/











































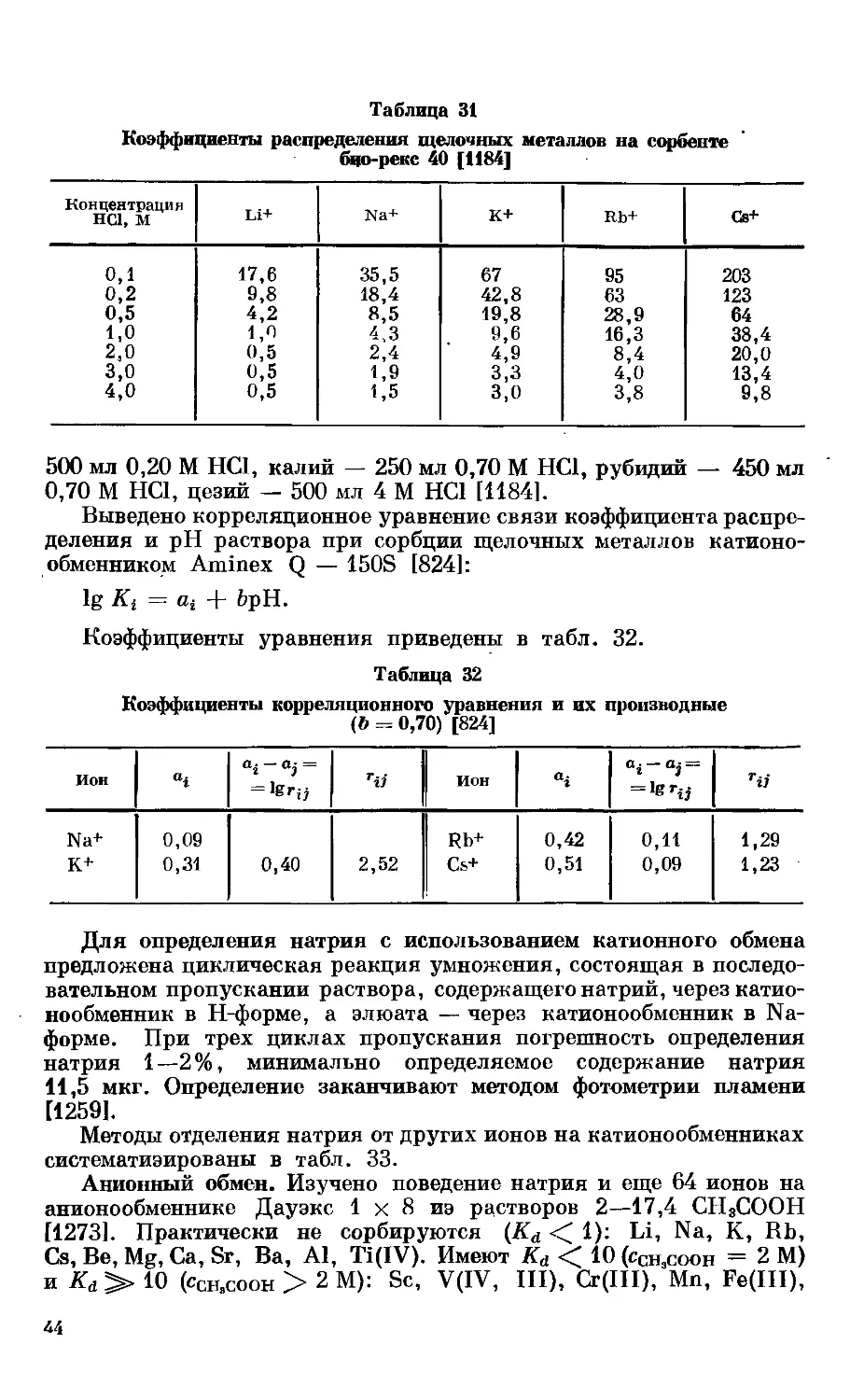


































































































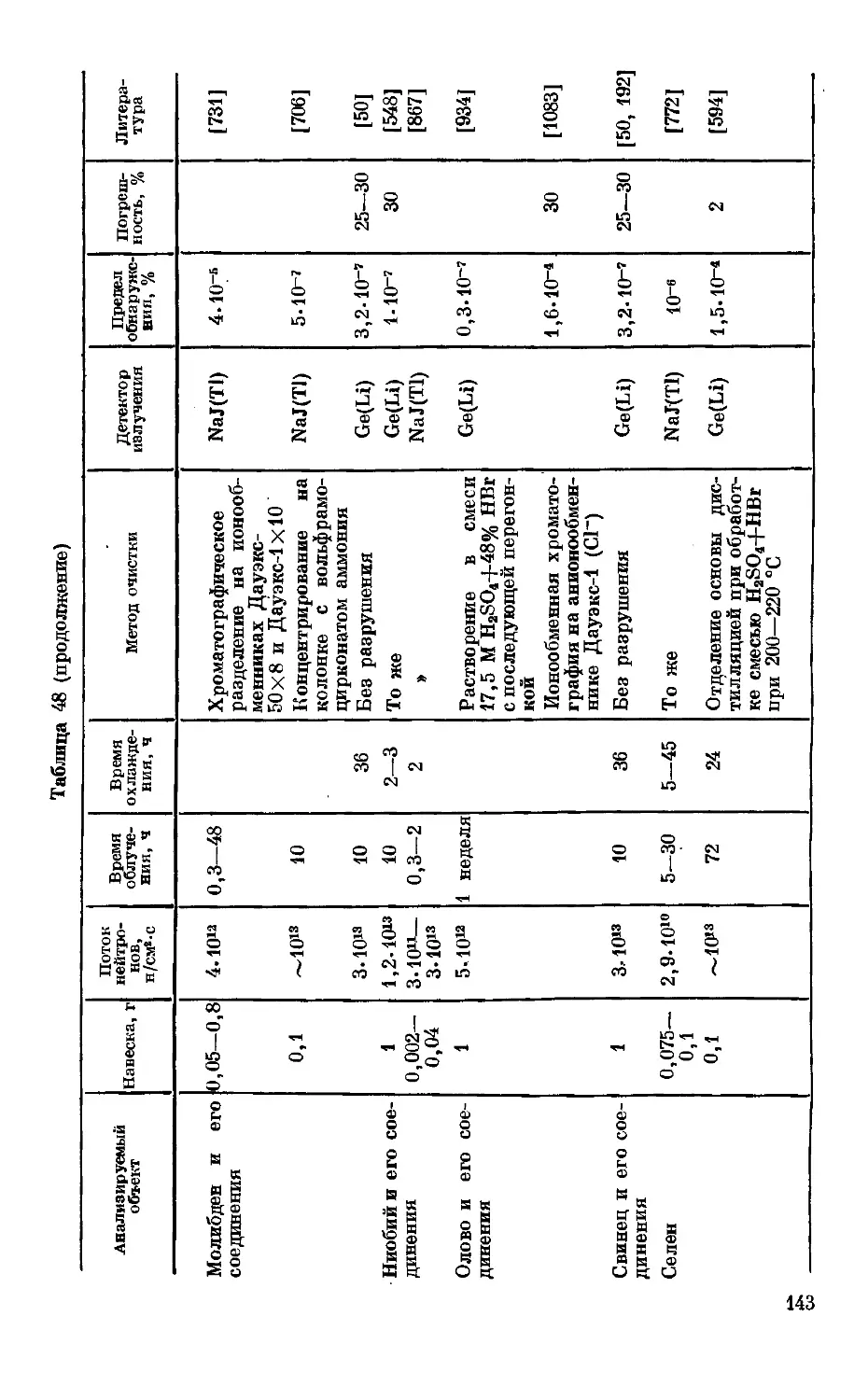

































































































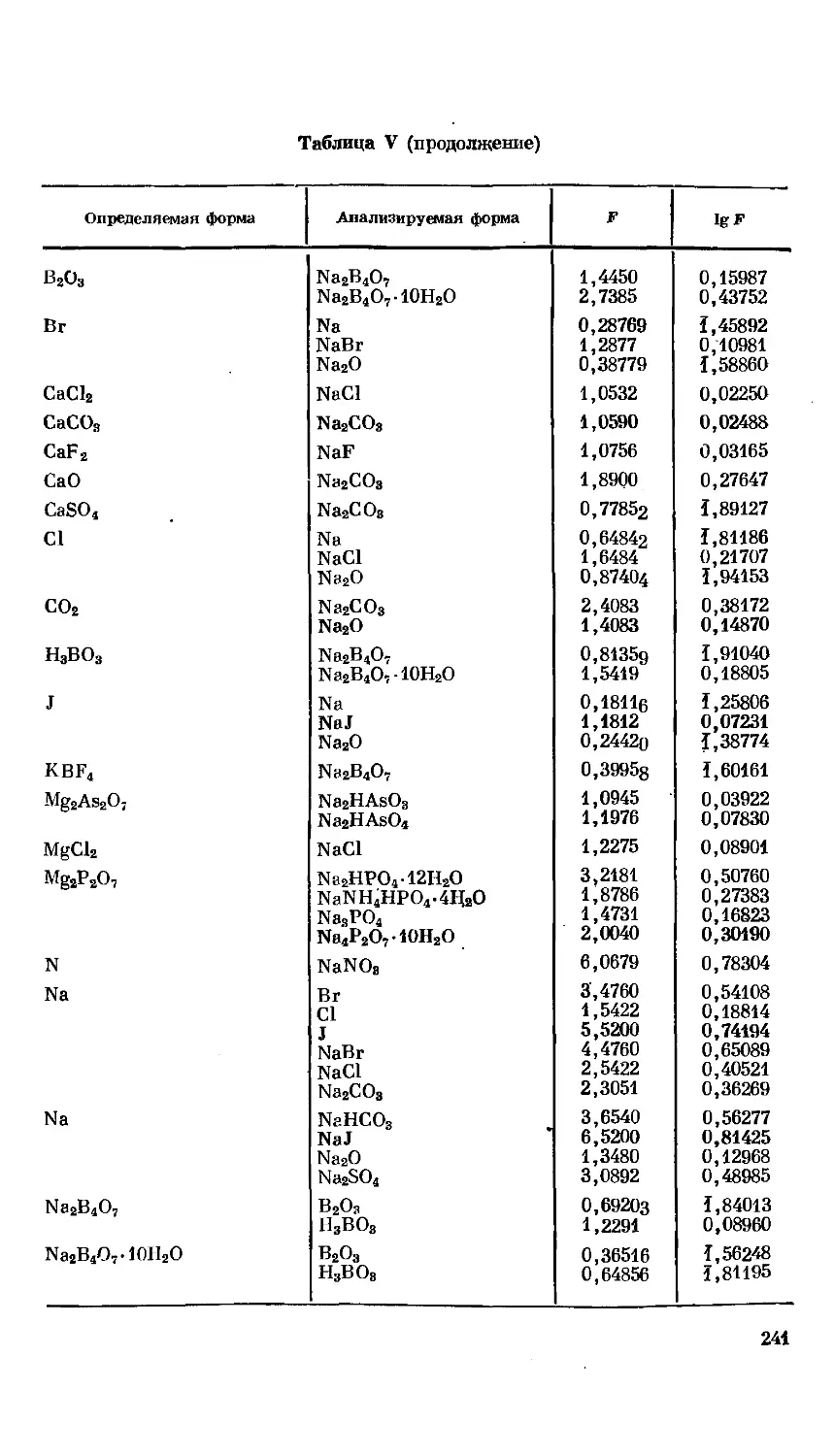



















Текст
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА И ОРДЕНА ОКТЯБРЬСКОЙ РЕВОЛЮЦИИ
ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ
ИМ. В. И. ВЕРНАДСКОГО
АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ
В. М. ИВАНОВ К. А. СЕМЕНЕНКО
Г. В.'ПРОХОРОВА Е.Ф. СИМОНОВ
НАТРИЙ
Ответственный редактор
кандидат технических наук
Ю. И. БЕЛЯЕВ
МОСКВА
«НАУКА»
1986
УДК 546.33 : 543
Иванов В. И., Семененко К. А., Прохорова Г. В., Симонов Е.Ф. Натрий.
—И.: Наука, 1986.—255 с. (Аналитическая химия элементов).
В монографии рассмотрено современное состояние аналитической химии натрия. При-
водятся физико-химические и химико-аналитические характеристики его простых и комп-
лексных соединений с неорганическими и органическими реагентами. Описаны основные
методы обнаружения, отделения и концентрирования натрия при анализе различных при-
родных и промышленных объектом, а также методы определения основы и примесей в
натрии и его соединениях. '
Книга предназначена для специалистов, работающих в области аналитической и
неорганической химии.
Табл. 86. Библиогр. 1292 назв.
Серия: «Аналитическая химия элементов»
Главный редактор
член-корреспондент АН СССР
Ю. А. Золотов
Редакционная коллегия:
И. П. Алимарин, Ю. А. Банковский, Ю. И. Беляев, М. И. Волынец,
В. М. Иванов, А. В. Карякин, Н. М. Кузьмин, Б. Ф. Мясоедов,
С. Б. Саввин, Е. И. Шумилова (ученый секретарь)
Рецензенты:
М. П. Волынец, И. М. Кузьмин
Адрес редколлегии: 117334» Москва, ул. Косыгина, 19,
Ордена Ленина и ордена Октябрьской Революции
Институт геохимии и аналитической химии им. В. И. Вернадского
Академии наук СССР
1'804000000-506
И б42(02)-86 ' 444-86'—г
© Издательство «Наука», 1986 г»
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернадско-
го АН СССР осуществляет издание серии монографий по аналитиче-
ской химии отдельных элементов. Эта серия — «Аналитическая хи-
мия элементов» — составит около 50 томов. Потребность в подобного
рода изданиях назрела давно. Вместе с тем у нас накопился огромный
опыт многочисленных лабораторий, и теперь стало возможным и
необходимым его подытожить. Таким образом возникло настоящее
издание — серия «Аналитическая химия элементов», которое осу-
ществляется впервые. Издание серии было начато по инициативе
академика А. П. Виноградова, который с 1958 по 1975 г. был ее
главным редактором.
Аналитическая химия любого элемента и его различных соеди-
нений в настоящее время представляется чрезвычайно разнообраз-
ной как вследствие сложности современных объектов исследования
и широты диапазона концентраций, которые бывает необходимо опре-
делить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий план как
в смысле содержания, так и последовательности изложения мате-
риала.
В монографиях содержатся общие сведения о свойствах элемен-
тов и их соединений. Затем рассматриваются химические реакции,
являющиеся основанием для аналитических методов. Методы, как
физические, так и физико-химические и химические, излагаются
применительно к количественному определению данного элемента,
начиная с анализа сырья, далее — типичных полупродуктов произ-
водства и, наконец, конечной продукции — металлов и сплавов,
оксидов, солей и других соединений и материалов. Как правило,
приводятся принципы определения и, где это необходимо, дается
точное описание всего процесса определения. Особое внимание уде-
ляется быстрым методам анализа. Самостоятельное место занимает
изложение методов определения так называемых элементов-примесей
в чистых материалах.
Обращается внимание на точность и чувствительность методов
в связи с общей тенденцией повышения чувствительности методов
определения следов элементов-примесей.
Монографии содержат обширную библиографию, доведенную до
последних лет; они рассчитаны на широкий круг химиков, в первую
очередь химиков-аналитиков исм£дддахшшиио—ияса»»у«ю»-»-9а®од-
ских лабораторий различных отр ^слейД^тщ^наяятпмекМбсими] :ов-
преподавателей и студентов хил г-----------—а------------
К составлению монографий при]
196084,г. Санкт-Петербург
Цветочная,18____________
3
циалисты, имеющие опыт работы в области аналитической химии того
или иного химического элемента.
Отдельные тома серии «Аналитическая химия элементов» выходят
самостоятельно по мере их подготовки. Вышли в свет монографии,
посвященные торию, таллию, урану, рутению, молибдену, калию,
бору, цирконию и гафнию, кобальту, бериллию, редкоземельным
элементам и иттрию, никелю, технецию, прометию, астатину и фран-
цию, ниобию и танталу, протактинию, галлию, фтору, селену и тел-
луру, алюминию, нептунию, трансплутониевым элементам, платино-
вым металлам, радию, кремнию, германию, рению, марганцу; кад-
мию, ртути, кальцию, фосфору, литию, олову, серебру, цинку, зо-
лоту, рубидию и цезию, вольфраму, мышьяку, сере, плутонию, ба-
рию, азоту, стронцию, сурьме, хрому, брому, ванадию, актинию,
хлору.
Готовится к печати монография, посвященная аналитической
химии Свинца.
Мы обращаемся с просьбой ко всем читателям присылать свои
замечания и отзывы о монографиях.
ПРЕДИСЛОВИЕ
Соединения натрия известны человеку с глубокой древности. За
последние годы металлический натрий и его соединения все шире
используют в науке и технике. В связи с этим возрастают требования
к чистоте натрия, к чувствительности методов определения примесей
в натрии и его соединениях.
Аналитическую химию натрия начали особенно широко изучать
и развивать начиная с 50-х годов XX столетия. Известные, став-
шие классическими гравиметрические методы определения натрия,
основанные на образовании малорастворимых тройных ацетатов,
были модифицированы за счет растворения осадка в кислотах и кос-
венного определения натрия титриметрическим определением ура-
Ha(VI) или двухвалентного катиона с использованием реакций окис-
ления-восстановления или комплексообразования. Вследствие этого
возросла экспрессность анализа.
Значительное развитие получил пламенный вариант атомно-
эмиссионного и атомно-абсорбционного анализа, применимый для
определения натрия практически в любых природных и промышлен-
ных объектах. К ставшим традиционными электротермическим и
пламенным способам атомизации добавились лазерные источники
возбуждения. Разработанные методы сочетают очень высокую чув-
ствительность (до 10-16 г натрия) с высокой селективностью, особен-
но при ступенчатой лазерной ионизации атомов.
Высокочувствительны активационные методы определения нат-
рия (до 10“10 г натрия), преимущественно применимые для опреде-
ления натрия в особо чистых химических элементах и их соедине-
ниях. Методы выделения радиоизотопов натрия С носителем требуют
дальнейшего усовершенствования.
В связи с развитием химии координационных-соединений натрия
появились новые электрохимические методы с использованием нат-
рий-селективных, в том числе мембранных, электродов. Сравнитель-
но невысокая чувствительность этих электродов (pNa ~ 6) компен-
сируется их высокой селективностью.
Следует подчеркнуть, что успех в развитии физико-химических и
физических методов применительно к определению натрия невозмо-
жен без изучения химических свойств натрия, поскольку задача зна-
чительного снижения пределов обнаружения натрия не может быть
решена без предварительного отделения и концентрирования натрия
либо отделения сопутствующих компонентов.
Литература по аналитической химии натрия чрезвычайно рассре-
доточена. Отсутствуют обзоры, систематизирующие сведения о новых
методах определения натрия. Единственная монография Р. Фрезе-
5
ниуса и Г. Яндера по аналитической химии натрия (Fresenius R.',
Jander G. Handbuch der analytischen Chemie. Berlin: Springer Ver-
lag, 1940. Vol. III. Bd. la) неполна и значительно устарела. Наша
задача сводилась к обобщению и оценке имеющегося материала.
Авторы рассмотрели большинство имеющихся методов обнаружения,
отделения и определения натрия. Подробно описаны наиболее надеж-
ные и проверенные методы. На основании литературных данных и
собственного опыта отмечены чувствительность, селективность, на-
дежность и перспективность методов. Особое внимание авторы обра-
щали на методы отделения натрия или сопутствующих ионов и на
методы устранения мешающего влияния сопутствующих ионов при
определении натрия в биологических объектах, учитывая специфич-
ность пробоотбора и пробоподготовки и особенности анализа этих
объектов. В большинстве случаев для определения натрия в этих
объектах можно использовать методы, проверенные при анализе
других объектов.
В список литературы включены работы в основном начиная
с 1950 г. При написании монографии была использована литература
до 1984 г.
Главы I—VI, XI написаны В. М. Ивановым, глава VII —
Г. В. Прохоровой, глава VIII — К. А. Семененко и В. М. Ивановым,
глава IX — Е. Ф. Симоновым, глава X — В. М. Ивановым и
К. А. Семененко.
Авторы выражают искреннюю благодарность профессору
Н. М. Кузьмину, доктору химических наук М. П. Волынец за рецен-
зирование монографии, полезные советы и замечания, кандидату
технических наук Ю. И. Беляеву за редактирование.
Критические замечания читателей по настоящей книге авторы
примут с большой благодарностью.
В. М. Иванов, К. А. Семененко,
Г. В. Прохорова, Е. Ф. Симонов
Глава 1
ФИЗИКО-ХИМИЧЕСКАЯ
И ХИМИКО-АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА
НАТРИЯ
ОБЩИЕ СВЕДЕНИЯ
Натрий в виде соединений известен человеку с глубокой древ-
ности. Металлический натрий впервые получен в 1807 г. Г. Дэви
электролизом NaOH.
Натрий — элемент главной подгруппы первой группы периоди-
ческой системы элементов Д. И. Менделеева. Его порядковый номер
11, атомная масса 22,9923 ± 0,0056 [604]; 22,9898 [506]; 22,997139
(по кислородной шкале) [506]. Естественный элемент содержит 100 %
2SNa. Известны радиоактивные изотопы натрия от 20Na до 26Na.
Изотопный состав натрия и характеристика изотопов подробно рас-
смотрены в главе IX «Активационные методы определения натрия».
Распространенность натрия в земной коре составляет 2,4% ат.
(2,64% мае.) [506]. По распространенности натрий занимает шестое
место среди других элементов. Натрий обнаружен в атмосфере Солн-
ца и в межзвездном пространстве. В гидросфере натрий содержится
в виде растворимых солей в количестве около 2,9% (при общей кон-
центрации солей в морской воде 3,5—3,7%). Абсолютное содержание
натрия в морской воде Составляет около 1,5-1016 т [226].
Вследствие своей большой реакционной способности натрий
в земных условиях находится только в виде солей. В сочетании с ще-
лочноземельными, редкоземельными элементами, магнием и алюми-
нием натрий входит в состав многих природных силикатов. Минералы,
в состав которых входит натрий, следующие .[46]: анальцим —
Na[AlSi2Oe]-H2O (14,07% Na2O); арфведсонит — Nas(Fe, Mg)4(Fe,
Al) [Si4Ou]2[OH]2; боронатрокальцит — NaCaB6O9-8H2O (7,7%Na2O);
бура — Na2B40?-10H20 (12,6% Na); галит — NaCl (39,4% Na);
гаюин — NaeCa[AlSiO4]e[SO4]; гейландит — (Ca, Na2)[AlSi3O8]2-
•5H2O; десмин — (Na2, Ca)[Al2SieOie]-6H2O; жадеит — Na,
Al [Si2Oe]; канкринит — Na3Ca[AlSiO4]3[CO3, SO4]-nH2O (15,6—
18,9% Na2O); криолит — Na3AlFe (32,8% Na); лазурит —
Na8[AlSiO12]e[SO4] (16,8% Na2O); лопарит — (Na, Ce, Ca)(Nb, Ti)Os
(7,8—9,0% Na2O); мирабилит — Na2S04-10H20 (19,3% Na2O);
натролит — Na2[Al2Si3O10]-2H2O (16,3% Na2O); нефелин —
Na[AlSiO4]; нозеан — Na8[AlSiO4]e[SO4]; пирохлор — (Na, Ca...)2-
•(Nb, Ti...)2Oe[F, OH] (1—6% Na2O); плагиоклаз — (100 — n)Na-
7
• [AlSigO8] • nCa[Al2Si2O8] (0,76—2,92% Na2O); роговая обманка —
Ca2Na(Mg, Fe(II))4(Al, Fe(III))[(Si, A1)4()11|2|()H]2; скаполит —
(75 — n)Na4[AlSi3O8]3Cl-nCa4[Al2Si2O8]3[SO4, CO3]; сода — Na2CO3-
• 10H2O (21,6% Na2O); содалит — Na8[AlSiOJeCl2 (25,5% Na2O);
тенардит — Na2SO4 (43,7% Na2O); чилийская селитра— NaNO3
(36,5% Na2O); шабазит — (Ca, Na2)[AlSi2Oe]2-6H2O; эвдиалит —
(Na, Ca)eZrSie0i8[0H, Cl] (11*6—17,3% Na2O). Кроме того, имеются
минералы: бериллонит — NaBePO4, виллиомит — NaF; глаубе-
рит— Na2Ca[SO4]2; катаплеит — Na2ZrSi3O9-2H2O; натрофи-
лит — NaMnPO4; рибекит — Na3Fe3Fe2[Si4On]2[O, ОН]2; трона —
NaH[CO3]2-2H2O.
Важнейшие из минералов натрия — галит, мирабилит, чилийская
селитра, криолит и бура.
Месторождения натрия широко распространены на Земле [226].
Богатые залежи каменной соли имеются в СССР, в том числе в соленых
озерах Эльтон и Баскунчак, в США (штаты Техас, Оклахома, Кан-
зас), в Австрии. Мирабилит и тенардит встречаются в СССР (залив
Кара-Богаз-Гол, Сибирь), в США (западные штаты), на острове Си-
цилия, в Испании, в Северной Африке. Залежи селитры имеются
в Южной Америке (Перу, Чили), богатые залежи троны — в Север-
ной Африке (АРЕ, Алжир, Судан, Ливан), а также в СССР (Армения,
Сибирь) и в США (штаты Невада, Калифорния).
Небольшие количества натрия находятся в растениях. Так,
в свежей траве тысячелистника обнаружено всего 6-10~4% натрия.
Гораздо богаче натрием морские растения. Свежая морская трава
содержит 0,547%, а ее зола — 16,78% натрия. Соединения натрия
входят в состав животных организмов главным образом в виде NaCl.
В плазме крови человека ионы натрия составляют 0,32%; в ко-
стях — 0,6%; в мышечной ткани — 0,6—1,5%.
Металлический натрий (чистый или в виде сплавов с другими ме-
таллами) находит разнообразное применение в качестве теплоноси-
теля в клапанах авиационных двигателей, в машинах для литья под
давлением (для охлаждения плунжера), а также в ряде химических
процессов, где возникает необходимость равномерного обогрева
в пределах 450—650° С. Особое место занимает применение натрия и
его сплава с калием в качестве жидкометаллического теплоносителя
в ядерных энергетических установках благодаря малым эффектив-
ным сечениям поглощения нейтронов, высокой температуре кипения,
высокому коэффициенту теплопередачи, хорошей теплостойкости,
а также отсутствию взаимодействия с обычными конструкционными
Материалами при высоких температурах, развиваемых в современ-
ных энергетических ядерных реакторах.
Характерное свечение паров натрия используют в специальных
светильниках. В металлургии натрий применяют как восстановитель,
а также для упрочнения сплавов. Например, сплав на основе свинца,
идущий на изготовление железнодорожных осевых подшипников,
содержащий 0,58% натрия, 0,04% лития и 0,73% кальция, имеет
твердость по Бринеллю 34 кг/мм2, прочность на сжатие 1800—
2200 кг/см2 и может сжиматься на 28% без растрескивания. Сплав,
8
содержащий 10% натрия и 90% свинца, применяют в производстве
тетраэтилсвинца — наиболее эффективного из антидетонаторов мо-
торных топлив [226].
Соединения натрия входят в состав многих лекарственных пре-
паратов (растворимый норсульфазол, салицилат натрия и др.), а так-
же физиологического раствора. Радиоактивный изотоп 24Na приме-
няют для лечения некоторых форм лейкемии и в диагностических
целях. В виде селитры натрий используют для удобрения почвы,
для консервирования (пищевая промышленность), в металлообраба-
тывающей промышленности как компонент солевых закалочных ванн,
как окислитель (производство стекла). Нитрит натрия применяют
в производстве красителей, иода, в пищевой промышленности и
медицине. Сульфат натрия используют в стекольном производстве,
при получении сульфатной целлюлозы, в текстильной, мыловарен-
ной и кожевенной промышленности, в цветной металлургии, в меди-
цине и ветеринарии. Тиосульфат натрия применяют в фотографии
как закрепитель, в текстильной, кожевенной промышленности, меди-
цине и ветеринарии. Фосфаты натрия применяют в качестве мою-
щих и водосмягчающих средств, при обогащении руд, в текстильной,
кожевенной, различных областях пищевой промышленности, фото-
графии, в электролитических процессах. Хлорид натрия — важный
пищевой продукт — служит также для консервирования мяса, рыбы,
применяется для кормления скота; является одним из главных
видов химического сырья и используется для получения едкого нат-
ра, хлора, соды, сульфата натрия [226].
По данным [784а], вклад различных стран в мировое производство
NaCl составляет (в %): США — 27; КНР — 10; СССР — 8; ФРГ —
7; Франция — 4; Индия— 4; Австралия — 3; Канада — 3; Италия—
3; Мексика— 3; ПНР — 2; остальные страны — 26. В 1974 г. США
произвели 42,5 млн. т NaCI. Его потребление в различных отраслях
хозяйства составило (в %): получение NaOH и металлического нат-
рия электролизом — 49,4; очистка дорог от снега— 16,6; получение
Na2COs — 9,8; получение остальных солей натрия — 7,6; на нужды
животноводства — 4,2; домашнее хозяйство — 2,7; смягчение воды —
1,8; мясная промышленность — 1,3; текстильная промышленность
и красильное производство — 0,4.
Гидрокарбонат натрия применяют в хлебопечении, в пищевой
промышленности, в медицине, а также при изготовлении зарядов
для огнетушителей. Безводный карбонат натрия применяют для про-,
изводства стекла, алюминия, мыла, едкого натра, моющих средств,
различных солей и красок, для обессеривания чугуна, очистки нефти,
мойки шерсти, стирки белья. Соду каустическую (технический NaOH)
потребляют для производства искусственного волокна, мыла, алю-
миния, красок, в писчебумажной и целлюлозной промышленности,
для отделки и мерсеризации хлопчатобумажных тканей, очистки
нефти [226].
Натрий металлический получают электролизом расплавов NaCl
с добавками КС1, NaF, СаС12 при температуре 575—585° С. Реже его
получают электролизом NaOH. Натрий можно получить также дей-
9
ствием на его соли углерода и других восстановителей при темпера-
турах выше температуры их плавления, однако этот способ не имеет
промышленного значения [12].
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА НАТРИЯ
Натрий — металл серебристо-белого (в очень чистом виде — ро-
зового) цвета с твердостью по минералогической шкале 0,5 [506].
Плотность натрия 0,97 г/см3; температура плавления 98° С; темпера-
тура кипения 890° С [330].
Имеются указания на возможность существования у натрия двух
аллотропических модификаций с точкой взаимного превращения
между —20° С и +20° С. Натрий очень мягок и легко режется ножом.
Пары натрия окрашены в пурпурно-красный цвет.
По разным источникам, размер радиуса иона натрия колеблется
от 0,095 до 0,099 нм [506]. Первый потенциал ионизации натрия равен
8,22-10~19 Дж (5,138 эВ), второй — 75,68-Ю"19 Дж (47,29 эВ). Низ-
кий потенциал ионизации внешнего электрона и тот факт, что обра-
зующийся ион Na+ имеет конфигурацию атома инертного газа и яв-
ляется, таким образом, сферическим и слабо поляризуемым, влияет
на химические свойства щелочных металлов, особенно на свойства их
ионов М+. Другие состояния окисления этих элементов неизвестны:
исходя из величин вторичной ионизации трудно предположить их
существ ов ание.
Некоторые свойства атомов натрия и других металлов I группы
сопоставлены в табл. 1.
Таблица 1
Некоторые свойства атомов металлов I группы [221]
Элемент Порядковый номер Электронная конфигура- ция Потенциал ионизации- 7l-10w, Дж Е°, В для реак- ции M+(aq) + е = = М(тв.)
1 и
Li 3 [He]2s 8,62 121,00 —3,02
Na 11 [Ne]3s 8,22 75,68 —2,71
К 19 [Аг] 4s 6,94 50,90 —2,92
Rb 37 [Кг] 5s 6,68 43,78 —2,99
Cs 55 [Xe]6s 6,23 37,44 —3,02
Fr 87 [Rn]7s
Давление паров натрия при различной температуре приведено
ниже [506]:
т, к р, Н/м2 т, К Р, Н/м2 т, к Р, Н/м2
454,8 6,56-10- з 958,6 1,17-10* 1327,6 3,95-10®
534,2 3,80-10- 1 1073,3 4,55-10» 1383,7 5,56. IO®
870 3,32 1118,3 7,2-10* 1408,2 6,57-10*
893,5 4,76-10® 1228,1 1,91-105
10
Вычислена энтропия натрия в газообразном состоянии [506]:
т, к 8® , Дж/(кг-ат-град) т, К 8°, Дж/(кг-ат-град)
298 153697,43 2000 193304,55
500 164457,50 2500 197951,90
1000 178901,96 3000 201803,76
1500 187317,43
Сродство к электрону у натрия 1,344-10-19 Дж (для сравнения
у лития и калия — 1,312-10"19 Дж) [506].
Теплота гидратации натрия равна 423,19 кДж/ион [571, с. 10].
В табл. 2 приведены некоторые физические константы по гидратации
щелочных элементов.
Таблица 2
Данные о гидратации ионов элементов I группы
в водных растворах [737]
Характеристика Li+ Na+ К+ Rb+ Cs+
Кристаллографический радиус *х 0,060 0,095 0,133 0,148 0,169
Радиус гидратированного иона, нм 0,340 0,276 0,232 0,228 0,228
Гидратное число *2 25,3 16,6 10,5 9,9
Энергия гидратации, ккал/моль 124,4 97,0 77,0 71,9 61,1
Подвижность ионов *3 33,5 43,5 64,6 67,5 68,0
** Для к. ч. 6. *» Приблизительное значение. *• При бесконечном разбавлении, 18° С.
Гидратное число натрия зависит от сорастворителя. Например,
при 25° С в 50—60%-ной HNOS обнаружен ион [Na(H2O)2]+ [737].
По данным ИК-спектроскопии в области 6000—7500 см-1 в водных
растворах числа гидратации возрастают в ряду NaCl NaBr <^ NaJ
[963]. Константы Скорости диссоциации воды в аква-ионах приведены
ниже [571, с. 48]:
Ион к, с-» Ион к, c-* Ион к, c-*
Na+ 8,8-107 Zn2+ 3-107 Co2+ S3-107
К+ 1,5.10s Cd2+ 3-10» Ni2+ 5-10»
Mg2+ 1.10s Mn2+ 2-10e Cu2+ 2-10s
Ca2+ 1-10® Fe2+ 2-10*
Приведены сольватные числа некоторых ионов в неводных
средах (табл. 3) и сольватные числа электролитов, определенные раз-
личными методами (табл. 4).
Энергия сольватации зависит от природы растворителя (табл. 5);
например, в ацетоне она на 42—63 кДж, а в ацетонитриле всего на
8—12 кДж ниже, чем в воде.
11
Таблица 3
Сольватные числа ионов, определенные методом измерения подвижности [658]
Растворитель Li+ Na+ к+ ci- вг- I-
Ацетон 2,9 2,6 2,0 1,0 1,0
С2Н5ОН 5,0 4,0 3,0 2,0 2,0 1,0
СН3ОН 5,0 5,0 4,0 1,5 1,0 1,0
ДМФА 3,2 3,0 2,0 0,5 0,5 0,5
ДМСО 4,3 2,3 2,4 0,6 0,5 0,4
Примечание. ДМФА — диметилформамид, ДМСО — диметилсульфоксид
Таблица 4
Сольватные числа электролитов, определенные различными методами [658]
Электролит По измерению сжимаемости В соответствии с правилом Робинсона—Стокса
метанол этанол метанол этанол
NaCl 4,7 3,7
NaBr 5,6 2,9 3,2 3,3
NaJ 6,2 3,2 2,2
КВг 5,2 2,7 2,5
Таблица 5
Энергии сольватации натрия в различных растворителях [181]
Растворитель Дирлектрическая проницаемость Энергия сольватации
кДж/г-ион ккал/г-ио н
нсоон 57,0 384,3 91,5
N2H4 51,7 386,4 92,0
CHSCN 36,7 380,1 90,5
C5HUOH 15,8 357,0 85,0
Ацетон 19,0 336,0 80,0
В метаноле при 25° С энергия сольватации натрия равна
436,4 кДж/г-ион (103,9 ккал/г-ион) [184].
Вычислены энтропийные характеристики ближней и дальней
сольватации натрия в одноатомных спиртах [234]: Досолы, -- изме-
нение энтропии при сольватации иона в одноатомных спиртах;
Д£п — изменение энтропии молекул одноатомных спиртов под дей-
ствием ионов; Д5дальн и ДДсл — соответственно изменение энтро-
пии в области дальней и ближней сольватации (табл. 6).
12
Таблица 6
Некоторые энтропийные характеристики (э. е.) ионов натрия
в одноатомных спиртах [234]
Спирт ~ Д®СОЛЬВ -ASjj “ДЯдальн -ДЯбл
CHjOH 36,5 14,8 9,9 17,1
С2НБОН 41,1 19,4 12,9 18,7
н-С4Н9ОН 22,9
Определены стандартные изменения энтальпии (Д2/раств) при
растворении некоторых солей натрия в ДМФА (табл. 7).
Таблица 7
Стандартные изменения энтальпии при растворении солей натрия
в ДМФА при 26° С [235]
Соль т, моль/ /кг Н2О Vm А#раств, ккал/моль Соль т, моль/ /кг Н,О Jm д траста, ккал/моль
NaBr NaJ 0,009425 0,01288 0,02543 0,03261 0,05357 0,01475 0,01652 0,0180 0,09709 0,1135 0,1595 0,1806 0,2315 0,1215 0,1275 0,1342 —6,720 —6,650 —6,530 —6,460 —6,290 —11,520 —11,450 —11,270 NaJ NaNOs 0,02212 0,02514 0,03003 0,02313 0,03003 0,0434 0,0470 0,05219 0,0650 0,1488 0,1585 0,1733 0,1521 0,1733 0,2083 0,2168 0,2285 0,2550 —11,130 —10,820 —10,730 —1,910 —1,790 —1,730 —1,790 —1,630 —1,635
В табл. 8 приведены коэффициенты активности NaNO3 в водных
растворах.
Метод расчета констант диссоциации в водноорганических сме-
сях с помощью ионного обмена применен для определения константы
Таблица 8
Средние коэффициенты активности NaNOa в водных растворах
при 298—623 К (7-10') [147]
Концентра- ция NaNO„ моль/кг Н2О 298 К 423 473 523 573 623 К
1,0 548 516 478 394 272 138
1,5 507 502 468 377 244 116
2,0 478 495 465 368 277 102
3,0 437 489 465 357 206 86
4,0 408 481 462 346 190 76
6,0 370 457 441 317 167 63
8,0 346 428 413 288 149 54
10,0 329 410 396- 271 138 49
13
устойчивости NaNOs в смесях воды с уксусной кислотой [1099].
Значения рК быстро уменьшаются с ростом содержания воды в смеси
от 4,8 до 2,5% мае.
Вычислены средние коэффициенты активности NaCl в интервале
температур 283—318 К в водно-спиртовых смесях, содержащих 0,7
мольных долей метанола, этанола, н-пропанола [447]. Концентра-
цию NaCl изменяли в пределах от 10~8 т до значений, близких к на-
сыщению раствора. Коэффициенты активности зависят от присут-
ствия в растворе газов воздуха. Кривые зависимости 1g y-j. от j/m
имеют четко выраженные экстремумы и прямолинейные участки.
НЕОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ НАТРИЯ
Простые соединения
Ионы натрия, как и остальных щелочных металлов, бесцветны.
Почти все соли, образуемые ими с обычными кислотами, хорошо рас-
творимы в воде. В противоположность выделяющимся обычно без
кристаллизационной воды солям калия, рубидия и цезия для солей
лития весьма характерно образование кристаллогидратов. Натрий
занимает промежуточное положение.
Гидроксид натрия. Как и гидроксиды остальных щелочных ме-
таллов, гидроксид натрия очень гигроскопичен. Взаимодействует
с большинством соприкасающихся с ним материалов; в расплавлен-
ном состоянии сильно разъедает стеклянную, фарфоровую, а при
доступе воздуха й платиновую посуду.
В табл. 9 сопоставлены свойства гидроксидов щелочных металлов.
Таблица 9
Некоторые свойства гидроксидов щелочных металлов [330]
Свойство LiOH NaOH КОН RbOH CsOH
Плотность, г/см3 2,5 2,1 2,0 3,2 3,7
Температура плавления, °C 450 328 360 300 275
Теплота образования из М2О, ккал/моль И,2 18,0 21,7 25,7 25,0
Теплота растворения, ккал/ /моль Растворимость в воде, моль/ /л Н2О 4,8 10,4 13,2 14,7 16,8
при 15° С ' 30° С 5,3 ; 26,4 19,1 17,9 25,8
5,4 29,8 22,6 16,9 20,2
В противоположность гидроксидам остальных металлов NaOH
и его аналоги не отщепляют воду даже при нагревании до своих вы-
соколежащих! температур кипения (NaOH — 1388° С; КОН —
1324° С). Исключение составляет LiOH, который начинает терять
воду уже око л о 600° С. Из растворов LiOH и NaOH выделяются при
14
обычных условиях с одной молекулой кристаллизационной воды,
КОН с двумя.
В воде и этаноле NaOH хорошо растворим, причем растворимость
сопровождается выделением большого количества тепла. Сведения
о плотности растворов NaOH приведены в табл. 10. В водных раство-
рах молекулы NaOH практически нацело диссоциированы.
Таблица 10
Плотность водных растворов едкого натра при 20° С [505]
ПЛОТНОСТЬ г/мп Количество NaOH, г Плотность г/мл Количество NaOH, г Плотность г/мл Количество NaOH, г
в 100 г Р-Ра в 1 л р-ра в 100 г Р-ра в 1 л [р-ра в 100 г Р-Ра в 1 л р-ра
1,02 1,938 19,77 1,19 17,35 206,4 1,36 33,07 449,7
1,03 2,838 29,23 1,20 18,26 219,1 1,37 34,04 466,3
1,04 3,746 38,96 1,21 19,17 231,9 1,38 35,02 483,3
1,05 4,655 48,88 1,22 20,08 245,0 1,39 36,00 500,4
1,06 5,564 58,98 1,23 20,99 258,2 1,40 37,00 517,9
1,07 6,473 69,26 1,24 21,90 271,6 1,41 37,99 535,7
1,08 7,378 79,68 1,25 22,82 285,2 1,42 39,00 553,7
1,09 8,282 90,27 1,26 23,73 299,0 1,43 40,00 572,0
1,10 9,191 101,1 1,27 24,65 313,0 1,44 41,03 590,8
1,11 10,10 112,1 1,28 25,56 327,2 1,45 42,06 609,9
1,12 11,01 123,3 1,29 26,48 341,6 1,46 43,11 629,4
1,13 11,92 134,7 1,30 27,41 356,3 1,47 44,16 649,2
1,14 12,82 146,2 1,31 28,34 371,2 1,48 45,22 669,3
1,15 13,73 157,9 1,32 29,27 386,3 1,49 46,28 689,6
1,16 14,63 169,7 1,33 30,20 401,6 1,50 47,32 709,8
1,17 15,54 181,8 1,34 31,15 417,4 1,51 48,37 730,4
1,18 16,44 194,0 1,35 32,10 433,3 1,52 49,44 751,4
Некоторые термодинамические характеристики неорганических
соединений натрия, их растворимость и плотность приведены
в табл. 11, 12. Термические константы неорганических соединений
натрия приведены в справочнике [71].
Из указанных солей в качестве первичных стандартов исполь-
зуют Na2B4O7-10H2O, Na2COs в кислотно-основном титровании,
Na2SO4 и NaCl — в титриметрии для приготовления стандартных рас-
творов сульфата и хлорида, в гравиметрии в качестве весовой формы
при определении натрия.
Тетраборат натрия является исходным веществом для установки
титра кислот [260]. Стехиометрические соотношения можно предста-
вить уравнением
В4О2-+ 2Н++ 5HaO=4H3BOs.
Следовательно, эквивалентная масса тетрабората натрия
Na2B4O7• 10Н20 составляет половину его молекулярной, массы и
равна 190,69. Водный раствор тетрабората натрия содержит анион
15
Таблица 11
Свободная энергия (AjF°), теплота образования (Д№),
энтропия (8°) и молярная теплоемкость (С°р)
неорганических соединений натрия [605]
Вещество Состояние ДГ«, ккал/моль АН*, ккал/моль S’, кал/град- •моль / кал-град- •моль
Na Крист. 0,000 0,000 12,2 6,79
Na2O » —90,0 —99,4 17,4 16,3
NaF » —129,3 —136,0 14,0 11,0
NaCl » —91,785 —98,232 17,30 11,88
NaBr —86,030 12,5
NaJ » —68,84 13,0
NaOH Крист., II —101,99 19,2
NaN03 » —87,45 —111,54 27,8 22,24
Na2SO4 » —302,78 —330,90 —35,73 30,50
Na2COs Крист. —250,4 —270,3 32,5 26,41
Na2CO3-10H2O » —975,6
NaHCO3 » —203,6 —226,5 24,4 20,94
Na2O2 » —120,6
NaClOg » —85,73
NaC104 Крист., II —92,18 24,1
Na2SO3-7H2O Крист. —753,4
Na2SO4-10H2O » —870,93 —1033,48 141,47 40,4
N&2S2O3 • 5H2O Крист., I —621,89 186,2
Na Крист., II —620,60
NaNO2 » —85,9
Na2HPO4-12H2O Крист. —1266,4 133,3
Na3PO4-12H2O » —1309,0
NaCHaCOO » —169,8
NaCN Крист., 1П —21,46
NaSCN Крист. —41,73.
NaaSiOs » —341 —363 27,2 26,72
Na2CrO4 » —317,6
Na2B4O7.10H2O » —1497,2 147
NaBH4 Крист., I —28,57 —43,82 25,02 20,7
Na2HPO4 —417,4
Na+ Гипот., т=1 —62,589 —57,279 14,4
Примечание. Гипот., m = 1—гипотетическая концентрация 1 моль на 1 кг воды;
1 кал =4,1840 абс. Дж.
тетрабората В4О?~, который взаимодействует с ионами водорода по
схеме
В4О2- + Н+ НВ4О,- 1/Да = 10%
НВ4О7~ 4- Н+ Н2В4О7, i/Kj. = 10%
16
Таблица 12
Некоторые характеристики неорганических соединении натрия [505]
Соединение *КИП» °C Плот- ность, г/мл Растворимость (г/100 мл> в воде
холодной горячей
Na3AsO4- 12Н2О 86,3 1,759 26,7/17°
NaCH3COO 324 1,528 119/0° 170/100°
Na2B4O7. ЮН2О 75 (—10Н20/200°) 1,73 1,3/0° 52,3/100°
NaBr 755 1390 3,205 116/50° 121/100°
NaaHPO4-2H2O (—НаО/93°) 2,06 82,5/50° 96,6/80°
NaOH 318,4 1390 2,130 42,0° 347/100°
NaJ 651 1300 3,667 158/0° 257/60°
NflgCOs 851 разл. 2,533
Na2CO3.10H2O 33,5 (разд.) (—9НаО/35°) 1,44 21,4/0° 420/100°
NaHCOs 270 (разл.) 2,20 6,9/0° 16,4/60°
NaNH4HPO4.4HaO 79 (разл.) 1,554 16,7 100
NaNOs 308 380 (разл.) 2,261 73/0° 180/100°
NaN02 271 320 (разл.) 2,168 81,5/15° 163/100°
N&gO^ разл. 2,805 р. разл. разл.
Na2SO4.10HaO 32,4 (разл.) 1,464 5,0/0° 42,5/100°
Na2SO8-7HaO разл. (—7НаО/150°) 1,561 32,8/0° 196/40°
Na2S2Os.5H2O 48,0 220 (разл.) 1,685 74,7/0° 301,5/60°
Na2HP04< 12H2O 34,6 (—12^0/100°) 1,52 9,3
NaCl 804 1413 2,163 35,7/0° 39,8/100°
^Примечание. разл. — разлагается; р — растворим.
Образовавшееся соединение подвергается деполимеризации:
H2B4O7-J-5H2O=4H3BO3, ЛГ = 2,7-10®.
Разбавленный водный раствор тетрабората натрия можно рас-
сматривать как раствор борной кислоты (Ка — 6,4-1СГ10), наполо-
вину оттитрованный сильным основанием. В точке эквивалентности
раствор представляет собой разбавленную борную кислоту, имею-
щую в 0,1 М растворе pH 5,1. Это значение точно согласуется с рК
метилового красного, который является в данном случае наилучшим
индикатором.
Преимуществом тетрабората натрия перед другими первичными
стандартами является его высокая эквивалентная масса, легкость
получения в чистом виде, возможность проведения титрования в одну
стадию. Основной недостаток применения заключается в необходи-
мости соблюдения ряда предосторожностей для обеспечения точного,
состава этого соединения, зависящего от степени гидратации.
Химически чистый безводный карбонат натрия получают из
коммерческого препарата — гидрокарбоната натрия, очищенного
перекристаллизацией.
1?
Соль NaHC03 растворяют в кипящей воде из расчета 15 г гидрокарбоната
натрия на 100 мл воды (при 20° С в 100 мл воды растворяется 9,6 г ИаНС03).
Горячий раствор фильтруют через складчатый фильтр, вставленный в воронку
со срезанным концом; лучше применять воронку для горячего фильтрования.
Фильтрат собирают в стакан, погруженный в холодную воду. Содержимое ста-
кана помешивают стеклянной палочкой. Выпавшие кристаллы гидрокарбоната
отделяют на фильтр воронки Бюхнера, затем отжимают между листами филь-
тровальной бумаги и высушивают в сушильном шкафу при температуре около
120° С. Высушенные кристаллы помещают в тигель (лучше платиновый); тол-
щина слоя кристаллов не должна превышать 2—3 см. Тигель на полорину его
высоты вставляют в вырезанное отверстие в асбестовом картоне и прокаливают
30—40 мин на небольшом пламени горелки. До красного каления нагревают
только дно тигля. Прокаливаемую массу время от времени помешивают толстой
платиновой проволокой. Температура в тигле не должна превышать 300° С.
Прокаливают до постоянной массы. Вместо прокаливания на горелке тигель
можно поместить в сушильный шкаф на 2—3 ч при 270—300° С. Полученный про-
дукт хранят в бюксе, помещенном в эксикатор.
Есть сведения [260], что даже свежепрокаленный образец карбо-
ната натрия содержит до 0,05% влаги. Последние следы влаги могут
быть удалены плавлением образца в токе чистого диоксида угле-
рода, который по мере охлаждения образца постепенно вытесняют
воздухом. При температуре выше 300° С соль разлагается с выделе-
нием диоксида углерода; так, нагревание в течение 1 ч при 310—
315° С приводит к ошибке более 1 %.
Плотность водных растворов Na2CO3 приведена в табл. 13.
Таблица 13
Плотность водных растворов карбоната натрия при 20° С [505]
Плот- ность, г/мл Количество Na2CO3 (г) Плот- ность, г/мл Количество Na2COs (г) ПЛОТ- НОСТЬ, г/мл Количество Na2CO3 (г)
в 100 г р-ра В 1 Л р-ра в 100 г Р-Ра в 1 л р-ра в 100 г Р-Ра в 1 л Р-ра
1,01 1,135 11,46 1,06 - 5,942 62,99 1,11 10,66 118,3
1,02 2,096 21,38 1,07 6,895 73,78 1,12 11,59 129,8
1,03 3,058 31,50 1,08 7,848 84,76 1,13 12,51 141,4
1,04 4,019 41,80 1,09 8,788 95,79 1,14 13,43 153,1
1,05 4,980 52,29 1,10 9,728 107,0 1,145 13,88 158,9
Перхлорат натрия по растворимости в воде и органических рас-
творителях отличается от перхлоратов остальных щелочных и щелоч-
ноземельных элементов (табл. 14).
Перхлорат натрия используют в качестве промежуточной формы
при гравиметрическом определении натрия в форме хлорида.
Арсенат натрия NasAsO4 количественно осаждается на холоду
из водно-этанольных растворов с pH 8—9 и при содержании этанола
75—80 % об. При нагревании осадок растворяется. Образование сое-
динения используют при разделении натрия и лития и их количест-
венном определении [762].
, Ванадат натрия NaVOs осаждается при pH 8—9 при действии
ванадата калия на раствор соли натрия. Реагент применяют для
18
Таблица 14
Растворимость (в г/100 мл раствора) перхлоратов щелочных,
щёлочноземельных элементов и аммония в различных растворителях
при 25° С [102]
Соль Вода Метанол Этанол н-Бутанол Этилацетат
NaC104 113,88 35,833 11,134 1,495 8,425
ЫС1О* 47,42 89,44 79,41 49,25 63,40
NH4C1O4 21,91 5,268 1,488 0,0137 0,0286
КС1О4 2,0394 0,0830 0,0094 0,0036 0,0013
RbC104 1,328 0,0472 0,0071 0,0016 0,0014
CsC104 1,961 0,0734 0,0086 0,0048 Не раств.
Ва(С1О4)3 128,99 119,85 78,543 41,716 80,812
Са(С1О4)2 112,34 113,68 89,551 68,419 57,377
Sr(C104)2 157,51 113,95 100,01 71,205 76,67
Mg(C104)a 73,453 37,749 18,398 44,638 54,173
отделения натрия от лития [761]. Соль KeV10O28 • 10Н20 приме-
няют для гравиметрического определения натрия в присутствии
104-кратных количеств калия [1142]. Растворимость соединения
K4NaHV10O28-10H2O уменьшается при замене калия на другие ще-
лочные металлы в ряду К > Rb > Cs.
Относительно состава соединений натрия с кислородсодержащими
соединениями сурьмы мнения противоречивы. В более ранних рабо-
тах им приписывали состав NaH2SbO4, Na2H2Sb2O7, в более позд-
них — NaSb(OH)e. Последний состав представляется более правдо-
подобным, поскольку сурьма даже в слабокислых растворах сильно
гидролизована, не говоря о нейтральных и щелочных средах. Раство-
римость гидроантимоната приведена в табл. 15, 16.
Таблица 15
Растворимость NaHgSbO^H/) (в г/100 мл раствора) [748]
Растворитель 18,0° С 20,0° 25,0° 33,5° С
Вода NaCl или KH2SbO4-2Н2О, 0,001 М раствор 0,0564 0,0518 0,0738 0,1018
NaCl или KHaSbO4-2H2O, 0,01 М раствор Ацетат натрия, 1%-ный раствор 0,0061 0,0148 0,0146 0,0205
Ацетат натрия, 2,5%-ный раствор 0,0031 0,0040 0,0094
Антимонат калия применяют для обнаружений натрия в система-
тическом ходе качественного анализа [284], для гравиметрического
[1076], титриметрического [94, 538], колориметрического [781] и
нефелометрического [219] определения натрия.
19
Таблица 16
Растворимость NaH2SbO4-2H2O (в г/100 мл спиртов) [748]
х
Растворитель 18,0° с 25,0° 33,5° С
Этанол 15,23 % мае. 27,33 % мае. 41,36 % мае. 0,0118 0,0038 0,0001 0,0150 0,0060 0,0004 0,0403 0,0140 0,0008
Метанол 20 % об. 33,3 % об. 50 % об. 0,0120 0,0044 0,0006 0,0193 0,0095 0,0031 0,0270 0,0150 0,0062
Комплексные соединения
Образование окрашенного тройного нитрита 6NaNO2-9CsNO2-
•5Bi(NO2)3 используют для колориметрического определения натрия
(1288].
Свойства неорганических соединений натрия рассмотрены в ра-
ботах [102, 226, 330, 748].
СОЕДИНЕНИЯ С ОРГАНИЧЕСКИМИ РЕАГЕНТАМИ
Карбоновые кислоты
В аналитической химии натрия широко используют образование
малорастворимых тройных ацетатов типа NaM(UO2)3(CH3COO)e •
•жН2О, где М = Си, Са, Zn, Cd, Мп, Fe(II), Со, Ni. Их некоторые
свойства приведены в табл. 17.
Таблица 17
Растворимость в воде тройных ацетатов типа NaM(UO2)3(CH3COO)#-а?Н2О
при 20° С [752]
М(П) Плотность, г/см’ Растворимость, моль/л М(П) ПЛОТНОСТЬ, г/см® Растворимость, моль/л
Си 2,15 0,70 Fe 2,09 0,44
Cd 2,58 0,97 Со 2,58 0,57
Мп 2,77 0,75 1 Ni 2,40 , 0,37
Установлено, что в тройных ацетатах магния, цинка и никеля
число молекул кристаллизационной воды равно 6 и отклоняется от
него со стандартным отклонением, не превышающим 0,02 моля 1985].
Условия осаждения натрия в виде тройных ацетатов уранила и Си,
Mg, Cd, Со, Ni изучены в работе [703].
Магнийуранилацетат используют для гравиметрического [985],
титриметрического [21, 834] и фотометрического [769] определения
натрия.
20
Цинкуранилацетат применяют для обнаружения [456,498,628,
1152], выделения [1052, 1116], гравиметрического [274, 436, 476,
552, 839, 1068, 1101], титриметрического [18, 19, 240, 245, 288, 289,
297, 338, 644, 670, 671, 813, 1147, 1164, 1227], фотометрического [22,-
206, 237, 238, 255, 901, 1219] и поляриметрического [244, 895] опре-
деления натрия.
Марганецуранилацетат применяют для гравиметрического и
фотометрического определения натрия [40]. В водно-этанольной
среде состав осадка NaMn(UO2)3(CH3COO)9-10H2O, молярная масса
1599,71; содержание натрия в осадке 1,437%.
Кобалътуранилацетат применяют для гравиметрического [752]
и титриметрического [703, 752, 1166] определения натрия.
Никелъуранилацетат применяют для гравиметрического [39,
239, 752], титриметрического [752] и фотометрического [39, 239, 528]
определения натрия. Осадок устойчив при высушивании до темпера-
туры 130° С; при высушивании в течение 2 ч при температуре 135—
140° С масса становится постоянной, состав осадка соответствует
формуле NaNi(UO2)3(CH3COO)B-6H2O, молярная масса 1531, 408;
содержание натрия 1,50% [39].
Фенил-а-метоксиуксусная кислота (I) образует .
с натрием малорастворимую кислую соль со- CH—СООН
става C6H5CH(OCH3)COOH-C6H5CH(OCH3)COONa Ч? ^СНз f
[1088] в виде белых кристаллов с молярной мас-
сой 354,33 и содержанием натрия 6,49 %. Растворимость соли (в г):
в 100 г воды при 0° С и 30°С составляет 0,56 и 2,66 соответствен-
но; в 100 мл абсолютного этанола при 29° С 0,18; в 100 мл ме-
танола при 25° С 1,3. При комнатной температуре в 100 мл высших
Спиртов, ацетона, диэтилового эфира или диоксана растворяется
<0,1 г соли. Реагент применяют для обнаружения натрия [7, 498,
1088].
м-Хлорфенил-а-метоксиуксусную кислоту применяют для титри-
метрического определения натрия [1089].
п-Хлорфенил-а-метоксиуксусная кислота образует с натрием оса-
док, растворимость которого при 1° С в 100 мл смеси (3 : 1) воды
с Метанолом составляет 316 мг, или 17 мг натрия. Реагент применяют
для гравиметрического и титриметрического определения натрия
[816].
2-Нафтцл-а-метоксиуксусная кислотпа(П)при CH—СООН
PH 3—4 образует осадок кислой малораство- L II 1 1
римой соли натрия (табл. 18), ^Пл = 242-4-243° С. ОСН3 п
Из нейтральной среды осаждается хорошо
растворимая средняя соль с 1ПЛ = 298 -4- 300° С.
Реагент применяют для гравиметрического [817, 818] и титри-
метрического определения натрия. Рекомендовано при гравиметри-
ческое определении использовать тетраметиламмонийную соль реа-
гента [817].
Оксалат 'натрия (Na2C2O4, эквивалентная масса 67,002) является
первичным стандартом и применяется в оксидиметрии, в частности
при установлении концентрации рабочих растворов КМпО4. Ней-
21
Таблица 18
Растворимость (в мг/100 мл) кислой натриевой соли
2-иафтил-а-метоксиуксусной кислоты [818]
Растворитель 20° С 2° С Растворитель 20° С 2° С
Вода 180 100 Вода—этанол 1 : 1 164
Метанол 560 362 Диоксан 205
Вода—метанол Вода—диоксан
5:1 201 3:1 558 272
3:1 167 102 1:1 1030
2:1 1: 1 256 451 Ацетон 18 7
Этанол 94 71
тральные растворы соли не отличаются большой устойчивостью, рас-
творы в 0,1 М НС104 устойчивы при хранении. Оксалат натрия
используют как первичный стандарт также в ацидиметрии: при про-
каливании точной навески Na2C2O4 в платиновом тигле получают
чистый карбонат натрия. Оксалат натрия мало растворим в этаноле
[236], что используют для определения натрия фототурбидиметричес-
ким титрованием. В водно-этанольной среде в присутствии ЭДТА
и триэтаноламина натрий избирательно осаждается оксалатом [466].
Метод применяют для отделения натрия.
Малоновая кислота СН2(СООН)2 образует с натрием в водной
среде соединение с соотношением компонентов 1 : 1 и константой
образования 1,1 + 0,3 (определена с помощью катионселективного
стеклянного электрода Beckman 78137V) [1084]; при ионной силе
0,28 на фоне (CH3)4NBr и хлорида эта константа рав- Н2С—СООН
на 2,8 + 0,3 (определена полярографически) [726].
Соединения натрия с винной кислотой НООС— °
(СНОН)2СООН имеют константы устойчивости с—С
lg Kr = 0,56 ± 0,02 и 1g К2 = 0,20 + 0,03 (р = 0,2) « \
[496]. - О
Лимонная кислота НООС—СН2—С(ОН)(СООН)— нс—
СН2СООН в среде уксусного ангидрида' взаимодей-
ствует с ионами натрия, при этом раствор окрашивает- О
ся в фиолетово-красный цвет; реакция количествен- Па
на, но не селективна. По-видимому, образуется соединение (Па)
[688]. Реагент применяют для фотометрического определения
натрия. ।
Нуклеиновые кислоты
Для комплекса натрия с дезоксирибонуклеиновой кислотой ]g р =
= -0,15 [571, с. 43].
Методом pH-метрического титрования (в атмосфере азота) вычис-
лена условная константа образования соединения натрия с водной
дисперсией фосфатидной кислоты, равная 9,7-103 (24 1° С)
[576].
22
Аминополикарбоновые кислоты
Константы устойчивости этилендиаминтетраацетатов натрия:
для NaH3Y — 1g ₽ = 1,66 (20° С; ц = 0,1) [1141]; 1,88 (25° С;
р, = 0,1) [1100]; для NaY3- 1g Р = 2,61; для предполагаемого соеди-
нения NaHY2- 1g Р = —0,03 [1038].
Для мононатриевой соли нитрилотриуксусной кислоты 1g Р =
= 2,1 (20° С; ц —> 0) [1141]; l-пропилендиаминтетрауксусной кис-
лоты (NaL3-) 1g Р = 2,55 + 0,06 (25° С; (х — 0,5) [668]; циклогексан-
диаминтетрауксусной кислоты 1g Р = 4,40 + 0,08 (25° С; (х = 0,1)
(667] и 3,79 + 0,12 (25° С) [53].
Ароматические сульфокислоты
1-Нафтиламин-8-сулъфокислота образует с натрием и некото-
рыми другими ионами соединения с различающейся растворимостью
(табл. 19). При 105° С соединение имеет постоянный состав и негиг-
роскопично.
Реагент применяют для обнаружения [143, 498], гравиметричес-
кого [142, 1155] и титриметрического [142] определения натрия.
Таблица 19
Растворимость 1-нафтиламин-8-сульфонатов некоторых металлов [143]
Ион Растворимость Ион Растворимость
г/л моль/л г/л моль/л
Na+ 6,3 0,03 К+ 26,3 0,10
NH+ 18,6 0,08 кь+ 49,4 0,16
Li+ 22,9 о,ю Mg2+ 111,0 0,24
Дибензофуран-2-сулъфокислота образует с натрием кристалли-
ческий осадок в форме блестящих призм. Реагент применяют для
микрокристаллоскопического обнаружения натрия [712].
Определен предел обнаружения натрия (в мг/мл) производными
антрахинон-1-сулъфокислоты (1-SO3H) и антрахинон-2-сулъфо-
кислоты (2-SO3H) [778]:
Реагент (R) R-l SO8H R-2-SO.H
З-Бензамидо- Не получен 15
5-Бензамидо- 2 0,10
8-Бензамидо- 20 12
5-Трифторацетил- Не получен 0,30
23
5-Бенаамидоантрахинон-2-сулъфокислоту (III) применяют для
обнаружения [498, 778], гравиметрического [778] и фотометричес-
кого [519] определения натрия.
О
н II so8H
HsC.-CO-N-QjQQj''
II
о
ш
Барбитуровая кислота и ее аналоги
Виолуровая (5-изонитрозобарбитуровая)
кислота (IV) образует с натрием в присут-
ствии H2S соединение красно-фиолетового
цвета, являющееся натриевой солью пур-
пурной кислоты [612]. Термогравиметричес-
ки изучены свойства виолурата и 1,3-диме-
тилвиолурата натрия на воздухе и в атмос-
фере гелия [1211]. Виолуровую кислоту
применяют для обнаружения натрия [612],
а 1,3-диметилвиолуровую кислоту — для
определения натрия спектрофотометричес-
ким титрованием [1212].
Оротовая (2,6-диоксотетрагидропирими-
дин-4-карбоновая кислота (V) и ее производ-
ные осаждают натрий в виде соединений, имеющих следующую рас-
творимость (в г/мл) в среде 80%-ного этанола при 25° С [922]:
Реагент
Оротовая кислота
У рацил-5-карбоновая кислота
5-Метилоротовая кислота
1-Фенилурацил-5-карбоновая кислота
Растворимость соли, г/л
0,08
0,08
0,91
1,25
Соединения изучены как реагенты для гравиметрического опре-
деления натрия. Отмечается, что в присутствии 90% об. зтанола
5-этилоротовая кислота осаждает только натрий.
5-Нитробарбитуровую кислоту применяют для обнаружения
натрия [744].
Определены константы устойчивости нат- О
риевых солейурамилдиуксусной кислоты (VI)
при р, = 0,1 и различной температуре методом 0=\ N(CHaCOO)2
потенциометрического титрования: 2,72 +
±0,01 (20°С); 2,54(27°С) и 2,42 (34°С) [830] ° vi
и полярографическими ± 0,13 (25° С) 53.
24
Азосоединения
1-{2-Оксинафтилазо)-2-нафтол (VII) в силь-
нощелочной среде является реагентом для
обнаружения натрия [1222]. Предел обнару-
жения в мкг/мл: Na 10; Mg 0,003; Са 0,2;
Sr 0,3; Li 0,5.
2-Окси-2'-сульфоазосоединения в присутствии
ОН НО
гидроксида тетрабутиламмония в водно-органических средах об-
разуют с натрием окрашенные соединения, пригодные для его фо-
тометрического определения [295]. Наилучшим оказался МФП—
АНИФЕСК (VIII). Свойства соединения с натрием в различных сре-
дах приведены в табл. 20.
Таблица 20
Свойства натриевой соли МФП—АНИФЕСК [295]
Среда 7., нм е-10-1 ^ОПТ» нм
Вода—ацетон—ДМФА - 485 3,13 550
Вода—диокс ан— ДМФА 495 2,93 550
Вода—этанол—ДМФА 480 2,67 530
Вода— изопропанол—ДМФА 485 2,67 530
Вода—ДМФА 530 2,86 550
Из группы азосоединений на основе пиразолина лучшим признан
нитроантранилазо (2-карбокси-4-нитрофенил-<1-азо-4'>-1-фенил-
3'-метил-5'-пиразолон), применяемый для фотометрического опре-
деления натрия [296].
5-Нитро-2-(3-метил-5-оксоизоксазол-4-ил-азо)бензолсульфокислоту
применяют для фотометрического определения натрия [38].
Производные 4-фенилазо-5-изоксазолонов [137]: 3-метил-4-(4-нит-
ро-2-карбоксифенилазо)-5-изоксазолон, 3-метил-4-(4-нитро-2-суль-
фофенилазо)-5-изоксазолон и 3-метил-4-(4-нитро-2-арсенофенилазо)-
5-изоксазолон образуют с натрием соединения, пригодные для его
фотометрического определения.
2-Окси-2-(2-оксиэтокси)азосоединения (типа IX) и бнс-азосоеди-
нения, содержащие оксиэтиленовую или полиоксиэтиленовую
25
цепочку (типа X), в водно-органической среде в присутствии гидро-
ксида тетраметиламмония образуют с натрием окрашенные соеди-
нения, пригодные для его фотометрического определения [510].
ОСН2СН2ОН
Максимальная контрастность реакции 15 нм, при комплексообра-
зовании наблюдается гипсохромный сдвиг, молярные коэффициенты
погашения изменяются от 1250 до 10 850.
Наиболее ценными на натрий признаны реагенты ряда [(4,5-
дигидро-3-метил-5-оксо-1-фенйл-1 Н-4-пиразолилиден ) гидразино ]-4'-
иитро-2'-(2Х)-бензола, где X = SO3H, СООН, ОСН2СООН,
ОСН2СН2ОН [5091.
Криптаты и краун-эфиры
Определены константы устойчивости соединений натрия с крип-
татами следующего строения [571, с. 46].
Криптат 1g ₽
(2, 1,1) т = 0, п = 1 2,8
(2, 2, 1) т = 1, п = 0 5,4
(2, 2, 2) т = 1, п = 1 3,9
(3,2,2) т = 1, п = 2 1,7
(3,3, 2) т — 2, п — 1 2,0
(3, 3, 3) т — 2, п = 2 2,0
2,2,1-Криптат селективен к ионам натрия. Для соединения нат-
рия с 2,1,1-криптатом вычислены термодинамические характеристи-
ки [1002] (при 10° С): 1g Р = 9,57; AG2e8 = —51,9 кДж/моль;
ДЯ2й8 = —53,8 кДж/моль; —TSW9 = 2,1 кДж/моль.
В качестве реагентов на натрий изучены краун-эфиры следую-
щего строения [571, с. 46]:
О(СН2СН2О);
О(СН2СН2О),
Константы устойчивости их соединений с натрием приведены в
табл. 21, 22.
26
Таблица 21
Константы устойчивости комплексов некоторых краун-эфиров
С натрием [571]
Краун-эфир тп п 1g В
Дибензо-18-краун-6 2 2 4,4
Дибензо-21-краун-7 2 3 2,4
Дибензо-24-краун-8 3 3 —
Дибензо-30-краун-10 4 4 2,0
Бензо-15-краун-5 3,7
Дициклогексил-14-краун-4 2,2
Таблица 22
Константы устойчивости комплексов натрия с краун-эфирамн
в водных и водно-органических средах при 25° С
и в присутствии ионов С1 [1057]
Краун-эфир Среда Константа
15-Краун-5 Вода Ki = 0,70+0,10
Бензо-15-краун-5 Вода Ig Р = 0,4
20% об. СН3ОН Ig Р = 0,72-1-0,03
40% об. СН3ОН lgp = 1,174:0,12
60% об. СНзОН 1g p = 1,64+0.04
70% об. СН3ОН Ig P = 1,994-0,10
80% об. СН3ОН 1g P = 2,26+0,02
18-Краун,-6 Вода Ig p = 0,80+0,10
70% об. СН3ОН Ig p = 2,764:0,02
Циклогексил-18-краун-б Вода * K1 = 6,3
Дибензо- 18-краун-6 То же Kt= 14,64;l,0
Дибензо-24-краун-8 70% об. СН3ОН Ig P = l,54±0,01
Дибензо-27-краун-9 То же Ig P = 1,5044),01
* В отсутствие ионов С1_.
На константу устойчивости влияет природа растворителя
(табл. 23).
Для ряда комплексов вычислены термодинамические характе-
ристики (табл. 24, 25).
Поли-, б1Ас-(бензо-15-краун-5) и бис-(бензо-18-краун-6)-эфиры об-
разуют с натрием в присутствии пикратов экстрагирующийся хлоро-
формом комплекс с максимумом светопоглощения при 374 нм; мо-
лярный коэффициент погашения 1,86-104; константа образования
Ig К = 6,02—6,72 [8761.
27
Таблица 23
Константы устойчивости (в единицах 1g fl) комплексов натрия
с краун-зфирами в различных растворителях [926]
Растворитель 18-Краун-б 15-Краун-5 Вензо-15-краун-5
Вода 0,82+0,05 0,44-1-0,05
Тетрагидрофуран >4 >4 >4
Ацетон >4
Нитрометан >4 >4 >4
Пиридин >3 2,68-4-0,08 2,6±0,1
дмсо 1,41+0,07 1,31+0,06 1,1+0,2
ДМФА 2,31±0,05 1,97±0,05 1.6±0,1
Таблица 24
Термодинамические характеристики комплексов натрия
с краун-зфирами в воде при 10° С [1002]
X арактеристика 15-Краун-5 18-Нраун-6 Дибензо-18-краун-6
1g ₽ 5,23 4,73 5,11
А<?2в8, КДЖ/МОЛЬ —28,4 —27,2 —28,6
Д//298’ КДЖ/МОЛЬ —24,1 1.6 —14,3
—Z<S®98, кДж/моль —4,5 —28,7 —14,2
Таблица 25
Термодинамические характеристики комплекса натрия с 18-краун-6
в метаноле или водном метаноле при 25° С [797]
Характеристика 100%-ный СН8ОН 90%-ный СН8ОН
АН, ккал/моль —TAS, ккал/моль 8,36±0,37 2,41 6,64+0,07 1,65
4'-Пикриламинобензо-15-краун-5, его нитро* и бромпроиэводные
в щелочной среде образуют анион L~, взаимодействующий с ионами
натрия. Получающийся комплекс имеет кроваво-красный цвет
(^тах = 550 н-560 нм), соотношение компонентов 1:1; комплекс
экстрагируется раствором триэтиламина в хлороформе. Константа
экстракции для натрия 1,0 М раствором триэтиламина в хлороформе
1g = —13 [1001]. Реагенты на практике не используют.
28
Краун-эфиры дициклогексил-18-краун-6 и 18-краун-6 приме-
няют для разделения Na, К, Rb и Cs [698] и их определения [10]»
Краун-эфиры: (2-окси-3,5-динитрофенил)оксиметил-15-краун-5,
(2-окси-3,5-динитрофенил)оксиметил-18-краун-6, [2-окси-5-(4-нит-
рофенилазо)фенил]оксиметил-15-краун-5, [2-окси-5-(4-нитрофенил-
азо)фенил]оксиметил-18-краун-6 применяют для фотометрического
определения натрия [999, 1000]. Максимумы светопоглощения нахо-
дятся при 423—571 нм, молярные коэффициенты погашения (1,3—
3,7)-104 (в дихлорэтане). По отношению к натрию селектив.ен [2-
окси-5-(4-нитрофенилазо)фенил]оксиметил-15-краун-5; в дихлор-
этане растворы имеют максимум светопоглощения при 560 нм, мо-
лярный коэффициент погашения 3,7-104.
Оксиоксосоединения
Кроконовая кислота (4,5-диоксицикл опен-
тен-4-трион-1,2,3) XI при упаривании с раство-
ром соли натрия обра&ует соль красного цвета.
Реагент применяют для обнаружения натрия
[7, 1063].
Натрий при pH 2—9 дает цветную реакцию с
нингидрином (XII), но только на бумаге, в то
время как аминокислоты реагируют и в раство-
ре. Реагент применяют для обнаружения натрия
в присутствии аминокислот [786].
О хи
Аминосоединения
Гексанитродифениламин (XIII)
образует с натрием соединение, име-
ющее два максимума светопоглоще-
ния в водных растворах: при 426 нм
(е = 2,69-104) и 210 нм (е = 3,24-104)
[1121]. Комплекс экстрагируется
no2 O2N
NO2 O2N хш
нитробензолом и применяется для фотометрического определения
натрия [908].
Прочие соединения
Тетра-{п-толил)борлитий количественно осаждает натрий в ви-
де соединения NaB(C6H4CH3)4, а также ионы калия, рубидия и
цезия [432].
Тетрафтороборат применяют для гравиметрического определе-
ния натрия [429].
Амид 5,6-дифенил-3-оксо-2,3-диоксипиридин-4-карбдновой кисло-
ты [883] применяют для обнаружения натрия.
Определены константы устойчивости ряда соединений натрия
с карбоновыми кислотами и аминокислотами [1085]:
29
Кислота К
'Пировиноградная кислота 2,7
Яблочная кислота 1,9
Молочная кислота 1,1
.и-Оксибензойная кислота 0,81
Кислота К
о-Оксибензойная кислота 0,78
Z-Лейцин 0,3
dZ-a-A ланин 0,2
Обнаружена прямая пропорциональность между константами
устойчивости и ширинами АЯ линий спинового резонанса на ядрах
23Na в растворах, содержащих анионы этих кислот.
Опубликованы обзоры по применению органических реагентов,;
дающих цветные реакции с натрием [1221], и по координационной
химии щелочных металлов [1057].
Аналитической химии натрия посвящены работы [102, 474, 748].
Глава II
МЕТОДЫ ОБНАРУЖЕНИЯ НАТРИЯ
Методы обнаружения натрия в настоящее время представлены
химическими и физическими методами. Реакции обнаружения нат-
рия малоселективны, требуется предварительное выделение натрия
или сопутствующих ионов. Поэтому большинство химических мето-
дов применяют после разделения ионов в систематическом ходе ана-
лиза. Более перспективны физические методы, основанные на спо-
собности солей натрия окрашивать пламя горелки в характерный
желтый цвет. Существуют способы устранения влияния других ще-
лочных металлов; основа этих методов описана в главе VIII «Спек-
тральные методы определения натрия». По чувствительности они
также превосходят химические методы.
ХИМИЧЕСКИЕ МЕТОДЫ ОБНАРУЖЕНИЯ
Пробирочные и капельные реакции
Схемы анализа с использованием сероводорода, основанные на
различии растворимости сульфидов и тиосолей, приведены, напри-
мер, в работах [1115, 1201]. Разработана [932] схема обнаружения
23 катионов (Li, Na, К, Mg, Са, Sr, Ba, Си, Ag, Zn, Cd, Hg,
Al, Sn, Pb, As, Sb, Bi, Cr, Mn, Fe, Co, Ni), использующая разли-
чие в растворимости сульфатов, сульфидов, нитратов, оксидов и
карбонатов и включающая применение ионообменников. Ионы
Li, Na, К, Mg и Са разделяют после отделения от сульфидов и суль-
фатов действием оксида серы(1У) и тиоацетамида; в качестве ионо-
•обменника используют Дауэкс 50 в Н-форме. Вначале 1М раствором
НС1 элюируют литий (25 мл), а затем натрий (40 мл).
Способы обнаружения натрия капельным методом рассмотрены
в книге [498], люминесцентным методом — в руководстве [457].
30
Основные реагенты, применяемые для обнаружения натрия^
приведены в табл. 26.
Таблица 26
Реагенты для обнаружения натрия [7]
Реагент Цвет осадка Предель- ное раз- бавление Мешающие ионы
Фторид аммония Белый 1:2-10* ЩЗЭ, Ag и др.
Гексагидроксостибанат ка- лия 1:5-103 NH+, Li, Mg
Нитрит цезия—калия—вис- мута Бледно-желтый 1:10* Li, Rb, Cs, NH+, Ag и др.
Ацетат магния—уранила Желто-зеленый 1:2,5-103 К, NH+, ЩЗЭ и др.
Ацетат цинка—уранила > 1:2,5-103 К, NH+, ЩЗЭ и др.
Пикролоновая кислота Белый 1:4-102 К, Rb, Cs, ЩЗЭ
Диоксивинная кислота 1:1,7-103 К, Rb, Cs, ЩЗЭ
Бромбензолсульфокислота Бледно-желтый 1:2,5-103 К
1-Нафтиламин-8-сульфо- кислота Телесный 1:4-103 ЩЗЭ и др.
Оротат NjN-диалкилэта- ноламмония Белый 1:2-10* К
Примечание. ЩЗЭ — щелочноземельные элементы.
Обнаружение дигидроантимонатом (гексагидроксостибанатом),
калия [284]. Реагент мало селективен на натрий, его применяют
после отделения натрия от остальных ионов по сероводородной
схеме.
Анализируемый раствор подкисляют соляной кислотой, выпаривают на во-
дяной бане досуха и прокаливают до прекращения выделения белых паров..
^Сильного прокаливания избегают. К сухому остатку прибавляют несколько
^миллилитров воды и нагревают. Полученный раствор не содержит ионов NH^r
И), Sb(III, V), Sn(II, IV) и многих катионов III и V аналитических групп
‘‘(по сероводородной схеме). К одной порции раствора прибавляют 3%-иый рас-
твор К2СО3 до явно щелочной реакции по лакмусу и кипятят несколько минут.
Смесь фильтруют, фильтрат охлаждают, пропускают углекислый газ для осаж-
дения гидроксида алюминия в течение 5—10 мин или прибавляют равный объем
втанола. В фильтрате или центрифугате обнаруживают натрий.
Селективность обнаружения повышают маскировкой сопутству-
ющих ионов ЭДТА или диоксиэтилглицином при pH 11 [1206].
Обнаружению не мешают ионы Си, Ni, Со, Fe(II, III), Мп, РЬ/
Sn(II), Al, Hg(II), Cd, Zn, Ba, Sr, Ca, Mg и Li; предел обнаружения
натрия 0,3 мг/мл. В присутствии большого избытка солей калия и
аммония и маскирующего реагента предел обнаружения возрастает.
Обнаружение в виде тройных ацетатов. Метод обнаружения нат-
рия в виде NaMg(UO2)3(CH3COO)e-a:H2O предложен в 1884 г. Штрен-
гом [752]. Реакцию обнаружения натрия в виде тройного ацетата
цинка можно выполнять капельным методом [498]. В этом случае
34
соли Си, Hg, Cd, Со, Ni, Al, Mn, Zn, Ca, Sr, Ba, Mg и NH* силь-
но влияют только при их содержании более 5 г/л. Литий осаждается
из растворов, содержащих 1 г/л лития, а калий — из растворов,
содержащих ^>5 г/л калия. Предел обнаружения снижается при
выполнении реакции в водно-этанольных растворах. В этом случае
натрий можно обнаружить в солях калия. Предел обнаружения
натрия 12,5 мкг; предельное разбавление 1 : 4-103.
Анализируемый раствор нейтрализуют аммиаком или оксидом цинка. На
темную капельную пластинку или черное часовое стекло помещают каплю про-
зрачного раствора и смешивают с 8 каплями раствора реагента. Образование
желтого помутнения или осадка свидетельствует о присутствии натрия.
Отмечается [456], что чувствительность обнаружения натрия
в виде натрийцинкуранилацетата повышается при выполнении
реакции на кварцевой пластинке и наблюдении желто-зеленой люми-
несценции осадка под микроскопом. Для возбуждения люминесцен-
ции используют источник света с длиной волны 313 нм. Предел об-
наружения натрия 0,03 мкг. Обнаружению не мешают ионы К и
NH4.
При отсутствии микроскопа люминесцентную реакцию можно
выполнять капельным методом [498]. Предел обнаружения выше
2,5 мкг натрия, предельное разбавление 1 : 2-104.
На полоску фильтровальной бумаги капилляром наносят каплю анализи-
руемого раствора. Предварительно бумагу обрабатывают дважды разб. НС1,
4—6 раз дистиллированной водой и высушивают. После впитывания капли бума-
гу тщательно высушивают. Затем каплю раствора реагента помещают в центр
высушенной капли, а другую — рядом. Влажное пятно облучают кварцевой
лампой. В присутствии больших количеств натрия желто-зеленая люминесцен-
ция наблюдается сразу же; при содержании натрия менее 10 мкг необходимо вы-
держивание в течение 1—4 мин.
Реагент. Раствор уранилацетата цинка, а) 10 г уранилацетата раство-
ряют при нагревании в 6 мл 30%-ной СН3СООН и разбавляют водой до 50 мл;
б) 30 г ацетата цинка перемешивают с 3 г 30%-ной СН3СООН и разбавляют во-
дой до 50 мл. Растворы «а» и «б», не охлаждая, смешивают, к полученному про-
зрачному раствору добавляют немного NaCl и оставляют стоять. Через 24 ч
•осадок натрийцинкуранилацетата отфильтровывают, а раствор реагента исполь-
зуют для обнаружения натрия.
Подробные сведения об обнаружении натрия по люминесценции
тройного натрийцинкуранилацетата приведены в монографии [57].
Обнаружение фенил-а-метоксиуксусной кислотой [1088]. Реакция
более селективна по сравнению с образованием тройных ацетатов.
Обнаружению не мешают NH4C1 «2 М), LiCl «4 М), КС1 «2,3 М),
MgCl2 «8 М). Сульфаты этих катионов мешают сильнее. Время
образования осадка зависит не только от концентрации натрия в рас-
' творе, но также от концентрации и природы других солей. Напри-
мер, при концентрации натрия 1,0 мг/мл в отсутствие посторонних
ионов при 15° С осадок образуется в течение 3 мин, а в присутствии
LiCl (4 М), MgCl2 (8 М) или КС1 (2,3 М) соответственно в течение 15,
•60 и 10 мин. При концентрации натрия 0,3 мг/мл даже в отсутствие
посторонних ионов осадок образуется в течение 6 ч.
По данным [7], предел обнаружения натрия составляет 1,83 мг/мл
<(20° С) и 1,28 мг/мл (0° С). Обнаружению натрия в нейтральном
32
водно-ацетоновом растворе не мешают СГ, NO3, РО4 , SO^~>
100-кратные количества ионов Li, К, Mg и 10-кратные — NH4.
При выполнении реакции в микропробирке предел обнаружения
натрия 30 мкг, предельное разбавление 1 : 1667. Обнаружению ме-
шают ионы Ba, Sr, Са и Cs [498].
В микропробирке смешивают каплю анализируемого раствора с тремя кап-
лями раствора реагента и выдерживают при 15° С приблизительно 15 мин. Появ-
ление белого осадка указывает на присутствие натрия.
Для приготовления раствора реагента 13,33 г (0,08 моля) фенил-а-метокси-
уксусной кислоты растворяют в 27,8 мл 1,08 М раствора оксида тетраметиламмо-
ния (0,03 моля) и разбавляют абсолютным этанолом до 100 мл. Растворы выдер-
живают при комнатной температуре 5 ч, иногда перемешивая, и затем еще 12 ч
при 0° С. Холодный раствор декантируют с осадка натриевой соли и фильтруют
с отсасыванием.
Обнаружение кроконатом калия. При упаривании соли натрия
с кроконатом калия появляется красное окрашивание. Предельное
разбавление 1 : 7,7-105 [1063]. Обнаруживаемый минимум натрия
составляет 0,03 мг/мл [7]. Обнаружению не мешают 70-кратные коли-
чества ионов Mg, 60-кратные — NH4, 80—100-кратные — Li, К,
Rb, Cs. В присутствии цезия предел обнаружения натрия понижает-
ся и составляет 0,015 мг/мл. Мешают обнаружению ионы и соли,
образующие при упаривании окрашенные осадки.
При выполнении реакции в микропробирке предел обнаружения
натрия 1,7 мкг; предельное разбавление 1 : 3,3-104 [498].
В микропробирке к капле водного раствора реагента добавляют каплю ана-
лизируемого раствора. Появление красного осадка свидетельствует о присутст-
вии в растворе натрия.
Обнаружение 5-бензамидоантрахинон-2-сульфокислотой [778].
Предел обнаружения натрия 0,0025 мг/мл, предельное разбавление
1 : 4-105. Предел обнаружения других ионов в присутствии 50%
об. этанола гораздо выше (в мг/мл): Li (0,60), К (0,28), Rb (0,10),
Cs (0,08), NHt (0,25).
1 мл анализируемого раствора в маленькой центрифужной пробирке смеши-
вают с 1 мл 3%-ного раствора реагента в 50%-ном этаноле и взбалтывают 1 мин.
Через 10 мин в присутствии ионов щелочных металлов выпадает осадок.
Обнаружение 1-нафтиламин-8-сульфонатцм магния [143]. Предел
обнаружения натрия 28 мкг, предельное разбавление 1 : 4-103.
Обнаружению натрия не мешают 4-кратные количества К, Li;
7-кратные — Rb; 15-кратные — Mg; 3-кратные — NHt; мешают
катионы остальных аналитических групп.
Обнаружение оротовой кислотой [1145]. Оротаты 1У,КГ-диметил-
или N,N-диэтилэтаноламмония позволяют обнаружить натрий при
концентрации не менее 50 мкг/мл.
Обнаружение 5,6-дифенил-3-оксо-2,3-дигидропиразин-4-карбоно-
вой кислотой [883]. По пределу обнаружения реакция близка
к реакциям обнаружения в виде натриймагнийуранилацетата или
гексагидроксостибаната. Реакция неселективна, мешают все ионы,
2 В. M. Иванов и др.
33
а из щелочных металлов — литий и аммоний. Допустимо присутст-
вие 5-кратных количеств ионов К, Rb и Cs. Белый кристаллический
осадок выпадает на холоду только в присутствии больших количеств
натрия.
Микрокристаллоскопические реакции
При обнаружении натрия в форме сульфата натрия—висмута
Bi2(SO4)3-3Na2SO4 предельное разбавление 1:2,5-104. Для повы-
шения чувствительности натрий сорбируют катионообменником, за-
тем зерно сорбента погружают в каплю раствора реагента (раствор
Bi2(SO4)3 в H2SO4) и наблюдают кристаллы под микроскопом. В этом
случае предельное разбавление 1 : 105 [205].
Натрий можно обнаружить в виде тройного роданида натрия—
цезия—висмута Cs2Na[Bi(SCN)6] [689]. Мешают ионы лития и калия.
В присутствии калия последний предварительно отделяют в форме
КС104. Предел обнаружения натрия 0,05 мкг, предельное разбавле-
ние 1 : 2>104.
К капле анализируемого раствора прибавляют каплю насыщен-
ного раствора CsBi(SCN)4 в 4%-ной HSCN. В присутствии натрия
образуются плоские шестиугольные кристаллы красного цвета.
Ионы натрия при взаимодействии с фосфорноватокислым калием
образуют кристаллы игольчатой формы; реакцию удобно выполнять
как микрокристаллоскопическую. Предел обнаружения натрия
0,04 мкг, предельное разбавление 1 : 2,5-104. Испытуемый раствор
предварительно обрабатывают 0,5 М раствором К2СО3 для удаления
ионов тяжелых и щелочноземельных элементов [452].
При взаимодействии ионов натрия с дибензофуран-2-сулъфокис-
лотой образуется кристаллический осадок в форме призм. Хотя
реагент взаимодействует еще с 28катионами (из числа исследованных),
только натрий образует осадок с такой формой кристаллов [712].
Предложено использовать 5-нитробарбитуровую кислоту для
микрокристаллоскопического обнаружения натрия. В аналогичных
условиях осадки образуют также ионы К, Li, Mg, Са, Sr и Ва
[744]. Реагент применяли для обнаружения натрия в водораствори-
мых полимерах, целлюлозе и известняке.
РЕАКЦИИ НА БУМАГЕ
Опубликован обзор по обнаружению элементов методом хромато-
графии на бумаге в активационном анализе [657].
В присутствии калия и магния натрий обнаруживают после раз-
деления хлоридов на бумаге Ватман № 4, используя метанол в ка-
честве элюента. Величины Rf. для NaCl 0,43; КС1 0,24; MgCl2 0,68.
Зоны проявляют раствором дихлорфлуоресцеина, а затем раствором
AgNO3. В присутствии указанных ионов на желтом или розовом фоне
зоны окрашиваются в синевато-красный цвет [578].
Для обнаружения натрия в присутствии аминокислот рекомен-
довано использовать нингидрин [786]. Кислотность раствора не
влияет на обнаружение в интервале pH 2,0—8,9. Авторы отмечают,
что реакцию можно выполнять только на бумаге.
94
РЕАКЦИИ ОКРАШИВАНИЯ ПЛАМЕНИ
Соединения натрия легко возбуждаются в низкотемпературном
пламени светильный газ—воздух (температура равна 1870° С),
окрашивая пламя в характерный желтый цвет. В аналогичных усло-
виях пламя окрашивается в различные цвета от присутствия лету-
чих соединений остальных щелочных и щелочноземельных элемен-
тов. В присутствии последних натрий удобнее обнаруживать с по-
мощью спектроскопа прямого зрения, наблюдая линию натрия при
590 нм. Предел обнаружения натрия данным методом очень низок,
поэтому натрий можно обнаруживать практически везде: в воде,
газе, реагентах.
Способность окрашивать пламя в характерный цвет широко ис-
пользуют при количественном определении натрия методом фото-
метрии пламени. Подробно возможности этого метода рассмотрены
в главе VIII «Спектральные методы».
Глава III
МЕТОДЫ ОТДЕЛЕНИЯ
И КОНЦЕНТРИРОВАНИЯ НАТРИЯ
ОТДЕЛЕНИЕ ХИМИЧЕСКИМИ МЕТОДАМИ
Осаждение неорганическими реагентами
Осаждение сопутствующих ионов. Число методов группового
отделения ряда ионов от натрия невелико. Более распространенным
был метод отделения натрия от 1—3 ионов. Разработан систематичес-
кий ход разделения многих ионов с использованием неорганических
и органических реагентов [1094]. Вначале в среде 6 М СН3СООН
в присутствии ацетоксима отделяют с помощью [Co(CN)6]3- ионы
двухвалентных металлов: Мп, Fe, Со, Ni, Си, Zn, Cd, Hg, а так-
же Ag; затем при pH 3 осаждают малорастворимые средние фосфаты
Ti, Cr(III), иОг+, Pb, Bi, Sn(IV), Sb(III), а в щелочной среде — сред-
ние фосфаты Mg, Са, Ba, Sr и РЗЭ. В фильтрат попадают калий и
натрий.
Ферроцианид никеля позволяет отделять микроколичества руби-
дия и цезия от основы — натрия [381]. Метод применяли при опре-
делении рубидия и цезия в хромате, фосфате и хлориде натрия.
Литий отделяют количественно от натрия в виде среднего фосфа-
та [120]. Прием используют при обнаружении натрия в системати-
ческом ходе анализа.
При pH 8—9 и содержании этанола 75—80% об. 5 %-ный раствор
KH2AsO4 осаждает литий и натрий, но при нагревании осадок
Na3AsO4 растворяется, а Ы3Р04 не растворяется [762].
Калий можно осаждать вместе с натрием в виде хлоридов [94].
При обработке осадка этанолом NaCl растворяется. Прием можно
2*
35
применять при гравиметрическом определении натрия в присутствии
равных или преобладающих количеств калия.
Большие количества калия отделяют осаждением в форме КС104
[436]. Из растворов, содержащих 75% об. этанола, осаждается 98,8%
калия. Перхлорат калия мало растворяется и в других органических
растворителях, что используют для отделения натрия от калия
(табл. 27).
Таблица 27
Растворимость (в г/100 мл раствора) перхлоратов калия
и натрия при 25° С [1264,1265]
Растворитель КСЮ* NaClO* Растворитель КСЮ* NaClO.
Вода - 2,04 113,9 н-Бутанол 0,0036 1,5
Метанол 0,083 35,8 Этилацетат 0,0013 8,4
Этанол 0,0094 11,1 Ацетон 0,118 36,6
Большое различие в растворимости перхлоратов калия и натрия
положено в основу методов отделения избирательным растворением
NaC104 в метаноле [1067], этаноле [900], н-бутаноле [855], пентаноле
[973] или смеси н-бутанола с этил ацетатом [215].
Тетрафторобораты калия и натрия в водно-этанольных средах
заметно различаются по растворимости при 25° С: фтороборат натрия
хорошо растворим, растворимость фторобората калия приведена ниже
[428]:
Содержание этанола,
% об.
0
40
50
60
Растворимость
KBF*, %
0,561
0,242
0,204
0,143
Содержание этанола,
% об.
70
80
96
Растворимость
KBF*, %
0,099
0,053
0,009
В присутствии этанола Ca2[Fe(CN)6] осаждает калий; соль натрия
хорошо растворима [763].
Свинец в форме нитрата осаждается из сильноазотнокислых сред
[491]. Например, при 23° С содержание остающегося в растворе
свинца изменяется от 330 г/л в нейтральной среде до 1,2 г/л в среде
конц. HNO3. Вместе со свинцом соосаждается 0,8—1,1% натрия.
В фильтрат, кроме натрия, переходят ионы Са, Ag, Sb, Cd, In, Те,
Fe, Co, Zn, As.
Основу — свинец — можно отделить от микроколичеств натрия
осаждением PbS04 [524]. С помощью радиоактивных индикаторов
показано, что большинство микрокомпонентов, в том числе и натрий,
соосаждается на 30—50% с PbSO4. Промыванием осадка 6М HNO3
можно практически полностью десорбировать натрий.
Осаждение натрия. От многих ионов натрий отделяли высалива-
нием хлористым водородом [1095]. Следовые количества ионов Sr,
36
Са, Со, Cr, Sc, Cd, Sb, Ра на 96% остаются в растворе, а при пере-
осаждении NaCl отделяются от натрия количественно. Прием целе-
сообразно применять при вскрытии объектов сплавлением с Na2O2
перед хроматографическим отделением примесей.
Осаждение органическими реагентами
Осаждение сопутствующих ионов. Микроколичества (^>3-10-8%)
Си, Ag, Ni, Ti, Мп, Сг, Bi, Pb, Sn, Mo, V, Co, Au, Al, Fe, Zn и
Sb отделяли от основы — NaOH — действием соли кадмия, серо-
водорода и диэтилдитиокарбамината натрия при pH 7,5. Метод при-
меняли при определении примесей в NaOH [374].
От примесей Fe, Си, Ni, Со, V, Мо натрий отделяли осаждением при-
месей диэтилдитиокарбаминатом кадмия и 8-оксихинолином [87].
Прием применяли при определении примёсей в метафосфате натрия.
Ионы Al, Bi, Ga, Ge, Fe, Au, In, Co, Mn, Cu, As, Ni, Sn, Pb,
Ag Q>2-10_®%) отделяли от основы — солей натрия — тиоацетами-
дом и диэтилдитиокарбаминатом [371]. Способ применен при химико-
спектральном определении примесей в солях натрия.
Вольфрам отделяли от малых количеств натрия (1,2-10-6 г)
осаждением а-бензоиноксимом [685]. Вместе с натрием в фильтрате
находятся ионы Си, As, Zn, Cr, Fe. Прием использовали при актива-
ционном определении натрия в вольфраме.
Осаждение натрия. Оксалат-ионы в водно-этанольной среде в при-
сутствии ЭДТА и триэтаноламина в качестве маскирователей осаж-
дают натрий [466]. Получены удовлетворительные результаты при
отделении 2,5—10 мг натрия от 0,1 мг Li, Mg, Са, Sr, Ba, Fe (II, HI),
Co, Ni, Mn, Cu, Zn, Cd, Hg (II), Al, Pb; 5 мг К и 2,5 мг NHj. Частич-
но осаждаются оксалаты и основные соли Sn(II, IV), Sb(III), Bi(III),
Cr(HI). В осадке можно определить содержание натрия пермангана-
тометрическим титрованием в среде серной кислоты.
Соосаждение на неорганических коллекторах
В качестве неорганического коллектора описан гидроксид желе-
за(Ш) [126], который при pH 9,5—10,0 количественно адсорбирует
микроколичества (2>3-1О-6%) никеля, а при pH 8,5—9,0 — кобаль-
та. Прием использован при определении примесей в NaCl.
Микроколичества германия количественно адсорбируются тем
же коллектором, что было использовано при определении германия
в NaCl [550].
Фосфат цирконила в натриевой форме селективно сорбирует
стронций из нейтральных растворов, содержащих большие количест-
ва натрия [592]. Константа обмена натрия на стронций составляет 440.
Ряд методов с использованием отделения на неорганических кол-
лекторах применен в колоночном варианте. Микроколичества нат-
рия (калия) отделяли от больших количеств хлорида лития методом
ионообменной хроматографии на колонке, заполненной сурьмяной
кислотой. Сорбированные натрий и калий десорбировали 5 М раство-
37
ром NH4NO3 в 1 М HNOs и определяли методом фотометрии пламе-
ни [574]. Из растворов минеральных кислот натрий выделяли про-
пусканием раствора пробы через колонку с катионообменником на
основе полисурьмяной кислоты. Натрий оставался на колонке при
элюировании других катионов растворами 10 М НС1, HN03 или 5 М
H2SO4.
Молибдаты и вольфраматы циркония и титана изучены [692]
в качестве ионообменников для щелочных и щелочноземельных эле-
ментов. Максимальная емкость 0,9—1,2 моль/г для щелочных и
0,4—0,45 моль/г для щелочноземельных элементов. В среде NH4C1
по коэффициентам распределения элементы располагаются в ряды
Na+ К+ < Rb+ Cs+ и Са2+ Sr2+ Ва2+. По пригодности для
разделения ионообменники располагаются в ряд: молибдат титана
молибдат циркония вольфрамат титана вольфрамат цирко-
ния. Вольфрамат циркония применяли [188] для разделения щелоч-
ных металлов при радиоактивационном определении их в кремнии.
Вначале 0,1 М раствором NH4C1 элюируют натрий, затем 0,3 М
NH4C1 — калий, 1 М NH4C1 — рубидий и 5 М NH4C1 — цезий.
Для разделения натрия, рубидия и цезия использовано различие
в коэффициентах распределения между NH4C1 и фосфатом, вольфра-
матом и молибдатом циркония в Ь1Н4-форме [603]. Наилучшие ре-
зультаты получены на молибдате циркония. Натрий вымывают
0,1 М раствором NH4C1, рубидий — 0,5 М NH4C1, цезий — 1 М
NH4C1.
Определены коэффициенты распределения ионов на 12-молибдо-
фосфате аммония из сред 0,1 М NH4NO3 [681]:
Катион Li+ Na+ К+ Rb+ Cs+ Н3О+
Kd 0,57 0,78 4,7 266 4024 0,84
Носитель предложен для отделения натрия от К, Bb, Cs, но не
от лития. Элюентом для натрия является 0,01 М HNO3. Получены
удовлетворительные результаты при отделении 0,25—0,5 мМ натрия
и 0,1—0,25 мМ калия от смеси, содержащей 0,1—0,4 мМ Mg, Са и
0,1-0,25 мМ Си.
На бумаге, импрегнированной 12-молибдофосфатом аммония,
в водно-этанольном растворе, 0,6 М по NH4N03 и 0,1 М по HNO3,
определены значения Rf ионов щелочных металлов [1151]:
Катион Ы+ Na+ К+ Rb+ Cs+
Rf 0,88 0,77 0,65 0,14 0,00
В отсутствие молибдофосфата эти ионы имеют близкие величины Rf:
0,81 (Li); 0,63 (Na); 0,62 (К); 0,61 (ВЬ) и 0,56 (Cs), не позволяющие
разделить щелочные металлы.
