/






























































































































































































































































































































































































Автор: Зырина Н Г. Орлова Д.С.
Теги: неорганическая химия почвоведение геодезия геодезические науки издательство мгу
Год: 1980




Текст
ФИЗИКО-ХИМИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ
ПОЧВ
Под ред Н Г. Зырина, Д С Орлова
Допущено Министерством высшего и сред-
него специального образования СССР в
качестве учебного пособия для студентов
почвенных и агрохимических специальностей
университетов и сельскохозяйственных
вузов
ИЗДАТЕЛЬСТВО
МОСКОВСКОГО
УНИВЕРСИТЕТА
1980
Авторы:
Л. А. Воробьева, Г. И. Глебова,
£. И. Горшкова, Н. М. Гриндель,
Н. Г. Зырин, Г. В. Мотузова,
А. И. Обухов, Д. С. Орлов, Н. Н. Осипова,
Т А. Соколова
Рецензенты:
кафедра почвоведения Воронежского ун-та;
докт. 1еолого-минералогических наук И. И. Горбунов
, мляая тятей
I им. Горького
I МГУ___________
6600- //-до ;Ь
Физико-химические методы исследования почв.
Под ред. Н. Г. Зырина, Д. С. Орлова. — М.: Изд-во
МГУ, 1980, 382 с. с ил.
В пособии из (агаются теоретические основы современных физи-
ко-химических методов исследования почв (потенциометрического,
кондуктометрического, потяро! рафического, спектрального, рентгенов-
ского, термического и др ) Дано потребное описание методов, оха-
рактеризованы области их применения в почвоведении
° 077(02)-80 8°~137 3802000000
© Издательство Московского университета, 1980 г.
Введение
Современное почвоведение и агрохимия используют широкий
набор методов исследования, в числе которых одно из централь-
ных мест занимают физико-химические, или инструментальные,
методы анализа. Это большая группа методов, в которую часто
включают все приемы определения вещественного состава почв,
пород, природных вод, органических остатков, приемы изучения
свойств почв и слагающих их компонентов, приемы определения
строения органических и минеральных компонентов почв, основан-
ные на количественном измерении физических свойств изучаемых
объектов. Во многих случаях измеряют физические свойства почв
пли пород непосредственно в их естественном состоянии (напри-
мер, спектры поглощения или отражения; потенциалы, возникаю-
щие на погруженном в почву электроде; магнитные свойства);
но широко распространены и такие методы, при использовании
которых на почву оказывается то или иное и часто весьма жесткое
воздействие (нагревание до 1000°С и более, облучение электрона-
ми, испарение пробы почвы в пламени вольтовой дуги и др.),
вызывающее изменение состояния вещества, а происходящие при
этом изменения свойств фиксируются измерительной аппаратурой.
В числе наиболее употребительных методов, чаще всего ис-
пользуемых для изучения состава, свойств и строения почв,
почвенных компонентов и сопряженных с почвами природных тел,
[следует назвать:
1 1. Потенциометрические методы, применяемые в почвоведе-
нии и агрохимии для определения pH, окислительно-восста-
новительных потенциалов, активностей ионов натрия, калия,
кальция, хлорид-ионов, нитрат-ионов и др.
2. Кондуктометрические методы, используемые для определе-
ния солесодержания в почвах и почвенных растворах.
3. Полярографические методы, широко применяемые для ко-
личественного определения многих катионов и анионов.
3
чаще — микроэлементов, а также для определения некото-
рых компонентов органических веществ почвы и изучения
их взаимодействия с ионами металлов.
4. Фотометрические и нефелометрические методы, позволяю-
щие количественно определить практически любые компо-
ненты почв и почвенных растворов.
5. Спектрофотометрические методы, используемые как для
определения количественного содержания различных эле-
ментов и веществ в почвах, так и для изучения структуры
гумусовых веществ, минералов тонкодисперсных фракции
и продуктов их взаимодействия. Изучение спектров отра-
жения открыло новые возможности для объективной коли-
чественной характеристики окраски почв.
6. Метод эмиссионной пламенной и атомно-абсорбционной
спектрофотометрии, используемые для количественного
определения различных элементов в вытяжках из почв,
в почвенных растворах и в растворах, полученных в ре-
зультате полного разложения почвы.
7. Эмиссионный спектральный анализ, успешно используемый
для количественного определения элементного состава почв
без их предварительного разложения.
8. Электронная просвечивающая и растровая микроскопия,
позволяющая изучать микростроение почв, органических и
минеральных составляющих почвы и идентифицировать
минералы топкодисперсной фракции почв.
9. Методы рентгеноструктурного анализа, широко используе-
мые для идентификации и полуколичественного определе-
ния минералов тонкодисперсной фракции почв и перспек-
тивные для изучения строения специфических гумусовых
веществ.
10. Методы термического анализа, применяемые в почвоведе-
нии для изучения минералогического состава почв и почвен-
ных коллоидов.
Кроме перечисленных в почвоведении применяются и многие
другие инструментальные методы. Значительные успехи достигну-
ты в анализе почв методами рентгеновской флуоресценции, ней-
тронно-активационного анализа, газожидкостной хроматографии,
гель-хроматографии, пиролиз-масс-спектрометрии, люминесценции
и др. В настоящем руководстве излагаются только те методы,
которые в настоящее время наиболее широко применяются в поч-
венно-химических производственных или исследовательских лабо-
раториях.
Физико-химические методы имеют ряд особенностей, отличаю-
щих их от классических методов химического анализа: грави-
метрического (весового) и титрометрического (объемного).
Конечно, граница между группами методов не абсолютна. Практи-
чески все физико-химические приемы исследования базируются
на предварительно изученной (установленной) зависимости сос-
4
тав — свойства. Причем свойство может характеризовать как на-
тивную почву, так и почву, переведенную в то или иное состояние.
Поэтому первым этапом разработки и применения любого физико-
химического метода является установление зависимости между
составом исследуемой пробы и тем или иным ее свойством, выра-
женным обычно математически в виде формулы или графика.
Так, в потенциометрии используется связь между активностью
иона в почвенном растворе и потенциалом электрода, в эмиссион-
ной спектроскопии — связь между интенсивностью излучений
определенных длин волн с содержанием отдельных элементов в
пробе почвы, переведенной в состояние плазмы.
Такие формулы имеют обычно универсальный характер, что
позволяет использовать их для анализа различных объектов;
специфичность свойств изучаемых веществ, характер реакций,
особенности изучаемых почв находят отражение в величинах пара-
метров уравнений. Это позволяет применять одни и те же при-
боры и способы исследования для различных объектов. Однако
было бы неверным считать, что любой физико-химический метод,
разработанный для изучения непочвенных объектов, может быть
механически перенесен в почвоведение. Почва очень сложный
объект исследования и анализа; в ее состав входят почти все эле-
менты периодической системы Д. И. Менделеева, причем соотно-
шение элементов с аналитической точки зрения часто бывает
неблагоприятным; почва содержит как растворимые, так и не-
растворимые соединения; в их состав входит большой набор
специфических органических соединений, многие из которых
трудно- или полностью нерастворимые в известных растворите-
лях. И органические и минеральные части почвы содержат обычно
серии близких по строению и свойствам веществ, что затрудняет
их идентификацию и препаративное разделение. Поэтому внедре-
ние новых инструментальных методов в почвоведение — процесс
творческий, требующий хорошей подготовки исследователя, тща-
тельной проверки условий эксперимента, смелости мышления.
Другая особенность физико-химических методов заключена в
том, что показатели, характеризующие свойства вещества или
системы в обычных условиях, не зависят от взятого объема ве-
щества. Потенциал электрода не зависит от того, в какой объем
раствора он погружен; интенсивность излучения электромагнит-
ных колебаний веществом, которое вводится в пламя горелки, не
зависит от общего объема введенного раствора, а определяется
только скоростью его подачи и концентрацией. Это вносит ряд
особенностей в технику работы по сравнению с обычными химиче-
скими методами и часто позволяет значительно упростить процеду-
ру исследования.
Очень большое преимущество и особенность физико-химических
методов состоит в том, что во многих случаях они позволяют изу-
чить состав, строение и свойства почв, не производя с ними ни-
каких химических операций. Почвоведы уже располагают электро-
5
дами для определения pH, рСа, рК, pNa, pNO3; такие электроды
можно погрузить в почву непосредственно в природной обстановке
и постоянно или периодически снимать показания, записывая
результат на диаграммную ленту. Методом инфракрасной спектро-
метрии можно получить подробную характеристику важнейших
атомных групп и химических связей в неизмененном образце почв,
идентифицируя исходные соли и минералы. Возможность работы
с ненарушенными образцами имеет значение в двух аспектах
Во-первых, с помощью этого приема мы получаем информацию об
истинном состоянии почвы и ее компонентов, тогда как при хими-
ческом анализе мы составляем предположительное заключение
об объекте на основе данных о составе растворов. Во-вторых,
именно такие методы позволяют осуществлять дистанционные
измерения как с помощью постоянно погруженных в почву датчи-
ков, так и путем измерения спектров отражения почв с помощью
приборов, установленных па самолетах или искусственных спут-
никах. Конечно, не все инструментальные методы обладают таки-
ми возможностями; в эмиссионном спектральном анализе проба
почвы полностью испаряется в пламени вольтовой дуги.
В большинстве случаев проведение анализов физико-химиче-
скими методами требует немного времени; это — экспрессные ме-
тоды. И хотя используется часто дорогостоящая аппаратура, все
же достигается большая экономия средств и сил благодаря быст-
роте определения. Вместе с тем большая часть методов обладает
и высокой чувствительностью. Это значительно расширяет возмож-
ности исследователя и одновременно позволяет снизить расходы
реактивов
Для проведения физико-химических анализов нужна специаль-
ная, зачастую не всегда легкодоступная аппаратура, безотказно
работающая только при умелом обращении с ней. Поэтому успех
и более широкое использование физико-химических методов в
почвоведении зависят прежде всего от подготовки почвоведов в
этой области. За последние 10—15 лет устройство приборов резко
усложнилось. Если, например, первые модели полярографов Гей-
ровского (1957—1960 гг.) почвовед-химик мог самостоятельно
отладить и даже выполнить несложный ремонт, то для ремонта
современных полярографов нужны специалисты по электронной
технике. Еще более сложные требования предъявляются к уста-
новке, наладке и ремонту дифрактометров, электронных микро-
скопов и т. п. Всю эту часть работы в лабораториях должны
выполнять специалисты-инженеры, в то время как почвовед-ис-
следователь должен хорошо знать принцип метода и принцип
устройства прибора, порядок работы на нем, возможности и не-
достатки аппаратуры, источники ошибок.
Особенно важно для почвоведа-исследователя глубоко пони-
мать реальное значение и пути использования различных мето-
дов, уметь правильно выбрать метод и извлечь из полученных
результатов максимум информации. Недопустимо использовать
6
сложные методы и дорогостоящую аппаратуру только для полу-
чения иллюстративного материала, не обязательного для решения
стоящей перед почвоведом задачи.
Данное руководство составлено с таким расчетом, чтобы озна-
комить читателя с основами теории методов, принципами дейст-
вия, с порядком работы на приборе и показать рациональные пути
применения отдельных методов. Руководство рассчитано на сту-
дентов университетов по специальности почвовед-агрохимик, а
также на сотрудников почвенных и сельскохозяйственных лабора-
торий, использующих в своей работе физико-химические методы
для исследования строения, свойств и состава почв.
i
Глава 1
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
ИОНОВ ВОДОРОДА, НАТРИЯ, КАЛИЯ,
КАЛЬЦИЯ, ХЛОРИД-ИОНА, НИТРАТ-ИОНА
ПОТЕНЦИОМЕТРИЧЕСКИМ МЕТОДОМ
Активности ионов и солей в почвах относятся к числу важней-
ших физико-химических характеристик почвы. В теории и прак-
тике почвоведения и агрохимии используются не только абсолют-
ные величины активностей различных ионов^ но и их логарифми-
ческое выражение-— pH, pNa, рК и т. п. С измерением величин pH
связаны определение буферности почв и расчет потребности кис-
лых почв в известковании. Эти же величины лежат в основе
понятий потенциалов элементов питания: калийного, известкового,
фосфатного.
Измерение этих показателей, как и окислительно-восстанови-
тельных потенциалов, наиболее просто и надежно выполняется с
помощью потенциометрии.
ПРИНЦИП
ПОТЕНЦИОМЕТРИЧЕСКОГО
МЕТОДА.
ЗНАЧЕНИЕ
И ОБЛАСТЬ ПРИМЕНЕНИЯ
В ПОЧВОВЕДЕНИИ
Потенциометрические методы исследования и анализа почв
основаны на использовании электродов, потенциал которых обус-
ловлен каким-либо свойством или концентрацией компонента
раствора, находящегося в равновесии с электродом. Для проведе-
ния анализа потенциометрическим методом исследователь должен
располагать индикаторным электродом и устройством для изме-
рения его потенциала (потенциометр).
Индикаторный электрод может быть выполнен из стекла, ме-
талла (с покрытием из окислов солей или без такового), ионооб-
менных смол. Рабочая поверхность электрода может быть твердой
или жидкой. Главная особенность и требование к индикаторному
9
лектроду в том, что при погружении его в испытуемую систему
почв}, почвенный раствор) на поверхности электрода должна
фотекать реакция, в результате которой электрод приобретает
тотенциал, обусловленный только изучаемым показателем.
Например, на поверхности платинового платинированного
электрода, погруженного в водный раствор, в присутствии молеку-
лярного водорода протекает реакция
Н2^2Н^2Н^ - 2е.
Скачок потенциала на границе раздела фаз платина — раствор
прямо зависит от концентрации водородных ионов. Это позволяет
с помощью такого электрода определять pH растворов или сус-
пензий. Чтобы найти потенциал индикаторного электрода, изме-
ряют электродвижущую силу (э. д. с.) элемента, составленного в
простейшем случае из индикаторного электрода и электрода
сравнения. Для измерения э.д. с. используют потенциометры с
компенсационной схемой или измерительные устройства с очень
высоким входным сопротивлением. Благодаря этому можно не
принимать во внимание влияние внутреннего сопротивления эле-
мента и в результате однократного измерения сразу находить не
падение напряжения во внешней цепи элемента, а его э. д. с., рав-
ную разности потенциалов на концах разомкнутого элемента.
В почвоведении и агрохимии потенциометрические методы
нашли применение для измерения концентрации или активности
ионов водорода, натрия, калия, кальция, хлора и некоторых
микроэлементов в почвенных вытяжках, суспензиях, пастах и для
измерения окислительно-восстановительного потенциала (ОВП)
почв. Впервые они были применены для определения активности
водородных ионов, или pH водных вытяжек и суспензий. Методы
определения pH почв были разработаны в начале XX в. Актуаль-
ная реакция почвы при помощи водородного электрода была
измерена Зайделем в 1913 г. С тех пор электрометрическое опре-
деление pH получило широкое распространение. Начиная с 30-х го-
дов в практике почвенных исследований широко используется
метод измерения ОВП. Первые полевые наблюдения ОВП вы-
полнены Н. П. Ремезовым в 1928 г. Он составил и первое руко-
водство «Физико-химические методы исследования почв» (1931 г.).
В конце 30-х годов С. Е. Маршалл применил глинистые мемб-
ранные электроды для определения активности некоторых катио-
нов в почвенных суспензиях. Исследования электродных функций,
выполненные Б. П. Никольским в 1937—1939 гг., положили нача-
ло работам П. А. Крюкова, Н. А. Комаровой, Н. О. Авакяна,
Д. С. Орлова по использованию в почвенных лабораториях стек-
лянных электродов, обратимых к металлическим катионам. Сна-
чала это были натриевые, а затем и калиевые электроды.
В настоящее время разработаны и внедрены новые ион-селек-
тивные электроды, позволяющие быстро и достаточно надежно
10
определять также активности ионов кальция, ряда анионов: хло-
рид-иона, сульфатов, сульфидов и др. Предложены электроды
селективные к микроэлементам: йоду, брому, фтору, кадмию, ме-
ди, свинцу, марганцу.
Значение потенциометрических методов в полевых и лабора-
торных почвенных исследованиях очень велико. Это обусловлено
несколькими причинами. Важное значение имеет то обстоятельст-
во, что потенциометрически определяются активности компонен-
тов, а не их концентрации. Почвы — сложные многофазные и
многокомпонентные системы, имеющие обычно невысокую влаж-
ность; твердые частицы почвы несут положительные и отрицатель-
ные заряды, а почвенный раствор содержит большой набор
растворенных органических и минеральных веществ и находится
в тонких порах (капиллярах), где сильно выражен пристенный
эффект. В таких условиях активность компонента может значи-
тельно отличаться от его концентрации, и для оценки реальных
свойств почв особое значение приобретают величины активности.
Не меньшее значение имеет также возможность изучать почвы
без изменения их свойств и состава. Ион-селектнвные электроды
и электроды для измерения ОВП можно погружать прямо в поч-
ву, а затем измерять э. д. с., не воздействуя на почву никакими
химическими реактивами и даже водой. Таким способбм удается
получать характеристики почв в их естественном состоянии, что
более правильно отражает природу и механизм происходящих в
почве процессов. Немаловажной является возможность исследо-
вания почв непосредственно в природе, в полевой обстановке.
Созданы достаточно портативные приборы, пригодные даже для
сложных экспедиционных условий.
Наконец, потенциометрические методы легко можно использо-
вать для дистанционных измерений свойств почв, регистрации
показателей с помощью самописцев, непрерывного контроля за
состоянием почвы в полевом или лабораторном опыте. Активность
ионов калия, кальция, фосфатов и других служит показателем
мгновенной обеспеченности растений теми или иными элементами
питания.
Таким образом, потенциометрические методы исследования
почв дают новую и часто принципиально иную информацию о
почвах, чем методы измерения концентрации. Потенциометриче-
ские методы отличаются быстротой исполнения и мог^т быть час-
тично или полностью автоматизированы.
Потенциал
индикаторного электрода
В основе потенциометрических методов лежит уравнение Нерн-
ста, связывающее потенциал электрода с составом равновесного
раствора. В случае, когда металлический электрод погружен в pa-
ll
створ соли, содержащей катион того же металла, то для равно-
весного состояния скачок потенциала Е между электродом и
раствором можно вычислить по уравнению Нернста:
Е = Ео Ч —— In ам,
nF
где ам—активность катионов металла М в равновесном раство-
ре, R — газовая постоянная, Т — абсолютная
F — число Фарадея, п — изменение
температура в °К,
заряда иона в результате
электрохимической реакции
и Ео — потенциал стандарт-
ного электрода, т. е. элект-
рода, находящегося в равно-
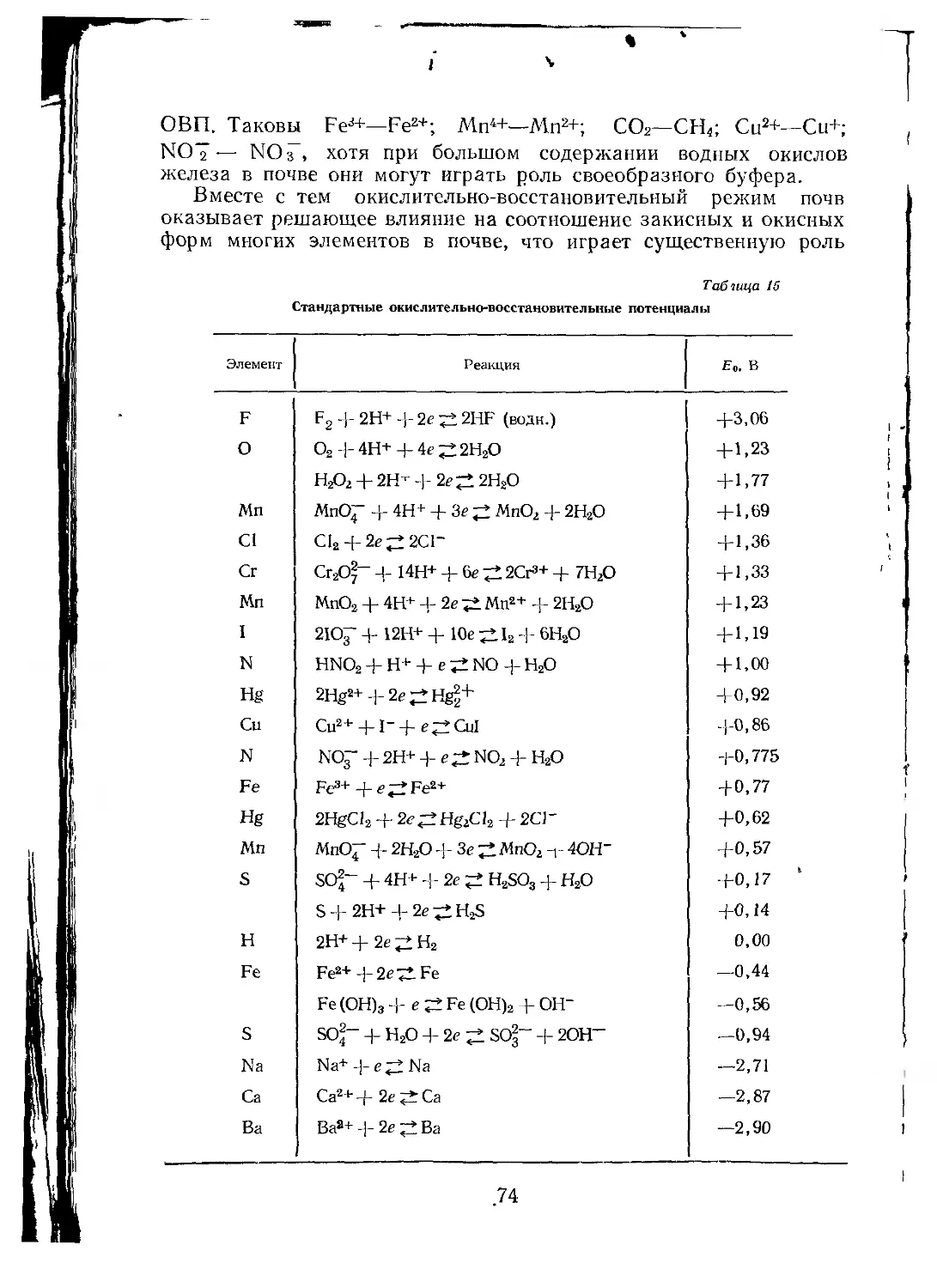
Таблица 1
RT
Значение коэффициента О — 2,303---
t, °C О t, °с О С, °C О весии с раствором, для кото- рого Ядг=1. Если Пд!=1, тог- да 1ппм=0 и Е=Е0. При
переходе от натуральных ло-
10 0,0561 17 0,0575 24 0,0589 гарифмов к десятичным по-
11 0,0563 18 0,0577 25 0,0591 лучим:
12 0,0565 19 0,0579 26 0,0593
13 14 0,0567 0,0569 20 21 0,0581 0,0583 27 28 0,0595 0,0597 Е =Е0 1 2,3031g ам.
15 0,0571 22 0,0585 29 0,0599
16 0,0573 23 0,0587 30 0,0601 Коэффициент уравнения обозначается буквой б («тэ-
та»). Если потенциал выра-
жен в вольтах, а п—1, тогда при 20°С (или 293,15° К) величина
Ф—0,0581 В, при 25°С 0=0,0591 В (табл. 1). При увеличении тем-
пературы на 1° величина 0- увеличивается в среднем на 0,0002 В.
Это уравнение справедливо для случая простой электродной реак-
ции восстановления катионов металла на
ния атомов металла до катионов:
элект
роу
е или окисле-
М М+ е.
Однако в электродной реакции может одновременно участво-
вать несколько веществ (А, В, С, D) в разных соотношениях,
тогда в общем виде можно записать, что если на электроде осу-
ществляется реакция
nA - 6В сС — cfD - не,
где а, Ь, с, d — стехиометрические коэффициенты, [е] — концент-
рация свободных электронов, то для вычисления потенциала не-
обходимо подставить в уравнение Нернста выражение константы
равновесия реакции; в данном примере константа (А) равна:
асс • adD • И"
А =----------------
Тогда
Й ,„аС • aD- И"
/2 „О
аА аВ
12
Принимая концентрацию свободных электронов, поступающих
из металла электрода, достаточно высокой и практически не из-
меняющейся в ходе реакции, мы можем считать [е] величиной
постоянной и объединить ее с константой уравнения Ео, не вводя
каких-либо обозначений.
В результате получим:
Чтобы электрод можно было использовать в аналитических
целях, потенциал электрода должен зависеть только от одного
из компонентов раствора. Свойство электрода изменять потенциал
под влиянием только одного (определяемого) компонента раство-
ра и не реагировать на изменение концентрации других компо-
нентов называется селективностью (избирательностью) электрода.
Следовательно, ион-селективные электроды — это электроды, из-
бирательно реагирующие только на ионы определенного вида.
Так, потенциал Н-стеклянных электродов практически не зависит
от присутствия ионов Na+, К+, Са2+, M.g2+; потенциал Na-стеклян-
ного электрода не зависит в определенных пределах от концентра-
ции ионов Н+, К+, Са2+. Перед использованием любых видов
электродов необходима проверка подчинения потенциала электро-
да уравнению Нернста и их селективности.
Электроды сравнения
Потенциал отдельно взятого индикаторного электрода невоз-
можно измерить обычными потенциометрическими методами.
На вход потенциометра, как во всякой электрической цепи, под-
ключаются два проводника. Один из них соединяет потенциометр
с индикаторным электродом 1, второй (рис. 1) служит для замы-
кания электрической цепи. Соединительный провод нельзя просто
опустить в испытуемый раствор. В противном случае в зависимости
от состава металла, из которого изготовлен проводник, и от сос-
тава раствора на таком проводнике возникнет тот или иной потен-
циал. Другими словами, проводник станет выполнять роль второ-
го электрода, потенциал которого остается неизвестным. Измерив
э. д. с. такой системы, невозможно вычислить потенциал индика-
торного электрода. Для измерения потенциала электрода состав-
ляется элемент из двух или более электродов, потенциалы кото-
рых (кроме индикаторного) известны п постоянны в ходе экспе-
римента.
Электроды с известным и постоянным скачком потенциала,
используемые в измерительных цепях, называют электродами
сравнения, или вспомогательными электродами (или стандартны-
ми полуэлементами). В качестве электродов сравнения выбирают
13
обычно так называемые неполяризующиеся электроды, особен-
ность которых заключается в том, что потенциал этих электродов
практически не изменяется (они не поляризуются) при прохож-
дении через них тока.
Рис. 1. Элемент для
определения потен-
циала индикаторного
Н-стеклянного элек-
трода:
1 — стеклянный элек-
трод с водородной
функцией, 2 — кало-
мельный электрод
сравнения, 3 — испы-
туемый раствор
Рис 2. Типы каломельных электродов:
1 — раствор КС1, 2 — кристаллы КС1, 3 — каломель,
4 — ртуть, 5 — платиновый контакт, 6 — соединитель-
ный отросток, 7 — бумажная пробка
Устройство и принцип действия неполяризующегося электрода
сравнения удобно рассмотреть на примере наиболее широко рас-
пространенного каломельного электрода. Предложены различные
конструкции каломельных электродов; две из них показаны на
рис. 2. Электродный сосуд (слева) имеет в нижней части шарооб-
разную форму. В дно шарообразной части сосуда впаяна платино-
вая проволока, через которую осуществляется контакт со ртутью.
Перед заполнением сосуд и платиновый контакт тщательно про-
14
мывают концентрированной серной кислотой и дистиллированной
водой. Часть платиновой проволоки, находящуюся внутри сосуда,
амальгамируют. Для этого в сосуд наливают раствор Hg2(NOs)2,
подкисленный азотной кислотой, а наружный конец платиновой
проволоки подсоединяют к отрицательному полюсу аккумулятора
на 2В. В качестве анода также используют платиновую проволо-
ку, которую подсоединяют к положительному полюсу того же
аккумулятора и погружают в раствор внутри сосуда. Через раствор
пропускают ток примерно в течение 1 мин, за это время плати-
новая проволока, впаянная в стенку сосуда, покрывается серым
налетом ртути По окончании амальгамирования раствор из сосу-
да выливают, а сосуд тщательно промывают дистиллированной
водой и высушивают.
В сухой сосуд наливают ртуть с таким расчетом, чтобы тонкий
слой ртути находился и в цилиндрической части сосуда. Поверх
ртути наливают немного суспензии Hg2Cl2 в растворе КО, кон-
центрация КС! должна соответствовать виду приготовляемого
электрода (насыщенный, нормальный или децинормальный,
табл. 2).
Т аб ища 2
Потенциалы каломельных электродов, В
°C Концентрация KQ в электроде °C Концентрация КС1 в этектроде
насыщен- ный 1,0 н 0,1 н насыщен- ный 1,0 Н 0 1 н
10 0,2536 0,2864 0,3374 21 0,2464 0,2838 0,3367
11 0,2529 0,2862 0,3373 22 0,2458 0,2835 0,3367
12 0,2523 0,2859 0,3373 23 0,2451 0,2833 0,3366
13 0,2516 0.2857 0,3372 24 0,2445 0,2830 0,3366
14 0,2510 0,2854 0,3372 25 0,2438 0,2828 0,3365
15 0,2503 0,2852 0,3371 26 0,2431 0,2826 0,3364
16 0,2497 0,2850 0,3370 27 0,2425 0,2823 0,3364
17 0,2490 0,2847 0,3370 28 0,2418 0,2821 0,3363
18 0,2483 0,2845 0,3369- 29 0,2412 0,2818 0,3363
19 20 0,2477 0,2471 0,2842 0,2840 0,3369 0,3368 30 0,2405 0,2816 0,3362
После отстаивания суспензии толщина слоя каломели па по-
верхности ртути должна быть 1—2 мм Затем отверстие соедини-
тельного отростка закрывают и наливают в сосуд раствор КО
Если готовится насыщенный каломельный электрод, то заливают
раствор КС1, насыщенный при 60°. Горлышко сосуда закрывают
притертой пробкой; осторожно приоткрывая пробку при открытом
отверстии отростка, заполняют его полностью и горлышко снова
закрывают пробкой. Затем в отверстие отростка вводят пробочку
из фильтровальной бумаги, как это описано для электролитиче-
ского ключа (см. с. 45). По охлаждении раствора из него выпа-
дают кристаллы КС1, оседающие поверх слоя каломели. Через
15
сутки после изготовления электрод можно использовать в работе.
Суспензию каломели для заполнения электрода готовят из пре-
парата квалификации «х. ч.». Препарат Hg2Cl2 растирают с насы-
щенным раствором КС1 и несколькими капельками ртути в ага-
товой ступке до получения гомогенной массы, переносят с по-
мощью насыщенного раствора КС1 в склянку и 6 ч взбалтывают
на ротаторе. Раствор КС1 над осадком Hg2Cl2 сливают, и осадок
несколько раз промывают свежими порциями насыщенного раство-
ра КС1. Хлорид калия перед работой дважды перекристаллизовы-
вают, используя зависимость растворимости КО от температуры:
при 20° —растворяется примерно 34,4 г КО в 100 мл, а при 60е —
около 46 г в 100 мл (очистка ртути описана в гл. 3).
Для приготовления каломельных электродов используют со-
суды и другой формы. В лаборатории удобен сосуд Михаэлиса
(см. рис. 2). Электрод готовится аналогично, несколько меняется
только порядок заполнения сосуда. Сначала на дно сосуда зали-
вается немного ртути, после чего он закрывается пробкой с впаян-
ным платиновым контактом. Ртуть должна полностью закрыть
платиновый контакт, затем в сосуд последовательно насасывают
суспензию Hg2Cl2 и раствор КС1 через длинный отросток с краном.
Промышленность выпускает и готовые каломельные электроды,
входящие в комплекты потенциометров (ЛПУ-01, ППМ-03) поля-
рографов и других приборов.
Каломельный электрод сохраняет постоянное значение потен-
циала независимо от прохождения через него сравнительно не-
} больших по силс токов. 'Неполяризуемость электрода объясняется
1 следующим. В электроде скачок потенциала возникает за счет
реакции
2HgJ±Hgo+- 2е.
Но концентрация ионов ртути определяется растворимостью
Hg2Cl2 и зависит от концентрации КС1 вследствие присутствия
одноименного иона С1~. При наложении на электрод положитель-
ного потенциала от внешнего источника тока металлическая ртуть
окисляется, и в растворе появляется дополнительное количество
ионов Hg2+, реакция сдвигается вправо. Но избыток ионов Hg^2
сразу связывается хлорид-ионом и выпадает в осадок в виде
Hg2Cl2; в результате концентрация катионов ртути в растворе не
меняется. Изменение направления тока вызывает обратный про-
цесс: восстановление Hg2+ до металлической ртути. Но и в этом
случае концентрация Hg2'1- в растворе и потенциал электрода не
изменяются благодаря дополнительному растворению Hg2Cl2.
Таким образом, потенциал электрода будет сохранять постоянное
значение до тех пор, пока не будут полностью израсходованы
металлическая ртуть, Hg2Cl2 или КС1. Если же направление тока,
проходящего через электролит, периодически меняется, тогда ста-
бильность потенциала сохраняется неопределенно долго.
16
Кроме каломельных электродов сравнения широко применяют
и некоторые другие. В комплектах к потенциометрам часто вхо-
дят хлорсеребряные электроды. Хлорсеребряный электрод со-
стоит из серебряной проволоки с губчатой поверхностью, покры-
той осадком хлористого серебра. Такая проволока погружена в
раствор хлористого калия (рис. 3).
Скачок потенциала возникает на электроде вследствие реакции
Ag°=ptAg+ + e и зависит, как и в случае каломельного электрода,,
от концентрации хлорид-ионов.
Потенциалы
хлорсеребряных
электродов, В
КС! насыщ. (20°) +0,202
1,0 н. КС1 +0,238
0,1 н. КС1 +0,290
0,1 н. НС1 +0,289
1,0 н. НС1 +0,218
Хлорсеребряный электрод может быть использован не только
как электрод сравнения, но и как индикаторный электрод. В этом
случае сам электрод — серебряную проволоку с покрытием AgCl
погружают не в раствор КС! известной и постоянной концентра-
ции, а в испытуемый раствор. По измеренному потенциалу элект-
рода можно найти актцваость хлорид-ионов, в этом растворе.
Строго говоря, любые электроды можно использовать либо
как индикаторные, либо как неполяризующиеся вспомогательные.
Чтобы превратить индикаторный электрод во вспомогательный, его
надо поместить в раствор с известной и достаточно стабильной
концентрацией деполяризатора. В качестве вспомогательных
употребляются даже такие электроды с водородной функцией, как
классический водородный электрод и хингидронный электрод.
Последний, благодаря простоте изготовления и хорошей вос-
производимости потенциала, удобен в полевых условиях.
В последнее время находит широкое применение комбинирован-
ный стеклянный электрод (рис. 4). Он удобен тем, что вместо
двух электродов — индикаторного и электрода сравнения — ис-
пользуется один комбинированный электрод. Конструкция комби-
нированного электрода предусматривает расположение двух элект-
родов в одном корпусе: стеклянного электрода с водородной
функцией и вспомогательного хлорсеребряного электрода сравне-
ния. Контакт стеклянного электрода с внешней средой осуществ-
ляется через стеклянный шарик; электрод сравнения сообщается
с внешней средой через асбестовую нить, впаянную в корпус
электрода.
1АЛЯАЯ ЕНБЛИ'ЛЬ+к
им. Горького
МГУ
17
Стеклянный шарик на конце комбинированного электрода вы-
полнен из специального электродного стекла с водородной функ-
цией. Шарик заполнен раствором НС1, в который опущена сереб-
ряная проволока, покрытая губчатым слоем хлористого серебра,
служащая контактным полуэлементом стеклянного электрода (см.
с. 36). Электрод сравнения расположен внутри стеклянного кор-
Рис. 3 Устрой-
ство хлорсереб-
ряного электро-
да ЭВЛ-1МЗ
1 — корпус, 2—
асб е с т о в а я
нить, 3 — рас-
твор КС1. 4 —
отверстие для
заливки раство-
ра КС1, 5 —
резиновая проб
ка, 6 — сереб-
ряная проволо
ка, 7 — слой
AgCl
Рис. 4 Комби-
нированный
стеклянный
электрод к по-
тенциометру
ппм-озм
1 — шарик из ,
электродного
стекла, 2 —
внутренний
вспомогатель-
ный хлорсерсб-
ряный электрод
в растворе НС1,
3 — хлорссреб>
ряный электрод
сравнения, 4 —
асбестовая
нить, 5 — поро-
шок AgCl и
КС1, 6 — насы-
щенный рас-
твор КО, 7 —
асбестовая
нить, 8 — от-
верстие с проб-
кой для долив-
ки раствора
КО, 9 — соеди-
нительный ка-
бель, 10 — кол-
пачок, 11 — по-
лиэтиленовая
трубка
пуса электрода и отделен от него полиэтиленовой трубкой. Ниж-
няя часть полости между полиэтиленовой трубкой и стеклянным
корпусом заполнена насыщенным раствором КС1. В верхней части
полости между двумя пробками расположен электрод сравнения,
который состоит из смеси порошкообразного хлористого серебра
и хлористого калия и помещенной в эту смесь серебряной про-
волоки. Контакт электрода сравнения с внешней средой осуществ-
ляется через асбестовую нить, опущенную в насыщенный раствор
КО, по которой раствор поднимается вверх до электрода сравне-
ния, а сам раствор через асбестовую нить, впаянную в корпус
электрода, сообщается с внешней средой.
18
На корпусе электрода имеется отверстие для заливки насыщен-
ного раствора КО, который со временем вытекает из электрода
через асбестовый контакт.
Комбинированный стеклянный электрод удобен при полевых
исследованиях, достаточно прочен, хорошо сохраняется во време-
ни, не требует дополнительного ухода, кроме заливки раствора
КС1, что нетрудно сделать в полевых условиях.
Э.д.с. цепи
и принципы ее измерения
Для определения потенциала индикаторного электрода состав-
ляют элемент, в который входят индикаторный электрод, погру-
женный в испытуемый раствор, и один или несколько вспомога-
тельных электродов. При определении pH индикаторным может
быть водородный электрод, состоящий из платиновой пластинки,
покрытой платиновой чернью и находящейся в растворе, насыщен-
ном газообразным водородом. Реакция на таком электроде соот-
ветствует уравнению
Н2 2Н 2Н+ -- 2е,
а потенциал электрода является функцией pH:
Ен = Е0 \ ftlgaH+ = £ь — й pH.
Потенциал стандартного водородного электрода Ео принят рав-
ным нулю при всех температурах и используется как отсчетная
точка в шкале потенциалов. Тогда
Ен =“ — й pH
или (при 20°С)
Ен — —0,0581 pH.
В качестве второго электрода можно использовать насыщен-
ный каломельный электрод. Составленный таким образом элемент
выглядит, как показано на рис. 5, и может быть записан следую-
щим образом:
Pt, н2
Водородный
электрод
Раствор, рНж
КС] насыщ.
Электролити-
ческий ключ
Hg2Cl2, | Hg
Каломельный
электрод
В этой записи вертикальная черта означает границу раздела
фаз. Задача нахождения потенциала индикаторного электрода сво-
дится к измерению разности потенциалов между точками В и К
при разомкнутой цепи. В этом случае найденная величина будет
равна э. д. с. элемента. Если между точками В и К включить
сопротивление R, тогда цепь будет замкнута и общая э. д. с. эле-
19
мента будет равна сумме падения напряжения во внешней цепи
BRK и внутреннего падения напряжения BRiK. Подключенный к
точкам ВК измерительный прибор покажет тогда величину, отли-
чающуюся от э. д. с. тем больше, чем меньше величина^/? и больше -
Ri. Контролировать внутреннее сопротивление элемента в прак-
В R К
Ri
Рис. 5. Элемент из индикаторного водородного и вспомо-
гательного каломельного электродов:
1 — водородный электрод, 2 — каломельный электрод, 3 —
сосуд с испытуемым раствором, Pi — сопротивление внут-
ренней цепи, Р — сопротивление внешней цепи
тике почвенных исследований практически невозможно, поскольку
концентрация и состав испытуемых растворен, от которых зависит
Ri, меняются в широких пределах. Чтобы избежать ошибок, свя-
занных с этим явлением, для измерения э.д. с. используют компен-
сационную схему Поггендорфа.
Простейшая компенсационная схема показана на рис. 6. Осно-
вой компенсационной схемы является потенциометрическая прово-
лока, или реохорд АВ. От внешнего источника тока Акк — акку-
мулятора или сухой батареи на эту проволоку подается напряже-
ние Е. Сопротивлением подводящих проводов можно пренебречь
20
и считать, что падение напряжения от аккумулятора полностью
приходится только на реохорд АВ. Если вдоль реохорда через рав-
ное расстояние нанести деления,
то при однородной проволоке на
каждый отрезок между делени-
ями придется одно и то же паде-
ние напряжения. Если выразить
длину реохорда АВ в мм, то на
каждый миллиметр придется па-",-
дение напряжения в EjAB В/ымА~
Элемент, э. д. с. которого Ех
надо измерить, также подсоеди-
няют к реохорду, но таким спосо-
бом, чтобы один полюс Ек был
присоединен к точке А. Обяза-
тельно, чтобы от АМ{ и от Ех к
точке А были подключены одно-
именные полюса. Второй полюс
изучаемого элемента Ех через пе-
реключатель П и ключ Д' соеди-
няют с подвижным контактом С,
Рис. 6. Компенсационная схема.
АВ — реохорд, С — скользящий кон-
такт, Акк—источник тока, R — бал-
ластное сопротивление, В к — выклю-
чатель, Г — гальванометр, К — ключ.
П — переключатель, Ев — элемент
Вестона, Ех —испытуемый элемент
который может свободно скользить вдоль реохордпой проволоки,
обеспечивая в то же время надежное точечное соединение. В эту
же цепь Ех—1\—С включен стрелочный гальванометр типа ГМП
или М-122 с чувствительностью порядка 10-5—10-7 а на мм шка-
лы (рис. 7).
В составленной таким образом схеме электродвижущие силы
аккумулятора и изучаемого элемента направлены навстречу друг
другу. Напряжение аккумулятора или батареи выбирается по
крайней мере в 2 раза большим, чем э. д. с. элемента. Тогда на
реохордной проволоке всегда найдется некоторая точка, потенци-
ал которой окажется равным потенциалу одноименно заряженно-
го полюса изучаемого элемента. Естественно, что при помещении
подвижного контакта С в эту точку, гальванометр покажет* отсут-
ствие тока на участке цепи А—П—К—Г—С. В цепи А—С—В ток,
естественно, обнаруживается при любом положении подвижного
контакта С.
Зная напряжение аккумулятора Еакк, длину реохорда АВ и
длину отрезка АС, нетрудно вычислить э.д.с. элемента. Очевидно,
э. д. с. элемента в момент компенсации (при отсутствии тока в
цепи А—П—К—Г—С) равна падению напряжения от аккумуля-
тора, приходящемуся на участок реохорда АС. Если па весь рео-
хорд приходится падение напряжения Еакк, то на одно деление
соответственно приходится Е&кк: АВ. Чтобы найти падение напря-
жения на участке АВ, надо последнюю величину умножить на
число делений от А до С.
Егкк ' АС
АВ
21
Напряжение аккумулятора или сухой батареи не остается
строго постоянным во времени и его приходится регулярно про-
верять С этой целью в компенсационную схему параллельно Ех
включают стандартный источник тока с точно известной величи-
ной э д с Обычно это стандартный элемент Вестона (рис 8).
Рис 7 Стрелочный гатьванометр Рис 8
ГМП для потенциометрических Нормальный элемент
схем Вестона
Включив в цепь элемент Вестона (Ев) с помощью переключа-
теля 77, замыкают кратковременно ключ К и наблюдают за поло-
жением стрелки гальванометра Отклонение стрелки говорит о на-
личии тока в малой цепи Перемещением скользящего контакта С
находят точку компенсации, при этом положении стрелка гальва-
нометра не отклоняется при замыкании цепи Правильность на-
хождения точки компенсации проверяют, несколько смещая впра-
во и влево контакт С Точка компенсации найдена правильно, если
небольшие смещения (на одно деление) контакта С вправо и вле-
во вызывают отклонения стрелки гальванометра в противополож-
ные стороны
22
В соответствии с формулой напряжение аккумулятора или
батареи Elimi вычисляют следующим образом:
Ев- АВ
Р — ,
АС,
где АС, — положение контакта С в точке компенсации.
Затем переключателем П в цепь вводят изучаемый элемент Ех
и с помощью тех же приемов находят его э. д. с.:
Еакк • АС Ев- АВ АС Ев АС
Е. =--------=-----------------------
АВ АС, АВ АС,
Э. д. с элемента Вестона близка к 1,0184 В и сравнительно
мало меняется с температурой (табл. 3). Это свойство можно
использовать для устройства схемы, в которой цена одного деле-
ния реохорда (потенцио-
метра) будет соответство-
вать точно 1 мВ. С этой
целью на реохорд АВ на-
носят 1018 делений, а эле-
мент Вестона через пере-
ключатель соединяют с кон-
цами реохорда. Согласно
выбранным условиям, на
каждое деление реохорда
от элемента Вестона прихо-
дится в этом случае точно
1 мВ. Аккумулятор подклю-
чают к точке Дик тысяч-
Таблица 3
Э. д. с. элемента Вестона при различных
температурах, в
°C э д С СС 3 д с °C э. Д с
10 1,01881 17 1,01842 24 1,01816
11 1,01874 18 1,01838 25 1,01813
12 1,01869 19 1,01834 26 1,01810
13 1,01863 20 1,01830 27 1,01807
14 1,01858 21 1,01826 28 1,01804
15 1,01850 22 1,01822 29 1,01802
10 1,01846 23 1,01818 30 1,01799
ному делению реохорда
Включив Ев переключателем П и замкнув ключ К, изменяют
поданное на реохорд напряжение от аккумулятора с помощью
реостата R, добиваясь, чтобы стрелка гальванометра показала от-
сутствие тока в цепи. При компенсации, очевидно, на 1000 делений
шкалы от аккумулятора было подано 1000 мВ; при последующей
работе с аккумулятором цена одного деления остается равной
1 мВ.
Если в такую схему включить изучаемый элемент и найти
точку компенсации, как описано выше, то отсчет по шкале реохор-
да в делениях шкалы будет соответствовать э. д. с. элемента в мВ.
Погрешность измерения э.Д. с. зависит от длины реохорда, его
однородности и питающего напряжения. При Е,1№=2В и
АВ=1000 мм погрешность измерения составляет около 2 мВ,
при EavK— 1 В погрешность снижается до 1 мВ.
При обычных измерениях pH и ОВП погрешность в 1—2 мВ
вполне допустима; в расчете на pH это не превышает 0,02—0,03
единиц pH. Однако изучение активностей компонентов почвенных
растворов часто требует более точных измерений.
23
Простые сборные компенсационные схемы (см. рис. б) просты
в обращении, их можно легко изготовить в лаборатории, они при-
годны и для полевых работ. При правильном обращении схема
устойчива и надежна в работе. Отказ в работе бывает связан со
следующими типичными неисправностями или ошибками монтажа:
1. При замыкании ключа стрелка гальванометра не отклоняет-
ся независимо от положения скользящего контакта С. При-
чина: нет контакта в цепи А—П—К—Г—С. Обнаружить
место разрыва можно, проверяя механическую прочность
контактов или последовательно исключая отдельные участки
цепи.
2. При замыкании ключа стрелка гальванометра не движется
или движется очень вяло — нарушено жидкостное соедине-
ние в испытуемом элементе. Установить причину можно ви-
зуальным осмотром или поставив переключатель П в поло-
. жение Ев.
3. При замыкании ключа стрелка гальванометра независимо
от положения скользящего контакта С отклоняется в одну
сторону. Причин может быть несколько. Простейшая причи-
на: разомкнут выключатель В^. Затем — напряжение, пода-
ваемое на реохорд от аккумулятора, меньше, чем э. д. с.
испытуемого элемента или элемента Вестона. Наконец, воз-
можно, при монтаже не соблюдено правило полярности и к
точке А от аккумулятора и испытуемого элемента подклю-
чены разноименные полюса.
Простая сборная компенсационная схема сейчас редко исполь-
зуется в лабораторной практике. Несмотря на это, она по-преж-
нему имеет принципиально важное значение и лежит в основе
потенциометров П-4, П-5, которые до сих пор удобны в экспеди-
ционной практике; на ее основе построены ламповые потенцио-
метры ЛП-5, Л П-6. Та же схема используется в полярографах,
электронных автоматических потенциометрах и других приборах.
Общий принцип компенсации сохранен и во многих электронных
потенциометрах и pH-метрах, даже если в них и не используется
реохорд со скользящим контактом. Изучение компенсационной
схемы необходимо для более правильного и глубокого понимания
устройства, принципа действия и порядка работы многих совре-
менных приборов.
Потенциометры
В последние годы наибольшее распространение получили элект-
ронные потенциометры и pH-метры. Их особенность и отличие от
более ранних потенциометров типа ЛП-5 заключаются в том, что
измеренные значения pH или э. д. с. считываются прямо со шкалы
стрелочного прибора. i
24
Все приборы этого типа имеют сложные электронные схемы,
ремонт и наладка которых могут производиться только специа-
листами. При эксплуатации подготовленных приборов производит-
ся только настройка pH-метров по буферным растворам. С этой
целью pH-метры обычно снабжены двумя настроечными потенцио-
метрами.
Ниже приводятся описание и порядок работы на некоторых по-
тенциометрах и рН-метрах.
Лабораторный рН-метр-милливольтметр pH-340. Этот прибор
предназначен для определения pH или э.д.с. какого-либо источ-
ника тока. При наличии электродов, обратимых к различным ви-
дам ионов, прибор может быть использован для измерения их
активности. В сочетании с блоком БАТ-12ЛМ прибор позволяет
проводить автоматическое титрование, а при подключении автома-
тических регистрирующих потенциометров (например, типа ЭПП)
записывать результаты в виде диаграмм на специальной ленте.
Прибор pH-340 позволяет измерять э.д.с. в пределах от —100
до +1400 мВ или от +100 до —1400 мВ. Этот интервал может
быть разбит на диапазоны по 300 мВ. Погрешность измерений при
работе на всей шкале +0,5 единицы pH и 29 мВ; при работе в
узких диапазонах погрешность снижается в 10 раз.
На передней панели прибора (рис. 9, А) выведены рукоятки
управления и показывающий прибор, шкала которого дана в мил-
ливольтах и в единицах pH. Гнезда для подключения датчиков,
выходные гнезда, зажим заземления и другие расположены на
задней стенке прибора (рис. 9, Б).
Чтобы начать работу на приборе pH-340, к нему подключают
измерительный (индикаторный) и вспомогательный электроды к
соответствующим гнездам на задней стенке. Провод заземления
датчика соединяют с соответствующей клеммой прибора и линией
заземления. Ручки переключателей «Род работы», «Размах»,
«Температурный компенсатор» устанавливают в соответствии с
выбранной схемой измерения и условиями работы. Прибор под-
ключают к сети 220 В, 50 Гц с помощью сетевого шнура; для
включения питания поворачивают ручку НИ по часовой стрелке.
Когда напряжение подано, загорается контрольная лампочка на
передней панели прибора. Перед началом работы устанавливают
механический нуль прибора; для этого отверткой поворачивают
корректор нуля, подводя стрелку прибора на начальную отметку
шкалы. Затем прибор включают в сеть, переключатель «Размах»
устанавливают в положение «15 pH» и дают прибору прогреться
в течение 60 мин. Стеклянный электрод и электрод сравнения
промывают дистиллированной водой и удаляют остатки воды
фильтровальной бумагой. Буферным раствором с pH 1,68 ополас-
кивают электроды и погружают их в свежую порцию того же
буферного раствора. Переключатель «Пределы измерения» устанав-
ливают в положение «—1—2», температурный корректор перево-
дят в положение, отвечающее температуре раствора, а переклю-
25
A
Рис 9 Потенциометр pH 340
А — передняя панель 1 — показывающий прибор, 2—переклю
чатель рода работ, 3 — переключатель пределов измерении, 4 —
ручной термокомпенсатор, 5—индикатор включения, 6 — потен
циометр настройки £м, 7 — потенциометр настройки S, 8 — по-
тенциометр установки нуля нуль индикатора и включатель сети,
9 — переключатель размаха шкалы
Б — задняя панель 1 — клеммы для автоматического термо
компенсатора, 2 — перемычка, 3 — переменный резистор для гру-
бой регулировки 4, 5—выходные гнезда, 6, 7—переменные
резисторы для грубой и плавной регулировки рНи, 8 — пере-
менные резисторы для заводской градуировки прибора, 9 —
гнездо для подключения индикаторного электрода, 10— гнездо
для вспомогательною электрода, 11—зажим заземления, 12 —
сетевой разъем, 13 — держатель предохранителя
чатель «Размах» — в положение «3 pH». Установив ручку потен
циометра Ей примерно в среднее положение, подводят стрелку
показывающего прибора к отметке 1,68 pH, используя последова-
тельно потенциометры «Ей грубо» (на задней стенке прибора) и
Ей. Затем переключатель «Размах» переводят в положение
«15 pH» и проверяют прибор по другим буферным растворам,
пользуясь соответствующими поддиапазонами шкалы Отклоне-
ния показаний прибора от pH буферных растворов не должны
превышать 0,05 pH на всех узких диапазонах шкалы и 0,5 pH
на диапазоне —1—44 pH.
Если отклонения превышают указанные допустимые ошибки,
то проводят дополнительную настройку по двум буферным раство-
рам, используя потенциометр S. С этой целью сначала проводят
настройку по раствору с pH 1,68, как указано выше
Затем переносят электрод в буферный раствор с pH 9,22; если
показания прибора отличаются от этого значения больше, чем на
0,05 pH, то ослабляют цанговый зажим потенциометра S и уста-
навливают ею положение так, чтобы прибор показал 9,22 pH. За-
тем повторно проверяют показания с буферным раствором 1,68 pH
и настройку продолжают, пока показания прибора окажутся в
допустимом интервале отклонений. После этого можно приступить
к измерениям pH исследуемых растворов или суспензий.
Для измерения pH испытуемого раствора сначала ополаски-
вают электроды этим раствором, затем погружают электроды в
свежую порцию тою же раствора Переключатель «Размах» ставят
в положение «15 pH», отмечают примерное значение pH испытуе-
мого раствора и переводят его в положение «3 pH», а на переклю-
чателе «Пределы измерения» устанавливают соответствующий
диапазон pH. Когда стрелка прибора займет устойчивое положе-
ние, записывают показания прибора
При использовании прибора в качестве милливольтметра к
нему можно подключить любые элементы, э д с которых не пре-
вышает 1400 мВ. При отсчете показаний знак, соответствующий
положению ручки переключателя «Род работы» ( + мВ или —мВ),
соответствует знаку' потенциала индикаторного электрода Если
переключатель «Размах» установлен в положение «1500 мВ», то
отсчет показаний производят по нижней шкале прибора, оцифро-
ванной от —1 до 14. Если переключатель установлен на один из
узких диапазонов, то отсчет берут по верхней шкале, принимая
во внимание положение переключателя. Например, если переклю
чатель диапазонов установлен в положение «5-?-8», а стрелка по-
казывающего прибора остановилась на делении 1,40, то измеряе-
мая э д. с. равна:
(5 । 1,40) • 100 - 6,40 • 100 640 мВ.
Множитель 100 вводится для переведения показания шкалы в
милливольты.
27
Лабораторный рН-метр-милливольтметр pH-121. Этот прибор
(рис. 10) предназначен для измерения активности ионов водоро-
да, а при наличии ион-селективных электродов и других ионов.
Имеющиеся в комплекте прибора платинированные электроды по-
зволяют использовать прибор для измерения окислительчо-восста-
Рис 10 Потенциометр pH 121
Д — Передняя панель 1 — показывающий прибор, 2—кнопки включения рода
работ, 3 — переключатель пределов измерений, 4 — ручной термокомпенсатор,
5 — клавиша включения ручного термокомпенсатора, 6 — клавиша включения
индикаторного электрода, 7— включатель сети и потенциометр устатовки пуля
нуль-индикатора при нажатой клавише, 8 — кнопка выбопа диапазона измене-
ний, 9 — переменные резисторы для регулировки pH, 10 — переменный резистор
«калибровка», 11 — переменный резистор «крутизна»
£ — задняя панель 1—гнезда для подключения индикаторною электрода, 2 —
гнездо для подключения вспомогательного электрода, 3—гнездо «ток поляри
зации», 4 — включатель ручной и автоматической термокомпенсации, 5—ро-
зетка подключения автоматического термокомпенсатора, 6 — перемычка, 7 —
розетка подключения потенциометра, 8— держатель предохранителя, 9— за-
жим заземления
28
новительного потенциала, кроме того, pH-метр может использо-
ваться как высокоомный нуль-индикатор
В отличие от рН-метра pH 340 несколько изменен внешний
вид прибора, все переключатели заменены на клавишные Весь
интервал измеряемых величин pH и мВ разбит на более широкие
диапазоны, что позволяет
проводить измерения в од-
ном диапазоне, не пере-
ключая прибора, вместе с
тем понижена абсолютная
погрешность измерений
Величины pH определя-
ют в пределах от —1 до
14 с абсолютной погреш-
ностью +0,4 pH, п в более
узких интервалах —1+4,
4+9, 9+14, рН-абсолют-
ная погрешность в каждом
интервале составляет 0,04
pH
Измерение э д с на при-
боре производят в пределах
от +100 мВ до +1400 мВ
При более точных измерени-
ях работают на диапазонах
по 500 мВ Абсолютная
погрешность при работе в
широком диапазоне измере-
ний ±40 мВ, в более уз-
ких— ±5 мВ
Измерения на рН-метре
pH-121 выполняются в та-
кой же последовательно-
сти, что и на pH-340, под-
робное описание хода вы-
полнения определений при-
ведено в заводской инст-
рукции, которая прилага-
ется к каждому прибору
Лабораторный рН-метр
ЛПУ-01. Этот прибор так-
Рис 11 Передняя панель pH метра
ЛПУ 01
1 — показывающий прибор, 2 - тумблер
включения прибора, 3— контрольная
лампочка включения прибора, 4 — пере-
ключатель пределов измерений, 5 — пе-
реключатель вида работ, 6 — гнездо
подключения датчика, 1 — переменное
сопротивление ручной температурной
компенсации, 8 — переменное сопротнв
ление настройки по буферному раство-
ру, 9 — переменное сопротивление на-
стройки крутизны, 10 — клемма зазем-
ления датчика
же может применяться
как многопредельный pH-метр, как высокоомный милли-
вольтметр и как нуль-индикатор (рис 11) Пределы измере-
ния pH от —2 до +14, шкала может быть разбита на диапазоны
от —2 до +2, от 2 до 6, от 6 до 10 и от 10 до 14 Соответственно,
пределы измерения э д с составляют от —200 до +1400 мВ или
от +200 до —1400 мВ
29
Все элементы измерительной схемы и усилителя рН-метра
смонтированы в металлическом корпусе, а органы управления’
выведены на переднюю панель (см. рис. 11). С левой стороны
прибора под крышкой помещены потенциометры, изменяющие
координаты пзопотенциальной точки (см. с. 38), на которую,
настроен прибор. На задней панели выведены клеммы для автома-j.
тического термокомпенсатора, клеммы для подключения регист-
рирующего прибора, для заземления, предохранитель и вход от
сети переменного тока 220 В.
Перед началом работы к pH-метру подключают электроды
датчика в клемму заземления прибора на задней стенке его сое-
диняют с линией заземления датчика. На подготовленном таким
образом pH-метре устанавливают «механический нуль» показы-
вающего прибора. Для этого поворачивают отверткой корректор
нуля, расположенный в нижней части показывающего прибора, до
тех пор, пока стрелка прибора не остановится точно на нулевом
делении шкалы. После этого прибор включают в сеть и настраи-
вают. Если предпола! ается измерение pH, то настройка ведется ,
по буферным растворам; если прибор используют как милливольт- '
метр, то его настраивают с помощью потенциометров типа ППТВ-1
или Р-307.
При работе в единицах pH рукоятка «Виды работ» па перед-
ней панели прибора ставится в положение «pH» и по нижней
шкале прибора отмечают примерное значение pH исследуемого
раствора. Затем переводят переключатель «Пределы измерений»
на соответствующий диапазон pH. После того как стрелка прибо-
ра займет устойчивое положение, записывают показания прибора.
Например, если переключатель пределов измерений установлен в
положении «2—6», а стрелка показывающего прибора останови- I
лась на делении 2,70 по верхней шкале, то измеряемая величина '
pH равняется 2+2,70=4,70.
При использовании прибора в качестве милливольтметра пере-
ключатель «Виды работ» устанавливают в положении «-(-2004---
1400 мВ» или «—200+4-1400 мВ» и снимают значение э. д. с. по
нижней шкале прибора. Пределы измерения потенциалов могут •
быть изменены, и вся шкала может охватывать диапазон 200 или
400 мВ. Для работы в таких диапазонах к прибору подключается
имеющееся в комплекте специальное сопротивление 160 или *
350 Ом. Более подробно использование прибора в качестве мил-
ливольтметра описано в заводской инструкции, прилагаемой к
прибору.
Для настройки прибора по буферным растворам включают
прибор в сеть, дают ему прогреться в течение 30 мин. Стандарт-
ным буферным раствором с pH 4,00 тщательно промывают элект-
роды и опускают их в свежую порцию буферного раствора. Уста-
навливают на приборе диапазон измерений pH от 2 до 6, рукоят-
ку температурного корректора устанавливают на температуру
раствора и через 1—2 мин снимают отсчет.
30
Если показания прибора отличаются от pH 4,00, то рукояткой
«Настройка по буферному раствору» устанавливают стрелку при-
бора точно на деление, отвечающее 4,00 pH. Аналогично прове-
ряют показания по другим буферным растворам для каждого диа-
пазона в отдельности.
' Расхождения по всем диапазонам не должно превышать
0,04 pH. Если расхождения оказались более высокими, то прово-
дят дополнительную настройку рукояткой «Крутизна». С этой
целью электроды помещают в буферный раствор с pH 1,68, пере-
ключатель пределов измерений устанавливают на диапазон от —2
до +2 и ручкой «Настройка по буферному раствору» устанавли-
вают стрелку на деление 1,68. Затем переносят электроды (после
предварительного ополаскивания) в буферный раствор с pH 9,22
и переключатель пределов устанавливают на диапазон «64-10».
Если показания прибора отличаются от 9,22 более чем на 0,04,
тогда с помощью специального ключа ослабляют цанговый зажим
переменного сопротивления «Крутизна» и поворачивают ось пере-
менного сопротивления на 5—10° по часовой стрелке, если пока-
зания прибора занижены, или против часовой стрелки, если по-
казания прибора завышены. Снова затягивают цанговый зажим
и повторяют настройку начиная с буферного раствора 1,68 pH до
тех пор, пока не будут найдены положения ручек «Крутизна» и
«Настройка по буферному раствору», при которых расхождения
показаний не будут превышать 0,02—0,04 pH.
После настройки прибора можно приступать к измерениям pH
или э. д. с. Для этого электроды датчика ополаскивают испытуе-
мым раствором или суспензией, погружают электроды в раствор
или суспензию, переключатели пределов измерений ставят в тре-
буемые положения и снимают отсчет по шкале приборов.
Переносной рН-метр-милливольтметр ППМ-03М1. Этот прибор
(рис. 12) размером 200X130X70 мм весом 1,3 кг и помещенный
в кожаный футляр на ремне может быть использован для поле-
вых исследований. Питание потенциометра осуществляется от эле-
мента «Сатурн» или от аккумуляторной батареи, которая подза-
ряжается от сети через специальное ' устройство, прилагаемое к
! прибору. Прибор снабжен двумя электродами: комбинированным
• стеклянным с водородной функцией и комбинированным платини-
1 рованным электродом для измерения ОВП. При выходе этих
рлектродов из строя можно пользоваться обычными, несколько
(изменив ввод электродов. Этот прибор позволяет измерять pH от 2
'до 12 с погрешностью не более 0,1 pH и э.д.с. от 0 до 1000 мВ
с погрешностью 10 мВ. Для обеспечения надежности в работе
перед выездом в поле прибор следует тщательно проверить и при
необходимости наладить.
Настройка pH-метров и градуировка электродов с водородной
функцией проводятся по буферным растворам. Используются
2 вида буферных растворов: образцовые стандарты и рабочие
буферные растворы.
о
в
я
31
В СССР принята шкала pH, основанная на пяти образцовых
буферных растворах (ГОСТ 10170—62 и 10171-62).
1. Раствор, содержащий в 1 л при 20°С 12,7+0,02 г тетра-
оксалата калия КНС2О4-Н2С2О4-2Н2О (0,05 моль/л).
II. Насыщенный при 25°С раствор калия виннокислого кисло-
го кс4н5о6.
Рис. 12. Переносной потенциометр
ППМ-03М1:
1 — показывающий прибор, 2 — ручка
установки нуля и включения прибора,
3 — ручка потенциометра температурной
компенсации, 4 — ось потенциометра
регулировки напряжения питания, 5 —
переключатель рода измерений, 6 — за-
глушка резистора «начало шкалы», 7 —
заглушка резистора «размах шкалы»,
8 — i нездо для подключения стеклянно-
го комбинированного электрода, 9, 10 —
гнезда для подключения комбинирован-
ного платинового электрода, 11 — кор-
ректор нуля
Таблица 4
Значения pH образцовых
буферных растворов
Темпе рату- ра, °C Номер образцового раствора
I И Hi IV V
0 1,67 . 4,01 6,98 9,46
5 1,67 ,— 4,01 6,95 9,39
10 1,67 —. 4,00 6,92 9,33
15 1,67 — 4,00 6,90 9,27
20 1,68 —. 4,00 6,88 9,22
25 1,69 3,56 4,01 6,86 9,18
30 1,69 3,55 4,01 6,84 9,14
35 1,69 3,55 4,02 6,84 9,10
40 1,70 3,54 4,03 6,84 9,07
45 1,70 3,55 4,04 6,83 9,04
50 1,71 3,55 4,06 6,83 9,01
55 1,72 3,56 4,08 6,84 8,99
60 1,73 3,57 4,10 6,84 8.96
70 1,75 3,59 4,12 6,85 8,92
80 1,77 3,61 4,16 6,86 8,88
90 1,80 3,64 4,20 6,88 8,85
95 1,81 3,65 4,22 6,89 8,83
III. Раствор, содержа-
щий в 1 л при 20°С 10,21 +
+0,02 г калия фтале-
вокпслого КС8Н5О4
(0,05 моль/л).
IV. Раствор, содержащий
в 1 л при 20°С 3,40 ±0,01 г калия фосфорнокислого однозамещен-
ного КН2РО4 (0,025 моль/л) и 3,55±0,01 г натрия фосфорнокис-
лого двузамещенного Na2HPO4 (0,025 моль/л)
V. Раствор, содержащий в 1 л при 20сС 3,81+0,01 г натрия
тетраборнокислого Na2B4O7-10 Н2О (0,01 моль/л).
Значения pH образцовых растворов приведены в табл. 4.
Для приготовления образцовых растворов применяют дистил-
лированную воду с удельной электропроводностью не более
2-10~6 Олт’ см-1. Стандартные вещества должны быть квалифи-
32
кации для pH-метрии; с>шат их до постоянного веса при следую-
щих условиях:
тетраоксалат калия.......................57+2°
калий фталевокислый.....................110+5°
калий фосфорнокислый однозамещенный . 110+5°
натрий фосфорнокислый двузамещенный . 120+5°
Натрий тетраборнокислый выдерживают до постоянного веса при
комнатной температуре в эксикаторе над смесью влажного хло-
ристого натрия и сахара. Калий виннокислый кислый предвари-
тельно не сушат.
Готовят и хранят образцовые буферные растворы так, чтобы
в них не попадала двуокись углерода из воздуха, пары аммиака
и т. п. Щелочные растворы хранят в предварительно парафини-
рованной посуде. Стандартные образцовые растворы можно хра-
нить не более трех месяцев, а фосфатные — не более двух. В на-
| сыщенный раствор виннокислого калия рекомендуют добавлять
I тимол для предупреждения развития микроорганизмов.
I
Электроды для измерения pH
г
Водородный электрод. Классическим электродом с водородной
функцией является так называемый водородный электрод, потен-
циал которого линейно зависит от pH раствора в диапазоне значе-
ний pH от 0 до 14. Потенциал водородного электрода обусловлен
реакцией
Н2 2НI- + 2е;
величина его может быть выражена уравнением
£н = — 2,303 pH, или при 20° С
Еи = —0,0581 pH.
Если имеется электрод сравнения с точно известным потенциа-
лом Еср, то измерение величины э. д. с. элемента, составленного из
водородного электрода с испытуемым раствором и электрода срав-
нения, позволяет непосредственно вычислить величину pH. При
20°С значение pH равно:
э. д. с. — £ср
0,0581
Для приготовления водородного электрода берут гладкую пла-
тиновую проволоку или пластинку, впаянную в стеклянную труб-
ку. Поверхность платины тщательно очищают и промывают горя-
чим раствором хромовой смеси, а затем дистиллированной водой.
2 Зак 284
33
Промытый электрод оставляют в чистой дистиллированной воде
на ночь. Затем приступают к платинированию электрода. С этой
целью в 100 мл воды растворяют 2—3 г PtCl4 и 0,02 г
(СН3СОО)2РЬ. Уксуснокислый свинец добавляют, чтобы получить
более прочное и равномерное покрытие. В раствор опускают под-
лежащий платинированию платиновый электрод и соединяют его
Рис. 13. Водородный электрод:
1 — платинированная платиновая
проволока, 2 — раствор, 3 — мед-
ная проволока, 4 — винтовой кон-
такт
с отрицательным полюсом аккуму-
лятора на 2—4 В. Анодом служит
также платиновая пластинка или
проволока. Через раствор пропус-
кают ток, регулируя его силу та-
ким образом, чтобы на аноде не-
прерывно, но не слишком быстро
выделялись пузырьки газа. Через
4—6 мин поверхность электрода
покрывается ровным бархатисто-
черным слоем губчатой платины.
Платинированный электрод промы-
вают дистиллированной водой и
снова подвергают катодной поля-
ризации, но уже в 0,05 н. H2SO4,
чтобы удалить адсорбированные
ионы С1~. Готовый электрод тща-
тельно промывают дистиллирован-
ной водой и хранят погруженным
в воду.
Для проведения измерений в
электродный сосуд (рис. 13) на-
ливают буферный раствор с из-
вестным значением pH или иссле-
дуемый раствор с неизвестным зна-
чением pH. Через раствор пропус-
кают чистый водород с такой
скоростью, чтобы можно было
считать пузырьки газа. После 10 мин пропускания водорода
электрод соединяют с вспомогательным электродом через нижний
отросток, подключают электроды к потенциометру и измеряют
э. д. с. Насыщение водородом и измерения э. д. с. повторяют до тех
пор, пока не будет получено постоянное значение результата.
Если сосуд водородного электрода был запол.нен буферным
раствором, то по результатам измерений можно решить и другую
задачу, а именно: установить точное значение потенциала вспомо-
гательного электрода сравнения. Так как
э. д. с. = Еср —Ен,
то Еср — э. д. с. Ен, но Ен = —0,0581 pH (20°),
и тогда Еср = э. д. с. —0,0581 pH.
34
Если в электродный сосуд был налит испытуемый раствор, то
значение pH этого раствора находят по формуле
pH =
0,0581
На потенциал водородного электрода очень сильно влияют
многие компоненты почв и почвенных растворов. Показания элект-
рода искажаются в присутствии сильных окислителей и восстано-
вителей. Отрицательно влияют соли азотной, хромовой, хлорно-
ватой кислот, соединения закисного и окисного железа, сера, суль-
фиды, сулема, аммиак, амины. Большие ошибки возникают в при-
сутствии поверхностно-активпых и коллоидных веществ. Особен-
но сильное влияние можно ожидать в почвенных суспензиях, всег-
да содержащих минеральные коллоиды и гумусовые вещества.
Поэтому водородный электрод используют главным образом
для целей стандартизации, но не для непосредственного измере-
ния pH почвенных вытяжек и суспензий.
Хингидронный электрод. Хингидронный электрод длительное
время был основным электродом при изучении реакции почв и в
свое время был рекомендован Химической комиссией Между-
народного общества почвоведов в качестве стандартного. В по-
следнее время он почти полностью вытеснен стеклянным электро-
дом, но все еще сохраняет определенное значение вследствие лег-
кости изготовления и простоты измерения потенциала. Конструк-
тивно хингидронный электрод представляет собой платиновую
проволоку или пластинку, погруженную в испытуемый раствор,
к которому добавлено небольшое количество хингидрона. Хингид-
рон С6Н4О2-СбН4(ОН)2 — труднорастворим и образует окислитель-
но-восстановительную систему СвН4О2СвН4Ог— 2е, равновесие
в которой определяется активностью водородных ионов. Измерив
окислительно-восстановительный потенциал такой системы с по-
мощью платинового электрода, можно рассчитать pH раствора,
поскольку потенциал хингидронного электрода Ехг линейно зави-
сит от pH:
Ехг = Ео 2,303 *2- 1g пня = Ео - 2,303 pH.
1 г
Потенциал стандартного хингидронного электрода (т. е. потен-
циал хингидронного электрода, активность водородных ионов в
котором равна единице) при 20° равен 0,7027 В; при 25°—0,6990 В.
Поскольку измеряемый скачок потенциала обусловлен окисли-
тельно-восстановительной системой, на результаты измерения pH
сильно влияют окислители и восстановители, а при pH более 8
происходит быстрое окисление гидрохинона кислородом воздуха.
Это не позволяет применять хингидронный электрод для анализа
заболоченных и щелочных почв.
Развитие промышленного производства электронных потенцио-
метров и высококачественных стеклянных электродов привело к
2*
35
практически полному вытеснению хингидронных электродов из
лабораторной практики и замене их на более простые в обраще-
нии и надежные стеклянные электроды с водородной функцией.
Несмотря на это, хингидронный электрод часто бывает полезен в
лабораториях как легко изготовляемый из подручных материалов
индикаторный электрод. В экспедиционных условиях он может
быть очень удобен как вспомогательный электрод при изучении
окислительно-восстановительных потенциалов почвы. Достоинст-
вом хингидронного электрода является невысокое внутреннее
сопротивление, благодаря чему для измерения потенциала элект-
рода пригодны простые неэлектронные потенциометры.
Стеклянный электрод. Он меньше других электродов зависит
от состава раствора: не отравляется как водородный электрод,
не боится присутствия окислителей и восстановителей и пригоден
для работы в широком интервале pH. Он прост в обращении и
удобен в работе. Изготовленный электрод требует только вымачи-
вания в дистиллированной воде или в воде, подкисленной НС1,
и практически не нуждается в иных подготовительных операциях.
Теория возникновения скачка потенциала на границе стекло —
раствор довольно сложна. Сейчас установлено, что стеклянные
электроды можно рассматривать как капюнообменньте мембраны.
высокосслективные к различньпд_кадионям В зависимости от сос-
тава стекло может обладать различными функциями: или
Н+-функцией и тогда электрод используется для измерения pH,
или функциями щелочных, щелочноземельных и других катионов.
Общая теория стеклянных электродов была разработана
Б. П. Никольским, а затем развита М. М. Шульцем и Дж. Эйзен-
маном. Согласно теории Б. П. Никольского, в простом случае по-
тенциал стеклянного электрода может быть выражен уравнением
Дст = Ео 4 (Ppj+ * ^°т\1а+) ’
где К—константа обмена ионов Н+ и Na+ между стеклом и
раствором. Если рассматривать стекло как ионообменник, то в
присутствии Н+- и На+-ионов будет происходить реакция
Нст 4 Na+ = NaCT i Н+,
где Нет и NaCT — ионы водорода и натрия в составе стекла Тогда
[NaCT] а .
К =---------—.
[Нс-Т] • oNa+
Если обеспечиваются такие условия, когда
то уравнение потенциала стеклянного электрода можно упростить
и записать в виде
Ест ~Е0 ' & 1g ант = Ео — е pH.
Иными словами, потенциал стеклянного электрода, как и дру-
гих электродов с водородной функцией, линейно зависит от pH
\
36
раствора. Формально возникновение скачка потенциала можно
связать с явлением отдиссоциации водородных ионов от силикат-
ного остова стекла. При диссоциации ионы водорода уносят в
раствор положительный заряд, на силикатном остове остается
В
Рис. 14. Стеклянные электроды:
А—электрод типа ЭСЛ-41Г-04, Б — электрод типа ЭСЛ-41Г-05, В —
электрод типа ЭСЛ-40-07: 1 — корпус, 2 — шарик из электродного стек-
ла, 3—внутренний электрод сравнения, 4 — раствор, 5—выводной ка-
бель, 6 — штеккер, 7—экран
заряд отрицательный. Увеличение концентрации водородных ионов
в растворе подавляет диссоциацию соединений, входящих в состав
стекла, и уменьшает заряд его поверхности.
К недостаткам стеклянных электродов’следует отнести потен-
циал асимметрии-, Или разность потенциалов между внешней и
внутренней поверхностью стеклянной мембраны при равенстве pH
внутреннего и—внешнего растворов. Потенциал асимметрии может
37
быть сведен до минимума длительным вымачиванием электрода.
Другой недостаток в том, что всякий стеклянный электрод до
некоторой степени полифункционален, и поэтому рекомендуется
опытным путем проверять пределы "его применимости. К числу
недостатков следует отнести и высокое омическое сопротивление
электрода, что вынуждает пользоваться электронными потенцио-
метрами с высокими коэффициентами усиления.
Потенциал стеклянного электрода изменяется при изменении
температуры. Для каждой температуры от 0 до 100°, в зависи-
мости от марки электрода, сохраняется прямолинейная зависи-
мость Е=Е0—ОрН, а угол наклона прямой, отвечающей этому
уравнению, обусловлен температурой. Однако все прямые для
данного электрода, соответствующие определенным температурам,
пересекаются в какой-то одной точке, т. е. в этой точке величина
потенциала стеклянного электрода, соответствующая определен-
ному значению pH, не зависит от температуры и является постоян-
ной характеристикой электрода. Эта точка называется изопотен-
циальной, а соответствующие ей величины pH и потенциалы — ее
координатами.
Промышленность выпускает большой набор стеклянных
электродов, устройство которых показано на рис. 14, а характе-
ристики приведены в табл. 5.
Таблица 5
Характеристики некоторых стеклянных электродов отечественного производства
Тип электрода Координаты изопотенци- ал ьной точки относительно насыщенного хлорсеребря- ного электрода сравнения Темпера- турный интервал, °C Пределы измерения pH при 20°С Электрическое сопротивление при 20°С, Мом Крутизна характеристик, мВ/pH
pH Е. мВ
ЭСЛ-41Г-04 ЭСЛ-41Г-05 3,28+0,2 —33^-24 —5++40 0—12,7 50+40 59,1±0,5
ЭСЛ-11Г-04 ЭСЛ-11Г-05 ЭСЛ-43-07 7,0+0,2 — 10+12 15—100 0—50 —0,5+ +12,7 0—12 500+250 50+40 59,1+0,5 59,1+0,5
ЭСЛ-63-07 7,0+0,2 —10±12 20—100 0—14 700+300 59,1+0,5
ЭСП-44Г-04 4,15 — 184+24 10—30 0—12,7 50+40 59,1+0,5
РЕАКЦИЯ ПОЧВЕННЫХ ВЫТЯЖЕК
, И СУСПЕНЗИЙ
v И ЕЕ ОПРЕДЕЛЕНИЕ
Охарактеризовать кислотность или щелочность почв можно
двумя путями. Первый из них заключается в том, что общий уро-
вень кислотности (щелочности) находят титрованием почвенных
вытяжек или суспензий раствором щелочей (кислот). Результаты
38
титрования указывают на общее содержание компонентов, способ-
ных реагировать с основаниями или с кислотами — при заданных
значениях pH.
В подзолистых, дерново-подзолистых, красноземах и других
кислых почвах такими компонентами могут быть свободные низко-
молекулярные органические кислоты, растворимые высокомолеку-
лярные гумусовые вещества с кислыми функциональными группа-
ми, гидролитически кислые минеральные соединения, особенно
соединения железа и алюминия, и, наконец, компоненты почвенно-
го поглощающего комплекса, если в составе поглощенных катио-
нов находятся ионы водорода, алюминия, железа. Принято разли-
чать актуальную кислотность, обусловленную водорастворимыми
компонентами почвы и определяемую титрованием водных вытя-
жек, и кислотность потенциальную, природа которой связана с
составом обменных катионов. Потенциальную кислотность подраз-
деляют обычно на обменную и гидролитическую.
Обменную кислотность находят титрованием солевой вытяжки
из почвы; для приготовления солевой вытяжки используют нейт-
ральный раствор 1 п. КС1. При взаимодействии КС1 с почвой из
поглощающего комплекса вытесняются ионы водорода
пн ; кс1^:пк на.
Образующаяся НО полностью диссоциирована, и в однократ-
ную вытяжку не вытесняются полностью все способные к обмену)
ионы водорода. Величину обменной кислотности находят по коли-
честву щелочи, пошедшей на титрование НС1.
При определении гидролитической кислотности обменные ка-
тионы вытесняют раствором гидролитически щелочной соли, а не
раствором гидролитически нейтральной соли КС1. Обычно упот-
ребляют CH3COONa. Тогда
ПН CH8COONa^± ПХа - СН8СООН.
В этом случае образуется слабая уксусная кислота, в форме
которой связываются вытесняемые из почвы ионы водорода. Свя-
зывание одного из продуктов реакции, а именно Н+-иона, обеспе-
чивает более полное протекание реакции. Поэтому на титрование
ацетатной вытяжки расходуется больше щелочи, чем на титрова-
ние KCl-вытяжки.
Кроме ионов водорода в этих реакциях могут участвовать об-
менные ионы А13+ и Fe3+. Алюминий практически всегда обнару-
живается в KCl-вытяжках из кислых почв. Его появление в вы-
тяжках можно объяснить двумя путями. Во-первых, прямой реак-
цией вытеснения обменного А13+:
па1 1 зкс1^пк3-- Aici3.
Хлористый алюминий в водных растворах неустойчив и гидро-
лизуется с образованием гидроокиси алюминия:
aici3 । зн2о^:знс1 А1(он)3.
39
*
Таким образом, А1С1з появляется лишь как промежуточное
соединение, а образовавшаяся НО оттитровывается щелочью.
Если pH солевой вытяжки будет ниже 4—4,5, то А1С13 остается
в растворе до начала титрования, а затем оттитровывается:
А1С1а - 3NaOH 3NaCl - Al (ОН)3.
Во-вторых, алюминий может появиться в вытяжке вследствие
вторичной реакции растворения некоторых соединений алюминия.
Схема реакции такова:
ПН8 - ЗКО^ПКз ЗНС1,
А1 (ОН)3 - ЗНС1 А1С13 ' ЗН2О.
Эти механизмы лежат в основе двух гипотез о природе обмен-
ной кислотности (обменного водорода и обменного алюминия).
Дискуссия о природе почвенной кислотности не завершена, но
можно полагать, что в сильногумусированных почвах может пре-
обладать обменный водород, а в минеральных горизонтах — об-
менный алюминий. В щелочных почвах реакция среды наиболее
часто обусловлена карбонатами и обменными катионами натрия.
Второй путь характеристики кислотности или щелочности почв
заключается в измерении pH почвенных паст, вытяжек и суспен-
зий. Согласно определению, величина pH равна отрицательному
десятичному логарифму активности водородных ионов:
pH = —Igo^..
Активность ионов водорода существенно отличается от общего
содержания в системе кислых функциональных групп, которые
могут быть оттитрованы щелочью. Рассмотрим простейший случай,
когда раствор содержит только сильную кислоту, например НС1.
Такая кислота полностью диссоциирована в водном растворе, и
активность ионов водорода отличается от их содержания, или
концентрации [Н+], только величиной коэффициента активности f:
Значение f зависит от заряда иона и ионной силы раствора.
Более сложен вариант, когда в растворе присутствуют непол-
ностью диссоциированные кислоты, такие как уксусная Кислота,
фульвокислоты, или изучается почвенная суспензия, содержащая
водородные ионы в поглощенном состоянии. Естественно, что не-
диссоциированные ионы водорода будут учтены при титровании
таких систем щелочью. Полнота учета зависит только от pH ко-
нечной точки титрования. В то же время активность ионов водо-
рода будет определяться только свободными водородными ионами.
Если обозначить водородные ионы, высвободившиеся при диссо-
циации кислот и почвенного поглощающего комплекса через [Н+Гв],
то
«н- = [нС0]7.
40
Таким образом, величина pH водных вытяжек и суспензии почв
является некоторой обобщенной функцией общего содержания
способных к диссоциации водородных ионов, степени диссоциации
соответствующих водородсодержащих соединений и ионной силы
раствора, от которой зависит величина коэффициента актив-
ности.
Для солевых вытяжек смысл величины pH, конечно, сохра-
няется, но количество свободных водородных ионов в солевой
вытяжке определяется не только диссоциацией кислых компонен-
тов почвы, но и реакцией вытеснения Н+-ионов из почвенного по-
глощающего комплекса.
Оценка кислотности или щелочности почв необходима в раз-
личных целях. При генетических исследованиях знание величин
pH почв или почвенных растворов позволяет определить направ-
ление многих элементарных микропроцессов: растворение или
осаждение гидроокисей, вероятность разложения почвенных мине-
ралов, условия миграции многих элементов в почвенном профиле.
Вместе с тем величина pH, как и титруемая кислотность и
щелочность, указывает на общий характер почвообразовательного
процесса, его итог, что особенно важно при формировании нена-
сыщенных основаниями почв, солонцов, содовых солончаков.
Большое значение показатели реакции среды имеют при диаг-
ностике и классификации почв. Степень насыщенности почв осно-
ваниями, обменная и гидролитическая кислотность, общая и част-
ная щелочность и величины pH водных и солевых вытяжек (сус-
пензий) относятся к числу важнейших классификационных при-
знаков почв. Реакция почв относится также к числу наиболее
важных показателей пригодности почв для выращивания различ-
ных сельскохозяйственных культур. Растения очень чувствительны
к реакции среды, и оптимальный диапазон pH бывает сравнитель-
но узким (табл. 6).
Реакция среды влияет на многие химические свойства почв.
В сильнокислых почвах появляется много токсичного для расте-
ний подвижного алюминия. В нейтральной и слабощелочной сре-
дах фосфаты переходят в нерастворимые и малодоступные расте-
ниям формы соединений. Поэтому регулирование реакции почв
является одной из важнейших задач сельскохозяйственной прак-
тики. Наиболее часто для устранения неблагоприятной кислот-
ности почв употребляется известкование, для снижения щелоч-
ности и мелиорации солонцовых почв — гипсование. В некоторых
случаях, например при заложении чайных плантаций, почвы при-
ходится кисловать, поскольку чайный куст хорошо растет только
на кислых почвах. Нельзя забывать, что с реакцией почв тесней-
шим образом связан их пищевой режим. Поэтому определение
доз извести, гипса и других химических мелиорантов надо про-
водить на строго научной основе. Избыточное известкование, на-
пример, не только экономически нецелесообразно, но может на-
нести и прямой ущерб сельскохозяйственному производству.
41
Отношение растений к кислотности почв и известкованию
(по Д Л Аскинази)
Таблица 6
Отношение к кислот- ности почв и извест- кованию Культуры
зерновые кормовые техничес- кие овощные и картофель плодово- ягодные сидераты
Переносят сла- бую, среднюю и сильную кислот- ность почв, отзы- ваются на извест- кование очень силь- нокислых почв — —- — карто- фель, редька малина люпин однолет- ний, се- раделла
Переносят сла- бую и среднюю кислотность почв, отзываются на из- весткование силь- нокислых почв овес, ози- мая рожь, кукуру- за, просо, гречиха турнепс, брюква, тимофеевка, овсяница подсол- нечник, лен морковь, огурцы, томаты, репа земляни- ка, гру- ша, яб- лоня
Переносят сла- бую кислотность почв, отзываются на известкование главным образом сильно- и средне- кислых почв яровая и особенно озимая пшеница, ячмень кормовая свекла, кор- мовая ка- пуста, ляд- венец конопля, кок-сагыз столовая свекла, капуста, лук вишня, слива, сморо- дина
Понижают уро- жаи и отзываются на известкование даже при слабой кислотности почв - клевер и особенно люцерна, донник, рапс сахарная свекла, горчица, мак
Измерение pH водных
суспензий почв
Измерение pH водных суспензий почв относится к числу наи-
более распространенных почвенных анализов. По величине pH
водных суспензий судят о пригодности почв для выращивания
различных культур, о характере и направлении протекающих в
ней процессов, о необходимости известкования или других прие-
мов регулирования реакции почвы. Значения pH водных суспензий
почв меняются в широком диапазоне. В подзолистых почвах, в
бурых лесных почвах, особенно развитых на кислых породах (гра-
ниты и т. п.), pH водных суспензий могут иногда опускаться до
3,0—3,5, более низкие значения встречаются на рекультивируемых
отвалах горных пород, терриконов. Если такие породы содержат
42
пирит, то его окисление кислородом воздуха приводит к образова-
нию серной кислоты, и реакция таких пород и рядом располо-
женных почв становится сильнокислой. Значение pH доходит до
1—2 и даже более низких величин.
Большая часть дерново-подзолистых, серых лесных почв ха-
рактеризуется умеренно кислой реакцией. В водных суспензиях
почв значения pH обычно находятся в интервале от 3—4,0 до 5,5—
6,5. В мошных типичных, обыкновенных, южных черноземах, каш-
тановых почвах значения pH постепенно нарастают и достигают
в карбонатных почвах и черноземах (в присутствии СаСОз) вели-
чин порядка 8,0—8,5.
Щелочную реакцию водных суспензий имеют солонцеватые
почвы, солонцы и содовые солончаки: в них pH может достигать
9—10.
Таким образом, методы определения pH почвенных суспензий
должны быть рассчитаны на широкий интервал величин от 2—3
до 10—11 единиц pH. Используемые для анализа водных суспен-
зий индикаторные электроды должны, следовательно, сохранять
водородную функцию в интервале pH 2—11, кроме того, они
должны быть нечувствительны к присутствию коллоидальных час-
тиц, а также не должны зависеть от присутствия восстановителей
(для оглеенных и заболоченных почв), легкорастворимых солей
(солончаки) и небольших количеств водорастворимых органиче-
ских веществ.
Этим требованиям в наибольшей степени удовлетворяют стек-
лянные электроды с водородной функцией, которые и используют-
ся практически во всех почвенных и агрохимических лабораториях
в сочетании с электронными потенциометрами (рН-метрами).
Среди других электродов сохраняет свое значение хингидрон-
ный электрод, дающий хорошие результаты для почв с pH в ин-
тервале 2—8 (за исключением сильнооглееных почв). Хингидрон-
ный электрод может быть очень легко изготовлен, не требует
сложной аппаратуры и специальной градуировки. Поэтому при
необходимости им можно легко заменить стеклянные электроды
как в лабораторных условиях, так и в экспедиционной обстановке.
Подготовка почвы к анализу. Были проведены специальные
исследования, в которых подбирались наилучшие способы подго-
товки почв к анализу: высушивание, хранение, характер измельче-
ния. Для получения сопоставимых результатов необходимо соблю-
дать стандартные условия: перед анализом принято разминать
воздушно-сухие образцы и просеивать их через сито с отверстия-
ми в 1 мм.
Наиболее правильное представление о реакции почвы в естест-
венном состоянии можно получить, используя для анализа свеже-
взятые образцы, не подвергавшиеся высушиванию. Этим обеспе-
чивается состояние образца, близкое к нативному.
Обычно для приготовления суспензии берется 25 мл воды на
10 г воздушно-сухой почвы, т. е. отношение почва : вода, равное
43
1 : 2,5. Если взять почву с естественной влажностью порядка 20%,
то объем воды в суспензии увеличивается вследствие этого на
1—1,5 мл (естественная влага минус гигроскопическая вода)
Тогда вместо отношения 1 : 2,5 получим отношение, равное при-
мерно 1 : 2,9. Чтобы устранить это расхождение, можно рекомен-
довать навеску почвы увеличить до 11 г, а объем добавляемой
воды уменьшить до 24 мл. Тогда соотношения почва : вода не бу-
дут сильно отличаться от принятого отношения 1 : 2,5, если брать
навеску почвы с естественной влажностью.
Несмотря на преимущества анализа образцов с естественной
влажностью, в практике лабораторных работ принято использо-
вать воздушно-сухие образцы, что значительно удобнее, поскольку
анализу обычно предшествует довольно длительная процедура
отбора и транспортировки проб почвы, не говоря уже о длитель-
ности их хранения. Поэтому на практике поступают следующим
образом.
Собранные в поле образцы почвы высушивают в тени на воз-
духе, этикетируют и затем хранят в специальном помещении с
чистым воздухом. В помещении для хранения образцов почв и в
лаборатории, где измеряются значения pH, не допускается хране-
ние или работа с летучими кислотами и аммиаком.
Для определения pH берут высушенный, как указано выше,
образец почвы и отбирают из него квартованием среднюю пробу.
Из средней пробы берут лабораторную пробу, масса которой
должна быть около 100 г. Из лабораторной пробы выбирают круп-
ные (хорошо видимые невооруженным глазом) корни и другие
органические остатки. Затем разминают почву в ступке пестиком
с резиновым наконечником и пропускают через сито с отверстиями
в 1 мм. Из подготовленной таким образом лабораторной пробы
берут на технических весах среднюю навеску точно 10 г. Навеску
переносят в конические или плоскодонные колбочки на 100 мл для
приготовления суспензии
Приготовление суспенши. При определении pH существенное
значение имеет соотношение между навеской почвы и количеством
воды, взятой для приготовления суспензии. Почвенные суспензии
не отличаются высокой буферностью, и поэтому каждое дополни-
тельное разбавление изменяет величину pH. Понятно, что при
очень большом разбавлении pH суспензии практически окажется
равным pH дистиллированной воды, взятой для опыта.
Решением II Международного конгресса почвоведов было при-
нято измерять pH водных вытяжек и суспензий при отношении
почва : вода, равном 1 : 2,5. Чтобы получить это отношение, к на-
вескам почвы по 10 г, взятым в конические или плоскодонные
колбы, добавляют 25 мл дистиллированной воды, освобожденной
от СО2 (pH около 5,6). Затем закрывают колбу пробкой и содер-
жимое колбы встряхивают на ротаторе 10 мин. После встряхива-
ния суспензии дают осесть крупным частицам почвы и через не-
сколько минут переливают частично отстоявшуюся суспензию в
44
стаканчик для измерения pH. Остатком суспензии ополаскивают
предварительно промытые электроды
В стаканчик с суспензией погружают индикаторный электрод
и комбинированный электрод сравнения. В тех случаях, ког-
да в лаборатории нет комбинированного электрода сравнения,
в суспензию погружают индикаторный электрод и электролитиче-
ский ключ, с помощью которого соединяют испытуемую суспен-
зию с электродом сравнения. На рис. 15 показаны оба варианта
составления элемента на примере стеклянного электрода и насы-
.шейного каломельного электрода сравнения.
Электролитический ключ заполняют горячим раствором агар-
агара в насыщенном КС1. После охлаждения агар-агар застудне-
вает и прочно удерживается в ключе. Можно приготовить электро-
литический ключ и без агар-агара. В этом случае готовят насы-
щенный (при комнатной температуре) раствор КС1 и заливают
'его в трубку электролитического ключа. Последнюю держат вер-
тикально, так, чтобы раствор заполнял трубку до краев. Из
фильтровальной бумаги туго свертывают маленькие пробочки по
диаметру отверстий ключа. Одну пробочку смачивают насыщен-
ным раствором КС1 и плотно закрывают ею один из концов клю-
ча. Если из второго конца ключа вылилось несколько капель
раствора КС1, то раствор доливают и затем этот конец тем же
способом закрывают второй пробочкой. Главное условие, чтобы
под пробочками не появились пузырьки воздуха. Лезвием бритвы
ровно срезают выступающие части пробочек, и электролитический
ключ готов к работе. Хранят ключи в банке с насыщенным раство-
ром КО.
Ход измерений. Включают в есть pH-метр, дают ему прогреться
указанное в описании прибора время. В стаканчик наливают
буферный раствор с pH 4,01, погружают в этот раствор электроды
и проверяют правильность показаний мрибора. Если прибор пока-
зывает значение pH, отличающееся от 4,01+0,02, его настраивают
по двум буферным растворам (см. с. 25).
Буферный раствор выливают, стаканчик и электроды ополас-
кивают дистиллированной водой, избыток воды снимают фильтро-
вальной бумагой. Затем электроды и стаканчик ополаскивают
приготовленной суспензией почвы и той же суспензией заполняют
стаканчик. Погружают в суспензию электроды и через 2—3 мин
берут отсчет по шкале прибора.
После измерения выливают суспензию, стаканчик и электроды
ополаскивают дистиллированной водой, слегка сушат их фильтро-
вальной бумагой и ополаскивают суспензией следующего образца
почвы. В стаканчик заливают подлежащую измерению суспензию
и далее поступают, как описано выше. Результаты измерений
записывают, как это показано в табл. 7.
Обработка результатов и анализ ошибок. Как правило,
значения pH считывают прямо со шкалы прибора. Иногда возни-
кает необходимость построить градуировочный график для элект-
45
рода. Такая необходимость возникает при неисправностях рН-мет-
ра, когда отказал в
функции электрода, проверке
работе блок настройки, или
при изучении
соответствия потенциала уравнению
Нернста и выявлении влияния со-
путствующих катионов на элект-
родную функцию.
Табища 7
Результаты измерений pH водных
суспензий почвы
Об
ра-
зец
7
8
9
Горизонт и
глубина,
см
Аь 2—11
11—40
В, 60—81
Отноше-
ние поч-
ва : вода
pH
1:2,5
1:2,5
1:2,5
4,90
4,94
5,18
1,26-10~6
1,15 10~6
0,66-10~5
Рис. 15. Составление эле-
мента из стеклянного ин-
дикаторного и насыщенного
каломельного электродов:
1 — стеклянный электрод,
2 — каломельный электрод,
3 — сосуд с испытуемым
раствором, 4 — сосуд с на-
сыщенным раствором КС1,
5 — гидролитический ключ
Рис 16. Зависимость э. д. с. цепи со
стеклянным электродом от величины
pH раствора
Для построения градуировоч-
ного графика используют серию
буферных растворов со значени-
ем pH от 2 до 11. С каждым из
буферных растворов проводят измерения, но показания снимают
не в единицах pH, а в вольтах, или условных единицах (делениях
46
шкалы). По результатам строят график зависимости э. д. с. —
pH (рис. 16).
Для элемента со стеклянным электродом э. д. с. складывается
из нескольких скачков потенциалов:
Э. Д. С. Ест ЕС1 Е£СС " £хс £кал - ДдИф,
где Ес? — скачок потенциала между стеклянным электродом и
испытуемым раствором;
Е ст — скачок потенциала между стеклом электрода и внут-
ренним раствором;
Дасс — потенциал асимметрии;
£хс — потенциал хлорсеребряного электрода, помещенного
внутри стеклянного электрода;
Дкал— потенциал каломельного электрода;
Ддпф— диффузионный потенциал, возникающий за счет жид-
костного соединения (электролитический ключ — ис-
пытуемый раствор).
При смене испытуемого раствора изменяются только величины
Ест и Ддиф, но последний потенциал мал, и его изменениями мож-
но пренебречь. Тогда
э. д. с. = £ст — const,
или
э. д. с. = Ео — ttpH , const = Eq — ttpH.
Таким образом, по градуировочному графику (см. рис. 16) или
уравнению можно найти pH испытуемого раствора. Так как в
уравнении величина Ео неизвестна, а & может отклоняться от тео-
ретического значения, то эти величины находят, измеряя э. д. с.
элементов с двумя буферными растворами и решая затем обыч-
ную систему двух уравнений с двумя неизвестными.
То же решение используется и при графическом методе нахож-
дения pH. Построив график (см. рис. 16), мы практически нахо-
дим неизвестные ранее величины Ео и величина Ео отвечает
отрезку, отсекаемому градуировочным графиком на ординате, а
•& равна тангенсу угла наклона (а) градуировочного графика:
&=tg а.
Для ясного представления о реакции среды недостаточно огра-
ничиться только величиной pH.
Поскольку pH — это логарифмическое выражение активности
Н+-ионов, то с величиной pH нельзя производить арифметические
действия. Более того, равным изменениям величин pH, но в раз-
ных диапазонах отвечают разные приращения величин активнос-
ти Н+. Например, для изменения pH от 3 до 2 надо к литру
раствора добавить около 9 мг-экв Н+, а для изменения pH от 6
до 5 — только около 0,009 мг-экв Н+. Поэтому более наглядное
представление дает величина ан+ или [Н+].
47
Чтобы найти йц, по величине pH, поступают следующим об-
разом. Согласно определению, рН = —1g йн+; или 1g ан4 =—pH.
Допустим, что водная суспензия гор. А дерново-подзолистой поч-
вы имела значение pH 5,7. Тогда 1g ан 4 =—5,7. Чтобы получить
положительную мантиссу логарифма, прибавляют к характеристи-
ке — 1, а к десятичным долям----(-1. Получим 6,3. По таблице
логарифмов находим, что мантиссе соответствует число 199, а ха-
рактеристика 6 указывает число нулей перед значащими числами.
Тогда получим, что
аи+ = 0,00600199, или 1,99- ЮЛ
Ошибки измерения pH складываются из ошибок взятия сред-
ней пробы и навески, электродной ошибки (отклонение от идеаль-
ной водородной функции плюс влияние сопутствующих компонен-
тов на потенциал электрода), ошибки измерения э.д.с. и погреш-
ности градуировки электрода с помощью буферных растворов.
Наибольший вклад в суммарную ошибку определения связан с
погрешностью измерения э. д. с., которая на многих приборах
достигает +5 мВ и практически никогда не бывает ниже +1 мВ.
Ошибка в 5 мВ отвечает погрешности нахождения pH примерно
на 0,1, ошибка в 1 мВ соответствует примерно 0,02 единицы pH.
Буферные растворы также могут вызвать ошибку порядка сотых
долей pH; ошибки взятия средней пробы, навески и состав раство-
ра (при работе со стеклянным электродом) на результаты изме-
рений существенно не влияют. В целом, очевидно, нельзя считать
достоверными различия между двумя образцами почв, если раз-
ность измеренных значений pH не превышает 0,05.
V Измерение pH водных
вытяжек из почв
Правильно приготовленная водная вытяжка отличается от вод-
ной суспензии только отсутствием взвешенных в водной среде
почвенных коллоидов. Для того чтобы вытяжка удовлетворяла
этому требованию, необходимо соблюсти определенные правила,
поскольку в ходе приготовления вытяжки могут произойти изме-
нения ее состава по двум причинам: 1) изменение состава в ре-
зультате взаимодействия вытяжки с фильтровальной бумагой при
фильтровании; 2) поглощение газов, особенно двуокиси углерода,
при длительном контакте с воздухом в процессе фильтрования.
Устранить первый фактор можно просто: достаточно отбросить
первые порции фильтрата. Устранить второй фактор сложнее
Кстати сказать, контакт с воздухом может повлиять и на pH
водных суспензий. Слабощелочные суспензии, слегка розовые от
фенолфталеина, при стоянии на воздухе могут потерять окраску.
При фильтровании такая потеря окраски происходит очень часто
48
В этом случае прибегают или к замене фильтрования на более
быстрое центрифугирование, или проводят все операции в среде
без СОг.
Величины pH водных вытяжек целесообразно определять
только в тех случаях, когда проводится общий анализ ионного
(солеаософ..состава. С этой целью готовят обычные водные вытяж-
ки из почв при отношении 1:5, как это описано в руководствах
по химическому анализу почв (Аринушкина, 1970). Из приготов-
ленной вытяжки берут 10—20 мл в специальный стаканчик и
измеряют pH, как это описано для водной суспензии почвы.
Наряду с этим рекомендуется измерять pH специально при-
готовленных водных вытяжек из почв. В этом случае вытяжки
готовятся уже при отношении почва : вода 1 : 2,5. Рекомендации
по определению pH в таких вытяжках возникли по двум причи-
нам. Во-первых, первоначально измерение pH почв проводилось
водородным или хингидронным электродами. Эти электроды чув-
ствительны к присутствию коллоидальных частиц, и поэтому для
правильного определения pH приходилось от этих частиц избав-
ляться, т. е. готовить водную вытяжку. С внедрением в практику
почвенных лабораторий стеклянных электродов такая необходи-
мость отпала. Во-вторых, долгое время считалось, что по крайней
мере в кислых почвах значения pH водных вытяжек и водных
суспензий неодинаковы вследствие так называемого суспензионно-
го эффекта, или эффекта Вигнера. Сущность этого эффекта в сле-
дующем. В хорошо перемешанной суспензии значения pH одина-
ковы, в каком бы объеме суспензии их ни измеряли. При стоянии
суспензии последняя постепенно расслаивается: взвешенные час-
тицы опускаются ближе ко дну сосуда, в верхней части остается
чистая дисперсионная среда. Процесс можно ускорить центрифу-
гированием. Опыты, проведенные по измерению pH в суспензии
и в надосадочной дисперсионной среде, показали, что в кислых
почвах pH суспензии обычно ниже, чем pH раствора. Это различие
объяснялось на основе Донанновского равновесия. Более нагляд-
ное объяснение сводилось к тому, что в суспензии Н+-ионы нахо-
дятся в двух формах: свободные Н+-ионы распределены равно-
мерно во всем объеме системы, тогда как поглощенные ионы Н+
удерживаются у поверхности коллоидных частиц, но также влияют
на показания электрода. Концентрация последних пропорциональ-
на количеству коллоидных частиц, что и вызывает снижение pH
суспензии по сравнению с вытяжкой.
Правильное решение вопроса дает химическая термодинамика.
