/



























































































































































Автор: Волынец В.Ф. Волынец М.П.
Теги: аналитическая химия общие сведения о металлоидах (неметаллах) химия
Год: 1977




Текст
АКАДЕМИЯ НАУК СССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ им. В. И. ВЕРНАДСКОГО
Серия: «АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ»
АНАЛИТИЧЕСКАЯ ХИМИЯ
АЗОТА
В. Ф. Волынец, М. П. Волынец
ИЗДАТЕЛЬСТВО «НАУКА»
МОСКВА 1977
УДК 543 : 546.17
Серия: «Аналитическая химия элементов»
Главный редактор
член-корреспондент АН СССР 10. А. Золотов
Редакционная коллегия:
И. П. Алимарин, Ю. И. Беляев, А. И. Бусев, М. П. Волынец, А. Н. Ермаков, В. М. Иванов, А. В. Карякин, Н. М. Кузьмин, С. Б. Саввин, Н. М. Ростоцкая (ученый секретарь)
Редактор тома «Аналитическая химия азота»
доктор химических наук
Ю. А. Карпов
Адрес редколлегии: 117334, Москва, Воробьевское шоссе, 47а Ордена Ленина Институт геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
20506-091
В 055(02)-77
103-77
© Издательство «Наука», 1977 г.
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернадского АН СССР осуществляет издание серии монографий по аналитической химии отдельных элементов. Эта серия — «Аналитическая химия элементов» — составит около 50 томов. Потребность в подобного рода издании назрела давно. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий и теперь стало возможным и необходимым его подытожить. Таким образом возникло настоящее издание—серия «Аналитическая химия элементов», которое осуществляется впервые. Издание серии было начато по инициативе академика А. П. Виноградова, который с 1958 по 1975 г. был ее главным редактором.
Аналитическая химия любого элемента и его различных соединений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазона концентраций, которые бывает необходимо определить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий план как в смысле содержания, так и последовательности изложения материала.
В монографиях содержатся общие сведения о свойствах элементов и их соединений. Затем рассматриваются химические реакции, являющиеся основанием для аналитических методов. Методы, как физические, так и физико-химические и химические, излагаются применительно для количественного определения данного элемента начиная с анализа сырья, далее — типичных полупродуктов производства и, наконец, конечной продукции — металлов и сплавов, окисей, солей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей.
Монографии содержат обширную библиографию, доведенную до последних лет; они рассчитаны на широкий круг
3
химиков, в первую очередь химиков-аналитиков исследовательских институтов и заводских лабораторий различных отраслей хозяйства, а также на химиков-преподавателей и студентов химических высших учебных заведений. К составлению монографий привлечены крупнейшие советские специалисты, имеющие опыт работы в области аналитической химии того или иного химического элемента.
Отдельные тома серии «Аналитическая химия элементов» будут выходить самостоятельно по мере их подготовки. Вышли в свет монографии, посвященные торию, таллию, урану, рутению, молибдену, калию, бору, цирконию и гафнию, кобальту, бериллию, редкоземельным элементам и иттрию, никелю, технецию, прометию, астатину и францию, ниобию и танталу, протактинию, галлию, фтору, селену и теллуру, алюминию, нептунию, трансплутониевым элементам, платиновым металлам, радию, кремнию, германию, магнию, рению, марганцу, кадмию, ртути, кальцию, фосфору, литию, олову, серебру, цинку, рубидию и цезию, вольфраму, мышьяку, сере и плутонию.
Готовятся к печати монографии по аналитической химии углерода, стронция, хрома, меди, бария и сурьмы.
Мы обращаемся с просьбой ко всем читателям присылать свои замечания и отзывы о монографиях.
ПРЕДИСЛОВИЕ
Азот — один из самых распространенных элементов, окружающих нас. Он входит в состав атмосферы, различных природных объектов и всего живого, существующего на Земле. Некоторые соединения — продукты технологических процессов, в состав которых входит азот (например, окислы азота, цианиды и др.),— являются вредными примесями, загрязняющими окружающую человека среду. Примеси азота в ряде продуктов металлургического производства (металлах, сплава?) существенно отражаются на их качестве.
Именно эти обстоятельства определяют важность методов определения содержания азота и его различных соединений в воздухе, водах, органических соединениях, удобрениях, а также многих других объектах.
В 1970 г. опубликована двухтомная монография «The Analytical Chemistry of Nitrogen and its Compounds» [868], которая посвящена обстоятельному описанию методов анализа в основном органических соединений азота. Однако, во-первых, это издание малодоступно для широкого круга советских читателей; во-вторых, в нем недостаточно полно приведены работы исследователей нашей страны. Кроме того, методы определения азота в неорганических соединениях не нашли в этой монографии, на наш взгляд, достаточно исчерпывающего отражения.
Целью настоящей книги, в которой мы стремились показать современное состояние аналитической химии азота и его соединений, была систематизация и критическая оценка самых различных инструментальных и классических химических методов применительно к анализу объектов неорганической природы. Монография охватывает опубликованные в отечественной и зарубежной литературе работы вплоть до 1974—1975 гг. К сожалению, ограниченный объем издания не позволил дать достаточно полного описания методов анализа широкого круга органических соединений. Так, здесь мы не касались функционального анализа, а привели лишь методы элементного анализа (этому вопросу посвящена самостоятельная глава).
Монография, являющаяся"очередным томом серии «Аналитическая химия элементов», написана в основном по схеме, принятой редколлегией для этой’серии. Но своеобразие химии азота, а также
5
особенность его соединений, существующих в различных агрегатных состояниях и во многих степенях окисления, потребовали, с одной стороны, введения дополнительных глав—«Газометрические методы» и «Органический элементный анализ», — а с другой — несколько усложнило построение книги. Для удобства читателей во всех главах при изложении материала мы старались придерживаться такой последовательности: сначала описывать методы определения молекулярного азота, затем его неорганических соединений с водородом, с кислородом, а далее — наиболее распространенных в неорганическом анализе соединений с другими элементами — серой и углеродом. В число неорганических соединений, выбранных объектами нашего описания, вошли следующие: аммиак (аммоний), гидразин, гидроксиламин, азотистоводородная кислота, окислы азота, нитраты, нитриты, цианиды, роданиды. Последовательно описываются химические методы для соединений азота в различных степенях окисления по мере их возрастания.
Современные требования к чувствительности, правильности и экспрессности методов анализа стимулируют в аналитической химии азота, так же как и других элементов, преимущественное развитие автоматических и инструментальных методов. Однако и традиционные, прекрасно зарекомендовавшие себя классические химические методы (например, Кьельдаля, Несслера и др.) имейт здесь огромное значение. Поэтому в настоящей монографии, наряду с описанием физических и физико-химических методов, довольно много внимания уделено титриметрическим и фотометрическим методам определения различных соединений азота.
В разделах книги, посвященных описанию методов анализа природных и промышленных объектов, для удобства и для компактности изложения многие методики систематизированы в сводных таблицах. Необходимо отметить, что характеристики, оценивающие правильность приведенных методов, авторы не унифицировали, так как оказалось практически невозможным пересчитать их. Поэтому всюду в таблицах в колонках под рубрикой «Точность» приведены оценки методов по первоисточникам.
Авторы выражают искреннюю признательность доктору химических наук Ю. А. Карпову, кандидатам химических наук Е. А. Терентьевой и А. М. Вассерману за прочтение рукописи и ценные советы, а также благодарны Ю. И. Беляеву, Ю. Н. Дуброву, О. Л. Кабановой, А. К. Лаврухиной, Г. В. Мясое-довой, А. А. Немодруку, И. G. Скляренко, Ю. В. Яковлеву, прочитавшим отдельные разделы рукописи и сделавшим критические замечания.
В. Ф. Волынец, М. П. Волынец
Глава I
ОБЩИЕ СВЕДЕНИЯ ОБ АЗОТЕ
ПОЛОЖЕНИЕ В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ ЭЛЕМЕНТОВ ИСТОРИЯ ОТКРЫТИЯ АЗОТА
Азот — химический элемент V группы периодической системы элементов. Атомный номер 7, атомная масса 14,0067. Это — бесцветный газ, не имеющий запаха и вкуса.
В 1772 г. Д. Резерфорд установил, что воздух, оставшийся под колпаком, где жила мышь, после сжигания в нем фосфора не поддерживает горения и дыхания. Этот газ он назвал «ядовитым воздухом». В этом же году Д. Пристли, получив «ядовитый воздух» иным путем, назвал его «флогистированным воздухом». В 1773 г. К. Шееле, шведский аптекарь из города Штральзунда, установил, что воздух состоит из двух газов, и назвал газ, не поддерживающий горения и дыхания, «дурным или испорченным воздухом». В 1776 г. известный французский ученый А. Лавуазье, подробно исследуя «ядовитый», «флогистированный» и «дурной» воздух, установил тождество между ними. И лет спустя, будучи членом комиссии по выработке новой химической номенклатуры, он предложил назвать эту часть воздуха азотом (от греческих слов «а» — означающего отрицание, и «zoos» — жизнь). Латинское название азота происходит от слова «нитрогениум», что значит «рождающий селитру» («селитрообразователь»). Этот термин введен в науку в 1790 г. Ж. Шапталом.
НАХОЖДЕНИЕ В ПРИРОДЕ
Азот вслед за водородом, гелием и кислородом является четвертым по распространенности элементом Солнечной системы. Азот обнаружен в спектрах звезд, в том числе в фотосфере Солнца, в метеоритах, кометах, солнечном ветре и в межзвездных облаках газа. Молекулярный азот наблюдается в атмосферах Венеры и Марса, а аммиак характерен для Юпитера и Сатурна. Во всех космических объектах азот встречается только в восстановленном состоянии.
В земной коре по распространенности азот занимает 20-е место. Подавляющая его часть сосредоточена в следующих основных ре
7
зервуарах: атмосфере (3,86- 10ls m), литосфере (1,7-1015 m), гидросфере (2,2-IO13 m) и биосфере (~ 1010 m). В атмосфере свободный азот в виде молекулярного Na составляет 78,09% по объему (или 75,6% по массе), не считая незначительных примесей его в виде аммиака и окислов.
В литосфере среднее содержание азота составляет 6-10~3 вес. %. Основная масса азота в силикатах находится в химически связанном состоянии в виде NH4, изоморфно замещающего ион калия в силикатной решетке. Кроме того, в природе встречаются и азотные минералы: нашатырь (NH4C1), выделяющийся из вулканов в довольно больших количествах, баддингтонит (NH4AlSi3O8-•0,5 Н2О) — единственный найденный аммониевый алюмосиликат с цеолитной водой. В самых приповерхностных областях литосферы обнаружен ряд минералов, состоящих в основном из нитратных солей. Среди них широкоизвестная селитра (NaNO3), крупные скопления которой характерны для сухого пустынного климата (Чили, Средняя Азия). Долгое время селитра была главным источником связанного азота. (Сейчас основное значение имеет промышленный синтез аммиака из азота воздуха и водорода.) В природе обнаружены и нитриды: сильвестрин (FeBNa) в лавах Везувия и осборнит (TiN), синоит (SiaNaO), карлсбергит (CrN) в метеоритах.
По сравнению с силикатными минералами ископаемое органическое вещество существенно обогащено азотом. Нефть содержит от 0,01 до 2% азота, а каменный уголь — от 0,2 до 3%. Как правило, повышенное содержание азота имеют алмазы (до 0,2%).
В гидросфере среднее содержание азота составляет 1,6-•10-3вес.%. Основную часть этого азота составляет молекулярный азот, растворенный в воде; химически связанный азот, которого примерно в 25 раз меньше, представлен нитратной и органической формами. В меньших количествах в воде содержится аммиачный и нитритный азот. Концентрация связанного азота в океане примерно в 104 раз меньше, чем в почвах, пригодных для сельскохозяйственного производства. Это ставит под сомнение оптимистические высказывания по поводу безграничных резервов Мирового океана.
Хотя название азота означает «не поддерживающий жизни», на самом деле это необходимый для жизнедеятельности элемент. В растительных организмах его содержится в среднем 3%, в живых организмах до 10% от сухого веса. Азот накапливается в почвах (в среднем 0,2 вес.%). В белке животных и человека среднее содержание азота составляет 16%. Человек и животные не могут синтезировать 8 незаменимых аминокислот (валин, изолейцин, лейцин, фенилаланин, триптофан, метионин, треонин, лизин), и поэтому для них основным источником этих аминокислот являются белки растений и микроорганизмов.
Между атмосферой, литосферой и биосферой происходит непрерывный обмен, с которым связана и смена химических форм азо
8
та. Этот обмен и определяет круговорот азота в природе. Обмен азота между атмосферой и биосферой получил название биохимического цикла азота.
Несмотря на то что за историю Земли выделились огромные (по сравнению с весом биосферы) количества азота и образовали атмосферу Земли, биосфера в своем развитии постоянно ощущает недостаток в азотном питании. Основным процессом движения азота в биосфере является его переход из одной химической формы в другую в замкнутом цикле. Постоянная смена химических форм азота является источником жизни для многих организмов начиная от микроорганизмов и кончая высокоорганизованными формами жизни. Накопленные в почве запасы связанного азота служат источником питания высших растений, откуда связанный азот может поступать и в организмы животных. Растения и животные, отмирая, дают начало органическому азоту, находящемуся главным образом в аминокислотах. В процессе амонификации органических остатков азот органических соединений переходит в аммонийную (аммиачную)' форму. Последняя с помощью микроорганизмов переходит в нитритную форму. При этом выделяется около 70 ккал/моль. Другая группа микроорганизмов завершает окисление аммиака до нитрата. Полученный в процессе нитрификации нитрат усваивается растениями, и цикл движения азота в биосфере замыкается.
Главными неорганическими соединениями азота в почвах являются нитрат, аммоний и в редко встречающихся условиях нитрит. Поведение первых двух компонентов в почве совершенно различно. Если нитрат является легкоподвижным соединением, не сорбируется минералами почвы и остается в растворенном в воде состоянии, то аммоний легко хемосорбируется глинистыми минералами, хотя это не мешает ему в определенных условиях легко окисляться до нитрата. Такое различие в подвижности нитрата и аммония предопределяет источники азотного питания растений. С энергетических позиций аммонийная форма азота более предпочтительна, так как валентность азота в ней одинакова с валентностью азота в аминокислотах.
Нитратная форма служит основным источником азотного питания растительности в силу своей подвижности, несмотря на необходимость траты дополнительной энергии, связанной с восстановлением нитрата растением.
Неиспользованные живым веществом запасы химически связанного азота под действием микроорганизмов непрерывно преобразовываются в формы, доступные для азотного питания растений. Так, фиксированный глинистыми минералами аммоний окисляется до нитратов. В определенных условиях при отсутствии свободного кислорода и наличии неиспользованного живым веществом нитрата может происходить обусловленное процессом денитрификации восстановление азота до молекулярного с уходом последнего в атмосферу.
9
Количества азота, выведенные денитрифицирующими бактериями из биосферы, компенсируются процессами фиксации азота из атмосферы азотфиксирующими бактериями. Последние подразделяются на две группы: живущие самостоятельно и живущие в симбиозе с высшими растениями или с насекомыми. Первая группа бактерий фиксирует примерно 10 кг!га. Симбионты высших растений фиксируют значительно большие количества азота. Так, симбионты бобовых культур фиксируют до 350 кг]га. С осадками выпадает азота порядка нескольких килограммов на гектар.
В балансе фиксируемого азота все большее значение приобретает искусственно синтезированный аммиак, причем его количество удваивается каждые 6 лет. Уже в ближайшее время это может вызвать дисбаланс между процессами фиксации и денитрификации в биосфере.
Следует отметить подцикл круговорота аммиака и окислов азота через атмосферу, особенно если учесть, что этот подцикл регулирует масштабы развития биосферы. Источниками атмосферного аммиака служат биохимические процессы в почве и, в первую очередь, аммонификация. Окисляясь, аммиак дает основную массу окислов азота в атмосфере. Получающаяся в процессе денитрификации закись азота ответственна за содержание окислов азота в стратосфере, которые каталитически разрушают озон, защищающий живое вещество биосферы от губительного действия жёсткого ультрафиолетового излучения. Таким образом в природе установились определенные пределы развития биосферы.
Деятельность человека грозит нарушить установившееся равновесие. Так, подсчет показал, что количества окиси азота, выделившиеся при планируемых полетах сверхзвуковых самолетов в стратосфере, будут сравнимы с поступлениями ее из природных источников.
Таким образом, завершается цикл движения молекулярного азота через биосферу. В этом геохимическом цикле само существование азотной атмосферы Земли определяется скоростями процессов фиксации и денитрификации. При резком разбалансе этих скоростей азотная атмосфера Земли может исчезнуть всего за несколько десятков миллионов лет.
Помимо атмосферы, биосфера определяет существование и другого крупного резервуара азота в земной коре — литосферы, так как именно взаимодействие живого вещества с молекулярным азотом играет главную роль в процессе круговорота поверхностного азота через земную Кору. Небольшая часть накопленного в биосфере связанного азота вместе с осадками увлекается в глубь земной коры. В резко восстановительных условиях из осадочных пород исчезает нитратная форма азота. С увеличением температуры и давления и выходом за границы биосферы реакции превращения органического вещества становятся односторонними и сдвигаются в сторону разрушения органической формы азота. Основной формой связанного азота становится ион аммония. С увеличением 10
степени метаморфизма осадочных пород начинает разрушаться и уходить из пород и аммонийный азот. На высоких ступенях метаморфизма этот процесс практически заканчивается, а с ним заканчивается цикл обращения поверхностного азота в земной коре. Время жизни азота в этом цикле составляет около 1 млрд. лет.
ВАЛЕНТНЫЕ СОСТОЯНИЯ АЗОТА
Внешняя электронная оболочка атома азота состоит из пяти электронов (одной неподелепной пары и трех неспаренных — конфигурация 2s22p3) (табл. 1).
Чаще всего азот в соединениях трехковалептен за счет неспаренных электронов (как в аммиаке NH3). Наличие неподеленной
Таблица 1
Электронные оболочки атомов элементов подгруппы азота
Номер оболочки
Элемент 1 2 3 4 5 6
8 sp spd spdf spd 8p
N 2 2, 3
Р 2 2, 6 2, 3
As 2 2, 6 2, 6, 10 2, 3
Sb 2 2, 6 2, 6, 10 2, 6, 10 2, 3
Bi 2 2, 6 2, 6, 10 2, 10, 14 2, 6, 10 2, 3
T а б ли ц а 2 Состояния окисления азота
Степень окисления Соединения азота
3- 2— 1— 1/э- 0 1+ 2+ 3+ 4+ 5+ NHa, амиды, имиды, нитриды, цианиды Гидразин NH2NH2 Гидроксиламия NH2OH Азотистоводородная кислота HNa, азиды Na n2o NO HNO2, N2Os, нитриты no2 HNOa, N2Os, нитраты
11
пары электронов может приводить к образованию еще одной ковалентной связи, и тогда азот становится четырехковалентным (как в ионе аммония NH^). Азот в химических соединениях практически никогда не образует пятиковалентных связей, в отличие от его ближайшего аналога — фосфора, так как возбуждение одного из 2х-электронов для его перехода на Зх-орбиту (2х22р3
2х2р3) является энергетически невыгодным.
Дальнейшее изложение химико-аналитической характеристики соединений азота удобно построить в зависимости от состояний окисления азота (табл. 2).
ИЗОТОПЫ АЗОТА
Азот природных объектов состоит из двух стабильных изотопов с массами 14 и 15. Искусственно могут быть получены 4 радиоактивных изотопа с массами 12, 13, 16, 17 (табл. 3). Существование у азота двух стабильных изотопов было впервые установлено С. Ноде в 1929 г., когда он открыл изотопе массой 15 в спектре окиси азота. А. Нир в 1950 г. измерил отношение 14N/15N в атмосферном азоте и получил величину 273 ± 1 [1106], что отвечает концентрациям изотопов 14N и 15N 99,635 и 0,365 ат. % соответственно. Эти цифры вошли во всю справочную литературу. Одна-
Табл ица 3
Изотопы азота
Изотоп Тип распада Период полураспада Метод получения
12n 3+, 3+,3a 0,0125 сек. 12С (р, в); “N (7, 2п)
13N 14N P+ Стабильный, pacn] ценность 99,634% Стабильный, paciif ценность 0,366% 10,08 мин. >остра-Атмосфер- >остра- ный Х°В (a, n); 12С (d, в); (р, 7); Х|С (р, пу, 14N (п,2п)- 14N (d, Ту, 14N (7, я); 4|О (в, рЗп); 13О (7, р2п)
|i~ 7,35 сек. “N (п, 7); 1«N (d, р); “О (п, Ру, “F (п, а)
|j~, п 4,15 сек. 14С (а, р); 13О (п, р); “О (7, РУ (7, 2р)
12
ко в 1958 г. эта величина была уточнена [894], и с одобрения Нира было рекомендовано значение отношения двух стабильных изотопов 272 ± 0,3. Поэтому содержание 14N и 15N в воздухе следует признать равным 99,634 и 0,366%, как и указано в табл. 3.
Азот — единственный элемент на Земле, у которого наиболее распространенными являются ядра изотопа 14N нечетно-нечетного типа (7 протонов, 7 нейтронов). Это единственное исключение из систематики 234 ядер стабильных изотопов. Другими словами, самым распространенным изотопом азота на Земле должен был бы быть 15N с ядром нечетно-четного типа (7 протонов, 8 нейтронов). Теоретические расчеты показывают, что на Солнце изотоп 14N является также самым распространенным. Это объясняется различными временами жизни изотопов азота в термоядерном цикле образования гелия:
12C + p-»13N; 13N13С + е+; 13С + р -+ UN; "N + р -+ 150;
150 _>15N _|_е+; 16N-)-р —» 12С + 4Не.
Измерение изотопного состава азота природных объектов показало, что отношение 15N/14N отличается от воздушного (стандарт) максимально на 3%.
В газообразном азоте существует равновесие между тремя видами изотопных молекул по реакции
15N2 _|_ un2 214N16N.
Константа равновесия этой реакции при комнатной температуре равна приблизительно 4.
В верхних слоях атмосферы под действием нейтронов космического излучения 14N превращается в радиоактивный изотоп 14С, на чем основана геохронологическая датировка геологических образцов, содержащих «древний» углерод.
В настоящее время возможно получение химических соединений азота, искусственно обогащенных тяжелым изотопом 15N до 99,9 атомн.%. Обогащенные по 15N образцы используются при исследованиях в биохимии, биологии, медицине, химии и физической химии, физике, в сельском хозяйстве, в технологии и химическом машиностроении, в аналитической химии и т. д.
Общие сведения об азоте, химико-аналитическую характеристику его соединений (см. главу II) можно найти в работах [54, 68, 156, 229, 232, 271, 278, 293а, 1285, 1293, 1310].
Глава II
ХИМИКО-АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА АЗОТА И ЕГО СОЕДИНЕНИЙ
МОЛЕКУЛЯРНЫЙ АЗОТ
Плотность обычного азота при стандартных температуре и давлении составляет 1,2505 г/л. Эта величина соответствует молекулярной формуле N2. Магнитные измерения в молекулярном азо- . те показывают, что он диамагнитен и характеризуется удельной магнитной восприимчивостью —0,430-10~6 па 1 г при 25° С.
Межъядерное расстояние в молекулярном азоте составляет 1,0945 А. Изучение колебательных и вращательных спектров молекулярного азота показывает, что имеется два типа молекул азота, а именно с симметричным и несимметричным ядерным спином. При обычных температурах обе эти формы присутствуют в соотношении 2:1. Вследствие симметрии молекулы азота и стабильности его электронных состояний межмолекулярные силы чрезвычайно малы.
В твердом состоянии азот образует две фазы — кубическую а и гексагональную р. Переход из одной фазы в другую происходит при —237,49° С; ДЯперехода = 54,74 кал/молъ.
Физико-химические свойства молекулярного азота приведены ниже.
Конфигурация внешней электронной оболочки Молекулярное состояние газа Атомный объем, .ил Плотность жидкого, г] см3 газообразного, г/л твердого, г/см,3 кубическая а-форма гексагональная 3-форма Температура плавления, °C Температура кипения, °C Атомный радиус, А 2«22рЗ n2 15,95 0,808 1,2505 1,0265(—252,2°С) 0,8792(—210°С) —209,86 —195,8 0,71
14
Радиус атома азота в химических соединениях, кристаллический, А
№"
N6+(NOs~)
Теплота плавления, ккал/молъ
Теплота испарения, ккал/молъ Теплота диссоциации, ккал/молъ Теплоемкость, кал/г град
при постоянном объеме С„
при постоянном давлении Ср
Растворимость в воде, см3 /н.у.)/мл
Ткр, °C
Ркр, атм
Энергия ионизации, эв
N+
№+
Ns+
N4+
N5+
Тройная точка температура, °C давление, мм рт. ст.
Диэлектрическая проницаемость (25°С, 760 мм рт. ст.)
Электроотрицательность
Стандартный электронный потенциал Е0(в) для
HNO2
HNOs
Поверхностное натяжение жидкого азота в контакте с воздухом, дин/см
Давление пара (amj рт. ст.) при
—226,1°С
- 214°С
— 200,9°С
— 195,8°С
1,48 0,15
0,1723(—209,86°С) 1,333(—195,8°С)
225,2
0,178
0,249
0,016(20°С)
-147,1
33,54
14,54
29,60
47,43
77,45
97,86
—210,24 94,01
1,000538
3,0
—1,44
-1,24
8,5(— 196°С)
1
40
400
760
Теплота диссоциации азота чрезвычайно велика: для реакции N2(r) = 2N(r) &Н = 225,8 ккал/молъ, К2$° = 10~120. Вследствие эндотермичности процесса диссоциации константа равновесия возрастает с увеличением температуры, но даже при 3000° С и обычном давлении все еще не заметна диссоциация. Большая прочность связи N = N в молекуле азота объясняет в принципе его химическую инертность.
До недавнего времени считалось, что при обычной температуре молекулярный азот инертен по отношению к большинству химических реагентов. В 1964 г. были открыты реакции восстановления молекулярного азота в неводных средах в присутствии ряда пере-
15
ходных металлов (титан, молибден, вольфрам и др.) при комнатной температуре и атмосферном давлении. Восстановителями в Этой реакции могут быть свободные металлы (литий, магний), гидриды металлов, металлоорганические соединения. Азот при.подобном восстановлении образует продукты, разлагаемые водой до аммиака [4181.
Существуют комплексы переходных металлов с Ns. Известно, что как — С=С-, так и —CsN-группы могут быть донорами за счет своих л-электронов. До настоящего времени такие связи для молекул N=N не были обнаружены. В последнее время получены комплексныесоединениярутения и осмия (например, [Ru(NH3)5N2] • •Х2, где X = Br~, J- и др.), в которых в качестве лигандов содержатся молекулы N2 [450]. В этом случае, вероятно, связь N2 с координационным центром осуществляется за счет л-связей.
При высоких температурах и давлениях молекулярный азот взаимодействует с кислородом, образуя окись азота, а в присутствии катализаторов — с водородом при синтезе аммиака. Он также реагирует при умеренных температурах с литием, кальцием, стронцием, барием, магнием, бериллием, бором, алюминием, титаном, кремнием и хромом с образованием нитридов. При температурах около 1800—1900° С смесь углерода, водорода и азота медленно реагирует с образованием цианистого водорода. Типичными для молекулярного азота реакциями являются следующие:
N2(r) + Зн2(г) = 2NH3(r), = 10-5 атм-2.
N2{r) + °2(Г) = 2NO(r), = 5-10-51;
N2(r) + СаС2(тВ) = С(тв) + CaCN(TBp
^2(г) + 3^1^(тв) ^18з^2(ТВ)’
Под воздействием тлеющего электрического разряда часть азота переходит в метастабильное активированное состояние. Этот газ имеет устойчивый золотисто-желтый цвет и химически очень активен. Недавно было установлено масс-спектрометрическим и другими методами, что активный азот состоит главным образом из атомов азота в их основном состоянии; они рекомбинируют сравнительно медленно, образуя возбужденные молекулы N2 в квин-тетных спиновых состояниях, которые затем испускают характерное излучение при переходе в основное состояние. Большое различие в электронном спине, когда два атома в спиновых квартетных состояниях (всего шесть неспаренных электронов) объединяются с образованием молекулы в квинтетном состоянии (четыре неспаренных электрона), объясняет медленность рекомбинации, в то время как присутствие атомарного азота объясняет высокую химическую активность азота в метастабильном состоянии.
Азот способен растворяться во многих металлах, образуя твердые растворы внедрения. При увеличении концентрации атомарного азота в металле он «выпадает в осадок» в виде соответствующего 16
нитрида. При соответствующих термодинамических условиях азот стремится выделиться из высокоазотистых нитридов и образовать отдельную газообразную фазу. Это приводит к возникновению дислокаций, границ зерен и в пустотах изолированных объемов газообразного азота, пребывающего нод высоким давлением сначала в атомарном, а затем в молекулярном состоянии. Когда молекулярный азот выделяется, на свободной поверхности образуется адсорбированный слой. Однако связь газообразного N2 с нитридом осуществляется только через растворенный азот. Так, например, в истинно-стабильной системе Fe—N равновесие имеет место между a-твердым раствором, у-твердым раствором и газообразным азотом, а не нитридом. Это не исключает возможности образования метастабильных нитридов, которые, хотя и являются, строго говоря, переходными фазами, па самом деле весьма стойки, что оказывается чрезвычайно полезным на практике. Так, высокая твердость, свойственная азотированному слою у сталей, может быть непосредственно связана с искажениями, возникающими в кристаллической решетке металла из-за принципиальной нестабильности положения атомов азота в занимаемых ими позициях.
Промышленный метод получения азота основан па фракционной разгонке жидкого воздуха. Полученный таким образом азот обычно содержит некоторое количество аргона и (в зависимости от качества) несколько более 0,003% кислорода. Последний можно удалить добавлением небольшого количества водорода и пропусканием смеси над платиновым катализатором, над раскаленной медью, барботированием через водные растворы солей Сг2+ или V2+. Спектрально чистый азот обычно получают термическим разложением азидов натрия или бария, например, по реакции
2NaN3 = 2Na 4- 3N2.
неорганические соединения азота с водородом
Физико-химические свойства некоторых неорганических соединений азота с водородом приведены в табл. 4.
Аммиак NH3 — бесцветный газ с едким запахом и жгучим вкусом. Он значительно легче воздуха (уд. вес. 0,5963 относительно воздуха). Сухая.смесь аммиака с воздухом способна взрываться; при комнатной температуре границы взрывоопасности лежат в пределах концентраций NH3 15,5 — 28%. Аммиак ядовит, он сильно раздражает слизистые оболочки. Жидкий аммиак имеет высокую теплоту испарения. Ниже приведены физические свойства аммиака.
Температура плавления, °C —77,72
Температура кипения, °C —33,42
Теплота плавления, кал/моль 13516
Теплота испарения, кал/молъ 5576
17
Критическая температура, °C Критическое давление, атм Диэлектрическая проницаемость (жидкий при —60°С)
Плотность, г/с.м3
газообразный, при нормальных условиях
жидкий при—70°С » » —30°С
Теплота образования при 25°С, ккал/молъ
Стандартная свободная энергия образования, AG298, ккал) молл.
Энтропия Д^дз, кал/град -моль
Теплоемкость (7° 2д8, кал(град-моль
Вязкость (жидкий при 25°С), пуаз Давления паров, мм рт. ст.
жидкий при—20° С
» » 0°С
» » 20°С
Дипольный момент газообразной молекулы р, (ед. CGS)
Поверхностное натяжение жидкости при Ткип, Эин/см
Константа Трутона
132,4
115,0
26,7
0,7714
0,7253
0,6777
—11,02
-3,993
46,04
8,50
0,00135
1426,8
3221,0
6428,5
1,43-10-1»
34,25
23,6
По физическим свойствам жидкий аммиак напоминает воду; сильная ассоциация молекул возникает вследствие их полярной природы и прочной водородной связи. Его диэлектрическая проницаемость достаточно высока, поэтому он может служить хорошим ионизирующим растворителем.
Химия водородных соединений азота во многих отношениях похожа на химию соединений кислорода, являющихся производными воды. Так, аммиак и вода имеют исходные уравнения для реакций ионизации:
2NHs = NH+ + NH.j, К^' = [NH+][NH2] = ~10-30;
2Н2О = Н3О+ + ОН", А'25» = [НзО+][ОН-] = Ю-14. ,
Металлы I группы периодической системы и в меньшей степени Са, Sr, Ва растворимы в жидком аммиаке с образованием синих растворов, содержащих ионы металлов и сольватированные электроны. Такие растворы устойчивы в течение длительного времени, и их используют в качестве эффективных восстановителей. Однако в таком растворе медленно протекает реакция образования амида; например, для натрия опа выражается уравнением
Na + NH3 = NaNH2 + Н2О.
18
Таблиц а 4
Физико-химические свойства некоторых неорганических соединений азота с водородом
Характеристика N2H4 HN« NHzOH
Стандартная энтальпия образования ккал/молъ
газ 22,77 70,3 —12,16
жидкость 12,07 63,1 -27,5
Стандартная свободная энергия образования кал/моль
газ 38,023 78,401 -0,866
жидкость 35,671 78,202 —4,161
Энтропия ‘$208’ кал/молъ-град
газ 57,0 57,08 56,3
жидкость 29,0 33,6 —
Теплоемкость С°, кал/моль-град
газ 11,6 10,44 11,2
жидкость 23,62 — —
Температура плавления, °C 1,54 -80 32,05
Температура кипения, °C 113,5 36 57
Теплота плавления Д7/Пл, ккал/молъ 3,025 — 3,94
Теплота испарения ДЯИСп, ккал/моль 9,7 7,1 —
Критическая температура, °C 380 —. —
Критическое давление, атм 145 —- —
Плотность (жидкость), г/см3 1,012(15°) 1,13 —
Эта реакция ускоряется солями переходных металлов, например FeCl3.
Наиболее важными являются реакции аммиака с кислородом и водой. Аммиак горит в чистом кислороде, но реакция протекает до разных конечных продуктов. Наиболее энергетически выгодно окисление аммиака до молекулярного азота:
4NH3(r) 4- ЗО2(Г) = 2N2(r) + 6Н2О(Г), = 10*4
Но в присутствии платинового или платино-родиевого катализатора реакция протекает по другой схеме:
NH3 + 5О2 = 4NO + 6Н2О, Л-26» = 1О168.
Растворимость аммиака в воде исключительно велика: при 0° С один объем воды поглощает 1200 объемов аммиака, при 20° С — около 700 объемов. Удается выделить два кристаллогидрата, устойчивых при низкой температуре—NH3-H2O (температура плавления
19
—79° G) и 2NH3-H2O (температура плавления — 78,8° С), в которых молекулы аммиака и воды связаны водородными связями. Оба кристаллогидрата не содержат ни ионов NH4 и ОН-, ни отдельных молекул NH40H. В водных растворах аммиак, вероятно, гидратирован подобным же образом. Хотя обычно считают, что водные растворы аммиака являются растворами слабого основания NH4OH, называемого «гидратом окиси аммония», это не соответст вует известным фактам, так как нет никаких данных, свидетельствующих о существовании недиссоцииров энного NH4OII. Взаимодействие аммиака с водой описывается формально уравнением
МН3(водн) + н2° - + ОН-, Х18° = 1,75.10-\
Появление небольшого количества гидроксил-ионов объясняет тот факт, что водные растворы аммиака обнаруживают щелочную реакцию и ведут себя как растворы слабых оснований. Сильные окислители, например перекись водорода, хромовая кислота или перманганат калия, окисляют аммиак. Аммиак энергично реагирует в водных растворах с хлором и бромом:
ЗС12 + 8NH3 == N2 + 6NH4CI.
Существует множество довольно устойчивых кристаллических солей тетраэдрического иона аммония; большинство из них растворимо в воде, подобно солям щелочных металлов. Соли силь-; ных кислот полностью ионизированы, и их растворы слабокислы:
NHiCI = NH+ + С1", К ж оо;
NH+ + Н2О = ГШ3(ВОДН) 4- Н3О+, K.lt° = 5,5.10-ю.
Вполне очевидной закономерности, определяющей состав и устойчивость аммиачных комплексов металлов, не наблюдается; известно, однако, что часто положительно однозарядные ионы металлов присоединяют две молекулы аммиака, двухзарядпые — четыре и трехзарядные — шесть молекул.
Аммиачные комплексы, как правило, разлагаются под действи-ет кислот с образованием иона аммония:
Ag (NH3)+ + Cl- + 2Н+ - AgCl + 2NH+.
Устойчивые аммиачные комплексы и их характеристики приведены в табл. 5.
В лабораторных условиях аммиак может быть получен вытеснением его сильными щелочами из аммониевых солей по схеме
2NH4C1 + Са(ОН)2 = 2NH3 + СаС12 -j- 2Н2О.
Старейший промышленный способ получения аммиака — выделение его из отходящих газов при коксовании угля. Основной современный способ промышленного получения аммиака — синтез из элементов — азота и водорода, предложенный в 1908 г. немецким химиком Ф. Габером.
20
Таблица 5
Константы устойчивости аммиачных комплексов металлов
Ион металла t, *c igK (lg(3)
Fe2+ 25 3» 3,7
Со2+ 25 3e 4,90
30 Ki 1,99; K2 1,51; K3 0,93; Ke 0,64; 0,06; Ks 0,74; 4,39
Со3+ 25 3e 33,66;
30 3e 34,36; Ki 7,3; K2 6,7; K3 6,1; Ke 5,6
Ni2+ 25 & 7,32; pe 7,68
30 Ki 2,67; K2 2,12; K3 1,61; Ke 1,07; Ke 0,63; Ke 0,09; J3e 8,01
Cu+ 18 Ki 5,93; K2 4,93; 3s 10,86
Cu2+ 18 Ki 4,25; K2 3,61; K3 2,98; Ke 2,24
30 kj 3,99; K2 3,34; K3 2,73; Ke 1,97; 3* 12,03
Ag+ 25 Ke 3,315; K2 3,915; 32 7,23
Au+ Комнатная 02 27
Zn2+ 30 Ke 2,18; K2 2,25; K3 2,31; Ke 1,96; 3i 8,70
Cd2+ 30 Ke 2,51; K2 1,96; K3 1,30; Ke 0,79; 3< 6,56
Hg2+ 22 Ki 8,8; K2 8,7; K3 1,00; Ke 0,78; 32 17,5; Pi 19,4
Металлические амиды, имиды, нитриды. Если в молекуле аммиака последовательно замещать атомы водорода ионами металлов, то образуются следующие химические соединения: амиды металлов (амидная группировка NH2-), имиды металлов (имидная группировка NH = ) и, наконец, нитриды металлов (нитридогруппа N=). Когда радикал NH2 — связан с определенным электроотрицательным атомом или группой, например С1~ или ОН-, то получающиеся соединения называются аминами (например, гидроксил-амин).
Амиды металлов могут быть разделены на две группы: амиды активных металлов и амиды тяжелых металлов. Амиды щелочных и щелочноземельных металлов получаются при нагревании металла или его гидрида с аммиаком по реакциям
M + NH3-MNH2 + i/2H2;
LiH + NH3 -> LiNH2 4- Н2.
Многие амиды могут быть получены действием KNH2 на соли соответствующих металлов в жидком аммиаке:
KNH2 + NaJ^”2NaNHa(TB) + KJ.
21
При взаимодействии амида металла с N20 образуются азиды металлов
2NaNH2 + N2O -» NaN3 -f- NaOH + NH3.
Амиды и имиды тяжелых металлов получаются по реакциям замещения в жидком аммиаке:
Cd(SCN)2 + 2KNHa н.нз(жидк) Cd(NH2)2(TB) + 2KSCN;
PbJ2 + 2КМН2-Нп(жИД2ЦрЬМН(тв) + NH3 + 2К J.
Имид лития Li3NH может быть приготовлен действием Li на NH2 при 400° С, причем вначале образуется LiNH2, а затем Li2NH. Амиды щелочноземельных металлов при нагревании переходят в имиды — Me<n>NH. Амиды и имиды металлов легко окисляются на воздухе и гидролизуются в воде
2KNH2 + ЗО2 — 2KNO3 + 2Н2О;
NaNHa + Н2О — NaOH + NH3.
Они являются слабоионизованными соединениями с очень нестабильным анионом [NHjl-, который тотчас реагирует с водой с образованием NH3.
В некоторых случаях амиды и имиды не являются устойчивыми фазами, и из аммиачного раствора выпадает нитридная фаза
3HgBr2 + + 6KBr + 4NH3.
Как правило, амиды и имиды тяжелых металлов, а также нитриды тяжелых металлов, полученные указанным выше путем, являются взрывчатыми.
Кроме этих взрывчатых нитридов имеется еще большой класс стабильных нитридов. Их можно разделить на три группы. Li, Mg, Ga, Ba, Gr, Zn, Cd, Th образуют ионные нитриды, о чем свидетельствует их легкий гидролиз до аммиака и гидроокиси металла. Они получаются либо взаимодействием азота или аммиака с металлом, либо термическим разложением амидов:
ЗВа (NH2)2G»Ba3N2-f-4NH3.
В, Al, Ga, In, Si, Ge, Sn образуют нитриды с ковалентной связью. И, наконец, переходные металлы, например Ti, Zr, V, Та, La, образуют нитриды со структурой внедрения. Такие нитриды часто не стехиометричны, а по внешнему виду, твердости и электропроводности напоминают металлы.
Гидразин N2H4. Чистый гидразин представляет собой бесцветную гигроскопическую жидкость, которая плавится при 1,5° С. Безводный гидразин обладает необычно высокой диэлектрической проницаемостью. С водой и низкомолекулярными спиртами смешивается во всех соотношениях. Одной из основных химических характеристик гидразина являются его сильновосстановительные
22
свойства. Он претерпевает самоокисление как в разбавленных, так и концентрированных водных растворах с образованием на промежуточной стадии Н2О2, а затем N2 и Н2О.
Гидразин энергично горит на воздухе с выделением значительного количества тепла:
N2Hj -|- О2 —> N2 4-2Н2О, А/7 = 148,6 ккал[молъ.
Он интенсивно окисляется галогенами, азотной кислотой и другими окислителями, чем объясняется проявляемый к нему интерес как к потенциальному ядерному топливу. Нитрат серебра и другие соли также восстанавливаются гидразином.
При взаимодействии его с кислотами образуются два типа солей, содержащих ионы N2Hg и N2H|+. Гидразин — более слабое основание, чем аммиак:
N.Jh + Н2О N2H4-H2O N2H+ + ОН-, К№° = 8,5-10-’;
N2H+ + Н2О N2HeOH+ Л N2H2+ + ОН-, К№° = 8,9 • Ю-™.
Соли гидразина с катионом N2Hg устойчивы в водных растворах, тогда как соли с катионом N2H|+ немедленно гидролизуются:
N2H,X2 + H2O = N2H+ + H3O++2X. Л/= 11.
Гидразин так же, как и аммиак, способен с ионами никеля, цинка, кадмия образовывать координационные соединения типа fNi(N2H4)e]2 + или [Zn(N2H4)4]5;+.
В промышленных масштабах гидразин получают по методу Ра-шига; суммарная реакция выглядит как
2NH3 + NaOCl = N2Ht + NaCl + H2O.
В настоящее время известны другие методы получения гидразина, являющиеся разновидностями процесса Рашига.
Гидроксиламин NH2OH. В свободном состоянии гидроксиламин представляет собой бесцветное кристаллическое вещество, которое плавится при ~ 33° С и образует жидкость с плотностью 1,204 г/см9.
В свободном состоянии гидроксиламин пе очень устойчив, и разложение жидкости на N2 и N2O и другие продукты ускоряется с повышением температуры. В вакууме (остаточное давление 22 лсл1 рт.ст.) жидкий гидроксиламин кипит при 57° С, но даже и в этих условиях он частично разлагается. Быстрый нагрев до температуры 100° С приводит к взрыву.
Гидроксиламин смешивается с водой и низкомолекулярными спиртами во всех соотношениях. Водные растворы гидроксиламииа вплоть до 60% NH2OH практически стабильны. Подобно гидразину, гидроксиламин — более слабое основание, чем аммиак:
МН2ОН(водн) + Н2О = NH3OH+ -J- ОН-, К№° = 6.6.10-».
23
Гидроксиламин, как и перекись водорода, может выступать либо в роли окислителя, либо восстановителя в зависимости от pH раствора. В кислом растворе гидроксиламин восстанавливает Ag+, Au3+, Hg2+, Мп07, Вг2 и другие окислители с выделением азота и окислов азота. Щелочные растворы гидроксиламина окисляют Fe(II) до Fe(III) (гидроокись), но восстанавливают в гидроокиси Сп(П) до окиси Сп(1). Соли Ti(III) и Сг(Ш) в кислом растворе восстанавливают гидроксиламмониевый ион NH3OH+ до иона аммония.
Гидроксиламин можно получить гидролизом нитропарафинов в растворе серной кислоты либо электролитическим восстановлением нитратов и нитритов в солянокислом или сернокислом растворе.
Азотистоводородная кислота и азиды. HN3 представляет собой бесцветную, обладающую острым запахом, жидкость с плотностью 1,13 г!смА при 20° С, кипящую при 36° С и затвердевающую при —80° С. Его ядовитые, разъедающие слизистые оболочки пары при соприкосновении с нагретыми предметами с большой силой взрываются:
2HN3(r) = 3N2 + Н8 + 141,8 ккал.
В водных растворах азотистоводородная кислота устойчива. Это слабая кислота, несколько слабее уксусной (константа диссоциации 1,8-10'в). В присутствии платины разлагается на азот и аммиак.
Азотистоводородная кислота реагирует с HCI и HNO2 по реакциям
3HN3 + HCI = NHiCl + 4N2,
HNS + HNO2 = N2 + H2O + N2O.
Она может выступать в окислительно-восстановительных реакциях как в качестве окислителя, так и в качестве восстановителя.
Получается азотистоводородная кислота разными методами, в том числе взаимодействием расплавленного амида натрия с закисью азота либо с азотистой кислотой:
2NaNH2(}K) + N2O(r) = NaN3(TB) + NaOH(Tn) + NH3(r).
Азиды — химические соединения, содержащие одну или несколько rpynnN3, производные азотистоводородной кислоты. Большинство неорганических азидов взрывается при легком ударе или трении даже во влажном состоянии; таков, например, азид свинца, применяющийся как инициирующее взрывчатое "'вещество. Исключение составляет азид натрия и соли других щелочных и щелочноземельных металлов. Исходным материалом для получения других солей, а также самой кислоты обычно служит азид натрия, получаемый пропусканием закиси азота через расплавленный амид натрия:
NaNH2 + N2O NaN3 + Н2О.
24
НЕОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ АЗОТА С КИСЛОРОДОМ
Физико-химические свойства соединений, которые будут рассмотрены ниже, приведены в табл. 6.
Закись азота N2O представляет собой бесцветный, почти без запаха, со сладковатым привкусом газ. Он хорошо растворим в воде и спирте. Закись азота сравнительно нереакционноспособна, хотя по термодинамическим данным она является более сильным окислителем, чем кислород. При комнатной температуре она инертна по отношению к галогенам, щелочным металлам и озону. При более высоких температурах разлагается на азот и кислород, реагирует со щелочными металлами и многими органическими соединениями и поддерживает горение. Закись азота в обычных условиях не окисляется кислородом до окисла азота. При вдыхании в незначительных количествах закись азота вызывает состояние легкого опьянения («веселящий газ»). При вдыхании в больших количествах он действует как наркотическое средство.
Окись азота NO — продукт соединения азота с кислородом при очень высоких температурах. Это бесцветный газ. Он мгновенно реагирует с кислородом, образуя бурую двуокись азота. Окись азота реагирует со фтором, хлором, бромом (но не иодом). Некоторыми сильными окислителями окись азота окисляется до HNO3. Реакция с перманганатом протекает количественно, и ее используют в анализе. Окись азота восстанавливается до N2O сернистым ангидридом и до NH2OH солями двухвалентного хрома в кислом растворе. Она термодинамически нестабильна при обычных условиях, а при высоком давлении легко разлагается при 30-50° С:
3NO = N2O+NO2.»
Трехокись азота N2O3, ангидрид азотистой кислоты — темносиняя жидкость, застывающая при сильном охлаждении в бледно-голубую массу. Этот окисел существует только в твердом состоянии при низких температурах: в виде жидкости и пара он в значительной степени диссоциирован на NO и NO2. Вследствие легкой обратимости реакции газообразная смесь равных объемов NO и NO2 при большинстве химических реакций ведет себя подобно соединению N2O3; при взаимодействии со щелочами в водных растворах образуются нитриты:
N2O3 + 2NaOH = 2NaNO2 + Н2О.
С водой вначале образуется азотистая кислота, которая быстро разлагается с образованием азотной кислоты:
N2O3 + Н2О = 2HNO2; 3HNO2 = HNO3 + 2NO + H2O.
Двуокись и четырехокись азота, NO2 и N2O4. Эти два окисла азота находятся в равновесии, сильно зависящем от температуры
25
Таблица 6
Физико-химические свойства основных неорганических соединений азота с кислородом
Водный раствор.
26
и давления:
2N02 s==s N2O4.
Бурый Бесцветный
парамаг- диамагнит-
нитный ный
В твердом состоянии окись существует исключительно в виде молекул N2O4. В жидкости происходит частичная диссоциация. В паре при 100° С содержание NO2 увеличивается до 90%, а полная диссоциация наступает при температуре выше 140° С. N02 очень токсична и энергично реагирует с металлами. Реакция с водой протекает по уравнению
2NO2 + Н2О = HNO3 + HNO2.
Азотистая кислота разлагается до азотной (см. выше). В водном растворе смесь окислов — довольно энергичный окислитель, по силе сравнимый с бромом:
N2O4 + 2Н+.+ 2е = 2HNO2> Е° = + 1,07 в.
Смеси окислов («нитрозные газы») используют в органической хи-ции в качестве селективного окислителя. Четырехокись азота была подробно изучена в качестве неводного растворителя.
Пятиокись азота N2O5, ангидрид азотной кислоты — бесцветное летучее кристаллическое вещество, возгоняющееся при 32,4° С при атмосферном давлении. Кристаллы N2OB гигроскопичны и легко растворимы в воде с образованием азотной кислоты.
Пятиокись азота является сильным окислителем и реагирует с металлами и различными органическими веществами. Твердая пятиокись азота в своей устойчивой форме представляет собой нитрат нитрония NO2NO3. Подобно N2O4, пятиокись азота в безводных серной, азотной или фосфорной кислотах образует ион NO2:
N2O6 + 3I-I2SO4 = 2NO2 + 3HSO; +ньо+.
Многие реакции в газовой фазе зависят от диссоциации N2OB на NO2 и NO3, причем последний далее реагирует в качестве окислителя.
Азотистая кислота HNO2 — слабая кислота (К25° — 6-10"6). В жидком состоянии кислота неизвестна, тем не менее ее можно получить в газовой фазе в виде смеси транс- и цис-форм. В газовой фазе быстро устанавливается следующее равновесие:
NO + NO2 + Н2О = 2HNO2, #2о° = 1,56 w'.
Нитриты щелочных металлов получают нагреванием нитратов с восстановителями, такими, как уголь, свинец, железо и др.
Азотная кислота HNO3 и ее соли принадлежат к наиболее важным кислородным соединениям азота. В настоящее время HNO3 получают превращением атмосферного азота в аммиак, каталитическим окислением последнего до NO и поглощением NO водой
27
в присутствии кислорода. Безводная азотная кислота — бесцветная жидкость, подверженная самоионизации:
2HNO3 = no+ + no; + Н2О.
В разбавленных водных растворах HNO3 — сильная кислота; в 0,1 М растворе она диссоциирована на ~93%. Обычный концентрированный раствор азотной кислоты (~70 вес. %) бесцветен, но бывает желтым вследствие фотохимического разложения с образованием NO2:
2hNO3^2NO2 + Н2О + V2O2.
Так называемая «дымящая азотная кислота» содержит растворенную NO2 (в количестве, большем, чем то, которое может быть гидратировано с образованием HNO3 + N.O).
Концентрированный водный раствор азотной кислоты является сильным окислителем; из металлов только Au, Pt, Rh и Ir устойчивы к ее действию. Другие металлы, такие, как Al, Fe и Сг, она «пассивирует». Природа процесса пассивирования неизвестна, но, вероятно, некоторую роль при этом играет образование труднопроницаемой пленки окислов.
Взаимодействие растворов азотной кислоты с металлами сопровождается не образованием свободного водорода, а восстановлением азота иона NO3- Причем, в зависимости от концентрации HNO3 и природы металла, восстановление протекает до различных продуктов. Золото и платиновые металлы растворяются в «царской водке» (3 ч. конц. НС1 1 ч. конц. HNO3), которая содержит свободный хлор и C1NO. Более эффективное действие царской водки по сравнению с азотной кислотой объясняется, по-видимому, комплексообразованием с ионами хлора.
Известны нитраты практически всех металлов. Часто они гидратированы и большей частью растворимы в воде. Многие нитраты можно получить в безводном состоянии, некоторые из них возгоняются без разложения. Нитраты щелочных металлов сублимируются в вакууме при 350—500° С; при более высоких температурах происходит разложение до нитритов, а при очень высоких температурах — до окислов (например, NH4NO3 дает N2O и Н2О). В нейтральном растворе нитраты могут восстанавливаться, но с трудом. Механизм восстановления еще неясен. Алюминий и цинк в щелочном растворе восстанавливают нитраты до NH3.
НЕОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ АЗОТА С ДРУГИМИ ЭЛЕМЕНТАМИ
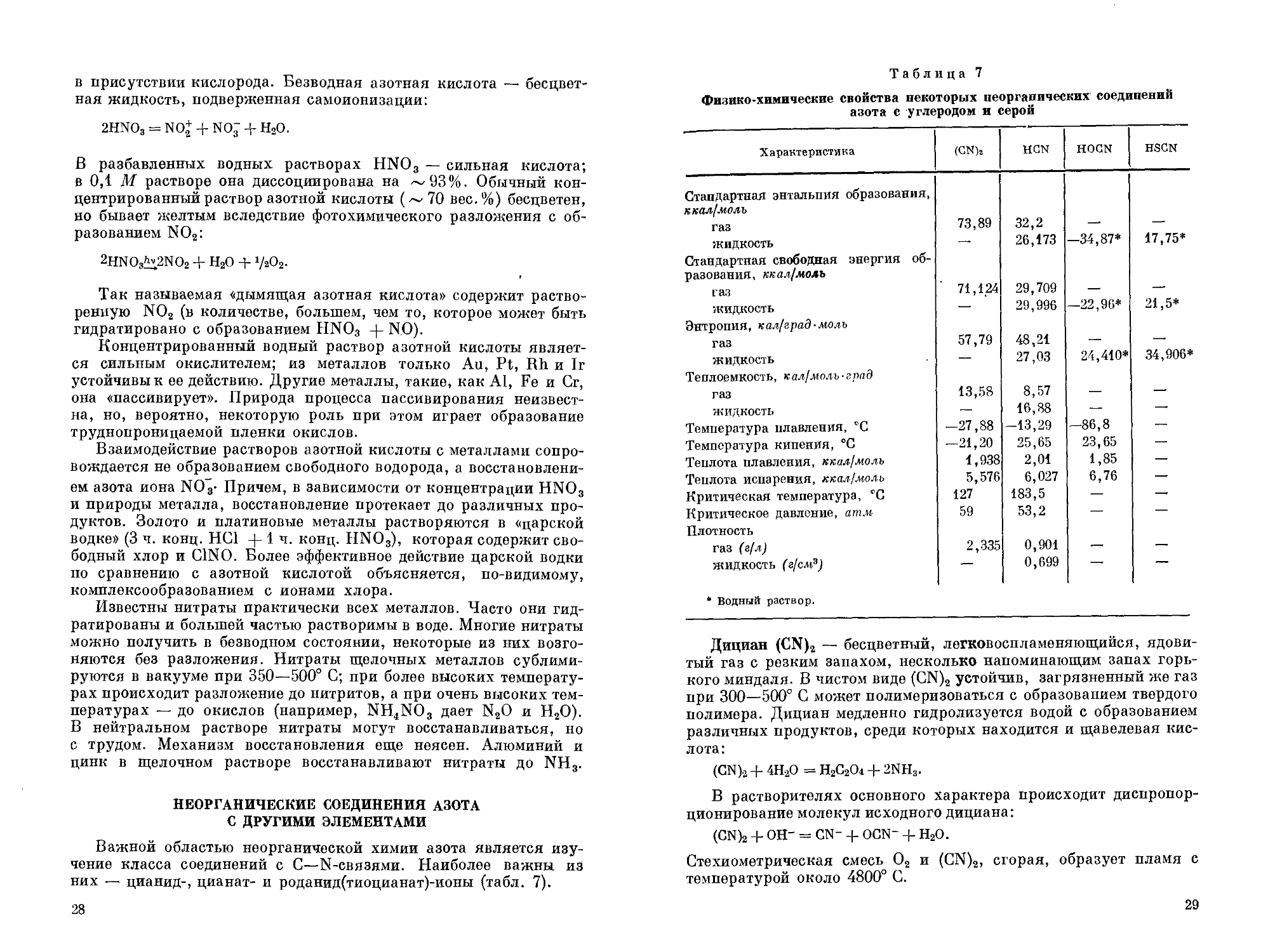
Важной областью неорганической химии азота является изучение класса соединений с С—N-связями. Наиболее важны из них — цианид-, цианат- и роданид(тиоцианат)-ионы (табл. 7).
28
Таблица 7
Физико-химические свойства некоторых неорганических соединений азота с углеродом и серой
Характеристика (GN), HGN HOCN HSCN
Стандартная энтальпия образования, ккал/моль
газ 73,89 32,2 —• —
жидкость Стандартная свободная энергия образования, ккал/моль —. 26,173 —34,87* 17,75*
газ 71,124 29,709 — —
жидкость Энтропия, колорад-моль — 29,996 —22,96* 21,5*
газ 57,79 48,21 — —'
жидкость Теплоемкость, к^моль-град — 27,03 24,410* 34,906*
газ 13,58 8,57 — —
жидкость — 16,88 — —'
Температура плавления, °C -27,88 —13,29 —86,8 —
Температура кипения, °C —21,20 25,65 23,65 —
Теплота плавления, ккол!молъ 1,938 2,01 1,85 —
Теплота испарения, ккы/моль 5,576 6,027 6,76 —•
Критическая температура, °C 127 183,5 — —'
Критическое давление, атм Плотность 59 53,2 — —
газ (г/л) 2,335 0,901 — —
жидкость (г/см3) — 0,699 — —
* Водный раствор.
Дициан (CN)2 — бесцветный, легковоспламеняющийся, ядовитый газ с резким запахом, несколько напоминающим запах горького миндаля. В чистом виде (CN)2 устойчив, загрязненный же газ при 300—500° С может полимеризоваться с образованием твердого полимера. Дициан медленно гидролизуется водой с образованием различных продуктов, среди которых находится и щавелевая кислота:
(CN)2 + 4Н2О = Н2С2О< + 2NH3.
В растворителях основного характера происходит диспропорционирование молекул исходного дициана:
(CN)2 + ОН" = CN- + OCN- + Н2О.
Стехиометрическая смесь О2 и (CN)2, сгорая, образует пламя с температурой около 4800° С.
29
Цианистый водород (синильная кислота) HCN — бесцветная, легкоподвижная жидкость с наркотическим запахом, в большом разведении напоминающим запах горького миндаля. Синильная кислота чрезвычайно ядовита (противоядие — перекись водорода). 50 мг — смертельная для человека доза. С водой, спиртом и эфиром цианистый водород смешивается во всех соотношениях. В водных растворах HCN является слабой кислотой (К — 2,1 • 10-9), вследствие чего растворимые цианиды гидролизуются в водном растворе:
CN- + Н2О = HCN + ОН-.
Цианиды щелочных и щелочноземельных металлов легко растворимы в воде, за исключением Ag(I), Hg(I). Цианиды тяжелых металлов большей частью труднорастворимы, за исключением Hg2CN2.
Цианид-ион имеет большое значение в качестве лиганда, и существует большое количество цианидных комплексов переходных металлов (табл. 8). Некоторые из них, подобно Ag(CN)2 и Au(CN)2 , имеют важное значение в технике. Помимо этого цианидные комплексы применяют в аналитической химии.
Таблица 8
Константы устойчивости цианидных комплексов металлов при 25 °C
Комппексо-образователь IgK(lgP) Комплексо-образователь IgK (lg 3)
Fe2+ Ре 24 Ag+ p2 18,42; 19,85
Fe3+ Ре 31 Au+ P2 38,3
Со2+ Be 19,09 Au3+ Pe~56
Соз+ Ре 64 Zn2+ Pe 16,76; 16,72
Ni2+ Ре 31,0 Cd2+ pe 18,24
Cu+ Р2 16 Hg2T Ai 5,18; K2 4,42; K3 4,32; Kt 3,19; Pi 17,11 Pe 41,51; Ki 18,00; K2 16,70; K3 3,83; Kt 2,98
Циановая кислота HOCN — чрезвычайно неустойчивая, в воде при 0° С быстро гидролизуется до NH3 и СО2. Это более сильная (Ко° = 1,2-10'4) по сравнению с уксусной (А = 1,8-10~5) кислота.
Соли циановой кислоты — цианаты — чрезвычайно устойчивы, особенно соли щелочных металлов. Они не разлагаются при нагревании в сухом виде до температуры красного каления. Но в воде или водных растворах спирта они быстро гидролизуются до карбоната и аммиака.
30
Роданистоводородная кислота HSCN — бесцветная, маслянистая, очень летучая, резко пахнущая, легко затвердевающая жидкость. При ее разложении образуются цианистый водород и твердый желтый остаток (H2C2N2S2). С водой роданистоводородная кислота смешивается во всех соотношениях. Водный раствор роданистого водорода — сильная кислота. В водном растворе она, подобно хлористоводородной кислоте, почти полностью диссоциирована.
Соли роданистоводородной кислоты — роданиды — легко получаются из цианидов путем присоединения серы. По химическим свойствам они напоминают хлориды. Большая часть роданидов хорошо растворима в воде. Нерастворимы роданиды серебра, ртути, меди и золота.
Техническое применение роданиды находят прежде всего при крашении тканей.
Глава III
МЕТОДЫ КАЧЕСТВЕННОГО ОБНАРУЖЕНИЯ
Методы качественного анализа, не требующие большой затраты времени, реактивов и анализируемого материала на их выполнение, позволяют аналитику быстро и просто оценить пределы содержания определяемого элемента (с целью последующего выбора количественного метода его определения), а также в ряде случаев получить сведения о формах его нахождения в исследуемом образце.
Подробную информацию о качественных методах обнаружения неорганических соединений азота можно найти в ряде руководств [6, 158, 334]. Методы обнаружения азота в органических материалах (органический качественный анализ) подробно излагаются в книге [868]. Здесь же описаны способы превращения общего азота в легкоизмеряемые формы. Вопросам систематической микроидентификации органических соединений, в том числе методам быстрого открытия азота с использованием кольцевой бани Вейсса (наряду с другими важнейшими гетероатомами), посвящена работа [412]. Открываемый минимум азота 0,01—1 мкг. Качественный элементный анализ органических веществ без предварительной их минерализации описан в работе [777]. Ультрамикрокапцллярно-му методу открытия азота в органических веществах посвящена работа [1237].
Для качественного обнаружения азотсодержащих ионов используются специфические химические и физические их свойства: цветные реакции в пробирках, капельные реакции, в том числе и на бумаге, микрокристаллоскопические реакции, сорбция на А12О3, электрофорез на бумаге, ИК-спектроскопия, флуоресценция, каталитические методы и т. д.
Ниже приводится краткое описание наиболее распространенных методов открытия ионов аммония, нитрат-, нитрит-, роданид-и цианид-ионов [6, 334].
Аммоний
Едкие щелочи (NaOH, КОН) выделяют из растворов солей аммония при нагревании газообразный аммиак, который обнаружи. 32
вается по запаху, с помощью лакмусовой или фенолфталеиновой бумаги.
Реактив Несслера, представляющий смесь комплексной соли KJHgJJ с КОН, образует с растворами солей аммония характерный красно-бурый осадок (или при очень малых количествах — желтое окрашивание). Чувствительность реакции 0,0003 мг в капле объемом 0,002 мл. Мешают ионы элементов Ag, Hg(II), Pb, ион S2-.
Нитрит-ионы
Кислоты разлагают все нитриты с образованием газообразной NO2, окрашенной в бурый цвет.
Иодид калия в присутствии H2SO4 окисляется нитритами до свободного J2 (так же действуют и другие окислители: МпО4> СгОГ, АзОГ).
Уксуснокислый раствор бензидина в присутствии ионов NO2 образует желтоокрашенное соединение.
Сульфаниловая кислота и ’ а-нафтиламин (реактив Грисса— Илосвая) в уксуснокислой среде образуют с нитрит-ионами окрашенный азокраситель.
Предложена микрокристаллоскопическая реакция для обнаружения нитрит-ионов: крупинку исследуемого вещества вносят в каплю раствора, содержащего ацетат калия, свинца и меди(П) и подкисленного СН3СООН. Выделяются черные кристаллы K2Pb[Cu(NO2)6]. Этим способом удается открыть до 0,75 мг NO2. Предельное разбавление 1 : 1500. Присутствие ионов NO3 не мешает реакции.
Реакция образования K3[Co(NO2)6]. При смешивании испытуемого раствора с растворами Co(NO3)2, разбавленной уксусной кислоты и К.С1 в присутствии NO2 появляется желтый кристаллический осадок.
Перманганат калия обесцвечивается в кислой среде при нагревании в присутствии нитрат-ионов в результате восстановления марганца до Мп2+.
о-Аминоанилид бензолсулъфоновой кислоты (сернокислый раствор) в кислой среде осаждает ионы NO2.
Нитрат-ионы
Для открытия нитрат-ионов применяются преимущественно реакции окисления—восстановления.
Реакция с медью и серной кислотой при нагревании приводит к выделению бурого газа NO2.
Реакция с FeSO4 в присутствии концентрированной H2SO4 приводит к образованию в пробирке бурого кольца в результате образования комплексного соединения [Fe(NO3)]SO4. Мешают ионы J -, Вг-, анионы-окислители, SCN-.
2 В. Ф. Волынец, М. П. Волынец
33
Реакция восстановления до аммиака в присутствии концентрированного раствора щелочи цинковой пылью, алюминиевым порошком или сплавом Деварда. Обнаруживают NH3 лакмусовой (посинение) или фенолфталеиновой (покраснение) бумагой. Мешают NHj, NO2, SCN-, [Fe(CN)6]4-, [Fe(CN)6]3-, также ионы, восстанавливающиеся алюминием до NH3.
Реакция с МпС]2. При нагревании исследуемого раствора нитрата с двойным объемом насыщенного раствора МпС12 в концентрированной НС1 раствор становится темно-бурым вследствие образования комплексных ионов [МпС16]2-. Мешают МпО4, СгО4-, NO2.
Реакция восстановления NO3 до NO2 при действии на нитраты металлического цинка в присутствии GH3GOOH. Далее NO2 обнаруживается по его характерным реакциям (см. выше).
Реакция с дифениламином (C6H5)2NH. На часовое стекло помещают 4—5 капель раствора дифениламина в концентрированной H2SO4. Вносят туда на кончике чистой стеклянной палочки немного анализируемого раствора и перемешивают. В присутствии NO3 появляется интенсивно-синяя окраска вследствие окисления дифениламина образующейся азотной кислотой. Мешают NO2, СгО|-, МпО;, [Fe(CN)6]3-, Fe3+ и другие окислители, а также J", окисляющийся в присутствии H2SO4 до J2.
Микрокристаллоскопическая реакция с нитроном. На каплю исследуемого раствора нитрата действуют каплей 10%-ного раствора нитрона в 5 %-ной GH3GOOH. Выпадают характерные пучки игл азотнокислого нитрона C20Hi6N4-HNO3. Реакция позволяет открыть 0,025 л<кг NO3. Предельное разбавление 1 : 120 000. Мешают NO2, SCN-, J-.
Родан ид-ионы
Нитрат серебра осаждает ионы SCN- в виде белого осадка роданида серебра, нерастворимого в разбавленных минеральных кислотах.
Нитрат ртути(1 Г) осаждает белый осадок роданида ртути(П), растворимый в избытке роданида с образованием комплексного соединения.
Соли <нселеза(11Г) в кислой среде (хлорид, сульфат, нитрат) дают с ионами SCN- красную (или розовую — при малых концентрациях SCN-) окраску вследствие образования роданида железа. Мешают [Fe(CN)e]8’_, J.
Соли меди(1Г) дают с роданидами сначала изумрудно-зеленую окраску; при избытке реагента образуется черный осадок Cu(SCN)2; осадок состава CuSCN белый.
Соли кобалъта{11) с роданидами в присутствии их большого избытка дают комплексные соединения, растворимые в амиловом спирте с образованием окрашенного в синий цвет кольца.
34
Нитрит натрия в кислом растворе дает темное окрашивание, исчезающее через некоторое время.
Металлический цинк в кислой среде разлагает роданиды с образованием H2S и HCN.
Реакция образования берлинской лазури Fe4[Fe(CN)6]3. В пробирку с испытуемым раствором, содержащим С№-ионы, вводят раствор NaOH и соли Fe(II), нагревают (образуется [Fe(CN)e]4-). Затем подкисляют щелочной раствор соляной кислотой и добавляют раствор соли Fe(III). Выпадает синий осадок берлинской лазури.
Реакция с пикриновой кислотой. Смачивают фильтровальную бумагу раствором пикриновой кислоты, Na2CO3 и каплей испытуемого раствора; высушивают. В присутствии CN--hohob появляется розовое окрашивание.
Цианид-ионы
Реакция образования цианистого водорода. При действии на цианид бикарбонат-ионов при нагревании получают цианистый водород HCN, последующая реакция которого ci солью меди(П) (ацетат или сульфат) и уксуснокислым бензидином приводит к окислению последнего с образованием окрашенных в синий цвет продуктов.
В ходе систематического анализа ионов аммоний открывают в первой аналитической группе катионов (первая подгруппа, осаждается реактивами Na3Co(NO2)e, NaHC4H4O6); анионы CN-, SCN-, NO2, NO3 — в первой группе анионов (образуют растворимые соли бария).
Качественному микроопределению аммонийного азота посвящена работа [729]. Существуют обзоры по методам открытия и определения небольших количеств цианидов [491], нитратов [885]. Микрокристаллоскопические реакции подробно описаны в работе [153].
В табл. 9 указаны микрокристаллоскопические реакции для NHt, NH3, CN-, SCN-, NO; и NO3~.
В табл. 10 приведены не упомянутые выше реакции качественного обнаружения азота в виде различных неорганических его соединений.’
? В работе [1296] предложен небольшой карманного типа колориметрический индикатор для обнаружения и грубой количественной оценки содержания двуокиси азота в атмосфере. Индикатор представляет собой прозрачный пластмассовый пакет одноразового использования, в котором запечатана таблетка или шарик из устойчивого при хранении порошка. При распечатывании пакета таблетка окрашивается от воздействия NO2, и по интенсивности окраски (сравнением ее с цветовой шкалой, подклеенной на этот же
2*
35
Таблица 9
Микрокристаллоскопические реакции обнаружении азота [153, стр. 356[
Ион или соединение Реактив Состав осадка Характеристика осадка Предел обнаружения, мкг Мешающие ионы
NH1+ NaHPOi, MgCl2, NaOH NH4MgPO4-6H2O Трапеции, прямоугольные призмы, звезды, дендриты 0,1 Rb+, Cs+
H2[PtCle] (NH4)2[PtClel Желтые октаэдры 0,1 К+, Rb+, Cs+, Т1+
H2[PtCle], NaJ (NH4)2[PtJe] Черные кристаллы — К+, Rb+, Cs+
Na[PbF3] NH4[PbJ3] Длинные иглы — K+, Rb+, Cs+
612(504)3 3(NH4)2SO4-Bi2(SO4)3 Шестиугольники, мелкие звезды — K+, Rb+, Cs+, Na+, LiT
Na3[Co(NO2)e] (МНа)з[Со(МО2)в] Желтые мелкие кубы, Друзы — K+, Rb+, Cs+
Na2Pb[Cu(NO2)e] (NH4).2Pb[Cu(NO2)e] Черные кубы и прямоугольники 0,15 K+, Rb+, Cs+, T1+
AgCl, AuCl3, HCI — Красные кристаллы — Rb+, Cs+
NaMnO4-3H2O NH4MnO4 Темно-фиолетовые кристаллы Малочувствительная реакция K+, Rb+, Cs+
CiHiOeHN a C4H40eHNH4 Крупные призмы — K+, Ca2+, Sr2+, Ba2T
Пикриновая кислота CeH2(NO2)3ONH4 Желтые длинные иглы 0,3 K+, Rb+, Cs+, T1+
Нафтоловый желтый CioH4(N02)2(ONH4)-S03H Желтые мелкие иглы 2 K+, Rb+, Cs+, Ca2+, Sr2+, Ba2*
NHS Na3[Co(NO2)6] (NH4)3[Co(NO2)a[ Желтые мелкие кубы, друзы — —
- HJO3 nh4j3o. Квадраты, Кресты — —
Таблица 9 (продолжение)
Ион или соединение Реактив Состав осадка Характеристика осадка Предел обнаружения, мкг Мешающие ионы
NHs GN- Винная кислота Пикриновая кислота Пикриновая кислота, CuSO4 Пикриновая кислота, формалин 5-Нитробарбитуровая кислота AgNO3, HNO3 C4HtOeHNH4 CeH2(NO3)3ONH4 (CeH2N3O7)2[Cu(NH3)4l Пикрат уротропина 5-НитробарбитураТ ам- мония AgCN Крупные призмы Желтые длинные иглы Желтовато-зеленые кристаллы Желтые шестиугольники, палочки Иглы, расширенные посредине Палочки, иглы, мелкие 0,3 2 0,06 С1~, Вг-> I-
SON" Cu(CH3COO)2, бензидин, СН3СООН CeHsHgOH CeH6CH2HgOH Окисление до CN-, осаждение раствором AgNO3 Нитрон Сульфат берберина CuSO4, пиридин CuSO4, HNO3, хинолин Продукты окисления бензидина CeHsHgCN CeHsCHaHgCN AgCN CaoHieN-HSCN C2oHi704N-HSCN [Cu(C5H5N)2](SCN)2 Комплексная соль звезды Синие иглы Палочки, розетки Иглы, пучки игл Иглы, палочки Иглы, пластинки Иглы, дендриты- Желтовато-зеленые розетки из палочек, Х-об-разные изогнутые кристаллы Кристаллический осадок 0,02 3 4 0,1 1 0,6 0,3 J-, SCN-, СггО,2- СГ, Вт', J-С1-, Вг-, J-CN- NO3-, NO2-, I- С1-, Br-, J~, NO3-Вг- Вг-
Таблица 9 (окончание)
Ион или соединение Реактив Состав осадка Характеристика осадка Предел обнаружения, МКВ Мешающие ионы
SCN- Co(NO3)2, акридин Комплексная соль Синие кристаллы
Сульфат ферродипири-дила [Fe(CioH8N2)3](SCN)2 Оранжево-красные шестиугольники, призмы 0,2 J-, МпОг, СгО42~, vo3-
(C2HsHg)3PO4 CaHsHgSCN Прямоугольники, треугольники, розетки 1 С1-, Вт-, Г
NOa~ CeHsHgOH CeHsHgSCN Квадраты, пирамиды, октаэдры 0,06 С1-, Вт-, J-, CN-
AgNO3 AgNO2 Длинные шестиугольники и параллелограммы 2 С1~, Br~, J-, CN-, SCN-
Ацетаты К+, Rb+, Cu2+, уксусная кислота K2Pb[Cu(NO2).[ Черные кубы 0,75 —
Нитрон C2oHieN4-HN02 Иглы, розетки 0,4 no3-, С1о3", сюг, j-
NOS- Ацетат 1,4-аминофенил-ртути CeH4(NH2)HgNO2 Розетки, призмы, Х-об-разные кристаллы — S2-
Нитрон CaoH1.N4.HNO3 Пучки тонких игл 0,012—0,025 no2-, сю3-, J-, SCN-
Сульфат берберина C2oH1704N-HN03 Желтые розетки из игл 0,005 С1-, Вт-, J-, SCN-
Хлорид цинхонамина Нитрат цинхонамина Квадраты, прямоугольники, шестиугольники — J-, SCN-
Ацетат 1,4-аминофенил-ртути C.H<(NH2)HgNO3 Розетки, призмы, Х-об-разные кристаллы 0,3 Cl", Вт-, Г, CN-, SCN-
Т а б л и ц а 10
Реакции качественного обнаружения азота
Ион или соединение Реактив или реакция Способ выполнения, метод Видимый эффект реакции Предел обнаружения, мкг Предельное разбавление Мешающие элементы или ионы Литература
NH1 Бумага Ватман № 54, растворитель (CaHjJO—СН3ОН — вода — конц. НС1 (50:30:15:4) ХБ, К Обнаружение зоны на хроматограмме обработкой реагентом, дающим с NH* цветную реакцию — — [1175]
15%-ный раствор NaOH К — — — — [333]
Ферроцианид кадмия Cd2Fe(CN)e, 0,3%-ный раствор в 5%-ной НС1 — Образование двойной соли постоянного состава 5Cd2Fe(CN)e • 4(NH1)1Fe(CN)e 1 мг 1:1000 Щелочные металлы [149]
Нингидрин ВЭБ Образование окрашенного соединения (нагревание до 80°С в течение 10 мин.) К.+ , Na+, Li+, Са2+, Mg2+ [786]
Фосфат натрия и хлорид магния Пикриновая кислота и сульфат меди Нитрикобальтиат натрия Раствор пикриновой кислоты в формамиде м м м м Образование характерных трапеций или прямоугольных призм Образование желтых кристаллов Образование желтых кубов, шестиугольников, звездочек Образование желтых шестиугольников, прямоугольников и палочек [264]
Тетраметилдипикриламин, 1%-ный раствор м Желтый кристаллический осадок 2 мг/мл [1079]
Таблица 10 (га родолжение)
Иои или соединение Реактив или реакция Способ выполнения, метод Видимый эффект реакции Предел обнаружения мкг Предельное разбавление Мешающие элементы или ионы Литература
NO' Реакция с Си2+ п, к Образование зеленого комплексного иона [Cu(NO2)4]2- — — [131]!