На колонке с фосфатом аммония—бериллия разделяли сравнимые
количества натрия и аммония [835].
Колонка, заполненная гексацианоферратом титана, пригодна
для разделения калия и натрия, а также натрия и лития [923]. При
разделении натрия и калия натрий элюируют 0,025 М раствором
NH4C1 со скоростью) 0,01 мл/мин; затем элюируют калий 0,05 М
38
раствором NH4C1. Для разделения лития и натрия литий элюируют
0,001 М НС1, а затем элюируют натрий 0,05 М раствором NH4C1.
Предложено [1179] хроматографическое разделение калия и нат-
рия в виде гидроксидов на суспензии силикагеля, пропитанного олеил-
амином. Амин снижает адсорбцию натрия.
Натриево-кальциевое стекло (73% SiO2, 12% СаО, 14% Na2O,
0,5% A12Os, 0,5% MgO) в виде порошка применяли для быстрого
выделения следов натрия [909]. Предварительно стекло обрабаты-
вали гидроксидом лития. Обменная емкость около 10-3 мг-экв/г.
ОТДЕЛЕНИЕ ДИСТИЛЛЯЦИЕЙ
Как правило, прием применяли для отгонки основы, при этом
натрий и другие микрокомпоненты остаются. Методы применяли для
определения натрия в чистых металлах и их соединениях: боре
[405], алюминии [1114], германии и диоксиде германия [1255], сурь-
ме [22], хроме [679] и соляной кислоте [405].
Большие количества бора отделяют от натрия О1-10"4%) от-
гонкой в форме борнометилового эфира после обработки пробы азот-
ной кислотой до полного окисления бора в борную кислоту [405].
Алюминий обрабатывают этилбромидом и образовавшийся три-
этилтрибромид алюминия отгоняют [1114]. Метод применен при
определении ±10-6% натрия в алюминии [1006].
Для концентрирования натрия в германии и его оксиде основу
отгоняют в виде тетрахлорида в кварцевом аппарате [1255]. Из
1 —5 г германия или диоксида германия основа полностью отгоняется
в течение 1—2 ч при обработке пробы 20 мл конц. НС1 и 2—5 мл
конц. HNO3.
Макроколичества сурьмы отгоняют в форме SbCl3 нагреванием
пробы с NH4C1 [22]. При отделении 100—500 мкг натрия от 4—5 г
сурьмы авторы определили 102—106 и 500—506 мкг натрия соответ-
ственно.
Хром отделяют от микроколичеств натрия, а также от К, Са,
Мп, Mg дистилляцией СгО2С12 после окисления до хрома(У1) хлор-
ной кислотой и прибавления НС1 (1:1) [679].
РАЗДЕЛЕНИЕ ЭКСТРАКЦИЕЙ
Изучена экстракция натрия из водного раствора нитробензолом
в присутствии иода, иодид-ионов и роданид-ионов (р 0,1) [1232].
Реакция описывается схемой
”J2(O)+ Х(в) + Na(B) (Ja)nX(o) + Na(o) >
где X = J~, SCN-. Из зависимости коэффициентов распределения от
концентрации иода в органической фазе и NaX в водной фазе найден
состав и вычислены константы образования комплексов: для NaJ3 —
^5,4 ±0,5)-10“2; Naif-20 ±1,5; NaJ4SCN4- -4,5 ±0,5. Метод
перспективен для эффективного выделения натрия и отделения его и
цезия из кислых сред.
39
Исследована экстракция натрия и других щелочных металлов
[750] растворами тетрацианодианилинохромата следующего состава:
[Cr(CeH6NH2)2(NCS)4]-(TAnCr-) и тетрацианодиамминхромата
(TAmCr-)[Cr(NH3)2(NCS)4]- (табл. 28).
Таблица 28
Степень извлечения щелочных металлов из 10~4 М растворов (pH 4—5) [750]
Реагент Растворитель R, %
Na+ K+ Rb+ Cs+
TAnCr- Нитрометан 95,2 98,9
TAmCr- » 26,6 82,3 94,6 98,2
TAnCr- Нитробензол 5,6 75,1 94,3 99,1
TAmCr- » 1,5 17,2 70,5 91,6
Метод применяли для отделения натрия от цезия.
На примере дициклогексил-18-краун-6 и 18-краун-6 показано,
что краун-эфиры мало пригодны для разделения натрия и щелочных
металлов [698]. Определены коэффициенты распределения натрия
(лития, калия, рубидия, цезия) между кислыми растворами и осад-
ком, образованным при встряхивании растворов бенэо-15-краун-5,
а-дибензо-24-краун-8, дибензо-18-краун-6 и 15-краун-5 в СН2С12 с
раствором молибдофосфорной гетерополикислоты в 2 М HNO3 [732].
В виде тетрафенилбората натрия натрий образует с дициклогек-
сил-18-краун-6 ионный ассоциат, экстрагирующийся хлороформом
[978]. Метод используют для выделения натрия при его нейтронно-
активационном определении в SiO2.
Изучена экстракция щелочных металлов в виде комплексов с кра-
ун-эфирами типа 4'-пикриламинобензо-15-краун-5, его нитро- и
бромэамещенными растворами триэтиламина или триэтаноламина
в хлороформе из сильнощелочных сред [1001]. Экстрагируемость
комплексов уменьшается в ряду K^>Rb^> Cs Na Li. Комплекс
лития не экстрагируется. Метод позволяет отделить натрий от 10—
800 мкг калия.
При изучении экстракции хлороформом комплексов пикратов
щелочных металлов с поли-, бпс-(бензо-15-краун-5) и бис-(бензо-
18-краун-6)-эфирами показано, что образуются соединения с соотно-
шением М:В = 1:1и1:2. Для натрия константы экстракции и
образования минимальны по сравнению с рубидием и цезием [876].
При исследовании экстракции ионов (по 5 мкг) метилизобутил-
кетоном найдено [190], что экстрагируется 1% натрия; другие эле-
менты экстрагируются из среды 7,5 М НС1 (в скобках приведена сте-
пень извлечения, %): As(V) (16); Ап(П1) (100); Ва (0); Bi (0); Со (1);
Сг(Ш) (1); Си (4); Fe(III) (100); Ga(100); Ge (93); Hf (9); In (67);
К (8); La (0); Mn(II) (0); Mo(VI) (92); Nd, Ni, Pr (0); Pb (5); Pt(IV)
(74); Re(VII) (73); Sb(V) (100); Sc (0); Se(IV) (39); Sn(IV) (63); Ta (0);
Te(IV) (75); W(VI) (61); Zn (10).
40
Натрий отделяют от ионов Си, Ag, Au, Pb, Zn, Fe, Cd, Ni, С ,
In, T1 (I, III), Bi, Mn, Se, Те, Sb(III) их экстракцией в форме диэти -
дитиокарбаминатов хлороформом при pH 5,5—6,5 [534]. Ионы V,
Ga, Pt экстрагируют при pH 2,5—3. От алюминия и магния натрий
отделяют экстракцией оксихинолинатов хлороформом или четырех-
хлористым углеродом.
Экстракцией три-н-бутилфосфатом из солянокислой среды можно
отделить натрий от Cr(VI), Fe(II, III), Ge(IV), Hg(II), Mo(VI), Pt(IV),
Sb(V), Te(VI), U(IV, VI), V(V), W(VI) [1237].
Изучена экстракция 22Na при комнатной температуре 0,01—1,0 М
раствором ди-(2-этилгексил)фосфорной кислоты в алифатических,
ароматических и полярных органических растворителях из водных
растворов хлоридов и нитратов с pH 0,5—3,0 (время контакта фаз
1 ч). Наклон зависимости 1g D—Ig [НА] равен 2 для гексана, октана,
декана, додекана, тетрадекана, циклогексана; 1,5 — для гексанола,
деканола, три-н-бутил фосфата, три-2-этилгексилфосфата. Из зависи-
мости 1g D —pH найдено уравнение экстракции
Mn+ -|- (п -|- p)/s(HA)S0 7^ (МАп-рНА)0 -|- пН+,
где s — степень полимеризации НА в органическом растворителе
(S); п + р — наклон функции lg D — lg[HA]; п — наклон функции
lg D — pH [709].
Натрий можно отделить от многих элементов их экстракцией из
азотно- или солянокислых сред 5%-ным раствором триоктилфосфин-
оксида в толуоле [831].
Отделение микроколичеств элементов от основы — натрия —
приведено в табл. 29.
Таблица 29
Условия отделения микрокомпонента от основы — натрия
Матрица Экстрагент Отделяемый микроном- понент Применение Лите- ратура
NaCl ДДТК в СНС1з (в присутствии ЭДТА, NH3, сульфосалицило- вой кислоты) Ag Полярографическое определение серебра в NaCl [461]
Цитрат натрия ТБФ + NCS- Са (5-10-з%) Определение кальция в цитрате натрия [256]
NaCl Азоазокси БН + 4-ТБФ в СС]4 Са (5-10-3- -2-10-*%) Экстракпионно-фото- метрическое определе- ние кальция в NaCl [110]
CH3COONa, C4H4OeKNa 8-(п-То луо лсуль- фониламино) хинолин Zn (1-10-5%) Флуориметрическое определение цинка в CH3COONa и C4H4OeKNa [440]
NaCl ДДТК в СНС13 Sb, Ag Полярографическое определение сурьмы в NaCl [198]
Примечание. ДДТК — диэтилдитиокарбаминат, ТБФ — три-н-бутилфосфат.
41
Отделение микроколичеств натрия экстракцией основы. Натрий
отделяют от суръмы(1Н) экстракцией бензолом галогенидов сурьмы
из среды 5 М H2SO4 + 0,01 М KJ или 5 М H2SO4 + 0,03 М НВг
[785]. В этих условиях в органическую фазу переходит 1 % натрия.
Большие количества урана (1 г UO2(NO3)2) отделяли от натрия
(Li, К, Са, Mg) экстракцией 5%-ным раствором три-н-бутилфосфата
в мепазине из среды 1,6 М HNO3 [939]. Прием применен для опреде-
ления натрия в уране методом фотометрии пламени. Экстракция
урана с целью его отделения от натрия изучена также в работе
[1008].
Железо отделяют от микроколичеств натрия экстракцией бутил-
ацетатом из среды 8 М НС1. Метод применяли при определении нат-
рия в железных сплавах [21].
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Ионообменная хроматография
Предложен метод расчета оптимальной концентрации элюентов
для щелочных и щелочноземельных элементов, основанный на вычис-
лении максимальной работы (Л) элюирования ионов при идеальном
обмене по следующему уравнению [455]:
А = 2,ЪПТ [— Ig С -f- п Ig Мн+ 4~ Ig MH+ -|- lg[Mм+],
где Мн+ — концентрация Н+ в элюенте; М'н > — равновесная концен-
трация Н+ в фазе ионообменника (для идеального обмена равна его
обменной емкости); Л/м+ — равновесная концентрация ионов металла
в исходном растворе; С — функция коэффициентов активности ионов
в обеих фазах и загрузки ионообменника. Величина линейного участка
изотермы сорбции, характеризующего идеальный обмен, резко су-
жается с увеличением атомной массы иона металла и повышением
степени сшивки катионообменника и расширяется с увеличением кон-
центрации элюирующего иона. Поскольку при элюировании концен-
трированными растворами кислот величина работы определяется
главным образом концентрациями этих кислот, значение индивиду-
альных свойств разделяемых ионов почти стирается, что затрудняет
и усложняет разделение. Поэтому, например, 0,7 М НС1 — неэффек-
тивный элюент для разделения натрия и лития; оптимальная кон-
центрация элюента — 0,065 М НС1.
Катионный обмен. Изучено влияние количества сорбированного
натрия при его исходных количествах 10~2—10-в г на положение его
вымывания 0,1—0,5 М раствором НС1 или нитрилотриуксусной кис-
лотой [125]. При использовании НС1 в качестве элюента положение
пика вымывания практически постоянно при содержании натрия
<^10-8 г; при содержании ^>10~3 г натрия пик смещается влево, т. е.
в сторону более быстрого вымывания. Иное поведение натрия при
использовании нитрилотриуксусной кислоты (pH 10,3). Положение
пика постоянно при содержании <Ч0~Б г натрия, при содержании
>10-Б г натрия пик смещается вправо, что авторы объясняют ком-
плексообразованием натрия с элюентом (1g |3 = 2,14).
42
Изучена кинетика ионного обмена из 0,01—1 М растворов NaGl
на катионообменниках СДВ-3, СМ-12, КУ-1 и СБС при температуре
6—35° С [354]. Для примера ниже приведены скорости обмена из
0,01 М растворов при 20° С:
Сорбент КУ-1 СБС СДВ-3 СМ-12
К, с"1 0,0028 0,0017 0,0009 0,0010
Изучена кинетика обмена ионов щелочных металлов (Na, Li, К)
в воде и 60%-ном метаноле на катионообменнике КУ-2 [116]. Ско-
рость ионного обмена хлоридов из 1 М растворов уменьшается в ряду
К+ Na+ Li+; в среде 60%-ного метанола обмен лития замедля-
ется, а обмен натрия и калия ускоряется по сравнению с водными
растворами. Ниже приведены константы равновесия обмена:
Катион Вода
Li+ 0,84
Na+ 1,49
К+ 2,35
60%-ный метанол
1,89
8,43
16,70
Определена свободная энергия обмена протон—катион на суль-
фированном вулканизате полибутадиена, содержащем 4% связанной
серы (СБР х 4), и на КУ-2, содержащем 4% дивинилбензола [454]:
Ионообменник
СБРХ4
КУ-2
—АГ, кал/г-экв
699
—62
Коэффициент распределения натрия на катионообменнике
AG50W X 8 зависит от содержания этанола в растворе (табл. 30).
Таблица 30
Коэффициенты распределения щелочных металлов в 0,50 М растворе
НС1 на катионообменнике AG50WX8 [1188]
Ион Содержание этанола, % об.
0 20 40 60 80
Cs+ 44,2 63 108 229 852
Rb+ 33,2 42,8 73 165 571
К+ 29,1 47,3 89 201 838
Li+ 8,1 10,8 17,1 28,8 50
Na+ 13,5 19,1 34,1 79 254 (0,50 М HCI)
6,9 9,7 16,6 36,5 138 (1,00М НС1)
3,8 5,5 8,9 24,5 (2,0 М НС1)
2,7 3,8 6,8 20,5 (3,0 М НС1)
Определены коэффициенты распределения щелочных металлов
на фенолоформальдегидной смоле био-рекс 40 (табл. 31).
Разработан метод разделения пар Li—Na, Na—К: литий элюи-
руют 500 мл раствора 1,00 М НС1 в 80%-ном этаноле, натрий,—
43
Таблица 31
Коэффициенты распределения щелочных металлов на сорбенте
бцо-рекс 40 [1184]
Концентрация HQ, M Li+ Na+ K+ Rb+ Cs+
0,1 17,6 35,5 67 95 203
0,2 9,8 18,4 42,8 63 123
0,5 4,2 8,5 19,8 28,9 64
1,0 1,0 4,3 9,6 16,3 38,4
2,0 0,5 2,4 4,9 8,4 20,0
3,0 0,5 1,9 3,3 4,0 13,4
4,0 0,5 1,5 3,0 3,8 9,8
500 мл 0,20 М НС1, калий — 250 мл 0,70 М НС1, рубидий — 450 мл
0,70 М НС1, цезий — 500 мл 4 М НС1 [1184].
Выведено корреляционное уравнение связи коэффициента распре-
деления и pH раствора при сорбции щелочных металлов катионо-
обменником Aminex Q — 150S [824]:
1g = at + ЬрН.
Коэффициенты уравнения приведены в табл. 32.
Таблица 32
Коэффициенты корреляционного уравнения и их производные
(Ъ = 0,70) [824]
Ион “i ai~ai = = lgri} rii Ион ai ai~aj = = *егО rii
Na+ K+ 0,09 0,31 0,40 2,52 Rb+ Cs+ 0,42 0,51 0,11 0,09 1,29 1,23
Для определения натрия с использованием катионного обмена
предложена циклическая реакция умножения, состоящая в последо-
вательном пропускании раствора, содержащего натрий, через катио-
нообменник в Н-форме, а элюата — через катионообменник в Na-
форме. При трех циклах пропускания погрешность определения
натрия 1—2%, минимально определяемое содержание натрия
11,5 мкг. Определение заканчивают методом фотометрии пламени
[1259].
Методы отделения натрия от других ионов на катионообменниках
систематизированы в табл. 33.
Анионный обмен. Изучено поведение натрия и еще 64 ионов на
анионообменнике Дауэкс 1 х 8 иэ растворов 2—17,4 СН3СООН
[1273]. Практически не сорбируются (Kd 1): Li, Na, К, Rb,
Cs, Be, Mg, Ca, Sr, Ba, Al, Ti(IV). Имеют Kd < 10 (ссщсоон = 2 M)
и Kd > 10 (cCHscooh > 2 M): Sc, V(IV, III), Cr(III), Mn, Fe(III),
44
Таблица 33
Использование катионообменников для отделения натрия
Катионообмен- ник Отделяемые эле- менты Элюент для натрия Применение метода Лите- ратура
Дауэкс 50 Изучение распреде- [881]
ления
Li 0,5М (NHjJaSO* Разделение [853]
Дауэкс 50x4 Li 12М НВт » [ЮН]
Дауэкс 50Wx8 Li Анализ минеральной воды [1015]
К 0,704М НС1 Разделение [1286]
Li, Rb Разделение [322]
Mg, Ca, Sr, Ba 0,1М HCOONHa » [1234]
Rb, Cs — » [846, 920]
К 0,4М НС1 Анализ силикатов [1055]
Амберлит Li, К Фенол—СН3ОН— Разделение [1243]
IR-120 к, Mg —НС1 0,12М НС1 Анализ горных пород и минералов [1090]
к • 0,5М НС1 Анализ КС1 [586]
Амберлит IR-1 Изучение распреде- [648]
ления
Амберлит Li, К 0,14М НС1 в 30%- Разделение [903]
GG-120 ном i-C3H7OH
КУ-2 Cs, Ca » [101]
Mg, Ca 0,001М НС1 [106]
Li, К » [247]
Al 0,01—0.02М НС1 [106]
NH+, K, Mg, 0,1М HNOs » [393]
Ca, Fe3*
Леватит S100 K, Mg Определение натрия [753]
Эспатит I, Концентрирование [25]
сульфоуголь К, калия и натрия при
Вофатит Р анализе водопровод- ной воды
СКВ, СБРХ4 Li, K, Rb, 0,025М НС1 Разделение [454]
Cs, Mg
Цёокарб-225 Cu, Sr, Y 1,1М НС1 Активационное опре- деление натрия в ли- [1168]
тии
Li, К 0,1М (NHjfoCOs Разделение [983]
Duolite 3 Cs Определение цезия в NaOH [1097]
KMZ К 0,5М НС1 Анализ стекла и по- левого пшата [976]
45
Таблица 33 (окончание)
КатионооСмей- ник Отделяемые эле- менты Элюент для натрия Применение метода Лите- ратура
Мерк IV КБ-4 П-2 и к Анализ урана Очистка KJ и КВг от натрия [589] [242]
РФ Ba, Zn, К, А1, Mg, Pb 0,1М НС1 Разделение [551]
Катекс S Катекс S8 к, Mg, Са Li 0,6М НС1 » » [1014] [1200]
Сорбент на ос- нове а-дибен- зо-24-краун-8 К 0,05М HNO3 * [732]
Биорад AG 50WX8 Be, Ba, Y, Zr 0,5М HNO3 0,5М HNOS » Анализ силикатов [1186] [1183]
Со, Ni, Cu, Zn, Ga, Ge, As, Se(IV), Y, In, Sn(IV), Sb(III), РЗЭ,
Pb, Np(VI); Kd > 10 (cCHscooh = 2ч-17,4 M) F~, POf, Cl", Cr(VI),
As(V), Br~, Zr, Mo, Tc(VI), Ru(III), Rb, Pd, Ag, Sb(V), Te(IV),
J-, Hf, Ta, W, Re(VII), Os(IV), Pt(IV), Au(III), Hg(II), Tl(III),
Bi, Po(IV), Th, U(VI). Разработан метод разделения смеси
Na—As—Cu.
На этом же анионообменнике можно отделить микроколичества
натрия (Ti, Sc, Cr, Ag) от микроколичеств As, Sb, W, Cu, Ta, Zn,
Co, Fe, Au, Sn, Mo. Из растворов 1 M HF + 9 M HC1 натрий и Ti,
Sc, Cr, Ag не сорбируются. Метод применен для выделения натрия
при его нейтронно-активационном определении [1010].
Для количественного отделения натрия от Li, К, Mg, Са, Sr,
Ba, Al, Ti, Mn(II), Cu, Со, Fe(III), Zn, Hg, Sb(III) и Bi раствор
7 M по HC1 в метаноле пропускают через колонку с амберлитом
CG400 в Cl-форме и элюируют Na, Li, К, Mg, Са, Sr, Ba, Ni,
Al и Ti 80 мл раствора 7 M НС1 в метаноле. Элюат выпаривают, оста-
ток растворяют в 0,3 М НС1, пропускают через колонку с амберлитом
и вымывают литий 170 мл раствора 0,3 М НС1, натрий — 190 мл
0,3 М НС1, калий — 360 мл 0,3 М НС1 [742]. В качестве элюентов
для натрия с анионообменника амберлит CG 120 изучены смеси со-
ляной кислоты с метанолом, этанолом или н-пропанолом (0—60%
об. органического растворителя). Отмечается, что коэффициенты рас-
пределения возрастают с увеличением содержания спиртов [743].
Для отделения натрия от щелочноземельных элементов его элюи-
руют смесью 0,3 М НС1 и 40% об. метанола и этанола.
Сильноосновной анионообменник АВ-17 в ОН-форме (R'OH)
используют для переведения хлорида натрия в NaOH и его титри-
метрического определения [431]:
R'OH + NaCl R'Cl + NaOH.
46
Определению не мешают катионы поливалентных металлов, обра-
зующие при ионном обмене гидроксиды, задерживаемые колонкой:.
3R'OH + МС13 3R'C1 + М(ОН)3 | .
На колонке с анионообменником варион AD в ОН-форме отделя-
ли натрий от Zn, Sn(II), Al, Fe(III), Ni; в фильтрате находится NaOH,
который можно оттитровать кислотой и определить содержание
натрия [918].
Методы отделения натрия от отдельных ионов с использованием
анионообменников рассмотрены ниже.
Натрий отделяют от алюминия на церолите FF в С2О4-форме
[1012]. Метод применяют при определении натрия в криолите.
Для отделения натрия от микроколичеств титана использовали
Дауэкс 1x8 (элюент 1 М HF) [1010] и Вофатит L 150 в ОН-форме
[1111]. Во втором случае сорбируют сульфосалицилатный комплекс
титана; метод применяют при определении натрия в титановых
белилах.
Малые количества натрия (0,016—0,095%) отделяют от больших
количеств хрома (1 г СгО3) сорбцией хрома анионообменником АВ-17
в Cl-форме после окисления хрома(Ш) до хрома(УТ) [79]. Метод при-
меняют при определении натрия методом фотометрии пламени в хро-
мовом ангидриде.
Разработан хроматографический метод отделения больших коли-
честв молибдена и вольфрама от калия и натрия на сильноосновном
анионообменнике Вофатит SBW из растворов, содержащих НС1—
Н2С2О4—Н2О2 [706]. В этих условиях коэффициенты распределения
молибдена и вольфрама достигают 104, в то время как для натрия и
калия они меньше единицы. Метод применен для отделения натрия и
калия при их активационном определении в молибдене и вольфраме.
Основу — вольфрам отделяют от микроколичеств натрия сорб-
цией вольфрама Сефадексом ДЕАЕ [898]. Метод применен для опре-
деления натрия в вольфраме и его соединениях.
Разработан метод отделения натрия от урана сорбцией урана ионо-
обменником Дауэкс-2 в Cl-форме из среды 8 М НС1 или 8О4-форме
из среды 0,025 М H2SO4 [8931. Вместе с натрием не сорбируются ионы
Th, La, Cr, Си, Со, Ni, Zn, Cd, Мп. Метод применяют при опреде-
лении натрия в уране активационным методом.
Определено содержание натрия в ионообменниках [705]. По мере
увеличения содержания натрия они располагаются в ряд: Вофатит
SBW Вофатит SBK<^ ЭДЭ 10П, но во всех сорбентах оно крайне
мало и сорбенты можно использовать для сорбции следов элементов
и отделения их от натрия. Содержание натрия в сорбентах не изме-
няется при пропускании NaCl или ЭДТА при различной их кислот-
ности.
Экстракционная хроматография
Метод экстракционной хроматографии на кизельгуре в качестве
носителя неподвижной фазы применен для разделения щелочных
металлов. В качестве экстрагента использованы ациклические по-
лиэфиры. При применении 1,13-бис-(8-хинолин)-1,4,7,10,13-пента-
47
оксатридекана коэффициенты распределения изменяются в последо-
вательности Cs Rb К Na Li, а при применении краун-
эфира дибензо-18-краун-6— в последовательности Li Na Rb ^>
> К Cs. Коэффициенты разделения соседних пар составляют
1,13—3,12 [1171].
Хроматография на бумаге
Из различных вариантов наиболее часто для разделения приме-
няют восходящую хроматографию (табл. 34).
Таблица 34
Условия разделения натрия и ряда элементов восходящей хроматографией
на бумаге
Бумага Подвижная фаза й/ Лите- ратура
Machery Nagel № 261 Катионообменник Ам- берлит IR 120 или суль- фоцеллюлоза Анионообменник IRA 400 Ватман № 1 Ватман № 1, импрегни- рованный молибдофос- фатом аммония Ватман № 3 Ватман № 540 Шлейхер и Шюлль Шлейхер и Шюлль 2045в 96%-ный С2НБОН—2М СНзСООН (8 : 2) Вода—ацетон—конц. НС1 (20 : 8 : 18) 0.0125М НТ А—ОДОМ NHg—О.О75М KCN 4М НС1 в С2НБОН 0,32М С2О*~—9,20М C4H4OI-—0,32М цитрат в 90%-ном С2НБОН 0,1М HNO3—0,2М NH4NO3 2-этилгексанол—СН3ОН (3:7) 94%-ный С2НБОН СНзОН-С2НБОН—Н2О (2 : 14 :4), насыщенные сер- нистым газом СН3ОН-Н2О (9 : 1) СН3ОН—С2НБОН— Н2О (2 : 14:4), насыщенные серо- водородом фенол, насыщенный 2М НС1 СН3ОН (95%-ный С2НБОН) Li 0,89; Na 0,56; К 0,35; Mg 0,85; Са 0,70; Ва 0,14; Sr 0,26 Li 0,78; Na 0,77; К 0,56; Rb 0,06; Cs 0,0 Li 0,65; Na 0,21; К 0,06 Na 0,62; Li 0,72; К 0,0 Li 0,44; Na 0,06; К 0,00; Rb 0,23; Cs 0,53; Fr 0,57 Li 0,71 (0,58); Na 0,46 (0,26); К 0,28 (0,10); Rb 0,28 (0,10); Ca 0,61 (0,32) [600] [1152] [1152] [1003] [1162] [582] [649] [1233] [612] [659] [951] [И57] [961]
48
Количества щелочных и щелочноземельных элементов на восхо-
дящих бумажных хроматограммах определяют после электролитиче-
ского выделения на ртутном катоде с большой поверхностью при
3—4 В [676]. Применяют ячейку из полиакриловой смолы, сообща-
ющуюся с ртутным резервуаром. Анодное и катодное пространства
разделяют фильтровальной бумагой, в качестве анода используют
графит. При определении 1,2—20 мкг натрия (в форме NaCl) отно-
сительное стандартное отклонение <0,007.
Методом радиальной хроматографии разделяли ионы Li, Na и К
[1241], используя в качестве подвижной фазы смесь (1 : 1) СН8ОН
и С2Н6ОН. Разделяемые элементы имеют следующие величины Rf
(при 27 1° С): Li 0,84; Na 0,61; К 0,39. В качестве проявителя
используют 0,2 %-ный раствор феносафранина в ацетоне, смешанный
с равным объемом 0,1 М раствора AgNO3.
Этим же методом разделяли Na, К, Са и Mg [624]. Элюентом была
смесь (5 : 1) С2Н5ОН и 2 М раствора масляной кислоты. После раз-
деления зон (18 ч) кальций и магний элюировали разб. НС1, а натрий
и калий — безводным диметилформамидом; натрий определяли спек-
трофотометрически виолуровой кислотой.
На бумаге Шлейхер и Шюлль разделяли щелочные металлы с ис-
пользованием фенола, насыщенного 2 М НС1 [1157]. За 6 мин фронт
растворителя перемещался на 14 см при исходной концентрации
щелочных металлов 3 мг/мл; при более высоких концентрациях ще-
лочных металлов авторы наблюдали перекрывание зон натрия и
калия.
Радиохроматографический метод позволяет разделить смесь,
содержащую по 80 мкг натрия и калия, используя нисходящий
вариант [1124]. Подвижным растворителем являлся метанол, ионы
разделяли в форме иодидов, меченых нуклидом 131J, который опре-
деляли авторадиографически или радиометрически.
Методом двумерной хроматографии на бумаге отделяли натрий от
щелочных и щелочноземельных элементов [482]. В качестве первого
растворителя использовали смесь (87 : 13) абсолютного этанола и
воды, в качестве второго — фенол, насыщенный водой. Разделяемые
ионы имеют следующие величины 7?/ (при 19° С): Li 0,17; Na 0,14;
К 0,19; Rb 0,26; Cs 0,40; NH4 0,25; Mg 0,09; Ca 0,08; Sr 0,80; Ba
0,07.
Тонкослойная хроматография
При изучении поведения иодидов натрия и других щелочных
металлов методом распределительной тонкослойной хроматографии
на силикагеле [843] найдены корреляции между величиной 7?/м
и ионным радиусом, свободной энергией гидратации или диэлектри-
ческой проницаемостью растворителя (нитробензол—бензол, нитро-
бензол—СС14, нитрометан—бензол, нитрометан—СС14). Эти законо-
мерности использованы для выбора условий разделения щелочных
металлов методом тонкослойной хроматографии.
Методом тонкослойной хроматографии на слое силикагеля можно
отделять натрий (/?f = 0,02) от Fe, Mo, Sn, Au, Sb, Те (Rf = 0,97),
49
Li (0,51), Мп (0,43), Mg (0,34) [1154]. Элюентом является смесь 20%
об. конц НС1,20% об. СН3ОН, 20% об. С2НБОН, 20% об. амилацетата,
20% об. метилизобутил кетона; вместе с натрием находятся ионы Ti,
К, Ва и Zn.
Изучалась роль олеиламина при хроматографическом разделении
калия и натрия на пластинках, покрытых силикагелем Н и пропи-
танных олеиламином (суспензию силикагеля в хлороформном раство-
ре амина испаряли под вакуумом) [1178]. Силикагель содержал
0,025 г амина/г сорбента. В качестве элюентов использованы вода и
0,1 М СН3СООН, в качестве проявителя — виолуровая кислота или
бромтимоловый синий.
Ионы Na, К, ВЬ и Cs можно разделить на стеклянных пластинках,
покрытых тонким слоем А12О3 марки G, содержащего 10% CaSO4,
активированного в течение 30 мин при 130° С и промытого 0,05 М
раствором НС1. В качестве элюента предложен фенол, насыщенный
6 М раствором НС1 (140—150 мин при 32° С). На высушенной хро-
матограмме натрий обнаруживают проявлением цинкуранилацетатом
и облучением пластинки УФ-светом [791].
На пластинках с тонким слоем Zn2[Fe(CN)6] разделены ионы Na,
К, ВЬ и Cs [866] в течение 16 мин с использованием 0,2 М раствора
NH4NO3 в качестве подвижной фазы. Метод пригоден для разделе-
ния хлоридов нуклидов щелочных металлов 24Na, 42К, 8вВЬ и 137Cs.
Методом восходящей хроматографии на тонком слое гипофосфита
циркония отделяли натрий от двухвалентных ионов Са, Sr, Ва, Со,
Ni, Си, Zn, Cd, Hg, Sn и Pb [896]; подвижной фазой являлась
0,1 М НС1.
Другие методы разделения
Разработан метод выделения натрия с использованием фронталь-
ной динамики [209]. При пропускании воды через колонку ионооб-
менника из-за эквивалентности обмена концентрация менее сорби-
руемого иона в фильтрате достигает суммарной концентрации катио-
нов в исходной смеси, после чего эффект вытеснения проявляется
в расширении зоны вытесняемого иона. Второй, более сорбируемый
компонент смеси (например, натрий) начинает проявляться в филь-
трате после того, как концентрация менее сорбируемого иона, после
достижения суммарной концентрации смеси, начинает снижаться.
Пробы для анализа следует брать на восходящей ветви выходной
кривой натрия, вблизи к максимуму концентрации. В сочетании
с изотопным разбавлением метод применен для определения натрия
в морской воде.
Многокомпонентные смеси электролитов разделяли методом дви-
жущейся границы в условиях гидродинамического противотока
в колонках-капиллярах. Под действием постоянного тока ионы
различаются по подвижности и разделяются. Подвижность ионов
в растворах хлоридов убывает в ряду Н+ > К+ Na+ Ва2+
> Са2+ > Nd3+ > Mg2+ > Th(IV) > Li+ > Co2+ > Al(III) >
> Mn2+ > Cu2+> Ti(IV)> Fe(III) > U(VI) > Zn2+ > In3+ > Cd2'
50
Растворы бромидов и иодидов показывают аналогичное уменьшение
подвижности ионов за небольшими исключениями [350].
Предложено уравнение для расчета скоростей движения зон при
электрохроматографическом разделении натрия и лития на сульфо-
катионите СДВ-3 [540].
Описан метод электрохроматографии на бумаге для разделения
натрия и калия [1056]. На фоне (NH4)2CO3 при напряжении 220—
360 В и силе тока 35—55 мА эа 40—100 мин отделено 0,11—250 мкг
калия от 0,03—236 мкг натрия. Погрешность при определении калия
<16,7% (0,12 мкг), при определении натрия <7,9% (38 мкг). После
высушивания полоски бумаги (Ватман № 4) катионы обнаруживали
бромтимоловым синим. Метод применим при определении щелочных
металлов в почве.
Ионы натрия и остальных щелочных металлов разделяли с по-
мощью колоночной распределительной хроматографии на носителе
политрифторхлорэтилене с обращенными фазами [579]. В качестве
неподвижной фазы использовали смесь иода, NH4J и нитробензола,
элюентами являлись вода, 1 М НС1 или 6 М НС1, содержащие иод и
NH4J. Элементы элюируются в последовательности Li Na < К^>
<-Rb < Cs. Метод позволяет отделять натрий от цезия.
Метод разностной ионообменной хроматографии применен для
наблюдений за изменением содержания натрия в морской воде
[942]. Использованы ионообменные мембраны AM FiOH, А-104В
и С-103С. Измерения проводили в камере, состоящей из анионной
й катионной мембран, соединенных через образец морской воды, ис-
пользуемой в качестве стандарта, с хлорсеребряным электродом.
Изменение содержания натрия в морской воде приводило к появле-
нию пика, характеризующего изменение концентрации.
Предложен [285] метод разделения ионов Na, Rb, Cs и Fr на си-
ликагеле КСК-2. Элементы сорбируются в последовательности
Na < Rb < Cs < Fr. Избирательным элюентом является 0,05—
0,2 М раствор NH4NO8 с pH 10.
Для разделения натрия и калия использован ионный обмен
в сочетании с электромиграцией на колонке с диаионом РК-228
в Ш14-форме без наложения и с наложением электрического поля
(150 В, 3,3 мА) [987]. В качестве элюента использован 0,1 М раствор
NH4C1. Содержание щелочных металлов во фракциях оценивали по
разности концентраций NHt в элюенте и в выходящих из колонки
фракциях. При наложении электрического поля калий перемещался
быстрее натрия, эффективность разделения повышалась.
Методом радиальной электрохроматографии на бумаге Ватман № 3
разделяли смесь, состоящую из 300—400 мкг лития, 100—150 мкг
натрия и 150—400 мкг калия [614]. Напряжение 450—500 В, сила
тока 2—2,5 мА; растворителем являлась смесь, содержащая 40% об.
пропиленгликоля, 20% об. изопропанола, 40% об. буферного рас-
твора (смесь 50 мл 0,2 М NH4C1 и 6,7 мл 0,2 М НС1 разбавлена водой
до 200 мл).
Микроколичества натрия отделяли от основы — магния — элек-
тролизом на ртутном катоде и последующей обработкой амальгамы
51
водой [941]. За 5 ч электролиза натрий выделялся количественно.
Метод применен при активационном определении натрия в магнии.
Электролизом на ртутной капле при силе тока около 3 мА на фоне
0,05 М иодида тетраметиламмония отделяли натрий [189]. Вместе
с натрием концентрируются калий, аммоний, барий и стронций.
Для концентрирования натрия из слабоминерализованных вод
предложено использовать электроосмос через электроотрицательные
политетрафторэтиленовые мембраны [318]. Коэффициент концентри-
рования для натрия достигает 1000; в то же время хлорид-ионы не
проникают сквозь мембрану.
Предложен метод разделения кобальта, марганца и натрия
электрофоретическим фокусированием ионов [770]. Полноту разде-
ления контролировали по нуклиду 22Na, в качестве элюента исполь-
зована смесь ЭДТА и NaOH (pH 5,8).
Щелочные металлы отделяли от T1(I), Ag, Hg(I) и многозарядных
катионов электрофорезом на бумаге Итон-Дайкмен № 301 [1153].
Натрий проявляли цинкуранилацетатом и наблюдали флуоресцен-
цию при облучении УФ-светом; количественно определяли методом
фотометрии пламени.
Изотахофорез (вытеснительный электрофорез) применен для раз-
деления смеси NHt, К+, Na+, Li+ и Fe(III) [606].
Методом гелъхроматографии на колонке, заполненной сефадек-
сом G 25, можно разделить натрий и другие щелочные металлы,
а также аммоний, используя в качестве элюента 0,01 М НС1 в
70%-ном водном растворе диоксана [1282].
Ниже приведена литература по отделению натрия от других
элементов.
Li [120, 247. 322, 399, 455, 466, 540, 574, 588, 600, 606, 614, 618, 628, 648,
649, 657, 659, 698, 741, 742 , 761, 762 , 815 , 853, 903, 923, 951, 982,
1003, 1004, 1011, 1033, 1034, 1049, 1105, 1152, 1154, 1157, 1171, 1184,
1187, 1188, 1200, 1233, 1241, 1243, 1264, 1265, 1282].
К [31, 54, 94, 97, 170, 188, 215, 241, 242, 247, 305, 393, 428, 433, 436,
475, 551, 578, 588, 600, 606, 614, 618, 624, 628, 648, 649, 659, 662,
676 , 681, 698, 732, 742, 763, 791, 815, 824, 843, 866, 885 , 903, 907, 923,
951, 953, 982, 987, 1001, 1003, 1004, 1009, 1014, 1029, 1033, 1034, 1048,
1049, 1124, 1157, 1165, 1170, 1171, 1178, 1179, 1181, 1183, 1184, 1185, 1233,
1241, 1243, 1253, 1264, 1265, 1282, 1286].
ВЬ [170, 188, 285, 322, 379, 381, 588, 603, 791, 824. 843, 846, 866, 920, 953,
1033, 1034, 1157, 1183].
Cs [188, 285, 379, 381, 579, 588, 603, 618, 791, 824, 843, 846, 866, 919, 920
1033, 1034, 1095, 1097, 1157, 1183]
Fr [285, 1157]
NH44 [393, 606, 691, 835, 1162, 1282]
Си [87, 100, 178, 223, 243, 259, 350, 370, 371, 373, 374, 377, 378, 466, 471, 534,
618, 681, 742, 896, 977, 1010, 1037, 1094, 1127, 1183, 1213, 1273]
Ag [178, 370, 371, 374, 461, 534, 618, 897, 1094, 1153, 1213]
Au [178, 370, 371, 374, 534, 618, 1010, 1154]
Be [243, 618, 1183, 1186]
Mg [8, 106, 223, 393, 466, 534, 578, 600, 618, 624, 691, 742, 941, 1014, 1048,
1094, 1154, 1162, 1183, 1185, 1233, 1234, 1279]
52
Ca [106, 110, 223, 256, 393, 466, 600, 618, 624, 681, 742, 743, 791, 879, 896,
897, 1014, 1094, 1127, 1128, 1183, 1185, 1233, 1234, 1244, 12791
Sr [384, 466, 592, 600, 618, 742, 743, 896, 1094, 1095, 1183, 1234]
Ба [466, 600, 618, 742, 743, 791, 896, 1094, 1183, 1186, 1234]
Zn [178, 243, 259, 370, 374, 440, 466, 534, 742, 896, 918, 1010, 1094, 1127,
12131
•Cd [178, 350, 466, 534, 897, 1037, 1094, 1095, 1127, 1183, 1213]
Hg [466, 742, 896, 897, 1094, 1153, 1237]
В [405, 459, 521, 618]
Al [106, 223, 243, 259, 342, 353, 370, 371,'373, 374, 466, 534, 618, 741, 742, 918,
962, 980, 1006, 1012, 1050, 1114, 1183, 1185, 1213, 1279]
Ga [370, 371, 534]
In [350, 370, 371, 534, 1213]
T1 [370, 371, 534, 1153]
Sc [178, 1095]
Y [618, 896, 1186]
РЗЭ [178, 350, 431, 896, 1094]
C [360]
Si [259, 360, 384, 1279]
Ge [370, 371, 550, 1237, 1255]
Sn [371, 373, 374, 618, 896, 918, 1010, 1154]
Pb [202, 243, 370, 373, 374, 383, 466, 491, 534, 618, 896, 1094, 1127, 1183]
Ti [350, 374, 618, 622, 742, 1094, 1111, 1127, 1185, 1279]
Zr [618, 1185, 1186]
Hf [1127]
Th [350, 897]
N [618]
P [458]
As [178, 370, 371, 1010, 1273]
Sb [178, 198, 370, 371, 374, 534, 742, 785, 1010, 1094, 1095, 1154, 1237]
Bi [243, 370, 371, 374, 385, 534, 618, 742, 1094, 1183]
V [87, 169, 223, 243, 374, 378, 534. 618, 977, 1127, 1185, 1237]
Nb [223, 1127]
Ta [96, 1010, 1127]
0 [618]
S [155]
Se [534, 626]
Те [140, 534, 1154, 1237]
Cr [79, 178, 223, 370, 371, 373, 374, 377, 378, 384, 618, 679, 823,884, 897, 977,
1037, 1094, 1095, 1127, 1237]
Mo [87, 178, 374, 618, 706, 862, 884, 1010, 1037, 1127, 1154, 12371
W [178, 685, 706, 898, 1010, 1237]
U [350, 589, 647, 893, 939, 1008, 1037, 1094, 1237]
F [618]
Cl [520, 618, 1127, 1205]
Br [1127]
J [1127]
Mn [95, 178, 223, 243, 259, 370, 371, 363, 374, 377, 378, 534, 618, 770, 823, 977,
1037, 1094, 1127, 1154, 1183, 1185, 1213]
53
Fe [21, 87, 178, 223, 243, 259, 342, 370, 371, 373, 374, 377, 378, 393, 466, 471,
534, 606, 618, 742, 823, 884, 897, 918, 977, 1010, 1037, 1094, 1127, 1154,
1183, 1185, 1237, 1279]
Co [87, 178,1 223, 243, 370, 371, 373, 374, 377, 378, 466, 534, 618, 742, 770, 884,
896, 897, 977, 1010, 1037, 1094, 1095, 1127, 1213].
Ni [87, 100, 178, 223, 243, 259, 370, 371, 373, 374, 377, 378, 466, 471, 534, 618,
823, 884, 896, 897, 918, 977, 1037, 1050, 1094, 1127, 1213]
Os [124]
Pt [1237]
He разработаны методы отделения натрия от Тс, Re, Ru, Rh, Pd, Ir.
Глава IV
гравиметрические; методы
ОПРЕДЕЛЕНИЯ НАТРИЯ
Аналитическая химия натрия бедна гравиметрическими методами,
поскольку большинство соединений натрия с неорганическими и
органическими реагентами растворимо в воде. Реагенты, образую-
щие малорастворимые соединения с натрием, как правило, недоста-
точно селективны, так как образуют осадки с большинством других
ионов. Поэтому чаще всего натрий отделяют, а затем определяют.
Значительная часть гравиметрических методов в первоначально раз-
работанном варианте со взвешиванием осадка представляет только
исторический интерес. Ряд методик изменен в настоящее время и со-
четает осаждение натрия с целью его отделения и выделения с кос-
венным определением натрия по ионам и реагентам, входящим в со-
став осадка (см., например, главу V «Титриметрические методы опре-
деления натрия»).
Большие количества натрия обычно определяют гравиметрически
в форме Na2SO4 или NaCl. Метод мало селективен и в присутствии
калия дает суммарное количество натрия и калия. В другой аликвот-
ной части раствора определяют гравиметрически калий в форме пер-
хлората и содержание натрия находят по разности. Средние коли-
чества натрия (не более 20 мг) определяют гравиметрически, осаждая
его в форме тройных ацетатов. Метод более чувствителен и гораздо
селективнее. Он позволяет не только оканчивать определение нат-
рия взвешиванием высушенного осадка, но и определять натрий кос-
венно титриметрическими или фотометрическими методами.
ПРЯМЫЕ МЕТОДЫ
Определение неорганическими реагентами
Предложен метод осаждения натрия из водно-ацетоновых раство-
ров в форме малорастворимого соединения состава Na2H2Sb2O7-H2O,
устойчивого при высушивании до 125° С. Определению не мешают
хлорид, карбонат, оксалат и щелочные металлы [1076].
Гравиметрически можно определять натрий в виде соединения
состава K4NaHVi0O28-10H2O, осаждая натрий избытком KeVi0O28-
54
• 10Н20 в присутствии КС1 для понижения растворимости осадка.
Осаждение проводят при pH 4,5 [1142].
При гравиметрическом определении натрия в присутствии калия
вначале определяют сумму хлоридов натрия и калия, а в другой
.аликвотной части осаждают тетрафторобораты и вытесняют натрий
из осадка аналогичной солью аммония; затем осадок высушивают
при 170—180° С [428]. Содержание натрия и калия рассчитывают
по уравнениям
mNaci = 5,2741 2 ТФБ — 8,908 2 хлоридов,
текш = 9,9085 2 хлоридов — 5,2741 2 ТФБ,
где ТФБ — тетрафенилоорат натрия или калия. При анализе смеси,
содержащей 5—95% КС1 и 95—5% NaCl,, получены удовлетвори-
тельные результаты; можно определять 382,2—24,1 мг натрия в при-
сутствии 20,4—467 мг калия.
Метод с переведением натрия в тетрафтороборат натрия применен
для определения натрия в его солях: тетраборате, фториде, хлориде,
бромиде, ио диде, нитрате и нитрите [427]. Отмечается легкость пере-
хода натрия из всех указанных солей в тетрафтороборат. Осадок
высушивали при 170—175° С.
Тетрафтороборатный метод определения натрия применен для его
определения во фтороборных флюсах, получаемых сплавлением
KBF4 и Na2B4O7 и не содержащих значительных количеств других
металлов, фосфатов или сульфатов [429].
При нагревании тетрафтороборат натрия разрушается (табл. 35).
Таблица 35
Термическая устойчивость тетрафторобората натрия [427]
t, °C Длительность нагревания, ч Уменьшение массы, % °C Длитель- ность нагре- вания, ч Уменьшение массы, %
200 4 0,00 0,00 260 4 0,61 0,62
220 2 0,08 0,08 300 2 0,66 0,62
220 260 4 2 0,16 0,31 0,20 0,31 300 4 1,34 1,34
При определении натрия в форме сульфата возможно образова-
ние двух форм. Глауберова соль Na2SO4*10H2O малопригодна в ка-
честве гравиметрической формы, так как кристаллизационная вода
выветривается и состав осадка изменяется. При температуре около
32° С декагидрат переходит в безводный сульфат Na2SO4. Эта соль
кристаллизуется в виде бесцветных гигроскопических кристаллов
ромбически-бипирамидальной формы. При нагревании до 240° С обра-
зуется гексагональная форма, устойчивая в интервале температур
400—700° С; при нагревании выше 700° С выделяется около 0,05 %
воды; при температуре около 900° С разложение соли еще незначи-
тельно. Оно заметно при температуре 1200—1220° С и особенно при
температуре выше 1330° С [748].
55
Предложен вариант метода, в котором вначале определяют
сумму сульфатов натрия и калия, затем в отдельной порции осажда-
ют гидротартрат калия и определяют калий в осадке алкалиметри-
ческим титрованием. Содержание натрия определяют по разности
[979].
Хлорид натрия после высушивания при 14D° С содержит еще
0,20% воды, а при температуре 550° — 0,07% воды. При осаждении
хлорида натрия из растворов НС1 соль содержит 0,11—0,25% гигро-
скопической воды. Соль устойчива при нагревании до температуры
не выше 820° С; уже при 1080° С разложение заметно, а при 1150° С
оно протекает очень быстро [748].
Хлорид натрия использован в качестве весовой формы при гра-
виметрическом определении натрия в присутствии щелочных метал-
лов [102]. Предварительно хлориды щелочных металлов превращают
в перхлораты, затем экстрагируют н-бутанолом и этилацетатом пер-
хлораты натрия и лития и отделяют их от калия, рубидия и цезия.
Экстракт упаривают до удаления этилацетата и осаждают натрий
в форме хлорида н-бутанолом, насыщенным хлористым водородом,
отделяя натрий от лития.
Смесь хлоридов растворяют в воде и переносят в стакан из стекла пирекс
вместимостью 150 мл. Раствор обрабатывают хлорной кислотой, взятой в 2—3-
кратном количестве по отношению к стехиометрическому (в любом случае не
менее 1 мл 60—70%-ной HClOi). Выпаривают досуха на горячей плитке (не
выше 350° С) и удаляют кислоту, сконденсировавшуюся на стенках стакана,
нагреванием пламенем горелки. По охлаждении растворяют соли в 3—5 мл воды
и снова выпаривают на плитке досуха. Охладив, прибавляют 10—20 мл смеси
(1 : 1) безводных и-бутанола и этилацетата, нагревают при температуре, близкой
к кипению, 2—3 мин и снова охлаждают до комнатной температуры. Отстояв-
шуюся жидкость декантируют в предварительно прокаленный и взвешенный
тигель Гуча и трижды промывают осадок декантацией и-бутанольно-этилацетат-
ной смесью (1 : 1) порциями по 5 мл. Фильтрат и промывной раствор сохраняют.
Остаток в тигле растворяют в минимальном количестве воды, собирая раствор
в стакан, в котором находится основная масса остатка, и снова выпаривают
досуха. По охлаждении вводят 10 мл растворителя, нагревают и охлаждают,
как прежде. Фильтруют в тот же тигель, остаток взмучивают в и-бутанольно-
этилацетатной смеси и тонкой струей переносят в тигель, а затем из промывалки
промывают 10—15 раз той же смесью, порциями по 0,1—1 мл. Объединяют
фильтраты и промывные воды, общий объем которых не должен превышать
55 мл, и выпаривают на горячей плитке до удаления этилацетата, что происхо-
дит после упаривания раствора по крайней мере до 40% его первоначального
объема. Упаривание продолжают до объема 20 мл. Нагревают до 80—90° С и
по каплям при перемешивании прибавляют 2 мл и-бутанола, содержащего 20%
хлористого водорода, а затем вводят еще 6 мл этого раствора. Охлаждают до ком-
натной температуры, переносят осадок в тигель Гуча и промывают 8—10 раз
порциями по 1—2 мл 6—7%-ного раствора (пл. 0,8425—0,8485) хлористого во-
дорода в и-бутаноле (получают прибавлением 100 мл и-бутанола к 40 мл 20%-
ного раствора хлористого водорода). Тигель с осадком сушат несколько минут
при 110° С, а затем прокаливают при 600° С в муфельной печи или до темно-крас-
ного каления на пламени горелки. Для гравиметрического определения натрия
тигель с загрязненным NaCl охлаждают в эксикаторе и взвешивают. Затем осадок
в тигле обрабатывают тонкой струей воды до растворения NaCl и тщательно
промывают тигель. Тигель с нерастворимым остатком сушат 1 ч при 110° С,
затем охлаждают и снова взвешивают. Потеря в массе соответствует содержанию
NaCl.
56
Перхлорат натрия NaC104 также известен как гравиметрическая
форма определения натрия. Соль кристаллизуется в форме моно-
гидрата из растворов с температурой ниже 50° С; при повышении
температуры кристаллизационная вода удаляется, образуется фор-
ма, устойчивая до температуры не выше 300° С; при нагревании выше
этой температуры NaC104 разлагается на NaCl, NaC10s и кислород.
Примеры использования данной формы при гравиметрическом опре-
делении натрия рассмотрены в монографии [748].
При гравиметрическом определении суммы щелочных металлов
в минералах и рудах микрохимическим методом навеску разлагают
фтористоводородной кислотой для удаления кремневой кислоты
[19]. Остаток фторидов нагревают с щавелевой кислотой, которая при
высокой температуре вытесняет фтор. Образовавшиеся оксалаты
металлов прокаливают при 800° С. При этом большинство металлов
образует оксиды, а щелочноземельные элементы, магний и щелочные
металлы — карбонаты. При обработке прокаленного остатка горя-
чей водой в раствор переходят карбонаты щелочных металлов, гид-
роксид магния и небольшое количество карбонатов щелочноземель-
ных элементов. Если образец содержит большие количества алюми-
ния, железа и хрома, последние при прокаливании могут образо-
вать алюминаты, ферраты и хромиты. Для их разложения раствор
с осадком нагревают на водяной бане и после охлаждения обраба-
тывают насыщенным раствором карбоната аммония. Небольшое
количество катионов, главным образом магния, оставшихся в рас-
творе, осаждают 8-оксихинолином. Осадок отфильтровывают, рас-
твор упаривают досуха и остаток прокаливают. Полученные карбо-
наты щелочных металлов переводят в сульфаты, которые взвешивают.
Умножая на фактор пересчета, находят сумму оксидов лития, нат-
рия, калия, рубидия и цезия.
Навеску 5—10 мг минерала или горной породы помещают в платиновый
тигель вместимостью 4—5 мл, смачивают 2 каплями дистиллированной воды и
затем прибавляют 2 мл 40%-ной HF. Тигель помещают на водяную баню и на-
крывают крышкой. Через 10 мин крышку снимают, обмывают водой и затем упа-
ривают содержимое тигля досуха, снова добавляют 2 мл HF и упаривают. Оста-
ток смачивают 2 каплями НС1 (пл. 1,12) и нагревают 1—2 мин, после чего при-
бавляют 2 мл насыщенного на холоду раствора щавелевой кислоты (свободной
от примеси щелочных металлов), которая должна покрывать все частицы, при-
ставшие к стенкам тигля.
Жидкость упаривают досуха, стенки тигля обмывают небольшим количест-
вом воды и опять упаривают досуха, остаток нагревают на водяной бане около
20 мин. Затем тигель вытирают снаружи фильтровальной бумагой, неплотно
закрывают крышкой и переносят в алюминиевый блок, где нагревают вначале
при 120° С, а потом медленно повышают температуру до 200—220° С для удаления
щавелевой кислоты. При осторожном нагревании около 200° С щавелевая кисло-
та теряет кристаллизационную воду и возгоняется. Более высокая температура
вызывает плавление солей и разбрызгивание.
Операцию удаления фтора щавелевой кислотой повторяют еще 1—2 раза,
после чего остаток прокаливают на пламени горелки или в электрической печи
при 700—750° С в течение 10 мин.
После охлаждения тигля снимают пинцетом крышку и обмывают стенки
тигля, а также внутреннюю сторону крышки небольшим количеством горячей
воды из промывалки. В тигель прибавляют 2—3 капли 25%-ного раствора ам-
миака и 2—3 капли насыщенного раствора карбоната аммония. Тигель накры-
57
вают крышкой, помещают на водяную баню и нагревают до прекращения выде-
ления углекислого газа, после чего снимают крышку, обмывают ее и стенки
тигля горячей водой и жидкость упаривают почти досуха. К остатку прибавляют
1 мл горячей воды и нагревают 2—3 мин на водяной бане для извлечения щелоч-
ных металлов. После охлаждения в тигель прибавляют 0,2—0,3 мл 5%-ного
этанольного раствора 8-оксихинолина, 0,5 мл разбавленного аммиака (1 часть
25%-ного аммиака на 10 частей воды). Смесь нагревают на водяной бане 5 мин
при 70° С. Осадок оксихинолинатов, а также оксидов и карбонатов перемешивают
платиновой проволокой и через 10 мин фильтруют на фильтровальную трубочку
с бумажной массой.
Фильтрат собирают в платиновую чашку вместимостью 10 мл, осадок про-
мывают 3—4 раза насыщенным водным раствором 8-оксихинолина. Одновремен-
но промывают платиновую проволоку, крышку тигля и фильтровальную тру-
бочку. Общий объем фильтрата обычно составляет 5—7 мл. Он должен быть
окрашен в желтый пвет, что указывает на избыток 8-оксихинолина.
Полученный раствор упаривают досуха на водяной бане, остаток высуши-
вают несколько минут на электрической плитке и осторожно прокаливают на
пламени горелки или в электрической печи при темно-красном калении.
Остаток растворяют в 1 мл горячей воды, раствор переносят в предваритель-
но взвешенный платиновый тигель и упаривают. К раствору карбонатов щелоч-
ных металлов прибавляют 4 капли H2SO4 (1 : 1), тигель накрывают крышкой»
помещают на водяную баню и нагревают до прекращения выделения углекисло-
го газа. Затем внутреннюю сторону крышки и стенки тигля обмывают водой и
содержимое тигля упаривают возможно более полно. Тигель переносят на слабо
нагретую электрическую плитку и удаляют избыток серной кислоты. Когда ее
выделение прекратится, тигель охлаждают, обтирают снаружи кусочком филь-
тровальной бумаги, смоченной НС1 (пл. 1,12), для очистки от слоя солей, обра-
зовавшихся во время упаривания на водяной бане, а потом влажной фильтро-
вальнох! бумагой или полотняной тряпочкой и помещают его в электрическую
печь, где прокаливают 10—15 мин при —700° С; температуру печи заранее регу-
лируют с помощью платиновой термопары.
Так как при упаривании с серной кислотой и прокаливании остатка могут
образоваться пиросульфаты щелочных металлов, то рекомендуется в тигель
с прокаленным остатком прибавить 1 каплю конц. NH3, упарить на водяной бане,
а потом снова прокалить в электрической печи. После охлаждения тигель поме-
щают в эксикатор на алюминиевый блок для охлаждения и переносят к весам.
Через 10 мин его вместе с блоком вынимают из эксикатора, помещают в шкафчик
весов и через 5—10 мин взвешивают. Из найденной массы- сульфатов щелочных
металлов вычитают результаты контрольного опыта, который проводят одно-
временно с определением. В большинстве случаев масса его не превышает 0,030мг.
Образующиеся сульфаты должны полностью раствориться в воде.
Иногда взвешенные сульфаты могут содержать примесь кальция, не пол-
ностью отделенного в первой стадии анализа. Поэтому рекомендуется прокален-
ные сульфаты растворить в 0,5 мл горячей воды, прибавить 2—3 капли конц.
NH3 и 1—2 капли насыщенного раствора оксалата аммония. Тигель с раствором
нагревают на водяной бане 1—2 мин и оставляют стоять на столе под колпаком.
Если через 1—2 ч выпадет осадок оксалата кальция, то его отфильтровывают на
фильтровальную трубочку с бумажной массой и промывают 3—4 раза по 0,5 мл
водой, содержащей немного оксалата аммония.
Фильтрат собирают в другой прокаленный и взвешенный тигель, упаривают
на водяной бане досуха, остаток прокаливают до удаления солей аммония, обра-
батывают 1—2 каплями H2SO4 (1 : 1) и упаривают на электрической плитке до
полного удаления серной кислоты. Далее поступают, как описано выше.
Сумму оксидов щелочных металлов находят умножением массы сульфатов
на фактор 0,4487, если предполагают, что в минерале преобладает К2О, и на
0,4364, если преобладает NaaO.
При анализе минералов, содержащих фосфор, ванадий или хром,
конечная стадия определения должна быть изменена следующим
образом.
58
К раствору карбонатов щелочных металлов, находящемуся в платиновой
чашке, после разрушения 8-оксихинолина прибавляют 2—3 капли раствора ок-
сида ртути в аммиаке (растворяют 1 г свежеосажденного HgO в 5 мл раствора,
содержащего 1,2 г карбоната аммония и 2 мл 25%-ного раствора аммиака) и
нагревают на водяной бане 5 мин, покрыв чашку часовым стеклом. Затем стек-
ло снимают, обмывают его небольшим количеством воды, осадок оксида ртути,
фосфата, ванадата или хромата ртути отфильтровывают на фильтровальную
трубочку и промывают 3—4 раза холодной водой, приливая каждый раз не
более 0,5 мл.
Фильтрат собирают во взвешенный тигель и далее поступают,
как описано выше.
Определение органическими реагентами
Самую многочисленную группу составляют методы, основанные
на образовании малорастворимых тройных ацетатов натрия, урани-
ла и катионов двухвалентных элементов.
Белявская [39] разработала гравиметрический метод определения
1 мг натрия в присутствии 6,3—20,2 мг магния или 25 мг калия.
Ошибка определения не превышает 0,9%. Отмечается, что удовлетво-
рительные результаты получены при осаждении натрия не только
из раствора хлорида, но и из раствора сульфата.
При использовании в качестве весовой формы тройных ацетатов
натрия, уранила и магния или цинка при неконтролируемом избытке
осадителя могут получаться завышенные результаты [985]. Пред-
почтительнее натрий осаждать в виде магниевой соли, менее раство-
римой по сравнению с тройным ацетатом цинка.
Присутствие в растворе ^>20-кратных по отношению к натрию
количеств урана вызывает положительную ошибку, которую можно
устранить введением перед осаждением небольших количеств уксус-
ной кислоты. При этом допустимо присутствие уже 50-кратных ко-
личеств урана [274].
Определению 2 мг натрия в форме натрийцинкуранилацетата не
мешают (в мг): А1 (6); К, Fe (5); Ti, Cr, Ми, Mg, Са (2) и NH4C1
(0,5 г) при осаждении из среды 0,2 М НС1 или НСЮ4 [476]. Время
выстаивания осадка и концентрация серной кислоты влияют на
полноту осаждения натрия (табл. 36).
Таблица 36
Влияние условий на полноту осаждения натрия
в форме натрийцинкуранилацетата [476]
Продолжитель- ность осажде- ния, ч Концентра- ция H,SO4, М Осаждено натрия, % Продолжи- тельность осаждения, ч Концентра- ция H,SOt, М Осаждено натрия, %
0,17 0,0025 48 8 0,05 94
1 0,0025 90 8 0,15 94
2 0,0025 94 8 0,25 94
4 0,0025 96 1 8 0,40 92
8 0,005 94 8 0,50 80
Примечание. Взято 0,5 мг натрия.
59
Для полного осаждения натрия рекомендован 6-кратный избыток
уранил-ионов и 6—10-кратный избыток цинка.
Метод применен для гравиметрического определения натрия
в плавах хлоридов титанового производства [476]. При содержании
натрия 1,92—4,94% погрешность <Д,20%. Метод применим для
определения натрия в других продуктах титанового производства,
содержащих натрий в виде хлорида: возгонах, расплаве хлоратора
и остатках после хлорирования.
Навеску образца 1 г растворяют в воде или 0,01 М HG1 при нагревании,
раствор охлаждают, разбавляют в мерной колбе вместимостью 100 мл водой до
метки и фильтруют через сухой фильтр. Первые порции фильтрата отбрасывают,
из следующих порций отбирают аликвотные части по 10 мл в кварцевые стаканы.
Добавляют аммиак до pH ~ 3 (по универсальному индикатору) и 1 мл 1 М НС1
или 1 М HCICU. Раствор упаривают до объема 5 мл и охлаждают до комнатной
температуры. Добавляют 20 мл осадителя и перемешивают плексигласовыми
палочками 3 мин (осадитель готовят смешиванием двух растворов: 1) 10 г ацетата
уранила растворяют в 49 мл воды и 6 мл 30%-ной СН3СООН; 2) 30 г ацетата цин-
ка растворяют в 32 мл воды и 3 мл 30%-ной СН3СООН). Осадки через 30 мин
отфильтровывают в фильтрующие тигли № 4, стаканы и тигли промывают 3 раза
раствором осадителя, затем 5—6 раз этанолом, насыщенным цинкуранилацета-
том натрия, и 10 мл эфира. В течение 3—5 мин просасывают через тигель с осад-
ком воздух и через 15—20 мин взвешивают.
Цинкуранилацетат применен для определения 0,1% натрия в ти-
тане [552]. Погрешность определения не превышает 10%.
Навеску пробы 1 г обрабатывают в платиновой чашке по каплям 25 мл HF
(1: 3). После растворения пробы окисляют титан(Ш) небольшим количеством
HNO3(1 : 1),добавляя ее по каплям, охлаждают и вводят 25мл H2SO4(1 : 4). Рас-
твор упаривают до появления паров HgSCU, охлаждают и разбавляют водой до
объема 100 мл в мерной колбе. Переносят в прежнюю платиновую чашку 25 мл
раствора, упаривают его до появления паров H2SOi и охлаждают. Вводят 2 мл
НС1 (1 : 4) и нагревают до растворения растворимых солей. Вводят 25 мл цинкура-
нилацетата, перемешивают, чтобы вызвать кристаллизацию натрийцинкуранил-
ацетата, затем дают отстояться 2 ч. Если осадок не образовался, оставляют рас-
твор стоять еще на 4 ч.
Для приготовления раствора осадителя растворяют 100 г соли
UO2(CH3COO)2-2H2O и 300 г гп(СН3СОО)2-2Н2Овсмеси 25 мл ледяной уксусной
кислоты и 850 мл воды. Нагревают раствор до кипения, добавляют 10 мг NaCl
и выдерживают при комнатной температуре не менее 24 ч. Непосредственно
перед использованием раствор фильтруют. Готовят раствор для промывания
осадка. Для этого к 2 мл 4%-ного раствора NaCl прибавляют 20 мл раствора
цинкуранилацетата и оставляют на 10 мин. Отфильтровывают осадок на фильтр
№ 3, промывают ацетоном и высушивают при 105° С в течение 30 мин. Заливают
2 г этого осадка 500 мл этанола. Подогревают раствор до 60° С на водяной бане
и дают раствору постоять при комнатной температуре не менее 24 ч. Непосред-
ственно перед использованием раствор фильтруют.
Полученный раствор с осадком натрийцинкуранилацетата отфильтровывают
через стеклянный фильтр № 3, переносят осадок на другой стеклянный фильтр,
используя для этого минимальное количество раствора цинкуранилацетата.
Трижды промывают осадок промывным этанольным раствором и дважды ацето-
ном (суммарный объем ацетона не должен превышать 10 мл). Высушивают
фильтр с осадком 30 мин при 105° С, охлаждают и взвешивают. Растворяют оса-
док на фильтре в теплой воде, промывают ацетоном, высушивают 30 мин при
105° С и снова взвешивают. Разность результатов двух взвешиваний равна массе
натрийцинкуранилацетата. Фактор пересчета на натрий равен 0,015.
Цинкуранилацетат применен для определения натрия в урановых
рудах и концентратах [1068], в продуктах производства поташа —
60
КС1, K2SO4, К2СОз, бардяном угле после отделения калия в форме
перхлората [436, 839].
Из водно-этанольного раствора количественно осаждается нат-
риймарганецуранилацетат [40]. При осаждении 0,1—1 мг натрия
ошибка определения не превышала 0,008 мг. Фактор пересчета на
натрий 0,01437. Определению не мешают 2-кратные количества ка-
лия, 20-кратные количества магния (по массе). Осадок имеет постоян-
ный состав при высушивании при температуре 50—60° С.
К 1 мл анализируемого раствора, содержащего около 1 мг натрия, прибав-
ляют 6 мл раствора реагента, энергично перемешивают до выпадения обильного
осадка, палочку ополаскивают 1 мл раствора реагента и удаляют из раствора.
Через 1 ч осадок отфильтровывают в тигель с пористым дном, промывают 6—8
раз порциями по 1 мл 96%-ного этанола, насыщенного натриймарганецуранил-
ацетатом, затем диэтиловым эфиром и высушивают до постоянной массы при
50—60° С. Для приготовления реагента смесь 7 г уранилацетата, 35 г ацетата
марганца, 6 мл лед. СН3СООН и 100 мл воды нагревают до полного растворения,
после охлаждения прибавляют 1/3 объема этанола и немного NaCl для насыщения
раствора натриймагнийуранилацетатом. Через 2 сут раствор отфильтровывают
и используют как раствор реагента.
Натрий определяли также осаждением в форме тройного ацетата
натрия—кобальта—уранила [752]. Погрешность определения 0,5—
4 мг натрия не превышала 0,5%. Фактор пересчета на натрий 0,01448.
Исследовано влияние температуры на полноту осаждения натрия
в форме натрийникельуранилацетата (табл. 37).
Таблица 37
Влияние температуры на полноту осаждения натрия
в форме NaNi(UO2)3(CHsCOO)e-6H2O [239]
Введено МагО, мг Найдено Na2O (в %) осаждением
50° С 40° С 30° с 20° С 10° С
о,1 8 81 100 118 120
0,2 45 86 100 107 108
0,3 58 91 100 104 105
0,4 65 92 100 102 102
В качестве оптимальной рекомендована температура 30° С и выстаи-
вание осадка под маточным раствором при этой температуре.
Особенности определения натрия в виде тройных ацетатов подроб-
но обсуждены в монографии [748].
Предложено осаждать и определять натрий в присутствии маг-
ния тетра-(п-толил)боратом лития [432]. Вместе с натрием осажда-
ются калий, рубидий и цезий; предварительно их рекомендуется
отделять тетрафенил боратом лития. Недостаток реагента — малая
устойчивость при хранении.
Разработан метод с использованием п-хлорфенил-а-метокси-
уксусной кислоты (I) или ее калиевой соли (II) [816]. Осаждают
61
из смеси воды с метанолом (3 : 1) при pH 3—5. Для осаждения 23 мг
натрия необходимо 401,2 мг I или 426,6 мг II. Температура 0—5° С.
Осадок промывают очень холодной водой и высушивают в вакуум-
эксикаторе. Затем промывают на фильтре безводным ацетоном,
хорошо его отсасывают и высушивают при 75—90° С.
Определению 92,8—150,7 мг NaCl с погрешностью <Д% с исполь-
зованием в качестве осадителя 2-нафтил-а-метоксиуксусной кислоты
не мешают сравнимые количества хлоридов щелочных металлов и их
смесей [818]. Осадок высушивают при 105—110°С; фактор пересчета
на натрий 0,05066. Селективность повышается при использовании
в качестве реагента 2-нафтил-а-метоксиацетата тетраметиламмония.
В этом случае определяли 21,4—36,3 мг натрия в виде хлорида,
перхлората, нитрата или сульфата с погрешностью <^1% в присут-
ствии 259,8 мг NH4C1, 298,2 мг LiCl, 441,9 мг RbCl, 291,2 мг CsBr,
134,8 мг MgCl2, 500,8 мг КС1 [817].
К анализируемому раствору, содержащему хлорид, нитрат, сульфат или
перхлорат натрия с концентрацией натрия 0,5—5 мг/мл, прибавляют 1 М НС1
до pH 3—4 из расчета 0,2 мл 1 М НС1 на 1 мл раствора реагента. Прибавляют
реагент в 75%-ном избытке для полноты осаждения натрия. Осадок выдерживают
при комнатной температуре 90—100 мин (в присутствии посторонних ионов—
дольше) и отфильтровывают на фильтр G-4. Стакан ополаскивают водно-этаноль-
ной смесью; осадок промывают трижды 15 мл промывной жидкости и высушивают
при 105—110° С до постоянной массы. В качестве промывной жидкости исполь-
зуют этанол, насыщенный кислой натриевой солью 2-нафтил-а-метоксиуксусной
кислоты. Для приготовления раствора реагента 129,7 г (0,6 моля) кислоты сме-
шивают в колбе вместимостью 1 л с 0,6 моля гидроксида тетраметиламмония и
разбавляют водой до 1 л. Раствор устойчив в течение месяца. В случае выпаде-
ния кремнекислоты осадок отфильтровывают.
1-Нафтиламин-8-сульфоновую кислоту применяли для гравимет
рического определения натрия в морской воде и рассоле [1155].
Определению 40—80 мг натрия не мешают 10 мг калия, 60 мг аммо-
ния; в присутствии 300 мг зтилендиаминтетраацетата магния не
мешает 10 мг калия, 60 мг аммония; в присутствии 300 мл этилен-
диаминтетраацетата магния не мешает 10 мг кальция.
Медленно приливают 10 мл 12%-ного раствора реагента к нейтрализован-
ному анализируемому раствору, содержащему 40—80 мг- натрия, выдерживают
30 мин при 10—15° С, отфильтровывают и промывают сначала 3 раза порциями
по 2—Змл насыщенного этанольного раствора 1-нафтиламин-8-сульфоната натрия,
а затем 2 раза порциями по 1 мл абсолютного этанола. Осадок высушивают до
постоянной массы при 105° С.
Определению натрия с 1-нафтиламин-8-сульфоновой кислотой
не мешают (в кратных количествах): К (2—2,5), Са (3), SO4- (4)
Mg (10) [142]. Фактор пересчета на натрий 0,09377. Продолжитель-
ность определения 60—80 мин, погрешность 0,5%.
Аликвотную часть раствора NaCl упаривают на водяной бане почти досуха.
К сухому остатку прибавляют трехкратный избыток реагента (на каждые 10 мг
натрия необходимо 7—8 мл раствора реагента) и такой же объем 96 %-него
этанола. Раствор перемешивают и через 30—60 мин отфильтровывают осадок
в тигель Шотта № 3 или № 4, промывают 6—7 порциями 96%-него этанола,
насыщенного 1-нафтиламин-8-сульфонатом натрия, и одной порцией чистого
этанола. Тигель с осадком высушивают до постоянной массы при 105° С. Осади-
62
телем для натрия является 10%-ный раствор 1-нафтиламин-8-сульфоната магния,
который готовят смешением горячей водой суспензии 1-нафтиламин-8-сульфоно-
вой кислоты и MgCO3. После прекращения выделения СО2 раствор охлаждают
до комнатной температуры. Выделившуюся соль дважды перекристаллизовы-
вают и высушивают на воздухе; она имеет состав (CloH6NH2SO3)2Mg-81I2O.
5-Бензамидоантрахинон-2-сульфоновая кислота рекомендована
для гравиметрического определения 1—25 мг натрия в растворах
чистых солей [778]. Реагент не селективен к натрию. Фактор пере-
счета на натрий 0,05358.
Для определения 1—10 мг натрия аликвотную часть раствора хлорида на-
трия переносят в стакан вместимостью 50 мл. Если анализируют навеску кри-
сталлического NaCl, его растворяют в 5 мл воды; прибавляют 5 мл этанола и
15 мл 3%-ного раствора реагента в 50%-ном этаноле. Смесь оставляют на 30 мин
при комнатной температуре, время от времени перемешивая. Осадок отфиль-
тровывают в сухой пористый тигель № 4, промывают трижды порциями по 1 мл
этанола, 6 раз — по 1 мл этанола, насыщенного натриевой солью реагента,
и затем 4 раза — по 1 мл сухого диэтилового эфира. Осадок оставляют на возду-
хе на 5 мин, а затем высушивают в шкафу до постоянной массы при НО—120° С.
Соли оротовой кислоты с NjN-диметилэтаноламином или N,N-
диэтилэтаноламином осаждают малорастворимое соединение
C5H3N204Na, пригодное для гравиметрического определения натрия
[1145]. Определению не мешают ионы NH4, Li, щелочноземельных
элементов, Fe(III), Al, Ni, Со и Си; мешает К+. Факторы пересчета
на натрий 0,12913; Na20 — 0,17406; NaCl — 0,32823.
К анализируемому раствору с pH 6,5—8,0 прибавляют в избытке 0,1 М
раствор осадителя (20—25 мл) и выдерживают 2 ч при 3° С. Осадок отфильтро-
вывают в стеклянный тигель, промывают дважды порциями по 2 мл 70%-него-
этанола, высушивают при 105° С до постоянной массы и взвешивают.
КОСВЕННЫЕ МЕТОДЫ
Метод с осаждением NaCl применяли для определения натрия
в минеральной воде [1015]. Предварительно натрий и литий отделяли
на катионообменнике. При определении 10—900 мг/л натрия погреш-
ность ~0,5%. Не мешают (в скобках указаны концентрации, мг/л):
К «130); NH, «10); Mg «220); Са«900); Sr «9); Ва «2,3);
Мн «5); Fe(III) «60); Н3ВО3 «250); НРО4“ «27).
Пробу воды, содержащую 35—45 мг-экв катионов, подкисляют соляной кис-
лотой и кипятят для удаления С02. Коллоидный осадок Fe(0H)3 не мешает раз-
делению. Пробы воды с малым минеральным составом предварительно упари-
вают в платиновой чашке до 200—400 мл. Пробу пропускают через колонку со
скоростью 25—30мл/ч-см2, заполненную катионообменником Дауэкс 50W X 8.
Колонку промывают 250 мл 35%-ного этанола. Литий элюируют 370 мл 0,6 М
НС1 в 60%-ном этаноле со скоростью 9—12мл/ч-см2. Затем для элюирования
натрия колонку промывают раствором 0,4 М НС1 в 40%-ном этаноле. При содер-
жании натрия меньше 100 мг необходимо 500 мл, для 100—400 мг натрия —
720 мл, для >400 мг натрия — 870 мл этанола. Фильтрат после пропускания
пробы через колонку вместе с промывным раствором упаривают досуха в пла-
тиновой чашке, прокаливают при 500—600° С и взвешивают NaCl.
Метод определения натрия в форме Na2SO4 применяли для ана-
лиза стекол и полевого шпата после хроматографического отделения
натрия [976]. На катионообменнике Леватит S-100 в калиевой фор-
63
ме ионы натрия количественно замещают ионы калия, которые затем
осаждают в форме тетрафенилбората калия и взвешивают. Фактор
пересчета 0,06418. При определении 8—16 мг натрия погрешность
определения не превышает 1,5% [992].
Глава V
ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
ОПРЕДЕЛЕНИЯ НАТРИЯ
Титриметрические методы определения натрия по химизму вклю-
чают все типы химических реакций: протолитические, окислительно-
восстановительные, реакции комплексообразования (в гомогенной
и гетерогенной системах). Это объясняется тем, что большинство
методов определения натрия — косвенные, основанные на образо-
вании малорастворимых комплексных соединений. После отделения
от раствора осадок растворяют и определяют натрий по компоненту,
входящему в состав комплекса. Ярким подтверждением преиму-
ществ таких методов перед прямыми, не характерными для аналити-
ческой химии натрия, является титриметрическое определение, ос-
нованное на образовании тройных ацетатов. В осадке можно опреде-
лить катионы двухвалентных металлов комплексиметрически, ионы
уранила — комплексиметрически или окси диметрически, ацетат-
ионы — ацидиметрически в неводной среде. Разнообразны и методы
установления конечной точки титрования — визуальные, оптичес-
кие, электрохимические.
Несмотря на разнообразие титриметрических методов определе-
ния натрия, актуальна разработка методов, сочетающих высокую
воспроизводимость с экспрессностью. Эти характеристики связаны
между собой селективностью определения. Поэтому актуален и поиск
новых селективных реакций на натрий.
КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Прямые методы
Методы определения натрия в виде гидроксида, карбоната или
гидрокарбоната описаны в большинстве учебников и практических
руководств и в данной монографии не рассматриваются. Использо-
вание этих методов для определения натрия в различных природных
и промышленных объектах весьма разнообразно и дополнительно
рассмотрено в главе X. Ниже приведены отдельные примеры.
Определение оксида натрия в стекле [865]. Тонко измельченную пробу
нагревают на водяной бане в платиновой чашке с 15 мл 40%-ной HF с добавле-
нием 0,5 мл конц. H2SO4; обработку смесью кислот повторяют еще раз, остаток
выпаривают досуха, по охлаждении прибавляют 20 мл насыщенного раствора
Ва(ОН)2, нагревают до кипения и выдерживают 20 мин (для выделения гидрокси-
дов магния, алюминия и других элементов). Пропускают углекислый газ для
64
осаждения избытка Ва(ОН)2 и кипятят 10 мин для разложения гидрокарбонатов.
Осадок отфильтровывают на стеклянную воронку с пористым дном с отсасыва-
нием и хорошо промывают. Фильтрат титруют 0,5 М раствором НС1 по бромфено-
ловому синему. Одновременно проводят контрольный опыт с тем же количест-
вом реагентов и вносят поправку. 1 мл 0,5 М НС1 соответствует 0,0155 г Na2O.
Продолжительность определения 8 ч.
Определение натрия в оксиде алюминия [1058]. Метод пригоден
для определения 0,1—1% натрия в А12О3 и основан на прокаливании
А12О3, выщелачивании едкого натра водой и титровании кислотой
по фенолфталеину. Погрешность метода 0,1%.
Навеску пробы 1 г прокаливают 6 ч при 600° С, переносят остаток в кониче-
скую колбу, прибавляют предварительно прокипяченную и освобожденную от
углекислого газа воду и титруют 0,01—0,05 М раствором НС1 или 0,005—0,025 М
раствором H2SO4 по фенолфталеину.
Разработан электрометрический некомпенсационный метод опре-
деления NaOH, Na2CO3, NaHCO3 и их смесей [539]. Из испытанных
индикаторных электродов (вольфрамовый, хингидронный, сурьмя-
ный, молибденовый, танталовый) наилучшим оказался вольфрамо-
вый, дающий минимальную погрешность определения: 0,5—1%.
В качестве электрода сравнения применяли сульфатно-медный
электрод.
Для определения перхлората натрия разработан метод потен-
циометрического титрования в среде пиридина раствором гидроксида
трибутилэтиламмония с использованием в качестве индикаторного
стеклянного электрода, электрода сравнения — насыщенного кало-
мельного электрода [597]. Определению мешают вода (в присутствии
1 % воды потенциал смещается на 65 мВ) и литий, титрующийся ана-
логично. Метод пригоден для определения NaC104 в присутствии
НС104 или СвН5СООН.
Около 2мМ NaClО4 растворяют в 250 мл безводного пиридина. Титруют али-
квотную часть раствора, содержащую 0,5—1,0 мМ NaClO4, разбавленную пири-
дином до объема 100 мл.
Безводный ацетат натрия титруют потенциометрически в среде
лед. СН3СООН 0,1 М раствором НСЮ4 или в диоксане [669]. При
содержании ацетата натрия в препарате 99,58 и 99,52% (определено
гравиметрически осаждением натриймагнийуранилацетата) найдено
предлагаемым методом 99,59%; 99,55% и 99,50%. Можно конечную
точку титрования определять визуально, используя в качестве ин-
дикатора кристаллический фиолетовый (1 капля 0,5%-ного раствора
индикатора на 55 мл анализируемого раствора).
К прямым методам можно отнести методы, основанные на осажде-
нии натрия фенил-а-метоксиуксусной [1087], ж-хлорфенил-а-меток-
сиуксусной [1089], п-хлорфенил-а-метоксиуксусной [816] и 2-нафтил-
а-метоксиуксусной [817] кислотами, растворении осадка в горячей
воде и титровании кислоты раствором NaOH в присутствии фенол-
фталеина [1087, 1089]. или тимолового синего [816, 817]. Осадок
имеет постоянный состав, например СвН5СН(ОСН3)СООН-СвН5СН-
• (OCH)3COONa [1087], поэтому погрешность метода «11%) не превы-
3 В. M. Иванов и др.
65
шает погрешности титриметрических методов определения. Во всех
случаях 1 мл 0,1 М раствора NaOH эквивалентен 2,3 мг натрия.
Метод с использованием лг-хлорфенил-а-метоксиуксусной кислоты
пригоден для определения натрия в форме хлоридов, нитратов и
карбонатов [1089]. Определению 88—118 мг NaCl или 110 мг Na2CO3
не мешают (в мг): LiCl (65), КС1 (614), RbCl (27), CsCl (15), MgCl2
(94), ВаС12 (56). Аналогична селективность метода с использованием
фенил-а-метоксиуксусной кислоты [1087]. В этом случае небольшие
количества ионов аммония не мешают. При экспрессном выполнении
анализа (при выдерживании осадка в течение 1 ч под маточным рас-
твором) рекомендуется вводить поправку на растворимость осадка.
Навеску пробы, содержащую около 2 мг-экв натрия, обрабатывают в колбе
3 мл воды до растворения, вводят 1 каплю раствора метилового оранжевого и
нейтрализуют 6М раствором КОН или НС1. Вводят 10 мл водно-этанольного
раствора реагента, перемешивают, оставляют на 30—60 мин при комнатной
температуре, а затем на 10—12 ч при 0—2° С. Осадок отфильтровывают, промы-
вают дважды ацетоном (20 и -45 мл) и растворяют в 250 мл нагретой до кипения
воды. По охлаждении титруют 0,1 М раствором NaOH в присутствии 20 капель
фенолфталеина.
Натрий титровали потенциометрически 0,5 М раствором НС104
в среде уксусной кислоты на фоне смеси нитробензола и бензола
[874]. Метод пригоден для определения натрия в присутствии калия
и лития [873].
К раствору, содержащему 0,5—5 мг-экв натрия, калия и лития (и не содер-
жащему катионов остальных аналитических групп),, прибавляют 10 мл 1 М
H2S04, упаривают до выделения белого дыма (в присутствии органических ве-
ществ остаток прокаливают 10—15 мин при 600° С), прибавляют 70 мл воды,
20 мл 0,5 М раствора Ва(СН3СОО)2 и разбавляют водой до 100 мл. 50 мл деканти-
рованной жидкости выпаривают досуха, остаток промывают безводным этанолом
5—7 раз порциями по 5 мл, фильтруют Через стеклянный фильтр № 4. Осадок на
фильтре растворяют в 1 мл лед. СН3СООН, содержащей 10% уксусного ангид-
рида (в присутствии ионов аммония раствор кипятят), вводят 30 мл смеси (-4:1)
бензола с нитробензолом и титруют 0,5 М раствором НС1О4 в лед. СН3С00Н
со стеклянным электродом (электрод сравнения — хлоридсеребряный).
Описано определение натрия в катализаторе, содержащем марга-
нец, титрованием 0,1 М НСЮ4 в метилэтилкетоне [20].
Суммарное содержание натрия и калия определяли потенциомет-
рическим титрованием раствором НС1 в ацетоне в среде СН3СООН:
уксусный ангидрид : ацетон = 1 : (0,5 ~ 10) [927].
Косвенные методы
Принципы, лежащие в основе данной группы методов, весьма
разнообразны. Их можно условно разделить на две большие группы:
основанные на переведении натрия в соединения, обладающие ос-
новными свойствами (борат, гидроксид), и на их ацидиметрическом
определении. Вторая группа методов включает использование ионо-
обменников, в основном катионообменников.
Методы, основанные на переведении натрия в соединения с ос-
новными свойствами. При совместном присутствии калия и натрия удо-
бен метод, заключающийся в превращении солей калия и натрия
66
в бораты, титровании хлорной кислотой в присутствии солянокислого
п-.этоксихризоидина, последующем извлечении NaC104 этанолом
(для гравиметрического определения калия в форме КС104), превра-
щении перхлората натрия в борат натрия и титровании последнего
раствором НС104 с тем же индикатором [1138].
Навеску, содержащую 0,1—1 мг-экв натрия и калия, взвешивают, выпари-
вают на водяной бане 2—3 раза с 1—2 мл 50%-ной HNO3 досуха, растворяют
в 2 мл воды и переносят в платиновую чашку. Вводят 0,5 г дважды перекристал-
лизованной борной кислоты, выпаривают на водяной бане, остаток высушивают
при 130—140° С и нагревают до плавления. Плав растворяют при нагревании
в 30—40 мл воды и по охлаждении титруют 0,1 М раствором НС104 в присутст-
вии 1—2 капель 0,2%-него этанольного раствора n-этоксихризоидина. Оттитро-
ванный раствор выпаривают на водяной бане досуха, остаток высушивают при
130—140° С, экстрагируют 5 порциями по 4 мл абсолютного этанола, фильтруют
через фильтр G3 и промывают абсолютным этанолом. Фильтр с КС104 высуши-
вают при 130° С в быстром токе воздуха и взвешивают. Этанольный раствор
выпаривают в платиновой чашке на водяной бане, вводят столько борной кис-
лоты, чтобы масса осадка была около 0,5 г, высушивают при 130—140° С и про-
каливают 10 мин при 800° С. Остаток растворяют при нагревании в 30—40 мл
воды и по охлаждении титруют с тем же индикатором 0,1 М раствором НС104.
КСЮ4 растворяют в небольшом количестве горячей воды, вводят 0,5 г Н3ВО3,
прокаливают и титруют 0,1 М раствором НС104.
Для титриметрического определения натрия и калия в присутствии суль-
фатов или фосфатов навеску растворяют в небольшом количестве воды и под-
кисляют соляной кислотой по метиловому красному [1138]. Полученный раствор
по каплям добавляют в кипящий раствор (0,5 г ВаС12-2Н2О и 1 мл 1 М НС1
в 50 мл раствора) со скоростью 1—2 капли в с. По охлаждении тонкой струей
добавляют 10 мл 5%-ного раствора (NH4)2CO3, содержащего 1% NH3. Осадок
фильтруют на бумажный фильтр и промывают аммиаком, фильтрат выпаривают
досуха, высушивают остаток при 150° С, долго нагревают, далее переводят
в бораты и поступают, как указано выше.
Для титриметрического определения натрия и калия в присутствии фосфа-
тов навеску растворяют в 10 мл воды, вводят 20 мл 5%-ного раствора Ва(ОН)2,
фенолфталеин, пропускают углекислый газ до исчезновения розовой окраски
раствора. Раствор с осадком выпаривают досуха на водяной бане, к остатку при-
бавляют 4—5 мл воды и снова упаривают досуха. Повторяют обработку водой
и высушивание 4 раза порциями по 2—3 мл воды, раствор фильтруют через
бумажный фильтр, фильтрат подкисляют азотной кислотой и выпаривают досу-
ха. Затем дважды обрабатывают порциями по 1 мл 30%-ной HNO3, упаривая
досуха каждый раз. Далее поступают, как указано при определении натрия и
калия.
Разработан метод определения хлоридов калия и натрия после
их переведения в гидроксиды обработкой оксидом серебра. Кондук-
тометрическое титрование МОН проводят этанольным раствором
НС1О4 [1025, 1028]. При анализе смесей 15,8—79,2 мг калия и 9,2—
46 мг натрия (соотношение К : Na (5 : 1)—(1 : 5)) погрешность не
превышает 2% по калию и натрию. При увеличении соотношения
К : Na до 1 : 10 или 10 : 1 погрешность возрастает до 5%. Для ко-
личественного перевода в оксид соотношение Cl : Н2О : Ag2O должно
быть 1 : 320 : 9.
5 мл водного раствора хлоридов калия и натрия смешивают в колбе Эрлен-
мейера (с пробкой) с эквивалентным количеством влажного оксида серебра.
Смесь взбалтывают 10 мин, затем осадок Ag2O и AgCl отфильтровывают на
стеклянный фильтр G4, раствор переносят в мерную колбу вместимостью 250 мл.
Для отсасыван'ия жидкости используют эксикатор с двумя отверстиями в одной
пробке. В одно отверстие помещают конусообразную воронку со стеклянным
3*
67
фильтром, другое отверстие служит для откачки воздуха. Оксид серебра на
фильтре и его остатки в колбе промывают небольшими порциями воды (всего
5 мл воды), затем этанолом. Мерную колбу заполняют этанолом до метки и
перемешивают. Аликвотную часть раствора (50 мл) титруют 0,1 М раствором
НС1О4 кондуктометрически.
Методы, основанные на использовании ионообменников. Аци-
диметрическое титрование. Предложен метод, ос-
нованный на пропускании анализируемого раствора,через колонку
с сильноосновным анионообменником АВ-17 в ОН-форме и титро-
вании в элюате NaOH [431]. Метод можно использовать для опреде-
ления натрия в присутствии 100-кратных количеств церия(Ш). При
определении 0,92—22,80 мг натрия в присутствии соответственно
91—0,3 мг церия погрешность 5—0,2%.
В колонку диаметром 10—15 мм помещают 5 мл набухшего анионообменни-
ка АВ-17 в ОН-форме. Навеску анализируемого вещества, содержащую не менее
1 мг натрия и не более 100 мг церия(Ш), переводят в солянокислый раствор
объемом 10 мл с pH 4—5 и концентрацией хлорида 0,1 М. Этот раствор дважды
пропускают через колонку со скоростью 0,05 мл/с и третий раз со скоростью
0,5 мл/мин. Затем промывают 30 мл дистиллированной воды, вытекающий из
колонки элюат и промывной раствор собирают в колбу, фильтруя через бумаж-
ный фильтр. В аликвотной части раствора титруют NaOH раствором НС1 с ме-
тиловым красным в качестве индикатора.
Титриметрический ацидиметрический метод применен для опреде-
ления натрия в синтетическом криолите [1012]. Метод основан на
разделении натрия и алюминия на анионообменнике церолит FF
в формах С2О4 и ОН (две колонки) и титровании NaOH раствором
НС104. Определению не мешают большие количества кальция и
железа.
Навеску криолита 1 г растворяют в платиновой чашке в 10 мл конц. НС1
и выпаривают досуха. Сухой остаток дважды обрабатывают раствором НС104
(по 1 мл), каждый раз упаривая смесь досуха, растворяют в 100 мл горячей
0,7%-ной НС1. Раствор охлаждают и разбавляют водой до 250 мл. Колонки,
установленные одна над другой, промывают 50 мл 50%-ного этанола. В верхнюю
колонку с церолитом FF в С2О4-форме вводят 20 мл исследуемого раствора со
скоростью примерно 1 мл/мин. Щелочной элюат со второй колонки собирают
в колбу, содержащую 25 мл 0,1 М НС1, и ее избыток оттитровывают раствором
NaOH.
Описан способ раздельного определения натрия и калия в суль-
фатной золе органических соединений [117].
Навеску пробы растворяют и пропускают через колонку с анионообменни-
ком АВ-17 в Cl-форме для перевода в хлориды, выпаривают досуха, растворяют
в смеси хлороформа и уксусной кислоты, обрабатывают раствором ацетата
ртути(П) в ледяной уксусной кислоте и титруют 0,01 М раствором НС104 в диок-
сане. Первый скачок соответствует титрованию КОН, второй — NaOH. По-
грешность при определении соизмеримых количеств натрия и калия ~ЬЗ%.
Метод пригоден для определения натрия при соотношении натрий : калий от
10 : 1 до 1 : 10. Мешают щелочноземельные металлы.
Алкалиметрическое титрование. Разработан
титриметрический метод определения калия и натрия в их солях
[736]. Образец после растворения пропускают через колонку с катио-
нообменником Вофатит KS и в элюате определяют сумму калия и
68
натрия титрованием вытесненной НС1 0,01 М раствором NaOH
со смешанным индикатором (метиловый красный + метиловый го-
лубой). В другой аликвотной части осаждают тетрафенилборат ка-
лия, затем переводят в КВО2, растворяют в избытке НС1 и избыток
НС1 оттитровывают. Количество натрия находят по разности.
Алкалиметрический косвенный метод применен для определения
натрия в природных водах; после отделения кальция и магния и
удаления солей аммония раствор пропускали через катионообменник
вофатит и титровали кислоту щелочью [478].
Косвенный алкалиметрический метод определения натрия осно-
ван на осаждении натрийцинкуранилацетата, растворении его и
пропускании раствора через катионообменник в Н-форме [670],
Образовавшуюся уксусную кислоту титруют щелочью. Достоинство
метода — в отсутствии мешающего влияния цинка и урана и в бла-
гоприятном соотношении ацетата к натрию — 9:1. Метод позволяет
определять 0,115—2,3 мг натрия и рекомендован для клинических
исследований.
К анализируемому раствору, содержащему 0,1—1 мг натрия,'в центрифужной
пробирке прибавляют цинкуранилацетат по общепринятой методике осаждения
натрия, осадок центрифугируют и 4—5 раз промывают ацетоном или этанолом,
насыщенным натрийцинкуранилацетатом, а затем 2—3 раза эфиром. Эфир хоро-
шо декантируют, испаряют на воздухе, растворяют осадок в воде и пропускают
через колонку диаметром 12 мм, заполненную вофатитом F в Н-форме (10 см3),
со скоростью 1 капля в 1с. Колонку промывают 20—75 мл воды, пропуская
ее со скоростью 2 капли в 1 с. К элюату добавляют фенолфталеин и титруют
0,02—0,1 М раствором бескарбонатвдй щелочи. 1 мл 0,02 М NaOH эквивален-
тен 0,05111 мг натрия.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ ТИТРОВАНИЕ
Известны только косвенные окислительно-восстановительные
методы определения натрия.
Арсенатно-иодометрическое определение натрия [288]. Метод осно-
ван на образовании натрийцинкуранилацетата, осаждении арсена-
тов цинка и уранила и их йодометрическом определении по арсена-
ту. Это повышает чувствительность определения, так как эквива-
лент по натрию равен 1/8 молярной массы натрия. 1 мл 0,1 М раст-
вора Na2S2O3 эквивалентен 0,29 мг натрия. При определении 0,25—
0,6 мг натрия погрешность определения 3—5%; при определениц
> 1,5 мг натрия 1%. Метод применим для определения натрия в не-
сильноминерализованных водах [289] и в силикатных горных поро-
дах [290]. В последнем случае при определении 0,3—5,0 мг натрия
абсолютная ошибка «^0,03 мг.
Раствор, содержащий 0,3—5 мг натрия, выпаривают до объема 1—2 мл,
прибавляют 1 мл этанола и осаждают натрий добавлением 10 мл раствора
цинкуранилацетата. Раствор с осадком перемешивают и оставляют на 30 мин,
изредка перемешивая. Осадок отделяют на стеклянном фильтре № 3 или № 4,
промывают 5 раз порциями по 2 мл этанола и растворяют в 20—25 мл воды.
К раствору прибавляют 15 мл 10%-него раствора арсената натрия. Осадок раст-
воряют в нескольких каплях конц. НС1 и нагревают раствор до кипения. К го-
рячему раствору при перемешивании прибавляют раствор NH3 (1 : 1) до слабого
запаха, затем продолжают нагревание на водяной бане до исчезновения запаха
69
аммиака (около 20 мин). Осадок отделяют на пористом фильтре № 3 или №-4
и промывают горячим 1%-ным раствором КС1 8—10 раз порциями по 5—7 мл.
Колбу Бунзена заменяют чистой и осадок растворяют в несколько приемов
в 25—30 мл HaSO4 (2:5). Первой порцией серной кислоты (5—6 мл) смачивают
стакан, в котором осаждали, и выливают в тигель. К фильтрату после растворе-
ния осадка прибавляют 20—25 мл бензола, ЗмлЗМ раствора KJ, взбалты-
вают несколько секунд, разбавляют равным водному слою объемом воды и
титруют 0,1 М раствором тиосульфата натрия до полного обесцвечивания слоя
бензола.
Определение натрия титрованием сурьмы в дигидроантимона-
те натрия. Рекомендовано осаждать дигидроантимонат натрия
Na2H2Sb2O7 и после растворения осадка в НС1 определять натрий
Косвенно йодометрическим титрованием сурьмы [94]. От преобла-
дающих количеств калия натрий отделяют обработкой смеси хлори-
дов этанолом. Метод применен для определения натрия в NaCl.
При анализе смеси 0,0218 г NaCl и 0,0219 г КС1 определено 100,44%
NaCl от введенного количества.
Смесь сухих хлоридов натрия и калия, содержащую не менее 0,1г NaCl
В мерной колбе вместимостью 100 мл, растворяют при нагревании в минималь-
ном количестве воды, разбавляют до 50 мл 95%-ным этанолом и 30 мин взбалты-
вают. Затем разбавляют до метки этанолом, взбалтывают и отстаивают. Фильтр
руют раствор в сухой сосуд, 20 мл раствора упаривают на водяной бане, сухой
остаток растворяют в небольшом количестве воды, вводят 40 мл раствора оса-
дителя, через 1ч — 20 мл этанола и далее определяют сурьму иодометрически.
При осаждении натрия из водно-этанольной среды повышается
селективность к натрию в присутствии калия [538]. В присутствии
24—25% об. этанола определению натрия не мешают 50-кратные,
а в присутствии 30% об. этанола — 100-кратные количества калия.
При определении 0,1—1,0 мг натрия погрешность не превышает 6%.
Селективность реагента повышают осаждением сопутствующих эле-
ментов смесью аммиака, карбоната аммония и 8-оксихинолина при
60° С в центрифужной пробирке с последующим осаждением натрия
в центрифугате. Осадок дигидроантимоната натрия промывают ох-
лажденным 35%-ным этанолом и растворяют в конц. НС1. Метод
применен для определения натрия в растениях, воде, продуктах
Животного происхождения, в отходах пищевой промышленности.
В другом варианте предложено растворять осадок Na2H2Sb2O7
в НС1, восстанавливать сурьму до трехвалентного состояния и оп-
ределять ее броматометрически [94]. 1 мл 0,05 N раствора КВгО8
Эквивалентен 0,0005755 г натрия или 0,001463 г NaCl. С удовлетво-
рительной точностью проанализированы смеси, содержащие 0,1 г
NaCl и 0,4—5,1 г КС1. Отмечается^ что в присутствии более чем 2-
кратных количеств калия необходимо разделение хлоридов натрия
и калия обработкой этанолом. Недостаток метода — в необходимости
предварительного восстановления сурьмы.
Пробу подготавливают, как описано выше при йодометрическом определении
сурьмы [94]. Осадок Na2IJ2Sb2O7 смывают в колбу Эрленмейера соляной кисло-
той и разбавляют водой до 250 мл в мерной колбе. В колбу вносят около 1г
Na2SO3, кипятят 20 мин для восстановления сурьмы и удаления SO2, охлаждают
до 60—70° С, вводят 2—3 капли раствора метилового оранжевого и титруют
0,05 N раствором КВгО3 до обесцвечивания окраски индикатора.
70
Определение натрия титрованием урана в натрийцинкуранилаце-
тате. Метод [240] основан на восстановлении урана до урана(1У)
и титровании раствором сульфата церия(1У). Он пригоден для опре-
деления натрия в природной воде. При определении 23 мкг/мл —
23 мг/мл натрия погрешность не превышает 1 %. Определению не ме-
шают Са, Mg, NH4 и не более 10-кратных количеств калия.
Пробу воды выпаривают досуха на водяной или воздушной бане, затем осаж-
дают натрий 5 мл раствора цинкуранилацетата и оставляют на 1 ч. Осадок от-
фильтровывают в тигель с пористым дном № 2, осадок и чашку для выпаривания
воды промывают 5—6 раз порциями по 2 мл лед. СН3СООН, насыщенной натрий-
цинкуранилацетатом. Затем воронку присоединяют к редуктору с электролити-
чески восстановленным кадмием и осадок в чашке и тигле растворяют в 10 мл
1 М H2SO4, прибавляемой по частям при медленном отсасывании. После этого
чашку, воронку и редуктор промывают 10 мл воды. К восстановленному раство-
ру прибавляют 2 мл 5%-ного раствора Fe2(SO4)3, через раствор продувают воздух
15 мин и при продолжающемся продувании титруют уран раствором Ce(S04)2
в присутствии фенилантрайиловой кислоты до слабо-розового окрашивания.,
Уран(1У) можно оттитровывать стандартным раствором ванадата
аммония в присутствии фенилантраниловой кислоты [19]. Уран реко-
мендовано восстанавливать металлическим кадмием или хромом(П).