Следует напомнит!, что активность любого компонента системы
является мерой его химического потенциала ц:
щ RT In at.
В равновесной системе химический потенциал каждого компо-
нента в любом объеме (фазе) системы имеет одно и то же значе-
49
ние. Этого требует состояние равновесия. Тогда и активность дан-
ного компонента во всех частях системы должна быть одинакова.
Отсюда следует, что значения pH в суспензии и в дисперсионной
среде не должны различаться. Исключение составляют системы,
в которых на границе раздела фаз возникает скачок потенциала.
Тогда уже говорят о равенстве не химических, а электрохимиче-
ских потенциалов. В этом случае в разных фазах могут быть не-
одинаковые величины активностей данного компонента, а разность
должна соответствовать по величине скачку потенциала между
фазами. Однако в почвенных суспензиях и в системах суспен-
зия— вытяжка скачок потенциала обнаруживается только
между коллоидной частицей и раствором. Обнаружить суспензион-
ный (точнее, коллоидный) эффект Вигнера можно было бы
только с помощью микроэлектродной техники, позволяющей из-
мерить pH непосредственно вблизи коллоидной1 частицы, т. е. в
неподвижном слое компенсирующих ионов.
Чем же можно объяснить практически наблюдаемую разность
измеряемых значений pH? Наиболее вероятно, что эта разность
вызвана диффузионным потенциалом, т. е. потенциалом жидкост-
ного соединения. В почвенную вытяжку или в суспензию погру-
жается обычно не сам электрод сравнения, а электролитический
ключ, вследствие чего неизбежно возникает, хотя и небольшой по
величине, диффузионный потенциал ЕДИф. Величина ЕДИф зависит
от электролита, который использован для приготовления электро-
литического ключа, от его концентрации и от состава испытуемого
раствора. При изменении состава раствора, а особенно при пере-
носе ключа из раствора в суспензию величина ЕДпф может изме-
няться. Этим и объясняется суспензионный эффект.
По изложенным причинам специальное приготовление водных
вытяжек для измерения pH надо признать нецелесообразным.
Вполне достаточно измерить pH в водной суспензии почвы.
о Измерение pH солевых
вытяжек из почв
Величины pH солевых вытяжек определяют только для почв с
кислой или нейтральной реакцией водной суспензии. Если pH сус-
пензии выше 7, то анализ солевой вытяжки теряет смысл или мо-
жет даже создать неверное представление о природе изучаемого
образца почвы.
При взаимодействии почвы с раствором КС1, как описывалось
выше (см. с. 39), из почвы вытесняются поглощенные катионы.
Если в числе последних присутствуют ионы Н+ или А13+, то вы-
тяжка приобретает кислую реакцию. Таким образом, pH солевой
вытяжки характеризует уровень потенциальной (обменной) кис-
лотности почв.
50
Зная pH солевой вытяжки и степень насыщенности почв осно-
ваниями, можно оценить потребность почв в извести (табл. 8).
Таблица 8
Потребность в извести различных почв в зависимости от pH солевой вытяжки
и степени насыщенности почв основаниями V
(no М. Ф. Корнилову)
Потребность в известковании
сильная средняя слабая отсутствует
Почвы pH в КС1 V, % pH в КС1 V, % pH в КС1 К % pH в КС1 И, %
Подзолистые
а) тяжело- и 5,0 45 5,0—5,5 45—60 5,5—6,0 60—70 6,0 70
среднее угли- 4,5 50 4,5—5,0 50—65 5,0—5,5 65—75 5,5 75
нистые 4,0 55 4,0—4,5 55—70 4,5—5,0 70—80 5,0 80
б) легкосугли- 5,0 35 5,0—5,5 35—55 5,5—6,0 55—65 6,0 65
нистые 4,5 40 4,5—5,0 ’40—60 5,0—5,5 60—70 5,5 70
4,0 45 4,0—4,5 45—55 4,5—5,0 65—75 5,0 75
в) супесчаные и 5,0 30 5,0—5,5 30—45 5,5—6,0 45—55 6,0 55
песчаные 4,5 35 4,5—5,0 35—50 5,0—5,5 50—60 5,5 60
4,0 40 4,0—4,5 40—55 4,5—5,0 55—65 5,0 65
Заболоченные торфянистые и тор- фянисто-болотные 3,5 35 3,5—4,2 35—55 4,2—4,8 55—65 4,8 65
Для приготовления солевых вытяжек принято использовать
1 н. раствор КС1. Такая концентрация КС! обеспечивает довольно
полное вытеснение поглощенных катионов даже при однократной
обработке навески почвы. Отношение почва : раствор, как и в
случае водной суспензии, берется равным 1 : 2,5.
Ход измерения сводится к следующему. Берут навеску средней
пробы почвы в 10 г, подготовленной, как при анализе водной
суспензии. Навеску помещают в коническую или плоскодонную
колбу на 100 мл, приливают 25 мл 1 н. КС1, закрывают колбу
пробкой и содержимое взбалтывают на встряхивателе 10 мин.
(Для приготовления 1 н. раствора КС! отвешивают 74,55 г хими-
чески чистого КС!, переносят в стакан, растворяют в небольшом
объеме дистиллированной воды, лишенной СО2, и фильтруют в
мерную колбу емкостью I л. Затем объем раствора в колбе дово-
дят дистиллированной водой без СО2 до метки.) В это же время
включают в сеть pH-метр и подготавливают его к работе, как
описано выше (см. с. 24 и сл.). После взбалтывания суспензии да-
ют отстояться. В солевом растворе частицы суспензии оседают до-
вольно быстро, и осветленную надосадочную жидкость сливают в
стаканчик потенциометра. В тот же стаканчик опускают индика-
торный стеклянный и вспомогательный каломельный электроды и
через 3 мин записывают показания прибора. При работе со стек-
51
Лянным электродом присутствие взвешенных почвенных частиц
не имеет значения, и поэтому фильтрование вытяжки не прово-
дится.
Расчет и обработка результатов те же, что и при анализе вод-
ной суспензии.
V
Измерение pH в почвенных пастах
и непосредственно в почве
Рис. 17. Конус Васильева-
1 — конус, 2 — насечка, 3 — балан-
сир с шарами, 4 — сосуд с почвой,
5 — подставка
Современная аналитическая техника позволяет проводить из-
мерения pH непосредственно в почвах при их естественной влаж-
ности. В этом случае не образец почвы извлекается из профиля
и транспортируется в лаборато-
рию, а, наоборот, исследователь
выходит в поле с рН-метром.
Для измерения pH индикаторный
электрод сравнения погружают в
толщу изучаемого почвенного го-
ризонта.
Измерение pH непосредствен-
но в почве наиболее правильно
отражает реакцию той среды, в
которой протекают почвенные
процессы, развиваются корни
растений и функционируют мик-
роорганизмы. К сожалению, одна-
ко, любые индикаторные электро-
ды работают, начиная лишь с оп-
ределенной минимальной влаж-
ности почвы. Поэтому некоторые
инструкции рекомендуют предва-
рительное иск}сственное увлаж-
нение почвы в поле. Такое увлажнение сдвигает, конечно, естест-
венную реакцию.
Предложен и другой способ — измерение pH в почвенной пас-
те, т. е. в почве, увлажненной искусственно до какого-то опреде-
ленного, заранее установленного состояния. С этой целью можно
выбрать какой-либо постоянный уровень содержания влаги в поч-
ве, или одно из стандартных состояний почвы. Последний способ
предложили Н. К. Крупский и А. М. Александрова (1969) в ме-
тоде определения pH стеклянным электродом в почвенной пасте,
увлажненной до состояния границы текучести. Чтобы получить
почву с такой влажностью, к 100 г почвы, подготовленной для
определения pH и помещенной в фарфоровую чашку с диаметром
5—7 см, добавляют небольшими порциями дистиллированную
воду без СОг и тщательно перемешивают шпателем до получения
52
однородной пасты Влажность, соответствующая границе текучес-
ти, контролируется балансирным конусом Васильева.
Конус Васильева (рис. 17) изготавливают из нержавеющей
стали, весом 76 г, с углом наклона боковой поверхности 30° и
высотой конуса 45 мм. Поверхность конуса отшлифована и отпо-
лирована. На расстоянии 10 см от острия конуса сделана насечка,
которая служит для наблюдения за глубиной погружения конуса
в почвенную массу. Для того чтобы конус погружался строго вер-
тикально, к нему припаивают балансирное коромысло, вес кото-
рого учитывается при изготовлении конуса.
Увлажненную и тщательно перемешанную в фарфоровой чаш-
ке почву выравнивают с поверхности и на поверхность почвы уста-
навливают конус и дают ему свободно погрузиться в почву. Если
конус погрузился в почву до уровня насечки, то влажность почвы
соответствует границе текучести. Если конус погрузился недоста-
точно глубоко и насечка осталась выше поверхности почвы, в
чашку следует добавить еще немного воды, почву тщательно пере-
мешать и вновь провести испытание с помощью конуса. Эти опе-
рации повторяют до тех пор, пока конус не будет погружаться
в почву точно до насечки Если конус погружается глубже насеч-
ки, то следует подсыпать сухой почвы и пасту вновь тщательно
перемешать, затем опять погрузить конус. При некотором навыке
в работе доведение влажности почвы до границы текучести зани-
мает несколько минут.
Через 5 мин после увлажнения в почву помещают стеклянный
электрод и электрод сравнения и определяют величину pH.
V БУФЕРНОСТЬ ПОЧВ
Буферность почвы — это способность почвы поддерживать
свойственную ей реакцию при добавлении к почве кислоты или
щелочи. Более общее определение буферности включает и незави-
симость реакции среды от разбавления. Зависимость реакции вод-
ной суспензии почвы от количества добавленной к ней кислоты
НС1 или щелочи NaOH (кривые буферности) показана на рис. 18.
При действии на чистый песок воды величина pH воды не из-
меняется (см. рис. 18,/). Добавление небольших количеств кисло-
ты резко изменяет значение pH до 1,5—2. Затем величина pH из-
меняется постепенно. При добавлении щелочи также наблюдается
резкое изменение pH. Значения pH на кривой буферности песка
практически соответствуют pH водных растворов кислоты или
щелочи. Иная картина наблюдается при изучении почв. Дерново-
подзолистая почва имеет уже более пологую, чем у песка, кривую
буферности (см. рис. 18, 2), т. е. при добавлении к почве кислоты
или щелочи реакция среды меняется не так резко. В щелочном
плече кривой различия выражены сильнее. Кривая буферности
чернозема (см. рис. 18, «3) имеет еще более пологий характер, что
53
говорит о незначительном изменении реакции почвы при дейст-
вии на нее как кислоты, так и щелочи.
Буферность почвы имеет большое практическое значение.
Малобуферные почвы имеют неустойчивую реакцию среды, быст-
Рис 18. Кривые буферности почв
1 — чистый песок, 2 — дерново-под-
золистая почва, гор Ai, 3 — черно-
зем типичный, гор Ai
ро и значительно изменяющую-
ся при выпадении дождей, поли-
вах, внесении удобрений и дру-
гих воздействиях на почвы. В ря-
де случаев это может отрица-
тельно влиять на растение. Реак-
цию малобуферных почв легче
сместить добавлением извести
или путем кислования, но после
мелиорации в таких почвах реак-
ция среды быстрее возвращается
к исходному состоянию, чем в
почвах высокобуферных.
Почвы с ' высокой буфер-
ностью обеспечивают стабильные
кислотно-основные условия для
сельскохозяйственных культур, но
при неблагоприятной реакции
среды они требуют внесения бо-
лее высоких доз мелиорирующих
средств.
Определение буферности почв
Все методы определения буферности почв основаны па опреде-
лении сдвига величин pH почв или почвенных суспензий при до-
бавлении к ним кислот или щелочей. Варианты существующих
ныне методов различаются по характеру используемых реактивов
(например, КОН, Са(ОН)г, СаСОз), времени и условиям взаимо-
действия с почвой и по выбранной базисной линии отсчета. В ка-
честве базисной линии часто берут кривую титрования суспензии
песка.
Определение буферности почвы по Аррениусу. Этот метод при-
годен для определения буферной способности кислых, нейтраль-
ных и щелочных почв. Для анализа” берут серию навесок почв и
приливают растворы кислоты или щелочи различной концентра-
ции. После наступления равновесия в суспензиях определяют ве-
личину pH. Количество прилитой кислоты или щелочи и соответ-
ствующие им значения pH наносят на график. Получают кривую
зависимости АС—pH, т. е. кривую буферности почв, и сравнивают
ее с кривой буферности чистого песка.
54
На технических весах отвешивают 7 навесок по 10 г почв (для
более точного построения кривой количество навесок следует уве-
личить) и переносят их в плоскодонные колбы емкостью 100 мл.
Общее количество раствора (т. е. вода и кислота или вода и
щелочь) должно быть постоянным, поэтому к навескам почвы
приливают различные количества дистиллированной воды без
СО2 В первую колб}' наливают только воду (25 мл), в следующие
три колбы — 13, 19 и 22 мл воды. В эти же колбы приливают 12, 6
и 3 мл 0,1 НС1 соответственно В оставшиеся 3 колбы приливают
13, 19 и 22 мл воды и соответственно 12, 6 и 3 мл 0,1 н. NaOH.
Те же операции проводят и с чистым песком. Колбы плотно за-
крывают пробками и встряхивают на качалке в течение часа, пос-
ле чего дают осесть крупным частицам суспензии и в надосадоч-
ной жидкости определяют величину pH Результаты измерения
записывают, как это показано в табл. 9.
Таблица 9
Результаты измерений буферности почв
Дерново-подзолистая почва
(гор At. 2—11)
№ об- разца Реактив Прилито мл
1 2 3 4 5 6 7
1 Н2О 0,1 н. NaOH 0,1 н. НС1 рн 16 9 8,42 19 6 7,66 22 3 6,74 25 4,90 22 3 3,50 19 6 2,96 16 9 2,25
По результатам измерений строят график, откладывая по оси
абсцисс количество миллилитров добавленной кислоты пли щело-
чи, а по оси ординат — соответствующие им величины pH. Для
сравнения буферной способности различных почв Товборг-Иенсен
предложил строить кривые в одном масштабе- в 1 см — 2 мл НС1
или NaOH; в 1 см — 1 ед — pH. Построив таким образом кри-
вые, оценивают .буферность почв по «площадямДэуферности», .ко-
торые определяют как площади между кривыми титрования поч-
вы и песка.__
'Определение буферности почвы по Н. П. Ремезову. Этот метод,
в отличие от метода Аррениуса, основан на изучении изменения ,
реакции кислых почв при действии на них гидроокисгГкальция.
Для выполнения анализа в 5—Дкбничсских колб емкостью"ТОСГшТ
помещают по 10 г исследуемого образца почвы, подготовленной,
как при анализе водной суспензии. К навескам почвы приливают
по 25 мл 1,0 н. раствора хлористого кальция, приготовленного из
кристаллической соли СаС12-6Н2О. Затем в первую колбу при-
ливают 5 мл 0,04 н. раствора Са(ОН)2, приготовленного из
7-ЧЪ* ’ • Г .,55 /
I (9 ч л
CaO !. Во вторую, третью и последующие колбы приливают 10,
15... мл Са(ОН)2. Колбы с суспензией плотно закрывают пробка-
ми, встряхивают и оставляют на 24 ч. После этого, не встряхивая
суспензии, часть надосадочной жидкости отливают в стаканчик
и измеряют pH раствора, как указано на с. 45. Те же операции
проделывают с предварительно промытым и высушенным песком,
который служит эталоном буферности. По результатам измерений
строят график, откладывая по оси .абсцисс количество .миллилит-
ров добавленного- раствора Са(ОИ)2 в масштабе 1 см — 6 мл
Са(ОН)2, а по оси ординат—Измеренные величины р1Г в масшта-
бе 1 см — 1 ед. pH. По характеру полученной кривой оценивают
буферность почв, как описано в методе Аррениуса.
По кривой буферности можно рассчитать и количество извести,
необходимое для известкования почвы до заданного значения pH.
Для этого на графике буферности (см. рис. 18) через заданное
значение pH проводят прямую, параллельную оси абсцис, до пе-
ресечения с кривой буферности. Из точки пересечения опускают
перпендикуляр на ось абсцисс. Точка на оси абсцисс соответст-
вует тому количеству дцел очи. которое нужно добавить к 10 г поч-
вы, чтобы получить искомос зпачсние pH. Если, например, к 10 г
почвы нужно добавить А мл 0,04 н. раствора щелочи, или
А-0,04 мг-экв основания, тогда в пересчете на СаСО3 к 1 г почвы
необходимо прибавить следующее количество (1 мг-экв СаСО3
соответствует 50 мг):
0,04. X 50
10
СаСО8 мг/Г почвы.
В практике обычно вычисляется количество извести, требуемое
для нейтрализации пахотного слоя на площа’ди 1 га; если вес па-
хотного слоя принять равным 3000 т, то для его нейтрализации
потребуется: 0.04-А-5-3-109 мг/^ТГли 0,04-А-15-10 ц СаСО3 на
1 га- . • > 'Ъ" А '
АКТИВНОСТИ HQHO0 НАТРИЯ' В ПОЧВАХ
f v и почв^ннЬ1х11с^спЕПа^ях^
Активность ионов натрия имеет исключительно важное значе-
ние для солонцов, солонцеватых и засоленных почв. Натрий отно-
сится к числу компонентов почвенных растворов и почвенного
1 Для приготовления раствора Са(ОН)2 на технических весах отвешивают
40—50 г СаО квалификации «х ч» Навеску помещают в фарфоровую чашку
и приливают равное по весу количество дистиллированной воды При гашении
СаО водой происходит сильное разогревание, поэтому чашку с суспензией
закрывают часовым стеклом или кристаллизатором и дают остыть. Через не-
которое время все содержимое чашки переносят в сосуд емкостью 10 л и
заливают водой. После того 'как раствор отстоится и станет прозрачным, его
сливают декантацией и определяют титр.
56
поглощающего комплекса, наиболее неблагоприятно влияющих на
свойства почв и развитие растений. В засоленных почвах натрие-
вые соли NaCI, NaHCO3, Na2CO3 обычно отрицательно влияют на
развитие сельскохозяйственных растений. При содержании обмен-
ного натрия 10—15% от емкости поглощения обнаруживается за-
метное угнетение культурных растений При содержании натрия
до 20—35% от емкости угнетение резко возрастает и требуется
химическая мелиорация почвы.
С поглощенными катионами натрия связывают и неблагоприят-
ные свойства солонцеватых почв и солонцов: легкая пептизируе-
мость, способность к набуханию, высокая липкость, дезагрегиро-
ванность. С угольной кислотой натрий образует щелочные соли
NaHCO3 и Na2CO3. Щелочная реакция может возникнуть и при
взаимодействии Na-содержащих почвенных коллоидов с водой.
Эти свойства натриевых
соединений лежат в основе таблица ю
гипотез происхождения со-
лонцов, высказанных
К. К. Гедройцем.
Измерение активности
ионов натрия в таких поч-
вах позволяет быстро полу-
чить данные для решения
многих важных вопросов.
Можно определить распре-
деление активностей нат-
рия по генетическим гори-
зонтам, что является важ-
Показатели солонцеватости почв
Почвы pNa G. + Na
Несо.топцеватые Слабосолонцевлтые Среднесолонцева- тые Сил ьносолонцеватые и солонцы >3,0 2,4—3,0 1,5—2,4 <1,5 <0,001 0,004—0,001 0,030- -0,004 >0,030
ной генетической характе-
ристикой почв. Активность натрия может служить показателем
уровня накопления вредных для растений натриевых солей. На-
конец, для незасоленных солонцеватых почв активность натрия
может служить важным диагностическим признаком степени их
солонцеватости. С этой целью удобно воспользоваться величиной
р\а, которая по аналогии с pH обозначает отрицательный лога-
рифм активности ионов натрия: pNa =—lg«.\<. Чем выше в поч-
вах активность ионов натрия, тем ниже величины pNa
На этой основе Н. Г. Зырин и Д. С. Орлов (1958) предложили
различать незаселенные солонцеватые и несолонцеватые почвы по
величинам pNa, измеренным в водных суспензиях (табл. 10).
Для определения активности ионов натрия используются стек-
лянные электроды с натриевой функцией. Внешний вид и устройст-
во их такие же, как и у стеклянных электродов с водородной
функцией (см. рис. 14). Особенностью является только состав
стекла, из которого изготовлена рабочая часть электрода. В на-
стоящее время наиболее распространены электроды марки
ЭСЛ-51Г-05, ЭСЛ-51Г-04 из стекла № 78 следующего состава:
SiO2 56,4—56,6%; Na2O 22,9—23,0; А12О3 14,1 — 14,2; В2О 6,4—6,5%.
Принцип действия Na-стеклянных электродов тот же, что i
электродов с водородной функцией. Но селективность по отношс
нию к ионам натрия или пределы применимости Na-стеклянных
электродов зависят от величины константы К в уравнении Николь-
ского, как это было показано на с. 36. Если КаКл+ ^>йн+ , то
можно считать, что электрод обладает только натриевой функцией,
и его потенциал определяется формулой
Ест = £о1 2,303-^1 lg nNa+ = Ео- (JpNa.
Для соблюдения указанного условия необходимо, чтобы значе-
ние константы К было достаточно велико. Это достигается подбо-
ром стекла специального состава. Конечно, в сильнокислых раство-
рах заметно проявляется и влияние водородных ионов. Поэтом}'
Na-стеклянные электроды обычно используются в слабокислой,
нейтральной и щелочной средах; минимально измеряемая величи-
на ам близка к 10~4 М. При меньшей активности ионов нат-
рия показания электрода уже не реагируют на изменение вели-
чины Пка+ •
Определение активности ионов натрия
в водных суспензиях солонцовых
и солончаковых почв
Лабораторную пробу почвы массой около 100 г разминают
в ступке пестиком с резиновым наконечником, отбирают крупные
корпи и почву просеивают через сито с отверстиями 1 мм. Из про-
сеянной почвы на технических весах берут среднюю навеску мас-
сой 10 г и переносят ее в коническую колбу, заливают дистилли-
рованной водой без СО2, исходя из соотношения: на 10 г почвы
50 мл воды. Содержимое колбы встряхивают 10 мин на ротаторе.
После встряхивания дают осесть крупным частицам суспензии
почвы и затем сливают ее в стаканчики.
В некоторых случаях требуется определить активность ионов
натрия в водной вытяжке. Для приготовления водной вытяжки
полученную суспензию после встряхивания на ротаторе центрифу-
гируют при 7000 об/мин в течение 10 мин. Сильносолонцеватые
почвы центрифугируют более длительное время. Надосадочную
жидкость сливают в стаканчики для измерения pNa-стеклянным
электродом.
Ход измерений. Включают в сеть pH-метр, дают ему прогреть-
ся в течение времени, указанного в описании прибора. Затем про-
изводят градуировку электрода по стандартным растворам NaCl
(табл. 11).
С этой целью измеряют э.д. с. цепи, составленной из натриево-
го стеклянного электрода и хлорсеребряпого или каломельного
электрода сравнения, погруженных в стандартные растворы. При
смене растворов электроды следует ополоснуть дистиллированной
58
водой, осушить фильтровальной бумагой, а затем ополоснуть ис-
следуемым раствором. После погружения электродов в раствор
показания прибора снимают через 5 мин.
Используя полученные данные, строят график зависимости ве-
личины э.д.с. от pNa (рис. 19). В течение дня зависимость
Таблица 11
Стандартные растворы NaCl (25°С)
Концентра- ция NaCl, М Т4- cNa+ pNa
0,4000 0,693 0,2772 0,56
0,2000 0,735 0,1470 0,83
0 1000 0 778 0,0780 1,11
о'0500 0,822 0,0410 1,39
0,0100 0,903 0,0090 2,05
0,0020 0,952 0,0019 2,72
0,0010 0,963 0,0010 3,00
0,0005 0,975 0,0005 3,30
0,0001 1,0 0,0001 4,00
э. д. с. — pNa может несколько
измениться, поэтом)' градуиро-
вочную кривую периодически
проверяют.
После градуировки электро-
да стаканчик и электроды спо-
Таблица 12
Результаты измерений pNa
водных суспензий почв
Горизонт и глубина, см э. д. с., мВ pNa aNa+
Ai 0—7 —24 3,08 0,0008
7-17 0 2,65 0,0022
17—27 4-27 2,20 0,0063
27—37 4-54 1,67 0,0214
Рис. 19. Зависимость э. д. с. цепи с
натриевым стеклянным электродом
от pNa раствора
ласкивают дистиллированной водой, осушают фильтровальной
бумагой, ополаскивают суспензией исследуемого образца почвы.
Наливают в стаканчик суспензию, погружают туда оба электрода
и через 5 мин снимают показания прибора. Результаты измере-
ний заносят в таблицу (табл. 12).
По градуировочному графику (см. рис. 19) находят величину
pNa и потенцированием рассчитывают активность иона натрия
в суспензии так же, как активность иона водорода (пример расче-
та см. с. 47—48). j .,/<
Определение содержания натрия
в водных вытяжках
из засоленных почв
В почвах и почвенных растворах, содержащих в составе раство-
римых солей преимущественно хлориды натрия, стеклянным элект-
родом с натриевой функцией можно определить не только актив-
59
ность, но и концентрацию ионов натрия, которую часто бывает
нужно знать для решения ряда мелиоративных вопросов. Для
этого достаточно принять, согласно Н. О. Авакяну, что коэффи-
циенты активности в стандартных и анализируемых растворах
равны. Тогда градуировочный график строят в координатах
Е—lg С, а весь ход определений остается прежним, как при опре-
делении активности ионов натрия. Результат получается в тех
единицах концентрации, которые были выбраны для построения
графика — гл-1, М, н.
В почвах с более сложным солевым составом, особенно при вы-
соком содержании двухвалентных ионов, а также в водных вытяж-
ках, содержащих много органических веществ или соединений
железа и алюминия, концентрацию натрия, как описано выше,
определить нельзя, не выровняв коэффициенты активности в стан-
дартных и испытуемых растворах. С этой целью было предложено
пользоваться избытком индифферентной соли (Д. С. Орлов и
Н Н. Цикурина), нивелирующей разницу ионных сил растворов.
Было установлено, что коэффициенты активности ионов натрия
остаются практически постоянными, если раствор содержит СаС12
в концентрации от 0,3 до 0,5 М. Это позволяет определить содер-
жание натрия в различных засоленных почвах, пользуясь следую-
щим методом.
Готовят три исходных раствора: 0,01 М NaCl, 1,0 М. NaCl и
раствор СаС12, содержащий 180 г соли в литре раствора. В колбы
на 100 мл вносят последовательно 3, 10 и 30 мл 0,01 М раствора
NaCl и далее— 1, 3, 10 и 30 мл 1,0 М раствора NaCl. В каждую
колбу добавляют по 25 мл раствора СаС12 и доливают до метки
дистиллированной водой, лишенной СО2. Получается серия стан-
дартных растворов, каждый из которых содержит СаС12 из расче-
та 45 г в литре и NaCl в следующей последовательности: 3-10~4,
1-Д0~3, 3-Ю-3, МО’2, 3-10-2, 1-Ю-1 и 3-Ю'1 М. По этим раство-
рам градуируют электрод и строят градуировочный график в коор-
динатах Е — lgCNa+-
Для приготовления водной вытяжки образцы засоленных почв
заливают водой в соотношении 1 : 5 и встряхивают 10 мин. Части-
цам почвы дают осесть, раствор над осадком сливают в центри-
фужные пробирки и центрифугируют до полного осветления при
скорости 6—7 тыс. об/мин. Отбирают 50 мл прозрачной вытяжки
и переносят в мерные колбы емкостью 100 мл, добавляют в каж-
дую колбу по 25 мл исходного раствора СаС12 п доливают водой
до метки. После перемешивания раствор наливают в стаканчик и
измеряют э. д. с. цепи. По графику находят содержание ионов
натрия в растворе.
Полученный результат выражает концентрацию ионов натрия
в анализируемом растворе в грамм-молях на литр и численно сов-
падает с содержанием грамм-молей . натрия в 100 г почвы. Это
вытекает из следующего расчета. Если вытяжку готовят при соот-
ношении почвы к раствору 1 : 5, то 100 г почвы отвечает 500 мл
60
вытяжки; последнюю разбавляют перед измерением активности
в два раза, следовательно, 100 г почвы отвечает 1000 мл раствора.
Таким образом, если концентрация ионов натрия в растворе ока-
залась равной 2-10~2М, то в 100 г почвы содержалось 2-10~2 г-моль
ионов натрия. Чтобы найти содержание натрия в граммах на 100 г
почвы, найденную величину надо умножить на атомную массу
натрия: 2-10 2-22,99=0,46 г/100 г почвы, или 0,46%.
Определение водорастворимого
и поглощенного натрия по Боуэру
Метод определения натрия в избытке СаС12 (см. с. 60) при-
годен для почв с реакцией, близкой к нейтральной. Если реакция
сильно отличается от нейтральной, то в качестве индифферентно-
го электролита применяют буферные растворы. В методе Боуэра
роль буфера и индифферентного электролита выполняет
Mg(CH3COO)2. Измерения э. д. с. осуществляют на ламповом по-
тенциометре с чувствительностью до ±1 мВ. Стеклянный электрод
с натриевой функцией градуируют по стандартам, содержащим 1,
10 и 100 мг-экв Na+ в 1 л 1,0 н. раствора Mg(CH3COO)2.
Порядок определения натрия в природных водах и водных вы-
тяжках из почв. 10 мл пробы воды или водной вытяжки из почвы
помещают в сдакан емкостью 50 мл. Добавляют 10 мл 2,0 н.
раствора Mg(CH3COO)2 и перемешивают. В раствор погружают
Na-стеклянный и каломельный электроды и измеряют э. д. с. цепи.
Содержание ионов Na+ (в мг-экв/л) находят по градуировочному
графику.
Порядок определения содержания поглощенного натрия в со-
лонцевых почвах. 5 г почвы помещают в круглодонную центри-
фужную пробирку емкостью 50 мл. Добавляют в пробирку 33 мл
1,0 н. раствора Mg (СН3СОО)2, закрывают пробкой и встряхивают
10 мин. После встряхивания снимают пробку и суспензию центри-
фугируют, при скорости вращения 1500—2000 об/мин до полного
осветления раствора. Прозрачную жидкость сливают в мерную
колбу на 100 мл, в центрифужную пробирку снова доливают
33 мл 1,0 н. раствора Mg(CH3COO)2, встряхивают и центрифуги-
руют. Такую обработку проводят 3 раза, сливая экстракты в мер-
ную колбу, после чего объем раствора в колбе доводят до 100 мл,
-добавляя 1,0 и. раствор Mg(CH3COO)2.
Полученный раствор переносят в стаканчик, погружают в ра-
створ Na-стеклянный и каломельный электроды и измеряют э. д. с.
цепи. Найденное по графику количество ионов натрия равно сум-
ме поглощенного натрия и водорастворимых форм его соединений.
Вычитая из найденной суммы количество натрия, входящего в
состав водорастворимых соединений, получают содержание погло-
щенного натрия. Результат, как и при других потенциометрических
измерениях, выражен в единицах концентрации (мг-экв/л); для
61
пересчета на 100 г почвы необходимо знать соотношение почвы и
раствора. В данном случае 5 г почвы отвечает 100 мл раствора,
следовательно, количество миллиграмм-эквивалентов натрия в
литре раствора соответствует содержанию натрия в 50 г почвы.
Для пересчета на 100 г почвы полученный результат умножают на
два.
АКТИВНОСТИ ИОНОВ КАЛИЯ в ПОЧВАХ
И ПОЧВЕННЫХ СУСПЕНЗИЯХ
Прямое определение величин рК в почвах и почвенных вытяж-
ках и суспензиях имеет то же значение, что и определение pH и
pNa. Кроме того, на измерении рК основано определение калий-
ного потенциала и потенциальной буферной способности почв в
отношении калия.
Прямое измерение активности ионов калия в почвенных суспен-
зиях впервые было осуществлено Д. С. Орловым и А. X. Альзу-
байди (1965). В почвах с невысокой ионной силой можно опре-
делять К-стеклянным электродом не только активность, но и кон-
центрацию ионов калия, поскольку коэффициенты активности в
таких почвенных суспензиях близки к единице.
Стеклянные электроды с калиевой функцией изготавливаются
из стекла, например, следующего состава (в молекулярных про-
центах) :
К2О —20,5, А12О3 —4,5, SiO2—75,0.
Потенциал калиевого стеклянного электрода определяется ак-
тивностью ионов в равновесном растворе:
Ек =Е0 —2,303-^ рК.
Проявление калиевой функции зависит от состава стекла и опре-
деляется прочностью связи ионов калия с силикатным остовом
стекла. Промышленность выпускает калиевые стеклянные электро-
ды типов ЭСЛ-96-10 и ЭСЛ-96-11.
Калиевые электроды позволяют измерить активность ионов ка-
лия до значений ак+ порядка п-104 моль/л; они достаточно селек-
тивны по отношению к ионам кальция и магния. Ионы натрия
и аммония сильно влияют на потенциал К-стеклянного электрода,
когда концентрация этих ионов становится соизмеримой с кон-
центрацией ионов калия. Но столь высокая концентрация иона
аммония в почвах возможна лишь в небольшие интервалы времени
после внесения аммонийных удобрений, а высокие концентрации
натрия свойственны только солонцам и солончакам. В остальных
случаях почвы исследуют с помощью К-стеклянного электрода.
62
Определение активности ионов
калия в почвенных суспензиях
по Орлову и Альзубайди
Навеску воздушно-сухой почвы разминают в ступке пестиком
с резиновым наконечником, отбирают крупные корни и просеивают
почву через сито с диаметром отверстий 1 мм. Из просеянной поч-
вы берут среднюю пробу 20 г, заливают ее в колбе 40 мл дистил-
лированной воды, перемешивают
и встряхивают на взбалтывателе
10 мин. После взбалтывания сус-
пензии дают отстояться и над-
осадочную жидкость сливают в
стаканчик. В жидкость погружа-
ют стеклянный калиевый элект-
род и электролитический ключ,
приготовленный на CH3COOLi.
Использование ключа с КС1 в
данном случае, естественно, не-
возможно. Другой отросток клю-
ча помещают в насыщенный
раствор КО, куда погружен
Таблица 13
Стандартные растворы КС1 (25°С)
Концентра- ция KCI, М V4- °К( РК
1,0000 0,604 0,604 0,22
0,1000 0,769 0,0769 1,11
0,0100 0,901 0,0090 2,05
0,0010 0,965 0,0010 3,00
0,0001 1,000 0,0001 4,0
и электрод сравнения. Элек-
троды соединяют с входными клеммами предварительно
подготовленного потенциометра. Устанавливают на потенцио-
метре требуемый диапазон измерений и через 5 мин берут отсчет
в мВ. Градуировочный график К-стеклянного электрода строят в
координатах э. д. с. — рК. Для градуировки используют стандарт-
ные растворы КС1, приготовленные из перекристаллизованной
соли квалификации «х. ч.» или из фиксанала (табл. 13). Правиль-
ность градуировочного графика периодически проверяют.
По градуировочному графику находят значение рК испытуемо-
го раствора и, если необходимо, потенцированием находят а^+,
Определение калийного потенциала
и потенциальной буферной способности
почв в отношении калия
Одной из важнейших задач химии почв и агрохимии является
определение обеспеченности растений элементами питания, в том
числе калием. Обычно с этой целью используют различные вытяж-
ки из образцов почв. Считается, что содержание того или иного
элемента в вытяжке является мерой обеспеченности растений этим
элементом. Этот прием оказался полезным для относительной ха-
рактеристики почв и определения доз вносимых удобрений. В то
же время разные вытяжки не могут рассматриваться как универ-
сальный прием. Эти вытяжки частично или полностью извлекают
63
только некоторые соединения калия и не отражают способность
почвы поддерживать на определенном уровне содержание доступ-
ного растениям калия в течение вегетационного периода.
Разработка универсальной теории обеспеченности растений
элементами питания основана на использовании двух важнейших
понятий: фактора емкости и фактора интенсивности. Под факто-
ром емкости понимается общее содержание элемента или не-
которых его соединений в почвах. Эта величина позволяет судить
о том, какое количество элемента в принципе может быть извле-
чено из почвы растениями. Фактор емкости находят путем анали-
за различных вытяжек; это может быть водорастворимый, обмен-
ный, необменный калий или другие его соединения. Фактор интен-
сивности свидетельствует о том, какое воздействие на растение
(или на другие объекты) может оказать калий в данный момент,
если рассматривать неизменную, нативную систему почва — рас-
тение. При характеристике калийного состояния почвы в качест-
ве фактора интенсивности используют «калийный потенциал».
Понятие калийного потенциала возникло на основе представле-
ний о том, что важнейшей формой доступного растениям калия
является обменный калий. Поступление К+ из поглощенного со-
стояния в почвенный раствор зависит от обмена ионов почвенного
раствора — Са2+, Mg2+ и других на поглощенные ионы К4. Если
принять, что в почвенном растворе преобладает из катионов Са2+,
тогда обменную реакцию можно записать так:
ПК2 Са'21ХПСа 2К“,
или
[ПСа] _ Са24
[ПК2] а^+
Вудруфф и Ульрих приняли, что изменение свободной энергии
системы AZ° в результате этой реакции можно выразить урав-
нением
AZ°-= — 2,ЗЯТ(рК —0,5 рСа),
или в более общей форме:
AZ° = — 2.3RT [рК — 0,5р(Са - Mg)] -
= — 1364[рК — 0,5р(Са Mg)].
Это выражение и принято для вычисления калийного потенциала.
При расчете по последнему уравнению калийный потенциал выра-
жается в калориях. По Вудруффу, при значениях потенциала
ниже 2000 кал — в почвах избыток калия; при потенциалах от —
2500 до — 3000 кал — оптимальные условия питания растений; при
потенциалах — 3500—4000 кал растения испытывают недостаток
калия. О. П. Медведева (1975) подчеркивает, что отечественные
авторы дают несколько отличающиеся градации обеспеченности.
64
Иногда формулу калийного потенциала (КП) упрощают и запи-
сывают ее в виде КП = рК—0,5 рСа. Ниже изложено определение
калийного потенциала и ПБС? по описанию О. П. Медведевой.
Определение калийного потенциала
в водной вытяжке по Ульриху
30 г воздушно-сухой почвы помешают в центрифужную про-
бирку емкостью 100 мл, добавляют 20 мл дистиллированной воды,
к которой добавлен тимол (на кончике ножа) или азид натрия
NaN3 (на 100 мл воды 100 мг NaN3) для подавления микробиоло-
гических процессов. Через суспензию 15 мин пропускают ток воз-
духа, затем суспензию центрифугируют 20 мин при 15000 об/мин
и фильтруют для удаления органических остатков, не осаждаю-
щихся при центрифугировании.
Затем, по Ульриху, в фильтрате определяют концентрации ка-
лия, кальция и магния. Калий определяют на пламенном фото-
метре. Сумму кальция и магния находят комплексонометрически,
титрованием аликвоты фильтрата 0,02 н. раствором трилона Б.
Для вычисления калийного потенциала по результатам изме-
рений необходимо знать не концентрации ионов, а их активности.
Рассчитать активность ионов по величинам концентрации можно,
зная значение коэффициентов активности /, которые вычисляют
по уравнению Дебая — Гюккеля:
. , 0,51 z2/|T
Ш f =---------
1 +
где z— валентность катионов, ц— ионная сила раствора. Для на-
хождения коэффициента активности по этому уравнению нужно
знать ионную силу раствора, которую можно рассчитать, если
принять, что в водной вытяжке преобладают только Са2+ и К+ и
эквивалентные им количества одновалентных анионов (например,
С1~), тогда р можно рассчитать согласно следующему уравнению:
CiZ| -|- С2г| -| ... -Ц С,
р. . -
ССа2+’22лСк+ 12 +Ссг’12
2
где С выражена в г-ионах/л.
Подставив значение р в уравнение Дебая — Гюккеля,
средний коэффициент активности иона калия:
находят
0,51 /р.
3 Зак 284
65
и иона кальция:
Отсюда находят
»
и/с1„ = __2ДШд
1 Б/н
flK+ = ClC /к+
и калийный потенциал КП:
Определение калия на пламенном
прямым измерением пк+ с помощью
Аналогично можно воспользоваться
функцией (см. с. 69) для нахождения
Рис 20 Определение калийного потенциа-
ла и ПБСК в почвах по методу Бекетта
КП = рК —0,5 рСа.
фотометре можно заменить
К-стеклянного электрода,
электродом с кальциевой
аСа2+. Искомую величину
КП можно вычислить сра-
зу, минуя стадию нахожде-
ния ионной силы и коэф-
фициентов активности.
Определение потенци-
альной буферной способно-
сти почв по отношению к
калию по Бекетту. Этот
метод позволяет сразу най-
ти калийный потенциал
почв, обозначаемый КП
или 1 (фактор интенсив-
ности), и общее количество подвижного калия в почве — Q
(фактор емкости). Потенциальная буферная способность почв в
отношении калия — ПВО выражает способность почвы противо-
стоять изменению калийного потенциала при выносе калия рас-
тениями или при внесении калия с удобрениями. Эта способность
выражается величиной отношения фактора емкости к фактору ин-
тенсивности:
ПБС* =
I
Для определения ПБС1' берут 6 навесок почвы с полевой влаж-
ностью из расчета, чтобы каждая навеска содержала 5 г воздуш-
но-сухой почвы. К навескам приливают по 50 мл 0,002 М раствора
СаС12, содержащего последовательно 0; 0,2; 0,4; 0,6; 0,8 и
1 мг-экв/л КО. Почвы с раствором взбалтывают 30 мин при
25±1°С и фильтруют. В фильтрате определяют К+ на пламенном
фотометре или измеряют активность ионов калия с помощью
К-стеклянного электрода. Сумму кальция и магния находят
комплексонометрически.
По результатам определения К+ и (Ca2+ + Mg2+) в растворах
находят отношение активностей °к+: °(Ca2+-i мк2+> , используя
те же приемы, что и при нахождении калийного потенциала (см.
66
с. 65). Эти величины откладывают на графике по осп абсцисс
(рис. 20). Затем вычисляют величину ЛК — то количество калия,
которое было поглощено почвой из раствора или, наоборот, отдано
в раствор почвой. При малых концентрациях калия в исходном
растворе почва отдает часть калия, его концентрация после
взаимодействия раствора с почвой увеличивается. Эти величины
обозначают — ЛК. При большом содержании К+ в растворе почва
поглощает некоторое его количество--|-Л К- Величины ±Д К от-
кладывают по оси ординат того же графика (см. рис. 20). Очевид-
но, что при некотором отношении ак+: °'cl2+< Mg2+> величина
Л К=0 Из изложенного следует, что величина ак 1 , найденная
при условии Л К=0, отвечает активности ионов калия в почве,
находящейся в равновесии с 0,002 М раствором СаСБ- Следова-
тельно, в этой точке отношение активностей : c\ct2+^Mg2+)
отвечает величине, свойственной неизмененной почве. Согласно оп-
ределению, эта величина отношения равна калийному потенциалу
почвы КП и является численным выражением фактора интенсив-
ности I Общее количество калия, которое в этих условиях может
быть отдано почвой в раствор, находят путем экстраполяции
прямолинейного участка графика до пересечения с осью ординат.
Эту величину можно обозначить Q=A Ко- Количество трудно-
доступного калия можно найти, экстраполируя график полностью
до пересечения с осью ординат в точке L.
Величина потенциальной буферной способности почв по отно-
шению к калию выражается, по Бекетту, тангенсом угла наклона
прямолинейного отрезка графика:
ПБС/г = tga =--------------- = -5-.
[°К+ -“(Ca^+Mg24)]. 1
Определение в почвах активностей других ионов
Кроме стеклянных электродов с водородной, калиевой и на-
триевой функциями в почвоведении достаточно широко исполь-
зуются ион-селективные электроды для определения хлорид-ионов,
кальция и нитрат-ионов.
Определение активности хлорид-ионов
Определение активности хлорид-ионов в почвах и почвенных
растворах производится хлорсеребряным электродом, который яв-
ляется электродом второго рода, обратимым к ионам С1~. Класси-
ческий хлорсеребряный электрод состоит из серебряной проволо-
ки, покрытой слоем AgCl. Такое устройство выражается формулой
Ag/AgCl. Потенциал электрода зависит от активности ионов Ag+,
3'
67
но поскольку AgCl — трудпорастворимое соединение, то при избыт-
ПР
ке хлорид-ионов величину czAg+ можно заменить на aAg+ =-----г
аа~ '
где ПР — произведение растворимости соли, и тогда потенциал
электрода выразится формулой
р г 2,3RT .
-----7--- 1g «сГ-
Такой электрод может быть изготовлен на платиновой основе.
когда платиновая проволока покрыта слоем серебра, поверх кото-
рого нанесен слой хлористого серебра; выпуска-
ются также тонкослойные хлорсеребряные элект-
роды на стеклянной основе. Последний электрод,
предложенный В. А. Долидзе и Э. П. Сарухано-
вой, обладает высокой механической прочностью,
что позволяет использовать его при изучении почв
в полевых условиях. Тонкослойный хлорсеребряный
электрод представляет собой стеклянную трубку
(рис. 21) с копьевидным концом. Поверхность
трубки покрыта слоем серебра, на который нане-
сен AgCl. Хлорсеребряный электрод обладает вы-
сокой чувствительностью и может быть применен
для исследования различных объектов с концент-
рацией хлорид-ионов от 1(Н до 10~3 М. Реакция
Таблица 14
Стандартные растворы NaCl для градуировки
хлорсеребряного электрода (25°С)
NaCl, М асГ рС1
0,2000 0,735 0,1470 0,83
0,1000 0,778 0,0778 1,11
0,0500 0,822 0,0411 1,39
0,0100 0,903 0,0090 2,05
0,0050 0,928 0,0046 2,34
0,0010 0,963 0,0010 3,00
0,0005 0,975 0,0005 3,30
Рис. 21. Тонкослойный хлорсеребряный электрод- 1 — стек-
лянный корпус, 2 — слой Ag, [AgCl, 3 — серебряная про-
волока, 4 — колпачок
среды не влияет на потенциал электрода. Электрод можно приме-
нять при исследовании всех почв, за исключением почв с хлоридно-
68
сульфатным засолением, так как функция электрода нарушается
при избытке сульфатов, когда соотношение ионов Cl- :SO<^ дости-
гает 1 : 5.
При определении активности хлорид-ионов в качестве электро-
да сравнения используют насыщенный каломельный электрод и
электролитический ключ, приготовленный из агар-агара в насы-
щенном растворе KNO3. Перед определением активности хлорид-
ионов в почвах или растворах строят градуировочный график в
координатах э. д. с. — рС1. Стандартные растворы с известным
значением рС1 (табл. 14) готовят из перекристаллизованных солей
КС1 или NaCl квалификации «х. ч.» или фпксаналов.
Для построения градуировочного графика измеряют э. д. с.
цепи, составленной из хлорсеребряного электрода, опущенного в
стаканчик со стандартным раствором, куда погружают также конец
электролитического ключа; другой конец ключа опускают в ста-
канчик с насыщенным раствором КС1, в который помещен элект-
род сравнения. Выводы электродов подсоединяют к рН-метру,
который может использоваться как милливольтметр. Отсчет на
приборе снимают через 5 мин после того, как электроды были по-
гружены и закреплены в растворе. При смене растворов ополаски-
вают дистиллированной водой хлорсеребряпый электрод и конец
электролитического ключа, после осушения фильтровальной бума-
гой их споласкивают следующим раствором. При измерении ак-
тивности хлорид-ионов в водных суспензиях, вытяжках поступают
так же, как со стандартными растворами.
По калибровочному графику находят значение рС1 и рассчиты-
вают величину активности хлорид-иона.
Хлорсеребряпый электрод, кроме того, удобен для измерения
активности хлорид-иона в естественной почве с влажностью не
ниже 13—15%. В данном случае для полевых исследований удоб-
но пользоваться полевым потенциометром ППМ-ОЗМ.
При работе в поле хлорсеребряный электрод и электрод срав-
нения погружают в почву на расстояние 8—10 см друг от друга,
электролитический ключ в данном случае не нужен. Отсчет по при-
бору снимают через 10—15 мин. Величину рС1 находят по градуи-
ровочному графику зависимости э. д. с. — рО и потенцированием
рассчитывают активность хлорид-ионов.
Определение активности ионов кальция
В практике почвенных исследований все более широкое при-
менение находят мембранные электроды на основе поливинил-
хлоридных мембран или мембран марки М.К-40, обратимых к
ионам Са2+. Мембрана МК-40 приготовлена на капроновой основе
с активной группой — SO3Na. Сам электрод представляет собой
трубку (рис. 22), заполненную 0,1 М раствором СаС12 и гермети-
чески закрытую с одной стороны мембраной, насыщенной ионами
69
Сз2+. В раствор СаС12 внутри электрода погружают хлорсеребря-
ный электрод сравнения, через который осуществляется контакт
электрода с регистрирующим прибором; внешним электродом
сравнения служит каломельный электрод. Для насыщения мембра-
ны ионом Са2+ ее выдерживают 2—3 сут в 0,5 М растворе СаСЬ:
затем проверяют соответствие кальциевой функции электрода
у уравнению Нернста. С этой целью строят график за-
/ ВИСНМОС1Н э. д. с. цепи с кальциевым электродом от
( величины рСа. Для построения графика измеряют
J| э. д. с. цепи, составленной из кальциевого мембранного
электрода и электрода сравнения, опущенного в стан-
дартный раствор СаС12. Значение потенциала электро-
да, опущенного в раствор, устанавливается через 5—
10 мин. Высокая селективность электрода, изготовлен-
ного на основе мембраны МК-40, позволяет использо-
2 вать его для изучения различных почв, за исключением
почв магниевого засоления, солонцов и содовых солон-
чаков.
" Этот электрод может быть использован также для
Д 2 определения активности ионов кальция в почве с нена-
. - рушенной структурой при влажности почвы не ниже
. Д . j '5—20%. Для определения потенциала кальциевого
Ы электрода металлическим цилиндром с заостренным.
Рис. 22.
Кальцие-
вый мем-
бранный
электрод’
1 — труб-
ка, 2 —•
раствор
СаС12,
3 — мем-
брана,
4 — внут-
ренний
хлорсе-
ребряный
Как у бура, краем и с диаметром, равным диаметру
электрода, делают в почве отверстие до глубины, на ко-
торой собираются производить измерения. В это отвер-
стие устанавливают электрод так, чтобы он плотно со-
прикасался с почвой, затем почву вокруг электрода уп-
лотняют. На расстоянии 5—8 см от кальциевого элек-
трода в почву погружают электрод сравнения. Через
30 мин подсоединяют оба электрода к потенциометру
и измеряют величину э. д. с. цепи. По градуировочному
графику зависимости э. д. с.—рСа находят значение
рСа и рассчитывают активность иона кальция в почве
в данный момент времени.
электрод
Определение активности нитрат-ионов
По такому же принципу, как кальциевый электрод, устроен
мембранный электрод с нитратной функцией. Мембраной служит
полиамидная пленка, насыщенная нитратными ионами. В качестве
внутреннего раствора употребляют смешанный раствор, состоя-
щий из 0,1 М KNO3 и 0,005 М КС1. Внутренним электродом также
служит хлорсеребряный электрод, внешним электродом сравне-
ния — каломельный. Соответствие нитратной функции электрода
уравнению Нернста сохраняется в пределах 0,0005—0,1 М кон-
70
центрации N03 . Рабочий интервал электрода составляет] 0,5—3,2_]
pNO3. Электроды обладают достаточной селективностью П'ТТри-
сутствии десятикратного избытка анионов Н2РО4~, НСО?, COl~
и SQj-. Хлорид-ионы мешают определению, начиная с соотношения,
равного =1:4.
ЛИТЕРАТУРА
Агрохимические методы исследования почв. М., «Наука», 1975.
Ар и Пушкина Е. В Руководство по химическому анализу почв. М. Изд-во
Моск, уи-та, 1970.
Бейтс Р. Определение pH. Теория и практика. Л., «Химия», 1968.
Ион-селективные электроды. М„ «Мир», 1972.
Скорчелетти В. В. Теоретическая электрохимия. Л., «Химия», 1974.
Глава 2
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЙ
ПОТЕНЦИАЛ ПОЧВ
И МЕТОДЫ ЕГО ИЗМЕРЕНИЯ
Окислительно-восстановительный потенциал (ОВП) какой-
либо системы, в том числе и почвы, характеризует ее способность
вступать в окислительно-восстановительные реакции. Уровень ОВП
отражает преобладание процессов окисления или восстановления,
протекающих в почве, или преобладающее содержание в ней окис-
ленных или восстановленных (Ьорм соединений. Любая почва со-
держит одновременно окисленные и восстановленные формы сое-
динений, отношение которых и обусловливает экспериментально
обнаруживаемый ОВП. Величину ОВП часто обозначают Ек, и ее
можно выразить отношением активностей окисленной (Ох) и вос-
становленной (Red) форм соединений.
Если в системе имеет место равновесие
Ох 1 /ze^Red,
то потенциал такой системы равен
i--^m
zjF «Red
F E
где Ео — стандартный потенциал данной окислительно-восстанови-
тельной системы, определяемый условием аох:оЦе<1=1; п— число
электронов, принимающих участие в окислительно-восстановитель-
ной реакции. Остальные обозначения прежние.
При 20° уравнение можно переписать в следующем виде:
Еа
0,0581 j «ох
« «Red
В более общем случае под знаком логарифма ставится не
только отношение активностей окисленной и восстановленной
форм, но выражение для константы равновесия данной окисли-
тельно-восстановительной реакции.
72
Величина ОВП почв зависит главным образом от режима
влажности, аэрации и интенсивности деятельности микрофлоры.
В автоморфных почвах потенциалы колеблются в довольно узких
пределах, например для дерново-подзолистых целинных и пахотных
почв характерны Eh порядка 450—500 мВ. Максимальные значе-
ния ОВП часто бывают приурочены к горизонтам Bi и В2, тогда
как в горизонтах А] или Anax величины несколько понижены.
Уменьшаются они и в более глубоких, чем гор. Вь слоях почвы.
Как абсолютные значения потенциалов, так и их профильные
распределения зависят от погодных условий. Длительное пере-
увлажнение снижает ОВП, особенно в оглеенных разностях почв.
Кратковременное увлажнение за счет проходящих дождей сущест-
венно не влияет на потенциал, но в периоды гроз возможны
скачкообразные снижения потенциалов. В почвах, занимающих
пониженные элементы рельефа, ОВП обычно уменьшается. Анало-
гичные показатели характерны для серых лесных, черноземных и
каштановых почв.
Восстановительные условия с потенциалами, опускающимися
до 200 мВ и ниже, свойственны торфяно-болотным, перегнойно-
глеевым почвам, торфяникам. В пойменных почвах возможны рез-
кие перепаян ОВП- дерновые и луговые умеренно увлажненные
пойменные почвы имеют в верхних горизонтах ОВП порядка 450—
500 мВ. Если эти почвы связаны с близко расположенными грун-
товыми водами, то ОВП в них понижается до 400—450 мВ, а из-
быток органического вещества на периферии торфяников пони-
жает потенциалы до 150—300 мВ.
Резко снижаются потенциалы во многих .орошаемых почвах.
При обычных вегетационных поливах эти снижения сравнительно
невелики, а если поливные воды обогащены кислородом, то в поч-
вах, имеющих склонность к образованию почвенной корки, ОВП
после полива может даже повышаться. При длительном затопле-
нии почв, как это имеет место под культурой риса, величина ОВП
сначала уменьшается до 350—400 мВ, а затем может приобретать
даже отрицательные значения.
Регулирование окислительно-восстановительного потенциала —
одна из важнейших задач при поливном земледелии и мелиора-
ции переувлажненных почв Нечерноземья. Воздействовать на
окислительно-восстановительный режим почв можно с помощью
агротехнических приемов (вспашка, рыхление), воздействием на
водный режим (полйВ, дрёйаж), Известкованием, внесением опг~
~нйЧеекиХ. Удоорений '
Колебания величин ОВ'П в почвах тесно связаны с динамикой
различных окислительно-восстановительных систем. Верхний пре-
дел возможных величин Eh в почвах определяется растворимостью
кислорода в почвенном растворе;11йя<ний -- накоплением восста-
новленных, соединений, в том числе водорода, в результате дея-
тельности микроорганизмов. Многие присутствующие в почвах
окислительно-восстановительные пары слабо влияют на уровень
73
ОВП. Таковы Fe3+—Fe2+; Mn4+—Mn2+; CO2—CH4; Cu2+—Cu+;
NOT— NO3 , хотя при большом содержании водных окислов
железа в почве они могут играть роль своеобразного буфера.
Вместе с тем окислительно-восстановительный режим почв
оказывает решающее влияние на соотношение закисных и окисных
форм многих элементов в почве, что играет существенную роль
Таблица 15
Стандартные окислительно-восстановительные потенциалы
Элемент Реакция E„. В
F F2 + 2Н+ + 2е Л 2HF (водн.) 4-3,06
О О2 + 4Н+ + 4е 2Н2О 4-1,23
Н2О2 + 2Н' + 2е 2Н2О 4-1,77
Мп МпО~ + 4Н+ + Зе МпО2 + 2Н2О 4-1,69
С1 С12 + 2е^2С1~ 4-1,36
Сг Сг2О2~ 4- 14Н+ + бе 2Cri+ + 7Н2О 4-1,33
Мп MnO2 + 4Н+ + 2е Мп2+ + 2Н2О 4-1,23
I 2Ю7 + 12Н+ + 1 Ое X12 + 6Н2О 4-1,19
N HNO2 + Н+ 4- е NO + Н2О 4-1,00
Hg 2Hg2+4-2e^Hg2+ 4-0,92
Си Cu2+ +I--F е^. Cui 4-0,86
N NOT + 2H+ 4- e NO2 + H2O 4-0,775
Fe Fe3+ -f- e Fe2+ 4-0,77
Hg 2HgCl2 + 2e^?Hg2Cl2 + 2C1~ 4-0,62
Мп MnOT 4- 2H2O 4- 3e MnO2 4- 4OH~ 4-0,57
S SO2~ 4- 4H+ 4- 2e H2SO3 4- H2O 4-0,17
S 4- 2H+ 4- 2e H2S 4-0,14
Н 2H+4- 2e^H2 0,00
Fe Fe2+ 4-2eTi.Fe —0,44
Fe (OH)3 4- e Fe (OH)2 4- OH- —0,56
S SO2- 4- H2O 4- 2e SO|" -J- 2OH~ —0,94
Na Na+ 4- e Na —2,71
Са Ca2+4- 2e j±Ca —2,87
Ва Baa+ 2e T Ba —2,90
.74
в питании растений. И. П. Сердобольскпй предложил схему гра
нимных условий нормального питания высшйх растений марган
нем, железом и нитратами "в зависимости от величий pH-и Eh
На графике (рис. 23) прямо-
угольник ABCD охватывает ус-
ловия, обычные для большинст-
ва культурных почв. Прямая
ограничивает область нормально-
го снабжения растений марган-
цем. Если Е/, и pH таковы, что
условия питания попадают в об-
ласть, расположенную выше^ и
правее прямой RS, то растения
могут испытывать ^марганцевое
голодание. Аналогично прямая
KL ограничивает область нор-
мального обеспечения ^железом.
Линия MN, проведенная н# уров-
не 0,34” В, отделяет область уси-
ленной . деритрификании. „Таким
образом, нагйболее благоприятные
для питания марганце^ и желе,;
5л, мВ
Рис 23. Граничные условия нор-
мального питания растений (по Сер-
добольскому):
1 — область нормального снабжения
железом и марганцем, 2-—область
нормального снабжения железом,
-марганцем и нитратами
зом условия ограничиваются за-
штрихованной площадью, а с учетом денихрификационных про-
цессов — площадью, отмеченной двойной штриховкой.
в Рассматривая поведение различных окислительно-восстанови-
тельных пар в почвах, следует иметь в виду, что такие пары, как
Ее3-*-—Fe2+, Мп3+—Мп2+, МОГ—NOT и другие, сравнительно редко
представлены простыми ионами. В большинстве случаев они нахо-
дятся в виде комплексных соединений. Поэтому стандартные по-
тенциалы таких пар отличаются от табличных значений для
простых ионов. Некоторые стандартные потенциалы окислитель-
но-восстановительных систем приведены в табл. 15.
f Электроды для измерения ОВП
tv?
s Дйя измерения окислительно-восстановительных потенциалов
нужны электршгообпатимыс индикаторные электроды и электроды
сравнения. Электроды сравнения употребляют-—обычные: кало-
мельные, хлорсеребряные, иногда хингидронные (см. с. 13).
В качестве индикаторных обычно применяют хи мински инерт-
ные платиновые, платинированные или__едцциальные" стеклянные
электроды. Иногда употребляют графитовые электроды, но они
имекуг~_'мтгото недостатков. Используют также методы бумажной
автографии, которые дают четкое представление о расположении
восстановительных зон в почве, и методы простых цветных реак-
ций. Но все же основным методом остается потенциометрическое
измерение ОВП с помощью платиновых электродов.
75
Платиновые электроды для измерения ОВП изготавливают в
различных вариантах. Это могут быть проволочные -фЬктроды,
пластинчатое электроды с Гладкой поверхностью, или стеклянные
платинированные электроды. Их устройство ясно из рис. 24.
* Рис. 24.
Электрода для • изме-
рения окислительно-
восстановительного
потенциала.
А — игольчатый, Б —
* копьевидный, В —
платинированный:
1 — платиновая про-
волока, 2 —Платино-
вая пластина, 3 —
медная проволока,
4 — пробка из менде-
леевской замазки,
5 — ртуть, 6 — ялати-
новое покрытие, 7 —
4® ватная пробка;
Г — датаик для из-
мерений ОВП к по-
тенциометру ППМ-
03М1: 1 — платино-
вая пленка, 2 — кор-
пус, 3 — асбестовая
нить, 4 — внутренний
полуэлемент., 5, —
резиновые пробки,
7 — насыщенный рас-
твор КО, 8 — асбе-
стовая нить, 9 — от-
верстие для заливки
раствора, закрытое
пробкой, 10 — колпа-
чок, 11 — соедини-
тельный кабель
Проволочные, или игольчатые, платиновые электроды иволь-
зуют в тех случаях, когда предстоит измерение ОВП.в небоЖщрй
пообъему пробе Для работц^с густыми почвенными пастами и в
полевых условиях более удобны- копьевидные ддатиновые элект-
"реды, Которые легко погружать даже в сравнительно твердые и
слабоувлажненные почвы. Перед измерениями ОВП все платино-
вые электроды должны быть одинаково обработаны, включая
время прокаливания и вид пламени.
Платиновые электроды требуют большого расхода дорогостоя-
щего металла. В целях экономии можно воспользоваться платини-
рованными электродами. Они выпускаются промышленностью (см.
pi?c. 24) и могут быть изготовлены в любой лаборатории. По ме-
тоду В. А. Рабинович и О. В. Дуровской для изготовления электро-
76
дов подбирают ^стеклянные трубки, длина которых, диаметр и
толшина стенок определяются задачами исследования. Практи-
чески удобны толстостенные стеклянные трубки с внешним диа-
метром 4—10 мм.и толщиной стенок 1—2 мм.
Для платинирования трубок готовят специальную смесь. С этой
целью канифоль растворяют в скипидаре при отношении 1:1,
нагревая эту смесь на песчаной бане. Канифоль в скипидар вносят
небольшими порциями при постоянном помешивании. После ох'7
лаждения полученную маслообразную коричневую жидкость сме-
шивают с равным количеством этилового спирта. Затем 1 г хлор-
* цой платины растворяют в 12 мл~с11йрта, добавляют равный объем
л насыщенного спиртового раствора борной кислоты и полученный
^раствор смешивают с 25 мл ранее приготовленного раствора кани-
фоли. Эту смесц используют для платинирования.
Берут очищенную етеклянную трубку, погружают ее конец па
2—3 см в смесь для платинирования, дают жидкости несколько
- стечь и вводят этот конец в коптящее пламя газовой горелки.
Трубку держат в наклонном положении платинируемым концом
вниз, а нагревание начинают несколько выше границы жидкости,
постепенно вводя в пламя всю платинируемую поверхность. При
обжиге трубку вращают, не давая жвдкости скапливаться на ниж-
нем срезе. После того как растворитель испарится и на внешней
поверхности трубки появится блестящий слой восстановленной
платины, продолжают обжиг, но уже в окислительном пламени
горелки до тех пор, пока внутренняя поверхность трубки не по-
кроется ровным серым налетом. Трубке дают остыть, снова погру-
жают в смесь для платинирования и повторяют обрабсГгку до тех
пор, пока наружный конец трубки не будет покрыт ровным слоем
металлической платины. Платинированная поверхность должна
быть ровной и блестящей.
Отверстие платинированного конца заливают менделеевской
замазкой или битумом, для чего подогретый плаФинированный
конец погружают в расплавленную, замазку на 5—7 мм и быстро
вынимают. По охлаждении образуется прочная пробка. Внутрь
электрода наливают немного ртути и в нее погружают медную
* проволоку для соединения с потенциометром.
Проверку дщдпкаторных электродов проводят по буферным
растворам с точно известными значениями Х5КП? _ Такой смесью
моЖет служить смесь красной п желтой_ к^овяных__ солей/—
Кз[Ре(СЫ)6] мол. масса 329,26 и 1\4[Рс(СГ\т)б]-ЗН2б, мол. масса
422,41. Для приготовления раствора берут 3,8018 г соли
K4[Fe(CN)6]-ЗН2О и 13,5001 г Кз[Ре(СМ)6], растворяют'в дистил-
лированной воде и доводят объем раствора до 1 л. В этом раство-
ре потенциал платинового электрода, измеренный относительно
насыщенного хлорсеребряного электрода при температуре 25°С,
• равен 272 +3 мВ.
МожНо для-ироверки воспользоваться и буферным раствором
с точно известным значением pH, насыщенным по отношению к
77
хингидрону.) Платиновая проволока, помещенная в такой раствор,
обрадует хингидронный электрод. Потенциал хингидронного
электрода вычисляют по формуле
Ехг = Ео — й pH или при 20° С '
ЕКГ = ЕО~ 0,0581 pH,
Тде Ео — потенциал стандартного хингидронного электрода, рав-
ный
Ео = 0,6990 — 7,4-10-4 (f° —25).
Лабораторные измерения ОВП,почв
Отобранные в поле образцы для -измерения ОВП необходимо
предохранить от потери влаги и от контакта с атмосферным__воз-
'духом. с этой целью отобранный из разреза образец почвы упа-
ковывают в мешочек из полиэтилена, стараясь как можно больше
сократить время контакта образца с атмосферным воздухом.
" необхо-
сократить время контакта образца с атмосферным
В лаборатории освобождаю^ часть поверхности образца,
к потенциометру
Рис 25. Измерение ОВП почвы.
д___измерение без электролитического ключа в образце почвы с ненарушенным
строением, Б — измерение в почвенной пасте с использованием электролитиче-
ского ключа 1 — индикаторный электрод, 2 — электрод сравнения, 3 — элек-
тролитический ключ, 4 — раствор КС1, 5 почва или почвенная паста, 6 по-
лиэтиленовый мешочек
78
димую для погружения электродов, срезают сверху слой почвы
1—2 см, а затем в образец погружают платиновый электрод и
электрод сравнения |нли электролитический ключ), как это пока-
зано на рис. 25. Платиновый электрод погружают так, чтобы почва
плотно прилегала к поверхности электрода и изолировала его от
воздуха. При погружении в почву электрода сравнения или
электролитического ключа необходимо следить, чтобы погружае-
мый отросток не был забит почвой и чтобы из него не вытекал
раствор КС1.
Измерения ОВП возможны и в почвенной пасте, т. е. в почве
с нарушенным строением и увлажнённой—да—заданного уровня.
Такие измерения обычно проводят при изучении динамики ОВП
~и~влияния на величину ОВП реакции среды, известкования, вне-
сения органического вещества и в других опытах. В этом случае
эксперимент ставится аналогично вышеописанному, но почву на-
сыпают в стаканчик и смачивают дистиллированной водой до же-
лаемого уровня (см. рис. 25).
Погруженные в почву электроды подсоединяют к потенцио-
метру с высоким входным сопротивлением и измеряют э. д. с. цепи.
Измерение э. д. с. на потенциометре pH-340 (см. с. 25) произво-
дят следующим образом: переключатель «Размах» ставят в поло-
жение «1500 мВ» и, подобрав переключателем «Род работы» знак
потенциала « + мВ» или «—мВ», определяют приблизительное
значение э. д. с. цепи. Более точное значение получают, установив
переключатель «Пределы измерений» в соответствующее положе-
ние и переключатель «Размах» в положение «300 мВ». Отсчет
производят по верхней шкале прибора с точностью до ±5 мВ.
Можно добиться более высокой точности измерений до 0,1 мВ,
подключив для этого к pH-метру дополнительный потенциометр
типа ППТВ-1 или Р-307; схема подключения описана в инструкции
к прибору pH-340.
' Найденное значение э.д.с. позволяет вычислить потенциал
индикаторного электрода, который принимается равным ОВП
почвы. Для вычисления ОВП надо знать полярность электродов,
которую определяют по положению переключателя на потенцио-
метре или по обозначениям у входных клемм. Если платиновый
электрод при измерении э.д. с. был соединен с клеммой « + », то
его потенциал был выше, чем потенциал электрода сравнения.
В этом случае
э. д. с. £л —£ср,
где Eh — потенциал платинового электрода и £tp — потенциал
электрода сравнения. Тогда
£^ = э. д. с. £ср.
Если индикаторный электрод имел знак «—», тогда
э. д. с. -£ср —£ft ф
79
E-h — -^cp A- c.
Кроме величины Eh часто вычисляют величину rH2:
Г"’ = “1“ Г2РН' } " 'i /
где Eh выражен в милливольтах. /
— ВеличиначгН2, по мнению ряда исследователей, выражает сум-
марное влияние окислительно-восстановительного потенциала и
реакции среды на почвенные и биологические процессы.
Полевые измерения ОВП почв
Более правильные представления об окислительно-восстанови-
тельном режиме почв дают прямые измерения ОВП непосредст-
венно в полевой обстановке. Обычно полевые измерения проводят
в двух вариантах: разовые измерения и наблюдения за динамикой.
Разовые измерения ОВП позволяют получить сравнительную ха-
рактеристику различных почв и горизонтов. В стационарных усло-
виях, кроме того, ведут наблюдения за суточной и сезонной дина-
микой ОВП.. При разовых измерениях пользуются переносными
электродами. В намеченной для измерений точке очищают поверх-
ность почвы от живых растений и растительных остатков и в поч-
ву погружают платиновый электрод и электрод сравнения. Через
7—10 мин измеряют э. д. с. с помощью полевого потенциометра.
Чтобы измерить ОВП по генетическим горизонтам, зачищают
переднюю стенку разреза и в середину генетического горизонта
погружают копьевидный платиновый электрод. На расстоянии
8—10 см погружают в почву электрод сравнения (для этого в
стенке разреза удобно сделать небольшую горизонтальную пло-
щадку). Затем измеряют э.д.с., как описано выше.
Для изучения динамики ОВД--в почву устанавливают стацио-
нарные платиновые пли платинированные электроды. Предвари-
телБно тонким буром (по диаметру электрода) подготавливают в
почве отверстие на глубину предполагаемой установки электрода.
Затем в это отверстие вставляют электрод, погружая платиновую
(или платинированную) часть его в ненарушенный слой почвы.
Почву около электрода слегка уплотняют (обжимают), чтобы изо-
лировать электрод от атмосферного воздуха. Электрод сравнения
погружают в почву на расстоянии 10—15 см; обычно используется
переносной электрод сравнения. Вывод от индикаторного электро-
да должен быть надежно изолирован от почвенной и атмосферной
влаги. Место расположения электрода отмечается хорошо замет-
ным репером. Измерения э. д. с. проводят, как описано выше.
ЛИТЕРАТУРА
Оксредметрия. Под ред. Б. П. Никольского и В. В. Пальчевского. Л., «Химия»,
1975.
80
Глава 3
Полярографические методы
в почвоведении
Полярографический метод был предложен в 1922 г. чешским
ученым Я- Гейровским, за разработку метода ему в 1959 г. была
присуждена Нобелевская премия.
Полярографическим методом принципиально возможно качест-
венно и количественно определять любые соединения, способные
электроокисляться или электровосстанавливаться; косвенными
приемами исследуют также электрохимически неактивные вещест-
ва. С помощью полярографии изучаются вопросы кинетики реак-
ции, строения вещества, адсорбция, комплексообразование и др.
При решении столь разнообразных аналитических и теоретических
задач может быть использована одна и та же аппаратура и близ-
кие технические приемы. В количественном анализе по одной
полярограмме удается определять содержание нескольких компо-
нентов при их совместном присутствии в растворе, а также кон-
центрацию одного или нескольких компонентов на фоне большого
числа сопутствующих веществ.
Полярографическая кривая
Полярографический метод основан на прямой зависимости
между величиной предельного диффузионного тока, возникающего
в результате электровосстановления или окисления исследуемого
вещества, и концентрацией вещества в растворе.
При полярографировании в раствор погружают два электрода,
на которые подается от внешнего источника тока постепенно воз-
растающее напряжение. Ток, возникающий при электролизе, фик-
сируется чувствительным гальванометром (рис. 26). Таким путем
получают кривую зависимости величины тока от приложенного
напряжения (рис. 27). Эту кривую называют полярограммой. Ее
используют при качественном и количественном определении ком-
понентов раствора. На полярограмме различают:
81
остаточный ток iOCT, соответствующий участку О А (см. рис. 27),
величина его мала и очень медленно возрастает с увеличением от-
рицательного потенциала;
потенциал восстановления Ев , при котором начинается элект-
рохимическая реакция, приводящая к резкому возрастанию тока;
АКК
Рис 26. Принципиальная схема
полярографа:
Р — резервуар со ртутью, РКЭ —
ртутный капельный электрод,
ЭС — электрод сравнения, АВ —
реохорд, Г — гальванометр, Ш —
шунт гальванометра, Акк — акку-
мулятор
предельный ток тПр, величина ко-
торого зависит от концентрации
электрохимически активного ве-
щества, но практически не ме-
няется при изменении потенци-
ала (участок ВС на рис. 27);
полярографическую волну —
участок кривой от начала увели-
чения тока (потенциала восста-
новления) до его предельного
значения.
Электрохимически активное
вещество качественно определяют
по положению середины поляро-
Рис 27 Полярографическая
кривая
графической волны на шкале по-
тенциалов, так называемому по-
тенциалу полуволны £1/?.
О количественном содержании
определяемого компонента судят
по предельному току, величина которого связана с концентра-
цией С согласно уравнению Ильковича (см. с. 84) и в простей-
шем случае прямо пропорциональна концентрации: ЕЛ.= КС.
Ртутный капельный электрод
и неполяризующиеся электроды сравнения
При полярографировании напряжение от внешнего источника
тока подается на два электрода, погруженные в анализируемый
раствор. Один из электродов — поляризуемый; на нем протекает
82
электрохимическая реакция окисления или восстановления. Вто-
рой электрод — нуполяриз} ющийся, его потенциал в процессе
электролиза остается практически постоянным.
В качестве поляризуемого в полярографии используют ртутный
электрод. Наиболее широкое применение находит ртутный капаю-
щий электрод. Он представляет собой стеклянный капилляр, из
которого капли ртути вытекают с интервалом около трех секунд.
Капающий электрод обладает рядом преимуществ. Особенно
важно, что при работе с ртутным капающим электродом электро-
химические процессы осуществляются на постоянно обновляющей-
ся чистой поверхности ртутной капли, состояние которой практи-
чески не зависит от предшествующих реакций. Постоянное обнов-
ление поверхности ртути позволяет избежать накопления продук-
тов реакции па поверхности электрода, стандартизовать условия
эксперимента и получить воспроизводимые результаты.
Ртутный капельный электрод идеально поляризуется. Это озна-
чает, что когда в растворе отсутствуют электрохимически актив-
ные в данной области потенциалов вещества, электрические за-
ряды, поступающие от внешнего источника тока, идут только на
изменение потенциала электрода. Поляризация, при которой
электроду сообщается положительный заряд, называется анодной;
придание электроду отрицательного заряда соответствует катодной
поляризации. Уровень анодной поляризации ртутной капли огра-
ничен окислением ртути, которое начинается при потенциале около
+ 0,55 В относительно насыщенного каломельного электрода
(нас. к. э.). Катодная поляризация ртутного электрода ограниче-
на восстановлением присутствующих в растворе катионов.
В качестве второго электрода может быть использован любой
неполяризующнйся электрод, потенциал которого очень мало из-
меняется при прохождении тока. Нередко с этой целью используют
большую поверхность ртути, налитой на дно сосуда (донную
ртуть). Площадь поверхности слоя ртути в несколько сот раз
больше площади капельного электрода, и потенциал донной ртути
остается практически постоянным в связи с очень малой плот-
ностью тока на ее поверхности. В аналитической практике часто
используют неполяризующиеся электроды второго рода (ртутно-
сульфатные, хлорсеребряные и каломельные). Наибольшее рас-
пространение в качестве неполяризующихся электродов сравнения
получили каломельные электроды (см. с. 14).
Потенциал ртутного капельного электрода можно считать рав-
ным разности между приложенным от внешнего источника напря-
жением и потенциалом неполяризующегося электрода сравнения.
Потенциал электрода сравнения обычно условно принимают рав-
ным нулю, и тогда потенциал капельного ртутного электрода
считают равным по абсолютной величине приложенному извне
напряжению.
83
Диффузионный ток
и уравнение Ильковича
Электродный процесс включает ряд стадий: подачу электрохи-
мически активного вещества к электроду, собственно электрохими-
ческую реакцию, сопровождающуюся переносом электронов, и
отвод продуктов реакции от поверхности электрода.
Если потенциал ртутного капельного электрода недостаточен
для осуществления электрохимической реакции, через полярогра-
фическую ячейку протекает очень небольшой ток заряжения, или
емкостный ток. Когда будет достигнут потенциал восстановления
или окисления одного из компонентов раствора, на электроде на-
чинается электрохимическая реакция, и величина тока резко воз-
растает. В ходе реакции раствор у электродной поверхности
обедняется электрохимически активным веществом; это создает
разность концентраций между приэлектродным слоем и основной
массой раствора. Разность концентраций вызывает диффузию —
направленное движение вещества из области с большей кон-
центрацией в область с меньшей его концентрацией, к электроду.
Величина тока, возникающего в результате электрохимической
реакции, ограничена скоростью поступления вещества к элект-
роду.
Поступление вещества к электроду может происходить как
путем диффузии, так и вследствие электростатического притяже-
ния ионов электродом. Ток, обусловленный электростатическим
притяжением, называют миграционным; в аналитической практике
миграционные токи устраняют введением избытка индифферентно-
го электролита.
Ток, обусловленный диффузией вещества к электроду, назы-
вают диффузионным током. Согласно первому закону Фика ско-
рость диффузии прямо пропорциональна разности концентраций
диффундирующего вещества и зависит от его природы.
Если диффузия происходит в одном направлении к электроду
с плоской поверхностью, то величина диффузионного тока i опре-
деляется уравнением
i — nFT ^ S(c — c3)
Уnt
где S — площадь поверхности электрода, t — время электролиза,
D — коэффициент диффузии, численно равный количеству молей
вещества, которое диффундирует через единицу площади в едини-
цу времени при градиенте концентрации, равном единице. Коэф-
фициент диффузии измеряется в см2/с и для ионов в водных
растворах имеет порядок 10-5—10~® см2/с.
При увеличении потенциала электрода скорость электрохими-
ческой реакции увеличивается, и поэтому концентрация вещества
в приэлектродном слое Са меняется в процессе электролиза, на-
84
чпная от исходного значения С практически до нуля. Когда Сэ
становится равной нулю, скорость диффузии, а следовательно, и
величина диффузионного тока оказываются прямо пропорциональ-
ными общей концентрации вещества в растворе С и достигают
предельного значения, т. е. не изменяются при дальнейшем уве-
личении потенциала. Этот ток в полярографии называют предель-
ным диффузионным током. Приведенное выше уравнение характе-
ризует величину диффузионного тока в любой момент времени
электролиза. Для предельного диффузионного тока справедливо
выражение
. _ nF-/~D S-C
1пР .--
у at
Величина диффузионного тока при постоянной величине прило-
женного напряжения непрерывно уменьшается обратно пропорцио-
нально корню квадратному из времени.
Ртутная капля имеет форму, близкую к форме шара. При
диффузии к электроду, имеющему форму шара, величина тока с
некоторыми допущениями определяется теми же параметрами, что
и при диффузии к плоской поверхности. Подставив в уравнение
величину поверхности ртутной капли 5=4лг2, получим
nFD^'2 4лг2С
1пр 7= •
у nt
Это уравнение определяет величину предельного тока в любой
момент существования ртутной капли. Величина t означает здесь
время, прошедшее от начала роста капли.
За время существования капли ее радиус непрерывно изме-
няется. Переменную величину г удобно заменить на постоянную —
скорость вытекания ртути из капилляра т. Исходя из того, что
/и=-у-, массу капли Q в любой момент времени можно выра-
зить произведением массы ртути т, вытекающей из капилляра за
1 с, на время t, прошедшее с начала образования капли:
Q = mt.
С другой стороны, массу капли можно выразить через ее объем
и плотность ртути:
О = — л г3 d — mt.
Это позволяет узнать радиус капли в любой момент времени:
85
Подставляя значение г в уравнение, получим
nFD^ 4л
^пр
Зт/
4л d
2/3
С
V nt
= 4 l/\rt ( —-— )2/3 пFD'''~ m2ri ti/& С.
\ 4л d )
В последнем уравнении не учтено, что, увеличиваясь в разме-
ре, капля движется навстречу фронту диффузии вещества. Это
движение приводит к увеличению тока в 1,53 раза. Принимая во
внимание указанную поправку и объединяя все числовые коэффи-
циенты, входящие в уравнение, получим
inp = 0,732 nFD1/2 т2/3 tl/6C.
В этом уравнении величина тока выражена в амперах, кон-
центрация — в моль/см3, коэффициент диффузии — в см2/с, масса
ртути, вытекающая из капилляра в единицу времени, — г/с, вре-
мя t в любой момент от начала существования капли — в секун-
дах, число Фарадея — в кулонах. Уравнение применимо для
7п
Рис. 28. Изменение тока на каждой капле ртути при
восстановлении никеля и цинка (А) и полярограмма,
построенная по максимальным токам (Б)
вычисления величины предельного диффузионного тока в любой
момент времени t от начала существования капли.
Из уравнения следует, что изменение величины тока в период
существования одной капли выражается параболой шестого по-
рядка. По мере роста капли величина тока увеличивается сначала
быстро, а затем все медленнее, при отрыве капель от капилляра
ток падает почти до нуля (рис. 28). Такое изменение тока повто-
ряется на каждой капле, и зависимость ток — потенциал
имеет вид не плавной, а осциллирующей кривой, где каждая
осцилляция соответствует изменению величины тока па одной кап-
ле ртути.
Если в уравнение подставить величину т, равную времени су-
ществования одной капли, или периоду капания ртути, получим
86
выражение для максимального предельного диффузионного тока
в период жизни капли:
1макс = 0,732hFD1/2/н2/3т1/6С.
Записать полностью кривую зависимости тока от времени в период
жизни одной капли можно с помощью осциллографа. В обычной
лабораторной практике пользуются самописцами, которые регист-
рируют лишь небольшие колебания (осцилляции) около среднего
значения тока. Расчет показывает, что средний ток связан с мак-
симальным значением тока соотношением
i = 'макс*
Учитывая это, можно записать:
7ПР = 0,627 п FDl/2 ш2/3 т1/6 С.
Это уравнение для среднего предельного диффузионного тока по-
лучило название уравнения Ильковича и является частным слу-
чаем уравнения для любых мгновенных диффузионных токов:
i = 0,627 riFD'1'2 т2/3 т1/6 (С — Сэ).
Если число Фарадея включить в числовую константу, концентра-
цию выразить в миллимолях на литр, то величина тока будет
представлена в микроамперах, а уравнения для максимального и
среднего значений предельного диффузионного тока примут вид:
г'макс = 706;г£)'/2т2/3т1/6С,
ln^ 605nDl/2m2/3rl/6C.
Уравнение Ильковича справедливо для ионов, независимо от их
заряда, и для нейтральных молекул, диффундирующих к капель-
ному электроду.
Из уравнения Ильковича следует, что при работе с одним и
тем же капилляром и при постоянной высоте столба ртути, т. е.
при постоянных т и т, диффузионный предельный ток пропорцио-
нален концентрации электрохимически активного вещества в
растворе:
'пр ~~ КС,
где К = 605нГ1/2т2/3т1/6.
Это уравнение лежит в основе количественного полярографи-
ческого анализа.
Факторы, влияющие на диффузионный ток. Диффузионный ток
пропорционален произведениюDi/2 т2/3 т1/6, поэтому все факторы,
которые влияют на период капания, скорость вытекания ртути из
капилляр.а и коэффициент диффузии, могут вызвать изменение ве-
личины тока и высоты полярографической волны.
87
Скорость вытекания ртути и период капания зависят от ради-
уса капилляра, его длины и высоты столба ртути, под действием
которой ртуть вытекает из капилляра, так называемой эффектив-
ной высоты ртутного столба Н.
Эффективная высота, под действием которой происходит выте-
кание ртути из капилляра, несколько меньше измеряемой. Это
обусловлено тем, что при образовании капель ртути поверхностное
натяжение <т на границе раздела ртуть — раствор препятствует
увеличению поверхности капли и возникают силы, направленные
в глубь растущей капли перпендикулярно к ее поверхности.
Скорость вытекания ртути из капилляра прямо пропорцио-
нальна, а период капания электрода обратно пропорционален
эффективной высоте ртутного столба:
ьп
т = klH, т =----,
Н
где k1 и k11 — коэффициенты пропорциональности, зависящие от
геометрических параметров капилляра.
Подставив выражения для т и т в уравнение Ильковича, по-
лучим, что величина тока прямо пропорциональна корню квадрат-
ному из высоты столба ртути:
tnp-605«D'/2 Ст2/3т,/6 =
= 605п£)1/2С(^//)2/3 = А1/7Г
Линейная зависимость между величиной диффузионного тока
и корнем квадратным из высоты ртутного столба дает возмож-
ность выявить природу предельного тока. При диффузионном
ограничении предельного тока зависимость между величиной тока
и высотой ртутного столба должна быть линейной и удовлетворять
приведенному уравнению.
Из этого уравнения следует также важный для аналитической
практики вывод: в количественном полярографическом анализе
при работе с одним и тем же капилляром необходимо сохранять
постоянной высоту ртутного столба.
Потенциал электрода практически не влияет па скорость выте-
кания ртути из капилляра, но период капания ртути зависит от
потенциала; графическое изображение этой зависимости соответст-
вует электрокапиллярной кривой (рис. 29). Электрокапиллярная
кривая выражает связь между потенциалом ртутного капельного
электрода и поверхностным натяжением ртути на границе раздела
ртуть — раствор.
В растворах электролитов не содержащих поверхностно-актив-
ных веществ (ПАВ), электрокапиллярная кривая имеет максимум
около — 0,56 В относительно насыщенного к. э. При потенциале
88
электрокапиллярного максимума, который соответствует потен-
циалу свсбоднокапающей ртути при разомкнутой электрической
цепи, заряд электрода в отсутствие специфической адсорбции
равен нулю. Потенциал электрокапиллярного максимума назы-
вают потенциалом нулевого заряда. При менее отрицательном по-
тенциале поверхность ртутного электрода имеет положительный
заряд, при более отрицательном — отрицательный.
Если в растворе присутствуют ПАВ, которые адсорбируются
на ртутной капле, то поверхностное натяжение ртути уменьшается
и тем сильнее, чем больше адсорбция вещества. Адсорбция ПАВ
зависит от потенциала электрода. Элек-
трокапиллярные кривые, выражающие
зависимость потенциал — поверхностное
натяжение или потенциал — период ка-
пания ртути для растворов, содержа-
щих ПАВ, позволяют определить об-
ласть потенциалов их адсорбции. Этот
прием использовался для изучения ад-
сорбционных свойств фульвокислот.
Температурный коэффициент диффу-
зионного тока равен 1,63 %/град, это зна-
чит, что при повышении температуры на
1° величина диффузионного тока возрас-
тает на 1,63%.
Природа растворителя в значительной
степени изменяет вязкость раствора, КО-
Рнс 29 Электрокапилляр-
ная кривая
торая влияет на период капания ртути и, что особенно важно,
изменяет коэффициент диффузии.
Все рассмотренные выше закономерности справедливы только
по отношению к так называемому нормальному диффузионному
току. Этот ток наблюдается в присутствии большого избытка ин-
дифферентного электролита, устраняющего миграционный ток, и
при условии, что поверхность растущей капли движется только в
радиальном направлении, а раствор искусственно не перемеши-
вается. Нормальный диффузионный ток может быть получен при
малых давлениях ртутного столба, когда скорость течения ртути
в капилляре не превышает 1,5—2 мг/с. При большей скорости
истечения ртути возникает увеличенный диффузионный ток.
Двойной электрический слой
и емкостный ток.
Остаточный ток.
На границе раздела электрод — раствор возникает двойной
электрический слой, который можно рассматривать как своеобраз-
ный конденсатор. Одна обкладка этого конденсатора — заряжен-
ная поверхность электрода, другая — ионы противоположного
знака, которые находятся вблизи поверхности электрода благодаря
89
действию электростатических сил. С изменением площади поверх-
ности ртутной капли меняется и емкость двойного слоя, поэтому
ртутный капающий электрод можно рассматривать как конденса-
тор с изменяющейся поверхностью. Если к такому конденсатору
подвести внешнее напряжение, то возникает емкостный ток. Поэто-
му даже в отсутствие электрохимически активных веществ через
полярографическую ячейку с ртутным капельным электродом про-
ходит ток. Направление емкостного тока зависит от потенциала
электрода; при потенциале нулевого заряда емкостный ток равен
нулю. Емкостный ток, в отличие от диффузионного тока, растет
при уменьшении периода капания ртути. Это обусловливает и
иную, чем для диффузионного тока, зависимость емкостного тока
от высоты ртутного столба; емкостный ток пропорционален высо-
те ртутного столба:
ic = КН.
Емкостный ток ограничивает чувствительность полярографиче-
ских методов, так как искажает полярографические волны особен-
но при определении малых концентраций. Величина емкостного
тока имеет тот же порядок, что и диффузионный ток, который
возникает при полярографировании растворов с концентрацией
электрохимически активного вещества 10-4—10-5 М. Поэтому
пределом обнаружения веществ классическими полярографически-
ми методами является концентрация 10~5 М.
Остаточный ток включает кроме емкостного (ic) фарадеевский
ток (г'ф), возникающий за счет электрохимического восстановления
примесей:
^ост 1 Ц)‘
Миграционный ток
и индифферентный электролит
В полярографии используется предельный диффузионный ток,
величина которого контролируется скоростью диффузии вещества
к электроду, пропорциональна концентрации электрохимически
активного вещества и практически не изменяется с ростом потен-
циала.
Когда в электрохимической реакции участвуют имеющие заряд
ионы, их движение к электроду может происходить не только
путем диффузии, но и за счет миграции под действием электриче-
ского поля. Катионы движутся к катоду, анионы — к аноду. Если
направление миграции иона, участвующего в электрохимической
реакции, совпадает с направлением его диффузии, величина тока
увеличивается по сравнению с предельным диффузионным током.
Если направления миграции и диффузии не совпадают, то пре-
дельный ток уменьшается. Так как величина тока обусловлена
90
общим количеством разряжающихся на электроде ионов, то вы-
деляют диффузионную и миграционную составляющие предель-
ного тока.
При восстановлении катионов направление миграции совпадает
с направлением диффузии, и предельный ток равен алгебраиче-
ской сумме миграционного и диффузионного токов:
1пр " 1д 1ыИГр-
При восстановлении анионов направление миграции противо-
положно направлению диффузии. За счет диффузии анионы
движутся к катоду, а под действием электрического поля отрица-
тельно заряженные анионы будут двигаться к положительному
электроду — аноду. В этом сл> чае
Др ~ 1д 1ыигр*
Если в электрохимической реакции участвуют электронейтральные
молекулы, то
Др ~ 1д-
Закономерности, выведенные для предельных диффузионных
токов, нельзя распространять на миграционные токи; последние
растут с увеличением приложенного напряжения. Поэтому в поля-
рографии миграционные токи сводят к нулю, добавляя в раствор
индифферентный электролит, или фон, концентрация которого в
50—100 раз выше концентрации электрохимически активного ве-
щества.
В качестве индифферентных (фоновых) электролитов исполь-
зуются вещества, которые восстанавливаются в более , отрица-
тельной области потенциалов, чем определяемые ионы.
Полярографические максимумы
Довольно часто полярографическая кривая искажается макси-
мумом, появление которого обусловлено увеличением и последую-
щим уменьшением величины тока (рис. 30). Увеличение тока и
появление максимума связаны с тангенциальным (перпендикуляр-
ным радиусу) движением поверхности ртутной капли. При этом
в движение вовлекаются прилежащие к капле слои раствора, что
приводит к дополнительной подаче электрохимически активного
вещества к электроду. В результате наблюдается увеличение тока
по сравнению со значениями соответствующих диффузионных
токов. В зависимости от причин, вызывающих движение поверх-
ности ртутной капли, различают максимумы первого и второго
родов.
Максимумы первого рода. Максимумы первого рода обычно
появляются в растворах с низкой концентрацией индифферентного
электролита и имеют форму пика (см. рис. 30). Величина тока
91
в максимуме зависит от концентрации вещества, участвующего в
электрохимической реакции.
Причина возникновения максимумов первого рода — неравно-
мерное распределение зарядов на поверхности ртутной капли. Не-
равномерное распределение зарядов связано с различным расстоя-
нием отдельных участков поверхности капли от электрода срав-
нения и с экранированием части по-
V верхности капли капилляром. В ре-
зультате возникает неодинаковое по-
верхностное натяжение вдоль поверх-
ности капли и, как следствие, дви-
жение поверхностного слоя ртути от
мест с малым поверхностным натя-
жением к местам с более высоким
поверхностным натяжением. Движе-
ние поверхности ртутной капли сопро-
вождается перемешиванием раствора
и дополнительной подачей электрохи-
мически активного вещества к элек-
троду. В результате ток увеличивает-
J . ся. Таким образом, максимумы пер-
( вого рода не связаны с вытеканием
J ртути из капилляра, а обусловлены
/ изменением поверхностного натяже-
.' ния вдоль капли. Поэтому они появ-
ляются как при работе с ртутным ка-
Рис. 30. Полярографический пающим, так и с неподвижным, или
максимум^на^волне^восстанов- стационарным, ртутным электродом.
В зависимости от потенциала, при ко-
тором появляется максимум, разли-
чают положительные и отрицательные максимумы первого рода.
Максимумы, появляющиеся при потенциалах восходящей вет-
ви электрокапиллярной кривой (см. рис. 29), положительные,
при потенциалах нисходящей ветви электрокапиллярной кривой—
отрицательные. Вблизи потенциала нулевого заряда максимумы
первого рода не появляются.
Появление максимумов создает дополнительные трудности для
количественного полярографического анализа, поэтому их, как
правило, устраняют добавлением ПАВ, адсорбирующихся на по-
верхности ртутной капли. Вследствие неоднородной поляризации
ртутной капли адсорбция ПАВ на ее поверхности происходит
также неравномерно, что в конечном итоге приводит к выравнива-
нию поверхностного натяжения на ртутной капле, и движение
ртути и раствора вблизи капли прекращается. ПАВ подавляют
максимумы в той области потенциалов, в которой они адсорби-
руются на ртути. Органические анионы адсорбируются на поло-
жительно заряженной поверхности ртути, катионы — на отрица-
тельно заряженной поверхности, а молекулярные вещества — воб-
92
ласти потенциалов нулевого заряда. Вещества амфотерного ха-
рактера, способные образовывать как катионы, так и анионы,
могут адсорбироваться при всех потенциалах. Это желатин, метил-
целлюлоза, агар-агар и др. Высокие концентрации ПАВ могут
привести к снижению предельного тока. Поэтому при работе с
ними находят такую минимальную их концентрацию, которая,
устраняя максимум, не влияет на высоту волны. Оптимальная
концентрация подбирается эмпирически, при выполнении анализов
или экспериментов одной серии ее следует сохранять постоянной.
Максимумы второго рода. Максимумы второго рода возникают
при перемешивании раствора в результате вытекания ртути из
капилляра. Эти максимумы появляются, если линейная скорость
течения ртути в капилляре превышает 2 мг/с, они специфичны для
капающих электродов. При определенных скоростях в образую-
щейся капле возникают вихревые движения, приводящие к раз-
мешиванию раствора и увеличению тока. Наибольшая скорость
движения наблюдается при потенциале нулевого заряда. Макси-
мумы второго рода появляются в растворах с высокой концентра-
цией индифферентного электролита и имеют пологий вид. Они
так же, как и максимумы первого рода, могут быть подавлены
добавлением ПАВ.
Способность подавлять максимумы используется для коли-
чественного определения ПАВ. О содержании таких веществ судят
в этом случае по снижению высоты максимума. В некоторых слу-
чаях по степени подавления максимума можно оценивать молеку-
лярную массу веществ: растворы веществ сходного строения при
одной и той же массовой концентрации подавляют максимум тем
сильнее, чем ниже их молекулярная масса. Изучена способность
подавлять максимумы гуминовыми кислотами и фульвокислотами.
Уравнение полярографической волны
Уравнение полярографической волны устанавливает соотноше-
ние между потенциалом электрода и величиной диффузионного
тока в любой точке полярографической волны. Оно было выведено
Гейровским и Илькевичем в 1935 г.
Потенциал ртутного капельного электрода задается внешним
источником напряжения, для обратимых процессов в соответствии
с уравнением Нернста он равен
Концентрацию окисленной и восстановленной форм электро-
химически активного вещества можно выразить через величину
диффузионного тока. Для процессов восстановления диффузион-
ный ток в любой точке волны определяется уравнением Илько-
вича. Тогда i = К([Ох] — [0хэ]) и [0хэ] = IMlL Где [Ох] —
93
концентрация окисленной формы электрохимически активного ве-
щества в основной части раствора, [Охэ] — ее концентрация в
приэлектродной части, /< — константа Ильковича, равная
605 n£>i/2m2/3 т1/6. Концентрацию восстановленной формы также
можно вычислить по величине тока. Если справедливо i=
= /([Ох] — [Оэ], то должно выполняться и i = Ki [Red3] —[Red],
где Ki — константа, равная 605 n£>l/2m2/3T|/6, a Dx — коэффи-
циент диффузии восстановленной формы электрохимически ак-
тивного вещества.
Когда в растворе находится только окисленная форма электро-
химически активного вещества, т. е. [Red]=0, выражение упро-
щается: I— ^[Reda] и тогда
[Red3] =
Выразив концентрации окисленной и восстановленной форм через
величину тока и подставив их в уравнение Нернста, получим
Поскольку предельный диффузионный ток равен inp=K[Ox], то,
подставляя это значение в уравнение и учитывая, что
К
Ki
получим
Е = Е0
D
Di
!пр 1
RT
nF (пр • i
где Е — потенциал электрода, i — соответствующая этому потен-
циалу величина тока, inp — предельный ток для данной волны.
Из уравнения следует, что с увеличением концентрации элект-
рохимически активного вещества потенциал его выделения сдви-
гается в более положительную область. Поэтому потенциал вос-
становления не может служить надежной качественной характе-
ристикой природы восстанавливающихся ионов. Для качественной
характеристики вещества используют потенциал полуволны или
такой потенциал катода, при котором величина диффузионного
тока равна половине своего предельного значения: i = 0,5inp. Вы-
ражение для потенциала полуволны Е1/2 легко получить, если в
уравнение волны подставить значение i=0,5 inp:
Е = Ео — -Е— In
nF
D
Di
nF f'np 0,5tnp
94
Таблица 16
Потенциалы полуволн ионов
Ион Состав фона Электрохимичес- кая реакция f>/2- в
1 2 3 4
А13+ Алюминий 0,1 M КС1 или NH4C1 3-М) —1,75
Ва2+ Барий 0,1 М КС1 или NHjCl 2—>0 — 1,92
vor Ванадий 1 м nh4ci, nh4oh 5->4 —0,97
V (III) 1 М KCNS 4->2 3->2 —1,26 —0,46
Bi3+ Висмут 2 М К2СО3 3->-0 —0,64
Bi3+ 1 М НС1 3-М) —0,09
Bi3+ 2 М СН3СООН + 2 М CH3COONH4 3-М) —0,25
Fe2+ Железо 0,25 М Н2С4Н4Ов (винная кислота) 2->3 —0,12
Fe3+ то же 3—>2 —0,12
Fe2+ 0,25 М Н2С2О4 ьо со 11 со о — 1,50 —0,23
Fe3+ то же 3->2 —0,23
ЮГ Йод 0,05 М КС1 5^(-1) — 1,28
Cd2+ Кадмий 1 М KNO3 2-М) —0,63
Cd2+ 1 М NH4OH, 1 М NH4CI 2->0 —0,81
Cd2+ 1 М Na ОН 2—>0 —0,78
Cd2+ 0,5 М H2SO4 2-М) —0,59
K+ Калий 0,1 М (CH3)4NC1 1-М) —2,13
Ca2+ Кальций растворы солей тетраметил аммония 2-М) —2,22
o2 Кислород 0,1 н. КС1 или НС1, NaOH Og- —0,1
Co2+ Кобальт 0,1 М КС1 н2о2-^н2о 2-М) —0,9 —1,20
Co2+ 1 м КОН 2-М) —1,46
Mg2t Магний 0,1 М (CH3)4NC1 2->0 —2,30
Mn2+ Марганец 1 М NaOH 2-М) —1,70
Mn2+ 0,1 М КС1 2—>0 —1,51
Мп (VII) » 7—>2 >0
Mn2+ 1 М NH4OH + 1 М NH4C1 2-+0 —1,66
Cu+ Медь 1 М NHjOH + 1 М NH4C1 1->2 —0,22
Cu2+ » » 1-М) 2—>1 —0,50 —0,24
Cu2+ 0,1 М КС1 о — t t CN СЧ —0,51 +0,04
2-»-0 —0,22
95
Продолжение табл. 16
> 2 3 4
Молибден
Mo (VI) 1 M NH4OH + 1 M NH4CI 6->-5 ' -1,71
Mo (VI) 3 M HClOt 6^5 —0,14
5->3 —0,79
Натрий
Na+ 0,1 M (CH3)4NC1 l->0 —2,1
Никель
Ni2+ 7,3 M H3PO4 2-<-0 —1,18
Ni2+ 0,1 M KC1 2->0 — 1,1
Ni2+ 5 M CaCl2 2->0 —0,56
Свинец
РЬ2 + 1 M HC1 2->0 —0,44
Pb2+ 0,1 M KC1 2->0 —0,40
7,3 M H3PO4 2->0 —0,53
Титан
Ti (IV) 2 M K2CO3 4->3 —1,5
Хром
Cr” 1 M HC1 3—>2 —0,99
3^0 —1,26
Cr(VI) » 6->3 —0,78
Цинк
Zn2+ 7,3 M H3POj 2—>-0 — 1,13
Zn2+ 0,1 M KC1 2—>0 —1,00
Zn2+ 1 M NH4C1 + 1 M NHjOH 2->0 —1,35
Zn2+ 0,25 M Н2С4Н4О6 (винная кислота) 2—>0 — 1,20
Последний член уравнения равен нулю, тогда
£=£0
Все величины, входящие в правую часть последнего уравнения,
можно считать постоянными, следовательно, потенциал электрода
при i = 0,5inp Для данной электрохимической реакции — величина
постоянная, не зависящая от тока и концентрации электрохими-
чески активного вещества в растворе, при условии, что состав
фонового раствора и температура постоянны. Правую часть урав-
нения, содержащую только постоянные величины, обозначают
символом Ецг:
,, с RT 1 1 / D
Е\!2 = £п-----------In 1/ --------
' 0 nF V Dr
Потенциал полуволны очень важная в полярографии величина,
дающая возможность установить природу вещества, участвующего
в электрохимической реакции. В табл. 16 приведены значения по-
тенциалов полуволн некоторых ионов в различных по составу ин-
дифферентных, или фоновых, электролитах.
96
Вводя потенциал полуволны в качестве константы в уравне-
ние волны, получим
г- rr RT '
Е = £1/2-------In--------.
nF 'пр—'
Подставив числовые значения констант для 20° и заменив нату-
ральные логарифмы десятичными, уравнение полярографической
волны можно представить в следующем виде:
г р 0,058 i
Е =-Ei/2----------1g—--------.
" 'пр '
Во всех обратимых системах потенциал полуволны не зависит
от концентрации электрохимически активного вещества, а опреде-
ляется его природой; значительное влияние на £1/2 оказывают
состав и концентрация индифферентного электролита. С увеличе-
нием ионной силы раствора потенциал полуволны сдвигается в сто-
рону более отрицательных значений.
Графическая зависимость между 1g--------—— и потенциалом выра-
'пр '
жается прямой линией с угловым коэффициентом (тангенсом угла
наклона), равным —-—:
0,058
Прямая проходит через нулевое значение
(рис. 31).
Как следует из уравнения, обратная
1g -7—---- когДа i=°)5inp
'пр '
величина углового коэф-
фициента для одноэлектронной злектгюхимической реакции равна
0,058 В, для двухэлектронной —
0,029 В и для трехэлектронной —
0,019 В (см. рис. 31). Таким обра-
зом, анализ полярографических
кривых с помощью уравнения вол-
ны дает возможность установить
число электронов п, участвующих в
электрохимической реакции.
В практике полярографического
анализа очень большое значение
имеют случаи восстановления ком-
плексных ионов. Введение комп-
лексообразующего реагента может
сильно изменить потенциал полу-
волны, и это используется при раз-
работке аналитических методов.
Рис. 31. Зависимость ~g--------
от потенциала электрода
Для обратимо восстанавливающегося комплексного соединения
в присутствии значительного избытка комплексообразующего ре-
4 Зак 284
97
агента уравнение волны имеет вид
Г D
Г? С ЕТ 1 '
Е = Е..-------In----
«£ 'пр
---^-ln[Xm-]P 1^-1П К,.ест
nF nF
где D — коэффициент диффузии комплексного иона, Хт~ — анион
комплексообразующего реагента, р — координационное число,
Кнест — константа нестойкости комплексного иона.
