Q = fe,S/m; C = klk2S/m = KS/m, (H. II)
где т — масса образца.
Площадь пика вычисляют как произведение высоты пика h
на его полуширину при высоте h/2 (в мм2). При
использовании метода абсолютной градуировки получают пирограммы
стандартных образцов с известной массой и строят график,
откладывая по оси ординат площадь пика, деленную на
навеску, а по оси абсцисс—массовую или молярную долю
определяемого мономера в образце (в %) по отношению ко всей
массе образца. В этом случае график обычно имеет линейный
или близкий к линейному характер, и если проходит через
начало координат, то описывается уравнением (11.11), а если не
проходит через начало координат, то уравнением:
C = a + KS/m. (IF. 12>
Метод абсолютной градуировки неудобен тем, что требует
точного определения навески образца, а это не всегда
возможно, поскольку она очень мала (1 мг и менее). Кроме того,
117
площадь пика, как известно, чувствительна к колебаниям
условий пиролиза и хроматографирования.
В связи с этим метод абсолютной градуировки применяется
сравнительно редко. Чаще для количественного анализа
используют различные вариации метода, когда расчет ведут по
отношению площадей характеристических пиков к площади
стандартного пика. Это позволяет исключить из расчетов массу
навески и уменьшить влияние факторов, связанных с изменением
условий пиролиза и хроматографирования.
Наибольшее распространение получил метод, при котором
для системы, состоящей из п компонентов за стандарт выбирают
пик, относящийся к одному из них xCt и выражают все
остальные концентрации Xi относительно него:
*^=т^=Ш=к'^ (П13)
/(^определяют по образцам известного состава, строя
графики в координатах СУССТ и 5,75ст. Уравнений типа (II. 13) для
системы из п компонентов получается п—1. Кроме того,
можно написать уравнение:
2*,- = Ю0%. (И.14)
Полученную систему уравнений (11.13) — (II. 14) решают
относительно Х(.
Для трехкомпонентной системы, состоящей из сомономеров
с концентрациями Х\, х2 и х3, система уравнений будет иметь
вид:
х,/х3 = KSt/S3; x2/x3 = KS2/S3; x, + х2 + х3 = 100 %.
А для двухкомпонентной системы:
x,/x2 = KSJSb х, + х2 = 100%. (П. 15)
Последний случай — анализ двухкомпонентной системы —
наиболее часто встречается в практике, поэтому уравнение
типа (II. 15) получило наиболее широкое распространение. Гра-
дуировочный график, построенный в координатах Ci/CCr и
Si/SCr — обычно имеет линейный характер.
При использовании метода внутреннего стандарта в образец
вводят известное количество определенного полимера, который
должен давать при пиролизе в основном только одно
соединение. В качестве таких полимеров чаще всего используют
полистирол и полиметилметакрилат. В этом случае уравнение
(II. 15) принимает вид:
Х\/лст = Ki^i/^crt ^2/*ст == Аг^г/^ст*
Однако при введении внутреннего стандарта увеличивается
продолжительность анализа и появляется опасность
перекрывания характеристических пиков пиками стандарта.
118
Метод внешнего стандарта [56] также не получил большого
распространения, так как анализ в этом случае становится
длительным и сложным.
11.2.3. Пиролиз полимеров
11.2.3.1. Полистирол
При пиролизе полистирола основным компонентом
продуктов пиролиза является стирол. Работы по изучению
зависимости выхода стирола от температуры пиролиза проводились на
нестандартных установках в различных условиях, и поэтому,
вероятно, полученные результаты отличаются большим
разнообразием. Интервал температур, при которых наблюдается
максимальное выделение стирола, составляет 400—600 °С.
Типичная зависимость выхода стирола от температуры
представлена на рис. 1.3. Выход может составлять от 40 до
100%, но чаще он колеблется около 80%. Помимо стирола
при пиролизе полистирола выделяются также метан, этан,
этилен, бензол, толуол, этилбензол, а-метилстирол и димеры. Хотя
эти соединения выделяются в очень малых количествах
(десятые и сотые доли процента), но они дают ценную информацию
о механизме пиролиза полистирола и о его строении. Ниже
указаны продукты пиролиза полистирола [136—138]:
Легкие углеводороды
Бензол CGHG
Толуол ССН5—СН3
Ксилолы Н3С—С0Н4—СН3
Этилбензол CGH4—С2Н5
Стирол СН2=СН—CGH5
Аллилбенэол СН2=СН—СН2—CGH5
а-Метилстирол СН2=С—ССН5
СИ,
(З-Метилстирол СНЭ—СН=СН—CGH5
1-Фенилбутан СН3—СН2—СН2—СН2—CGH5
1-Фенилпентан СН3—СН2—СН2—СН2—СН2—С0Н5,
2-Фенилбутен-2 СН3—С=СН—СН3
CGH5
4-Фенилбутадиен-1,3 СН2=СН—СН=СН—CGH5
Дифенилэтан С0Н5—СН2—СН2—С0Н5
1,2-Дифенилпропан ССН5—СН2—СН—СН3
CGH5
1,3-Дифенилпропан CGH5—СН2—СН2—СН2—CGH5
1,2-Дифенилпропен CGH5—СН2—С=СН2
СсН5
119>
1,3-Дифенилбута н CGH5—СН2—СН2—СН—СН3
cgh5
1,3-Дифенилбутен-1 CGH5—СН=СН—СН—СНа
CGH5
1,3-Дифенилбутен-2 CGH5—СН2—СН=С—СН3
СвН5
2,4-Дифенилбутен-1 СН2=С—СН2—СН2—СвН5
CGH5
1,4-Дифенилбутадиен-1,3 С6Н5—СН=СН—СН=СН—С0Н5
2,4,6-Трифенилгексен-1 СН2=С—СН2—СН—СН2—СН2—С6Н5
СсН5 С0Н5
/°
Бенэальдегид С6Н5—С
ХН
Индан, 1-метилиндан I II 1 [ I]1
СН,
Инден
С аналитической точки зрения интерес представляют
обнаруженные в ряде работ дифенилэтан (0,3%) [136—138], 1,4-
дифенилбутадиен-1,3 [136] и 1,2-дифенилпропан [138]. Было
естественно предположить, что эти соединения могут
образоваться из полистирола только при наличии в нем структур
«голова к голове». Действительно, в продуктах пиролиза
модельного образца, состоящего в основном из таких структур,
содержание дифенилэтана составляло 1,3% [137]. В работе
[136] выделение продуктов пиролиза, связанных с наличием
структур «голова к голове», было использовано для
относительной оценки влияния способа полимеризации на образование
этих структур.
Наиболее распространенной насадкой при исследовании
продуктов пиролиза полистирола является полиэтиленгликоль, по-
липропиленгликоль или их эфиры на целите или хромосорбе.
II. 2.3.2. Поли-а-метилстирол
Повышенный выход мономера при пиролизе поли-а-метил-
стирола по сравнению с полистиролом, можно объяснить
наличием в поли-а-метилстироле четвертичного атома углерода.
В широком интервале температур выход превышает 90 % и при
120
температуре 770 °С (пиролизер по точке Кюри) достигает
практически 100% [139]. Кроме мономера — а-метилстирола
было идентифицировано 20 соединений. Многие из них
совпадают с продуктами пиролиза полистирола [138].
11.2.3.3. Гомополимеры эфиров метакриловой кислоты
Из полиметакрилатов наиболее широко применяется поли-
метилметакрилат. На его примере обычно изучают механизм
пиролиза полимеров, которые при разложении дают выход
мономера, близкий к 100%. Выход метилметакрилата при
пиролизе полиметилметакрилата в пиролизере по точке Кюри в
интервале температур 450—650 °С составляет 90% [140]. При
использовании пиролизера филаментного типа такой же выход
наблюдается в интервале температур 550—650 °С.
Выход мономеров при пиролизе других полиэфиров
метакриловой кислоты также составляет не менее 96% (табл. II. 6).
Исключение составляет поли-2-гидроксиэтилметакрилат, для
которого при 650 °С выход мономера равен только 26%, а
помимо него выделяются еще ацетальдегид, метакриловая кислота,
этилен- и диэтиленгликоль и др. Такое, не типичное для метак-
рилатов, поведение этого полимера, вероятно объясняется
наличием гидроксигруппы.
11.2.3.4. Гомополимеры эфиров акриловой кислоты
Полиакрилаты, в отличие от полиметакрилатов, при
пиролизе дают небольшой выход мономеров [140]. С повышением
температуры выход мономера растет (см. табл. П. 6). Кроме
мономера все полиакрилаты при пиролизе образуют
соответствующие спирты, которые являются основными продуктами, димеры
и С02. Соотношение выхода спирта и мономера меняется
с температурой. Так, для полиэтилакрилата с увеличением
температуры от 350 до 500 °С отношение этиловый спирт : этил-
Таблиц a II.6. Выход мономеров (в %) при пиролизе по точке
Кюри полиалкилакрилатов и полиалкилметакрилатов
при различных температурах [140]
Алнил
Метил
Этил
Пропил
Бутил
Амил
Гексил
2-Этилгексил
480-0
9,2
6,4
6,8
10,1
8,8
9,6
4,9
Акрилать!
610 °С
14
11,3
10,8
10,7
10,7
12,0
6,8
770 °С
17,1
14
12,5
14,0
12,6
13,0
8,4
Метакрилаты
480 °С
92
89,0
90,0
90,6
90,1
96,0
86,0
610 °С
98,3
98,0
98,5
98,0
97,0
97,3
96,4
770 "С
91,8
90,0
86,2
84,0
87,0
90,2
83,2
121
.•акрилат меняется от 14,5 до 5,3. Это происходит от того, что
выход этилакрилата с повышением температуры пиролиза
растет быстрее, чем выход этилового спирта.
Выбор насадки при хроматографировании продуктов
пиролиза полиакрилатов и полиметакрилатов не представляет
проблемы. Применяются разнообразные насадки, наиболее широко,
например, силоксановые жидкости на хромосорбе W или ди-
этиленгликоль и его производные на диатомитовых носителях.
II. 2.3.5. Полиакрилонитрил
При пиролизе полиакрилонитрила образуется мономер, но
зыход его невелик. Помимо мономера при пиролизе при 510°С
в сравнимых количествах образуются димеры и более
высокомолекулярные соединения [141]:
Cri2^=C—СНо—C=CH2j СН2—СН2—C^CHoj
II I I .
C=N C=N C=N C=N
Ori2"~~Cri2"~~Cri2J CH2^=C CH2 CH Cri2~~C^=Ori2l
I I 111
C=N C=N C=N C=N C=N
CHj—CH2—CH—CH2—СН=СНг; CH2—CH2—CH—CH=CH;
I I I I I I
Ce=N C^N C=N C=N C=eN C=N
CH2—CH2—CH—CH2—CH2.
I I I
Ch=N C=N CsN
Кроме летучих продуктов при пиролизе могут
образовываться нелетучие соединения, состоящие из структур циклического
типа с сопряженными связями:
СН2 СН2 СН2
/ \о" \с/ \с/
I I I
\ ^Cv, ^Cv, ^Cv,
NN N
Эти структуры более термостабильны и их пиролиз требует
более высокой температуры.
II. 2.3.6. Полиэтилен
При пиролизе полиэтилена образуется большое число самых
разнообразных предельных и непредельных углеводородов как
нормального, так и изостроения. Хроматографический анализ
такой смеси представляет экспериментальные трудности,
поэтому, чтобы упростить картину и более надежно разделить и
идентифицировать продукты, часто используют метод
гидрирования [142—144]. Для этого продукты пиролиза перед вводом
в хроматограф пропускают через колонку, заполненную
катализатором, которым служит металлическая платина, нанесенная
122
на твердый носитель. В качестве гидрирующего агента
используют водород, который является одновременно и
газом-носителем. После .гидрирования состав продуктов упрощается, так как
непредельные углеводороды превращаются в предельные. Таким
образом, при гидрировании часть информации исчезает. Знание-
состава непредельных углеводородов существенно для
понимания механизма пиролиза, но для аналитической задачи —
изучения разветвленности полиэтилена — они существенного
значения не имеют.
Состав продуктов пиролиза сильно зависит от температуры.
Обычно пиролиз проводят при 600—650 °С. Но и после
гидрирования число продуктов пиролиза велико — представлено
несколькими десятками пиков, поэтому рекомендуется
использовать капиллярные колонки [142—144].
Деструкция линейного полиэтилена происходит по закону
случая с образованием алканов нормального строения, но прет
наличии боковых коротких групп—метальной, этильной или'
бутильной — разрыв макроцепи происходит в р-положении к
разветвлению с образованием 2-, 3- и 5-метилалканов
соответственно. Например, образование 3-метилгептана можно
представить следующим образом:
н н
I ! , I
R—СН2—СНг— СН2—СН2—С—СНг-rR —»- R—СН2—СН2—СН2—СН2—С— СН3«—>-
I ' I
СН2 СНг
I I
СИ3 СНЭ
н н н
^К-СНг-СН-СНг-СНа-С-СНэ — СН2 = С-СН2-СН2-С-СН3 Г««ЖР<»»"»¥1
сн2 сн2
СНЭ СНз
н
—*- СНз—(СН2)з—С—СНз
сн2
сн3
Более длинные разветвления склонны к разрыву —С—С-
связей в а- и р-положении к основной цепи [142].
Сопоставление продуктов пиролиза полиэтилена высокой плотности
(ПЭВП) и полиэтилена низкой плотности (ПЭНП) показало,
что качественный состав их идентичен, но имеются различия в
количественном распределении линейных (рис. 11.21) и
особенно разветвленных (рис. 11.22) фрагментов. В продуктах
пиролиза ПЭНП 3-метилалканов в несколько раз больше, чем в
продуктах пиролиза ПЭВП, что соответствует данным ИКС.
Особенно это заметно для соединений от 3-метилпентана до
3-метилнонана. Очевидно, содержание 2-, 3- и 5-метилалканов
123
9 13 17 21 25 29
Число атомов углерода
33 37
Рис. 11.21. Распределение линейных алканов, образующихся при пиролизе полиэтилена
(с последующим гидрированием), в зависимости от числа углеродных атомов С..
О4*
■p-i
со
0,6
0,5
0,4
И1о,з
Я
0,2
0,965 «^ 0,951
д 10 12 74 13 18 20
Числе атомов углерода
2?. 24
Рис. 11.22. Распределение Э-метилалканов, образующихся при пиролизе полиэтилена
равной плотности (с последующим гидрированием), в зависимости от числа углеродных
атомов Cj.
в гидрированных продуктах пиролиза может быть использовано
для сравнительной характеристики разветвленности полиэтиле-
нов, полученных разными способами.
Для насадочных колонок чаще всего используют апиезон
L на хромосорбе W, для капиллярных колонок — апиезон L,
сквалан, полифениловый эфир OS-138.
II. 2.3.7. Полипропилен
При пиролизе полипропилена образуется соединений еще
больше, чем из линейного полиэтилена. Например, в работе
[145] на пирограмме полипропилена было идентифицировано
до 40 продуктов (последним зафиксированным на пирограмме
продуктом был гранб,-гракс-2,4,6,8|Ю-пентаметилундекан).
Продукты после пиролиза гидрировали. Основные пики относились к
продуктам с составом Зп, где п — число мономерных единиц в
фрагменте. Наиболее интенсивные пики принадлежали
фрагментам с п = 1 и п = 3. Наряду с соединениями с числом
124
С-атомов Зп образуются также фрагменты состава Зп±1.
В частности, на пирограмме полипропилена имеются
интенсивные пики, относящиеся к н-пентану и 2,4-диметилгептану. Их
образование можно представить как результат
внутримолекулярного переноса атома Н с пятого углеродного атома на
образующийся при статическом разрыве цепи радикал.
Основными продуктами пиролиза являются изоалканы, но
наблюдалось также в небольшом количестве образование цик-
лоалканов.
Как известно, полипропилен находится в двух стереоформах:
изотактический и синдиотактический изомеры. Промышленный
полипропилен является в основном атактическим, который
можно рассматривать как сополимер изо- и синдиотактического.
Продукты, образующиеся при пиролизе из изотактической
части макромолекулы, представляют собой изоалканы цис-формы,
а из синдиотактической части—изоалканы транс-формы, кроме
того из атактического полипропилена образуются смешанные
^ыс-гранс-формы. Были сделаны попытки использовать этот
факт для оценки тактичности полипропилена [146]. Однако
такая оценка носит скорее качественный, сравнительный
характер.
Как и для полиэтилена, для анализа используются
капиллярные колонки с такими же насадками. Температура
пиролиза —600—650 °С.
II. 2.3.8. Поливинилацетат
При пиролизе поливинилацетата выделяются уксусная
кислота, бензол и легколетучие углеводороды. Уксусная кислота
образуется в результате отщепления ацетатной группы через
промежуточный шестичленный цикл. При этом в полимерной
цепи образуется двойная связь:
н н н
I I I
—С—С—С * —СН=СН—СН2—+ СНзСООН.
I I I
нон
6=С—СН3
Двойные связи, накапливаясь в полимере, образуют полие-
новые системы, которые в результате реакции циклизации
образуют бензол. Низкокипящие углеводороды являются
продуктами более глубокого разложения. Более детальное изучение
пиролиза поливинилацетата позволило обнаружить 25
соединений в основном ароматического характера и мономер [138].
11.2.3.9. Политетрафторэтилен
Политетрафторэтилен при пиролизе распадается на тетра-
фторэтилен, гексафторпропилен и октафторциклобутан. Два
последних продукта являются вторичными — они образуются из
125
тетрафторэтилена в результате вторичных реакций и их
количество в пиролизате может меняться очень значительно в
зависимости от условий пиролиза, в первую очередь от давления:
чем выше давление, тем больше выход этих продуктов, так как
давление мешает быстрому удалению первичных продуктов из
зоны реакции. Когда пиролиз проводят в динамическом режиме,
газ-носитель, способствует их удалению, и в этих условиях
выход тетрафторэтилена составляет 90 % и больше [147].
Температура пиролиза мало влияет на качественный и
количественный состав продуктов. Чаще всего пиролиз проводят
в интервале температур 650—750°С. Максимум выделения
тетрафторэтилена наблюдался при 600 °С [147]. При пиролизе
ПТФЭ всегда появляется белый налет на холодных частях
установки. Это, по мнению некоторых исследователей, олигомеры
тетрафторэтилена, которые образуются в результате вторичной
полимеризации выделяющегося при пиролизе мономера.
Однако, эта точка зрения не является общепринятой.
В качестве насадки в основном используется порапак Q
при длине колонки 1,5—2 м и температуре колонки 100—
150 °С. Хорошее разрешение было также получено на триэти-
ленгликольдибутирате, нанесенном на диатомитовый носитель
ИНЗ-600.
II. 2.3.10. Другие фторсодержащие гомополимеры
Для поливинилиденфторида основными продуктами
пиролиза при температуре ниже 500 °С являются фтороводород и бен-
590'С
485°С1^1
740°С
-1
бЗО'С
-1
ю о
Время,мин
У
V^л^_
10
Рис. 11.23. Зависимость выхода продуктов пиролиза поливинилиденфторида от
температуры пиролиза:
/ — пнк винилнденфторида.
126
зол. С повышением температуры выход этих соединений
падает и увеличивается выход мономера — винилиденфторида
(рис. 11.23). Кроме винилиденфторида образуются также три-
фторбензол и фторолефины, количество которых зависит от
температуры.
Насадка для колонки используется та же, что и при
исследовании продуктов пиролиза ПТФЭ.
При пиролизе политрифторхлорэтилена в основном
образуются мономер — трифторхлорэтилен, летучие
низкомолекулярные продукты и в небольшом количестве пентафторхлорпропан.
Температура пиролиза 590 °С, насадка — 10% диоктилфталата
на диазолиде М.
Для поливинилфторида в литературе приводятся
противоречивые данные. Основными продуктами пиролиза называют
бензол, в небольшом количестве толуол и октан. Были найдены
также фторпроизводные метана, винилфторид и фрагменты,
содержащие три атома С [148, 149]. В указанных работах
использовали пиролизер по точке Кюри; температура пиролиза
составляла 590 °С [148] и700°С [149].
(1.2.3.11. Поливини лхлорид
Для пиролиза поливинилхлорида (ПВХ) не характерны
реакции разрыва основной цепи. Как правило, основными
продуктами пиролиза ПВХ являются хлороводород и бензол.
Первичная реакция пиролиза — отщепление НС1 и образование по-
лиеновых структур:
~СН2— CHCI—СН2—СНС1—СН2—СНС1 *■
—>. ~СН=СН—СН=СН—СН=СН~ + raHCI.
В результате циклизации эти структуры образуют бензол и
некоторые другие ароматические соединения. Выход хлороводо-
рода достигает 58 %. что близко к теоретически возможному.
Кроме хлороводорода и бензола при изучении продуктов
пиролиза ПВХ были обнаружены и алифатические углеводороды,
но ароматические всегда преобладали. Среди последних
помимо бензола в значительных количествах присутствуют также
голуол и нафталин.
Некоторые продукты могут образовываться в результате
обратной реакции присоединения НС1 по двойным связям.
Наличие оксидов металлов может приводить к изменению
соотношения между алифатическими и ароматическими углеводородами
[150]. Для более детального изучения структуры ПВХ
проводили [151] его дегидрохлорирование в атмосфере водорода.
Пирограммы дегидрохлорированного в таких условиях ПВХ
оказались близкими к пирограммам гидрированного
полиэтилена. Это показывает, что ПВХ также имеет разветвленность,
оценить характер которой можно таким же образом, что и в
случае полиэтилена.
127
11.2.4. Определение состава сополимеров различных классов
11.2.4.1. Сополимеры стирола
Сополимеры стирола с эфирами метакриловой и акриловой
кислот. Наибольшее число работ посвящено изучению состава
сополимеров стирола с метилметакрилатом (ММА). Во всех
работах за характеристические пики были выбраны пики
мономеров — стирола и ММА. Соотношение выходов стирола и ММА
из сополимера сильно зависит от температуры (рис. 11.24). Это
легко объяснить, если учесть, что деполимеризация полистирола
достигает максимума при 600 °С, а ПММА — при 400 °С
[152]. Из приведенных на рис. 11.24 зависимостей следует,
что температура пиролиза сополимера должна быть не ниже
550°С, т. е. такой, при которой выход продуктов пиролиза
мало зависит от ее изменения. Имеющиеся в литературе
данные подтверждают этот вывод—большинство авторов получали
хорошо воспроизводимые результаты при температурах 560—
600 "С. Только в двух работах анализ проводили при 500 °С.
В смесях гомополимеров выход мономеров всегда
пропорционален их содержанию. Для сополимеров выход стирола растет с
увеличением содержания ММА в сополимере, а выход ММА
падает с увеличением содержания стирола. Это особенно
заметно проявляется при температуре 500°С, при 600—700"С —
гораздо меньше [139].
Для определения состава сополимеров или строили графики
в координатах Сст/Смма и Sct/Smma [154, 155], или выражали
связь между площадями пиков и составом с помощью
следующих уравнений (где Смма и Сст — содержание ММА и
стирола в сополимере соответственно, %):
Смма = 30,99 + 33,56SMMA/SCt [152]; (II. 19)
Сдша = 14.76 + 97,79 SMMA/(SCT + SMMA) [152]; (11.-20)
CCt=60 + 99,1SCt/(SCt + Smma) [156]. (11.21)
Уравнения (11.20) и (П. 21) показывают, что анализ
сополимера стирола с ММА протекает в оптимальных условиях:
содержание мономеров
практически совпадает с отношением
площади соответствующих
пиков к сумме площадей пиков,
умноженным на 100. Это
объясняется тем, что выход обоих
мономеров из гомополимеров
близок к 100 % и
коэффициенты чувствительности почти
Рис. 11.24. Зависимость выхода продуктов
400 500 В00 700 800 пиролиза сополимера стирола с
метилметакрилатом от температуры пиролиза:
Температура пиропиза'С / — стирол; 2—метилметакрилат.
128
равны. Высокий выход мономеров подтверждается и данными
работы [153, с. 12], где содержание ММА в сополимере
определяют по уравнению:
^м.ма = 0>93jcj\ima»
где Л'мид — содержание ММА в летучих продуктах пиролиза, определенное
хроматографическнм методом.
При сопоставлении выхода мономеров из статистического
сополимера с выходом мономеров из смеси гомополимеров или
блоксополимера того же состава, было показано, что они могут
значительно различаться [108, 139, 154]. Определив в
уравнении Сст/Смма = К So/Smma значение К для сополимеров,
блоксополимера и механической смеси и зная состав анализируемых
образцов, можно отличить их друг от друга. Рекомендуемая
для этого температура пиролиза 480°С. При других
температурах различия меньше [154].
Сополимеры стирола с другими эфирами метакриловой
кислоты изучены меньше. Имеются данные по сополимерам
стирола с бутилметакрилатом [139]. В качестве характеристических
полос были использованы также пики мономеров. Условия хро-
матографирования аналогичны условиям хроматографирования
сополимеров стирола с ММА. Данных о сополимерах стирола с
эфирами акриловой кислоты практически нет, вероятно,
поскольку они не находят большого практического применения.
Такие сополимеры упоминаются только наряду с другими
сополимерами стирола.
В подавляющем большинстве работ использовалась
температура 550—600 °С. В качестве насадки для колонок применяли
полиэтиленгликоль или полиэтиленгликольадипинат на целите.
В некоторых работах [152, 156] использовали диметилсилок-
сановый каучук на диатомитовом носителе.
Сополимер стирола с акрилонитрилом. Во всех работах в
качестве характеристических пиков были выбраны пики
мономеров— стирола и акрилонитрила. Зависимость выхода
мономеров от температуры изучена мало, но на основании
имеющихся работ можно сказать, что при работе с пиролизером по
точке Кюри максимальный выход акрилонитрила наблюдается
около 700 °С, максимальный выход стирола — при более
низкой температуре [157, 158]. Выход акрилонитрила меньше
выхода стирола, поэтому важно знать, как меняется этот выход
от состава сополимера. Из рис. 11.25 видно, что выход
акрилонитрила с увеличением его содержания в сополимере растет,
однако не превышает 20% [159]. В литературе приводятся
данные по определению состава сополимеров с содержанием
акрилонитрила от 20 до 60%. На рис. 11.26 показан пример
градуировочного графика. Количество акрилонитрила для
стандартных образцов обычно определяют по содержанию азота
методом Кьельдаля.
5 Зак. 632
129
Вызывает удивление разнообразие температур, при которых
изучали сополимеры методом ПГХ. В различных работах они
варьируются от 470 до 980°С, и из приведенных результатов
следует, что хорошая воспроизводимость и точность
результатов достигаются как при 480 °С, так и при 770 °С. Выход
акрилонитрила для механической смеси по отношению к его
содержанию остается постоянным в отличие от сополимеров и
составляет менее 10%. Этот факт можно использовать для
того, чтобы отличать смеси от сополимеров в тех случаях, когда
известен их состав, определенный другим методом [157]. Для
этой же цели можно использовать коэффициент К:
^Ст/^АН
к =
W.
АН
для которого получены следующие значения:
610 °С
0,024
0,47
980 °С
0,048
0,37
Механические смеси
Статистические сополимеры
В работе [158] предложено использовать для
характеристики образцов неизвестного строения (смесь или статистический
сополимер) количество выделяющегося пропионитрила.
Очень разнообразны насадки для колонок, используемые при
определении состава сополимеров стирола с акрилонитрилом:
полиизопропиленгликоль UCLB-550X на хромосорбе Р, апие-
зон L на диконите, полиэтиленгликольсукцинат на хромосорбе
W; ОК.-101, полидиметилсилоксан (капиллярная колонка).
Сополимеры стирола с бутадиеном. В качестве
характеристических пиков чаще всего используют пики стирола и
бутадиена [160—162]. Изучено влияние температуры пиролиза на
выход сомономеров и показано, что при использовании пироли-
700
а?
«о- 80
8.
1 so
о
5
<ъ 40
О
К
* 20
V
\
' \
\
\
\
\/'
V
\
V
/^
^ ,
2
1—
\
\
. ^~
О 0,2 0,4 0,В 0,8 1,0
Молярная доля акрилонитрила
6 сополимере *" ""*
Рис. 11.25. Зависимость выхода продуктов пиролиза сополимера стирола с акрилонитрилом
от его состава (температура пиролиза 400 °С):
/—стирол; 2 —акрнлонитрил.
Рис. П. 2в. Зависимость отношения концентраций мономеров в составе статистических
сополимеров от отношения площадей пиков стирола и акрилонитрила (пиролиэер по
Точке Кюри, температура пиролиза 770 °С).
130
Рис. 11.27. Зависимость отношенин
площадей пиков стирола и бутадиена от
температуры пиролиза статистического
сополимера стирола с бутадиеном (/)
и механической смеси соответствующих
гомополимеров (2) (пиролизер по точке
Кюри).
зера по точке Кюри для
статистического сополимера
отношение Scr/Sb остается
постоянным в широком интер- *>о воо воо юоо
Вале (рис. 11.27). Для ПИрО- Температура пиролиза,"С
лизера филаментного типа
характер зависимости
аналогичен. Пиролиз проводят обычно при температуре около
700°С. Кроме бутадиена при пиролизе полибутадиена
образуется его димер — 4-винилциклогексен, по мнению авторов
работы [160] только при использовании пиролиэера по точке
Кюри. Воспроизводимость результатов при использовании для
определения состава отношения площадей пиков стирола и
бутадиена составляет 8—10%, а при использовании отношения
площадей пиков стирола и 4-винилциклогексена — 1—2'%. Это,
вероятно, объясняется тем, что бутадиен имеет низкую
температуру кипения и время удерживания для него близко к
временам удерживания легких углеводородов, которые обычно
образуются при пиролизе.
Смеси гомополимеров и блоксополимеры имеют максимум
отношения площадей пиков стирола и бутадиена при 770 °С.
Не понятно, почему авторы работы [160] считают эту
температуру оптимальной для анализа. Для пиролизера печного типа
максимум выхода стирола при массовой доле бутадиена около
7% наблюдается при 700°С (выход стирола при пиролизе
полистирола достигает максимума при 600 °С). Температуры
кипения стирола и бутадиена сильно различаются, поэтому при
хроматографическом разделении этих мономеров не возникает
трудностей; сложнее отделить бутадиен от других легколетучих
соединений.
Используемые насадки очень разнообразны: карбовакс на
диатомитовом носителе, трикрезилфосфат на хромосорбе Р, по-
лиэтиленгликольадипинат на хезосорбе, полипропиленгликоль
на целите 545.
С помощью ПГХ можно выяснить, является ли данный
образец статистическим сополимером или блоксополимером.
Отношения площадей пиков стирола и бутадиена для таких
сополимеров различны. При пиролизе блоксополимеров и
механических смесей гомополимеров выход стирола больше, чем в
случае статистических сополимеров (см. рис. 11.27). Имеются
данные об определении 1,4- и 1,2-изомерных структур, а также
1,4-^ис- и 1,4-гранс-изомеров в статистических и блоксополи-
мерах [161]. Согласно работе [160], 1,2- и 1,4-изомерные струк-
4
5*
131
туры можно различить только при использовании пиролизера
по точке Кюри.
Сополимеры стирола с а-метилстиролом. При пиролизе
сополимеров стирола с а-метилстиролом в основном образуются
стирол и а-метилстирол [163]. Гомополимеры в смеси пиро-
лизуются независимо друг от друга; выход стирола при
пиролизе сополимеров гораздо выше, чем при пиролизе смеси гомо-
полимеров того же состава. Чем больше содержание в
сополимере а-метилстирола, тем больше выход стирола.
Максимальный выход обоих мономеров в работе [139] наблюдался при
770°С при использовании пиролизера по точке Кюри.
АБС-пластики. В качестве^ характеристических пиков при
анализе АБС-пластиков используют пики стирола, акрилонитри-
ла и бутадиена [164]. Состав стандартных образцов
АБС-пластиков определяют химическими методами (см. раздел. II. 4)
или используют смеси бутадиенового каучука с сополимерами
стирола с акрилонитрилом. Типичная хроматограмма
приведена на рис. I. 16. Пиролиз проводили при температуре 500—
600°С. В качестве насадки для колонок использовали
полиэтилен- или полипропиленгликоль на дпатомитовом носителе или
на полисорбе-1.
II. 2Д.2. Сополимеры эфиров метакриловой и акриловой кислот
Сополимеры различных метакрилатов. Гомополимеры
эфиров метакриловой кислоты деструктпруют при пиролизе с
выходом мономеров, близком к 100% [140], поэтому естественно
ожидать, что сополимеры метакрилатов поведут себя
аналогичным образом. Многочисленными исследованиями было
показано, что при пиролизе действительно в основном выделяются
исходные мономеры, а другие продукты, в частности спирты,
образуются в очень небольших количествах. Пирограммы
сополимеров являются наложением пирограмм соответствующих гомо-
полимеров.
При определении состава сополимеров различных эфиров
метакриловой кислоты возникают трудности с получением
стандартных образцов, необходимых для построения градуировочно-
го графика. Вследствие структурного сходства сомономеров
оказывается затруднительно использовать химический и другие
независимые методы для определения состава стандартных
образцов, поэтому приходится использовать для этой цели
механические смеси гомополимеров, что всегда нежелательно. Более
целесообразно строить градуировочный график не по смесям
гомополимеров, а по смесям мономеров и определять их
содержание в пнролизатах. Было показано [165], что
относительное содержание мономеров в пиролизатах, как правило,
совпадает с таковым в сополимере и в исходной полимеризацион-
ной смеси. В данном случае не наблюдается взаимного влияния
сополимеров друг на друга, поэтому методом ПГХ нельзя отли-
132
Таблица II. 7. Выход мономеров из сополимеров и смесей
гомополимеров акрилатов и метакрилатов (состав 1 : 1), % [144]
Mi
Мг
Сополимеры
М,
М2
Гомополнмеры
Mi
М2
Метилметакрилат
Этилметакрилат
Бутилметакрилат
Метилакрилат
Метилакрилат
Этилакрилат
Бутилакрилат
Гексилакрилат
2-Этилгексил акрилат
Бутилметакрилат
Метилакрилат
»
Бутилакрилат
98
98
98
97
97
98
98
96
13
40
28
27
23
20
97
38
40
9
98
98
98
97
97
98
96
95
12
14
11
10
8
7
96
15
15
10
чить эти сополимеры от механической смеси. Температура
пиролиза в различных работах варьируется от 400 до 700°С.
Сополимеры метакрилатов с акрилатами. При пиролизе
сополимеров метакрилатов с акрилатами помимо мономеров
выделяются также спирты, соответствующие акрилатам, СО, СОг,
низкомолекулярные углеводороды, димеры акрилатов и
смешанные димеры, а также такие акрилаты и метакрилаты, которые
не входят в качестве мономеров в сополимер, а образуются в
результате перегруппировки [166]. Например, на пирограмме
сополимера метилметакрплата с этилакрилатом имеются пики,
соответствующие этилметакрилату и метилакрилату.
Наибольший интерес с аналитической точки зрения представляют
основные продукты — мономеры и спирты.
Выход метакрилатов из их сополимеров с акрилатами
зависит от состава очень мало и совпадает с выходом из
гомополимеров. Выход метилакрилата при пиролизе его сополимеров
возрастает с увеличением его содержания в сополимере. Так,
метилакрилат выделяется из гомополимера в количестве 14 %,
а из сополимера с метилметакрилатом — 40% (табл. П.7).
В работе [140] были рассчитаны индексы, представляющие
собой отношение содержания мономера в смеси гомополимеров,
к выходу мономера из этой смеси /г и отношение содержания
мономера в сополимере к выходу этого мономера из
сополимера /с. Для всех метакрилатов оба индекса, как и следовало
ожидать, практически равны единице (1,02), а для сополимеров
акрилатов с метилметакрилатом существенно различаются в
зависимости от химической структуры акрилатов:
Метилакрилат
Этилакрилат
Пропилакрилат
Бутилакрилат
Амилакрилат
Гексилакрилат
2-Этилгексилакрилат
г
7
8,9
9,4
10,0
9,8
8,0
14,0
'с
2,5
3,7
3,9
4,1
4,2
4,3
5,0
133
Из сополимеров в виде мономера выделяется от 20 до 40 °/о
содержащихся в них акрилатных звеньев, а из гомополимеров-
в смеси —только 7—15%. Зная качественный состав образца,
можно с помощью приведенных выше индексов рассчитать
выход мономеров и, сравнив его с определенным
экспериментально, установить, является ли этот образец с'месью гомополимеров
или сополимером. Можно решить и обратную задачу: зная
выход мономеров и характер образца (сополимер или смесь
гомополимеров) определить его количественный состав.
Выход спирта из сополимеров акрилатов с метакрилата'ми,
так же, как и выход акрилата, сильно зависит от состава, на
для спирта эта зависимость имеет противоположный характер:
выход спирта падает при переходе от гомополимеров к
сополимерам. В сополимерах отношение выхода спирта к выходу
акрилата CVoh/Crakp зависит от температуры, в то время как их
сумма остается величиной постоянной. Если количество спирта
и акрилата выражать как отношение (в %) к количеству
акрилата в сополимере, то для сополимеров метилметакрилата
с метил-, этил- и бутилакрилатами сум'ма Croh + CRAKp будет
близка к 21 %. Способ получения, тип акрилата, состав
сополимера мало влияют на эту величину при условии проведения
пиролиза в близких условиях. Отношение Croh/CRAKp для
сополимеров акрилатов, как и для их гомополимеров, зависит от
температуры пиролиза и для сополимера этилакрилата с 'ме-
тилметакрилатом (50:50) меняется от 1,56 до 0,34 при
температурах пиролиза от 300 до 500°С. Но особенно
чувствительно это отношение к способу получения сополимера, к его
микроструктуре. Например, для гомополи'мера этилакрилата это
отношение колеблется в интервале от 3 до 5, для блоксополи-
меров — от 2 до 3, а для статистических сополимеров — от 0,2
до 0,5. Чувствительна эта величина и к конверсии: чем выше
конверсия, тем выше отношение. Из всего сказанного можно
сделать вывод, что выход спирта тем больше, чем больше
блочность сополимера, а так как источник образования спирта
и 'мономера акрилата один и тот же, то с ростом выхода
спирта выход акрилата неизбежно падает. Можно предположить,
что для образования спирта требуется наличие доступного
атома водорода. Таким атомом может быть третичный водород
акрилатов, но не из того же звена, по которому происходит
разрыв, а из расположенного на определенном расстоянии,
поэтому из изолированной молекулы акрилата спирт
образоваться не может.
Таким образом, для определения состава сополимеров мета-
крилатов с акрилатами в качестве характеристических пиков
целесообразнее использовать пики метакрилатов и сумму
пиков, относящихся к акрилату и спирту, а не пики метакрилата
и акрилата, как это делают в некоторых работах. Чтобы
отличить механические смеси гомополимеров от статистических
сополимеров, 'можно использовать отношение выхода спирта к
134
выходу акрилата или судить об этом по наличию смешанных
димеров и тримеров.
Сополимеры метакрилатов и акрилатов с другими
мономерами. Для сополимеров различных акрилатов характерно, что
отношение выходов спирта и мономера для них не отличается
от такового для смеси гомополимеров. В качестве
характеристических пиков 'можно использовать пики как акрилатов, так
и спиртов.
Сополимеры метакрилатов с акрилонитрилом анализировали
по пикам акрилонитрила и метакрилата. Учитывая низкий
выход акрилонитрила по сравнению с выходом метакрилатов, в
случае малого содержания последних погрешность
определения очень велика при использовании метода отношения
площадей пиков. Поэтому рекомендуется в этом случае применять
метод абсолютной градуировки [164].
Сополимеры винилхлорида с метилметакрилатом при
пиролизе дают метилхлорид, метилметакрилат, метиловый спирт и
углеводороды. Метилхлорид выделяется в результате реакции
лактонизации между группа'ми двух соседних звеньев сомономе-
ров, при которой образуется у-бутиролактон:
н сн3 н сн3
II II
~СН2—С—СН2— С—СН2~ —> ~СН2—С—СН2— С—СН2~ + С1СН3.
II II
С1 С=0 О С=0
I
НзС-0
Степень этой циклизации зависит от распределения
мономерных звеньев в сополимере. Статистический сополимер винил-
хлорида с метилметакрилатом выделяет метилметакрилат как
основной продукт пиролиза, но альтернатные сополи'меры
совсем не образуют метилметакрилата [167]. Очевидно, что
определять состав этих сополимеров методом ПГХ невозможно,
но зато легко получить информацию о чередовании звеньев.
Аналогичная картина наблюдается для сополимеров метил'ме-
такрилата с метакриловой кислотой.
Температура пиролиза для всех сополимеров акрилатов и
метакрилатов в различных работах варьируется в широком
интервале— от 300 до 700 °С, однако на практике чаще всего
используют температуру 450—500 °С. В качестве насадки
применяют снлоксаны различного строения на хромосорбе и
целите, а также полиэтиленгликоль.
II. 2.4.3. Сополимеры олефинов
Метод ПГХ не очень пригоден для определения состава
сополимеров олефинов. Довольно многочисленные работы по
исследованию полиолефинов методом ПГХ в основном посвящены
установлению механизма термического распада сополимеров и
чередования звеньев, а не определению состава.
135
Сополимер этилена с пропиленом. Пирограммы
сополимеров этилена с пропиленом (СЭП) представляют собой
наложение пирограмм полиэтилена, полипропилена и ряда пиков,
принадлежащих смешанным димерам, тримерам и т. д. При
пиролизе статистического СЭП, содержащего 45,8 % пропилена, при
использовании капиллярной колонки с последующим
гидрированием была получена пирогра'мма, на которой
идентифицировано 72 соединения [146]. Обычно в качестве
характеристических пиков выбирают наиболее интенсивные пики, но в данном
случае этим принципом было трудно воспользоваться, так как
в хроматограмме имеется много пиков, сравнимых по
интенсивности, и 'многие из них перекрываются с пиками полиэтилена.
В связи с этим при выборе характеристических пиков для СЭП
нет единого мнения и рекомендации очень разнообразны:
этилен и пропилен, 1-гептен и тримеры пропилена, 1-октен и 4-ме-
тил-1-гептен, нормальные алканы и изоалканы. Относительная
погрешность определения составляла 3 %, а предел
обнаружения пропилена — 1 %.
Более надежные результаты были получены при
использовании в качестве характеристического не одного отдельного
пика, а нескольких пиков [146]. При гидрировании продуктов
пиролиза СЭП за характеристические пики для пропилена были
выбраны 2-метилпентен, 2,4-диметилпентен и 2,4,6-триметилпен-
тен, а для этилена — нормальные гексан, гептан, октан, нонан,.
декан, ундекан, додекан и тридекан. Если продукты пиролиза
не гидрировали, то за характеристические пики для пропилена
принимали пики 2-метил-2-пентена (димер), 2,4,6-триметил-1-
нонена (тетрамер, сум'ма двух стереоформ), 2,4-диметил-1-геп-
тена(тример), для этилена — пики нормальных 1-алкенов
Сб—Си. Для всех возможных пар пиков строили зависимости
Si\/Si2 от Ci/Сг и определяли соответствующие
коэффициенты пропорциональности Ki,i,2, где i — номер пика, 1 —
указывает на отношение пика к пропилену, а 2 — к этилену. Получив
пирограмму анализируемого образца и определив состав по
все'м парам, вычисляют среднее арифметическое значение,
которое и принимают за результат анализа.
Для блочного сополимера и механических смесей градуиро-
вочные зависимости носят линейный характер, а для
статистического сополимера было показано, что Ki и Кг меняются от
состава и их надо определять для каждого интервала
концентраций отдельно. Абсолютная погрешность определения состава
по нескольким парам пиков составляет ±0,2 %, относительное
стандартное отклонение 0,002 -=- 0,02, предел обнаружения
содержания моно'мера 0,3—1,0 %• Эти результаты были получены
при использовании пиролизера печного типа на таких же
капиллярных колонках, что и при анализе полиэтилена.
Сополимер пропилена с винилциклогексаном. Особенностью
методики, примененной для определения состава сополимера
пропилена с винилциклогексаном, является то, что авторы не-
136
использовали капиллярной колонки и, вероятно, поэтому не
смогли обнаружить на пирограм'ме пиков, характерных для
полипропилена [168]. Поливинилциклогексан (ПВЦГ) дает
несколько пиков, отсутствующих на пирограмме полипропилена,
а все пики полипропилена совпадают с пиками ПВЦГ. В связи
с этим для определения пропиленовых звеньев в сополимере
был выбран пик, представляющий собой сумму пиков,
характерных как для полипропилена, так и для ПВЦГ,— пик 1, а для
определения звеньев винилциклогексана — пик, принадлежащий
бензолу,— пик 2. Определив отношение высот пиков а —
= h2/hi = 1,63 для ПВЦГ, а затем получив хроматограмму
механической смеси, вычисляют вклад в пик 1 пика ПВЦГ:
Л?ыч = ft2K%J- По разности между экспериментально найденной
высотой пика 1 h*KC и рассчитанной по уравнению высотой пика
Л?ыч находят высоту пика 1, соответствующую содержанию
полипропилена (ПП). Отношение этой высоты к общей высоте пика 1
Л"п//г?"с дает молярную долю пропилена СПп в смеси. В
качестве насадки использовали порапак Q.
II. 2.4.4. Сополимеры винилацетата
Сополимеры винилацетата с этиленом. При пиролизе из
сополимеров этилена с винилацетатом образуются низкокипящие
углеводороды, бензол и уксусная кислота [169, 170].
Углеводороды могут образовываться как из этиленовых, так и из винил-
ацетатных звеньев сополимера, а уксусная кислота и бензол —
только из винилацетатных. Для сополимеров с высоким
содержанием винилацетата отмечено быстрое падение выхода
уксусной кислоты с повышением температуры и образование
продуктов вторичных реакций: бензола, толуола, стирола и др. (при
использовании печного пиролизера). При низких температурах
(ниже 500 °С) выход уксусной кислоты высокий, но скорость
пиролиза низкая, поэтому наблюдается размывание пиков.
В работе [170] рекомендуется температура пиролиза 550°С.
Зависимость между массовым выходом уксусной кислотой X
(в %) и составом сополимера имеет линейный характер и может
быть выражена формулой:
*ВА= U90.Y —4,27,
где дгва — массовая доля винилацетата в сополимере, %.
Средняя относительная погрешность определения 1,44%.
Сополимеры винилацетата с метакрилатами и акрилатами.
При пиролизе сополимера винилацетата с эфирами метакрило-
вой кислоты выделяются уксусная кислота и эфиры метакрило-
вой кислоты [171]. Если эфирные группы содержат большой
алкильный заместитель, то образуются еще легколетучие
соединения, количество которых растет с ростом температуры.
137
Определение состава этих сополимеров методом ПГХ не
представляет трудностей.
Пирограммы сополимеров винилацетата с эфира'ми
акриловой кислоты имеют более сложный состав. Изучение
характера пирограмм в зависимости от температуры пиролиза и
состава сополимеров показало, что часть сополимера, которая
относится к эфира'м акриловой кислоты, качественно
разлагается по механизму, характерному для распада акрилатных
звеньев и других сополимерах, но имеет свои количественные
особенности, которые надо учитывать.
II. 2.4.5. Сополимеры фтормономеров
Сополимеры тетрафторэтилена с гексафторпропиленом.
При пиролизе сополимеров тетрафторэтилена (ТФЭ) с
гексафторпропиленом (ГФП) выделяются ТФЭ, ГФП, октафторцик-
лооктан и более легколетучие продукты, например, тетрафтор-
метан. Таким образом, качественный состав продуктов
пиролиза сополимера ТФЭ—ГФП подобен составу продуктов
пиролиза ПТФЭ, но количество выделяющегося из сополимера ГФП
больше, чем из ПТФЭ. Характеристическими пиками для
определения состава сополимера являются только пики ТФЭ и ГФП.
Тот факт, что ГФП образуется и из тетрафторэтиленовых
звеньев, создает трудности при разработке методики. При
использовании пиролизера печного типа для ПТФЭ максимум
выделения ТФЭ находится при 600 °С, а ГФП при 750 "С. Для
сополимера ТФЭ—ГФП температура максимума выделения ТФЭ
сохраняется, а для ТФП она смещается к 700°С [147].
Следует отметить, что деструкция сополимера начинается раньше,
чем деструкция ПТФЭ на 100 °С — при 550 °С.
Описаны две 'методики определения состава сополимера
ТФЭ—ГФП в интервале молярных концентраций ГФП от 5
до 20 % [172] и от 4 до 13 % [173]. В первой методике [172]
использовали пиролизер печного типа, температура пиролиза
650 °С, относительное стандартное отклонение 0,05—0,1. Во
второй методике [173] применяли пиролизер по точке Кюри,
температура пиролиза 920°С, относительное стандартное
отклонение 0,12. Такая высокая температура была выбрана
авторами работы потому, что при более низких температурах не
получались воспроизводимые результаты. В обеих методиках
навеска составляла 1 мг, насадкой для колонки служил пора-
пак Q (2 м), но в первой методике в качестве детектора был
использован катарометр, а во второй — ДИП. Сравнение гра-
дуировочных графиков для этих методик (рис. 11.28)
показывает, что чувствительность и воспроизводимость второй методики
выше, чем первой, так как график имеет более крутой наклон.
Авторы работы [173] считают, что ПГХ дает завышенные,
плохо воспроизводимые результаты, поэтому они не рекоменду-
138
Рис. 11.28. Градуировочные графики для опреде- 5ГФП
ления состава сополимера тетрафторэтиле на с гек- ■?
сафторпропнленом по данным работ [172] (/) и °тфэ
[>73] (2).
0,8
5 10 15 20 25
Молярная доля ГФП^
сополимере, %
ют эту методику для определения
состава сополимеров ТФЭ — ГФП. 07
Сополимеры тетрафторэтилена с
этиленом и пропиленом. Фторполи- о,6
меры, имеющие в составе атомы
водорода, при пиролизе образуют 0,5
очень небольшое количество хрома-
тографируемых летучих соединений. ^
Для таких фторполимеров харак- 03
терно образование при пиролизе на '
холодных частях установки белого Q2
налета, состоящего из
низкомолекулярных фрагментов полимера, ИК- о,1
спектры которых сходны со
спектром исходного полимера [172, 174].
Для сополимеров ТФЭ с этиленом
количество летучих продуктов
пиролиза составляет всего несколько
процентов от массы сополимера. Состав летучих продуктов
сополимера ТФЭ с этиленом представлен в табл. П. 8 [175]. При
пиролизе сополимера ТФЭ с этиленом в наибольшем
количестве выделяется винилиденфторид (ВДФ) — 18,5 %. выход
мономера ТФЭ составляет 5 %, а этилен вообще не был
идентифицирован (см. табл. II. 8). При пиролизе сополимера такого
же состава в пиролизере печного типа при 650 °С также
выделяется ВДФ, но его выход составляет 45% [176]. Помимо
ВДФ в этом случае были идентифицированы этилен, ТФЭ и
фтороводород, но в малых количествах (всего несколько
процентов). Выход ВДФ быстро возрастает с повышением
температуры, тогда как выход ТФЭ и этилена меняется
сравнительно мало (рис. II. 29, а). Зависимость выхода ВДФ, ТФЭ и
этилена от состава сополимера имеет сложный характер
(рис. 11.29, б). Это обусловлено тем, что сополимер ТФЭ с
этиленом распадается в основном не на исходные сомоно'меры,
как это наблюдается для большинства ранее рассмотренных
сополимеров, и дает гибридные продукты за счет разрыва
внутримолекулярных связей сомономеров:
-СН2—СН2—CF2-f-CF2—CH2-fCH2—CF2-bCF2—СН2
Тот факт, что основным продуктом, выделяющемся при
пиролизе, является ВДФ, свидетельствует о высокой степени
регулярности чередования звеньев этилена и ТФЭ в сополимере
молярного состава 1 : 1. Из рис. 11.29, б следует, что при моляр-
139
Таблица II. 8. Выход продуктов пиролиза сополимерои
тетрафторэтилена с этиленом и пропиленом, % [175].
Пиролизер по точке Кюри, температура 740 °С, навеска образца 0,5 мг
Номер пика
на пирогрэм-
ме
1
2
3
4
5
6
7
8
9
10
11
12
13, 14
15
16
17
18
19
20
21, 22
23
24
25
Соединение
CF3H
C2F4
C2F2H2
C2FH3 и C2F3H
C2F2H4
C2F4H2
C3H6
C3F4H2
C3H2F4
C3F4H4
C3F6H2 (или
C4F2H6 (или
C4F4H4
C3F6H2 (или
CeF4H4 (или
C5F0H4
C5F4H6
C4F6H2 (или
C5F6H4
C6F6H6
CeFeH4
C6FcH6
C„F4H2)
C4F2H4)
C3F7H)
C6F4H6)
C5F5H3)
Неидентифицнрованные
продукты
Сополимер
этилена
1,8
5,0
18,5
0,8
2,1
2,8
0,6
1,3
1,6
4,4
3,0
—
4,4
1,5
3,1
8,7
2,6
1,5
—
3,8
2,9
4,6
25,0
Сополимер
пропилена
9,5
5,0
11,2
0,5
1,5
3,4
1,8
0,4
4,3
3,3
1,9
1,6
3,4
—
3,2
1,2
7,8
—
3,2
11,5
—
—
25,3
ном соотношении сомономеров, близком к 1, для определения
состава можно использовать соотношение площадей пиков ТФЭ
.» б
к
',см
15
10
5
п
\ /
. /
. /
/^ ^°
&г -у- *-—*з
600 700
Температура пиролиза,°с
0,3 0,5 0,7
Молярная доля
этилена д сополимере
Рис. II. 29. Зависимость выхода основных продуктов пиролиза сополимера
тетрафторэтилена с этиленом (ft и 5 получены при массе образца 1 мг) от температуры пиролиза при
составе сополимера I : I (а) и от состава сополимера при температуре пиролиза 650 °С (б):
1—винилнденфторид; 2—этилен; 3 — тетрафторэтнлен.
140
и этилена, при большем содержании этилена — этилена и ВДФ,
а при меньшем содержании этилена — ТФЭ и ВДФ [172].
Следует иметь в виду, что на эти соотношения очень сильно
влияет микроструктура .сополимера, поэтому определять по ним
состав можно только продуктов, получае'мых строго по одной
технологии.
Состав продуктов пиролиза сополимера ТФЭ с пропиленом
качественно во многом аналогичен составу продуктов пиролиза
сополимера ТФЭ с этиленом, но существенно отличается в
количественном отношении (см. табл. II. 8), что вытекает из его
строения:
-CF2-r CF2—CH2-j-CH2—CF2—CF2-i-CH2—CH
ICH3 CH
i №2—\п-г'
3
CF2 — CF2-
Данных по определению состава этого сополимера 'методом
ПГХ в литературе нет, но очевидно, что в данном случае
будут наблюдаться те же закономерности, что и для сополимеров
ТФЭ с этиленом.
Сополимер тетрафторэтилена с перфторметиловым эфиром.
Основными продуктами пиролиза сополимера ТФЭ с перфтор-
метилвиниловым эфиро'м являются тетрафторметан (ТФМ),
октафторциклобутан и сомономеры. Выход ТФМ больше
суммарного выхода октафторциклобутана и перфторметилового
эфира. В качестве характеристических пиков были выбраны
пики ТФМ и ТФЭ. Градуировочный график построен на
основании данных ИК-спектроскопии [177]. Молярный состав
анализируемого сополимера менялся от 10 до 36 %. Для
определения использовали пиролизер печного типа, те'мпература
пиролиза 650 °С, насадка для колонки — порапак Q.
Другие фторсодержащие сополимеры. Имеются данные по
определению состава сополимера ГФП с винилиденфторидом
[178]. При пиролизе этого сополимера образуется три
основных соединения: винилиденфторид, трифторметан и ГФП.
Предполагается, что трифторметан образуется в результате
отщепления трифторметильной группы, которая затем отрывает атом
водорода от полимерной цепи. Для определения состава этого
сополимера использовали два типа градуировочных графиков:
один выражал зависимость суммы площадей пиков трифторме-
тана и ГФП от массовой доли ГФП (в %) в сополимере,
определенной методом ЯМР; второй — зависимость
относительной площади пика винилиденфторида от его массовой доли
(в %) в сополимере, определенной также методом ЯМР. Оба
градуировочных графика представляют собой прямую с
тангенсом угла наклона в обоих случаях, близким к 1. Средние
различия в массовом содержании мономеров в сополимерах,
полученные методом ПГХ и ЯМР, составляют для ГФП ±0,6%
141
(по первому графику) и для винилиденфторида ±0,85 % (по
второму графику).
Следует отметить, что анализ методом ПГХ сополимеров,
содержащих наряду с атомами фтора и атомы водорода,
представляет большие трудности, так как механизм пиролиза таких
сополимеров сильно зависит от состава и чередования звеньев.
II. 3. ЭЛЕМЕНТНЫЙ АНАЛИЗ
Определить состав сополимеров методом элементного
анализа 'можно в том случае, если один из сомономеров содержит
атом, отсутствующий в другом сомономере. Так, этим методом
можно определить состав сополимеров стирола или этилена с
винилхлоридом, винилацетато'м, тетрафторэтиленом и акрило-
нитрилом.
Содержание элемента в навеске рассчитывают по
содержанию образовавшихся при разложении образца веществ. Зная
содержание элемента в навеске рассчитывают содержание
мономера в сополимере по формуле:
где Хм — массовая доля мономера Mi в сополимере, %; Э — массовая доля
элемента Э в навеске, %; ЛМ[ — атомная масса звена мономера М.; Аэ —
атомная масса элемента Э в звене Mi.
Методы элементного анализа полимеров, как и других
органических веществ, в подавляюще'м большинстве основаны на
предварительном разложении их в атмосфере кислорода,
аммиака, диоксида углерода или инертных газов до конечных
стабильных продуктов, пригодных для дальнейшего химического
или физико-химического анализа. Чаще других при анализе
высокомолекулярных соединений проводят сжигание в атмосфере
чистого кислорода. В результате сгорания образцов
сополимеров, состоящих только из атомов С, Н и О, образуются С02
и Н20. При наличии в составе сополимера атома N в
продуктах сгорания присутствуют оксиды азота, при наличии атома
S — оксиды серы. При сжигании в атмосфере кислорода га-
логенсодержащих соединений образуются соответствующие га-
логенид-ионы.