Для получения точных результатов необходимо, чтобы соотношение
объемов анализируемого раствора и раствора осадителя равнялось
1 : 10. Метод применен для определения натрия в минералах и ру-
дах в микрохимическом варианте. Рекомендуется осадок тройного
ацетата промывать 95%-ным этанолом. Для уменьшения раствори-
мости осадка спирт для промывания предварительно насыщают,
комплексной солью. Определению) натрия мешают ионы AsO4~ и
РО4 . Если содержание калия в 50 раз превышает содержание натрия,
то калий рекомендуется отделить в форме КС1О4. 10-кратные коли-
чества ионов аммония не мешают определению натрия. Метод дает
удовлетворительные результаты при содержании натрия в анализи-
руемой навеске не более 0,5 мг. При наличии больших количеств
натрия получаются завышенные результаты вследствие трудности
промывания большого количества осадка. В таких случаях берут
меньшую навеску минерала или определяют натрий из аликвотной
части раствора сульфатов.
Взвешенные сульфаты натрия и калия, полученным способом, описанным
ранее (см. главу IV «Гравиметрические методы определения натрия»), раство-
ряют в минимальном количестве воды и переносят в стакан вместимостью 8 —.
10 мл. Тигель несколько раз обмывают горячей водой. Раствор упаривают до
объема 0,5 мл на водяной бане. После охлаждения прибавляют при помешива-
нии 5 мл раствора цинкуранилацетата и продолжают помешивание палочкой
в течение нескольких минут. Через 1,5—2 ч осадок отфильтровывают на фильтро-
вальную трубочку с бумажной массой. Осадок, стакан и трубочку вначале про-
мывают 2 мл реагента-осадителя; после удаления всей промывной жидкости пу-
тем отсасывания осадок промывают 4 раза порциями по 1 мл 95%-ного этанола,
насыщенного тройной солью. Затем стакан, в котором проводили осаждение,
вместе с осадком помещают под колокол фильтровального прибора, а фильтро-
вальную бумагу с приставшим осадком опускают в стакан, в который наливают
1 мл 1%-ной горячей серной кислоты. Через несколько минут осадок на поверх-
ности фильтровальной трубочки и в порах бумажного фильтра растворяется;
71
тогда раствор серной кислоты медленно пропускают через трубочку, собирая его
в стакан, в котором] проводили осаждение.
В стакан опять наливают около 1 мл 1%-ной горячей серной кислоты, и ее
снова пропускают через трубочку. Наконец, стакан и фильтр промывают 4—
5 раз горячей водой, приливая ее каждый раз по 0,5 мл. К раствору в стакане
прибавляют 1,5 мл H2SO4 (1 : 1) и упаривают до выделения паров серной кис-
лоты. После охлаждения стенки стакана промывают водой, раствор разбавляют
приблизительно до 5 мл и переносят в воронку редуктора, заполненного амальга-
мированным кадмием. В коническую колбу вместимостью 25—50 мл, служащую
приемником, наливают 10 мл (1 : 1) и колбу присоединяют к редуктору.
Затем анализируемый раствор пропускают через редуктор со скоростью 1 капля
в 2 с и собирают жидкость в колбу-приемник. Стакан промывают несколько раз
из промывалки 2 мл 2,5 М H2SO4 и этот раствор также пропускают через редук-
тор. Затем промывают редуктор 4 раза 2,5 М раствором H2SO4 порциями по 2 мл,,
давая каждый раз стекать кислоте до верхнего уровня металла. После этого
колбу отсоединяют от редуктора и конец трубки редуктора промывают 2,5 М
раствором H2SO4.
В колбу прибавляют 2 капли 0,02%-ного раствора фенилантраниловой
кислоты и уран(1У) оттитровывают 0,01 М раствором ванадата аммония до
Появления бледно-фиолетовой окраски, которая усиливается спустя некоторое
время.
При восстановлении ypana(VI) сульфатом хрома(П) сернокислый раствор
комплексной соли переливают из стакана в коническую колбу вместимостью
25 мл, прибавляют 3—4 мл H2SO4 (1 : 1) и после перемешивания прибавляют
из бюретки редуктора по каплям необходимое количество восстановителя и сверх
того небольшой избыток. По расчету на 1 мг Na2S04 необходимо около ' 30 мг
CrS04. Так как раствор хрома(П) может содержать небольшое количество хро-
ма(Ш), то практически к исследуемому раствору прибавляют на каждый мил-
лиграмм Na2SO4 1 мл восстановленного раствора сульфата хрома, содержащего
50 мг CrS04. После 5—6-минутного взбалтывания раствора или пропускания,
воздуха через отрезок стеклянной трубки избыток хрома(П) окисляется кислоро-
дом воздуха до хрома(Ш); уран(1 V) оттитровывают раствором ванадата аммония.
Йодометрическое титрование 1-нафтиламин-8-сульфоната натрия
[1421. Определение натрия после его осаждения в форме 1-нафтил-
амин-8-сульфоната можно закончить титриметрически. Продолжи-
тельность определения не более 2 ч, погрешность 0,8 %. Определению
не мешают (в -кратных количествах): К (2,5), SO4~ (7), Са (3),
Mg (10).
Осадок, полученный описанным выше способом (см. главу IV «Гравиметриче-
ские методы»), растворяют в тигле Шотта в горячей воде и разбавляют раствор
водой до 250 мл. Аликвотную часть раствора 20 мл переносят в склянку с при-
тертой пробкой и обрабатывают избытком (^100%) 0,1 N КВгО3 в присутствии
1г КВг и 5 мл конц. НС1. При этом образуется трибромпроизводное состава
C10H3Br3NH2SO3H. Через 15—25 мин вводят 1 г KJ и выделившийся иод оттитро-
вывают 0,1 М раствором Na2S2O3.
Прочие методы. Весьма селективен метод, включающий осаждение
оксалата натрия в водно-этанольной среде, его отделение и титрова-
ние стандартным раствором КМпО4. В присутствии ЭДТА и три-
этаноламина предложенным методом отделяют и определяют натрий
в присутствии ионов Li, Mg, Са, Sr, Ba, Fe, Co, Ni, Mn, Cu, Cd,
Zn, Hg, Al, Pb, К и NHt.
.72
КОМПЛЕКСИМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Прямые методы
Известен прямой метод, основанный на титровании соединений
натрия стандартным раствором K3A1F6 [1113]. Теплоту, выделяю-
щуюся в результате реакции и образования малорастворимого осад-
ка K2NaAlFe, измеряют прибором (термометрическое титрование).
Определению натрия не мешают большие количества аммония и
калия. По химизму реакцию можно отнести к комплексиметрической.
Натрий, а также калий и литий титруют, осаждая в виде окса-
латов в среде изопропанола раствором щавелевой кислоты в изопро-
паноле, используя биамперометрическую индикацию конечной точки
титрования (медный амальгамированный вращающийся катод и
медный большой перфорированный анод, ДЕ = 1 В) [64]. Перегиб
на кривой титрования обусловлен изменением электропроводности.
Предел обнаружения 1 • 10-4 М натрия, погрешность не превышает
0,7%. Метод позволяет определять натрий в смеси с калием в соот-
ношениях от 10 : 1 до 1 : 10 и натрий в присутствии калия и лития
в соотношениях от 10 : 10 : 1 до 1 : 1 : 10. При титровании смесей
сначала титруется натрий (растворимость оксалата натрия 0,475 М),
затем калий (1,85 М) и литий (2,31 М).
Готовят 0,1—0,01 М раствор натрия в изопропаноле. В стакан для титрова-
ния вместимостью 30 мл вводят 1—5 мл этого раствора, 10—15 мл изопропанола,
опускают электрод, устанавливают потенциал 1 В и титруют, прибавляя по 0,1—
0,2 мл 0,05 М раствора щавелевой кислоты в изопропаноле. На кривой титрова-
ния наблюдается два перегиба, первый из которых соответствует половине отти-
трованного количества натрия, второй — точке эквивалентности.
Фототурбидиметрическое титрование солей натрия щавелевой
кислотой в среде этанола применяли для определения натрия в его
солях с салициловой, n-аминосалициловой, каприловой кислотами
и с сульфотиазолом [236]. При определении 8,3—14,4% натрия стан-
дартное отклонение 0,07—0,13%.
Образование достаточно устойчивого комплекса натрия с цикло-
гександиаминтетрауксусной кислотой использовано для прямого
титриметрического определения натрия в пиперидиновом буферном
растворе (pH 12,7) с применением стеклянного ионоселективного
электрода модели Coming 476210 [667]. Определению натрия не ме-
шают остальные щелочные, а также щелочноземельные, переходные
и редкоземельные элементы.
Косвенные методы
Разработан метод, включающий осаждение натрия в виде трой-
ного ацетата с цинком и уранилом и титрование цинка дитизоном
[297]. Определению 0,1—0,5 мг натрия не мешают 50-кратные коли-,
чества кадия. Метод имеет существенный недостаток, связанный
с малой устойчивостью стандартных растворов дитизона и трудно-
стью наблюдения зеленой окраски дитизона на фоне интенсивно-
малиновой окраски дитизоната цинка.
73
Предложен кондуктометрический метод определения натрия и
калия титрованием их гидроксидов этанольным раствором H2PtCle
в этанольной среде [10261. Предварительно получают хлориды калия
и натрия, затем их переводят в гидроксиды обработкой оксидом се-
ребра. При определении калия и натрия при совместном присутствии
на кривой титрования получают два излома.
Косвенный амперометрический метод определения натрия основан
iia осаждении в этанольной среде оксалата натрия избытком щаве-
левой кислоты и титрования этого избытка этанольным раствором
нитрата кадмия. В качестве индикаторного использован ртутный
капающий электрод. Метод применен для определения натрия в со-
лях салициловой, n-аминосалициловой, муравьиной, уксусной, бен-
зойной, капроновой кислот, этилате, феноляте натрия. При их
навеске 8,60—12,00 мг погрешность определения не превышает 0,8%.
При определении натрия в морской воде его осаждают в форме
натрийцинкуранилацетата, осадок после отделения растворяют
в воде и титруют цинк амперометрически раствором K4[Fe(CN)e]
1245].
КОМПЛЕКСОНОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Описаны косвенные методы, включающие осаждение натрия
в виде тройных ацетатов с уранилом и магнием, цинком, кобальтом
или никелем и комплексонометрическое титрование магния [21,
834], цинка [338, 644, 813, 1147, 1164], кобальта [752, 1166], никеля
[752] или урана [21, 703]. В качестве металлоиндикаторов исполь-
зуют ксиленоловый оранжевый [21, 703, 1164], дитизон [813], муре-
ксид [1166] и эриохромовый черный Т (в остальных случаях).
Определение натрия титрованием магния [834]. Метод применен
Для определения 4—11 % натрия в рассолах, содержащих природные
газы.
К 1 мл раствора пробы прибавляют 50 мл раствора осадителя (смешивают
раствор, содержащий 25 г UOa(CH3COO)2-2H2O и 17 мл лед. СН3С00Н в 280 мл
Горячей воды, с раствором, содержащим 170 г Mg(CH3C00)2 и 17 мл лед. СН3СООН
в 280 мл горячей воды, выдерживают около 12 ч и фильтруют). Осадок периоди-
чески перемешивают в течение 1ч, отфильтровывают в тигель IG4, промывают
5 раз порциями по 2 мл промывной жидкости (к раствору NaCl прибавляют
раствор осадителя для полного осаждения натрия и взбалтывают полученный
Осадок с 95%-ным этанолом до насыщения; перед употреблением фильтруют) и
растворяют в 5 мл буферного раствора (67 г NH4C1 и 570 мл конц. NH3 в 1 л
воды) при перемешивании палочкой в течение 15 мин. К полученному раствору
прибавляют 3 капли раствора эриохромового черного Т (0,5 г индикатора и
4,5 г гидроксиламина в 100 мл этанола) и титруют 0,01 М раствором ЭДТА.
Определение натрия титрованием цинка. Видимо, наиболее чув-
ствителен метод, позволяющий определять 80—380 мкг натрия за
счет использования высокочувствительного металлоиндикатора на
Цинк — дитизона и Ю-8 М раствора ЭДТА [813]. Метод [1164] при-
менен для определения натрия в стеклах и других силикатных ма-
териалах. Определению не мешают Са, Ba, Sr, Мп (^240 мг), К,
Ti, Fe(III), Al, SOt, РОГ, AsOf-
74
Навеску пробы 0,5 г растворяют в смеси 10 мл HF и 5 мл НС1О4 и нагревают
до начала выделения белого дыма НС1О4. Затем опять добавляют 10 мл HF и
повторяют упаривание. Остаток растворяют в воде и разбавляют водой до 100 мл.
Аликвотную часть раствора, содержащую не более 40 мг натрия, упаривают до
сиропообразного состояния, разбавляют водой до 2—4 мл и при перемешиваний
осаждают натрий добавлением 15-кратного избытка в виде раствора, содержа-
щего 100 г ацетата уранила, 300 г ацетата цинка и 35 мл лед. СН3СООН в 1 л
раствора. Через 30—40 мин осадок отфильтровывают в стеклянный фильтр № 4,
промывают 2—3 раза раствором осадителя порциями по 5 мл, затем 8—10 раз
промывным раствором (400 мл абсолютного этанола, содержащего 4,5 мл 30%-ной
СН3СООН, насыщают осадком натрийцинкуранилацетата при 25° С) и 2—3 раза
ацетоном. Осадок растворяют в 50 мл воды, прибавляют 20 мл ацетатного буфер-
ного раствора с pH 5,3, 10 мл 10%-ного раствора NH4F для маскирования
ypana(VI), 6 капель раствора ксиленолового оранжевого и титруют 0,025 М
раствором ЭДТА.
Предложен косвенный титриметрический метод определения 0,7—
5,5 мг натрия осаждением его в форме натрийцинкуранилацетата и
комплексонометрическим титрованием цинка с радиохимическим
контролем конца титрования [644]. Уран маскируют карбонатом
аммония. В качестве радиоактивного изотопа применяют noAg
в форме соединения AgJO3. При титровании в среде аммиачно-карбо-
натного буферного раствора (pH 10) в избытке ЭДТА осадок AgJO3
растворяется. Избыток ЭДТА оттитровывают стандартным раство-
ром цинка при pH 5. Погрешность метода <5,7%.
Аналогичный метод с титрованием цинка раствором ЭДТА в при-
сутствии эриохромового черного Т применен для определения нат-
рия в природных водах [338]. Влияние урана(У1) устраняют введе-
нием (NH4)2CO3.
Метод [1147] основан на осаждении натрийцинкуранилацетата
и комплексонометрическом титровании цинка с эриохромовым
черным Т. 1 мл 0,01 М раствора ЭДТА эквивалентен 0,5845 мг
NaCl.
К анализируемому раствору в стакане вместимостью 150 мл прибавляют
20 мл раствора осадителя: 20 г уранилацетата растворяют в 12 г 30%-ной
СН3СООН и разбавляют водой до 130 г (раствор «а»); 60 г ацетата цинка раство-
ряют при слабом нагревании в 6 г 30% -ной СН3СООН и разбавляют водой до
130 г (раствор «б»). Оба раствора смешивают, оставляют на 24 ч и фильтруют
в склянку из стекла пирекс со стеклянной пробкой. Раствор перемешивают и
оставляют на 1 ч. Осадок отфильтровывают, декантируя жидкость на фильтр
Ватман № 40. Осадок промывают 8 раз порциями по 2 мл декантацией (для при-
готовления промывной жидкости 95 мл этанола насыщают патрийципкуранил-
ацетатом, для последнего промывания применяют абсолютный этанол). Затем оса-
док промывают декантацией 5 раз порциями по 2 мл абсолютного этанола.
После этого фильтр вынимают и все кристаллы смывают горячей водой в стакан,
в котором проводили осаждение. Общий объем раствора должен составлять 50—
75 мл. К раствору прибавляют 1 г карбоната аммония и 3 мл аммонийхлоридно-
го буферного раствора. После растворения карбоната аммония прибавляют ин-
дикатор и титруют 0,01 М раствором ЭДТА до перехода красно-фиолетовой
окраски в голубую.
Определение натрия титрованием кобальта. Кобальт титровали
при pH 9 в присутствии эриохромового черного Т [752]. При опреде-
лении 0,5—4 мг натрия погрешность не превышала 0,8%.
Косвенный метод предложен для определения Ю мг натрия
в присутствии больших количеств кобальта [1166].
75’
Анализируемый раствор, содержащий не более 10 мг натрия, упаривают
примерно до 1 мл, прибавляют 10 мл осадителя (100 г ацетата уранила растворя-
ют в 200 мл воды; 290 г ацетата кобальта растворяют в 200 мл воды; оба раство-
ра смешивают, добавляют 30 мл лед. СН3СООН, 5 г CH3COONa-3H2O и через
24 ч фильтруют от выпавшего осадка тройной соли. Полученный раствор насыщен
по отношению к тройной соли). Через 2 ч осадок отфильтровывают в тигель G4
с вложенным в него бумажным фильтром. Осадок промывают порциями по 2 мл
3 раза раствором осадителя, 5 раз этанолом и растворяют в 20 мл воды. К раство-
ру прибавляют воду до объема около 100 мл, вводят 0,5 г CH3COONa-3H2O и
(NH4)2COs до исчезновения образующегося осадка (раствор может опалесци-
ровать), 5 капель насыщенного раствора мурексида и титруют кобальт 0,05 М
раствором ЭДТА до перехода желтой окраски в оранжево-красную.
Определение натрия титрованием никеля. Условия титрования
никеля: pH 9, индикатор — эриохромовый черный Т; при определе-
нии 0,5—4 мг натрия погрешность не превышает 1,0% [752].
Определение натрия титрованием урана. В минеральных водах,
натриевых солях некоторых органических кислот, используемых
в фармации (цитрат, салицилат, лактат), физиологических растворах
натрий определяли осаждением в форме ацетата натрия—кобальта—
уранила, восстановлением урана(У1) аскорбиновой кислотой до ура-
на(1У) и комплексонометрическим титрованием последнего обратным
титрованием избытка ЭДТА раствором Th(NO3)4 в присутствии
ксиленолового оранжевого [703]. Отмечается, что осаждение натрия
в видя тройной соли кобальта предпочтительнее осаждения в виде
тройных солей меди, цинка, кадмия, магния или никеля. При опре-
делении 115—2300 мкг натрия погрешность составляет 0,69—1,13%
соответственно. Определению 230 мкг натрия не мешают (в мг):
Zn (65), Mg (24), Cd (112), Мп (54), а определению 690 мкг натрия
не мешают (в мг): NH4 (15),! Fe(II) (55), Са (40), Sr (87), Ва (137),
Hg (200).
К 0,05—1 мл пробы, содержащей 10—250 мкг натрия, прибавляют 5—25 мл
раствора осадителя, через 1—2 ч осадок отфильтровывают в фильтрующий ти-
гель А1 и промывают уксусной кислотой, насыщенной взбалтыванием в течение
8 ч с осадком натрийкобальтуранилацетата. При применении водного раствора
осадителя для промывания используют 10—30 мл промывной жидкости, при
использовании этанольного раствора — 2—3 мл. Осадок растворяют в 25—
100 мл воды, вводят в избытке измеренный объем 0,01 М стандартного раствора
ЭДТА, 0,2—0,5 г аскорбиновой кислоты и кипятят не менее 15 мин. После охлаж-
дения до комнатной температуры вводят 2—3 капли 0,1 %-него раствора ксиле-
нолового оранжевого и оттитровывают избыток ЭДТА 0,01 М раствором Th(NO3)4
до изменения окраски раствора из желтой в красно-фиолетовую. Наиболее отчет-
ливо изменение окраски раствора в конечной точке титрования при содержании
5 мг урана в 50—100 мл раствора; большие количества определять труднее из-за
наложения зеленой окраски ионов ypana(IV).
Раствор осадителя готовят смешением 2,5 г ацетата уранила с 10 г ацетата
кобальта и 10 мл ледяной уксусной кислоты, разбавлением 500 мл воды и 250 мл
Этанола. Раствор нагревают на водяной бане до растворения солей, через 24 ч
отфильтровывают и хранят в склянке из темного стекла.
Косвенный комплексонометрический метод, основанный на осаж-
дении натриймагнийуранилацетата, восстановлении урана(У1), вве-
дении избытка ЭДТА и титровании его раствором Bi(NO3)3 в при-
сутствии ксиленолового оранжевого применен для определения
0,02 г натрия в железных сплавах [21]. Предварительно отделяют
76
основу экстракцией бутилацетатом из среды 8 М НС1. Погрешность
метода 6,8%.
Для определения 0,02—0,10% натрия в железных сплавах 1 г пробы рас-
творяют в 20 мл НС1 (1 : 1), раствор выпаривают досуха и отделяют Si02 и графит.
В фильтрате окисляют железо азотной кислотой, упаривают раствор до объема
8—10 мл, переносят в делительную воронку 15—20 мл 8 М НС1, добавляют рав-
ный объем бутилацетата и экстрагируют железо несколько секунд. Экстракцию
повторяют. К водной фазе прибавляют 1 мл 30%-ного Н2О2, выпаривают точти
досуха, добавляют 10—15 мл воды и выпаривают до небольшого объема. Раствор
переносят в полиэтиленовый стакан с помощью 10%-ной уксусной кислоты, вво-
дят 2—Змл 96%-ного этанола и осаждают натрий 15—20 мл раствора осадителя
(16,8 г аммонийуранилацетата и 50 г MgCO3 растворяют в смеси 10 мл лед.
СН3СООН, 250 мл этанола и 150 мл воды при нагревании, после чего разбавля-
ют водой до объема 500 мл) и выдерживают 2 ч. Осадок отфильтровывают в ти-
гель № 4, промывают 10 раз СН3СООН (порциями по 1 мл), растворяют в 50 мл
горячей воды и вводят 10—15 мл 0,01 М раствора ЭДТА. Для восстановления
ypana(VI) вводят 0,1—0,2 г аскорбиновой кислоты и кипятят 20—25 мин.
По охлаждении раствор разбавляют примерно до 80 мл водой, добавляют по
каплям HNO3 до pH 2 по индикаторной бумаге и титруют избыток ЭДТА 0,01 М
раствором Bi(NO3)3 в присутствии ксиленолового оранжевого (смесь 1 : 100 с
KNO3) до перехода желтой окраски раствора в красную.
Глава VI
ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
НАТРИЯ
Фотометрические методы определения натрия представлены в на-
стоящее время прямыми и косвенными методами с использованием
неорганических и органических реагентов. Следует подчеркнуть,
что число прямых методов, даже с применением органических реа-
гентов, весьма невелико. Несмотря на высокую чувствительность
определения натрия, они находят ограниченное применение из-за
низкой селективности, поэтому натрий перед определением отделяют.
Косвенные методы весьма разнообразны, хотя в большинстве случаев
и основаны на выделении натрия в виде тройных ацетатов, в которых
натрий определяют косвенно по одному из ионов, входящих в состав
тройного ацетата.
ПРЯМЫЕ МЕТОДЫ
Определение неорганическими реагентами
В водно-этанольной среде, содержащей 50% об. этанола, реагент
K2H2Sb2O7 образует с солями натрия устойчивую суспензию, пригод-
ную для нефелометрического определения натрия (219]. Метод при-
меняли для определения 1 мг натрия в осадочных породах.
Образование малорастворимого натрийцезийвисмутнитрита и ин-
тенсивное окрашивание раствора после растворения соли в серной
или соляной кислотах использованы при колориметрическом опре-
делении 0,1—0,8 мг натрия в растворах чистых солей (1288].
77
К анализируемому раствору, охлажденному до 5° С и не содержащему ме-
шающих ионов, прибавляют раствор осадителя из расчета 3,5 мл на 1 мг натрия.
Сосуд закрывают пробкой с отводной трубкой и пропускают порциями с интер-
валом в несколько секунд светильный газ, промытый щелочью. Колбу закрывают
пробкой и оставляют на 4—6 ч в холодильнике. Желтый кристаллический оса-
док быстро отфильтровывают на пористую пластинку, быстро промывают рас-
твором натрийцезийвисмутнитрита, азатем 50%-ным ацетоном (около 15мл)..
Осадок смывают в стакан, прибавляют 2 мл 50 % -ной H2SO4 и разбавляют водой
до 250 мл. К 1 мл этого раствора прибавляют 100 мл воды, 0,2 мл раствора нит-
ритного реактива и 1 мл конц. НС1. Через 15 мин сравнивают окраску с окраской
стандартного раствора, полученного аналогично (взят 1 мл раствора NaNO2).
Для получения раствора реагента 100 мл 40%-ного раствора KNO2 смешивают
с 3,2 г нитрата висмута, растворенного в 5 мл 2 М HNO3. Образующийся осадок
растворяют в HNO3 (1 : 1). Затем прибавляют 16 мл 10%-ного раствора CsNO3.
Если раствор мутнеет, снова прибавляют HNO3 (1:1). Раствор должен быть
прозрачным и иметь оранжево-желтый цвет. Образующийся позже (из-за при-
сутствия натрия в стекле посуды и в воде) осадок отфильтровывают. Раствор
хранят на холоду в темноте.
Для приготовления стандартного раствора сравнения 30 мг чис-
того NaNO2 растворяют в 1 л воды. 1 мл содержит 10 мкг натрия.
Определение органическими реагентами
Продукт взаимодействия лимонной кислоты с уксусным ангидри-
дом реагирует с ионами натрия и других щелочных металлов, обра-
зуя соединение фиолетово-красного цвета с максимумом светопогло-
щения при 560 нм [688]. Определению мешают соли других щелочных
металлов и даже 1 мкг четвертичных аминов. Реакцию полностью
ингибируют (в мкг): Fe(III) (0,5); Н3РО4 (40,0) и (в г): вода (0,03);
СН3СООН (0,1); СН3ОН (0,05); не мешают (в мкг): Са (32,6);
Си(313,0); Fe(II) (13,0); Hg(II) (68,6)ищавелевая кислота (0,03 г).
Селективность реакции повышают введением ацетилацетона. Закон
Бера соблюдается до концентраций натрия 5-10-7 М.
Прибавляют 0,3 мл раствора лимонной кислоты (2,5 г лимонной кислоты
растворяют при нагревании в 100 мл ацетилацетона) пипеткой на 1,0 мл в три
градуированных цилиндра вместимостью по 25 мл, закрывающихся стеклянны-
ми пробками. Один цилиндр оставляют в запасе. В каждый цилиндр вводят
аликвотную часть анализируемого раствора, приготовленного на уксусном ан-
гидриде и содержащего не более 5-10-7 М натрия. Растворы разбавляют уксус-
ным ангидридом до 5 мл. Вводят по 5 мл толуола, закрывают пробкой и встря-
хивают. Выпускают лишний воздух из цилиндров, закрывают их пробками
и помещают на 25 мин в водяную баню, нагретую до 98° С. После этого цилиндры
охлаждают до комнатной температуры и измеряют оптическую плотность экст-
ракта при 560 нм (Z = 1 см). Содержание натрия находят по градуировочному
графику.
Определению натрия с помощью 5-нитро-2-(3-метил-5-оксоизо-
ксазол-4-ил-азо)бензолсульфокислоты не мешают (в кратных количе-
ствах): Rb, Cs, Ba, Mg (12,5); Ca (10);.К, Sr (2,5); Li (0,05) [38].
Метод применен для определения натрия в минеральной воде (в мг/л)
Дилижан (1033), Ессентуки (3000), Московской (370) и Нарзан (280).
Реагент нйтроантранилазо[2-карбокси-4-нитрофенил-<Д-азо-4'^>-
1'-фенил-3'-метил-5'-пиразолон] в водной среде, содержащей ДМФА
и ацетон (1,2 : 0,2 : 8,6), в присутствии гидроксида тетраметиламмо-
ния образует с ионами натрия соединение с максимумом светопогло-
78
щения при 540 нм, е = (1,10 ± 0,02)-IO4. Реагент применен для
определения натрия в минеральной воде Боржоми. Предел обнару-
жения натрия 0,5 мкг/мл, определению не мешают (в кратных коли-
чествах): К, Cs, Ва, Cd, Pb, Са, Sr (5); Mg, Со (2,5); Zn (1); Li (2-
• 10-2) [296]. При определении 1,50 ± 0,06 мг/мл натрия погрешность
определения 4,2%.
1,0 мл воды в колбе вместимостью 100 мл разбавляют до метки бидистилля-
том, отбирают 1,0 мл для определения, прибавляют 0,1 мл бидистиллята, 0,4 мл
0,002 М раствора реагента в ДМФА, 8,4 мл ацетона и 0,1 мл 0,33 М раствора
гидроксида тетраметиламмония. Сразу измеряют оптическую плотность раство-
ра при 540 нм относительно воды. Градуировочный график строят в аналогич-
ных условиях для содержания натрия 5—40 мкг.
Реагент 3-метил-1-фепил-5-пираэолон-4-азо-1 ',4'-нитрофенил-2'~
сульфокислота (МФП-АНИФЕСК) образует с ионами натрия в при-
сутствии гидроксида тетрабутиламмония в среде вода—ацетон—
ДМФА (1,2 : 8,65 : 0,15, общий объем 10 мл) соединение с молярным
коэффициентом погашения 9,5-IO3 (XonT = 550 нм) [295]. Предел
обнаружения натрия 0,1 мкг/мл. Определению 5 мкг натрия не ме-
шают 2-кратные количества ионов Си, Mg, Fe(III), Со; 5-кратные
количества ионов К, Са, Zn, Cd, Ni; 10-кратные — Rb, Cs, Ba; 20-
кратные — Sr; 200-кратные — Li. Метод применен для определения
1,7 мг/мл натрия в минеральной воде Боржоми, погрешность опреде-
ления не превышает 7,1%.
Из производных 4-фенил-5-изоксазолов для фотометрического
определения натрия предложен 3-метил-4-(4-нитро-2-сульфофенила-
зо)-5-изоксазолон [137]. Определению натрия не мешают 12,5-крат-
ные количества Rb, Cs, Ва; 10-кратные — Са; 2,5-кратные — К,
Sr; 1,25-кратные — Mg; 5 • 10-2-кратные — Li.
К анализируемому раствору, содержащему менее 8 мкг натрия и объемом
не более 1,1 мл, прибавляют воду до объема 1,1 мл, 0,1 мл 0,05 М раствора гид-
роксида тетрабутиламмония; 8.8 мл раствора реагента, приготовленного смеше-
нием 2 мл 0,004 М раствора реагента с 86 мл ацетона, и измеряют оптическую
плотность при 510 нм.]
1,3-Диметил виол у ровая кислота образует с натрием соединение
с максимумом светопоглощения при 538 нм [1212]. В аналогичных
условиях образуют соединения также ионы Li, К, Bb, Cs, Mg и Be,
которые необходимо отделять. Реагент применен для фотометриче-
ского титрования натрия. В присутствии калия вначале оттитровы-
вают сумму калия и натрия, а в отдельной аликвотной части раство-
ра определяют калий гравиметрически осаждением тетрафенилбо-
ратом натрия. Аналогичные приемы можно использовать при раз-
дельном определении натрия в присутствии какого-либо одного ще-
лочного металла.
КОСВЕННЫЕ МЕТОДЫ
В отличие от прямых эта группа методов часто сочетает использо-
вание неорганических реагентов для осаждения натрия в виде
малорастворимого соединения и отделения его от сопутствующих
ионов с применением органических реагентов для повышения чувстви-
79
тельности определения. Методы требуют получения индивидуальных
соединений строго стехиометрического состава.
Значительную часть составляют методы, основанные на образо-
вании тройных ацетатов натрия, урана(У1) и двухвалентных элемен-
тов: цинка [22, 237, 238, 255, 901, 1219], марганца [40], никеля [39,
239, 528]. Сравнительно редко определяют натрий косвенно по двух-
валентному элементу, чаще определяют по урану(У1), для которого
известно много цветных высокочувствительных реакций. Кроме того,
содержание урана в осадке (в молях) в 3 раза превышает содержание
натрия или двухвалентного элемента.
Большие количества натрия определяют дифференциально-фото-
метрическим методом после осаждения натрийцинкуранилацетата,
его растворения и измерения оптической плотности, обусловленной
поглощением урана(УТ) либо его комплекса с Н2О2 [255]. Второй ва-
риант точнее: относительное стандартное отклонение 0,005.
К анализируемому раствору с pH 3—4 прибавляют 4 мл теплого раствора
цинкуранилацетата в 3%-ной СН3СООН, насыщенного натрийцинкуранилацета-
том, и оставляют на ночь в холодном месте. Осадок отфильтровывают на стеклян-
ный фильтр № 2, промывают 1,5 мл раствора осадителя, тремя порциями по 2 мл
этанола, насыщенного натрийцинкуранилацетатом, несколькими миллилитрами
ацетона и растворяют в горячей воде. К полученному раствору прибавляют
20 мл насыщенного раствора Na2C03, 10 мл 3%-ного раствора Н202, разбавляют
водой до 200 мл и измеряют оптическую плотность по отношению к раствору,
приготовленному аналогично, с аликвотной частью стандартного раствора, со-
держащей 2 мг натрия. В качестве раствора сравнения можно применять также
раствор К2Сг2О7 в 0,0025 М H2SO4.
Никель в тройном ацетате определяли диметилглиоксимом в ще-
лочной среде в присутствии окислителя. При определении 0,5—2 мг
натрия погрешность <0,028 мг натрия [39]. Меньшие количества
натрия (0,02—1 мг) определяли с погрешностью 4% после свя-
зывания урана в цитратный или тартратный комплекс [528]. В на-
стоящее время эти методы практически не используют.
Для определения урана в тройном ацетате применяют роданид
[206], Н2О2 в среде карбоната аммония [22, 255], ферроцианид [40,
238], цитрат [901, 1219]. Использование неорганических реагентов
нецелесообразно ввиду их малой чувствительности и стабильности.
В то же время эти методы интересны в историческом аспекте и при-
менительно к разнообразным объектам. Например, при определении
0,1—1 мг натрия косвенным методом определения урана(У1) ферро-
цианидом погрешность не превышала 0,002 мг натрия [40]. Этот
же метод применяли для определения натрия в стекле, по-
левом шпате, криолите [238]. Определению 0,1 мг натрия не
мешают 5-кратные количества ионов К, Mg, Са, Ba, А1 и РЬ.
При определении [0,043—0,293 мг натрия погрешность не пре-
вышает 2,5%. При использовании роданида введен эмпирический
коэффициент пересчета урана на натрий 0,0339, в то время как
теоретический коэффициент равен 0,0322; 0,2—0,9 мг натрия опре-
деляли с погрешностью <0,5% [206]. Косвенный метод определе-
ния натрия по реакции урана(У1) с Н2О2 в среде карбоната аммония
80
применен для определения натрия в металлической сурьме после ее
отделения возгонкой при нагревании с NH4C1 [22].
Растворы натрийцинкуранилацетата в цитрате натрия имеют два
максимума светопоглощения в ультрафиолетовой части спектра —
при 224 и 244 нм [901]. На оптическую плотность влияет концентра-
ция цитрата натрия. Растворы чувствительны к свету. Закон Бера
соблюдается в интервале концентраций натрия 0,23—3,2 мкг/мл»
воспроизводимость метода 1,5%.
0,5—1 мл раствора пробы, 0,01 М по НС1, содержащего 6—80 мкг/мл NaCl,
упаривают досуха на водяной бане в градуированной центрифужной пробирке
вместимостью 15 мл. После охлаждения прибавляют 0,08 мл 7 %-него раствора
трихлоруксусной кислоты, с этого момента рекомендуется работать при рассеян-
ном электрическом освещении. Прибавляют 1 мл раствора цинкуранилацетата 1
и 0,5 мл абсолютного этанола. Затем раствор встряхивают на механическом виб-
раторе 2—3 ч, оставляют до следующего дня, затем центрифугируют 30 мин
при 3500 об/мин и декантируют. Из пипетки в пробирку вводят при взбалтывании
5 мл ацетона 2 и центрифугируют 20 мин. Декантируют и повторяют промыва-
ние и декантацию еще 2 раза. Затем остаток высушивают 12 мин при 90—95° С,
охлаждают и смешивают с 6 мл 0,4%-ного раствора цитрата натрия (0,4 г
Na3CeH8O7-5,5H2O растворяют в бидистилляте и разбавляют им до метки в мер-
ной колбе вместимостью 100 мл). Через 2 мин раствор переносят в мерную кол-
бу вместимостью 10 мл, пробирку споласкивают 4 мл раствора цитрата натрия
и растворы объединяют в мерной колбе, разбавив объем до метки раствором цит-
рата натрия. Через 2,5 ч измеряют оптическую^плотность при 224 нм или 244 нм
относительно 0,4%-ного раствора цитрата натрия. После каждого промывания
жидкость с фильтра тщательно отсасывают. Осадок переносят в мерную колбу
вместимостью 500 мл со стеклянной пробкой и разбавляют до метки ацетоном.
Раствор можно использовать через сутки. Рекомендуется хранить в темноте.
Определению мешают ионы Li, К, взаимодействующие с осадите-
лем, арсенаты, оксалаты, большие количества фосфатов, реагирую-
щие с отдельными компонентами осадителя [1219]. Метод применя-
ли для определения натрия в морской воде.
Предложен косвенный колориметрический метод определения
натрия, основанный на его осаждении в форме Na[Sb(OH)6], растворе-
нии осадка в НС1 и определении сурьмы по реакции с тиомочевиной
[781]. Метод применен для определения натрия в фармацевтических
препаратах (NaCl, натриевые соли и-аминосалйциловой, бензойной
и салициловой кислот).
Навеску 0,1—0,2 г анализируемого вещества растворяют в воде, охлаждают
до 0° С и прибавляют избыток раствора реагента (10 г K[Sb(OH)e] в 500 мл воды +
1 Раствор цинкуранилацетата готовят следующим образом: а) 7,7 г соли
UO2(CH3COO)2-2H2O смешивают с 40 мл бидистиллята, содержащего 1,4 мл
конц. СН3СООН. Полученный раствор слегка нагревают в мерной колбе вмес-
тимостью 50 мл и разбавляют водой до метки; б) 23,1 г Zd(CH3COO)2,3H2O
растворяют при нагревании на водяной бане в 40 мл бидистиллята, содержа-
щего 0,7 мл конц. СН3С00Н. После растворения еще немного нагревают в мер-
ной колбе вместимостью 50 мл и разбавляют до метки водой. Оба раствора сли-
вают в склянку из коричневого стекла со стеклянной пробкой, оставляют на
сутки, затем фильтруют через пористый стеклянный фильтр.
2 7,5 мл раствора цинкуранилацетата смешивают с 1,5 мл 5%-ного раствора
NaCl и 2,5 мл абсолютного этанола.Через 0,5 ч фильтруют, 5 раз промывают
на стеклянном фильтре маленькими порциями 95%-ного этанола и диэтиловым
эфиром.
81
— 15 мл 10%-ного] раствора КОН) и 20—25 мл 95%-ного] этанола. Через
30—40 мин осадок отфильтровывают, промывают 30%-ным этанолом, растворя-
ют в НС1 (пл. 1,15) и разбавляют до 50 или 100 мл насыщенным раствором тио-
мочевины в смеси (1 : 2) НС1 и воды. Возникшую окраску сравнивают с окраской
стандартного раствора, приготовленного в аналогичных условиях.
Разработан косвенный фотометрический метод определения нат-
рия и калия при их совместном присутствии, основанный на экстра-
гировании дипикриламинатов этих элементов нитробензолом [908].
Вначале строят градуировочные графики в координатах A — IgCR/cNa,
причем концентрации калия и натрия варьируют, оставляя постоян-
ной их сумму и соотношение (ск + ска)/сдпА- Получены удовлетво-
рительные результаты при определении 0,0276—5,7300 мг натрия
в присутствии 0,5865—1,0320 мг калия.
Натрий определяют косвенно с помощью 5-бензамидоантрахинон-
2-сульфокислоты после осаждения реагентом и измерения оптической
плотности избытка реагента [519]. Определению не мешают <^0,3 мг
калия и аммония, мешают щелочноземельные элементы.
К анализируемому раствору, содержащему <СЗ мг натрия, прибавляют 1 мл
95%-ного этанола, 5 мл 3%-ного раствора реагента в 50%-ном этаноле, энергич-
но перемешивают, разбавляют 50%-ным этанолом до 10 мл и выдерживают 30 мин
при 0—5° С. Жидкость фильтруют, 1 мл фильтрата разбавляют до 250 мл водой
и измеряют его оптическую плотность при 425 нм.
ПРОЧИЕ МЕТОДЫ
Разработан косвенный поляриметрический метод определения
натрия, основанный на его отделении осаждением в форме натрий-
тщнкуранилацетата, растворении осадка в d-винной кислоте и изме-
рении оптического вращения, пропорционального содержанию урана.
Метод применен для определения натрия в рассолах [895] и морской
воде [244, 895].
Предложен метод, основанный на изменении вращения плоскости
поляризации света от длины волны при прибавлении соли натрия
к раствору комплекса лития с реагентом Чирал в метиленхлориде
11275].
Глава VII
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
НАТРИЯ
Удельный вес электрохимических методов в аналитической хи-
мии натрия сравнительно невелик. Важнейшим методом определения
натрия является прямая потенциометрия (ионометрия). Известно
довольно много работ, посвященных полярографическому определе-
нию натрия, но с развитием ионометрии они заметно утратили прак-
тическую значимость. Реже для определения натрия применяют
кондуктометрическое титрование и кулонометрию.
82
ИОНОМЕТРИЯ
Для прямого потенциометрического определения натрия обычно
применяют стеклянные натрий-селективные электроды. Разработкой
стеклянных электродов, обладающих металлической функцией, за-
нимались многие исследователи [37, 333]. Опубликован обзор по при-
менению стеклянных электродов для определения натрия [957].
Селективность электродов. Важнейшей характеристикой ионосе-
лективного электрода является коэффициент селективности КАВ —
мера относительной чувствительности электрода к изучаемому иону
А и постороннему иону В. Если КАв < 1» электрод обладает повы-
шенной селективностью относительно иона А, при Аав > 1 —
повышенной селективностью относительно иона В.
Коэффициент селективности можно рассчитать из результатов из-
мерения потенциала электрода в растворах, содержащих только один
ион (чистые растворы, содержащие только изучаемый или посторон-
ний ион) либо смесь изучаемого и постороннего иона (смешанные рас-
творы). Расхождения между величинами коэффициентов селективно-
сти, определенных различными способами, могут достигать почти
порядка [334]. Хотя чаще применяют метод смешанных растворов,,
во многих исследованиях способ определения коэффициента селек-
тивности не указан и поэтому характеристики одного и того жа
электрода сильно различаются [1269].
Для изготовления натрий-селективных электродов применяют
алюмосиликатные или боросиликатные стекла, например стекло,
содержащее 11% Na2O, 18% А12О3, 71 % SiO2 [720] (электрод, изго-
товленный из такого стекла, известен под маркой NAS 1118) или 25%
Na2O, 3% А12О3,61 % SiO2nll % В2О3 [112]; 24% Na2O, 3% А12О3,12%
В2О3, 61 % SiO2 [50]. Предложено изготовлять натрий-селективный
электрод из стекла с добавкой 2—8% Та205 (сплавляется стекло при
1620—1670° С) и до 3% оксидов Са, Ва и РЗЭ [838]. При pH 12 этот
электрод характеризуется А\аК = 1,4-10~3 и XNaNH4+ = 8-10~3.
Показано, что замена натрия на литий резко увеличивает селектив-
ность электрода по отношению к натрию, но потенциалы электродов
из литиевых стекол устанавливаются медленно. Кроме того, эти
электроды характеризуются довольно широкой областью водородной
функции, что делает невозможным их применение для работы с
разбавленными растворами натрия. Эти недостатки отмечены для
электрода из стекла, содержащего 10,4% Li2O, 22,6% А12О3и 67 %SiO2,
обладающего высокой селективностью относительно натрия [1269].
Сопоставлены девять сортов стекол, применяемых для изготов-
ления натрий-селективных электродов [634, 635]. Лучшие из них
обладают пределом обнаружения pNa ~6 и характеризуются вы-
полнением электродной функции в интервале pNa 0—4. Время от-
клика колеблется от 80 до 300 с при уменьшении концентрации
натрия от 10 3 до 10~6 М. При концентрации натрия в интервале от
10“5 М до 1 М воспроизводимость результатов составляет 0,01—0,02
pNa. Теоретическая электродная функция выполняется при условии
pH > pNa + 3; XNaI< = 1 -10~2.
83.
Характеристики некоторых электродов промышленного изготов-
ления. В настоящее время известны электроды Гомельского завода
ЭСЛ-51-05 [4701, ЭСЛ-51-04 [88], зарубежных фирм Orion 96-11 [617],
Orion 94-11 А [580, 617], Beckman 39278 [617] и др.
Высокой специфичностью обладает электрод Beckman 39278, но
для его изготовления применяют исключительно тугоплавкое и труд-
нообрабатываемое стекло. Кроме того, этот электрод характеризует-
ся довольно большим временем отклика и заметной чувствитель-
ностью к ионам водорода. Охарактеризована селективность электрода
Beckman 39278 относительно хлоридов К, NH4, Са, Ba, Mg и суль-
фата магния [1229]. В интервале pNa от 1 до 2 при ионной силе,
равной 1, наклон градуировочного графика составляет 33, 56,7,
57,3, 57,8, 58,8 и 58 мВ соответственно в присутствии хлоридов К,
NHt, Са, Ba, Mg и сульфата магния.
По степени влияния на натриевую функцию электрода ионы мож-
но расположить в ряд Н > Ag > К > Li > Rb > NH4 > Mg >
>• Ca [302, 335—337]. Изучена селективность четырех типов стеклян-
ных натрий-селективных электродов (Beckman 39278 (I), EILGEA
ЗЗС (II), Orion 94-11 (III), Radiometer G 502 (IV)) относительно
ионов водорода [1269]. Перед измерением электроды 5 дней выдержи-
вают в 0,1 М растворе NaCl, а затем последовательно по 5 мин в рас-
творах, содержащих от 1-10-6 до 0,1 MNaCL Установлены интерва-
лы pH, в которых показания электродов не зависят от концентра-
ции ионов водорода для растворов с различными концентрациями
натрия (табл. 38). При pH 9,54 электродная функция выполняется
Таблица 38
Интервалы pH, в которых в зависимости от концентрации натрия
выполняется электродная функция для электродов I—IV [1269]
cNaCb м Электрод
I II III IV
10~1 5,5—10,0 4,5—10,0 4,5—10,0 4,0—10,0
10-2 7,0—10,0 5,0—10,0 5,5-10,0 4,5—10,0
IO"3 8,0—10,0 6,0—10,0 7,0—10,0 5,5—10,0
1Q-4 9,0—10,0 7,0—10,0 8,0—10,0 6,5-10,0
для электродов I—IV в интервалах концентраций натрия 10-1—1СГ4,
10-1—5-10-5, 10-1—10-6 и 10-1—5-10'8М соответственно.
Изучена селективность перечисленных выше четырех электродов
относительно К, Ag и NH4 [1270]. Отмечено, что А’Как увеличивает-
ся при увеличении концентрации ионов калия. Так, для электрода
Radiometer G502 ANaK соответственно 2-10"4 и 1-10-4 в 0,1 мМ и
0,5 М растворах КС1.
Стеклянный натрий-селективный электрод Orion 96—11 сохра-
няет электродную функцию в интервале pNa от 1 до 3 при pH ~ 10.
Ионную силу рекомендуется регулировать с помощью сульфата маг-
ния [889].
Потенциал натрий-селективного электрода Orion 94-11 А устанав-
ливается быстрее, чем для других натрий-селективных электродов,
выпускаемых промышленностью [580]. В интервале pNa 3—4 на-
блюдается хорошая корреляция теоретического и экспериментально-
го градуировочного графиков. Недостатком электрода является то,
что определению натрия мешают эквивалентные количества калия
(погрешность ~6%), хотя ANaK = 1,4-10-3.
Довольно высокой воспроизводимостью результатов характери-
зуется электрод GNA 33 [793]. Для 1-10-3М раствора NaCl (20° С)
значение pNa через 10 ч равно 3,000, через 18 ч — 2,993. Величина
pH не влияет на результаты определения pNa при условии pH
pNa + 4. В этом случае погрешность не превышает 1 %. В щелоч-
ной среде линейная зависимость сохраняется в интервале pNa 0—5.
Определению натрия не мешают 1000-кратные количества Fe3+, Fe2+,
Mg2+, Hg2+, Cu2+, NH4, 100-кратное количество кальция, 10-кратное
количество калия, эквивалентное количество лития.
Электроды ЭСЛ-51 гомельского завода характеризуются умерен-
ной селективностью, но они отличаются низким сопротивлением, ма-
лым временем установления потенциала, хорошей воспроизводимо-
стью потенциала во времени для одного электрода и от электрода
к электроду.
Электрод ЭСЛ-51-05 пригоден для определения натрия в интер-
вале pNa 0—4 [470]. Наклон градуировочного графика составляет
53—55 мВ, относительное стандартное отклонение 0,06. Время уста-
новления постоянного значения потенциала 5 мин. Определение реко-
мендуется проводить при pH > 7, кислые растворы предварительно
обрабатывать с помощью MgO или СаО. Определению не мешают
100-кратные количества Са, Mg, Ва, 10-кратные количества Li,
nh^, сг, Вг-, г, no;, сю;, нсо;, cot, sot, н2во;, он-,
эквивалентное количество калия.
Электроды ЭСЛ-51-04 и ЭСЛ-51-05 применены для определения
натрия в карналлите и продуктах его обезвоживания [441]. Для
повышения селективности в раствор вводили стандартный раствор
NaOH, создающий требуемую величину pH 10—12, и NaCl. Из испы-
танных вариантов определения натрия — градуировочного графика
и добавок — более точным оказался второй: при определении 7,8 —
23,9% NaCl относительное стандартное отклонение составило
0,059—0,021 соответственно при навеске карналлита 0,2 г.
Сравнена правильность результатов трех способов прямого по-
тенциометрического определения pNa с помощью стеклянного элек-
трода [955]. Сравнивали метод градуировочного графика, модифици-
рованный метод «нулевой точки» и дифференциальный потенциоме-
трический метод. Лучшим при определении натрия в интервале pNa
1—4 оказался последний, худшим — первый способ. Электродная
функция выполняется при условии pH = pNa + 4,5- Нарушение
электродной функции замечено в присутствии более чем 20-кратного
избытка калия, аммония и более чем 1000-кратного избытка кальция.
85
Показано, что стандартное отклонение при определении натрий,
характеризуемое отношением стандартного отклонения потенциала
электрода к коэффициенту наклона градуировочного графика, ли-
митируется погрешностью измерительного прибора [799].
Величина минимально определяемой концентрации натрия с по-
мощью стеклянного электрода обусловлена переходом катионов ще-
лочных металлов (главным образом лития) из стекла [774]. Для пяти
видов серийных электродов отклонение от нернстовской функции
электрода наблюдается при концентрации натрия <20 мкг/л. Влия-
ние pH незначительно сказывается при pH 10,5 и использовании
диэтиламина для создания щелочной среды. За счет применения
аминов создается фон ~0,1 мкг/л натрия. Время отклика электрода
зависит от температуры: при понижении температуры от 45 до 8° С
время отклика увеличивается вдвое, но при этом уменьшаются
помехи за счет присутствия лития. При правильном выборе условий
можно определять натрий при концентрациях 0,07 мкг/л.
Изучено влияние температуры на результаты определения натрия
в 0,02 М растворе диизопропиламина [716]. Аномальная зависимость
результатов измерения при концентрации натрия 1 нг/мл при повы-
шении температуры до 60° С объяснена откликом электрода на резкое
изменение активности протона при повышении температуры. Для
получения правильных результатов рекомендуется проводить изме-
рения при низких температурах (желательно охладить раствор до-
0° С), при этом одновременно уменьшается влияние ионов щелочных
металлов, переходящих в раствор из стекла.
Другие мембранные натрий-селективные электроды. В последние
годы предложен ряд других мембранных натрий-селективных электро-
дов. Описан мембранный электрод из поливинилхлорида и трикре-
зилфосфата [927]. Предложен проволочный ионоселективный элек-
трод, пригодный для определения натрия с помощью полевого-
транзистора [1218]. Электрод работает аналогично биологическим
мембранам. Устойчивые показания наблюдаются в течение недели.
Наклон градуировочного графика составляет 19,5 ± 0,4 мВ/pNa.
Платиновую проволочку покрывают ионоселективной мембраной (25—
100 мг моненсина, 0,33 г поливинилхлорида и 0,89 мл ди-н-октил-
адипината или ди-н-октилфталата растворяют в 13 мл тетрагидрофу-
рана). Толщина образующейся пленки 100—300 мкм.
Описана конструкция натрий-селективного электрода с гетеро-
генной катионселективной мембраной, в которой в качестве связую-
щего агента для Амберлита IR 120 использована эпоксидная смола
[852]. Постоянное значение потенциала электрода достигается через
5 мин и сохраняется в течение 6 ч. Можно определять натрий при
концентрации <0,2 М.
Известен натрий-селективный электрод на основе мембраноак-
тивного комплексона (I) [585]. Электрод селективен относительно
натрия в присутствии ионов щелочных и щелочноземельных металлов.
Электродная функция выполняется в интервале pNa 1—4. Недостат-
ком электрода является малая продолжительность жизни — от 2 не-
дель до 2 месяцев.
86
В дальнейшем на основе этого лиганда, но с использованием дру-
гих растворителей — пластификаторов — были созданы электроды
с лучшими характеристиками [13, 301]. В качестве пластификатора
использовали дибутилфталат. Состав мембраны: 24% поливинилхло-
рида, 71,2% дибутилфталата, 3,9% лиганда. Срок жизни электрода
6 месяцев. Характеристики мембран позволили использовать элек-
трод в биологических анализах сыворотки крови и цельной крови.
Постоянное значение потенциала электрода при перенесении его из
водного раствора хлорида натрия в сыворотку устанавливается
в течение 5—7 мин и не изменяется более чем на 1—2 мВ в течение
двух первых часов.
Для определения натрия в сыворотке крови или цельной крови
электрод выдерживают в анализируемом образце в течение 20—
30 мин, затем градуируют его по водным растворам хлорида натрия
с моляльной концентрацией 0,05; 0,1 и 0,2. По полученным данным
строят график зависимости Е = / (1g mNaci) и, измерив потенциал
электрода в сыворотке или крови, находят содержание натрия.
Разработали электрод [301] на основе мембраноактивного комп-
лексона другого строения (II), впервые использованного в рабо-
те [787]. Концентрация лиганда в мембране была равна 0,05 и
0,1 моль/кг растворителя. В качестве растворителей-пластификато-
ров использовали дибутилфталат, дигексилсебацинат и 2-нитро-н-
цимол. Время установления постоянного значения потенциала
2—3 мин при концентрации NaCl от 0,01 до 4,5 т и 5—
10 мин при более низких концентрациях. Коэффициент селектив-
ности An0m f (М+ = К, NH£, Li) мало зависит от природы пластифи-
катора, практически одинаков в присутствии ионов калия (5-10-1)
и аммония (2,5-10-1) и несколько ниже (1,5-10-2) в присутствии ли-
тия. Из двухзарядных катионов наименьшее влияние оказывает
кальций (/CNaca = 3-10-3). Электроды с мембраной на основе дибу-
тилфталата и дигексилсебацината пригодны для работы в интервале
pH 2,5—10, а электрод на основе 2-нитро-н-цимола — в интервале
pH 3—10.
Особых преимуществ по сравнению со стеклянными эти электро-
ды не имеют, но их можно применять в тех случаях, где исключено
использование стеклянного электрода.
Применение ионометрии. Ионометрию применяют для определе-
ния натрия в воздухе над океаном [146], природных водах [14, 28,
87
88, 112—114, 759, 776, 796], особо чистой воде [1256], сточных водах
животноводческих комплексов с высокой степенью загрязненности
(2—136 г/л органических и 0,6—31 г/л неорганических веществ)
[332], воде, циркулирующей в промышленной установке [1263],
почвах [16, 210, 346] и почвенных растворах [346], солях натрия
[927], карналлите [441], глиноземе [746, 1217], алюмосиликатах и
кремневой кислоте [1182], минералах-ортофосфатах [710], солях ор-
ганических кислот [580], многокомпонентных солевых растворах
[473], двойной аммонийной соли вольфрама [303], сульфатных щело-
ках целлюлозно-бумажной промышленности при реальных темпера-
турах без охлаждения [141], в поглотительных растворах сероочистки
[90], в черной металлургии [812]. Довольно подробно освещается
применение натрий-селективных электродов для определения натрия
в биологических объектах [182, 334].
Натрий определяют в золе кремневой кислоты с помощью кати-
онселективного электрода 78137V [1209]. При pH 6,0 ± 0,2 (26 ±
± 1° С) между измеренным потенциалом ячейки Е (мВ) и логариф-
мом концентрации натрия (S) в мкг/мл существует зависимость
S =0,0203 Е 4-2,914. Метод пригоден для определения натрия
в присутствии 2-кратного избытка кальция и железа(Ш). Мешают,
вызывая погрешность до400%, аммоний и калий, слабо мешают (по-
грешность 2—10%) 2-кратные избытки алюминия и лития.
К 10,0 г золя с pH 2,0—3,0 прибавляют около 40 мл воды, смесь пропускают
через колонку емкостью 50 мл, заполненную Дауэкс 1 X 8 (20—50 меш) в;
ОН-форме, со скоростью 4 мл/(см2-мин). Смывают 150 мл воды, повышая ско-
рость до 14—15 мл/(см2-мин), вводят воду до получения 230 мл элюата. Если
pH > 6,2, то добавляют 0,01—0,05 М H2SO4 до pH 6,2 + 0,2 и разбавляют во-
дой до 250 мл. Аликвотную часть раствора используют для измерения потенциа-
ла электрода. Расхождение результатов определения 12,1—333,8 мкг/мл натрия
с данными фотометрии пламени 5,7% отн.
Натрий определяли в концентрированных растворах, исполь-
зуемых в автоматических проточных системах [617]. Типичный со-
став растворов: 2,1—2,6 М NaOH; 0,4—0,7 М Na2S; 0,2—0,5 М
Na2CO3 и небольшие количества Na2S2O3 и Na2SO4. Из изученных
авторами электродов (Orion 94-11 A, Radiometer G502, Beckman
39278, Philips G 15 и электрод с катионообменной мембраной Рег-
maplex С20) наиболее пригодным оказался натрий-селективный
электрод Radidmeter G 502. На его основе сконструирована проточ-
ная ячейка для определения натрия в указанных выше растворах
после разбавления 1 М раствором карбоната аммония в соотношении
1 : 20. Относительное стандартное отклонение при определении от
1,00 до 5,00 М натрия составляет 0,007—0,005.
В природных водах с pH 4> 6 (более кислые воды предварительно
нейтрализуют газообразным аммиаком по нейтральному красному)
при соотношении числа эквивалентов (Са + Mg)/Na </6 можно опре-
делить от 3,67 до 163,7 мг-экв/л натрия [114]. Ионную силу рассчи-
тывают по результатам сокращенного анализа воды. Среднее откло-
нение от результатов стандартного гравиметрического метода опре-
деления натрия в форме натрийцинкуранилацетата ±2%.
88
Для определения натрия в А12О3 (и материалах на его основе) [1039] навес-
ку пробы растворяют в платиновом тигле в Н3РО4, нагревают до появления бе-
лых паров, переносят в колбу вместимостью 100 мл, прибавляют 10 мл 60%-ного
этаноламина, разбавляют водой до метки. Оптимальное значение pH 9,6.
При содержании натрия до 0,1% относительное стандартное отклонение 0,01—
0,04.
Предложена методика быстрого определения свободного и свя-
занного оксида натрия в высокочистом реактивном глиноземе [623].
Навеску пробы 1 г обрабатывают 100 мл воды, кипятят при перемешивании
5 мин, прибавляют 10—15 капель триэтаноламина для предотвращения взаимо-
действия натрия и алюминия и разбавляют до 100 мл. После перемешивания
и отстаивания прозрачный раствор переносят в стаканчик и измеряют pNa с по-
мощью стеклянного натрий-селективного электрода. Для определения общего
содержания оксида натрия 0,2—0,3 г измельченной пробы обрабатывают смесью
НС1О4 и HF, затем дистиллированной водой, прибавляют 1—2 капли конц. НС1,
встряхивают, нагревают 10 мин, разбавляют до 100 мл. К 10 мл полученного
раствора прибавляют триэтаноламин, разбавляют до 25 мл и измеряют pNa.
Содержание связанного оксида натрия находят по разности.
Метод применен для образцов с общим содержанием оксида нат-
рия от 0,0065 до 0,2540% и содержанием свободного оксида натрия
от 0,0021 до 0,0480%. Результаты согласуются с данными фотомет-
рии пламени.
Проточные ячейки для определения натрия. На основе ионосе-
лективных электродов сконструированы проточные ячейки для опре-
деления натрия.
Описана проточная ячейка, снабженная компьютером, для одно-
временного определения натрия и калия [751]. Для определения нат-
рия используют стеклянный электрод Orion 94-11. Определение про-
водят при температуре 24,5° С, создавая ионную силу 0,50 М рас-
твором MgSO4.
Для определения 10~3—5% натрия в потоке в присутствии 0,25%
полиакриламида и 0,5% ПАВ на основе сульфонатов использовали
электрод фирмы Orion [693]. За 1 ч можно выполнить до 20 определе-
ний. Коэффициент корреляции с данными фотометрии пламени 0,98.
Установлено, что ошибки определения низких концентраций
натрия в потоке уменьшаются при увеличении скорости потока непо-
средственно вблизи детектора [718]. Предложена конструкция детек-
тора со специальной формой канала для ввода пробы, позволяющая
резко увеличить скорость потока вблизи чувствительного элемента без
увеличения общей обменной скорости протекания пробы. Предел
обнаружения натрия 0,13 нг/мл при скорости потока выше
150 мл/мин. С увеличением скорости потока при определении на-
трия в проточной ячейке уменьшается также ошибка, связанная
с выщелачиванием лития из стекла электрода [774].
На основе электрода Orion 96-11 разработано автоматическое
устройство для определения натрия в сточных и природных водах
с производительностью 20 проб в ч. Предел обнаружения натрия
0,1 мкг/мл, стандартное отклонение <10% [1144].
Автоанализатор с натрий-селективным электродом Beckman 39278
использовали для определения натрия в речных и минеральных
89
водах [1274]. При скорости потока 3,90 л/мин и отношении объемов
проба : промывной раствор = 1:3 производительность 20 проб в ч.
Стандартное отклонение <2,2% для интервала концентраций натрия
1,4—1400 мкг/мл. Определению мешает более чем 10-кратный избы-
ток калия. Для устранения мешающего влияния К и Н в качестве
промывного раствора рекомендуется смесь 2,5% NH3 и 5% ВаС12.
При анализе сильноминерализованных вод мешающее влияние маг-
ния устраняют с помощью промывного раствора, содержащего
NH3, NH4NO3 и ЭДТА.
Стеклянный натрий-сёлективный электрод ЭСЛ-51-04 применен
в датчике автоматической станции и в переносном датчике для опре-
деления натрия в поверхностных водах. Градуировочный график
устойчив в течение месяца, хотя уже через 3 дня электрод покры-
вается солями. Электрод рекомендуется градуировать по раствору
хлорида натрия с pNa 1—3. Коэффициент автокорреляции через
1ч — 0,99, через 2 ч — 1,00, через 3 ч — 0,20. Максимальное рас-
хождение с данными фотометрии пламени — 17%, минимальное —
2% [88]. Установлено, что при определении 1—10 мэкв/л натрия
с помощью электрода ЭСЛ-51-Г-04, предварительно выдержанного
в течение 8 ч в 0,1 М растворе NaCl, стандартное отклонение
±0,11 мэкв/л для метода эталонных растворов и ±0,16 мэкв/л для
метода добавок [759].
Для автоматического определения pNa после подщелачивания
кислых водных растворов пропусканием через анионообменные
фильтры (АВ-17), регенерируемые растворами NH3 и Ва(ОН)2,
применен электрод ЭСЛ 51-05 [183].
Потенциометрическое титрование. Натрий-селективные электро-
ды применяются и для потенциометрического титрования. Потен-
циометрическим титрованием с мембранным электродом на основе
поливинилхлорида или трикрезилфосфата определяли натрий
с погрешностью <1% в NaOH, Na2CO3, NaHCO3, Na2SiO3 и
Na2B4O7 [927].
Показана возможность потенциометрического титрования милли-
граммовых количеств натрия раствором цинкуранилацетата в 85—
95 %-ном этаноле с использованием натрий-селективного стеклянно-
го электрода Jena 20 [1227]. Определению не мешают эквивалентные
количества калия.
ВОЛЬТАМПЕРОМЕТРИЧЕСКИЕ МЕТОДЫ
. Полярография. На ртутном капающем электроде четкие волны
восстановления ионов натрия можно наблюдать только на фоне солей
или гидроксидов тетраал кил аммония. Впервые это было показано
Я. Гейровским еще в 1935 г. Гидроксиды рекомендуется получать
перекристаллизацией соответствующих солей и пропусканием через
колонку с анионитом Дауэкс-1 в ОН-форме [629] или обработкой
оксидом серебра(1) [749].
Пропорциональная зависимость между высотой волны натрия и
его концентрацией наблюдается, например, на фоне 0,1 М раствора
(CH3)4NOH (pH 3-13) в интервале 2-10~5—1 -Ю"3 М [630].
90
Потенциалы полуволны на различных фонах приведены в табл.
39, 40 (отн. нас. к. э.).
Восстановление иона натрия на ртутном электроде протекает
практически обратимо, предельный ток имеет диффузионный харак-
тер. Это показано результатами измерений с помощью классической,
Таблица 39
Величины потенциалов полуволн натрия на различных фонах
Фон е1/2- в Литература
0,1М [(C2H6)3CeH5CH2] NC1 —2,12 [463]
0.1М (C2H5)4NJ —2,11 [464]
0,1М (CH3)4NJ —2,11 [464]
0,1М (C2H5)4NC1O4 —2,11 [682]
0,1М (C2H5)4NOH или 0,1М (CHs)4NOH в 50%-ном этаноле —2,08 [464]
0,1М (C2H5)4NC1O4 в ацетонитриле —1,92 [682]
О,1М (C2H5)4NC1O4 в диметилформамиде —2,07 [1125]
0,1М (C2H5)4NC1O4 в диметилсульфоксиде —2,09 [1125]
Таблица 40
Потенциалы полуволны натрия на фоне (CH3)tNOH
в присутствии различных комплексонов [53]
Фон п1/2, В мВ * Фон £i/2. В Д£1/2’ мВ*
0,1М (CH3)4NOH —2,125 0,1М (CH3)4NOH4- —2,140 —15
О,1М (CH3)4NOH + —2,250 —125 4-0,02 М ЭДТА —2,125
4-0,02М ЦДТА 0,1М (CH3)4NOH4- 0
0,1 M(CH3)4NOH + 4-0.02М УДА —2,205 -89 4- 0,02 М ЭГТА 0,1М (CH3)4NOH4- | 4- 0,02 М НТА -2,125 0
Примечание. УДА — урамил-М.К-диуксусная кислота; ЭГТА — этиленгликоль-ди
-(2-аминоэтил)тстрауксусная кислота; НТА — нитрилотриуксусная кислота; * разность по-
тенциалов полуволны до и после введения комплексона.
переменнотоковой полярографии, циклической хроновольтамперо-
метрии [630] и осциллографической полярографии [189, 666, 857].
В табл. 39 приведены величины Еуг натрия на различных фонах.
Как видно, природа фона практически не влияет на величину Е1/г.
В присутствии органических растворителей (этанол, ацетонитрил
и др.) наблюдается небольшое смещение Ei/2 в область более поло-
жительных значений,. Очень близкие значения Еуг имеют ионы и
других щелочных металлов. Так, например, на фоне 0,1 М
(C2H5)4NC1O4 в диметилсульфоксиде рубидий имеет Ег/г—2,09 В,
а калий — 2,12 В [1125]. Исключение составляет ион лития, который
91
восстанавливается при потенциалах, примерно на 0,2 В более от-
рицательных. Это приводит к тому, что в реальных объектах возмож-
но определение только суммы натрия и калия. Из-за различия коэф-
фициентов диффузии точность определения невысока.
Суммарное содержание натрия и калия (при известном соотно-
шении их концентраций) предлагается определять на фоне гидрокси-
да фенил триметил аммония [699]. На этом фоне натрий образует волну
при тех же потенциалах, что и калий. В соответствии с величинами
коэффициентов диффузии высота волны натрия в 1,22 раза меньше
высоты волны калия. При равных концентрациях (мг/мл) высота
волны калия составляет 72 % от высоты волны натрия. Это позволяет
провести расчет суммарного содержания натрия и калия по общей
волне.
Иногда лучшие результаты получаются при применении спирто-
вых или диоксановых растворов фонов, так как в этом случае в при-
сутствии эквивалентных количеств щелочных металлов образуются
волны равной высоты. Предприняты попытки получить одинаковые
диффузионные токи натрия и калия в смешанных растворителях
[445]. В 80 %-ном этаноле отношение диффузионных токов состав-
ляет 0,76; 0,70, а в 80%-ном диоксане диффузионные токи эквива-
лентных количеств натрия и калия равны, но волны плохо выра-
жены.
Методами классической и переменнотоковой полярографии изу-
чено поведение натрия и других щелочных металлов на фоне 0,1 М
(CH8)4NOH в присутствии нитрилотриуксусной кислоты, ЭДТА,
циклогексан-диамин-К,М,М,К-тетрауксусной кислоты (ЦДТА) и
урамилдиуксусной кислоты [53]. В присутствии всех указанных ли-
гандов восстановление натрия протекает обратимо, волна имеет
диффузионный характер. Установлено, что образуются комплексы
1:1. Наиболее прочные комплексы образует ЦДТА, для натрия
1g К = 3,79 ± 0,12. В этих условиях наблюдается разделение пи-
ков натрия (—2,25 В) и калия (—2,16 В) на переменнотоковых по-
лярограммах, а литий до разряда фона не восстанавливается. Пока-
зана возможность определения при совместном присутствии натрия
и калия, натрия и цезия (—2,14 В), натрия и рубидия (—2,11 В).
К сожалению, интервал допустимых соотношений концентраций не
указан, но, судя по величинам Ei/t, он весьма невелик.
Определение 10~4 М натрия в присутствии 10-кратного избытка
калия возможно на фоне (C2H5)4NJ, содержащем ЫВ(С6Н5)4 [180].
Для определения натрия в присутствии Al, Са, Fe и других тя-
желых металлов в качестве фона рекомендуется 0,1 М (CHa)4NOH
или (C2H5)4NOH в 50 %-ном этаноле [464]. Образующийся осадок гид-
роксидов можно не отфильтровывать. Высота волны натрия с Ei/t =
— —2,08 В пропорциональна концентрации натрия в интервале
2. 10-4—8-Ю-3 М.
Этот же фон применен для полярографического определения
^0,1 % натрия в природных и технических силикатах [1240].
0,05—0,35 г тонкоизмельченной пробы, содержащей около 0,005 г суммы
оксидов^ натрия и калия, смачивают водой в платиновом тигле, прибавляют 3—
92
4 мл 40%-ной HF, 5—10 капель НС1О4 или конц. H2SO4 и 5—10 капель 55%-ной
Н3РО4. Смесь выпаривают, не допуская кипения, на песчаной бане до исчезно-
вения белых паров, затем прокаливают для полного удаления Р2О6. По охлаж-
дении к остатку прибавляют 4—5 мл горячей воды, нагревают 5 мин, затем при-
бавляют еще 4—5 мл воды, переносят в мерную колбу вместимостью 25 мл и раз-
бавляют до метки водой. При содержании Fe2O3 С 0,5% пробу разлагают без
Н3РО4 и осадок не прокаливают, только перед окончательным разбавлением
до 25 мл вводят 2—3 капли HNO3. К 10 мл полученного раствора прибавляют
0,5—1 мл гидроксида тетраметиламмония (pH среды 10—12), перемешивают
и полярографируют от —1,5 до —2,2 В. Содержание суммы натрия и калия
определяют методом добавок. Другие 10 мл раствора упаривают в платиновом тиг-
ле с 4—5 каплями конц. НСЮ4 и 2—3 каплями гидроксида тетраметиламмония
до появления белых паров НСЮ4, по охлаждении остаток переносят, этанолом
в пробирку, центрифугируют, осадок перхлоратов калия и тетраметиламмония
промывают этанолом, содержащим 10% НСЮ4, затем растворяют в горячей воде,
по охлаждении разбавляют до 10 мл водой и полярографируют. Натрий опреде-
ляют по разности. (Реагент гидроксид тетраметиламмония очищают от натрия,,
калия и кальция электролизом на ртутном катоде при 4 В и хранят в пластмас-
совой посуде.)
Для определения NaNO2 в лекарственных формах используют
волну восстановления NO2 на капающем ртутном электроде на фоне
0,2 М раствора хлорида калия в присутствии 0,02 М НС1 (pH 1,8)
[881]. Высота волны пропорциональна концентрации нитрита натрия
в интервале 2-10-4—2-10~3М. Погрешность определения 2,4—
2,7% (п = 10).
Полярографически определяли натрий в каучуках [462], природ-
ных и технических силикатах, содержащих железо [1240], воде,
циркулирующей в промышленной установке [1263]. Сумму натрия
и калия определяли в огнеупорных материалах [151], алюминатных
растворах без предварительного отделения алюминия, нефелино-
вых концентратах, снеках и шламах [382]. Результаты согласуются
с данными гравиметрического анализа и эмиссионной фотометрии
пламени. Сумму натрия и калия определяли также в водах [445,
768, 1070—1072], сумму обменных натрия и калия определяли
в почвах [3, 444, 445], стеклах [749].
Инверсионные методы. Высокая электроотрицательность натрия
делает невозможным применение твердых электродов, поэтому ин-
версионные методы для определения натрия используются редко.
Однако следует заметить, что первые попытки определения натрия
по току растворения амальгамы появились на заре инверсионных
методов [189, 857].
Принципиальная возможность определения щелочных металлов
методом инверсионной вольтамперометрии основана на восстановле-
нии в неводной среде до металла и сравнительно высокой раствори-
мости в ртути [460]. Анодные пики всех металлов, кроме лития, сов-
падают. Разработаны методики определения суммы натрия и калия
(10~5—10-в%) в HNO3, HF, а также методики определения до
10~7—10~8 г этих металлов в пленках SiO2.
Анодный пик натрия при —1,68 В (относительно Hg-анода)
наблюдали при растворении амальгамы, полученной электролизом
при —2,2 В на фоне 0,025 М раствора (C4He)4NJ в ДМФА [173].
Потенциалы анодных пиков других металлов имеют весьма близкие
93
величины К —1,67, Rb —1,60 В, Cs —1,58 В, Li —1,94 В [173].
Аналогичные результаты получены в работе [174].
Амперометрическое титрование. Описана методика биамперомет-
рического титрования натрия с двумя поляризованными электродами
по реакции осаждения его в виде оксалата в среде изопропинола
[64]. В качестве индикаторных электродов использовали медный
амальгамированный вращающийся микрокатод и медный перфори-
рованный анод с большой поверхностью. Четкие перегибы на кри-
вых титрования наблюдали при потенциале 1 В. Возникновение тока
в цепи обусловлено переходом ионов меди с анода в раствор и вос-
становлением их на катоде, перегибы на кривых титрования возни-
кают за счет изменения электропроводности раствора вследствие
осаждения определяемых ионов.
Раствор определяемого вещества (0,1—0,01 М) готовят в колбе вместимостью
50 мл; В стакан для титрования помещают 1—5 мл этого раствора, 15—10 мл
изопропанола, вводят электроды, устанавливают напряжение 1 В и титруют
порциями по 0,1—0,2 мл 0,05 М раствором щавелевой кислоты в изопропаноле.
Эта методика пригодна для титрования смесей солей натрия и
калия при соотношении 10 : 1— 1 : 10, а также смесей солей натрия,
калия и лития при соотношениях 10 : 10 : 1, 5:5:1, 1:1:2,
1:1 :.5 и 1:1:10. При титровании смесей первым титруется натрий.
Погрешность при титровании натрия не превышает 1 % для бинарных
смесей и 2 % — для тройных смесей.
ДРУГИЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Кондуктометрическое титрование. Описан метод кондуктометри-
ческого титрования, пригодный для определения натрия и калия
при совместном присутствии, основанный на переведении их хлори-
дов в гидроксиды с помощью Ag2O и последующем титровании спир-
товым раствором НС104 [1027].
К 5 мл прибавляют влажный Ag2O в количестве (в г), равном числу милли-
грамм-эквивалентов натрия и калия, гстряхивают 10 мин, фильтруют, промы-
вают 5 мл воды, 5 мл этанола и фильтрат разбавляют до 250 мл этанолом. 50 мл
полученного раствора переносят в ячейку и титруют 0,1 М раствором НСЮ4
в этаноле.
На кривой титрования наблюдается два скачка. Первый скачок
соответствует титрованию NaOH, второй — КОН. При этом, хотя
сила обоих оснований в этаноле одинакова, NaOH титруется как сла-
бое основание, а КОН как сильное, что обусловлено одновременным
протеканием реакции осаждения и кислотно-основной, вследствие
чего электропроводность быстро уменьшается. Для уменьшения
ошибок следует учитывать, что NaOH плохо вымывается спиртом,
а при долгом промывании на фильтре может протекать реакция
с углекислым газом воздуха. Поэтому рекомендуется в одной пор-
ции пробы определить общее содержание NaOH и КОН в фильтрате
0,1 М раствором НС1О4 по метиловому оранжевому. Во второй части
пробы кондуктометрически определяют только калий, а содержание
натрия затем находят по разности. При соотношении концентраций
94
натрий : калий от 1 : 5 до 5 : 1 максимальная погрешность ±2%
отн., при соотношении от 1 : 10 до 10 : 1 ±5% отн.
Кондуктометрически можно титровать NaJ (или KJ) в ацетоне
или ацетонитриле раствором хлорида лития в этаноле или ацетоне
[801]. Из-за близкой растворимости хлоридов натрия и калия (в аце-
тоне 4-10“5 и 8-10-5 М соответственно, в ацетонитриле 1,5 -10~4
и 2,4-10-3 М) определить натрий в присутствии калия невозможно.
Титрованию не мешает кальций, мешает барий.
Разработана методика кондуктометрического титрования
0,046—2,3 мг натрия, основанная на осаждении в виде цинкуранил-
ацетата, растворении осадка в воде и титровании раствором НС1
[671]. Преимущество этого способа перед другими заключается в том,
что соотношение натрий : ацетат равно 1 : 9, поскольку в основе
титрования лежит реакция 9 СН3СОО“ + 9 Н+ — 9 СН3СООН.
К 1 мл пробы прибавляют 3 мл раствора реагента (10 г кристаллического
цинкуранилацетата и 6 г 30%-ноп СН3СООН в 50 мл воды или 30 г препарата
и 3 г 30%-ной СН3СООН в 50 мл воды), оставляют на 2—3 ч (при малых содер-
жаниях на 4—6 ч), центрифугируют 15 мин, промывают осадок 4—5 раз пор-
циями по Змл 96%-ного этанола, насыщенного реагентом, 2—3 раза по 3 мл
эфира. Растворяют в 50 мл воды и титруют.
Предложен микрокондуктометрический метод титрования и*
• 10-3 М натрия в виде натрийцинкуранилацетата 0,2 М раствором
НС1 [18]. Осадок получают и растворяют в специальном микрососуде,
2—4 мкл полученного раствора переносят в капиллярную ячейку
с вплавленным Pt-электродом и титруют. В бидистилляте хорошо
растворяется осадок, несколько раз промытый спиртом и эфиром,
предварительно насыщенным растворяемой солью, и высушенный на
воздухе в течение 1 —2 ч. Ошибка титрования десятых и сотых долей
микрограмма натрия ~3% отн. (п = 20).
Методом обратного кондуктометрического титрования избытка
(NH4)3AlFij раствором NaCl определяли натрий в природных водах
после отделения сопутствующих катионов в виде карбонатов (карбо-
нат аммония) в спиртовой среде [355].
Кулонометрия. Микрограммовые количества натрия можно опре-
делить кулонометрически при восстановлении на ртутном катоде
в 0,1 М (C2H5)4NC1O4 в ацетонитриле [683].
Раствор хлорида или нитрата натрия выпаривают с несколькими каплями
60%-ной НСЮ4 при 200—250° С на масляной бане. Сухой остаток растворяют
в 10 мл сухого очищенного ацетонитрила. Часть раствора (~1 мл) переносят
в электролизер, прибавляют 5 мл фона, пропускают 5 мин азот и проводят вос-
становление иона натрия до амальгамы (на процесс восстановления требуется
5 мин), используя 3—5 мл ртути, при —2,3 В (относительно внешнего нас. к. э.).
Содержание натрия можно также определить по току окисления образовавшейся
амальгамы при —0,1 В в течение 5—7 мин.
При контролируемом потенциале ртутного катода —2,0 В опре-
деляют натрий на фоне (C2H5)4NC1O4 в ацетонитриле [621]. Процесс
восстановления иона натрия осложняется каталитическим током,
вызываемым присутствием в растворе следов кислорода. Метод при-
годен для определения до 2 мкг-экв натрия с погрешностью 1 %.
Продолжительность определения 23 мин.
95
Для кулонометрического определения натрия (а также Li, К,
Sr и Ва) предложена двойная ячейка, в которой верхняя и нижняя
части разделены слоем ртути, налитой на катионообменную мембрану
1323]. В верхней части ячейки происходит восстановление иона ме-
талла с образованием амальгамы, в нижней — растворение амаль-
гамы. Содержание определяемого металла находят, интегрируя ток
окисления! амальгамы при контролируемом потенциале.
Кулонометрическое титрование. Определение металлического на-
трия в алюмогидриде натрия проводили с помощью 1-бром-н-бутана
на лабораторной полуавтоматической установке в ячейке с серебря-
ными электродами [220]. Конечную точку титрования устанавливали
амперометрически с помощью двух поляризованных серебряных
электродов (20 мкА). Предел обнаружения натрия 0,02%. Относи-
тельное стандартное отклонение не превышает 0,08 при содержании
натрия от 0,5 до 1,5%.
Навеску алюмогидрида натрия 0,1—0,2 г отбирают в герметизированной
камере в токе азота и помещают в коническую колбу вместимостью 100 мл, за-
полненную 20—30 мл 1-бром-н-бутана. В течение 10 мин проводят реакцию при
50—80° С при непрерывном перемешивании магнитной мешалкой. По охлажде-
нии прибавляют 20—50 мл 2,4 М раствора HNO3, встряхивают 3 мин и остав-
ляют для расслаивания. Электролитическую ячейку заполняют 35 мл 0,1 М
раствора HNO3 в 10%-ной СН3СООН, прибавляют 10 капель 0,5%-ного раство-
ра желатина и проводят генерацию ионов серебра, затем вводят 0,1—1 мл вод-
ного экстракта и вновь проводят генерацию ионов серебра.
Электродиализ. Видоизмененный способ Паппа применен для оп-
ределения натрия в криолите [1060]. Метод пригоден при содержании
кальция <^1%.
Навеску пробы 0,1 г дважды выпаривают досуха' с H2SO4, остаток прокали-
вают при температуре ~700° С, суспендируют в воде (натрий переходит в рас-
твор), помечают в анодное отделение электродиализатора Паппа, проводят элект-
ролиз при постоянном токе (110 В). Католит несколько раз отбирают, объединя-
ют и, предохраняя от атмосферного углекислого газа, титруют 0,1 М раствором
НС1 через 1,5 ч.
Глава VIII
СПЕКТРАЛЬНЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
НАТРИЯ
Спектральные методы — наиболее селективные и чувствительные
из имеющихся методов определения натрия. Их можно классифици-
ровать по способу подготовки пробы к анализу (прямые и химико-
спектральные), по способу регистрации сигнала (спектрографические
и фотоэлектрические), по способу возбуждения (пламенные, элек-
тротермические, флуоресцентные, рентгеноспектральные и др.) Спек-
трографические прямые и химико-спектральные методы применяют
для обнаружения натрия при групповом определении примесей, на-
пример в веществах особой чистоты. По пределам обнаружения, экс-
86
прессности, высокой воспроизводимости и простоте аппаратуры особое
место занимает метод атомно-эмиссионной пламенной спектро-
метрии, он находит самое широкое применение при анализе практи-
чески любых объектов.
Примеры определения натрия спектральными методами в раз-
нообразных объектах приведены в главе X «Определение натрия
в объектах».
Соединения натрия атомизируются даже в низкотемпературных
пламенах вследствие невысокой температуры кипения. В спектре
натрия наиболее яркими являются резонансные дублеты, по которым
проводят анализ (табл. 41).
Таблица 41
Аналитические линии натрия
Переход Длина вол- ны, нм Энергия воз- буждения, эВ
589,6 2,10
589,0 2,11
n2SVs-(n + l№/2 пад1/2-(п + 1)2ро,2 330,3 330,2 3,75
п2р°4,, г/. — '’8£>3/2, '/г и2?®, — n?D,. .. 3/г. Чг 818,3 819,5 3,61
Кажущийся потенциал ионизации натрия в интервале температур
5000—7000 К больше на 0,3—0,4 эВ истинного потенциала иониза-
ции [640]. Использование высокотемпературных источников не-
желательно, поскольку ионные линии натрия находятся в далекой
УФ-области спектра, для их регистрации необходима специальная
аппаратура из-за поглощения света воздухом. Линии, соответствую-
щие переходам с 5-уровней на P-уровень, расположены в ИК-обла-
сти (1113,1—1140,4 нм), и их использование требует также специаль-
ной техники.
СПЕКТРОГРАФИЧЕСКИЕ МЕТОДЫ
Количественно натрий определяют по дублетам 589,6 и 589,0 нм
или 330,3 и 330,2 нм, используя фотографические пластинки «пан-
хром» в первом случае и «спектрографические» (тип I или II) — во
втором. При определении натрия на фоне группы элементов исполь-
зуют УФ-область спектра. Предел обнаружения натрия в видимой
области (1—3)-10-4%, в УФ-области — (1—3)-10-2%.
Основными источниками света являются электрические дуги,
искра, высокочастотный разряд, разряд в полом катоде, плазмотроны;
4 В. М. Иванов и др.
97
их применяют при групповом определении элементов. При опреде-
лении натрия в ряде объектов археологии, медицины, криминалис-
тики, минералогии, металлургии используют лазерный локальный
микроспектральный анализ [984].
При спектрографическом определении натрия пробу разбавляют
буфером, в состав которого часто входит элемент, являющийся внут-
ренним стандартом: кобальт [512, 715, 1105], кадмий [560], медь [535],
свинец [511], алюминий [211, 1289], литий [968], стронций [357],
кремний [387], железо [391], цезий или рубидий [65]. Иногда исполь-
зуют фон вблизи натрия [121, 1289]. Эффективность влияния состава
буфера показана на примере GeO2, Li2B4O7, Li2CO3, SiO2 и SrC03
[701]. При спектрографическом определении натрия в присутствии
калия используют линии, расположенные в различных областях
спектра: Na — 330,23 нм и К — 404,41 нм [148]. Градуировочный
график строят в координатах (Sns—kSK)—сиа/ск, где к — коэффи-
циент, зависящий от свойств фотоэмульсии пластинки. Погрешность
определения натрия в большой степени связана с близкими физико-
химическими свойствами солей натрия и калия.
Предел обнаружения натрия зависит от природы объекта, поло-
жения аналитической линии, типа спектрографа и может достигать
при прямом определении 10~4% [224]. Проецирование на щель спект-
рографа прикатодного слоя дуги постоянного тока позволяет значи-
тельно снизить предел обнаружения натрия [492, 590, 591]. При ис-
пользовании аэрозольно-искрового метода анализа для определения
до 5-10-5% натрия в алюминиевых сплавах относительная погреш-
ность составляла 10% [121]. При высоком коэффициенте концентри-
рования предел обнаружения натрия достигает 10-7—10-6% и в ряде
определений ограничен содержанием натрия в контрольных опы-
тах [360, 492].
Электрические источники света. В рядах летучести Русанова соли
натрия расположены в группе летучих соединений [424]. Работ по
изучению влияния других элементов на интенсивность спектраль-
ных линий натрия в дугах и искре мало. В основном решается обрат-
ная задача, имеющая большое теоретическое и прикладное значение.
Описан эффект прикатодного усиления интенсивности спектраль-
ных линий элементов с низкими потенциалами ионизации [944].
Использование прикатодной области плазмы дуги постоянного тока
позволяет значительно снизить предел обнаружения натрия. Так,
при определении натрия в материалах на основе урана (пробу поме-
щали в анод) он равен 5-10-6% [590]. Такой же метод используют
при анализе фосфатов [591]. Дуговой разряд стабилизируют с по-
мощью КОН [43] или К2СО3 [132]. В последней работе имеются све-
дения о влиянии количества К2СОз на интенсивность линий натрия.
Изучено влияние хлоридов, фторидов и иодидов на определение нат-
рия в А12О3 [1189].
Изучено распределение интенсивности спектральных линий нат-
рия по радиусу дугового разряда для различных соединений натрия,
и найдена корреляция между энергией связи галогенидов натрия
и изменением числа свободных атомов в более холодных частях стол-
98
<5а дуги [107, 148, 416]. На примере анализа среднего фосфата каль-
ция в дуге постоянного тока изучено влияние параметров дугового
разряда — концентрации электронов, времени пребывания вещества-
в разряде, градиента напряженности электрического поля — на от-
ношение интенсивности спектральной линии примеси к линии осно-
вы [591]. Среднее время пребывания атомов натрия в свободно горя-
щей дуге постоянного и переменного тока при 10 А соответственно
равно 1,7-10-3 и 4,1 -10~3 с [286].
При изучении распределения интенсивности спектральных линий
натрия по горизонтальной оси факела в методе искрового анализа
аэрозолей было показано [317], что максимум интенсивности прихо-
дится на центральную часть факела. При использовании медного
.электрода раствор соединения натрия наносят на торец этого элект-
рода («метод медного электрода») [118].
При анализе глин, гранитоидов и других силикатных пород с
различным содержанием основных компонентов: кремния, алюминия,
железа, кальция и магния и содержанием натрия от 0,5 до несколь-
ких десятков процентов установлено, что кинетика испарения нат-
рия из пробы в дуге переменного тока 5 А, положение градуировоч-
ных графиков и точность определения не зависят от валового состава
пробы [89]. Не обнаружено также взаимного влияния натрия и ка-
лия. При относительно малом содержании щелочных металлов в со-
став буфера вводят карбонат лития, оксид меди и угольный поро-
шок. При определении натрия в силикатах с содержанием щелочных
металлов свыше 8% применяют метод ширины спектральных линий.
При анализе микропроб применяют высокоточную импульсную
аргоновую дугу, предел обнаружения достигает 0,5—1 нг натрия
[1196]. В атмосфере азота предел обнаружения натрия понижается
на полтора порядка [954]. Приведены полные сведения об определе-
нии натрия методом локального лазерного микроспектрального ана-
лиза в различных объектах с использованием двухступенчатой схе-
мы: лазерного пробоотбора с последующим возбуждением спектров
в электрическом источнике; предел обнаружения натрия 10-6 г
[984].
Прямой спектрографический метод применяют для определения
натрия в минералах и горных породах [810], глиноземе [349], осадоч-
ных породах [201], глинах [89, 208, 217, 472], силикатах [91], слюде
[391], алюмосиликатах [387], природных фосфатах [591], водах [523,
756, 850, 917], атмосферных осадках [960], при анализе особо чистых
веществ [194, 360]. Очень часто метод применяют для определения
натрия в элементах (табл. 42).
Метод применяют также для определения натрия в соединениях
лития [610], солях калия [356], К2СО3 [484], КОН [211, 212, 483], ВеО
[359, 403, 1069], MgO [372, 376], CaF2 [375], солях стронция [357],
сплавах алюминия [121, 185], СеО2 [665], бориде титана [132], бабби-
тах [177, 389, 511], ванадиевой кислоте [1091], солях и оксиде висму-
та [225], WO3 [714], U3O8 [877], материалах на основе урана [590],
соединениях плутония [674], МпО2 [968], СоО [268], CoSO4 и NiSO4
(кристаллогидраты) [535], а также в золе угля [861], смазочных
4#
99
Таблица 42
Применение прямого спектрографического метода
для определения натрия в элементах
Эле- мент Источник возбуждения спектра Аналитические линии натрия, нм Определяемые коли- чества, % (cmjn, %) Литера- тура
Li Дуга, 6—7 А [610]
Rb Дуга переменного тока 589,0-589,6 1-Ю-3 [65]
Cs То же 589,0—589,6 sr = 0,06 [65]
Au — 589,0—589,6 (1-Ю-3) [131]
Be Дуга постоянного тока 589,0—589,6 5.10-3—0,3 [403]
Ga То же [1064]
Ti Дуга переменного тока (2—5)-10-2 [118]
Zr То же 1-Ю-3 [450]
Pb » 2-Ю-5 [327]
588,9 3,8-10-4—2,1-10-2 [258]
330,22 2-10-3—0,1 [556]
[69]
Sn Дуга постоянного тока [557]
Sb То же 5-10-3—1 [449]
Bi — [530]
Mo Дуга постоянного тока 588,99 4.10-4—10-2 [715]
Se — (1 мкг) [1047]
Дуга переменного тока [122]
Те То же [55, 122,
560]
U Дуга постоянного тока 330,299 sr = 0,065 [1043]
Pu То же (5-Ю-3) [674]
маслах [799J, цементе [792], катализаторах [47], золотом электролите
[43] и во флюсах [512].
Определение натрия в рубидии и цезии [65]' Предварительно ме-
таллы переводят в раствор в виде хлоридов. В качестве аналитиче-
ских используют линии Na 588,995 нм—Rb 615,962 нм; Na
589,592 нм—Rb 615,962 нм; Na 588,995 нм—Rb 620,631 нм; Na
589,592 нм—Rb 620,631 нм; Na 588,995 нм—Cs 603,409 нм; Na
589,592 нм—Cs 603,409 нм. Эти линии свободны от наложения ли-
ний калия, лития и других элементов. Спектр возбуждают в активи-
зированной дуге переменного тока, раствор вводят в пламя дуги при
помощи фульгуратора. Сила тока 2 А, напряжение 120 В, пластинки
панхром, ширина щели 0,014 мм. При' определении натрия в руби-
дии градуировочные графики строят в координатах AN—1g с; при
определении в цезии — в координатах 1g (Ц/Ц)—1g с. Нижняя гра-
ница определяемых содержаний натрия 1 • 10~3%, погрешность опре-
деления <8%; при увеличении экспозиции предел обнаружения
натрия можно значительно снизить.
100
Определение натрия в золоте [131]. Метод применен для определе-
ния натрия в золоте после его регенерации химическим путем. Его
можно использовать также для контроля чистоты золота при его до-
быче и очистке. Предел обнаружения натрия 1-10~3% при исполь-
зовании аналитических линий Na 589,592 нм — фони Na 588,995 нм —
фон. Кроме натрия, можно определить данным методом также Си,
Ag, Pt, Be, Mn, Ca, Mg, Cd, Ba, Si, Fe, Cr, Ni, Al, Sb, Bi, Co, Pb,
Mo, К и Sn. Спектры возбуждают в руге переменного тока силой
6 А и снимают одновременно спектрографами ИСП-22 и ИСП-51,
оптические оси которых располагают под прямым углом друг к
ДРУГУ- Спектр для натрия регистрируют на пластинку панхром
(65 ед. ГОСТ), линию натрия ослабляют пленочным фильтром про-
пускаемостью 20%, укрепленным в пазу на выдвижной бленде
в кассетной части спектрографа ИСП-51.
Из проб и эталонного материала готовят 10%-ный раствор золота. Для это"
го порошок, корольки или губчатое золото растворяют в смеси (3 : 1) НС1 и HNO3
и добавляют равный объем бидистиллята. Спектрально-чистое золото для эта-
лонов получают экстракцией его диэтиловым эфиром из среды 1 М НС1 с после-
дующим восстановлением до металлического сернистым газом. Примеси опреде-
ляемых элементов добавляют в эталонные растворы в виде водного раствора нит-
ратов, натрий и кремний вводят в виде Na2SiO3. Электродами служат угольные
стержни длиной 30 мм с плоским торцом.
На плоскую поверхность угля наносят каплю смеси (3 : 1) НС1 и HNO3
н после впитывания ее обжигают электрод в дуге постоянного тока силой 20—
30 А в течение 30—45 с. Торцы электродов покрывают полистироловым лаком
(3,3 г полистирола и 100 мл бензола), погружая их в раствор полистирола на
10 с, а затем просушивая на воздухе в течение 1 мин. Затем на поверхность элект-
родов наносят пипеткой 0,04 мл анализируемого раствора. Электроды с раство-
ром погружают в гнезда латунного держателя, нагретого до 80—90° С, а через
15 мин переносят в гнезда другого держателя, нагретого до 160—170° С. Одно-
временно под действием горячей смеси (3 : 1) ПС1 и HNO3 разрушается полисти-
роловая пленка, при этом соль золота проникает в глубь электрода и закрепля-
ется там в поверхностном слое толщиной 0,7—1 мм. Толщину слоя соли можно
изменять, регулируя температуру прокаливания.
Расстояние между электродами 1,5 мм. Соль золота наносят на оба электро-
да. Экспозиция 60 с. Градуировочный график строят в интервале концентраций
натрия 10-3—2-10~2% в координатах —1g с. Спектры эталонов и проб фото-
графируют по 3—4 раза.
Определение натрия в бериллии и оксиде бериллия [195]. Натрий
определяют в пробах оксида, возбуждая спектры в дуге переменного
тока. При использовании линии натрия 588,995 нм предел обнаруже-
ния составляет 2-1(Г3%. В отдельной пробе можно определить Mg,
Са, Ba, Al, Ti, V, Cr, Mo, Mn, Fe, Со, Ni, Cu, Ag, Zn, Cd, Sn, Pb, Sb,
Bi, Те, Ga, In, TI, Ge, Au, Pt, Li, B, Si, Sm с пределом обнаруже-
ния^ l-10~7 %.
Навеску 0,2 г металлического бериллия (или 0,56 г ВеО) растворяют в квар-
цевой чашке в 10 мл НС1 (1 : 1) с добавлением 1 мл конц. HNO3. Упаривая раст-
вор до сиропообразного состояния, после трехкратной обработки порциями конц.
HNO3 (8, 3 и 3 мл) получают Be(NO3)2. Осаждают Ве(ОН)2 в виде кашицы конц.
аммиаком, высушивают и прокаливают при 300° С, а затем при 500° С. Получен-
ную стекловидную массу измельчают кварцевой палочкой-пестиком и помещают
в угольный электрод в виде графитового стержня с отверстием на торце диамет-
ром 4 мм и глубиной 6 мм.
101
Определение натрия в титане и оксиде титана (IV) [196]. Метод
позволяет определять натрий из отдельной навески с пределом обна-
ружения 1-1СГ3% после перевода титана в оксид. Спектры фотогра-
фируют на спектрографе ИСП-51 в дуге переменного тока; сила тока
5 А, экспозиция 1 мин. Используют угольные электроды; нижний
электрод — стержень с отверстием диаметром 4 мм, глубиной 4 мм,
аналитическая линия натрия 588,995 нм. Градуировочный график
строят в интервале концентраций натрия 5-10-3—1-10~2%.
Предварительно титан переводят в оксид титана(1У) одним из
способов: непосредственным сжиганием пробы металла (0,25 г) в виде
порошка, опилок или очень мелкой стружки в платиновом тигле
при постепенном повышении температуры в электропечи до 1000° С
и прокаливанием в течение нескольких часов до полного окисления.
При химическом способе растворяют 0,25 г металла в конц. НС1,
выпаривают с HNO3 для перевода хлоридов в нитраты, которые про-
каливают в течение 30 мин при 600° С. Порошок TiO2 смешивают
с носителем — AgCl в соотношении 4 : 1 по массе.
В отдельной пробе в дуге постоянного тока можно определить Mg,
Са, Ba, Al, V, Сг, Mo, Мп, Fe, Со, Ni,Cu, Zn,Cd, In, Sn, Pb, Sb и Bic
пределом обнаружения ^>l-10~4% и относительной погрешностью
-20%.
Определение натрия в цирконии [450]. Метод позволяет определять
натрий из отдельной навески по линии 588,995 нм с пределом обна-
ружения 1-Ю-3 %. Спектры проб и эталонов возбуждают (спектро-
граф ИСП-51) в дуге переменного тока; сила тока 5 А, экспозиция
1 мин. Нижний электрод — угольный стержень диаметром 4 мм,
глубиной 4 мм. Предварительно металлический цирконий перево-
дят в ZrO2 прокаливанием в платиновой посуде. Если проба посту-
пает в виде порошка металла, то ее сжигают по частям. Первую пор-
цию помещают в холодную печь и окисляют, постепенно повышая
температуру до 1000° С. Полученный оксид ZrO2 смешивают со сле-
дующими порциями порошкообразного металла и прокаливают эти
смеси. Эталоны готовят на основе чистого Zr02 исходя из сульфата
циркония спектральной чистоты, полученного многократным пере-
осаждением из насыщенного водного раствора при добавлении сер-
ной кислоты. Пробы и эталоны Zr02 смешивают с носителем AgCl
в соотношении 4 : 1 по массе.
В отдельной порции можно определить Mg, Ba, Al, Ti, V, Сг, Мо,
Мп, Fe, Со, Ni, Си, Cd, In, Sn, Pb, Bi, Li, Si, Be, Ca с пределом обна-
ружения \>l-10-6%.
Определение натрия в висмуте [227]. Метод пригоден для опреде-
ления натрия в висмуте и его соединениях после перевода в Bi2O3.
Спектры проб и эталонов фотографируют на спектрографе ИСП-51.
Электроды угольные; условия съемки: щель 10 мк, источник — дуга
переменного тока, сила тока 5 А, экспозиция 40 с, фотопластинки
панхром. Предел обнаружения натрия по линии 588,995 нм состав-
ляет 5-10~4%. В отдельной пробе можно определить Mg, Са, Ba, А1,
Ti, V, Сг,' Мо, Мп, Fe, Со, Ni, Pt, Си, Ag, Aii, Zn, Cd, In, Tl, Sn, Pb,
Sb, Те с пределом обнаружения J>2-10~6%.
102
Навеску висмута 3—5 г растворяют в кварцевой чашке в 10—15 мл 7 М
HNO3, выпаривают досуха и прокаливают 30 мин при 550° С. Прокаленный ок-
сид Bi2O3 смешивают со спектрально-чистым ВеО в соотношении 5 : 1 и помеща-
ют в угольный электрод в виде угольного стержня с отверстием диаметром
4,5 мм и глубиной 6 мм.