Реакция с KJ в кислой среде к Выделение J2 (коричневое) окрашивание) 0,01— 0,1 м — — [1154]
а-Нафтиламин (диазотирование) к Образование красного осадка или окрашивание раствора в фиолетовый цвет 0,01-5 — [237]
Диметиланилин (в среде СН3СООН, НСООН) КФБ Образование темно-зеленого га-нитрозодиметиланилина 10 1:4000 — [120]
N .N-Диметилбензидин (реакция диазотирования в среде СН3СООН) ЦР Образование диазония красного цвета 0,2 — — [1044]
Смеси диметилсульфоксида с ангидридами кислот (уксусной, пропионовой, масляной) в присутствии КЛ ЦР Образование окрашенного соединения — — [1416]
Смесь диметилсульфоксида и SOCh ЦР Появление розово-красной окраски 5 мг — — [885]
Дифенилоранж, 0,1—0,01%-ный раствор (в кисл. среде) ЦР Переход красной окраски в желтую 0,07 — — [Ю2]
Малахитовый зеленый ЦР, к Появление красной или красно-пурпурной окраски, исчезающей со временем (в присутствии NOp; появление желтой или желто-зеле-нойокраски (в OTCyTCTBneNOg) 75 мкг! /0,25 мг [886]
Таблица 10 (продолжение)
Иои или соединение Реактив или реакция Способ выполнения, метод Видимый эффект реакции Предел обнаружения мкг Предельное разбавление Мешающие элементы или ионы Литература
NO" Конго красный ЦР, к Появление фиолетовой или пурпурной окраски или осадка 550 мкг/ /0,5 мл J- [886]
Антипирин в присутствии К2СгО4 ЦР Выпадение желтого осадка, последующее его растворение и окрашивание раствора в интенсивно-красный цвет 0,5 J-, S2-, s2o32-, SO2- [533]
Каталитическая реакция окисления комплекса Мп2+ с ЭДТА с помощью Н2О2 в присутствии HNO2 ЦР Образование промежуточного продукта — надазотистой кислоты 0,01 — [135]
UO2(NO3)2 и аскорбиновая кислота ЦР Появление красновато-бурой или оранжево-бурой окраски, постепенно переходящей в светло-желтую 5 мг — [885]
Сульфатиазол и А13+ (или Ag+) — Выделение оранжевого или оранжево-желтого осадка 5 — — [885]
no; Смесь H2SO4 с FeSO4 ЦР Возникновение красной окраски («бурого кольца»). При избытке NO; окраска исчезает 15 s2o2-, cio;, ci-, Bro;, Br-, jo;, Cr2O2- [1313]
Меркаптогидрохинон ЦР — — 1:1,5-ю5 — [581]
Бруцин, 1%-ный раствор сульфата ЦР — 0,5—10 — no; [1271]
Окись алюминия К, последу-ощая ЦР Микрограммо-вые количества — — [395]
£
Таблица 10 (продолжение)
Иои или соединение Реактив или реакция Способ выполнения, метод Видимый эффект реакции Предел обнаружения, мкг Предельное разбавление Мешающие элементы или ионы Литература
NO- NO" Бруцин КБФ Появление желтого окрашивания <0,02 и 0,5 соответственно — Свободные галогены, сильные восстановители [780J
KJ и уксусная кислота Восстановление магнием Выделение J2 (бурое окрашивание) — — — [487]
Восстановление водородом NOj и NOg и идентификация продукта с феноловым красным ЦР Образование NH3 NO;-5 мкг/мл NO--25 мкг/мл NO’-l: : 2-106 NO’-l : : 4-Ю4 [1009]
SCN- Молибдат Sn(IV) к Ионообменная реакция с образованием окрашенного соединения — — — [1194]
Цианид ртути (кислый раствор) к Выделение HCN и его идентификация 0,5 мкг KSCN — — [694]
Эффект Велера — осаждение BaSO4 в присутствии больших количеств КМпО4; обработка восстановителем (NaNOa) к Розовая или фиолетовая окраска кристаллов BaSO4, не исчезающая при действии восстановителей 5-8 — [567, 695]
Хромат серебра Ag2CrO4 (с применением бумаги, пропитанной реагентом) к Образование белых пятен AgSGN на красном фоне бумаги (анализируемый раствор нейтральный) Микрограм-мовые количества (10— 150) — Hg+, РЬ2+, Вг-, CN", s2o*- [335, 1238]
Образование берлинской лазури к Образование красного FeSCN 0,001—0,005 ммоль — — [1172]
Таблица 10 (продолжение)
Иои или соединение Реактив или реакция Способ выполнения, метод Видимый эффект реакции Предел обнаружения, мкг Предельное разбавление Мешающие элемен^ы^ илилбны Литература
SON" Комплекс Hg с 5-нитрозо-8-оксихинолином Косвенный метод Разрушение ионами SCN" комплексного соединения ртути 0,5 мкг/мл — — [2221
— икс Появление в спектре характеристических полос поглощения — — [795]
Комплекс Си с анилином или толуидином м Образование желто-коричневого и коричневого осадка соответственно [Fe(GN)e]*-, [Fe(GN)e]3-, NO’, S2O*-, sot 1158]
Комплекс Си с пирамидоном или а-нафтиламином ЦР Появление фиолетовой окраски раствора и выделение фиолетово-синего осадка соответственно — — J-, S2O|- [158]
GN" Молибденовая синь к Образование комплексного иона Gu(GN)2~ и восстановление им МоОд~ до молибденовой сини 0,15/0,1 мл 1: 30 000 — [1349]
га-Нитробензальдегид (I) и о-динитробензол (П) ЦР Образование промежуточного продукта с (I) — цианогидрина, который в свою очередь реагирует с (II) с образованием интенсивно окрашенного пурпурного соединения 1,3-10-9г/.ил (при СФ-оп-ределении) [788]
со
Таблица 10 (окончание)
Литература [1007] [747] [1235]
Мешающие элементы или ионы 5 | । СО
Предельное разбавление 1 1 1
Предел • обнаружения, мкг 1 1Л 1 о"
Видимый эффект реакции Изменение окраски комплекса Hg2+ с(1) из оранжево-красной в желтую в присутствии HCN (разрушение комплекса) Появление максимума интенсивности флуоресценции продуктов реакции с CN- в области 500 нм Образование медью (II) с замещенными га-диаминоди-фенила комплексных катионов, реагирующих с CN~, SCN~ и дающих голубоватозеленоватую окраску 1
Способ выполнения, метод । 1 га д И аб н з, ы S О, Я Ц й S и АН В S и а н И СС ах й ® Ц Я н Ф SgSaSgg'w* е д' g лЗ И Svo ЗЙ е g ‘ и и и н g
Реактив или реакция Комплекс Hg2+ с пиридилазорезорцином (I) Производные хинона Смеси, содержащие замещенные га-диаминодифенила и соли Си (CuSO4, CuCh, СиСО3); pH 4,5-5,0
SsE-S а ч § и Я и о ® О щ 1 ** 1 Й5 L, z • и
Примечание. ХБ — хроматография на [бумаге; К—капельная реакция; КФБ — капельная реакция на фильтровальной бумаге; ВЭБ — высоковольтный электрофорез на бумаге; М — микрокристаллоскопическая реакция; П — пробирочная реакция; ЦР — цветНад реакция; X — хроматографический метод; ИКС — инфракрасная спектроскопия; Ф — флуоресцентная реакция; КЛ — кислоты Льюиса (CEhCQCl,
Aids, ZnCl2, MgCIj, HgCl2, FeCli).
44
пакет) определяют содержание NO2 в воздухе. Таблетка-индикатор содержит 0,25 г сульфаниловой кислоты (диазотирующий реагент) 0,25v дигидрохлорида М-(1-нафтил)этилендиамина (связующий реагент) и различные добавки (MgSO4, глицерин, борная кислота, тальк, гель кремневой кислоты, гипс, вода). Здесь же указан способ изготовления индикаторных таблеток.
В работе [1008] рассмотрены методы открытия микро- и ультра-микроко^ичеств анионов (в том числе NO2, NO3, SCN“, CN-) в капиллярах с использованием специфических реагентов. Для открытия NO2~ и NO; используют дифениламин и Fe(II) 4- Fe(III) (цветная реакция); для SCN- — Fe(II) Fe(III) (цветная реакция); для CN-— Fe(II) 4-Fe(III) и AgNO3 (образование осадка) и NaHCO3 HgBr2 4 метиловый красный (цветная реакция).
Интересен метод открытия микроколичеств нитрит-ионов с помощью малахитового зеленого или конго красного [886].
Открытие с малахито вы м зеленым (М3). К 0,25 мл анализируемого нейтрального раствора прибавляют 2 капли 0,1%-ного водного раствора М3 и 1—2 капли НС1 (1 : 3). В присутствии NO; появляется красная или красно-пурпурная окраска, интенсивность которой сначала увеличивается, а затем через 1—3 мин. исчезает (в отсутствие NO; раствор имеет желтую или желто-зеленую окраску). Открываемый минимум 75 мкг NO2 в 0,25 мл.
Открытие с конго красным (КК). К 0,5 мл анализируемого нейтрального раствора прибавляют 2 капли 0,1%-ного водного раствора КК, 1 каплю НС1 (1 : 1), 2—3 капли ацетона и перемешивают. В присутствии NO; появляется фиолетовая или пурпурная окраска или осадок (в отсутствие NO; окраска раствора голубая). Открываемый минимум 550 мкг NO; в 0,5 мл. Мешают азиды и иодиды.
Предложена новая капельная проба на цианид-ион и газообразный циан (CN)2 [1349], основанная на образовании комплексного иона Cu(CN)4- и восстановлении им МоО1- до молибденовой сини.
Одну каплю исследуемого раствора обрабатывают одной каплей 0,001%-ного раствора CuSO4 и 1—2 каплями 5%-ного раствора Na2MoO4 в конц. HCI. В присутствии CN~ сразу же развивается синее окрашивание. Предел обнаружения 0,15 мкг CN" в 0,1 мл, предельное разбавление 1 : 30 000.
Для обнаружения (CN)2 испытуемый газ пропускают через смесь, 0,01 %-ную по CuSO4 и 1%-ную по NaOH, и прибавляют 2—5 капель 5%-ного раствора Na2MoO4 в конц. HG1. В присутствии (GN)2 развивается синее окрашивание.
Обнаружению CN~ и (CN)2He мешают CNO“, NO;, SO3-, РО|-, AsO®-, SON", GeO^-; мешают Fe£+, Sn2+, Sb3"1', S2O3-, J~.
Глава IV
ГРАВИМЕТРИЧЕСКИЕ МЕТОДЫ
НЕОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ АЗОТА С ВОДОРОДОМ
Аммоний
Гравиметрические методы для определения аммония используются относительно редко. Известные способы основаны на осаждении комплексных солей.
Одним из самых старейших методов гравиметрического определения аммония является метод, основанный на осаждении хлороплатината аммония. Чтобы обеспечить получение точных результатов при использовании платинохлористоводородной кислоты в качестве осадителя, требуется соблюдение специальных условий: соли аммония должны присутствовать в растворе в форме хлоридов; в раствор должны быть добавлены органические реагенты для обеспечения минимальной растворимости; другие катионы (Rb+, Cs+), образующие также с этим осадителем труднорастворимые соединения, должны отсутствовать. В настоящее время этот метод не имеет практического значения вследствие замены его более простыми и точными методами, в частности, например, методами ацидиметрического титрования.
Тетрафенилбор также служит осадителем для иона аммония, правда, не специфичным. Поэтому требуется предварительная отгонка аммиака для отделения от мешающих ионов. Методика осаждения тетрафенилбората аммония заключается в добавлении к анализируемому раствору (pH 2), содержащему ионы аммония, 100%-ного избытка тетрафенилбора, отделении осадка, промывании его спиртом, высушивании при 100° С. Растворимость осадка существенно зависит от ионной силы раствора в диапазоне pH 1,5—3,1. При pH < 1 происходит разложение реагента, а при pH 5,5 аммиак может быть потерян вследствие улетучивания.
В качестве реагента для гравиметрического определения иона аммония (наряду с Cs+, Ва2+, Zn2+, Mg2+) предложена нитробар-битуровая кислота [1058], с которой NH^ образует малорастворимый, легкофильтрующийся кристаллический осадок. Чувствительность метода 1 мкг/мл.
46
0,1—0,2 г пробы растворяют в 25—30 мл воды, добавляют равный объем 95%-иого этанола и по каплям 2%-ный раствор нитробарбитуровой кислоты в 50%•ном этаноле до желтоватой окраски раствора над осадком. Осадок отфильтровывают через фильтр № 4, промывают этанолом и эфиром, взвешивают. Фактор пересчета на NH* равен 0,0949.
Изучено также термогравиметрическое поведение нитробарбитуратов ряда катионов и выведены формулы, позволяющие по весу исходных осадков и продуктов их разложения определять содержание каждого элемента в бинарных смесях. Удовлетворительные результаты получены, в частности, при анализе смесей Zn-ВД, Cd —NHJ.
Для гравиметрического определения иона NH4+ в водной среде использован реактив Несслера [436]. Получающийся осадок имеет cocTaBNH4Hg2Jс очень низким фактором пересчета на азот(0,02584).
К 25 мл анализируемого раствора прибавляют двойной избыток реактива Несслера и раствор с осадком выдерживают 10 час. при 20° С и еще 3 часа при нагревании на водяной бане. Осадок отфильтровывают через плотный стеклянный фильтр (с величиной пор 1 мк), промывают деионизованной водой и высушивают при 150° С.
При определении иона аммония в растворе хлорида аммония с содержанием от 2 до 4 мг N ошибка определения составила 2,1 %. Мешают определению комплексообразующие соединения: KCN, KSCN, этилендиамин, пиридин, а также восстановители: NH2OH и N2H4.
Гидразин
Косвенное гравиметрическое определение с нитратом серебра [473]. Предложенный метод основан на действии определенного количества нитрата серебра в присутствии аммиака на раствор гидразина по реакции
6N2H4-HNOb+6HNO3 + 21AgNO3 + 32NH3 = 21Ag + UN + 33NH4NO3.
Оставшиеся в растворе ионы серебра осаждают соляной кислотой и осадок взвешивают.. Метод не имеет практического значения.
Азотистоводородная кислота
Из-за взрывоопасности большинства осажденных азидов (например, серебра, ртути) гравиметрические методики для определения азотистоводородной кислоты применяются крайне редко. При этом должны быть обеспечены меры предосторожности. Например, в методе [646] предпочитают для надежности высушивать не азид серебра, а промытый, влажный осадок азида обработать азотной кислотой, осадить соляной кислотой хлорид и этот осадок взвешивать.
47
К 10 мл раствора, содержащего около 0,03 г свободной азотистоводйрод" ной кислоты, добавляют в небольшом избытке едкий кали, нейтрализуют по лакмусу азотной кислотой (1 : 4), добавляют избыток в 1 каплю азотной кислоты (1 : 100) и осаждают нитратом серебра. Осажденный азид серебра промывают декантацией водой, осадок растворяют в разбавленной азотнай кислоте (1:4), отгоняют кипячением азотистоводородную кислоту, осаждают избытком соляной кислоты хлорид серебра, собирают осадок, как обычно, и взвешивают. 1 г HN3 соответствует 0,03002 г AgCl.
НЕОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ АЗОТА С КИСЛОРОДОМ
нитрат-ионы
Одним из наиболее широко и давно (с 1905 г.) употребляемых для гравиметрического определения азота реактивов является нитрон — органическое основание, образующее малорастворимые соединения с некоторыми кислотами [582]. Нитрон применяется в основном для определения азотной, хлорной и пикриновой кислот, органических нитратов, азотнокислых эфиров, нитросоединений и окислов азота. Формула соединения
Н СвН6
СвН5
Nz N - CeHs
ХХ = n
или CjoHieNi.
Азотнокислый нитрон (весовая форма) имеет формулу C2oH16N4-•HNO3 (мол. вес 375,40).
По порядку уменьшения растворимости анионы могут быть расположены в следующий ряд: бромид нитрит хлорат хромат роданид иодид^> нитрат перхлорат пикрат. Имеется указание, что щавелевая и лимонная кислоты также образуют более или менее труднорастворимые соли с нитроном.
Мешают определению кислоты, образующие нерастворимые соли: НВг (1 : 800), HJ (1 : 20 000), HNO2 (1 : 4000), H2CrO4 (1 : : 6000), НСЮ3 (1 : 4000), НС1О4 (1 : 50 000), HSCN (1 : 15 000), гексацианоферраты(П) и (III), большие количества хлоридов. При определении от 0,1 до 0,15 г KNO3 результаты бывают занижены на 2—5%.
Не мешают определению следующие соединения: йодаты, алюминий, сульфаты магния и аммония, фосфат калия, хлорид магния. Серная, соляная, фосфорная, борная, муравьиная, уксусная, бензойная и винная кислоты образуют с нитроном растворимые соли [61].
К анализируемому раствору (80—100 мл), содержащему < 1 мг!мл NO3 и свободному от перечисленных выше веществ, добавляют 1 каплю концен-48
триррванной H2SO4 или 1 мл ледяной уксусной кислоты и нагревают до кипения. Й один прием вводят 10—12 мл 10%-ного раствора ацетата нитрона в 5%-ной уксусной кислоте (хранится в склянке из темного стекла в прохладном месте), Перемешивают и оставляют стоять в течение ~ 1 часа. После охлаждения в течение 1,5 час. в ледяной воде осадок отфильтровывают через взвешенный тигелф при умеренном отсасывании, используя фильтрат для перенесения осадкй.в тигель и ополаскивания стакана. Затем промывают осадок 5 раз ледяной водой порциями по 2 мл. Чтобы удосторериться в полноте осаждения, нагревают фильтрат до кипения, прибавляют еще немного осадителя и охлаждают, как прежде. Тигель с осадком сушат при 105° С в течение 1 часа и по охлаждении в эксикаторе взвешивают. Эмпирический фактор пересчета на HNO3 равен 0,1679, на NaNO3 — 0,2264.
Для отфильтровывания нитрата нитрона в [744] рекомендуют использовать фильтры из стекловолокна. Эти фильтры при работе с водными растворами не набухают, не изменяют размеров пор в результате контакта с кислыми или умереннощелочпыми растворами, устойчивы к органическим растворителям. Вес фильтра из стекловолокна диаметром 5 см составляет около 150 мг, что позволяет применять их для гравиметрического определения небольшого осадка.
В работе [1018] после осаждения NO3 в виде азотнокислой соли нитрона осадок используют для определения нитратов с помощью ИК-спектроскопии.
При определении нитратов в присутствии нитритов мешающее действие последних устраняют обработкой пробы сульфатом гидразина.
0,2 г пробы растворяют в 5—6 мл воды и при охлаждении медленно добавляют 0,25 г измельченного в порошок сульфата гидразина. После окончания выделения газа раствор разбавляют до 100 мл и далее работают, как описано выше.
При равных соотношениях нитрат- и нитрит-ионов результаты определения нитратов бывают завышены на 5%.
Нитриты могут быть переведены в нитраты с помощью Н2О2.
Растворяют вещество, содержащее от 0,1 до 0,2 г нитрита и нитрата, в 50 мл воды, добавляют' 20 мл 3%-ного раствора перекиси водорода и нагревают’ до 70 С. Затем приливают из капельной воронки 20 мл 2%-ной серной кислоты, нагревают до кипения и осаждают нитрат, как описано выше, с помощью 12 мл ацетатного раствора нитрона.
Гравиметрический метод определения нитратов осаждением нитроном представляет собой относительно селективный и точный метод, хотя и сравнительно дорогостоящий и, как все гравиметрические методы, трудоемкий. Поэтому в сегодняшней практике он используется довольно редко.
В других известных методах осаждения нитратов работают с еще более дорогостоящими осадителями, причем иногда они менее специфичны для нитрат-ионов, что требует предварительного уда
49
ления мешающих ионов. Эти методы представляют поэтому только исторический и теоретический интерес.
Осаждение а-динафтодиметиламином [1236]. Формула этого основания следующая:
CH2-NH—СН2
СО 00
С нитрат-ионами образует труднорастворимое соединение, используемое в качестве весовой формы при гравиметрическом определении нитратов. Состав осадка C22H19N • HNO3, мол. вес 360,42. 1 г нитрата динафтодиметиламина соответствует 0,1748 г HNO3 или 0,2358 г NaNO3.
В качестве осадителя используют 10%-ный раствор основания в 50 %-ной уксусной кислоте. Ацетат кристаллизуется при комнатной температуре, а при нагревании снова переходит в раствор. Для получения крупного, легкофильтруемого и хорошо отмываемого осадка осаждение проводят при температуре кипения.
Пробу растворяют в 150—200 мл воды, добавляют 2—5 мл 10%-ного раствора серной кислоты, 10 мл горячего 10%-ного раствора основания в 50%-ной уксусной кислоте. Охлаждают, фильтруют, промывают холодной водой и высушивают при 110° С. Точность метода + 0,2—0,4%.
Мешают определению галогенид-, нитрит-, хлорат-, бромат- • и перхлорат-ионы, образующие также труднорастворимые осадки. Эти ионы должны быть предварительно отделены.
Осаждение п-толилизомочевиной [466]. Формула осадителя C7H7-S-C(NH)(NH2). Это вещество в присутствии серной кислоты образует с нитрат-ионами очень трудно растворимую двойную соль, которая выпадает при разбавлении 1 : 100 000. Она содержит на каждые 3 моля основания 1 моль серной кислоты и 1 моль азотной кислоты. В качестве осадителя используют ~ 0,75 N раствор основания (1 ч. ацетата n-толилизомочевины на 5 или 6 ч. холодной воды).
Растворяют ~ 0,15 г нитрата калия в 10 мл воды, добавляют 10 мл 2N НС1 и осаждают с помощью 20 мл указанного выше раствора осадителя; выдерживают осадок в течение 0,5 часа в ледяной воде, отфильтровывают через стеклянный тигель, промывают несколько раз разбавленным раствором осадителя и один раз небольшим количеством ледяной воды, после чего, наконец, многократно — охлажденным спиртом. Затем высушивают при НО—120° С и взвешивают. При расчете'1’г'осадка'соответствует 0,0955 г HNO3 или 0,1288 г NaNO3. Результаты получаются обычно завышенными на 0,5—1%.
Хотя хлорид-ионы образуют соответствующую двойную соль, примерно в 100 раз более растворимую, чем нитрат, даже небольшие количества хлорида мешают определению и должны быть предварительно отделены, например, с помощью сульфата серебра. Мешают также другие галогенид-ионы, нитрит-, хлорат-, перман-50
ганат-, роданид-ионы и гексацианоферрат(П), в отличие от фосфата, цианида и гексацианоферрата(Ш).
Осаждение цинхонамином [465]. Брутто-формула C19H24N2O. Это соединение образует с 1 молем HNO3 труднорастворимый осадок, который используется для гравиметрического определения нитрат-иоцрв. Так как в 1 л растворяется примерно 2 г нитрата цинхонамийа, осаждение следует проводить с помощью насыщенного водного раствора.
К нейтральному нитратному раствору (содержащему ~ 0,1 г нитрата), свободному от хлорид-ионов, добавляют 1 каплю уксусной кислоты, нагревают до кипения и обрабатывают теплым раствором сульфата цинхонамина. Выдерживают 12 час. на холоду, фильтруют и промывают насыщенным водным раствором нитрата цинхонамина, затем небольшим количеством воды, высушивают при 100° С и взвешивают. 1 г осадка (мол. вес 359,43) соответствует 0,1753 г HNO3 или 0,2365 г NaNO3. Полученные результаты бывают примерно на 1% занижены.
Мешают определению иодиды, хлориды; не мешают — хлораты.
Осаждение ионами дициклогексилталлия(Ш) [816]. Этот метод имеет гораздо меньшее значение, чем нитронный. Реагент не дешевле.
50—100 мл анализируемого раствора, содержащего от 50 до 100 мг NOg , нагревают до кипения и приливают 50—100 мл 0,05 N раствора карбоната дициклогексилталлия(Ш) в 0,2 N H2SO4. Охлаждают, фильтруют через стеклянный фильтр Гуча, промывают 2—5 раз по 10 мл ледяной воды, высушивают в сушильном шкафу при температуре не более 150° С. Формула осадка C12H22T1NO3 (мол. вес 432,7), фактор пересчета на NO“ равен 0,1433, на HNO3 —0,1456. Точность метода составляет ± 5%.
Хлорид-, бромид-, иодид-ионы также образуют труднорастворимые комплексные соли с реагентом и должны быть предварительно удалены с помощью фторида или ацетата серебра.
Для определения нитрат-ионов более перспективными (по сравнению с нитроном) оказываются N-замещенные 1-нафтилметилами-нов [506] вследствие меньшей растворимости их нитратов. Например, предельное разбавление для нитрата М-(4-хлорбензил)наф-тилметиламина составляет 1 : 70 000 [854]. Растворимость этого нитрата примерно в 4 раза меньше растворимости нитрата нитрона.
Замещенными бензил-1-пафтилметиламина не осаждаются ВгОз, Вг~, JO3, SO2-, Н2РО7, НС4Н40ё, S20g~ при их концентрации ^5 мг!мл. Эти соединения образуют осадки с СЮ3, С1О4, Fe(CN)t, Fe(CN)t, J", NO2’, MnO;, SCN", WO42~, C2O2~ и MoO2’.
При использовании М-бензил-1-нафтилметиламина и его М-(4-метилбензил)-иМ-(4-хлорбензил) производных ошибка опреде-
51
ленияот 1до 50 jttsNOg составляет <1,2; <1,0% соответ-
ственно.
М-(4-хлорбензил)-1-нафтилметиламин применен для определения нитрат-ионов в смеси с перхлорат-ионами после предварительного осаждения СЮ^-ионов с помощью хлорида тетрафеиилфосфо-ния [855].
Нитрит-ионы
Для количественного определения нитрит-ионов гравиметрические методы имеют меньшее значение по сравнению 0 гравиметрическими методами определения азотной кислоты. Азотистая кислота образует с 2,4-диамино-6-оксипиримидином труднорастворимое в воде нитросоединение. Но точных гравиметрических методов на основе этой реакции не разработано.
Косвенные гравиметрические методы основаны на весовом определении продуктов реакций, протекающих количественно с нитритами. Такой реакцией может быть взаимодействие бромата серебра с азотистой кислотой; после проведения этой реакции образующийся в результате восстановления бромид серебра может быть взвешен.
Косвенное определение нитрит-ионов осаждением в виде галогенида серебра основано на реакции
3HNO2 + AgBrO3 -* 3HNO3 ± AgBr.
По методу [583] подкисленная уксусной кислотой проба, содержащая нитриты, обрабатывается горячим раствором бромата серебра и получающийся бромид серебра взвешивается.
В колбе Эрленмейера емкостью 750 мл растворяют при нагревании до ~80° С от 1,4 до 1,5 а бромата серебра в 100 мл воды и ПО мл 2N уксусной кислоты. Затем при этой же температуре приливают по каплям из капельной воронки нитритный раствор (1 г в 200 мл воды) при помешивании. При этом образуется слегка окрашенный зеленоватый осадок бромида серебра. После промывания воронки медленно добавляют еще 30 мл серной кислоты (1 : 4) при 85° С. При взбалтывании и дальнейшем нагревании до 90° С раствор осветляется. Сразу же желтый осадок отфильтровывается через стеклянный фильтр Гуча, отмывается от избытка бромата примерно 1 л нагретой до кипения воды, высушивается при 130° С и взвешивается. 1 г AgBr соответствует 0,7350 г NO^•
Параллельные определения выполняются с воспроизводимостью в пределах ±0,1%. Галогениды реагируют подобным же образом. Их содержание должно быть определено отдельно и вычтено.
Можно обрабатывать нитритный раствор амидосульфоновой кислотой и образующуюся при этом серную кислоту осаждать в виде сульфата бария или определять взвешиванием возникшую потерю в весе.
52
Было установлено, что нитриты образуют с солью 2,4-диамино-6-оксипиримидипа трудпорастворимое соединение — 2,4-диами-но-5-нш;розо-6-оксипиридин, однако при определении веса этого соединения, высушенного при 120—140° С,’получаются значения, различающиеся на 0,4—2% и более. Поэтому на основе этой весовой формы не было предложено точного гравиметрического метода. Этот метод предлагается для качественного обнаружения или отделения азотистой кислоты перед последующим обнаружением в виде нитрата. 0,05 мг NO2 в 1 мл образует отчетливый осадок.
Окислы азота
Гравиметрические методы для определения окислов азота применяются редко и в настоящее время не имеют практического значения .
Гравиметрическое определение окиси азота по методу [536] основано на полном поглощении NO хромовой кислотой в азотнокислом растворе (10 г СгО3 в- 15 мл 12%-ного раствора HNO3), который помещен во взвешенный сосуд, соединенный с также взвешенной хлоркальциевой трубкой. По увеличению веса сосуда определяют содержание окиси азота. В качестве газа-носителя используют чистую двуокись углерода, свободную от примесей воздуха. Растворимость двуокиси углерода в поглощающей жидкости обусловливает неконтролируемую ошибку определения.
При пропускании закиси азота через раскаленную докрасна железную спираль NaO разлагается на азот и кислород, причем последний связывается железом. Определяют увеличение в весе последнего [721]. Из-за малой специфичности метод не имеет большого значения.
Можно пропускать закись азота в смеси с водородом через све-жевосстановленную медную спираль в маленькой трубке для сжигания при красном калении и собирать образовавшуюся воду во взвешенную хлоркальциевую трубку.
Роданид-ионы
В работе [1258] предложен гравиметрический метод определения роданида (в медноцианидных гальванических ваннах), основанный на окислении SCN~ с помощью последовательной обработки образца рядом окислителей (HNO3, Н2О2) и осаждении образующихся ионов SO^- в виде BaSO4. Мешают определению сульфаты и серусодержащие соединения.
Глава V
ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
Титриметрические методы применяются более широко, чем гравиметрические, для определения азота и его соединений. Эти методы основаны на различных реакциях: нейтрализации, осаждения, окисления — восстановления и комплексообразования. Во всех случаях реакция должна протекать строго стехиометрически и до конца.
Конечная точка титрования (КТТ или точка эквивалентности) устанавливается визуально, фотометрически или с использованием электрохимических методов.
В качестве титрантов используют как неорганические, так и органические соединения.
КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
Аммоний
Наиболее часто используемым методом определения азота в водных и неводных средах является прямое титрование после поглощения аммиака избытком кислоты. Титрование проводится с внутренними индикаторами, изменяющими окраску в интервале значений pH от 4 до 5 (в водной среде). В неводной среде чувствительность обнаружения КТТ увеличивается. Так, при использовании ледяной уксусной кислоты в качестве поглощающего раствора и титровании хлорной кислотой с кристаллическим фиолетовым как внутренним индикатором (либо с потенциометрической фиксацией КТТ) получаются хорошо воспроизводимые результаты [730, 949, 1168].
Избирательность прямого метода титрования может быть существенно улучшена путем предварительной отгонки аммиака, так как после этого мешают только некоторые летучие амины, мочевина и амиды.
Метод Кьельдаля. Наиболее распространенный титриметриче-ский метод определения аммонийных солей был предложен Кьель-далем [931]. По первому варианту этого метода проба, содержащая неорганические и органические соединения азота, разлагается ки-54
пяченйем с серной кислотой в присутствии катализаторов. Азот при этом переходит в сульфат аммония. После подщелачивания образовавшейся смеси гидроокисью натрия аммиак отгоняется, поглощается известным количеством титрованной серной кислоты, избыток которой оттитровывается щелочью.
Удобным поглотителем аммиака является 4%-ный раствор борной кислоты. Раствор борной кислоты не изменяется при хранении в сосуде из стекла пирекс. При поглощении аммиака не требуется точно отмерять объем поглощающего раствора и использовать титрованный раствор едкой щелочи. Образующийся по реакции
NH3 + НВО2 — NH4BO2
борат аммония титруют кислотой:
NH4BO2 + Н+ — NH+ + НВО2.
В качестве внутреннего индикатора рекомендуют метиленовый голубой или метиловый красный [949, стр. 168]. Окраска изменяется от зеленой через бесцветную (конец титрования) до пурпурной. Состав поглотительных растворов приведен в работах [1364, 1408].
В настоящее время не известен вариант метода, который был бы применим для любого азотсодержащего соединения. Варианты этого метода с необходимыми изменениями для анализа различных классов соединений азота изложены в соответствующих разделах настоящей монографии.
Модификация метода Кьельдаля применительно к определению небольших концентраций аммиака заключается в титровании системы аммиак—борная кислота сульфаминовой кислотой [1067].
Известна модификация метода определения азота по Кьельда-лю без применения отгонки [817а]. Ионы аммония окисляют до азота гипогалогенитами, причем регулирования температуры и значения pH раствора NaBrO не требуется. Вредное влияние катализаторов окисления устраняют прибавлением NaBr, H2SO4 и Na2S.
0,1 г вещества, содержащего 2—10 мг N, кипятят в колбе Кьельдаля с 7 г K2SO4, 20 мл концентрированной H2SO4 и соответствующим количеством катализатора (0,1 г HgO, HgSO4 и Se или 0,05 г CuSO4). Смесь охлаждают, нейтрализуют 10 N раствором NaOH в присутствии метилового красного, прибавляют еще 2 капли раствора щелочи, 75 мл ОДУ раствора NaBrO (16 г NaOH растворяют в 40 мл воды, прибавляют 4 мл брома и доводят раствор водой до объема 1 л; хранят в закупоренной склянке из желтого стекла), через 5 мин. приливают 20 мл 10%-ного раствора КД и 50 мл 2N H2SO4. Выделившийся иод титруют раствором Na2S2O3 в присутствии крахмала. 1 мл ОДУ раствора Na2S2O3 соответствует 0,4666 мг N.
Если в качестве катализатора употребляют Se, то раствор, полученный после разложения вещества, разбавляют водой до 500 мл, прибавляют 10 мл
55
5 %-ной H3SO3, кипятят смесь до обесцвечивания, фильтруют, фильтрат нейтрализуют 1OJV раствором NaOH и заканчивают анализ, как описайо выше.
При применении в качестве катализатора CuSO4 раствор, полученный после разложения вещества, разбавляют водой до 20 мл, прибавляют 5 мл 1%-ного раствора Na2S, осадок CuS отфильтровывают, фильтрат кипятят с 5 мл H2SO4 до тех пор, пока он не перестанет давать желтый осадок с CdCl2, и заканчивают анализ, как описано выше.
В качестве растворителя для титриметрического определения аммония и его замещенных солей предложено [495] использовать диметилсульфоксид, в котором хорошо растворимы многие аммонийные соли. Титрованию в этой среде не мешают небольшие количества воды (<^11%).
При определении аммиака (так же, как и NO2) путем кислотно-основного титрования можно использовать как вторичные стандарты гексамминникельбромид (или хлорид) и тетрамминникель-нитрит. Оба вещества могут быть синтезированы с содержанием основного вещества ~99,9% и устойчивы длительное время при хранении в закрытой посуде [598].
Конечная точка титрования в кислотно-основном методе определения иона аммония хорошо может быть установлена с помощью стеклянного электрода [1281].
К кислотно-основным методам относится формальдегидный метод [1232, 1393], в основе которого лежит следующая реакция:
4NH+ + 6НСНО (CH,)eN4 4- 4Н+ + 6Н2О.
Реакция протекает быстро и при высоких значениях pH. Образовавшаяся в результате этой реакции кислота титруется щелочью и затем рассчитывается содержание аммония. Предусмотрен метод обратного титрования, гарантирующий полноту протекания реакции ионов аммония с формальдегидом [1317].
Гидразин
Гидразин может быть превращен в аммиак действием цинка и хлорной кислоты и определен по описанному выше методу Кьель-даля. Этот процесс не специфичен для гидразина, так как подобным образом превращаются в аммиак также многие другие азотсодержащие соединения.
Гидразин можно определить в водных и неводных средах прямым титрованием. В неводных средах (ледяная уксусная кислота) хорошо выражена конечная точка при титровании хлорной кислотой. В водных растворах для получения лучших результатов необходимо определять КТТ потенциометрическим методом [1160]. При использовании кристаллического фиолетового в качестве внутреннего индикатора или потенциометрической индикации КТТ получаются хорошо воспроизводимые результаты [460, 578].
56
Гидразин можно определять ацидиметрически титрованием в водной среде или в среде ледяной уксусной кислоты с помощью фенилгидразипа или бензальдегидсульфоиовой кислоты [1055].
Смеси гидразина с аммиаком также могут быть проанализированы титриметрическим методом. Если требуется определить небольшие количества аммиака в присутствии гидразина, то основную массу последнего предварительно удаляют осаждением с помощью сульфата меди из кислых растворов, фильтрат обрабатывают избытком щелочного окисляющего раствора, а затем отгоняют аммиак и определяют его ацидиметрически.
Азотистоводородная кислота
Азотистоводородную кислоту можно определять прямым титрованием щелочью, фиксируя КТТ либо потенциометрически, либо с помощью внутреннего индикатора — метилового красного [577]. При расчете следует вносить поправки на влияние мешающих примесей.
Нитрат-ионы и азотная кислота
После поглощения окислов азота нейтральным 3%-ным раствором перекиси водорода образовавшаяся азотная кислота может быть определена методом ацидиметрического титрования [453, 647, 1190] стандартным раствором гидроокиси натрия с потенциометрическим или визуальным обнаружением КТТ. Этот метод обеспечивает большую точность определения.
Чистая безводная азотная кислота также может быть определена методом кислотно-основного титрования с большой надежностью, однако следует иметь в виду, что этот метод дает оценку общей кислотности, поэтому либо примеси других кислот должны отсутствовать, либо необходимо вносить соответствующие коррективы на их содержание при расчетах содержания азотной кислоты [441, 1110].
Дымящую азотную кислоту определяют после поглощения ее избытком стандартного раствора щелочи методом обратного титрования 0,1 N серной кислотой, используя фенолфталеин в качестве индикатора. Необходимо учитывать содержание двуокиси азота в дымящей азотной кислоте при ее анализе.
Анализ смесей азотной и серной кислот может быть выполнен титрованием в неводных средах [630, 792, 1026]. Морфолин является эффективным титрантом в среде ацетонитрила. При потенциометрической фиксации КТТ серной и азотной кислот четко различаются. Для этой же цели с успехом может быть применена гидроокись тетраалкиламмония в случае титрования в среде пиридина или ацетона. На потенциометрической кривой титрования первый излом соответствует оттитровыванию азотной кислоты и одного иона водорода серной кислоты. Разница между
57
этой конечной точкой и вторым изломом соответствует оттитровы-ванию второго протона серной кислоты. Раствор гидроокиси тет-рабутилметиламмония в ацетоне был использован для титримет-рического анализа смеси указанных кислот с автоматической индикацией точек эквивалентности потенциометрическим и спектрофотометрическим методами.
В работе [220] предложено применение борида никеля для восстановления малых количеств NOg до NH3 при определении 10— 100 мг NOg.
В колбу для отгонки NHg вводят 25 мл анализируемого раствора, прибавляют 100—150 мл бидистиллята, 20 мл 20%-ного раствора NaOH, 0,25— 1,5 г борида никеля (получают прибавлением 1 М NaBH4 к 1 М NiCl2 и промыванием выпавшего черного осадка борида никеля бидистиллятом, не содержащим NH3) и отгоняют NHg (30—40 мин.), поглощая его раствором Н3ВО3. Добавляют к дистилляту 2—3 капли раствора смешанного индикатора^! ч. метилового красного и 5 ч. бромкрезолового зеленого) и титруют раствором НС1 до перехода зеленой окраски раствора в винно-красную.
При содержании в пробе 0,1—1 г выделяющийся аммиак поглощают разбавленным раствором соляной кислоты и определяют фотометрически с реактивом Несслера.
В методе [393, 458] для определения 0,1—10 мг NOg после переведения в аммиак последний отгоняют с водяным паром из щелочного раствора (в присутствии FeSO4 или CuSO4) и титруют соляной кислотой. Авторы указывают, что при определении 0,1 — 1 мг и 5—lOjitaNOg ошибка составляет 1—Зи0,5% соответственно.
Нитрит-ионы
Нитриты можно определить ацидиметрически, используя титрование в неводных растворителях [1144]. Достаточно хорошая воспроизводимость результатов была достигнута при титровании нитрита в смеси этиленгликоля и пропанола (или хлороформа). Титрантом являлся раствор 0,1 N хлорной кислоты в той же смеси. КТТ устанавливали визуально (с внутренним индикатором тимоловым голубым) или потенциометрически со стеклянным и каломельным электродами. С теми же электродами нитрит-ионы могут быть определены (и в присутствии нитратов) потенциометрическим титрованием гидроокисью тетрабутиламмония в бензол-метаноловой смеси (10 : 1) [633].
Разработаны методы определения нитратов, нитритов и их смесей, а также смесей нитратов элементов различных групп периодической системы титрованием в неводных растворах [27]. Эти методы основаны на различном поведении нитрат- и нитрит-ионов при титровании в неводных средах. Определение NO3 основа
58
но на прямом потенциометрическом титровании в среде метанола (или смеси метанола с ацетоном) неводными растворами кислот, в частности метаноловым или метилэтилкетоновым раствором НС104. При определении нитратов элементов первой и второй групп периодической системы элементов предварительно проводят ионный обмен на катионите СДВ-3 или КУ-2 в Н-форме и затем титруют потенциометрически полученную HNO3, так как указанные нитраты в среде метанола или кетонов — нейтральные соединения. В случае определения нитратов аммония, элементов третьей и последующих групп проводят прямое титрование их метанольных или метанольно-кетоновых растворов неводными растворами оснований.
Предложен титриметрический метод определения HNO3 и НС104 при совместном присутствии, основанный на титровании их в среде метилэтилкетона метанольно-кетоновым 0,05 N раствором КОН с потенциометрической регистрацией КТТ [76]. Точность определения ± 10 отн.%.
Цианид-ионы
Макро- и микроколичества цианид-ионов определяют ацидиметрическим методом, основанным на реакции образования в щелочной среде нитрила гликолевой кислоты (в присутствии формальдегида), гидролизующегося в щелочной среде при кипячении с образованием NH3. Последний отгоняют и определяют, титруя стандартным раствором НС1 [1032]. Метод позволяет определить 0,004— 0,01 г-ион CN~.
Методы кислотно-основного титрования применяют для определения азота в ряде природных и промышленных объектов. Например, 0,1 мг!мл (и более) NHt, NO3 или NOg, содержащиеся в почвенных и растительных вытяжках, могут быть определены с ошибкой 0,3—0,5% [561]. Метод основан на отгонке NH3 с водяным паром и последующем титровании NH4 стандартными растворами НС1 или H2SO4.
Ацидиметрически определяют азот в нитридах бора и кремния, причем получающиеся результаты хорошо согласуются с данными, получаемыми по методу Дюма [324].
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ
ТИТРОВАНИЕ
В титриметрических методах определения азота оксидиметри-ческие методы наиболее распространены.
Аммоний
Для определения аммония наиболее общей реакцией окисления-восстановления, представляющей интерес в аналитической практике, является титрование растворами гипохлорита или гипо-
59
бромита (OX):
2NH+ + ЗОХ _ N2 + 2H+ + ЗХ- + 3H2O.
Гипохлорит более предпочтителен вследствие большей стабильности раствора. Избыток гипохлорита определяют иодометрически или титрованием раствором арсенита [943, 948]. Содержание иона аммония рассчитывают по указанному выше уравнению.
Гидразин
Гидразин — сильный восстановитель и количественно окисляется до азота по реакции
N2H« = N2 + 4Н+ + 4е (Е° = 0,22 в)
многими окислителями. Поэтому для него разработано много методик оксидиметрического титрования с использованием различных окислителей, указанных ниже.
Титрование йодатом и иодом. В наиболее общепринятом методе в качестве титранта используют иодат калия в сильнокислой среде с образованием монохлориода в КТТ [1160, 1189] по реакции
N2H« + JO~ + Cl- + 2Н+ N2 + JC1 + ЗН2О.
По этой методике к анализируемому раствору, 3—5 N по HCI, добавляют несколько миллилитров хлороформа или четыреххлористого углерода в качестве несмешивающихся с водой растворителей для поглощения иода, который образуется по реакции как промежуточный продукт. Смесь титруется стандартным раствором йодата калия. Точка эквивалентности четко устанавливается по исчезновению фиолетового цвета в слое органического растворителя. При титровании могут быть использованы и внутренние индикаторы [1298].
Гидразин можно определять прямым титрованием раствором иода в нейтральной среде [460, 942] по уравнению реакции
N2H4 4- 2J2 N2 + 4Н+ + 4J-.
Наилучшие результаты получаются, если поддерживать значение pH раствора между 7,0 и 7,4 (бикарбонатный буфер) и быстро заканчивать титрование. При pH 7 реакция протекает количественно, но очень медленно; при pH 7,5 получаются заниженные результаты.
Титрование броматом. Бромат калия был успешно использован для количественного определения гидразина в кислой среде [943, стр. 524] по реакции
3N2H4 + 2ВгО; 3N2 + 2Вг- + 6Н2О
с индиго и фосфорномолибденовой кислотой как внутренними индикаторами. Для устранения образования ряда побочных продуктов должна поддерживаться высокая кислотность раствора.
60
Титрование гипохлоритом. Гипохлорит количественно окисляет гидразин до азота в растворе с pH 7 [558] в соответствии с уравнением
N2H4 + 2С1О- N2 + 2С1- + 2Н2О.
Избыток гипохлорита реагирует с иодидом, раствор подкисляется и затем титруется тиосульфатом. КТТ определяется в присутствии крахмала.
О титровании гидразина гипохлоритом см. также [753].
Титрование перманганатом. С помощью перманганата гидразин определяют в щелочной среде, используя избыток окислителя, который оттитровывают обратно раствором двухвалентного железа [871, 943, стр. 68].
Титрование хлорамином Т. Гидразин окисляется до молекулярного азота при титровании раствором хлорамина Т в щелочных или кислых средах по реакции
N2H4 + 2C7H7SO2NC1- N2 + 2C7H7SO2NH2 ± 2Cl“.
Точность метода определения азота титрованием этим реагентом составляет ±0,1 % [611].
Титрование ванадатом. Гидразин окисляется количественно до молекулярного азота в случае титрования растворами пятивалентного ванадия [1290] в кислой среде:
N2H4 + 4 VO.; + 12Н+ - N2 ± 4VO2+ ± 8Н2О.
Использование ванадата вместо йодата более перспективно, так как он не улетучивается.
Титрование нитритом. От 0,1 до 100 мг гидразина можно количественно определить титрованием в среде 7—10%-ной НС1, 15— 30%-ной НС104, 10—12%-ной H2SO4 раствором нитрита калия или натрия [514]. При этом протекает следующая реакция:
N2H4 + 2NO“ + 2Н+ N2 + N2O + 3H2O.
Для получения стабильных результатов растворы титранта хранят без доступа воздуха в темных склянках.
Титрование сульфатом церия. Гидразин сначала количественно окисляется до азота феррицианидом в щелочной среде в соответствии с уравнением
N2H4 + 4Fe (CN± 4ОН~ N3 + 4Fe (GN )*' + 4H2O,
а затем полученный ферроцианид титруется сульфатом церия в сильнокислой среде (1,8 N НС1) [650] до перехода зеленой окраски раствора в коричневую. Избыток неиспользованного феррицианида можно оттитровать стандартным раствором роданида калия [634].
Титрование монохлориодом. Для количественного определения гидразина можно проводить титрование в кислой среде либо в при
61
сутствии ацетатного буфера, используя раствор монохлориода [1292]:
N2H4 4JC1 —► Ng -|- 2Jg -[ 4НС1.
КТТ обнаруживается потенциометрически. В нейтральной или щелочной среде монохлориод переходит в иодид, и точка эквивалентности устанавливается потенциометрически либо визуально.
Титрование солью железа(Ш). В качестве титранта можно применять раствор железных квасцов, которые окисляют гидразин до азота [580]. Избыток железа(Ш) оттитровывают нитратом ртути, используя роданид калия как индикатор.
Гидроксиламин
Обычно гидроксиламин определяют методом окислительно-восстановительного титрования, поскольку он легко переходит из одного состояния окисления в другое в зависимости от условий эксперимента.
Окисление солями железа(Ш). Окисление сульфатом трехвалентного железа происходит в среде 2 N H2SO4 при температуре кипения [559]:
2NH2OH + 4Fe(III) -> N2O + 4Fe(II) + H2O + 4H+.
После охлаждения раствора железо(П) оттитровывают перманганатом. Не мешают соли аммония и небольшие количества хлорид-ионов. При обратном титровании избыток железа(Ш) можно оттитровать нитратом ртути, используя роданид калия в качестве индикатора.
Феррицианидом гидроксиламин окисляется до молекулярного азота в буферированном растворе при pH 8—9:
2NH2OH + 2Fe (CN)|" N2 + 2Fe(CN)|* + 2HaO + 2H+.
Образовавшийся ферроцианид титруется сульфатом церия [1247] в кислом растворе с ферроином в качестве индикатора.
Окисление броматом и гипобромитом. Гидроксиламин окисляется этими реагентами до азотной кислоты. Окисление броматом калия в присутствии НС1 протекает по реакции [943]:
nh2oh + Bro; no; 4- Вг- + н,о + н+.
Избыток бромата определяется иодометрически при добавлении раствора иодида калия и титровании стандартным раствором тиосульфата до конечной точки, устанавливаемой с помощью крахмала.
Метод с гипобромитом калия [613] требует более тщательного Соблюдения необходимых условий проведения реакции.
Окисление солями церия(1У) протекает количественно по реакции
2NH2OH + 4Ce(IV) —N2O + 4Се(Ш) + Н2О + 4Н+
62
с четырехокисью осмия как катализатором. Прямое титрование не применяют из-за малой скорости окисления. Гидроксиламин кипятят с раствором церия в течение 3 мин., раствор охлаждают и избыток церия(1У) определяют титрованием раствором мышьяковистой кислоты с ферроином в качестве индикатора конечной точки.
Титрование монохлориодом. Гидроксил амин окисляется до нитрита этим реагентом по реакции [513]
NH2OH + 4JC1 + Н2О No; + 2,Т2 + 4СГ + 5Н+.
Избыток монохлориода определяют обратным титрованием раствором сульфата гидразина.
Восстановление хлоридом титана(Ш). Гидроксиламин может быть восстановлен количественно до аммиака растворами Ti(III) [934]:
NH2OH 2Ti(III) + 2Н+ NH3 + 2Ti(IV) + H2O.
Избыток хлорида трехвалентного титана оттитровывают стандартным раствором железа(Ш), используя роданид как индикатор.
Азотистоводородная кислота
Окисление солями церия(1У). Азотистоводородная кислота окисляется количественно церием(1У) до азота по реакции
2HN3 + 2Ce(lV) -» 3N2 + 2Ce(III) + 2H+.
В качестве окислителя используют нитрат аммония-церия(1У) [467], избыток которого определяют титрованием стандартным раствором сульфата железа(П), используя ферроин в качестве индикатора.
Обработка перманганатом. К анализируемому раствору, 1 М по H2SO4, 1 М по Н3РО4 и 1 М по MnSO4, добавляют избыток перманганата; смесь встряхивают в течение 5 мин. В этих условиях образуются промежуточные соединения марганца, которые в кислой среде количественно окисляют азиды. После окончания выделения газа избыток непрореагировавшего перманганата оттитровывают иодометрически.
В общем виде принципиальная схема ре акции может быть представлена следующим уравнением:
10HN3 + 2MnO; + 6Н+ — 15N2 + 2Мп2+ + 8Н2О.
Титрование нитритом в кислых растворах проходит в соответствии с уравнением
HN3 + HNO2 -» N2 + N2O + H2O.
Прямое титрование проводят либо с внешним индикатором (крахмал—иодид), либо с внутренним индикатором — ионами желе-за(Ш), которые с азид-ионами дают красную окраску [1203].
63
Можно провести обратное титрование, добавив избыток нитрита и оттитровывая непрореагировавшее его количество стандартным раствором азотистоводородной кислоты, применяя Fe2(SO4)3 в качестве индикатора. При таком способе КТТ обнаруживается более четко.
Двуокись азота. Азотная кислота
Окисление перманганатом. После поглощения двуокиси азота серной кислотой образующаяся нитрозилсерная кислота может быть оттитрована перманганатом калия [613]. Для получения более надежных результатов рекомендуется обратное титрование избытка перманганата, например оксалатом натрия. Разбавленные растворы подвергаются обратному титрованию по йодометрической методике. Этот классический метод применяется редко.
Цериметрия — достаточно быстрый метод для определения растворенных окислов азота(1У), в том числе и в концентрированной азотной кислоте.
Газообразная двуокись азота поглощается избытком нитрата аммония-церия [1323] и реагирует с Ce(IV) по следующему уравнению:
NO2 + Ce(IV) + Н2О -» NO; + Ce(III) + 2Н+.
Избыток церия(1У) оттитровывается обратно.
Восстановление сульфатом железа(П). Это наиболее широко применяемый метод определения азотной кислоты [554]. Реакция протекает в сильнокислой среде
HNOS + 2Fe(II) + 2Н+ HNO2 + 2Fe(I II) + H2O.
Для стабилизации в раствор добавляют некоторое количество серной кислоты, и, когда нитрат полностью восстановится до нитрита, нитрозилсерная кислота реагирует с избытком титранта — сульфата железа(П) с образованием комплекса, имеющего розовато-красную окраску.
Нитрат-ионы
Методы окислительно-восстановительного титрования при анализе нитратов основаны на восстановлении их с помощью стандартных реагентов до соединений, в которых азот находится в более низких степенях окисления, и на последующем прямом титровании полученных восстановленных соединений либо на обратном окислении избытка восстановителя.
Титрование солями железа(П) превосходный метод определения нитратов. Избыток железа(П) оттитровывают бихроматом с ферроином в качестве индикатора.
NO; + 3Fe(I I) + 4Н+ -» NO + 3Fe(I II) + 2H2O;
6Fe(II) + 2Cr2O*- + 14H+ 6Fe(III) + 2Cr(III) + 7H2O.
64
В конечной точке титрования наблюдается резкое изменение окраски от коричневой до голубовато-зеленой. Реакцию титрования раствором железа(П) лучше всего проводить при температуре кипения раствора (в течение 3 мин.) в присутствии больших количеств серной кислоты (до 65 вес. %). Следовые количества молибдена оказывают сильное катализирующее влияние на скорость реакции [947]. В сосуде для титрования должна поддерживаться инертная атмосфера (например, СО2).
Этот метод с успехом использовался для определения как больших [576], так и ультрамалых (0,002—0,1 мг) количеств нитрата [9951.
В работе [996] описан новый улучшенный вариант метода окси-диметрического определения нитратов с помощью железа(П), по которому анализируемый раствор вносят в предварительно нагретую до кипения реакционную смесь, содержащую известное количество FeSO4, H2SO4 и немного НС1, и оттитровывают избыток Fe(II) раствором КМпО4 или К2Сг2О7.
Предложено 3 варианта метода определения микроколичеств нитратов органических оснований (НО), неорганических нитратов (НН) и нитроаминов (НА), основанного на восстановлении желе-зом(П), титаном(111) и их смесью [471].
Вариант 1. 2—5 мг вещества растворяют в ацетоне, прибавляют 5 мл концентрированной НС1 и в течение 5 мин. вытесняют из колбы воздух струей СО2. Прибавляют 10 мл 0,04 М раствора Fe(NH4)2(SO4)2 и вносят 2 капли 10%-ного раствора (NH4)2MoO4. Кипятят 5 мин., охлаждают, прибавляют 30 мл 1 М НС1, 3 мл 75%-ного раствора Н3РО4 и 3 капли индикатора (1%-ного раствора дифенилбензидина в H2SO4) и титруют избыток Fe(II) 0,05 N раствором К2Сг2О7 до появления неисчезающей фиолетовой окраски. На 1 моль НН и НО расходуется 3 моля Fe(II).
Вариант 2. 2—5 мг вещества растворяют в ацетоне, прибавляют 5 мл концентрированной НС1 и после вытеснения воздуха струей СО2 — 10 мл 0,04 М раствора Ti2(SO4)3. Кипятят 2 мин., охлаждают и титруют избыток Ti(III) 0,04 М Fe(NH4)2(SO4)2. Незадолго до конца титрования прибавляют 0,5 мл 10%-ного раствора NH4SCN. Одновременно проводят холостой опыт. На 1 моль НН или НО расходуется 3 моля Ti(III).
Вариант 3. 3—5 мг НН, НО, НА растворяют в 2—4 мл смеси салициловой кислоты, 450 мл концентрированной H2SO4 и 50 мл ледяной уксусной кислоты, прибавляют 5 мг безводного А1С13 и осторожно подогревают до полного растворения. Затем прибавляют 10 мл 75%-ного раствора цитрата натрия и 10 мл 0,04 М раствора Ti2(SO4)3, кипятят 3 мин., охлаждают, прибавляют 10 мл 6 М H2SO4 и титруют избыток Ti(III), как в варианте 2. На 1 моль НН, НО или НА расходуется 6 молей Ti(III).
Методы проверены при анализе чистых препаратов KNO3, ряда органических соединений. Органические нитро-, нитрозо-, азо- и гидразосоедипения не мешают при определении нитратов по варианту 1, но мешают при анализе по вариантам 2 и 3.
3 В. Ф. Волынец, М. П. Волынец
65
Восстановление оксалатом. Щавелевая кислота редко используется для определения нитрата. Однако в присутствии солей марганца в кислой среде она может восстановить нитрат до окиси либо закиси азота.
При концентрации H2SO4 )> 1 : 5 протекает реакция 1943]
2NO~ + СаО®“ + 8Н+ 2NO + 2СО2 + 4Н2О.
Избыток щавелевой кислоты затем титруется перманганатом, а содержание нитрата рассчитывается по приведенной выше реакции.
Восстановление солями олова(П). Нитрат количественно восстанавливается хлоридом олова до гидроксиламина в инертной атмосфере (углекислота) в соответствии с реакцией
NO; + 3Sn(I I) + 7Н+ NH2OH + 3Su(IV) + 2H2O
при температуре кипения в присутствии избытка 0,2 N SnCl2 в среде HG1. Избыток хлорида олова определяют обратным титрованием хлоридом железа(Ш), используя либо потенциометрическое, либо фотометрическое (с роданидом) определение КТТ [1092].
Восстановление солями титана(Ш). Действием раствора солей титана(Ш) в щелочной среде нитрат восстанавливается при нагревании в инертной атмосфере до аммония:
NO; + 8Ti(II I) + 6ОН- NH+ + 8Ti(IV) + Н2О-
Избыток стандартного раствора титана оттитровывают солями же-леза(Ш), используя роданид как внутренний индикатор [934].
В нейтральной среде нитрат восстанавливается до окиси азота [1394]:
no; + ЗТ1(Ш) + 4Н2О -> NO 4- ЗТЮ2 + 8Н+.
Проводится прямое титрование при температуре кипения с ализарином в качестве внутреннего индикатора, причем в процессе титрования индикатор адсорбируется выпадающей двуокисью титана, меняя цвет от красного до серовато-зеленого в КТТ.
Восстановление солями хрома(11) проводят как в кислой, так и в щелочной средах [1004]. Рекомендуемый метод заключается во взаимодействии избытка ионов хрома(П) с нитратом, добавлении избытка раствора Fe(III) и оттитровывании избытка последнего стандартным раствором хрома(П) с потенциометрической фиксацией КТТ. Хорошие результаты получаются при добавлении небольших количеств сульфата титана(ГУ) для ускорения реакции взаимодействия хрома(П) с нитратом.
Восстановление сульфатом ванадия(П). Определение нитрата основано на реакции
no; + 8 V( II) + ЮН+ NH+ + 8V(II I) + ЗН2О, которая протекает в кислой среде. Избыток ионов ванадия(П) ти-66
труют растворами перманганата или Fe (III), используя потенциометрическое обнаружение КТТ [485, 673].
Восстановление металлами. Одним из наиболее известных методов определения нитратов является восстановление сплавом Де-варда, который состоит из 45%А1, 50%Cu, 5%Zn [655]. Восстанавливающим агентом служит также алюминий, который переводит нитрат в аммиак:
3NO; + 8А1 + 5ОН- + 2Н2О 3NH3 + 8АЮ~
Аммиак отгоняют в щелочной среде, улавливают в приемник с кислотой и титруют методом Кьельдаля.
Для более эффективного восстановления применяют сплав меди с магнием и проводят отгонку из раствора, насыщенного гидроокисью магния [454, 455]. Метод не специфичен для нитратов. Нитриты восстанавливаются также, и требуется введение поправки на их содержание. Метод успешно применяется для микроопределений [922].
При низких концентрациях нитрата метод, использующий сплав Деварда, оказался неэффективным [1384]. Нельзя получить количественных результатов, применяя в качестве восстановителя цинковую амальгаму, так как при этом нитрат восстанавливается до гидроксиламина и других соединений.
Восстановление сульфаминовой кислотой. В сильнокислых растворах нитрат можно определить титриметрически по реакции с сульфаминовой кислотой, избыток которой оттитровывают стандартным раствором нитрита натрия, используя внешний индикатор (крахмал—иодид) для обнаружения КТТ. От 5 до 100 мг нитрата калия определяют этим методом с отклонением от среднего + 0,6 отн.% и средней ошибкой <Т),1%. Этот метод применен для одновременного определения нитратов и нитритов [768].
Восстановление ураном (IV). В работе [1376] нитрат был определен титриметрически восстановлением избытком сульфата ура-на(1У) при кипячении. Избыток последнего оттитровывали раствором железа(Ш). Практического значения этот метод не приобрел.
Нитрит-ионы
При определении содержания нитритов методы окислительно-восстановительного титрования могут быть основаны как на окислении нитрита сильноокисляющим агентом, так и на восстановлении его до ряда продуктов в зависимости от природы восстановителя. Правда, восстановление нитрита в аналитической практике используется редко.
Окисление гипохлоритом кальция [596] применяется для косвенного определения нитритов. Прямое титрование растворов нитритов затруднено из-за малой скорости реакции. Рекомендуемый метод заключается в окислении NO2-hohob раствором гипохлори-
3*
67
та, прибавлении избытка раствора Na3AsO3 и титровании последнего раствором NaOCI в присутствии бромтимолового синего как индикатора.
К избытку 0,01—0,05 JV раствора Са(ОС1)3 прибавляют анализируемый раствор, содержащий 1,5—30 мг NaNO2, 2 мл насыщенного раствора NaHCO3 и через 15 мин. 1 мл раствора КВт (50 г в 100 мл), определенное количество 0,01—0,05 W раствора Na3AsO3, 5 мл раствора буры (8 г в 100 мл) и титруют 0,01—0,05 N раствором NaOCI до зеленовато-желтого окрашивания. Ошибка определения NO^^C 0,2/6.
Окисление броматом калия проводят по реакции
3NO~ + Вго; - 3NO” 4- Вг-
в среде HCI без добавления бромида. Избыток раствора бромата обрабатывают иодидом калия и титруют раствором тиосульфата до КТТ по крахмалу [943, стр. 526].
Можно обработать раствор, содержащий нитрит, бромом (в избытке) в присутствии пиридина, который ускоряет реакцию. После выдерживания раствора следует добавить иодид калия и оттитровать образовавшийся иод тиосульфатом.
Восстановление иодидом в случае йодометрического определения нитрита требует тщательного соблюдения условий эксперимента для получения надежных результатов. Реакция азотистой кислоты с иодидом в кислом растворе может быть описана уравнением
2HNO2 -j- 2Г + 2Н+ -> 2NO + J2 + 2Н2О.
Должны быть приняты меры для предотвращения побочных реакций, в частности, необходимо обеспечить отсутствие кислорода в реакционном сосуде. Наиболее надежная методика заключается в следующем [439].
К смеси избытка бикарбоната натрия, иодида калия и амилового спирта добавляют известное количество анализируемого раствора, содержащего нитрит-ионы. Полученный раствор перемешивают вращением в колбе, добавляют небольшое количество уксусной кислоты. После начала выделения СО2 почти прекращают вращать колбу и осторожно добавляют избыток серной кислоты (1 : 1). Сосуд сразу же плотно закрывают и начинают энергично вращать для перемешивания раствора до прекращения выделения СО2. Затем титруют стандартным раствором тиосульфата, используя крахмал в качестве внутреннего индикатора.
Окисление перманганатом. Нитрит количественно окисляется перманганатом только в кислом растворе:
5NO- + 2МнО; + 6Н+ 5NO; + 2Мп2+ + ЗН2О.
Прямое титрование приводит к неудовлетворительным результатам. Поэтому рекомендуется обработка нейтрального или щелоч-
68
кого раствора нитрита избытком перманганата, подкисление полученного раствора и последующее йодометрическое титрование [946].
Другой косвенный метод определения нитрита титрованием перманганатом включает нагревание раствора (1—5 М по HNO3) до 40° С с избытком КМпО4 и обратное титрование не вошедшего в реакцию КМпО4 раствором KNO2. Используемая перегнанная HNO3 не должна содержать окислов азота [1245].
Титрование раствором перекиси водорода. Показано, что 0,1 N раствор Н2О2 в 17V H2SO4, содержащий Ti(SO4)2 (для стабилизации), не меняет своего титра в течение 12 месяцев и пригоден для прямого и косвенного оксидиметрического определения нитрита [593].
1—5 мл 0,1 N раствора Н2О2 разбавляют равным объемомводы и титруют при энергичном перемешивании анализируемым раствором, содержащим 2,5% С2Н5ОН, до обесцвечивания реакционной смеси.
При косвенном определении вводят в разбавленный раствор перекиси водорода 2 капли 1 раствора молибдата аммония (20 г (NH4)2MoO4-4H2O в 100 мл} в качестве катализатора, через 2—3 мин. после добавления анализируемого раствора титруют избыток Н2О2 иодометрически. Для этого прибавляют 2 г KJ и 4 JV H2SO4 до ее концентрации, равной 1 IV, выдерживают 15 мин. в темноте и титруют 0,1 раствором Na2S2O3 в присутствии крахмала.
Полученные этим методом результаты хорошо согласуются с данными йодометрического определения NO2.
Титрование сульфатом церия(1У) [1402]. Количественные результаты могут быть получены обработкой раствора нитритов 0,17V раствором сульфата церия(ТУ) в кислой среде и последующим обратным титрованием избытка последнего стандартным раствором оксалата натрия.
Титрование тетраацетатом свинца( IV). Нитрит можно количественно оттитровать раствором тетраацетата свинца(1У), который получают растворением РЬ3О4 (красный свинец) в ледяной уксусной кислоте при умеренной температуре. Прямое титрование проводится в 1 М растворе NaCl по реакции
NO” + РЬ‘.+ + Н2О -» no; 4- РЬ2+ + 2Н+.
Титрование феррицианидом проводится в нейтральном растворе:
NO; + 2Fe (CN)®’ + Н2О _ NO; Д- 2Fe (CN)f Д-2Н+.
Полученный по реакции ферроцианид титруется стандартным раствором сульфата церия.
Титрование раствором хлорамина Т. Прямое титрование не дает удовлетворительных результатов. Однако превосходные результаты получаются’в’случае'использования избытка хлорамина Т в растворе бикарбоната или уксусной кислоты и обратного титрования тиосульфатом после добавления раствора иодида калия
69
[943, стр. 641; 1289]. Использование диметилформамида в качестве растворителя улучшает метод обратного титрования [923].
Тиомеркуриметрическое определение — чувствительный метод определения нитритов [1423]. Установлено, что реакция между NO2 и HSCH2COOH протекает довольно быстро и количественно с образованием ONSCH2COOH — достаточно прочного соединения в кислых, а также в не слишком щелочных растворах. Это дает возможность определять NO; титрованием избытка HSCH2COOH, не вошедшего в реакцию, раствором о-оксимеркури-бензоата.
К пробе, содержащей 0,2—50 мг NaNO2, добавляют 1 мл 1 N NaOH, раствор HSCH2COOH (в 50%-ном избытке), смесь разбавляют до ~40 мл и прибавляют 5 мл 1 W H2SO4. Через ^20 сек. (при комнатной температуре реакция между HSCH2COOH и NO2 за это время заканчивается) добавляют 10 мл 1 N NH4OH и оттитровывают избыток HSCH2COOH 0,005—0,05 # раствором о-оксимеркурибензоата в присутствии тиофлуоресцеина в качестве индикатора.
При определении 0,45—28 мг NaNO2 полученные описанным методом результаты хорошо совпадают с данными, найденными перманганатометрическим методом. Определению NO2 этим методом не мешает присутствие многих органических веществ, реагирующих с КМпО4.
Восстановление солями титана(Ш) [934]. При использовании раствора титана(Ш) в качестве восстановителя необходимо соблюдать особенно тщательно условия его хранения, и тогда можно быстро получить надежные результаты при титровании нитрит-ионов:
NO; + 6Ti(IH) + 4Н2О — NH3 + 6TiO2+ + 5Н+.
Нитрит восстанавливается количественно, а избыток хлорида титана определяется обратным титрованием стандартным раствором Fe(III) с роданидом в качестве внутреннего индикатора.
Восстановление гидразином. Метод заключается в добавлении раствора нитрита к избытку сульфата гидразина в разбавленной H2SO4 и последующем обратном титровании избытка гидразина стандартным раствором иода [934, стр. 647]. Лучшие результаты были получены при потенциометрическом титровании гидразина раствором нитрита натрия [934, стр. 187].
Восстановление гидроксиламином. Избыток стандартного раствора гидрохлорида гидроксиламина смешивают с анализируемым раствором. При этом происходит восстановление нитрита до окиси азота и азота [873]:
2NO; + 2NH2OH; 2NO + N2 + 4Н2О.
Избыток гидроксиламина определяют обратным титрованием стандартным раствором гидроокиси натрия со смешанным индикатором — метиленовым голубым и фениловым красным (в спирте).
70
Роданид-, цианид-ионь/
ОписаИ Метод прямого оксидиметрического определения роданида аммония с использованием в качестве окислителя бихромата калия [1200]. Титрование проводится в среде 7—18 N H2SO4 без добавления катализатора с потенциометрической индикацией точки эквивалентности.
К определенному объему 0,025 М титрованного раствора К2Сг2О7 прибавляют необходимое количество (10—20 мл) концентрированной H2SO4, разбавляют до 40 мл, охлаждают до комнатной температуры и титруют анализируемым раствором при перемешивании электромагнитной мешалкой, измеряя э.д.с. с помощью потенциометра с прямым отсчетом через каждые 1—2 мин. после добавления очередной порции раствора.
При определении 1—40 мг NH4SCN средняя ошибка составляет ±0,5%. Не мешают определению до 200 мг С1~ и CN- (последний немного замедляет реакцию). Мешают иодиды и бромиды.
Цианиды можно определять титриметрически с помощью стандартного раствора серы в неводной среде [750].
Для этого анализируемый раствор, содержащий цианиды, смешивают с четырехкратным количеством изопропилового спирта, добавляют 10 мл ацетона, вводят 3—4 капли 0,2 %-ного раствора титанового желтого, нагревают до 50—60° С, пропускают через раствор N2 и титруют 0,1 М раствором серы в смеси бензола и ацетона (4 : 1) до изменения розовой окраски в желтую.
КТТ можно установить потенциометрически, используя стеклянный и нас.к.э. и проводя титрование в термостатируемой ячейке. Ошибка определения ~0,1%.
Разработаны условия количественного определения SCN_-ho-нов титрованием гипохлоритом натрия [753]. Реакция протекает по схеме
4KSCN + 6NaC102 + 4Н2О — 6NaCl ± 4H2SO4 4- 4KCN.
Ошибка определения составляет ~ 0,12%.
КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Эти методы основаны на образовании комплексных соединений с азотсодержащими ионами. Большое место в этой группе методов занимают методы комплексонометрического титрования, связанные с использованием в качестве титрантов растворов ЭДТА и ее солей.
Аммоний
Способность аммиака давать комплексы с медью в водных растворах была положена в основу гетерогенного титрования при определении аммиака [527]. В этом методе аммиак в форме карбо
71
ната аммония используется в качестве титранта при титровании свежеосажденного карбоната меди в соответствии с реакцией
СиСО3 + 2NH+4 —»Cu(NH3)2+ + Н2СО3.
К этой группе методов относится комплексонометрическое титрование раствора, содержащего ион аммония, комплексонатом ртути(П) [1239]. Эта реакция может быть описана следующим уравнением:
HY2- + NH4 -» HYNH2- -|- Н+,
где Y — анион соответствующего комплексона (ЭДТА, гидрокси-этилэтилендиаминтетрауксусной или циклогександиаминтетраук-сусной кислоты).
В случае использования этилендиаминтетраацетата ртути(П) (pH 6,8) для определения ионов аммония и аммиака количество комплексоната должно быть выбрано таким образом, чтобы оно было больше эквивалентного суммарному содержанию иона аммония и аммиака в анализируемом образце. К раствору комплексоната добавляют образец, и получившийся раствор титруют щелочью до pH 6,8.
Роданид-ионы
В работе [244] предложен титриметрический метод определения роданида с помощью комплексоната ртути(П) с применением в качестве индикатора метилтимолового синего. Ионы SCN- реагируют с комплексом Hg(II) с метилтимоловым синим (состава [HgMtb]4-) с образованием смешанного комплекса синего цвета состава [HgMtb(SCN)]5-, а с комплексонатом Hg(II) ([HgY]2-) — с образованием бесцветного комплекса состава (HgY(SCN)]3-. Первый комплекс в кислой среде в присутствии избытка SCN-неустойчив. При титровании раствора, содержащего SCN_-hohh и небольшие количества индикатора (HgMtb)4-, стандартным раствором HgY2- сначала образуются Hg(SCN)2 и [H2Mtb]4-, которые при дальнейшем добавлении титранта переходят в [HgMtb](SCN)5- по реакции
Hg(SCN)2 + H2Mtb«" + HgY2- -> [HgMtb](SCN)5- + [HgY](SCN)3- + 2H+, окрашивая раствор в точке эквивалентности в синий цвет при небольшом избытке HgY2-.
На этой основе разработан титриметрический метод определения 0,5—14 мг SCN~ со стандартным отклонением 0,012—0,066. Предельно допустимые количества ионов (в мкг)-. Со3+ 3, Zn2+ 5, Cd2+6, Ni2+ и82“ 9, Cu2+ 10, Mn2+ 15, CN“ 28, Bi3+ 100, Ca2+ 120, Ag+ 150, Br~ 180, J- 220, Fe3+ 250.
72
Цианяд-ионы
Предложены методы комплексонометрического определения CN“-hohob с помощью ряда комплексонов [121,147,245]. Косвенный метод титрования раствором нитрилотриуксусной кислоты (НТА) основан на взаимодействии CN- с Ni2+ (с образованием комплекса Ni(CN)4-) и титровании избытка никеля раствором НТА при pH 8—9 с применением мурексида в качестве индикатора [147].
Комплексонометрическое определение цианида с помощью этилендиаминтетраацетата ртути(П) проводится с применением пиридилазонафтола [121] или крезолфталеинкомплексоната ртути [245] в качестве индикаторов. В первом случае [121] CN--noHbi взаимодействуют при pH 4—8 с Na2HgY -4Н2О (Y4-— анион ЭДТА) с образованием устойчивого комплекса состава [HgY(CN)]3-, который при pH 6,0—7,7 количественно взаимодействует с CN-с выделением эквивалентного количества ЭДТА по реакции
[HgY(CN)]3- + CN- - Hg(CN)2 + Y4-.
Титрование выделившихся Y4--hohob раствором CuSO4 позволяет косвенно определить содержание CN-.
К анализируемому раствору с pH 7,1 прибавляют 10 мл 0,01 М раствора [HgY(CN)]3-, разбавляют до 50 мл и выдерживают 2—3 мин.; затем вводят 20 мл СН3ОН, несколько капель раствора индикатора и титруют 0,005 М раствором CuSO4.
Метод позволяет определить 0,025—2 мг CN- с ошибкой <4%. Определению мешают Си, Ag, Ni, Со, Hg, VOg, S2-.
Второй метод определения [245] основан на том, что CN^-ионы реагируют с HgY2- и с комплексом Hg(II) + крезолфталеинком-плексон (HgZ4-) с образованием бесцветных комплексов состава [HgY(CN)]3- и [HgZ(CN)]5- соответственно (Y и Z — анионы), причем первый комплекс устойчивее второго. Индикатор HgZ4-окрашен в красно-фиолетовый цвет.
В анализируемом растворе, содержащем 0,2—6 мг CN-, устанавливают pH И с помощью фосфатного буфера (0,1 N NaOH + 0,1 N Na2HPO4), добавляют 2 мл 5-10-4 М раствора Hg(NO3)2 и 6 капель 0,2%-ного метанольного раствора крезолфталеинкомплексона, разбавляют до 50 мл и титруют 0,02 М раствором HgY2- до появления красно-фиолетовой окраски (HgY2- вытесняет HgZ4- из его комплекса с CN-). Содержание CN- находят по калибровочному графику.
Определению CN- мешают J-, S2- и некоторые ионы металлов, но их влияние (за исключением серы) можно устранить отгонкой CN- из подкисленного HNO3 анализируемого раствора.
73
МЕТОДЫ ТИТРОВАНИЯ, ОСНОВАННЫЕ НА РЕАКЦИЯХ ОСАЖДЕНИЯ
Титриметрические методы этой группы основаны на реакциях, сопровождающихся образованием малорастворимых соединений. Наиболее важное значение здесь имеют реакции образования солей серебра (аргентометрия), которые положены в основу титри-метрических методов определения роданид- и цианид-ионов.
Роданид- и цианид-ионы
Серебро образует с SCN~ и СМ“-ионами малорастворимые соли. При титровании растворов цианидов солью серебра вначале происходит комплексообразование по реакции
2CN- + Ag+ -, [Ag(CN)2]-,
а при дальнейшем прибавлении раствора AgNO3 образуется осадок цианида серебра:
[Ag(CN)2]- + Ag+ Ag[Ag(CN).2] |.