Процесс разложения полимеров осуществляется
значительно труднее, чем низкомолекулярных органических соединений.
Это является одной из основных причин обычно более
высокой погрешности определения С, Н и других элементов в
полимерах сравнительно с низкомолекулярными веществами.
Низкомолекулярные соединения при высокой температуре (900—
950°С) сначала плавятся, а затем испаряются в объеме
реактора. Полимер при введении в горячую зону вначале деструк-
тирует до низкомолекулярных соединений, которые затем уже
взаимодействуют с кислородом. Термоокислительный распад
142
полимера в условиях горения весьма сложен и многостадиен
[114]. Различия в терморазложении низкомолекулярных
веществ (обычно применяемых в качестве эталонных при
определении С, Н и N на анализаторах CHN) и полимеров
иллюстрирует рис. II. 30.
При проведении элементного анализа высокомолекулярных
соединений с предварительным разложением навески образца
серьезной проблемой является достижение полноты разложения
и выделения элементов в виде, пригодном для последующего
анализа.
Для определения углерода и водорода в полимерах
применяются классические методы с ручным сжиганием образца
[115]. Однако промежуточные продукты разложения полимеров
не успевают окислиться до С02 и Н20 при сжигании в пустой
трубке. Поэтому для анализа полимеров используют сжигание
навески образца в трубке с каталитическим наполнением
(например, последовательные слои оксида кобальта(II) и (III),
серебра, осажденного на А1203, и серебряной проволоки [179]),
которое способствует более полному протеканию процессов
окисления. Если полимеры содержат значительное количество
галогенов, то наполнение трубки не только должно обладать
каталитическими свойствами, но и поглощать образующиеся при
разложении галогенводороды, мешающие определению С и Н
[180]. При определении С и Н в полимерах температура и
время разложения образца, скорость подачи кислорода влияют
на результаты анализа в большей степени, чем при анализе
100
100 300 500
Температура, "С
300 500
Температура'С
Рис. 11.30. Кривая ТГА ( ) и ДТА ( ) низкомолекулярных (а) и
высокомолекулярных (б) органических соединений:
/, 4—ацетанилид; 2, 5 —фенацетин; 3, 6—цистин; 7,10 — полиэтилен; 8. 11 — полистирол;
9. 12 — поливнннлхлорид.
143
низкомолекулярных веществ. Поэтому необходимо строго
соблюдать условия анализа, которые подбирают индивидуально
Для каждого полимера (см. п. II. 5.2).
В последние годы для определения С и Н, а также N, S и
О в основном используют CHN- и С,Н,Ы,0,8-анализаторы, в
которых количество продуктов разложения определяют хромато-
графическим методом (рис. 11.31). Подробно применение
автоматических приборов для определения С, H,N, О, S описано в
обзоре [181]. При использовании CHN-анализаторов для
определения элементов в полимерах необходимо в качестве
эталонных использовать не низкомолекулярные вещества, а полимеры,
близкие по химической природе к анализируемым. Это
значительно повышает воспроизводимость результатов [182].
По-видимому, специфика окислительного высокотемпературного
разложения полимеров учитывается при использовании для расчета
содержания С, Н и N (в %) коэффициентов чувствительности,
определенных по полимерным эталонам:
[С], [Н], [N] = Кс_ н. N (5с, н, N - 4. н. N)/«!
[С, Н, N]T. эттэт
К
С, Н, N
{SC. Н, N ~ 5С, Н, n)
где Kq и к — коэффициент чувствительности каждого элемента; Sc> H N(
^СТ Н № ^С Н N ~ пл°ЩаДь пиков С02, Н?0 или Щ анализируемого,
эталонного вещества и контрольного опыта соответственно; т, тэт — навеска
анализируемого и эталонного вещества соответственно, мг; [С, Н, N]T. Эт —
теоретически рассчитанное содержание С, Н или N в эталонном веществе, %.
'С02
со2
Н,0
N,
Рис. 11.31. Типичные хроматограммы продуктов пиролиза, полученные на CHN-анализа-
торе фирмы „Covo" (ЧССР) (а) и отечественном (б).
144
Высокая термостойкость фторполимеров является основным
источником низкой воспроизводимости результатов при
определении фтора в этих полимерах по методу Шёнигера. Занижение
результатов наблюдается даже при полном сгорании образца,
что связано, по-видимому, с образованием в зоне пламени
термодинамически стабильных и довольно устойчивых к окислению
продуктов COF2 и CF4, а также атомарного фтора, наличие
которого в пламени подтверждено экспериментально. Иногда в
процессе сжигания политетрафторэтилена и некоторых других
фторполимеров не весь образец переходит в газообразное
состояние, поскольку частицы полимера при разогревании
разлетаются за пределы зоны горения, при этом на холодных частях
платиновой проволоки образуется белый налет. При неполном
сгорании поливинилиденфторида, в отличие от ПТФЭ,
образуется карбонизированный остаток.
Для обеспечения воспроизводимого сжигания фторсодержа-
щих полимеров в колбе с кислородом в качестве горючих
добавок предлагались сахароза, глюкоза, парафин, полиэтилен, а
окислительных — пероксид натрия, хлорат калия, нитраты
аммония и калия. Для фторполимеров лучшей горючей добавкой
оказалась полиэтиленовая пленка в виде кусочка 10X15 мм,
толщиной 0,1 мм в сочетании с окислительной добавкой —
нитратом натрия, 2'%-ным раствором которого предварительно
пропитывались бумажные фильтры для сжигания образцов
[183] (см. п. II. 5.3).
При анализе полимерных композиционных материалов,
например полнстирольных пластиков с антипиренами, также
применяются бумажные фильтры, пропитанные 2%-ным раствором
нитрата натрия, что позволяет устранять нежелательные
процессы сажеобразования [184].
Кроме добавок, улучшающих процесс сгорания полимеров,
в ходе элементного анализа используют добавки, которые
устраняют мешающее влияние одних элементов на определение
других. Например, влияние фтора на определение серы во
фторсодержащих полимерах устранялось путем сжигания
образцов в бумажных фильтрах, предварительно пропитанных
0,5 %-ным раствором борной кислоты (после пропитки фильтры
высушивали при 100 °С). При сгорании образцов в этом
случае фтор большей частью удаляется в виде летучего
соединения BF3 и не мешает титрнметрическому определению серы
[185] (см. п.П.5.4).
Определение азота проводят не только на CHN-анализато-
рах и на автоматических анализаторах азота, но также
микрометодом Дюма и полумпкрометодом Кьельдаля. При невысоких
содержаниях (0,5—3%) азот в сополимерах стирола с акри-
лонитрилом и в АБС-пластиках определяется на CHN-анализа-
торах с недостаточной воспроизводимостью. Заниженные
результаты получаются при анализе поли-Ы-винилпирролидона и
его сополимеров [186].
145
Метод Дюма универсален и позволяет определять азот в
соединениях любого химического строения. При анализе
сополимеров стирола с акрилонитрилом, с метилметакрилатом и
акрилонитрплом, АБС-пластиков и различных композиционных
материалов на их основе микрометодом Дюма получаются
плохо воспроизводимые результаты. Это, по-видимому, связано с
особенностями высокотемпературного разложения полистироль-
ной части сополимеров в атмосфере диоксида углерода,
при котором может образоваться некоторое количество
метана, искажающее результаты газометрического определения
азота.
Метод Кьельдаля более прост и удобен, и хотя круг
соединений, которые успешно анализируются этим методом без
применения дополнительного восстановления, ограничен аминами,
амидами и нитрилами, для анализа полимеризационных
пластиков он пригоден и в модифицированном виде применяется чаще,
чем метод Дюма. Для ускорения разложения полимерного
образца при нагревании с концентрированной серной кислотой
добавляется пероксид водорода и каталитическая смесь,
состоящая из персульфата калия и сульфата меди (см.
п. II.5.5). Этим методом при увеличении времени разложения
до 90 мин получены вполне удовлетворительные результаты
определения азота в поли-Ы-винилпирролидоне и его
сополимерах. Однако при анализе на азот этих весьма гигроскопичных
полимеров следует определять содержание воды методом
Фишера и учитывать его при расчете содержания азота [186].
Кислород в полимерных материалах часто определяют по
разности между 100 % и суммарным содержанием элементов
С, Н, N и др. Метод прямого микроопределения кислорода
основан на пиролитическом разложении навески полимера в
течение 2—3 мин при 1000 °С в замкнутой системе. Продукты
пиролиза с током аргона проходят над слоем
гранулированного технического углерода, нагретым до 1125°С. При этом весь
кислород превращается в оксид углерода, который затем
количественно окисляется до диоксида углерода с помощью
оксида меди(П), нагретого до 500°С. Количество
образующегося кислорода определяют гравиметрически и
рассчитывают молярный состав сополимеров: стирола с винилацетатом,
стирола с метакриловой кислотой, с метилметакрилатом и др.
[187].
Для количественного определения гетероэлементов в
полимерных образцах после сжигания применяются различные
методы, чаще всего титриметрические, фотометрические и
электрохимические. Среди электрохимических методов для определения
галогенов следует отметить ионометрический метод с
использованием селективных электродов, обладающих высокой
чувствительностью и избирательностью. Этот метод прост и удобен в
работе, если применяется серийная измерительная аппарату-
146
pa [183]. В последние годы все более широкое применение
находит ионная хроматография, которая позволяет определять с
высокой чувствительностью несколько ионов одновременно в
одной пробе.
II. 4. ФУНКЦИОНАЛЬНЫЙ АНАЛИЗ
Функциональные группы, входящие в состав полимерной
молекулы, в принципе способны вступать в те же химические
реакции, в которые они вступают, находясь в составе
низкомолекулярных соединений. При функциональном анализе обычно
используются реакции полимераналогичных превращений
полимерной молекулы с участием низкомолекулярных соединений.
Протекание таких реакций имеет свою специфику, которая
делает разработку методик количественного функционального
анализа применительно к полимерам достаточно сложной
задачей.
Условия, при которых можно переносить представления и
закономерности, известные для реакций низкомолекулярных
органических соединений, на реакции высокомолекулярных
соединений с низкомолекулярными можно сформулировать"
следующим образом [188]:
1) реакция должна протекать в гомогенной жидкой среде
и все исходные вещества, промежуточные и конечные продукты
реакций должны быть растворимы в этой среде;
2) в каждом элементарном акте реакции должна
участвовать лишь одна функциональная группа полимера.
Следует иметь в виду также, что на скорость реакции
может оказывать влияние тот факт, что реакция протекает
в вязком растворе и возможны надмолекулярные эффекты,
связанные с ассоциацией и агрегацией макромолекул. Кроме того,
скорость реакции может меняться в процессе реакции с
изменением конверсии, так как меняется химическая природа
соседнего звена и это может изменять реакционную способность
анализируемых функциональных групп. Поэтому при
разработке методик определения состава сополимеров методами
функционального анализа необходимо уделять большое внимание
подбору растворителей, в среде которых протекает реакция, и
продолжительности проведения реакции.
Из полимеризационных пластмасс функциональный анализ
применяется для определения состава сополимеров винилацета-
та, акрилатов и ацеталей.
II. 4.1. Сополимеры винилацетата
Если в сополимере винилацетата другой сомономер не
подвергается омылению, то определение состава сополимера
основывается на его щелочном омылении с последующим потенцио-
147
метрическим титрованием избытка щелочи соляной кислотой:
~СН2—СН— СН2—СН— СН2 СН—СН2~ + NaOH —>
I
О—СО—СНЭ X
—v ~СН2—СН—СН2-СН—СН2—СН—СН2~ + КаОСОСНэ
X ОН X
NaOH + HCl —>• NaCl + H20.
К таким сополимерам относятся сополимеры винилацетата
с виниловым спиртом, с этиленом, поливинилацетали. Для
анализа поливинилового спирта в качестве растворителя
используют воду, для поливинплацеталей — смесь этилового
спирта с бензолом (1 : 1), а для сополимера винилацетата с
этиленом— смесь ксилола с гекснловым и этиловым спиртами.
Растворы полимеров обычно готовят концентрацией не выше 2—
3 %• Растворитель желательно выбирать более высококипящий,
так как это позволяет вести реакцию омыления при более
высокой температуре, значит сокращать ее продолжительность, а
следовательно, и длительность всего анализа.
Содержание ацетатных групп в сополимере X (в %)
вычисляют по формуле:
X = (£/,- U2) A- 100/т,
где Vi и V2 — объем раствора соляной кислоты, затраченный на титрование
контрольной и анализируемой пробы, соответственно, мл; А — количество
винилацетата, соответствующее 1 мл раствора соляной кислоты, используемой
для титрования, г; т — навеска сополимера, г.
Для титрования используют обычно 0,5 н. раствор соляной
кислоты; в этом случае значение А составляет 0,0295 г.
Анализ сополимеров, в которых второй сомопомер также
может хотя бы частично омыляться, значительно усложняется,
и вследствие его трудоемкости применяется теперь редко,—
его вытесняют другие методы. К таким сополимерам относятся
сополимеры винилацетата с вннилхлоридом, винилпропионатом,
вннплбутнратом [12, с. 245, 458].
11.4.2. Полиацетали
Метод определения ацетальных групп в полиацеталях
основан на разложении полиацеталей серной кислотой с
последующей отгонкой паром образовавшихся альдегидов. Гидролиз по-
ливпнилацеталей протекает по следующей реакции:
■£>
H2SO4 Н20 ^
~СН2—СН—СН2—СН—СН2~ *■ R—С +
I \н
-СН—О
А-с
R
+ ~СН2—СН—СН2—СН—СН2~.
I I
он он
148
Образовавшиеся альдегиды в отгоне можно определять
различными методами. Если поливинилацетат содержит только
ацетальные группы одного типа, то количество образовавшихся
альдегидов определяют методом оксимирования, основанным на
взаимодействии альдегидов с хлоридом гидроксиламиния:
R—С + NH2OH-HCl —► R—Ч +НС1 + Н20
\н хн
НС1 + NaOH —► NaCl + Н20.
Образовавшуюся соляную кислоту затем оттитровывают
щелочью потенциометрически или в присутствии индикатора. Если
поливинилацетали смешанные, то образуется не один, а два
различных альдегида, которые требуется определять раздельно.
В этом случае для определения альдегидов используют
полярографический [ГОСТ 15874—81] или хроматографнческий
анализ [189, 190]. Подобное описание установки для разложения
поливинилацетата, получения отгона и анализа методом
оксимирования приводится в [12, с. 443].
II. 4.3. Сополимеры метакрилатов и акрилатов
Содержание связанных алкилметакрилатов и алкилакрила-
тов в их сополимерах со стиролом и с акрилонитрилом можно
определять методом Фпбека.
Метод Фнбека основан на взаимодействии алкоксильных
групп с иодоводородной кислотой. Образующийся алкилиодид
при действии брома переходит в алкнлбромид и иодбром;
последний окисляется бромом до йодноватой кислоты, которая
определяется иодометрически [12, с. 374]:
СН3
I
~СН2—СН—СН2—СН— СН2~ + «HI —>
I I
X COOR
СНз
I
—► raRI + ~СН2—СН—СН2— СН— СН2~
I I
СООН X
RI + Br2 —*■ RBr + IBr
1Вг + 2Вг2 + ЗН20 —► НЮз + 5НВг
HI03 + 5HI —> 3I2 + 3H20
3I2 + 6Na2S203 —>■ 6NaI + 3Na2S4Oe.
149
Содержание алкилметакрилата или алкилакрилата в
сополимере X (в %) вычисляют по формуле:
X = (Vl-V0)Ka-lOO/m,
где V] и Va — объем 0,1 н. раствора Na2S203, затраченный на титрование
анализируемой и контрольной проб, мл; К — поправочный коэффициент 0,1 н.
раствора Na2S2CV, а — количество акрилата, соответствующее 1 мл точно
0,1 н. раствора Na2S203, для метилметакрилата а = 0,00166 г, для метил-
акрилата а = 0,00143 г; т — навеска сополимера, г.
Следует иметь в виду, что для алкилметакрилатов и алкил-
акрилатов, в алкильных заместителях которых больше 4
атомов углерода, реакция Фибека протекает не количественно.
II. 4.4. Ударопрочные полистирольные пластики
Задача определения состава ударопрочных полистирольных
пластиков отличается от остальных аналогичных задач тем, что
они являются гетерогенными системами и состоят из
полимерной матрицы, привитого сополимера и сшитой полимерной
части (гель-фракции). На практике бывает необходимо
определить количество и состав каждой из этих частей в отдельности.
Для этого необходимо перед анализом разделить сополимер на
составные части. Разделение проводят методом селективного
фракционирования.
Растворители подбирают таким образом, чтобы вначале
отделить полимерную матрицу, не содержащую каучука, от
привитой части и гель-фракции. Затем привитую часть отделяют
от гель-фракции. Для этого ее растворяют и раствор
центрифугируют. Выделенные части анализируют по соответствующим
методикам.
Растворимая полимерная матрица может представлять собой
полистирол или его сополимеры. В последнем случае ее состав
определяют по методикам, применяемым для определения
состава сополимеров стирола.
Привитая часть представляет собой полимер, как правило,
того же состава, что и полимерная матрица, привитый к
каучуку (бутадиеновому, этилен-пропиленовому, бутилакрилат-
ному).
Гель-фракция по составу отличается от привитой части
только в количественном отношении — она содержит меньшее
количество каучука, чем привитая. В привитой части и
гель-фракции определяют содержание каучука: бутадиенового (см.
п.II. 5.11) и бутилакрилатного прямым методом, а этилен-про-
пиленового—по разности (см. п.II. 5.10).
Следует иметь в виду, что разделение на фракции является
условным, так как зависит от природы выбранных
растворителей, и поэтому можно сравнивать только результаты,
полученные по одной и той же методике.
150
11.5. МЕТОДИКИ ОПРЕДЕЛЕНИЯ СОСТАВА СОПОЛИМЕРОВ
МЕТОДАМИ ЭЛЕМЕНТНОГО
И ФУНКЦИОНАЛЬНОГО АНАЛИЗА
II. 5.1. Одновременное определение содержания углерода,
водорода и кремния
Метод основан на пиролитическом разложении полимера в
токе кислорода с использованием для каталитического
наполнения трубки сжигания оксида кобальта (II) и (III). При
800°С происходит полное окисление углерода и водорода до
диоксида углерода и воды, а азота — до диоксида азота.
Наличие галогенов, серы и азота не мешает определению, так как
продукты окисления серы и галогены полностью задерживаются
слоем губчатого серебра, а оксиды азота улавливаются
диоксидом марганца вне трубки. Диоксид углерода и воду определяют
по привесу поглотительных аппаратов, наполненных аскаритом
и ангидроном соответственно; кремний (при анализе кремний-
органических соединений)—-по привесу оксида кремния,
адсорбированного на кварце в стаканчике для разложения
полимера.
Аппаратура, реактивы
Стандартная установка для определения углерода, водорода и кремния.
Ацетальдегид.
Нитрат алюминия.
Карбонат аммония.
Перманганат калия.
Азотная кислота.
Щавелевая кислота.
Нитрат кобальта.
Сульфат марганца.
Гидроксид натрия.
Нитрат серебра.
Метиловый красный.
Этиловый спирт ректификованный технический.
Приготовление катализатора оксида кобальта(П) и (III)
Соз04. Оксалат кобальта осаждают из раствора хлорида или
нитрата кобальта раствором щавелевой кислоты. 88 г
щавелевой кислоты растворяют в 500 мл дистиллированной воды при
слабом подогреве и смешивают с раствором 145 г Co(N03)2-
•6Н20 в 200 мл воды. Выпавший в осадок оксалат кобальта
переносят на воронку Бюхнера и промывают на ней
дистиллированной водой до исчезновения реакции на щавелевую кислоту
(с метиловым красным). Затем осадок промывают спиртом,
после чего кусками или пластом переносят в чашки Петри,
высушивают в течение 12 ч в сушильном шкафу при 110—120 °С
и прокаливают в фарфоровой чашке при 550—600°С 4—5 ч.
Из слегка смоченного порошка Соз04 делают гранулы,
протирая его через сито № 1. Высушенные при 100°С в сушильном
шкафу гранулы Со304 прокаливают в токе кислорода в течение
151
20 ч при 800 °С. Готовый катализатор хранят в склянках с
пришлифованными пробками.
Приготовление губчатого серебра, обработанного оксидом
алюминия. Растворы, содержащие 85 г AgN03 в 100 мл
дистиллированной воды и 31 г NaOH в 120 мл воды, осторожно
сливают и перемешивают. Образующийся осадок оксида серебра
при перемешивании восстанавливают в 150 мл раствора,
содержащего 14,5 мл ацетальдегида. Образуется губчатое
серебро, которое промывают декантацией до исчезновения щелочной
реакции (по лакмусовой бумаге). Полученную водную
суспензию обрабатывают вначале раствором 13,2 г А1(Г\10з)з-9Н20 в
200 мл воды (5%-ный раствор), а затем раствором 1 г
(NH4)2C03-H20 в 50 мл воды. В стакан с осадком наливают
дистиллированную воду, дают осадку отстояться, после чего
дважды декантируют и фильтруют через стеклянную воронку
с бумажным фильтром. Осадок высушивают в сушильном
шкафу при 150°С, измельчают до размеров частиц 0,3—1,2 мм
и прокаливают в токе кислорода при 500°С в течение 5 ч.
Готовое губчатое серебро ссыпают в склянку темного стекла с
пришлифованной пробкой и хранят в эксикаторе.
Приготовление диоксида марганца Мп02 для поглощения
оксидов азота. 31,5 г перманганата калия растворяют в 600 мл
горячей дистиллированной воды (50—60°С) и этот раствор
понемногу, при интенсивном перемешивании вносят в раствор
68 г MnS04-4H20 в 200 мл воды. Выпавший осадок Мп02
декантируют 3—4 раза водой, переносят на пористый стеклянный
фильтр № 3, промывают водой до отрицательной реакции на
ион SOi'h сушат 5—10 ч сначала на воздухе, а затем в
сушильном шкафу при 80°С до содержания воды 16—18%.
Высушенный Мп02 измельчают до размеров частиц 1—1,5 мм, ссыпают
в склянку с пришлифованной пробкой и хранят в эксикаторе.
Подготовка кварца для определения кремния. Кварцевое
стекло разбивают в ступке до размеров частиц 1—2 мм,
обрабатывают их сначала 20 %-ным раствором гидроксида натрия в
течение суток, затем раствором Н1МОз (1:1) и отмывают
дистиллированной водой до нейтральной реакции по метиловому
красному. Полученный кварц сушат, прокаливают в токе
кислорода при 900°С 3—7 ч, охлаждают в токе кислорода, затем
ссыпают в склянку с пришлифованной пробкой и хранят в
эксикаторе.
Подготовка трубки для сжигания. В носик трубки
помещают кусок серебряной проволоки длиной 30—40 мм, затем
прокладку из прокаленного асбеста и слой губчатого серебра,
длиной 50—70 мм, затем между прокладками из прокаленного
асбеста слой Со304 длиной 100—120 мм. Через вновь
собранную установку при нагретых печах в течение 1 ч пропускают
кислород со скоростью 35—50 мл/мин, затем трубку
отсоединяют, ставят в вертикальное положение и легким
постукиванием деревянной линейкой уплотняют слой катализатора.
152
Трубку присоединяют к установке и пропускают кислород еще
в течение 30 мин, прокаливая трубку электрогорелкой. После
этого установка готова к работе.
Выполнение анализа. Определение углерода и водорода. 3—
5 мг анализируемого полимера взвешивают в кварцевом
стаканчике с точностью до 0,02 мг. Стаканчик помещают в трубку
для сжигания, закрывают ее, на стаканчик надвигают
электрогорелку и быстро (в течение ~5 мин) проводят сжигание
полимера при температуре 950 °С. Продукты разложения в
течение 15—20 мин вытесняют током кислорода, пропускаемого со
скоростью 15—20 мл/мин, т. е. за 25—30 мин через трубку
проходит около 500 мл кислорода. По окончании сжигания
поглотительные аппараты для диоксида углерода и воды
отсоединяют, переносят к весам и взвешивают — через 10 мин аппарат
для поглощения диоксида углерода и через 15 мин аппарат
для поглощения воды. По привесу поглотительных аппаратов
вычисляют содержание углерода и водорода.
В аналогичных условиях проводят контрольный опыт (как
правило, на сахарозе) и устанавливают поправки (если они
есть) на углерод и водород.
При анализе полиолефинов устанавливают температуру
печи разложения 880°С, продолжительность разложения 7 мин,
скорость пропускаемого кислорода 17 мл/мин. Поправки
определяют по полиэтилену.
Определение углерода, водорода и кремния. 3—5 мг
анализируемого полимера взвешивают с точностью до 0,02 г в
кварцевом стаканчике длиной 90 мм, диаметром 7 мм и засыпают
на 3Д объема свежепрокаленным кварцем. Затем стаканчик
вновь взвешивают и помещают в трубку сжигания; трубку
закрывают и проводят разложение, медленно продвигая
электрогорелку от открытого конца стаканчика к навеске, но не
надвигая ее непосредственно на навеску. Необходимо следить за
тем, чтобы разложение проходило медленно и заканчивалось в
стаканчике на кварце.
Скорость разложения полимера контролируют по изменению
цвета кварца под влиянием продуктов разложения от желтого
к черному, а затем к постепенному обесцвечиванию. В общей
сложности процесс разложения и вытеснения длится 40 мин
(для трудносгорающих полимеров — до 50 мин). Скорость
кислорода и температуру разложения устанавливают в
зависимости от химической природы анализируемого продукта:
при анализе кремнийорганических соединений скорость
кислорода 8—12 мл/мин, температура разложения 950 °С;
при анализе ударопрочных сополимеров стирола на основе
силоксановых каучуков скорость кислорода 5—6 мл/мин,
температура разложения 1000 °С.
По окончании разложения поглотительные аппараты для
диоксида углерода и воды отсоединяют и переносят к весам.
Кварцевый стаканчик с кварцем осторожно достают из трубки
153
сжигания, ставят на подставку, накрывают колпачком и
переносят к весам. Взвешивают аппарат для поглощения
диоксида углерода через 10 мин, аппарат для поглощения воды через
15 мин, кварцевый стаканчик — через 25 мин.
В аналогичных условиях проводят контрольный опыт на
эталонных кремнийорганических соединениях и устанавливают
поправки (если они есть) на углерод, водород и кремний.
Расчет. Содержание углерода [С], водорода [Н] и
кремния [Si] (в %) вычисляют по формулам:
[С] = mCQ2Mc ■ Ю0/(тМСО2) = 27,29mC02/m
[Н] = mHi0MH7 ■ Ю0/(тМН2О) = 11.19m„iQ/m
[Si] = ihs10sAJs, • 100/(mMSiO2) = 46,75mSIOs/m,
где тсо, «гн,0 и т5Ю2 — масса образовавшихся СО„, Н,0 и SiO,
соответственно, мг; т — масса образца, мг; Мс, Мн, MSi, МСОг, МНг0 и AfSi0, —
молекулярные массы углерода, водорода, кремния, СОг, НгО и S1O2
соответственно.
П. 5.2. Определение углерода и водорода
в высокогалогенированных полимерах
Метод основан на пиролитическом разложении полимеров,
жидких мономеров и других органических веществ с высоким
содержанием фтора (до 76%), хлора или брома (до 75%).
содержащих также серу и азот, в токе кислорода в
присутствии платины и смешанного катализатора, полностью
окисляющего продукты разложения и поглощающего фтор, хлор, серу
и бром. Оксиды азота улавливаются вне трубки сжигания
диоксидом марганца при комнатной температуре. Диоксид
углерода и воду определяют по привесу поглотительных аппаратов.
Аппаратура, реактивы
Стандартная установка для определения углерода и водорода.
Нитрат серебра.
Вольфрамат натрия.
Оксид магния.
Нитрат церия.
Этиловый спирт ректификованный, технический.
Метиловый красный.
Щавелевая кислота.
Перманганат калия.
Сульфаг марганца.
Приготовление катализатора. К раствору вольфрамата
натрия (29,3 г/л) при перемешивании приливают равный объем
0,1 н. раствора нитрата серебра и смесь кипятят 10 мин.
Осадок отфильтровывают, промывают сначала горячей, потом
холодной водой до удаления ионов серебра, после чего сушат в
течение 2 ч при 105°С. К раствору щавелевой кислоты (80 г
щавелевой кислоты растворяют в 500 мл подогретой
дистиллированной воды) добавляют при перемешивании раствор нитрата
154
церия (150 г нитрата церия в 200 мл дистиллированной воды).
Выпавший белый осадок оксалата церия переносят на воронку
Бюхнера, промывают на ней дистиллированной водой до
исчезновения реакции на щавелевую кислоту (с метиловым
красным). Затем осадок промывают спиртом, переносят в чашки
Петри, высушивают в течение 3—5 ч в сушильном шкафу при
110°С, после чего прокаливают в фарфоровой чашке при
550—600 °С в течение 4—5 ч. Диоксид церия и оксид магния
перед применением прокаливают в муфеле при 800°С в
течение 3 ч. Вольфрамат серебра, диоксид церия, оксид магния
смешивают в соотношении 2 : 14: 1, хорошо растирают в
ступке с небольшим количеством воды до получения однородной
густой массы, которую продавливают через сито с диаметром
отверстий 1,5 мм, высушивают в течение 2 ч при 120°С и
отсеивают от пыли. После заполнения трубки катализатор
прокаливают в токе кислорода при 850 °С 4 ч.
Приготовление диоксида марганца Мп02 для поглощения
оксидов азота, см. в II. 5.1.
Подготовка кварца для определения кремния, см. в II. 5.1.
Подготовка трубки для сжигания. В трубку, начиная от
носика, помещают слой губчатого серебра длиной 50 мм, слой
универсального катализатора длиной 80 мм — смесь вольфрама-
та серебра, оксида церия и оксида магния в соотношении
2:14:1. Слои губчатого серебра и катализатора разделяют
прокладками из прокаленного асбеста. После слоя
катализатора в трубку помещают кварцевый патрон с дырчатым дном,
заполненный мелкораздробленным кварцем, который заменяют
новым по мере отработки. Окислительная зона нагревается
электропечью МА-2/14, отрегулированной на . 680—700°С
(катализатор), и электропечью МА-Г/6Р, отрегулированной на
400—450°С (губчатое серебро).
Выполнение анализа. Через вновь собранную установку при
нагретых печах пропускают в течение 30 мин кислород со
скоростью 35—50 мл/мин, прокаливая в это время трубку для
сжигания электрогорелкой (900—1000°С). После этого
установка готова к работе.
Взвешенные поглотительные аппараты присоединяют к
установке. 3—5 мг анализируемого полимера взвешивают с
точностью до 0,02 мг в кварцевом стаканчике и навеску засыпают
кварцем на '/5 длины стаканчика (при анализе хлор- и бром-
содержащих продуктов кварц не добавляют). В стаканчик
вносят платиновый контакт—свернутую в спираль проволоку или
свернутую в рулон сетку из платины (длиной около 20 мм) и
помещают его в трубку для сжигания открытым концом против
тока кислорода. Надвигают на стаканчик электрогорелку и
проводят сжигание, начиная от открытого конца стаканчика и
постепенно приближаясь к навеске. Для фторполимеров,
содержащих водород, скорость кислорода 10—15 мл/мин (т. е. за
20—25 мин проходит 250—300 мл кислорода), для фторполи-
155
адеров, не содержащих водорода, скорость кислорода снижают
до 3—5 мл/мин.
По окончании сжигания поглотительные аппараты для
диоксида углерода и воды отсоединяют, переносят к весам и
взвешивают аппарат для поглощения диоксида углерода через
10 мин, аппарат для поглощения воды через 15 мин. По
привесу поглотительных аппаратов вычисляют содержание углерода
и водорода.
В аналогичных условиях проводят контрольный опыт и
устанавливают поправки (если они есть) на углерод и водород.
Содержание углерода [С] и водорода [Н] (в %) вычисляют
по формулам, приведенным в II. 5.1.
П.5.3. Определение фтора во фторполимерах
Методика основана на переведении органически связанного
фтора в ионное состояние путем сжигания навески фторполи-
мера в атмосфере кислорода в присутствии платины.
Последующее определение ионов фтора проводится потенциометриче-
ским титрованием с использованием фторселективного
электрода.
Аппаратура, реактивы
рН-метр или иоиомер универсальный.
Хлорсеребряный проточный электрод ЭВЛ-ШЗ.
Фторселективный электрод.
Мешалка магнитная, лабораторная.
Муфельная печь.
Бюретки вместнмотгью 10 мл с ценой деления 0,02 мл.
Колбы мерные вместимостью 100 мл и 1 л.
Пипетки градуированные на 20 мл.
Колбы конические со шлифом из термостойкого стекла вместимостью
100 мл, 500 мл н 1 л.
Пробки пришлифованные со впаянными в них платиновыми проволоками
(длиной 130—150 мм) и сетками на конце проволоки (40X20 мм).
"Стаканы химические вместимостью 500 мл и 1 л.
Тигли фарфоровые.
Колонка стеклянная высотой 1000 мм и диаметром 30 мм.
Катионит марки КУ-2-8.
Анионит марки АВ-17-8.
Кварц дробленый.
Пленка полиэтиленовая толщиной 100 мк.
Вода дистиллированная.
Этиловый спирт ректификованный технический.
Нитрат лантана.
Оксалат аммония.
Нитрат натрия.
Фторид натрия.
■Соляная кислота.
Аммиак водный.
Гидроксид натрия, 0,1 н. и 4%-ный водный раствори.
Азотная кислота, 0,3 н. водный раствор.
Серная кислота, 5 %-ный водный раствор.
Метиловый красный
Метиловый оранжевый.
156
Фенолфталеин.
Фильтры беззольные.
Кислород газообразный.
Получение деионизированной воды. Для приготовления
растворов используется только деионизированная вода, которую
получают на стеклянной колонке, заполненной катионитом
КУ-2-8 и анионитом АВ-17-8. Подготовку ионитов проводят
следующим образом. Катпонит и анионит по 200 мл помещают в
стаканы вместимостью 1 л, заливают дистиллированной водой и
оставляют для набухания 6—8 ч. Затем воду сливают, катио-
нит заливают 500 мл 5%-ного раствора серной кислоты,
анионит— 500 мл 4 %-ного раствора едкого натра и выдерживают
2—3 ч при помешивании.
Для ионитов, не использовавшихся ранее, необходимо
дважды повторять регенерацию. Затем иониты отмывают
дистиллированной водой до нейтральной реакции промывных вод: катио-
нит —по метиловому оранжевому, анионит —по фенолфталеину.
Иониты загружают в колонку в смеси с водой во избежание
образования воздушных пузырьков между зернами.
Дистиллированную воду пропускают со скоростью 55—60 мл/с.
Загрузку колонки начинают, помещая в суженную часть
колонки тампон из стекловаты, затем слой (2—3 см) дробленого
кварца, предварительно промытого горячей дистиллированной
водой. На дробленое кварцевое стекло помещают слой аниони-
та, покрывают его тампоном из стекловаты, затем помещают
слой катионита, вновь слой (2—3 см) дробленого кварца и
покрывают тампоном из стекловаты. По мере обработки
иониты выгружают из колонки и регенерируют, как описано выше.
Приготовление раствора нитрата лантана и определение его
титра. Для титрования используют 0,004 М раствор нитрата
лантана, который готовят разбавлением 0,027И раствора.
Для приготовления 0.02М раствора нитрата лантана
навеску 8,658 г La(N03)3-6H20, взвешенную с точностью до
0,0002 г, растворяют в 200—300 мл деионизированной воды в
мерной колбе вместимостью 1 л и доводят раствор этой же
водой до метки при 20°С.
Для определения титра 0,02М раствора нитрата лантана
мерной пипеткой берут 50 мл приготовленного раствора,
переносят в стакан вместимостью 500 мл и добавляют мерным
цилиндром 21 мл 0,5 н. раствора оксалата аммония для
осаждения оксалата лантана. Выпавший осадок растворяют в
концентрированной соляной кислоте, прибавляя ее в стакан по
каплям и избегая большого избытка. Содержимое стакана
разбавляют водой до 100 мл, нагревают на плитке до 70—80 °С
и вновь осаждают оксалат лантана при энергичном
размешивании, прибавляя по 1—2 капли в секунду с некоторыми
интервалами разбавленный водой 1:1 10%-ный раствор аммиака.
Аммиак прибавляют до ощутимого, но не очень сильного
запаха. Осадок отфильтровывают не ранее, чем через 6 ч после
157
Рис. 11.32. Колба (а), фильтр-
„Флажок" (6") н капилляр (в) для
проведения анализа по методу
Шёиигера.
осаждения, чтобы он
успел полностью
созреть.
Отфильтрованный осадок (с
фильтром) помещают в
фарфоровый тигель, озоля-
_ ют и прокаливают при.
а 6 в температуре 1100°С в-
муфеле до постоянной
массы.
Поправочный коэффициент К 0.02М раствора нитрата
лантана вычисляют по формуле:
К = т/0,1630,
где т — определенная масса осадка ЬагОз, г; 0,1630 — теоретически
рассчитанная масса осадка La2Oa, образующегося из 50 мл точно 0,02 М раствора
нитрата лантана, г.
0,004М раствор нитрата лантана готовят, разбавляя в
мерной колбе вместимостью 100 мл 20 мл 0.027W раствора нитрата
лантана и доводя раствор деионизированной водой до метки
при 20 °С. Вычисленный по приведенной выше формуле
поправочный коэффициент К сохраняется и для 0,004/W раствора.
Приготовление бумажных фильтров для сжигания навески-
полимера. Из беззольных фильтров вырезают по шаблону
размером 20X15X40 мм «флажки» (рис. 11.32,б), которые
пропитывают 2'%-ным раствором нитрата натрия таким образом,
чтобы длинный конец «флажка», оставляемый для поджигания,
не был пропитан раствором. Фильтры помещают на чашку
Петри и высушивают в сушильном шкафу при 100 °С. Сухие
фильтры хранят в эксикаторе.
Выполнение анализа. Подготовку рН-метра (или иономера)
к измерениям проводят в соответствии с его паспортом. Фтор-
селективный электрод до проведения измерений выдерживают
не менее 30 мин в растворе фторида натрия с содержанием
ионов фтора 0,001 моль/л.
Навеску фторполимера в количестве 3—5 мг (в
зависимости от содержания фтора) взвешивают с точностью до 0,02 мг,
помещают на середину фильтра, накрывают кусочком
полиэтиленовой пленки размером 10 X 15 мм, заворачивают в пакетик
и зажимают не очень туго (для обеспечения большего доступа
кислорода к горящему образцу) в платиновую сетку, оставляя
свободным нижний конец фильтра для поджигания. В колбу,
помещенную в обрешетку из стальной сетки, наливают 10 мл
деионизированной воды и наполняют кислородом в течение 3—
5 мин, затем шлиф колбы смачивают водой с помощью
стеклянной палочки и плотно закрывают пробкой с подожженным
образцом.
158
После сжигания навески колбу встряхивают и выдерживают
30—40 мин для поглощения продуктов сжигания. Затем колбу
открывают, обмывают пробку с платиновой сеткой над колбой
водой и содержимое колбы количественно переносят в
коническую колбу из термостойкого стекла вместимостью 100 мл
таким образом, чтобы общий объем был 75—80 мл. В колбу
добавляют 3 мл 0,1 н. раствора едкого натра и упаривают на
плитке с асбестовой сеткой до объема 25 мл (метка
предварительно наносится на колбу). Раствор охлаждают, приливают
40 мл этилового спирта и из бюретки по каплям нейтрализуют
0,5 н. раствором азотной кислоты до рН 5,7—6,2 по стеклянному
электроду или по метиловому красному (3 капли).
Колбу помещают на магнитную мешалку, опускают фторсе-
лективный и хлорсеребряный электроды и титруют постепенно
из микробюретки 0,004М раствором нитрата лантана, пока
стрелка прибора не достигнет значений 140—150' мВ (область
перед началом скачка титрования). Далее раствор нитрата
лантана прибавляют по 0,1 мл, регистрируя после каждого
прибавления значение ЭДС (в мВ) при полной остановке
стрелки прибора (через 2—3 мин). Титрование считается
законченным при получении практически одинаковой разности
между двумя последующими измерениями ЭДС.
Расчет. По полученным результатам строят кривую потен-
циометричского титрования в координатах ЭДС (мВ) — объем
раствора нитрата лантана (мл). Эквивалентный объем,
пошедший на титрование, можно определять любым из известных
методов — графически, по первой или второй производной или
расчетом.
Содержание фтора в образце [F] (в %) вычисляют по
формуле:
F = VK- 0,004- 57- 100/m,
где V—объем 0,004 М раствора нитрата лантана, пошедший на титрование,
мл; /( — поправочный коэффициент 0,004 М раствора нитрата лантана;
0,004—молярность раствора нитрата лантана; 57— масса фтора в реакции
титрования (LaFs); m — масса анализируемого образца, мг.
II. 5.4. Определение серы в высокофторированных
серусодержащих полимерах
Метод основан на сжигании серуфторсодержащего полимера
или мономера в колбе, наполненной кислородом, в присутствии
платинового катализатора и борной кислоты. Продукты
сгорания окисляются пероксидом водорода, а образовавшийся
сульфат-ион титруют нитратом бария в присутствии нитхромазо в
качестве индикатора. Мешающее влияние фторид-иона
устраняют действием на него борной кислоты и упариванием раствора.
Аппаратура, реактивы
Колбы конические со шлифом из термостойкого стекла вместимостью
500 мл. Пробки пришлифованные со впаянными в них платиновыми прово-
159
локами (длиной 130—150 мм) и сетками на концах проволок (40x20 мм).
Колбы конические из термостойкого стекла вместимостью 100 мл.
Колбы мерные вместимостью 100, 500 мл и 1 л.
Ампулы и капилляры для анализа жидкостей.
Микробюретка вместимостью 10 мл с ценой деления 0,02 мл.
Пипетки вместимостью 2, 10 и 15 мл.
Обеззоленные фильтры, вырезанные по шаблону «флажок» (см. рис. 32,6)
размером 20X15X40 мм; фильтры пропитывают 0,5 %-иым раствором
борной кислоты и высушивают в сушильном шкафу при 90—100 °С.
Пероксид водорода, 6 %-ный раствор.
Соляная кислота, 0,1 н. раствор.
Серная кислота, 0,03 н. раствор, приготовленный из фиксанала.
Борная кислота, 0,5 %-иый раствор.
Нитрат бария, 0,03 н. раствор.
Нитхромазо, 0,2 %-ный раствор.
Этиловый спирт ректификованный технический.
Баллон с кислородом.
Установление поправочного коэффициента к титру
0,03 н.раствора нитрата бария. В коническую колбу вносят
пипеткой 3—5 мл 0,03 н. раствора серной кислоты,
приготовленного из фиксанала, добавляют 8—10 мл дистиллированной
воды, 2 мл 0,1 н. раствора соляной кислоты, 25 мл этилового
спирта, 3 капли индикатора и титруют 0,03 н. раствором
нитрата бария до перехода фиолетовой окраски в голубую.
Поправочный коэффициент К 0,03 н. раствора нитрата бария
вычисляют по формуле:
K=V/Vlt
где V—объем 0,03 и. раствора серной кислоты, взятый для определения, мл;
Vi — объем 0,03 н. раствора нитрата бария, израсходованный на
титрование, мл.
Выполнение анализа*. 3—10 мг (в зависимости от
ожидаемого содержания серы) полимера взвешивают с точностью до
0,02 мг, высыпают кучкой на середину фильтра и аккуратно
завертывают в «пакетик». Навеску летучих жидкостей берут в
ампулу, нелетучих жидкостей—в капилляр, который также
завертывают в фильтровальную бумагу так, чтобы открытый
конец его был обращен вниз и покрыт несколькими слоями
бумаги. Пакетик с навеской зажимают в платиновую сетку, при
этом узкий длинный конец фильтровальной бумаги должен
быть обращен вниз.
В колбу, помещенную в обрешетку из стальной сетки,
наливают 10 мл 6 %-ного раствора пероксида водорода и наполняют
кислородом. Выступающий конец фильтровальной бумаги
пакетика поджигают и быстро закрывают колбу пробкой. Через
40—50 мин колбу открывают, пробку и платиновую сетку
тщательно промывают дистиллированной водой над колбой, рас-
* Для полимеров и веществ, не содержащих фтора, анализ проводят,
сжигая навеску в обычных (иепропитаиных раствором борной кислоты)
фильтрах; раствор, перенесенный из колбы сжигания, выпаривают иа
электроплитке до объема примерно 10 мл и далее титруют, как описано в методике.
160
твор переносят в коническую колбу вместимостью 100 мл и
упаривают до объема 3—5 мл на электроплитке с асбестовой
сеткой, а затем досуха на водяной бане. Колбу охлаждают,
сухой остаток растворяют в 10 мл дистиллированной воды,
добавляют 2 мл 0,1 н. раствора соляной кислоты (рН 1,7—2,0),
25 мл спирта, 3 капли 0,2 %-ного водного раствора нитхромазо
и титруют 0,03 н. раствором нитрата бария до перехода
фиолетовой окраски в голубую. В аналогичных условиях проводят
контрольный опыт.
Расчет. Содержание серы [S] (в °/о) вычисляют по формуле:
S = (V2 - V3) К • 0,48099 • 100/m,
где У % и Vj — объем 0,03 н. раствора нитрата бария, израсходованный на
титрование анализируемой и контрольной проб, соответственно, мл;
К—поправочный коэффициент 0,03 н. раствора нитрата бария; 0,48099 —
количество серы, соответствующее 1 мл точно 0,03 н. раствора нитрата бария, мг;
т — навеска полимера, мг.
П.5.5. Определение азота
модифицированным полумикрометодом Кьельдаля
Метод основан на разложении анализируемого полимера
концентрированной серной кислотой в присутствии
каталитической смеси с количественным переходом азота в сульфат
аммония. Под действием щелочи сульфат аммония выделяет аммиак,
который отгоняется в борную кислоту. В результате
взаимодействия аммиака и борной кислоты образуется борат аммония,
который титруют стандартным раствором серной кислоты.
При анализе органических и полимерных продуктов,
содержащих нитро-, нитрозо-, гидразо-, диазо- и оксимные группы,
необходимо их предварительное восстановление, поскольку
данный метод дает заниженные результаты из-за образования
газообразного азота в процессе разложения. Наиболее успешно
анализируются этим методом полимеры и другие органические
вещества, содержащие аминный, амидный и нитрильный азот.
Для остальных азотсодержащих соединений предпочтительнее
пользоваться методом Дюма.
Аппаратура, реактивы
Колбы Кьельдаля вместимостью 60—70 мл со стеклянным полым
шариком.
Микробюретка вместимостью 10 мл с цеиой деления 0,02 мл.
Вода дистиллированная.
Серная кислота, концентрированная и 0,025 н. раствор.
Иодоводородная кислота (плотность 1,7 г/см3).
Борная кислота, насыщенный раствор (~4 %-ный).
Персульфат калия.
Сульфат меди.
Пероксид водорода, 30—35 %-ный раствор.
Гидр оксид натрия.
Индикаторы — метиленовый красный и метиленовый голубой.
Фосфор красный.
Этиловый спирт ректификованный технический.
6 Зак. 632
161
Подготовка образцов к анализу. 1. В образцах
гигроскопичных органических веществ (добавок) и полимеров (поли-Ы-ви-
нилпирролидон и сополимеры N-винилпирролидона) необходимо
определять воду по методу Фишера и учитывать ее количество
при расчете содержания азота.
2. Органические вещества и полимеры, содержащие нитро-,
нитрозо-, гидразо-, азо-, диазо- и оксимные группы, подвергают
предварительному восстановлению. В колбу Кьельдаля
помещают 20—45 мг анализируемого продукта, взвешенного с
точностью до 0,02 мг, добавляют 0,1 г красного фосфора и 4 мл
иодоводородной кислоты. Колбу закрывают полым шариком,
нагревают на плитке до кипения и кипятят 30 мин. Затем по
стенке колбы добавляют 3 мл воды, 2 мл концентрированной
серной кислоты и нагревают смесь в открытой колбе до
удаления воды и иодоводородной кислоты. Далее проводят анализ,
добавляя каталитическую смесь и серную кислоту, как
описано ниже.
Выполнение анализа. В колбу Кьельдаля вносят 23—45 мг
анализируемого вещества, 6 г персульфата калия, 0,1 г
сульфата меди, взвешенных с точностью до 0,0002 г, добавляют
3 мл концентрированной серной кислоты и 6 мл пероксида
водорода. Колбу закрывают полым шариком и нагревают,
сначала высоко (на 6—8 см) подняв ее над источником нагрева
(из-за сильного вспенивания), а затем, постепенно опуская в
течение 30 мин, еще до обесцвечивания 15 мин, после чего
кипятят еще 5 мин. (Для поли-Ы-винилпирролидона,
N-винилпирролидона и его сополимеров время нагревания соответственно
45 мин, 20 мин и 25 мин.)
После охлаждения содержимое колбы разбавляют 15 мл
дистиллированной воды и колбу 2 присоединяют к установке
для перегонки с водяным паром (рис. 11.33). Конец дистилля-
ционной трубки погружают в колбу-приемник 7, содержащую
10 мл насыщенного раствора борной кислоты.
В колбу 2 с навеской разложенного вещества через
патрубок 3 вводят 15 мл 40 %-ного раствора щелочи и пропускают
пар из парообразователя 5. Одновременно включают колбона-
Рис. 11.33. Прибор для определения азота ло методу Кьельдаля:
/ —колбонагревателн; 2 —колба Кьельдаля; 3 — патрубок для введения 40 %-ного раствора
щелочи; 4—холодильник; 5 —парообразователь; 6—промежуточный приемник водяного
пара; 7—приемная колба.
162
греватели / и проводят отгонку аммиака со скоростью 3—■
4 мл/мин. Собирают 25—30 мл отгона, затем приемник
опускают так, чтобы конец дистилляционной трубки не касался
жидкости, и продолжают отгонку еще 2—3 мин. Содержимое
приемника оттитровывают 0,025 н. раствором серной кислоты в
присутствии 6—7 капель смешанного индикатора (0,125 г ме-
тиленового красного и 0,083 г метиленового голубого
растворяют в 50 мл этилового спирта каждый отдельно, перед
титрованием смешивают растворы 1 : 1 по 10 мл) до появления
фиолетовой окраски раствора.
Расчет. Содержание азота [N] (в %) вычисляют по
формуле:
(У-У,)-0,35-loo
1 J (т — mg/\00) |
где V и Vt — объем 0,025 н. раствора серной кислоты, израсходованного на
титрование анализируемой и контрольной проб соответственно, мл; 0,35 —
количество азота, соответствующее 1 мл точно 0,025 н. раствора серной кислоты,
мг; т — навеска анализируемого вещества, мг; g — содержание воды в
веществе, найденное методом Фишера, %
Для пересчета результатов анализа по азоту на
азотсодержащее вещество в сополимерах необходимо в числитель
формулы ввести дополнительно коэффициент К = 100/а, где а —
теоретическое содержание азота в данном веществе. Например,
для расчета содержания акрилонитрила в сополимерах со
стиролом и АБС-пластиках К= 100 : 26,4 = 3,79; для расчета
содержания N-винилпирролидона в сополимерах К = 100: 12,61 =
= 7,93.
II. 5.6. Определение галогенов (хлора, брома и иода)
Метод основан на сжигании вещества в атмосфере
кислорода в присутствии платинового катализатора с последующим
поглощением продуктов сжигания щелочью и определением
образовавшихся галогенид-ионов меркуриметрическим титрованием
с дифенилкарбазоном в качестве индикатора.
Аппаратура, реактивы
Колбы конические со шлифом из термостойкого стекла вместимостью
500 мл.
Пробки пришлифованные со впаяниымн в них платиновыми проволоками
(длиной 130—150 мм) и сетками на концах проволок (40X20 мм).
Фильтры беззольные «белая лента», вырезанные по шаблону «флажок»,
размером 20 X 15 X 40 мм (см. рис. II. 32, б).
Ампулы и капилляры для анализа жидкостей. Капилляры можно
применять обычные и с небольшим расширением посредине, запаянные с одного
конца (см. рис. П. 32, а).
Стакан вместимостью 50 мл.
Колбы конические из термостойкого стекла вместимостью 100—150 мл.
Бюретка вместимостью 10 мл с ценой деления 0,02 мл.
Пипетки градуированные на 1 мл.
Колба мерная вместимостью 1000 мл.
Гидроксид калия.
6*
163
Перокснд водорода, 6 %-ный раствор.
Вода дистиллированная.
Азотная кислота, концентрированная (плотность 1,4 г/см') и 0,5 н.
раствор.
Этиловый спирт ректификованный технический.
Дифенилкарбазон, 1 %-ный спиртовый раствор.
Бромфеноловый синий 0,05 %-ный раствор.
Нитрат ртутн(П), 0,03 н. раствор.
Хлорид натрия.
Кислород газообразный.
Установление поправочного коэффициента к титру
0,03 н. раствора нитрата ртути. Навеску 5,01 г нитрата ртути,
взвешенную с точностью до 0,0002 г, растворяют в стакане в
2 мл концентрированной азотной кислоты, количественно
переносят в мерную колбу вместимостью 1 л и доводят
дистиллированной водой до метки.
Поправочный коэффициент устанавливают по высушенному
при температуре 120 °С химически чистому хлориду натрия.
3—5 мг хлорида натрия, взвешенного с точностью до 0,0002 г
растворяют в 10 мл дистиллированной воды, добавляют 20 мл
этилового спирта, 3 капли 0,05%-ного раствора бромфенолово-
го синего, 2 капли раствора гидроксида калия, нейтрализуют
0,5 н. раствором азотной кислоты до желтого окрашивания и
дают избыток (0,5 мл) азотной кислоты (рН 2,3—2,5). Затем
добавляют 5—7 капель 1 %-ного раствора дифенилкарбазона и
титруют 0,03 н. раствором нитрата ртути до слабо-розового
окрашивания.
Поправочный коэффициент К 0,03 н. раствора нитрата
ртути вычисляют по формуле:
К = m/(V • 1,7535),
где т — навеска хлорида натрия, мг; V — объем 0,03 н. раствора нитрата
ртути, израсходованный на титрование, мл; 1,7535 — количество хлорида
натрия, соответствующее 1 мл точно 0,03 н. раствора нитрата ртути, мг.
Выполнение анализа. 3—5 мг анализируемого вещества
взвешивают по разности в трубке для взятия навесок с
точностью до 0,02 мг, высыпают на фильтр и заворачивают в
«пакетик», который зажимают в платиновую сетку.
В колбу наливают 10 мл дистиллированной воды, 1 мл
2 н. раствора гидроксида калия и 3 капли раствора пероксида
водорода. Колбу помещают в обрешетку из стальной сетки и
заполняют кислородом в течение 3—5 мин. Выступающий конец
фильтровальной бумаги пакетика поджигают и колбу быстро
закрывают пробкой.
Через 20—30 мин колбу открывают, пробку и платиновую
сетку тщательно промывают дистиллированной водой над
колбой. Содержимое переносят в колбу вместимостью 100 мл и
выпаривают до 10 мл, охлаждают, добавляют 20 мл этилового
спирта, 3 капли 0,05 %-ного раствора бромфенолового синего,
нейтрализуют 0,5 н. раствором азотной кислоты до желтого
164
окрашивания и дают избыток (0,5 мл) азотной кислоты
(рН 2,3—2,5), после чего титруют 0,03 н. раствором нитрата
ртути в присутствии 5—7 капель дифенилкарбазона до
слаборозового окрашивания. В аналогичных условиях проводят
контрольный опыт.
Расчет. Содержание галогенов [Hal] (в %) вычисляют по
формуле
[Hal] = {V-V:)-K-M- 100/m,
где V и Vi — объем 0,03 н. раствора нитрата ртути, израсходованный на
титрование анализируемой и контрольной проб соответственно, мл; К —
поправочный коэффициент 0,03 н. раствора нитрата ртути; М — количество
галогенов, соответствующее 1 мл точно 0,03 н. раствора нитрата ртути, мг
(для С1 М= 1,06371 мг, для Вг М = 2,3976 мг, для 1 М = 3,8071 мг);
т — навеска анализируемого вещества, мг.
II. 5.7. Определение связанного винилацетата в сополимере
винилацетата с этиленом
Методика позволяет определять в сополимере массовую
долю винилацетата от 0,5 до 100 %.
Аппаратура, реактивы
рН-метр. Согласно инструкции по эксплуатации присоединяют
измерительный стеклянный электрод сравнения, заполненный 4 %-ным раствором
хлорида лития в этиловом спирте.
Соединительный мостик, заполненный 4 %-ным раствором хлорида лития
в этиловом спирте.
Стакан вместимостью 100 мл.
Магнитная мешалка.
Колба вместимостью 100 мл.
Холодильник шариковый.
Пипетка на 5 мл.
Мерный цилиндр вместимостью 50 мл.
Бюретка вместимостью 10 мл с цеиой деления 0,02 мл.
Ксилол или и-ксилол.
Гексиловый спирт.
Этиловый спирт гидролизный.
Соляная кислота 0,3 н. раствор.
Гидроксид калия.
Ацетон.
Хлорид лития, 4 %-ный раствор в этиловом спирте.
Выполнение анализа. Навеску сополимера, измельченного
до частиц размером не более 3X3 мм, в количестве 0,1—1 г
(в зависимости от содержания винилацетата), взвешенную с
точностью до 0,0002 г, вносят в колбу вместимостью 100 мл.
Затем вливают 25 мл ксилола, 10 мл - гексилового спирта и
5 мл 0,5 н. спиртового раствора гидроксида калия. Колбу
присоединяют к холодильнику, ставят на горячую песчаную баню
и нагревают ее содержимое до кипения в течение 30 мин.
В аналогичных условиях ставят контрольный опыт.
Непосредственно перед титрованием, но не раньше чем
через 5—6 мин после прекращения нагревания, через верх
холодильника вводят 35 мл ацетона, после чего колбу отсоединяют
165
от холодильника. Затем колбу ставят на магнитную мешалку.