Подставив в уравнение i=0,5inp, получим выражение для по-
тенциала полуволны комплексного иона металла:
£1/2 = Ео —1П 1/------------1П рГ-]₽ Ь Ь Япест-
nF у Dj. nF nF
Согласно уравнению потенциал полуволны комплексного иона
металла не зависит от концентрации этого иона в растворе, а из-
меняется в зависимости от концентрации комплексообразующего
реагента. Потенциал полуволны тем более отрицателен, чем мень-
ше константа нестойкости комплекса, т. е. чем устойчивее комп-
лекс.
Потенциал полуволны свободного иона металла, как было пока-
зано выше, равен
Вычитая почленно последнее уравнение из уравнения потенциала
полуволны комплексного иона, найдем сдвиг потенциала полувол-
ны при образовании комплексных ионов:
£1/2 - £1/2 = Д £1/2 = In л/~ ---------
nF V D
---*Zl_ln[Xm-]P ; -^1п£нест.
Для приближенных расчетов эго выражение можно упростить:
аг4 0»058 1 rz 0,058 t г у-щ—-I
Д £,/2 = — -------- 1g Хнест — р----------lg [X ].
п п
Графически зависимость между Д £,/2 и lg [Xm ] выражается пря-
0,058
мои линиеи, тангенс угла наклона которой равен р —-—, а отре-
зок, отсекаемый на оси ординат, —
0,058 1гг
------- Лнест-
п
98
Это позволяет использовать уравнение для изучения комплексных
А Aj М
соединений. Если известно п и Е i/2 , то по отношению----— =
A 1g [Х"г-]
0,058
— —р--------можно рассчитать координационное число р, а затем
п
И величину Кпест-
Таким образом, полярографический метод дает возможность
определить константу нестойкости комплексного иона.
Мы рассмотрели уравнение волны для восстановления комп-
лексных ионов металла в самом простом случае, когда металл
восстанавливается с образованием амальгамы. Могут иметь место
более сложные варианты, когда восстановление комплекса проис-
ходит ступенчато или с изменением валентности катиона-комп-
лексообразователя.
Сдвиг потенциала полуволны ионов металлов в присутствии
комплексообразующих веществ имеет очень важное практическое
значение. В анализируемых растворах часто присутствуют одно-
временно ионы нескольких металлов. Если потенциалы полуволн
восстанавливающихся веществ различаются более чем на 0,2 В,
то при полярографировании такого раствора получают хорошо
выраженную ступенчатую кривую, состоящую из нескольких поля-
рографических волн. Но если потенциалы полуволн близки, то на
полярограмме вместо нескольких ступеней получается одна сум-
марная волна от восстановления нескольких ионов. В этом случае
часть ионов удобно перевести в комплексные соединения для
того, чтобы сдвинуть их потенциалы полуволн в область более
отрицательных значений. Если сдвиг потенциалов достаточно
велик, удается проводить раздельное определение ионов при их
совместном присутствии в растворе.
Для раздельного определения нескольких элементов часто
применяются фоновые растворы довольно сложного состава.
Кинетические токи
В предыдущих разделах рассматривались преимущественно
такие электродные процессы, при которых величина предельного
тока определялась скоростью диффузии электрохимически актив-
ного вещества к электроду. Однако электродный процесс состоит
из нескольких стадий; в простейшем случае этими стадиями яв-
ляются подача вещества к поверхности элеырода, собственно
электрохимическая реакция (перенос электронов) и отвод продук-
та реакции от электродной поверхности. Скорость электродного
процесса в целом определяется скоростью его отдельных стадий.
Существует большое число электродных процессов, при кото-
рых величина тока определяется кинетикой химических реакций,
протекающих у поверхности электрода. Соответствующие им токи
и полярографические волны называют кинетическими.
4
99
Различают кинетические волны с предшествующими, после-
дующими и параллельными химическими реакциями.
В настоящее время известно, что многие электродные процес-
сы с участием органических веществ включают химическую
стадию.
Величина чисто кинетических токов не зависит от высоты ртут-
ного столба, пропорциональна т2/3 и имеет высокий температур-
ный коэффициент.
В количественном полярографическом анализе неорганических
компонентов большое значение имеют так называемые каталити-
ческие токи. Они возникают, когда продукт электрохимической
реакции, взаимодействуя с компонентами раствора, частично или
полностью регенерирует исходное электрохимически активное ве-
щество, которое вновь вступает в электрохимическую реакцию.
Высота каталитической волны определяется скоростью химической
реакции. Примером может служить схема, включающая восста-
новление железа в присутствии перекиси водорода.
Свободные ионы Fe (П1) в кислой среде восстанавливаются,
и возникающий диффузионный ток подчиняется уравнению Иль-
кевича. Добавление перекиси водорода повышает величину пре-
дельного гока. Увеличение предельного тока вызвано тем, что
ионы железа (II), возникающие в результате электрохимической
реакции, окисляются вблизи электрода перекисью водорода до
Fe (Ш), а образовавшиеся химическим путем ионы Fe (III) вновь
электрохимически восстанавливаются на электроде. При избытке
перекиси водорода увеличение тока определяется скоростью хи-
мической реакции между ионами Fe (II) и перекисью водорода.
Процесс можно представить следующим образом:
Fe3+ F Fe2+,
2Fe2+-H2O2 2H+->2Fe3+ I 2H,O.
Кроме перекиси водорода в каталитических процессах могут
участвовать другие окислители. При определении молибдена в
почвенно-биологических объектах широко используются катали-
тические токи перхлоратов и нитратов. В случае полярографирэ-
вания молибдена на фоне хлорной кислоты процесс можно пред-
ставить следующим образом:
Mo(V) -e^Mo(IV),
8 Mo (IV) ЬСЮ? H8H+->8Mo(V) I С1-у4Н2О.
В данном случае увеличение предельного тока вызвано реге-
нерацией ионов Mo (V) из продукта электродной реакции — Мо
(IV). Каталитические токи, увеличенные за счет регенерации де-
поляризатора, могут в 10—100 раз превышать токи диффузионные.
100
Полярографические методы
Зависимость величины тока от потенциала регистрируют с по-
мощью специальных приборов — полярографов. Полярографы обес-
печивают автоматическое изменение напряжения между электро-
дами и запись полярограмм. По виду поляризующего напряжения
выделяют несколько групп полярографических методов.
Классическая полярография. Основы теории классической по-
лярографии изложены в предыдущих разделах. В классической
полярографии поляризующее линейно-возрастающее во времени
напряжение подают на электроды от источника постоянного тока.
Скорость изменения напряжения составляет сотые или десятые
доли вольта в секунду. Полярографические кривые записывают
с помощью гальванометров или самописцев. Классическим поля-
рографическим методом по диффузионным токам можно анали-
зировать растворы с концентрацией электрохимически активного
вещества не ниже 1 • 10 5 М.
Осциллографическая полярография. Более низкий предел об-
наружения электрохимически активных веществ (1-10~6М) удает-
ся получить методом осциллографической полярографии. В осцил-
лографической полярографии используют высокие скорости изме-
нения поляризующего напряжения (0,1—50 В/с), а полярограммы
регистрируют с помощью электронно-лучевых трубок. Величина
максимального тока обратимых процессов зависит от скорости
изменения поляризующего напряжения в степени 1/2:
i = 2,344- 105-п3/2 О1/2 (щ/)2/3 VI/2C,
где V — скорость развертки напряжения.
Чувствительность осциллографической полярографии ограни-
чивает емкостный ток, который растет пропорционально скорости
изменения напряжения (ic=AV), т. е. быстрее, чем максимальный
ток, обусловленный электрохимической реакцией.
Высокая скорость изменения напряжения позволяет получить
полярограмму в период существования одной капли, обычно по-
лярограмма регистрируется в конце жизни капли, когда ее поверх-
ность мало изменяется. Осциллографическая полярограмма отли-
чается от классической и имеет форму максимума (pjic. 32).
Производная, или дифференциальная, полярография. Для уве-
личения разрешающей способности полярографических методов
вместо обычных интегральных кривых зависимости величины тока
от потенциала
рируют кривые
помощью соответствующего устройства регист-
—-----Е, так называемые производные, или диф-
dE
ференциальные, полярограммы. Дифференциальные полярограм-
мы имеют форму пиков, потенциалы которых соответствуют
потенциалам полуволн, так как в этих точках наблюдается макси-
мальное изменение тока с ростом потенциала.
101
С помощью дифференциальной полярографии можно опреде-
лять компоненты, потенциалы полуволн которых различаются на
~50мВ. Однако предел обнаружения, получаемый с помощью
методов дифференциальной полярографии, выше соответствующих
интегральных методов.
Переменно-токовая полярография. В переменно-токовой поля-
рографии на электроды кроме поляризующего линейно-нарастаю-
щего во
времени напряжения
Рис. 32. Осциллографические поля-
рограммы:
А — простая, Б — производная
подают переменное напряжение
малой амплитуды (5—50 мВ).
Под действием переменного на-
пряжения возникает переменная
составляющая тока, величина ко-
торой пропорциональна концент-
рации вещества. В переменно-
токовой полярографии регистри-
руют зависимость переменной
составляющей тока от поляризу-
ющего напряжения. Поляро-
грамма имеет форму пика, потен-
циал которого для обратимых
процессов совпадает с потенци-
алом полуволны в классической
полярографии.
Большие аналитические возможности переменно-токовой поля-
рографии обусловлены тем, что законы формирования тока элект-
рохимической реакции и емкостной составляющей переменного
тока различны. Это дает возможность отделить полезный сигнал —
ток электрохимической реакции — от основного мешающего сиг-
нала в полярографии — емкостного тока.
Метод переменно-токовой полярографии дает возможность ана-
лизировать растворы с концентрацией определяемого компонента
до 1-10~7М и позволяет селективно определять компоненты, по-
тенциалы пиков которых различаются всего на 40 мВ.
Инверсионная вольтамперометрия. Одним из приемов повыше-
ния чувствительности полярографических методов является на-
копление определяемого компонента на электроде и регистрация
токов анодного растворения. При восстановлении ионов металлов
на стационарной ртутной капле могут образовываться амальгамы.
Инверсионная полярография, или амальгамная полярография с
накоплением, исследует токи анодного растворения металла из
амальгам. Если зависимость ток — потенциал изучают с помощью
твердых электродов, то такие исследования называют вольтампер-
метрическими (полярография — частный случай вольтамперомет-
рии). Концентрирование определяемого компонента на твердом
электроде с последующей регистрацией токов анодного растворе-
ния называют инверсионной вольтамперометрией твердых фаз,
можно анализировать растворы с концентрацией определяемого
компонента до 10-8— 10~10 М.
102
Количественные измерения
в полярографическом анализе
Полярографическая кривая выражает зависимость между ве-
личиной тока и потенциалом электрода. Поэтому любые виды
работ можно свести к измерениям этих двух показателен. Точные
измерения потенциала электрода необходимы для идентификации
вещества, участвующего в электрохимической реакции; ток или
высоту волны измеряют при количественном анализе.
Измерение высоты волны и величины предельного тока. Абсо-
лютные величины тока в количественном полярографическом ана-
лизе используются довольно редко. Величина тока бывает нужна
при вычислении концентрации по уравнению Ильковича. Практи-
чески концентрацию определяют по градуировочному графику,
методом стандартов или методом добавок. В этих случаях доста-
точно измерить высоту волны или, что то же самое, выразить
предельный ток в условных единицах. Чтобы найти абсолютные
значения тока, сначала также надо измерить высоту волны, а за-
тем, зная чувствительность прибора, можно вычислить ток в мил-
лиамперах или микроамперах.
Для измерения высоты волны используют различные графи-
ческие приемы, но во всех случаях измерение ведут строго по
вертикали, т. е. в направлении, параллельном оси ординат. До-
пустимо измерять расстояние по вертикали между площадкой
предельного тока и линией, продолжающей плошадку остаточного
тока.
Наиболее точные и воспроизводимые результаты получаются
путем графического построения трех касательных: одна из них
проводится вдоль площадки остаточного тока, вторая служит про-
должением площадки предельного тока, а третья проводится
по направлению волны. Расстояние по вертикали между точка-
ми пересечения этих трех прямых линий принимается равным вы-
соте волны (рис. 33). На полярограммах с осцилляциями высоты
волн можно измерять по минимальным или максимальным зна-
чениям осцилляций или по их среднему значению. Очевидно, для
получения сопоставимых результатов способ измерения высоты
волны в серии экспериментов должен быть постоянным.
Найденное значение высоты волны h\ выражают в миллимет-
рах, а затем приводят к постоянной чувствительности (см. с. 108)
регистрирующей системы. Удобней всего вести расчет на макси-
мальную чувствительность. Если, например, полярограмма была
снята при чувствительности f, равной —— от максимальной чувст-
вительности, то высота волны, приведенная к полной чувстви-
тельности прибора, или просто приведенная высота волны h равна
1
50
=50 ht.
103
Пользуясь приведенной высотой волны, можно сравнивать по-
лярограммы, снятые при любых значениях чувствительности. При
сравнении приведенных высот волн следует иметь в виду, чтэ
после пересчета абсолютные ошибки измерения увеличиваются
пропорционально чувствительности.
Измерение высот максимумов, пиков на производных и пере-
менно-токовых полярограммах и на полярограммах анодного
растворения несколько отличается от измерений обычной волны.
Способы измерения показаны на рис. 34.
Рис. 34. Измерение высот поляро-
графических пиков
Рис. 33. Измерение высот полярогра-
фических волн
В современных приборах предусмотрена автоматическая за-
пись полярографических кривых пером на диаграммной ленте.
Чувствительность, или цена деления, в этом случае может быть
выражена величиной тока, вызывающего отклонение пера на одно
деление диаграммной ленты пли на 1 мм. Умножая приведенную
высоту волны на цену деления, находят абсолютную величину
предельного тока.
Способы определения концентрации. Уравнение Ильковича
редко используется для вычисления концентрации, так как в этом
случае необходимо измерение всех параметров уравнения.
Период капания и массу ртути, вытекающую из капилляра в
единицу времени, можно легко найти с требуемой для анализа
точностью, но коэффициент диффузии зависит от состава раство-
ра, и его точное значение не всегда известно.
Широко используются для количественного анализа метод
градуировочного графика и метод добавок.
Градуировочный график строят по результатам полярографи-
рования стандартных растворов. Стандартные растворы и иссле-
дуемые пробы полярографпруют на идентичных фонах, и пред-
полагается, что зависимость между током и концентрацией ве-
щества в обоих случаях одинакова. В действительности исследуе-
мые растворы, полученные при обработке образцов природных
объектов, могут отличаться от стандартов, что влияет на характер
полярограммы и результаты количественных определений. Для
104
построения графика готовят серию стандартных растворов, кон-
центрации которых несколько перекрывают диапазон концентра-
ций, ожидаемых при анализе образцов. После получения поляро-
грамм измеряют высоты волн, вычисляют приведенные значения
высот волн и строят график в координатах /г — С.
По графику находят концентрацию определяемого вещества
в полярографируемом растворе, выраженную в тех единицах, ко-
торые были приняты для стандартов (чаще всего в мг/мл). Для
пересчета на содержание в почве принимают во внимание навес-
ку, объем вытяжки и все разбавления или объем аликвот. Вполне
допустимо на оси абсцисс откладывать непосредственно содержа-
ние определяемого элемента в анализируемом объекте (почве,
растении и др.), если все условия анализа сохраняются стабиль-
ными. Способы и приемы пересчета концентрации раствора на со-
держание элемента в пробе общие для всех методов количествен-
ного анализа.
Метод добавок применяют в тех случаях, когда состав анали-
зируемого раствора точно не известен, а его компоненты влияют
на характер полярограммы. По методу добавок снимают минимум
две полярограммы. Сначала полярографируют исследуемый раст-
вор, а затем — тот же раствор, но после добавления определен-
ного объема стандартного раствора. Предполагается, что прира-
щение высоты волны пропорционально количеству внесенного
стандарта. Если объем добавленного стандартного раствора мал
по сравнению с общим объемом и не превышает 1%, то измене-
нием объема испытуемого раствора можно пренебречь и считать
отношение высот волн прямо пропорциональным отношению кон-
центраций. Тогда
С _ hCvt
* — (// — h)v ’
где h и h' — исходная высота волны и высота волны после введе-
ния добавки, С — концентрация стандартного раствора, Vi и v —
объем добавки и испытуемого раствора.
Методом добавок можно пользоваться и тогда, когда добавка
значительно изменяет объем раствора; расчет проводят по фор-
муле
Q __ _____Cvji_____
х (/г' — h) v + h’vi
Метод добавок, как явствует из условия, также основан на пря-
мой пропорциональности между концентрацией и высотой волны.
Для нахождения концентрации методом добавок можно дать
и графическое решение. Для этого на графике вправо от оси ор-
динат откладывают приращение концентрации за счет добавки,
влево — в том же масштабе концентрацию анализируемого раст-
вора. Первоначальную высоту волны (без добавки) откладывают
по оси ординат против абсциссы, равной нулю (АС=0). Высоту
105
волны после внесения добавки (с поправкой на разбавление) от-
кладывают против соответствующего значения АС. Через полу-
ченные две точки проводят прямую до пересечения с осью абсцисс
и по отрезку, отсекаемому прямой на левой части графика, нахо-
дят искомую концентрацию. В правомочности построения легко
убедиться путем переноса начала координат в точку пересечения
экспериментальной прямой с осью концентрации.
Иногда для графического решения используют две последова-
тельные добавки. Вторая (как и последующие) добавка уточняет
угол наклона прямой h — С, но не изменяет принцип решения:
прямая, построенная только по двум точкам, не всегда достаточно
надежна.
Для снижения помех за счет нестабильности условий поляро-
графирования иногда применяют метод внутреннего стандарта,
при котором можно пользоваться всеми приведенными выше
приемами вычислений, беря вместо высоты волны определяемого
электрохимически активною вещества отношение высот волн оп-
ределяемого и стандартного элемента, который был введен в раст-
вор в качестве внутреннего стандарта.
Полярографическая аппаратура
Любая полярографическая установка включает ячейку, источ-
ник поляризующего напряжения, преобразователь напряжения и
регистрирующее устройство.
Полярографическая ячейка. Она включает индикаторный элект-
род, электрод сравнения и электролизер. Наиболее широко исполь-
зуют ртутный капающий электрод. Это — толстостенный капилляр
с внутренним диаметром 0,05— 0,08 мм, который с помощью труб-
ки соединен с резервуаром, заполненным очищенной ртутью. Уро-
вень ртути устанавливают на такой высоте, чтобы из капилляра
ртуть вытекала со скоростью 1 капля за 2—4 с. В качестве элект-
родов сравнения используют донную ртуть или неполяризующиеся
электроды второго рода: каломельные, ртутно-сульфатные, хлор-
серебряные.
Полярограф Л П-60. Этот полярограф работает в классическом
режиме; с его помощью можно получить интегральные (обычные),
производные и инверсионные полярограммы.
Источником поляризующего напряжения служат аккумуляторы
или в более поздних моделях (ЛП-7) — специальные выпрямите-
ли переменного тока. В качестве преобразователя напряжения,
обеспечивающего медленное возрастание потенциала электрода
во времени, используется реохорд. Полярограммы записываются
на диаграммной ленте с помощью электронного самопишущего
устройства.
На переднюю панель полярографа выведены рукоятки управ-
ления (рис. 35). Питание от сети включается тумблером /; вводы
106
для подключения штепсельной вилки, для соединения с регистри-
рующим устройством и для подключения аккумулятора располо-
жены на задней стенке.
Реохорд полярографа соединен с рукояткой 19, которая позво-
ляет воучную устанавливать требуемое напряжение. Поляризую-
щее напряжение считывается по шкале в смотровом окошке 13.
Рис. 35. Полярограф ЛП-60.
А — собственно полярограф: 1 — вы-
ключатель полярографа, 2 — пере-
ключатель для изменения направле-
ния поляризации, 3 — переключатель
изменения скорости поляризации,
4 — выключатель двигателя поляро-
графа, 5 — кнопка включения элек-
тродвигателя, 6 — контрольная лам-
па, 7, 8 — выключатели подзарядки
аккумулятора, 9 — выключатель ак-
кумулятора, 10 — кнопка и рукоятка
для точной установки напряжения
по элементу Вестона, 11, 12 — гру-
бая и точная компенсации емкост-
ных токов, 13 — редуктор чувстви-
тельности, 14 — переключатель диа-
пазона напряжения, 15—вольтметр,
16 — кнопка для измерения потен-
циала ртутного электрода, 17—уста-
новка предела поляризации, 18—
смотровое окошко, 19 — рукоятка
потенциометра.
Б — электронный потенциометр: 20 —
переключатель демпфирования ос-
цилляций и записи производных по-
лярограмм, 21, 22 — установки пера,
25 — ручная протяжка диаграммной
ленты, 24 — выключатель автомати-
ческой протяжки диаграммной ленты,
рычажок для опускания пера
Грубый отсчет напряжения ведется по шкале в смотровом окошке,
точный — по шкале реохорда. В смотровом окошке расположена
также вспомогательная шкала, по которой устанавливают пре-
дельное напряжение поляризации; для перемещения шкалы слу-
жит рукоятка 17. При достижении установленного на этой шкале
напряжения запись полярограммы автоматически прекращается.
Автоматическое вращение реохорда и запись полярограммы
включают кнопкой 5, а переключатели 2 и 3 изменяют направле-
ние и скорость поляризации. Переключатель 4 позволяет записы-
вать ток при разных режимах; если он находится в верхнем по-
ложении, то синхронно с реохордом движется диаграммная лента
на регистрирующем приборе и записывается кривая: ток — потен-
циал. В нижнем положении переключатель 4 исключает движение
1.07
реохорда, и тогда можно записать изменение тока по времени
при постоянном напряжении.
Диапазон поляризующего напряжения устанавливается рукоят-
кой 14 в следующих пределах: от -{-2 В до —2 В, от -}-1 В до
—3 В, от 0 до —4 В, от -}-2 В до 0, от -}-1 В* до —1 В, от 0 до
—2 В, от —1 В до —3 В и от —2 В до —4 В. Редуктор чувстви-
тельности 13 имеет 20 положений, которые позволяют использо-
вать от максимальной 1/1 до 1/10000 чувствительности прибора.
Для подачи напряжения от аккумулятора на реохорд служит
рукоятка а величина поданного напряжения регулируется ру-
кояткой 10 и контролируется вольтметром 15.
Остаточный ток компенсируется с помощью рукоятки 11 (гру-
бо) от 0 до 1 мкА/B и рукоятки 12 (точно) —от 0 до 0,1 мкА/В,
при чувствительностях меньших 1/50 компенсирующим устройст-
вом не пользуются.
На самопишущем приборе (рис. 35,Б) имеются выключатели
прибора 25 и диаграммной ленты 24, рукоятка 26 для опускания
пера на диаграммную ленту, рукоятки установки нулевой линии
21, 22 и переключатель демпфирования 20. При положении пере-
ключателя демпфирования, обозначенном D, регистрируется про-
изводная полярограмма.
Скорость передвижения диаграммной ленты может изменяться
при помощи редуктора от 10 мм/ч до 180 мм/мин.
Запись полярограмм. Чтобы получить полярограмму ис-
пытуемого раствора, полярограф подключают к сети переменного
тока тумблером 1, переключателями 25 и 24 включают самописец,
рукояткой 9 подают от аккумулятора напряжение на реохорд пли
включают выпрямитель переменного тока. Переключателем 14 ус-
танавливают нужный диапазон напряжения, а переключателями 2
и 3— нужное направление и скорость поляризации (400 мВ/мин).
В анализируемый раствор спускают капилляр ртутного капель-
ного электрода и электрод сравнения. Электролизер с раствором
изолируют от внешней среды, например, прижимая края электро-
лизера с помощью кремальеры к неподвижной крышке, нижняя
плоскость которой оклеена вакуумной резиной и надежно изоли-
рует раствор от атмосферного воздуха. Через раствор 5 мин про-
пускают инертный газ. Затем поднимают резервуар со ртутью и
убеждаются, что капли ртути вытекают из капилляра. Рукояткой
19 устанавливают потенциал начала поляризации, а рукоятками
20 и 21 устанавливают перо самописца в начале шкалы. Редуктор
чувствительности устанавливают в такое положение, чтобы при
вращении рукоятки 19 и изменении потенциала от начального дэ
конечного значений перо самописца переместилось к концу шкалы,
но не достигло упора. Затем переключатель 4 ставят в положение,
обеспечивающее перемещение рукоятки 19 и изменение потенциа-
ла электрода, кнопкой 5 включают электродвигатель и записыва-
ют полярограмму. Затем с помощью редуктора 13 уменьшают
чувствительность на одну градацию, ставят начальное значение
108
потенциала рукояткой 19, перо перемещают к- началу шкалы и
снова записывают полярограмму. Съемку повторяют еще один
раз при меньшей чувствительности прибора. Если концентрацию
предполагается определять методом добавок, рукоятку 13 ставят
в положение 1/10 000, опускают резервуар со ртутью и в поляро-
графпруемый раствор добавляют строго отмеренный объем стан-
дартного раствора определяемого компонента, несколько минут
пропускают инертный газ, поднимают резервуар со ртутью, под-
бирают чувствительность прибора и полярографируют. Затем при-
ступают к анализу следующего раствора. После окончания работы
рукоятку 13 ставят в положение 1/10 000, обеспечивающее наи-
меньшую чувствительность прибора, и выключают полярограф и
самописец тумблерами 1 и 25. Резервуар со ртутью опускают, со-
держимое электролизера выливают в склянку для отработанной
ртути, капилляр и электрод сравнения промывают из промывалки
дистиллированной водой, собирая воду в стаканчик или чашку.
Затем капилляр опускают в стаканчик с дистиллированной водой,
а насыщенный каломельный электрод — в стаканчик с насыщен-
ным раствором хлорида калия.
Внимание! Пары ртути — опасный кумулятивный яд.
Нельзя оставлять поверхность ртути открытой, ртуть должна
находиться под слоем жидкости. Нельзя пользоваться нагре-
вательными приборами вблизи ячеек со ртутью или склянок,
в которых хранится ртуть. Нельзя ртуть выливать в ракови-
ну; отработанную ртуть из электролизера выливают в спе-
циальные склянки, которые хранятся в вытяжном шкафу.
Работу со ртутью необходимо проводить в специальных кю-
ветах. Если обнаружены капли ртути, необходимо сообщить
об этом лаборанту.
На полярограммах измеряют высоты полярографических волн,
рассчитывают среднее приведенное значение h и находят концент-
рацию определяемого компонента по калибровочному графику
или по формуле, используемой в методе добавок.
Полярограф ЛП-7е. В полярографе ЛП-7е применяется элект-
ронная развертка напряжения. Прибор состоит из собственно по-
лярографа А, самописца Б (рис. 36, Л, Б) и полярографической
ячейки.
Полярограф подключают к сети переменного тока кнопкой 1
за 30 мин до начала работы (см. рис. 36). Перед присоединением
электродов переключатель 5 ставят в положение «стоп», на потен-
циометре устанавливают нулевое значение потенциала и включа-
ют кнопку 10. При работе с двумя электродами клеммы 12 и 14
соединяют перемычкой, ртутный капельный электрод присоеди-
няют к клемме 13, неполяризующийся электрод — к клемме 14.
При работе с тремя электродами перемычку устраняют, и третий
электрод присоединяют к клемме 12.
109
Управление работой полярографа осуществляется переключа-
телями, выведенными на переднюю панель прибора Начальное
напряжение поляризации устанавливают ручкой потенциометра 3.
Цена деления внешнего круга потенциометра составляет 250 мВ,
внутреннего — 25 мВ Потенциал может быть изменен от +2,5 до
—2,5 2 Интервал изменения потенциала, или диапазон развертки
Рис 36 Полярограф ЛП-7е
А — полярограф 1 — выключатель, 2 — переключатель скорости развертки на-
пряжения, 3—потенциометр для установки начального напряжения, 4 — по-
тенциометр для установки диапазона развертки пилообразного напряжения,
5 — переключатель режима работы, 6 — переключатель чувствительности, 7 —
переключатель подавления осцилляций, 8 — потенциометр для компенсации ем-
костных токов, 9 — кнопка ускорения развертки напряжения, 10 — кнопка, воз-
вращающая потенциал к исходному значению, 11 — вольтметр, 12, 13, 14—
гнезда для присоединения электродов
Б — самописец 15 — выключатель, 16 — кнопка управления хронометрического
узла, 17 — переключатель скорости и направления движения бумаги, 18 — ру-
коятка установки нулевой линии
напряжения, устанавливают с помощью рукоятки 4, максималь-
ная развертка напряжения 3 В. Например, если начальное напря-
жение рукояткой 3 установить равным +1 В, а диапазон разверч-
ки напряжения равным 1,5 В, то в процессе полйрографированпя
потенциал электрода будет изменяться от +1 до £-0,5 В Скорость
изменения потенциала устанавливается переключателем 2. Зна-
чение потенциала приблизительно можно оценить по панельному
вольтметру 11 или по шкале на регистрационной бумаге Точное
значение потенциала можно измерить цифровым вольтметром, кз-
110
торыи присоединяют к клеммам на задней стенке полярографа.
Ручкой 5 устанавливают режим работы полярографа. положение
«вверх» обеспечивает рост отрицательного потенциала, положение
«вниз» — падение потенциала, прекращение изменения потенциа-
ла обеспечивается положением «стоп» Положение «реверс» обес-
печивает циклическое изменение потенциала. Кнопка 9 позволяет
ускорить изменение потенциала в ус-
тановленном направлении. Кнопка 10
возвращает поляризационное напря-
жение к заданному начальному зна-
чению Ручка 6 переключает чувстви-
тельность прибора. Переключатель 7
позволяет регулировать степень по-
давления осцилляций; в положении О
полярографический фильтр не дейст-
вует, в положение D осуществляется
запись дифференциальных кривых
Самописец (см. рис. 36, Б) вклю-
чают кнопкой 15, одновременно перо
опускается на бумагу; самописец мо-
жет быть использован в работе спу-
стя 10 с после включения. Движение
бумаги обеспечивается нажатием
кнопки 16, скорость и направление
движения бумаги устанавливают руч-
кой 17. Рукоятка 18 позволяет переме-
щать перо. Порядок работы на прибо-
ре тот же, что и на полярографе
ЛП-60.
Осциллографический полярограф
ЦЛА. Кривые ток — напряжение или
производные тока по напряжению ре-
гистрируются на экране электронно-
лучевой трубки. Экран трубки облада-
ет длительным послесвечением, и по-
Рис 37 Полярограммы свин-
ца, полученные в непрерывном
(слева) и таст-режимах (спра-
ва) записи на фоне 3 н НС1
и аскорбиновой кислоты
лярограммы можно зарисовать на каль-
ку, наложенную на экран, или сфотографировать с помощью
фотоприставки. Полярограф пригоден не только для анализа со-
става раствора, но для исследования процессов в ячейке, в част-
ности для съемки i—^-кривых
Осциллографический полярограф состоит из нескольких бло-
ков стабилизатора напряжения, силового блока, измерительного
блока и датчика В измерительный блок осциллографического
полярографа входят электронно лучевая трубка, генераторы и вы-
прямители, с помощью которых осуществляется ее питание. В из-
мерительный блок включен ламповый источник поляризующего
напряжения, который подает на ячейку линеино-возрастающеэ
напряжение В составе измерительного блока имеется синхрони-
111
затор, который позволяет согласовать развертку напряжения с
отрывом ртутной капли. Развертка напряжения включается через
определенное время после отрыва ртутной капли (задержка;.
В блоке имеются усилители напряжения по горизонтали и верти-
кали электронно-лучевой трубки. Ручки управления выведены на
переднюю панель измерительного блока.
Полярограф ППТ-1. Примером переменно-токового полярогра-
фа может служить выпускаемый нашей промышленностью прибор
ППТ-1. Это универсальный многоцелевой прибор. Он может рабо-
тать в классическом и осциллографическом режимах с записью
полярограмм на диаграммной ленте, в переменно-токовых режи-
мах с трапецеидальной и синусоидальной формами переменного
напряжения и в осциллографическом переменно-токовом режиме.
Полярограф обеспечивает непрерывную запись полярограмм, ко-
торые имеют вид осциллирующих кривых, и регистрирует так
называемые таст-полярограммы, на которых осцилляции отсутст-
вуют. В этом случае сигнал регистрируют не непрерывно, а один
раз за период жизни капли в течение короткого промежутка вре-
мени. В результате получают ступенчатые кривые (рис. 37), где
число ступеней соответствует количеству капель, на которых ре-
гистрировалась полярограмма.
Полярограф ППТ-1 выполнен в виде стойки с четырьмя встав-
ными блоками.
1. Блок поляризующих напряжений вырабатывает 3 вида на-
пряжений для поляризации рабочего электрода: начальное
постоянное калиброванное напряжение; напряжение, изме-
няющееся по линейному закону с заданной скоростью (раз-
вертка напряжения); переменное калиброванное синусои-
дальное или трапецеидальное напряжение.
2. Блок компенсации и усиления обеспечивает заданную вели-
чину потенциала рабочего электрода, возможность компен-
сации емкостного тока и формирует импульсы напряжения
в моменты отрыва ртутных капель.
3. Блок синхронизации формирует переменное трапецеидаль-
ное или синусоидальное напряжение поляризации рабочего
электрода, обеспечивает регистрацию выходного напряже-
ния полярографа в заданный промежуток жизни капли и
другие функции.
4. Блок питания обеспечивает все блоки полярографа необхо-
димыми постоянными и переменными напряжениями.
Автоматическая запись полярограмм осуществляется с по-
мощью электронного автоматического самопишущего потенцио-
метра.
Электролитическая ячейка, электромеханическая мешалка и
другие вспомогательные элементы объединены в полярографичес-
ком датчике.
112
К полярографу придается имитатор электрохимической ячей-
ки, предназначенный для проверки, настройки и регулировки по-
лярографа, а также стабилизатор напряжения 220 В для питания
полярографа.
Области применения полярографии
в почвоведении
В почвоведении полярографический метод применяют для изу-
чения состава и свойств минеральных и органических компонен-
тов почв и исследования механизмов их взаимодействия.
Рис. 38. Элементы, определяемые в почвах полярографическим методом (за-
штрихованы)
Полярографический анализ минеральной части почв. Предло-
жены полярографические методы определения 27 элементов, вхо-
дящих в состав почв (рис. 38). Однако разработанные методы
не равноценны по значимости. Методы определения щелочных и
щелочноземельных элементов (К, Na, Са, Mg) малоперспективны,
поскольку катионы этих металлов восстанавливаются в области
113
высоких отрицательных потенциалов и образуют близко располо-
женные или сливающиеся полярографические волны. Предложены
лишь косвенные полярографические методы определения бора,
алюминия, фосфора, сульфатов.
Относительно легко полярографическим методом определяют-
ся элементы IV, V и VI периодов, именно для них предложено
наибольшее число удовлетворительных методов. Эффективность
применения полярографии для определения этих элементов обус-
ловлена невысокими потенциалами восстановления и склонностью
переходных элементов к комплексообразованию, что позволяет
добиваться четкого разделения полярографических волн даже в
таких сложных по составу растворах, какие получаются в ходе
анализа почв. При изучении ионов переменной валентности перс-
пективно применение полярографии для определения различных
валентных состояний элементов.
Электрохимическая активность гумусовых веществ. Первые
попытки применить полярографию для изучения и количественного
определения гумусовых кислот относятся еще к 1930-м годам.
Токуока и Ружичка в 1934 г. установили инертность гуминовой
кислоты и электрохимическую активность гиматомелановой кис-
лоты и разработали полярографический метод определения гима-
томелановой кислоты в торфах.
К настоящему времени установлено, что фульвокислоты об-
разуют полярографические волны при потенциале—1,5 В относи-
тельно нас. к. э. на фоне 1 М КС1. Волна обусловлена восстанов-
лением Н+-карбоксильных групп, образуется только при поляро-
графировании фульвокислот и исчезает, если их переводят в фор-
му фульватов.
Методом переменно-токовой полярографии показано, что гуму-
совые кислоты восстанавливаются на фоне гидроксида лития при
потенциале —• 1,3В относительно ртутного дна. Появление пика
на полярограммах, вероятно, обусловлено восстановлением фур-
фурола.
Поверхностная активность гумусовых веществ. Гумусовые ве-
щества почв легко адсорбируются на поверхности ртутной капли.
Адсорбируясь, они изменяют поверхностное натяжение, период
капания ртути, емкость двойного слоя, подавляют полярографи-
ческие максимумы. Исследование перечисленных показателей в
зависимости от содержания гумусовых веществ позволяет оценить
поверхностную активность последних.
Установлено, что адсорбция гумусовых кислот происходит в
широком диапазоне потенциалов как при положительном, так и
при отрицательном заряде ртутной поверхности. Максимальная
адсорбция наблюдается при положительном заряде ртутной кап-
ли. Она связана с высоким содержанием кислотных функциональ-
ных групп, которые вследствие диссоциации придают гумусовым
кислотам значительный отрицательный заряд. Адсорбция на отри-
цательно заряженной поверхности свидетельствует о наличии по-
114
ложительно заряженных участков в гумусовых кислотах; ими
могут быть, например, аминогруппы.
Адсорбция гумусовых кислот зависит от pH. В кислой среде
фульвокислоты адсорбируются в широком интервале потенциалов
от __|_о,1 до —1,4 В. По мере подщелачивания раствора поверх-
ностная активность уменьшается, в щелочной среде (pH 10) уже
при потенциале —0,3 В фульвокислота десорбируется с поверхно-
сти капли.
Взаимодействие гумусовых кислот с катионами металлов. Вза-
имодействие ионов металлов с гумусовыми кислотами и устойчи-
вость образующихся комплексных соединений можно оценить по
сдвигу потенциала полуволны (см. с. 98).
Гуминовые кислоты не сдвигают потенциалы полуволн ионов
металлов, но уменьшают высоты их полярографических волн.
Фульвокислоты уменьшают высоты волн и сдвигают потенциалы
полуволн меди, свинца, никеля, кобальта, железа; волна восста-
новления цинка уменьшается при добавлении фульвокислоты, но
не изменяет своего положения. Однако полученные результаты
не могут однозначно интерпретироваться с точки зрения явлений
комплексообразования. И уменьшение высот волн, и сдвиг потен-
циала полуволны могут быть вызваны простой адсорбцией фуль-
вокислоты на поверхности ртутного электрода.
Вопрос о возможности применения полярографии для изучения
комплексообразующей способности природных органических ве-
ществ требует дополнительных исследований. Перспективным нам
кажется использование неводных растворителей, применение ко-
торых позволит исключить адсорбцию органических веществ на
поверхности ртутной капли.
Подготовка почв к анализу
и условия полярографирования
Полярографические методы разработаны для анализа раство-
ров. Поэтому при исследовании почв определяемые компоненты
одним из принятых в почвоведении способов переводят в раствор.
Для этой цели применяют разложение почв кислотами, сплавле-
ние, спекание, (валовой анализ почв), извлечение определяемых
компонентов из почв различными вытяжками (при изучении под-
вижных форм соединений элементов и равновесий твердые фазы
почв — раствор), растворение гумусовых веществ' в различных
растворителях.
Техника анализа полученных растворов может быть различной
в зависимости от электрохимических свойств определяемых и со-
путствующих компонентов, от имеющейся в распоряжении иссле-
дователя аппаратуры (см. с. 101).
При определении минеральных компонентов необходимо учи-
тывать, что органические вещества почв, адсорбируясь на поверх-
115
ности ртутного капельного электрода, влияют на величину пре-
дельного тока и измеряемые высоты полярографических волн.
Наиболее резко влияние ПАВ проявляется при анализе низких
концентраций. Поэтому в большинстве методик предусмотрено
предварительное разрушение органических веществ в вытяжках
из почв.
Если с помощью имеющейся полярографической аппаратуры не
удается провести количественный анализ полученного раствора
либо из-за низкой концентрации определяемого элемента, либо
из-за присутствия мешающих элементов, проводят концентриро-
вание определяемых или отделение мешающих элементов. Для
этой цели при анализе почв могут быть использованы осаждение,
соосаждение, хроматография, экстракция, цементация, электролиз.
После соответствующей подготовки пробы к аликвоте получен-
ного анализируемого раствора добавляют определенный объем
фонового электролита; можно также в точно отмеренном объеме
фонового электролита растворить сухой остаток, полученный в
процессе подготовки пробы к анализу. Раствор переносят в элект-
ролизер. В том случае, если концентрацию определяют по калиб-
ровочному графику, в электролизер помещают произвольный объ-
ем раствора, если концентрацию находят методом добавок — точно
отмеренный объем раствора. Если необходимо, из раствора удаля-
ют растворенный кислород химическим методом или пропуская
через раствор инертный газ (водород, азот, аргон).
Кислород восстанавливается на ртутном капельном электроде,
образуя две полярографические волны и мешая проведению ана-
лиза. Из щелочных растворов кислород удаляют, восстанавливая
его сульфитом натрия. С помощью инертных газов кислород можег
быть удален из растворов независимо от их состава.
Затем, подобрав экспериментально чувствительность прибора,
записывают полярограмму. Полярографирование повторяют еще
два раза, при измерении высот волн берут среднее из трех значе-
ний. Полезно начинать работу с полярографирования раствора
фонового электролита.
ЛАБОРАТОРНЫЕ РАБОТЫ
Определение чувствительности прибора
и характеристик капилляра
Качество работы классического полярографа может быть оце-
нено по кривым зависимости величины тока от напряжения. Если
кривые регистрировать, подключив вместо ячейки сопротивление,
которое является проводником первого рода, то зависимость бу-
дет выражаться прямой линией, в соответствии с законом Ома.
Проверка работы полярографа. Вместо полярографической
ячейки к клеммам прибора ЛП-60 подключают магазин сопротив-
116
лений. Зависимость ток — напряжение записывают при разных
сопротивлениях внешней цепи и различных положениях редуктора
чувствительности прибора.
В каждой серии опытов, например сопротивление 10 000 Ом
и чувствительности 1/70, 1/100, 1/150, 1/200, запись начинают из
одной точки на диаграммной ленте. При правильной работе при-
бора будет получена серия прямых линий.
Определение чувствительности прибора по току. К полярогра-
фу ЛП-60 вместо ячейки подключают магазин сопротивлений и
ставят сопротивление, равное 50 000 0м. При положении редукто-
ра чувствительности 1/100 измеряют отклонение стрелки самопис-
ца при изменении напряжения от 0 до 0,4 В. Записывают
показание прибора X в миллиметрах, поданное на реохорд напря-
жение Ei=0,4 В, сопротивление jRi и — положение редукто-
ра чувствительности. Изменяют сопротивление в два раза, увели-
чивают (или уменьшают) подаваемое на реохорд напряжение до
тех пор, пока стрелка не вернется в прежнее положение или,
другими словами, пока ток не достигнет прежнего значения. За-
писывают величины Е2 и R2 и рассчитывают величину тока, вызы-
вающую отклонение стрелки самописца на 1 мм, или цену 1 мм
шкалы при полной чувствительности прибора:
i° =
(Jh-EJR
(Ri—R2)X
Л/мм.
Чтобы вычислить по полученным результатам абсолютную ве-
личину предельного тока, достаточно умножить приведенную вы-
соту волны на цену' деления:
inP = (i° Л/мм) (й мм).
Определение характеристик капилляра. Период капания ртути
и скорость вытекания ртути из капилляра, входят в уравнение
Ильковича. Эти показатели необходимы при расчете концентра-
ции электрохимически активного вещества по уравнению Илько-
вича.
В электролизер помещают бидистиллированную воду, поднима-
ют резервуар со ртутью и на электроды подают напряжение
—0,5 В. С помощью секундомера измеряют период капания ртути.
Затем определяют массу ртути, вытекающую из капилляра в еди-
ницу времени. Для этого в электролизер, содержащий только би-
дистиллированную воду, собирают 10—20 капель ртути. Вылива-
ют воду, сушат электролизер и ртуть фильтровальной бумагой,
взвешивают электролизер со ртутью. Выливают ртуть в специаль-
ную склянку, электролизер взвешивают и по разности находят
массу ртути.
Рассчитывают т, т и константу тУ3 г'1,.
117
Определение концентрации
никеля и цинка
по уравнению Ильковича
Одним из способов определения концентрации вещества в
растворе является расчет по уравнению Ильковича. В этом случае
необходимо знать характеристики капилляра: период капания
ртути (т, с); массу ртути, вытекающую из капилляра в единицу
времени (т, мг/с); чувствительность прибора по току (Г, А/мм);
* коэффициент диффузии ве-
Рис. 39. Полярографические вол-
ны никеля и цинка на фоне
0,05 М KNaC4H4O6 и 0,1 М KCNS
щества (D, см2 с— ) и чис-
ло электронов, участвую-
щих в электрохимической
реакции (п).
Определение цинка и ни-
келя проводят на фоне рас-
твора, содержащего сегне-
тову соль и роданистый ка-
лий. Коэффициент диффу-
зии иона никеля принимают
равным 0,69-IO-5 см2 с1,
иона цинка — 0,72-10~&
см2 с-1. Электрохимическое
восстановление ионов нике-
ля и цинка протекает с уча-
стием двух электронов.
Сначала готовят фоно-
вый раствор для этого в
мерную колбу емкостью
100 мл помещают 10 мл 1 М раствора сегнетовой соли (калия —
натрия виннокислого) и 10 мл 2 М раствора роданида калия,
объем жидкости в колбе доводят до метки бидистиллированной
водой. При проведении анализа этот раствор разбавляется
в два раза. На фоне 0,05 М раствора сегнетовой соли и 0,1 М
KCNS никель и цинк восстанавливаются в интервале потенциалов
от —0,2 до —1,6 В относительно нас. к. э. В этом же интервале
потенциалов восстанавливается кислород, образуя две полярогра-
фические волны при потенциалах около —0,2 и 1,1 В. Эти волны
мешают определению никеля и цинка.
При выполнении лабораторной работы полярографируют фо-
новый раствор до и после удаления кислорода. Для этого в элект-
ролизер помещают 2 мл приготовленного раствора фона и 2 мл
бидистиллированной воды, подбирают чувствительность и раствор
полярографируют от —0,2 до 1,6 В. Затем через раствор 5 мин
пропускают инертный газ — водород, который удаляет из раствора
растворенный кислород, и раствор снова полярографируют. Поля-
рограмму снимают дважды, при той же и более высокой чувстви-
тельности прибора.
118
Водород для удаления кислорода подводят к электролизеру
от генератора водорода СГС-2 по резиновым трубкам. Действие
генератора основано на электролизе 25%-ного раствора КОН. плот-
ностью 1,14—1,24 г/см2 при 20°. Конструкция генератора предус-
матривает контроль и автоматическое регулирование давления
водорода 1,1 атм. Включение и выключение генератора произво-
дятся строго по инструкции, прилагаемой к прибору.
Для определения цинка и никеля в электролизер приливают
2 мл фонового раствора и 2 мл анализируемого раствора, содержа-
щего цинк и никель. Затем через раствор 5 мин пропускают водо-
род. После этого снимают полярограмму от потенциала —0,2 В до
1,6 В.
Первая волна на полярограмме (рис. 39) обусловлена восста-
новлением никеля (£1/2 =0,73 В), вторая волна соответствует
восстановлению цинка (£1/2 = 1,08 В). Меняя чувствительность,
снимают отдельно волны N1 и Zn в трех повторностях. Измеряют
высоты волн для каждой полярограммы и рассчитывают приве-
денные 'высоты волн h=(hi:fi) мм, где h — приведенная высота
волны элемента в мм, hi — высота волны в мм, fi — чувствитель-
ность полярографа, при которой шла съемка. Затем, зная чувст-
вительность прибора по току, находят величину среднего предель-
ного диффузионного тока. Для этого приведенную высоту волны
умножают на чувствительность прибора по току:
inp (мк Л) = h (мм) • i° (Л/мм) 106.
Далее по уравнению Ильковича (с. 87) рассчитывают концент-
рацию никеля и цинка в полярографируемом растворе.
Например, в опыте было найдено, что т=3с; i°=3,6-10~10
А/мм; т=2,3мг/с. Высота волны Ni при Д=-^- равна 34 мм, при
fn = ---70 мм при /щ = -----101 мм. Средняя приведенная
высота волны h равна 696 мм, тогда концентрация никеля в поля-
рографируемом растворе равна
„ 696-3,6-10~10-106 п лоо ,
CNi = -------------------По-----57ч-Пк— ~ 0,038 ммоль/л.
605-2-(0,69- 10-Б)1/2-(2,3)2/3-3|/б
Концентрация анализируемого раствора в два раза выше, по-
скольку он разбавлялся фоном в два раза.
Определение емкости
катионного обмена почв
При оценке емкости катионного обмена почв (ЕКО) поляро-
графический метод используют для количественного определения
насыщающего катиона. Применение полярографии позволяет
119
\
уменьшить навески почв, расход реактивов, но не вносит принци-
пиальных изменений в методику изучения почвенного поглощаю-
щего комплекса. В вариантах полярографического метода опре-
деления ЕКО, предложенных И. Л. Абарбарчуком и Е. М. Скоб-
цом, в качестве насыщающего катиона используют Ва2+, Mn2J_,
Ni2+, Cd2+, Pb2+.
Определение ЕКО в некарбонатных почвах по Ni2+. Из лабора-
торной пробы почвы, пропущенной через сито с диаметром отвер-
стий 1 мм, на аналитических весах в стаканах емкостью 50 мл
берут навеску почвы 0,5 г. В стаканы приливают 10—15 мл 0,5 н.
раствора NiCl2 (pH 6—6,5), суспензию тщательно перемешивают
стеклянной палочкой и содержимое стакана фильтруют. Почву
количественно переносят на фильтр раствором NiCl2. Насыщение
почвы никелем продолжают, обрабатывая почву на фильтре 12—
15 раз раствором NiCh порциями по 10 мл. Раствор NiCl2 при-
ливают после того, как предыдущая порция раствора полностью
профильтруется. При полном насыщении поглощающего комплек-
са почвы никелем pH фильтрата должен соответствовать pH ис-
ходного раствора NiCl2.
Затем из почвы удаляют избыток Ni2+, промывая ее ди-
стиллированной водой до потери реакции на Ni2+ (проба с диме-
тилглиоксимом в присутствии ацетата натрия) или на С1~ (проба
с AgNO3). Если в трубке воронки появится мутный раствор, про-
мывание прекращают. Затем поглощенный почвой Ni2+ вытесня-
ют, обрабатывая почву на фильтре 60—70 мл 0,5 н. НС1. Фильтрат
собирают в мерные колбы емкостью 100 мл. В колбы добавляют
NHaOH до слабощелочной реакции и объем жидкости доводят
водой до метки. Содержимое колб тщательно перемешивают.
После того как выпавший осадок гидроксидов осядет на дно
колбы, 10 мл прозрачного раствора помещают в электролизер,
в раствор с помощью шпателя добавляют небольшое количество
сульфита натрия, содержимое электролизера перемешивают и по-
лярографируют от потенциала —0,7 В. Затем в электролизер до-
бавляют 0,3 мл 0,05 н. раствора NiCl2 и снова полярографируют.
Измеряют высоты волн и рассчитывают содержание никеля в ана-
лизируемом растворе, как это принято в методе добавок (с. 105).
Зная навеску почвы, объшм солянокислого раствора и концен-
трацию никеля в нем, рассчитывают величину ЕКО.
Определение кальция в почвах
Ион кальция восстанавливается на ртутном капельном элект-
роде в области потенциалов от —2,0 до 2,2 В на фоне гидроксид-
тетраметиламмония N(CH3)4OH или хлорида тетраметиламмония
N(CH3)4C1. В той же области потенциалов восстанавливаются К1',
Li+ и Na+. Это осложняет прямое полярографическое определение
кальция в почвах, природных водах и образцах растений. Поэтому
120
были предложены косвенные методы определения кальция, в част-
ности с применением ЭДТА.
Методика полярографического определения кальция с примене-
нием ЭДТА описана Р. Пршибилом, а затем применена
Е. М. Скобцом для анализа почв. Метод основан на способности
кальция количественно вытеснять цинк из комплексоната цинка
в сильноаммиачной среде. Если обозначить двухзарядный ион
этилендиаминтетрауксусной кислоты символом НгУ2-, то образо-
вание комплексоната цинка можно представить по схеме:
Н2У2" ! Zn2+->-ZnY2- . 2Н+.
Константа устойчивости комплексоната цинка (Ig KznY2—
= 16,6) значительно выше константы устойчивости комплексоната.
кальция (Ig KcaY2-= 10,7), поэтому полное количественное вы-
теснение цинка из комплекса происходит только в среде с кон-
центрацией NH4OH не менее, чем 4 М.
Вытесненное из комплекса количество цинка эквивалентно
содержанию кальция в анализируемом растворе и легко может
быть определено полярографически. Определению мешает магний,
поскольку последний реагирует с комплексонатом цинка анало-
гично кальцию. Поэтому магний удаляют из раствора осаждением
в виде MgNH4PO4.
Раствор комплексоната цинка готовят растворением 1,9170 г
ZnSO4-7H2O и 2,4800 г комплексона III в 1 л 8,0 М NH4OH. При
полярографировании разбавленного 1 : 1 раствора получают хоро-
шо выраженную волну цинка. Для построения градуировочного
графика берут разность высот волн цинка в смеси испытуемого
раствора с раствором комплексоната цинка и разбавленном 1 : 1
растворе комплексоната цинка.
Выполнение анализа сводится к следующему. В электролизер
наливают 2 мл раствора комп'тексоната цинка, добавляют 2 мл
бидистиллированной воды, 0,2 мл 70%-ного раствора (Тх’^ДНРСД,
5 капель 0,25%-ного раствора желатина и 5 мин пропускают
инертный газ. Затем раствор полярографируют, съемку повторяют
еще два раза при других положениях редуктора чувствительности.
Те же операции проводят со стандартными растворами, содержа-
щими 0,4; 0,8; 1,2; 1,4; 1,6ммоль/л кальция, добавляя их в электро-
лизер вместо воды. По этим данным строят градуировочный гра-
фик в координатах: приращение высоты волны цинка, пропорци-
ональное содержанию кальция, — концентрация кальция в поля-
рографируемом растворе. Затем готовят вытяжку из почвы и
таким же образом снимают полярограммы исследуемых раство-
ров, содержащих кальций. Если растворы содержат много каль-
ция. их надо разбавить, чтобы концентрация кальция не превыша-
ла 1,6 ммоль/л. По градуировочному графику находят концентра-
цию кальция в исследуемом растворе. Этим методом удобно
определять в почвах содержание обменного и водорастворимою
кальция.
121
Для определения обменного Са2+ берут навеску почвы 5 г, по-
мещают ее в колбу емкостью 100 мл, приливают 50 мл 1 н. раство-
ра NH4C1 и взбалтывают на ротаторе в течение 30 мин. Суспензию
фильтруют и в фильтрате определяют содержание кальция.
Определение железа в почвах
Полярографическим методом железо может быть определено
по волнам восстановления Fe (III) и Fe (II) и по волне окисле-
ния Fe (II).
В отсутствие комплексообразующих веществ восстановление
Fe (III) начинается при нулевом значении потенциала. Однако
Ре(Ш)
Fe(H)
Рис 40. Анодно-катодная
полярографическая волна
Fe (II)—Fe (III) на фоне
оксалата калия
для определения железа, как правило,
применяют фоновые электролиты, содер-
жащие комплексообразующие реагенты,
которые сдвигают волну восстановле-
ния железа в область более отрица-
тельных потенциалов. Полярографи-
ческим методом можно раздельно опре-
делить Fe (III) и Fe (II). Разные ва-
лентные формы элементов определяют
- методом классической полярографии,
которая позволяет различать направле-
ние тока: катодный ток, возникающий в
результате электрохимического восста-
новления вещества на электроде, и
анодный ток, имеющий противоположное
направление и обусловленный окисле-
нием электрохимически активных ве-
ществ. Разные валентные формы желе-
за определяют на фоне оксалата калия,
Fe (III) и Fe (II) образуют анодно-
катодную волну с потенциалом полувол-
ны —0,24 В относительно нас. к. э. Кон-
центрацию Fe (III) находят по катодно-
му участку волны, лежащему выше ну-
левой линии регистрирующего прибора,
а концентрацию Fe (II)—по анодному
участку, лежащему ниже нулевой линии (рис. 40). По суммар-
ной высоте волны определяют общее содержание железа в раст-
воре.
Определение Fe (HI) и Fe (II) в почвах. При определении раз-
ных валентных форм железа в почвах необходимо соблюдать ус-
ловия, при которых соотношение Fe (III)/Fe (II) не будет менять-
ся в течение анализа. Отношение Fe (Ш) к Fe (II) наиболее
устойчиво в 1 н. растворе H2SO4. Оксалат-ион способствует окис-
лению железа (II), поэтому К2С2О4 добавляют в сернокислый
полярографируемый раствор после удаления из него кислорода,
122
непосредственно перед полярографированием. Соединения железа
извлекают из почвы 1 н. раствором H2SO4. Сернокислые вытяжки
из почв необходимо анализировать сразу же после их получения.
При хранении вытяжек происходит окисление двухвалентного
железа; например, отношение Fe (III) /Fe (II) при суточном вы-
держивании 1 н. Н28О4-вытяжки из дерново-глеевой почвы увели-
чилось в четыре раза.
Содержание железа находят по калибровочному графику. Так
как окисление Fe (II) и восстановление Fe (III) происходят с
участием одного электрона, определение разных валентных форм
железа проводят по одному и тому же калибровочному графику.
Для его построения могут быть использованы стандартные раст-
воры с любым соотношением Fe (III) и Fe (II).
Определение Fe fill) и Fe (II) в 1 н. Н25О4-вытяжках из почв.
На технических весах взвешивают 5 г почвы в состоянии естест-
венной влажности и помещают в колбу емкостью 50 мл. В колбу
приливают 50 мл 1 н. H2SO4, закрывают пробкой и взбалтывают
2ч. Суспензию фильтруют через рыхлый фильтр и 5мл фильтрата
помещают в электролизер. Через раствор в электролизере 5 мин
пропускают водород, добавляют около 1 г оксалата калия и снова
15 мин пропускают водород. Затем раствор полярографируют от
+0,15 В. Содержание железа находят по калибровочному графи-
ку. Для его построения полярографируют растворы с концентра-
цией железа 5; 10; 25; 50; 75 мкг/мл.
Определение никеля и цинка в почвах
Никель и цинк образуют полярографические волны на фоне
разнообразных по составу электролитов. При анализе почв никель
определяют как по диффузионным токам, так и по каталитичес-
ким волнам, которые возникают в присутствии диметилглиоксима.
Для определения цинка в почвах предложены методы инверсион-
ной полярографии.
При анализе почв и растений никель и цинк часто определяют
на фоне NH4C1 и NH4OH; никель и цинк присутствуют в растворе
в форме аммиакатов и образуют хорошо выраженные полярогра-
фические волны. Кобальт может мешать определению цинка, так
как восстанавливается в той же области потенциалов.
Я. Г. Баркан с сотрудниками при анализе солянокислых вытя-
жек из почв определял медь, никель, цинк и марганец на хлорид-
но-аммиачном фоне методом переменно-токовой полярографии.
Н. А. Чеботарева и О. И. Локалина показали, что методом клас-
сической полярографии на фоне NH4C1, NH4OH удается получить
удовлетворительные результаты при определении никеля и цинка.
Определение никеля и цинка в 1 н. HCl-вытяжках из почв.
К 5 г почвы приливают 25 мл 1 н. НС1, взбалтывают 30 -мин и
суспензию фильтруют. К 8 мл фильтрата приливают 4 мл разбав-
123
ленного 1 : 1 раствора аммиака и на кончике шпателя добавляют
сульфит натрия для восстановления растворенного кислорода.
Через 3—5 мин осадок отфильтровывают, фильтрат собирают в
электролизер и полярографируют от потенциала —0,7 В. Одновре-
менно к другой порции вытяжки объемом 8 мл добавляют 0,1 мл
стандартного раствора, содержащего по 100 мкг/мл никеля и цин-
ка, содержимое стаканчика перемешивают, добавляют аммиак и
сульфит натрия, осадок отфильтровывают, а фильтрат полярогра-
фируют. Измерив высоты волн никеля и цинка на полярограммах
исследуемых вытяжек и вытяжек с добавкой стандартного раство-
ра по формуле (с. 105), рассчитывают концентрацию никеля и
цинка в полярографируемом растворе.
Определение свинца в почвах
Свинец образует хорошо выраженные полярографические вол-
ны на фоне кислых, нейтральных и щелочных растворов. Методом
классической полярографии свинец в почвах, как правило, опре-
деляют после предварительного концентрирования. Для опреде-
ления свинца в почвах и других природных объектах используют
инверсионную полярографию. Переменно-токовая полярография
позволяет определять общее содержание и содержание подвиж-
ных форм соединений свинца в почвах без применения концент-
рирования.
Определение свинца в почвах методом переменно-токовой по-
лярографии. Этим методом свинец в почвах определяют на фоне
3 н. НС1 и аскорбиновой кислоты. Аскорбиновая кислота восста-
навливает железо (III) и растворенный кислород, что позволяет
проводить анализ без удаления кислорода инертным газом. На
фоне 3 н. НС1 свинец восстанавливается обратимо с участием двух
электронов (см. рис. 37).
На высоту пика свинца влияют некоторые сопутствующие эле-
менты. Медь, в количествах превышающих свинец более чем в
50 раз, уменьшает пик свинца и искажает его форму. Таллий,
хотя и восстанавливается при том же потенциале, что и свинец,
при равном содержании на пик свинца пе влияет, так как восста-
навливается с участием одного электрона. Увеличивает пик свинца
олово, но оно удаляется в- форме тетрахлорида при выпаривании
растворов, содержащих НС1, в процессе подготовки пробы к ана-
лизу. Железо, алюминий, титан, марганец, ванадий, хром, в коли-
чествах значительно превышающих среднее содержание элемен-
тов в почвах, не влияют на пик свинца.
Методика определения общего содержания
свинца в почвах. Навеску почвы около 1 г прокаливают, раз-
лагают фтористоводородной кислотой в присутствии серной кисло-
ты, сухой остаток растворяют в 1 н. НС1, раствор фильтруют через
плотный фильтр в мерную колбу емкостью 100 мл, фильтр и пла-
124
типовую чашку промывают подогретым раствором HCI до отри-
цательной реакции на железо. Объем жидкости в колбе доводят
до метки 1 н. HCI.
Аликвоту раствора (15 мл) помещают в кварцевую чашку, до-
бавляют 0,5 мл концентрированной HNO3 и 1,5 мл концентриро-
ванной НС1 и выпаривают на водяной бане досуха. К остатку при-
ливают 3 мл концентрированной НС1, 2—3 капли Н2О2 и содержи-
мое чашек выпаривают досуха; эту операцию повторяют еще раз.
К сухому остатку приливают 15 мл 3 н. НС1, содержимое чашек
перемешивают круговыми движениями и добавляют около 0,25 г
аскорбиновой кислоты. Спустя 5 мин аликвоту раствора помещают
в электролизер и полярографируют от потенциала —0,38 В отно-
сительно ртутного дна в следующем режиме работы полярографа
ППТ-1: амплитуда переменного напряжения — 4 мВ, скорость
развертки — 2,5 мВ/с, период капания — 3,5 с, время задержки —
2,5 с.
Раствор полярографируют еще два раза и добавляют в элект-
ролизер стандартный раствор с содержанием свинца 2,5-10-4 мг/
/мл с таким расчетом, чтобы высота пика увеличилась приблизи-
тельно в два раза. Концентрацию свинца находят, как это принято
в методе добавок.
Методика определения подвижных форм со-
единений свинца. Аликвоту солянокислой или ацетатно-ам-
монийной вытяжки из почв (15 мл) помещают в кварцевую чаш-
ку, добавляют 0,5 мл концентрированной HNO3, 1,5 мл концентри-
рованной НС1 и выпаривают досуха на водяной бане, чтобы раз-
рушить перешедшее в вытяжку органическое вещество почв. Затем
остаток обрабатывают НС1 в присутствии Н2О2 и полярографиру-
ют, как указано в методике для определения валового содержания1
свинца в почвах.
ЛИТЕРАТУРА
Виноградова Е. Н., Г а л л ай 3. А., Финогенова 3. М. Методы поля-
рографического и амперометрического анализа. М., Изд-во Моск, ун-та,
1963.
Воробьева Л. А., Орлов Д. С Полярографические методы исследования'
почв. М., Изд-во Моск, ун-та, 1972.
М а й р а н о в с к ий С Г., С т р а д ы н ь Я- П„ Бе з у г л ы й В. Д. Полярогра-
фия в органической химии. Л., «Химия», 1975.
Глава 4
СПЕКТРОФОТОМЕТРИЧЕСКОЕ
ИЗУЧЕНИЕ СОСТАВА
И СВОЙСТВ ПОЧВЕННЫХ КОМПОНЕНТОВ
Оптические методы исследования природных объектов дают
разностороннюю и обширную информацию о составе и свойствах
почв и почвенных компонентов, они позволяют решать широкий
круг вопросов в почвоведении. Оптические методы основаны на
в з а и модействии спета и вещества Наиболее часто~~в~-анал'ит1тчет>-
кой практике используют следующие виды взаимодействия: погло-
щение CBeia, отражение света, рассеяние, преломление, вращение
плоскости поляризации.
^Спектрофонтмегр^. юснована на .явлениях поглощения и от-
ражения—саутаГ^&Веювой (видимый глазом) диапазон электро-
магнитных колеоаний сравнительно узок и лежит в интервале
длин волн 400—750 нм. Кроме видимой области спектра в спект-
рофотометрической анализе широко используется ультрафиолето-
вая область (УФ) спектра от 200 до 400 нм и область инфракрас-
ного излучения (ИК) от 1—2 до 20—25 мк. Общие законы вза-
имодействия электромагнитных колебаний и вещества применимы
не только к видимой, но и к УФ- и ИК-областям. Однако характер
взаимодействия и конкретные механизмы изменяются в зависи-
мости от длины волны.
Абсорбционная спектрофотометрия, устанавливающая зависи-
мость м ежду погл ощением свег а и концентр ацией вещества, ш и -
роко’ттутймснястся для количественных определений в почвахУрас-
тениях, природных водах различных макроэлементовП(Ы, Р7С, А1,
Fe, Са, Mg, Si) и микроэлементов (Си, Мп, Со, Mg, Zn, В). Метод
позволяет вести определение в растворе нескольких элементов по
смешанной окраске.
Абсорбционная спектрофотометрия широко используется так-
же для определения гумусовых веществ почвы — гуминовых и
фульвокислот, а также неспецифических органических соедине-
ний — аминокисло!» пуриновых оснований, хлорофилла, фракции
Pg-
126
Спектрофотометрические исследования в ультрафиолетовой и
инфракрасной областях спектра дают возможность решать вопро-
сы о строении органических и минеральных веществ в почве.
Изучение спектров отражения почв позволяет объективно оце-
нивать окраску почв, распределение основных почвенных компо-
нентов в почвенном профиле, а также быстро проводить качест-
венные и полуколичественные определения ряда минеральных и
органических компонентов почвы.
Взаимодействие света и вещества
Поглощение света веществом сопровождается процессами, при-
водящими к изменению энергии атомов и молекул. Световая энер-
гия—частично переходит гГтёплсвую энергию, а частично расхо-
дуется на~изменение~внутрвИнёиГэнергии молекул. При этом воз-
можньГ'слёдуюгцие процессы: 1) изменение вращательного дви-
жения молекул и отдельных групп атомов внутри молекулы;
2) изменение характера колебаний ядер атомов в молекуле;
3) изменение характера колебаний электронов по отношению к
ядру.
Частота электромагнитных колебаний не зависит от среды,
в которой распространяется излучение, и выражается числом ко-
лебаний, совершающихся в одну секунду. Обозначается частота
символом v и измеряется в с1. В спектрометрии редко пользуют-
ся значениями частоты, так как она имеет очень большие числен-
ные значения. Гораздо чаще вместо частот пользуются длинами......
волн и волновыми числами. Водное число показывает число волн,
укладывающихся на 1 см. Обозначается вол'повое число симво-
лом v и выражается в см-1. Длину волны К видимой и ультра-
фиолетовой областей выражают в нанометрах (1нм=10-9м) или
в ангстремах (1 А= 10“’° м); в инфракрасной области принято
выражать длины волн в микрометрах (1 мкм=10~6м). Если длины
волн выражены в нанометрах, то волновое число v = —— 107 см-1.
При поглощении света веществом внутренняя энергия атома
или молекулы повышается'от нормального уровня Ео до более
высокого уровня Е|, отвсчающето'возбуждснному состоянию. Воз-
буждённоесостояниЕ-а том а или молекулы неустойчиво и длится
около 10~8с, после чего атом или молекула переходит в нормаль-
ное состояние, выделяя избыток энергии в виде_элрктромагнитно-
го колебания. Поглощение и выделение энергии происходят пор-
циями, или кванта_ми /1у. Уравнение, связывающее изменение энер-
гии атома при "сто возбуждении с частотой или длиной волны,
называется условием частот Бора:
Е± — Ео = h v =
127
где v — частота электромагнитных колебаний, h — постоянная
Планка, равная 6,62-10~27 эрг/с, К — длина волны, С — скорость
света, равная 3-1010 см/с (или 3-1017 нм/с).
Переход молекулы или атома под действием световой энергии
из основного состояния в возбужденное может быть обусловлен
поглощением кванта света с различной частотой. Наиболее проч-
ные связи в молекулах возбуждаются при воздействии электро-
магнитных колебаний с высокой частотой — далекая ультрафио-
летовая область спектра (200—300 нм). Энергий квантов видимой
и ближней ультрафиолетовой областей спектра (300—750 нм) спо-
собна переводить в возбужденное состояние электроны, связываю-
щие атомы в молекуле. Излучение менее высоких частот (следова-
тельно, кванты меньшей величины) способно изменить энергию
колебания атомов в молекуле (ближняя инфракрасная область
800—20000 нм). Наконец, кванты малой величины, поглощенные
веществом, вызывают вращательное движение молекулы, это на-
блюдается при поглощении далекой инфракрасной области. По-
глощение инфракрасного участка спектра имеет большее значение
для изучения строения вещества и в меньшей степени использует-
ся для аналитических задач. Энергия квантов ультрафиолетового
излучения настолько велика, что часто вызывает фотохимические
реакции.
Понятие «спектр поглощения»
Спектром поглощения вещества называют сложную кривую
зависимости энергии светопоглощения от длины волны. Макси-
мальное поглощение электромагнитных колебаний веществом про-
исходит только при некоторых значениях длин волн. При других
длинах волн поглощение ослаблено или вовсе не происходит. По-
следовательное расположение по длинам
.Рис. 41. Спектр светопоглощения пер-
манганат-иона, Z2—Zi — величина полу-
ширины полосы поглощения в нм
волн участков интенсив-
ного поглощения и участков,
где световая энергия не пог-
лощается или поглощается
незначительно, образует
спектр поглощения вещества
(рис. 41). Спектры поглоще-
ния обычно изображают гра-
фически: по оси абсцисс от-
кладывают длину волны, по
оси ординат -— уменьшение
энергии светового потока.
Уменьшение энергии света
можно выражать различными
способами: в виде величин оп-
тической плотности D, пропус-
кания Т, величины молярного
коэффициента погашения в к
128
(см. с 131). Длина волны, при которой светопоглощение до-
стигает наибольшей величины, обозначается Z.MaKr. Участки с по-
вышенным поглощением световой энергии образуют полосы погло-
щения, положение которых на шкале длин волн и интенсивность
являются основой соответственно для качественного и количест-
венного спектрофотометрического анализа.
Для характеристики полосы поглощения пользуются понятием
полуширины полосы, которая равна интервалу длин волн Zi — л2,
соответствующему половине максимальной оптической плотности
полосы в целом (см. рис. 41). В большинстве случаев для моле-
кулы с одной хромофорной группой полуширина полосы поглоще-
ния в видимой области составляет 80—100 нм.
Количественные закономерности
поглощения света веществом
Закон Бугера—Бера
Количественные закономерности поглощения света веществом
выражаются законом Бугера—Бера для „монохроматического из-
лучения. Закон может быть выражен в экспоненциальнои“’шпГ
логарифмической форме: -6 0
/z = /0.io~evC/, ~
4Hg-^=Ez.C-/, Jo п - -
где /0 и /( — поток энергии электромагнитного излучения соответ-
ственно до и после прохождения через раствор; С — концентра-
ция поглощающего свет вещества в растворе (или в той среде,
через которую проходит излучение); / — толщина поглощающего
слоя (ширина кюветы); ю. — коэффициент погашения, завися-
щий от длины волны и природы вещества; индекс 7. указывает
длину волны электромагнитного колебания.
Отношение энергии электромагнитного колебания, прошедшего
через раствор, к энергии первоначального потока называют про-
пусканием (или прозрачностью) и обозначают Т. Та же величина,
выраженная в процентах, называется коэффициентом пропуска-
ния:
т =г и т % = — • 100.
1« 10
Отрицательный логарифм пропускания называют оптической
плотностью и обозначают О:
D = — \gT = ^С4.
5 Зак 284
129
Оптическая плотность прямо пропорциональна концентраций
вещества в растворе и толщине поглощающего слоя. Для моно-
хроматического потока света закон Бугера—Бера выражается
jp’ ! прямой линией, проходящей че-
\] у рез начало координат, выража-
/ У— а ющих зависимость Z)==f(C)
// (рис. 42).
Как видно из уравнения, вели-
чина оптической плотности без-
размерна. Ширину кювет обычно
выражают в см. 'Если концентра-
- ция выражена в молях на литр, то
/ ех называют молярным ко-
ZAd эффициентом погашения или мо-
V-—1лярным коэффициентом экстинк-
ции и он имеет размерность
Рис 42. Кривая зависимости оп- л/моль-см.
тической плотности D от концен- Молярный коэффициент погаше-
о-при подаинении = закону Бу- НВВ ~ - Равен 0ПТИ~
гера—Бера, б — при неподчине- ческой ^плотности раствора с кон-
нии закону Бугера—Бера; а— цсйтрацисй 1 моль/л при ТОЛЩИНе
угол наклона прямой поглощающего слоя в 1 см.
В тех случаях, когда концентрация раствора выражена в про-
центах, вмеЁтсГ величины ел, используют величину удельной экс-
тинкиии Ei'cm (или Е-величину), численно равную оптическойГ
плотности 1%-ного" "раствора” при ширине поглощающего слоя
в 1 см. Величину Е можно вычислить для растворов другой кон-
центрации — 0,1% или 0,01%. В этом случае в индексе указывает-
ся концентрация, при которой определялась оптическая плотность:
. г-0,01 % г/
например, Ei см -
Если в растворе находится смесь соединений, подчиняющихся
закону Бугера — Бера и не вступающих между собой в химиче-
скую реакцию, то оптическая плотность смеси будет равна сумме
оптических плотностей этих соединений:
2 Е> = e1C,-Z | е? -С2 / К ... е?"
где е?., Ех. ... , Ех —"эделярные коэффициенты погашения компо-
нентов раствора при длине волны X, а Сь С2, ..., Сп — их концен-
трации. Это уравнение носит название закона Фирордта или за-
кона аддитивности оптических плотностей. Если для одного из
компонентов смеси закон Бугера—Бера не выполняется, уравне-
ние Фирордта будет справедливо дрлько для смеси с постоянной
концентрацией данного компонента.
Закон этот имеет большое практическое значение, так как он
лежит в основе спектрофотометрического анализа многокомпо-
нентных смесей.
130
Отклонения
от закона Бугера—Бера
Причины отклонения от закона Бугера—Бера можно разделить
на две группы: 1) химические и фимвд^имииеские^-обусдовлеа-
ные свойствами раствора или анализируемого вещества; 2) ин-
струментальные, .зависящие-рт используемого прибора.
Химические и физико-химические причины отклонения вызва-
ны главным образом реакциями ионизации, полимеризации, асссь.
циации вещества в растворе'. 'Вследствие этих процессов величина
концентрации" окрашенного компонента может не соответствовать
заданной концентрации вещества в растворе. Это несоответствие
можно устранить путем подбора оптимальных условий, при кото-
рых указанные явления лишь минимально влияют на состояние
вещества.
Одной из причин отклонения от закона Бугера—Бера может
быть также явление флуоресценции вещества, которое приводит
к положительному отклонению оптической плотности от линейной
зависимости от концентрации (см. рис. 42).
К инструу1£цхаль.цым причинам относится любая неисправность
фотоэлектрического устройства. Эти причины легко обнаружить
и исправить. Более важной причиной является немонохроматич-
ность светового потока, падающего на анализируемый раствор,
а также присутствие рассеянного света. Влияние рассеянного све-
та возрастает на тех участках спектра, где мала эмиссия источ-
ника излучения. В наибольшей степени возможно присутствие
рассеянного света в УФ-области (190—220 нм) В этой области
рассеянный свет может даже вызвать ложные максимумы; анало-
гичный эффект возможен при изучении систем с высокой оптичес-
кой плотностью растворителя.
Молярный коэффициент погашения
и предел обнаружения
окрашенного компонента
_Молярный коэффициент погашения характеризует внутренние
индивидуальные свойства поглощающего свет вещества. Его абсо-
лютная""" величина зависит от молекулярного строения вещества,
длины волны поглощаемого излучения;- тсмпературьГ._ Всличипу
молярного коэффициента погашения в максимум’е' палосы погло-
щения обозначают ехмакс, и у различных веществ она колеблет-
ся в широких пределах. Наименьшие абсолютные величины
ех-маКс в пределах нескольких единиц л/моль-см свойственны
окрашенным ионам и аквакомплексам металлов, например Си (II),
Fe (III), Ni (II). Комплексные ионы металлов имеют е^макс
5*
131
порядка 102 — 103 л/моль-см. Наибольшие величины елмакс свой-
ственны комплексным соединениям металлов с органическими
лигандами — п-104 — п-105 л/моль-см1. Таким образом, величина
е х позволяет дать сравнительную характеристику предела обна-
Рис. 43. Приложение закона Буге-
ра—Бера к растворам гумата нат-
рия:
а — оптическая плотность раствора
гумата натрия и величины е , б •—
условия количественных определений
в различных участках спектра
ружения веществ фотометриче-
ским методом.
Чувствительность фотометри-
ческой реакции можно выразить
величиной угла наклона графика
зависимости D от С. Тангенс
угла наклона этого графика ра-
вен молярному коэффициенту по-
гашения, умноженному на тол-
щину поглощающего слоя:
ел -/ = tga. При /=1 см — ел =
= tga. Пользуясь этим соотно-
шением, можно графически по-
казать необходимость монохро-
матизации света при количест-
венных определениях. В качест-
ве примера приведены два гра-
фика (рис. 43,а и б). На рис.
43, а показана кривая светопо-
глощения раствора гумата нат-
рия. Если толщина слоя раство-
ра 1 см, а концентрация
1 г-моль/л, то вертикальные от-
резки, опущенные из любой точки
этой кривой на ось абсцисс, рав-
ны молярному коэффициенту по-
гашения при данной длине вол-
ны. Если построить гра-
фик зависимости D от С для двух длин волн
Xi и Ха, то получим две прямые, причем lgai = £.?.l >tga2=eA2
(см. рис. 43,6). Прямая с большим углом наклона (т. е. обуслов-
ленная большей величиной молярного коэффициента погашения)
позволяем с большей точностью учитывать величины D и обеспе-
чивает меньший предел обнаружения.
Численную величину ел можно определить экспериментально
толькр при высокой монохроматичности светового потока — при
работе на спектрофотометрах (см. с. 146). Для этого берут
йеск^/ько растворов определяемого вещества с концентрацией,
выраженной в молях на литр. При постоянной величине I измеря-
ют их оптические плотности в области Хмакс- Расчет ведут по
1 В''дальнейшем в тексте будут даны только абсолютные величины молярных
коэффициентов погашения без указания их размерности.
132
формуле ex = Рмакс-, а затем находят среднее значение. Если
при изменении концентрации определяемого вещества величина
е? непостоянна, то это означает, что в растворе идут побочные
реакции, в результате которых происходит изменение структуры
вещества, и эти процессы приводят к нарушению линейной зави-
симости D от С.
Зная величину молярного коэффициента погашения, можно
рассчитать предел обнаружения фотометрического метода для
анализируемого вещества. Минимальная концентрация, которую
можно найти, определяется уравнением
н ^мин • (®^макс ^маЛс)‘
Минимальная оптическая плотность, которую можно измерить,
на фотоэлектроколориметрах — 0,05—0,02, на спектрофотомет-
рах — 0,01. Максимальная длина кюветы 5—10 см. Подставляя
эти величины в уравнение, получим:
С = °’01 _ 0.001
МИН- -10 ~
макс “макс
Пользуясь этой формулой, можно рассчитать предел обнару-
жения любого спектрофотометрического метода, например опре-
деления алюминия с применением хромазурола-S; молярный ко-
эффициент погашения в максимуме полосы поглощения комплекса
алюминия с хромазуролом-S равен 5-104, атомная масса алю-
миния — 26,98.
0,01-26,98-1000 а лплг/1 /
С„,,„ = ~1---------= 0,00054 мкг/мл;
w н 50 000-10
ч
коэффициент 1000 приведен для пересчета величины, на мкг/мл.
Не всегда целесообразно для фотометрического определения
применять методы с наименьшим пределом обнаружения. Напри-
мер, при высоком содержании элемента в образце метод с малым
пределом обнаружения может дать ошибки за счет сильного раз-
бавления раствора для анализа. Применению метода может пре-
пятствовать также присутствие сопутствующих компонентов, ме-
шающих определению.
Количественный анализ
вещества
Чтобы определить концентрацию вещества в растворе, измеря-
ют его оптическую плотность при определенной длине волны.
Обязательным условием определения концентрации фотометри-
ческим методом является идентичность спектров анализируемого
133
и стандартного вещества. При анализе соблюдают следующий
порядок работы.
1. Выбирают оптимальную длину волны. Выбор X должен обес-
печить наименьшую величину «предела обнаружения» и хо-
рошую воспроизводимость определения. Для выбора длины
волны снимают спектр определяемого соединения и находят
интервал для волн, в котором величины ех или D имеют
наибольшие значения. В тех случаях, когда соединение име-
ет несколько максимумов поглощения при разных длинах
волн, рекомендуется использовать максимум с большей по-
лушириной полосы поглощения в более длинноволновой об-
ласти спектра; последнее условие снижает влияние рассеян-
ного света при работе на приборе.
2. Выбирают кювету для определения оптической плотности.
При выборе необходимо придерживаться допустимого рабо-
чего интервала оптических плотностей (см. с. 137).
3. Готовят серию стандартных растворов определяемого соеди-
нения с известной концентрацией — калибровочную шкалу.
Интервал концентраций калибровочной шкалы должен пе-
рекрывать диапазон концентраций анализируемых раство-
ров, но не выходить из пределов подчинения закону Бугера—
Бера. Далее, измеряют оптические плотности этих растворов
относительно растворителя или «нулевого» раствора, т. е.
раствора, в который введены все реактивы, кроме опреде-
ляемого вещества, пользуясь кюветами с одной шириной
поглощающего слоя. По величинам оптических плотностей
стандартных растворов строят график зависимости D от С.
4 Измеряют оптическую плотность анализируемого раствора
(в кювете той же ширины) относительно растворителя или
«нулевого» раствора и по графику находят его концентрацию.
5. Зная концентрацию анализируемого раствора (например,
А1 в мг/100мл), рассчитывают содержание элемента в образ-
це в процентах по формуле
А1% = . C-IV100 . .
1<2-навеска-1000
где С — концентрация алюминия в мг/100 мл, рассчитанная
по графику; Vi — общий объем раствора; V2 — аликвота,
взятая для фотометрического определения; навеска образца
в граммах; 100 — коэффициент для пересчета на 100 г об-
разца; 1000 — коэффициент' пересчета миллиграммов в
граммы.
6. Если известна величина ех, то концентрацию можно рас-
считать по формуле С = —-у , не прибегая к калибровоч-
еХ‘
ному графику.
134
Анализ двухкомпонентных смесей
Рис 44. Спектры поглощения ме-
тилового оранжевого, R-, его со-
пряженной кислоты — HR и их
смеси:
/-Си =100%, Син = 0; 2 —
Сии=100%, Си =0; 3 —Си =
=35%, Син=65%
на шкале длин
находится достаточно
соединения. В этом
колебаний только одним
Присутствие в растворе двух окрашенных компонентов (двух
окрашенных комплексов или окрашенного комплекса и избытка
окрашенного реагента), не реагирующих между собой, но погло-
щающих свет в различных областях спектра, приводит к образо-
ванию смешанной окраски. В спектре смеси полосы индивидуаль-
ных компонентов могут лежать в различных интервалах длин волн,
но могут образовывать и одну
сложную полосу (рис. 44). Макси-
мумы поглощения света при этом
могут быть смещены. При рас-
смотрении двухкомпонентной смеси
можно выделить несколько случаев.
1. Спектры компонентов не пе-
рекрываются, и максимумы
полос не смещаются; случай,
когда полоса определяемого
компонента
волн
далеко от полосы сопутству-
ющего
случае фотометрическое оп-
ределение обоих компонен-
тов возможно без их предва-
рительного разделения.
2. Перекрывание и смещение
полос происходят, но не во
всех областях спектра. На
кривой светопоглощения име-
ется участок, где происходит
поглощение электромагнитных
компонентом. Практически количественное определение это-
го компонента возможно, если расстояние между максиму-
мами поглощения этих двух компонентов будет больше или
равно 100 нм.
3. Полное перекрывание спектров поглощения и искажение
формы кривой при сложении спектров происходит, если рас-
стояние между максимумами поглощения света обоих ком-
понентов менее 100 нм. В этом случае количественное опре-
деление одного из компонентов возможно только после пред-
варительного разделения компонентов или маскирования
примеси. Однако если известны величины молярных коэф-
фициентов погашения компонентов смеси, для ее анализа
можно применить расчетный метод.
Первый этап работы при анализе двухкомпонентных систем —
изучение спектродш1С7Ъ1х_са£Д2щх^1нй_и 1-щ£месей. На основании
изучения спектров выбирают аналшшческнедлинй волн. Для оп-
135
ределения оцтических плотностей смеси необходима высокая мд-
н охремдт и з_а цид^ИЗ^чд^ м^котбр а я позволяет использовать узкий
участок спектра. Количественный анализ состава раствора со сме-
шанной окраской основан на следующем расчете.
Если раствор содержит два компонента Л и В, то по принципу
аддитивности (см. с. 130) оптические плотности смеси этих двух
компонентов при двух длинах волн Xi и 7.2 будут равны:
I
I
I
(
ef -Св-1,
Г*2
где Сл и Св — концентрации компонентов; е^, и е®, е®, —
их молярные коэффициенты погашения при длинах волн У., и /.2;
I — ширина кюветы (постоянная). После решения системы двух
уравнений относительно СА и Св формулы для вычисления кон-
центраций имеют следующий вид.
п ~ ez, п ~ ех. ’
с а —- 1 ---—----——Св — -—• ———-—-———
-е?—е?)-/ (е^1 е?—ef-e?)-l
Определение концентраций будет тем точнее, чем больше разность
отношений молярных коэффициентов погашения компонентов, изме-
ренных при и ?.2: (еД/е£ —«£/<) или (</< — е£/8*).
В тех случаях, когда в спектре имеется область, в которой
поглощает свет только один компонент, расчет двухкомпонентной
системы упрощается, так как г области 7.2 величина =о.
Концентпации компонентов в таком случае рассчитывают по фор-
мулам:
Если спектры не перекрываются и для каждого из компонен-
тов существуют полосы индивидуального поглощения света, то
анализ такой системы сводится к определению каждого из компо-
нентов по уравнениям.
рв -I
Сл I
Л2
Ошибки при спектрофотометрических
измерениях
Ошибки при определении концентрации, а также величин е?.
или Е-величин могут быть обусловлены как систематическими,
так и случайными причинами
136
При фотометрических определениях систематические ошибки
могут быть вызваны несколькими причинами, например ошибкой
при нахождении величины е?,, связанной’со смещением Хмакс при
отклонении от закона Бугера — Бера. При измерении оптической
плотности в коротковолновой области спектра систематические
ошибки могут возникать за счет большой доли рассеянного света
в монохроматическом пучке. Систематические ошибки могут быть
связаны и с5неточной_калибров_кой прибора, на котором ведут из-
мерение. Проверку калибровки шкал спектрофотометров лучше
всего проводить по спектру излучения ртутной лампы (в области
250—1100 нм). Контроль шкалы длин волн иногда проводят с по-
мощью растворов неорганических соединений (0,4%-ного КгСгО4
в 0,05 н. КОН или 0,006006%-кого К2СГ2О7 в 0,01 н. H2SO4).
Воспроизводимость фотометрических измерений также зависит
от многих причин. Возможны погрешности за *счет_приготовления
стандартных п анализируемых растворов, влияния мутности, флуо-
ресценции, присутствия примесей. Неодинаковые положения кю-
вет в кюветодержателе, чистота кювет, различная толщина пог-
лощающего слоя приводят к кюветным ошибкам. Часто погреш-
ности бывают вызваны загрязнением реактивов. Эту погрешность
легко учесть путем измерения оптической плотности анализируе-
мого раствора относительно раствора холостого опыта. Случайный
характер могут иметь погрешности при установлении длины_волны_
на приборе, при настройке^ прибора на «нуль», при отсчете на
шкале потенциометра.
Воспроизводимость полученных данных в большой степени за-
висит также от правильно выбранного интервала оптических плот-
ностей при проведении измерений. Связано это с тем, что при
малых величинах оптической плотности относительная ошибка
измерения резко возрастает. Вследствие этого при малых концен-
трациях вещества в растворе воспроизводимость измерений ста-
новится невысокой. Оптимальный интервал для измерения опти-
ческой плотности выбирают таким образом, чтобы во всем интер-
вале измерении стандартное отклонение не превышало удвоенного
минимального стандартного отклонения. По литературным дан-
ным, для приборов с оптической компенсацией величина оптималь-
ной оптической плотности (£>Опт), удовлетворяющая этому требо-
ванию, равна 0,43 (7’опт=30)8%), а оптимальный рабочий интер-
вал лежит в области оптических плотностей от 0,2 до 0,7. При
малых значениях оптической плотности (£)<0,1) или больших
(£>> 1,2) ошибка измерения резко возрастает.
СПОСОБЫ КОЛИЧЕСТВЕННОГО АНАЛИЗА.
АППАРАТУРА
В основе количественного фотометрического анализа лежит
определение ослабления светового потока после прохождения чс-
137
рез раствор. Поэтому фотометрический анализ состоит из двух
операций: первая — переведение вещества в соединение, погло-
щающее свет, вторая — измерение его светопоглощения. Способы
измерения могут быть визуальные и фотоэлектрические. Закон Бу-
гера—Бера, связывающий оптическую плотность раствора с его
концентрацией, является основой для количественного определе-
ния вещества как в визуальных, так и фотоэлектрических методах.
Монохроматизация света, необходимая для спектрофотометричес-
ких определений, расширяет возможности и визуальных опреде-
лений.
Светофильтры, монохроматоры, кюветы
Рис. 45. Оптическая схема характе-
ристики светофильтра (по Полуэк-
тову)
Для выделения из светового потока излучения с определенной
шириной спектрального интервала служат монохроматоры и све-
тофильтры. Идеальное монохроматическое излучение получить
невозможно, но можно из линейчатого или сплошного спектра
выделить более или менее узкий интервал длин волн ДХ=Л2—Ц.
Светофильтры. Цвет светофильтра обычно совпадает с тем
участком спектра, который этот светофильтр пропускает. Свето-
фильтры широко применяются
почти во всех фотометрических
приборах; главным образом ис-
пользуются полосовые или моно-
хроматические абсорбционные
светофильтры, которые сильно
поглощают весь, спектр за иск- _
лючением одной или нескольких
полос пропускания. "В качестве
светофильтров такого рода ис-
пользуют твердые окрашенные
среды: окрашенные стекла, стек-
ла с прослойкой желатина, окра-
шенного органическими красите-
лями. Вместо светофильтров
можно использовать окрашенные
растворы различных веществ,
налитые в кюветы и поставлен-
ные перед кюветой с анализиру-
емым раствором и растворителем. Более высокая монохроматиза-
ция света достигается интерференционными светофильтрами.
Оптическая характеристика светофильтра характеризуется ве-
личиной пропускания света в .максимуме пропускания Тмакс и
полушириной пропускания 7.2— М прй~ 1/2 Тыаис (рис. 45). Чем
меньше абсолютная величина полуширины пропускания, тем более
узкий участок спектра можно выделить этим светофильтром. По-
луширина пропускания светофильтров у фотоэлектроколориметров
модели ФЭК-М в пределе 100 нм; в приборах более поздней кон-
138
струкции ФЭК-56, ФЭК-60, в которые вмонтированы интерферен-
ционные светофильтры, она не выше 20—40 нм.
Выбранный для работы светофильтр должен пропускать лучи
-.ой области спектра, которую наиболее полно поглощает анали-
зируемое вещество. Неправильно выбранный светофильтр снижает
воспроизводимость и правильность определений. В тех случаях,
когда спектральная характеристика определяемого вещества не-
известна, светофильтр подбирают эмпирическим путем. Для этого
измеряют оптическую плотность анализируемого вещества со все-
ми имеющимися в приборе светофильтрами. Для работы использу-
ют гот светофильтр, при котором оптическая плотность будет наи-
большая. Более быстрый, но менее точный способ выбора свето-
фильтра основан на предварительной визуальной констатации
цвета раствора: цвет выбранного светофильтра должен быть до-
полнительным к цвету фотометрируемого вещества.
Применение светофильтров повышает чувствительность опре-
деления лишь до определенного предела концентрации раствора,
различной в каждом конкретном случае. Это свойство связано с
высокой полушириной пропускания светофильтров, благодаря
чему участки спектра, «оптически не реагирующие» с окрашенным
раствором, создают «фон». Влияние этого фона в большей степе-
ни сказывается при высокой концентрации раствора, что приводит
к отклонению от линейной зависимости между D и С. Это явление
может не наблюдаться при фотометрировании этих же растворов
на спектрофотометрах с высокой монохроматизацией излучения.
Монохроматоры. Монохроматоры построены на принципе дис-
персии света, т. е. разложения белого излучения на монохрома-
тические составляющие с помощью призм или дифракционных
решеток. Ширина выделяемого спектрального интервала зависит
от конструкции прибора и в первую очередь от дисперсии призмы
и ширины щели, которая ограничивает выходящий пучок моно-
хроматического излучения. В приборах СФ-8, СФ-18 цена деления
. шкалы длин волн достигает соответственно 0,2 нм и 0,6 нм.
* Различные участки спектра диспергируются призмами, изготов-
ленными из различных материалов: для видимой области спектра
используются стеклянные призмы, для ультрафиолетовой области—
кварцевые, для инфпакрасной — призмы из кристаллов солей
NaCl, КВг, КС1, LiF.'
Кюветы. В комплекте с приборами всегда имеются наборы кю-
вет. Кюветы изготовляются различной формы и различной шири-
ны поглощающего слоя (от 0,2 до 100 мм). Точная ширина погло-
щающего слоя (длина кюветы) обозначена на ее рабочей грани,
которую при работе располагают перпендикулярно направлению
светового потока в приборе. Крепление кюветы в держателе долж-
но обеспечивать неподвижность и точную воспроизводимость уста-
новки кювет. При работе с испаряющимися жидкостями кюветы
закрывают крышками из шлифованного стекла, которые приложе-
ны к набору кювет.
139
Для фотометрирования в видимой области спектра кюветы
изготовляют из шлифованного оптического стекла, для ультра-
фиолетовой области — из шлифованного кварца, для инфракрас-
ной области используют разборные кюветы из шлифованных соле-
вых пластинок NaCl или КВг.
Стенки кювет должны быть чистыми и прозрачными. Перед
работой и после работы их необходимо промыть, ополоснуть ди-
стиллированной водой и обтереть снаружи фильтровальной бума-
гой. Если кюветы сильно загрязнены, их моют концентрированной
НС1 пли HNO3, а затем тщательно промывают дистиллированной
водой. Внутреннюю поверхность кювет сушат только при работе
с органическими растворителями, не смешивающимися с водой.
В других случаях кюветы перед работой ополаскивают тем раст-
вором, оптическую плотность которого будут измерять.
Л/ Подбор кювет для работ проводят следующим образом: в одну
из кювет набора наливают раствор средней концентрации (из
серии приготовленных анализируемых растворов) и измеряют его
оптическую плотность с предварительно выбранным светофильт--]
ром. Если оптическая плотность лежит в пределе 0,3—0,5, кювету
оставляют для работы. Если измеренная оптическая плотность
выходит из этого интервала, то берут кювету большей или мепь-^
шей длины.
Визуальные методы измерения
интенсивности окраски
Визуальные методы основаны на сравнении или уравнивании
окрасок стандартного и испытуемого растворов. Количественную
оценку степени различия или сходства окрасок эти методы не
дают. Уравнивания окрасок можно добиться несколькими путями:
1) изменением концентрации раствора (метод стандартных серий,
метод разбавления, колориметрическое титрование); 2) изменением
толщины поглощающего слоя (колориметрия); 3) изменением ин-
тенсивности светового потока (путем диафрагмирования). Не все
визуальные методы в одинаковой степени применяются в анали-
тической практике.
Метод стандартных серий. Метод заключается в сравнивании
интенсивностей окрасок стандартных и испытуемых раствороз.
В серию однотипных измерительных пробирок (цилиндров или
мерных колб) приливают возрастающие количества стандартного
раствора определяемого компонента. В такую же пробирку поме-
щают аликвоту анализируемого раствора. Во всех пробирках про-
водят реакцию возбуждения окраски и уравнивают объемы раст-,
воров. После этого сравнивают окраску серии стандартных раст-
воров (шкалу) с окраской испытуемого раствора; подбирают
такой стандартный раствор, который имеет одинаковую интенсив-
ность окраски с испытуемым раствором. Концентрацию испытуе-
140
мого раствора считают равной концентрации данного раствора
шкалы.
Метод прост в исполнении, не требует приборов. Для работы
необходим только набор хорошей химической посуды. Применяет-
ся метод для массовых однотипных анализов в полевых агрохими-
ческих лабораториях. Часто в качестве стандартных шкал исполь-
зуют устойчивые растворы солей, анилиновых красителей, имити-
рующих окраску определяемого соединения. Такие шкалы пред-
ложены для определения кремневой кислоты, pH почвенных
вытяжек, подвижных форм соединений фосфора, меди, цинка,
марганца, бора, кобальта в почвах.
Метод уравнивания поглощающего слоя. Метод можно исполь-
зовать только для растворов, подчиняющихся закону Бугера —
Бера. Для определения концентрации с помощью этого метода
применяют колориметры погружения К0Л-1М, микроколориметр
КОЛ-52, иногда одну из первых моделей — колориметр Дюбоска.
Приборы К0Л-1М и КОЛ-52 снабжены набором светофильтров,
поэтому перед работой необходимо выбрать оптимальную длину
волны.
Принцип работы всех колориметров одинаков и основан на
следующем. Если имеются два окрашенных раствора одного и
того же соединения с концентрациями С] (известная) и Сх (не-
известная), то, изменяя величины Ц и 1Х, можно найти такие их
значения, при которых оптические плотности этих растворов ока-
жутся равными:
Dl = е/- Сх • = Dx = • Сх • 1Х.
Так как величина ех для обоих растворов одна и та же, то
Ci •/1 = С\ -/х. Решая равенство относительно Сх, получим
Сх = ^
1х
При работе на колориметрах погружения в две кюветы нали-
вают окрашенные растворы: в одну — стандартный раствор с из-
вестной концентрацией, в другую — анализируемый раствор.
В кювету погружают бесцветные стеклянные цилиндры, через ко-
торые наблюдают окраску растворов. Чем глубже погружен
цилиндр, тем меньше толщина слоя раствора, через который про-
ходит световой поток. Таким образом, перемещение цилиндров
позволяет уравнивать интенсивность наблюдаемых окрасок, а зная
глубину погружения цилиндра, можно рассчитать концентрацию
испытуемого раствора.
Метод диафрагмирования. При визуальном сравнивании окра-
сок метод применяется в фотометрах типа ФМ, в некоторых не-
фелометрах. Метод основан на уравнивании интенсивности двух
световых потоков, прошедших через эталонный и испытуемый
растворы одного и того же окрашенного соединения. Уравнение
потоков достигается гашением части светового потока диафрагма-
141
л
ми, которые соединены с барабанами, градуированными в значе-
ниях D и Т%. Приборы снабжены светофильтрами, поэтому перед
измерением оптической плотности необходимо выбрать длину
волны, при которой значение D максимально.
ФОТОЭЛЕКТРИЧЕСКИЕ МЕТОДЫ
ИЗМЕРЕНИЯ ПОГЛОЩЕНИЯ
Фотоэлектрические методы отличаются от визуальных тем, чго
сравнение интенсивностей световых потоков производится не «на
глаз», а путем преобразования световой энергии в электрическую
с последующим измерением (уравнением) силы тока в цепи фото-
элемента.
Рис. 46. Оптическая схема фотоэлектроколоримстра ФЭК-60:
Л — источник света, Ki, Кг— конденсоры, 3it З2, З3, З4 — зеркала,
01, 02 и 0'1, 0'2 — линзы, П — призма, Ф — фотокатод фотоэле-
мента, Цюв1, К/ов2 — кюветы, Д\ — диафрагма измерительного ба-
рабана, Д2 — диафрагма компенсационного барабана, Ci, С2— све-
тофильтры
В фотометрических приборах применяют вентильные фотоэле-
менты (селеновые, сернисгосеребряные), эмиссионные фотоэле-
менты с внешним фотоэффектом (газонаполненные или вакуум-
ные) и фотоэлементы с внутренним фотоэффектом. С течением
времени спектральная чувствительность фотоэлементов падает.
В этих случаях фотоэлементы необходимо менять. Фотоэлементам
свойственна также «утомляемость», которая сказывается в изме-
142
нении чувствительности при длительном ярком освещении. Чтобы
избежать ошибок за счет утомляемости фотоэлемента, рекомен-
дуется освещать его до начала работы в течение 10—15 мин и
только после этого, когда фототок стабилизируется, приступать
к работе. В ходе работы необходимо периодически проверите
правильность показаний прибора по эталонному раствору.
Фотоэлектроколориметры. Принцип работы фотоэлектроколори-
метров различных моделей одинаков. Он основан на компенсаци-
онном принципе уравнивания интенсивностей двух монохромати-
ческих потоков света с по-
мощью измерительной ще-
левой диафрагмы. Луч све-
та от источника при помо-
щи линз и зеркал (рис. 46)
разделяется на два пучка.
Один световой пучок (пра-
вый от наблюдателя) явля-
ется измерительным, другой
(левый от наблюдателя) —-
компенсационным. В изме-
рительный пучок света поме-
щают последовательно оди-
наковые кюветы: сначала, с
растворителем, затем с раст-
вором, в компенсационный
пучок света — кювету с
растворителем. Световые
потоки, пройдя через свето-
фильтры и кюветы, попада-
ют на фотокатоды фотоэле-
ментов и возбуждают элект-
рический ток, пропорцио-
нальный разности интенсив-
ностей левого и правого по-
токов. О наличии и направ-
Рис 47 Фотоэлектроколориметр ФЭК-60:
1 — нулевой гальванометр, 2 — барабан
измерительной диафрагмы, 3 — барабан
компенсационной диафрагмы, 4 — рукоятка
потенциометра, 5—рукоятка переключения
светофильтров, 6 — отделение для свето-
фильтров, 7 — кнопка для включения вы-
сокой чувствительности, 8 — отделение для
фотоэлементов
лении тока судят по отклонению стрелки гальванометра. Уравни-
вание световых потоков проводят путем диафрагмирования пра-
вого светового пучка. Если потоки выравнены, то гальванометр
прибора показывает «нуль». Диафрагма соединена с измеритель-
ным барабаном, на котором нанесены величины оптической плот-
ности D и величины пропускания Т (в %).
Фотоэлектроколориметр ФЭК-60. Прибор позволяет проводить
измерения в широком диапазоне длин волн от 360 до 980 нм. Ис-
точником света служит лампа накаливания СЦ-61. Приемники
энергии — два сменных фотоэлемента: сурьмяно-цезиевый СЦВ-4
для области 360—620 нм и кислородно-цезиевый ЦВ-4 для области
620—980 нм, которые включены через усилитель. В приборе имеет-
ся 9 пар светофильтров. Светофильтры № 1 и № 2 абсорбцион-
143
ные, оиальные интерференционные Общий вид прибора показан
на рис. 47
Порядок работы на приборе Прибор включают
в сеть, дают прогреться и через 15—20 мин приступают к работе
Для этого сначала проверяют «электрический нуль» закрыв изме-
рительную 2 и компенсационную 3 диафрагмы, проверяют показа
ния на гальванометре 1 Если стрелка отклоняется от нулевого
деления, то рукояткой потенциоиетра 4 подводят стрелку к нуле
вому положению сначала грубо, а затем более точно, при нажа-
тии кнопки 7 После этого приступают к измерению оптической
плотности или пропускания раствора
Измерительный барабан 2 устанавливают на деление 100%
пропускания В правый пучок света (от наблюдателя) помещаю!
кювету с исс щдуемым раствором, в левый помещают кювету с
растворителем Вращением компенсационного барабана (5) ус-
танавливают сгретку гальванометра на путь с точностью ±1 де-
ление Затем нанимают кнопку высокой чувствительности 7 п
более точно подводят стрелку к нулю В правый пучок света по-
мещают, вместо раствора, такую же кювету с растворителем (и щ
с «нулевым» раствором) и, вращая измерительный барабан 2,
доводят стрелку гальванометра снова к нулю, сначала грубо, а за-
тем точно, при нажатии кнопки 7 Величину оптической плотности
или пропускания отсчитывают по шкале измерительного бараба-
на 2
Для определения оптической плотности применяют также
спектроколориметры, у которые вместо светофильтров дифрак-
ционные решетки
Спектроколориметр комбинированный СФК-601. Прибор пред-
назначен д 1я измерения оптической плотности, пропускания, не-
фелометрии, турбодиметрии, флуорометрии жидких сред Рабочий
диапазон прибора от 250 до 800 нм Источники излучения смен-
ные— ртутная лампа Д1М-100-2 и лампа накаливания К12-30-2.