Определение натрия в теллуре 1122]. Метод применен для опре-
деления 5-10-4—2-10~2% натрия в техническом теллуре, предел об-
наружения натрия 3-10'4%. Эталоны готовят механическим смеши-
ванием мелкодисперсных порошков чистых металлов с теллуром по-
вышенной чистоты. Вначале примеси вводят в концентрации 0,1 —
1%, затем разбавляют в 10 раз чистым теллуром. После тщательного
перемешивания и растирания стандартную смесь порошков приме-
няют в дальнейшем для приготовления эталонов. Пробы и эталоны
теллура смешивают с графитовым порошком в соотношении 5 : 1
и помещают в тонкостенные графитовые электроды диаметром 3 мм
и глубиной 2 мм. Помимо натрия, метод позволяет определять (с пре-
делом обнаружения 10-6—10-3%) Ag, Rh, Mg, Cu, Pt, Au, Si, Al,
Fe, Ni, Bi, Sn, Sb, Pb, Ir, Ru, Са, Ba и Se. При определении 5-
•10-3—2-1СГ2% натрия выбрана аналитическая линия натрия
330,23 нм, линия сравнения теллура 317,51 нм; при определении 5-
•1СГ4—2-10-2% натрия интенсивность линии 588,995 нм измеряют
относительно фона. Спектры фотографируют на спектрографе ИСП-28
с трехлинзовой системой освещения, источник возбуждения — дуга
переменного тока, сила тока 2 А, экспозиция 30 с. Используют пан-
хроматические пластинки или негативную фотопленку.
Если содержание палладия в пробах <^0,01 %, натрий определяют
вместе с другими примесями по линии 330,23 нм. Влияние палладия,
имеющего линию 330,21 нм, исключают с помощью расчетной линей-
ки. Предварительно определяют содержание палладия по линии
342,12 нм и вносят поправку на его содержание. При содержании
палладия >0,01% для определения натрия используют линию
588,995 нм и отдельную навеску пробы.
Всю систему, включая щель спектрографа и штатив, помещают
в бокс из плексигласа. Каждый эталон и пробу готовят в четырех
экземплярах.
Определение натрия в солях калия [356]. Метод применен для оп-
ределения 10~3—10% натрия в растворах КС1, KNO3, K2SO4. Ана-
лиз проводят в дуге переменного тока с напряжением 180—200 В,
силой тока 8—9 А, применяя активизатор Свентицкого. Спектры сни-
мают на пластинки изоорто или изопанхром, ширина щели 0,0015—
0,01 мм, экспозиция 8—15 с. При определении 10-3—10-1% натрия
аналитическая пара линий Na 588,995 нм—К 580,10 нм, относитель-
ная погрешность 14%. При определении 10-1—10% натрия измеряют
почернение линий натрия 330,23 нм, калия 344,64 нм и почернение
фона вблизи этих линий; относительная погрешность 5%.
Определение натрия в солях стронция [357]. Метод применен для
определения 10-3—10-1% натрия в SrCl2-6H2O и 10-2—10"г% нат-
рия в Sr(NOs)2. Спектры снимают при светосиле 1 : 10 в активизи-
рованной дуге переменного тока; напряжение 200 В, сила тока 6—
103
7 А. Нижний электрод имеет цилиндрический канал (диаметр 3 мм,
глубина 6—7 мм), куда после обжига электродов в течение 1 мин
вносят пипеткой несколько капель испытуемого или эталонного
раствора. Каждую пару электродов используют для съемки одного
спектра. Дуга находится на расстоянии 20 см от щели спектрографа.
Щель при анализе открывают до зажигания дуги. Используют спо-
соб трех эталонов. Аналитическая пара линий Na 588,995 нм—Sr
532,98 нм.
Определение натрия в оксиде кобальта(11) [268]. Метод применен
для определения 5-10~:|—10-1% натрия. Спектр возбуждают дугой
переменного тока, сила тока 10 А. Спектр регистрируют на спектро-
графе КСА-1 со стеклянной оптикой, трехлинзовым конденсором
и трехступенчатым ослабителем. Для снижения эффекта фракциони-
рования тонкий слой анализируемого оксида кобальта наносят на
медный электрод (марки М-0 или М-1). Спектр фотографируют на
перемещающуюся пластинку. Верхний медный электрод диамет-
ром 5 мм заточен на усеченный конус, нижний электрод диаметром
3 мм заточен на плоскость и имеет рифленую поверхность. Оксид ко-
бальта втирают тонким слоем на торец рифленого электрода так, что-
бы бороздки насечки были заполнены пробой, а верхние кромки сво-
бодны от нее.
Стандартные образцы порошковых проб готовят синтетически из
оксидов определяемых элементов (Ni, Fe, Мп), от кальция и натрия
отмывают декантированием горячей водой (10—15 раз). Остаточные
содержания определяют методом добавок. Градуировочный график
строят в координатах AN—1g с, аналитическая пара линий Na
589,592 нм-Со 599,188 нм.
Широко применяют химико-спектральные методы после концент-
рирования микрокомпонента или отделения основы. Химические ос-
новы методов весьма разнообразны, равно как и способы отделения.
Используют физические и химические методы концентрирования
примесей, в том числе и натрия: методы фракционной дистилляции
[161, 517, 665], отделение основы осаждением [195] или экстракцией
[492]. Более полные сведения о применении химико-спектрального
анализа для определения натрия в числе других элементов приведе-
ны в обзорах [195, 196]. В большинстве случаев используют резо-
нансный дублет 589,6—589,0 нм; дублет 330,23—330,30 нм использу-
ют редко [130, 405, 493]. Метод применим к анализу органических
веществ после постепенного упаривания с угольным порошком [536],
ароматических кремнийорганических соединений, диэтиламина и
тетратиурамдисульфида после упаривания с сульфатом стронция
(предел обнаружения натрия 3-10-6 %) [386]. Некоторые примеры
применения химико-спектральных методов приведены в табл. 43.
Определение натрия в кадмии [492]. Метод применен для опреде-
ления 3-10~5—3-10-3% натрия в кадмии высокой чистоты, предел
обнаружения натрия 1 • 10~6 % при использовании аналитической ли-
нии 589,50 нм. Предварительно основу отделяют экстракцией хло-
роформом пиридин-иодидного комплекса. Вместе с натрием концент-
рируются и могут быть определены Al, Со, Са, Fe, Bi, In, Ga, Mg,
104
Таблица 43
Применение химико-спектральных методов для определения натрия
в элементах и их соединениях
Объект Метод концентрирования натрия Источник возбуж- дения Определяемые ко- личества, % (сш|п, %) Лите- ратура
Ве, ВеО Осаждение основного ацетата бериллия Дуга переменно- го тока (1 КГ4) | [195]
В Отгонка борнометило- вого эфира То же (МО-4), sr<0,3 [405]
Cd Отделение кадмия экстракцией Дуга постоянного тока (1-10-’) [492]
Ti, TiO2 Отгонка с носителем AgCl Дуга переменно- го тока (1-10-4) [196]
Zr Хлорирование основы с помощью AgCl То же (1-10-4) [450]
ZrO2 Испарение в вакууме Конденсирован- ная искра З.Ю-4—1.10-2, sr = 0,20 [163]
CeO2 Фракционная дистил- ляция Дуга постоянного тока 5-10-5—5-10-2 [665]
ThO2 Вакуумное испарение примесей Конденсирован- ная искра (1-Ю-3) [161]
NH3, AsHe, SbH8 Отгонка основы, уголь- ный порошок Дуга постоянного тока (2-10-e—l-10-4) [130]
Bi и его соедине- ния Осаждение BiJ3 (3-10-5) [227, 314]
Pb Концентрирование при- месей на РЬ(ОН)2 (3-10-5) [494]
Соедине- ния свинца Осаждение PbSO4 Растворение серы в па- рафине Дуга постоянного тока sr = 0,15 -т- 0,40 IO”3 [60] [155]
Те Осаждение ТеО2 (5-10-5) [493]
Мо Осаждение а-бензоин- оксимом Дуга переменно- го тока (2,8—3,2)-10-з (4-10-4) [197]
и Хроматографическое отделение (3—5). IO"1 [647]
Ри Испарение в вакууме, концентрирование на медном электроде [160]
Ni, Sb, Pb, Sn и Zn. Спектр возбуждают в дуге постоянного тока си-
лой 12 А и фотографируют на спектрографе ДФС-8 с дифракционной
решеткой 600 штр/мм. Щель спектрографа освещают через кварце-
вую линзу F = 75 мм, проектируя изображение электродов. Фото-
графируют область дугового промежутка у катода. Применяют фото-
пластинки ЭС-СП и панхром. Эталоны готовят введением растворов
определяемых элементов в навеску спектрально-чистого угольного
порошка и 5% CdO с последующим прокаливанием при 400—450° С.
105
Навеску кадмия 5 г растворяют в 30 мл HJ, упаривают раствор досуха,
приливают 50 мл воды, 1 мл 0,025 М раствора аммонийной соли ЭДТА и перено-
сят в делительную воронку. Вводят смесь, состоящую из 20 мл пиридина и 60 мл
хлороформа и экстрагируют 1—2 мин. Органический слой отделяют и повторно
экстрагируют 5 мл смеси пиридина с хлороформом. Раствор переносят в чашку
для выпаривания, добавляют 1—2 мл HNO3 пл. 1,40 г/см8, 50 мг угольного по-
рошка и выпаривают досуха. Полученный осадок прокаливают при 400—450° С.
Осадок в чашке перемешивают и по 20 мг переносят в кратер угольных электро-
дов глубиной и диаметром 4 мм с толщиной стенок 0,5 мм. Верхний конус зата-
чивают на усеченный конус.
Определение натрия в боре [405]. Метод основан на удалении бора
в виде борнометилового эфира с. последующим спектральным опре-
делением в растворе азотной кислоты. Используют аналитические
линии натрия 330,30 или 330,23 нм, предел обнаружения натрия
составляет 1-10'4%, относительное стандартное отклонение равно
0,30. Для приготовления эталонных растворов берут растворимые
в воде соли квалификации х. ч. Головной эталон содержит все опре-
деляемые примеси в концентрации 1 -10“2% каждого элемента в от-
дельности. Последовательным разбавлением головного эталона во-
дой готовят серию эталонов, отличающихся по содержанию примесей
в два раза. Метод позволяет определять также Mg, Si, Al, Си, Pb,
Fe, P, As, Mo с пределом обнаружения 1>1-10-6%.
Навеску бора 0,1 г помещают в кварцевую коническую колбу вместимостью
100 мл и по каплям добавляют конц. HNO3. Для полного окисления бора до
борной кислоты необходимо 1,5—2 мл HNO3. Раствор выпаривают досуха при
осторожном нагревании на электроплитке или газовой горелке, не допуская
плавления. Затем вводят 1—2 мл перегнанного сухого метанола и отгоняют бор-
нометиловый эфир, осторожно нагревая колбу до 100—150° С. Отгонку ведут до
полного удаления борной кислоты, что легко контролируется визуально. Ос-
тавшиеся примеси растворяют в 0,5 мл конц. HNO3 и раствор выпаривают (при
нанесении каплями) на специальном графитовом электроде, обточенном в виде
шляпки гвоздя, который нагревают до 200—250° С при помощи нихромовой спи-
рали, в которую помещена нижняя, необточенная часть электрода.
Графитовая шляпка полностью сжигается в пламени дуги пере-
менного тока в течение 20—30 с. Сила тока 5 А, ширина щели спект-
рографа 0,015 мм. Источник света расположен на расстоянии 100 мм
от конденсора. Расстояние между конденсором и щелью равно
500 мм. Градуировочный график строят в координатах А5—1g с.
Определение натрия в титане [196]. Метод основан на концентри-
ровании примесей удалением основы отгонкой после перевода в TiCl4
хлорированием. Коэффициент концентрирования составляет 20—
50, предел обнаружения натрия по линии 588,995 нм равен 1 • 10-4%.
Спектры проб и эталонов фотографируют на спектрографе ИСП-51
в дуге переменного тока силой 5 А, экспозиция 1 мин. Натрий оп-
ределяют из отдельной навески.
Навеску металлического титана 4 г в виде порошка, стружки или кусочков
во взвешенной кварцевой лодочке помещают в кварцевую трубку прибора для
хлорирования. Током углекислого газа удаляют из системы воздух, прибор мед-
ленно нагревают электронагревателем до 160—180° С. Не прекращая тока угле-
кислого газа, постепенно подают хлор. После начала реакции снижают темпера-
туру до НО—120° С и поддерживают ее в этих пределах в течение всего времени
хлорирования (4—6 ч). После окончания хлорирования систему промывают уг-
106
лекислым газом. Остаток в лодочке, содержащий нелетучие примеси, обрабаты-
вают конц. HNO3, высушивают, прокаливают при 600° С в течение 30 мин и
взвешивают для расчета коэффициента концентрирования. Пробу смешивают
с AgCl в соотношении 4 : 1 и-помещают в угольный электрод в виде стержня с от-
верстием диаметром 4 мм и глубиной 4 мм.
Метод позволяет из отдельной пробы определять по одной спект-
рограмме Mg, Са, Ba, Al, V, Cr, Mo, Mn, Fe, Со, Ni, Си, Zn, Cd, In,
Sn, Pb, Sb, Bi с пределом обнаружения ^>1-1СГ5%.
Определение натрия в цирконии [450]. Предложено два варианта
концентрирования микропримесей. В первом варианте навеску ме-
талла 4 г хлорируют в кварцевом аппарате при 350—370° С в тече-
ние 2—3 ч. При этом основная часть циркония сублимируется в виде
ZrCl4 и уносится током хлора. Остаток после хлорирования, содержа-
щий нелетучие примеси и небольшое количество ZrOCl2 и ZrO2, об-
работкой 5 мл. конц. HNO3 и прокаливанием переводят в ZrO2. По-
лученный концентрат массой 200—300 мг (коэффициент концентри-
рования 20—30) смешивают с носителем AgCl в соотношении 4:1.
Предел обнаружения натрия 1 • 10~4 %. В отдельной пробе можно оп-
ределить Mg, Ba, Mn, Со, Ni, Cu, Cd, In, Pb, Bi, Са с пределом об-
наружения >5-10~® %.
Во втором варианте элемент-основу отделяют от примесей осажде-
нием миндальной кислотой из солянокислого раствора с последую-
щей количественной флотацией осадка миндалята циркония изоами-
ловым спиртом и введением примесей в новую основу PbSO4. В от-
дельной пробе можно определить Mg, Ba, Al, Ti, V, Cr, Mn, Fe, Со,
Ni, Cu, Ag, Zn, Cd, In, Sb, Bi с пределом обнаружения 3-10~Б%.
Отмечается, что натрий в основе PbSO4 не определяли.
Навеску циркония 0,3 г растворяют в кварцевом стакане в 10 мл конц.
H2SO4 при кипячении. Избыток серной кислоты удаляют упариванием. Остаток
солей растворяют в 15 мл воды при нагревании и вводят 10 мл НС1 пл. 1,19 г/см3.
Прибавляют 25 мл 16%-ного раствора миндальной кислоты. Концентрация НС1
при осаждении должна соответствовать —2,5 М. Раствор с осадком выдерживают
1—2 ч при 80—90° С, охлаждают, переносят в делительную воронку вмести-
мостью 100 мл и вводят 20 мл изоамилового спирта. Встряхивают 2 мин, соля-
нокислый раствор сливают в кварцевый стакан и после повторного осаждения
и флотирования миндалята циркония упаривают с добавкой спектрально-чис-
того PbSO4.
Спектры возбуждают (спектрограф ИСП-51) в дуге переменно-
го тока, сила тока 5 А, экспозиция 1 мин, аналитическая линия нат-
рия 588,995 нм.
Определение натрия в свинце [60]. Метод позволяет определять
натрий в свинце и его соединениях после отделения основы осажде-
нием в форме PbSO4. Вместе с натрием концентрируются и могут быть
определены Li, Mg, Са, Al, Ti, V, Cr, Mo, Mn, Fe, Co, Ni, Cu, Ag,
Zn, Cd, In, Sn, Sb, As и Bi. Натрий определяют в отдельной пор-
ции, используя аналитическую линию 588,995 нм. С учетом коэффи-
циента концентрирования, равного 100, предел обнаружения натрия
составляет 3-10-Б%. Спектр возбуждают в дуге переменного тока*
сила тока 5 А, экспозиция 1,5 мин. Регистрируют излучение на спект-
рографе ИСП-51, фотопластинка «инфра 7бО». Угольные электроды;
107
анод — стержень с торцовым кратером диаметром 4 мм и глубиной
4 мм, верхний электрод — стержень, отточенный на конус. Предва-
рительно электроды обжигают в дуге постоянного тока в течение
15 с. В каждый электрод помещают по 50 мг пробы или эталонов.
Навеску свинца 10 г растворяют при нагревании в 45 мл HNO3 (1 : 1) в ко-
нической кварцевой колбе'вместимостью 200 мл. После полного растворения
пробы прибавляют 100 мл воды, нагревают почти до кипения и осторожно по
каплям прибавляют 7 мл H2S04 (1 : 1). После полного осаждения PbSO4 (прове-
ряют полноту осаждения, добавляя 2—3 капли конц. H2SO4) осадок оставляют
на 2—4 ч. Затем маточный раствор, содержащий все примеси, отдельными пор-
циями сливают во взвешенную кварцевую чашку вместимостью 75 мл и упари-
вают его до малого объема.
Отделенный от маточного раствора осадок сульфата свинца подвергают
очистке в колбе, в которой проводилось осаждение. Для этого осадок кипятят
с 40 мл HNO8 (1 : 1) в течение 2 ч, закрыв колбу кварцевым стеклом. После ох-
лаждения добавляют 0,5 мл конц. H2SO4 и, охладив раствор, декантируют в
кварцевую чашку, в которой выпаривали маточный раствор. При сливании с пос-
ледней порции сливают 3—4 капли суспензии PbSO4 и содержимое чашки упа-
ривают до полного удаления паров серной кислоты. Полученный концентрат
примесей, основой которого является сульфат свинца, прокаливают 30 мин
при 550° С, взвешивают и смешивают с угольным порошком в соотношении 1 : 1
по массе.
Для приготовления первого эталона в навеску спектрально-чистого PbSO4,
помещенную в кварцевую чашку вместимостью 100 мл, вводят стандартный рас-
твор элементов с концентрацией 1 мг/мл. Сульфат свинца с введенным стандарт-
ным раствором соли натрия упаривают при перемешивании и прокаливают при
550° С в течение 1 ч. К приготовленному эталону прибавляют спектрально-чис-
тый угольный порошок в соотношении по массе PbSO4 : С = 5 : 1 и тщательно
перемешивают в агатовой ступке. Аналогично готовят еще три серии эталонов
смешением со спектрально-чистым сульфатом свинца и угольным порошком.
Для этого первый эталон разбавляют указанным способом в 3, 10, 30 и 100 раз.
К полученному комплекту эталонов присоединяют спектрально-чистую основу —
сульфат свинца,— смешанную с углем в соотношении 5:1, которая служит
нулевым эталоном. Готовые эталоны пересыпают в баночки с притертыми проб-
ками и хранят в закрытой коробке из органического стекла.
Угольные электроды предварительно обжигают в дуге постоян-
ного тока в течение 15 с.
Определение натрия в висмуте и его солях [227]. Метод основан
на концентрировании примесей осаждением основы в виде BiJs-
Спектры возбуждают в дуге переменного тока, сила тока 5 А, экспо-
зиция 40 с, фотопластинки панхром. Предел обнаружения натрия
3-10“6% при использовании аналитической линии натрия 588,995 нм.
В отдельной пробе можно определить Mg, Са, Ba, Al, Ti, V, Сг, Мо,
Мп, Fe, Со, Ni, Zn, Cd, In, Sb, Те с пределом обнаружения ^>2-
•10~6%. Коэффициент концентрирования примесей составляет
25—30.
Навеску металлического висмута 10 г растворяют в 50 мл 7 М HNO3 в квар-
цевой конической колбе вместимостью 450 мл. Раствор нагревают для удаления
оксидов азота, а затем разбавляют 0,3 М раствором HNO3 до объема около
300 мл. Висмут осаждают при комнатной температуре добавлением стехиомет-
рического количества свежеприготовленного раствора HJ (концентрацию рас-
твора устанавливают титрованием 1 М раствором NaOH) с небольшим избытком
~0,2 мл. Через 4 ч измеренный объем (около 275 мл) маточного раствора отде-
ляют от осадка BiJ3 декантацией. Кислый раствор выпаривают во взвешенной
кварцевой чашке и сухой остаток нитрата висмута, в котором сконцентрированы
108
примеси определяемых элементов, прокаливают 30 мин при 550° С до получения
В1203. (Прокаленный концентрат взвешивают, смешивают со спектрально-чис-
тым ВеО в соотношении 5 : 1 и помещают в электрод в виде угольного стержня
с отверстием в торце диаметром 4,5 мм и глубиной 6 мм.
Определение натрия в теллуре [493]. Метод позволяет определять
1 • 1СГ4—1-1СГ2% натрия в теллуре после отделения основы в виде
ТеОг в среде 0,5—1 М HNO3. Вместе с натрием концентрируются
и могут быть определены Fe, Al, Си, Ag, As, Pb, Mg, Ca, Zn, Cd, In,
Tl, Mn, Co, Ni и Cr. Предел обнаружения зависит от прибора, он
равен 5-10-6 и 1,5-10-6% при регистрации спектров на спектрогра-
фах ИСП-28 и ДФС-8 соответственно при использовании аналитиче-
ской линии натрия 588,995 нм и возбуждении спектра в дуге пере-
менного тока силой 12 А. Относительное стандартное отклонение
равно 0,15—0,30.
Навеску теллура 0,5—1 г растворяют в HNO3, затем выпариватот досуха
и прокаливают при температуре до 300° С. Для обогащения примесей навеску
теллура 2 г растворяют в 30 мл HNO3 пл. 1,40 г/см3 при нагревании и выпари-
вании до влажных солей. К остатку прибавляют 50 мл горячей воды и кипятят
3—5 мин. Раствор в течение 30—60 мин охлаждают в проточной воде, затем сли-
вают его в чашку для выпаривания, а осадок дважды промывают 5 мл холодной
воды. Раствор выпаривают, полученный осадок прокаливают и взвешивают.
Одновременно проводят контрольный опыт на основе чистого ТеО2. Подготов-
ленные пробы загружают по 50 мг в угольные электроды формы «рюмка» (глу-
бина 5 мм, диаметр 4 мм, диаметр шейки 2 мм, длина 3 мм). Верхний электрод
заточен на полусферу. Для удаления поверхностных загрязнений электроды
предварительно обжигают в дуге переменного тока (сила тока 10 А). Спектры
снимают ла спектрографах ИСП-28 и ДФС-8 или ДФС-13 с трехлинзовыми систе-
мами освещения, генератор ПС-39, сила тока 12 А, экспозиция 1 мин. Фотоплас-
тинки тип II. Концентрацию примесей определяют по градуировочным графикам,
построенным в координатах S—1g с.
Эталоны готовят введением растворов определяемых элементов в навеску
спектрально-чистого ТеО2 с концентрациями 1-10-1; 3-10-4; 1-10~3 и 1-10-2%.
Для получения чистого ТеО2 навеску теллура 50 г растворяют.в конц. HNO3,
упаривают до небольшого объема и разбавляют водой. Осадок отстаивают и де-
кантируют, затем трижды промывают горячим 1 М раствором HNO3. Осадок
ТеО2 растворяют в НС1 пл. 1,19 г/см3 и раствор упаривают до сиропообразного
состояния для удаления Sb, Sn, Au. Прибавляют HNO3 для разрушения ТеС14,
разбавляют водой и кипятят 1—2 мин. Осадок ТеО2 отстаивают, декантируют,
промывают 1 М раствором HN03 и переносят в полиэтиленовую посуду. Для
удаления кремния прибавляют 5—10 мл HF и перемешивают 1 ч. Затем разбав-
ляют горячим 1 М раствором HNO3, перемешивают, отстаивают, декантируют
и промывают горячей водой 5—6 раз. Осадок высушивают во фторопластовой
посуде и прокаливают при температуре до 300° С.
Определение натрия в молибдене и его соединениях [197]. Метод
основан на отделении основы осаждением а-бензоиноксимом и опре-
делении натрия из отдельной пробы. Источником возбуждения явля-
ется дуга переменного тока, сила тока 5 А, экспозиция 1 мин, плас-
тинки панхром. При использовании линии натрия 588,995 нм пре-
дел обнаружения натрия составляет 4-10-4%, коэффициент концент-
рирования натрия равен 5. В отдельной пробе можно определить
Mg, Са, Ва, Al, Ti, V, Cr, Mn, Fe, Со, Ni, Си, Ag, Zn, Cd, In, Sn,
Sb, Pb, Bi с пределом обнаружения Z>2-10-6%.
Навеску молибдена 0,5 г растворяют при нагревании в 15 мл смеси (3 : 1)
НС1 и HNO3, раствор разбавляют водой до 50 мл, переносят в делительную во-
109
ронку и прибавляют 10 мл насыщенного этанольного раствора а-бензоиноксима-
Растворы смешивают и оставляют на несколько минут для полного образования
осадка. Затем добавляют 25 мл хлороформа и встряхивают. Органический слой
с суспензией осадка сливают в колбу, а операцию осаждения и флотации повто-
ряют снова, добавляя по 10 мл раствора а-бензоиноксима до полного осаждения
молибдена, что замечают по прекращению образования белого осадка а-бензо-
иноксимата молибдена. Кислый раствор, содержащий примеси, упаривают с добав-
лением нескольких порций по 1 мл конц. HNO3 для разрушения избытка реаген-
та. К азотнокислому раствору примесей приливают раствор спектрально-чистого
нитрата бериллия, содержащий 100,0 мг бериллия. Раствор упаривают с добав-
лением избытка аммиака при перемешивании. Осадок Ве(ОН)2 высушивают и
прокаливают сначала при 250° С, а затем при 500° С в течение 30 мин. Серию
эталонов для спектрального анализа готовят также на основе ВеО введением
известных количеств стандартных растворов в навески спектрально-чистого
нитрата бериллия так, чтобы рабочий интервал охватывал концентрации элемен-
тов 10-4—10-1%.
Определение натрия в вольфраме [533]. Метод позволяет опреде-
лять 5-10~3—10“1% натрия в вольфраме и сплавах вольфрам—рений,
предел обнаружения натрия равен 2-1СГ3%. Спектры возбуждают
в дуге постоянного тока при силе тока 15 А, фотографируют с по-
мощью спектрографа ДФС-8 на спектральные пластинки тип II,
экспозиция 1 мин. Аналитическая линия натрия 330,30 нм, линия
сравнения — минимум фона в сторону коротких волн. Основу отго-
няют фракционной дистилляцией после перевода в WO3 окислением
в муфельной печи при 800° С либо после растворения в 30 %-ном
растворе Н2О2. Остаток прокаливают, смешивают с угольным порош-
ком в соотношении 2 : 1, в который предварительно вводят 1,2%
Си О в качестве внутреннего стандарта. Пробу помещают в анод с
кратером диаметром 4 мм и глубиной 6 мм.
Навеску сплава вольфрам—рений 1 г помещают в кварцевый стакан разме-
ром 25 X 30 мм, вводят 10 мл 3—5%-ного раствора Н2О2 и нагревают на элект-
роплитке. Через 5—10 мин после гачала реакции Н2О2 сливают, а пробу про-
мывают водой. Затем Н2О2 прибавляют по частям во избежание бурной реакции.
После окончания реакции раствор упаривают досуха и прокаливают в муфеле
при 550—600° С для удаления рения. Полученный остаток хранят в эксикаторе.
Пробу растирают в ступке из органического стекла и смешивают с угольной
смесью (угольный порошок -|- 2% СпО) в соотношении 2 : 1 (400 мг пробы и
200 мг угольной смеси). Перемешивают с помощью механической мешалки или
в ступке из органического стекла в течение 5—8 мин и набивают электроды с по-
мощью трамбовки из органического стекла[так, чтобы поверхность пробы была
ниже края электрода на 1,5—2,0 мм. |
Разряд в полом катоде. Особое место среди источйиков света
в спектрографическом анализе веществ особой чистоты занимает раз-
ряд в полом катоде, позволяющий понизить пределы обнаружения
на несколько порядков [162, 165, 361, 367, 1163]. Показана эффектив-
ность применения полого катода для определения многих примесей,
в том числе натрия, в труднолетучих основах и особо чистых вещест-
вах [386]. Изучено влияние различных факторов на интенсивность
линий натрия: химических свойств газа-носителя, геометрических
размеров полости, величины разрядного тока [358], давления газа
[176, 358, 661], способа введения раствора в полый катод [366], на-
ложения магнитного поля [423, 541]. Исследовано распределение
интенсивности спектральных линий натрия по поперечному сечению
110
разрядной трубки без [44, 661] и при наложении магнитного поля
[423]. Так, при использовании железного катода и введении в источ-
ник NaJ интенсивность спектральных линий натрия была постоян-
ной по всему сечению и уменьшалась к стенкам. В спектре короны
у катода линии натрия наблюдаются вблизи открытого конца трубки.
Вдоль оси катода интенсивность спектральных линий уменьшается.
Поскольку групповое определение примесей проводят в средней
УФ-области, то чаще всего используют резонансный дублет натрия
330, 23—330, 30 нм; с изменением давления в разрядной трубке ин-
тенсивность линий натрия возрастает к краям тлеющего разряда.
При увеличении разрядного тока интенсивность убывает [661]. При
анализе соединений элементов со сложным спектром при силе тока
выше оптимального значения наблюдается сильное возрастание фо-
на [218]. Отмечалось влияние матрицы на интенсивность спектраль-
ных линий натрия [165].
Пределы обнаружения натрия при использовании полого катода
в качестве источника света составляют 3-1СГ11 [176] и 1СГ10—1-1СГ® г
натрия в капле раствора при анализе соединений кремния с приме-
нением молибденовых электродов [366]. Так определяют 4-ICT® г
натрия в растворе, содержащем 0,1 мг/л америция, после выпарива-
ния раствора досуха [165], 10-6% натрия в алюмоаммонийных квас-
цах [358], 2-10~8% натрия в воде [361]. При анализе ряда трудноле-
тучих соединений предел обнаружения лимитируется чистотой осно-
вы [162].
При изучении влияния электрических характеристик разрядной
трубки с полым катодом обращают внимание на оптимальное значе-
ние силы тока. Поскольку температура пробы связана с силой раз-
рядного тока, то при определении примеси натрия в труднолетучих
основах с целью разделения спектров примеси и матрицы анализ
проводят при сравнительно малой силе разрядного тока [218].
При использовании в качестве источника света разряда с полым
катодом микроколичества натрия в труднолетучих основах или ве-
ществах особой чистоты определяются наряду с большим числом при-
месей других элементов [218, 358] прямым методом или методом хи-
мико-спектрального анализа. Предел обнаружения натрия при этом
может измениться на несколько порядков.
Описано определение натрия в оксиде и солях алюминия из на-
вески 25 мг с чувствительностью (1—5) •10“®% после перевода солей
в оксиды [133, 358, 363, 368] в америции с пределом обнаружения
4-10“® г [165], в оксиде бериллия из брикетированной навески [359,
364], в оксиде ванадия(У) [321], соединениях кремния после перевода
их в раствор (предел обнаружения натрия 10~10—10"® г) [366], в хло-
риде титана после концентрирования (предел обнаружения 3 • 10“6 %)
[362, 365, 367], в оксиде циркония с тем же пределом обнаруже-
ния [218].
Определение натрия в тетрахлориде титана [365]. Метод основан
на концентрировании примесей из тетрахлорида титана на коллекто-
ре упариванием и анализе концентрата в разрядной трубке с полым
катодом. Вместе с натрием концентрируются и могут быть определе-
111
ны Al, Fe, К, Са, Со, Mg, Mn, Cj, Ni, Pb, Cr; предел обнаружения
натрия 3-10~6% (лимитирован загрязнением угольного порошка),
остальных элементов ^>3-10~’%. Спектр возбуждают на установке
«полый катод» и регистрируют на спектрографе ИСП-28 с однолин-
зовой системой освещения щели и трехступенчатым ослабителем.
Электроды угольные, спектрально-чистые, диаметром 4 и 6 мм.
В электродах диаметром 6 мм высверливают цилиндрический канал
диаметром 4 мм и глубиной 15 мм; приготовленные электроды об-
жигают в разряде при токе 1 А в течение 2 мин, а спектр контроли-
руют на отсутствие линий определяемых элементов. Из электродов
с диаметром 4 мм на токарном станке отрезают диски толщиной
0,5 мм. Используют фотопластинки микрочувствительностью 22 ед.
ГОСТ. Аналитическая линия натрия 330,23 нм. Ниже приведена ме-
тодика, позволяющая определять все указанные примеси.
Растворяют в воде 10 г AgNO3, рассчитанным количеством НС1 осаждают
AgCl, промывают несколько раз водой и растворяют в аммиаке при нагревании.
Из полученного раствора вновь осаждают AgCl действием НС1. Осадок промыва-
ют декантацией, а затем на воронке Бюхнера водой до исчезновения реакции на
хлорид-ион и затем этанолом. Осадок высушивают в сушильном шкафу при 80° С
в течение 1 ч, растирают в ступке и далее снова высушивают. Полученный пре-
парат хранят в склянке из темного стекла.
Для подготовки пробы в кварцевую чашку помещают с помощью пипетки
2,5 мл Т1С14 и насыпают 25 мг угольного порошка. Чашку помещают в устройст-
во для упаривания, накрывают фторопластовой пленкой и упаривают досуха под
инфракрасной лампой.
Эталоны готовят на основе угольного порошка. Головной эталон с содержа-
нием каждой примеси 1 % готовят тщательным растиранием в ступке из органи-
ческого стекла 52 мг NaCl, 40 мг КС1, 28 мг NiO, 34 мг MgO, 21 мг РЬО, 28 мг
Fe2Os, 30 мг Сг2О3, 32 мг МпО2, 25 мг СиО, 28 мг СоО, 28 мг СаО, 38 мг А12О3
и 1,62 г угольного порошка. Эталоны с содержанием 10-1, 10-2, 10-8 и 10-4%
примесей готовят разбавлением головного эталона чистым угольным порошком,
растирая 1 вес. ч. каждого предыдущего эталона с 9 вес. ч. угольного порошка.
В каждый эталон добавляют 5% продукта полного гидролиза Т1С14 с 5 вес. ч.
воды и последующим высушиванием под инфракрасной лампой. В качестве
контрольного опыта используют угольный порошок с введением 5% продукта
гидролиза ИСЦ.
На дно обожженного угольного электрода помещают 1 мг AgCl, набивают
25 мг эталона (угольный порошок с продуктом гидролиза TiClj) или концентрат
примесей и далее в электрод вставляют угольный диск так, чтобы он плотно
прилегал к пробе. Электроды с пробами и эталонами помещают в разрядную
трубку, создают давление 5-10-2 мм рт. ст., анализируют при давлении гелия
20 мм рт. ст., силе тока 0,5 А (30 с), 1 А (30 с) и ширине щели 0,015 мм. Перед
экспозицией проводят отжиг при токе 0,05 А в течение 1 мин. На одной фотоплас-
тинке в одинаковых условиях фотографируют по 3 раза спектры проб и эталонов.
Градуировочный график строят в координатах — 1g с.
Другие источники возбуждения. В многоэлементном анализе в
качестве источников возбуждения при определении натрия в природ-
ных водах применяли плазмотрон постоянного тока, работающий
в атмосфере аргона [850]. Изучены спектральные характеристики фа-
кела плазменной горелки и влияние различных факторов (ток разря-
да, скорость вдувания образца в разряд и тангенциального потока
газа) на интенсивность спектральных линий [707, 777, 878]. Для нат-
рия предел обнаружения равен 0,5 мкг/мл.
112
В ВЧ-искровом разряде натрий определяли при следующих пара-
метрах высокочастотной плазмы: рабочий газ — азот, давление
0,7 атм, расход 0,7—3 л/ч, магнето — 850 Вт, 2,4 Гц, интегрирова-
ние 15 с, предел обнаружения 1,8-10“в% 11149]. В работе [390]
0,002—0,5% натрия определяли методом высокочастотного искро-
вого разряда (sr = 0,05).
В последние годы все большее применение в химико-спектраль-
ном анализе находит индукционный высокочастотный разряд
(ICP-плазма), который стабилен и имеет высокую температуру ана-
литической зоны разряда. С использованием этого источника натрий
определяли в смазочных маслах [970], а также при серийном испыта-
нии качества воды (предел обнаружения натрия 20 мкг/л) [756].
Показано отсутствие влияния поверхностно-активных веществ на
интенсивность спектральных линий [970]. При определении натрия
в смазочных маслах стандартными растворами служили растворы
металлоорганических соединений [861].
ПЛАМЕННЫЙ АТОМНО-ЭМИССИОННЫЙ
И АТОМНО-АБСОРБЦИОННЫЙ АНАЛИЗ
При определении натрия в пламенах предпочтительно использо-
вать метод атомно-эмиссионной спектрометрии. Поскольку современ-
ные спектрофотометры позволяют регистрировать абсорбционный
и эмиссионный сигналы, при определении большого числа элементов
в сложных объектах атомно-абсорбционным методом натрий (калий)
определяют в режиме эмиссии. На сигналы эмиссии и абсорбции зна-
чительно влияют физико-химические процессы в пламенах, опреде-
ляющие механизм и степень атомизации вещества, поэтому в этом
разделе рассматриваются помехи, общие для обоих методов [397].
Особенности каждого метода оговорены или вынесены в специальный
раздел.
Проведено сравнение условий определения щелочных элементов,
в том числе натрия, методами пламенной атомно-эмиссионной и атом-
но-абсорбционной спектрометрии [410], Использована установка на
основе монохроматора УМ-2, источниками света в атомно-абсорбцион-
ном анализе служили высокочастотные лампы (безэлектродные ша-
риковые). Изучено влияние различных условий проведения анализа,
а также влияние кислот (соляной, серной) , органических растворите-
лей (метанол, этанол) разных концентраций. Из результатов экспе-
римента сделан вывод, что по чувствительности и уровню помех атом-
но-абсорбционный метод определения натрия не имеет преимуществ
перед атомно-эмиссионным. При оценке современного состояния
атомно-абсорбционного анализа и его роли в современном анализе
самых разнообразных объектов отмечается несомненное преимущест-
во атомно-эмиссионного метода определения натрия (калия и лития)
перед атомно-абсорбционным [67].
Сопоставлены пределы обнаружения натрия, полученные разны-
ми исследователями, при определении натрия по эмиссионному и аб-
сорбционному сигналам [1104].
113
Атомно-абсорбционный метод анализа рекомендуют применять
при определении относительно больших количеств натрия [67]. Пре-
дел обнаружения натрия не снижается при применении двухлучево-
го атомно-абсорбционного спектрофотометра.
Влияние различных факторов на эмиссию и абсорбцию натрия.
Изучено влияние различных пламен [68, 70, 84, 99, 324, 326, 397,
401, 419, 453, 497, 567, 607, 678, 807, 819, 1107, 1133, 1244, 1284;
12851; зоны пламени [30, 420, 803, 1284]; конструкции горелки и рас-
пылителя [134, 602, 910,1238]; других инструментальных условий
[9, 30, 85, 409, 755, 819, 870, 935, 1210, 1232, 1238]. Предложены
уравнения, которые учитывают влияния различного вида [76, 419,
567, 892, 1244]. Отмечено, что использование низкотемпературных
пламен при определении натрия приводит к снижению помех, в част-
ности, в присутствии щелочноземельных элементов, особенно каль-
ция [397].
Для пламен светильный газ—воздух и ацетилен—воздух изучена
зависимость логарифма интенсивности от логарифма концентрации
щелочного элемента [401]. Угол наклона градуировочных графиков
согласуется с теоретически рассчитанным, зависящим от абсорбцион-
ного параметра а (а равно отношению лоренцовской и допплеровской
полуширин спектральных линий). Отмечено, что в воздушно-пропа-
новом пламени влияние калия и кальция на определение натрия со-
ответствует уровню случайных ошибок измерений [1133].
Изучено влияние ряда пламен, а также зон пламени ацетилен—
воздух на испускание натрия [324, 497, 803]. Для трех пламен: аце-
тилен—воздух, пропан—воздух и природный газ—воздух интенсив-
ность линий натрия при измерениях в одних и тех же условиях зави-
сит от температуры пламени и в интервале температур 1840—2115 К
возрастает в 4 раза. Натрий вводили в пламя в виде хлорида.
Рекомендуется использовать пламя ацетилен—воздух, в котором
интенсивность линий натрия не изменяется в присутствии элементов
с низким потенциалом ионизации [324]. Зона максимального свече-
ния натрия в этом пламени не зависит от введения раствора сульфата
натрия в качестве буферного с концентрацией 2,5 мг/мл. Оптималь-
ная зона для натрия отличается от зон для других щелочных элемен-
тов. Это объясняют изменением степени атомизации натрия и обра-
зованием гидроксидов в пламени. В работе использован спектрофото-
метр на основе спектрографа ИСП-51 с фотоэлектрической приставкой
ФЭП-1. Применение низкотемпературного пламени водород—
воздух приводит к уменьшению ионизационных помех и ослаблению
фона по сравнению с высокотемпературным пламенем ацетилен—
воздух и ацетилен—оксид азота(1) [1107]. В качестве буфера предло-
жены соли лития. Рассматривается [419] аммиачно-кислородное пла-
мя с температурой 1720° (1993 К). Отмечается, что кальций (до
500 мкг/мл) не мешает определению натрия; интенсивность линии
натрия возрастает в присутствии калия, что предлагается учитывать
расчетным способом. Использование резонансных линий натрия (и
других щелочных элементов) приводит к искривлению градуировоч-
ного графика за счет самопоглощения. При определении натрия в пла-
114
мени ацетилен—оксид азота(1) [807] спектральные и химические по-
мехи не выше, чем в пламени ацетилен—воздух. Отмечается, что
в новом пламени — смеси паров этанола с воздухом — ионизация
натрия низка. Натрий можно определять в сложной смеси после раз-
ложения силикатных пород смесью HF и H2SO4.. Температура пла-
мени 1150° С (1423 К) [99].
Низкотемпературное пламя бензин—воздух применено при опре-
делении натрия в присутствии 10-кратных количеств щелочноземель-
ных элементов [453]. Изучено влияние температуры на эмиссию нат-
рия [1285]. Изменение температуры на 10% приводит к погрешности
определения 3%. Использован фильтровый фотометр с визуальной
регистрацией сигнала. Изучены характеристики водородно-кисло-
родного пламени при применении комбинированной горелки-распы-
лителя, работающей в турбулентном режиме [68]. Показано, что
собственный фон пламени уменьшается и натрий можно определять
с пределом обнаружения 10~4 мкг/мл.
Реабсорбция линий натрия заметна, начиная с его концентрации
1 мг/л. При использовании резонансного дублета 330,23—330,00 нм
•линейность наблюдается до содержания натрия 1 г/л. Отмечается,
что при применении комбинированной горелки-распылителя для
пламени водород—кислород и шлейфового осциллографа абсорб-
ционный сигнал повышается в 25 раз [910].
При анализе суспензий изучалась зависимость интенсивности из-
лучения натрия от концентрации кремния [263]. Кремний увеличива-
ет эмиссию натрия, однако характер влияния не установлен. Если
вводить в пламя суспензию в растворе хлорида кальция, интенсив-
ность спектральных линий щелочных элементов возрастает в 1,7—
2 раза, что объясняется снижением влияния дисперсности частиц на
эмиссию натрия (и других щелочных металлов). Интенсивность ис-
пускания натрия постоянна во времени.
При изучении зависимости интенсивности от концентрации нат-
рия в растворе выведено уравнение, учитывающее изменение скорос-
ти поступления раствора и различные влияния в пламени [1244].
Получены сравнительные теоретические и экспериментальные дан-
ные для различных пламен. Предполагается, что выведенные урав-
нения можно применить к другим методам пламенной спектрометрии:
атомной абсорбции и атомной флуоресценции.
Весьма интересна работа [567], в которой предложено учитывать
контрольное содержание натрия в газах с помощью расчетной по-
правки. Для пламени пропан—воздух поправку рассчитывали с уче-
том формулы Больцмана по температурам в условиях эксперимента
(2183 К) и «сухого» пламени (2230 К). Детальное изучение влияния
дисперсности аэрозоля на сигнал эмиссии натрия позволило разра-
ботать метод атомно-эмиссионного определения натрия по одному
эталону по зависимости частоты подачи раствора от концентрации
натрия в растворе [602]. Для усовершенствования метода генерации
аэрозоля предложен специальный генератор. Метод применен для
определения натрия в крови с относительным стандартным отклоне-
нием 0,001. Изучено влияние размера капель на интенсивность спект-
115.
ральных линий натрия [134]. Применение метода ультразвукового
распыления растворов позволило уменьшить размер капель до
1 мкм, что привело к возрастанию интенсивности испускания нат-
рия в 10 раз. Показано, что повышение эмиссионного сигнала связа-
но с уменьшением температуры пламени и возрастанием эффектив-
ности распыления аэрозоля.
Описан метод определения натрия, позволяющий учесть мешаю-
щее влияние различных факторов на интенсивность линий натрия
и контрольную пробу [1238]. Для этой цели применяют трехканаль-
ный спектрофотометр и два распылителя — обычный и V-образный.
Натрий определяют по эмиссии в пламени воздух—оксид азота(1)—
ацетилен. Для подавления ионизации используют соли калия, рас-
твор инжектируют через обычный распылитель током оксида азота(1).
Воздух вводят через V-образный распылитель. Через ветви этого
распылителя вводят растворы анализируемой и контрольной проб.
Концентрацию натрия определяют с помощью программ по фотото-
кам растворов, содержащих натрий и контрольную пробу.
Для снижения фона предложено охлаждать фотоэлектрический
умножитель с помощью электронной морозильной камеры [819]. Ис-
пользование прямоточной горелки для пламени водород—кислород
также позволяет получить при определении натрия лучшее отноше-
ние сигнал/помеха и достичь предела обнаружения натрия 3-10~10%
при определении его в полупроводниковых материалах в специаль-
ных условиях: анализ проводят в очень чистых помещениях и приме-
няют воду с содержанием натрия <2-1СГ6%.
Изучено изменение относительной интенсивности излучения нат-
рия в пламени пропан—бутан—воздух в зависимости от размера ка-
пель аэрозоля, поверхностного натяжения и вязкости раствора, его
расхода и концентрации соляной кислоты и натрия [76]. Предложено
уравнение, которое описывает связь уменьшения относительной ин-
тенсивности излучения натрия с указанными параметрами.
При определении натрия атомно-абсорбционным методом изучено
влияние условий измерения и различных параметров на величину
абсорбции и наклон градуировочных графиков [935]. Применяли
спектрофотометр фирмы «Перкин-Элмер» (модель 303), пламена аце-
тилен—воздух и ацетилен—оксид азота(1). Предложена новая мо-
дель многоэлементного пламенного спектрометра с детектором-види-
коном, оснащенным ЭВМ, Предусмотрены программы, позволяющие
исключить наложения спектров мешающих элементов, корректиро-
вать фон, проводить коррекцию с помощью внутреннего стандарта,
измерять аналитический сигнал по отношению к усредненному фону.
Прибор используют для одновременного определения натрия, калия,
лития и кальция [755].
Из автоматизированных приборов, в которых определение натрия
возможно по искривленным графикам (зависимость I — с), можно
отметить установку с автоматическим пробоотбором, разбавлением
и расчетом результатов с учетом кривизны графиков с погрешностью
<1 % [1210]. Излучение натрия измеряют в направлении, перпен-
дикулярном оси щелевой горелки.
116
Можно значительно повысить чувствительность определения
элементов при импульсном испарении вещества в пламени с поверх-
ности микрозонда, выполненного из графита, пирографита, стекло-
углерода или металла [280, 413, 414, 728].
Предлагается определять натрий в пламени кислород—воздух—
ацетилен при импульсном испарении его соли с графитового микро-
зонда [413]. Время импульса <1 с, чувствительность <1СГ10 г.
Используется пламенно-фотометрическая установка на основе моно-
хроматора ДФС-12. Предлагается [728] эмиссионный метод опреде-
ления натрия в диффузионном пламени азот—водород при исполь-
зовании графитовой нити. Применяют адаптер пламени — медную
трубку. Изучение ее полости проецируется на цель монохроматора
SP-900II.
Атомизация соединений натрия в пламенах. Степень атомизации
соединений натрия в различных пламенах стали оценивать сравни-
тельно недавно [200, 347, 583, 638, 694, 789, 911, 1045, 1080, 1268].
Во всех более ранних монографиях отмечали термическую нестой-
кость соединений натрия в пламенах [397]. В работе [1268] рассчи-
тана концентрация атомов натрия в изолированном воздушно-аце-
тиленовом пламени горелки Меккера, равная 1,17-Ю11 ат/см3 при
следующих параметрах распылительной системы: скорость подачи
раствора 3,85 мл/мин, эффективность распыления 4,9 мл/мин, расход
воздуха 166 см3/с, ацетилена 23 см3/с, воды 3,14-1СГ3 мл/с, темпера-
тура пламени 2320 К, начальная концентрация натрия в растворе
1СГ4 М. Проверена концентрация свободных атомов натрия с исполь-
зованием в качестве источника света сплошного излучения. Экспери-
ментально полученные близкие значения указывают на полноту ато-
мизации. Расхождения с результатами Ранна объяснены неучетом
сверхтонкой структуры линии с линейчатым источником [1080].
Концентрацию свободных атомов определяли методом атомной аб-
сорбции.
Отмечается, что для щелочных металлов в пламенах водород—
кислород и ацетилен—воздух вероятным устойчивым соединением
является гидроксид МОН [397, 401]. Натрий не образует такого со-
единения. Для пламен ацетилен—воздух и пропан—-бутан—воздух
рассчитаны парциальные давления паров Рм с учетом эффективности
распыления (горелка с распылительной камерой) [401]. При кон-
центрации соли натрия 1 • 10-4 М получено Рм = 10~3>7 атм для пла-
мени светильный газ—воздух.
Эффективность атомизации натрия (|3) в воздушно-ацетиленовом
пламени в зависимости от состава газовой смеси изучал Халл [789].
Полученное значение [3 = 0,98 мало зависит от состава горючей сме-
си. Изучен механизм атомизации соединений натрия в водородных
пламенах. Для этого установлена и сопоставлена с теоретическими
расчетами зависимость концентрации ОН- и воды от температуры
пламени. Установлено, что в пламени может присутствовать гид-
роксид натрия, а единственной реакцией, ответственной за атомиза-
цию натрия (а также лития и калия), является реакция типа
МОН + Н М + НгО.
117
Эффективность образования соединений щелочных металлов в
пламени уменьшается от лития к натрию. Возможные равновесия
в пламенах для соединений натрия рассматривались в ряде работ,
результаты которых обобщены [583, 789]. Из теоретических вычис-
лений сделано заключение, что в обогащенном горючим водородном
пламени при 2300 К образование NaOH и Na+ незначительно. Это
заключение справедливо и для углеводородных пламен. В дальней-
шем данная методика была использована для диагностики пламен
[583].
При изучении профилей натрия для различных пламен установ-
лено, что в более низкотемпературном пламени эмиссия увеличива-
ется с ростом высоты рабочей зоны [1284]. Сделано предположение,
что в присутствии соляной кислоты интенсивность испускания нат-
рия в пламени ацетилен—воздух падает вследствие образования
молекул NaCl, т. е. снижения доли свободных атомов.
Указано, что натрий (медь, серебро) можно рассматривать как
полностью атомизированный стандартный элемент [583]. Методом
интегральной абсорбции вычислено, что натрий полностью атомизи-
рован в обогащенных пламенах ацетилен—оксид азота(1) (Т =
= 2950 К), водород—оксид азота(1) (Т = 2900 К), ацетилен—воз-
дух (Т = 2450 К) и водород—воздух (Т = 2000 К). Такое же за-
ключение сделано для натрия при его определении в пламени ацети-
лен—оксид азота(1) с отношением окислителя к горючему, равным
1,95—2,8. Вычисления показали, что при более низком отношении
образуется карбид натрия, при более высоком — моногидроксид
и монооксид. Образование молекул Na2 исключено.
Изучение механизма атомизации соединений натрия в пламенах
позволило разработать оптические методы определения абсолютных
и относительных концентраций некоторых радикалов, особенно в
обогащенных и нестехиометрических водородных пламенах. Эти ме-
тоды детально описаны [583]. Например, для определения абсолютной
и относительной концентраций Н-радикала используют несколько
методов: LiOH-метод, при реализации которого измеряют отно-
шение [Li]/[Na]; NaCl-метод, основанный на измерении атомной кон-
центрации натрия, при известном количестве хлорида натрия, вве-
денного в пламя; NaOH-метод измерения относительной концентра-
ции радикала ОН. Этот метод основан на эмиссии континуума
(область 300—600 нм), происходящей при реакции рекомбинации
Na + ОН NaOH + hv.
Ионизация атомов натрия в пламени. Константа ионизации, рас-
считанная по методу Саха при 1270 К, равна 6,0-10~20 атм. Отмеча-
ется, что в пламени ацетилен—воздух изменение степени ионизации
заметно для растворов с концентрацией натрия <10~4 М, поэтому
при определении натрия в высокотемпературных пламенах предло-
жен метод смещения равновесия ионизации при введении в раствор
солей щелочных металлов с более низкими потенциалами Ионизации
[397].
118
По вопросу влияния ионизационных помех в пламени на опреде-
ление натрия единого мнения нет. В ряде работ отмечено взаимное
влияние натрия и калия, причиной которого является смещение
равновесия ионизации [419, 938, 991]. Показано, что при введении
сульфата калия в качестве буфера в растворы хлорида натрия в пла-
менах ацетилен—воздух и пропан—воздух повышается интенсив-
ность излучения натрия (использован пламенный фотометр фирмы
«К. Цейсс» [326]. Предложено уравнение, учитывающее влияние
ионизации при определении интенсивности излучения натрия в за-
висимости от концентрации натрия [1244]. Отмечено взаимное влия-
ние калия и натрия в пламени аммиак—воздух и аммиак—кислород
[419]. Рассмотрены преимущества низкотемпературного пламени во-
дород—воздух в снижении ионизационных помех [1107]. Отмечено,
что литий стабилизирует равновесие ионизации атомов натрия и что
интенсивность излучения натрия не изменяется в присутствии эле-
ментов с низким потенциалом ионизации [324]. В то же время авторы
работы пришли к выводу, что при определении натрия в пламени
ацетилен—воздух сульфат калия не является буферным раствором.
Расчетным методом показано, что при концентрации натрия в раст-
воре 10-4—1СГ1 М равновесие ионизации натрия в пламени смещено
влево [401]. Логарифм константы ионизации равен —11,38 и —9,0
в пламенах светильный газ—воздух (1970 К) и ацетилен—воздух
(2360 К) соответственно.
Равновесие ионизации натрия в пламенах более детально рас-
смотрено в обзорных работах [583, 789], где сопоставляются расчет-
ные и экспериментальные данные по оценке степени ионизации нат-
рия за 10 лет. Имеются сведения, что при определении степени ато-
мизации натрия методом интегрального поглощения в присутствии
избытка хлорида цезия величина 0 изменяется от 0,5 до 1,0, что сви-
детельствует о заметной ионизации натрия при введении 10-2 М
раствора соли натрия в пламя горелки ацетилен—воздух. В то же
время многие исследователи отрицают возможность образования
ионов натрия в пламенах. Видимо, различие мнений о механизме
ионизации натрия в пламенах связано с разными экспериментальны-
ми условиями и неполной информацией 6 механизме процессов в
пламенах.
Изучение равновесных состояний соединений натрия в различ-
ных пламенах имеет практическое значение не только для оптимиза-
ции условий определения натрия по сигналам эмиссии или абсорб-
ции, но и для определения ряда термодинамических констант и ре-
шения других проблем.
Метрологические характеристики пламенно-фотометрических
методов определения натрия. Чувствительность, предел обнаружения
и другие метрологические характеристики пламенно-фотометриче-
ских методов определения натрия изучали в работах [68, 167, 264,
411-413, 415, 425, 660, 677, 678; 684, 735, 760, 794, 933, 1054, 1073,
1078, 1271, 1272]. Исследовано влияние различных факторов на мет-
рологические характеристики [167, 677, 1271, 1272]. Предложено
[1078] математическое выражение для «динамической области» кон-
119
центраций с учетом чувствительности и максимально определяемой
концентрации. Предел обнаружения натрия 0,002 мкг/мл, верхняя
граница определяемых концентраций 2,8 мкг/мл (при использова-
нии спектрофотометра).
Предел обнаружения натрия в пламени кислород—водород —
0,0002 мкг/мл [1054], в пламени оксид азота(1)—ацетилен —
0,0004 мкг/мл при использовании щелевой горелки в оптимальных
условиях работы спектрофотометра IL-153 [677]. Изучено влияние
различных факторов на величину предела обнаружения: оптимальной
зоны пламени оксид азота(1)—ацетилен, ширины щели спектрофото-
метра, напряжения на ФЭУ, ионизационных помех и органических
растворителей [677]. В турбулентном пламени кислород—водород
при применении комбинированной горелки-распылителя предел об-
наружения натрия составил 0,0001 мкг/мл [68]. Предложено урав-
нение для определения предела обнаружения в различных пламенах
при оптимальной ширине щели спектрофотометра в эмиссионном
варианте пламенной спектрометрии [1271], которая учитывает также
температуру пламени, флуктуации источника и элемент для абсорб-
ционного варианта [1272].
Показано, что в пламени воздух—пропан—бутан чувствитель-
ность определения натрия повышается в 10 раз при подогреве рас-
пылительной камеры до 200° С [167]. Сопоставлены пределы обнару-
жения натрия методом эмиссионной и абсорбционной спектрометрии
при использовании одной и той же аппаратуры [678]. Приведены
пределы обнаружения натрия при испарении его солей с зонда [412,
413]. В пламени оксид азота(1)—ацетилен предел обнаружения нат-
рия составляет 1 • 10“6 мкг/мл по Зх-критерию и 10-10 г при опре-
делении его эмиссионным методом. При использовании графитовой
печи HGA-72 предел обнаружения натрия составил 10-Х1 г [660].
Применение графитовой кюветы и лазера на красителе родамин 6Ж
снижает предел обнаружения натрия до 3-109 ат/см3 [933].
Особое место занимают лазерные методы определения субмикро-
количеств натрия с регистрацией сигналов атомно-флуоресцентным
методом и методом ступенчатой фотоионизации, которые позволяют
регистрировать единичные атомы. Подробные сведения приведены
в соответствующем разделе.
За меру селективности определения элементов методом атомно-
эмиссионного анализа Полуэктов и сотр. [402] предлагают прини-
мать «факторы специфичности», которые являются характеристика-
ми прибора, позволяющими оценить спектральные помехи при опре-
делении элемента в присутствии посторонних солей. В табл. 44
приведены «факторы специфичности» при определении натрия в при-
сутствии солей калия, лития, стронция, кальция и бария для раз-
личных пламен в зависимости от класса прибора.
Несмотря на условность «факторов специфичности», сведения о
них имеют большое практическое значение. Так, при решении во-
проса о возможности определения натрия в присутствии кальция
и железа в шламах с помощью пламенного фотометра найдены «фак-
торы специфичности» в присутствии кальция (12,5) и железа (150),
120
Таблица 44
Приблизительные значения «факторов специфичности»
при определении натрия
Пламя «Факторы специфичности» в присутствии солей
калия ЛИТИЯ кальция стронция
Спектрофотометр на основе монохроматора типа УМ-2 ФЭУ-19 [402]
Светильный газ — воз- дух Ацетилен — воздух 2,75-10® 1,5-10® 4-10* 1,5-10* 1,1-10* 3,8-10® 2-10*
Фильтровый фотометр [491] Ацетилен — воздух 1 2,1-IO2— | (2—3)-10* | 15—6-10® 1 1 7,8-10® | 1 1 Спекрофотометр на основе монохроматора СД-2, ДФС-12 [408]
Ацетилен — воздух 3-10® 5-107 5-107 (барий)
что позволило правильно оценить оптимальные условия выполнения
•анализа [294].
Влияние катионов на эмиссию и абсорбцию натрия (катионный
эффект). Влияние различных катионов описано в работах [15, 26, 61,
62, 73, 99, 150, 168, 171, 203; 213, 263, 269, 300, 311, 324, 406, 419,
438, 453, 468, 555,575,599,636,730, 780,798,821,844,947,948,974,
1013, 1054, 1077, 1098, 1106, 1107, 1137, 1207, 1208, 1215, 1280]. При-
чиной влияния могут быть: изменение степени ионизации натрия в
присутствии катионов щелочных элементов (К, Li, Cs) [821, 991,
1107, 1284] — так называемое взаимное влияние элементов; спект-
ральные помехи за счет наложения постороннего излучения, напри-
мер, Са, Fe, Мп [15, 61, 62, 115, 150, 203, 213, 555, 599, 636, 798,
1106]. В некоторых случаях посторонний элемент снижает аналити-
ческий сигнал, видимо, за счет изменения условий испарения частиц
в пламени. Так, отмечено [1207], что кальций уменьшает поглощение
натрия.
Большое значение при анализе сложных объектов имеет тип прибо-
ра и температура пламени. Так, в низкотемпературном пламени влия-
ние кальция на эмиссию натрия снижается. Отмечено, что в пла-
мени водород—воздух литий стабилизирует равновесие ионизации
[1107]. В пламени водород—кислород определению 10 мкг/мл натрия
не мешает 5 мг/мл калия и лития при использовании атомно-абсорб-
ционного метода [1098]. При определении натрия в молибдокремне-
вой, вольфрамокремневой, молибдофосфорной кислотах интенсив-
ность излучения натрия снижается в интервале концентраций !•
• 10-4—1-10”1 М при использовании пламенного фотометра фирмы
«К. Цейсс» с интерференционными светофильтрами [300]. Мешающее
влияние железа, марганца и кальция устраняют введением в раствор
соли алюминия [555, 1106, 1137], серной кислоты [636] или смеси
121
соли алюминия и серной кислоты. Иногда хром и железо осаждают
в виде гидроксидов [1273], либо железо и частично марганец отде-
ляют экстракцией [26, 438] перед определением натрия с помощью
фильтрового пламенного фотометра.
В новом пламени — смеси этанола и воздуха — натрий можно
определять сразу же после разложения силикатов смесью HF и
H2SO4, так как не обнаружено влияния железа, кальция и других
элементов [99]. В пламени кислород—водород при определении на-
трия по линии 589,6 нм не наблюдалось влияние лития, магния,
меди, бария, стронция, алюминия, циркония и ванадия [1207].
Влияние ванадия не наблюдали также при его содержании до
2,5 мг/мл с использованием спектрофотометра на основе монохрома-
тора УМ-2 в пламени светильный газ—воздух [168]. Отмечается, что
при концентрации 0,9 г/л на эмиссию натрия не влияют Mg, Си,
Sr, Ba, Ag, Cd, Bi, Сг, Fe, Мп, Ni, Al, Ti [73]. При использовании
пламенного спектрофотометра на основе спектрографа ИСП-51
с фотоэлектрической приставкой ФЭП в пламени ацетилен—воздух
не наблюдалось влияния железа на эмиссию натрия [310]. На пла-
менном фотометре ФПФ-58 натрий можно определить на фоне
500 г/л хрома, 50 г/л железа, 2 г/л алюминия [15]. При больших
содержаниях хрома и железа необходимо учитывать создаваемый
ими фон. Мешающее влияние марганца и железа [62], калия, каль-
ция и магния [150], алюминия [844, 974] учитывают введением солей
этих металлов в эталонные растворы. Влияние алюминия (а также
фосфата) можно устранить фотометрированием высокой зоны пла-
мени — 12 мм над срезом горелки [1208].
Не обнаружено влияния алюминия на эмиссию натрия при опре-
делении 10-3—10~2% натрия в металлическом алюминии в пламени
водород—кислород с использованием фильтрового фотометра [12151;
не обнаружено влияния 140—220 мкг/мл алюминия при определении
0,4—5 мкг/мл натрия на пламенном фотометре фирмы «К. Цейсс»
(модел ь(Ш)) [269].
При изучении влияния платины на поглощение натрия установ-
лено их соотношение, которое соответствует максимальному взаим-
ному влиянию [280]. При определении натрия в соединениях воль-
фрама и молибдена [300, 468, 798, 1013] отмечается влияние фонового
излучения вольфрама [798], а также возможное образование молибда-
тов и вольфраматов натрия, термически устойчивых в пламени, что
приводит к снижению эмиссии натрия. Исследования проводили
в различных пламенах: светильный газ—воздух, светильный газ—
воздух—кислород, воздух—ацетилен, водород—кислород [468].
Показано, что при определении натрия методом пламенной эмис-
сионной спектрометрии в техническом порошке хрома и продуктах
его выщелачивания с использованием пламени воздух—пропан—
бутан и фильтрового фотометра эмиссия хрома(Ш) при 589 нм моно-
тонно возрастает с увеличением содержания хрома в пробе [40(?1.
Хром увеличивает аналитический сигнал натрия на 20—25 %,
поэтому при анализе хромсодержащих объектов необходимо готовить
стандартные растворы на основе хрома.
122
Влияние анионов на эмиссию и абсорбцию натрия (анионный
эффект). Этот вопрос имеет большое практическое значение для
правильной подготовки пробы к анализу [32—34, 72, 74—76, 99,
149, 403 , 453, 486, 488, 497, 545, 584, 620, 713, 728, 872, 875, 1031,
1208, 1284]. Механизм взаимного влияния при определении элемен-
тов атомно-эмиссионным и атомно-абсорбционным методами в пла-
менах трактуется по-разному: с точки зрения физических свойств
раствора, особенно при введении органических кислот; с позиций
изменения условий атомизации за счет образования новых термиче-
ски более устойчивых соединений натрия при десольватации частиц
аэрозоля; смещения равновесия атомизации в пламени за счет иони-
зационных процессов с участием анионов.
Влияние анионов при определении натрия в пламенах проявляет-
ся в снижении эмиссии или абсорбции, что объясняют изменением
степени ионизации натрия. Специальными исследованиями показано,
что при введении 1М раствора фосфорной кислоты в 0,001 М раствор
соли натрия электропроводность пламени повышается примерно
в 4 раза, что связано со смещением равновесия ионизации в присутст-
вии кислотного остатка по уравнениям
М+ + X s МХ+, е + х Х-.
Использование метода двух распылителей [397] показало, что
соляная, серная и фосфорная кислоты снижают эмиссию натрия
как при введении их в пламя в одном растворе, так и при раздельном
способе — через второй распылитель, однако степень влияния раз-
лична — больше в первом случае, и тем больше, чем ниже темпера-
тура пламени. Причиной влияния кислот на сигнал эмиссии являет-
ся, видимо, влияние на стадии испарения частиц аэрозоля.
Показано, что гасящее влияние кислот на эмиссию натрия усили-
вается в ряду кислот: лимонная, азотная, борная, серная, соляная
и фосфорная [488]. По данным работы [713], муравьиная и уксусная
кислоты повышают интенсивность испускания натрия, винная и
лимонная кислоты — снижают. Объясняется это изменением поверх-
ностного натяжения раствора и его влиянием на размер капель аэро-
золя. В присутствии 100%-ной уксусной кислоты чувствительность
повышается в 5—10 раз. При атомно-абсорбционном определении
натрия в силикатах в пламени ацетилен—воздух борная кислота
устраняет все влияния [620].
Изучено влияние кислот на определение натрия в пламени про--
пан—бутан—воздух [486]. В присутствии кислот изменяются такие
физические свойства раствора, как вязкость и поверхностное натя-
жение, что вызывает изменение скорости распыления и расхода раст-
вора. По степени влияния кислоты расположены в ряд: СН3СООН <
< Н3РО4 < НС1 Н3РО3 < H2SO4. В то же время при определе-
нии натрия в удобрениях не отмечено влияния фосфат- и сульфат-
ионов на поглощение натрия [1223]. Изучено влияние сульфат- и
хлорид-ионов на абсорбцию натрия, а также на электросопротивле-
ние пламени [1031].
123
Исследованием влияния серной кислоты на интенсивность излу-
чения натрия в пламени пропан—бутан—воздух показано, что в сре-
де 4М H2SO4 кинематическая вязкость возрастает на 35%, поверх-
ностное натяжение — на 8% [34]. Депрессирующее действие серной
кислоты на эмиссионный сигнал натрия уменьшается с возрастани-
ем давления в распылителе. Аналогичное исследование проведено для
НС1 [74]. Депрессирующее влияние НС1, HNOS, H2SO4 и HsP04
также связывают с увеличением диаметра частиц аэрозоля. Это про-
верено двумя методами — атомно-эмиссионными атомно-абсорбцион-
ным. Уксусная кислота, наоборот, вызывает уменьшение диаметра
частиц на 30%. Высказывается и другое предположение, что НС1
уменьшает эмиссию натрия за счет смещения равновесия в сторону
образования молекул NaCl [1284].
При использовании метода импульсного испарения вещества
с платинового зонда [545] снижается роль термически устойчивых
соединений, т. е. влияние кислот снижается. При испарении вещества
с угольной нити в диффузионном пламени азот—водород хлорид не
влияет на эмиссию натрия, сульфат снижает эмиссию [728].
В ряде работ предлагаются эмпирические формулы для учета
влияния различных кислот на определение натрия: H2SO4 и НС1
[76, 488, 584, 892], HNOS [32]; выведена формула, учитывающая
большое количество физических факторов [75].
При введении в раствор анионов в виде'различных солей нельзя
не учитывать возможного влияния катионов.
Влияние физических свойств раствора на атомно-эмиссионное
и атомно-абсорбционное определение натрия. В ряде исследований
отмечается изменение физических свойств раствора при определении
натрия в присутствии некоторых органических и неорганических
кислот и органических растворителей [33, 248, 351, 409, 410 , 453,
486—488, 497, 559, 713, 803, 910]. Влияние органических раствори-
телей на результаты определения натрия методами пламенной фото-
метрии обусловлено многими причинами: изменением эффективности
распыления раствора и увеличением его количества в пламени, изме-
нением диаметра частиц аэрозоля, повышением эффективности ато-
мизации вещества в пламени за счет восстановительных свойств
углерода органического растворителя в пламени и реакций хемилю-
минесценции.
Изучено влияние соляной, серной, борной, фосфорной, уксусной,
лимонной кислот на процесс испарения частиц аэрозоля и реакции
в газовой фазе при определении натрия [486]. Измерялись вязкость,
поверхностное натяжение, скорость поступления растворов в пла-
мя, применялась техника двух распылителей. Показано, что присут-
ствие уксусной, лимонной и серной кислот влияет на дисперсность
капель аэрозоля и скорость испарения частиц. Фосфорная, соляная
и борная кислоты влияют также на процесс испарения и равновесные
реакции в газовой фазе.
При исследовании поведения азотнокислых растворов натрия
в пламени установлено, что коэффициент вязкости раствора возраста-
ет, поверхностное натяжение снижается и диаметр капель аэрозоля
124
уменьшается [33]. Наблюдалось увеличение скорости поступления
натрия с поверхности частиц аэрозоля, и отмечено, что влияние вяз-
кости проявляется сильнее.
Изучена абсорбция ряда элементов, и в том числе натрия, в вод-
ных растворах в присутствии уксусной кислоты, этилацетата, ацето-
на и этанола при введении их в раствор до 5% об. В интервале кон-
центраций 0,5—1% об. поглощение возрастает [803]. Поверхностно-
активные вещества повышают эффективность определения натрия
в пламенах. При изучении действия добавок (25% об. этанола и
5% об. бутанола) на интенсивность излучения натрия показано, что
спирты повышают эффективность распыления [397]. Изучено влия-
ние метанола, этанола и неорганических кислот на интенсивность
спектральных линий натрия (табл. 45).
Таблица 45
Влияние спиртов на относительную интенсивность
спектральных линий натрия [410]
Содержание спирта, % об. Относительная интенсивность в присутствии
метанола этанола
25 1,61 2,1
50 2,19 2,7
75 3,11 3,75
100 5,0 4,6
Большое значение имеет конструкция распылителя и горелки.
Так, при применении распылителей с камерами распыления и комби-
нированных горелок-распылителей механизм влияния органических
растворителей различен. Отмечена неоднозначность результатов
влияния органических растворителей на интенсивность спектраль-
ных линий натрия, полученных разными авторами в различных экс-
периментальных условиях [248]. Использована пламенно-фотоме-
трическая установка на основе спектрографа ИСП-51. Сравнивалось
влияние метанола, этанола, пропанола, бутанола, муравьиной и
уксусной кислот, диоксана, ацетилацетона и водных растворов на
эмиссию щелочных элементов в пламени ацетилен—воздух. Отмечено
полное соответствие между увеличением скорости распыления раство-
ра, уменьшением вязкости в ряду спиртов и ростом интенсивности
спектральных линий натрия. Для кислот изменение интенсивности
коррелирует с уменьшением вязкости и увеличением поверхностного
натяжения. Все органические растворители практически не изме-
няют скорость распыления. Сделано предположение, что влияние ор-
ганических растворителей связано с изменением диаметра капли
аэрозоля. Из общей схемы выпадает ацетил ацетон. Спирты в зависимо-
сти от их концентрации в растворе позволяют повысить чувствитель-
ность определения щелочных металлов (натрия) в 4—12 раз.
125
При определении натрия в пламени пропан—воздух заметно влия-
ние метанола, этанола, пропанола, этиленгликоля и глицерина на
интенсивность спектральных линий натрия [487]. Для первых трех
спиртов изменение интенсивности коррелирует с изменением вязко-
сти раствора. Сделано обоснованное предположение об изменении
условий распыления растворов. Для пламени ацетилен—воздух изу-
чено влияние метанола, этанола', глицерина и неорганических кислот
на эмиссию и абсорбцию натрия [409].
Отмечается [713], что при пламенно-фотометрическом определе-
нии натрия с помощью фильтрового фотометра «К. Цейсс» (модель III)
этанол снижает интенсивность излучения натрия за счет увели-
чения самопоглощения, изменения температуры пламени и кинетики
процессов, несмотря на увеличение эффективности распыления рас-
твора. При изучении влияния муравьиной, уксусной, винной и ли-
монной кислот на определение натрия с помощью спектрофотометра
на основе спектрографа ИСП-51 установлено повышение чувствитель-
ности определения натрия в 5—10 раз в присутствии 100%-ной
уксусной кислоты и в 1,5—2 раза для 2 М раствора кислоты [713].
В несколько меньшей степени влияет муравьиная кислота. Винная и
лимонная кислоты снижают интенсивность излучения натрия. Ос-
новное значение придается роли поверхностного натяжения раствора.
Отмечается, что уксусная кислота увеличивает эмиссию и абсорб-
цию натрия за счет уменьшения диаметра частиц аэрозоля [497].
Изучено влияние метанола, этанола, бутанола и уксусной кислоты
на распределение свободных атомов в пламени ацетилен—воздух и
на температуру [559]. Для этой цели применяли пламенно-фотометри-
ческую установку на основе спектрографа ИСП-51, комбинирован-
ную горелку-распылитель. При концентрации органического раство-
рителя 1 М температура пламени повышается на 100° С. Интенсив-
ность линий натрия в присутствии органических растворителей
максимальна в более высокой зоне пламени по сравнению с водным
раствором. Общий объем пламени возрастает. Аналогичные резуль-
таты получены в работе [397].
Применение метода атомно-абсорбционной спектрометрии с пла-
менной атомизацией. Определению натрия в различных объектах
методом атомно-абсорбционного анализа посвящено много работ [30,
77, 78, 171, 420, 663, 847, 872, 910, 914, 935, 940, 991, 1193, 1223,
1246]. Заслуживают внимания монографии [67, 407].
В теории атомно-абсорбционного метода анализа некоторые тео-
ретические модели рассматриваются на примере элементов с высокой
степенью атомизации в пламенах, в частности натрия [845, 1080].
Так, в работе [1080] дается обоснование атомно-абсорбционного
метода определения концентрации вещества в пламени без примене-
ния стандартных растворов. При расчете концентрации свободных
атомов в пламени рассматривают количество вещества, попадающее
в пламя в виде аэрозоля, распределение атомов в рабочей зоне,
скорость прохождения газов через поглощающий слой. Вычисленные
значения величины поглощения света для натрия (меди и серебра)
сравнены с экспериментальными. Экспериментальные данные исполь-
126
зованы для создания модели безэталонного атомно-абсорбционного-
метода анализа. Математическая модель была основана на исполь-
зовании уравнения Фойхта. Основной неизвестный параметр — это
степень диссоциации соединений или коэффициент, учитывающий
влияние лоренцовского уширения в общем контуре поглощения
линии. При оценке эффективности метода для определения натрия
принималось, что соединения натрия при температуре 2273 К дис-
социируют на 40%. Получено хорошее совпадение рассчитанных и
экспериментальных данных. Таким образом, сделан вывод о воз-
можности определения одного из указанных параметров в пламени,
если другой известен.
Проведено сравнение расчетных величин, полученных для зависи-
мости интегрального поглощения линии от концентрации атомов
в пламени с известными экспериментальными данными для линий
натрия [845]. Показано, что при малых концентрациях свободных
атомов в пламени интегральное поглощение пропорционально кон-
центрации атомов и не зависит от параметров спектральной линии,
влияющих на поглощение лишь при высоких концентрациях.
Обсуждено влияние источника света, атомизатора [30, 420, 870,
1193], конструкции распылителей и горелок [872, 910], состава
раствора^ на результаты анализа, сравниваются пределы обнару-
жения.
Показано, что реабсорбция при определении натрия по первому
резонансному дублету наблюдается при малых концентрациях на-
трия, поэтому для повышения концентрационной чувствительности
метода рекомендуется использовать второй резонансный дублет
[1040]. Это особенно характерно для турбулентного пламени.
При использовании спектрофотометров фирмы «Бекман» (модель
979 и ДИ-2) для натрия и еще 23 элементов с помощью математиче-
ского выражения определена «динамическая область» концентраций,
в которой рекомендуется проводить измерения: (2,0—2,8)-10~3 мкг/мл
[1078].
Для уменьшения расхода раствора предложено применять комби-
нированную горелку-распылитель со скоростью подачи раствора
25 мл/с [910]. Атомизатор — пламя водород—кислород, предел обна-
ружения натрия 0,008 мкг/мл. В работе [77] толщину поглощающего
слоя увеличили втягиванием пламени пропан—бутан—воздух при
помощи насоса в абсорбционную кювету. Обсуждено влияние раз-
личных факторов на градуировочные графики при определении
натрия методом атомно-абсорбционного анализа [935, 991].
Подробно обсуждено влияние органических растворителей на
результаты атомно-абсорбционного анализа сточных вод [803]. Пока-
зано, что если концентрация органических растворителей много мень-
ше 0,1 %, то нет влияния на абсорбцию натрия в интервале концент-
раций 0,1—5 мкг/мл. Приведены данные об определении натрия мето-
дом атомной абсорбции при применении монохроматора высокого
разрешения Span 101 [870]. Пределы обнаружения натрия при исполь-
зовании линейчатого источника света — лампы с полым катодом —
и источника сплошного излучения'—дуговой ксеноновой лампы —
'127
0,04Г/и 0,025 мкг/мл соответственно. Описано применение спектро-
фотометра с детектором-видиконом, оснащенным ЭВМ, позволяю-
щим определять одновременно несколько элементов. Экран видикона
установлен в фокальной плоскости монохроматора; сигнал с видико-
на поступает в оптический многоканальный анализатор, который
формирует 500-канальный спектр. В работе прибора предусмотрены
программы для учета мешающих влияний, коррекции фона, внутрен-
него стандарта, оценки полиномиальных моделей методом наимень-
ших квадратов и т. д. Приведены результаты одновременного опреде-
ления натрия, калия, лития и кальция [755].
Определение натрия во фториде магния [504]. Метод позволяет
определять 1-10~2—3-10-1% натрия после растворения образца
в смеси борной и соляной кислот. Измеряют интенсивность излуче-
ния резонансной линии натрия 589,0—589,9 нм и фона спектра с обе-
их сторон этой линии (7$;, 1^). Среднее арифметическое двух послед-
них величин (Ат,) вычитают из значений интенсивности излучения
линии натрия (1л+ф)- По полученным для эталонов данным строят
градуировочный график в координатах: величина фототока (1Л+Ф —
— 7ф) — концентрация натрия (в % мае.). Для определения натрия
в контрольной пробе используют метод добавок.
Навеску фторида магния 0,5 г растворяют в 10 мл боратного растворителя
{растворяют 30 г борной кислоты в 317 мл HF пл. 1,34 г/см3 и разбавляют водой
до метки при нагревании в платиновом тигле). Одновременно проводят контроль-
ный опыт. Раствор переносят в мерную колбу вместимостью 50 мл и разбавляют
водой до метки. Раствор разбавляют водой в 10 раз, вводят в пламя при постоян-
ном давлении воздуха 0,6—0,9 ати и фотометрируют.
Для получения эталонных растворов 0,5 г препарата помещают в платино-
вый тигель, вводят различные количества соли натрия до концентрации в конеч-
ном объеме 0,01—0,3% мае. по отношению к фториду магния, прибавляют 10 мл
боратного растворителя и осторожно нагревают до полного растворения. Раство-
ры переносят в мерные колбы вместимостью 50 мл и разбавляют водой до метки.
Для определения натрия эти растворы разбавляют водой в 10 раз.
Определение натрия в солях цинка [514]. Метод применен для опре-
деления 5-1O3—5-10-2% натрия в сульфате и хлориде цинка.
Спектр возбуждают в воздушно-ацетиленовом пламени при исполь-
зовании пламенного фотометра «К. Цейсс» (модель III) с интерфе-
ренционными светофильтрами для выделения характеристического
излучения натрия. Давление воздуха 0,4 кг/см2, давление ацетилена
35—40 мм вод. ст. Метод позволяет определять также калий и каль-
ций.:
Стандартные растворы готовят добавлением известных количеств натрия
к раствору хлорида или сульфата цинка, не содержащему определяемых эле-
ментов. Раствор хлорида цинка готовят растворением люминофорного оксида
цинка в разб. НС1. Раствор ZnSCb готовят растворением трижды перекристалли-
зованной соли в бидистилляте. Используемые в ходе анализа растворы готовят
разбавлением головных растворов, содержащих 100 мг/л натрия (в пересчете на
металл) и 100 г/л соответствующей соли цинка (в расчете на соль). Используемые
в ходе анализа растворы содержат 1; 3; 6 и 10 мг/л натрия и 20 г/л соли цинка.
Анализируемые пробы растворяют в бидистилляте. Фотометрирование про-
водят по способу ограничивающих растворов. Содержание натрия в пробе рас-
128
считывают по формуле
[Со — Ci ”1
-----— (п — пх) + Q ,
rtg — ri>l J
где q и са — содержание натрия в стандартных растворах (в мг/л), наиболее
близко находящихся к содержанию натрия в образце; п, щ и м2 — отклонения
гальванометра для раствора анализируемой пробы и стандартных растворов со-
ответственно.
Определение натрия в хлориде бария [270]. Метод применен для
определения 0,01—0,05% натрия (и калия) в хлориде бария, предел
обнаружения натрия 0,1 мкг/мл. Спектр возбуждают в пламени
воздух—пропан—бутан и регистрируют на пламенном фотометре
«Цейсс» (модель III) с интерференционными светофильтрами. Ме-
шающее влияние фона бария устраняют добавлением в пробу нитра-
та алюминия.
Навеску хлорида бария 0,2 г помещают в мерную колбу вместимостью
100 мл, растворяют в воде, добавляют 10 мл 20%-ного раствора A1(NO3)S, разбав-
ляют до метки водой и перемешивают. В мерные колбы вместимостью 100 мл
помещают по 20 мл воды, добавляют 1—2—4—6—8—12 мл раствора с содержа-
нием натрия 0,01 мг/мл, разбавляют водой до метки и перемешивают. Через со-
ответствующий интерференционный светофильтр фотометрируют излучение эта-
лонных растворов в порядке возрастания концентраций и испытуемые пробы.
При фотометрировании эталонных растворов вводят поправку иа фон пламени
при распылении воды. При фотометрировании пробы учитывают содержание
определяемой примеси в нитрате алюминия. Содержание натрия находят методом
ограничивающих растворов.
Определение натрия в солях алюминия [2691. Метод применен для
определения 0,05—0,25% натрия в солях алюминия AlClg-6Н2О,
A1(NOS)S-9Н2О и A12(SO4)s- 18Н2О. Спектр возбуждают низкотем-
пературным пламенем пропан—бутан—воздух при использовании
пламенного фотометра «Цейсс» (модель III) с интерференционными
светофильтрами. Определению 0,04—0,5 мг/100 мл натрия не ме-
шает алюминий в интервале концентраций 14—22 мг/100 мл и анио-
ны NO3, SO4” и С1_ при’ содержании <0,1 М. Натрий определяют
методом ограничивающих растворов.
Навеску соли алюминия 0,2 г растворяют в воде в мерной колбе вмести-
мостью 100 мл и разбавляют водой до метки. Через соответствующий интерферен-
ционный светофильтр фотометрируют серию эталонных водных растворов, содер-
жащих 0,04—0,25 мг/100 мл натрия в порядке возрастания концентраций,
и испытуемые растворы. Для уменьшения инструментальной ошибки повторяют
фотометрирование в обратной последовательности начиная с высшей концентра-
ции. Вычисляют среднее арифметическое из этих показаний. При фотометриро-
вании испытуемой пробы и эталонных растворов вводят поправку на фон пламе-
ни при распылении воды. По данным фотометрирования находят, между какими
эталонами заключен испытуемый раствор.
Содержание натрия определяют по формуле
Na, % = I et4
(са — ci) (А — Ai)
А2 — Ai
100
g.1000 ’
где Aj и А2 — показания гальванометра для двух эталонных растворов; А —
показание гальванометра для испытуемого раствора; q и са — концентрации
натрия в двух, эталонных растворах (в мг/100 мл); g — навеска пробы (в г).
5 В. М. Иванов и др.
129
Определение натрия в пентаоксиде ванадия [271]. Метод применен
для определения 2-10“3—2’10-2% натрия (калия, кальция) в
пентаоксиде ванадия; предел обнаружения натрия составляет
0,05 мкг/мл, относительная погрешность определения 10—12%.
Спектр возбуждают в пламени воздух—ацетилен и регистрируют
спектрофотометром на основе спектрографа ИСП-51 с фотоэлектри-
ческой приставкой ФЭП-1. Используют резонансную линию натрия
588,995-589,593 нм.
Навеску пентаоксида ванадия 0,5 г с точностью до 0,01 г растворяют в ко-
нической кварцевой колбе в 12 мл НС1 (2 : 3) при нагревании до кипения. Раствор
охлаждают, переносят количественно в мерную колбу вместимостью 100 мл,
вводят 8 мл 20%-ного раствора 8-оксихинолина в ледяной уксусной кислоте, раз-
бавляют водой до метки и перемешивают.
Для приготовления эталонных растворов в мерные колбы вместимостью
100 мл помещают по 25 мл воды, 12 мл НС1 (2 : 3), 8 мл раствора 8-оксихинолина
и добавляют 1—2—4—6—8—10 мл стандартного раствора с содержанием натрия
0,01 мг/мл. Растворы разбавляют водой до метки и перемешивают. Кроме эталон-
ных, готовят раствор, содержащий только 12 мл НС1 (2:3) и 8 мл раствора
8-оксихинолина в 100 мл воды (контрольный раствор).
Фотометрируют испытуемый, контрольный и эталонные растворы в порядке
возрастания концентраций, вводят поправку на фон с обеих сторон аналитичес-
кой линии и поправку на содержание примесей в контрольном растворе. Содер-
жание натрия в анализируемом растворе находят методом ограничивающих
растворов.
Определение натрия в карбонате марганца [515]. Метод применен
для определения 0,1—1% натрия. Спектр возбуждают воздупшо-
ацетиленовым пламенем при использовании пламенного фотометра
«К. Цейсс» (модель III) с интерференционными светофильтрами для
выделения характеристического излучения натрия. Давление воз-
духа 0,4 кг/сма, давление ацетилена 35—40 мм вод. ст.
Стандартные растворы готовят добавлением известных количеств натрия
к раствору МпС12, не содержащему натрия. Очистку соли марганца проводят
следующим образом. Готовят раствор M11SO4 с плотностью 1,12 г/см®. К нагре-
тому до кипения раствору прибавляют небольшими порциями при интенсивном
механическом перемешивании свежеприготовленный раствор полисульфида
аммония до зеленого окрашивания выпадающего в осадок MnS. Осадок отфиль-
тровывают на воронку Бюхнера и промывают на фильтре горячей дистиллиро-
ванной водой. Затем промывание продолжают декантацией при кипячении. Опе-
рацию декантации повторяют 6—8 раз. Отмытый осадок переносят в стакан и
растворяют в НС1. Полученный раствор МпС12 разбавляют бидистиллятом до
требуемой концентрации. Содержание марганца в растворе определяют комп-
лексонометрически.
Используемые в ходе анализа растворы готовят разбавлением головных рас-
творов, содержащих 100 мг/л натрия (в расчете на металл) и 10 г/л марганца
(в расчете на карбонат марганца). Используемые в ходе анализа растворы содер-
жат 1; 3; 6 и 10 мг/л натрия и 1 г/л марганпа.
Навеску пробы 0,5 г помещают в мерную колбу вместимостью 500 мл, до-
бавляют 3 мл бидистиллята, 1,5—2 мл конц. НС1 и несколько капель 30%-ного
раствора Н2О2. После растворения пробы раствор разбавляют до метки бидистил-
лятом. Фотометрируют по способу ограничивающих растворов. Расчет приведен
в разделе «Определение натрия в солях цинка».
Определение натрия в породах [99]. Метод прим енен для опреде-
ления натрия в глинистых фракциях (0,5—2%), флогопите (0,8%)
и глауконите (1,5% ). Селективность определения повышают исполь-
130
зованием низкотемпературного пламени (1120—1160° С) этанол—воз-
дух, поскольку ионизация натрия в этом пламени (1,26-10-8%)
значительно ниже, чем при использовании пламен ацетилен—кисло-
род (26,50%) или водород—кислород (5,23%). При фотометрирова-
нии используют резонансную линию натрия 588,995—589,6 нм,
градуировочные графики строят в координатах интенсивность излу-
чения—концентрация в интервале концентраций натрия 1 —
80 мкг/мл. В стандартные растворы вводят литий и калий в соот-
ношении по массе Li : Na : К = 0,1 : 2 : 1. Определению не мешают
1 Обратные избытки ионов Fe, Al, Ti, Саи Mg, до 2% об. NHS, НСЮ4,
сильно мешают анионы NO3, SO4, С1~, РО|~.
Тонкоизмельченную навеску пробы 0,1 г помещают в платиновую чашку,
смачивают несколькими каплями воды, прибавляют 5—7 мл конц. HF, 3—5 мл.
конц. H2SO4 и полностью разлагают при нагревании на песчаной бане. После уда-
ления паров серной кислоты пробу обрабатывают небольшими порциями горя-
чей воды и фильтруют в мерную колбу вместимостью 100 мл. Раствор подкисля-
ют 1—2 мл H2SO4 (1 :1), разбавляют водой до метки и тщательно перемешивают.
Определяют содержание натрия.
Определение натрия в чугуне [26]. Метод применен для определе-
ния натрия в чугунах — передельном (0,004%), ковком (0,02%)
и литейном (0,045%). Влияние железа устраняют экстракцией ди-
этиловым эфиром. Спектры возбуждают в пламени пропан—воздух,
градуировочный график строят в интервале концентраций натрия
0,01—60 мкг/мл. Предел обнаружения натрия 10-4 мкг/мл. Исполь-
зован метод ограничивающих растворов.
Навеску чугуна 4 г помещают в колбу вместимостью 300 мл, прибавляют
75 мл НС1 (1 : 1), нагревают до полного растворения навески, охлаждают и ос-
торожно прибавляют 7 мл конц. HNOS. Содержимое колбы нагревают, избегая
кипения, прибавляют 10 мл конц. НС1 и выпаривают до получения сухого сстат-
ка. Соли растворяют в 50 мл НС1 (1 : 1). прибавляют 25 мл воды, охлаждают
и фильтруют в колбу вместимостью 150—200 мл. Вводят 20 мл 1%-ного раствора
НС1, 1 мл 30%-ного раствора Н2О2, раствор нагревают и упаривают до сиропо-
образного состояния. Колбу снимают с плитки, прибавляют 5 мл конц. НС1,
взбалтывают и охлаждают. Содержимое колбы переливают в делительную во-
ронку вместимостью 500 мл, ополаскивая стенки колбы 2 раза по 10 мл конц.
НС1, содержащей диэтиловый эфир и 140 мл безводного диэтилового эфира. Со-
держимое воронки охлаждают под током воды в течение 2 мин при постоянном
перемешивании. Затем добавляют дистиллированной воды и еще раз взбалтывают.
После прибавления воды и взбалтывания нижний слой желтого цвета переливают
в коническую колбу, а верхний слой зеленого цвета отбрасывают. Раствор
в колбе упаривают при медленном и слабом подогревании до сиропообразного
состояния, прибавляют 5 мл воды и 1 мл НС1 (1 : 2). Нагревают до полного рас-
творения, фильтруют в мерную колбу вместимостью 50 мл, разбавляют водой до
метки и фотометрируют.
Определение натрия в ПАН-лаках [435]. Метод применен для оп-
ределения натрия (и калия) в ПАН-лаках (10%-ный раствор полиак-
рилнитрила в диметил формамиде), в ДМФА, смесях ПАН + иод,
ПАН + хинон. Предел обнаружения натрия в ДМФА 1-10~6%,
в ПАН-лаке — 2-10-Б%. При определении 1-10~4% натрия относи-
тельное стандартное отклонение составляет 0,01—0,02. Спектры
возбуждают в пламени воздух—пропан—бутан, регистрируют на
5*
131
спектрофотометре «Перкин-Элмер» (модель 403). Иод и хинон силь-
но влияют на интенсивность излучения натрия, поэтому анализ
проводят методом добавок, вводят натрий в виде хлорида в дважды
перегнанный ДМФА. Наиболее удобным объектом анализа является
ДМФА, обладающий малой вязкостью и поверхностным натяжением.
После однократной перегонки в кварцевом аппарате содержание на-
трия в ДМФА не превышает 1-10~6%. Другие объекты и композиции
из-за большой вязкости перед анализом разбавляют в соотношении
1 : 4.
Градуировочный график строят в интервале концентраций натрия
5-10-6—1-10~4% в координатах I—с; для концентраций натрия
1-10-4—1-10-3 — в координатах IgZ—Igc. От наиболее чистой по
натрию анализируемой пробы отбирают несколько раз по 1 мл, вво-
дят в них по 4 мл одного из стандартных образцов. Приготовленные
таким образом рабочие стандартные образцы содержат в пересчете
на анализируемую пробу количество примесей натрия (калия) в че-
тыре раза больше их содержания в стандартных образцах на основе
ДМФА.
Определение натрия в хроме [406]. Метод применен для определе-
ния 0,1—1% натрия в технических порошках хрома и растворах их
выщелачивания, содержащих <Д5—20 г/л магния и <Z5 г/л хрома.
Порошки хрома и полупродукты металлотермического восстановле-
ния хлоридов содержали (в %): Сг 50—95; Mg < 6; Мп < 8; Na «С 1.
Спектр испускания возбуждали в пламени воздух—пропан—бутан,
эмиссию натрия измеряли при 589 нм. Определению натрия не ме-
шают до 2,5 г/л марганца, 20 г/л магния. В присутствии 2,0—2,5 г/л
хрома (соответствует навеске хромового порошка 0,25 г) результаты
определения натрия завышены на 0,25—0,30%. Поэтому эталонные
растворы готовят на хромовой основе. При определении 0,1—0,5%
и 0,5—1,0% натрия погрешность определения составляет 0,03 и
0,5% соответственно. Анализ рекомендовано проводить методом огра-
ничивающих растворов.
Применение метода атомно-абсорбционной спектрометрии с не-
пламенной атомизацией. Этот метод существенно снижает предел об-
наружения элементов и влияние состава раствора на результаты ана-
лиза.
Метод применен для определения следов натрия в воде [760]. Ис-
пользована графитовая кювета HGA-74; газ-носитель — аргон.
Градуировочные графики прямолинейны в интервале концентраций
натрия (1,3—9,2)-10~8%, предел обнаружения 1,1-10~9%. При уве-
личении объема раствора в 10 раз предел обнаружения может быть
снижен в 8 раз. Отмечается, что хлориды, сульфаты, аммиак и гид-
роксиды не влияют на результаты определения при использовании
спектрофотометра «Перкин-Элмер» (модель 305В).
Описан атомно-абсорбционный метод определения натрия с пре-
делом обнаружения 3-1011 г по линии 589,0 нм с непламенной ато-
мизацией вещества [704]. Анализируемое вещество помещают в ци-
линдрическую кварцевую камеру, которую устанавливают в лодочку
из графита, танталовой или вольфрамовой фольги. Лодочку нагре-
132
вают током 30—50 А до температуры 2200° С в течение 0,1 с. Изуче-
но влияние атмосферы и давления в камере, химического состава
раствора, материала лодочки, температуры нагревания.
При определении натрия и калия предел обнаружения лимитиру-
ется чаще всего чистотой реактивов, а не возможностями метода
определения. Поэтому весьма интересна работа [1261], в которой
детально рассмотрены источники загрязнений и пути их снижения
при атомно-абсорбционном анализе жидких проб с использованием
способа непламенной атомизации.
Метод внутрирезонаторной атомно-абсорбционной спектрометрии.
Внутрирезонаторная спектрометрия.— новый вариант атомно-аб-
сорбционного анализа с использованием лазерной техники. Этот
метод применен для определения натрия с непламенной атомизацией
пробы [933]. Кювету помещают внутрь резонатора — лазера на кра-
сителе родамин 6Ж. Концентрация красителя соответствует макси-
мальной генерации в области линейного поглощения натрия для
резонансного дублета 589,6—588,6 нм. Для определения натрия
используют дифракционный спектрограф. Изучено влияние темпе-
ратуры кюветы и длительности накачки на предел обнаружения.
Сравнивают данные для четырех лазеров, различающихся длитель-
ностью импульсов, полушириной светового импульса лампы накачки,
областью генерации и длиной кюветы. При изменении температуры
кюветы от 100 до 155° С предел обнаружения натрия изменялся от
12-10—6 до 82-10-6 мм рт. ст. Если кювета находится вне лазерного
резонатора, то предел обнаружения натрия возрастает в 200 раз.
Внутрирезонаторная атомно-абсорбционная спектрометрия является
перспективным методом снижения предела обнаружения элементов.
ДРУГИЕ СПЕКТРАЛЬНЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ НАТРИЯ
Для определения субмикроколичеств натрия используют флуо-
ресцентные [656], атомно-флуоресцентные [59, 108, 109, 119, 158,
193, 775, 783, 848, 906, 1195] методы, метод резонансно-ионизацион-
ной или ступенчатой фотоионизации [109, 775, 860, 1046, 1235, 1236,
1258], оптико-гальванический [489, 782] метод, метод спектрометрии
ядерного магнитного резонанса [1239], масс-спектрометрию [902,
964, 1156], рентгенофлуоресцентную спектроскопию [304, 664, 851,
1051, 1247].
Атомно-флуоресцентный метод позволяет определять 10-8—10-14 г
вещества в самых разнообразных объектах, а также локальные
концентрации в светящемся облаке [158, 159]. В этом методе может
быть использована бездисперсионная аппаратура. Для получения
атомного пара применяют пламенные и непламенные атомизаторы,
в качестве источника света — ксеноновые лампы СВД (предел обна-
ружения натрия 8-10-7 г). Лазерное возбуждение атомов натрия
в пламени позволило определить на фоне загрязнений атмосферы
104 атомов в 1 см-8. Для наблюдения флуоресценции натрия исполь-
зуют чаще всего резонансные дублеты 589,0—589,6 и 330,23—
330,30 нм.
133
Рассмотрены условия определения натрия с пределом обнаруже-
ния 10_®% методами атомно-абсорбционного и атомно-флуоресцент-
ного анализа в оксидах редкоземельных элементов (иттрия, лантана,
неодима, празеодима и тербия) [119]. Применялся метод импульсно-
го электротермического испарения вещества из графитового тигля
при пропускании тока 200—400 А. Спектрофотометр сконструирован
на базе монохроматора МДР-2, детектор — фотоумножитель ФЭУ-18.
Помехи уменьшаются при применении модулированного первичного
излучения на частоте 756 Гц. Эталонирование осуществляли на осно-
ве графитового порошка.
Рассчитан [1195] квантовый выход для натрия в зависимости от
типа пламени и чистоты аргона при 3000° С.
Особенно перспективно применение в качестве источника света
лазеров на красителях непрерывного действия, а также лазеров
с оптической накачкой с длительностью импульсов 10~10—10-11 с.
При определении натрия атомно-флуоресцентным методом (предел
обнаружения 0,2 мгк/л) с применением лазеров на красителях
с перестраиваемой частотой линейность градуировочного графика
наблюдается в пределах 5 порядков [906]. На аналитический сигнал
не влияют флуктуации интенсивности лазера благодаря насыщению
электронных переходов.
Изучены [783] возможности применения резонансной пламенной
атомно-флуоресцентной спектроскопии при возбуждении непрерыв-
ным спектром лазера на красителях с двоякопреломляющим филь-
тром со сканирующим механизмом на выходе.
На предел обнаружения натрия влияют дробовой и фликкер-
шумы пламени и рассеяние света лазера с непрерывным спектром.
В этом методе предел обнаружения приближается к пределу обна-
ружения в атомно-эмиссионном методе. Для резонансной флуорес-
ценции отношение сигнал/шум не возрастает при повышении мощ-
ности лазера до 1 кВт. Для нерезонансной флуоресценции высокая
мощность приводит к снижению тушения флуоресценции.
На примере определения натрия в графитовом порошке проверена
возможность использования резонансной атомной флуоресценции при
использовании перестраивающегося импульсного лазера [59]. При-
менялся лазер на основе красителя родамин 6Ж, накачку проводи-
ли излучением второй гармоники неодимо-кадмиевого лазера. Дли-
тельность импульса составляла 2-10~8 с, мощность 10Б Вт, ширина
линии генерации 0,1 нм. Атомизацию натрия проводили в атмосфере
аргона, температура проволоки 1000° С, концентрация натрия была
равна 1,2-108 ат/см3. Минимальный регистрируемый сигнал флуо-
ресценции 5-1017 Дж. Предел обнаружения ограничивался флуктуа-
циями релеевского рассеяния.
Для определения 10-10 г/л натрия в пламени использован лазер
на красителе родамин 6Ж в метаноле с концентрацией 50 мг/л, имею-
щий широкую полосу пропускания 80 нм [887]. Описан атомно-
флуоресцентный метод определения ряда примесей, в том числе и
натрия, в водопроводной воде. Предел обнаружения натрия 2-
•10'4 мкг/мл [193]. Сравнивались аналитические возможности атом-
134
но-флуоресцентного метода определения натрия при использовании
двух источников: непрерывного линейчатого (высокочастотная без-
электродная лампа ВСБ-2) и импульсного лазера на красителе рода-
мин 6Ж с атомизацией натрия в пламени. Применение в качестве ис-
точника света лазера на красителе на три порядка снижает предел
обнаружения натрия по сравнению с использованием лампы ВСБ-2.
Отмечено, что при определении натрия методом атомной флуоресцен-
ции лучше использовать пламя пропан—бутан—воздух. Метод при-
менен для определения натрия в воде.
Большие возможности заложены в новом методе определения суб-
микроколичеств элементов — методе ступенчатой фотоионизации
атомов в пламени с помощью лазеров на красителях (СФА) или СЛИ—
ступенчатой лазерной ионизации атомов.. Он основан на регистрации
в просвечиваемом лазером объеме газа электрических зарядов, кото-
рые возникают вследствие селективной ионизации атомов. В качестве
атомизаторов применяют различные пламена, можно использовать
непламенный вариант. Метод позволяет детектировать единичные
атомы.