В колбу опускают стеклянный электрод и конец
соединительного мостика, другой конец которого опущен в стакан с 4 %-ным
спиртовым раствором хлорида лития. В этот же стакан
вставлен конец хлорсеребряного электрода сравнения.
Проводят потенциометрическое титрование 0,3 н. раствором
соляной кислоты до первого скачка потенциала. Сначала ти-
трант приливают из бюретки по каплям быстро, а по мере
приближения к точке эквивалентности — по 0,05 мл,
допускается приливание по 0,04 мл или 0,06 мл. Каждый раз после
установления равновесного потенциала записывают показания
потенциометра в милливольтах против прилитого объема.
Скачок устанавливают по наибольшему изменению потенциала от
введения вышеуказанного объема титранта. При двух
одинаковых наибольших значениях изменения за скачок принимают
первое значение.
Расчет. Содержание связанного винилацетата в сополимере
X (в %) рассчитывают по формуле:
X = (V, — V2) • 0,0258 • 100/от,
где Vi — объем 0,3 н. раствора соляной кислоты, израсходованный на
титрование контрольной пробы, мл; Кг — объем 0,3 н. раствора соляной кислоты,
израсходованный на титрование рабочей пробы, мл; 0,0258 — количество
винилацетата, соответствующее 1 мл точно 0,3 и. раствора соляной кислоты, г;
т — навеска сополимера.
Ниже указаны навески сополимера в зависимости от
содержания в нем связанного винилацетата:
Навеска сополимера, г 1 0,5 0,2 0,1
Содержание связанного винил- 0—2 2—5 5—30 > 30
ацетата, %
Допускаемое расхождение ме- 0,3 0,5 1,5 2,0
жду параллельными
определениями, %
За результат принимается среднее арифметическое между
двумя параллельными определениями, допускаемое
расхождение между которыми не должно превышать указанного выше.
П.5.8. Определение состава ударопрочного полистирола
на основе бутадиенового каучука
Аппаратура и реактивы
Центрифуга с частотой вращения ротора не менее 8000 об/мин.
Пробирка центрифужная (металлическая) вместимостью ие менее 10 мл.
Встряхивающая машина.
Чашки Коха или Петри.
Вакуум-сушильный шкаф.
Колбы плоскодонные со шлифом вместимостью 100 мл.
Метилэтилкетон.
Этиловый спирт ректификованный технический.
166
Выполнение анализа. Навеску ударопрочного полистирола
1 г, взвешенную с точностью до 0,01 г, помещают в колбу
вместимостью 100 мл, заливают 50 мл метилэтилкетона и
растворяют на встряхивающей машине в течение 2—2,5 ч. После
такой обработки гомополистирол и небольшое количество
привитого растворимого сополимера переходят в раствор, а
основная масса привитого растворимого сополимера и гель-фракции
остаются в осадке.
Раствор с осадком разливают в 3 или 4 центрифужные
пробирки (в зависимсти от комплектности центрифуги) и
центрифугируют в течение 30 мин (2 раза по 15 мин). Раствор над
осадком осторожно по стеклянной палочке сливают в чашку
Коха или Петри, а в пробирку вновь добавляют свежую
порцию раствора и продолжают центрифугирование.
После разделения раствора осадок промывают, добавляя в
лробирки по 5 мл метилэтилкетона и тщательно размешивая
стеклянной палочкой. Остаток на палочке после размешивания
смывают в ту же пробирку небольшим количеством
растворителя (до э/4 вместимости пробирки) и центрифугируют 15 мин.
Операцию повторяют до полного удаления гомополистирола.
Осадок считается отмытым, если при добавлении 2—3 капель
раствора к 2 мл этилового спирта отсутствует помутнение.
Промывные жидкости собирают в ту же чашку Коха. После
центрифугирования в чашке Коха остается раствор, содержащий
гомополистирол и небольшое количество привитого растворимого
■сополимера; в центрифужных пробирках — осадок, содержащий
привитый нерастворимый сополимер (гель-фракция), привитый
растворимый сополимер.
Для выделения из раствора гомополистирола и привитого
растворимого сополимера, перешедшего в раствор, растворитель
из чашки Коха испаряют при комнатной температуре, остаток
сушат в вакуум-сушильном шкафу при комнатной температуре
и остаточном давлении 666 Па (5 мм рт. ст.) до постоянной
массы (в течение 1,5—2 ч).
Для выделения сшитого сополимера — гель-фракции осадок
из центрифужных пробирок количественно переносят, смывая
метилэтилкетоном в чашку Коха или Петри, предварительно
взвешенную с точностью до 0,0002 г (mi).
Растворитель из чашки Коха испаряют при комнатной
температуре, остаток сушат в вакуум-сушильном шкафу при
комнатной температуре и остаточном давлении 666 Па до
постоянной массы. Чашку Коха с высушенным остатком взвешивают
с точностью до 0,0002 г (т2).
Навески для определения содержания каучука в привитых
сополимерах (растворимом и сшитом) после высушивания и
взвешивания заливают растворителем в тот же день во
избежание окисления каучука.
Для определения содержания каучука в привитом
сшитом сополимере Ki берут навеску сухого остатка 40—60 мг,
167
взвешенную с точностью до 0,0002 г, а для определения
содержания привитого растворимого сополимера, перешедшего в
раствор вместе с гомополистиролом, определяют содержание
каучука Кг, для чего берут навеску образца 0,5 г, взвешенную с
точностью до 0,0002 г. В качестве растворителя можно
использовать хлороформ, бензол или толуол. Определение каучука
проводят по методике II. 5.11.
Содержание привитого растворимого сополимера или
привитого сшитого сополимера, перешедшего вместе с
гомополистиролом в раствор, рассчитывают, принимая его состав таким же,
как и состав привитого растворимого сополимера или гель-
фракции.
Расчет. Содержание (массовую долю) выделенных фракций,
вычисляют по следующим формулам.
Содержание привитого сшитого сополимера (гель-фракции)
X (в %):
Х = (т2— т,)-100/т.
Содержание гомополистирола и привитого сшитого
сополимера, перешедшего в раствор вместе с гомополистиролом, Х\
(в %):
Х1 = \т — Х.
Содержание привитого сшитого сополимера (гель-фракции),
перешедшего в раствор вместе с гомополистиролом, Х2 (в %):
л 2 = K2^l/Kl-
Содержание привитого сшитого сополимера (общее) Хз
(в %):
X э = X + Л2-
Содержание гомополистирола X* (в %):
Xt = 100 — ЛГ3,
где т — навеска сополимера, взятого на фракционирование, г; /яг — масса-
чашки Коха или Петры с образцом после испарения растворителя, г; т1 —
масса пустой чашки Коха или Петри, г; Ki — содержание каучука,
полученное по результатам анализа в привитом сшитом сополимере, %; Кг —
содержание каучука, полученное по результатам анализа в гомополистироле, %.
II. 5.9. Определение состава АБС-пластиков
АБС-пластик представляет собой смесь привитого
растворимого сополимера, привитого нерастворимого сополимера (гель-
фракция), сополимера стирола с акрилонитрилом, а также
свободного бутадиенового каучука.
Анализ проводится аналогично анализу ударопрочного
полистирола на основе бутадиенового каучука (см. методику II. 5.8).
Во всех случаях взамен метилэтилкетона следует использовать,
ацетон.
168
Содержание фракций рассчитывают по формулам,
приведенным в методике П. 5.8, содержание каучука во фракциях — в
методике II. 5.11, содержание акрилонитрила во фракциях и
исходном сополимере — в методике II. 5.5.
II.5.10. Определение состава атмосферостойкого ударопрочного
полистирола на основе СКЭПТ
Атмосферостойкий ударопрочный сополимер на основе
стирола и этиленпропиленового каучука представляет собой смесь
привитого сополимера, гомополистирола, а также свободного
этиленпропиленового каучука (СКЭПТ).
Аппаратура и реактивы
Центрифуга с частотой вращения ротора 16 000 об/мин (допускается
работа на центрифуге с частотой вращения ротора 8000 об/мин).
Гептан.
Остальное см. в методике II. 5.8.
Выполнение анализа. Навеску сополимера (примерно 1 г)
взвешивают с точностью до 0,0004 г, помещают в колбу
вместимостью 100 мл, заливают 50 мл метилэтилкетона и
выдерживают 15—18 ч для растворения. Гомополистирол переходит в
раствор, а привитый сополимер и свободный этиленпропилено-
вый каучук остаются в осадке.
Содержимое колбы взбалтывают, разливают в пробирки и
центрифугируют в течение 30 мин (в два приема по 15 мин).
По окончании центрифугирования раствор над осадком
осторожно по палочке сливают в чашку Коха, а в пробирки вновь
добавляют оставшийся в колбе раствор и снова
центрифугируют.
После разделения раствора из колбы осадок промывают,
добавляя в пробирки по 5 мл метилэтилкетона и тщательно
размешивая смывают в ту же пробирку небольшим
количеством метилэтилкетона (до 3Д вместимости пробирки) и
центрифугируют. Промывные жидкости сливают в ту же чашку
Коха, осадок промывают до полного удаления
гомополистирола. Осадок считается отмытым, если при добавлении 2—3
капель раствора к 2 мл этилового спирта отсутствует помутнение.
После центрифугирования в чашке Коха находится раствор
гомополистирола, а в центрифужных пробирках — осадок,
содержащий привитый сополимер и этиленпропиленовый каучук.
Для выделения гомополистирола растворитель из чашки
Коха испаряют при комнатной температуре, остаток сушат в
вакуум-сушильном шкафу при комнатной температуре до
постоянной массы. В чашке Коха остается гомополистирол.
Для выделения свободного этиленпропиленового каучука
осадок в пробирках заливают гептаном на 3Д вместимости
пробирки и выдерживают 15—-18 ч. Затем содержимое пробирки
тщательно перемешивают стеклянной палочкой и оставляют
169
раствор на 1 ч. Свободный этиленпропиленовый каучук
переходит в раствор, а привитый сополимер остается в осадке.
Раствор с осадком центрифугируют в течение 15 мин, после чего
раствор над осадком осторожно по палочке сливают в
предварительно взвешенную (mi) с точностью до 0,0004 г чашку
Коха (I), а в пробирки вновь добавляют гептан, раствор
перемешивают и центрифугируют.-После центрифугирования в чашке
Коха находится свободный этиленпропиленовый каучук, а в
центрифужных пробирках — осадок, содержащий привитый
сополимер.
Растворитель из чашки Коха (I) испаряют при комнатной
температуре, остаток сушат в вакуум-сушильном шкафу при
комнатной температуре до постоянной массы (т2).
Для выделения привитого сополимера осадок из
центрифужных пробирок количественно переносят гептаном в чашку
Коха (II), предварительно взвешенную с точностью до 0,0004 г
(т3). Растворитель из чашки Коха испаряют при комнатной
температуре до постоянной массы (т4). В чашке Коха (II)
остается привитый сополимер.
Расчет. Содержание выделенных фракций определяют по
следующим формулам.
Содержание свободного этиленпропиленового каучука X
(в %):
Х = (т2 — m,)-100/m.
Содержание привитого сополимера Х\ (в %):
X, = (т4 — т3) • 100/т.
Содержание гомополистирола Х2 (в %):
Х2=100-Х-Х„
где т — навеска сополимера, взятого на фракционирование, г; ffti — масса
пустой чашки Коха (I), г; т.г — масса чашки Коха (I) с высушенным
осадком, г; Шъ — масса пустой чашки Коха (II), г; mt—масса чашки Коха (Щ
с высушенным осадком, г.
За результат анализа принимается среднеарифметическое
значение двух параллельных определений, допускаемое
расхождение между которыми не должно превышать 0,5 %.
II.5.11. Определение содержания бутадиенового каучука
в ударопрочном полистироле, АБС- и МБС-пластиках,
в гель-фракциях этих пластиков и в исходном каучуке
Метод основан на бромировании каучука раствором
бромида иода в тетрахлорметане с последующим иодометрическим
титрованием избытка бромида иода.
Аппаратура, реактивы
Колба плоскодонная с пришлифованной пробкой вместимостью 250 мл>.
Холодильник шариковый.
Баня песчаная.
170
Пипетки градуированные на 5, 10 и 20 мл.
Мерные цилиндры вместимостью 50 и 1000 мл.
Иод металлический.
Бром.
Тетрахлорметан.
Иодид калия, 10%-ный раствор.
Крахмал, 0,5 %-ный раствор.
Тиосульфат натрия, 0,1 н. раствор.
Хлороформ.
Этиловый спирт ректификованный технический.
Бромид иода, 0,2 н. раствор в тетрахлорметане.
Приготовление раствора бромида иода. 13 г измельченного
иода растворяют в 1 л тетрахлорметана. Раствор фильтруют и
определяют его титр (10 мл раствора оттитровывают 0,1 н.
раствором тиосульфата натрия). В полученный раствор иода
вводят 8 г, или 2,56 мл, брома и вновь определяют титр
(к 10 мл раствора добавляют 10 мл 10 %-ного раствора иодида
калия и оттитровывают 0,1 н. раствором тиосульфата натрия).
Титр при вторичном титровании должен быть по сравнению с
первым титрованием несколько менее удвоенного.
Выполнение анализа. В колбу вместимостью 250 мл вносят
навеску ударопрочного полистирола 0,5—0,2 г либо АБС- или
МБС-пластика (в зависимости от количества каучука,
введенного при сополимеризации — 5—25%). взвешенную с точностью
до 0,0002 г. При определении каучука в гель-фракции и
исходном каучуке навески в количестве 60—80 мг и 30—40 мг
соответственно взвешивают с точностью до 0,005 мг. Навески
заливают 25 мл хлороформа, выдерживают 12 ч при комнатной
температуре или нагревают в течение 1 ч на песчаной бане,
присоединив к колбе холодильник. Затем пипеткой добавляют
20 мл 0,2 н. раствора бромида иода в тетрахлорметане. Колбы
помещают в темное место на 30 мин, после чего наливают
15 мл 10 %-ного раствора иодида калия, 50 мл воды, 20 мл
этилового спирта и титруют 0,1 н. раствором тиосульфата
натрия, прибавляя к концу титрования раствор крахмала. При
титровании необходимо энергично встряхивать содержимое
колбы для предотвращения образования эмульсии. Титрование
продолжают до обесцвечивания раствора. В аналогичных условиях
проводят два контрольных опыта.
Расчет. Содержание бутадиена Б (в %) вычисляют по
формуле:
B = (V—Vi)K- 0,0027 • 100/m,
где V и Vi—объем 0,1 н. раствора тиосульфата натрия, израсходованный
на титрование контрольной и анализируемой проб соответственно, мл; К —
поправочный коэффициент к титру 0,1 н. раствора тиосульфата натрия;
0,0027 — количество бутадиена, соответствующее 1 мл точно 0,1 н. раствора
тиосульфата натрия, г; т — навеска анализруемого сополимера, г.
За результат испытания принимают среднее арифметическое
двух параллельных определений, допускаемое расхождение
171
между которыми не должно превышать 0,5 % при определении
содержания каучука в привитом сшитом сополимере и 0,2 %
при определении содержания каучука в растворимой части.
ГЛАВА III
ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ МАССЫ
И МОЛЕКУЛЯРНО-МАССОВОГО РАСПРЕДЕЛЕНИЯ
ПОЛИМЕРОВ
Определение молекулярной массы (ММ) и молекулярно-мас-
сового распределения (ММР) полимеров является
специфическим разделом в физико-химическом анализе веществ.
Используемые для этого методы существенным образом отличаются от
методов аналитической химии. Действительно, такие методы
анализа веществ, как спектральные (ЯМР, ИКС, УФС), тепло-
физические (ДТА, ДТГ, ДСК), газовая хроматография, пиролиз
и другие широко используются в анализе металлов,
неорганических и органических низкомолекулярных веществ.
Применение их к полимерам связано с перекалибровкой прибора. В
литературе довольно подробно описаны теоретические основы
методов, приборы фактически являются однотипными для
низко- и высокомолекулярных веществ.
Совершенно иная ситуация сложилась с определением ММ
и ММР полимеров. Полимеры отличаются от других классов-
веществ именно значением ММ. Перед исследователем,
занимающимся анализом низкомолекулярных веществ, вопросы
определения ММ и ММР вообще не стоят. Имеется чрезвычайно
мало книг, посвященных теоретическим вопросам физической
химии растворов полимеров, которая является основой для
разработки методов определения ММ и ММР. Это же касается
описания методов и методик анализа.
Другая специфика этого раздела аналитической химии
связана с особенностями интерпретации данных о ММ и ММР.
ММР является в своеобразной форме отражением
статистического характера полимер из ационного процесса, особенностей
температурного и гидродинамического режимов работы
реакторов, специфического применения реагирующих веществ. Малые
добавки компонентов полимеризационной системы могут
существенно изменить ММР, не меняя химическую природу (состав)
полимера. В связи с этим анализ информации о ММР требует
достаточно широкой эрудиции исследователя в области
математической статистики, химии полимеризационных процессов,
широкого использования вычислительной техники.
Существует достаточно большое число методов определения
ММ [191, с. 205; 192, с. 86]. При этом часть методов
определяют значение ММ полимера вне зависимости от
предположения о структуре макромолекулы. Такие методы получили на-
172
звание абсолютных. Это осмометрия, крио- и эбулиоскопия,
рассеяние света, приближение к седиментационному
равновесию. Более широкое распространение получили относительные
методы определения ММ и ММР — вискозиметрия, скоростная
седиментация, гель-проникающая хроматография. Информация,
получаемая этими методами, зависит от предположений о
структуре макромолекулы. Поэтому обычно эти методы «калибруют»,
т. е. на специальных образцах полимеров исследуемого класса
получают зависимость измеряемой величины от ММ. Учитывая
ограниченный объем главы и тот факт, что абсолютные методы
не дают возможности определять ММР, основное внимание
здесь уделяется относительным методам анализа ММ и ММР.
III. 1. НЕКОТОРЫЕ ПРЕДСТАВЛЕНИЯ О МОЛЕКУЛЯРНОЙ МАССЕ
И МОЛЕКУЛЯРНО-МАССОВОМ РАСПРЕДЕЛЕНИИ
Понятие о молекуле и молекулярной массе для
высокомолекулярных соединений имеет ряд существенных отличий от
таковых для низкомолекулярных веществ. Полимерная молекула
представляет собой цепную структуру, состоящую из большого
числа групп атомов, соединенных между собой химическими
связями. Наличие различных конфигураций и конформаций
макромолекул обусловливает различия в свойствах полимерных
материалов и их поведении под действием внешних
воздействий. Особенность полимеров, имеющих одинаковое стереохи-
мическое расположение атомов, заключается в том, что они
представлены широким набором полимергомологов — цепей
одинакового химического строения, но различной длины. Из
этого набора полимергомологов невозможно выделить
молекулы точно определенной молекулярной массы. Так, если гексан
по свойствам значительно отличается от гептана или декана
(например, температуры плавления 177,8; 216,4 и 243,5СС
соответственно), то для полиэтиленов с MM 5-104 и 2-105 в
пределах экспериментальных возможностей не зафиксировано
различия в температурах плавления.
Полимерные вещества всегда полидисперсны * (полимоле-
кулярны) — обладают распределением по ММ—ММР.
Необходимо отметить, что кроме полимолекулярности полимеры
характеризуются большим набором различного вида неоднородностей
(полидисперсности): чередование звеньев различной природы
(сочетание «голова — хвост», сомономеры), ориентация боковых
групп, строение цепи — разветвленные цепи и т. д. Однако
обычно под полидисперсностью полимера понимают его поли-
молекулярность.
Хотя полимер нельзя разделить на отдельные гомологи, тем не
менее ММ является важнейшей молекулярной характеристикой
* Здесь не рассматривается класс высокомолекулярных биологических
объектов, некоторые из которых мономолекулярны.
173
образца. Действительно, именно ММ определяет способы
переработки полимера, его физико-механические свойства. При
этом важно отметить, что свойства полимерного материала по-
разному зависят от ММ. Например, вязкость расплава
полимера определяется ММ в соответствии с формулой:
где Ki, <ii — эмпирические константы, при этом а\ = 2,5 -г- 5.
При изменении ММ вязкость расплава может измениться в
105_Ю'° раз.
Температура хрупкости полимера Тхр, определяющая
возможность использования его в виде конструкционного
материала, связана с ММ соотношением типа:
Тхр = А1 + В1/М,
где А,, В, —эмпирические константы.
Приведенные соотношения показывают примеры очень
сильной и слабой зависимости свойств полимерного материала от
ММ. Естественно, что свойства полимеров зависят и от
полидисперсности образца. Некоторые свойства определяются
низкомолекулярными компонентами, другие —
высокомолекулярными, а следовательно, в общем молекулярно-массовым
распределением.
ММ и полидисперсность полимера зависят от условий
проведения реакций полимеризации, аппаратурного оформления
процессов, его технологических параметров. Поэтому
существует ряд «обратных» кинетических задач, когда по ММ и ММР
определяют константы химических реакций, изучают наличие
тех или иных реакций в процессе полимеризации. В связи с
этим значимость определения ММ возрастает.
В последнее десятилетие развитие инструментальной базы
представило возможности в полной мере удовлетворить
запросы науки и промышленности в определении ММ. и ММР. Это
связано со становлением нового экспресс-метода определения
ММ и ММР — гель-проникающей хроматографии [192, с. 247;
193, с. 81]. Современные жидкостные хроматографы позволяют
автоматизировать процесс измерения, применять ЭВМ. При
серийных измерениях результаты можно получить за 10—30 мин.
III. 1.1. Средняя молекулярная масса и ММР
В связи с тем, что полимер характеризуется наличием
молекул с разными ММ, для описания такого полимера
необходимо знать, сколько имеется молекул ni данной молекулярной
массы Mi. Получают ряд дискретных значений «,-, которые в
совокупности задают численную функцию распределения по
ММ (степени полимеризации Р). Достаточно задать степень
полимеризации Pk, чтобы узнать соответствующее число моле-
174
кул tik- Поскольку интервал изменений Р очень велик (от 10
до 106), то считают Р непрерывно изменяющейся величиной.
Ордината в этом случае равна -тр •
Обычно исследователь не интересуется общим числом
молекул По, его интересует доля молекул ni/tio, имеющая заданную
массу Mi (или Pi). Для непрерывного распределения вводят
среднечисленную функцию распределения по Р — -гр- или сред-
нечисленную функцию распределения по MM qn:
*•<">-£-&■ <ШЛ)
Величина qn(M)dM характеризует численную долю молекул
(относительное число молекул), лежащих в интервале от М до
M-\-dM. Функция qn(M) нормирована на единицу:
оо
[qn{M)dM = \. (III. 2)
о
Зная распределения (III. 1) и (III. 2), легко оценить и
средние молекулярные массы. Так, среднечисленная ММ Мп равна:
оо
Мп=\ Mqn dM. (III. 3)
о
Величина Мп определяет среднюю массу (среднюю длину)
макромолекулы.
Очень часто рассматривают среднемассовые распределения.
Дифференциальное среднемассовое распределение (именно это
распределение и называют обычно ММР) определяется
следующим образом:
qw(M) = qn{M)M/Mn. (III. 4)
Если подставить (III. 1) в (III. 4), то становится ясным
физический смысл qw(M): qwdM дает относительную массу (мас-
М dn\
совую долю -т; ) макромолекул, молекулярная масса ко-
торых заключена в интервале от М до М -f- dM.
Наравне с дифференциальным ММР часто используют и
интегральное ММР W(M):
м
W(M)=[qw(M)dM. (III. 5)
о
Интегральное ММР W{M) характеризует массовую долю
макромолекул, ММ которых меньше или равна М.
Максимальное значение W(M) равно 1.
На рис. III. 1 приведены примеры qw{M) и W(M).
Величина qw имеет очень наглядный вид: она может иметь несколько
175
W,QW Рис. III. 1. Дифференциальные (/, 3) qw и
интегральные (2, 4) W молекулярно-массовые
распределения:
/, 2—унимодальные; 3,4—бимодальные
распределения.
//V
У /
yf '
-Л
\
4м
максимумов [би-, три- (см. рис.
III. 1) и мультимодальные
распределения] и легко позволяет
определить ММ в максимуме.
Величина W(M) имеет большее значение при анализе полимера
и оценке взаимосвязи свойства — ММ, так как позволяет легко
оценивать доли низко- или высокомолекулярных «хвостов»
распределения. Так, массовая доля макромолекул с молекулярной
массой больше М равна 1— W{M).
III. 1.2. Количественные характеристики ММР
Наиболее полное описание ММР — это описание кривых
qw(M) или W(M). Поэтому желательно (и некоторые
зарубежные фирмы уже делают это) в качестве «паспорта» марки
полимера приводить распределение по ММ (как правило, это
хроматограмма). Однако это делается еще не для всех
полимеров. Кинетический анализ полимеризационного процесса
опирается, как правило, на значение Мп. При установлении
корреляции свойства — молекулярные характеристики также
необходимо пользоваться количественными параметрами ММР.
В качестве основной характеристики ММР принимают
моменты распределения (Mq}:
оо
(М") = [ Mlqn dM. (III. 6)
о
Как правило, экспериментально определяют отношения
моментов распределения. Среднечисленная ММ Мп равна:
Мп = (М1) = \ Mqn dM = ! .
\ (QwIM) dM
Среднемассовая MM Mw равна:
Mw = (M2)/(M') = \ Mqw dM.
Если Мп определяет обычно низкомолекулярная часть
распределения, а Мш — среднюю часть ММР, то для
характеристики высокомолекулярной части вводят обычно Мг:
Мг = (М3)/(М2).
Аналогично вводятся ММ и другой степени усреднения.
Однако экспериментальные функции ММР редко определяются с
176
достаточной точностью (особенно «хвосты»), чтобы
рассчитывать другие ММ (более высокой степени усреднения).
Прежде чем перейти к дальнейшим способам
количественной характеристики ММР, необходимо сделать одно очень
важное замечание. Хотя в качестве количественной характеристики
можно использовать ММ разной степени усреднения (или
другие параметры — см. ниже), однако смысл эти характеристики
имею только для унимодального распределения (см. рис. III. 1).
Для би- или мультимодальных распределений рассчитать эти
характеристики также можно, однако установление корректных
взаимоотношений между молекулярными характеристиками и
свойствами (или параметрами технологии), как правило,
невозможно. Единственно разумный путь к характеристике
мультимодальных распределений — это разделение ММР на
составляющие, т. е. унимодальные распределения, и оценка отдельных
распределений обычным образом, а также их массовых долей
wt. Так, если мы имеем бимодальное распределение с
массовыми долями первого W\ и второго Дог = 1 — W\ компонентов
смеси, то для всего образца будут справедливы соотношения:
Mw = wtM, + w2M2, \IMn = w1/Ml + wiIMi. (HI-7)
Из приведенных соотношений наглядно видны
ограниченность и неоднозначность средних характеристик Мп и Mw\
кроме ММ компонентов они зависят также и от их долей и>,-.
Соотношения (III. 7) легко обобщаются на случай
мультимодальных распределений.
Пример. Пусть Мт:Мт = Мп%: Мщ = 10. Тогда MJMWi = ai, +
+ 10а; • MnJMn = w. + 0,1 а>2. Видно, что наличие высокомолекулярного
компонента шг резко увеличивает Mw и фактически ие сказывается на Мп, и,
наоборот, наличие низкомолекулярного компонента Wi существенным образом
уменьшает М„. Если w2 = 0,1, то MwjMWx = 1,9; MnjMni= 1,1 и даже, если
компоненты монодисперсны, полидисперсность всего образца равна 1,8. При
варьировании w2 Мш1Мп будет меняться, хотя ММ компонентов остаются
прежними.
В общем виде для полной характеристики ММР необходимо
знать все моменты {Mi) распределения или их соотношения.
Сопоставляя рассчитанные и экспериментальные значения
Мп, Mw, Мг, можно оценить адекватность принятой
кинетической схемы.
Так же, как это принято в теории вероятности, ММР может
быть охарактеризовано с помощью других величин: моды,
дисперсии, медианы. Модой называется значение ММ Мт в
максимуме дифференциального распределения, т. е. наиболее
вероятное значение ММ. Под медианой Mol5 распределения
понимается такое значение ММ, для которого вероятность того, что
М ^ М0,5, равна 0,5, т. е. W(M0is/) = 0,5. Дисперсия среднечис-
ленного распределения ап дается соотношением:
а2„ = J (M - Mnf qn dM = Ml (MwjMn - 1).
177
Здесь о„ означает среднечисленное отклонение ММ от
Мп и прямо связано с полидисперсностью Mw/Mn образца:
Vn/Mn = л/мш/Мп — 1.
Аналогично вводится и стандартное отклонение ош для сред-
немассового распределения qw:
a2w = Mw(Mz-Mw).
III. 1.3. Аналитическое описание ММР
В некоторых случаях исследователь сталкивается с задачей
аналитического описания ММР. Аналитическое описание ММР,
если оно найдено, позволяет провести детальную проверку
кинетических схем процессов полимеризации, а также уточнить
доли «хвостов» распределений. Для математического описания
экспериментальной кривой ММР подбирается несколько
известных модельных функций. Методики подбора заключаются: а) в
исследовании соотношения между моментами распределения,,
экспериментальными и рассчитанными для той или иной
модельной функции; б) в подборе модельной функции по всей
экспериментальной кривой с использованием ЭВМ. Рассмотрим,
несколько наиболее употребительных модельных функций
распределения.
Для описания достаточно узких ММР (с малым отношением
Мш/Мп) применяют обычно распределение Гаусса (нормальное
распределение).
Очень широкое распространение получило
гамма-распределение (в физической химии полимеров это распределение ввел
в практику Г. Шульц)*:
' e_aiM, (III. 8)
чп Г (Л" + 1
где cti, К — параметры функции; Т(К) —гамма-функция.
ai и К определяют значение ММ:
М„=*-±!; Л*Ш = А±2-; М2 = Л±1. (III. 9)
При К = 0 получаем наиболее вероятное распределение,
введенное в химию полимеров П. Флори **. Для
гамма-распределения характерны следующие соотношения между ММ:
Мш - Мп = Мг - Mw или Mz/Mw = 2-Mn/Ww. (III. 10)
* При графическом сопоставлении необходимо помнить, что «хвосты»
экспериментальной кривой <7л определяются с малой точностью, поэтому
естественны отклонения экспериментальных и теоретических кривых на «концах»
распределения.
** Распределение Флори имеет полимер, полученный при постоянных
константах химических реакций, постоянных концентрациях реагентов и
случайном обрыве цепи (за исключением обрыва рекомбинаций радикалов).
178
Рис. III. 2. Зависимость М Z\!AW от Mw/Mn Mz/Mw
для логарифмически нормального (I) и гам- У
ма-(2) распределений. 4 - Ж
С ростом полидисперсности 3 - уГ
Mz/Mw-+2. Это характерная / ^^^— '
черта гамма-распределения. Для 2 - /^**
гамма-распределения Мп совпа- Л*"^
дает с медианой М0,ь и Мт < 7Lr^ ■
<Мп. 3 5 м„/М„
Очень важное распределение — логарифмически-нормальное,
введенное в химию полимеров В. Лансингом и Е. Крамером.
Оно обычно рассматривается для массового дифференциального
распределения:
1 1 Г \п2(уМ) )
qw=—= expi ——-К, (III. 11)
V*m I -р2 у
где Р и у — параметры.
Интегральное массовое распределение дается соотношением:
И7 = 0,5[1 + Фв(1пУМ/р)],
где Фв — интеграл вероятности.
Средние ММ вычисляются по формулам:
М„ = —е~р2/4; Мш = —ер2/4. (III. 12)
V V
Соотношения между ММ даются уравнениями:
Mz:Mw:Mn = e^:e°^':l; Mz/MW = MW/Mn. (III. 13)
Соотношения (III. 10) и (III. 13) позволяют легко
идентифицировать вид ММР [194] (рис. III. 2). В области малых
значений Mw/Mn вид распределения найти трудно, так как требуется
очень высокая точность определения экспериментальных
данных. При Mw/Mn > 2 различия в экспериментальной
зависимости Mz/Mw от Mw/Mn достаточны, чтобы выбрать вид функции.
Из расчетов следует, что Мп совпадает со значением моды Мт
для среднемассового гамма-распределения. Для
логарифмически нормального распределения Мп > Мт, Значение медианы
-М0,5 совпадает со среднемассовым значением MM Mw для
гамма-распределения, в то время как для
логарифмически-нормального распределения jW0,5 совпадает с (MwMn)0'5.
Для описания экспериментальной кривой ММР модельной
функцией распределения можно использовать следующую
процедуру с использованием ЭВМ. Анализируя соотношения для
моментов распределения, выбирают класс модельных
(теоретических) функций распределения. Экспериментальное ММР
при этом рассматривается в качестве эмпирического
распределения. Дальнейшая задача состоит в проверке гипотезы о
согласии эмпирического распределения с некоторым теоретическим.
179
В качестве критериев согласия обычно используются
критерии Стьюдента, Колмогорова — Смирнова, причем критерий
Колмогорова — Смирнова является наиболее мощным.
При использовании критерия Колмогорова — Смирнова для
выборки из К значений случайной величины X оценивается
максимальное значение выражения:
Г = /С0'5! /=■. (X)-F,(X)\,
где Fi(X) и F2{X)—соответственно эмпирическая и теоретическая функции
распределения величины X.
Согласно условиям применимости критерия Колмогорова —
Смирнова наименьший объем выборки должен быть К ^ 50.
Увеличение числа выборки К > 100 не приводит к значимому
улучшению оценки параметров функции распределения.
После оценки величины у по таблицам * определяется
значение вероятности В принятия гипотезы о том, что
экспериментальное распределение согласуется с соответствующим
теоретическим распределением. На выбранном классе модельных
(теоретических) функций выделяется функция с максимальным
значением В и, соответственно, она применяется для описания
экспериментальной кривой. При этом, однако, следует помнить,
что для удовлетворительного согласия экспериментальной и
теоретических функций распределения должно быть В ^95%.
III. 2. МЕТОДЫ ОПРЕДЕЛЕНИЯ МОЛЕКУЛЯРНЫХ МАСС ПОЛИМЕРОВ
Существует большое число задач, когда достаточно знать
какую-либо среднюю ММ (Мп, Мш). Кроме того, для
калибровки относительных методов измерения ММ требуется знание ММ
фракций полимеров. Поэтому в практике лабораторий,
занимающихся целенаправленно измерением ММ и ММР,
необходимо иметь абсолютные методы измерения ММ.
Наибольшее значение, с нашей точки зрения, имеют методы
светорассеяния, приближения к седиментационному равновесию
и сочетания седиментации и диффузии. Эти методы позволяют
как правило, определять Мп и Mw. Меньшее значение имеют
методы измерения Мп: осмометрия, крио- и эбулиоскопия,
газовая осмометрия — измерение тепловых эффектов
конденсации. Это связано с тем, что последние методы существенно
ограничены по интервалу измерения ММ, очень чувствительны
к низкомолекулярным примесям.
В этом параграфе основное внимание мы будем уделять
двум методам: вискозиметрии и седиментации. Укажем
литературу, где можно найти описание методов измерения Mw —
светорассеяние [195] и Мп — осмометрия и др. [192, с. 90].
* В математическом обеспечении практически всех современных ЭВМ
имеются программы, позволяющие определять величину В для заданного
эмпирического распределения.
180
Интересной модификацией применения метода светорассеяния
является метод спектротурбодиметрии [196, с. 8].
В некоторых случаях относительные значения ММ можно
получать, и не растворяя полимер: исследование реологических
свойств полимеров предоставляет такую возможность.
III. 2.1. Метод седиментации — приближение
к седиментационному равновесию
Характеристики седиментационных процессов в растворах
полимеров измеряют в специальных
приборах—ультрацентрифугах.
Наиболее широкое распространение в СССР получили
ультрацентрифуги фирмы MOM (Венгрия). Они снабжены ротором
с электрическим приводом. На серийных приборах может быть
установлена оптика Фильпота-Свенсона, интерференционная и
ультрафиолетовая абсорбционная [197, с. 38; 198, с. 82].
Роторы современных аналитических ультрацентрифуг имеют
приблизительно одинаковые размеры и форму. В роторе имеется два
отверстия для размещения кювет: рабочей и балансировочной.
Расстояние от оси вращения х составляет примерно 6,5 см.
Вращение ротора происходит в вакууме, сам ротор термостати-
руется, обычно в интервале 0—50 °С. В некоторых случаях
ультрацентрифуги снабжены титановыми роторами с
высокотемпературной приставкой, позволяющей проводить измерения
до 150°С.
Аналитическая кювета состоит из цилиндрического
вкладыша с секторной полостью. Полость герметически закрывается
кварцевыми (или сапфировыми) окошками. В настоящее время
разработано несколько типов кювет (вкладышей): односектор-
ные, двухсекторные, с искусственным граничным слоем и т. д.
Конструкции кювет и работа с ними подробно освещена в
инструкции к ультрацентрифугам.
Подготовленную, промытую и высушенную кювету при
помощи шприца с затупленной иглой заполняют раствором
полимера. Обычно в кювету входит 0,4—0,8 мл раствора.
Собранную ячейку уравновешивают на аналитических весах
противовесом (или другой кюветой). Затем кюветы устанавливают в
ротор и термостатируют.
Одним из важных показателей работы ультрацентрифуги
является постоянство температуры ротора во время опыта.
Перепад температуры вызывает тепловую конвекцию раствора,
искажает нулевую линию и распределение концентрации в
кювете, может привести к появлению паразитных пиков.
Особенно тщательно надо термостатировать при работе с «плохими»
растворителями. Небольшие колебания температур и большие
давления (до 10—20 МПа), развивающиеся в кювете, могут
привести к осаждению полимера.
181.
Таблица III. 1. Значения инкрементов показателей преломления
и удельных парциальных объемов
Полимер
Полиэтилен
Полипропилен
Полистирол
Полиакрилонитрил
ТГолим ети лм етакрилат
Полиметилметакрилат
Полнвинилацетат
Полибутадиен
Поливиниловый спирт
Поливинилхлорид
ТГолидиметилсилоксан
Этнлцеллюлоза
Тринитрат целлюлозы
Растворитель
Хлорнафталин
>
Толуол
Диметилформамид
Ацетон
Бензол
Ацетон
Гексан
Вода
Тетрагидрофуран
Толуол
Метиловый спирт
Ацетон
dn
dC
—0,199
—0,189
0,109
0,087
0,134
0,01
0,1
0,15
0,17
0,11
0,094
0,13
0,105
V
0,903
0,835
0,798
0,625
1,075
0,765
0,705
—
0,86
~
Приготовление раствора является обычной операцией.
Существенным является выбор растворителя. Растворитель должен
удовлетворять двум требованиям. Во-первых, необходимо
подобрать растворитель с показателем преломления, максимально
отличающимся от показателя преломления полимера.
В табл. III. 1 приведены инкременты показателей преломления
для некоторых систем. Более подробные таблицы имеются в
работе [199]. Вторым обстоятельством является получение
максимального архимедова множителя 1 — vp. Здесь v —
удельный парциальный объем, р — плотность растворителя (см.
табл. III. 1).
Как правило, величину v определяют независимо.
Физический смысл v — это изменение объема, происходящее при
добавлении 1 г вещества к бесконечно большому объему
растворителя. Обычно v определяют пикнометрическим способом.
Берут пикнометр объемом 50 мл. Приготовляют раствор полимера
(обычно переосажденного) с концентрацией С « 1 г/дл.
Архимедов множитель определяется по формуле:
1 — бр = (т — т0) р/тС, (III. 14)
где т—масса раствора; т0 — масса растворителя в пикнометре.
Пример. Концентрация раствора полистирола в бензоле 1 г/дл. Масса
пикнометра 25,5754 г, масса пикнометра с растворителем 70,7838 г, масса
пикнометра с раствором полимера 70,8646. Подставляя в формулу (III. 14),
получаем:
(45,2897 — 44,2084) • 0,8736 • 100 . ,_*
1-"Р = 4W897T1 = 0'156-
Таким образом, подготовив раствор и заполнив кювету,
производят вращение ротора и наблюдают смещение х кривых в
кювете.
Смещение границы (профиль распределения концентрации
или градиента концентрации) фиксируется, как правило, на
фотопленке (рис. III. 3).
182
Движение макромолекул в ячейке ультрацентрифуги
описывается, исходя из баланса сил трения, центробежных сил с
учетом архимедова множителя и сил случайного,
диффузионного движения. В условиях равновесия Г —тт = О J можно по вели-
определить молекулярную массу. Однако в обычных
чине
dx
условиях равновесие в кювете достигается только за
длительное время, поэтому этот метод определения ММ используется
редко.
Наибольшее распространение получил абсолютный метод
определения ММ: метод приближения к седиментационному
равновесию (метод Арчибальда). Действительно, на
поверхности мениска и на дне кюветы не наблюдается движения
макромолекул. Тогда на этих границах соблюдаются условия
-п- = 0, т. е. имеет место равенство диффузионного и седимен-
тационного потоков. Наблюдается характерное распределение
концентрации и ее градиента в кювете (см. рис. III. 3): в
середине кюветы имеется область плато, соответствующего
исходной концентрации. Форма градиента концентрация — кривая с
минимумом. Фиксируя на фотопленку наблюдаемую картину
распределения концентрации, определяя -?г-jjr У дна (или ме"
ниска) кюветы, вычисляют кажущуюся молекулярную массу Мл:
Мк =
2RT
din С
(1 — бр) со2 х dx
(III. 15)
где со — частота вращения ротора; R—газовая постоянная; Т — абсолютная
температура.
По данным эксперимента определяется величина -т- на
линии мениска (дна). Для этого находят высоту Zm кривой
градиента концентрации у мениска в относительных единицах и
площадь Дт! под ней. В независимом эксперименте подсчитывают
в тех же единицах площадь пика Ш\ для того же раствора,.
а б В
Г
3L
Va
А
Рис. III. 3. Зависимость концентрации (а) и градиента концентрации (б, в) полимера от-
расстояния от мениска т в кювете ультрацентрифуги при приближении к равновесию
(<х, о") и в опытах по скоростной седиментации (в):
/ — начальное распределение; 2—при t^O.
183-
когда наблюдается полная кривая распределения концентрации
в кювете. Тогда
d\nC Z
dx тх — Д^1
Далее по формуле (III. 15) и рассчитывают Мк.
Пример. Необходимые данные для расчета Мк в случае раствора по-
.листирола в бе
нзоле приведены ниже:
t, мин 4
Zm 0,75
Д/и, 0,06
/и, 0,93
Мк ■ 105 0,89
16
0,8
0,08
0,93
0,97
34
0,9
0,1
0,93
1,12
Частота вращения ротора 15 000 об/мин.
Мх рассчитывают по формуле (111. 15). Приведем расчет для одного из
значений времени:
м mTZm 10-2,47- 10". 0,8
к (1-бр)аАетСт ~~ 0,1558-2,465-106.6,26-0,8 ' '
где 10 — фактор полного увеличения оптической системы ультрацентрифуги и
фотоувеличителя.
Для получения Mw проводят экстраполяцию Мк на С " 0
и / * 0. Эти процедуры связаны с неидеальностью растворов и
полидисперсностью образцов. Действительно, концентрация
более тяжелых компонентов вследствие седиментации будет
увеличиваться у дна ячейки и уменьшаться у мениска. Поэтому со
временем эксперимента. Мк у мениска будет уменьшаться, а у
дна кюветы —увеличиваться. Противоположное влияние
оказывает неидеальность растворителя. Поэтому для
полидисперсного образца в хорошем растворителе может наблюдаться очень
слабая зависимость Мк от t.
Таким образом, указанным методом определяют М„. В прин-
d"C
ципе, рассмотрев более высокие производные —[-q-> можно
определить и ММ более высокой степени усреднения.
III. 2.2. Сочетание коэффициентов седиментации и диффузии
Ультрацентрифуга позволяет определять еще одним
абсолютным методом ММ. Это метод сочетания коэффициентов
седиментации S и диффузии D.
В условиях скоростной седиментации формируется полная
кривая градиента концентрации (см. рис. III.3,в). Как
правило, рассматривают смещения х пика этой кривой во времени.
Тогда можно рассчитать коэффициент седиментации 5:
s—^-4^-- <ш-16)
to2 dt
Пример. Пусть частота вращения ротора ультрацентрифуги
40 000 об/мин. Отсюда (о2 = 17,53-106 рад/с. Значения Igx равны 0,7889 и
184
0,7972 при временах эксперимента 10 и 60 с. Подставляя этн данные в
формулу (III. 16), получаем:
0 _ 2,303(0,7972-0,7889) 13
S- 17,53.10^.300 = 3-63-10 ■
Для более точного определения —-тт— строят зависимость lgx
от t и определяют наклон этой прямой.
Поскольку седиментация происходит в замкнутом сосуде,
возникают сложные гидродинамические эффекты. В связи с
этим 5 зависит от ряда факторов: давления, секториального
разбавления, концентрации. Поэтому обычно величину 5
экстраполируют на С "0 и учитывают изменение вязкости
растворителя и архимедова множителя (по таблицам
физико-химических свойств низкомолекулярных веществ) при р = 0,1 МПа.
Такое значение 5 обозначают через S0- Обычные значения 5
имеют порядок Ю-13 с.
Для экспериментального определения D обычно применяют
специальные установки [191, с. 357; 200]. Иногда для этой
цели можно использовать ультра центрифуги со специальной
кюветой, создающей искусственную резкую границу между
раствором и растворителем с помощью подслаивания (наслоения)
одной жидкости под другой. Полученный коэффициент D также
экстраполируют на С—"0. По этим величинам (So и D0)
рассчитывают ММ:
М**-ШГ*Ы- (ШЛ7)
Пример. Пусть значение 5 = 5-10->э, (1 — tip) =0,1238, Z)=0,3-10-e.
Тогда Msd равно:
^Д=5,10"3'62'477'10'0-3.3.103.
SD 0,3-Ю-6-0,1238
Обычно Msd больше Мп, но меньше Mw.
III. 2.3. Определение молекулярных масс
по коэффициентам седиментации
Коэффициент седиментации S0 связан с коэффициентом
трения f соотношением:
S0 = M(\-vp)/(NAf), (III. 18)
где NA — число Авогадро.
Коэффициент трения, в свою очередь, зависит от ММ: для
гибкоцепных макромолекул / « М0,5""0,6; для жесткоцепных
макромолекул / = УИ0'8^0,9. В связи с этим для гибкоцепных
макромолекул 5о « М°'ъ -=- УМ0'4, тогда как для жесткоцепных So да
~ М°-2-ь М0-1. Таким образом, для палочкообразных частиц
наблюдается очень слабая зависимость So от М. Это
существенный недостаток метода седиментации при анализе
жесткоцепных макромолекул.
185
Таблица III. 2. Эмпирические константы Ks и Ь
в уравнении (III.19)
Полимер
Полиэтилен
Полиизобутилен
Полибутадиен
Полиизоггрен
Полистирол
Поливииилацетат
Поливиниловый спирт
Полиметилметакрилат
Полиакрилонитрил
Этилцеллюлоза
Растворитель
Бромнафталин
Циклогексан
Октан
»
Циклогексан
Метилэтилкетон
Ацетон
Ацетон
Вода
Бензол
Диметилформамид
Этилацетат
Температура, °С
ПО
20
20
20
35
20
20
20
20
20
25
20
Ks-io'5
7,7
1,42
15,9
6,1
1,35
2,6
6,76
1,33
3,8
1,07
2,14
4,6
*
0,656
0,595
0,7
0,62
0,49
0,5
0,534
0,59
0,6
0,57
0,61
0,63
В общем случае между S0 и М существует соотношение:
S0 = KsMl-b, (HI. 19)
где Ks и Ь — эмпирические константы (табл. III. 2; см. также [192, с. 233;
197, с. 78; 198, с. 187].
Зная зависимость (III. 19) для конкретной системы
полимер— растворитель, т. е. зная калибровочную кривую, можно
по 5о рассчитать ММ, обращая (III. 19).
Прим ер. Пусть S = 5-Ю-'3, Ks = 4-10-">, Ь = 0,4, тогда
т. е. М=4,0-10s.
Таким образом, рассчитанное значение Ms меньше Mw, но
больше Мп-
III. 2.4. Метод вискозиметрии
Одним из самых распространенных методов определения ММ
полимеров является измерение вязкости их разбавленных
растворов (вискозиметрия). Простота оборудования (стеклянный
вискозиметр, термостат), легкость измерения времени течения
раствора через капилляр обеспечили широкое распространение
этого метода, однако он не лишен и ряда недостатков.
Во-первых, это относительный метод, т. е. требуется предварительная
калибровка вискозиметра. Во-вторых, этим методом
определяется средневязкостная ММ—М„. Для обычных гибкоцепных
полимеров существует соотношение М„ < М^ < Мш.
Взаимосвязь между Мп, Мц> Mw зависит от формы ММР и
полидисперсности полимера. И третье, вязкость сильно зависит от
концентрации, температуры, качества растворителя, иногда и от
186
градиента скорости. К сожалению, все эти факторы
недостаточно учитывают при проведении эксперимента, что приводит к
путанице данных и некорректности выводов.
III. 2.4.1. Определение характеристической вязкости
Вначале рассмотрим просто определение характеристической
вязкости [т|] для обычных растворов полимеров, т. е. гибко-
цепного полимера средней ММ (~105) в хорошем
растворителе. Впоследствии рассмотрим особенности определения и
влияние различных параметров на вязкость разбавленных
растворов.
При растворении полимера в растворителе вязкость
раствора увеличивается. В качестве меры увеличения вязкости вводят
характеристическую вязкость (или предельное число вязкости)
[т]], которая измеряется в см3/г (или в дл/г):
h]= lim (^^-). (Ш. 20)
с->о V Ло^ )
где г), т|о — вязкость раствора и растворителя.
Тот факт, что проводится экстраполяция на С—*■(),
предполагает, что полученная величина характеризует индивидуальную
макромолекулу. В случае агрегирования молекул, образования
комплексов, когда операция разбавления не приводит к
разрушению таких образований, [ц] характеризует размер ассоци-
ата молекул.
Настоящая методика основана на ГОСТ 18249—72 «Метод
измерения вязкости разбавленных растворов полимеров». Для
определения вязкости разбавленных растворов полимеров
применяют, как правило, капиллярные вискозиметры *,
помещенные в термостат. При этом колебания температуры не должны
превышать 0,05 °С, а при повышенных температурах 0,15°С.
Вискозиметр устанавливают вертикально. Термостатирование
проводят не менее 15 мин. Время истечения то чистого
растворителя должно находиться в пределах 80—150 с. Время
истечения раствора для правильного определения [i\] должно
составлять (1,2-f- 1,6)то. Соответственно выбирают концентрацию
С полимер, т. е. С[к\] должно лежать в пределах 0,15—0,5
(ориентировочно).
Концентрацию раствора С (в г/см3) вычисляют по
формуле:
С = G/V,
где G — навеска полимера, г; V — объем раствора, см3.
Растворы не должны содержать нерастворившиеся частицы
полимера, поэтому растворы (и растворитель) фильтруют,
* Обычно используются вискозиметры с висячим уровнем.
187
Концентрацию раствора, полученную разбавлением в
вискозиметре, вычисляют по формуле:
Ci=Ci-iVi-I/(Vi-1 + Vo),
где C;_i и V,- 1—концентрация и объем раствора, залитого в вискозиметр;
V0 — объем добавленного растворителя.
За результат измерения принимают среднее арифметическое
не менее трех измерений. Как правило, разброс не должен
превышать 0,4 с.
При концентрации раствора не более 0,01 г/см3 плотность
раствора практически равна плотности растворителя и
отношение вязкостен равно отношению времен истечения:
"П/По = т/т0.
Инкремент вязкости вычисляют по формуле: i\/n\o— 1, а
число вязкости — по формуле (т)— Цо)1(г\оС). Иногда
рассчитывают логарифмическое число вязкости: 1п(г)/г]о)/С.
Предельное число вязкости, или характеристическую
вязкость [ц] определяют графической экстраполяцией числа
вязкости или логарифмического числа вязкости на нулевую
концентрацию, по крайней мере, по четырем точкам
(концентрациям).
Кроме [tj] по этим данным вычисляют константу Хаггинса
Кх из соотношений:
JL=^ = [l1] + .KxC[Ti]a, (III. 21)
^^ = bl]-(y-*x)c№ (Ш. 22)
Обычно Кх = 0,2 -=- 0,4, для плохих растворителей Кх =
=0,5 -г- 0,8; в случае агрегирования значение Кх может
достигать 1—2. Для олигомеров значение [ц] может быть
отрицательным, а Кх — как положительным, так и отрицательным
(до ±10) (см. п. III. 2.33).
Пример. Ниже приведены экспериментальные значения времени
истечения для растворов полистирола в бензоле при 25 °С, а также рассчитанные
значения относительной вязкости, числа вязкости, логарифмического числа
вязкости и константы Хаггинса:
In (т)/л«)
С
0,37
0,38
0,37
0,36
0,37
0,36
0,35
*Х
0,4
0,4
0,42
0,49
0,45
0,48
Из анализа этих данных видно, что для определения [г\] можно
использовать только первые четыре точки. Использование последних двух точек
приведет к занижению [r\] и завышению Кх- Для большей точности удобно ис-
г/дл
0
0,25
0,5
1,0
1,25
1,5
2,0
2,5
т, с
80
88
97
116
126
140
165
194
_П_
"По
1
1,10
1,21
1,45
1,58
1,75
2,06
2,42
Т1 — Т1,
—
0,4
0,42
0,45
0,47
0,5
0,53
0,57
188
пользовать построения по уравнениям (III. 21) и (III. 22). Видно также, что
погрешность определения числа вязкости зависит не только от точности
определения т] и Tio (т и То), но и от величины т|/т|о—1. Действительно, если
погрешность определения т составляет 0,5 %, а погрешность определения
относительной вязкости около 1 %, то погрешность определения числа
вязкости (г\ — *1°)/(г1<><-') может составлять уже 10 — 2% для растворов с
увеличивающейся концентрацией. Это означает, что для раствора с минимальной
концентрацией величина (г) — г\о)1(г)оС) = 0,4 ± 0,04 г/дл. Поэтому
использование растворов с меньшей концентрацией (т.е. г|/т)о < 1.1) приводит к еще
большей погрешности. Полученное значение [iq] для указанного примера
составляет 0,38 г/дл.
Еще большая погрешность возникает при расчете константы Хаггинса.
Рассуждая аналогичным образом, приходим к выводу, что для раствора с
минимальной концентрацией (0,25 г/дл) погрешность определения Кх
достигает 100 %. Поэтому эту точку в дальнейшем анализе Кх можно вообще не
учитывать. Приведенные выше данные показывают некоторую тенденцию к
росту Кх- Это естественная зависимость, так как мы не учитываем более
высокие слагаемые в зависимости вязкости от концентрации (см. п. III. 2.3.4).
Поэтому необходимо по данным для Кх провести экстраполяцию на С-+0.
Недостаточно четкий учет концентрационной зависимости Кх приводит к
завышению ее значения на 20—50 %. Приводимые в литературе данные Кх,
как правило, действительно завышены.
Здесь мы должны сделать одно замечание относительно
погрешности вычисления величин. Действительно, погрешность
единичного измерения (например, для раствора С = 0,25 г/см3)
может быть велика, особенно для Кх- Однако, поскольку мы
знаем вид функциональной зависимости (tj— Цо)/{т\оС) от С
(см. п. III. 2.3.4), использование вычислительной техники или
«на глаз» графической экстраполяции значительно увеличивает
точность определения величин. В связи с этим очень важно
знать функциональную связь вязкости и концентрации (см.
п. III. 2.3.4). Тогда значимость погрешности точек при малых
концентрациях существенно уменьшается.
К сожалению, в практике физико-химических лабораторий
эти вопросы учитываются недостаточно.
III. 2.4.2. Взаимосвязь [т|] с ММ и структурой макромолекулы
Для линейной цепной макромолекулы рассматривают соот-
ношение между [ц] и ММ в виде:
hl-^M"4, (Ш. 23)
где К и ап—эмпирические константы (табл. III. 3), более подробные
таблицы см. [192, с. 306; 201, IV-7].
Важно знать, особенно для полидисперсных полимеров,
каким методом определяем ММ при нахождении констант
уравнения (III. 23) для конкретного полимера. Если это
соотношение получено для узких фракций, а сопоставляются два разных
по полидисперсности образца, то рассчитанные по (III. 23) сред-
невязкостные ММ могут существенно различаться, даже если
Мп (или Mw) для этих образцов одинаковы. Это
обстоятельство необходимо учитывать при установлении корреляции
ММ — свойства или технология процесса.
189
Таблица III. 3. Эмпирические константы Кц и а.
в уравнении (III.23)
Полимер
Полиэтилен
Полипропилен
Полиизобутилен
Бутилкаучук
Полибутадиен
Полиизопрен
Полистирол
Поливинилацетат
Поливиниловый спирт
Поливинилхлорид
Полиметилметакрилат
Полиакриламид
Полиакрилонитрил
Этилцеллюлоза
Растворитель
Декалин
»
Толуол
Бензол
Толуол
Бензол
Бензол
Ацетон
Вода
Тетрагидрофуран
Толуол
Вода
Диметилформамид
Этилацетат
Кт,-10<
3,87
9,6
4,0
13,4
11,0
5,02
4,17
1,9
5,9
2,19
0,71
0,63
3,0
1,07
°Ч
0,738
0,63
0,6
0,63
0,62
0,675.
0,6
0,66
0,67
0,54
0,73
0,8
0,767
0,89
Так, для логарифмически нормального распределения
(III. 11) соотношение между Мц и Mw дается уравнением.
(III.24), а для гамма-распределения (III.8)—(III.25):
К + 1 + av
Ма
К + 2
(III. 24)
(III. 25)
Пусть сравниваемые образцы имеют одинаковую
полидисперсность: Mw/Mn = 5, но первый образец имеет
логарифмически нормальное, а второй — гамма-распределение. Тогда (при-
ац = 0,5) для первого образца имеем Мп = 0,45 Mw, для
второго Мг\ = OfiMw Исследователь может прийти к выводу, что
образцы различаются по ММ, хотя для этих образцов могут
быть одинаковые Mw. В этом заключается одна из трудностей
характеристики полидисперсных полимеров. Для полимеров,
имеющих мультимодальное распределение, как уже
указывалось в п. III. 1.2, ситуация еще более сложная.