Приемником энергии служит фотоумножитель ФЗУ-4
Спектроколориметр «СПЕКОЛ» (ГДР) СПЕКОЛ — однолуче-
вой прибор с гальванометром для прямого отсчета оптической плот-
ности по отклонению стрелки Рабочий диапазон прибора от 365
до 850 нм Прибор изготовляется в двух вариантах — «Основной
СПЕКОЛ» и «Универсальный СПЕКОЛ» В обоих приборах ис-
точники излучения сменные ртутная лампа и лампа накаливания
Приемниками энергии в «Основном СПЕКОЛЕ» служит селено-
вый фотоэлемент с транзисторным усилителем, в «Универсальном
СПЕКОЛЕ» приемники энергии сменные для области 340-
630 нм — газонаполненная фотоячейка 494-Д для области 630—
850 нм — газонаполненная фотоячейка 494-z/ Фотоэлементы снаб-
жены дополнительным усилителем с переключателем для регулн
ровки чувствительности Преимущество прибора «СПЕКОЛ» в том,
что в комплекте его имеются 16 специальных измерительных пр i-
ставок Они позволяют повышать чувствительность прибора Ме-
144
няя их, можно проводить кроме измерения оптической плотности
и пропускания также измерение помутнения, флуоресценции, от-
ражения, блеска, вести фотометрическое титрование, флуоромет-
рическое титрование и титрование мутности Общий вид прибора
изображен на рис 48
Рис 48 Спектроколориметр «СПЕКОЛ»
1 — шкала отсчетного устройства, 2 — рукоятка для установки прибо-
ра на «нуль» оптической плотности, 3 — рукоятка для установки при-
бора на максимум пропускания (100%), 4 — кюветное отделение, 5 —
переключатель длин волн, 6 — рукоятка шторки фотоэлемента (два
положения), 7 — держатель кювет передвижной
Порядок работы
1 Включить прибор в сеть через стабилизатор напряжения и
дать npoiреться 1 ч
2 Рукояткой (5) установить необходимый диапазон длин волн.
3 Поместить в кюветодержатель (7) кювету с растворителем
и с помощью винтов (2) «нуль» и (5) «100%» установить
стрелку на шкале отсчетного устройства сначала на нуль
оптической плотности, а затем на 100% пропускания Для
этого рукоятку шторки фотоэлементов (6) устанавливают в
положение «0» (шторка закрыта) и, вращая винт (2), уста-
навливают на шкале отсчетного устройства стрелку в по-
ложение «нуль оптической плотности» Затем рукоятку (6)
переключают в положение «1» и, вращая винт (3), перево-
дят стрелку отсчетною устройства в положение 100% про-
пускания
4 В кюветодержатель (7) помещают кювету с окрашенным
раствором (стандартным или анализируемым) и снимают
~ у"”' '',!я показания оптической плот-
145
ности. (Нулевое положение прибора в процессе работы не<
обходимо проверять несколько раз.)
Спектрофотометры для видимой и ультрафиолетовой областей
спектра.
Спектрофотометр для любой области спектра состоит из сле-
дующих важнейших узлов (рис. 49): источник излучения Л, мо-
нохроматор М, приемник излучения Ф, регистрирующее устрой-
ство Р.
М
Рис. 49. Принципиальная схема спектрофотометра:
Л — источник излучения, М — монохроматор, Щ — выходная щель
прибора, Эт — эталонный раствор; Обр — образец, Лин — линзы,
Ф •— приемник излучения, Р — регистрирующее устройство
Промышленность выпускает спектрофотометры, работающие
как в широком диапазоне длин волн от 200 до 1100 нм, так и в
более узкой области спектра (в видимой, инфракрасной или ульт-
рафиолетовой). Приборы бывают с самопишущей автоматической
регистрацией показаний оптической плотности и пропускания при
различных длинах волн, а также с ручной регулировкой показа-
ний на отсчетном устройстве. Ниже дается кр’аткая характеристи-
ка некоторых спектрофотометров отечественного производства и
импортных.
Спектрофотометр СФ-18. Прибор предназначен для измерения
оптической плотности и пропускания прозрачдых--и мутных сред,
а также для измерения коэффициентов диффузного отражения
твердых и порошкообразных веществ. Рабочий диапазон 400—
750 нм. Общий вид прибора изображен на рис. 50.
' Измерение оптической плотности и пропускания на приборе
происходит следующим образом. Два монохроматических потока
света проходят через анализируемый раствор и растворитель, по-
падают в интегрирующую сферу 8, 9, в которой потоки света пре-
терпевают многократное отражение от стенок и попадают на фото-
элемент. Освещенность последнего в каждый момент времени
определяется суммой потоков света, прошедших через анализи-
руемый раствор и растворитель. Если световые потоки равны, то
сигнала в усилительной системе не будет. Если же анализируе-
мый раствор поглощает некоторое количество света, то в усили-
теле появится сигнал. Возникшее в цепи усилителя напряжение
подается на электродвигатель отработки, который с помощью фо-
146
тометрического кулачка открывает диафрагму до тех пор, пока
не исчезнет разность световых потоков. Одновременно подается
сигнал на механизм перемещения пера записывающего устрой-
ства, который фиксирует на бланке величину оптической плот-
ности или пропускания анализируемого раствора.
Порядок работы на приборе. Подключить прибор
к сети, а на пульте управления включить тумблер «сеть» 17. Пр i
этом должна загореться сигнальная лампа 16. Дать прибору про-
греться 30 мин. Включить тумблер «лампа» 13. Для фокусирова-
ния изображения нити лампы служит рукоятка, расположенная
у основания штатива лампы. Выключить тумблер 13. Включить
электродвигатель развертки спектра тумблером «запись» 12 и ус-
тановить на счетчике длин both 11 отсчет 400 нм. Выключить
тумблер 12. Установить необходимую скорость развертки спектра.
Для этого одновременно нажимают две кнопки: 7 — кнопку х-1
или х-3 и кнопку «10» или «90». Сочетание кнопок позволяет по-
лучить следующие скорости:
Сочетание включен-
ных кнопок
Скорость записи,
нм/мин н мм/мин
10 и х—1
10 и х- 3
90 и х—1
90 и х—3
10
30
90
270
Установить бланк на барабане. Для этого, придерживая рукой
барабан, отвернуть кольцо (/), поднять планку 3 и, нажав на
фиксатор 4, установить бланк для записи D или Т. Опустить план-
ам с помощью фиксатора 4, опустить перо 2 и повернуть рукой
барабан до совмещения пера с линией на бланке, соответствующей
длине волны 400 нм. После этого завернуть зажимное кольцо (/).
Вдвинуть до упора рукоятки, расположенные слева от интегри-
рующей сферы.
Запись спектров оптической плотности. Запол-
нить две кюветы растворителем и поставить в держателях в кю-
ветную камеру. Поставить фотометрический кулачок в положение
«Д» или «СМ» (по риске, расположенной на левой стенке при-
бора). Включить тумблеры «лампа» 13 и «отработка» 15. Уста-
новить перо так, чтобы оно совместилось с линией «0» оптической
плотности на бланке. Если перо движется в противоположную
сторону, переключить отработку в положение «обратный ход»
тумблером 14. Включить тумблер «запись» 12 и провести запись
«нулевой линии». После этого выключить тумблеры 13 и 15. Для
возвращения бланка в первоначальное положение включить раз-
вертку спектра тумблером 14, поставив положение отработки
«обратный ход».
В кюветное отделение поставить кювету с анализируемым
раствором. Рукоятку верхнего экрана слева от интегрирующей
147
сферы оттянуть влево от упора. Включить тумблеры 13 и 15.
Когда перо остановится, включить тумблер «запись» 12 и записать
спектр.
Запись спектров отражения. Установить фотометри-
ческий кулачок в положение «Т» или «СМ». Вдвинуть до упора
рукоятки, расположенные слева от сферы. Включить тумблеры
Рис 50 Спектрофотометр СФ-18
1 — зажимное кольцо, 2 — перо, 3—планка для зажима бумажного
бланка, 4 — фиксатор, 5 — кнопка для включения записи спектра по Дли-
нам волн, 6 — кнопка включения записи на определенной длине волны,
7 — кнопки для регулирования скорости записи, 8 — верхние стандарт-
ные отражатели интегрирующей сферы, 9— нижние стандартные отра-
жатели, 10—рукоятка для регулирования усиления, 11 — счетчик длин
воли, 12 — тумблер для включения электродвигателя развертки спектра,
13 — тумблер для включения лампы, 14 — тумблер для включения мото-
ра отработки в положение «обратный ход», 15 — тумблер для включения
мотора отработки, 16— сигнальная лампа, 17 тумблер включения при-
бора в сеть
«лампа» 13 и «отработка» 15. Перо должно перемещаться к линии
100%-ного пропускания, если перо передвигается в противополож-
ную сторону, включить тумблер «обратный ход» 14. Установить
перо на деление 100%-ного пропускания и записать линию
100%-ного пропускания. Выключить тумблеры 13 и 15. Включить
тумблер «обратный ход» 14 и вернуть бланк в первоначальное
положение. Нижнюю рукоятку экрана сферы оттянуть влево до
упора. Твердый образец поместить в держатель и укрепить в пра-
вом окне в нижней части интегрирующей сферы 9. В левом
окне сферы 8 должен находиться стандартный отражатель.
Включить тумблеры 13 и 15. Когда перо остановится, включить
тумблер «запись» 12 и записать спектр отражения.
148
По окончании работы выключить все тумблеры, отключить
прибор от сети. Привести в порядок кюветы и держатели. Снять
бланк.
Спектрофотометр СФ-14. Рабочий диапазон прибора 400—
750 нм. Оптическая схема и принцип проведения измерений те же,
как на спектрофотометре СФ-18. Отличается прибор тем, что у
него отсутствует счетчик длин волн и меньше количество скоростей
развертки спектра.
Спектрофотометр СФ-18-М. Рабочий диапазон прибора 400—
750 нм. Прибор малогабаритный транзисторный. Оптическая схе-
ма и принцип проведения измерений те же, как на спектрофото-
метре СФ-18.
Спектрофотометр СФ-26. Однолучевой прибор; предназначен
для измерения пропускания и оптической плотности жидких и
твердых веществ. Рабочий диапазон 186—1100 нм. Источники све-
та: для области 186—350 нм — дейтериевая лампа ДДС-30, для
области 340—1100 нм — лампа накаливания ОП-ЗЗ-ОЗ. Приемни-
ки энергии: сурьмяно-цезиевый фотоэлемент — для области 186—
600 нм; кислородно-цезиевый фотоэлемент — для области 600—
1100 нм. Отсчет показаний пропускания и оптической плотности
по шкале измерительного прибора с ручной регулировкой.
Измерение оптической плотности на приборе происходит сле-
дующим образом. В монохроматический пучок поочередно вводят
растворитель (или стандартный раствор вещества) и анализируе-
мый раствор. При введении растворителя стрелку измерительного
прибора устанавливают на 100% пропускания путем регулирова-
ния ширины щели. Затем в поток света вводят анализируемый
раствор. Стрелка измерительного прибора отклоняется пропор-
ционально изменению интенсивности потока света. Величины ко-
эффициента пропускания или оптической плотности отсчитываю г
по шкале, отградуированной в процентах пропускания и едини-
цах оптической плотности.
Преимуществом прибора СФ-26 является возможность путем
включения резисторов компенсации растянуть любой 1О°/о-ный
диапазон пропускания на всю шкалу измерительного прибора.
Резисторы компенсации имеют 10 положений, обеспечивающих
работу в диапазонах коэффициента пропускания от 10 до 100%
и от 10 до 0%. Общий вид прибора изображен на рис. 51.
Порядок работы на приборе.
1. Установить в рабочее положение фотоэлементы рукояткой
16 (если положение Ф — сурьмяно-цезиевый, если положе-
ние К — кислородно-цезиевый) и источники излучения со-
ответственно выбранному диапазону измерения. Для вклю-
чения дейтериевой лампы переключатель находится на зад-
ней стенке прибора.
2. Шторку фотоэлементов рукояткой 13 поставить в положение
«закрыто». Рукояткой 11 установить ширину щели прибора
0,1 мм.
149
3. Включить тумблер «сеть» 6, после чего должна загореться
сигнальная лампа «сеть» и сигнальная лампа «Д» 7 или «Н»
8 в соответствии с выбранным источником света.
4. Стабильная работа прибора обеспечивается только через 1 ч
после включения.
5. Выключение прибора производится тумблером «сеть» (6).
6. В кюветную камеру 3 поставить кюветы со стандартным
и анализируемым образцом.
Рис 51 Спектрофотометр СФ-26
1 — отделение для монохроматора. 2 — измерительный прибор со шкалой,
3 — кюветное отделение, в котором расположены держатели кювет, 4 — ка-
мера фотоприемников с усилителем, 5—рукоятка установки Длин волн, 6 —
тумблер «Сеть», 7 — сигнальная лампа «Дейтерий», 8 — сигнальная лампа
«Накал», 9 — переключатель регистров компенсации для растяжки диапазо-
нов (имеет 10 положений), 10 — переключатель шкалы измерений (имеет
4 положения XU Х0,1, х0,01, «калибр»), 11 — рукоятка механизма изме-
нения ширины щели, 12 — рукоятка кюветодержателя, 13 — рукоятка штор-
ки фотоэлементов, 14 — рукоятка установки «нуля», 15— рукоятка включе-
ния прибора в рабочее положение (имеет 4 положения), 16 — переключа-
тель смены фотоэлементов, 17 — шкала длин волн
7. Поставить рукоятку 15 в положение «1» (рабочее положе-
ние). Если поток излучения недостаточен и измеряемый или
стандартный образец сильно поглощают свет, то рукоятку 15
установить в положение «2», «3» или «4» и оставить в этом
положении до конца работы. (Это касается и работы при
измерении с повышенной чувствительностью, которое изло-
жено ниже )
8. Рукояткой 5 установить нужную длину волны. Поворот про-
водить в сторону увеличения длин волн. Если при установке
шкала длин волн повернется на большую величину, то воз-
вращают ее назад на 3—5 нм и снова подводят к нужному
делению.
Измерение коэффициента пропускания и опти-
ческой ПЛОТНОСТ 11
1) перемещая каретку, в которой установлены кюветы, рукоя!-
кой 12 па пути излучения установить растворитель (или
стандартный образец),
150
2) рукояткой 14 установить стрелку измерительного прибора
на «нуль»;
3) рукояткой 10 установить резистор компенсации в положе-
ние «х1»;
4) поставить шторку фотоэлемента в положение «открыто» ру-
кояткой 13;
5) установить стрелку измерительного прибора на деление
100%. Для этого вращают рукоятку 11 механического изме-
нения ширины щели;
6) перемещая каретку кювет, рукояткой 12 устанавливают на
пути излучения анализируемый образец. Снимают на шкале
измерительного прибора показания 7% или D.
Проверка измерения: для этого, перемещая каретку кювет
рукояткой 12, на пути излучения снова ставят растворитель.
Стрелка измерительного прибора должна вернуться к делению
100%.
Измерение с повышенной чувствительностью.
В диапазоне от 10 до 100%:
1) провести последовательно все операции пунктов от 1 до 6
и получить на шкале измерительного прибора отсчет;
2) переключателем компенсатора 9 компенсировать отсчет до
тех пор, пока показания на шкале не станут менее 10% Г;
3) рукоятку 10 установить в положение «х0,1» и снять отсчет
на шкале;
4) для получения величины пропускания снятый отсчет необ-
ходимо умножить на 0,1 и прибавить число процентов, со-
ответствующее положению рукоятки компенсатора 9.
Например: отсчет на шкале прибора 82,6%; положение руко-
ятки компенсатора 40. Следовательно, величина коэффициента
пропускания:
Т = 82,6 -0,1 1-40 = 48,3%,
D = — 1g 0,483 = 0,32.
Измерение с повышенной чувствительностью
в диапазоне от 0 до 10%:
1) провести все операции пунктов от 1 до 4;
2) установить рукоятку 10 в положение «калибр» и установить
на шкале отсчет 100%;
3) ввести в поток света анализируемый образец и компенси-
ровать отсчет рукояткой 9 до тех пор, пока на шкале изме-
рительного прибора показания станут менее 1%;
4) установить рукоятку 10 в положение «х0,01»;
5) отсчет, полученный по шкале измерительного прибора, ум-
ножить на 0,01 и прибавить число процентов, соответствую-
щее положению рукоятки компенсатора 9, умноженное на
0,1.
151
Например: отсчет на шкале прибора 34,9%; положение руко-
ятки компенсатора 20. Следовательно, величина коэффициента
пропускания:
7 = 34,9-0,01 , 20-0,1 = 2,35%,
О = —1g 0.0235 = 1,63.
Спектрофотометр СФД-2. Рабочий диапазон прибора от 220
до 1000 нм. Монохроматизация света осуществляется при помощи
дифракционной решетки. Источник излучения сменный: лампа
накаливания и водородная лампа. Способ отсчета по шкале из-
мерительного прибора с ручной регулировкой.
Спектрофотометр Specord — UVVIS саморегистрирующий
(ГДР). Рабочий диапазон прибора 170—800 нм. Источник излу-
чения сменный: д'ля области 170—530 нм — дейтериевая лампа
(типа D2E), для области 530—800 нм — лампа накаливания.
Запись спектра проводится на регистрационных листах, где по
оси ординат отложена оптическая плотность (и пропускание), по
оси абсцисс — волновые числа.
Спектрофотометр «СПЕКТРОМОМ-203» (ВНР). Рабочий диа-
пазон прибора 190—1100 нм. Источник излучения: для области
190—350 нм — дейтериевая лампа, для области 320—1100 нм —
лампа накаливания. Метод отсчета измерений — по шкале изме-
рительного прибора с ручной регулировкой.
СПЕКТРОФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
ОПРЕДЕЛЕНИЯ ОБЩЕГО СОДЕРЖАНИЯ
ФОСФОРА, ЖЕЛЕЗА
И ОРГАНИЧЕСКОГО УГЛЕРОДА В ПОЧВЕ
В фотометрическом анализе одной из главных операций яв-
ляется получение в растворе окрашенного соединения определяе-
мого элемента. Поэтому для определения валового содержания
в почве фосфора и железа нужно предварительно перевести все
элементы твердой фазы почвы в растворимые соединения. Для
этой цели используют сплавление со смесью карбонатов калия
и натрия или обработку почвы фтористоводородной кислотой.
Затем элементы переводят в раствор, в котором проводят реакцию
образования окрашенного комплекса с определяемым элементом.
Определение общего содержания углерода органических соедине-
ний почвы фотометрическим методом основано на образовании
окрашенного соединения Cr (III) в процессе кипячения образца
почвы с окислительной смесью.
152
Определение общего содержания
фосфора в почве методом
молибдено-фосфорной сини
В кислой среде в присутствии избытка молибдат-ионов орто-
фосфорная кислота образует молибдено-фосфорную гетерополи-
кислоту желтого цвета Н3[Р(МозО10)4]- При добавлении к желто-
му молибдено-фосфорному комплексу аскорбиновой кислоты в
смеси с сурьмяновиннокислым калием (антимонилтартрат калия
К (SbO) С4Н4О6-0,5Н2О) возникает голубая с фиолетовым оттен-
ком окраска восстановленной формы гетерополикислоты. Опти-
мальные условия образования комплекса создаются в среде 0,1—
0,5 н. H2SO4 или НС1. Сурьма в этой реакции не является ката-
лизатором, а входит в состав комплекса в соотношении Sb:P = 2:l.
Устойчивая окраска возникает через 15 мин после введения вос-
становителя и сохраняется не менее 2 ч. Образующаяся «синь»
имеет два максимума поглощения: при 680 и при 882 нм, моляр-
ные коэффициенты погашения их равны 3-104 и 5-104 соответ-
ственно.
Порядок работы. Почву, растертую в пудру, разлагают
сплавлением с углекислыми щелочами, как описано в руководстве
Е. В. Аринушкиной. Аликвоту раствора после отделения кремне-
вой кислоты, соответствующую 30—40 мг почвы и содержащую
до 0,04 мг Р, переносят в мерную колбу емкостью 100 мл. Раствор
разбавляют водой до объема 70—75 мл и медленно, при переме-
шивании круговыми движениями, добавляют 16 мл молибдено-
сурьмяного реактива. Местное пересыщение при введении реакти-
ва может вызвать выпадение осадка гидроксида сурьмы. Колбу
доливают водой до метки, раствор перемешивают и оставляют
на 15 мин. Оптическую плотность измеряют при 880 или 680 нм
относительно нулевого раствора.
Калибровочную шкалу готовят в интервале концентраций от 0
(нулевой раствор) до 0,04 мг Р в 100мл раствора. Для этого
в серию мерных колб емкостью 100 мл наливают 0,0; 0,5; 1,0; 2,0;
4,0; 6,0; 8,0 мл стандартного раствора фосфата с содержанием
0,005 мг/мл Р. Доливают 0,025 н. НС1 до объема 70—75 мл и при
перемешивании приливают 16 мл реактива. Доводят объем водой
до метки, перемешивают и оставляют на 15 мин. Оптическую плот-
ность измеряют, как указано выше при описании метода. По ве-
личинам оптических плотностей стандартных растворов строят
график в координатах D и С. По графику находят концентрацию
фосфора в растворе и рассчитывают содержание фосфора в почве
(см. с. 134).
Реактивы: 1) стандартный раствор фосфата КН2РО4 с содер-
жанием 0,005 мг/мл Р; 2) молибдено-сурьмяный реактив с аскор-
биновой кислотой.
Для приготовления реактива смешивают 200 мл 3%-ного раст-
вора молибденовокислого аммония и 100 мл 0,15%-ного раствора
153
антимонилтартрата калия. К смеси добавляют 500 мл 5 н. раствора
H2SO4 и разбавляют водой до 1 л. Перед работой к литру раст-
вора добавляют 5,3 г сухого препарата аскорбиновой кислоты.
Определение общего содержания
железа в почве
с применением сульфосалициловой кислоты
Сульфосалициловая кислота, в зависимости от реакции среды,
дает окрашенные комплексные соединения с Fe (III) с различным
соотношением металл : реагент. В кислой среде при pH 1—2 об-
разуется соединение с соотношением металл : реагент 1 : 1 красно-
фиолетового цвета с максимумом поглощения при 500 нм и моляр-
ным коэффициентом погашения, равным 2,4-103. Меньшая вели-
чина предела обнаружения реакции в аммиачной среде при pH
8—10. В этих условиях сульфосалициловая кислота образует
комплекс с Fe (Ш), с соотношением металл : реагент 1 : 3 как с
ионами Fe (HI), так и с ионами Fe (II), которые в процессе
реакции окисляются. Молярный коэффициент погашения трисуль-
фосалицилатного комплекса железа при максимуме поглощения
420 нм равен 5,8-103. При pH более 11 соединение разрушается
с выпадением гидроксида железа. Для фотометрического опреде-
ления железа чаще используют трисульфосалицилатный комплекс.
Реакция идет при избытке реагента, и скорость ее возрастает
с увеличением концентрации аммиака. Окраска устойчива во вре-
мени. Закон Бугера—Бера соблюдается в широком интервале
концентраций до 0,75 мг Fe в 100 мл раствора.
Определению мешает высокая концентрация алюминия, каль-
ция и магния, которые образуют бесцветные комплексы с сульфэ-
салициловой кислотой и связывают большие количества реактива.
Мешает также Мп (II), который в аммиачной среде окисляется
до Мп (IV) и выпадает в осадок в виде бурого МпО(ОН)2. Для
устранения влияния марганца в раствор вводят восстановитель —
гидроксиламин солянокислый NH2OH-HC1.
Порядок работы. Навеску почвы, растертую в пудру,
сплавляют с углекислыми щелочами, как описано в руководстве
Е. В. Аринушкиной. Аликвоту фильтрата (5 мл) после отделения
кремневой кислоты переносят в мерную колбу емкостью 100 мл
и разбавляют дистиллированной водой до 30—40 мл. Приливают
10 мл 25%-ного раствора сульфосалициловой кислоты, вводят 0,5 г
кристаллического гидроксиламина солянокислого и перемешивают
до полного растворения кристаллов. Затем, при перемешивании,
добавляют 25%-ный NH4OH до достижения раствором устойчивой
желтой окраски, после чего добавляют еще 1 мл аммиака. Если
при введении сульфосалициловой кислоты в растворе появляется
осадок, то объем сульфосалициловой кислоты нужно увеличить
до исчезновения осадка (объем реагента для всей партии анали-
154
зируемых растворов должен быть одинаков). Объем раствора в
колбе доводят дистиллированной водой до метки, перемешивают
и через 10 мин измеряют оптическую плотность при 420—430 нм
относительно воды.
Калибровочную шкалу готовят в интервале концентраций от
0,05 до 0,5 мг Fe в 100 мл. Для этого в серию мерных колб на
100 мл наливают из бюретки 1,0; 2,0; 4,0; 6,0; 8,0; 10,0 мл стан-
дартного раствора железоаммонийных квасцов с содержанием
0,05 мг Fe в 1 мл. Раствор разбавляют дистиллированной водой
до 30—40 мл и далее проводят псе операции, как изложено выше
при описании метода. По измеренным величинам оптических плот-
ностей стандартных растворов строят график в координатах D—
С. По графику находят концентрацию железа в испытуемом раст-
воре и рассчитывают содержание железа в почве.
Реактивы: 1) стандартный раствор (МНДгЗСД-Рег^ОДз-бНгО
с содержанием 0,05 мг Fe в 1 мл; 2) сульфосалициловая кислота
(2-окси-5-сульфобензойная кислота), 25%-ный водный раствор.
Определение содержания
в почве углерода
органических соединений (гумуса)
Окисление углерода гумуса почвы раствором бихромата калия
в 0,4 н. H2SO4 приводит к образованию в растворе ионов трех-
валентного хрома зеленого цвета в количестве, эквиватентном
окисленному углероду. Максимум поглощения раствора Cr (III)
находится в области 584—594 нм, молярный коэффициент пога-
шения при 590 нм равен 2,0- 103.
Избыток бихромат-иона в этой области практически не по-
глощает свет, так как максимум полосы поглощения Сг2О5Д
находится в ближней ультрафиолетовой области при 350 нм, а в
более длинноволновой области поглощение резко падает, достигая
минимума при 500 нм. Это позволяет, измерив оптическую плот-
ность раствора смеси в области 580—590 нм, определить концент-
рацию трехвалентного хрома и рассчитать содержание углерода.
После проведения реакции окисления гумуса почвы бихрома-
том раствор разбавляют и оставляют на несколько часов. Это
необходимо потому, что в растворе серной кислоты Cr (VI) может
находиться не только в форме бихромат-иона, но также и в фор-
мах Сг3О^~ и Сг4О1з—, которые поглощают в более длинно-
волновой области спектра. При разбавлении раствора происходи г
их деполимеризация до иона Сг2О|~, окраска раствора становит-
ся более устойчивой. Кроме того, в то же время происходит
оседание частичек минерального остатка почвы. Раствор становит-
ся прозрачным и не требуется его фильтрования или центрифуги-
рования.
155
Ход определения углерода органических соединений почвы
включает два этапа: окисление гумуса по методу Тюрина (при
фотометрическом окончании избыток бихромата не влияет на пра-
вильность определения Cr (III), поэтому окислитель можно при-
ливать мерным цилиндром); фотометрирование полученного раст-
вора после разбавления его до определенного объема.
При измерении оптической плотности на спектрофотометрах
(СФ-14, СФ-18) наиболее благоприятна область 588—592 нм, где
на кривой светопоглощения хрома (Ш) имеется небольшой гори-
зонтальный участок. При работе на фотоэлектроколориметрах
можно использовать область длин волн от 588 до 660 нм, учитывая
набор светофильтров прибора: в приборе ФЭК-60 - светофильтр
с эффективной длиной волны в максимуме пропускания 590 нм,
ФЭК-Н-52, ФЭК-Н-54, ФЭК-Н-57 — 610 нм, ФЭК-56 — 597 нм.
После окончания измерения оптической плотности содержание
углерода в почве находят по калибровочному графику.
Порядок работы. Навеску почвы 0,3 г, из которой отобра-
ны корни и которая измельчена до 0,25 мм, помещают в коничес-
кую .кодбу емкостью на 100 мл из термостойкого стекла (эта на-
веска пригодна при содержании в почве от 0,6 до 13% гумуса).
В колбу мерным цилиндром приливают 20 мл 0,4 н. раствора
К.2Сг2О7, приготовленного на серной кислоте в соотношении вод-
ный раствор бихромата : кислота 1:1. Суспензию осторожно пере-
мешивают, колбу закрывают маленькой воронкой и кипятят точно
5 мин, отмечая время с момента закипания. Охлаждают колбу
до комнатной температуры и переносят раствор, вместе с остат-
ком почвы, в мерный цилиндр емкостью на 100 мл с притертой
пробкой. Колбу и воронку ополаскивают несколько раз дистилли-
рованной водой, перенося промывные воды в цилиндр. Объем
раствора в цилиндре доводят до метки, перемешивают и оставля-
ют на ночь. Одновременно ведут холостое определение, которое
проводят через все стадии анализа, но в конические колбы вносят
вместо почвы небольшое количество прокаленных песка или
пемзы.
Прозрачный отстоявшийся раствор переносят в кювету и из-
меряют оптическую плотность на спектрофотометре при 590 нм
относительно холостого раствора.
Калибровочную шкалу готовят из точно, по навеске, приготов-
ленного 0,2000 н. раствора соли Мора и бихромата калия.
Для приготовления шкалы в 6 мерных колб емкостью 100 мл
наливают по 20 мл 0,4 н. окислительной смеси и бюреткой отмеря-
ют 5,0; 10,0; 15,0; 20,0; 25,0; 30,0 мл 0,2 н. раствора соли Мора.
Растворы доливают до метки водой, перемешивают и измеряют их
оптическую плотность при гой длине волны, при которой проводи-
ли измерение при определении гумуса почвы. По величинам опти-
ческих плотностей стандартных растворов строят график в коор-
динатах D—С, считая, что концентрация трехвалентного хрома
в стандартных растворах эквивалентна 1,0; 2,0; 3,0; 4,0; 5,0; 6,0%
156
углерода в почве при условии, что была взята навеска почвы для
определения гумуса точно 0,3000 г.
ОПРЕДЕЛЕНИЕ В ПОЧВАХ
НЕКОТОРЫХ ПОДВИЖНЫХ ФОРМ
СОЕДИНЕНИИ ЭЛЕМЕНТОВ
Подвижные формы соединений элементов извлекаются из поч-
вы с помощью различных химических экстрагентов. Определение
степени подвижности этих элементов при таком способе перевода
их в раствор во многом зависит как от свойств соединений (раст-
воримости. форм связи, степени окисленности элемента и т. п.),
так и от свойств и степени воздействия на почву выбранного экст-
рагента. Элементы, извлекаемые таким способом из почвы, в боль-
шинстве случаев не сохраняют степень окисленности, которую они
имели в естественном состоянии. Однако для некоторых элементов
можно подобрать условия, когда элемент в вытяжке сохраняет
природную степень окисленности. Например, вытяжка 0,1 н. H2SO4
позволяет определить с раствопе раздельно содержание Fe (II)
и Fe (III).
Определение подвижного
двухвалентного железа в почвенной вытяжке
с применением 1,10-фенантролина
Содержание в почве подвижных форм Fe (II) очень мобильно
и тесно связано с pH почвенного раствора, окислительно-восста-
новительными условиями и биохимическими процессами. Наиболь-
шее содержание Fe (II) можно обнаружить в почвах с повышен-
ной увлажненностью и неблагоприятными условиями аэрации.
Изучение динамики содержания Fe (II) в почве позволяет про-
следить направленность почвообразовательного процесса.
Ион двухвалентного железа образует с 1,10-фенантролином
водорастворимое комплексное соединение [Fe(Phen)3]2+ оранже-
во-красного цвета. Максимум полосы поглощения находится при
512 нм, молярный коэффициент погашения равен 1,1 -104, подчине-
ние закону Бугера—Бера соблюдается в интервале концентраций
0,01—0,1 мг Fe в 50 мл раствора. Реакция идет в широком интер-
вале pH — от 2 до 9; оптимальные условия создаются при pH
4—5 и пяти-шестикратном избытке реагента. Устойчивая окраска
появляется через 10 мин и сохраняется очень долго, а железо,
связанное в комплекс, не окисляется кислородом воздуха.
Определению мешает Fe(III), которое необходимо замаскиро-
вать. Для этого используют фторид-ион, который связывает его
в бесцветный комплекс. Избыток фторид-иона, в свою очередь,
157
связывают в комплексный ион BF~ борной кислотой. Для устра-
нения влияния алюминия и титана, гидроксиды которых выпадают
в осадок при pH 5, определение железа ведут .в цитратном или
тартратном буферном растворе.
Порядок работы. 5г почвы с естественной влажностью
взбалтывают 5 мин с 50 мл 0,1 н. H2SO4. Суспензию фильтруют
через сухой беззольный фильтр в сухой приемник. Перед тем как
проводить определение, делают качественную пробу на Fe (II).
Для этого в пробирку помещают 1—2 мл вытяжки, 1 мл цитрата
натрия и 2 капли 1,10-фенантролина. По интенсивности окраски
судят о содержании Fe (II) в вытяжке.
Для определения железа (II) из прозрачного фильтрата берут
аликвоту с содержанием не выше 0,2 мг Fe (II), переносят в мер-
ную колбу емкостью 100 мл и разбавляют водой до 20 мл. В колбу
добавляю! 1 мл 1%-ного раствора NaF, перемешивают и через
1 мин приливают 1 мл 1%-ной Н3ВО3. Через 2 мин добавляют 1 —
2 капли индикатора тимолового синего и нейтрализуют раствор
0,1 н. NaOH до изменения окраски индикатора из красной в жел-
тую (pH 3). Затем, чтобы довести pH до 4,6, приливают 10%-ный
цитрат натрия почти до метки колбы. Добавляют 2 мл 0,5%-ного
раствора 1,10-фенантролина, доводят до метки водой и переме-
шивают. Оптическую плотность измеряют через 10 мин при 510 нм
относительно холостого раствора, в который добавлены все реак-
тивы, кроме железа.
Калибровочную шкалу готовят в интервале концентраций от
0,02 до 0,2 мг Fe в 100 мл раствора. Для этого в серию мерных
колб емкостью 100мл приливают 2,0; 5,0; 10,0; 15,0; 20,0 мл стан-
дартного раствора соли Мора с содержанием 0,01 мг/мл Fe. Раст-
воры доливают водой до 20 мл и далее проводят все операции,
как изложено выше при описании метода. По измеренным вели-
чинам оптических плотностей стандартных растворов строят гра-
фик в координатах D и С. По графику находят концентрацию
железа в растворе и рассчитывают содержание его в почве (см.
<с. 134).
Реактивы:
1) стандартный раствор соли Мора (NHiKSOvFeSO/,-бНгО
с содержанием 0,01 мг/мл Fe;
2) 1,10-фенантролин, 0,5%-ный раствор в 0,1 н. H2SO4.
Определение подвижных форм
железа в вытяжке Тамма
с применением нитрозо-И-соли
Содержание подвижных форм железа в почве тесно связано с
pH и окислительно-восстановительными условиями. К подвижным
формам железа в почве относят аморфные и слабо окристаллизо-
158
ванные водные окисные формы Fe (II) и Fe (III) и соединения
железа с органическим веществом почвы. Часть соединений такого
типа может находиться в почвенном растворе в виде золя, а так-
же истинных растворов, представленных катионами Fe(OH)2+,
Fe(OH)^» молекулами Fe(OH)3 и комплексными соединениями
железа с органическим веществом. Эти соединения переходят в
раствор при обработке почвы буферным раствором Тамма
с pH 3,3.
Нитрозо-К-соль (1-нитрозо-2-гидрокси-3,6-нафталин-динатрий-
сульфонат) реагирует с Fe (II), образуя водорастворимое комп-
лексное соединение темно-зеленого цвета с максимумом полосы
поглощения при 710 им; молярный коэффициент погашения равен
2,2-104. При концентрации Fe (II) ниже 5-10~5М образуется
преимущественно устойчивое соединение с соотношением Fe (II) :
реагент 1 :3. Подчинение закону Бугера—Бера до 100 мкг Fe (II)
в 100 мл раствора. Избыток реактива не мешает определению,
так как в области 710 нм нитрозо-Е-соль не поглощает свет. Об-
разование комплекса идет при широком интервале pH — от 5 до
11. Устойчивая окраска возникает через 30 мин после добавления
всех реактивов и постоянна несколько часов. При pH 10—11 об-
разованию комплекса не мешает высокая концентрация в растворе
щавелевой кислоты. Для перевода соединений Fe (III) в валент-
ную форму Fe (II) применяют гидразинсульфат NH2-NH2-H2SO4.
С нитрозо-Е-солью образуют окрашенные комплексы также Сех
(II), Си (И) и Ni (II), но максимумы поглощения этих комплек-
сов находятся в более коротковолновой области спектра. Кроме
того, содержание подвижных форм этих соединений в почве много
меньше, чем соединений Fe (II) и Fe (Ш).
Порядок работы. Готовят вытяжку Тамма из почвы по
методике, описанной в руководстве Е. В. Аринушкиной (1970).
Из прозрачного фильтрата вытяжки берут пипеткой 1—2 мл,
переносят в мерную колбу емкостью 100 мл и разбавляют дистил-
лированной водой до 20 мл. Добавляют 2 мл 5%-кого раствора
гидразинсульфата и оставляют на 5 мин. Затем приливают 10 мл
раствора нитрозо-Е-соли, 50 мл боратного буферного раствора с
pH 11. Раствор в колбе разбавляют водой до метки, перемешива-
ют и оставляют на 30 мин. Оптическую плотность измеряют при
710 нм (или на ФЭК-60 со светофильтром №6 с максимумом про-
пускания 670 нм) относительно «нулевого раствора», в который
введены все реактивы, кроме Fe (II).
Калибровочную шкалу готовят в интервале концентраций от
10 до 100 мкг Fe (II) в 100 мл раствора. Для этого в серию мер-
ных колб на 100 мл приливают 0,0; 2,0; 6,0; 10,0; 15,0; 20,0 мл
стандартного раствора железа, содержащего 5 мкг/мл Fe. В каж-
дую колбу добавляют по 2 мл раствора гидразинсульфата, урав-
нивают объемы во всех колбах водой и проводят все операции,
изложенные выше при описании метода. По величинам оптических
плотностей стандартных растворов строят график в, координатах
159
D и С. По графику находят концентрацию анализируемого раст-
вора и рассчитывают содержание подвижного железа в почве (см
с. 134).
Реактивы:
1) стандартный раствор соли Мора (NH4)2SO4-FeSO4-6H2O
с содержанием 5 мкг/мл Fe;
2) нитрозо-И-соль, 0,02 М водный раствор. 4,75 г препарата
растворяют в небольшом количестве дистиллированной во-
ды и разбавляют водой до 500 мл (если необходимо, то
перед разбавлением фильтруют);
3) боратный буферный раствор с pH 11. Смешивают равные
объемы (по 500 мл) 0,1 н. раствора Na2B4O?-ЮН2О и 0,1 н.
раствора NaOH. Реакцию проверяют потенциометрически.
Определение подвижного марганца
в почве с применением формальдоксима
Из-за поливалентности марганца динамика его соединений в
почве выражена очень сильно. Изменение гидроморфности и
окислительно-восстановительных условий в почве ведет к измене-
нию форм его соединений. Соединения высоковалентного марганца
Мп (III), Мп (IV) представлены в почвах малорастворимыми
соединениями, окисями и гидроксидами; обменный и водораство
римый марганец представлен Мп (II).
Формальдоксим CH2NOH в щелочной среде образует с ионом
Мп (II) водорастворимое бесцветное соединение. Под действием
кислорода воздуха Мп (II) в комплексе быстро окисляется, и в
растворе образуется комплексное соединение формальдоксима
с Мп (IV) — [Mn(CH2NO)6]2~, окрашенное в винно-красный цвет.
Максимум полосы поглощения комплекса находится при 455 нм,
молярный коэффициент погашения равен 1,12-104, подчинение
закону Бугера—Бера в интервале концентраций от 0,1 до
30 мкг/мл Мп.
Оптимальные условия образования комплекса создаются при
pH 8—10 ч большом избытке реагентов. Очень важна очередность
добавления реактивов — избыток щелочи необходимо добавлять
только после введения в раствор формальдоксима. Устойчивая
окраска возникает через 10 мин после создания щелочной среды.
При определении марганца в почве определению мешает толь-
ко железо. Никель, кобальт, хром и ванадий, которые также об-
разуют окрашенные соединения с реактивом, в почве находятся
в количествах ниже предела обнаружения формальдоксимового
метода. Определению мешают также катионы, образующие осадки
в щелочной среде (Al, Ti, Fe), поэтому их необходимо или уда-
лять из раствора, или (при малом содержании) маскировать
комплексоном III с гидроксиламином солянокислым. Если в почве
много магния, то для создания щелочной среды лучше использо-
160
вать аммонийные буферные смеси, мешающие выпадению гидро-
ксида магния.
Порядок работы (по С. Г. Самохвалову и Н. А. Чебота-
ревой). Навеску почвы в 10 г (измельченной до 1 мм) взбалтывают
с 50 мл 0,1 н. H2SO4 в течение 30 мин. Суспензию фильтруют че-
рез сухой беззольный фильтр в сухой приемник.
Аликвоту фильтрата в 5 мл помещают в мерную колбу ем-
костью 50мл, добавляют 2 мл 10%-ного свежеприготовленного
водного раствора аскорбиновой кислоты, перемешивают и разбав-
ляют раствор до половины колбы. Затем приливают 3 мл формаль-
доксима, 5 мл аммиачного буферного раствора и оставляют на
2 мин. Приливают Змл 0,1 М. раствора комплексона III и Змл
20%-ного гидроксиламина солянокислого для маскирования со-
путствующих элементов, доливают до метки колбы водой и пере-
мешивают. Оптическую плотность измеряют при 455—490 нм от-
носительно воды не позднее, чем через 15 мин после введения
в раствор комплексона.
Калибровочную шкалу готовят в интервале концентраций до
0,3 мг Мп в 50 мл раствора. Для этого в серию мерных колб на
50 мл наливают 0,0; 0,5; 1,0; 1,5; 2,0; 2,5; 3,0 мл стандартного
раствора марганца с содержанием 0,1 мг/мл-Мп. Разбавляют
водой до половины колбы и далее проводят все операции, как
изложено выше при описании метода. По величинам оптических
плотностей стандартных растворов строят график в координатах
D и С, определяют концентрацию испытуемого раствора и рассчи-
тывают содержание подвижного марганца в почве (см. с. 134).
Реактивы:
1) стандартный раствор марганца с содержанием 0,10 мг/мл
Мп. Приготавливают из точно 0,1 н. раствора КМпО4;
2) формальдоксим (свежеприготовленный раствор) — 20 г
кристаллического гидроксиламина солянокислого NH2OH-
НС1 растворяют в 90 мл дистиллированной воды и добав-
ляют 10мл 36%-ного формальдегида (формалина, СН2О);
3) аммиачный буферный раствор — 70 г NH4C1 растворяют в
800 мл 25%-ного NH4OH и доливают водой до 1 л.
Определение подвижного алюминия
в почве с применением хромазурола- S
При обработке почвы растворами нейтральных солей в вытяж-
ку переходит некоторая часть подвижных форм соединений алю-
миния. Содержание обменного алюминия в почве увеличивается
с возрастанием кислотности. По лабораторным данным, из кислых
почв с pH 3,5 в вытяжку 1 н. КС1 переходит до 250 мкг/мл А1,
из менее кислых почв с pH 5,5 — около 0,04 мкг/мл А1.
z Хромазурол-S (натриевая соль 2,"6"-дихлор-5',5-дикарбокси-
3 ,3-диметил-4'-оксифуксон(4)-5"-сульфокислоты) принадлежит к
6 Зак 284
161
трифенилметановым красителям, которые образуют с ионом алю-
миния окрашенные комплексные соединения, находящиеся в вод-
ном растворе в виде золя. Введение защитных коллоидор (поли-
винилового спирта, желатина) стабилизируют эти коллоидные
растворы.
В слабокислой среде хромазурол-S образует с ионом алюми-
ния комплексное соединение сине-фиолетового цвета с максиму-
мом полосы поглощения при 545 нм и молярным коэффициентом
погашения 5-104. Растворы подчиняются закону Бугера—Бера до
концентрации 0,025 мг А1 в 50 мл раствора.
Узкий интервал кислотности, при котором образуются устой-
чивые комплексы с трифенилметановыми красителями, является
недостатком метода. При различных условиях кислотности хром-
азурол-S образует комплексы с различным соотношением алюми-
ний : реагент. При pH 3 образуется комплекс с соотношением 1 : 1,
в интервале pH 4—5,8 — комплекс с соотношением 1:2, при
рН>6 идет разрушение комплекса и интенсивность окраски па-
дает. Это затрудняет проведение реакции и требует обязательного
применения буферных растворов. Для фотометрического опреде-
ления алюминия используется комплекс с соотношением 1 :2. Для
проведения реакции применяют ацетатные буферные растворы с
pH 5,7—5,8. В оптимальных условиях кислотности устойчивая
окраска возникает через 10 мин при комнатной температуре и
остается постоянной до 50 мин.
Определению мешает Fe (III), который образует с хромазуро-
лом-S окрашенное соединение. Для маскирования невысоких кон-
центраций железа (III) применяют восстановители: аскорбиновую
кислоту или гидроксиламин солянокислый.
Порядок работы. 40г почвы, просеянной через сито с от-
верстиями в 1 мм (или в состоянии полевой влажности), помеща-
ют в плоскодонную колбу и встряхивают 1 ч с 100 мл 1 н. КС1.
Вытяжку фильтруют через сухой фильтр в сухой приемник.
Аликвоту вытяжки (не более 25 мл) с содержанием алюминия
менее 25 мкг помещают в мерную колбу емкостью 50 мл. В колбу
приливают последовательно, при перемешивании, 0,5 мл 1 % -ного
свежеприготовленного раствора аскорбиновой кислоты, через 3—-
4 мин 2 мл 2%-ного поливинилового спирта, 5 мл 0,04%-ного раст-
вора хромазурола-S, приготовленного на буферном растворе с
pH 6. Разбавляют водой до метки колбы и оставляют на 10—
15 мин. Оптическую плотность измеряют при 545 нм относительно
нулевого раствора.
Калибровочную шкалу готовят в интервале концентраций до
25 мкг А1 в 50 мл раствора. Для этого в серию мерных колб
емкостью 50мл приливают 0,0 (нулевой раствор); 1,0; 2,0; 3,0;
4,0; 5,0; 6,0 мл стандартного раствора алюминия с содержанием
4 мкг/мл AJ Растворы во всех колбах уравнивают 1 н. КС1 до
объема взятой аликвоты почвенной вытяжки. Затем проводят все
операции, как изложено выше при описании метода. По величи-
162
нам оптических плотностей стандартных растворов строят график
зависимости D от С. По графику находят концентрацию испытуе-
мого раствора, а затем рассчитывают содержание подвижного
алюминия в почве (см. с. 134).
Реактивы.
1) стандартный раствор алюминия с содержанием 4 мкг/мл А1;
2) ацетатный буферный раствор с pH 6 — 326 г CH3COONa-
ЗН2О растворяют в дистиллированной воде и разбавляют
водой до 800 мл. В этот раствор вливают 42 мл 2,5 н.
СНзСООН;
3) хромазурол-S 1 — 0,1%-ный в буферном растворе с pH 6
(запасной) Приготовленный раствор оставляют на сутки,
а затем фильтруют. Хранят запасной раствор в темном
месте. Рабочий раствор реагента (0,04%-ный) готовят раз-
бавлением запасного дистиллированной водой. После раз-
бавления pH должен остаться равным 6,0.
Определение в почве подвижного магния
с применением титанового желтого
В верхнем горизонте незасоленных почв содержание магния
может колебаться от 0,5 до 2,4%. Большое количество магния
почвы принадлежит к легкоподвижным, обменным формам, пере-
ходящим в раствор при обработке почвы растворами нейтральных
солей.
В разбавленных щелочных растворах гидроксид магния обра-
зует с титановым желтым адсорбционное соединение розового
цвета. Введение защитного коллоида (гуммиарабика, поливини-
лового спирта) стабилизирует образующийся золь Максимум по-
лосы поглощения окрашенного псевдораствора магния с титано-
вым желтым находится при 540 нм, молярный коэффициент пога-
шения равен 3,6-103. В этой области реагент почти не поглощает
свет. Оптимальные условия для образования окрашенного золя
создаются при pH 12,5 и концентрации магния не выше 0,15 мг
в 50 мл раствора. Устойчивая окраска возникает через 5 мин
после введения всех реактивов и сохраняется до 2 ч. Кальций не
образует окрашенных соединений с титановым желтым, но при-
сутствие 4—6 мг Са2+ в 50 мл раствора даже увеличивает интен-
сивность окраски адсорбционного соединения магния. Для маски-
рования алюминия, железа, марганца применяют комплексон III
или триэтаноламин N (СН2СН2ОН)3.
Порядок работы. 40г почвы, просеянной через сито с
отверстиями в 1 мм, помещают в плоскодонную колбу и встря-
Продажные препараты хромазурола-S содержат кроме основного вещества
также и примеси, поэтому пользоваться лучше переосажденными в хлористо-
водородной кислоте, очищенными препаратами.
6*
163
хивают 1 мин с 100мл 1 н. КС1 (по Самохвалову). Суспензию
оставляют на сутки, затем перемешивают и фильтруют через су-
хой складчатый фильтр в сухой приемник. Аликвоту прозрачного
фильтрата (для суглинистых почв — 2 мл, для песчаных — 5 мл;
с содержанием магния не выше 0,16 мг переносят в конические
колбы емкостью на 100 мл. К раствору пипеткой добавляют точно
50 мл реактива на магний и перемешивают. Затем из бюретки
по каплям при перемешивании (чтобы не было местного пересы-
щения) добавляют 5 мл 2 н. раствора NaOH и оставляют на 5—
10 мин. Оптическую плотность измеряют в кювете 3 см при 540 нм
относительно нуле0ого раствора.
Калибровочные шкалы для аликвот в 2 и 5 мл готовят отдель-
но. Сначала готовят две серии стандартных растворов: первую
для аликвоты в 2 мл в пределе концентраций от 0 до 8 мг Mg в
100 мл; вторую для аликвоты 5 мл — от 0 до 3,2 мг Mg в 100 мл.
Для этого в мерные колбы емкостью 100 мл приливают из бюрет-
ки стандартный раствор магния с содержанием 0,4 мг/мл Mg
в следующих количествах: 0,0 (нулевой раствор); 2,5; 5,0; 10,0;
12,5; 15,0; 20,0 мл и 0,0; 1,0; 2,0; 3,0; 4,0; 5,0; 6,0; 8,0 мл. Растворы
разбавляют 1 н. КС1 до метки колбы. Для приготовления калиб-
ровочных шкал из каждой колбы соответствующей серии берут
по 2 мл (или по 5мл), переносят в конические колбы емкостью
на 100 мл и далее проводят все операции, как изложено выше
при описании метода. По величинам оптических плотностей стан-
дартных растворов строят график зависимости D от С. По графи-
ку находят концентрацию магния в испытуемом растворе, а затем
рассчитывают содержание подвижного магния в почве (см. с. 134).
Реактивы:
1) стандартный раствор магния с содержанием 0,4 мг/мл Mg;
2) комбинированный реактив на магний. К 5 мл 1 н. раствора
CaCls добавляют 700 мл дистиллированной воды, 12 мл
5%-ного раствора солянокислого гидроксиламина, 25 мл
разбавленного (1 :4) триэтаноламина, 50 мл 0,05%-ного вод-
ного раствора титанового желтого, 5 мл 2%-ного раствора
поливинилового спирта. Хорошо перемешивают и разбавля-
ют водой до 1 л. Готовят в день анализа.
Определение подвижного цинка
в почве эстракционно-фотометрическим
методом с применением дитизона
Среднее содержание цинка в почвах, по А. П. Виноградову,
составляет 7,5-10-3% при колебании в различных почвах от 3-10_4
до 1,2-10~2%. Наименьшая подвижность цинка в почве в области
164
pH от 5 до 8. Повышение или понижение pH приводит к увели-
чению подвижности цинка почвы.
В слабокислой и нейтральной средах дитизон C6H5N =
NC(SH) =N-NHC6H5 образует с ионом цинка внутрикомплекс-
ное соединение — однозамещенный дитизонат цинка. Соединение
не растворимо в водных растворах, но экстрагируется органиче-
скими растворителями: хлороформом и четыреххлористым угле-
родом. Растворы комплекса в органических растворителях ярко-
малинового цвета; максимум полосы поглощения комплекса на-
ходится при 538 нм, молярный коэффициент погашения равен
9,6-104; раствор подчиняется закону Бугера — Бера до концентра-
ции цинка 0,8 мгк/мл.
Оптимальные условия образования и экстрагирования комп-
лекса создаются при pH 5—6,5; в более кислой среде реакция
протекает не полностью, в щелочной среде, при pH более 8, комп-
лекс разрушается.
Фотометрическое определение цинка можно проводить методо.ч
одноцветной и смешанной окраски. Метод смешанной окраски
основан на фотометрировании экстракта, в котором находится
дитизонат цинка и избыток реактива. Оптическую плотность в этом
случае измеряют при 538 нм относительно раствора дитизона в
органическом растворителе (при той концентрации, которая ис-
пользовалась в анализе). Сам дитизон имеет два максимума по-
глощения — при 450 и 620 нм, а в области 538 нм имеет низкий
молярный коэффициент погашения.
При введении в раствор тиосульфат-нона определению цинка
не мешают Си, Ag, Hg, Pb, Cd, которые с дитизоном также об-
разуют окрашенные комплексы. Небольшие количества Fe (III),
Al, Ti маскируют цитрат-нопом.
Порядок работы. Юг почвы, просеянной через сито с от-
верстиями в 1 мм, помещают в плоскодонную колбу и взбалтыва-
ют на ротаторе 1 ч со 100 мл 1 н. КС1, предварительно очищенного
от следов цинка дитизоном в СС14. Суспензию фильтруют через
сухой беззольный фильтр в сухой приемник; первую порцию
фильтрата отбрасывают.
Аликвоту вытяжки (50 мл) переносят в делительную воронку
на 200 мл. Добавляют 10 мл маскирующего реагента в буферном
растворе и тщательно перемешивают. Затем приливают из бюрет-
ки точно 10 мл раствора дитизона в СС14 и встряхивают воронку
1 мин. После разделения фаз органический слой сливают в кювегу
с шириной поглощающего слоя 1 см. Измеряют оптическую плот-
ность экстракта при 538 нм относительно нулевого раствора, кото-,
рый содержит все реагенты, кроме цинка.
Калибровочную шкалу готовят в интервале концентраций цин-
ка от 0 (нулевой раствор) до 5 мкг в 10 мл СС14. Для этого
в серию делительных воронок приливают 0,0; 1,0; 2,0; 3,0; 4,0;
5,0 мл стандартного раствора цинка с содержанием 1 мгк/мл Zn.
Разбавляют до 50 мл 1 н. КС1, а затем проводят все операции,
165
изложенные выше при описании метода. По величинам оптичес-
ких плотностей строят график в координатах D—С. Находят
концентрацию цинка и рассчитывают содержание его в почве (см
с. 134).
Реактивы:
1) стандартный раствор цинка с содержанием 1 мкг/мл Zn;
2) разбавленный, свежеприготовленный раствор дитизона в
СС14. Готовят разбавлением 15 мл запасного (0,04%-ного
раствора дитизона в ССЦ) до 500 мл перегнанным СС14;
3) маскирующий раствор. 500 мл ацетатного буферного раст-
вора с pH 5 (136 г CH3COONa-3H2O растворяют в биди-
стиллированной воде, добавляют 29 мл ледяной СНзСООН
и доливают водой до 500 мл) смешивают с 50 мл 50%-кого
раствора Na2S2O3-5H2O. Приготовленный раствор очищают
от следов цинга экстракцией дитизоном в СС14.
Определение подвижной меди
в почве экстракционно-фотометрическим методом
с применением диэтилдитиокарбамината свинца
Среднее содержание меди в почвах, по А. П. Виноградову,
2-10-3%. На степень подвижности соединений меди в почве боль-
шое влияние имеют pH, содержание органического вещества и
биологическая активность почвы.
Диэтнлдитиокарбаминат меди [(C2Hs)2N—CS2]2-Cu окрашен в
желто-коричневый цвет, не растворим в водных растворах и экст-
рагируется органическими растворителями (этилацетатом, хлоро-
формом, четыреххлористым углеродом). Максимум полосы погло-
щения комплекса в органическом растворителе находится при
436 нм, молярный коэффициент погашения равен 1,4-104. Закону
Бугера—Бера подчиняются растворы с содержанием от 0,1 до
50 мкг Си в 10 мл органического растворителя. Реакция высоко-
селективна, если в качестве реагента для определения меди ис-
пользовать бесцветный раствор диэтилдитиокарбамината свинца
в органическом растворителе. Метод основан на реакции обмена
между ионом меди, находящимся в водном растворе, и ионом
свинца — в органической фазе. В результате реакции происходит
экстракция более устойчивого комплексного соединения — диэтил-
дитиокарбамината меди, которое окрашивает органическую фазу
в желто-коричневый цвет. Оптимальные условия образования и
экстрагирования комплекса меди обеспечиваются при pH 4—8.
Порядок работы. Навеску почвы 5г, просеянной через
сито с отвестиями 1 мм, помещают в плоскодонные колбы и взбал-
тывают на ротаторе 1 ч с 50 мл 1 н. НС1. Суспензию фильтруют
через беззольный сухой фильтр в сухой приемник. На определение
меди берут аликвоту объемом в 25 мл и переносят в делительную
воронку емкостью 100 мл. Приливают 5 мл лимоннокислого ам-
166
моння, одну каплю индикатора фенолфталеина и нейтрализуют
раствор разбавленным (1:1) аммиаком до pH 8,2 (розовая окрас-
ка индикатора). Затем из бюретки приливают точно 15мл раст-
вора диэтилдитиокарбамината свинца в СС14 и встряхивают смесь
2 мин. После разделения фаз сливают окрашенный органический
слой. Перед тем как поместить экстракт в кювету, его фильтруют
через сухой беззольный фильтр, чтобы осушить от мельчайших
капель воды, придающих экстракту небольшую мутность. Опти-
ческую плотность экстракта измеряют при 436 нм относительно
чистого СС14.
Калибровочную шкалу готовят в интервале концентраций от
2 до 20 мкг Си в 15 мл экстракта. Для этого в серию делительных
воронок наливают из микробюретки 0,2; 0,5; 1,0; 1,5; 2,0 мл стан-
дартного раствора меди с содержанием 10 мкг/мл Си. Разбавляют
1 н. НС1 до объема взятой аликвоты вытяжки и далее проводят
все операции, как изложено выше при описании метода. По ве-
личинам оптических плотностей стандартных растворов строят
график зависимости D от С. По графику находят концентрацию
анализируемого раствора и далее рассчитывают содержание под-
вижной меди в почве (см. с. 134).
Реактивы:
1) стандартный раствор меди с содержанием 10 мкг/мл Си
(готовят в день работы из стандарта с содержанием 1 мг/мл
Си);
2) раствор диэтилдитиокарбамината свинца в СС14: 664 мг ди-
этилдитиокарбамината натрия помещают в делительную во-
ронку, приливают 1л СС14 и добавляют 486 мг РЬ(ЫОз)2,
растворенного в 100 мл бидистнллированной воды. Смесь
встряхивают 5 мин и после разделения фаз органическую
фазу фильтруют через сухой беззольный фильтр в сухую
склянку темного стекла. Раствор должен быть бесцветным.
Хранят его в холодильнике;
3) лимоннокислый аммоний СзН4(ОН) • (СОО)з(ЫН4)з, 5%-ный
водный раствор, очищенный от следов меди дитизоном.
ИЗУЧЕНИЕ ГУМУСОВЫХ ВЕЩЕСТВ
Гумусовые вещества характеризуются интенсивным поглоще-
нием электромагнитных колебаний в ультрафиолетовой области.
С увеличением длины волны поглощение монотонно убывает, при-
ближаясь в области 750 нм к минимальному значению. Специфи-
ческих полос поглощения (максимумов) в спектре гумусовых ве-
ществ нет (рис. 52). Это лишает спектры поглощения гумусовых
веществ в ультрафиолетовой и видимой областях специфичности
и делает их малопригодными для изучения химических свойств
и структуры вещества. Однако интенсивность поглощения света
гумусовыми веществами (коэффициенты экстинкции) хорошо увя-
зывается с генетическими особенностями почв, что позволяет ис-
167
Рис 52 Спектры поглощения гу-
мата натрия
1 — чернозем, 2 — горно-луговая
альпийская почва с высоким со-
держанием Pg
пользовать спектрофотометрические методы при диагностике гу-
миновых кислот и фульвокислот и при сравнительной характери-
стике гумусовых веществ различного происхождения.
В спектрах некоторых препаратов гуминовых кислот обнару-
живаются небольшие максимумы в области 230—240, 265, 450 и
610 нм. Эти .максимумы обусловлены присутствием в препаратах
особой фракции зеленого цвета,
для обозначения которой К- Ку-
мада предложил символ Pg. Эта
фракция широко распространена
в различных почвах дерново-подзо-
листой зоны и гидроморфных
почвах других зон и является, ви-
димо, зеленым пигментом, проду-
цируемым некоторыми почвенными
грибами (см. рис. 52).
Кроме функции Pg в почвен-
ных гумусовых веществах присут-
ствуют и другие пигменты крас-
ного, ^бурого, зеленого ,и_черного
цвета. Среди них наибольшее зна-
чение имеет хлорофилл, дающий, в
спекхдах—сппртобепзольных—вытя-
жек максимум при 660 нм Присут-
ствие хлорофилла в спнртобензоль-
ной вытяжке из гумусовых веществ является важным диагности-
ческпм признаком при оценке интенсивности процессов вторичных
биогеохимических преобразований органического вещества почв.
При изучении спектров поглощения гумусовых кислот важное
значение имеют распределение оптических плотностей по длинам
волн и абсолютная интенсивность светопоглощения. Максималь-
ные значения ^-величин гуминовых кислот находятся в области
200—210.НМ. Величина £210 превышает значение Е450 в шесть и
более раз. Высокие значения Е позволяют считать, что окраска
гуминовых кислот, очевидно, обусловлена системой сопряженных
двойных связей в сочетании с кислородсодержащими хромофорны-
ми -Груштами. Система сопряжения не распространяется, видимо,
на всю частицу гуминовой кислоты (средневесовая молекулярная
масса которой 50 000 — 70 000), но ограничивается отдельными
изолированными фрагментами. Каждая частица гуминовой кисло-
ты может содержать несколько таких участков, которые ведут
себя независимо по отношению к электромагнитным колебаниям.
Благодаря этому спектр гуминовой кислоты является суммой аб-
сорбционных кривых изолированных участков.
Растворы гумусовых веществ подчиняются закону Бугера—
Бера при всех значениях длин волн до абсолютных величин оп-
тической плотности 1,0—1,2. Отклонение от закона Бугера—Бера
при высоких концентрациях вещества обусловлено рассеянием
168
света. Для количественной оценки интенсивности светопоглоще-
йия (окраски) гумусовых веществ используют запись закона Бу-
гера— Бера в следующем выражении:
D = £«€•/,
1см ’
где Е™®к> — Е-величина. Размерность ее зависит от выбранной
концентрации гуминовой или фульвокислоты (см. с. 130).
По смыслу определения Е-величины молярный коэффициент
погашения равен Е-величине (ЕГК<Ф1'>), умноженной на молекуляр-
ную массу, ex = ErhWI')-Mu, если Е-величина выражена в
расчете на йШ5центрацййП г/л. Если Е-величина рассчитана на
концентрацию гуминовой кислоты, равную 0,001%, тогда =
= 100 Егь • М,с или Егк ———-. Исходя из этого величину Егк
100-Мш
можно рассматривать как условный коэффициент погашения, рас-
считанный на условную единицу молекулярной массы.
Е-величину гумусовых кислот (или фульвокислот) рассчиты-
вают двумя способами. В первом способе расчета измеренную ве-
личину ’оптической плотности гуминовой кислоты или фульво-
кислоты относят к содержанию органического углерода в вещест-
ве. Эту величину можно обозначить Ес. При втором способе расче-
та ^измеренную оптическую плотность., относят, к концентрации
вещества (гуминовой кислоты или фульвокислоты) и сравнивают
растворы с равным содержанием вещества. Эту величину обозна-
чают Е1К или Е*к. Второй способ вычисления Е-величин более
объективен и более близок по смыслу к величинам молярного
коэффициента погашения (см. с. 131). В этом случае Е-величина
характеризует вещество в целом, поскольку оптические свойства
органических соединений зависят от общей молекулярной струк-
туры, а не только от содержания в ней углерода.
Распределение величин оптических плотностей по длинам волн
характеризуют коэффициентами цветности или крутизной спект-
рофотометрической кривой. Коэффициентами цветности называют
отношение величин Е46э: Е650 (или Е4:Е6); иногда используют
Е-величины, измеренные при других длинах волн. Коэффициенты
цветности, как и Е-величины, характеризуют природу гумусовых
веществ, но этот показатель значительно менее надежен, чем
Е-величины. Малая надежность коэффициентов цветности может
быть вызвана несколькими причинами. Например, при измерении
оптических плотностей в области 650 нм, где поглощение гумусо-
вых веществ мало, возникают большие ошибки. При оптической
плотности порядка 0,01—0,05 ошибка измерения можег достигать
50—100%. Это ведет к колебаниям величин коэффициента цвет-
ности в 1,5—2 раза. Снизить погрешность можно путем подбора
оптимальных концентраций или ширины поглощающего слоя (кю-
веты) с таким расчетом, чтобы измеренная оптическая плотность
169
при 650 нм была не менее 0,2 (см. с. 137). Ошибку измере-
ния коэффициента цветности вызывает мутность растворов гуму-
совых веществ. Это может быть обусловлено присутствием в вы-
тяжке минеральных коллоидов. Особенно большая ошибка-наблю-
дается при малых величинах оптических плотностей—Минеральные
кадлоиды можно удалить из вытяжки центрифугированием или
высаливанием сернокислым натрИеМ| Последний Спосоо,"несмотря
"на полйбе осаждение минеральных коллоидов, может привести
к новым ошибкам, так как вместе с минеральными коллоидами
происходит коагуляция и гумусовых веществ. Наконец, величина
оптической плотности гумусовых веществ зависит и от реакции
среды, это особенно важно для фульвокнслот. Спектры фульво-
кислот не меняют свойхарактёрв широком диапазоне pH — от
1—2 до 12—13, но интенсивность поглощения в щелочной среде
резко возрастает. Так, при 450 нм увеличение pH на единицу
вызывает увеличение оптической плотности примерно на 0,025
единицы. Это обстоятельство обусловливает необходимость стро-
гого соблюдения идентичности условий при спектрофотометричес-
ких исследованиям гумусовых веществ.
Надо иметь в виду, что величина оптической плотности раст-
воров гумусовых веществ изменяется при хранении растворов.
Уменьшение оптической плотности растворов гуматов и фульватов
происходит довольно быстро уже в течение первых 3—5 мес хра-
нения. За 15 мес. хранения оптическая плотность раствора гумата
может снижаться на 36—72% от первоначального значения. Сухие
препараты гуминовых кислот можно хранить длительное время,
не опасаясь изменения их свойств.
Чтобы изучить спектры поглощения гумусовых веществ, не-
обходимо подготовить почву и извлечь из нее важнейшие группы
почвенного гумуса.
Подготовка образца. Из воздушно-сухого образца почвы
отбирают корни и другие органические включения. Образец раз-
мельчают и просеивают через сито с отверстиями в 1 мм.
Декальцирование почвы. Почву в количестве 10—50 г поме-
щают в химический стакан и заливают 0,05 н. H2SO4 из расчета
80—100 мл кислоты на 10 г почвы. Вскипающие почвы предва-
рительно обрабатывают 10%-ной НС1 до полного разрушения кар-
бонатов, а затем уже начинают декальцирование. Почву, зали-
тую кислотой, перемешивают и дают хорошо отстояться, затем
декантируют прозрачную надосадочную жидкость и снова залива-
ют тем же количеством 0,05 н. H2SO4. Декальцирование продол-
жают до потери реакции на кальций с хромогеном черным; де-
кальцннат отбрасывают.
Выделение фракции I гумусовых веществ. Декальцированную
почву промывают водой и заливают 100 мл 0,1 н. NaOH. Суспен-
зию перемешивают и оставляют для настаивания на 3—4 ч. Затем
в стакан добавляют 50 г кристаллического ЫагЗСД-ЮНгО и пере-
мешивают до полного растворения соли. После осаждения мине-
170
ральных коллоидов отстоявшийся окрашенный раствор фильтруют
через рыхлый складчатый фильтр (чтобы фильтр не прорвался,
в воронку вкладывают плотный фильтр малого диаметра). Филь-
трат должен быть прозрачным; если необходимо, раствор пере-
фильтровывают несколько раз через тот же фильтр или прибегают
к центрифугированию. К остатку почвы снова приливают 0,1 н.
NaOH и тем же способом продолжают извлечение гумусовых ве-
ществ из почвы до тех пор, пока раствор над почвой не станет
лишь слабоокрашенным. Темноокрашенные фильтраты объединя-
ют и осаждают гуминовую кислоту. Для этого в раствор добавля-
ют 0,5—0,7 мл концентрированной H2SO4 и перемешивают, а затем
добавляют ту же кислоту по каплям до кислотности раствора
pH 2—3 (проверяют индикаторной бумагой или потенциометри-
чески). Гуминовая кислота коагулирует и выпадает в осадок.
Осадку дают осесть на дно сосуда, а затем надосадочную жид-
кость сливают на фильтр, не взмучивая осадка. Осадок в стакане
промывают 2—3 раза водой, подкисленной H2SO4 до pH 2—3.
Промывную жидкость сливают на фильтр и присоединяют к ос-
новному фильтрату. В осадке остается гуминовая кислота, а в
фильтрате — фульвокислоты. Осадок гуминовой кислоты промы-
вают несколько раз дистиллированной водой, если при этом на-
чинается пептизация осадка, то промывание прекращают.
Осадок гуминовой кислоты растворяют в небольшом объеме
0,1 н. NaOH, раствор переносят в мерную колбу емкостью 100 мл
и разбавляют до метки водой. В аликвоте измеряют оптическую
плотность или снимают спектр поглощения раствора гумата.
Кислый фильтрат, содержащий фульвокислоты, нейтрализуют
и упаривают (без кипячения) на песчаной бане. При упаривании
недопустимо появление осадка, если он появляется, то упаривание
прекращают. Упаренный раствор переносят в мерную колбу ем-
костью 100 мл, доводят pH до И—12 и измеряют оптическую плот-
ность (или снимают спектр поглощения) раствора фульвата
натрия.
Аналогичными приемами извлекают, если это необходимо, дру-
гие фракции гумусовых кислот из почвы (Орлов и др., 1969).
Спектрометрические характеристики растворов гумусовых кис-
лот, полученных в ходе анализа группового и фракционного
состава гумуса, используют для характеристики типа гумуса и
гумусного состояния почв. При структурно-химических исследо-
ваниях гумусовых веществ обычно выделяют из почвы препараты
гуминовых кислот и фульвокислот, очищают их, высушивают и
хранят в сухом темном месте. Для съемки спектров поглощения
берут навески гумусовых кислот от 5 до 20 мг, растворяют их
в 0,1 н. NaOH, доводят объем до 100 мл в мерных колбах. Затем
снимают спектры поглощения.
Съемка спектров поглощения растворов гумусовых веществ.
Подготавливают к работе спектрофотометр, как описано выше,
и записывают на бланке линию нулевой оптической плотности.
171
Одну кювету заполняют полученным раствором гумата натрия
или фульвата натрия и ставят кювету на пути левого пучка спект-
рофотометра. На пути правого пучка помещают кювету с водой.
Устанавливают на барабане длин волн значение 400 нм, включа-
ют мотор обработки, и корда перо достигнет предельной отметки,
включают запись спектра тумблером «Развертка» (см. с. 147). j
Обработка спектров. Внимательно изучают ход кривой свето-
поглощения и отмечают наличие максимумов или минимумов в
спектре. Для каждого максимума или минимума находят и запи-
сывают длину волны в максимуме (минимуме) поглощения. До-
вольно часто полосы поглощения могут быть представлены не г
максимумами на кривой, а уступами (спрямленными или гори-
зонтальными площадками). Для каждой площадки находят зна-
чения длин волн в начале площадки, в ее конце и в середине.
Найденные значении заносят в таблицу и по набору полос погло-
щения делают вывод о присутствии в изучаемом препарате фрак-
ции Pg или других сопутствующих компонентов.
Затем измеряют оптические плотности при длинах волн 465 и
650 нм. Зная концентрацию гумусовых веществ, рассчитывают ‘
Е-величины по следующим формулам.
Для раствора препарата гуминовой кислоты (фульвокислоты)
в 0,1 н. NaOH:
'СМ /-Сгк(фк)
где СГК(фК) — концентрация гуминовой кислоты (фульвокислоты)
в мг/100 мл.
Для раствора, полученного в ходе анализа группового и фрак-
ционного состава гумуса:
ГО, 001'X,С -
1СМ 1-Сс ’
где Сс — концентрация органического углерода в вытяжке (раст-
воре в мг/100 мл).
Чтобы перейти от одного значения Е к другому, надо знать
содержание органического углерода в препарате гуминовой кис-
лоты (фульвокислоты).
По найденным значениям Е-величин определяют принадлеж-
ность гумусовых кислот к подгруппам черных или бурых гумино-
вых кислот и используют эти значения для сравнительной харак-
теристики изучаемых почв.
При анализе спектров препаратов гумусовых кислот можно
вычислить и коэффициент цветности Е465 Eeso- Поскольку это от-
ношение не зависит от концентрации раствора, то для его нахож-
дения достаточно взять отношение оптических плотностей, изме-
ренных при длинах волн 465 и 650 нм. Более точное нахождение
требует специального измерения оптической плотности при 650 нм
в кюветах с большой толщиной поглощающего слоя (см. с. 169).
172
Исследование влияния
pH раствора на ЕФК
Порядок работы. Взять две одинаковые навески сухого
препарата фульвокислоты (40 мг) и растворить каждую в неболь-
шом объеме 0,1 н. NaOH. Перенести растворы в две мерные колбы
емкостью в 100 мл. Один раствор довести 0,1 н. НС1 до
pH 3, другой довести до pH 12 0,1 н. NaOH. Разбавить растворы
до метки водой и проверить pH по универсальному индикатору.
Снять спектры поглощения приготовленных растворов. Изме-
рить оптическую плотность при 465 нм и рассчитать величину
•^4650имФК °боих растворов.
Спектрофотометрическое
определение хлорофилла
и (или) его производных в почвах
В ходе анализа группового состава гумуса из почв выделяют
группу воскосмол. или липидов, действием спиртобензольной сме-
си в аппарате Сокслета. В этот экстракт наряду с другими ве-
ществами переходит также хлорофилл или его производные, ко-
торые дают в спектрах хорошо выраженные полосы с максимума-
ми при 660—670 нм. Е. В. Фрндланд с соавторами (1976) описала
метод прямого количественного определения хлорофилла по ин-
тенсивности полосы поглощения в указанной области. Максимум
поглощения может несколько варьировать по длинам волн, что
объясняется присутствием в почве разнообразных продуктов транс-
формации хлорофиллов а и Ь (феофитин, феофорбид и др.). По-
этому в спектре находят полосу, лежащую в области 650—670 нм,
и измеряют оптическую плотность в максимуме этой полосы.
Порядок работы. Из пробы воздушно-сухой почвы отбира-
ют корни, почву разминают и пропускают через сито с отверстия-
ми в 1 мм. Навеску подготовленной почвы экстрагируют в аппара-
те Сокслета спиртобензольной смесью по обычной методике
(Орлов и др., 1969). Полученный экстракт фильтруют через бу-
мажный фильтр. Если раствор слабо окрашен, его несколько упа-
ривают и количественно переносят в мерную колбу емкостью
50 мл. Объем раствора доводят в колбе до метки спиртобензоль-
ной смесью (1:1), перемешивают и затем снимают спектр погло-
щения в области 600—700 нм. Интенсивность светопоглощения
вычисляют как разность между оптической плотностью найденной
кривой и оптической плотностью фоновой кривой при той же
дтине волны. Фоновую кривую получают интерполяцией участков
спектра, лежащих вне полос поглощения хлорофилла.
173
Содержание
муле
хлорофилла в экстракте рассчитывают по фор-
„ 13,26-ADx-V
Схл =--------t------мкг,
I
где Д£Ц — приращение оптической плотности по сравнению с
фоновой кривой при длине волны в максимуме поглощения, V —
объем мерной колбы в мл, / — толщина кюветы в см, 13,26 —•
постоянная величина.
Содержание хлорофилла в почве рассчитывают по формуле
~ 13,26-ADx-V
Схл =------—------мкг/г,
Z-P
где р — навеска почвы в г, соответствующая объему раствора V
в мл.
АНАЛИЗ ПОЧВ ПО КРИВЫМ
СПЕКТРАЛЬНОЙ ЯРКОСТИ
Цвет почвы — важнейший диагностический признак, с по-
мощью которого удается надежно различать почвенные генети-
ческие горизонты одного почвенного профиля или даже одинако-
вые горизонты различных типов почв. Но значение цвета не огра-
ничивается только возможностями диагностики. По окраске почвы
можно сделать предварительное заключение о ее химическом со-
ставе, особенно о содержании гумуса и различных форм соедине-
ний железа. От окраски почв зависит в значительной мере и теп-
ловой режим почв, поскольку хорошо гумусированные темно-
окрашенные почвы поглощают значительную долю падающей на
них световой энергии, они обычно быстрее и в большей степени
нагреваются, чем почвы светлые, бедные гумусом или имеющие
белесый оттенок из-за большого скопления карбонатов.
Объективной мерой оценки цвета почв могут служить коэффи-
циенты спектральной яркости или коэффициенты отражения света
почвами.
Падающая на поверхность почвы световая энергия частично
поглощается почвой, а частично рассеивается, диффузно отра-
жается. Отраженная почвой энергия распределяется в простран-
стве неравномерно; в одном направлении почва может отражать
больше энергии, чем в другом. Чтобы найти общее количество
отраженной энергии, применяют интегрирующие сферы, имеющие-
ся в спектрофотометрах типа СФ-14, СФ-18. Такая сфера пред-
ставляет собой полый шар, внутренняя поверхность которого по-
крыта слоем вещества, отражающего свет практически на 100%.
Это может быть окись магния. В нижней части сферы делается
небольшое окно, в котором с помощью специальных держателей
укрепляется кювета с почвой. Поверхность почвы сглаживают
174
часовым стеклом так, чтобы она составляла как бы продолжение
внутренней поверхности сферы. Во втором окне помещают стан-
дартный отражатель. В процессе измерения поверхность почвы
и поверхность стандартного отражателя освещают идентичными
световыми пучками, как это описано выше на примере спектро-
фотометра СФ-18 (см. с. 148). Отраженный от поверхности почвы
во всех^направлениях свет многократно отражается от внуТПГЙней
поверхности сферы, и затем его ннтейсивноеть- йзмерпотеп с по._
мощыо фотоэлемента. В результате спектрофотометр 'измеряет
ко шчес1 во отраженной почвой световой энер-ршгГ^выраженной
в процентах-к-световой энергии исходного светбвбпГпотока. Эту"
величину, или коэффициент отражения, обозначаюТ~ги51вблЬм R.
Если коэффициент отражения измерен только при одной длине
волны X, тогда его значение записывают как R-,, Измерив коэф-
фициенты отражения при различных длинах волн и построив гра-
фик зависимости R& от длины волны %, получают кривую спект-
ральной яркости почвы. На спектрофотометрах типа СФ-18 эта
кривая записывается автоматически.
Спектр отражения наиболее полно и наиболее объективно
характеризует цвет почвы. Ни цветовые шкалы, ни цветовые ин-
дексы не могут в полной мере заменить спектр отражения; такая
замена особенно нежелательна в почвенных исследованиях, по-
скольку многие оттенки цвета почв, заметные при сопоставлении
спектров отражения, утрачиваются при переходе на цветовые ин-
дексы.
По общему характеру кривых спектры отражения почв можно
объединить в три группы. В первую группу входят спектры отра-
жения верхних, достаточно гумусированных горизонтов почв.
Спектры этой группы представлены кривыми, очень полого под-
нимающимися от синей (400 нм) к красной (750 нм) части спект-
ра. В наибольшей мере спектры этого типа присущи черноземам,
для которых отражение в области 400—450 нм не превышает не-
скольких процентов, а в области 700—750 нм может достигать
10—20%.
Спектры второй группы также не имеют сколько-нибудь сильно
выраженных максимумов или минимумов на кривых, нет на этих
кривых и значительных перегибов. Но в отличие от кривых первой
группы коэффициенты отражения света в несколько раз выше и
в области 400 нм могут составлять 10—20%, а около 750 нм — до
40—70%. Такие спектры свойственны белесым горизонтам почв,
как, например, элювиальным горизонтам подзолистых почв А2
или горизонтам, обогащенным солями белого цвета (особенно
карбонаты). Наконец, в третью группу входят спектры отражения
почв или отдельных генетических горизонтов, которые значитель-
но обогащены несиликатными соединениями железа. Спектры
таких почв имеют на кривой отражения ясно выраженный перегиб
(подъем) в интервале длин волн от 480—500 до 600—700 нм. По
величине этого перегиба, или разности коэффициентов отражения
175
при 480 и 650 нм: ДД=Д650— /?480, можно судить об относителв-
ном содержании несиликатного железа в почвах. Если образец
почвы перед съемкой спектра отражения прокалить при 800—
900°, то это приводит к разрушению органического вещества, раз-
ложению силикатов до окислов и в результате окраска прокален-
ного образца уже обусловлена только соединениями железа. По
спектру отражения такого образца можно судить об относитель-
ном валовом содержании железа в почвенном профиле.
Об отражении почвы в целом судят по величине(Тинтёг~рального,
< отражения Величина интегрального отражения представляет
собой общее количество с.нетовой энергии в интервале длин волн
от 400 до 750 нм, отраженное почвой и выраженноё~в~процентах
от^бщавд-д<бличества^исходной энергии,в/гом~Жё интервале длин
-ВЩЩ^Величину интегрального отражения можно найти разными
путями. Наиболее точное значение находят следующим образом:
.подсчитывают площадь, „ограниченную..кривой спектральной яр-
-Крсти, _линией__нулевого отражения света и вертикальными осями
(расиможенными дш спектре отражения вещества, снятого на
спектрТ5фотометре). Эчу площадь относят к общей площади гра-
фика, ограниченного вертикальными линиями с абсциссами 400—
750 нм и горизонтальными линиями нулевого и стопроцентного от-
ражения света. Величину интегрального отражения выражают в
процентах Этот способ вычисления величины интегрального от-
ражения очень трудоемок. Для сравнительно пологих кривых, не
имеющих значительных максимумов, подсчет можно упростить.
Находят значения коэффициентов отражения при нескольких
длинах волн, расположенных через равные интервалы, например
при 400, 440, 480, 52и, 560, 600, 640, 680, 720, 760 нм. Суммируют
эти величины и делят сумму на общее число использованных для
подсчета величин
__ #400 + Rw Г + #760
п
где п — общее число суммируемых коэффициентов отражения.
Метод спектральной отражательной способности пригоден и
для изучения распределения различных окрашенных компонентов
в толще почвенной массы. Если съемку спектров отражения сов-
мещать с предварительной экстракцией почв различными реаген-
тами, извлекающими из почв различные вещества: гумус, соеди-
нения железа, марганца и т. д., можно составить представление
о том, какие вещества обусловливают окраску почв и как они
пространственно расположены (переслаиваются) в почвенной мас-
се. Если снять спектры фракций структурных агрегатов, выделен-
ных из почв, а затем тех же агрегатов, но растертых и просеян-
ных через сито, то можно решить вопрос о химической однород-
ности или неоднородности почвы в пределах почвенного агрегата.
Отражательная способность почв зависит не только от содер-
жания важнейших красящих компонентов, таких как гумус или
476
окислы железа, но также от влажности почв и их структурного
состояния. Увеличение влажности почв до уровня наибольшей
влагоемкости обычно снижает стражательную способность почв
в 2—3 раза. На этой основе были предложены методы определе-
ния влажности почв. Зависимость интегрального коэффициента
отражения от влажности почвы можно выразить в виде функцио-
нальной зависимости
где — интегральное отражение, А и В — постоянные величи-
ны, характерные для типа почв и их генетических горизонтов,
W — влажность в процентах. Взаимосвязь этих величин для
гор. А] различных почв представлена в табл. 17.
Табгица 17
Значение постоянных в уравнениях, связывающих интегральное отражение
с влажностью почв и размером агрегатов
Почва, юризонт Интервал вчажности, % Постоянные уравнений
А В п к «оо
Дерново-подзолистая, А] 2,1 — полная капил- 18,6 0,25 0,22 10,0 33,0
^-2 л яркая влагоемкость - 0,27 12,0 38,5
В1 —. .—. .—. 0,26 6,0 35,5
Серая лесная, '\г 1,5—20% 26,8 0,57 —. —_ —
Чернозем, Аг 1,7 — полная капил- 11,1 0,16 — — —
Мощный чернозем, Лг лярная влагоемкость .—. 0,60 2,0 15,5
Обыкновенный чернозем, Ai .—. 1,22 3,6 13,0
Каштановая, А, 3,1 — полная капил- 12,3 0,09 .—. .— —-
Темно-каштановая, Ах лярная вчагоемкость .— 0,38 4,0 18,0
Краснозем, Ах 5,4—21% 31,2 0,92 —- — •—*
Отражательная способность почв зависит от размера частиц
и формы их поверхности. При величине почвенных агрегатов бо-
лее 2—3 мм отражательная способность сравнительно постоянна.
Дальнейшее размельчение агрегатов приводит к возрастанию
отражательной способности образца, и при величине диаметра
частиц =С0,25 мм коэффициент отражения возрастает в 1,2—
2 раза. Это объясняется характером расположения (упаковок)
агрегатов. Мелкие частицы образуют более выровненную поверх-
ность без пор и трещин, присутствие которых в образце приво-
дит к гашению части световой энергии светового потока. Хими-
ческий состав агрегатов может влиять на отражательную спо-
собность поверхности размельченного образца только таких
177
почв, у которых химико-минералогический состав внутрен-
ней части агрегата отличен от состава поверхностной части.
Зависимость коэффициента отражения R от размера агрегатов d
можно выразить экспоненциальным уравнением:
R = К-10-'7'
где п, К. и — постоянные уравнения и характерны для типа
почв и их генетических горизонтов. К и п — константы, выра-
жающие форму кривой отражения, Rx — коэффициент отраже-
ния агрегата максимального размера (см. табл. 17).
Вопрос об изменении отражательной способности почв в за-
висимости от влажности и размера частиц имеет практическое
значение, так как он тесно связан с вопросом о стандартизации
анализа, с вопросом способа подготовки образца для получения
воспроизводимых и правильных результатов при определении
величин коэффициентов отражения почвенных образцов.
Получение спектров отражения почв
Пор яд ок работы. Из воздушно-сухого образца почвы
«отобрать крупные корни, размять почву в ступке пестиком с ре-
зиновым наконечником и пропустить через сито с диаметром
отверстий в 1 мм. Просеянную почву насыпать ровным слоем в
специальные плоские кюветы, слегка пригладить выпуклым часо-
вым стеклом, чтобы придать ее поверхности вогнутую форму с
радиусом, близким к радиусу интегрирующей сферы. Затем кю-
вету с почвой прижать к левому окну интегрирующей сферы и
провести запись спектра на предварительно подготовленном при-
•боре (см. с. 148).
Обработка результатов измерений. Найти на кривых спект-
ральной яркости области резкого изменения коэффициента отра-
жения (перегибы) и отметить длины волн начала и конца пере-
гибов.
Эти величины служат для качественного сопоставления спект-
ров различных почв или различных генетических горизонтов
одной почвенной разности. По кривым отражения найти следую-
щие величины:
1. Коэффициент отражения при длине волны 700 или 750 нм.
Его показания считывают прямо по графику. По величине
/?75о обосновать относительную обогащенность почвы (ря-
да почв) гумусом.
2. Вычислить разность коэффициентов отражения при 650 -и
480 НМ Д/? = /?б50 ^480 (величины /?650 И /?480 считывают
по графику). По величинам Д/? охарактеризовать распре-
деление железа по генетическому профилю.
178
3. Рассчитать величину интегрального отражения Дг. Для
расчета использовать формулу или способ расчета по пло-
щади, ограниченной кривой спектральной яркости (см.
с. 176).
ЛИТЕРАТУРА
Аринушкина Е. В. Руководство по химическому анализу почв. М., Изд-во
Моск, ун-та, 1970.
Бабко А. К-, П и л и п е н к о А. Т. Фотометрический анализ. Общие сведения
н аппаратура. М, «Химия», 1968.
Карманов И И. Спектральная отражательная способность и цвет почв
как показатели их свойств. М, «Колос», 1974.
Марченко 3. Фотометрическое определение элементов. М., «Мир», 1971.
Орлов Д. С., Гришина Л. А, Ерошичева Н. Л. Практикум по биохи-
мии гумуса. М, Изд-во Моск, ун-та, 1969.
Методы изучения минералогического состава и органического вещества почв.
Ашхабад, 1975.
Глава 5
ИДЕНТИФИКАЦИЯ ОРГАНИЧЕСКИХ
И МИНЕРАЛЬНЫХ КОМПОНЕНТОВ ПОЧВЫ
МЕТОДОМ ИНФРАКРАСНОЙ СПЕКТРОСКОПИИ
Инфракрасные спектры поглощения применяются в почвове-
дении при решении различных задач. Метод инфракрасной спек-
троскопии (ИКС) позволяет установить присутствие важнейших
атомных групп и типов связей в гумусовых веществах, иденти-
фицировать отдельные индивидуальные соединения, изучать ме-
ханизмы органо-минерального взаимодействия, адсорбцию и де-
сорбцию влаги, определять присутствие карбонатов, сульфатов,
некоторых минералов в почве. Этот метод применим для иссле-
дования трех основных фаз почвы: твердой, жидкой и газообраз-
ной. Во многих случаях метод ИКС позволяет работать с почвой
или ее компонентами без применения каких-либо химических опе-
раций и тем самым исключить возможную денатурацию иссле-
дуемых веществ в ходе анализа. Как большинство физических
методов, метод ИКС дает сведения как о качественных особен-
ностях (строении) вещества, так и о его количественном содер-
жании в образце.
Инфракрасная спектроскопия как один из разделов спектро-
скопии включает получение, исследование и применение спект-
ров испускания, поглощения и отражения в инфракрасной обла-
сти спектра. Инфракрасная область спектра условно разделяется
на ближнюю (Z, от 0,74 до 2,5 мкм), среднюю (2,5—50 мкм) и
далекую (50—2000 мкм); в этой области расположено большин-
ство колебательных и вращательных спектров молекул, поэтому
инфракрасная спектроскопия использует главным образом моле-
кулярные спектры.
Наиболее широкое распространение получило исследование
ИК-спектров поглощения в ближней и средней областях спект-
ра, позволяющее определять структуру молекул, их химический
состав, идентифицировать важнейшие атомные группы и типы
связей, а также обнаруживать индивидуальные соединения в со-
ставе смесей веществ.
180
Метод ИКС основан на взаимодействии исследуемого вещест-
ва с электромагнитными колебаниями в диапазоне длин волн от
0,75 до 50—100 мкм. Практически используется более узкий уча-
сток спектра — от 2 до 25 мкм, а в практике почвенных иссле-
довании пока преимущественно ограничиваются интервалом от
2,5 до 12—15 мкм. В результате поглощения колебаний с опре-
деленными п специфичными для каждого вещества значениями
длин волн формируется спектр поглощения. Набор длин волн и
максимумов полос поглощения характеризует качественные осо-
бенности вещества. По степени поглощения колебаний при опре-
деленных значениях длин волн судят о количестве вещества.
Наиболее интенсивное поглощение веществом электромагнит-
ных колебаний наблюдается в том случае, когда энергия кванта
точно отвечает разности двух возможных энергетических уровней
молекулы. В ИК-области изменение энергии молекулы связано
с колебаниями отдельных атомных групп или вращением моле-
кулы в целом, поэтому ПК-спектры часто называют молекуляр-
ными спектрами в отличие от электронных спектров поглощения,
обусловленных электронными переходами и проявляющимися в
видимой и ультрафиолетовой областях.
Для качественной характеристики инфракрасного спектра
пользуются или длиной волны, обычно выражая ее в микрометрах,
или волновым числом v, которое выражают в обратных санти-
метрах —см-1. Соотношение между длиной волны и волновым
числом выражается равенством:
10000 _ 10000
длина волны (мкм) = =—или v = д(мкм)'
Отсюда следует, что волновое число показывает, какое число
длин волн укладывается на отрезке длиной 1 см. Для быстрого
перевода длин волн в волновые числа пользуются таблицей
обратных величин. В литературе иногда встречается неправиль-
ное использование понятия «частота», когда частотой называют
волновые числа. Это разные понятия, имеющие неодинаковое
численное выражение, и смешивать их недопустимо
Колебания атомов даже в относительно простых молекулах
весьма разнообразны. Они могут совершаться в направлении
валентной связи, как бы растягивая ее; такие колебания назы-
вают валентными, и бывают они симметричными (два атома дви-
жутся в одном направлении по отношению к третьему) и асим-
метричными (атомы движутся в разных направлениях). Колеба-
ния с изменением угла между направлениями связей называют
деформационными: в зависимости от характера движений атомов
их делят на ножничные, веерные, крутильные и маятниковые
колебания. Такая характеристика колебаний в некоторой мере
условна. Практически всегда одновременно происходит как из-
менение угла между связями, так и расстояния между атомами,
поэтому деление на Ьалентные и деформационные колебания
отражает лишь проявление наиболее выраженного эффекта.
181
Большой набор возможных колебаний обусловливает соответ-
ственно большое число полос в спектрах даже относительно про-
стых молекул.
Количественная характеристика спектров дается, как и для
видимой области, на основе закона Бугера — Бера (см. с. 129).
Рис. 53. ИК-спектр бензойной кислоты: А, В, С — максимумы по-
лос поглощения
Инфракрасные спектры обычно изображают в виде графика
(рис. 53), откладывая по оси ординат пропускание, поглощение
или оптическую плотность, а по оси абсцисс — частоту колеба-
ний, длину волны или волновые числа. В большинстве работ
используют пропускание в процентах и волновые числа. Для
удобства сравнения целесообразно получать спектры при равных
концентрациях сравниваемых веществ или при построении гра-
фика откладывать по оси ординат величины е>, (Е). Иногда
удобно строить спектр в координатах lg Б—X (или 1g £>—v).
182
В этом случае глубина полос поглощения не зависит от концен-
трации вещества, как это вытекает из следующего. Поскольку
D = e\-cl, то lgI)=lge7,+lgcZ. Очевидно, что изменение cl вы-
зовет только смещение всего спектра вдоль оси D, а наблюдае-
мая интенсивность полос будет пропорциональна 1g ек- Этот
способ выигрышен для сравнительной характеристики отдельных
полос.
ОПТИКА
И ИСТОЧНИКИ ИЗЛУЧЕНИЯ
Для изучения ИК-спектров поглощения применяются ИК-
спектрометры двух типов. У одних в качестве диспергирующего
элемента используют призмы, изготовленные из кварца, NaCl,
КВг, LiF, CsI (призменные спектрометры), у других — дифрак-
ционные решетки (дифракционные спектрометры).
В призменных приборах используется солевая оптика, про-
зрачная для ИК-лучей, и только в самой коротковолновой обла-
сти применяют призмы из специального стекла, пригодные для
работы в области волновых чисел более 2850 см-1. Обычно
.используют следующие призмы: CaF2 — в диапазоне 4200—
1300 см-1, LiF — в диапазоне 4000—1700 см-1, NaCl — 5000—
650 см~!, КВг — 1100—385 см-1, CsI — от 1000 до 200 см-1. Из
пластин КВг изготавливают также защитные стекла, вкладыши
для кювет, некоторые линзы. Солевая оптика чувствительна к
парам воды, а из-за растворяющего действия воды на материал
кювет ограничивается возможность анализа водных растворов.
Последнее возможно только в специальных кюветах, например с
окнами из плавленого AgCl. Кроме того, жидкая вода имеет
сильные полосы поглощения в областях 3650—2930, 1750—1580,
1400—1300, 930—650 см-1 и менее сильные в других диапазонах.
Это также ограничивает применение воды как растворителя.
В качестве источника излучения используют штифты Нерн-
ста (стержни из окисей циркония, тория и церия), глобары
(стержни из карбида кремния), платиновые ленты и другие
излучатели, разогреваемые при прохождении тока до 1100—
1300°.
Более перспективно использование дифракционных спектро-
метров, так как последние по разрешающей способности превос-
ходят призменные приборы; они также более удобны и стабиль-
ны в работе.
Методы подготовки образцов
к съемке ИК-спектров
Для подготовки образцов к съемке ИК-спектров существуют
различные способы в зависимости от свойств и агрегатного со-
стояния образца. В газовой фазе можно определять СО2, пары
183
воды, аммиака, органических растворителей и др. Для этих це-
лей приборы снабжены специальными газовыми кюветами.
Поскольку вода малопригодна для работы в ИК-области, а
важнейшие компоненты почвы плохо растворимы или вовсе не
растворимы в органических растворителях и в воде, то обычно
снимают ИК-спектры сухих (твердых) препаратов и почв. Ис-
пользуют метод нанесения тонких слоев вещества на пластинки
из стекла KRS-5 (синтетический монокристалл бромйодида тал-
лия). Эти стекла малорастворимы в воде и прозрачны для ИК-
лучей в широкой области спектра.
Некоторые вещества можно снимать после нанесения их тон-
ким слоем на солевые пластинки. Этот метод удобен для иссле-
дования почвенных воскосмол. Для приготовления объекта ис-
пользуют прозрачную пластинку толщиной 1—2 мм, отколотую
по плоскостям спайности от кристалла NaCl; вполне пригодны
кристаллы из природных месторождений. Экстракт воскосмол
(или фракцию в спиртовом, бензольном, спиртобензольном ра-
створах) упаривают и несколько капель концентрированного
раствора наносят на поверхность солевой пластинки. Дают уле-
тучиться растворителю и слегка подогревают пластинку в су-
шильном шкафу. При этом слой битумов равномерно распреде-
ляется по поверхности. После охлаждения образец готов для
анализа.
В почвенных работах наибольшее значение имеют методы
подготовки к съемке твердых образцов.
Простейший способ съемки в твердом состоянии тех образцов,
которые легко образуют прозрачные пластинки или пленки. Так.
из различных слюд нетрудно получить тонкие пластинки, даю-
щие вполне удовлетворительные спектры. Иногда удается при-
менить тонкие срезы (шлифы) различных минералов, стекол.
В твердом состоянии снимают спектры пленок органических по-
лимеров, например полистирола, который используется для гра-
дуировки спектрометров.
Значительно труднее получить хорошие спектры порошков,
которыми обычно представлены все важнейшие почвенные объ-
екты: гуминовые кислоты, фульвокислоты, почва в целом, ее
механические фракции, образцы глинистых минералов. Любые
порошки так сильно рассеивают ИК-излучение, что съемка спек-
тров часто становится практически невозможной, если не исполь-
зовать специальные иммерсионные среды.
В качестве иммерсионной среды можно использовать парафи-
новое масло (нужоль или нуйоль). Для съемки спектров препа-
раты гумусовых веществ растирают с парафиновым маслом и
каплю суспензии помещают в разборную кювету, состоящую из
двух полированных солевых пластин, разделенных тонкой коль-
цеобразной прокладкой из алюминиевой или свинцовой фольги.
Этим методом сейчас пользуются редко, но иногда ему прихо-
дится отдавать предпочтение перед КВг-техникои, особенно при
184
исследовании валентных колебаний ОН-групп. Дело в том, что
использование суспензии вещества в нужоле позволяет избавить-
ся от помех за счет адсорбированной воды.
Известны попытки введения порошков в различные пленки,
например агаровые. Однако наиболее эффективным оказалось
запрессовывание порошков в таблетки из NaCl или КВг (так
называемая КВг-техника).
Для приготовления таблеток
исследуемый образец почвы или
ее компоненты растирают до
пудры в агатовой ступке или виб-
ромельнице, таким же образом
готовят тонкорастертый порошок
КВт (ОСЧ). Затем подготовлен-
ные порошки сушат в вакууме
в течение 3—5 ч: препараты ГК
и ФК при температуре 60—70°,
механические фракции почв —
при 105° и КВг — при 200°. За-
тем берут навеску высушенного
образца от 0,5 до 1,5 мг и тща-
тельно смешивают его с 200—
300 мг порошка КВг. Соотноше-
Рис. 54. Пресс-форма для приготов-
ления солевых пластин:
1 — головка пресс-формы, 2 — сталь-
ной шарик, 3 — штуцер для отсоса
воздуха, 4 — разъемная форма для
прессования (полированные сталь-
ные пластинки), 5— винт зажима
разъемной формы, 6 — корпус, 7 —
резиновое кольцо, 8 — подпятник
Рис. 55. Пресс гидравлический
ПСУ-10:
1 — нижняя плита, 2 — крышка
пресс-формы, 3 — стержень, 4 —
верхняя плита, 5 — ходовой впит,
6 — вакуумметр, 7 — шкала нагру-
зок, 8 — рукоятка силовозбудителя,
9 — вакуумный насос, 10 — рукоятка
включения вакуумного насоса
185
ние между навеской образца и количеством КВг сохраняют строго
постоянным в течение всей серии экспериментов.
Смесь, подготовленную для прессования, помещают равномер-
ным слоем в специальную пресс-форму (рис. 54) между полиро-
ванными стальными пластинками 4. Для прессования исполь-
зуется пресс гидравлический ПСУ-10 отечественного производ-
ства (рис. 55). Корпус пресс-формы закрывают крышкой 2 и
устанавливают на нижней плите пресса 1. Для отсоса воздуха из
пресс-формы включают вакуумный насос 9 рукояткой 10. После
достижения стрелкой вакуумметра значения 680 мм рт. ст. про-
должают откачку воздуха в течение 3 мин. Не прекращая отка-
чивание воздуха, включают реле электромотора, находящееся на
правой стенке корпуса пресса, и электромотор кнопкой «Пуск».
Равномерно вращая рукоятку силовозбудителя 8 в сторону ука-
зателя «больше», устанавливают стрелку на круговой шкале
нагрузок 7 до значения 9700—9800 кгс (килограмм-силы). Пресс-
форму выдерживают под давлением в течение 3 мин, не прекра-
щая откачивание воздуха. Затем выключают электромотор прес-
са кнопкой «Стоп» и вакуумный насос рукояткой 10. Вращая
рукоятку сброса масла против часовой стрелки, равномерно
сбрасывают давление до значения «0» на шкале нагрузок прес-
са 7. Ходовой винт 5 вручную повернуть влево, приподнимая
верхнюю плиту пресса 4 таким образом, чтобы освободить пресс-
форму. Далее впускают воздух в пресс-форму плавным вытя-
гиванием металлического стержня 3 из крышки пресс-формы,
снимают крышку 2 и с помощью гаечного ключа отворачивают
винты зажима разъемной формы. Пресс-форму разбирают и вы-
нимают готовую пластину, в толще которой равномерно распре-
делен образец исследуемого препарата; если же образец пред-
ставлен в толще пластинки отдельными крупными скоплениями
или видны мутные пятна, пластины выбраковывают. Приготов-
ленная пластинка должна быть прозрачной или окрашенной в
зависимости от цвета исходного вещества, однородной по толщи-
не с гладкой блестящей поверхностью. Параллельно прессуют
таблетку из чистого КВг, которую используют в качестве холо-
стого опыта. На двухлучевых приборах снимается разностный
спектр между таблеткой с образцом и таблеткой из чистого
КВг. Это почти полностью компенсирует отражение излучения от
поверхности таблетки, рассеяние света и исключает наложение
примесей, особенно воды, которые могут быть в КВг.
Градуировка прибора
и получение спектров
Прежде чем приступить к съемке ИК-спектров исследуемых
образцов, производят градуировку прибора. Для градуировки
используют стандартные вещества: полистирол, бензол, пары
186
аммиака, воды и др. На приборах типа ИКС-14, ИКС-14А преду-
смотрена линейная развертка спектра по длинам волн или по
волновым числам, но отметочные точки наносятся в условных
единицах, отвечающих показаниям барабана длин волн. Чтобы
перевести эти показания в волновые числа, снимают спектр по-
листирола (рис. 56), строят график, откладывая по оси ординат
Рис. 56. Спектр пленки полистирола (1=0,025 мм), у максимумов полос
указаны волновые числа (в см-1)
1£7
волновые числа, известные для полос полистирола, а по оси
абсцисс — показания барабана длин волн. Этим графиком впо-
следствии пользуются для нахождения волновых чисел, соответ-
ствующих полосам поглощения исследуемого вещества. На мно-
гих приборах (ИКС-29, UR-20 и др.) шкала волновых чисел на-
носится автоматически или используют заранее градуированные
бланки. В этом случае предварительная съемка стандартов по-
зволяет выявить приборные ошибки.
Следующей операцией является проверка чистоты иммерсион-
ной среды (растворителей, матрицы, кювет). Для этого снача-
ла снимают спектр кюветы с растворителем или пластинки
КВг против воздуха. Если в спектре матрицы обнаруживаются
посторонние полосы, то определяют их положение на шкале вол-
новых чисел и в зависимости от интересующего исследователя
участка спектра решают вопрос о пригодности матрицы для
дальнейших работ. Затем проводят проверочную съемку
двух одинаковых кювет с растворителем или пластинок
КВг. Если запись спектра близка в этом случае к линии 100%-
ного пропускания, то сравниваемые объекты одинаковы по спект-
ральным характеристикам и, следовательно, можно приступить
к анализу почвенных образцов.
Для получения качественного спектра существенное значение
имее! выбор скорости записи. На приборе ИКС-14 можно в ши-
роком диапазоне одновременно варьировать и скорость измене-
ния длин волн, и скорость протяжки диаграммной ленты. По-
скольку прибор обладает определенной инерционностью, не сле-
дует вести запись при слишком высокой^ скорости изменения длин
волн. В противном случае может иметь место смещение' макси-
мумов полос поглощения в сторону меньших волновых чисел.
Наибольшая точность измерений достигается при минимальной
скорости сканирования. Скорость протяжки диаграммной ленты
влияет на масштаб записи и точность отсчета волновых чисел.
При съемке почвенных объектов на ИКС-14 оптимальное соотно-
шение скоростей определяется установкой редуктора мотора длин
волн в положение 5, а редуктора мотора протяжки бумаги —
в положение 3.
Ширина щели подбирается с таким расчетом, чтобы получить
максимальное разрешение спектра. При слишком большом рас-
крытии щели близко расположенные полосы могут сливаться
или с образованием одного нового максимума, или без измене-
ния частоты наиболее сильной полосы. Однако слишком узкая
щель также может быть источником ошибок, поскольку умень-
шение светового потока снижает чувствительность прибора в це-
лом. Если шум усилителя не препятствует, то выгоднее умень-
шить ширину щели, увеличивая соответственно коэффициент уси-
ления.