Метод СФА применен для определения натрия с пределом обна-
ружения 12 мкг/мл (775]. Установлено, что сигнал СФА обусловлен
двухступенчатым процессом, который происходит под воздействием
лазеров на красителе и на неодиме. Для накачки лазера на красите-
ле использовали вторую гармонику (632 нм) лазера на неодиме мощ-
ностью 300 кВт. Детектор ионов — нихромовая проволока диамет-
ром 0,6 мм, находящаяся под напряжением 600 В. Предложен новый
метод регистрации усиления ионизации элементов в пламени под
действием лазера на красителе с перестраиваемой частотой; на высоте
1 см над наконечником горелки устанавливались параллельно друг
другу на расстоянии 1 см два вольфрамовых стержня, к которым при-
кладывалось напряжение 500—1500 В [1236]. Луч пульсирующего
лазера проходит перпендикулярно стержням. Аналитический сигнал—
импульс тока, протекающего между стержнями и наконечником го-
релки. Линейная зависимость сигнала от концентрации элемента на-
блюдается в пределах двух порядков. Предел обнаружения натрия
0,05 мкг/мл. Чувствительность зависит от разности энергии возбуж-
дения основного уровня и потенциала ионизации элемента. Изучено
влияние мощности лазера, спектральной ширины пучка лазерного
излучения и приложенного потенциала.
Разработаны методы лазерной ступенчатой фотоионизации и ре-
зонансной флуоресценции для определения субмикроколичеств
натрия с пределами обнаружения 10-1Б и 6-10-14 г соответственно
при атомизации хлорида натрия непламенным методом в графитовом
стаканчике [109]. Для осуществления ступенчатой фотоионизации
натрия использовали излучение азотного лазера и одного лазера на
красителях или первой гармоники неодимового лазера и двух лазеров
на красителях: азотный лазер мощностью 100 кВт с частотой повторе-
ния импульсов 10 нс. Мощность второго лазера на неодим-алюми-
ний-иттриевом гранате в первой гармонике 1,2 мВт, во второй
300 кВт, частота повторения импульсов 12,5 Гц. Ширина линии ла-
135
зеров на красителях 0,03 нм. Кювета атомизатора наполнялась ар-
гоном при атмосферном давлении. Излучение лазера с диаметром луча
2 нм направляли в кювету атомизатора и фокусировали над графи-
товым стаканчиком. Для изучения сигнала флуоресценции натрия
использовали монохроматор SPM-1 и ФЭУ-19, сигнал регистриро-
вали осциллографически после усиления с помощью широкополос-
ного усилителя УИС-2М. Ионы регистрировали с помощью зонда, на
который подавали положительный потенциал 0,6—2 кВ. Градуи-
ровочный график при определении натрия методом лазерной атом-
ной флуоресценции линеен в] пределах 3-10~12—10~9 г натрия, пре-
дел обнаружения по 2в-критерию равен 6-10~14г. Оценка предела
обнаружения для СФА с помощью градуировочного графика невоз-
можна из-за высокого фонового фотоионизационного сигнала. Предел
обнаружения равен (1—3)-10~1Бг.
При использовании двухступенчатой схемы ионизации натрия
возникает фотоэффект за счет попадания рассеянного излучения
азотного лазера на графитовый стаканчик. Флуктуации сигнала от
фотоэффекта ограничивают величину cmin, поэтому использовали
трехступенчатую схему ионизации с применением неодим-алюминий-
иттриевого лазера. Это позволило исключить шумы, связанные с фо-
тоэффектом на поверхности атомизатора, а также шумовые эффекты
многофотонной ионизации. Детально изучено влияние температуры
нагревания графитового стаканчика, материала зонда, его полярно-
сти и напряжения на зонде на сигнал фотоионизации. Лучшие усло-
вия для соотношения сигнал/шум получены для зонда из нержавею-
щей стали (d = 1,1 мм), на который накладывают напряжение
600 В, температура стаканчика в конечной стадии нагревания 1900° С.
В оптико-гальваническом методе (ОГ) селективную ионизацию
атомов в пламени и плазме осуществляют при использовании только
одноступенчатого возбуждения высоколежащих состояний атомов.
Для этой цели необходимо использовать лазеры, дающие излучение
в вакуумной ультрафиолетовой области. ОГ-эффект применен для
определения различных элементов в пламени [782].
Натрий определяли по резонансной линии 589 нм в пламени аце-
тилен—воздух [185]. Облучение проводили модулированным излу-
чением (2 кГц) перестраиваемого лазера на красителях с АХ =
= 0,003 нм мощностью 500 Вт/см2-нм. Для концентрации натрия
10 мкг/л изменение тока составляло 2%. Градуировочный график
в билогарифмических координатах близок к линейному в диапазоне
концентраций натрия 0,01—1 мг/л. Отношение сигнал/шум при кон-
центрации натрия 2 мг/л не больше 20.
Флуоресцентный метод позволяет определять 1,9-10-7—3,8-
•10-4% натрия с относительным стандартным отклонением 0,02 для
концентрации натрия 1,9-10~6% и 0,06 — для концентрации натрия
1,9-IO-7%; предел обнаружения натрия 8-10“8% [656].
Описано применение рентгенофлуоресцентного метода для опре-
деления 0,11—0,98% натрия в А12О3 (стандартное отклонение 0,03%)
[329], глиноземах и продуктах стекольного производства [2], в гор-
ных породах [11, 641, 800, 1260] и высокочисгых реактивах [977].
136
Рентгеноспектральный метод применяли для определения боль-
ших количеств натрия (1—10%) с использованием прибора БАРС-1
с трубкой БХ-3, счетчиком СРПП-22М с селективным фильтром [1].
В качестве анода использовали алюминиевую фольгу. Предел обна-
ружения натрия 0,15%. В горных породах в числе основных породо-
образующих элементов определяли 2,7—5,4% Na2O на квантометре
АРЛ-72000 [66].
Масс-спектрометрическим методом натрий и многие элементы оп-
ределяли с низкими пределами обнаружения в деионированной воде
[306, 994], кислотах [994], UF6 [822], магнии и его соединениях
[261, 306], ниобии и тантале особой чистоты [1194], кремнии [1036],
полупроводниковых материалах [105], атмосферных твердых части-
цах [965]. Пределы обнаружения натрия ^>10-5%.
Глава IX
АКТИВАЦИОННЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
НАТРИЯ
Активационный анализ — метод определения ничтожно малых
количеств вещества, основанный на образовании радионуклидов
в результате облучения анализируемого вещества однородным
потоком ядерных частиц. По типу частиц, используемых для облу-
чения, различают нейтронно-активационный анализ, активационный
анализ с помощью заряженных частиц и фотонно-активационный
анализ, основанный на реакции (у, п). В настоящее время наиболь-
шее значение имеет нейтронно-активационный анализ, в котором
используют поток медленных нейтронов (£’<0,001 МэВ) ядерного
реактора или поток быстрых нейтронов (£ = 1—14 МэВ) нейтронно-
го генератора. Для быстрых нейтронов характерны реакции типа
п, р; п, а и п, 2и; при облучении медленными нейтронами протекает
только реакция п, у. Вследствие высоких сечений реакций (1 —
1000 барн) медленные нейтроны обеспечивают большую чувствитель-
ность анализа, нежели быстрые нейтроны, для которых сечение реак-
ций существенно ниже (0,001—0,1 барн).
Чувствительность активационного анализа обусловлена интен-
сивностью потока ядерных частиц, сечением ядерной реакции, перио-
дом полураспада и характером излучения образующегося радио-
нуклида.
НЕЙТРОННО-АКТИВАЦИОННЫЙ МЕТОД
В табл. 46 приведены данные по определению некоторых элемен-
тов нейтронно-активационным методом. Благодаря отчетливо выра-
женным различиям в свойствах отдельных радионуклидов активацион-
ное определение данного элемента в значительной мере свободно от
мешающего влияния других элементов.
137
Таблица 46
Предел обнаружения активационного анализа при облучении образцов
потоком нейтронов интенсивностью 1012 н/см2-с [502]
Определяемый элемент Продукт активации Предел обна- ружения, г Определяе- мый элемент Продукт активации Предел обна- ружения, г
Германий 76Ge 5-10-’ Мышьяк 76 As 5-10-и
Железо s’Fe 10-» Натрий MNa 10-ю
Золото 198 Au 5-10-12 Никель «'Ni IO’8
Индий nein 10-и Олово iziSn 5-10-e
Иод 128 J io-» Ртуть 203Hg IO”9
Кадмий 116Cd 10-» Свинец гоерь 5-Ю-6
Калий 42K 10-8 Селен siSe 5-Ю-8
Кальций «Са 10-’ Сера S2S 5-Ю-8
Кислород «О 5.10-’ Серебро noAg 5-Ю-о
Кобальт «’Co 5-10-1° Сурьма 122Sb 10-ю
Кремний 3JSi IO-8 Фосфор 32p 5-10-ю
Марганец 56Mn 10-и Хлор 38C1 5-Ю-о
Медь MCu 10-ю Хром “Cr IO"8
Молибден 8<JMo IO-» Цинк 69Zn 5-10-0
Таблица 47
Характеристика ядерных реакций. MNa с нейтронами [17, 525, 1220]
Реакция Образую- щийся радио- нуклид Сечение реакции, барн Г1/2 Энергия излучения, МэВ Радио- токсич- ность
(3-излучение у-излучение
n, 7 2*Na 0,534 15,03 ч 14,97+0,02 ч [928] 1,390(100%) 1,368 (100%); 2,754(100%) Слабо- опасен
п, 2п 22Na 6,0-Ю-о 2,603 года 0,546(90,5%) 0,511; 1,275 (100%) Средне- опасен
п, Р 23Ne 0,7-10-s 37,6 с 2,4 (1%); 3,95 (32%) 4,39 (67%) 0,44 (32%) 1,63 (0,8%)
п, а 2JJ? 0,47-10-s 11,56 с 5,42 (99%) 1,631
Выгодные ядерные характеристики радионуклидов натрия
(табл. 47), 100%-ное содержание a3Na в природной смеси, достаточно
высокое сечение активации для медленных нейтронов обусловливают
высокую чувствительность активационного определения натрия даже
при малых временах облучения. Следует, однако, иметь в виду, что
при использовании ядерной реакции (и, у) немедленной регистрации
24Na по у-пикам с энергией 1,368 и 2,754 МэВ мешают соответственно
41Аг с периодом полураспада 1,827 ч и энергией у-излучения
138
1,370 МэВ и 5вМп (Т112 = 2,578 ч, Еу — 2,657 МэВ). Поэтому обыч-
но рекомендуется измерять радиоактивность 24Na через 10—15 ч
после активации, когда распадутся короткоживущие радионуклиды.
При использовании в активационном анализе быстрых нейтронов
необходимо учитывать вклад параллельно протекающих ядерных
реакций 24Mg(n, p)24Na и 27Al(n, a)24Na в образование радионуклида
24Na; сечение этих реакций — 1,2-10“3и0,56-10~3 барн соответствен-
но. Экспериментальное изучение мешающего влияния пороговых
ядерных реакций на активационное определение натрия в Mg и А1
с использованием реакции (и, у) показало, что степень влияния
зависит от доли быстрых нейтронов в общем нейтронном потоке [442].
Количественный активационный анализ может быть выполнен
абсолютным или относительным способом. Радиоактивность образую-
щегося радионуклида (расп/с) пропорциональна содержанию опреде-
ляемого в образце элемента, потоку ядерных частиц и сечению ядер-
ной реакции:
А = (6,02.10» траЦМ) [1 — ехр (— 0,693</Т1/г)],
где m — количество определяемого элемента, г; р — доля активи-
руемого нуклида в элементе; о — сечение ядерной реакции, см2;
/ — плотность потока ядерных частиц, частиц/см2-с; М — атомная
масса; t — время облучения, с; Г1/2 — период полураспада, с.
Так как радиоактивность облученного образца (AJ измеряют
через некоторое время t после окончания облучения, а Аг =
то формула для определения содержания искомого элемента прини-
мает вид
АгМ
m~ 6,02-10а3ро/[1 —ехр(—0,693t/7’1/!)]exp(—0,693t1/7’1/!) 1
Реализация абсолютного способа связана с рядом трудностей (необ-
ходима точная оценка значений /, о, р) и не может обеспечить погреш-
ность ниже 20—50%.
При относительных измерениях, когда вместе с анализируемым
образцом облучают один или несколько стандартных образцов с из-
вестным содержанием определяемого элемента, исключаются ошибки,
связанные с определением величин /, о, р. В этом случае количество
определяемого элемента в образце находят по формуле
m = (А/А ст) ^ст,
где А и Аст — радиоактивности анализируемого и стандартного об-
разцов, а тпст — содержание элемента в стандартном образце. По-
грешность относительного способа оценивается в 10%.
В табл. 48 приведены самые разнообразные примеры использо-
вания нейтронно-активационного анализа для определения натрия
в различных объектах.
Предложен активационный метод определения примеси натрия
в металлическом магнии, устраняющий мешающее влияние ядерной
реакции 24Mg(n, p)24Na путем предварительного электролитического
отделения натрия от магния перед облучением [873]. Чувствитель-
139
Таблица 48
Нейтронно-активационное определение натрия в различных объектах
Анализируемый объект Навеска, г Поток нейтро- нов, н/см8-с Время облуче- ния, ч Время охлажде-' НИЯ, ч Метод очистки Детектор излучения Предел обнаруже- ния, % Погреш- ность, % Лите- ратура
Алюминий и его соединения 1 0,05 В потоке 1 0,02 2.10*8 4-1013 2-Ю8 1,5-1013 0,3 1 неделя 4—60 мин 20 Экстракционно-хрома- тографическое отделе- ние натрия Без разрушения То же » Осаждение цинкуранил- аце'тата натрия Без разрушения NaJ(Tl) Торцовый счетчик Ge(Li) IO"7 2-10-3 1,4.10"* 5-—10 1—2 1 1,5 [700, 921] [467] [613] [199] [1052] [950]
Бериллии и его соединения Бор и его соеди- нения 0,5 1 0,15 0,1 5-1012 ~101з 1,2.1013 1,8-1013 0,5 3 100 2 >24 ’ 96 То же » Ионообменная хромато- графия ' Без разрушения NaJ(Tl) Ge Торцовый счетчик NaJ(Tl) 1,7.10-з 4-10-* i.io-’ 6,2.10~® [916] [63] [437] [277]
Вольфрам 0,3 0,1 0,03-0,1 0,02—0,1 8,7-1012 ~1013 5-1012 5-1012 20 10 400с 200 с 8—30 То же Концентрирование на колонке с вольфрамо- цирконатом аммония Хроматографическое отделение на анионооб- меннике Дауэкс 1x8 Без разрушения NaJ(Tl) NaJ(Tl) NaJ(Tl) NaJ(Tl) 1,7.10-3 5-10-’ 1,5.10-3 10-3 15 [451] [706] [553] [1074]
Галлий (арсенид) 0,1 1,8-1013 9 12 Экстракционное отделе- ние NaJ(Tl) 2,6-10-1 10 [24]
Анализируемый объект Навеска, г Поток нейтро- нов, н/см*-с Время облуче- ния, ч
Галлий (арсенид) 0,1 1,8.10м 1
Железо 0,5—1 5-1032 1
Калий (соли) 0,02 1,5.IO33 4—60 мин
0,005 6-1013 0,5
~1 2-108 2 мин
1012 62
Кремний и его 0,005—1 2-10Ю 2—24
соединения 1-2 —IO33 24—72
1 100
0,5-2 ЗЛО32 24—48
1 ЗЛО»3 0,3
0,1-8,1
~10» 4 с
0,01—0,2 ~1013 1—200
5.10-5 ~1013 12—20
1 ~10м 24
1—1,5 1,2-10м 15 мин
Таблица 48 (продолжение)
Время охлажде- ния, ч Метод очистки Детектор излучения Предел обнаруже- ния, % Погреш- ность, % 'Литера- тура
Экстракционное отделе- ние основы и тяжелых элементов NaJ(TI) IO-» 10 [103]
Без разрушения Ge(Li) io-® 5—10 [929]
20 То же Ge(Li) 1,5 [950]
24 » Ge(Li) 3-Ю-3 [127]
2 мин Р-Жид кост- ной сцин- 20 [993]
тилляцион- ный счетчик
Ионообменная' хромато- графия 5.10-4 5 [648]
1—30 Без разрушения NaJ(Tl) 5.10-s 7 [871, 998]
То же NaJ(TI) 6-10-5 [633, 996]
20 NaJ(Tl) 5-10-7 50 [1109]
12 » NaJ(Tl) 2.10-7 10—40 [152]
24 » Ge(Li) 5.10-е [827]
» Ge(Li) 10-’ [562]
0,5 NaJ(Tl) [788]
25—30 » NaJ(Tl) 10—40 [298]
Растворение в конц. HF и отгонка SiF4 Ge(Li) 10—15 [894]
8—10 Хроматографическое Торцовый зло-» 15 [188]
отделение на КУ-2, АВ-17 и вольфрамате счетчик
циркония Без разрушения Ge(Li) 2.10-е [1102]
Анализируемый объект Навеска, г Поток нейтро- нов. н/см**с Время облуче- ния, ч
Кремний и его 5,2—5,3 ~1013
соединения 5.10х3 ~10И 1 неделя
3 2- 10м 24
1,2-1013 70
0,3—3 1.10х3 24
1,5-10х3 14
Литий и его —1012
соединения 0,1 4. IO33 10—15
0,05—0,1 2-1012 12-15
Магний 0,15 5-1012 10 мин
1 3- 1012
Медь 0,2 3- IO3’ 2
Таблица 48 (продолжение)
Время охлажде- ния, ч Метод очистки Детектор излучения Предел обнаруже- ния, % Погреш- ность. %' Литера- тура
Разложение пробы смесью HF-[-HN.O8 с последующим отделе- нием компонентов с носителями Без разрушения Ge(Li) 1,5.10-’ 1,1.10-’ [319] [1018]
Ионообменная хромато- графия Стандартные аналити- ческие операции Хроматографическое Торцовый счетчик NaI(Tl) 9.10-8 4-10-8 IO”8 15 [312] [1216] [178]
6—7 разделение на ионооб- менниках КУ-2х8 и АВ-17 х 8 Без разрушения Ge(Li) IO"8 10 [1035]
36 15 То же Ионообменная хромато- графия Без разрушения Ge(Li) NaJ(Tl) 5.10-1 4-10-8 5 20 [869] [648] [129, 204]
2—3 Ионный обмен на ионо- Торцовый 2.10-е [1168]
Несколь- обменнике цеокарб-225 Электролиз на ртутном счетчик NaJ(Tl) ЗЛО-* [941]
ко часов катоде Экстрагирование основы Без разрушения NaJ(Tl) 2-10-’ [921] [328]
Анализируемый объект Навеска, г Поток нейтро- нов, н/см!-с Время облуче- ния, ч
Молибден и соединения его 0,05—0,8 4-1012 0,3—48
0,1 ~1013 10
З.Юй 10
Ниобий и его сое- 1 1,2-1013 10
динения 0,002— 3-10U— 0,3—2
0,04 3-1013
Олово и его динения сое- 1 5-1012 1 неделя
Свинец и его динения сое- 1 3-1013 10
Селен 0,075— 2,9-101° 5-30
0,1
0,1 ~101з 72
Таблица 48 (продолжение)
Время охлажде- ния, ч Метод очистки детектор излучения Предал обнаруже- ния, % Погреш- ность, % Литера- тура
Хроматографическое разделение на ионооб- менниках Дауэкс- NaJ(Tl) 4.10-5 [731]
50x8 и Дауэкс-1хЮ
Концентрирование на колонке с вольфраме- NaJ(Tl) 5-10-’ (706]
цирконатом аммония
36 Без разрушения Ge(Li) 3,2-10-’ 25—30 [50]
2—3 То же Ge(Li) 1-10-’ 30 [548]
2 » NaJ(Tl) [867]
Растворение в смеси 17,5 М H2SO4+48% НВг Ge(Li) 0,3-10-’ [934]
с последующей перегон- кой
Ионообменная хромато- графия на анионообмен- нике Дауэкс-1 (С1~) 1,6-10-* 30 [1083]
36 Без разрушения Ge(Li) 3,2-10-’ 25—30 [50, 192]
5—45 То же NaJ(Tl) IO"6 [772]
24 Отделение основы дис- тилляцией при обработ- ке смесью H2SO4-f-HBr при 200—220 °C Ge(Li) 1,5.10-* 2 [594]
Анализируемый объект Навеска, г Поток нейтро- нов, н/см2-с Время облуче- ния, ч
Селен 0,1—0,2 1,8-101? 8
7- 10м 0,3
Тантал и его сое- 0,03 5-1013 24
динения 0,002— 0,04 3- Юн— 3-1013 0,3—2
0,1 1,2-1013 0,5
Теллур (теллуро- вая кислота) 1 0,005— 0,04 6,3-1012 1,8-1013 0,25 0,3
0,1 ~101« 15
Углерод (графит, алмаз, уголь) 0,005— о,1 1,2-1012 5-1012 72 0,5
1 ~10м 24
0,004— 0,305 2-1013 70
0,06—0,1 1,2-1013 2
Таблица 48 (продолжение)
Время охлажде- ния, ч Метод очистки Детектор излучения Предел обнаруже- ния, % Погреш- ность, % Литера- тура
14 Экстракционное рааде- NaJ(Tl) зло-9 15—20 [104]
2 ление элементов Растворение в конц. NaJ(Tl) io-’ [1281]
2 HNO3 и перевод в соля- нокислый раствор Хроматографическое разделение на колон- ках с фторопластом и АВ-17ХЙ Без разрушения Экстракционное отделе- NaJ(Tl) NaJ(Tl) NaJ(Tl) 7.10-e 10—15 [5071 [867] [96]
15 ние основы Без разрушения NaJ(Tl) 1,4.10-a [938]
1 То же NaJ(Tl) зло-» 3—12 [48, 49]
24—72 Хроматографическое Ge(Li) 2,1.10-s 20 [1010]
0,5 разделение на ионооб- меннике Дауэкс-1х8 Без разрушения NaJ(Tl) 5.10-6 [925]
То же АРГ Ge(Li) 1.10-5 l,3.10-2 20 [858, 958] [729]
96 Ge(Li) 6,2-10-5 [1079,
0,5 Ge(Li) 2-Ю-5 10-15 1146] [309]
Анализируемый объект Навеска, г Поток нейтро- нов, н/см«-с Время облуче- ния, ч
Углерод (графит, алмаз, уголь) 5.1012 2 мин— 100 ч
0,5 1.1013 20
1 0,5
5 4-1012 0,3
Фосфор 0,4—0,5 5-1012 15 мин
2,6 ~1012 42 дня
Хром 0,5—1 5-1012 65
Цинк 7-1013 0,3
Цирконий 1 —1012 200
Кислоты (НС1, HNOa, аскорбино- вая) 0,4—2 —Ю12 0,3—10
Атмосферная пыль 0,1 4.1012 5 мин
Почвы 0,04-3 4.1011— 1,8-1013 5—20 мин
&
Таблица 48 (продолжение)
Время охлажде- ния. ч Метод очистки Детектор излучения Предел обнаруже- ния, % Погреш- ность, % Литера- тура
0,5 с—30 Без разрушения NaJ(Tl) [207]
суток 7—30 То же NaJ(Tl) 6.10-e [111]
Ge(Li) 4- КГ® [191]
» NaJ(Tl) 2,5.10-6 [960]
» NaJ(Tl) 1-10-5 [1040]
34 Выделение натрия с но- Торцовый 1-10-5 [766]
сителем в форме NaCl после обработки пробы смесью HN03-[-Br2 счетчик
Ионообменная хромато- графия на ионообмен- Ge(Li) IO"8 5—10 [929]
нике Дауэкс-1х8
2 Выделение натрия в форме NaCl NaJ(Tl) IO"7 [1281]
Ионообменная хромато- графия на ионообменни- кё Дауэкс-50х8 10-8 [740]
10—15 Без разрушения NaJ(Tl) io-’ 20 [508, 577, 997]
2 мин То же Ge(Li) 2,5-10-6 20 [811, 868]
0,5—24 Ge(Li) 1-10-5 10 [4, 426, 637, 771]
146
Анализируемый объект Навеска, г Поток нейтро- нов,. н/см*-с Время облуче- ния, ч
Горные породы, минералы, метео- риты 0,01 Ю10—10.12 1—Змин
0,Ol- О.02 1,6.10м 30 с
0,1—0,3 1010—10» 0,5 мин— 2 ч
0,1—3 10U_i0M 8—10
Вода (почвенная, минеральная, во- допроводная) 2—10 10м 5 мин— 14 ч
16 5.10U 10
200 ~10м 24
Нефть и нефтепро- 1 ~1012 4
дукты 0}1—0,5 10U—1013 5 мин— 5 ч
Марганцево-цин- ковые ферриты 0,1 5.10’2 15 мин
Керамические ма- териалы 0,02 1,5-1013 4—60 мин
Таблица 48 (продолжение)
Время охлажде- ния, ч Метод очистки Детектор излучения Предел обнаруже- ния, % Погреш- ность, % Литера- тура
24 1—6 мин Без разрушения То же Торцовый счетчик NaJ(Tl) 1,7.10-2 5—6 2 [267, 1116] [637,
20—24 » Ge (Li), 2.10-e 5—20 1132, 1277] [278, 733,
2—3 » NaJ(Tl) Ge(Li) IO"3 3-IO”5 2 1109, 1126] [287, 655, 771, 1086, 1030, 1134] [526, 653,
Сорбция натрия на гид- Торцовый 7.10-s 15 680J [806]
24 24—48 5—20 ратированном Sb2O5 Без разрушения То же » счетчик Ge(Li) NaJ(Tl) 10_® 6-10-e IO-5 8—13 [924] [616] [45, 490,
30 20 » NaJ(Tl) Ge(Li) IO"3 1,5 1150] [1278] [950]
Анализируемый объект Навеска, г Поток нейтро- нов, н/см>-с Время облуче- ния, ч
Серебряные изде- 0,01— 1,8-10м 16
ЛИЯ 1 мг
Лазерные к рис- 0,05—0,1 1,5-101® 2
таллы 2,3-1013 0,75
Стекла 1,5—3 2-10* До насы- щения
Апатиты 0,1 1,7-lOU 2 мин
Синтетические 0,04— 10U—1013 30 с—24 ч
органические ма- териалы 0,25
Биологические 0,1—0,3 1012— Ю13 2—8 мин
объекты и расти- тельный материал
Ионообменники ~101з 10—15
0,1 5-10-1» 6
Лунный грунт 0,001— —1013 2 мин
0,002
0,1 2-1012 1 мин
Таблица 48 (окончание)
Время охлажде- ния, ч Метод очистки Детектор излучения Предел обнаруже- ния, % Погреш- ность, % Литера- тура
3 Химическое выделение в присутствии носителя Ge (Li) i.io-6 5—10 [971]
0,5 Без разрушения NaJ(Tl) [1032]
20 То нее Ge(Li) 3,6.10-8 3,5 [417]
NaJ(Tl) 1,8-10-3 [1117, . 1118]
3—30 дней Ge(Li) 4 [654]
1 с—72 ч NaJ(Tl) io-» 10 [446, 467, 825, 1041, 1140]
2—4 мин » Ge (Li) 1.10-s 10—50 [426, 645]
12—24 Ge(Li) 1.10-5 [808]
36 » Ge(Li) [946]
10 » Ge (Li) 15 [529, 767]
4—12 Ge (Li) <10-4 6—10 [988]
ность метода ограничена высокой поправкой на контрольный опыт
(14 мкг Na). Воспроизводимость результатов определения ~15%.
Предложенный метод пригоден также для определения примеси на-
трия в алюминии.
0,5 г пробы растворяют в 3,6 мл конц. НС1, раствор разбавляют до 30 мл
13%-ным раствором лимонной кислоты и проводят в течение 4 ч электролитичес-
кое выделение натрия на ртутном катоде с одновременным разложением образую-
щейся амальгамы натрия водой в электролитической ячейке с диафрагмой при
напряжении 30 В (начальная и конечная сила тока — 400 и 200—250 мА соот-
ветственно). Химический выход натрия 93+5%. 10 мл полученного раствора
натрия облучают 10 мин в ядерном реакторе (поток тепловых и быстрых нейтронов
5-1012 и 1,5-1012 н/см2-с соответственно) и затем измеряют активность образо-
вавшегося 24Na по у-пику 2,76 МэВ при помощи 200-канального сцинтилляцион-
ного у-спектрометра.
Определение натрия в трихлориде бора [437].
Пробу ВС13 гидролизуют во фторопластовом сосуде, раствор выпаривают
под ИК-лампой и остаток прокаливают 2—3 ч в платиновой чашке при 400—
500° С. 0,15 г полученного ВоО3 и такое же количество эталона облучают 100 ч
потоком нейтронов 1,2-1013 н/см2.с. Облученные ампулы кипятят в царской вод-
ке для удаления поверхностных загрязнений, вскрывают, отделяют бор отгон-
кой борнометилового эфира (трехкратное добавление 10 мл СН3ОН к В2О3),
остаток нагревают несколько раз с конц. НС1 или HNO3 в присутствии носителей
определяемых элементов и раствор фильтруют. Фильтрат выпаривают, произво-
дят разделение определяемых элементов с помощью ионообменной хроматогра-
фии. Полученные фракции выпаривают досуха под ИК-лампой на фторопласто-
вых подложках и измеряют радиоактивность на установке ПП-8 с торцовым
счетчиком МСТ-17. Радиохимическую очистку выделенных нуклидов контро-
лируют по у-спектру с помощью анализатора АИ-100 и кривым распада.
Определение натрия в арсениде галлия [24]. 0,1 г пробы промывают раство-
ром НС1, помещают в кварцевый бокс, который вместе с эталонами запаивают
в кварцевую ампулу, облучают 9 ч в нейтронном потоке 1,8 -1018 н/см2 -с и выдер-
живают 12 ч. Облученную пробу протравливают раствором НС1, растворяют
в 3—4 мл конц. НС1 в присутствии носителей определяемых элементов (по 10 мкг)
при добавлении по каплям брома. Раствор упаривают почти досуха, остаток
растворяют в НС1 с добавлением брома, упаривают и повторяют эту операцию
еще раз с добавлением «обратного» носителя — мышьяка (отгонка бромидов As,
Sn, Se, Sb и частично Ga, Те и Мо). Остаток растворяют в смеси (3 : 1) ЙС1 и
HNO3, раствор упаривают, прибавляют 8—10 мл 10—11 М НС1 и экстрагируют
Ga, Au, Fe, Т1 и большую часть Pt, Те и Мо 4 раза равными объемами 2,2-дихлор-
диэтилового эфира. Водный слой упаривают почти досуха, остаток растворяют
в 0,01 М НС1, экстрагируют Cu, Zn,Cd, Ag и In (а также остатки Se и Теи ~40%Sc)
5%-ным раствором ди-и-бутилтиофосфорной кислоты (ДБТФК) в ССЦ, экс-
тракт промывают 0,1 М раствором НС1 для удаления скандия, реэкстрагируют
Zn и In 1 М раствором НС1, a Cu, Cd и Ag — 10 М НС1. Последний реэкстракт
упаривают почти до.суха, остаток растворяют в 0,2 М растворе НС1 и экстраги-
руют Cd и Ag 0,05 М раствором хлорида диметилбензилалкиламмония (ЧАО)
в 1,2-дихлорэтане (медь остается в водной фазе). Водный раствор, оставшийся
после экстракции раствором ДБТФК, подкисляют до 0,2 М по НС1, экстрагируют
Re, Pd, In и W с использованием ЧАО, реэкстрагируют вольфрам 3 М раствором
НС1, a Re, Pd и 1г — конц. HNO3. Последний реэкстракт выпаривают, остаток
растворяют в 1 М НС1 и затем избирательно экстрагируют палладий с помощью
ДБТФК (Re и 1г остаются в водном растворе). Раствор, оставшийся после экст-
ракции Re, Pd, 1г и W, упаривают, прибавляют 9 М НС1, экстрагируют Со по-
средством ЧАО и оставшийся водный слой используют для определения Na и К.
В полученных фракциях измеряют радиоактивность нуклидов определяемых
элементов при помощи 100-канального сцинтилляционного у-спектрометра. Вос-
производимость результатов анализа составляет +10%.
148
Определение натрия в олове [934]. Одновременно можно опреде-
лять несколько примесей. Предел их обнаружения для образцов
олова высокой чистоты изменяется от 2-10~9% (для скандия) до
2-10“Б% (для железа) при облучении 1 г олова в течение 1 недели
потоком тепловых нейтронов 5-1012 н/см2-с.
Облученный образец олова трижды протравливают в НС1, взвешивают и
растворяют в 20 мл 17,5 М H2SOj и 2 мл 14 М HNO3 в колбе вместимостью 100 мл
с обратным холодильником. Перед растворением добавляют по 10—100 мкг
носителей определяемых элементов. К полученному раствору добавляют 10 мг
носителя — золота, растворенного в 1 мл смеси (3 : 1) НС1 и HNO3, и смесь пе-
реносят в перегонную колбу. Отгоняют олово по общепринятой методике с ис-
пользованием 100 мл 48%-ной НВг. Остаток от дистилляции переносят в колбу
Эрленмейера вместимостью 100 мл. Осадок золота, образовавшийся во время
дистилляции и осевший на стенках колбы, растворяют в 10 мл смеси (3 : 1) НС1
и HNO3, которую также переносят в колбу Эрленмейера. Через 16 ч снимают
у-спектр с помощью Ое(Ы)-детектора.
Определение натрия в этилате тантала [96]. Интервал определяе-
мых концентраций п-10~Б—и-10-8% по массе. Предел обнаружения,
рассчитанный по Зе-критерию, для образца массой 0,1 г равен
7-10~6%. Относительное стандартное отклонение 10—15%.
Этилат тантала (—0,1 г) облучают в запаянной кварцевой ампуле одновре-
менно с эталоном натрия в вертикальном канале ядерного реактора в потоке
нейтронов 1,2-1013 н/см2-с в течение 30 мин. После вскрытия ампулы этилат тан-
тала с помощью микропипетки переносят в стеклянный бюкс и взвешивают.
К анализируемому образцу приливают 3 мл HNO3 (1 :10), содержащей изотопный
носитель натрия (10-4г), и нагревают при 50° С в течение 10 мин. Раствор с осад-
ком центрифугируют, осадок отбрасывают, а к центрифугату добавляют 3 мл 2 М
HF и 6 мл три-к-бутилфосфата. После встряхивания в делительной воронке в те-
чение 10 мин водную фазу переносят в измерительную пробирку. у-Излучение
радионуклида 24Na измеряют на сцинтилляционном у-спектрометре, состоящем
из детектора NaJ(Tl) размером 80 х 80 мм и 256-канального амплитудного ана-
лизатора. Интенсивность излучения “Na определяют по площади фотопика в об-
ласти 2,75 МэВ.
При определении натрия в геологических образцах и минеральных
водах можно использовать изотопные источники нейтронов неболь-
шой интенсивности. Так с помощью Ро—Be-источника определяли
натрий в вулканических породах; погрешность определения состав-
ляла 15% [650]. Калифорниевый источник (0,4 мг 252Cf) применяли
для оценки содержания натрия в пробах минеральных вод. Предел
обнаружения натрия составлял 10~3% [316, 581]. Описано использо-
вание магнитного p-сепаратора для активационного определения
натрия в различных образцах. Для повышения чувствительности оп-
ределения разделяют суммарную активность образца на р~ и у-состав-
ляющие, а также сортируют заряженные компоненты излучения по
энергиям. Предел обнаружения натрия в воде составил 1-10“3г/л,
а в горной породе — 1-10~3% [276].
Предложено проводить нейтронно-активационный анализ эле-
ментов после их разделения методом бумажной хроматографии
[657]. Полученные хроматограммы облучают в течение 10 мин потоком
нейтронов 1012 н/см2-с, выдерживают 5 мин после облучения и фикси-
руют распределение радиоактивности вдоль хроматограммы с помо-
щью у-спектрометрии, авторадиографии или путем сканирования. Эта
149
методика дает возможность определить <Ю-8 моля натрия. Следует,
однако, иметь в виду, что хроматографическая бумага может быть
загрязнена следами металлов, что вызовет наведенную радиоактив-
ность (нужна тщательная предварительная очистка), и что длитель-
ное облучение может привести к нарушению хроматографических
свойств бумаги.
ФОТОННО-АКТИВАЦИОННЫЙ МЕТОД
Фотонно-активационный метод определения натрия по ядерной
реакции 23Na(y, n)22Na предусматривает использование тормозного
у-излучения, возникающего в линейном ускорителе электронов при
выводе пучка электронов различной энергии на платиновую или тан-
таловую мишень. Наведенную радиоактивность, как правило, реги-
стрируют спектрометрически. Для определения натрия в некоторых
металлургических и археологических образцах вместо тормозного
у-излучения предложено использовать жесткое у-излучение 21Na,
образующегося при облучении образцов потоком тепловых нейтронов
[584, 943]. Погрешность отдельного определения 3%, предел обна-
ружения 0,4 мкг. Фотонно-активационный анализ менее чувствите-
лен, чем нейтронно-активационный (табл. 49), но он может быть
Таблица 49
Фотонно-активационное определение натрия
Объект Энергия электронов, МэВ Интенсив- ность пучка, мкА Предел обна- ружения, мкг Литера- тура
Na2SO4 35 100 0,03 [722]
40 100 0,017
Стандартные горные породы 30 70 [1119]
Руды и донные отложения 300 110 [952]
105-115 10—15А [1092]
Атмосферные аэрозоли 35 50 2 нг,'м3 [587]
с успехом использован для определения легких элементов (С, N, О),
нуклиды которых плохо активируются нейтронами.
Чувствительность химических методов контроля содержания С,
N и О в щелочных металлах обычно не превышает 10-3% мае., ис-
пользование же ядерной реакции (у, и) позволяет повысить избира-
тельность, а также чувствительность определения этих элементов
до 10-6% мае.
ПРОТОННО-АКТИВАЦИОННЫЙ МЕТОД
В ряде случаев удовлетворительные результаты дает активацион-
ный анализ с использованием заряженных частиц. Так, для натрия
разработан протонно-активационный метод определения, основанный
на регистрации мгновенного у-излучения, возникающего при ядер-
150
ных реакциях 23Na (р, py)23Na и 23Na (р, pa)20Ne (табл. 50). При-
менение протонов с низкой энергией позволяет значительно снизить
фон от матрицы и исключить влияние реакции (р, п), которая для
большинства элементов невозможна при Ер 1 МэВ.
Таблица 50
Протонно-активационное определение натрия
Объект Энергия протонов, МэВ Энергия ре- гистрируемых у-квантов, МэВ Предел обна- ружения, % Литера- тура
NaCl + SiOg 1 1,45—1,80 1,7-10-? [135, 310]
NaCl + КС1 1 1,45—1,80 6,2-10-» [ЗЮ]
Горные породы, стекла, Li—Mg- 1,8 1,33 ю-« [639]
сплавы
Биологические и минералоги- ческие объекты 2,7 0,439 1.10-4 [632]
Геологические образцы <3 0,439 10~4 [697]
Природные воды 14,7 0,439 4-Ю-5 [598]
Высокой чувствительности определения натрия в сверхчистом
алюминии удалось достигнуть сочетанием протонно-активационного
анализа с масс-спектрометрией [1017]. 80 мг образца облучали на
синхроциклотроне протонами с энергией 155 МэВ, измеряли 0-ра-
диоактивность газопроточным 4л-счетчиком и рассчитывали коли-
чество нуклида 22Na, образовавшегося по реакции 27А1 (р, 3p3n)22Na.
Возгонкой в вакууме натрий из облученного образца наносили на
металлическую нить, с помощью масс-спектрометра определяли
отношение 23Na/22Na и рассчитывали количество натрия в анализи-
руемом образце. Для определения не надо знать ни эффективного
сечения реакции, ни интенсивности пучка протонов. Чувствитель-
ность метода может достигать 10“®%.
ДЕЙТРОННО-АКТИВАЦИОННЫЙ МЕТОД
Разработана методика недеструктивного дейтронно-активацион-
ного определения натрия в минералах и горных породах [1065].
Образцы облучают дейтронами с энергией 5,5 МэВ, которые получа-
ют в ускорителе Ванде—Граафа. Методика рекомендуется для опре-
деления основных элементов геологических образцов с содержанием
меньше 0,1 % мае. Проведено дейтронно-активационное определение
натрия в марганцевых конкрециях [1174].
ВАРИАНТЫ АКТИВАЦИОННЫХ МЕТОДОВ
Существует два основных варианта проведения активационного
анализа: радиометрический (инструментальный) и радиохимический.
Инструментальный активационный анализ проводится без разруше-
151
ния образца и заключается в исследовании излучения образовавших-
ся радионуклидов с помощью различной радиометрической аппара-
туры (у-спектрометры, многоканальные анализаторы, полупровод-
никовые детекторы). Он отличается быстротой и экономичностью,
допускает использование ЭВМ для анализа многокомпонентных у-
спектров на основе решения системы соответствующих уравнений [45],
но уступает, как правило, по чувствительности радиохимическому
варианту метода.
Радиохимический активационный анализ основан на разложении
образца с последующим разделением активированных элементов и
определением их радиоактивности. Предложена схема разделения
30 элементов на группы, пригодные для ^-спектрометрического опре-
деления радионуклидов, образующихся в процессе облучения проб
различных веществ [764, 765]. В эти группы входят: 1) элементы,
отделяемые дистилляцией (Ge, As, Sb, Sn, Se, Hg); 2) элементы,
образующие труднорастворимые оксигидраты (W, Si); 3) благород-
ные металлы (Ag, Au, Pt, Ir); 4) щелочноземельные металлы (Са,
Sr, Ba); 5) щелочные металлы (Na, К, Cs); 6) редкоземельные элементы
и 7) прочие элементы (Си, Cd, Fe, Cr, Ga, In, Ni, Zn, Mo, Zr, P).
Схематически описана методика определения элементов в каждой
группе. Показано, что предложенная схема обеспечивает достаточное
разделение. В качестве иллюстрации радиохимического варианта
анализа можно привести определение натрия в тантале высокой
чистоты [507].
Образец (~30 мг) и эталоны определяемых элементов (10~®—10-7 г) облуча-
ют в течение 24 ч потоком тепловых нейтронов 5 -1013 н/см2-с. Облученный обра-
зец растворяют в 3—5 каплях HF и 1 капле HNO3 в присутствии носителей
(10-5 г). Раствор выпаривают досуха и остаток растворяют в 0,5 М HF. Пропус-
канием полученного раствора через колонку (18 X 40 мм) с пористым фторо-
пластом, пропитанным циклогексаном, отделяют основу (Та). Колонку промы-
вают 0,5 М раствором HF (6 свободных объемов) и из элюата выделяют примеси
на трех колонках с анионообменником АВ-17 X 8, находящимся в различных
формах. На первой колонке (4 X 180 мм, F-форма) сорбируют Zr, W, Мо и Nb
из среды 6 М HF и элюируют эти элементы смешанными растворами HF и HCI
различной концентрации. Раствор элементов, не сорбировавшихся на первой
колонке, выпаривают досуха, остаток растворяют в 4—5 каплях 8 М раствора
НС1, содержащего 30% этанола, и полученный раствор пропускают через вторую
колонку (2 X 150 мм, Cl-форма). На этой колонке сорбируются Со, Си, Ga, Fe
и Zn. Эти элементы элюируют растворами НС1 понижающейся концентрации.
Фильтрат со второй колонки выпаривают досуха, остаток растворяют в воде и
раствор пропускают через третью колонку (1,5 X 50 мм, ОН-форма). В филь-
трате определяют натрий. Радиоактивность регистрируют сцинтилляционным
у-спектрометром. Химический выход составляет 85—95%. Продолжительность
анализа не превышает 7 ч.
Радиохимический активационный анализ требует больших времен-
ных затрат для выполнения химических операций и нахождения вы-
хода анализируемых элементов, но отличается высокой чувствитель-
ностью (до 10-9%) и позволяет одновременно определять большое
число разных элементов.
Практически все элементы периодической системы могут быть
определены с помощью различных вариантов активационного анали-
за, результаты которого не зависят ни от химической формы анали-
152
зируемого элемента, ни от чистоты применяемых реактивов, а связа-
ны только со свойствами образующихся ядер. Следует также отметить,
что в настоящее время не существует другого метода, , который мог
бы конкурировать с активационным анализом по числу одновременно
определяемых элементов.
ДРУГИЕ МЕТОДЫ
При количественном анализе сходных, но трудно разделяемых
веществ может, быть с успехом применен метод изотопного разбавле-
ния. При этом не требуется количественного выделения исследуемых
веществ, нужно лишь выделить пробу с максимальной степенью
чистоты. Выбор варианта метода изотопного разбавления опреде-
ляется в основном видом и составом анализируемого образца.
В простейшем случае количество анализируемого вещества тх
определяют по формуле
тх = (/(/А — A то,
где т0— количество добавленного вещества с молярной радиоактив-
ностью 70; 1г — молярная радиоактивность выделенного вещества.
Так как в формулу входит отношение молярных радиоактивностей,
не требуется абсолютного определения их, а нужно лишь проводить
измерения двух образцов в идентичных условиях.
Для определения содержания натрия в смеси с другими щелоч-
ными металлами рекомендован метод изотопного разбавления с экст-
рагированием натрия из среды НС1О4 этилацетатом [885]. При зна-
чительном содержании лития в смеси необходима повторная экстрак-
ция натрия изобутанолом, или диэтиловым эфиром. Метод позволяет
определять <0,1 % натрия в КОН или КС1.
Сочетанием метода изотопного разбавления и дистилляции ниже
температуры кипения определено содержание натрия в особо чистых
кислотах (НС1, HF, НС1О4, H2SO4, HNO3) и воде [904]. Содержание
натрия в одном из анализированных образцов не превышало 1 •
•10-8 г/л.
Метод изотопного разбавления использован для определения
натрия в морской воде. Погрешность определения ~3% [209].
Разработаны две радиоизотопные методики анализа смеси четырех
щелочных металлов, в которых содержание легких элементов (Na,
К) значительно превосходит содержание тяжелых (Rb, Cs) [543].
В основе обеих методик лежит использование лишь одного радиону-
клида (86Rb или 137Cs) для определения всех четырех элементов.
Следует, однако, иметь в виду, что методом изотопного разбав-
ления можно повысить лишь точность, но не чувствительность анали-
тических методов.
Показано [1093], что при облучении анализируемого образца ио-
нами 3Не увеличение их энергии с 9 до 18 МэВ не приводит к сущест-
венному повышению чувствительности определения натрия.
153
Глава X
ОПРЕДЕЛЕНИЕ НАТРИЯ В ОБЪЕКТАХ
Объекты, в которых необходимо определять натрий, весьма раз-
нообразны как по происхождению, составу, так и по диапазону кон-
центраций натрия и сопутствующих ионов. В связи с этим нет уни-
фицированных способов вскрытия натрийсодержащих объектов и
подготовки их к анализу. В данной главе приведены объекты, опре-
деляемые содержания натрия, методы определения и литература.
В зависимости от поставленной задачи аналитик может выбрать наи-
более приемлемый метод и воспроизвести его по оригиналу.
ПОЧВЫ, СИЛИКАТЫ, ГОРНЫЕ ПОРОДЫ
Некоторые методы рассмотрены в табл. 51.
Рассмотрены методы вскрытия силикатных пород [912]. Наиболее
целесообразным признано растворение в HF.
Количество обменных калия и натрия в почвах определяют мето-
дом классической полярографии [3].
Таблица 51
Определение натрия в почвах, силикатах, горных породах, пыли
Объект Навеска, г 1 Определяемые количест- ва, % (Ст£д) Метод определения Литера- тура
Почва 0,04—0,06 1,42—2,95 Нейтронно-актива- ционный [4]
(1-10-5%) » [426]
0,05 1,1 (10~*г) » [202]
Глинозем Полярографический [3, 445]
Ионоселективный элек- трод [1217]
Силикаты Полярографический [1240]
Горные породы о,1 (10-зо/о) Нейтронно-актива- ционный [1086]
» [655]
Вулканические породы » [650]
Дуниты 0,01 » [1116]
Биотит (Сопоставление мето- дов) [481]
Осадочные породы Нефелометрический [219]
Атмосферная пыль Нейтронно-ак тива- ционный [868]
154
Навеску 1 г хорошо растертой, пропущенной через сито (1 мм) почвы обра-
батывают 100 мл насыщенного раствора Са(ОН)2, к которому добавлен СаС12.
Взбалтывают в течение 2 ч, 20 мл вытяжки переносят в электролизер, продувают
водородом и полярографируют, определяя сумму натрия и калия по градуиро-
вочному графику или методом добавок.
Проведено сравнение способов разложения пробы на примере
чистых NaCl и КС1 [779].
При сухом способе 4 г пробы озоляют в течение 3 ч при 600° С, смачивают
раствором НС1 (1 : 1), высушивают, повторно обрабатывают НС1 и вновь высу-
шивают. Остаток высушивают при 110° С в течение 1 ч, растворяют в НС1 (1 : 1)
и воде, разбавляют до определенного объема водой и определяют натрий (калий).
При кислотном способе 0,5 г пробы растворяют в 5 мл смеси (2 :1) HNO# и HClOi,
выпаривают на песчаной бане до удаления оксидов азота и белого дыма НС1О«.
К остатку прибавляют 0,1 М раствор НС1, фильтруют, разбавляют до опреде-
ленного объема и определяют натрий (калий).
Результаты определения натрия и калия методами эмиссионной
фотометрии пламени и абсорбционной фотометрии пламени показали,
что при сухом способе вскрытия наблюдаются значительные потери
калия и натрия. Поэтому рекомендован мокрый способ вскрытия.
МИНЕРАЛЫ, РУДЫ, КОНЦЕНТРАТЫ
В табл. 52 систематизированы некоторые методы определения
натрия в минералах, рудах и концентратах.
Как было отмечено в главе VIII, при определении большой груп-
пы элементов, особенно в веществах высокой чистоты, широко исполь-
зуют спектрографические методы — прямые и химико-спектральные.
Основными являются методы пламенной спектрометрии, особенно
в атомно-эмиссионном варианте (метод фотометрии пламени). Поэто-
му основные методические сведения, относящиеся к переводу объек-
тов в раствор и подготовке его к анализу, будут приведены ниже
именно для этих методов анализа.
Таблица 52
Определение натрия в минералах, рудах и концентратах
Объект Определяемые количества, % 1 Метод определения Литера- тура
Минералы, руды Гравиметрический (см. главу IV) Титриметрический (см. главу V) [19] [19]
Апатит ;о, 09—0,23 Нейтронно-активационный [654]
Урановые руды и кон- центраты 0,5—5 Гравиметрический [1068]
Калийные руды Потенциометрическое титро- вание [186]
Нефелиновые концент- раты 0,5—7 Полярографический (см. гла- ву VII) [382]
Марганцевые конкре- ции Дейтронно-активационный [1174]
155
Применение различных методов спектрального анализа для опре-
деления натрия описано в ряде монографий, например [84, 162, 397,.
424], обзорах [804, 805], систематизировано в библиографических
указателях [947, 948].
Определение натрия в горных породах, минералах и рудах мето-
дами пламенной спектрофотометрии рассмотрено в работах [41, 83,
'92, 153, 166, 169, 187, 262, 263, 313, 315, 316, 402, 458, 508, 566,
573, 611, 620, 625, 627, 686, 696, 702, 747, 807, 828, 829, 840, 847,
912—915, 966, 1044, 1055, 1066, 1075, 1106, 1135, 1167, 1192, 1199,
1252, 1266, 1283].
Используют различные схемы разложения силикатных пород:
а) классическую схему с применением смесей кислот HF + H2SO4,
HF + НС1, HF + HN03 + HC1, HF + HNO3, + HC1O4 [83, 92,
262, 315, 611, 620, 807, 1199]; с последующим отделением мешающих
компонентов осаждением или хроматографией либо без отделения;
б) разложение в герметической камере (автоклаве) в присутствии
борной кислоты [316, 620, 915]; в) сплавление с боратами лития [573,
966, 1252, 1283] или стронция [847]; г) без растворений образца
[263, 611].
Чаще всего определение натрия проводят из растворов, исполь-
зуя пламенный способ атомизации в атомно-абсорбционном методе
и низкотемпературные пламена в качестве источников возбуждения
в атомно-эмиссионном методе анализа.
Современный анализ силикатов на присутствие всех компонентов
проводят из одного раствора атомно-абсорбционным методом. Сили-
каты и стекла переводят в раствор без удаления кремния. Натрий
определяют в пламени пропан—бутан—воздух или ацетилен—воз-
дух. Широко обсуждаются влияние других компонентов пород и
минералов на величину эмиссии и абсорбции натрия и вопросы, свя-
занные с эталонированием при анализе силикатных горных пород
[696, 702, 847, 1106, 1199, 1283].
Для анализа силикатов используют пламенный фотометр фирмы
«К. Цейсс»(модель III) [483]. Натрий определяют в пламени ацетилен—
воздух. Изучено влияние кальция на эмиссию натрия и установлено,
что при содержании кальция ^>4% необходимо вводить поправку
или вводить кальций в стандартные растворы. Изучено также взаим-
ное влияние натрия и калия. Метод перевода силикатов в раствор
стандартен, однако не рекомендуется оставлять растворы надолго.
Приводятся величины факторов избирательности при определении
натрия в минералах эмиссионным методом [402].
При определении натрия в силикатных породах (гнейсах, грани-
тах, сиенитах) с содержанием не менее 10-3% используют метод
Аренса для концентрирования щелочных и щелочноземельных эле-
ментов с последующим определением натрия атомно-эмиссионным
методом по линии 588,9 нм в воздушно-ацетиленовом пламени [424].
Пламенно-фотометрическая установка сконструирована на основе
двойного стеклянного монохроматора ДМ. Фотоэлектрическое уст-
ройство состоит из фотоумножителя ФЭУ-17, выпрямителя ВВС-1
и зеркального гальванометра М-21.
156
Навеску 0,7—1,0 г тщательно измельченного силиката в платиновой чашке!
смачивают несколькими каплями воды, прибавляют 5—10 мл HClOi, 15—20 мл
HF и выпаривают до появления паров хлорной кислоты. Если необходимо, по-
вторяют выпаривание с 5—10 мл HF. После разложения пробы удаляют фторид,
добавлением нескольких капель воды и НС1О« и упариванием смеси досуха дваж-
ды. Сухие остатки перхлоратов прокаливают в течение часа при 500—550° С,
дважды обрабатывают при нагревании 50 мл 2 М НС1, а затем водой, каждый раз.
собирая раствор в стакан. Незначительную часть железа и алюминия, перешед-
ших в раствор, осаждают аммиаком, раствор после фильтрования и промывания
осадка упаривают и переносят в мерную колбу вместимостью 50 мл. Для опре-
деления натрия раствор разбавляют до концентрации 5—50 мкг/мл, стандартные-
растворы должны содержать калий и кальций в количествах, соответствующих
содержанию их в анализируемой породе. Погрешность определения не более-
5-6%.
Мешающие натрию катионы предложено отделять ионообменной
хроматографией [1055].
Навеску 0,1 г силикатной породы в платиновой чашке растворяют в смеси
HF и НС1, раствор выпаривают с НС1. Сухой остаток хлоридов растворяют-
в воде и пропускают через колонку с катионообменциком Дауэкс 50W х 2
в Н-форме. Натрий десорбируют 0,4 М раствором НС1, элюат разбавляют водой
до 250 мл. Кальций остается на колонке.
Методика анализа силикатных пород упрощена [611]. После раз-
ложения пробы смесью HF и HN03 прибавляли НС1О4, к выпарен-
ному досуха остатку прибавляли конц. НС1 и воду и раствор пере-
носили в мерную колбу вместимостью 50 мл. Натрий определяли
в аликвотной части раствора по резонансному дуплету 330,299 и
330,222 нм в пламени ацетилен—воздух атомно-абсорбционным ме-
тодом.
Предложен способ разложения силикатов, заключающийся в об-
работке образцов смесью кислот в герметически закрытых сосудах
с последующим введением борной кислоты для связывания фторид-
иона и определением всех компонентов силикатной породы атомно-
абсорбционным методом. Так, например, используют сосуд из фто-
ропласта диаметром 30 мм и высотой 40 мм в виде тигля, запрессо-
ванного в железный корпус [620]. Разложение пробы проводят при
полной герметизации: 50 мг образца обрабатывают 0,5 мл воды, при-
бавляют 3 мл HF и нагревают в течение 30—40 мин при температуре
110° С. Натрий определяют в пламени ацетилен—воздух.
Предложен измененный метод разложения образцов [316].
В герметический фторопластовый сосуд помещают 0,1 г силиката, обраба-
тывают 0,5 мл смеси (3 : 1) НС1 и HNOS и 5 мл HF при температуре 105—110° С
в течение 45 мин. После охлаждения прибавляют 2,8 г борной кислоты и разбав-
ляют до 100 мл водой. Аликвотную часть раствора разбавляют в 10 раз и опре-
деляют натрий в пламени воздух—пропан на спектрофотометре фирмы «Перкин-
Элмер» (модель 303).
Рассмотрены методы разложения силикатных пород сплавлением,
с боратами [573, 847, 966, 1252, 1283]. В частности, предложена сле-
дующая методика [1283].
В платиновом тигле сплавляют 0,1 г пробы с 0,6 г бората лития в течение-
15 мин при температуре 750° С. Плав растворяют в горячей воде в присутствии
НС1, переносят раствор в колбу вместимостью 200 мл и разбавляют водой до-
157
метки. После пятикратного разбавления раствора определяют натрий (и другие
основные компоненты) методом атомно-абсорбционного анализа. В стандартные
растворы вводят все определяемые элементы.
Предложена следующая методика подготовки пробы к анализу
[966].
Навеску силиката 0,1 г в графитовом тигле сплавляют с 0,5 г бората лития
в течение 10 мин при температуре 1000° С. Плав обрабатывают 40 мл HNOS.
Натрий определяют из исходного раствора после прибавления 20-кратного из-
бытка воды в пламени ацетилен—воздух на спектрофотометре «Перкин-Элмер».
В горных породах, почвах, цементе натрий определяют в пламени
оксид азота(1)—ацетилен (спектрофотометр Техтрон АА-5) после’
разложения пробы сплавлением с боратом стронция [847].
В цилиндрическом графитовом тигле диаметром 20 мм и высотой 20 мм
сплавляют 0,1 г пробы с 1,0 г бората стронция при температуре 1100—1200° С
в течение 20 мин. Плав выщелачивают 100 мл воды, прибавляют 1 мл HNO3,
перемешивают магнитной мешалкой, доводят pH раствора до 2, разбавляют во-
дой до 200 мл и анализируют раствор.
Трудноразлагаемые алюмосиликаты — минералы группы дистен-
силиманита — не разлагаются классическими методами, применяе-
мыми в силикатном анализе. Предложен оригинальный метод введе-
ния вещества в пламя [398].
Навеску 0,1 г пробы предварительно брикетируют со смесью (1 : 8) СаСО3
и NHiCl (диаметр брикета 4 мм, масса 50 мг). На специальной подставке брикеты
вводят в пламя'ацетилен—воздух и регистрируют эмиссионный сигнал натрия
по резонансной линии 589 нм. Градуировочные графики.строят по искусствен-
ным стандартным смесям. Линейность сохраняется до 0,1% натрия. Смесь-
основу готовят из А12О3 и кварца. На нее наносят растворы хлорида натрия.
Стандартные смеси растирают с хлорирующей смесью в соотношении 1 : 4 и из-
готавливают брикеты. Предел обнаружения натрия ограничен величиной кон-
трольного опыта и равен 0,02% натрия.
Второй способ анализа минералов группы дистен-силиманита
заключается в сплавлении 0,5 г пробы с боратом лития в платино-
вой чашке, на которую насыпают сверху 0,1 г карбоната лития. Про-
должительность сплавления 10 мин при 900—950° С. Для определе-
ния натрия используют спектрофотометр на основе монохроматора
УМ-2 и пламя светильный газ—воздух.
При определении натрия в алуните [29] пробу предварительно обжигают
в течение 1 ч при температуре 600° С. Навеску кипятят с 5 мл конц. НС1 и 45 мл
воды, через 30 мин осадок отфильтровывают, промывают, разбавляют до 200 мл
водой. Натрий определяют в пламени ацетилен—воздух .по резонансному дуп-
лету 589,0—589,6 нм. Соляная кислота не влияет до концентрации 0,3 М, сер-
ная — 2М.
При определении натрия в каолине вязкость раствора повышали
прибавлением глицерина [899]. В работе [32] навеску глинозема рас-
творяли в смеси фосфорной и серной кислот. Натрий определяли
атомно-эмиссионным методом (линия натрия 589 нм). При определе-
нии натрия в глиноземе высокой чистоты использовали пламя кис-
лород—водород [622]. При применении пламенного фотометра фирмы
«К. Цейсс» (модель III) для определения натрия в цеолитах влияние
458
"рльция на эмиссию натрия в присутствии алюминия не наблюдали
[945].
Для определения натрия в эмалях и сырье предлагается исполь-
зовать пламенный фотометр ПАЖ-1 в пламени пропан—бутан—воз-
дух [348[.
Навеску полевого шпата 0,1 г обрабатывают в платиновой чашке смесью
4 мл H2SC>4 (1 : 4) и 20 мл HF и нагревают до прекращения выделения паров
серной кислоты. Остаток растворяют в 1 мл конц. НС1 и разбавляют до 250 мл
водой. Навеску криолита 0,1 г разлагают смесью 6 мл HjSOa (1 : 1) и 5 мл HF,
остаток растворяют в 1 мл конц. НС1 и разбавляют до 500 мл водой. Навеску
эмали 0,2 г райлагают смесью 3 мл H2SO4 (1 : 1) и 25 мл HF, упаривают досуха,
остаток растворяют в 1 мл конц. НС1 и разбавляют до 200 мл водой. Остаток ти-
тановых эмалей растворяют в 1 мл 30%-ного раствора Н2О2.
В стекловолокне определяют натрий атомно-абсорбционным ме-
тодом на спектрофотометре фирмы «Перкин-Элмер» (модель 5000)
с коррозионно-стойким распылителем [1061].
К 0,25 г пробы, измельченной в вибромельнице, прибавляют 2—3 мл воды,
через 5 мин 11 мл 49%-ной HF. Через несколько минут после разложения пробы
добавляют 5 мл конц. HNOS, 1 мл конц. НС1 и растворяют пробу в течение 5—
10 мин. Затем осторожно прибавляют 25 мл воды и через несколько минут 4,4 г
борной кислоты. После полного растворения раствор разбавляют водой до 250
мл. При определении натрия аликвотную часть раствора разбавляют в 100 раз.
При определении натрия в золе угля методом атомно-эмиссион-
ной спектрометрии изучено влияние Al, Fe, Mg, силикатов и боратов.
Влияние кальция устраняют введением алюминия [840].
Предложены методы определения натрия без предварительного
растворения пробы [1196]. В пламя вводят суспензии. Отмечается,
что точность определения натрия не хуже, чем в обычном методе,
так как нет химической подготовки.
При анализе различных - железных руд и шлаков разложение
проводят фтористоводородной кислотой в различных герметических
камерах и определяют все компоненты методом атомно-абсорбцион-
ного анализа в различных условиях.
Предложена следующая методика разложения железных руд
и шлаков [914].
Навеску пробы 0,2 г помещают в герметически закрывающийся сосуд из
пластика, прибавляют смесь кислот, состоящую из 0,75 мл конц. НС1, 0,25 мл
конц. HNOS и 5 мл HF. Разложение проводят в течение 30 мин при температуре
110° С. Перед определением раствор разбавляют водой до объема 100 мл.
Для анализа лимонитов, гематитов, сидеритов и стандартных
образцов предлагается следующая методика подготовки проб [1228].
Навеску пробы 0,2 г помещают в автоклав из алюминия, защищенного из-
нутри пленкой, прибавляют смесь кислот, состоящую из 0,75 мл конц. НС1,
0,25 мл конц. HNOS и 5 мл HF, и нагревают при давлении 8 атм при непрерывном
перемешивании магнитной мешалкой в течение 30 мин при температуре 145—
155° С. К раствору добавляют 15 мл насыщенного раствора борной кислоты и раз-
бавляют до 100 мл водой. Определение проводят на спектрофотометре фирмы
«Перкин-Элмер» (модель 403) в пламени ацетилен—воздух.
При определении натрия в марганцевых рудах в пламени светиль-
ный газ—воздух использовали два метода: отделяли марганец осаж-
159
дением 8-оксихинолином и экстрагировали смесью хлороформа и
бутилцеллозольва или снижали спектральные помехи введением в рас-
твор нитрата алюминия. Эти меры необходимы при применении пла-
менного фотометра модели EEL [1106]. Отмечается, что при работе
на спектрофотометре фирмы «Бекман» помех при определении натрия
не наблюдается.
Навеску пробы разлагают в 15 мл. конц. НС1, выпаривают досуха, остаток
растворяют в 20 мл воды и разбавляют в мерной колбе до метки. Осадок отфиль-
тровывают, 20 мл полученного раствора смешивают с 2 г солянокислого гидро-
ксиламина, прибавляют 10 мл 8%-ного раствора 8-оксихинолина в этаноле, до-
водят pH раствора до 7—9 и экстрагируют смесью 15 мл хлороформа и 2 мл бу-
тилцеллозольва, а затем повторно 5 мл хлороформа. После экстракции раствор
упаривают с HNO3 в присутствии НСЮ«, остаток растворяют в воде, разбавляют
водой в мерной колбе вместимостью 50 мл и фотометрируют на фотометре со све-
тофильтрами.
Если не отделять мешающие элементы и марганец экстракцией, то для ни-
велирования влияния железа и марганца вводят раствор 713 г нитрата алюминия
в 1 л воды. Навеску руды растворяют в НС1, раствор упаривают с серной кисло-
той, разбавляют до 30 мл, фильтруют, и фильтрат смешивают с 10 мл раствора
нитрата алюминия и фотометрируют.
Для выбора способа перевода спековых и отвалочных шлаков
в раствор с целью определения натрия методом атомно-эмиссионного
анализа с помощью фильтрового фотометра изучено влияние кислот,
железа, алюминия и титана [291].
Навеску шлака 0,2 г помещают в стакан, слегка смачивают водой и залива-
ют смесью (4 : 1) H2S04 и Н3РО4. Раствор нагревают до выделения паров серной
кислоты, охлаждают, прибавляют 50 мл горячей воды, подогревают на песчаной
•бане в течение 10 мин, переносят в мерную колбу вместимостью 200 мл и после
охлаждения разбавляют водой до метки. Перемешивают и фильтруют раствор
через сухой фильтр. Стандартные растворы готовят на основе хлорида натрия,
поэтому для получения точных результатов вводят поправочный коэффициент
(1,5—1,13).
В почвах обменный натрий в основном определяют методом атом-
но-эмиссионного анализа [340, 341, 695, 1062, 1238, 1267]. Мешаю-
щее влияние кальция устраняют введением в раствор солей алюми-
ния и фосфата [1267].
Предложена следующая методика подготовки проб [1062].
Навеску почвы 10—20 г обрабатывают 1 М раствором ацетата аммония. Рас-
твор упаривают досуха, остаток прокаливают при температуре 550° С в течение
15 мин. Остаток растворяют в 1—5 мл 1 М НС1 и 10 мл воды при нагревании,
остаток отфильтровывают, раствор разбавляют водой до 100 мл. Сульфаты и фос-
фаты удаляют хроматографически. Для этого аликвотную часть раствора про-
пускают через колонку, наполненную анионообменником Дауэкс I в С1-форме.
К 5—20 мл элюата добавляют 5 мл раствора хлорида магния, содержащего
8,5 мг/мл магния, и разбавляют до 25 мл водой. Натрий определяют в пламени
кислород—водород.
В почвенных экстрактах и растворах растительной золы натрий
«определяют методом атомно-эмиссионной спектрометрии пламени
в смеси газов воздух—оксид азота(1У)—ацетилен с помощью трех-
канального спектрофотометра [1238]. Концентрацию натрия опре-
деляют по сравнению с фототоком контрольного раствора.
160
В удобрениях, сырье и продуктах калийной промышленности нат-
рий определяют методом атомно-эмиссионной [554, 570, 754, 880,
1133] и атомно-абсорбционной [1223] спектрометрии пламени. При
использовании воздушно-пропанового пламени на определение нат-
рия не влияют калий и кальций [1133]. Для одновременного опреде-
ления калия и натрия в сильвините, щелоках, хвостах флотации,
шламах применяют автоматизированную двухканальную пламенно-
фотометрическую установку системы «КЛИНА» (фирма «Бекман»).
ВОДЫ
Различные методы определения натрия в водах систематизирова-
ны в табл. 53. Опубликован обзор методов определения натрия в во-
дах [734]. Предложен гравиметрический метод определения натрия
в морской воде и суммы натрия и калия [784].
30 мл воды (при содержании хлорида 19%) пропускают через колонку
(0,7 X 18,5 см), заполненную Амберлитом CG 120 X 8 в Н-форме. Промывают
колонку 5 мл дистиллированной воды и элюируют натрий и калий 220 мл 0,15 М
раствора NH4CI. Сумму калия и натрия определяют гравиметрически. Для этого
элюат выпаривают досуха, подкислив 0,6 мл конц. H2S04, и прокаливают 1 ч
при 800° С. Затем определяют калий гравиметрически в форме тетрафенилбората
калия. Для этого сухой остаток растворяют в 40 мл воды, прибавляют 2,2 мл
1 М H2SO4, охлаждают до 0° С и прибавляют 20 мл 1,13%-ного раствора
(CeH5)4BNa. Осадок отфильтровывают, промывают 2 раза порциями по 5 мл
насыщенного раствора осадителя и высушивают при 120° С в течение 3 ч. Содер-
жание натрия определяют по разности.
Методом спектрометрии пламени натрий определяли в природных
водах [150, 164, 272, 279, 299, 318, 513, 527, 646, 719, 802, 888, 949,
986, 1031, 1053, 1081, 1242], в рассолах [1197], минеральной пита-
тельной среде [439] и промышленных сбросовых водах [803], в вы-
сокочистой воде [279, 567, 1257], в атмосферных осадках [995]. Для
определения ультраследовых количеств натрия в воде с пределом
обнаружения 1 -10—8 % использовали атомно-абсорбционный метод
с непламенной атомизацией [760].
В природных и минеральных водах натрий определяли в пламени
водород—кислород методом атомно-эмиссионной спектрофотомет-
рии с пределом обнаружения 2-10_^%, рассчитанным по Зз-крите-
рию [1053]. Подготовка пробы заключалась в удалении СО2 нагрева-
нием 1 л воды, подкисленной 10 мл конц. НС1, разбавлении до 1 л
депонированной водой и удалении анионов на колонке с ионообмен-
ником Дауэкс 2x8.
Показано [949], что при использовании спектрофотометра фирмы
«Бекман» на определение натрия не влияют магний и железо. Опре-
деление проводили по линии 589,0 нм в интервале концентраций нат-
рия 5-10~4—2-10“3%. Эталонные растворы готовили из хлорида
натрия.
В кислых источниках натрий определяли атомно-эмиссионным
методом после сорбции ионов Амберлитом IR-120 и элюированием
натрия 0,5 М НС1 [1242]. Железо и алюминий отделяли аммиаком,
кальций — оксалатом аммония, фильтрат упаривали досуха, затем
6 В. М. Иванов и др.
161
Таблица 53
Определение натрия в водах
Вода Навес- ка Определяемые коли- честна, % (cmin, %) Метод определения Литера- тура
Морская, рассол 30 мл Гравиметрический » Фотометрический Поляриметрический Амперометрическое тит- рование [1155] [784] [1219] [244, 895] [245]
Минеральная «Боржоми» Минеральная 1,7±0,1 Г/Л 0,015—0,036 г/л (0,5 мг/л) 2,3—23 мг/л 10—900 мг/л Фотометрический » » (см. главу VI) Ионоселективный элект- род Полярографический Титриметрический Гравиметрический (см. главу IV) [295] [769] [38, 296] [776] [445] [1138] [1015]
Речная, озерная Природная 200 мл (10-в) Нейтронно-активацион- ный Полярографический Титриметрический [924] [1072] [338,478, 672] [240] [237] [88] [1070, 1071] [1139]
Поверхностная Питьевая 200 мл 0,023—23 мг/л » (см. главу V) Фотометрический Ионоселективный элект- род Полярографиче ский Титриметрический
Водопроводная 11—230 мг/л » [25]
Чистая 16 г (7.10-») Нейтронно-активацион- ный [806]
Особо чистая Ю-e—io-? Ионоселективный элект- род [717]
Подземная 2 г (IO-) Нейтронно-активацион- ный [489]
Пресная 200 мл Нейтронно-активацион- ный [1276]
Рассолы Сточная 4—11 Титриметрический (см. главу V) Ионоселективный элект- род [834] [332]
162
растворяли остаток в небольшом количестве воды, подкисленной
НС1. В морской воде натрий определяли из объема 1 мл в пламени
пропан—кислород [527]. Внутренним стандартом служил литий. По-
грешность определения 4 %.
Используя пламенный фотометр фирмы «К. Цейсс» (модель III)
в пламени пропан—бутан—воздух, определяли натрий в пресной
поде с погрешностью 1,7% [150]. Изучено влияние калия, кальция
и магния, соли которых вводят в стандартные растворы. Влияние
этих элементов обнаружено для малых концентраций натрия
(<^ 5 мг/л).
Атомно-абсорбционный метод применен для определения натрия
в солончаковых и подпочвенных водах с использованием спектрофото-
метра «Varian-Techtron АА-120» [1031]. Источник света — лампа с
полым катодом. При электросопротивлении воды ^>5-103 МОм-см
пробы разбавляли в 5 раз. Изучено взаимное влияние элементов
и анионов — сульфата и хлорида. В интервале концентраций натрия
5-10“3—4-10“2% определение проводили по линии 330,2 нм; 1-10-8 —
5-10“4% — по линии 589,6 нм (погрешность 4%). Этот же метод при-
менен без разделения и концентрирования [646]. В слабоминерали-
зованной воде натрий определяли после концентрирования в 1000 раз
методом электроосмоса [318]. В речной воде определяли натрий без
дополнительного разбавления с использованием спектрофотометра,
сконструированного на основе спектрографа ИСП-51 с приставкой
ФЭП-1 и записью спектра на потенциометре ЭПП-09 в турбулентном
пламени пропан—бутан—воздух [164].
Обзор по анализу воды атомно-абсорбционным методом представ-
лен в работе [719], приведены методики определения многих элемен-
тов. Рассмотрены прямые методы с различными способами атомиза-
ции, в том числе и пламенный вариант, а также методы с предвари-
тельным концентрированием, например упариванием и ионообменной
хроматографией.
МЕТАЛЛЫ И НЕМЕТАЛЛЫ
Для этих целей в основном используют нейтронно-активацион-
ный (табл. 54) и спектральные методы. Разработан также фотомет-
рический метод определения 8-10-3—1 • 10“2 % натрия в сурьме [22].
Многие спектральные методы, разработанные для определения
натрия в элементах, применимы для определения натрия в сплавах
и соединениях этих элементов. Поэтому такие методы также рассмот-
рены в данном разделе. Спектральные методы применяют для опре-
деления натрия в рубидии [42, 421], магнии [1112], кальции [485],
алюминии [537, 690, 820, 844, 956, 974, 1006, 1112, 1114, 1208, 1215],
графите [936], кремнии [138], олове [388], свинце [495, 522, 773],
ванадии [78], мышьяке [1007], сурьме [115, 149, 1007], ниобии [35],
тантале [129], селене [123, 969, ИЗО], теллуре [123, 140, 1198], хро-
ме [406, 679], молибдене [179, 469, 862], вольфраме [35, 469, 798,
898, 1013], уране [156, 589, 1054], осмии [124, плутонии [1245].
Предложен ряд методик анализа металлического рубидия.
6*
163
Таблица 54
Определение натрия в металлах и неметаллах
нейтронно-активационным методом
Объект Навеска, г Определяемое содержание, % (cmin> %) Литература
Li 0,05—0,1 ЗЛО"»—1,3-10-;!(2-10-6) [1168]
0,1 (4-10-4) [129, 204]
Бе 0,5 (1,7.10-3) [916]
1 (4-10-4) [63]
Mg 0,15—0,2 (4—10). 10-з(3. IO-*) [941]
Zn (10-’) [1281]
В 0,15 (1-10-’) [437]
0,1 (6,2.10-e) [277]
Al 1 (10-’) [700, 921]
0,05 (2-10-3) [613]
1 (1,4-40-4) [1052]
С 5 (б-io-») 111]
1 (0,4—230). 10-4(4-IO-®) 191]
0,005—0,10 (5.10-6) 925]
1 (1,3-IO"2) 729]
5 ’ (2,5-10-6) [960]
Si 1—2 (6-Ю-6) [633, 996, 1109]
0,005—1,0 5.10-8—1,2-16-3 [871]
0,5—2 2.10-’—3,4-10-6 [152]
4-10-3—9.10-8 [1216]
1 (3-Ю-8) [188]
(З-10-з мкг) [998]
1 (5-10-’) [1019]
1 (5-10-6) [827]
1—1,5 (2-10-8) [1102]
0,3—3,0 (IO-6) [1035]
Sn 1 (ЗЛО"8) [934]
[491]
Pb 1 (3,2-10-’) [50, 192]
Ti 0,005—0,020 (ЗЛО"6) [48]
0,1 2,1.10-6 [1010]
Zr 1 (IO-®) [740]
P 2,6 1.10-6 [766]
0,4—0,5 1-10-6 [1040]
Nb 0,002—0,04 (1-Ю"’) [548, 867]
Ta 0,002—0,04 [867]
0,1 (7-10-6) [96]
Se 0,08—0,1 (1.10-6) [772]
(10-’) [1281]
0,1 (1,6—2,5). 10-4(1,5.10-4) 594]
0,1—0,2 (ЗЛО"») [104]
Cr 0,5—1 2-10-5(10-8) [929]
Mo 0,1 1,3-10-3(5-10-’) [706]
(3,2-10-’) [50]
164
Таблица 54 (окончание)
Обтект Навеска, г Определяемсе сгдержание, % <ст1П’ %) Литература
W Fe Rb, Pd, Pt 0,1 0,3 0,03—0,1 0,02—0,1 0,5 1,8.1(Н(5.10-;) (1,7-10-=) (1,5-10-5) (IO" 5) 1,5.10-«(1-10-«) [766] [451 [553 [1074] [929] [959]
1. После гскрытия ампулы металл растворяют в этаноле, затем раство-
ряют осадок вводе и доводят концентрацию раствора по рубидию до 1%, прибав-
ляя смесь (1 : 1) воды с этанолом. Для анализа используют атомно-абсорбционный
метод на основе монохроматора ЗМР-З с приемником излучения ФЭУ-22, пламя —
смесь пропана с воздухом. Источник света — безэлектродные ВЧ-лампы типа
ВСБ-2. Рубидий не влияет на определение натрия. Чувствительность анализа
повышается за счет применения органического растворителя и нагревания аэрс-
золя [421].
2. Пробу в ампуле взвешивают, ампулу вскрывают под слоем петролейиого
эфира. Металл растворяют в этаноле, раствор подкисляют НС1, после удаления
эфира и спирта упариванием раствора в кварцевом стакане разбавляют до кон-
центрации рубидия 1,0—1,5 мг/мл. Натрий определяют пламенным атомно-
эмиссионным методом, используя спектрофотометр на основе УМ-2, фотоумно-
жителя ФЭУ-38, пламя — ацетилен—воздух. Эталонные растворы содержат
50 мг/мл RbCl и 0,5—10 мкг/мл натрия. Предел обнаружения натрия 0,02 мкг/мл
[42].
Для определения натрия в алюминии и его сплавах в основном
используют пламенный атомно-змиссионный метод в пламенах про-
пан—бутан—воздух [269], водород—воздух [1215], ацетилен—воз-
дух [537]. В абсорбционной спектрофотометрии используют пламя
ацетилен—воздух [844] или ацетилен—кислород. В эталонные рас-
творы вводят соли алюминия [690]. При применении пламени ацети-
лен—кислород в раствор вводят 40% об. метанола [956]. Предел об-
наружения натрия — 10-3%. Основу отделяют добавлением аммиа-
ка [920], высаливанием А1С13 [1114] или отгонкой триэтилтрибромида
алюминия [1114]. Отмечено, что алюминий в интервале концентраций
140—220 мкг/мл не мешает определению натрия при использовании
фильтрового фотометра [269].
При определении 10-3—10-2% натрия в алюминиевой проволоке
кондиции 4н стандарты готовили на основе хлорида натрия. Приме-
няли фильтровый пламенный фотометр, пламя водород—кислород
[1215].
Образец растворяют в 2 мл HaSO4 и 7 мл HF при нагревании. Избыток HF
удаляют, дважды упаривая раствор до паров серной кислоты. Остаток раство-
ряют в воде, переносят в мерную колбу вместимостью 100 мл и разбавляют до
метки водой.
Для учета влияния серной кислоты на излучение натрия ее до-
бавляют в стандартные растворы [537].
165
При определении натрия в селене и теллуре высокой чистоты при-
меняют различные способы отделения основы. Большие количества
селена отгоняют в форме SeBr4 [ИЗО]. Натрий определяют атомно-
эмиссионным методом в пламени водород—кислород. После отгонки
селена в остатке остаются К, Li, Cu, Cd, Fe, Al, TI, Bi, Hg, Ca
и Mg. Для уменьшения влияния элементов (например, Са) в раствор
вводят буферную добавку — нитрат алюминия (25 г/л). Присутствие
щелочных металлов — калия и лития — определению не мешает.
Предел обнаружения натрия 10-8 %.
Описано три варианта определения теллура: при анализе теллура
чистоты 99,8% рекомендуют осаждать ТеО2, натрий определять в
фильтрате методом атомно-эмиссионного анализа [1198]. Подготовка
образца к анализу заключается в растворении пробы в смеси (3:1)
HCI и HNO3, многократном упаривании раствора до удаления азот-
ной кислоты с НС1, растворении остатка в воде и определении нат-
рия в фильтрате.
При анализе теллура чистоты 99,999% его отгоняют в кварцевой
лодочке при пропускании тока газообразного хлора при 420° С.
Натрий остается в остатке, который растворяют в 2 М НС1 и после
разбавления фотометрируют [1198].
Предложен экстракционно-пламенно-фотометрический метод оп-
ределения натрия в теллуре [140]. Теллур экстрагируют смесью
(3 : 7) трибутилфосфата с хлороформом из среды 12 М НС1. Метод
позволяет определить 10-3% натрия.
В цветных металлах (свинец, олово, сурьма) натрий определяют
методом атомно-эмиссионного анализа [115, 149, 249, 388] после пе-
ревода металлов в раствор. В стандартные растворы вводят соответ-
ствующее количество кислоты или соли основы, например олова
[388] или сурьмы [115]. Предложена простая методика определения
3-1СГ4—2-10~3% натрия с помощью фильтрового фотометра без от-
деления основы [115].
Навеску 3 г растворяют в 10—12 мл конц. НС1 при постепенном прибавле-
нии 30%-ного Н2О2. Затем вводят 25 мл 50%-ного раствора винной кислоты.
Раствор перед фотометрированием разбавляют водой до 50 мл. В растворы стан-
дартного ряда (1—10 мкг/мл натрия) вводят сурьму. Относительное стандарт-
ное отклонение при определении натрия 0,014—0,046.
В металлической сурьме и продуктах сурьмяного производства из
навески 0,5 г после растворения ее в смеси (3 : 1) НС1 и HNO3 и раз-
бавления раствора до 25 мл 5 М НС1 натрий определяют в пламени
ацетилен—воздух с погрешностью 3—5% [149].
При анализе особо чистых металлов основу отгоняют в виде ле-
тучих хлоридов (например, сурьмы [1007]), осаждают в виде мало-
растворимых соединений (PbS04 [522, 773]) или отделяют амальга-
мированием [495, 773].
В металлическом молибдене, вольфраме и их сплавах натрий оп-
ределяют методами пламенной атомно-эмиссионной и атомно-абсорб-
ционной спектрометрии [35, 82, 179, 443, 469, 790, 798, 862, 898,
1013]. Молибден и вольфрам в пламени излучают сплошной спектр,
который мешает определению малых количеств натрия, поэтому пред-
166
ложены методики с предварительным отделением основы методом
ионной хроматографии [862, 898], отгонкой хлоридов [798], осажде-
нием 8-оксихинолином [82], осаждением вольфрамовой кислоты.
Вольфрам и молибден растворяют в пероксиде водорода при
определении 10-3% натрия в пламени светильный газ—воздух [179,
469]. При увеличении навески до 2 г предел обнаружения нат-
рия 10~4% [179]. Рекомендован следующий ход анализа [1013].
Навеску пробы 1 г растворяют в 10 мл 30%-ного раствора Н2О2, содержаще-
го 1 г лимонной кислоты, вводят 5 мл раствора хлорида цезия и разбавляют до
50 мл водой: Натрий определяют атомно-абсорбционным методом на спектро-
фотометре фирмы «Перкин-Элмер» (модель 303) в пламени ацетилен—воздух;
предел обнаружения 3-10~4% . Хлорид цезия смещает равновесие ионизации ато-
мов натрия.
При определении натрия в вольфраме (триоксиде вольфрама)
вольфрам хлорируют насыщенными парами СС14 в токе воздуха [798].
Образец помещают в лодочку. После отгонки основы остаток рас-
творяют в 5 мл 0,1 раствора НС1и раствор фотометрируют на фото-
метре фирмы «К. Цейсс» (модель III).
Анализ хромсодержащих объектов (хром и его соли) выполняют
с помощью фильтровых фотометров в пламени пропан—бутан—воз-
дух или ацетилен—воздух [80, 81, 406, 679]. При анализе хрома вы-
сокой чистоты хром отгоняют в форме СгО2С12. Изучено влияние хро-
ма на эмиссию натрия и показано, что хром с концентрацией больше
450 мг/л создает фон в области 540—700 нм. Это влияние учитывают
введением хрома в эталонные растворы [81, 406].
В уране натрий определяют методом пламенной атомно-эмиссион-
ной спектрометрии в пламенах ацетилен—воздух, водород—кисло-
род после отделения от основы [156, 589, 939, 1054, 1237]: уран экст-
рагируют трибутилфосфатом из солянокислой [1237] или азотнокис-
лой [1054] среды. Описан метод выделения примеси натрия сорбцией
его катионообменником [589].
При анализе реакторного урана рекомендован следующий метод
[1054].
1—3 г пробы растворяют в HNO3, после упаривания досуха остаток раство-
ряют в 20 мл 5 М HNO3. Для экстракции урана используют раствор трибутил-
фосфата в керосине. Для определения применяют установку на основе монохро-
матора D222 фирмы Хильгер, пламя водород—кислород. Изучено влияние ка-
тионов на определение натрия. Предел обнаружения натрия 2-10-4%, sr = 0,03.
СПЛАВЫ
При анализе различных сплавов успешно применяют методы пла-
менной спектрофотометрии. В объектах сталелитейного производ-
ства натрий определяют с помощью пламенных фотометров и спектро-
фотометров. В последнем случае фоновое излучение железа, хрома
и других компонентов в меньшей степени влияет на результаты ана-
лиза. Отмечается, что при использовании пламенного фотометра
ФПФ-58 определению натрия не мешают 500 г/л хрома, 50 г/л же-
леза, 2 г/л алюминия [15]. При больших содержаниях железа и хро-
ма вводят поправку на фоновое излучение.
167
Для определения состава включений в поликристаллических фер-
ритах железо и хром отделяют в виде гидроксидов [213]. Влияние
алюминия и марганца на эмиссию натрия устраняют введением в фо-
тометрируемый раствор нитрата алюминия [1106]. Определен «фак-
тор специфичности» при определении натрия в пламени пропан—
бутан—воздух с помощью фильтрового фотометра в присутствии
железа, равный 150 [294].
При определении натрия в сырье для ферритов и в ферритах
изучали влияние Fe, Мп, Mg, Zn, Сг, Ni на эмиссию натрия [61, 213,
438]. При использовании спектрофотометра на основе монохромато-
ра УМ-2 в пламени ацетилен—воздух на интенсивность излучения
натрия не влияют цинк, магний и никель при их концентрации
в растворе до 10 мкг/мл. Предложена следующая методика [61].
Навеску феррита 1 г растворяют в 10 мл НС1 (1 : 3), раствор переносят в
мерную колбу вместимостью 100 мл, разбавляют до метки водой и фотометри-
руют. В стандартные растворы вводят соли марганца и железа для учета влия-
ния излучения монооксидов железа и марганца.
Для определения натрия (и других элементов) в ферритах желе-
зо—марганец—цинк или железо—марганец—магний использовали
пламенный фотометр ФПЛ-1. Изучено влияние железа, марганца,
цинка и магния на эмиссию натрия. Железо отделяли экстракцией
хлоридного комплекса 30%-ныМ раствором трибутилфосфата в ди-
этиловом эфире. Цинк и магний не влияют на определение натрия,
марганец вводят в эталонные растворы [438].
Описана методика анализа хромоникелевой стали на содержание
щелочных элементов и кальция [1173].
Навеску стали 1 г растворяют в 2 мл 20%-ной НС1, раствор фильтруют, под-
кисляют НС1 и разбавляют до метки водой в мерной колбе вместимостью 100 мл.
Отбирают пробу 20—50 мл, разбавляют 1 М раствором НС1 и проводят электро-
лиз при силе тока 1,5 А в течение 1,5—2 ч для осаждения на ртутном катоде
железа, никеля, хрома и частично марганца. Ток снижают до 0,2 А и удаляют
ртуть, не разрывая цепи. Раствор нагревают до 60° С, осаждают титан и марга-
нец аммиаком при pH 10. Фильтрат упаривают до 8 мл, подкисляют до pH 4
и разбавляют водой до 10 мл. Определяют калий, натрий, литий и кальций на
фотометре.
При определении натрия в чугунах используют практически одну
и ту же методику перевода образца в раствор; железо в виде хлорид-
ного комплекса экстрагируют диэтиловым эфиром [26, 905]. Предло-
жена следующая методика [905].
Навеску 4 г стружки растворяют в 75 мл НС1 (1 : 1) при нагревании. После
удаления газов кипячением прибавляют 7 мл HNO3 для окисления железа. Оса-
док кремневой кислоты отфильтровывают, к фильтрату прибавляют 1 мл 30%-но-
го раствора Н2О2 и 3—4 мл НС1 (1:1). Экстрагируют железо смесью (1 : 1) ди-
этилового эфира и НС1. Для удаления эфира водную фазу упаривают до сиропо-
образной массы, обрабатывают 5 мл воды и 1 мл НС1 (1:2), переносят в мерную
колбу вместимостью 50 мл, разбавляют до метки водой. Используют спектрофото-
метр «Бекман» (модель ДИ). '
В ферросилиции определяют до 10-3% натрия, используя эмис-
сионный вариант анализа в пламени ацетилен—воздух [311]. Отме_
168
чается, что при использовании спектрофотометра железо не влияет
на определение натрия.
Некоторые примеры определения натрия в сплавах другими ме-
тодами приведены в табл. 55, а также в работах [273, 533, 568, 1287].
/
- Таблица 55
Определение натрия в сплавах
Объект Определяемые количества, % Метод определения Литера- тура
Сплав алюминия 0,01—0,04 0,2—20 >10-5 Нейтронно-а ктивационный Атомно-аб сорбционный » [1052] [1208 [1006 ,
Спл^в РЬ—Na 9,3—11,2 Волюмометрический, титри- метрический [456]
Сплав РЬ—Ва (Са)-—Na 1,5—2,3 Гравиметрический [1143]
Феррит Мп—Zn 0,01—0,02 Нейтронно-активационный [1278]
Сплав железа Сплав Ni—Al 0,02—0,10 Комплексонометрическое тит- рование (см. главу V) Спектральный [21] [1050]
Бронза Сплав родия IO-3 Фотометрия пламени Атомно-абсорб ционный [344] [561]
ХИМИЧЕСКИЕ СОЕДИНЕНИЯ
При определении натрия в оксиде алюминия навеску переводят
в раствор сплавлением с карбонатом лития и оксидом бора [642,
643] либо обработкой пробы в кварцевом тигле соляной кислотой
[1224]. Для определения натрия используют спектрофотометр «Пай
Юникем SP-20» и пламя ацетилен—воздух. Спектральные методы
применяли для определения натрия в У203 [832, 833], SiO2 [320,
601], V2O5 [2711, NiO [863, 864].
При анализе V2O5 предложен следующий способ разложения
пробы [168].
Навеску 0,125 г обрабатывают 18 мл конц. HNO3 и 54 мл воды и нагре-
вают. Раствор разбавляют до 100 мл водой и фотометрируют. Изучено влияние
ванадия на интенсивность излучения натрия при использовании спектрофото-
метра на основе монохроматора УМ-2. До концентрации 2,5 мг/мл ванадия в при-
сутствии 2 М раствора HNO3 влияния не обнаружено. Стандартные растворы со-
держат 2 М HNO3.
Предложен атомно-абсорбционный метод определения натрия в
'пентаоксиде ванадия [77, 78].
Навеску пробы 0,1—0,2 г растворяют в 70 мл HNO3 (1 : 3) при нагревании,
разбавляют до 100 мл водой и определяют натрий с пределом обнаружения 10~3%
и относительной погрешностью 5% с помощью атомно-абсорбционного спектро-
фотометра на основе монохроматора УМ-2 в пламени пропан—бутан—воздух.
169
Толщина поглощающего слоя увеличивается благодаря тому,
что пламя втягивают в кювету с помощью насоса [77].
При определении натрия в оксиде никеля в стандартные растворы
вводят хлорид никеля (2 мг/мл), используют фильтровый фотометр
фирмы «К. Цейсс» (модель III) и пламя ацетилен—воздух [1108].
Анализ титановых белид и оксида титана проводят после отделения
титана отгонкой тетрафторида титана [516] или сорбцией сульфоса-
лицилатного комплекса титана анионообменником [1111]. Оксиды
цинка, железа, магния, никеля переводят в раствор с помощью НС1
[62]. Натрий определяют атомно-эмиссионным методом в пламени
ацетилен—воздух с помощью пламенно-фотометрической установки
монохроматора УМ-2 с фотоумножителем ФЭУ-38. Основные пара-
метры установки: напряжение на ФЭУ 1200 В, расход ацетилена
2 л/мин, воздуха 8 л/мин. Эталонные растворы готовят в интервале
концентраций натрия 5-10~5—1-10-3%. Изучено влияние НС1, К,
Са, Fe и Мп на интенсивность резонансных линий натрия. Погреш-
ность определения — sr = 0,03 0,05 [79].
На определение натрия влияет фон хрома в пламени, поэтому
хром отделяют хроматографически сорбцией на анионообменнике
или вводят в холостой раствор [62].
В вольфрамовой кислоте определяли 0,006—0,033% Na2O [443,
790]. При определении натрия в тетрахлориде германия рекомендо-
вана следующая методика [420].
Навеску пробы 2 г помещают в платиновый тигель и отгоняют GeCl4 при тем-
пературе 70° С в токе неона или аргона. Остаток растворяют в 6 М НС1, высуши-
вают и растворяют в воде. Для определения натрия используют атомно-абсорб-
ционный метод, спектрофотометр на основе монохроматора ЗМР-З, источник све-
та — безэлектродные ВЧ-лампы ВСБ-2, пламя пропан—воздух. Предел обна-
ружения натрид 5-10~6%. При содержании натрия 0,0002 мг/мл относительное
стандартное отклонение 0,05.
При анализе ниобата бария—натрия—лития натрий определен
методом атомно-эмиссионного анализа после отделения BaSO4 [214].
Натрий определяют с помощью пламенного фотометра ФПФ-58 в пла-
мени ацетилен—воздух после следующей подготовки пробы [35].
Навеску образца 0,1 г растворяют в 15 мл HF при добавлении нескольких
капель конц. HNO3. После выпаривания раствора остаток растворяют в 20 мл
смеси 3%-ных растворов оксалата и цитрата аммония и 0,4 г борной кислоты,
переносят в мерную колбу вместимостью 50 мл, разбавляют водой до метки и фо-
тометрируют. Определению не мешает основа образца — ниобий, тантал или
вольфрам.
Методы определения натрия в химических соединениях система-
тизированы в табл. 56.
В криолите и расплаве криолита натрий определяют электродиа-
лизом [1060]. Метод применим при содержании кальция <1%; по
правильности он сравним с гравиметрическим.
Навеску 0,1 г криолита или его расплава дважды упаривают досуха с сер-
ной кислотой; остаток кратковременно прокаливают при <^700° С, суспендиру-
ют в воде и переводят в анодное пространство электродиализатора Поппа. Элект-
родиализ ведут при постоянном напряжении 110 В; во избежание разогревания
католита раствор несколькд раз отбирают, объединяют порции, защищая от уг-
лекислого газа, и через 1,5 ч титруют 0,1 М раствором НС1.
170
Таблица 56
Определение натрия в химических соединениях
Объект Навеска, г Определяемье количества, % (сгшп> %) Метод определения Лите- ратура
HNO3 2 5.10-5—5.10-е Нейтронно-активационный [5081
(8-10-8 г)
НС1 0,4—0,5 (10--) » [577J
HNO3, hf,h2o2 [977].
СНзСООН, NH, » [977]
A12O3 0,05 (2.10-;) » [613]
0,27 Атомно-абсорбционный |859]
1 • Титриметрический (см. главу V) [10581
5-10-3 Атомно-смиссионный [7391
SiO2 1 2,4.10-5-4.10-* Нейтронно-активационный [978J
1,2.10-4-1,3.10-« » [178]
TiO2 0,04 (3-10-5) » [48]
0,1 2,1-10-5 » [1010]
Фосфат Потенциометрическое титро- [6]
ванне
Nb2O5 (1-107) Нейтронно-активационный [548]
uo2 (4—8). 10-* То же [893|
LiF 0,1 (4-10-*) » [204]
LiNtO3 (1.10-7) » [548]
NaCl, беизоат, Фотометрический (см. главу [781]
салицилат нат- рия VI)
KgCOg, K2S04, KC1 0,13—0,5 Гравиметрический [436]
KBr (5-Ю-4 % мол.) Радиолюминесцентный [241]
NaAlH4 0,1—0,2 0,5—1,5 Кулонометрическое титрова- ние (см. главу VII) [220]
Na3AlHe Электродиализ [1060]
Na2TiO3 Ионоселективный электрод [345]
TiCl4 1,92—4,94 Г равиметрический (см. главу IV) [476]
Этилат тантала 0,1 п.10-5—п-10-8 Нейтро нно-а кти вационный (см. главу IX) [96]
(CH3COO)2UO2 9.IO-4 То же [893]
Соединения урана » [1037]
171
Основным методом при анализе солей щелочных и щелочнозе-
мельных элементов является метод пламенного атомно-эмиссион-
ного анализа [157, 172, 175, 249, 250, 252-254, 270, 394, 395, 400,
414, 503, 563-565, 572, 586, 636, 826, 1107, 1136, 1230, 1231]. При
определении натрия в солях щелочных и щелочноземельных элемен-
тов методами пламенной спектрометрии могут проявляться особен-
ности влияния матриц, заключающиеся в смещении равновесных
состояний натрия в пламенах, а также может возрастать роль спект-
ральных влияний при применении метода атомно-эмиссионного ана-
лиза. Из-за специфических особенностей матриц в отдельный под-
раздел выделен анализ солей щелочных и щелочноземельных Ме-
таллов.
В солях цезия определяют п-10-4% натрия в пламени пропан—
бутан—воздух [172, 400]. Отмечается, что при определении натрия
в бихромате цезия в пламени ацетилен—воздух цезий является спект-
роскопическим буфером [826]. Нуль прибора устанавливают по рас-
твору бихромата цезия, содержащему 2500 мг/л соли. При примене-
нии низкотемпературного пламени водород—воздух снижается фон
по сравнению с пламенами ацетилен—воздух и ацетилен—оксид
азота(1) [1107]. Предлагается при анализе КС1 сп. ч. раствор КС1
наносить на микрозонд, определение проводить в пламени ацетилен—
воздух [414]. Этим методом определяли из навески 100—200 мкг КС1
8-10~4% натрия с использованием спектрофотометра на основе мо-
нохроматора ДФС-12. Сообщается о расширении возможностей пла-
менно-фотометрического определения микроколичеств щелочных
элементов, в том числе и натрия, в особо чистых реактивах и препара-
тах, если применять монохроматор с высокой разрешающей способ-
ностью (R = 10 000), например СД-2 или ДФС-12 [408]: в 1 %-ном
растворе исследуемого препарата можно определять (2—5)-10“5 %
примеси. Из 5%-ного раствора иодида кальция методом пламенного
атомно-абсорбционного анализа с использованием спектрофотометра
«Спектр-1» в пламени пропан—бутан—воздух определяют 5-10-3%
натрия. Источник света — ВЧ-безэлектродные шариковые лампы
[503]. При мешающем влиянии основы, например кальция, послед-
ний осаждали в виде оксалата [636]. При определении 1-10-4—1-
•10~3% натрия в хлориде натрия готовили растворы эталонов и об-
разцов, содержащие 2,5% СаС12 [572]. При фотометрировании 1 %-ных
растворов солей лития, калия, магния, стронция, бария и кальция
определяли 0,1 мкг/мл натрия [175].
Метод атомно-абсорбционного анализа применяли для определе-
ния натрия в хлориде натрия [596], иодиде калия [171], фториде маг-
ния [504], карбонате бария [339], фториде кальция [505], хлориде
цинка [1225], а метод атомно-эмиссионного анализа — для определе-
ния натрия в бихромате [251] и перманганате [396] цезия, перрена-
тах аммония, калия, магния и щелочноземельных элементов [563],
карбонате марганца [515], сульфате и хлориде цинка [514], препара-
тах рения [430], солях магния [480], полый катод — для определе-
ния натрия в алюмоаммонийных квасцах [369].
172
НЕФТЕПРОДУКТЫ, ОРГАНИЧЕСКИЕ РАСТВОРИТЕЛИ,
СОЛИ ОРГАНИЧЕСКИХ КИСЛОТ
Методы атомно-эмиссионного и атомно-абсорбционного анализа
применяют для анализа нефтепродуктов и топлива [882, 1148, 1246],
органических растворителей [608, 609], продуктов органического
производства [279], солей органических кислот [558, 564, 565].
В нефти, керосине, бензине натрий в числе 23 элементов опреде-
ляли атомно-абсорбционным методом в интервале концентраций
10-7—10-4% ,с использованием спектрофотометра «Перкин-Элмер»
(модель 403) [1246].
При подготовке пробы к анализу 2—75 г пробы озоляют в платиновом тиг-
ле. Жидкую пробу адсорбируют на сульфонате магния или калия, прибавляют
5 капель конц. H2SO4, нагревают до выделения паров серной кислоты и озоляют
в муфельной печи в течение 0,5—4 ч. Остаток растворяют в НС1, HNO3 или
H2SO4. Стандартные растворы готовят на основе сульфоната магния или калия.
Относительное стандартное отклонение 0,15—0,20.
При атомно-абсорбционном определении натрия в слабощелоч-
ном реактивном топливе при подготовке пробы удаляют углеводоро-
ды упариванием пробы со смесью HNO3 и НС1О4 [882]. Интервал оп-
ределяемых содержаний натрия 10 ~3—2-10“3%.
Рекомендована методика определения натрия в ацетате целлюло-
зы [279].
Навеску 10 г пробы прокаливают в муфельной печи в фарфоровом тигле
сначала при 300° С, а затем при 600° С в течение 4 ч. Остаток растворяют в 1 мл
конц. НС1 и разбавляют до 50 мл в мерной колбе дистиллированной водой. При
фотометрировании пробу разбавляют в соотношении 1:5. Определение прово-
дят по эмиссии натрия в пламени пропан—воздух.