Рассмотрим теперь соотношения между [ц] и структурой,
формой цепи [192, с. 167], которые и определяют значения
эмпирических констант в уравнении (III. 23) (рис. III. 4).
Характеристическая вязкость раствора жестких сплошных
частиц определяется их формой. Для частиц, имеющих форму
сферы [т]] = 2,5£7 (формула Эйнштейна) и [т)] не зависит от
ММ. Поэтому, если определяемая характеристическая вязкость
для олигомеров меньше 2,5у, это свидетельствует о наличии
специфических взаимодействий (см. п. III.2.3.3).
Для растворов асимметричных частиц (стержень,
эллипсоиды вращения с осевым соотношением р)
[т\] = б [2,5 + 0,4075 (р - I)1'508] (формула Куна)
190
Рис. III. 4. Схематичная логарифмическая
зависимость [л] от молекулярной массы для гибкоцеп-
пых (/, 2) и жесткоцепных (3) макромолекул вб-
растворителе (/) и в „хорошем" растворителе (2). ^^а= 0,5
И При р > 10 [т]] ~ J01'508, Т. в.
[т,] ~ М1-508 (см. рис. III. 4). В этом
случае влияние полидисперсности
на [т]] особенно велико и Мп > Мш.
Соответствующие расчеты можно
провести, используя соотношения
(III. 8) и (III. 11).
Для растворов гибкоцепных полимеров значения an лежат
в пределах 0,5—0,8 и определяются качеством растворителя.
Действительно:
гДе [ilia ~ характеристическая вязкость в 0-растворителе, пропорциональная
jVfo.s. a — коэффициент набухания.
Коэффициент набухания а макромолекулы зависит от Ж и
температуры:
аг=1 + СмУм /(Г-в,, %-Т),
где См — константа; f — функция качества растворителя, определяемая
удаленностью температуры эксперимента от верхней и нижней 0-температур
[192, с. 183].
Этим и определяется сложная форма зависимости [tj] от М
(см. рис. III. 4).
Кстати, из приведенных соотношений следуют очень
полезные правила, помогающие исследователю при работе с новым
полимером и при отсутствии калибровочной зависимости типа
(III. 23). Действительно, между /Сл и ац существует (для гибко-
цепных полимеров) определенная корреляция [192, с. 167].
Если a2 ~ Ме, то:
Kn = Ke(e/.y/M^)e, А„ = 0,5+ 1,5е,
где Мс — молекулярная масса сегмента; е — эмпирический коэффициент.
Отсюда следует, что между Кц и а,, имеется соотношение:
In Кц = 1п (Кв ч/Щ) - ! + ац (1п мс ~ 2У
Действительно, такая корреляция между Кг\ и а^ была
найдена экспериментально еще в работе [202] и получено простое
соотношение для [ц]:
м4(щГ' (IIL26)
где М0 — молекулярная масса мономера.
191
Таким образом, для расчета М неизвестного полимера по
значению [г\] необходимо знать только ац.
Здесь надо отметить, что найдено эмпирически [44, с. 272]
и обосновано [203] еще одно простое правило, которое
позволяет упростить и нахождение значения а„. Действительно, было
показано, что для широкого класса гибкоцепных полимеров
ац + Кх=\,\ + \,2. (III. 27)
Таким образом, для растворов неизвестного полимера по
вязкости разбавленных растворов можно определить значения
[tj] и Кх. Используя соотношение (III. 27), рассчитывают ал,
а далее по формуле (III. 26) определяют ММ. Несомненно,
подобные определения ММ носят несколько ориентировочный
характер. Однако эта методика выручает, когда по каким-либо
причинам неизвестны константы в уравнении (III. 23) и у
экспериментатора нет фракций исследуемого полимера.
Определение структурных параметров макромолекул
подробно рассматривается в работе [192, с. 167]. Здесь мы только
заметим, что влияние качества растворителя и негауссовости
цепной молекулы в в-растворителе приводят к однотипным
качественным эффектам (формально, увеличение ац). Поэтому
интерпретация данных должна проводиться с учетом всех
указанных обстоятельств и постановки дополнительных опытов.
Необходимо также отметить, что появление длинноцепной
разветвленности макромолекул также сказывается на значении
[т]] при одной и той же ММ. Этот вопрос подробно разобран"
в [192, с. 270] и частично — в применении к методу
гель-проникающей хроматографии в разделе III. 3.
III. 2.4.3. Влияние неоднородности строения цепи
и специфических взаимодействий на вязкость разбавленных растворов
Для растворов олигомеров, когда существенно влияние
концевых групп и других неоднородностей строения цепи, или
для растворов полимеров со специфическими взаимодействиями
(водные растворы полимеров, поверхностно-активные вещества)
иногда получают отрицательные значения [ц] и очень большие
значения Кх. Этот эффект связан с тем, что внедрение
молекулы полимера между молекулами растворителя приводит к
существенному уменьшению (увеличению) вязкости
растворителя вблизи молекулы полимера. В то же время формально для
расчета [г\] используется исходное значение вязкости
растворителя т]о. Теория этого эффекта рассмотрена в работе [204].
Было показано, что экспериментально определяемое значение-
[г)]э описывается формулой:
[ч]э = 1ч] + Лт).
где [ti] — обычное значение характеристической вязкости (см. п. III. 2.3.2);
величина Ац определяется изменением «локальной» подвижности молекул
растворителя, в простейшем случае Ац можно рассчитать по [204].
192
Рис. III. 5. Зависимость времени истечении т
раствора полимера от концентрации в случае
специфического взаимодействия полимера с
растворителем:
Т0 — время истечения растворителя;
Тд—исправленное т0.
Для учета этого эффекта при
проведении эксперимента можно
рекомендовать следующую
процедуру. Строят зависимость времени
истечения т. раствора полимера от С
концентрации С. При наличии
обсуждаемого эффекта в области малых концентраций
(С<;1/[т]]) наблюдается искривление зависимости т = /(С)
(рис. III.5). Проведя экстраполяцию т на С-»-0, получают
исправленное время течения т^ растворителя (т„ может быть
как больше, так и меньше то; как правило V0 < т0). Тогда [г\]
рассчитывается по соотношению:
[r|] = lim
т —тп
с-»о т0С
Далее исследователь уже проводит анализ [ц] описанными
выше методами.
В случае водных растворов полиэлектролитов также
наблюдается искажение концентрационной зависимости числа
вязкости (т] — Ло)/(т1оС)- Действительно, в растворе полиэлектролитов
кроме полиионов находятся низкомолекулярные противоионы.
При изменении концентрации происходит перераспределение
противоионов внутри макромолекулярного клубка.
Поскольку цепная молекула гибкая, то при уменьшении концентрации
полимера наблюдается «полиэлектролитное набухание» цепи из-
за появления некомпенсированных зарядов на макромолекуле:
низкомолекулярные противоионы уходят из объема
макромолекулы. Теория концентрационной зависимости вязкости
растворов полиэлектролитов отсутствует, поэтому в практике
подавляют полиэлектролитное набухание. Как правило, это достигается
с помощью растворения в растворе полиэлектролита
определенного количества низкомолекулярной соли, например NaCl,
LiBr и т. д. Наличие добавочных ионов в растворе экранирует
заряды на цепи. При этом, как правило, ухудшается качество
растворителя и может наблюдаться осаждение полимера из
раствора. Экспериментатор опытным путем подбирает
необходимую концентрацию низкомолекулярной соли таким
образом, чтобы наблюдались закономерности, описанные в
п.Ш. 2.4.1.
7 Зак. 632
193
III. 2.4.4. Влияние скорости сдвига и концентрации на вязкость
растворов полимеров
Для высокомолекулярных объектов даже вязкость
разбавленных растворов может зависеть от скорости сдвига. Этот
эффект связан с тем, что с ростом градиента скорости сдвига g
из-за неравномерного вращения макромолекул происходит
ориентация макромолекул вдоль потока. Это приводит к
уменьшению вязких потерь при деформировании раствора. В
качестве характеристики меры ориентации рассматривают
величину
В случае р л; 1 необходимо учитывать уже градиентную
зависимость вязкости раствора. Для обычных вискозиметров при
[т]] > 4 дл/г также необходимо вводить попавку на
зависимость [т]] от g.
Для учета зависимости вязкости разбавленных растворов от
g обычно проводят измерения в вискозиметрах с различными
значениями g. Обычные вискозиметры имеют g » 103 с-1.
Увеличение длины капилляра пропорционально увеличивает время
истечения т, т. е. уменьшает значение g. Для уменьшения g
также несколько увеличивают радиус капилляра г.
Существенное увеличение г неудобно, так как приводит к резкому
уменьшению т или увеличению V. Для очень малых g используют
вискозиметр Зимма [192, с. 185], который можно изготовить е
лаборатории. Он крепится на магнитной мешалке и состоит из
неподвижного внешнего цилиндра с термостатируемой
рубашкой и ротора, плавающего в исследуемой жидкости.
Учет влияния концентрации полимера на вязкость его
раствора — один из важнейших факторов, определяющих
правильность определения [т]]. Хотя этот вопрос обсуждается во
многих публикациях и даже введен в ГОСТ 18249—72, тем не
менее практика показывает, что во многих лабораториях
неправильно учитывают этот эффект (см. п. III. 2.4.1). Кроме того,
при проведении стандартизованных испытаний часто вместо
[г\] определяют зависимость вязкости концентрированных
растворов от М и по вязкости сопоставляют образцы между
собой. В общем виде вязкость концентрированных растворов гиб-
коцепных полимеров описывается соотношением [205, с. 95]:
_i_
r^rHl+vCh^expj^^^}, (III.28)
где у — коэффициент, характеризующий межмолекулярное термодинамическое
взаимодействие: К\ = (1—Y)/2j Н — энергия активации растворителя;
а = 0,5 -и 0,8 и зависит от удаленности температуры эксперимента от
температуры стеклования полимера; <р — объемная доля полимера в растворе.
194
При ф<0,3 третьим сомножителем в (111.28) можно
пренебречь. Тогда для числа вязкости имеем:
-^г-тЛ = [л] + КХС W + Кх —+ СЧг,]3 + ...
Отсюда видно, что наиболее точно определяется величина
Кх только тогда, когда Кх ~ 0,25. В случае плохого
растворителя, когда Кх ~ 0,5 и более, погрешность в определении Кх
максимальна.
Используя полученное соотношение (III. 28), можно
разработать программу для ЭВМ вычисления вязкостных констант
[т]] и Кх по экспериментальным данным в широком интервале
концентраций.
Таким образом, правильный выбор концентрации раствора
для определения ММ по т) определяется соотношением
значений второго и третьего сомножителей в уравнении (III. 28).
Если температура эксперимента близка к температуре
стеклования системы, то относительный вклад последнего
сомножителя является определяющим. В связи с этим можно ожидать
небольшой чувствительности вязкости к изменению ММ и очень
большой к изменению температуры. При не очень надежном
термостатировании точность определения ММ невелика.
III. 2.4.5. Определение ММ с использованием гидродинамических
инвариантов
Как мы уже говорили, определение ММ по [г\] возможно,
если есть калибровочная зависимость [т]]=/(М).
Аналогичная ситуация прослеживается, когда измеряются
коэффициенты диффузии D и седиментации S. Однако сочетание [т]] и D
(или [т]] и 5) позволяет определить ММ вне зависимости от
модели цепи (абсолютный метод определения ММ).
Многочисленными экспериментальными данными и
теоретическими расчетами показано, что выполняется
гидродинамический инвариант Флори — Манделькерна — Цветкова [191,
с. 354; 206]:
А Solo hi'7' =JMjL(M[ ])Чз (IIL2g)
(l-op)Af/3 kT
Для гауссовых цепных молекул /4г=1,26-107 или 2,7-106,
если [т]] выражена соответственно в см3/г или дл/г.
Гидродинамический инвариант не зависит от качества
растворителя, фактически не зависит от гауссовости цепной
макромолекулы, несколько зависит от полидисперсности [207].
Выполнение гидродинамического инварианта приводит к
соотношению
36=1+0,, (III. 30)
7*
195
для коэффициентов в уравнениях (III. 19), (111.23) и D=
= KdM-K
Таким образом, зная значения констант в этих уравнениях,
можно рассчитать ММ.
III. 2.5. Определение молекулярных масс
нерастворимых полимеров
Для большинства полимеров достаточно точное измерение
их средних ММ и ММР основано на исследовании свойств
разбавленных растворов. Однако многие полимеры невозможно
анализировать ни одним из этих методов вследствие их
нерастворимости. Ряд методов оценки средних молекулярных масс
и вида ММР плавких линейных полимеров может быть
предложен на основе исследования взаимосвязи молекулярно-мас-
совых и реологических характеристик расплавов полимеров.
Основными факторами, определяющими вязкостные и вязко-
упругие свойства расплавов полимеров, являются ММ и ММР.
Как правило, вязкость расплавов полимеров зависит от
скорости сдвига g.
Известно [208, с. 179], что зависимость вязкости растворов
и расплавов узких фракций линейных полимеров от
молекулярной массы описывается степенным законом. Показатель
степени имеет различные значения для двух областей молекулярных
масс, разделенных критическим значением Мкр *:
( tf ,ма'
4<M)H пр„м<мкр, (шз1)
при М > Мкр,
где К\ и Кг — эмпирические константы полимергомологического ряда; а.\ tsz \;
Р, « 3,4 Ч- 3,5.
Для полидисперсных полимеров анализ литературных
данных показал, что соотношение (III. 31) также может быть
применено, если М заменить среднемассовой молекулярной
массой Mw- Хотя в общем случае вязкость является функцией
полидисперсности полимера, оцениваемой обычно отношением
Mw/Mn, а вид этой функции зависит от формы ММР [209]
полимера, для приблизительных расчетов можно принять т) ~
~Мщ5. Определив значение т) по кривой течения расплава
полимера и зная из независимых данных значение Ki для данного
гомологического ряда полимеров и выбранных условий
измерения, можно по соотношению (III. 31) оценить значение Mw.
* Принято считать, что критическая молекулярная масса Мк? является
граничной величиной, которая отвечает образованию сетки зацеплений
макромолекул, ведущей к возникновению большого сопротивления течению.
Современные теории молекулярного течения показывают, что в этой области ММ
происходит переход к «рептелевидному» движению, т. е. когда возможно
движение только в направлении концов цепи. Ограничения к поперечному
движению цепи равномерно распределены вдоль цепи.
196
В том случае, когда значение Ki неизвестно, для
относительной оценки изменения Мт нерастворимых линейных полимеров
при их переработке, термостарении и других процессах, а
также для сопоставления Мш различных образцов используется
соотношение:
lg <> - lg Ajg» = Jg- (lg г,'1» - lg r,<2>), (Ш. 32)
где индексы (1) и (2) относятся к различным образцам одного и того же
полимера.
На основании измерения наибольшей ньютоновской вязкости
расплава можно оценить только Mw. Однако приведенные в
монографии [208, с. 179] данные свидетельствуют, что при
молекулярных массах выше критической неньютоновское течение
расплава полимера имеет место при тем меньших скоростях
сдвига, чем больше молекулярная масса и чем шире ММР.
Это говорит о связи ММР с формой и расположением кривой
течения расплава. В настоящее время установлено, что
получить инвариантную зависимость v\ = f{g, M) для всех типов
ММР и любой полидисперсности невозможно. Тем не менее,
исследование зависимости градиентной вязкости расплава
линейных полидисперсных полимеров от Мп и вида ММР
позволило получить аналитические соотношения, на основе которых
возможен количественный анализ ММР нерастворимых
полимеров.
Суть метода [209] сводится к расчету по этим
соотношениям зависимости
,е(лМ)р_,(1г^)
для различных значений Мп и Mw/Mn и сопоставлении
рассчитанных значений с экспериментальной зависимостью
Для реализации данного метода необходимо выполнение
следующих условий: а) исследуемый полимер должен иметь
линейные гибкоцепные макромолекулы с ММ, превышающей
критическое значение Мкр; б) между макромолекулами в
расплаве не должно существовать никакого специфического
взаимодействия, приводящего к структурированию, образованию
агрегатов и т. п.; в) из априорных представлений должен быть
известен вид функции распределения макромолекул по ММ,
наиболее близкой к реальному ММР; г) кривая течения
расплава полимера должна быть определена в возможно более
широком интервале скоростей (напряжений) сдвига, причем
особое внимание следует обратить на точное измерение участка
наибольшей ньютоновской вязкости расплава полимера.
При измерениях вязкости расплава на ротационных
вискозиметрах можно использовать режим периодического деформиро-
197
йЧсМп
Рис. III. б. Номограмма дли расчета ММ
по вязкости полимера (логарифмически
нормальное распределение):
1—5— расчетные кривые с MwJMn =
= 1, 2. 5, 10, 20 соответственно: 6,7 —
моноднсперсные полистнролы; 8—10—
полиэтилены с М Шп = 3,6; 6,7; 7,8.
соответственно.
вания расплава полимера.
При этом кривая течения
получается в координатах
к, »£ = /Н (здесь т£ — абсо-
*2 лютное значение
комплексна ной динамической вязкости
vZ расплава полимера, ш —
частота деформирования).
Такую зависимость можно
применять аналогично
f\(g) = f(g), предположив, что для исследуемых полимеров
соблюдается правило Кокса — Менца, согласно которому
Л» = 4 (2) при co = g.
Связь молекулярно-массовых и реологических характеристик
дается системой уравнений, приведенных в [209, 210]. В
процессе расчета в качестве функции распределения gn(M)
целесообразно для линейных полиэтиленов использовать нормальное
логарифмическое распределение, а для полимеров,
синтезируемых радикальной полимеризацией,— гамма-распределение (см.
п. III. 1.3).
Наиболее точные значения Mw и Мп по предлагаемой
методике можно получить, используя ЭВМ. Для этого Mw и Мп.
для каждого образца находят методом последовательных
приближений путем минимизации функционала Ф:
где N — число экспериментальных точек на кривой течения расплава; ( I
\ Л S3.
И (2«Ш
V ч h
экспериментальное и расчетное значения градиентной
вязкости расплава при скорости сдвига g.
Критерием окончания расчета служит совпадение расчетных
и экспериментальных значений градиентной вязкости в
пределах погрешности эксперимента. Описанный метод
определениями, и М„ был успешно реализован при анализе ММР
сополимера тетрафторэтилена с этиленом [210].
Для менее точной, но более быстрой и простой оценки М-^
и Мп целесообразно использовать графический метод анализа
реологических данных (рис. III. 6). Для этого зависимость
198
рассчитанную с использованием ЭВМ для различных qn{M) и
значений Mw/Mn, изображают в виде графика (рис. III. 6).
Экспериментальную кривую течения r\ (g) = f (g) [или "% = f (со)]
перестраивают в координатах
-(■TX-'W - *т.-'№)-
изображая ее на кальке в том же масштабе, что и расчетные
зависимости. Для оценки значений Мп и Mw/M„ исследуемого
полимера накладывают график экспериментальной зависимости
на расчетные кривые так, чтобы оси абсцисс совместились, и
сдвигают его вдоль оси абсцисс до совпадения
экспериментальной кривой с одной из расчетных. Совпадение форм кривых
означает, что экспериментальное значение MwjMn исследуемого
образца полимера соответствует расчетному. Значение Мп,ъ.
определяют по соотношению:
где первая часть уравнения представляет собой разность
значений абсцисс произвольной точки на совпавших
экспериментальной и расчетной кривых. Определив таким образом значение
Мп и зная Mw/Mn, легко найти Мш. Примером исследования
графического метода оценки Mw и Мп нерастворимых
полимеров может служить анализ ММР сополимера тетрафторэтилена
с гексафторпропиленом [210].
Изложенные выше методы анализа ММР нерастворимых
линейных полимеров были основаны на взаимосвязи молекуляр-
но-массовых характеристик с чисто вязкостными свойствами
расплавов полимеров. Существует также метод оценки ММ
монодисперсных полимеров или полимеров с узким ММР по вяз-
коупругим характеристистикам расплава. Известно [208, 236],
что кривая зависимости модуля упругости G^ монодисперсного
линейного полимера от частоты деформирования при
измерениях в режиме периодического деформирования в области ММ
(более 5-^-10 Мкр) имеет характерные плато высокоэластично-
сти, ограниченные начальной и конечной частотами cuh и о)н.
При этом как высота G'a, так и протяженность плато высоко-
эластичности Д lg со = lg шк — lg сон связаны с ММ полимера
определенными соотношениями. ММ вычисляют по уравнению:
W = ^+l^), (Ш. 33)
где р — плотность полимера.
Из приведенного нами краткого обзора методов определения
молекулярно-массовых характеристик нерастворимых полимеров
по реологическим данным ясно, что в настоящее время это
выполнимо лишь для частичного случая линейных полимеров с
узким ММР и известной функцией ММР. Распространение анали-
199
за ММР линейных полимеров на общий случай произвольного
вида ММР, а также оценка разветвленное™ макромолекул по
реологическим данным являются целью дальнейших
исследований.
В работе [211] была предложена методика определения Мт
и полидисперсности в низкомолекулярной (Мш/М„) и
высокомолекулярной (Mz/Mw) области ММР с использованием
только одного прибора—реометра постоянного давления. Эта
методика является относительной, т. е. требует предварительной,
калибровки (или позволяет только сравнивать образцы между
собой). Как было сказано ранее, характер кривых течения
определяется параметрами Mw и Mw/M„. При выходе расплава
из капилляра прибора вследствие запасенной им
высокоэластической деформации наблюдается разбухание Р полимерной
струи, которое определяется значением Мг\Мщ. По известным
образцам проводят калибровку показателя текучести расплава
при двух нагрузках и значений р. С помощью ЭВМ находят
регрессионные соотношения. Так, для полистирола показатель,
текучести расплава / определяли по ГОСТ 11645—73 на экстру-
зионном пластометре типа ИИРТ-М с внутренним диаметром
капилляра 2,095 мм при 200°С и массе груза 1,2 кг (/]) и
12,5 кг (h)- На этом же приборе находили р в идентичных
вязкосдвиговых условиях. Было получено:
lg Mw = 5,36 - 0,2 lg /, + 0,02 lg (/„//,),
Ig Mw/Mn = -1,1 + 0,17 lg /, + 0,89 lg (/,//,).
По разработанным моделям можно рассчитать номограмму,,
позволяющую быстро находить Mw, Mw/Mn, Mz/Mw.
III. 3. МЕТОДЫ ОПРЕДЕЛЕНИЯ МОЛЕКУЛЯРНО-МАССОВОГО
РАСПРЕДЕЛЕНИЯ
Полную информацию о молекулярных характеристиках
полимерных материалов можно получить, только имея ММР,.
которое дает представление о наличии (доли) тех или иных
гомологов в образце. Как мы уже неоднократно говорили,
характеристика ММР теми или иными ММ (Мп, Мц, Mw) дает
недостаточную информацию об образце. Особенно это важно^
учитывать при установлении корреляции свойства — ММ, так
как некоторые свойства определяются либо высоко-, либо
низкомолекулярными хвостами. ММР наиболее часто определяют
двумя методами: скоростной седиментацией и гельпроникающей
хроматографией.
III. 3.1. Метод скоростной седиментации
Как мы уже говорили (п. III. 2.1), при вращении ротора-
ультрацентрифуги развиваются большие центробежные
ускорения. Под действием этих центробежных сил макромолекулы!
200
смещаются. Скорость смещения определяется коэффициентом
седиментации S [см. уравнение (III. 16)]. Коэффициент
седиментации, в свою очередь, определяется формой и ММ
макромолекулы [уравнения (III. 18) и (III. 19)]. Таким образом,
более массивные молекулы двигаются быстрее и с помощью
оптических методов можно наблюдать распределение по S (или
ММ).
III. 3.1.1. Определение распределения по коэффициентам седиментации
Поскольку макромолекуле с заданной ММ соответствует
свой коэффициент седиментации S, а он, в свою очередь,
определяет скорость движения и, следовательно, положение
границы х, то в кювете ультрацентрифуги в каждый момент времени
устанавливается распределением по х:
х = хме ,
где Хм — расстояние от оси вращения ротора до мениска кюветы (~6 см).
С помощью оптических схем регистрируют распределение
CdC , .
или градиента концентрации ~j— = q (х) как
■функции х. Мгновенная картина, например q(x), дает
возможность перейти к распределению по S:
q (S, t) = q(x,t)-£- — =q (x, t) ■£- 0>2t. (III. 34)
*м dS xl
Здесь уже произведен учет секториального разбавления при
движении макромолекул в кювете. Видно, что q(S) является
функцией как х, так и t.
Существуют две методики обработки экспериментальных
данных: методика «фиксированного» времени и методика
«фиксированной» координаты.
При определении q(x) по первой методике на фотопленках
регистрируют седиментограммы в различные моменты времени.
Существенным здесь является то, что фотографирование
начинается тогда, когда образуется кривая распределения (см.
рис. III. 3), т. е. у мениска имеется «свободная» от полимера
область. Далее обработку кривой распределения h(x) no x (в
данный момент времени) ведут традиционным образом.
Кривую разбивают равноотстоящими вертикальными линиями на
ряд участков (Дд; = *,+i — х,) с соответствующими высотами
hi(xi). Число точек не менее 10, для полидисперсных
образцов— не менее 20. Каждой точке приписывается свой
коэффициент седиментации Si:
Si = S±/ASf; i=i, 2, 3 ....
где 5 — значение S в некоторой выбранной точке х: для симметричных
кривых — это максимум кривой, в других случаях — середина кривой.
201
Значения S и Д5,- рассчитывают по формулам:
^ _ 1п (х/хн) т д5 = .Дл;
а>4 ' ц>Чх~
Предполагается, что Ах <С х. Ординаты при S,-
рассчитывают по формулам:
А (5;) = A (Jtf)—|- а>4; xt = x± /Дат.
Для получения q(S) проводят нормировку полученной
кривой:
(III. 35)
\h(S)
dS
Поскольку такая обработка данных требует проведения
однотипных расчетов, удобно составить соответствующую
программу для ЭВМ.
Важным моментом в обработке данных является выбор
времени t. Обычно граница отрывается уже при разгоне ротора,
еще до установления стационарной частоты вращения. Поэтому
t рассчитывают, как правило, по соотношению:
t-r + ±bt.
где V — время, отсчитываемое от момента достижения стационарной частоты
вращения; At — время разгона.
Однако правильнее для каждой ультрацентрифуги выбирать
свое эмпирическое соотношение для t. Для этого At
определяют из графика зависимости \пх от / при экстраполяции х на
Хы-
Вторая методика обработки данных [212] заключается в
том, что фиксируется на изображении кюветы координата
отсчета хс. Анализируется изменение высоты седиментограммы h
как функции времени: ft (xc, t). Тогда:
х3
Л (S) = А {хс, о -f- аЧ. (III. 36)
Эта методика значительно упрощает расчеты.
Действительно, {хЦх^а?— постоянная величина, поэтому переход от ft (xc, /)
к ft(5) очень прост. Величина 5 рассчитывается по
соотношению:
1 1п (хс/х„)
S =
t
В то же время эта методика требует некоторого изменения
условий проведения эксперимента. Съемка седиментации или
202
слежение за изменением h (xc, t) со временем с
использованием стекла с нанесенными калиброванными делениями
начинается значительно раньше: как только наблюдается изменение
нулевой линии. Если пропустить этот момент, то можно не
заметить высокомолекулярную часть распределения. Строится
зависимость h(xi, t) и далее по формуле (III.36) переходят к
Л(5) и по формуле (III.35) —к q(S).
Выбор координаты произволен, однако лучше ее выбирать
вблизи мениска. Недостатком методики является иногда
наблюдаемое изменение базовой линии, что сказывается на величине
h (хс, t). Поэтому использование бисекториальной кюветы
является обязательным. Для контроля можно провести обработку
данных при другом значении хс. Существенное достоинство этой
методики в том, что она позволяет проводить анализ очень
полидисперсных образцов, а также содержащих гелеобразные
частицы. Действительно, отсчет времени начинается сразу,
когда изменяется базовая линия, т. е. когда начинают седименти-
ровать высокомолекулярные фракции образца. В методике
«фиксированного» времени отсчет начинается тогда, когда в
области мениска появляется зона, свободная от полимера. Для
полидисперсного образца к этому времени уже
высокомолекулярные фракции «осели» на дно кюветы и не фиксируются на
фотопленке. Таким образом, методика «фиксированной»
координаты дает более широкое распределение, чем методика
«фиксированного» времени.
Полученные распределения по q (S) еще не являются
истинными. Необходимо также учесть поправки на диффузионное
размывание кривой, на концентрационную зависимость S [192,
с. 233; 198, с. 197].
Эффект диффузионного размывания проявляется для
образцов с достаточно узким ММР. В этом случае наблюдается
непрерывное «заострение» кривых q (S) с течением времени.
Влияние диффузии принято исключать экстраполяцией данных
на t-*-oo. Если экспериментальные кривые q{S) [или /i(S)],
полученные в разные моменты времени, совпадают, то
диффузионным уширением можно пренебречь.
Более существенным является поправка на
концентрационную зависимость 5. Действительно, было показано, что
величина 5 зависит от концентрации:
l/S=l/S0(l + /CsC[i|]+...), (III. 37)
где So = S при С^-0; Ks — численный коэффициент, равный 1,4-г-1,7 для
хороших растворителей и 0 -=- 0,6 — для плохих растворителей.
Концентрационная зависимость 5 приводит к «сужению»
кривой распределения. Особенно этот эффект сказывается на
высокомолекулярном хвосте распределения. Для учета этого
эффекта проводят экстраполяцию на C-vO по следующей
методике. От дифференциального распределения q(S) переходят
203
Рис. III. 7. Зависимость W от S при разных
концентрациях раствора полимера.
к интегральному распределению:
s
о
(S)=\q(S)dS.
Строят графики зависимости
If от S для растворов разных
концентраций (рис. III. 7). Далее для массовых долей ©,•
графическим фракционированием определяют значения Si при
нескольких концентрациях и находят экстраполяционное значение
Si о (С-*-0). Теперь уже строят зависимость W (5,-, 0) и нахо-
„ п .„. dw (S0)
дят проэкстраполированное на С-*-0 q(S0) =—-т$—.
111.3.1.2. Переход к распределению по ММ
Используя соотношение (III. 19), легко получить q{M) или
W(M) по данным q(S) или W(S). Интегральное распределение
W(M) получается просто заменой переменной на оси абсцисс:
S-*-M. Распределение q(M) рассчитывается по соотношению:
qw(M)=-.q(S0)-^- = q(S0)
So (1 - Ь)
М
(III. 38>
Отсюда видно, что проводить эксперимент предпочтительнее
в 0-растворителе (6 = 0,5); происходит более четкое
разделение по ММ. Кроме того, в в-растворителе уменьшается и
концентрационная зависимость S, что позволяет работать с
высокими концентрациями.
При проведении однотипных анализов иногда достаточно
только первоначально проделать указанные выше операции.
В дальнейшем, используя данные о концентрационной
зависимости S(C), можно определять q(S0) no q(S) при заданной
концентрации, либо построив монограмму, либо с помощью
ЭВМ, введя в нее соответствующие коэффициенты и
соотношения типа (III. 37). При однотипных экспериментах (один и тот
же полимер и растворитель) подобная операция значительно
сокращает время обработки данных.
Пример. Рассмотрим расчет ММР сополимера стирола с акрилонитри-
лом. Экспериментальные измерения проведены на ультрацеитрифуге Г-180,
снабженной оптикой Фильпота—Свенсона. Растворитель—метилэтилкетои,
исходная концентрация раствора полимера 3 мг/мл, частота вращения ротора
45 000 об/мин
На рис. III. 8 представлена зависимость h(xc) от t.
Для фиксированной координаты хс формулы расчета существенно
упрощаются, так как величины
1п (хс/хм)
= А
-ш2 = В
204
40 tfi
Рис. III. 8. Развертка седиментационных диаграмм во времени для фиксированной
координаты лгс=6,7 см.
для данного эксперимента постоянны. Таким образом:
S = A/t; qw = h (xc) Bt.
В нашем примере хк = 6,6 см, хс = 6,7 см. Тогда
1п (*с/*м)
со2-60
= 112,9-Ю-13; В = -^-ш2.60 = 92.108.
Таблица III. 4. Расчетные данные при обработке седиментографи-
ческих результатов методом фиксированной координаты
t, мин
8
10
12
14
16
18
20
22
24
S,
единицы Свед-
берга
14,1
11,3
9,4
8,1
7,1
6,3
5,6
5,1
4,7
h (xc)
2
18
40
38
35
30
18
20
13
^(SHO"
2,2
16,6
44,2
48,9
51,5
49,7
33,1
40,5
28,7
t, мин
26
28
30
32
34
36
38
40
41
S,
единицы Свед-
берга
4,3
4,0
3,8
3,5
3,3
3,1
3,0
2,8
2,7
ММ
12
10
9
8
3
3
1
1
0
Qw(S)-\0"
28,7
25,7
24,8
23,5
9,4
9,0
3,7
3,5
0
Таблица III. 5. Интегральное распределение по константам
седиментацви и ММ сополимера стирола с акрилонитрнлом
W
0,1
0,2
0,3
0,4
0,5
S{C).
единицы
Сведберга
3,7
4,6
5,5
6,2
6,9
So,
единицы
Сведберга
4,7
5,9
7,3
8,3
9,3
ЛМО-4
4,5
8,7
16,0
23,0
31,0
W
0,6
0,7
0,8
0,9
1.0
S(.C),
единицы
Сведберга
7,6
8,2
8,8
9,7
14,1
S„,
едииииы
Сведберга
10,4
11,3
12,0
13,5
20,5
М-10-4
42,0
55,0
62,0
88,0
290,0
205
Распределение по коэффициентам седиментации qw{S)
представлены в табл. III. 4. Учет концентрационных эффектов
проводится обычным образом. Нормированные интегральные
распределения по константам седиментации преобразуем в ММР
с помощью соотношения (III. 38) (табл. III. 5) и рассчитываем
средние значения ММ.
III. 3.2. Метод гель-проникающей хроматографии
III. 3.2.1. Теоретические основы метода
Физические основы этого метода Очень просты и наглядны.
Исследуемый раствор полимера протекает через колонку,
наполненную пористым сорбентом. Разделение смесей
компонентов основано на распределении определенного компонента
между подвижной (текущий растворитель) и неподвижной
(растворитель в порах) фазами. Таким образом, определение ММР
методом гель-проникающей хроматографии основано на разной
способности макромолекул проникать в поры гранул геля,
отсюда и принятое название метода — гель-проникающая
хроматография.
Рассмотрим увеличенный разрез зерна пористого сорбента
(рис. III. 9). Поверхность гранулы покрыта множеством
каналов, углублений и других неровностей, условно называемых
порами. Объем, недоступный для растворителя, называют
«мертвым» объемом, объем пор — Vn. Пусть мимо такой
поверхности протекает раствор с молекулами, размеры которых
соизмеримы с размерами пор (или меньше их). Часть таких
молекул проникает в поры, если концентрация их в
движущемся растворителе больше, чем в порах. Когда зона
растворенного вещества покидает данный участок насадки,
концентрация молекул внутри пор геля больше, чем снаружи, и
молекулы вновь диффундируют в поток подвижной фазы. Если
же размер макромолекул больше размеров пор, то такая
молекула проходит мимо гранулы геля, не задерживаясь, или
«исключается» из порового пространства *. Таким образом,
макромолекулы большего размера протекают через колонку
быстрее. Это означает, что различные макромолекулы
полидисперсного образца будут выходить из колонки в разное время при
различном объеме вымывания V:
v = v0 + Kvva,
где Vo — объем подвижной фазы (текущий растворитель); Kv— коэффициент
распределения пор по объему; для больших, полностью исключаемых из пор
макромолекул Kv = 0, для молекул растворителя Kv = 1 **.
* Часто этот метод называется эксклюэионной хроматографией
(exclude — исключить из ...). Иногда метод называют поровой, ситовой
хроматографией.
** Kv > 1 — адсорбционный режим хроматографа, когда разделение идет
не только по размерам, но и по числу функциональных групп (см. п. III. 4.3).
206
Рис. III. 9. Разрез колонки хроматографа вблизи гранулы геля: • # ©а
а — положение макромолекул разного размера до входа в колонку;
б—после выхода нз колонки.
Теория хроматографического процесса
построена на решении задач для своеобразных
диффузионных процессов. Как для любого
стохастического процесса наряду с разделением £ g
компонентов происходит и размывание зон. Кро- ф
ме того, необходимо также учитывать и
специфическое строение цепных макромолекул, а также динамику
сорбции — десорбции [193, с. 52].
Поведение макромолекулы в полимерном растворе легко
поддается детальному анализу, если определить ее энергию
Гиббса AG. Если макромолекула попадает в пору, ее энтропия
уменьшается. При наличии взаимодействия сегментов
макромолекулы со стенками поры происходит изменение энтальпии. При
притяжении энтальпия уменьшается и наоборот. Поэтому при
отсутствии адсорбции AG > 0, при сильной адсорбции
макромолекул на стенках поры AG ■< 0. Соответственно в первом
случае — эксклюзионная хроматография (хроматография по
размерам), во втором — адсорбционная. Условия, когда AG =
= 0, называются критическими.
В области AG >. 0 происходит разделение макромолекул по
размерам, т. е. по ММ в случае линейных полимеров. В
случае разветвленных полимеров процесс разделения усложняется
и зависит от типа и числа ветвлений (см. п. III.4.1), в случае
сополимеров зависит также от состава и блочности цепи (см.
п. III.4.2).
Для набухающих сорбентов описание хроматографического
процесса осложняется. Учет неравновесности
хроматографического процесса позволяет рассчитать и «размывание»
хроматографического пика (см. п. III. 3.2.4).
111.3.2.2. Жидкостные хроматографы
Жидкостные хроматографы для анализа ММР полимеров
выпускаются во всех промышленно развитых странах. В
Советском Союзе наиболее известны хроматографы серии ХЖ.
Общая блок-схема хроматографа приведена на рис. III. 10.
Блок дегазатора служит для удаления газов из растворителя и
способствует поддержанию одинакового качества растворителя
в течение продолжительного времени. Блок дозатора позволяет
в удобное время вводить пробу заданного объема и работать
в автоматическом режиме. Электрическая схема прибора
включает электронные блоки, управляющие работой насоса,
дегазатора, дозатора, рефрактометра и расходомера, потенциометра,
записывающего сигнал рефрактометра, терморегуляторов
блоков прибора. Современные хроматографы снабжены
специальными ЭВМ, позволяющими по принятым программам проводить
207
Рис. III. 10. Блок-схема хроматографа:
/—сосуд с растворителем; 2 — дегазатор; 3 — насос для
подачи элюеита; 4—демпфер; 5—край; 6 — дозатор;
7 — колонки, заполненные пористым сорбентом; 8 —
рефрактометр; 9 — расходомер.
расчет ММР и ММ, учитывать
погрешности работы прибора • (например,
дрейф «нулевой линии»), проводить
учет поправки на размывание и т. д.
Специальные микропроцессоры
управляют работой блоков прибора по
заданной программе.
Блок рефрактометра может быть
заменен другими блоками,
позволяющими записывать концентрацию протекающего через кювету
раствора. Часто используют измерение поглощения в УФ-об-
ласти спектра, проточный вискозиметр, проточный нефелометр.
Сочетание двух детекторов существенно при анализе
макромолекул сложной структуры (см. п. III.4.1 и III.4.2).
Рассмотрим схему микрогель-хроматографа ХЖ 1309.
Основой прибора является высокочувствительный рефрактометр,
позволяющий регистрировать изменения состава жидкости по
различию в показателях преломления в интервале An = 5 н- Ю-8
при сверхмалом объеме кюветы детектора 0,07 мкл.
Уникальные параметры лазерного детектора позволили использовать
микроколонки диаметром всего лишь 0,5 мм. Для
высокоскоростных анализов достаточно 10—50 нг полимера и 100 мкл
растворителя, анализ занимает всего лишь 5 мин. Особые
элементы конструкции прибора полностью исключают возможность
контакта используемого растворителя с атмосферой
лаборатории. Это позволяет использовать в качестве растворителя
пожароопасные и токсичные вещества. Удобство и быстрота
обработки информации обеспечиваются вычислительным
комплексом.
III. 3.2.3. Выбор и калибровка колонок
Важнейший блок прибора, определяющий эффективность
разделения,— колонки. Колонки могут быть разной длины (от
100 до 20 мм), выполнены в виде трубок из нержавеющей
стали, стекла или другого материала, индифферентного к
растворителю и полимеру. Диаметр колонок от 8 до 2 мм. Они
заполнены пористым сорбентом.
Наибольшее применение получили гели гидрофобных
материалов, например полистирола, сшитого дивинилбензолом. В
таких гелях практически полностью отсутствуют эффекты
адсорбции анализируемых проб. В последнее время широкое
распространение получают макропористые стекла, которые обладают
по сравнению с полимерным сорбентом как преимуществами,
так и недостатками. Жесткость частиц, размер, варьирование
208
размеров пор, химическая стабильность — несомненные
преимущества макропористых стекол; к недостаткам относится
повышенная сорбция на них полимеров. Чтобы уменьшить
сорбцию полимеров, поверхность стекол химически модифицируют
специальными веществами. В качестве экстренной меры
подавления адсорбции иногда вводят в растворитель вещества,
активно взаимодействующие с сорбентом, или применяют
соответствующий растворитель. Сорбент должен совмещаться с
анализируемым полимером, полимер должен хорошо растворяться в
растворителе. Растворитель и полимер должны иметь разные
показатели преломления (рефрактометрический детектор).
Поскольку метод ГПХ не является абсолютным, то каждую
заполненную колонку калибруют, т. е. пропускают через нее
полимерные образцы с известной ММ. На рис. III. 11 приведены
типичные кривые \gM = f(V). Видно, что при V < V0 не
происходит разделения по ММ образца. V = V0 + V„, где Vn —
объем пор. Таким образом, колонка с указанной
калибровочной кривой может быть эффективно использована только для
разделения фракций с ММ: М[ < М < М*2. В случае, если
образец полимера содержит фракции с большей или меньшей ММ,
то эти фракции выходят в области V0 (или V"), существенно
искажая форму хроматограммы вплоть до появления
«паразитных» пиков распределения (рис. III.12). Наблюдается
появление паразитного пика (или искажение формы хроматограммы) в
области малых V с характерным резким фронтом. И хотя
определение молекулярных характеристик таких частиц остается
еще нерешенной проблемой, тем не менее исследователь четко
видит долю таких образований в материале. В связи с этим,
как правило, анализ проводят на наборе колонок,
фракционирующих в ожидаемой области (см. на рис. III.11 кривую
1-\-2). Сочетая колонки, мы проигрываем в длительности
анализа или должны использовать большие скорости элюирования,
а это, в свою очередь, связано с необходимостью применения
очень высоких давлений, что вызывает определенные требования
к насосам и другому оборудованию.
В некоторых случаях используют колонки с очень малым
размером зерна сорбента, так как они обладают высокой
разрешающей способностью, требуют малого количества
растворителя и полимера. Однако многие промышленные полимеры
имеют так называемый высокомолекулярный хвост
распределения — «сшитые» частицы, сильно разветвленные
макромолекулы, глобулы. Колонки с микросферическим сорбентом
моментально забиваются этими компонентами и выходят из строя.
При фильтровании раствора такого полимера, естественно, эта
* На колонках с размером пор сорбента ~6 нм обычно делятся
макромолекулы с М = 104 -г- 105, с размером пор сорбента ~60 нм — М =
= 105Ч-10в, при М > 10е используют колонки с размером пор сорбента
>200 нм.
209
lgMa
Рис. III.11. Калибровочные кривые колонок:
/—иизкомолекуляриая; 2—высокомолекулярная; 2+1 — набор колонок.
Рис. III. 12. Хроматограмма для колонки с плохим разделением высокомолекулярной:
области (области малых V).
часть полимера теряется, а она может сказываться на
свойствах материала. В этом случае предпочтительнее применение
колонок с большим размером частиц сорбента *.
Действительно, большие частицы проходят по большим межчастичным
каналам и не забивают колонок.
Таким образом, имея стандарты образцов, можно получить
калибровочную кривую, которая обычно выражается в виде:
1gM = C,-C2V; V = c\ — c'2\gM,
где С,, С2, Cj, C2 — эмпирические коэффициенты (линейная калибровка).
В общем случае (как видно из рис. III.11) наблюдается
сложная зависимость М от V.
Полимеры различной структуры на одной и той же колонке
дают различающиеся калибровочные зависимости M(V); это же
наблюдается и при переходе от одного растворителя к другому.
Для различных линейных полимеров и для различных
растворителей можно получить единую зависимость между V и
произведением М [г\] [213]:
In M [ц] = В, - B2V.
(III. 39)
Этот факт связан с тем, что величина М [т)]
пропорциональна R3, где R — радиус инерции макромолекулы. Наличие
универсальной зависимости свидетельствует о том, что
разделение идет по размерам макромолекул.
Между коэффициентами С\, С2 и Ви В2 можно установить
соотношение:
В,=1п/Сч + (1+ал)С,; В2 = С2(\+ац), (III. 40)
где *„ и V
коэффициенты в уравнении (III. 23).
* Можно использовать и метод скоростной седиментации (методика
фиксированной координаты см. п. III. 3.1).
210
Наличие универсальной зависимости У(-М[г)]) значительно
облегчает получение калибровочной зависимости V(M) для
конкретного полимера *. Действительно, прокалибровав колонки
по доступным образцам одного полимера, легко рассчитать
калибровку для другого, зная коэффициенты Кц и ац для первого
{К-ци avl) и второго (/Сц2, а^) полимеров:
1 /Г_, 1 + а.
^Мг = Т1Г7-^1г- + Т1-г^М1, (III. 41)
где Mi и ЛЬ— ММ соответствующих полимеров.
В некоторых случаях отсутствует информация о /(, и а„
анализируемого образца или не имеется узкодисперсных
стандартов. Тогда привлекают добавочную информацию об
образцах. Так, если калибровочные образцы полидисперсные (но не
очень, т. е. Mw/M„ < 2), то максимум пика хроматограммы
соответствует ММ = л/МтМп. Когда известно ММР
полидисперсного образца, калибровку колонки проводят следующим
образом. Строят интегральную хроматограмму и сопоставляют
значения ММ и V при одинаковых интегральных долях.
Нередко встречаются случаи, когда отсутствуют какие-либо
данные о полимерных образцах. В этих случаях параллельно с
хроматографическим анализом проводят измерение каким-либо
методом интегральных параметров образцов Mn,Mw, [ц].
Значения этих параметров могут быть независимо рассчитаны с
использованием универсальной калибровки (III.39) и данных хро-
матографического анализа по соотношениям:
1
М = <+°ч \ ехр { -ррЬ- / (V) } С (V) dV,
м~1 = к^ \^p{-TTr}C{V)dV' (IIL42)
где f(V)—конкретный вид универсальной калибровки (III. 39).
С помощью ЭВМ решают систему уравнений типа 11.42, где
искомыми переменными являются коэффициенты Си С2 (или
В{, В2). Ясно, что необходимо иметь по крайней мере два
образца с известным Мп (или Мш), а также и [г\], если
известны Кг\ и а„. В общем случае число образцов должно быть
больше, а также необходимо учесть возможность и
нелинейность калибровки.
* Хотя и отмечается наличие универсальной калибровочной зависимости,
в некоторых случаях она не выполняется. Поэтому при сопоставлении ММ,
получаемых в разных лабораториях, необходимо обязательно получать
истинную калибровку.
211
Кстати, точно так же можно найти и значения Кц и а„ при
знании калибровочной зависимости. Если исследователь
располагает достаточно большим количеством образцов (более 10),
то, используя вышеперечисленные подходы, можно определить
как значения /С,,, а„, так и параметры нелинейной калибровки
хроматографа.
Интересный прием предложен [203] для работы с
неизвестными полимерами. Измеряя вязкость разбавленных растворов
[(см. п. III.2.3.2) и уравнения (111.23) и (111.24)], можно по
величине Кх определить av и /(„. Тогда уже с использованием
универсальной калибровки III.39 можно рассчитать конкретную
калибровку III.38 для данной системы полимер — растворитель.
Знание распределения по элюационному объему C(V) и
калибровочной кривой V(M) позволяет уже получить
интегральное распределение по ММ:
v м
W(M) = W(V) = \. С (V) dV= [qw (M) dM. (III. 43>
Таким образом, переход от интегрального распределения по
V к интегральному распределению по М очень прост: он
сводится к замене оси абсцисс V на ось IgM по соотношению III.38.
Для qw справедливо выражение
и в случае линейной калибровки:
qw = С (V) 0,4343/(С2М). (III. 44)
Обычно в низкомолекулярной области часто появляются
побочные пики: пики стабилизатора, «растворителя» и т. д.
Поэтому при расчете ММР вводится нижняя граница
распределения М.
Существенным при калибровке является учет
концентрационной зависимости V, особенно для высокомолекулярных
стандартов. Необходимо помнить, что стандартные образцы
узкодисперсные, выходят они в более узком интервале элюационных
объемов, поэтому концентрационная зависимость V пика для
них является более сильной, чем для полидисперсных
образцов. При очень больших концентрациях происходит перегрузка
колонок, что может привести даже к искажению формы хрома-
тограмм.
III. 3.2.4. Погрешности и оценка эффективности
хроматографического процесса
Как мы уже указывали, неравномерность и стохастичность
процесса приводят к размыванию хроматографического пика.
В связи с этим существенно оценить эффективность прибора,
его разделяющую способность. Отличительные черты хромато-
212
графического процесса тесно связаны с такими
характеристиками как эффективность N, разрешение kk = AV/o,
производительность N/t, время анализа t, чувствительность 6.
Эффективность анализа обычно характеризуется числом теоретических
тарелок N или высотой теоретической тарелки Н:
N = V2/<j2v; N=a2L/Lt
где ov, ol — дисперсии хроматографического пика.
При проведении анализа необходимо также учитывать и
экстраколоночное размывание (вне колонки). Величину аэ
можно определить экспериментально как дисперсию размывания
при введении пробы в хроматограф, минуя колонку. Таким
образом, величина а является одной из важнейших
характеристик процесса.
Под чувствительностью понимают отношение сигнала
прибора к количеству q анализируемой пробы. Сигнал
пропорционален концентрации в центре хроматограммы:
С = q = q ^
<splu,ov фл V
Таким образом, повышение эффективности приводит к росту
чувствительности прибора. Хроматографический процесс
является оптимальным по чувствительности анализа, если требуемая
эффективность N достигается на колонке, имеющей
минимальную длину L (минимальное V).
Специфика размывания хроматографической зоны в
хроматографии полимеров определяется малой диффузионной
подвижность макромолекул. Обычно диффузионное размывание в
каналах подвижной фазы, а также вклад профиля скорости
можно не учитывать. Основной вклад в величину Н дает
неравновесный массообмен. Тогда высота эффективной теоретической
тарелки равна:
А_ 2nkvvn г2
L * Wo 15 '
где D — коэффициент диффузии в неподвижной фазе; г — радиус поры
сорбента.
Поскольку D и kv уменьшаются с ростом ММ, то Н
экстремально зависит от V (или ММ). Этот факт получил как
экспериментальное, так и теоретическое подтверждение.
Оптимальным видом калибровочной зависимости является
линейная зависимость:
V = С,- c'2\gM.
Для оценки эффективности высококачественной
хроматографической системы удобно воспользоваться [214] значениями
ММ, скорректированных (ЛЬ(О)) и нескорректированных
213
{Mk(a)) на размывание:
( a2, In2 ю 1
Мк (а) = Мк (0) exp j (3 - Ik) j±- J , (III. 45)
где Mk — k-я MM.
Задаваясь погрешностью в определении ММ примерно 5%.
находим C'Ja^^l,3. Учитывая, что N = V2ja\, получаем
AT>5,0V2/(Q2.
Очевидно, что с шириной интервала разделяемых ММ
{М\ < М < Мг) тесно связано отношение C'JC'r Так, при
2 ^ lgM < 6, C\jC2 -10 и JV должно быть более 103.
Для визуального разделения двух- пиков с одинаковыми
амплитудами kk = kV/a должно быть примерно 0,5 и для М{ х
т 104 и М-2 ~ Ю5 требуется эффективность N^1400, а для
Mi «105иЛ}2«2-105#= 10\
Погрешность, допускаемую при определении ММ методом
ГПХ, можно оценить следующим образом. Во-первых, в нее
вносят вклад погрешности абсолютных методов, используемых
для характеристики стандартов, во-вторых, погрешности самого
метода ГПХ, связанные с эффективностью системы,
воспроизводимостью результатов.
Обычно точность (A7W/M) определения ММ стандартных
образцов не превышает 5%. Она может быть уменьшена в
2—3 раза в условиях линейной калибровки и при большом
числе образцов стандартов.
При высоком уровне работы погрешность на
воспроизводимость хроматографических данных может составлять 0,5 % от
объема удерживания. В табл. III.6 приведены [215] данные о
положении пика стандартного образца полиэтилена, полученные
при высокой температуре (130 °С) в течение года на колонках,
наполненных макропористым стеклом на приборе Вотерс 200.
Основная погрешность в определении ММ и
полидисперсности не очень полидисперсных образцов связана с размыванием
соотношением:
ДМ/М = а/С2.
Ориентировочная оценка погрешностей свидетельствует, что
АМ/М может составить от 5 до 20 %.
В зависимости от задач, стоящих перед исследователем,
можно рассматривать несколько уровней имтрепретации
данных. Для сопоставительного анализа промышленных образцов
достаточно сравнить хроматограммы, рассчитать М„, Мш и
полидисперсность образца, не проводя коррекции на приборное
уширение.
При установлении корреляции между физико-химическими и
механическими свойствами и его ММ требуется более точный
расчет ММ: учет приборного уширения по простейшим
соотношениям (III. 45).
214
Таблица III.6. Данные по долговременной воспроизводимости
начала (V0), максимума (VMaKC) и конца (VK) хроматограмм
образца ПЭВП NBS 1475
Порядковый номер
анализа
V0 (счет)
^макс <счет>
V (счет)
1267—02.1979
1291
1305
1346
1471
1645
1646
1648
1706
1711
2007
2008
2010
2011
2043
2078
2083—09.1980
15,0
16,0
15,9
16,3
15,9
14,9
15,4
15,0
15,3
15,4
15,0
15,8
15,3
15,4
15,0
15,3
15,7
19,7
19,7
20,0
19,7
19,9
19,9
19,3
20,0
19,7
19,9
20,0
19,9
19,6
19,8
19,8
20,0
19,8
25,0
24,3
24,6
24,7
23,9
25,9
24,8
25,7
24,7
24,4
24,3
24,8
24,9
24,7
25,3
25,3
25,3
Средние значения:
15,4±0,3
19,8±0,1
24,8±0,4
Обычно коррекция на приборное размывание существенна
при исследовании сравнительно узких фракций (или образцов)
полимеров (Mw/My < 1,5), а также для уточнения наличия
мультимодальности ММР. При N > 2000, как правило,
поправку на размывание пика уже не проводят. В случае достаточно
узких ММР можно проводить поправку по соотношениям
(III.45). В прецизионных случаях учитывается поправка на
размывание с помощью специальных программ для ЭВМ.
Коррекция на приборное уширение при количественных
расчетах ММ и ММР для не очень полидисперсных образцов
является важной операцией, которой должен владеть любой
исследователь. Пусть имеется функция G(V, M), характеризующая
размывание хроматографического пика. Тогда эксперименталь
ная хроматограмма будет равна [193, с. 40]:
ль
F(V)= [ q (M) G (V, Af) dM, (III. 46>
Mi
где q(M) —искомое ММР.
Задача сводится к решению уравнения (III.46), т. е. к
нахождению q(M) по экспериментально заданной F(V).
Подобный класс задач примыкает к классу «некорректно»
поставленных задач, так как любая экспериментальная погрешность
может привести в общем-то к неверному решению.
Стандартные методы решения таких уравнений приводят к
появлению «осцилляции», паразитных пиков и т. д.
215
В простейших случаях можно считать, что G(V, M)
задается гауссовой функцией с дисперсией сг. Тогда отношение
скорректированных и нескорректированных Мк(а) ММ ^-усреднения
дается уравнением (111.45) [214].
Определив а с использованием стандартных образцов,
можно рассчитать и «скорректированные» на уширение значения
ММ изучаемого образца.
Развиты многочисленные приближенные методы решения
уравнения (111.45). Все они в той или иной степени приводят
к появлению осцилляции на концах распределения. В работе
[216] был предложен хотя и трудоемкий аппроксимационный
метод решения уравнения (111.45), свободный от этих
недостатков. Он сводится к тому, что как экспериментальную хромато-
грамму, так и экспериментально определенную функцию
размывания a(V,M) описывают с помощью функции Р(х):
f G\ exp {G'2x}
Р{*) = \ Рп (*)
{ G'3 exp {G'4x}
где х — переменная; Р„ — полином степени п.
Коэффициенты полинома, а также G[ находят из
согласования с экспериментальными данными. Такой вид функции
позволяет проводить вычисления с любой точностью и любым
количеством итераций, так как возникающие на концах интервала
осцилляции подавляются выбранным видом функции. Даже в
условиях большого размывания предложенная методика
позволяет получить адекватные истинному распределению данные.
Число итераций достигает 100—200, тогда как в обычных
программах уже при 3—5 итерациях начинаются осцилляции.
Таким образом, проведя необходимую калибровку
хроматографа, разработав программы расчета данных хроматографи-
ческого эксперимента, можно приступить к серийному анализу
ММР полимерных образцов. К сожалению, здесь
экспериментатора поджидает несколько неожиданных трудностей. Основная
трудность — выбор колонок для хроматографирования. Стиро-
гелевые колонки пригодны для обычных органических
растворителей. При использовании специфически растворителей
(ДМФА, ДМАА) срок службы колонок резко сокращается.
Особенно быстро стирогелевые колонки изменяют свои
свойства при повышенных температурах. Изменение свойств насадок
происходит постепенно. Поэтому исследователь должен
регулярно проверять правильность работы колонок, проводить
калибровку, оценивать эффективность колонок.