Реакции осаждения роданида и цианида серебра протекают количественно, растворимость этих соединений очень мала (например, для AgSCN ПР ~ 1-10-12). Это дает возможность титриметрически определять роданиды и цианиды, используя в качестве титранта стандартные растворы серебра в присутствии различных индикаторов, фиксирующих КТТ. Наибольшее распространение нашли адсорбционные индикаторы, например, некоторые красители, сорбирующиеся образующимися в процессе титрования осадками и изменяющие при этом свою окраску. К таким адсорбционным индикаторам в случае аргентометрического определения роданида относятся металлохромные индикаторы — пирокатехиновый фиолетовый (I), пирогалловый красный (II), фталеинкомплексон (III), ксиленоловый оранжевый (IV), кальциен (V), мурексид (VI). Они используются в виде 0,2%-ных водных растворов (2 капли раствора индикатора на 10 мл титруемого раствора). Минимально определяемые при этом концентрации (в г-экв!л SCNr) составляют: 0,002 (для II, III), 0,005 (I, IV), 0,01 (V, VI) [1341].
Одновременное определение роданида в смеси с тиомочевиной, основанное на реакции с нитратом серебра, описано в [519].
Установлена пригодность бромхлорфенолового синего, бромфенолового красного, о- и л-крезолового красного как адсорбционных индикаторов для аргентометрического определения роданидов; эти индикаторы могут быть применены в случае титрования SCN_-hohob при pH 3,0; 7,5; 9,0 соответственно [449]. Хорошие результаты получаются при использовании основного азокрасителя — n-аминоазобензола в случае аргентометрического титрования роданидных растворов [773]. Оптимальное значение pH 3—4, ошибка определения ± 0,5%.
74
Роданид Определяют количественно в присутствии Цианида с применением хромата калия в качестве индикатора [1354].
В цикле работ венгерского исследователя Л. Легради изучена возможность применения и механизм действия ряда адсорбционных индикаторов при аргентометрических титрованиях роданидных и цианидных растворов [992, 992а]. В качестве универсального индикатора им предложен 4-(4'-нитрофенилазо)-1-нафтол, получаемый азосочетанием диазотированного n-нитроанилина с а-нафтолом. Предложенный этим исследователем индикатор особенно эффективен, так как способен к ионизации оксигруппы в условиях выполнения определения с резким изменением окраски. При аргентометрическом определении CN~-hohob его окраска изменяется в КТТ из красной в фиолетовую, а при анализе роданидных растворов — из желтой в розовую. Ошибка титрования 0,12V растворов указанных анионов 0,01—0,1 N раствором AgNO3 с этим индикатором <Т %.
В работе [6351 макро- и микроколичества цианид-ионов успешно определяют аргентометрически с использованием трехкомпонентных комплексных соединений (в частности, системы 1,10-фе-нантролин— бромпирогалловый красный) в качестве индикаторов при визуальном и фотометрическом определениях CN~ в случае титрования раствором AgNO3. Индикаторное действие в данном случае основано на том, что тройной комплекс [Ag(phen)B] (где phen — молекула 1,10-фенантролина, В — анион бромпиро-галлового красного) образуется с переходом окраски только в отсутствие CN~-hohob.
При визуальном титровании к 25 мл анализируемого раствора (10-1 — 10-‘л1оль/л CN") прибавляют 1 мл 0,2%-ного водного раствора 1,10-фенантролина, 5 мл 20%-ного раствора CH3COONH4,1 мл 0,1 М раствора комплексона!II (маскирующий агент для многих катионов), 1 мл раствора бромпирогал-лового красного (3 мг красителя -ф 0,5 г CH3COONH4 в 50 мл воды) и титруют стандартным раствором AgNO3 (10-1—10-1 М) до перехода красной окраски в синюю.
При фотометрическом титровании к 25 мл анализируемого раствора (10-1—10-5 молъ!л CN~) в фотометрической кювете емкостью 50 мл прибавляют 1 мл 0,2%-ного раствора 1,10-фенантролина, 5 мл 20%-ного раствора CH3COONH4, 1 мл 0,1 М раствора комплексона!!!, 1 мл раствора бромпиро-галлового красного и титруют 10~3—10-1 М раствором AgNO3, измеряя оптическую плотность при 600 нм.
С методом осаждения тесно связан титриметрический метод, основанный на образовании малодиссоциирующих солей, например Hg(SCN)2 (меркуриметрия). Роданид- и цианид-ионы могут быть определены меркуриметрическим титрованием с использованием 5-(8,1-сульфонафтилазо)-8-оксихинолина в качестве индикатора [297]. Индикаторный переход окраски мгновенен и достаточно контрастен (из оранжевой в фиолетовую). Используют насы
75
щенный водно-этанольный раствор сульфонафтазоксина и титруют 0,01 М раствором Hg(NO3)2 в среде HNO3 (pH 1—2). Определению не мешают нитраты, сульфаты, фосфаты, а также Na+, К+, Са2+, Mg2+, А13+ и умеренные количества Fe3+.
В качестве меркуриметрического индикатора для определения цианида предложено применять 2-(2-тиазолилазо)-4-метоксифенол [974].
Нитрат-ионы
Предложен способ количественного титриметрического определения нитратов путем осаждения в виде Pb(NO3)2 в среде безводной уксусной кислоты с амперометрической или визуальной оценкой КТТ [159].
Вариант 1. 0,02—0,07 г анализируемого нитрата (калия, натрия, кадмия, аммония и др.) растворяют в 10 мл 0,1 М раствора CH3COOLi в безводной СН3СООН (при необходимости нагревают) и титруют амперометрически 0,2 N раствором (СН3СОО)2РЬ в безводной СН3СООН при —1,0 в с капельным ртутным электродом (относительно донной ртути).
Вариант 2. К определенному количеству (1—10 л1л) 0,1 N раствора (СН3СОО)2РЬ в безводной СН3СООН добавляют 10—15 капель свежеприготовленного насыщенного раствора К2СгО4 в безводной СН3СООН и титруют анализируемым раствором (0,15 Лг) нитрата в присутствии 15—20 капель 2%-ного раствора дифениламина в СН3СООН (добавляемого перед концом титрования) до появления темно-зеленой окраски.
МЕТОДЫ, ОСНОВАННЫЕ НА ЭЛЕКТРОХИМИЧЕСКОЙ РЕГИСТРАЦИИ КОНЕЧНОЙ ТОЧКИ ТИТРОВАНИЯ
В основе методов этой группы лежат также химические реакции нейтрализации, окисления—восстановления, осаждения и комплексообразования, но фиксирование КТТ проводится одним из электрохимических методов: потенциометрическим, амперометрическим, кондуктометрическим (в том числе с помощью высокочастотного титрования), кулонометрическим и т. д.
Потенциометрическое титрование
Методы потенциометрического титрования несколько более трудоемки, чем прямое потенциометрическое измерение, но более точны и дают более воспроизводимые результаты.
При анализе азотсодержащих материалов содержание газообразного N2 предлагается определять путем переведения азота в аммиак и автоматического оттитровывания последнего в электрохимической ячейке [1040].
Нитрат-ионы могут быть определены методом потенциометрического осадительного титрования с анионитовыми мембранными
76
электродами (АВ-17, АН-2Ф) в NO3-форме. В качестве титранта используют раствор нитрона в 5%-ной уксусной кислоте. По перегибу на кривой титрования удается довольно точно определить КТТ [196].
В работе [628] описана простая ячейка для титрования нитрат-ионов с жидким ионообменным селективным электродом.
Методом дифференциального титрования в неводных средах (в смеси метанола с ацетоном) определяют нитраты различных элементов. Водный насыщенный раствор KG1 в каломельном элементе заменяют насыщенным раствором KG1 в метаноле. В качестве титранта используют гидрат окиси тетраэтиламмония. Ошибка определения составляет около 1% [160а].
В смеси (СП3СО)2О—СН3СООН—-СНС13 нитраты элементов различных групп периодической системы элементов являются слабыми основаниями одинаковой силы и могут быть оттитрованы потенциометрически ацетоновым раствором НС1О4 [435].
В качестве индикаторного электрода при меркуриметрическом определении роданидов в сыворотке крови был использован ртутно-графитовый стержень [92].
Предложен метод определения свободного и связанного цианида в пробе анализируемого электролита потенциометрическим титрованием раствором соли Hg(II) с индикаторным Ag-электро-дом и Hg—HgO-электродом сравнения [369]. Потенциометрическое титрование цианида с использованием серебряного индикаторного ионоселективного электрода описано в [620].
При аргентометрическом титровании цианида в качестве индикаторных используют стеклянные электроды, обладающие катионной функцией. Показано, что природа катиона практически не влияет на точность получаемых результатов. В случае титрования С№-ионов в присутствии окислителей (Н2О2, КМпО4) электрод с катионной функцией дает более точные результаты, чем Ag-элек-трод. Присутствие органических растворителей практически не влияет на измерения [740]. Применение стеклянного электрода в качестве электрода сравнения при аргентометрическом титровании GN- с потенциометрической индикацией КТТ проще и удобнее, чем обычное применение насыщенного каломельного электро -да [623].
Были проведены титрования CN электролитически генерированным Ag+ в эвтектическом расплаве LiNO3 -|- NaNO3 при 150° С с применением потенциометрической и амперометрической индикации КТТ. Для индикации использовали Ag- и AgHal-электроды (Hal — галогенид-ион). Ошибка определения 1—2%. При титровании CN" происходит окисление этого иона нитратом лития; [Ag(GN)2]_ в растворе устойчив [541].
Описан ультрамикрометод определения азота при анализе органических соединений (навески 30—50 мкг) с потенциометрической индикацией КТТ [504]. Вещество разлагают концентрированной серной кислотой в присутствии ртутного катализатора. К смеси по
77
Сле разложения прибавляют щелочь и избыток раствора гипохлорита для окисления образовавшегося NH3. Избыток гипохлорита титруют иодометрически с использованием специальной аппаратуры.
Предложенный в [488] метод количественного хронопотенцио-метрического титрования гидразина в растворе серной кислоты имеет ограниченное применение вследствие возможного протекания ряда побочных реакций.
Амперометрическое титрование
При амперометрическом титровании КТТ устанавливают по изменению величины предельного диффузионного тока. Метод амперометрического титрования применяется довольно широко для определения нитрит-, роданид-ионов, а также для определения цианидов. Работ по определению нитрат-ионов и аммония этим методом значительно меньше [525, 661а, 814, 950, 983].
При амперометрическом титровании аммиака гипобромитом измеряют ток при 0,2 в с использованием насыщенного каломельного электрода и вращающегося платинового электрода [950]. Более стабильные результаты получаются при использовании гипохлорита в качестве титранта [983], добавляемого к аммоний-содержащему раствору вместе с бромидом калия. При этом в растворе образуется гипобромит, который и окисляет аммоний.
Гидроксиламин определяется амперометрическим титрованием стандартным раствором титана в присутствии щавелевой кислоты при 50° С с капельным ртутным электродом [525].
Для амперометрического определения некоторых неорганических азотсодержащих соединений (гидразин, гидроксиламин, азотистоводородная кислота, нитрит) можно применить метод титрования при помощи Ce(IV) без наложения э.д.с. с использованием платинового и насыщенного каломельного электрода [671]. Метод пригоден для прямого титрования N3, NH2OH и обратного титрования NO2, N2H4.
Для определения N2H4 и НН2ОНвсмеси пхсумму определяют обратным титрованием после выдерживания раствора с избытком Ce(IV) в течение 15 мин. Титрование проводят раствором С2С)Д на фоне 5 N НСЮ4.
К другой порции раствора прибавляют 25 мл боратного буферного раствора (pH 8—9), 20 мл 0,1 N K3Fe.(CN)e, через 30 мин. прибавляют 5 'мл раствора Cd, 10 мл 60%-ной НС1О4, 10 мл 0,1 N Се(С1О4)4 и через 2—3 мин. избыток церия титруют раствором С2О4“. Содержание N2H4 и NH2OH находят из количеств миллиграмм-эквивалентов Се, пошедших на оба титрования.
Определение нитрит-иона основано на измерении тока окисления NO2 при использовании различных окислителей (сульфаминовой кислоты [1316], перманганата калия [1291], хлорамина Т [652] и др.), а также на использовании вращающегося платинового
78
электрода [652, 1316], на применении поляризуемого Pt-микро-электрода и Ag/AgCl-электрода для четкого определения точки эквивалентности [1291]. Пределы определяемых концентраций NO 2-ионов составляют 10-5—10-2 г-ион/л. Ошибка определения в интервале 10-2—10-3 г-ион/л составляет в среднем 1 %; при концентрации NO2 Ю“4—10“5 г-ион/л она возрастает до 21—3%. Ионы NO3 не мешают определению.
Титрованием в среде безводной уксусной кислоты с применением амальгамированных серебряных, а также платиновых и медных электродов определяют нитрат- и роданид-ионы [34]. Определение нитритов в присутствии нитратов методом амперометрического титрования описано в [603].
При определении 10-5—10'1 М роданид-ионов методом иодато-метрического титрования точка эквивалентности может быть зарегистрирована биамперометрическим методом [719]. Ошибка определения при этом составляет около 1%.
Описан метод амперометрического титрования 0,1—0,6 ммоль/л SCN~ раствором AgNO3 по току Ag+ при 0 в (отн. нас.к.э.) на стационарном Pt-электроде в перемешиваемом растворе [483]. Ошибка определения также не превышает 1 %.
Точный метод последовательного амперометрического титрования селепоцианат- и роданид-ионов раствором AgNO3 по току восстановления Ag+ на фоне 0,01A NHjOH ОДА КОН предложен в работе [858]. В качестве индикаторного электрода использован вращающийся Pt-электрод (1000 об/мин), электродом сравнения служит Hg/HgJ2-3neKTpofl. Ионы SCN” титруют при -j-0,247 в.
Определение смеси селеноцианат- и цианид-ионов методом амперометрического титрования раствором AgNO3 описано в [859].
Одной из разновидностей метода биамперометрического титрования является предложенный для определения ряда анионов, в том числе и SCN-, достаточно точный и чувствительный метод титрования с амальгамированным Gu-катодом и Cu-анодом в среде безводной уксусной кислоты [34]. Возникновение тока связано с переходом в раствор ионов Си+ с анода и восстановлением их на катоде.
Для определения роданида калия предложен метод амперометрического титрования растворами окислителей (J2, JC1, КМпОД с помощью вращающегося платинового электрода по току восстановления реагента при потенциале —0,1 в [88]. Титрование иодом и JC1 проводят на фоне 0,3 М раствора NaHCO3, а раствором КМпО4 — на фоне 1А H2SO4. Нижний предел титруемых концентраций KSCN ~ 0,05 мкг/мл. Определению не мешают 100-кратные количества сульфатов, нитратов, фосфатов. Мешает присутствие сильных восстановителей.
При совместном присутствии цианида, роданида и цианата методом амперометрического титрования раствором AgNO3 последовательно определяют смесь CN' SCN~, а затем OCN- [223, 224].
79
Вначале при температуре >10° С на фоне KNO3 или Ba(NO3)2 + 0,02% желатина смесь CN~ + SCN- титруют с использованием вращающегося Pt-микроэлектрода раствором AgNO3 (вследствие малой растворимости осадки AgCN и AgSCN не деполяризуют электрод) до появления тока восстановления AgOCN, который значительно лучше растворим в водной среде. Затем добавляют 25% j СНдОН, охлаждают до <5° С (для понижения растворимости AgOCN) и титруют OCN- раствором AgNO3.
Для определения SCN- в отдельной порции раствора маскируют CN-формальдегидом, избыток НСНО и OCN- разрушают добавлением HNO3 до pH <3 и титруют SCN- раствором AgNO3. Содержание CN- находят по разности.
Ошибка составляет 0,5; 1; 2% при определении 0,001 — 0,01 моль!л SCN~, 0,001—0,01 молъ/л CN~ и 0,05—0,01 моль/л OCN-соответственно.
Показана возможность прямого амперометрического титрования при pH 1—2 0,005—0,02 жо,гь/.г нитрат-ионов раствором дифе-нилталлийсульфата [661а] с использованием капельного Hg-элек-трода; ионы F", SO4“ и РО4“, эквимолярные количества С1О4 не мешают. Ион NO2 в условиях титрования диспропорационирует с образованием NO3 и поэтому должен быть удален восстановлением. Галогенид-ионы следует удалять осаждением. Точность определения ±1 %
Малые концентрации нитрат-иона определяют амперометрическим титрованием перманганатом в среде 0,05 М H2SO4 при -[-1,10 в по току окисления NO2 [1315]. Используют вращающийся Pt-электрод и нас. к.э. Титрование проводят при комнатной температуре ~ 0,25 М раствором КМпО4; анализируемый раствор и раствор КМпО4 предварительно деаэрируют. При концентрации 1 • • IO'3—1 - 1(У4 г-ион/л ошибка определения <Д°/6.
Кулонометрическое титрование
Метод кулонометрического титрования был применен для определения аммонийных солей, гидразина, нитритов, роданидов и цианидов.
Анализируемый раствор аммонийных солей пропускают через катионообменник в Н-форме и кулонометрически титруют выделившуюся кислоту электрогенерированными ионами ОН- с установлением точки эквивалентности по переходу окраски метилового красного или фенолфталеина (в зависимости от аниона) [1109]. При определении 10 мг аммонийных солей ошибка составляет 0,3— 0,5%.
Определение гидразина кулонометрически генерированным бромом может быть использовано в качестве аналитической методики [1132]. Для количественного определения можно использовать либо смешанный раствор, содержащий уксусную кислоту,
80
метанол, воду и ацетат ртути, либо водный раствор, содержащий 3 М НС1 и 0,1 М НВт. КТТ устанавливают амперометрически. От 3 до 5 мг гидразина можно определить с точностью ±1%.
Гидроксиламин также определяют кулонометрически генерированным бромом в кислой среде [1330]. При этом определении генерированный бром окисляет гидроксиламин до нитрат-ионов.
Разработаны условия кулонометрического титрования нитрит-ионов с помощью электрогенерированного брома [1019]. Рабочий электрод — Pt-пластинка с поверхностью ~ 8 см2; противоэлек-трод — Pt-спираль — отделен от исследуемого раствора перегородкой из пористого стекла.
Титрование ведут в растворе, 1,6 М по КС1 (для стабилизации Сп+), 0,1 М по КВг, 2,5>10-2 М по CuSO4; с помощью НС1 устанавливают pH 3,5. В этот раствор добавляют известный объем анализируемой пробы (общий объем раствора в ячейке должен быть —100 мл), при 5 ма генерируют ~ 2-кратный избыток брома. По окончании реакции (через 5—40 мин. в зависимости от концентрации ионов NO”) генерируют Сп+ путем изменения полярности рабочих электродов и титруют оставшийся бром биамперометрически.
Микроколичества нитрит-ионов определяют кулонометрически посредством окисления NO2 избытком электролитически генерированного Мп3+ и обратного кулонометрического титрования последнего электролитически генерированным Fe2+ [571]. Генерирование проводят на платиновом или угольном электроде. Кислород предварительно удаляют из раствора продуванием инертного газа. При определении 0,3—65 мкг-экв NO2 стандартное отклонение равно 0,2-1,6 %.
Кулонометрическое титрование нитрит- и нитрат-ионов (раздельное и при совместном присутствии) генерированным Ti3+ со спектрофотометрической индикацией КТТ при 480 нм выполнено в работе [604].
Кулонометрическое титрование роданида [701] проводят электролитически генерированным серебром с биамперометрической индикацией КТТ при помощи Ag-электродов. Ошибка титрования составляет /—0,5—0,6% при концентрации титруемого раствора роданида ~ 10-47V. Титрование проводят в кислом растворе во избежание осаждения Ag2O. Для ускоренного автоматического кулонометрического титрования может быть использован серийно выпускаемый автотитратор [1220].
Метод кулонометрического титрования электрогенерированным ВгСГ с установлением КТТ при помощи двух поляризуемых индикаторных электродов применен для определения 25—110 мкг SCN- с ошибкой <2% [794].
Недавно была показана возможность кулонометрического бромометрического определения свободных, а также координационно
31
связанных цианид-ионов [1074]. Определение проводят в слабощелочной среде в присутствии боратного буфера. Точку эквивалентности устанавливают потенциометрически с использованием двух поляризуемых платиновых электродов. Титруют в атмосфере инертного газа. При определении 2020; 776 и 94,8 мкг KCN доверительный интервал составляет ±10,1; ±17,5 и ±1,3 мкг KCN при надежности 95%. Метод рекомендуется для определения CN~ в присутствии С1--, Вг~-ионов (до 2,7 -Ю4- и 5,9-104-кратного избытка этих ионов соответственно).
Кондуктометрическое титрование
По изменению электропроводности и диэлектрической проницаемости анализируемых растворов в процессе титрования соответствующими титрантами возможно кондуктометрическое определение некоторых соединений азота. Так, в работе [1246] кондуктометрическим титрованием определяли 5—120 мг нитрат-ионов, осаждая их в виде Ba(NO3)2 действием 0,27V Ва(СН3СОО)2-Н2О в ледяной уксусной кислоте (степень диссоциации соли в этой среде мала).
Роданид-ионы при их концентрации 2-10-2—5-10~3 моль/л могут быть определены кондуктометрически титрованием 2-Ю-1— 1-ИГ2 М раствором лактата 2-этокси-6,9-диаминоакридина [665] с ошибкой около ±2%.
Кондуктометрический метод применен для определения содержания аммиачного и нитратного азота в удобрении — сульфат-нитрате аммония [1233]. Установлено, что смесь ацетон—вода (94,5 : 5,5) экстрагирует NH4NO3 количественно из его смеси с (NH4)2SO4. Сначала определяют содержание общего аммиачного азота в удобрении кондуктометрическим титрованием раствором NaOH. Затем после экстракции NH4NO3'* смесью ацетон—вода определяют содержание аммиачного азота в сульфате. Разница между обоими значениями дает содержание аммиачного азота или нитратного азота в NH4NO3.
Для раздельного определения концентрации азотной и азотистой кислот в растворах их смесей (0,002—2 М HNO2 и 0,002— 8 М HNO3) предложен осциллометрический метод, основанный на резком изменении ВЧ-электропроводпости раствора в точке эквивалентности при титровании его стандартным раствором NaOH [290]. Первый перегиб на кривой титрования соответствует отти-тровыванию HNO3, второй — HNO2. Продолжительность анализа 2—3 мин. Средняя ошибка -—1,5%.
Метод высокочастотного титрования азотной кислоты (в смеси с серной) основан на связывании HNO3 в реакции нитрования о-нитро толуол а [225].
Для измерения концентрации азотной кислоты используют кондуктометрическое титрование 1,3-дифенилгуанидином в среде пиридина [1090].
82
Нитрит натрия определяется высокочастотным титрованием в среде 30 вес.% этанола 0,17V раствором НС1 [959]. КТТ измеряется при 130 Мгц li соответствует образованию неионизован-ной азотистой кислоты. На точность определения 10—50 мг нитрита натрия (—2 отн. %) не влияет присутствие 50—200 мг нитрата калия.
Метод деполяризации
При аргентометрическом титровании цианид-ионов с концентрацией 5-10*4—5-10“57V в работе [892] для определения КТТ был применен метод деполяризации Pt-электрода.
Индикаторная схема состоит из Pt-электрода, пас. к. э., гальванометра и внешнего источника э.д.с. Pt-электрод (пластинку размером 5 X 10 мм) перед использованием погружают на 1 час в холодную НС1 и затем отмывают водой. В анализируемый раствор (~80 мл) перед началом определения вводят КОН, балансируют индикаторную схему до нулевого показания гальванометра и титруют стандартным раствором AgNO3, добавляя раствор титранта по каплям. По достижении КТТ в результате деполяризации Pt-электрода и разбалансирования индикаторной схемы в последней возникает ток, фиксируемый гальванометром.
На кривой титрования наблюдается два перегиба, первый из которых отвечает полному связыванию CN- в Ag(CN)2, а второй — окончанию осаждения Ag[Ag(CN)2]. При определении 5,228 и и 0,5 мкг KCN средняя ошибка определения составляет 0,3 и 3,5% соответственно.
Описано применение титриметрических методов для определения азота при анализе различных объектов: нитридов бора и кремния [324, 1178], удобрений [932, 997, 1029], органических веществ [715, 1352], почв и растительных вытяжек [561] и др.
Глава VI
ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
Фотометрические методы широко применяются для определения малых количеств азота в виде различных его соединений; особенно многочисленны методы фотометрического определения нитритного, аммиачного, нитратного азота. В основном это методы, основанные на измерении поглощения света при определенных длинах волн.
Известно немного способов определения азота, в основе которых лежат реакции, сопровождаемые излучением (например, флуориметрический метод определения цианидов, определение нитрит-ионов с 2,3-диаминонафталином).
Реакции, положенные в основу фотометрических и спектрофотометрических методов определения азота, могут быть классифицированы следующим образом:
— образование окрашенных золей и коллоидов (реакция Несслера);
— образование нитрозосоединений, нитрование, образование окрашенных комплексных соединений (с бруцином, риванолом и др.);
— образование окрашенных продуктов окисления реагентов (дифениламин, дифениламинсульфоновая кислота, дифенилбен-зидин, стрихнин и др.);
— образование азокрасителей (реакция Грисса и ее модификации);
— образование ионных ассоциатов (с красителями, хелантами и т. д.).
Для фотометрического определения азота используются как неорганические, так и органические реагенты. Круг первых значительно ограничен (гипохлорит, иодид калия и некоторые другие). Очевидно, наиболее чувствительные методы — с применением органических реагентов.
Для повышения чувствительности фотометрических определений используют экстракционно-фотометрические методы, позволяющие сконцентрировать окрашенные продукты реакции в небольшом объеме органического растворителя и фотометрировать непосредственно экстракт. Значительно повышают чувствитель
84
ность фотометрических методов каталитические реакции с фотометрической регистрацией, но число их в аналитической химии азота невелико.
Из большого числа фотометрических методов следует особенно отметить следующие как наиболее широко употребляемые и хорошо зарекомендовавшие себя: метод Несслера (для определения аммония), индофенольная реакция (аммоний), реакция с бруцином (нитрат-ионы), с 2,4-ксиленолом в щелочной среде (нитрат-ионы), метод Грисса (нитраты, гидроксиламин) и реакция с а-наф-тиламином (нитрит-ионы).
Аммоний
Метод Несслера — наиболее общепринятый и распространенный для определения содержания аммиака и аммонийных солей [720, 1065, 1346]. Он основан на образовании коллоида, окрашенного в красно-бурый цвет, при взаимодействии аммиака и аммонийных солей с реактивом Несслера — щелочным раствором мер-курииодида калия (K2[HgJ4l) по следующей реакции:
2NH3 + 2HgJf -> NH2Hg2J3 + 5 J~.
Возникшая окраска поглощает излучение в широком диапазоне длин волн и в определенных условиях интенсивность окраски пропорциональна количеству аммония. Фотометрируют при 400— 425 нм (для количеств 0,2 мг NH3) и при 550—580 нм (для ' 1 мг). При измерении на спектрофотометре предел концентраций может быть расширен до 1,25 мг (при 580 нм). Молярный коэффициент погашения равен 2-106 [1075]. При определении микро-граммовых количеств азота оптическая плотность растворов сохраняется постоянной в течение 6 час. после вливания реагентов. Колебания температуры в пределах 20—40° С нй влияют на результаты Определения [1075].
Если реакция протекает дольше 15—20 мин., то при содержании азота )>1 мг может наблюдаться опалесценция или помутнение раствора, что исказит результаты. Одной из причин, вызывающих помутнение окрашенных растворов, являются пары органических растворителей (особенно кетонов, спиртов, хлороформа) [1081]. Поэтому рекомендуется определять азот по методу Несслера в помещениях, не содержащих органических растворителей. Для получения воспроизводимых результатов необходимо строго соблюдать условия проведения реакции и приготовления реактива Несслера. Концентрация щелочи в анализируемом растворе должна быть в пределах 0,5—0,6 N, порядок прибавления растворов унифицирован, время протекания реакции до момента измерения окраски постоянно; температура не должна быть слишком повышена, так как иначе вместе с ускорением образования окраски ускоряется и процесс помутнения раствора.
85
Существуют различные модификации метода и различные способы приготовления реактива Несслера [720]. Ниже приведен один из распространенных способов.
Приготовление реактива Несслера. 10 г KJ растворяют в 10 мл воды, к раствору приливают при постоянном перемешивании насыщенный водный раствор HgCl2 до появления осадка. Затем прибавляют 30 г КОН, перемешивают до его растворения, приливают 1 мл насыщенного раствора HgCl2, раствор разбавляют водой до 200 мл, дают ему отстояться и сливают с осадка. Хранят раствор в хорошо закупоренной посуде из темного стекла.
Растворы реактива Несслера, приготовленные как из металлической ртути, так и из HgJa, устойчивы длительное время, если их защитить от действия СОа воздуха патронной известью. Для приготовления реактива Несслера следует использовать воду, свободную от СО2. Оптимальная концентрация реактива при фотометрическом определении NH3 составляет 2 мл раствора реактива на 30 мл анализируемого раствора, содержащего 3—100 мкг аммиачного азота. При этом стандартное отклонение составляет 0,06 жкгХт. Минимально определяемое количество аммиачного азота 1,4 .мкг N [1075].
Мешают определению алифатические и ароматические окрашенные соединения, амины, альдегиды, ацетон.
Следует учесть, что при определении этим методом могут происходить потери азота вследствие неполного превращения его в NH3 (при минерализации), а также в результате частичного осаждения окрашенного соединения или улетучивания NH3 при проведении цветной реакции Несслера [284].
Индофенольный метод. По чувствительности он в 10 раз превосходит метод с реактивом Несслера. Оптимальный диапазон концентраций азота 0,3—3 ч. на млн. Этот спектрофотометрический метод основан на реакции аммиака с фенолом и гипохлоритом с образованием индофенола, имеющего интенсивную голубую окраску в щелочной среде [538, 1227, 1262]. Максимальное развитие окраски наблюдается в растворе, содержащем 0,3 М NaOH и 0,8% фенола. Измеряют интенсивность окраски при 625 нм. При определении аммиака в растворе с содержанием 1 ч. па млн. квадратичное отклонение составляет 0,03 ч. на млн.
В работе [1227] приведен несколько модифицированный метод, характеризующийся большей чувствительностью и воспроизводимостью результатов. Этим экстракционно-спектрофотометрическим методом удается определять 0,05—20 мкг N. Реакция проводится в слабокислой среде и при пониженной температуре во избежание возможных потерь аммиака. В качестве окислителя применяют хлорамин Т, устойчивый к хранению (несколько месяцев).
Раствор, содержащий 0,05—20 .икг азота (в форме NH+), помещают в 50-миллилитровую мерную колбу, добавляют 5,0 мл 5%-ного раствора 86
борной кислоты и разбавляют до 25 мл водой. Сосуд помещают в ледяную баню и охлаждают раствор до —3° С. Затем вынимают колбу и вводят в нее пипеткой при помешивании 4,0 мл 5%-ного водного раствора хлорамина Т. Через 20 сек. (±2 сек.) добавляют быстро при помешивании 10 мл 8%-ного водного раствора фенола и помещают сосуд в водяную баню (60° С), нагревая раствор в течение 16 мин. (±30 сек.). Извлекают сосуд и прибавляют пипеткой при помешивании 5,0 мл 3 М NaOH. Охлаждают до комнатной температуры под струей воды, разбавляют раствор и измеряют оптическую плотность при 625 нм.
В экстракционно фотометрическом варианте (для определения 0,05— 10 мкг N в 50 мл) полученный окрашенный раствор переносят в делительную воронку на 100 мл, споласкивают склянку 10 мл воды, присоединяя ее к основному раствору. Добавляют пипеткой 10 мл изобутанола и взбалтывают 1 мин. Отстаивают и удаляют водную фазу. Отфильтровывают через бумажный фильтр органический слой в ячейку прибора и измеряют светопоглощение при 625 нм.
Предложен быстрый и простой метод спектрофотометрического определения азота, основанный на взаимодействии иона аммония с гипохлоритом в слабощелочной среде с образованием монохлорамина, при реакции которого с а-нафтолом получается краситель индофенольного типа, имеющий максимум светопоглощения при 720 нм [216, 218].
К 5 мл анализируемого раствора (^25 мкг N в виде NH^) прибавляют 1 мл раствора NaClO (0,1%-ного по активному хлору), 3 мл 0,2 N NaOH и 15 мл 5%-ного ацетонового раствора а-нафтола, разбавляют водой до 25 мл и через 5—10 мин. фотометрируют при 720 нм.
Метод чувствителен (до 1 мкг N/мл), точен и воспроизводим. Если образовавшиеся окрашенные соединения экстрагировать органическими растворителями, то максимум поглощения переместится в спиртовых растворах к 740 нм, а в растворах эфиров, кетонов, галогенированных алифатических или ароматических углеводородов — в область 540—550 нм.
Окрашенный комплекс типа индооксихинолина, пригодный для фотометрического определения аммонийного азота, образует NHJ в слабощелочной среде с С1О~ и 2-метил-8-оксихинолином [217]. Метод отличается от описанного выше большей устойчивостью реагента, хотя и уступает ему в отношении стабильности окраски комплекса.
К анализируемому раствору «ДО мкг N) добавляют 5 мл раствора гипохлорита (0,1%-ного по активному хлору), через 2 мин. вводят 3 мл раствора реагента (смесь 1ч. 0,1 М этанольного раствора 2-метил-8-оксихи-нолииа, содержащего 15% ацетона, с 1 ч. 0,2 М раствора NaOH) и спустя 10 мин. измеряют оптическую плотность при 710 нм (максимум поглощения).
87
В работах японских ученых [430] для колориметрического определения аммония используют цветные реакции с гваяколом и .м-крезолом. Показано, что использование .м-крезола улучшает воспроизводимость результатов индофенольного метода, снижает значение поправки холостого опыта по сравнению с другими вариантами этого метода. При этом получена линейная зависимость между оптической плотностью и концентрацией NHJ в пределах 0— 30 мкг!мл. Применяемые растворы реагентов устойчивы около 3 дней.
Для определения аммиачного азота используется цветная реакция NHJ с GeH6ONa и С1О~ с образованием интенсивно окрашенного фенолиндофенолята [1419]. В качестве катализатора этой реакции рекомендуется смесь растворов K4[Fe(GN)e] и водорастворимых солей ртути.
Готовят смесь равных объемов фенольного реагента (смесь 1,0—1,2 г С6Н6ОН и 0,004—0,04 г KJFe(CN)e] в 100мл раствора) и гипохлоритного реагента (0,05—0,1 г ЫСЮ, 0,5—1,0 г NaOH и 0,01-0,05 г CH3COOHgCeH6, C6H5OHgNO3, Hg(NO3)2 или HgCl2 в 100 мл раствора), добавляют ее к анализируемому раствору, нагревают 3—5 мин. при 100° С и по охлаждении измеряют оптическую плотность при 630 нм. Ошибка ^7%.
В работах [74, 75] предложена фенолят-гипобромитная реакция для определения аммонийного азота в прозрачных бесцветных, а также мутных и окрашенных растворах и для определения органического азота. В основе методов также лежит образование индофенола в щелочной среде. Окраска устойчива 3 часа при 20—25° С. При фотометрическом определении 0,01—0,1 мг NH4/ju в прозрачных растворах ошибка составляет 10%, при содержании 1 мг!мл— 5 %. Индофенол может быть экстрагирован с помощью СНС13.
Индотимоловый метод [375, 384]. Для спектрофотометрического определения следов аммонийного азота применяют экстракционнофотометрический метод, основанный на образовании индотимола в результате реакции NH4 с тимолом.
В анализируемом растворе « 30 мл), содержащем 0,4—0,5 мкг NH4, устанавливают рН~ 10, добавляют 2,5 мл карбонатного буферного раствора с рНЮ, содержащего 0,03% NaClO; через 2 мин. вводят раствор тимола (приготовленного перед употреблением путем смешивания его 10%-ного раствора в ацетоне с равным объемом 0,7 N раствора NaOH) и устанавливают pH 11,4—11,9. Через 2 часа раствор переводят в полиэтиленовую делительную воронку, разбавляют до 50 .ил водой, прибавляют 5 мл 4 N раствора NaOH, 18 г NaCl, 10 мл изобутанола, экстрагируют 1 мин. и измеряют оптическую плотность экстракта при 675 нм.
В работе [72] индотимоловый метод применен для анализа стали. Окислителем служит хлорамин Т в щелочной среде. Экстрагируют индотимол амиловым спиртом.
88
Тимол-гипобромитная реакция описана в [165].
Пиридин-пиразолоновый метод [963, 1185] применяют обычно для определения 0,05 ч. на млн. аммиака. После взаимодействия аммиака с хлорамином Т при pH 3,7 добавляют пиридин-пиразолоновый реагент. Через 60 сек. пурпурный комплекс (рубазоновая кислота) экстрагируют четыреххлористым углеродом и измеряют интенсивность окраски при 450 нм. В [1185] применена смесь бис-пиразолона и пиразолона в буфере при pH 5,8; экстрагируют окрашенное соединение трихлорэтиленом. Это повысило чувствительность метода и позволило определить 1—500 мкг NHJ/л (0,001—0,5 ч. на млн.) с точностью + 3%.
Фотометрический метод с пиридин-пиразолоновым реагентом применен при определении азота в органических соединениях [905].
По рациональному ассортименту органических реактивов для определения неорганических ионов [387] для фотометрического определения малых количеств аммонийного азота предложен бис-пиразолоновый реактив, содержащий в своем составе 2 вещества: бнс-(1-фенил-3-метил-5-пиразолон) и 1-фенил-3-метил-5-пиразолон. Это высокочувствительный и достаточно селективный реактив, позволяющий определять азот в присутствии большинства обычно встречающихся, наиболее распространенных элементов периодической системы.
Определению ионов аммония с этим реагентом не мешают 100-кратные количества Fe(III), ацетамида и тиомочевины, 10-кратные количества Си, Zn, Ag, SO4“, равные количества CN- и SCN--ионов, а-аминокислот, метиламина, триметиламина. Мешают Fe(II) и значительные количества SO^. Для их предварительного окисления в раствор вводят КМнО4 при pH — 2. Значительные количества CN-, SON" можно удалить ионообменным методом, пропуская анализируемый раствор через Амберлит IRA-400. При высоком содержании Со, Си, Ag, Fe они могут быть удалены в виде гидроокисей.
Косвенный спектрофотометрический метод [847] основан на окислении аммиака гипобромитом до N2 и последующем фотометрическом определении избытка ВгО- при 330 нм (pH 11,1). Определению не мешают 1000—2000-кратные количества Na+, К+, F-, Cl-, Br-, CH3COO-, СЮз, сю;, SOf, С2ОГ, ВО33-, РОГ, AsOt и 10—100-кратные количества Zn2+, А13+, СЮ;, NO;, NO; и цитратов. Мешают ионы, образующие в условиях определения осадки (Mg2+, Са2+, Мн2+, Fe2+, Со2+, Ni2+, Cu2+, Cd2+, Fe3+), а также J-, CN-, анилин, гидроксиламин и др.
Другие методы. Для количественного определения аммиака (2—12 мг/мл) был использован его комплекс с медью. Голубая окраска развивается при реакции солей аммония с избытком карбоната меди и измеряется при 700 нм.
Низкие концентрации аммиака в воздухе определяют реакцией с нингидрином [1403], который наносят на силикагель, помещенный
89
в стеклянную трубку. При данной скорости потока (или общего объема воздуха, содержащего аммиак) сопоставляют длину окрашенной полосы с концентрацией аммиака в воздухе. Метод пригоден для полевых условий.
Спектрофотометрический метод с использованием комплекса аммиак—кобальт(П)—диоксан применен для определения аммиака после отгонки его методом Кьельдаля [1325].
В работе [1336] предложен кинетический метод определения микрограммовых количеств аммиака в природных водах, основанный на его каталитическом влиянии на скорость замещения лиганда в комплексе Hg2+ с 3,3-бнс-Н,Н-ди(карбоксиметил)аминоме-тил-о-крезолфталеином и 1,2-диаминоциклогексан-Н,Н,Н',Н'-тет-рауксусной кислотой. За скоростью реакции наблюдают фотометрически по уменьшению окраски комплекса при 583 нм.
Гидразин
n-Диметиламинобензальдегид образует с гидразином окрашенное в желтый цвет соединение, которое в присутствии сильной кислоты переходит в хиноидную структуру с интенсивной краснооранжевой окраской [1385] с максимумом поглощения при 458 нм. Закон Бера соблюдается для концентраций гидразина до 0,77 ч. на млн. Окраска устойчива в течение 12 час.
Пикрилхлорид реагирует с гидразином с образованием продукта, окрашенного в коричневый цвет. К раствору, содержащему гидразин, добавляют хлороформный раствор пикрилхлорида, этанольный раствор ацетата калия и измеряют оптическую плотность. При концентрациях до 40 ч. на млн. соблюдается закон Бера.
Гидроксиламин
Предложенный в [1115] спектрофотометрический метод определения NH2OH основан на образовании в щелочной среде (pH 8—12) фиолетового комплекса в присутствии V5+ и триэтаноламина.
В мерную колбу емкостью 50 мл вносят 20 мл 2 N Na2CO3, 5 мл 50%-ного раствора триэтаноламина, 4,5 мл 0,1 М NH4VO4, 5—10 мл анализируемого раствора и разбавляют до метки. Смесь нагревают до кипения и по охлаждении измеряют оптическую плотность в 2-сантиметровой кювете при 510 нм относительно раствора холостого опыта. Определению не мешают SO2-, SaO~ и N3H4. Ошибка <2,6%.
Азотистоводородная кислота
При реакции ионов Fe(III) с азотистоводородной кислотой
Fe (III) + HN3 — FeNf + Н+
возникает соединение интенсивной красной окраски [461, 668,
90
1217], которое может быть использовано при фотометрическом определении HN3 в интервале концентраций 0,1—2-10-3 М- Максимум поглощения комплексного иона азида железа(Ш) при 460 нм.
Окислы азота
Большинство методов определения окислов азота связано с определением закиси, окиси, двуокиси азота или их смесей. Вследствие легкости их взаимодействия с загрязняющими газами и другими побочными продуктами, с водой из-за коррозионных свойств необходимо соблюдать специальные меры предосторожности при отборе проб для анализа, чтобы быть уверенным в представительности анализируемого материала и получить надежные результаты. Токсичность окислов NO и NaO4 накладывает дополнительные требования.
Если раньше для определения газообразных окислов азота чаще применяли титриметрические методы, то в последнее время шире стали использовать физические и физико-химические методы, такие, как фотометрия, ИК-спектроскопия, масс-спектрометрия, распределительная хроматография и др. Их применение обусловлено простотой, специфичностью и возможностью анализировать малые количества веществ быстро и с большой точностью.
Окись азота. Окись азота не поглощает света в видимой области спектра, но может быть определена спектрофотометрически при 320 — 500 нм после окисления в паровой фазе до окрашенной двуокиси азота [1105].
Фотометрические методы, используемые для определения окиси азота, основаны на измерении содержания нитратов или нитритов, образующихся при проглощении окиси азота соответствующими растворами. Эти методы не избирательны, так как при этом мешает двуокись азота. Наиболее широко используемый колориметрический метод определения окиси азота основан на окислении ее до двуокиси и определении нитрита, образующегося при поглощении в щелочной среде, по реакции Грисса — Илосвая (см. ниже).
И, наоборот, окись азота может быть превращена в нитрат окислением в растворе перекиси водорода и определена в виде нитрата специфичными для него методами, подобными методу с фенолди-сульфоповой кислотой.
При поглощении окиси азота щелочными растворами сульфита натрия образуется Н-нитрозогидроксиламин-Н-сульфонат. Измеряя светопоглощение этого раствора при 258 нм, можно определить следовые количества окиси азота. Другие вещества не мешают измерению при этой длине волны [1382]. Фотометрический метод определения окиси азота описан в работе [66].
Интересен спектрофотометрический метод определения окиси азота в двуокиси азота, предложенный в работе [1421]. Растворы NO в жидкой N2O4 имеют темпо-зеленую окраску, переходящую в изумрудно-зеленую при охлаждении раствора до 0° С в результате
91
образования N2O3. На этой основе разработан быстрый и точный метод определения <1 1,5% NO в жидкой N2O4. Фотометрируют пробы в специальной металлической кювете с кварцевыми окошками, охлаждаемой ледяной водой, циркулирующей в охладительной рубашке. В процессе измерения окна кюветы обдувают сухим воздухом или азотом для устранения запотевания. Калибровочный график строят для интервала 0—1,5% NO в координатах «разница оптической плотности фотометрируемого раствора при 700 и 550 нм — процентное содержание NO в N2O4». В связи с трудностями приготовления стандартных растворов NO для калибровки используют фильтры-эталоны, соответствующие определенной концентрации NO (например, зеленые стеклянные фильтры Корнинг-4015). Попадание в пробу воды вызывает отрицательную ошибку. Небольшие количества О2, С12 и Fe не мешают.
Окись азота определяют в воздухе с чувствительностью от 40— 3 ч. на блн. (при —25° С) до 100—3 ч. на блн. (при комнатной температуре) хемилюминесцентным методом [865].
Двуокись и четырехокись азота обычно анализируются в двух физических состояниях: в виде газа или в виде жидкости. Ниже приведены методы их определения.
Коричневый цвет двуокиси азота часто используется как качественный метод ее обнаружения. Эта окраска была использована и для количественного определения двуокиси азота в воздухе [809] измерением поглощения в видимой области спектра с абсолютной точностью + 0,05 мм рт. ст. Калибровочные кривые строили в координатах поглощение—парциальное давление двуокиси азота.
Двуокись или четырехокись азота при поглощении водой или другими растворителями образуют нитрат- или нитрит-ионы, которые могут быть определены методами, описанными в соответствующих разделах. Например, предложен способ определения двуокиси азота в воздухе [188], основанный на поглощении NO2 водой и последующем фотометрировании окрашенного соединения с тиокетоном Михлера при 656 нм в 1-сантиметровой кювете.
Для фотометрического определения двуокиси азота после растворения применяются различные методы, описанные ниже, в частности методы Грисса — Илосвая, Грисса — Зальцмана и др.
Предложены визуальные колориметрические методы определения двуокиси азота в воздухе (сравнением со стандартами) по изменению окраски силикагеля при пропускании анализируемой пробы через трубку с силикагелем, на котором предварительно сорбированы реагенты. В таком варианте при окислении двуокиси азота до нитратных солей с последующим нитрованием дифенил-бензидина удается определять 0,005—0,5 ч. на млн. NO2 в 1 л образца [866].
Схожий метод, основанный на реакции двуокиси азота с риванолом, нанесенным на силикагель-бисульфатный носитель, предложен в работе [366] для определения 0,002—0,05 ч. на млн. NO2. Небольшие количества двуокиси азота дают бледно-розовую, вы
92
сокие концентрации — красную окраску. Хлор, иод, сероводород, двуокись серы, двуокись углерода, окись углерода и аммиак, как было установлено, не мешают определению.
При измерении светопоглощения в ультрафиолетовой области существенную роль играет равновесие между двуокисью и четы-рехокисью азота. Количественные данные могут быть получены лишь тогда, когда возможен тщательный контроль за температурой и давлением. В работе [1088] предложен спектрофотометрический метод, в котором двуокись азота определяется при 394 нм, четырехокись — при 240 нм и их смесь — при 353 нм. Было найдено, что в исследованном диапазоне давлений (0—35 мм рт. ст.) эффекты уширения полосы с увеличением давления незначительны.
Флуоресцентное определение двуокиси азота с чувствительностью 1 ч. на блн. в атмосферном воздухе описано в работе [739] (автоматическое и непрерывное определение).
Концентрацию NO2 в воздухе определяют в работе [820] после барботирования анализируемой пробы через 0,1 N NaOH; при этом образуется раствор нитрата натрия. Затем нитрат-ионы взаимодействуют в кислой среде с сульфаниламидом и N-1-нафтилэтиленди-амином, образуя азокраситель, который определяется спектрофотометрически при 540 нм.
Нитрит-ионы
Чаще всего для определения нитрит-ионов используют методики, основанные на образовании азокрасителей. Нитрит реагирует с первичными ароматическими аминами в кислом растворе с образованием промежуточной диазониевой соли, которая после обработки соединением, содержащим аминогруппу или гидроксил-ион, образует соответствующий азокраситель, пригодный как для визуального, так и для фотометрического определения.
Кроме того, для фотометрического определения нитритов используются реакции образования соли диазония, нитрозосоединений, комплексных соединений, ионных ассоциатов с красителями.
Обзор методов, главным образом спектрофотометрических, определения нитритов, а также соединений, восстанавливающихся или окисляющихся до NO, (нитраты, окислы азота, азотсодержащие органические соединения), дан в [1256].
Образование азокрасителя
Метод с сульфаниловой кислотой (метод Грисса — Илосвая) [783, 861]. Образование промежуточной соли диазония при взаимодействии нитрита с сульфаниловой кислотой происходит по реакции
NO2 + 2Н+ + HO3S-^2/-nh2 HO3S—+ 2Н2О.
Сульфаниловая кислота Соль диазония
93
Для сочетания с образовавшейся солью диазония с целью получения азокрасителя используются многие первичные ароматические амины; наиболее часто — 1-нафтиламин:
nh2
Образуется 1-(4-аминонафтилазо)-бензол-4-сульфоновая кислота — краситель красного цвета. Реакция весьма специфична и чувствительна, позволяет обнаруживать 3 мкг NO2/^. Условия проведения этих реакций уточнялись [214, 390, 1212]: диазотирование должно проводиться в сильнокислых растворах на холоду, сочетание должно быть проведено только после того, как полностью закончится диазотирование и при возможно более низкой кислотности. Это должно быть учтено при подготовке реактивов (сульфаниловой кислоты и 1-нафтиламина). Точное установление pH для реакции сочетания достигается использованием раствора ацетата натрия. Оптическую плотность образовавшегося красителя измеряют при 520 нм в кювете с толщиной слоя 1 см. Подчинение закону Бера наблюдается вплоть до 0,5 мг ИО2/л.
Кроме реактива Грисса — Илосвая (растворы 0,6 г сульфаниловой кислоты в 100 мл 20%-ной НС1, 0,48 г 1-нафтиламина в 100 мл 13%-ной НС1 и 272 г CH3COONa-ЗН2О в 1 л воды) используют реактив Грисса — Ромийна, представляющий собой сухую смесь (1,0 : 0,1 : 8,9) сульфаниловой кислоты, 1-нафтиламина и винной кислоты [239, 684]. В работе [239] установлены оптимальные условия для развития окраски при фотометрическом определении NO2 с реактивом Грисса — Ромийна: при pH 1,9—3,0 в интервале температур 15—30° С окраска устойчива 20—80 мин. Для определения NO2 при его концентрации 0,1 ч. на млн. к 20 мл анализируемого раствора с pH 2,3 добавляют 0,1 г реактива. Мешающее влияние соединений серы (до 50—100 мкг/мл), иода (до 100 мкг/мл) легко устраняется добавлением 50 мг HgCl2 к 20 мл раствора. При выделении осадка его удаляют фильтрованием. Добавление HgCl2 эффективно также при использовании других реагентов, образующих азокрасители с NO2.
В методе образования азокрасителей используются также многие производные сульфаниловой кислоты и 1-нафтиламина и другие соединения: диметил-1-нафтиламип [742], .и-фенилепдиамини бен
94
зидин [12, 85, 540], N-пафтилэтилендиамин, бензидин, диметилаии-лин, 2-нафтол и др. [296, 540], смесь тг-диаминодифенилсульфопа с дифениламином [1333], п-диаминодифенилсульфон и дифениламин [1331], п-нитроанилин и азулен [736], 8-аминохинолин [710], сульфаниламид [494], фенилазоанилин [1257] и др.
В работе [710] предложен экстракционно-спектрофотометрический вариант метода с 8-аминохинолином для определения малых количеств нитрита (0,5—16 мкг NO?).
К 40 мл нейтральной пробы, помещенной в склянку с завинчивающейся пробкой, прибавляют 8 мл 2%-ного раствора 8-аминохинолина в 50%-ной СН3СООН (pH раствора при этом 0,4—1,9). Через 10 мин. прибавляют 4 М CH3COONa до pH 2,2—2,3 и нагревают 20 мин. при 60° С. После охлаждения раствора добавляют ЮМ NaOH (~6 мл) до pH 5,25—6,0, вводят 5 мл н-гептана, встряхивают склянку 2 мин. и после разделения фаз органическую фазу спектрофотоМетрируют при 465 нм. Чувствительность 0,151 мкг NOjj мешают Cu(II), Fe(III), NH*.
Образование соли диазония
Растворы солей диазония, образующихся в методе с сульфаниловой кислотой, чрезвычайно сильно поглощают в УФ-области, что используется для определения нитрита [1148]. Измерения проводятся при 270 нм в 1-сантиметровой кювете. Закон Бера соблюдается в интервале концентраций 0,2—3,2 мг NOj/.z.
Соли диазония, образовавшиеся из фенилендиамина и многих его производных, были использованы для определения нитритов. Чувствительность при этом в 3—4 раза больше, чем с,солями, образованными из сульфаниловой кислоты. Хлор-п-фенилендиамин особенно пригоден, так как дает хорошо воспроизводимые результаты и стабилен во времени. В диапазоне длин волн 320 —400 нм, в котором производятся измерения, сам реагент существенно не поглощает.
Нитрование, образование нитрозосоединений, окрашенных и. флуоресцирующих комплексных соединений
Реакция с М,]Ч-диметиланилином [1062]. При реакции с N,N-диметиланилином азокрасителя кислота превращается в п-нитро-зо-Х,Х-диметиланилин в соответствии с уравнением
Н3С СН3 Н3С СН3
I N=O
Желтая окраска нитрозосоединения используется для фотометри-
95
ческого определения нитрита. Метод позволяет определять до 1 ч. на млн. NO.J.
Реакция с 2,6-ксиленолом [812]. В растворах с оптимальной кислотностью [серная кислота — вода — уксусная кислота (5 : 4 : : 1)1 при реакции 2,6-ксиленола с HNO2 образуется 4-нитрозо-2,6-ксиленол, светопоглощение которого при 307 нм пропорционально концентрации NO2 в диапазоне 0—10 мкг!мл. Мешают галогениды и их необходимо удалять осаждением раствором Ag2SO4.
Определение в отсутствие NOg. 1 мл анализируемого раствора, содержащего 8 мкг!мл NO~, смешивают с 8 мл H2SO4 (5 : 3), прибавляют 1 мл 0,01 М раствора 2,6-ксиленола и через 5 мин. фотометри-руют при 307 нм.
Определение в присутствии NOg. 1 мл анализируемого раствора обрабатывают и фотометрируют так же, как и в отсутствие NO~. В другой такой же порции анализируемого раствора разрушают NOg сульфаниловой кислотой, полученный раствор обрабатывают, как указано выше, и фотометрируют при 307 и 324 нм. Значение оптической плотности при 307 нм вычитают из оптической плотности первой порции раствора и по разности оптических плотностей находят концентрацию NOg. По значению оптической плотности второй порции раствора при 324 нм находят концентрацию N Од .
Реакция с тиогликолевой кислотой [1431]. При взаимодействии нитрита с тиогликолевой кислотой в слабокислой среде образуется нитрозотиогликолевая кислота:
NO" + СН2 - СООН + Н+ СН2 — СООН + Н2О.
I I
SH S - NO
Это нитрозосоединение используется для колориметрического определения NO2 в интервале концентраций 0—100 мг)л. Оптическая плотность измеряется при 355 нм. Мешают ионы Fe(II) (при отношении Fe : NO2 50), Go(II), MnO4, WO4~-
Реакция с бруцином [281, 704]. Разработан быстрый колориметрический метод с использованием бруцина в качестве хромофорного реагента в растворе серной кислоты. Ионы NO2 дают окрашенные соединения в более разбавленной H2SO4, чем NOg (1:5 и 1 : 1 соответственно). На этой основе разработан метод фотометрического определения NO2 и NOg в щелочных растворах.
Для определения NOg 1 лм анализируемого раствора разбавляют и нейтрализуют раствором H2SO4 (1 : 8) так, чтобы объем раствора после нейтрализации составлял 25 мл. К полученному раствору добавляют 1 мл 2%-ного раствора бруцина, 5 мл концентрированной H2SO4, охлаждают до 30° С (в термостате) и измеряют оптическую плотность раствора на ФЭК-М с синим светофильтром (№ 3) в 2-сантиметровой кювете относительно
96
раствора, состоящего из 25 мл воды, 1 мл раствора бруцина и 5 мл концентрированной H2SO4.
Для определения NO^ и NOg 1—2 мл анализируемого раствора разбавляют и нейтрализуют раствором H2SO4 (1 : 8), так, чтобы объем раствора после нейтрализации составил 15 мл, затем добавляют 1 мл 2%-ного раствора бруцина, 5 мл концентрированной H2SO4 и перемешивают. Через 3 мин. прибавляют еще 10 мл концентрированной H2SO4, охлаждают до 30° С и фотометрируют, как и при определении NO^. Раствор сравнения содержит 15 мл воды, 1 мл раствора бр.уцина, 15 мл концентрированной H2SO4.
Содержание NOJ рассчитывают по разности.
Реакция с риванолом [14]. Нитрит-ионы образуют интенсивную окраску при реакции с риванолом (лактатом 2-этокси-6,9-диамино-акридина) в 1,8 М HG1. Соответствие с законом Бера наблюдается при 515 нм для диапазона концентраций 0—1,2 мкг NO^/мл. Максимальное поглощение при 505 нм. Чувствительность 0,1 мг/л (в пересчете на N). Молярный коэффициент погашения 4,1 • 103. Не мешают ионы NOg, ВО|“.
Реакция с тиомочевиной [853]. При реакции азотистой кислоты с тиомочевиной
S II
HNO2 + NH2,-C-NH2->HSCN-|-2H2O-|-N2
образуется роданистоводородная кислота, которая при взаимодействии с введенным Fe(III) дает окрашенный комплекс. Оптическая плотность раствора измеряется при 455 нм. Метод применен для определения 2—12 мкг №02/жл. Хотя этот метод менее чувствителен, чем классический метод Грисса с сульфаниловой кислотой, но простота методики является ее определенным преимуществом.
Реакция с 2,3-диаминонафталином [1400]. Этот реагент использован для спектрофотометрического и флуориметрического определения нитрит-иона. Он реагирует в — 1 N HG1 с образованием 2,3-нафтотриазола, хорошо экстрагирующегося из водных растворов 1,1,2,2-тетрахлорэтаном. Экстракт используют для спектрофотометрического и флуориметрического измерения.
К 50 мл анализируемого раствора приливают 10 мл 0,05%-ного раствора 2,3-диаминонафталина в 6 TV НС1 и через 2 мин. дважды экстрагируют порциями по 5 мл тетрахлорэтана, встряхивая по 30 сек. Объединенные экстракты фотометрируют в 1-сантиметровой кювете при 355 нм или измеряют их флуоресценцию со светофильтрами Корнинг-7-54 (первичный) и 47В Раттен (вторичный).
Определению мешают Sn(II) и Se — в первом случае, и Al, Bi, Gr(III), Gu, Ni, Se, Sn(II), Fe — во втором. Ионы NH4 и NOg не мешают. Чувствительность спектрофотометрического определения NO2 равна 1 мкг!мл, флуориметрического — 0,0065 мкг/мл.
4 В. Ф. Волынец, М. П. Волынец
97
В качестве аналитических реагентов для фотометрического определения нитрит-ионов используют некоторые красители. Разработаны методы косвенного спектрофотометрического определения NOJ с бриллиантовым зеленым и кристаллическим фиолето* вым [2771. Используется окислительно-восстановительная реакция, происходящая в сильнокислой среде (HCI) между NO2 и KJ, взятым в избытке, и красителями. Эквивалентное количество образующегося окрашенного продукта окисления красителей определяют при pH 3,6—3,9. Для этого реакционную смесь нейтрализуют раствором CH3COONa. Этим методом определяют 5-10-5 М NO-Д
Предложен экстракционно-фотометрический метод определения NO2 в воде с использованием основного синего К [267].
2 мл анализируемой воды помещают в пробирку емкостью 25 мл (с меткой на 10 мл) и прибавляют последовательно 1мл 5%-ного раствора КВт, 0,5 мл 20 N H2SO4, 0,5 мл 0,3%-ного раствора красителя в смеси этанол —вода (1 : 4), 6 мл бензола и встряхивают 1 мин. Через 15 мин. экстракт переводят в 1-сантиметровую кювету и измеряют на ФЭК-Н-57 оптическую плотность со светофильтром № 8. Калибровочный график строят для аналогично приготовленной серии стандартных растворов, содержащих от 2,5 до 80 мг ЬЮ~Лг.
Для фотометрического определения ионов NO2 успешно используются также цветные реакции с рядом других органических реагентов (в скобках указана чувствительность реакций в мкг ШО^/мл): 1,2,3-трпс-(2-диэтиламиноэтокси)-бензолом [1124] (1,15—11,5); диметилсульфоксидом и соединениями ацилсульфок-сония [605, 1300] (2—100 мг в пробе); хлористоводородным хлорпромазином [921] (0,023); п-розанилинметансульфокислотой [1017] (0,019-1).
Реакция с сульфидом железа(П) [1093]. Восстановление нитрита железом (II) в 2,5 М фосфорной кислоте было успешно использовано для количественного определения нитрита. Образующийся комплексный ион нитрозил-железо(П) поглощает в этой среде при 455—460 нм. Чувствительность реакции 2,5 мкг1мл.
От 0,2 до 40 мкг ^ОЦмл определяют экстракционно-фотометри-' ческим методом с помощью а-тиолуксусной кислоты [518]. В качестве экстрагента используют амилацетат, ТБФ. Фотометрируют экстракт при 232, 332 и 547 нм.
Кинетический метод
Для определения нитрит-ионов предложен фотометрический метод, основанный на окислении комплексоната марганца(П) до комплексоната марганца(Ш) посредством Н2О2, катализируемом нитритом [134].
98
К 0,5 мл анализируемого раствора добавляют 8 мл раствора комплексоната марганца(П), 0,5 мл 0,22 N раствора Н2О2 и через 2 мин. измеряют оптическую плотность при 18° С с зеленым светофильтром.
Для приготовления комплексоната марганца(П) растворяют 10 г комплексона III в 150 мл дистиллированной воды, раствор нагревают, добавляют 200 г МпС12-4Н2О, кипятят и охлаждают.
Метод позволяет определить 1 • 10~8 г!мл NO2 с ошибкой — 10 % и 3- 10~1в г!мл с ошибкой 20%.
Нитрат-ионы
Фотометрические методы определения нитратов основываются на следующих реакциях: методы нитрования и образования нитрозосоединений; методы, основанные на окислении реагента нитратом; образование иовных ассоциатов; восстановление нитрата. Из перечисленных способов метод нитрования (и особенно методика с фенолдисульфоновой кислотой) является доминирующим.
Методы нитрования и образования нитрозосоединений
Реакция с фенолдисульфокислотой [883]. По реакции нитрата с аммиачным раствором 2,4-фенолдисульфокислоты образуется соединение интенсивно желтого цвета:
ОН ONH4
I S03H 02N I SO3NH4
+3NH4OH-> \/\Z +oh- + 3h2o.
I 03H SO3NH4
Реагент приготовляют обработкой фенола дымящей серной кислотой и сохраняют его в кислой форме. Образец испаряют досуха, добавляют реагент и нагревают. Затем смесь разбавляют водой и для развития окраски добавляют концентрированный раствор NH3.
Наибольшую чувствительность и точность определения можно достигнуть при измерении оптической плотности при 410 нм (максимальное поглощение). Мешают нитрит-, хлорид-ионы и органические вещества.
Хлориды удаляют осаждением солями серебра, органическое вещество разрушают обработкой перекисью водорода, а нитриты или удаляют, или окисляют до нитратов.
Реакция с бруцином [689, 1242, 1271]. Нитрат- и нитрит-ионы взаимодействуют с бруцином в среде серной кислоты при различной кислотности: нитрит-ионы при более низкой концентрации
4*
99
(17 вес.%), нитрат-ионы при более высокой (50 вес. %). Нитрат-ионы образуют с бруцином сначала соединение красного цвета, но затем окраска быстро изменяется на желтую, сильно поглощающую в области 400—410 нм. Чувствительность метода — 0,1 мкг 'NOJma. Наилучшие результаты получаются в диапазоне 1—4 мкг!’мл, когда кривая поглощение—концентрация NO3 близка к линейной.
В смеси H2SO4 и НС1О4 следует спектрофотометрировать раствор при 430 нм [1242]. Ошибка определения составляет + 1,5%. Мешают Fe, Gu, Cd, К, Na, Мп, Mg, Zn, Al, Cl-, F-, Pb. В растворах, содержащих NO3 и NO2, нитриты предварительно окисляют до NO3 с помощью КМпО4; непрореагировавший КМпО4 восстанавливают щавелевой кислотой и затем определяют суммарное содержание NO3 и NO2.
Определение NO3 в присутствии NO2 можно проводить также в более кислой среде (^>6,5 М), причем к анализируемому раствору добавляют KNO2, так как специальными опытами установлено [689], что присутствие 2—10 мкг NO2 дает постоянное, легко учитываемое завышение оптической плотности фотометрируемого раствора.
Предложен микрометод определения нитратов с помощью солей бруцина [1271].
К 2 мл анализируемого раствора (0,5—10 мкг NO3) прибавляют 1 мл 1%-ного раствора сульфата бруцина, 10 мл концентрированной H2SO4 и через 30 мин. спектрофотометрируют при 412 нм. Мешающее влияние NO” устраняют добавлением мочевины.J
Метод определения высоких концентраций нитратов с помощью бруцина описан в работе [845].
Реакция с ксиленолами [811, 1003]. В реакции нитрования используют ряд изомеров ксиленола: 3,4-ксиленол, при взаимодействии с которым образуется нитро-3,4-ксиленол [1003]; 2,6-ксиле-нол, из которого образуется 4-нитро-2,6-ксиленол [811]. В первом варианте оптическая плотность измеряется при 430—450 нм-, чувствительность ^0,02 мкг/мл. Во втором случае подчинение закону Бера соблюдается в диапазоне 320—324 нм.
Реакция с и-диаминодифенилсульфоном [1332]. Метод основан на нитровании 4,4'-диаминодифенилсульфона с образованием 3,3'-динитро-4,4'-диаминодифенилсульфона’ (максимум светопоглоще-ния при 410 нм). Молярный коэффициент погашения составляет 2,4-10’.
К 1 мл анализируемого раствора (30—300 мкг NO”), не содержащего галогенидов, добавляют по каплям при непрерывном перемешивании 4 мл раствора п-диаминодифенилсульфона (2 г реагента растворяют в 1 л концентрированной H2SO4), нагревают на водяной бане до кипения и кипятят 2 мин.
100
По охлаждении до комнатной температуры разбавляют до 25 мл и измеряют оптическую плотность при 410 нм в 1-сантиметровой кювете относительно раствора холостого опыта.
Реакция с хромотроповой кислотой [496]. Предложено спектрофотометрическое определение нитрата при 400 нм. Закон Бера соблюдается для концентраций NOS 6 мкг!мл.
3 мл анализируемого раствора (6,5 мкг1мл NO3) охлаждают льдом, медленно добавляют 5 мл концентрированной H2SO4 и 1 мл раствора хромотроповой кислоты (0,1 г реагента в 25 мл концентрированной H2SO4). Нагревают 15 мин. на водяной бане при 60° С, охлаждают льдом, осторожно разбавляют раствором H2SO4 до 10 мл и спектрофотометрируют в 4-сантиметровой кювете, применяя холостую пробу в качестве раствора сравнения.
Предложен спектрофотометрический метод определения малых количеств нитрата и нитрита превращением в нитротолуол и экстракцией толуолом [520].
Методы, использующие нитрование других соединений (1,3-бензолдисульфоновая кислота, 1-нафтол-5-сульфоновая кислвта), не обладают какими-либо преимуществами по сравнению с методами нитрования фенолдисульфоновой кислоты и 2,6-ксиленола.
Окисление реагентов нитратом
В качестве реагентов,’ образующих окрашенные продукты окисления при взаимодействии с нитратом, используют дифениламин, дифениламинсульфоновую кислоту, дифенилбензидин и восстановленный стрихнин.
Сульфат стрихнина восстанавливают в растворе НС1 [1259]. При оптимальных условиях образуется розовая окраска с NO3-ионами. Чувствительность 0,02 мкг/мл. Хлориды не мешают, но мешают другие окисляющие агенты. Необходима очень тщательная обработка при восстановлении, чтобы получить воспроизводимые результаты. Это затрудняет широкое' использование метода . В случае предварительного ионообменного концентрирования NO3 чувствительность метода повышается [1398].
Голубая окраска, характерная для окисленного дифениламина, образующегося в растворе концентрированной H2SO4 (после дополнительного введения хлорида), используется для определения NO3 с чувствительностью 0,05 мкг!мл [1218]. Необходим тщательный контроль условий анализа, поскольку окраска зависит от температуры, времени, количества хлорида и серной кислоты.
Такие же трудности и в методе с дифенилсульфоновой кислотой [945], дифенилбензидином [1214], пирогаллолсульфоновой кислотой [1100].
101
Образование ионных ассоциатов
В работе [1129] предложен экстракционно-спектрофотометрический метод, использующий образование ионных ассоциатов, возникающих при взаимодействии нитрата с катионными комплексами меди(1) с 2,2'-дихинолилом и 2,3-диметил-1,10-фенантролином. Наилучшие экстрагенты — хлороформ (наибольшая чувствительность) и хлорбензол (лучшая воспроизводимость). Этим методом удается определить в воде 8 • 10~6 — 4-10"5 молъ/л NO3 с точностью 0,7-1,7 %.
В качестве реагентов для определения нитрат-ионов применяют нильский голубой А [1173] (^ 250 мкг NO”), кристаллический фиолетовый [106, 1426] (4-10-6 — 2-10"5 моль/л), метиленовый голубой [1415] (микрограммовые количества). Экстрагентами для образующихся ионных ассоциатов являются соответственно 1,2-ди-хлорбензол, хлорбензол, 1,2-дихлорэтан. Оптическую плотность растворов измеряют при 650, 595 и 655 нм, соответственно.
В методе с кристаллическим фиолетовым определению не мешают 1000-кратные количества РО1-, SO*- (мешает 10-кратный избыток СГ); в методе с метиленовым голубым — не мешают 1000 мкг/мл SO1- (мешают С1О7, SCN-, CN", С1“, J-, Вг~, фтат латы).
Образование азокрасителя
Предложен метод разложения нитрит-ионов сульфаниловой кислотой и определения нитрат-ионов с помощью реактива Грисса — Ромийна [213].
Восстановление нитрата
Фотометрические методы определения нитрата после его восстановления могут быть разделены на 3 группы в зависимости от степени восстановления нитрата (до аммиака, окиси азота или нитрита). Для конечного определения каждой из восстановленных форм применяют специфические фотометрические методы.
Восстановление до аммиака. Восстанавливать нитрат до аммиака можно хлоридом закиси титана [1415], алюминием [387], сплавом Деварда [452], сульфатом железа(П) в щелочных растворах [1147]. Получившийся аммиак определяют реактивом Несслера [452],пиридин-пиразолоновым [1147] или бнс-пиразолоновым реагентом' [387] (см. выше).
Восстановление до окиси азота [1114]. Нитратсодержащий раствор обрабатывают раствором сульфата Fe(II) в серной кислоте. При этом нитрат восстанавливается до окиси азота, ’которая"реагирует с избытком Fe(ll) с образованием нитрозилжелезногр комплекса красно-пурнурного цвета. Фотометрирование проводят при 520—525 нм.
102
Восстановление до нитрита может выполняться с помощью смеси Zn + NaCl [332], металлическим цинком в присутствии CH3COONa и сульфаниловой кислоты [214], гидразинохм [1254]. Образовавшийся нитрит может быть определен по реакции Грисса после диазотирования и азосочетания с 1-нафтиламином [332], по реакции с и-нафтилэтилендиамином [214]. Ниже приведен один из спектрофотометрических методов [214].
К 25 мл анализируемого раствора последовательно прибавляют 15 мл 30%-ного раствора CH3COONa, 0,07 г сульфаниловой кислоты и 0,3 г Zn-порошка, перемешивают 10 мин. и фильтруют. Затем 10 мл фильтрата смешивают с 1,5 мл 35%-ной НС1 и 1 мл 0,1%-ного раствора п-нафтилэтилендиамипа и через 20 мин. измеряют оптическую плотность образовавшегося азокрасителя при 550 нм.
В присутствии NO” этим методом определяется сумма NO3 и NO.7- Для определения одного NO~ в смеси с NO2 к раствору пробы прибавляют сульфаниловую кислоту, нагревают 10 мин. в кипящей водяной бане (NO2 при этом разрушается) и далее продолжают анализ, как описано выше.
Чувствительность метода 0,1 мкг!мл нитратного азота; ошибка определения 0,2 мкг NO3 /мл составляет + 2,5%..
Другие методы
Для определения NO3 использовано его ингибирующее влияние на комплексообразование рения с а-фурилдиоксимом [529].
«
Раствор, содержащий 3—15 мкг NOg, разбавляют до 2мл, добавляют 0,2 мл 0,02%-ного раствора КВеО4, 0,45 мл концентрированной НС1, 3 мл метанола и 1 мл 0,85%-ного раствора SnCl2-2H2O, перемешивают, точно через 10 мин. добавляют 1 мл 0,35%-ного раствора а-фурилдиоксима и разбавляют до 10 мл. Через 10 мин. к раствору добавляют 1мгК.1ЧО3 в капле воды для стабилизации окраски и спектрофотометрируют при 532 нм. Влияние NO2 незначительно.
Определению NO3 мешают SCN-, С1О3, WOj'. Метод позволяет определять 3—35 мкг ^03/мл со стандартным отклонением + 0,19 мкг. Чувствительность метода может быть повышена экстракцией комплекса рения хлороформом и спектрофотометри-рованием экстракта.
Цианид-ионы
Критический обзор классических (визуальных) и инструментальных методов открытия и определения цианидов приведен в работах [82, 221, 491, 778]. Наряду с методами осаждения, потенциометрии, полярографии, амперометрии, визуальной титриметрии для определения малых количеств цианид-ионов применяют спектрофотометрические и флуориметрические методы. В большинстве
103
случаев проводится косвенное спектрофотометрическое определение CN'-ионов на основании уменьшения поглощения различных комплексных соединений в присутствии цианидов. Но, кроме того, используют и прямые спектрофотометрические и флуориметриче-ские методы; некоторые из них приведены ниже.
Косвенные методы
Ослабление поглощения комплексных солей ртути. Разработан спектрофотометрический метод определения CN_-hohob, основанный на уменьшении поглощения этилендиаминтетраацетата ртути [151] при 250 нм. Уменьшение оптической плотности в присутствии цианидов пропорционально концентрации CN- в интервале 0,2—24 мкг/мл (молярный коэффициент погашения —1260). Реакция завершается за 2—3 мин. в фосфатной буферной среде с pH 6—8. Мешают определению SCN”, S2“, Вг“, J~, Сн2+, Ni2+, Со2+.
Косвенное спектрофотометрическое определение цианида в ультрафиолетовой области выполняется также с помощью иодид-ного комплекса состава [HgY(J)]3- (где Y — ЭДТА) [241]. Устойчивость цианидного комплекса [HgY(CN)]3~ значительно выше устойчивости иодидного комплекса, и поэтому CN- количественно вытесняет J-. По уменьшению оптической плотности раствора [HgY(J)]3' при 260 нм после добавления CN“ определяют содержание цианида (в пределах 2,5—280 мкг CNY50 мл).
К анализируемому раствору с pH 5—8 добавляют известный избыток 10-3 М раствора [HgY(J)]3-, разбавляют до 50 мл и через несколько минут фо-тометрируют относительно воды. Определению мешают S2-, VO3, In, Си, Со, Ni, Hg(II), Ag.
Комплекс ртути с фталеинкомплексоном, образующийся при pH 9,6—10,3 и значительно поглощающий при 583 нм, также используется для косвенного определения CN- [242]. При реакции этого комплекса с цианидом образуется комплекс, не поглощающий свет при указанной длине волны. На основании этого по уменьшению оптической плотности первого комплекса определяют фотометрически содержание цианид-ионов. Определению мешают S2~, J- и некоторые катионы.
Разработан спектрофотометрический метод определения 0,2— 6 мкг CN- с помощью дифенилкарбазона в присутствии Hg(II) и KJ (по ослаблению окраски смеси реагентов [250]).
К 10 мл анализируемого раствора прибавляют 1 мл 0,05 N HNO3 (в присутствии С1~ вместо HNO3 добавляют буферный раствор с pH 7), 1 мл смешанного реагента, содержащего по 2,5-10-4 моль!л Hg(NO3)2 и KJ, 10 мл СвНв, 1 мл 0,05%-ного раствора дифенилкарбазона и экстрагируют 1 мин. Через 1—2 мин. измеряют оптическую плотность экстракта при 562 нм в 1-санти-метровой кювете.
104
Предложен косвенный метод определения микроколичеств CN~, заключающийся в образовании прочного соединения CN~ с Hg(II) с последующей экстракцией и спектрофотометрированием избытка Hg(II) в виде дитизоната четыреххлористым углеродом И07].
К 10 Л4Л. анализируемого раствора (1—10 мкг СИ'Лил) прибавляют 2 мл •10~4 М Hg(NO3)2, 1 мл 2 N HNO3, разбавляют водой до 20 мл и встряхивают гв течение 30 сек. В полученную смесь вводят 10 мл раствора дитизона в СС14 ‘(3,5 мл 0,01%-ного раствора дитизона в СС14 смешивают с 96,5 мл СС14) и встряхивают в течение 6 сек. После 1 мин. отстаивания нижний органический «слой отделяют и через 15 мин. фотометрируют при 495 нм в 1-сантиметро-иой кювете.
Комплекс ртути(П) с тиофенолтрифторацетоном применен в косвенном экстракционно-фотометрическом методе определения CN'-ионов [1427]. Метод основан на подавлении реакции образования этого комплекса в присутствии цианида. Закон Бера соблюдается в пределах концентраций 0,48—4,32 лкг/100 мл.