В солях щелочных металлов муравьиной, уксусной, лимонной
и винной кислот определяли 10-3—10-1% натрия [558]. Для повыше-
ния чувствительности определения натрия соли растворяют в ук-
сусной кислоте или используют 2 М растворы солей.
В нефти определяли 10-Б—10-4% натрия из навески 0,2—1 г ней-
тронно-активационным методом [45]. Этот же метод позволил опре-
делить натрий с пределом обнаружения 10-Б% из навески 0,1—0,15 г
[490].
ПРОЧИЕ ОБЪЕКТЫ
Методы, применяемые для этих целей, приведены в табл. 57.
Метод эмиссионной фотометрии пламени применен для определе-
ния натрия (и калия) в железохромовых катализаторах после отделе-
ния железа электролизом [1190]. Другие методы отделения железа,
например осаждением пиридином гидроксида железа(Ш), непри-
годны вследствие мешающего влияния на определение натрия. При
определении 0,15—6,2% Na2O погрешность не превышает 3%.
Навеску пробы 1 г нагревают с 20 мл 60%-ной НС1О4 в закрытой чашке
в течение 30—45 мин, упаривают до 2—4 мл, разбавляют водой до объема 100 мл
и отфильтровывают кремнекислоту. Упаривают примерно до 100 мл, переносят
в электролизер и отделяют железо на ртутном катоде при силе тока 3,5 А. По-
173
Таблица 57
Определение натрия н прочих объектах
Объект Навеска, г Определяе- мые количе- ства, % (cmin> %> Метод определения Литера- тура
Стекло Стекло, эмаль Алмаз Лазерные кристаллы типа MgAl2O4:Cr Огнеупорные мате- риалы Катализаторы Ее20з — Сг20з Продукты калийной промышленности Амальгама Археологические образцы Растения Электровакуумные материалы Бораты Органические мате- риалы 0,2 0,004- 0,305 0,05-0,1 1 20,5 1 (6,2.10-5) 0,15-6,2% Na2O 5,8-20 0,37 0,1 — 0,5 мг Фотометрия пламени Гравиметрический Полярографический Титриметрический » (см. главу V) Нейтронно-актива- ционный То же Полярографический Фотометрия пламени То же Волюмометрический Нейтронно-актива- ционный Фотометрия пламени То же » Нейтронно-актива- ционный Гравиметрический [569] [551} [749} [976J [1164] [1146] [1032] [151] [1190] [1133] [687] [943] [854} [9431 [459] [825] [981}
лученный раствор разбавляют до 100 мл водой и из аликвотных частей опреде-
ляют натрий и калий методом эмиссионной фотометрии пламени. При построе-
нии градуировочного графика в стандартные растворы вводят то же количество
хлорной кислоты, а соотношение концентрации калия и натрия поддерживают
примерно равным их соотношению в катализаторе.
При волюмометрическом определении натрия в амальгаме посту-
пают следующим образом [687]. Вначале получают электролизом
раствора NaCl с ртутным катодом амальгаму натрия, затем ее разру-
шают серной кислотой и измеряют объем водорода, эквивалентный
содержанию натрия в амальгаме. Продолжительность анализа 1—
2 мин, погрешность определения 1%.
Анализ проводят в колбе Бунзена вместимостью 300 мл, наполовину запол-
ненной 30%-ной H2SO4 я снабженной воронкой с двумя кранами. Отводную
трубку колбы соединяют резиновой трубкой с бюреткой Бунже вместимостью
50 мл, заполненной окрашенной водой и соединенной в свою очередь с уравни-
тельной склянкой. В воронку вносят 20,5 г амальгамы, которая заполняет про-
странство между двумя кранами. Если первый кран закрыт, а второй открыт,
то проба стекает в колбу и присутствующие в ней щелочные и щелочноземельные
174
металлы реагируют с cepHOii кислотой. Объем выделившегося водорода измеря-
ют бюреткой.
Разработаны методы определения натрия в катализаторах [544,
549, 989, 1226], атмосферной пйли [1191], аэрозолях [1214], стеклах
[154], керамических материалах [266], шламах [418], полупроводни-
ковых материалах [434], лаках [435], продуктах производства пиг-
ментов [222], щелоках [547].
Глава XI
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ В НАТРИИ
И ЕГО СОЕДИНЕНИЯХ
К натрию реактивной квалификации, а также к натрию, предна-
значенному для некоторых специальных целей, предъявляются вы-
сокие требования по чистоте (табл. 58).
Таблица 58
Критерии чистоты натрия реактивной квалификации (в %) [12]
(ТУ МХП 1664-50)
Компонент ч. ч.д.а. Компонент ч. ч.д.а.
Металлический нат- >99,8 >99,9 Кремнекислота и <0,007 <0,005
рий Хлориды Сульфаты <0,01 <0,01 <0,005 <0,005 вещества, осаж- даемые аммиаком Кальций <0,08 <0,05
Железо <0,01 <0,005 Калий Качественное
Тяжелые металлы группы сероводоро- да, считая на свинец <0,007 <0,005 Азот Фосфаты йены гание
В техническом натрии общее содержание примесей не превышает
0,1%. Из металлов, которые образуют сплавы с натрием и смешива-
ются с ним во всех отношениях, наиболее значительно содержание
калия (0,01—0,045%). Содержание Pb, Cd, Cs, Ag, также образую-
щих сплавы с натрием, не превышает 0,001%. Из металлов, смеши-
вающихся с натрием ограниченно (Са, Li, Zn, Pd, La, Се), в натрии
содержится главным образом кальций (до 0,019%). Металлы, не об-
разующие с натрием сплавов (Fe, Al, Cr, Mo, Ni, Ta, U), содержат-
ся в нем на уровне <^0,01 %. Наиболее вредной примесью является
кислород в виде Na2O и частично в виде Na2CO3.
ОПРЕДЕЛЕНИЕ ОСНОВНЫХ КОМПОНЕНТОВ
Наиболее чистый натрий получали зонной плавкой [708]. Его
чистоту проверяли по величине остаточного сопротивления и спект-
ральным методом. Эффективность очистки увеличивается с умень-
175
шением скорости перемещения расплавленной зоны вплоть до
3,5 мм/ч.
Погрешность определения Na2O в натрии экстракцией ртутью
можно уменьшить несколькими приемами: 1) увеличением чистоты
газа пропусканием его через барботер с KNa; 2) обогреванием экст-
рактора газовым пламенем на стадии амальгамирования вместо
охлаждения воздуходувкой. Na2CO3 в натрии определяют кондукто-
метрическим титрованием раствором Ва(ОН)2 после гидролитическо-
го разложения образца водяным паром, растворением получившейся
смеси NaOH + Na2CO3 в разб. H2SO4 и поглощения выделяющегося
СО2 [856]. Метод позволяет определять >10-3% Na2CO3.
В оксиде Na2O определяют натрий волюмометрически по выделе-
нию водорода, получившегося после взаимодействия смеси с водой
[967]. Примеси других элементов не определяли.
В перекисных соединениях натрия определяли раздельно натрий
и Na2O2 волюмометрически [343]. При обработке пероксида натрия
с примесью натрия этанольным раствором щелочи (t < 0° С) обра-
зуется алкоголят натрия и выделяется водород в количестве, экви-
валентном содержанию натрия. При этом образуется кислая натрие-
вая соль пероксида водорода, устойчивая в данных условиях. При
повышении температуры и в присутствии солей кобальта она разла-
гается при действии разбавленных кислот, выделяя эквивалентное
количество кислорода. Метод применяют в контроле производства
Na2O2 окислением металлического натрия.
В гидриде натрия натрий определяют титриметрически раствором
Hg(NOs)2 в присутствии дифенилкарбазона [281]. Предварительно
проводят реакцию
2Na + 2C4HeBr=2NaBr + CbHiE.
Затем добавляют воду, при этомNaBr резкстрагируется в водную фазу,
гидрид натрия образует с водой гидроксид и не мешает определению.
Из навески 0,5—4 г гидрида натрия определяют 0,2—20% мае. нат-
рия с относительным стандартным отклонением 0,07 при содержании
натрия до 4%.
Гидрид натрия (0,5—4 г) отбирают в герметизированной камере и помещают
в токе азота в коническую колбу вместимостью 100 мл, заполненную 20—30 мл
и-бромистого бутила, очищенного от непредельных соединений промыванием
конц. H2SO4 и перегнанного. Реакцию ведут при 67—71° С в течение 10 мин при
непрерывном перемешивании магнитной мешалкой. Затем колбу охлаждают,
добавляют 40—50 мл воды и встряхивают в течение 1 мин. После расслаивания
фаз отбирают аликвотные части водного раствора, содержащие 1—20 мг натрия,
разбавляют водой до 50 мл и нейтрализуют по универсальному индикатору до
pH 7 вначале конц. HNO3, а затем 0,1 М раствором HNO3. Добавляют 0,6 мл
0,1 М раствора HNO3, 8 капель 1%-ного раствора дифенилкарбазона и титруют
0,1 М раствором Hg(NO3)2 до появления розовой окраски.
В хлориде, бромиде, нитрате и нитрите натрия определяют нат-
рий гравиметрически в форме тетрафторобората [427]. При нагрева-
нии соли с раствором HBF4 и высушивании при 170—175° С HF
и Н3ВО3 полностью испаряются, образующийся NaBF4 термически
устойчив при 200° С. Погрешность определения натрия не превышает
0,33%.
176
В натриевых солях лимонной, молочной и салициловой кислот
натрий определяли косвенным комплекеонометрическим методом
[703] (см. главу V). В Na2S0s определяли Na2CO3 потенциометриче-
ски [546]. \
В гидросернистом натрии определяли Na2CO3 титриметрически
косвенным алкалиметрическим Методом [98].
25 г пробы Na2S2O4 растворяют в воде, нейтрализуют 2 М раствором NaOH
по метиловому красному, вводят избыток 10 мл, разбавляют водой до 250 мл
и фильтруют'. К 50 мл раствора прибавляют 15 мл 30%-него раствора Н202 для
окисления серы до сульфата, нейтрализуют 0,2 М раствором НС1 по смешанному
индикатору (1%-ный раствор тимолового синего смешивают с 0,1%-ным раство-
ром крезолового красного в соотношении 6:1), причем вводят 5 капель раствора
индикатора в начале титрования и 2 капли — в конце. Конец титрования оп-
ределяют по свидетелю, для приготовления которого к 70 мл воды прибавляют
20 мл 0,05 М раствора Na2CO3, 10 мл 0,1 М раствора НС1 и 7 капель смешанного
индикатора. В нейтрализованный раствор вводят 25 мл 0,1 М раствора НС1,
кипятят 5—10 мин и титруют избыток HCI (не должен быть меньше 10 мл) 0,1 М
раствором NaOH по метиловому красному.
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ
В металлическом натрии определяют 46 примесей:: Н [144, 283,
727, 795, 841, 1005, 1202, 1249, 1251], Li [618, 841], К [216, 618, 841,
1009, 1160], Rb [1176], Cs [618, 1175, 1176], Cu [243, 534, 618, 1082,
1127], Ag [618, 1176, 1177], Au [615], Mg [534, 618, 841, 1131, 1160],
Ca [595, 618, 791, 841, 1127, 1128, 1129, 1160], Sr [618], Ba [618, 791,
1127, 1129], Zn [243, 534, 1127, 1176, 1177], Cd [534, 841, 1127], Hg
[1176, 1177], В [521, 618, 841, 1110], Al [243, 534, 618, 841],
Ga [534], In [534], Y [618], C [51, 145, 246, 618, 631, 721, 723, 725,
841, 856, 930, 972, 990, 1020, 1022—1024, 1131, 1161, 1249], Si [618,
1131], Sn [618, 1176], Pb [243, 534, 618, 1127], Ti [618, 1127], Zr [618],
Hf [1127], N [51, 145, 246, 282, 283, 532, 618, 631, 675, 723, 1131,
1254], Sb [534, 1176, 1177], Bi [243, 534, 618], V [243, 534, 618, 841,
1127], Nb [1120, 1127, 1131], Ta [1127], О [23, 51, 139, 145, 246, 275,
283, 307, 308, 531, 532 , 615, 618, 651, 652 , 673, 675, 711, 723—725,
809, 814, 836, 841, 842, 931, 975, 1016, 1020, 1022, 1024, 1103, 1123,
1159, 1169, 1172, 1203, 1204, 1249, 1250, 1262], S [36, 1021], Cr [23,
618, 823, 841, 884, 1127, 1131, 1177], Mo [618, 841, 884, 1127], U [841],
F [618, 1020], Cl [520, 618, 837, 841, 1020, 1127, 1205], Br [1127], J
[1127], Mn [243, 534, 618, 823, 841, 1127], Fe [23, 243, 534, 618, 823,
841, 884,1127, 1131, 1176, 1177, 1248], Co [243, 534, 841, 1127, 1158,
1176, 1177], Ni [23, 243, 534, 618, 738, 823, 841, 884, 1127, 1131,1176].
Основу не определяют, но определяют примеси соединений натрия.
Опубликован обзор по методам определения Mg, С, Si [1131].
В едком натре определяют воду [477], К [1160], Си [374, 849,
886, 1082], Mg [1160], Hg [1059], Al [518], Cl [520], а также Ag, Ni,
Ti, Mn, Cr, Bi, Pb, Sn, Mo, V, Co, Au, Fe, Zn, Sb [374].
В галогенидах натрия (фторид, хлорид, бромид, иодид), нитри-
тах, нитратах и тетраборате натрий определяют гравиметрически
в форме тетрафторобората с погрешностью не выше 0,25% [427].
В хлориде натрия определяют К [408, 1160], Rb [126, 408], Cs [408],
177
Си [126, 370], Ag [370, 461], Au [370], Са [110, 479], Zn [126, 128,
370], Hg [126], Ga [126, 370], In [126, 128], Ge [370, 550], Sb [126,
128, 198, 370], V [228], Se [126, 128], Cr [370, 758], Br [126], J [126],
Mn [126, 230, 370], Fe [126, 128, 229, 370, 758], Co [126, 128, 370], Ni
[370, 758], Ir [126, 128], а также Bi, Pb, Tl, Al, As [370]. В бромиде
натрия определяют марганец [352]. В иодиде натрия определяют Rb,
Си, Zn, Hg, Ga, In, Eu, Sb, Se, Br, J, Mn, Fe, Co, Ir [126], Ca [58,
1279], Ba [27], Mn [352, 373], а также Al, Cr, Fe, Ni, Co, Mn, Си, Sn,
Pb [373], Fe, Al, Mg, Si, Ti [1279]. В гипофосфите натрия определяют
C i [232, 292], Zn [500], Pb [448], Zr [501], Bi [292, 499], Mn [231],
а также Ag, Au, Fe, Ti, Cr, V [292] и Fe [233]. В ацетате натрия опре-
деляют Zn [440], Al [56], а также Cj, Ni, Co, Fe, Mn, Cr, V [977].
В тартрате натрия—калия определяют Zn [440], в цитрате натрия —
Са [256], в азиде натрия — Са, Sr, Ва [1096], в сульфате натрия Ст,
Fe, Ni [758], Pb [202, 331], в нитрате натрия — Ag, Bi, An, Cu, Mn,
Ni, Pb, Tl, Mn, Ga, Fe, Al, In, Zn, Ge, Sb, As, Co, Cr [370], Ca [879],
Al, Y, Fe, Cu, Co, Mg, Mn, Ca, Ni. Nb, Cr [223], в карбонате натрия —
Ag, Bi, Au, Cu, Mn, Ni, Sb, As, Co, Cr, Pb, Tl, Mn, Ga, Fe, Al, In,
Zn, Ge [370], Cu, Ni, Co, Fe, Mn, Cr, V [977], в фосфате натрия —
Cs, Rb [379, 381], Al [353], в метафосфате натрия — Mn, C i, Mo,
Fe, V, Cr, Pb, Ni, Co, Ti[86], Fe, Ni, C>, Co, V, Mo [87], в ниобате,
титанате и танталате натрия — Fe, Si, Al, Mn, C1, Zn, Ni [259],
в ниобате натрия-бария — Si, Fe, Co, Ni, Bi, Mn [293], в молибдате
натрия-лантана — С\ Mn, Со, Fe, Cr, Ni [377], в молибдате натрия-
гадолиния и натрия-неодима — С’, Fe, Мп, Ni, Со, Cr, V [378],
в перренате натрия — Fe, Мп, С ’, Al, Mg, Ni, Mo, Si, Pb [542],
в диэтилдитиокарбаминате натрия — Мп, Bi, Ti, Со, Pb, Cr, In, Ag,
Ni, Cu, Sn [291].
МЕТОДЫ МНОГОЭЛЕМЕНТНОГО АНАЛИЗА
Из методов определения примесей наибольший интерес представ-
ляют физические и физико-химические методы, позволяющие одно-
временно определять несколько элементов..Например, в натрии при-
меси Rb, Cs, Ag, Zn, Hg, Sn, Sb, Cr, Fe и Co определяли недеструк-
тивным нейтронно-активационным методом [1176]. В хлоридах и
иодидах натрия недеструктивным нейтронно-активационным мето-
дом определяли примеси Rb, Zn, In, Eu, Sb, Se, Br, J, Fe, Co, Ir,
а примеси Cu, Zn, Hg, Ga, In и Mn — с радиохимическим выделени-
ем [126, 128].
В методическом отношении активационное определение примесей
в натрии и его соединениях мало чем отличается от определения нат-
рия в различных объектах. Выбор варианта или метода активацион-
ного анализа зависит прежде всего от характера определяемых при-
месей. Так, при определении С, N и О в металлическом натрии ис-
пользован фотоактивационный метод, основанный на фотоядерных
реакциях 12С(у, п) UC, 14N(y, п) 18N и 16О(у, п) 15О с последующими
химическими превращениями (например, С -► СО2 -> Na2CO3) и из-
мерением активности аннигиляционного у-излучения на сцинтилля-
178
ционном у-спектрометре [631, 723]. Предел обнаружения углерода
и азота — 10-6%, кислорода — 3-10~®%.
В случаях определения элементов, имеющих достаточно высокие
сечения активации, пробу облучают нейтронами. При этом для од-
них и тех же элементов возможно определение как с отделением ос-
новы (радиохимический вариант), так и без отделения (инструмен-
тальный вариант). Очень мало работ по определению одних и тех же
примесей различными методами, по метрологической оценке этих
методов и их возможностей применительно к объектам. Например,,
в натрии определяют фотометрическим методом В, Al, V, Сг, Мо, U,
Мп, Fe, Ni; атомно-абсорбционным — Li, К, Mg, Са, Cd, Сг, Мп,
Fe, Со, Ni; пламенно-фотометрическим — К; масс-спектральным —
U; вакуумной дистилляцией — О; сожжением — С; амальгамирова-
нием — Ни потенциометрическим титрованием — С1 [841].
Наиболее многочисленны спектральные методы, позволяющие раз-
дельно определять много примесей в натрии и его соединениях. При-
меняют пламенный атомно-эмиссионный, прямой спектральный, хи-
мико-спектральный, рентгенофлуоресцентный, масс-спектральный,
лазерный микроспектральный методы. Влияние матрицы, как пра-
вило, снимают предварительным концентрированием примесей раз-
личными методами: экстракцией, соосаждением, сорбцией.
Определение примесей в натрии. Для определения примесей при-
меняют химико-спектральные [243, 534, 884, 1009], рентгенофлуо-
ресцентные [791, 1127, 1128], масс-спектральные [618, 619], атомно-
абсорбционные [757, 823, 1122] методы.
Методом атомной абсорбции с использованием графитовой печи
определяли Сг, Fe, Мп, Ni. [823]. Предварительно основу отгоняли
в глубоком вакууме из молибденовых или танталовых тиглей. Из на-
вески 4 г определяли (в скобках приведена длина волны в нм, и пре-
дел обнаружения, в %): Мп (279,5; 1-10“7); Сг (357,9; 2,5-10-7);
Fe (248,8; 5-10-7); Ni (232,0; 1,5-10-6) с относительным стандартным
отклонением 0,01—0,04.
В чистом натрии определяли 15 примесей химико-спектральным
методом, концентрируя примеси экстракцией в форме диэтилдитио-
карбаминатов и 8-оксихинолинатов. Некоторые результаты и харак-
теристики приведены в табл. 59.
Рентгенофлуоресцентным методом определяли многие примеси
в металлическом натрии из навески 4 г со следующими пределами
обнаружения [1127]: (1—2)-10-6% Са, Сг, Ni, Zn, Nb, Та, Hf, Мо,
Вг, J; (0,5—1)-10“4% Ba, Fe, Pb, Cd, Cl; (l-5)-10~6 % Cu, Mn, Co,
Ti, V.
Определение примесей в едком натре. В методе [374] определяли
17 примесей после предварительного концентрирования соосажде-
нием на сульфиде кадмия с помощью тиоацетамида и диэтилдитио-
карбамината. При навеске щелочи 50 г коэффициент концентрирова-
ния примесей 103. Примеси определяли спектральным методом в дуге
постоянного тока с использованием в качестве коллектора графито-
вого порошка, в качестве носителя — 5%-ного NaCl. Аналитиче-
ские линии определяемых элементов и пределы обнаружения приве-
179
Таблица 69
Примеси, определяемые в натрии
химико-спектральным методом [534]
Таблица 60
Аналитические линии и
пределы обнаружения
примесей в едком натре
Примесь Аналитичес- кая линия, нм % Определяемое содержание, % Элемент Аналитическая линия, нм Предел обнару- жения, %
А1 308,216 0,35 5.10-5—5-10-* Си 324,75; 327,40 3-Ю-8
V 318,540 0,20 (2—5). IO"5 Ag 328,07 3- 10-s
Bi 306,772 0,15 (2—5)-IO-5 Ni 305,08 3-10-8
Ti 308,80; 323,45 3-10-s
Ga 294,364 0,15 (2—5)-10-5 Mn 280,11 3-1C-8
Fe 248,327 0,35 (1-5)-10-« Cr 283,56 3- 10-s
In 325,609 0,20 (2— 5)-Ю-s Bi 306,77 3-10-3
Cd 228,802 0,30 (2—5)-IO"5 Pb 283,31 3-10-8
Co 304,401 0,15 (2-5). 10-5 Sn 283,99 3-10-’
Mo 317,03; 313,26 3-1C-’
Mg 277,983 0,35 5.10-5— 5. IO-* V 318,54; 318,39; 1 • 10-5
Mn 280,106 0,20 5.10-5—2-10-5 318,34
Cu 324,754 0,25 5.10-8—1.10-5 Co 345,35 1-10-5
Ni 305,082 0,20 (2-5)-10-5 Au 267,59 1-10-5
Al 308,22 1.10-5
Pb 283,307 0,20 (2—5). 10-5 Fe 302,C6 1-10-5
Sb 259,806 0,15 (2—5). 10-5 Zn 334,50 3-10-f
Zn 213,856 0,30 5.10-5—5-10-4 Sb 259,81 3-10-5
дены в табл. 60. Относительное стандартное отклонение не превыша-
ет 0,15.
Раствор, содержащий 50 г щелочи (в пересчете на 100%-ную), нейтрализу”
ют в полиэтиленовом стакане соляной кислотой до pH 7,0 по универсальной ин-
дикаторной бумаге. После охлаждения к раствору добавляют 1 мл раствора соли
кадмия, свежеперегнанный раствор аммиака до pH 7,5, 50 мг угольного порош-
ка и все перемешивают. Затем добавляют 1 мл раствора диэтилдитиокарбамина-
та натрия и снова перемешивают. Через 30 мин в раствор пропускают очищенный
сероводород из аппарата Киппа в течение 10 мин. Через 2 ч полученный осадок
отфильтровывают на фильтр синяя лента на воронке Бюхнера, промывают 5 мл
сероводородной воды, разбавленной аммиаком до pH 7,5. К сухому остатку
прибавляют 2,5 мг хлорида натрия и тщательно перемешивают. Параллельно
проводят контрольный опыт. Для этого соляную кислоту в количестве, равном
израсходованному на нейтрализацию щелочи, в платиновой чашке упаривают
в присутствии 50 мг угольного порошка почти досуха. Затем приливают такое
количество воды, какое было добавлено при подготовке пробы, добавляют 1 мл
раствора кадмия и нейтрализуют аммиаком до pH 7,5. Затем добавляют 1 мл
раствора диэтилдитиокарбамината натрия. Остальные операции те же, что и при
подготовке проб.
Готовят эталоны с содержанием 1% каждого определяемого элемента и за-
тем разбавляют их до необходимых концентраций. Для молибдена и ванадия
готовят отдельные эталоны, так как при совместном определении их с другими
элементами возможно наложение линий. Для приготовления эталонов, содер-
жащих по 1% молибдена и ванадия и 0,1% золота, сначала прибавляют к 0,9565 г
180
угольного порошка 1 мл стандартного раствора, содержащего 1 мг золота в 1 мл,
осторожно подсушивают, избегая перегрева, под инфракрасной лампой, тща-
тельно перемешивают в агатовой ступке, затем добавляют 0,0257 г молибдата ам-
мония и 0,0178 г пентаоксида ванадия и снова перемешивают. Для облегчения
растирания смеси добавляют этанол (2 мл на 1 г смеси), затем этанол удаляют
высушиванием приготовленного эталона в сушильном шкафу при 60—70° С.
Остальные эталоны с убывающим содержанием примеси (до 1-10“4%) готовят
последовательным тщательным растиранием одной весовой части каждого пре-
дыдущего эталона с девятью весовыми частями угольного порошка. Готовят
также промежуточные эталоны, содержащие по 3,33-10'”% примеси. К 1г
каждого эталона прибавляют по 0,05 г хлорида натрия и по 20 мл сульфата
кадмия, после чего все тщательно перемешивают.
Пробу или 50 мг эталона помещают в канал нижнего электрода и зажига-
ют дугу постоянного тока. На одну фотопластинку в одинаковых условиях фото-
графируют по 2—3 раза спектры угольных концентратов, эталонов и контроль-
ной пробы. Каждый раз ставят новую пару электродов. Щель открывают до за-
жигания дуги.
Содержание примесей в концентрате находят по градуировочным графикам.
Рекомендуемые условия анализа: ширина щели 0,015 мм; время экспозиции
1,5 мин; высота диафрагмы на средней линзе конденсорной системы 5 мм; на-
пряжение 220 В; сила тока 12 А; фотопластинки спектральные, тип I, чувстви-
тельность 3 ед. ГОСТ. Угольные электроды: нижний (анод) с цилиндрическим
каналом диаметром 4,5 мм и глубиной 6 мм, верхний электрод заточен на конус.
Определение примесей в солях натрия. Разработан химико-спект-
ральный метод определения 19 примесей в солях натрия на уровне
^2-10~9% с предварительным концентрированием примесей на кол-
лекторе с использованием тиоацетамида и диэтилдитиокарбамината.
Примеси определяют с использованием катода или дуги постоянного
тока (табл. 61).
Таблица 61
Аналитические линии и пределы обнаружения при определении примесей
в солях натрия [371 ]
Элемент •Аналитическая линия, нм Предел обнаруже- ния (%) при использовании Элемент Аналитическая линия, нм Предел обнаруже- ния (%) при использовании
полого катода дуги постоян- ного тока ПОЛОГО катода дуги постоян- ного тока
А1 396,15 1-Ю-7 1 • 10’7 As 234,98 2-10“6 1-10~5
Bi 306,77 1 • 10'8 1-10~7 Ni 341,48 2-10-7 з-ю-7
Ga 294,36 2-10-8 1 • ю-6 Sn 317,50 1-1 о-7 7-10”'
. Ge 265,12 8-10"7 1-10"* Pb 283,31 4-10-3 3-Ю'7
Fe 371,97 2-10 ~8 1-Ю"7 Ag 328,01 2-10“9 1-10"7
Au 267,59 2-Ю'8 1-1(Г* Sb 287,79 1-10'6 1-10“
. In 325,61 2-10-8 1-10“ Tl 337,57 1-10“’ i-icr®
Co 345,35 2-Ю-7 3-10“е Cr 357,87 2-Ю"7 5-10-7
Mn Cu 280,11 324,75 1 10~7 2-10~8 1-10~7 1-Ю"7 Zn 334,50 4-Ю'7 3-10“
В кварцевой колбе с притертой пробкой растворяют 25 г анализируемой со-
ли в 80 мл бидистиллята. Затем прибавляют 1 мг-ион кадмия в виде соли, Снеже-
-Приготовленный раствор аммиака до pH 7,5 (внешний индикатор — универсаль-
181
ная индикаторная бумага) и 0,05 г угольного порошка. Раствор перемешивают,
добавляют 0,25 мл 1%-ного раствора диэтилдитиокарбамината натрия, снова
тщательно перемешивают встряхиванием и через 30 мин постепенно добавляют
0,07 г очищенного тиоацетамида при вращательном движении колбы. Через 2 ч
осадок отфильтровывают на плотный беззольный фильтр синяя лента диаметром
1,5 см на воронке Бюхнера и промывают 5 мл 0,07%-ного водного раствора тио-
ацетамида, нейтрализованного аммиаком до pH 7,5. Параллельно проводят конт-
рольный опыт со всеми реактивами.
Для приготовления головного эталона, содержащего по 0,01% каждой из
перечисленных примесей, в кварцевой колбе вместимостью 50 м i 1 мл раствора,
содержащего по 0,01 мг/мл каждой из определяемых примесей, разбавляют во-
дой до 30 мл, нейтрализуют до pH 7,5, добавляют 0,1 г угольного порошка,
0,5 мл 1%-ного раствора диэтилдитиокарбамината натрия и пропускают очи-
щенный водород в течение 20 мин. Через 2 ч осадок отфильтровывают и высу-
шивают. Остальные эталоны с убывающим содержанием каждой примеси готовят
последовательными разбавлениями головного эталона. К 1г каждого эталона
перед анализом добавляют 20 мг сульфида кадмия и тщательно перемешивают.
Внутреннюю поверхность разрядной трубки протирают ватным тампоном,
смоченным этанолом. В канал электрода помещают 0,05 г эталона или концент-
рат примесей и вставляют в электрод угольный диск так, чтобы он плотно приле-
гал к пробе. Электрод помещают в разрядную трубку, создают давление 5-
• 10~2 мм рт. ст. и анализируют пробы при давлении гедия 20 мм рт. ст. Сила то-
ка 0,5 А (30 с) и 1 А (30 с), ширина щели 0,020 мм. Спектр фотографируют на
одну и ту же пластинку. Перед экспозицией проводят отжиг образца при силе
тока 0,1 А в течение 60 с.
Используют фотопластинки изо хром чувствительностью 5 ед. ГОСТ и УФШ-0
для регистрации спектра в дальнем ультрафиолете. Угольные электро-
ды обожженные, с диаметром канала 4 мм и глубиной 15 мм и электроды диа-
метром 4 мм, из которых изготовляют диски толщиной 0,5 мм.
При использовании дуги постоянного тока можно определять
следующие примеси: 3-10~6% V; 5-IO-7% Ti; 1 • 10"6% Мо на спект-
рографе ИСП-28 с трехлинзовой системой освещения щели и трех-
ступенчатым ослабителем. Сила тока 12 А, время экспозиции 1 мин.
Угольные электроды спектрально-чистые; нижний — с диаметром
кратера 4,5 мм и глубиной 6 мм; верхний заточен на конус. К кон-
центрату или эталону добавляют 2,5 мг хлорида натрия. Пластин-
ки — спектральные тип I чувствительностью 3 ед. ГОСТ для обла-
стей 450—330 нм и изохром нормальные чувствительностью 45 ед.
ГОСТ для областей' 330—240 нм; ширина щели 0,015 мм. На одной
фотопластинке фотографируют по 3 раза спектры эталонов, конт-
рольных проб и образцов.
Для указанных дополнительно элементов используют следующие
аналитические линии (в нм): Ti 308,80; V 318,54; Мо 317,06.
В гипофосфите натрия определяли 18 примесей [292]. Примеси
Мп, Са, Mg и А1 определяли прямым спектральным методом после
смешивания препарата с графитовым порошком. Микропримеси Ag,
С , Bi, Ач, Fe, Ti, Сг и V электролитически выделяли на графитовый
диск и далее определяли спектральным методом. Определение микро-
примесей In, Zr, Zn, Со, Ga и Pb основано на предварительном
концентрировании примесей на графитовом коллекторе с использо-
ванием в качестве осадителя уксуснокислого раствора 8-оксихиноли-
на. Все примеси определяют в дуге постоянного тока. Пределы об-
наружения (в %): Со, In, Ag, Ga и Pb 1 • 10-6; Си и Bi 3-10~e; Au
5-1СГ6; Fe, Ti, Сг, V, Zn, Мп и Al 1-1СГ5: Ca и Mg 3-10"6; Zr l-10~4.
182
Относительное стандартное отклонение при использовании прямого
спектрального анализа равно 0,10—0,15; при использовании химико-
спектральных методов — 0,20—0,30.
Методика определения микропримесей Mn, Bi, Ti, Со, РЬ, Сг,
In, Ag, Ni, Си и Sn в диэтилдитиокарбаминате натрия основана на
концентрировании их на сульфидно-графитовом коллекторе из рас-
твора реагента и спектральном анализе концентрата примесей в дуге
постоянного тока. Предел обнаружения 1-10“7—1-10-6%; относи-
тельное стандартное отклонение 0,20—0,25.
В метафосфате натрия примеси Mn, Cu, Мо, Fe, V, Ст, РЬ, Ni, Со
и Ti определяют прямым спектральным методом после смешения
пробы с графитовым порошком в соотношении 1:1. Предел обна-
ружения (в %): Мп, Ст 1-10"5; Мо 5-10's; Fe, V, Cr, Pb 1 -10-4; Ni,
Co 5-IO-4; Ti l-10~3. Относительное стандартное отклонение 0,10—
0,15 [86]. Примеси Fe, Cu, Ni, Co, V и Mo определяли химико-спект-
ральным методом после их концентрирования на смешанном кол-
Таблица 62
Определение примесей в перренате натрия [642]
Элемент Аналитическая линия, нм Предел обна- ружения, %, п-103 Элемент Аналитическая линия, нм Предел обна- ружения, %, п-103
Fe 248,32; 302,66 1 Ni 305,08; 341,48 3
Си 327,40 1 Мо 317,03 3
А1 309,27 3 Мп 279,48 5
Mg 279,55 3 Si 288,16 5
Pb 283,31 5
Примечание. Навеска 90 мг, дуга постоянного тока.
Таблица 63
Определение примесей в титанате, ниобате и танталате натрия [259]
Элемент Аналитическая линия, нм Определяемые содержания, %
Fe 309,11 — Со 303,44 0,01-0,2
Si 288,15 —фон 0,01—0,2
Al 308,22 —Со 303,44 0,005—0,1
Мп 294,92 — Со 303,44 0,005—0,1
Cu 327,40 —• фон 0,005—0,1
Ni 305,08— Со 303,44 0,005-0,1
Zn 334,50 —- фон 0,005—0,1
Примечание. Навеска 10мг, дуга постоянного тока, относительное стандартное
; отклонение 0,2—0,3.
183
лекторе из диэтилдитиокарбамината кадмия, 8-оксихинолина и гра-
фитового порошка [87]. Предел обнаружения 5-1СГ6—1-10-Б%; от-
носительное стандартное отклонение 0,25.
Помимо прямых и химико-спектральных, используют рентгено-
флуоресцентные и масс-спектральные методы. Возможности некото-
рых методов приведены в табл. 62, 63.
Анализируемые объекты, определяемые примеси и методы их об-
наружения систематизированы в табл. 64.
Остальные методы анализа (фотометрические, электрохимические,
люминесцентные, кинетические) применяют, как правило, для оп-
ределения одной или небольшого числа примесей. Эти методы, а так-
же радиоактивационные методы анализа приведены ниже для всех
определяемых элементов.
ОПРЕДЕЛЕНИЕ ОТДЕЛЬНЫХ ЭЛЕМЕНТОВ
Водород определяли методом амальгамирования, позволяющим
раздельно определять ^>5-10-6% негидроксильного и ^>1-10-5%
гидроксильного водорода: при 200° С в вакууме при давлении 10~3 мм
освобождается негидроксильный, а при 400° С в токе аргона — гид-
роксильный водород [1005, 1202].
Разработан диффузионно-волюмометрический метод [795]. В ни-
келевых капсулах диффузия водорода заканчивается за 5—10 мин,
в то время как в стальных капсулах — зД 1 ч. Метод позволяет оп-
ределять 10'4% водорода с погрешностью не выше 0,12%. Предло-
жен метод изотопного разбавления тритием, позволяющий опреде-
лять 2-10~4% водорода с погрешностью 15% [727]. Описан метод
вакуум-плавления и поглощения водорода оловянной ванной, после
чего водород откачивают через палладиевый фильтр и определяют
по разности давления [144, 283]. При навеске натрия 0,5 г предел
обнаружения водорода 2-10'4%. При определении 5-10-3% водоро-
да относительное стандартное отклонение зт = 0,05.
Сопоставлены различные методы определения водорода в натрии
[1249].
Калий определяли гравиметрически в форме КС1 после отделе-
ния основы [1160]. Предложен радиометрический метод определения,
основанный на осаждении K2Na[Co(NO2)6] в присутствии радионук-
лида 60Со и измерении активности осадка. При определении калия
в NaF, NaCl и Na2CO3 измеряют естественную радиоактивность ра-
дионуклида “К, входящего в состав осадка [216].
Цезий определяют нейтронно-активационным методом [1175].
В работе [1180] цезий отделяли от основы и концентрировали ионо-
обменным методом, а затем определяли по рентгеновскому у-пику
134”’Cs с энергией 31 кэВ при помощи у-спектрометра с тонким (1 мм)
кристаллом NaJ(Tl). Предел обнаружения цезия 8-10“®%, погреш-
ность определения не превышает 12%.
Медь (1—100 мкг) в едком натре определяли кинетическим мето-
дом по ее каталитическому действию на реакцию
2Fe34 + 2S2O|''==2Fe24 + SaO|“
1S4
Таблица 64
Спектральные методы определения примесей в натрии и его соединениях
Объект Навес- ка, г Определяемое элементы Метод определения Литера- тура
Натрий 20 Cu, Mn, Bi, Cd, Co, Ga, In, Ni, Pb, Sb, V, Al, Mg, Fe Химико-спектральный [1009]
4 Zn, Fe, Al, V, Ni, Cu, Bi, Co, Be, Pb, Mn » [243]
1 Ca Рентгенофлуоресцент- ный [1128]
Ca Масс-спектрометричес- кий [619]
Натрий Li, Be, В, C, N, O, F, » [618]
(реактор- Mg, Al, Si, Cl, K, Ca,
ный) Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Sr, Y, Zr, Mo, Ag, Sn, Cs, Ba, Au, Pb, Bi
Fe, Ni, Cr, Mo, Co Химико -сп ект ра льный [884]
4 Ca, Ba Рентгенофлуоресцент- [791]
НЫЙ
4 Ca, Cr, Ni, Zn, Nb, Ta, Hf, Mo, Br, J, Ba, Fe, Pb, Cd, Cl, Cu, Mn, Co, Ti, V » [1127]
0,2- К Пламенная эмиссионная [1009]
0,7 фотометрия
4 Mn, Cr, Fe, Ni Атомно-абсорбционный [823]
Bi, Ga, Fe, In, Cd, Co, Mg, Mn, Cu, Ni, Pb, Химико-спектральный [534]
Sb, Zn, Al, V
NaOH 50 Cu, Ag, Ni, Ti, Mn, Cr, Bi, Pb, Sn, Mo, V, Co, Au, Al, Fe, Zn, Sb [374]
NaCl K, Rb, Cs Пламенная эмиссионная фотометрия [408]
NaCl, 25 Ag, Bi, Au, Cu, Mn, Химико-спектральный [370]
NaNO3, Ni, Pb, Tl, Mg, Ga, Fe,
Nagcog Al, In, Zn, Ge, Sb, As, Co, Cr
NaCl, 0,5 Cu, Ni, Co, Fe, Mn, Рентгенофлуоресцент- [977]
Na2C03, CHgCOONa Cr, V ный
NaCl, NaH2P04, Na2CrO4 1 Rb, Cs Химико -спе ктральный [379,381]
NaJ 10 Al, Cr, Fe, Ni, Co, Mn, Cu, Sn, Ph » [373]
Fe, Ca, Al, Mg, Si, Ti Лазерный микроспёкт- [1279]
ральный
185
Таблица 64 (окончание)
Объект Навес- ка, г Определяемые элементы Метод определения Литера- тура
NaNO3 Са Масс-спектрометричес- кий [879]
NaNO3 (расплав) Al, Fe, V, Си, Со, Mg, Мп, Са, Ni, Nb, Сг Химико-спектральный [223]
Фосфат натрия Гипофосфит натрия 1 Al Ag, Си, Bi, Au, Fe, Ti, Сг, V, Mn, Ca, Mg, Al, In, Zr, Zn, Co, Ga, Pb Люминесцентный Химико-спектральный [353] [292]
Метафосфат 4 Fe, Си, Co, Ni, V, Mo [87]
натрия 1 Мп, Си, Mo, Fe, V, Cr, Pb, Ni, Co, TI Спектральный [86]
Na2SO4 0,1- 0,6 Pb Рентгенофлуоресцент- ный . [202]
Ниобат, ти- танат, тан- талат нат- рия 0,010 Fe, Si, Al, Мп, Си, Zn, Ni Спектральный [259]
Ниобат ба- рия-нат- рия 1 Si, Fe, Co, Ni, Bi, Mn [293]
Молибдат Na — Gd, Na—Nd 1 Си, Fe, Mn, Ni, Cr, Co, V » (фракционная дистил- ляция) [378]
Молибдат Na —La 0,24 Си, Mn, Co, Fe, Cr, Ni » [377]
Na2CrO4 Fe, Си, Cd, Ba Спектральный [380]
NaReO4 0,09 Fe, Mn, Си, Al, Mg, Ni, Mo, Si, Pb [542]
Соли нат- рия К Масс-спектрометричес- кий [305]
25 Al, Bi, Ga, Ge, Fe, Au, In, Co, Mn, Си, As, Ni, Sn, Pb, Ag, Sb, TI, Cr, Zn Химико-спектральный [371]
K, Rb Пламенная эмиссионная фотометрия [170J
Диэтилди- тиокарба- минат нат- рия 2 Mn, Bi, Ti, Co, Pb, Cr, In, Ag, Ni, Си, Sn Химико-спектральный [291]
186
и уменьшению оптической плотности раствора роданидного комплек-
са железа [886].
Менее чувствителен фотометрический метод с использованием
ДДТК-натрия в безэкстракционном варианте [849]. Мешающие оп-
ределению ионы Mn, Fe, Со, Ni маскируют ЭДТА, избыток послед-
ней связывают магнием. Погрешность определения 3%. Мешают оп-
ределению Bi, Hg2+ и Ag.
К порциям стандартного раствора, содержащего 10 мкг/мл меди, прибавляют
около 50 мл воды, 5 мл 20%-ного раствора лимонной кислоты, 10 мл 5 М раство-
ра NH3, 2 мл 1%-ного раствора гуммиарабика, 10 мл 0,1%-ного раствора диэтил-
дитиокарбамината натрия, разбавляют водой до 100 мл и измеряют оптическую
плотность (1 = 4 см). Около 10 г технического NaOH растворяют в 25 мл воды,
накрывают стеклом, осторожно прибавляют 50 мл 5 М НС1, упаривают до 50—
60 мл. К охлажденному раствору прибавляют при перемешивании 5 мл 20%-но-
го раствора лимонной кислоты, 10 мл 5 М NH3, 2 мл 1%-ного раствора гуммиара-
бика, 10 мл 0,01 М раствора этилендиаминтетраацетата натрия и 2,5 мл раствора
MgSO4 (1 мг/мл магния; 5,07 г MgSO4-7H2O и 5 г NH4C1 растворяют в воде и раз-
бавляют водой до 500 мл). Разбавляют раствор.водой до 90 мл и делят на 2 рав-
ные части. К первой порции прибавляют 4 мл раствора диэтилдитиокарбамина-
та натрия и оба раствора разбавляют водой до 50 мл. Измеряют оптическую
плотность первого раствора относительно второго.
В гипофосфите натрия медь определяли кинетическим методом
по реакции окисления гидрохинона пероксидом водорода при pH 5—
8 [232]. Из навески 1 г авторы определяли 1,7-10'®% меди с погреш-
ностью 25—30%. Продукты реакции поглощают свет при 460—
500 нм. Пиридин увеличивает скорость каталитической реакции,
гипофосфит в количестве 0,1—0,5 г снижает скорость реакции в 2—
3 раза, поэтому определение проводят методом добавок. Мешающее
определению железо маскируют фторидом.
Навеску 1,0 г гипофосфита натрия растворяют в 10 мл воды. В две пробир-
ки приливают по 0,5—1,0 мл анализируемого раствора, в одну из них вводят
0,1 мкг меди. В две другие пробирки приливают по 0,02 мл анализируемого раст-
вора (контрольный опыт); в одну из них вводят 0,1 мл раствора с содержанием
меди 1 мкг/мл. Во все пробирки приливают по 0,5 мл 0,5%-ного раствора пири-
дина, 0,2 мл 0,5 М раствора NH4F, 0,5 мл 3%-ного Н2О2 и воду до 4,5 мл. Затем
последовательно измеряют скорость реакции в каждом из четырех растворов,
для чего в пробирку приливают 0,5 мл 0,5%-ного раствора гидрохинона, встря-
хивают, пускают секундомер, переливают раствор в кювету с толщиной слоя
1 см и измеряют оптическую плотность при 536 нм в течение 6 мин через мину-
ту. По полученным данным строят график с координатами оптическая плот-
ность — время и по нему рассчитывают тангенс угла наклона. Вычисляют со-
держание меди в гипофосфите натрия.
Серебро определяли методом амальгамной полярографии с накоп-
лением после экстракции диэтилдитиокарбамината серебра и его ре-
экстракции роданидом калия [461]. При навеске 0,58 г NaCl достиг-
нут предел обнаружения серебра 1 -10-8%.
К 100 мл 0,1 М раствора NaCl прибавляют 5 мл 0,05 М раствора ЭДТА,
5,0 мл 50%-ного раствора сульфосалициловой кислоты, 10 мл 25%-ного раство-
ра NH3. Раствор переносят в делительную воронку, прибавляют 1 мл 10%-ного
раствора диэтилдитиокарбамината натрия, 2—3 мл хлороформа и экстрагируют
серебро встряхиванием (3—4 раза в минуту). Затем серебро реэкстрагируют 5 мл
1 М раствора KSCN (pH 2 по H2SO4), сливают в стаканчик для полярографиро-
вания, продувают азотом для удаления кислорода и полярографируют. Потен-
187
циал электролиза —0,6 В (нас. ртутно-серебряный электрод). Концентрацию се
ребра определяют методом добавок.
Кальций (6-10-4—10"3%) определяли фотометрически после осаж-
дения в форме нафтилгидроксамата и растворения его в ЭДТА [595].
Молярный коэффициент погашения 1,25-IO® (410 нм) и 1,4-10Б
(339 нм). При навеске металлического натрия 1—1,5 г определению
кальция не мешают (в мкг): А1, Си, Fe, Mn, Sn, V (100); Ba, Mg (300);
5 мг К и по 5 мг-ион F-, SO®-, NOg, CrO4-, РО3-. Фотометрический
метод с использованием арсеназо III позволяет определить >3-10-3 %
кальция в NaCl [479].
Экстракционно-фотометрический метод определения кальция с ис-
пользованием азоазокси БН позволяет определять 5-10-5—2-10-4%
кальция в NaCl с погрешностью 5—20%. Экстрагентом комплекса
является 20%-ный раствор ТБФ в СС14 [110].
В делительную воронку помещают 20 мл анализируемого раствора хлорида
натрия, содержащего 2—8 мкг кальция. Добавляют 1 мл 5 М NaOH и разбав-
ляют водой до 25 мл. Вводят 25 мл экстрагента АТ и экстрагируют кальций встря-
хиванием в течение 2 мин. После расслаивания органический слой переносят
в центрифужную пробирку и центрифугируют 5 мин (3000 об/мин). Прозрачный
раствор помещают в кювету (1=5 см) и фотометрируют на фотоэлектроколорй-
метре (434 нм) или спектрофотометре (440 нм) относительно контрольного раст-
вора на реактивы. Содержание кальция находят по градуировочному графику.
Для построения градуировочного графика помещают в делительную ворон-
ку 20 мл раствора NaCl, очищенного от кальция экстракцией экстрагентом АТ
(концентрация раствора NaCl должна примерно соответствовать концентрации
его в анализируемых растворах). Вводят 1 мл 5 М раствора NaOH и 2—4—5—6—
7—8 мкг кальция, разбавляют водой до 25 мл и добавляют 25 мл экстрагента
АТ. Далее поступают, как при построении градуировочного графика.
Экстрагентом АТ является 0,04%-ный (1-10~3М ) раствор азоазокси БН
в смеси СС14 с 20% об. трибутилфосфата. Для приготовления экстрагента 0,4 г
азоазокси БН в мерной колбе вместимостью 1 л растворяют в СС14. Затем вводят
200 мл трибутилфосфата и 500—600 мл СС14. Общий объем растворителей в кол-
бе 700—900 мл. Содержимое колбы слегка подогревают на водяной бане и пе-
риодически взбалтывают до полного растворения реагента. Затем раствор ох-
лаждают и прибавляют до метки СС14. Раствор частями переносят в делительную
воронку вместимостью 500 мл, добавляют равный объем 0,1 М НС1 и встряхива-
ют 5 мин. Водный слой отбрасывают, а органический встряхивают еще раз с НС1.
.Органический слой дважды промывают катионированной водой. Водный слой
удаляют, а посветлевший органический помещают в склянку из оранжевого
стекла и используют в работе. Растворы реагента меньшей концентрации гото-
вят из .исходного разбавлением в день работы.
Флуориметрический метод с использованием 8-хинолилгидразо-
на 8-оксихинальдинового альдегида позволяет определить 5-10-6%
кальция в NaJ [58]. Предел обнаружения 0,1 мкг кальция в 5 мл
раствора при навеске 1 г NaJ. Определению 0,2 мкг кальция не ме-
шают соли щелочных металлов, а также 10-кратный избыток строн-
ция, 100-кратный — бария и магния. Равные количества Pb, Fe(III),
Mn, Си, Cd, Zn, In, 10-кратный избыток Со, Fe(II), Та, 100-кратный
избыток Be, Cr(VI), 1000-кратный избыток W, Pt(IV); Ni, Bi гасят
люминесценцию комплекса.
Растворяют 0,1 г NaJ при предполагаемом -содержании кальция 5-10-Б%
или 0,01 г при предполагаемом содержании кальция 5-10-4% в 5 мл 0,1 М КОН.
Раствор помещают в делительную воронку, приливают 5 мл раствора азоазокси
188
БН (0,02%-ный раствор в смеси трибутилфосфата с СС14) и встряхивают 1 мий.
После расслоения фаз экстракт сливают в другую делительную воронку, вводят
5 мл 0,1 М НС1 для реэкстракции кальция и встряхивают 1 мин. Отделяют ре-
экстракт, прибавляют к нему 0,5 мл 2 М КОН, 0,25 мл 0,02% -него ацетонового раст-.
вора 8-хинолилгидразона 8-оксихинальдинового альдегида и через 20 мин изме-
ряют интенсивность флуоресценции. Содержание кальция находят по градуиро-
вочному графику. Для его построения в четыре пробирки наливают по 5 мл 0,1 М
НС1, по 0,5 мл 2 М КОН, вводят соответственно 0,00; 0,05; 0,07; 0,1 мкг кальция,
по 0,25 мл раствора реагента и через 20 мин измеряют интенсивность флуорес-
ценции.
В цитрате натрия определяли 5-10—3% кальция из навески 10 г
комплексонометрическим титрованием после экстрагирования рода-
нидного комплекса кальция трибутилфосфатом [256]. Погрешность
определения 7%.
Навеску препарата 10 г помещают в мерную колбу вместимостью 100 мл,
растворяют в воде и разбавляют водой до метки. В делительную воронку вмес-
тимостью 100 мл помещают 10 мл полученного раствора, добавляют конц. НС1
до pH 2—3 по универсальной индикаторной бумаге, прибавляют 10 мл 4 М раст-
вора роданида аммония, 20 мл трибутилфосфата и встряхивают в течение 1 мин.
После расслоения нижний водный слой отбрасывают, а к органическому слою
приливают 20 мл смеси (80 г роданида натрия растворяют в 200 мл воды, при-
бавляют 5 мл конц. НС1 и разбавляют водой до 500 мл) и встряхивают снова
(1 мин). Водный слой после расслаивания отбрасывают, а к органическому слою
прибавляют равный объем 1 М раствора НС1 и реэкстрагируют кальций 1 мин.
После расслаивания фаз реэкстракт помещаютв коническую колбу вместимостью’
250 мл, вводят 10 мл 25%-ного раствора NH3, около 0,1 г метилтимолового си-
него (сухая смесь с KNO3 в соотношении 1 : 50) и титруют из микробюретки
0,005 М раствором ЭДТА до изменения окраски из голубой в бесцветную иди се-
роватую. Параллельно титруют раствор контрольного опыта.
Барий определяют химико-спектральным методом в дуге пере-
менного тока после концентрирования соосаждением с хроматом
свинца [27]. Предел обнаружения бария 2-10~5% в NaJ при навеске
образца 5 г. В качестве аналитической использована линия 230,4 нм.
Из навески 1 г тартрата калия—натрия, CHgCOONa [440] или
гипофосфита натрия [500] определяли 1-10-5% цинка флуориметри-
чески 8-(п-толуолсульфониламино)хинолином. Погрешность метода
20—30%; предел обнаружения цинка 0,01 мкг/мл [500].
Оптимальную кислотность pH 8,0—8,3 создают тетраборатным
буферным раствором. Комплекс флуоресцирует зеленым светом,
в отсутствие цинка флуоресценция имеет голубой оттенок. Спектр
флуоресценции комплекса представляет собой бесструктурную по-
лосу в интервале 440—640 нм с максимумом при 520—530 нм. При
экстракции комплекса хлороформом предел обнаружения цинка по-
нижается и составляет 0,002 мкг/мл. Кроме цинка, в оптимальных
условиях взаимодействует кадмий; 10-кратные количества Со, С ,
Ni и Fe гасят флуоресценцию. Аналогичный эффект вызывает присут-
ствие 100-кратных количеств Li, Fe(II), Gd, Sb(III), Hg(II), Zr, Ga,
Sn(II) и 1000-кратных количеств Be и Mn(VII).
Для определения цинка в гипофосфите натрия в три пробирки помещают
по 1,0 г гипофосфита натрия, во вторую пробирку добавляют 0,3 мкг, а в
третью — 0,5 мкг цинка. Если анализируемый образец представляет собой раст-
вор, то предварительно досуха выпаривают раствор с добавками цинка. Оста-
ток после выпаривания смывают 5 мл буферного раствора с pH ~8 (смешивают
189
55,9 мл 0,05 М раствора тетрабората натрия с 44,1 мл 0,1 М НС1) и переносят
в три пробирки, в которые добавляют по 0,2 мл 0,01%-ного ацетонового раство-
ра 8-(п-толуолсульфониламино)хинолина. Одновременно готовят контрольную
пробу: 5 мл буферного раствора и 0,2 мл раствора реагента. Через 5 мин во все
пробирки наливают по 2 мл хлороформа и встряхивают’ в течение 1 мин. Через
10 мин измеряют интенсивность люминесценции анализируемого и стандартных
растворов.
При определении цинка в ацетате натрия и в сегнетовой соли по-
ступают аналогично, но оптимальную кислотность создают гликоко-
левым буферным раствором.
Ртуть определяют экстракционно-фотометрически кадионом А,
.диазоаминобензолом или п, п'-динитродиазоаминобензолом [1059].
Метод применен для определения 0,62—1,37 мкг/мл ртути в NaOH;
относительное стандартное отклонение 0,043—0,22.
Бор определяют с помощью куркумина [521, 1110] или диантри-
мида [521]. Предложено концентрировать бор на анионообменнике,
а натрий сорбировать катионообменником [521]. Прием пригоден для
определения бора в металлическом натрии и NaOH. В методе [1110]
после растворения пробы натрий переводят в хлорид и экстрагируют
бор этанолом.
Малые количества алюминия (5-10~7—5-10~5%) определяли с по
грешностью 15—20% в CHgCOONa флуориметрически с использова-
нием салицилаль-о-аминофенола из навески 3—30 г [56].
Алюминий образует комплекс, флуоресцирующий зеленым све-
том. Оптимальную кислотность (pH 5,6—6,2) создают ацетатным бу-
ферным раствором. Спектр флуоресценции представляет собой бес-
структурную полосу в интервале длин волн 440—640 нм с максиму-
мом интенсивности флуоресценции при 520—530 нм. Предел обна-
ружения алюминия 0,0005 мкг/мл. Определению 0,0025 мкг алюми-
ния не мешает ни один катион в количествах до 25 .мкг в 5 мл
раствора. Гасят флуоресценцию только катионы меди и железа в
количествах, превышающих 0,1 мкг в 5 мл.
В мерную колбу вместимостью 100 мл помещают 3—30 г анализируемого
ацетата натрия, растворяют в бидистилляте и разбавляют им же до метки. На-
веску 3 г используют в случае, если предполагаемое содержание алюминия в пре-
парате 5-10“®—5-10-6%. Если предполагаемое'содержание алюминия 5-Ю'7—
5-10~®%, то навеска составляет 30 г. Отбирают 10 мл полученного раствора
в колбу, добавляют 2—3 капли раствора ализарина и нейтрализуют азотной кис-
лотой (1 : 8) до изменения окраски раствора из красной в желтую. При этом
считают число капель кислоты, израсходованной на нейтрализацию. К оставшим-
ся 90 мл раствора ацетата натрия добавляют столько же азотно кислоты, но не-
разбавленной, и тщательно перемешивают. Этой операцией достигают оптималь-
ной кислотности (pH 5,7—5,9) анализируемого раствора.
В четыре колбы вместимостью по 25—50 мл вводят по 9 мл полученного
раствора. В одну из них добавляют 0,5 мл стандартного раствора (что соответ-
ствует содержанию алюминия 0,05 мкг), во вторую — 0,8 мл того же раствора
(0,08 мкг алюминия) и в третью — 2 капли 0,05 М раствора ЭДТА. Объемы раст-
воров во всех четырех колбах доводят до 10 мл дистиллированной водой, прили-
вают по 0,3 мл 0,01 %-него ацетонового раствора салицилаль-о-аминофенола
и оставляют на 45—50 мин для образования комплекса.
Растворы из каждой колбы последовательно наливают в кювету вмести-
мостью 10 мл и измеряют относительную интенсивность флуоресценции при
520 нм.
190
Менее чувствителен фотометрический метод с использованием
стильбазо, примененный для определения 1-10-2—1-10-3% алюми-
ния в NaOH. Предел обнаружения алюминия 0,2 мкг в 5 мл раство-
ра, погрешность 7%. Влияние железа(Ш) устраняли восстановле-
нием его аскорбиновой кислотой [518].
Общее содержание углерода в натрии определяли после окисле-
ния натрия при 600° С кислородом, разбавленным аргоном, раство-
рения смеси Na2O + Na2COs в разбавленной серной кислоте, погло-
щения СО2 [856]. Далее проводили кондуктометрическое титрование
раствором Ва(ОН)2. Метод позволяет определять ^>10-3% углерода.
Методом газовой хроматографии определяли углерод в виде С02
после сожжения навески 0,2—0,5 г натрия в смеси азота и воздуха
и концентрирования СО2 в ловушке, охлаждаемой жидким азотом
[990].
Нейтронно-активационным методом определяли (1—5)-10~4 %
углерода [51], ау-активационным — ^>3-10~4% углерода [725] в ме-
таллическом натрии.
Сопоставлены методы определения углерода в натрии и его сое-
динениях [1249].
В металлическом натрии определяли 0,1—0,9 мкг цианида
с погрешностью 5—10% фотометрическим методом по реакции обра-
зования красителя полиметанового фиолетового из барбитуровой
кислоты и хлорамина Т [1042].
Навеску образца помещают в кварцевую колбу, охлаждаемую до 0° С, и в
течение 15 мин пропускают аргон со скоростью 70 мл/мин. В качестве сосуда для
сбора цианида используют мерную колбу вместимостью 25 мл, которую также
помещают в охладительную смесь. В эту колбу наливают 5—7 мл воды и до-
бавляют 2 капли 1 М раствора NaOH. В колбу с натрием вводят 10 мл воды со
скоростью 1 капля в 20—30 с, а затем около 6 мл 25%-ной H2S04. Затем колбу
нагревают и отгоняют половину ее содержимого. В мерную колбу добавляют
5 мл фосфатного буферного раствора с pH 5,5; 1 мл 1%-ного раствора хлорами-
на Т, а затем 3 мл раствора реагента (к 3 г барбитуровой кислоты добавляют
15 мл пиридина, 3 мл конц. НС1 и разбавляют водой до 50 мл). Измеряют опти-
ческую плотность раствора при 580 нм.
Для определения (3—15) -1СГ7 % германия в NaCl из навески 1—
300 г рекомендован фотометрический метод с использованием фенил-
флуорона [550]. Предварительно германий концентрируют соосажде-
нием с гидроксидом железа(Ш).
Взвешивают 1—300 г NaCl (в зависимости от содержания германия) с точ-
ностью до 0,01 г, помещают в стакан вместимостью 1 л и растворяют в 0,5—
1,0 л воды. В раствор вносят 2 мл 20%-ного раствора FeCl8, 2 мл 30%-ного раст-
вора Н2О2 и соосаждают германий с гидроксидом железа(Ш), добавляя в раст-
вор небольшими порциями 25%-ный раствор NHS до устойчивого запаха. Раст-
вор нагревают до кипения и оставляют до полной коагуляции осадка. Жидкость
над осадком осторожно декантируют, а осадок фильтруют на фильтр красная
лента и промывают тремя порциями горячего 1%-ного раствора NH4C1. Осадок
на фильтре растворяют в 9М растворе НС1, собирая раствор в мерную колбу
вместимостью 50 мл, и разбавляют до метки 9 М раствором НС1. Содержимое
колбы помещают в делительную воронку и экстрагируют германий двумя пор-
циями СС14 (10 и 15 мл) в течение 3 мин. Органический слой сливают в делитель-
ную воронку, промывают 5—10 мл 9 М НС1, вносят туда 0,3 мл 0,1%-ного раст-
вора фенилфлуорона в смеси (35 :15) СС14 и этанола, 1 мл этанола, перемешивают
и оставляют стоять на 5 мин. Затем вносят 2 мл смеси (4 : 1) диметилформ-
191
амида и воды и энергично встряхивают. После расслоения верхнюю (органичес-
кую) фазу сливают в центрифужные пробирки, споласкивают воронку 6 мл воды,
сливая промывную воду в ту же пробирку. Раствор центрифугируют в течение
10 мин при 4000 об/мин. Жидкость над осадком осторожно декантируют, а оса-
док растворяют в 2,5 мл диметилформамида. Перемет вают, разбавляют раст-
вор 2,5 мл воды и добавляют еще 1 мл диметилформамида. Тщательно переме-
шивают и центрифугируют в течение 15—20 мин при 6000 об/мин. Жидкость над
осадком осторожно декантируют, по возможности полностью удаляя ее остатки.
Осадок растворяют в смеси 6 мл 0,1 М NaOH и 2 мл диметилформамида. Комп-
лекс растворяют непосредственно перед измерением оптической плотности. Оп-
тическую плотность измеряют относительно воды при толщине слоя кюветы
2 см, содержимое германия находят по градуировочному графику, полученному
для 1—5 мкг германия, проведенного через все операции.
Флуориметрический метод определения свинца в виде хлоридно-
го комплекса, замороженного при температуре жидкого азота и раз-
мораживаемого при комнатной температуре, позволил определить
1-10~5% свинца в гипофосфите натрия из навески 0,05 г со стандарт-
ным отклонением 6-10_’% [448]..
Спектры люминесценции хлоридных комплексов свинца при тем-
пературе жидкого азота лежат в синей области спектра с максиму-
мом люминесценции при 385 нм (синее свечение), а при разморажи-
вании растворов спектр люминесценции сдвигается в длинноволновую
область с максимумом люминесценции при 490 нм (зеленое све-
чение). Интенсивность всплеска зеленой люминесценции пропор-
циональна количеству свинца, присутствующего в растворе. Опре-
делению свинца не мешают 1000-кратные количества Na, К, Са,
Ва, В, Mg, Sr, Cd, Sn, Be, Co, Ni, Cr, In, Al, Sb, Bi, Se, Те, Ca, Fe,
As, J-, F_, PO®-, SOt и SCN~. Железо и медь несколько гасят ин-
тенсивность всплеска, но при применении метода добавок их дейст-
вие нивелируется.
Растворяют 0,05 г гипофосфита натрия в 1 мл 4М НС1. Помещают 0,2 мл
полученного раствора в кварцевую трубочку, замораживают в жидком азоте, а
затем помещают в кварцевую пробирку. Записывают всплеск интенсивности лю-
минесценции раствора при размораживании от —196 до 0° С. Аналогично изме-
ряют интенсивность всплеска люминесценции анализируемого образца с добав-
кой 0,1 мл стандартного раствора свинца (0,1 мкг свинца). Количество свинца
рассчитывают по формуле для метода добавок.
Цирконий (1-Ю“4%) в гипофосфите натрия определяли флуори-
метрически с использованием морина [501]. При навеске образца
0,05 г предел обнаружения циркония составляет 0,02 мкг в 5 мл
раствора; погрешность определения 20—30%.
Спектр люминесценции комплекса представляет собой бесструк-
турную полосу с максимумом люминесценции при 520 нм. Время
развития люминесценции составляет 15 мин. В условиях определе-
ния ионы Al, Be, Ga, Ge, Nb и другие также флуоресцируют, но
при значительно большем содержании по сравнению с цирконием.
Введение ЭДТА полностью уничтожает люминесценцию только цир-
кония.
В три пробирки помещают по 50 мг анализируемого гипофосфита натрия
и растворяют в 5 мл 2 М НС1; во вторую пробирку добавляют 0,05 мкг, а в
третью — 0,2 мкг циркония. Все растворы помещают в кипящую водяную баню
192
на 30 мин. После охлаждения вводят по 0,15 мл 0,04%-ного этанольного раство-
ра морина и через 15 мин измеряют интенсивность люминесценции анализируе-
мых и стандартных растворов. Затем во все колбы добавляют по 0,5 мл 5%-ного
раствора ЭДТА и в тех же условиях снова измеряют интенсивность люминесцен-
ции.
Азот определяют активационным и фотометрическим методами,
у-активационный метод позволил определить 5-1СГ6—9,7-10-4%
азота в натрии из навески 0,9 г. После облучения образец растворяют
в воде, отгоняют с парами воды 13NHs, поглощают его кислотой и из-
меряют активность [5321. Предел обнаружения азота 2-10~®% при
навеске образца 0,8—0,9 г [283, 675]. Нейтронно-активационным ме-
тодом определяли (1,4—2,3)-10-3% азота в металлическом натрии
[51]. Фотометрическим методом с использованием тимол-гипобромит-
ной реакции определяли 6-10-4% азота в металлическом натрии [282].
Предел обнаружения 5-10-6% при навеске 1 г; стандартное отклоне-
ние 7,5-К) 5%. Разработан фотометрический метод, основанный на
образовании NH^, его реакции с фенолом в присутствии NaOBr
и экстракции комплекса амиловым спиртом [283]. Предел обнаруже-
ния азота 5-10“®% при навеске 1 г металлического натрия.
Нитридный азот (1-1СГ4—1-10~2%) определяли в металлическом
натрии методом Кьельдаля с относительным стандартным отклоне-
нием 0,025 [1254].
Для ванадия предложен кинетический метод определения по реак-
ции окисления Н-кислоты броматом калия, катализируемой вана-
дием [228]. Из навески 2 г NaCl определяли (2,4 ± 0,6)-10_®% ва-
надия с пределом обнаружения 1-10-6%.
Определению ванадия в 5 мл раствора не мешают Са, Ва, РЬ,
Cd, Sr в количестве 100 мкг и In, Th, La, Fe(III), Ge, Al, Co, Mn,
Hg(II), Sn(IV), Mg, PO|-, Be, Cr(III, VI) в количестве 10 мкг. Уско-
ряют реакцию ионы WO|”, MnOj, замедляют фториды, хлориды,-
сульфаты, цитраты. При температуре 60—80° С предел обнаружения
понижается и составляет 5-10~4 мкг/мл.
Навеску 2 г NaCl растворяют в 10 мл воды и для анализа отбирают в три
пробирки по 1—3 мл раствора. В первую пробирку ничего не наливают, во вто-
рую пробирку вводят 0,02 мкг, в третью — 0,04 мкг ванадия. Одновременно
проводят контрольный опыт, для чего в три пробирки наливают соответственно
0,00; 0,20 и ,40 мл стандартного раствора с содержанием ванадия 0,1 мкг/мл.
Вводят по 0,4 мл 1 %-него раствора КВгО8 и разбавляют до 5,0 мл уксусной кис-
лоты (1 : 10Э). Затем вводят по 0,20 мл 0,01 М раствора Н-кислоты, пробирки
встряхивают, помещают на 10 мин в термоста ” с температурой 80° С, затем для
прекращения реакции вводят по 0,2 мл 10-3 М раствора ЭДТА и после охлажде-
ния измеряют оптическую плотность на фотоэлектроколориметре ФЭК-Н-57
(светофильтр № 9). Строят градуировочные графики в координатах оптическая
плотность—количество введенного ванадия для анализируемого и контрольных
растворов и по графику определяют содержание ванадия в анализируемом объеме.
Ниобий определяли в металлическом натрии фотометрическим
методом в форме роданидного комплекса [1120]. Предел обнаруже-
ния ниобия 0,040 мкг/мл.
Сурьму определяли методом амальгамной полярографии с накоп-
лением после экстрагирования диэтилдитиокарбамината сурьмы
7 В. М. Иванов и др.
193
хлороформом [198]. При навеске 0,4 г NaCl предел обнаружения
сурьмы составил 5-10-6 %; погрешность определения '10—12%.
Помещают 50 мл анализируемого' раствора хлорида натрия в делительную
воронку вместимостью 150 мл, вводят 6 мл 5%гного раствора ЭДТА, 6 мл 5%-но-
го раствора KJ, 8 мл 0,5%-ного раствора диэтилдитиокарбамината натрия, тща-
тельно перемешивают и дважды экстрагируют 1,5 мл хлороформа встряхиванием
в течение 1 мин каждый раз. Объединенный экстракт переносят в ячейку для по-
лярографирования, смешивают с 6 мл 0,2 М раствора NH4N03 в этаноле и пос-
ле предварительной очистки фонового раствора от кислорода полярографируют.
Потенциал анодного пика сурьмы на данном этанольном фоне равен —0,1 В.
Условия анализа: потенциал электролиза —0,8 В, время электролиза 5 мин,
чувствительность по прибору 6-10-8 А/мм, скорость изменения потенциала 1,6-
10“2 В/с. Содержание сурьмы определяют методом добавок стандартгого раст-
вора (для его приготовления используют SbCl8, который растворяют в этаноле
с подкислением изопиестической соляной кислотой; раствор содержит 0,01 мг/л
сурьмы).
В гипофосфите натрия определяли 5-10-6% висмута люминес-
центным методом при температуре жидкого азота по образованию
и свечению кристалл офосфора. Предел обнаружения висмута
0,005 мкг [499].
Для определения кислорода предложено много методов. Основные
затруднения при определении кислорода в натрии (и других щелоч-
ных металлах) заключаются в способе отбора проб и в отделении
оксида щелочного металла от суммы выделенных примесей (гидри-
дов, нитридов, гидроксидов, карбонатов, карбидов). Классический
метод основан на отделении натрия от Na2O амальгамированием
ртутью и его ацидиметрическом титровании [308, 673, 978]. Из на-
вески 2 г металлического натрия можно определить 16 мкг кислоро-
да с погрешностью 5% [673]. Более совершенны методы, основанные
на амальгамировании натрия и его определении методом фотометрии
пламени [308, 673, 978]. При определении (5—30)-10-3% кислорода
в натрии стандартное отклонение ^13-10“4% [308]. Указывается,
что при амальгамировании в ячейке определенной конструкции ва-
куум составляет 10-5 мм рт. ст. [673]. В методе определения кисло-
рода амальгамированием учтены различные поправки на контроль-
ный опыт, обусловленные чистотой атмосферы в боксе, размерами
и чистотой площади внутренней поверхности реактора, методом
очистки ртути и поверхности ампулы для образца [836], удалось
значительно снизить поправку на контрольный опыт.
В металлическом натрии определяли кислород [651, 652] методом
ИК-спектроскопии при 11,38 мк. Предварительно проводили реак-
цию и-амилхлорида с натрием; при этом кислород давал с натрием
оксид, который в присутствии СО2 превращался в Na2CO3, послед-
ний определяли ИК-спектроскопически [651]. Минимально опреде-
ляемое содержание кислорода 2-10-3%, погрешность 20%. При оп-
ределении 0,006—0,01% кислорода погрешность 5%.
Предложен [1159, 1172, 1262] бутилбромидный метод, основанный
на взаимодействии бутилбромида с натрием, растворении продукта
реакции в этаноле и титровании алкоголята натрия 0,05 М НС1 по
метиловому красному [1172] либо титровании 10-3 М раствором
194
HNO3 co стеклянным электродом [1262]. При определении 0,001—
0,1% кислорода стандартное отклонение 0,003—0,005% [1262].
В расплавленном натрии определяли кислород полярографически
на фоне расплава эвтектики LiCl + CsCl (40 и 60% мол. соответст-
венно) при 400° С [1103].
Наиболее представительна группа радиохимических методов оп-
ределения кислорода. Применяли нейтронно-активационный [(1,8—
2,6)-10-2%] [51], у-активационный [283, 531, 532, 675, 725], спектро-
метрии у,у-совпадений [814] методы и метод активации фотонами
[931]. При навеске металлического натрия 1 г пределы обнаружения
кислорода составляют 1-10-4% [532], 3*10-4% [725], 2«10~®% [283,
531].
При определении кислорода в натрии по реакции 16O(4He, pn)18F
радионуклид 18F может образоваться также по реакции
23Na(a, 2n)18F, порог которой 20,9 мэВ. Поэтому при определении
кислорода энергия ионов 4Не должна быть ниже порога реакции
на изотопе 23Na [145]. Определению кислорода по реакции 16О(у, п)
15О мешают радиоизотопы 22Na и 18F, образующиеся по реакциям
23Na(y, n)22Na и 23Na(y,an)18F.
Сопоставлены методы определения кислорода в натрии [1249,
1250], при сравнении 2 методов (у-активационного и вакуумной дис-
тилляции) отдано предпочтение первому методу вследствие более
высокой чувствительности и точности: метод позволяет определить
(1,3—17,3)-10“4% об. кислорода [809]. Опубликован обзор [1016]
методов определения кислорода в натрии, указана их сущность,
оценена чувствительность и источники возможных ошибок. Методы
имеют следующие пределы обнаружения кислорода (в %): бутил-
бромидный 5*10~3; ртутной экстракции 1 • 10"4; вакуумной дистил-
ляции 1-10-3; акваметрический с использованием реактива Фишера
1 • 10“3; изотопного разбавления 1СГ3; нейтронно-активационный
<10'3.
Серу определяют протонно-активационным методом по нуклиду
34WC1, образующемуся в результате реакций MS(p, у)34тС1 и 34S(p,
п)34гаС1 [1021]. При навеске 1 г металлического натрия предел обнару-
жения серы 10-6%.
Хром в металлическом натрии определяют фотометрически ди-
фенилкарбазидом [23]. Хром определяют также методом атомной
абсорбции с электротермической атомизацией после отгонки основы
при 1050—1400° С [758]. Аналитическая линия хрома 357,9 нм. Пре-
дел обнаружения зависит от анализируемого объекта: 0,062 мкг/мл
при анализе NaCl и 0,08 мкг/мл при анализе Na2SO4. При определе-
нии 5-10“3% хрома относительное стандартное отклонение 0,01—
0,056.
В диуранате натрия определяют уран оксидиметрическим титро-
ванием раствором К2Сг2О7 в присутствии дифениламинсульфоната
бария после восстановления урана(У1) до урана(1У) раствором FeSO4
в среде Н3РО4 [890].
В металлическом натрии, полученном вакуумной дистилляцией,
определяют уран методом изотопного разбавления (предел обнару-
7*
195
жения 1 • 10~7 % по изотопу 2SBU) и фотометрически с использованием:
арсеназоШ (предел обнаружения 1-10-6% при навеске 5 г) [1203L
Хлор определяют фотометрически в виде хлорида по ослаблению-
интенсивности окраски хлоранилата ртути. Предел обнаружения
2 -1СГ6 % (в металлическом натрии) и 1-1СГ6 % (в NaOH) [520]. Про-
должительность анализа 3,5—4,5 ч. В среде 3—8 М NaOH коэффи-
циент распределения на анионообменнике Амберлит CG400 состав-
ляет: J" > 2-Ю3; Вг~ > 80; СГ > 50; S»t, РОГ, AsOf > 3.
Пробу анализируемого NaOH растворяют в воде и пропускают-
через колонку с сильноосновным анионообменником Амберлит
CG400. Колонку промывают водой, затем сорбированный хлорид
элюируют 0,5 М раствором NH4NO3 и определяют в элюате спект-
рофотометрически по реакции с хлоранилатом ртути.
Для определения хлорида в металлическом натрии предваритель-
но отделяют натрий амальгамированием (или вакуумной дистилля-
цией), а в остатке титруют кулонометрически натрий ионами рту-
ти(П), электрогенерированными на амальгамированном золотом
электроде на фоне 0,1 М НС1О4 + 5 М NaClO4 в этаноле [1205]. Ко-
нечную точку титрования обнаруживают потенциометрически. Ме-
тод позволяет определять 1-10“4% хлора из навески 3—5 г натрия.
В металлическом натрии хлор определяют также косвенным
экстракционно-фотометрическим методом по ослаблению интенсив-
ности окраски комплекса ртути с дифенилкарбазоном [837].
Марганец (3,6-1СГБ%) определяют в гипофосфите. натрия кине-
тическим методом по реакции окисления люмогаллиона пероксидом
водорода при pH 10—11 [231]. При навеске образца 1 г предел об-
наружения марганца составляет 1-10"6%.
При pH 10—11 определению 0,02 мкг марганца не мешает 100—
500-кратный избыток ионов Ni, Mg, Cu, Zn, Co, а также 0,5 г фосфа-
та аммония. Скорость реакции определяют дифференциальным ме-
тодом по снижению оптической плотности растворов при 536 нм в
первые 3—4 мин после приливания раствора Н2Ог.
Навеску гипофосфита натрия 1,0 г растворяют в 10 мл воды и ставят в эк-
сикатор, на дно которого налит раствор NH3. Через 30—40 мин, к< гда будет дос-
тигнут pH > 8 (проба на вынос с индикаторной бумагой), чашку вынимают и от-
бирают в две пробирки по 0,2 мл раствора. В одну пробирку прибавляют 0,1 мл
25%-ного раствора NH3, в обе пробирки — по 0,1 мл раствора фенолфталеина
и разбавляют растворы водой до 5,0 мл. Измеряют оптическую плотность полу-
ченных растворов Аг и А 2 на фотоэлектроколориметре ФЭК-Н-57 со светофильт-
ром № 5 (536 нм). Если отношение (А, — А2)/А1 < 0,25, то к раствору в чашке
прибавляют 0,1—0,2 мл раствора аммиака, снова отбирают 0,2 мл, вводят фе-
нолфталеин и воду до 5,0 мл, измеряют оптическую плотность и рассчитывают
новое значение (Лг — Л2)/Л,. При достижении величины этого отношения ^0,25
раствор имеет pH 9,5. Этот раствор анализируют. В две пробирки (контроль-
ный опыт) вводят по 0,02 мл анализируемого раствора, в одну из них вводят
0,3 мл раствора, содержащего 0,1 мкг марганца в 1 мл. В две другие пробирки
при ивают по 0,5—1,0 мл анализируемого раствора, в одну из них вводят
0,03 мкг марганца. Во все четыре пробирки вводят по 0,5 мл 0,3%-ного Н2О2,
1 мл буферного раствора с pH 10,0 (смешивают 50 мл 0,1 N раствора Na2B40T
с 50 мл 0,1 М NaOH) и разбавляют водой до 4,5 мл. Затем последовательно изме-
ряют скорость реакции в каждом иэ четырех растворов, для чего в пробирку
приливают по 0,15 мл 0,01%-ного раствора люмогаллиона, встряхивают и пус-
196
кают секундомер, переливают раствор в кювету (1=1 см) и измеряют оптиче-
скую плотность через каждые 15 с в течение 3 мин. Полученные данные наносят
на график в координатах оптическая плотность—время и по графику определяют
тангенс угла наклона прямых К = tg а = ДА/Ат, мин-1. Содержание марган-
ца в анализируемом растворе X (в %) за вычетом контрольного опыта рассчиты-
вают по формуле
Ка1(К* - Ка) - Кк/(К* ~ К )
Х = 0,33-10е-Г »
где индексы: а — анализируемый, к — контрольный, д — с добавкой 0,03 Мкг
марганца; Г — навеска, отвечающая аликвотной части раствора, взятой для ана-
лиза, г.
Кинетический метод определения марганца по реакции окисле-
ния люмомагнезона пероксидом водорода при pH 11 позволил опре-
делить 1,4-10-6 % марганца из навески 1 г с относительным стандарт-
ным отклонением 0,12. Предел обнаружения марганца 1 • 10“6% [230],
В галогенидах натрия (NaBr, NaJ) марганец определяют кине-
тическим термометрическим методом по реакции окисления 1,10-фе-
нантролина пероксидом водорода, катализируемой марганцем [352].
Метод позволяет определять 5-10-6—1-10-5% марганца из навески
0,5—1,0 г. Определению не мешают 200-кратные количества щелоч-
ноземельных элементов, Со, Al, Cr, Zn, Cd, Pb, Hg, Cu, Ni, Ag, Mg,
V, W и Mo. Тяжелые металлы предварительно экстрагируют хлоро-
формом в виде комплексов с 1-(2-пиридилазо)-2-нафтолом.
Навеску 0,5—1,0 г NaJ обрабатывают 5 мл конц. HNO3 до полного удале-
ния иода, упаривают с водой, растворяют в 5 мл воды и переносят в делительную
воронку. Вводят 5 мл ацетатного буферного раствора с pH 4,5 и 2 мл 0,01%-ного
ацетонового раствора ПАН. Через 15 мин экстрагируют комплексы ПАН с же-
лезом, кобальтом и другими тяжелыми металлами хлороформом двумя порциями
по 5 мл. Водную фазу сливают в кварцевый тигель, воронку споласкивают 5 мл
воды, прибавляют ее к раствору в тигле и выпаривают досуха под инфракрасной
лампой. Остаток переводят с помощью 1 мл воды и 1 мл 1-10-3 М раствора
1,10-фенантролина в один из отростков сосуда-смесителя. В два других отростка
помещают 5 мл 2,5 М раствора Н2О2 и 3 мл 0,4 М раствора КОН. Сосуд поме-
щают в термостат и выдерживают в нем в течение 30 мин при температуре 25,0
-f. 0,1° С. Затем растворы в смесителе перемешивают, засекая при этом время,
и переводят в сосуд Дьюара. Туда же помещают термисторный датчик и записы-
вают изменение температуры в ходе реакции.
Для определения железа предложены кинетические методы, осно-
ванные вГа катализе реакций окисления Н-кислоты пероксидом во-
дорода [229] или вариаминового голубого пероксидом водорода при
pH 2—5 [233]. Методы позволили определить 2-10-5% железа (на-
веска гипофосфита натрия 0,05—0,1 г, погрешность 20%) [233]
и 1-10“5—1-10-4% железа (навеска солей натрия 0,05—0,1 г) [229].
Н-кислота окисляется пероксидом водорода в присутствии же-
леза. При окислении образуются продукты, окрашенные в красный
цвет (максимум светопоглощения при 500 нм). Скорость реакции
максимальна в уксуснокислых растворах при pH 3. Оптимальная
концентрация Н2О2 0,01 М. Реакция избирательна. Соли уменьшают
скорость реакции, но их влияние учитывают применением метода
добавок.
197
Навеску гипофосфита натрия 1,0 г растворяют в 10 мл воды в мерном ци-
линдре. В три пробирки наливают по 0,1—1,0 мл полученного раствора и соот-
ветственно 0,0; 0,1 и 0,2 мл стандартного раствора железа, содержащего
1,0 мкг/мг железа. Одновременно проводят контрольный опыт, для чего в три
другие пробирки наливают по 1 мл воды и такие же количества раствора железа.
Во все пробирки наливают по 0,5 мл 0,3%-ного Н2О2, уксусную кислоту (1 i
: 100) до объема 4,5 мл и с интервалом 1 мин — по 0,5 мл 0,18%-ного раствора
Н-кислоты, встряхивая пробирки. Через 15 мин измеряют оптические плотности
растворов в той же последовательности, что и при добавлении Н-кислоты. Строят
график зависимости оптической плотности от количества введенного железа и по
графику определяют содержание железа в анализируемом и контрольном раст-
ворах.
Технология очистки гипофосфита натрия обусловливает наличие
в нем ЭДТА, которая мешает определению железа вариаминовым
голубым, маскируя железо [233]. Это резко снижает чувствительность
метода. Влияние ЭДТА снимают введением большого избытка соли
никеля. Предварительно методом каталиметрицеского титрования
устанавливают содержание ЭДТА в соли. Определению железа пред-
ложенным методом мешают окислители (хроматы, ванадаты, перман-
ганат и др.) и вещества, связывающие железо в прочный комплекс.
Скорость реакции окисления вариаминового голубого солями желе-
за в присутствии Н2О2 пропорциональна концентрации железа (III)
и вариаминового голубого и максимальна при pH растворов 4,5—5,0
и концентрации Н2О2 0,06—0,08 М.
При анализе гипофосфита, не содержащего ЭДТА, навеску Соли 1 г раство-
ряют в 10 мл воды в мерном цилиндре. В три пробирки наливают по 1 мл полу-
ченного раствора и соответственно 0; 0,5 и 1,0 мл раствора, содержащего 0,1 мкг
железа в 1 мл. В три другие пробирки (контрольный опыт) приливают по 1,0 мл
воды и такие же количества раствора железа. Во все пробирки приливают по
0,5 мл 3%-ного Н2О2, 1,0 мл ацетатного буферного раствора с pH 4,5 (смешивают
42 мл 1 М раствора ацетата натрия с 58 мл 1 М СЙ3СООН) и разбавляют водой
до объема 4,5 мл. Затем в одну из пробирок вводят 0,5 мл 0,1%-ного раствора
вариаминового голубого, встряхивают, переливают содержимое пробирки в кю-
вету с толщиной слоя 1 см и записывают кинетическую кривую при 536 нм. По
полученным данным строят график в координатах оптическая плотность—время
и определяют величину прироста оптической плотности за единицу времени как
тангенс угла наклона прямой (tg а = ДА/Дт). Полученные шесть значений tg а
наносят на другой график, где по оси абсцисс откладывают количества введенно-
го железа. По графику, слева от нуля на оси абсцисс, определяют количество же-
леза в контрольном опыте (с0) и в аликвотной части (с) анализируемого раствора
(в мкг). Содержание железа в соли X (в %) рассчитывают по формуле
X = (с — ео).1О-4-У1/ГУ2,
где У] — объем раствора, в котором растворена навеска, мл; У2 — объем алик-
вотной части раствора, мл; Г — навеска соли, г.
Если в гипофосфите натрия обнаружены следы ЭДТА, то анализ проводят
следующим образом. В три пробирки приливают по 1,0 мл раствора гипофосфита
натрия и раствор, содержащий 0,05 г хлорида никеля. В три другие пробирки
(контрольный опыт) прибавляют по 1,0 мл воды и те же количества хлорида ни-
келя. Во все пробирки приливают по 1,0 мл буферного раствора и нагревают
в течение 10 мин в горячей воде. Охлаждают, прибавляют недостающие реакти-
вы и проводят анализ, как указано выше.
Разработан фотометрический метод определения железа 1,10-фе-
нантролином [23]. В NaCl и Na2SO4 определяли 5-10~3% железа ме-
тодом атомной абсорбции с электротермической атомизацией, ис-
198
польауя длину волны 248,3 нм [758]. Предварительно основу отго-
няли при 1050—1400° С. Предел обнаружения железа 0,056 мкг/мл
при анализе NaCl и 0,20 мкг/мл при анализе Na2SO4.
Кобальт в металлическом натрии определяли экстракционно-
фотометрическим методом с использованием 2-нитрозо-1-нафтола
[1158]. Из навески 1 г можно определить кобальт с пределом обна-
ружения 1-10~6%. Для получения надежных результатов рекомен-
довано при построении градуировочного графика вводить в раство-
ры по 25 г NaCl.
Фотометрический метод с использованием комплекса железа(Ш)
с 1,10-фенантролином позволяет определять 1>10“4% кобальта с по-
грешностью 3% [1248]. Метод основан на окислении кобальта комп-
лексом и измерении оптической плотности тпрпс-фенантролината же-
леза(П). На этом же принципе основан метод потенциометрического
титрования кобальта в присутствии 1,10-фенантролината раствором
железа(Ш). Метод позволяет определять JH0-3 % кобальта с погреш-
ностью 2,5%. Скачок потенциала в конечной точке тйтрования со-
ставляет 120 мВ на 0,05 мл 10~3 М раствора FeCl3.
В металлическом натрии определяли никель фотометрически в
форме цианидного комплекса, устраняя влияние ионов Fe(III) и
Сг(Ш) методом гомогенного осаждения мочевиной, а влияние ме-
ди — восстановлением гидроксиламином [738]. Метод применим для
определения никеля в различных солях натрия. Можно определять
никель также диметилглиоксимом в щелочной среде в присутствии
окислителя — бромной воды [23].
Для определения никеля в NaCl и Na2S04 предложен атомно-
абсорбционный метод с электротермической атомизацией [758]. Пре-
дел обнаружения никеля 0,093 и 0,028 мкг/мл при анализе NaCl
и Na2SO4 соответственно. Предварительно основу отгоняют при
1050—1400° С.
Воду определяли вольтамперометрически в NaOH или эвтектиче-
ской смеси NaOH—КОН [477]. В качестве реагента использован
пероксид натрия, взаимодействующий даже со следами воды. Предло-
жен простой способ построения градуировочного графика для оп-
ределения <Д,3% воды.
ЛИТЕРАТУРА
1. А б Э. А., Глинский Е. Е.— В кн.: Аппаратура и методы рентгеновского
анализа. Л.: Машиностроение, 1973, вып. 12, с. 297—298.
2. Аб Э. А., Глинский Е. Е., Федорова П. М.— В кн.: Аппаратура и методы
рентгеновского анализа. Л.: Машиностроение, 1974, вып. 14, с. 3—6.
3. Абарбарчук И. Л.— В кн.: Теория и практика полярографического ана-
лиза. Кишинев: Штиинца, 1962, с. 7—11.
4. Абдуллаев А. А., Анищенко ГО. М., Грахов В. А., Захидов А . ШХаитов
Б. К.— В кн.: Активационный анализ горных пород и других объектов.
Ташкент: Фан, 1967, с. 90—94.
5. Авербух М. А.— Завод, лаб., 1961, т. 27, с. 358.
6. Агасян П. К., Стенина Я. И., Шарапова Г. Н.— Завод, лаб., 1974, т. 40,
с. 1069—1071.
7. Адамович Л. П., Кравченко М. С., Смоляр В. С.— Ж урн. аналит. химии,
1969, т. 24, с. 782—785.
8. Азизов X. Ф., Асламханова X.— В кн.: Новые виды сложных удобрений.
Ташкент: Фан, 1970, с. 92—93.
9. Айдаров Т. К., Дорофеев В. С., Разяпов А. 3., Чупахин М. С.— Журн.
прикл. спектроскопии, 1975, т. 23, с. 522—527.
10. Акахори Саданори.— Коге есуйн, 1976, № 208, с. 39—46; РЖХим, 1976,
14Г18.
11. Акишев С. Н., Изотова Е. М.— Завод, лаб., 1977, т. 43, с. 450—451.
12. Алабышев А. Ф., Грачев К. Я., Зарецкий С. А., Лантратов М. Ф. Натрий
и калий. Л.: Госхимиздат, 1959. 391 с.
13. Алагова 3. С., Матерова Е. А., Шумилова Г. И.— Вести. ЛГУ, 1978,
вып. 22, с. 112—115.
14. Алагова 3. С., Матерова Е. А., Шумилова Г. И.— Вести. ЛГУ, 1980,
вып. 22, с. 134.
15. Алешечкина А. Е., Булычева И. Б.— Сб. тр./Урал. н.-и. хим, ин-т, 1971,
вып. 26, с. 7—11.
16. Алешин С. Я., Бодырев А. И.— Почвоведение, 1962, № 1, с. 114—121.
17. Алиев А. И., Дрынкин В. И., Лейпунская Д.-И., Касаткин В. А. Ядерно-
физические константы для нейтронного активационного анализа. М.:
Атомиздат, 1969. 324 с.
18. Алимарин И. П., Петрикова М. Н.— Журн. аналит. химии, 1969, т. 24,.
с. 935—936.
19. Алимарин И. Я., Фрид Б. И. Количественный микрохимический анализ
минералов и руд. М.: Госхимиздат, 1961, с. 275, 280.
20. Алябьева Т. М., Русинов И. Е.— Нефтепереработка и нефтехимия: Реф.
сб., 1975, № 9, с. 41.
21. Амшеева А. А.— Вести. Харьк. политехи, ин-та, 1968, № 32(80), с. 45—
47.
22. Андреев А. С.— Тр./Ленингр. политехи, ин-т, 1959,’№ 201, с. 35—39.
23. Андреев А . С., Шматова А. К.— В кн.: Жидкие металлы. М.: Госатомиз-
дат, 1963, с. 270—285.
24. Артюхин П. И., Гильберт Э. Н., Пронин В. А.— Радиохимия, 1976,
т. 9, с. 341—-346.
25. Архангельская 3. В.— Завод, лаб., 1953, т. 19, с. 423—424.
26. Асылбеков Н. А., Май И. И., Яковлев И. Я.— В кн.: Новые методы хими-
ческого анализа материалов. М.: МДНТП им. Ф. 3. Дзержинского, 1971,
№ 1, с. 45—48.
АЛЛ
27. АфанасиадиЛ. ИГлушкова Л. В.— Сб. науч. тр./ВНИИ монокристаллов,
сцинтилляц. материалов и особо чистых хим. веществ, 1980, № 5, с. 130.
28. Афанасьева Н. А., Орадовский С. Г.— Тр./Гос. океаногр. ин-т, 1972,
вып. 113, с. 66—71.
29. Багбанлы И. Л., Аллахвердиева Э. Г.— В кн.: Исследования в области
неорганической и физической химии. Баку: Элм, 1970, с. 157—162.
30. Бажов А. С., Кока И. А.— В кн.: Исследования в области химических и
физических методов анализа минерального сырья. Алма-Ата: Наука, 1971,
158—164.
31. Банных 3. С., Сачко А. П.— Тр./Урал. н.-и. хим. ин-т, 1957, вып. 4,
с. 209—217.
32. Барсуков В. И., Букреев Ю. Ф., Григорьев В. Р.— Завод, лаб., 1972, т. 38,
с. 422—425.
33. Барсуков В. И., Букреев Ю. Ф., Золотавин В. Л.— Тр. Моск, ин-та хим.
машиностроения, 1972, вып. 4, с. 79—84.
34. Барсуков В. И., Букреев Ю. Ф., Золотавин В. Л.— Тр. Моск, ин-та хим.
машиностроения, 1972, вып. 8, с. 91—98,-
35. Баусова Н. В., Решетникова Г. А.— Тр./Ин-т химии УФАНСССР, 1966,
вып. 10, с. 103—106.
36. Бегагян И. В., Куликова О. И., Ильичева И. А .— Завод, лаб., 1975, т. 41,
с. 934—935.
37. Бейтс Р. Определение pH. Теория и практика. Л.: Химия, 1968. 319 с.
38. Беленький С. М., Дулънева Т. И., Клячко Ю. А., Зеличенок С. Л.— Завод,
лаб., 1976, т. 42, с. 1317—1318.
39. Белявская Т. А.— Вести. МГУ. Сер. физ.-мат. и естеств. наук, 1949,
№ 2, с. 93—98.
40. Белявская Т. А.— Вести. МГУ. Сер. физ.-мат. и естеств. наук, 1950, № 5,
с. 59—62.
41. Беляевский А. Т.,'Протас Н. С,— В кн.: Методы анализа минерального
сырья. Апатиты, 1971, с. 233—236.
42. Беляевский А. Т., Протас Н, С.— Завод, лаб., 1972, т. 38, с. 175—177.
43. Бе'ненсон Н. М., Виноградова Л. С., Попова 3. И.— В кн.: Обмен опытом
в радиопромышленности. М., 1973, вып. 6, с. 35—36.
44. Берегин И. А.— В кн.: Спектроскопия. М.: Наука, 1964, с. 60—62.
45. Беркутова И. Д., Вьюнова Е. С., Жалнина Т. И., Злотова И. М., Якуб-
сон К .И.— В кн.: Активационный анализ в народном хозяйстве. Ташкент:
Фан, 1974, с. 68—73.
46. Бетехтин А. Г. Курс минералогии. М.: Госгеолтехиздат, 1961. 539 с.
47. Биктимирова Т. Г., Байбазаров А. А.— Тр./Башк.НИИ по переработке
нефти, 1964, вып. 7, с. 141—143.
48. Блинков Д. И., Лобанов Е. М.— В кн.: Ядерно-физические методы анализа
вещества. М.: Атомиздат, 1971, с. 90.
49. Блинков Д. И., Лобанов Е. М.— Тр. по химии и хим .технологии, Горький,
1969, вып. 3(24), с. 108—109.
50. Блинов В. А., Гаврилов В. М., Жерехов Р. Г. и др.— Прикладная ядер,
спектроскопия. 1975, вып. 5, с. 159—162.
51. Блохин В. И., Ластов А. И., Мссеев Л. И.— В кн.: Активационный ана-
лиз. Ташкент. Фан, 1971, с. 219—225.
52. Боброва Л. А.— В кн.: Методы анализа вещественного состава горных
пород и вод при геохимических исследованиях. Минск, 1978, с. 82—90.
53. Бобровски А., Зарембски Ю.— Журн. аналит. химии, 1972, т. 27, с. 1472—
1479.
54. Богатырев К. С., Шамрицкая И. П., Мелешко В. П., Золотарева Л. В.—
В кн.: Теория и практика сорбционных процессов, Воронеж: Изд-во Во-
ронеж. ун-та, 1975, вып. 10, с. 88—91.
55. Богоявленская А. Н., Молоствова Н. И., Переверзева Е. Ф., Поляко-
ва В. В.— Науч. тр./НИИ цветных металлов, 1977, № 43, с. 50—55.
56. Божевольнов Е. А.— Методы анализа хим. реактивов и препаратов, 1969,
вып. 16, с. 146—147.
57. Божевольнов Е. А. Люминесцентный анализ неорганических веществ. М.,
Химия, 1966. 237 с.
201
58. Вожееолъное Е. А., Федорова Л. Ф., Бодрова Н. И.— Методы анализа
хим. реактивов и препаратов. 1069, вып. 16, с. 148—151.
59. Большов М. А., Гузеев И. Д., Зыбин А. В., Колошников В. Г., Майоров
И. А., Педлер В. В., Мандельштам С. Л., Тимофеев Е. Ф., Филимонов
Л. НЖурн. прикл. спектроскопии, 1973, т. 19, с. 821—824.
60. Бондаренко Л. С., Пейзулаев Ш. И., Карабаш А.Г.— В кн.: Методы ана-
лиза веществ высокой чистоты. М.: Наука, 1965, с. 315—319.
61. Борзов В. П., Мальцев М. Г., Плющ Г. В., Семенова М. В., Шарыгина
И. Н., Лукьянова Т. Ф,— В кн.: Прикладная спектроскопия. М.: Наука,
1969, т. 1, с. 219—222.
62. Борзов В. 11., Мальцев М. Г., Плющ Г. В., Янклович Н. Г.— В кн.: При-
кладная спектроскопия. М.: Наука, 1969, т. 1, с. 255—257.,
63. Борисов Г. И., Демидов А. М., Захаров Е. А.— Атом, энергия, 1969,
т. 26, с. 14—19.
64. Борк В. А., Швыркова Л. А., Ким Л. Б.— Журн. аналит. химии, 1971,
т. 26, с. 269—272.
65. Боровик-Романова Т. Ф., Фарафонов М. М.— Тр./Комис. по аналит.
химии АН СССР, 1960, т. 12, с. 322—330.
66. Борходоее В. Я., Серый В. Г.— Колыма, 1976, № 6, с. 42—43.
67. Брицке М. Э. Атомно-абсорбционный спектрохимический анализ. М.:
Химия, 1982. 223 с.
68. Брицке М. Э.— Завод, лаб., 1964, т. 30, с. 1465—1469.
69. Брицке М. Э., Зданоеич И. ДИванцов Л. ММалинина В. И., Поляко-
ва В. В.— В кн.: Обогащение и металлургия цветных металлов. М.: Ме-
таллургиздат, 1953, с. 99—112.
70. Брицке М. Э., Савельева А. Н.— Завод, лаб., 1966, т. 32, с. 1480.
71. Брицке Э. ВКапустинский А. ф., Веселовский Б. К., Шамовский Л. М.,
Ченцова Л. Г., Анеаер Б. И. Термические константы неорганических ве-
ществ. М.; Л.: Изд-во АН СССР, 1949. 1011 с.
.72 . Букреев Ю. Ф.— Тр./Моск. ин-т хим. машиностроения, 1972, вып. 44,
с. 84—87.
73. Букреев Ю. Ф.— Тр./Моск. ин-т хим. машиностроения, 1972, Вып. 46,
с. 102—105.
74. Букреев Ю. Ф., Барсуков В. И., Золотавин В. Л., Федулов А. К.— Журн.
прикл. спектроскопии, 1972, т. 16, с. 210—215.
75. Букреев Ю. Ф., Григорьев В. Ф., Барсуков В. И.— Тр./Моск. ин-т хим.
машиностроения, 1972, вып. 44, с. 87—93.
76. Букреев Ю. Ф., Григорьев В. Ф., Барсуков В. И., Федулов А. К.— В кн.:
Урай. конф, по спектроскопии, 1971. Свердловск: Наука, 1971, вЫп. 1,
с. 218—222.
77. Букреев Ю. Ф., Золотавин В. Л., Безрукое И. Я., Толстов Л. К.— Завод,
лаб., 1966, т. 31, с. 1085.
78. Букреев Ю. Ф., Толстов Л. К., Золотавин В. Л.— Тр./Ин-т химии УФАН
СССР, 1967, вып. 14, с. 159—162.
79. Булычева И. Б., Алешечкина А. Е.— Завод, лаб., 1971, т. 37, с. 435—436.
80. Булычева И. Б., Алешечкина А, Е.— Тр./Урал. н.-и. хим. ии-т, 1970,
вып. 19, с. 180—184.
81. Булычева И. Б., Алешечкина А. Е.— Тр./Урал. н.-и. хим. ин-т, 1973,
вып. 27, с. 242—244.
82. Булычева И. Б., Ульянова М. С.— Тр./Урал. н.-и.хим.ин-т, 1964, вып. 11,
с. 98—101.