Качество спектра можно признать удовлетворительным, если
в области минимального поглощения пропускание образца со-
188
ставляет 60—90%, а в максимумах наиболее интенсивных полос
оно не превышает 10—30%. Если обнаруживаются очень силь-
ные полосы с пропусканием менее 5—10%, то следует повторить
съемку с меньшей навеской образца. Для очень слабых полос
навеску соответственно увеличивают. В хорошо снятом спектре
полосы поглощения обычно имеют вид острых или плавных ми-
нимумов, симметричных, с постепенными переходами между
соседними полосами. Если полосы тупые, заканчиваются про-
должительными горизонтальными участками, а переходы между
ними резкие, в виде уступов, то вероятно, что чувствительность
прибора недостаточна. При съемке разностных спектров иногда
наряду с минимумами полос поглощения можно наблюдать рез-
кие максимумы, всплески, когда пропускание быстро увеличи-
вается и может превышать 100%- Это так называемое отрица-
тельное. поглощение, обычно обусловленное неравенством толщи-
ны кювет или толщины таблеток КВг. Если толщина таблетки
с образцом оказалась меньше толщины контрольной таблетки,
то в области даже слабых собственных полос примесей может
наблюдаться отрицательное поглощение. Надо очень вниматель-
но анализировать получаемые спектры, особенно при анализе
таких сложных объектов, как почва и ее компоненты, чтобы из-
бежать ошибок за счет указанных причин.
На полученной и принятой к обработке спектрограмме запи-
сывают название образца, способ подготовки к съемке, условия
съемки, дату, а у максимумов полос поглощения надписывают
волновые числа. Если полоса широкая, то указывают волновое
число центра полосы и интервал от ее начала до конца. Кроме1
полос отмечают уступы (плечи) на крыльях полосы и перегибы;
они обычно отвечают отдельным полосам, не проявившимся пол-
ностью из-за недостаточной разрешающей способности прибора.
Для сравнительной характеристики, а также при количественных
анализах измеряют интенсивности полос. В случае относительно-
простых спектров с небольшим набором узких полос их интенсив-
ность можно определить по максимуму поглощения, ведя отсчет
от линии 100%-ного пропускания. Для почвенных объектов этот
способ практически не применим из-за большого набора широ-
ких составных полос. В этом случае интенсивность оценивают
по глубинам пиков.
Оформленный спектр служит основным документом для даль-
нейшей работы. Предварительная расшифровка спектра произ-
водится по таблицам частот и атласам стандартных веществ
(Беллами, 1963; Наканпси, 1965). Таблицы волновых чисел поч-
венных компонентов можно найти в работах Д. С. Орлова (1973)
и И. С. Степанова (1974).
При расшифровке учитывают волновые числа в максимумах
поглощения, число и набор полос, их конфигурацию и относи-
тельные интенсивности. Расшифровка по таблицам и атласам
позволяет в большинстве случаев осуществить более или менее-
189
точное отнесение так называемых характеристических полос, т. е.
полос отдельных атомных групп, положение которых мало ’зави-
сит от общего строения молекул. Уточнение результатов и под-
робное определение атомных групп и связей требуют специаль-
ных исследований.
В некоторых случаях не стремятся к полной расшифровке
спектра. Для установления идентичности сравниваемых веществ
достаточно бывает сравнить спектры в широком интервале вол-
новых чисел; внешние изменения спектра могут служить доказа-
тельством происшедших изменений. Однако такие сравнения
допустимы только при полной идентичности способов подготов-
ки препаратов к съемке.
Спектрофотометр ИКС-14
Двухлучевой инфракрасный призменный спектрофотометр
ИКС-14 предназначен для регистрации спектров поглощения
твердых, жидких и газообразных веществ в области спектра от
13330 до 400 см-1 (0,75—25 мкм). Прибор обеспечивает линей-
ную развертку спектра по волновым числам и регистрирует
спектр в процентах пропускания. Запись спектра проводится пе-
ром на рулонной бумажной ленте шириной 275 мм (без перфо-
рации). Прибор удобен в работе и прост в обращении, имеет
высокую чувствительность и достаточное разрешение. Некото-
рым недостатком является необходимость замены призм при
работе в широком диапазоне длин волн.
Прибор ИКС-14 состоит из монохроматора, двухлучевого
осветителя, входного блока, выходного блока и записывающего
устройства (рис. 57). Принцип действия прибора по двухлучевой
схеме основан на нулевом методе. Радиация от источника излу-
чения (глобар) направляется по двум каналам: в одном канале
помещается исследуемый образец, в другом — фотометрический
клин и образец сравнения. С помощью прерывателя пучки света
из обоих каналов попеременно проходят через монохроматор,
разлагаются в спектр и поступают на приемник радиации —
болометр. Когда интенсивность пучков в обоих каналах одина-
кова (что при отсутствии поглощающего образца обеспечивается
оптической схемой осветителя), на болометр поступает не изме-
няющаяся во времени тепловая радиация, и сигнал на входе
усилительной системы не в_рзникает. При введении поглощающе-
го излучение образца на болометр попадают попеременно пучки
различной интенсивности; в результате на входе усилителя появ-
ляется переменный сигнал, частота которого равна частоте пре-
рывания пучков. Этот сигнал после усиления и преобразования
подается на обмотку электродвигателя отработки, который пере-
мещает фотометрический клин, уменьшая до нуля возникшую
разность интенсивностей пучков. Фотометрический клин механи-
190
чески связан с пером записывающего устройства; перемещение
пера пропорционально перемещению клина и показывает погло-
щение излучения исследуемым образцом (в %).
Рис. 67. Спектрофотометр ИКС-14:
1 — рукоятка включения глобара, 2 — рукоятка регулировки скорости диа-
граммной ленты, 3 — сигнальная лампа, 4 — кнопка пуска электродвигателя
мотора длин волн, 5 — кнопка пуска электродвигателя диаграммной ленты,
6 — кнопка пуска электродвигателя прерывателя, 7 — рукоятка регулировки
фотометрического клина, 8 — окна, 9 — рукоятка регулировки усиления, 10 —
экран индикатора, 11 — рукоятка регулировки баланса, 12— тумблер включе-
ния разбаланса моста, 13 — тумблер включения усилителя, 14 — рукоятка ре-
гулировки угла теневого сектора, 15— тумблер включения мотора длин волн,
16 — тумблер включения мотора диаграммной ленты, 17 — тумблер включения
мотора прерывателя, 18 — тумблер включения мотора отработки, 19 — перо,
20 — диаграммная лента, 21 — микрометрический винт для регулировки шири-
ны щели, 22 — барабан длин волн
Порядок работы.
1. Подключить прибор к сети переменного тока, включить
усилитель тумблером 13, переключатель 12 поставить в
положение «Разбаланс моста» и дать усилителю прогреть-
ся 15 мнн.
2. На экране индикатора 10 установить угол теневого сек-
тора примерно 45°, вращая рукоятку 14.
3. Включить воду для охлаждения глобара.
4. Вынуть заслонку из окон блока источника излучения и
включить глобар рукояткой 1.
При достаточном напоре воды в системе охлаждения
глобар разогревается и начинает светиться. Если свечение
отсутствует, а на пульте загорается сигнальная лампа Зг
надо увеличить подачу воды.
5. Установить микрометрический винт 21 на деление «11».
191
Рекомендуется раскрывать щель до такой величины, при
которой еще возможно получить требуемое разрешение.
6. Включить электродвигатель прерывателя тумблером 17
и кнопкой 6.
7. Включить электродвигатель отработки тумблером (18).
8. Повернуть рукоятку усиления 9 на 2—4 деления шкалы.
Открыть оба окна 8. Последовательно перекрывая окна
на короткий промежуток времени так, чтобы перо отхо-
дило от положения равновесия приблизительно на 20%,
подбирают такое усиление, при котором перо будет воз-
вращаться на одно и то же место с разбросом 0,5%. При
недостаточном усилении перо будет медленно возвра-
щаться к положению равновесия; при чрезмерном усиле-
нии могут возникнуть автоколебания, нарушающие нор-
мальную работу прибора.
9. Закрыть оба окна заслонками 6. Перо записывающею
устройства 19 должно остановиться на середине шкалы.
Если перо перемещается в одну сторону, то вращением
рукоятки «Баланс» 11 следует добиться, чтобы оно коле-
балось относительно какого-то среднего положения.
10. Вынуть заслонки из обоих окон. Перо должно двигаться
к отметке 100%-ного пропускания. Если перо не останав-
ливается на делении 100%, отрегулировать его положение
рукояткой фотометрического клина 7.
11. Ушановпть барабан длин волн 22 на деление, с которо-
го будет производиться запись Барабан поворачивается
вручную, если рукоятку скорости развертки спектра поста-
вить между делениями шкалы коробки скоростей.
12. Установить требуемую скорость развертки спектра (на
определенное деление шкалы коробки скоростей) и ско-
рость движения диаграммной ленты рукояткой 2.
13. Закрепить исследуемый образец и образец сравнения в
держатели. Держатели представляют собой две парал-
лельно скрепленные между прижимными винтами метал-
лические пластины. В центре пластины имеются прямо-
угольные окна, соответствующие размеру отпрессованных
пластинок. Пластинки вставляются в окна держателей и
фиксируются с помощью прижимных винтов. Держатели
установить в окна прибора (образец — в левом, конт-
роль — в правом по ходу световых потоков).
14. Включить электродвигатель отработки тумблером 18 и
дать перу остановиться.
15. Включить электродвигатели: диаграммной ленты тумб-
лером 16 и кнопкой 5 и мотора длин волн тумблером
15 и кнопкой 4.
16. Записать спектр, отмечая в ходе записи деления отсчет-
ного барабана при прохождении пера через максимумы
полос поглощения.
192
17 По окончании записи проверить угол теневого сектора,
установив его примерно на 45° рукояткой 14
18. По окончании работы выключить все тумблеры на панели
прибора, поставив их в положение «Выключено», выклю-
чить глобар и усилитель. Затем отключить воду, отсоеди-
нить прибор от сети, закрыть окна заслонками
Рис 58 Дв^хлучсвой автоматический инфракрасный спектрофото
метр ИКС 29
В настоящее время отечественной промышленностью выпус-
каются автоматические инфракрасные спектрофотометры ИКС-29
(рис 58) на дифракционных решетках с регистрацией спектров
поглощения по пропусканию и волновым числам для области
4200—400 см'1, модель ИКС-27 — для области спектра 4000—
650 см-' и ИКС-31 — для области спектра 1000—200 см-1
Для расшифровки полученных ИК-спектров можно восполь-
зоваться приведенными ниже характеристиками минеральных и
органических компонентов почв
ИК-спектры гумусовых веществ
Средь почвенных органических компонентов методом ИКС
наиболее подробно изучены гуминовые кислоты и фульвокисло-
ты Другим классам органических веществ уделено значительно
меньше внимания
7 Зак 284
193
775 600
7
Рис. 59. ИК-спектры гумусовых кислот, выделенных из гор. Ai южного черно-
зема и лигнина:
1 — гуминовая кислота, 2 — фульвоскислота, 3 — гиматомелановая кислота, 4 — лигнчя
В некоторых интервалах волновых чисел спектры многих поч-
венных компонентов очень схожи (рис. 59 и 60). Причем это вы-
является не только при сравнении спектров гуминовых кислот и
фульвокпслот, но и других составляющих. И минеральные и орга-
нические части почвы имеют широкие полосы в области 3200—
3500, 1600—1700 и около 1000 см-1. Такая общность вызвана
значительным содержанием гидроксильных групп в органических
веществах, глинистых минералах и гидроксидах. В то же время
в спектрах отдельных минералов и гумусовых веществ обнару-
живаются специфические особенности, которые позволяют ана-
лизировать не только фракционированные образцы, но и доволь-
но сложные по составу смеси. Например, при съемке разностных
спектров удалось установить наличие полос С=С-двойных свя-
7*
195
зей непосредственно в почве без какой-либо химической обра-
ботки.
Гуминовые кислоты и фульвокислоты имеют широкие и ин-
тенсивные полосы поглощения при JJ51H1—3300 см-1, обусловлен-
ные гидроксильными группами, в той пли иной мере связанными
водородными связями. В том же интервале поглощают и группы
NH, но их содержание меньше, чем групп ОН, и отдельные по-
лосы, как правило, не выявляются. На длинноволновом крыле
упомянутой полосы появляется
Рис. 61. ИК-спектры смеси фульво-
кислоты и фульвата натрия до (У)
и после (2) катионирования
одна или две полосы с максиму-
мами при 2920 п 2860—2850 см-1;
они обусловлены метиленовы-
ми группами СН2 и частично
метильными группами СН3. По-
лосы последних при 2960 и
2870 см-1 обычно лучше выраже-
ны в спектрах почвенных липи-
дов (воскосмол), чем гумусовых
кислот.
Следующая интенсивная по-
лоса обнаруживается в интерва-
ле 1720—1700 см-1 и обусловлена
преимущественно карбонилом
карбоксильных групп СООН. Ин-*
тенсивность этой полосы может
меняться в больших пределах,
что давало повод для суждения
о неодинаковом содержании кар-
боксильных групп в гумусовых
кислотах различного происхож-
дения. Такие выводы следует де-
лать крайне осторожно, посколь-
ку' исследуемые препараты часто
содержат заметные количества
золы и поэтому часть карбок-
сильных групп может в них нахо-
диться в ионизированной фор-
ме. Истинное значение интенсивности можно получить только
в том случае, когда препарат на 100% представлен кислотной
формой. Например, в спектре препарата, представленного сме-
сью фульвокислоты и фульвата натрия, полоса карбоксильной
группы обнаруживается только в виде уступа (рис. 61). После
пропускания фульвокислоты через кагионит в Н+-форме все
ионы натрия были замещены на Н4-, т. е. R—COONa—>-RCOOH,
и выявилась интенсивная полоса СООН-групп при 1700 см-1.
Одновременно практически полностью исчезла полоса около
1400 см-1, свойственная ионизированной группе СОО~, а полоса
с максимумом 1595 см-1 сместилась до 1610 см-1. Очевидно, что
полоса при 1595 см-1 была сложной и включала поглощение
196
1
ионизированной карбоксильной группой около 1600 см-1. После
катпонпрования эта полоса также исчезла, и выявилось истин-
ное значение положения полосы 1610 см~'. Этот пример одно-
временно подчеркивает необходимость осторожной интерпрета-
ции спектров в тех случаях, когда можно ожидать взаимное
наложение полос. При недостаточном разрешении спектра нало-
жение полос может вызвать кажущееся смещение одной из по-
лос и ошибочный вывод о прошедших химических (структурных)
изменениях.
В области 1600—1650 см-1 проявляются полосы С=С-связей
алифатических и ароматических систем, однако здесь же влияет
вода, а также азотсодержащие группировки. Последние вообще
сопутствуют почти всем важнейшим полосам в спектрах гумусо-
вых кислот, поскольку азотсодержание группы поглощают коле-
бания в тех же интервалах частот, что и аналогичные углерод-
содержащие группы.
Вид спектра в области 1600—1650 см-1 может быть различен
в зависимости от состава гумусовых кислот. При высоком содер-
жании азота и хорошем разрешении выявляется полоса амид I
около 1650—1640 см-1; ей обычно сопутствует полоса амид II
при 1550—1540 см-1. Обе полосы исчезают после кислотного гид-
ролиза.
Дополнительным подтверждением ароматичности гумусовых
веществ может служить сравнительно слабая полоса с максиму-
мом 1510 см -1.
Полосы метильных и метиленовых групп в спектрах гумусо-
вых кислот, как отмечалось, легко идентифицируются в области
валентных колебаний С—Н при 2920—2860 см-1. Они же обна-
руживаются и по деформационным колебаниям в интервале
1480— 136Q см~‘, (несколько полос). Однако поглощение в по-
следнем диапазоне осложняется за счет С—О-связей и полосы
ионизированной карбоксильной группы около 1400 смД
С карбоксильными группами в значительной мере связано
происхождение полосы и в области 1260—1200 см Д о чем гово-
рит резкое снижение ее интенсивности в результате замещения
водорода и образования гуматов и фульватов.
Сильное поглощение в области 1100—1000 см-1 обычно свя-
зывают с гидроксильными спиртовыми группами, но интерпрета-
ция полос в этой области должна проводиться с максимальной
осторожностью из-за влияния полисахаридов, имеющих сильную
полосу в той же области спектра, а также из-за сильных полос
групп S:—О в спектрах гумусовых веществ с высокой зольно-
стью.
Общая сводка важнейших полос поглощения гумусовых кис-
лот дана в табл. 18.
Очень характерны ИК-спектры липидной фракции, экстраги-
руемой из почв спиртобензольной смесью («битумы», или воско-
смолы). Около 3000 см-1 в их спектрах проявляются полосы ОН-
197
Таблица 18
Полосы поглощения гумусовых кислот и некоторых сопутствующих компонентов
Волновое число, см"1 Интенсивность полосы Группа (колебание)
3600 средняя или слабая несвязанная ОН (валентное)
3500—3300 сильная ОН, связанные межмолекулярными водород-
3200 слабая ными связями, частично NH (валентное) NH (валентное)
2920 и 2860 средняя или слабая СН2 (СН3) (валентное)
2960 и 2850- СН3 (валентное), СН2 (валентное)
2600 слабая, уступ ОН карбоксилов в димерах карбоновых кислот
1850 и 1785 — (валентное) 5- и 7-членные циклические ангидриды
1835 и 1765 слабая ангидриды кислот
1775 ацетатная группировка
1745 С=О в СН3СОО-
1725—1700 сильная С=О в СООН, частично другие С=О и слож-
1690—1630 — ные эфиры (валентное) C=N (валентное)
1650 переменная амид I
1625—1600 сильная сложная полоса, обусловленная группами
1610 средняя С=С, СОО-, полосой амид I и частично гигроскопической водой. Обнаруживается и в глинистых минералах С=С, ароматических соединений
1590—1580 и переменная СОО-
1400—1390 1540 переменная амид II (возможно, нитрогруппа в ГК, окис-
1510—1500 слабая ленных азотной кислотой) С=С, ароматических соединений СН в СН2 (или СН3) (деформационное)
1460—1440 слабая, иногда сред-
1400—1390 няя средняя сложная полоса, СН, СОО-, ОН
1375” С—СН3
1325 и 780 — оксалаты кальция
1250—1225 переменная карбоксильные группы
(1260—1200) (С—О, частично ОН)
1150 переменная третичные спирты
1100 переменная вторичные спирты
1050 переменная первичные спирты
1080—1050 переменная полисахариды
900—860 слабая, часто от- СНаром> деформационное
860—730 с у тс тв у ет слабая, часто от- СНаром, деформационное
750 и 490 сутствует слабая С—С и С—О полисахаридов
730 и 720 слабая (СН2)П при п > 4
групп п валентных С—Н-колебаннй. В диапазоне 2000—700 см-1
(призма NaCl) выявляется большое число узких, хорошо очер-
ченных полос с четкими максимумами. Серия интенсивных полос
в интервале 1480—1380 см-1 принадлежит симметричным дефор-
мационным колебаниям CHs- и СНг-групп. Сильная полоса око-
198
Таблица 19
Волновые числа максимумов полос поглощения некоторых солей н минералов
Соль, минерал Волновые числа, см""1 (интенсивность полосы)
1 2
N а2СО3, К2СО3 MgCOs СаСО3 Кальцит (Саяк, Казахская ССР) Арагонит (Улухо-Хонлу), Армян- ская ССР) Доломит (Приморский край) Сидерит (КМА) карбонаты 1750—1760 (ср), 1450 (с), 1375 (с), 880 (с), 700—705 (ср) 1640 (ср), 1520 (с), 1420 (с), 855 (с), 705 (сл) 1790 (ср), 1430 (с), 875 (с), 710 (ср) 1790 (ср), 1425 (c), 1160 (сл), 875 (с), 710(c) 1780 (сл), 1430(c), И60(сл), 880(c), 715 (ср) 1620 (сл), 1420 (ср), 1065 (с), 1020 (сл), 930 (с), 690 (ср) 1430 (с), 1170 (ср), 895 (ср), 795 (ср)
Бикарбонаты
КНСО3 1850 (сл), 1640 (ср), 1400(c), 1370(c), 1000(c), 980 (с), 840 (с), 705 (с)
NaHCO3 1765 (сл), 1375 (с), 1140 (сл), 835 (с), 715 (сл) Сульфаты
Na2SO4, K2SO4 Тенардит (Усун-Су, Туркменская ССР) Мирабилит (Евпатория) CaSO4 Гипс (Кунгур, Урал) 1380 (сл), ИЗО (с), 980 (сл), 785 (сл), 615(c) 1620 (сл), ИЗО (с), 795 (сл) 1620 (сл), ИЗО (с), 780 (сл), 1625 (ср), 1150 (с), 1015 (сл), 670 (с) 1695 (ср), 1620(c), 1415 (сл), 1150(c), 1115(c), 1015 (сл), 780 (сл), 670 (с) Фосфаты
СаНРОг2Н2О 1645 (с), 1380 (ср), 1125 (с), 1060 (с), 985 (с), 870 (с)
Апатит (Хибины) Вивианит (Керчь) 1620 (ср), 1095 (с), 1045 (с), 960 (сл), 740 (сл) 1615 (ср), 1370 (сл), 1045 (с), 1005 (с), 960 (с), 945 (с), 790 (ср) Нитраты
KNO3 NH4NO3 1820 (сл), 1370, (с), 890 (с) 1760 (ср), 1365 (с), 935 (ср), 835 (с), 715 (ср) Силикаты
SiO2-nH2O Кварц (Дульдурга) Опал (Чиатура, ГрузССР) Микроклин (Мама, Иркутская обл.) Мусковит (Мама, Иркутская обл.) Биотит (Вишневые горы, Урал) Флагопит (Слюдянка, Байкал) Вермикулит (Ковдор, Кольский п-ов) 1150 (с), 1090 (с), 795 (ср) 1170 (с), 1090 (с), 795 и 770 (с), 695 (ср) 1615 (ср), ИЗО (ср), 1080(c), 795 (ср), 695 (ср) 1620 (ср), ИЗО (с), 980 (с), 770 (с), 720 (с) 1620 (ср), 1070—970 (с), 830 (сл), 750 (ср) 1615 (сл), 1020—960 (с), 720 (ср), 680 (ср) 1625 (ср), 1010—960 (с), 720 (ср), 690 (ср) 1615 (сл), 1065—1015 (с), 825 (сл), 796 (сл), 745 (ср)
199
Продолжение табл. 19
! |
Хлорит (Урал) 1610 (сл), 1430 (сл), 1010—960 (с), 880 (сл), 760 (ср), 725 (сл)
Глауконит (ЭССР) 1615 (сл), 1430 (сл), 10/0—960 (с), 805 (ср), 670 (ср)
Каолинит (УССР) 1620 (сл), 1420 (сл), 1100(c), 1030 (c), 1015(c), 935—910 (с), 790 (ср), 750 (ср), 695 (с)
Иллит 1630 (ср), 1460 (ср), 1190 (сл), 1160 (сл), 1060—1000 (с), 910 (ср), 825 (с), 745 (с), 670 (ср)
Нонтронит 1620 (ср), 1170 (сл), 1040—960 (с), 790 (сл)
Монтмориллонит (ГрузССР) 1640 (сл), 1050 (с), 1015 (с), 915 (ср), 795 (ср), 685 (сл)
Примечание, с — сильная полоса, ср — средняя, сл — слабая.
ло 880 cm j, вероятно, принадлежит концевым группам С = СНг,
полоса 1760 см Л возможно, ее обертон. Поглощение около
1670—1640 см-1 связано с несопряженнымн двойными связями,
но здесь же возможно влияние групп СО альдегидов и кетонов.
Между 1100 п 1030 см-1 наблюдается сильное поглощение, пред-
ставленное тройной полосой, что может быть вызвано колеба-
ниями OH-ipynn первичных, вторичных и третичных спиртов.
Внешний вид спектров «битумов» и гумусовых кислот резко
различен, и по этому признаку их можно легко отличить друг о г
друга. Но ге и другие содержат целый ряд одинаковых полос,
потому в смешанных препаратах не всегда удается установить,
какие из полос принадлежат «битумам», а какие — гумусовым
кислотам. Для контроля за чистотой гумусовых кислот, вероят-
но, могут быть использованы полосы «битумов» при 650, 680,
880, 1766 и 1820 см-1. О влиянии минеральных примесей на
спектры гумусовых веществ можно судить по материалам
табл. 19.
ИК-спектры минеральных
компонентов почвы
Среди минеральных компонентов почвы важнейшую ррль
играют глинистые минералы. Все глинистые „минералы имеют
полосы поглощения в области валентных колебаний ОН-групп
при 3700—3400 см'1. Полосы валентных колебаний ОН- сме-
щаются в область меньших частот до 3400 см'1 при образова-
нии водородных связей; в том же диапазоне проявляются поло-
сы, обусловленные адсорбированной водой (рис. 62). Идентифи-
цировать минералы по этой области спектра не всегда просто
200
из-за близкого расположения полос и их смещения под влиянием
разной степени обводненности.
В длинноволновой области спектра глинистые минералы
обладают также довольно близким набором полос, что, естест-
венно, обусловлено близким набором атомных групп и' связей.
Перечень важнейших полос приведен в табл. 19; в ряде случаев
в этот перечень включены по-
лосы и адсорбированной воды,
поскольку спектры получены
для воздушно-сухих образцов.
Наиболее сильные полосы у
всех силикатов обнаружива-
ются около 1000 см-1. Эти по-
лосы обусловлены валентны-
ми колебаниями связей Si—О
или —О—Si—О— и у отдель-
ных минералов несколько раз-
личаются по частоте и конфи-
гурации. У аморфной кремне-
кислоты и кварца это двойная
полоса с максимумами около
1150—1170 и 1090 см-1; для
—।----1_______ _____I—।
3625 MOO err’ 36953620 см~г
Рис. 62. Полосы валентных колебаний
ОН-групп в спектрах монтмориллонита
(/) и каолинита (2):
а — выдержан в атмосфере влажного
воздуха, б—высушен при 100°, 24 часа,
в — высушен при 100°, 36 часов
каолинита характерны макси-
мумы при 1090, 1030 и 1010 см-1, для монтмориллонита — 1050
и 1015 см-1. Некоторые из этих максимумов, как и в других об-
ластях спектра, проявляются только в виде уступа, и их конфигу-
рация зависит от концентрации вещества и ширины щели. Дру-
гая характерная полоса группы —Si—О— относится к деформа-
ционным колебаниям и проявляется в области 470—430 см-1.
Связями Н—О—А1 (или вообще Н—О—Х3+) обусловлены
полосы около 900 см-1. У каолинита и диккита это дублет с вол-
новыми числами 910 и 935 см-’, у галлуазита единственный пик
при 910 см"1; Al-бейделлит имеет полосу при 935, а нонтронит
при 830 см-’. Группам Si—О—А1 свойственно поглощение около
540 см-’. Таким образом, спектры чистых минералов имеют ряд
специфических особенностей, вполне пригодных для их диагно-
стики. Анализ образцов, содержащих одновременно несколько
минералов, как это характерно для почв и почвенных коллоидов,
значительно труднее.
Кварц легко обнаруживается по дублету 770—795 см-1;
аморфные модификации кремнезема вместо дублета обнаружи-
вают одну полосу с максимумом при 795 см-1. Характерные
спектры ряда минералов показаны на рис. 63.
Характерным поглощением обладают карбонаты (см.
табл. 19). Все нормальные карбонаты имеют сильную полосу
при 1450—1410 см~> и при 880—850 см-1, причем частота цент-
ра полосы 880—850 см-' у многих карбонатов линейно зависит
201
7
Рис 64. ИК-спектры солей:
1 — Na2SO4, 2 — СаСОз
от логарифмов массы катионов. Для различения кристаллических
форм СаСО3 метод ИКС более чувствителен, чем рентгеногра-
фия. По характеристическим полосам карбонаты легко обнару-
живаются, как и кварц, в глинистых минералах и почвах.
Бикарбонаты поглощают при 1420—1400, 1000—990, 840—
830 и 705—695 см-1, ион аммония интенсивно поглощает излуче-
ние при 3335—3030 и 1485—-1390 см-1, а нитрат-ион — при 1410—
1340 и 860—800 см-1; различные фосфаты характеризуются ин-
тенсивными полосами поглощения при 1100—950 см-"1, сульфа-
ты — при ИЗО—1080 и 680—610 см 1 (табл. 19; рис. 64).
203
Рис. 65. ИК-спектры почв:
1 — сильноподзолистая, гор. А2, 2 — дерново-подзолистая, гор. А>, 3 — чернозем обык-
новенный, гор. Ai, 4 — серозем, гор. Ai, S — краснозем, гор. В1
Практически все почвенные объекты содержат разные формы
воды, дающие интенсивное поглощение вблизи 3500—3400 и
1650 см-1.
ИК-спектры почв
Почвы различных типов имеют характерный набор общих
полос, а также ряд полос, свойственных только некоторым груп-
пам почв (рис. 65). Во всех случаях основу спектров составляют
полосы силикатов: сильные и средние по интенсивности полосы
Si—О пр и^ 680—690 и 1000—1100 см-1, дублет при J70—795
(кварц), слабые полосы Si—О—Mg-связей при 910—91S см-1 и
полосы поглощения воды и ОН-групп при 1600—1660 и 3400—
3550 см-1.
В карбонатных почвах четко выявляются полосы СО32~ при
705—707, 870—875 и 1430—1450 см-1.
Изучение ИК-спектров
почвенных компонентов
Перед тем как приступить к изучению почв, необходимо про-
вести градуировку прибора. С этой целью выполняют следующие
операции.
1. Прибор ИКС-14 подключают к сети, включают тумблер и
прогревают усилитель 15 мин. Производят настройку при-
бора, как указано на с. 191.
2. Держатель с пленкой полистирола помещают в левое окно
спектрофотометра (по ходу луча).
3. Записывают спектр полистирола (против воздуха), отме-
чают на диаграммной ленте показания барабана длин волн
для контрольных полос поглощения, указанных в атласе,
прилагаемом к прибору (см. рис. 56).
4. После этого строят график, откладывая по оси ординат
волновые числа, а по оси абсцисс — показания барабана
длин волн.
После того, как прибор был проградуирован, можно присту-
пить к съемке спектров образцов почв, илистых фракций или
гумусовых веществ. Спектры илистой фракции получают следую-
щим образом.
1. 10—15 мг образца илистой фракции растирают до состоя-
ния пудры в агатовой ступке и высушивают в вакуумном
шкафу при 105° в течение трех часов.
2. 0,6—1 мг высушенного образца смешивают с 200 мг пред-
варительно высушенного и тонкорастертого порошка КВг.
Смесо тщательно растирают в ступке или вибромельнице.
3. Смесь переносят в пресс-форму и прессуют (см. с. 185).
Одновременно прессуют две контрольные пластины из чи-
стого КВг.
205
4. Закрепляют пластинку чистого КВг в держателе и поме-
щают ее в левое окно прибора (по ходу луча).
5. Снимают спектр пластины КВг против воздуха, на спектре
отмечают полосы, которые могут быть обусловлены при-
месями в КВг. В зависимости ог наличия полос решают
вопрос о пригодности КВг к работе.
6. Закрепляют вторую пластинку чистого КВг в держателе
и помещают ее в правое окно прибора (по ходу луча).
Производят проверочную съемку разностного спектра двух
одинаковых пластинок КВг. Если запись спектра близка к
линии 100%-ного пропускания, приступают к съемке спек-
тра образца илистой фракции.
7. Пластинку КВг, содержащую навеску образца илистой
фракции, закрепляют в держателе и помещают ее в левое
окно прибора (по ходу луча). Записывают спектр образца
против чистого КВг, отмечая на диаграммной ленте макси-
мумы полос поглощения. При наличии в спектре широких
полос отмечают интервалы начала, конца и середины их.
При записи спектров ведут регистрацию не только от-
дельных четко выраженных полос поглощения, но и отме-
чают уступы (плечи) на крыльях полос и перегибы; они
обычно отвечают отдельным полосам, не проявившимся
полностью из-за недостаточной разрешающей силы при-
бора.
8. Находят максимумы полос поглощения в см-1, используя
калибровочный график.
9. Определяют, каким атомным группам и типам связей при-
надлежат максимумы полос поглощения. По числу и на-
бору полос, их конфигурации и относительной интенсивно-
сти определяют входящие в состав образца минералы, ис-
пользуя атласы и сводные таблицы (см. табл. 18—19).
10. На спектрограмме записывают название образца, способ
подготовки к съемке, условия съемки, дату, а у максиму-
мов полос поглощения надписывают волновые числа и
атомные группы.
Спектры гумусовых веществ получают аналогичным способом.
Некоторые особенности связаны только с подготовкой образца,
поскольку высушивание при 105° может вызвать разложение пре-
паратов Ход получения спектров следующий.
1. 10—15 мг препарата растирают до состояния пудры в
агатовой ступке и высушивают в вакуумном» шкафу при
60—70° в течение 3—5 ч.
2. 1 мг высушенного препарата смешивают с 200 мг предва-
рительно высушенного и тонкорастертого порошка КВг.
Смесь тщательно растирают в ступке или вибромельнице:
3. Смесь переносят в пресс-форму и прессуют пластину, как
указано на с. 185. Одновременно прессуют две пластины
из чистого КВг.
206
Далее поступают, как описано на с. 206, начиная с п, 4,
4. Предварительная расшифровка спектра производится по
таблицам частот и атласам стандартных веществ (Белла-
ми, 1963; Наканиси, 1965), а также по таблице волновых
чисел гумусовых кислот (см. табл. 18).
ЛИТЕРАТУРА
Беллами Л. Инфракрасные спектры сложных молекул. М., ИЛ, 1963.
Наканиси К. Инфракрасные спектры и строение органических соединений.
М., «Мир», 1965.
Орлов Д. С. Гумусовые кислоты почв. М., Изд-во Моск, ун-та, 1974.
Плюснина И И. Инфракрасные спектры минералов. М., Изд-во Моск, ун-та,
1977.
ОПРЕДЕЛЕНИЕ В ПОЧВЕ
СОДЕРЖАНИЯ МАКРО- И МИКРОЭЛЕМЕНТОВ
МЕТОДОМ ЭМИССИОННОГО
СПЕКТРАЛЬНОГО АНАЛИЗА
Назначение и возможности метода
в почвенных исследованиях
Под спектральным эмиссионным анализом понимают физиче-
ский метод анализа химического состава вещества, основанный
на наблюдении и измерении оптических’ спектров излучения ато-
мов, ионов и, реже молекул. Излучаемые атомами и ионами
эмиссионные линейчатые спектры не зависят от вида химических
соединений, из которых состоит исследуемое вещество. Поэтому
этот вид анализа применяется для определения элементного
(атомного) состава почв.
Как и все другие методы исследования, эмиссионный спект-
ральный анализ имеет свои преимущества и недостатки, ограни-
чивающие его возможности применительно к исследованию почв.
Основные преимущества спектрального метода перед другими
состоят в его универсальности, высокой чувствительности, точно-
сти и скорости определения. Метод дает возможность одновре-
менно в одной навеске почв без предварительной подготовки
определить до 30 элементов. Многие элементы в почве могут
быть определены при содержании 10-3—10-4%. При определении
малых концентраций (10~3—10~5%) точность эмиссионного спек-
трального метода достигает 10—30%.
Применение фотоэлектрических методов регистрации спектра
при определении высоких концентраций элементов (от 1 % и вы-
ше) дает возможность увеличить точность до 1—2%. Поэтому
наибольшее распространение эмиссионный спектральный анализ
получил для определения щелочных, щелочноземельных элемен-
тов в почвах (К, Ка, Са и др.), а также для определения микро-
элементов.
Сопоставление среднего содержания микроэлементов в почвах
и чувствительности их определения спектральным методом пока-
зывает, что, используя простую технику определения, одновре-
менно в почве можно определить большинство из наиболее важ-
ных микроэлементов. В их число входят биолошчески незамени-
208
мне В, Мп, Си. Наряду с ними определяются Ba, Ti, Zr, Sr, Сг,
V, Ni, Li, Rb, иногда Co. Средние концентрации Zn, Mo, некото-
рых других тяжелых металлов (Hg, Cd, Pb, As) в почвах ниже
чувствительности их прямого определения. Для гЛнтролирования
содержания их в почвах необходима прецизионная техника опре-
деления или приемы предварительного концентрирования. Мето-
ды определения Si, Al, Fe, Mg в почвах пока недостаточно раз-
работаны и мало применяются в почвоведении.
Методы эмиссионного спектрального анализа можно разде-
лить на две большие группы, различающиеся техникой выполне-
ния. Первую группу составляют методы пламенной фотометрии,
требующие, как правило, перевода пробы в раствор. Растворы
с помощью распылителей вводят в сравнительно низкотемпера-
турное газовое пламя; интенсивность излучения элементов изме-
ряется фотоэлектрическими приборами.
Вторая группа спектральных методов применяется для опре-
деления макро- и микроэлементов непосредственно в почвах (без
их специальной подготовки и разложения) с фотографической
регистрацией спектра, использованием электрической дуги в ка-
честве источника возбуждения. Это деление условное и сделано
лишь для простоты рассмотрения наиболее часто применяемых
методик в почвенно-аналитической практике. В реальной дейст-
вительности известны многие варианты спектральных методов,
где почвы анализируют на макро- и микрокомпоненты с предва-
рительным химическим разложением и фотоэлектрической реги-
страцией спектра.
Основы теории эмиссионного
спектрального анализа
В методическом отношении эмиссионный спектральный ана-
лиз состоит из следующих трех основных, связанных между со-
бой процессов и операций (рис. 66).
1. Возбуждение спектров при сжигании некоторою количест-
ва исследуемого вещества в пламени, электрической дуге,
искре или в другом подходящем источнике. При сжигании
проб происходит испарение, молекулярные соединения
обычно диссоциируют на атомы, которые возбуждаются и
дают свечение.
2. Разложение сложного излучения возбужденных атомов в
спектр с помощью спектрального прибора, который про-
странственно разделяет монохроматические составляющие
(спектральные линии) и располагает их в упорядоченную
систему по длинам волн (спектр).
Под спектральной линией понимают монохроматическое
излучение света, соответствующее одному определенному
переходу электрона в атоме из квантового состояния с
209
большей энергией в состояние с меньшей энергией. Полный
набор упорядоченного расположения по длинам волн спект-
ральных линий, возникающих в условиях данного способа
возбуждения, составляет спектр вещества.
14
Рис. 66. Принципиальная сх-ема эмиссионного спектрального анализа:
1—источник света (проба), 2, 3, 4 — конденсорные линзы, 5 — диафрагма
Гартмана, 6 — входная щель спектрального аппарата, 7 — зеркальный объек-
тив, 8 — кварцевая призма, 9 — кварцевый объектив, 10 — зеркало, 11— фо-
кальная плоскость (регистрация спектра), 12—визуальное наблюдение види-
мой области спектра прн помощи окуляра, 13 — фотографический метод реги-
страции спектра, 14 — фотоэлектрический метод регистрации спектра (а — фо-
тоумножитель, б — усилитель, в — самописец): А — амперметр, V — вольт-
метр, R — балластное сопротивление
3. Спектр исследуемого вещества регистрируется визуально
(глазом), фотографически или с помощью фотоэлектриче-
ской регистрации (фотоэлементы, фотоумножители). В за-
висимости от типа применяемого аппарата и способа реги-
страции спектра спектральные приборы подразделяют на
стилоскопы и стилометры, предназначенные для визуаль-
ного наблюдения спектра, спектрографы, у которых спект-
ры регистрируются на фотографическую пластинку, фото-
метры и квантометры с фотоэлектрической регистрацией
спектра.
Происхождение спектров
Атомы Сионы) всех элементов могут находиться в нормаль-
ном (основном) и возбужденном состояниях. В нормальном со-
стоянии атомы обладают минимальной энергией Ео и в этом слу-
210
чае не излучают. Поглощение и излучение световой энергии
обусловлены переходом атомов из нормального состояния в воз-
бужденное и обратно. Возбуждение атомов можно осуществить
различными способами.
1. Быстролетящие электрон, ион, атом, молекула при столкно-
вении (или в результате действия электрического поля)
передают частице вещества энергию, которая трансформи-
руе"ся в энергию возбуждения атомов, ионов, молекул и,
далее, в квант излучаемой ею световой энергии.
2. Возбужденный атом или ион при столкновении или взаи-
модействии с невозбужденными частицами вещества может
передать им свою энергию.
3. Путем поглощения квантов лучистой энергией.
4. Химическая энергия реакции служит в ряде случаев при-
чиной возбуждения атомов и ионов, молекул вещества и
излучения света.
Возбужденный атом по истечении некоторого времени (по-
рядка 10-Б с) самостоятельно (спонтанно) возвращается в нор-
мальное или какое-либо промежуточное возбужденное состояние.
Освобождающаяся при таком переходе энергия излучается в ви-
де кванта hv и наблюдается в виде одной спектральной линии
с длиной волны X. Длина волны измеряется в ангстремах (1А—
= 10_8 см) иль в нанометрах.
Длина волны излучения связана с частотой соотношением Л = —,
V
где К — длина волны, с — скорость света, v — частота излучения. Вы-
ражая процесс излучения формулой, получаем ДЕ = Ег — Ео = hv =
К
где Ех и Efl — энергия исходного возбужденного и конечного состоя-
ния атома, h—постоянная Планка.
Число возможных энергетических переходов, соответственно
и число спектральных линий в спектре зависит от строения ато-
ма каждого элемента. На практике имеет место одновременное
излучение миллиардов атомов и наблюдаются излучения, соот-
ветствующие комбинациям всех возможных энергетических со-
стояний атомов данного элемента, т. е. излучение всего спектра
элемента. Число возможных энергетических переходов и, следо-
вательно, число спектральных линий в спектре определяется
строением и динамикой электронных уровней атома каждого
элемента.
Спектры сложных веществ, к которым принадлежат и почвы,
состоящие из атомов многих элементов, представляют собой сум-
му спектров этих элементов. В условиях идеальной разрешаю-
щей способности спектральных аппаратов это позволяет прово-
дить качественный анализ проб любого химического состава.
Для количественных определений выбирается одна из линий
спектра, которая легко обнаруживается уже при минимальном
количестве вещества. Такие линии получили название «поспщ1-
211
них», аналитических линий, так как в спектре они наиболее ин-
тенсивны и исчезают последними при уменьшении концентрации.
Измерив интенсивность этой линии, устанавливают количество
атомов данного элемента во взятой пробе.
Интенсивность спектральных линий
Интенсивность I спектральных линий в общей форме выра-
жается следующим уравнением:
I = frv -А N е кт ,
mi k тп птц zv0c
ёп
где 1тп— интенсивность спектральной линии, Атп— вероятность
перехода с уровня т на уровень п, g,„ и gn — статистические
веса верхнего и нижнего состояний энергии, No — общее число
атомов, Ет — энергия верхнего возбужденного состояния, К —
константа Больцмана, Т — температура источника возбуждения.
Из этой формулы следует, что температура источника воз-
буждения определяет соотношение интенсивностей отдельных ли-
ний п весь спектр в целом.
С увеличением температуры число возбужденных атомов бы-
стро растет, количество переходов, совершаемых в единицу вре-
мени, увеличивается и интенсивность излучения данной линии
спектра также возрастает, поэтому повышение температуры зна-
чительно увеличивает чувствительность метода, однако роль тем-
пературного фактора весьма ограничена, так как с повышением
температуры усложняется спектр, а при использовании атомных
линий чувствительность может даже понизиться вследствие иони-
зации атомов.
Энергия верхнего уровня атома является основным фактором,
определяющим интенсивность спектральных линий. Чем меньше
энергия, затрачиваемая на возбуждение агома до нужного уров-
ня, тем легче совершаются электронные переходы и тем выше
интенсивность излучения. Энергию, которую нужно затратить для
перемещения валентного электрона с нормального уровня на
более высокий уровень, называют потенциалом возбуждения и
выражают в электрон-вольтах. Потенциалы возбуждения раз-
личных элементов находятся в пределах 1,5—20 эВ. Наимень-
шие потенциалы возбуждения имеют щелочные элементы, наи-
большие — инертные газы.
В спектральном анализе используют спектры излучения
некоторых молекул. Например, спектр излучения радикала CN
используют для обнаружения углерода, a CaF — для количест-
венного определения фтора в почвах.
При сообщении атому достаточно высокой энергии валент-
ный электрон может быть удален из атома. Величину энергии
отрыва электрона называют потенциалом ионизации и выражают
212
в электрон-вольтах. При использовании источников возбуждения
с большой энергией линейчатые спектры дают одно- и много-
кратно ионизованные атомы. Механизм излучения иона анало-
гичен механизму излучения атома.
Источники возбуждения
Для проведения эмиссионного спектрального анализа необ-
ходимы такие источники, которые способны перевести твердые
вещества в газ, а образовавшееся газовое облако нагреть до тем-
пературы, обеспечивающей диссоциацию молекул до атомов, воз-
буждение последних с последующим излучением спектра. Роль
источника возбуждения в спектральном анализе гораздо шире,
чем простое сообщение атому энергии, достаточной для возбуж-
дения, так как процессу возбуждения в большинстве источни-
ков предшествует плавление пробы, ее испарение, диссоциация
на атомы, -затем возбуждение атомов и их возможная ионизация.
От применяемого источника возбуждения, режима его работы
в значительной степени зависят число определяемых элементов,
точности, чувствительность и скорость определения элементов.
При анализе почв наиболее широкое применение получили пла-
мя, дут’а постоянного и переменного тока между угольными (ре-
же металлическими) электродами.
Пламя. Пламена получили широкое применение в эмиссион-
ном спектральном анализе и являются наиболее старыми источ-
никами возбуждения спектра. Высокая стабильность горения
пламени, возможность регулирования температуры за счет изме-
нения состава газовой смеси и небольшое количество линий в
спектре позволили занять пламени основное место в качестве
источника света при пламенно-спектрофотометрическом анализе.
Н. Люндегард впервые применил пламя к анализу почв, вы-
тяжек растений и природных вод на содержание натрия, калия,
кальция, магния и железа.
Для получения пламени используют горючий газ и окисли-
тель. Горючим газом могут служить водород, пропан, бутан, аце-
тилен, окислителем — чаще всего воздух или кислород. Можно
также применять пламена, получаемые сжиганием паров горю-
чих жидкостей: спирта, ацетона, бензина и др. Находят приме-
нение и другие горючие газы, например аммиак, дициан — C2N2
в смеси с кислородом, закись азота — ацетилен и др. В зависи-
мости от состава смеси и соотношения компонентов получают
пламена с различной температурой, значения которой приведе-
ны в табл. 20.
Температура является важной характеристикой пламени. Воз-
буждение спектров в пламени носит в основном термический
характер. Температура пламени непостоянна в разных зонах
(рис. 67). Четкое разделение пламени на зоны характерно толь-
213
ко для ламинарного (спокойно горящего) пламени. Деление ла-
минарного пламени на зоны и непостоянство температуры в раз-
ных зонах широко используют в спектрально-аналитической
практике, так как концентрация свободных атомов в разных зо-
нах пламени для элементов различна. Используя разные зоны
свечения, можно получить оптимальную чувствительность опре-
деления или избавиться от возможных помех.
Рис. 67. Распределение темпера-
туры по зонам ламинарного пла-
мени:
1 — наконечник горелки, 2 — вну-
тренний конус, 3 — промежуточ-
ная зона, 4 — внешний конус
Таблица 20
Температура некоторых пламен,
используемых в спектральном анализе
Горючая смесь Темпера- тура пла- мени, °C
Светильный газ — воздух 1700—1840
Карбюрированный газ — воз- 1920
дух
Бутан — воздух—| Не 1800—1900
Пропан — воздухД « 1900—2000
Водород — воздух 2000—2100
Ацетилен — воздух 2100—2400
Водород — кислород 2500—2700
Водород — закись азота 2690
Светильный газ — кислород 2700—2800
Окись углерода •— кислород 2700
Пропан — кислород 2900—3000
Ацетилен — закись азота 3000
Ацетилен — кислород 3100—3200
Дициаи — киелррод 5000—5200
Меняя температуру пламени в широких пределах, удается
подобрать условия возбуждения так, что в спектре будут преоб-
ладать аналитические линии элементов, на которые не наклады-
вается спектр основного компонента, не возбуждающегося в пла-
мени.
Наибольшую чувствительность определения элементов можно
получить в участке пламени, расположенном непосредственно
под внутренним конусом. Щелочные металлы успешно опреде-
ляют в низкотемпературном пламени смеси пропан (бутан) —
воздух. В воздушно-ацетиленовом пламени набор определяемых
элементов можно значительно расширить. Следует отметить, чем
выше температура пламени, тем выше чувствительность, однако
увеличение чувствительности происходит до некоторого предела.
Отдельные элементы (алюминий, кремний, молибден, ванадий и
др.) могут давать в пламени термически устойчивые соединения
с компонентами пламени или с элементами анализируемой про-
бы, степень диссоциации таких соединений в низкотемператур-
ных пламенах незначительна. Для определения таких элементов
необходимо применять более горячие или восстановленные пла-
мена.
214
Существенным для получения стабильных и воспроизводимых
условии-воз бу жден ия спектра при работе с пламенем является
поддержание постоянства Сдавления горючего,,паза! или окислш
теля и^объема горючей смесиДчто довольно легко обеспечить.
Это делает применение пламен удобным для решения многих
задач.
Анализируемое вещество обычно вводится в пламя в виде
растворов. Для равномерного введения раствора в зону пламени
применяются специальные распылители, действие которых осно-
вано на всасывании жидкости и распылении ее струей газового
потока. Почвы можно вводить в пламя в виде мелко измельчен-
ных порошков или суспензий, вдувая их с помощью специаль-
ных приспособлений.
При хорошо отрегулированном давлении газов, питающих го-
релку, яркость излучения линии атомных спектров колеблется
в пределах десятых долей процента, и при рациональном выборе
условий работы точность количественных анализов в пламени
может быть доведена до 2—3%. Все это расширяет возможности
использования пламен в качестве источников возбуждения при
спектральном анализе почв, растений и других объектов био-
сферы.
Дуговые разряды между электродами. При анализе тугоплав-
ких и токонепроводящих материалов, какими являются почвы,
применяют разряд между угольными, графитовыми и, реже,
металлическими электродами. Температура дугового разряда в
основном зависит от состава плазмы в разрядном промежутке
и варьирует от 4000 до 7000° К- Благодаря высокой температуре
в дуге испаряются все элементы почв и возбуждаются спектры
преимущественно нейтральных атомов (дуговые спектры). В прак-
тике спектрального анализа для питания дугового разряда
используют как постоянный, так и переменный ток. Дуга постоян-
ного тока питается от динамо-машины или ртутных и полупро-
водниковых выпрямителей. Спектральные линии многих элемен-
тов в дуге постоянного тока появляются при содержании послед-
них в пробе 10-3—10“4%, причем можно повысить чувствитель-
ность обнаружения, используя катодное излучение атомов и
ионов. Серьезным недостатком дуги постоянного тока являются
нестабильность ее работы, блуждание анодного и катодного пя-
тен по поверхности электродов. Это значительно снижает вос-
производимость определений. Более широкое применение для
возбуждения спектров почв, растений получила дуга переменно-
го тока. Для питания дуги переменным током применяют спе-
циальные генераторы ДГ-1, ДГ-2 или генераторы серии ИВС
—21, ИВС—27.
В основе работы дуги переменного тока лежит активизи-
рование междугового промежутка искровым разрядом малой
мощности. Этот разряд получают с помощью активизатора, схе-
ма которого показана на рис. 68.
215
Дуга обладает падающей характеристикой, увеличение силы
тока в дуге ведет к уменьшению разности потенциалов на элект-
родах. Это приводит к нестабильности горения дуги без после-
довательно включенного балластного сопротивления. Без балласт-
ного сопротивления, ограничивающего силу тока, спонтанно рас-
тет сила тока дуги, и режим последней будет нестабильным.
Рис. 68. Электрическая схема активизированной дуги переменного тока:
1 — источник переменного тока, 2 — реостат повышающего трансформатора,
3 — повышающий трансформатор, 4 — разрядный промежуток активизатора,
5 — конденсатор активизатора, 6 — первичная катушка высокочастотного транс-
форматора, 7 — реостат питания дуги, 8 — блокировочный конденсатор, 9 —
амперметр дуги, 10— рабочий дуговой промежуток, И—вторичная катушка
высокочастотного трансформатора
Если обозначить через V — напряжение источника тока,
Ra и Rb — балластное сопротивление и сопротивление дугового
промежутка, то сила тока /, текущего через дугу, по закону
Ома равна /= —------—, когда Ra^Rt.- При Ra^>Rb сила тока в
ixb
дуге определяется главным образом величиной балластного со-
противления, увеличение сопротивления дугового промежутка
при случайных колебаниях пламени не будет вызывать значи-
тельного снижения силы тока и разрыва дуги. Таким образом,
сила тока, а значит, и температура разряда становятся более
стабильными. Питание дуги напряжением 220—250 В дает зна-
чительно большую стабильность возбуждения, чем напряжение
НО—120 В, так как с увеличением Ra для поддержания необ-
ходимой силы тока нужно увеличить напряжение V. Балластное
сопротивление (реостат, генератор) должно обеспечивать как
можно более плавное регулирование силы тока от 0 до 25 А.
Для повышения стабильности горения дуги и соответственно
точности анализа желательно использовать источники с постоян-
ными и контролируемыми характеристиками по току и напряже-
216
нию. Средняя температура разряда активизированной дуги пе-
ременною тока зависит от момента проскакивания и длительно-
сти активизирующих искр. Варьировать условиями возбуждения
спектров можно изменением режима активизирующего контура,
а также изменением величины дугового промежутка. Эти пара-
метры оказывают также влияние и на процессы поступления
проб в столб дуги. Количество вещества, испаряющегося с элек-
тродов, зависит как от мощности дуги, так и от отношения дли-
тельности вспышек к паузам тока. Чем больше это отношение,
тем интенсивнее идет испарение вещества.
Активизированная дуга переменного тока вследствие перио-
дических пауз тока имеет значительно меньшую температуру
нагрева электродов по сравнению с дугой постоянного тока.
В связи с этим испарение труднолетучих элементов из почв и
пород происходит медленнее, иногда менее полно или требует
значительно больше времени. Этот недостаток можно устранить,
если работать с дугой переменного тока при больших силах тока
(20 А;. Генераторы ДГ-1 и ДГ-2 легко можно приспособить для
работы с большими силами переменного тока, подключив к ним
дополнительные мощные реостаты.
В спектральном анализе почв применяют и другие источники
возбуждения спектра. К их числу относятся высоковольтная дуга
переменного тока, искровой разряд, импульсные источники воз-
буждения, плазмотроны (плазменные горелки, представляющие
собой генераторы потока плазмы, образующегося при нагревании
инертного газа электрической дугой), оптические квантовые гене-
раторы (лазеры), остро сфокусированное излучение которых спо-
собно испарять небольшие порции вещества, а также возбудить
свечение их паров. Луч лазера можно направить на пробу и
сфокусировать в пятно диаметром 50 мк. Комбинированные ла-
зерно-искровые установки, известные под названием лазерного
зонда, начинают широко применяться для локального анализа
поверхностей почв, шлифов, включений и отдельных зерен мине-
ралов. Однако количественные методы спектрального анализа
почв с применением этих источников еще недостаточно разрабо-
таны.
Спектральные приборы
для эмиссионного анализа
Спектральные приборы обеспечивают разложение идущего от
источника света излучения в спектр по длинам волн. Разложе-
ние излучения источников света в спектр производится с помо-
щью преломляющей призмы, дифракционной решетки или интер-
ференционного устройства.
Спектрографы. Основными, наиболее массовыми спектраль-
ными приборами в почвенно-агрохимической практике являются
217
спектрографы и пламенные фотометры. Спектрограф имеет кол
лиматорную (от S до D) п камерную части (D—Р) (рис. 69).
Главнейшими узлами прибора являются щель (S), линза кол-
лиматорной части (L]), диспергирующее устройство (£>), линза
(линзы) камеры L2, в фокальной плоскости камеры (Р) располо-
жены кассета с фотографической пластинкой или другой прием-
ник света. Входная щель пропускает узкий параллельный пучок
света, ширину и высоту щели можно изменять. Обычная шири-
на щели — сотые доли миллиметра, высота — доли или целые
миллиметры в зависимости от задач анализа.
Рис 69 Принципиальная схема спектрального при-
бора
Линза Li, в фокусе которой расположена щель, подает на
диспергирующее устройство параллельный пучок света. Диспер-
гируюшее устройство расчленяет сложную совокупность световых
колебаний на составляющие (Х1 + Х2 + Х3 ...) и далее такие моно-
хроматические лучи идут параллельно друг другу и собираются
линзой камерного объекта (L2) на фокальной плоскости (Р).
Таким образом, в фокальной плоскости камеры получают
спектр — сочетание монохроматических несколько увеличенных
изображений входной щели спектрографа.
На фэтопластинке, поставленной в фокальной плоскости, пос-
ле экспозиции, проявления и закрепления появятся темные па-
раллельные линии, составляющие эмиссионный спектр вещества.
Большинство задач эмиссионного спектрального анализа почв
решается при использовании спектральных линий, расположен-
ных в видимом, ближнем ультрафиолетовом и инфракрасном
участках спектра. В соответствии с этим чаще всего применяют
спектрографы, работающие в интервале длин волн 200—1000 нм.
Спектральные приборы с кварцевой оптикой позволяют работать
в области 200—700 нм, со стеклянной — в области 400—1000 нм.
Разрешающая способность призменных приборов меняется по
спектру. Приборы с дифракционной решеткой могут дать спектр
обеих указанных областей с практически одинаковым разреше-
нием по спектру. Это существенное преимущество дифракцион-
ных приборов перед призменными.
В почвенно-агрохимических лабораториях используют чаще
всего кварцевый спектрогпаф ИСП-30 со средней разрешающей 1
I I
218
способностью и дифракционный спектрограф ДФС-8 с высокой
разрешающей способностью.
Спектрограф ИСП-30 (рис. 70) позволяет получить спектр в
диапазоне 200—600 нм. Хотя рабочий диапазон у данного спект-
рографа довольно широк, практически работать можно только
в области 2о0—330 нм. Линейная дисперсия (расстояние в фо-
Рис. 70. Общий вид спектрографа ИСП-30:
1 — кожух спектрографа, 2 — клиновой зажим кассеты, 3 — кассета, 4 —
шкала подъема кассеты, 5 — сигнальная лампочка подсветки шкалы, 6 —
реле экспозиции, 7 — реле обжига, 8 — переключатель «пуск» — «стоп»,
9— переключатель ручной подвижки кассеты, 10 — переключатель уста-
новки шага подъема кассеты, 11 — выключатель ручного открытия затво-
ра, 12 — сигнальная лампочка включения сети, 13 — выключатель сети
калькой плоскости прибора между двумя линиями, отнесенное
к разности длин волн этих линий) спектрографа ИСП-30 в диа-
пазоне 250—310 нм составляет 0,9- 1,6 нм/мм, в то время как
в диапазоне 360 нм она уже 2,5 нм/мм. Так что разрешающая
способность приборов данного типа в области <300 нм чаще
всего достаточна для разделения линий в спектре элементов,
составляющих все типы почв, но в области >300 нм дисперсия
снижается и не всегда удовлетворяет требованиям почвенно-агро-
химического анализа. Встречаются породы и почвы (например,
с высоким содержанием железа, титана, марганца), имеющие
сложные многолинейные спектры, и возможны помехи за счет
наложения и перекрытия линий одних элементов линиями дру-
219
гих. В этих случаях требуется применение спектрографов с боль-
шим разрешением.
Прибор ИСП-30 снабжен реле времени, которое управляет
работой генератора, дает возможность автоматически выдержи-
вать время обжига и экспозиции до 3 мин и перемещать кассе-
ту. Перемещение кассеты за один цикл работы возможно на
1 или 2 мм.
твора
Рис 71. Общий
вид дифракцион-
ного спектрогра-
фа ДФС-8-
1 — кожух спек-
трографа, 2 — ос-
нование спектро-
графа, 3— рельс
для установки
осветительной си-
стемы и источни-
ка, 4 — кассета,
5 — клиновой за-
жим кассеты, 6 —.
щель спектрогра-
фа, 7 — барабан
раскрытия щели,
8 — ручка переме-
щения кассеты,
9 — ручка для по-
ворота решетки и
перемещения шка-
лы, 10 — окно для
контроля установ-
ки решетки по
длинам волн, 11 —
тумблер для включения лампочки освещения шкалы длин волн, 12 — контроль-
ная лампочка освещения шкалы длин волн, 13 — рукоятка для включения за-
Наиболее совершенен дифракционный спектрограф ДФС-8
(рис. 71). Прибор работает в диапазоне 200—1000 нм. В каче-
стве диспергирующего элемента в приборе используется дифрак-
ционная решетка 600 штр/мм (ДФС-8-1) пли решетка
1200 штр/мм (ДФС-8-2). Линейная дисперсия спектрографа
ДФС-8 с решеткой 600 штр/мм — 0,6 нм/мм, а с решеткой
1200 штр/мм — 0,3 нм/мм. Спектры фотографируют отдельными
участками (по 108 пли 54 нм) на фотопластинке размером
13Х 18 см.
Дифракционные приборы с высокой разрешающей способно-
стью целесообразно применять в первую очередь для определе-
ния отдельных элементов пли групп элементов с близко распо-
ложенными аналитическими линиями. Одним из главных преи-
муществ дифракционных спектрографов высокой разрешающей
220
способности является повышение чувствительности определения
большинства элементов за
рывного
фона
тех
областях
. Повышение
спектра, где
счет снижения интенсивности непре-
чувствительностп особенно заметно в
разрешение призменных приборов
Рис. 72. Общий вид фотометра пламенного ПФМ:
1— ручка регулятора расхода газа, 2— ручка регулятора
давления ^воздуха, 3 — кнопка поджига пламени горелки, 4 —
ручка изменения раскрытия ирисовой диафрагмы, 5—ручка
установки светофильтров, 6—ручка переключения диска с
постоянными диафрагмами, 7—ручка потенциометра регу-
лирования микроамперметра на нуль «точно», 8 — ручка по-
тенциометра регулирования микроамперметра на нуль «гру-
бо», 9 — ручка регулятора чувствительности, 10 — тумблер
включения компрессора, 11 — тумблер включения прибора,
12 — окно сигнальной лампочки, 13 — ротаметр для газа,
14 — oki.o наблюдения за пламенем, 15 — манометр для воз-
духа, 16— микроамперметр
невелико. Благодаря большой разрешающей способности при-
бора во многих случаях происходит разделение аналитических
линий мешающих элементов, что не только облегчает проведение
анализа, но может также повысить чувствительность.
Для многих элементов чувствительность определения на диф-
ракционных приборах с большим разрешением на полпорядка
221
или целый порядок выше, чем на призменных приборах средней
дисперсии. В почвенно-агрохимических исследованиях для мас-
совых анализов выгодно использовать приборы с линейной дис-
персией 0,4—0,6 нм/мм, так как они позволяют путем сжигания
одной пробы получить участок спектра от 245,0 до 350,0 нм, где
находятся аналитические линии большинства микроэлементов.
Такая дисперсия достаточна и для определения микроэлементов
в других областях спектра.
Для фотоэлектрической регистрации спектра используют 36-
канальную установку ДФС-10М (квантометр). Этот прибор обес-
печивает одновременное определение 12 элементов. Он чувстви-
телен к температурным изменениям и требует кондиционирован-
ного помещения. На базе квантометра ДФС-10М сконструирован
универсальный аппарат — квантометр ДФС-36. Прибор позво-
ляет выполнять анализы с более высокой чувствительностью и
воспроизводимостью, но главное преимущество приборов этого
типа заключается в повышении скорости определений.
Пламенные фотометры. Для целей фотометрического анализа
почв широкое применение получили отечественные пламенные
фотометры ФПЛ-1 (фотометр пламенный лабораторный) и ПФМ-
(фотометр фотоэлектрический пламенный) (рис. 72).
Фотометр пламенный лабораторный предназначен для коли-
чественного определения Na, К, Са в растворах. Его горелка
обеспечивает работу на горючей смеси пропан — бутан — воз-
дух. Допустимый суммарный дрейф электрического и фотомет-
рического нуля в течение 1 ч не превышает 3% от верхнего зна-
чения шкалы. Нижний предел измерений составляет для Na и
К — 0,5 мг/л, для Са — 5 мг/л. Верхний предел измерений для
этих элементов — около 100 мг/л.
Более совершенен фотоэлектрический фотометр ПФМ, он
дает возможность определить Na, К, Са, Sr, Li, Rb, Cs, Ba, Mn.
Чувствительность определения щелочных металлов на этом при-
боре почти на два порядка выше, чем на ФПЛ-1. Прибор комп-
лектуется интерференционными светофильтрами для выделения
излучения Na Umax — 589±5 нм); К (ЛШах — 766±5 нм);
Са Umax — 620±5 нм) II Li Umax — 671 ±5 нм).
Принципиальная схема пламенного фотометра показана на
рис. 73. Анализируемый раствор из стаканчика 1 засасывается в
распылитель 2 током воздуха, который поступает из компрессо-
ра 3 через фильтр 4. Давление воздуха измеряется маномет-
ром 5. Распыленный раствор проходит через смеситель 6 и по-
дается в горелку 7. Горючий газ также проходит через редуктор
и фильтр и смешивается с аэрозолем в смесителе 6. Крупные
капли осаждаются сепаратором, а мелкие вместе с газом и воз-
духом поступают в горелку прибора и испаряются в пламени.
Оптическая система состоит из следующих элементов: отража-
тельного зеркала 8 и конденсора 9, проектирующих изображение
источника эмиссии (пламени) на фотоэлемент. Излучение про-
222
ходит через тепловой фильтр, предохраняющий оптические узлы
и фотоэлемент, адсорбционные и интерференционные светофиль-
тры 10, 11, 12 для монохроматпзации эмиссии исследуемых эле-
ментов. Сигнал с фотоэлемента поступает на усилитель 14 и
фиксируется микроамперметром 15.
Рис. 73. Принципиальная схема пламенного фотометра:
1 — испытуемый раствор, 2 — распылитель, 3 — компрессор, 4 — фильтр для
воздуха, 5— манометр для воздуха, 5а— манометр для газа, 6—камера сме-
сителя, 7 — горелка, 8 — зеркало, 9 — конденсор, 10, 11, 12 — светофильтры,
13— фотоэлемент, 14 — усилитель, 15— гальванометр, 16 — стабилизатор, 17 —
баллон для газа
Широкое применение при анализе почв получили пламенные
фотометры, выпускаемые в ГДР, наиболее совершенным из них
является Флафо-4. Во многих лабораториях успешно используют
пламенные фотометры самодельного изготовления.
Регистрация спектров
и определение интенсивности
спектральных линий
В эмиссионном спектральном анализе распространен фотогра-
фический метод регистрации. Он позволяет получить достаточно
полный спектр сложного материала, расположенный от близкой
инфракрасной до ультрафиолетовой области, определить длины
волн и интеиспвносгп спектральных линий и фона на спектро-
223
грамме. Фотографический метод фиксирует возможные помехи и
наложения мешающих линий, и, кроме этого, полученная спектро-
грамма является объективным документом, который может хра-
ниться длительное время и в необходимых случаях подвергаться
дополнительному и повторному изучению и измерению.
При выборе фотоматериалов для фотографирования спектров
в первую очередь необходимо учитывать их спектральную чувст-
вительность и область спектра, где лежат чувствительные ли-
нии определяемых элементов. Действующим началом в фотома-
териалах является светочувствительный слой, называемый
фотографической эмульсией. Фотографическая эмульсия состоит
из водного раствора желатина и равномерно распределенного 'в
нем галоидного серебра. Под действием излучения галоидное
серебро восстанавливается до металлического серебра. Восста-
новленное серебро образует скрытое изображение распределения
освещенности спектра. При специальной химической обработке
(проявлении) серебро вступает в соединения, которые дают ви-
димое изображение (черный цвет), и на фотопластинке фикси-
руется распределение почернений.
Почернение зависит о г световой энергии, времени воздейст-
вия ее на эмульсию, от состава проявителя и других условий
проявления.
Для оценки величин оптической плотности почернения поль-
зуются выражением
Ig A = lg^-,
где S — плотность почернения, 10 — интенсивность света, про-
шедшего через прозрачное место (или вуаль) на фотопластинке,
/ — интенсивность света, прошедшего через почернение или
изображение линии. Почернение спектральных линий измеряют с
помощью микрофотометров, тогда А(1 и А — величины отсчетов,
получаемые на шкале микрофотометра.
Отечественная промышленность выпускает несколько моделей
микрофотометров (ИФО-451, ИФО-460, МФ-2, МФ-4). В почвен-
но-агрохимических лабораториях наибольшее распространение
получил нерегпстрирующий микрофотометр МФ-2. С его харак-
теристикой и методикой работы можно ознакомиться по прила-
гаемой к нему заводской инструкции.
Чувствительность фотопластинок различна для разных длин
волн, в зависимости от чувствительности они подразделяются на
типы I, II, III, ЭС и др Для целей спектрального анализа при-
меняют фотопластинки типа I и II, чувствительные в диапазоне
225—450 нм; тип III — чувствительные в области 200—450 нм.
Для регистрации аналитических линий щелочных и щелочнозе-
мельных металлов используют фотопластинки панхром и инфра-
хром, чувствительные в видимой и ближней инфракрасной обла-
стях спектра.
224
Фотоэлектрические методы измерения интенсивности спект-
ральных линий. Фотографические методы в ряде случаев не обес-
печивают необходимой точности анализа, значительная часть
ошибок обусловлена неоднородностью эмульсии фотопластинки,
нестабильностью процесса проявления, фиксирования и т. д.
Замена фотографического процесса фотоэлектрической реги-
страцией спектра открывает новые возможности в совершенство-
вании техники спектрального анализа, значительно повышает его
быстроту и точность. Существует ряд приемов фотоэлектрических
измерений интенсивностей, но фактически все они основаны на
том, что спектральные линии попадают на фотоэлектрический
приемник света (фотоэлемент, фотоумножитель), который пре-
образует лучистую энергию в электрическую, измеряемую с по-
мощью гальванометра или других приборов. Величина тока на
выходе фотоумножителя или фотоэлемента пропорциональна
световому потоку, освещающему фотокатод.
В настоящее время имеется значительное количество различ-
ных фотоэлектрических установок для спектрального анализа.
В почвенно-агрохимических лабораториях целесообразно приме-
нять недорогие упрощенные фотоэлектрические приборы или фо-
тоэлектрические приставки к имеющимся спектрографам типа
ФЭП-1, ФЭП-2.
Основные положения
количественного
спектрального анализа
Для количественного определения элементов в пробе поч-
вы используют эмпирическую зависимость 1=аСь или
lg/=blgC + lgcz,
где I — интенсивность атомной (ионной) аналитической линии,
С — концентрация атомов (ионов) в пробе, а и b — постоянные
величины. Эта зависимость экспериментально была установлена
в 1930 г. советским ученым Б. А. Ломакиным. Физический смысл
величин а и b был раскрыт С. Л. Мандельштамом. Величина а
зависит от условий перевода анализируемого вещества в пар и
возбуждения соответствующей линии спектра. Для однородных
веществ и при неменяющихся условиях опыта эта величина оста-
ется постоянной. Величина b зависит от поглощения излучения
внутри плазмы. При небольшой концентрации элемента в про-
бе линия не испытывает самопоглощения и Ь=1, а с увеличе-
нием интенсивности излучения начинает сказываться самопогло-
щение и величина b становится меньше единицы.
Сравнивая интенсивность линий в спектре пробы и эталонов
с известным содержанием определяемого элемента, можно легко
определить концентрацию элемента в пробе. В практике спект-
8 Зак 284
225
рального анализа чаще всего пользуются графическим методом.
По оси ординат указывают интенсивность (логарифм интенсив-
ности) аналитической линии (при фотографической регистра-
ции — почернение), а по оси абсцисс — соответствующие дм
концентрации элемента в эталонах. Полученный график назы-
вают аналитическим или градуировочным (рис. 74). Наклон
прямой к оси абсцисс определяется коэффициентом Ь. При Ь,
равном единице, угол наклона
0,0006 Ц0010,005 0,010,06 0,1 0,3 С°0
-3,5 -3,0 -2,5 -2,0 -1,5 -1,0 -О,51дС,%
Рис. 74. Градуировочный график
равен 45°. Градуировочный гра-
фик применим для образцов,
сфотографированных только на
данной пластинке.
Для улучшения точности оп-,
ределений измеряют относитель-
ную интенсивность спектральных
линий определяемого элемента
и элемента сравнения, находя-
щегося или введенного в данную
пробу. Все случайные изменения
в условиях анализа влияют на
интенсивность обеих линий, и
погрешности снижаются. Естест-
венно, что подбирается такая па-
ра линий, которые одинаково
реагируют на изменение усло-
вий испарения и возбуждения.
Такие линии получили название
гомологических. Относительные
интенсивности аналитических линий определяемого элемента и
внутреннего стандарта связаны с концентрацией элемента в про-
бе сравнительно простым соотношением.
Обозначим интенсивность линии определяемого элемента /л,
тогда lg I„=blg C + lg а, соответственно для линии элемента
сравнения /ср
1g /ср = b 1g Сср ' 1g цср,
lg/л—lg4P = blgC “ Iga —6 lg Ccp — lg acp,
при равенстве а и acp
te-^=b(lgC-lgCcp)
'cp
Концентрация элемента сравнения постоянна в пробах и эта-
I С
лонах, тогда уравнение примет вид lg —=b 1g----.
Др Сср
Основное затруднение при выполнении спектрального анали-
за почв состоит в том, что интенсивность аналитических линий
определяемых элементов зависит не только от их концентрации
в пробе, но и от общего состава пробы почвы, химических и фи-
зических свойств ее.
226
1
Причины, нарушающие теоретическую связь между концен-
трацией элемента в пробе и интенсивностью его линии, могут
быть различными. Они зависят от характера источника возбуж-
дения, приемов введения проб в источник света, процессов, кото-
рые происходят с пробой в источнике, а также от индивидуаль-
ных свойств элементов и пробы в целом.
При возбуждении спектра в пламени или дуге одновременно
могут существовать небольшие количества свободных электро-
нов, ионы, атомы и молекулы Равновесие между этими форма-
ми зависит от температуры пламени или дуги, состава испаряе-
мого раствора и окислительно-восстановительных условий. Этим
и объясняется сильное влияние состава проб на интенсивность
излучения. Поэтому, приготавливая эталоны, стремятся к наи-
большему совпадению их состава с составом проб. При анализе
водных, солевых и кислотных вытяжек из почв пробы отличаются
переменным составом, и не удается приготовить эталоны, пол-
ностью идентичные вытяжкам почв.
В этих случаях наиболее существенно, чтобы пробы и этало-
ны были максимально близки по содержанию компонентов, ока-
зывающих существенное влияние на интенсивность линий. С этой
целью при анализе солевых и кислотных вытяжек из почв стан-
дарты готовят на тех же растворах, которые были использованы
для получения вытяжек из почвы. Большой избыток кислоты или
соли нивелирует условия возбуждения и позволяет получить
сравнимые результаты. Труднее анализировать водные вытяжки
и почвенные растворы, особенно из засоленных почв, ибо состав
солей и их содержание резко колеблются даже в пределах одного
почвенного профиля, что сильно влияет на качество определения
по методу обычного градуировочного графика.
Состав растворов оказывает воздействие не только на про-
цессы, происходящие в пламени, но и на качество работы рас-
пылителя. Оценивая в целом влияние состава на результаты ана-
лизов, следует иметь в виду следующее. Температура, вязкость
и поверхностное натяжение раствора изменяют его распыление
и поступление в пламя горелки. В результате яркость свечения
пламени меняется. Несовпадение по этим параметрам анализи-
руемых и эталонных растворов вызывает ошибки определения
концентрации.
Большой избыток катионов сдвигает равновесие в реакциях
диссоциации молекул и ионизации ионов. Например, интенсив-
ность излучения кальция и других щелочноземельных металлов
снижается в присутствии алюминия, образующего трудно диссо-
циирующие соединения состава п-СаО-AI2O3. В меньшей степе-
ни А1 снижает интенсивность излучения щелочных металлов.
Щелочные катионы усиливают излучение друг друга, при
этом влияние катиона тем больше, чем меньше величина иониза- •аЛ3 _
ционного потенциала атомов элемента. Влияние щелочных ка-
тионов убывает в ряду. Cs>Rb>K>Na>Li, что связано с раз-
8* ^27^ М
ным ионизационным потенциалом их атомов. При ионизации,
отщеплении электрона от атома, образуются две частицы — ион
металла и электрон:
е,
где М° — нейтральный атом, М+ — однократно ионизованный
атом и е — электрон.
Реакция ионизации носит обратимый характер и характери-
зуется константой
Л _ [М+1 • [е]
[Мо] ‘
Присутствие в пробе легко ионизирующихся атомов приводит
к образованию, дополнительного количества электронов, которые
подавляют ионизацию определяемого элемента, увеличивают
концентрацию его атомов и соответственно интенсивность излу-
чения. Это и приводит к ошибкам в определении концентрации
щелочного металла. При почвенных исследованиях прежде всего
нужно учитывать взаимное влияние натрия и калия при анализе
водных вытяжек из засоленных почв и определения калия (или
натрия) в солевых вытяжках. Существенные помехи возможны
при определении лития, рубидия и цезия, содержащихся в почве
в микроколичествах.
Степень ионизации падает с увеличением общей концентрации
элементов в источнике, но она увеличивается с возрастанием
температуры. Щелочноземельные металлы не оказывают влия-
ния на ионизацию щелочных металлов; даже при молярном соот-
ношении Ca(Mg) : К (Na) = 100 интенсивность излучения щелоч-
ных элементов не меняется.
Не все если одинаково хорошо диссоциируют в пламени, по-
этому в присутствии некоторых анионов снижается концентрация
и интенсивность излучения атомов металла. Анион NOF меньше
других влияет на излучение щелочных металлов. Степень влия-
ния растет в ряду: сульфат-ион < хлорид-ион < фосфат-ион.
Для кальциевых солей интенсивность излучения при одной и
той же концентрации располагается в следующий ряд:
СаС12 > Са (NO3)2 > CaSO4 > Са3 (РОЩ.
Многие примеси, присутствующие в пробе, часто сами дают
излучение с длиной волны, близкой к аналитической линии
определяемого элемента. Наложение излучения примеси на излу-
чение определяемого элемента при недостаточной монохромаги-
зации приводит к завышенным результатам.
Полоса излучения СаО накладывается на аналитическую ли-
нию натрия (589,0—589,6 нм) и тем создает затруднения в точ-
ном опредс 1ении натрия. Очень близки по длинам волн линии
калия, лития и рубидия, их совместное присутствие может внес-
228
ти ошибки в определение каждого из этих элементов при плохом
разрешении спектра.
Если пробы почв вводят в дуговой источник света из канала
угольного электрода, то большое влияние на интенсивность ли-
ний оказывают условия их испарения. Входящие в состав почв
элементы по относительной скорости испарения можно объеди-
нить в три главные группы, в которых они расположены по убы-
ванию летучести.
Легколетучие элементы: Zn, Pb > Sn, К, Na > Li > Rb >
> Cs > Mg > Си, Mg, Ga.
Среднелетучие элементы: Co, Ni, Cr, V, Fe, Ba, Sr, Cd > Si > Al,
Mo, B.
Труднолетучие элементы: B>Be->Mo2>Sc2>Zr>-Ti.
Химические реакции, протекающие в канале электрода при
испарении проб, влияют на последовательность поступления эле-
ментов. Изменение химического состава почв может изменить
последовательность расположения элементов в группах, но с по-
зиций количественного анализа благоприятно то, что обычные
колебания химического состава в пределах главнейших типов
почв практически мало сказываются на интенсивности линий
микроэлементов. Сказанное справедливо при полном испарении
пробы тонкорастертых почв из канала угольного электрода в ду-
ге переменного тока. Для определения микроэлементов в почвах
это позволяет использовать для эталонов основу одного состава,
соответствующую по содержанию макроэлементов некоторому
«среднему» составу зональных типов почв. В тех случаях, когда
анализируемые почвы резко отличаются по составу от эталонов,
необходимо применять методы, снижающие возможные погреш-
ности (например, буферирование, метод добавок), или готовить
эталоны, точно отражающие состав исследуемых почв.
В зависимости от способа построения градуировочных графи-
ков, числа применяемых для этого эталонов, частоты их съемки
разработано несколько методов количественного спектрального
анализа.
Метод трех эталонов. Он заключается в фотографировании
трех пли большего числа эталонов и анализируемых проб на
одной фотопластинке и в последующем построении градуировоч-
ного графика для каждой фотопластинки в координатах 5—lg С
или AS—lg С, где AS — разность почернений аналитической ли-
нии и внутреннего стандарта. Метод трех эталонов универсален,
прост и относительно точен, его удобно применять для одновре-
менного анализа большого количества проб. Недостатком этого
метода являются большие затраты времени на фотографирование
спектров эталонов, построение для каждой пластинки градуиро-
вочных графиков.
Метод твердого графика. Градуировочную кривую строят с
учетом коэффициента контрастности фотопластинки и исполь-
зуют длительное время. В зависимости от способа учета свойств
229
фотопластинки данный метод имеет несколько разновидностей.
В одном из них вместо S (или AS) по оси ординат откладывают
AS
величину----, где у — коэффициент контрастности пластинок,
Y
который определяют по спектру пробы, снятому через ступенча-
тый ослабитель. Измерив разность почернений аналитической
линии и внутреннего стандарта в спектрах нескольких эталонов
и поделив ее на у, можно нанести на график полученные значе-
, ASt AS2 AS3
ния----, ---, ——и т. д. и соответствующие им значения кон-
у у у
центрации элементов в эталонах (1g Сь lg С2, 1g С3 и т. д.). Это
дает твердый график, с помощью которого можно по вычислен-
AS
ному значению-----в спектре проб определить искомую концент-
Y
рацию элемента. Для учета смещения градуировочного графика
рекомендуется вместе с образцами при строго стандартных усло-
виях получения спектрограмм снимать один контрольный эталон,
гт AS
По величине----контрольного эталона при необходимости произ-
Y
водится коррекция твердого графика. Достоинство метода твер-
дого графика состоит в том, что он позволяет сократить
количество и расход эталонов, а также время, затрачиваемое на
фотографирование и измерение линий в эталонах.
Метод добавок. Он рентабелен для анализа единичных проб
сложного состава, когда изготовление эталонов оказывается
трудоемким. Метод дает возможность частично исключить влия-
ние состава пробы на результаты анализа, однако полностью
исключить влияние химического состава проб на точность опреде-
ления элементов вряд ли возможно, поскольку практически нель-
зя внести в пробу добавляемый элемент в форме того же хими-
ческого соединения, в каком оно присутствует в почве.
При работе по методу добавок берут три-четыре навески ана-
лизируемой пробы, содержащей Сх определяемого элемента, и
вводят в них различные добавки определяемого элемента. Пер-
вая добавка должна быть по порядку величины близка к содер-
жанию элемента в исследуемой пробе. Добавки в каждой после-
дующей навеске должны быть в 2—3 раза больше, чем в преды-
дущей. Обозначим их как Сц С2, С3. Пробы с добавками хоро-
шо перемешивают и перетирают в агатовой ступке. В строго
идентичных условиях фотографируют спектры как исходной про-
бы, так и с добавками элемента; определяют интенсивность ли-
ний определяемого элемента. Существуют различные варианты
построения градуировочного графика. По одному из них график
строят в координатах /—С, предполагая b равным единице (что
справедливо при небольших концентрациях элемента). Тогда по
оси ординат при значении С=0 откладывают величину /, най-
денную для пробы, в которую не был добавлен определяемый
элемент. Затем откладывают значения интенсивностей, соответ-
230
ствующие добавкам элемента Сь С2, С3. Строят градуировочную
прямую. Отрезок, отсекаемый продолжением прямой по оси
абсцисс, численно равен искомой концентрации Сх.
Метод ограничивающих эталонов. Его используют в пламен-
ной фотометры!. Градуировочный график строят по двум этало-
нам с концентрациями определяемого элемента Сг и С2. Концен-
трация элемента в пробе должна лежать в диапазоне С1<СЖ<С2.
В пламя вводят анализируемый раствор и фиксируют соответ-
ствующие ему показания гальванометра 1Х- Подбирают два близ-
ких эталона, дающих показания Ц и /2, соответственно больше
и меньше 1Х-
Концентрацию определяемого элемента в пробе находят по
формуле
сЛ = с± i
'2—*1
Метод справедлив для прямолинейных участков графика.
ОПРЕДЕЛЕНИЕ В ПОЧВАХ
ЩЕЛОЧНЫХ МЕТАЛЛОВ
МЕТОДОМ ПЛАМЕННОЙ ФОТОМЕТРИИ
В зависимости от уровня концентрации щелочные металлы в
почвах подразделяются на макроэлементы — натрий, калий и
микроэлементы — литий, рубидий и цезий. Содержание щелоч-
ных макроэлементов превышает концентрацию микроэлементов
почти в 1000 раз. Микроэлементы не оказывают влияния на опре-
деление натрия и калия в почвах, в то время как макроэлемен-
ты могут существенно повлиять на определение лития, рубидия,
цезия, как правило, завышая результаты анализа. Определение
этих элементов требует применения специальных методов.
Содержание и соотношение натрия и калия в почвах, как
правило, колеблются в узком интервале, поэтому их взаимное
влияние на точность определения не может иметь существенного
значения.
При определении натрия и калия ь почвах выгодно исполь-
зовать низкотемпературное пламя, поскольку при этом снижают-
ся помехи, вызываемые щелочноземельными элементами, в пер-
вую очередь кальцием. Наличие в растворе кальция может су-
щественно влиять на точность определения натрия и калия в воз-
душно-ацетиленовом пламени, особенно при анализе карбонат-
ных почв. В воздушно-ацетиленовом пламени натрий и калий
дают по нескоЛоку линий, различающихся по чувствительности,
но наибольшая чувствительность свойственна калию по линии
766,5—769,9 нм (чувствительность 0,01 мкг/мл) и Na — 589,6—
589,0 нм (чувствительность 0,0002 мкг/мл). Литий, рубидий и
цезии определяют с применением воздушно-пропанового или воз-
231
душно-ацетиленового пламени по линиям: Li — 670,8 нм, Rb —
789,0 нм, Cs — 852,1 нм, чувствительность определения их соот-
ветственно равна 0,001, 0,01 п 0,1 мкг/мл. Количественное опре-
деление щелочных микроэлементов в почвах на пламенных фото-
метрах ненадежно, поэтому здесь эти методы не рассматри-
ваются.
Определение общего
содержания калия и натрия
в почвах
Валовое содержание калия и натрия в почвах определяют
после разложения почвы следующими методами:
1) спеканием почв со смесью СаСОз и NH4C1;
2) разложением HF в платиновых чашках в присутствии
H2SO4, HNO3 или НС1;
3) разложением почв во фторопластовой посуде под давле-
нием смесью HF и H2SO4.
Разложение почв и подготовка раствора к анализу. При раз-
ложении почв методом спекания тонкоизмельченную навеску 0,1 г
спекают со смесью СаСО3 и NH4C1. Спек выщелачивают горя-
чей водой. В водной вытяжке основную массу кальция осаждают
карбонатом аммония при кипячении. Осадок отфильтровывают.
Фильтрат и промывные воды помещают в мерную колбу на
250 мл, объем раствора доводят до метки водой и на пламенном
фотометре определяют в нем содержание калия и натрия. В ка-
честве стандартных растворов применяют растворы с известным
содержанием калия и натрия с добавкой карбоната аммония в
количестве, соответствующем его содержанию в испытуемом
растворе.
В большинстве случаев определение калия и натрия в почвах
проводят после разложения их HF в присутствии H2SO4. В этом
случае в полученном растворе можно определить калий, натрий,
кальций, магний и ряд других элементов.
Навеску 0,1—0,2 г тонкопзмельченной почвы (до «пудры»)
помещают в платиновую чашку. Органическое вещество окис-
ляют предварительно или непосредственно в платиновой чашке
концентрированной азотной кислотой или путем прокаливания
почв при 600—700° в течение двух часов. Затем навеску почвы
в платиновой чашке смачивают несколькими каплями дистилли-
рованной воды, добавляют 1 мл концентрированной H2SO4 и 5—
10 мл 40%-ной HF. Чашку ставят на электрическую плитку с
закрытой спиралью и постепенно нагревают до появления густых
паров серного ангидрида (SO3).
В случае неполного разложения почвы, что узнается по нали-
чию своеобразного «хрустения» при помешивании смеси тефло-
новым (платиновым) шпателем, в чашку прибавляют еще 5 мл
232
HF и вновь упаривают до паров серного ангидрида. По оконча-
нии разложения усиливают нагревание чашки с целью полного
удаления паров серного ангидрида. Окончательную отгонку SO3
более быстро можно осуществить в муфельной печн при 400°,
установленной под вытяжным шкафом.
К сухому остатку в чашке добавляют 5 мл HCI (1 : 1), смесь
нагревают несколько минут до полного растворения остатка.
Затем добавляют 10—15 мл дистиллированной воды и переносят
содержимое чашки подкисленной (НС1) горячей водой через
фильтр в мерную колбу емкостью 100—250 мл. Чашку и фильтр
на воронке несколько раз тщательно промывают горячей подкис-
ленной дистиллированной водой, колбы охлаждают и доводят до
метки водой. Содержимое перемешивают, калий и натрий в ра-
створе определяют в пламени пропан—бутан—воздух на пламен-
ном фотометре, как описано ниже на с. 236. Введение кислоты
в стандартные растворы (эталоны) необязательно, так как ре-
зультаты определения калия и натрия в нейтральных растворах
и в растворах слабых кислот совпадают между собой.
Хорошие результаты дает разложение почв фтористоводород-
ной кислотой з присутствии «царской водки» во фторопластовой
посуде. Для этого 0,1—0,2 г тонкоизмельченной почвы («до пуд-
ры») помещают в чашку (или стаканчик) из фторопласта-4, сма-
чивают несколькими каплями дистиллированной воды, добавляют
2 мл «царской водки» и затем приливают 10 мл HF. Чашку ста-
вят на закрытую этернитовую плитку и постепенно нагревают.
Нагревание и выпаривание продолжают до получения сстпгка
из влажных солей, после чего в чашку добавляют еще 10 мл HF
и нагревают до полного разрушения почвенных силикатов. После
последнего приливания HF и полного выпаривания содержимого
нагревание чашки усиливают. Температура поверхности плитки
не должна превышать 23р—240°. После того как полностью уда-
лена фтористоводородная кислота, сухой остаток растворяют,
как описано выше, а раствор используют для пламенно-фотомет-
рического определения калия и натрия, как описано ниже.
Определение водорастворимых
соединений калия и натрия
в почвах
Методом пламенной фотометрии можно определить К и Na
в водной вытяжке без ее обогащения, когда содержание водо-
растворимых соединений этих элементов не менее 1 мг/кг
почвы (10-4%). Для приготовления водной вытяжки образец
почвы доводят до воздушно-сухого состояния, растирают пести-
ком с резиновым наконечником и пропускают через сито с отвер-
233
стаями 1 мм. Из приготовленной почвы берут среднюю пробу в
количестве 10 г (в расчете на абсолютно сухой вес).
Навеску почвы помещают в колбу емкостью 100 мл, прили-
вают 50 мл дистиллированной воды, лишенной СО2, и взбалты-
вают на ротаторе 5 мин. После взбалтывания дают почвенным
частицам осесть на дно колбы и осторожно сливают раствор над
осадком в центрифужные пробирки. Пробирки с раствором урав-
новешивают попарно на весах и размещают в гнездах центри-
фуги так, чтобы в противоположных гнездах находились пробир-
ки со строго одинаковым весом. В случае нечетного числа образ-
цов одну из пробирок заполняют водой. Растворы центрифуги-
руют в течение 30 мин при 6000—8000 об/мин, после чего раствор
над осадком сливают в стаканчик. Если после центрифугирова-
ния в растворе обнаруживаются какие-либо взвешенные частицы
(часто в верхней части пробирки собираются мелкие корешки),
то необходимо дополнительное фильтрование, поскольку даже
мелкие частицы могут засорить капилляр распылителя и вывести
прибор из строя. Первые порции фильтрата в этом случае отбра-
сывают.
Прозрачную вытяжку используют для определения К и Na
на пламенном фотометре (см. с. 236).
Водные вытяжки из засоленных почв, содержащих большие
количества натрия и калия, необходимо перед анализом разбав-
лять до концентрации 10—50 мг/л. В качестве стандартных раст-
воров применяют водные растворы хлоридов натрия и калия.
Для получения правильных результатов необходимо следить за
соотношением содержания калия и натрия в водных вытяжках,
так как оно может сильно варьировать, особенно в засоленных
почвах. Непостоянство этого соотношения служит причиной из-
менения ионизационного эффекта. При очень резком преоблада-
нии одного из элементов в растворе необходимо внести соответ-
ствующие поправки и в эталоны. Сильноокрашенные органиче-
ским веществом водные вытяжки необходимо разбавлять водой,
если позволяют концентрации определяемых элементов. Если
разбавление невозможно, то водную вытяжку выпаривают, окис-
ляют органические вещества, прокаливая остаток при 500—
600° С, а оставшиеся соли снова растворяют и прозрачные ра-
створы используют для анализа.
Определение содержания
обменных калия и натрия
в почвах
Для определения обменных калия и натрия пользуются ра-
створами солей или кислот, обеспечивающими полноту вытесне-
ния катионов из почв. Одним из лучших методов вытеснения
обменных катионов из почв является обработка их 1 н.
234
CH3COONH4. Для этого 5—10 г почвы, просеянной через снто
1 мм, промывают декантацией 1 н. раствором CH3COONH4 при
pH около 6,5 до прекращения реакции на ион Са2+. Фильтрат
собирают в мерную колбу, доводят до метки 1 н. раствором
CH3COONH4, перемешивают и затем определяют концентрацию
ионов К+ и Na+ (см. с. 236). При малом содержании Na+ и К4-
фильтрат предварительно концентрируют выпариванием, сухой
остаток прокаливают и растворяют в подкисленной дистиллиро-
ванной воде в аликвоте в 10—50 раз меньшей, чем объем исход-
ного фильтрата. Эталонные растворы для анализа вытяжек без
какой-либо предварительной подготовки готовят на основе ра-
створа 1 н. CH3COONH4. Для фильтратов, которые концентриро-
вали упариванием, эталоны готовят на водных растворах NaCl и
КС1.
Определение доступного калия
в почвах
Для определения доступного калия в некарбонатных почвах
(дерново-подзолистых, серых лесных, черноземах, красноземах и
желтоземах) рекомендован 1 н. раствор CH3COONH4. Соотноше-
ние почва : раствор 1 : 10, одночасовое взбалтывание на ротато-
ре. 5 г воздушно-сухой почвы, просеянной через сито с отвер-
стиями 1 мм, помещают в колбу емкостью 100 мл, добавляют
50 мл 1 н. раствора CH3COONH4 (pH 7,0) и ставят на ротатор.
После взбалтывания вытяжку фильтруют через складчатый
фильтр, первые порции фильтрата отбрасывают, в следующих
порциях прозрачного фильтрата определяют калий на пламен-
ном фотометре. Эталонные растворы готовят на 1 н. растворе
CH3COONH4 из водных растворов хлорида калия.
В случае определения доступного калия в карбонатных поч-
вах для извлечения применяют 1%-ный раствор (NH4)2CO3
(pH 9,0) при соотношении почва : раствор 1 : 20. Вытяжку гото-
вят аналогично вышеописанному, время взбалтывания — 5 мин,
настаивание — 20 ч. Шкалу эталонных растворов готовят на
1%-ном раттворе (КН4)2СОз.
Определение емкости
поглощения почв
При определении емкости поглощения по универсальному
методу Гедройца почву насыщают раствором NaCl, а затем ион
Na+ вытесняют раствором Са(НСО3)2 в токе СО2. Вытесненный
из почвы Na определяют на пламенном фотометре. Для этого
5—10 г почвы, просеянной через сито 1 мм, помещают в фарфо-
ровую чашку или стакан, добавляют 1 п. раствор NaCl (pH 6,5),
235
перемешивают содержимое стеклянной палочкой. Почву постепен-
но переносят на фильтр методом декантации и промывают ра-
створом NaCl до исчезновения реакции на Са2+-ион. Чем дольше
промывают почву декантацией, чем меньше добавляемые порции
раствора NaCl, тем меньше будет истрачено насыщающего ра-
створа NaCl и скорее произойдет насыщение почвы Na+-iiOHOM.
После полного насыщения почвы ионом Na+ избыток NaCl
удаляют, промывая фильтр с почвой вначале небольшими пор-
циями воды, а затем спиртом. Промывание прекращают при ис-
чезновении реакции на хлор. Затем почву вместе с фильтром пе-
реносят в колбу, добавляют 1 г мела, приливают 1 л воды и в
течение 3 час пропускают через систему ток СО2. Каждый 15 мин
содержимое колбы встряхивают. После окончания этой процеду-
ры раствор фильтруют, в фильтрате определяют Na пламенно-
фотометрическим методом. Эталонные растворы готовят на ди-
стиллированной воде.
Приготовление стандартных
и эталонных растворов
Стандартные растворы калия и натрия готовят растворением
1,907 г и 2,542 г соответственно их хлористых солей (квалифи-
кации «х. ч.») в 1 л дистиллированной воды. Полученные таким
образом стандартные растворы содержат 1 мг ионов металла в
1 мл раствора. Растворы на калий и натрий можно приготовить
в одной мерной колбе. Эти растворы считают запасными и ис-
пользуют для приготовления рабочих стандартных растворов с
концентрацией по ионам К+ и Na+ 50—100 мкг/мл. Рабочие
растворы готовят также в мерных колбах на дистиллированной
воде или растворителе, соответствующем применяемому экстра-
генту (см. выше). Эталонные (градуировочные) растворы гото-
вят обязательно в мерных колбах (100—250 мл), приливая из
бюреткь необходимое количество стандартных рабочих раство-
ров калия и натрия. Удобна следующая шкала концентрации
эталонных растворов (в мкг/мл): 0,5; 1,0; 2,0; 4,0; 8,0; 15,0; 30,0
и 60,0. КолбБ1 доводят до метки растворителем, соответствующим
определяемой задаче (см. выше).
Ход анализа на пламенном
фотометре ПФМ
К работе на пламенном фотометре допускаются операторы
только после прохождения технического обучения по безопасно-
му обращению и работе с газовыми баллонами.
Внешний вид прибора показан на рис. 72. До начала работы
прибор следует тщательно проверить и убедиться, что шлан-
236
ги для подачи газа и воздуха плотно надеты на соответствующие
штуцеры и в этих местах нет утечек газа и воздуха; шланги не
имеют трещин, проколов, резких перегибов пли других повреж-
дений, допускающих утечку газа. Ручка регулировки подачи газа
должна быто повернута против часовой стрелки. Необходимо
следить за наличием жидкости в сифоне (водяной затвор), от-
сутствие жидкости можег вызвать проскок пламени в корпусе
горелки.
Подготовка прибора к работе включает следующие операции.
1. Установить на горелку фотометра колпачок, соответствую-
щий применяемому газу.
2. Залить сифон для удаления конденсата из горелки водой
до верхнего уровня колена стеклянной трубки.
3. Заземлить прибор и включить штепсельную вилку в сеть
соотзетствующего напряжения.
4. Включить прибор (тумблер И, рис. 72), прогреть прибор
в течение 15 мин.
5. Перекрыть конденсор фотометра заслонкой (ручка 5 в по-
ложении 5), установить минимальную чувствительность
прибора (ручка 9 в положении 0), ручками 7 и 8 «грубо»
и «точно» установить стрелку микроамперметра на нуль.
6. Включить компрессор и регулятором 2 «воздух» установить
давление по манометру 15 в пределах 0,4 атм. Убедиться,
что распылитель всасывает жидкость из стаканчика.
7. Открыть вентиль на редукторе баллона и подать газ в при-
бор давлением 0,4—0,5 атм. Ручкой, плавно открывая вен-
тиль для газа на приборе, подать газ в горелку. Расход
пропан-бутана по шкале ротаметра 13 должен составлять
5—10 делений. Одновременно с подачей газа в горелку при-
бора нажимать кнопку 3 «Поджиг» до тех пор, пока не
произойдет воспламенение газа.
8. После того как загорелось пламя, регулируя расход газа
и давление воздуха вентилями прибора газ — воздух, до-
биться, чтобы фон пламени был минимальным, конусы
пламени над отверстиями колпачка не были сильно вытя-
нуты, имели голубое свечение, были ярко очерчены и го-
рели спокойно, без мерцания.
9. Перед началом измерений промыть горелку дистиллиро-
вание т водой через распылитель при включенном пламени
в течение 5—10 мин. О работе распылителя можно судить
по изменению уровня жидкости в стакане.
Порядок работы.
1. Включить в рабочее положение светофильтр, соответствую-
щий измеряемому элементу (переключатель 5).
2. Подобрать диапазон чувствительности, начиная с наиме-
нее чувствительного — 5-го (переключатель 9). Постоян-
ную диафрагму 6 установить в положение 1. Для умень-
шения погрешности измерений рекомендуется работать при
237
чувствительности прибора, вызывающей отклонение стрел-
ки микроамперметра на 80—100 делений. Диапазон изме-
рений должен быть выбран так, чтобы изменением ирисо-
вой диафрагмы (переключатель 4) при введении в пламя
эталонного раствора с максимальной концентрацией было
возможно установить стрелку микроамперметра на деление
80—100. С уменьшением концентрации элемента в раство-
рах чувствительность прибора может быть увеличена пере-
ключением ручки 9 в положение 4—3—2—1. Ручка пере-
ключателя 9 уменьшает чувствительность прибора в отно-
шении 1 : 2 : 10 : 100 : 1000.
3. Установить под капилляр распылителя стаканчик с дистил-
лированной водой и, распыляя воду ручками потенцио-
метра 8, 7 «грубо» и «точно», установить стрелку микро-
амперметра на нуль. Затем, распыляя эталонные растворы
с последовательно возрастающей концентрацией элемента,
фиксируют отсчеты на шкале микроамперметра. Число эта-
лонных растворов берут таким, чтобы получить достаточ-
ное число точек для построения градиуровочной кривой.
После каждого раствора, отличающегося концентрацией
элемента, в пламя необходимо распылять дистиллирован-
ную воду для промывания горелки и проверять, не измени-
лось ли положение стрелки микроамперметра относительно
нуля. Если стрелка отклонилась от нулевого положения, то
потенциометром ее снова устанавливают на нуль. Во время
измерений давление воздуха и расход газа в приборе долж-
ны быть постоянными. Зная концентрации элементов в
эталонных растворах и показания микроамперметра по
каждой концентрации, строят градуировочную кривую для
данного элемента в координатах показания прибора — кон-
центрация. Затем в пламя горелки распыляют испытуемый
раствор, снимают для него отсчеты по микроамперметру и
по градуировочному графику определяют концентрацию
элемента в растворе.
Через каждый час работы на приборе необходимо проверять
положение крайних точек градуировочною графика распылением
эталонных растворов с минимальной и максимальной концентра-
циями определяемого элемента.
В случае изменения положений крайних точек необходимо про-
извести соответствующую корректировку путем изменения рас-
крытия ирисовой диафрагмы 4.
Выключение прибора.
1. По окончании работы промывают распылитель дистиллиро--
ванной водой в течение 5—10 мин.
2. Перекрывают вентиль сначала на газовых баллонах, а за-
тем на фотометре.
3. Отключают компрессор и питание прибора от сети.
238
Вычисление результатов. По градуировочному графику ре-
зультаты получают обычно в мгк/мл или мг/л (в пересчете на
металл). Данные пересчитывают в общепринятые в почвенно-
агрохимической практике единицы измерении.
Валовое содержание К (или Na) в почвах в
Р 10 000 ’
где С — найденная концентрация К (Na) в мкг/мл; V — объем
колбы в мл; Р — навеска образца почвы в г; 10000 — коэффи-
циент пересчета.
Содержание водорастворимых соединений Na (К) в почвах
в
. С•V
мц 100 г =------.
Р • 10
С • V
Содержание обменного К в мг-экв/100 г почвы = р 1Q 39 t °б-
С • V
менного Na в мг-экв/100 г почвы =----------. доступного К з поч-
С V
вах в мг/кг =-----.
Р
ОПРЕДЕЛЕНИЕ ОБЩЕГО
СОДЕРЖАНИЯ МИКРОЭЛЕМЕНТОВ
В ПОЧВАХ
Общее содержание микроэлементов в почвах определяют ме-
тодом трех эталонов, испаряя пробу в дуте переменного тока из
канала угольного электрода. Регистрацию спектров одновремен-
но от одного источника проводят на кварцевом спектрографе
ИСП-30 и дифракционном ДФС-8-2 (с решеткой 1200 штр/мм).
Сочетание высокой разрешающей способности спектрографа
ДФС-8-2 и широкой области одновременного фотографирования
спектра на ИСП-30 позволяет с высокой производительностью
определять в почве 15—20 микроэлементов (Be, В, Ti, V, Сг,
Мп, Со, Ni, Си, Zn, Ga, У, Sr, Zr, Mo, Sn, Yb, Ba, Pb, Sc и др.).
Подготовка почв. Навеску 5—10 г представительного образца
почв растирают в агатовой ступке до состояния пудры. Из рас-
тертой пробы берут навеску около Зги помещают ее в фарфо-
ровый или кварцевый тигель. Тигель с почвой ставят в муфель-
ную печь и прокаливают 2 ч при 450—500° для удаления орга-
нического вещества и воды. Определяют, при необходимости,
потерю веса от прокаливания. Прокаленную почву сохраняют в
пакетиках из кальки в эксикаторах и используют по мере надоб-
ности для спектрального анализа.
239
Приготовление эталонов. В качестве эталонов используют ис-
кусственно приготовленные смеси, имитирующие средний состав
почв. Содержание макрокомпонентов в этой смеси (назовем ее
основой) следующее: SiO2 — 75%, А12О3 — 15,0, Fe2O3 — 4,5,
СаО — 1,5, Mg —• 1,5, 1\2СО3 — 1,5, Na2CO3— 1,0%. Контроль за
точностью определений проводят по стандартным (контрольным)
образцам почв, аттестованным Госстандартом СССР по валовому
содержанию макро- и микроэлементов (СП-1—типичный черно-
зем, СП-2 — дерново-подзолистая и СП-3 — каштановая; см.
табл. 22).