К аликвотной части исследуемого раствора добавляют 2 мл буферного раствора с pH 11,5, разбавляют до 10 мл водой, вводят 10 мл раствора ртути (содержащего 40 мг Hg(II) в 10 мл) в СС14; встряхивают 2 мин. и измеряют оптическую плотность комплекса при 360 нм относительно растворителя.
Многие анионы не мешают определению CN'; ионы SO|~ мешают в количестве ~ V4 от содержания CN~. Ошибка метода 2,8 и 1% для содержаний 1,44 и 3,84 мкг CN~ соответственно.
Ослабление светопоглощения комплексных соединений серебра. Косвенный фотометрический метод определения микроколичеств CN“ основан на ингибиторном действии цианида при образовании трехкомпонентного комплекса серебра с 1,10-фенантро-лином и бромпирогалловым красным в нейтральной среде (pH 6—8) 1636]. Максимум поглощения трехкомпонентного комплекса находится при 635 нм (молярный коэффициент погашения составляет 5,1 • 104), окраска устойчива 30 мин. Оптическая плотность пропорциональна концентрации CN” в пределах 0,26—2,6 мкг/мл.
К анализируемому раствору (— 10~6 М по CN~), прибавляют 10 мл 10~5 М раствора AgNO3, 1 мл 3%-ного раствора комплексона III, 1 мл 20%-ного раствора CH3COONH4, 1 мл 10~3 М раствора 1,10-фенантролина и 4 мл 10“4 М раствора бромпирогаллового красного, разбавляют до 50 мл и измеряют оптическую плотность при 635 нм в 1-сантиметровой кювете относительно такого же раствора, не содержащего С№ или AgNO3.
При определении 5,2 мкг CN“ стандартное отклонение составляет 3,4%.
Ослабление светопоглощения окрашенных комплексных соединений тяжелых металлов (Zn, Си, Ni, Со) [1170]. Описан фото-
105
метрический метод, состоящий в отгонке HGN в щелочной раствор, содержащий ионы тяжелых металлов в виде окрашенных комплексов. Концентрацию CN~ находят по уменьшению оптической плотности раствора в приемнике дистиллята. Ослабление окраски поглощающего раствора по мере поступления в него GN~ измеряют на промышленных анализаторах в случае непрерывных определений или путем сравнения со стандартом.
В качестве комплексообразующих реагентов могут использоваться дитизон, диэтилдитиокарбаминат натрия, диметилдиоксим, бнс-циклогексаноноксалилдигидразон, цинкон, хромчерный Т.
Измерение оптической плотности окрашенного фенантролината железа(П) [973]. Метод основан на вымывании ионами GN~ окрашенного фенантролината Fe(II) из его малорастворимого трииоди-да и последующем фотометрировании фенантролината железа. Этим методом определяют 0—8 мкг С№~/мл. Не мешают значительные количества Na+, К+, NH*, Са2+, О2, J~, Cl~, NO3, СО1" и равные количества F~; мешают Fe(III) f>10 мкг/мл).
Установлено, что вариаминовый синий с Си(II) в присутствии CN- или SGN~ приобретает синюю окраску, интенсивность которой пропорциональна концентрации CN-- или SCN'-ионов [574]. Предполагается, что механизм этой реакции заключается во взаимодействии Сп(П) с CN~ с образованием соединения Cu(CN)a, которое переходит в Cu2(CN)2 с выделением (CN)2, окисляющего вариаминовый синий до окрашенной окисленной формы [n-(N-анизил)хинондиимина].
Образование пиридиниевых красителей
Для фотометрического определения малых количеств цианид-ионов по реакции образования пиридиниевых красителей в работе [492] рекомендованы следующие ароматические амины: о-анизидин, n-фенетидин, о-дианизидин, n-aMHHO-N,N'-диметил анилин, пиперазин, бензидин, n-фенилендиамин и n-аминодифениламин. Три последних являются лучшими. При определении 0,1—1 мкг С№~7мл с применением 0,2%-ного раствора амина в 0,5 М НС1 лучшие результаты дает n-фенилендиамин. Для более высоких концентраций GN- ( ~ 2 л»кг/лсл)’и с применением 1 %-ного раствора амина лучшим является бензидин.
Определение цианидов с помощью пиридина и пиразолона описано в [1070, 1138].
Реакция с органическими дисульфидами
Цианид-ионы реагируют с органическими дисульфидами (2,2'-дитиопиридином, 4,4'-дитиопиридином, 5,-5'-дитио-бнс-(2-нитро-бензойной кислотой) при pH 9 с образованием окрашенных тиольных анионов. В работе [852] изучено влияние диметилформ-амида (ДМФА) на скорость этой реакции и показано, что в присут-106
ствии ДМ ФА скорость реакции значительно увеличивается: время, необходимое для спектрофотометрического определения С№-ио-нов, уменьшается с 2 до <^0,5 час.
Реакции с барбитуровой киелотом
В работах [87, 182] показана возможность определения CN“-ионов с барбитуровой кислотой.
Анализируемую пробу, содержащую 0,25 мг CN-, помещают в колбу Кьельдаля, прибавляют дистиллированную воду до объема 50 мл, 20 мл 17V раствора Pb(NO3)a и через капельную воронку приливают 0,1 N HaSO4 до кислой реакции по метилоранжу. В приемник предварительно наливают 15 мл 1%-ного раствора NaOH. Отгоняют 75 мл дистиллята, доводят объем отгона до 100 мл. 5 или 10 мл отгона (в зависимости от концентрации цианидов) нейтрализуют в отдельной пробе 0,1 N раствором НС1 по бромкрезоловому красному; отгон помещают в мерную колбу емкостью 25 мл, приливают необходимое для нейтрализации количество 0,1 У раствора НС1, доводят объем до метки, переносят в коническую колбу, прибавляют 1 мл 1%-ного раствора хлорамина и через 1 мин. 3 мл смешанного реактива. (Последний готовят следующим образом: в мерную колбу емкостью 50 мл помещают 1 г барбитуровой кислоты, 5 мл пиридина, растворяют в воде, приливают 1 мл концентрированной НС1 и доводят водой до метки.) В зависимости от интенсивности полученной окраски определяют светопоглощение на фотоколориметре с зеленым светофильтром (для концентрации CN- «С 0,2 мг/л) через 8 мин. после прибавления реактива или с синим светофильтром (для концентрации CN-О.2—1 мг!л) через 30 мин. после прибавления реактива. Концентрацию CN-рассчитывают по калибровочным кривым.
Флуориметрические методы
Предложен снецифический флуориметрический метод определения малых количеств CN“ с бензохинонмонооксимным эфиром бензолсульфокислоты [787]. С этим реагентом CN~ взаимодействует с образованием соединения, обладающего интенсивной зеленой флуоресценцией при возбуждении светом с длиной волны 440 нм.
К 3 мл 3,4-Ю-4 М раствора указанного реагента в диметилсульфоксиде прибавляют 0,1 мл анализируемого раствора и измеряют интенсивность флуоресценции при 500 нм на спектрофлуориметре, возбуждая раствор светом с длиной волны 440 нм.
Метод позволяет определять >0,5 мкг GN- в 1 мл раствора; не мешают J~, Cl-, Br~, CIO-, GIO;, GlOj, JO3, SiO|“, SOV, SCN-, тартрат, BrOg, NO;, PO3', S2O3', цитрат, PO’*, F-.
J О;, AsOt, SOt, Fe(CN)J", GO^, HGO;, ВОГ. TeOg', GraOh Bi Oh Fe(GN)e“, S2- реагируют с образованием розовато-коричневого соеди-
107
нения, которое не влияет на флуоресценцию продукта реакции с CN-. (
Быстрый косвенный флуориметрический метод определения цианид-ионов предложен в работе [1051]. Он основан на измерении флуоресценции экстрагированного гексаном комплекса селена с 2,3-диаминонафталином (L), образующегося в количестве, эквивалентном содержанию CN'-ионов в растворе, по реакции
Pd2 (LSe) Cl4 + 8CN- 2Pd (CN)f + 4С1~ + 2LSe.
Мешают ионы Hg(II), S2'. Органическую фазу флуориметри-руют при 520 нм при возбуждении светом с длиной волны 377 нм.
Роданид-ионы
Спектрофотометрические методы определения роданид-ионов не столь многочисленны. Они либо основаны на использовании реакций с красителями, либо являются косвенными методами. Некоторые из них приведены ниже.
Реакции с красителями
Предложен экстракционно-фотометрический метод определения SCN'-ионов (и NOg), которые с кристаллическим фиолетовым (КФ) образуют ионные ассоциаты, хорошо экстрагируемые хлорбензолом из фосфатных буферных растворов [1359]. Экстракционное извлечение оптимально в интервале pH 2,5—10,5 (лучше всего при pH 5,8) и при соотношении КФ : SCN' = 25 : 1. Спектрофо-тометрирование экстракта проводят при 595 нм. Закон Бера выполняется для концентраций (0,8—8)-10~6 М SCN'. Ошибка определения 1—2%. Не мешают определению Cl', SO^, РО|~.
Для спектрофотометрического определения микроколичеств роданид-ионов используется реакция образования трехкомпонентного комплекса (с максимумом светопоглощения при 620 нм, молярный коэффициент погашения 1,4- 104), возникающего при реакции комплекса Hg(II) — метилтимоловый синий с SCN'-ионами при pH 6,9—7,4 [243, 1111]. Закон Бера выполняется при концентрации SCN' 0,1—2 мкг!мл.
При определении 58,5 мкг SGN' стандартное отклонение и коэффициент вариации оптической плотности составляют 0,0033 и 1,42% соответственно. Определению SGN' этим методом мешают многие ионы (CN~, S2', J~, Br“, Cl', NH4, Ni2+, Со2+, Bi3+, Cd2+, Mn2+, Cu2+, Zn2+, Al3+, Fe3+), которые необходимо предварительно отделять методом ионообменной хроматографии.
Метиленовый голубой и SGN'-ионы образуют комплекс состава (1 : 1), экстрагируемый дихлорэтаном [941] в кислой среде с получением окрашенного экстракта. Максимум оптической плотности при 675 нм. Закон Бера выполняется при концентрации SCN” 108
2-10“7—1 • Ю-s м. Определению мешают ^>100-кратные количества С1_, >4-кратные количества Вг~, CIO3, BrOg, NO3, JO7, NO2, равные количества J- и С1О4.
Косвенные методы
Для спектрофотометрического определения SCN “-ионов (а также NO3 и NO2) предложен метод, основанный на реакции замещения ими бромфенолового синего (БФС) в комплексе с тетра-фениларсонием (R4As+), растворенным в хлороформе [1253].
SCN“-hohh, содержащиеся в анализируемом растворе, при встряхивании с хлороформным раствором комплекса 2(R4As) с БФС вытесняют анион [БФС]2-, переходящий при этом в водную фазу в количестве, эквивалентном содержанию в ней определяемого аниона. Затем измеряют оптическую плотность водного раствора и по соответствующему калибровочному графику находят содержание SCN“ в анализируемом растворе. Значение pH анализируемого водного раствора перед встряхиванием с хлороформным раствором реагента в пределах 7,0—11,5 является оптимальным. Ошибка определения <^5%.
Предложены косвенные спектрофотометрический и атомно-абсорбционный методы, основанные на образовании роданидного комплекса меди, экстрагируемого хлороформом, и определении в полученном экстракте меди методом атомной абсорбции или спектрофотометрическим методом при 407 нм [639]. Чувствительность определения SCN- равна 0,05 мкг!'мл. Для повышения чувствительности определения рекомендуют выпарить экстракт досуха и остаток растворить в 25 мл этилацетата. Стандартное отклонение при значении оптической плотности 0,005 равно 1,87%. Присутствие 5 мг AsO4~, Вт-, СО|“, Cl~, NO3, 3 мг CIO3, 2 мг SO4”, С1О7, РО^-, 1 мг СгО4-, WO4~ не влияет на определение SCN-.
В работе [1097] предложен косвенный спектрофотометрический метод определения родяЬид-ионов с диэтилдитиокарбаминатом меди и ионом одновалентного серебра. Сущность метода заключается в осаждении AgSCN при добавлении известного избытка Ag+. После отфильтровывания осадка к фильтрату добавляют раствор диэтилдитиокарбамината меди в СС14 и по уменьшению окраски раствора (в результате реакции замещения меди в комплексе ионом Ag+) определяют концентрацию серебра, которую пересчитывают на содержание SCN-. Величина стандартного отклонения и коэффициента вариации для содержания 45,3 мкг SCN- составляет 3,7-10-3 и 0,63% соответственно.
Глава VII
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Электрохимические методы, используемые для определения соединений азота, представлены следующими вариантами вольтамперометрия (полярография) — прямая и инверсионная; кондуктометрия; кулонометрия — определение при контролируемом потенциале или контролируемом токе; хронопотенциометрия; потенциометрия — определение по величине потенциала электрода (в том числе с использованием ионоселективных электродов).
ПОЛЯРОГРАФИЧЕСКИЙ МЕТОД (ВОЛЬТАМПЕРОМЕТРИЯ)
Полярографический метод отличается высокой чувствительностью, экспрессностью, возможностью определения нескольких веществ в одном растворе. Имеются методы прямого и косвенного полярографического определения азотсодержащих веществ.
Аммоний
Из способов полярографического определения аммония перспективен метод, предложенный в [3531 и уточненный в [428]. Этот метод основан на образовании полярографически активных продуктов реакции между ионом аммония и избытком формальдегида. Он был использован для определения аммония в азотной кислоте. При избыточной и постоянной концентрации формальдегида, при постоянном значении pH раствора величина предельного тока зависит прямо пропорционально от концентрации аммония. Удовлетворительные результаты получены при содержаниях 6—15 ммоль/л.
В варианте осциллографической полярографии эта реакция была использована для определения сотых долей процента азота в стали и сплавах [421] с точностью 0,002%. Предложены два спо-
1 В настоящую главу не включены титриметрические методы с электрохимической регистрацией конечной точки титрования; о них см. соответствующий раздел в главе V «Титриметрические методы».
110
соба: с отгонкой и без отгонки аммиака. В последнем случае непосредственному полярографическому определению иона аммония предшествует введение в раствор ’лимоннокислого калия или комплексона III, которые сдвигают потенциалы восстановления ионов, мешающих этому определению элементов (Fe, Ст, Ni), в более отрицательную область.
0,05 г образца растворяют при нагревании в 1 мл H2SO4 (1 : 1) с добавлением нескольких капель перекиси водорода. Раствор переносят в мерную колбу емкостью 25 мл, прибавляют 10 мл iN раствора лимоннокислого калия или 0,4 N раствор комплексона III. Введением едкого натра pH смеси устанавливают равным 5,0. Затем добавляют 3 мл формалина и объем раствора доводят до метки дистиллированной водой. Из раствора отбирают аликвотную часть, равную 2 мл, и переносят в полярографическую ячейку. Полярографирование проводят при скорости развертки потенциала 0,25 в/сек в интервале напряжений (—0,5) -г- (—1,5) е. Снимают дифференциальные кривые. Содержание азота определяют методом добавок.
Для определения малых количеств азота, когда требуются большие навески и концентрации мешающих элементов в растворе высоки, используют метод с предварительной отгонкой аммиака.
0,2 г стружки стали растворяют при нагревании в H2SO4 (1 : 1) с добавлением Н2О2. Раствор помещают в сосуд для отгонки аммиака и добавляют щелочно-глицериновую смесь для создания сильнощелочной реакции. Присутствие глицерина ускоряет отгонку аммиака, понижая его растворимость. Аммиак отгоняют при пропускании через раствор тока водорода или какого-либо инертного газа, не содержащего кислород. Выделившийся аммиак через газоотводную трубку поступает в полярографическую ячейку, содержащую определенный объем 5%-ного формальдегида в универсальном буферном растворе с pH 5. Отгонка считается законченной, когда высота полярографического пика остается постоянной. Для ускорения отгонки рекомендуется раствор нагревать в сосуде с обратным холодильником.
Аммиак определяют количественно, используя анодное окисление ртути в растворе 0,1 М нитрата калия без буфера [1118]. Электродная реакция выражается уравнением
Hg + 2NH3 + 2Н2О = Hg (ОН)2 + 2NH* + 2е.
Предельный диффузионный ток прямо пропорционален концентрации аммиака в‘ диапазоне 1 • 10-4—1 • 10-3 М;' измерения проводят при +0,3 в отн. нас.к.э.
Полярографический метод, предложенный в [1113], основан на конденсации аммиака с фталевым альдегидом в карбонатном буфере с pH 10,4. При избытке фталевого альдегида не образуется никаких восстанавливаемых соединений, и в этом случае уменьшение тока прямо пропорционально количеству аммиака, находящегося в растворе. Метод дает хорошие результаты (ошибка 0,3—
111
3,6%) в диапазоне концентраций аммиака 10~3—10~5 М (или 0,2—20 мкг!мл полярографируемого раствора). /
Косвенный полярографический метод [6131 основан н4 осаждении аммония реактивом Несслера и последующем вольтамперометрическом определении ртути в осадке после превращения в тиосульфатный комплекс. Этот метод презназначен для высоких концентраций солей аммония в мутных и коллоидных растворах, в которых затруднено фотометрическое определение.
Гидроксилам ин
Гидроксиламин восстанавливается обратимо в двухэлектронном процессе в растворе с pH 6—7 [1430]. 0,05—1,0 ммоль гидроксиламина могут быть определены с обычной точностью, характерной для полярографического метода.
При анодном окислении гидроксиламина на платинированном-платиновом вращающемся электроде при pH 2,0 наблюдается хорошая линейная зависимость концентрация — предельный ток в диапазоне 1-10~®—5-10*4 М [1005].
Нитрат-ионы
Обзор методов полярографического определения нитрат-ио-нов по каталитической волне восстановления NO3 в присутствии ряда катионов и по участию в реакциях нитрования органических соединений см. в работе [976].
В нейтральных и щелочных растворах на ртутном катоде не наблюдается волны восстановления нитрата. Однако если в растворе присутствуют некоторые высокозаряженные ионы — лантана, уранила, молибдата, церия,— то потенциал восстановления нитрата сдвигается в более положительную область.
Таблица 11
Полярографическое определение нитратов [868, гл. 4]
Среда Катализатор Потенциал, в (отн. нас. к. э.) Диапазон концентра- ций NOg, ммоль Литература
0,ШНС14-0,14/ КС) 0,2Л/ UO2C12 -1,2 0,05—0,40 [944]
0,lMH2SO44- 4-0,2M Na2SO4 0,0875 ммоль Na2MoO4 — 0,2—50 [887]
0,054/ КСЮ44-0,004% желатина (pH 3,4— 7,5) 0,5—2,5 ммоль Се(Ш) —глицерин — 0,01-2,5 [798]
0,044/ LaC)3 Комплексные ионы -1,58 — [618]
0,1,1/ СеС13 — —1,60 — [618]
112
В работе [1350] рекомендуют использовать раствор хлорида лантана (потенциал восстановления —1,22 в), а в [856, 944, 1077] — уранил-ион для ускорения восстановления нитрата. Используют также каталитическое действие нитрата, вызывающее увеличение высоты второй волны при восстановлении ионов молибдена [887].
Для полярографического определения 0,02—1,0 ммоля нитрата успешно используют его реакцию с 2,6-ксиленолом (диметилфенолом) и получение полярографической волны восстановления продукта [806]. Полярографически определяют NOg в фосфорной кислоте и ее солях [349].
В табл. 11 приведены сведения о других полярографических методах определения нитрат-ионов.
Нитрит-ионы
Нитрит-ионы ведут себя в описанных выше методах аналогично нитратам, но их диффузионные константы различны. Количественно определить сумму нитрит- и нитрат-ионов можно измерением общего предельного диффузионного тока, за которым следует окисление в полярографической ячейке нитрита до нитрата перекисью водорода в кислом растворе и измерение возрастания диффузионного тока нитрата. Решение двух независимых уравнений позволяет вычислить содержание нитрита и нитрата [914].
Нитрит-ионы можно определять в присутствии нитрат-ионов вольтамперометрически с применением Pt-электрода. Для концентраций NO2 в пределах 10-в—10-3 М хорошо выраженная волна окисления NO2 получается при pH 0—8. При pH 3,5—8 потенциал полуволны не зависит от pH, причем на электродах протекает реакция NO7 NO2 + е; при pH 0—3 протекает реакция HNO2 NO2 + Н+ 4- е [1204].
В случае окисления NO2-ионов на вращающемся микродиско-вом Pt-аноде высота волны пропорциональна концентрации ионов; реакция необратима [186]. Описан модифицированный метод полярографического определения нитрита в присутствии восстановленной формы аскорбината [226] на фоне, содержащем комплекс Сг3+ с глицерином, при pH 6,0—7,0. Высота волны пропорциональна концентрации NO2 до 20 ч. на млн.
Достаточно быстрый (10 мин.) и точный (ошибка Порядка 1 %) косвенный полярографический метод определения нитрита основан на восстановлении 4-нитрозо-2,6-ксиленола, образующегося при реакции NO2 с 2,6-ксиленолом в смеси (5:4:1) H2SO4 с водой и уксусной кислотой. На этом фоне наблюдается четкая 'двух-электронная волна с Е^г — —0,05 в (отн. электрода Hg/HgSO4). Предельный ток, измеряемый при —0,15 в, пропорционален концентрации NO, в интервале 0—14 мкг/мл. 100-кратный избыток
ИЗ
NOg определению NO2 не мешает, так как 4-нитрозо-2,6Лксиленол образует волну с Е^г = —0,27 в [813]. Изучению вольтамперометрического поведения нитрит-ионов (наряду с другими анионами) в расплавах посвящена работа [187]. 1
Окислы азота
Было изучено поведение окислов азота (NO2, NO, N2O3hN2O) на капельном ртутном электроде в среде безводного диметилсульф-оксида [785]. NO2 образует две четкие необратимые волны восстановления с Ец2 — —1,06 и —1,53 в (отн. нас.к.э.). Предельный ток первой волны является диффузионным и пропорционален концентрации NO2. При концентрации NO2 5-10“3 М на обеих волнах появляются максимумы 1-го рода.
NO образует необратимую волну с Ft|2 — —1,44 в. N2O3 образует волну при —1,18 в. При анализе смеси NO2 и NO сначала определяют NO2 по волне с Еу2 = —1,06 в, затем NO окисляют до NO2 и вновь полярографирутот; содержание NO находят по разности. N2O дает необратимую волну восстановления при -2,22 в.
Роданид-ионы
Для определения роданид-ионов в элюатах хроматографических колонок малого диаметра (2—4 мм) был предложен полярографический метод [1358].
Главная часть полярографической ячейки представляла собой толстостенную капиллярную трубку диаметром 1,5 мм. Элюат из колонки поступал через шланг к капилляру, в вертикальный отросток которого был введен капилляр с капающим электродом. Ячейка была соединена через агар-агаровую пробку (раствор агар-агара в 0,2 М KNO3) и через раствор электролита (0,2 М KNO3) с выносным нас. к.э. в качестве электрода сравнения.
При использовании полярографа большой чувствительности этим методом удавалось определять концентрацию роданид-ионов в пределах 0,5—3 ммоля при линейной скорости потока элюата 20—210 см!час.
Цианид-ионы
Полярографическое восстановление цианид-ионов было исследовано методом осциллографической полярографии [835] на фоне 1 М KNO3 в водно-пиридиновом растворе (10% пиридина). В этих условиях на катодной ветви возникает зубец, высота которого пропорциональна концентрации ионов CN- в интервале 5— 50 мг!л. Зубец отвечает восстановлению цианида ртути, причем ртуть растворяется при анодной поляризации.
Следовые количества CN- можно концентрировать на висящей ртутной капле путем восстановления добавляемой в раствор Спа+ 114
в 0,1 7lfKNO3 при — 0,2 в (отн. нас.к.э.) [511]. Определение следовых количеств GN“ проводится далее методом инверсионной вольтамперометрии при окислении выделившейся на поверхности капли ртути пленки CuCN; высота пика при +0,17 в линейно зависит от концентрации CN~ до 10~6 М. При концентрации CN“ 5-10~7—1 • 10“6 М воспроизводимость определения ±4%. Определению мешают 10-кратный избыток Вг~ и J~, не мешают SCN-, С1~ и катионы металлов.
В работе [951] описан полярографический метод определения цианид-ионов в случае анализа сточных вод с наложением прямоугольного напряжения. HCN выделяют дистилляцией и поглощают раствором КаСО3 или КОН. Полярографируют относительно насыщенного каломельного электрода.
Было установлено [347], что в растворах комплексных соединений Ni(ll) с этилендиамином в присутствии цианид-ионов образуется каталитическая волна (при потенциале —1,8 в отн. нас.к.э.), высота которой пропорциональна концентрации GN“-ионов в интервале 10-7—10~в г-ион/л. Эта волна может быть использована для определения малых концентраций цианида (до 5-10~7 г-ион/л). Полярографические измерения проводили на полярогра-фе с ртутно-капельным электродом. Электродом сравнения служил насыщенный каломельный электрод. Электролитическая ячейка термостатировалась при 25° С. Измерения pH проводились на pH-метре ЛПУ-01.
При восстановлении гидроксиламинового комплекса никеля(П) на ртутном капельном электроде в аналогичных условиях [348] образуется двухэлектронная волна с Ец, = —1,2 в. В присутствии CN~-hohob появляется новая волна при —1,5 в, высота которой также зависит от концентрации цианид-ионов. Прямолинейная зависимость сохраняется в интервале 2,7-10-’—1,4-10“6 г-ион/л. Каталитически активный комплекс существует в ограниченной области pH (8—10). При одинаковых условиях каталитический эффект в данной системе выше, чем в системе Ni — этилендиамин — CN-. Определение CN“-hohob в чистых растворах проводилось при концентрации никеля 1,8-10-3 М и гидроксиламина 0,4 М при pH 8,9 (боратный буфер) на фоне 1 М NaCl.
Описана методика определения концентрации цианид-ионов при помощи двух короткозамкнутых электродов разных размеров, больший из которых является электродом сравнения [870]. Если потенциал поляризующегося электрода отвечает области предельного диффузионного тока электроактивного иона, то ток, возникающий в цепи, пропорционален концентрации деполяризатора.
115
КОНДУКТОМЕТРИЧЕСКИЙ МЕТОД
Аммиак
Кондуктометрический метод был успешно использован для определения аммиака в коксовом газе в присутствии углекислоты [489]. Метод основан на непрерывном измерении электропроводности раствора, поглощающего выделяющийся газ, который обрабатывают уксусной кислотой с целью подавления диссоциации угольной кислоты и уменьшения влияния ее на общую электропроводность.
Предложен селективный высокочувствительный детектор по электропроводности для детектирования азотсодержащих органических веществ, разделенных на компоненты методом газо-жидкостной хроматографии; метод основан на определении аммиака, образующегося при гидрировании веществ на Ni-катализаторе [627].
КУЛОНОМЕТРИЧЕСКИЙ МЕТОД
Аммиак
Кулонометрическое определение аммиака, основанное на окислении его гипобромитом (который образуется в растворе) до азота, было предметом многих исследований. Установлено, что при этом для получения воспроизводимых результатов необходимо точно регулировать pH раствора (8,5), иначе при определенных условиях аммиак окисляется не до молекулярного азота, а до закиси азота; кроме того, гипобромит может диспропорционировать до бромида и бромата. При высоких значениях pH аммиак может улетучиться.
Гидразин
В растворах сульфата аммония гидразин определяют автоматически кулонометрическим методом с контролируемым потенциалом [543]. Платиновый электрод предварительно подвергают анодной поляризации при 1,3 в отн. Hg/HgSO4 в течение 2 мин., а затем — катодной поляризации при —0,7 в в течение 5 мин. в 0,1 М H2SO4. Гидразин окисляют в ячейке при стабилизированном напряжении -Ц-0,41 в. Показания прибора записывают в виде функции тока от времени. При вычислении содержания гидразина вносят поправку на фон.
Гидроксиламин
Гидроксиламин может быть определен кулонометрически титрованием электрогенерированным бромом в кислой среде. При этом бром окисляет гидроксиламин до нитрат-иопа [1330].
116
Двуокись азота
При автоматическом контроле состава газовых смесей (выхлопных газов автомобилей) применяли кулонометрический метод определения двуокиси азота [383]. В этом методе NO2 реагировала с электрогенерированным бромом. Использовали электрохимическую ячейку, работающую в потенциостатическом режиме.
Предложена также методика кулонометрического определения >0,02 мг/л двуокиси азота в воздухе с использованием ячейки с непроточным электродом. Метод основан на реакции взаимодействия NO2 с NaBr и последующем восстановлении образовавшегося брома на платиновом (или графитовом) электроде кулонометрической ячейки. Электрод сравнения — активированный уголь марки Б. Электролит — 1%-ная H2SO4 + 3%-ный NaBr. Мешают определению H2S, SO2 (должны быть предварительно удалены).
ХРОНОПОТЕНЦИОМЕТРИЧЕСКИЙ МЕТОД 2
Предложен хронопотенциометрический метод количественного определения гидразина [488]. Для получения воспроизводимых результатов требуется точное соблюдение условий предварительной обработки электродов и проведения анализа.
Хронопотенциометрически определяли гидроксиламин (~ 1,4 ммоля) [643] или его смеси с гидразином (50 : 1) и (1 : 50) [1087] с точностью ±5 отн.%. Происходит ступенчатое окисление этих двух компонентов, наблюдаемое на хронопотенциограмме.
ИОНОСЕЛЕКТИВНЫЕ ЭЛЕКТРОДЫ
В последние годы все больше внимания уделяется использованию ионоселективных электродов при потенциометрических измерениях. В настоящее время применение ионоселективных электродов в аналитической практике многочисленно и разнообразно. Эти электроды используются для определения содержания десятков ионов при решении задач в самых различных областях науки и техники: от обычных научных исследований в химии до практического применения в сельском хозяйстве, геологии, океанографии, при контроле загрязнения воды и воздуха и т. п.
Существуют ионоселективные мембранные электроды различных типов — с мембранами из твердых и жидких ионообменников, а также с гетерогенными (осадочными) мембранами. Потенциометрическое определение нитрат-ионов и многих других до последнего времени не проводилось из-за отсутствия подходящих электродов 1-го и 2-го рода. Теперь появилась принципиальная возможность создания электродов, обратимых по отношению к лю
2 О потенциометрическом титровании см. соответствующий раздел в главе V «Титриметрические методы».
117
бому иону. Они позволяют с успехом определять общую концентрацию ионов в растворах.
О теории ионоселективных мембранных электродов, механизме их действия и применении в практике научных исследований и производственном контроле см. [112, 585].
Потенциометрические измерения с ионоселективными электродами обладают большими достоинствами: проводятся относительно быстро, время установления равновесного потенциала иногда составляет сотые доли секунды. При соответствующей коп-струкпии электродов можно анализировать очень малые объемы растворов (до сотых долей миллилитра), автоматизировать процесс измерения. Приборы для подобных измерений сравнительно просты по конструкции и недороги.
Однако существуют и трудности, связанные с проведением потенциометрических измерений ионоселективными электродами. Например, разработанные в настоящее время электроды не отличаются высокой точностью измерений, поэтому требуется периодическая проверка градуировки электрода.
В случае анализа растворов, содержащих соединения азота, применяются анионо- и катионоселективные электроды.
Аммоний
Для определения ионов аммония используются аммонийселек--тивные электроды как с твердой мембраной [626, 1420], так и с жидкой мембраной, содержащей антибиотик [1265]. Для потенциометрического определения используются также ионоселективные электроды со стеклянной мембраной. Так, в искусственных растворах определяли NH4 (и косвенно мочевину) с помощью электрода Бекман 39137. Относительная ошибка определения 10~3моль/л NHJ составляет 3 % [912]. Определяют энзимы по количеству NHJ, образующегося при ферментативном гидролизе соответствующей аминокислоты, с помощью стеклянного электрода Бекман 39047 или 39137 с погрешностью ±2,5%. Чувствительность ±20 мв при концентрации NHJ 10-4—10~5 М [789].
Аммонийселективный стеклянный электрод был применен при определении концентрации аммиака (10—1000 мкг/л) в водах, питающих паровые котлы [763]. Стандартные отклонения при определении 10, 100, 500 и 1000 мкг!л NH£ при помощи этого электрода соответственно составили 2, 7, 17 и 33 мкг/л NHJ. О быстром методе определения аммония в воде с применением стеклянного электрода см. [490].
Нитрат-ионы
Для определения нитрат-ионов используют анионоселективные жидкие мембраны. Так как жидкая фаза при этом находится в контакте с водными растворами, она должна быть нерастворима в воде
118
и иметь достаточно низкое давление паров, чтобы избежать интенсивного ее испарения. Этим требованиям удовлетворяют жидкие органические вещества, обладающие сравнительно большим молекулярным весом и низкими диэлектрическими проницаемо-стями. Электрод, селективный относительно данного иона, должен иметь активную группу ионита, которая обеспечивала бы требуемую селективность по отношению к интересующему иону и способна была бы к быстрому установлению ионообменного равновесия.
В работе [1270] предложена конструкция электрода для прямого потенциометрического определения нитрат-ионов. Концентрационный элемент, имеющийся в этой конструкции, состоит из двух полуэлементов, соединенных солевым мостом. Один из них заполнен ртутью, которая покрывает платиновую проволоку, затем слоем пасты Hg2(NO3)2-H2O и бензольным раствором алкиламмо-нийнитрата. Второй полуэлемент и мост заполнены бензольным раствором той же соли, но большей концентрации. Измеряют зависимость э.д.с. такого элемента от логарифма электропроводностей растворов, заполняющих полуэлементы.
Для определения нитрат-ионов применяют жидкие мембраны с активными группами — комплексами переходных металлов (Fe, Ni) с органическими лигандами, обладающими разветвленными углеродными цепями, содержащими о-фенантролиновую хелатную группу. Комплексные ионы этих металлов достаточно стабильны, и скорость катионного обмена переходного металла на поверхности мембраны настолько мала по сравнению со скоростью анионного обмена, что не оказывает заметного влияния на электродный потенциал. Например, для определения NO3 используют жидкую мембрану с ионитом, имеющим активную группу следующего строения: j
Коэффициенты селективности электродной функции имеют следующие значения для различных анионов: СЮ4 10®, J~ 20, С1О3 2, Вг- 0,9, S2- 0,57, NO2 6-Ю-2, GN’2.10-2, НСО3’2-10-2, С1-6-10-3, ОАс- 6-Ю-3, S20t 6-Ю-3, SO23~ 6-10-3, F- 9-Ю-4, SO2f 6-ЮЛ Н2РО; 3-10% ВОГ З Ю-4, НРОГ 8- 10-s [112, стр. 76].
Предложен потенциометрический метод с использованием системы жидкость—мембрана—жидкость для определения нитрат-
119
ионов (и других анионов) и мембранного электрода Орион 92-20, разделяющего 10%-ный раствор соли метилтрикаприламмония в 1-деканоле и анализируемый водный раствор. Предварительно электрод выдерживают в контакте с 0,1 М водным раствором определяемого аниона в течение 2—3 час. Электродная функпия линейна в интервале 10-1—10-3 М; воспроизводимость значений э.д.с. ±0,2 мв; время достижения равновесия от 5 мин. (10-3 М растворы) до ~1 сек. (10-1 М растворы). В интервале концентраций 10-1—10-5 М ошибка определения NO» составляет ±0,7 % [616].
В работе [1030] отмечается, что для определения концентрации нитрат-ионов по методу добавок в электрической цепи с нитрат-селективным мембранным электродом очень удобен в качестве электрода сравнения мембранный электрод, селективный к F--ионам. К анализируемому раствору добавляют постоянное количество NaF, измеряют потенциал нитратного мембранного электрода, затем добавляют из бюретки стандартный раствор NO3 до изменения э.д.с. на ~10 мв, делают второй замер и повторяют добавку до отклонения э.д.с. на ~15 мв, а затем на 20 мв. При концентрации NOg 10-2—10-4 г-ион/л ошибка составляет <Г1%.
О параметрах электродов, избирательно чувствительных к ни-трат-ионам, с мембраной из поливинилхлорида с введенными жидкими ионообменниками Корнинг 477316 или Орион 92-07-02 см. [642]. Для них электродная функция линейна в интервале концентраций NO3 от 10-1 до 10~4 М.
В работе [70] при определении до 10-3Л/ нитрат-ионов в качестве жидкой мембраны применены растворы нитрата нитрона в анилине, хлороформе, бензиловом спирте; этот селективный мембранный электрод с нитратной функцией может быть использован для определения нитратов в присутствии ряда посторонних электролитов, например KF, К3РО4, K2SO4 и др. Определение нитрата и нитрита в смеси с помощью нитратного электрода Орион 92-07 описано в работе [1083].
Нитратный электрод будет иметь большое значение в агротехнических анализах и контроле загрязнения окружающей среды. Он используется для определения нитрата в биологических материалах [477, 1060], удобрениях [737а], почвах [1020, 1094, 1192], воздухе [913, 977, 1277], продуктах сгорания топлива [666]; разработана методика определения концентрации нитрата в водных вытяжках высушенных стеблей растений со стандартным отклонением 3,1% [1155].
Исследование характеристик нитратного селективного электрода см. в [1182]; использование ионоселективного электрода для определения HNO3 в олеуме см. в [1211].
'Нитратный электрод можно использовать для определения нитрит-ионов в тех случаях, когда последние удается окислить до нитратов (прямые измерения нитрита не рекомендуются).
120
Метод прямого определения водорастворимого нитрата В почве 6 помощью электрода более удобен, чем стандартный колориметрический бруциновый метод 1654]. Он пригоден как для лабораторных, так и для полевых анализов, так как определения можно выполнять непосредственно в пульпах. При содержании NO3 (в пересчете на азот) в пробах (2 30) МО-4 % ошибка определе-
ния составляет ~5%. Для лабораторных определений стандартное отклонение составляет 12,5%, а при прямых определениях в пульпах ~25%.
Цианид-ионы
Цианидный электрод, который по существу является иодо-серебряным мембранным электродом, рекомендуется использовать при концентрации CN- от 10~3 до 10~5М, хотя он дает воспроизводимые результаты в пределах 10—10-6 М. Серьезное влияние на показания электрода оказывают только сульфид и иодид. Чтобы предотвратить снижение концентрации свободных ионов цианида в результате образования HCN, величина pH в измеряемых растворах должна быть >10.
Определение малых концентраций цианида с ионоселективным серебряным электродом выполнено в [707]. Этот электрод может быть использован для контроля отходов промышленности, измерения концентрации свободного цианида в гальванических растворах и для определения содержания цианидов в пищевых продуктах.
Для быстрого потенциометрического определения цианидов в растворах предложено использовать иодидселективный резиновый мембранный электрод, приготовленный из силиконового каучука и суспензии AgJ [380,821], с использованием как прямого, так и косвенного метода. При прямом определении С№ мембранный электрод предварительно выдерживают 2 часа в 0,1 М KCN и строят калибровочную кривую по стандартным растворам KCN. Чувствительность методики в этом случае 10-5 г-ион CN*. В случае косвенного определения CN~ анализируемый раствор титруют раствором AgNO3 с иодидным мембранным электродом в качестве индикаторного. Ошибка определения 10~4 М CN- составляет ±0,5%.
Косвенным методом с помощью Ag-селективного электрода (Орион 94-16) определяют малые количества CN~ (10~5—10 2 г-ион/л) [713].
Применение ионоселективных электродов для определения цианид-ионов описано также в работах [549, 706, 1036, 1041, 1159, 1288].
Роданид-ионы
Роданидный электрод относится к электродам с твердой мембраной и обладает ионной функцией до концентрации SGN~ ~ 10-s М. Электрод нельзя использовать в растворах, содержа
121
щих восстановители или вещества, которые образуют комплекс' ные соединения или труднорастворимые соли в используемой системе.
Показана возможность применения бромидселективного мембранного электрода для потенциометрического определения рода-нид-ионов [952]. Линейная зависимость между потенциалом и концентрацией SCN- сохраняется в области от 10'1 до 10~5 М. Не оказывают влияния на потенциал мембранного электрода F-, NO3, SOf, СОз~- Незначительное влияние оказывают ионы [Fe(CN)6]3- и [Fe(CN)e]4~.
Для определения роданид- и цианид-ионов используются электродные системы с твердыми мембранами на основе смесей соединений Ag2S и AgX (X — галогенид). Хлорид и бромид серебра являются соединениями с ионной проводимостью, в которых перенос заряда осуществляется ионами серебра. Мембрана из смеси AgSCN—Ag2S применяется для определения SCN~ (мешающие ионы: Вг_, J-, S2-, NHt и CN-), из смеси AgJ —Ag2S — для определения CN" (мешают J-, S2-) [112, стр. 84].
Электроды второго рода, состоящие из металла, покрытого слоем его труднорастворимой соли, были предложены для потенциометрического определения концентрации роданид- и цианид-ионов [1126]. В работе [31] для потенциометрического измерения концентрации цианид-ионов предложены индикаторные электроды — сульфиды металлов вместо соответствующих металлических электродов.
Глава VIII
ФИЗИЧЕСКИЕ МЕТОДЫ
В аналитической’ химии азота наряду с химическими используются также физические методы анализа, к которым относятся спектральные методы (оптические и рентгеноспектральные), масс-спектральные, методы изотопного разбавления, радиоактивацион-ные и радиохимические методы-, резонансные методы (ЭПР, ЯМР).
СПЕКТРАЛЬНЫЕ МЕТОДЫ
Для определения азота применяются методы как атомной, так и молекулярной спектроскопии, причем первые из них наиболее распространены. Методы атомного спектрального анализа основаны на излучении или поглощении света атомами азота. В оптических методах (эмиссионные, атомно-флуоресцентные, пламеннофотометрические, атомно-абсорбционные) регистрируются атомные спектры азота в видимой и УФ-областях. Рентгеноспектральные методы основаны на исследовании характеристического рентгеновского спектра (эмиссионный, флуоресцентный, микрорентгеноспектральный анализ).
Спектральные методы применяют главным образом для определения азота в металлах и сплавах, а также при анализе газовых смесей. При определении связанного азота (например, в форме нитридов и др.) необходима предварительная атомизация азота. Газовые смеси анализируются по молекулярным спектрам.
Данные о спектре азота в области от ПК- до вакуумной УФ-области приведены в [98, стр. 549]. Там же [98, стр. 757] указаны чувствительные линии (5679,6; 5676,0; 4110,0; 4103,4; 4099,9; 4097,ЗА). Наиболее чувствительной линией для азота является линия N II 3995,00 А.
В эмиссионных спектральных методах определения азота применяются различные типы источников света, основанных на газовом разряде: искровой разряд (с температурой в десятки тысяч градусов), дуговой разряд (4000—8000° С), пламенная фотометрия (1900-3100° С).
Обзор по спектрометрическим методам определения азота приведен в работе [1050].
123
Эмиссионный атомный спектральный анализ
Вакуумная спектроскопия. Азот является трудновозбудимым элементом, и его последние линии лежат в далеком или вакуумном ультрафиолете (до 2000 А)- Чувствительность прямого эмиссионного спектрального определения азота в вакуумной области возрастает по сравнению с близкой УФ- или видимой областями.
Таблица 12
Спектральные линии азота [99, стр. 246]
Длина волны, А Яркость * Ион Длина волны, А. Яркость * Ион Длина волны, A Яркость * Йон
1745,249 8 NI 1084,580 15 Nil 915,612 4 Nil
1742,724 15 NI 1084,562 5 Nil 775,965 20 Nil
1718,52 5 NIV 1083,990 7 Nil 772,385 3 N III
1494,668 10 N I 991,579 30 N III 771,901 2 N III
1492,812 2 NI 991,514 3 N III 771,544 1 N III
1492,615 20 NI 989,790 15 N III 765,140 30 N IV
1242,79 15 N V 955,335 3 N IV 764,357 10 NII1
1238,80 30 N V 924,274 5 NIV 763,340 5 N III
1200,710 10 N I 923,669 4 NIV 686,335 6 N III
1200,224 20 N I 923,211 15 N IV 685,816 30 N III
1199,550 30 N I 923,045 3 NIV 685,513 12 N III
1134,981 30 N I 922,507 4 NIV 684,996 6 N III
1134,415 20 N I 921,982 5 NIV 660,286 5 Nil
1134,166 10 N I 916,703 20 Nil 645,178 5 Nil
1085,701 30 Nil 916,015 8 N II 644,837 3 N II
1085,546 5 Nil 915,962 4 Nil 644,634 1 Nil
* Значения, полученные с использованием газоразрядной трубки.
В табл. 12 приведены резонансные линии азота в далекой УФ-области [99]. Использование этих чувствительных линий лимитируется сложностью их инструментального измерения.
В качестве примера можно указать определение азота в титане эмиссионным методом в вакуумной области спектра с использованием скользящей искры как источника возбуждения. Анализ выполняется по аналитическим линиям N IV 7651А и N V 1238,8А в области концентраций 0,003—0,004%. Минимально определяемая концентрация 3-10-3%. Метод регистрации спектра — фотографический.
Для определения азота в металлах применяется низковольтная искра, в которой легко возбуждаются линии N III, N IV. Технически последний способ возбуждения легче осуществить, чем скользящую искру. В работе [125] приведена схема генератора 124
низковольтных импульсов для возбуждения спектра элементов во всей вакуумной области спектра вплоть до рентгеновской области.
Искровой разряд. В спектральном анализе трудновозбудимого азота искровой разряд наиболее перспективен, так как дает наиболее высокую температуру. Быстрые процессы, происходящие на электродах в высоковольтном искровом разряде, и мгновенные спектры N I, N II, N III изучены с помощью спектрометра с временной разверткой в работе [1381].
Работы Скотникова и др. [307—310] посвящены использованию эмиссионного спектрального метода для определения азота в металлах и окислах металлов. Используется зависимость логарифма отношения интенсивности аналитической линии N II 3995 А к интенсивности фона, расположенного близ нее, от концентрации при различных условиях проведения анализа и объектов различной природы. Установлено, что оптимальными условиями для определения азота являются следующие: емкость искрового контура 300—400 мкф, индуктивность 5—10 мкгн, давление СО2 в разрядной камере 200—300 мм рт.ст. Сделаны попытки создать единые условия определения азота для широкого круга металлов (Cr, Ti, V) и их сплавов с использованием нестандартного генератора низковольтной искры и вакуумной камеры [308].
Для определения азота в окислах металлов [309] (WO3, TiO2, Nb2O5) необходимо обеспечить идентичное для всех образцов поступление материала пробы в плазму разряда и возбуждение спектральной линии азота N II 3995 А. Предполагается, что механизм поступления состоит в прямом испарении под воздействием плазменного нагрева.
В этом методе пробу окисла вводят путем набивки в калиброванное отверстие чистого по азоту металлического электрода (медного, пруткового) диаметром 5 мм с фульгуратором диаметром 0,3 мм на конце, глубиной 0,3 мм. Анализ проводят в стандартных условиях возбуждения в атмосфере СО2 и гелия с помощью искрового разряда. Эталоны приготовлены из нитридов промышленного производства.
Для определения азота в хроме [363] применяют спектрограф ИСП-51 (F =270лме).
Источником возбуждения служит низковольтная искра (емкость 400 мк$, индуктивность 9 мкгн, рабочий промежуток 0,35 мм, экспозиция 0,44 сек.). Вспомогательный электрод — вольфрамовый стержень диаметром 6 мм, заточенный на полусферу. Анализ проводят в герметичной камере (с остаточным давлением 1-Ю-1 мм рт. ст.), наполненной гелием при давлении 350 мм рт.ст. Аналитическая линия 3994,99 А, фон используется в качестве внутреннего стандарта. Освещение щели однолинзовое. Анализ проводят по методу трех эталонов. Аналитические графики строят в координатах AS — 1g С.
125
Интервалы определяемых концентраций 0,2—1,35% х. Воспроизводимость 10—30%. Метод может быть применен для определения азота в никелевом сплаве (индуктивность 27 мкгн, рабочий промежуток 0,75 мм, экспозиция 0,22 сек.). Воспроизводимость анализа 5—9%.
Скорость поступления примесей в разряд при спектральном определении азота в металлах и сплавах зависит от формы его нахождения, что требует выбора оптимального режима возбуждения спектра для получения воспроизводимых результатов при анализе различных материалов [39].
Предложен количественный спектральный метод анализа смеси Н2, СО и N2 с использованием искрового разряда [379]. Спектральный эмиссионный детектор для обнаружения ряда газов, в том числе N2, NO, NO2, NH3, описан в работе [544]. С его помощью возбуждается спектр газа-носителя (гелия) с примесями обнаруживаемых элементов. Спектр изучен в области 2000—4000 А. Предел обнаружения (в ч. на млн.): NH3 1,9; NO 0,4; NO2 0,3; N2 1.
Тлеющий разряд. В случае анализа газовых смесей используют молекулярные эмиссионные спектры испускания. При спектральном определении азота в гелии [358] используется аналитическая линия 3998 А. В случае анализа смесей Аг—N определяют азот в интервале концентраций 10~4—1% по полосе молекулярного спектра 3371 А, используя трубку с тлеющим разрядом, работающую при атмосферном или повышенном давлении [692, 693]. В области концентраций 2-10~1 2% среднее отклонение составляет 5-10~3%. Спектральное определение азота в аргоне и гелии в потоке при атмосферном давлении с чувствительностью 1-10-4— 1-10~*•% описано в [36а]. Коэффициент вариации 10%.
Дуговой разряд используется значительно реже. В работе [532] показана возможность использования аргоновой дуги большой силы тока для эмиссионного спектрального анализа, в частности, и для количественного определения примесей азота в аргоне.
Электрическая дуга постоянного тока (14,5 в, 80 а) поджигается высокочастотной искрой (50 кв) между катодом в виде заостренного стержня из вольфрама (с добавкой 1% окиси тория), омываемого аргоном (600 л/час\ и анодом в виде полой полусферы из меди, охлаждаемой водой. Расстояние между электродами 5 мм. Температура в центре дуги 26 700° К, на периферии 16 000° К.
Пламенная фотометрия. В работе [908] идентификация азота в газовой смеси проводится методом пламенной фотометрии в пламенах со щелочными металлами после выделения его методом газовой хроматографии. В газовом хроматографе используют детектор с двумя пламенами. Над нижним пламенем находится Pt-сетка
1 При определении 0,01—0,2% N емкость составляет 800 мк$, давление Не
300 мм рт. ст.
126
с нанесенными на нее солями щелочных элементов. Верхнее пламя помещают на оптической оси пламенного спектрофотометра Перкин — Эльмер (модель 202). Анализируемая газовая смесь поступает в нижнее пламя детектора и изменяет режим его горения, что вызывает изменение испарения солей щелочных элементов с Pt-сетки. Ионизация их в верхнем пламени способствует, в свою очередь, изменению тока ионизации и, следовательно, изменению интенсивности линий щелочного элемента. Таким образом косвенно определяют содержание азота. Введение паров щелочного элемента способствует увеличению чувствительности и специфичности определения азота.
Разработана методика определения 0,2% азота в гелии по интенсивности свечения в высокочастотном разряде, измеряемой с помощью пламенного фотометра [674]. В спектре азота обнаружено 11 пиков излучения, характеристическими из которых являются пики при 337 и 358 нм. Присутствие более 0,04% О2 мешает определению азота этим методом, и поэтому необходимо предварительное удаление избытка кислорода.
Абсорбционный спектральный анализ
Атомная абсорбция
В последние годы в практике спектрального анализа все большее распространение получает атомно-абсорбционный спектральный анализ, который в ряде случаев предпочтительнее эмиссионного метода анализа [185]. Он заключается в измерении поглощения в центре атомной линии при просвечивании паров.
В отличие от широких полос поглощения, наблюдаемых в молекулярной абсорбционной спектроскопии, поглощение света атомами от источника со сплошным спектром происходит в очень узких интервалах спектра, порядка сотых долей ангстрема. Поэтому атомное поглощение проявляется на спектрограммах в виде отдельных тонких линий. Атомно-абсорбционный метод более точен, чем эмиссионный и рентгеноспектральный. Чаще всего наиболее чувствительными в поглощении являются линии, соответствующие переходам в нижнее невозбужденное состояние. В качестве просвечивающих источников света используются лампы с полыми катодами, высокочастотные и парометаллические лампы.
Применение атомно-абсорбционного метода для спектрального анализа газовых смесей проще, чем в случае анализа остальных веществ. Но резонансные линии газов лежат в вакуумной УФ-области спектра, труднодоступной для измерения. Основным затруднением при измерении атомной абсорбции азота является необходимость эффективной диссоциации молекул (потенциал ионизации 14,53 в). Схема уровней энергии молекул азота приведена в [99, стр. 295].
127
В вакуумной УФ-облаСти есть возможность определять газы по спектрам поглощения. Изучению спектра поглощения азота посвящено большое число экспериментальных работ. Это объясняется тем, что азот является основной составной частью воздуха, и поэтому его поглощение влияет на состав излучения многих исследуемых объектов. Спектральные приборы должны быть откачаны до определенных давлений, чтобы поглощение излучения азотом воздуха не искажало яркости источника.
Для определения NO в атмосфере в присутствии паров воды используют специальную абсорбционную кювету [1342], представляющую собой цилиндр длиной 21 м и диаметром 0,76 м, в которой луч благодаря многократному отражению может проходить путь до 1 км и далее направляется в спектрометр. При длине пробега луча до 560 м возможно определение NO до 0,06 ч. на млн. Продолжительность анализа 10 мин., точность ~15%.
Спектр поглощения молекулярного азота можно разбить на 3 области: 1450-1000 А; 1000-796 А; 796-160 А [99, стр. 343]. Поглощение в области 1450—1000 А очень слабое, поэтому для фотографирования спектра поглощения при атмосферном давлении необходим слой азота толщиной в несколько сантиметров. В спектральной области 1000—796 А наблюдается сильное поглощение в отдельных полосах, а сплошное поглощение отсутствует. Длины волн абсорбционных полос азота (в А), полученные с использованием конденсированного разряда в аргоне, приведены ниже:
1450,4 1298,6 1185,2
1416,1 1273,4 1165,9
1384,0 1249,4 1147,6
1353,8 1226,9 1130,4
1325,4 1205,6 1114,2
С применением вакуумной УФ-области разработаны только методы определения азота в рудах, металлах и сплавах, а методы анализа газовых смесей не разработаны.
Анализ растворов на содержание ионов NH4, NO3, SCN-проводят косвенными атомно-абсорбционными методами. Так, при определении аммония используется возрастание поглощения света цирконием на линии 3601 А в присутствии азотсодержащих веществ [542]. Увеличение поглощения света (линейно зависящее от возрастания концентрации аммония) можно объяснить большей летучестью соединений циркония с азотсодержащим веществом. При определении 0,001—0,01 М аммония раствор должен содержать 0,02 М циркония в 0,1—3 М НС1. Стандартные растворы готовят на основе NH4C1. Для подавления ионизации пиркония в пламени в растворы добавляют КС1 (до 0,06 М). Влияние нитратов и сульфатов заметно при их 100-кратном избытке. Фосфаты необходимо удалять.
128
Нитрат-ионы определяют косвенным атомно-абсорбционным методом с помощью экстракции метилизобутилкетоном (при pH 3,5— 5,5) ионного ассоциата бис-неокупроината двухвалентной меди с нитрат-ионами [432, 9681. Экстракт распыляют в воз-душно-ацетиленовом пламени горелки и измеряют поглощение парами меди излучения разрядной трубки с полым Си-катодом (линия Си 3247 А). Интенсивность поглощения света экстрактом линейно зависит от концентрации NOgB исходном водном растворе в интервале 1 • 10~5 —7-10~5 М. Мешают равные количества CN-, Вг~, J~, GIO;, NO;, SCN~ и 10-кратные количества С1~.
Косвенный атомно-абсорбционный метод определения роданид-ионов основан на образовании роданидного комплекса меди, экстрагируемого хлороформом, и определении в полученном экстракте меди. Чувствительность определения SCN- 0,05 мкг!мл. Для повышения чувствительности определения рекомендуют выпарить экстракт досуха и остаток растворить в 25 мл этилацетата. Присутствие 5 мг AsO|“, Вг~, СО|~, СГ, NOg, 3 .мг СЮ;, 2 мг ЗОГ, СЮ4, РО®-, 1 мг СгО^- и WO2f" не влияет на определение SCN". Стандартное отклонение 1,87% при значении оптической плотности 0,005 [639].
Молекулярная абсорбция
К этой области относится УФ-, КР- и ИК-спектроскопия. Просвечивающими источниками света в этих методах анализа служат нагретые твердые тела при 1000—1500° С (ИК-спектроскопия) или газовый разряд (водородные лампы, УФ-спектроскопия).
Атомная флуоресценция. Безэлектродные разрядные трубки, возбуждаемые высокочастотным полем, используются как источники спектров для атомно-флуоресцентной и атомно-абсорбционной спектроскопии [637], в том числе и для определения азота наряду с большим числом других элементов.
УФ-спектроскопия. Поглощение аммиаком в УФ-области (при 204,3 нм и атмосферном давлении) было использовано для прямого определения низких его концентраций в воздухе [793]. Коэффициент погашения при этой длине волны составляет 3127 л!моль-см. Возможно определение 7—1000 ч. на млн. NH3 в воздухе.
Азотистоводородная кислота проявляется в УФ-области спектра, которая используется для прямых определений азида и NH3 в водных средах спектрофотометрическим методом [577]. Азотистоводородная кислота имеет максимум молярного коэффициента погашения при 260 нм (46,9 л/моль-см).
В поглощении света окислами азота в УФ-области существенную роль играет равновесие между двуокисью и четырехокисью азота. Количественные данные могут быть получены лишь при
5 В. Ф. Волынец, М. П. Волынец
129
тщательном контроле за температурой и давлением [797]. В работе [1088] предложен спектрофотометрический метод, в котором двуокись азота определяется при 394 нм, четырехокись — при 240 нм, их смесь — при 353 нм.
С помощью УФ-спектрофотометрии проводят экспресс-анализ окислов азота в производстве азотной кислоты с достаточно высокой точностью при небольшом числе измерений на простых и точных приборах [283].
Для определения нитрат-ионов в разбавленных растворах измеряют поглощение в области 200—230 нм [1365]. В присутствии COg“, NO2 измерения проводят при 220 нм и pH 1—3. В некоторых случаях поглощение измеряют при 275 нм, чтобы избежать влияния органических компонентов. Добавление к раствору, содержащему нитрат, концентрированной серной кислоты, так же как и присутствие хлорида, вызывает смещение максимума до 230 нм.
УФ-спектроскопическое определение нитрит-ионов описано в работах [171, 1365, 1390]. В отсутствие NO3 ионы NO2 опреде--ляют при 220—230 нм и pH 6.
Предложен быстрый метод дифференциально-спектрофотометрического определения больших концентраций NO2 по светопо-глощению при-355 ил/ [171]. Закон Бера соблюдается при концентрации NOj в анализируемом растворе до 7,5 мг!мл. Определению-не мешают 10-кратные количества NOg.
Метод комбинационного рассеяния использован для определения NO3, SO|", РО1" в воде в пределах 50—75 мг!л [479]. Спектр возбуждают излучением аргонового лазера и записывают на монохроматоре Спекса (модель 1401).
ИК-спектроскопия. ИК-спектры ряда неорганических веществ, содержащих в своем составе многие анионы, в том числе SCN", NO2, NO3, CN-, изучены в работе [329].
Все неорганические нитраты обнаруживают характеристические полосы поглощения в интервалах 1380—1350 см'1 и 840— 815 см"1 [21]. Нитриты также имеют полосы поглощения от слабой до средней интенсивности в области 840—815 см'1, но их легко отличить от нитратов по наличию сильного поглощения в области 1250—1230 см'1.
Для солей аммония найдены две характеристические полосы поглощения. Одна из них соответствует валентным колебаниям N—Н и проявляется в области 3200—3100 см'1, тогда как другая, также интенсивная полоса, находится в области 1430—1390 см'1.
Нитрат аммония имеет 3 полосы поглощения с волновыми числами 3160, 3060 и 3030 см'1. Полоса поглощения с частотой 1410 см'1 довольно интенсивна. Цианиды могут быть идентифицированы по поглощению в области 2200—2000 см'1. Роданиды также поглощают в области 2090—2020 см'1.
130
Частоты колебаний молекул азота , цианидов, нитратов, окислов азота, гидразина и гидроксиламина приведены также в [228, стр. 106, 109, 132, 143, 145].
Ионы аммония обнаружены с помощью ИК-спектроскопии при реакциях термического разложения парамолибдата и паравольфрамата (UR-10) [929]. Содержание NH$ в сульфате гидроксиламина предложено определять по измерению интенсивности полос поглощения при 1400 см'1. Поглощение обусловлено деформационными колебаниями связи N—Н.
К пробе (10 мг) прибавляют 390 мг КВт, смесь размалывают и 300 мг прессуют под вакуумом в таблетку. Таблетку сразу же помещают в спектрофотометр Перкин — Эльмер IR-8. Интенсивность пучка сравнения регистрируют таким образом, чтобы минимальная оптическая плотность при 1850 см~х составляла 0,1—0,2; спектр записывают в интервале 1200—2000 см-1. Следы NO“, обусловливающие появление слабой полосы при 1390 с-и-1, не мешают. Оптическая плотность линейно зависит от концентрации NH*. Чувствитель. ность 0,1% NHJ.
Закись азота имеет три основные характеристические полосы поглощения, одна из которых при 2224 см"1 является специфичной и не может быть приписана другим газам. Эта полоса была эффективно использована при количественных определениях закиси азота во многих газовых смесях [1241].
ИК-спектр окиси азота имеет несколько полос, которые используются при анализе (1850 и 1925 см'1) [1164]. Точность определений ±3 %.
Двуокись (и четырехокись) азота имеет набор специфических инфракрасных полос [1164]; количественные определения обычно проводят, используя поглощение при 1625 см'1. Содержание NO2 при концентрациях порядка нескольких частей на миллион определяют при помощи бездисперсионного инфракрасного анализа [1163].
Газы, выделяющиеся из кипящей царской водки до и после добавления к ней нержавеющей стали (в том числе N2O, NO, NO2, NOCI, HNO3, N2), определены после выделения с помощью газовой хроматографии методом инфракрасной спектрофотометрии на приборе Перкин — Эльмер модели 21 с призмой из NaCl [1136].
Водные растворы нитратов и нитритов также могут быть проанализированы с помощью ИК-спектроскопии [1018, 1363] при концентрации 10—50 мг/мл. Ошибка определения ~3%. Определению не мешают катионы металловДно мешают большинство органических соединений, бораты, карбонаты, фосфаты, перхлораты, аммонийные соли, поглощающие в той же области длин волн.
5*
131
В работе [1018] определение нитрат-ионов проводят с предварительным отделением NO3~ осаждением в виде азотнокислой соли нитрона. Далее используют технику прессования в диски. Спектры поглощения регистрируют и по градуировочному графику в координатах 1g — количество NO3 определяют содержание нитрата со средней квадратичной ошибкой ±5%.
Анализ неорганических нитратов методом ИК-спектроскопии с использованием техники КВг-таблеток выполнен в работе [602]. Спектр записывали на спектрометре Перкин— Эльмер модели 621. В качестве аналитических частот для анализа NaNO3, KNO3, Ba(NO3)2, Sr(NO3)2 выбраны соответственно 836, 825, 730 и 737 ел-1. При исследовании смесей NaNO3 и KNO3 использовались аналитические полосы 836 и 825 см-1', при определении Ba(NO3)2 и Sr(NO3)2 — 730 и 737 см-1 соответственно. Погрешность определения ~2% (при соотношении компонентов 1:20). Присутствие сульфата не мешает.
Для определения азота в нитроцеллюлозе (—12—13,6%) методом ИК-спектрометрии использовали полосу поглощения при 1666 см-1 [1002].
О применении спектральных методов для определения изо-тоцного состава азота см. стр. 142 — 144.
РЕНТГЕНОСПЕКТРАЛЬНЫЕ МЕТОДЫ
Анализ рентгеноспектральным методом осуществляется по линиям характеристического рентгеновского спектра. Анализ может быть проведен без разложения или разрушения образца, что является большим преимуществом метода.
Рентгеноспектральный метод используется для объектов со средним и высоким содержанием азота.
Хорошими информационными возможностями характеризуется электронно-зондовый микроанализ [1025]. Он основан на взаимодействии высокоэнергетического электронного пучка с веществом, приводящем к возникновению рентгеновского характеристического спектра атомов. Электронный пучок диаметром до 1 мкм не изменяет пробы. В работе [1388] подробно описаны принцип действия и устройство стандартного микроанализатора с электронным зондом типа АМХ. Точность локального анализа ~1-2%.
С помощью рентгеновского микроанализа с использованием электронного зонда обнаружены примеси 00,1 %) легких элементов (в том числе и азота) в стали [302]. Определению легких элементов (и азота) с помощью электронного микрозонда в стали и чугуне посвящены работы [301, 971].
Рассмотрены преимущества и недостатки двух способов диспергирования рентгеновских лучей при определении легких элементов (В, С, N, О) — кристального и бескристального рент-132
геновского микроанализа [924]. Бескристальный анализ позволяет определять каждый из этих элементов с большей чувствительностью, но исключает возможность определения двух соседних элементов при их одновременном присутствии.
Содержание легких элементов, в том числе и азота, можно определить методом рентгеновского анализа с помощью многослойных пленочных анализаторов [345, 398] с достаточно высокой чувствительностью.
В работе [970] описана аппаратура и приемы рентгеноспектрального анализа; рассмотрено влияние основы, обусловленное поглощением и третичным возбуждением; указаны пути подавления или учета влияния посторонних элементов.
Определены параметры распределения импульсов азота по экспериментально найденному распределению' импульсов BN [1279].
Рентгенофазовым методом [8] проведен количественный анализ смешанных удобрений. В число определяемых компонентов входят NOg, NHj. Внутренним стандартом служит шпинель.
МАСС-СПЕКТР АЛЬНЫЕ МЕТОДЫ
В аналитической химии азота существуют две основные области, в которых используется масс-спектрометрия: определение содержания азота в конкретных объектах и измерение его изотопного состава.
Изотопный анализ
Масс-спектрометрия является в настоящее время одним из основных методов определения изотопного состава азота, а для необогащенных образцов с природной распространенностью 15N — это единственный метод. (Определение изотопного состава оптическими методами, активационным анализом, ЯМР и методами газовой хроматографии см. в соответствующих разделах.) Перед масс-спектральным анализом азот образца переводится в измеряемую газообразную форму. Наиболее распространенной формой для изотопного измерения азота на масс-спектрометре является N2 (табл. 13). Для газообразных образцов вся подготовка сводится к очистке N2 от примесей, мешающих определению. Обычно зто окись углерода, метан, вода, кислород и двуокись углерода [699]. Другими газообразными формами, используемыми для изотопных измерений, служат окись азота и атомарный азот. Азот аммонийных, нитратных, нитритных и органических соединений в общем случае переводится в N2 либо по методу Дюма [1188], либо по методу Риттенберга окислением аммонийного азота гипобромитом натрия [1309]. Окисленные формы азота предварительно восстанавливаются до аммиака сплавом Деварда. При использовании последних методов возможно достижение точности изотопного определения +0,01 % [1278].
133
Таблица 13
Масс-спектрометрическое определение изотопного состава азота в объектах
Литература 1- ОТ от S rS °°гм г— Г- чн о „ ю от £ Ьг о о о оз од од -ч-ч Ю «чН О <2 Ю Ю тн СО X? -чН «н от — —1 й —1 — — —От — — —
Примечание Разложение в электрическом тлеющем разряде Химическая ионизация Метод Дюма То же
I Точность, отн. % 2 11 0,5 3 0,5 0,05 0,05 0,05 0,03 о; 5 0,01 1
Предел обнаружения И z й г г г г s а г « г 1 w 3 1 w ® 1 1 1 1 й « II 1 55 1 7 1 ю . . о- 1 sf ОД од
Мешающие ионы или молекулы ± о +» 8 и .О « и 3? +ч* +ч* * +'1' +—+*+<* 3 « © Мй !=«% % tg,ww _ +-и О О Щ О и О и и ООО 4* О , * л ” - »• Г- - - - * - г- - « <О + + + Ml + ГЧ +++ + + + + И 3?5 о о М о Z о о о о ооо О КО ООО OS О О О О ООО
ё Ю ОТ ОТ От ОТ ОТ^НОТОТООТ от от от оз го од ем од од од од СО СО со СО СО Ф СО СО СО 00 ОС ОС ОС -гЧ^ОДОДОД ем со од од од од од од од
Объект исследования »• 1 1 Ф 1 II л~> го lh*f6 о S S« S S S 3 « £ §« £ а И = « И О S m o3=f gf«S д- зыйз « §S Л'и o-S ! sfS S shH -£3 £ £g §§§ S. a sgsies;SggSz; ° g | g | 1|? й® и«” - § 1 s K- § g s § g § I S. g g. 3 «Jig g я g. Й 5 1-:Ог“9ОиОгрККй ЩСьннЩиянОС'Б
Соединение азота, вводимое в ионный источник г к £ g £ г г
134
Особенности аппаратуры И методик для перевода аммонййноГо азота образца в молекулярный приведены в работах [148, 481, 1046, 1260]. Помимо отгонки аммиака с паром предлагается метод диффузионного поглощения аммиака раствором серной кислоты в замкнутом объеме при подщелачивании исходных растворов [1046].
Изотопный состав аммонийного азота может быть определен и без перевода в N2, прямо в виде иона NHJ при химической ионизации [1010]. Азот из HNO3 и смеси окислов азота переводят в N2 либо разложением в электрическом разряде, либо термическим разложением в присутствии металлической меди при 800—900° С [1156]. Количества, необходимые для анализа, представлены в табл. 13. В случае анализа образцов, обогащенных по 15N, требуется меньшая точность анализа и меньшее количество образца, измеряемое микрограммами. Для измерения проб с природной распространенностью необходима точность 0,01 — 0,05 отн. %. При использовании компенсационного метода измерения на одно измерение. требуется 0,5—1 мг N.
Определение 15N в биологических материалах при медицинских и биологических экспериментах подробно описано в работах [515, 624, 731]. Об использовании масс-спектральных методов для определения азота методом изотопного разбавления см. стр. 142.
Элементный анализ
Достоинством масс-спектрального метода является сочетание в нем возможности разделения компонентов смеси в результате электромагнитной сепарации с количественным измерением химического состава путем измерения ионных токов отдельных ионов. Этот метод характеризуется большой чувствительностью и производительностью. Он позволяет измерять небольшие содержания азота в газовых смесях без предварительного обогащения (табл. 14).
Как правило, и для элементного, и для изотопного анализа применяют одинаковые методы выделения азота из образцов и методы его определения, за исключением определения азота в металлах искровой масс-спектрометрией.
АКТИВАЦИОННЫЙ АНАЛИЗ
Одним из самых чувствительных методов определения малых количеств азота является активационный анализ. Этим методом удается определять до 10~4—10-5% азота в металлах и газах. Определение азота происходит часто без разрушения образца.
Сложность определения азота с помощью радиоактивационного метода по п,у-реакции на тепловых нейтронах состоит в том, что реакция имеет малое сечение, а радиоактивные продукты возникают из малораспространенных изотопов и обладают малым
135
Таблица 14
Масс-спектральное определение содержания азота в различных объектах
Соединение азота, вводимое в ионный источник Объект исследования т|е Мешающие ионы или молекулы Предел обнаружения Точность, отн. % Примечание Литература
N2 Атмосфера Земли Атмосфера [космического корабля Газы кислородных конверторов Газы включений в стекле 28 28 — 105 частиц/см3 10-7 см3 1 Прибор работает в автоматическом режиме [1306] [1234], [615]! [696]'
Остаточные газы вакуумных систем Смесь СО + N2 — Парциальное давление N2 до 10-10 мм рт. ст. 2-3 Окисление СО происходит на катализаторе Шютце [864,. 1202] [1021]!
Примесь N2 в газовых смесях 28 — 1-10-2 объемн. % — — [641]
Примесь N2b чистом О2 28 — 1-10-3 объемн. % — Концентрируют из пробы 50 см3 над металлической медью [331]
|Примесь N3 в аргоне и водороде Примесь N3 в серебрен олове 28 28 — 1-10-2 объемн. 0,8 -10~2 мкг % 10 10 Прибор с высоким разрешением (1 : 2500); разделяются мультиплеты СО+, N2+, Сан,,4- с вакуумным плавлением [409]' [191, 622];
Таблица 14 (окончание)
Соединение азота, вводимое в ионный источник Объект исследования т|е Мешающие ионы или молекулы Предел обнаружения Точность, отн. % Примечание Литература
N Примесь N в аргоне и 14 8-10-4 объемн. % 3,5 Концентрация опреде- [938]
гелии ляется по калибровочной кривой
Примесь N в металлах 14 56Fq4+ 2-10-4 вес. % . 20 Прибор высокого раз- [64, 551,
решения с искровым 953,
источником; разделяется мультиплет 56Fe4+ и 1123]
14N+
NO Органическое вещество 30 — 1-10-4 вес. % — Окисление органического вещества в токе О2 [1367]
N2O Атмосфера 44 со2+, ch4n2+, с2н4о+, c2h6n+, 1-10-3 см3 20 — [1295]
ь с3н8+
Почва 44 — 3-10-4 см3 10 — [1344]
NO, N2O, Смесь окислов азота в 30 — 0,01 ат. % Требуется предвари- [722—
no2 атмосфере; азотная кис- 44 — тельная обработка ва- 724]
лота 46 С16О18О+, CHSN2+, 1—4 куумной системы масс-
С2Н6О+, c2h8n+ спектрометра парами no2
периодом полураспада. Поэтому для определения содержания азота используются ядерные реакции на быстрых нейтронах, ^-квантах и заряженных частицах (протонах, дейтронах).
Обзор по применению активационного анализа для определения ряда элементов, в том числе и азота, приведен в работах [246, 445].
Инструментальные методы
Активация нейтронами. В методе активации быстрыми нейтронами используется ядерная реакция 14N (и, 2ra)13N. Исследуемый образец облучают на нейтронном генераторе потоком нейтронов (107—109 нейтр/см1 • сек) с энергией 14—14,5 Мэв. Регистрируется аннигиляционное у-излучение (энергия 0,511 Мэв) образующегося радиоизотопа 13N (Тц, = 10 мин.) , например, на многоканальном сцинтилляционном у-спектрометре с кристаллом NaJ(Tl) [230, 459]. Анализ занимает 10—20 мин. Относительная ошибка метода ~2—5%. Точность и чувствительность метода могут быть повышены вращением ампулы с -пробой вокруг двух взаимно перпендикулярных осей при облучении нейтронами и тщательным подбором стандарта сравнения [1272]. В качестве внутреннего стандарта используют Си, Al, SrCO3 [114, 396].
Нейтронно-активационный метод используют при определении азота в органических материалах [416, 1348], в каменном угле и смоле [36], в образцах нефти [200, 790], в металлах [29, 164, 711], в газах [46], взрывчатых веществах [1272].
Влияние насыпной плотности пробы на определение азота методом активации быстрыми нейтронами изучено в работе [459] при облучении нитратов лантана, натрия, никеля, бария, свинца. В качестве стандарта сравнения использовались тиомочевина с насыпным весом 1 г/см9. Установлено, что удельная активность 13N в различных солях, облученных в одинаковых условиях, уменьшается с ростом насыпной плотности пробы.
Для нейтронно-активационного определения легких элементов в различных материалах без разложения образца применен портативный источник быстрых нейтронов с энергией 14 Мэв, состоящий из высоковольтного нейтронного генератора, позволяющего получать поток 108 нейтр/сек [1140]. В работе [249] показана возможность определения некоторых химических элементов, в том числе и азота, по у-излучению, сопровождающему нейтронный захват с использованием нейтронных источников с низким потоком нейтронов (Ra—Ве-источник, 50—250 мкюри) и сцинтилляционного у-спектрометра.
Активация протонами происходит по ядерной реакции 14N (р, а) ПС (Т»|2 = 20 мин.) на циклотроне с энергией протонов 18 Мэв [189, 190]. Возникающая позитронная активность может регистрироваться спектрометром у—у-совпадений. Мешающей определению азота реакций является ИВ (р, п) иС. Чувствительность определения азота в тантале этим методом достигает 1 • 10-в %,
138
Активация дейтронами. Анализ путем активации ускоренными дейтронами по ядерной реакции 14N (d, п) 15О при энергии дейтронов до 3,2 Мэв позволяет определить 5-Ю”11 г N [1414].
Активация ионами 3Не. Теоретические основы метода изложены в работе [1208]. Этот способ позволяет определять азот с большей чувствительностью, чем нейтронно-активационный метод. При активации пучком ионов 3Не (ток 0,005—0,01 мка) с энергией 5—18 Мэв удается определять азот до концентрации 1 ч. на млн. [1207] по реакции 14N(3He, a)13N. Определение проводится по короткоживущим радиоактивным изотопам у-спектромет-рическим методом без химического разрушения пробы.
При активационном анализе по очень короткоживущим изотопам (например, 17N с Тц, = 4,16 сек.) измеряют активность запаздывающих нейтронов с использованием «челночного» контейнера пневмопочты (быстродействующая пневмопочта) [456].
Активация у-квантами. Метод отличается большой простотой и экспрессностью выполнения. В некоторых случаях может выполняться без химического разрушения пробы. Используется ядер-ная реакция 14N (у, п) 13N. Чувствительность определения азота 1 ч. на млн. [446].
При использовании у-излучения с максимальной энергией 28 Мэв удается количественно определять азот с чувствительностью 10“2—10-3 ч. на млн. [677]. Метод особенно полезен при анализе материалов высокой чистоты. Например, в редких металлах и полупроводниковых материалах у-активационным методом определяют азот с чувствительностью 10"4—2-10~5% [275].
Об использовании радиоактивационного метода для определения азота в металлах высокой чистоты см. также главу XII.
Недостатком прямых инструментальных методов является небольшая селективность в связи с тем, что при облучении элементов с Z = 4 -г- 20 (от Be до Са) образуются радиоизотопы, обладающие аннигиляционным у-излучением. Это затруднение может быть устранено использованием ЭВМ для анализа графиков распада аннигиляционной активности, а также применением в ряде случаев радиохимических методов.
Радиохимические методы
Химические методы выделения азота после облучения используются редко.