83. Бурова Т. А.— Тр./Ин-т геологии руд. месторождений, петрографии, ми-
нералогии и геохимии АН СССР, 1961, вып. 64, с. 86—90.
84. Бурриель-Марти Ф., Рамирес-Муньос X. Фотометрия пламени. М.:
Изд-во иностр, лит., 1962. 520 с.
85. Буянов Н. В., Иванова Л. А., Полякова Р. С., Сухова Н. П.— Сб. тр'./
ЦНИИ чер. металлургии, 1964, вып. 37, с. 89—97.
86. Быкова И. Н., Манова Т. Г., Федорович А. А.— Методы анализа хим. ре-
активов и препаратов, 1973, вып. 21, с. 28—30.
87. Быкова И. И., Манова Т. Г., Федорович А . А.— Методы анализа хим. ре-
активов и препаратов, 1973, вып. 21, с. 76—78.
202
88. Быстрова Л. Ф., Страдомский В. Б., Назарова А. А.— Гидрохим. мате-
риалы, Л., 1979, т. 73, с. 49—59.
89. Бэчко И. М.— В кн.: Применение спектрального анализа в народном хо-
зяйстве и научных исследованиях. Б. м., 1974, с. 140—142.
90. Вайль Е. И., Халаимова А. М.— Кокс и химия, 1967, № 5, с. 47-—50.
91. Вайнштейн Э. Е., Королев В. В.— Журн. аналит. химии, 1956, т. 11,
с. 627—633.
92. Вайнштейн Э. Е., Лебедев В. И.— Геохимия, 1961, № 4, с. 362—-363.
93. Вахобов А. В., Пачаджанов Д. Н-, Худайбердиев В. Т.— Изв. АН Тадж.
ССР. Отд. физ.-мат. и геол.-хим. наук, 1970, № 1(35), с. 43—46.
94. Виноградов А. В., Дундур Е. И.— Журн. аналит. химии, 1949, т. 4,
с. 117—121.
95. Витько Н. Д.,БрилеваЛ. Г., Коник М. А., ЯременеикоЗ .В.— Завод, лаб.,
1976, т. 42, с. 619—620.
96. Власов В. С., Пережогин Г. А.— Завод, лаб., 1978, т. 44, с. 720—721.
97. Вольхин В. В., Колесова С. А., Кощеева Е, А., Затонская Т. П.— Тр./
НИИ хроматографии Воронеж, ун-та, 1968, вып. 2, с. 175—179.
98. Ворончихина А. П., Масалович В. М., Лапшаноеа Н. С.— Сб. тр./Урал.
н.-и. хим. ин-т, 1971, вып. 26, с. 85—88.
99. Ворсин А. И., Гусев Г. М.— В кн.: Количественный'анализ минералов
и горных пород физическими методами. Новосибирск: Наука, 1965,
вып. 32, с. 3—15.
100. Гаврилюк А. Т., Чуйко В. Т.— В1сник Льв1в. ун-ту. Сер. xiM., 1971,
вип. 12, с. 42—44.
101. Галкина Н. К., Рубинштейн Р. Н., Сенявин М. М.— Журн. физ. химии,
1962, т. 36, с. 1860—1869.
102. Гиллебранд В. Ф., Ленделъ Г. Э., Брайт Г. А., Гофман Д. И. Практи-
ческое руководство по неорганическому анализу. М.: Госхимиздат, 1960.
667 с.
103. Гильберт Э. Н., Артюхин П. И.— Изв. СО АН СССР. Сер. хим. наук,
1967, № 4, вып. 2, с. 90—94.
104. Гильберт 3. Н., Пронин В. А.~ Изв. СО АН СССР. Сер. хим. наук, 1969,
вып. 2, № 4, с. 168—170.
105. Главин Г. Г., Горюшина В. Г., Каплан Б. Я., НоткинаМ. А., Солодов-
ник С. М., Хотин Б. А.— В кн.: Методы анализа веществ высокой чистоты.
М.: Наука, 1965, с. 127—135.
106. Головатый Р. Н., Новосельская М. И., Ощаповский В. В,— Укр. хим.
журн. 1962, т. 28, с. 112—115.
'IO?. Гольдфарб В. М., Ильина Е. В.— В кн.: Прикл. спектроскопия. М.:
Наука, 1969, т. 1, с. 459—463.
108. Гончаков А. С., Зоров Н. Б., Кувяков Ю. Я.— Журн. аналит. химии, 1979,
т. 34, с. 2057—2060.
109. Гончаков А. С., Зоров Н. Б., Кувяков Ю. Я., Матвеев О, И.— Журн.
аналит. химии, 1979, т. 34, с. 2312—2315.
110. Горбенко Ф. П., Вашунь 3. М., Хорошайлова И. И.— Методы анализа
хим. реактивов и препаратов, 1973, вы . 21, с. 143—145.
111. Горбунова Л. Б., Кутейников А. Ф., Марунина И. И., Сухов Г. В.—
Журн. аналит. химии, 1976, т. 31, с. 2061—2064.
112. Горемыкин В. Э.— В кн.: Современные методы анализа природных вод.
М.: Изд-во АН СССР, 1962, с. 22—31.
113. Горемъкин В. Э., Крюков П. А.— Гидрохим. материалы, Л., 1959, т. 28,
с. 170—179.
114. Горемыкин В. Э., Крюков П. А.— Изв. АН СССР. ОХН, 1957, № 11,
с. 1387—1389.
115. Городенцееа Т. Б.— Тр./ВНИИ стандарт, образцов и спектр, эталонов,
1968, т. 4, с. 171—174.
116. Горшков В. И., Панченков Г. М., Иванова Т. В.— Жури. физ. химии,
1962, т. 36, с. 1690—1694.
117. Грибова Е.А., Баврина Ю.П.— Завод, лаб., 1976, т. 42, с. 925—926.
118. Грикит И. А., Гула Л. А.— Сб. тр./ВНИИ проект, ин-т титана, 1968»
т. 2, с. 362—365.
203
119. Гузеев И. Д., Блинова Э. С., Майоров И. А., Педлер В. В.— Завод, лаб.,
1973, т. 39, с. 165—167.
120. Гуревич В. Г., Астахина Н. С:— Тр./Харьк. фарм. ин-т, 1957, вып. 1,
с. 106—109.
121. Гусарский В. В., Тарасевич Н. И.— Завод, лаб., 1962, т. 28, с. 183—184.
122. Гутько А. Д., Панкратова Н. И., Козяева 3. Н., Ткачева Г. В. —,В кн.:-
Спектроскопия. М.: Наука, 1964, с. 88—91.
123. Гутько А. Д., Ткачева Г. В., Панкратова Н. И., Козяева 3. П., Во-
робьев Н. К.— В кн.: Анализ благородных металлов. М.: Наука, 1965,
с. 114—118.
124. Гутько А.Д., Фирсова И.Н., Козяева 3. И.— В кн.: Спектральный
анализ в геологии и геохимии. М.: Наука, 1967, с. 243—245.
125. Гэинар И., Гэинар Е.— Радиохимия, 1962, т. 4, с. 175—180.
126. Дамбург Н. А., Вееерис О. Э., Вирцавс М. В., Микельсон Г. Г.— В кн.:
Активационный анализ. Рига: Зинатне, 1976, с. 69—84.
127. Дамбург Н. А., Вееерис О. Э., Микельсон Г. Г.— Изв. АН ЛатвССР.
Сер. физ. и техн, наук, 1975, № 2, с. 5—7.
128. Дамбург Н. А., Микельсон Г. Г., Вирцавс М.В.— Изв. АН ЛатвССР.
Сер. физ. и техн, наук, 1975, № 3, с. 3—7.
129. Дамбург Н.А., Пелекис З.Э., Пелекис JI.JI.— В кн.: Радиационная
физика. Рига: Зинатне, 1965, № 3, с. 33—38.
130. Дееят х Г. Г., Шишов В. Н.— Журн. аналит. химии, 1977, т. 32,
с. 1578—1582.
131. Дегтярева О. Ф., Островская М. Ф.— Завод, лаб., 1960, т. 26, с. 564—566.
132. Дегтярева О. Ф., Синиц на Л. Г., Барихина Т. А.— Журн. аналит.
химии, 1973, т. 28, с. 1164—1168.
133. Дегтярева О. Ф., Федяева Н. В., Островская М. Ф,— Завод, лаб., 1961,
т. 27, с. 842—844.
134. Десятков В. Д., Антонов А. В., Абрамов Ю.А.— Науч. тр. н.-и. и.
проект, ин-т редкометал. пром-сти, 1972, т. 42, с. 155—157.
135. Джемардьян Ю. А., Михайлов Г. И., Старчик Л. П.— Завод, лаб., 1971,
т. 37, с. 555—560.
136. Дзиомко В. М., Зеличенок С. Л., Маркович И. С.— Журн. аналит. хи-
мии, 1968, т. 23, с. 170—173; РЖХим, 1977, 9Г83.
137. Дзиомко В. М., Маркович И. С:, Зеличенок С. Л., Агзибекова Л. О.,
Круглова Н. В.— Тр./ВНИИ хим. реактивов и особо чистых веществ,
1974, вып. 36, с. 6—10.
138. Дицман С. А., Куприянова Т.А., Селезнева М. А.— Изв. АН СССР.
Неорган. материалы, 1975, т._ 2, с. 537—538.
139. Дмитриева И. Б., Коновалов Э. Е.— В кн.: Методы определения и иссле-
дования состояния газов в металлах. М.: Наука, 1968, с. 195—202.
140. ДойковаР., ЦветановК,— Металлургия (НРБ), 1976, т. 31, № 6, с. 28—
29.
141. ДолиЗзе В. А., Крунчак В. Г., Парфенов А. И., Саруханова Э. М., Сер-
геев А. С., Тарасова В. М.— В кн.: Материалы Всесоюз. науч-техн. совещ.
«Аналитическое приборостроение. Методы и приборы для анализа жид-
ких сред». Тбилиси, 1975, т. 3, ч. 2, с. 217—225.
142. Драницкая Р. М.— Тр./Одес. ун-т. Сб. хим. фак., 1953, т. 3, с. 89—97.
143. Драницкая Р. М., Дремлюк Р. Л.— Укр. хим. журн., 1956, т. 22, с. 821 —
823.
144. Евжанов X., Маликова Е. Д., Кунин Л. Л.— Журн. аналит. химии, 1973,
т. 28, с. 235—239.
145. Евжанов X., Чапыжников Б. А., Маликова Е.Д., Кунин Л. Л.— Журн.
аналит. химии, 1971, т. 26, с. 1373—1377.
146. Егоров В. В., Краснопевцев Ю. В., Малахов С. Г.— Тр./Ин-т эксперим.
метеорологии. Гл. упр. гидрометеорол. службы при Сов. Мин. СССР,
1970, вып. 5, с. 38—41.
147. Егоров В. Я., Зарембо В. И., Соболева П. Г., Пучков Л. В.— Журн-
। прикл. химии, 1981, т. 54, с. 1247—1250.
148. Егоров Н. П., Ковалев И. А.— Журн. аналит. химии, 1959, т. 14, с. 489 —
490.
204
449. Емельянов С. X.— Учен. зап. ЦНИИ оловян. пром-сти, 1965, № 1, с. 74 —
75.
150. Енаки И. Г.— Гидробиол. журн., 1966, т. 2, № 5, с. 89—95.
151. Ермолаева Е. В., Коробка Л. А.— Бюл. науч.-техн. информ. ВНИИ
огнеупоров, 1957, т. 2, с. 89—93.
152. Ерохина К. И., Лемберг И. X., Макашева И. Е., Маслов И. А., Обу-
хов А. П.— Завод, лаб., 1960, т. 26, с. 821—827.
153. Есиков А.Д., Каленчук Г.Е.— В кн.: Химический анализ минералов
и их химический состав. М.: Наука, 1964, с. 3—15.
154. Железцов В. А., Ляшенко С. П., Шеломенок М. Е.— Стекло и керамика,
1963, № 1, с. 26—28.
455. Журавлев А. 4., Аминова М. М.~ В кн.: Прикладная ядерная физика.
Ташкент: Фан, 1973, кн. 2, с. 143—146.
456. Журавлев Г. И., Гаврилова И. А.— Журн. аналит. химии, 1964, т. 19,
с. 54—58.
457. Забияко В. И., Булычевой. Б.— Тр./Урал. н.-и. хим. ин-т, 1964, вып. 11,
с. 16—18.
158. Зайдель А.Н. Атомно-флуоресцентный анализ. М.: Наука, 1980. 187 с-
459. Зайдель А.Н. Атомно-флуоресцентный анализ. М.: Наука, 1983. 126с.
160. Зайдель А. И., Калитеевский Н. И., Липис Л. В., Тараканов В. М.—
Оптика и спектроскопия, 1957, т. 3, с. 16—20.
161. Зайдель А. Н., Калитеевский Н. И., Липис Л. В., Чайка М. П.— Журн.
аналит. химии, 1957, т. 12, с. 17—22.
462. Зайдель А. Н., Калитеевский Н. И., Липис Л. В., Чайка М. П. Эмиссион-
ный анализ атомных материалов. М.: Физматгиз, 1960, 686 с.
163. Зайдель А. Н., Липис Л. В., Петров К. И.— Журн. аналит. химии, 1959,
т. 14, с. 197.
164. Заславская М.Б.,Цыцарин Г. В.— Вод. ресурсы, 1975, № 5, с. 33—42-
465. Захаров Е. А., Мясоедов Б. Ф., Карякин А. В.— Журн. аналит. химии,
1973, т. 28, с. 879—885.
166. Зобнина Н. А., Камаева Л. В., Шавку нова Н. Н. — Стандарт, о'разцы
в чер.металлургии, 1979, № 8, с. 41—44.
467. Золотавин В. Л., Барсуков В. И.. Букреев Ю. Ф.— Тр./Тамбов. ия-т хим.
машиностроения, 1972, вып. 8, с. 105—111.
168. Золотавин В. Л., Букреев Ю. Ф., Толстов Л. К., Безруков И. Я.—
Журн. прикл. спектроскопии, 1965, т. 2, с. 461—462.
469. Золотавин В. Л., Букреев Ю. Ф., Толстов Л. К., Безруков И. Я.— Тр./
Урал, политехн. ин-т, 1966, сб. 148, с. 139—142.
170. Золотовицкая Э. С., Фидельман Б. М., Булгакова А. М.— В кн.: Получе-
ние и анализ вещества особой чистоты. М.: Наука, 1966, с. 155—157.
171. Золотовицкая Э. С., Фидельман Б. М., Чепурная В. Г.— В кн.: [Моно-
кристаллы, сцинтилляторы и органические люминофоры. Черкассы,
1972, вып. 6, ч. 1, с. 66—70.
172. Золотовицкая Э. С., Фидельман Б. М., Чепурная В. Г., Бондарева Н. В.—
В кн.: Методы анализа галогенидов щелочных и щелочноземельных
металлов высокой чистоты. Харьков: ВНИИ монокристаллов, 1971, ч. 2,
с. 3.
173. Иванов В. К., Стромберг А. Г.— В кн.: Редкие щелочные элементы.
Пермь: Политехн. ин-т, 1969, с. 458—460.
474. Иванов В. К., Стромберг А. Г., Каплин А. А.— Журн. аналит. химии,
1970, т. 25, с. 584—585.
175. Иванов Н. П., Козырева Н. А.— Тр. ВНИИ хим. реактивов и особо чистых
хим. веществ, 1966, вып. 23, с. 137—142.
176. Иванов Н. И., Красильщик В. 3.— Методы анализа хим. реактивов и
препаратов, 1963, вып. 7, с. 5—68.
177. Иванова Е. И., Андреева В. А., Клычева В. И.—• Завод, лаб., 1951,
т. 17, с. 498—500.
178. Иванова М. М.— Журн. аналит. химии, 1977, т. 32, с. 1066—1070.
179. Игнатьева М. Я., Прилепская В. А., Чеботарев В, Е.— В кн.: Методы
химического анализа. М.: Химия, 1969, с. 171—175.
205.
180. Исибаси Н., Кохара X., Харагути Т.— Бунсэки кагаку, Jap. Analyst,
1965, vol. 14, р. 62—68; РЖХим, 1966, 2Г73.
181. Измайлов Н. А.— Докл. АН СССР, 1963, т. 149, с. 1103—1106.
182. Ионоселективные электроды/Под ред. Р. Дарста. М.: Мир, 1972, с. 14—16.
183. Ипатов П. Ф., Семочкина Л. А.— Тр./ВНИИ водоснабжения, канализац.,
гидротехн. сооружений и инженер, гидрогеологии, 1975, вып. 48, с. 8—9.
184. 1змайлов М. А., Рибк1н Ю. Ф.— Доноввд АН УРСР, 1962, с. 1071—1075.
185. Казаченко В. И.— Технология легких сплавов. Науч.-техн. бюл. ВИЛС,
1975, № 3, с. 67—68.
186. Калак И. С., Комарова К. А., Горбачев В. П., Данилина В. М.— Тр./
Моск, хим.-технол. ин-т, 1974, вып. 82, с. 141—143.
187. Каленчук Г. В.— В кн.: Химический анализ минералов и их химический
состав. М.: Наука, 1964, с. 16—32.
188. Калинин А. И., Кузнецов Р. А.— В кн.: Методы анализа веществ высокой
чистоты. М.: Наука, 1965, с. 93—100.
189. Калъвода Р.— Завод, лаб., 1958, т. 24, с. 1319—1326.
190. Каммори О., Такахари Г., Вандо С.— Бунсэки кагаку, Jap. Analyst,
1967, vol. 16, р. 826—829;.РЖХим, 1968, 21Г101.
191. Камэмото Ю., Ямагиси С.— Nippon kagaku zasshi. J. Chem. Soc. Japan.
Pure Chem. Sec., 1962, vol. 83, p. 572—573; РЖХим, 1963, 8Г137.
192. Камэмото Ю., Ямагиси С.— Nippon kagaku zasshi. J. Chem. Soc. Japan.
Pure Chem. Sec., 1962, vol. 83, p. 887—888; РЖХим, 1963, 8Г149.
193. Канунникова В. И., Майзилъ Э. Е.— Тр./Всёсоюз. н.-и. и конструкт, ин-т
«Цветметавтоматика», 1975, вып. 8, с. 65—67.
194. Карабаш А. Г., Пейзулаев Ш.И.— Тр./Физ.-энерг. ин-т, 1974, с. 482—
492.
195. Карабаш А. Г., Пейзулаев Ш.И., Слюсарева Р.Л., Липатова В. М,—
Тр./Комис. по аналит. химии АН СССР, 1960, т. 12, с. 331—340.
196. Карабаш А. Г., Пейзулаев Ш.И., Сотникова Н. П., Сазанова С. К.—•
Тр./Комис. по аналит. химии АН СССР, 1960, т. 12, с. 108—116.
197. Карабаш А. Г., Самсонова 3. Н., Смирнова-Аверина Н . И., Пейзу-
лаев Ш. И.— Тр./Комис. по аналит. химии АН СССР, 1960, т. 12,
с. 255—264.
198. Карбаинов 10, А., Карбаинова С. Н.— Методы анализа хим. реактивов и
препаратов, 1971, вып. 20, с. 63—64.
199. Карташев Е. Р., Маркун Н.Ю., Чулкин В. Л., Штанъ А. С.— Тр./
ВНИИ радиац. техники, 1972, вып. 7, с. 114—118.
200. Кацков Д.А., Гринштейн И. Л.— Журн. прикл. спектроскопии, 1978,
т. 28, с. 968—974.
201. Кветкус К. К., Навицкас В. С., Шакалис И. А., Шопаускас К. К., Ясю-
ленис Р. Ю.— Тр./Ин-т эксперим. метеорологии Гл. упр. гидромет-
службы при Сов. Мин. СССР, 1976, вып. 4(56), с. 145—152.
202. Кист А. А., Орестова И. И., Лобанов Е. М.— Вкн.: Нейтронно-акти-
вационной анализ. Ташкент: Фан, 1971, с. 88—95.
203. Клинский Г.Д., Иванов С, А., Князев Д.А Кудрявцева А. П.— Изв.
ТСХА, 1978, с. 196—200.
204. Клишане Д. А., Пелекис 3. Э., Пелекис Л. Л.— В кн.: Нейтронно-акти-
вационный анализ. Рига: Зинатне, 1966, с. 77—81.
205. Коблянский А. Г.— Тр./Комис. по аналит. химии АН СССР, 1956, т. 7(10),
с. 89—95.
206. Коваленко П. И., Теньковцев В. В.— Укр. хим. жури., 1954, т. 20,
с. 411—416.
207. Кодочигов П. Н., Глазунов М. П., Меднис И. В., Спицын В. И.— В кн.:
Нейтронно-активационный анализ. Рига: Зинатне, 1966, с. 115—134.
208. Колечкова А. Ф., Останина И. М.— Тр./Всесоюз. гос. ин-т н.-и. и проект,
работ огнеупор, пром-сти, 1961, вып. 31, с. 188—194.
209. Колосова Г. М., Богачева М. П., Варсанова 3. А.— Журн. аналит. химии,
1968, т. 23, с. 1403—1404.
210. Колтунов Ю. В.— Почвоведение, 1964, № 7, с. 110—111.
211. Комиссаренко В. С.— Завод, лаб., 1952, т. 18, с. 1110.
212. Комиссаренко В. С.— Завод, лаб., 1958, т. 24, с. 462—463.
206
213. Комиссарова Т. Е., Баскакова И. И., Николаева Л. Н.— Завод, лаб., 1971,
т. 37, с. 1175—1176.1
214. Конькова О. В., Парташникова М. 3., Юркина Н. Г., Зайкова Н. Н.,
Денискина Г. Н., Никифорова Т. Е.— В кн.: Материалы IV межотрасл.
конф, по методам получения и анализа ферритовых, сегнето- и пьезоэлек-
трических и конденсаторных материалов и сырья для них. М.: ИРЕА,
1973, с. 229—236.
215. Коренман И. М. Аналитическая химия калия. М.: Наука, 1964. 255 с.
216. Коренман И. М., Шеянова Ф. Р., Туманов А. А., Глазунова 3. М., Де-
мин О. И.— Тр. по химии и хим. технологии, Горький, 1959, вып. 1,
с. 94—101.
217. Корж П. Д., Штутман М. Н.— Завод, лаб., 1947, т. 13, с. 441—447.
218. Коровин Ю. И., Липис Л. В.— Оптика и спектроскопия, 1958, т. 5,
с. 334—337.
219. Котенева Т. В.— Тр./НИИ геологии Арктики, 1959, т. 98, с. 130—138.
220. Котова В.Н., Ларикова Г. Г., Шимичева Н.А., Тагирова Н. А., Ан-
типова О. А.— Журн. аналит. химии, 1981, т. 36, с. 880—883.
221. Коттон Ф., Уилкинсон Дж. Современная неорганическая химия. М.:
Мир, 1969, ч. 2, 494 с.
222. Красильникова М. Ф., Жолин А. В.— Лакокрасоч. материалы и их при-
менение, 1976, № 3, с. 40—41.
223. Красильщик В. 3., Воропаев Е. И.— Завод, лаб., 1980, т. 46, с. 224—226.
224. Красильщик В. 3., Штейнберг Г. А.— Методы анализа хим. реактивов и
препаратов, 1973, вып. 21, с. 105—108.
225. Краснобаева Н., Задгорска 3.— Химия и индустрия (НРБ), 1970, т. 42,
№ 4, с. 165—168.
226. Краткая химическая энциклопедия. М.: Сов. энциклопедия, 1964, т. 3,
с. 367.
227. Крауз Л. С., Карабаш А. Т., Пейзулаев Ш. И., Липатова^. М., Ма-
лева В. С,— Тр./Комис, по аналит. химии АН СССР, 1960, т. 12, с. 175—
. 186.
228. Крейнголъд С. У., Божееольнов Е. А.— Методы анализа хим. реактивов
и препаратов, 1969, вып. 16, с. 187.
229. Крейнгольд С. У., Божееольнов Е. А., Антонов В-.Н.— Методы анали-
за хим. реактцвов и препаратов, 1969, вып. 16, с. 187—188.
230. Крейнголъд С. У., Божееольнов Е. А., Антонов В. Н.~ Методы анализа
хим. реактивов и препаратов, 1969, вып. 16, с. 192—193.
231. Крейнгольд С. У., Божееольнов Е. А., Антонов В. Н., Сосенкова Л. И.—
Методы анализа хим. реактивов и препаратов, 1971, вып. 18, с. 26—28.
232. Крейнголъд С. У., Божееольнов Е. А., Антонов В. Н., Сосенкова Л. И.—
Методы анализа хим. реактивов и препаратов, 1971, вып. 18, с. 36—38.
233. Крейнгольд С. У., Сосенкова Л. И.— Методы анализа хим. реактивов
и препаратов, 1973, вып. 21, с. 175—179.
234. Крестов Г. А.— Изв. вузов. Химия и хим. технология, 1968, т. 11,
с. 762—765.
235. Крестов Г. А., Зверев В. А.— Изв. вузов. Химия и хим. технология,
1968, т. 11, с. 990—995.
236. Крешкое А. П., Лебедева М. И., Исаева Б. И.— Журн. аналит. химии,
1970, т. 25, с. 1710—1713.
237. Кристалееа Л. Б.— Тр./ Том. ун-т, 1954, т. 126, с. 191—212.
238. Крупкин А. И.— Завод, лаб., 1950, т. 16, с. 31—34.
239. Крупкин А. И., Лорина Г. А.— Журн. аналит. химии, 1956, т. 11, с. 30—
32.
240. Крюков П.А., Горемыкин В. Э.— В кн.: Современные методы химиче-
ского анализа природной воды. М.: Изд-во АН СССР, 1955, с. 14—18.
241. Кузин И. А., Нечаев А. Ф., Плаченов Б. Т., Похитонов Ю. A., Тауш-
канав В. П.~ В кн.: Получение и анализ чистых веществ. Горький: На-
ука, 1976, вып. 1 (45), с. 42—44.
242. Кузин И. А., Таушканов В. П., Блохин А. А.— Жури, прикл, химии,
1976, т. 49, с. 2518-2520.
207
243. Кузьмин Н. М., Дубровина Т. П., Шемшук О. М.— Журн. аналит. хи-
мии, 1973, т. 28, с. 364—367.
244. Кулев И. И., Бакалов В. Д.— Докл. Болг. АН, 1973, т. 26, с. 787—789.
245. Кулев И. И., Станев Д. С., Бакалов В. Д.— Khim. Ind. (Sofia), 1974,
vol. 46, p. 65—66.
246. Кунин Л. Л., Маликова Е. Д., Чапыжников Б. А. Определение кисло-
рода, углерода, азота и водорода в щелочных и щелочноземельных метал-
лах. М.: Атомиздат, 1972. 177 с.
247. Курин М. Н., Голышев С. И., Тихомиров И. А.— Изв. СО АН СССР.-
Сер. хим. наук, 1964, вып. 2, № 7, с. 89—93.
248. Кустас В. Л., Мамонтова Л. И., Шепета Н. Г., Юделевич И. Г. —
В кн.: Прикладная спектроскопия. М.: Наука, 1969, т. 1, с. 252—255.
249. Кустас В. Л., Мамонтова Л. И., Шепета Н. Г., Юделевич И. Г.—
В кн.: Синтез, очистка и анализ неорганических материалов. Новосибирск:
Наука, 1971, с.’ 259—265.
250. Кустас В. Л., Палева Г. В., Чучуева Р. С., Юделевич И. Г.— Методы
анализа хим. реактивов и препаратов, 1971, вып.л20, с. 32—34.
251. Кустас В. Л., Палева Г. В., Юделевич И. Г.— Методы анализа хим.
реактивов и препаратов, 1971, вып. 20, с. 45—46.
252. Кустас В. Л., Палева Г. В., Юделевич И. Г., Шепета Н. Г.— Методы
анализа хим. реактивов и препаратов, 1971, вып. 20, с. 39—41.
253. Кустас В. Л., Шепета Н. Г., Полева Г. В., Юделевич И. Г.— Методы
анализа хим. реактивов и препаратов, 1971, вып. 20, с. 36—38.
254. Кустас В. Л., Яковлева И. А., Мамонтова Л. И., Юделевич И. Г.—
Методы анализа хим. реактивов и препаратов, 1971, вып. 20, с. 47—50.
255. Куус X., Лонб Э.— Учен. зап. /Тарт, ун-т, 1969, вып. 235, с. 128—133.
256. Кучкина Е. Д., Горбенко Ф. П., Назарова Н. Н.— Методы анализа хим.
реактивов и препаратов, 1973, вып. 21, с. 285—286.
257. Кэй Дж., Лэби Т. Таблицы физических и химических постоянных. М.:
Физмйтгиз, 1962. 247 с.
258. Лаврова Е. А., Кузнецова А. В.— Завод, лаб., 1965, т. 31, с. 50—54.
259. Лазебная Г. В., Москальчук Э. К., Полева Г. В.— Методы анализа'хим.
реактивов и препаратов, 1971, вып. 20, с. 11—13.
260. Лайтинен Г. А. Химический анализ. М.: Химия, 1966. 656 с.
261. Ларин Н. В., Андон В. М.— Тр. по химии и хим. технологии, Горький,
1975, вып. 1 (40), с. 35—37.
262. Лебедев В. И.— Журн. аналит. химии, 1959, т. 14, с. 283—287.
263. Лебедев В. И.— Журн. аналит. химии, 1969, т. 24, с. 337—341.
264. Лебедев В. И., Вайнштейн Э. Е.— Журн. аналит. химии, 1961, т. 16,
с. 124—128.
265. Лебедева М. И., Исаева Б. И.— Тр./Тамбов, ин-т хим. машиностроения,
1970, вып. 4, с. 105—108.
266. Левин Д. И., Запорожец А. С., Басин Е. В.— Тр./Гос. н.-и. керам,
ин-т, 1959, вып. 1, с. 52—66.
267. Лейпунская Д. И., Путятина Н. Д.— В кн.: Труды I Всесоюз. коорди-
нац. совещ. по активационному анализу. Ташкент: Фан, 1964, с. 87—90.
268. Лерман Г. М., Корнеев В. А., Стоянова Т. К.— Завод, лаб., 1961,
т. 27, с. 838—839.
269. Лерман Р. В., Полонская Ц. С.— Методы анализа хим. реактивов и пре~
паратов, 1966, вып. 12, с. 9—16.
270. Лерман Р. В.,. Полонская Ц. С.— Методы анализа хим. реактивов и пре-
паратов, 1971, вып. 18, с. 70—72.
271. Лерман Р. В., Полонская Ц- С.— Методы анализа хим. реактивов и пре-
паратов, 1971, вып. 18, с. 73—75.
272. Ли Лань-син, Хуан Цзе-шэн.— Хуасюэ тунбао, Chemistry, 1964, № 1,
- р. 50—52; РЖХим, 1965, 1Г171.
273. Ливанов В. А., Горохов В. П., Голофаев Т. И., Малявкина В. П.— Изв.
АН СССР, Сер. физ., 1962, т. 26, с. 914—918.
274. Линдваль Р. В., Семина 3'. Г., Филимонова И. Г.— Тр./Казан. хим.-
теХнол. ин-т, 1957 (1958), вып. 22, ч. 1, с. 88—90.
208
275. Литвинова Н. Ф., Малышев В. И., Туровцева 3. М.— Тр./Комис. по>
аналит. химии АН СССР, 1960, т. 10, с. 97—102.
276. Лобанов Е. М., Акбаров У.— В кн.: Активационный анализ горных по-
род и других объектов. Ташкент: Фан, 1967, с. 140—146.
277. Лобанов Е. М., Валиева М. Г., Усманова М. М.— В кн.: Нейтронно-ак-
тивационный анализ. Ташкент: Фан, 1971, с. 72—75.
278. Лобанов Е. М., Хайдаров А.— Докл. АН ТаджССР, 1974, т. 17,-№ 4,
с. 28—35.
279. Майзельс Д. М., Тулъчук 3. Д., Гусев В. К.— Хим. волокна, 1973, № 5,
с. 70—7.1.
280. Макаров Д. Ф., Кукушкин Ю. Н., Ерошевич Т. А., Мошкова Н. Г.—
Журн. прикл. химии, 1971, т. 44, с. 2107—2109.
281. Маликова Е. Д., Котова В. И., Шимичева И. А., Ларикова Г. Г., Цио-
мо С. Н.— Журн. аналит. химии, 1979, т. 34, с. 805—807.
282. Маликова Е. Д., Кунин Л. Л., Урицкая Т. П.— Журн. аналит. химии,.
1977, т. 32, с. 88—91.
283. Маликова Е. Д., Чапыжников Б. А., Кунин Л. Л.— В кн.: Проблемы
технологии и коррозии металлов в натриевом теплоносителе и защитном
газе. Дрезден, 1977, т. 1, с. 180—185.
284. Малиновская Г. Н.— Сб. тр./Пензеи. с.-х. ин-т, 1958, вып. 2, с. 459—465.
285. Мальцева Н. С., Щегловски 3. — Радиохимия, 1970, т. 12, с. 622—626.
286. Малых В. Д., Серд М. А.— Оптика и спектроскопия, 1964, т. 16,
с. 368—369.
287. Мамаджанов Ф. И., Гулямов М.— Стекло и керамика, 1976, № 2,
с. 34—35.
288. Мамедов И. А., Эмирджанова А. А.— Азерб. хим. журн., 1963, № 3,
с. 113—117.
289. Мамедов И. А., Эмирджанова А. А.— Изв. вузов. Нефть и газ, 1963,
№ 11, с. 84.
290. Мамедов И. А., Эмирджанова А. А., Гусейнова 3. А.— Изв. вузов. Хи-
мия и хим. технология, 1968, т. 11, с. 1326—1328.
291. Манова Т. Г., Бобыкина Н. А.— Методы анализа хим. реактивов и пре-
паратов, 1973, вып. 21, с. 79—81.
292. Манова Т. Г., Красильщик В. 3., Рогинская Л. К., Яковлева А. Ф., Бе-
ликова И. П., Кузнецова О. С.— Методы анализа Хим. реактивов и пре-
паратов, 1973, вып. 21, с. 89—93.
293. Манова Т. Г., Рогинская Л. К., Астахова В. М.— Методы анализа хим.
реактивов й препаратов, 1973, вып. 21, с. 49—50.
294. Марголис Л. Д., Краченко А. П., Музыченко В. В.— Завод, лаб., 1962,
т. 28, с. 1072—1075.
295. Маркович И. С., Зеличенок С. Л., Филягина Н. А., Дзиомко В. М.,—
Журн. аналит. химии, 1973, т. 28, с. 227—230.
296. Маркович И. С., Зеличенок С. Л., Филягина Н. А., Чубарова Э. В., Дзи-
омко В. М.— Журн. аналит. химии, 1971, т. 26, с. 1097—1100.
297. Маркова Л. В., Клейнер К. Е.— Завод, лаб., 1959, т. 25, с. 144—145.
298. Марунина Я. И., Сухов Г. В., Ионов В. И., Прокофьева Н. К., Рейф-
ман М. Б.— В кн.: III Всесоюз. конф, по полупроводниковому карбиду
кремния, 1968. М., 1970, с. 258—267.
299. Масико — Якугаку дзасси., J. Pharmacol. Soc. Jap., 1956, vol. 76,
p. 1272—1275; РЖХим, 1957, № 48280.
300. Маслов Л. П., Карина Л. М., Цветков Н. А.— В кн.: Методы химиче-
ского анализа. М.: Химия, 1969, с. 180—183.
301. Матерова Е. А., Алагова 3. С., Шумилова Г. И., Ватлина Л. П., Сте-
колъникова И. К.— Вести. ЛГУ, 1980, вып. 22, с. 72—75.
302. Матерова Е.А., Моисеев В. В., Шмитт-Фогелевич С. П.— Журн. физ.
химии, 1959, т. 33, с. 893—901.
303. Матюшин А. Г., Горячева В. П., Суворова С. Н., Денисов И. Б.— Науч.
тр./Всесоюз. н.-и. и проект, ин-т тугоплав. металлов и твердых сплавов,
1976, № 16, с. 194—196.
304. Межевич А. Н.-— В кн.: Рентгеновские и эмиссионные спектральные ме-
тоды анализа. М.: 1975, с. 50—53.
209
-305. Миллер Ю. М., Попонова Р. В., Иванов Н. П., Певцов Г. А.-- В кн.:
Получение и анализ веществ особой чистоты. М.: Наука, 1966, с. 99—101..
306. Миллер Ю. М., Чупахин М. С., Зотов Н. П.— Журн. аналит. химии,
1968, т. 23, с. 1765—1768.
307. Минчевски Е., Данцевич Д., Вонсович С.— Acta chim. Acad. sci. hung.,
1962, vol. 33, p. 51—57.
308. РЖХим, 1962, 17Д83.
309. Мирзаев M. P., Хабиров H. А.— Изв. АН УзССР. Сер. физ.-мат. наук,
1972, № 1, с. 91—93.
310. Михайлов Г. И., Старчик Л. П.— В кн.; Ядерно-физические методы ана-
лиза вещества. М.: Атомиздат, 1971, с. 188—206.
311. Михайлова Т. П., Печорина С. И., Левченко Л. М.— Завод, лаб., 1973,
т. 39, с. 1463—1465.
312. Миякава Й., Камэмото Ю.— Nippon kagaku zasshi, J. Chem. Soc. Jap.
Pure Chem. Sect., 1962, vol. 83, p. 1029—1032; РЖХим, 1963, 10Г188.
.313 . Мойжес И. Б., Столярова И. А., Шувалова Н. И.— Тр./Всесоюз. н.-и.
геол, ин-т, 1966, т. 125, с. 186—194.
314. Мслева В. С., Пейзулаее Ш. И.— В кн.: Методы анализа веществ высокой
чистоты. М.: Наука, 1965, с. 331—335.
315. Молот М. А., Мустафин И. С., Иванова А. И., Петрикова К. Г., Ко-
валева В. С., Аграновская Л. Л.— В кн.: Передовые методы химической
технологии и контроля производства. Ростов н/Д: Изд-во Рост, ун-та, 1964,
с. 329—333.
316. Морозова И. Ф., Филимонова Г. П.— Тр. /ВНИИ золота и ред. металлов,
1977, т. 36, с. 137—148.
317. Мосели М., Тарасевич И. И.— Вести. МГУ. Сер. Химия II, 1966, № 4,
с. 92—95.
318. Москвин А. И., Калинин Н. Н., Гордон Л. А.— Журн. аналит. химии,
1977, т. 32, с. 1899—1903.
319. Мотодзима К., Банто С., Никаяма Р.— Бунсэки кики, Anal, and Inst-
rum., 1968, vol. 6, p. 567—572; РЖХим, 1969, 17Г161.
320. Мошкович Г. Н.— Тр./Ленингр. н-и. и проект, ин-т хим. пром-сти, 1971,
вып. 4, с. 276—278.
321. Музгин В. Н., Золотавин В. Л., Гаврилов Ф. Ф., Улыбышева Л. В.—
Тр./Всесоюз. н.-и. ин-т стандарт, образцов и спектр, эталонов, 1964, т. 1,
с. 127—133.
322. Мураками Т., Уэсуги К.— Rep. Himeji Inst. Technol., 1967, № 20A,
p. 79—83; РЖХим, 1968, 19Г185.
323. Муто Г., Нодзаки К., Нонака Т.— Бунсэки кагаку, Jap. Anal., 1971,
vol. 20, р. 180—187, РЖХим, 1971, 20Г59.
324. Мушко' О. Л.— В кн.: Методы анализа редкометальных минералов, руд
и горных пород. М., 1971, вып. 1, с. 66—74.
325. Мушко О. Л., Фабрикова Е. А.— В кн.: Методы анализа редкометальных
минералов, руд и горных пород. М., 1973, вып. 4, с. 38—42.
326. Мушко О. Л., Фабрикова Е. А.— В кн.: Редкие щелочные элементы.
Пермь: Политехн. ин-т, 1969, с. 425—429.
327. Наймарк Л. Э.— Тр./Ин-т металлургии и обогащения АН КазССР, 1968,
т. 31, с. 72—77.
328. Накаи Т., Камэмото Ю., Вэй Мин Тун.— Nippon kagaku zasshi, J.
Chem. Soc. Jap. Pure Chem. Sect., 1962, vol. 83, p. 1194—1197; РЖХим,
1963, 13Г169.
329. Натано H., Оно К., Фудзии Т.— Бунсэки кагаку, Jap. Anal., 1968,
vol. 17, р. 560—565; РЖХим, 1969, ЗГ148.
330. Некрасов Б. В. Курс общей химии. 14-е изд. М.: Госхимиздат, 1962,
668с.
331. Николаева К. И., Божевольнов Е. А., Нафикова Н. Г.— Методы анализа
хим. реактивов и препаратов, 1971, вып. 18, с. 19—20.
332. Никольская Е. Б., Шор Н. Б.— Сб. науч. тр./Ленингр. вет. ин-т, 1977,
вып. 49, с. 19—24.
333. Никольский Б. П., Матерова Е. А.— Вести. ЛГУ, 1975, вып. 22, с. 84—
92.
210
334. Никольский Б. П., Матерова Е. А. Ионоселективные электроды. Л.:
Химия, 1980, с. 48.
335. Никольский Б. П., Шульц М. М., Пешехонова Н. В.— Журн. физ. химии,
1958, т. 32, с. 19—26.
336. Никольский Б. П., Щульц М. М., Пешехонова Н. В.— Журн. физ. химии,
1958, т. 32, с. 262—269.
337. Никольский Б. П., Шульц М. М., Пешехонова Н. В.— Журн. физ. химии,
1959, т. 33, с. 1922—1927.
338. Ногина А. А., Кобяк Г. Г.— Учен. зап. Перм. ун-та, 1964, вып. 111,
с. 122—125.
339. Обозная Л. И., Николаев М. Ф-, Ефименко Н. П.— Тр./Н.-и. и проект,
ин-т основ, химии, 1976, т. 42, с. 74—77.
340. Оболенская Л. И.— Почвоведение, 1951, № 7, с. 420—425.
341. Оболенская Л. И.— Почвоведение, 1952, № 3, с. 278—286.
342. Оки Я., Оки С., Сибата X., Сакакибара Ю.— J. Geol.Soc. Jap., 1962,
vol. 68, р. 329—333; РЖХим, 1963, 7Г50.
343. Олейник Т. В.— Тр./Н.-и. и проект, ин-т основ, химии, 1976, т. 42,
с. 84—91.
344. Ольхоеич П. Ф., Воропай В. П.— Укр. хим. журн., 1974, т. 40, с. 1336.
345. Ониси К.— Бунсэки кагаку, Jap. Anal., 1967, vol. 16, р. 1152—1155,
РЖХим, 1969, 1Г61.
346. Орлов Д. С., Цикурина Н. Н.— Вести. МГУ. Сер. биология, почвоведение,
1962, № 2, С; 57—62.
347. Орлов Н. А., Победоносцев В. А Савельев Ю. А .— Журн. прикл. спектро-
скопии, 1976, т. 24, с. 214—218.
348. Остальцева Р. Н.— Тр./Урал. НИИ чер. металлургии, 1977, т. 30, с. 87—
88.
349. Останина Н. М., Задворнова Е. Г., Колечкова А. Ф.— Тр./Всесоюз. ин-т
н.-и. и проект, работ огнеупор, пром-сти, 1974, № 2 (45), с. 102—108.
350. Ошуркова О. В., Лядов Н. С., Кожуркина И. А.— Журн. прикл. химии,
1973, т. 46, с. 776—779.
351. Павлюченко М. М., Ульянова Т. М.— В кн.: Калийные соли и методы их
переработки. Минск: Изд-во АН БССР, 1963, с. 126—133.
352. Панталер Р. П., Алфимова Л. Д., Булгакова А. М.— Журн. аналит.
химии, 1975, т. 30, с. 1584—1589.
353. Панталер Р. П., Пуляева И. В.— В кн.: Монокристаллы и техника. Харь-
ков: ВНИИ монокристаллов, 1976, вып. 14, с. 196—199.
354. Панченков Г. М., Горшков В.И., Слоеецкий В. И.— Кинетика и катализ,
1963, т. 4, с. 82—87.
355. Пасоеская Г. Б.— Изв. АН ТССР. Сер. физ.-техн., хим. и геол, наук,
1960, № 3, с. 57—63.
356. Певцов Г. А.— Тр./Всесоюз. н.-и. ин-т хим. реактивов, 1951, вып. 20,
с. 233—236.
357. Певцов Г. А.— Тр./Всесоюз. н.-и. ин-т хим. реактивов, 1956, вып. 21,
с. 62—63.
358. Певцов Г. А., Красильщик В. 3.— В кн.: Прикладная спектроскопия. М.:
Наука, 1969, т. 1, с. 243—246.
359. Певцов Г. А., Красильщик В. 3.— Журн. аналит. химии, 1964, т. 19,
с. 1106—1109.
360. Певцов Г. А., Красильщик В. 3., Манова Т. Г.— Методы анализа хим.
реактивов и препаратов, 1969, вып. 16, с. 10—25.
361. Певцов Г. А., Красильщик В. 3., Яковлева А. Ф.— Методы анализа хим.
реактивов и препаратов, 1969, вып. 16, с. 25—27.
362. Певцов Г. А., Красильщик В. 3., Яковлева А. Ф.— Методы анализа хим.
реактивов и препаратов, 1969, вып. 16, с. 70—71.
363. Певцов Г. А., Красильщик В. 3., Яковлева А. Ф.— Методы анализа хим.
реактивов и препаратов, 1969, вып. 16, с. 76—78.
364. Певцов Г. А., Красильщик В. 3. Яковлева А. Ф.~ Методы анализа хим.
реактивов и препаратов, 1969, вып. 16, с. 78—80.
365. Певцов Г. А., Красильщик В. 3., Яковлева А. Ф. Методы анализ.а хим.
реактивов и препаратов, 1971, вып. 18, с. 55—58.
21:1
366. Певцов Г. А., Красильщик В. 3., Яковлева А. Ф.— В кн.: Спектроскопия
атомов и молекул. Киев: Наук, думка, 1969, с. 56—58.
367. Певцов Г. А., Красильщик В. 3., Яковлева А. Ф.— Журн. аналит. химии,
1970, т. 25, с. 580—581.
368. Певцов Г. А ., Красильщик В. 3., Яковлева А . Ф.— Завод, лаб., 1969, т. 25,
с. 1340—1343.
369. Певцов Г. А ., Красильщик В. 3., Яковлева А . Ф., Егорова И. И.— Методы •
анализа хим. реактивов и препаратов, 1969, вып. 16, с. 53—55.
370. Певцов Г. А ., Манова Т. Г.— Тр./Всесоюз. н.-и. ин-т хим. реактивов и осо-
бо чистых хим. веществ, 1966, вып. 29, с. 7—22.
371. Певцов Г. А., Манова Т. Г., Красильщик В. 3.— Методы анализа хим. ре-
активов и препаратов, 1969, вып. 16, с. 74—76.
372. Певцов Г. А., Манова Т. Г., Рагинская Л. К., Бавыкина Н. А.— Методы
анализа хим. реактивов и препаратов, 1969, вып. 16, с. 84—86.
373. Певцов Г. А., Манова Т. Рагинская Л. К., Герасимова Т. С.— Методы
анализа хим. реактивов и препаратов, 1969, вып. 16, с. 71—73.
374. Певцов Г. А ., Манова Т.Г.,РагинскаяЛ.К., ГожаяЛ.Д., Хасянова С. Ф.—
Методы анализа хим. реактивов и препаратов, 1969, вып. 16, с. 101—103.
375. Певцов Г. А., Манова Т.Г., Рагинская О. К., Лебединская К. Л.— Методы
анализа хим. реактивов и препаратов, 1969, вып. 16, с. 63—64.
376. Певцов Г. А., Рагинская Л. К., Манова Т. Г., Красильщик В. 3.— Завод,
лаб., 1969, т. 35, с. 1073—1074.
377. Певцов Г. А ., Рагинская Л. К., Манова Т. Г., Лебединская К. Л.— Методы
анализа хим. реактивов л препаратов, 1969, вып. 16, с. 64—66.
378. Певцов Г. А., Рагинская Л. К., Манова Т. Г., Сотникова В. С., Астахо-
ва В. И.— Методы анализа хим. реактивов и препаратов, 1971, вып. 18,
с. 52—54.
379. Певцов Г. А., Широкова М. Д., Манова Т. Г., Михеева И. П.— Методы
анализа хим. реактивов и препаратов, 1966, вып. 12, с. 25—28.
380. Певцов Г. А., Широкова М. Д., Михеева И. П.— Методы анализа хим.
реактивов и препаратов, 1969, вып. 16, с. 66—68.
381. Певцов Г. А., Широкова М. Д., Михеева И. П.— Методы анализа хим.
реактивов и препаратов, 1969, вып. 16, с. 68—70.
382. Педанова В. Г.— В кн.: Проблемы большой металлургии и физической
химии новых сплавов:. М.: Наука, 1965, с. 290—294.
383. Пейзулаев Ш. И.— Журн. аналит. химии, 1960, т. 15, с. 623.
384. Пейзулаев Ш. И.— Завод лаб., 1958, т. 24, с. 723—724.
385. Пейзулаев Ш. И.— Тр./Комис. по аналит. химии АН СССР, 1960, т. 12,
с. 108, 151, 331.
386. Пейзулаев Ш. И., Попова Л. К., Слюсарева Р. Л.— Завод, лаб., 1960,
т. 26, с. 552—553.
387. Песочинская Э. М.— Завод, лаб., 1952, т. 18, С. 1375—1376.
388. Петренко О. Н., Заворотнова Г. И.— Учен. зап./ЦНИИ оловян. пром-
сти, 1970, № 3, с. 80—83.
389. Писарев В. Д., Иванова Т. А.— Завод, лаб., 1951, т. 17, с. 497—498.
390. Писарев В. Д., Иванова Т. А.— Завод, лаб., 1954, т. 20, с. 586—587.
391. Пластинин В. В.— Завод, лаб., 1961, т. 27, с. 856—857.
392. Подгорный Л. Н., Павлова Н. А ., Терехов П. В.— Завод, лаб., 1976,
т. 42, с. 789—790..
393. Покровская Е. И., Терещенко А. П.— В кн.: Иониты и ионный обмен.
М.: Наука, 1966, с. 103—107.
394. Полева Г. В., Ку стае В. Л.— Методы анализа хим. реактивов и препара-
тов, 1971, вып. 20, с. 35.
395. Полева Г. В., Кустас В. Л.— Методы анализа хим. реактивов и препара-
тов, 1971, вып. 20, с. 42.
396. Полева Г. В., Кустас В. Л., Шепета Н. Г.— Методы анализа хим. реак-
тивов и препаратов, 1971, вып. 20, с. 43—44.
397. Полуэктов Н. С. Методы анализа по фотометрии пламени. М.: Химия,
1967. 307 с.
398. Полуэктов Н. С., Мешкова С. Б., Никонова М. И.— Завод, лаб. 1969,
т. 35, с. 166—169.
212
399. Полуэктов Н. С., Мешкова С. Б., Полуэктова Е. Н. Аналитическая химия
лития. М.: Наука, 1975. 203 с.
400. Полуэктов Н. C.t Никонова М. П.— Завод, лаб., 1958, т. 24, с. 528—531.
401. Полуэктов Н. С., Никонова М. Н.— Завод, лаб., 1959, т. 25, с. 263—-268.
402. Полуэктов И- С., Никонова М. П., ВиткунР.А .— Журн. аналит. химии,
1958, т. 13, с. 48—55.
403. Поляков П. МРусанов А. К., Блох И. М.— Завод.лаб., 1957, т. 23,
с. 1320—1323.
404. Полякова Р. С., Соловьева 3. В.— Сб. тр./ЦНИИ чер. металлургии, 1964,
вып. 37, с. 118—120.
405. Попков К. К., Кудрявцева А . С.— В кн.: Методы анализа веществ высокой
чистоты. М.: Наука, 1965, с. 486—487.
406. Попова Т. В., Михайленко А. С.— Завод, лаб., 1978, т. 44, с. 1093—1094.
407. Прайс В. Аналитическая атомно-абсорбционная спектроскопия. М.:
Мир., 1976. 355 с.
408. Прудников Е. Д.— В кн.: Редкие щелочные элементы. Пермь: Политехи,
ин-т, 1969, с. 430—432.
409. Прудников Е. Д.— Вести. ЛГУ. Биология, 1965, № 10, с. .125—127.
410. Прудников Е. Д.— Журн. аналит. химии, 1965, т. 20, с. 40—43.
411. Прудников Е. Д.— Журн. аналит. химии, 1972, т. 27, с. 2129—2133.
412. Прудников Ё. Д.— Журн. аналит. химии, 1972, т. 27, с. 2327—2332.
413. Прудников Е. Д.— Журн. прикл. спектроскопии, 1971, т. 14, с. 145—147.
414. Прудников Е. Д.— Завод, лаб., 1972, т. 38, с. 183—184.
415. Прудников Е. Д., Шапкина О. С.— Журн. прикл. спектроскопии, 1978,
т. 28, с. 139—142.
416. Райхбаум Я. Д., Костюкова Е. С.— В кн.: Спектральный анализ в гео-
логии и геохимии. М.: Недра, 1967, с. 6—10.
417. Резчиков В. И., Голубев А. Н.— Тр./НИИ ядер, физики, электроники и
автоматики при Том. политехи, ин-те, 1977, № 7, с. 81—84.
418. Розова О. Ф., Базыкина Е. Н., Смагунова А . НЖаркова Г. А., Заруби-
на Г. Д.~ В кн.: Аппаратура и методы рентгеновского анализа. Л.:
Машиностроение, 1975, вып. 17, с. 102—111.
419. Рубинштейн М. М., Григорьев И. Г., Узнадзе Э. Д., Гельман О. Я.,
. Лашхи Б. А.— Сообщ. АН ГрузССР, 1960, т. 24, с. 683—690.
420. Руднееский Н. К., Демарин В. Т.— В кн.: Сб. VII. Урал. конф, по спект-
роскопии, 1971, Свердловск, 1971, вып. 1, с. 205—207.
421. Руднееский Н. К., Демарин В. Т., Склемина Л. В., Ефремов А. В.— Тр.
по химии и хим. технологии, Горький, 1975, вып. 1(40), с. 103—104.
422. Руднееский Н. К., Демарин В. Г., Склемина Л. В., Туманова А. Н.—
Тр. по химии и хим. технологии. Горький, 1975, вып. 7(40), с. 106—107.
423. Руднееский Н. К., Максимов Д. Е., Лазарева Л. П.— Журн. аналит. хи-
мии, 1974, т. 29, с. 1422—1424.
424. Русанов А. К. Основы количественного спектрального анализа руд и ми-
нералов. М.: Недра, 1978. 400 с.
425. Русанов А . К., Гусяцкая Э. В., Ильясова Н. В.— Завод, лаб., 1950, т. 16,
с. 447-453.
426. Рустамов Р., Хатамов Ш., Орестова И. И.— В кн.: Активационный ана-
лиз в народном хозяйстве. Ташкент: Фан, 1974, с. 107—109.
427. Рисе И. Г., Пилус Э. Л.— Журн. аналит. химии, 1957, т. 12, с. 64—69.
428. Рысс И. Г., Пилус 3. Л.— Завод, лаб., 1958, т. 24, с. 1349—1352.
429. Рысс И. Г., Пилус Э. Л.— Тр./Днепропетр. металлург, ин-т, 1961,вып. 44,
с. 197—300.
430. Рябчиков Д. И., Герлит Ю. Б.— В кн.: Методы определения и анализа
редких элементов. М.: Изд-во АН СССР, 1961, с. 629—662.
431. Рябинин А. И., Афанасьев Ю. А., Аксенов И. А.— Журн. аналит.химии,
1966, т. 21, с. 1258—1260.
432. Сазонова В. А., Леонов В. Н.— Журн. аналит. химии, 1959, т. 14, с. 483—
484.
433. Саэки, Фудзии — Кэмикару эндзиниярингу, Chem. Engng., 1957, vol. 12,
р. 814—818; РЖХим, 1958, № 81283.
213
434. Свердлина О. А., Кузнецов Ю. И., Кузовлев И. А.— В кн.: Получение ь
анализ веществ особой чистоты М.: Наука, 1978, с. 233—235.
435. Свердлина О. А., Кузнецов Ю. И., Кузовлев И. А.— Завод, лаб., 1975»
т. 41, с. 1344—1345.
. 436. Свешников А. Т., Навасартоеа К. А.— Завод, лаб., 1941, т. 10, с. 425—
437. Севрюгова Н. И., Ионов В. П., Атанов И. Г., Кудинова А. А., Жаворон-
ков Н. М.— Вкн.: Методы получения и анализа веществ особой чистоты.
М.: Наука, 1970, с. 64—70.
438. Семененко К. А., ГулъкоН.И.,ЛобановФ.И.— Рукописьдеп.в ВИНИТИ»
№ 2525—74.
439. Семененко К. А ., Тарасевич Н. И., Гулько Н. И., Пронина Е. В.— Вести.
МГУ. Сер. 2, Химия, 1974, № 6, с. 690—692.
440. Серебрякова Г. В., Божевольнов Е. А., Монахова А. Г.— Методы анализа
хим. реактивов и препаратов, 1969, вып. 16, с. 160.
441. Сериков Ю. А., Пальникоеа Т. И-—Журн. аналит. химии, 1983, т. 38,
с. 162—164.
442. Сиката Э.— J. Atom. Energy Soc. Jap., 1964, vol. 6, p. 579—582; РЖХим,
1965, 13Г38.
443. Симова Л., Данчееа Р., Пешкова Л., Наумова С.— Б юл. науч.-техн. ин-
формации Нипроруда, 1973, № 1, с. 65—69; РЖХим, 1974, 17Г176.
444. Скобец В. Д., Абарбарчук И. Л., Скобец Е. М.— Науч. докл. высш, шко-
лы. Биол. науки, 1962, № 3, с. 189—193.
445. Скобец В. Д., Абарбарчук И. Л., Скобец Е. М-— Укр. хим. журн., 1962,
т. 26, с. 251—259.
446. Смахтин Л. А., Филиппова Н. В., Бровцын В. К.— В кн.: Труды 1-го
Всесоюз. совещ. по активационному анализу, 1962. Ташкент: Фан, 1964,
с. 63—68.
447. Смирное В. Д., Овчинникова В. Д., Крестов Г. А.— Изв. вузов. Химия
и хим. технология, 1981, т. 24, с. 440—444.
448. Соловьев Е. А., Божевольнов Е. А., ЛебедеваН.А.— Вкн.: Методы анализа
химических реактивов и препаратов. М.: ИРЕА, 1973, вып. 21, с. 167—
169.
449. Солодовник С. М.— Изв. АН СССР. Сер. физ., 1955, т. 19, с. 182—184.
450. Сотникова Н. П., Романович Л. С., Пейзулаев Ш. И., Карабаш А. Г.—
Тр./Комис, по аналит. химии АН СССР. 1960, т. 12, с. 151—159.
451. Спицын В. И., Глазунов М. П., Кодочигов П. Н., Ионов В. П.— Журн.
аналит. химии, 1963, т. 18, с. 1272—1273.
452. Способная А. И.— В кн.: Сборник работ студ. науч, о-ва Ленингр. тех-
нол. ин-та пищ. пром-сти, 1956, вып. 1, с. 74—77.
453. Станчев Н.— Химия и индустрия (НРБ), 1966, т. 38, с. 311—314.
454. Старобинец Г. Л., Мартинчик Г. С.— Журн. аналит. химии, 1961, т. 16,
с. 538—543.
455. Старобинец Г. Л., Седнев М. П., Дубовик Т. Л.— Тр./Комис. по аналит.
химии АН СССР, 1965, т. 15, с. 323—330.
456. Столяров К. П.— Журн. аналит. химии, 1952, т. 7, с. 195—200.
457. Столяров К. П. Методы люминесцентного открытия и определения не-
органических ионов. Л.: ЛДНТП, 1961. 46 с.
458. Столярова И. А., Акельева А. С.— Тр./Всесоюз. н.-и. геол, ин-т, 1966,
т. 125, с. 179—185.
459. Столярова И. А., Мойжес И. Б.— Тр./Всесоюз. н.-и. геол, ин-т, 1966,
т. 125, с. 157—169.
460. Стромберг А. Г., Каплин А. А.~ Изв. СО АН СССР. Сер. хим. наук,
1975, вып. 2, № 4, с. 58—73.
461. Стромберг А. Г., Колпакова И. А., Каплин А. А., Немтинова Г. М.,
Белоусова Н. И.— Методы анализа хим. реактивов и препаратов, 1971,
вып. 20, с. 78—81.
462. Супин Г. С.— В кн.: Теория и практика полярографического анализа.
Кишинев: Штиинца, 1962, с. 348—351.
463. Супин С. Г.— Журн. аналит. химии, 1962, т. 17, с. 258—259.
464. Супин С. Г.— Журн. аналит. химии, 1963, т. 18, с. 318—322.
214
•465. Сявцилло С. В.— Завод, лаб. 1952, т. 18, с. 407—408.
466. Такамото С., Окамото М.— Nippon kagaku zasshi, J.Ghem. Soc. Jap.
Pure Chem. Sect., 1962, vol. 83, p. 1102—1104; РЖХим, 1963, 18Г54.
467. Такэути Ц., Исии Д.^~ Kogyo kagaku zasshi, J. Chem. Soc. Jap. Industr.
Chem. Sect. 1964, vol. 67, p. 1835—1838; РЖХим, 1965, 11Г148.
468. Тарасевич H. И., Чеботарев В. Е., Бочкарева И. И.— В кн.: Оптические
методы^^контроля химического состава материалов. М.: МДНТП, 1974,
469. Тарасевич Н. И., Чеботарев В. Е., Семененко К. А., Бочкарева И. И.—
В кн.: Новые методы контроля материалов на остаточные элементы и
микропримеси. М.: МДНТП, 1975, с. 56—60.
470. Тарасянц Р. РБондаревская Е. А., Шелудякова Л. Л.— Журн. аналит.
химии, 1970, т. 25, с. 1876—1879.
471. Тарковская И. А., Гращенкова Л. Н., Завьялов А. Н., Пичугин Б. И.,
Желин Б. И.— Укр. хим. журн., 1976, т. 42, с. 698—702.
472. Тенчева Р., Салчева М.— Годипшик н.-и. и проект, ин-та рудодоб. и
обогатит., 1968, т. 6, с. 93—99; РЖХим, 1969, 24Г121.
473. Тимофеев В. И., Загорских Г. И.— В кн.: Методы и средства аналити-
ческого контроля в калийной промышленности. Л.: ЛДНТП, 1979, с. 39—
474. Титов В. И.-, Цветков Н. А ., Рысев А . П.— Ассортимент реактивов на ли-
тий и натрий. М.: НИИТЭХим, 1969, с. 27—38.
475. Тихомиров И. А., Гофман Э. Р., Тихонов Г. С., Бокор М. И. — Журн.
физ. химии, 1976, т. 50, с. 485—487.
476. Тихонов В. Н., Гранкина М. Я.— Журн. аналит. химии, 1963, т. le-
с. 900—902.
477. Ткаленко Д. А., Сажин С. В., Власова М. А., Антропов Л. И.— Жури,
аналит. химии, 1978, т. 33, с. 1281—1284.
478. Товбин М. В., Дятлоеицкая Ф. Г.— Укр. хим. журн., 1952, т. 18, с. 657—
659.
479. Третъяк 3. А., Шкоп Т. Н.— РЖХим, 1981, 23Г196Деп.
480. Трикит И. А ., Макаренко В. С., Солдатова А . Г., Галушко Е. Г., Бубырь
С. И., Тула Л. А., Карнаухова 3. А,— Сб. тр./Всесоюз. н.-и. и проект,
ин-т титана, 1968, т. 2, с. 349—355.
481. Тренева Н. В., Цепин А. И.— В кн.: Эксперимент в области технологии
минералообразования. М.: Наука, 1975, с. 51—59.
482. Tpehan Миленко Б.— Гласник хем. друштва, 1956, т. 21, с. 222—228;
РЖХим,1957, № 74660.
'483 . Угнячев Н. Я., Тютюнникова Т. И.— Тр./ВНИИ содовой пром-сги, 1956,
т. 9, с. 113—116.
484. Угнячев Я. Я., Тютюнникова Т. И.— Тр./ВНИИ содовой пром-сти, 1956,
т. 9, с. 117—119.
485. Угнячев Н. Я., Тютюнникова Т. И., Дубровина Т. П.— Тр./НИИ основ,
химии, 1958, т. 11, с. 332—333.
486. Ульянова Т. М., Павлюченко М. М.— Изв. АН БССР. Сер. хим. наук,
1967, № 2, с. 10—14.
487. Ульянова Т. М., Павлюченко М. М.— Изв. АН БССР. Сер. хим. наук,
1967, № 4, с. 25—28.
488. Ульянова Т. М., Павлюченко М. М., Акулович В. М.— Изв. АН БССР,
Сер. хим. наук, 1978, № 2, с. 61—64.
489. Умаров М. У., Исаматов Э. Е.— Докл. АН УзССР, 1977, № 2, с. 24—25.
490. Умаров М. У., Кавтанюк В. Э., Хасанов А. С.~ В кн.: Вопросы гидро-
геохимии Средней Азии. Ташкент: Фан, 1974, с. 79—84.
491. Устимов А. М., Тембер Г. А.— Завод, лаб., 1969, т. 35, с. 1440.
492. Устимов А. М., Тембер Г. А., Чалков Н. Я.— Завод, лаб., 1971, т. 37,
с. 660—662.
493. Устимов А. М., Чалков И. Я.— Завод, лаб., 1969, т. 35, с. 177—178.
494. Устимов А. М., Чалков. Н. Я.— Завод, лаб., 1971, т. 37, с. 931—932.
495. Устимов А. М., Чалков Н. Я., Яковлева А. В.— Завод, лаб., 1965, т. 31,
с. 1090.
215
496. У Цаин-гуан, Сюй Гуан-сянъ.— Kexue tongbao, Scientia, 1959, № 10,
р. 330; РЖХим., 1960, № 2604».
497. Фабрикоеа Е. А., Мушко О. П.— В кн.: Новые методы исследования по-
анализу редкометальных минералов, руд и горных пород. М.: Наука, 1970,
с. 83—89.
498. Файгль Ф., Ангер В. Капельный анализ неорганических веществ. М.г
Мир, 1976. Т. 1. 390 с.
499. Факеееа О. А., Божевольное Е. А.— Методы анализа хим. реактивов и
препаратов, 1971, вып. 18, с. 12—13.
500. Факеееа О. А., Божевольное Е. А., Комлева В. И.— Методы анализа хим.
реактивов и препаратов, 1973, вып. 21, с. 164—166.
501. Факеееа О. А., Божевольнов Е. А., Шмакова Н. И.— Методы анализа
хим. реактивов и препаратов, 1973, вып. 21, с. 158—160.
502. Фиалкое Ю. Я. Применение изотопов в химии и химической промышлен-
ности. Киев: Технша, 1975, с. 167.
503. Фиделъман Б. М., Золотоеицкая Э. С.— В кн.: Методы анализа галоге-
нидов щелочных и щелочноземельных металлов высокой чистоты. Харь-
ков: ВНИИ монокристаллов, 1971, ч. 2, с. 11—14.
504. Фиделъман Б. М., Золотоеицкая Э. С., Бондарева Н. В.— В кн.: Мето-
ды анализа галогенидов щелочных и щелочноземельных металлов высокой
чистоты. Харьков: ВНИИ монокристаллов, 1971, ч. 2, с. 15—16.
505. Фиделъман Б. М., Чепурная В. Г., Золотоеицкая Э. С.— В кн.: Методы
анализа галогенидов щелочных и щелочноземельных металлов высокой
чистоты. Харьков: ВНИИ монокристаллов, 1971, ч. 2, с. 19.
506. Физико-химические свойства элементов: Справочник/Под ред. Г. В. Сам-
сонова. Киев: Наук, думка, 1965. 807 с.
507. Филатов Т. Я., Викторов Б. В., Чернова М. А., Ярмаркин В. К.—
Электрон, техника. Науч.-техн. сб. «Радиодетали», 1972, вып. 1(26), с. 55—
58.
508. Филиппова Я. В., Смахтин Л. А., Миглина Н. В.— Журн. аналит. хи-
мии, 1970, т. 25, с. 2327—2331.
509. Филягина Я. А., Агзибекоеа Л. О.— Тр./ВНИИ хим. реактивов и особо
чистых хим. веществ, 1979, вып. 41, с. 165—166.
510. Филягина Я. А., Маркович Я. С., Дзиомко В. М., Зеличенок С. Л.*
Блохина Л. Я., Агзибекоеа Л. О.— Тр./ВНИИ хим. реактивов и особо
чистых хим. веществ, 1979, вып. 41, с. 3—9.
511. Фишкоеа Я. Л.— Завод, лаб., 1957, т. 23, с. 591—592.
512. Фишкоеа Я. Л., Комлев Л. Я.— Завод, лаб., 1960, т. 26, с. 566—567.
513. Флис Я. Е., Туманова Т. А., Зубова Г. М., Никитина Я. Ф.— Тр./Ле-
нингр. технол. ин-т целлюлозно-бум. пром-сти, 1964, вып. 13, с. 57—61.
514. Фраткин 3. Г., Мошкович Г. Я.— Методы анализа хим. реактивов и
препаратов, 1966, вып. 12, с. 5—6.
515. Фраткин 3. Г., Мошкович Г. Я.— Методы анализа хим. реактивов и
препаратов, 1966, вып. 12, с. 7—8.
516. Фраткин 3. Г., Мошкович Г. Я., Филиппова Ж. А.— Завод, лаб., 1965,
т. 31, с. 1090—1091.
517. Фраткин 3. Г., Шебунин В. С.— В кн.: Получение и анализ веществ
особой чистоты. М.: Наука, 1966, с. 142—146.
518. Фрегер С. В., Иванова Е. И.— Методы анализа хим. реактивов и препа-
ратов, 1973, вып. 21, с. 152—154.
519. Фудзинага Т.,Нисида Т.— Nippon kagaku zasshi, J. Chem. Soc. Jap. Pure
Chem. Sect., 1964, vol. 85, p. 547—549; РЖХим, 1965, 20Г68.
520. Фукадзава Ц., Кано С., Оно X., Мидауике А.— Бунсэки кагаку, Jap.
Anal., 1970, vol. 19, р. 1417—1423; РЖХим, 1971, 11Г191.
521. Фукасава Ц., Ядзима К., Кано С., Мидзуикэ А.— Бунсэки кагаку, Jap.
Anal., 1971, vol. 20, р. 193—200; РЖХим, 1971, 19Г128.
522. Фукисима, Ивата, Кума, Сигэмото — Бунсэки кагаку, Jap. Anal., 1956,
vol. 5, р. 704—707.
523. Харизаное Ю., Трендафилоеа Я.— Изв. хим. Бълг. АН, 1975 (1976), т. 8,
с. 751—755; РЖХим, 1976, 21Г189.
216
524. Химай Н.А., Артюхин П.И., Каплин А. А.— Изв. /Том. политехн.
ин-т, 1975, т. 197, с. 49—50.
525. Хольнов Ю. В., Чечев В, П., Камынов Ш. В., Кузьменко Н. К., Недо-
весов В. Г. Характеристики излучений радиоактивных нуклидов, приме-
няемых в народном хозяйстве: Оцененные данные. М.: Атомиздат, 1980,
с. 19—34.
526. Хоригути Я., Окано Я., Судзуки Т.— РЖХим, 1976, 14Г199.
527. Хосокава И., Хонда С., Масаки И.— Бунсэки кагаку, Jap. Anal., 1964,
vol. 13, р. 1044—1045; РЖХим, 1965, 19Г36.
528. Цывина Б. С,— Завод, лаб., 1949, т. 15, с. 139—142.
529. Чайка М., Сабо Э.— В кн.: Лунный грунт из Моря Изобилия. М.: Наука,
1974, с. 287—289.
530. Чалков Я. Я., Юделевич И. Г., Устимова А. М., Яковлева А. В.—
Журн. аналит. химии, 1973, т. 28, с. 1471—1475.
531. Чапыжников Б. А., Бежанов X., Маликова Е. Д., Кунин Л. Л., Само-
ток В. Н.— Журн. аналит. химии, 1973, т. 28, с. 57—60.
532. Чапыжников Б. А., Маликова Е. Д., Евжанов X., Самоток В. И.—
В кн: Активационный анализ в народном хозяйстве. Ташкент: Фан, 1974,
с. 120—123.
533. Чеботарев В. Е., Прилепская В. А.— В кн.: Определение микропримесей.
М.: МДНТП, 1968, № 1, с. 39—47.
534. Чернигов Ю. А., Черкашина Т. В., Ноткина М. А., Петрова Е. И.,
Меньшова Н. Я., Луговская В. И., Горянская Г. П.— Журн. аналит.
химии, 1966, т. 21, с. 714—717.
535. Черницкая Р. Е.— Завод, лаб., 1951, т. 17, с. 441—443.
536. Чернова А. И.— Завод, лаб., 1967, т. 33, с. 972.
537. Черных Л. Н., Булычева И. Б.— Тр./Урал. н.-и. хим. ин-т, 1973, вып. 30,
с. 88—93.
538. Чешев К. С.— Журн. аналит. химии, 1954, т. 9, с. 239—244.
539. Чирков С. К.— Журн. аналит. химии, 1959, т. 14, с. 370—372.
540. Шабанов А. А., Горшков В. И., Панченков Г. М.— Журн. физ. химии,
1962, т. 36, с. 1695—1697. '
541. Шабанова Т. М., Лазарева Л. П., Максимов Л. Е., Рудневский Н. К.—
Тр. по химии и хим. технологии, Горький, 1973, вып. 3(34), с, 79—80.
542. Шайдурова Г. В.— Методы анализа хим. реактивов и препаратов, 1971,
вып. 20, с. 14—16.
543. Шамаев В. И., Дьячкова В. Г., Каретникова И. Л.— Журн. аналит.
химии, 1981, т. 36, с. 1284—1289.
544. Шамсиев С. М., Абдурахимова М. К.— Узб. хим. журн., 1971, № 5,
с. 12-14.
545. Шапкина Ю. С., Прудников Е. Д.— Изв. вузов. Химия и хим. техно-
логия, 1975, т. 18, с. 889—892.
546. Шарапова Г. Н., Степина Н. И.— Тр./Урал. н.-и. хим. ин-т, 1977, вып. 43,
с. 69—72.
547. Шафранская В. А.— Бумаж. пром-сть, 1969, J'S 6, с. 12—13.
548. Шманенкова Г. И., Фирсов В. И., Щелкова В. П., Шулепников М. Н.—
Журн. аналит. химии, 1973, т. 28, с. 323—327.
549. Шмуляковский Я. Э.— В кн.: Методы исследования продуктов нефтепе-
реработки и нефтехимического синтеза. Л.: Гостоптехиздат, 1962, с. 11—16.
550. Шпак А. В., Йсаенко Н. И.— Методы анализа хим. реактивов и препара-
тов, 1971, вып. 20, с. 101—103.
551. Щипакина Н. К., Немировская Е. М., Сенявин М. М.— Журн. аналит.
химии, 1957, т. 12, с. 70—77.
552. Элвелл В. Т., Вуд Д. Ф. Анализ новых металлов. М.: Химия, 1970, с. 91.
553. Элек А., Сабо Э.— Acta chim. Acad. sci. hung., 1968, vol. 57, p. 385—389.
554. Эпштейн T. Б.— Тр./НИИ по удобрениям и инсектофунгицидам, 1978,
вып. 232, с. 44—47.
555. Эпштейн Т. Б.— Тр./НИИ по удобрениям и инсектофунгицидам, 1978,
вып. 232, с. 47—48.
556. Юделевич И. Г., Ковалева В. Г., Левитина А. Л.— Завод лаб., 1956,
т. 22, с. 1310—1312.
217
557. Юделевич И. Г., Кондратенко Л. А., Протопопова II. П.— Изв. СО АН
СССР. Сер. хим. наук, 1974, вып. 5, №12, с. 85—89.
558. Юделевич И. Г., Кустас В. Д., Illenema Н. Г., Мамонтова Л. И.—
Журн. аналит. химии, 1965, т. 21, с. 542—547.
559. Юделевич И. Г., Кустас В. Д., Яковлева Я. А., Полева Г. В.— В кн.:
Спектроскопия: Методы и применения. М.: Наука, 1973, с. 92—94.
560. Юделевич И. Г., Шелпакова И. Р., Сосновская Т. И., Бортник Л. С.—
Завод, лаб., 1959, т. 25, с. 959—961.
561. Юнусов 3. Т., Агиевский Д: А.— Химия и технология топлив и масел,
1975, № 11, с. 46—48
562. Якимова Е. Е., Бибин В. Ф., Богатиков Б. Ф., Дад^кин А. А., Черно-
ва А. И.— Изв. АН СССР. Неорган. материалы; 1979, т. 15, с. 1900—1901.
563. Яковлева Я. А., Кустас В. Л.— Методы анализа хим. реактивов и препа-
ратов, 1971, вып. 20, с. 56—57.
564. Яковлева И. А., Кустас В. Л., Юделевич И. Г.— Методы анализа хим.
реактивов и препаратов, 1971, вып. 20, с. 51—52.
565. Яковлева Я. А., Кустас В. Л., Юделевич И. Г.— Методы анализа хим.
реактивов и препаратов, 1971, вып. 20, с. 53.
566. Ямаути Ф., Ота-а И.— Бунсэки кагаку, 1968, vol. 17, р. 1384—1387;
РЖХим, 1969,. 10Г146.
567. Якубов А. М., Ризаев К. У., Саидахмедов У. С., Зинатуллин X. А.—
Узб. хим. журн., 1980, № 1, с. 15—17.
568. Ямамото Ю., Кумамару Т., Хаяси ЯОтани Ю., Муцунобу Ц.— Коге
кагакудзасси, J.Chem.Soc. Jap. Industr.Chem. Sect., 1971, vol. 74, p.779—
781; РЖХим, 1971, 22Г215.
569. Ян Бао-тун, Сян Ци-ган, Ху Минь-цзе, Се Цин-чжун, Чэнь Пэнфэй.—
Хуасюэ шицзе, 1966, vol. 20, т. 276—278; РЖХим, 1967, 11Г191.
570. Янковский В. Р., Грибанова Р. Я., Винокурова М. ЯМуратова А. X.—
В кн.: Методы и средства аналитического контроля в калийной промышлен-
ности. Л.: ЛДНТП, 1979, с. 75—82.
571. Яцимирский К. Б. Введение в бионеорганическую химию. Киев: Наук,
думка, 1976. 144 с.
572. Aasness Я.— Medd. Norsk parmac. selskap., 1957, bd 19, N 4, s. 53—63;
РЖХим, 1958, № 24884.
573. Abbey S.— Pap. Geol. Surv. Canada, 1970, N 23, p. 20; РЖХим, 1971,
1Г147.
574. Abe M.— Separ. Sci. and Technol., 1978, vol. 13, p. 347—365.
575. Abe M., Hayashi K.— Бунсэки кагаку, Jap. Anal., 1979, vol. 28, p. 700—
702; РЖХим, 1980, 8Г113.
576. Abramson M. B., Katzman B., Gregor M., Curci B.— Biochemistry, 1966,
vol. 6, p. 2207—2213.
577. Adamski L., В ouzyk J,,JozefowiczK.— Nukleonika, 1960, vol.5, s. 317—327.
578. Ainley D.— In: Chromatography. L., 1973, p. 26—28; РЖХим, 1975,
17Г37.
579. Akaza I.— Bull. Chem. Soc. Jap., 1966, vol. 39, p. 585—596; РЖХим,
1967, 19Г47.
580. Akimoto N., Hozumi А,—Бунсэки кагаку, Jap. Anal., 1972, vol. 21,
p. 1490—1497; РЖХим, 1973, 11Г74.
581. Al-Jobori S. MSzegedi S., Pazsit A.— Radiochem. and Radioanal. Lett.,
1977, vol. 30, p. 45—52.
582. Alberti G., Grassini G.— J. Chromatogr., 1960, vol. 4, p. 423—425.
583. Alkemade C. Th. J., Hollander T., Snelleman W., Ziegers P. J. Th.— Metal
vapours in flames. Oxford; N. Y. etc.: Pergamon Press, 1982.
584. Amiel S., PeisachM.— Anal. Chem., 1963, vol. 35, p. 1072—1076.
585. Amman I)., Pretsch E., Simon W.— Anal. Lett., 1974, vol. 7, p. 23—32.
586. Angot J.— Mikrochim. acta, 1959, N 3, S. 346—356.
587. Aras N. K., Zoller W. H., Gordon G. E., Lutz G. J.— Anal. Chem., 1973,
vol. 45, p. 1481—1490.
588. Arnikar H. J.— Nature, 1958, vol. 182, p. 1230.
589. Athavale V. T., Krishnan С. V.— J. Sci. and Industr. Res., 1962, vol. B21,
p. 339—340; РЖХим, 1963, 6Г52.
218
590. Avnl R.— Spectrochim. acta B, 1968, vol. 23, p. 619—634.
591. Avni R., Boukobza A.— Isr. J. Chem., 1968, vol. 6, N 25: РЖХим, 1969,
6Г115.
592. Baetsle L., Ruys D.— J. Inorg. and Nucl. Chem., 1961 (1962), vol. 21,
p. 133—140.
593. Balis E. W., Bronk L. B., Pfeiffer H. G., Welbon W. W., Winslow E. H.,
Zemany P. D.— Anal. Chem., 1962, vol. 34, p. 1731—1733.
594. Ballaux C., Dams R., Hoste J.— Anal. chim. acta, 1969, vol. 47, p. 397—
405.
595. Banerjee D. K., Budke С. C., Miller F. D.— Anal. Chem., 1961, vol. 33,
p. 418-421.
596. Banerjee G.— Ztschr. anal. Chem., 1974, Bd. 270, S. 287.
597. Banick W.'M., jun., Holzer R. A.— Anal. Chem., 1963, vol. 35, p. 1413—
1415,
598. Bankert S. F., Bloom S. D., Sauter G. D.— Anal. Chem., 1973, vol. 45,
p. 692—697.
599. РЖХим, 1958, № 64171.
600. Barreto H. S. R., Barreto R. C. R.— J. Chromatogr., 1960, vol. 4, p. 153—
155.
601. Barry J. E., Donega H. M., Burgess T. F.— J. Electrochem. Soc., 1969,
vol. 116, p. 257—259.
602. Bastiaan G. J., Rieftje G. M.— Anal. Chem., 1973, vol. 45, p. 1994—2001.
603. Batanero P. S., May S.— Bull. Soc. chim. France, 1971, N 5, p. 1935—
1939.
604. Batuecas T., Carreira M.— An. Real soc. esp. fis. у quim. B, 1956, vol. 52,
p. 519—521.
605. Bauman Л,— Mikrochim. acta, 1977, Bd. 1, S. 69—72.
606. Beckers J. L., Everaerts F. M.— J. Chromatogr., 1970, vol. 51, p. 339—342.
607. Bedrosian A. J., Lerner M: W.— Anal. Chem., 1968, vol. 40, p. 1104—1107.
608. Bejan C., Catuneanu R., Niculescu I.— Rev. chim. (RSR), 1975, vol. 26,
p. 335—337.
609. Bejan C., Niculescu I., Catuneanu R.— Rev. chim. (RSR), 1976, vol. 27,
p. 535—537; РЖХим, 1977, 37312.
610. Веке E.~ Rev. chim. (RSR), 1974, vol. 25, p. 155—156.
611. Belt С. B., jun.— Anal. Chem., 1967, vol. 39, p. 676—678.
612. Beltran J., Faus J.— Inform, quim anal., 1972, vol. 26, p. 63—70.
613. Bereznai T., Keomley G., Miskei M., Vigvari M.— Magy kem. folyoirat,
1976, k. 82, old. 233—237.
614. Bergamini C., Rapi G.— Ann chim. (Ital.), 1960, vol. 50, p. 1314—4320.
615. Berge P., Oberlin C., Saint P. P.— J. Nucl. Mater., 1975, vol. 57, p. 283—
286.
616. Bergerioux C., Galinier J. L., Zikorsky L.— J. Radioanal. Chem., 1979,
vol. 54, p. 255—265.
617. Bergner K.— Anal. chim. acta, 1976, vol. 87, p. 1—6.
618. Berkey E., Sweeney G. G., Hickam W. M.— Nucl. Appl., 1968, vol. 5,
p. 344—353; РЖХим, 1969, 15Г127.
619. Berkey E., Sweeney G. G., Hickam W. M.— Trans. Amer. Nucl. Soc., 1968,
vol. 11, p. 113; РЖХим, 1969, 15Г127.
620. Bernas B.— Anal. Chem., 1968, vol. 40, p. 1682—1686.
621. Bessette R. R., Olver J. W.— J. Electroanal. Chem., 1968, vol. 17, p. 327—
334.
622. Bettison W. A.— J. Brit. Ceram. Soc., 1968, vol. 5, p. 173—181; РЖХим,
1969, 4Г149.
623. Bhattacharyya A. P., Guha S. K.— Trans. Ind. Ceram. Soc., 1982, vol. 41,
N 4, p. 94—96.
624. Bhatnagar M. L., Mathur N. K., Dalchand.— J. Sci. and Industr. Res.,
1960, vol. BC19, p. B501; РЖХим, 1961, 16Д38.
625. Biagi L., Pirani R., Simboli G.— Miner, et petrogr. acta, 1963, vol. 9,
p. 111—162; РЖХим, 1965, 6Г115.
626. Bilal B. A., Bratter P., Keim M.— J. Radioanal. Chem., 1974, vol. 21,
p. 437—443.
219
627. Blanchet M. L., Malaprade L.— Chim. anal., 1967, vol. 49, p. 11—27.
628. Blasius E., Gottling W.— Ztschr. anal. Chem., 1958, Bd. 162, S. 423—429.
629. РЖХим, 1977, 2Г98.
630. РЖХим, 1977, 10Г83.
631. Bock P:, Engelmann C., Hatterer A.— J. Radioanal. Chem, 1977, vol. 38,
p. 97—106.
632. Bodart F., Deconninck G., Demortier C.— J. Radional. Chem., 1977, vol. 35,
p. 95—108.
633. Bogancs J., Quittner P,, Szabo E.— Magy. kem. folyoirat, 1967, k. 73,
old.. 346—349; РЖХим, 1968, 7Г132.
634. Boksay Z., Czakvari B., Havas J., Patko M.— Hung. Sci. Instrum., 1977,
N 41, p. 41-45; РЖХим, 1978, 20Г105.
635. Boksay Z., Czakvari B., Havas J., Patko M.— In: Ion-select, electrod. conf.,
Bp., 1977, Bp.', 1978, p. 269—280; РЖХим, 1978, 19Г115.
636. Bond R. D., Hutton J. T.— Analyst, 1958, vol. 83, p. 684—686.
637. Bondy C., Rica J. C.— Bull. Soc. chim. France, 1968, p. 4227—4279.
638. Boss С. B., Hieftje G. MAnal. Chem., 1979, vol. 51, p. 1897—1905.
639. Boulton R. B., Ewan G. T.— Anal. Chem., 1977, vol. 49, p. 1297—1304.
640. Boumans P. IV. J. M.— Spectrochim. acta B, 1968, vol. 23, p. 559—566.
641. Bouvier J. L., Lachance G. R.— Pap. Geol. Surv. Canada, 1975, vol. 75,
p. 1; РЖХим, 1975, 22Г92.
642. Braicovich L., Landi M. F.— Met. ital., 1957, vol. 49, p. 465—474; РЖХим,
1958, 17535.
643. Braicovich L., Landi M. F.— In: Proc. VI colloq. spectroscopicum intern.
L.: Pargamon press, 1957, p. 51—61; РЖХим, 1958, 43017.
644. Braun T.— Acta chim. Acad. sci. hung., 1964, vol. 41, p. 199—207.
645. Bressan D. J., Carr R. A., Hannan P. J., Wilkniss P. E.— J. Radioanal.
Chem., 1974, vol. 19, p. 373—381.
646. Brodtmann N. V., Naughton I.— Water and Sewage Works, 1975, vol. 122,
N 4, p. 64—66; РЖХим, 1975, 23Г161.
647. Brody J. K., Faris J. P., Buchanan R. F.— Anal. Chem., 1958, vol. 30,
p. 1909—1912.
648. Brooksbank W. A., Leddicotte G. W.— J.Phys. Chem., 1953, vol. 57, p. 819—
823.
649. Broomhead J. A., Gibson N. A.— Anal. chim. acta, 1961, vol. 24, p.446—
450.
650. Brownell G. M., Bramadat K., Knutson R. A., Turnosck A. C.— Trans. Roy.
Soc. Canad. Sect. 4, 1957, vol. 51, N 6, p. 19—31; РЖХим, 1958, 67315.
651. Bruin H. J. de.-— Anal. Chem., I960, vol. 31, p. 360—362.
652. Bruin H. J. de, Smythe L. E.— Nature, 1958, vol. 182, p. 387.
653. Brune D., Landstrom O.— Radiochim. acta, 1966, vol. 5, p. 228—230.
654. Brunfelt A. O., Roelandts I.— Taianta, 1974, vol. 21, p. 513—521.
655. Brunfelt A. O., Steinnes E.— Geochim. et cosmochim. acta, 1966, vol. 30,
p. 921—928; РЖХим, 1967, 7Г108.
656. Bryant M. F., O'Keefe K., Malmstadt H. V.— Anal. Chem., 1975, vol. 47,
p. 2324—2326.
657. Budzynski A. Z., Beer J. Z.— Anal. chim. acta, 1969, vol. 46, p. 281—306.
658. Burger K.— In: Solvation, ionic and complex formation reactions in nona-
queous solvents. Bp.: Akad. Kiado, 1983, p. 37.
659. Burma D. P.— Anal. chim. acta, 1953, vol. 9, p. 513—517.
660. Buttgereit G.— Ztschr. anal. Chem., 1973, Bd. 267, S. 81—88.
661. Bilger P. A., Fink W.— Ztschr. anal. Chem, 1969, Bd. 244, S. 121—122.
662. Caletka R., Konecny C., Simkova M.— J. Radioanal. Chem., 1972, vol. 10,
p. 5—15.
€63. Campbell D. E., Passmore W. O.— Anal. chim. acta, 1975, vol. 76, p. 355—
360.
664. Campbell J. A., Laul J. C., Nielson К. K., Smith R. D.— Anal. Chem.,
1978, vol. 50, p. 1032—1040.
665. Capdevila P. C., Alvarez G. F.— An. Real soc. esp. fis. у quim., 1962,
vol. B58, p. 13—18; РЖХим, 1963, ЗГ126.
666. Cappellina F., Millefiori S.— Ann. chim. (Ital.), 1965, vol. 55, p. 926—934.
22C
667. Carr J. D., Swartzfager D. G.— Anal. Chem., 1970, vol. 42, p. 1238—1241'.
668. Carr J. D., Swartzfager D. G.— Anal. Chem., 1971, vol. 43, p. 583—585-
669. Casey A. TStarke K.— Anal. Chem., 1969, vol. 31, p. 1060—1062.
670. Ceausescu D.— Ztschr anal. Chem., 1960, Bd. 176, S. 1—3.
671. Ceausescu D., Pirvu I., Pirvu F.— Taianta, 1963, vol. 10, p. 861—864.
672. Ceausescu D., Vlad L.— Rev. chim. (HSR), 1966, vol. 17, p. 694—696.
673. Champeix L., Darras R., Duflo J.— J. Nucl. Mater., 1959, vol. 1, p. 113—
119; РЖХим, 1960, 30516.
674. Chaput M., Robichet J.— Rapp. CEA, 1965, N 2869, p. 1—35; РЖХим,
1966, 10Г162.
675. Chapyzhnikov B. A., Malikova E. D., Kunin L. L., Samosyuk V. N., Vas-
serman A.M., Frolov E. V., Evganov K. N.— J. Radioanal. Chem., 1973,.
vol. 17, p. 275—290.
676. Chikui S.— Бунсэки кагаку, Jap. Anal., 1972, vol. 21, p. 1569—1574-
РЖХим, 1973, 12Г48.
677. Christian G. D., Feldman F. J.— Anal. Chem., 1972, vol. 43, p. 611—613-
678. Christian G. D., Feldman F. J.— Appl. Spectrosc., 1971, vol. 25, p. 660—
663.
679. РЖХим, 1963, 16Г162.
680. Clemente G. F., Mastinu G. G.~~ J. Radioanal. Chem., 1974, vol. 20, p. 707—
714.
681. Coetzee C. J., Rohwer E. F. С. H.— Anal. chim. acta, 1969, vol. 44, p. 293—
299.
682. Coetzee C. J., Siao Wei-San.— Inorg. Chem., 1963, vol. 2, p. 14—19.
683. Cokal E. J., Wise E. N.~- Anal. Chem., 1963, vol. 35, p. 914—915.
684. Cook T. E., Pardue H. L., Santini R. E.~ Anal. Chem., 1976, vol. 48,.
p. 451—452.
685. Cosgrove J. F., Morrison G. H.— Anal. Chem., 1957, vol. 29, p. 1017—1019-
686. Coutinho C. A., Teixeira P. H., Pereira С. MMetallurgia ABM, 1975,.
vol. 31, p. 817—822; РЖХим, 1976, 16Г208.
687. Creanga L.— Rev. chim. (RSR), 1958, vol. 9, p. 219—221.
688. Critchfield F. E., Johnson J. B.— Taianta, 1960, vol. 5, p. 58—61.
689. РЖХим, 1967, 24Г53.
690. Czajka Z.~ Magy. kem. folyoirat, 1962, kl 68, old. 519—523.
691. Czerwenka G., Scheuheek E.— Ztschr. anal. Chem, 1975, Bd. 276, S. 37—40.
692. Cziboly C.t Zsinka L., Szirtes L.— Magy. kem. lapja, 1969, k. 24, old. 470—
472; РЖХим, 1970, 6Г79.
693. Dahnke K. F., lorns T. V.— In: Abstr. Pittsburgh conf. anal. chem. and
appl. spectrosc.,Cleveland, Ohio, 1977. Pittsburgh, 1977, p. Ill; РЖХим,
1978, 16Г145.
694. Daidoji H.— Бунсэки кагаку, Jap. Anal., 1980, vol. 29, p. 64—69; РЖХим,.
1980, 18Г26.
695. David D. J.— Analyst, 1960, vol. 85, p. 495—503.
696. Debras-Guedon J.— Bull. Soc. franf. ceram., 1975, N 106, p. 59—68.
697. Deconninck G.~ J. Radioanal. Chem., 1972, vol. 12, p. 157—169.
698. Delphin W. H., Hbrwetz E. P.— Anal. Chem., 1978, vol. 50, p. 843—848.
6991 Dempir J.— Stavivo, 1963, vol. 41, N 2, p. 66—67; РЖХим, 1963, 23Г35.
700. Deschamps NLoeillot A., Albert P.— C. r. Acad, sci., 1962, vol. 254,.
p. 682—684.
701. Diaz-Guerra J. P.~ An. Real soc. esp. fis. у quim., 1971, vol. 67, p. 135—
146; РЖХим, 1973, 1Г32.
702. Doerfjel K„ Geyer R., Muller G.~ РЖХим., 1962, 24Д71.
703. Dolezal J., Novozamsky I., Zyka J.— Coll. Czechosl. Chem. Commun.,
1962, vol. 27, p. 1830—1834.
704. Donega H. M., Burgess T. E-— Anal. Chem., 1970, vol. 42, p. 1521—1524.
705. Donev I. Y., Baloushev B. S., Marichkova L. M.— Докл. Волг. АН, 1974,.
T. 27, с. 1549—1552.
706. Doge Н. G.— Anal. chim. acta, 1967, vol. 38, p. 207—211.
707. Dube G„ Boulos M. I.— Canad. J. Spectrosc., 1977, vol. 22, p. 68—76.
708. Dubowy J., Holzhauser W.— Zesz. nauk. UJPr. Chem., 1971, vol. 264,.
p. 291—297; РЖХим, 1972, ЗГ13.
221
709. Dubuowoy C., Guillaumont R., Boussieres G.— Radiochim. acta, 1967,
vol. 8, p. 49—57.
710. Duff E. J., Stuart J. L.— Taianta, 1975, vol. 22, p. 823—826.
711. Dumoulin E., Hatterer A.— Analusis, 1976, vol. 4, p. 185—188.
712. Dunbar R. E , Ferrin F. J.— Microchem. J., 1961, vol. 5, p. 145—157.
713. Dvorak J., Krejcarova M., Kucerova J., Zizka B.— Coll.Czechosl. Chem.
Common., 1966, vol. 31, p. 1881—1885.
714. РЖХим, 1963, ЗГ127.
715. Dyck R., Veleker T. J.— Anal. Chem., 1959, vol. 31, p. 1640—1643.
716. Eckfeldt Е,— Anal. Chem., 1975, vol. 47, p. 2309—2310.
717. Eckfeldt E. L., Proctor W. E., fun.— Anal. Chem., 1971, vol. 43, p. 332—
337.
718. Eckfeldt E. L., Proctor W. E.— Anal. Chem., 1975, vol. 47, p. 2307—2309.
719. Ediger R. A.— Atom. Absorpt. Newslett., 1973, vol. 12, p. 151—157.
720. Eisenman G.,Rudin D. O.,Casby J. £.—Science, 1957, vol. 126, p. 831—834.
721. 'Eng K. Y., Meyer R. A., Bingham C. D.— Anal. Chem., 1964, vol. 36,
p. 1832—1834.
722. Engelmann C.— J. Radioanal. Chem., 1970, vol. 6, p. 399—412.
723. Engelmann C., Gosset J., Grumet C.— J. Radioanal. Chem., 1975, vol. 28,
p. 185—199.
724. Engelmann C., Loeuillet M.— Bull. Soc. chim. France, 1969, N 2, p. 680—
683.
725. Engelmann C., Nordmann F., Tinelli G.— In: Liquid alkali metals. Proc.
Intern, conf., Nottingham, 1973. L., 1973, p. 71—75; РЖХим, 1975, 11Г124.
726. Erickson L. E., Denbo J. A.— J. Phys. Chem., 1963, vol. 67, p. 707—709.
727. Evans C., Herrington J.— Anal. Chem., 1963, vol. 35, p. 1907—1910.
728. Everett G. L., West T. S., Williams R. W.— Spectrosc. Lett., 1973, vol. 6,
p. 767—770.
729. Fallyu M. P., AndersonD. L., Zoller W. H., Gordon G. E., Lindstrom R. M.—
Anal. Chem., 1979, vol. 51, p. 2209—2211.
730. Farrow R. N. P., Hill A. G.— Taianta, 1961, vol. 8, p. 116—128.
731. Fedoroff M.— Bull. Soc. chim. France, 1970, N 3, p. 1233—1236.
732. Fernando L. A., Miles Mf. L., Bowen L. H.— Anal. Chem., 1980, vol. 52,
p. 1115—1119.
733. Filby R. H., Haller W. A., Shah K. R.— J. Radional. Chem., 1970, vol. 5,
p. 277—289.
734. Fishman M. J., Erdman D. E.— Anal. Chem., 1975, vol. 47, p. R334—R361.
735. Fitzgerald J. J., Chester T. L., Winefordner J. D.— Anal. Chem., 1975,
vol. 47, p. 2330—2339.
736. Flaschka H.. Amin A. M — Chem. Anal., 1953, vol. 42, N 4, p. 78—80.
.737. Flatz R., Benquerel F.— Helv. chim. acta, 1962, vol. 45, p. 1777—1785.
738. Florence T. M.— Anal. chim. acta, 1958, vol. 19, p. 548—551.
739. Foner H. A.— Isr. J. Chem., 1970, vol. 8, p. 541—544.
740. Fournet L., Albert PC. r. Acad, sci., 1962, vol. 254, p. 1076—1078.
741. Frache R., Dadone A. — Chromatographia, 1972, vol. 5, p. 449—453.
742. Frache R.. Dadone A.— Chromatographia, 1972, vol. 5, p. 581—586.
743. Frache R., Dadone A., Baffi F.— Ann. chim. (Ital.), 1974, vol. 64, p. 387—
396.
744. Francis С. V.— Microchem. J., 1963, vol. 7, p. 375—382.
745. Frank M. J., Johnson J. B., Rubin S. H.— J. Pnarm. Sci., 1976, vol. 65,
p. 44—48.
746. Fray D.J.— Met. Trans., 1977, vol. B8, p. 153—156.
747. French W. J., Adams S. J.— Analyst, 1974, vol. 99, p. 551—554.
748. Fresenius R., JanderG.— In: Handbuch der analytischen Chemie. B.:
Springer Verlag, 1940, Bd. la, S. 16—112.
749. Friedrich M.— Sklar a keram., 1955, sv. 5, N 4, s. 74—77.
750. Fujinaga T., Koyama M., Tochiyama O.— Anal. chim. acta, 1968, vol. 42,
p. 219—224.
751. Gaarenstroom P., English J. C., Perone S. P., Bixler J. W.— Anal. Chem.,
1978, vol. 50, p. 811—817.
752. Gagliardi E., Reimers H.— Mikrochim. acta, 1957, N 6, S. 784—795.
222
753. Gagliardi E., Reimers H.— Ztschr. anal. Chem., 1958, Bd. 160, S. 1—6.
754. Gana R. S., Philipp.— J. Sci., 1970 (1972), vol. 99, p. 191—194;
755. Ganjei I.D., Howell N.G., Roth I. R., Morrison G.H.— Anal. Chem.,.
1976, vol. 48, p. 505—510.
756. Garbartno J. R., Taylor H. E.— Appl. Spectrosc., 1979, vol. 33, p. 220—
226.
757. Garbett K., Goodfellow G. I., Marshall G. B.— Anal. chim. acta, 1981,
vol. 126, p. 135—145. '
758. Garbett K., Goodfellow G. I., Marshall G. B.— Anal. chim. acta, 1981,
vol. 126, p. 147—156.
759. Garcia J. M., Gutierrez J., Fernandez C.-— Volun. hidraul., 1977, vol. 14,.
p. 20—25; РЖХим, 1979, 11Г173.
760. Gardner D.J., Pritchard J. A., Sadler M. A.— Analyst, 1976, vol. 101,
p. 278—285.
761. Gaspary A. T.— Chim. anal., 1962, vol. 44, p. 67—69.
762. Gaspar у Arnal A. T.— Chim. et industr., 1960, vol. 83, p. 406—410;. ’
РЖХим, 1960, 73008.
763. Gaspar у Arnal T.— Chim. et industr., 1960, vol. 84, p. 523—528; РЖХим,
1961, 9Д39.
764. Gebauhr W.— Radiochim. acta, 1965, vol. 4, p. 191—197.
765. Gebauhr W.— Radiochim. acta, 1966, vol. 5, p. 8—14.
766. Gebauhr W., Martin J.— Intern. J. Appl. Radiat. and Isotop., 1959, vol. 4,.
p. 173—178; РЖХим, 1959, 86224.
767. Geisler M.— J. Radioanal. Chem., 1975, vol. 28, p. 209—219.
768. Ghelberg N. W., Puscasiu M.— Iciena, 1964, vol. 13, p. 347—352; РЖХим,.
1.966, 1Г206.
769. Ghimicescu G., Ghimicescu C., Dumbrava E.— Stud. Univ. Babes-Bolyai.
Ser. chim., 1963, vol. 8, p. 231—244; РЖХим, 1965, 10Г190.
770. Gijbels R., Meyer L., Hoste J.— Anal. chim. acta, 1964, vol. 31, p. 159—
165.
771. Gladney E. S., Curtis D. B., Jurney E. T.— J. Radioanal. Chem., 1978,
vol. 46, p. 299—308.