Более устойчивы насадки с макропористым стеклом. Однако
активные центры на поверхности стекла обладают повышенной
адсорбцией макромолекул, что приводит к искажению формы
хроматограммы.
216
Растворы полимеров для анализа готовят обычным
способом: после растворения полимера раствор фильтруют,
желательно фильтрование (или какая-либо очистка) растворителя.
Часто, особенно при анализе при высоких температурах,
необходимо в раствор добавлять стабилизатор. Все эти особенности
анализа являются специфическими для той или иной системы
полимер — растворитель — насадка колонок. Укажем некоторые
работы, где подробно описаны методики: для полиэтилена
[217], сополимеров винилиденфторида [218], блоксополиме-
ров [219].
Рассмотрим пример определения ММР для ПЭВД.
Характерной особенностью высокотемпературной ГПХ полиолефинов
является необходимость подавления окислительных процессов,,
быстро развивающихся при повышенных температурах и
приводящих к изменению ММР полимеров, для подавления
термоокислительной деструкции из стабилизаторов наиболее широко
применяется ионол (2,6-ди-7,рег-бутил-4-метилдиенол). В случае
приготовления растворов полиолефинов в о-дихлорбензоле с
использованием ионола эффект стабилизации сохраняется не
менее 3 сут, причем изменение концентрации стабилизатора от
0,1 до 4 % не влияет на результат измерений.
Концентрация исследуемых растворов полиолефинов
составляет 0,1—0,3 % в зависимости от молекулярной массы и ММР
полимера. В случае гранулированного полимера для учета
возможной неоднородности гранул по свойствам раствор готовят
не менее, чем из 10 гранул. Растворение проводят в
ультратермостате при 135—150°С. В процессе растворения,
длящегося в зависимости от вида полимера от 30 мин до нескольких
часов, раствор необходимо несколько раз взбалтывать для
интенсификации перемешивания. Готовый раствор фильтруют на
высокотемпературном фильтре при 140—150°С, продавливая
его аргоном (для предупреждения окисления) через
асбестовый фильтр с эффективным диаметром пор около 15 мкм.
При расчете ММР полимера по гель-хроматограмме прежде-
всего необходимо прокоординировать экспериментальную
кривую. Для этого проводят нулевую линию, на ней определяют
начало и конец хроматограммы, интервал между ними делят
на 25—40 равных промежутков AV и в каждой точке деления
Vi определяют высоту хроматограммы А,-. Затем для каждого
из выделенных удерживаемых объемов V; на основании
заранее известной калибровочной зависимости рассчитывают ММ
соответствующего полимер-гомолога Mi и его долю ш( во всем
полимере [соотношения (III. 39), (III. 43), (11.44)]. Далее
рассчитывают значения средних молекулярных масс: Мп, Мш, Мг,
Mv и т. д., а также характеристическую вязкость образца [г]]
ГПХ. Если макромолекулы исследуемого полимера линейные,
то при правильно построенной калибровочной зависимости
[т|] гпх= [г|]э и рассчитанное значение ММР является
истинным. Если макромолекулы имеют длинноцепные разветвления,
217
Таблица Ш.7. Экспериментальные (V, hi) и расчетные
(wi, Mi) значения ММР для растворов ПЭВД в о-дихлор-
бензоле
V
hi
Wj
мгю~3
линейный
полимер
разветвленный
полимер
38,8982
38,5649
38,2316
37,8983
37,5650
37,2317
.36,8984
36,5651
36,2318
35,8985
35,5652
35,2319
34,8986
34,5653
34,2320
33,8987
35,5654
33,2351
32,8988
32,5655
32,2322
31,8989
31,5656
31,2323
30,8990
30,5657
30,2324
29,8991
29,5658
29,2325
28,8992
28,5659
28,2327
27,8994
27,5661
27,2328
26,8995
26,5662
26,2329
25,8996
25,5663
25,2330
24,8997
24,5664
24,2331
23,8998
0
2,43
14,08
29,57
41,27
49,93
52,48
49,15
45,71
40,50
35,69
31,41
25,59
22,38
21,31
18,39
17,05
15,74
15,27
14,39
13,94
14,10
14,45
13,81
12,97
11,8633
11,8045
10,70
9,9
9,8
9,0
9,5
8,8
8,0
5,6
4,8
4,4
3,4
1,1
3,1
1,9
2,0
0,9
0,6
0,01
0
0
0,010
0,058
0,122
0,170
0,205
0,216
0,202
0,188
0,167
0,147
0,129
0,105
0,092
0,088
0,076
0,070
0,0648
0,0628
0,0592
0,0573
0,058
0,0595
0,0568
0,0534
0,0534
0,0488
0,0486
0,0440
0,0407
0,0403
0,0370
0,0389
0,0364
0,0328
0,0232
0,0198
0,0183
0,0138
0,0044
0,0124
0,0080
0,0083
0,0038
0,0026
0
0,353
0,5-20
0,754
1,076
1,511
2,090
2,850
3,832
5,083
6,653
8,600
11,000
13,900
17,300
21,3
26,1
31,6
37,8
44,9
52,9
61,8
71,7
82,5
94,3
107,2
121,0
136,0
152,0
169,0
188,0
207,0
228,0
251,0
274,0
300,0
327,0
357,0
389,0
424,0
462,0
503,0
549,0
600,0
656,0
720,0
792,0
0,352
0,518
0,752
1,07
1,50
2,09
2,86
3,8
5,1
6,7
8,7
П.2
14,2
17,8
22,1
27,2
33,2
40,1
48,0
57,0
67,2
78,6
91,2
105,1
120,4
137,1
155,2
174,9
196,0
218,9
243,4
270,0
298,4
329,2
362,5
399,0
438,0
481,0
528,0
581,0
639,0
704,5
778,2
862,0
958,0
1068,5
218
то [tilmx > [ilia и расчет ММР полимера следует вести по
специальной программе.
Пример. В приведенных в табл. III.7 данных AV = 0,333 счета,
калибровка прибора:
lgM = 21,9759 — 1,58782V+ 5,20663- \0~2V — 5,08213- 10~3V3
Расчет по калибровке для линейного полиэтилена:
М„ = 4,47- 10э; Мш = 54,7-103; М„ = 38,Ы03; Mz = 221,6 • 10э;
h]rnx = 0-73; Mw/Mn= \2,2; MzjMw=W
Расчет по калибровке для разветвленного полиэтилена:
М„ = 4,49-Ш3; Mw = 63,7-10Э; М„ = 43,2 • 103; Мг = 280,6 - 103;
[т,]э = 0,64; Мш1Мп = 14,2; Mz/Mw = 4,41; т. = ХМ; % = 0,63 • 10~5.
Таким образом, учет разветвленности макромолекул практически не
меняет М„, увеличивает Мш и М2.
III. 3.3. Особенности интерпретации данных о ММР в терминах
констант элементарных реакций
Основной задачей экспериментального исследования
является либо установление взаимосвязи между молекулярными
характеристиками и свойствами, либо определение вида
кинетической схемы полимеризации и вычисления констант
элементарных реакций. Если первая задача не представляет особых
трудностей, то вторая — требует определенных приемов и
методик. Рассмотрим некоторые из них. При этом сначала
рассмотрим применение этих методик для анализа продуктов
радикальной полимеризации, а далее — ионно-координационной
полимеризации. Необходимо отметить, что такое деление методик
условно и связано в излагаемом ниже материале только с
использованием конкретных соотношений.
III.3.3.I. Градиентный метод
Градиентный метод, или метод «производных» моментов
[220], основан на решении системы алгебраических уравнений,
следующих из классической схемы радикальной полимеризации.
Для случая рекомбинации радикалов: *
(1 -*)'-" dQn С+ 0,5 (я + \)А
М0я! dx (C+A)n
л = 0, 1, ... (III. 47)
где
А (*) = Д*^]о V2/*pac [I] + mkT (1 - х)т ; (III. 48)
Qo = x/*n> Qi = x> Q2 = xPw; Q3 = xPwPz;
CS = *S/V CI=W Cm = *m/V
* Вычисления можно упростить, введя переменные С + А = у и А.
219
где ftp, ks, h\, ku —константы роста и передачи на 5-агент, инициатор,
мономер соответственно; kp2C — константа распада инициатора; kT — константа
термического инициирования; Р„, Pw, Рг — среднечнсленная, ореднемассовая,
г-средняя степени полимеризации соответственно; Мо — молекулярная масса
мономера; / — эффективность инициирования; х— конверсия; т —
эмпирический множитель.
Для случая диспропорционирования радикалов система
принимает вид:
U-x?-"dQn_ С + 0,5„Л „ = 0(1>2... (ш.49)
М0п\ dx (C+ А)п '
Решить систему уравнений (III. 47), (III. 49) относительно С
и А можно только при определенном выборе условий
эксперимента (отсутствие передачи цепи на полимер или ее
независимое определение). Важно отметить, что в уравнение входят две
переменные Л и С, в то время как уравнений (число моментов
ММР) может быть больше. Однако в правых частях стоят
экспериментальные величины, определенные с погрешностью.
Кроме того, некоторые моменты вычисляются по «хвостам» ММР,
т. е. определяются с большей погрешностью. Поэтому
необходима проверка получаемых решений с привлечением не двух,
а большего числа моментов.
Для случая разветвленной радикальной полимеризации,
протекающей путем передачи цепи на полимер за счет отрыва
атома водорода (ее скорость равна W), получено следующее
уравнение [221]:
1 dQ, 1 + wPn Г 1 + шРщ ]
M0(l-x) dx C + A + w\_C + A + wy
Аналогично могут быть получены соотношения и для случая
присоединения макрорадикалов к ненасыщенным группам и к
концевым С=С-группам полимера.
Решение системы дифференциальных уравнений вида
(III.47), (III.49) содержит ряд характерных методических
особенностей. Во-первых, решением системы являются не искомые
значения констант скоростей, а значения А, С, ш, связанные с
таковыми соотношениями (III. 48). Тогда искомые отношения
констант скоростей определяют путем анализа зависимостей А
и С от концентраций реагентов в системе. Так, путем анализа
зависимости Л от концентрации инициатора в степени 0,5 при
заданной конверсии можно определить значение отношения
kp[ л/2/ftpac^o' а ПРИ анализе зависимости С от [S]/[M] или
[1]/[М] при постоянном х—значения Cs или Ст. Кроме того,
разделение отношения kv' -\f2fkpack0 на йр' V^o" и 2fkp!1c может
"р
быть выполнено с использованием данных о скорости процесса
v при данном х по следующим формулам:
vA=2fkpac[I]; v/A=k0[M]/k2p.
220
Во-вторых, при решении системы уравнений (III. 47) —
(III. 49) используются значения производных моментов ММР,
определяемых путем дифференцирования экспериментальных
конверсионных (временных) зависимостей Q по х. В связи с
этим такая задача относится к классу так называемых
некорректно поставленных задач.
Для повышения точности расчета производных моментов
разработан метод, позволяющий повышать достоверность их
значений. Сущность этого метода состоит в том, что для
определения производных моментов используется сглаженная
экспериментальная зависимость моментов от х. При этом
значения производных моментов, рассчитываемых по этой
зависимости, характеризуют скорость изменения моментов ММР с х в
некоторых постоянных инкрементах приращения Лх
Окончательно искомую зависимость производных моментов от х
находят путем усреднения сглаживания приводимой через
соответствующие значения производных моментов при постоянном
приращении по х. Также к повышению достоверности решения
таких задач приводит использование дополнительной
информации о искомых функциях: они являются гладкими и, более того,
имеется некоторая количественная информация (см. методы
«разностных» хроматограмм и аппроксимационный, а также
значения констант в области малых конверсии). В табл. III. 8
приведены примеры результатов расчетов.
III. 3.3.2. Метод разностных хроматограмм
Метод разностных хроматограмм [222] представляет собой
прямой экспериментальный метод определения производных
моментов ММР полимера по х или по времени полимеризации
путем вычитания гель-хроматограммы полимера, образующегося
к моменту времени M*i), из гель-хроматограммы полимера,
образующегося к последующему моменту времени t2{x2). При
этом, если площади под этими гель-хроматограммами отнорми-
рованы так, что их отношение равно отношению массовых
выходов полимера к этим моментам времени (х\ и х2), то
результат представляет собой гель-хроматограмму полимера,
образовавшегося в интервале времени t2—t\. Если этот интервал мал
и изменение параметров полимеризационной системы за этот
период времени незначительно, то «разностная» гель-хромато-
грамма описывает «мгновенное» ММР и может быть
использована для определения отношений констант скоростей
полимеризации при t[ < t < t2. Расчет проводится по уравнению (III. 47),
что в силу аддитивности моментов ММР полимерного образца
по отношению к ММР его составных частей соответствует п-у
моменту ММР фракции полимера, образовавшегося за
промежуток времени t2— ^1 (см. табл. III. 8).
Эту методику можно использовать при исследовании не
только линейной радикальной, но и ионной полимеризации.
221
Таблица III.8. Результаты расчетов констант определяющих реакций
при полимеризации стирола
Метод
Градиентный [220]
Разностных
хроматограмм [222]
Аппроксимационнын
[2231
* Данные при х->0 в
X
0,1
0,4
0,5
0,6
0,7
0*
0,1
0,3
0,5
0,6
0,1
0,2
0*
ЗПТЫ ИЗ
V*7
kp
29,4
13,4
10,9
10,6
3,8
26±5
29
15
10
4
8,3
8
fcT-104 с"1
3,7
3,6
4,7
8,6
8,3
8±5
—
—
—
—
—
—
8,3±0,4 —
работы (2241.
См-ю4
1,6
1,25
1,24
1,25
1,35
1,6±0,1
—
—
—
—
—
—
с1
0,15
0,16
0,12
0,11
0,11
0,125 ±0,05
0,13
0,13
0,12
0,11
—
—
cs
—
—
—
—
—
—
—
—
19'
17
15
Таким образом, метод «разностных» хроматограмм дает
альтернативные возможности использовать экспериментальные
данные о ММР для расчета производных моментов путем
дифференцирования конверсионных зависимостей Q по х или
путем вычитания хроматограмм.
Предпочтение той или иной методики определяется
качеством накопленного экспериментального материала. Здесь
имеется в виду прежде всего размер инкрементов по х и
погрешности хроматографического анализа.
III.3.3.3. Степень достоверности решения обратной задачи
кинетики радикальной полимеризации виниловых мономеров
При решении обратной задачи существуют значительные
трудности в связи с погрешностью используемых данных ММР.
Влияние погрешностей экспериментальных данных на точность
определения А, С и до из уравнений (III. 44), (III. 46) изучалось
в [220, 221] с помощью численного эксперимента. Для этого
были заданы возможные отклонения от параметров ММР и их
производных, моделирующие экспериментальные погрешности.
Установлено, что случайные погрешности в определении
параметров ММР вызывают соизмеримые относительные
погрешности в рассчитываемых константах. Однако систематические
экспериментальные погрешности в определении Рп и Pw, особенно
если одна из них завышена, а другая занижена, на начальных
участках полимеризации приводят к погрешостям в значениях
С и А, значительно превосходящим экспериментальные [221].
С целью повышения точности расчета С и А были
разработаны критерии, позволяющие корректировать менее достоверные
значения этих величин в начале полимеризации по их более
222
достоверным значениям в конце процесса. Применение этих
критериев при исследовании изотермической полимеризации
позволяет значительно увеличить достоверность рассчитываемых
параметров.
В [225] рассмотрена проблема достоверности решения
обратной задачи с использованием данных ММР в более общем
виде. Показано, что источником погрешности искомых
параметров являются не только погрешности используемых
экспериментальных данных (как это предполагали в [220, 221] при
численном эксперименте), но и погрешности результатов анализа
экспериментальных данных (вторичные экспериментальные
результаты), являющиеся результатом обработки их в
соответствии с теоретическими функциональными соотношениями.
Ниже сопоставлены значения коэффициентов вариации v *
искомых параметров, определенных с использованием развитых
методов и прямых методов химической кинетики (ЭПР,
вращающийся сектор):
v. %
^о'5Ар 10—90 Кинетический метод
10—25 Градиентный метод
Cs = ksjkp 45 Метод ММР
10—100 Градиентный метод
При расчете использованы методы статистического и
регрессионного анализа экспериментальных данных.
Приведенные данные показывают, что значения v для kj\/k0
я Cs в достаточной степени велики, однако все другие
используемые методы определения этих величин, в том числе и
прямые, характеризуются также высокими значениями
коэффициентов вариации и даже в некоторых случаях еще более
высокими [например, методы ЭПР (~100%), вращающегося
сектора (~40%), кинетический, если конверсию определяли
гравиметрическим методом].
Основной рекомендацией, следующей из проведенного
анализа, является правильная программа исследований (переменный
шаг по конверсии, времени, вариация начальных условий) и
методика обработки данных (использование специальных
процедур «сглаживания», шаг дифференцирования и т. д.).
III. 3.3.4. Определение констант скоростей основных химических
реакций процесса ионно-координационной полимеризации '
Среднечисленная степень полимеризации Рп в любой момент
процесса полимеризации определяется как отношение числа
* Вариация v определялась по следующим соотношениям:
S Та, Т (а, - а)2
где <а> — среднее арифметическое значение отдельных экспериментальных
значений а\, й2, ... из выборки объема т.
223
заполимеризовавшихся мономерных звеньев к суммарному числу
растущих и «мертвых» макромолекул:
[ wdt
<Я„) = Ц= , (III. 50)
я (0 + \ w0 dt
где па — скорость полимеризации; wo — скорость (суммарная) обрыва и
переноса цепи на различных агентах; < —продолжительность полимеризации:;
n(t) — суммарное число активных центров в момент времени t.
В общем случае справедливы следующие соотношения:
w = Z kPini tMi= wo = Z Z coikoiini> «(o = Z ni>
где л,- — концентрация активных центров 1-го типа; kvi — константа скорости
реакции роста макроцепи для »-го типа активных центров; [М] —
концентрация мономера в реакционной среде; Аол— константа скорости реакции обрыва
(или переноса цепи) j-m агентом обрыва иа /-м активном центре; Со,- —
концентрация /-го агента переноса цепи.
Наиболее простой вид соотношение (III. 50) принимает в
случае идеализированного процесса полимеризации, когда
константы скоростей реакций роста и обрыва цепи не зависят от
продолжительности полимеризации, концентрации реагентов
поддерживаются постоянными, а активные центры
каталитической системы представлены одним типом и их концентрация
не меняется во времени:
1 _ 1 ZCoi koi
Т7 ~ kp [M] t + kp [M] '
Построив график зависимости l/Pn = f(t~l), по его наклону
можно определить абсолютное значение константы скорости
роста макроцепи kp, а по отрезку, отсекаемому на оси
ординат,— отношение скоростей реакций обрыва и роста макроцепи.
Если продолжительность процесса полимеризации велика
настолько, что первым членом в правой части соотношения
(III.. 50) можно пренебречь, тогда среднечисленная степень
полимеризации получаемого полимера практически зависит лишь
от соотношения скоростей реакций обрыва и роста
макроцепи: *
1 _*" ^ос[АОС]-М0н[Н2]-Мс0"
Рп kp + АР[М]
где k0 , k0 ,k0, kg" — соответственно константы скоростей реакции
передачи цепи на мономер, алюминийорганическое соединение (АОС), водород, а
также спонтанного обрыва цепи; [АОС], [Нг]—концентрации АОС и
водорода.
* Здесь приводятся конкретные соотношения для полимеризации этилена.
224
Проведя серию опытов при различных концентрациях одного
из агентов передачи цепи, можно определить относительную
константу koilk? скорости реакции передачи цепи для этого
агента. Например, при варьировании концентрации водорода из
графика зависимости \/Рп = f([H2)) относительная константа
скорости реакции передачи цепи на водород определяется по
его наклону, а отрезок, отсекаемый прямой на оси ординат,
определяет отношение суммы скоростей передачи цепи на
мономер, на АОС и спонтанного обрыва цепи к скорости роста
макроцепи.
Если в•каталитической системе содержится два или более
типов активных центров, причем концентрация каждого типа
не зависит от времени, то соотношение (III. 50) принимает вид:
Рп [М] £ kpin, t [M] £ V/
В этом случае, построив график зависимости 1/Р„ =
= f(t~l) из его наклона можно определить «среднечисленную»
константу скорости реакции роста макроцепи:
(*P)„=£Vi/5>r
Если каталитическая система содержит несколько типов
активных центров с разными константами скоростей реакций
роста и обрыва цепи, причем концентрации активных центров
разных типов меняются во времени, выражение для среднечис-
ленной степени полимеризации получаемого полимера в общем
случае упрощению не подлежит и рассчитать какие-либо
кинетические параметры по среднечисленной степени полимеризации
не представляется возможным. Необходимо привлекать
дополнительную информацию о Pw, W и т. д.
Для полицентровых каталитических систем анализ
особенностей кинетики процесса полимеризации можно провести на
основании данных о ММР синтезируемого полимера. Известно,
что для полиолефинов, получаемых по анионно-координацион-
ному механизму, ММР синтезируемого полимера определяется
выражением [226]:
<//*> = С ф (а) а еХр (- аМ) da, (III. 51)
где а — вероятность обрыва растущей макроцепи, при скорости роста
полимерной цепи w и суммарной скорости обрыва цепи w0a = w<,/(M0w); Ma —
молекулярная масса мономера; <р(а)—функция распределения активных
центров по вероятности обрыва на них растущей макроцепи; величина g>(os)da
характеризует численную долю активных центров с вероятностью обрыва на
них растущей цепи от а до a + da.
Методика расчета распределения ф(а) из данных о ММР
полимера без дополнительных предположений о характере по-
лицентровости катализатора приведена в работе [226].
8 Зак. 632
225
fi (V) a W*> 6
Рис. III. 13. Хроматограмма (а) и функция распределения по вероятности обрыва (б) для
образца полиэтилена.
В ряде случаев на графиках (рис. III. 13) рассчитанных
распределений проявляются два или более субмаксимумов, что
указывает на присутствие в системе соответствующего числа
заметно различающихся по среднему значению щ типов
активных центров. Поскольку для каждого типа активных центров
в условиях стационарной полимеризации вероятность обрыва
макроцепи определяется выражением, аналогичным
- _ 2i Colkoll
1 VM] '
по изменению а» каждого из субмаксимумов распределения tp(a)
при варьировании концентрации одного из агентов переноса
цепи можно определить соответствующие относительные
константы скоростей реакций передачи цепи на этот агент для
каждого типа активных центров. В табл. III. 9 приведены константы
скоростей химических реакций полимеризации этилена.
В области больших конверсии при условии постоянства
констант скоростей удобно анализировать следующие соотношения
(один тип активного центра):
•^ ™п —knx
-я- = п + -г- [Щх + ах; к=\~е Р ,
Рп «р
где а — коэффициент, характеризующий передачу на какой-либо агент.
Таблица III. 9. Константы химических реакций полимеризации
этилена [215]
Способ определения
0 / Р
к»/к
0 / р
По Мп 3,2-10~4 2,6 -1СГ2
По ф(а) (АЦ 1) 6,3-10~5 1,2-10~3
Поф(а)(АЦ2) 5,9- \0~* 1,4-10 2
226
Построив зависимости х/Рп от х (или времени), из отрезков
и наклонов можно определить основные константы скоростей
химических реакций.
II 1.4. ПРИМЕНЕНИЕ МЕТОДА ГПХ К ОПРЕДЕЛЕНИЮ
ОСОБЕННОСТЕЙ СТРУКТУРЫ МАКРОМОЛЕКУЛ
Выше были описаны методики применения ГПХ к
исследованию ММР линейных макромолекул гомополимеров. Однако
большинство полимеров имеет более сложное строение.
Применение ГПХ к анализу таких полимеров имеет ряд особенностей
и связано в основном с тем, что используется дополнительная
экспериментальная информация. Это может быть либо
интегральная величина какой-либо характеристики полимера {[ц],
Мп, Mw), либо значения характеристик по «фракциям»
(использование проточных вискозиметров, нефелометров, скоростной
седиментации и т. д.).
III.4.1. Определение параметров длинноцепной разветвленности
макромолекул
Многие полимеры — каучуки, полиолефины, фторопласты —
имеют разветвленные макромолекулы, т. е. макромолекулы,
цепи которых имеют более двух концевых элементов. Здесь
нас будут интересовать разветвленные цепи, длина ветвей
которых сопоставима с длиной всей макромолекулы—длинноцеп-
ная разветвленность (ДЦР). При этом макромолекула* имеет
случайно расположенные узлы ветвления, длины ветвей также
распределены случайно. Такие цепи обладают статистически
или хаотически разветвленными структурами. Модели
статистически разветвленных структур рассматривались уже давно
[192, с. 272]. Однако в работе [227] было показано, что эти
модели не отражают некоторые черты, характерные для
реальных разветвлений цепей. Новый класс моделей имеет общее
название — хаотически разветвленные цепи, в них отражена и
специфика роста разветвлений макромолекулы.
Основной характеристикой разветвленной цепи является
число узлов т, функциональность узла f, общая ММ. Для числа
звеньев п в разветвленной макромолекуле имеем:
n = m(f-\)P'n + p'n,
где Рп — среднечисленная степень полимеризации ветви.
Обычно сопоставляют размеры макромолекулы
разветвленной /?р и линейной /?л при одинаковой ММ и статистической
жесткости А:
&=# •
. кл м, А
* Мы не будем рассматривать звездо- и гребнеобразные макромолекулы.
8*
227
Для монодисперсных статистически разветвленных цепей
(трифункциональный узел ветвления) имеем:
1
[4т/(9я) + (1 + т/7)
'/а 1 'Л"
Для хаотически разветвленных структур важно учесть
также очередность и условия роста ветвей. Если считать, что эти
условия постоянны, т. е. длина растущих ветвей всегда
одинакова *, то
т + \
5 3(т+2) у> 1 Чтг _ 3 In от
ё~ т+\ + (m+1)2 Zj k (от+1)3 * т '
1
Видно, что одно и то же значение т приводит к различным
значениям g. Поэтому определяемое по той или иной методике
число узлов ветвления является эффективной величиной,
позволяющей количественно сравнивать полимеры, полученные по
одной и той же технологии, и только качественно — по разной
технологии.
Наиболее принятыми являются методики определения т по
зависимости [г\] (или D, или S) от М [192, с. 283], а также
сочетающие ГПХ и скоростную седиментацию, ГПХ и
вискозиметрию.
III. 4.1.1. Сочетание ГПХ и вискозиметрии
Обычно предполагается, что существует универсальная
калибровочная зависимость V = f] (M [ц]), которая не связана
со степенью разветвленное™ макромолекул. Если хроматограф
оснащен проточным вискозиметром [193, с. 230], то
экспериментатор измеряет две зависимости: C(V) от V и [т)] от V.
Используя также универсальную калибровку, имеем:
lgAffo] = f(V); lg[T)] = <p(lO. (Ш. 52)
Отсюда можно получить калибровку хроматографа по ММ
для данного полимерного образца:
lg М = f (V) - Ф (V) = * (V).
Далее, имея зависимость C(V), можно получить
распределение по ММ для образца разветвленного полимера. Зная
распределение по ММ и распределение по [ц] [<p(V)]> можно
рассчитать и число ветвлений. Для этого сопоставляют значения
[г\] реальной фракции (при заданном значении V) с [г\]
линейного образца той же ММ:
hip
Мл
= а. (Ш. 53)
М, А
* Даже если длина вырастающих ветвей одинакова, то все-такн
распределение длин между узлами ветвления не является таким, как в модели
статистически разветвленной цепи.
228
Определив G, можно рассчитать и т, используя зависимость:
где е = 0,5 -г- 1,5.
Конкретное значение е выбирается для данного
полимера.* Нам кажется предпочтительнее е ж 1,33.
Если отсутствует проточный вискозиметр (или проточный
нефелометр), параметры длинноцепных разветвлений (ДЦР)
определяют [192, с. 283; 193, с. 290] с помощью расчетов на
ЭВМ и предположений о структуре цепи. В качестве исходного
предположения принимают, что исследуемый полимер
линейный. Зная C(V) и калибровку хроматографа для линейного
полимера, рассчитывают значения [ц]Р. Сопоставление
рассчитанного значения [ri]p с 'Экспериментально измеренным [ц]э
позволяет судить о разветвленное™ образца. Если [л]э<
< [ц]Р, то образец разветвленный. Предполагая модель
ветвления т = КМ, по специальной программе для ЭВМ методом
последовательных приближений подбирают К. Таким образом
можно рассчитать и ММР, и число ветвлений т.
Аналогичная процедура применяется, когда вместо [ц]
имеется значение Мш или Мп, или какой-либо другой величины.
III. 4.1.2. Сочетание ГПХ и скоростной седиментации
Сочетание ГПХ и скоростной седиментации так же, как и
сочетание гель-хроматографа и проточного вискозиметра,
позволяет наиболее полно оценить ММР и ДЦР разветвленного
образца (два «фракционирующих» метода [217]). Соотношение
между 5Л и Ул для линейного образца имеет вид**:
^5л = Кзу-(\-Ь)С2Ул. (III. 54)
Таким образом, зависимость IgSj, от V выражается прямой
линией. Для разветвленных полимеров соотношение lgSP =
= f(Vp) имеет вид (при постоянном числе ветвлений):
'8 SP = Ksv ~ С1 - *) C2VP ~ »В Bsv ("L 55>
где Bsv является известной функцией от [192, с. 280; 217].
При проведении расчетов удобно построить номограммы
lgS— V (рис. III. 14): строят зависимость для линейного
полимера, а потом для различного числа т рассчитывают Bsv и
строят зависимости lgSp—Vp при заданном значении Bsv(m).
Получив экспериментально интегральные распределения по
S ц V, строят зависимость lgS от V при интегральных долях
0,1; 0,2 и т. д. Нанося экспериментальную зависимость lgS от
* Точной теории связи между g и G не имеется, существуют указания
о влиянии качества растворителя, структуры цепи на взаимосвязь G и g.
** Имеется в виду линейная калибровка для хроматографа. В общем виде
см. [214].
229
Рис. III.14. Зависимость lg S от V дли
растворов разветвленного полиэтилена. Прямые
линии — теоретические.
Лт V на рассчитанную, определяют
т для указанных «фракций».
Для определения ММ
«фракции» проводят через точку с
заданной интегральной долей
прямую с наклоном ЬС2 до пересе-
~Т~ чения ее с прямой \gSa = f(Kn).
По полученному значению Sr
(или V) определяют ММ. Таким образом можно определить
ММР и зависимость т от ММ для реального образца.
III.4.2. Определение композиционной неоднородности
сополимеров
Известно, что гидродинамический объем макромолекул
сополимера зависит не только от ММ, но и от состава. Поэтому
даже самые узкие по удерживаемому объему ГПХ-фракции
гетерогенного по составу сополимера могут, в принципе,
содержать макромолекулы разной массы и разного состава. В этом
случае определение кривых молекулярной (ММР) и
композиционной гетерогенности сополимера по данным ГПХ возможно,
если указанные характеристики известны для любого
удерживаемого объема, т. е. для всех ГПХ-фракций образца. Такую
задачу можно решить при использовании так называемой
ортогональной или кросс-хроматографии.
Реализация условий кросс-хроматографии технически
сложна, поэтому обычно для определения молекулярной и
композиционной неоднородности сополимера используют мультидетек-
торную ГПХ. Особенно перспективно применение таких
детекторов, как проточный фотометр малоуглового рассеяния света
или проточный вискозиметр, наряду с традиционными —
дифференциальным рефрактометром и УФ-,
ПК-спектрофотометрами. С помощью первых детекторов можно определить истинную
молекулярную массу ГПХ-фракций сополимера, а с помощью
последних — их средний состав.
Композиционную неоднородность гетерогенных по составу
сополимеров (например, статистических сополимеров стирола и
метилметакрилата, стирола и акрилонитрила) рекомендуется
[228] определять в режиме соответственно адсорбционной и
гидрофобной (обратнофазной) хроматографии. В этом случае
нужно только предварительно убедиться, что процессы
адсорбции или гидрофобных взаимодействий сополимера с
поверхностью сорбента не зависят от молекулярной массы сополимера,
а целиком определяются его составом.
Для определения точного ММР блоксополимера ММ можно-
находить из принципа универсальной калибровки в ГПХ [219].
230
lgS
Истинную молекулярную массу ГПХ-фракции блоксополимера
рассчитывают на основании ее гидродинамического объема,
констант Марка — Куна — Хувинка для составляющих
сополимер гомополимеров и состава фракции, определяемого по
сигналам от двух детекторов. Этот подход был развит в работе
[229] и на случай статистических сополимеров. Было показано,
что гидродинамический объем Vn макромолекулы сополимера из
т компонентов с соответствующими массовыми долями связан
с молекулярной массой М сополимера и константами Марка —
Куна — Хувинка a^i и /(„,■ для составляющих гомополимеров
соотношением:
С другой стороны
где а^, Ку) — константы Марка—Куна—Хувинка для сополимера состава р.
Зная зависимость К^(р) и аТ)(р), можно рассчитать и ММР
сополимера.
III.4.3. Хроматографический анализ
гюлимеризационно-способных олигомеров
В последние два десятилетия широкое распространение
получили синтетические низкомолекулярные полимеры с
концевыми функциональными группами, известные под названием
полимеризационно-способных или реакционноспособных
олигомеров [230, с. 13]. Поскольку степень полимеризации этих
соединений составляет обычно 10—50, термин «олигомеры» в
данном случае носит чисто условный характер и лишь подчеркивает
их низкую, по сравнению с обычными полимерами,
молекулярную массу.
В зависимости от степени полимеризации, размеров
мономерного звена, а также эффективности используемых колонок
хроматограммы олигомеров при их разделении методом ГПХ
могут быть либо разрешены относительно присутствующих в них
полимер-гомологов (или их части), либо чаще, как это имеет
место при анализе высокомолекулярных соединений, быть
полностью неразрешенными. Калибровочная процедура, методы
коррекции и интерпретации неразрешенных хроматограмм при
определении ММР олигомеров в целом аналогичны методам,
используемым в случае высокомолекулярных соединений. Вместе
с тем малая молекулярная масса, наличие полярных
функциональных групп, а главное, характерное для этих соединений
наличие нескольких типов неоднородности (по функциональности,
231
топологии, микроструктуре) вносят определенную специфику в
их анализ и в ряде случаев требуют учета некоторых
факторов, отсутствующих или несущественных в анализе высокопо-
лимеров.
Поскольку элюентный объем в ГПХ является функцией не
только ММ, но и ряда других параметров макромолекул,
используемая для количественной обработки хроматограмм
калибровочная зависимость справедлива лишь при условии химической
и структурной однородности калибровочного и анализируемого
образцов во всем исследуемом интервале молекулярных масс
или, во всяком случае, при одинаковой зависимости параметров
неоднородности от ММ.
Если для данного олигомера это условие выполняется, то
калибровка колонок может быть осуществлена любым методом,
в том числе и с помощью универсальной калибровочной
зависимости [231]. В противном случае следует избегать применения
косвенных методов и проводить калибровку с помощью узких
фракций исследуемого образца, охарактеризованных по
молекулярной массе. Однако и в этом случае условие идентичного
распределения неоднородностей в калибровочном и
анализируемом образцах должно выполняться. Необходимо также
отметить, что вследствие зависимости элюентного объема от
микроструктуры, топологии [232], а в низкомолекулярной области и
от функциональности [230,233] макромолекул калибровочная
зависимость для олигомеров, обладающих каким-либо типом
неоднородности, может отличаться от линейной даже на тех
колонках, на которых для других типов олигомеров она обладала
линейностью.
Другим фактором, который необходимо учитывать при ГПХ-
анализе олигомеров, является их высокая адсорбируемость.
В зависимости от характера распределения полярных групп
(адсорбционных центров) в молекулах адсорбция может
оказывать различное влияние на эксклюзионное разделение. Если
адсорбционные центры статистически распределены по цепи, то
энергия взаимодействия макромолекул с поверхностью
адсорбента возрастает с ростом ММ, и адсорбция приводит к
ухудшению эксклюзионного разделения, а в пределе—к разделению
по адсорбционному механизму, сопровождающемуся инверсией
порядка элюирования. Если же адсорбционные центры
сосредоточены на концах макромолекул, то при неизменности энергии
адсорбции изменение энергии Гиббса из-за уменьшения
энтропии увеличивается с уменьшением ММ, что приводит к
дополнительному, по сравнению с чисто эксклюзионным разделением,
возрастанию значений kv. При этом слабая адсорбция, вообще
говоря, не препятствует анализу и, более того, несколько
увеличивает селективность в низкомолекулярной области. Однако
даже в этом случае возможные нарушения калибровки, влияние
неоднородности по функциональности, а также наблюдаемая
иногда потеря части образца вследствие необратимой сорбции
232
не позволяют рекомендовать проведение анализа в условиях,
допускающих проявление адсорбционных эффектов.
Внешне адсорбционные эффекты в ГПХ проявляются в
зависимости формы хроматограмм от полярности растворителя и
значениях kv для всего образца или его отдельных фракций,
больших единицы. Исключить их обычно удается применением
в качестве сорбентов органических гелей, а в качестве
подвижных фаз — растворителей достаточно высокой полярности.
Концентрационные эффекты в ГПХ олигомеров проявляются,
как и в случае высокомолекулярных соединений, в сдвиге
хроматограмм в сторону больших элюентных объемов и
наблюдаются при концентрациях анализируемого раствора,
превышающих некоторое пороговое значение, зависящее от ММ и
коэффициента полидисперсности образца. В общем случае эта
величина выше для высокополимеров и для олигомеров с М =
= 103ч- 104 составляет около 10 мг/мл.
Определенные трудности при хроматографнческом анализе
олигомеров может вызвать использование в качестве
детектирующего устройства дифференциального рефрактометра, сигнал
которого пропорционален не только концентрации
анализируемого вещества в элюате, но и разности показателей преломления
вещества п и растворителя ns:
h~C(n — ns). (III. 56)
В свою очередь, зависимость показателя преломления
полимера от молекулярной массы может быть описана уравнением
п = А + В/М, (III. 57)
где А и В — константы.
Из уравнений (III.56) и (III.57) следует, что влияние
зависимости показателя преломления, а следовательно, и сигнала
рефрактометра от ММ возрастает по мере уменьшения ММ.
При анализе ММР олигомеров, вследствие их низкой
молекулярной массы, эта зависимость может привести к искажению
формы хроматограмм, завышению (при «>«,) значений Мп
и занижению (при п < ns) коэффициента полидисперсности.
В принципе, учет рефрактометрической ошибки в случае
незначительного вклада приборного уширения может быть
осуществлен с помощью относительно простой корректировочной
процедуры, описанной в работе [234], а совместно с учетом при-
бор-ного уширения — по методу, приведенному в работе [235].
Однако вследствие наличия в олигомерах различных,
указанных выше типов неоднородности соотношение (III. 57) для них
редко выполняется, и учет рефрактометрической ошибки может
превратиться в довольно сложную проблему. Единственным
практически приемлемым выходом из этого положения
является использование при анализе олигомеров в качестве
подвижной фазы растворителя с показателем преломления,
максимально отличающимся от показателя преломления образца. Из
?33
уравнения (III. 56) очевидно, что отношение сигналов
рефрактометра при прохождении через кювету двух равных по
концентрации растворов полимер-гомологов разной молекулярной
массы будет тем ближе к единице, чем больше абсолютное
значение.
Таким образом, при хроматографическом анализе олигоме-
ров растворитель, используемый в качестве подвижной фазы,
должен обладать достаточно высокой полярностью,
обеспечивающей отсутствие адсорбционных эффектов, и иметь
показатель преломления, максимально отличающийся от показателя
преломления анализируемого образца. Выполнение последнего
требования позволит не только избежать рефрактометрической
ошибки, но и, вследствие повышения при этом
чувствительности детектирования, исключить концентрационные эффекты.
Указанные требования не являются взаимоисключающими,
поскольку растворители с большей полярностью, благодаря
наличию в них кислород- или азотсодержащих функциональных
групп (спирты, эфиры, кетоны, амиды и т. п.), обладают
меньшими показателями преломления по сравнению с менее
полярными углеводородами и их галогензамещенными.
Одной из важнейших молекулярных характеристик полиме-
ризационноспособных олнгомеров является распределение по>
типу функциональности (РТФ), характеризующее содержание в
олигомере молекул с различным числом функциональных групп.
Наряду с ММР РТФ олигомеров играет определяющую роль
в формировании комплекса физико-механических свойств вул-
канизатов, полученных на их основе [229]. РТФ олигомеров
определяют методом жидкостной адсорбционной
хроматографии. Определение основано на различной адсорбируемости
олигомерных молекул разной функциональности. Разделение
обычно проводят на кремнеземных адсорбентах (силикагель, си-
лохром) в элюенте постоянной или переменной полярности.
Теоретические вопросы хроматографического разделения
олигомеров по функциональности подробно рассмотрено в работах
[236, 237].
Как при хроматографическом определении ММР олигомеров
осложняющее влияние оказывает наличие в них иных типов
неоднородности (в том числе и РТФ), так и при определении
РТФ основным мешающим фактором является дисперсность
олигомеров по ММ. Поскольку адсорбируемость молекул данной
функциональности зависит от их ММ, разделить олигомеры,
обладающие более или менее широким ММР, часто не удается
вследствие значительного перекрывания хроматографических
полос, отвечающих молекулам разной функциональности.
Однако в настоящее время теоретически и экспериментально
показано [238, 239], что при определенной энергии
взаимодействия звеньев цепи макромолекул с адсорбентом, которая
может быть реализована путем использования соответствующих
подвижной и неподвижной фаз (критические условия) зависи-
234
мость удерживания олигомерных молекул от ММ исчезает.
Используя высокоэффективные колонки и проводя разделение
в критических условиях, удалось определить РТФ ряда гидр-
оксилсодержащих олигомеров. Вместе с тем анализ в
критических условиях возможен лишь в том случае, когда энергия
адсорбции звеньев цепи олигомера сопоставима с энергией
адсорбции концевых групп, по которым ведется разделение. Если
же она значительно меньше, то в критических условиях
энергия взаимодействия концевых функциональных групп с
адсорбентом оказывается настолько большой, что приводит к
практически необратимой адсорбции олигомера в колонке. Так, до
настоящего времени не удалось осуществить разделение в
критических условиях углеводородных олигомеров с концевыми
гидроксильными и карбоксильными группами. Анализ таких
образцов, обладающих узким ММР, может быть проведен в
условиях градиентного элюирования. Образцы с более широким
ММР необходимо предварительно расфракционировать по ММ
с последующим анализом РТФ каждой фракции. Вопросы
оценки активности бинарных элюентов и выбора оптимального
профиля элюирования при хроматографнческом определении РТФ
олигомеров рассмотрены в работах [238, 239].
ГЛАВА IV
ОПРЕДЕЛЕНИЕ ДОБАВОК И ПРИМЕСЕЙ
В ПОЛИМЕРНЫХ МАТЕРИАЛАХ
Необходимость количественного определения добавок и
примесей в полимерных материалах возникает при контроле
содержания добавок, способных расходоваться в процессе
полимеризации и переработки полимера, примесей, влияющих на
свойства полимера и его перерабатываемость, а также диктуется
санитарными нормами на допуск полимеров к применению в
быту, пищевой промышленности и медицине. Методики
количественного определения необходимы также для изучения
превращений добавок в полимерах и с целью выбора
оптимального количества добавки при разработке полимерных композиций.
В общем случае задача количественного определения
добавок и примесей может решаться двумя путями: прямым
анализом полимера или его раствора или с предварительным
отделением добавок от полимерной части, а в сложных полимерных
композициях — и от других низкомолекулярных веществ [240].
Можно определять органические добавки, содержащие гетеро-
атомы, и неорганические добавки и примеси после
предварительной минерализации полимера.
Метод прямого анализа очень привлекателен для аналитика,
но имеет ограниченную применимость, особенно в отношении
образцов неизвестного состава, а также в силу сложности
235
современных полимерных композиций. Прямой анализ
полимеров и их растворов чаще всего осуществляется спектральными
методами анализа.
Вопросы выделения добавок из полимеров для целей
качественного анализа рассматривались в гл. I. В случае
количественного анализа для выделения добавок и примесей помимо
методов, о которых говорилось в разделе 1.2, применяют
вакуум-термическую экстракцию, включающую комбинированное
использование вакуума и теплоты, газовую экстракцию,
основанную на вытеснении летучих соединений из нагретой пробы
потоком инертного газа, и метод ГПХ.
При выборе метода и условий выделения добавок и
примесей из полимерной матрицы для целей количественного
анализа основным критерием правильности подхода является полнота
их извлечения (за исключением дискретной газовой экстракции
в парофазном анализе полимеров, применяемой для
определения летучих примесей). Проще всего доказать полноту
извлечения можно, используя стандартные образцы, содержание
определяемого вещества в которых известно. Однако для
большинства примесей это практически исключено и составляет
сложность для ряда добавок, которые могут в процессе
получения полимерной композиции и подготовки ее к анализу
претерпевать превращения, разлагаться или частично испаряться.
Полноту экстракции для всех видов исчерпывающей
экстракции определяют обычно по кинетической кривой зависимости
содержания определяемого компонента в экстракте от
продолжительности экстракции. Выход на плато в кинетической
кривой считается окончанием экстракции. Отсутствие этого
компонента в экстракте при повторном экстрагировании образца
подтверждает правильность выбранных условий.
При применении методов растворения — высаждения и ГПХ
полноту извлечения в случае отсутствия стандартных образцов
можно проверить только методом добавок.
Специальными опытами при разработке методик
количественного анализа необходимо проверять стойкость добавки или
примеси в процессе выделения ее из полимера, поскольку
длительное температурное воздействие на этой стадии создает
потенциальную возможность разложения и окисления добавок в
ходе аналитического процесса. Для предотвращения
возможного окисления выделение добавок и примесей иногда проводят
в атмосфере инертного газа.
Наиболее распространенным методом выделения
органических добавок для их количественного определения является
экстракция органическим растворителем. Это обусловлено тем,
что использование вакуум-термической и газовой экстракции
ограничено летучестью определяемого соединения, метод ГПХ
применим только к растворимым полимерам, а в методе
растворения— высаждения всегда возможно захватывание
низкомолекулярных веществ осаждающимся полимером, что особен-
236
но существенно при низких концентрациях определяемого
соединения.
Скорость экстракции в основном определяется диффузией
вещества из полимера и, следовательно, его концентрацией.
Зависит она также от структуры полимера, его плотности и от
строения определяемой добавки, т. е. от ее растворимости в
применяемом экстрагенте и степени взаимодействия добавки с
матрицей полимер — наполнитель. Примером влияния
последнего фактора может служить увеличение продолжительности
экстракции добавок из полиэтилена, наполненного техническим
углеродом, по сравнению с образцами без наполнителя [241].
При прочих равных условиях увеличение скорости
экстракции может быть достигнуто за счет повышения температуры
процесса, а также увеличения отношения поверхности
экстрагируемого образца к его объему. Поэтому ускорению
экстракции способствует измельчение полимера, которое может быть
выполнено различными средствами: с помощью шаровых и
режущих мельниц, микротомов, перетиранием образца при
температуре жидкого азота, применением ультразвуковых диспер-
гаторов.
Отделенные от полимера добавки и примеси или
минерализованный полимер можно анализировать непосредственно, или
предварительно разделив низкомолекулярные вещества каким-
либо подходящим методом. В случае органических добавок для
этих целей в основном используют различные виды
хроматографии. Наибольшее применение находит жидкостная
хроматография. Это объясняется тем, что условия разделения в ней более
мягкие, чем в газожидкостной хроматографии. Применение
высоких температур в последней, которое вызвано низким
давлением паров большинства органических добавок, создает
возможность разложения добавок в процессе хроматографирования.
При использовании рпубликованных в литературе методик
определения добавок и примесей в полимерах аналитик должен
помнить, что они разработаны для конкретных образцов,
полученных по определенной технологии и рецептуре, и изменения
в них могут сказаться на результатах анализа по этим
методикам непредвиденным образом.
Дальнейшее рассмотрение аналитических проблем
количественного определения добавок и примесей и путей их решения
будет проведено на конкретных примерах.
IV. I. ОПРЕДЕЛЕНИЕ ДОБАВОК
IV. 1.1. Стабилизаторы
Количественное определение стабилизаторов необходимо
прежде всего для подбора стабилизирующих систем: одни
стабилизаторы предназначены для защиты полимера на стадиях его
получения и переработки и поэтому должны израсходоваться
237
в процессе производства, другие должны обладать
долговременным действием и увеличивать срок службы полимера в
условиях его эксплуатации. Количественное определение
стабилизаторов необходимо также для контроля их загрузки.
IV. 1.1.1. Аналитические проблемы количественного определения
стабилизаторов
Количественное определение стабилизаторов осложняется
следующими факторами:
1) малые концентрации стабилизаторов в полимере —
0,01-1,0%;
2) наличие в используемых стабилизаторах побочных
продуктов их синтеза;
3) высокая реакционная способность и соответственно
низкая стойкость ряда стабилизаторов;
4) необходимость анализа смесей близких по свойствам
соединений;
5) присутствие в полимере сложных продуктов разложения
стабилизаторов и других добавок;
6) трудность получения стандартных образцов.
Работы по определению продуктов превращения
стабилизаторов в полимерах в настоящее время единичны и носят полу-
колнчественный характер, тогда как разработка методик
определения этих продуктов очень важна, поскольку некоторые из
них также обладают стабилизирующим действием [242].
Большое значение имеет вопрос приготовления стандартных
образцов, особенно для прямых методов анализа полимеров,
которые основаны на использовании спектральных
характеристик стабилизаторов [39, с. 140] и заключаются в измерении
поглощения пленок полимера в УФ- или ИК-областн спектра
относительно нсстабилизировапного полимера. По градуировоч-
ному графику, построенному по стандартным образцам с
известным количеством введенного стабилизатора, определяют его
содержание в анализируемом образце. Для построения градуи-
ровочного графика используют пленки полимера,
приготовленные путем введения в полимер известного количества
стабилизатора перемешиванием на вальцах в течение 15 мин при
160°С [39, с. 140] или горячим прессованием смесей
стабилизатора с порошком полимера [153, с. 51].
Как свидетельствуют данные ряда авторов [153, с. 51; 243],
потери стабилизаторов на стадиях термической обработки
образцов (вальцевание, формование, горячее прессование пленок)
могут достигать 30 %. Поэтому при использовании таких
«стандартных» образцов возникают систематические погрешности из-
за неконтролируемых потерь добавки в процессе получения
пленки. Точное содержание стабилизатора в стандартных
образцах необходимо определять после его экстрагирования из
пленки.
238
Не менее важно правильно подобрать применяемый для
построения градуировочных графиков стандарт стабилизатора.
Выбор стандарта определяется стоящей перед аналитиком
задачей. Если необходимо контролировать загрузку стабилизатора,
то желательно в качестве стандарта использовать ту партию
стабилизатора, которую вводили в анализируемый полимер,
поскольку состав и содержание примесей в промышленных
стабилизаторах может меняться от партии к партии. Если
изучается превращение стабилизатора и контролируется его расход,
то градуировочный график следует строить по очищенному
стандарту, а анализ проводить хроматографическим методом.
IV. 1.1.2. Выделение стабилизаторов из полимеров
Экстракция растворителем. В табл. IV. 1 приведены условия
количественной экстракции стабилизаторов из ряда полимеров.
Использование для экстракции низкокипящих растворителей
имеет два преимущества: сведение к минимуму потерь
экстрагируемых добавок вследствие термического разложения и
выигрыш во времени анализа за счет большей скорости
испарения высоколетучих растворителей на стадии концентрирования
экстракта.
Наиболее употребительным экстрагентом для полиолефпнов
является хлороформ, используемый для экстракции как при
комнатной температуре, так и при температуре кипения
растворителя. Эффективными экстрагентами являются также те-
трагидрофуран, тетрахлорметан, метнленхлорид. При
использовании всех перечисленных в табл. IV. 1 растворителей, за
исключением тетрагидрофурана, наблюдается частичный переход в
экстракт олигомеров олефинов. Если использовать в качестве
экстрагентов углеводородные растворители, то в экстракт
переходит и низкомолекулярный полимер. Экстракт освобождают
от олигомеров, если это необходимо для последующих стадий
анализа, путем высаждения их метиловым спиртом.
Для экстракции стабилизаторов из полистирола и поливи-
нилхлорида чаще используют полярные растворители,
например, ацетон, диэтиловый эфир.
Применяемое соотношение полимер: растворитель (г/мл),
как правило, меняется в пределах от 1 : 5 до 1 : 10.
Полнота выделения стабилизатора из анализируемого
образца при подборе условий экстракции контролируется по
кинетическим кривым. Одним из вариантов построения такой
кривой при анализе пленок является прямой анализ пленок
спектральным методом. В этом случае по уменьшению поглощения
пробы в максимуме аналитической полосы контролируют
исчезновение добавки из полимера; выход на плато на этой
кинетической кривой также можно считать окончанием экстракции.
Полнота выделения,стабилизатора из полимера может быть
подтверждена качественным анализом проэкстрагированного
239
Таблица IV.1. Условия количественной экстракции
стабилизаторов из полимеров
Полимер
Полиолефи-
ны
Полиэтилен фирмы
BASF, Lu-
polen 2400F
ПЭНП,
полипропилен
Полиэтилен,
полипропилен
Полипропилен
Полипропилен
Полипропилен изотак-
тический
Полипропилеи,
наполненный
тальком
Повннил-
хлорид
Стабилизатор
Ионол, сантонокс R,
топанол СА
Ионол, антиоксидант
НГ-2246, дидодецил-
тиодипропионат
Ионол, ирганокс
1010, ирганокс 1076,
сантоиокс-R, ионокс
330
Ионол. ирганокс
1076, циасорб 531,
типувин 327
Дилаурилтиодипро-
пиопат, дистеарил-
тиодипропионат
Тинувин 770, хоста-
вин TMN 20
Дилаурилтиодипро-
пиопат
Тонанол СА,
тинувин 327, ирганокс
1076, дастиб 242,
дндодецнлтиодипро-
пиопат
Ионол, дистеарил-
тиодипропионат,
топанол СА.
сантонокс R, сантовайт
—
Растворитель
Метиленхло-
рид или
хлороформ
Ацетон
Хлороформ
Тетрахлор-
метан для
полиэтилена,
тетрагидро-
фуран для
полипропилена
Хлороформ
Хлороформ
Хлороформ:
этиловый
спирт : гек-
саи = 1:1:4
Ацетон
Тетрагидро-
фуран
Ди этиловый
эфир, затем
пература,
"С
'кип
'кип
' кнп
Г*
КИП
'кнп
22
* кип
* кнп
22
' кип
жительность
экстракции, ч
8
4
6
2
2
16
—
8
24
8
Навеска,
г
2-5
10
5
2
50
10
1
4
2-5
Литера тур-
ный
источник
[244]
[245]
[246]
[243]
[247]
[248]
[249]
[250]
[251]
[252]
Стирол-бу- Аминные антиокси-
тадненовый данты
каучук
азеотроп тет-
рахлорметан:
метиловый
спирт = 2:1
Ацетон
'кип
16
[253]
* Кипячеш'е в колбе с обратным холодильником, в остальных случаях — экстракция
аппарате Сокслетл.
240
ооразца на азот в случае стабилизаторов аминного типа; на
фосфор, серу, олово в случае соответствующих элементоргани-
ческнх стабилизаторов.
Обычно экстракцию проводят в аппарате Сокслета. При
анализе полиэтилена эффективность экстракции снижается при
всплывании образца. Для лучшего контакта полимера с
растворителем рекомендуется насыпать поверх навески полимера
слой стеклянных бусинок толщиной 1 см [247]. При
кипячении навески полимера с растворителем в колбе с обратным
холодильником полимер по окончании экстракции отделяют
декантацией, центрифугированием, а чаще — фильтрованием через
микропористые фильтры.
Метод растворения — высаждения. Выделение
стабилизаторов из растворимых полимеров проводят обычно путем
высаждения полимера из его раствора равным объемом осадителя —
метилового или этилового спирта. Основной проблемой анализа
при этом является захватывание добавок высаждаемым
полимером.
При анализе полиолефннов, которые растворяются при
температуре выше 100 °С, приходится учитывать термическую
стойкость стабилизаторов и их летучесть. Для растворения
полиолефннов используют толуол, декалин и ксилол, т. е. вы-
сококипящие углеводородные растворители. По одной из
методик полимер растворяют в кипящем толуоле {Ткпп = 110—
111 °С) кипячением навески образца не менее 1 ч в колбе с
обратным холодильником и высаждают высокомолекулярную
фракцию этиловым или метиловым спиртом. При таких
жестких условиях экстракции может происходить частичное
разложение некоторых стабилизаторов [251], во всяком случае
наблюдается их потеря.
В работах [254—256] в качестве растворителя используется
декалин (Ткт1 = 190°С)—25 мл растворителя на 1 г
полимера. Растворение полимера проводят при 110°С; обычно оно
занимает 0,5—1 ч. Сравнительные опыты по выделению 2,6-ди-
гр<?х-бутил-4-метилфенола (ионола) и ирганокса 1010 из
раствора полиэтилена показали, что при высаждении полимера
бутнлцеллозольвом или этиловым спиртом происходит
захватывание добавок полимером, в то время как при высаждении
путем охлаждения раствора при перемешивании результаты
получаются количественными [254]. Полимер при этом выпадает
в форме губчатого геля, который легко можно удалить из
раствора, например, шпателем. Предложено [254] удобное
приспособление для фильтрования раствора, состоящее из
микропористого фильтра, соединенного со шприцем фторопластовой
трубкой через иглу с металлическим наконечником, плотно
входящим в эту трубку. Часть экстракта затягивается в шприц
и используется для дальнейшего анализа. Хорошее открытие
добавок показывает, что в декалине добавки распределены
241
100 200 300
Температура'°С
Рис. IV.1. Термогравиметрические кривые аитиоксн-
даитов:
/ — иомол; 2 — тинувпи 327; 3 — циасорб 53U 4 — ирга-
нокс 1076.
равномерно как внутри, так и
снаружи осаждаемой «губки» полимера.
Авторы работы [257] в
качестве растворителя применяли я-ксилол
(Гкип = 140 С)—150 мл на 3 г
полимера, полимер высаждали равным
объемом диоксана при охлаждении на
ледяной бане. После
концентрирования экстракта на роторном
испарителе при 45—50°С до объема 1 мл проводилось повторное вы-
саждение полимера 5 мл диоксана.