Активационное определение азота (наряду с углеродом) в арсениде галлия, кремния, титана с применением радиохимического выделения описано в работе [101].
Анализируемые образцы облучают тормозным у-излучением с максимальной энергией 25 Мэв, получаемым на линейном ускорителе электронов, или потоком нейтронов с энергией 14 Мэв от нейтронного генератора. При определении азота в кремнии и германии облученную пробу сплавляют при
139
900°—1000° С с NaOH в присутствий нитрида кремния в качестве неактивного носителя. Выделяющийся аммиак поглощают 0,1 N раствором H2SO4, осаждают NH* в виде тетрафенилбората и в осадке измеряют активность 13N на спектрометре у—у-совпадений. Химический выход азота равен 90—95% .
При определении азота в титане облученную пробу протравливают смесью НС1 и HF (1 : 1), растворяют при слабом нагревании в смеси НС1 и HF (10 : 1) в присутствии 5 мг носителя (в виде нитрида алюминия или NH4C1), раствор подщелачивают, отгоняют аммиак с водяным паром, поглощают его 0,1 jV H2SO4, осаждают тетрафенилборат аммония и измеряют в нем активность 13N-
Чувствительность определения азота по (у, п)-реакции составляет 5 • 10"5 %, чувствительность нейтронно-активационного определения азота равна 2-10’4%.
В упоминавшемся выше методе определения азота на быстрых нейтронах [230] по измерению активности образовавшегося изотопа 13N определению мешает медь. В ее присутствии предварительно выделяют 13N (после облучения в течение 40 мин.) отгонкой в виде NH3, затем осаждают (NH4)2PtGle и измеряют активность осадка. Чувствительность определения при этом снижается в 10 раз по сравнению с прямым инструментальным определением.
При определении азота активационным методом по ядерной реакции 14N (d, п) 15О [1047] разработана методика экспрессного радиохимического выделения 15О из облученной пробы с использованием восстановительной плавки. После облучения пробу быстро протравливают, плавят в графитовом тигле с добавлением соответствующего плавня или без него, образующийся СО окисляют до СО2, поглощают СО2 аскаритом и измеряют его радиоактивность. Продолжительность выделения 2 мин., химический выход 15О равен 70%. Чувствительность определения азота 1 ч. на блн.
Известны косвенные радиометрические методы, используемые для определения азота и его соединений.
Метод отражения p-излучения применен для определения 0,03—0,18 г NHt с максимальным отклонением 0,0016 г [1035]. Метод основан на осаждении при 0° С NHt при помощи избытка Na2Pb[M(NO2)e] (где М = Cu2+, Ni2+, или Со2+) и получении кристаллов (NH4)2Pb[M(NO2)e]. Образовавшийся осадок отделяют фильтрованием и измеряют интенсивность отраженного от фильтра p-излучения источника 2C4T1NO3 активностью 1—10 лгкю-ри при помощи двух параллельно соединенных счетчиков Гейгера — Мюллера типа СТС-6 или сцинтилляционного счетчика «Numedit-714». Источник 2J4T1NO3 располагают на расстоянии 1,2—1,5 см от поверхности фильтра. Содержание NHJ (Сх) находят по формуле
Сх = [С4 (Л - 1Х) 4- (Ix - / j] / (Iz-Ij,
где Cj и С2 - концентрации двух эталонных растворов; Ix, Ilt 140
I2 — интенсивности отраженного излучения анализируемой и эталонных проб соответственно.
Метод поглощения 0-излучения использован в работе [1034] для косвенного радиометрического определения ионов аммония в солянокислых растворах. После осаждения NH4 способом, указанным выше, осадок отфильтровывают, фильтрат переносят в цилиндрическую плексигласовую кювету с толщиной дна 0,03, диаметром 5,5 и высотой 1,3 см\ устанавливают кювету под радиоактивном источником 204Т1 (2 мкюри) и регистрируют прошедшее через раствор 0-излучение при помощи счетчика Гейгера —Мюллера типа GTG-6, расположенного на расстоянии 1,5 см над поверхностью раствора. По степени поглощения 0-излучения раствором рассчитывают содержание NHJ в пробе с помощью калибровочного графика.
По другому варианту метода отфильтрованный осадок растворяют в HNO3, раствор разбавляют водой до 25 мл и измеряют поглощение в нем 0-излучения 204Т1. Получено хорошее совпадение результатов определения NHJ обоими методами.
Описан простой радиохимический метод косвенного определения цианидов (0,5 мкг — 10 мг), основанный на обработке исследуемым раствором осадка AgJ, меченного радиоизотопом 1KmAg [553].
Анализируемый раствор пропускают через колонку, наполненную AgJ, меченным радиоактивным изотопом (активность 0,01 мккюри). В фильтрате измеряют активность no^Ag по фотопику с энергией 0,66 Мэв на сцинтилляционном у-спектрометре с кристаллом NaJ. Вводят поправку на холостую пробу и рассчитывают содержание CN- в пробе с помощью калибровочного графика. Определению мешают 82Од_-ионы, почти не мешают SCN-, [Fe(CN)e]4-, [Fe (CN)6]3-.
Метод газового анализа, основанного на ослаблении амплитуды импульсов в счетчике Гейгера, описан в работе [1405]. Исследованием влияния примесей в газе, которым заполняют счетчики Гейгера (смесь 1,3% бутана ± 98,7% гелия) на амплитуду импульсов показано, что ее уменьшение находится в линейной зависимости от количества примеси (в том числе и азота). Это явление использовано для анализа газов. Если возможно определение уменьшения амплитуды импульса с точностью до 1 в, то можно определить азот с ошибкой ±4,4% в бинарной смеси с СН4. Калибровочный график имеет линейный характер.
ИЗОТОПНОЕ РАЗБАВЛЕНИЕ СТАБИЛЬНЫМИ ИЗОТОПАМИ
Метод изотопного разбавления осуществляется с применением элементов или соединений, меченных стабильными изотопами. Основы метода и техника работы описаны ь [263а, 1352а]. При
141
определении азота стабильным индикатором является изотоп iSN.
Общая схема проведения анализа изотопным разбавлением стабильными изотопами состоит во взвешивании и растворении анализируемого образца; добавлении к раствору известного количества обогащенного изотопом-индикатором компонента; уравновешивании индикатора с образцом; выделении определяемого элемента, представляющего собой смесь нормального изотопа и изотопа-разбавителя, в чистом виде; измерении изотопного отношения в выделенном препарате. Этот метод не требует полного извлечения элемента и учета потерь в процессе анализа. Условием правильности метода является полнота изотопного обмена между всеми фазами.
С помощью стабильных изотопов азота и других элементов анализировали различные органические соединения [773а, 1264а, 1364а]. Однако наиболее важным практическим применением метода является определение газов, в том числе и азота, в металлах, сплавах и сталях [129, 254, 731а, 1042а, 1158]. В зависимо-мости от метода выделения азота из металла изотопный индикатор добавляется либо в виде нитрида металла при вакуум-плав-лении в Ni-ванне [62], либо в виде соли аммония при кислотном разложении. Чувствительность обнаружения азота этим методом составляет 5-10~5%.
Точность метода изотопного разбавления может быть повышена, если используемый плавень предназначен не только для создания легкоплавкой эвтектики с анализируемым металлом, но и для введения изотопного индикатора [51].
Химический метод разложения металла в сочетании с изотопным разбавлением описан в [1311]. При растворении образца в кислоте добавляют обогащенный изотопом 15N сульфат аммония. Весь присутствующий азот окисляют до N2, в котором затем определяют изменение изотопного состава. Метод может быть применен для анализа образцов, содержащих от 2-10—1 до 6% азота, со средней ошибкой менее ±5%.
Изменение изотопного состава смеси в результате разбавления индикатора азотом, содержащимся в анализируемом образце и имеющим изотопный состав, близкий к атмосферному, может быть установлено методом искровой масс-спектрометрии, как это было сделано при определении содержания азота в титане, молибдене и никеле [64]. Проведено сравнение трех методов — изотопного разбавления, Кьельдаля и вакуум-плавления — и показано, что два первых метода дают сходные результаты, в то время как по третьему методу получаются, как правило, заниженные результаты.
Относительный изотопный состав азота в уравновешенной газовой фазе может быть определен также спектроскопически. Спектрально-изотопным методом оценивался изотопный состав азота при определении его в металлах до содержания 10-1% [86, 97].
142
Анализируемый образец помещают в предварительно обезгаженный реакционный сосуд известного объема. После откачки воздуха впускают азот, обогащенный изотопом 15N. Изотопное уравновешивание производят при температуре, превышающей температуру разложения нитридов данного металла. Определение равновесной концентрации 16N в газовой фазе производят спектроскопическим методом. Свечение молекул Na возбуждается в кварцевом капилляре при помощи высокочастотного генератора. Для получения спектра служит зеркальный спектрограф-монохроматор с дифракционной решеткой. Регистрация спектра производится фотометром, состоящим из фотоэлектронного умножителя, усилителя фототока и самописца типа ЭПП-09. Развертка спектра осуществляется посредством возвратно-вращательного передвижения диффракционной решетки. Аналитическая полоса азота 2976,8 А.
При спектрально-изотопном методе исследования материалов в зависимости от величины растворимости азота металлы могут быть разделены на две группы: металлы с низкой растворимостью азота (10~3—10“4вес. %) — к ним относятся железо, молибден, вольфрам, кобальт, никель и другие — и металлы с повышенной растворимостью— титан, цирконий, гафний, торий, ванадий, ниобий, тантал. Для разложения некоторых металлов первой группы достаточны температуры низкотемпературного варианта уравновешивания (1100—1200° С). Нитриды ряда металлов столь устойчивы, что их эффективное разложение затруднительно даже в условиях высокотемпературной установки (1600—1900° С). Например, анализ титана, циркония требует специальных мер для их растворения в ванне. Скорость изотопического уравновешивания для систем азот — металл меньше, чем для систем водород — металл.
Низкотемпературный вариант спектрально-изотопного метода характеризуется воспроизводимостью ~10 отн. % при концентрациях азота выше 10-1вес.% и чувствительностью лучшей, чем 10-1вес.%. Время уравновешивания составляет 2—4 часа. Воспроизводимость высокотемпературных определений при анализе металлов с низкой сорбционной способностью находится на том же уровне. Достигнутая чувствительность определений ЗЛО-8— 3- 10-в вес.% из навески 1 и 10 а соответственно [263а].
Анализ металлов с высокой сорбционной способностью связан с затруднениями, обусловленными малым равновесным давлением газа над металлом, а также возможностью образования устойчивых соединений азота типа карбонитридов. Преодолеть эти трудности можно с помощью ряда приемов: например, применения металлической ванны с последующим компрессированием уравновешенного газа в разрядную трубку; предварительной накачки уравновешенного газа из обменника в компрессор и т. д. В случае анализа титана достигается воспроизводимость определений ~15 отн. % из навесок 0,05—0,1 г при концентрации азота ~10-2вес.%; воспроизводимость анализов ниобия и тантала составляет 4—
143
5отн.% при содержании азота ~10-3 вес. % при навеске 0,05—0,1 г [263а].
Спектрально-изотопный анализ многокомпонентных газовых смесей с использованием изотопного разбавления (в том числе и определение азота) выполнен в работах [95, 1162]. Определяют этим методом 15N в пределах 0,36—2 ат. % [993].
Обзор литературы по оценке содержания 1SN в природе, физическим свойствам 15N, методам его концентрирования, измерения, по применению и синтезу соединений, обогащенных 15N, см. [601].
РЕЗОНАНСНЫЕ МЕТОДЫ
Электронный парамагнитный (ЭПР) и ядерный магнитный резонанс (ЯМР) используются как методы исследования азотсодержащих соединений (алмазы, органические вещества) и для определения азота.
Метод ЭПР применяют для исследования структуры и электронного строения частиц (молекул, атомов, ионов, дефектов в структуре твердого тела), которые содержат неспаренные электроны. Он позволяет получить сведения о содержании парамагнитных частиц в веществе, об их ориентации в кристаллах, о характере химических связей (степени делокализации неспаренного электрона), о достаточно быстром движении парамагнитных частиц.
Хорошо изучено простейшее парамагнитное соединение азота — атомарный азот в газовой фазе. Спектр ЭПР атомарного азота в основном состоянии состоит из трех линий, обусловленных сверхтонким взаимодействием электронов с ядрами 14N (ядерный спин 1=1) и двух слабых линий, которые отнесены в работе [1362] к сверхтонкой структуре атомов изотопа 1SN (I = ’/г).Отношение констант сверхтонкой структуры a =A(14N) / A (1SN) = 1,388 находится в хорошем согласии с результатами Рамзея (а = 1,402). Константы сверхтонкой структуры для изотопов 14N и 1SN соответственно составляют 10,46 и 14,63 Мгц.
Влияние спиновой релаксации на спектр ЭПР газообразных атомов азота описано в работе [569]. Спектр парамагнитного резонанса атомов азота в метастабильном состоянии приведен в работе [1195].
Метод ЭПР нашел широкое применение для исследования основной примеси в алмазах — азота. Результатам исследований ЭПР в природных алмазах посвящены обзорные работы [526, 670]. Кроме линий от парамагнитных центров, содержащих азот, в тонкоизмельченных порошках алмазов наблюдалась одиночная линия с g-фактором, равным 2,0027 ± 0,0002 и шириной АН = = 5,5 э [1383], которая отнесена к поверхностным центрам.
При оценке концентрации азота в синтетических алмазах следует учитывать влияние ферромагнитных включений на уширение линий ЭПР азота [268]. Прочность синтетических алмазов серийных марок может быть установлена на основании замеченной
144
корреляции с интенсивностью линии N2: степень прочности возрастает с ростом концентрации азота.
Обзор работ по ЭПР на примесях азота в синтетическом алмазе представлен в [422].
Спектры ЭПР раздробленных природных алмазов с высоким содержанием азота, полученные при комнатной температуре в 3-сантиметровом диапазоне, исследованы в [286]. Средний размер частиц менялся от 54±7 до 1 мк. В спектрах обнаружены линии типа а, обусловленные донорным азотом; концентрация неспаренных спинов, обусловливающих этот тип спектра, равна 1015 см~3. Интенсивность спектра другого вида, состоящего из линий типа б, зависит от размера кристаллов, а сам спектр вызван центрами на поверхности, число которых соответствует по порядку величины числу атомов углерода на поверхности образца. На образцах с размером кристаллов 1 .чк обнаружен новый тип линий, который обусловлен сверхтонким взаимодействием неспаренного электрона с магнитным моментом ядра 13С.
Высокотемпературные спектры ЭПР азота в естественном монокристалле алмаза и синтетическом порошке исследованы в области температур от 600 до 1230° К [1006]. Слабые сателлиты в спектре ЭПР естественного монокристалла, обусловленные 13С, исчезают приблизительно при 680° К. При 900° К ненормальное уширение азотных линий в спектре монокристалла и широкая ферромагнитная фоновая линия исчезают. При 1200° К спектр ЭПР синтетического порошка является полностью изотропным.
Методом ЭПР определяют концентрации окиси и двуокиси азота, молекулы которых парамагнитны, в загрязненном воздухе [1360], при анализе газов [1169]. Количество NO2 в загрязненном воздухе, в том числе и в выхлопных газах, определяют при давлении 1 амт, используя зеемановское расщепление уровней электронной энергии в магнитном поле. Концентрацию NO в загрязненном воздухе определяют при давлении ~0,1.чл рт. ст., применяя эффект штарковского расщепления уровней энергии в электрическом поле. Одновременное присутствие в системе NO и NO2 не влияет на точность определения этих примесей. Чувствительность определения NO2 составляет 10 ч. на млн.
Для определения более низких содержаний NO и NO2 (до 30 ч. на бин.) предложен метод обогащения проб, основанный на захвате молекул NO и NO2 при низких температурах.
При анализе газов методом ЭПР на серийных радиоспектрометрах фирмы «Вариан» установлено, что максимальное отношение интенсивности сигнала к шуму достигается для NО при 4 мм рт. ст., а для NO2 непрерывно растет с увеличением давления и максимально при 760 3tjn рт. ст. [1169].
Оба встречающиеся в природе изотопа азота (14N, содержание 99,634% и 1SN, содержание 0,366%) имеют спин, отличный от нуля, и, следовательно, могут изучаться методом ядерного магнитного резонанса [425, стр. 343—350]. Однако низкое природное
145
содержание изотопа 15N препятствует наблюдению его сигнала при высоком разрешении.
Изотоп 14N имеет спиновое число I = 1 и в соответствии с этим обладает квадрупольным моментом. Его низкий магнитный момент (р =0,40357рв) является причиной того, что характеристическая чувствительность для ЯМР 14N очень низка (1,01 10-3 от чувствительности для протонов при постоянной напряженности поля).
Малая чувствительность и влияние квадрупольного уширения линий объясняют относительно малый интерес к исследованиям ядра 14N методом ядерного резонанса.
Химические сдвиги 14N для ряда азотсодержащих органических и неорганических соединений (в том числе аммония, нитрат-, нитрит-, роданид-, цианид- и азид-ионов) приведены в [425, стр. 344]. По величине химического сдвига и относительной ширине линий поглощения судят об изменении ионного характера химических связей атома азота в различных соединениях. По спектрам ядерного магнитного резонанса на ядрах 14N определяют строение молекул.
Аналитического значения исследования соединений азота методом ЯМР не имеют.
Спектры ЯМР соединений азота с фтором приведены в [563], ряда органических соединений — в [1011], метилпроизводных кремния — в [1263].
Глава IX
ГАЗОМЕТРИЧЕСКИЕ МЕТОДЫ
СПОСОБЫ ПЕРЕВЕДЕНИЯ РАЗЛИЧНЫХ СОЕДИНЕНИЙ АЗОТА В ИЗМЕРЯЕМУЮ МОЛЕКУЛЯРНУЮ ФОРМУ
Наличие таких природных объектов, как атмосфера и природные газы, способствовало созданию и развитию газовых методов анализа. В дальнейшем оказалось удобным переводить химически связанный азот соединений в газообразные формы и измерять их с помощью методов газового анализа. Основой этих методов служит выделение молекулярного азота в процессе разложения образца при нагревании. На этом основан появившийся в 1831 г. метод Дюма, предложенный им в основном для азотсодержащих органических соединений. В дальнейшем этот метод оказался пригодным и для других химических соединений азота. С увеличением температуры удалось выделить молекулярный азот при плавлении металлов. Эти методы нашли широкое применение при определении азота в металлах и сплавах. К этой группе методов перевода связанного азота в молекулярную форму примыкает и метод количественного окисления аммонийного азота до молекулярного.
Кроме выделения молекулярного азота широко используется в практике и перевод химически связанного азота в аммиак [1477]. На этом, в частности, основан метод Кьельдаля [326, 8001; более подробно о переведении соединений азота в аммиак см. главу XI.
Меньшее развитие получили методы перевода азота химических соединений в окислы азота. Однако в связи с бурным развитием методов определения окислов азота в различных газовых средах следует предполагать прогресс и в методах выделения азота в виде окислов.
Методы разложения образца при нагревании с окислителем (метод Дюма)
Этот метод достаточно прост в аппаратурном оформлении и получил широкое распространение. Он довольно детально описан в ряде книг [868, 1458, 1478, 14801. Сущность метода заключается
147
fi сожжений азотсодержащего органического ьещестьа за счет кислорода окисла металла, добавляемого к образцу, поглощении получаемых продуктов реакции (СО2 и Н2О) раствором КОН и измерении оставшегося газообразного азота объемным методом в азотометре. Сожжение проводится в токе углекислого газа, который в предложенном Дюма методе получался при разложении карбоната свинца. Б случае определения N, С, Н, S и галогенов в неорганических материалах [247], азота в нитридах металлов [916], карборундах [276], нитратах [918], взрывчатых веществах [522] лучше говорить о разложении образца, чем о сожжении или пиролизе.
В 1912 г. Р. Преглем был предложен микровариант метода Дюма. Это позволило устранить основную ошибку, свойственную макрометоду Дюма — неполноту разложения больших навесок образца. В современных исследованиях этот вариант используется наиболее широко [1447]. Обзор этих методов приведен в [385, 394]. Дальнейшее развитие метода пошло по пути еще большего уменьшения навески и в настоящее время созданы варианты ул'ьтрамикрометода (3—500 мкг образца) [926, 927]. В отдельных случаях, когда величина навески [111, 796, 918] достигает 100 мкг и выше, используют полумикровариант [896]. Основными недостатками всех этих методик являются: 1) возможность появления в измеряемом объеме посторонних газообразных примесей, не абсорбируемых раствором КОН; 2) натекание азота воздуха вследствие негерметичности аппаратуры; 3) образование окислов азота, окиси углерода и метана в ходе разложения образца; 4) примеси в газе-носителе; 5) выделение газообразных продуктов из вакуумной смазки шлифов. Особенно тщательно должны контролироваться все эти возможные помехи при минимальных навесках.
Аппаратура для методов Дюма состоит из трех основных узлов: источника углекислого газа, устройства для разложения образца и абсорбционно-измерительного блока — азотометра. Обзор всех этих устройств приведен в [920].
Источники углекислого газа. В настощее время разложение карбоната свинца для получения тока СО2 не используется, так как при этом методе невозможно получить равномерного тока газа. Применяются три источника СО2: аппарат Киппа [2, 1134], сосуд Дьюара с сухим льдом и баллоны со сжатым углекислым газом. Загрязнения, вносимые в прибор с СО2, устраняют специальной обработкой мрамора, используемого в аппарате Киппа [1334]. Измельченный мрамор после обработки раствором HG1 и кипячения с водой (10 мин.) охлаждают, вносят в эксикатор, наполненный СаС12, и обезгаживают под вакуумом. При использовании в качестве источника СОа сухого льда последний помещают в стеклянную колбу, присоединенную к прибору и охлаждаемую жидким воздухом [916]. Во время анализа колбу со льдом охлаждают при помощи бани со смесью метанола и сухого льда с температурой 148
—65° С. Вследствие постоянного испарения С02 температура охлаждающей смеси автоматически регулируется так, что в приборе создается равномерный ток СО2.
Устройство для разложения образца. Блок представляет собой трубку из пирекса, если температура разложения не превышает 500° С, или из кварца, если разложение идет при более высоких температурах. В трубку помещают образец и два вида наполнения. На трубку для сжигания надвигают одну или несколько печей сопротивления для создания по длине трубки зон с различной температурой.
В этом блоке осуществляются две основные стадии процесса. Первая стадия — испарение образца, разложение основой массы образца и перевод вещества пробы в газообразные продукты; все эти процессы происходят с участием переменного наполнения трубки для сожжения, сюда же вводятся дополнительные катализаторы для окисления трудноразлагаемых веществ. Во второй стадии протекают процессы окончательного окисления вещества пробы и удаление из газовой смеси нежелательных компонентов, таких, как СО, О2, СН4, и восстановление окислов азота до элементного азота на постоянном наполнении трубки для сжигания, которое не меняется в течение серии анализов.
Постоянное наполнение состоит в общем случае из чередующихся слоев металлической меди и ее окисла. Подобная последовательность заполнения трубки для сожжения определяется многоцелевым назначением постоянного наполнения. Сжигание метана возможно на окиси меди при температурах 800—900° С. При температуре 700° С окисление метана не происходит. Однако при таких высоких температурах происходит термическое разложение окиси меди с выделением свободного кислорода, который в свою очередь поглощается металлической медью. Температуру металлической меди не следует поднимать выше 500° С, так как при более высоких температурах происходит частичное восстановление СО2 до СО. С другой стороны, недостаточный нагрев металлической меди может привести к тому, что часть окиси азота не восстанавливается на меди и попадает в азотометр. Поэтому вслед за металлической медью в трубку для сжигания помещают еще один слой окиси меди при температуре 200° С [209, 402, 522] для окисления образовавшегося СО.
С целью улучшения свойств постоянного наполнения .окись меди активируют добавлением Со3О4 при температуре 450— 500° С [1054]. Сделана также попытка использовать Со3О4 в качестве окислительного наполнения трубки для сожжения [1369]. При выборе рабочей температуры слоя этого катализатора следует учитывать перегрев (на 70° С и выше) начала слоя за счет тепла реакции окисления и то, что рабочая температура катализатора должна быть примерно на 200° С ниже температуры, при которой начинается его разложение (930° С). Улучшению условий разложения образца способствует также замена окиси меди на окись ни
149
келя [572, 1167], так как при окислении образца на этом наполнении не образуются окислы азота. Кроме того, окись никеля, в отличие от СиО, не спекается даже при 1000—1100° С и не разлагается с выделением кислорода. Поэтому окисление паров образца на окиси никеля проводят при 1000° С [1167].
Для разложения Р-, F- и Sn-содержащих веществ в качестве наполнения использована смесь Си с MgO и Ag либо Си и смесь ,СеО2 с пемзой при 700—800° С [904].
Для удаления воды из продуктов сжигания используют ангидрон [714, 1071], а для поглощения СО2 — аскарит [714]. Использование в качестве временного наполнения вместо СиО других окислов позволяет в некоторых случаях поднимать температуру разложения образца до 1000—1100° С. В таких условиях используемая в качестве газа-носителя углекислота будет частично диссоциировать на СО и О2. Для того чтобы перевести эти продукты обратно в СО2, в состав постоянного наполнения вводят слой гопкалита при 130—150° С [103, 1167]. Если в продуктах разложения образца отсутствуют сера и галогены, то слой гопкалита служит достаточно долго.
Переменное наполнение представляет собой окислитель, который смешивается непосредственно с образцом, либо располагается в непосредственной близости от него. В первоначальном методе Дюма это была окись меди при температурах, близких к 900° С. Однако при таких высоких температурах окись меди обладает помимо перечисленных выше недостатков еще одним: начиная с 800° С окись меди спекается с материалом стенок трубки для сжигания. Поэтому образцы стали вводить в трубку для сжигания либо в платиновой [904, 916, 1071], никелевой [586, 1134], алюминиевой [918], фарфоровой лодочке [1167], либо в стаканчиках [103, 209, 404], либо в кварцевых лодочках [1, 2].
Исследования, проведенные с использованием газохроматографического окончания, показали, что при сожжении в лодочке часть азота выделяется в виде окислов, в то время как при пиролитическом разложении вещества в стаканчике весь азот, независимо от его связи в молекуле, выделяется в виде N2 [404], и, следовательно, отпадает необходимость иметь в составе постоянного наполнения металлическую медь. Этот вариант метода пригоден и для восстановления азота неорганических нитратов, однако для полноты выделения N2 необходимо добавлять к переменному наполнению органическое вещество с высоким содержанием углерода, например антрацен [918] или сахарозу [404]. По-видимому, выделяющиеся в момент пиролитического разложения органического вещества уголь и продукты пиролиза при 900— 950° С восстанавливают окислы азота до N2. По другой методике при работе со стаканчиками разложение проводится без постоянного наполнения; остается только слой гопкалита [111].
При необходимости удаления мешающих примесей к переменному наполнению добавляют окись магния, например, для 150
связывания фтора [58], МпО2 для поглощения хлора, выделяющегося при термическом разложении [1151; окись марганца используется также для удаления SO2 [716].
При анализе веществ, не разлагающихся полностью в условиях обычного варианта метода Дюма, рассмотренного выше, пользуются вариантами метода с более жесткими окислительными условиями. Это достигается тремя способами: увеличением температуры разложения образца; добавлением катализаторов, активирующих окисление без повышения температуры; использованием в качестве газа-носителя не чистого углекислого газа, а смеси СО2 и О2 [247, 586, 904, 926, 1334, 1451, 1467].
В случае работы при повышенных температурах приходится отказаться от окиси меди, которая при этом спекается. В таких случаях переменное наполнение состоит либо из окиси никеля [58, 402, 572, 1167, 1334], либо из РЬО + РЬСгО4 (до 1200° С) [276], либо из WO3 (до 1300° С) [247, 1467].
Более низкоплавкие катализаторы при нагревании с образцом образуют расплавленный окислительный флюс, обеспечивая тесный контакт образца с источником кислорода. Наиболее широко в качестве катализатора используется окись кобальта [209, 586, 1054, 1071, 1134, 1334]. Применяются также хромат свинца [209, 276], пятиокись ванадия [209], перманганат калия [2], хромат бария либо ВаСгО4 4- Ag [586].
Использование свободного кислорода в качестве дополнительного окислительного агента осложняется возможностью образования окислов азота. Этой опасности можно избежать, используя достаточно низкие парциальные давления кислорода. Смесь СО2 и О2 получают электролизом насыщенного раствора КНСО3 [904] либо из перекиси водорода [1049].
Азотометр представляет собой градуированную бюретку (микробюретку) с измеренным объемом (обычно в 3t3t3). В классическом варианте метода Дюма вся бюретка до верха заполняется 50%-ным раствором КОН, под которым находится ртутный затвор. Бюретка соединена с помощью гибкого шланга с уравнительным сосудом, также заполненным ртутью. Выделившийся азот вытесняет часть раствора КОН из бюретки, а СО2 поглощается этим же раствором. По объему вытесненной жидкости судят о количестве выделившегося газа. Однако полное вытеснение раствора в азотометрах данного типа — задача трудно выполнимая. Часть раствора, остающуюся в самом верху бюретки под запорным краном, осторожно вытесняют с помощью уравнительного сосуда через кран в воронку над бюреткой. А на объем, занимаемый пленкой раствора КОН на стенках бюретки, вводится поправочный коэффициент. Вводятся поправочные коэффициенты также на барометрическое давление, температуру, давление паров воды над раствором КОН и холостой опыт. Все эти поправки н измеренному объему, сведенные в одно уравнение, в виде таблиц представлены в работе [1134].
151
Другим недостатком этих азотометров является то, что при перемещениях уравнительного сосуда и, следовательно, изменении давления внутри бюретки часть азота растворяется или выделяется смазкой запорного крана. Эти объемы азота достигают 0,1 см3 и могут внести значительные ошибки при измерении выделившегося N2.
Ошибки, свойственные рассмотренным азотометрам, были устранены при создании азотометров улучшенного типа [434, 534, 716, 1069, 1134, 1334, 13571, в которых раствор КОН не заполняет бюретку, а подводится к отметке, расположенной в самом низу бюретки, ниже калиброванной части. Другой конец бюретки не имеет запорного крана и соединен с ртутным поршнем, ход штока которого калиброван в единицах объема. При заполнении бюретки азотом, полученным в ходе разложения образца, уровень раствора понижается. Перемещением поршня уровень раствора КОН возвращается к исходной отметке, а объем непосредственно вычисляется по ходу штока поршня. После выпуска измеренного газа азотометр готов к следующему измерению без какой-либо дополнительной операции. При объеме раствора КОН 450 мл на азотометре подобной конструкции можно произвести 150 определений без замены реактива [1069].
Точность определения по микрометоду Дюма—Прегля не превышает в среднем ±0,2% при значении холостого опыта < 0,001 мл!час N2 при ошибке отсчета температуры <0,5° С, давления <1 мм рт. ст., объема <0,001 мл. При навесках, составляющих 3—9 3tK2 или 100 мг и выше, ошибка возрастает и равняется ±2—3%. Время анализа составляет в среднем 20 мин.
Достоинством микрометода Дюма—Прегля является то, что на его основе возможна автоматизация определения не только азота, но и одновременного определения углерода, водорода и азота. В обзоре [534] рассмотрены автоматические приборы, применяющие различные приемы измерения выделяющихся газов. Так, анализатор Колемана [1319] использует газометрическое определение одного азота. Приборы фирмы «Техников» (метод Валиша), фирмы «Перкин—Эльмер» (метод Симона) и фирмы «F and М» (метод Дерге) используют газохроматографическое определение углерода, водорода и азота. Для анализа требуется от 0,05 до 1 мг вещества. Заполнение обычное (СиО и Си), газ-носитель — Не + О2. Выделившийся N2 отделяют от СО и СН4 и количественно определяют методом газовой хроматографии. Продолжительность анализа в среднем 10 мин. Ошибка составляет ~0,2%. В автоматическом приборе Мерца [1467] вместо СиО в качестве окислителя предложено использовать смесь окислов кобальта и вольфрама, которые улучшают условия сгорания, способствуя уменьшению выделения угля и продуктов крекинга на внутренней поверхности трубки для сжигания.
Варианты метода. Ниже рассмотрены группы газометрических методов, несколько отличающихся от классического варианта 152
метода Дюма—Прегля. В первую ойёрёдь это группа методов, основанных на «мокром» сжигании вещества пробы в аппаратуре по методу Дюма. Для разложения пробы используют смесь Сп2О и раствора HG1 в платиновой лодочке (200° С) [1661, либо смесь 57V раствора хромовой кислоты и концентрированной серной кислоты (150° С) [833, 8961. Содержание азота в гидразине определяют при окислении последнего в щелочной среде K3[Fe(CN)e], КМпО4, K2HgJ4 до молекулярного азота [1464]. До NO восстанавливается азот нитратов и нитраминов ртутью, железом(П), титаном(Ш) и гидрохиноном в кислой среде (H2SO4, НС1) [471]. Во всех методах в качестве газа-носителя использовали СО2; выделившиеся N2 или NO измеряли азотометром. Ошибки методов не превышали 0,2%.
Вторая группа методов основана на разложении вещества пробы в запаянной трубке с окислительным наполнением [714, 716, 926, 9271.
В откачанную до остаточного давления 10-3 мм рт. ст. трубку помещают от 0,5 до 3—5 мг сухой пробы илй 0,15 мл раствора, содержащего 1 мг!мл вещества. К твердому образцу добавляют СиО и Си, запаивают и проводят разложение при 900—950° С. В специальном устройстве запаянную трубку вскрывают, галоиды и SO2, образующиеся при разложении, поглощают соответственно металлической медью и серебром (325° С) [714] или амальгамой меди [716] и МпО2 (135° С). NO восстанавливают на металлическом уране (140° С). Жидкую пробу замораживают [716] вместе с 0,1%-ным раствором полиэтиленгликоля сухим льдом, добавляют металлическую медь и твердый NaOH, заполняют газообразным кислородом и запаивают. После проведения разложения трубку вскрывают под слоем ртути и по изменению уровня ртути определяют объем выделившегося азота [926].
И наконец, методы одновременного определения содержания азота в нескольких пробах за один анализ [103, 528, 10711. Для этого пробы помещают в трубку для сожжения в отдельных стаканчиках [103, 1071], либо вносят смесь веществ и используют различные скорости реакций разложения этих веществ при нагревании их с СиО [528].
Методы выделения азота из металлов и сплавов описаны в главе XII, химические методы перевода восстановленных соединений азота в молекулярную форму — в главе V.
МЕТОДЫ ОПРЕДЕЛЕНИЯ ГАЗООБРАЗНЫХ СОЕДИНЕНИЙ АЗОТА
Газообразные соединения азота могут быть измерены либо в виде чистого компонента, либо прямо в газовой смеси. В силу своей большой химической инертности молекулярный азот, в отличие от других своих газообразных химических соединений,
153
МОЖёт быть хорошо оЧиЩей of йримёсей химическими Методами. Поэтому он часто определяется в виде чистого компонента. Подобные определения проводятся двумя способами: определяется объем газа при постоянном давлении, температуре и числе молей газа (объемный метод) или фиксируется изменение давления при постоянном объеме, температуре и числе молей газа (манометрический метод). Другие газообразные химические соединения азота, если они выделены достаточно чисто, могут быть также определены одним из данных методов. Если же разделение газовой смеси на отдельные компоненты — задача трудновыполнимая, то для определения всех газовых компонентов в смеси служат такие методы, как газовая хромотография, эмиссионная спектроскопия, масс-спектроскопия и другие физические методы.
Манометрический и объемный методы
При измерении манометрическим методом выделившиеся газы переводят в измеренный объем автоматическим насосом Теппера или диффузионным' вакуумным насосом. Давление смеси измеряется манометром Мак-Леода. Затем по мере уменьшения числа молей газа измеряется падение давления. Вода удаляется из газовой смеси вымораживанием в ловушке при —78° С. СО2 выводится из газовой смеси охлаждением до —145° С. Кислород количественно поглощается металлической медью при 500° С. СО переводится над СпО в СО2. Водород окисляется окисью меди до воды. В конце анализа в газовой среде остается только газообразный азот с примесью инертных газов, если они присутствовали в составе исходной газовой смеси. Парциальное давление азота в данной смеси может быть измерено связыванием азота металлическим кальцием при 900° С [54].
^Объемныйметод был описан выше при изложении метода Дюма (см. стр. 147).
Газохроматографический метод
; Газовая^хроматография как метод анализа начала развиваться сравнительно^ недавно, в 50-е годы. В настоящее время газовая хроматография является наиболее универсальным методом газового анализа.
Теория процесса, основные принципы работы и устройство} основных блоков хроматографа приведены в многочисленных монографиях и обзорах] [16, 23, 67, 91,128,192,199,368,415,476,874,1176]. Высокие эффективность, чувствительность и большая экспресснозть этого метода обусловили его широкое применение в научных исследованиях и*, в промышленности.
Аппаратура для газохроматографического анализа разбивается на отдельные блоки, которые обеспечивают проведение следующих операций: 1) подготовка и отбор пробы; 2) газохроматографическое разделение пробы на составляющие компоненты» 154
3) детектирование этих компонентов; 4) преобразование сигналов детектора в цифровые данные.
Блок подготовки пробы. Это блок многоцелевого назначения, позволяющий проводить целый ряд операций. Они включают в себя системы: отбора пробы, калибровки детектора, очистки газов от нежелательных примесей, предварительного концентрирования измеряемого компонента, перевода компонента газовой смеси в удобную для измерения форму.
Отбор пробы из металлических контейнеров, баллонов и пластиковых мешков не представляет большой сложности. Более сложным представляется отбор пробы растворенного в воде газа. Выделение растворенных газов проводят двумя методами. По первому [1078] газ-носитель (обычно водород) циркулирует через исследуемый раствор до установления равновесия с растворенным газом. После этого пробу газа отбирают на газохроматографический анализ. По второму методу [469] газ-носитель и проба воды разделены фильтром, проницаемым для растворенных в воде газов.
Калибровку детекторов для расшифровки хроматограмм осуществляют либо по чистому компоненту, в данном случае по азоту [313, 357, 1224, 1409], либо путем приготовления калибровочных смесей методом парциальных давлений [79]. Наиболее хорошие результаты были получены при кулонометрической калибровке [1409]. Калибровочные кривые, построенные с помощью N2, выделенного прямо в хроматографе при электролизе 0,1 М раствора сульфата гидразина, показали хорошую линейность (<±1%) в диапазоне 0,25—5 молей азота.
При газохроматографическом определении молекулярного азота и его окислов мешают, как правило, углекислый газ и вода [24, 179, 969, 1366, 1391]. Для удаления СО2 используют раствор NaOH [24] или колонку с карбосорбом [1366], для Н2О — Mg(C104)2 [1082].
В некоторых случаях (при малых концентрациях определяемого компонента) приходится его предварительно концентрировать. Таким образом удалось определить в газовых смесях 1 • 10-5% NO2 при его концентрировании на угле АГ-3 и СКТ [24]. Древесный уголь использовался для накопления газов, выделенных при плавлении металлов [909]. Метод предварительного накопления применим для определения примеси азота в кислороде, когда основной компонент смеси (О2) удаляется либо металлической медью [424], либо по реакции 2Н2 + О2 = 2Н2О с Pd в качестве катализатора [756, 822]. Было определено до 0,001% N2 в пробе кислорода объемом 100 см3 [424] и >0,03% в водороде высокой чистоты [288].
Для определения суммарного содержания в газовой смеси окислов азота проводят предварительное восстановление их до молекулярного азота над Pt—Pd- и Си—Сг-катализаторами при 300-500° С [313].
155
Блок хроматографических колонок служит для разделения газовой смеси на отдельные компоненты. Для выделения азота и его соединений используется во всех случаях газоадсорбционная хроматография. В качестве сорбентов используют молекулярные сита 5А [872, 880, 969, 972, 1012, 1033, 14491, 13Х [7001, силохром [509], природный морденит [3461, алюмосиликат [1791, алюмогель [265], силикагель [599, 1091], пористый полимер типа порапак [610], графитизированную сажу [1196], цеолит СаА [1139] и активированные угли. При определении NO2 в газовых смесях используют газо-жидкостную хроматографию с применением в качестве неподвижной фазы органических фосфинов и фосфитов [664]. Наилучшие результаты дает реакция NO2 с трифенил-фосфитом, так как при этом NO2 целиком переводится в NO. При разделении NO2 и NO используют в качестве неподвижной фазы политетрафторэтилен и политрифтормонохлорэтилен [663]. Анализ продуктов разложения гидразина (N2, Н2О, NH3) проводят также с помощью газо-жидкостной хроматографии. Неподвижные фазы в этом анализе состоят из триэтиламина и тетраэтиленпентаамина на хромосорбе и 10% триэтаноламина на порапаке Т [1248]. При совместном определении аэота и его соединений с углеводородными газами сочетают газоадсорбционную и газо-жидкостную хроматографию [71, 79, 357, 377, 656, 1091]. В этих работах неподвижная фаза состоит из 10% диметилсебазита на КУ-2 [80], 15% динонилфталата на кирпиче ИНЗ-600 [71], 0,5% полиэтиленгликоля на стеклянных шариках [357], диметилсульфолана [656] и силиконового масла на флуоропаке [984]. Пропитка сорбентов молекулярных сит 5А и силикагеля н-гептаном после их промывки водой, спиртом и эфиром уменьшает специфическую адсорбционную активность зерен молекулярных сит по отношению к окислам азота и делает более равномерным свойства сорбента по длине колонки [915].
Компонентный состав разделяемых газовых смесей, размеры колонок, виды сорбентов и детекторов представлены в табл. 15. Хроматографические колонки, как правило, работают в изотермическом режиме, для чего их помещают в термостат с заданной температурой. В некоторых случаях для лучшего разделения компонентов используют программирование температуры [846, 888].
При определении молекулярного азота на катарометре в качестве газа-носителя лучше всего использовать гелий, обладающий максимальной теплопроводностью. Однако гелий, особенно гелий высокой чистоты, является дорогим реактивом и его заменяют на аргон или водород. В последнее время для проявления хроматограмм используют неон. Применение его в ионизационных детекторах вместо аргона втрое повышает чувствительность определения молекулярного аэота [754, 757, 758, 759, 999]. Чувствительность определения повышается, если пропускать через хроматографическую систему смесь газов: Аг + пары парафинового углеводорода [397], Аг + н-толуидин [756], Ne -|- 0,1 —
ю ч-I
Сб
Я Ч Ю еб Н
Газохроматографический метод I Литература г—< cq ио мн о о о СЧ СЧ Г— г— Г- Г- КГ СЧ ОО Ю — —• Ч-I со
Объект Ф СЙ Д S- S >» и 1 S 5 S 2 Ч 5 g й 2 S ® 1 1 ° 1 “S «и «И - В “ ч 5 я щ «Я о § £ 2 о ss е-I д» eg
к м 2 ® И ти « S с> III | 00 1 7 ю
Проба газа, мл * « м 1 1 1 и 1^4 « и 1 и к
Ошибка, % М- о ф О I кГ Ф 1 Ю 1 00 t? 1 о" * 1 ’
Газ-носитель; скорость, мл\мин со” ® ф м S g <о « щ щ ® ” Ь" ф j? ® 2? д К Я д Щ Д дЩ
Колонки и ® 1Л й 1Ф О О о СО О ОО Я сч 2 кг СЧ кГ СЧ О £ о О Ю | хк iMeq с© со 1 ф 1 ® ОО
сорбент N . Ч — — ® о ф « g « ъ § «Хи ” си g. » “tc н ь <-у о- + а и Н Н О' О' д <; О ю О ю « « Lj ю 2 « Z ой О ИИ . • ! 3 S != й- Кч г-1 ГН 2 Z0 ем eSesaasSss 2 «к s о s s < s
размеры X X X X X XX ® ° S S ш ш У X X = X X« о X X X СЧ 'TO О UO IO Ю S g 2 М 4- M <= <= M CO t- 4 4 Д Д co Д ° СЧ CO СЧ о о °
Состав газа J? «Г - СЛ 6« - ° 8 3 я о « - - й X1 о ® A® ° X О о < z 6s A® c? Z ОЗ О M 03 (Я О ОД ~Г1 O1 ZOO Z ZQ ZU ZU z
156
157
Таблица 15 (продолжение)
Состав газа Колонки Газ-носитель; скорость, МЛ\МЫН Ошибка, % Проба газа, мл Время анализа, мин. Объект Литература
размеры сорбент (, °C
n2, н2, о2, сн4, со, СО2, С2Н4, СдНб, С3н8 2,25.и 2м м. с. 5А Силикагель 50 20 Аг; 60 Не; 38 — 1 20 Сырой газ коксовых печей [78]
n2, со2, со, О2, Н2, Н2О, сн4, С2Н4, С2н6 4м 2м Порапак Q м. с. 5А — Не; 41 — — — — [463]
n2, со2, со, cos, so2, 02 2,15м X 4мм 1 ,5л4 X 4мм Порапак R и. с. 5А 160 Н2; 40 — 0,5-1 — Газовая атмосфера печей металлургического производства [476]
N2, О2, Аг, СО, СО2, H2S, COS, SO2 1,5м X 1,17мм 7,5м X 1 ,17л«л4 Порапак Q То же 75 -65 Не; 0,01 % so2 — •— 9 — [1119, 1120]
n2, co2, ch4, 02 1,8.и X 5мм 4,5ai X 5мм Порапак S м. с. 5А Разделе 50 ние н Н2; 45 а 1 кож )нк е 0,2 — Газы анаэробного разложения [1366]
NO, NO2, N2O, CO, ch4, o2, n2 Зм X 4мм а. у. СКТ 150 Не; 40 8 — 11 — [25]
N2 (следы H§, Ne и H2) — То же — N2; 50 2,2 — — Примеси в баллонном азоте [90]
NO, N2, H2, CO Зм X 6мм и. с. NaX 130 Аг; 100 4 — — — [170]
Таблица 15 (окончание)
Состав газа Колонки Газ-носи-телъ; скорость, мл{мин Ошибка, % Проба газа, мл Время анализа, мин. Объект Литература
размеры сорбент (°, с
n2, н2, о2, сн4, СО 1,2м X 4м м. с. 13Х 30 Не(Аг); 40 — 1,5 — Продукты оксосинтеза [231]
N2 (примесь в гелии) — То же 40 Не — 200 — Гелий марки А [1191]
Примеси Аг и О2 в азоте 0,5л« X 3,5л«Л4 м. с. 5А —78 Не; 30 — — Примеси в газах-теплоносителях [468]
n2, н2, о2, сн4, СО2, Кг 7м X 1,5мм Порапак Q + 4-6,01% Н3РО4 20 Не; 15 — . — — — [588]
N2 (примесь в О2) 1м X 6мм м. с. 5А или 13А+ +0,5% Pd — н2 0,05 — — — [756]
N2 в воздухе 1,8м X 6мм м. с. 5А 60 Не; 60 .— 10-3-10-2 — — [761]
Зм X ЗлШ То же -40 48 Н2; 30 0,054 0,16 0,25 1 — — [1121]
Ng, Н2О, СО 2,54л« X 6,8л4л« м. с. 13Х — Аг — — 6-7 Вакуумное плавление стали [803]
NO2 в газе 0,4-и X 2мм Силикагель 50-60 Н2; 30 — — — — [969]
NO, N2, Н2, СО Зм X 6мм м. с. NaX 130 Аг; 100 4 — — — [1449]
n2, о2, н2, сн4, СО 1,2м X 6мм м. с. 5А 100 Не; 120 5 — 3 [207]
N2O в воздухе 0,3м X 6,4л«л4 Порапак Q 25 Не; 50 2 — — — [9721
Примечание: а. у. — активированный уголь; м. с. — молекулярные сита.
сл
СО --------------------------------------------------------------------------------------------------------------------------------
0,3 объемн. % Ar, Кг, Хе [757, 758], Не + 0,1% Н2 [888]. При необходимости кондиционировать колонки по какому-либо компоненту последний добавляется к газу-носителю (Не 4- 0,01 % SO2) [1119].
Блок детектора используется для преобразования изменения химического состава газовой смеси в электрический сигнал, который затем записывается на ленте самописца. Наиболее часто для детектирования молекулярного азота и его соединений используют катарометр [40, 341, 419, 566, 640, 898, 975, 1033]. Он применяется в очень широком диапазоне концентраций азота от 20 мкг [898] до 80 объемн. %. Чувствительность определения с катарометром можно повысить до 1 нг при использовании криптона в качестве внутреннего стандарта [588]. Чувствительность катарометра повышается также при работе по методике приведения к нулю, когда на самописец накладывается напряжение, примерно равное и противоположное по знаку показанию катарометра [1374]. Катарометр довольно стабилен в работе. При измерении чувствительности его работы в течение 92 дней было показано, что она изменилась в среднем на 0,46% [1472].
Для определения следовых количеств азота и азотсодержащих функциональных групп применяются и другие виды детекторов.
1. Ионизационные детекторы с Не [60, 758, 761, 1224] и Аг [377, 548, 735, 754, 980] как газами-носителями. В качестве источника излучения используются RaD [397, 1001], 85Кг [548,-757], ®°Sr [757, 761], №Ni [1395] и тритий [757, 1299].
2. Кулонометрический детектор (чувствительность для аммиака и окислов азота 10-6—10-4%) [162].
3. Разрядный детектор, работающий в области отрицательного коронного разряда (чувствительность для N2 3,6-10-11 молъ/сек) [261, 298, 799, 1406, 1424].
4. Импульсный метод детектирования газов, при котором в качестве детектора используется проточный пропорциональный счетчик малого объема [880].
5. Детектор, основанный на измерении электропроводности водных растворов аммиака, полученного восстановлением N2 водородом над катализатором [890].
6. Детектор по изменению люминесцентных спектров разряда (чувствительность для N2 и N2O от 50 до 500 мкг) [979].
7. Детектор по захвату электронов [1224] (чувствительность определения в воздухе NO и NO2 > 10-3 объемн. % [984], N2O 10-7% [1395]).
8. Спектральные детекторы [1142, 1179]. Чувствительность пламенно-фотометрического детектора при определении N2 составляет <1 нг [1179].
9. Модифицированный пламенно-ионизационный детектор [1216].
10. Детектор Ридмана, обладающий высоким уровнем селективности к соединениям, содержащим азот, серу и галогены [1213].
160
11. Газовые весы Мартина, которые используются при измерении изотопного состава азота [1396].
12. Использование в качестве детектора масс-спектрометра [752].
Возможно раздельное определение S, С1 и N с помощью комбинированного пламенно-ионизационного и пламенно-фотометрического детектора [1274], причем при выходе из детектора можно отобрать отдельные хроматографические фракции [837].
В последние годы газовая хроматография использовалась для полного разделения изотопов азота [342, 570] и измерения изотопного состава азота природных объектов [1396[.
Химические методы определения газообразных соединений азота нашли применение в основном только для окислов азота. Это обусловлено их высокой реакционной способностью, которая препятствует использованию для анализа окислов азота таких методов, как газовая хроматография (принимая во внимание большую поверхность сорбента) и масс-спектрометрия (абсорбция на стенках аппаратуры). Обычно химическим методом определяют сумму окислов (NOX). При необходимости раздельного определения сначала определяют NO2, а затем окисляют NO до NO2.
Достоинства, недостатки и возможности применения химических и инструментальных методов определения азота в различных объектах приведены в обзорах [1454, 1473]. NO окисляют до NO2 в воздухе 0- и УФ-излучениями, высокочастотным электрическим разрядом [1444], 3%-ным раствором двухромовокислого калия в 2,5%-ном растворе H2SO4 [1444], озоном [1463]. NОж определяют на фотоколориметре [1438], с помощью нитратного ионоселективного электрода [1466]. Содержание NO2 измеряют в виде нитрит-иона реакция с 8%-ным раствором иодида калия) [1442] в кулонометрической гальванической ячейке [1463], хемилюминесценцией (10“7—10~4%) [1471]. Учитывая способность озона окислять NO, необходимо обратить особое внимание на методику отбора пробы при совместном присутствии этих компонентов и, в частности, на время нахождения этих газов в пробоотборнике [1455].
Молекулярный азот определяется химически после поглощения его титановой губкой при 900—1000° С [1460] либо активационным анализом в газовой смеси (чувствительность 10-8 г! см1-') [1469].
6 В. Ф. Волынец, М. П. Волынец
<0
СМ
оГ
Глава X
МЕТОДЫ РАЗДЕЛЕНИЯ СОЕДИНЕНИЙ АЗОТА В РАСТВОРАХ
Разделение различных соединений азота в растворах осуществляется главным образом методами хроматографии, экстракции, электрофореза.
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Хроматографические методы, используемые для выделения и разделения азота и его соединений, основаны на принципах адсорбции, ионного обмена, распределения.
По технике выполнения эти методы являются либо колоночными, либо плоскостными (листовыми) вариантами — тонкослойная, бумажная хроматография.
Ионообменная хроматография
Методы ионного обмена довольно широко используются в аналитической химии азота и его соединений. Существуют ионообменные методы выделения и разделения различных азотсодержащих ионов в растворах и методы разделения стабильных изотопов азота.
В настоящем разделе рассмотрены в основном способы разделения соединений азота методом жидкостной ионообменной хроматографии в гетерогенных системах в динамических условиях при течении потока элюента через колонку с зерненым сорбентом (колоночный вариант). В качестве сорбентов используют как анионо-, так и катионообменники.
В табл. 16 приведены сведения по ионообменным методам разделения соединений азота в колонках. Ионообменные методы разделения на ионообменных бумагах, на бумагах, импрегнирован-ных жидкими ионообменниками, и в тонких слоях ионитов на пластинке см. ниже в соответствующих разделах («Хроматография на бумаге и в тонком слое сорбента»).
Ионообменное разделение стабильных изотопов 14N и 1SN на катионите КУ-2X9 описано в [71а], на flayaKC-50WX4 и Дау-3KC-50WX2 и др.- в [589, 590, 1222].
162
S И св
g р. и
ф я о
я м и
о fc*
о S
1 со Рч
и о
§ Z сб сб S 1
сб и &
g п 3
st о и
ф о >&
и н
3 и о й s SK п я сб О
Д’ о И
ф рч 2Z
и Рч
о о сб S3
S И Е-> Р.
S о
Промывной раствор 0,2М HNO3 Вода л А 1 1 1 1 1
Обменник (название, тип) КУ-2Х20 в Н-форме, К Амберлит IRA-400 в ОН , а затем в 1~-форме Амберлит IRA-410 в ОН-форме, А МК-2 в Н-форме, К Вофатит-F и вофатит-KRS-200 в Н-форме, К Амберлит А-29 в ОН-фор-ме, А ЭДЭ-10П, А КУ-2., К «Srafion» СЮ(К) и амберлит IR-45, А
Объект анализа Смеси разновалент- ных катионов Органические соединения Азотные удобрения (Na- и Са-селитра) То же NH4NO3 Щелочные растворы Водные растворы I Двухкомпонентные электролиты на основе NaCl и NaNO3 Сточные воды
и 2 Я Сб й
« ф
s S Ф £ № X 58 § и
о
к
6* 163
Таблица 16 (окончание)
Промывной _ раствор Примечание 22 и i i й g g А и ® a CL Рч м S Д Ч S S CQ*" ЗЙег* ф м о И и о фя а и о 2 S 2 м нсо но м 2 о 2 ч о и 2£йя2Яо§Зйа°>е< ” 2 « 5 §5 s к s ч еъ ф w r_. сХ ? J л Е ф а мЫ я й 2 *7 Й о Й X ? со Лев Я 10 И о « 2 р< я . - « ф л >> в я д и g. о & g св kj rL, Н Ф Я JR Д q Я ® м фФр^НфДЗсв И Д Ф »- 5Н S н Ф >а -< О .>> ,м О иИо1«ёпИЙивииии Ф« и и § й§ 3 § й Ug Z §§ g§g=§gg| 5 SS IsfW 1.& м m М К S u Ч ВИф^и sJ“ о Ей £оофЯ!Й св о Д ® ф иКИфМДм^^я н S д 5 Д ф ес Д10 о Ий &g t £ 83 ВИЩ.и “ g»s & Е g g § и S5 §§Ч-2ни|>§‘«^я>и° и 8и§ § н о И §й иВ§££S£&5g§§gк wSsSsg О§
t а о д §н§ "pss , as*?* г . , §3?., 1 gigSS is 1 11 о и и 3 и Я 5 н Й и 5 и§|£§о-д М ₽< 5J о д.
Обменник (название, тип) йй § < а° Ф * И S 0<Ч 1 |& л < § 4* 4 XI ? g 1 ®g < о * i §згя5 a н И о Щ Д со * ° оо< § О <? g <С S £ в- £ м § £ She < о Ч Д ef । Ф 84 ф z—к ФО т Ф Ф СО w g й И -г И W g g®s гв и । К ” Л-J. re й © L оз О ^2- О ' Ч «И& g Й
Объект анализа Й L и .-3 ю ? 1 д > 3 о 1^4 1 со И j1 Q1 S § «э2|-»иоы8§ s । « । । ^ । а § 5 1 со 1 Г со М Q св ex ® 5 УО^ОиаИ ф св >Я Н’т’Гл'^ЧЙФф Ч И Ф Я’Т'Ч'^мЯЯм Д § и Ч ф S ф Т й и J §g ёй 05 й|§^§ § S ОЧ О ЧдОЯВ О
Si о*
8 3 * й ф 5 «ч й
I d О Z
Id I со
о о
а к
Примечание. А — анионообменник; К — катионообменник; Н — неорганический; Ж — жидкий ионообменник на твердом носителе.
164
Хроматография на бумаге и в тонком слое сорбента
Листовые варианты хроматографии — бумажная и тонкослойная хроматография — обладают общими преимуществами по сравнению с колоночным вариантом хроматографии [41, 55, 381]: экспрессностью, простотой осуществления процесса и отсутствием необходимости дорогостоящего оборудования, возможностью разделения и идентификации субмикрограммовых количеств смесей веществ, универсальностью (вследствие возможности варьирования выбора сорбента, особенно в случае тонкослойной хроматографии, ТСХ), простотой детектирования разделенных зон на хроматограмме, возможностью быстрой количественной оценки содержания элементов в зонах различными высокочувствительными методами и др.
Общий принцип разделения веществ методами ТСХ и бумажной хроматографии заключается в различной скорости перемещения компонентов по слою сорбента при пропускании через него подвижной фазы в результате различия коэффициентов распределения этих компонентов в данной хроматографической системе между стационарной и нестационарной фазами.
Механизм разделения смесей, в зависимости от выбора условий хроматографического процесса, может быть различным: адсорбционным, распределительным, ионообменным и т. д. Однако как бумажной, так и ТСХ присущи и свои особенности.
Бумажная хроматография
Большим преимуществом бумажной хроматографии (по сравнению с ТСХ) является постоянство значений R* компонентов в выбранных условиях, а также простота отделения определяемых компонентов от целлюлозной основы путем сухого или мокрого сжигания.
Для разделения различных азотсодержащих ионов применяют бумагу Ватман 3 ММ, Ватман № 1, ионообменные и модифицированные бумаги (табл. 17).
Одной из разновидностей хроматографии на бумаге является метод кольцевой бани. В работе [819] описана микроидентификация в одной капле 20 анионов (в том числе NOg, NOg, SCN-) с применением кольцевой бани с нихромовой проволокой. Сущность метода сводится к следующему.
Аппаратура состоит из круговой камеры и капельной пипетки для подачи растворителя. В центр листа фильтровальной бумаги Ватман № 1 с помощью капиллярной пипетки наносят 1—2 капли анализируемого раствора (растворы калиевых или натриевых солей указанных анионов в дистилли-
* Отношение расстояния, пройденного на хроматограмме веществом, к рас. стоянию, пройденному фронтом растворителя. Характеризует хроматогра. фическое поведение соединения в данной хроматографической системе.
165
Таблица 17
Разделение и выделение ионов методом хроматографии на бумаге
Выделяемые (разделяемые) ионы) Сорт бумаги Подвижная фаза Способ обнаружения иона «/ Примечание Литература
NH+ (от К+, Cs+, Rb+) Ватман № 4 Фенол, насыщенный 20%-нойНС1 Опрыскивание 10%-ным раствором натрийко-бальтнитрата в 5%-ной уксусной кислоте 0,11 0,19; 0,43; 0,27) Раствор содержал по 50 мкг каждого элемента [1312]
NH+(ot К+, Na+, Mg2*, Li+) Тойо Роши № 50 Пропанол + метанол (9:1) Опрыскивание оксихинолином 0,33 (0,08; 0,17; 0,47; 0,56) — [862]
NHj (от K+, Na+) Бумажные фильтры с белой лен-той Метанол 4- этанол (1:1) — — Качественное обнаружение [382]
no;, NO" scn-и др. Ватман ЗММ и катионообменные бумаги SA-2 (SO3H-rpynna) и WA-2 (СООН-группа) Растворы (NH4)2SO4 и H2SO4 [988]
no;, no;, scn-(и 29 других анионов) Ватман № 1 Изопропанол -|-+ вода(различные соотношения) Круговая хроматография (возможно разделение смесей NO;—NO;; С1_(Вг")— -SON"; С1--ВГ-- -J--SCN-; S2-—SCN-и ряда других) [194]
Таблица 17 {продолжение)
Выделяемые (разделяемые) ионы Сорт бумаги Подвижная фаза Способ обнаружения иона «/ Примечание Литература
SON"; NO; (и 19 других анионов) SON" (от Г ) no; (от сю;, Bro;, jo;, jo;) no;, no;(ot сю;) SCN" (от Cl", J") SCN" (от галогенидов) Бумага, импрег-нированиая анионитами (Амберлитом IRC-50 и Амберлитом IRA-400) Ватман № 1 » » Бумага, импрег-нированная Ag2CrO4 Анионообменная бумага в ОН-фор-ме 1А KNO3 Вода Опрыскивание смесью Fe(NO3)3+ 4- Н2О2 (красное окрашивание) Опрыскивание раствором KJ, подкисленным НС1 (бурое пятно на белом фоне)_ Опрыскивание раствором Ti8+ в H2SO4 (белые пятна на фиолетовом фоне) По бесцветному пятну AgSCN 0,10; 0,63 AgJ> >AgSCN> >AgCl Определяют 10— 150и<кг SCN"; мешают Вгу, CN", S2O|" [1141] [1228] [1228] [1228[ [56] [258]
Таблица 17 (окончание)
’3/2
' о
168
рованной воде). Затем полосу бумаги нагревают горячим воздухом, чтобы пятно «не расплывалось», помещают ее на подставку из твердого картона таким образом, чтобы пятно оказалось в центре круглого отверстия на картоне. Круговая камера помещается на фильтровальную бумагу и после регулирования силы тока и напряжения через нихромовую проволоку (в виде кольца) подается ток в круговую камеру.
Капельная пипетка, содержащая подвижный растворитель (ацетон + конц. NH4OH, смесь 1:1), вводится внутрь круговой камеры таким образом, чтобы немного не прикасаться к поверхности бумаги и чтобы капли с пипетки попадали в центр пятна с нанесенным анализируемым раствором. Подвижный растворитель подается в течение 1—2 мин., после чего пипетку удаляют и питание током прекращают. Полоску фильтровальной бумаги извлекают, разрезают на 4 равных сектора и опрыскивают различными реагентами для идентификации анионов. Анализ длится 1 час.
В табл. 18 приведены реагенты и чувствительность обнаружения.
Таблица 18 Метод кольцевой бани [819]
Анион Обнаруживающий реагент и окраска Предел обнаружения, Л4КЗ
NO“ Розово-красная; 1%-ный раствор сульфаниловой кислоты в 30%-ной ледяной уксусной кислоте, затем 0,3%-ный раствор а-наф-тиламина в 10%-ной СН3СООН 0,1
NO" То же 0,2
SON' Голубовато-зеленая; 1%-ный раствор сульфата кобальта 2,0
Тонкослойная хроматография
Техника работы и использование ТСХ в неорганическом анализе описаны в [55]. Метод ТСХ позволяет за более короткое время (по сравнению с бумажной хроматографией) разделять и идентифицировать с большей чувствительностью различные ионы (табл. 19). При разделении анионов подвижные растворители должны быть более полярными, чем при разделении катионов. Чаще всего применяют системы, содержащие различные спирты (метанол, этанол, бутанол и др.), ацетон, воду, NH4OH. В качестве сорбентов используют силикагель, крахмал, А12О3, ионообменные материалы.
В качестве примера приведены прописи двух методов разделения большого числа анионов.
Разделение на тонком слое силикагеля [732]. На слое силикагеля (с величиной зерен 5—25 мк), закрепленного гипсом,
169
Таблица 19
Разделение ионов методом тонкослойной хроматографии
Разделяемые ионы Исходный раствор Сорбент Подвижный растворитель Способ обнаружения иона Rf Время разделения, мин. Литература
NH+ Li+, Na+, К+ Rb+, Cs+ 0,4M растворы полииодидов Силикагель (воздушно-сухой) Нитрометан + бензол — — 25—45 1875]
NHj, Na+, K+, Li+, Ba2+, Sr2+, Ca2+, Mg2+ — Силикагель 4- -(-крахмал (5%) Метанол + н-бу-танол + 35%-ная НС1 (80 :10 : 10) — — — [323]
NO' S2O|~, Or Of, Ng, CN-, SCN-, BO|~, S2-, AsO|~, AsOf, NO" SO*", POI- SON-, NO;, C1-, [Fe(CN)e]8-, [Fe(CN)e]4~, CIO;, BrOs-, /Од" Смеси 1М растворов К- и Na-co-лей Кукурузный крахмал Силикагель + -|-крахмал (5%) Ацетон + ЗМ NH4OH (1:1) Ацетон -|- вода (10:1) NO;, опрыскивание 1%-ным раствором K.J в 0,17V НС1; CN-, SCN'— раствором FeCl3; NO;, опрыскивание индикаторным раствором, содержащим метиловый синий, бромтимоловый синий, фенолфталеин iVVVVVVVVS । V VVVVV 3 у v у 90 [587] [И7]
Таблица 19 (окончание)
Разделяемые ионы Исходный раствор Сорбент Подвижный растворитель Зпособ обнаружения иона Время разделения, мин. I Литера- I | тура I
no;,f-,s2o83-, SO2', CrO2", PO»-, — То же Метанол 4-и-бута-нол -|- вода (3:1:1) — — — [И7]
AsO®~, AsOg-
CN-, SCN', n;, F-, C1-, Br“, J-, [Fe(CN)e]4-[Fe(CN)6]3' Круговая TCX; SCN", [FeCN)6]‘-, [Fe(CN)6]3- CN~, SCN-, [Fe(CN)el4“, [Fe(CN)«]’- Растворы К- и Na-солей Силикагель 4-4- гипс А12О3 марки DS-5 и-Бутанол + пропанол-1 + ди-н-бутиламнн (45 : : 45 : 10); н-бута-нол-1 + бензиламин (90 : 10); пропанол-1 + + СНС1а + бензиламин (60:30:10) и-Бутанол + вода 4- пиридин Ц-+ конц. NH3 (8 : 8 : 4 :1) 3MNaCl С помощью подходящих веществ в видимой и УФ-частях спектра Опрыскивание 10%-ным раствором FeCl3 I- > SCN" > > Вг-> Cl' > >n;> >[Fe(CN)e]3-> >[Fe(CN)e]4-> > CN'> F' 55-95 [732] [818] [512]
методом восходящей ТСХ были разделены анионы: Cl-, Br~, J-, С№, SCN , [Fe(CN)e]3-, [Fe(CN)e]*-, N”
На пластинку наносили по 1 мкл анализируемых растворов в форме калиевых и натриевых солей. Подвижными растворителями были следующие смеси: н-бутанол + пропанол + ди-н-бутиламин (45 : 45 : 10) (I); н-бута-нол + бензиламин (90 : 10) (II); пропанол + хлороформ + бензиламин
(60 : 30 : 10) (III). Время подъема этих фаз на высоту 10 см (в мин.): 95 (для I); 90 (для II); 55 (для III). Для всех трех систем ряд последовательности подъема анионов на хроматограмме одинаков:
J- > SCN- > Вг~ > Cl- > N“ > [Fe (CN)e]3- > [Fe(CN)e]1- > CN- > F-.
Значения Rf практически совпадают для отдельно взятых ионов и для их смесей. Чувствительность обнаружения ионов (в мкг) после опрыскивания цветными и люминесцентными реагентами следующая: F- 0,5; С1~ 0,5; Вт- 0,1; J- 0,1; CN" 2,0; SCN- 0,1; Ng 10,0; [Fe(CN)e]3- 0,1; [Fe(CN)e]4- 0,3.
Разделение на тонком слое маисового крахмала [587]. С использованием подвижного растворителя, состоящего из смеси ацетона и 3 М NH4OH (1 : 1), за 90 мин. удалось разделить различные комбинации из 13 анионов (NO”, S2O3-, СгО3-, Ng, CN-, SCN-, ВО3-, S2-, AsO3-, AsO|”, NO”, SO3”, PO|”), взятых в форме калиевых и натриевых солей (и Н3ВО3), и идентифицировать их по цветным реакциям. Значения Rf для этих анионов располагаются вряд: SCN-> NO3 > NO3 > N3 > S2o|” > > S2- > SO3- > CrO3- > BO3- > AsO3- > PO3- > AsO3- > CN-. Чувствительность определения (в мкг) равна соответственно 0,046; 0,112; 0,116; 4,2; 2,6; 0,058; 11,8; 0,32; 12,3; 1,39; 6,2; 9,6; 0,95.
ЭЛЕКТРОФОРЕТИЧЕСКИЕ МЕТОДЫ
При проведении хроматографического процесса с наложением электрического поля (электрохроматография) имеет значение выбор подходящего электролита и сорта носителя (в случае электрофореза на бумаге и в тонком слое).
Для разделения и выделения азотсодержащих ионов электрофоретические методы немногочисленны (табл. 20), однако представлены различными видами электрофореза: электрофорезом на бумаге, тонкослойным электрофорезом и вытеснительным электрофорезом в трубке. В качестве примера укажем способ количественного определения роданидов после их электрофоретического выделения [893].
Используют бумагу Ватман № 1 (300 X 10 мм). В качестве электролита применяют 0,1 М НС1О4. При градиенте потенциала 8,82 в!см в течение 20 мин. ионы SCN- мигрируют на расстояние 2,00 ± 0,05 см. Для их обнаружения электрофореграмму опрыскивают 0,1 М раствором Fe(C104)3 или Fe(CN)3 с pH 1,0 (для получения окрашенного комплекса [FeSCN]2+). В случае прямого определения измеряют интенсивность окраски пятен или их поверхность и определяют содержание по этим параметрам (используя калибровочный график) с ошибкой 3,5—5% (для 0,01—0,2 М растворов).
Литература Ю со [893] [1039, 1338[
Способ обнаружения Опрыскивание раствором 0,1 М Fe(C104)3 или Fe(NO3)3 с pH 1,0
Сб Продолжительность, мин. 8 0 ез 1
О сч сб Д' ф Ф-О е* о е- х ф я Я О о Градиент потенциала, плотность тока 1 zvco 0Z 8,82 в/см | 100 в/см, 80 мха) см?
И Я ю сб И Разделение ионов мет Электролит Фосфатные и ацетатные буферные растворы с pH 4 и 7 соответственно 0,1 М НС1О4 Растворы NaCl и ок-сиэтилце л лю лозы
Носитель Анионообменная бумага3,1 Амберлит WB-2 и SB-2 Бумага Ватман № I*1 См. *2
Разделяемые ионы N02-, NO3- SCN- О ©» О 1 * 1 - 00 .0 оя zo
»i На бумаге. *« В тонком слое.
172
173
Таблица 21
Экстракционные методы выделения и способы определения азота и его соединений
Определяемое соединение яли иои Объект анализа Экстрагент Условия экстракционного выделения и способ определения Литература
Ns Карбид титана Бутанол [132]
NH<+ Морская вода — Экстракция в виде индофенола [1045]
HNOS Окрашенные и мутные воды Хлороформ Экстракция красителя, образовавшегося при фенолгипобромитной реакции с аммиаком; фотометрирова-ние экстракта [123]
— ТБФ — [771]
NOg м-Гептанол Экстракция окрашенного соединения с 8-аминохинолином и спектрофотометрическое определение NO3_ при 465 нм [7Ю]
HNOg — Кетоны (циклогексанон, метилизобут илкетон) Образование в органической фазе дисольватов [371]
Кетоны (ацетофенон, метил-этилкетон, метилизобутилке-тон, метилгексилкетон, неразбавленные растворы) — [373]
— Дибутиловый эфир, 0,6—6 М растворы в различных разбавителях 3,5 и 7 М HNO3; степень сольватации равна 1 [45а]
— ТБФ, 0,1 М раствор в бензоле Образование НХО3-ТБФорг (0,45—3,5 М HNO3) и (HNO3)3-ТБФорг(>3,5 М HNO3) [370]
-- ТБФ Изучение взаимодействия методом физико-химического анализа [235]
Таблица 21 (продолжение)
Определяемое соединение или ион Объект анализа Экстрагент Условия экстракционного выделения и способ определения Литература
HNOg ТБФ, 3,3 М раствор в СС14 Диалкилфосфорные кислоты Трифенилфосфиноксид, три-н-октилфосфиноксид, растворы в 1,2-дихлорэтане Метилен-бис-(ди-м-гексилфос-финоксид), растворы в СС14 Изучение влияния строения фосфорорганических кислот на экстракцию HNO3 При выделении 0,05 М растворами экстрагентов (R) в органической фазе существуют главным образом соединение HNO3-R или (HNO3)2-R [236] [279] [1273] [1131]
—' Триизоамилфосфиноксид, 0,003—0,1 М раствор в бензоле и з«-ксилоле При <1 М HNO3 экстрагируется моносольват HNO3-ТИАФО с константой экстракции 17,0±0,6 (бензол) и 17,9±0,7 (.и-ксилол) [206]
Амины (ТОА), растворы в С6Н6 и СС14 — [372]
Амины (ТОА, ТИОА и др.), растворы в ксилоле При низкой концентрации HNOg экстрагируется с небольшим количеством воды; в области умеренных концентраций экстрагируется HNO3 в безводной форме; в области высоких концентраций [H2O]o/[HNO3]o< <0,5 [751]
— Диалкилсульфиды (ди-н-октил-и ди-н-амилсульфоксид) В области <2 М HNOg экстрагируется 'безводный моносольват [208]
Лнтерату pa СО СО о О’ СО со сч о л е тн • 1—> CQ (Л сч —• CQ —' ю
Условия экстракционного выделения и способ определения О 1 2 :И И Д Im • iio 4is О § Л е о S5gz 1g ц ® «о is. i Вв§.й Su-fflK S>e<z § »§Sg »в| Z g Е«§ §вй § ggg* Ss! S Sg g “I sjl ® S U g Svl« S'g.H * s тЛио g’g.S ago S&o н ® S м=вйй>2 ils> Л « 8 M ВЛ 8 H Bggs в« — ® ФОЭ м В м ф <_« Я О Ч м В ° м Z S s е“ 5 о ? s. £ >• 5 S и 5 о 52» В =? И- Ви и я Bft £ и о ° В и Вес U о фййфа U в S £ н S 2 tf—* w « s § a w ч н m w£ » и о ° w § 2 м Л ® И ф и «5 § £J£ 5 Сб св § Ь «Л н В 5 ф ф н ф о w н г* о og р ft2 М s A S $ М = в 3 S Ею $ S К О Ф И св Я *• О ft W W<J W ф В W Bo. Мд Ф М Ч о о*& И Ф ГО • Ф р н Я Л ф ft со Ф Л
Экстрагент S о A w “ л w 5 C_J Ф “ Д со - 2 в Л g я И •§• и в ь- л sB м 11 « 2 “ <*'. 2 И л 5 И о г И о 'о ?? в? м 2 » а о Е 9 а " о AJ со св < 5 И 1 § ft§ Вн К" Н н Я Т > й о । лфьи К <5 ч сч Str® S ОЙ
Объект анализа Щелочные растворы (0,01—0,2% NaOH, 0,02—0,3о/о NaCN)
Определяемое! соединение 1 или ион । 1» 6 Z 1 9 53 0 z Z M co 0
176
При косвенном определении из полученной электрофореграммы вырезают зону, содержащую SCN~-hohh, минерализуют нагреванием с 2 мл дымящей азотной кислоты в колбе с охлаждением до полного окисления серы, содержащейся в SCN-, до SO^“. В полученном растворе определяют SO4" нефелометрически в виде BaSO4 или восстанавливают SO4” до S2- с помощью Ti(III) в среде Н3РО4. В последнем случае образовавшийся H2S отгоняют в раствор NaOH и титруют S2- раствором (CH3COO)2Hg в присутствии дитизона в качестве индикатора. Ошибка определения при нефелометрическом и тит-риметрическом окончании составляет 3,3—5,2 и 2,7—4,7% соответственно»
Предложенный в работе [1039] метод вытеснительного электрофореза, в частности для разделения смеси ацетат-, нитрат-, хлорид- и оксалат-ионов, относительно прост и требует всего 10-7 моля каждого из анионов.
Прибор состоит из длинного (1м) тонкого (внутренний диаметр 0,4 мм, внешний диаметр 0,7 мм) стеклянного капилляра, соединяющегося двумя четырехходовыми кранами с электродными сосудами, наполненными кислотой (—) и щелочью (+). В самом капилляре находится раствор NaCl и растворимый полимер — оксиэтилцеллюлоза, не содержащая ионогенных групп. После введения пробы через кран включают ток, и ионы движутся по капилляру (в соответствии с их подвижностями в данных условиях) с резкими границами между ними. Так как подвижность ионов в зонах разная, то различен и градиент потенциала, что обусловливает разное количество выделяющегося в зонах тепла. Измеряя двумя термопарами температуру и градиент потенциала в зонах, регистрируют на самописце вид электрофореграмм.
ЭКСТРАКЦИОННЫЕ МЕТОДЫ
Используемые в аналитической химии неорганических соединений азота экстракционные методы чаще всего связаны с выделением в органическую фазу кислот (азотной, азотистой, роданистоводородной) либо с косвенными методами определения (в основном фотометрическими) соединений азота при помощи экстракции продуктов различных реакций. Некоторые из подобных методов приведены в табл. 21.
Кроме того, сведения по экстракции окрашенных продуктов реакпии соединений азота с различными реагентами см. в разделе «Экстракционно-фотометрические методы» (глава VI «Фотометрические методы»).