Основа для приготовления эталонов при ана-
лизе почв. Большинство макроэлементов вводят в основу в
виде окислов особой чистоты; используются следующие реакти-
вы и соединения: SiO2 — спектрально-чистые кварц или квар-
цит, окись кремния особой чистоты; А12О3 — спектрально-чистая
или особой чистоты окись алюминия; Fe2O3 — спектрально-чи-
стая или особой чистоты окись железа; СаО и MgO — в виде
соответствующих спектрально-чистых, предварительно прокален-
ных окислов или углекислых солей; К и Na — в составе карбо-
натов, не содержащих определяемых элементов (соли особой чи-
стоты, спектрально-чистые или химически чистые).
Техника изготовления основы эталонов. Состав-
ные части основы, каждую в отдельности, тщательно перетирают
в ступке. Материал, из которого сделана ступка, не должен за-
грязнять реактивы. Использование ступок из нержавеющей ста-
ли ведет к загрязнению элементами семейства железа и другими
тяжелыми металлами, однако оно не повлияет на содержание
в пробах и эталонах бора, цинка, щелочных и щелочноземельных
элементов.
Универсальными и наиболее удобными для почвенно-агрохи-
мических исследований являются агатовые и кварцевые (халце-
доновые) ступки. В соответствии с составом основы берут навес-
ки компонентов и помещают их в банку-смеситель. Общая масса
основы должна быть 150—200 г. В банке-смесителе основу тща-
тельно встряхивают, после чего перетирают небольшими порция-
ми (по 10—15 г в зависимости от диаметра агатовой ступки).
Хорошо перемешанную и перетертую основу помещают в квар-
цевые чашки и 10—12 ч прокаливают в муфельной печи при
1100—1200°. После охлаждения основу снова растирают и про-
веряют на загрязнение примесями определяемых микроэлемен- 1
тов. Проверку проводят методом эмиссионного спектральногоi
анализа, принятым для почв. При отсутствии загрязнений основу '
используют для приготовления эталонов. j
Введение микроэлементов в основу. Исследуемые ,
микроэлементы вводят в основу сухим способом из их окислов
или солей. Содержание микроэлементов в рабочих эталонах
должно перекрывать уровни содержания их в наиболее типичных
почвах и должно быть также согласовано с чувствительностью
240
их определения. Наиболее удобные уровни содержания микро-
элементов в рабочих эталонах показаны в табл. 21.
Так как сухим способом трудно приготовить концентрации
микроэлементов, показанные в первом рабочем эталоне, то пер-
воначально готовят концентрированный и запасной эталоны (см.
табл. 21). Масса запасного эталона должна быть 20 г. Произ-
водят необходимые расчеты для получения заданной концентра-
ции микроэлементов в запасном эталоне. Взвешивают расчетные
количества окислов (солей) микроэлементов на аналитических
весах и смешивают их с основой. Смеси тщательно перетирают
в агатовой ступке. Затем запасной эталон разбавляют чистой
основой в 10 раз и получают концентрированный эталон, из ко-
торого последовательным разбавлением основой в 5—2 раза
готовят серию рабочих эталонов с различным содержанием опре-
деляемых микроэлементов (см. табл. 21).
Таблица 21
Содержание микроэлементов в эталонах для анализа почв, %
Эталонируемые элементы
Эталоны Mo, Yb Be, Со Li, В, Sc, Ni, Си, Zn, Ga, Rb, Y, Sn, Cs V, Cr Sr, Zr Ba Ti Mu
Запасной эталон 0,20 0,30 0,50 1,50 2,50 4,0 . .
Концентрирован- ный эталон Рабочие эталоны: 0,02 0,03 0,05 0,15 0,25 0,40 —— —
№ 1 0,0040 0,0060 0,0100 0,0300 0,0500 0,0800 1,5 0,2
№ 2 0,0020 0,0030 0,0050 0,0150 0,0250 0,0400 0,7500 0,1000
№ 3 0,0010 0,0015 0,0025 0,0075 0,0125 0,0200 0,3750 0,0500
№ 4 0,0005 0,00075 0,00125 0,0037 0,0062 0,0100 0,1875 0,0250
№ 5 0,00025 0,00037 0,000625 0,00185 0,0031 0,0050 0,0937 0,0125
№ 6 0,000125 0,000185 0,000312 0,0009 0,0015 0,0025 0,0470 0,0062
Приготовленные таким образом эталоны проверяют на одно-
родность и качество приготовления, а однородность каждого эта-
лона — повторным спектральным анализом. Для этого из всего
объема каждого эталона берут 10—20 проб, каждую весом около-
30 мг. Этими пробами набивают стандартные угольные электро-
ды и снимают их спектры (см. с. 243). Спектры расшифровывают
и промеряют абсолютные (или относительные по отношению
к фону или линиям элемента сравнения) почернения аналитиче-
ских линий микроэлементов, которые вносили в эталон. В хоро-
шо перемешанных эталонах колебания почернений за счет неод-
нородности эталона не должны превышать ошибку воспроизво-
димости метода. Практически разница в почернениях линий
241
эталонов между повторными определениями не должна превы-
шать (0,1S). В противном случае необходимо дополнительное
перетирание эталонов.
Наилучшими способами контроля концентрации микроэлемен-
тов в эталоне являются химические (колориметрические), поля-
рографические и спектральные (метод добавок) методы опреде-
ления микроэлементов. Прямолинейность градуировочного гра-
фика служит лишь косвенным подтверждением отсутствия грубых
ошибок в разбавлении, но не исключает возможности систе-
матических ошибок в концентрациях микроэлементов, повторяю-
щихся во всех эталонах.
После того как внесены коррективы в состав эталонов, их
снова следует проверить спектральным методом и убедиться в
прямолинейности градуировочных графиков уже с учетом истин-
ных концентраций микроэлементов. Только после получения пря-
молинейных, хорошо воспроизводимых графиков эталоны при-
меняют для анализа. Отсутствие систематических ошибок в при-
готовлении эталонов проверяют, анализируя по ним стандартные
контрольные образцы почв.
Форма электродов и их подготовка. В этом методе почвы
вводят в источник света из канала угольного электрода. Дтя
этого используют угольные безборные электроды марки С-1 или
МГ-ОСЧ (особо чистые, содержание бора ниже 1-10_5О/о) диа-
метром 6 мм. Нижний угольный электрод имеет фигурную фор-
му с концентрической канавкой и стерженьком посередине.
Оптимальная глубина концентрической канавки для определения
большой группы разнолетучих микроэлементов в почвах — 2 мм,
толщина наружной стенки электрода — 0,5 мм, диаметр внут-
реннего стержня — 2,5 мм, ширина концентрической канавки
»1 мм. Для быстрого высверливания углубления в электродах
строго заданных размеров используют специальную фрезу, укреп-
ленную на валу электромотора. Верхние электроды изготавли-
вают из аналогичных угольных стержней в форме усеченного
конуса, диаметр заточенного конца ~ 1 мм.
Набивка пробы в электрод. Пробу (эталоны, почвы)
набивают в концентрическую канавку электрода по объему.
Заполнение канавки пробой производят вычерпыванием материа-
ла торцом электрода в несколько приемов из различных мест
пробы, находящейся на листе кальки. Пробу уплотняют в канав-
ке электрода специальным уплотнителем (набивалкой). Набивал-
ка представляет собой цилиндрическую стальную трубку с про- (
дольными вырезами, впрессованную в ручку из оргстекла. После
уплотнения пробы набивалкой торец электрода снова прижимают
к порошку на бумаге, вращением электрода канавка заполняется
доверху пробой. После этого торец электрода прислоняется к
кальке, проба как бы втирается в электрод, поверхность запол-
няющего слоя выравнивается с поверхностью торца. Приемы
заполнения электрода должны быть стандартными.
242
Проба почвы заполняет углубление в электроде вровень с
поверхностью торца. На его поверхности почвы не должно быть.
Наличие поивы на торцевой поверхности стенок и внутреннего
стержня приводит к беганию шнура дуги. Каждой пробой почвы
(эталоном) набивают 3—5 электродов и устанавливают их в спе-
циальный штатив — деревянный (или пластмассовый) брусок с
отверстиями.
Фотографирование спектров
почв и эталонов
Приступая к выполнению практических работ, необходимо-
ознакомиться с заводской инструкцией к приборам, а также с
инструкцией о мерах предосторожности и по технике безопасно-
сти при работе в лаборатории спектрального анализа.
Работу проводят на хорошо отъюстированных и исправных
спектрографах. Подключают приборы к сети. Устанавливают
условия фотографирования спектра. Ширина щели спектрографа
ИСП-30 0,008—0,01 мм, ДФС-8 — 0,018 мм, высота щели 1 мм
(устанавливается при помощи диафрагмы Гартмана). Источник
возбуждения — генератор дуги переменного тока ДГ-2 в дуго-
вом режиме. Трехлинзовая конденсорная система, вырез револь-
верной диафрагмы на втором конденсоре — 3,2 мм. В темной
комнате заряжают кассету спектрографа фотопластинкой. Для
спектрографа ИСП-30 используют спектральные фотопластинки
«тип-1» чувствительностью 3—4 ед. ГОСТ. Для этого фотопла-
стинку (9Х 12 см) помещают в кассету так, чтобы правый край
ее на 4—5 см не доходил до правой рамки кассеты. При этом
положении фотопластинки фотографируют область спектра 230—
440 нм. Эмульсия фотопластинки должна быть обращена к ис-
точнику света (внутрь спектрографа). Кассета спектрографа
ДФС-8 заряжается фотографической пластинкой (13X18 см)
«спектральная тип-1» чувствительностью 5—6 ед. ГОСТ. На спек-
трографе ДФС-8 фотографируется область спектра 300—350 нм.
Заряженные кассеты закрепляют в спектрографе. При помощи
ходового винта кассету устанавливают в нулевое положение, на
спектрографе ДФС-8 это делают вручную, перемещая ручку 8
(см. рис. 71), а на ИСП-30 — с помощью электромотора, вклю-
чая тумблер 9 вниз (см. рис. 70). Открывают кассеты и при же-
лании впечатывают миллиметровую шкалу спектрографа ИСП-30
и шкалу длины волн на ДФС-8. Для этого на спектрографе
ДФС-8 включают тумблер 11 (см. рис. 71) на 2 мин, а на
ИСП-30 проворачивают рукоятку шкалы спектрограммы, после
чего перемещают кассету вверх на 4—5 делений.
Устанавливают в штативе для электродов железные электро-
ды. Контроль за правильным положением электродов производят
по проекционному изображению их концов на револьверной диа-
243
фрагме. Расстояние между электродами 2 мм отмечается штри-
хами на револьверной диафрагме. Реле обжига и экспозиции на
спектрографе ИСП-30 устанавливают па 10 с. Открывают щель
спектрографа ДФС-8 (снять крышку 6 и повернуть ручку 13,
рис. 71), тумблером 8 (см. рис. 70) зажигают дугу. Величину
тока дуги с помощью реостата устанавливают на 10 А. Световое
пятно от разряда электродов должно полностью попадать в вы-
рез револьверной диафрагмы, если этого не происходит, то по-
правляют положение электродов винтами держателя электродов.
По окончании фотографирования спектра реле выключает гене-
ратор и на спектрографе ИСП-30 перемещает кассету на 1 мм
вверх. На спектрографе ДФС-8 это необходимо сделать вручную.
Ручку реостата дуги выводят на минимальную величину тока.
Затем устанавливают в держатель угольные электроды с про-
бой, реле обжига ставят на нулевое положение, а реле экспози-
ции на 3 мин (см. рис. 70). Включают дугу, во избежание выбро-
са пробы из электрода в момент включения дуги величина тока
не должна превышать 3—5 А, в последующие 5—7 с величину
тока повышают до 20 А.
Контроль за положением электродов производят по проекции
их концов на экране револьверной диафрагмы. В процессе горе-
ния дуги расстояние между электродами поддерживают постоян-
ным с помощью винтов держателей.
Фотографирование спектра ведут до полного испарения иссле-
дуемых микроэлементов из канавки электрода, на что требуется
около 3 мин. После каждого снимка спектра перемещают кассету
и выводят реостат дуги на минимальный ток. Вначале снимают
спектры одной повторности эталонов, затем почв, после чего пе-
ремещают кассету вверх на 1 мм и повторяют съемку эталонов
и почв второй повторности, аналогичным образом — третьей
и т. д.
По окончании фотографирования спектров кассету закрывают,
переносят ее в темную комнату и проявляют.
Проявление и фиксирование. Экспонированную фотопластин-
ку вынимают из кассеты и погружают ее эмульсией вверх в
проявитель, налитый в кювету. Для проявления используют проя-
витель Д-19, при температуре 20° проявление продолжается 3—
4 мин (в загисимости от типа фотоэмульсии). Для равномерно-
го проявления всей поверхности фотоэмульсии необходимо вре-
мя от времени покачивать кювету. По окончании проявления
фотопластинку вынимают и быстро промывают в воде одновре-
менно по всей поверхности. Промытую пластинку переносят в
кювету с фиксирующим раствором (250 г гипосульфита натрия
растворяют в 1 л воды). Процесс фиксирования длится 10—
15 мин, после чего фотопластинка промывается 30 мин в проточ-
ной холодной воде. Промытые фотопластинки помещают в спе-
циальный штатив и сушат в помещении, свободном от пыли.
'244
Расшифровка спектрограммы
и измерение почернений
аналитических линий
Полученную спектрограмму помещают эмульсией вверх на
столик спектропроектора ПС-18 (или СПП-2). Проектируют чет-
кое изображение спектра на экран спектропроектора. С помощью
атласа линий отождествляют аналитические линии исследуемых
микроэлементов. Для этого один из планшетов атласа наклады-
вают на соответствующую область спектрограммы на экране
спектропроектора так, чтобы линии железа в проекции спектра
совпали с соответствующими линиями атласа. Штриховые ука-
затели линий на атласе покажут нахождение аналитических
линий определяемых микроэлементов. Для определения микро-
элементов в почвах используют наиболее чувствительные анали-
тические линии (табл. 22).
Таблица 22
Аналитические линии, чувствительность и содержание микроэлементов
в аттестованных Госстандартом СССР почвах, мг/кг
Эле- менты Аналити- ческая линия, нм Чувстви- тельность определе- ния. мг/кг СП-1 СП-2 сп-з
аттесто- вано найдено аттесто- вано найдено аттес- товано найдено
Ве 234,86 10 2,0 Н. О. 1,5 н. О. 4,2 и. о.
В 249,77 5 53 45 43 40 71 65
Т1 295,61 10 450 400 503 650 437 500
V 318,53 10 77 60 64 66 ПО 125
Сг 301,47 10 82 78 84 90 140 160
Мп 294,92 10 596 600 542 500 712 650
Со 345,35 5 10 8,5 10 9,5 14 15
N1 341,47 10 33 34 25 27 56 50
Си 327,39 1,0 22 25 17 20 30 28
Zn 334,50 100 52 65* 45 44* 73 83
Ga 294,36 10 10 Н. О. 8,5 Н. О. 13 Н. О.
Sr 346,44 100 130 Н. О. 120 Н. О. 160 Н. О.
Y 332,78 10 39 30 27 32 28 30
Zr 339,19 10 450 500 540 500 300 250
Mo 317,03 0,5 1,0 1,2 1,0 1,2 1,1 1,6
Sn 317,50 1,0 3,9 3,3 2,8 3,3 4,9 4,6
Yb 328,93 1,0 4,0 4,3 3,1 4,0 3,2 3,5
Ba 233,52 30 430 530 530 500 470 450
Pb 283,30 10 16 12 14 11 16 18
Sc 335,37 10 12 10 9,4 9,0 14 13
* Определено атомно-абсорбционным методом.
Измерение почернений линий проводят на микрофотометре
МФ-2 (с устройством и работой микрофотометра следует озна-
комиться заранее по инструкции, прилагаемой к прибору). При-
245
ступая к фотометрированию, следует убедиться, что прибор уста-
новлен правильно. Через 10—15 мин после включения лампы
микрофотометра приступают-к измерению, для этого указатель
на экране микрофотометра совмещают с началом отсчетной шка-
лы (с делением оо) при закрытом фотоэлементе. Наводят на
щель микрофотометра измеряемую линию, включают фотоэле-
мент и с помощью микрометрического винта проводят спектраль-
ную линию через щель микрофотометра. Отсчет берут в момент
максимального значения почернений. Затем переходят к измере-
нию той же линии, но в другом спектре и т. д. Результаты фото-
метрирования записывают в таблицу.
Расчеты. Полученные результаты фотометрирования усред-
няют. По найденным величинам почернений линий в эталонах
строят градуировочный график (см. рис. 74) в координатах
5 — lgС (или AS — lgС), где AS — разность между почерне-
нием аналитической линии и внутреннего стандарта или фона
спектрограммы. По градуировочному графику определяют кон-
центрации микроэлементов в исследуемых почвах. Результаты
выражают в % или мг/кг почвы.
Чувствительность определения микроэлементов по изложенно-
му выше методу показана в табл. 22. Воспроизводимость опре-
деления в зависимости от элемента и уровня концентрации
варьирует от 10 до 20%. Такие микроэлементы, как Zn, Mo, Sn,
в некоторых типах почв определить не удается из-за недостаточ-
ной чувствительности изложенного метода. В этих случаях необ-
ходимо применять методы концентрирования или другие приемы,
повышающие чувствительность определения.
ЛИТЕРАТУРА
3 ы р и н Н. Г., О б у х о в А. И. Спектральный анализ почв, растении и других
биологических объектов. М., Изд-во Моск, ун-та, 1977.
Тарасевич Н. И., Семененко К. А, Хлыстова А. Д. Методы спек-
трального и химико-спектрального анализа. М., Изд-во Моск, ун-та, 1973.
Глава 7
ОПРЕДЕЛЕНИЕ В ПОЧВЕ СОДЕРЖАНИЯ
МАКРО- И МИКРОЭЛЕМЕНТОВ
МЕТОДОМ АТОМНО-АБСОРБЦИОННОГО
СПЕКТРАЛЬНОГО АНАЛИЗА
Принцип метода
Атомно-абсорбционный спектральный анализ основан на ис-
пользовании способности свободных атомов определяемых эле-
ментов селективно поглощать* резонанслгск^’йз'лучеттй'е определен-
ной для каждого~эЛёмёнта длины АбдтптТ’ТГрииципиальная схема
атомно-абсорбционного метода показана на рис. 75. В классиче-
ском варианте метода для измерения поглощения анализируемый
раствор 9 в виде аэрозоля вводят в пламя горелки 3. В пла-
мени происходят испарение растворителя, плавление и испарение
Рис. 75. Принципиальная схема атомно-абсорбционного спектрофо-
тометра:
1 — источник питания лампы, 2 — лампа с полым катодом, 3 —
пламя, 4 — монохроматор, 5 — фотоэлектрический детектор, 6 —
усилитель, 7 — гальванометр, 8 — распылитель, 9 — анализируемый
раствор
пробы, термическая диссоциация молекул и образование свобод-
ных атомов. Большинство образующихся при этом атомов нахо-
дится в нормальном, невозбужденном состоянии. Они могут
поглощать излучение внешнего стандартного источника света 2,
если энергия кванта соответствует энергии перехода атома
с нижнего энергетического состояния (основного) на более высо-
кий уровень В качестве источника излучения чаще всего служит
247
лампа с полым катодом из одного или нескольких определяемых
элементов.
< Световой поток от лампы пропускают через пламя горелки 3
I и монохроматор 4. Далее измеряют поглощение света атомами
исследуемого элемента. Выходящий световой поток регистрируют
фотоэлектрическим детектором 5. Сигнал с детектора усиливает-
ся с помощью усилителя 6, регистрируется гальванометром 7
лдц ленточным самописцем.
Назначение
и возможности метода
в почвенных исследованиях
Как метод аналитической химии атомно-абсорбционный ана-
лиз впервые был предложен в 1955 г. и сразу же нашел самое
широкое применение в почвенно-агрохимических исследованиях.
Этот метод выгодно отличается от традиционных аналитиче-
ских методов универсальностью, простотой выполнения анализа
и высокой производительностью. Атомно-абсорбционный метод
анализа обеспечивает предел обнаружения многих элементов по-
рядка 0,1—0,01 мкг/мл и ниже, что практически оказывается
достаточным в почвенных исследованиях. Воспроизводимость
метода 1—3%. В настоящее время этим методом можно опреде-
лить более 70 химических элементов в разных объектах.
В почвенно-агрохимической практике разработаны методы
определения около 35 элементов для решения самых различных
проблем. Предложены методы определения валового содержания
Si, Al, Fe, Са, Mg, К, Na, Мп, Ti в почвах, многих биологически
важных микроэлементов (валовое содержание и подвижные фор-
мы) — Zn, Си, Со, Ni, Cr, V и др. Атомно-абсорбционные методы
позволяют определить обменные основания и емкость поглоще-
ния, исследовать состав и количество водорастворимых катио-
нов в почве.
При контроле загрязнения окружающей среды тяжелыми ме-
таллами атомно-абсорбционный метод занимает ведущее поло-
жение, особенно для таких металлов, как Pb, Zn, Cd, Hg, As, Se.
Однако атомно-абсорбционный метод на сегодняшний день еще
не обеспечивает определения ряда элементов, и в частности очень
важных для почвоведения — азота, углерода, хлора, бора и йода.
» Основы теории
атомно-абсорбционного метода
Поглощение света атомом. При условии термического равно-
весия величина поглощения света в пламени зависит от соотно-
шения атомов, находящихся в возбужденном и основном состоя-
248
пнях. Последнее при заданных
ляется формулой Больцмана:
температурных условиях опреде-
— (Ет Еп)
т ёт „---------
где Nm и Nn — число атомов в возбужденном и основном со-
стояниях; gm и gn — статистические веса рассматриваемых со-
стояний; К-— постоянная Больцмана, равная 1,38032-10~23 Дж;
£„г и Еп — соответствующие энергетические состояния атомов;
Т — абсолютная температура пара.
При температурах от 2000 до 5000°К отношение числа ато-
мов, находящихся в возбужденном состоянии, к числу атомов в
основном состоянии очень мало, т. е. число атомов, способных
поглощать резонансные линии, будет практически равно общему
числу атомов определяемого элемента.
В отличие от эмиссионного спектрального анализа, где не-
большое колебание температуры значительно изменяет концент-
рацию возбужденных атомов, в атомно-абсорбционном методе
число атомов, способных поглощать энергию первичного источни-
ка излучения, мало изменяется с температурой. Это ценное преи-
мущество атомно-абсорбционного метода анализа перед эмис-
сионным.
Закон атомного поглощения в пламени аналогичен закону
светопоглощения в молекулярной спектрофотометрии (см. с. 129)
и характеризуется экспоненциальным убыванием интенсивности
проходящего света I в зависимости от дгшны пламени I и кон-
центрации С. В определенном интервале концентраций поглошн—
ние света атомами подчиняется закону Бугера—Eepaj I = Ine~Ki-c'1
где /0 — интенсивность излучения до''вЗДтЖбдёиствия;'7 — ин-
тенсивность излучения после его взаимодействия с атомами;
Кк — коэффициент поглощения света, зависящий от длины вол-
ны и линии поглощения; С — концентрация или число Nn по-
глощающих атомов; I — длина пламени (столб паров поглощаю-
щих атомов Л-„ или зона поглощения). —---------
' Коэффициент—поглощения является~~важной характеристикой,
описывающей свойства линий поглощения. Величины коэффи-
циентов атомного поглощения некоторых элементов измеряются
порядком п-108, так для Na — 1,46- 108; для Mg и Cd —
2,4-108. Этим объясняется высокая чувствительность атомно-
абсорбционного метода по сравнению с молекулярной спектро-
фотометрией, где для цветных реакций в водных растворах мак-
симальные величины молярных коэффициентов светопоглощения
выражаются порядком п-105.
В атомно-абсорбционном методе для аналитических целей исполь-
зуют те же понятия, что и в молекулярной спектрофотометрии, а
именно: пропускание Т = — 1ОО°о и поглощение 1—Т, а также
/о
249
оптическая плотность D — lg -j- или D = К.к-1-С. Величину T вира
жают в процентах, тогда D = 1g— • 100 или D = 2 — lg Т.
Теоретический расчет концентрации элемента по измеренной
оптической плотности атомного пара пока невозможен из-за не-
полного испарения растворенного вещества и неполноты диссо-
циации на атомы окислов и гидроокислов большинства элемен-
тов. Поэтому при измерении концентрации элементов в растворе
предварительно строят градуировочный график в координатах
D—С, исходя из экспериментальных данных по серии стандарт-
ных растворов. Прямолинейная зависимость оптической плотно-
сти от концентрации атомов сохраняется при отсутствии влияния
посторонних элементов на поглощение, а также при низких кон-
центрациях определяемых элементов в сравнительно небольшой
области концентраций. По мере увеличения концентрации гра-
дуировочный график искривляется к оси абсцисс.
Получение поглощающего
слоя атомов.
Атомизаторы
Так как атомно-абсорбционный спектральный анализ основан
на измерении поглощения света свободными атомами, то наибо-
лее важным звеном является «атомизация» анализируемого
образца, т. е. переведение вещества в состояние, при котором
определяемые элементы находятся в виде свободных атомов, спо-
собных поглощать свет. От способа атомизации зависит чувстви-
тельность и правильность метода. Известные способы получения
поглощающих слоев можно подразделить на две основные груп-
пы: к первой относятся равновесные методы (пламя), ко второй
группе — импульсные методы (графитовая кювета, импульсные
источники, лазерные испарители).
Наиболее универсальным, удобным и стабильным источником
получения свободных атомов является пламя. Пробы (чаше §
виде растворов) вводят в пламя с помощью распылителя. В пла-
мени при 2000—3000° К происходит испарение растворителя, раст-
воренные вещества превращаются в мелкие твердые частицы,
которые далее плавятся и испаряются. Образующиеся парь' со-
держат смесь различных химических соединений, частично рас-
падающиеся на отдельные атомы. Атомы получают в пламени
дополнительную энергию и могут переходить в возбужденное и
ионизированное состояния. Степень атомизации различных эле-
ментов зависит от вида и температуры пламени, от химического
состава пробы и радикалов, образующихся в пламени.
На первых этапах развития атомно-абсорбционного метода,
особенно применительно к почвенно-агрохимическим объектам, в
качестве источника атомизации использовали пламЯ1 'воздух —
250
пропан — бутан. .Эту смесь использовали для элементов, кото-
рые легко поддаются атомизации, — щелочные металлы, кадмий,
медь, свинец, серебро, цинк. Однако в настоящее время эта смесь
используется уже не так широко, как раньше, из-за помех, кото-
рые обнаружили в процессе атомизации Помехи обусловлены
низкой температурой этого пламени и неполной атомизацией
некоторых элементов, особенно в таких сложных объектах, как
почвы. Стабильность горения этого пламени несколько меньше,
чем пламени ацетилен — воздух.
Наиболее эффективным^способом атомизации является пламя
ацетилен — воздух, Эта смесь используется для определения
большинства элементов, не образующих термостойких окислов
(Са, Mg, Со, Fe, Cr, Ni). Для металлов, склонных образовывать
термостойкие окислы, чувствительность определения может быть
повышена путем применения горючей смеси, обогащенной топли-
вом (восстановленное пламя). Низка чувствительность определе-
ния в пламени ацетилен — воздух алюминия, титана, циркония
и некоторых других элементов, где энергия связи металл — кис-
лород превышает 5 эВ.
Использование смеси закиси азота (в качестве газа окислите-
ля) с ацетиленом, значительно расширило возможности атомно-
абсорбционного анализа определения элементов, образующих
прочные комплексы непосредственно в растворе или в зоне ато-
мизации. Появилась возможность определять в почвах Al, Si, Ti,
Zr, Са, Mg, Sr, Ba без применения -специальных буферов. Повы-
шение температуры пламени устраняет химические помехи при
определении многих элементов, о которых будет сказано ниже.
Некоторые пламена (воздушно-ацетиленовые, закись азота —
ацетилен) в дальнейшем ультрафиолете дают интенсивное соб-
ственное излучение, что увеличивает «шумы» при анализе, осо-
бенно для таких элементов, как мышьяк, чселен, олово, для опре-
деления которых используют смесь роз Дух — водород. Равновес-
ная концентрация определяемого элемента^ЭгтлшЯешГ~достигается
за счет непрерывного потока распыляемого раствора через
пламя. Для превращения раствора в аэрозоль и далее в атомный
пар применяют специальный узел, состоящий из распылительной
камеры и горелки. От работы этого узла зависит чувствитель-
ность и точность анализа, возникновение многих помех при ана-
лизе связано с недостаточно эффективной работой распылителя.
Использование аэрозоля с очень меткими частицами ослабляет
или полностью устраняет химические помехи, поскольку в этом
случае для перевода аэрозоля в атомный пар требуется меньше
энергии, т. е. атомизация будет более полной.
Поскольку оптическая плотность поглощающего слоя пропор-
циональна его длине, то чем длиннее слой пламени, тем больше
чувствительность анализа. Для увеличения чувствительности
применяют насадки на горелки и специальные адаптеры пла-
мени.
В качестве импульсных атомизаторов используются также
графитовые кюветы, графитовые трубчатые кюветы Массмана,
угольная нить, носящие общее название «иепламенные атомиза-
торы». Непламенные атомизаторы повышают чувствительность и
пределы обнаружения элементов на 2—3 порядка, поэтому их
применяют для определения следов элементов (на уровне
Ю~5—10-7%) и при анализе микрообразцов. Увеличение чувстви-
тельности обусловлено увеличением средней продолжительности
пребывания атомов в графитовой кювете до 1 с, в то время как
в пламени оно на 3—4 порядка меньше. При использовании кю-
вет в поглощении света участвует подавляющее большинство
атомов внесенной пробы, тогда как в пламени — всего лишь
несколько процентов. Непламенные атомизаторы исключают
помехи, возникающие в пламени за счет химических реакций, в
частности образование термостойких окислов некоторых элемен-
тов. При анализе почв особо ценное преимущество непламенных
атомизаторов заключается в принципиальной возможности опре-
деления некоторых элементов непосредственно в почве без пере-
ведения ее в раствор.
Графитовые кюветы представляют собой трубку длиной 8—
10 см, диаметром 6—8 мм с круглым отверстием (1—2 мм) по-
середине. Внутренняя часть трубки покрыта пиролитическим
особо чистым графитом. Пробу (раствор) с помощью микро-
шприна помещают через центральное отверстие в трубку и на-
гревают электрическим током от специального источника (сила
тока до 400—500 А) до температуры 2000—2700°.
С помощью источника нагрева графитовой кюветы задают и
регулируют температуру атомизации в зависимости от опреде-
ляемого элемента, время и температуру термической подготовки
проб в зависимости от количества и состава взятого для иссле-
дования раствора. Нагрев графитовой кювегы обычно осуществ-
ляют в инертной атмосфере (аргон, азот), поэтому образование
термостойких окислов в этом случае не происходит. Инертный
газ предохраняет от окисления и разрушения саму графитовую
кювету, уменьшает диффузию паров пробы через ее стенки, а
также способствует быстрому и полному удалению паров, обра-
зующихся во время термической подготовки пробы.
В качестве атомизаторов применяют и комбинированные уст-
ройства, например нагреваемую электрическим током графито-
вую трубку и пламя. При атомизации пробы таким способом
пары ее диффундируют через стенки трубки и попадают в пла-
мя горелки. При таком способе атомизации наряду с высокой
чувствительностью помехи от состава пробы сводятся к миниму-
му. Другие методы атомизации проб (лазер, плазменные источ-
ники) мало используют для анализа почв, но они являются пер-
спективными. Современные графитовые атомизаторы обеспечи-
вают точность определений 2—5%.
Г
252
Оптические системы
в атомно-абсорбционном анализе
В общей схеме атомно-абсорбционных. измерений оптическая
система представлена четырьмя основными узлами (рис. 76):
1) источник первичного излучения; 2) предщелевая оптика —
система линз, проектирующих излучение источника на середину
атомизатора и далее на щель монохроматора; 3) монохроматор;
4) детектор излучения.
Рис. 76. Однолучевая осветительная система
1 — источник первичного излучения, 2 — зона атомизации, 3—мо-
нохроматор, Li и Lj — линзы
Источники первичного излучения. Источник первичного излу-
чения должен излучать узкие интенсивные резонансные линии
определяемых элементов. Излучение должно быть стабильным
во времени. Такими источниками служат лампы с полым като-
дом Современные лампы с полым катодом изготавливают в виде
стеклянного баллона с впаянными в него электродами. Баллон
заполнен инертным газом (аргон, неон, гелий). Входное окно
лампы сделано из материала, пропускающего ультрафиолетовое
излучение. Чтобы первичный эмиссионный спектр был свободен
от посторонних линий, полый катод лампы должен изготовляться
из металла (или сплавов в случае многоэлементных ламп) высо-
кой чистоты.
Для питания лампы требуется постоянное напряжение не ме-
нее 500 В, рабочий ток лампы 10—30 мА. Для получения ста-
бильного излучения лампы с полым катодом применяют выпря-
мители с высокой степенью стабилизации тока. Показано, что
для того чтобы колебания интенсивности линии Mg 2852 А не
превосходили 1%, колебания разрядного тока не должны быть
больше ±0,1 мА.
Аналитические характеристики и срок службы ламп с полым
катодом зависят от режима питания, рода наполняющего газа и
его давления, конструкции полого катода. Учитывая это, можно
значительно воздействовать на чувствительность и точность атом-
но-абсорбционного анализа. В результате оптимизации ряда па-
раметров удалось достигнуть увеличения срока службы ламп
(до 1000—3000 ч) и повысить их аналитические характеристики
Широкое применение нашли лампы с повышенной яркостью
излучения. Они отличаются от обычных ламп с полым катодом
253
наличием дополнительного разряда на выходе из полости като-
да, где находится облако невозбужденных паров. Под действием
второго разряда это облако паров также становится источником
излучения. С помощью таких ламп удалось улучшить отношение
сигнал — шум и увеличить прямолинейную часть калибровочных
кривых благодаря снижению самопоглошенпя резонансных лп-
ЛИЙ-В-самой_лампе. Высокоинтенсивные лампы позволяют также
работать с узкими щелями монохроматора, т. е. более «чисто»
монохроматизпровать необходимое излучение и регистрировать
его в относительно мягких и стабильных условиях.
Для почвенно-агрохимических исследований удобны многоэле-
ментные лампы с полым катодом, наиболее рентабельны лампы
на 4—5 элементов.
Кроме ламп с полым катодом в атомно-абсорбционном ана-
лизе применяются высокочастотные безэлектродные (шариковые)
штампы. Они представляют собой кварцевый шарик (диаметром
~20 мм), в который введено несколько миллиграммов соответ-
ствующего металла (или его соединения) и инертный газ. Тако-
вы лампы ВСБ-2. Высокочастотные безэлектродные лампы могут
использоваться для возбуждения спектров относительно боль-
шого числа элементов (до 50), но выгоднее их использовать для
тех элементов, лампы с полым катодом для которых трудно
изготовить или они ненадежны в работе. К таким элементам
относятся мышьяКт- еуръм-а... висмут, уеден, теллур. Стабильность
работы этих ламп сравнима и даже лучше, чем ламп с полым
катодом на эги элементы. В других случаях предпочтение нуж-
но отдать лампам с полым катодом, хотя шариковые лампы
сравнительно дешевы. Разряд в шариковой лампе возникает при
введении_ее_в—высокочастотное электромагнитное поле. Для пп-
Тгшия"~ламп такого типа промышленность выпускает прибор
Г1Г1БЛ-3, генератор высокой частоты.
Предщелевая оптика. Для получения максимально возможной
чувствительности определения элементов от источника излучения
нужно пропустить как можно больший световой поток через
зону атомизации. Для этого в зависимости от конструкции при-
бора используют различные осветительные системы, которые
должны уменьшить количество рассеянного света на пути от
лампы до~~Детсктора. Наиболее" типичной является оптическая
схема, показанная на рис. 76. Это двухлпнзовая система с про-
межуточным изображением источника в поглощающей ячейке,
она обеспечивает хорошую светосилу при условии полного за-
полнения коллиматора спектрофотометра.
Размеры источника первичного излучения, атомизатора и
оптическая апертура монохроматора должны быть строго согла-
сованными. Для удлинения пути поглощения излучения часто
применяют различные зеркальные системы, обеспечивающие мно-
гократное прохождение светового луча источника через зону
атомизации.
254
Чтобы снизить ошибки за счет колебаний интенсивности пер-
вичного излучения, применяют двухлучевую схему (см. рис. 77).
В такой схеме луч от источника падает на зеркальный сектор 2Г
который поочередно направляет луч либо через пламя, либо, ми-
нуя поглощающий слой, прямо на зеркало 4 и монохроматор 5.
После прохождения пламени оба луча совмещаются с помощью
полупрозрачного зеркала 4. Благодаря сдвигу лучей по фазе вы-
ходные сигналы детектора регистрируют и усиливают раздельно,
а затем сравнивают друг с другом с помощью мостовой схемы.
Рис 77. Двухлучевая схема:
1 — источник первичного излучения, 2 — зеркальный сектор, 3 —
горелка, 4 — полупрозрачное зеркало, 5 — монохроматор
Возникающий на усилителе сигнал разбаланса компенсируют с
помощью потенциометра вручную или автоматически с помощью
электронной схемы. С помощью аналоговых схем можно преоб-
разовать сигнал в логарифмическую форму (оптическая плот-
ность). ----—-------— - -______________
.. ^Монохряматорд Монохроматор в атомно-абсорбционном ана^
лизе выделяет резонансную аналитическую линию и отделяет ее
от молекулярные спектров и сплошного фовд.^ излучаемых ато^.
-мизаторами. Д1д5этой причине монохроматор в классической
схеме анализа всегда расположен после атомизатора. В принци-
пе от монохроматора не требуется высокая разрешающая спо-
собность. При одноэлементном спектре излучения и определении
щелочных и щелочноземельных элементов в почвах хорошие
результаты можно получить даже с помощью простых стеклян-
ных или интерференционных светофильтров.
В некоторых случаях можно обойтись вовсе без монохрома-
тора (бездисперсионные анализаторы). Для этого необходимо
детектор строго согласовывать по спектральной чувствительности
с источником излучения, т. е. использовать приемники света, чув-
ствительные в нужной и узкой областях спектра. Однако при
анализе сложных объектов и при использовании многоэлемент-
255
ных источников излучения необходимо использовать монохрома-
тор.
Монохроматор при минимальной эффективной ширине щели
практически должен разделять две линии, различающиеся по
длине волны на 0,1 нм. В качестве диспергирующего элемента
в монохроматоре может быть призма или дифракционная решет-
ка. Для лабораторного изготовления атомно-абсорбционного
спектрофотометра из отечественных монохроматоров подходящи-
ми являются СФ-4, СФД-2, ДМР-3 и др. Предпочтение следует
отдать дифракционным монохроматорам, обеспечивающим равно-
мерную дисперсию по всему спектру и в широком диапазоне.
Наиболее совершенные приборы обычно комплектуют двумя
дифракционными решетками, обеспечивающими хорошее разре-
шение и светосилу в ультрафиолете и в видимой части спектра.
Детектор излучения. В качестве детектора излучения в основ-
ном используют фотоэлектронные умножители ФЭУ. ФЭУ долж-
ны обладать достаточной чувствительностью в широкой области
спектра (от линии мышьяка /,= 193,7 нм до цезия — 852,1 нм),
хорошей интегральной чувствительностью и стабильностью рабо-
ты. С точки зрения спектрального диапазона чувствительности
наиболее универсальными являются ФЭУ с сурьмяно-цезиевым
фотокатодом и еще лучше трехщелочной катод (сурьмяно-нат-
рий-калий-цезиевый), чувствительный до 850 нм.
Для приборов лабораторного изготовления можно использо-
вать широко распространенный отечественный ФЭУ-19А, позво-
ляющий решить многие задачи почвенно-агрохимического ана-
лиза.
Системы регистрации и пересчета. Фототок с фотоумножите-
ля поступает на регистрирующий прибор. Величина фототок.?
пропорциональна пропусканию пламени. Рёгистрирование фото-
тока (усиленного или неусиленного) с помощью гальванометра,
отградуированного в линейных единицах, обеспечивает измере-
ние сигнала в линейной шкале пропускания. Переход от пропу-
скания Т к оптической плотности D, согласно вышеприведенно-
му уравнению, несложен. При работе на простых приборах лабо-
раторного изготовления для этого можно также воспользоваться
предварительно составленной таблицей связи D и Т. Почти все
современные атомно-абсорбционные спектрофотометры оснащают-
ся специальным устройством, превращающим линейный выход с
усилителя в логарифмический. Таким образом регистрируется
величина D.
В настоящее время ряд приборов уже оснащен цифровой
индикацией, имеет выход на цифропечать, телетайп и ЭВМ.
Самые современные приборы имеют электротехническое устрой-
ство или небольшую ЭВМ, позволяющее переходить от оптиче-
ской плотности к концентрациям, электротехнически или матема-
тически (с помощью ЭВМ) спрямлять нелинейные калибровоч-
ные функции. Сигнал, поступающий на экран прибора или в циф-
256
ропечать, может суммироваться и выдаваться как средний ре-
зультат за определенный промежуток времени (от 10 до 100 с)
в процессе распыления образца.
Помехи, влияющие
на результаты атомно-абсорбционного анализа
Помехи, возникающие в ходе атомно-абсорбционного анализа
почв, можно подразделить на пять групп: 1) спектральные поме-
хи; 2) химические помехи; 3) помехи за счет ионизации; 4) по-
мехи из-за различий в составе проб (и эталонов); 5) фоновые
помехи (неселективное поглощение). Причиной спектральных
помех является поглощение излучения не только резонансной
линией интересуемого элемента, но также и атбмами’другнх'Ъле-
ментов с близкой длиной волны. При атомно-абсорбционных
определениях вероятность взаимного наложения спектральных
линий элементов практически исключается. Это, безусловно, до-
стоинство метода по сравнению с эмиссионным анализом.
Наибольшее значение при анализе почв имеют химические и
ионизационные помехи, обусловленные химическими эффектами,
протекающими в растворе или в атомизаторе. Они возникают при
образовании соединений исследуемых элементов, обладающих
повышенной устойчивостью к атомизации^ Соединения или ра-
дпкалътг^—сслггав~тцпюрыТ^ходит~Ъпрёд^яемь1Й элемент, могут
не распадаться на отдельные атомы при температуре используе-
мого пламени. В ряде случаев такие соединения могут образо-
зоваться непосредственно в пламени при распылении в него ана-
лизируемого раствора. В результате число свободных атомов в
зоне атомизации, способных к поглощению резонансного излуче-
ния, меняется, следовательно, меняется и интенсивность резо-
нансного поглощения. Если таких изменений нет в этало-
нах, используемых для градуировки, то аналитические результа-
ты будут содержать ошибку.
Типичным примером такого рода помех является понижение
абсорбции при определении в почве щелочноземельных элемен-
тов в присутствии алюминия, кремния, фосфора, а также элемен-
тов, образующих устойчивые окислы (алюминий, ванадий, бор,
молибден и др.).
Устранить влияние химических помех можно двумя путями:
1) использовать высокотемпературные пламена, энергия которых
достаточно высока, чтобы разрушить комплексы и атомизпровать
пробу; 2) добавлять к анализируемому раствору (эталонам)
маскирующие вещества, которые реагируют с мешающими эле-
ментами и снимают возможные химические помехи. В качестве
примера можно привести добавление к * почвенному раствору
лантана, который при определении Са, Mg, Sr и Ва устраняет
депрессирующее действие некоторых элементов (Al, Р, Si, S)
9 Зак. 284
257
в пламени ацетилен — воздух. Объяснение устраняющего дейст-
вия определенных химических реагентов получают из положения
о химическом равновесии:
Ме-О-Х - RT-——R-O-X Me.
Если R — химический реагент, образующий устойчивое соедине-
ние (R—О—X), и если этот реагент вводят в пламя в избытке,
то равновесие реакции будет смещено вправо, т. е. в сторону уве-
личения свободных атомов Me.
Использование высокотемпературного пламени закись азота —
ацетилен позволяет за счет дополнительной энергии разрушить
устойяивые соединения и избежать возможных помех без введе-
ния лантана или других химических реагентов. В почвенно-агро-
химической практике лантан^как правило, не является анализи-
руемым элементом и его' часто используют в качестве защитного
реагента при определении щелочноземельных элементов в обще-
доступном пламени ацетилен -— воздух. Для элементов, склонных
к образованию окислов, эффективным защитным агентом являет-
ся .углерод пламени или углеродсодержащие радикалы. Для
этого пламя специально обогащают топливом, и тогда избыток
кислорода устраняется путем связывания его в моноокись угле-
рода.
Ионизационные помехи, как и в эмиссионном анализе, возни-
кают тогда, когда температура пламени достаточна для удаления
электрона от нейтрального атома и превращения его в положи-
тельно заряженный ион. В результате количество нейтральных
атомов в зоне атомизации падает, что уменьшает интенсивность
поглощения. Контролировать ионизацию в пламени можно путем
добавления к исследуемым растворам и стандартам избытка
.легкоиоццзируемых элементов. В этом случае наиболее часто
применяют, ^целочные элементы (Cs, Kb, К, Li), которые легко
ионизуются в пламени ацетилен — воздух или закись азота —
ацетилен. Например, прибавление к пробе избытка лития может
стать дополнительным источником электронов в пламени, что
будет препятствовать ионизации калия по схеме: Li^tLi+-|-e и
К+ + е^К. Часто бывает достаточно добавить к раствору 1500—
2000 мкг/мл солей щелочных металлов.
’Помехи из-за различий в составе проб (эталонов) проявляют-
ся через изменение вязкости, поверхностного натяжения или дру-
гих физических свойств раствора. Влияние состава растворов
можно контролировать, применяя разбавление (если это возмож-
но) или забуферивапие. КбТда сближ'&тщщ, состава растворов
(эталон&в)—-обячийми приемами невозможно, то для точного
определения концентрации элемента используют .метод добавок.
Установлено, чем эффективнее работает распылитеЙдг-чемТтень'-
ше получаемые капли, тем меньше возникает помех из-за раз-
личий в составе проб.
258
Неселективное поглощение излучения возникает в результате 1
светорассеяния,” молекулярногб-поглощения, а также поглощения/
его пламенем. Рассеяние света возникает из-за присутствия не-
больших твёрдых частиц в зоне просвечивающего пучка света.
Частицы могут появляться в пламени при распылении концентри-
рованных растворов вследствие неполного испарения веществ
или могут образовываться непосредственно в самом пламени.
Величина светорассеяния очень сильно зависит от длины волны
света, на которой происходит абсорбция. Она сильно возрастает
с уменьшением длины волны резонансного излучения, и особен-
но этот эффект сказывается на определении небольших концен-
траций элементов, аналитические линии которых расположены в
коротковолновой области, особенно в случае определения мышья-
ка, селена, кадмия, цинка и свинца. Рассеяние приводит к силь-
ному завышению поглощения, так как в итоге добавляется
ложный сигнал к истинному резонансному поглощению Ложный
сигнал сопряжен со значительными шумами, и на уровне преде-
ла обнаружения величина таких шумов может быть в несколько
раз больше истинного сигнала. Неселективные помехи особенно
могут быть значительными при определении элементов на уров-
не 1-10~5—1-10~70/о в графитовой кювеге.
Величину светорассеяния можно измерить и, таким образом,
вычленить из полезного сигнала путем измерения интенсивности
любой другой линии, испускаемой источником света, располо-
женной вблизи аналитической линии, но не поглощаемой данным
элементом.
Чаще всего для этих целей используют сплошной спектр дей-
териевой лампы. С помощью луча дейтериевой лампы измеряют
неселективное поглощение (ложный сигнал) в единицах погло-
щения и вычитают этот сигнал из результатов, полученных с ли-
нейчатым источником. Значительно труднее избавиться от шумов,
связанных со светорассеянием, поэтому в аналитической практи-
ке чаще предпочитают уменьшать уровень светорассеяния. Это
можно сделать путем отделения элемента от основы (экстракция,
концентрирование) пли путем другой специальной подготовки
проб к анализу. Если рассеяние связано с самим пламенем, то
его уменьшают, изменяя режим горения.
Помехи за счет молекулярного поглощения можно ликвидиро-
вать, применяя высокотемпературные пламена, с энергией, до-
статочной для разрушения ”молекулярных~соединений, пли с по-
мощью той же дейтериевой лампы. В последние годы для устра-
нения неспецифических помех в основном применяют дейтерие-
вый корректор. Очевидно, без учета неспецифических помех
нельзя проводить атомно-абсорбционный анализ почв на элемен-
ты, содержащиеся на уровне 10-5—10~70/о, особенно работая с
графитовой кюветой.
9*
259
Определение в почвах макро-
и микроэлементов
Методы определения валового содержания макроэлементов в
почвах разработаны еще недостаточно полно. Лучше они разра-
ботаны для анализа силикатных горных пород. Главные трудно-
сти определения валового состава связаны с разложением проб
и переведением их в раствор, с межэлементными влияниями.
Для большинства резонансных линии макроэлементов характер-
на высокая чувствительность, поэтому необходимо предусматри-
вать разбавление проб или другие меры, уменьшающие чувст-
вительность.
Для разложения почв и переведения их в раствор используют
две группы методов: сплавление и кислотную обработку. При
определении кремения дшзместно с другими элементами пред-
почитают сплавление почв с метаборатом, стронция (лития) или
со смесью буры и соды.
Разложение почв и подготовка
раствора к анализу
Сплавление почв с тетраборатом стронция по Дженруа. Для
почв, горных пород и минералов Дженруа предложил метод
сплавления проб с Sr(BO2)2. Присутствие в растворе стронция
в сочетании с высокотемпературным пламенем закись азота —
ацетилен приводит к подавлению взаимных влияний элементов.
Плавень Sr(BO2)2 получают путем смешивания тонкоизмельчен-
ных солей SrCO3' и Н3ВО3 и нагревания в муфеле до 300°, с по-
следующим выдерживанием в течение часа при температуре 700°.
Тонкоизмельченную пробу 0,1 и 0,5 г плавня смешивают и по-
мещают в графитовый тигель. Тигель помещают в высокочастот-
ную индукционную печь п сплавляют содержимое при темпера-
туре 1100° в течение 15 мин до полной прозрачности плава. Плав
выливают в разбавленную HNO3 (1 : 100) и перемешивают с
помощью магнитной мешалки. Раствор разбавляют до 200 мл и
используют для атомно-абсорбционного анализа. Определение
Si, Al, Ti, Fe, Ca, Mg производят в пламени закись азота — аце-
тилен, для определения Na, К, Мп, Си, N1 и других элементов
используют пламя .ацетил ен-=^^оздух. Эталонные растворы го-
товят разбавлением исходных растворов элементов раствором,
содержащим 50 г Sr(BO2)2 и 50 мл 65%-ной HNO3 в 1 л. Усло-
вия и техника определения приводятся ниже.
Кислотное разложение почв и подготовка раствора к анализу.
Выбор кислот п применяемая техника разложения почв обуслов-
ливаются аналитической задачей. Для определения в почвах
макро- и микроэлементов, за исключением кремния, пробу раз-
ла1ают смесью HNO3, НС1 и HF. Разложение почв этими кпе-
260
лотами в замкнутой системе (автоклавы, бомбы из фторопласта)
обеспечивает возможность определения кремния.
1—2 г тонкорастертой (до пудры) и прокаленной при 450—
500° почвы помещают во фторопластовую чашку. При необ-
ходимости определяют потерю веса после прокаливания.
Прокаленную почву смачивают несколькими каплями биди-
стиллированной воды. Карбонатные почвы предварительно обра-
батывают 10 мл 10%-ного раствора НС1 при нагревании для рас-
творения карбонатов. Надосадочную жидкость, содержащую
большую часть щелочноземельных металлов почв, сливают
в мерную колбу емкостью 100 мл, а остаток почв подвер-
гают дальнейшей обработке. К некарбонатной почве (или
остатку после удаления карбонатов) добавляют 5 мл цар-
ской водки и 15 мл 48%-ной HF, чашку медленно нагревают на
этернитовой плитке, выпаривая содержимое почти досуха. Затем
снова добавляют в чашку 10—15 мл HF и выпаривают содержи-
мое до сухого остатка. К сухому остатку прибавляют 5 мл кон-
центрированной НС1, и смесь снова выпаривают досуха до пол-
ного удаления фтора. К остатку приливают 10 мл разбавленной
(1 : 1) НС1, нагревают на плитке до растворения остатка и филь-
труют через фильтр средней плотности в колбу емкостью 100 мл.
Фильтрат карбонатных почв объединяют с фильтратом после
растворения карбонатов. Фильтр и чашку промывают несколько
раз 0,5 н. НС1 до отсутствия реакции на железо с роданидом
калия. Колбу доводят до метки 0,5 н. НС1, полученный'раиЖф
используют для определения макро- й микроэлементов.
Аликвоту этого раствора разбавляют бидистиллированной во-
дой в 100 раз, добавляют буферный раствор (хлориды лантана
или стронция), концентрация буферирующего элемента должна
быть 0,15—0,20%. В полученном растворе определяют макроэле-
менты Al, Fe, Са, Mg, К, Na. Титан определяют в отдельной али-
квоте этого раствора, предварительно добавив в него алюминий
(хлорид) в качестве буфера до концентрации «1%. Алюминий и
титан определяют в газовой смеси закись азота — ацетилен, все
другие элементы — в смеси ацетилен — воздух.
Эталонные растворы для определения макроэлементов гото-
вят на основе 0,5 н. НС1, добавляя в них такое же количество
буфера. Некоторое расхождение в кислотности анализируемых и
эталонных растворов не оказывает существенного влияния на
результаты определений. В неразбавленных растворах опреде-
ляют валовое содержание Мп, Zn, Си, Со, Ni, Pb, Ва и Sr. При
высоком содержании Мп и Zn в почвах анализируемые растворы
разбавляют в 10 раз бидистиллированной водой. В качестве эта-
лонных растворов используют солянокислые растворы этих эле-
ментов. Барий и стронций определяют после буферирования
раствора лантаном (1500—2000 мг/л). Определение кобальта
(иногда меди, свинца) в почвах с низким их содержанием про-
водят после предварительного концентрирования.
261
Лучшим методом концентрирования в атомной адсорбции
является экстракция. В качестве комплексообразователей ис-
пользуют дитизон или дизтилдитиокарбаминат натрия, а в каче-
стве растворителей — амилацетат или метилизобутилкетон.
Для концентрирования к 25—50 мл анализируемого раствора
добавляют 10—20 мл 1 М раствора тартрата аммония (в зависи-
мости от возможной концентрации железа). Устанавливают pH
раствора аммиаком (при экстракции амилацетатом pH 7,0, ме-
тилизобутил кетоном pH 7,5—8,0). Добавляют 10 мл 0,1%-ного
раствора дитизона в амилацетате или метилизобутилкетоне.
Встряхивают в делительной воронке в течение 10 мин. Органи-
ческую фазу используют для анализа атомно-абсорбционным ме-
тодом. Эталонные растворы готовят аналогичным образом, про-
водя стадию концентрирования.
Определение содержания обменного
кальция и магния в почвах
Обменные кальций и магний в почвах определяют после вы-
теснения их 1 н. раствором CH3COONH4. Техника вытеснения
обменных оснований описана выше (см. с. 234). Для подавления
депрессирующего влияния алюминия, фосфора на определение
кальция и магния в полученный раствор добавляют буфер (0,15%-
ный хлорид лантана или стронция). Эталонные растворы готовят
на 1 н. растворе CH3COONH4 с добавлением соответствующих
количеств буферного раствора. В пламени закись азотщ—-ацети-
лен кальций и магний можно определить без применения буфера.
Определение водорастворимых
соединений кальция
и магния в почвах
Водную вытяжку из почв готовят, как описано на с. 233.
К аликвоте водной вытяжки (10—20 мл) добавляют буферный
раствор (0,15%-ный хлорид лантана или стронция). В получен-
ном растворе определяют содержание Са и Mg атомно-абсорб-
ционным методом. Эталонные растворы готовят из слабокислых
растворов хлорида кальция и магния на бидистиллированной
воде с добавлением соответствующих количеств буферного ра-
створа.
Определение доступного растениям
магния по Шахтшабелю
5 г воздушно-сухой почвы, пропущенной через сито диамет-
ром 1 мм, помещают в колбу на 100 мл, приливают 50 мл
0,025 н. раствора СаС12 встряхивают на механическом встряхп-
262
вателе 2 часа. Фильтруют через плотный фильтр, свободный от
следов магния. Аликвоту этого фильтрата (в зависимости от со-
держания магния) разбавляют бидистиллированной водой в 10—
50 раз, добавляют буферный раствор (0,15%-ного лантана или
стронция) и используют для анализа. Эталонные растворы гото-
вят на основе 0,025 н. СаС12 с добавлением буфера.
Ниже приводятся данные, характеризующие обеспеченность
(в мг/100 г) почвы магнием в зависимости от их механического
состава по Шахтшабелю.
Содержание
Легкие
Суглинистые
Глинистые
Высокое
Среднее
Низкое
5,0
2,5-5,0
2,5
7,0
3,5—7,0
3,5
12,0
7,0—12,0
7,0
Определение подвижных форм
соединений железа, алюминия
и кремния в почвах
(вытяжка Тамма)
Растворителем служит смесь щавелевой кислоты и щавелево-
кислого аммония (31,5 г щавелевой кислоты и 62,1 щавелево-
кислого аммония растворяют в 2,5 л дистиллированной воды,
pH такого раствора 3,2—3,3). К 2 г почвы приливают 100 мл
раствора Тамма, взбалтывают 1 ч на ротаторе и фильтруют че-
рез фильтр «белая лента». Затем фильтр с почвой снова поме-
щают в ту же колбу, приливают еще 100 мл раствора Тамма,
снова взбалтывают 1 ч, фильтруют и промывают почву холодной
водой. Фильтраты и промывные воды собирают вместе, доводят
объем до метки в мерной колбе емкостью 250 мл. В фильтрате
определяют кремний и алюминий в пламени закись азота — аце-
тилен, железо можно определить без помех в пламени ацети-
лен — воздух.
Эталонные растворы на Si, Al и Fe готовят на растворе
Тамма.
Определение несиликатного железа
в почвах по методу
Мера — Джексона
Навеску почв в 1 г помещают в центрифужную пробирку,
обрабатывают 3—4 раза пергидролем для окисления органиче-
ского вещества, добавляют'"!!) МЛ ’ 0?3' М раствора лимоннокис-
лого натрия и 1,5 мл 1 М раствора КаНСОз. Пробирку поме-
263
щают в специальный штатив, который ставят в широкий стакан,
заполненный водой на 30 мм. В стакан помешают также термо-
метр. Стакан ставят на водяную баню и нагревают до 80°. Затем
в пробирку вносят 1 г твердого дитионита (гидросульфита) натрия
(Na2S2O4), смесь периодически перемешивают в течение 15 мин,
после чего в пробирку добавляют 2 мл концентрированного
раствора NaCl. Смесь снова перемешивают, подогревают на во-
дяной бане и центрифугируют 5 мин при 2000 об/мин. Прозрач-
ную жидкость декантируют в мерную колбу емкостью 100 мл.
К осадку в пробирке снова добавляют 10 мл 0,3 М раствора ли-
моннокислого натрия и 1,5 мл 1 М раствора NaHCO3. Содержи-
мое тщательно перемешивают стеклянной палочкой, также нагре-
вают, добавляют дитионпт, концентрированный раствор NaCl и
центрифугируют.
Для почв, содержащих больше 5% экстрагируемого железа,
обработку перечисленными реактивами повторяют 2—3 раза,
причем все порции центрпфугатов объединяют в одной и той же
колбе. По окончании обработки промывают осадок (два раза)
раствором лимоннокислого натрия, содержимое пробирки цент-
рифугируют, центрифугат объединяют с предыдущими, объем
доводят до метки колбы водой. Для определения железа аликво-
ту раствора разбавляют в 50—100 раз дистиллированной водой.
Определение железа проводят в пламени ацетилен — воздух, гра-
дуировочный график строят по эталонным слабокпслым (НС1)
растворам железа, содержащим ионы натрия в количествах,
близких к содержанию в испытуемых растворах.
Определение подвижных форм
соединений Мп, Zn, Си, Со в почвах
Подвижные формы соединений Мп, Zn, Си и Со извлекают
ацетатно-аммонийным буферным раствором с pH 4,8. Отноше-
ние почвы к“ раствору 'ТПО, время взаимодействия 1 ч при
взбалтывании на ротаторе или кратковременное встряхивание и
настаивание в течение суток. Метод пригоден как для некарбо-
натных, так и для карбонатных почв и принят агрохимической
службой для оценки обеспеченности почв микроэлементами.
Пробу почвы массой 5 г, растертой и пропущенной через сито
с отверстиями 1 мм, помещают в колбу емкостью 100 мл, при-
ливают 50 мл ацетатно-аммонийного буферного раствора с
pH 4,8. Суспензию взбалтывают 1 ч (пли настаивают в течение
суток). Вытяжку фильтруют через сухой складчатый фильтр в
пробирку, первыми порциями споласкивают пробирку и их вы-
брасывают. В полученном фильтрате определяют микроэлементы
атомно-абсорбционным методом в пламени ацетилен — воздда
или в смеси пропан — бутан — воздух. Градуировочные графики
264
строят по эталонным растворам соответствующих микроэлемен-
тов, приготовленных на ацетатно-аммонийном буфере.
Реактивы для приготовления буфера должны быть квалифи-
кации «особой чистоты» или в крайнем случае «х. ч.», вода долж-
йа быть бидпстиллированной. Одновременно ставят холостой
опыт, проводя ею через все стадии анализа, исключая взятие
пробы почвы. Для приготовления 1 л буферного раствора с
pH 4,8 необходимо 108 мл 98%-ной СН3СООН и 75 мл 25%-ного
раствора NH4OH добавить к 800—900 мл бидистиллированной
воды, перемешать, измерить pH и, если необходимо, довести его
до 4,8, после чего раствор водой довести до 1 л.
Определение кобальта и меди в некоторых почвах в ацетатно-
аммонийном буферном растворе прямым методом невозможно
из-за очень низких концентраций. В этих случаях применяют
предварительное концентрирование. Для определения меди доста-
точным бывает простое выпаривание вытяжки и растворение
осадка в меньшем объеме (10-кратное концентрирование). Уни-
версальным методом концентрирования является экстракция
дитизонатов меди и кобальта из вытяжки объемом 100—150 мл.
Ход экстракции такой же, как указано выше (см. с. 262).
Для определения обеспеченности почв подвижными формами
микроэлементов в зависимости от потребности в них растений
принята следующая шкала градаций (табл. 23).
Таблица 23
^Группировка почв по обеспеченности растений микроэлементами
(экстрагент — ацетатно-аммонийный буфер с pH 4,8)
Обеспеченность Содержание микроэлементов, мг/кг почвы
Мп Си Zn Со
1-я группа растений невысокого потребления
Низкая <5 <0,1 <0,5 <0,05
Средняя 5—10 0,1—0,2 0,5—1,0 0,05—0,1
Высокая >ю >0,2 >1,0 >0,1
2-я группа растений повышенного потребления
Низкая <10 <0,2 <1,0 <0,1
Средняя , 10—20 0,2—0,5 1,0—2,0 0,1—0,2
Высокая >20 >0,5 >2,0 >0,2
3-я группа растений высокого потребления
<2,0
2,0—5,0
>5,0
<0,2
0,2—0,5
>0,5
Низкая <20
Средняя 20—40
Высокая >40
<0,5
0,5—1,0
>1,0
265
v Определение подвижных форм
микроэлементов в почвах
Нечерноземной зоны
по Пейве-Ринькису
Метод основан на извлечении нз почв: Мп — 0,1 н. H2SO4,
Си — 1 н. НС1, Со — 1 н. HNO3 и Zn — 1 н. КС1. Отношение
почвы к раствору 1 : 10, время взаимодействия 1 ч при взбалты-
вании на ротаторе. Пробу почвы массой 5 г помещают в колбу
на 100 мл. К пробе добавляют 50 мл соответствующего экстра-
гента и взбалтывают суспензию на ротаторе в течение 1 ч. Для
предупреждения контакта раствора с резиной пробки последнюю
обертывают полиэтиленовой пленкой iijiii целлофаном. Вытяжку
фильтруют через складчатый фильтр. Первые порции фильтрата
отбрасывают, в последующих определяют микроэлементы, в пла-
мени ацетилен — воздух или пропан — бутан — воздух.) В случае
использования пламени пропан — бутан — воздух для определе-
ния Мп в анализируемый раствор (и в эталонные растворы) до-
бавляют хлорид лантана (или стронция) до конечной концентра-
ции в растворе 0,2%• Высокая концентрация КС1 в растворе при
определении Zn нарушает нормальное распыление раствора и
горение пламени. Для устранения указанного явления необхо-
димо производить разбавление вытяжки, использовать трехщеле-
вую горелку на спектрофотометре. Необходимо также при этом
учитывать неселективные помехи (см. с. 259).
Таблица 24
/ Группировка почв по обеспеченности растений Микроэлементами
(экстрагены ко Пейве—Ринькису)
Обеспеченность Содержание микроэлементов, мг/кг почвы
Мп Си Zn Со
1-я группа растений невысокого потребления
Низкая <15 <0,5 <0,3 <0,3
Средняя 15—30 0,5—1,5 0,3—1,5 0,3—1,0
Высокая >30 >1,5 >1,5 >1,0
2-я группа растений повышенного потребления
Низкая <45 <2,0 <1,5 <1.0 I
Средняя 45—70 2,0—4,0 1,5—3,0 1,0—3,0 1
Высокая >70 >4,0 >3.0 >3,0 'li
3-я группа растений высокого потребления
Низкая <100 <5,0 <3,0 <3,0 «
Средняя 100—150 5,0—7,0 3,0—5.0 3,0—5,0
Высокая >150 >7,0 >5,0 >5,0
266
Градуировочные графики строят по эталонным растворам
определяемых микроэлементов на соответствующих экстрагентах.
Данный метод принят для некарбонатных почв Нечернозем-
ной полосы и красноземов. Для суждения об обеспеченности почв
микроэлементами принята следующая группировка (табл. 24).
Приготовление
стандартных растворов
Для построения градуировочного графика готовят шкалу ра-
бочих стандартных растворов (см. табл. 25). Для этого готовят
запасные растворы, содержащие 1 мг иона металла в 1 мл раст-
ворителя. Стандартный раствор кремния готовят сплавлением
навески 1,070 г предварительно прокаленного при 900° квар-
ца (или двуокиси кремния марки «ос. ч.») с 6-кратным ко-
личеством смеси буры и соды (1:2) в платиновой чашке в му-
феле при 950—1000°. Охлажденный тигель с плавнем помещают
в стакан емкостью 500 мл, добавляют 200 мл кипящей воды и
нагревают на водяной бане до полного выщелачивания плава.
Приливают 100 мл разбавленной НС1 (1:3), раствор пере-
мешивают и переносят в мерную колбу емкостью 500 мл, после
охлаждения доводят водой до метки. Раствор хранят в полиэти-
леновой посуде. Запасной стандартный раствор алюминия гото-
вят растворением 0,500 г металлического алюминия в 100 мл
НС1 (1:1) при нагревании. Раствор переносят в колбу емкостью
0,5 л и доводят водой до метки.
Запасной раствор титана готовят растворением 0,500 г метал-
лического титана в 100 мл HCI (1 : 1), кипятят в течение 3 ч
до полного растворения, переносят в колбу емкостью 0,5 л и до-
водят до метки 6 н. НС1.
Запасной раствор железа готовят растворением 0,500 г метал-
лического железа (проволока) в 100 мл НС1 (1:1) при нагре-
вании, приливают 0,5 мл HNO3, кипятят несколько минут, пере-
носят в колбу емкостью 0,5 л и доводят водой объем до метки.
Запасные стандартные растворы на Са и Mg готовят раство-
рением соответствующих навесок карбонатов этих металлов в
разбавленной НС1. Раствор кипятят для удаления СОг, по охлаж-
дении переводят в мерную колбу емкостью 1 л и доводят до
метки водой.
Стандартные растворы на К и Na готовят из их хлористых
солей, как указано выше (см. с. 236).,.
Запасные стандартные растворы Мп, Си, Zn, Со, Ni, Pb гото-
вят растворением соответствующей навески металла в HNO3
(1:1) сначала на холоду, а затем при нагревании. Растворы
переносят в мерные колбы и доводят водой до метки.
Запасной стандартный раствор стронция готовят растворением
в разбавленной НС1 навески ЗгСОз. Раствор кипятят для удале-
267
ния С02, охлаждают, переводят в мерную колбу и доводят до
метки водой.
Запасной раствор Ва готовят растворением соответствующей
навески нитрата бария в дистиллированной воде, раствор под-
кисляют HNO3.
Для определения микроэлементов из запасных стандартных
растворов путем разбавления готовят промежуточные стандарт-
ные растворы с концентрацией 100—50 мкг/мл, а из последних —
рабочие стандартные растворы. Элементы в зависимости от усло-
вий определения, их концентрации в рабочих стандартах груп-
пируют и готовят стандарты в одной колбе. Одну группу
составляют Fe, Мп, Zn, Со, Ni, Си, РЬ, К, Na. Вторую группу
составляют щелочноземельные элементы (Са, Mg, Sr, Ва), для
правильного определения которых необходимо в рабочие стан-
дарты (также и в пробы) добавлять буферный раствор хлорида
лантана. Рабочие стандартные растворы для третьей группы
элементов (Si, Al, Ti) готовятся индивидуально в отдельных кол-
бах, куда также вносят буферный раствор.
Таблица 25
Шкала рабочих стандартных растворов для определения
макро- и микроэлементов в почвах, мкг/мл
Стан- дарт Si AI Ti Fe, Мп Ca, Mg. Zn, Co. Ni, Cu, Pb Sr, Ba K. Na
I 10 1 3 0,1 0,1 0,2 0,5
II 20 5 5 0,2 0,2 0,5 1
III 50 10 10 0,5 0,5 1 2
IV 100 20 20 1 1 2 5
V 200 50 50 5 2 5 10
VI 400 100 100 10 5 10 15
Для приготовления рабочих стандартных растворов в мер-
ные колбы емкостью 100 мл бюреткой отмеряют расчетные коли-
чества промежуточных стандартных растворов (табл. 25), после-
довательно, начиная с меньшей концентрации, объем доводят до
метки растворителем, соответствующим целям исследования.
Подготовка прибора к работе
и техника измерения поглощения
»
Прежде чем приступить к выполнению определения, необхо-
димо:
1) ознакомиться с техническим описанием прибора и мето-
дикой работы с ним;
268
2) детально ознакомиться с конкретным описанием всего хо-
да выполнения аналитической задачи;
3) при накале лампы с полым катодом избегать силы тока,
превышающей оптимальный режим, указанный в паспорте
данной лампы;
4) при питании ФЭУ избегать подачи напряжения выше пре-
дельного;
5) приступая к работе с баллонами, содержащими сжиженный
или растворенный газ, необходимо ознакомиться с инструк-
цией по технике безопасности.
Аппаратура
и ее подготовка к работе
В настоящее время промышленность ряда стран выпускает
значительное количество атомно-абсорбционных спектрофотомет-
ров разных типов, успешно используемых в почвенно-агрохимиче-
ских исследованиях. Более совершенными являются двухлучевые
спектрофотометры «Сатурн-2» и С-112, позволяющие работать на
Рис. 18. Общий вид спектрофотометра AAS-1
высоком уровне автоматизации как с различными пламенами, так
и с графитовой кюветой. Более простыми, построенными по одно-
лучевой схеме являются спектрофотометры С-302 и AAS-1, они
предназначены для работы в двух режимах: абсорбции и эмиссии,
т. е. эти приборы можно использовать и для работ по методу эмис-
сионной фотометрии пламени.
269
На рис. 78 показан общий вид спектрофотометра AAS-1. Пе-
ред включением прибора в сеть необходимо проверить заземле-
ние корпуса прибора, высоковольтного выпрямителя, кожуха фо-
тоумножителя. Проверить прочность соединения шлангов, подво-
дящих горючие газы и окислитель, проверить наличие воды в во-
дяных затворах. Убедиться, что установлен минимальный ток на
лампе с полым катодом, напряжение на ФЭУ, минимальная чув-
ствительность гальванометра. Для этого рукоятки регулирования
тока лампы в рабочей цепи повернуть налево до упора, а ступен-
чатый переключатель напряжения ФЭУ перевести в положение 1.
Ступенчатый переключатель установки постоянной времени по-
ставить в положение 0,5 с. Ступенчатый переключатель коэффи-
циентов усиления поставить в положение 0. Переключатель диа-
пазонов измерения установить на «0—100». Ширину щели уста-
новить около 0,1 мм.
s Включение прибора. Установить нужный источник света, для
этого необходимую для данного анализа лампу с полым катодом
ставят в держатель ламп, переключатель ламп устанавливают
на номер места крепления ламп с полым катодом на устройстве
смены ламп. Устанавливают желаемую длину волны (табл. 26),
которая отсчитывается по индикатору длины волн.
Включают прибор в сеть и нажимают клавишу сети, при этом
должна загореться контрольная лампочка. Устанавливают ра-
бочий ток лампы с помощью необходимых рукояток. Одно-
временно нажимают на клавишу, относящуюся к выбран-
ной рабочей цепи, и отсчитывают ток лампы по нижней
шкале показывающего прибора. При соответствующей уста-
новке длины волны выбирают ток лампы с полым катодом, шири-
ну щели, усиление и напряжение на ФЭУ так, чтобы на показыва-
ющем гальванометре при минимальных значениях вышепере-
численных параметров получилось отклонение стрелки около 100
делений шкалы. Если необходимо/то производят для этого юсти-
ровку ламп.
Прогревают прибор в течение 20—30 мин. От компрессора по-
дают сжатый воздух, открывают редуктор воздуха и, вращая ру-
кояткой для точной регулировки воздуха, устанавливают необ-
ходимый расход сжатого воздуха по левому реометру. На бал-
лоне с ацетиленом открывают редукторы, устанавливают давле-
ние на выходном редукторе около 1 атм, открывают запорный кла-
пан на приборе и вращением рукоятки устанавливают расход
ацетилена ~ 100 л/ч, который контролируется по реометру. За-
тем следует кратковременно закрыть запорный клапан, чтобы
газ, попавший в камеру распылителя и горелки во время процесса
наладки, мог утечь через отводящее устройство. Потом снова от-
крывают запорный клапан и одновременно зажигают пламя. При
первом зажигании горелки воспламенение произойдет лишь спус-
тя 1—2 мин, так как шланги должны наполниться газом. Затем
следует вставить сверху защитную решетку, экранирующую ка-
270
Таблица %
Аналитические линии, Чувствительность и правильность атомно-абсорбциоНного определения ьаЛевого содержания
макро- и микроэлементов в почвах
Элементы Линия, HM Газовая смесь Чувствитель- ность опреде- ления, мкг/мл Оптимальная область кои- центрации, мкг/мл Типичный чернозем СП-1 Дерново подзолистая СП-2 Светло-каштанова я СП-3
аттесто- вано найдено аттесто- вано найдено аттесто- вано найдено
Si 251,6 n2o— c2h2 2,0 70—200 32,47 32,29* 36,58 36,37* 30,69 30,66*
Al 309,2 n2o— c2h2 1.0 40—125 5,48 5,78 5,06 5,34 6,67 7,09
Ti 364,3 N2O —C2H2 3,0 60- 200 0,45 0,43 0,50 0,44 0,44 0,50
Fe 248,3 воздух — C2H2 0,1 2-5 2,66 2,42 2,08 1,94 3,43 3,24
Са 422,7 воздух — C2H2 0,01 0,5—3 1,16 1,10 0,58 0,55 2,04 2,00
КЗ Mg 285,2 воздух — C2H2 0,001 0,1—0,5 0,62 0,60 0,47 0,43 1,18 1.Ю
►—* Мп 279,5 воздух — C2H2 0,05 0,5—3 0,059 0,057 0,054 0,049 0,071 0,065
К 766,5 воздух — пропан 0,01 0,5—3 1,90 1,80 2,05 2,00 2,08 2,00
Na 589,6 воздух — пропан 0,01 0,1—1 0,59 0,63 0,85 0,90 0,86 0,90
Sr 460,7 n2o — с2н2 0,05 2—10 130 н. о. 120 н. о. 160 н. о.
Ba 553,6 n2o — С2Н2 0,5 10—30 430 н. о. 530 н. о. 470 н. о.
Cu 324,7 воздух — С2Н2 0,05 2—5 22 28 17 20 30 36
Zn 213,8 воздух — С2И2 0,01 0,4—1,5 52 65 45 44 73 83
Co 240,7 воздух — С2Н2 0,1 3—5 10 13 10 11 14 16
Ni 232,0 воздух — С2Н2 0,1 3—5 33 38 25 28 56 64
Pb 217,0 воздух — С2Н2 0,1 5—20 16 13 14 10 16 14
Примечание* Si, Al, Ti, Fe, Ca, Mg, К, Na, Мп в %; Другие элементы в мг/кг.
* Найдено химическим методом.
меру распылителя и горелки. В течение 10 мин распыляют в го-
релку дистиллированную воду или растворитель, после чего на-
жимают клавишу автоматики нулевого уравновешивания, т. е.
приводят оптическую плотность пламени к нулю.
Распыляя один из стандартных растворов, оптимизируют пара-
метры определения для получения максимальной чувствительности
определения. Регулируют усиление так, чтобы на измеритель-
ном приборе было показано среднее значение. Затем варьиру-
ют расход газов до тех пор, пока не будет достигнуто максималь-
ное отклонение стрелки показывающего прибора. Юстируют го-
ловку горелки также по максимальному отклонению стрелки по-
казывающего прибора. После этого фиксируют ее положение.
Чаще всего просвечивают область пламени, непосредственно
прилегающую к его зеленому конусу (высота 1—2 мм от основа-
ния горелки). Для разных элементов зона максимального погло-
щения различна.
Некоторые особенности имеет эксплуатация спектрофотомет-
ров, предусматривающих работу с пламенем закись азота—аце-
тилен. Прежде всего необходимо строго придерживаться инструк-
ции, приложенной к прибору. При несоблюдении мер по технике
безопасности возможен проскок пламени, т. е. образование взрыв-
ной волны за счет сгорания горючей смеси в камере предвари-
тельного смешения газов. Для исключения возможности проскока
пламени при работе со смесью закись азота—ацетилен вначале за-
жигают смесь воздух—ацетилен, затем, создав избыток ацетиле-
на, с помощью специального крана переключают воздух на закись
азота и устанавливают нужные расходы закиси азота и ацети-
лена.
При выключении пламени смеси закись азота — ацетилен сно-
ва создают избыток ацетилена, затем производят переключение
с закиси азота на воздух. При анализе используют горелку, пред-
назначенную для пламени закись азота—ацетилен.
Полное выключение спектрофотометра осуществляют в обрат-
ной последовательности. Прерывают подачу горючего газа, после
того как пламя погасло, прекращают подачу сжатого воздуха.
Затем выключают прибор с помощью выключателя и отключа-
ют кабель питания от сети.
Измерения начинают только при хорошо настроенном спектро-
фотометре, для чего производят проверку. Распыляют в пламя ра-
створитель и устанавливают отсчет 100 делений по шкале измери-
тельного прибора или нуль оптической плотности. Распыляют
стандартный раствор с минимальной концентрацией, проверяют
чувствительность определения, затем распыляют стандартные ра-
створы с увеличивающейся концентрацией элемента, подбирают,
оптимальную область концентраций, устанавливают прямолиней-
ный участок градуировочной кривой. Только при хорошо настро-
енном приборе приступают к измерениям поглощения.
272
Измерение поглощения
и определение концентрации
При стабилизировавшемся режиме работы прибора в пламя
распыляют растворитель, устанавливают нулевую точку, нажи-
мая на клавишу, вводят в пламя стандартные растворы и запи-
сывают соответствующие им величины пропускания или оптиче-
ской плотности.
По найденным значениям оптической плотности пламени и со-
ответствующим им значениям концентрации элемента в стандар-
тах строят градуировочный график в координатах D—С. Вводят
в пламя анализируемые растворы и соответствующим образом оп-
ределяют Т или D для данных растворов. По значениям оптиче-
ской плотности и градуировочному графику находят соответству-
ющие им концентрации данного элемента. Стандартные и анали-
зируемые растворы распыляют в пламени при постоянстве усло-
вий работы (расход горючего газа, окислителя, электрические па-
раметры). Нулевой отсчет и полученную калибровочную кривую
периодически проверяют. Регистрация отсчетных сигналов может
быть выполнена визуально или с помощью подключенного к
спектрофотометру самописца. Современные спектрофотометры
для атомной абсорбции могут выдавать результаты непосредст-
венно в концентрациях. Для этого их калибруют по стандартным
растворам, при необходимости в определенном диапазоне кон-
центраций вводят коррекцию на нелинейность зависимости опти-
ческой плотности от концентрации.
Некоторые самые последние модели атомно-абсорбционных
спектрофотометров снабжены ЭВМ, где для калибровки прибора
необходимо лишь задать концентрацию стандартов (одного, двух
или трех в зависимости от степени кривизны графика) на цифро-
вой распределительной панели и провести их анализ. ЭВМ, при-
нимая показания сигналов от стандартов, автоматически рассчи-
тывает надлежащую калибровочную кривую.
Иногда для компенсации химических помех и также физиче-
ских свойств растворов, условий их распыления применяют метод
добавок. Для получения правильных результатов достаточным
является проведение только двух измерений: определение плотно-
сти от неизвестного раствора без добавки элемента £)0 при Сх и
раствора того же образца, но с добавленным количеством опреде-
ляемого элемента (£>| при CJ. Расчет искомой концентрации про-
изводят графически или по следующей формуле:
Если оптические плотности испытуемых растворов выходят за
пределы оптимальной области, то их необходимо разбавить. При
определении некоторых микроэлементов, особенно Zn, Со, Pb, Cd„
273
когда концентрация их в растворе близка к чувствительности оп-
ределения, возникают неселективные помехи.
Учет неселективного поглощения производят по непоглощае-
мым линиям (для Со — 245,6 или 239,3 нм; Ni — 231,6; Си —
296,1; РЬ — 220,4; Zn — 210,4; Cd — 228,9 нм) или с помощью дей-
териевой лампы. Для этого измеряют оптическую плотность дваж-
ды. Вначале определяют оптическую плотность по резонансной
линии Dy. Полученное значение представляет собой сумму опти-
ческих плотностей от истинной концентрации атомов определяе-
мого элемента (D) и от неселективных помех (£>2). Затем выво-
дят на пламя непоглощаемую линию или ставят дейтериевый ис-
точник и в аналогичных условиях определяют £)2 от того же
раствора. Истинная оптическая плотность будет равна D = DX—D2.
В приборах со встроенным дейтериевым корректором фона исклю-
чение неселективных помех может производиться в автоматиче-
ском режиме.
Чувствительность и точность атомно-абсорбционного определе-
ния элементов во многом определяются конструкцией и характе-
ристикой прибора. Ориентировочные величины чувствительности
(в мкг/мл на 1% поглощения), наиболее характерные аналитиче-
ские линии элементов, а также возможная точность определений
показаны в табл. 26. Воспроизводимость результатов зависит от
уровня концентраций определяемых элементов. При работе в оп-
тимальном диапазоне концентраций коэффициент вариации мо-
жет составлять 1—2%, а при измерении небольших оптических
плотностей этот коэффициент может возрастать до 20—30%.
В этих случаях для повышения точности определений необходимо
предварительное концентрирование элемента.
В заключение следует отметить, что в настоящее время усилен-
но разрабатываются методы атомно-абсорбционного анализа почв
с непламенными атомизаторами (графитовой кюветой), позволя-
ющие увеличить чувствительность определения на два порядка.
Опыт показывает, что некоторые виды почвенных анализов, не-
выполнимые или трудновыполнимые пламенным вариантом ме-
тода, могут быть успешно разрешены с помощью графитовой
кюветы, гидридной техники или другими специфическими при-
емами.
ЛИТЕРАТУРА
Иванов Д. Н. Спектральный анализ почв. М., «Колос», 1974.
Прайс А. Аналитическая атомно-абсорбционная спектрофотометрия. М., I
«Мир», 1976. ’>«
Глава 8
ИЗУЧЕНИЕ ПОЧВЕННЫХ
КОМПОНЕНТОВ МЕТОДАМИ
РЕНТГЕНОВСКОГО АНАЛИЗА
Рентгеновские методы исследования широко используются в
почвоведении для определения минералогического состава почв,
для изучения органических и органо-минеральных соединений.
Они совершенно незаменимы при исследовании тонкодисперсных
почвенных фракций, которые не могут быть изучены с помощью
обычных оптических микроскопов из-за малого размера частиц.
Значение этих фракций в почвах чрезвычайно велико. В силу
большой дисперсности и огромной удельной поверхности, измеряе-
мой десятками и сотнями квадратных метров на грамм вещества,
илистая фракция — наиболее активная и реакционноспособная
часть минеральной почвенной массы. На поверхности глинистых
минералов в почвах происходят реакции сорбции, десорбции и
обмена катионов, реакции гидратации и дегидратации, реакции
взаимодействия с органическими веществами специфической и не-
специфической природы, в том числе с гербицидами.
Минералогический состав илистой фракции оказывает сущест-
венное влияние на ряд почвенных свойств: емкость поглощения,
водно-физические характеристики, обеспеченность растений до-
ступными формами калия, способность почвы к необменной фик-
сации ионов калия и аммония. Поэтому определение минералоги-
ческого состава тонкодисперсных фракций имеет большое теоре-
тическое и практическое значение. Рентгеновские методы позво-
ляют диагностировать минералы, входящие в состав этих фрак-
ций, определять их количественное содержание и получать све-
дения об изменениях в структуре кристаллической решетки ми-
нералов, возникающих в процессе выветривания и почвообразо-
вания.
Достоинствами метода являются быстрота получения, надеж*
ность и -хорошая воспроизводимость результатов. К числу недо-
статков можно отнести сложность и высокую стоимость необхо-
275
димой аппаратуры и ограниченные возможности приложения к
плохо окристаллизованным и аморфным соединениям.
В практике почвенных минералогических исследований наи-
большее распространение имеет метод фазового рентгеновского
анализа. Принцип метода заключается в получении дифракцион-
ной картины, которая возникает при прохождении рентгеновских
лучей через изучаемый объект и в сопоставлении положения и
интенсивности дифракционных эффектов с эталонами.
ПРИРОДА И ПОЛУЧЕНИЕ
РЕНТГЕНОВСКИХ ЛУЧЕЙ
Рентгеновское излучение, подобно радиоволнам и видимому
свету, является волновым электромагнитным процессом. Длины
волн рентгеновских лучей измеряются в ангстремах (А). В общем
спектре электромагнитных излучений рентгеновские лучи нахо-
дятся между гамма-излучением (со стороны более коротких волн)
и ультрафиолетовым излучением (со стороны более длинных
волн). В дифракционных методах используются рентгеновские
лучи с длиной волны, соизмеримой с расстояниями между атома-
ми в кристаллах, т. е. около 1 А или несколько больше.
Рентгеновское излучение возникает в рентгеновской трубке
при взаимодействии потока электронов с веществом анода. Это
излучение состоит из двух налагающйхсяГдруг на друга спект-
ров — сплошного (или~белого) с широкой непрерывной полосой
различных длин волн и линейчатого(или характеристического),
состоящего из отдельных линий с определенной длиной волны.
Причины появления и закономерности изменения этих спектров в
зависимости от приложенного напряжения различны. Сплошное
(белое) излучение возникает за счет потери электронами энергии
при торможении и ударо~~бб~анод рентгеновской труоки. Иосколь-
ку электрон теряет свою энергию не сразу, а постепенно, сталки-
ваясь с~несколькими атомами, возникает ряд фотонов с разными
длинами волн. Возникает сплошной (или белый) спектр рентге-
новского излучения. Если на полюсах трубки увеличить разность
потенциалов, для всех значений длин волн возрастет интенсив-
ность белого излучения (рис. 79, а), поскольку возрастает как
число столкновений электронов с зеркалом анода, так и энергия
каждого отдельного электрона.
Характеристический (или линейчатый) спектр имеет в сотни
раз большую интенсивность чем белый спектр, и возникает при
достижении потенциала Возбуждения, т. е. определенного напря-
жения на полюсах трубки специфического для каждого вещества
анода. Длины волн линии, из которых состоит характеристиче-
ский спектрТзависят от вещества анода (см. рис. 79, б). Возникно-
вение и закономерности поведения характеристических спектров
хорошо объясняются на основе представлений о строении атома.
276
Известно, что электроны в атоме находятся на определенных
энергетических уровнях К, L, М, N (рис. 80). Если в электриче-
ское поле атома попадает электрон с достаточно большой кине-
Рис. 79. Сплошной (белый) спектр от вольфрамового анода при раз-
ных напряжениях на трубке (а) и характеристический (линейчатый)
К-спектр от медного анода прн напряжении 35 кВ (б)
3
Рис. 80. Переходы между энерге-
тическими уровнями, порождаю-
щие рентгеновские спектры
тической энергией, как это происходит на аноде рентгеновской
трубки, он может вызвать переход того или иного электрона ато-
ма на один из вышележащих энергетических уровней. При этом
атом приходит в возбужденное сос-
тояние, которое прекращается
после самопроизвольного перехода
одного из внешних электронов
на освободившееся место во внут-
ренней оболочке. Этот переход
сопровождается выделением кванта
рентгеновского излучения. Часто-
та излучения определяется фор-
мулой: hv=E2—Elt где h — по-
стоянная Планка, v — частота
излучения, Е2 и £i — энергия
атома в нормальном и возбужден-
ном состоянии.
Поскольку в атоме такие разно-
сти дискретны и их значения
меняются при переходе к другим
атомам, характеристическое из-
лучение состоит из линий с отдельными дискретными значениями
длин волн, характерными для вещества, из которого сделан анод
трубки. При повышении напряжения на трубке растет количество
электронов, переходящих с внутренних орбит атомов анода на бо-
277
лее внешние и, следовательно, возрастает число квантов рентге-
новского излучения, т. е. растет его интенсивность, но величины
длин волн характеристического излучения остаются неизменными,
так как разность Ё2—Е[ не зависит от напряжения на трубке, а
зависит только от вещества анода.
При возвращении электронов на /(-уровень возникают /(-линии
спектра: Ка — для электронов, переходящих с L-уровня, Кр —
для электронов, переходящих с /И-уровня, и т. д. При возвращении
электронов на L-, М-, /V-оболочки возникают соответственно се-
рии L-, М-, N- и т. д. линий. Поскольку даже на одном уровне
электрон может находиться в нескольких энергетических состоя-
ниях и при переходах излучать разную энергию, возникают серии
линий с близкими, но неодинаковыми длинами волн (например,
/Са-линия состоит из двух — /Са, и /Са2, /Ср-линия тоже представ-
лена несколькими; (см. рис. 80).
Таблица 27
Порядковый номер элемента, длина волны К^-излучения, потенциал
возбуждения и вещества фильтров для различных анодов
Вещество анода Порядковый номер эле- мента Длина волны /< -излуче- о ния, А Потенциал возбуждения, kV Вещество фильтра и по- рядковый но- мер элемента
Fe 26 1,936 6,54 Мп (25)
Си 29 1,541 8,86 Ni (28)
Мо 42 0,712 20,0 Zn (30)
В практике рентгеновского анализа почвенных объектов чаще
всего используют медный, железный и, реже, молибденовый ано-
ды. В табл. 27 представлены порядковые номера элементов, дли-
ны волн Ка-серии и величины потенциала возбуждения, т. е. раз-
ности потенциалов, при которых возникает характеристическое
излучение.
Чаще всего используют /Са-излучение, поскольку оно обладает
максимальной интенсивностью, /(р,-линии рентгеновского излуче-
ния в ряде случаев. являются помехой при расшифровке рентге-
нограмм и их обычно отфильтровывают, ставя на пути рентгенов-
ского луча тонкую пластинку (фольгу) металла, атомный номер
которого примерно на единицу меньше атомного номера вещест-
ва анода.
Дифракция рентгеновских
лучей кристаллами
При взаимодействии атомов кристаллических веществ с про-
ходящим через них рентгеновским излучением имеет место ди-
фракционный эффект, при котором по определенным направлени-
278
ям возникают вторичные («дифрагированные») лучи. Чтобы по-
нять, по каким именно направлениям распространяются дифраги-
рованные лучи, рассмотрим явление дифракции как отражение от
определенных серий плоскостей.
Поскольку во всех кристаллических веществах атомы располо-
жены не хаотично, а чередуются по определенному для каждого
вещества закону, в кристалле можно провести бесконечное коли-
чество серии параллельных пло-
скостей, отстоящих на равном
расстоянии друг от друга и про-
ходящих через идентичные ато-
мы. Геометрически это показано
на рис. 81. Пусть А, В и т. д.—
серия параллельных идентичных
плоскостей (атомных сеток) кри-
сталла. Поскольку расстояние от
источника излучения до кристал-
ла и от кристалла до наблюдате-
Рис 81. Отражение лучей от двух
соседних параллельных идентичных
плоскостей
ля несоизмеримо с расстояния-
ми между атомами и плоскостями в кристалле, то падающие и
отраженные лучи можно считать параллельными. Разность хода,
т. е. разница в длине пройденного пути для лучей, отраженных
соседними плоскостями, составит, очевидно, сумму отрезков
DF+FG, если ED и EG — перпендикуляры, опущенные из точки
Е на направление падающего и отраженного луча. Если расстоя-
ние между плоскостями равно d, а угол, под которым рентгенов-
ский луч падает на плоскость и отражается от нее, равен 0, то
из треугольников DFE и EFG находим: DF+FG = 2d sin 0.
Если разность хода лучей, отраженных соседними параллель-
ными плоскостями составит целое число длин волн, все лучи, от-
раженные этой серией плоскостей, придут к наблюдателю в одной
и той же фазе, их амплитуды суммируются и возникнет вторич-
ный (дифрагированный) луч.
При разности хода лучей, неравной целому числу длин волн,у
лучи, отраженные параллельными плоскостями, гасят друг друга. '
Разность хода лучей, отраженных двумя соседними параллельны-
ми плоскостями, как было показано выше, равна 2d sin б. Если
А — длина волны, ап — целое число, условие возникновения от-
раженного (дифрагированного) излучения выглядит следующим
образом:
2d sin 0 = пХ.
Эта формула лежит в основе рентгеновского анализа и извест-
на как закон Брегга—Вульфа. Число п в этой формуле называет-
ся порядком отражения.
От одной *и той же серии плоскостей можно получать отраже-
279
ния разных порядков, которые соответствуют кратным величинам
межплоскостных расстояний. Так, например, если отражения пер-
вого порядка соответствуют величине межплоскостного расстояния
10 А, то отражение второго, третьего и т. д. порядков будут соот-
ветствовать d/л, равным 5, 3,33 А и т. д.
Каждому дифрагированному лучу присваиваются определен-
ные дифракционные индексы, которые вычисляются следующим
образом. В любой кристаллической решетке можно провести 3
координатные оси, из которых ось а (х) идет на наблюдателя, ось
b (у) идет вправо от наблюдателя, а ось с (z) проводится в вер-
тикальном направлении. Индексы плоскостей представляют собой
величины, обратные величинам отрезков, отсекаемых этой плос-
костью на координатных осях. Индекс плоскости, параллельной
какой-либо из осей, равен, очевидно, нулю. Если индекс плоско-
сти выражается, например, цифрами 002, это значиц что плос-
кость параллельна осям а и b и отсекает по оси с отрезок, равный
1/2 параметра кристаллической решетки по этому направлению.
Дифракционные индексы вычисляются как произведение индексов
плоскостей, попавших в отражающее положение, на величину п—
порядок отражения. Например, отражение второго порядка от се-
рии плоскостей с индексами 001 будет иметь дифракционный ин-
декс 002.
Величина длины волны рентгеновского излучения К всегда бы-
вает известна исследователю, так как она постоянна для каждого
анода. Угол между падающим лучом и отражающей плоскостью
0 может быть измерен, причем техника измерения зависит от кон-
струкции аппарата. Зная величины X и 0, на основании формулы
Брегга—Вульфа можно вычислить величины d/п. Так как каждое
кристаллическое вещество характеризуется своим собственным
законом пространственного расположения атомов, набор межплос-
костных расстояний, а следовательно, и величин d/n будет специ-
фичным для каждого индивидуального кристаллического веще-
ства.
Основной задачей фазового рентгеновского анализа является
идентификация входящих в исследуемый образец кристаллических
веществ на основании набора межплоскостных расстояний. При
выполнении этой задачи очень важно найти максимально полный
набор межплоскостных расстояний. С этой целью в практике рент-
геновского анализа почвенных объектов, особенно тонкодисперс-
ных почвенных фракций, применяют так называемый метод по-
рошка, или метод Дебая—Шеррера. Он заключается в том, что
рентгеновский луч направляют на объект, состоящий из множест-
ва мелких кристаллов. Поскольку число таких кристаллов в об-
разце бесконечно велико и расположены они хаотично (в общем
случае), среди этого множества кристаллов найдутся и такие,
в которых соответствующие серии плоскостей удовлетворяют ус-
ловию Брегга—Вульфа.
280
Устройство рентгеновских аппаратов
Конструкции аппаратов, используемых в настоящее время для
фазового рентгеновского анализа, сложны и многообразны. Но
принципиальные электрические схемы и основные узлы, предназ-
наченные для получения рентгеновских лучей, сходны в аппара-
тах различных конструкций (рис. 82, о).
Рис 82. Схема рентгеновской установки с кенотроном (а) и электронной рент-
геновской трубки (б)
Одним из основных элементов электрической схемы рентге-
новской установки является высоковольтный масляный трансфор-
матор, который повышает входное напряжение сети питания до
20—60 kV. Высокое напряжение подается на кенотрон или группу
кенотронов, которые включены последовательно с рентгеновской
трхбкой и играют роль выпрямителей. Выпрямленное высокое на-
пряжение подается на рентгеновскую трубку.
В рентгеновских аппаратах заземляется один из полюсов вто-
ричной обмотки трансформатора и анод рентгеновской трубки,
цепь замыкается через землю. Напряжение, подаваемое на труб-
ку, регулируется изменением напряжения, идущего на первичную
обмотку трансформатора. В первичной цепи высоковольтного
трансформатора ставится вольтметр. Зная показания вольтмет-
ра и коэффициент трансформации, можно определить напряжение
во вторичной цепи, т. е. напряжение, подаваемое на трубку. Сила
тока в трубке определяется накалом нити катода и регулируется
изменением силы тока в цепи накала. Контроль за силой тока в
трубке осуществляется с помощью миллиамперметра.
В настоящее время в практике рентгеновского анализа исполь-
зуются преимущественно запаянные электронные рентгеновские
трубки (см. рис. 82, б). Электронная рентгеновская трубка изго-
281
товляется в виде запаянного стеклянного цилиндра, из которого
откачан воздух до 10-5—Ю'6 мм рт. ст., внутри цилиндра поме-
щены электроды. Катод представляет собой вольфрамовую спи-
раль. Анод (антикатод) трубки — металлическая пластинка ив
Cr, Fe, Со, Ni, Си, Мо и других материалов, напаянная на массив-
ное медное основание (на
Рис. 83. Схема рентгеновской ка-
меры типа РКД:
1 — винт центрировки столика
для образцов, 2 — образец, 3 —
столик для образца, 4 — передняя
диафрагма (коллиматор), 5 —
задняя диафрагма («ловушка»),
6 — корпус камеры, 7 — устано-
вочные винты
рис. 82, б зачерненная часть анода).
Она называется зеркалом анода.
Поскольку при торможении элек-
тронов о поверхность анода выде-
ляется большое количество тепла,
анод при работе трубки охлажда-
ют непрерывной циркуляцией хо-
лодной воды в основании анода.
Часть зеркала анода, бомбарди-
руемая пучком электронов, назы-
вается фокусным пятном. В кор-
пусе трубки напротив анода распо-
ложены окна из слабо погло-
щающего рентгеновское излучение
материала (алюминий, берил-
лий или слюда). Через эти окошки
проходит возникающее на ано-
де рентгеновское излучение и по-
падает на образец.
В зависимости ог способа реги-
страции отраженных дифрагиро-
ванных лучей рентгеновские аппа-
раты делятся на две группы:
а) аппараты с фотографической
регистрацией; б) аппараты с ис-
пользованием счетчиков излучения — рентгеновские дифракто-
метры.
В комплект аппаратов с фоторегистрацией входит одна или
несколько рентгеновских камер. При работе с почвенными объек-
тами чаще всего используются камеры типа РКД и РКУ. Они
представляют собой металлические цилиндры с плотно прилегаю-
щей крышкой. В отверстии в стенке цилиндра вмонтирована диа-
фрагма (коллиматор) (рис. 83). В центре камеры на специальном
держателе помещается образец в виде цилиндрического столбика
или вытянутого параллелепипеда длиной около 1 см и толщиной
0,2—0,3 мм. Специальное устройство (центрирные и юстирные
салазки) позволяет устанавливать образец точно в центре камеры
таким образом, чтобы он находился в середине рентгеновского лу-
ча, проходящего через коллиматор.
Позади образца в корпусе камеры монтируется так называе-
мая «задняя диафрагма», или «ловушка», -— полый металличе-
ский цилиндр (или щель), в который первичный пучок попадает
сразу же после прохождения через образец. Это приспособление
282
препятствует почернению пленки от первичного пучка и понижает
уровень фона, возникающего при рассеянии рентгеновских лучей
воздухом. Рентгеновская пленка (имеющая форму узкой полосы)
помещается вдоль внутренней стенки корпуса камеры. После
съемки и проявления на пленке обнаруживается система симмет-
ричных дуг, формирование дифракционной картины при этом мож-
но представить следующим образом. В столбике образца тонко-
дисперсных почвенных фракций кристаллики минералов распола-
гаются хаотически, поэтому осуществляются все возможные ори-
ентации самих кристалликов и слагающих их серий атомных плос-
костей по отношению к направлению рентгеновского луча. Среди
Рис. 84. Отклонение рентгеновских лучей на угол 2 6 от первоначального
направления (а) и связь величины 6, I и R (б)
этих ориентаций будут и такие, которые удовлетворяют закону
Брегга—Вульфа: 2dsin0 = nL Луч, отраженный какой-то опреде-
ленной серией плоскостей, отклоняется от исходного направления
на угол 2 0 (рис. 84, а). Луч, отраженный от одной серии плоско-
стей в одном индивидуальном кристаллите, дает на пленке одно
Дифракционное пятно. Поскольку в образце присутствует доста-
точно большое количество кристаллитов, ориентация которых удов-
летворяет одному и тому же условию, отраженные от них лучи
образуют конус, осью которого является направление первичного
луча, а угол полураствора равен 2 0; пленка отсекает две симмет-
ричные дуги, угловое расстояние между которыми равно 4 0 (см.
рис. 84, б). Если расстояние между симметричными дугами в мм
I 45/
равно /, то 0 =---- (в радианах) и 0 =------ (в градусах), где
4Rk nRk
“к — радиус камеры. Для точного определения величины произ-
водят калибровку камеры по какому-либо веществу с известными
283
межплоскостными расстояниями. Такими веществами может быть
поваренная соль, медная проволока. При работе с почвенными объ-
ектами удобно калибровать камеры по величине межплоскостного
расстояния кварца 3,343 А, линия которого присутствует на
рентгенограммах илистых фракций почв. Пользуясь формулой
Брегга—Вульфа или вычисленными на ее основе таблицами меж-
плоскостных расстояний находим величину d/n, которая соответ-
ствует межплоскостному расстоянию 3,343 А и для медного анода
составляет 13°19'.
45/ 45/
Преобразовав приведенную выше формулу 6 -------вА =------,
nRk 6л
измерив на рентгенограмме величину / в мм (с точностью до де-
сятых долей мм, трехкратно и взяв среднюю из трех измерений
величину) и подставив в преобразованную формулу значения I и
0, находят радиус камеры.
45
Поскольку величина —— для каждой камеры есть величина
постоянная, ее удобно вычислить и пользоваться ей в дальнейших
расчетах. Обозначают ее буквой А.
При промере рентгенограмм удобно пользоваться табл. 28.
В первой графе таблицы отмечается порядковый номер линии,
во второй — интенсивность линии (т. е. степень почернения рент-
Таблица 28 Результаты промера и расчета рентгенограмм геновской пленки), измеря- емая визуально в десяти- балльной шкале. В третьей графе для каждой пары дуг пишется значение 1 (среднее из трех промеров)
№ линии 1 1 / .< к (6) d/n Примеча- ния
1 2 3 8 30,0 7,42 7,50 диф. с точностью до десятой до- ли мм. Для удобства про- мера линий на рентгено- граммах существуют специ- альные световые столики и
приборы — компараторы,
снабженные оптической
системой для более точного промера линий. В четвертой
графе помещены значения углов 0, которые вычисляются как
произведение /ХА. В пятой графе пишутся значения межплоскост-
ных расстояний, определенные по таблицам Гиллера. В шестой
графе примечаний описывается характер линии (острая, широкая,
диффузная, асимметричная и т. п.). По набору межплоскостных
расстояний диагностируются присутствующие в образце кристал-
лические вещества, т. е. выполняется основная задача рентгенов-
ского фазового анализа.
В настоящее время в практике рентгеновского анализа поч-
венных объектов, особенно тонкодисперсных фракций, все шире
используются рентгеновские дифрактометры. Работа ведется так-
же методом Дебая—Шеррера. В отличие от аппаратов, в которых
используются рентгеновские камеры и дифракционная картина ре-
284
гистрируется на пленке фотоспособом, в рентгеновских дифракто-
метрах отраженное излучение регистрируется счетчиками кван-
тов.
Необходимой составной частью дифрактометра является го-
ниометрическое устройство, которое обеспечивает вращение об-
разца и счетчика вокруг общей вертикальной оси. В процессе вра-
щения образца серии плоскостей, удовлетворяющие уравнению
Брегга—Вульфа, поочередно попадают в отражающее положение.
Отраженный луч воспринимается счетчиком квантов. Импульсы
отраженного рентгеновского излучения регистрируются пересчет-
ным и цифропечатающим устройством или на двигающейся диаг-
раммной ленте. В последнем случае на диаграммной ленте запи-
сывается рентгенодифрактограмма — серия дифракционных пиков
на линии фона.
Измерение углов 0 на рентгенодифрактограммах значительно
проще, чем на рентгенограммах, полученных фотоспособом. Спе-
циальное устройство (отметчик углов), вмонтированное в гонио-
метр, отмечает каждый градус (или каждую десятую долю гра-
дуса) вертикальным штрихом на дифрактограмме. Если дифрак-
ционный пик попадает между двумя отметками угла, то, зная рас-
стояние между этими отметками в миллиметрах и зная расстоя-
ние от вертикальной линии, проведенной через вершину пика, до
ближайшей отметки, несложно точно высчитать угол 0, соответ-
ствующий дифракционному максимуму. Зная угол 0 и материал
анода в рентгеновской трубке, по таблице Гиллера находят меж-
плоскостные расстояния и по набору межплоскостных расстояний
определяют присутствующие в образце кристаллические веще-
ства.