Определение широко распространенного стабилизатора —
ионола осложняется потерями его при высоких температурах.
Согласно термогравиметрическим данным [243], ионол
начинает разлагаться уже при 100 °С, однако наблюдаемую при
ТГА потерю массы образца (рис. IV. 1) ,следует отнести скорее
к испарению ионола. Это подтверждается определением в
полиэтилене продуктов термического превращения ионола [246].
Методом высокоэффективной жидкостной хроматографии
анализировали хлороформенный экстракт полимера, полученный в
условиях, исключающих термическое разложение
стабилизатора в ходе экстракции. При выдерживании образца
полиэтилена, содержащего ионол, в течение 15 мин при 200°С было
обнаружено 80 % ионола от содержания его в непрогретом
образце и следовые количества продуктов термического
превращения ионола. Через 90 мин выдерживания образца при 200 °С
ионол исчезал полностью, в то время как содержание
продуктов его термического разложения увеличивалось только до 20 %•
Таким образом, потери ионола при высоких температурах
происходят в первую очередь благодаря его испарению.
Потери термически неустойчивых и летучих стабилизаторов
при выделении их из полиолефинов методом растворения — вы-
саждения могут быть учтены путем пропускания через те же
стадии анализа стандарта стабилизатора, используемого для
калибровки на конечной стадии анализа. Именно таким
образом были получены количественные результаты в работах
[254—256].
Вакуум-термическая экстракция. При вакуумировании
полимера, в частности полиэтилена, многие стабилизаторы
улетучиваются из него с достаточно большой скоростью уже при
комнатной температуре. Наибольшей летучестью обладают
низкомолекулярные антиоксиданты (мол. масса менее 300). Авторы
работы [258] расположили исследованные ими антиоксиданты
в порядке уменьшения летучести в следующий ряд: 2,6-ди-
грег-бутил-4-метилфенол > 2,4,6-три-грет-бутилфенол = фенил-
242
Рис. IV.2. Прибор для выделения добавок из полимера
методом вакуум-термической эстракции:
1 — вкладыш; 2—пробирка; Л —стаканчик с сухим льдом; 4 —
шлиф; 5 — воронка для смывания стабилизатора с внутренних
стенок пробирки.
бензоат = дифениламин > фенил-а-нафтил-
амин > фенил-р-нафтиламин > 2,2'-мети-
ленбис (4-метил-6-грег-бутилфенол). Это
различие можно использовать для
раздельного выделения добавок из полимера.
Метод вакуум-термической экстракции,
предложенный советскими
исследователями [259], позволяет менее чем за 1 ч
количественно выделить антиоксиданты из
полимера. 20—100 мг полимера в виде
гранул, порошка или пленки помещают на дно
стеклянной пробирки со шлифом. В
пробирку вставляют вкладыш — стеклянную трубку с выступом в
верхней части (рис. IV. 2). Снаружи на среднюю часть
пробирки надевают стаканчик с сухим льдом. В пробирке создают
глубокий вакуум и погружают ее в нагретый термостат на
глубину 1—2 см, где выдерживают в течение 20—40 мин.
Антиоксидант, содержащийся в полимере, оседает на
внутренней поверхности пробирки и на вкладыше, откуда его смывают
растворителем в мерную колбу.
Этот метод был применен для раздельного определения
ионола и антиоксиданта НГ-2246 [2,2'-метиленбис(4-метил-6-
грег-бутилфенола)], введенных в ПЭНП в суммарном
количестве 1 %• Ионол отгоняли из полимера при комнатной
температуре (22 °С, 2 ч), антиоксидант НГ-2246 начинает отгоняться
только при 50 °С, поэтому его отгоняли при 200 °С в течение
30 мин. Открытие введенных добавок достигает 94—100%.
Позднее было изучено испарение из ПЭНП «тяжелых» ан-
тиоксидантов (мол. масса более 350)—топанола СА, ирганокса
1010, диафена НН, нонокса WSP, дилаурилтиодипропионата
[260]. Уже при 160 °С все антиоксиданты заметно испаряются.
Минимальной летучестью при температуре меньше 210°С
обладает ирганокс 1010. Количественное испарение таких антиок-
сидантов требует длительной (~2 ч) термообработки примерно
при 220 °С при соблюдении точного температурного и
временного режимов испарения.
Гель-проникающая хроматография. Для растворимых
полимеров привлекательной техникой разделения является
гель-проникающая хроматография. Качество разделения зависит от
молекулярной массы отделяемых добавок, в ряде случаев — от их
структуры, а также от применяемой нагрузки на колонку.
В работе [261] показано, что на колонку размерами 330 X
X 15 мм, заполненную сефадексом LH-60, можно наносить до
180 мг полимера (до 5 мл раствора), чтобы отделить
высокомолекулярную часть от низкомолекулярных добавок с молеку-
243
лярной массой не более 1000. Низкомолекулярные соединения
на такой колонке выходят в объеме 50—100 мл. Из растворов
поливинилхлорида и полистирола (мол. масса ~ 105) за
0,5—2,5 ч в зависимости от скорости элюента — тетрагидрофу-
рана (1—5 мл/мин) выделяли низкомолекулярные добавки,
среди которых из стабилизаторов былитинувин Р и полигард.
Тетрагидрофуран является наиболее часто используемым
элюентом в ГПХ, поскольку гели прекрасно в нем набухают, а
анализируемые добавки хорошо растворяются. К тому же
тетрагидрофуран обладает слабым поглощением в УФ-области и
имеет низкий показатель преломления, что делает возможным
использование УФ- и рефрактометрического детектора. Однако
с помощью данного растворителя не удается количественно
выделить методом ГПХ такие антиоксиданты, как ионол, олово-
органические стабилизаторы, что, возможно, связано [261] с
их стабилизирующим действием на этот растворитель.
Концентрирование экстрактов. Выделенная методом
экстракции, растворения — высаждения или ГПХ фракция
низкомолекулярных веществ, содержащая определяемые
стабилизаторы, чаще всего подвергается концентрированию.
Необходимость этой операции может быть вызвана как низкой
концентрацией стабилизаторов в полученном экстракте, что не
позволяет использовать выбраный метод из-за недостаточной
его чувствительности, так и несовместимостью применяемого на
стадии выделения растворителя с последующими
аналитическими процессами. В последнем случае растворитель испаряют
досуха. Обычно концентрирование проводят при разрежении,
создаваемом водоструйным насосом, в инертной атмосфере при
температуре не выше 45 °С; для этой же цели можно
использовать ротационные испарители.
IV. 1.1.3. Методы количественного определения стабилизаторов
Для определения стабилизаторов наиболее широко
применяются спектральные методы анализа, при этом испытанию
подвергаются пленки полимеров, их растворы и экстракты.
Возможность проведения спектрального анализа в первую очередь
определяется прозрачностью полимера и растворителя в
аналитической области спектра и отсутствием примесей, мешающих
определению.
Большинство стабилизаторов являются ароматическими
соединениями и имеют полосы поглощения в УФ-области спектра.
Наиболее интенсивные полосы поглощения характерны для
азотсодержащих стабилизаторов. Дополнительные возможности
для анализа в УФ-области спектра дает использование бато-
хромного эффекта, наблюдаемого при добавлении к растворам
некоторых стабилизаторов спиртового раствора КОН [39,
с. 129, 140].
Применение ИК-спектроскопии в основном ограничено
определением карбонилсодержащих стабилизаторов по интенсивной
244
полосе поглощения валентных колебаний связи С=0 в области
1625—1750 см-1 [39, с. 131, 142].
Спектрофотометрический анализ экстрактов основан на
высокой реакционной способности стабилизаторов, которые легко
вступают в различные реакции с образованием окрашенных
соединений.
Часть антиоксидантов и термостабилизаторов и большинство
светостабилизаторов проявляют заметную флуоресценцию. Флу-
ориметрические методы анализа намного чувствительнее спект-
рофотометрических, но, к сожалению, не нашли столь широкого
применения.
Из титриметрических методов наибольшее значение имеет
восстановление антиоксидантов нитратом серебра с
растворением осажденного серебра и титрованием его раствором этилен-
диаминтетраацетата натрия; обработка стабилизаторов феноль-
ного типа бромид-броматной смесью и титрование
неизрасходованного бромида раствором тиосульфата натрия; окисление
производных фенилендиамина хлоранилом и титрование
образующегося основания хлорной кислотой [262].
Полярографический анализ стабилизаторов основан на
измерении высоты волны, обусловленной восстановлением
растворенного кислорода [262]. Однако пользоваться этим методом
нецелесообразно, когда доступны более простые методы.
Хроматографические методы анализа применяют в основном
к экстрактам полимеров. Для разделения экстрактов широко
используют все виды жидкостной хроматографии.
Доминирующим хроматографическим методом определения стабилизаторов
до настоящего времени остается тонкослойная хроматография
(ТСХ), благодаря малой продолжительности анализа, высокой
чувствительности метода, несложности оборудования и
возможности одновременного анализа многих компонентов, которые
могут быть разделены в одном хроматографическом процессе.
Распространению метода способствовал промышленный выпуск
готовых пластинок. Хотя основную ценность этот метод имеет
для идентификации добавок, он позволяет получать и
количественные результаты, причем не с меньшей точностью, чем
другие хроматографические методы. Количественное определение
можно проводить после снятия пятен с пластинки и экстракции
их полярным растворителем или непосредственно на пластинке
с использованием сканирующих денситометров.
Наиболее широко применяемым вариантом количественной
ТСХ является спектрофотометрическое определение вещества
после элюции его с пластинки. Хотя это более трудоемкий
процесс, чем денситометрия, однако он завоевал популярность
благодаря тому, что спектрофотометры стали стандартным
оборудованием аналитических лабораторий. Погрешность определения
в этом методе обусловлена в основном неполнотой элюции
вещества с сорбента и влиянием примесей в стационарной фазе.
Второй фактор больше сказывается в УФ-, чем в видимой об-
245
ласти спектра, уменьшаясь по мере увеличения длины волны
до практически незначимого при % = 400 нм. Полезно
проводить предварительное вымывание примесей с сорбента путем
пропускания через пластинку полярного растворителя. Обычно
же для учета влияния этих примесей измерения проводят
относительно контрольной пробы — экстракта с участка адсорбента
равной площади, находящегося на уровне анализируемого
вещества. Менее распространенным, но также позволяющим
учитывать влияние примесей сорбента, является снятие спектра
поглощения экстрагированной добавки и измерение поглощения в
максимуме спектра до базисной линии, проводимой через
основание пика поглощения; в качестве контрольной пробы в этом
случае вместо экстракта применяют растворитель. И наконец,
как уже упоминалось ранее, для стабилизаторов кроме
измерения их поглощения в УФ-области проводят измерение
оптической плотности в более длинноволновой области, применяя
для этого различные химические реакции. Эти реакции можно
проводить как до нанесения экстракта на пластинку, так и
после элюцин вещества с сорбента.
В случае применения сканирующих денситометров строят
зависимость площади пятна от количества нанесенного на
пластинку стабилизатора. Линейной является зависимость корня
квадратного площади пятна от логарифма массы вещества в
пятне [240, с. 66].
Полноту элюции вещества с пластинки проверяют
предварительными опытами со стандартными растворами
стабилизаторов. Для ускорения отработки условий экстракции опыты
проводят со смесями стабилизаторов, используя в качестве
оценочного критерия массу вещества в пятне, а не содержание (в %)
стабилизатора в смеси.
Чувствительность метода ТСХ при определении
стабилизаторов составляет примерно 1 мкг. Обычно экстракт
концентрируют до получения 0,02—0,5 %-ных растворов стабилизаторов и
на пластинку с толщиной слоя сорбента 0,2—0,3 мм полоской
или в точку наносят от 1 до 40 мкл экстракта, в котором
содержится 1—20 мкг стабилизатора. Ниже приведены системы
растворителей, применяемые для разделения стабилизаторов
методом ТСХ на силикагеле:
Бензол
Бензол : гексан =1:1
Бензол : гексан = 1:8
Бензол : диэтнловый эфир =
= 3:2
Бензол : этиловый спирт =95 : 5
Бензол : ацетон : раствор
аммиака (концентрированный)
100:5 :0,1
100:5:2
Толуол: изопропиловый
спирт = 88 : 12
Фенольные антиоксиданты [263, 264]
Смесь стабилизаторов [253]
Пространственно затрудненные [263, 264]
фенолы
Смесь стабилизаторов [253]
Аминные антиоксиданты [253]
Аминные антиоксаданты [263]
Фенольные антиоксиданты [252]
Смесь стабилизаторов [263]
246
Циклогексан : метиловый
спирт = 96 : 4
Циклогексан : этилацетат =
= 4:1
Гексан : изопропиловый
спирт = 88: 12
Петролейный эфир :
этилацетат = 5:1
Хлороформ
Хлороформ : гексан = 2:1
Бутиловый спирт : ледяная
уксусная кислота = 97 : 3
Смесь стабилизаторов [265]
Производные бензофенона [265]
Аминные светостабилизаторы [248]
Сантонокс R в полиэтилене [265]
Фенольные антиоксиданты [244]
Светостабилизаторы [252]
Оловоорганические стабилиза- [252]
торы
Значения Rf некоторых стабилизаторов при разделении их
на силуфоле-УФ254 с применением в качестве растворителя
смеси толуола с гексаном (1:1) и проявлением пятен в УФ-све-
те и парах иода указаны в табл. IV. 2.
Для проявления стабилизаторов пластинку можно
поместить в пары иода, облучить УФ-светом или опрыскать
раствором подходящего реагента. В качестве реагентов используют
не проявители общего характера типа перманганата калия, а
соединения, дающие характерное окрашивание с данным
классом стабилизаторов. Наибольшее распространение получили
3%-ный раствор фосфорномолибденовой кислоты [265] и 2 %-
ный раствор 2,6-дихлорхинон-4-хлоримина в этиловом спирте
[263]. После опрыскивания фосфорномолибденовой кислотой
пластинку выдерживают в парах аммиака, при использовании
2,6-дихлорхинон-4-хлоримина пластинку через 15 мин
обрабатывают 2 %-ным раствором буры. Для лучшего развития
Таблица IV.2. Спектральные и хроматографические характеристики
некоторых стабилизаторов
Спектры стабилизаторов в растворе хлороформа. При ТСХ на пластинку наносили
20 мкг (1 мкл) стабилизатора.
Стабилизатор
Я, нм
Е-102,
л/(м-г)
основное
вещество
примеси
Ионол
НГ-2246
Агидол-2 А
Агидол-2 Б
Фенозан 23
Ирганокс 1010
Фосфит НФ
Иргафос 168
Фау-13*
280
280
285
285
285
276
277
277
270
271
281
9,7
9,5
14,5
14,9
14,2
5,3
6,7
6,9
3,7
6.4
6,4
0,83
0,83
0,38
0,39
0,39
0
0
0
0,94**
0,94**
0
0,13; 0
0,13; 0,06
0,89; 0,13
0,37; 0.19
0,46; 0,07; 0,05
0 —
* На пластинку наносили 1 мкг.
** Белые пятиа в парах иода.
247
окраски в некоторых случаях, особенно при анализе
пространственно затрудненных фенолов, пластинку нагревают в течение
5 мин при 120°С. Для аминных антиоксидантов
специфическими проявителями являются также 4 %-ный раствор бензо-
илпер'оксида в бензоле [263] и 20 %-ный раствор хлорида
сурьмы (V) _в тетрахлорметане. Оловоорганические стабилизаторы
можно отличить от остальных по характерному голубому
окрашиванию этих соединений после опрыскивания пластинок
0,1 %-ным раствором катехолового фиолетового в 95 %-ном
этиловом спирте [252]. Окраска появляется после 10-минутного
облучения пластинки в УФ-свете и повторного опрыскивания
тем же реагентом.
Перечисленные проявители можно использовать как для
детекции стабилизаторов на пластинке, так и для
количественного определения. Чтобы исключить неравномерность
разбрызгивания проявителя, анализ проводят методом сравнения со
стандартным раствором, замеряя оптическую плотность
нанесенного на пластинку известного количества стандарта после
экстракции его с пластинки или сканируя пятно стандарта
денситометром.
Для определения стабилизаторов используется и колоночная
жидкостная хроматография. С 1970-х годов, когда технология
получения сорбентов достигла высокого уровня, в основном
применяется метод высокоэффективной жидкостной
хроматографии (ВЭЖХ), который позволяет быстро и с высокой
точностью проводить количественное определение стабилизаторов.
В 4—5 раз можно сократить длительность анализа при
улучшении эффективности хроматографического разделения,
используя мелкодисперсные набивки (3—5 нм) для колонок и
малоинерционные детекторы при увеличении скорости потока
подвижной фазы до 2—5 мл/мин. Более высокая
чувствительность ВЭЖХ исключает в ряде случаев необходимость
предварительного концентрирования экстрактов.
В табл. IV. 3 приведены условия определения стабилизаторов
методами ВЭЖХ. При разделении соединений, близких по
полярности, применение элюента постоянного состава (изократи-
ческая система), дает большую точность по сравнению с
градиентным элюированием. Для разделения веществ, сильно
различающихся по полярности, обычно применяют градиентную
элюцию.
Авторы приведенных в табл. IV. 3 работ отмечают ряд
практических моментов, важных для проведения анализа методом
ВЭЖХ. Например, при использовании в подвижной фазе
градиента хлороформа небольшие количества этилового спирта,
присутствующего в растворителе в качестве консерванта, могут
изменять активность поверхности силикагеля (д.-порасила).
ВЭЖХ с обращаемой фазой требует предварительного
испарения растворителя из экстракта, поскольку введение фенолов в
колонку в таком растворителе для экстракции, как хлороформ,
248
Таблица IV.3. Условия определения стабилизаторов
методом ВЭЖХ
Стационарная фаза
Сили-
кагель
(размер
частиц
5 нм)
Лихро-
сорб-
NHJ,
(размер
частиц
10 нм)
Зорбакс-
сил
ц-Бонда-
пак
ц-Порасил
ц-Порасил
ц-Пора-
сил,
(размер
частиц
10 нм)
* Предк
Размеры
колонки.
мм
250X4
250X4
250X4
600X3,9
300X3,9
300X3,9
300X3,9
олоика 20>
Подвижная фаза,
состав, скорость
Гексан 3 мин;
градиент до 30 %
метиленхлорида
в гексане
(2%/мин)
Ацетонитрил: во-
да=99,5:0,5;
0,2 мл/мин
Гексан с 0,2 %
или 25 %
метиленхлорида;
градиент от 0,9 %
до 70 %
метиленхлорида
(10%/мин);
1 мл/мин
Метиловый
спирт: вода:
ТГФ=63:7:30;
0,2 мл/мин
Градиент от
гептана к метилен-
хлориду
1,2-Дихлорэтан
Метиленхлорид;
градиент от
гептана к метилен-
хлориду;
2 мл/мин
<4 мм, заполненная ли
Детектор
УФ
(242 нм)
УФ
(208 нм)
УФ и
рефрак-
томет-
ричес-
кий
—
УФ
(283 нм)
УФ
(230 нм)
УФ
(280 нм)
Разделяемые
стабилизаторы
Ионол, ирганокс
1010, ирганокс
1076, сантонокс R,
ионокс 330
Тинувин 770, хо-
ставин
Ионол, топанол
СА, сантонокс R,
дистеарилтио-
дипропионат,
сантовайт
Ионол, ирганокс
1076, тинувин 327,
циасорб 531
Полигард, нонил-
фенол, ионол
Дистеарилтиоди-
пропионат, дилау-
рилтиодипропио-
нат
Ионол, ирганокс,
1010, ирганокс
1076, сантонокс,
топанол СА, этил
лжитель-
разделе-
ин
Про до
ность ;
ния, м
30
5
15
120
40
15
330, гудрайт 3114
хропрепом-ЫНг с размером частиц 25 —
1
й£
Литер;
нсточн
[246]
[248]
[251]
[243]
[266]
[247]
[256]
40 нм.
может вызвать появление разорванных пиков [254] .-При
работе на ц-порасиле с декалиновыми экстрактами полиолефинов
возможно увеличение давления из-за присутствующей в
экстракте олигомерной фракции. Олигомеры можно удалить из
раствора подходящим растворителем. Чтобы не уменьшать
концентрацию стабилизаторов в экстракте за счет разбавления
осадителем и не прибегать затем к концентрированию раствора,
а также для удлинения срока службы колонки можно
применять предколонку. Обычные ее размеры 300 X 4 мм. Полимер
249
и олигомеры удаляются в этом случае в результате сильного
взаимодействия со стационарной фазой предколонки. Колонки
заполняют, например, корасилом Ci8 с размером частиц 30—
40 нм [256].
Некоторые авторы [261, 266] отмечают специфическое
взаимодействие между антиоксидантами и веществами,
присутствующими в растворителях. Так, при определении полигарда [266]
наблюдалась зависимость высоты пика стабилизатора от
качества применяемого в подвижной фазе гептана. Поэтому для
приготовления и при анализе растворов стандартов следует
использовать растворитель той же партии, что и для
анализируемых проб. Во всех случаях чистоте растворителя в ВЭЖХ
придается большее значение, чем в спектральных методах
анализа. Используемые в качестве элюентов растворители перед
проведением анализа пропускают через микропористые
фильтры [256, 266], подвергают специальной очистке.
Некоторые авторы для определения стабилизаторов в
экстрактах предпочитают гель-проникающую хроматографию.
Обычно для хроматографирования из концентрированного экстракта
приготавливают 2—5 %-ный раствор. Применение двух
детекторов, рефрактометрического п УФ-, позволяет анализировать
смеси алифатических и ароматических соединений, имеющих
близкие объемы элюции. Поскольку молекулярная масса
веществ, обычно применяемых в качестве стабилизаторов,
находится в интервале 200—800, то для фракционирования
выбирают гели с оптимальными разделяющими свойствами в этом
интервале молекулярных масс.
При разделении веществ близкой молекулярной массы на
обычных мягких гелях трудно получить хорошее разрешение за
короткое время, поэтому обычно применяют длинные колонки
в системах рецикла и серии колонок [245, 250, 251]. Как и в
случае ВЭЖХ, применение мелкосферических частиц
полужесткого геля намного увеличивает разрешение.
Несмотря на ограничения газожидкостной хроматографии,
возможность определения ряда стабилизаторов этим методом
показана в работах [257, 262]. Основными особенностями
процесса хроматографирования стабилизаторов является
применение высоких температур, больших скоростей газа-носителя и
малого количества нанесенной неподвижной фазы с целью
снижения времен удерживания определяемых стабилизаторов.
Во многих работах количественное определение проводится
на колонках с SE-30. Максимальная рабочая температура этой
фазы составляет примерно 350 °С, что на 50°С выше, чем у
других промышленных жидких фаз. На медной колонке
размерами 1500x6 мм, заполненной хромосорбом (размер частиц
0,25—-0,35 мм) с 10 % нанесенной на него жидкой неподвижной
фазы SE-30, удалось [257] без разложения проанализировать
13 соединений, применяемых для стабилизации полипропилена.
Анализ проводился при программировании температуры от 100
250
до 340°С со скоростью 15°С/мин с удерживанием
температуры на верхнем пределе в течение 1 ч; в качестве детектора
использовался ДИП. В таких условиях анализировался ксилоль-
ный экстракт полипропилена с целью определения в нем ионо-
ла и 4-додецилгидрокси-2-гидроксибензофенона.
Один из вариантов определения стабилизаторов методом
ГЖХ—превращение их в легкохроматографируемые продукты.
В работе [249] дилаурил-р,р'-тиодипропионат (ДЛТДП) гид-
ролизовали до лаурилового спирта. Для анализа
полипропилена, содержащего 0,02—0,3 % ДЛТДП, необходим 1 г образца.
Условия экстракции приведены в табл. IV. 1. ДЛТДП отделяли
от продуктов его окисления и других добавок методом ТСХ на
силнкагеле, экстрагировали с сорбента, гидролизовали 5 н.
раствором КОН в течение 30 мин, продукты гидролиза
экстрагировали хлороформом и хроматографировали при 165 °С на
стеклянной колонке, заполненной хромосорбом с 1,5 % фтор-
силиконового масла FS-1265, применяя в качестве
газа-носителя аргон. Введение дополнительной стадии делает и без того
многостадийный анализ более трудоемким. Такие методы редко
используются для аналитического контроля.
Чтобы судить с помощью описанного варианта анализа об
изменении концентрации стабилизатора в процессе
производства полимера или при его старении, необходимо уточнить
исходную концентрацию стабилизатора путем анализа
конкретной партии, введенной в полимер. Это необходимо потому,
что стабилизатор содержит примеси других эфиров тиодипропи-
оновой кислоты, о чем свидетельствует более низкий выход
лаурилового спирта при гидролизе промышленных образцов
ДЛТДП по сравнению с очищенным стандартом, для которого
выход соответствует теоретическому (две молекулы
лаурилового спирта на молекулу ДЛТДП).
Определение полигарда. В последние годы повысился
интерес к жидким стабилизаторам вследствие их более легкого
дозирования. К ним относятся фосфорорганические соединения
типа полигарда
(c9h,9-Q-o-)3P,
который вводят для предотвращения деструкции полимера в
процессе его переработки.
Методы определения полигарда основаны на определении
фосфора [261, 267, с. 38, 268], измерении поглощения в УФ-
свете [269], переводе его в фенолят-ион в щелочной среде
[270], образовании окрашенных продуктов [271]. Они
включают разделение с применением ГПХ [261], ТСХ (British
Standard 2782, part 4, method 434A), ВЭЖХ [266] и ГЖХ [272]
рис. IV. 3).
Наибольшее распространение получил деструктивный метод,
основанный на определении фосфора, при котором проводят
25!
Рис. IV.3. Хроматограмма разделения смеси аити-
оксидантов методом ГЖХ (колонка 750 X 6 мм
с 10 % Е-30 иа диатомите CQ; температура
колонки 250 °С, температура детектора 380 °С, скорость
газа-иосителя аргона 45 мл/мии):
/— топанол ОС; 2, 7 — полпгард; 3 — нргаиокс 1010;
4— тииувии Р; 5, 6 — нонокс ДСР; 8 — тинувин 326;
9 — тинувин 327; 10 — сантонокс.
мокрое сжигание полимера или
предварительно отделяют
полимерную часть методом ГПХ. Для
минерализации полимера используют
, смеси концентрированных кислот
15 12 9 s з о (азотной и хлорной, серной и азот-
время,мин ной) или концентрированную
серную кислоту в присутствии перокси-
да водорода или персульфата аммония. Для ускорения процесса
минерализации и исключения при этом потерь фосфора за счет
образования летучих продуктов типа РН3 был предложен
вариант проведения минерализации в два этапа [268]: серной
кислотой с небольшим количеством азотной, а затем с
добавлением пероксида водорода. Полигард, выделенный методом ГПХ,
окисляют серной кислотой в присутствии пероксида водорода
[261]. Содержащийся в полигарде фосфор в результате
окисления переходит в фосфат, количественное определение
которого во всех работах проводят спектрофотометрическим
методом.
Определение фосфора в полимере дает суммарное
содержание полигарда и его фосфорсодержащих продуктов
разложения, поэтому данный метод можно применять для контроля
загрузки стабилизатора. Недостатком определения полигарда по
фосфору является трудоемкость стадии минерализации и
использование концентрированных кипящих кислот.
Более привлекательным является выделение полигарда
методом экстракции. В качестве экстрагента для извлечения его
из стирол-бутадиенового сополимера использовали изооктан
[270] (трехкратная экстракция 1,5 г полимера 80 мл
растворителя)— эффективный растворитель полигарда, мало
растворяющий при этом сополимер. Из ударопрочного полистирола
полпгард экстрагировали этиловым спиртом [269] кипячением 1 г
полимера с 15 мл спирта в колбе с обратным холодильником
в течение 1 ч, а затем еще дважды по 30 мин и уменьшением
объема экстрагента для повторных экстракций до 10 мл.
Применение ультразвука на стадии выделения позволяет сократить
продолжительность экстракции в 3 раза.
Количественное определение полигарда в экстрактах
проводилось одним из перечисленных выше методов. Использование
батохромного сдвига в спектре фенола устраняет влияние на
количественное определение примеси нонилфенола,
присутствующего в полимере. В качестве щелочного реагента в работе
[270] использовали 0,2 н. раствор гидроксида тетрабутиламмо-
252
ния, получаемый пропусканием через анионит в ОН-форме
спиртового раствора тетрабутиламмонинподида. 3 мл
щелочного раствора добавляли к 25 мл экстракта, объем раствора
доводили изооктапом до 50 мл и замеряли поглощение раствора
при 299 им относительно экстракта, к которому вместо
раствора гидроксида тетрабутиламмоння добавляли 3 мл этилового
спирта.
Раздельное определение полигарда и нонилфенола можно
провести хроматографическим методом. В случае бутадиен-сти-
рольных сополимеров хроматографировали [266] 25 мкл 4 %-
ного раствора полимера в циклогексане на колонке размерами
300 X 3,9 мм, заполненной ц-порасилом, с применением
градиента от 100 % гептана к 100 % метиленхлорида и УФ-детекто-
ра (280 нм). Присутствие в некоторых образцах ионола не
влияло на точность определения.
Очевидно, что наибольшую информацию о превращениях
полигарда в полимере может дать хроматографпческнй метод, в
частности ТСХ. Ниже приведены значения Rf полигарда при
хроматографировании в тонком слое силикагеля с
использованием различных систем растворителей [252, 263, 264]:
*t
Бензол : этилацетат : ацетон = 0,52; 0,61
= 100:5:2
Хлороформ : гексан = 2:1 0,53
Бутиловый спирт: уксусная 1,0
кислота = 97 : 3
Бензол 0,63; 0,52; 0,48; 0,39; 0,22; 0,12
0,92; 0,77; 0,63; 0,48; 0,26
Толуол : петролейный эфир = 0,53; 0,35; 0,07
= 1:1
Различное число примесей объясняется, вероятно, не только
различным поведением полигарда в разных системах
растворителей, но и различием образцов, анализируемых авторами, а
также присутствием следов воды в некоторых растворителях.
IV. 1.2. Пластификаторы
Пластификаторы можно определять без отделения
полимерной части и с предварительной экстракцией пластификаторов.
Спектральный анализ пленки полимера для этой цели проводят
редко, поскольку соединения, относящиеся к пластификаторам,
обладают интенсивными полосами поглощения, что в сочетании
с их большим содержанием в полимере (до десятков
процентов) требует приготовления для анализа пленок очень малой
толщины, что весьма затруднительно. Поэтому для прямого
анализа обычно используют растворы полимеров.
IV. 1.2.1. Анализ растворов полимеров
Необходимым условием проведения прямого спектрального
анализа раствора полимера является прозрачность
применяемого растворителя и полимера в аналитической области спектра.
253
При определении сложных эфиров, наиболее часто
используемых в качестве пластификаторов, аналитической полосой в
ИК-спектрах обычно является полоса поглощения валентных
колебаний связи С=0 (1725 — 1735 см-1) сложноэфирной
группы. Эту полосу можно использовать, например, для
определения пластификатора в растворах поливинилхлорида, поливи-
нилбутираля, но нельзя—в растворах поливинилацетата и по-
лиметилметакрилата, содержащих карбонильные группы. Для
фосфатов аналитической полосой обычно служит интенсивная
полоса поглощения, относящаяся к валентным колебаниям
связи Р = 0 (1270—1300 см-1) или Р—О—С. Количественное
определение пластификатора в растворе полимера возможно не по
измерению абсолютного поглощения в максимуме
характеристической полосы, а методом внутреннего стандарта, когда в
качестве эталона используется полоса поглощения полимера.
Преимущество этого метода заключается в уменьшении случайных
инструментальных погрешностей.
УФ-область спектра может быть использована для
определения пластификаторов, содержащих в молекуле бензольное
кольцо. Это, прежде всего, широко применяемые фталаты, а также
арилфосфаты. В УФ-области поглощения фталатов и арилфос-
фатов прозрачен поливпнилацетат, полиметилметакрилат, поли-
винилхлорид, поливинилбутираль, полиэтилен. Выбор
органических растворителей, прозрачных в аналитической области
спектра, в данном случае богаче по сравнению с ИК-спектроско-
пией.
Калибровочные графики для количественного определения
пластификаторов спектральными методами следует строить по
растворам пластификаторов, содержащим полимер.
В некоторых случаях, когда известен состав полимерной
композиции, для определения пластификаторов в ее растворе
можно применить спектрофотометрнческий метод, основанный
на предварительном проведении цветной реакции (см.
n.IV.3.3).
Находит применение также метод газожидкостной
хроматографии. При анализе растворов полимеров, содержащих
пластификаторы, возможно термическое разложение полимера в хро-
матографической колонке. Поэтому рекомендуется применять
форколонку или предварительно высаждать полимер нз его
раствора.
Наибольшую сложность для газохроматографического
анализа, как и для любого другого метода, представляют
неоднородные пластификаторы, получающиеся на основе смешанной
спиртовой фракции и содержащие иногда до 10 компонентов.
В работе [273] изучали газохроматографическое поведение
такого рода пластификаторов [диизооктилфталата, диизодецил-
фталата, трис(фенилизопропил)фенилфосфата и др.] на хромо-
сорбе W с различным количеством нанесенной на него
стационарной фазы, в качестве которой применяли SE-30, OV-17,
254
OV-25, ПЭГ, поливинилпирролидон, поли(неопентилгликоль)-
адипинат и др. Был предложен метод количественного
экспресс-анализа образцов поливинилхлорида, содержащих один из
указанных пластификаторов: на колонке с 10 % OV-25 хромато-
графировали 5%-ные растворы полимера в тетрагидрофуране;
температура колонки 260 °С, продолжительность анализа 1 ч.
К числу нехроматографируемых пластификаторов относятся
трикрезилфосфат и «немигрирующие» пластификаторы типа
полиэфиров.
От недостатков, присущих газожидкостной хроматографии
(необходимость применения высоких температур, возможность
разложения полимера), свободен метод ГПХ. Он наиболее
перспективен для определения пластификаторов в растворах
полимеров, особенно при содержании в полимере только одного
пластификатора. Этим методом можно определять
пластификаторы в полистироле, используя УФ-детектор. В работе [274]
методом внутреннего стандарта определяли триэтиленгликоль-
дибензоат и трициклогексилцитрат в их смеси с полистиролом
при содержании 5—30 %. 7—10%-ные растворы полимера хро-
матографировали на последовательных колонках с
наполнителем разной пористости.
Методом ГПХ легче определять в полимерах неоднородные
пластификаторы. Если количественное определение таких
пластификаторов в газожидкостной и жидкостной адсорбционной
хроматографии затруднено тем, что они дают мультиплеты, то
в гель-проникающей хроматографии эти пластификаторы
выходят одним пиком. Данным методом в полистироле определяли
[275] полнхлорированные бифенилы, используемые в качестве
вторичных пластификаторов полимеров.
IV. 1.2.2. Анализ экстрактов полимеров
Определение пластификаторов в экстрактах полимеров более
распространено вследствие ограничений прямого анализа
растворов полимеров.
Наиболее подробно описана экстракция пластификаторов
из поливинилхлорида. Наилучшим растворителем является ди-
этиловый эфир, пригодный для экстракции пластификаторов с
молекулярной массой 1000 и меньше. В некоторых случаях для
полноты выделения пластификатора требуется дополнительная
экстракция другим растворителем, а чаще смесью
растворителей с низкой температурой кипения, в которой поливинилхлорид
хорошо набухает. В этом случае из полимера экстрагируются
и полиэфирные пластификаторы. Из таких смесей следует
прежде всего назвать азеотропы метилового спирта с
минимальной температурой кипения — с тетрахлорметаном,
хлороформом и ацетоном, содержащие на 1 ч. метилового спирта
соответственно 2, 4 и 7 ч. указанных растворителей [240, с. 69].
В применении к сополимерам винилхлорида недостаток Этих
255
4 Рис. IV.4. Хроматограмма разделения смеси
фталатов в условиях ГПХ высокого
разрешения (колонка 600 X 8 мм, заполненная полисти-
рольным гелем с номинальной пористостью
10 нм; температура колонки 45 °С; давление
9 МПа: скорость элюента тетрагидрофураиа
2 мл/мин, объем пробы 50 мкл, концентрация
раствора 20 мг/мл; рефрактометрический
детектор):
/—диоктплфталат; 2 — дибутнлфталат; 3 — ди-
этилфталат; 4 — диметилфталат.
I 1 I 1
10 д в 7 систем в том, что они заметно
Время,мин растворяют полимер. Хорошим
экстрагентом пластификаторов
для выделения их из поливинилхлорида является также тетра-
хлорметан, однако он также вызывает частичную экстракцию
низкомолекулярного поливинилхлорида, солей кадмия и бария.
Присутствие полимера в экстракте может быть обнаружено
добавлением к нему метилового спирта, а низкомолекулярного
поливинилхлорида — анализом на хлор, если последний не
содержится в молекуле экстрагента.
Применяемое соотношение полимер : растворитель
варьируется в интервале 1 : 10 до 1 : 25. Для пластификаторов обычно
требуется более длительная экстракция, чем для
стабилизаторов. Поскольку содержание пластификаторов в полимере
довольно высоко, то экстракцию можно проводить с точностью
до 0,5%.
При устранении попадания в экстракт полимера и солей
можно определять пластификатор в экстракте
гравиметрическим методом без учета захваченных в экстракт стабилизаторов
и микропримесей. Из экстракта испаряют растворитель,
высушивают остаток до постоянной массы и взвешивают. В
настоящее время этот метод мало применим вследствие использования
смесей пластификаторов и сложности полимерных композиций
вообще.
Если не существует ограничений, экстракт анализируют
спектральными методами (см. раздел IV. 1.2.1), в остальных
случаях-—хроматографическими методами. Наиболее полного
разделения можно достичь, применяя к экстракту
высокоэффективные виды хроматографии. Гель-проникающая
хроматография высокого разрешения позволяет, например, на колонке
размерами 60 X 8 мм, заполненной полистирольным гелем
(размер частиц 5 нм), всего за 10 мин проанализировать смесь
фталатов. Этим методом разделяли смесь фталатов, часто
используемых в качестве пластификаторов : диоктилфталата, ди-
бутил-, диэтил- и диметилтерефталатов (рис. IV. 4) [276].
Анализ смесей пластификаторов методом тонкослойной
хроматографии не столь экспрессен и более трудоемок, но в
отличие от высокоэффективной хроматографии он доступен любой
лаборатории. Схема анализа не отличается от определения
стабилизаторов в экстрактах полимеров, исключая не нужную в
данном случае стадию концентрирования экстракта.
МК
256
Методом тонкослойной хроматографии определяли
пластификаторы в пленках поливиннлхлорида пищевого назначения
[277]. Пластификаторы экстрагировали гексаном в аппарате
Сокслета в течение 6 ч, при этом 0,2 г измельченного
полимера заливали 20 мл растворителя. 0,1 мл полученного экстракта
наносили на закрепленный слой силикагеля в виде пятна
диаметром не более 5 мм. Разделение экстракта проводили в
бензоле, толуоле, гептане или хлороформе в зависимости от
состава анализируемого экстракта. Затем пластинку высушивали в
течение 5 мин в сушильном шкафу при 130—150 °С и
проявляли разделенные пластификаторы, опрыскивая пластинку
проявляющим реагентом.
Сложные эфиры гидролизовали концентрированной серной
кислотой до высших спиртов с последующей их дегидратацией
в ненасыщенные соединения, которые с я-диметиламинобензаль-
дегидом образуют окрашенные продукты реакции. Для этого
пластинку опрыскивали 0,5 %-ным раствором и-диметиламино-
бензальдегида в смеси концентрированной серной кислоты с
диэтиловым эфиром (1:1) и снова выдерживали в сушильном
шкафу при 130—150 °С 5—10 мин. На сероватом фоне
появлялись красно-бурые пятна пластификаторов. Количественное
определение проводили по измерению площади пятен. При
использовании денситометров погрешность определения
снижается с 10 до 5 %. Чувствительность такого проявителя по
отношению к сложным эфирам 0,5—4 мкг.
Подробнее о хроматографическом поведении
пластификаторов в тонком слое сорбента см. [278, с. 464].
IV. 1.3. Наполнители и порообразователи
Условно методы количественного определения наполнителей
в полимерных материалах можно разделить на деструктивные
и недеструктивные, подразумевая под деструктивными методы
с предварительным озолением полимера.
К недеструктивным методам определения неорганических
наполнителей относится прямой анализ пленок полимера
спектральным методом. Большинство оксидов, используемых в
качестве наполнителей, обладают характерным поглощением в
ИК-области спектра. Наличие характеристических интенсивных
полос поглощения позволяет проводить количественное
определение даже в смеси наполнителей [279], используя
калибровочные графики. Однако прямой анализ пленок полимера
возможен практически только для полиэтилена, обладающего
большой прозрачностью в ИК-области спектра [39, с. 147].
Аналитическими полосами для количественного определения
наполнителей могут служить, например, узкая изолированная
полоса карбоната при 880 см-1 для определения мела СаСОз,
полоса 1020 см-1 или 670 см-1 — для определения талька
Mg3[Si4Oio] (ОН) 2, 1100 см-1 — для диоксида кремния Si02,
9 За к. 632
257
1000 см-1—для слюды. Для определения диоксида титана ТЮг
(рутила) можно воспользоваться его широкой полосой 500—
700 см-1.
Для остальных полимеров, а также при наличии
органических добавок, осложняющих спектральный анализ
наполнителей, определение проводят после выделения наполнителей. Для
проведения количественного анализа достаточно 0,3—2 мг
выделенного наполнителя. Из него приготавливают таблетку с
КВг и записывают спектр в области 1500—650 см-1.
Вопросы выделения минеральных наполнителей довольно
подробно изложены в гл. I. Отделение полимерной части
необходимо практически для всех остальных вариантов
количественного определения наполнителей.
Самым простым методом количественного определения
выделенного неорганического наполнителя является
гравиметрический метод, который естественно может быть применен только
тогда, когда можно ручаться за индивидуальность выделенного
наполнителя.
В большинстве случаев гравиметрическое определение
наполнителя сводится к сжиганию полимера и определению его
зольности (ГОСТ 15973—82) в пересчете на образующиеся при
этом оксиды. Примером гравиметрического определения
наполнителя в полимере с включением стадии его выделения может
служить определение диоксида титана в самозатухающих
композициях полиэтилена [267, с. 45]. Для анализа навеску
измельченного полимера (1 г) растворяют в 50 мл горячего
ксилола. После растворения добавляют 10 мл концентрированной
соляной кислоты для извлечения Sb203, раствор перемешивают
и добавляют 20 мл воды. Выпавший полимер отфильтровывают
на воронке Бюхнера, осадок последовательно промывают
подогретой кислотой и 50 мл дистиллированной воды, высушивают,
озоляют без воспламенения и прокаливают в фарфоровом тигле
при 500 °С в муфельной печи до постоянной массы.
При сложном составе неорганической части полимерной
композиции для количественного определения неорганического
наполнителя можно использовать метод эмиссионного
спектрального анализа золы полимера.
Конечное определение можно вести также ионометриче-
скнм методом. Так, ионометрическим методом определяли
содержание фторсодержащего наполнителя (апатита) в
полиэтилене (см. п. IV. 3.4) [73].
Термогравиметрический метод определения наполнителей,
разложение которых сопровождается характерным экзо- или
эндотермическим эффектом, также нашел применение для
количественного анализа. Этот метод не требует обязательного
предварительного разделения минеральной части полимера.
Данным методом в полимере определяли, например, каолин
[280]. Для этого 600 мг золы, полученной из полимера,
помещали в платиновый тигель и записывали дериватограмму. Со-
258
держание каолина в полимере рассчитывали по градуировочно-
му графику по высоте пика на кривой ДТА при 950—1000°С.
К широко применяемым наполнителям относится
технический углерод. Способ определения технического углерода в
полимере разработан только для полиэтилена (см. раздел 1.2.2).
Поскольку технический углерод выполняет и функцию
стабилизатора, то на его количественном определении,
проводимом гравиметрическим методом, следует остановиться
подробнее.
Основной проблемой при гравиметрическом определении
технического углерода является захват его частиц продуктами
деструкции полиэтилена. Особенно велик вклад этой ошибки при
малом содержании технического углерода в полимере. Для
подбора условий количественного определения технического
углерода при малом его содержании в полимере было исследовано
влияние временного фактора и температурных условий на
деструкцию полиэтилена в инертной атмосфере [71]. Было
показано, что время, за которое происходит полная деструкция
полимера, зависит от температуры. При этом при температуре
выше 550 °С происходит слишком быстрое удаление
фрагментов деструкции полиэтилена и наблюдается унос частиц
технического углерода; при 700°С технический углерод
начинает взаимодействовать с примесями кислорода и воды в
инертном газе. Таким образом термическая деструкция
полиэтилена при температуре выше 550 °С, по данным автора
работы [71], происходит с потерей некоторого количества
введенного в полимер технического углерода. Была предложена
методика, при которой навеску полиэтилена, содержащую 1—30 мг
технического углерода, помещают в предварительно
прокаленной кварцевой лодочке в центральную часть трубчатой
электропечи, перед ней в зоне нагрева располагают катушку из
медной проволоки и трубку выдерживают при 500 =F 10 °С в
течение 30 мин, продувая трубку азотом с постоянной скоростью
(второй конец трубки остается при этом открытым). После
этого лодочку помещают в эксикатор и через 30 мин взвешивают.
Для определения зольности полимера пробу дожигают при
900°С в присутствии кислорода воздуха. Зольность полимера
можно не учитывать при расчете результата анализа, если
она составляет менее 2 % от содержания технического
углерода.
В качестве вспенивающего агента (порообразователя) для
вспенивающегося полистирола обычно используют пентановую
фракцию углеводородов. Количественное определение
порообразователя во вспенивающемся полистироле проводят газохрома-
тографическим методом, причем наиболее перспективным
следует считать парофазный анализ, поскольку выделение
порообразователя из полимера происходит очень легко при
незначительном повышении температуры.
9*
259
IV. 1.4. Антипирены
В качестве антипиренов наиболее часто используют
галоген- и фосфорсодержащие органические соединения, которые
иногда применяют в сочетании с оксидом сурьмы. Поэтому
основными методами определения огнегасящих добавок являются
методы, основанные на определении гетероатомов, входящих в
органическую молекулу, и сурьмы, вводимой в виде SD2O3 [281].
Предварительной стадией анализа является минерализация
полимера за исключением случаев поверхностного нанесения
антипиренов, когда выделение добавки может быть
осуществлено смыванием антипирена с поверхности полимера подходящим
растворителем.
Наиболее широко в промышленности применяют бромсодер-
жащие антипирены, в молекуле которых присутствует 60—
80 % брома. Одним из достаточно экспрессных методов выде-
ленения брома для дальнейшего аналитического определения
является сжигание полимера в колбе с кислородом в
присутствии платинового катализатора — так называемый метод Шё-
нигера. Основной проблемой при этом становится достижение
полноты сжигания анализируемых образцов, поскольку при
сжигании в колбе с кислородом происходит вынос обугленных
частиц полимера (сажи) из зоны горения. Наиболее склонны
к сажеобразованию полимеры, при пиролизе которых
образуются ароматические углеводороды. Это, в первую очередь, по-
листирольные пластики, которые, например, в США
составляют 80 % всех огнестойких пластмасс. Оптическими методами
доказано [282], что в результате столкновения сажевых частиц
в пламени происходит образование агломератов, которые
скапливаясь в верхней части пламени, понижают его температуру,
что в свою очередь замедляет процесс горения полистирола и
способствует выносу сажи. Введенные в полимерную композицию
антипирены промотируют этот эффект.
В условиях сжигания полимера в колбе с кислородом, т. е.
в замкнутом пространстве, для подавления сажеобразования
при горении полимеров наиболее целесообразно введение
химических добавок. В работе [184] методом совмещенного
термического анализа ДТА и ТГА исследовалось влияние
антисажевых реагентов на термическое разложение образцов
полистирола, содержащего в качестве антипирена гексабромциклододе-
кан, при сгорании которого отмечалось наиболее обильное
образование сажи.
Введение антипирена в молекулу полистирола уменьшает
эндотермических эффект при разложении полимера.
Добавление окислителей — солей азотной кислоты KN03, NaN03,
NH4NO3 и молибдата аммония — способствует возрастанию
эндотермического эффекта разложения самозатухающего
полистирола. Самым эффективным из испытанных добавок оказался
нитрат натрия, действие которого связывают с выделением при
260
его разложении под действием высокой температуры
относительно больших количеств 02 и N0. Выделяющиеся в газовую
фазу окислители подавляют сажеобразование, что приводит к
повышению температуры пламени и более быстрому сгоранию
образца.
На качество сгорания полимера в колбе с кислородом
влияет способ введения неорганической соли: при добавлении ее к
навеске полимера, как правило, остается зола, которая может
содержать карбонизированные остатки полимера. Лучший
результат дает пропитка беззольного фильтра, в который
помещается навеска анализируемого полимера, раствором соли.
Наиболее эффективным по антисажевому действию является
2 %-ный раствор нитрата натрия.
После поглощения продуктов сгорания раствором щелочи с
добавлением пероксида водорода определяют в растворе
бромид любым доступным методом, чаще всего методом потенцио-
метрического титрования нитратом серебра. Измерительным
электродом служит сульфидсеребряный или бромселективный
электрод. Наблюдаемый скачок потенциала при титровании
увеличивает применение солевого мостика, заполненного
электролитом типа 0,1 н. раствора KNOs или NH4N03.
IV. 1.5. Пер оксиды
Устойчивые пероксиды применяют как синергические
добавки к бром- и фосфорорганическим антипиренам, а также как
источник образования свободных радикалов, ускоряющих
скорость газификации и плавления полимера, предназначенного
для изготовления моделей для литья металлов. Поскольку в
полимере могут также присутствовать остатки пероксидов,
используемых в качестве инициаторов полимеризации,
применение классического иодометрического определения пероксидов,
основанного на определении «активного кислорода»,
оказывается в данном случае малоэффективным, так как не позволяет
дифференцировать пероксиды. Побочное действие на реакцию
могут оказывать и другие примеси и добавки полимера.
В ряде случаев можно применять полярографический
анализ раствора или экстракта полимера [240, с. 52]. Наиболее
хорошо изучено восстановление пероксидов на ртутном
капающем электроде. Основные классы пероксидов обладают
способностью восстанавливаться на нем, давая полярографические
волны в разных областях потенциалов. Применение
вращающихся дисковых электродов расширяет возможности
полярографического анализа пероксидов.
Спектральные методы применяются в основном при
исследовании процессов разложения пероксидов.
Наиболее достоверные результаты дает введение в анализ
разделяющей стадии. Как и для стабилизаторов, идеальной для
этого класса добавок является жидкостная хроматография.
261
Предварительно пероксид отделяют от полимерной части путем
экстрагирования пероксида или высаждения полимера из
раствора. Например, в работе [283] раствор полистирола в ме-
тиленхлориде разбавляли в 100 раз смесью ацетонитрила с
дистиллированной водой (75:25). Было показано, что высаждае-
мый полимер захватывал при этом только незначительное
количество пероксида. 100 мкл полученного раствора хроматогра-
фпровали на колонке размерами 250 X 4,5 мм, заполненной
зорбаксом Ci8, применяя в качестве подвижной фазы ту же
смесь растворителей, подаваемую со скоростью 1 мл/мин, и
используя УФ-детектор. Предел обнаружения методом ВЭЖХ
составляет 0,001% пероксида в полимере [283], что на
порядок выше чувствительности метода ТСХ, применение которого
будет проиллюстрировано ниже на примере определения
пероксида дикумнла в полистироле.
Подбор экстрагента для выделения пероксида из полимера
проводится обычным способом с контролем термической
стойкости и полноты экстракции пероксида из полимера.
Эффективность экстрагента для одного и того же пероксида
зависит не только от класса полимера, но и от его марки. В
работе [284] для выделения пероксида дикумила из полистирола
рекомендована 8-кратная экстракция 25 г полимера порциями
ацетона по 50 мл при комнатной температуре путем
встряхивания навески образца с растворителем в течение 5 мин.
Применение этого способа экстракции к выделению пероксида из
вспенивающегося полистирола [153, с. 40] оказалось
неэффективным: экстракция при комнатной температуре протекает
очень медленно. При проведении экстракции ацетоном в
аппарате Сокслета в раствор частично переходит полимер, который
осложняет хроматографическое разделение экстракта. Из
большого числа низкокипящих растворителей для экстракции
пероксида дикумила из образцов вспенивающегося полистирола
марок ПСВ-С, ПСВ-СВ и ПСВ-Л в наибольшей степени
подходит углеводородный растворитель, который обеспечивает
идеальные условия для экстракции при проведении ее в аппарате
Сокслета: полимер в нем не растворяется и хорошо набухает.
В качестве такого растворителя можно использовать гексан и
петролейный эфир, причем углеводородный экстрагент
обеспечивает полноту экстракции пероксида из бисера полимера с
диаметром до 5 мм за 2 ч без предварительного измельчения
полимера.
При экстракции пероксида дикумила из сополимера
стирола с акрилонитрилом (САН), получаемого методом
суспензионной полимеризации, углеводородный растворитель оказывается
неэффективным. И только добавление к нему одного из
растворителей, в котором полимер растворим (бензол, толуол, ксилол,
метилэтилкетон, диметилформамид, хлороформ, ацетон),
позволяет достичь цели: полимер в смеси растворителей набухает,
что способствует экстракции. В качестве такой добавки наибо-
262
Рис. IV. 5. Кинетика экстракции пероксида sc qj
ликумила из САН. §j^ '
<и „
лее удобным для последующих ^| 0„
стадий анализа является ацетон. §|^ '
Было найдено, что для САН *^§
оптимальным является примене- ^^ о,1
ние смеси петролейного эфира и 3^
ацетона в сотношении 1 :3 для о
бисера и 1 :4 для гранул. Кине- $ ' ? ' 2~
тика экстракции пероксида ди- время, ч
кумила из этого полимера
приведена на рис. IV. 5. Полнота экстракции пероксида дикумила,
экстрагируемого из сополимера в течение 2 ч, подтверждалась
повторным экстрагированием того же образца полимера в
аппарате Сокслета еще в течение 2 ч. В повторном экстракте перок-
сид не был обнаружен.
При применении в качестве разделяющей стадии ТСХ
экстракт предварительно концентрируют. Для этого растворитель
полностью удаляют при разрежении, создаваемом
водоструйным насосом, а сухой экстракт повторно растворяют в
фиксированном объеме растворителя. Минимальный объем, в который
можно без потерь перевести экстракт, равен 0,2 мл. На
пластинку наносят от 5 до 200 мкл сконцентрированного экстракта в
виде полоски длиной от 1 до 15 см [284], минимальное
содержание пероксида в которой в случае пероксида дикумила
равно 40 мкг. Для разделения экстракта используют
следующие системы растворителей: толуол : тетрахлорметан = 1:2
[153, с. 40], толуол : тетрахлорметан = 2:1 [284], гексан :
тетрахлорметан = 1 : 1 [153, с. 40] и дважды перегнанный
хлороформ [283]. Хроматографирование экстракта в двух системах
растворителей, в которых определяемый пероксид имеет разные
значения Rf, обеспечивает дополнительную его идентификацию.
Специфическим проявителем пероксидов на пластинке
является раствор дигидрохлорида 1\Г,1\Г'-диметил-п-фенилендиамина,
приготавливаемый растворением 1,5 г реактива в смеси 128 мл
метилового спирта, 25 мл воды и 1 мл уксусной кислоты, при
опрыскивании которым пероксиды дают пурпурно-красные
пятна. При использовании пластинок с флуоресцентным
индикатором типа силуфол УФ254 пероксиды, содержащие
ароматические ядра, становятся видимыми при кратковременном
облучении пластинки источником УФ-излучения. В обоих случаях для
идентификации пероксидов 0,5%-ные растворы
соответствующих стандартов наносят на линию старта рядом с
анализируемой пробой на боковой части пластинки на расстоянии 1,5 см
от края.
Для количественного определения пероксида участок
сорбента с находящимся на нем пероксидом снимают с пластинки,
пероксид экстрагируют с сорбента и проводят реакцию с
иодид-ионом с применением микротитрования выделившегося
263
Таблица IV. 4. Сравнительные данные определения содержания
пероксида дикумила в полистирольных пластиках
Полимер
Введено,
Образец
Найдено, "л
с
применением
тех
иодомет-
рический
метод
Максимальная
температура
процесса, вС
САН
САН
ПСВ-СВ
псв-с
0,15
0,15
0,30
0,30
0,20
0,20
0,40
0,30
Бисер
Гранулы
Бисер
Гранулы
Бисер
«
«
«
0,09
0,045
0,26
0,09
0,18
0,21
—
0,26
0,15
0,19
0,07
0,34
0,17
0,21
0,26
0,16
0,36
0,10
140
200—220
120
200—220
110
ПО
ПО
135
135
иода разбавленным раствором тиосульфата натрия [284].
Такой способ количественного определения является общим для
всех пероксидов: условия иодометрического определения
зависят от реакционной способности пероксида. Однако для
пероксидов, имеющих в составе молекулы ароматическое ядро,
количественное определение может быть проведено путем
измерения поглощения пероксида в УФ-области спектра после
экстракции его с сорбента. Такой способ количественного
определения пероксида является наиболее простым и экспрессным.
Влияние примесей, вымываемых с сорбента и бумажных
фильтров и способных поглощать в той же области спектра,
учитывается проведением контрольного опыта.
Обсуждаемая схема анализа с применением ТСХ является
общей для всех пероксидов и полимеров при условии
обеспечения полноты экстракции пероксида из полимера без его
термического разложения, а также исключения возможности
разложения пероксида в процессе хроматографирования. В этом
отношении предпочтительнее разделять экстракты на силика-
геле, а не на оксиде алюминия; в частности, хорошо
зарекомендовали себя пластинки силуфола (ЧССР). В табл. IV.4
приведены сравнительные данные определения пероксида
дикумила во вспенивающемся полистироле и САН,
иллюстрирующие преимущества метода с применением ТСХ над иодометри-
ческим определением пероксида в растворе полимера.