Глава XI
ОРГАНИЧЕСКИЙ ЭЛЕМЕНТНЫЙ АНАЛИЗ
Наиболее исчерпывающую информацию о качественном и количественном определении азота и азотсодержащих функциональных групп в различных органических соединениях можно найти в фундаментальном издании [868]. В настоящем разделе будет идти речь только об элементном анализе органических соединений на содержание азота. Обзор соответствующих методов см. [210, 391]. Обзору методов функционального анализа посвящена работа [215]. Основные методы микроэлементного и функционального анализа органических соединений описаны в книге [133].
Элементный анализ органических соединений может выполняться различными методами. Классическими являются методы КьеЛьдаля и Дюма. Большое внимание в последние годы уделяется разработке и использованию методов микро- и полумикроопределения азота и применению для этих целей специальных автоматических приборов, чаще всего основанных на применении газо-жидкостной хроматографии.
ВЫДЕЛЕНИЕ АЗОТА В ВИДЕ АММИАКА
Методы выделения аммиака с последующей его отгонкой имеют большое значение в аналитической химии азота, так как при анализе различных соединений, содержащих связанный азот (органические соединения, биологические материалы, воды, нитраты и др.), последний предварительно может быть переведен тем или иным способом в эту легковыделяемую и легкоотделяемую форму.
Метод отгонки аммиака для количественного определения содержания азота в органических соединениях и материалах растительного и животного происхождения был предложен Кьель-далем еще в 1883 г. Варианты этого метода изложены в главе V. Он основан на разложении органического вещества концентрированной серной кислотой при нагревании до температуры кипения в присутствии катализаторов — передатчиков кислорода (ртути и ее соли), окислителей (К2Сг2О7, Н2О2 и др.) и сульфата калия, повышающего температуру кипения кислот. При этом азот превращается в аммиак, дающий с серной кислотой бисульфат аммо
178
ния. Реакционную смесь охлаждают, разбавляют водой, прибавляют избыток щелочи, немного тиосульфата или сульфида натрия и цинковой пыли (для восстановления азотистых соединений) и отгоняют аммиак, поглощая его титрованным раствором кислоты (НС1, H2SO4, HSBOS). (Титриметрическое окончание метода см. в главе V.)
Азот выделяется количественно в виде аммиака только из аминов и их производных. Другие азотсодержащие вещества (нитро-, нитрозо-, азосоединения и т. п.) образуют наряду с аммиаком также молекулярный азот, что приводит к получению заниженных результатов. Для определения азота в этих веществах их предварительно восстанавливают до аминов, нагревая с цинковой пылью и кислотой, или разлагают в смеси H2SO4 и Н3РО4 (3:1), содержащей небольшие количества селена и сульфата меди, или в среде H2SO4 в присутствии глюкозы и сульфата калия.
Для уменьшения вспенивания сернокислой реакционной массы при определении азота по методу Кьельдаля в пищевых продуктах, органических удобрениях и других органических веществах рекомендуется прибавлять полиэтилен в виде кусочков пленки толщиной 0,06 мм или вносить в реакционную массу смесь катализаторов в полиэтиленовом пакете [1086].
Разложение органических соединений окислением смесью хлорной и иодной кислот с последующей отгонкой аммиака по методу Кьельдаля дает также хорошие результаты [1080].
Усовершенствованная аппаратура для определения азота этим методом описана в [1319]. Методы отделения аммиака отгонкой подробно описаны в [720, стр. 274—316].
Отгонка миллиграммовых количеств аммиака
Для перегонки аммиака могут быть использованы перегонные аппараты различных конструкций, широко описанные в литературе. Эти аппараты состоят из перегонной колбы Кьельдаля, холодильника и приемника.
Классическая методика работы по [1282] заключается в следующем.
После разбавления пробы безаммиачной дистиллированной водой вносят в колбу прокаленные кусочки гранулированной пемзы, обмазывают шлиф чистейшим парафиновым маслом и собирают перегонный аппарат. В качестве приемника служит колба Эрленмейера емкостью 100 мл, которая предварительно наполняется известным количеством титрованной 0,1 или 0,02 N борной кислоты. Трубка холодильника должна прикасаться к дну приемника, чтобы во время дистилляции в ней находился столбик жидкости. Нагревать начинают небольшим пламенем. Одновременно из припаянной воронки по каплям добавляют в колбу столько прокипяченного 15%-ного раствора едкого натра, чтобы после достижения нейтральной точки был еще избыток щелочи 10 капель (для небольших количеств азота достаточно 1 капли),
179
Отгоняют две трети первоначального объема жидкости, вынимают конец трубки дистилляционного аппарата из отогнанного раствора, споласкивают небольшим количеством воды.
Несущественно отличается аппаратура и техника работы в [458].
При титриметрическом определении отогнанного (по описанной выше методике) аммиака из раствора сульфата аммония ошибка определения составляла 0,005; 0,12 и 1% при исходном содержании азота в пробе 0,5; 3 и 20 мг соответственно.
Для количественного разрушения солей аммония при подобном способе отгонки оптимальной является концентрация ионов водорода, соответствующая pH 7,4. В приемнике для полного поглощения отогнанного аммиака наливают избыток борной кислоты. Раствор в приемнике должен быть не нагретым, чтобы избежать потерь аммиака от улетучивания. Вся аппаратура должна быть изготовлена из специального стекла, устойчивого к щелочным средам.
Серусодержащие органические вещества также переходят в дистиллят и мешают последующему колориметрическому определению аммиака. Мешают также соединения ртути, нитрит-, цианид-и роданид-ионы.
Отгонка микрограммовых количеств аммиака
Микроопределение аммиака имеет большое значение при анализе органических материалов, вод, пищевых продуктов, физиологических материалов, в которых аммиак содержится в микроконцентрациях. Особое значение имеет отделение аммиака в присутствии органических азотсодержащих соединений. Очень важно в этих случаях использование особочистых (безаммиачных) реагентов, дистиллированной воды. Атмосфера лаборатории также должна контролироваться.
При микроопределении аммиака используют простую отгонку, отгонку с водяным паром, способ аэрации при обычном и пониженном давлении, диффузионные методы. Подробно об этом см. [720, стр. 292].
Метод отгонки микрограммовых количеств аммиака по [966] заключается в следующем.
350—400 мл водного раствора соли аммония, содержащего 0,01—0,2 мг азота, подщелачивают с помощью 20 мл безаммиачного 33%-ного раствора едкого натра. Из этого раствора отгоняют 90 мл в приемник, содержащий 5 мл 0,01 N НС1.
Этим методом удается определить (в опытах с хлоридом аммония) от 0,01 до 0,2 мг азота с отклонением в результатах до 1,6 мкг.
180
Метод отгонки с водяным паром [1184] ускоряет выделение и позволяет работать с малыми количествами жидкости.
Метод аэрации см. [1404], диффузионный метод — [621].
Условия отгонки аммиака при анализе различных объектов см. в соответствующих разделах главы XII. В качестве примера использования дистилляции как метода отделения от мешающих компонентов можно привести работу [717], в которой после отгонки аммиака определяют микроколичества азота методом спектрофотометрии в потоке, обрабатывая полученные данные с помощью компьютера.
ТИТРИМЕТРИЧЕСКОЕ И ФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ АММИАКА
В дистилляте аммиак определяют чаще всего титриметрически или фотометрически.
К группе титриметрических методов принадлежат прежде всего различные варианты метода Кьельдаля, пригодные для анализа разнообразных объектов [557]. Обзор методик микроопределения азота в органических веществах методом Кьельдаля приведен в работе [1335]. Подробно о титриметрических методах определения аммиака см. в главе V.
Микро- и полумикроопределение азота в органических веществах может быть осуществлено Йодометрическим титрованием избытка КВгО3 после разложения вещества по Кьельдалю, окисления полученного (NH4)2SO4 гипобромитом калия, образующимся при взаимодействии КВгО3 и КВг [1284].
5—10 мг органического вещества (30—50 мг при полумикроопределении) вносят в круглодонную колбу с обратным холодильником, прибавляют 0,7 г смеси 0,625 г K2SO4 и 0,075 г HgSO4 (2,8 г при полумикроопределении) и 3 мл концентрированной H2SO4 (6 мл при полумикроопределении). В случае нитро-, нитрозо- и азосоединений прибавляют 0,2—0,3 г глюкозы и 4 мл H2SO4 (1 г глюкозы и 10 мл H2SO4 при полумикроопределении). 1—2 часа кипятят смесь, охлаждают и споласкивают обратный холодильник 25 мл воды. Колбу помещают в холодную воду, приливают 20 мл 0,04 А раствора КВгО3 (20 мл 0,1 N раствора при полумикроопределении), прибавляют 0,5 г КВг (1 г при полумикроопределении) и закрывают колбу пробкой, в которую вставлена закрытая пробкой N-образная трубка с 25 мл 25%-ного раствора NaOH (25 .ил 50%-ной щелочи при полумикроопределении). Легким встряхиванием колбы раствор щелочи постепенно вливают в нее, содержимое колбы перемешивают и оставляют на 20 мин. Прибавляют в колбу 6 мл 7N НС1 (12 мл при полумикроопределении) и закрывают ее пробкой с N-образной трубкой, содержащей 10 мл 10%-ного раствора KJ (20%-ного раствора при полумикроопределении). Через 5 мин. после прибавления кислоты сливают в колбу раствор из N-образной трубки. Еще через 5 мин. титруют выделившийся иод 0,02 JV раствором Na2S2O3 (0,05 TV при полумикроопределении) в присутствии крахмала. В тех же условиях выполняют контрольный опыт.
181
Для определения сантимиллиграммовых количеств азота в органических соединениях предложен титриметрический метод, состоящий в разложении вещества серной кислотой в запаянной кварцевой трубке при нагревании, пропускании реакционной смеси через ионообменную смолу основного характера и титровании образовавшегося NH4OH раствором НС1О4 с потенциометрической регистрацией КТТ [782]. Продолжительность анализа 12 мин. Определяемый минимум 30 мкг N. Абсолютная ошибка +0,25%. Металлы мешают определению.
Предложен фотометрический метод определения азота в органических веществах, основанный на сожжении их в атмосфере О2, взаимодействии образующихся окислов с хлоргидратом а-нафтилэтилендиамина в присутствии тг-аминобензолсульфамида и на фотометрировании окраски получающегося азокрасителя [285].
В работе [20] описана модификация микроопределения азота в ароматических нитро- и полинитросоединениях методом Кьельдаля, включающая их предварительное восстановление. В [172] микрометод Кьельдаля усовершенствован применительно к анализу полимерных соединений: для разложения анализируемого вещества используют не концентрированную H2SO4, а дымящую H2SO4 (в смеси с K2SO4 и селеном), что позволяет сократить продолжительность разложения полимерных образцов. Образующийся NH3 определяют фотометрически (без перегонки) с реактивом Несслера. Ошибка определения не превышает ± 0,3%.
ГАЗОХРОМАТОГРАФИЧЕСКОЕ ОПРЕДЕЛЕНИЕ АЗОТА
С помощью приборов для автоматического элементного анализа азот обычно определяют методами газовой и газо-жидкостной хроматографии одновременно с углеродом и водородом. Подробно эти методы .описаны в главе «Газометрические методы».
Предварительно анализируемое органическое соединение сжигают за счет кислорода окислителя под действием катализатора [337] или разлагают пиролитически. При этом весь азот переходит в N2 [84, 404]. В качестве окислителя наиболее эффективной оказывается окись меди; используют для этой цели также окиси никеля, марганца. Органические вещества сжигают по принципу Дюма в автоматическом анализаторе Колемана, использующем простое газометрическое измерение N2 [1314.] Быстрый метод сожжения органических веществ для количественного определения в них азота предложен в работе [1153]. Разложение проводят в атмосфере гелия с использованием индукционной высокочастотной печи.
Продукты минерализации затем поступают в газовый хроматограф для разделения и количественного определения. По площади 182
пика N2 с помощью калибровочного графика определяют содержание азота с точностью до ±0,2 абс. %.
Сорбентами служат молекулярные сита 5А [337, 902, 1107, 1153, 1209, 1314], 13Х [562], карбовакс 20М [648], селектосорбен [755], хромосорб [1161], активированный уголь [404], силикагель [84, 404, 1401] и др.
Методом газо-жидкостной хроматографии определяют азотсодержащие летучие компоненты в твердых веществах и высоко-кипящих органических соединениях [1161, 1209]. Обзор методов определения азота, водорода и углерода в органических соединениях методом газо-жидкостной хроматографии см. [155].
В качестве газа-носителя чаще всего используют гелий, иногда — неон [755]. Детектируют N2 после хроматографического выделения с помощью детектора по теплопроводности [84, 648, 902, 1061, 1153, 1161, 1209], ионизационного аргонового детектора [755], водородно-воздушного пламенного детектора [815] и др. Предложенный в работе [337] метод одновременного определения кислорода и азота в органических соединениях состоит в сожжении вещества с образованием N2 и одновременном превращении кислорода в СО над слоем сажи. Последующее определение N2 и СО осуществляется методом газовой хроматографии.
Навеску 0,005—0,010 г анализируемого вещества в кварцевой или платиновой лодочке помещают в кварцевую трубку диаметром 0,6—0,7 см и объемом 10 мл, содержащую слой «никелированной» сажи (способ приготовления см. ниже), нагретой до 900° С. В трубке создают разрежение до остаточного давления 2 мм рт. ст., выдерживают 5 мин., затем в течение 3 мин. вводят гелий, очищенный от О2. Снова создают разрежение на 5 мин. для окончательного удаления воздуха, затем разлагают навеску в атмосфере гелия в течение 5 мин. при помощи газовой горелки и вытесняют продукты разложения в течение 10 мин. током гелия со скоростью 5 мл/мин в хроматографическую колонку (60,0 X 0,4 см), заполненную молекулярными ситами 5А с величиной частиц 0,05—0,10 см, высушенными в течение 2 час. в вакууме при 300° С, при скорости газа-носителя (гелия) 50 мл/мин. Расчет производят по величине площадей соответствующих пиков N2 и СО при помощи калибровочных кривых.
Способ приготовления «никелированной» с а -ж и: смешивают раствор формиата никеля с сажей (Ni: С — 1 : 1) и затем нагревают смесь до получения пасты, которую сушат и прокаливают в токе N2 или Не при 900° С в течение 3—4 час.
В качестве примера можно привести работы по определению азота методом газовой хроматографии в газах, образующихся при радиолизе органических соединений [1401]^ в аминокислотах и других органических соединениях [815], в газах над закупоренным пивом [562].
В связи с тем, что газовая хроматография позволяет иметь дело с довольно малыми количествами анализируемых материалов,
183
особое значение этот метод имеет для микро- и ультрамикроопределений азота из милли- и микрограммовых навесок [648, 755, 902, 1061, 1107].
Для одновременного ультрамикроопределения азота, углерода и водорода применим следующий метод [902].
2—3 мг органического вещества в лодочке вносят в трубку для сожжения, имеющую камеру сожжения и часть, содержащую слой окислителя, нагретого до 700° С, слой Ag-ваты (500° С) и слой меди (500° С), полученной термическим разложением оксалата меди в инертной атмосфере. За трубкой для сожжения следуют две поглотительные трубки (для поглощения воды и СО2), содержащие соответственно смесь СаС12 с кварцем и молекулярные сита 5А. В каждую из поглотительных трубок помещен также слой кварца той же длины, что и слой сорбента. Обе трубки соединены друг с другом и каждая — с детектором по теплопроводности, термостатированным при 60° С и снабженным интегратором и двумя сосудами для смешения продуктов сгорания с гелием. Перед началом сожжения вещества установку продувают током гелия (50 мл/мин), на слой кварца в поглотительной трубке надвигают печи, нагретые до 300°С, ток гелия прекращают, в трубку для сожжения вводят 50 мл О2, при помощи магнита лодочку с веществом вводят в камеру сожжения, нагретую до 900— 1000° С, и за 30—60 сек. сжигают вещество. Продукты сожжения вытесняют током гелия: азот — прямо в ячейку детектора, а Н2О и СО2 — в разделительные трубки. После отсчета азота сдвигают печь, нагретую до 300° С, со слоя кварца на слой молекулярных сит 5А, за 1—2 мин. десорбируют СО2 и током гелия подают его в ячейку. После снятия отсчета углерода аналогично десорбируют воду и током гелия подают ее в ячейку.
При вычислении умножают соответствующие значения площадей пиков на калибровочные факторы, т. е. значения площадей, соответствующие 1 мг N, С, Н, которые определяют, анализируя стандартные вещества.
Эффективно для микроопределений использование автоматического CHN-анализатора, с помощью которого за 6 час. можно проанализировать 24 пробы [648]. Сообщается об автоматическом CHNO-микроанализе с помощью нового анализатора модели 1102 («Carlo Erba», Милан), снабженного электронными весами и небольшим компьютером [1318]. Расчетный метод оценки данных анализатора по поправочным факторам, получаемым с использованием специальных устройств с программной системой, приведен в работе [766].
ВОЛЮМОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ АЗОТА
Классическим волюмометрическим методом определения азота в органических соединениях является метод Дюма, заключающийся в сожжении вещества в атмосфере СО2 нагреванием с СиО, восстановлении металлической медью до N2 и в измерении объема выделяющегося элементного азота. Подробно об этом методе см. главу IX «Газометрические методы».
184
Обзор по количественному органическому элементному анализу, в том числе и по определению азота методом Дюма, приведен в [385, 534].
Описан объемный метод определения азота в органических соединениях сожжением в атмосфере получаемого электролитически кислорода [1328]. Абсорбция окислов азота в слое СиО устранена посредством восстановления последней парами органического растворителя, например бензола, и выделения при этом азота из СиО.
1—4 мг вещества в Pt-лодочке вносят в трубку для сожжения, содержащую (начиная от носика) слой Си (7 см), СиО (8 см) и снова Си (13 см), нагретые до 720—750° С. (Слой Си длиной 4 см выступает из печи.) При сожжении галоид- и серусодержащих веществ в трубку сожжения помещают серебро.
Трубку для сожжения 3—5 мин. продувают током СО2 (30—35 мл/мин), включают электролизер, устанавливают скорость тока О2 6 мл/мин и медленно сжигают вещество в печи, нагретой до 800—850° С. По окончании сожжения ток О2 прекращают и пропускают через трубку для сожжения СО2. После появления в азотометре микропузырьков ток СО2 пропускают через барботер с СвНв со скоростью 20—30 мл1мин. Через несколько минут, когда останется невосстановленным слой СиО длиной ~2 см (в печи есть щель, через которую видно изменение цвета наполнения трубки для сожжения), СО2 пропускают в трубку для сожжения, минуя барботер. Через 5—10 мин. после появления микропузырьков отсчитывают объем N2.
Предложена автоматическая двухтрубчатая аппаратура [1076], позволяющая за 11—13 мин. выполнить два параллельных определения С, N, О или одновременное определение СиО или N и Н.
В последнем случае в конец одной из трубок помещают слой (1—2 см) СаН2. Образовавшуюся при сожжении вещества в этой, трубке смесь N2 и Н2 измеряют в азотометре. Во второй трубке (без СаН2) при сожжении образуется только N2. Содержание Н вычисляют по разности первого и второго объемов.
Для автоматического непрерывного определения ультрамикроколичеств С, Н, N в органических смесях существует устройство, позволяющее определять одновременно три элемента (400 мкг и более) [1380]. Оно основано на использовании метода сжигания исследуемого вещества в токе Не -|- О2 и дальнейшем последовательном исследовании продуктов сгорания.
Полуавтоматический микрогазоволюмометрический метод определения азота в органических веществах при применении в качестве катализатора окисления Со3О4 описан в работе [1368].
Особое внимание уделяется микрометодам определения азота в органических соединениях: например, разработана модификация метода Дюма для анализа трудносгорающих веществ со стандартным отклонением + (0,19—0,24)% [1167]; микрометод определения азота диазоаминосоединений (длительность анализа
185
Таблица 22
Методы элементного анализа органических соединений на содержание азота
Метод определения Объект анализа Предел обнаружения • Точность Примечание Литература
Г азометрический, Различные органические (5s-7). Навеска 3—15 мг; определяют N по [37]
по теплопроводно- соединения •IO-8 теплопроводности смеси Не -Ь N2 пос-
сти ле пиролиза и поглощения СО2 и Н2О
То же — Стандартное откло- Использован самоинтегрирующий ав- [614]
нение 0,18% тематический анализатор с катарометром
» — — N2 определяют по теплопроводности в токе Не [901]
» •— — Используют катарометры с W-нитя-ми; навеска 2—2,5 мг [849]
» — Стандартное откло- Используют катарометр с W-нитью; [1392]
нение 0,3% навеска 0,1—0,9 мг
» -— — Разлагают анализируемое вещество над смесью СиО и Си в токе Не и О2 [672]
.и-Динитробензол, уголь, полученный пиролизом сахарозы] — — Навеска ~0,5 мг [1133]
Газометр ический, Низкокипящие жидкие — — Используют автоматический CHN- [632]
по измерению объ- вещества (г^яп 35-290°С) анализатор Перкин — Эльмер, модель
ема n-Нитроанилин, дифе- — Среднеквадратич- 240; навеска 0,5—3 мг Навеска 1,5—5,0 мг [1166]
нилтиокарбазон, дифе-нилкарбазид, дифенил-карбазон, фенилсемикарбазид, 2,4-динитрофе-нилгидразин, уротропин, нал ошибка ±0,09% Средне квадр этичная ошибка ±0,13% Навеска 0,5—2,5 мг [1166]
меламин
Таблица 22 (продолжение)
Метод определения Объект анализа Предел обнаружения Точность Примечание Литература
Газометрический, по измерению объема Гетероциклические, алифатические и монофтор-органические соединения — — Использован метод сожжения без постоянного окислительного наполнения в трубке [414]
Гетероциклические, многоядерные ароматические, алифатические соединения с длинными цепями, полимеры, высо-кофторированные соединения Метод Дюма с окислителем Со3О4; сжигают образец при 700°С без подачи кислорода [413]
Многие трудносжигае-мые вещества (паранитроанилин, фенилтиомочевина, ацетанилид и др.) — Отклонения от теоретической величины ±0,2% Микрометод Дюма (сожжение в атмосфере О2) [1049]
Водные растворы органических соединении 1 мг/мл Абсолютная ошибка ±0,2% Объем анализируемого раствора 150 мкл (ультрамикро- и микромето-ды) Навеска 30—90 л«кг Навеска 3—9 мкг [926]
Твердые вещества — Абсолютная ошибка ± 0,3% Абсолютная ошибка ± 2% [926] [926]
Легко- и трудносжи-гаемые органические соединения — Абсолютная ошибка 0,1—0,2% Модификация микрометода Дюма— Прегля; пиролитическое разложение навески [401]
00 Азотсодержащие органические вещества — — Сочетание газообъемного метода с ГХ; пиролитическое сожжение [404]
Таблица 22 (продолжение)
Метод определения Объект анализа Предел обнаружения Точность Примечание Литература
Газометрический, по измерению объема Органические соединения любых классов — Погрешность метода ± 0,0—0,20% Скоростной метод с пиролитическим разложением вещества; навеска 3—5 мг [405]
Пикриновая и пикрами-новая кислоты, азобензол, карбазол, кофеин Ошибка 0,ll-о.83% (при содержании 8,38— 28,85%) Упрощенный вариант метода Дюма— Прегля; навеска 2—4 мг [35]
Различные органические соединения — Ошибка ± 0,05% Сожжение в токе СО2 + О2; катализатор ВаСгОа [586]
То же Децимилли-граммовые количества Стандартное отклонение от ± 0,05 до ± 0,03% Сожжение в заплавленной трубке [392]
» — Ошибка 0,14% Микроопределение методом Дюма одновременно в 8 навесках веществ [1071]
» — Средняя абсолютная ошибка ± 0,06% Автоматическое определение N сожжением органического вещества в токе чистого О2 [728]
» — — Субмикрометод определения N сожжением в атмосфере О2 в запаянной трубке; навеска 0,3—0,5 мг [848]
» Децимилли-граммовые количества Абсолютная ошибка ± 0,2—0,3% Навеска от 1—3 до 0,3—0,6 мг\ используют ультрамикроазотометр [1165]
» Абсолютная ошибка 0,2% Микрогазометрический метод; навеска 1,5—4 мг [1368]
Таблица 22 (продолжение)
Метод определения Объект анализа Предел обнаружения Точность Примечание Литература
Газометрический, по измерению объема Газохроматогр афи-ческйй Различные органические соединения То же Трудносжигаемые вещества То же Сельскохозяйственные материалы (почвы, подсолнечник, пшеница, овес, казеин, молоко и ДР-) Различны® органические соединейия То же Сантимил-лиграммо-вые количества Стандартное отклонение 0,17% Стандартное отклонение ± 0,1— 0,14% Средняя абсолютная ошибка ± 0,02 -0,05%, средняя относительная ошибка ± 0,2-0,5% Абсолютная ошибка ± 0,2% Абсолютная погрешность 0,2— 0,3% Полумикрометод; навеска 15—25 мг; сочетают с титриметрическим определением NHa-групп после превращения в NH3 Метод сожжения в запаянной трубке; навеска 30—80 мкг; измеряют изменение давления газа Метод Дюма — Прегля; навеска от 3—10 до 0,4—1 мг Сочетают с методом ГХ; навеска 50—100 мг Метод Дюма; навеска 100—300 мг Окисление, в замкнутой системе кислородом окиси никеля при 900— 1000°С; навеска 0,3—1,5 мг Разложение в замкнутой системе в атмосфере инертного газа; сорбент — активированный уголь БАУ-2; навеска 1—1,5 мг [895] [895] [1054] [796] [1085] [262, 407] [403, 406]
ОО СО
s
Таблица 22 (nродолжение}
Метод определения Объект анализа Предел обнаружения Точность Примечание Литература
Газохроматографический Различные органические соединения — — Навеска 1—5 мг\ измерения с помощью катарометра [234]
То же — Навеска 4—6 мг; сжигают в вакуумированной трубке над СиО; сорбент—25% триэтаноламина на тефлоне; детектор по теплопроводности [219]
» — — Сорбент—карбовакс-1500 и активированная сажа на тефлоне; применяют катарометр [839]
Различные группы гомологов Предел определений 3,5% — Используют CHN-анализатор «F and М» (модель 180) [330]
Различные органические соединения — Абсолютная ошибка ± 0,4—0,5% Сорбент—силикагель; используют катарометр [505]
То же — — Навеска 40—80 мкг; сорбент—силикагель; используют микрокатарометр [508]
» Навеска 0,6—0,8 мг; сорбент—пора-пак; используют анализатор «Хьюлетт—Паккард» (модель 185) с катарометром и электронным интегратором [774]
» — Абсолютная ошибка ± 0,3% Навеска 0,6—1 мг; используют детектор по теплопроводности [649]
» 0,1—0,005% Разложение по методу Кьельдаля; навеска 4 мг; используют катарометр [712]
Таблица 22 (продолжение}
Метод определения Объект анализа]! Предел обнаружения Точность Примечание Литература
Метод Кьельдаля Гетероциклические соединения (д-пиколин, акридин, индол, бензимидазол, о-оксихинолин и ДР-) — Относительная ошибка 0,7% Навеска 0,05—0,20 г; дистиллят улавливают 0,1 N H2SO4 и избыток кислоты титруют 0,1 N NaOH [426]
Пищевые продукты, органические удобрения — — Для уменьшения вспенивания реакционной сернокислой массы добавляют полиэтилен [1086]
Различные органические соединения — Абсолютная ошибка ± 0,3% Навеска 2—4 мг; с помощью ионного обмена (NH4)iSO4—>NH4OH-»NH4J; иод оттитровывают раствором Na2S2O3 [1276]
То же — — Субмикрометод (навески 30—80 мкг}; титрование 0,01 W Na2S2O3 [507]
» — — Улавливают NH3 раствором Н3ВО3 и титруют борат аммония раствором кислоты [967]
» — — Минерализация хромовой смесью [336]
Ацетанилид, триоксиметил аминометан , пшени- — Абсолютная ошибка 0,3% Окисление смесью НС1О4 и HJO4 [1080]
Фотометрический ца Различные органические соединения — — Микрометод; навеска 3—5 мг; фото-метрирование без отгонки NH3 с реактивом Несслера [172]
То же Использование автоматических установок с фотоэлектроколориметрами в качестве анализаторов [521]
Таблица 22 (продолжение)
Метод определения j Объект анализа Предел обнаружения Точность Примечание Литература
Фотометрический Различные органические соединения От 2,5 мг до 5 мкг — Разложение с помощью НСЮ<; фото-метрирование NH3 с реактивом Несслера [733]
Вода — — После отгонки из пробы NH3 определяют с реактивом Несслера [960]
Хлоразид, гидрохлор-тиазид, ацетазоламид, триазурол — Абсолютная ошиб-ка< ± 0,3% Разложение пробы по методу Кьельдаля и фотометрирование (без отгонки NH3) с пиридин-пиразолоновым реактивом [905]
Нитросоединенин, амиды, нитрилы,амины, гетероциклические соединении 0,005— 1,0% — Реакция с сульфамииовой кислотой и 1-нафтиламином (после .поглощения продуктов сожжении пробы) [1412]
Титриметрический Различные органические соединения Органические вещества разлагают в присутствии порошка металлического Ti; из нитрида Ti при подщелачивании выделяют NH3, отгоняют и определяют титрованием [365]
То же Разложение пробы при помощи K2S2O8 и Н2О2 в кислом растворе в присутствии Ag2SOa, отгонка NH3 из щелочной среды и ацидиметрическое титрование [743]
» Термическое разложение вещества в присутствии СиО в вакуумированной системе, восстановление окислов азота до N2, связывание его в Mg3N2, разложение последнего кислотой, титрование образовавшегося NH3 иодометрически [364]
7 В. Ф. Волынец, М. П. Волынец
Таб лица 22 (окончание)
Метод определения Объект анализа Предел обнаружения Точность Примечание Литература
Гр авиметр иче ский Органические НИЯ соедине- — Абсолютная ошибка ± 0,3% Определяют NH3 после разложения пробы (40—50 мг) по методу Кьельдаля (без дистилляции) в виде тет-рафенилбората аммония; мешают К, Hg, Си [317]
Активационный анализ То же 5-10-5 г — Активация быстрыми нейтронами по реакции 14N (п, 2n)uN; определению мешает Вт [416]
» 0,0001— 0,01 % Точность до 2% Анализ занимает 5—10 мин. [790]
» 0,2-1,2 г Относительная ошибка 3,3% Облучение потоком ЗЛО7 нейтр!см2^ • сек [114]
Регистрируется излучение с энергией 10,82 М-эв, образующееся при ндерной реакции (п, у) на ядрах N [1348]
25 мин.) [166]. Одновременное полуультрамикро- и ультрамикроопределение С, Н и N в органических веществах газометрическим методом выполнено в работах [926, 927]. При навеске анализируемого вещества 3—9 мкг абсолютная ошибка определения достигает ± 2%.
Для определения азота в гетероциклических соединениях предложена модификация метода Дюма с улучшенной техникой микросожжения [1249]. Элементный микроанализ органических веществ по методу Дюма—Прегля описан в работе [1334].
Различные методы анализа объектов органического происхождения (угли, нефти и нефтепродукты, корма, пищевые продукты и др.) более подробно описаны в соответствующих разделах главы XII.
Сводка методов анализа некоторых органических веществ на содержание азота приведена в табл. 22.
Глава XII
ОПРЕДЕЛЕНИЕ АЗОТА
В ПРИРОДНЫХ И ПРОМЫШЛЕННЫХ ОБЪЕКТАХ
Объекты, которые анализируются на содержание азота, очень многочисленны и разнообразны по своему характеру: объекты окружающей среды (воздух, почвы, воды, растения); минеральное сырье (минералы, руды, породы); горючие материалы (каменный уголь, торф, нефть, нефтепродукты, битумы); полупродукты и продукты металлургического производства (металлы, стали, сплавы, нитриды, карбиды, окислы, технологические газы); удобрения; органические соединения различного происхождения (в том числе пищевые продукты и биологические материалы) и т. д.
Многообразны и методы анализа. Здесь используется практически весь арсенал существующих методов определения азота — физические, физико-химические и химические.
Применяемые методы разложения, разделения и концентрирования варьируют в зависимости от специфики анализируемого объекта, от его агрегатного состояния, от содержания азота и др.
Ниже приведены методы анализа различных групп объектов. Здесь не уделяется внимания обсуждению представительности проб, хотя это очень важное обстоятельство. Но это — специальный и достаточно общий аналитический вопрос, который выходит за рамки тематики настоящей книги.
В связи с большим объемом опубликованного материала по определению азота в различных природных и промышленных объектах мы сочли целесообразным систематизировать его и по ряду разделов представить в виде таблиц. Отдельные, наиболее интересные, с нашей точки зрения, методики приведены в виде прописей.
Поскольку анализу органических соединений посвящена отдельная глава, в настоящей главе будут описаны методы определения азота лишь в отдельных конкретных веществах органической природы: угле, нефти и нефтепродуктах, растениях, пищевых продуктах и биологических материалах.
7*
195
МИНЕРАЛЫ, РУДЫ, ПОРОДЫ, ПОЧВЫ, АЛМАЗЫ, МЕТЕОРИТЫ
Особенности методов определения азота в почвах и силикатах определяются формами его нахождения в этих объектах. Если в почвах подавляющая часть азота находится в слабосвязанном состоянии и легко экстрагируется с помощью вытяжек, то в силикатных породах и минералах основная часть азота входит в состав кристаллической решетки силиката и может быть выделена только при полном разрушении последней. Поэтому в случае анализа почв и силикатных пород используются различные методические подходы. При анализе почв азот определяют в вытяжках, а для минералов и пород методики анализа основаны на предварительном разложении силикатов.
В таблице 23 приведены методы определения азота в минералах, рудах, почвах, алмазах и метеоритах.
Сводка методов определения общего, нитратного, нитритного, аммонийного и органического азота в почвах приведена в [352, стр. 64]. Универсальный метод определения всех неорганических форм азота в почвах (NH4, NO3, NCK?) приведен ниже [561]. Необходимость разработки этого метода была обусловлена тем, что существовавшие до этого многочисленные способы определения неорганических форм азота оказались непригодными для анализа почв, содержащих биологический материал. Для этого необ--ходимы быстрые, избирательные и точные методы определения NH^, NO3 и NOa.B мутных и окрашенных почвенных экстрактах, которые позволяли бы измерять содержание этих форм в количестве от 0,1 до 2 мг и избегать влияния органических веществ.
В описываемых ниже методиках все формы азота переводятся в аммоний и определяются в виде NH3. Аммиак отделяют отгонкой с паром, собирают в раствор борной кислоты и определяют титрованием стандартным раствором кислоты. Влияние азотсодержащих органических соединений, подвижных в щелочной среде, устраняется использованием небольших количеств окиси магния вместо КОН и уменьшением времени отгонки. При отгонке с MgO только аммиак является подвижным компонентом. Можно сократить время, требуемое для количественного восстановления в растворе нитрата и нитрита до аммиака, применяя сплав Де-варда в тонкораздробленном виде. Определение нитрата в присутствии нитрита возможно с использованием сульфаминовой кислоты, которая количественно разлагает нитрит до молекулярного азота при комнатной температуре. Сульфаминовая кислота не реагирует ни с аммонием, ни с нитратом и не мешает их определению с MgO и сплавом Деварда в условиях отгонки с паром.
Определение аммонийного азота. Наливают 5 мл раствора смеси борной кислоты с индикатором в приемник — колбу Эрлен-мейера объемом 50 мл, на которой отмечен объем 30 мл, и помещают колбу 196
Методы определения азота в минералах, рудах, породах, почвах, алмазах, метеоритах
Литера* тура 1О СО* иэ 00* со с© t“— *4* tO sr *ч* М*
Примечание 1 Определяют суммарный азот из навесок 50—300 мкг\ детектор по теплопроводности Определяют общий азот Дистилляцию NH3 проводят перегретым водяным паром за 5—7 мин. из 150 мл Определяют К Н4+, NO3~ и NO2_ Определяют NH4+ и NO3_ (проба 2 г) Определяют NH4+ с реактивом Несслера Используют индофенольную реакцию Автоматическое определение NH4+ я NO3“ индофенольным методом Определяют NO3~ нитрованием 7-окси-4,8-диметилкумарина
Точность 1 хдЭ । и • я ю н Я ё о g © 1 © © 00 ф z-о. , zi- ю И со W 4 1 о 1 1 1 1 5 |?"я OSS О" О
Предел 1 обнаружения z S 1 <54 в । о- § । । । । i Ю ° Г-Г О ° V/
Метод определения « 0 g Ф sH ее о г f и g g .5. « к >и И = О >Q< Ч о s 2 и u «s 3 ф м ч ь © ft и в< д в И ь- л И о g а Й 2 я с“< » » в1 и О S S и § и S и Я & 2 ы © ы ф ч и & ч 8 ct я °>е<а> о=« £ й * * 2 £ Ф св S4 со § ф _ 5 q нк Q
Анализируемый объект св Ы Ф -^ © © ESI Ё § м S Я s § св *3 ф J5 ИИ M м 2 о. Ь ч Йч § 5 S я &S и ° 3 5 z = «в 3 нд и 5 a s 3*®*св иРч § «Зв 3 g g S Sg, & & й w Я S га 2 я ®\о Ъ Й и оgя и“ §g gg ч - ф 5 о а® м И т?- |gs3g|§3 S3 а| 5s? и g^§g gg§ gg Sg g aggrS ё! ё
197
Таблица 23 (продолжение)
Анализируемый объект Метод определения Предел обнаружения Точность Примечание Литература
Почвы Фотометрический Микрограммо-вые количества (50—1750 мкг NO3/e) Ошибка определения < ± 5% Определяют NO3~ с применением хромотроповой кислоты [1440]
Почвы, растения, дождевая вода » -— — Определяют NO3_ по реакции окисления им дифенилбензиди-на или дифениламина [1481]
Почвенные экстракты » 0,02 мкг NO3~/мл — То же [1457]
» ^1 мг NNq3_ Определяют NO3_ путем предварительного восстановления до NH4+ и последующего определения по фенолятгипохло-ритной реакции [1445, 1474]
Почвенные растворы и экстракты Спектрофотометрический (УФ) 0,5 ч. на млн. N Стандартное отклонение ± 0,480 мкг Определяют нитритный азот с сульфаминовой кислотой [1456]
Минеральное сырье Фотометрический — — Определяют NO2~ с синим основным К [1452]
Почвенные вытяжки Полярографический — — Определяют NO3" на фоне ацетата уранила [1470]
Горные породы и минералы (газовая фаза) Г азохроматографический — — Определяют N2 на колонках с молекулярными ситами [879]
Включения в минералах (до 0,03 мм) Спектральный эмиссионный 10-8—10-9 г N Полуколичественный анализ; температура плазмы искрового разряда ~20 000° К, навеска 0,001 мг [1439]
Таблица 23 (окончание)
Анализируемый объект Метод определения Предел обнаружения Точность Примечание Литература
Почвы Масс-спектральный >0,1 мг — Определяют 16N по формуле А — 100/?/(2 -]- R), где R — отношение пиков 28/29 ]791]
Горные породы и РУДЫ Т-Активационный '—- — Облучают порошковые пробы жестким тормозным 7-излуче-нием с регистрацией излучения радиоактивных продуктов [1441]
Алмазы природные, графит Газохроматографический n-10-2% N Средняя относительная ошибка 10-15% Пробу (20—100 мг) сжигают при импульсной подаче О2 в потоке инертного газа-носителя; сорбент — цеолит СаА; детектор по теплопроводности [1443]
Алмазы природные ИК-спектроскопия, люминесценция Радиоактивационный 0,2 ч. на млн. N Ошибка определения ±20% Появление осцилляций в спектрах возбуждения люминесценции при наличии в алмазе примесного азота Фотоядерная реакция 14N('f, n)13N; поток 2-Ю10 фотон/сек-*см^ [1444а] [1476]
Алмазы синтетические | ЭПР — — 1422]
Метеориты Активационный — — Нейтронная активация [1461, 1479]
под холодильник дистилляционного аппарата. Аликвотную часть анализируемого раствора, содержащую до 2 мг неорганического азота, помещают в колбу Кьельдаля, доводят водой объем до ~ 20 мл и добавляют 0,2 г окиси магния. Присоединяют колбу к перегонному аппарату и подключают к системе источник пара. Через 3—5 мин., когда количество дистиллята достигнет в приемнике 30-миллилптровой отметки, перегонку прекращают, открыв выход пара в атмосферу, обмывают конец трубки холодильника и определяют азот в дистилляте титрованием 0,005 N серной кислотой. В конце титрования окраска раствора меняется от зеленой до светло-розовой.
Определение суммы нитратного и нитритного азота. После удаления аммония из образца, как описано выше, быстро добавляют 0,2 г сплава Деварда через дополнительное отверстие в пробке колбы Кьельдаля, тотчас закрывают пробкой это отверстие и определяют аммонийный азот отгонкой с паром по методике, описанной выше.
Определение суммы аммонийного, нитратного и нитритного азота. Действуют по прописи метода определения аммонийного азота, но одновременно с окисью магния добавляют сплав Деварда.
Определение суммы аммонийного и нитратного азота. Следуют методике определения суммы аммонийного, нитратного и нитритного азота, но перед добавлением окиси магния и сплава Деварда обрабатывают раствор в колбе Кьельдаля одним миллилитром 2%-ного раствора сульфаминовой кислоты и встряхивают содержимое колбы в течение нескольких секунд для разложения нитрита.
Определение нитратного азота. Выполняется пропись метода для определения нитратного и нитритного азота, но перед выполнением анализа следует обработать раствор сульфамииовой кислотой для удаления нитрита аналогично методике, описанной для определения суммы аммонийного и нитратного азота.
Приготовление реагентов. Окись магния. Нагревают окись магния (ч.д.а.) в электрической муфельной; печи при 600—700° С в течение 2 час. Охлаждают продукт в эксикаторе над гранулами КОН и сохраняют в тщательно закрытом сосуде.
Раствор борной кислоты с индикатором. Растворяют 20 г борной кислоты (х. ч.) в ~ 700 мл горячей воды и переносят охлажденный раствор в литровую колбу, содержащую 200 мл 95%-ного этанола и 20 мл раствора смешанного индикатора, приготовленного растворением 0,330 г бромкрезолового зеленого и 0,165 г метилового красного в 500 мл 95 %-него этанола. После смешивания содержимого колбы осторожно добавляют 0,05 N NaOH до тех пор, пока при смешивании 1 мл этого раствора с 1 мл воды окраска не изменится от розовой до бледно-зеленой. Затем добавляют в колбу воды до метки и тщательно перемешивают.
Сплав Деварда. Истирают сплав (х.ч.) до тех пор, пока он не пройдет через сито 100 меш и, по крайней мере, 75% — через сито 300 меш. Сохраняется в тщательно закрытой посуде.
Сулъфаминовая кислота. Очищают сульфаминовую кислоту (х.ч.) перекристаллизацией из горячего раствора. Растирают 0,2 г очищенного препарата в 100 мл воды. Сохраняют раствор в холодильнике.
200
Стандартный раствор (аммонийный + нитратный азот). Растворяют О 236 г сульфата аммония и 0,361 г нитрата калия в воде, доводят раствор до метки в двухлитровой колбе и тщательно перемешивают. При использовании чистых и сухих реагентов этот раствор содержит 25 мкг аммонийного и 25 мкг нитратного азота в 1 мл. Сохраняют раствор в холодильнике.
Стандартный раствор (аммонийный + нитратный + нитритный азот). Растворяют 0,236 г сульфата аммония, 0,246 г нитрата натрия и 0,361 г нитрата калия в воде, доводят раствор до метки в двухлитровой колбе и тщательно перемешивают. Если использовать чистые и сухие реагенты, этот раствор содержит 25 мкг аммонийного, 25 мкг нитритного и 25 мкг нитратного азота в 1 мл. Сохраняют раствор в холодильнике.
Методы разложения пород и минералов можно разделить на две группы: кислотное разложение (чаще всего фтористоводородной или пирофосфорной кислотой) и выделение азота из силикатных расплавов при высоких температурах. Первая группа методов по технике выполнения более проста и не требует специального аппаратурного оформления, но предел обнаружения азота при этом определяется чистотой (по азоту) используемых реактивов и окружающей атмосферы. Вторая группа методов разложения анализируемого образца используется чаще всего перед последующим высокочувствительным инструментальным методом определения — масс-спектрометрическим, газохроматографическим и т. д.
Методы разложения горных пород и минералов см. [81].
Мокрый способ разложения, на наш взгляд, практически удобнее осуществлять с помощью пирофосфорной кислоты, чем с агрессивной HF. Кроме того, при использовании фтористоводородной кислоты сложность метода заключается в трудности исключения потерь азота в результате возможной сублимации фтористого аммония. Ниже приводится пропись метода анализа пород с разложением пирофосфорной кислотой [49, 53], по которому определяется содержание связанного азота. Молекулярный азот, входящий в состав включений и адсорбированный на поверхности пород и минералов, методами кислотного разложения не определяется. Поэтому эти методы дают заниженные значения содержания азота в породах и минералах по сравнению с методом плавления в токе инертного газа, которым определяют суммарный азот.
В настоящее время до конца не ясен механизм разложения силикатов пирофосфорной кислотой. По-видимому, легкая растворимость силикатных минералов в пирофосфорной кислоте (за 12— 14 мин.) может быть объяснена тем, что фосфорные кислоты образуют водорастворимые комплексы как с кремнеземом, так и с окислами металлов.
Из куска породы отбивают внутреннюю часть, промывают безаммиачной водой и сушат. Это помогает освободиться от возможных загрязнений образца посторонними аммонийсодержащими соединениями. Затем этот кусок породы дробят в ступке (до ~ 0,05 мм) и тщательно растирают в агатовой ступке.
201
Последнее особенно важно для кварцсодержащих пород: увеличение степени измельчения образца резко сокращает время его разложения.
0,5 г тонкорастертого образца помещают в кварцевую пробирку и заливают 4 мл смеси пиро- и ортофосфорной кислот. Выбор материала для реактора сильно усложняется тем, что при нагревании > 200° С эта смесь очень агрессивна. Так, платиновая посуда в этих условиях выдерживает нагрев только до 300° С, при более высокой температуре можно использовать сплав платины с золотом с преобладающим содержанием последнего. Подобная посуда слишком дорога для серийных анализов. В описываемом методе была использована кварцевая посуда, так как большинство силикатных минералов растворяется смесью фосфорных кислот за 12—14 мин., и кварц не успевает сильно корродировать.
Кварцевую трубку с образцом помещают в трубчатую печь сопротивления и устанавливают температуру (240° С) по хромель-алюмелевой термопаре. Время, необходимое для полного разложения образца, определяется целым рядом факторов и, в первую очередь, содержанием и размерами зерен кварца в породе; для однотипных пород оно колеблется в незначительных пределах, что позволяет легко установить его практически.
После окончания разложения раствор охлаждают до 50° С, разбавляют безаммиачной водой и количественно переносят в колбу Кьельдаля по возможности минимальным количеством дистиллированной воды, ибо разбавление образца ухудшает условия последующей отгонки аммиака с паром. Колбу присоединяют к системе для отгонки с паром. В приемник заливают 40%-ный раствор КОН (в количестве, достаточном для получения pH конечного раствора в пределах 9—10). К холодильнику подставляют коническую колбу с 5 мл 0,01 .А НС1 таким образом, чтобы его конец находился ниже уровня раствора НС1. При осторожном покачивании колбы Кьельдаля в нее через кран заливают щелочь и проводят реакцию нейтрализации. Щелочь выливают не до конца, и над краном всегда остается столбик жидкости, препятствующий случайному прорыву аммиака через кран из замкнутого объема. Затем через исследуемый раствор пропускают пар, который уносит с собой выделяющийся аммиак в приемник с кислотой. Скорость пропускания пара подбирается таким образом, чтобы получить ~ 25 мл конденсата за 3—5 мин. Полнота отгонки определяется повторным отбором конденсата в новый приемник с 5 мл 0,01 .А НС1 непосредственно после выделения всего аммиака из исходного раствора (контрольная проба). Отсутствие аммиака в контрольной пробе свидетельствует о полноте отгонки.
Содержание связанного азота в конденсате определяют фотоколоримет-рическим методом на ФЭК-56М по цветной реакции с реактивом Несслера. Для этого к 25 мл полученного конденсата добавляют 2 мл реактива Несслера, смесь встряхивают и оставляют стоять в течение 15 мин., чтобы дать возможность развиться окраске комплекса. Затем измеряют оптическую плотность с синим светофильтром в 50-миллиметровой кювете (X = 410 нм). Содержание азота определяют по градуировочному графику, построенному по стандартным растворам хлористого аммония. Аналогичным образом определяют содержание азота и в контрольной пробе.
Приготовление реактивов. Стандартный раствор NH4C1. 3,819 г NH4C1 (х. ч.), предварительно дважды перекристаллизованного и вы-202
сушенного при 105° С, растворяют в мерной колбе емкостью 1 л примерно в 700 мл безаммиачной воды, добавляют для консервирования 2 мл хлороформа и затем доводят объем раствора до метки водой. В 1 мл этого раствора содержится 1,0 мг азота (1,29 мг NH*). Рабочий стандартный раствор хлористого аммония получают разбавлением 10 мл исходного стандартного раствора безаммиачной водой в 100 раз. В 1 мл этого раствора содержится 10 мкг азота (0,0129 мг NHj).
Реактив Несслера. К 15 г йодистого калия и 11 г иода прибавляют сначала 10 мл воды и затем 15 г металлической ртути. Энергично встряхивают и оставляют на 10—15 мин. отстояться до просветления. Горячий раствор охлаждают и взбалтывают до приобретения им зеленоватого оттенка. После этого раствор сливают, а оставшуюся ртуть промывают водой, которую присоединяют к первой порции раствора. Раствор доводят до 200 мл. Из этого раствора отбирают 75 мл, прибавляют 75 мл безаммиачной воды и 350 мл 10%-ного раствора NaOH, не содержащего следов Na2CO3. Раствор после перемешивания должен отстояться, пока не станет совершенно прозрачным.
Смесь пиро- и ортофосфорной кислот. К 85%-ной ортофосфорной кислоте (х. ч., пл. 1,68) добавляют Р2О5 (х. ч.) до насыщения. Затем проводят обезгаживание 100%-ной ортофосфорной кислоты при нагревании и доводят удельный вес до величины 1,9 (при 25° С) и выше. Этот процесс довольно длительный: доведение плотности раствора до 1,98 (пирофосфорная кислота) завершается после 30-часового нагревания при 200° С.
При мокром способе разложения пород и минералов, как уже отмечалось, могут быть определены только формы азота в химически связанном состоянии. Термическое же разложение дает возможность определить общий азот, в том числе и молекулярный. Один из подобных методов, основанный на плавлении образца породы или метеорита в токе инертного газа и газохроматографическим окончании, описан в работе [745]. В настоящее время это наиболее надежный метод определения содержания общего азота в силикатном материале. Трудность анализа этих объектов определяется тем, что азот в них находится в нескольких химических формах, по-разному связанных с кристаллической решеткой минералов. Так, молекулярный азот полностью теряется при химических методах разложения силикатного вещества. Неизбежны потери части химически связанного азота при вакуумном плавлении в результате осаждения галогеноводородных солей аммония на холодных стенках аппаратуры.
Плавление образца в токе инертного газа в графитовом тигле при заполнении всего рабочего объема сажей позволило избежать потерь, характерных для других методов. Недостатком этого метода является образование огромных количеств окиси углерода, в Ю4 — 105 раз превышающих содержание азота. Появление таких количеств СО мешает определению азота хроматографическим или масс-спектральным методом. Поэтому в описанной ниже методике предусмотрено количественное окисление СО до СО2. Содержание азота определяют по калибровочным графикам, построен-
203
ным по стандартным образцам металлов и химических соединений. Наблюдается линейная зависимость между площадью пика и концентрацией азота (в мкг) в сталях.
Графитовый тигель помещают в цилиндр, заполненный сажей, и дегазируют при 2400° С в течение 3 час., в результате чего удаляются газы, абсорбированные на тигле и саже. Измельченные образцы (от 50 до 300 мг) приготавливают для анализа взвешиванием прямо в оловянных капсулах, не содержащих азот. Затем образцы помещают на 48 час. в вакуум и герметизируют в боксе с избыточным давлением гелия. Перед анализом капсулы с образцами помещают в приемник для образцов, расположенный выше тигля, и промывают током очищенного гелия в течение 30 мин. Гелий очищают при прохождении над циркониевой губкой, нагретой до 200° С, для удаления следов азота и кислорода.
После измерения фона образец сбрасывают в графитовый тигель, нагретый до 2400° С. Выделившиеся продукты пропускают над катализатором, состоящим из смеси окиси меди и окислов редких земель, при 400° С для окисления СО до СО2. Азотсодержащие соединения и любой вид растворенного в образце азота превращаются в молекулярный азот. Ловушки с аска-ритом и ангидроном служат для удаления двуокиси углерода и воды. Оставшиеся непоглощенными газообразные продукты собираются в течение 5 мин. в ловушке, заполненной молекулярными ситами, при температуре жидкого азота. После этого ловушку нагревают и сконденсированные газы одновременно попадают в колонку (длина 61 см, диаметр наружный 6,5 мм), заполненную молекулярными ситами 5А (0,35—0,28 мм) при 45° С. По мере выхода из колонки газы регистрируются катарометром. После окончания анализа образца опять замеряют фоновую концентрацию азота. Среднее из двух фоновых определений дает концентрацию азота в холостом опыте.
Предел обнаружения азота в силикатных образцах составляет 2—4 мкг. Поправка на фон колеблется от 4 до 6 мкг азота, в зависимости от состояния графитового тигля. После добавления 5— 8 г силикатного материала (12—25 образцов, в зависимости от их размера) поправка на фон начинает возрастать. Когда это произойдет, в установке заменяют тигель на новый. «Время жизни» графитового тигля можно увеличить добавлением 8—10 кусочков-разломанного нового тигля (3 — 5 мм). Это позволяет расплавить дополнительно 2—3 г материала образца (8—12 дополнительных образцов).
Изучение искусственно приготовленных стандартов показало, что при таком способе работы происходит количественный переход аммония, нитрата и нитрида в молекулярный азот.
УГЛИ, ГОРЮЧИЕ ИСКОПАЕМЫЕ, НЕФТЬ И НЕФТЕПРОДУКТЫ
Первая стадия анализа заключается либо в сожжении анализируемого образца в токе чистого кислорода в присутстви катализатора (с целью переведения азота в окислы) — чаще всего пе-204
ред газохроматографичесцим определением, а иногда перед спек-трофотометрированием подце поглощения NO2, — либо в мокрой минерализации (например, пр методу Кьельдаля, с помощью НС1О4) — перед титриметричесЬщм или спектрофотометрическим определением.
В последние годы все чаще испдльзуют высокочувствительный радиоактивационный метод, не требующий разрушения анализируемого образца.
В табл. 24 приведена характеристика методов, используемых для определения азота в различных горючих ископаемых.
ВОЗДУХ, ПРИРОДНЫЕ И ПРОМЫШЛЕННЫЕ ГАЗЫ, ГАЗОВЫЕ СМЕСИ
Определение вредных примесей, загрязняющих атмосферу, токсичных веществ в промышленных газах — одна из актуальных задач аналитической химии, в том числе аналитической химии азота и его соединений. Не менее важна задача определения азота и его окислов в различных газовых смесях. Можно отметить несколько обзоров, посвященных методам изучения загрязнения атмосферы, в частности, методам определения окислов азота в окружающем воздухе [204, 353а, 1145], в дымовых газах [1428], в выхлопных газах автомобилей [661]. Сравнительный обзор методов определения инертных газов в природных газах дан в [584]. Методы определения следов газов и микроанализа газов и паров приведены в [807].
Широко используются автоматические методы анализа газов, например, для непрерывной регистрации концентрации образующихся в воздухе ионов [1053] по его проводимости, регистрируемой с помощью самопишущего потенциометра. Этот метод использован для регистрации ионов на американской атомной подводной лодке, а также при облучении чистых газов N2, О2 и СО2 радиоактивным источником. Описаны автоматические приборы для определения миллионных долей примесных газов (от сожжения тощ лив) в воздухе [658].
Отмечена возможность определения загрязненности воздуха с помощью анализа проб снега, что дает возможность легко обследовать большие площади [936]. Содержание NO2, NO3, NH3 определялось методами, принятыми при исследовании питьевой воды.
Успехи современной науки и техники позволили исследовать химический состав атмосферы Венеры [48, 320]. На автоматических межпланетных станциях «Венера-5» и «Венера-6» было проведено прецизионное определение СО2, Н2О, N2, О2. На каждой станции было установлено по два газоанализатора. Измерение состава производилось на участке парашютного спуска станций* при давлениях от 0,6 до 10 атм и температурах от 25 до 225° С. Для
205
Таблица 24
Метод определения азота в углях, горючих ископаемых, нефти и нефтепродуктах
Анализируемый объект Метод определения Предел обнаружения Точность Примечание Литература
Горючие ископаемые Газометрический Абсолютная ошиб- Продолжительность определе- [1347]
Твердое топливо Титриметрический — ка 0,2—0,3% ния 30—40 мин. Навеска 100—150 мг ]260]
Газохроматографический — — Сорбент — цеолит СаА [126]
Угли Фотометрический — — Навеска 0,25 г; используют реактив Несслера Продолжительность анализа [240]
Каменный уголь и Р адиоактивационный -0,1% Среднеквадратич- [36]
смола Т-Активационный пая ошибка 5-12 % Относительная 0,5—1,5 часа; навеска 1 г; внутренний стандарт алюминиевая фольга Продолжительность облучения [5]
Уголь, кокс Различные ошибка <5% 5 мин. (25 Мэв) Обзор стандартных методов [1280]
Каменный уголь Метод Кьельдаля — — Минерализация образца 70%- [1183]
Уголь, кокс, нефть, Метод восстановительно- — Ошибка до 10% ной НС1О4 Сплавление пробы с металли- [965]
мазут Неф^ь и нефтепродук- го плавления с последующим титрованием Модификация метода Дю- . (результаты занижены) ческим калием и перевод азота в форму KCN; титрование KCN раствором NiSO4 с индикатором Навеска образца до 200 мг [734]
ты ма и метод Кьельдаля Метод Кьельдаля До 1% Абсолютная Навески от 0,2—0,3 до 0,02 г [497]
То же — 0,002—0,004% Автоматический метод [827]
Нефтяные дистилляты, бензины Нефтепродукты Метод Кьельдаля Ацидиметрический или фотометрический 0,1—10 мг/кг ю-4% Средняя относительная ошибка 3—10% Дистиллят собирают в водный раствор борной кислоты и титруют амидосульфоновой кислотой Предварительно превращают азотсодержащие соединения в NH3 [1327] [691]
Нефтяные фракции Спектрофотометрическ ий Микрограммо-вые количества Среднее абсолютное отклонение от + Г,4 мкг (в диапазоне содержания 0—40 мкг N) до ±8 мкг (для 100—800 мкг N) Органический N превращают в NO2 сожжением, поглощают окись и определяют NO2“ с реактивом Грисса—Илосвая; продолжительность определения <1 часа [1112]
Нефти Неводное титрование — — Проба нефти —10 г [ИЗИ ^03]
Нефть и остаточное печное топливо Микрокулонометрический Следовые количества — Титрование по Дорману
Фракции нефти Газо-жидкостная хроматография — — Используют хромосорб W; поноситель — Н2; объем пробы 30—50 мкл-, кулонометрический детектор [448]
Нефти из различных Активационный — — — [200]
месторождений Западной Сибири » — — АктивацияТ-квартами (18,5 М эв) в течение 2 мин. [201]
Нефти, органические вещества » 0,0001—0,1% 2% Продолжительность 5—10 мин. [790]
Нефть, нефтепро- дукты — — Нейтронная активация (14 Мэв) [1339, 1340]
Автоматический 0,05—0,1% Стандартное отклонение 0,12% Использован CHN-анализатор Перкин — Эльмер, модель 240 [1297]
Нефть » — — — [376]
определения содержания основных компонентов атмосферы применялся преимущественно манометрический метод, отличающийся простотой и надежностью. Азот определяли двумя методами. На Станции «Венера-4» — по разности давлений между двумя отсеками газоанализатора, в одном из которых поглощались все активные газы вместе с азотом, а в другом — углекислый газ, кислород и пары воды. На станциях «Венера-5» и «Венера-6» — по остаточному давлению после поглощения основных компонентов атмосферы — СО2, О2 и Н2О. Содержание азота в атмосфере Венеры оказалось равным около 2%.
На основании обсуждения предполагаемых путей эволюции азота в атмосфере Венеры была обоснована возможность, а результатами измерения, проведенными с помощью автоматической межпланетной станции «Венера-8», подтверждено наличие в атмосфере Венеры аммиака [320]. На станции был установлен прибор, основанный на дифференциальном линейно-колористическом методе определения, т. е. регистрации изменения цвета химического реактива под воздействием на него аммиака. В качестве такого индикатора был использован тетрабромфенолсуль-фофталеин на прокаленном силикагеле. По команде программновременного устройства станции прибор разгерметизировался, и атмосфера поступала через рабочий отсек в калиброванную камеру. Во время спуска станции газоанализатор был открыт, и газ непрерывно проходил под действием внешнего давления через рабочий отсек кюветы, где происходило определение аммиака. Объемное содержание газа на участке измерения (на двух уровнях при давлении ~ 2'и ~ 8 атм) может быть оценено как 0,01 — 0,1%.
Анализ литературного материала, посвященного методам определения азота и его соединений в воздухе, природных и промышленных газах, различных газовых смесях (табл. 25), показывает, что наиболее часто для этих целей используются газохроматографические, масс-спектральные и фотометрические методы. Подробно эти методы описаны в соответствующих главах.
ПРИРОДНЫЕ И ПРОИЗВОДСТВЕННЫЕ СТОЧНЫЕ ВОДЫ
Унифицированные методы исследования качества вод, принятые на совещании руководителей водохозяйственных органов стран-членов СЭВ [355], рекомендуется применять для анализа поверхностных вод. Химическому анализу производственных сточных вод посвящена книга [183]. В этих руководствах отмечается, что отбор, консервирование и хранение пробы воды является важной частью ее анализа, необходимой гарантией правильности получаемых результатов исследований и применимости их на практике [355, стр. 16—39].
Обзоры по методам анализа вод: по химическим методам [374, 851, 989, 1379], по определению CN~ и NO2 в сточных водах [545, 208
Таблица 25
Методы определения азота в природных и промышленных газах, газовых смесях
209
Таблица 25 (продолжение)
Метод определения Объект анализа Определяемый компонент Предел обнаружения Точность Примечание Литература
Г азохроматографический Промышленные газы (продукты горения) N2 . — — Две колонки с молекулярными ситами 5А и силикагелем; газ-носитель — гелий [433]
Дымовые, топочные газы N2 — — Сорбент — молекулярные сита 5А [1264, 1301]
Доменные газы (содержат до 50% N2) n2 — Относительная ошибка < 0,5% Газ-носитель—гелий; сорбент— молекулярные сита, активированный уголь [566, 878, 1012, 1361]
Колошниковый газ n2 — Применяют молекулярные сита 10Х [417]
Газы нефтеперегонных заводов n2, nh3 — — Две колонки с порапаком, детектор — катарометр [888]
Отработанные газы автомобильных двигателей n2 — — — [195]
Газы реактивных двигателей n2 — . ±1% Применена автоматическая система [1410]
NO + NO2 + C02-J-+ О2 + Н2О + n2 n2 — — Двойные и тройные газовые смеси [915]
NO + NO2 + СО2+ + О2 + Н2О + n2 n2 — — Анализируют продукты разложения взрывчатых веществ [1355]
NO + NO2+CO2+ + О2+н2о + n2 n2 Используют молекулярные сита; СН4 и NO не могут быть определены при их совместном присутствии; NO2 сорбируется необратимо [660]
Таблица 25 (продолжение)
Метод определения Объект анализа Определяемый компонент Предел обнаружения Точность Примечание Литература
Газохроматографический N2O -|- N2*, N2O + NO n2o — — Адсорбент — силикагель КСК-2,5; детектор — катарометр; время удерживания N2 — 90 сек., N2O — 216 сек. [280]
n2 + 02 n2 — — Сорбент — молекулярные сита 13Х [1219]
N2O -|- СО -|- СН4 n2o — — Сорбент—порапак Q [1219]
N2+ О2 + N2O + 4- СО2 + СН4 n2 — Относительная ошибка зНЗ.3% Две колонки с порапаком Q и молекулярными ситами 13Х; газ-носитель — гелий [1302]
Ne + Аг + N2; Аг -J- Н2 И- N2 n2 — — Используют молекулярные сита 5А, охлаждаемые жидким N2 [1370]
О2 -|- n2 + со + + со2 + н2 + + Н2О + сн4 n2 — — Используют хроматограф фирмы DAM с молекулярными ситами [656]
Ат n2 — — Используют ионизационный детектор [962]
Не (высокой чистоты) n2 1,25-IO"5 объемн. % — Используют разрядный детектор [203]
Не n2 — 2% Применяют газовый хроматограф Бекман с катарометром, две колонки с силикагелем и молекулярными ситами 5А [470]
Не n2 0,2 ч. на млн. — Использует радиоактивный ионизационный детектор [552]
• Не n2 1,02 ч. на млн. — Применяют хроматограф Бекман CG-2A; сорбент — молекулярные сита 13Х [1191]
Ь5
Таблица 25 (продолжение)
ьэ . Метод определения Объект анализа Определяемый компонент Предел обнаружения Точность Примечание Литература
Газохроматографический Н2 (технический) N2 — — Сорбент — молекулярные сита 5А [1014]
Газы, применяемые для синтеза аммиака n2 — —" Система из двух колонок с силикагелем и молекулярными ситами 5А; газ-носитель — гелий [325]
Масс-спектр ал ь-ный Атмосфера Земли n2 — — Масс-спектрометр с двойной фокусировкой [1306]
Природные и инертные газы n2 0,0001% Относительная ошибка несколько десятков процентов Изотопное разбавление, масс-спектральный вариант [89]
Доменные газы n2 —, — Используют модифицированный масс-спектрометр CSF-SM102 [828]
Газы конверторов металлургического производства n2 — —• Применяют малогабаритный четырехканальный масс-спектрометр типа MAT-GDI 50/4 [1267]
н2 n2 5-IO-6 объемн.% — Используют масс-спектрометр МХ-1302; проводят адсорбционное обогащение примесей [42]
Кислород сверхчистый n2 ~l-10~5% — Вредные остаточные газы удаляют поглощением Na—К-спла-вом (коэффициент обогащения >106) [1221]
То же n2 — —- Используют масс-спектрометр СЕС-21-103 [429]
Таблица 25 (продолжёние)
Метод определения Объект анализа Определяемый компонент Предел обнаружения Точность Примечание Литература
Масс-спектраль-ный Двуокись углерода N2 2-10-5% — Обогащение примесей конденсацией СО2 на стенках стеклянной охлаждаемой трубки [1152]
n2 + co n2 — — Рассмотрены три метода [1322]
Остаточные газы в вакуумных системах n2 Используют полюсные наконечники для концентрирования магнитного поля в области радиальной траектории ионного пучка [1202]
Не (сверхчистый) n2 — — • — [227, 1326]
Ат n2 0,05—0,01% Абсолютная ошибка з-ю-“% n2 Используют масс-спектрометр Хитачи [388]
Аг и Не n2 7,7-10-“% Коэффициент вариации 5,9% (концентрация n2 2-10%) Используют масс-спектрометр типа МС-10 фирмы AEI (Англия) [938]
Ат и Н2 n2 n2 IO-3 4-IO"3 объемн. % — Используют масс-спектрометр типа МИ-1305 и МХ-1302 [408] [409]
Спектральный N2 GO2 -f- О2 Совместное определение N2 и H2 0,5% Ошибка 7—8% Используется изотопное разбавление; аналитическая линия азота 3159,ЗА [233]
Ne n2 5-10-“ объемн. % — Используют спектрограф ИСП-51 [17]
« Не n2 7-10-5 % — Используют монохроматор Хильгер D-285 [1320]
to
Таблица 25 (продолжение)
Метод определения Объект анализа Определяемый компонент Предел обнаружения Точность Примечание Литература
Спектральный Ne N2 6-10-4% — — [1320]
Не n2 0,03—3% Ошибка 2—5% Используют вакуумную установку, спектрограф ИСП-51; аналитическая линия N2 4278? А [263]
Аг n2 1 • 10-4 абс. % (по линии 3804,9А для содержания 0,005% N2) Коэффициент вариации 8—10% при концентрации азота 5-10-3% Наиболее интенсивная линия N 3371,9 А [154]
Аг n2 — — — [532]
Аг + Не n2 4 IO-3— 4-10-2% — Аналитическая линия N 8216.46А [1198]
О2 + N2J N2 + Не*, N2 + Ar; N24-O2 (второй газ — основной компонент смеси) n2 Применяют разряд в неохлажденном поле Си-катода [573]
Пламенно-фотометрический Не n2 0,04—1 объем н. % Абсолютная ошибка 4;0,03% По интенсивности свечения в высокочастотном разряде, измеряемой с помощью пламенного фотометра; характеристические пики N2 337 и 358 нм [674]
Хемилюминесцентный Газы поршневых и турбинных двигателей NO — — — [1108]
ИК-спектроскопи-ческий Атмосфера NH3, NO, no2 В ч. наблн.: NH3 0,4; NO 0,4; NO2 0,1 Точность 0,1% Используют лазер на основе СО и СО2 [961]
Таблица 25 (продолжение)
Метод определения Объект анализа Определяемый компонент Предел обнаружения Точность Примечание Литература
Фотометрический Атмосфера nh4+, no2 NO, NO2 NO, NO2 Окислы азота Окислы азота N03_ и no3-+no2- HCN 0,5 .ws/л (1,8-10в) Несколько частей на 1 блн. 0,14-1,6 мкг N Ошибка +5% Ошибка ±4% NH4+ определяют с реактивом Несслера Используют реактив Грисса — Зальцмана для определения no2 По окраске п-оксиазобензол-сульфамида Используют фотометр, предназначенный для непрерывного определения содержания частиц аэрозолей Использован модифицированный метод Грисса — Илосвая Реагент 1-аминопирен, спектро-фотометрируют при 456 нм Используют индикатор на носителе — фильтровальной бумаге или гранулированном силикагеле [991] [550] [1177] [607] [617] [1255] [1048]
Выхлопной газ дизельных двигателей Окислы азота — — — [28]
Выхлопные газы автомобилей То же — — Модифицированный метод Зальцмана [703]
Электрохимический Воздух NH3 Окислы азота и пары HNO3 1-10“4% 0,0003— 20 мг)л — По изменению электропроводности поглощающего раствора То же [675] [Н]
§8 S?
СО
00
CSJ
М ев
1-9 съ
3
В” я
сб Й
Я я ев В* Ф S Я ft а
2 И
"&S
Л
сб я
§....
>> к
S S 2 2 и и И м М м й । я
5 Й*ё я о s KS Ю со §§.« £««
Я я S
я и я ф ч и
я я я ф ч
И
*О S;g °£ н < 2^ сб о Во
я ео Я Я О И
я сб
я я к §
6 о и
§ н ао ф О ч
йЗ <м сб о «Se
A i g g §•2 S § w ® tri H
Ф ,2. ф sfri
Й ео Я
'а «
Й.в
S о Я ЕГ Я
я ф л
S ео Я
я я я ф ч
S о ч
S я ,и
§
со Я
я я ф В о ч
г?
Ж Ф - ••• S» 3'5'М &g 5 й*
ГТ* сб о сб g 2>в< я s S С ч ч
Таблица 25 (окончание)
ев Я Я о « ° я Я S о Я w Р5
Л'ё
Л я
\D
1 о о
° 1 ?
я Л
я Я >Р \о
я »
o+l Э ° 'О О сб
я я ю
я э о
g я
а
я
§
Я
В
с
л
я
а
£ Й Я О £ >» ° Я 6 Л&С
я Я
с
я
а
£s о w я
Я А Я
Л
и
И
с а
е
я
Л я
я я я ф »я «я я в я О «г !« О<£ я 5 и “
я ф ч ф ft я § ф я я Л S 5 a S м & Я н 2 л §« я Ф о
я Е еич Л
о ч о о< ы и S я Ch изме ни у ф ф S 5 га а н га к
ч я о о о я я
(X) Ен С аС аи
216
560, 645, 781, 935, 1000, 1052, 1128, 12871; по определению нитратов в природных водах [1137]; по полярографическим методам определения нитратов в природных и сточных водах [480]; по методам определения нитратов в питьевой и столовых водах [535].
В таблице 26 приведены некоторые описанные в литературе методы определения соединений азота в различных типах вод. Наиболее часто для этого используются химические методы [355].
Аммоний и аммиак
Ионы аммония и аммиак появляются в грунтовых водах в результате жизнедеятельности микроорганизмов. Соотношение концентраций свободного аммиака (NH3) и ионов аммония (NH4) зависит от концентрации водородных ионов.
Для качественного обнаружения аммиака к 10 мл пробы прибавляют несколько кристалликов сегнетовой соли и 0,5 мл реактива Несслера. Желтое окрашивание раствора, помутнение или выпадение желто-коричневого осадка указывает на присутствие аммиака. В присутствии повышенного количества органических веществ, особенно гуминовых кислот, вызывающих усиление коричневой окраски после подщелачивания, необходимо провести холостой опыт, добавив сегнетову соль и 0,5 мл 15%-ного раствора едкого натра.
Для количественного определения аммиака используются: 1) метод непосредственного колориметрического определения в питьевых и поверхностных водах с реактивом Несслера; 2) метод отгонки с колориметрическим или объемным окончанием в зависимости от концентрации аммиака в пробе (определение в поверхностных и особенно в сточных водах); 3) метод фотометрического определения с гипохлоритом и фенолом.
Если пробы для определения аммиака взяты не сразу же после отбора, их консервируют прибавлением 1 мл концентрированной серной кислоты или 2—4 мл хлороформа на 1 л пробы.
По найденному общему содержанию аммиака можно рассчитать концентрацию аммиака свободного и присутствующего в виде ионов NH^, если известно значение pH воды (табл. 27) [355, стр. 224].
Ниже приведено описание фотометрического определения с реактивом Несслера (нижний предел определения равен 0,05 мг NHt в 1 л; без разбавления можно определять не более 4 мг NHt в 1 л воды [355, стр. 211—215].