772. GobrechtH., Bock-Werthmann W., Tausend A., BratterP., Willers G.—
Intern. J. Apll. Radiat. and Isotop., 1965, vol. 16, p. 655—659; РЖХим,
1966, 9Г169.
773. Gomez C.A., Dorado L.M. T., Jimenez S.J.L.— Rev. met. CENIM,..
1971, vol. 7, p. 390—406; РЖХим, 1972, 9Г146.
774. Goodfellow G.I., Midgley D., Webber H. W.— Analyst, 1976, vol. 101,.
p. 848—855.
775. Gonchakov A. S., Zorov N. B., KyzyakovYu. Ya., MatveevO. I.— Anal. Lett.,.
1979, vol. A12, p. 1037—1048.
776. GorencB., Katie M., Gomiscek S.— Acta pharm. jugosl., 1976, vol. 26,.
p. 309—312; РЖХим, 1977, 17Г206.
777. GotoH., Atsuya I.— Ztschr. anal. Chem., 1967, Bd. 225, S. 121—129.
778. Gowda H.-S., Stephen W. I.— Anal. Chem., 1961, vol. 25, p. 153—158.
779. Grabner E.— Mikrochim. acta, 1975, N 1, S. 65—68.
780. Grba V.— Analusis, 1975, vol. 3, p. 363—367.
781. Grecul., Вucerzan I.— Farmacia (RSR), 1957, vol. 6, p. 528—533;.
РЖХим, 1958, 49985.
782. Green R. B., Keller R. A., Schenk P.-К., Travis J. C., Luther G. G.—
J. Amer. Chem. Soc., 1976, vol. 98, p. 8517—8518.
783. Green R.B., Travis J.C., Keller R.A.— Anal. Chem., 1976, vol. 48,.
p. 1954—1959.
784. Greenhalgh R., Riley J. P., Tongudai M.— Anal. chim. acta, 1966, vol. 36,
p. 439—448.
784a. Greenwood N. N., Earnshaw A. Chemistry of the elements. Oxford etc.:
Pergamon press, 1984. 1542 p.
785. Grimanis A. P., Hadzistelios I.— Anal. chim. acta, 1968, vol. 41, p. 15—21.
786. GrossD.— J. Chromatogr., 1963, vol. 10, p. 233—234.
787. Giiggi M., Ochme M., Pretsch E.—• Helv. chim. acta, 1976, vol. 59, p. 2417—
2420.
223-
788. Haerdi W., Daniel R.— J. Radioanal. Chem., 1970, vol. 5, p. 353—358.
789. Halls D. J.— Spectrochim. acta B, 1977, vol. 32, p. 397—412.
'790. Hamernik R.— Hutn. listy, 1959, sv. 14, s. 142—144.
791. Handa A.C., John K. N.— Chromatographia, 1971,. vol. 4, p. 530—531.
"792. Hanna Z. G., Ibrahim J. M.— Mikrochim. acta, 1964, N 6, S. 990—997.
793. Hanns M., Heaulme M., Morin P.— Bull. Soc. chim. France, 1963,
p. 2658—2661.
794. Haragucht H., Fukuta N., Yoshimura E., Fuwa K.— Бунсэки кагаку, Jap.
Anal., 1975, vol. 24, p. 733—735; РЖХим, 1976, 11Г30.
795. Haubold W — DEMAG-Nachr., 1964, N 174, S. 17412—17413; РЖХим,
1965, 2Г141.
796. Hawthorn D., Ray N. J.— Analyst, 1968, vol. 93, p. 158—165.
797. Haymore B. L., Lamb J. D., Izatt R. M., Christensen J. J.— Inorg. Chem.,
1982, vol. 21, p. 1598—1602.
798. Hegediis A. J., Neugebauer J., Dvorszky M.— Mikrochim. acta, 1959, N 2,
g 282________293.
799. Neideke G., Stork G., Schindler J. G., Gulich M. V., Schmid W., Maier H.,
Lindt H. O., Sailer D — Ztschr. anal. Chem., 1980, Bd. 301, S. 406—409.
800. Henke B. L.— In: Advances of X-ray analysis. Denver (Col.), 1964, vol. 7,
p. 460—488; РЖХим, 1966, ЗГ73.
801. Henrion G., Pungor E.— Anal. chim. acta, 1967, vol. 38, p. 357—362.
"802. Heres A., Girard-Devasson O., Gaudet J., Sputg J. C.— Analusis, 1972,
vol. 1, p. 408—412; РЖХим, 1973, 8Г143.
803. Hicks J. E., McPherson R. T., Salyer J. W.— Anal. chim. *acta, 1972,
vol. 61, p. 441—448.
804. Hieftje G. MCopeland T. R — Anal. Chem., 1978, vol. 50, p. 300R—327R.
805. Hieftje G. M., Copeland T. R., Olivares D. R.— Anal. Chem., 1976, vol. 48,
p. 142R—174R.
806. Higuchi H., Nonaka NHamagucht H., Tomura K.— J. Radioanal. Chem.,
1977, vol. 36, p. 457—467.
807. Hildon M.A., Allen W. J.-E — Analyst, 1971, vol. 96, p. 480—487.
808. Hirose A., AshiiD.— J. Radioanal. Chem., 1978, vol. 45, p. 305—315.
809. Hislop J.S., Stevens J. R., Wood D.A.— J. Radioanal. Chem., 1977,
vol. 30, p. 409—419.
810. Hodges R. J., Belcher С. B.— Appl. Spectrosc., 1975, vol. 29, p. 163—167.
811. Hoffman F. L., Walsh P. R., Doyle M. P.— Anal. Chem., 1974, vol. 46,
p. 492—496.
812. Hofton M. E.— In: Proc. 28th chem. conf., Scarborough, 1975. L., 1976,
p. 72—79; РЖХим, 1978, 16Г191.
"813. Holasek A., Dugandzic M.— Mikrochim. acta, 1959, N 4, S. 488—489.
814. Holm D.M., Sanders M. W.— NucL Appl., 1967, vol. 3, p. 308—313.
815. Holzapfel H., EhthardtH., Ttscher W.— J. prakt. Chem., 1962, Bd. 18,
S. 62—71.
'816. Holzapfel H., Nenning P.— Ztschr. Chem., 1964, Bd. 4, S. 151—152.
• 817. Holzapfel H., Nenning P., Kramer P.— Anal. chim. acta, 1967, vol. 37,
p. 535—538.
818. Holzapfel H., Nenning P., Schlegel R.— Ztschr. anal. Chem., 1965, Bd.
213, S. 401—404.
819. Honma N., Kawashima I.— Бунсэки кагаку, Jap. Anal., 1977, vol. 26,
p. 429—433; РЖХим, 1978, 2Г192.
820. Hourigan H. F., Robinson J. W.— Anal. chim. acta, 1955, vol. 13,
p. 179—182.
821. Howard A.G., Greenhalgh D. A.—Anal. chim. acta, 1979, vol. 106,
p. 361—364.
822: Howard О. H.— Anal. Chem., 1972, vol. 44, p. 1482—1484.
823. Huber I., Schreinlechner I., Benischek F.— Atom. Absorpt. Newslett.,
1977, vol. 16, N 3, p. 64—66.
824. Huber J.F.K., Urk-Schoen A. M.— Anal. chim. acta, 1972, vol. 58,
p. 395—409.
825. Iddings F. A., Wade TIn: Proc. Intern, conf. mod. trends, activat. ana-
lysis, College Station, Tex., 1965. s. a., p. 149—151; РЖХим, 1967, 7Г117.
•99Л
826. Illner E.— Ztschr. Chem., 1969, Bd. 9, S. 69—70.
827. Imahashi T., Fujiki I., Enomoto S.— Radioisotopes, 1976, vol. 25, p. 37—38.
828. Ingamells С. O. — Taianta, 1962, vol. 9, p. 781—783.
829. losof V., Neacsu V.— Rev. roum. chim., 1980, vol. 25, p. 589—597.
830. Irving H., Silva J. J. R. F.— J. Chem. Soc., 1963, p. 448—457.
831. Ishimori T., Kimura K., Fujino T., Murakami H.— J. Atom. Energy Soc.
Jap., 1962, vol. 4, p. 117—126; РЖХим, 1963, 10B52.
832. IshizukaT., SunaharaH.—Бунсэки кагаку', Jap. Anal., 1972, vol. 21,
p. 264—270; РЖХим, 1972, 16Г156.
833. Ishizuka T., Sunahara H., Tanaka K.— Нагоя коге гидзюцу сикэнсе хо-
коку, Rep. Gov. Industr. Res. Inst. Nagoya, 1972, vol. 21, N 6, p. 131—
136; РЖХим, 1973, ЗГ113.
834. Iwata S.— Бунсэки кагаку, Jap. Anal., 1960, vol. 9, p. 178—179; РЖХим,
2Д38. ’
835. Iyer R. K., Krishnamoorthy K. R., Das M.S.—Anal. chim. acta, 1972,
vol. 60, p. 454—457.
836. Izawa K., AoyagiH., Yoshida Z., Takahashi M.— Бунсэки кагаку, Jap.
Anal., 1972, vol. 21, p. 860—867; РЖХим, 1973, 1Г122.
837. Izawa K., Aoyagi H., Yoshida Z., Takahashi M.— Бунсэки кагаку, Jap.
Anal., 1973, vol. 22, p. 1046—1051; РЖХим, 1974, ЗГ137.
838. Jaber A. M. V., Moody G. I., Thomas I.D.R.— Analyst, 1977, vol. 102,
p. 943—948.
839. Jackson C.— Analyst, 1953, vol. 78, p. 599—602.
840. Jackson P.J., Smith A. C.— J. Appl. Chem., 1956, vol. 6, p. 547—559.
841. JAERI, 1973, N 1225. 114 p.; РЖХим, 1978, 4Г167.
842. Jahns IV., Weismann G.— Nukleonika, 1959, wol. 1, s. 189—190.
843. Janauer G. E., Johnston R. C., Oliveri A. J., Carrano J.— Anal. chim. acta,
1967, vol. 38, p. 127—135.
844. Janousek I.— Chem. listy, 1973, sv. 67, s, 982—985.
845. Jansen B. J., Hollander T. J.— Spectrochim. acta, B, 1977, vol. 32,
p. 165—172.
846. Jasz A., Lengyel T.— Acta chim. Acad. sci. hung., 1964, vol. 33, p. 395—
398.
847. Jeanroy E.— Analusis, 1974, vol. 2, p. 703—712.
848. Jennings D. A., Keller R. A.— J. Amer. Chem. Soc., 1972, vol. 94,
p. 9249—9250.
849. Jewsbury A.— Analyst, 1953, vol. 78, p. 363—367.
850. Johnson G. IV., Taylor H.E., Skogerboe R. K.— Spectrochim. acta B,
1979, vol. 34, p. 197—212.
851. Johnson IV.— In: Analysis Calcareous Mater. L.: Soc. Chem. Industr.,
1964, p. 306—315; РЖХим, 1965, 5Г124.
852. Joshi К. M., Ganu G. M.— Ind. J. Technol., 1978, vol. 16, N 2, p. 53—55.
853. JouyA.D., CousierJ.— Bull. Soc. chim. France, 1958, N 3, p. 323—325.
854. Jurion R.— Chim. anal., 1968, vol. 50, p. 325—333.
855. Kallmann S.— Industr. Eng. Chem. Anal. Ed., 1944, vol. 16, p. 712.
856. Kallmann S., Liu R.— Anal. Chem., 1964, vol. 36, p. 590—592.
857. Kalvoda R.— Anal. chim. acta, 1958, vol. 18, p. 132—139.
858. Kamemoto Y., Yamagishi S.— Bull. Chem. Soc. Jap., 1963, vol. 36,
p. 132—136; РЖХим, 1963, 21Г133.
859. Kantor T., Bezur L., Pungor E.— Mikrochim. acta, 1981, N 1, S. 289—307.
860. Kantor T., Poles L., FodorP., Pungor E.— Taianta, 1976, vol. 23, p. 585—
586.
861. Karachi S. S., Corcoran F. L.— Appl. Spectrosc., 1973, vol. 27, p. 41—42.
862. Karajannis S., Ortner H. M., Spitzy H.— Taianta, 1972, vol. 19, p. 903—
914.
863. Karanjikar N. P., Saksena M. D.— Gov. India Atom. Energy Commis,
Rep., 1973, N 678, p. 7; РЖХим, 1974, 14Г163.
864. Karanjikar N. P., Saksena M.D.— Taianta, 1974, vol. 21, p. 652—654.
865. Karch Z.— Szklo i ceram., 1959, wol. 10, s. 102—105.
866. Kawamura S., Kurotaki K., Kuraki H., Izawa M.— J. Chromatogr., 1967»
vol. 26, p. 557—560.
8 В. M. Иванов и др.
225
867. Kawashima T.— In: Proc. Intern, conf. mod. trends activat. analysis,
College Station, Tex. 1965, s. a., p. 61—65; РЖХим, 1967, 7Г163.
868. Keane J. R., Fisher E. M. R.— Atoms. Environ., 1968, vol. 2, p. 603—614;
РЖХим, 1969, 9Г190.
869. Keenan J. A., Larrabee G. B.— Chem. Instrum., 1971, vol. 3, p. 125—140.
870. Keliher P. N., Wohlers С. C.— Anal. Chem., 1974, vol. 46, p. 682—687.
871. Кеппа В. T., Conrad F. J.— Taianta, 1968, vol. 15, p. 418—420.
872. Kerbyson J.D., Ratzkowski C.— Canad. Spectrosc.' 1968, vol. 13, p. 102—
106.
873. РЖХИМ, 1967, 24Г67.
874. РЖХИМ, 1968, ЗГ54.
875. Kilroy W. P., Moynihan С. T.— Anal. chim. acta, 1976, vol. 83, p. 389—
392.
876. Kimura K., Maeda T., Shono T.— Taianta, 1979, vol. 26, p. 945—949.
877. King H. G., Neff С. M.— Appl. Spectrosc., 1963, vol. 17, N 2, p. 51—53.
878. Kitagawa K., Takeuchi T.— Kagaku, Chemistry, 1975, vol. 30, p. 892—
895; РЖХим, 1976, 8Г35.
879. Klaus G., Klbppel H., Kubassek E.— Mikrochim. acta, 1977, N 2, S. 551—
564.
880. Klenert G.— Kali und Steinsalz, 1966, Bd. 4, S. 229—239; РЖХим, 1967,
21Г146.
881. РЖХим, 1977, 2Г203.
882. Knight D. M.— Atom. Absorpt. Newslett., 1970, vol. 9, p. 108; РЖХим,
1971, 7Г151.
883. Knotz F., Raber H., Huber H.— Mikrochim. acta, 1974, N 1, S. 81—84.
884. Ko R., Anderson P.— Anal. Chem., 1969, vol. 41, p. 177—178.
885. Koch H.— Kernenergie, 1960, Bd. 3, S. 315—320.
886. РЖХим, 1954, № 13232.
887. Konjevic R., Konjevic N.— Spectrosc. Lett., 1973, vol. 6, p. 177—181.
888. Korba G.A., Veung E.S.— Anal. chim. acta, 1978, vol. 99, p. 209—216.
889. Korhonen J., Lumme P. ОKinnunen L. J.— Pap. ja pun, 1973, n. 55,
• s. 559—561, 564—566, 569—570; РЖХим, 1974, 6Г175.
890. Korkisch J., Abascal F., Hernandez A., Braico E.— Mikrochim. acta, 1976,
N 2 S. 291________298
891. Korkisch J., Orlandini K.A.— Anal. Chem., 1968, vol. 40, p. 1952—1955.
892. Koscielniak P., Parczewski A., Walas S.— Ztschr. anal. Chem., 1979, Bd.
297 S 156____158
893. Kosta L., Cook G. B.— Taianta, 1965, vol. 12, p. 977—987.
894. Kotos P., Kukula F.— Radioisotopy, 1972, sv. 13, s. 253—254.
895. Koulev I. I., Bakalov V.D.— Докл. Болг. АН, 1973, т. 26, с. 787—789.
896. Kbnig К.Н., Demel К.— J. Chromatogr., 1969, vol. 39, p. 101—102.
897. Konig K.H., Graf H.— J. Chromatogr., 1972, vol. 67, p. 200—202.
898. Krainer H., Ortner H. M., Muller K., Spitzy H.— Taianta, 1974, vol. 21,
p. 933—943.
899. Krause В,— Beckman Rep., 1966, N 3/4, p. 9—11; РЖХим, 1967, 14Г104.
900. Kreider D. A., Breckenridge J. E.— Amer. J. Sci., 1896, vol. 2, p. 263.
901. Kreisky F.—Mikrochim. acta, 1959, N 2, S. 243—248.
902. Kremer J.— Ztschr. Phys., 1967, Bd. 199, S. 94—97.
903. Krishnan С. V., Venkateswarlu Ch., Athavale V. T.— Ind. J. Appl. Chem.,
1968, vol. 31, p. 21—26; РЖХим, 1969, 8Г57.
904. Kuchner E., Alvarez R., Paulsen P. J., Murphy T. J.— Anal. Chem.,
1972, vol. 44, p. 2050—2056.
905. KuemmelD.F., KarlH.L.— Anal. Chem., 1954, vol. 26, p. 386—391.
906. Kuhl J., Neumann S., Kriese M.— Laser -j- Elek. = Opt., 1974, Bd. 6,
S.46—50.
907. KyrS M., Halova J.— J. Radioanal. Chem., 1979, vol. 52, p. 53—67.
908. Kyrs M., Pivonkova M., Selucky P.— Anal. chim. acta, 1968, vol. 43,
p. 132—134.
909. Lakhoua N., Haerdi W.~ Chimia, 1971, vol. 25, p. 125—127.
910. Lang W., Herrmann' R.— Mikrochim. acta, 1963, N 5/6, S.872—879.
911. Langmyhr F.J.— Analyst, 1979, vol. 104, p. 993—1016.
226
912. Langmyhr F. J., Paus P. E.— Anal. chim. acta, 1968, vol. 43, p. 397—
408.
913. Langmyhr F. J., Paus P. E.— Anal. chim. acta, 1968, vol. 43, p. 508—510.
914. Langmyhr F. J., Paus P. E.— Anal. chim. acta, 1969, vol. 45, p. 157—162.
915. Langmyhr F. J., Paus P. E.— Atom. Absorpt. Newslett., 1968, vol. 7.
p. 103—106.
916. Laverlochere J., May S.— Bull. Soc. chim. France, 1963, p. 457—462.
917. Le Roy V. M., Lincoln A. J.— Anal. Chem., 1974, vol. 46, p. 369—373.
918. Legradi L.— Magy. kem. lapja, 1964, k. 19, old. 167—168; РЖХим, 1964,
23Г71.
919. Lengyel T.— Acta chim. Acad. sci. hung., 1962, vol, 34, p. 29—33.
920. Lengyel T., Jasz A.— Acta chim. Acad. sci. hung., 1962, vol. 34, p. 19—
28.
921. Lesbats A.— Ann. chim., 1969, vol. 4, p. 29—42.
922. Lewis В. C., Stephen W. I.— Anal. chim. acta, 1966, vol. 36, p. 234—237.
923. Lieser K.H., Bastian J., Hecker A. В.— Ztschr. anal. Chem., 1967, Bd.
228, S. 98—106.
924. Lieser К. H., Neitzert V.— J. Radioanal. Chem., 1976, vol. 31, p. 397—
405.
925. Lightowlers E.C.— Anal. Chem., 1963, vol. 35, p. 1285—1291.
926. Lin J.D., Popov A. I.— J, Amer. Chem. Soc., 1981, vol. 103, p. 3773—
3777. .
927. Liteanu C., Hopirtean E.— Rev. roum. chim., 1971, vol. 16, p. 55—62.
928. Lockett E. E.,Thomas R. H.— Nucleonics, 1953, vol. 11, N 3, p. 14—17.
929. Loos-Neskovic Ch., Fedoroff M., Revel G.— Anal. chim. acta, 1976, vol. 85,
p. 95—102.
930. Lutz G. J., Soete D. A.— Anal. Chem., 1968, vol. 40, p. 820—822.
931. Lutz G. /.— Anal. Chem., 1970, vol. 42, p. 531—532.
932. Machtroux R., Merciny E., Schreiber A.— Mikrochim. acta, 1973, N 5,
S. 829—840.
933. Maeda M., Ishitsuka F., Miyazoe Y.— Кюсю дайгаку когаку сюхо,
Technol. Rep. Kyushu Univ., 1975, vol. 48, p. 29—34; РЖХим, 1975, 16Г15.
934. Maenhaut W., Adams F., Hoste J.— J. Radioanal. Chem., 1973, vol. 14,
p. 295—316.
935. Magyar B., Widmer G.— Taianta, 1976, vol. 23, p. 693—700.
936. -Maillard P., Ades C.— Canad. Spectrosc., 1969, vol. 14, p. 17—20.
937. Mairesse-Ducarmois C. A., Patriarch# G.J., Vandenbalck J. I.— Anal,
chim. acta, 1975, vol. 79, p. 69—78.
938. РЖХим, 1962, 4Д63.
939. РЖХим, 1963, 15Г169.
940. Malmstadt H. P., Chambers W. E.— Anal. Chem., 1960, vol. 32, p. 225—
232.
941. Malvano R.— Anal. chim. acta, 1967, vol. 38, p. 341—347.
942. Mangelsdorf P. C., jun.— Anal. Chem., 1966, vol. 38, p. 1540—1544.
943. Mani К. P., Sankar D. M.— Ind. J. Technol., 1972, vol. 10, p. 218—220.
944. Mannkopf R., Peters C.— Ztschr. Phys., 1931, Bd. 70, S. 444.
945. Manoliu C., Toni B., Panovict I., Popescu O.— Chim. anal., 1970, vol.
52, p. 1270—1275.
946. Marichkova L. M., Donev T. Y., Baloushev B. S.— Докл. Волг. АН, 1975,
т. 28, с. 367—370.
947. Marrodineanu R.-~ Appl. Spectrosc., 1959, vol. 13, p. 131—133.
948. Marrodineanu R.— Appl. Spectrosc., 1960, vol. 14, p. 17—21.
949. Marsh G. E.— Appl. Spectrosc., 1956, vol. 10, p. 8—10.
950. Marsh R. H„ Allie W„ jun.— Anal. Chem., 1968, vol. 40, p. 2037—2040.
951. Martinez J. B., Paya J. F.t~ J. Chromatogr., 1974, vol. 93, p. 485—486.
952. Masumoto K., Suzuki N.— J. Radioanal. Chem., 1978,' vol. 46, p. 121—
135.
953. Mathew J., Rajcev-Tandon S. N.— Chromatographia, 1977, N 1, p. 45.
954. Matocha C., Petit J.~ Appl. Spectrosc., 1968, vol. 22, pt 1, p. 562—571.
955. Matsushita H., Hironaka H.— Нихон кагаку кайси, J. Chem. Soc. Jap-
Chem. and Industr. Chem., 1974, N 3, p. 502—504; РЖХим, 1974, 18Г59.
8*
227
956. Matteli G., Attini E.— Alluminio, 1960, vol. 29, p. 227—230: РЖХим,
1961, 8Д69.
957. Mattock G.— Chimia, 1967, vol. 21, p. 209—217.
958. May S.— Bull. Soc. chim. France, 1961, N 12, p. 2203—2208.
959. May S., Piccot D., Pinte G.— Analusis, 1978, vol. 6, p. 246—254.
960. May S., Pinte G.— J. Radioanal. Chem., 1969, vol. 3, p. 329—343.
961. Mazza L., Sardo L.— Gazz. chim. ital., 1963, vol. 93, p. 1155—1164;
РЖХим, 1965, 6Г63.
962. Mazzucotelli A., Franche R., Dadone A., Baffi F.— Taianta, 1976, vol. 23,
p. 879—882.
963. McCabe W. C„ Fischer H. F.— J. Phys. Chem., 1970, vol. 74, p. 2990—
2998.
964. McCrea J. M.— Appl. Spectrosc., 1969, vol. 23, p. 55—62.
965. McHugh J. A., Stevens J. F.— Anal. Chem., 1972, vol. 44, p. 2187—2192.
966. Medlint J. H., Suhr N. H., Bodkin J. B.— Atom. Absorpt. Newslett.,
1969, vol. 8, p.,25—29; РЖХим, 1969, 22Г138.
967. Meffert A., Meier-Ewert Н,— Ztschr. anal. Chem., 1963, Bd. 198, S. 407—
410.
968. Mellichamp J. W.— Anal. Chem., 1954, vol. 26, p. 977—979.
969. Mellichamp J. W.— Appl. Spectrosc., 1954, vol. 8, p. 114—117.
970. Merryfield R. N., Loyd R. C.— Anal. Chem., 1979, vol. 51, p. 1965—1968.
971. Meyers P., Zelst L., Sayre E. V.— J. Radioanal. Chem., 1973, vol. 16,
p. 67—78.
972. Miles С. C.— Anal. Chem., 1969, vol. 41, p. 1041—1048.
973. Miller С. C., Traves F.— J. Chem. Soc., 1936, vol. 139, p. 1390.
974, РЖХим, 1963, 4Г128.
975. РЖХим, 1962, 18Д119.
976. Miscicka M.— Szklo i ceram., 1965, wol. 16, s. 243—249.
977. Mitchell J. W., Luke C. L., Norfhover W. R.— Anal. Chem., 1973, vol. 45,
p. 1503—1506.
978. Mitchell J. W., Shanks D. L.— Anal. Chem., 1975, vol. 47, p. 642—646.
979. Mituzas J.— Науч. тр. высш, учебн. заведений ЛитССР. Химия и хим.
технол., 1963, № 3, с. 33—37. На лит. яз.
980. Mizohata A., Mamuro Т., Tsujimoto Т.— J. Radioanal. Chem., 1978, vol.
42, р. 143—152.
981. Mizukami S., leki T.— Mikrochim. acta, 1966, N 1/2, S. 147—153.
982. Modreanu F.— J. Chromatogr., 1958, vol. 1, p. 554—555.
983. Mody M. C., Baxi D. R.— Ind. J. Technol., 1976, vol. 14, p. 517—519.
984. Moenke H., Moenke-Blankenburg L., Quillfeldt W.— MiKrochim. acta,
1968, N 3, S. 221—241.
985. Monk R. G — Taianta, 1968, vol. 15, p. 1259—1265.
986. Montaser A., Menrabzadeh A. A.— Anal. chim. acta, 1979, vol. Ill,
p. 297—300.
987. Mori Y., Ooyasi T., Matsuda H.— Chem. Lett., 1973, p. 875—876.
988. Morrison G.H., Nadkarni R.A.— J. Radioanal. Chem., 1973, vol. 18,
p. 153—167.
989. Mosescu N., Kalmutchi G., Badea S.— Rev. chim. (RSR), 1975, vol. 26,
p. 677—679.
990. Mungall T. G., Mitchen J. H., Johnson D. E.~ Anal. Chem., 1964, vol. 36,
p. 70—72.
991. Murata M., Kitani S.— J. Nucl. Sci. and Technol., 1972, vol. 9, p. 622—
’623; РЖХим, 1973, 10Г52.
992. Musil A., Reimers 7/.—Ztschr. anal. Chem., 1956, Bd. 152, S. 154—158.
993. MustafaM. A.,OldhamG.— J. Radioanal.Chem., 1980, vol. 59, p. 629—633.
994. Mykytiuk A., Russell D. S., Boyko V.— Anal. Chem., 1976, vol. 48,
p. 1462—1464.
995. Nagourney S. I., Bogen D. C.— Canad. J. Spectrosc., 1978, vol. 23,
p. 101—104.
996. Nagy L. G.— Acta chim. Acad. sci. hung., 1967, vol. 52, p. 365—374.
997. Nagy L.G., Mester Z., Takacs G.— Magy. kem. folyoirat, 1967, k. 73,
old. 137—138; РЖХим, 1967, 21Г223.
228
998. Nakai T., Yajima S., Fujii I., Okada M.— Бунсэки кагаку, Jap., Anal.,
1959, vol. 8, p. 367—372; РЖХим, 1960, № 34553.
999. Nakamura H., Nishida H., Takagi M., Ueno K.— Anal. chim. acta, 1982,
vol. 139, p. 217—227.
1000. Nakamura H., Nishida H., Takagi M., Ueno K.— Бунсэки кагаку Jap,
Anal., 1982, vol. 31, p. E131—E134; РЖХим, 1982, 20Г70.
1001. Nakamura H., Takagi M., Ueno K.— Taianta, 1979, vol. 26, p. 921—927.
1002. Nakamuta T., Yumoto У., Izutsu K.— Bull. Chem. Soc. Jap., 1982
vol. 55, p. 1850—1853.
1003. Nascutiu T.— Rev. room, chim., 1973, vol. 18, p. 161—174.
1004. Nascutiu T — Stud, si cercet. chim., 1965, vol. 13, p. 1025—1033; РЖХим,
1967, 1Г50.
1005. Naud G., Sannier J.— Bull. Soc. chim. France, 1963, N 12, p.'2735—2737.
1006. Neeb К. H.— Ztschr. anal. Chem., 1960, Bd. 174, S. 328—333.
1007. Neeb К. H.— Ztschr. anal. Chem., 1964, Bd. 200, S. 278—286.
1008. Neeb К. H., Duda R., Franke R., Seyfarth R., Stockert H.— Ztschr. anal,
chem., 1966, Bd. 215, S. 161—175.
1009. Neeb К. H., Gebauhr W.— Ztschr. anal. Chem., 1968, Bd. 162, S. 167—174.
1010. Neirinckx R., Adams F., Hoste J.— Anal. chim. acta, 1968, vol. 43, p. 369—
380.
1011. Nelson F., Michelson D. C.— J. Chromatogr., 1966, vol. 25, p. 414—441.
1012. Neubauer L., Padel J.— Chem. jrum., 1975, sv. 25, s. 236—237.
1013. Neumann G. M.— Taianta, 1971, vol. 18, p. 1047—1051.
1014. Nevoral V., Hajkova G.— Chem. listy, 1965, sv. 59, s. 222—224.
1015. Nevoral V., Okac A.— Ceskosl. tarmac., 1965, sv. 14, s. 342—346.
1016. Newman L.— In: NASA-AEC liquid metals corros., meet. Brookhaven,
’ 1961, p. 1, 141—142; РЖХим, 1966, 9Г147.
1017. Nguyen Long-Den, Borot M., Albert P.— C. r. Acad, sci., 1961, vol. 253,
p. 2067—2068.
1018. Niese S.— J. Radioanal. Chem., 1977, vol. 38, p. 37—41.
1019. Niese S.— Kernenergie, 1964, Bd. 7, S. 105—108.
1020. Nordmann F.— Rapp. CEA, 1974, N 4539, p. 89; РЖХим, 1976, 4Г186.
1021. Nordmann F., Fluhr A., Tinelli G., Engelmann C.— Analusis, 1975, vol. 3,
p. 171—176.
1022. Nordmann F., Tinelli G., Engelmann C.— Analusis, 1973, vol. 2, p. 96—
105.
1023. Nordmann F., TinelliG., EngelmannC.— Analusis, 1973, vol. 2, p. 739—745.
1024. Nordmann F., Tinelli G., Engelmann C.— J. Radioanal. Chem., 1973,
vol. 17, p. 255—273.
1025. Novak V.— Sb. ved. pr. VSCHT Pardubicich, 1959, s. 121—138; РЖХим,
1960, № 77014.
1026. Novak V., Kozak P.— Sb. ved. pr. VSCHT Pardubicich, 1962 (1963), N 1/2,
s. 41—47; РЖХим, 1964, 18Г48.
1027. Novak V., Krajina A.— Chem. listy, 1958, sv. 52, s. 1506—1512.
1028. Novak V., Krajina A.— Coll. Czechosl. Chem. Commun., 1959, vol. 24,
p. 1808—1814.
1029. Oehlmann F., Heimer M.— Chem. Techn., 1958, Bd. 10, S. 296—299.
1030. Ohde S., Ohta N., Tomira R.— J. Radioanal. Chem., 1978, vol. 42, p. 159—
167.
1031. Ohir R. P'., Kohalkar A. S., Bhola S. N.— Ind. J. Technol., 1976, vol. 14,
p. 198—200.
1032. Ortega R. F.— US Dep. Commer. Nat. Bur. Stand. Spec. Puhi., 1969,
N 312/1, p. 536—540; РЖХим, 1974, 15Г159.
1033. Ortner H., Spitzy H.— Ztschr. anal. Chem., 1968, Bd. 238, S. 167—182,
1034. Ortner H., Spitzy H.— Ztschr. anal. Chem.,*1968, Bd. 238, S. 251—267.
1035. Osborne J. F., Larrabee G. B., Harrap V.— Anal. Chem., 1967, vol. 39,
p. 1144—1148.
1036. Oshima M., Seki M., Kawashima I.— Jap. J. Appl. Phys., 1978, vol. 17,
p. 1697—1698; РЖХим; 1979, ЗГ167.
1037. Ordogh M., Upor-Juvavcz V.— J. Chromatogr., 1966, vol. 25, p. 464—470.
1038. Palaty V.— Canad. J. Chem., 1963, vol. 41, p. 18—20.
229
1039. Palmer T. A., Winkler J. M.— Anal. chim. acta, 1980, vol. 113, p. 301—
306.
1040. Patek P., Sorantin H.— Ztschr. anal. Chem., 1967, Bd. 226, S. 338—346.
1041. Pauly J., Girardi F.— Bull. Soc. chim. France, 1963, p. 244—247.
1042. Pelerin J., Hatterer A.— Anal. chim. acta, 1975, vol. 74, p. 212—215.
1043. Pereira G. R., Brito J.— Publ. TEA, 1978, N 497, p. 10; РЖХим, 1978,
21Г217.
1044. Perrin R. M. S.~ In: Analysis Calcareous Mater. L.: Soc. Chem. Industr.,
1964, p. 207—221; РЖХим, 1965, 5Г111.
1045. Perry Ct J., Keyworth D. A.— Canad. Spectrosc., 1967, vol. 12, p. 47—50.
1046. Perry J. A., Bryant M. F., Malmstadt H. V.— Anal. Chem., 1977, vol. 49,
p. 1702—1710.
1047. Peterson G. E., Currier E. W.— Appl. Spectrosc., 1956, vol. 10, p. 1—4.
1048. Peuschel G., Hagedorn F.— Ztschr. anal. Chem., 1975, Bd. 277, S. 177—
182.
1049. Pfeil E.— Oster. Chem. Ztg, 1964, Bd. 65, S. 177—179; РЖХим, 1965,
ЗГ14.
1050. Pitt E. E. H.— Analyst, 1963, vol. 88, p. 399—403.
1051. Plesch R.— Amer. Lab., 1977, vol. 9, N 3, p. 87—92.
1052. Plumb R. C., Stiverman R. H.— Nucleonics, 1954, vol. 12, N 12, p. 29—
31.
1053. Podobnik B., Dular M., Korosin J.— Mikrochim. acta, 1965, N 5/6,
S. 1073—1080.
1054. Podobnik B., Korosin J., Kosta L.— Ztschr. anal. Chem., 1966, Bd. 218,
S. 184—192.
1055. Polakovicova J.— Acta geol. et geogr. Univ. Comen. Geol., 1968, N 15,
p. 185—187.
1056. РЖХим? 1959, № 86237.
1057. Poonia N. S., Bajaj А. V,— Chem. Rev., 1979, vol. 79, p. 389—445.
1058. Pop A., Dumitrescu P.— Rev. chim. (RSR), 1977, vol. 28, p. 1201—1202.
1059. Popa G., Popescu M., Danet A.— Rev. roum. chim., 1980, vol. 25,
p. 1427—1432.
1060. Posgay G.— Kohasz. lapok, 1955, vol. 10, p. 231—232; РЖХим, 1957,
№ 4714.
1061. Prake L.— Chem. N. Z., 1980, vol. 44, N 3, p. 98—99; РЖХим, 1980,
23Г139.
1062. Pratt P. F., Bradford G. R.— Soil Sci., 1960, vol. 89, p. 342—346; РЖХим,
1QK1 эти 22
1063. РЖХим, 1958, № 24735.
1064. Preis H.,' Esenwein A.— Schweiz. Arch, angew. Wiss. und Techn., 1966,
Bd. 32, S. 79—86; РЖХим, 1966, 22Г133.
1065. Pretorius R., Odendaal F., Peisach M.— J. Radioanal. Chem.., 1972, vol.
12, p. 139—149.
1066. Price W. J., Whiteside P. J.— Analyst, 1977, vol. 102, p. 664—671.
1067. Primbsch E. O.— Emailwaren Industr., 1940, Bd. 17, S. 97; C. A., 1941,
vol. 35, p. 1955.
1068. Product, group U. K. Atomic Energy Author. Rep. 1961, N 3. 5 p.;
РЖХим, 196’, 21Д67.
1069. Product, group U. K. Atomic Energy Author. Rep., 1961. N 58 (S.). 10 p.;
РЖХим, 1963, 2Г44.
1070. Proszt J., Gyorbiro K.— Anal. chim. acta, 1956, vol. 15, p. 585—591.
1071. Proszt J., Gyorbiro K.— Chem. zvesti, 1957, sv. 11, s. 198—'204.
1072. Proszt J., Gyorbiro K.— Magy. kem. folyoirat, 1956, k. 62, old. 342—345;
РЖХим, 1957, № 60877.
1073. Pruiksma R., Ziemer J., Yeung E. S.— Anal. Chem., 1976, vol. 48,
p. 667—670.
1074. Quittner P., Simonits A., Elek A.— Taianta, 1967, vol. 14, p. 417—-420.
1075. Radmacher W., Schmitz W.— Brennst.-Chem., 1965, Bd. 46, S. 21—27;
РЖХим, 1965, 13Г145.
1076. Radulescu M. F., Bentea A.— Rev. chim. (RSR), 1961, vol. 12, p. 100—
101; РЖХим, 1961, 20Д50.
230
1077. Ragone S. E., Finelli R.— Atom. Absorpt. Newslett., 1971, vol. IP-
р. 126—127.
1078. Ramirez-Munos J., Rrace R. O.— Appl. Spectrosc., 1968, vol. 22, p. 736—
74!.
1079. Randa Z., Benada J., Kuncif J., Kouzimsky J.— J. Radioanal. Chem.,
1973, vol. 14, p. 437—443.
1080. Rann C. S.— Spectrochim. acta B, 1968, vol. 23, p’. 827—847.
1081. Ranwiler L. E., Moyers J. L.— Environ. Sci. and Technol., 1974, vol. 8,
p. 152—156; РЖХим, 1974, 18Г225.
1082. Raspi G., Zanello P., Cinquantini A., Corti P.— Anal. chim. acta, 1973,
vol. 66, p. 435—442.
1083. Rauscher H. E.— Anal. Chem., 1966, vol. 38, p. 519—520.
1084. Rechnitz G. A., Brauner J.— Taianta, 1964, vol. 11, p. 617—620.
1085. Rechnitz G. A., Zamochnick S. B.— J. Amer. Chem Soc., 1964, vol. 86,
p. 2953—2954.
1086. Reddy G. R., Pant D. R., Rao B. L., Sankar D. M.— J. Radioanal. Chem.,
1976, vol. 33, p. 39—51.
1087. Reeve W.— Anal. Chem., 1959, vol. 31, p. 1066—1068.
1088. Reeve W„ Christoffel 7.—Anal. Chem., 1957, vol. 29, p. 102—105.
1089. Reeve W., Peiffer J- S.— Anal. chim. acta, 1968, vol. 43, p. 150—153.
1090. Reichen L. E.— Anal. Chem., 1958, vol. 30, p. 1948—1950.
1091. Ricard R., Ricard J., Robin J.— In: 19е congr. GAMS, 1956. P.: Meru
(Oise), 1958, p. 195—199; РЖХим, 1960, N 61027.
1092. Ricci E.— Anal. Chem., 1974, vol. 26, p. 615—618.
1093. Ricci E., Hahn R. L.— Trans. Amer. Nucl. Soc., 1966, vol. 9, p. 104—105;
РЖХим, 1967, 2Г39.
1094. Rich R.— J. Chem. Educ., 1962, vol. 39, p. 403—406.
1095. Ricq J. C.— Anal. chim. acta, 1966, vol. 35, p. 263—264.
1096. Riepe W., Kaiser H.— Ztschr. anal. Chem., 1966, Bd. 223, S. 321—335.
1097. Ring S. A.— Anal. Chem., 1956, vol. 28, p. 1200—1201.
1098. Robinson J. IV.— Anal. chim. acta, 1960, vol. 23, p. 458—461.
1099. Rodriquez A. R., Poitrenaud C.— Anal. chim. acta, 1977, vol. 93, p. 359—362.
1100. Rorabacher D. B., MacKellar W. J., Shu F. R., Bonavita S. M.— Anal.
Chem., 1971, vol. 43, p. 561—573.
1101. Ross W. J.— Anal. Chem., 1965, vol. 37, p. 168—169.
1102. РЖХим, 1980, 11Г190.
1103. Rowlands D. L. G., Watson P. G. U. K. Atom. Energy Author Res. group.,
NAERE R 6855. 1972. 41 p.; РЖХим, 1973, 19Г142.
1104. Rubeska J.— Progr. Anal. Atom. Spectrosc., 1979, vol. 2, p. 309—353.
1105. Ruch R. R., Tera F., Morrison G. N.— Anal. Chem., 1964, vol. 36, p. 2311 —
2315.
1106. Russel B. G.— Analyst, 1966, vol. 91, p. 511—519.
1107. РЖХим, 1977, ЗГ217.
1108. РЖХим, 1962, 20Д35.
1109. Rybach L.— Radiochim. acta, 1964, vol. 2, p. 138—140.
1110. Rynasiewicz J., Sleeper M. P., Ryan J. W.— Anal. Chem., 1954, vol. 26,
p. 935—936.
1111. Rezac Z., Dovrak J.— Chem. prum., 1964, sv. 14, s. 485—487; РЖХим,
1965, 7Г172.
1112. Sajo F. M., Klug O. N., Bellomo A.— Atti Soc. pelorit. sci. fis., mat.
e natur., 1964, vol. 10, p. 255—267; РЖХим, 1965, 19Г116.
1113. Sajo I.— Magy. kern, folyoirat, 1969, k. 75,- old. 1—3; РЖХим, 1969, 21Г85.
1114. Sajo I., Posgay G., Horvath M.— Femipari kut. intez. kozl., 1966, k. 6,
old. 307—314; РЖХим, 1968, 6Г137.
1115. Salaria G. B. S.— Anal. chim. acta, 1955, vol. 13, p. 513—517.
1116. Salmon L.— Rep. Atom. Energy Res. Establ., 1957, N G/M 323, 6 p.;
РЖХим, 1958, № 57146.
1117. Sanchez M. L., Casanova J.— Ion (Esp.),1975, vol. 35, p. 725—729.
1118. Sanchez M. L., Casanova J. L., Casanova J.— An. Real. soc. esp. fis.
у quim., 1975, vol. 71, p. 208—213; РЖХим, 1976, 21Г93.
1119. Sato N., Kato T., Suzuki N.— Radiochim. acta, 1974, vol. 21, p. 63—68.
231
1120. Sattler P. F., Schreinlechner I. E.— Anal. Chem., 1977, vol. 49, p. 80—82.
1121. Savage H. R., Butt J. B., Tallmadge J. A.— Analvst, 1966, vol. 91,
p. 714—718.
1122. Scarborough J. M., Bingham C. D., DeVries P. F.— Anal. Chem., 1967,
vol. 39, p. 1394—1397.
1123. Scarborough J. M., DeVries P. F.— Anal. Chem., 1967, vol. 39, p. 826 —
829.
1124. Schiller P., Tolgyessy J— Chem. zvesti, 1957, sv. 11, s. 508—510.
1125. Schmid R., Gutmann V.— Chem. zvesti, 1970, sv. 23, s.-746—758.
1126. Schmitt R. A., Linn T. A., jun., Wakita H.— Radiochim. acta, 1970,
vol. 13, p. 200—212.
1127. Schneider H., Borgstedt H.-U.— In: Liquid alkali metals. Proc. Intern,
conf., Nottingham, 1973. L., 1973, p. 77—80; РЖХим, 1975, 13Г111.
1128. Schneider H., Schonwald D., Schumann H.— Ztschr. anal. Chem., 1970,
Bd. 249, S. 175—176.
1129. Schneider H., Schumann H.— Ztschr. anal. Chem., 1971, Bd. 256, S. 337—
340.
1130. Schreiber E.— Ztschr. anal. Chem., 1965, Bd. 210, S. 93—106.
1131. Schreinlechner I., Sattler P., Kozuh J.— Mikrochim. acta, 1980, N 2,
S. 423—433.
1132. Schroeder G. L., Winchester J. W.— Anal. Chem., 1962, vol. 34, p. 96—99.
1133. Schubert K. D., Petzold M., Heine S.— Bergakademie, 1967, Bd. 19,
S. 147—150; РЖХим, 1967, 21Г74.
1134. Schubiger A., Wyttenbach A.— Chem. Rdsch. (Schweiz.), 1977, Bd. 30,
N 20, S. 16.
1135. Schuknecht W., Schinkel H.— Brennst.-Chem., 1957, Bd. 38, S. 275—277.
1136. Schuknecht W., Schinkel H.— Ztschr. anal. Chem., 1954, Bd. 143, S. 321—
330.
1137. Schuknecht W., Schinkel H.— Ztschr. anal. Chem., 1963, Bd. 194, S. 161—
. 183.
1138. Schulek E., Koros E.— Acta chim. Acad. sci. hung., 1953, vol. 3, p. 281—
287.
1139. Schulek E., Koros E.— Ann. Univ. sci. budapest. Sect, chim., 1960, vol. 2,
p. 561—566; РЖХим, 1961, 10Д39.
1140. Schulte К. E., Henke G., Tjan K. S.— Ztschr. anal. Chem., 1970, Bd.
252, S. 358—366.
1141. Schwarzenbach G.— Helv. chim. acta, 1967, vol. 50, p. 2333.
1142. Schwarzenbach G., Geier G.— Helv. chim. acta, 1961, vol. 44, p. 859—869.
1143. РЖХим, 1960, № 92090.
1144. Sekerka I., Lechner J. F.— Anal. Lett., 1974, vol. 7, p. 463—472.
1145. Selleri R., Caldini O.— Anal. Chem., 1961, vol. 33, p. 1944—1945.
1146. Sellschop J. P. F., Bibby D. M., Erasmus C. S., Mingay D. W.— Dia-
mond Res., 1974, p. 43—50; РЖХим, 1976, 9Г173.
1147. Sen B.— Ztschr. anal. Chem., 1957, Bd. 157, S. 2—6.
1148. Serbanescu A., Banateanu G., Fedin T., Badea S.— Rev. chim. (RSR),
1975, vol. 26, p. 863—869.
1149. Sermin M.— Analusis, 1973, N 3, p. 186—189.
1150. Shah K. R., Filby R. H., Haller W. A.— J. Radioanal. Chem., 1970,
vol. 6, p. 185—192.
1151. Shen Shih-nien, Zhang Zhu-jun.— Хуасюэ сюэбао: Acta chim. sinica, 1964,
vol. 30, p. 21—24; РЖХим, 1965, 13Г48.
1152. Sherma J., Goldstein D. A., Gutai J. P.— Separ. Sci., 1967, vol. 2, p. 691—
\₽95.
1153. Sherma J., Strain H.— Anal. Chem., 1962, vol. 34, p. 76—80.
1154. Sherwood A. E.— Met. and Metal Form., 1972, vol. 39, p. 216—217;
РЖХим, 1972, 22Г29.
1155. Shimizu K., Takemoto N.— Bull. Soc. Salt Sci. Jap., 1958, vol. 12, p. 316—
322; РЖХим, 1960, № 51644.
1156. Shiraiwa T., Fujuno N., Murayama J.— Сумитомо киндзоку, Sumitomo
Metals, 1974, vol. 26, p. 470—479; РЖХим, 1976, 8Г52.
232
1157. Shukla S. К.— Chromatographia, 1973, vol. 6, p. 371—374.
1158. Silverman L., Seitz R.L.— Anal. chim. acta, 1959, vol. 20, p. 340—343.
1159. Silverman L., Shideler M.— Anal. Chem., 1955, vol. 27, p. 1660—1662.
1160. Silverman L., Trego K.— Analyst, 1953, vol. 78, p. 717—721.
1161. Sinclair V. M., Drummond J.L., Smith A. W.— Analyst, 1966, vol. 91,
p. 582—586.
1162. Singh E. J., Dey A. K.— Anal. chim. acta, 1961, vol. 25, p. 57—58.
1163. Singhal К. C., Benerjee В. K.— Technology, 1975, vol. 12, p. 145—147.
1164. Sinha B. S., Boy S. K.— Taianta, 1979, vol. 26, p. 596—598.
1165. Skafi M., Lieser К. H.— Ztschr. anal. Chem., 1970, Bd. 251, S. 177—180,
1166. РЖХим, 1971, 12Г93.
1167. Skrivanek V., Klein P.— Rudy, 1963, sv. 11, s. 89—92; РЖХим, 1963,-
20Г94.
1168. Smales A . A ., Lovaridge B. A.— Anal. chim. acta, 1955, vol. 13, p. 566—
573.
1169. Smith D.L.— J. Nucl. Mater., 1974, vol. 51, p. 280—282.
1170 .. Smits R., Massari D., Juillard J., Morel J. P.— Anal. Chem., 1976, vol. 48,-
p. 458—464.
1171. Smulek W., Lada W.— Radioanal. Lett., 1977, vol. 30, p. 199—207.
1172. Smythe L. E., Bruin H. J.— Analyst, 1918, vol. 83, p. 242—244.
1173. Sobkowska A., Basinska M.— Mikrochim. acta, 1975, N 2, S. 227—234.
1174. Sparks R. J., Glasby G. P.— N. Z. Sci. Rev., 1973, vol. 16, p. 643—655;:
РЖХим, 1974, 15Г181.
1175. Stamm H. H.— J. Radioanal. Chem., 1973, vol. 14,.p. 367—373.
1176. Stamm H. H.— In: Liquid alkali metals. Proc. Intern, conf., Nottingham,
1973. L., 1973, p. 65—69; РЖХим, 1975, 11Г125.
1177. Stamm H.H., Clauss H., Nolte K.— Ztschr. anal. Chem., 1973, Bd. 266,
S. 337—340.
1178. Starer M. H.— C. r. Acad. sci. C., 1971, vol. 273, p. 342—345.
1179. Starer M. H.— C. r. Acad. sci. C, 1971, vol. 273, p. 1573—1576.
1180. Sterlinski S., Dybszynski R.— Nukleonika, 1966, sv. 11, s. 533—553.
1181. Steyn W. M., Wet W.J.— Separ. Sci., 1972, vol. 7, p. 457—464.
1182. Strauss H., Rutkowski R.— Chem. Techn., 1974, Bd. 26, S. 718—720.
1183. Strelow F. W. E., Coetzee J.H-.J., Van Zyl C. R.— Anal. Chem., 1968,
vol. 40, p. 196—199.
1184. Strelow F. W. E., Liebenberg C. J., Toerien F. S.— Anal. chim. acta,
1968, vol. 43, p. 465—473.
1185. Strelow F. W.E., Liebenberg C.J., Victor A. H.— Anal. Chem., 1974,
vol. 46, p. 1409—1414.
1186. Strelow F. W. E., Rethemeyer R.., Bothnia C. J.— Anal. Chem., 1965,
vol. 37, p. 106—111.
1187. Strelow F. W. E., Weinert С. H. S. W., Walt T. N.— Anal. chim. acta,
1974, vol. 71, p. 123—132.
1188. Strelow F. W. E., Zyl C. R., Bothma J. C.— Anal. chim. acta, 1969, vol.
45, p. 81—92.
1189. РЖХим, 1971, 18Г47.
1190. Sucha L.— Sb. VSCHT. Praze B, 1966, sv. 9, s. 147—155.
1191. Sugimae A., Skogerboe R. K.— Anal. chim. acta, 1978, vol. 97, p. 1—11.
1192. Suhr N.H., Ingamells С. O.— Anal. Chem., 1966, vol. 38, p. 730—734.
1193. Sullivan J. V., Walsh A.— Spectrochim. acta, 1966, vol. 22, p. 1843—
1852.
1194. Surowiec J.— Pr. nauk. Inst. chem. nieorg. met. pierwiast. rzadkich PWr.,
1975, N 32, c. 103—113; РЖХим, 1976, 4Г198.
1195. Svoboda V., Browner R. F., Winefordner J. D.— АррГ. Spectrosc., 1972,
vol. 26, p. 505—520.
1196. Svoboda V., Kleinmann I.— Anal. Chem., 1968, vol. 40, p. 1534—1540.
1197. РЖХим, 1976, 17Г262.
1198. РЖХим, 1974, 5Г129.
1199. Sauman Z.— Chem. zvesti, 1957, sv. 11, s. 168—174.
1200. Sulcek Z., Povondra P., 'Stangl R.— Coll. Czechosl. Chem. Commun., 1965,
vol. 30, p. 380—387.
233
1201. Taimni I. К., Agarwal R. P.— Anal. chim. acta, 1953, vol. 9, p. 216—222.
1202. Takahashi M.— J. Nucl. Sci. and Technol., 1973, vol. 10, p. 54—60.
1203. Takahashi M., Matsuda Y., Toita Y., Ouchi M., Komori T.— Бунсэки ка-
гаку, Jap. Anal., 1971, vol. 20, p. 1085—1091; РЖХим, 1972, ЗГ134.
1204. Takahashi M., Ouchi M.— Бунсэки кагаку, Jap. Anal., 1972, vol. 21,
p. 936—938; РЖХим, 1973, 1Г120.
1205. Takahashi M., Yoshida Z., Aoyagi H., Izawa K.— Mikrochim. acta, 1974,
N 2, S. 329—338.
1206. Takamoto S.— Бунсэки кагаку; Jap. Anal., 1960, vol. 9, p. 281—284;
РЖХим, 1960, № 88277,
1207. Takeuchi T., Suzuki M.— Taianta, 1964, vol. 11, p. 1391—1397.
1208. Tamnev В. K., Havezov I., lotova L. K.— Ztschr. anal. Chem., 1974, Bd.
271 S. 349__351.
1209. Taulli T.A.— Anal. Chem., 1960, vol. 32, p. 186—189.
1210. Tausch W., Gausmann H., Bottger K.— Ztschr. anal. Chem., 1976, Bd. 279,
S. 102—103.
1211. Taylor M.E., Berlin A., Robinson R.J.— Anal. chim. acta, 1965, vol.
33, p. 102—107.
1212. Taylor M. E., Robinson R. J.— Anal. Chem., 1962, vol. 34, p. 533—536.
1213. Tera F., Ruch R. R., Morrison G. Anal.Chem., 1965, vol. 37, p. 358—
360.
1214. Tesarova I.— Chem. listy, 1974, sv. 68, s. 12—22.
1215. Than E., Konig H.— Ztschr. Chem., 1972, Bd. 12, S. 344—345.
1216. Thompson B. A., StrauseB.M., Leboeuf M.B.— Anal. Chem., 1958,
vol. 30, p. 1023-1027.
1217. Thompson D. E., Danchik R. S.— Anal. Lett., 1975, vol. 18, p. 699—708.
1218. Thompson M., Krull U. J., Worsfold P. J.— Taianta, 1979, vol. 26,
p. 1015—1018.
1219. Tiberiu SBui. Inst, cercet. piscicob., 1957, vol. 16, p. 61—64; РЖХим,
1959, № 19082.
1220. Tobailem J.— Ann. Phys. (US), 1955, vol. 10, p. 783—829.
1221. Toei K'., Iwachido -TСайсин-но бунсэки кагаку, 1975, № 26, p. 1—20;
РЖХим, 1975, 20Г60.
1222. Toei К., Kobatake TTaianta, 1966, vol. 13, p. 1194—1196.
1223. Tokres E. T., Lopez M.— Rev. Soc. quim.Mex., 1976, vol. 20, p. 217—222;
РЖХим, 1977, 9Г154.
1224. Toma O., Crisan T.— Rev. chim. (RSR), 1970, vol. 21, p. 334—335.
1225. Toma O., Crisan T.— Rev. chim., (RSR), 1972, vol. 23, N 10; Chim. anal.,
1972, N 3, p. 189—192; РЖХим, 1973, 11Г176.
1226. Tomi B., Manoliu C.— Rev. chim. (RSR), 1976, vol. 27, p. 1082—1084.
1227. Tomicek O., Pulpan R.~ Chem. listy, 1955, sv. 49, s. 497—502.
1228. Tomlianovic M., Grobenski Z.— Atom. Absorpt. Newslett., 1975, vol, 14,
p. 52—56.
1229. Townsing P. C., Posner A. M., Quirk j. P.— Anal. chim. acta, 1967, vol.
38, p. 464—467.
1230. Toyama I., Takauchi K.— Бунсэки кагаку; Jap. Anal., 1960, vol. 9,
p. 123—127; РЖХим, 1960, № 84400.
1231. Toyama I'., Takauchi K.— J. Spectrosc. Soc. Jap., 1958, vol. 7, N 2,
p. 22—25; РЖХим, 1960, № 4586.
1232. Tribalat S., Grail M.— C. r. Acad. sci. C, 1966,". vol. 263, p. 399—402.
1233. Tristram D. R., Phillips C. S. G.— J. Chem. Soc., 1955, N 2, p. 580—581.
1234. Tsubota H., Kitano Y.— Bull. Chem. Soc. Jap., 1960, vol. 33, p. 770—775.
1235. Turk G. S„ Mallard W. G„ Schenck P. K„ Smyth K. C.~ Anal. Chem.,
1979, vol. 51, p. 2410—2412.
1236. Turk G. S., Travis J. C., de Voa J. R., O'Haver T. C.— Anal. Chem.,
1978, vol. 50, p. 817—820.
1237. Upor E., Gorbicz L., Nagy G.— Magy. kem. folyoirat., 1969, k. 75, old.
549—553; РЖХим, 1970, 13Г50.
1238. Ure A. M., Ewen G. J., Mitchell M. C. A.— Anal. chim. acta, 1980, vol.
118, p. 1—9.
234
•1239. Van Geet A. L.-, Templeman G. J.— Anal. Chem., 1975, vol. 47, p. 1448—
1449.
1240. Vancea M., Berindeanu G.— Ind. isoara, 1960, vol. 7, p. 399—404; РЖХим,
1961, 16Д45.
1241. Vasudeva M.A.R., Narayan V. A.— J. Ind. Inst. Sci., 1960, vol. 42,,
N.3, p. 51—55; РЖХим, 1961, 12Д40.
1242. Vendange J.— Bull. Dir. mines et geol. Gouv. gen. A. E. F., 1957, N 8,
p. 151—156; РЖХим, 1958, № 64174.
1243. Venturello G., Gualandi C., Mazzei I.— Ann. chim. (Ital.), 1959, vol. 49,
p. 149—168.
1244. Vickers T. J., Remington L. D ,,Winefordner J. D.— Anal. chim. acta, 1966,-
vol. 36, p. 42—56.
1245. Vienney J.— Anal. chim. acta, 1971, vol. 55, p. 37—46.
1246. Vigler M. S., Gaylor V. F.— Appl. Spectrosc., 1974, vol. 28, p. 342—344.
1247. Vulli M., Janghorbani M., Starke K.— Anal. chim. acta, 1976, vol. 82,
p. 121—135.
1248. Vydra F., Pribil R.~ Ztschr. anal. Chem., 1962, Bd. 188, S. 273—276.
1249. Walker J. A . J., France E. D., Drummond J. L., Smith A. W.— In:
Alkali metals. L.: Chem. Soc., 1967, p. 393—400; РЖХим, 1969, 4Г134.
1250. Walker J. A. J., France E.D., Edwards W. T.— Analyst, 1965, vol. 90 y
p. 727—731.
1251. Walker J: A. J., Seed H.— Analyst, 1965, vol. 90, p. 19—23.
1252. Walsh I. N.— Spectrochim. acta B, 1980, vol. 35, p. 107—111.
1253. Wanner D. E., Conrad F. J.— Appl. Spectrosc., 1967, vol. 21, p. 177—178'.
1254. Ward С. H., Gardner R.D., Ashley W. H., Zerwekh A.— Sci. and Techn.
Aerospace Rep., 1963, vol. 1, N 16, p. 1153; РЖХим, 1966, ЗГ131.
1255. РЖХим, 1967, 4Г135.
1256. Webber H. M., Wilson A.L.— Analyst, 1969, vol. 94, p. 209—220.
1257. Webber H. M., Wilson A. L.— Analyst, 1969, vol. 94, p. 569—574.
1258. Weeks S. J., Heraguchl H., Winefordner J.D.— Anal. Chem., 1978, vol.
50, p. 360—368.
1259. Weisz H., Fritsche U.— Mikrochim. acta, 1973, N 3, S. 361—372.
1260. Welday E. E., Baird A. K., McIntyre D. B., Madlem K. W.— Amer. Mi-
ner., 1964, vol. 49, p. 889—903; РЖХим, 1965, 12Г115.
1261. Welz B.~ Ztschr. anal. Chem., 1976, Bd. 279, S. 103—104.
1262. White J. C., Ross. W. J., Rowan R.— Anal. Chem., 1954, vol. 26, p. 210—
213.
1263. Wild P. W.— Galvanotechnik, 1976, vol. 67, p. 553—554.
1264. Willard H. H., Smith G.F.— J. Amer. Chem. Soc., 1922, vol. 44, p.,2816.
1265. Willard H. H., Smith G. F.— J. Amer. Chem., 1923, vol. 45, p. 286.
1266. Willgalis A.— Ztschr. anal. Chem., 1957, Bd. 157, S. 249—257.
1267. Williams С. H.— Anal. chim. acta, 1960, vol. 23, p. 183—185.
1268. Willis J. B.— Spectrochim. acta B, 1971, vol. 26, p. 177—189.
1269. Wilson M. F., Haikala E., Kivavo P.— Anal. chim. acta, 1975, vol. 74,
p. 395—410.
1270. Wilson M. F., Haikala E., Kivavo F.— Anal. chim. acta, 1975, vol. 74,
p. 411—421.
1271. Winefordner J.D., Vickers T.J.— Anal. Chem., 1964, vol. 36, p. 1939—
1946.
1272. Winefordner J. D., Vickers-T.J.— Anal. Chem., 1964, vol. 36, p. 1947—
1954.
1273. Winkel P., De Corte F., Hoste J.— Anal. chim. acta, 1971, vol. 56, p. 241—
259.
1274. Winkel P., Mertens J., Baenst G., Massart D. L.— Anal. Lett., 1972, vol. 5,
. p. 567—577.
1275. Wudl F.— J. Chem. Soc. Chem. Common., 1972, N 22, p. 1229—1230.
1276. Wyttenbach A., Bajo S., Farrenkothen K.— Radiochem. and Radioanal.
Lett., 1980, vol. 42, p. 307—317.
1277. Wyttenbach A., Dulakas H.— Chimia, 1968, vol. 22, p. 484—487.
1278. Yamamoto Y., Kumamaru T., Hayashi Y.— Radioisotopes, 1969, vol. 18,
N 2, p. 50—51; РЖХим, 1969, 15Г149.
235
1279. Yamane TYamada NSpectrochim. acta B, 1975, vol. 30, p. 305—307»
1280. Yasuda S., Kakiyama H.— Бунсэки кагаку, Jap. Anal., 1975, vol. 24,
p. 377—382; РЖХим, 1976, 2Г34.
1281. Yoshida H., Yohezawa C.— Бунсэки кагаку..Jap. Anal., 1973, vol. 22,
p. 929—931; РЖХим, 1974, 1Г153.
1282. Yoshimura K., Sakata K., Tarutani T.— Mem. Fac. Sci. Kyushu Univ.
C, 1979, vol. 12, p. 77—82; РЖХим, 1980, 10Г77.
1283. Yule J. W., SwansonG. A.— Atom. Absorpt. Newslett., 1969, vol. 8, N 2,
p. 30—33; РЖХим, 1969, 22Г137.
1284. Zama A., Dokiya Y., Fukami M., Aizawa S., Hamachi M., Toda S
Fuwa K.— Spectrosc. Lett., 1976, vol. 9, p. 653—661.
1285. РЖХим, 1965, ЗГ43.
1286. РЖХим, 1969, 6Г50.
1287. Zemek О., Zemek I., Hudnik V., Gomiscek S.— Vestn. Slov. kem. drust.,
1975, sv. 21, N 3/4, s. 81—88; РЖХим, 1976, 1Г159.
1288. Zimmer H.— Ztschr. anal. Chem., 1957, Bd. 155, S. 337—340.
1289. Zimmer K., Robertne B. Z.— Magy. kem. folyoirat, 1970, k. 76, old. 170—
173.
ПРИЛОЖЕНИЕ
Таблица 1
Растворимость неорганических соединений натрия (безводное соединение, г/100 г воды) в воде при различной
температуре [1, с. 1601]
Соединение 0= c 10° 20’ 30° 40° 50° 60° 70° 80’ 90° 100° c
NaBr-2H2O* NaBr * 44,3 47,5 49,4 51,4 53,7 54,2' 54,8
Na2B4u7. 10н2и 1,3 1,6 2,7 3,9 10,5 20,3
Na2B4O,.5H2O 24,4 31,5 41 52,5
NaBrO3 27,5 34,5 50,2 62,5 75'7 90 j 9
N a C2H3O2 • 3H2O . 36,3 40,8 46,5 54,5 65,5. 83 139
NaC2H3O2 119 121 123,5 126 129,5 134 139,5 146 153 161 170
Na2C20a 3,7 6,33
NaCl 35,7 35,8 36,0 36,3 36,6 37,0 37,3 37,8 38,4 39,0 39,8
NaC103 79 89 101 113 126 140 155 172 189 230'
Na2CO3.10H2O Na2CO3«H2O 7 12,5 21,5 38,8 50,5 48,5 46,4
45,8 45,5
Na2CrO4.10H2O* 24,07 33,4 47
Na2CrO4.4H2O * Na2CrO4* 47 48,97 51 53,4 55,15 55,53 55,74
Na2Cr2O7.2H2O* 61,98 64 71 76 79
Na2Cr2O7 Na4 [Fe (CN)6J 17,9 30 59 81 63
NaHCO3 6,9 8,15 9,6 11,1 12,7 14,45 16,4
NaH2PO4.2H2O 57,9 69,9 85,2 106,5 138,2
NaH2PO4-H2O NaH2PO4 158,6 179,3 190,3 207,3 225,3 246,6
Na2HAsOa 7,3 15,5 26,5 37 47 65 85 '
Na2HPO4.12H2O 1,67 3,6 7,7 20,8
8SZ
Приведена растворимость безводного соединения, г/100 г насыщ. раствора.
222 2 22512 й) И р 65 Р ^яьэ ьэ ьэ ьэ СП СП СП СР О ф ф “ о » о орк' щ » 2 о я ьэ о рэ ьэ СП о £ ьэ о 222 22222 рэ рэ рэ рэ сэ ЬЭ ЬЭ ЬЭ А со СП СИСЛ • • • ЬЭ Л 03 сл соо£-> ьэ ?-Jb3 .. н* °»Ио2я *о «Як3 Я 3 И 2! 2! 2! 2!« д 3 3 Соединение
РЭ д> ьэ ьэ 33 РР (S3 и* що о» * о * aVO3 рэ рэ ьэ ьэ О) СП оо A W о И ьэ о рэ ЬЭ СП О W S ЬЭ О рэ рэ рэ рэ рэ рэ рэ 000022!: ИДДДООр • • • со ьэ рэ р> рэ рэ S3 C_jb3 ьэ ьэ ИЯД
3,5Н2О Н20 * Я ьэ о ьэ о О4.7Н2О 04.2Н20 0*
* О® /
СП СО ►А СО СП сю СО на СП ЬЭСО^АССОО О
СО СП О СО Си 03 СО СП -3 О
61,0 со со о* о 20 ИА СО СО 4S со СО ИА 03 сл 51,5 43,8 80 168,6 о о
СО 70,0 4S 19,4 26,9 и 6,23 15,8 109 9 45,8 .88 178,7 ЬЭ о
84,7 44,05 4S О 00 СО сз 20 9,95 18,4 । 119 47,8 96 190,3 со о
26,23 30,2 48,8 102,6 28,0 31 13,50 22,2 I 129 15 49,6 104 205,0 51,8 £> О о
44,5 46,7 169,7 28,2 28,48 26,7 43 17,45 145 51 114 227,8 80,2 СП О
32,97 68,4 45,3 206,7 28,8 29,92 28,1 55 21,83 174 СО со 256,8 82,9 О ’О
36,9 31,38 30,22 294 00 09 НА о
47,7 43,7 248,8 28,3 33,95 32,95 81 30,04 4к»СПСО 00 *q -Л 296 92,4 оо о
0J © CD
JJ "со
49,3 СО 42,5 266,0 347 108 40,26 00 оз со О СО JS 302 102,2 □ oOOl
Таблица I (окончание)
Таблица II
Растворимость (в г/100 мл) хлоридов лития, натрия и калия
в органических растворителях [2, с. 52]
Растворитель LiCl NaCl КС1
Этанол 2,54 0,0649 0,0294
н-Бутанол 12,98 0,0050 0,0030
н-Бутанол’+ 6% НС1 4-0,5% НС104 10,61 0,0007 0,0005
Изобутанол 10,57 0,0005 0,0008
Пентанол 9,03 0,0020 0,0007
Изопентанол 7,30 0,0016 0,0006
н-Гексанол 7,20 0,0010 0,00005
2-Этйлгексанол 3,70 0,0001
Бензиловый спирт 1,43 0,0170 0,0160
Ацетон 3,86
Пиридин 13,39
Диоксан - « 2,8
Таблица III
Коэффициенты распределения щелочных металлов
в среде неорганических кислот
на сильнокислом катионообменнике био—рад AG-50WX8 [2, с. 63]
Кислота, N Li Na+ K+ Rb+ Cs+
НС1 0,1 33,0 52,0 106,0 120,0 182,0
0,2 19,0 28,3 64,0 72,0 99,0
1,0 4,0 5,6 14,0 15,4 19,4
2,0 2.5 3,6 7,4 8,1 10,4
4,0 1,0 1,0 1,0 1,0 1,0
HN03 0,1 33,1 54,0 99,0 118,0 148,0
0,2 18,6 29,4 59,0 68,0 81,0
1,0 4,0 6,3 11,4 13,4 16,8
2,0 2,5 3,4 5,7 6,6 7,6
4,0 1,1 1,3 2,6 2,9 3,4
H2SO4 0,1 48,0 81,0 138,0 148,0 175,0
0,2 28,2 47,7 86,0 91,0 108,0
1,0 5,8 8,9 19,4 ’ 21,3 24,7
2,0 3,0 3,7 7,4 8,3 9,1
4,0 1,1 1,7 2,9 3,1 3,5
239
Таблица IV
Разделение лития, натрия и калия
мета дом распределительной хроматографии на .бумаге [2, с. 71]
Подвижный растворитель
L1+ Na+ К+
Метанол 0,71 0,46 0,28
Метанол—вода (9:1) 0,72 0,62 0,0
Метанол + 5% 15М NH3 0,74 0,30 0,12
Метанол—СНзСООН (98:2) 0,66 0,48 0,32
Метанол—этанол (1:1) 0,65 0,23 . 0,08
Метанол—бутанол (7:3) 0,62 0,26 0,11
Метанол—пентанол (7:3) 0,63 0,25 0,09
Метанол—ацетон (3:2) 0,50 0,25 0,15
Метанол—метилэтилкетон (3:2) 0,62 0,30 0,14
Этанол 0,58 0,26 0,10
Этанол—2М СНзСООН (8:2) 0,89 0,56 0,35
Этанол—вода—пропионовая кислота 0,68 0,31 0,17
Моноэтилцеллосольв—вода—конц. НС1 (7:2:1) 0,31 0,46 0,61
Фенол, насыщенный водой 0,17 0,14 0,19
Фенол—метанол—конц. НС1(57,5:22,5:20) 0,32 0,13 0,14
Коллидин—0,4 MHN03(l:l) 0,54 0,45 0,3
Таблица V
Факторы пересчета (F). определяемой формы
на анализируемую форму [1, с. 1686]
масса анализируемой формы
масса определяемой формы
Определяемая форма Анализируемая форма F lg F
Ag NaBr NaCl NaJ 0,95390 0,54179 1,3895 1,97950 1,73383 0,14286
AgBr NaBr 0,54797 1,73876
AgCl NaCl NaC103 NaC104 0,40777 0,74264 0,85427 1,61041 1,87078 1,93159
AgJ NaJ 0,63845 1,80512
BaSO4 NaHSO4 NaHSO4-H2O Na2S Na2SO3 Na2SO4-7H2O Na2SO4 Na2S04-10H20 0,51436 0,59154 0,33436 0,53999 1,0803 0,60854 1,3803 1,71127 1,77198 1,52421 1,73239 0,03353 1,78429 0,13999
240
Таблица V (продолжение)
Определяемая форма Анализируемая форма F Ig F
В2О3 МагВдО? 1,4450 0,15987
Na2B4O7-10H2O 2,7385 0,43752
Вг Na 0,28769 1,45892
NaBr 1,2877 0,10981
Na2O 0,38779 1,58860
CaClz NaCl 1,0532 0,02250
СаС03 Na2C0s 1,0590 0,02488
CaF2 NaF 1,0756 0,03165
СаО ГТагСОз 1,8900 0,27647
CaSO4 Na2CO8 0,77852 1,89127
Cl Na 0,64842 1,81186
NaCl 1,6484 0,21707
Na2O 0,87404 1,94153
СО2 Na2CO8 2,4083 0,38172
Na2O 1,4083 0,14870
Н3ВО3 Na2B4O7 0,8135g 1,91040
Na2B4O,-10H2O 1,5419 0,18805
J Na 0,18116 1,25806
NaJ 1,1812 0,07231
Na2O 0,2442o 1,38774
kbf4 Na2B4O7 0,3995g 1,60161
Mg2As2O7 Na2HAsO3 1,0945 0,03922
Na2HAsO4 1,1976 0,07830
MgCl2 NaCl 1,2275 0,08901
Mg2P2O7 Na2HP0412H20 3,2181 0,50760
NaNH4HPO4.4E[2O 1,8786 0,27383
Na8PO4 1,4731 0,16823
Na4P267-10H20 2,0040 0,30190
N NaNOs 6,0679 0,78304
Na Br 3,4760 0,54108
Cl 1,5422 0,18814
J 5,5200 0,74194
NaBr 4,4760 0,65089
NaCl 2,5422 0,40521
Наг^Оз 2,3051 0,36269
Na NaHCO3 3,6540 0,56277
NaJ 6,5200 0,81425
Na2O 1,3480 0,12968
Na2SO4 3,0892 0,48985
Na2B4O7 B2O3 0,69203 1,84013
H3BO3 1,2291 0,08960
Na2B4O7.WH2O B2O3 0,36516 1,56248
H3BO8 0,64856 1,81195
241
Таблица V (продолжение)
Определяемая форма Анализируемая форма F lg F
NaBr Ag 1,0483 0,02050
AgBr 1,8249 0,26124
Br 0,77658 1,89019
Na 0,22342 1,34911
Na2O 0,30116 1,47879
NaC2H3O2 • Mg(C2H3O2)2 • Na 0,015266 2,18372
• 30 О2(С2Н3О2)2 6,5Н2О NaBr 0,068329 2,83461
Na2CO3 0,035189 2,54641
NaCl 0,038809 2,58893
NaF 0,027882 2,44532
NaHC03 0,055782 2,74649
NaJ 0,099533 2,99797
NaNO3 0,056439 2,75158
Na2O 0,020578 2,31340
NaOH 0,026559 2,42421
Na2SO4 0,047159 2,67357
NaC2H3O2' Zn(C2H3O2)2 • Na 0,014948 2,17458
• 30 О2(С2Н3О2)2 • 6Н2О NaBr 0,066906 2,82547
Na2CO3 0,034456 2,53727
NaCl 0,038001 2,57979
NaF 0,027301 2,43618
NaHCO3 0,054620 2,73735
NaJ 0,097460 2,-98883
NaN03 0,055263 2,74243
NaOH 0,026006 2,41507
Na2O 0,020149 2,30426
Na2SO4 0,046177 2,66443
Ма2СО3 CaC03 0,94432 1,97512
CaO 0,52909 1,72353
CaS04 1,2855 0,10873
co2 0,41523 1,61828
Na 0,43382 1,63731
NaCl 1,1029 0,04252
NaHCO3 1,5852 0,20008
NaOH 0,75475 1,87780
Na2O 0,58477 1,76699
Na2SO4 1,3402 0,12716
Na2CO3-10H2O Na2SO4 0,49641 1,69584
NaCl Ag , 1,8457 0,26617
AgCl 2,4524 0,38959
Cl 0,60664 1,78293
Na 0,39336 1,59479
NaC103 1,8212 0,26037
NaC104 2,0950 0,32118
Ma2C03 0,90673 1,95748
NaHCO3 1,4373 0,15756
Na2HPO4 1,2145 0,08438
Na2SO4 1,2152 0,08464
NaC10s AgCl 1,3465 0,12922
NaCl 0,54908 1,73963
242
Таблица. V (окончание)
Определяемая форма Анализируемая форма F 1g F
NaC104 AgCl 1,1706 0,06841
NaCl 0,47733 1,67882
NaF CaF2 0,92972 1,96835
Na2HAsO3 Mg2As2O7 0,91366 1,96078
NaHCOs Na 0,27367 1,43723
NaCl 0,69573 1,84244
0,63084 1,79992
Na2O 0,36890 1,56690
Na2HPO4 NaCl 0,82341 1,91562
Na2O 0,43660 1,64008
P2O6 0,49995 1,69892
NaHS03 SO2 0,61563 1,78932
NaHS04 BaSO4 1,9442 0,28873
NaHSO4-H2O BaS04 1,6905 0,22802
NaJ AgJ 1,5663 0,19488
J 0,84663 1,92769
Na 0,15337 1,18575
Na2O 0,20674 1,31543
NaNH4HPO4-4H2O NH3 0,081462 2,91096
NaNO3 N 0,16480 1,21696
NH3 0,20038 1,30185
NaOH Na2CO3 1,3249 0,12220
Na4P2O7 Na2HPO4-12H2O 2,6938 0,43036
Na2S BaS04 2,9908 0,47579
Na2SO4 BaSO4 1,8519 0,26761
Na2S04-10H20 BaS04 0,72446 1,86001
Таблица VI
Плотность растворов арсената натрия при 14° С [1, с. 1882]
Содержание Na2HAsO4 Плотность, г/мл Содержание NajHAsO* Плотность, г/мл
% г/л % г/л
1 10,08 1,0083 10 109,8 1,0980
2 20,35 1,0175 12 134,4 1,1197
4 41,46 1,0365 14 159,9 1,1419
6 63,38 1,0563 16 186,3 1,1645
8. 86,14 1,0768
Примечание. В табл. VI—XXI плотность растворов солей отнесена к плотности во-
ды при 4° С.
243
Таблица VII
Плотность растворов кристаллогидрата арсената натрия при 14° С [1, с. 1882]
Содержание NaJHA8Ot-12HBO Плотность, г/мл Содержание NaaHAsO*-1211,0 Плотность, г/мл
% г/л % г/л
2,163 21,80 1,0083 21,63 237,4 1,0980
4,325 44,01 1,0175 25,95 290,6 - 1,1197
8,650 89,66 1,0365 30,28 345,7 1,1419
12,98 137,1 1,0563 34,60 402,9 1,1645
17,30 186,3 1,0768
Таблица VIII
Плотность растворов карбоната
натрия при 20° С
[1, с. 1886]
Таблица IX
Плотность растворов
кристаллогидрата карбоната натрия
при 20° С [1, с. 1886]
Содержание Na,CO3 Плотность, г/мл Содержание NasCOa-lOH/) Плотность, г/мл
% г/л % г/л
1 10,09 1,0086 2,70 27,23 1,0086
2 20,38 1,0190 5,40 55,02 1,0190
4 41,59 1,0398 10,80 112,3 1,0398
6 63,64 1,0606 16,20 171,8 1,0606
8 86,53 1,0816 21,60 233,6 1,0816
10 110,3 1,1029 27,00 297,7 1,1029
12 134,9 1,1244 32,40 364,3 1,1244
14 160,5 1,1463 37,80 433,3 1,1463
Таблица X
Плотность растворов бромида натрия при 20° С [1, с. 1886]
Содержание NaBr Плотность, г/мл Содержание NaBr Плотность, г/мл
% г/л % г/л
1 10,06 1,0060 18 207,8 1,1546
2. ' 20,28 1,0139 20 234,9 1,1745
4 41,19 1,0298 22 262,9 1,1951
6 62,77 1,0462 24 291,9 1,2163
„8 85,05 1,0631 26 321,9 1,2382
10 108,0 1,0803 28 353,0 1,2608
12 131,8 1,0981 30 385,2 1,2841
14 156,3 1,1164 35 471,2 1,3462
16 181,6 1,1352 40 565,5 1,4138
244
Таблица XI
Плотность растворов хлорида натрия при 20° С [1, с. 1887]
Содержание NaCl Плотность, г/мл Содержание NaCl Плотность, г/мл
% г/л % г/л
1 10,05 1,0053 14 154,1 1,1009
2 20,25 1,0125 16 178,6 1,1162
4 41,07 1,0268 18 203,7 1,1319
6 62,48 1,0413 20 229,6 1,1478
8 84,47 1,0559 22 256,1 1,1640
10 107,1 1,0707 24 283,3 1,1804
12 1зо ;з 1,0857 26 311,3 1,1972
Таблица ХП
Плотность растворов нитрата натрия при 20° С [1, с. 1890]
Содержание NaNO3 Плотность, I г/мл Содержание NaNO3 Плотность, г/мл
% г/л % г/л
1 10,05 1,0049 20 228,6 1,1429
2 20,23 1,0117 22 255,0 1,1589
4 41,02 1,0254 24 282,0 1,1752
6 62,35 1,0392 26 309,8 1,1917
8 84,26 1,0532 28 338,4 1,2085
10 106,7 1,0674 30 367,7 1,2256
12 129,8 1,0819 35 444,5 1,2701
14 153,5 1,0967 40 527,0 1,3175
16 177,9 1,1118 45 615,7 1,3683
18 202,9 1,1272
Таблица XIII
Плотность растворов нитрита натрия при 16° С [1, с. 1890]
Содержание NaNO, Плотность, г/мл Содержание NaNO3 Плотность, г/мл
% г/л % г/л
1 10,06 1,0058 12 129,8 1,0816
2 20,25 1,0125 14 153,4 1,6959
4 41,04 1,0260 16 177,6 1,1103
6 62,38 1,0397 18 202,5 1,1248
8 10 84,28 106,8 1,0535 20 227,9 1,1394
245
Таблица XIV
Плотность растворов тартрата натрия — калия при 20е С [1, с. 1891]
Содержание NaKC1II4O„ Плотность, г/мл Содержание NaKCjH^O, Плотность, г/мл
% г/л % г/л
1 10,05 1,0049 20 228,4 1,1419
2 20,23 1,0116 22 254,7 1,1576
4 41,01 1,0252 24 281,6 1,1735
6 62,34 1,0390 26 309,3 1,1896
8 84,24 1,0530 28 337,7 1,2059
10 106,7 1;0673 30 366,8 1,2225
12 129,8 1,0818 32 396,6 1,2394
14 153,5 1,0965 34 427,2 1,2566
16 177,8 1,1114 36 458,7 1,2742
18 202,8 1,4265
Таблица XV
Плотность растворов кристаллогидрата тартрата натрия—калия
при 20° С [1, с. 1891]
Содержание NaKCJHjO, х X 4Н,О Плотность, г/мл Содержание NaKCJLO, X X 4Н2О ПЛОТНОСТЬ, г/мл
% г/л % г/л
1,343 13,50 1,0049 26,86 306,7 1,1419
2,686 27,17 1,0116 29,55 342,0 1,1576
5,372 8,058 55,07 1,0252 32,23 378,2 1,1735
83,72 1,0390 34,92 415,4 1,1896
10,74 113,1 1,0530 37,60 453,5 1,2059
13,43 143,3 1,0673 40,29 492,5 1,2225
16,12 174,3 1,0818 42,98 532,6 1,2394
18,80 206,2 1,0965 45,66 573,8 1,2566
21,49 238,8 1,1114 48,35 616,1 1,2742
24,17 272,3 1,1265
Таблица XVI
Плотность растворов сульфата натрия при 20° С [1, с. 1895]
Содержание Na2SO4 Плотность, г/мл Содержание Na2SO4 Плотность, г/мл
% г/л % г/л
1 10,07 1,0073 14 158,3 1,1306
2 20,33 1,0164 16 184,1 1,1506
4 41,39 1,0348 18 210,8 1,1709
6 63,21 1,0535 20 238,3 1,1915
8 85,79 1,0724 22 266,7 1,2124
10 109,2 1,0915 24 296,1 1,2336
12 133,3 1,1109
246
Таблица XVII
Плотность растворов кристаллогидрата сульфата натрия при 20s С [1, с. 189а]
Содержание Na^Ot-10HsO Плотность, г/мл Содержание NajSOjlOHjO Плотность, г/мл
% г/л % г/л
2,268 22,85 1,0073 31,75 359,0 1,1306
4,536 46,11 1,0164 36,29 417,6 1,1506
9,073 93,88 1,0348 40,83 478,1 1,1709
13,61 143,4 1,0535 45,36 540,5 1,1915
18,15 194,6 1,0724 49,90 605,0 1,2124
22,68 27,22 247,6 302,4 1,0915 1,1109 54,44 671,5 1,2336
Таблица XVIII
Плотность растворов тартрата натрия при 20° С [1, с. 1901]
Содержание NajC^O, Плотность, г/мл Содержание Na^HjO, Плотность, г/мл
% г/л % г/л
1 10,05 1,0052 16 178,5 1,1156
2 20,25 1,0123 18 203,6 1,1313
4 41,06 1,0266 20 229,4 1,1471
6 62,46 1,0410 22 255,9 1,1633
8 84,44 1,0555 24 283,1 1,1797
10 107,0 1,0702 26 311,0 1,1963
12 14 130,2 154,0 1,0851 1,1002 28 339,7 1,2132
Таблица XIX
Плотность растворов кристаллогидрата тартрата натрия при 20° С
[1, с. 1901]
Содержание NajC^Oj^HjO Плотность, г/мл Содержание Na2C1H1Oe-2H,O Плотность, г/мл
% г/л % г/л
1,186 11,92 1,0052 18,97 211,6 1,1156
2,371 24,01 1,0123 21,34 241,4 1,1313
4,743 .48,69 1,0266 23,71 272,0 1,1471
7,114 74,06 1,0410 26,09 303,5 1,1633
9,486 100,1 1,0555 28,46 335,7 1,1797
11,86 126,9 1,0702 30,83 368,8 1,1963
14,23 16,60 154,4 182,6 1,0851 1,1002 33,20 402,8 1,2132
247
Таблица XX
Плотность растворов тиосульфата натрия при 20° С [1, с. 1902]
Содержание Na£S,Os Содержание Na£S£O£ Плотность, г/мл
% г/л % г/л
1 10,07 1,0065 18 207,9 1,1551
2 20,30 1,0148 20 234,8 1,1740
4 41,26 1,0315 22 262,5 1,1932
6 62,90 1,0483 24 291,1 1,2128
8 85,23 1,0654 26 320,5 1,2328
10 108,3 1,0827 28 350,9 1,2532
12 132,0 1,1003 30 382,2 1,2739
14 156,5 1,1182 35 464,6 1,3273
16 181,8 1,1365 40 553,1 1,3827
Таблица XXI
Плотность растворов кристаллогидрата тиосульфата натрия при 20° С
[1, с. 1902]
Содержание Ка28гОг-!>Н2О Плотность, г/мл Содержание Na£S2Oa-5H,O Плотность, г/мл
% Г/л % г/л
1,570 15,80 1,0165 28,25 326,4 1,1551
3,139 31,86 1,0148 31,39 368,6 1,1740
6,279 64,77 1,0315 34,53 412,1 1,1932
9,418 98,73 1,0483 37,67 456,9 1,2128
12,56 133,8 1,0654 40,81 503,1 1,2328
15,70 170,0 1,0827 43,95 550,8 1,2532
18,84 207,3 1,1003 47,09 599,9 1,2739
21,98 245,7 1,1.182 54,94 729,2 1,3273
25,12 285,4 1,1365 62,79 868,2 1,3827
Таблица XXII
Спектральные линии натрия [3, с. 567—569]
Длина волны, нм Яркость Энергия возбужде- ния, эВ Длина волны, нм Яркость Энергия возбужде- ния, эВ
I 2337,913 4,28 I 1476,748 4,59
1 2334,841 4,28 I 1267,917 4,59
1 2208,367 3,75 I 1140,378 К 12 3,20
I 2205,644 3,75 I 1138,145 К 11 3,20
I 1846,525 4,29 I 1119,721 К 2 4,86
I 1638,885 4,51 I 1119,019 К 1 4,86
I 1637,385 4,51 I- 1083,487 К 8 4,76
I 1477,973 4,59 I 1074,929 К 9 4,35
248
Таблица XXII (продолжение)
Длина волны, нм Яркость Энергия возбужде- ния, эВ Длина волны, нм Яркость энергия возбужде- ния, эВ
I 1074,644 I 1057,228 I 1056,600 I 996,128 I 946,594 I 915,388 I 894,296 I 865,089 I 864,992 I 819,482 I 818,326 I 781,024 I 780,978 I 737,349 I 737,323 I 616,074 I 615,422 I 589,592 I 588,995 I 568,822 I 568,263 I 515,340 I 514,884 I 498,281 I 497,854 I 475,182 I 474,794 I 466,856 I 466,481 I 454,519 1 454,163 I 449,765 I 449,418 I 442,325 I 441,989 I 439,334 I 439,003 I 434,474 I 434,149 I 432,462 I 432,140 К 10 К 3 К 1 К 7 К 6 К 4 К 2 К 6 К 7 220 110 К 3 К 1 К 1 К 2 6 . 3 1000 2000 14 7 Д 2 Д 1 Д 2 Д 1 Д 2 Д 1 Д 2 Д I К 8 К 7 К 11 К 10 К 7 К 6 К 9 К 8 К 5 К 4 К 7 К 6 4,35 4,93 4,93 4,86 4,93 4,97 5,00 4,62 4,62 3,61 3,61 4,78 4,78 4,87 4,87 4,12 4,12 2,10 2,11 4,29 4,29 4,51 4,51 4,59 4,59 4,71 4,71 4,76 4,76 4,83 4,83 4,86 4,86 4,91 4,91 4,92 4,92 4,96 4,96 4,97 4,97 I 429,101 I 428,784 I 427,679 I 427,364 I 425,252 I 424,941 I 424,208 I 423,899 II 412,307 II 371,107 II 363,127 II 353,301 I 342,686 I 330,298 I 330,237 II 328,575 II 327,422 II 325,797 II 321,219 II 318,978 II 317,906 II 316,373 II 314,927 II 313,548 II 312,937 II 309,273 II 307,832 II 307,433 II 305,616 II 305,366 II 304,559 II 303,707 II 302,907 II 301,540 II 298,418 II 297,966 II 297,499 II 295,123 К 3 К 2 К 5 К 4 К 2 К 1 К 3 К 2 К 3 К 6 К 8 К ю К 6 15 30 К 8 К 5 К 6 К 6 К 6 К 5 К 6 К 6 К 5 К 6 К 10 К 6 К 6 К 6 К 6 К 5 К 5 К 6 К 6 К 7 К 5 К 6 К 8 4,99 4,99 5,00 5,00 5,02 5,02 5,03 5,03 41,28 36,34 36,34 36,34 3,62 3,75 3,75 37,09 40,98 41,00 37,17 37,20 40,99 41,00 37,25 36,96 36,89 36,85 41,10; 36,96 41,11. 36,89 41,25 41,23 41,24 41,29; 41,00 41,00 37,12; 41,10 41,06 37,17 41,04
249
Таблица ХХП (окончание)
Длина волны, нм Яркость Энергия возбужде- ния, эВ Длина волны, нм Яркость Энергия возбужде- ния, эВ
II 294,744 К 5 41,29 111249,315 К 5 38,29
II 293,772 К 5 41,11 I 249,073 К 3 4,98
II 291,905 К 5 41,09 111247,470 Е 40 50,40
II 291,752 К 5 37,09 111246,888 Е 30 51,47
II 290,491 К 7 37,20 . 111245,940 Е 45 51,36
II 289,395 К 6 41,24 III 230,998 Е 30 51,68
I 289,362 К 1 4,28 111228,572 Е 35 51,87
II 288,114 К 6 37,24 111227,848 Е 40 51,89
II 287,127 К 5 37,25 111225,144 Е 45 51,10i
II 285,948 К 5 37,18 III 224,667 Е 40 51,02
I 285,301 К 15 4,35 111223,944 Е 45 51,10
I 285,281 К 16 4,35 111223,217 Е 40 51,87
II 284,172 К 7 37.20 111223,030 Е 50 50,95
II 280,951 К 5 37,25 111222,590 Е 45 51,07
I 268,043 К 7 4,62 III 221,417 Е 25 51,10
I 268,034’ К 8 4,62 111220,278 Е 40 51,02
II 267,809 К 5 40,98 111202,856 Е 25 57,06
II 267,183 К 6 40,99 III 201,188 Е 30 57,52
II 266,100 К 7 41,00 111200,524 Е 30 51,74;
II 261,182 К 7 41,08 57,65
I 259,392 К 3 4,78 III 198,558 Е 30 51,74
I 259,387 К 3 4,78 111195,121 Е 40 51,74
111256,332 Е 25 50,40 111194,643 Е 20 61,03
111255,362 Е 25 50,35 III 193,387 Е 30 57,43
I 254,387 4,86 III 192,627 Е 45 57,38
I 254,384 К 1 4,86 III 185,673 Е 20 57,08
II 253,155 К 6 41,24 III 185,039 Е 18 57,10
I 251,221 К 4 4,94 III 184,958 Е 35 57,06
I 251,213 К 2 4,94 III 184,436 Е 20 57,08;
111251,038 Е 20 50,44 57,74
111249,705 Е 50- 50,35 III 183,522 Е 15 61,41
Примечание. Римская цифра перед длиной волны указывает степень ионизации
натрия; К — трубка с полым катодом; Д — дуга; Е — Сезэлектродный разряд.
ЛЛТЕРАТУРА
1. Handbook of chemistry and physics. Ed. 37, Р. 2. Cleveland: Chem. Rubber
PubL Co., 1955—1956, 3156 p.
2. Полуэктов П. С., Мешкова С. Б., Полуэктова E. H. Аналитическая хи-
мия лития. M.: Наука, 1975. 203 с.
3. Зайдель А.Н., Прокофьев В. К., Райский С. М.,„ Славный В. А., Шрей-
дер Е.Я.— В кн.: Таблицы спектральных линий. 4-е изд. М.: Наука,
1977, с. 567—569.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азосоединения 25
Активационные методы 137
Аминополикарбоновые кислоты 23
Аминосоединения 29
Антрахинон-1-сульфокислота 23
Антрахинон-2-сульфокислота 23
Ароматические сульфокислоты 23
Атомно-абсорбционная спектромет-
рия 113
Барбитуровая кислота 24
5-Бензамидоантрахинон-2-сульфо-
кислота 24, 33, 63, 82
Винная кислота 22, 178
Виолуровая кислота 24, 79
Вольтамперометрия 90
Гексанитродифениламин .29
Гравиметрические методы 54
Дезоксирибонуклеиновая кислота 22
Дейтронно-активационный метод
150
Дибензофуран-2-сульфокислота 23
Ионометрия 83
Кислотно-основное титрование 64
Кобальтуранилацетат 21, 75
Комплексиметрическое титрование
73
Комплексонометрическое титрова-
ние 74
Концентрирование натрия 35 .
Краун-эфиры 26
К риптаты 26
Кроконовая кислота 29, 33
Лимонная кислота 22, 78, 178
Магнийуранилацетат 20, 59, 76
Малоновая кислота 22
Марганецуранилацетат 21
Натрий
месторождения 8
методы
концентрирования 35
обнаружения 30
определения 54
активационные 137
гравиметрические 54
спектральные 96
титриметрические 64
фотометрические 77
электрохимические 82
отделения 35
минералы 7
неорганические соединения 14, 35,
54, 77
арсенат 18
ванадат 18
гидроксид 14
карбонат 17
перхлорат 18
тетраборат 15
определение в
водах 161
горных породах 154
металлах 163
неметаллах 163
нефтепродуктах 173
органических растворителях
173
почвах 154
силикатах 154
солях органических кислот 173
сплавах 167
251
химических соединениях 169
органические соединения с
азосоединениями 25
аминополикарбоновыми кисло-
тами 23
аминосоединениями 29
антрахинон-1-сульфокислотой
23
антрахинон-2-сульфокислотой 23
ароматическими сульфокисло-
тами 23
барбитуровой кислотой 24
5-бензамидоантрахинон-2-суль-
фокислотой 24, 33, 63, 82
винной кислотой 22
виолуровой кислотой 24, 79
гексанитродифениламином 29
дезоксирибонуклеиновой кис-
лотой 22
дибензофур ан-2-сульфокисло-
той 23
краун-эфирами 26
криптатами 26
кроконовой кислотой 29, 33
лимонной кислотой 22
малоновой кислотой 22
2-нафтил-а-метоксиуксусной
кислотой 21, 62
1-нафтил амин-8-сульфокисло-
той 23, 33, 62, 72
нингидрином 29
нитрилотриуксусной кислотой
23
нитроантранилазо 25
5-нитробарбитуровой кислотой
24
5-нитро-2-(3-метил-5-оксоизо-
ксазол-4-ил-азо)бензолсуль-
фокислотой 25, 78
нуклеиновыми кислотами 22
оксалатом 21
1-(2-оксинафтилазо)-2-нафтолом
25
оксиоксосоединениями 29
оротовой кислотой 24, 33, 63
Z-пропилендидминтетрауксус-
ной кислотой 23
тетра-(п-толил)борлитием 29, 61
урамилдиуксусной кислотой 24
фенил-а-метоксиуксусной кис-
лотой 21, 32
фосфатидной кислотой 22
.и-хлорфенил-а-метоксиуксус-
ной кислотой 21, 65
п-хлорфенил-а-метоксиуксус-
ной кислотой 21, 61, 65
циклогександиаминтетрауксус-
ной кислотой 23, 73
этилендиаминтетрауксусной
кислотой 23
5-этилоротовой кислотой 24
отделение 35
дистилляцией 39
соосаждением 37
химическими методами 35
хроматографическими методами
42
экстракцией 39
применение 8
тройные ацетаты 20, 31, 59, 74,
80
кобальтуранилацетат 21, 75
магнийуранилацетат 20, 59, 76
марганецуранилацетат 21, 61
никельуранилацетат 21, 61, 76
цинкуранилацетат 21, 31, 59,
71, 81
физико-химическая характерис-
тика 7, 10
химико-аналитическая характе-
ристика 7
2-Нафтил-а-метоксиуксусная кисло-
та 21, 62
1-Нафтиламин-8-сульфокислота 23,
33, 62, 72
Нейтронно-активационный метод
137
Никельуранилацетат 21, 61, 76
Нингидрин 29
Нитрилотриуксусная кислота 23
Нитроантранилазо 25
5-Нитро-2-(3-метил-5-оксоизоксазол-
4-ил-азо)бензолсульфокислота 25,
78
5-Нитробарбитуровая кислота 24
Нуклеиновые кислоты 22
252
Обнаружение натрия 30
Окислительно-восстановительное
титрование 69
1-(2-Оксинафтилазо)-2-нафтол 25
Оксиоксосоединения 29
Оротовая кислота 24, 33, 63
Отделение натрия 35
Пламенный атомно-абсорбционный
анализ 113
Пламенный атомно-эмиссионный
анализ 113
Z-П ропил ендиаминтетр ауксусная
кислота 23
Протонно-активационный метод 150
Резонаторная атомно-абсорбцион-
ная спектрометрия 133
Спектральные методы 96
Тетра-(п-толил)борлитий 29, 61
Тетрафтороборат 29, 55
Титриметрические методы 64
Урамилдиуксусная кислота 24
Фенил-а-метоксиуксусная кислота
21, 32
Фосфатидная кислота 22
Фотометрические методы 77
Фотонно-активационный метод 150
л1-Хлорфенил-а-метоксиуксусная
кислота 21, 65
п-Хлорфенил-а-метоксиуксусная
кислота 21, 65
Ц иклогександи аминтетр ауксусная
кислота 23, 73
Цинкуранилацетат 21, 31, 69, 71,
81
Электрохимические методы 82
Этилендиаминтетрауксусная кисло-
та 23
5-Этилоротовая кислота 24
ОГЛАВЛЕНИЕ
От редколлегии.............................................
Предисловие................................................
Глава I
Физико-химическая и химико-аналитическая характеристика натрия
Общие сведения.............................................
Физико-химические свойства натрия..........................
Неорганические соединения натрия...........................
Соединения с органическими реагентами......................
Глава II
Методы обнаружения натрия..................................
Химические методы обнаружения..............................
Реакции на бумаге..........................................
Реакции окрашивания пламени................................
Глава III
Методы отделения и концентрирования натрия.................
Отделение химическими методами.............................
Отделение дистилляцией...................................
Разделение экстракцией.....................................
Хроматографические методы..................................
Глава IV
Гравиметрические методы определения натрия ................
Прямые методы............................................
Косвенные методы...........................................
3
5
7
7
10
14
20
30
30
34
35
35
35
39
39
42
54
54
63
Глава V
Титриметрические методы определения натрия..................
Кислотно-основное титрование..............................
Окислительно-восстановительное титрование...................
Комплексиметрическое титрование................................ 73
Комплексонометрическое титрование.............................. 74
Глава VI
'Фотометрические методы определения натрия..................... 77
Прямые методы................................................ 77
Косвенные методы............................................. 79
Прочие методы................................................ 82
Глава VII
Электрохимические методы определения натрия.................. 82
Ионометрия................................................... 83
254
Вольтамперометрические методы............................... 90
Другие электрохимические методы............................. 94
Глава VIII
Спектральные методы определения натрия....................... 96
Спектрографические методы.................................... 97
Пламенный атомно-эмиссионный и атомно-абсорбционный анализ 113
Другие спектральные методы определения натрия............... 133
Глава IX
Активационные методы определения натрия..................... 137
Нейтронно-активационный метод............................... 137
Фотонно-активационный метод . . . .......................... 150
Протонно-активационный метод................................ 150
Дейтронно-активационный метод............................... 151
Варианты активационных методов............................. 151
Другие методы............................................... 153
Глава X
Определение натрия в объектах............................... 154
Почвы, силикаты, горные породы.............................. 154
Минералы, руды, концентраты................................. 155
Воды........................................................ 161
Металлы и неметаллы......................................'. 163
Сплавы...................................................... 167
Химические соединения....................................... 169
Нефтепродукты, органические растворители, соли ’ органических
кислот...................................................... 173
Прочие объекты.............................................. 173
Глава XI
Определение примесей в натрии и его соединениях............. 175
Определение основных компонентов............................ 175
Определение примесей........................................ 177
Методы многоэлементного анализа ............................ 178
Определение отдельных элементов............................. 184
Литература.................................................. 200
Приложение.................................................. 237
Предметный указатель........................................ 251
ИСПРАВЛЕНИЯ
К стр. Таблица Напечатано Должно быть
138 46, 2-ой столбец слева, 6 сн. 1SQ 18JT
138 46, 2-ой столбец справа, 7 сн. 32S тар
138 47, 2-ой столбец справа, 5 сн. 0,511 0,511(181%)
В. м. Иванов и др.