Использование ГЖХ для определения пероксидов
ограничивается их термической нестойкостью.
IV. 2. ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ
IV. 2.1. Остаточные мономеры
В соответствии с санитарными нормами содержание
остаточных мономеров в полимерах строго регламентируется. В
зависимости от назначения полимера допустимое содержание оста-
264
точных мономеров изменяется в пределах от 10~4 до 0,5%.
Для контроля таких концентраций требуются чувствительные
аналитические методы.
В настоящее время газохроматографический метод вытеснил
практически все другие методы контроля остаточных
мономеров и вошел в действующие стандарты. Чаще всего анализу
подвергается раствор полимера. Так определяют остаточный
стирол, акрилонитрил, бутадиен, винилхлорид, метилметакри-
лат. Для предотвращения загрязнения испарителя
хроматографа полимером либо проводят предварительное высаждение
полимера спиртом или водой, либо применяют форколонки,
заполненные стекловолокном. Иногда анализируют экстракты
полимеров или выделяют мономер методом газовой экстракции.
Парофазный анализ, при котором в хроматограф вводят
не непосредственно анализируемый образец или его раствор, а
находящуюся с ним в контакте газовую фазу, обладает явным
преимуществом перед прямым методом. Расчетные формулы
для определения результатов парофазного анализа основаны на
законе распределения:
С = КСТ,
где С — концентрация мономера в полимере или в его растворе; Сг —
концентрация мономера в равновесной газовой фазе; К — коэффициент
распределения.
Наиболее важные преимущества метода: отсутствие
загрязнения колонки полимером, малые помехи, вызываемые
введением больших доз растворителя, высокая чувствительность.
При использовании этого метода для определения
остаточных мономеров анализируется газовая фаза либо над твердым
образцом, либо над раствором полимера. При этом анализ
можно проводить как в равновесных, так и в неравновесных
условиях.
При анализе равновесной газовой фазы в простейшем
варианте навеску анализируемого образца помещают в пеницил-
линовый флакон с фторопластовой пробкой, уплотненной
прокладкой из силиконовой резины, который затем вставляют в
патрон с завинчивающейся крышкой. Патрон выдерживают в
термостате до равновесного распределения мономера между
твердой или жидкой и газовой фазами. Нагретым шприцем
отбирают 1—3 см3 газовой фазы и вводят в хроматограф.
Определив концентрацию мономера в газовой фазе, рассчитывают
его первоначальную концентрацию в полимере, используя
коэффициенты распределения для анализируемой марки полимера.
Время установления равновесия в газовой фазе,
находящейся в контакте с полимером, зависит от температуры,
соотношения объемов фаз, летучести мономера. Хотя анализ газовой
фазы над твердым образцом примерно в 10 раз чувствительнее
растворного метода, он может быть применен только к
образцам, для которых система быстро приближается к равновесию.
265
Этот метод получил наибольшее распространение при
определении остаточного мономера в полимерных пленках,
предназначенных для упаковки пищевых продуктов, в порошках поли-
винилхлорида рыхлой структуры [285, 286]. Однако к образцам
суспензионного поливинилхлорида, содержащим крупные
монолитные зоны, этот метод уже неприменим [285]. В случае
крупных гранул равновесие твердая фаза—газ
устанавливается даже при высоких- температурах слишком долго. Например,
остаточный стирол в гранулах полистирола не достигает
равновесия с газовой фазой даже через 20 ч выдерживания при
75 °С [287]. Растворный метод имеет более широкое
применение, поскольку равновесие в этом случае устанавливается
быстрее и упрощается процесс калибровки. При выборе
растворителя кроме растворяющей способности по отношению к
анализируемому полимеру учитывается легкость очистки
растворителя. Предпочтительными для парофазного анализа
являются высококипящие растворители, имеющие большие, чем у
остаточного мономера, времена удерживания. Чаще всего в
качестве растворителей полимеров используют диметилацет-
амид (ДМАА), диметилформамид (ДМФА), диметилсульфок-
сид (ДМСО). Для ускорения анализа растворитель и другие
высококипящие примеси удаляют из колонки обратной
продувкой.
Чувствительность растворного метода зависит от давления
паров мономера, т. е. относительной летучести мономера, его
растворимости в растворителе и концентрации полимера в
растворе. Таким образом менее летучие мономеры, такие, как,
например, стирол, нельзя определить с той же чувствительностью,
что и более летучие типа винилхлорида. Чтобы увеличить
равновесную концентрацию мономера в газовой фазе, можно
уменьшить растворимость мономера, изменяя состав фазы
растворителя. Для этой цели используют растворители,
смешивающиеся с применяемым для растворения полимера
растворителем и кипящие при температуре не ниже 100 °С. Так, по
данным работы [287], из нескольких испытанных растворителей,
наиболее эффективной добавкой оказалась вода, добавление
которой к раствору полимера дает резкое увеличение
чувствительности метода (табл. IV. 5). Поскольку при парофазном
анализе для определения остаточных мономеров используют ДИП,
то применение воды для уменьшения растворимости
мономера имеет и то преимущество, что ДИП ее не чувствует и
таким образом не вносится дополнительных осложнений в
анализ.
Присутствие полимера в растворе влияет на равновесную
концентрацию мономера в газовой фазе. Для компенсации
этого эффекта при абсолютной калибровке используют растворы
полимера с низким содерхонием остаточного мономера, в
которые вводится известное количество стандарта. Влияние
полимера проявляется не в равной степени у всех полимеров.
266
Таблица IV. 5. Предел обнаружения остаточных мономеров
различными хроматографическими методами
Мономер '
ГКИП' °С
Предел обнаружения *, %
•
II
III
Вииилхлорид -13 10 * 5-10 * 2« 10
Бутадиен -4 5 ■ 10_, 5 • 10 f 2 • 10
Акрилоиитрил 76 10 , 5-10 , 2 - 10
Стирол 145 10 ' 2 -10_3 10
2-Этилгексилакрилат 214 2-10 10 * 5-10
* I—хроматографироваиие 10 %-ного раствора полимера; II — парофазнып анализ
раствора полимера; III — модифицированный метод парофазного анализа с добавлением
к раствору полимера воды.
Варьирование навески полимера имеет особенно большое
значение для определения остаточного мономера в случае стирола
и 2-этнлгексилакрилата по сравнению с винилхлоридом, акри-
лонитрнлом и бутадиеном [287].
Для приготовления стандартов для калибровочной кривой
используют полимер с минимальным содержанием остаточного
мономера (меньшим того, которое соответствует первой точке
калибровочной кривой). Навеску полимера рстворяют в пени-
циллиновом флаконе с известным количеством ДМАА, нагревая
его для ускорения растворения до 90 °С. Раствор полимера
охлаждают и вводят в него 5—40 мкл стандартного раствора
мономера, раствор перемешивают и вводят в каждый раствор
порцию дистиллированной воды. Флакон встряхивают, чтобы
вода полностью смешалась с органической фазой и чтобы
помешать высаждающемуся полимеру образовать пленку на
поверхности раствора. Для достижения равновесия в системе
флакон перед анализом выдерживают в течение 1 ч при
90 °С.
Трудность анализа твердых образцов, особенно
нерастворимых полимеров, для которых невозможно определить
коэффициент распределения, привела к использованию кинетических
принципов в парофазном методе анализа, реализовавшихся в
варианте дискретной газовой экстракции (ДГЭ). Основные
вопросы теории парофазного газохроматографического анализа
методом ДГЭ рассмотрены в работе [288].
Количество перешедшего в газовую фазу вещества зависит
от продолжительности экстракции и скорости испарения,
которая в свою очередь зависит от коэффициента диффузии и
концентрации летучего вещества в конденсированной фазе, а также
от размера и формы частиц. Часто решающим фактором
является правильный выбор температуры. Прямой парофазный
анализ в случае ДГЭ, как правило, проводят при максимально
возможной температуре, при которой еще сохраняются
структура полимера и постоянство его объема.
267
Важнейшей характеристикой процесса ДГЭ является
отношение количеств компонента, извлекаемых на
последовательных стадиях экстракции. Для аналитических целей подходят
системы, характеризующиеся постоянством этого отношения
(В — буферный коэффициент) независимо от концентрации.
В этом случае содержащиеся в экстрактах количества вещества
образуют убывающую геометрическую прогрессию и общее
количество экстрагируемого компонента 5 может быть определено
по формуле суммы ее членов без осуществления
исчерпывающей экстракции:
S = /n,/(l-B),
где т,\— количество компонента, полученное при первой экстракции.
Для систем, удовлетворяющих указанному выше условию,
парофазный анализ сводится к определению количества вещества
в газовой фазе после первой и второй экстракции.
Ограничением метода является требование к значению буферного
коэффициента: для уменьшения погрешности метода должны быть
выбраны оптимальные условия анализа, обеспечивающие
достаточно малое его значение (0,5—0,6). Разработка условий
определения остаточного мономера методом ДГЭ состоит таким
образом в подборе условий анализа, обеспечивающих требуемое
значение буферного коэффициента. Затраты на поиски таких
условий оправдывают себя при проведении серийных
анализов.
Погрешность анализа методом ДГЭ во многом зависит от
способа его проведения и техники дозирования газовой фазы.
Предложены различные варианты осуществления процесса
экстракции с полной и частичной заменой газовой фазы и
показано, что проведение ДГЭ в условиях частичной замены газовой
фазы путем пневматического дозирования приводит к
повышению точности анализа по сравнению с ДГЭ в трубках при
полной замене газовой фазы за счет резкого увеличения
воспроизводимости измерения буферного коэффициента.
Ниже приведены сравнительные данные определения
остаточного стирола (в %) в полистироле методом ДГЭ (I) и хро-
матографнческим методом по ГОСТ 15820—78 (II) [288]:
Полистирол УПМ-0503 листовой с толщиной листа
20 мкм
100 мкм
150 мкм
Полистирол ППС-А, конденсаторная пленка
толщиной
20 мкм
45 мкм
0,028
0,061
0,068
0,066
0,126
0,030
0,056
0,062
0,070
0,122
На рис. IV.6 приведена хроматограмма летучих примесей
полистирола.
268
5 0 10 5 О
Время, мин
Рис. IV. б.'Хроматограмма примесей полистирола, полученная методом ДГЭ при 130 °С
для четырех последовательных экстракций (колонка с 5 % апиеэона на хромосорбе,
температура колонки 80 °С, температура испарителя 160 °С, ДИП; скорость газа-носителя
азота* 50>1л/мин, шкала электрометра 2Х 10—"* А, шкала самописца Ю-2 мВ,
дозирование через 30 мин):
/—бензол; 2—толуол; 3 — этилбеиэол; 4—стирол.
IV. 2.2. Вода
При проведении полимеризации суспензионным или
эмульсионным методом после отмывки полимера даже в высушенном
продукте остается какое-то количество воды. Присутствие ее в
избыточном количестве отрицательно сказывается в первую
очередь при переработке полимера. Этим определяется
необходимость аналитического контроля содержания воды в
полимерах. В зависимости от температуры, влажности воздуха и
сродства полимера к воде содержание воды в полимере может
изменяться, что следует принимать во внимание при хранении
образцов, подлежащих испытанию. Например, быстро
испаряется вода, присутствующая на поверхности полимера, и это
необходимо учитывать при контроле влажности бисера перед
переработкой его в гранулы; весьма гигроскопичными являются
порошкообразные полиакрилаты и т. д.
Существует очень много методов определения воды в
полимерах, как простых и непосредственных, так и требующих
269
предварительной подготовки образца, проведения химических
реакций [289—291]. Они позволяют определять воду при ее
содержании от миллионных долей до десятков процентов,
в данном разделе будут рассмотрены только основные,
получившие наибольшее распространение методы определения воды
в полимерных материалах. Выбор метода, помимо его
применимости к конкретному полимеру, должен определяться
экономической целесообразностью и, к сожалению, часто ограничен
аппаратурными возможностями лаборатории.
Для определения воды в полимере обычно требуется
предварительное ее извлечение. Это достигается термическим
выделением, экстракцией растворителем или дистилляцией, которая
заключается в отгонке воды из диспергированного в подходящем
растворителе полимера или из его раствора в виде азеотропа
[289]. При отгонке воды с носителем, в котором вода при
обычных условиях растворяется незначительно, определение
можно провести объемным методом. Этот метод очень прост и
ему можно отдать предпочтение, если в полимере содержится
несколько процентов воды.
В настоящее время универсальным методом определения
воды, принятым н узаконенным стандартами ряда стран, в том
числе ГОСТ 11736—78, является метод иодометрического
титрования— метод Фишера. Основным его преимуществом является
высокая селективность реакции, положенной в основу метода.
Традиционный реактив Фишера состоит из иода, диоксида
серы, пиридина и метилового спирта. В этой системе
взаимодействие реактива Фишера с водой представляет собой двух-
стадийную реакцию:
C5H5N • 12 + C5H5N • S02 + C5H6N + Н20 —■> 2C5H5N • HI + C5H5N • S03,
C5H5N • S03 + CH3OH —-> C5H5N(H)S04CH3.
Конечную точку титрования можно оценивать визуально, по-
тенциометрически, кулонометрически, фотометрически,
термометрически и с помощью биамперометрического метода. Чаще
всего для определения воды в полимерах используется метод
электрометрического титрования. Предел обнаружения воды
этим методом составляет 0,5 мг. Основой большинства
автоматических титраторов служит биамперометрический метод
титрования, который позволяет снизить предел обнаружения воды
до 0,3 мг.
Реактив Фишера с метиловым спиртом является
устойчивым, если водный эквивалент не больше 1 мг/мл. Для
сохранения чувствительности метода требуется тщательное
исключение доступа в аппаратуру воды из окружающего воздуха.
Предварительное термическое выделение воды из полимера
(газовая экстракция) при определении ее по методу Фишера
применяется в том случае, если возникают трудности в
подборе растворителя, извлекающего воду из полимера полностью
(гранулы низкой растворимости, плавленые порошки), а также.
270
если в полимере присутствуют примеси или добавки, способные
реагировать с реактивом Фишера.
В зависимости от способа выделения воды этим методом
иногда можно провести различие между внутренней и
поверхностной водой в полимере [153, с. 27]. Это относится
например, к различным способам экстракции воды. Применяя
растворитель, в котором полимер набухает, можно определить
методом Фишера общее содержание воды в полимере; при
использовании растворителя, смешивающегося с водой, в котором
полимер не набухает, экстрагируется только поверхностная вода.
Такие данные бывают полезными при определении воды для
оценки эффективности сушки бисера полимера.
Последние достижения в развитии метода Фишера связаны
со стабилизацией реагента и улучшением фиксации конечной
точки титрования. Большинство модификаций направлены на
исключение неудобного для применения пиридина и повышение
стабильности реактива, благодаря замене метилового спирта
на этилцеллозольв, этиленгликоль или формамид.
Основным недостатком метода является его трудоемкость-,
которая определяется подготовкой растворителей и самого
образца к определению.
Одним из самых старых методов определения воды в
полимерах является гравиметрический метод, заключающийся в
определении потери массы образца при его сушке.
Традиционная сушка взвешенного образца проводится в термостате при
атмосферном давлении. Температуру и продолжительность
сушки обычно устанавливают эмпирически, доводя навеску
образца после сушки до постоянной массы, и строго
контролируют. Точность такого метода определяется полнотой удаления
воды из образца и наличием других летучих соединений. Если
этот метод применим, он является наиболее простым и
экономичным.
При сушке полимерных образцов в качестве источника
теплоты можно использовать ИК-лампу. Диффузия воды из
образца происходит до тех пор, пока давление паров воды в
образце превышает давление паров воды в окружающей среде.
Более интенсивная вентиляция в случае сушки ИК-лампой
способствует созданию более низкого давления водяного пара,
поэтому продолжительность сушки под ИК-лампой намного
меньше, чем при использовании термостатов.
Выделение воды из нагретого образца — начальная стадия
многих инструментальных методов: электрохимического,
манометрического, хроматографического. В электрохимическом
методе определения воды, основанном на ее электролизе, вначале
происходит термическое выделение воды из образца в потоке
инертного газа-носителя, затем ее количественное поглощение
гигроскопическим веществом и на заключительной стадии —
электролиз воды на кислород и водород. Количество тока,
израсходованного на электролиз, является показателем содержа-
271
ния воды в полимере. На электрохимическом методе основаны
конструкции приборов для определения воды в полимерах, в
том числе советского кулонометрического анализатора «Бай-
кал-3» [292]. К анализируемым данным методом полимерам
относятся прежде всего полиолефины [290]. Метод позволяет
определять низкие концентрации воды в полимере вплоть до
ю-«%.
Манометрический метод, основанный на измерении давления
водяных паров, может осуществляться в нескольких вариантах.
Наиболее распространен вариант, при котором анализируемый
образец помещают в отвакуумированный сосуд и нагревают
до определенной температуры (150—200 °С). Через некоторое
время измеряют давление, получающаяся разность давлений
зависит от содержания воды в образце. Содержание воды в
полимере подсчитывают по калибровочной кривой, построенной по
известным навескам воды. Для поправки на другие
летучие соединения в пробе давление измеряют до и после
прохождения их через слой гидрида кальция. Продолжительность
и температуру нагревания подбирают для каждого полимера
эмпирически. В работе [293] гранулы полимера смешивали с
полисилоксаном, содержащим активный водород, смесь
нагревали до 170 °С и через 7—10 мин по разнице уровней
манометра, соединенного с трубкой, в которой находится смесь,
замеряли количество водорода, выделившегося в результате
взаимодействия с водой. Минимально детектируемое
манометрическим методом содержание воды в полимере равно Ю~3%.
Прямые хроматографические методы определения воды с
использованием в качестве детектора катарометра
заменили старые методы, заключавшиеся в хроматографировании
летучих продуктов взаимодействия экстрагированной из полимера
воды с реагентом — карбидом кальция, тетраацетатом свинца,
гидридом кальция или диметоксипропаном. В хроматографиче-
ских методах определения воды летучие продукты извлекают из
полимера в испарителе, конструкции которого очень
разнообразны, и сдувают инертным газом непосредственно в начальную
часть хроматографической колонки или предварительно
поглощают гигроскопичным веществом, способным высвобождать
воду при высоких температурах.
Для большинства полимеров отсутствует влияние
температуры на результат определения воды при 70—160 °С [289].
Однако сополимер винилхлорида и винилацетата выделяет
максимальное количество воды при 130—145 °С [294];
дальнейшее увеличение температуры приводит к снижению
содержания воды, что связано с гидролизом при повышенных
температурах остаточного мономера винилацетата, о чем
свидетельствует появление ацетальдегида и увеличение его количества с
ростом температуры. Другим исключением являются
полиолефины, вся вода из которых не освобождается до тех пор, пока
не наступает плавление полимера в случае порошкообразных
272
образцов; при анализе гранул температура испарителя должна
быть даже несколько выше температуры плавления полимера
[294]. Для полипропилена, например, выбрана температура
нагревания образца 240°С.
Скорость извлечения воды из полимера определяется во
многом структурой полимерной матрицы и степенью
кристалличности полимера. Интересен, например, такой факт [294],
что вода из поливинилхлорида высвобождается более легко,
чем остаточный мономер, который кипит при —13 °С.
Нагревание образца в испарителе для многих полимеров,
гомополимеров и сополимеров винилового и акрилового ряда,
можно проводить в присутствии воздуха. Однако полиолефины
следует нагревать только в инертной атмосфере [294], так как
в присутствии воздуха происходит их окисление, что дает
дополнительное количество воды. Поэтому образец до его
нагревания продувают инертным газом в течение 15 с. Нагревание
полимера в испарителе продолжается 2—5 мин.
Хроматографическое определение воды может
осуществляться как в адсорбционном, так и в газожидкостном варианте.
В первом случае наибольшее распространение из сорбентов
получили порапак Q и полисорб. В качестве неподвижной
фазы чаще других используют карбовакс.
Калибровку при газохроматографическом определении воды
в том случае, если она выделяется из полимера до его
плавления, можно проводить введением известного количества воды в
помещенный в испаритель высушенный порошкообразный
образец полимера. Для полиолефинов, которые нагревают для
испарения воды выше точки плавления, калибровку лучше
строить по навескам ВаС12-2Н20, взвешенным в пустой трубке
для образца. Хлорид бария теряет кристаллизационную воду
при 115°С (14,75%). При прямом введении воды в пустую
трубку во время первичной продувки инертным газом часть
воды сдувается в колонку до нагревания трубки, поэтому хро-
матограммы получаются непохожими на хроматограммы
образцов полимера.
Для лучшей воспроизводимости результатов хроматографи-
ческого определения воды предлагается [294] газ-носитель
пропускать через слой кристаллов CuS04-5H20,
«увлажняющих» газ с целью предохранения линии газа от высыхания, что
может приводить к адсорбции воды и последующей ее
десорбции при проведении анализа. Воспроизводимость газохромато-
графического метода для содержания воды 0,02—0,5 %
составляет 5%. Точность метода определить труднее из-за
отсутствия стандартных образцов.
Одним из вариантов газохроматографического определения
воды в полимерах является метод парофазного анализа. При
проведении анализа на анализаторе фирмы Perkin Elmer
(США) рекомендуется анализировать газовую фазу над 1 мл
10 %-ного раствора полимера в ДМФА. Для ускорения анализа
Ю Зак. 632
273
большой пик растворителя, который элюируется после пика
воды, удаляют обратной продувкой. Разделение проводят на
колонке размерами 2000 X 1,1 мм с порапаком Q,
газ-носитель— гелий; температура колонки 160°С, температура
испарителя 200 °С. Пробу предварительно термостатируют при
80 °С. Продолжительность анализа 8 мин, обратной продувки
10 мин.
Чувствительность определения содержания воды методом
парофазного анализа не ограничивается чувствительностью
детектора, а зависит от контрольного опыта. Хроматограмма
контрольного опыта дает пик воды, которая включает воду на
внутренней поверхности бутылочек для образцов, воду,
содержащуюся в воздухе, который остается в бутылочке, и воду из
растворителя. При условии, что пик воды такого контрольного
опыта достаточно воспроизводим, пик воды из анализируемого
образца легко скорректировать на этот контрольный и в этом
случае вариация в пике контрольного опыта, а не он сам
определяет предел детекции.
Количественное определение при парофазном анализе
осуществляется обычно методом добавки. Для этого образец
разделяют на две части: одну анализируют обычным способом, а к
другой добавляют известное количество воды. После поправки
пиков воды на обеих хроматограммах разницу в высотах или
площадях пиков соотносят с добавкой и таким образом
используют для калибровки.
Инструментальные методы на основе анализаторов с
термическим выделением воды из полимеров весьма перспективны
для контроля содержания воды в полимерах, однако они еще
не получили широкого распространения.
Среди редко применяемых методов количественного
определения воды следует назвать фотометрический метод,
основанный на сдвиге в спектре поглощения хлорида кобальта [289,
290] при переходе от органического растворителя к водному,
и метод ИК-спектроскопии. В последнем случае для полиоле-
финов и полиакрилатов аналитической полосой может служить
полоса поглощения при 5260 см-1. ИК-Спектроскопия
позволяет определять десятые доли процента воды в полимере [289].
IV.2.3. Прочие примеси
Помимо остаточных мономеров и воды в полимере
накапливаются также низкомолекулярные неполимеризующиеся
примеси, попадающие из сырья, а также остатки инициаторов,
катализаторов, поверхностно-активных веществ, следы
растворителей, продуктов деструкции полимера. Примеси могут
попадать в полимер из аппаратуры, привноситься с добавками.
Для их контроля используют различные методы, выбор
которых определяется природой примеси и чувствительностью
анализа. Необходимость такого контроля возникает для полимеров
274
специального назначения вследствие влияния примесей на
свойства полимера или диктуется санитарными нормами на
применение полимеров.
Остатки инициаторов могут быть определены, например,
методом ТСХ, описанным в разделе IV. 1.5.
ПАВ, применяемые в качестве эмульгаторов, определяют в
основном спектральными методами [39, с. 155], в том числе
спектрофотометрически по окрашенным продуктам реакции,
характерным для данного класса ПАВ. В ряде случаев такое
определение можно провести непосредственно в растворе
полимера (см. п. IV. 3.8).
Определение примесей металлов в полимере проводят
обычно путем анализа золы, получаемой сжиганием образца и
прокаливанием остатка до постоянной массы при температуре
800—900°С (ГОСТ 15973—82). Для устранения потерь
оптимизируются условия озоления, для легколетучих элементов
предпочтение отдается химической минерализации полимера.
Минерализованный полимер можно анализировать спектрофотомет-
рическим и эмиссионным спектральным методами.
Спектрофотометрические методы очень чувствительны и
позволяют обнаруживать металлы при содержании их в
полимере до 2-10-5 %. В работе [295] золу, полученную при
сжигании поливинилового спирта, растворяли в соляной кислоте и
в аликвотной части раствора определяли примеси железа, меди,
свинца, никеля и кобальта по образованию окрашенных
комплексов этих металлов с органическими реагентами: железа —
с 1,10-фенантролином, никеля — с диметилглиоксимом,
кобальта— с нитрозо-И-солью, меди — с дитизоном после экстракции
комплекса хлороформом при рН = 1 ч- 4; свинец образует
комплекс с дитизоном при рН = 6,5-f- 8.
Для количественного эмиссионного спектрального анализа
необходимо не менее 10 мг золы. Техника эксперимента и
способы количественного анализа описаны во многих руководствах
по спектральному анализу полимеров (например, [39, с. 157]).
Метод атомно-абсорбционной спектроскопии, позволяющий
анализировать образцы, содержание золы в которых не
определить взвешиванием, не нашел, к сожалению, широкого
применения в практике аналитического контроля пластмасс.
В ряде случае примеси металлов в полимере можно
определить прямым методом. Например, в работе [296] для
определения меди в сополимере N-винилпирролидона с кротоновым
альдегидом комплексонометрический метод применен к
раствору полимера. В качестве титранта авторы использовали этилен-
диаминтетраацетат в присутствии 1-(2-пиридилазо)-4-циклопен-
тилрезорцина. При прямом эмиссионном анализе полимерного
порошка или раствора полимера увеличивается погрешность
определения.
Важной задачей аналитического контроля является
определение летучих примесей в полимере, обусловливающих его
Ю*
5>7П
запах. Определение летучих соединений, многие из которых
являются токсичными, входит в оценку санитарных
характеристик и гигиенических свойств полимеров. Наиболее
подходящим методом для их определения является газохроматографи-
ческий, который, как и в случае определения остаточных
мономеров, может проводиться путем анализа раствора полимера,
его экстракта или газовой фазы над полимером или его
раствором. Находят применение и другие методы. Например,
остаточные меркаптаны можно определять ионометрическим
методом, используя в качестве индикаторного сульфидсеребряный
электрод.
Из летучих примесей в полимере чаще всего приходится
определять остаточные растворители, применяемые при
получении полимеров или полимерных покрытий. Для этой цели
широко используется газовая экстракция растворителей из
полимеров. Так, в работе [297] газохроматографическим методом
определяли остаточные растворители в пленках и порошках
фторсополимеров и сополимеров винилового спирта, в работе
[298]—в пленках и листах из полиэтилена и полипропилена,
покрытых поливинилиденхлоридом, а в работе [299]
исследовалось выделение летучих веществ из полимерных покрытий, в
том числе на основе поливинилиденхлорида.
Одной из причин появления примесей, как уже говорилось,
может быть частичная деструкция полимера в условиях его
технологической обработки. Например, количественный анализ
летучих примесей полиэтилена, проводимый на насадочных
колонках размерами 3000 X 3 мм, заполненных 5 % апиезона L
на хромосорбе G AW при 100 °С [300], показал, что
основными компонентами летучих примесей полиэтилена являются не
остатки углеводородных растворителей, применяемых при его
получении, а алканы С4—С|2 и олигомеры этилена.
Обнаружены и следовые количества кислородсодержащих соединений,
также относящихся к продуктам термоокислительной
деструкции полиэтилена. Аналитическая процедура сводилась к
периодическому дозированию через 1 ч газовой фазы, находящейся
в контакте с полимером при 100 °С, в хроматографическую
колонку. Результаты определения данным методом совпали (в
пределах 2 %) с результатами, полученными исчерпывающей
экстракцией.
Как известно, определяющим фактором в выборе
температуры выделения примесей из полимера является температура
разложения полимера. Но иногда интерес представляет
исследование полимера при более высоких температурах. При
проведении газовой экстракции при повышенной температуре
(200—400 °С) по характеру кинетических кривых выделения
летучих продуктов можно отличить летучие примеси полимера
от продуктов его термического разложения, протекающего при
температуре анализа. Данные о составе и количестве выделяю-
276
щихся из полимера соединений при повышенных температурах
необходимы для уточнения условий его переработки.
Предел чувствительности газохроматографического метода
определения примесей летучих соединений в полимере можно
снизить, применяя концентрирование криогенным или сорбцион-
ным способом. Для сорбции летучих соединений используют
термический технический углерод, силохром-80 с 5 % ПМС-100,
тенакс. Заполненный сорбентом концентратор, изготовленный,
например, из стальной трубки, можно для последующей
десорбции поместить непосредственно в гнездо испарителя
хроматографа. Благодаря концентрированию удается определять до
10^6 °/о летучих примесей.
Приведенные примеры не исчерпывают задач, стоящих
перед аналитиками при контроле примесей в полимерах, а
иллюстрируют определение только наиболее распространенных
примесей. В настоящее время проблему определения примесей в
полимерах нельзя считать решенной, поскольку она в
принципе сложнее, чем определение добавок. Самыми сложными для
анализа являются примеси, появляющиеся в результате
химических превращений, контроль которых практически
отсутствует.
IV. 3. МЕТОДИКИ ОПРЕДЕЛЕНИЯ ДОБАВОК И ПРИМЕСЕЙ
В ПОЛИМЕРАХ
IV.3.1. Определение ирганокса 1010 (фенозана 23)
в композициях полиэтилена низкого давления,
наполненных техническим углеродом
Определение основано на экстракции стабилизатора
ирганокса 1010 (фенозана 23) из полимера кипящим хлороформом с
последующим определением концентрации стабилизатора в
экстракте по УФ-спектру поглощения.
Аппаратура и реактивы
Мельница для измельчения полимера.
Спектрофотометр для УФ-областн спектра.
Колбы круглодонные вместимостью 100 мл.
Холодильники шариковые.
Водяная баня.
Мерные колбы вместимостью 25 и 50 мл.
Пипетка на 2 мл градуированная.
Хлороформ, очищенный перегонкой с дефлегматором.
Ирганокс 1010 (фенозан 23), стандартный раствор в хлороформе с
концентрацией ~2 мг/мл, приготовленный путем растворения и хлороформе
навески 100 ±5 мг, взвешенной с точностью до 0,0002 г, в мерной колбе
вместимостью 50 мл.
Построение градуировочного графика. В 5 мерных колбах
вместимостью 25 мл приготавливают растворы стабилизатора с
концентрацией в интервале 0,02—0,1 мг/мл путем разбавления
277
хлороформом 0,25; 0,50; 0,75; 1,0 и 1,25 мл стандартного
раствора ирганокса 1010. УФ-спектры полученных растворов
снимают по отношению к хлороформу в интервале длин волн
220—360 нм, используя кюветы с толщиной слоя 10 мм. По
методу наименьших квадратов строят градуировочный график
зависимости оптической плотности растворов от концентрации
стабилизатора в растворе. Оптическую плотность замеряют в
максимуме поглощения стабилизатора (£>2вз) по отношению к
базисной линии, проводимой между основаниями пика (255 и
295 нм).
Выполнение анализа. Гранулы анализируемой композиции
полиэтилена измельчают до порошкообразного состояния.
Навеску композиции около 2 г взвешивают с точностью до
0,0002 г, заливают 40 мл хлороформа и проводят экстракцию
стабилизатора кипящим растворителем в колбе с обратным
холодильником, нагреваемой на водяной бане, в течение 2 ч.
Экстракт отфильтровывают от полимера в мерную колбу
вместимостью 50 мл, промывают полимер тремя порциями
растворителя по 3 мл и доводят объем раствора до метки.
Раствор хорошо перемешивают и замеряют его оптическую
плотность, как при построении градуировочного графика.
Расчет. Массовую долю стабилизатора в композиции X
(в %) рассчитывают по формуле:
Х = С50- 100/(т-1000),
где С — концентрация стабилизатора в экстракте, найденная по градуировоч-
ному графику, мг/мл; т — навеска полимера, взятая для экстракции, г.
Относительная погрешность определения не превышает 12 %.
IV.3.2. Определение фосфорсодержащих стабилизаторов
в полиэтилене
Определение основано на разложении полимера серной,
азотной кислотами и пероксидом водорода с переведением
фосфора стабилизатора в фосфат-ион и фотометрическом определении
последнего по образованию молибденовой сини.
Аппаратура и реактивы
Спектрофотометр или фотоэлектроколориметр с красным фильтром.
Плитка электрическая с переключателем мощности.
Стаканы термостойкие вместимостью 400 мл.
Колбы мерные вместимостью 100 мл.
Пипетки на 5, 10, 15 и 20 мл.
Цилиндры вместимостью 100 и 25 мл.
Серная кислота концентрированная.
Азотная кислота.
Аммиак, водный раствор.
Пероксид водорода, 16—25 °/о-ный раствор.
Этиловый спирт ректификованный технический.
Фенолфталеин, 1 %-ный спиртовый раствор.
Молибдат аммония. 50 г реактива растворяют в 500 мл горячей
дистиллированной воды и вливают в охлажденный раствор серной кислоты, приго-
278
товленный в мерной колбе вместимостью 1 л добавлением 125 мл
концентрированной серной кислоты к 350 мл дистиллированной воды; раствор
охлаждают, доводят до метки дистиллированной водой и перемешивают.
Соль Мора, свежеприготовленный раствор. 10 г солн Мора растворяют в
дистиллированной воде, добавляют 1 мл серной кислоты (1:7) и разбавляют
дистиллированной водой до 100 мл.
Стандартный раствор стабилизатора с концентрацией ~2 мг/мл (спир-
товый раствор полигарда, хлороформенный раствор иргафоса 168),
приготавливается растворением точной навески стабилизатора в мерной колбе
вместимостью 50 мл.
Выполнение анализа. Полимер измельчают, навеску (0,2—
0,5 г), содержащую 0,4—0,7 мг стабилизатора, взвешивают с
точностью до 0,0002 г и помещают в стакан вместимостью
400 мл. К навеске приливают 15 мл серной кислоты, стакан
ставят на электроплитку, включенную на мощность 400—
500 Вт и нагревают до появления густых белых паров S03 и
почернения раствора. После того как раствор станет вязким,
стакан снимают, охлаждают до прекращения дымления и в
еще горячий раствор приливают 5 мл азотной кислоты, а
стакан снова помещают на плитку. Кислоту приливают по стенкам,
смывая попавшие на них частицы полимера. После активного
образования паров оксидов азота снова выделяются белые
пары S03. Для лучшего разложения полимера стакан снова
снимают, дают немного остыть и добавляют 1 мл серной кислоты.
Стакан опять ставят на плитку и нагревают. Когда в стакане
останется 5—6 мл раствора, его снимают с плитки, охлаждают
до 50—60 °С и по каплям прибавляют пероксид водорода до
осветления раствора и прекращения выделения бурых паров
оксидов азота.
Осветленный раствор снова помещают на плитку и
нагревают до появления белых паров; раствор при этом
обесцвечивается. Раствору дают закипеть и выпаривают при слабом
кипении до объема 4—5 мл.
После выпаривания в охлажденный раствор добавляют 50 мл
дистиллированной воды и для гидролиза частично
образовавшегося дифосфата до фосфата раствор кипятят в течение 5—
10 мин, оставляя в стакане 30—35 мл раствора. Стакан
снимают с плитки, охлаждают и при перемешивании из бюретки
приливают водный раствор аммиака до прекращения бурной
реакции нейтрализации. Затем к раствору прибавляют каплю
фенолфталеина и продолжают нейтрализацию раствором
аммиака до появления слабо-розового окрашивания.
Нейтрализованный раствор переносят в мерную колбу вместимостью 100 мл,
стакан промывают 10 мл дистиллированной воды и промывную
воду присоединяют к основному раствору.
В мерную колбу при перемешивании добавляют 5 мл
концентрированной серной кислоты и 20 мл раствора молибдата
аммония. Через 5 мин к анализируемому раствору добавляют
10 мл раствора соли Мора, перемешивают раствор и
разбавляют до 100 мл дистиллированной водой. Мерные колбы для
279
развития окраски помещают в кипящую водяную баню на
10 мин. После охлаждения под струей холодной воды раствор
фотометрируют на спектрофотометре при % = 650 нм или на
фотоэлектроколориметре с красным светофильтром в кюветах
с толщиной слоя 30—50 мм. Раствором сравнения служит
контрольный опыт, проведенный с образцом, близким по массе и
составу к анализируемому, но не содержащим фосфороргани-
ческого стабилизатора.
Построение градуировочного графика. В 6 стаканов
вместимостью 350 мл помещают от 0,2 до 2 мл стандартного
раствора стабилизатора и 15 мл серной кислоты. Стаканы ставят
на плитку и нагревают до появления густых белых паров БОз.
Затем стаканы снимают и после остывания приливают 5 мл
азотной кислоты, далее проводят все операции, как указано
при выполнении анализа.
Раствором сравнения служит контрольный опыт, который
готовят проведением через все операции 2 мл растворителя, в
котором готовился стандартный раствор. По полученным данным
строят градуировочный график зависимости оптической
плотности от содержания стабилизатора в 100 мл раствора (в мг).
Расчет. Массовую долю стабилизатора X (в %) в полимере
вычисляют по формуле:
Х = т1 ■ 100/(1000-га),
где mi — количество стабилизатора в анализируемой навеске полимера,
найденное по градуировочному графику, мг; га— навеска полимера, г.
Относительная погрешность определения не превышает 10 %
для интервала концентраций стабилизатора 0,1—0,3%.
IV.3.3. Определение глицерина в антисептических
поливинилспиртовых пленках
Методика предназначена для определения содержания
глицерина в интервале 3—10 % в антисептических
поливинилспиртовых пленках, пластифицированных глицерином и содержащих
в качестве антисептика до 5 % иода или катапола.
Определение основано на растворении пленки в
дистиллированной воде при нагревании, окислении глицерина йодной
кислотой и последующем взаимодействии образующегося при этом
формальдегида с флороглюцином при комнатной температуре
с образованием фиолетово-розового продукта реакции, который
определяют фотометрически.
Аппаратура и реактивы
Весы аналитические.
Ультратермостат жидкостный любой марки для поддержания
температуры 70—75 °С.
Спектрофотометр любого типа для видимой области.
Секундомер.
Колбы конические с пришлифованной пробкой вместимостью 100 мл.
280
Колбы мерные вместимостью 100 и 50 мл.
Воронка.
Пипетки градуированные на 1,5 и 10 мл.
Цилиндр мерный вместимостью 25 мл.
Фильтры бумажные.
Глицерин.
Йодная кислота, 0,05 М раствор в 0,05 н. серной кислоте, (срок годности
раствора 7 сут).
Гидроксид натрия, 1 н. раствор.
Этиловый спирт ректификованный технический.
Флороглюцин, 0,2 %-ный раствор.
Приготовление стандартных растворов глицерина. В мерной
колбе вместимостью 100 мл взвешивают 250 ± 10 мг глицерина
с точностью до 0,0002 г. Навеску глицерина растворяют в
дистиллированной воде и доводят объем до метки, раствор
хорошо перемешивают.
Для построения градуировочного графика приготавливают
ряд разбавленных растворов. Для этого в колбы вместимостью
100 мл с пришлифованной пробкой отбирают 0; 0,2; 0,3; 0,4;
0,5 и 0,6 мл приготовленного раствора глицерина и доводят
объем жидкости до 10 мл, добавляя пипеткой вместимостью
10 мл соответствующий объем дистиллированной воды. Ко всем
растворам приливают по 5 мл дистиллированной воды.
Построение градуировочного графика. Цветную реакцию
проводят с каждым раствором в отдельности. К раствору
прибавляют 10 мл 0,05 М раствора йодной кислоты, колбу
закрывают пробкой, осторожно взбалтывают содержимое и
оставляют для окисления на 5 мин. После этого
последовательно приливают 10 мл 1 н. раствора гидроксида натрия, 5 мл
этилового спирта и 10 мл 0,2 %-ного раствора флороглюцина.
Реактивы прибавляют быстро, раствор перемешивают и через
3 ± 0,25 мин после добавления флороглюцина измеряют
оптическую плотность раствора в кювете с толщиной слоя 10 мм
при X = 480 нм. Измерение проводят относительно
дистиллированной воды.
По полученным данным строят градуировочный график
зависимости оптической плотности от содержания глицерина в
растворе (в мг).
Выполнение анализа. 0,05—0,12 г пленки, содержащей 4—
6 мг глицерина, взвешивают с точностью до 0,0002 г,
измельчают ножницами, помещают в мерную колбу вместимостью
50 мл, приливают 25 мл дистиллированной воды и колбу
закрывают пробкой. Растворение навески проводят в водяной
бане при 70—75°С, периодически перемешивая раствор
круговым движением колбы. Растворение занимает 15—20 мин.
После полного растворения пленки колбу охлаждают и доводят
объем до метки дистиллированной водой. Раствор
перемешивают и фильтруют через бумажный фильтр. На анализ отбирают
10 мл отфильтрованного раствора, приливают 5 мл дистилли-
281
рованной воды и проводят определение, как описано при
построении градуировочного графика.
Расчет. Массовую долю глицерина в пленке X (в %)
вычисляют по формуле:
Х = а-50- 100/(10т-1000),
где а — количество глицерина, соответствующее измеренной оптической
плотности и иайдеииое по градуировочиому графику, мг; т — навеска образца, г;
50 — объем колбы для растворения образца, мл; 10—объем раствора
образца, взятый иа анализ, мл.
Относительная погрешность определения для интервала
концентраций глицерина 4—10 % не превышает 4 %.
IV.3.4. Определение фторсодержащего наполнителя (апатита)
в полиэтилене
Определение основано на выделении фтора из минерального-
остатка после озоления образца дистилляцией с водяным паром
в присутствии кислот и Si02 и ионометрическом анализе
полученного раствора с использованием фторселективного
электрода.
Аппаратура и реактивы
рН-метр или иоиомер.
Электрод хлорсеребряиый ЭВЛ-1МЗ.
Электрод фторселективиый.
Мешалка магнитная.
Колбы мерные вместимостью 250, 500 и 1000 мл.
Пипетки иа 25 и 100 мл.
Стаканы химические вместимостью 50 мл.
Лодочка кварцевая.
Тигель фарфоровый.
Прибор для дистилляции с водяным паром.
Чашка платиновая вместимостью 300 мл.
Муфель.
Плитка электрическая.
Серная кислота, концентрированная.
Фосфорная кислота.
Кварцевый песок.
Фторид натрия.
Гидроксид натрия, 0,5 и. раствор.
Лимонная кислота одиоводиая.
Фенолфталеин, 0,5 %-иый раствор в 60 %-иом этиловом спирте.
Буферный раствор рН = 6. 26,65 г лимонной кислоты растворяют в
386 мл 1 и. раствора гидроксида натрия и доводят объем раствора до 1 л
дистиллированной водой.
Стандартный раствор фторида натрия. Навеску 4,1988 г высушенного
при 150 °С и доведенного до постоянной массы фторида натрия, взвешенную
с точностью до 0,0002 г, растворяют в мерной колбе вместимостью 1 л
в дистиллированой воде и доводят объем раствора до метки. Раствор
содержит Ю-1 моль/л исиов фтора (—lg CF = I).
Построение градуировочного графика. Для построения
градуировочного графика приготавливают три разбавленных
раствора с концентрацией 5; 1 и 0,5 ммоль/л ионов фтора путем;
282
разбавления основного стандартного раствора с добавлением
200 мл буферного раствора (рН = 6) к 1 л разбавленного
раствора.
Измерение потенциала растворов проводят на рН-метре или
иономере согласно инструкции к прибору, начиная с самого
разбавленного раствора и используя в качестве измерительного
электрода фторселективный, а в качестве электрода
сравнения— хлорсеребряный. Исследуемый раствор наливают в
химический стакан и помещают на включенную магнитную
мешалку, опускают в него электроды и через 5 мин снимают
показания прибора. После каждого измерения электроды
промывают дистиллированной водой и осторожно осушают
фильтровальной бумагой, стараясь не повредить мембрану ионселек-
тивного электрода.
По полученным данным строят градуировочный график на
бумаге с полулогарифмической сеткой в координатах Е (мВ) —
С (ммоль/л). В день анализа график проверяют по двум
точкам в области концентраций, близких к ожидаемой.
Выполнение анализа. Навеску гранул полимера взвешивают
с точностью до 0,0002 г и помещают в прокаленный и
доведенный до постоянной массы фарфоровый тигель. Тигель
вносят в предварительно нагретый до 450±50°С муфель и
осторожно продвигают в глубь муфеля, не допуская вспышки
образца. Температуру муфеля постепенно увеличивают вначале
до 650 °С, а затем до 850±50°С и тигель с образцом
выдерживают при этой температуре 30 мин. Вынимают тигель из
муфеля, помещают в эксикатор и через 1 ч взвешивают.
Затем тигель с содержимым доводят до постоянной массы,
прокаливая по 20 мин в муфеле при 850±50°С с последующим
охлаждением в эксикаторе в течение 1 ч.
Навеску золы около 0,1 г помещают в кварцевую лодочку и
взвешивают с точностью до 0,0002 г. Лодочку с навеской
переносят в перегонную колбу установки для дистилляции с паром.
Туда же вносят 0,1—0,2 г кварцевого песка и добавляют 30 мл
раствора серной кислоты (2:1) и 2 мл концентрированной
фосфорной кислоты. В приемник установки для дистилляции с
паром наливают 2,5 мл 0,5 н. раствора гидроксида натрия и
50 мл дистиллированной воды. При закрытом зажиме,
соединяющем перегонную колбу с парообразователем, начинают
нагрев перегонной колбы. Когда температура в колбе достигнет
120—130°С, этот зажим открывают и закрывают зажим,
соединяющий парообразователь с атмосферой, и ведут отгонку
H2SiF6 и HF при 140±5°С со скоростью 4 мл/мин. Собирают
"250 мл дистиллята. По окончании перегонки прекращают
нагрев перегонной колбы и парообразователя и открывают зажим,
соединяющий парообразователь с атмосферой.
Содержимое приемника переливают в платиновую чашку,
колбу ополаскивают 20 мл дистиллированной воды, которую
сливают в ту же чашку, и выпаривают раствор на электриче-
283
ской плитке до объема 100—120 мл. Раствор охлаждают и
количественно переносят в мерную колбу вместимостью 250 мл.
Раствор в колбе нейтрализуют 0,5 н. раствором гидроксида
натрия по фенолфталеину. Затем в колбу приливают 50 мл
буферного раствора и доводят дистиллированной водой до
метки.
Полученный раствор наливают в стакан, который
помещают на магнитную мешалку. В стакан опускают электроды,
включают мешалку и через 5 мин замеряют потенциал раствора.
Расчет. Содержание фтора в полимере X (в %) вычисляют
по формуле:
X = т,С ■ 19 • 250 • 100/(mm2 • 1000),
где т.! — масса золы, полученной при сжигании образца, г; С — концентрация
фтора, найденная по градуировочиому графику, моль/л; 19 — молекулярная
масса фтора; 250 — объем дистиллята, мл; т — иавеска образца, г; т2 —
навеска золы для дистилляции, г.
Относительная погрешность определения не превышает 6%.
IV.3.5. Определение бромсодержащих антипиренов
в полистироле
Методика предназначена для определения, бромсодержащих
антипиренов в ударопрочном и вспенивающемся полистиролах.
Определение основано на переводе органических связанного
брома, введенного в полистирол в составе антипирена, в
ионное состояние путем сжигания навески образца (3—20 мг)
в колбе с кислородом с применением платинового
катализатора. Продукты сгорания поглощаются раствором щелочи в
присутствии пероксида водорода, образовавшийся бромид
определяют методом потенциометрического титрования разбавленным
раствором нитрата серебра с индикаторным
сульфид-серебряным электродом. Определению мешают хлориды и иодиды.
Аппаратура и реактивы
Иономер или рН-метр любой марки.
Электрод сульфидсеребряиый ЭСС-01.
Электрод хлорсеребряиый ЭВЛ-1МЗ.
Мешалка магнитная.
Колбы конические иа шлифах из термостойкого стекла вместимостью
500 мл.
Пробки пришлифованные (Ш-29) с впаяииыми в них платиновыми
проволоками длиной 130—150 мм с сетками (40X20 мм) иа конце.
Бюретка вместимостью 10 мл с цеиой деления 0,02 мл.
Мостик солевой, заполненный 0,1 и. раствором нитрата калия.
Гидроксид калия, 2 и. раствор.
Пероксид водорода, концентрированный раствор.
Азотная кислота, 1 и. раствор.
Бромфеиоловый синий, 0,05 %-иый раствор.
Нитрат серебра, 0,001 и. свежеприготовленный (из 0,1 и. раствор)
раствор.
Нитрат натрия, 2 %-иый раствор.
Баллои с кислородом.
284
Подготовка к определению. Из беззольных фильтров
вырезают по шаблону «флажки» (см. рис. П. 32, б), пропитывают их
2 %-ным раствором нитрата натрия, оставляя конец для
поджигания непропитанным, и высушивают при 100 °С.
Выполнение анализа. Навеску полимера выбирают из
расчета содержания в ней 0,15—0,3 мг брома.
В стеклянную трубку с длинной ручкой помещают
измельченный до частиц размером 1 мм анализируемый полимер.
Трубку взвешивают с точностью до 0,02 мг и высыпают
содержимое на середину подготовленного фильтра «флажок». С той
же точностью взвешивают трубку после высыпания навески и
навеску образца определяют по разности этих двух
взвешиваний. Навеску полимера заворачивают в тугой пакетик, сгибая
фильтр по пунктирным линиям (см. рис. 11.32, б), при этом
узкий конец фильтровальной бумаги должен оставаться
свободным. Пакет с навеской зажимают в платиновую сетку,
прикрепленную к пробке, так, чтобы узкий конец фильтровальной
бумаги был обращен кверху и оставался свободным.
В колбу наливают 10 мл дистиллированной воды, 1 мл
2 н. раствора гидроксида калия и 1 мл пероксида водорода.
Для защиты от осколков стекла в случае разложения вещества
со взрывом колбу помещают в обрешетку из стальной сетки, а
лицо закрывают маской из органического стекла. Горло колбы
смачивают небольшим количеством дистиллированной воды и
заполняют колбу кислородом, продувая его через шланг,
опущенный в колбу, в течение 3—5 мин так, чтобы не
разбрызгивался поглотительный раствор. Затем вынимают из колбы
шланг, берут полотенцем пробку и поджигают выступающий
конец фильтровальной бумаги. Колбу быстро закрывают
пробкой и плотно прижимают ее, пока горение не закончится, во
избежание выталкивания ее образующимися газами.
После сжигания образца горло колбы смачивают
несколькими каплями воды для предотвращения заедания пробки и
встряхивают колбу 10—15 мин или оставляют стоять в течение
30—40 мин до полного исчезновения в ней тумана, т! е.
полного поглощения продуктов сгорания.
Затем колбу открывают, пробку и платиновую сетку
промывают над колбой дистиллированной водой. Колбу помещают
на плитку, кипятят ее содержимое 5—7 мин для разложения
избытка пероксида водорода и охлаждают. Содержимое колбы
количественно переносят в стакан так, чтобы общий объем
поглотительного раствора и промывных вод составил 50 мл.
В стакан добавляют 3 капли индикатора бромфенолового
синего и раствор нейтрализуют 1 н. азотной кислотой до
перехода голубой окраски индикатора в желтую. Стакан помещают
на магнитную мешалку, опускают в него сульфидсеребряный
электрод и конец солевого мостика, другой конец солевого
мостика и электрод сравнения (хлорсеребряный) помещают в
стакан с насыщенным раствором хлорида калия.
285
Титрование проводят 0,001 н. раствором нитрата серебра
при постоянном перемешивании раствора, приливая титрант из
бюретки порциями по 0,2 мл. Значения ЭДС (в мВ) снимают
после полной остановки стрелки на шкале прибора.
Титрование заканчивают, когда объем титранта после наибольшего
скачка потенциала будет примерно равен объему титранта,
пошедшего на титрование до скачка потенциала.
Расчет. По полученным экспериментальным данным строят
кривую потенциометрического титрования в координатах ЭДС
(мВ)—объем титранта (мл).
Объем раствора нитрата серебра, израсходованный на
титрование бромида, находят следующим образом. Проводят
касательную к прямолинейному участку скачка потенциалов,
делят расстояние между точками касания пополам и из этой
точки опускают перпендикуляр на ось абсцисс.
Содержание антипирена в полимере X (в %) вычисляют
по формуле:
X = (V - V0) • 0,0000799 • 100 • 100/(m/f),
где V, V0 — объем точно 0,001 и. раствора нитрата серебра, израсходованный
на титрование соответственно анализируемой пробы и контрольного опыта,
найденный графически, мл; 0,0000799 — количество брома, соответствующее
1 мл точно 0,001 н. раствора нитрата серебра, г; т — навеска образца, г;
К — содержание брома в молекуле антипирена,
Соотносительная погрешность определения для интервала
концентраций антипирена 1—15 % не превышает 3 %.
IV.3.6. Определение пероксида дикумила
в полистирольных пластиках
Методика предназначена для определения пероксида
дикумила в различных марках вспенивающегося полистирола и
сополимере стирола с акрилоннтрилом (АН).
Определение основано на экстракции пероксида дикумила
из полимера, отделении его от других экстрагируемых
примесей и добавок методом ТСХ и количественном определении по
поглощению в УФ-свете при 258 нм.
Другие пероксиды определению не мешают.
Аппаратура и реактивы
Спектрофотометр для УФ-области.
Термостат жидкостного типа.
Источник УФ-иэлучения.
Камера для хроматографироваиия.
Мнкрошприц МШ-10.
Экстрактор Сокслета.
Насос водоструйный.
Установка для удаления растворителя в вакууме (рис. IV. 7).
Цилиндр вместимостью 25 мл.
Колба мерная вместимостью 5 мл.
Колба коническая с пришлифованной пробкой вместимостью 100 мл.
Холодильник спиралевидный на шлифе.
286
w
присоединяют
Рис. IV.7. Установка для удаления растворителя:
/ — пробирка вместимостью 10 мл; 2 — переход; 3 — пипетка на шлифе
с оттянутым в виде^длинного капилляра концом.
Пипетка градуированная на 1 мл.
Гексан.
Толуол.
Тетрахлорметаи.
Хлороформ.
Петролейный эфир (Тк„и 40—70 °С).
Ацетон.
Пластины силуфол УФ254 размерами 15 X 15 см.
Выполнение анализа. Навеску полимера 2—
5 г, сответствующую 10—15 мг пероксида ди-
кумила, взвешивают с точностью до 0,0002 г
и помещают в экстрактор Сокслета. К образцу
приливают 45 мл растворителя, экстрактор
к обратному холодильнику и помещают в жидкостный
термостат с температурой 70—80 °С. Экстракцию проводят в
течение 2 ч. Для экстракции пероксида дикумила из
вспенивающегося полистирола используют гексан, из САН — смесь петро-
лейного эфира и ацетона в соотношении 1 :3 для бисера и
1 :4 для гранул. По окончании экстракции экстрактор удаляют
из термостата, пинцетом вынимают горячий набухший
полимер, а экстракт сливают в коническую колбу. Экстрактор
ополаскивают 5 мл растворителя, которые присоединяют к
экстракту.
Из полученного экстракта удаляют полностью растворитель
при разрежении, создаваемом водоструйным насосом при 30 °С,
доливая экстракт порциями в пробирку установки,
изображенной на рис. IV. 7. Сухой экстракт растворяют в 0,25 мл
хлороформа, приливая растворитель по каплям и ополаскивая при
этом стенки пробирки и капилляр. Раствор перемешивают и
дают стечь на дно пробирки.
10 мкл полученного раствора наносят микрошприцем на
пластинку силуфола, края которой предварительно обрезаны
на 1 см для предотвращения краевого эффекта. Пробу
наносят полоской длиной 1 см. Ширина полоски не должна
превышать 3—4 мм. На стартовую линию наносят также 5 мкл
раствора пероксида дикумила в хлороформе (~ 5 мг/мл) для
фиксации местоположения пероксида дикумила на хромато-
грамме экстракта. Пластинку помещают в камеру для хрома-
тографирования, в которую налито 30 мл смеси тетрахлормета-
на и толуола в соотношении 2: 1. После подъема растворителя
на высоту пластинки ее вынимают из камеры, дают
растворителю испариться и помещают пластинку под источник УФ-из-
лучения. Вещества, поглощающие УФ-свет, проявляются на
пластинке в виде темных пятен на светящемся фоне. Типичная
хроматограмма приведена на рис. IV. 8. На хроматограмме чк-
стракта отмечают пятно, отвечающее пероксиду дикумила.
VM/
Рис. IV.8. Разделение гексанового экстрата из
вспенивающегося лолнстнрола ПСВ-СВ
методом ТСХ:
/, 2 — пробы экстракта: 3 — стандарт пероксида
дикумила; пунктиром обозначены участки
снимаемого силикагеля при количественном
определении перокснда.
Прямоугольный участок
силикагеля с находящимся на нем
пероксидом переносят в мерную
колбу вместимостью 5 мл. Для
этого силикагель тщательно
счищают шпателем на кальку, а
затем количественно переносят в
мерную колбу, пользуясь
химической воронкой.
На свободном участке пластинки на том же уровне
снимают участок силикагеля равной площади, с которым
проделывают те же операции, что и с анализируемой пробой
(контрольный опыт).
К силикагелю приливают 3 мл хлороформа и экстрагируют
пероксид дикумила, интенсивно встряхивая колбу в течение
2 мин. Затем доливают хлороформ до метки, раствор
тщательно перемешивают и сразу фильтруют через воронку с
бумажным фильтром в кювету с толщиной слоя 10 мм. Измерение
поглощения полученного раствора пероксида дикумила
проводят при 258 нм относительно контрольной пробы,
которая отфильтрована через фильтр той же марки и того же
размера.