К 50 мл первоначальной пробы, или к 50 мл осветленной пробы, или к меньшему ее объему, доведенному до 50 мл бидистиллятом, прибавляют 1— 2 капли раствора комплексона III или сегнетовой соли и смесь тщательно перемешивают. При анализе очень жестких вод нужно прибавить 0,5—1 мл раствора сегнетовой соли или комплексона III. Затем прибавляют 1 мл
217
Таблица 26
Методы определения азота и его соединений в природных и промышленных сточных водах
Определяемый компонент Метод определения Предел обнаружения Точность Примечание Литература
NH4+ Фотометрический Природны 0,01—0,1 мг ЫН4+/л е пресные воды Ошибка 15% Индофенольная реакция; [74]
0,1— 5 мг ИН4+/л Ошибка 2—3% • объект — бесцветные и прозрачные воды [1261]
1—50 мкг NH4+ Ошибка 10% Фенолят-гипобромитная ре- [124]
акция; объект — окрашенные и мутные воды С реактивом Несслера [930]
1 мкг N/л Стандартное отклоне- NH4+ окисляют гипохлори- [1210]
ние 3—1,1 мкг/мг Ошибка 1,3—5,7% том натрия до NO2_ и фо-тометрируют окраску продукта, образующегося с сульфаниламидом в присутствии И-(1-иафтил)эти-лендиамина Фенол-гипохлоритный ме- [817]
N03' Колориметриче- (при содержании 10— 1 мкг N соответственно) ТОД Метод основан на реакции [594, 1027,
ское титрование Фотометрический 0,01 мг N/л Ошибка ~0,5% окисления индигокармина NO3_ восстанавливают гид- 1197] [52, 705,
разинем и фотометрируют образовавшийся NO2“ по реакции Грисса — Илосвая и др. 776, 830]
Таблица 26 (продолжение)
Определяемый компонент Метод определения Предел обнаружения Точность Примечание Литература
NO3- Фотометрический ~1 мг NO3 /л — Используют в качестве реагента фенол-2,4-дисульфо-кислоту и о-толидин [1016, 1240]
<1 мг ИО3~/л Ошибка ~1— 2% Определяют с бруцином [564, 688, 881, 1324]
• Спектрофотометрия в УФ-области Спектрофотометрический Экстракционнофотометрический 0,03- 3 мг NO3"/.w8 Коэффициент вариации 4% (для 1 мг NOS~/мл) Стандартное отклонение ±0,05 Ошибка ~1,7% Определяют с хромотроповой кислотой Измеряют светопог лощение при 210 нм Метод основан на способности нитратов подавлять окраску комплекса рения с син-фенил-2-пиридил'кет-оксимом Экстрагируют NO3_ с катионами хелатов Си+ с нео-купроином в виде ионных ассоциатов и спектрофото-метрируют при 456 нм [1397] [464, 1101] [530] [1425]
Полярографический 0,2 мг N03_/4 Точность ±5% (при ^2 мг НО3“/л) Восстанавливают NO3," на ртутном электроде в слабокислой среде в присутствии уранил-ионов; мешают нитриты Определяют нитрофенолдисульфоновую кислоту в 0,1М растворе N.H4C1 при pH 5 [718] [832]
Таблица 26 (продолжение)
Определяемый компонент Метод определения Предел обнаружения Точность Примечание Литература
NO3- Г р авиметрический — Относительная ошибка <3% С помощью нитрона [1193, 1250]
NOr • Электролиз в капиллярах Расхождение результатов 2,6—23,5% при содержании NO3~ 17—2068 мг)л Разделение проводят при напряженности поля 10— 20 в1ем в течение 1,5—3 час.; определяют содержание по длине зоны [152]
Фотометрический Ошибка ±10% В качестве реагента применяют 0,2%-ный раствор риванола; не мешают 5000-кратные количества NO3_ и других анионов; мешают J-, F-, SO32", S2O82~, SCN- [13]
0,02—10 мг КО2~/л Абсолютная ошибка 0,005—0,05 мг[л Метод Грисса — Илосвая и др. [657, 764, 1122]
— — Определяют с индигокармином [1028]
GN- » — Ошибка ~1,2% (при ~40 мкг)л) Вначале отгоняют HCN из пробы [3, 770]
Следовые количества Ошибка ±1% По реакции с п-фенилен-диамином в присутствии пиридина и др. [493, 1068]
—* — Используют бумагу, пропитанную дифенилкарбази-дом ртути [303]
Полярографический <1 мг CN“pi — Объект — речная вода [882]
Таблица 26 (продолжение)
Определяемый компонент Метод определения Предел обнаружения Точность Примечание Литература
Органический азот Метод Кьельдаля — — Органические вещества экстрагируют изобутанолом, упаривают до постоянного веса при 70° С [69]
Колориметрический Минера льные воды По фенолят-гипобромитной реакции [74]
n2 Волюмометриче-ский — Ошибка ±2% Сочетают выделение газа с вакуумным кипячением воды [77]
nh4+ • Фотометрический — — Объект — сернистые воды; отгоняют NH3 и определн-ют с реактивом Несслера [546]
NO3" Спектрофотометрический в УФ-области 5-10-*—5-10-Зг NO3-/a — Измеряют светопоглощение при 210—220 нм [834]
Фотометрический — — Восстанавливают N03_ до NO2~ амальгамой кадмия и определяют с реактивом Грисса — Илосвая и др. [ИЗ, 282, 1418]
Морские и Стандартное отклонение <0,01 мг NO3-/» океанские воды Определяют с бруцином на автоанализаторе [900]
Растворенный N, Газо хроматографический —‘ Относительная ошибка 0,5% — [469]
NH4+ и орга-gg нический азот Микро метод Кьельдаля —‘ Ошибка ±0,5—5% Объект — жидкая и твердая фазы морских осадков [314]
Таблица 26 (продолжение)
Определяемый компонент Метод определения Предел обнаружения Точность Примечание Литература
nh4+ Фотометр ический — — По индофенольной реакции [193]
» — Относительное стандартное отклонение 1,6% Гипохлоритный метод [13561
Кулонометрическое титрование Конец титрования устанавливают потенциометрическим способом со стеклянным индикаторным электродом [253[
nh4+, NO,-, рог Фотометрический — — — [9851
Автоматический метод на основании фотометрических измерений — 12 анализов в час с высокой точностью и хорошей воспроизводимостью результатов [7751
NO," Фотометрический 5—100 «NO3'/.«3 Средняя ошибка 5% При помощи дифенилбен-зидинсульфоната натрия [1781
» Среднее отклонение 2% Восстанавливают NO3_ до NO2~ и определяют последний с ^(1-нафтил)этилен-диамином спектрофотометрически при 543 нм [10841
«^0,1 мкг N/мл Максимальное отклонение ±3 мкг/мл Определяют с дифениламином и дифенилбензидином [3221
NO," Полярографический — Ошибка ±4% Полярографируют при 25° С и при —1,1 в [11491
Фотометрическ ий — — — [251, 300, 6971 [2991
Органический азот » 0,15—0,25 мг/л — —
Таблица 26 (продолжение)
Определяемый компонент Метод определения Предел обнаружения Точность Примечание Литература
Сточные и технологические воды
NM NH3 Газохроматографический — — — [7841
nh4+ » — — Дистилляция NH3 в присутствии магнезиального молока [319]
NOr Фотометрический — — Используют 0,2%-ный раствор профлавина [15]
» Окрашенные сточные воды предварительно пропускают через колонку с анионитом Дауэкс 1X8; NO3“ элюируют 0,1А НС1 [386]
CN- Титриметрическ ий » 50—2000 мг/л — Прямое титрование в присутствии S2-, Cl" и SCN- После осаждения S2~ добавлением раствора CdSO4 к щелочному анализируемому раствору титриметрически определяют CN“ с использованием диметиламинобен-зилиденроданина [83] [1345]
При определении простых цианидов отгоняют HCN и определяют иодо- или аргентометрически или другим методом [555, 708, 836, 1252]
Таблица 26 (продолжение)
Определяемый компонент Метод определения Предел обнаружения Точность Примечание Литература
CN- Фотометрический 0,01 мг CN~M — С сульфаниловой и барбитуровой кислотами [182, 1436]
» Миллиграммовые количества Относительная ошибка 11% С ферроином [940]
» — — С бензидином [457 , 838, 1435]
» Микрограммовые количества — С пиридином; в присутствии SCN- (SCN- : Ctf- < 60) отгоняют HCN [181, 475]
Полярографический — — С наложением прямоугольного напряжения [779, 951]
Амперометрический По изменению тока, протекающего между электродами, погруженными в сточные воды; измерительный электрод — из графита^ никеля, амальгамированного цинка, золота; электрод сравнения — каломельный [1269]
Потенциометрический — — С ионоселективным электродом [810, 1125]
» Следовые количества цианидов — С пассивированным Bi-электродом [1171]
» — — G серебряно-ртутным электродом [212, 682]
8 В. Ф. Волынец, М. П. Волынец
Таблица 26 (окончание)
Определяемый компонент Метод определения Предел обнаружения Точность Примечание Литература
NO3~ Титриметрический
Органический азот Фотометр ический » У Ф-спектрофото-метр ический Автоматический
Питьевые воды
— Абсолютная ошибка 0,02% Алкалиметрическое титрование после отгонки NH3 [1181]
— — С хромотроповой кислотой [746]
— — С 2,6-ксиленолом [1095]
— — Измеряют поглощение при 206 нм в кислой среде [1102]
Стандартное отклонение ±8 мкг (при содержании 125—700 мкг N/л) Фотоокисление органических веществ под действием УФ-лучей до NO3~ и NO2~ [831]
Конденсат высокочистая вода, атмосферные осадки
NHt+ Потенциометрический 0,5 мкг NH4+/am Стандартное отклонение <10% Объект — конденсат и высокочистая вода паросиловых установок; непрерыв ное автоматическое определение [1057]
NO,-, NO2-NH*+ Фотометрический Объект — атмосферные осадки; определяют сумму NO3" и NO2_ с дифениламином, NH4+— с реактивом Несслера, NO2“ — с реактивом Грисса [44]
Таблица 27
Относительное содержание NH3 в воде (%)
pH
1, °C 6 7 8 8,5 9 9,5 10 10,5 11
25 0,05 0,49 4,7 13,4 32,9 60,7 83,1 93,9 98,0
15 0,02 0,23 2,3 6,7 19,0 42,6 70,1 88,1 96,0
5 0,01 0,11 0,9 3,3 9,7 25,3 51,7 77,0 91,5
реактива Несслера (растворяют 100 г HgJ ч.д.а. и 70 г KJ ч.д.а. в небольшом количестве бидистиллята и смешивают с раствором едкого натра, приготовленным растворением 160 г NaOH ч.д.а. в 500 мл дистиллированной воды. Смесь доводят бидистиллятом до 1 л. Применяется прозрачный раствор после отстаивания в течение по крайней мере 4 час.) и снова перемешивают. По истечении 10 мин. колориметрируют на фотометре с фиолетовым светофильтром (X = 400 -S- 425 нм) или сравнивают с серией стандартов, приготовленных в цилиндрах Несслера. Окраска смеси не изменяется в течение 30 мин. Из величины оптической плотности вычитают оптическую плотность холостого опыта. Если нужно, вычитают и оптическую плотность пробы, к которой вместо реактива Несслера прибавлен 1 мл 15%-ного раствора едкого натра, и по калибровочному графику находят содержание аммиака.
Содержание NH4+ в мг/л (х) и в мг-экв/л (у) вычисляют по формулам
С-50 С-50 2,77-С
X — . \ V = - ==__-___ .
V 18,04-7 V ’
где С — найденная концентрация NH4+, мг!л; V — объем пробы, взятой для анализа, мл; 18,4 — эквивалент NH4; 50 — объем пробы, мл.
Определению мешают амины, хлорамины, ацетон, альдегиды спирты и некоторые другие органические соединения, реагирующие с реактивом Несслера. В их присутствии определяют аммиак с отгонкой. Определению мешают компоненты, обусловливающие жесткость воды, железо, сульфиды, хлор, а также мутность. Мешающее влияние жесткости устраняют прибавлением раствора сегнетовой соли или комплексона III. Большое количество железа, сульфиды и муть удаляют осветлением воды цинковой солью.
К 100 мл пробы прибавляют 1 мл раствора сульфата цинка (100 г ZnSO4-•7Н2О ч. д. а. растворяют в бидистилляте и разбавляют до 1 л) и смесь тщательно перемешивают. Затем pH смеси доводят до 10,5 добавлением 25%-ного раствора едкого кали или едкого натра. Проверяют pH стеклянным электродом. После взбалтывания и образования хлопьев осадок отделяют центрифугированием или отфильтровыванием через стеклянный фильтр. Увеличение объема жидкости необходимо учесть при расчете.
226
Мешающее влияние хлора устраняют добавлением раствора тиосульфата или арсенита натрия (растворяют в бидистилляте 3,5 г Na2S2O8-5H2O ч.д.а. или 1,0 г Na2AsO8 ч.д.а. и доводят до 1 л). Для удаления 0,5 мг хлора достаточно прибавить 1 мл одного из указанных растворов.
Определение аммиака с перегонкой см. [355, стр. 215—220], фотометрическое определение в виде индофенолового синего — в [355, стр. 221-224].
Нитрат-ионы
Нитраты встречаются почти во всех видах вод. В поверхностных и родниковых водах количество их обычно незначительно. Большое количество нитратов содержат некоторые промышленные сточные воды.
Для качественного обнаружения нитратов к 5 мл концентрированной серной кислоты в пробирке при постоянном перемешивании прибавляют по каплям 2 мл испытуемой воды. После этого вводят незначительное количество твердого бруцина (Осторожно, сильный яд!) и смесь снова перемешивают. Появившееся желтое или коричнево-красное окрашивание указывает на присутствие нитратов. Предел обнаружения реакции 1 мг NO3~ в 1 л и более. Реакцию можно использовать для ориентировочного определения необходимого последующего разбавления пробы. Применяемая серная кислота не должна содержать нитратов.
Для определения нитратов хорошие результаты дает фотометрический метод с салицилатом натрия при концентрациях 0,1 — 20 мг!л. Для серийных (анализов пробе концентрацией 5—30 мг NOg в 1 л можно применять полярографический метод [355, стр. 442-447].
Метод восстановления нитрата сплавом Деварда до аммиака с последующей перегонкой может быть использован при анализе сточных вод, содержащих нитраты в количествах, превышающих 5 мг/л.
Если проба не была обработана в день отбора, ее хранят в холодильнике или консервируют.
Фотометрическое определение с салицилатом натрия [355, стр. 439—442] основано на реакции нитратов с салицилатом натрия в концентрированной трихлоруксусной кислоте с образованием соли нитросалициловой кислоты, окрашенной в желтый цвет. В среде трихлоруксусной кислоты устраняется мешающее влияние неустойчивых органических веществ, которое наблюдается при применении серной кислоты. Интенсив-ность окраски прямо пропорциональна концентрации нитратов.
Отмеренное количество пробы, содержащей от 0,01 до 0,50 мг NOS~, фильтруют через колонку катионита со скоростью 0,1 мл!сек. Затем колонку промывают трижды дистиллированной водой в количестве 10 мл и к фильтрату прибавляют по каплям едкий натр до щелочной реакции и 1,0 мл
8*
227
1%-ного свежеприготовленного раствора салицилата натрия. Раствор выпаривают на водяной бане досуха, к горячему сухому остатку прибавляют 2,0 мл трихлоруксусной кислоты (к 240 г СС13СООН прибавляют 25 мл дистиллированной воды) так, чтобы он весь был смочен, и оставляют стоять в течение 2—3 мин. на водяной бане. После охлаждения прибавляют немного дистиллированной воды и 5,0 мл едкого натра, раствор перемешивают, переливают в мерную колбу емкостью 50 мл и доливают дистиллированной водой до метки. Возникшая окраска устойчива в течение нескольких часов. Измеряют оптическую плотность при 410 нм (фиолетовый светофильтр) в кюветах толщиной 1—5 см, вводят поправку на холостой опыт и по калибровочному графику находят содержание нитрат-ионов; расчет ведут по формуле х = = С -50/ V, где С — концентрация нитратов, найденная по калибровочной кривой, мг1л; V — объем пробы, взятой для определения, мл; 50 — объем, до которого доведена проба перед измерением.
Определению мешают нитриты, так как они увеличивают интенсивность окраски. Для их устранения к пробе воды перед фильтрованием через катионит прибавляют небольшое количество азида натрия. Ионы железа и другие мешающие катионы устраняют, обрабатывая воду катионитом. Йодиды и бромиды мешают определению, ослабляя окраску; их содержание в обработанной пробе не должно превышать 0,01 мг. Не мешают анализу содержащиеся в пробе воды хлориды (до 50 мг), фториды (до 25 мг), сульфаты (до 25 мг), фосфаты (до 25 мг), сульфиты (до 10 мг) и бораты (до-1 мг ВОа).
Нитраты могут быть восстановлены амальгамой кадмия до нитритов, которые определяют колориметрическим методом, указанным ниже.
Нитрит-ионы
Нитриты являются промежуточным продуктом биохимического окисления аммиака или восстановления нитратов. В поверхностных водах нитриты быстро переходят в нитраты. Они присутствуют в концентрациях от нескольких микрограммов до десятых долей микрограмма в 1 л. В большом количестве они находятся в некоторых промышленных и биологически очищенных сточных водах.
Для качественного обнаружения нитритов к 10 мл пробы прибавляют 1 мл раствора сульфаниловой кислоты и 1 мл раствора а-нафтиламина (приготовление см. ниже). В присутствии нитритов появляется розовая или красно-фиолетовая окраска. Чувствительность определения составляет 0,01 мг NO2“ в 1 л воды.
Вследствие нестойкости нитритов их надо определять сразу же после отбора пробы. Если это невозможно, пробу консервируют. Можно также охлаждать пробу до 3—4° С.
228
Для определения нитритов в питьевых, поверхностных и сточных водах предлагается фотометрический метод с сульфаниловой кислотой и а-нафтиламином [355, стр. 454—458].
Для определения берут 50 мл или меньшее количество профильтрованной пробы и доводят до 50 мл дистиллированной водой. Прибавляют 1 мл раствора сульфаниловой кислоты (растворяют 6,0 г сульфаниловой кислоты ч. д. а. в 750 мл горячей дистиллированной воды. К полученному раствору прибавляют 250 мл горячей дистиллированной воды) и смесь тщательно перемешивают. После 5-минутного стояния прибавляют 1 мл раствора а-наф-тиламина (растворяют 1,2 г а-нафтиламина ч. д. а. в дистиллированной воде, прибавляют 50 мл ледяной уксусной кислоты и доводят дистиллированной водой до 200 мл. При образовании мути раствор фильтруют через хлопчатобумажную ткань, промытую дистиллированной водой. Раствор сохраняют 2—3 месяца) и смесь снова перемешивают. Пробу фотометрируют при 520 нм (зеленый светофильтр) или сравнивают с серией стандартов, приготовленных в цилиндрах Несслера, через 40 мин. после прибавления раствора а-нафтиламина и по калибровочной кривой находят содержание нитритов. Расчет содержания нитрит-ионов в мг!л (х) или в мг-экв!л (у) ведут по формулам
ж = С-50/7; у = <7-50/7-46 = <7-1,087/7,
где С — концентрация нитрит-ионов, найденная по калибровочному графику или по шкале стандартов, мг'л; V — объем пробы, взятой для определения, мл; 50 — объем, до которого разбавлена проба, мл; 46 — эквивалент нитрит-иона.
Определению мешают взвешенные вещества и мутность воды. Поэтому перед анализом пробу необходимо профильтровать. В анализируемой пробе не должны присутствовать сильные окислители или восстановители. Cu(II), Fe(III), Hg(II), Ag, Bi, Sb(III), Pb, Au(IIl), хлороплатинаты, метаванадаты мешают определению. Определению мешает также окраска воды. Слабое окрашивание и незначительная мутность питьевых и поверхностных вод при определении оптической плотности компенсируются холостым опытом, в котором к пробе прибавляют только раствор сульфаниловой кислоты.
Цианид-ионы
Цианиды встречаются в водах в форме ионов или в виде слабо-диссоциированной и весьма летучей токсичной синильной кислоты, что зависит от реакции среды. Они могут находиться также в комплексных соединениях с некоторыми металлами [например, Zn, Cd, Си, Ni, Со, Fe(III), Fe(II)]. Иногда в воде могут находиться и нерастворимые простые цианиды металлов. Для полного разделения отдельных форм цианидов пока не имеется подходящих химических методов.
В природных водах цианиды не встречаются. В поверхностные воды они могут попасть только при загрязнении сточными водами.
229
Для качественного обнаружения цианидов щелочных металлов, цианида аммония и комплексных цианидов цинка, кадмия и меди в пробирку, содержащую 10 мл пробы, вносят каплю раствора метилового оранжевого, затем по каплям добавляют 5%-ный раствор ортофосфорной кислоты до кислой реакции, после чего прибавляют еше 2 капли этого раствора. После добавления нескольких капель бромной воды (насыщенный раствор) пробирку встряхивают и это повторяют до тех пор, пока окраска раствора не станет желтой. Потом по каплям вносят 2%-ный раствор мышьяковистой кислоты до обесцвечивания и добавляют сверх того еще одну каплю. Вносят 10 мл w-бутилового, изобутилового, и-амилового или изоамилового спирта и пиридин-бензидиновый реактив, приготовленный смешением 5 мл 25%-ного раствора пиридина с 0,5 мл 2%-ного раствора солянокислого бензидина. При наличии цианидов после встряхивания пробирки слой спирта в ней окрашивается в красно-желтый цвет. Этим методом можно обнаружить цианиды даже при концентрации 0,05 мг в 1 л пробы.
Пробы вод, предназначенные для определения в них содержания цианидов, необходимо консервировать едким натром, доводя pH до величины не меньше И, и хранить при 3—4°С или же анализировать непосредственно после их отбора. При наличии окисляющих веществ (при положительной реакции пробы на иодо-крахмальную бумажку) необходимо перед подщелачиванием пробы обработать ее свежеприготовленным 1%-ным раствором сульфита натрия до появления отрицательной реакции на иодо7 крахмальную бумажку. Если проба содержит сульфиды или сероводород, необходимо ее обработать немедленно или осадить сульфиды карбонатом свинца или сульфатом кадмия и отделить их.
С аналитической точки зрения различают общие, «свободные» и «связанные» цианиды. В группу свободных цианидов входят соединения, из которых в нейтральной среде легко выделяется цианистый водород (синильная кислота, цианиды щелочных металлов, аммония, комплексные цианиды цинка, кадмия). Понятие связанные цианиды охватывает все остальные комплексные соединения цианидов. Общие цианиды — это свободные и связанные цианиды, содержащиеся в анализируемой пробе.
В условиях рационально проводимой перегонки можно отделить свободные цианиды от связанных или же получить дистиллят, . содержащий все цианиды или только связанные. Свободные цианиды определяют в дистилляте, когда перегонку производят из раствора, pH которого был доведен до 7. При дальнейшей перегонке из сернокислой среды можно собрать в приемник и определить цианистый водород, выделившийся из связанных цианидов. Общее содержание цианидов определяют после перегонки свободных и связанных цианидов из сернокислой среды в присутствии солей ртути и магния, содействующих разложению труднорас-щепляемых комплексных цианидов железа.
Для анализа поверхностных и сточных вод могут быть рекомендованы три метода. Для определения цианидов в концентра
230
циях от 0,05 до 1 мг в 1 л описан экстракционно-колориметрический метод с применением пиридина и бензидина [355, стр. 540— 549], а также пиридина и барбитуровой кислоты [355, стр. 549— 552]. При анализе прозрачных проб можно пользоваться методом и без перегонки. Для определения цианидов, концентрация которых превышает 2 мг/л, приведен аргентометрический метод [355, стр. 552—555], применяемый в большинстве случаев после предварительной перегонки. Ход перегонки одинаков при колориметрическом и аргентометрическом окончании определения.
Фотометрически с пиридином и бензидином в экстракционном варианте можно определить, не разбавляя пробу, от 0,05 до 1 мг/л цианидов. Прямое колориметрическое определение без предварительной перегонки не может быть проведено, если проба содержит сульфиды, роданиды, красители, окислители, восстановители и амины.
Фотометрическому определению с барбитуровой кислотой и пиридином не мешают хлориды, бораты, нитриты, нитраты, фосфаты, оксалаты, тартраты, ацетаты, бикарбонаты, сульфаты и фенолы до концентрации 1000 мг/л. Определению мешают в свободном виде щавелевая кислота и цианат в концентрациях 100 мг/л, сульфиды и Fe(III) (^> 20 мг/л), сероводород (^> 10 мг/л). Не мешают до 10 мг/л ферро- и до 5 мг/л феррицианиды, до 3 мг/л — перекись водорода. Одинаково взаимодействуют роданиды.
Аргентометрическое определение цианидов применяется для анализа дистиллята из 250 мл пробы, содержащей от 2 до 40 мг GN~ в 1 л, с применением 0,01 N титрованного раствора нитрата серебра, а при концентрации от 10 до 200 мг в 1 л — с 0,05 N титрованным раствором.
Общие, свободные и связанные цианиды, содержащиеся в пробе, перегоняют с целью их разделения. Применяют два поглотительных сосуда (промывалки) с 50 мл 1 N раствора NaOH. (При прямой обработке пробы без перегонки прибавляют раствор едкого натра до pH 11.)
Весь полученный дистиллят или его аликвотную часть переносят в колбу для титрования и доводят объем дистиллированной водой до 150— 200 мл. Титруемый раствор должен иметь pH не менее 11. Затем прибавляют 0,5 мл индикатора (0,03%-ный раствор n-диметилбензилиденроданина в ацетоне) и титруют 0,01 или 0,05 N титрованным раствором нитрата серебра до перехода желтой окраски в розовато-красную.
Параллельно с этим проводят холостое определение с применением поглощающего раствора едкого натра, в который добавляют индикатор, и по каплям приливают титрованный раствор нитрата серебра той же концентрации, как и при титровании пробы, до перехода окраски. Содержание цианид-ионов (х) в мг/л вычисляют по формуле
ж= (« — &)*W2-52,04-lG00 _ 52040(а — b)kNV2
VJ, vtv,
где а — расход титрованного раствора нитрата серебра, израсходованного на
титрование дистиллята, мл; b — расход тйтройанного раствора на холостое определение, мл; к — поправка для приведения концентрации раствора серебра к требуемой нормальности; N — нормальность титрованного раствора; Fj — объем дистиллята, взятого для титрования, мл; lz2 — общий объем дистиллята, мл; V3 — объем пробы, взятой для перегонки, мл; 52,04 — эквивалент цианид-ионов.
Определению цианидов титрованием мешает присутствие большого количества жирных кислот, улетучивающихся с водяным паром и вызывающих нечеткий переход окраски индикатора. Их перед перегонкой экстрагируют изооктаном, гексаном или хлороформом (200 мл на 1 л пробы) после предварительного доведения pH до 6—7.
Простые и экспрессные методы определения цианидов в природных и сточных водах с помощью индикаторных трубок предложены в [143, 547]. Эти методы могут быть рекомендованы для анализа вод в полевых условиях.
Роданид-ионы
Роданиды не являются естественной составной частью вод. Они имеются в некоторых сточных водах химической промышленности, где они встречаются почти так же часто, как цианиды, и обычно вместе с ними.
Для качественного обнаружения роданидов к 10 мл пробы прибавляют несколько капель концентрированной соляной кислоты и несколько капель 10%-ного раствора хлорного железа. По появлению красной окраски судят о присутствии роданид-ионов.
Пробы не консервируют, их следует анализировать в течение суток с момента отбора.
Для определения роданидов в концентрациях выше 10 мг/л применяют аргентометрический метод после отделения нерастворимого роданида меди [355, стр. 561—563]. Используют также чувствительные фотометрические методы с бензидином и пиридином (от 0,01 до 3,0 мг/л) [355, стр. 564—568], а также с барбитуровой кислотой и пиридином (более 0,05 мг/л} [355, стр. 568— 569]. Цианиды реагируют так же, как и роданиды. Их предварительно удаляют отгонкой в виде цианистого водорода или разрушают добавлением формальдегида. Небольшие количества окислителей или восстановителей не мешают определению, так как в ходе анализа сточную воду поочередно окисляют бромной водой и восстанавливают мышьяковистой кислотой.
232
Азот органических веществ («органический азот»)
Азот, содержащийся в аминокислотах, пептидах, белках и других естественных и синтетических органических соединениях, определяется суммарно.
Органический азот в поверхностных и сточных водах определяют в виде аммиака после разложения органических веществ пробы нагреванием с серной кислотой при каталитическом действии соли ртути. Если органический азот не определяют при отборе пробы, то ее консервируют.
Отгонкой при pH 7,4 отделяют аммиак. Кубовый остаток затем нагревают со смесью серной кислоты и сульфата калия при 345— 370° С. Органически связанный азот переходит в гидросульфат аммония. После подщелачивания пробы аммиак отгоняют в колбу с кислотой и определяют его титрованием (не менее 1 мг]л) или фотометрически (менее 2 мг/л) [355, стр. 596].
Содержание органического азота (х) в мг/л вычисляют по формуле х = С-0,78, где С — концентрация NH^, найденная титрованием или фотометрически после минерализации пробы, мг/л\ 0,78 — коэффициент пересчета NH^ на N.
Общее содержание азота («общий азот»)
В материальных балансах водоемов и очистных сооружений учитываются соотношения между содержанием различных соединений азота. За основную величину в этих расчетах принимается суммарное содержание всех присутствующих соединений азота, выражаемое величиной общего содержания азота. Содержание азота можно определить расчетом или непосредственно после перевода всех соединений азота в аммиак.
При определении общего азота расчетом содержание нитратов, нитритов и аммиака, выраженное в миллиграммах азота в 1 л, умножают на соответствующие пересчетные коэффициенты и прибавляют к содержанию органического азота. Коэффициенты для расчета приведены в табл. 28.
Таблица 28
Весовые соотношения различных соединений азота (мг) [355, стр. 601]
Форма нахождения N N03 no2 nh+ Форма нахождения N NOg N02 nh+
N 1,00 4,43 3,28 1,29 NO" 0,30 1,35 1,00 0,39
no; *» 0,23 1,00 0,74 0,29 NH+ 0,78 3,44 2,56 1,00
233
Содержание общего азота (х) в мг/л вычисляют по формуле x = a-\-b-\-C-\-d, где а — концентрация нитратов, мг N/л', b — концентрация нитритов, мг^/л', С — концентрация аммиака, мг N/л; d — концентрация органического азота, мгУ/л.
В случае определения «общего азота» после перевода в аммиак нитраты и нитриты, находящиеся в пробе, восстанавливают водородом в момент выделения до аммиака. После восстановления смесь минерализуют серной кислотой с сульфатом калия при 345— 370° С при каталитическом действии сульфата ртути. Этим способом все азотсодержащие соединения переводят в гидросульфат аммония. Минерализованную пробу подщелачивают, отгоняют аммиак, собирая его в раствор кислоты, и определяют титрованием.
В последние годы все шире используются ионоселективные электроды для определения различных ионных форм азота в водах, например: для определения аммония в океанской и морской воде [748], нитрат-ионов в поверхностных и грунтовых водах [913, 1304, 1389]. Использование таких электродов позволяет автоматизировать методы контроля качества вод.
Автоматические методы анализа вод с использованием различных установок описаны в [312, 600, 1037, 1127, 1429].
МЕТАЛЛЫ, СПЛАВЫ, СТАЛИ
Азот (наряду с кислородом, водородом и углеродом) относится к числу «газообразующих примесей» в металлах и сплавах, которые оказывают сильное влияние на свойства металлических материалов. В связи с тем, что уже незначительное содержание этих примесей (</ п-10-3 вес. %) в ряде случаев определяет возможность или невозможность использования металлов в различных областях новой техники, возникает необходимость в экспрессных, чувствительных и точных методах определения газообразных примесей.
Существует много методов определения азота в металлических материалах, и выбор того или иного метода выделения азота и его определения в каждом конкретном случае зависит от ряда факторов: требуемой чувствительности, точности/наличия необходимой аппаратуры, природы анализируемого объекта, характера связи азота-примеси с металлом-основой и т. д. Известные методы определения азота в металлах систематизированы в ряде монографий [167, 292, 351, 867] и обзоров [110, 274, 360, 702, 725, 749, 760, 804, 863, 1199].
Ряд работ посвящен сравнительной характеристике различных используемых методов анализа металлов и сплавов на содержание азота [176, 327, 484, 669, 726, 876, 910, 939, 1043, 1157, 1229, 1329, 1337, 1462]. На основании сравнения ряда методов в работе [176] делается заключение, что если компоненты сплавов не образуют устойчивых нитридов, то вакуум-плавление и химические методы дают хорошо сопоставимые результаты. В этих случаях 234
для определения малых содержаний азота предпочтителен метод вакуум-плавления. Если же в сплаве присутствуют компоненты, образующие устойчивые нитриды или карбонитриды, следует предпочесть химический метод, уделяя особое внимание полноте разложения пробы.
При определении азота в уране [327] хорошо согласуются результаты, полученные методами Дюма и Кьельдаля. При сравнении трех методов определения азота в сталях [876] (восстановительное плавление, окислительное плавление, химический метод) указывается, что результаты, полученные первым методом, не всегда совпадают с данными двух других методов. Вместо широко распространенного способа определения азота в смеси экстрагированных из образца газов по разности давлений рекомендуется для повышения точности анализа измерять непосредственно парциальное давление азота. В работе [1229] критически рассмотрены преимущества и недостатки масс-спектрометрического, активационного методов и метода изотопного разбавления при определении многих примесей, в том числе и азота, в бериллии. В области концентраций 0,003—0,2% N в железе и стали наблюдается хорошее согласие между результатами, полученными методами Кьельдаля и изотопного разбавления; метод вакуум-плавления дает хорошие результаты лишь при 2100— 2240° С. По данным работы [1157], при определении азота в сплавах Fe—Si наиболее удобным и точным оказался метод изотопного уравновешивания по сравнению с химическим методом и вакуум-плавлением.
Для определения газов в металлах все чаще используются автоматические методы с применением специальных приборов-анализаторов: например, эксхалографа EA-I фирмы «Бальцере» [109, 556, 597, 606], японского прибора «Нитромат» [168], эволо-графа VH-8 фирмы «Heraous — Feichting» [1307] и др. [957, 958, 1303].
В металлических материалах азот находится в различных формах: адсорбированный на поверхности, растворенный, связанный (нитриды, карбонитриды и др.). Необходимость определения суммарного содержания азота или связанной формы обусловливает выбор того или иного метода выделения азота из образца.
Методы выделения азота
Методы выделения азота могут быть разделены на несколько групп: высокотемпературная экстракция, сплавление, химическое растворение, анодное растворение.
Высокотемпературная экстракция осуществляется в различных вариантах: вакуумное плавление; плавление в атмосфере инертного газа; окислительное плавление в атмосфере активного газа. Одним из вариантов способа плавления в атмосфере инертного газа является плавка во взвешенном состоянии в электромагнитном поле.
235
Этот способ выделения приводит к определению валового содержания азота при выборе оптимальных режимов.
Для определения малых количеств азота в мартеновских шлаках предложен метод окислительного плавления в вакууме со смесью (1:1:1) РЬО, РЬО2, РЬСг2О3 [38]. В присутствии РЬ3О4 в атмосфере гелия плавят образцы с помощью ВЧ-генератора при анализе железа и стали [431]. В качестве окислителя применяют смеси РЬО + NaPO3; РЬО + NaP03 + В2О3 при окислительной плавке в токе СО2 в случае анализа сталей, циркония, урана [537]; в присутствии РЬО по методу окислительного плавления анализируют металлический цирконий [709]; ускоренный метод определения азота в стали при окислительном плавлении предложен в работе [289]. Плавка в аргоне использована для определения азота в сталях [805], в токе гелия — для анализа урана [698] и других металлов [683].
Метод восстановительного плавления используется для определения азота с чувствительностью 10~3—10~4 вес.%. В комбинации с методом активационного анализа, когда восстановительное плавление используется для выделения короткоживущего радиоактивного изотопа азота, достигается чувствительность '—1О6 вес. % [45].
Аппаратура для определения азота (и других газообразующих примесей) в металлах и неорганических материалах методами окислительного и восстановительного плавления описана в обзоре [168]..
При высокотемпературном спекании вольфрамовых штабиков в вакууме азот хорошо удаляется из образца [198]. Методом плавки во взвешенном состоянии в электромагнитном поле определяют азот в чистом железе при 1700—2600° С [294].
Метод выделения азота анодным растворением используется при фазовом анализе — определении азота, связанного в виде нитридов; при этом способе связанный азот переходит в электролит, а сорбированный молекулярный азот выделяется [139, 142, 484].
Различные химические методы «мокрого» разложения образца приводят к определению связанного азота. Эти методы разложения состоят чаще всего в переводе азота в аммонийные соли, отгонке аммиака (иногда с барботированием анализируемого раствора водяным паром) и в определении его по методу Кьельдаля, Несслера и др. Адсорбированный или находящийся в порах молекулярный азот при этом не учитывается.
Химические методы разложения образца связаны с растворением пробы в кислотах — H2SO4, НС1, реже — НС1О4 (разложение нитридов). Труднорастворимые нитриды, образующиеся из V, Nb, Mo, Ti, Si, С, переводят в раствор сплавлением с кислым сернокислым калием, нагреванием с H2SO4, K2SO4, CuSO4 (катализатор) или растворением в хлорной кислоте. Метод простого растворения для легированных сталей неприемлем, так как большая часть азота остается в нерастворимом остатке.
236
Для большинства сталей, многих ферросплавов и других продуктов металлургического производства применим метод последовательного разложения (в одной колбе): 1) растворением металлической части навески в НС1 или H2SO4; 2) разрушением карбидов надсернокислым калием; 3) разложением нитридов сплавлением со средним сернокислым калием [59].
Методы определения азота
Для определения газообразующих примесей в металлических материалах применяется комплекс современных методов: химические, активационные, спектральные, высокотемпературная экстракция и др.
Химические методы в настоящее время являются наиболее распространенными. К ним относятся прежде всего различные модификации метода Кьельдаля с титриметрическим, кондуктометрическим или фотометрическим (в основном с реактивом Несслера) окончанием. Одним из недостатков химического метода является существенная поправка холостого опыта [до (3 -5- 5) • 10-3 вес. % при навеске 1 г], связанная с загрязнением реактивов, посуды, окружающей атмосферы аммонийными солями и другими соединениями азота.
Подробное описание метода Кьельдаля и фотометрического метода Несслера см. в соответствующих главах.
Спектральные методы просты и экспрессны. Однако они требуют использования вакуумных спектрографов, специализированных источников возбуждения спектров, качественных эталонов.
Методы эмиссионной спектроскопии осуществляются в трех вариантах: 1) полное выделение газа из металла и последующий их спектральный анализ (определение кислорода и азота); чувствительность такого метода п-10-3%; основные погрешности связаны с несовершенством условий выделения газов из образца; 2) спектральное окончание метода изотопного разбавления; 3) экстракция газов из металлов с одновременным возбуждением их спектров (например, локальный и послойный анализ).
Большинство работ по спектральному определению азота посвящено анализу сталей в видимой и ИК-области спектра с чувствительностью п-10-2 вес. %, а также определению азота по линиям вакуумной УФ-области спектра [177, 293, 304].
Масс-спектральные методы, так же как в спектральном анализе, для определения газообразующих примесей применяются для трех целей: 1) после предварительной вакуумной экстракции для окончательного определения; 2) для измерения изотопных отношений в газовой фазе при анализе методом изотопного разбавления (см. ниже); 3) использование искровой масс-спектрометрии в случае совмещения процесса экстракции с ионизацией и последующим определением примеси. Обзор масс-спектральных методов определения газообразующих примесей сделан в работах [274, 410].
237
Искровая масс-спектрометрия позволяет определять азог с чувствительностью п-10-4 вес.%. Затруднения при использовании этого метода связаны с поверхностными загрязнениями Образца, остаточными газами в вакуумной системе, недостаточно ры-сокими коэффициентами ионизации азота по сравнению с другими примесями, перекрыванием аналитических линий азота линцями многозарядных ионов и т. д.
Особенно перспективно применение масс-спектрального метода при локальном анализе, анализе микрообъемов образца, изотопном анализе.
Для надежного определения микропримесей азота необходима стабилизация параметров искрового источника.
Активационные методы — высокочувствительны (до 10-3 — 10-5%) и селективны. Еще одно их преимущество — возможность анализа без разрушения образца. Обзор активационных методов определения азота приведен в работе [163].
Снятие поверхностного слоя после облучения пробы позволяет исключить влияние поверхностных загрязнений на результаты анализа. В этих методах используют для активации заряженные частицы, быстрые нейтроны и у-кванты.
При нейтронно-активационном определении следовых содержаний азота в большинстве материалов по реакции 14N (п, 2п) 13N (Тцг = 10 мин.) необходимо радиохимическое выделение 13N, что усложняет и удлиняет ход анализа. Кроме того, необходимо довольно длительное облучение для определения азота с высокой чувствительностью. Однако возможно определение азота в ряде металлов и без химического выделения — в ниобии и тантале [164], сталях [427]. Нейтронно-активационный метод анализа металлического натрия на содержание азота с отгонкой NHS по методу Кьельдаля после облучения и растворения пробы описан в [164, стр. 219].
у-Активационное определение азота в металлах проводят с помощью реакции 14N (у, n)13N. Максимальная чувствительность (до 10~4—10-5 вес. %) достигается только при радиохимическом выделении изотопа 13N. Например, при анализе металлических циркония, ниобия, бериллия — по методу Кьельдаля [447], при определении до п-10-5 вес. % аэота в германии — методом окислительного плавления с последующим улавливанием N2 на молекулярных ситах [680]. Определение азота в редких металлах и полупроводниковых материалах у-активационным методом описано в [275].
При активации протонами определяют примесь азота в тантале [190], бериллии, кремнии [679]. Активация пучком ускоренных дейтронов используется при определении примеси азота на поверхности металлических бериллия, молибдена и вольфрама [510, 523] по реакции 14N (d, п)15О, при анализе стали [1305].
Вакуум-плавление — один из современных простых и универсальных методов определения азота в металлах. Иначе его называ-238
яхт методом высокотемпературной вакуумной экстракции, восстановительным плавлением в вакууме, диффузионной экстракцией.
В принципе методы вакуумной экстракции не отличаются от методов экстракции в атмосфере инертного газа-носителя, но позволяет определять элементы с повышенной чувствительностью (до п*10~4%) 1805а]. Подробно методы вакуумной экстракции рассмотрены в работе [127]. Обзоры работ по этому методу приведены в [702,’ 760, 772].
Эти методы основаны на полном выделении примеси иэ образца в виде газа с последующей идентификацией и измерением количества определяемого элемента.
Высокотемпературная вакуумная экстракция азота в нейтральной среде (без окислителя и восстановителя) в безуглеродис-том никелевом расплаве (с масс-спектральным окончанием) позволяет определять азот с чувствительностью (1 -ь 2)• 10_4 вес. %, в то время как метод восстановительного плавления характеризуется чувствительностью 10-2 — 10-4 вес. %.
Комбинированные методы, сочетающие вакуумную экстракцию с нейтронно-активационным определением, позволяют определять в газовой фазе (это их преимущество) азот с чувствительностью 1-Ю-8 вес.% путем измерения активности выделенного радиоактивного изотопа (после облучения образца у-квантами и травления поверхности).
Возможно сочетание радиоактивационного метода с плавлением (импульсный нагрев) в инертной атмосфере.
Основные источники ошибок метода вакуумной экстракции обусловлены неполнотой извлечения азота, поглощением выделившегося газа на возгонах, поверхностными загрязнениями, колебаниями поправки холостого опыта и т. д.
Определению азота в металлах методом вакуум-плавления посвящено большое число работ. Количественно определяют выделенный этим методом азот измерением давления газов (манометрически), измерением его объема, сочетанием с газовой хроматографией, масс-спектрометрией и активационным анализом (как уже упоминалось).
If Ниже приведены описания двух методов вакуум-плавления для определения азота в тугоплавких металлах.
Определение азота в металлах методом вакуум-плавления в безуглеродистом расплаве [127, стр. 239]. В тигель (из корунда) экстракционной кварцевой печи помещают ~ 20 г никеля вакуумной плавки, загружают в установку пробы весом 0,15—0,4 г, систему вакуумируют, расплавляют никель, постепенно повышая температуру до 1800° С, дегазируют расплав 40—50 мин., затем сбрасывают в расплав образец и анализируют по обычной процедуре. После каждого определения обновляют ванну, добавляя 1—3 г никеля. В одном тигле анализируют 3—4 образца.
239
Стандартное отклонение составляет 1-10-4 вес. % в расчете на исходную навеску. Воспроизводимость метода в зависимости от концентрации азота 10—20 отн. % при определении 1(к2— 10~4 вес. % Экспериментальная гарантируемая чувствительность 1-10"4 вес.%.
Нейтронно-активационный метод определения азота с выделением 13N вакуумнрй экстракцией [127, стр. 241]. Пробу весом 1—3 г облучают быстрыми нейтронами с помощью нейтронных генераторов ИГ-150 и Т-400 в течение 5—10 мин., затем подвергают травлению в быстродействующих травителях [конц. HNO3 или смесь конц. HNO3 и HF (1 : 1)]. После травления образцы загружают в установку вакуум-плавления. Используют методику с применением графитового тигля (никелевая ванна, 1700—1750° С, газосодержащие добавки — никель, железо, гидрид ниобия). Расчет концентрации азота в пробе проводят по формуле
с = тст'Сст . ^Обр . (1 _е-Х(ст)е-^з.ст еТ 1оо ,
х тобр (1 _ е~х(з.0бр ер7ср
где Сх — искомое содержание азота в образце, вес. %; Сст — содержание определяемого элемента в стандарте (стандарты из полиамидной пленки с содержанием азота 12,8 вес. % располагали по обе стороны образца и облучали одновременно с ним); Л^обр — счет от образца, имп; 7VCT — счет от стандарта, имп.\ тобр, мст — массы образца и стандарта, г; t3 обр, t3 ст — соответственно время задержки между концом облучения и началом измерения активности образца и стандарта, мин.; к— эффективность переноса радиоизотопа из экстракционной части в детектор; еу — поправка на разницу в эффективности ер измерения активности образца и стандарта; Л — постоянная распада определяемого изотопа (Л = In 2/7’,^); — период полураспада опреде-
ляемого изотопа 13N; р — радиохимический выход при выделении 13N.
Сочетание активационного метода с вакуумной экстракцией повыщает чувствительность определения азота за счет снижения поправки холостого опыта, устранения поверхностных загрязнений. Добавление в расплав носителей в виде кусочков никеля и гидрида ниобия ускоряет экстракцию и увеличивает степень извлечения азота. Основная часть азота выделяется за первые 3—4 мин.
Метод изотопного разбавления (уравновешивания) целесообразно применять для тех объектов, для которых не удается достигнуть полной экстракции азота или избежать значительных потерь на возгонах. Принцип этого метода был подробно изложен на стр. 141.
К образцу добавляют точно измеренное количество стабильного изотопа 15N, проводят операции, обеспечивающие полноту изотопного обмена, выделяют фракцию азота и измеряют его изотопный состав, а затем по этим данным рассчитывают исходное содержание примеси.
240
Изотопный обмен в жидкой фазе и измерение изотопных отношений на масс-спектрометре выполнены в работах [51, 254, 925L Этим способом определяют азот в Fe, Ni, Со, Сг, Мп, В, Ti, Zr, La, Се, Рг, Та, Mo, Nb и их сплавах. Чувствительность анализа 10-4—10~5 вес.%.
Изотопный обмен может протекать между находящимися в газовой фазе индикатором и газовыми примесями расплавленной пробы |1158]. После установления равновесия газ анализируют на масс-спектрометре. Так определяют азот в сталях.
В работе [96] предложено проводить обмен между нагретым образцом в твердой фазе и индикатором в газовой фазе. При измерении изотопных отношений используют методы эмиссионного спектрального анализа. Этот вариант относительно прост и универсален. Авторами разработаны методы определения газов более чем в 20 металлах. Для ускорения скорости диффузии газов в твердых образцах предложены варианты высокотемпературного изотопного обмена с применением полого катода, используемого одновременно для уравновешивания изотопов и возбуждения свечения их спектров, а также с применением квантового генератора [87а] и искрового масс-спектрометра [64, стр. 97].
Недостатком метода изотопного разбавления является его длительность, сложность, большое влияние поверхностных загрязнений на результаты анализа при определении малых концентраций азота.
В качестве примера приводится описание спектрально-изотопного метода определения азота (и кислорода) в титане и его сплавах [86].
Образец титана или сплава на его основе (0,1—0,4 г) помещают в графитовый тигель с твердой никелевой ванной (8—10 г). Тигель и ванна дегазированы. В обменник вводят изотопсодержащий газ (1SN2), который при нагреве до 1000—1100° С поглощается анализируемым образцом. Затем при 1600— 1700° С титан растворяется в никелевой ванне и в течение 10—15 мин. осуществляется изотопическое уравновешивание.
Уравновешенный газ, выделившийся в объем обменника, попадает в разрядную трубку, где с помощью высокочастотного генератора возбуждается его свечение. Фотоэлектрически измеряют относительную интенсивность полос N2 (3804 А). Содержание газа рассчитывают по обычным формулам спектрально-изотопного метода.
Воспроизводимость результатов определения при содержании азота ~ 0,01 вес.% составляет ~20 отн. %.
Таким образом, валовое содержание азота в металлах лучше всего определять, как видно из приведенного выше материала, активационным методом и методом вакуум-плавления. Химические и спектральные методы менее чувствительны, хотя и проще в осуществлении. Однако бесспорно преимущество спектральных
241
методов для локального анализа веществ. Масс-спектрометрия используется в основном для обнаружения стабильных изотонов, но это — уникальный метод.
В таблице 29 приведены характеристики различных методов определения азота в металлах, сплавах, сталях.
Методы анализа, описанные выше для металлов и сплавов, используются также для определения азота в окислах, карбидах и нитридах металлов. Иногда эти методы несколько видоизменяются в связи со спецификой объекта. Обзор химических методов определения азота в нитридах бора, титана, циркония, хрома, молибдена, ниобия, тантала и ванадия приведен в работе [825].
В качестве примера ниже приводятся ссылки на работы, посвященные титриметрическим методам определения азота в нитридах бора и кремния [202, 323, 1146, 1150], нитридах урана и плутония [826, 1063, 1064], нитридах титана [4], ванадия [539], циркония и ниобия [295], алюминия [9]; газометрическим методам определения азота в нитридах урана [1066, 1343], нитридах ниобия и тантала [916], нитридах кремния, титана и ванадия [411], в карбидах кремния [619], карбидах и карбонитридах Ti, Zr, V, Nb, Та, Gr, Mo, W, Mn, Fe, U [1231], в окислах, нитридах и гидридах металлов [1143]; газохроматографическим методам определения азота в тугоплавких материалах — карбидах, нитридах, окислах, фосфидах и силицидах [857, 1056]; спектрально-муи спектрально-изотопному методу определения азота в окислах, карбидах и нитридах W, Nb, Ti, Si [105, 306] и др.
ПРОЧИЕ ОБЪЕКТЫ
Материалы высокой чистоты. Полупроводниковые материалы
Для определения примесей азота в материалах высокой чистоты используются в основном активационные методы. Обзор таких методов дан в [273]. Активация пробы ускоренными дейтронами применяется для определения ~10-8 г азота в пробах различной природы [676]. Активационными методами определяют примеси азота в полупроводниковом кремнии и карбиде кремния [180, 1117], в материалах атомных реакторов [765], в пиролитическом графите [1103, 1116], в высокочистых бериллии, кальции, литии, натрии и боре [678].
Методом искровой масс-спектрометрии с ионным источником с чувствительностью 3-10~5 ат. % определены примеси азота в арсениде галлия и антимониде индия [63].
В монокристаллах карбида кремния (a-SiG), полученных сублимацией из газовой фазы, определено содержание азота методом электронного парамагнитного резонанса [108].
242
Таблица 29
Методы определения азота в металлах, сплавах, стали, чугуне
| Литература <—। <—। 1—। <—। ’ *2 <—> чч <—о ' О Г- С5 СЭ Q О Г* «н М* чЧ с© чЧ 04 04 СО СО Ю чЧ чЧ »—• ео 04 чЧ 04 —> - чЧ чЧ чч ' чч ~ со — “ - § £ 3 —1 ио
1 । Примечание Эффективна для разложения смесь HaS04 с селеном (в виде HgSeOa, SeOCls, SeOa) В [294] использована плавка во взвешенном состоянии в электромагнитном поле । Стружку металла растворяют в НС1 (1:1) Навеску растворяют в смеси H2SO4 (1 : 5) и 1 %-ного раствора NaF с последующим добавлением 1г K.CIO4 Используют кулонометрическое титрование
Точность > Ф > Ф «из S L .L й ® И в Я g Я «< » И ЙЙ Ш ® S И И О ® Я . й<§ VO Я Q, я ° н -р Я Оф И о ф Ю м S EQ 03 js.s h- "i71 t’" " g «В 3 5 0 ® '(£§£[§• 2 5 33g йч ОЦчч н © Рч со Д К Оч К Рч
Предел обнаружения Я 1 ? 1 1 1 gg 1 ^§11 II 1 0 . О\ 0 о "о ф ЧИ ° О И • «. гб е о я
Анализируемый объект й ч ч S сб сб И И Й л я 2 * О< Н go >» сб Ф , Н *Й Сб сб « И 3 й Н и g 5 g S & Й 5 Й й ’S н § К ® ® П >Н О ° Н й и м- Й И ч » ояй я S В и *5 Я 2 и8я н л S | § «5* 1 ® Е-< >» ииИй ь о о о
Метод определения । Метод Кьельдаля с титриметриче-ским окончанием
243
Таблица 29 (п родолжение)
Метод определения Анализируемый объект Предел обнаружения Точность Примечание Литература
Метод Кьельдаля с титриметриче-ским окончанием Метод Кьельдаля с фотометрическим окончанием Стали Чугуны » Тугоплавкие металлы и сплавы Ниобий, тантал и их сплавы Цирконий и его сплавы То же Вольфрам Калий Литий Магний До 0,1— 0,02 мкг N2 0,005% N2 0,003— 0,01 % >1 ч. на млн. 0,003% N п-10-3 % 0,001 % N (при навеске 1 г) Ошибка ~10% (при навеске 1 г) Абсолютная ошибка ± 0,0004% Ошибка <25% Ошибка ±Ю% Коэффициент вариации ±(5—10)% Средняя ошибка <5% Ошибка 2% Используют кондуктометрический метод Разложение проводят в две стадии: сначала растворяют в НС1 (1:1), а затем нерастворимый остаток разлагают в смеси K.HSO4, CuSO4 и H2SO4 Количество азота определяют В Ч-титрованием Индофенольный метод; разлагают образцы смесью HF ± ± Н3РО4 ± К2СГ2О7 С реактивом Несслера То же Определение азота в поверхностном слое по индофенольной реакции С реактивом Несслера; пробу растворяют в 30%-ной Н2О2 С реактивом Несслера; растворяют образец в разб. Н3РО4 Индофенольная реакция С реактивом Несслера; пробу растворяют в НС1; индофенольная реакция без отгонки [65, 420] [174[ [9101 [9061 [122, 877] [565] [1286] [427J [928] [1041 [339]
Т а & л и ц а 29 (продолжение)
Метод определения Анализируемый объект Предел обнаружения Точность Примечание Литература
Метод Кьельдаля с фотометрическим окончанием Бериллий Железо и стали <0,2 мкг N (0,0004% N) Абсолютная ошибка п-10-5 % Фотометрируют после экстракции индофенольного соединения [12291 [9071
Хром Стали, чугун, железо и металлический алюми- — С реактивом Несслера [7371
3-1 о-4— — Индофенольная реакция с по- [1098]
3-10~2% N следующей экстракцией окра-
шейного продукта [1451
Кремнистые черные ме- — — фенол-гипобромитный метод
таллы Чугун — Абсолютная ошибка ±0,0005% С реактивом Несслера [175]
Сталь автолистовая — — То же [272]
Стали углеродистые, — — Реакция с тимолом и хлор- [7671
хромистые и марганце- амином Т
вые Стали и феррохром — — Индофенольная реакция и фо-тометрирование экстракта [1461
Стали — — Реакция с нитропруссидом и индофенольная реакция [14111 [НО]
Сплавы, содержащие титан Резистивные сплавы — — Улучшен процесс разложения нитридной фазы
1-10-*% N — С реактивом Несслера [3501
Осциллографическая полярография Стали и сплавы С отгонкой и без- отгонки NH3; полярографируют по методу добавок [4211
is
Таблица 29 (продолжение)
Метод определения Анализируемый объект Предел обнаружения ТОЧНОСТЬ Примечание Литература
Спектральный эмиссионный Различные металлы — — Используют генератор низковольтной искры и вакуумную камеру; фотометрируют линию N II3995 А [307 J
То же -— Определяют азот в фольге металлов; спектр возбуждают низковольтной искрой [305J
» 3—1000 мкг N Точность ~10% Источник возбуждения — ВЧ-разряд в аргоновой плазме; аналитическая линия N 4216,0 А [440J
» 0,001% N Средняя ошибка — несколько процентов Используют вакуумный спектрограф; спектры возбуждают конденсированной искрой; аналитическая линия N IV 765,14А [1225]
Железо и низколегированные стали 2-10-3— 1-10-2% Коэффициент вариации 11% Аналитические пары линий N 1745/Fel876 А и N 1742/Fe 1876 А; используют низковольтный искровой разряд (841]
Хром 0,2—1,35 % N Воспроизводимость 5—30% Источник возбуждения — низковольтная искра; аналитическая линия 3994,99 А [363]
Никель l-10-з вес. % Средняя квадратичная погрешность ~12% Аналитическая линия 3925,00 А; возбуждают спектр в низковольтном исКровом разряде (26J
Мо—W-сплавы и стали — — Спектр возбуждают в искровом [362]
Титан я его сплавы 0,01—0,6 вес. % Среднеарифметическая ошибка ±10 отн. % разряде (321]
Таблица 29 (п родолжение)
Метод определения Анализируемый объект Предел обнаружения Точность Примечание Литература
Спектральный эмиссионный Вакуумная УФ- и ИК-спектроскопия Искровая масс- спектрометрия Стали, карбид урана, медь, цирконий Стали я сплавы Стали » » » » Стали, металлы Ниобий, тантал, молиб- ден я их сплавы Различные металлы 0,55—2,5 мкг N 5-Ю-4— 5-10-2 о/о п-10-в ат. % Коэффициент вариации 7—25% Точность 10—20 отн. % Возбуждают спектры в разрядной трубке с полым катодом; аналитическая линия 3914,4 А Определение проводят в низковольтной искре в атмосфере СО2; возможно локальное определение азота Послойный спектральный анализ Исследовано влияние третьих элементов Спектры возбуждают в безэлек-тродном ВЧ-разряде; аналитическая линия N 2 3371 А Используют вакуумный искровой источник Возбуждают спектр в угольной дуге постоянного тока в атмосфере аргона Далекая УФ-область (500— 1500 А); методика позволяет проводить локальный анализ и наблюдать распределение компонентов в различных сплавах Источник возбуждения ВЧ-искра в вакууме; используют масс-спектрометр с двойной фокусировкой [1387] [391 (ЗЮ1 (ЗН1 [5001 (5161 [840, 8601 [197, 501, 1226] [10241 [6251
ts3 as Таблица 29 (продолжение)
Метод определения Анализируемый объект Предел обнаружения Точность Примечание Литература
Искровая масс-спектрометрия Различные металлы — — Установлена возможность использования отрицательных ионов, образующихся в ВЧ-искровом разряде [1268]
Чистые металлы 5.10-3— 7-10-<% — Отмечена необходимость стабилизации параметров искрового источника (СЮ%) [653]
Железо, вольфрам, медь, серебро Следовые количества — Может быть проведен локальный анализ [808]
Стали, золото, платина и др. 1 -10-4aT.%N — — [994]
Ниобий n-10-1% N Коэффициент вариации —50% — [1372]
Титан и его сплавы п* 10~4 вес.'% N — — [1373]
Вольфрам — Для повышения разрешающей способности предложено уменьшать величину магнитного поля анализатора [198, 591]
Железо (1-ь4)-10-1 вес. % N —- — [1371]
Активационный Цирконий — — Нейтронное облучение (711]
Тантал 1 -IO-® % —. — [190]
Ниобий -1 -10-2 % — Нейтронное облучение (181
Ниобий, тантал, титан — — То же (164]
Ниобий и тантал, бериллий, литий 2М0-3вес. % — Активация быстрыми нейтронами (19]
Таблица 29 (продолжение)
Метод определения Анализируемый объект Предел обнаружения Точность Примечание Литература
Актив ационный Редкие металлы и полу- 10-i—10-s % у-Активационный метод [275]
проводники Молибден, вольфрам, — — (681}
ниобий, тантал Бериллий, молибден, Определение азота в поверх- [510, 523]
вольфрам Алюминий, бериллий, —1 ч. на постном слое по реакции “N (d, п)16О Определение азота по реакции [446]
цирконий Бериллий, кремнии, таи- МЛН. — UN(y, n)uN Облучение тормозным у-излу- [679]
тал Натрий чением Определение азота по реакции (164]
Стали 0,01% Стандартное откло- “N(n, 2n)13N Использована реакция [1305]
Изотопное разбав- Ниобий, сталь,вольфрам, 1-10-1% нение ±0,004% Ошибка 2—30% py)15N Масс-спектральный вариант [256]
ление молибден, лантан, церий, хром, никель Титан, нитрид титана 5-10-4вес. % То же; изотопный обмен про- [257]
Ниобий, тантал, молиб- 10Г-1—ю-2 Относительная водят в жидком металле; изотоп 16N вводят в виде металла, насыщенного меченым “N То же [255]
ден, вольфрам, кремний, кобальт, никель, железо, стали Стали вес. % ошибка 3—5% — [378]
Таблица 29 (иродолжение)
Метод определения Анализируемый объект Предел обнаружения Точность Примечание Литература
Изотопное разбавление Хром, цирконий, титан 1 -IO”3 % Точность до 10% Изотопическое уравновешивание в горячем полом катоде [100]
Молибден, вольфрам, хром, никель 5Л0-«% Относительная ошибка 10% (при содержании >10-’% N) Спектроскопический вариант [94]
Вакуум-плавление Стали 5.10-2—10-4 вес. % Среднеквадратичная ошибка 13% (для 10-’% N) и 30% (для 10-*% N) Аналитические полосы 3159,3 и 3943,0 А [32, 93, 361]
Различные металлы До 2 мкг N2 — Манометрическое определение выделенного азота [844,1186, 1303]
Уран, плутоний 2-10-4% — Плавление в Pt-ванне [1377,1453а]
Тантал — — Плавление в графитовом тигле с Pt-баней [389]
Вольфрам — — Используется Со-ванна [136—138, 824] [498]
Ванадий — — —
Титан — — — [644, 1059]
Хром — — — [140, 998]
Сплавы железа, стали, железо — — Используют Ni-ванну с добавлением Sn [176, 418а 437, 1157]
Стали, никель, кобальт 10-’% N — Применяют эксхалограф EA-I (азот находят по разности) [338, 399, 400, 597]
Стали — — Плавление в Pt-ванне [690]
» — — Плавление в графитовом тигле [727, 741, 1089, 1275]
Таблица 29 {продолжение)
Метод определения Анализируемый объект Предел обнаружения Точность Примечание Литература
Газохроматографический Различные металлы 0,0009 вес. % — Детектор — катарометр; газ-носитель — аргон; сорбент — молекулярные сита 5А [318]
То же — Выделяют газы вакуум-плавле-нием; сорбент — молекулярные сита 5А; детектор — манометрическая лампа Л Т-2 [141]
— — Выделяют газы по методу вакуум-плавления; используют ионизационный детектор, плотномер [801, 1001, 1223, 1224, 1308]
3-10-*% — Плавят в присутствии восстановителя в токе инертного газа; газ-носитель — гелий; сорбент— молекулярные сита 5А и 4А [683]
Металлы и сплавы — — Сорбент — молекулярные сита I3X [802]
Железо высокой чистоты 1 - 10-е— l-10-’вес. % Пробы' сплавляют в графитовых тиглях в токе инертного газа-носителя (гелий); сорбент — молекулярные сита I3X; детектор — катарометр [462]
Примечание Выделяют газы вакуум-плав-лением; сорбент — молекулярные сита; ионизационный детектор; газ-носитель—гелий Плавят в инертном газе с последующей ГХ Вакуум-плавлением выделяют газы; сорбент—молекулярные сита I3X; детектор по теплопроводности Выделяют азот при восстановительной плавке в аргоне Экстракция газов в печи сопротивления при ВЧ-нагреве Выделяют газы с применением угольной дуги
Таблица 29 (окончание)
О о № ЕГ О
ез
252
Взрывчатые вещества
Взрывчатые вещества анализируют на содержание азота в основном газометрическими методами после сжигания [1399], по микрометоду Дюма — Прегля (и его модификациям) [522, 937, 1422] и в анализаторе Колемана [1399].
Методы нейтронной активации служат для быстрого и точного (абсолютная ошибка <Д,1% N) количественного определения азота во взрывчатых веществах и порохах [1215, 1272].
Стекла
Для анализа газов, присутствующих в виде пузырьков в стекле, лучшим методом является газовая хроматография [612, 829, 1023, 1321, 1433]. Предельная чувствительность определения азота составляет 0,003—0,005 мкл. При анализе проб объемом ~90 мкл средняя ошибка определения N2 равна 2,16%.
Кислоты, соли, минеральные удобрения
Азотная кислота. Разработан полярографический метод определения иона аммония (в интервале 10-3—10~5 М) в азотной кислоте, основанный на образовании полярографически активного CH2NH или NH2CH2OH при реакции NH4 с избытком СН2О [353, 428]. Метод прост и нетрудоемок.
На основании измерения степени поглощения света окислами азота и водой, содержащимися в отбеленной концентрированной азотной кислоте, предложен спектрофотометрический метод определения этих компонентов. Сконструирован специальный фотоэлектрический концентратомер окислов азота — PKAO-I [43].
Серная кислота. Окислы азота в серной кислоте определяют фотометрически по методу Грисса [359], нитрованием салициловой кислоты [1206]. Предложен косвенный метод определения NO2 (0,2—0,3%) в серной кислоте мокрого катализа с йодометрическим окончанием [291]. Амперометрический метод использован для определения азота в серной кислоте [486]. Метод высокочастотного титрования предложен для определения азотной кислоты в олеумных растворах 3%HNO3b 103%-ном олеуме) с применением о-нитротолуола [252, 423].
Соли, минеральные удобрения. В неорганических нитратах Ag, Ba, Pd, Cd, Со, Fe, Na, Ni, Zn определяют N методом Дюма [918]. Для определения азота в чистом нитрате натрия и натриевой селитре используют титриметрический метод после растворения пробы и пропускания ее через колонку с катионитом [955]. В нитратах La, Na, Ni, Ba, Pb определяют азот методом нейтронной активации [459]. Быстрый фотометрический метод определения нитратов (<5%) в нитритах щелочных металлов основан на нитровании салициловой кислоты [899]. В 0,01—1,0 М растворах NH4C1,
253
CH3COONH4, (NH4)2SO4, NH4NO3 алкалиметрически определяют NH£ с ошибкой (для наиболее разбавленных растворов) [499]. Общий азот, ионы NH4 и NO2 определяют фотометрически в солях NaJ, LiF [27а].
Минеральные удобрения анализируют на содержание в них NH^, NO3, азота органических соединений и т. д. Методы определения общего азота для анализа всех видов удобрений описаны в [685, 1205]. Предложены автоматические методы определения содержания азота в минеральных удобрениях [662, 738]. Ионы NH4 и NO3 определяют титриметрически [160, 592, 595, 911, 954, 997, 1233]. Показана возможность нейтронно-активационного определения азота в удобрениях [1135]; использования рентгеновского дифракционного метода для определения NO3, NH4 в различных типах смешанных удобрений [8]; ИК-спектроскопии для определения NHJ, NO3 [769]; термометрического метода, основанного на измерении теплоты, выделяющейся при специфической реакции с азотом аммиака, мочевины, нитрата [1243]; кулонометрического метода, основанного на кислотном разложении вещества и дальнейшем окислении NH3 до N2 посредством гипо-бромита, электрогенерированного на Pt-электроде при амперометрическом определении КТТ [609]. Разработан спектрофотометрический метод определения NO3 в смешанных и сложных удобрениях, основанный на измерении оптической плотности при 310 нм [367].'
Растения, корма, пищевые продукты, биологические материалы
Обзор работ по определению NO3, NO2 и других компонентов в пищевых продуктах дан в [1266]. Колориметрически определяют общий азот, нитраты и нитриты в кормах [1407, 1437], шпинате [442, 823], сахаре [119], табаке [1283]. Титриметрически определяют содержание азота в растительных материалах, пыльце растений [478, 981]. Кулонометрический метод использован для определения азота в растительных материалах после минерализации пробы по методу Кьельдаля [897].
В биологических материалах (например, сыворотке крови, золе волос) определяют азот фотометрически [638, 1031, 1096], активационным методом [869].
Глава XIII
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ В АЗОТЕ И ЕГО СОЕДИНЕНИЯХ
ГАЗООБРАЗНЫЙ АЗОТ
Обзор инструментальных методов определения Н2О, СО2 и углеводородов в полученных при воздушной сепарации газах (в том числе — в азоте) приведен в [238]. В работе [667] предложено удобное устройство для отбора пробы при определении следовых количеств примесей в газах, в частности в азоте.
Микроколичества кислорода в азоте определяют ф о т о м е т« рическим методом, основанным на взаимодействии О2 с парами серы с образованием (при 360° С) SO2, дающей окрашенное соединение с фуксинформальдегидным реактивом [205]. Чувствительность метода ~ 10"5 объемн. %, но практически она ограничивается величиной холостой пробы (5-10~4 объемн.%).
При анализе азота как газа, заполняющего вакуумную систему, принят ампульный вариант [205], заключающийся в следующем.
Ампулу с 20—40 мг S напаивают на установку, откачивают ее до 10-2jhjh рт. ст., нагреванием испаряют серу на холодные стенки ампулы, отключают насос и соединяют ампулу с пробой анализируемого газа. Затем ампулу отпаивают и помещают в печь с температурой 400—450° С и выдерживают 30— 35 мин. После охлаждения ампулу погружают в смесь 7 мл 0,009%-ного раствора фуксина, 7 мл 0,0045%-ного раствора СН2О и 7 мл 1,2 N H2SO4 и вскрывают в этом растворе. Через 30—40 мин. раствор разбавляют до 25 мл 1,2 N H2SO4 и фотометрируют в 5-сантиметровой кювете на ФЭК М-Н со светофильтром с максимумом пропускания 584 нм. Содержание О2 находят по калибровочной кривой, построенной по раствору Na2SOs, содержащему 10 мкг ЗО21мл.
Простой переносной прибор для определения кислорода в газах по цветной реакции (до 1-10~3%) с точностью 20% описан в [356].
Высокочувствительный электрохимический метод, основанный на обратимом катодном восстановлении О2, растворенного н воде, по реакции
О2— 4е4-2Н2О =4ОН-,
255
применен для определения кислорода в «защитном» газе (особочистых гелии и азоте) [1187].
Анализируемый газ просасывают через специальный электролизер, содержащий 15%-ный раствор КОН, и затем через измерительную ячейку, в которой находится Ag-сетка, служащая катодом. При этом следят за показаниями микроамперметра, включенного в цепь. После установления стабильного положения стрелки микроамперметра отсчитывают ее показания и находят содержание О2 в анализируемом газе по калибровочной кривой, построенной в тех же условиях на стандартных смесях газов с известным содержанием О2.
Этим методом можно анализировать газы, содержащие 1-10-4—1% О2. При определении 0,01% О2 ошибка составляет 1—5%. Продолжительность одного определения 10—15 мин.
Непрерывное количественное определение кислорода в азоте (в пределах 0,05—3 объемй.%) возможно радиолюминесцентным методом по тушению радиолюминесценции люминофоров [150].
Автоматический газоанализатор ГЛ-5108 позволяет определять микроконцентрации кислорода в азоте до 0,0001 объемн.% [184].
Для определения водорода в азоте используют эмиссионную спектроскопию [659]. Анализируемый газ непрерывно пропускают через разрядную кварцевую трубку под давлением 2 мм рт. ст. Исследовано возбуждение спектров при различных давлениях газа и разных диаметрах разрядной трубки. Спектры регистрируют на спектрометре. Анализ проводят по линии Н 4861,3/N4863 А в координатах Ц/Ц — С. Чувствительность определения равна 0,05%. Средняя квадратичная ошибка 2,5%.
С помощью кулонометрического влагомера электрохимическим методом определяют 1-10-3—1 вес.% воды в техническом азоте [270].
Исследована возможность масс-спектрометрического анализа газовых смесей (в частности, для определения окиси углерода в азоте) путем избирательной ионизации отдельных компонентов монохроматическим светом [1180]. Для устранения влияния фона, обусловленного фотоэлектронами, образующимися в камере монохроматора, между выходной щелью монохроматора и ионным источником устанавливают отклоняющие пластинки. Источником света служит искровой разряд в аргоне при давлении 2 мм рт. ст., улучшающий группу интенсивных линий в области 879 А. С применением Ar-фильтра предел обнаружения примеси СО в N2 составляет 0,01% по пику иг/е 28. В случае металлического In-фильтра предел обнаружения равен 0,05%. Основным недостатком метода является сравнительно низкое отношение' сигнала к фону.
256
Описан экспрессный кулонометрический метод определения субмикроконцентраций окиси азота в азоте (>=10~8%). Метод включает предварительную сорбцию NO на кварце, пропитанном FeSO4, десорбцию ее нагреванием при 350° С и окисление до NO2 раствором КМнО4 в 10%-ной H2SO4 [161]. Двуокись азота, взаимодействуя с J~ в проточной кулонометрической ячейке, выделяет J2, предельный ток восстановления которого регистрируется детектором. Время анализа 15 мин. Определяемая концентрация NO в азоте составляет 5-10~9%. Ошибка определения <23%.
Метод газовой хроматографии применяют для количественного определения закиси азота и исследования ее чистоты [1022]. В качестве сорбента используют молекулярные сита 5А, газ-носитель — водород (40 мл/мин). Указанным методом можно определять (в ч. на млн.): О2 4; N2 4; СО 4; общее количество N2O и NOa 8; СО2 0,5.
Газохроматографический метод использован в [90] для определения легких микропримесей (гелия, неона, водорода) в баллонном азоте с чувствительностью 8-10~5, 1,5-10~4,2,5-10-5% соответственно.
В [1151] описан масс-спектрометрическийме-тод определения гелия в азоте с чувствительностью до 1*10-8 объемн.%.
АММИАК (ЖИДКИЙ)
Жидкий аммиак получают из водорода и азота путем реакции на катализаторах. Исходные продукты для синтеза должны быть очищены. В безводном продукте (жидком аммиаке) могут содержаться примеси — углекислота, вода, растворенные газы и следовые количества ионов меди (после стадии очистки). Последние определяют методом фотометрии по окраске медно-аммиачного комплекса [568]. Углекислота и вода — наиболее часто встречающиеся примеси в жидком аммиаке, которые поглощаются последним из атмосферы во время транспортировки и разгрузки.
Малые количества углекислоты (несколько ч. на млн.) определить очень трудно, так как в воздухе содержится значительное количество СО2. Поэтому химический анализ связан с трудностью определения поправки холостого опыта. Вследствие того, что аммиак и СО2 образуют химическое соединение, обычные методы газового анализа здесь оказываются неприемлемыми. В работе [443] предложен титриметрический метод определения СО2 в безводном аммиаке, основанный на связывании углекислоты в виде карбамата аммония, отделении аммиака отгонкой при комнатной температуре, обработке остатка разбавленной серной кислотой, поглощении выделяющейся СО2 гидроокисью бария и титровании избытка последнего щавелевой кислотой. Этим методом при содержании углекислоты 0—50 ч. на млн. абсолютная ошибка равна ±1,5 ч. на млн.
9 В. Ф. Волынец, М. П. Волынец
257
Для определения низких концентраций воды в жидком аммиаке используется кулонометрический метод [933], основанный на реакции воды с калием, образующимся «in si/u» при электролизе хлорида калия, растворенного в образце.
Жидкий аммиак пропускают через трубку, заполненную КС1, и подают в стеклянную электролитическую ячейку с пористой перегородкой. Калий, выделяющийся на’ Pt-катоде, реагирует с водой, содержащейся в образце. Конечная точка устанавливается по увеличению электропроводности раствора, и этот момент служит указателем для прекращения электролиза.
Расчет содержания Н2О проводят по формуле'
Н20 (ч. на млн.) =
18(10)в it
96500 ' Vd
где 18 — мол. вес воды; 96500 — число Фарадея, пул; i — сила тока, а; t — время электролиза, сек.; V — объем образца в катодной секции, см3; d —• плотность жидкого аммиака при температуре измерения.
В результат вносят необходимые поправки.
Чувствительность описанного метода равна ~ 1 ч. на млн. Н2О; наилучшие результаты получаются для содержания ~100 ч. на млн.
ЧЕТЫРЕХОКИСЬ АЗОТА (ЖИДКАЯ)
Окислы азота(1У) существуют в виде мономера — двуокиси азота (NO2) и в виде димера — четырехокиси азота (NaO^. Эти две формы существуют в равновесии, которое зависит от температуры и давления окружающей среды. В жидкой форме окисел азота(1У) существует в виде тетраокиси. Последняя используется в виде окислителя, как ракетное топливо. К ней предъявляются особые требования в отношении чистоты, так как, например, продукты реакции, образующиеся при контакте лишь нескольких десятых долей процента воды с N2O4, существенно увеличивают ее коррозионные свойства. Тетраокись азота реагирует с водой с образованием нескольких молекулярных и ионных форм, в конечном счете — смеси HNO2, HNO3 и NaO3. Предложен газо-хроматографический метод определения воды в четырехокиси азота в сочетании с предварительным восстанов-лением анализируемых веществ [631]. Для пропускания легколетучей N2O4 над медью (800° С) используется поток гелия. При этом N2O4 превращается bN2, aHNO3 hHNO2 — bN2hH2O. Полученные продукты поступают в газовый хроматограф и разделяются на колонке с Порапаком-Q. Интенсивность пиков прямо пропорциональна содержанию азота и воды в образце N2O4. Чувстви-Тельндсть метода -~-0,1 мкг Н2О (10~® вес.%).
Органические вещества или растворенная двуокись углерода могут быть определены так же.
258
Загрязнения, состоящие из ионов металлов, с успехом можно определять методом атомной абсорбции после разбавления образца водой и нейтрализации непосредственно перед анализом.
Окись азота NO добавляют к четырехокиси для уменьшения ее корродирующего действия и понижения точки замерзания. Для определения малых содержаний окиси азота в N2O4 применяют спектрофотометрический метод [1421], основанный на измерении интенсивности зеленой окраски N2O3, образующейся по реакциям
N2O4^2NO2, no2 + no-»n2o,.
Измерения проводят на спектрофотометре Бекмана при температуре около 0° С и при 700 и 550 нм в ячейках специальной конструкции и из материала, устойчивого к воздействию N2O4. Оптимальный интервал 0—1,5% NO.
АЗОТНАЯ КИСЛОТА ВЫСОКОЙ ЧИСТОТЫ
Чистая азотная кислота находится в обратимом равновесии с продуктами разложения согласно уравнению
2HNO, 2NO2 + Н2О -+ //гОг.
Нагревание и воздействие света сдвигает равновесие вправо. Образовавшаяся двуокись азота, растворяясь в азотной кислоте, придает ей характерные цвета от желтого до красного. Азотная кислота, насыщенная двуокисью азота, красного цвета и поэтому называется красной дымящей азотной кислотой. Продажная азотная кислота содержит от 5 до 22 вес. % NO2. Так называемая белая дымящая азотная кислота содержит 97,5—99,8 вес. % азотной кислоты и не более 0,5% NO2, 2 вес.% Н2О. Иногда к красной дымящей азотной кислоте, используемой в качестве окислителя для определенных видов ракетного топлива, добавляют 0,6% HF для ингибирования коррозионного действия кислоты. Безводная азотная кислота содержит от 99,8 до 100% азотной кислоты и <0,1 вес.% СО2 и Н2О.
Анализ азотной кислоты относительно прост, но нужно соблюдать некоторые предосторожности при отборе пробы, взвешивании и т. д. ввиду ее большой химической активности и летучести. Образцы хранят в склянках с притертыми пробками в холодном и темном месте. Подробно об анализе азотной кислоты см. [868].
Хорошие результаты при определении воды в безводной и белой азотной кислоте получаются^ при титровании реагентом Фишера [1073].
Взвешенный образец кислоты сначала нейтрализуют с помощью раствора пиридинформамида. Затем добавляют избыток реагента Фишера и проводят обратное титрование стандартным водно-метанольным раствором.
9*
259
Точность метода ~ 1 % (в отсутствие растворенных солей) и ~5% (в присутствии солей). Количества < 1,5 вес.% NO2 не мешают определению.
Определение воды в красной дымящей азотной кислоте более сложно из-за более высокой окисляющей способности кислоты и побочных эффектов, вызываемых присутствием двуокиси азота. Предложен метод прямого титрования с реактивом Фишера с предварительным количественным превращением двуокиси азота в пиридиновые соли азотистой и азотной кислот [579]. Этим методом определяют содержание воды с точностью ±0,04 абс.%. Измерением электропроводности красной дымящей азотной кислоты, которая насыщается безводным нитратом калия, удается определить содержание воды с точностью ±0,3 абс.% [1042].