Особенности получения
дифракционной картины
илистых фракций почв
Глинистые минералы почв в большинстве случаев представле-
ны очень мелкими частицами пластинчатой формы, несовершен-
ной кристаллической структурой и разнообразного химического
состава,- По сравнению с другими группами минералов глины об-
ладают значительно большими межплоскостными расстояниями,
особенно по одному из кристаллографических направлений — по
оси С.
Для регистрации больших межплоскостных расстояний прово-
дят съемку в области малых углов, так как из формулы Брегга—
Вульфа следует, что чем больше величина d, тем меньше угол от-
ражения 0. В связи с этим большие круглые «ловушки» («задние
диафрагмы») камер заменяют тонкими щелевыми диафрагмами
и вносят изменения в коллиматор («переднюю диафрагму»). При
285
работе с рентгенодифрактометрами отражения в малоугловой об-
ласти можно получить при хорошей юстировке прибора, исполь-
зовании системы диафрагм и специальных приставок для малоуг-
ловой съемки. Идентификация глинистых минералов производит-
ся преимущественно по отражениям от так называемых базальных
плоскостей, т е. плоскостей, параллельных осям а и Ь. Такие
плоскости имеют индексы 001. Поскольку глинистые минералы
обычно тонкодисперсны и часто плохо окристаллизованы, очень
важно, чтобы максимальное число кристаллитов попало в отра-
жающее положение в малоугловой области, в которой регистриру-
ется большинство базальных рефлексов. Для этого готовят так
называемые ориентированные препараты, т. е. такие образцы, в
которых максимальное число частиц ориентировано базальными
гранями в одном направлении. Ниже изложены способы приготов-
ления ориентированных препаратов для камерной съемки и рент-
генодифрактометрии.
В ряде случаев бывает необходимо получать от глинистых ми-
нералов не только базальные, но и другие рефлексы. При работе
методом камерной съемки небазальные отражения получают при
съемке неориентированных препаратов, а также при вращении
столбика образца в камере вокруг своей оси.
При диагностике глинистых минералов по рентгенограммам
возникает еще одна трудность, ряд глинистых минералов дает
сходный набор рефлексов. Для того чтобы правильно идентифи-
цировать эти минералы, применяют систему обработок: насыщение
минералов глицерином или этиленгликолем, прокаливание при раз-
личных температурах и т. п.
Поскольку кристаллические решетки различных глинистых ми-
нералов по-разному реагируют на указанные обработки, этот при-
ем позволяет более точно диагностировать глинистые минералы в
смеси.
Методы подготовки илистых фракций
и приготовление ориентированных препаратов
для камерной съемки
и рентгенодифрактометрии
Чтобы получить хорошие ориентированные препараты илис-
тых фракций, необходимо удалить из образца все вещества, кото-
рые способствуют образованию агрегатов из частиц глинистых
минералов. Такими веществами являются карбонаты, гипс, легко-
растворимые соли, органическое вещество, несиликатные формы
железа. Обычно карбонаты, гипс и легкорастворимые соли уда-
ляются из образца еще перед выделением из него илистой фрак-
ции. Органическое вещество удаляют обработкой навески илистой
.фракции (2—3 г) 10%-ной Н2Ог в фарфоровой чашке на водяной
286
бане при нагревании. Если в образце содержится много органи-
ческого вещества, обработку повторяют несколько раз до посвет-
ления образца. После обработки навеску многократно промывают
дистиллированной водой и высушивают на воздухе.
Несиликатное железо удаляют из образца илистой фракции по
методу Мера — Джексона (см. с. 263).
После обработки по методу Мера—Джексона приступают к
насыщению образцов илистых фракций определенными катиона-
ми. Обычно рентгеновский анализ выполняют для образцов, на-
сыщенных Mg2+. Некоторые определения выполняются при насы-
щении илистых фракций К+ и Li+ из однонормальных растворов
соответствующих хлоридов. Насыщение можно проводить диали-
зом в целлофановых пакетах, помещаемых в большие сосуды
(стеклянные стаканы на 3—4 л или большие кристаллизаторы),
наполненные 1 н. растворами MgCl2, КС1 или LiCl. Навеску илис-
той фракции (200—300 мг для насыщения Mg2+ и 100—200 мг
для насыщения К+ и Li+) насыпают на кусочек целлофана, пред-
варительно замоченного в воде, размером 6X6 см, после чего края
целлофана стягивают ниткой таким образом, чтобы илистая фрак-
ция оказалась в целлофановом пакетике. Пакеты с фракциями
привязывают к стеклянной палочке, лежащей поперек стакана или
кристаллизатора таким образом, чтобы они оказались погружен-
ными в раствор соли. Для насыщения достаточно сменить раст-
вор соли в сосуде 4—5 раз с интервалом в сутки. После насыще-
ния образец отмывают дистиллированной водой от избытка солей
в тех же сосудах. Для этого в сосуды заливают дистиллирован-
ную воду и многократно меняют ее до отсутствия реакции на
Cl-нон.
Насыщение катионами и отмывку фракций можно проводить
также на фильтре или центрифугированием.
Отмытые от избытка солей илистые фракции высушивают на
воздухе, слегка растирают в агатовой ступке для получения од-
нородной смеси и помещают в пакетик из кальки или стеклянный
бюкс.
Ориентированные образцы для камерной съемки готовят сле-
дующим образом. Навеску илистой фракции подготавливают, как
описано выше, смачивают водой до состояния крутой пасты и по-
мещают в тиски между двумя полированными металлическими
поверхностями (брусками, пластинами и т. п.). Чтобы паста не
прилипала к металлу, можно использовать прокладки из кальки,
смоченной машинным маслом. Тиски зажимают, в результате чего-
кусок пасты превращается в пластинку. Пластинку режут попо-
лам, половинки кладут одна на другую и снова помещают в тис-
ки; эту операцию повторяют 4—5 раз для лучшей ориентации
глинистых частиц. Толщину пластинки доводят до 0,1—0,2 мм.
После этого пластинку режут лезвием безопасной бритвы на иго-
лочки длиной 8—10 мм, шириной «0,2 мм и толщиной, равной
толщине пластинки. Следует срезать пластинку в слегка влажном
287
состоянии. В противном случае она будет ломаться и крошиться.
Полученные иголочки закрепляют в центре столика для образца
в рентгеновской камере.
Ориентированные препараты для рентгенодифрактометра гото-
вят следующим образом: 100 мг подготовленного ила помещают в
агатовую ступку и смешивают с 1—2 каплями дистиллированной
воды так, чтобы получилась однородная крутая паста. После этого
добавляют 2,1 мл дистиллированной воды и очень осторожно рас-
тирают пасту (пальцем или мягким пестиком). Такое соотношение
ила и воды обеспечивает концентрацию суспензии, близкую к оп-
тимальной величине около 5%. Очень важно при растирании пас-
ты в воде получить совершенно однородную суспензию, без комоч-
ков на дне ступки; 7 капель такой суспензии равномерно пипет-
кой наносят на стекло размером 25x25 мм. Чтобы суспензия хо-
рошо легла на стекло, оно должно быть тщательно вымыто горя-
чей хромовой смесью, протерто спиртом и высушено. Если пред-
полагается провести количественную или полуколичественную
оценку содержания компонентов по данным рентгенодифракто-
метрии, недопустимо отклонение размеров стекол от указанной
величины. После приготовления исходных образцов (на 1—2 стек-
лах) в оставшуюся суспензию добавляют одну каплю глицери-
на (это количество может быть изменено в зависимости от степе-
ни дисперсности и минералогического состава образца), после
чего суспензия перемешивается, и на такое же стекло наносится
7 капель суспензии с глицерином. Рентгеновская съемка насыщен-
ных глицерином препаратов необходима для идентификации ми-
нералов группы монтмориллонита.
Затем нанесенную на стекло суспензию высушивают. Высуши-
вание препаратов следует проводить медленно: это способствует
лучшей ориентации глинистых частичек параллельно поверхности
стекла. Высушивание проводят на воздухе, стекла с суспензией
неплотно прикрывают часовыми стеклами или чашками Петри.
Между поверхностью стола и краем часового стекла или краем
чашки Петри должен оставаться зазор 4—5 мм. Такой прием обес-
печивает высыхание препаратов при комнатной температуре в те-
чение 16—20 ч. Не следует помещать исходные и насыщенные гли-
церином препараты под одно часовое стекло (чашку Петри). По-
верхность стола, на которой будут сушиться препараты, должна
быть строго горизонтальной (это необходимо проверить по уров-
ню). В противном случае суспензии осядут на стекла не равно- f
мерно, а стекут к одному краю.
Прокаливание ориентированных образцов проводится в муфе-
ле с терморегулятором при температуре 350 и 550° в течение 2 ч.
Для равномерного нагрева желательно помещать образцы в центр
муфельной печи. Прокаливание образцов необходимо для правиль-
ной диагностики минералов групп монтмориллонита и вермикули-
та по данным рентгеновского анализа.
288
Строение кристаллических решеток
и рентгеновская диагностика
основных групп
глинистых минералов
Чтобы понять принципы диагностики глинистых минералов по
данным рентгеновского анализа, необходимо иметь представле-
ние об особенностях строения их кристаллических решеток. В со-
временной кристаллографии принято описывать решетки различ-
ных кристаллических веществ как сочетания многогранников, по-
строенных определенными атомами и сочленяющихся по опреде-
ленному закону.
Рис. 85. Тетраэдр («), сочленение тетраэдров (б)
В основе строения кристаллических решеток значительной ча-
сти глинистых минералов лежат два многогранника — тетраэдр
и октаэдр. Тетраэдр (рис. 85, а) представляет собой четырехгран-
ник, грани которого — равносторонние треугольники; в вершинах
находятся атомы кислорода, а в центре атом кремния (или алю-
миния). Сочленяясь в пространстве своими вершинами, тетраэд-
ры образуют слои — двухмерную гексагональную сетку (см.
рис. 85, б). Такой характер сочленения тетраэдров дает основание
относить почти все глинистые минералы к подклассу слоистых си-
ликатов. Октаэдр представляет собой восьмигранник, грани ко-
торого — тоже равносторонние треугольники (рис. 86); в верши-
нах октаэдра находятся атомы кислорода и группы ОН, а в цент-
ре — различные катионы. Октаэдры сочленяются в пространстве
через общие ребра и тоже образуют двухмерную гексагональную
сетку. В зависимости от характера заполнения октаэдров все
слоистые силикаты делятся на ди- и триоктаэдрические. В диокта-
эдрических структурах катионы заполняют 2/3 возможных окта-
эдрических позиций, причем катионы всегда трехвалентные, чаще
всего А13+. В триоктаэдрических слоистых силикатах катионы за-
нимают все возможные октаэдрические позиции, причем катионы
двухвалентные, чаще всего Mg2+ в изоморфной смеси с Fe2+. Зна-
10 Зак 284
289
чительная часть глинистых минералов несет отрицательный заряд,
возникающий за счет изоморфных замещений в тетраэдрической
или октаэдрической сетке. Заряд компенсируется катионами, рас-
Рис. 86. Октаэдр (а), сочленение октаэдров (б)
положенными в межслоевых промежутках. Отдельные группы
глинистых минералов выделяются по соотношению в структуре
количеств октаэдрические ' тетраэдрических сеток, по характеру
заполнения октаэдрических пус-
тот и по величине и положению
заряда в кристаллической ре-
\ хА */ V/ шетке.
\ (Zn _\Г/д>- \ пи Минералы каолинитовой
группы имеют кристалличес-
Л/\®\ 2/ кую РешеткУ типа 1:1, т. е. в
J \ М \ них на один тетраэдрический
в слой приходится один слой окта-
эдров, причем сетки сочленяют-
Рис. 87. Строение кристаллической ся друг с другом через общие
решетки каолинита вершинки, в которых находится
кислород (рис. 87). Таким
образом, в основе структуры минералов группы каоли-
нита лежит двухслойный пакет. Наиболее широко распрост-
раненными представителями этой группы минералов являются
собственно каолинит и галлуазит. Оба минерала относятся к диок-
таэдрическим и характеризуются незначительными изоморфными
замещениями в решетке.
В собственно каолинитах — A14(OH)g [Si-jOio] — между ато-
мами кислорода тетраэдрической сетки одного пакета и гидро-
ксильными группами октаэдрической сетки соседнего пакета воз-
никает прочная водородная связь. Толщина двухслойного пакета
каолинита стабильна и составляет 7,15 А. Благодаря прочной во-
дородной связи в межпакетпые промежутки каолинита не могу?
проникнуть вода и глицерин. Решетка каолинита не изменяется
при прокаливании до 350°, но полностью разрушается при прока-
ливании до 550°, при этой температуре минерал теряет гидро-
ксильные группы. На основании указанных свойств производится
290
1
Диагностически важные значения межплоскостных расстояний для основных групп глинистых минералов (в А)
и дифракционные индексы отражений
Таблица 29
Минерал Препарат
насыщен Mg2+, исходный насыщен Mg, обра- ботан глицерином насыщен Mg, прокален прн 350° насыщен Mg, прокален прн 550° насыщен К+, исход- ный насыщен Li, прока- лен прн 300°, обрабо- тан глицерином
Каолинит Галлуазит ю СО »—•* Диок таэдрический иллит Т риоктаэдрический иллит Диоктаэдрический вермикулит 7,15 (001) 3,58 (002) 1,48 (060) 10,0 (001) 5,0 (002) 1,48 (060) 10,00 (001) 5,00 (002) 3,33 (003) 1,50 (060) 10,00 (001) 3,34 (003) 1,53 (060) 14,4 (001) 7,2 (002) 4,8 (003) 3,60 (004) 1,50 (060) 7,15 (001) 3,58 (002) 1,48 (060) >10 10,00 (001) 5,00 (002) 3,33 (003) 1,50 (060) 10,00 (001) 3,34 (003) 1,53 (060) 14,4 (001) 7,2 (002) 4,8 (003) 3,60 (004) 1,50 (060) 7,15 (001) 3,58 (002) 1,48 (060) 7,6 (001) 3,8 (002) 1,48 (060) 10,00 1001) 5,00 (002) 3,33 (003) 1,50 (060) 10,00 (001) 3,34 (003) 1,53 (060) 10,00 (001) 5,0 (002) 3,34 (003) 1,50 (060) отражения отсутствуют отражения отсутствуют 10,00 (001) 5,00 (002) 3,33 (003) 1,50 (060) 10,00 (001) 3,34 (003) 1,53 (060) 10,00 (001) 5,0 (002) 3,33 (003) 1,50 (060) не применяется то же » » » » » » » » » » 10,00 (001) 5,0 (002) 3,33 (003) 1,50 (060) не применяется то же » » » » » » » » . » »
Продолжение табл. 29
Препарат
Минерал насыщен Mg2+, ИСХОДНЫ!! насыщен Mg, обра- ботав глицерином насыщен Mg, прокален при 350° насыщен Mg, прокален при 550° насыщен К+> исход- ный насыщен Li, прока- лен при 300°, обрабо- тан глицерином
Диоктаэдрический МО нт морилЛОНИ т Бейделлит ьо to Хлорит триоктаэд- рический Почвенный хлорит 14,4 (001) 7,2 (002) 4,7 (003) 3,60 (004) 1,50 (060) то же » » » 14,4 (001) 7,2 (002) 4,8 (003) 3,6 (004) 1,53 (060) 14,4 (001) 7,2 (002) 4,8 (003) 3,6 (004) 17,7 (001) 8,8 (002) 4,5 (004) 1,50 (060) то же » » 14,4^(001) 7,2 (002) 4,8 (003) 3,6 (004) 1,53 (060) 14,4 (001) 7,2 (002) 4,8 (003) 3,6 (004) 10,0 (001) 5,0 (002) 3,33 (003) 1,50 (060) то же » » 14,4 (001) 7,2 (002) 4,8 (003) 3,6 (004) 1,53 (060) 10—14 (001) 9,6 (001) 4,8 (002) 3,2 (003) 1,50 (060) то же » » 14,4 (001) 7,2 (002) 4,8 (003) 3,6 (004) 1,53 (060) >10 (001) * 10,0 (001) 5,0 (002) 3,33 (003) 1,50 (060) не применяется то же » » » >ю 10,0 (001) 5,0 (002) 3,34 (003) 1,50 (060) 17,7 (001) 8,8 (002) 4,5 (004) 1,5 (060) не применяется то же » » » »
Сжатие решетки до 10 А при насыщении калием наблюдается только у высокозарядных монтмориллонитов
идентификация каолинитовых минералов по данным ренпеновско-
го анализа. Каолинитам свойственны межплоскостные расстояния
7,15 и 3,57 А — соответственно первого и второго порядков от од-
ной и той же сетки базальных плоскостей (табл. 29). Эти отраже-
ния не меняются при насыщении каолинита глицерином и прока-
ливании при 350°, но полностью исчезают -на рентгенограммах
образцов, прокаленных при 550° (рис. 88), поскольку при этой
температуре происходит полное разрушение кристаллической ре-
шетки.
Некоторые трудности возникают при идентификации каолини-
товых минералов по рентгенограммам в том случае, если в соста-
ве изучаемых образцов присутствуют хлориты, поскольку указан-
ные отражения каолинита совпадают с рефлексами второго и чет-
вертого порядков хлоритовых минералов. В этом случае рекомен-
дуется обработать образец разбавленной НС1 в течение несколь-
ких часов при 80°. Такая обработка приводит к растворению хло-
ритовых минералов и соответственно к исчезновению рефлексов
хлорита на рентгенограммах. Следует иметь в виду, однако, что в
теплой разбавленной НС1 растворяются не все хлориты, а только
тонкозернистые железистомагнезиальные разновидности этой груп-
пы минералов.
В почвенных объектах каолинит может быть представлен раз-
новидностями с различной степенью упорядоченности кристалли-
ческой решетки. Неупорядоченность решетки чаще всего бывает
связана с беспорядочным смещением слоев параллельно оси b и
с изменением ориентировки двухслойных пакетов относительно
друг друга.
Удобным приемом для определения степени упорядоченности
каолинита является получение небазальных отражений в области
углов от 6,5 до 12° (для Си Аа-излучения). Хорошо окристалли- /
зованные каолиниты дают в этой области набор рефлексов, со- /
ответствующий межплоскостным расстояниям 4,46, 4,36, 4,18, /
4,13 и 3,85 А. Каолиниты с разупорядоченной решеткой не дают
ни одного из этих отражений, кроме широкой диффузной полосы
в области 4,48 А.
В галлуазитовых минералах сила связи между соседними си-
ликатными пакетами значительно меньше, чем в собственно као-
линитах, и в межпакетных промежутках содержится молекуляр-
ная вода. Поэтому межплоскостные расстояния по оси с в крис-
таллической решетке галлуазита больше, чем в каолините, и со-
ставляют 10,1 А. Второй особенностью галлуазитовых минералов
является крайне беспорядочное наложение друг на друга двух-
слойных силикатных пакетов. Галлуазит — AU(OH)8[Si'tO^X
Х4Н2О — диагностируется по широким отражениям I и III поряд-
ков от базальной серии плоскостей, соответствующих межплоскост-
ным расстояниям 10,1 и 3,4 А и по широкому диффузному отра-
жению (02), соответствующему межплоскостному расстоянию
4,46 А. Некоторые образцы галлуазитов обладают способностью
293
Рис 88 Рентгено-дифрактограммы насыщенных Mg образцов наибо.ш
I— каолинит, II— галлуазит, III — диоктаэдрическая слюда, IV— тр1
хлорит, VIII — почвенный хлорит, а — исходный образец, б — обработа,
цифры на кривых — А
распространенных глинистых минералов:
октаэдрическая слюда, V— вермикулит, VI — монтмориллонит, VII—
ный глицерином, в — прокаленный при 350°, г — прокаленный при 550°,
1
к расширению решетки по оси с после обработки глицерином или
этиленгликолем.
Значительная часть присутствующей в межпакетных промежут-
ках молекулярной воды может быть удалена уже при температу-
ре 60°, что приводит к уменьшению величины межплоскостного
расстояния до 7,2—7,5 А. Прокаливание при 550° вызывает пол-
ное исчезновение отражений как гидратированного, так и дегидра-
тированного галлуазита, поскольку при этой температуре крис-
таллическая решетка галлуазита разрушается с потерей! ОН-
группы.
Рис. 89. Строение кристаллической решетки слюды (а) и монтморилло-
нита (б)
Минералы группы слюд и гидрослюд обладают трех-
слойной кристаллической решеткой типа 2:1. Это значит, что па-
кет состоит из двух тетраэдрических сеток, обращенных вершина-
ми тетраэдров навстречу друг другу, и заключенной между ними
октаэдрической сетки (рис. 89, а). Для всех слюдистых минера-
лов характерен высокий отрицательный заряд силикатного слоя,
равный —1 и возникающий за счет изоморфного замещения 1/4
части атомов Si в тетраэдрах на А1. Отрицательный заряд ком-
пенсируется ионами калия, который размещается в межпакетных
промежутках. В собственно слюдистых минералах силы электро-
статического взаимодействия между отрицательно заряженными
силикатными слоями и положительно заряженными межпакетны-
ми ионами К+ достаточно велики, чтобы обеспечить прочность и
стабильность кристаллической решетки и невозможность межпа-
кетной адсорбции воды и органических веществ. В гидрослюдис-
тых минералах, которые называются еще иллитами, заряд и силы
электростатического взаимодействия между слоями и катионами
296
обычно бывают несколько меньше, и часть ионов К+ замещается
другими катионами. Слюдистые и гидрослюдистые минералы де-
лятся на ди- и триоктаэдрические структуры. Идеальная формула
диоктаэдрической (белой) слюды — мусковита выглядит следую-
щим образом: KAl2(OH)2[AlSi3Oi0], а триоктаэдрические (цвет-
ные) слюды биотит — флогопитового изоморфного ряда имеют
формулу: К (Mg, Fe2+)3 (OH)2[AlSi3Oio].
На рентгенодифрактограммах собственно слюдистые минера-
лы диагностируются по отражениям I и III порядков от се-
рии базальных плоскостей, равным соответственно 10 и 3,33 А.
Эти отражения не изменяются при прокаливании и насыщении
минералов глицерином, так как слюды обладают прочной стабиль-
ной решеткой и не способны к
межпакетной адсорбции воды и
полярных жидкостей (см.
рис. 89, б). Гидрослюдистые (ил-
литовые) минералы за счет де-
Таблица 30
Отношение интенсивностей рефлексов
первого и второго порядка для
гидро с люд истых минералов с различными
катионами в октаэдрах
фектов структуры могут иметь
несколько большие межплоско-
стные расстояния и не вполне
целочисленную серию отражений.
Это значит, что отражения раз-
ных порядков от одной и той же
серии плоскостей будут не впол-
не кратными друг другу. При
прокаливании базальные меж-
плоскостные расстояния иллитов
несколько уменьшатся вследствие
потери воды гидратированными
Состав октаэдрического
слоя
4 А1
3 Al + 1 Fe
2 А1+ 2 Fe
1 Al -J- 3 Fe
6 Mg
5 Mg + 1 Fe
2 Mg + 4 Fe
6 Fe
1qoi
1 002
0,8
2,9
10,9
68,1
7,2
38,7
50,0
16,2
межслоевыми катионами.
Характер заполнения октаэдров в слюдистых и гидрослюдис-
тых минералах может быть определен на основании соотношения
интенсивностей рефлексов первого и второго порядков от серии
базальных плоскостей. Как видно из табл. 30, в диоктаэдрических
слюдах отношение интенсивности отражения 10 А к интенсивно-
сти рефлекса 5 А равно 2 или меньше. При заполнении октаэдров
только Mg2+ оно приближается к 7, двухвалентным Fe2+ — к 16,
а при наличии в октаэдрах трехвалентного Fe3+ это отношение
возрастает до бесконечности из-за отсутствия отражения второго
порядка на рентгенограммах.
Характер заполнения октаэдрического пространства можно ори-
ентировочно определить по отражениям 020 и 060, снимая ориен-
тированные препараты в камерах или снимая рентгенодифракто-
граммы ориентированных образцов «на просвет», ставя рабочую
плоскость образца под 90° к падающему рентгеновскому лучу.
В соответствии с параметром решетки по оси b диоктаэдрические
слюды дают отражение 020 и 060, соответственно 4,46 и 1,50 А, а
триоктаэдрические слюды — соответственно 4,58 и 1,53 А.
297
Минералы группы вермикулита тоже имеют кристалличе-
скую решетку типа 2:1, т. е. состоящую из двух тетраэдрических
сеток, между которыми заключен слой октаэдров. Отрицательный
заряд силикатного слоя, так же как и в слюдистых минералах,
возникает за счет изоморфного замещения Si на А1 в тетраэдрах,
но имеет несколько меньшую величину, чем в слюдах и гидрослю-
дах. Заряд равен —0,6—0,7 единицы по сравнению с •—1 в слю-
дах. Отрицательный заряд компенсируется обменными катионами,
находящимися в межпакетном пространстве. Вследствие более
низкой, по сравнению со слюдами, величины заряда силы элек-
тростатического взаимодействия между силикатными слоями и об-
менными катионами в вермикулитах меньше, чем в слюдах, и в
межслоевом пространстве воздушно-сухих образцов вермикулита
находятся 2 мономолекулярных слоя воды. Такая структура име-
ет межплоскостное расстояние по оси с, равное 14,4 А. Молекулы
органических жидкостей в межслоевые промежутки вермикулита
входить, как правило, не могут. Так же как и слюдистые й илли-
товые минералы, вермикулиты могут быть ди- и триоктаэдриче-
скими.
На рентгенограммах исходных воздушно-сухих образцов вер-
микулиты дают интенсивное отражение 1-го порядка от базальной
серии плоскостей, соответствующее межплоскостному расстоянию
14,4 А и серию базальных рефлексов, из которых наиболее интен-
сивными являются рефлексы 4-го и 5-го порядков, соответствую-
щие величинам d/n 3,60 и 2,88 А. Насыщение глицерином не из-
меняет величины базальных межплоскостных расстояний (см.
рис. 88), а прокаливание при 350 и 550° вызывает сжатие решет-
ки по оси с до 10 А вследствие выхода молекулярной воды из
межпакетных позиций. Поэтому на рентгенограммах прокаленных
образцов вермикулит характеризуется величинам din 10 А или
даже несколько меньше (до 9,02 А). Ди- и триоктаэдрические
вермикулиты хорошо различаются на дифрактограммах по отра-
жениям 060, которые соответствуют межплоскостным расстояниям
1,50 А для диоктаэдрических и 1,53 А для триоктаэдрических
структур.
Значительная часть вермикулитовых минералов обладает спо-
собностью к необменной сорбции из растворов ионов К+ и NHf-
При этом наблюдается образование слюдоподобных структур и
сжатие решетки по оси С до величины 10 А.
Минералы группы монтмориллонита имеют трехслой-
ный пакет 2:1, состоящий из двух тетраэдрических сеток, между
которыми заключен слой октаэдров (см. рис. 89, б). Всем мине-
ралам монтмориллонитовой группы свойственна низкая величина
заряда, равная 0,3—0,4 единицы.
Отрицательный заряд слоя компенсируется обменными меж-
слоевыми катионами. Вследствие низкой величины заряда силы
электростатического взаимодействия между силикатными слоями
и обменными межслоевыми катионами очень невелики, и в меж-
298
слоевые промежутки монтмориллонитовых минералов могут
внедряться молекулы органических веществ и воды. Другими сло-
вами, минералы монтмориллонитовой группы обладают ярко вы-
раженной способностью к межпакетной адсорбции различных ве-
ществ. Характерной особенностью строения кристаллической ре-
шетки монтмориллонитов является беспорядочное наложение друг
на друга силикатных слоев. Монтмориллонитовые минералы, как
слюдистые и вермикулитовые, могут быть ди- и триоктаэдрически-
ми. Положение заряда различно в разных минералах этой груп-
пы. В собственно монтмориллонитах заряд возникает за счет изо-
морфного замещения А1 на Mg в октаэдрах и, следовательно, лока-
лизован главным образом в октаэдрическом слое. Кроме того, к
монтмориллонитовой группе принадлежат бейделлиты — низкоза-
рядные минералы, в которых заряд локализован преимуществен-
но в тетраэдрическом слое и возникает, так же как и у слюд и
вермикулитов, за счет изоморфного замещения кремния на алю-
миний.
Благодаря низкому заряду базальные межплоскостные рас-
стояния минералов монтмориллонитовой группы лабильны. Они
могут- изменяться в зависимости от обменного катиона, степени
гидратации образна; расстояния увеличиваются при насыщении
образца глицерином или этиленгликолем. В воздушно-сухом со-
стоянии насыщенные Mg2+ образцы минералов монтмориллонито-
вой группы дают отражение, соответствующее межплоскостному
расстоянию 15 А и целочисленную серию рефлексов. После обра-
ботки образца глицерином межплоскостное расстояние увеличи-
вается до 17.7 А и возникает новая целочисленная серия отраже-
ний, кратных 17,7 А.
При прокаливании образца молекулярная вода выходит из
межслоевых промежутков, что приводит к сокращению межплос-
костных расстояний по оси с до 9,6—9,7 А. Ди- и триоктаэдриче-
ские монтмориллониты различаются так же, как слюдистые и
вермикулитовые минералы, по отражениям, равным 1,50 и 1,53
соответственно для ди- и триоктаэдрических монтмориллонитов.
Существует прием, который позволяет установить, в каких кри-
сталлографических позициях имеют место изоморфные замещения,
приводящие к появлению отрицательного заряда силикатного слоя,
т. е. установить принадлежность изучаемого минерала монтморил-
лонитовой группы к собственно монтмориллонитам или бейделли-
там. Этот прием заключается в следующем. Испытуемый ориен-
тированный образец насыщают литием и прокаливают при 200—•
300° в течение 10—12 ч. Затем его насыщают глицерином и сни-
мают рентгенограмму. Если после такой обработки минерал со-
храняет способность к набуханию, т. е. дает на рентгенограмме
отражение 17,7 А, минерал относится к бейделлитам. Если же
минерал сжимается необратимо и после обработки глицерином на
рентгенограмме обнаруживается отражение, соответствующее ве-
личине межплоскостного расстояния 9,5 А, то этот минерал отно-
299
сится к собственно монтмориллонитам с зарядом в октаэдриче-
ском слое. Отсутствие целочисленной серии отражений свидетель-
ствует о промежуточном характере минерала.
Минералы группы хлоритов имеют кристаллическую решет-
ку типа 2:1:1. Это значит, что трехслойные 2 : 1 пакеты череду-
ются в хлоритах с добавочным октаэдрическим слоем. В вершинах
©О ©ОН e5i 9Mg,Fe2^l
Рис. 90. Строение кристаллической
решетки хлорита
октаэдров добавочного октаэдри-
ческого слоя находятся гидро-
ксильные группы ОН (рис. 90).
Так же как и трехслойные сили-
каты, хлориты могут быть пред-
ставлены структурами с преиму-
щественным заполнением окта-
эдрических пустот по ди- или
триоктаэдрическому закону. Хло-
ритовые минералы хорошо иден-
тифицируются рентгенографиче-
ски по базальному и межплоско-
стному расстоянию 14 А и чис-
ленной серии рефлексов. В отли-
чие от монтмориллонитов и вер-
микулитов, хлориты характери-
зуются стабильной решеткой, и
свойственные им межплоскост-
ные расстояния не изменяются
при насыщении образца глице-
рином или после прокаливания.
Так же как и трехслойные си-
ликаты, ди- и триоктаэдрические
хлориты различаются по величи-
не сЦп, равной соответственно 1,50 Ав диоктаэдрических и 1,53—
1,54 А в триоктаэдрических структурах.
Почвенные хлориты имеют структуру, переходную от 2: 1 к
2:1:1. В кристаллической решетке этой группы минералов трех-
слойные пакеты разделяются прослойками гидроокиси А1 (иногда
Fe), которые в структурном отношении представляют собой фраг-
менты («острова») добавочного октаэдрического слоя. Исходны-
ми 2 : 1 структурами почвенных хлоритов могут быть пакеты вер-
микулита, монтмориллонита или выветривающегося иллита. В не-
которых случаях межслоевые пространства заполняются фрагмен-
тами («островами») добавочного октаэдрического слоя, а частич-
но — совершенными октаэдрическими слоями, такими, как в ис-
тинных хлоритах.
На указанных особенностях строения кристаллической решет-
ки основана диагностика почвенных хлоритов по данным рентге-
новского анализа. В исходном состоянии почвенные хлориты на
рентгенограммах дают отражение 14 А и серию рефлексов более
300
высоких порядков. В большинстве случаев межплоскостные
расстояния по оси с не изменяются при насыщении глице-
рином.
Наиболее важным диагностическим показателем для почвен-
ных хлоритов является положение рефлекса l-ro порядка на ди-
фрактограммах прокаленных образцов. В обшей форме можно
сказать, что все минералы группы почвенных хлоритов дают на
дифрактограммах прокаленных образцов отражение в промежут-
ке между 10 и 14 А, поскольку межпакетные прослойки гидрооки-
сей R2O3 и тем более совершенные октаэдрические слои (если они
есть) препятствуют полному сжатию решетки до 10 А при прока-
ливании образца. При этом отражение первого порядка от поч-
венных хлоритов при прокаливании может быть представлено ин-
дивидуальным пиком или диффузионным рассеянием в области
10—14 А.
Смешанослойные минералы представляют собой обшир-
ную группу минералов, которые широко распространены в почвах
и в которых слои, принадлежащие разным индивидуальным ми-
нералам, чередуются в различных пропорциях и по различным
законам. Смешанослойные минералы делятся на две большие груп-
пы: упорядоченные и неупорядоченные. Упорядоченные смешано-
слойные структуры характеризуются определенной периодично-
стью по оси с, так как слои индивидуальных минералов череду-
ются в них по определенным законам. Диагностика упорядочен-
ных смешаннослойных структур по данным рентгеновского анали-
за не представляет затруднений, так как их базальные межплос-
костиые расстояния равны сумме межплоскостных расстояний
слагающих компонентов. Например, отражение dOoi от упорядо-
ченного смешанослойного слюдисто-вермикулитового минерала,
состоящего из 50% слюды и 50% вермикулита, будет равно 24,4 А
(10 А+14,4 А).
Неупорядоченные смешанослойные образования характеризу-
ются отсутствием какого-либо закона в последовательности че-
редующихся слоев. В полностью неупорядоченных структурах, на-
пример двухкомпонентных, состоящих из чередующихся пакетов
минералов А и В, вероятность встретить слой А или слой В в
любом данном положении точно равна вероятности, в которой
они встречаются вообще. Положение дифракционных пиков от не-
упорядоченных смешанослойных структур и их изменение при на-
сыщении глицерином и прокаливании зависят от природы и соот-
ношения слагающих компонентов. Например, от неупорядоченно-
го смешанослойного слюдисто-вермикулитового минерала первый
дифракционный максимум будет находиться в промежут-
ке между 10 и 14 А, причем если в этом минерале преобладают
слюдистые пакеты, пик будет ближе к 10 А, а при преобладании
вермикулитовых пакетов — ближе к 14 А.
301
Расшифровка рентгенодифрактограмм
илистых фракций почв
На рис. 91 в качестве примера приведены рентгенодифракто-
граммы илистой фракции, выделенной из гор. Аг подзолистой нл-
лювиально-железисто-гумусовои
74,2
почвы на граните. На днфракто-
грамме образца в исходном со-
стоянии видны острый симмет-
ричный интенсивный пик, соот-
ветствующий межплоскостному
расстоянию 14,2 А, и кратное ему
отражение III порядка 4,83 А.
Эти рефлексы могут принадле-
жать трем минералам: монтмо-
риллониту, вермикулиту или хло-
риту. Поскольку при насыщении
глицерином межплоскостное рас-
стояние возрастает до 18 А, а
при прокаливании сжимается до
10 А, можно диагностировать
присутствие в образце разбухаю-
щего минерала монтмориллони-
товой группы. Более точно его
следует определить как бейдел-
лит, т. е. несущий заряд преиму-
щественно в тетраэдрическом
Рис. 91. Рентгеноднфрактограммы или-
стой фракции, выделенной из гор. А2
иллювиально-железистого подзола'
а — насыщенный Mg2+ исходный обра-
зец, б — насыщенный Mg2+ обработан-
ный глицерином, в — насыщенный Mg2'
образец после прокаливания при 350°,
г — насыщенный Mg2+ образец после
прокаливания при 550°, е — насыщен-
ный Li+ образец после прокаливания
при 300° в течение 12 час и обработки
глицерином; цифры на кривых — А
слое, так как насыщенные Li+ и прокаленные при 300° образцы
сохраняют способность к набуханию. Вермикулит в образце, оче-
видно, отсутствует, так как на днфрактограмме образца, насы-
щенного глицерином, нет дифракционного пика, соответствующего
межплоскостному расстоянию 14 А. По отсутствию того же пика
на днфрактограммах прокаленных образцов можно также кон-
статировать отсутствие хлорита. На днфрактограммах исходных,
насыщенных глицерином и прокаленных при 350° образцов хоро-
302
шо заметны отражения, соответствующие величинам din 7,15
и 3,57 А. При прокаливании образца до 550° эти отражения ис-
чезают. По указанным межплоскостным расстояниям можно уста-
новить присутствие в образце минералов группы каолинита. При
этом диагностировать каолинит следует главным образом по от-
ражениям на дифрактограммах образцов, насыщенных глицерином
или прокаленных при 350°, так как в исходном состоянии отраже-
ния каолинита 7,15 и 3,57 А совпадают с рефлексами второго и
четвертого порядков 14 А минералов. Кроме того, на рентгено-
дифрактограммах образца при всех обработках имеются дифрак-
ционные пики, соответствующие межплоскостным расстояниям
10, 4,92 и 3,34 А, по которым устанавливают наличие в образце
диоктаэдрических гидрослюдистых минералов. Небольшой дифрак-
ционный максимум 4,26 А свидетельствует о наличии в образце
кварца. Таким образом, в исследованном образце илистой фрак-
ции содержатся бейделлит, каолинит, диоктаэдрический иллит и
кварц.
Определение минералогического состава
илистой фракции
на приборе ДРОН-2
Для выделения илистой фракции берут навеску почвы, рав-
ную 20—100 г, в зависимости от содержания ила. Илистую фрак-
цию отделяют методом отмучивания по Горбунову. Препараты
для рентгенодифрактометрической съемки готовят из навески
илистой фракции ~1 г после удаления органического вещества
и несиликатного железа.
Из насыщенных Mg2+ образцов готовят три препарата из вод-
ной суспензии и один — из водной суспензии с добавлением гли-
церина. Из насыщенных К+ и Li+ образцов готовят по одному пре-
парату из водной суспензии. Один из насыщенных магнием пре-
паратов илистой фракции снимается в исходном состоянии, вто-
рой — после прокаливания при 350°, третий — после прокалива-
ния при 550°. Препараты Mg-фракций с добавлением глицерина и
фракций, насыщенных калием, снимают в исходном состоянии,
а Li-фракций — после прокаливания в течение 12 ч при 300° и
обработки глицерином. Препараты устанавливают в держатель
образца гониометрического устройства рентгенодифрактометра.
Включение прибора (дифрактометр ДРОН-2) осуществляется в
последовательности: высоковольтный источник питания (ВИП),
электронное вычислительное устройство (ЭВУ), блок автоматиче-
ского управления (БАУ). После установки образца тщательно за-
крывают защитную шторку из просвинцованной резины и включа-
ют высокое напряжение и механизм, обеспечивающий вращение
образца и счетчика квантов вокруг оси гониометра.
При съемке илистых фракций почв на дифрактометре ДРОН-2
используют трубки с Си-анодом, излучение фильтруют Ni; напря-
303
жение на трубке 40 kV, сила тока 20 mA, скорость вращения об-
разца 1° в минуту, диапазон счета 400 или 1000 имп/с. Съемку
образцов ведут в области от 1 до 15° углов 8.
Обработка дифрактограмм. По таблицам Гиллера
находят величины межплоскостных расстояний, соответствующие
пикам на дифрактограммах, и после этого приступают к опреде-
лению минералогического состава образцов на основании полу-
ченных рентгенодифрактограмм, пользуясь табл. 29.
Изучение гумусовых кислот почв
методом рентгеновского анализа
Гумусовые кислоты не были получены в кристаллическом со-
стоянии, тем не менее возможно изучение их строения с помощью
метода рентгеновского анализа. Рентгеновский метод исследования
применим для изучения как кристаллических веществ, так и твер-
дых аморфных и квазикристаллических веществ, а также веществ
в других агрегатных состояниях.
Дифракционная картина может быть получена от конденсиро-
ванных аморфных веществ, от жидкостей и газов, ибо любое ве-
щество в своем строении обладает той или иной степенью поряд-
ка в отношении взаимного расположения частиц в нем. Степень
упорядоченности в строении вещества может быть различна, на-
чиная с веществ, имеющих лишь ближний порядок в своем строе-
нии, выражающийся в том, что расстояния между атомами одного
вида или молекулами одного вида чаще имеют одни значения,
чем другие, и кончая веществами, имеющими кристаллическое
строение, т. е. такими веществами, которые обладают строгим по-
рядком в отношении расположения в них атомов и молекул. Меж-
ду этими двумя крайними состояниями имеется множество ве-
ществ, обладающих различной степенью порядка в отношении рас-
положения частиц в них.
Дифракционная картина, получаемая от вещества с различ-
ной степенью упорядоченности, естественно, будет различной. За-
висимость интенсивности расстояния I дифрагируемых рентгенов-
ских лучей от длины волны излучения К и угла рассеяния рент-
геновских лучей 6, выраженная как функция I = f- -5-п®- (рис. 92),
для одноатомных газов имеет характер плавно нисходящей кри
вой. В случае молекулярных газов на этой кривой возникает уже
несколько размытых максимумов, их число, расположение и вы-
сота зависят от сложности структуры молекулы газа. Жидкости
и аморфные твердые вещества, в которых существует определен-
ная степень дальнего порядка (флуктуирующая статистическая'
упорядоченность размещения структурных элементов), дают кри-
вые, на которых количество максимумов и их резкость еще более
возрастают. Наконец, в случае поликристаллического тела рассея-
304
ние рентгеновского излучения имеет дискретный характер, макси-
мумы превращаются в резкие острые пики.
Как и при исследовании кристаллических веществ, дифракци-
онный эффект от аморфных тел и тел с той или иной степенью
дальнего порядка (дальний порядок — это наличие периодично-
сти в распределении вещества) может быть использован не толь-
ко для нахождения межплоскостных расстояний, но и для более
глубоких структурных исследований —• задачи чрезвычайно слож-
ной и трудоемкой, требующей специальных условий и методов
Рис. 92 Зависимость интенсивности рассеяния дифрагируемых рентге-
новских лучей от угла рассеяния и длины волны излучения:
А— для одноатомных газов (/) и молекулярных газов (2); Б — для
жидкостей и стекол (3) и монокристаллов (4)
съемки и большого математического аппарата для рентгенострук-
турных расчетов с применением вычисления рядов Фурье. Однако-
необходимо подчеркнуть принципиальную разницу между рентге-
ноструктурным анализом, который применяется для исследования
кристаллов, и диффракционными методами, которые используют-
ся для изучения аморфных веществ, со структурой, в определен-
ной степени упорядоченной, но несовершенной, а также для жидко-
стей и газов. Ориентационная неупорядоченность некристалличес-
кого строения позволяет получить из дифракционных данных лишь
картину строения, усредненную по всем возможным ориентациям.
В случае кристалла метод преобразований Фурье позволяет по-
лучить трехмерную систему расположения атомов, в случае не-
кристаллического вещества аналогичное преобразование дает воз-
можность получить сведения лишь о наборе различных (преобла-
дающих) межатомных расстояний; пространственное расположе-
ние атомов остается неизвестным.
Рентгеновский метод исследования был применен при исследо-
вании структуры многих органических веществ, например каучу-
ка, пластмасс, целлюлозы, протеинов, углеводов, жиров.
Органические вещества почв начали изучать рентгеновским ме-
тодом еще в 1930-е годы, но до сих пор в экспериментальной
практике преобладает лишь первый, наиболее легко осуществи-
мый этап исследования — изучение зависимости интенсивности
305
рассеяния дифрагируемых лучей от угла рассеяния с последую-
щим вычислением межплоскостных расстояний.
В 1935 г. И. Д. Седлецкий предпринял попытку рассчитать па-
раметры элементарной ячейки гуминовой кислоты, считая гуми-
новую кислоту веществом кристаллическим и отождествляя ее
структуру со структурой графита. В 1951 г. В. И. Касаточкин осу-
ществил попытку расчета параметров пакетов гуминовой кисло-
ты. В большинстве исследований на рентгенограммах гумусовых
кислот отмечают два диффузных максимума (гало) — дифрак-
ционный максимум, отвечающий периоду (/002 = 3,4 А и у-дифрак-
ционный максимум, отвечающий периоду 4,6—5,5 А, а также до-
полнительный очень слабовыраженный дифракционный максимум,
отвечающий периоду (/ю0=2,1 А. Межплоскостное расстояние
^002=3,4 А рассматривают как свидетельство упорядоченности се-
ток циклически полимеризованного углерода по оси с, период
(/юо = 2,1 А — как следствие порядка внутри этих сеток, а период
4,6—5,5 А истолковывается как указание на наличие ближнего по-
рядка в расположении боковых алифатических цепей. В настоя-
щее время большинство исследователей как в почвоведении, так
и в области химии углей и торфов, придерживаются того же тол-
кования дифракционных максимумов. Однако считается, что сет-
ки циклически полимеризованного углерода не имеют сходства с
графитовыми; более вероятна структура сеток нерегулярная, рых-
лая, периодически нарушаемая.
Хорошо прослеживается общая закономерность для гумусовых
кислот как почв, так и торфов и углей: чем меньше в молекуле гу-
мусовой кислоты доля периферической части, построенной из али-
фатических цепей, и чем больше доля «ядра» молекул, тем более
ярко проявляется дифракционный максимум, отвечающий периоду
</оо2=3,4 А, и ослабевает дифракционный максимум, отвечающий
периоду 4,6—5,5 А. Например, фульвокислоты имеют сильнее вы-
раженный дифракционный максимум, отвечающий периоду 4,6—
5,5 А, и слабый дифракционный максимум, отвечающий периоду
(/оо2=3,4 А, тогда как у гуминовых кислот значительно интенсив-
нее дифракционный максимум, отвечающий периоду (/оо2 = 3,4 А.
Характерно, что гуминовые кислоты черноземов имеют ярче вы-
раженный дифракционный максимум, отвечающий периоду d002 =
= 3,4 А, и слабее выраженный дифракционный максимум, отве-
чающий периоду 4,6—5,5 А, чем те же дифракционные максиму-
мы у гуминовых кислот подзолистого типа.
Первая попытка более сложного анализа структуры гумусо-
вых кислот почв путем построения кривых радиального распреде-
ления электронной плотности (для фульвокислот) и расчет меж-,
атомных расстояний в молекуле были сделаны Кодами и Шнитце-’
ром в 1967 г. Считая, что состав фульвокислоты можно выразитй
формулой С28Н23О19 и приняв молекулярную массу фульвокисло-
ты равной 670, а плотность ее 1,61 г/см3, Кодами и Шнитцер на/
основании расчета распределения электронной плотности в моле-.
306
куле пришли к следующему выводу. В фульвокислоте у любого
углеродного атома число ближайших соседей — углеродных ато-
мов на расстоянии 1,6 А (1-я координационная сфера) — должно
быть около двух, тогда как в графите и саже их три. Это приводит
к выводу о значительной доле нециклических углеродных атомов
в составе молекулы фульвокислоты. Вторая координационная сфе-
ра включает 6—7 углеродных атомов — вторых соседей на рас-
стоянии 2,9 А, тогда как в саже число их равно 9. Эти данные по-
зволили авторам сделать заключение о том, что углеродный скелет
фульвокйслот скорее является прерывистой сетью, чем непрерыв-
ным сетчатым листом графита.
Теоретических работ, посвященных расчету картины дифрак-
ционного эффекта гумусовых кислот, чрезвычайно мало. Это на-
правление делает лишь первые шаги.
Характеристика гумусовых кислот
рентгенодифрактометрическим методом
Сравнительная характеристика гумусовых кислот различного
происхождения может быть получена путем съемки рентгеноди-
фрактограмм препаратов гуминовых кислот и фульвокйслот, вы-
деленных из почв. Гуминовые кислоты и
фульвокислоты извлекают из образца почвы
и очищают, как это описано в специальных
руководствах (Орлов и др., 1969). Сухой
препарат осторожно измельчают раздавлива-
нием в агатовой ступке, насыпают в кювету,
заполняя ее полностью, и уплотняют. Кюве- J
ту делают из стекла, которое не дает диф-
ракционных максимумов при рентгеновской
съемке. Из пластинки этого стекла размером
18X24 мм изготавливают открытую кювету,
имеющую с трех сторон бортики высо-
той 1—1,5 мм и шириной 4 мм (рис. 93). Кю-~^&мм
1,5 мм
Рис. 93. Кювета для
съемки спектра гуму-
совых кислот рентге-
нодифрактометриче-
ским методом
1Ъмм
вету с препаратом устанавливают в держа-
тель образца гониометрического устрой-
ства рентгеноднфрактометра ДРОН-2, вклю-
чают прибор и ведут съемку в области от 1
до 25° углов 0. При съемке гумусовых кис-
лот на дифрактометре используют рентгенов-
ские трубки с Cu-анодом, излучение фильтру-
ют через фильтр из никелевой фольги, режим
порядок включения и работы ни нем описаны выше.
На рентгенодифрактограммах гумусовых кислот, записываемых
на диаграммной ленте, имеются диффузные дифракционные мак-
симумы (рис. 94), необходимо найти углы 0, которым соответству-
ют вершины дифракционных максимумов. Углы 0 отмечаются вер-
работы прибора и
307
тикальными штрихами на диаграммной ленте специальным уст-
ройством, вмонтированным в гониометр.
Зная угол 0 и материал анода в рентгеновской трубке, по
таблицам Гиллера находят межплоскостные расстояния d/n, выра-
женные в ангстремах. Относительные высоты дифракционных мак-
симумов на дифрактограмме и значения периодов, которым они
Рис 94 Рентгено-дифрактограмма гуминовой кислоты из гор. А] чернозема ти-
пичного мощного
отвечают, помогают судить о строении гумусовых кислот, о соот-
ношении в их молекулах доли периферических алифатических
структур и «ядерной» части.
ПРАВИЛА ТЕХНИКИ БЕЗОПАСНОСТИ
ПРИ РАБОТЕ С РЕНТГЕНОВСКИМИ УСТАНОВКАМИ
Допустимый уровень рентгеновского излучения в рентгенов-
ской лаборатории не должен превышать 100 мр в неделю, что не-
обходимо проверять дозиметрическими наблюдениями.
Все рентгеновские установки должны быть снабжены надеж-
ной защитой. На рентгенодифрактометрах, помимо заводской за-
щиты, рекомендуется устанавливать защиты из просвинцованной
резины или просвинцованного стекла вокруг всего гониометриче-
ского устройства. При работе с аппаратами для камерной съемки
следует оградить весь столик с трубкой и камерами специальны-
ми экранами из металла с вмонтированными листами свинца или
из просвинцованной резины. Юстировку прибора и установку ка-
мер следует производить при минимальных возможных напряже-
нии и силе тока. Необходимо следить за тем, чтобы все неисполь-
зованные в работе окошки рентгеновской трубки были закрыты
свинцовыми заглушками. Необходимо тщательно закрывать гиб-
кими свинцовыми пластинками места сочленения окошек рент-
геновских трубок и входных коллиматоров камер. Присутствие
308
посторонних лиц в рентгеновской лаборатории не допускается.
При включенных приборах оператор может находиться в лабо-
ратории только, если необходима смена образца или юстировка
прибора. Желательно выносить пульт управления рентгеновски-
ми установками в другое помещение (для тех установок, для ко-
торых это возможно).
ЛИТЕРАТУРА
Горбунов Н. И. Минералогия и физическая химия почв. М., «Наука», 1978.
Методы изучения минералогического состава и органического вещества почв.
Ашхабад, 1975.
Методы минералогического и микроморфологического изучения почв. М, «Нау-
ка», 1971.
Рентгеновские методы изучения и структура глинистых минералов. М, «Мир»,
1965.
Орлов Д. С., Гришина Л. А., Е р о ш и ч е в а Н. Л. Практикум по биохи-
мии гумуса. М, Изд-во Моск, ун-та, 1969.
Глава 9
ТЕРМИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ МИНЕРАЛЬНЫХ
И ОРГАНИЧЕСКИХ КОМПОНЕНТОВ ПОЧВ
Термические методы исследования, так же как и рентгенов-
ские, широко используются в почвоведении главным образом для
определения минералогического состава тонкодисперсных фрак-
ций — илистой и коллоидной. Значение исследования состава этих
фракций обсуждалось в предыдущей главе. Преимуществом тер-
мических методов является простота и низкая стоимость приборов
для проведения термического анализа, а также возможность при-
ложения метода к тонкокристаллическим, плохо окристаллизо-
ванным и аморфным веществам, часто входящим в состав тонко-
дисперсных фракций почв. Среди таких веществ следует упомя-
нуть минералы гидроокислов железа и аллофаны.
Недостатком термических методов исследования тонкодисперс-
ных фракций является невозможность однозначно диагностировать
индивидуальные глинистые минералы в полиминеральных образ-
цах, так как термические эффекты отдельных глинистых минера-
лов могут накладываться друг на друга, а положение и форма
этих эффектов могут меняться в зависимости от состава образца
и условий эксперимента.
Термические методы используются в почвоведении также для
количественного определения содержания таких компонентов почв
и отдельных фракций, как гиббсит и кварц, химического состава
некоторых карбонатных минералов и легкорастворимых солей.
В последние десятилетия термический метод широко использует-
ся для анализа почвенных органических и органо-минеральнц’х
соединений, -для изучения продуктов взаимодействия глинистых
минералов с органическими веществами специфической и неспе-
цифической природы.
Термический анализ основан на изучении фазовых превраще
ний, которые происходят при нагревании или охлаждении веще-
ства. Известно, что, фаза — это совокупность однородных частей
системы, одинаковых""пб термодинамическим свойствам и отгра-
310
ничейных от других частей поверхностью раздела. Понятие фазо-
вых превращений достаточно широко и включает переходы из од-
ного агрегатного состояния в другое (плавление, затвердение, ис-
парение и т. п.), химические превращения, а также более сложные
процессы, при которых химические превращения сопровождаются
изменением агрегатного состояния.
При проведении термического анализа проводят регистрацию
изменений, которые имеют место при фазовых превращениях, про-
исходящих в образце при нагревании или охлаждении. Эти изме-
нения касаются таких параметров образца, как его температура,
вес, объем, химический состав, структура.
СУЩНОСТЬ МЕТОДА
И ОСНОВНЫЕ УЗЛЫ ПРИБОРОЙ
ДЛЯ ТЕРМИЧЕСКОГО АНАЛИЗА
При работе с почвенными объектами чаще всего используют •
модификации термического анализа, связанные с регистрацией
изменений температуры (дифференциальный термический ана-
лиз — ДТА) или веса образца (термовесовой анализ — ТВ).
Дифференциальный термический анализ. Этот анализ заклю-
чается в регистрации разницы температур между исследуемым об-
разцом и образцом эталонного термоинертного вещества при на~
грёвании или охлаждении. Разница температур возникает за счет
того, что в эталонном веществе при нагревании или охлаждении
никаких реакций не происходит, а в испытуемом веществе проте-
кают физические переходы или химические реакции, которые со-
провождаются выделением или поглощением тепла. К ним отно-
сятся плавление, перестройка кристаллической решетки, кипение,
испарение, реакции окисления, восстановления, разрушения ре-
шетки, дегидратации, кристаллизации новых фаз из продуктов
разложения и т. п. Дегидратации, восстановление и некоторые у,-
реакции разложения, 'ffa'fc правило, являются эндотермическими,
т. е.Дфотекают'Д^ипго'Щениём-тепла, а кристаллизация, окисле-
ние и_ некоторые процессы разложения происходят с выделением
теп та т. е. сопровождаются экзотермическим эффектом.
Технически дифференциальный термический анализ выполняет-
ся следующим образом (рис. 95). Испытуемое и термоинертное
вещество помещают в одинаковые платиновые или корундовые
тигли, в которые вводят рабегчие спаи термопар для регистрации
изменений температуры. Термопары соединяются так, что возни-
кающие в лих то.уП идут навстречу друг другу. Такое устройство
гывается дифференциальной термопарой. Свободные концы тер-
мопар замкнутЩчёрнз"' чувствйтеяьНЫй гальванометр, который на-
зывается дифференциальным, так_как^он предназначен для реги-
страции разности температур между~испытуемы~м Образцом ТГэтш
лоном' Гермопара введенная в испытуемый образец, замкнута
параллельно еще на один гальванометр (пирометрический), пред-
назначенный для регистрации температуры образца. Тигли с ис-
следуемым веществом и эталоном с введенными в них спаями
термопар помещают в печь и нагревают в диапазоне температур
от 20 до 1000°. По мере увеличения температуры образца стрелка
(или световой сигнал)
Эталон
Рис. 95. Схема прибора для дифференциаль-
ного термического анализа:
ДГ — дифференциальный гальванометр, НГ —
нормальный гальванометр
пирометрического гальванометра отклоня-
ется от нулевого положе-
ния. По отклонению
стрелки (сигнала) мож-
но судить о температуре
Образец образца. Поскольку тер-
мопары в эталоне и об-
разце соединены так,
что возникающие в них
токи идут навстречу друг
другу, световой сигнал
дифференциального галь-
ванометра остается в ну-
левом положении, если
образец и эталон нагре-
ваются с одинаковой ско-
ростью. Исследуемые ме-
тодом термического ана-
лиза вещества, в том чис-
ле и компоненты почвы,
/относятся к термоактив-
/ным веществам. Это зна-
чит, что в процессе нагре-
вания они претерпевают
различные
связанные,
потерей воды и других
• компонентов из кристал-
лической решетки, с поли-
морфными превращения-
ми с разрушением кристаллической решетки и кристаллизацией
новых фаз из продуктов распада. Указанные изменения могуг
сопровождаться поглощением (эндотермические реакции) или вы-
делением (экзотермические реакции) тепла. Очевидно, что при эк-
зотермических реакциях образец будет нагреваться, с большей ско-
ростью, а при эндотермических~рёакциях ^^^с- меньшей скоростью,
чем эталонное вещество? Разница-в скоростях нагрева, соответст-
венно и в температурах между исследуемым веществом и эталоном,
регистрируется дифференциальным гальванометром: его световой ,
сигнал отклоняется в разные стороны при эндо- и экзотермических
реакциях. Регистрируя на фотобумаге или на самописце передви-
жение световых сигналов от зеркальных гальванометров — нор-
мального и дифференциального, — имеется возможность записать
изменения,
например, с
312
термограмму, на которой будут две кривые: кривая нагрева образ-
ца и дифференциальная термическая кривая (ДТА), которая фик-
сирует все эндо- и экзотермические реакции, происходящие в ис-
пытуемом веществе при нагревании в виде набора эндо- и экзо-
термических эффектов. Дифференциальная термопара переклю-
чается обычно таким образом, что эндотермическим и экзотерми-
ческим эффектам отвечает отклонение сигнала гальванометра со-
ответственно вниз и вверх от нулевой линии. Температуры эффек-
Рис 96. Термограммы солей для калибровки термопар:
1 — K2SO4, 2 — Na2SO4- ЮН2О
тов определяют по кривой нагрева образца с помощью специаль-
ного градуировочного графика. Для построения графика снимают
термограммы веществ с точно известными температурами эффек-
тов. Удобными веществами для этой цели являются K2SO4, на
кривой ДТА которого имеются два ярко выраженных эндоэффек-
та при 585 и 1060°, и Na2SO4-ЮН2О, дающий при нагревании эн-
доэффекты при 240 и 884°. Через вершины эффектов проводят
перпендикуляры к оси абсцисс до пересечения с кривой нагрева
образца. Затем через точки пересечения проводят горизонтальные
линии до пересечения с осью ординат (рис. 96). Отмечают на оси
ординат температуры каждого эндоэффекта, а расстояние между
соседними точками, соответствующими точно известным темпера-
турам эндоэффектов, делят на равные отрезки (через 50 или
100°), условно принимая, что в промежутках между эндоэффек-
тами образец каждый раз нагревался равномерно. Размеченная
таким образом ось ординат и является градуировочным графиком.
По ней изготовляется линейка с такой же разграфкой. Для опре-
деления температуры эффектов на кривых ДТА испытуемых ве-
ществ линейку прикладывают к оси ординат термограммы. Через
вершины эффектов проводят перпендикуляры к оси абсцисс до
пересечения с кривой нагрева образца. Через точки пересечения
проводят горизонтальные линии, до градуировочной линейки, по
313
которой и определяют, при какой температуре произошел тот или
иной эффект. Современные приборы для термического анализа
обычно не требуют изготовления градуировочного графика и ли-
нейки, так как калибровка термопар производится на заводе.
В приборе имеется специальное устройство, с помощью которого
на фотобумагу, непосредственно перед проведением термического
анализа, наносится штриховая сетка (шаблон), по которой и оп-
ределяют температуру термоэффектов.
Поскольку многие вещества, входящие в состав твердой фазы
почвы, характеризуются специфическим набором экзо-’и эндотер-
мических эффектов, термический метод позволяет диагностировать
эти вещества по кривым ДТА.
Термовесовой анализ. Современные приборы для термического
970°
92О"
Рис. 97. Термограмма кал.щита:
ДТА — кривая дифференциально-
го термического анализа, ТВ —
термовесовая кривая, ДТВ — кри-
вая изменения скорости потери
веса
анализа дают возможность регистрировать не только температу-
ру образца и разницу температур испытуемого образца и этало-
на, но и потерю веса образцом в
процессе нагревания. Этот вид
анализа называется термовесовым,
а кривая потери веса — термо-
весовой кривой (ТВ). В приборах
для термовесового анализа ти-
гель с испытуемым образцом
ДТА УкРеплен на коромысле весов, и
регистрация кривых ДТА и ТВ
осуществляется одновременно. 11е-
редвижение коромысла весов за
счет потери веса образцом при
нагревании регистрируется оптиче-
ской системой, которая посыла-
ет свой световой сигнал на ту же
фотобумагу или на ту же ленту
самописца, где регистрируются кри-
вые нагрева образца и ДТА.
По данным термовесоього анализа
Тд в сочетании с ДТА можно не
только диагностировать присутст-
вующие в образце вещества, но и
делать количественные сопоставле-
ния содержания того или иного
компонента, руководствуясь пло-
щадью пика на кривой ДТА и вели-
чиной потери веса в соответствую-
щей области температур.
На рис. 97 приведены результаты термического анализа каль-
цита. На кривой ДТА виден интенсивный! эндотермический эф-
фект при 920°, вызванный реакцией разложения кальцита на СаО .
и СО?. Поскольку эта реакция сопровождается потерей газооб- I
разной СО2, на кривой потере веса (ТВ) регистрируется заметный |
314
перегиб, а на кривой ДТВ — интенсивный пик в той же области
температур.
Дериватограф. Наиболее совершенными приборами для терми-
ческого анализа являются дериватографы (рис 98), на термо-
граммах которых одновременно регистрируются температура об-
разца, разность температур между образцом и эталоном, потеря
Рис 98 Обший вид дериватографа
1 — блок управления, 2 — печи, 3— вспоми ательный стол, 4 —
рукоятка барабана, 5 — регулятор скорости вращения бараба-
на, 6 — тумблер лампы штриховки, 7 — тумблер включения мо-
тора, 8 — вилка весов, 9— тумблер гальванометра 1В, 10—
тумблер нормального гальванометра, 11—тумблер дифферен-
циального гальванометра, 12— тумблер гальванометра ДТВ,
13 — регулятор чувствительности ДТА, 14 — регулятор чувстви-
тельности ДТВ, 15—регулятор чувствительности весов, 16—
общее включение всех i альванометров
веса и скорость потери веса Кроме того, многие дериватографы
снабжены приспособлением для регистрации и31Менения объема
образца при нагревании и для улавливания и последующего ана-
лиза газообразных продуктов, выделяющихся из образца при на-
гревании. Скорость потери веса регистрируется на дериватографе
следующим образом На свободном от образца коромысле весов
имеется металлический стержень, на конце которого находится
315
катушка с большим количеством витков проволоки. Катушка по-
мещена между двумя подковообразными магнитами, а концы про-
волоки замкнуты на зеркальный гальванометр (рис. 99). По мере
того, как образец теряет вес, металлический стержень вместе с
катушкой приходит в движение. При перемещении катушки в по-
ле постоянного магнита в ней! индуцируется ток, напряжение ко-
торого тем выше, чем больше скорость движения, т. е. чем больше
Рис 99. Схема устройства дериватографа:
1 — печь, 2 — тигель с образцом, 3 — тигель с эталонным веществом, 4 — тер-
мопары, 5 — керамические трубки для термопар, 6 — линза, 7 — щель, 8—лам-
пы подсветки гальванометров, 9—магнит, 10 — катушка, 11 — гальванометры:
DTA — дифференциальный, Т — нормальный, DTT — скорости потери веса;
12 — барабан с фотобумагой
скорость потери веса образцом. Величина напряжения тока ре-
гистрируется гальванометром, световой сигнал которого тоже по-
падает на фотобумагу. Записанная, таким образом, кривая скоро-
сти потери веса (ДТВ) благодаря высокой чувствительности дает
более детальную информацию о процессах, которые сопровожда-
ются потерей веса, чем просто термовесовая кривая.
Режим работы всех термических аппаратов, в том числе и де-
риватографов, подбирается таким образом, чтобы при сохранении
максимальной чувствительности записи все кривые полностью ук-
ладывались в пределах фотобумаги. Изменение чувствительности
316
достигается включением дополнительных сопротивлении в цепи
зеркальных гальванометров.
Подготовка образцов илистых фракций
к термическому анализу
Для получения хороших термических эффектов от глинистых
минералов необходимо удалить из образцов органическое вещест-
во, поскольку его сгорание приводит к появлению интенсивного
экзотермического эффекта и потере веса в той области темпера-
тур, где сосредоточены термические эффекты от многих глинис-
тых минералов (250—700°). Для удаления органического вещест-
ва навеску илистой фракции обрабатывают Н2О2 на водяной ба-
не при 50—60°. Для этого 2—3 г илистой фракции помещают в
фарфоровые чашки, закрывают их большими часовыми стеклами*
ставят на нагретую водяную баню и приливают в чашки 4—5 мл
10%-ного раствора Н2О2. При высоком содержании органического
вещества реакция его окисления может сопровождаться разбрыз-
гиванием находящейся в чашках суспензии. Когда разбрызгива-
ние прекратится, капельки суспензии смывают в чашку с часового'
стекла, с краев чашки и досуха выпаривают суспензию. Такую
обработку повторяют 2—3 раза, после чего 4—5 раз промывают
навеску дистиллированной водой, чтобы удалить водорастворимые
органические вещества, образовавшиеся при окислении гумуса.
После этого образец подсушивают на воздухе, слегка растирают
и выдерживают 4—5 дней в эксикаторе над насыщенным раство-
ром Ca(NO3)2. Присутствие насыщенного раствора Ca(NO3)2
обеспечивает в эксикаторе относительную влажность воздуха,
равную 55%. Эта операция необходима для того, чтобы привести
все исследованные образцы к одинаковым стандартным условиям
влажности, так как величина влажности воздуха, при которой на-
ходится образец перед анализом, оказывает большое влияние на
интенсивность низкотемпературного (20—200°) эндотермического
эффекта и на величину потери веса илистыми фракциями в этой
области температур.
Поскольку величина и форма низкотемпературных эндотер-
мических эффектов зависят от состава обменных катионов, насы-
щающих глинистые минералы, желательно использовать при тер-
мическом анализе образцы, насыщенные определенными катиона-
ми. Рекомендуется насыщать илистые фракции перед термичес-
ким анализом Са2+ или Mg2+.
Термические характеристики
основных групп илистых минералов
Минералы группы каолинита. Наиболее широко распростра-
ненными в почвах представителями этой группы минералов явля-
ются собственно каолинит и галлуазит.
317
Каолинит при нагревании дает характерный набор термиче-
ских эффектов. На кривой ДТА наблюдается слабый низкотемпе-
ратурный эндотермический эффект, обусловленный удалением
сорбированной воды (рис. 100, табл. 31). Поскольку каолинит, как
Рис 100 Термограммы некоторых
минералов, широко распространен-
ных в почвах
правило, бывает представлен
довольно крупными частицами и
неспособен к межпакетной ад-
сорбции воды, низкотемператур-
ный эндотермический эффект
имеет незначительную интенсив-
ность, соответствующая ему ве-
личина потери веса очень мала
и измеряется десятыми долями
процента от веса образца. При
500—600° происходит распад
кристаллической решетки као-
линита, сопровождающийся по-
терей гидроксильных групп. По-
теря гидроксильных групп при
нагревании называется реакци-
ей дегидроксилации Эта реак-
ция идет с поглощением тепла, и
на кривой ДТА ей соответствует
интенсивный эндотермический
эффект при 575—625°. Потеря
веса в этом температурном ин-
тервале за счет потери ОН-груп-
пы составляет около 10%. В ди-
апазоне температур 951)—1000°
происходит экзотермический ~ф-
фект. Этот эффект соответствует
реакции кристаллизации новых
фаз из продуктов распада кри-
сталлической решетки каолинита.
По кривым ДТА можно не толь-
ко диагностировать каолинит в смеси, но и определить степень
упорядоченности его кристаллической решетки. Чем больше упо-
рядочена кристаллическая решетка каолинита, тем выше (в пре
делах указанных диапазонов) температура эндотермического эф-
фекта, соответствующего реакции потери ОН-групп и экзотерми-
ческого эффекта кристаллизации, и тем больше интенсивность и
симметричность экзотермического пика. С возрастанием степени
упорядоченности решетки каолинита уменьшается интенсивность
низкотемпературного эндотермического эффекта, вызванного, уда-
лением адсорбированной воды, и уменьшается потеря веса в диа-
пазоне температур от 20 до 200°.
Полностью гидратированная разновидность галлуазита, имею-
щая формулу Al^OHJsISiiOn)]-4НгО, также характеризуется ег.е-
318
Таблица 31
Температура и характер термических эффектов для некоторых лайнера лов
Минерал Диапазон - температуры эффекта Характер эффекта
Каолинит 140—160° 575—625° 950-=ЛЮ02_ эндотермический слабый эндотермический сильный экзотермический средний
,, Галлуазит 1 СО сл Са5 О 4*. .ООО 1 1 1 СО Сл •— Оо СО О ООО 10 0 0 эндотермический сильный эндотермический сильный экзотермический средний
7 т> Гидрослюда 140—160° эндотермический слабый
— 350—700° 850—900° 900 1000° эндотермический средний эндотермический слабый экзотермический слабый эндотермический сильный (иногда двухвершинный) эндотермический слабый эндотермический слабый экзотермический слабый эндотермический слабый эндотермический средний эндотермический средний экзотермический слабый эндотермический средний
120—520*^
О7 -Монтмориллонит 650—750° 850—900° 900°
„ -ту Хлорит 140—160° 550—700° 780—820° 820 -840°
Гетит грубодисперсный 380—420° ,
Гетит тонкодисперсный 280—350° эндотермический слабый эндотермический средний
Лепидокрокит Гель гидроокиси железа свежеприготовленный ' 50—200° 280—350° 450—550° ' 100—200° 250—350— эндотермический слабый эндотермический средний экзотермический средний эндотермический средний экзотермический сильный эндотермический сильный эндотермический сильный
Гиббсит Кальцит OZ5-_35Q°_ -800—1000°
Магнезит -580— Б80°Д_ эндотермический сильный
Кварц " 573° эндотермический слабый
цифьческим набором эффектов на термических кривых. При на-
гревании до 140—160° теряется адсорбированная вода, находящая-
ся в межпакетных промежутках. Этот процесс требует затрат
тепла, и ему отвечает эндотермический эффект в низкотемпера-
турной области (см. рис. 100), и потеря веса достигает в этом диа-
пазоне температур 9—10% от веса образца. Реакция потери ОН-
групп и связанный с ней эндотермический эффект и потеря веса
происходят при 560—590°. При этом теряется до 10—11% от веса
образца. Экзотермический эффект, вызванный кристаллизацией
новых фаз из продуктов распада кристаллической решетки, на-
блюдается при 930—960°.
Минералы группы слюд и гидрослюд (иллитов). Минералы
группы слюд и гидрослюд сильно варьируют по химитескому со-
319
Ч
ставу, степени и составу изоморфных замещений в октаэдрических
и тетраэдрических позициях и по степени дисперсности. Поэтому
трудно выявить набор термических эффектов, который был бы
свойствен этой большой группе минералов в целом. Но некоторые
термические эффекты считаются типичными для термических кри-
вых тонкодисперсных слюд и иллитов.
На кривой ДТА имеется эндотермический—эффект в области
температур 20—200°, связанный с удалением адсорбированной
воды, интенсивность которого и соответствующая потеря веса за-
висят от степени дисперсности и гидратированности материала
(см. рис. 100). Второй эндотермический эффект наблюдается в
широком диапазоне температур от 350 до 700°. Появление второ-
го эндотермического эффекта связано с потерей ОН-группы из
кристаллической решетки. Величина потери веса за счет потери
группы ОН варьирует в зависимости от многих факторов, но в
среднем составляет 2—4% от веса образца. Третий эндотермиче-
ский эффект наблюдается при 850—900° и связан с полным разру-
шением кристаллической решетки. Кристаллизация новых фаз из
продуктов распада и соответствующий ей экзотермический эф-
фект наблюдаются при 900—1000°, причем эта температура тем
выше, чем больше магния содержится в кристаллической решет-
ке слюдистых минералов. Степень выраженности третьего эндо-
термического и экзотермического эффектов на термограммах воз-
растает с увеличением количества Mg и Fe, изоморфно замещаю-
щих AI в октаэдрических позициях.
Минералы группы монтмориллонита. Все представители этой
группы минералов дают на кривой ДТА интенсивный эндотерми-
ческий эффект в диапазоне температур 20—250°, связанный с уда-
лением воды, адсорбированной на поверхности частиц и в меж-
пакетных промежутках (см. рис. 100). Форма, температура и
размеры первого эндотермического пика и связанная с этой эндо-
термической реакцией потеря веса зависят от природы насыщаю-
щего катиона и влажности воздуха, в котором сохранялся обра-
зец перед термическим анализом. Образцы смектитов, насыщенных
Na+, К+ или Cs+, характеризуются одновершинной формой низко-
температурного эндотермического эффекта, максимум которого
соответствует 180—190°. Насыщение образца Li+ или каким-либо
двухвалентным катионом — Ва2+, Cr2+, Са2+, Mg2+ — приводит
к появлению двухвершинного эндоэффекта, причем первая вер-
шина соответствует температуре 160—180°, а вторая — 210—220°.
На термограммах образцов, насыщенных Li, лучше выражен вто-
рой высокотемпературный максимум, а на термограммах образ-
цов, насыщенных двухвалентными катионами, большую интенсив-
ность имеет первый из двух максимумов при температуре 160—
180°.
Величина потери веса, соответствующая потери адсорбиро-
ванной воды, составляет 11—16% для собственно монтмориллони-
тов и 6—9% для бейделлитов, если, конечно, образец перед тер-
320
мпческим анализом находится в стандартных условиях относи-
тельной влажности, равной 55% /
Второй и третий эндотермические эффекты7 на кривой ДТА
обусловлены соответственно потерей ОН-групп кристаллической
решетки и полным разрушением ее. При этом температура и ин-
тенсивность высокотемпературных эндотермических эффектов за-
висят от_хямич£Щштих1£хакащ1ЩШ^ щ_кристалличе-
ской решетке монтмориллонитовых минералов. Так, для собствен-
но монтмориллонитов с зарядом в октаэдрическом слое характер-
ны эндотермические эффекты при 700 и 900°, а бейделлиты, т. е.
минералы монтмориллонитовой группы с зарядом в тетраэдрах,
дают эндоэффекты при более низких температурах — 550 и 850°.
В том и другом случае второй эндотермический эффект переходит
непосредственно в слабый экзотермический пик-,-соответствующий
кристаллизации новых фаз из продуктов распада. Присутствие
больших количеств железа в кристаллической решетке смектитов
существенно понижает температуру реакции потери ОН-групп.
Наличие магния в октаэдрических позициях 'повышает температу-
ру и интенсивность эндотермического эффекта потери ОН-групп.
Потеря веса минералами монтмориллонитовой группы, вызван-
ная потерей ОН-групп, достигает 5—6%
Минералы группы хлоритов. 'Ота разнообразны по химическо-
му составу, поскольку в их кристаллических решетках возможны
изоморфные замещения в тетраэдрических и октаэдрических по-
зициях трехслойного пакета и в.октаэдрах однослойного пакета.
В связи с этим варьируют и термические характеристики хлори-
тов. Низкотемпературный эндотермический эффект выражен
очень слабо.
Магнезиальные и железистомагнезиальные триоктаэдрпческие
хлориты дают на кривых ДТА интенсивный эндотермический эф-
фект при 550—700°, вызванный потерей ОН-групп из однослойно-
го пакета, т. е. добавочного окта.эдрического слоя (см. гл. 8). Эта
реакция сопровождается потерей 8% веса образца. Второй, мё'нее
значительный эндотермический эффект наблюдается в диапазоне
температур от 780 до 820° и соответствует потере ОН-группы из
трехслойного пакета, что сопровождается потерей 3—4% веса об-
разца. Второй эйдотермический эффект переходит непосредствен-
но в акзотерлдческий пик при 820—8402,-Обусловленный кристал-
лизацией новых фазПГнтенсивность этого пика тем выше, чем
больше содержание в хлоритах магния. Железистые триоктаэдри-
ческне хлориты дают несколько йную термическую картину. На
кривых ДТА этих минералов имеется экзотермический эффект
при 300—500°, соответствующий частичному окислению двухва-
лентного железа. Этот экзотермический эффект переходит непо-
средственно в эндотермический пик при 500—650°, вызванный
разрушением решетки и потерей ОН-групп.
П Зак 284
321
Термические характеристики
некоторых неглинистых
компонентов почв
Минералы гидроокисей Fe и А1. Минералы гидроокиси железа
представлены в почвах чаще всего гетитом и лепидокрокитом,
имеющими одинаковую формулу FeOOH, но несколько разное рас-
положение атомов в кристаллической решетке. На кривой ДТА
грубодисперсного хорошо окристаллизованного гетита имеется
заметный эндотермический эффект при 380—420°, соответствую-
щий дегидратации и переходу в а — Fe2O3. В почвах, особенно в
илистых фракциях, чаще встречается тонкодисперсная скрыто-
кристаллическая разновидность гетита, которая дает небольшой
эндотермический эффект в диапазоне температур 50—200°, вы-
званный потерей адсорбционной воды, и эндотермический эффект
при 280—350°, соответствующий потере гидроксильных групп из
кристаллической решетки. При этом теряется до 8,5% веса образ-
ца. Лепидокрокит дает термическую картину, сходную с тонко-
дисперсным скрытокристаллическим гетитом, но на кривой ДТА
этого минерала иногда наблюдается экзотермический эффект в
области 450—550°, связанный с полиморфными превращениями.
Свежеприготовленный гель гидроокиси железа дает интенсив-
ный эндотермический эффект обезвоживания при температуре
около 100°, который непосредственно переходит в острый интен-
сивный экзотермический пик, соответствующий реакции кристал-
лизации.
Минералы гидроокиси А1 в почвенных объектах бывают чаще
всего представлены гиббситом А1(ОН)3. На кривой ДТА гиббсит
дает характерный острый эндотермический эффект при 275—
350°, соответствующий потере ОН-групп. Этот процесс сопровож-
дается потерей до 35% веса образца.
Карбонаты. В почвенных объектах чаще всего они представ-
лены кальцитом, магнезитом и доломитом. Кальцит СаСО3 на
кривой ДТА дает интенсивный^ эндотермический эффект, темпе-
ратура которого варьирует от 800 до 1000° в зависимости от чис-
тоты образца. Этот эффект вызван разложением кальцита в соот-
ветствии с реакцией: СаСО3=СаО + СО2. Магнезит MgCO3 дает
эндотермический эффект при 580—680°, связанный с разложением
минерала на MgO и СО2. На кривой ДТА доломита наблюдаются
два эндотермических эффекта при 720—870° и 870—1000°, обус-
ловленных разложением карбонатов Са и Mg соответственно.
Поскольку при разложении карбонатов выделяется газообраз-
ная СО2, этот процесс сопровождается потерей 30—50% веса
образца.
Кварц. На кривой ДТА он дает отчетливый эндотермический
эффект при 573°, вызванный полиморфным превращением а-квар-
ца в р-кварц.
322
Факторы, влияющие на величину,
форму и температуру эффектов
Величина, форма и температура термических эффектов зави-
сят от условий эксперимента. Наибольшее влияние оказывают
скорость нагрева печи, расположение и чувствительность термо-
пар, масса и некоторые другие характеристики самого образца.
Увеличение скорости нагрева
печи приводит к _ возрастанию
интенсивности и температуры
термических эффектов.
С увеличением степени дис-
персности и с ухудшением окрис-
таллизованности образца снижа-
ются температуры и уменьшается
площадь многих термических эф-
фектов на кривых ДТА. Площадь
эффектов возрастает с увеличе-
нием массы образца. Температу-
ры термических эффектов мине-
ралов могут изменяться в зависи-
мости о» присутствия в образце
других компонентов
Пример расшифровки термограмм,
илистых фракций почв
На рис. 101 даны кривые
ДТА и ДТВ насыщенной Са2+
илистой фракции, выделенной из
горизонта ВС желтоземной почвы
андезитов. Ниже представлены ве.
Рис 101. Термограммы насыщенной
Са2+ илистой фракции, выделенной
из гор. ВС желтозема на древней
коре выветривания андезитов:
/ — кривая ДТА, 2 —кривая ДТВ
на древней коре выветривания
ичины потери веса по темпера-
турным интервалам для того же
вании кривой потери веса (ТВ).
образца, рассчитанные на осно-
20—250° 250—400° 400—1000° 2
6,0 1,2 8,0 15,2
На кривой ДТА хорошо виден интенсивный эндотермический
эффект при 16Ф. Пользуясь рис. 100 и табл. 31, находим, что этот
эффект может принадлежать либо галлуазиту, либо монтморилло-
ниту. Некоторые другие минералы (каолинит, гидрослюда, хло-
рит, тонкодисперсный гетит) тоже дают при нагревании эндотер-
мический эффект в той же температурной области, но значительно
меньшейинтенсивности. Монтмориллонита в исследуемом образ-
це нет. Это подтверждается отсутствием высокотемпературных
эндотермических эффектов при 650—750° и 850—900°. Следова-
тельно, можно предполагать, что образец содержит галлуазит.
Это подтверждается и другими, присущими галлуазиту эффекта-
ми —• интенсивным эндотермическим при 580° и экзоэффектом
11*
323
средней интенсивности при 930°. Учитывая, что других слоистых
силикатов в исследуемом образце не обнаружено и что галлуази-
товые минералы характеризуются величинами потери веса 9—10%
в низкотемпературном интервале и 10—11% в интервале темпера-
тур 400—1000°, можно ориентировочно оценить содержание гал-
луазита в образце.
Пользуясь данными потери веса в интервалах температур 20—
250° и 400—1000°, равными соответственно 6 и 8%, приблизи-
тельно определяем содержание галлуазита в образце ~80%-
Кроме указанных эффектов галлуазита на кривых ДТА и ДТВ
имеется' слабый эндотермический эффект при 300°, который, по
всей вероятности, принадлежит одному из минералов гидрооки-
сей железа — плохо окристаллизованному гетиту или лепидокро-
киту. Их содержание можно приблизительно определить, исходя
из величины потери веса образца в области температур 250—-
400°, равной 1,2%, и учитывая, что для плохо окристаллизованного
гетита оно составляет 8—8,5% от веса образца. Содержание гети-
та составляет ~14—-15%.
Таким образом, исследованный образец илистой фракции со-
стоит из галлуазита и плохо окристаллизованного гетита.
Задача: определить минералогический состав илистой фрак-
ции термическим методом на дериватографе *. Приступая к вы-
полнению термического анализа на дериватографе, прежде всего
включают тумблер на задней стенке блока управления (см.
рис. 98). После этого заряжают барабан фотобумагой. Для этого
его извлекают из вспомогательного стола и в фотокомнате при
красном свете вставляют в него фотобумагу размером 430Х
Х230 мм, после чего помещают барабан на место. Специальной
рукояткой устанавливают барабан в исходное (0) положение. Пос-
ле этого наносят на бумагу штриховую сетку, по которой после
съемки определяется температура эффектов. Сетка наносится по-
следовательным переключением скорости вращения барабана 5
на 6 мин и включением лампы штриховки и мотора барабана (со-
ответственно рукоятки 6 и 7 на рис. 98). После нанесения сетки
приступают к взвешиванию образца. Перед этим необходимо по-
чистить и взвесить пустой тигель. Платиновый или корундовый
тигель ставят на конец фарфоровой трубки — держателя термо-
пары; с помощью противовеса в форме полой гири с дробью
добиваются свободного положения трубки термопары в отверстии
основания печи (чтобы она не касалась стенок) и накрывают тиг-
ли с термопарами кварцевым стаканом. Последний необходим для
равномерного нагрева обеих термопар. Включают гальванометр
ТВ, устанавливают требуемую чувствительность. Затем при помо-
щи дроби, помещаемой в полую гирю (грубая доводка), и вил*,
кой на передней панели весов (тонкая доводка), устанавливают
нулевое положение светового зайчика гальванометра ТВ. Ука-М
занную операцию взвешивания необходимо проводить при вклю-я
* Раздел написан М. И. Гусевой. »
324
ченных весах. Затем илистую фракцию, подготовленную, как ука-
зано выше, помещают в тигель, а в другой тигель насыпают
термоинертное вещество, в качестве которого могут быть исполь-
зованы прокаленные окись алюминия или окись магния. Объем
окиси алюминия примерно равняется объему испытуемого ве-
щества.
Илистую фракцию почвы удобно анализировать в тиглях сред-
него размера, работая с навеской 500 и 1000 мг. Инертное вещест-
во и образец плотно и равномерно утрамбовываются стеклянной
палочкой. Тигель ставят на конец фарфоровой трубки (термопа-
ры), заполненный илистой фракцией. При помощи механизма ав-
томатической накладки гирь на плечо весов устанавливается груз,
соответствующий необходимой навеске, и засыпкой вещества в
тигель зайчик гальванометра ТВ доводят до нулевого положения.
После выполнения указанных операций приступают к съемке тер-
мограммы. Для этого последовательно включают гальванометры
(нормальный, дифференциальный и ДТВ, соответственно тумбле-
рами 9, 10, 11, 12-, рис. 98), весы и систему, обеспечивающую за-
пись кривой ДТВ (соответствующие рукоятки находятся на пра-
вой стенке прибора), и мотор барабана (тумблер 7). Съемка ве-
дется в режиме: чувствительность ДТА—1/5, ДТВ—1/3, весов—•
200 мг (чувствительности устанавливают рукоятками соответст-
венно 13, 14, 15 на рис. 98). После окончания съемки, которая
длится 100 мин, фотобумагу извлекают в фотокомнате при крас-
ном свете из барабана, проявляют, закрепляют, промывают в те-
чение 10—15 мин в проточной воде, после чего высушивают и при-
ступают к расшифровке, пользуясь табл. 31 и рис. 100.
Расчет энергии
активации отдельных реакций
при термической деструкции
органического вещества почвы *
Энергия активации твердых тел, в том числе почвы и ее ком-
понентов, — сложная величина, не имеющая точного определе-
ния с позиций общей физики, как, например, для газовых реак-
ций, ибо процесс термической деструкции обсуждаемого объекта
слагается из большого числа отдельных реакций, которые проте-
кают в динамике при близких или даже одних и тех же темпера-
турах. В общем случае можно считать, что энергия активации
определяет суммарные энергетические связи, которые разруша-
ются при пиролизе почвы или ее компонентов. Значение энергии
активации выражают количеством тепла, которое поглощается или
выделяется анализируемым веществом в ходе термодеструкции.
Показатель энергии активации следует рассматривать как услов-
* Этот раздел написан старшим научным сотрудником Почвенного института
имени В. В. Докучаева И. С. Степановым.
325
ную величину, которая позволяет дать сравнительную оценку энер-
гетических уровней сложных эндо- и экзотермических реакций.
Этот показатель можно использовать при характеристике анали-
зируемого объекта только в том случае, если для расчета приме-
няют один и тот же способ, а техника подготовки и проведения
термоанализа являются идентичными.
Согласно теории кинетики пиролиза, разработанной Ван Кре-
веленом с соавторами для углей, эндо- и экзотермические реакции
могут быть описаны уравнениями:
— = К-с", К = k0-e —,
dt ° frT
где с — концентрация анализируемого вещества, t — время реак-
ции, п — порядок реакции, К — константа скорости реакции, k0 —
предэкспоненциальный фактор, R — газовая постоянная, Т — аб-
солютная температура (по шкале Кельвина), А—температурный
фактор скорости.
В основном решение задачи сводится к нахождению значения
А по экспериментальным данным, согласно формуле
lg/1 = \gTm-Ig^- - 0,37-[2kг
* m 21 J
где Tm — температура в максимуме термоэффекта, АТ — диапа-
зон температур термоэффекта. В уравнении четвертый член опус-
кают при Тт : А больше 0,2.
Значение энергии активации рассчитывают по формуле
Е = R- А = 1,986 А кал/моль.
Расчет энергии активации. Чтобы найти значение
А, необходимо получить дериватограмму анализируемого веще-
ства, на которой записаны кривые ДТВ и Т.
В качестве примера рассмотрим экспериментальные данные
по пиролизу органических веществ, заимствованные из работы
Тарнера, Шнитцера (рис. 102). На оси ординат отложена шкала
потери веса вещества (мг/градус). Ось абсцисс — базисная, или
нулевая, линия теоретической кривой ДТВ. На ней для удобства
отложены значения температуры в градусах шкалы Кельвина,
снятые с экспериментальной кривой Т.
Для решения задачи необходимо определить максимумы тер-
моэффектов и их температурные диапазоны. На эксперименталь-
ной кривой ДТВ четко выражены только два термоэффекта: пер-
вый и четвертый, имеющие соответственно максимумы 550 и 795 К.
Два других термоэффекта проявляются в виде уступов. Чтобы оп-
ределить параметры этих эффектов, необходимо уточнить их кон-
фигурацию. Теоретические кривые ДТВ второго и третьего эффек-
тов проводят при соблюдении следующих условий.
Точка пересечения теоретических кривых двух эффектов долж-
на находиться на половине высоты, заключенной между базисной
326
линией и экспериментальной кривой. Точка максимума эффекта
теоретической кривой должна располагаться выше эксперимен-
тальной на отрезок такой длины, который равен расстоянию от
базисной линии до вновь проведенной теоретической кривой, про-
ходящей через площадь данного эффекта. Боковые ветви теоре-
тической кривой эффекта следует проводить близкими к зеркаль-
но подобным.
Рис. 102. ДТВ— кривая органических веществ, экстрагиро-
ванных из гор Ао подзола супесчаного 0,5 н. раствором
NaOH в отношении 10: 1 (Канада).
Сплошная линия — экспериментальная кривая; пунктирная —
теоретическая кривая; /, II, III, IV — номера термоэффектов;
Та+Тв=Т — температурный диапазон термоэффекта
Выполнив указанные требования, проводят теоретические кри-
вые ДТВ второго и третьего эффектов так, как показано на ри-
сунке. Их максимумы соответственно равны 620 и 685 К.
Следующий этап в решении задачи — определение значений
АТ, т. е. температурных диапазонов термоэффектов. За темпера-
турный диапазон термоэффекта принимают разность температур,
соответствующую полуширине контура эффекта. В данном приме-
ре АТ= Та+Ть = 60°+45°= 105°. Имея данные по значению Тт и
АТ, рассчитываем величину температурного фактора скорости ре-
акции А по формуле. Следует отметить, что при TmjA меньше
0,2, величину А рассчитывают дважды: сначала приближенно,
опуская последний член уравнения, а затем уточняя его, подста-
вив в этот член уравнения приближенное значение А.
Для первого термоэффекта имеем
lg4 = lg550 — 1g— । 0,37=3,8297.
327
г
Тогда Д = 6755 (приближенное значение). Четвертый член урав-
нения равен (550:6755)3/2=0,081423/2=0,0232. Отсюда 1g А =
=3,8297—0,0232 = 3,8065. Уточненное Л = 6404. Следовательно,
Е— 1,986-6404= 12718 кал/моль= 12,7 ккал/моль.
Таким же путем рассчитываем значения А и Е для трех других
термоэффектов, наблюдаемых на экспериментальной ДТВ-кривой
(табл. 32).
Обсуждение результатов. Величина энергии актива-
ции указывает на характер структуры анализируемого объекта.
Например, энергия активации
физической десорбции воды
3—5 ккал/моль, энергия акти-
Таблица 32
Значения энергии активации (Е)
Ke термо- эффекта rm, к AT A E, ккал/моль
I 550 105 6 404 12,7
II 620 45 19 770 39,3
III 685 60 18 030 35,8
IV 795 70 20 810 41,3
меныпе энергии активации дру:
вации при пиролизе лигни-
на — 27, целлюлозы и каль-
цита •— 40, каолинита —>
41,8, циклогексана — 50,0,
бентонита — 78,0, аромати-
ческих структур — около
200 ккал/моль.
Энергия активации перво-
го эффекта примерно в 3 раза
термоэффектов (табл. 32). Это
позволяет заключить, что при низких температурах разрушаются
периферические цепочки и боковые радикалы исследованного ор-
ганического вещества, а при высоких (II—IV эффекты)—цикличе-
ские группировки, в том числе ароматические, гетероциклы и др.
Таким образом, энергия активации четырех термоэффектов ис-
следованного органического вещества подзола найдена соответ-
ственно равной 12,7; 39,3; 35,8 и 41,3 ккал/моль, суммарная энер-
гия активации — 129,6 ккал/моль. Исследованное органическое
вещество имеет сложный характер строения. Оно включает пери-
ферические цепочки и радикалы, а также циклические группиров-
ки, которые гетерогенны по своему составу и составляют боль-
шую часть исследованного препарата.
ЛИТЕРАТУРА
Дубин В. Н. Термовесовая характеристика и кинетические параметры термо-
деструкции гумусовых кислот основных типов почв Молдавии.— «Почвове-
дение», 1970, № 9.
Методы изучения минералогического состава и органического вещества почв.
Ашхабад, 1975.
Термический анализ минералов и горных пород. Л, «Недра», 1974.
Уэндландт У. Термические методы анализа. М., «Мир», 1978
Turner R С. and S с h и i t z е г М. Thermogravimetry of the organic matter
of a Podzol — «Soil Science», 1962, 93, N 4, p. 225—232.
Глава 10
ИССЛЕДОВАНИЕ НЕКОТОРБ1Х
КОМПОНЕНТОВ ПОЧВ
электронно-микроскопическим
МЕТОДОМ
НАЗНАЧЕНИЕ И ВОЗМОЖНОСТИ
МЕТОДА В ПОЧВОВЕДЕНИИ
Электронно-микроскопический метод применяется в почвоведе-
нии для изучения объектов, линейные размеры которых столь ма-
лы, что не позволяют наблюдать их в световом микроскопе. Это
главным образом минеральные и органические тонкодисперсные
фракции почв п частично микробиологическое население почв.
Преимущество этого метода перед другими в том, что он позво-
ляет получить изображение объекта при высоком увеличении, до-
статочно хорошо «соответствующее» объекту, понятное наблюда-
телю и не требующее для своего осмысливания дальнейших преоб-
разований или математического анализа. В современных элек-
тронных микроскопах существует также возможность наблюде-
ния электронограмм, которые дают большую информацию об объ-
ектах, чем реальное их изображение, но для осмысливания элек-
тронограммы необходимо уже осуществлять дальнейшие преобра-
зования полученных результатов.
Электронно-микроскопический метод без привлечения других
методов позволяет решить лишь ограниченный круг вопросов. Ис-
следования бывают особенно успешны при разумном сочетании
этого метода с другими. Так, при исследовании минералогиче-
ского состава тонкодисперсной фракции почв электронно-микро-
скопический метод позволяет идентифицировать минералы груп-
пы каолинита, если они не сильно разрушены, в противном случае,
а также при идентификации других минералов необходимо сопо-
ставлять данные электронно-микроскопические с данными рент-
геновского, термического и химического анализов. При исследова-
нии органического вещества почвы интересно не только наблю-
дать формы, образуемые органическим веществом, но и, применяя
различные химические обработки, изучить различное поведение
молекул гумусовых кислот в условиях кислой, нейтральной, ще-
лочной сред и их способность ассоциировать под влиянием катио-
329
нов. Необходимо и тут данные электронно-микроскопического ана-
лиза сопоставлять с данными химического, рентгеновского, гель-
хроматографического анализов, данными ПК-спектров. Такой ком-
плексный подход дает не только сумму сведений, но и приводит
к качественному росту информации.
I
ОСНОВЫ ТЕОРИИ МЕТОДА
Важнейшей характеристикой микроскопа является его предель-
ное разрешение — наименьшее расстояние между двумя близле-
жащими точками, при котором эти точки еще воспринимаются
глазом наблюдателя раздельно. Изображение точек, расположен-
ных на меньшем расстоянии, воспринимается глазом как одна
Рис. 1U3. Кривая распределения ин-
тенсивности дифракционной картины
от двух точечных источников света:
1—от 1-го точечного источника све-
та, 2 — от 2-го точечного источника
света, 3 — суммарное распределение
интенсивности, а — расстояние меж-
ду центральным максимумом и пер-
вым минимумом (в масштабах объ-
екта)
изображениях кружки
рассеяния
точка. Чем меньше это крити-
ческое расстояние, тем выше раз-
решающая способность микро-
скопа. При формировании изо-
бражения как в световом, так и
в электронном микроскопах воз-
никает явление дифракции как
на краях изучаемого объекта,
так и на краях диафрагмы объ-
ективной линзы, это приводит к
потере абсолютной резкости те-
ней и ограничивает разрешение
микроскопа. Вследствие дифрак-
ции, обусловленной волновой
природой световых и электрон-
ных лучей, изображение любой
точки объекта выглядит как кру-
жок, окаймленный тонкими коль-
цами— диск Эри. Если две
точки располагаются очень близ-
ко друг к другу, то в их
перекрываются и, наконец,
при весьма малом удалении друг от друга две различные точки
объекта вообще невозможно идентифицировать как раздельные.
Суммарное распределение интенсивностей дифракционных колец
от двух точечных источников света представлено на рис. 103. На-
блюдать раздельно эти две точки оказывается возможным, если
максимум дифракционной картины изображения одной из них на-
ходится по отношению к максимуму дифракционной картины со-
седней точки на расстоянии не ближе чем до первого минимума
дифракционной картины. Расстояние (d) между центральным мак-
симумом и первым минимумом дифракционной картины равно
0,61Х
и sin а
330
'i
где X — длина волны; п — показатель преломления среды, находя-
щейся между линзой и объектом; а — апертурный угол линзы-
объектива, т. е. половина угла 6, образованного линиями, соеди-
няющими края линзы, или ограничивающей ее апертурной диаф-
рагмы, с точкой объекта (рис. 104).
Если пренебречь интенсивностью дифракционной картины вне
радиуса d, то можно d представить как радиус кружка рассея-
ния вследствие дифракции. Из уравнения следует, что наиболь-
шее разрешение, т. е. наименьшее значение d, может быть достиг-
нуто при минимальной длине волны, в то время как значения п и
Рис. 104. Ограничение апертурного угла линзой (а) или диафрагмой (6):
/—точка на объекте, 2 — линза, 3 — апертурная диафрагма
а должны быть по возможности больше. В воздухе или вакууме
п=1, при а, близком к 90°, sina=l, тогда разрешение равно при-
мерно половине длины волны. Единственным ресурсом в этих
условиях остается длина волны. Длины волн в видимой области
спектра находятся в пределах 4000—7000 А. Следовательно, наи-
лучшее разрешение, которого можно достигнуть с помощью све-
тового микроскопа, равно приблизительно 2000 А. Это означает
практическую невозможность исследования с помощью световой
микроскопии макромолекул, клеточных структур, тонкодисперс-
ных минеральных и органических частиц, линейные размеры кото-
рых менее длины волны видимого света. Увеличить разрешающую
способность микроскопов позволило применение электронной оп-
тики.
При напряжениях, используемых в современных электронных
микроскопах просвечивающего типа и равных 60—100 кВ, длина
волны движущегося электрона приблизительно равна 0,04—0,05 А.
Поскольку длина волны электронного пучка значительно меньше
длин волн в видимой области спектра, то и размеры диска Эри
будут значительно меньше, а разрешающая способность электрон-
ного микроскопа значительно выше. Теоретически поток электро-
нов с длиной волны 0,04—0,05 А мог бы обеспечить разрешение
электронного микроскопа порядка 0,025—0,03 А. Однако такое
предельное разрешение, приблизительно равное половине длины
331
волны электронного потока, не может быть достигнуто, так как в
электронных микроскопах используются очень малые апертурные
углы. Дело в том, что при формировании изображения в электрон-
ном микроскопе возникают явления аберраций и дифракции. Сфе-
рическая аберрация возникает вследствие того, что участки маг-
нитного поля вблизи оси линзы и удаленные от нее имеют различ-
ную преломляющую способность. В результате фокус получается
расплывчатым, изображение смазывается, снижается разрешаю-
щая способность микроскопа. Влияние сферической аберрации
можно свести к минимуму, если использовать узкую центральную
часть пучка электронов, проходящих через линзу. С этой целью в
канал линзы-объектива помещается металлический диск с неболь-
шим отверстием — апертурная диафрагма. Однако на краях дис-
ка возникает явление дифракции, которое также снижает чет-
кость изображения, ограничивает разрешающую способность мик-
роскопа, и чем меньше отверстие диска, тем меньше сферическая
аберрация, но тем больше дифракция. Компромиссное решение,
направленное на снижение сферической аберрации и уменьшение
дифракции, достигается применением апертурных диафрагм с
диаметром отверстия 25—50 мкм. В этом случае апертурный угол
равен примерно 36'. Кроме того, на разрешение электронного мик-
роскопа влияет множество других причин, как, например, трудно-
сти изготовления деталей микроскопа с необходимой точностью,
нестабильность высокого напряжения и тока в обмотках линз. Раз-
решение зависит не только от прибора, но и от физических свойств
исследуемого объекта Наибольшее разрешение — 2 А удается
получить для кристаллических препаратов, для биологических
объектов предельное разрешение примерно равно 10—20 А.
Схема конструкции
электронного микроскопа
Электронный микроскоп состоит из 3 основных узлов: колон-
ны микроскопа с электронной пушкой, электромагнитными линза-
ми, экраном и фотоустройством; блока питания и вакуумной си-
стемы. На рис. 105 изображен внешний вид электронного микро-
скопа. Исследуемый образец, помещенный на специальную сеточ-
ку, вставляют в объектодержатель и помещают в камеру объекта
1, в которой затем создают вакуум. В окошко камеры наблюде-
ния 2 на люминесцентном экране, который помещен в камере,
можно видеть изображение объекта. Объект рассматривают в би-
нокуляр 3. Ручками 4 можно перемещать сеточку с объектом в
горизонтальном направлении и искать наиболее интересные об-
ласти. На панели микроскопа 5 помещены рукоятки, с помощью
которых можно центрировать пучок электронов, отрегулировать
фокус, освещение объекта, изменить увеличение, сфотографиро-
вать объект. Камера с фотопластинками 6 помещается под экра-
ном.
332
Колонна микроскопа представляет собой цилиндр, в котором
поддерживается высокий вакуум и где последовательно располо-
жены катод (источник электронов) — вольфрамовая нить, анод —
металлическая пластина с отвер-
стием посередине, ряд магнит-
ных линз, люминесцирующий эк-
ран и фотографическое устрой-
ство (рис 106)
Для получения электронов
используется явление термоэлек-
тронной эмиссии V-образная
тонкая вольфрамовая проволока
4, нагреваемая переменным то-
ком до температуры 2000°, ис-
пускает электроны Между като-
дом 4 и анодом 5 приложено вы-
сокое напряжение в несколько
десятков тысяч вольт — уско-
ряющее напряжение Ускоренные
этим напряжением электроны
устремляются к аноду и через
отверстие анода — центральную
апертурную диафрагму — про-
ходят вниз по колонне микро-
скопа Далее электронный пучок
попадает в магнитное поле лин-
зы конденсора 8, которая су-
жает и фокусирует пучок на ис-
следуемом объекте Пучок элек-
тронов проходит через исследуе-
мый объект 9, как бы «просвечи-
вая» его Проходя через объект,
пучок электронов взаимодейст-
вует с атомами вещества; при
этом взаимодействии электроны
отклоняются от первоначальных
траекторий, рассеиваются Чем
большей рассеивающей способ-
ностью обладает тот или иной
участок, тем более темным будет
его изображение на экране, так
Рис 105 Электронный микроскоп
JEM-200
1 — камера объекта, 2 — окно каме-
ры наблюдения, 3 — бинокуляр, 4 —
ручки для перемещения объекта, 5 —
панель микроскопа с рукоятками ре-
гулировок, 6 — фотокамера
как в этом случае лишь небольшое число электронов достигает
экрана Участок объекта, слабо рассеивающий электроны, будет,
напротив, выглядеть на экране светлым. Если объект рассеивает
менее 5% электронного луча, его нельзя увидеть на экране Про-
шедшие через объект и апертурную диафрагму электроны фоку-
сируются второй магнитной линзой объективом 10 Линза-объек-
тив формирует первое увеличенное изображение 11. Его можно
333
наблюдать в тех микроскопах, где есть «промежуточный» экрат^-
Качество изображения, формируемого линзой-объективом, опреде-
ляет качество окончательного изображения на экране. Изображе-
ние, сформированное линзой-объективом, далее увеличивается
третьей магнитной линзой-проектором
12 и проектируется на экран 13. Ёс-
--------------------—ли убрать экран, то изображение по-
Рис. 106. Схематический раз-
рез колонны простейшего элек-
тронного микроскопа:
1 — к вакуумному насосу, 2 —
вакуумная трубка, 3 — к высо-
ковольтному источнику пита-
ния нити накала, 4 — катод,
5 — анод, 6 — заземление, 7 —
схематически изображенный
ход электронного луча (точеч-
ная линия), 8 — линза-конден-
сор, 9 — объект, 10 — линза-
объектив, 11—увеличенное
изображение объекта, 12 —
линза проектор, 13 — люми-
несцентный экран, 14 — фото-
графическая пластинка, 15 —
к источнику питания линз
/У
падает на фотопластинку 14, распо-
ложенную под экраном, и может быть
сфотографировано. В современных
микроскопах имеются две линзы кон-
денсора и две линзы проектора.