Расчет. Содержание пероксида дикумила в полимере X
(в %) вычисляют по формуле:
X = D • 0,58 • 5 • 0,25 • 100/(0,01 т • 1000),
где D— оптическая плотность анализируемого раствора; 0,58 — концентрация
раствора пероксида дикумила в хлороформе, соответствующая оптической
плотности 1, мг/мл; 5 — объем раствора пероксида дикумила в хлороформе
после экстракции с пластинки, мл; 0,25 — объем сконцентрированного
экстракта, мл; 0,01 —объем сконцентрированного экстракта, нанесенный на
пластинку, мл; т — навеска полимера, г.
Относительная погрешность определения не превышает
10%.
IV.3.7. Определение остаточного метилметакрилата
в сополимере метилметакрилата. с акриловой кислотой
Определение заключается в хроматографировании в
изотермическом режиме 10—15%-ного раствора сополимера на
сорбенте, состоящем из 5% полпэтиленгликольадипината (ПЭГА),
нанесенного на полихром-1, и количественном расчете хромато-
грамм методом внутреннего стандарта.
г«:::
Гс5>1
о
CD
С»
О
S3.
2
288
Аппаратура и реактивы
Хроматограф с детектором ионизации в пламени.
Форколонка, устанавливаемая перед испарителем, которая представляет
собой обогреваемую металлическую трубку (40X4 мм), имеющую у нижнего
основания резьбу для соединения с испарителем, в верхней части сбоку —
штуцер для подачи газа-носителя. Температура обогрева регулируется
изменением напряжения автотрансформатора, градуировка которого проводится
путем измерения температуры форколонки термопарой.
Микрошприц МШ-10.
Термопара хромель-алюмелевая.
Лупа со шкалой с ценой деления 0,1 мм.
Колба плоскодонная с пришлифованной пробкой вместимостью 50 мл.
Полиэтиленгликольадипииат.
Диметилформамид.
Ацетон.
Метилметакрилат.
Тетрахлорметаи.
Аргои.
Водород.
Воздух сжатый, очищенный от пыли и примесей.
Стекловолокно.
Приготовление насадки. Полихром-1 отсеивают на ситах,
отбирая фракцию 0,16—0,315 мм, и заливают раствором ПЭГА
в тетрахлорметане в таком количестве, чтобы сорбент
находился под слоем жидкости. Навеску ПЭГА берут из расчета
5 % к массе полихрома. Содержимое колбы периодически
перемешивают в течение 1,5 ч, затем растворитель выпаривают на
водяной бане при 50—60 °С, осторожно перемешивая
насадку. Высушенную насадку повторно просеивают и заполняют ею
две хроматографические колонки размерами 1000X3 мм.
Подготовка к анализу. Установленные в термостат
хроматографа колонки продувают в потоке газа-носителя в течение
20—30 ч при 200 °С, после чего свободные концы колонок
соединяют с детектором хроматографа.
Форколонку заполняют стекловолокном и присоединяют
к испарителю хроматографа при помощи резьбового
соединения.
Относительный коэффициент чувствительности метилметак-
рилата определяют путем хроматографирования 5—6
искусственных смесей, которые приготовлены из растворенных в ди-
метилформамиде мономера и ацетона и содержат стандарты
примерно в равных количествах в интервале концентраций
0,001—0,003%. Каждую искусственную смесь хроматографиру-
ют 3 раза при условиях анализа. Условия
хроматографирования:
Температура форколонки, °С 150
Температура термостата с колонками, °С 50
Скорость газа-носителя аргона, л/ч 2
Скорость водорода, л/ч 2
Скорость воздуха, л/ч 20
Скорость движения диаграммной ленты, мм/ч '^_ю
Предел измерения по току, А 2,5-10
289
JO 15 О
Время,мин
Рис. IV.9. Хроматограмма раствора сополимера метилметакрила-
та с акриловой кислотой:
/ — ацетон (внутренний стандарт); 2 — метплметакрилат; 3—диме-
тилформамид.
Выполнение анализа. 2—3 г сополимера
взвешивают в колбе вместимостью 50 мл с
точностью до 0,0002 г, заливают 10—15 г ди-
метилформамида и добавляют 1 %-ный
раствор ацетона в диметилформамиде так, чтобы
концентрация внутреннего стандарта
(ацетона) в растворе сополимера была примерно
равна предполагаемой концентрации
остаточного мономера. Сополимер растворяют при
комнатной температуре, перемешивая раствор
в течение 5 мин. 2—5 мкл раствора
сополимера вводят микрошприцом в хроматографиче-
скую колонку через форколонку и хромато-
графируют при условиях, указанных выше.
Общий вид хроматограммы приведен на рис. IV. 9.
Расчет. Количественный расчет хроматограмм проводят
методом внутреннего стандарта по площадям пиков компонентов,
которые вычисляют как произведение высоты пика и ширины
на половине его высоты. Содержание остаточного метилметак-
рилата в сополимере X (в %) вычисляют по формуле:
X = 5ArCCTmi/(SCTm),
где 5 — площадь пика метилметакрилата, мм2; 5СТ — площадь пика
внутреннего стандарта, мм2; К—коэффициент относительной чувствительности
метилметакрилата; Сет — концентрация внутреннего стандарта в хроматографируе-
мом растворе, %; т — навеска полимера, г; mi — масса раствора полимера с
внутренним стандартом, г.
Относительная погрешность определения не превышает
7,5%.
IV.3.8. Определение примеси волгоната в поливинилбутирале
Определение основано на взаимодействии волгоната *,
переведенного в раствор путем растворения навески полимера в
хлороформе, с метиленовым голубым и фотометрическом
определении образовавшегося комплекса при 656 нм.
Аппаратура и реактивы
Спектрофотометр.
Кюветы.
Воронка делительная вместимостью 250 мл.
Вороика химическая.
Бумажный фильтр «белая лента».
* Волгонат — синтетическое поверхностно-активное вещество,
представляющее собой смесь изомерных натриевых солей алкилсульфоновых кислот.
290
Колбы мерные вместимостью 50, 100 и 1000 мл.
Колба плоскодонная с пришлифованной пробкой вместимостью 100 мл.
Пипетки градуированные на 2 и 10 мл.
Цилиндры мерные вместимостью 5, 50 и 100 мл.
Кислота серная, 20 %-иын и 0,1 н. растворы.
Хлороформ.
Метнленовый голубой. 0,36 г метиленового голубого растворяют в 100 мл
0,1 н. раствора серной кислоты и доводят объем дистиллированной водой до
1 л.
Приготовление стандартного водного раствора волгоната.
Навеску волгоната в количестве 0,1 г, взвешенную с точностью
до 0,0002 г, растворяют в дистиллированной воде в мерной
колбе вместимостью 100 мл (раствор А). 10 мл раствора А
вносят в мерную колбу вместимостью 100 мл, доливают
дистиллированной водой до метки и перемешивают (раствор Б).
1 мл этого раствора содержит 0,1 мг волгоната.
Построение градуировочного графика. Для построения гра-
дуировочного графика в делительную воронку вносят 100 мл
дистиллированной воды, 2 мл 20 %-ного раствора серной
кислоты, от 0,2 до 1 мл стандартного раствора Б с интервалом
0,2 мл, 10 мл раствора метиленового голубого и 10 мл
хлороформа. Содержимое воронки перемешивают в течение 2 мин.
После отстаивания нижний слой сливают через бумажный
фильтр в мерную колбу вместимостью 50 мл. Экстрагирование
хлороформом повторяют 3 раза порциями по 10 мл при
встряхивании смеси в течение 1 мин.
Экстракты сливают через фильтр в ту же мерную колбу,
доводят объем хлороформом до метки и раствор
перемешивают. Оптическую плотность растворов измеряют на
спектрофотометре в кювете с толщиной слоя 20 мм при длине волны
656 нм относительно контрольного раствора, приготовленного
как указано выше, но без добавления стандартного раствора
волгоната. По полученным данным строят градуировочный
график зависимости оптической плотности от количества
волгоната (в мг).
Выполнение анализа. В колбу вместимостью 100 мл
помещают навеску анализируемого образца поливинилбутираля в
количестве 0,1—0,12 г, взвешенную с точностью до 0,0002 г,
приливают 10—15 мл хлороформа и растворяют при
нагревании на водяной бане в течение §—10 мин. Раствор переносят
в мерную колбу вместимостью 100 мл. Колбу ополаскивают
несколько раз хлороформом, который сливают в ту же мерную
колбу. Объем раствора доводят до метки и тщательно
перемешивают. 10 мл полученного раствора вносят в делительную
воронку, содержащую 100 мл дистиллированной воды, 2 мл
20 %-ного раствора серной кислоты и добавляют 10 мл
раствора метиленового голубого. Экстракцию хлороформом и
определение волгоната проводят так, как указано при
построении градуировочного графика.
291
Расчет. Содержание волгоната X (в °/о) вычисляют по
формуле:
X = a-\00- 100/(m-1000-10),
где а—содержание волгоната, найденное по градуировочному графику, мг;
т— навеска испытуемого поливинилбутираля, г.
Относительная погрешность определения не превышает 4 %.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Хмельницкий Р. А., Лукашенко И. М., Бродский Е. С. Пиролитическая
масс-спектрометрия высокомолекулярных соединений. М: Химия, 1980.
279 с.
2. Слоним И. #.//Современные физические методы исследования полимеров/
Под ред Г. Л. Слонимского. М.: Химия, 1982. С. 106—121.
3. Schmolke R., Kimmer W., Sauer W.I I Acta Polym. 1979. Bd. 30. № 7.
S 432
4. Pierre J., Van Bree /.//Kunststoffe. 1983. Bd. 73. № 6. S. 319.
5. Ryichiro /.//Kobunshi. High. Polym. Jap. 1980. V. 30. № 5. P. 357.
6. Haslam J.. Willis H. A., Squirrel D. С. М. Identification and analysis of
plastics. London e. a.: Heyden, 1980. 748 p.
7. Эляшберг М. E., Грибов Л. А., Серов В. В. Молекулярный спектральный
анализ и ЭВМ. М.: Наука, 1980. 307 с.
8. Попова Г. С, Рябикова В. М.//Пласт. массы. 1986. № 2. С. 46.
9. Фадеева Т. А., Рябикова В. М., Иванова Т. Л., Топоркова Л. В.//Пласт.
массы. 1986. № 7. С. 40.
10. Poomalai P.//Pop. Plast. 1983. V. 28. № 10. Р. 11.
11. Jorgen K7/Dan. kemi. 1984. V. 65. № 2. P. 42.
12. Анализ поли.меризационных пластмасс//В. А. Баландина, Д. Б. Гурвич,
М. С. Клещева и др. Л.: Химия, 1967. 512 с.
13. Калинина Л. С. Качественный анализ пластмасс. М.: Химия,1975. 248 с.
14. Анализ конденсационных полимеров/Л. С. Калинина, М. А. Моторина,
Н. И. Никитина, Н. А. Хачапуридзе. М.: Химия, 1984. 296 с.
15. Фиалков Ю. #., Житомирский А. Н. Физическая химия неводных
растворов. Л.: Химия, 1973. 375 с.
16. Аскадский А. А., Колмакова Л. К., Тагер А. А. и 5р.//Высокомол. соед.
1977. Т. А.19. С. 1004.
17. Хаслам Дж. Виллис Г. А. Идентификация и анализ полимеров: Пер. с
англ. М.: Химия, 1971. 431 с.
18. Sullivan J. F., Grob R. L.J/J. Chromatogr. 1983. V. 268. № 2. P. 219.
19. Буяновская А. Г., Володина М. А.ЦЖ. аналнт. химии, 1980. Т. 35. № 11.
С. 2100.
20. Lance R. С, Barnard A. J., Jou E. F.//Microchem. J. 1975. V. 20. P. 103.
21. Полюдек-Фабини Р., Бейрих Т. Органический анализ: Пер. с нем. Л.:
Химия. 1981. 624 с.
22. Siggia Sidney, Schulter D. D.//Anal. Chem. 1974. V. 46. № 6. P. 773.
23. Hoim R. D., Barksdale S. A.//Jon Chromatogr. Anal. Environ. Pollutans:
Ann Arbor. Mich., 1978. P. 99.
24. Шпигун О. А., Золотое Ю. А//Зав. лаб. 1982. Т. 48. № 9. С. 4.
25. Сотников Е. Е.//5-Я Всес. конф. по аналит. химии орг. соед. М. 1984:
Тез. докл. М.: Наука, 1984. С. 24.
26. Петере Д., Хайес Дж., Хифтье Г. Химическое разделение и измерение.
Теория и практика аналитической химии: Пер. с англ. В 2 т. М.: Химия.
1978. 816 с.
27. Химические добавки к полимерам: Справочник/Под ред. И. П. Масловой.
М.: Химия, 1981. 264 с.
292
28. Справочник по пластическим массам/Под ред. В. М. Катаева и др.
В 2 т. М.: Химия, 1975. Т. 1, 448 е.; т. 2, 568 с.
29. Поверхностно-активные вещества: Справочник/Под ред. А. А. Абрамэопа.
Л.: Химия, 1979. 376 с.
30. Vinson J. A., Grabowski W. T.//3. Chem. Educ. 1977. V. 54. № 3. P. 187.
31. Наполнители для полимерных композиционных материалов: Пер. с англ.
М.: Химия, 1981. 736 с.
32. Мальцев А. А. Молекулярная спектроскопия. М.: Изд. МГУ, 1980. 353 с.
33. Смит А. Прикладная ИК-спектроскопия: Пер. с англ. М.: Мир, 1982.
328 с.
34. Дехант Н., Данц Р., Киммер В., Шмольке Р. Инфракрасная
спектроскопия полимеров: Пер. с нем. М.: Химия, 1976. 471 с.
35. Грибов Л. А. Теория инфракрасных спектров полимеров. М.: Наука, 1977.
240 с.
36. Hummel D. О., Scholl F. К. Infrared Analysis of Polymers, Resins and
Additives: an Atlas. New York: Wiley-Interscience. V. 1. Plastics,
Elastomers, Fibers and Resins. 1969. V. 2. Additives and Processing Aids. 1973.
37. Атлас спектров химических продуктов/Под ред. В. А. Коптюга.
Новосибирск: Изд. СО АН СССР, 1975. Вып. 2. 378 с.
38. Инфракрасные спектры поглощения полимеров и вспомогательных
веществ/Под ред. В. М. Чулановского. Л.: Химия, 1969. 356 с.
39. Тарутина Л. И., Позднякова Ф. О. Спектральный анализ полимеров. Л.:
Химия, 1986. 248 с.
40. Малышев A.M., Помогайбо А. С. Анализ резин. М.: Химия, 1977. 232 с.
41. Иванова Т. Л., Рябикова В. М., Малахова Г. П. и <Эр.//Пласт. массы.
1986, № 9. С. 43; Иванова Т. Л., Рябикова В. М., Гольденберг А. Л.
и др.ЦЖ. аналит. химии. 1987. Т. 42. № 12. С. 2281.
42. Малышев А. И., Котов Ю. И., Черников А. Я. и др. Атлас спектров
химических продуктов: Выпуск 2/Под ред. В. А. Коптюга. Новосибирск:
Изд. СО АН СССР, 1984. 377 с.
43." Schroder £.//Plast. und Kautsch. 1981. Bd. 28. № 8. S. 438.
44. Ван Кревелен Д. В. Свойства и химическое строение полимеров: Пер.
с англ. М.: Химия, 1976. 414 с.
45. May R. W., Pearson Е. F., Scothem D. Pyrolysis Gas Chromatography.
London: Chem. Soc. 1977. 109 p.
46. Алексеева К. В. Пиролитическая газовая хроматография. М.: Химия,
1985. 256 с.
47. Folmer О. F.//Anal. Chem. 1971. V. 43. № 8. P. 1065.
48. Березкин В. Г., Алишоев В. Я.//Итоги науки и техники. Хроматография.
Т. 3. М.: ВИНИТИ, 1980. С. 3—28.
49. Гальченко А. Г., Халтуринский Н. А., Берлин А. Д.//Высокомол. соед.
1980. Т. А22. № 1. С. 16.
50. Руденко Б. А. Капиллярная хроматография. М.: Наука, 1978. 221 с.
51. Супина В. Насадочные колонки в газовой хроматографии: Пер. с англ.
М.: Мир, 1977. 255 с.
52. Богословский Ю. В., Анваер Б. И., Вигдергауз М. С. Хроматографиче-
ские постоянные в газовой хроматографии. М.: Изд. стандартов, 1978.
192 с.
53. Walker J. G.//J. Chromatogr. Sci. 1977. V. 15. № 7. P. 267.
54. Сотников E. E., Торосян Ж. К.//Ж. аналит. химии. 1985. Т. 40. № 10.
С. 1887.
55. Seisler H. W.//3. Mol. Struct. 1980. V. 59, Р. 15.
56. Алишоев В. Р., Березкин В. Г., Лашова С. М. и др.//Высокомол. соед.
1971. А13. С. 2777.
57. Стоев Г., Младенов //.//Химия и индустрия (НРБ). 1979. Т. 51. № з.
С 119.
58. Hallgass A., Mezzana M., Pecci G. е. a.//Po!ymer Testing. 1982. V. 3. № 1.
P. 55.
59. Алишоев В. Р., Березкин В. Г., Тинт Л. С, Мирзабаев Г. Л.//Высокомол.
соед. 1971. Т. А13. С. 2815.
293
60. Зигель А. Н., Рябикова В. М., Иванова Т. Л.//Применение хроматографии
в хим. и нефтехим. пром. Пермь, 1985: Тез. докл. Пермь, 1985. С. 70.
61. Зигель А. Н., Рябикова В. М., Малахова Г. П., Фадеева Т. А.//Пласт.
массы. 1986. № 8. С. 37.
€2. Тагер А. А., Колмакова Л. /(.//Высокомол. соед. 1980. А22. № 3.
С. 483-496.
63. Аскадский А. А., Колмакова Л. К., Тагер А. А. и 5р.//ДАН СССР. 1976.
Т. 226. № 4. С. 857.
64. Шрайнер Р., Фыозон Р., Кёртин Д., Моррил Т. Идентификация
органических соединений: Пер. с англ. М.: Мнр, 1983. 704 с.
■65. Сигги С, Ханна Дж. Г. Количественный органический анализ по
функциональным группам: Пер. с англ. М.: Химия, 1984. 671 с.
■66. Vargo J. D., Olson К. L. /Anal. Chem. 1985. V. 57. № 3. P. 672.
67. Milina R.Hi. Anal. Appl. Pyrolysis. 1981. V. 3. P. 179.
68. Squirrel D. С M.//Analyst. 1981. V. 106. № 1267. P. 1042.
■69. Hanchett V. £., Geiss R. Я.//1ВМ J. Res. and Dev. 1983. V. 27. № 4.
P. 348.
70. Пат. 4388410 США.
71. Новикова E. //.//Пласт, массы. 1982. № 4. С. 59.
72. Debal Е., Levy R//Talanta. 1978. V. 25. № 3. P. 183.
73. Свердлова С. И., Рябикова В. М., Зигель А. Н., Василенок Ю. //.//Охрана
окружающей среды при производстве пластмасс. Л.: ОНПО. «Пластпо-
лимер», 1983. С. 78.
74. Методы исследования ударопрочных полистиролов/Под ред. В. М.
Гальперина. Л.: Химия, 1975. 76 с.
75. А. с. 1060975 СССР.
76. А. с. 1010558 СССР.
77. Limouzin-Maire Y., Durocher A.//J. Marrot. Spectra 2000. 1983. V. П.
№ 84. P. 21.
78. Клещева M. С, Завьялов Ю. М., Коржова И. Т. Газохроматографический
анализ в производстве полимеризационных пластмасс. Л.: Химия, 1978.
224 с.
79. Эллиот А. Инфракрасные спектры и структура полимеров. М.: Мир, 1972.
150 с.
SO. Mullet G., Schroder E., Kludas £.//Acta Polym. 1980. V. 31. № 4. P. 239—
242.
81. Гольденберг А. Л.//Ж. прикл. спектр. 1973. Т. 19. № 3. С. 510—515.
82. Bald G., Markert H.//Z. Analyt. Chem. 1974. Bd. 268. № 5. S. 360—366.
83. Nerheim A. G.//Anal. Chem. 1975. V. 47. P. 1128—1130.
84. Danliovics A., Fares L.//J. Appl. Polym. Sci. 1983. V. 28. № 12. P. 3707—
3714.
85. Rueda D. R., Baltd Calleja F. J., Hidalgo -4.//Spectrochim. acta, 1979.
V. A35. № 7. P. 847—849.
86. Ereche P., Grenier-Loustalot M. F., Cascoin Д.//Макгото1. Chem. 1982.
Bd. 183. №. 4. S. 883—893.
87. Willbourne /.//J. Pol. Sci. 1959. V. 34. P. 569—572.
88. Badilescu S., loader M., Oprea M., Badilescu J. /.//Mater. Plast., 1970.
V. 7. P. 9.
89. Левин А. А., Муровцева Г. П., Ициксон Л. Б., Иванов В. //.//Методы
аналитических исследований и испытаний нефтей и нефтепродуктов. М.
1984. С. 199—202.
■90. Мс Rae M. A., Maddanec W. f.//Makromol. Chem. 1976. Bd. 177. № 2.
S. 461—471.
91. Воронин Н. И., Патрушева Л. Л., Филимошкин А. Г.//Синтез и
реакционная способность веществ. Томск. 1984. С. 228—231.
92. Luongo J. P.//J. Appl. Pol. Sci. 1960. V. 3. № 9. S. 302—309.
93. Киссин Ю. В., Цветкова В. И., Чирков Н. #.//Високомол. соед. 1968.
Т. А10. № 5. С. 1092
94. Gardner J. J., Cozewith C, Ver Sirade G.//Rubber Chem. and Technol.
1971. V. 44. № 4. С 1015—1024.
95. Tosi C, Simonaz?.i 7".//Angew. Makromol. Chem. 1973. Bd. 32. С 153—161.
294
96 Kimmer W, Smolke Я//Plast. und Kautsch, 1968. Bd. 15. № 11. S. 807—
811.
97. Crompton T. R. The Analysis of Plastics. London e. a.: Pergamon Press,
1984. 450 p.
98. Smith D. C.//Ind. Eng. Chem. 1956. V. 48. S. 1161.
99 Эфиндиева T 3., Ибрагимов X. Д., Гусейнов Ф. О., Портянский А. Е.Ц
Высокомол. соед. 1979. Т. A2I. № 10. С. 2392—2395.
100 Юрьева Г. А., Гусейнов Ф. 0.//Высокомол. соед. 1977. Т. A20. № 10.
С. 2401.
101 Badilescu S., Beregic S., Constantinescu V., Casadjicov D.//Mater. Plast.
1983. V. 20. № 2. P. 104—105.
102. Гольденберг А. Л., Заплетняк В. M., Ильченко П. А.//Спектроскопия
полимеров. Киев: Наукова Думка, 1968. С. 195—199.
103. Tosi С. Lachi М. P., Pinto Д.//Макгото1. Chem. 1968. Bd. 120. С. 225.
104. Попов В. П., Дуванова А. П.//Ж. прикл. спектр. 1975. Т. 23. № 6.
С. 1115
105. Koopmans R. J., Van der Linden R., Vansaut E. ^.//Polymer Eng. and
Sci. 1982. T. 22. № 14. C. 878—882.
106. Позднякова Ф. О., Попова Г. С.//Производство и переработка пластмасс
и синтетических смол. М.: НИИТЭХим, 1972. № 10. С. 23—26.
107. Ларин П. П., Ташмухамедов С. А., Ташпулатов Ю. Т., Тиллаев Р. С.If
Высокомол. соед. 1968. Т. А10. № 2. С. 310.
108. Mirabella F. M.//J. Polym. Sci.: Polym. Phys. Ed. 1982. V. 20. № 12.
P. 2309—2315.
109. Mayor J., Sodomka /.//Vyzkumny Makromol. Chem. (Brno) 1975. V. 26.
P. 601.
110. Koopmans R. J., Dommisse R., Vansant E. F., Alderweireldt F.//J.
Adhesion. 1983. V. 15. P. 117—124.
111. Munieanu D., Toader M., Laiber Af.//Mater. Plast. 1976. V. 13. № 2.
*p ay on
112. Hogea J.,' Andrei K.//Mater. Plast. 1975. V. 12. № 1. P. 17—19.
113. Czaja KJ/Zzsz. nauk. WSP Opolu Chern. 1981. V. 5. P. 83—88.
114. Позднякова Ф. О., Попова Г. С, Успенская 3. П.//Производство и
переработка пластмасс. 1976. Л» 10. С. 22—24.
115. Kimmer W., Schmolke #.//Plast. und Rautsch. 1972. Bd. 19. № 4. C. 260—
262.
116. Contreras A., Qareia-Escolar L., Acosla J. L., Sastre i?.//Revista de Plasti-
cos Modernos. 1978. V. 36. № 269. P. 633—640.
117. Takeuchi Т., Tsugo S., Sugimura Y.//J. Polym. Sci. 1968. P. Al. V. 6.
№ 12. S. 3415.
118. Gross D.l/Z. Anal. Chem. 1969. Bd. 248. № 1—2. S. 40.
119. Караснёв С, Костов Г., Милана Р., Михайлов М.//Высокомол. соед. 1974
Т. А16. № 9. С. 2163—2165.
120. Данкович А., Киссин Ю. В.//Высокомол. соед. 1970. Т. А12. № 4. С. 802—
809.
121. Forette J. E., Rozek A. L.//J. Appl. Polym. Sci. 1974. V. 18. P. 2973.
122. Barholin M., Boissier G., Dubois /.//Makromol. Chem. 1981. Bd. 182. № 7.
S 2075 2085
123. Kostkova V., Reckova G.//Petrochemia. 1979. V. 19. № 1—2. P. 1—4.
124. Serbanesku /lr.//Mater. Plast. 1974. V. 11. № 3. P. 137—139.
125. Перелыгин И. С, Пескова М. 3., Закиров 3. 3., Глинкин И. М.//Ж.
прикл. спектр. 1976. Т. 24. № 6. С. 1027—1030.
126. Андреева Н. Ю., Паншин Ю. А., Андреева М. А. и др.//Высокомол.
соед. 1981. Т. 23А. № 11. С. 2560—2566.
127. Пат. 2946763 США.
128. Пирожная Л. Н., Зубкова О. Б., Грибов Л. А.ЦЖ- прнкл. спектр. 1987.
Т. 40. № 4. С. 440.
129. Тарутина Л. И.ЦЖ- прикл. спектр. 1968. Т. 8. № 4. С. 653—656.
130. Пирожная Л. Н., Тарутина Л. И.ЦЖ. прикл. спектр. 1981. Т. 34 № 5.
С. 862-865.
295
131. Гиндин Л. Н., Абкин А. Д., Медведев С. С.//ДАН СССР. 1947. Т. 56.
С. 177—180.
132. Гольденберг A. Л., Литвинова М. А., Дунтов Ф. #.//Высокомол. соед.
1967. Т. А9. № 12. С. 2553—2554.
133. Берёзкин В. Г., Алишоев В. Р., Немировская И. Б. Газовая
хроматография в химии полимеров. М.: Наука. 1972. 287 с.
134. Mori Sadaol/Chromatographic determination of copolymer composition.
New York —Basel, 1983. V. 21. P. 187—241.
135. Cascaval С N. //Mater. Plast. 1975. V. 12. № 2. P. 88—92.
136. Cascaval С N.//Mater. Plast., 1979. V. 16. № 2. P. 120—122.
137. Sugimura Y., Nagaya Т., Tsuge 5.//Macromolecules. 1981. V. 14. № 3.
p. 520—523.
138. Alajbeg A., Arpino P., Deus-Siftar D., Guiochon G. J.US. Anal, and Appl.
Pyrol. 1980. V. 1. P. 203—212.
139. Haken J. K-, Ho D. K- M.//J. Chromatogr. 1976. V. 126. P. 239—247.
140. Haken J. K., Mc Kay T. R.//Ana\. Chem. 1973. V. 45. № 7. P. 1251—1257.
141. Nagaya Т., Sigimura Y., Tsuge S.//Macromolecules. 1980. V. 13. № 2.
P. 353—357.
142. Haney M. A., Johnston D. W., Clampitt B. //.//Macromolecules. 1983. V. 16.
№ 11. P. 1775—1783.
143. Sigimura Y., Tsuge S.//MacromoIecules. 1979. V. 12. № 3. P. 512—514.
144. Братчиков А. В., Берендеев Б. А., Иванчёв С. С.//Ж- аиалит. химии. 1984.
Т. 39. № 5. С. 903.
145. Братчиков А. В., Берендеев Б. Л.//Пласт. массы 1985. № 2. С. 54—56.
146. Братчиков А. В., Берендеев Б. А., Радионов А. Г-//Выскомол. соед. 1985.
Т. А27. № 5. С. 1107-1112.
147. Morisaki S.//Thermochim. acta. 1978. V. 25. № 2. P. 171—173.
148. Kretzschmor H. J., Gross D., Kelm /.//Analyt. Pyrolysis. Amsterdam e. a.
1977. P. 373—382.
149. Chatfield Dale A.//J. Polym. Sci.: Polym. Chem. Ed. 1983. V. 21. № 6.
P. 1681—1691.
150. Jida Т., Nakanishi M., Goto K-lli- Polym. Sci.: Polym. Chem. Ed. 1974.
V. 12. № 4. P. 737.
151. Ahlstrom D. H., Liebman S. A, Abbas K. B.//J. Polym. Sci., Polym. Chem.
Ed. 1976. V. 14. № 10. P. 2479.
152. Милина Р., Димов Я.//Нефт. и хим. 1982. Т. 16. № 1. С. 23-25.
153. Анализ полимерных материалов, сырья и сточных вод в производстве
полимеризациоииых пластмасс. Л.: ОНПО «Пластполимер». 1986. 188 с.
J54. Туркова Л. Д.. Беленький Б. Г.//Высокомол. соед. 1970. Т. 12А. № 2.
С. 467.
155. Evans D. L., Wiaver J. L., Mukherji A. K.//Anal. Chem. 1978. V. 50. P. 857.
156. Simono Г., Tanaka M., Shono T.//J. Anal, and Appl. Pyrol. 1979. V. 1.
№ LP. 77—84.
157. Туркова Л. Д., Ганичева С. Н., Нестеров В. В., Беленький Б. Г.//Ж.
аиалит. химии. 1979. Т. 34. № 5. С. 959—963.
158. Vukovil R., Onjatovic V.//J. Pol. Sci. 1970. V. 8. № I. C. 139—146.
159. Blazso M., Varhegyi G., Jakab E.//J. Anal, and Appl. Pyrol. 1980. V. 2.
№ 3. P. 177.
160. Алексеева К- В.//Ж. аналит. химии. 1972. Т. 27. № 2. С. 387.
161. Hausler К- G., Schroder E., Muting P.//Plast. und Kautsch. 1977. Bd. 24.
S 554 559
162. Krishen Л.//Апа1. chem. 1972. V. 44. № 3. P. 494—497.
163. Рапопорт Э. Л., Лопатина И. £.//Методы анализа и контроля качества
продукции в химической промышленности. М.: НИИТЭХИМ, 1978.
Вып. 10. С. 9.
164. Димов Н., Милина Р.//Высокомол. соед. 1981. Т. А23. № 11. С. 2486—
2493.
165. Musha Y., Sato Y., Katayata M.//J. Coll. Eng. Nihon Univ. 1980. V. A21.
P. 257—265.
166. Wallish K. L.HS. Appl. Polym. Sci. 1974. V. 18. № 1. P. 203.
296
167 Tanaka M., Nishimura F., Shono Г.//Апа1. Chim. acta. 1975. V. 74. № 1.
P. 119—124.
168. Корнева К- П., Америк В. В., Якобсон Ф. И./'/Пласт, массы. 1974. № 3.
С. 75—77.
169 Koopmans R. J., van der Linden R., Vansant E. F.//Polym. Eng. and Sci.
1982. V. 22. № 14. P. 878—882.
170. Okymoto Т., Takeuschi C, Cyge C.//Koge kagaku jassi. 1970. V. 73. № 4.
P. 702—704.
171. Daniel T. C, Michel J. H.//J. Gas. Chromatog. 1967. Bd. 5. № 9. S. 437—
446.
172. Шадрина Н. E., Клещёва M. С, Логинова Н. Н. и др.//Ж. прикл.
химии 1981. Т. 36. № 6. С. 1125—1129.
173 Akatsuka J., Noshiro М., Jitsugiri /.//Repts. Res. Lab./Asahi Glass. Co.
1976. V. 26. P. 111—118.
174. Cascaval С N.. Florin R. E.//J. Fluor. Chem. 1979. V. 14. № I. P. 65—70.
175. Morisaki S.//J. Chem. Soc. Japan. 1979. № 3. P. 364—369.
176. Попова Г. С., Сажин В. И., Шадрина Н. Е./УВысокомол. соед. 1979.
Т. Б21. № 10. С. 758-761.
177. Шадрина Н. Е., Клещёва М. С, Смирнова Г. М.//Пласт. массы. 1984.
№ 2. С. 44.
178. Blackwell J. 7\//Anal. Chem. 1976. V. 48. № 13. P. 1883—1885.
179. Зигель А. П., Рябикова В. М., Гольдин П. О., Свердлова С. //.//Пласт,
массы. 1983. № 5. С. 44—46.
180. Рябикова В. М., Зигель А. #.//Аиализ мономеров, полимеров,
промежуточных продуктов и сопутствующих веществ. Саратов: Изд. Саратовского
университета, 1977. С. 119.
181. Гельман Н. Э., Кшаренко Л. М.//ЖВХО им. Д. И. Менделеева. 1980.
Т. 5. № 6. С. 641.
182. Зигель А. Н., Рябикова В. М., Уткина О. Г., Коржова И. Г.//Пласт.
массы. 1985. № 6. С. 43—45.
183. Свердлова С. И., Рябикова В. М., Карпенко Г. В. и др.//Пласт. массы.
1985. № 11. С. 54—56.
184. Свердлова С. И., Рябикова В. М., Уткина О. Г., Марголина Е. 5.//Сиитез
и свойства полистнрольиых пластиков. Л.: Химия, 1985. С. 169—176.
185. Рябикова В. М., Николаева А. /7.//Ж. аиалит. химии. 1976. Т. 31. № 6.
С 1214—1216.
186. Зигель А. #., Рябикова В. М., Богомольный В. Я-, Свердлова С. И.//
Пласт, массы. 1983. № 7. С. 37—38.
187. Федорова Е. Ф., Калита О. С, Ворошилова Т. А., Платонова Н.
^.//Методы анализа и контроля качества продукции в химической
промышленности. М: НИИТЭХИМ. 1978. Вып. 2. С. 69—72.
188. Платэ И. А., Литманович А. Д., Ноа О. В. Макромолекуляриые реакции.
М: Химия, 1977. 256 с.
189. Худобина Г. В., Мальцева Л. £.//Методы анализа и контроля качества
продукции. М.: НИИТЭХИМ, 1982. № 9. С. 4—6.
190. Худобина Г. В., Мальцева Л. £.//Пласт. массы, 1983. № 6. С. 39—40.
191. Цветков В. И., Эскин В. Е., Френкель С. Я- Структура макромолекул в
растворах. М.: Наука, 1964. 712 с.
192. Рафиков С. Р., Будтов В. П., Монаков Ю. Б. Введение в физикохнмию
растворов полимеров. М.: Химия, 1978. 320 с.
193. Беленький В. Г., Виленчик Л. 3. Хроматография полимеров. М., Химия,
1978. 344 с.'
194. Будтов В. П., Подосенова Н. Г., Егорова Е. //.//Высокомол. соед. 1977.
Т. А19. № 9. С. 2160.
195. Эскин В. Е. Рассеяние света растворами полимеров. М.: Наука, 1986.
350 с.
196. Кленин В. И., Щеголев С. Ю., Лаврушин В. И. Характеристические
функции светорассеяния дисперсных систем. Саратов: Изд. СГУ, 1977. 176 с.
197. Практическое руководство по исследованию полимеров. Метод ультра-
цеитрифугирования/Под ред. Е. А. Бектурова. Алма-Ата: Мектеп, 1983.
84 с.
297
198. Сказка В. С. Седимеитационно-диффузионный анализ полимеров в
растворе. Л.: Изд. ЛГУ. 1985. 252 с.
199. Huglin /.//J. Appl. Pol. Sci. 1965. V. 9. № 12. P. 3963.
200. A. c. 966307 СССР.
201. Polymer Handbuch. New York e. a.: J. Wiley, 1966.
202. Павлова С. А., Рафиков С. Р.//Высокомол. соед. 1959. Т. 1. № 4. С. 623.
203. Виленчик Л. 3., Будтое В. Я.//Высокомол. соед. 1988. Т. АЗО. № 3. С. 501.
204. Будтое В. П., Арефьев В. В.//Высокомол. соед. 1976. Т. А18. № 6. С. 1353.
205. Будтое В. П., Консетов В. В. Тепломассоперенос в полимеризациоиных
процессах. Л.: Химия, 1983. 256 с.
206. Цветков В. Н., Лавренко П. #., Бушин С. В.//Успехи химии, 1982, 51.
№ 10. С. 1698.
207. Будтое В. Я.//Высокомол. соед. 1976. Т. 18А. № 1. С. 2606.
208. Виноградов Г. В., Вагин Ю. Н. Реология полимеров. М.: Химия, 1977.
438 с.
209. Виноградов Е. А., Малкин Ю. Л., Будтое В. Я.//Высокомол. соед. 1974.
Т. Б16. № 3. С. 250.
210. Вагин Ю Л., Григоров А. О., Малевская И. В.//Новое в реологии
полимеров. М.: Ин-т нефтехим. синтеза АН СССР, 1981. С. 203.
211. Шехмейстер И. Э., Мазин А. X., Никитин Ю. В. и др.ЦЖ. прикл. химии,
1985. № 5. С. 1169.
212. Беляев В. М., Френкель С. Я., Платонов М. Л.//Высокомол. соед. 1973.
Т. А15. № 7. С. 1913.
213. Grubistic Z., Rempp P., Benoit H.//J. Polym. Sci. 196-7. V. 5. № 2. P. 753.
214. Hamielec A., Ray W.//J. Appl. Polym. Sci. 1964. V. 13. № 6. P. 1319.
215. Пономарева Е. Л. Исследование молекулярно-массовых характеристик
полиэтилена в связи с технологией получения и свойствами: Автореф.
дис. канд. техн. наук: 01.04.19. Л.: ОНПО «Пластполимер», 1984. 24 с.
216. Кислое Е. Н., Зотиков Э. Г., Подосенова Н. Г. и <Эр.//Высокомол. соед.
1978. Т. А20. № 7. С. 1910.
217. Будтое В. П., Беляев В. М., Пономарева Е. Л.//Высокомол. соед. 1979.
Т. А21. № 12. С. 2152.
218. Мадорская А. Я., Будтое В. П., Отрадина Г. А. и <Эр.//Высокомол. соед.
1986. Т. А28. № 5. С. 952.
219. Нестеров В. В., Красиков В. Д., Изгонников В. Н. и <9р.//Высокомол.
соед. 1983. Т. А25. № 12. С. 2568.
220. Будтое В. П., Подосенова Н. Г., Зотиков Э. Г.//Высокомол. соед. 1981.
Т. А23. № 5. С. 1453.
221. Таганов И. Г.//Высокомол. соед. 1981. Т. А23. № 12. С. 2772.
222. Будтое В. П., Подосенова Н. Г.//Высокомол. соед. 1977. Т. А19. № 7.
С. 1643.
223. Будтое В. П., Подосенова Н. Г., Баллова Г. Д.//Ж. прикл. химии. 1980.
Т. 53. № 9 С. 2055.
224. Багдасарьян X. С. Теория радикальной полимеризации. М.: Наука. 1966.
300 с.
225. Будтое В. П., Подосенова Н. Г.//Известия вузов. 1988. Т. 31. Вып. 1.С. 113.
226. Будтое В. П., Зотиков Э. Г., Пономарева Е. Л., Гондельсман Е. #.//Вы-
сокомол. соед. 1985. Т. А27. № 5. С. 1094.
227. Коган С. И., Гандельсман М. И., Будтое В. Я.//Высокомол. соед. 1982.
Т. А24. № 6. С. 1148.
228. Danielewicz M., Kubin M., Vozkas S.//J. Appl. Pol. Sci. 1982. V. 27. № 9.
P. 3629.
'>29. Coldwasser J., Rudin A.//J. Lig. Chrom. 1983. V. 6. S. 2433.
230. Энтелис С. Г., Евреинов В. В., Кузаев А. И. Реакциоиноспособные олиго-
меры. М.: Химия, 1985. 304 с.
231. Беленький Б. Г., Бахтина И. А., Тараканов О. Г.//Высокомол. соед. 1974.
Т. Б16, № 7. С. 507.
232. Тютюджан И. П., Цветковский И. Б., Шляхтер Р. /(.//Високомол. соед.
1977. Т. Б19. № 2. С. 138.
233. Кузаев А Я.//Высокомол. соед. 1980. Т. Б22. № з С. 202.
298
234. Цветковский И. Б., Валуев В. И., Евдокимов В. Ф., Шляхтер Р.
Л.//Каучук и резина. 1976. № 7. С. 57.
235 Таганов II. Г., Новиков Д. Д., Коровина Г. Л.//Высокомол. сосд. 1971.
Т. Б13. № 6. С. 537.
236. Скворцова А. М., Жулана Е. Б., Горбунов А. А//Высокомол. сосд. 1981.
Т. А26. № 5. С. 946.
237. Горшков А. В., Евреинов В. В., Энтелис С. Г.//Высокомол. сосд. 1982.
Т. Л24. № 3. С. 525.
238. Цветковский И. Б., Шляхтер Р. Л.//Высокомол. соед. 1984. Т. А26. Л» 8.
С. 1777.
239 Гурьянова В. В., Алексеев О. Ф., Меланет С. £.//Высокомол. соед. 1983.
Т. Л25. № 2. С. 375.
240. Crompton Т. R. Chemical Analysis of Additives in Plastics, New York:
Pergamon Press, 1971. 162 p.
241 Walter R. W., Johnson I. F.//J. Polym. Sci.: Makromol. Reviews. 1980.
V. 15. P. 29.
242. Попова Г С.ЦЖ. прикл. химии. 1973. Т. 46. С. 419.
243. Francis V. С, Sharma Y. N., Bhardway I. S.//Angew. Makromol. Chem.
1983. V. 113 P 219.
244. Miles D. 7".//Analyst. 1974. V. 99. P. 724.
245. Protivova J., Pospisil J., Zikmund L.//J. Polym. Sci. Symp. 1973. V. 40.
P 233
246. Lichtenthaler R. G., Ranfelt F.//J. Chromatogr. 1978. V. 149. P. 533.
247. Schabron J. F.//J. Lig. Chromatogr. 1982. V. 5. № 7. P. 1269.
248. Franco S., Bruno M.//i. Chromatogr. 1983. V. 260. № 2. P. 507.
249. Sedlaf J., Foniokova E., Рас /.//Analyst. 1974. V. 91. P. 90.
250. Coupek J., Pokorny S., Protivova J. e.a.//J. Chromatogr. 1972. V. 65.
P. 279.
251. Wims A. M., Swarin S. /.//J. Appl. Polym. Sci. 1975. V. 19. № 5. P. 1243.
252. Simpson D., Currel B. ^.//Analyst. 1971. V. 96. P. 515.
253. Protivova /., Pospisil J., Holcik J.I 13. Chromatogr. 1974. V. 92. P. 361
254. Schabron J. F., Fenska L. £.//Anal. Chem. 1980. V. 52. P. I4ll.
255. Schabron J. F., Bradfild D. Z.//J. Appl. Polym. Sci. 1981. V. 26. P. 2479.
256. Schabron J. F., Smith V. J., Ware J. L.//J. Lig. Chromatogr. 1982. V. 5.
P. 613.
257. Lappin G. R., Zannucci J. S.//Anal. Chem. 1969. V. 41. № 14. P. 2076.
258. Ливанов Н. M., Ершов Ю. А., Миллер В. Б., Петухов С. И.//Ж, фи i
химии. 1977. Т. 51. № 2. С. 402.
259. Юшкевичюте С. С, Шляпников Ю. Д.//Пласт. массы. 1966. № 12. С. 62
260. Гедрайтите Г. Б., Юшкевичюте С. С.//Пласт. массы. 1975. № 4. С. ТА
261. Shepherd М. /., Gilbert /.//J. Chromatogr. 1979. V. 178. № 2. P. 435.
262. Wheeler D. 0.//Talanta. 1968. V. 15. P. 1315.
263. Kreiner J. G., Warner W. С Hi. Chromatogr. 1969. V. 44. P. 315.
264. Airaud Ch. В., Gayte-Sorhier A., Laurent P., Creuseneu R.//J. Chroniiilni'i
1984. V. 314. P. 349.
265. Amos fl.//Talanta. 1973. V. 20. P. 1231.
266. Schabron J. E., Bradfild D. Z.//Anal. Chim. Acta. 1981. V. 129. P 'li
267. Методы анализа и контроля качества продукции. М.: НИИТЭХим. 1 *•?*'
№ 7.
268. Новикова Е. И., Филиппова В. А.//Пласт, массы. 1985. № 10. С. \'\
269. Баландина В. А.. Корсаков В. Г., Малкина Н. И. и 5р.//ПлП( i m,i- ■•■
1967. № 10. С. 53.
270. Brandt H. /.//Anal. Chem. 1961. V. 33. № 10 P. 1390.
271. Parks С. A. Anal. Chem., 1961. V. 33. P. 140.
272. Denning J. A., Marshale J. /(.//Analyst. 1972. V. 97. № Ц58 1' /hi
273. Burns D. Т., Hayes W. P., Steele P.Hi. Chromatogr. 1975 V Ни
P. 241.
274. Alliet D. F., Pacco J. M.//Separation Sci. 1971. V. 6. № 1. 1' Г. i
275. Pacco J. M., Mukherji A. K.//i. Chromatogr. 1977. V. 144 I' Hi
276. Kato Y., Kido S., Watanabe H. e.a.Ui. Appl. Polym. Sci Г'/. \ ' •
№ 2. P. 629.
277. Катаева С. Е., Кофанов В. //.//Методы анализа и контроля производства
в хим. промышл. М.: НИИТЭХим, 1973. № 6. С. 15.
278. Кирхнер Ю. Тонкослойная хроматография. М.: Мир, 1981. Т. 2. 524 С.
279. Sklenarova М., Kostkova V.//Petrochimia. 1982. V. 22. № 3. P. 75.
280. Рарр /.//Papiripar, 1979. V. 23. № 4. P. 132.
281. Немодрук А. А. Аналитическая химия сурьмы. М.: Изд. АН СССР, 1978.
248 с.
282. Jagoda I. J., Prado G., Zahaye /.//Combust, and Flame. 1980. V. 37. № 3.
P. 261.
283. Read Я.//Апа1. Proc. 1984. V. 21. № 6. P. 197.
284. Brammer J. A., Frost S., Reid V. W.//Analyst. 1967. V. 92. P. 91.
285. Калмыкова Т. А.. Лазарис А. #., Константинова Е. И.//Ж. аналит.
химии. 1985. Т. 40. № 5. С. 915.
286. Иоффе Б. В., Резник Т. Л.//Ж. аналит. химии. 1980. Т. 35. № 7. С. 1410.
287. Steichen R. /.//Anal. Chem. 1976. V. 48. № 9. P. 1398.
288. Резник Т. Л. Гаэохроматографическое определение летучих примесей в
полимерных материалах методом дискретной газовой экстракции: Авто-
реф. дис. канд. хим. наук: 02.00.02. Л.: ЛГУ, 1982. 16 с.
289. Mitchell /.//Anal. Chim. Acta. 1976. V. 81. № 2. P. 231.
290. Dworak Z.//Wlok. Chem. 1980. V. 6. № 1. P. 29. № 2. P. 154.
291. Ничуговский Г. Ф. Определение влажности химических веществ. Л.:
Химия, 1977. 200 с.
292. Ларикова Т. Г., Котова В. Н., Горланова О. В., Князева Л. М.ЦЖ-
аналит. химии, 1980 Т. 35. Вып. 9. С. 1777.
293. Dinh M.//C. А. 1974. V. 81. 106648е.
294. Jeffs A. /^//Analyst. 1969. V. 94. № 1117. Р. 249.
295. Новикова Е. И., Пайкина Л. М., Комарова Г. Ф. Спектрофотометриче-
ское определение содержания примесей железа, меди, свинца, никеля,
кобальта в поливиниловом спирте для кинескопов. Л. 1974. 13 с. Деп. в
Черкассы: ОНИИТЭХим 13.09.74, № 323/74.
296. Ахрарходжаев С. С, Зимакова Е. В., Уринов Э. У. и др. Комплексоно-
метрическое определение меди в полимерных комплексных соединениях.
Ташкент. 1985, 6 с. Деп. в Ред. Узб. хим. ж. № 1220/85.
297. Макеева Н. М., Мальцева Л. Е., Попова Г. С.//Применение
хроматографии в химической и нефтехимической промышленности: Тез. докл. конф.,
Пермь, 1985 С. 71.
298. Davies J. Т. Bishop J. /^.//Analyst. 1971. V. 96. P. 55.
299. Решетникова Л. Е., Слюсарева Л. И., Шувалова Т. Н. и др. Физ.-хим.
методы аналчза Горький. 1978. № 3. С. 98.
300. Зенкевич И. Г.. Резник Т. Л., Иоффе Б. В.//Вестник ЛГУ. 1981. № 16.
С. 119.
ОГЛАВЛЕНИЕ
Предисловие 3
Глава I. ИДЕНТИФИКАЦИЯ ПОЛИМЕРНЫХ КОМПОЗИЦИЙ. 5
I. 1. Идентификация полимерной основы 6
I. 1.1. Внешний вид и физические свойства .... 6
I. 1.2. Воздействие пламени и высокой температуры 9
I. 1.3. Растворимость 13
I. 1.4. Качественное обнаружение элементов 19
I. 1.5. Цветные реакции полимеров . 26
I. 1.6. Инфракрасная спектроскопия .... 35
I. 1.7. Пиролитическая газовая хроматография 41
I. 1.8. Другие инструментальные методы 55
I. 2. Выделение и идентификация добавок и наполнителей ... 56
1.2.1. Характеристика добавок и наполнителей в полимерных
композициях 57
1.2.2. Органические добавки и наполнители . . . . 59
I. 2.3. Неорганические наполнители и добавки 63
1.3. Методики выделения и идентификации полимерной основы 66
1.3.1. Идентификация полимерной основы по продуктам пиролиза
методом ИК-спектроскопии 66
1.3.2. Идентификация полимерной матрицы методом пиролитиче-
ской газовой хроматографии - . . 70
1.3.3. Идентификация каучуков в ударопрочных полистирольных
пластиках 80
1.3.4. Выделение полиолефинов из композиционных материалов на
основе полиолефинов .... . . .82
Глава II. ОПРЕДЕЛЕНИЕ СОСТАВА СОПОЛИМЕРОВ . 83
И. 1. Инфракрасная спектроскопия 85
II. 1.1. Специфика анализа состава сополимеров методом
ИК-спектроскопии 85
II. 1.2. Определение состава сололимеров различных классов . . 90
П. 2. Пиролитическая газовая хроматография . 112
II. 2.1. Выбор условий количественного анализа . . .112
II. 2.2. Методы расчета состава сополимеров . . .116
П. 2.3. Пиролиз полимеров .119
II. 2.4. Определение состава сополимеров различных классов . . 128
II. 3. Элементный анализ ... 142
II. 4. Функциональный анализ . . 147
П. 4.1. Сополимеры винилацетата . 147
II. 4.2 Полиацетали 148
II. 4.3. Сополимеры метакрилатов и акрилатов . 149
II. 4.4. Ударопрочные полистирольные пластики . 150
II. 5. Методики определения состава сополимеров методами
элементного и функционального анализа 151
II. 5.1. Одновременное определение содержания углерода, водорода
и кремния IM
II. 5.2. Определение углерода и водорода в высокогалогенироваиных
полимерах . |!il
П. 5.3. Определение фтора во фторполимерах )Мг
II. 5.4. Определение серы в высокофторированных серусодержашич
полимерах |vi
II. 5.5. Определение азота модифицированным полумикрометаиш
Кьельдаля . . , I• • I
II. 5.6. Определение гааогенов (хлора, брома и иода) . . Ии
Ml
I]. 5.7. Определение связанного винплацетата в сополимере винил-
ацетата с этиленом 165
II. 5.8. Определение состава ударопрочного полистирола на основе
бутадиенового каучука . . I6S
II. 5.9. Определение состава АБС-пластиков 168
II. 5.10. Определение состава атмосферостойкого ударопрочного
полистирола на основе СКЭПТ 169
II. 5.11. Определение содержания бутадиенового каучука в
ударопрочном полистироле, АБС- и МБС-пластиках, в
гель-фракциях этих пластиков и в исходном каучуке . . 170
Глава III. ОПРЕДЕЛЕНИЕ МОЛЕКУЛЯРНОЙ МАССЫ И МОЛЕКУ-
ЛЯРНО-МАССОВОГО РАСПРЕДЕЛЕНИЯ ПОЛИМЕРОВ 172
III. 1. Некоторые представления о молекулярной массе и молеку-
лярно-массовом распределении ... . 175
III. 1.1. Средняя молекулярная масса и ММР , . 174
III. 1.2. Количественные характеристики ММР . . 176
III. 1.3. Аналитическое описание ММР , . 178
III. 2. Методы определения молекулярных масс полимеров .... 180
III. 2.1. Метод седиментации — приближение к седиментациониому
равновесию 181
Ш.2.2. Сочетание коэффициентов седиментации и диффузии .... 184
III. 2.3. Определение молекулярных масс по коэффициентам
седиментации ....... ... . 185
III. 2.4. Метод вискозиметрии 186
III. 2.5. Определение молекулярных масс нерастворимых полимеров 196
III. 3. Методы определения молекулярно-массового распределения 200
III. 3.1. Метод скоростной седиментации .... . 20О
III. 3.2. Метод гель-проникающей хроматографии 206
III. 3.3. Особенности интерпретации данных о ММР в терминах
констант элементарных реакций 219
III. 4. Применение метода ГПХ к определению особенностей
структуры макромолекул 227
III. 4.1. Определение параметров длинноцепной разветвленности
макромолекул 227
III. 4.2. Определение композиционной неоднородности сополимеров 230
III. 4.3. Хроматографический анализ полимеризационноспособных оли-
гомеров . . .... 231
Глава IV. ОПРЕДЕЛЕНИЕ ДОБАВОК И ПРИМЕСЕЙ В
ПОЛИМЕРНЫХ МАТЕРИАЛАХ . 235
VI. 1. Определение добавок . 237
IV. 1.1. Стабилизаторы . . 237
IV. 1.2. Пластификаторы . 253-
IV. 1.3. Наполнители и порообразователи . 257
IV. 1.4. Антипирены . . 260
IV. 1.5. Пероксиды .... .261
IV. 2. Определение примесей . ■ 264
IV. 2.1. Остаточные мономеры . . 264
IV. 2.2. Вода . . .269
IV. 2.3. Прочие примеси ■ 274
IV. 3. Методики определения добавок и примесей в полимерах . . 277
IV. 3.1. Определение ирганокса 1010 (фенозана 23) в композициях
полиэтилена низкого давления, наполненных техническим
углеродом . 277
IV. 3.2. Определение фосфорсодержащих стабилизаторов в
полиэтилене 278
IV. 3.3. Определение глицерина в антисептических поливинилспирто-
вых пленках ... ■ . * i . . . > s • i 280
302
IV. 3.4. Определение фторсодержащего наполнителя (апатита) п
полиэтилене .... . 282
IV. 3.5. Определение бромсодержащих антипиренов d полистироле . 284
IV. 3.6. Определение пероксида дикумила в полистирольных
пластиках Й8С»
IV. 3.7. Определение остаточного метилметакрилата в сополимеро МО-
тилметакрилата с акриловой кислотой ....... . WH
IV. 3.8. Определение примеси волгоната в поливинилбутирале . . 1'!И1
Библиографический список . . '.'0;1
ПРОИЗВОДСТВЕННОЕ ИЗДАНИЕ
Галина Сергеевна ПОПОВА
Владилен Петрович БУДТОВ
Валентина Михайловна РЯБИКОВА
Галина Вячеславовна ХУДОБИНА
АНАЛИЗ
ПОЛИМЕРИЗАЦИОННЫХ
ПЛАСТМАСС
Редактор А. Е. П и н ч у к
Переплет художника Ю. Б. Осенчакова
Техн. редактор 3. Е. Маркова
Корректор Л. С. Александрова
ИБ № 1899
Сдано в набор 25.06.87. Подписано в печать 01.12.87. М-24343. Формат бумаги
60X90'/ie. Бумага тип. № 2. Литературная гарнитура. Высокая печать.
Усл. печ. л. 19,0. Усл. кр.-отт. 19,0. Уч.-изд. л. 20,94. Тираж 5700 экз.
Зак. 632. Цена 1 р. 40 к. Изд. Ш 2741.
Ордена «Знак Почета» издательство «Химия», Ленинградское отделенге
191186, Ленинград, Д-186. Невский пр., 28.
Ленинградская типография № 2 головное предприятие ордена Трудового
Красного Знамени Ленинградского объединения «Техническая книга*
им. Евгении Соколовой Союзполиграфпрома при Государственном комитете
СССР по делам издательств, полиграфии и книжной торговли. 198052,
г. Ленинград, Л-52, Измайловский проспект, 29.
Ленинградская типография Кг 4 ордена Трудового Красного Знамени
Ленинградского объединения «Техническая книга» им. Евгении Соколовой
Союзполиграфпрома прн Государственном комитете СССР по делам
издательств, полиграфии н книжной торговли. 191126, Ленинград,
Социалистическая ул., |4, эак. 45.
АНАЛИЗ
П0ЛЙМЕШЗА1ДИ0ННЫХ
кйЯ
■ ■
| Ш ] ••■
1 i
В ■■■
•■•":-•-
- мм
3 й
. •*>•:•:- -^л ; ■ ?• ■ 1Ш ■:"--- ■ SB