Люминесцентный экран использу-
ется в электронном микроскопе для
того, чтобы сделать видимым элек-
тронное изображение. Под влиянием
бомбардировки электронными лучами
вещество, которым покрыт экран, из-
лучает видимый свет. Электроны, рас-
сеянные некоторыми атомами объек-
та и вышедшие из состава электрон-
ного луча, не достигают экрана, и эти
места, не подвергшиеся бомбардиров-
ке электронами, остаются темными.
Разрешение люминесцентного экрана
ограничивается размерами частиц
люминесцирующего вещества, а так-
же рассеянием света в слое это-
го вещества. При фотографировании
объектов используют фотопластинки
с мелкозернистой эмульсией, чтобы
получить на фото изображение всех
деталей электронного изображения.
В электронном микроскопе необхо-
димо поддерживать высокий вакуум,
чтобы электроны на своем пути от
электронной пушки до экрана не
сталкивались с молекулами воздуха.
В воздухе электроны могут проходить
лишь несколько микрометров, так как
при столкновении с молекулами га-
зов воздуха они либо теряют ско-
рость, либо останавливаются. По-
скольку расстояние между электронной пушкой и фотопластинкой
приблизительно 1 м, внутри микроскопа создают высокий вакуум
порядка 10~4 мм рт. ст. В этих условиях электрон может пройти
в среднем 2,5 м, прежде чем столкнется с молекулой газа. Для
создания и поддержания высокого вакуума в электронном микро-
334
скопе имеются насосы двух типов: ротационный механический
форвакуумный насос создает предварительный вакуум порядка
10у мм рт. ст., а диффузионный насос, включающийся после соз-
давая предварительного вакуума, обеспечивает высокий вакуум
порядка 10~4 мм рт. ci.
Блок питания обеспечивает энергией электронную пушку, все
линзы и вакуумную систему электронного микроскопа.
Просмотр образцов
в электронном микроскопе
После того как электронный микроскоп включен, настроен,
компенсирован астигматизм линз-конденсоров и линзы-объекти-
ва (астигматизм — явление, вызванное асимметрией магнитного
поля и проявляющееся в том, что изображение электронного пуч-
ка или точки объекта на экране будет не круглым, а вытянутым,
искаженным), включено ускоряющее напряжение, а ток нити на-
кала выключен и микроскоп, следовательно, находится в режиме
холостого хода, в него можно вносить изучаемый образец.
Сеточка с образцом (см. с. 337) вкладывается в объектодержа-
тель, который не следует трогать руками, так как жир и влага
с пальцев будут загрязнять колонну микроскопа. Сеточку берут
тонким пинцетом, а объектодержатель — рукой в белой нейло-
новой перчатке или неворсистой тканью. Для того чтобы объек-
тодержатель с образцом ввести теперь в камеру объекта микро-
скопа, надо отключить последнюю от остальной колонны микро-
скопа, перекрыв соответствующий клапан, затем в камеру объек-
та впустить воздух, и когда давление воздуха в камере достигнет
атмосферного, можно открыть заслонку камеры и установить объ-
ектодержатель с исследуемым образцом в камеру. Затем заслонку
закрывают и из камеры откачивают воздух. Когда индикаторная
лампа, установленная на панели микроскопа, изменив розовое
свечение на голубое, затем погаснет и этим укажет на создание в
камере необходимого вакуума, можно открывать клапан и соеди-
нять камеру объекта с колонной микроскопа. Включают ток нити
накала и осторожно его увеличивают только до того момента,
когда будет достигнута максимальная яркость сфокусированного
электронного луча на экране. С помощью ручек центровки луча
на панели микроскопа луч центрируют, и затем ручкой тонкого
контроля луч может быть расфокусирован, чтобы создать равно-
мерное освещение образца. Сначала полезно просмотреть сеточку
с образцом при малом увеличении и выбрать наиболее интерес-
ные участки. После этого перейти на большое увеличение с по-
мощью соответствующей ручки на панели микроскопа. При изме-
нении увеличения может возникнуть необходимость повторной
центровки луча. Изучая образец при большом увеличении, рас-
сматривают его в бинокуляр, смонтированный возле смотровой
335
камеры. Необходимо расстояние между тубусами бинокулярной
лупы установить точно по вашим глазам и сфокусировать на плос-
кость люминесцентного экрана. Просматривая сеточку с образцом,
сразу фотографируют те объекты, которые представляют интерес.
Фотографировать надо как можно скорее, тщательно фокусируя
изображение с помощью ручек фокусировок, вначале грубо^ и
средней, затем тонкой фокусировки. Под электронным лучом объ-
ект нагревается и может изменяться, иногда происходит возгонка
вещества образца; поэтому, работая с большими увеличениями,
рекомендуется проводить настройку фокуса по соседним участ-
кам и лишь затем поставить в центр интересующую вас область,
быстро поправить тонкий фокус и фотографировать желаемый объ-
ект. Прежде чем начать фотографировать, необходимо убедиться
в том, что изображение электронного луча на экране сохраняет
круглую форму и освещенность экрана равномерна. Затем яр-
кость немного снижают и фотографируют объект. Чтобы умень-
шение яркости луча не вызвало теплового смещения объекта, бо-
кового его сдвига, фокусировать объект рекомендуется при наи-
меньшей яркости, при которой еще возможно отрегулировать фо-
кус. Способы фотографирования различны в зависимости от кон-
струкции микроскопа, но все они состоят в том, что изображение,
после того как убирают экран, экспонируется на фотопластинку,
помещенную предварительно под экраном. По истечении време-
ни экспозиции экран возвращают на место. Движение экрана мо-
жет вызвать нежелательную вибрацию. Значительно удобнее кон-
струкции микроскопов, снабженные автоматическим затвором, ко-
торый позволяет получить точное время экспонирования и кото-
рый не срабатывает, пока не прекратится вибрация, вызванная
подъемом экрана. После того как снимок сделан, экспонирован-
ную пластинку возвращают в кассету и кассету убирают, приго-
тавливают следующую пластинку. Эти операции осуществляют с
помощью ручек или кнопок возле панели микроскопа, расположе-
ние их зависит от конструкции микроскопа. Данные, относящиеся
к снимку, а именно: номер пластинки, обозначение сфотографиро-
ванной структуры и увеличение, должны быть записаны в рабо-
чий журнал. Когда все пластинки в камере экспонированы, дефо-
кусируют конденсор, снижают ток луча, выключают ток нити на-
кала и устанавливают минимальное увеличение. Перекрывая со-
ответствующий клапан, отсоединяют фотокамеру от колонны мик-
роскопа и заполняют ее воздухом, открыв соответствующий кла-
пан. После этого фотокамеру извлекают, открывают, фотопластин-
ки проявляют, а камеру заряжают новыми пластинками, предва-
рительно тщательно высушенными, чтобы не вносить влагу в ко-
лонну микроскопа. Вставляют фотокамеру в микроскоп, откачи-
вают из нее воздух до достижения вакуума 10-2 мм рт. ст., после
чего фотокамеру можно соединить с колонной микроскопа и, до-
ждавшись, когда приборы зарегистрируют высокий вакуум в мик-
роскопе 10-4 мм рт. ст., можно снова включить ток накала нити.
336
\ Приготовление сеток
( и подложек
Исследуемые объекты помещаются на пленку-подложку, кото-
рая! крепится на специальных сетках, изготовленных из меди элек-
тролитическим способом или сплетенных из тонкой проволоки.
Плетеные сетки, для выравнивания их толщины, прокатывают ме-
таллическим валиком на ровной металлической плитке или отби-
вают молоточком.
Выровненные сетки очищают, опуская на 5—10 с в 5%-ную
азотную кислоту и затем тщательно промывают дистиллирован-
ной водой и спиртом. Промытые сетки высушивают на фильтре
под колпаком для предохранения от пыли.
Пленки-подложки должны удовлетворять следующим требова-
ниям: быть «прозрачными» для электронов, не проявлять собст-
венной структуры при просмотре пленки в электронном микро-
скопе, быть механически прочными, термостойкими, не заряжать-
ся под воздействием электронного пучка. Наиболее часто приме-
няются подложки, изготовленные из коллодия (нитроцеллюлоза),
формвара (поливинилформальдегид) или напыленного в высоком
вакууме углерода.
Для получения коллодиевых пленок используют 10%-ный ра-
створ коллодия в амилацетате; для его приготовления берут 0,1 г
сухого коллодия и растворяют в 13 мл амилацетата в закрытом
бюксе. Амилацетат — вещество летучее, пары его могут вызвать
поражение печени, если их длительно вдыхать, поэтому надо ра-
ботать с закрывающимися сосудами и в вытяжном шкафу. Раст-
ворение длится 6 ч, затем раствор фильтруют. Для приготовления
пленки-подложки в мелкую фарфоровую чашку наливают дистил-
лированную воду, на поверхность которой наносят из пипетки 2
капли раствора коллодия в амилацетате; раствор равномерно рас-
пределяется по поверхности воды, растворитель испаряется, и на
поверхности воды остается тонкая коллодиевая пленка, предназ-
наченная для очистки поверхности воды от частиц пыли. Ее уда-
ляют с помощью препаровальной иглы и выбрасывают. Эту опе-
рацию повторяют 2—3 раза, затем на очищенной поверхности
воды готовят такую же пленку и монтируют ее на сетки. Другой
способ позволяет получить более прочные пленки. С этой целью
небольшую каплю типола или сходного с ним детергента наносят
на одну сторону предметного стекла и растирают пальцем по всей
поверхности стекла, чтобы образовался тонкий слой этого веще-
ства. Излишки детергента удаляют чистой тряпочкой. На стекле
должен остаться тонкий слой детергента, он облегчит снятие плен-
ки. Стекло погружают в 0,5%-ный раствор коллодия в амилаце-
тате, затем извлекают, дают стечь избытку раствора и сушат в
вертикальном положении в защищенном от пыли месте. Лезвием
бритвы делают надрез, параллельный краю стекла на той сторо-
337
не, где под коллодиевой пленкой находится слой детергента ! и,
подышав на эту сторону стекла и наклонив его на 30° по отноше-
нию к горизонтальной поверхности воды, медленно погружают в .
чашку с дистиллированной водой, поверхность которой должна
быть предварительно очищена от пыли с помощью коллодиевой
пленки, как указано выше. Пленка коллодия должна отслоиться
на воду. Тонкая коллодиевая пленка кажется в отраженном све-
те серой или серебристо-серой. Золотистая пленка слишком тол-
ста. Чтобы получить требуемую толщину пленки, нужно менять
процентное содержание коллодия в растворе. Чтобы смонтировать
пленку на опорные сетки, на плавающую пленку опускают сетки,
слегка надавливают на сетки стеклянной оплавленной палочкой,
чтобы пленка коллодия прилипла к сеткам. Затем берут чистое
прямоугольное предметное стекло и короткой его стороной каса-
ются края пленки, наклонив стекло к поверхности воды под углом
примерно в 30°, сетки при этом должны быть расположены под
стеклом. Слегка нажимая на стекло, плавно погружают его в
воду под тем же углом до тех пор, пока все сетки с пленкой не
прилипнут к стеклу и не окажутся заключенными между стеклом
и пленкой. Тогда угол между стеклом и поверхностью воды по-
степенно увеличивают, поворачивая стекло, и когда погружаемая
сторона стекла направится к противоположной стороне сосуда и
пленка’ с сетками окажется на верхней его стороне, стекло осто-
рожно вытаскивают и ставят вертикально на фильтр для удале-
ния избытка воды, а затем помещают под стеклянный колпак для
просушки примерно на 40 мин. Высушенные сетки с пленками
можно отделить от стекла, обрезав пленку вокруг сеток острой
иглой или тонко заточенным пинцетом и подвинув сетку вдоль
стекла. Коллодиевые и формваровые пленки можно сделать бо-
лее прочными, нанеся на них тонкий слой углерода путем его ис-
парения в вакууме.
Углеродные пленки прочнее и устойчивее к действию электрон-
ного пучка, чем коллодиевые и формваровые, но менее «прозрач-
ны» для электронного луча. Можно получить прочную углерод-
ную пленку толщиной всего в 20 А, достаточно «прозрачную». Уг-
леродную пленку напыляют на стекло в высоком вакууме и отде-
ляют затем на поверхность подогретой до 50—60° дистиллирован-
ной воды. Можно отделить пленку с помощью желатины, для это-
го на нее наносят 20%-ный раствор желатины в воде, нагретой
до 60°. Через 6—8 ч желатина высыхает, и ее вместе с угольной
пленкой отделяют от стекла. Желатину удаляют с угольной плен-
ки растворением в дистиллированной воде, нагретой до 80—90°.
Пленку промывают в чистой дистиллированной воде и монтируют
на сетки.
\ Подготовка объекта
1 к исследованию
I Нанесение образца
на сетку с подложкой
При исследованиях в электронном микроскопе объект иссле-
дования — минеральные или органические частицы, микроорга-
низмы — наносится на пленку-подложку, прикрепленную к сетке-
держателю, в виде суспензии (раствора) или в виде сухих твер-
дых частиц (при распылении их). Чаще объект готовят в виде
суспензии или раствора, каплю которых наносят на сетку с под-
ложкой, высушивают и затем объект контрастируют и изучают в
электронном микроскопе.
Концентрация вещества в суспензии или растворе не должна
быть слишком высокой, так как при больших концентрациях в
поле зрения, на экране микроскопа, окажется множество частиц,
громоздящихся друг на друга. Но при очень малых концентраци-
ях в поле зрения может оказаться слишком мало частиц, и мы по-
лучим небольшую информацию. Оптимальные концентрации под-
бираются обычно опытным путем. При исследовании тонкодисперс-
ных минеральных почвенных фракций приготавливают слабо опа-
лесцирующие водные суспензии их. При изучении гумусовых ве-
ществ целесообразно приготавливать растворы гуминовых или
фульвокислот с концентрацией 0,005% (см. с. 350).
Раствор или суспензию набирают в пипетку с тонким носиком
и затем наносят одну каплю на сетку с подложкой. Сетку при этом
держат пинцетом с острыми концами. Для высушивания капель
на сетках их помещают на специальные дырчатые подставки или
прикрепляют в наклонном положении одним краем к клейкой лен-
те-лейкопластырю, туго натянутой на стеклянной пластинке. Про-
сушивание можно ускорить, используя тепло от обычной настоль-
ной лампы. Лампу помещают на расстоянии 50—60 см от сеток,
чтобы избежать перегрева, при котором может произойти разрыв
тонких пленок-подложек, смонтированных на сетках. После про-
сушивания сеточки переносят на чистое предметное стекло и конт-
растируют напылением (см. с. 340).
При высушивании суспензий или растворов частицы могут под-
вергаться деформации за счет возрастания сил поверхностного
натяжения у высыхающей капли. Существуют способы, направлен-
ные на снижение сил поверхностного натяжения и уменьшение
деформации частиц. Они трудоемки, требуют специальной аппа-
ратуры и применяются лишь в специальных исследованиях. Один
из них состоит в мгновенном замораживании капли раствора с по-
мощью жидкого азота (/°=—150°) с последующим испарением
(возгонкой) растворителя при низкой температуре в вакууме.
В другом способе используют последовательную смену раствори-
телей, обладающих все более и более низким поверхностным на-
339
тяжением. Наиболее эффективным является способ мгновенного
высушивания раствора при критической температуре и давлении
(критическая точка).. При этом способе исследуемое вещество
растворяют в спирте, а затем спирт постепенно вытесняют жид-
кой углекислотой под давлением. Когда спирт полностью вытес-
нится, повышают температуру в баллоне, где происходила эта
операция, и при определенном значении температуры и давления
углекислота мгновенно превращается в газообразную. При таком
способе высушивания силы поверхностного натяжения в критиче-
ской точке (момент перехода жидкой углекислоты в газообраз-
ную) минимальны, и объекты хорошо сохраняют свою форму.
Контрастирование объекта
«оттенением»
Трудности электронно-микроскопического изучения почвенных
компонентов, особенно органического вещества почв, связаны с
тем, что они состоят в основном из атомов с малыми атомными
номерами: водорода, углерода, азота, кислорода, фосфора и серы.
Объекты, состоящие из атомов с малой атомной массой, слабо
рассеивают электронный пучок и на экране и фотографиях выгля-
дят малоконтрастными. Атомы тем сильнее рассеивают электрон-
ный пучок, чем больше в ядре атома содержится протонов и чем
больше электронов окружает ядро атома. Минеральные частицы
большей частью содержат более тяжелые атомы и вследствие это-
го сильнее рассеивают электроны и более четко видны на экране
микроскопа, но при малой толщине даже минеральная частица
плохо видна в электронном микроскопе. Чтобы изображение сде-
лать более четким, приходится изучаемые объекты контрастиро-
вать, для чего их покрывают тонким слоем металла, атомы кото-
рого способны сильно рассеивать электроны («оттенение» напы-
лением, реплики), или вводят эти атомы в состав молекул объек-
та («крашение»).
При контрастировании объекта методом «оттенения» исполь-
зуют тяжелые металлы или углерод, испарение которых произ-
водят в вакуумных установках. Оттеняемый препарат помещается
под колокол вакуумной установки, где имеется испаритель из
вольфрамовой проволоки —• коническая спираль. Испаряемый ме-
талл помещают в испаритель, который нагревают током до не-
обходимой температуры (температура плавления металла). К мо-
менту испарения металла должен быть достигнут хороший вакуум
10"4— IO-5 мм рт. ст. При более высоком давлении увеличивается
рассеивание атомов металла на молекулах газа, атомы теряют
прямолинейные траектории и попадают на участки теней, что ве-
дет к снижению контрастности. При хорошем вакууме атомы ме-
талла при испарении разлетаются по прямолинейным траектори-
ям, и если под некоторым острым углом к направлению потока
Л
340
распыляемых частиц металла поместить исследуемый объект, то
на его поверхности будет осаждаться слой металла. На той сто-
роне объекта, которая обращена к источнику напыления, будет
скапливаться много атомов металла, на противоположной сторо-
не объекта их количество будет меньшим (рис. 107). За объектом
образуется участок, куда частицы металла не попадают, этот уча-
сток выглядит как «тень» от объекта. Поэтому этот метод кон-
ч I ,
/|\
I
Рис. 107. Схематическое изображение «оттенения» объекта:
1 — источник испаряющегося металла или углерода, 2 — объект, 3 —
пленка-подложка, 4 — оттеняющий напыленный слой; h — высота объ-
екта, I — длина «тени», г — радиус сферической частицы, 0 — угол
«оттенения»
341
трастирования называют методом «оттенения». На позитивном
фотоотпечатке оттененного объекта будут видны темные участки
на стороне объекта, обращенной к источнику напыления (скопле-
ния металла), и светлые «тени» с противоположной стороны. Ес-
ли известны угол напыления 6 (его измеряют) и длина тени /, ко-
торую находят по фотоотпечатку, то высоту объекта h можно вы-
числить по формуле: /i = /-tg6. Для сферической или цилиндриче-
_______________________________________________. , 6
ской частицы радиус г определяют по формуле: — - При
небольших углах оттенения практически можно воспользоваться
формулой: £>«s/-tg6, где D — диаметр частицы. Измеряя на фо-
тоотпечатке длину и ширину объекта, необходимо учитывать при-
рост размеров за счет толщины напыляемого слоя. Недостаток
этого метода заключается в том, что при напылении толстого слоя
металла создается хорошая контрастность, но атомы металла,
соосаждаясь, образуют агрегаты, которые растут, превращаясь
в кристаллы металла, величина их может превышать величину
деталей изучаемого объекта. Это снижает разрешение. Если на-
пылять тонкий слой металла, то контрастность изображения мо-
жет быть низкой. Хорошую контрастность при тонком слое напы-
ляемого металла позволяют получить платина, палладий, уран,
вольфрам, золото; из неметаллов — углерод. Чтобы испарить уг-
лерод, в вакуумной установке пропускают электрический ток си-
лой около 100 А через два графитовых стержня, к которым при-
ложено напряжение около 12 В. Стержни контактируют между со-
бой. Один стержень заточен на конус, в другом сделано углубле-
ние для контакта. Вследствие высокого локального сопротивле-
ния в месте соприкосновения графитовые стержни нагреваются
до температуры, достаточной для испарения углерода. Испарен-
ные частицы углерода будут осаждаться на холодной поверхности
объекта и контрастировать его.
Контрастирование «крашением»
Контраст изображения можно повысить не только напылением
металла на поверхность объекта, но и введением атомов или ионов
металла с большой атомной массой в структуру объекта. Катио-
ны тяжелых металлов образуют комплексы с отрицательно заря-
женными группами молекул изучаемого объекта. Ионы свинца,
урана, индия, висмута, железа, входящие в состав солей «краси-
телей», могут образовывать связи с кислородом. Многие «краси-
тели» обладают избирательным действием, например соли урана,
особенно уранил ацетат, являются универсальными «красителями»
нуклеиновых кислот и белков. В почвоведении метод крашения
применяется чаще при микробиологических исследованиях и изуче-
нии органических веществ почв, в биологии метод крашения уль-
тратонких срезов применяется особенно широко.
342
Существуют методы позитивного и негативного крашения.
Суть метода позитивного крашения заключается в том, что меж-
ду солями тяжелых металлов и молекулами объекта происходят
химические реакции, в результате которых ионы тяжелых ме-
таллов закрепляются в веществе объекта и делают его контраст-
ным при просмотре в электронном микроскопе. При негативном
способе крашения, напротив, вещество исследуемого объекта не
вступает в реакцию с раствором «красителя», раствор заполняет
свободные места на подложке и внутри объекта, и тогда изобра-
жение объекта на экране будет светлым на темном фоне «краси-
теля», который хорошо рассеивает электроны.
Метод реплик для исследования
поверхности твердых частиц
Если требуется исследовать почвенный агрегат, структурную
отдельность или твердый препарат гумусовых кислот, не расчле-
няя их на отдельные составляющие, то провести такое исследо-
вание в электронном микроскопе на просвет невозможно, так
как такой массивный объект «непрозрачен» для электронного
пучка. В этом случае прибегают к методу реплик, т. е. получе-
нию отпечатков с поверхности объекта. Метод реплик заклю-
чается в том, что на поверхность изучаемого объекта наносят
вещество, образующее тонкую пленку, детально повторяющую
рельеф поверхности. Затем пленку отделяют от объекта и иссле-
дуют в электронном микроскопе. Реплика может быть получе-
на с поверхности объекта, представляющей естественный скол,
но часто полезно провести «травление» поверхности твердого
объекта, позволяющее выявить рельеф поверхности, обусловлен-
ный структурным строением объекта. При травлении используют
растворы веществ, являющиеся хорошими растворителями изучае-
мого объекта. Раствор наносят на поверхность объекта и раство-
ряют вещество объекта. Растворенный материал смывают с по-
верхности или тщательно удаляют с помощью желатины, так же
как в методе получения углеродных напыленных реплик отслаи-
вают реплику с помощью желатины (см. с. 344). С обнаженной
поверхности далее делают реплику. Существует множество раз-
личных способов получения реплик.
Изготавливают одноступенчатые, двухступенчатые и много-
ступенчатые реплики. Чтобы изготовить одноступенчатую репли-
ку, исследуемый объект покрывают слоем вещества, передаю-
щим рельеф поверхности, и, отделив пленку вещества от объек-
та, получают реплику. Для получения двухступенчатой реплики
с исследуемой поверхности объекта вначале изготавливают тол-
стые непрозрачные первичные реплики, так называемые матри-
цы, с которых делают вторичные реплики, прозрачные для элект-
ронного пучка.
343
Одноступенчатый способ изготовления реплик нашел широкое
применение при исследовании почвенных, биологических, геоло-
гических объектов. Материалом, из которого готовится реплика,
может служить коллодий, формвар и некоторые пластмассы.
Если необходима реплика с высоким разрешением, используют
напыленные в вакууме слои углерода или кварца.
Наиболее часто работают с углеродными репликами. Они до-
статочно прочные, хорошо передают рельеф поверхности, не-
сложны в изготовлении. Для изготовления напыленных реплик
используют те же вакуумные установки, что и для контрастиро-
вания оттенением (см. с. 340). Если при напылении реплики
плоскость, на которой лежит объект, расположена под прямым
углом к потоку летящих при испарении углеродных частиц, то
напыленная реплика будет иметь примерно равномерную тол-
щину и будет лишена контраста при просмотре в электронном
микроскопе. Контраст у реплики можно создать, если напыле-
ние углеродом вести не под прямым, а под острым углом к по-
верхности, тогда получают так называемую самооттененную
угольную реплику. Оттенение под острым углом создает слой не-
равномерной толщины, и реплика приобретает контраст.
Полученную напылением пленку надо отделить от объекта.
Существует химический и механический способы отделения плен-
ки от объекта. При химическом способе объект с пленкой погру-
жают в жидкость, которая должна растворить объект, но не дол-
жна растворять или изменять пленку. Для растворения породы
или почвы используют плавиковую кислоту, карбонатную породу
растворяют в соляной кислоте, препарат гуминовой кислоты мо-
жет быть растворен в щелочи. Всплывшую реплику вылавлива-
ют на сетку и неоднократно промывают дистиллированной во-
дой, а затем спиртом. Промывание спиртом предохранит пленку
от разрывов при высыхании. Механическим способом отделяют
пленку в том случае, если образец трудно растворяется или же-
лательно его сохранить. В этом случае на пленку равномерно
тонким слоем наносят 30—40%-ный раствор желатины в го-
рячей воде (45—50°) и оставляют до полного высыхания. При-
мерно через 2 ч слой желатины вместе с угольной репликой сни-
мают с помощью бритвы и погружают в теплую дистиллирован-
ную воду, чтобы растворить желатину. Воду постепенно
нагревают до 80—90°. Сразу погружать пленку в горячую воду
нельзя, так как при этом пленка коробится и растрескивается.
После растворения желатины всплывшую пленку дважды про-
мывают в горячей дистиллированной воде, затем в спирте, вы-
лавливают на сетку, сушат и просматривают в электронном
микроскопе.
Лаковые реплики (коллодиевые, формваровые) готовят нане-
сением на изучаемую поверхность раствора коллодия в амилаце-]
тате или формвара в диоксане. Раствор заполняет все углубле-
ния микрорельефа поверхности образца, а внешняя поверхность
344
его выравнивается за счет растекания раствора. После высуши-
вания реплику отделяют от объекта. Формваровые реплики при
погружении в дистиллированную воду легко отделяются и всплы-
вают. Коллодиевые реплики можно отделить с помощью жела-
тины. Лаковая реплика имеет в различных участках неодинако-
вую толщину, отражающую неровность микрорельефа поверхно-
сти объекта, что создает контраст реплики в электронно-микро-
скопическом изображении. Для увеличения контрастности ее
можно напылить металлом или углеродом.
Двухступенчатые реплики приготавливают в тех случаях, ко-
гда необходимо сохранить исследуемую поверхность для повтор-
ного изучения или когда приходится изучать нерастворяющиеся
объекты. Двухступенчатые реплики могут изготавливаться из
различных материалов. Для приготовления матрицы используют
раствор полистирола в бензоле или трихлорэтилене, раствор
плексигласа в бензоле, поливиниловый спирт, раствор карбокси-
метилцеллюлозы или желатины в воде; а также легко полимери-
зующиеся вещества, например эфиры метакриловой кислоты, и
в частности техновит — быстро затвердевающая пластмасса на
основе метакриловой кислоты. Раствор одного из указанных ве-
ществ или легко полимеризующаяся пластмасса наносится на
исследуемую поверхность объекта, создаются условия для испа-
| рения или полимеризации (в случае пластмасс) и полученные
пленки матрицы отделяют от объекта. С твердых и достаточно
вязких объектов матрицу можно получить методом тиснения, ко-
гда поверхность объекта вдавливают в предварительно размяг-
ченный пластический материал. Таким материалом может слу^
жить пластмассовая пленка, полученная при высыхании на
стеклянной пластинке раствора полистирола в бензоле, или рент-
геновские пленки марок Agfa Rontgen Durv Film, Agfa Diapositiv,
Kodak Rontgen Film, Pholotechnische Platte С., отмытые от
эмульсии горячей водой. Для создания необходимой пластично-
сти рентгеновскую пленку вымачивают в ацетоне, а полистиро-
ловую пленку подогревают (термопластическая реплика). После
полного высыхания пленок они легко отделяются от вдавленного
в них объекта. Термопластиковая матрица легче отделяется при
охлаждении объекта с матрицей. С матрицы делают оттененную
угольную реплику и отделяют ее ог матрицы, растворяя послед-
нюю в ацетоне (рентгеновскую пленку) или бензоле (полисти-
роловую пленку). Реплику промывают в дистиллированной воде
и спирте, монтируют на сетку, сушат и просматривают в элек-
тронном микроскопе.
Диагностика минералов
илистой фракции почв
При' изучении минералогического состава илистой фракции
почв первым этапом подготовки образца является при-
345
готовлейие водной суспензии. Небольшое количество почвы
(0,3—0,5 г суглинистой или 0,5—1 г супесчаной почвы) помеща-
ют в фарфоровую чашку и заливают бидистиллированной водой.
Через сутки верхний прозрачный слой жидкости с всплывшими
органическими остатками сливают, добавляют к суспензии 4—
5 капель 5%-него раствора NH4OH и растирают почву пестиком
с резиновым наконечником. Затем добавляют в чашку бидистил-
лированной воды примерно на 3/4 ее объема, смесь размешивают
и через некоторое время, когда осядут наиболее крупные части-
цы, верхнюю часть суспензии сливают в пробирку. Оставшеюся
массу снова растирают, добавляют воду, размешивают, даюг
осесть крупным частицам и верхнюю часть суспензии опять сли-
вают в ту же пробирку. Растирание повторяют до тех пор, пока
все комочки, агрегаты не будут разрушены, а на дне чашки
останутся песчаные частицы. Содержимое пробирки взбалтывают
и оставляют отстаиваться. Через сутки пипеткой осторожно за-
бирают часть суспензии с глубины 6—7 см, помещая конец пи-
петки в среднюю часть пробирки. В этой части суспензии содер-
жатся частицы менее 1 мкм. В ряд пробирок с бидистиллирован-
ной водой добавляют из пипетки различное количество капель
суспензии, чтобы получить серию пробирок со слабо опалесци-
рующими суспензиями различных концентраций. Капли суспен-
зий из этих пробирок наносят на различные сетки с подложками
и высушивают. Сухие сетки с препаратами контрастируют напы-
лением углерода или металла под углом 35°. Просматривая сет-
ки в электронном микроскопе, устанавливают, какая концентра-
ция суспензии была оптимальной. Дальнейшие исследования
проводят, работая с суспензией этой концентрации.
Использовать высушенную илистую фракцию для приготовле-
ния суспензии не рекомендуется (при высушивании фракции ча-
стично происходит необратимая коагуляция). Можно приготовить
препарат не только из илистой фракции почв, но при необходи-
мости и из почвы в целом. В этом случае суспензию отбирают
пипеткой сразу же после взбалтывания и каплю ее наносят на
сетку с подложкой. Если почвы богаты карбонатами, раствори-
мыми солями, органическим веществом, гидроокислами железа
или алюминия, то перед приготовлением суспензии необходимо
освободиться от этих веществ, так как они затушевывают элект-
ронно-микроскопическую картину, образуют аморфные пленки,
соединяют частицы в агрегаты, в результате изображение частиц
минералов теряет четкость очертаний и диагностика становится
затруднительной. Для освобождения от растворимых солей поч-
ву промывают дистиллированной водой; карбонаты удаляют про-
мыванием слабыми кислотами — уксусной, щавелевой, разбав-
ленной соляной; от гидроокислов железа почву освобождают,
обрабатывая ее дитионитлимоннокислой смесью с буферным
раствором бикарбоната натрия по методу Мера и Джексона (см.
гл. 8), органическое вещество разрушают обработкой почвы 6%-
346
ной Н2О2 на горячей водяной бане Для лучшей диспергации ми-
нералов в суспензии используют также ультразвуковые диспер-
гаторы типа УЗДН-1, которые позволяют получить устойчивые
суспензии при определенном режиме работы диспергатора при
частоте колебаний 15—22 кгц в течение 3—5 мин
Изучение суспензии почв в электронном микроскопе дает све-
дения о морфологических особенностях частиц о их форме, ха-
рактере контуров изображения, плотности изображения, что ука-
зывает па толщину час
тиц, о размерах частиц
длине, ширине и высоте,
последнюю находят по тан-
генсу угла оттенения и дли-
не тени (см с 342), и о рас-
пределении частиц по раз-
мерам в изучаемой суспен-
зии Морфологические осо-
бенности частиц позво-
ляют диагностировать их,
определять их минералоги-
ческую принадлежность
Наиболее достоверно среди
глинистых минералов ус-
Рис 108 Каолинит, X 15 000
танавливается принадлежность к группе каолинита Для као
линита (рис 108) характерны форма правильного шестигранни-
ка в плоскости подложки и четкие контуры частиш Толщина их
может варьировать от непрозрачных для электронного луча (чер-
ное изображение) до почти «прозрачных» тончайших листочков,
едва видимых на экране В почвах кристаллы каолинита с хо-
рошо выраженными гексагональными очертаниями встречаются
редко Обычно у них хорошо выражено лишь несколько граней,
двух граней и угла между ними вполне достаточно для отнесе-
ния частицы к каолиниту Другой минерал группы каолинита —
галлуазит (рис 109) имеет вытянутую палочковидную трубча-
тую форму и хорошо диагностируется в электронном микроскопе
Минералы группы гидрослюд (рис НО) обычно выглядят как
светлые или темные (значит, тонкие или толстые) пластинчатые
изометрические частицы с четкими контурами, угловатыми или
сглаженными, реже они представлены вытянутыми, удлиненны-
ми, щепковидными (глауконит, селадонит). Минералы группы
монтмориллонита (рис 111) в электронном микроскопе выгля-
дят как пластинчатые образования с нечеткими размытыми кон-
турами, дающие хлопьевидные агрегаты, более толстые в центре
и тонкие к периферии частицы Насыщенные натрием монтморил-
лонитовые минералы образуют очень мелкие частицы, тогда как
при насыщении кальцием или магнием образуются крупные не-
прозрачные агрегаты Железистый минерал группы монтморил-
лонита — нонтронит имеет удлиненную вытянутую форму. Маг-
347
мезиальные силикаты (рис. 112) имеют игольчатую или волокни-
стую форму. Хлориты и смешаннослойные минералы (рис. 113)
чрезвычайно сложно диагностировать электронно-микроскопиче-
ским методом, так как они не имеют четко выраженных морфо-
логических особенностей. Неглинистые минералы — кварц, по-
Рис. 109. Галлуазит (трубочки) и каолинит, у 15 000
Рис. НО. Гидрослюды, X15 000
348
левые шпаты, гидроокислы железа, кароонаты, гипс, хлориды —-
имеют объемную форму, и изображение их будет темным с чет-
кими контурами вследствие большой толщины частиц минерала
или большой атомной массы, как в случае с гидроокислами же-
леза. Гидроокислы железа выглядят на экране мелкими точечны-
ми частицами с изрезанными контурами. Растворимые соли чр-
Pttc. 111. Монтмориллонит, х 15 000
Рис. 112. Магнезиальный силикат—-
полыгорскит (игольчатые кристал-
лы), XI5 000
Рис. 113. Смешанослойный минерал — слюда-монтмориллонит, Х15 00'>
349
сто образуют при высыхании суспензии на подложке кристаллы
правильной формы. Электронный микроскоп позволяет достаточ-
но точно диагностировать минералы с хорошо выраженными
морфологическими особенностями — минералы группы каолини-
та, магнезиальные силикаты, растворимые соли и т. п.; другие
минералы диагностируются лишь ориентировочно.
Электронно-микроскопическая
характеристика гумусовых кислот
Электронно-микроскопическое исследование гумусовых кис-
лот было осуществлено рядом исследователей с целью наблю-
дать форму и размеры молекул гумусовых кислот Однако при
приготовлении препаратов, в процессе высушивания капли на
подложке возможны коагуляция молекул и образование слож-
ных ассоциатов, и в электронном микроскопе мы наблюдаем не
молекулы гумусовых кислот, а надмолекулярные структуры.
Для наблюдения гумусовых кислот в электронном микроско-
пе просвечивающего типа сначала готовят растворы гумусовых
кислот Гумусовые кислоты должны быть предварительно полу-
чены в виде хорошо очищенных от растворимых солей и мине-
ральных частиц препаратов, кристаллы солей и минеральные
частицы затрудняют рассмотрение органического вещества, ибо
частицы последнего имеют значительно меньшие размеры, чем
частицы глинистых минералов и кристаллы солей. Кроме того,
присутствие растворимых солей даже в незначительных количе-
ствах оказывает коагулирующее действие на гумусовые кислоты
Для приготовления 0,0025—0,005 %-ных растворов 2,5—5 мг
гуминовой кислоты помещают в стакан на 100 мл и растворяют
в наименьшем количестве 2 %-него раствора КОН до полного ра-
створения препарата, затем приливают немного бидистиллиро-
ванной воды и фильтруют раствор через бумажный фильтр в
мерную колбу на 100 мл. С помощью 2 %-ных растворов НС1 и
КОН создают нужный pH раствора гуминовой кислоты и доли-
вают мерную колбу до метки бидистиллированной водой. Раст-
вор фульвокислоты готовят подобным же образом, но растворяют
ее сразу в бидистиллированной воде. Каплю раствора гумата ка-
лия или фульвокислоты наносят на медную сетку с подложкой
и высушивают.
Используемый раствор КОН должен быть предварительно
проверен на содержание органических веществ; для этого гото-
вят контрольную сеточку с подложкой, наносят на нее каплю ра-
створа щелочи, высушивают, напыляют углеродом и просматри-
вают в электронном микроскопе Часто даже химически чистые
щелочи содержат значительные количества органических ве-
ществ. Присутствие даже сравнительно небольших количеств ор-
350
ганических веществ делает щелочь непригодной для электронно-
микроскопических исследований. Необходимо обратить внимание
на подбор концентраций минеральных компонентов, используе-
мых для приготовления растворов. Если для приготовления ра-
створа гуминовой кислоты и нейтрализации растворов гумусовых
кислот использовать не 2%-ные, а 10%-ные растворы щелочей и.
соляной кислоты, то в поле зрения электронного микроскопа по-
являются игольчатые скопления кристаллов щелочи, кубические
кристаллы солей.
Рис. 114 Тангенс угла напыле-
ния ЛВ ВС
А — источник испаряющегося ме-
талла или углерода, В — основа-
ние перпендикуляра А В к плос-
кости ВС, ВС — схематически
изображенная плоскость, на кото-
рую помещен образец, С — обра-
зец; 0 — угол напыления («отте-
нения»), АВ : ВС=Щ 0 j
статочно большое количество
розненных частиц. На этих
ки измерить длину и ширину
Рис. 115 Нейтральный раствор гу-
миновой кислоты, концентрация
0,005%, Х75 500
При просмотре препаратов в
электронном микроскопе запи-
сывают наблюдения и фотогра-
фируют объекты исследования.
Затем из полученных фотографий
отбирают те, где имеется до-
равномерно расположенных раз-
фотографиях надо с помощью линей-
каждой частицы в мм. Зная
увеличение, полученное в микроскопе, и фотоувеличенпе, можно
рассчитать размеры частиц. Разберем один пример такого рас-
чета размеров частицы гуминовой кислоты. Частицы гуминовых
кислот (как и фульвокйслот) имеют округлую форму на фото-
графиях. Измеренные на фотографии длина и ширина частицы
были одинаковы и равны 2,5 мм. Увеличение микроскопа -—
Х30200, фотоувеличенпе — Х2,5, общее увеличение — Х75 500.
Следовательно, 1 мм на фотографии в действительности соответ-
ствует 1 мм : 75 500^0,0000130 мм, или 130 А (1 мм=107 А).
Итак, диаметр нашей частицы будет равен 2,5-130 А=325 А, а
радиус ее 162,5 А. Далее надо рассчитать высоту частицы. Для
этого воспользуемся формулой: /i=/-tgO, где I — длина тени,
351
в нашем примере измеренная на фотографии длина тени равня-
лась 1 мм, а угол 6 — это угот, под которым производилось
напытенпе образца Если расстояние (рис 114) от источника на
лыляемого углерода (точка А) до плоскости, на которой лежит
напыляемый образец (отрезок АВ), бы то равно 40 см, а рас-
стояние от точки В до точки С, где помещен напыляемый обра-
зец (отрезок ВС), равно 60 см, то tg0=40 60=06667, а угол
0=33°42' Высота частицы в масштабах фотографии будет рав-
на h=\ мм 0,6667=0,6667 мм Так как 1 мм на фотографии
соответствует в действительности 130 А, истинная высока части-
цы равна й=0,6667 130 А=86,67 А Соотношение между диа-
метром и высотой частицы равно 325 А 86,67 А«4, т е высота
частицы в 4 раза меньше ее диаметра, и, следовательно, частица
имеет не сферическую, а уплощенную форму Поскольку части
ца осаждалась из раствора на подложку, то, вероятно, ее форма
соответствхет форме сегмента шара Объем сегмента шара равен
у = _1_лй(/г2 З/2),
тогда объем частицы равен
V = 1/6 3 14 86,671(86,67)2 3 (162 5)2]= 3 933858,5 А3,
или V = 3933858 5 10“исм3, так как 1 А3 = 10- 4см3
Если принять удетьную массу гуминовой кислоты равной
1,5 г/см3, то масса частицы будет равна ее объему, умноженно-
му на удельную массу 3 933 858,5 10-24 см3 1,5 г/см3=
= 5 900 787 75 10-24 г
Теперь можно вычислить «молекулярную массу» такой части
цы, разделив массу частицы на 1/12 массы атома углерода
(1 662 10~24 г) — 5900787,75 10-24 1,662 10~24=3550414 Эта
«молекулярная масса» частицы на 2 порядка больше, чем сред-
невесовые молекулярные массы, найденные для гуминовых кис-
лот методами светорассеяния, ультрацентрифугирования, гель-
фильтрации Подобное сопоставление данных показывает, что
частицы, видимые в электронном микроскопе, представляют со-
бой не молекулу гуминовой кислоты, а надмолекулярную струк-
туру — ассоциат, агрегат, циаметр которого равен 325 А, высо-
та которого в 4 раза менее диаметра, следовательно, он имеет
уплощенную округлую форму Просчитав размеры всех частиц
на фотографии, можно составить представление о их размерах и
форме
Гуминовые кислоты, видимые в поле зрения электронного
микроскопа (рис 115), образуют округлые частицы с диаметром
130—500 А и более при реакции среды нейтральной и близкой к
нейтральной Препараты, приготовленные из кислых растворов
гуминовой кислоты (pH 3) или из нейтральных растворов, но с
352
Рис 116 Гуминовая кислота скоагулированная сульфатом меди X26 000
концентрация
0,005%,
Рис 117 Щелочной раствор гуминовой кис юты,
Х75 ООО
12 Зак 284
добавлением 0,02%-кого раствора CuSO4, представляют иную
картину: на фотографии (рис. 116) видно, как округлые части-
цы (ассоциаты) коагулируют, образуя ажурные цепочки и сетки.
В препаратах, приготовленных из щелочных растворов гуматов
калия, основная масса частиц имеет значительно меньшие раз-
меры и заполняет все поле зрения электронного микроскопа, об-
разуя сплошной слабый фон (рис. 117). Таким образом, изменяя
реакцию среды растворов гумусовых кислот, можно наблюдать
различное поведение молекул в зависимости от реакции среды.
Картина, наблюдаемая при щелочной среде, дает еще одно до-
казательство, что молекулы имеют размер, значительно мень-
ший, чем размер частиц, наблюдаемых в препаратах с нейтраль-
ной реакцией среды, и что при нейтральной среде мы видим ас-
социаты молекул.
В расчеты необходимо вводить поправку на искажение раз-
меров частиц при напылении на них углерода или металла. Для
выявления размеров этого искажения нужно приготовить препа-
раты из вещества, частицы которого имеют постоянные и извест-
ные параметры. Один препарат контрастировать напылением уг-
лерода или металла, другой не подвергать контрастированию;
изучить эти два препарата в электронном микроскопе (проме-
рить по фотографиям диаметр частиц и рассчитать их высоту)
и выявить, какие искажения в размерах частиц мы получаем при
том или ином режиме напыления. Подходящим объектом для ре-
шения этой задачи могут служить частицы латекса или микро-
организмы с постоянными известными параметрами.
Сканирующий электронный микроскоп
В отличие от просвечивающего электронного микроскопа в
электронном микроскопе сканирующего типа получают изобра-
жение поверхности объекта благодаря отражению от него элект-
ронов. Пучок электронов с катода проходит через систему маг-
нитных и электростатических линз и фокусируется в плоскости
образца. Здесь он обегает (сканирует) образец по квадрату
строку за строкой. Отраженные от образца и вторичные испус-
каемые образцом электроны воспринимаются коллектором, обра-
зуют сигнал, который после усиления передается в электронно-
лучевую трубку, покрытую люминофором с длительным
послесвечением. На экране этой трубки исследователь наблюдает
изображение объекта. Вторая электронно-лучевая трубка с корот-
ким послесвечением и высокой разрешающей способностью ис-
пользуется для получения фотографий образца.
Пределы полезного -увеличения сканирующего микроскопа
составляют 10—50 тыс., что несколько ниже, чем в микроскопах
просвечивающего типа. В современных сканирующих микроско-
пах удается, при оптимальных условиях, добиться выявления
354
тонких деталей поверхности с разрешением 100 А. Это примерно
на порядок ниже, чем разрешение, получаемое в электронных
микроскопах просвечивающего типа. Но сканирующий микро-
скоп незаменим при исследовании рельефа поверхности, так как
этот микроскоп обладает большой глубиной резкости и создает
изображение изучаемой поверхности как бы объемное, рще
большую рельефность изображения можно получить, сочетая
исследования в сканирующем микроскопе со стереографирова-
нием, когда с помощью сканирующего микроскопа получают два
снимка под углом не меньшим, чем 5—6°, а затем проецируют
их стереопроектором на экран и рассматривают изображение
через специальные очки.
Объект для изучения в сканирующем микроскопе готовят в
виде суспензии или раствора, которые затем высушивают на
тонком покровном стекле, или в виде твердых частиц со свежи-
ми сколами или с поверхностями травления, эти частицы при-
крепляют клеем к покровному стеклу. Сухой прикрепленный к
покровному стеклу образец напыляют в вакуумной установке
равномерно тонким слоем металла, вращая образец. Для напы-
ления используют чаще золото, но также платину, палладий, се-
ребро, химически чистую медь. Тонкий слой металла делает
поверхность образца электропроводной, что позволяет избежать
накопления электрического заряда, который может отрицательно
сказаться на разрешении. После напыления образец готов для
просмотра в микроскопе.
В настоящее время существуют электронные микроскопы, поз-
воляющие осуществлять переход от сканирования к просвечива-
ющей микроскопии и наоборот в течение всего нескольких минут,
так что один и тот же образец может быть исследован обоими
методами.
НЕКОТОРЫЕ ПРАВИЛА РАБОТЫ
ПРИ ЭЛЕКТРОННО-МИКРОСКОПИЧЕСКИХ
ИССЛЕДОВАНИЯХ
Работа в электронно-микроскопической лаборатории требует
строгого соблюдения чистоты. Посуда должна быть хорошо вы-
мыта хромовой смесью, затем водой и дистиллированной водой.
Пинцеты и пипетки моют дистиллированной водой после приго-
товления каждого препарата. Рабочее место должно быть тща-
тельно убрано. Сетки с препаратами сушат под стеклянным кол-
паком для предохранения от попадания пыли. Все реактивы го-
товят на бидистиллированной воде. Чистоту реактивов проверя-
ют изготовлением контрольных препаратов.
Многие вещества, используемые в электронной микроскопии,
ядовиты и обращаться с ними надо осторожно. Так, амилацетат,
используемый при приготовлении пленок-подложек, обладает
наркотическим действием, и при длительном его вдыхании воз-
12’
355
можны поражения печени. Свинец, содержащийся в растворах-
«красителях», способен проникать через неповрежденную кожу,
пыль свинцовых солей, образующаяся при высыхании случайно
пролитого раствора, может попасть в дыхательные пути. Дли-
тельное воздействие свинца может вызвать бесплодие и привести
к очень опасным хроническим отравлениям. Поэтому при работе
со свинцовыми «красителями» надо тщательно мыть руки и ра-
бочий стол. Глутаральдегид и четырехокись осмия, используемые
при крашении препаратов, токсичны и летучи; первый при по-
падании на кожу может вызвать дерматит, вторая является
сильным раздражителем дыхательных путей, вызывает ожогн на
коже, а также может привести к тяжелым поражениям глаз.
Ацетон и ксилол легко воспламеняются, а при длительном вды-
хании вызывают поражения печени.
При работе электронного микроскопа, когда ускоренные
электроны бомбардируют различные препятствия, возникают
рентгеновские лучи. Для защиты от рентгеновского излучения
производится тщательное экранирование колонны микроскопа, а
в смотровые люки вставляют свинцовые стекла. При хорошем
экранировании и нормальной работе прибора опасности рентге-
новского облучения фактически не существует. Однако вновь ус-
тановленный микроскоп должен быть проверен с помощью счет-
чика Гейгера, и первые 100 ч работы обслуживающий его персо-
нал должен носить на себе индикаторы излучения для проверки
надежности экранирования микроскопа.
ЛИТЕРАТУРА
Г р и ц а е н к о Г. С , Звягин Б. Б., Боярская Р. В. и др. Методы элек-
тронной микроскопии минералов. М, «Наука», 1969.
Методы изучения минералогического состава и органического вещества почв.
Ашхабад, 1975.
Сергеева Н Е. Введение в электронную микроскопию минералов. М.,
Изд-во Моск, ун-та, 1977.
Уикли Б. Электронная микроскопия для начинающих. М., «Мир», 1975.
Глава 11
МАТЕМАТИЧЕСКАЯ ОБРАБОТКА
РЕЗУЛЬТАТОВ НАБЛЮДЕНИЙ
Определение физико-химических свойств почвы состоит из
анализа и измерений в процессе аналитической работы и вычис-
ления полученных результатов. Исследователь, помимо аналити-
ческих навыков, должен владеть понятиями метрологии — нау-
ки об измерениях и методами математической обработки резуль-
татов наблюдений.
Цель эксперимента — получение количественных характери-
стик объекта, как можно более близких к истинным. Но любое
измерение неизбежно содержит некоторую погрешность. Полу-
ченные характеристики в разной мере приближаются к истинным.
Анализ источников погрешностей необходим для правильной
оценки достоверности полученных результатов.
Оценка методов анализа
Погрешность измерений. Погрешностью измерений е называ-
ют статистически значимую разность между полученным при из-
мерении х, и действительным значением а измеряемой величины:
е=х,—а. За действительное значение может быть принято содер-
жание определяемого элемента в стандартном образце, много-
кратно проанализированном разными методами или искусствен-
но приготовленном.
Погрешность является результатом несовершенства средств и
методов измерения, а также способностей экспериментатора. По-
грешности условно делятся на систематические и случайные.
В общую погрешность постоянно вносит вклад погрешность,
обусловленная чувствительностью прибора. Последняя называет-
ся погрешностью шкалы прибора (еШк-пр). Ее можно определить
по классу точности прибора, указанному на приборе или в инст-
357
рукции к нему. Класс точности показывает погрешность в про-
центах от максимального значения шкалы (ctmax):
Е-тк.пр' 100
----------= класс точности.
Ctmax
Если амперметр класса точности 0,5 имеет максимальное
значение шкалы 10,0 А, то абсолютная погрешность шкалы при-
бора равна 0,05 А.
Пример 1. При измерении силы тока правильно действую-
щим амперметром класса точности 0,5 получено значение 2,10 А.
Погрешность прибора, обусловленная его чувствительностью,
равна 0,05 А. Результат измерения с учетом только этого вида
погрешности равен (2,10±0,05) А.
Величина погрешности, определяемая чувствительностью при-
бора, ограничивает число верных значащих цифр в результатах
непосредственных измерений (предполагается, что прибор не
имеет дефекта). Результат измерения записывается так, чтобы
предпоследняя цифра результата была верной, а последняя —
сомнительной. В вышеприведенном примере вторая цифра после
запятой является сомнительной, так как погрешность шкалы при-
бора равна 0,05 А. Следовательно, и результат измерения дол-
жен быть записан с точностью до второго знака после запятой,
большее число цифр не имеет смысла.
Электроизмерительные приборы обычно характеризуются
классом точности от 0,05 до 4. Если класс точности не указан, за
величину чувствительности прибора принимают цену деления на
шкале прибора. Чем больше погрешность шкалы прибора, тем
ниже его чувствительность.
Систематическая погрешность (еСист) обусловлена дефектом
прибора, приближенным характером расчетных уравнений, не-
точным значением используемых в уравнении констант. Система-
тическая погрешность при повторных измерениях остается по-
стоянной по величине и по знаку или меняется по определенному
закону. Величину систематической погрешности мы чаще всего
не можем изменить, не переделывая прибор, не меняя вид ис-
пользуемого уравнения. Но учесть систематическую погрешность
можно. Если установлено, что прибор имеет дефект, то ошибки,
обусловленные этим дефектом, находят сравнением показаний
данного прибора с правильно действующим. При этом опреде-
ляется величина отклонения, и в дальнейшем в результаты из-
мерений вносится соответствующая поправка.
Так как систематические погрешности обусловлены индиви-
дуальными особенностями эксперимента и спецификой аппарату-
ры, то общей теории систематических погрешностей быть не мо-
жет. Обычно систематическая погрешность меньше случайной, но
возможно и обратное.
Случайная погрешность (еСЛуч) возникает в результате влия-
ния множества неучтенных факторов, которые при повторных из-
358
•»
мерениях появляются и исчезают нерегулярно, проявляются с ин-
тенсивностью, которую трудно предвидеть. Случайная погреш-
ность принимает случайные, взаимно независимые значения.
Чтобы знать, как их вычислить, следует познакомиться с зако-
ном распределения случайной погрешности.
При выполнении параллельных анализов одной и той же про-
бы в практически одинаковых условиях мы получаем ряд близ-
ких, но не одинаковых значений измеряемой величины. Разли-
чаются результаты измерения потому, что на них влияет
множество трудноконтролируе-
мых факторов, например вибра-
ция прибора, колебания силы
тока в сети, изменение внимания
оператора, условий температуры,
давления, трения частей прибора
и т. д. Можно сказать, что ре-
зультаты измерения содержат
случайную погрешность, вели-
чину которой определяют, поль-
зуясь методами математической
статистики, науки, применяющей
для обработки результатов на-
блюдений методы теории веро-
Рис. 118. Кривая распределения
_ Стьюдента:
х — среднее арифметическое, х<—•
ятности. ' частные значения признака, р — ве-
На измеряемую величину Роятность получения значений при-
действует множество разнона- знака
правленных факторов. Влия-
ние каждого невелико по сравнению с общим воз-
действием всех остальных. Поэтому при повторных ана-
лизах одной и той же пробы наиболее часто получаются резуль-
таты, близкие к среднему арифметическому из всех полученных
результатов. Очень малые или очень большие, по сравнению со
средним, значения встречаются редко.
Закон распределения случайных значений результатов парал-
лельных определений, который показывает, как часто встречают-
ся те или иные значения, известен — это закон Стьюдента, кото-
рый графически изображается кривой распределения Стьюдента
(рис. 118).
Если по оси абсцисс откладывать переменные значения ре-
зультатов параллельных определений х2, ..., х{, то вероятность
получения значения признака от Xi до хг определяется площадью
под кривой распределения. Очевидно, что наиболее вероятно по-
лучение значений, близких к среднему арифметическому X. Если
мы получим значение, равное среднему арифметическому, зна-
чит, случайная погрешность определения равна нулю. Кривая
распределения симметрична относительно среднего арифметиче-
ского, т. е. случайные погрешности, равные по величине и проти-
воположные по знаку, встречаются одинаково часто.
359
Итак, случайная погрешность — это отклонение единичного,
частного хг результата измерения от среднего арифметического
из результатов параллельных определений X, еСлУч=х,—X. Одна-
ко среднее значение может в разной степени приближаться к
действительному значению измеряемой величины. Систематиче-
ская погрешность — это отклонение среднего значения измеряе-
мой величины от действительного значения его, еСпст = *—а.
Теперь мы можем познакомиться с важными метрологически-
ми понятиями: воспроизводимостью и правильностью результа-
тов. Воспроизводимость — это показатель степени близо-
сти результатов параллельных определений измеряемой величи-
ны. О ней можно судить по величине случайной погрешности.
Правильность — показатель близости полученного при из-
мерении среднего и действительного значений измеряемой вели-
чины, т. е. показатель близости к нулю систематической погреш-
ности. Воспроизводимость •— необходимое, но недостаточное ус-
ловие правильности. Воспроизводимость не гарантирует пра-
вильность определения. При сравнительной близости результатов
параллельных определений среднее значение может отличаться
от действительного из-за систематической погрешности.
Как найти случайную погрешность? Мерой разброса или варь-
ирования результатов параллельных определений относительно
среднего служит среднее квадратичное отклонение, которое так-
же называют стандартным отклонением (S). Оно вычисляется
по формуле
S = l/ 2 —%)2
V п-1
где х{ — случайное значение измеряемой величины; х — среднее
арифметическое результатов параллельных определений; п —
число параллельных определений.
Например, из одной средней пробы взяли 4 аналитические и
проанализировали одним и тем же методом. Полученные резуль-
таты измерения варьируют около среднего арифметического, и
мера их разброса — стандартное отклонение S.
Средние арифметические и стандартные отклонения, получен-
ные по ограниченному числу параллельных определений, не яв-
ляются неизменными, постоянными. Если мы в той же анализи-
руемой пробе 5 раз выполним по 4 параллельных определения,
вычислим средние для каждой партии из 4 параллельных (т. е.
частные средние х), то эти средние будут близки, но не одинако-
вы. Они будут колебаться около общего среднего, найденного по
результатам всех 20 определений. Мерой их разброса будет вели-
S
чина —где 5 •— стандартное отклонение, вычисленное по ре-
у п
зультатам 20 определений; п — число параллельных (в нашем
примере н=4).
360
Итак, среднее, полеченное по какому-то числу параллельных
определений, есть сам^ по себе величина случайная, способная
принимать переменны^ значения. Поэтому мало назвать лишь
одно из возможных значений. Следует определить весь интервал
возможных значений среднего.
_ Интервал возможных значений среднего, содержащего слу-
чайную погрешность, м^жно записать так:
Л Л- Слу
Этот интервал иначе Называют довери-
тельным интервалом с
ляют по формуле
х + ' , а
|>еднего и вычис-
е ______ ip.f-S
c«y ~---------
У п
порученное по
Таблица 33
Значения критерия Стьюдента t
при Р = 0,95 и числе
параллельных т
/ f=m— 1 i
1 12,71 11 2,20
2 4,30 12 2,18
3 3,18 13 2,16
4 2,78 14 2,14
5 2,57 15 2,13
6 2,45 16 2,12
7 2,36 17 2,11
8 2,31 18 2,10
9 2,26 19 2,09
10 2,23 20 2,09
где х — среднее,
зультатам параллельных определений;
S — „ стандартное отклонение; tp,f — кри-
терий Стьюдента, числс^ которое показы-
вает, на сколько стандартных отклоне-
ний среднее арифметич^ское отличается
от действительного знцдения (предпола-
гается отсутствие систематической по-
грешности). Закон распределения t из-
вестен, возможные зна^рния его табули-
рованы и могут быть найдены по табл. 33.
Значение t берется таблице при f=m—1, т — число парал-
лельных определений, }10 результатам которых рассчитывали S.
Доверительная вероятность выбирается исследователем обычно
равной 0,95. Доверительная вероятность 0,95 показывает, что в
95 случаях из 100 результаты параллельных определений попа-
— tp.i s
дут в интервал х ±а большие и меньшие значения, вы-
г а
ре-
ходящие за пределы названного интервала, можно получить
5 раз из 100; п — чис^0 параллельных определений, рекомендо-
ванных по методике; н п могут совпадать, если стандартное
отклонение вычисляли по результатам параллельных определе-
ний текущих анализовх выполненных с двух- или трехкратной
повторностью. Но т м^жет быть и не равно п, если стандартное
отклонение находили ^о результатам большого числа (10-—20)
специально для этого доставленных параллельных опытов. Ведь
стандартное отклонение тем надежнее охарактеризовано, чем по
большему числу параллельных оно определено.
При mJ>.20 и P=04i)5 доверительный интервал среднего опре-
деляется по формуле
х +
25
Уп
361
Иногда можно встретить рекомендации определять довери-
тельный интервал среднего по формуле: х + — , где—— назы-
Г п 1' п
вается ошибкой среднего. Этот способ расчета предполагает ис-
пользование той же вышеприведенной формулы при условии, что
£=1. Такое значение t соответствует вероятности 0,68 и обозна-
чает, что при повторных исследованиях результат попадает в
— 5
заданный интервал х + в 68 случаях из 100, а в остальных
у п
32 случаях будет получено значение, выходящее за пределы это-
го интервала. Для аналитических работ такая вероятность ошиб-
ки велика, и определять, таким образом, доверительный интервал
среднего не следует.
Величину случайной погрешности можно сделать как угодно
малой, увеличивая число параллельных определений. Величина
доверительного интервала среднего существенно зависит от чис-
ла параллельных. Тем надежнее определено среднее и меньше
случайная погрешность, чем больше число параллельных т и п.
Это связано с положением п в расчетной формуле и с тем, что
значение t уменьшается с увеличением числа параллельных т.
Если предстоит анализировать много образцов одним методом,
целесообразно не жалеть времени и надежно определить стан-
дартное отклонение при большом числе параллельных т. Бла-
годаря этому текущие измерения можно выполнять с малым чис-
лом параллельных, чаще оно равно 2. Если же число образцов,
которые надо проанализировать, невелико, то предварительные
испытания для нахождения стандартного отклонения не прово-
дятся, а повышается число параллельных в текущих измерениях,
п. Особенно резко сужается доверительный интервал и умень-
шается случайная погрешность при переходе от числа парал-
лельных п, равного 2—3 к 4, дальнейшее повышение числа па-
раллельных дает незначительный эффект.
Общая погрешность измерения равна сумме систематической,
случайной погрешностей и погрешности шкалы прибора: еОбщ=
= Ешкпр+Есист + вслуч- Если при увеличении числа повторных оп-
ределений случайная погрешность будет стремиться к нулю, то
общая погрешность будет приближаться к сумме погрешностей
систематической и шкалы прибора.
Соотношение составляющих общей погрешности может быть
различным. При использовании приборов с низкой чувствитель-
ностью, т. е. с относительно высокой погрешностью шкалы при-
бора, результаты повторных измерений обычно совпадают. Слу-
чайная погрешность в этом случае мала, и повторные многократ-
ные измерения не имеют смысла. При использовании высокочув-
ствительных приборов уменьшается величина погрешности шка-
лы прибора и возрастает случайная погрешность. Многократны®'
измерения необходимы для более правильного определения слу-1
чайной погрешности. Надо отметить, что математическая обра4
362
ботка позволяет количественно оценить и исправить влияние слу-
чайной погрешности, но никакие математические приемы не
помогут, если измерения выполнялись неверно, без соблюдения
правил методики.
Кроме названных погрешностей возможны промахи и грлбые
ошибки, которые существенно превышают систематические и
случайные погрешности, связанные с условиями измерения. Со
способами их обнаружения мы познакомимся позже. Промахи
и грубые ошибки возникают из-за недостаточного внимания ис-
следователя и серьезной нейуправностн аппаратуры; результаты,
полученные при этом, невернк и должны быть отброшены.
Чувствительность определения. В физико-химическом анализе
почв все измерения относятся к косвенным. Это значит, что по-
сле выполнения ряда последовательных операций мы получаем
результат — аналитический сигнал у. Им может быть оптиче-
ская плотность в колориметрическом анализе, высота полярогра-
фической волны в полярографии, интенсивность спектральной
линии в эмиссионном спектральном анализе. Далее аналитиче-
ский сигнал пересчитывается на содержание определяемого
компонента. Связь между аналитическим сигналом и концентра-
цией устанавливается чувствительностью b = —. Математиче-
Ах
ская зависимость, связывающая аналитический сигнал и
концентрацию, называется градуировочной функцией, y=f(x).
Чувствительность — это значение первой производной градуиро-
вочной функции. Зависимость между аналитическим сигналом и
концентрацией может быть представлена и в виде градуировоч-
ного графика. Чувствительность равна угловому коэффициенту
градуировочного графика, построенного без преобразования ана-
литического сигнала и концентрации. Следует обратить внима-
ние, что нельзя определять чувствительность как формальный
тангенс угла наклона нарисованного градуировочного графика,
найденный по отношению длины (в см пли мм) отрезков, соот-
ветствующих сигналу и концентрации.
Прямолинейный градуировочный график имеет общий вид
уравнения прямой у~а-]-Ьх, где b — угловой коэффициент. Он
показывает изменение аналитического сигнала у при изменении
концентрации на единицу. Если при построении градуировочного
графика для k стандартных образцов с переменными концентра-
циями Xi, х2, > были получены соответствующие аналитиче-
ские сигналы уь у2, ..., уь, то точное значение углового коэффи-
циента (или чувствительности) может быть найдено по формуле
_k~£x-y— Ъх-Ъу
~ /гУх2 — (2х)2 ’
где Sx — сумма всех концентраций стандартных растворов; Sy —
сумма всех аналитических сигналов, полученных для всех ана-
лизируемых стандартных образцов; Sx-у — сумма произведений
363
к
концентраций стандартных растворов на соответствующие анали-
тические сигналы; Sx2 — сумма квадратов концентраций стан-
дартных растворов.
Например, в колориметрии, согласно закону Бугера — Бера,
оптическая плотность окрашенного раствора пропорциональна
концентрации в растворе элемента, образующего это окрашенное
соединение. Градуировочная функция имеет вид: D—et.-1-с, где
D — оптическая плотность; е к — коэффициент молярного погло-
щения; I — толщина поглощающего слоя; с — концентрация.
Первая производная градуировочной функции равна ел •/. Мож-
но определить чувствительность и по угловому коэффициенту
градуировочного графика, т. е. по отношению приращения ана-
литического сигнала к приращению концентрации. Эта величина
может быть различной для разного интервала концентраций.
Чтобы найти угловой коэффициент, надо при определенной дли-
не волны определить оптические плотности растворов с различ-
ной концентрацией. Разность оптических плотностей равна
ДП=Ел-/-Дс, а чувствительность соответственно елЕ Если
использовать вышеприведенную формулу вычисления чувстви-
тельности, то для колориметрического метода она будет иметь
вид
, ZjSC-D —ZC-SD
о =----------------.
/гХС2 — (2Z?)3
Воспроизводимость и правильность определения. Продолжим
знакомство с основными метрологическими характеристиками.
Уже названные характеристики воспроизводимость и пра-
вильность составляют содержание понятия точность. Общепри-
нятой единой численной характеристики для точности не имеется.
В литературе часто употребляется термин точность без объясне-
ния, о чем идет речь: о воспроизводимости или о правильности.
Поэтому Научным советом по аналитической химии АН СССР
не рекомендуется заменять термины воспроизводимость и пра-
вильность термином точность. Правильность определения содер-
жания элемента данным методом, как уже отмечалось, можно
оценить, сравнив полученное при анализе среднее содержание
элемента в стандартном образце (х) с действительным содержа-
нием элемента в этом же стандартном образце (а). Определение
правильно в том случае, если разница |х—«| не превышает слу-
чайную погрешность. Проверить это можно, вычислив эмпириче-
ское значение t:
f [x-al-Уп
1 — S
Если оно превышает значение t Стьюдента при f=m—1, то ре-
зультаты анализа считаются неправильными, они содержат систе-
364
матическую ошибку. Если /Выч<^ст, то можно считать, что систе-
матическая ошибка отсутствует.
Для оценки воспроизводимости можно использовать величину
стандартного отклонения результатов параллельных определений
е S
или относительное стандартное отклонение = -=г-, т. е. стан-
х
дартное отклонение, отнесенное к среднему и выраженное в до-
лях единицы или в процентах. В последнем случае эта величи-
на называется коэффициентом варьирования (V=Sr-100%).
В настоящее время для количественной оценки воспроизводимо-
сти рекомендуется величина, обратная относительному стандарт-
1 гг
ному отклонению,----.При выполнении анализа двумя разными
Sf
методами бывает необходимо оценить, какой метод обеспечивает
лучшую воспроизводимость. Для этого анализ одной пробы вы-
Значения критерия Фишера F
Таблица 34
П1—1
п,—1 «2—1 2 3 4 5 6 7 8 9 10
2 19,0 19,2 19,2 19,3 19,3 19,4 19,4 19,4 19,4
3 9,6 9,3 9,1 9,0 8,9 8,9 8,8 8,8 8,8
4 6,9 6,6 6,4 6,3 6,2 6,1 6,0 6,0 6,0
5 5,8 5,4 5,2 5,1 5,0 4,9 4,8 4,8 4,7
6 5,1 4,8 4,5 4,4 4,3 4,2 4,1 4,1 4,1
7 4,7 4,4 4,1 4,0 3,9 3,8 3,7 3,7 3,6
8 4,6 4,1 3,8 3,7 3,6 3,5 3,4 3,4 3,3
9 4,3 3,6 3,6 3,5 3,4 3,3 3,2 3,2 3,1
10 4,1 3,7 3,5 3,3 3,2 3,1 3,1 3,0 3,0
Примечание
Р — 0,65. число параллельных для наибольшего St, пг —
число параллельных для наименьшего S2
полняется тем и другим методом с числом параллельных, кото-
рое определяется возможностями каждого метода. Вычисляются
стандартные отклонения для каждого метода: Si и S2. Далее на-
ходится отношение квадрата стандартного отклонения (эта ве-
личина называется дисперсией), имеющего наибольшее значение,
S*
к наименьшей дисперсии:------,(S2 > S~). Полученное отношение
$2
сравнивается с табличным значением критерия Фишера F
(табл. 34). Если вычисленное значение отношения .меньше таб-
личного при вероятности 0,95 и том числе параллельных, которые
были взяты для анализа тем и другим методом (fi = ni—1 и
J:2=n2^-1), то у нас нет оснований считать дисперсии различны-
ми. Следовательно, нет оснований считать, что какой-то из про-
365
веряемых методов обеспечивает лучшую воспроизводимость ре-
зультатов. Если полученное отношение больше табличного, зна-
чит, дисперсии значимо различаются, а тот метод, который имеет
меньшее стандартное отклонение, характеризуется лучшей вос-
производимостью.
Предел обнаружения. Предел обнаружения — наименьшая
концентрация, по которой при заданной доверительной вероятно-
сти по данной методике можно обнаружить присутствие компо-
нента. Не рекомендуется называть предел обнаружения откры-
ваемым минимумом, чувствительностью, пороговой концентра-
цией.
Следует принимать во внимание, что почти в любом физико-
химическом анализе холостой опыт дает сигнал, т. е. аналитиче-
ский сигнал на приборе отмечается и при отсутствии определяе-
мого компонента. Значит, существует опасность, получив такой
сигнал в пробе без элемента, сделать вывод о том, что элемент
присутствует. Значение холостого сигнала непостоянно. Холостой
сигнал может принимать переменные значения, мерой разброса
их является стандартное отклонение фона So. Вычисляется оно,
как обычно, по ряду значений холостого сигнала. Наименьший
аналитический сигнал, который вызван наличием в пробе опреде-
ляемого элемента и который надежно может быть отличен ог
сигнала фона, равен 6-S0. Вероятность того, что этот сигнал вы-
зван определяемым элементом, равна 0,997, т. е. получить такой
сигнал в отсутствие элемента можно 3 раза из тысячи.
Для вычисления предела обнаружения нужно от надежно
улавливаемого аналитического сигнала, равного 6-S0, перейти к
наименьшей определяемой по этому сигналу концентрации, Cmm.
Для этого надо найти отношение-6"-1-, где b — чувствительность
Ь
данного метода анализа. Итак, предел обнаружения Cmm равен
6S0
Ь
Обработка результатов
измерений
Главными статистическими параметрами являются среднее
арифметическое и стандартное отклонения. Вычислить их можно,
используя классические методы параметрической статистики, ос-
нованные на участии в расчетах всех полученных значений, а
также методы непараметрической статистики. Последние не тре-j
буют обязательного применения всех без исключения результат
тов параллельных определений. Они позволяют быстрее и легча]
выполнить расчеты. Стандартное отклонение можно вычислить
но результатам специально для этого выполненных многократе
ных (10—20) параллельных анализов одной и той же пробы, $
f
366
целесообразно преобра-
Таб шца 35
Значения критерия хх при
Р — 0,95 в зависимости от числа
исходных данных п
п тл п тх
4 0,96 14 0,40
5 0,81 15 0,38
6 0,69 16 0,37
7 0,61 17 0,36
8 0,54 18 0,35
9 0,51 19 0,34
10 0,48 20 0,33
И 0,45 21 0,33
12 0,43 22 0,32
13 0,41 23 0,31
также по результатам текущих анализов нескольких разных об-
разцов с небольшим числом параллельных определений, рекомен-
дованных методикой, которое обычно равно 2—3.
Преобразования исходных значений признака. Прежде чем
приступить к вычислениям, часто бывает
зовать значения признака и упростить
их для того, чтобы избежать грубых
ошибок и ускорить вычисления. Если
исходные данные — дробные числа, их
следует умножить на постоянную вели-
чину так, чтобы дальше оперировать
целыми числами. Если данные различа-
ются в последних знаках, можно от-
бросить постоянную часть этих чисел и
проводить все необходимые вычисления,
а по окончании их сделать обратные
преобразования для того, чтобы опреде-
лить искомые значения среднего ариф-
метического и стандартного отклоне-
ний.
Пример 2. При определении окис-
лительно-восстановительного потенциала
в образце чернозема были получены сле-
дующие результаты 6 параллельных оп-
ределений (мВ): 427, 431, 432, 432,
433, 443. Такой ряд значений можно представить как сумму 430 +
-\-а, а имеет значения —3, 1, 2, 2, 3, 13. Этот упрощенный ряд и
следует дальше использовать для вычисления среднего арифме-
тического и стандартного отклонений.
Перед вычислением статистических параметров сначала на-
до проверить, не допущены ли при анализе грубьщ ошибки или
промахи, можно ли использовать в расчетах все исходные дан-
ные. Для этого все полученные значения располагают в ранжи-
рованный ряд в порядке возрастания х2, х3, ..., xn-i, хп, где
х, — наименьшее, а хп — наибольшее значение. Сомнение могут
вызвать крайние значения ряда xt и хп. Для подозрительного
наибольшего значения хп вычисляют отношение: тх — - Хп ~ Х,'~'
П ХП- х2
для наименьшего Xj: их =—————. Полученные значения
1 Х„_1 — Л).
Ххп и сопоставляют с табличным тх (табл. 35).
Если оно окажется больше табличного, проверяемый резуль-
тат следует исключить, как содержащий грубую ошибку. Такая
операция называется выбраковкой данных. Если полученное зна-
чение меньше табличного, у нас нет оснований отбрасывать про-
веряемое число.
В вышеприведенном (пример 2) ряду значений —3, 1, 2, 2, 3,
13 нужно проверить наибольшее (13) и наименьшее (—3) зна-
367
ка-
чения:
тх = = 0,83; тх = -Ш- = 0,67.
п 13 1 1 Зт3
При числе определений 6 и доверительной вероятности 0,95 таб-
личное значение равно 0,69 (см. табл. 35). Так как 0,67<0,69,
у нас нет оснований исключить число —3. Так как 0,83>0,69,
число 13 следует отбросить.
Этот пример иллюстрирует применение методов непараметри-
ческой статистики, так как в вычислениях участвовали не все ре-
зультаты, а лишь занимающие крайние положения в ранжиро-
ванном ряду. К подобным методам непараметрической статисти-
ки относятся все те, которые основаны на использовании величи-
ны размаха варьирования R. Размах варьирования — разность
между наибольшим и наименьшим значениями: R=xn—лх.
Если число параллельных невелико, то, прежде чем усреднять
полученные результаты, следует оценить, допустимы ли расхож-
дения между результатами параллельных определений. Допусти-
мые расхождения параллельных определений имеют статистиче-
ский смысл максимально допустимого размаха между ними.
По Пирсону, эта величина определяется по формуле R<Dp,nS,
где S — стандартное отклонение (часто полученное в предвари-
тельных исследованиях), Dp,n — табличное значение при вы-
бранной доверительной вероятности Р и числе параллельных п.
При доверительной вероятности 0,95 и двух параллельных До,95 ,2
равно 2,77, для трех параллельных — 3,31, для четырех — 3,65.
П ример 3. В 5 разных образцах чернозема, проанализиро-
ванных в двукратной повторности, получены следующие значения
окислительно-восстановительного потенциала (мВ):
563 465 487 526 510
568 461 498 520 515
По результатам предварительных исследований известно, что
стандартное отклонение S равно 3,6 мВ. Допустимое расхожде-
ние параллельных результатов равно 2,77-3,6» 10 мВ. Разность
между параллельными для третьего образца равна 11. Так как
11>10, такое расхождение следует признать недопустимым. Этот
образец должен быть заново проанализирован. Разность между
параллельными остальных образцов меньше 10. Эти расхожде-
ния допустимы.
Вычисление главных статистических параметров. Среднее
арифметическое х находится как частное от деления суммы ре-
зультатов параллельных определений на число определений:
—
х =—Усреднять можно и аналитические сигналы, и получен-
п
ные результаты, например концентрации. Вычислим среднее
368
арифметическое в примере 2:
- —3 + 1+ 2 4-2 + 3
~ 5
Так как среднее вычисляли по преобразованным значениям,
нужно выполнить_ обратные операции: 430+1=431. Искомое
среднее значение х равно 431 мВ.
В общем виде стандартное отклонение рассчитывается по
формуле
5 =
где Хг — отдельные значения аналитического сигнала или содер-
жания; х — среднее арифметическое.
В примере 2 стандартное отклонение для преобразованных
значений признака равно
(—3—1)2+(1—1)2 + (1 — 1)2 + (2—1)2+(2—1)а+(3— 1)а
4
= 2,4.
4
Этот способ расчета особенно удобен в том случае, когда данных
немного, и они представлены сильно различающимися числами.
Если числа невелики (одно- или двузначные), целесообразно рас-
считывать стандартное отклонение по следующим формулам:
9 (2+)2
2х? —-5—
* п
п — 1 ’
п —1
(_ 3)2 4- 12 22 + 22 + З2 — 5-12
Sx(2— п-х2
= 2,4:
' = 2’4-
Стандартное отклонение в конечном результате обычно приво-
дится с точностью до первой значащей цифры, в промежуточном
результате — до второй значащей цифры.
369
Если вычисляли стандартное отклонение по преобразованным
значениям после прибавления или вычитания из исходных како-
го-то числа, то для перехода от Sa к Sx обратные преобразова-
ния не требуются: Sa=Sx. В нашем примере стандартное откло-
нение и для преобразованных, и для исходных данных равно
2,4 мВ.
Стандартное отклонение тем правильнее вычислено, чем боль-
ше параллельных определений выполнено. Но 10—20 параллель-
ных может быть лишь в специально проведенном исследовании.
Однако для оценки стандартного отклонения можно использо-
вать и результаты текущих измерений, выполненных с малым чи-
слом параллельных.
Если п образцов анализировались в двукратной повторности,
то стандартное отклонение можно вычислить по формуле
s =
f 2-п
где di — разность результатов параллельных определений.
Пример 4. После исключения одного из результатов (при-
мер 3) разность между параллельными результатами определе-
ния Eh в 4 образцах равна 5, 4, 6, 5:
S = ]/ 52 42 -|-62 -|-52 = 3 6
У 2-4
Если число параллельных невелико (2—3), а образцов анали-
зировали много, стандартное отклонение можно найти по разма-
ху варьирования. При числе образцов п и размахе варьирования
между повторными результатами для каждого образца Ri сред-
ний размах R равен-----Между средним размахом и стандарт-
п
ным отклонением существует пропорциональность: <S =^= . Зна-
dm
чения коэффициента dm для числа параллельных в каждом образ-
це т и числа образцов п приведены в табл. 36.
В примере 4 размах варьирования для каждого образца ра-
вен 5, 4, 6, 5. Средний размах R равен —i—~—= 5- Значение
d для 4 образцов с двумя параллельными равно 1,21. Стандартное
•отклонение равно 5:1,21=4,1. Стандартное отклонение, найден-
ное по размаху варьирования, всегда выше найденного класси-
ческим способом.
Вычислять стандартное отклонение по обобщенным результа-
там анализа разных образцов, в том числе выполненных разны-
ми аналитиками, можно при условии, что стандартное отклоне-
ние не зависит от численного значения определяемой величины.
В этом случае отношение наибольшего значения этой величины
к наименьшей должно быть меньше 3. В примере это отношение
370
Таблица 36-
Значения коэффициента dm в зависимости от числа образцов п и числа
параллельных для каждого образца т
т п 1 2 3 1 5 6 7 8 9 10
9 1,41 1,28 1,23 1,21 1,19 1,18 1,17 1,16 1,15 1,14
3 1,91 1,81 1,77 1,75 1,74 1,73 1,72 1,71 1,70 1,69
равно 1,2 (568:461 = 1,2). Результаты анализа всех 4 образцов-
можно использовать для вычисления 5.
Определение воспроизводимости результатов анализа. При-
мер 5. При определении валового содержания Мп в образце,
выполненном химическим методом в 9-кратной повторности, по-
лучено среднее содержание 600 мг/кг и стандартное отклонение
30 мг/кг. При определении валового содержания Мп в этом же
образце спектральным методом в 8-кратной повторности получе-
но среднее содержание 651 мг/кг и стандартное отклонение
54 мг/кг. Оценить воспроизводимость результатов, полученных
тем и другим методом.
Относительное стандартное отклонение Sr, характеризующее восп-
роизводимость, для химического метода определения Мп равно
30 54
= 0,05, или 5%; для спектрального метода = 0,083, или
600--------------------------------------------651
8,3%. Величина, обратная Sr, для химического метода равна
-^-^- = 20, для спектрального —~ 12. Использование для
оценки воспроизводимости логически оправдано. Ведь воспроизводи-
1
мость — положительное качество анализа, показатель ее - должен
Sr
быть тем больше, чем лучше воспроизводимость результатов.
Но нельзя сказать, что один метод обеспечивает лучшую вос-
производимость, чем другой, только на том основании, что вели-
чины S, или Sr, или—— для этих методов не совпадают полно-
Sr
стью. Надо проверить, является ли различие между ними стати-
стически значимым. Вычислим отношение дисперсий:
S? 542
= = 3,2. Сравним это отношение с критерием Фишера.
$2-----30
При доверительной вероятности 0,95, nt—1=7, п2—1=8 крите-
рий Фишера (см. табл. 34) равен 3,5. Так как 3,2<3,5 нет осно-
ваний считать, что дисперсии статистически значимо различают-
ся. Следовательно, нет оснований считать, что химический и
спектральный методы определения валового содержания Мп в
почве значимо различаются по воспроизводимости результатов.
371
Определение чувствительности анализа. В качестве примера
рассмотрим, как можно определить чувствительность фотометри-
ческого определения Мп в вытяжке 1 н. H2SO4 из почв. Это кос-
венное измерение, так как на фотоколориметре мы измеряем оп-
тическую плотность окрашенного раствора и по ней определяем
концентрацию Мп в растворе.
Пример 6. Стандартные растворы имели известную кон-
центрацию Мп: 1, 2, 3, 4, 5, 10 мкг/мл. Оптическая плотность ок-
рашенных растворов равна соответственно 0,04; 0,14; 0,21; 0,28;
0,36; 0,72. Если анализируемые растворы сравнительно однооб-
разны по содержанию Мп, например концентрация элемента ко-
леблется от 2 до 5 мкг/мл, то целесообразно определить чувстви-
тельность именно в этом интервале. Чувствительность в интерва-
ле концентраций 2-—5 мкг/мл, найденная по формуле
, &D 0,36 — 0,14 „
о =---- равна —--------— = 0,073.
ДС 5 — 2
Чувствительность анализа в интервале концентраций 1 — 10мкг/мл,
найденная по формуле b =-------------, равна
feSC2 —(2С)а
[6(1-0,04 | 2-0,14 1-3-0,21 1-4-0,28 1-5-0,36 г 10-0,72) —
— (1 12 1-3141-51 10) (0,04 t 0,14 4-0,21 р 0,28 1 0,36 Р 0,72)]:
:[6(12 |- 22 + З2 4 42 1- 52 4- 102) — (1 4 2 Р 3 Р 4 Р 5 F 10)2] =
= [6 • 11,07 — 25 • 1,75]: [6 • 155 — 625] = 0,073.
Определение правильности анализа. Пример 7. Образец,
результаты которого приведены в примере 5, был искусственно
приготовленным стандартным образцом с известным содержани-
ем Мп 620 мг/кг. Определить, имелись ли систематические погреш-
ности при анализе его химическим и спектральным методами.
Другими словами, определить, правильны ли полученные резуль-
таты. Находим эмпирическое значение i:
для химического метода t = '600 ~620* = 2,00;
30
для спектрального метода / = . I651 ~б2Р I..P 8. _ j 52
Сравнив Л и t2 с tct (см. табл. 33) и убедившись, что 2,00<2,31
и 1,62<2,36, можем сделать вывод, что определение Мп и спек-
тральным и химическим методами было выполнено правильно.
Определение предела обнаружения. Пример 8. При опреде-
лении Мп фотоколориметрическим методом была получена опти-
ческая плотность «холостых» растворов: 0,005; 0,010; 0,010; 0,005;
0,010. Стандартное отклонение для этого ряда So равно
Г (5+10+10 + 5+10)2
)/ 5 + 10 + 10 + 5 + 10— -2-С - X.. JН]
/----------------------------------5--------- = 0,00271'.
4 I’
372 |
При чувствительности b = 0,073 (пример 6) предел обнаружения
бчлпп равен
6*0,0027 „ ол ,
---:---= 0,22 мгк/мл.
0,073
Определение погрешностей измерения. Рассчитаем случайную
погрешность прямых измерений по исходным данным примеров
3 и 4 по формуле: еСлуч = ^4--. Значение t при доверительной
вероятности 0,95 и f=8—1 равно 2,36 (см. табл. 33). При S=3,6
случайная погрешность равна —— =5,98.
У 2
Далее следует вычислить общую погрешность прямых изме-
рений, равную сумме случайной погрешности и погрешности шка-
лы прибора (систематическая погрешность предполагается рав-
ной 0). Погрешность шкалы прибора, найденная по цене деле-
ния прибора, равна 0,05. Следовательно, общая погрешность
равна 5,984-0,05=6,03.
При записи конечного результата следует помнить правила
округления результатов вычисления. Погрешность конечного ре-
зультата определяется наибольшей погрешностью. Конечный ре-
зультат не может быть более точным, чем наименее точное зна-
чение в цепи вычислений. Значит, результат должен оканчиваться
значащей цифрой того же разряда, что и наибольшая погреш-
ность в составе общей погрешности; числа, приведенные с боль-
шей точностью, приходится округлять. При этом лишние цифры
в целых числах заменяются нулями, а в десятичных отбрасы-
ваются. Если десятичная дробь оканчивается нулями, то нули
отбрасываются до того разряда, который соответствует разряду
погрешности. Если первая (слева направо) из заменяемых и от-
брасываемых цифр <5, то остающаяся цифра не меняется, если
она >5, то последующая увеличив.ается на 1.
В нашем примере наибольшей погрешностью в составе общей
является случайная погрешность. Она должна быть приведена с
точностью до первой значащей цифры, в нашем примере с точ-
ностью до единиц мВ. Значит, конечный результат вычисления
должен быть приведен с точностью до единиц. Для 4 проанали-
зированных в двукратной повторности образцов (пример 3)
усредненные результаты с учетом общей погрешности должны
быть записаны так:
(566±6) мВ, (523±6) мВ, (438+6) мВ, (513+6) мВ.
Рассчитаем случайную погрешность косвенных измерений.
Прежде всего нужно найти случайную погрешность аналитиче-
ского сигнала.
Пример 9. При анализе на Мп вытяжек 1 н. H2SO4 из
8 почв, выполненных в двукратной повторности, получены сле-
373
дующие оптические плотности:
0,15 0,22 0,14 0,30 0,19 0,23 0,34 0,16
0,17 0,19 0,14 0,32 0,18 0,21 0,36 0,19.
Разность между параллельными составляет:
0,02 0,03 0 0,02 0,01 0,02 0,02 0,03.
После преобразования по формуле х=0,01-а, а имеет значе-
ния: 2, 3, 0, 2, 1, 2, 2, 3.
Стандартное отклонение преобразованных значений признака
равно
22 + З2 + 02 + 22 + I2 + 22 4- 22 + 32 _ j 4
2-8
Стандартное отклонение исходных значений аналитического сиг-
нала равно 1,4 • 0,01 = 0,014.
Если вычисление стандартного отклонения вели по преобра-
зованным значениям после умножения или деления на какое-то
число, то для перехода от Sa к Sx выполняются обратные опера-
ции деления или умножения. Так и было сделано в вышеприве-
денном примере. Если при преобразованиях проводились одновре-
менно и сложение-вычитание и умножение-деление, то для обрат-
ных вычислений производится только умножение или деление.
Случайная погрешность аналитического сигнала равна
<0,95; 15-0,014 __2,13-0,014
у<2 — <2
От случайной погрешности аналитического сигнала через ве-
личину чувствительности (Ь = 0,073, пример 6) можно перейти к
случайной погрешности результата определения концентрации
Мп: -^- = 0 29.
0,073
Допустим, что систематическая погрешность аналитического
сигнала на фотоколориметре равна 0. Погрешность шкалы при-
бора, найденная по цене деления шкалы фотоколориметра, в еди-
ницах аналитического сигнала (в единицах оптической плотно-
Погрешность шкалы прибора в единицах кон-
0~(У73 = 0’14- Следовательно, общая погрешность
сти) равна 0,01.
центрации равна
определения Мп в вытяжке фотоколориметрическим методом рав-
на 0,29+0,14 = 0,43. Округленное значение общей погрешности
равно 0,4 мкг/мл.
По градуировочному графику найдено, что усредненным оп-
тическим плотностям растворов (пример 6) соответствуют кон-
центрации: 2,5; 3,1; 2,2; 4,5; 2,7; 3,3; 5,0; 2,6 мкг/мл. С учетом
найденной общей погрешности результаты должны быть записа-
374
ны так: (2,5+0,4) мкг/мл, (3,1 ±0,4) мкг/мл, (2,2+0,4) мкг/мл.
Сравнение средних арифметических. В практике анализа ча-
сто возникает задача оценки значимости различий между сред-
ними значениями. Этими средними могут быть результаты анали-
зов одной и той же пробы, выполненные в разных условиях или
разными методами, или результаты анализа различных проб.
Для оценки значимости различий между средними приме-
няются две разные формулы в зависимости от того, одинаковы
или различны дисперсии сравниваемых средних.
Если после оценки по критерию Фишера оказалось, что Si2 =
~S22, то вычисляется средневзвешенная дисперсия S2:
— («1 — 1) S] + («2 — 1) ^2
X ----------------------.
ni + п2 — 2
!8
0
2
Далее находится значение Т:
T = tp,rS
t берется при Р=0,95 и f=ti\ + n2—2.
Если разность сравниваемых средних |%i—х2|>Л то с веро-
ятностью 0,95 можно утверждать, что средние значимо разли-
чаются. Если |Xi—х21<Л то считать сравниваемые средние раз-
личными у нас нет оснований. Если после оценки по критерию
Фишера оказалось, что X? =#Х2, то значение Т вычисляется сле-
дующим образом:
где Хь Х2 — стандартные отклонения для сравниваемых средних,
Л = «1—1, f2=«2—1.
Далее, как и в первом случае, разность средних сравнивается с
вычисленным Т.
Оценим значимость разницы концентраций Мп в одной и той
же почве: 600 и 651 мг/кг, полученных спектральным и химиче-
ским методами (пример 6):
-А. = = 3,2. При /’=0,95 /1=9—1=8 и /2 = 8—1=7. Кри-
^2
терий Фишера равен 3,5 (см. табл. 34). Так как 3,2<?3,5, счита-
ем, что дисперсии значимо не различаются. Вычислим средне-
взвешенную дисперсию Хг:
<^-±-30!+7.54* ^84^44.
9 + 8—2
375
Находим значение Т при /0,95-,15=2,13:
7 = 2,13-44 I/ 1±1=43.
Г 9-8
Разность сравниваемых средних равна 651—600=51. Она боль-
ше вычисленного 7. Таким образом, с вероятностью 0,95 можно
утверждать, что средние концентрации Мп в почве, определен-
ные химическим и спектральным методами, значимо разли-
чаются.
Рассмотрим случай, когда стандартное отклонение для ре-
зультатов, полученных спектральным методом, было больше, а
sf 602
именно 60 мг/кг:—— =—-=4,0. Критерий Фишера равен 3,5.
S2
Так как 4,0>3,5, считать дисперсии одинаковыми не можем.
Вычислим значение 7 по второй формуле при tOi95;8=2,31 и
^0,95;7 = 2,36:
30а 60а
— -2,31 + —.2,36
= 56.
Разность средних, 51 мг/кг, меньше вычисленного значения Г.
С вероятностью 0,95 мы можем утверждать, что концентрации
Мп в почве, определенные химическим и спектральным метода-
ми, значимо не различаются. Это и понятно. Варьирование ре-
зультатов определения Мп спектральным методом увеличилось,
доверительный интервал среднего расширился и стал перекры-
вать доверительный интервал среднего для химического метода.
Это явление и служит доказательством отсутствия различий меж-
ду средними.
ЛИТЕРАТУРА
Дмитриев Е. А. Математическая статистика в почвоведении. М., Изд-во
Моск, ун-та, 1972.
ДоевФель К. Статистика в аналитической химии. М., «Мир», 1969.
Спиридонов В. П., Лопаткин А. А. Математическая обработка физико-
химических данных. М., Изд-во Моск, уи-та, 1970.
Оглавление
Введение............................................................... 3
Глава 1
ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ИОНОВ
ВОДОРОДА, НАТРИЯ, КАЛИЯ, КАЛЬЦИЯ,
ХЛОРИД-ИОНА, НИТРАТ-ИОНА
ПОТЕНЦИОМЕТРИЧЕСКИМ МЕТОДОМ
138
140
142
152
153
154
155
157
157
158
60
61
63
54
56
57
73
3
4
8
Принцип потенциометрического метода.................................... 9
Значение и область применения в почвоведении . . ,_... 9
Потенциал индикаторного электрода................................11
Электроды сравнения..............................................13
Э. д. с. ц^пи и принципы ее измерения............................19
Потенциометры....................................................24
Электроды для измерения pH.......................................33
Реакция почвенных вытяжек и суспензий и ее определение .... 38
Измерение pH водных суспензий почв 42
Измерение pH водных вытяжек из почв..............................48
Измерение pH солевых вытяжек из почв....................... . 50
f Измерение pH в почвенных пастах и непосредственно в почве . 52
Буферность почв........................................................53
Определение буферности почв.................................... 54
Активности ионов иатрия в почвах и почвенных суспензиях .... 56
Определение активности ионов натрия в водных суспензиях солон-
цовых и солончаковых почв........................................58
Определение содержания иатрия в водных вытяжках из засолен-
ных почв.........................................................59
Определение водорастворимого и поглощенного иатрия по Боуэру 61
Активности ионов калия в почвах и почвенных суспензиях .... 62
Определение активности ионов калия в почвенных суспензиях по
Орлову и Альзубайди..............................................63
Определение калийного потенциала и потенциальной буферной
способности почв в отношении калия...............................63
Определение калийного потенциала в водной вытяжке по Ульриху 65
Определение в почвах активностей других ионов ........................ 67
Определение активности хлорид-ионов...........................67
Определение активности ионов кальция ........................ 69
Определение активности нитрат-ионов ......................... 70
377
Глава 2
ОКЙСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫИ
ПОТЕНЦИАЛ ПОЧВ
И МЕТОДЫ ЕГО ИЗМЕРЕНИЯ
Электроды для измерения ОВП 7!»
Лабораторные измерения ОВП почв......................78
Полевые измерения ОВП почв...........................80
Глава 3
ПОЛЯРОГРАФИЧЕСКИЕ МЕТОДЫ
В ПОЧВОВЕДЕНИИ
Полярографическая кривая.......................................81
Ртутный капельный электрод и неполяризующнеся электроды
сравнения.......................................................82
Диффузионный ток и уравнение Ильковича ...... 84
Двойной электрический слой и емкостный ток.
Остаточный ток .................................................89
Миграционный ток и индифферентный электролит...............90
Полярографические максимумы................................... 91
Уравнение полярографической волны..........................93
Кинетические токи.............................................. 99
Полярографические методы..................................101
Количественные измерения в полярографическом анализе . . 103
Полярографическая аппаратура....................... . . 106
Области применения полярографии в почвоведении............113
Подготовка почв к анализу и условия полярографирования . . 115
Лабораторные работы
Определение чувствительности прибора и характеристик капилляра 116
Определение концентрации никеля и цинка по уравнению Иль-
ковича ...................................................... .118
Определение емкости катионного обмена почв ................... 119
Определение кальция в почвах ................................. 120
Определение железа в почвах...............................122
Определение никеля и цинка в почвах .......................... 123
Определение свинца в почвах ................................. ' 124
Глава 4
СПЕКТРОФОТОМЕТРИЧЕСКОЕ ИЗУЧЕНИЕ
СОСТАВА И СВОЙСТВ
ПОЧВЕННЫХ КОМПОНЕНТОВ
Взаимодействие света и вещества.......................127
Понятие «спектр поглощения»...........................128
Количественные закономерности поглощения света веществом
Закон Бугера—Бера.....................................129
Отклонения от закона Бугера — Бера................. . 131
Молярный коэффициент погашения и предел обнаружения вещества 131
Количественный анализ окрашенного компонента..........133
Анализ двухкомпонеитных смесей........................ 135
Ошибки при спектрофотометрических измерениях . . . 136
Способы количественного анализа. Аппаратура ........ 137
378
Светофильтры, монохроматоры, кюветы..............................138
Визуальные методы измерения интенсивности окраски . . . . 140
Фотоэлектрические методы измерения поглощения .... 142
Спектрофотометрические методы определения общего содержания фос-
фора, железа и органического углерода в почве ........................ 152
Определение общего содержания фосфора в почве методом молиб-
дено-фосфорной сини ... ... ..............153
Определение общего содержания железа в почве с применением
сульфосалициловой кислоты . ..............................154
Определение содержания в почве углерода органических соедине-
ний (гумуса).....................................................155
Определение в почвах некоторых подвижных форм соединений элементов 157
Определение подвижного двухвалентного железа в почвенной вы-
тяжке с применением 1,10-фенантролина ....... 157
Определение подвижных форм железа в вытяжке Тамма с приме-
нением нитрозо-К-соли............................................158
Определение подвижного марганца в почве с применением форм-
альдоксима ......................................................160
Определение подвижного алюминия в почве с применением хром-
азу рола-S ......................................................161
Определение в почве подвижного магния с применением титаново-
го желтого.......................................................163
Определение подвижного цинка в почве экстракционно-фотометри-
, ческим методом с применением дитизона................................164
Определение подвижной меди в почве экстракционно-фотометри-
ческим методом с применением диэт.илдитиокарбамината свинца . 166
Изучение гумусовых веществ . ................ ... 167
Исследование влияния pH раствора на ..........................' 173
Спектрофотометрическое определение хлорофилла и (или) его
производных в почвах.............................................173
Анализ почв по кривым спектральной яркости . . . . 174
Получение спектров отражения почв............................... 178
Глава 5
ИДЕНТИФИКАЦИЯ ОРГАНИЧЕСКИХ
И МИНЕРАЛЬНЫХ КОМПОНЕНТОВ ПОЧВЫ
МЕТОДОМ ИНФРАКРАСНОЙ СПЕКТРОСКОПИИ
Оптика и источники излучения...............................183
Методы подготовки образцов к съемке ИК-спектров .... 183
Градуировка прибора и получение спектров ................. 186
Спектрофотометр ИКС-14.....................................190
ИК-спектры гумусовых веществ.............................. 193
ИК-спектры минеральных компонентов почвы.................. 200
ПК-спектры почв.......................................... 205
Изучение ИК-спектров почвенных компонентов.................205
Глава 6
ОПРЕДЕЛЕНИЕ В ПОЧВЕ
СОДЕРЖАНИЯ МАКРО- И МИКРОЭЛЕМЕНТОВ
МЕТОДОМ ЭМИССИОННОГО СПЕКТРАЛЬНОГО АНАЛИЗА
Назначение и возможности метода в почвенных исследованиях ". 208
Основы теории эмиссионного спектрального анализа .... 209
Происхождение спектров.....................................210
Интенсивность спектральных линий...........................212
379
Источники возбуждения . . ......................
Спектральные приборы для эмиссионного анализа . . . . .
Регистрация спектров и определение интенсивности спектральных
линий ........................................................
Основные положения количественного спектрального анализа .
Определение в почвах щелочных металлов методом пламенной фото-1
метрии .......................................................
Определение общего содержания калия и натрия в почвах
Определение водорастворимых соединений калия и натрия в
почвах ............................. .........................
Определение содержания обменных калия и иатрия в почвах .
Определение доступного калия в почвах ..................
Определение емкости поглощения почв.....................
Приготовление стандартных и эталонных растворов
Ход анализа на пламенном фотометре ПФМ . . ,
Определение общего содержания микроэлементов в почвах
Фотографирование спектров почв и эталонов..................
Расшифровка спектрограммы и измерение почернений аналитичг
ских линий.............................................. -
Глава 7
ОПРЕДЕЛЕНИЕ В ПОЧВЕ
СОДЕРЖАНИЯ МАКРО- И МИКРОЭЛЕМЕНТОВ
МЕТОДОМ АТОМНО-АБСОРБЦИОННОГО
СПЕКТРАЛЬНОГО АНАЛИЗА
* Принцип метода.........................................
• Назначение и возможности метода в почвенных исследованиях
• Основы теории атомно-абсорбционного метода ...
Получение поглощающего слоя атомов. Атомизаторы
Оптические системы в атомно-абсорбционном анализе .
• Помехи, влияющие на результаты атомно-абсорбционного анализ
Определение в почвах макро- и микроэлементов ....
Разложение почв и подготовка раствора к анализу ....
Определение содержания обменною кальция и магния в почвах
Определение водорастворимых соединений кальция и магния
почвах ..................................................
Определение доступного растениям магния по Шахтшабслю
Определение подвижных форм соединений железа, алюминия
кремния в почвах (вытяжка Тамма).........................
Определение несиликатного железа в почвах по методу Мера -
Джексона.................................................
Определение подвижных форм соединений Мп, Zn, Си, Со
почвах ..................................................
Определение подвижных форм микроэлементов в почвах Нечерн
земной зоны по Пейве-Ринькису............................
Приготовление стандартных растворов.....................
Подготовка прибора к работе и техника измерения поглощения
Аппаратура и ее подготовка к работе .....................
Измерение поглощения и определение концентрации
Глава 8
ИЗУЧЕНИЕ ПОЧВЕННЫХ КОМПОНЕНТОВ
МЕТОДАМИ РЕНТГЕНОВСКОГО АНАЛИЗА
Природа и получение рентгеновских лучей ......
Дифракция рентгеновских лучей кристаллами
380
ентгеновских аппаратов ........................
получения дифракционной картины илистых фракций
281
285
ловки илистых фракций и приготовление ориентиро-
ратов для камерной съемки и рентгенодифракто-
сталлических решеток и рентгеновская диагностика
пп глинистых минералов ......
рентгенодифрактограмм илистых фракции почв
минералогического состава илистой фракции на при-
> .................................................
усовых кислот почв методом рентгеновского анализа
ха гумусовых кислот рентгенодифрактометрическим
техники безопасности при работе с рентгеновскими
ми ............. .......................
286
289
302
303
304
307
308
Глава 9
4ЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
>1Х И ОРГАНИЧЕСКИХ КОМПОНЕНТОВ ПОЧВ
основные узлы приборов для термического анализа 311
бразцов илистых фракций к термическому анализу . 317
характеристики основных групп илистых минералов . 317
«арактеристики некоторых неглинистых компонентов
..................................................322
яющие на величину, форму и температуру эффектов 323
и активации отдельных реакций при термической де-
шического вещества почвы..............................325
Глава 10
ШИЕ НЕКОТОРЫХ КОМПОНЕНТОВ ПОЧВ
РОННО-МИКРОСКОПИЧЕСКИМ МЕТОДОМ
жности метода в почвоведении.......................329
>да.............................................. 330
'кции электронного микроскопа .................... 332
азцов в электронном микроскопе.....................335
сеток и подложек.................................337
ъекта к исследованию. Нанесение образца на сетку
................................................339
ние объекта «оттенением»...........................340
нис «крашением»....................................343
для исследования поверхности твердых частиц . . 343
лов илистой фракции почв...........................345
тическая характеристика гумусовых кислот . . 350
онный микроскоп........................... 354
правила работы прн электронно-микроскопических
1ях..............................................355
Глава 11
МАТЕМАТИЧЕСКАЯ ОБРАБОТКА
РЕЗУЛЬТАТОВ НАБЛЮДЕНИИ
а
анализа....................................... 357
льтатов измерений..........................# 366
381
ФИЗИКО-
ХИМИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ
ПОЧВ
Заведующая редакцией
Л М Глазкова
Редактор Н А Жук
Художник А А Кущенко
Художественный редактор
М Ф Евстафьева
Технический редактор
3 С Кондрашова
Корректоры
Л А Костылева,
Т С Милякова
Тематически» тан 1980 г № 137
ИБ № 960
Сдано в набор 01 02 80 Подписано
к печати 27 08 80 Л 113063 Фор-
мат ЬОХЭО’/и Бумага тип № 1
Гарнитура Литературная Высокая
печать Уст п^ч л 24 0 Уч-изд
л 26,27 Тираж 3730 эчз Зак 281
Цена 1 р^б Изд № 775
Издательство
Московского университета
Москва К о, ул Герцена 5/7
Типография Изд-ва МГУ
Москва, Ленинские горы
ФИЗИКО-
ХИМИЧЕСКИЕ
МЕТОДЫ
ИССЛЕДОВАНИЯ
ПОЧВ