При анализе дымящей азотной кислоты используют также спектрофотометрические методы в ИК-области (для определения воды), видимой области (СО2) и УФ-области (NO3) [891, 1013]. > ) ; ; > >
Для определения фтористоводородной кислоты, добавляемой в дымящую азотную кислоту для уменьшения коррозии материалов контейнеров, предложено несколько методов. Если нет необходимости в получении высокоточных результатов, то пригодным оказывается простой и быстрый электрохимический метод, основанный на измерении тока от самопроизвольного электролиза разбавленной азотной кислоты между А1-анодом и Pt-катодом [474]. Метод позволяет определять 0,6% HF в течение нескольких минут с абсолютной ошибкой ±0,05%.
Могут быть использованы колориметрические методы. Однако кислота, хранящаяся в алюминиевой или стальной таре, может содержать ионы Al, Fe и другие, которые мешают в обычных колориметрических методах. Поэтому их необходимо предварительно отделять. В работе [1386] при определении фтористоводородной кислоты в азотной кислоте эти мешающие ионы удаляют ионообменным методом на колонке с катионитом Амберлит IR-120 в Н-форме. Дальше используется колориметрический метод, основанный на обесцвечивающем действии фторид-иона на окрашенный комплекс (например, тория с ализариновым красным). Метод быстр и точен. Для содержания 8,00 мг HF в аликвоте ошибка составляет ±0,1 мг (±1,25%).
ЛИТЕРАТУРА
1. Абрамян А. А., Кочарян А. А., Карапетян А. Т. Изв. АН АрмССР отд. хим. наук, 15, 225 (1962).
2. Абрамян А. А., Погосян Л. Е. Арм. хим. ж., 19, 188 (1966).
3. Адзаками Н., Ивакура Т., Сасаки Г. Когё ёсуй (Ind. Water), № 77, 17 (1965); РЖХим, 1966, 15Г191.
4. Адзума Н., Араи Д., Хаяси X. Когё гидзюцу сикэнсе хококу. (Repts. Govt. Industr. Res. Inst. Nagoya), 13, 388 (1964); РЖХим, 1965, 10Г168.
5. Азимов С. А., Казимов А. К., Миллер Р. А., Хакимов М. Научи, труды Ташкентского ун-та, № 388, 97 (1970).
6. Алексеев В. Н. Качественный анализ. М., Госхимиздат, 1960.
7. Амирханова Н. А., Журавский А. К., Ускова Н. Т., Потоцкая А. Е. Зав. лаб., 38, 922 (1972).
8. Андо Д., Акияма Т. Бунсэки кагаку (Japan Analyst), 13, 717 (1964); РЖХим, 1965, 4Г158.
9. Андреева И. Ю. Зав. лаб., 34, 32 (1968).
10. Антипов В. С., Витолъ Э. П., Николаев Г. И., Орлова К. Б. Сб. «Металлургия», вып. 14. Л.,«Судостроение», 1971, стр. 149.
И. Антошечкин А. Г. Гигиена и санитария, № 2, 65 (1965).|
12. Аулешева М., Саибова М. Т., Рустамов X. Р. Узб. хим. ж., № 3, 18 (1973).
13. Бабкин М. П. Гидрохим. материалы, 47, 157 (1968).
14. Багдасаров К. Н., Коваленко П. Н., Ивахненко П. Н. Изв. вузов, Химия и хим. технология, 7, 736 (1964).
15. Багдасаров К. Н., Коваленко П. И., Ивахненко П. Н., Чу
мак С. В. Электрохимические и оптические методы анализа сточных вод и электролитов. Ростов-на-Дону, 1967, стр. 206.
16. Байэр Э. Хроматография газов. М„ ИЛ, 1961.
17. Барабанщикова Н. Н., Дыхно Н. М., Котлик Л. Л., Галанин О. Г. Зав. лаб., 35, 1347 (1969).
18. Барит И. Я., Кудинов Б. С., Мусаелян Р. М., Пронман И. М. Зав. лаб., 33, 1108 (1967).
19. Барит И. Я., Кудинов Б. С., Мусаелян Р. М., Пронман И. М. Сб. «Методы определения и исследования состояния газов в металлах». М., «Наука», 1968, стр. 115.
20. Безингер Н. Н., Овечкина Т. И., Гальперн Г. Д. Ж. аналит. химии, 17, 1027 (1962).
21. Беллами Л. Инфракрасные спектры молекул. М., ИЛ, 1957, стр. 400—402.
22. Березкин В. Г., Гавричев В. С. Зав. лаб., 37, 901 (1971).
23. Березкин В. Г., Татарский В. С. Газохроматографические методы анализа примесей.М..«Наука», 1970.
24. Бескова Г. С., Канторович Л. М., Руссо Л. П., Боброва В. П., Тимохина 3. П. Сб. «Газовая хроматография», вып. 2. М., «Наука», 1964, стр. 65.
25. Бескова Г. С., Филиппов В. С. Зав. лаб., 38, 154 (1972).
26. Бессонова Г. А., Галилова А. В., Жарова Н. И., Золотарева Н. М., Корсунская М. К., Шаевич А. Б., Шубина С. Б. Сб. «Методы определения и исследования состояния газов в металлах». М., «Наука», 1968, стр. 146.
27. Бирук А. М., Комарова К. А., Крешкова Е. К., Яровенко А. Н. Изв. вузов, Химия и хим. технология, 9, 546 (1966).
261
27а. Бланк А. Б., Николенко А. Я., Сухомлинова Л. П. Сб. «Методы анализа галогенидов щелочных и щелочноземельных металлов высокой чистоты», ч. 2. Харьков, 1971, стр. 167.
28. Блинова Л. Л., Левинсон 3. Ф. Сборник трудов Карагандинского научно-исследовательского, проектно-конструкторского и экспериментального института, вып. 1 266 (1964); РЖХим, 1965, 19И336.
29. Блохин В. И., Ластов А. И., Моисеев Л. Н. Сб. «Активационный анализ». Ташкент, «Фан», 1971, стр. 219.
30. Богомолец И. Г. Тезисы докладов II конференции молодых специалистов Украинского н.-и. ин-та природных газов. Харьков, 1966, стр. 35; РЖХим, 1967, 10П206.
31. Борисов Е. М., Машевский Г. Н., Рой Н. И. Научные труды Ленинградского горного ин-та, вып. 4, 10 (1972).
32. Борисов В. П., Немец В. М., Петров А. А. Ж. прикл. спектроскопии, 7, 305 (1967).
33. Борисова Н. И., Родионов В. Н. Почвоведение, № 7, 123 (1970).
34. Борк В. А., Сальникова Н. С. Ж. аналит химии, 23, 901 (1968).
35. Бостоганашвили В. С., Турабе-лидзе Д. Г. Труды Ин-та фармакохимии АН ГрузССР, серия 1, вып.
10, 36 (1967).
36. Ботвина А. Я., Сокольский В. В., Файзрахманова Н. Р. Сб. «Активационный анализ». Ташкент, «Фан», 1971, стр. 189.
36a.Бочкова О. П., Гардашников А.Е., Михайлов С. К., Туркин Ю. И. Сб.
«Спектроскопия. Методы и применения». М., «Наука», 1973, стр.
37. Бреслер П. И., Рузин Б. И. Авт. свид. СССР, № 168046 (1965); Бюлл. изобр., № 3 (1965).
38. Бурмасов С. Л., Курочкин К. Т., Умрихин П. В. Зав. лаб., 33, 820 (1967).
39. Буянов Н. В., Федорова Л. М. Сборник трудов ЦНИИчермет, вып. 24, 105 (1962); вып. 60, 122 (1968).
40. Вагин Е. В., Петухов С. С. Сб. «Газовая хроматография», вып. 2. М., «Наука», 1964, стр. 125.
41. Варшал Г. М. Ж. аналит. химии, 27, 904 (1972).
262
42. Васильева Е. К., Закомарный А. Г Зав. лаб., 33, 471 (1967).
43. Васильева Н. Л., Рудько Б. Ф. Приборы промышленного контроля и средства автоматизации. Киев, Гостехиздат УССР, 1963, стр. 183.
44. Васильченко Л. А., Дроздова В. М. Труды Главной геофиз. обсерв., вып. 141, 99 (1963).
45. Вассерман А. М., Кунин Л. Л., Суровой Ю. Н. Определение газов в металлах. Метод восстановительного плавления в атмосфере газа-носителя. М., «Наука», 1976.
45а. Вдовенко В. М., Кривохатский А. С. Радиохимия, 1, 454(1959).
46. Венков С. Н., Еглазаров Б. Г., Зюбко В. А., Корытко А. А. Сб. «Активационный анализ». Ташкент, «Фан», 1971, стр. 323.
47. Винаров И. В., Орлова А. И., Кислица И. Ф. Укр. хим. ж., 28, 789 (1962).
48. Виноградов А. Л., Сурков Ю. А., Андрейчиков Б. М., Калинкина О. М., Гречищева И. М. Космические исследования, 8, 578
(1970).
49. Виноградов А. Л., Флоренский К. П., Волынец В. Ф. Геохимия, № 10, 875 (1963).
50. Витоль Э. Н., Орлова К. Б., Иваненко М. А. Зав. лаб., 37, 1466 (1971).
51. Витоль Э. Н., Орлова К. Б., Николаева Г. И., Антипов В. С. Ж. аналит. химии, 26, 777 (1971).
52. Волохонский А. Г. Рыбохоз. изучение внутр, водоемов (Л.), № 3, 51 (1970); РЖХим, 1970, 19Г157.
53. Волынец В. Ф. Канд. дисс. М., ГЕОХИ АН СССР, 1972.
54. Волынец В. Ф. Сб. «Вопросы разделения элементов и изотопов в геохимических процессах». М., «Наука» (в печати).
55. Волынец М. П. Тонкослойная хроматография в неорганическом анализе. М., «Наука», 1974.
56. Ганчев Н., Коев К. Труды Висш. ин-т народно столанство (Варна), 37, 25 (1965); РЖХим, 1966, 17Г70.
57. Гатова М. М., Кирсанова А. И., Нефедова Ф. М., Шварев В. С. Научные труды Гиредмет, 42, 196 (1972).
58. Гельман Я. 3., Ларина Я. И. Ж. аналит. химии, 18, 1100 (1963).
59. Генерозов Б. А. Зав. лаб., 13, 314 (1947).
60. Генкин Ю. М., Вагин Е. В., Самуилов В. И. Газовое дело, № 12, 20 (1970).
61. Виллебранд В. Ф., Лвндель Г. Э., Брайт Г. А., Гофман Д. И. Практическое руководство по неорганическому анализу. М., Госхимиздат, 1957, стр. 795.
62. Главин Г. Г., Завьялов О. В., Хохрин В. М., Бочкарев Е. Н. Научные труды Гиредмет, 47, 191 (1973).
63. Главин Г. Г., Кормилицын Д. В., Потоцкий А. Г. Сб. «Методы определения и исследования состояния газов в металлах». М., «Наука», 1968, стр. 156.
64. Главин Г. Г., Сихарулидзе Г. Г. Сб. «Методы определения газов в металлах и сплавах». М., изд. МДНТП им. Ф. Э. Дзержинского, 1971, стр. 97.
65. Глушко Ю. В., Шмелев Б. А., Зав. лаб., 32, 14 (1966).
66. Гоголь Н. А., Смирнова Г. Д. Изв. АН КазССР, № 1, 60 (1972).
67. Гольберт К. А., Вигдергауз М. С. Курс газовой хроматографии. М., «Химия», 1974.
68. Гольдшмидт X. Дж. Сплавы внедрения, вып. 1. М., «Мир», 1971, стр. 300.
69. Гончарова И. А., Дацко В. Г. Сб. «Современные методы анализа природных вод». М., изд-во АН СССР, 1962, стр. 88.
70. Гордиевский А. В., Штерман В. С., Сырченков А. Я., Саввин Н. И., Жуков А. Ф. Ж. аналит. химии, 27, 772 (1972).
71. Горшков А. П., Орлов А. Н., Колчин И. К. Газовая хроматография, № 10, 123 (1969).
71а. Горшков В. И., Чумаков В. А. Ж. физ. химии, 42, 3077 (1968).
72. Гото X., Какита Я., Яцуя И. Бунсэки кагаку (Japan Analyst), 12, 727 (1963); РЖХим, 1964, 6Г123.
73. Давыдов А. Т., Толмачева Ю. А. Ж. физ. химии, 36, 2347 (1962).
74. Дацко В. Г., Каплин В. Т. Со. «Современные методы анализа природных вод». М., Изд-во АН СССР, 1962. стр. 118.
75. Дацко В. Г., Фесенко Н. Г. Там же, стр. 123.
76. Денисова Н. И., Голубева А. А. Ж. аналит. химии, 27, 1221 (1972).
77. Джаппуев М. И. Труды Новочеркасского политехнического ин-та, 234, 5 (1971).
78. Две X., Миура И., Киносита С. Тэцу то хаганэ (J. Iron and Steel Japan), 48, 533 (1962); РЖХим, 1963, 5Г131.
79. Доброчивер И. Г., Акчурина С. Я., Зузыкина И. Я., Зорина Г. П., Шишкин Л. Я. Зав. лаб., 37, 158 (1971).
80. Доброчивер И. Г., Жарова С. Р., Зузыкина Я. Я. Зав. лаб., 35, 1053 (1969).
81. Долежал Я., Повондра Я., Шулъцек 3. Методы разложения горных пород и минералов. М., «Мир», 1968.
82. Должанская Ю. Б., Вайль Е. И. Сборник научных трудов Украинского н.-и. углехимического ин-та, вып. 24 (46), 148 (1971); РЖХим, 1972, 24И735.
83. Должанская Ю. Б., Эдельман И. И. Кокс и химия, № 8, 37 (1969).
84. Доманина О. Я., Непряхи-наА. В., Чудакова И. К., Константинов А. А., Крашенинников С. К., Новикова Г. А., Радикова Г. Г. Проблемы аналитической химии, т. 1. М., «Наука», 1970, стр. 24.
85. Дубинский Р. А., Филиппов В. А. Изв. АН СССР, серия хим., 1968, 2835.
86. Дудич Г. К., Петров А. А., Фаворская М. Я. Зав. лаб., 37, 436 (1971).
87. Дятловицкая Ф. Г. Вопросы гигиены населенных мест, т. 4. Киев, Госмедиздат, 1963, стр. 208.
87а. Евжанов X., Чапыжников Б. А., Маликова Е. Д., Кунин Л. Л. Методы определения газов в металлах. М., изд. МДНТП им. Ф. Э. Дзержинского, 1971, стр. 140.
88. Жданов А. К., Ахмедов Г., Розенблат М. М. Изв. вузов, Химия и хим. технология, 15, 503 (1972).
89. Журов Ю. А. Научно-технический сборник по геологии, разработке, транспортировке и использованию природного газа, вып. 5, 163 (1965); РЖХим, 1966, 10Г49.
90. Жуховицкий А. А., Свзв-
Й«3
нов M. Л., Шварцман В. П., Морозова С. Н., Хацкевич М. М. Зав. лаб., 33, 796 (1967).
91. Жуховицкий А. А., Туркелъта-уб Н. М. Газовая хроматография. М„ Гостоптехиздат, 1962.
92. Завгородний С. Ф., Камышников И. Ф. Сб. «Современные методы химического и технологического контроля производства». Ростов-на-Дону, 1968, стр. 51; РЖХим, 1969, 6Д66.
93. Зайдель А. Н., Иванова Т. Ф., Лазеева Г. С., Петров А. А., Федоров В. В. Материалы 4-го Уральского совещания по спектроскопии, 1963. М., «Металлургия», 1965, стр. 92.
94. Зайдель А. Н., Лазеева Г. С., Петров А. А., Сб. «Прикладная спектроскопия», т. 1. М., «Наука», 1969, стр. 218.
95. Зайдель А. И., Немец В. М., Петров А. А. Там же, стр. 202.
96. Зайдель А. Н., Петров А. 4., Ж. теор. физ., 25, 71 (1955).
97. Зайдель А. Н., Петров А. А. Зав. лаб., 28, 552 (1962).
98. Зайдель А. Н., Прокофьев В. К., Райский С. М., Славный В. А., Шрейдер Е. Я. Таблицы спектральных линий. М., «Наука», 1969, стр. 549.
99. Зайдель А. И., Шрейдер Е. Я. Спектроскопия вакуумного ультрафиолета. М., «Наука», 1967, стр. 246.
100. Закорина Н. А., Лазеева Г. С., Петров А. А. Ж. аналит. химии, 23, 1688 (1968); Методы определения и исследования состояния газов в металлах. М., «Наука», 1968, стр. 93.
101. Захарова Н. 4., Каплан Б. Я., Кудинов Б. С., Кучинская О. И., Пронман И. М., Урицкая Т. П. Научи, труды Гиредмет, 47, 121 (1973).
102. Зеленина Е. Н., Краснов В. А. Авт. свид. СССР, № 194401 (1967); Бюлл. изобр., № 8 (1967).
103. Золотухина К. Г., Домбровский А. В. Научи, ежегодник за 1959 г. Черновицкого ун-та, 1960, 640; РЖХим, 1964, 5Г192.
104. Иванова И. Л., Улицкий Р. И., Шматова А. К. Сб. «Методы определения и исследования состояния газов в металлах». М., «Наука», 1968, стр. 190.
264
105. Иванова Т. Ф., Федорова В. В. Ж. аналит. химии, 23, 1750 (1968).
106. ИеасакиИ.,ОдзаваГ., Омори Г., Муракава И., Такаги М. Когё ёсуй (Ind. Water), № 81, 38, 54 (1965); РЖХим, 1966, 16Г95.
107. Иеасаки И., Одзава Т., Ямал К., Омори Т. Когё ёсуй (Ind. Water), № 96, 40 (1966); РЖХим, 1967, 19Г90.
108. Иглицын М. И., Иванова И. И., Константинова Г. Е., Косагано-ва М. Г., Павлов Н. М. Зав. лаб., 31, 1450 (1965).
109. Изманова Т. А., Чистякова Е. М. Бессмертная А. Я. Сборник трудов ЦНИИчермет, вып. 49, 89 (1966).
110. Икэда С., Касима Д., Суда Э. Бунсэки Кагаку (Japan Analyst), 16, 135 (1967); РЖХим, 1967, 19Г112.
111. Иноуз Т., Саяма К., Таката Т. Бунсэки кагаку (Japan Analyst), И, 771 (1962); РЖХим, 1963, 5Г149.
112. Ионоселективные электроды. Под ред. Р. Дарста. М., «Мир»,
113. Исаева А. Б., Богоявленский А. И. Океанология, 8, 539 (1968).
114. Исии Д., Дзинно К. Когё кагаку дзасси (J. Chem. Soc. Jap., Ind. Chem. Sec.), 72, 656, 2684 (1969); РЖХим, 1970, 15Г107.
115. Ито Я., Узда Т. Нагоя-сирицу дайгоку якугакубу киё. (Bull. Nagoya Univ. Pharmac. School), 11, 34 (1964); РЖХим, 1965, 11Г169.
116. Йосимори T., Араи М., Икзда Й. Нихон киндзоку гаккайси (J. Japan Inst. Metals), 31, 1258 (1967); РЖХим, 1968, 12Г134.
117. Каванаб» К., Такитани С., Миядзаки М. Бунсэки кагаку (Japan Analyst), 13, 976 (1964); РЖХим, 1965, 14Г50.
118. Кагасое В. М., Жаркова И. А. Кокс и химия, № 9, 29 (1966).
119. Кагая А. Сэйто гидзюцу кэнкю кайси (Proc. Res. Japan Sugar Refin. Technol.), 12, № 12, 35 (1963); РЖХим, 1964, 3P296.
120. Казаринова Г. И. Уч. зап. Марийского гос. пед. ин-та, 30, 67 (1968).
121. Комацу С., Номура Т. Нихон кагаку дзасси (J. Chem. Soc. Ja
pan, Pure Chem. Sec.), 87, 1060 (1966); РЖХим, 1967, 14Г92.
122. Каплан Б. Я., Ревякина Г. Н. Зав. лаб., 29, 1427 (1963).
123. Каплин В. Т. Гидрохим. материалы, 32, 148 (1961).
124. Каплин В. Т., Фесенко Н. Т. Сб. «Современные методы анализа природных вод». М., Изд-во АН СССР, 1962, стр. 123.
125. Капорский Л. И., Свентиц-кий И. С. Материалы] 4-го Уральского совещания по спектроскопии, 1963. М., «Металлургия», 1965, стр. 81.
126. Карагодин Г. М. Химия твердого топлива, № 6, 102 (1968).
127. Карпов Ю. А. Докт. дисс. М., ГЕОХИ АН СССР, 1973.
128. Кейлеманс А. Хроматография газов. М., ИЛ, 1959.
129. Кикути М. Бунсэки кики (Anal. Instrum.), № 10, 685 (1972); РЖХим, 1973, 5Г147.
130. Кикути К., И май X., Сиота М. Фудзи дзихо (Fuji Electr. J.), 35, 455 (1962); РЖХим, 1963, 11Г180.
131. Киселев Н. П. Уч. зап. Кировского гос. пед. ин-та, вып. 13, 40 (1968).
132. Клибус А. X., Лазарчук Т. Н. Ж. аналит. химии, 16, 79 (1961).
133. Климова В. А. Основные микрометоды анализа органических соединений, изд. 2-е. М., «Химия», 1975.
134. Клочковский С. П., Чистова В. Д. Авт. свид. СССР, № 254191 (1970); Бюлл. изобр., № 31 (1970).
135. Клочковский С. П.,Чистова В. Д. Сборник научных трудов Магнитогорского горно-металлург. ин-та, вып. 87, 70 (1971).
136. Клячко Ю. А., Изманова Т. А., Чистякова Е. М. Зав. лаб., 29, 923 (1963).
137. Клячко Ю. А., Изманова Т^А., ЧистяковаЕ. М. Там же, стр. 1425.
138. Клячко Ю. А., Изманова Т. А., Чистякова Е. М. Сборник трудов ЦНИИчермет, вып. 31, 133 (1963).
139. Клячко Ю. А., Леви Л. И., Борисова О. М. Зав. лаб., 32, 273 (1966).
140. Клячко Ю. А., Чистякова Е. М. Сборник трудов ЦНИИчермет, вып. 31, 114 (1963).
141. Клячко Ю. А., Чистякова Е. М., Лабутьев Ю. Д. Там же, стр. 87.
142. Клячко Ю. А., Шапиро М. М, Сборник трудов ЦНИИчермет-вып. 37, 150 (1964).
143. Кобаяси И., Такэно X., Кики-гава Т., С угона С., Тани Т., Цу-тпия, С., Фукуи С. Когё ёсуй (Ind. Water), № 95, 56 (1966); Бунсэки кагаку (Japan Analyst), 15, 1245 (1966); РЖХим, 1967, 19Г140, 11Г200.
144. Ковтун М. С. Сб. «Новые методы анализа на металлургических и металлообрабатывающих заводах». М., «Металлургия», 1964, стр. 161.
145. Ковтун М. С., Алуева Т. М. Там же, стр. 165.
146. Ковтун М. С., Гаухман М. С. Сб. «Комплексообразование, мсж-молекулярвое взаимодействие п соосаждевие в некоторых системах». Днепропетровск, 1970, стр. 188.
147. Кодами М., Хабэ И. Ибараки дайгаку бунри гакубу киё (Сид-зен кагаку), № 13, 17 (1962); РЖХим, 1964, 1Г61.
148. Кожевников В. С. Изв. СО АН СССР, серия биол. наук, № 3, 158 (1970).
149. Козлов А. С., Мижидийн Ю. Сб. «Редкие щелочные элементы». Новосибирск, «Наука», 1967, стр. 300.
150. Кокоулин В. Г. Ж. аналит. химии, 21, 334 (1966).
151. Колаад С., Номура Т. Нихон । кагаку дзасси (Chem. Soc. Japan, Г Pure Chem. Sec.), 87, 845, A46 F (1966); РЖХим, 1967, 11Г126.
152. Константинов Б. И., Ошуркова О. В., Старобина И. 3. Ж. прикл. химии, 40, 1260 (1967).
153. Коренман И. М. Микрокристаллоскопия. М., Госхимиздат, 1955, стр. 127, 237, 241, 243, 249.
154. Королев В. В. Ж. аналит. химии, 23, 912 (1968); Зав. лаб., 34,. 1320 (1968).
155. Косу X. Кагаку-но рёики (J. Japan Chem.), прилож. № 64, ч. 6, 1 (1964); РЖХим, 1966,. 7Г181.
156. Коттон Ф., Уилкинсон Дж. Современная неорганическая химия, ч. 2. М., «Мир», 1969, стр. 155..
157. Кочеткова Е. А., С мир нова Г. И. Зав. лаб., 32, 1536 (1966).
158. Крешков А. П. Основы аналити
265
ческой химии, кн. 1. М., «Химия», 1970.
159. Крешков А. П., Борк В. А., Швыркова Л. А., Апаршева М. И. Авт. свид. СССР, № 181367 (1966); Бюлл. изобр., № 9 (1966).
160. Крешков А. П„ Яровенко А. Я., Невская В. Н. Ж. аналит. химии, 19, 725 (1964).
160а. Крешков А. И., Яровенко А. Н Саюшкина Е. Н., Зеленина Л. Н. Изв. вузов, Химия и хим. технология, 8, 196 (1965).
161. Кригмар С. И., Галан Т. М. Зав. лаб., 38, 1193 (1972).
162. Кригмар С. И., Степаненко В. Е., Чалан Т. М. Ж. аналит. химии, 26, 1340 (1971).
163. Кудинов Б. С. Канд. дисс. М., Ин-т биофизики АМН СССР, 1973.
164. Кудинов Б. С., Пронман И. М., Щулепников М. Н. Сб. «Активационный анализ». Ташкент, «Фан», 1971, стр. 180.
165. Куленок М. И. Изв. вузов, Химия и хим. технология, 4, 558 (1961).
166. Кунимин» Н., Угадзи X., Накамару М. Якугаку дзасси (J. Pharm. Soc. Japan), 83, № 1, 56 (1963); РЖХим, 1963, 24Г256.
167. Кунин Л. Л., Маликова Е. Д., Чапыжников Б. А. Определение кислорода, углерода, азота и водорода в щелочных и щелочноземельных металлах. М., Атомиздат, 1972.
168. Кунин Л. Л., Фролов Е. В., Вассерман А. М. Ж. аналит. химии, 27, 547 (1972).
169. Курбатова В. И., Сташкова Н. В., Макагонова Л. Н. Определение микропримесей (сб.), вып. 2, 86 (1968).
170. Курода Д. Кагаку то коге (Sci. a. Ind.). 45, 291 (1971); РЖХим, 1972, 4Г128.
171. Курода Р., Кавабути К. Бун-сэки кагаку (Japan Analyst), 11,841 (1962); РЖХим, 1963,17Г55.
172. Лебедев А. И., Новожилова И. В. Ж. аналит. химии, 22, 914, (1967).
173. Лебедев К. Б., Отто Д. Д., Байбатыров Е. Н. Труды н.-и. и проектного ин-та по обогащению руд цветных металлов «Казмехан-обр», вып. 10, 126, 156 (1972).
174. Леви Л. И., Борисова О. М. Зав. лаб., 32, 414 (1966).
266
175. Леви Л. И., Китаев Я. Я. Зав. лаб., 35, 1438 (1969).
176. Леви Л. И., Клячко Ю. А., Борисова О. М. Сб. «Методы определения и исследования состояния газов в металлах». М., «Наука», 1968, стр. 172.
177. Линдстрем И. С., Свентицкий Н. С., Шлепкова 3. И. Сб. «Спектральный анализ металлов и сплавов». М., изд. ОНТИ ВИАМ, 1965, стр. 44.
178. Ли Янь. Oceanol. et limnol. sinica, 5, 115 (1963); РЖХим, 1964, 12Г169.
179. Лобанов Г. А., Голод Л. П. Нефтепереработка и нефтехимия, научно-техн, сб., 4, 28 (1968).
180. Лобанов Е. М., Леушкина Г. В. Ядерная физика и ее применение, ч. 1. Ташкент, «ФАН», 1966, стр. 89.
181. Лурье Ю. Ю., Панова В. А. Зав. лаб., 31, 420 (1965).
182. Лурье Ю. Ю., Панова В. А. Научи, сообщ. ВНИИ водоснабжения, канализации, гидротехнических сооружений и инженерной гидрогеологии. Очистка промышленных сточных вод. М., 1963, стр. 31.
183. Лурье Ю. Ю., Рыбникова А. И. Химический анализ производственных сточных вод. М., «Химия», 1974.
184. Лындин Е. А., Л юту зова Т. П. Приборостроение, № 1, 24 (1966).
185. Львов Б. В. Атомно-абсорбционный спектральный анализ. М., «Наука», 1966.
186. Ляликов Ю. С., Мухамедназа-рова О. М. Бул. Акад. ШтииНце РСС Молд.; Изв. АН МолдССР, серия биол. и хим. наук, № 11, 38 (1964).
187. Ляликов Ю. С., Новик Р. М. Сб. «Полярография расплавленных солей». Киев, Изд-во АН УССР, 1962, стр. 41.
188. Лямин И. А., Полетаев А. И., Пушко А. Т. Авт. свид. СССР, № 298874 (1971); Бюлл. изобр. № 11 (1971).
189. Мазитов В. С., Муминов В. А., Мухаммедов С. В., Султанов В. В. Сб. «Прикладная ядерная спектроскопия», вып. 3. М., Атомиздат, 1972, стр. 62.
190. Мазитов В. С., Мухаммедов С. В. Сб. «Активационный анализ». Ташкент, «ФАН», 1971, стр. 212.
191. Макаров К. В., Березин Л. Ф., Барановский В. Е., Швецов Б. В. Сб. «Методы и приборы для анализа состава вещества». Киев, «Техника», 1971, стр. 45.
192. Макеев Е. Е., Смирнова Г, Д. Изв. АН КазССР, серия хим., 19, 87 (1969).
192а. Мак-Нейр Г., Бонелли Э. Введение в газовую хроматографию. М., «Мир», 1970.
193. Манабз Т. Нихон суайсаи гакайси (Bull. Japan Soc. Sci. Fish.), 35, 897 (1969); РЖХим, 1970, 97202.
194. Мао Чжи-сян. Хуасюэ сюэбао (Acta chim. Sinica), 31, 514 (1965); РЖХим, 1966, 23Г58.
195. Масленковский Л. Г., Маст В. С., Фомичева Н. И. Токсичность двигателей внутреннего сгорания и пути ее снижения. М., 1966, стр. 381.
196. Матер о еа Е. А., Гринберг Г. Л. Сб. «Ионный обмен». Изд-во МГУ, 1965, стр. 99.
197. Мацумото Т. Бунко кэнкю (J. Spectroscop. Soc. Japan), 15, 193 (1967); РЖХим, 1968, 7Г118.
198. Меерсон Г. А., Кормилицын Д. В., Абелева Л. Г., Амосов В. М. Лискович В. А. Изв. АН СССР, Неорган. материалы, 2, 592 (1966).
199. Методы-спутники в газовой хроматографии. Перевод с англ, под ред. В. Г. Березкина. М., «Мир», 1972.
200. Мещеряков Р. Л., Яковлев Б. М. Глухое Г. Г. Сб. «Активационный анализ». Ташкент, «Фан», 1971, стр. 230.
201. Мещеряков Р. П., Яковлев Б. М. Тропов Г. И. Изв. Томского политехи. ин-та, 216, 25 (1971).
202. Миненко Н. Н., Лазарчук Т. Я. Порошковая металлургия, № 6, 53 (1965).
203. Мирзоянов В. С., Охотников Б. И. Ж. аналит. химии, 21, 985 (1966).
204. Митани К. Когай то тайсаку (J. Environ. Pollut. Contr.), 8, 654 (1972); РЖХим, 1973, 5И543.
205. Митрофанова Т. П., Орлов Ю. Ф., Плоткина Е. И. Зав. лаб., 36, 161 (1970).
206. Михайличенко А. И., Дож-дева Я. М. Радиохимия, 15, 756 (1973).
207. Михайличенко А. И., Марков
В. К., Долгов А. С., Рудаков Т.В., Косых В. Г. Зав. лаб., 39, 13 (1973).
208. Михайличенко А. И., Соколова Н. Л., Су лайма нкулова С. К., Тетерин Э. Г. Радиохимия, 15, 693 (1973).
209. Михайлова Т. А., Хромов-Борисов И. В. Ж. аналит. химии, 20, 359 (1965).
210. Мияхар К. Бунсэки кагаку (Japan Analyst), 15, 727 (1966); РЖХим, 1967, ЗГ183.
211. Молчанове. М., Образцова А. X., Равдолъкина В. И., Куковврова В. Я. Табак (М.), 30, № 4,34 (1969).
212. Монастыренко Е. С. Материалы к совещанию молодых специалистов ВОДГЕО, 1966. Очистка сточных вод. М., изд. ВОДГЕО, 1966, стр. 57; РЖХим, 1967, 5И228.
213. Маримото М., Хиракобв А. Бунсэки кагаку (Japan Analyst), 13, 466 (1964); РЖХим, 1965, 2Г107.
214. Моримото М., Хиракобо А., Ясибаси Р. Бунсэки кагаку (Japan Analyst), 16, 1335 (1967); РЖХим, 1969, 4Г103.
215. Морита Я., Когур» Ю. Кагаку когё сирё (Chem. Engrs. Digest.), 32, 231 (1964); РЖХим, 1965, 23Г181.
216. Морита Я., Когур» Ю. Нихон кагаку дзасси (J. Chem. Soc. Japan, Pure Chem. Sec.), 84, 816, A-56 (1963); РЖХим, 1964, 13Г93.
217. Морита Я., Когур» Ю. Нихои кагаку дзасси (J. Chem. Soc. Japan, Pure Chem. Sec.), 91, 653, A-37 (1970); РЖХим, 1971, 5Г163.
218. Морита Я., Окурз К. Когё гидзюцу (Ind. Engrs. Techn.), № 8, 22 (1964); РЖХим, 1966, 6Г171.
219. Москвина А. А., Кузнецова Л. В.. Добычин С. Л., Розова М. И. Ж. аналит. химии, 19, 749 (1964).
220. Мочалов К. Я., Тремасова И. В. Труды Казанского хим.-технол. ин-та, вып. 34, 90 (1965).
221. Мулинова Р. А., Ташпулатов Ю. Т., Росина С. 3. Узб. хим. ж. Рукопись депонирована в ВИНИТИ, № 4255—72 (1972); РЖХим, 1972, 16Г116 Деп.
222. Мустафин И. С., Сивакова О. В Ж. аналит. химии, 19, 163 (1964).
223. Муся С., Икэда С. Нихои кагаку дзасси (J. Chem. Soc. Japan, Pure Chem. Sec.), 89, 767 (1968); РЖХим, 1969, 11Г107.
217
224. Муся С., Нисимура Т. Бунсэки кагаку (Japan Analyst), 14, 795 (1965); РЖХим, 1966, 13Г84.
225. Мутов Г. М., Берг В. К., Чиркова Р. Т., Орлова Е. Ю. Ж. аналит. химии, 27, 807 (1972).
226. Нагама Ю., Андо Н. Эйё то секурё (J. Japan Soc. Food a. Mitr.), 19, 282 (1966); РЖХим, 1967, 22Г90.
227. Накагава X., Иноуэ Н., Ути-да Т. Синку (J. Vacuum Soc. Japan), 9, № 10, 398 (1966); РЖХим, 1967, 10Д35.
228. Накамото К. Инфракрасные спектры неорганических и координационных соединений. М., «Мир», 1966, стр. 106, 109, 132, 143, 145.
229. Наумов Г. Б., Рыженко В. Н., Ходаковский И. Л. Справочник термодинамических величин. М., Атомиздат, 1971.
230. Наумова И. И. Зав. лаб., 31, 1205 (1965).
231. Наумова Т. И., Василевская М. В. Сб. «Методы исследования продуктов нефтепереработки и нефтехимического синтеза». Л., Гостоптехиздат, 1962, стр. 196.
232. Некрассв Б. В. Основы общей химии, т. 1. М., «Химия», 1973.
233. Немец В. М., Петров А. А. Ж. прикл. спектроскопии, 4, 101, (1966).
234. Непряхина А. В., Чудакова И. К Константинов А. А., Крашенинников С. К. Авт. свид. СССР, № 191877 (1967); Билл, изобр., № 4 (1967).
235. Николаев А. В., Миронов К. Е., Карасева Э. В. Докл. АН СССР, 147, 380 (1962).
236. Ниси Т., Асано М. Когё кагаку даасси (J. Chem. Soc. Japan, Industr. Chem. Sec.), № 10, 1424, A90 (1963); РЖХим, 1964, 13Б494.
237. Нисида X. Бунсэки кагаку (Japan Analyst), 13, 361 (1964); РЖХим, 1965, 2Г108.
238. Нисикаеа К. Когё кагаку двас-си (J. Chem. Soc. Japan, Industr. Chem. Sec.), 67, 1765 (1964); РЖХим, 1965, 13Г169.
239. Нисимура M., Мацу нага К., Канадзава X. Бунсэки кагаку (Japan Analyst), 18, 1372 (1969); РЖХим, 1970, 12Г121.
240. Новицкий Н. В., Хромых Г. И. Химия твердого топлива, № 6, 143 (1971).
268
241. Номура Т. Нихон кагаку дзас-си (J. Chem. Soc. Japan, Pure-Chem. Sec.), 88, 473, A29 (1967); РЖХим, 1968, ЗГ100.
242. Номура T. Там же, стр. 635, А39; РЖХим 1968, 7Г74.
243. Номура Т. Там же, стр. 961; РЖХим, 1968, 9Г113.
244. Номура Т., Комацу С. Бунсэки кагаку (Japan Analyst), 17, 1406-(1968); РЖХим, 1969, 16Г97.
245. Номура Т., Такэути К., Комацу С. Нихон кагаку дзасси (J. Chem. Soc. Japan, Pure Chem. Sec.), 89, 291, A16-A17 (1968); РЖХим, 1969, 6Г105.
246. Нэдзу X., Синодеаки Д. Кагаку [Chemistry (Japan)], 18, 659 (1963); РЖХим, 1965, 2Б669.
247. Ода Н., Кубо М. Бунсэки кагаку (Japan Analyst), 11, 406,. 411 (1962); РЖХим, 1963, 4Г52.
248. Одзава Т. Нихон кагаку дзасси (J. Chem. Soc. Japan, Pure Chem. Sec.), 87, 853, 959 (1966); РЖХим, 1967, 7Г123, 9Г127.
249. Окада M. Tore когё сиконсё кококу (Repts. Govt. Chem. Industr. Res. Inst. Tokyo), 61, 448 (1966); РЖХим, 1967, 16Г47.
250. Окутани T., Ицуми С. Нихон кагаку дзасси (J. Chem. Soc. Japan, Pure Chem. Sec.), 87, 444 (1966); РЖХим, 1967, 9Г108.
251. Орадовский С. Г., Токуев Ю. С. Материалы XXII Гидрохимического совещания, вып. 3. Новочеркасск, 1969, стр. 50.
252. Орлова Е. Ю. Авт. свид. СССР, № 260951 (1968); Бюлл. изобр., № 4 (1970).
253. Орлова И. Г. Труды Гос. океанографического ин-та, вып. 113, 47 (1972).
254. Орлова К. Б. Канд. дисс. М., ГЕОХИ АН СССР, 1972.
255. Орлова К. Б. Сб. «Методы опре- ' деления и исследования состояния газов в металлах». М., «Наука», 1968, стр. 74.
256. Орлова К. Б., Витоль Э. Н. Ж. аналит. химии, 20, 694 (1965).
257. Орлова К. Б., Витоль Э. Н. Ж. аналит. химии, 21, 1263 (1966).
258. Осима Т., Иосино Т. Кагаку то когё (Sci. a. Ind.), 39, 356 (1965); РЖХим 1966, 10Г56.
259. Осима Т., Иосино Т. Кагаку то когё (Sci. a. Ind.), 40, 440 (1966); РЖХим, 1967, 15Г120.
.260. Охапкина Л. Л., Быкова А. П., Евстратова Г. А. Изв. НИИ нефте- и углехимического синтеза при Иркутском ун-те, 8, 10 (1966); Зав. лаб., 31, 277 (1965).
261. Охотников Б. П., Бондаренко И. В., Шварцман В. П. Зав. лаб., 31, 895 (1965).
262. Пахомова И. Е., Чумаченко М. Н. Изв. АН СССР, серия хим., 1965, 1138.
263. Перминова Ж. Г. Зав. лаб., 33, 1096 (1967).
263а. Петров А. А. Спектральноизотопный метод исследования материалов. Изд-во ЛГУ, 1974, стр. 114, 173.
264. Петрова В. В. Сб. «Лекарственные сырьевые ресурсы Иркутской области», вып. 3. Иркутск, 1961, стр. 174.
265. Петряев Е. П., Коляда Л. Г., Ковалева Е. П., Баранчик Г. Н. Вестей Акад. Навук Беларус. ССР, серия хим. навук, № 1, 57 (1971).
266. Плаксин И. Н., Шиврин Г. Н. Докл. АН СССР, 150, 1104 (1963).
267. Подберезская Н. К. Сб. «Исследование и разработка фотометрических методов для определения микроколичеств элементов в минеральном сырье». Алма-Ата, 1967, стр. 105.
268. Подзярей Г. А., Зарицкий И. М. Теор. и экспер. химия, вып. 2, 281 (1968).
269. Покровская Е. И., Терещенко А. П. Сб. «Иониты и ионный обмен». М., «Наука», стр. 103.
270. Полищук И. М., Суворов В. В. Сб. «Автоматизация химических, нефтеперерабатывающих и целлюлозно-бумажных производств». Пермь, 1968, стр. 114.
271. Поллинг Л. Общая химия. М., «Мир», 1974, стр. 534, 578.
272. Попов В. А., Яковлев П. Я., Руденко Э. Н. Сб. «Методы химического анализа». М., «Металлургия», 1969, стр. 95.
273. Пронман И. М., Андреев А. В., Кудинов В. С. Сб. «Радиационная физика неметаллических кристаллов». Киев, «Наукова думка», 1967, стр. 424.
274. Пронман И. М., Главин Г. Г., Карпов Ю. А., Кормилицын В. Д., Андреев А. В., Кудинов Б. С. Сб. «Методы аналитического кон
троля в промышленности редких металлов и полупроводниковых материалов». М., «Металлургия», 1968, стр. 11.
275. Пронман И. М., Кудинов Б. С. Сб. «Ядернофизические методы анализа веществ». М., Атомиздат, 1971, стр. 222.
276. Разживина А. Л. Труды Всесоюзного гос. ин-та н.-и. и проектн. работ огнеупорной промышленности, вып. 29, 191 (1960); РЖХим, 1963, 16Г177.
277. Раманаускас Э. И., Б у нике не Л. В., Сапрагонене М. С.у Шулю-нене А. К., Жиленайте М. В. Ж. аналит. химии, 24, 244 (1969).
278. Реми Г. Курс неорганической химии, т. 1. М., ИЛ, 1963, стр. 625.
279. Розен А. М., Мартынов Б. В., Аникин В. И. Радиохимия, 14, 748 (1972).
280. Розенберг Г. И., Кузнецов-Фетисов Л. И. Труды Казанского хим.-технол. ин-та, вып. 36, 16 (1967).
281. Розенблюм Е. Н., Ваучскгя Р. В., Сорокина М. Н. Сборник работ по химическим источникам тока. М.— Л., «Энергия», 1966, стр. 273.
282. Романов А. С., Новоселова А. А. Гидрофизические исследования Тихого и Атлантического океанов в кругосветном плавании НИС «Михаил Ломоносов» (20-й рейс). Севастополь, 1967, стр. 82; РЖХим, 1968, 15Г179.
283. Рудько Б. Ф., Васильева Н. Л. Хим. пром-сть Украины. Научно-произв. сб., № 2 (32), 53 (1967),
284. Русанов Е. Л., Балевска П. Изв. Ин-та физиол. Бълг. АН, 6, 307 (1963); РЖХим, 1964, 10Г162.
285. Ряднина А. М., Неменьший И. Д. Авт. свид. СССР, № 166847 (1964); Бюлл. изобр., № 23 (1964).
286. Самсоненко Н. Л., Соболев Е. В. Письма в редакцию ЖЭТФ, 5, 304 (1967).
287. Сапогин Л. Г. Зав. лаб., 28, 1197 (1962).
288. Сато Т., Такахаси Т., Ого» С. Бунсэки кики (Anal. Instrum), 9, 465 (1971); РЖХим, 1972, 4Г208.
289. Camo С., Тати К. Дэнки Сэйко (Electr. Fum. Steel), 40, 222 (1969); РЖХим, 1970, 7Г152.
269
299. Сафин Р. М., Махоткин А. Ф., Галеев А. Ф. Сборник аспирантских работ Казанского хим.-техиол. ин-та, вып. 1, 173 (1970).
291. Сафонова А. А., Лукаш Е. В. Кокс и химия, № 8, 51 (1963).
292. Сб. «Анализ газов в металлах». Труды Комиссии по аналит. химии АН СССР», т. 10. М., Изд-во АН СССР, 1960.
293. Сб. «Теория металлургических процессов». Сборник трудов ЦНИИчермет. М., «Металлургия», вып. 24 (1962); вып. 60 (1968); вып. 79 (1972).
294. Свяжин А. Г., Чурсин Г. М., Вишкарев А. Ф., Явойский В. И. Сб. «Методы определения и исследования состояния газов в металлах». М., «Наука», 1968, стр. 225.
295. Селезнева Н. А., Плотникова О. М. Изв. АН КазССР, серия хим., № 6, 80 (1968).
296. Семавин Б. А., Волкова Е. В. Авт. свид. СССР, № 311185 (1971); Бюлл. изобр., № 24 (1971).
297. Сиванова О. В., Мустафин И. С. Ж. аналит. химии, 21, 242 (1966).
298. Симин В. Б., Маркевич А. В., Добычин С. Л. Ж. прикл. химии, 40, 1303 (1967).
299. Симолин А. В., Зеленина Г. Ю. Труды морского гидрофизического ин-та АН УССР, 38, 153 (1967).
300. Синюков В. В., Скопинцев Б. А. Океанология, 3, 127 (1963).
301. Сираива Т., Нобукацу Ф. Тэцу то хаганэ (J. Iron a. Steel Inst. Japan), 52, 1713 (1966); РЖХим, 1967, 10Г113.
302. Сираива Т., Фудзино Н. Сумитомо киндзоку (Sumitomo Metals), 18, 509 (1966); РЖХим, 1967, 12Г58.
303. Сираяма Ц. Суидо кёкай дзасси (J. Water Works a. sewer. Assoc.), № 351, 26 (1963); РЖХим, 1964, 16И97.
304. Скотников С. А. Автореф. канд. дисс. М., ИМЕТ АН СССР, 1963.
305. Скотников С. А. Ж. прикл. спектроскопии, 9, 316 (1968).
306. Скотников С. А. Сб. «Методы определения газов в металлах и сплавах». М., изд. МДНТП им. Ф. Э. Дзержинского, 1971, стр. 150.
307. Скотников С. А. Сб. «Спектральные и химические методы анализа материалов». М., «Металлургия», 1964, стр. 55.
270
308. Скотников С. А. Труды Ин-та металлургии АН СССР, вып. 15, 43 (1963).
309. Скотников С. А., Лазарев Я. Ю. Зав. лаб., 36, 420 (1970).
310. Скотников С. А., Утлинский Г. Г. Зав. лаб., 35, 1346 (1969).
311. Скотников С. А., Федорова Л. М. Зав. лаб., 28 , 555 (1962).
312. Смирнов Д. Н., Монастырен-ко Е. С. Водоснабжение, канали-вация, гидротехнические сооружения. Межведомств, республ. сб., вып. 1, 95 (1966); вып. 5, 29 (1967).
313. Сокольский Д. В., Макеев Е. Е., Алексеева Г. В. Вести. АН КазССР, 25, 62 (1969).
314. Старикова Н. Д., Яблокова О. Г. Труды ин-та океанологии АН СССР, 67, 157 (1964).
315. Старобинец Г. Л., Дубов1к Т. Л. Весц! АН БССР. Серия ф!з-тэхн. н., № 2, 48 (1963); РЖХим, 1964, 4Б691.
316. Старобинец Г. Л., Мечков-ский С. А. Ж. аналит. химии, 18, 298 (1963).
317. Струкова М. П., Федорова Г. А.
Ж. аналит. химии, 21, 509 (1966).-
318. Судзуки С. Бунсэки кагаку (Japan Analyst), 11, 618 (1962); РЖХим, 1963, 8Г143.
319. Судзуки Н., Ито М. Суидо кекай дзасси (J. Water Work а. sewer. Assoc.), № 343, 44 (1963); РЖХим, 1964, 6И165.
320. Сурков Ю. А., Андрейчиков Б. М. Геохимия, № 10, 1435 (1973).
321. Сухенко К. А., Григорова В. C.t Линдстрем И. С. Сб. «Методы определения и исследования состояния газов в металлах». М., «Наука», 1968, стр. 142.
322. Сюн Сяо-сянь. Хайян юй хуч-жао (Oceanol. е. limnol. sinica), 7, ИЗ (1965); РЖХим, 1966,12Г186.
323. Сяецилло С. В., Никольская А. М.,МашкоТ.Е. Сб. «Высокотемпературные неорганические соединения». Киев, «Наукова думка», 1965, стр. 395.
324. Сяецилло С. В., Никольская А. М., Машка Т. Е. Зав. лаб., 29, 806 (1963).
325. Тагаки Т., Аиура М. Тоё сода кэнкю хококу (Sci. Rept. Toyo Soda Manufact. Comp., Ltd.), 7,
№ 1, 16 (1963); РЖХим, 1964, 11Г160.
326. Танако С. Когё ёсуй (Ind. Water), № 110, 61 (1967); РЖХим, 1968, 18И291.
327. Такахаси Я. Бунсэки кагаку (Japan Analyst), 14, 939 (1965); РЖХим, 1966, 20Г137.
328. Такитани С., Фукадзава М., Хасэгава X. Бунсэки кагаку (Japan Analyst), 12, 1156 (1963); РЖХим, 1964, 17Г27.
329. Такэмото К. Когё то сэйхин. Когё сэйхин гидзюцу кёкай (Ind. a. Industr. Prod.), № 40, 45 (1967); РЖХим, 1968, 19Б222.
330. Такэути Ц., Мочи С. Бунсэки кагаку (Japan Analyst), 14, 622 (1965); РЖХим, 1966, 5Г177.
331. Талакин О. Г., Головин Ю. И., Дыхно Н. М. Ж. аналит. химии, 26, 1220 (1971).
332. Танака С., Сюда Я. Есуй то хайсуй (J. Water a. Waste),- 9, 186 (1967); РЖХим, 1967, 20Г68.
333. Танака Й., Танака Ю. Бунсэки кагаку (Japan Analyst), 13, 623 (1964); РЖХим, 1965, 7Г66.
334. Тананаев Н. А. Капельный метод. М., Госхимиздат, 1954.
335. Танчев Н., Коев К. Тр. Висш. ин-т народно стопанство (Варна), 37, № 4, 25 (1965).
336. Терентьев А. П., Л у скина Б. М. Труды Комиссии по аналит. химии АН СССР, 13, 20 (1963).
337. Терентьев А. П., Туркельта-уб А. М., Бондаревская Е.А., До-мочкина Л. А. Докл. АН СССР, 148, 1316 (1963).
338. Тимошенко Н. Н., Изманова Т. А., Чистякова Е. М. Зав. лаб., 31, 1068 (1965).
339. Тихонова А. П., Морозова А. А., Рудякин В. В. Сборник трудов ВНИИ титана, № 1, 293 (1967).
340. Ткаченко Г. В., Кригмар С. И., Степаненко В. Е. Азотная промышленность, № 1, 32 (1972).
341. Токуев Ю. С. Труды Гос. океанограф. ин-та, вып. 113, 30 (1972).
342. Толмачев А. М., Зотова Т. В., Гришина Л. Н. Ж. физ. химии, 42, 1263 (1968).
343. Толмачева Ю. А., Давыдов А. Т. Ж. физ. химии, 36, 2653 (1962).
344. Толмачева Ю. А., Давыдов А. Т. Изв. вузов, Химия и хим. технология, 5, 579 (1962).
345. Топорков С. А. Со. «Аппаратура
и методы рентгеновского анализа»,, вып. 5. Л., 1969, стр. 154.
346. Торин К., Асака М., Ямадзаки X. Когё кагаку дзасси (J. Chem. Soc. Japan, Ind. Chem. Sec.), 72, 664(1969); РЖХим, 1969, 18Г62.
347. Торопова В. Ф., А вер ко-Антонович А. А. Ж. аналит. химии, 26, 2437 (1971).
348. Торопова В. Ф., Аверко-Антоно-вич А. А. Ж. аналит. химии, 27, 116 (1972).
349. Трухачева В. А., Захарчук Н. Ф., Юделевич И. Г. Изв. СО АН СССР, серия хим. наук, № 2, вып. 1, 157 (1970).
350. Туманов А. А., Глазунова 3. И., Кулакова К. А. Труды по химии и хим. технологии (Горький), вып. 2, 138 (1970).
351. Туровцева 3. М., Кунин Л. Л. Анализ газов в металлах. М., Изд-во АН СССР, 1959, стр. 217.
352. Турчин Ф. В. Сб. «Агрохимические методы исследования почв».. М., «Наука», 1965, стр. 64.
353. Турьян Я. И., Жанталай Б. В. Зав. лаб., 12, 1431 (1962).
353а. Тэрабэ М. Бэссацу кагаку когё, 10, № 2, 1 (1966); РЖХим, 1967, 5И403.
354. У Ли-минь, Гао Ай-жу, Ван Цзун-сяо. Цзылинь гиида сюзбао< (Jilin shida xuebao), № 2, 61 (1964); РЖХим, 1966, 6Г99.
355. Унифицированные методы исследования качества вод, ч. 1. Методы химического анализа вод, изд. 2-е. М., изд. СЭВ, 1974.
356. Усачева В. И. Зав. лаб., 31, 627 (1965).
357. Устиновская И. А., Емельянова О. А., Сазонова Н. Н., Чиркова В. М., Малахов В. В. Изв. СО АН СССР, серия хим. наук, вып. 1, № 2, 103 (1972).
358. У Чжень-цю. Ули сюэбао (Acta, phys. sinica), 17, 500 (1961);. РЖХим, 1964, 7Г99.
359. Файнгольд С. Г., Леонова Н. А. Кокс и химия, № 7, 43 (1963).
360. Федоров А. А., Кричевская А. М. Зав. лаб., 34, 1425 (1968).
361. Федоров В. В., Лазеева Г. С., Петров А. А. Зав. лаб., 30, 545 (1964).
362. Федорова Л. М., Занина Е. П., Корнеенко В. П. Зав. лаб., 31, 1347 (1965).
274
363. Федорова Л. М. Сборник трудов ЦНИИчермет, вып. 31, 83 (1963).
364. Федосеев Д. НИгнатенко Л. С., Соколовская 3. Н. Изв. вузов, Химия и хим. технология, 10, 185 (1967).
365. Федосеев Д. И., Ученъ М. Т. Авт. свид. СССР, № 168040 (1965); Бюлл. изобр., .№ 3 (1965).
366. Федотов Ф. П. Гигиена и санитария, 28, 85 (1956).
367. Филиппов М. И., Каванский И. М., Панченко В. С., Куценко В. П. Зав. лаб., 30, 1444(1964).
368. Филипс К. Хроматография газов. М., ИЛ, 1958.
369. Финкелъштейнайте М. Л. Авт. свид. СССР, № 183470 (1966); Бюлл. изобр., № 13 (1966).
370. Фомин В. В., Майорова Е. П. Ж. неорг. химии, 1, 1703 (1956); 3, 1540 (1958).
371. Фомин В. В., Моргунов А. Ф., Коробов И. В. Ж. неорг. химии, 5, 1846 (1960).
372. Фомин В. В., Потапова В. Т. Ж. неорг. химии, 8, 990 (1963).
373. Фомин В. В., Соломатин В. С. Радиохимия, 9, 310 (1967).
374. Фудзинага Т., Мории Ф. Кагаку (Chemistry), 21, 652 (1966);
РЖХим, 1957, 7Г88.
375. Фудзинага Г., Симада Й., Хи-рано С. Бунсэки кагаку (Japan Analyst), 20, 131 (1971); РЖХим, 1971, 19Г80.
376. Фудзита М., Кумамото М. Бунсэки кики (Anal. a. Instrum.), 7, 243 (1969); РЖХим, 1970, ЗП291.
377. /ФунасакиВ., Кодзима Ц., Сэо И. Бунсэки кагаку (Japan Analyst), 17, 464 (1968); РЖХим, 1969, 4Г181.
378. Фуруя К., Камада X. Бунсэки кагаку (Japan Analyst), 14, 336 (1965); РЖХим, 1955, 24Г189.
379. Фуру я М. Бунсэки кагаку (Japan Analyst), 12, 923 (1963); РЖХим, 1964, 13Г63.
380. Хаваш И., Хубер М., Сабо И., Пунгор Э. Венг. исслед. оборудование, ноябрь, 45 (1967); РЖХим, 1968, 11Г54.
381. Хайе И. М., Мацек К. Хроматография на бумаге. М., ИЛ, 1962.
382. Халтурина К. А., Танцыре-ва Л., Сергеева А., Потылицына М. Труды Сибирского технол. ин-та, вып. 36, 87 (1963).
272 '
383. Хамракулов Т. К., Ильясов Д. А., АгасянП. К., Ивниц-кий Д. М. Зав. лаб., 38, 1447 (1972).
384. Хаситмни X., Йосида X. Бунсэки кагаку (Japan Analyst), 17, 1136 (1968); РЖХим, 1969, 11Г100.
385. Хе Линъ-син, Се Сю-хун. Хуа-сюэ тунбао (Huaxue tongbao), № 4, 7 (1963); РЖХим, 1964, 5Г194.
386. Хигасиура Л. Кагаку то когё (Sci. a. Ind.), 36, 453 (1962); РЖХим, 1963, 21И107.
387. Химические реактивы и препараты, вып. 30. М., изд. ИРЕ А, 1970, стр. 145.
388. Хирамацу К. Сицурё бунсэки (Mass Spectrosc.), 12, № 27, 145 (1964); РЖХим, 1966, 10Г167.
389. Хирано С., Огахара И. Сою сикэнсеиэмно (Ann. Rept. Engng. Res. Inst. Fac. Engn. Univ. Tokyo), 20, 11 (1962); РЖХим, 1963, 13Г133.
390. Хироси И. Бунсэки кагаку (Japan Analyst), 13, 361 (1964); РЖХим, 1965, 2Г108.
391. Ходзуми К. Кагаку (Chemistry), 19, 249 (1964); РЖХим, 1965, 2Г172.
392. Ходзуми К., Умэмото К. Бун>-сэки кагаку (Japan Analyst), 16, 800 (1967); РЖХим, 1968, 23Г157.
393. Ху Кэ-юань, Чжу Ли-чжи. Юаньцзынэн, № 11, 976 (1965); РЖХим, 1966, 15Г104.
394. Хэ Линь-син, Се Сю-хуан, Хуа-сюэ тунбао (Huaxue tongbao), № 4, 7 (1963); РЖХим, 1964, 5Г194.
395. Цитович И. К. Труды Кубанского с.-х. ин-та, вып. 9 (34), 204 (1964).
396. Цудзи X. Бунсэки кагаку (Japan Analyst), 15, 263 (1966); РЖХим, 21Г135.
397. Цутия М., Ясуда Й., Камада X. Бунсэки кагаку (Japan-Analyst), 14, 155 (1965); РЖХим, 1965, 21Г100.
398. Чернобережский Ю. М., Ян-к.гович А. И., Кузьмина Т. А., Соловьев А. М., Топорков С. А. Вести. ЛГУ, № 4, 163 (1970).
399. Чистякова Е. М., Ивмано-ва Т. А., Тимошенко Н. Н. Зав. лаб., 37, 889 (1971).
400. Чистякова Е. М., И гм ано-ва Т. А., Тимошенко Н. И. Сб. «Методы определения газов в металлах и сплавах». М., изд.
МДНТП им. Ф. Э. Дзержинского, 1971, стр. 21.
401. Чумаченко М. Н. Изв. АН СССР, серия хим., 1963, 1893.
402. Чумаченко М. Н., Левина Н. Б. Докл. АН СССР, 180, 894 (1968).
403. Чумаченко М. Н., Пахомова И. Е. Докл. АН СССР, 170, 125 (1966).
404. Чумаченко М. Н., Пахомова И. Е. Изв. АН СССР, серия хим., 1963, 2090.
405. Чумаченко М. Н., Пахомова И. Е. Изв. АН СССР, серия хим., 1964, 1561.
406. Чумаченко М. II., Пахомова И. Е. Изв. АН СССР, серия хим., 1968, 235.
407. Чумаченко М. Н., Пахомова И. Е., Левина II. Б. Авт. свид. СССР, К» 204014 (1967); Бюлл. изобр., № 21 (1967).
408. Чупахин М. С. Труды Комиссии по аналит. химии АН СССР, 16, 175 (1968).
409. Чупахин М. С., Дуев Л. Т. Ж. аналит. химии, 22, 1072 (1967).
410. Чупахин М. С., Крючкова О. И., Рамендик Г. И. Аналитические возможности искровой масс-спектрометрии. М., Атомиздат, 1972, стр. 129.
411. Чучмарев С. К., Камышов В. М. Зав. лаб., 30, 1068 (1964).
412. Чень Яо-цзу, У Ман-янь, Ли Кан-цзун. Ланьчжоу дасюэ сюэ-бао. Цзыжань кэсюэ (J. Lanchow Univ.), № 2, 23 (1964); РЖХим, 1965„13Г170.
413. Ша И-сянь, Ван Чан-и. Хуа-сюэ сюэбао. (Acta chim. sinica), 30, 513 (1964); РЖХим, 1965, 14Г168.
414. Ша И-сянъ, Ван Чан-и. Хуасюэ сюэбао (Acta chim. sinica), 31, 436 (1965); РЖХим, 1966, 12Г204.
415. Ill ай Г. Теоретические основы газовой хроматографии. М., ИЛ, 1963.
416. ШамаевВ.И. Труды I Всесоюзного координационного совещания по активационному анализу,1962. Ташкент, «Фан», 1964, стр. 45.
417. Шевцов В. Е., Лизунов Г. И. Изв. вузов, Черн. металлургия, № 5, 204 (1965).
418. Шилов А. Е. Вести. АН СССР, № 3, 61 (1972).
418а. ШитиковВ.С.,ГедеревичН.А. Зав. лаб., 33, 1503 (1967).
419. Шляховер И. В., Вельская Л. И.
Хим. пром-сть, № 4, 307 (1972).
420. Шмелев Б. А., Глушко Ю. В. Сб. «Методы определения и исследования состояния газов в металлах». М., «Наука», 1968, стр. 184.
421. Шмелев Б. А., Рютина Т. В. Зав. лаб., 35, 422 (1969).
422. Шульман Л. А. Синтетические алмазы. Научно-производств. сб., вып. 1, 8 (1969).
423. Шутов Г. М., Берг В. К., Чиркова Р. Г., Орлова Е. Ю. Ж. аналит. химии, 27, 807 (1972).
424. Щепотъева В. И., Воротынце-ва М. И., Зверев К. Г. Ж. аналит. химии, 28, 1021 (1973).
425. Эмсли Дж., Финей Дж., Сатклиф Л. Спектроскопия ЯМР высокого разрешения, т. 2. М., «Мир», 1969, стр. 343—350.
426. Эрдман М. Э., Маль С. С., Раковский В. Е. Зав. лаб., 29, 1058 (1963).
427. Ядерно-физические методы анализа веществ. Труды Конференции по применению радиоактивных изотопов и излучений в народном хозяйстве. М., Атомиздат, 1971.
428. Яковлев П. Я., Малинина Р. Д. Зав. лаб., 28, 1434 (1962).
429. Ямагути Н., Араки С. Бунсэки кагаку (Japan Analyst), 16, 348 (1967); РЖХим, 1968, 18Г164.
430. Ямагути Н., Матида Т., Узки М. Якугаку дзасси (J. Phar-mac. Soc. Japan), 89, 804, 1534 (1969); РЖХим, 1970, 7Г125, 14Г121.
431. Ямагути И., Суда Т., Каммори О. Бунсэки кагаку (Japan Analyst), 18, 13 (1969); РЖХим, 1969, 14Г121.
432. Ямамото Ю., Кимамору Т., Отани Ю. Бунсэки кагаку (Japan Analyst), 18, 359 (1969); РЖХим, 1970 , 2Г125.
433. Яначихора С., Синохара С. Дзидося гидзюцу (J. Soc. Automat. Engrs. Japan, Ine.), 18, № 3, 202 (1965); РЖХим, 1966, 16Г183.
434. Яо Шоу-пин. Хуасюэ шицзе (Huaxue shyie), 19, 456 (1965); РЖХим, 1966, 15Д62.
435. Яровенко А. Я., Бондарева Л. А. Труды МХТИ им. Д. И. Менделеева, вып. 58, 298 (1968).
436. Abe Sh., Mochlzukl Т., Uemura Т. Japan Analyst, 20, 1044 (1971).
437. Abe Y., Takazawa Sh. J. Iron a. Steel Inst. Japan, 57, 1350 (1971).
!/a10 В Ф. Волынец, M. П. Волынец
273
438. Abel К. Analyt. Chem., 38, 758 (1966).
439. Abeledo C. A., Kolthoff I. M. J. Am. Chem. Soc., 53, 2893 (1931).
440. Abravan^ll G. Rev. Group, methods avancem. spectrogr., oct.— dec., 289 (1966); РЖХим, 1968, 5Г155.
441. Acid Nitric (for ordnance Use). Toint Army — Navy Specification JAN-A-183, Amendment 2. U. S. Goverment Printing Office, Washington, D. C., February 28, 1945.
442. Acttzehn H. K., Hawart H. Le-bensmittel-Ind., 19, 482 (1972).
443. Adam A. R., Syputa R., Stephenson W. E. Analyt. Chem., 32, 1319 (1960).
444. Albein I. I. M., Elbakry A., Aly M. M. Chemist-Analyst, 56, 48 (1967).
445. Albert Ph. Chimia, 21, 116 (1967)-
446. Albert Ph. Proc. Intern. Conf. Mod. Trends Activat. Analysis. College Station Texas, 1961, p. 78; РЖХим, 1963, 10Г171.
447. Albert P., Engelmann Ch., May S., Petit J. C. r., 254, 119 (1962).
448. Albert D. K. Analyt. Chem., 39, 113 (1967).
449. Altotta G., Casal A. R. An. Asoc. quim. argent., 52, 223 (1964).
450. Allen A. D., Senoff C. W. Chem. Commun., No 24, 621 (1965).
451. Allenden M. Methods Phys. Anal., Oct.— Dec., 213 (1967); C. A., 68, 119003x.
452. Allerton F. W. Analyst, 72, 349 (1947).
453. Allied Chemical Corporation. Nitrogen Tetraoxide. New York, 1958.
454. Arndt T. Z. angew. Chem., 30, 169 (1917).
455. Arndt T. Z. angew, Chem., 33, 296 (1920).
456. Amiel S., Wiemik M. Israel Atomic Energy Commis (Repts.), No 1168, 73 (1968), РЖХим, 1968, 24Г59.
4J>7. Ammon F. von. Metalloberfla-che, 18, 136 (1964).
458. Andersen A. C., Jensen G. N. Fr., 83, 114 (1931).
459. Andersen G. H., Algots J. M. J. Radioanal. Chem., 3, 261 (1969).
460. Andrieth L. F., Ogg B. A.
The Chemistry Hydrazine. New York, Wiley, 1951, p. 155.
461. Anton A., Dodd I. G., Harvey A. E. Analyt. Chem. 32, 1209 (I960).
462. Antoniucci P., Durand J.-C., Rondot B., Montuelle J. Bull. Soc. chim. France, 1970, 1209.
463. Archer J. S. J. Inst. Fuel, 43, 56 (1970).
464. Armstrong F. A. J. Analyt. Chem., 35, 1292(1963).
465. Arnaud A., Pade L. C. r., 98, 1488 (1884); 99, 190 (1884).
466. Arndt F. Ann., 384, 322 (1911).
467. Arnold J. W. Ind. Eng. Chem., Anal. Educ., 17, 215 (1945).
468. Aubeau R., Reiss J., Cham-peix L., Ravnik V. J. Nucl. Mater., 6, 271 (1962).
469. Atkinson L. P. Analyt. Chem., 44, 885 (1972).
470. Attrill J. E., Boyd С. M., Meyer A. S. Analyt. Chem., 37, 1543 (1965).
471. Awad W. I., Hassan S. S. M. Taianta, 16, 1383, 1939 (1969).
472. Awasthi S. P., Sahasranamen S., Sundaresan M. Analyst, 92, 650 (1967).
473. Bach R. Phys. Chem., 9, 254' (1892); цит. no [868], S. 47.
474. Backer В. В. Analyt. Chem., 30, 1085 (1958).
475. Bahenersky VI., Chalupa J. Gal-vanotecbnik, 55, 709 (1964).
476. Bailey J. B. W., Brown N. S., Phillips С. V. Analyst, 96, 447 (1971).
477. Backer A. S., Smith R. J. Agr. Food Chem., 17, 1284 (1969).
478. Balcerek W. Hadowla rosl., aklimat. i. nasienn., 9, 151 (1965); РЖХим, 1966, 8Г168.
479. Baldwin S. F., Brown Ch. W. Water Res., 6, 1601 (1972).
480. Ballinger D. G. J. Water Pol-lut. Control. Federat., 35, 116 (1963).
481. Balstrieri C. Life Sci., 7, 269 (1968).
482. Bandi W. R., Straub W. A., Buy ok E. G., Melnik L. M. Analyt. Chem., 38, 1336 (1966).
483. Banerjea D., Das Gupta T. D. Indian J. Chem., 4, 91 (1966).
484. Banerjee N. D. Indian Ind., 9, 29 (1965).
485. Banerjee P. C. J. Indian Chem. Soc., 13, 301 (1936).
274
486. Bar H. J., Berndt Th., Hel-big H., Steinhauer H., Schwabe K. Wiss. Z. Techn. Hochschule Chem. Leuna-Merseburg, 6, 103 (1964); РЖХим, 1965, 12Г165.
487. Barcza L., Czuszy P. Mikro-chim. acta, 1963, 293.
488. Bard A. J. Analyt. Chem., 35, 1602 (1963).
489. Barendrecht E., Janssen L. M. Analyt. Chem., 33, 199 (1961).
490. Barica J., Fisch J. Res. Board. Can., 28, 759 (1971); РЖХим, 1972, 6Г203.
491. Bark L. S., Higson H. G. Analyst, 88, 751 (1963).
492. Bark L. S., Higson H. G. Ta-lanta, 11, 471 (1964).
493. Bark L. S., Higson H. G. Ibid., p. 621.
494. Barnes H., Folkard A. R. Analyst, 76, 599 (1951).
495. Barnes K., Mann Ch. K. Analyt. Chem., 36, 2502 (1964).
496. Batten J. J. Ibid., p. 939.
497. Battles W. R., Ellertson W. P., Hoggan G. D., Walker H. B., Wattenbarger C. W. 1. Inst. Petrol., 49, No 479, 340 (1963).
498. Baudin G., Deszcumaux J., Fontain R. Analyt. chim. acta, 38, 537 (1967).
499. Baudot Ph. Chim. analyt., 51, 425 (1969).
500. Baum W., Eskhard S. Z. anal. Chem., 209, 188 (1965).
501. Beanjard L., Cawalier G., Roche R., Berneron R. Rev. metallurgie, 62, 675 (1965).
502. Behrens W., Eichmann R., Plate A., Kroepelin H. Erdol, Kohle, Erdgas, Petrochem., 24, 76 (1971).
503. Behrens W., Eichmann R., Plate A., Kroepelin H. Ibid., p. 141.
504. Belcher R. Chem.-Ingr.-Techn., 36, 993, A1681, A1685 (1964).
505. Belcher R. Lab. Pract., 16, 578 (1967).
506. Belcher R. Magyar tud. akad. Kem. tud. oszt. kozl., 20, 231 (1963).
507. Belcher R., Campbell A. D., Gouverneuer P. I. Chem. Soc., 1963, 531.
508. Belcher R., Dryhurst G., McDonald A. M. G., Majer J. R., Roberts G. J. Analyt. chim. acta, 43, 441 (1968).
509, Banes M., Male chord M., Ma-
lecha J., Myslivecek J. Chem. lis-ty, 63, 703 (1969).
510. Benjamin R. W., England L.D., Blake K. R., Morgan I. L., Houston C. D. Trans. Am. Nucl. Soc., 9, 104 (1966).
511. Berge H., Jeroschewski P. Z. analyt. Chem., 228, 9 (1967).
512. Berger J. A., Meyniel G., Petit J. J. Chromategr., 29, 190 (1967).
513. Berka A., Vulterin J., Zyka J. Newer Redox Titrants. New York, Pergamon Press, 1965, p. 65.
514. Berka A., Vulterin J., Zyka J. Ibid., p. 189.
515. Bernat I., Cornides I. Honvedor-vos, 22, 201 (1970); C. A., 75, 92280b.
516. Berneron R., Romand J. Rev. metallurgie, 66, 695 (1969).
517. Bes R., Lacoste G., Mahenc J. Methods Phys. AnaL, 6, 109 (1970); C. A., 73, 71138u.
518. Bhachar V. M., Amar V. K. Indian J. Technol., 10, 433 (1972).
519. Bhatti A. M., Ralhan N. K. Z. analyt. Chem., 260, 370 (1972).
520. Bhatty M. K., Townshend A. Analyt. chim. acta, 56, 55 (1971).
521. Blaedel W. J., Olson C. L. J. Chem. Educ., 40, A549—A550; A552; A554; A556-A558 (1963).
522. Blais M. Microchem J., 6, 211 (1962).
523. Blake К. B., Martin T. C., Morgan I. L., Houston C. D. Proc. Intern. Conf. Mod. Trends Acti-vat. Analysis. College Station Texas, 1965, p. 76.
524. Blazejak-D itges D. Radex Rundschau, No 1, 335 (1963); РЖХим, 1963, 16Г144.
525. Blazek A. Chem. listy, 47, 269 (1953).
526. Bleaney B., Owen J. Diamond Res., No 15, 1965.
527. Blinn R. C., Gunther F. A. Analyt. Chem., 29, 1882 (1957).
528. Block J., Morgan E., Siggia S. Analyt. Chem., 35, 573 (1963).
529. Bloomfields R. A., Guyon J. C., Murmann R. K. Analyt. Chem., 37, 248 (1965).
530. Bloomfields R. A., Guyon J. C., Murmann R. К. J. Am. Water Work Assoc., 57, 935 (1965).
531. Bock R. Z. analyt. Chem., 133, 110 (1951).
532. Bagershausen W., Roelot van
10*
275
der Walt H. Z. analyt. Chem., 213, 161 (1965).
533. Bogs U., Gothe W. Z. analyt. Chem., 196, 87 (1963).
534. Bohle F. J. Mitt, dtsch. phar-maz. Ges., 38, No 12, 229 (1968).
535. Bohm E. Dtsch. Lebensmitt.-Rundschau, 62, No 10, 293 (1966).
536. Bohmer C. Fr., 22, (1883); цит. no [868], S. 78.
537. Boillot P., Boulin R., Jaudon E. Chim. analyt., 47, 120 (1965).
538. Bolleter W. T., Bushman C. J., Tidwell P. W. Analyt. Chem., 33, 592 (1961).
539. Bollman D. H. Analyt. Chem., 43, 154 (1971).
540. Boltz D. F. Colorimetric Determination of Non-Metals. Now York, Intersci. Publ., 1958, p. 131.
541. Bombi G. G., Fiorani M., Maz-zocchin G. A. J. Electroanalyt. Chem., 9, 457 (1965).
542. Bond A. M., Willis J. B. Analyt. Chem., 40 , 2087 (1968).
543. Booman G. L., Holbrook W. B. Analyt. Chem., 35, 1982 (1963).
544. Boos L. E., Winepordner J. D. Analyt. Chem., 44, 1020 (1972).
545. Borchert O. Metalloberflache, 19, 310 (1965).
546. Borgioli N., Talenti M. Nuo-viann igiene e microbiol., 12, 285 (1961).
547. Bortlisz T. Haus Techn.. Vort-ragsveroff., No 283, 9 (1972); РЖХим, 1973, 1И272.
548. Bothe H. K., Leonhardt J. J. Chromatogr., 19, 1 (1965).
549. Bound G. P.t Fleet B., Storp II. von, Evans D. II. Analyt. Chem., 45, 788 (1973).
550. Bourbon P., Alary J., Ecclassan J., Alengrin F. Inform, chim., No 105, 61, 71 (1972).
551. Bourguillot R., Canard A., Corun A., Robin A. M., Stefani R. Bull. Soc. chim. France, 1966, 2621.
552. Bourke P. J., Dawson R. W., Denton W. H. J. Chromatogr., 14, 387 (1964).
553. Bowen H. J. M. Analyst, 97, 728 (1972).
554. Bownan F. C., Scott W. W. J. Ind. Eng. Chem., 7, 766 (1915).
555. Boye E. Z. analyt. Chem., 207, 260 (1965).
556. Bozsai I., Hafdu, A. Csepeli miisz-kozgazd szamle, 10, No 2,
23, 35, 70, 71, 72 (1971); РЖХим, 1973, 9Г110.
557. Brandstreet R. B. Am. Book Publ. Rec. New York, Acad. Press, 6, No 11, 59 (1965).
558. Bray W. С., Сиу E. J. J. Am. Chem. Soc., 46, 858 (1924).
559. Bray W. C., Simpson M. E., Mackenzie A. A. J. Am. Chem. Soc., 41, 1363 (1919).
560. Brebion G., Cabriden R., Huriet B. Eau, 53, 463 (1966); РЖХим, 1967, 6И408.
561. Bremner J. M., Keeney D. R. Analyt. chim. acta, 32, 485 (1965).
562. Brenner M. IF., Vigilante C., Arthurs M. J. Am. Soc. Brew. Chemists Proc., 1963, p. 158.
563. Brey W. S., Hynes J- B. Fluorine Chem. Revs, v. 2. New York, 1968, p. 111-159.
564. Brezenski F. T. Environ. Sci. a. Technol., 1, 488 (1967).
565. Brhdbek L., Kalandra O. Chem. prumysl., 18, 417 (1968).
566. Brhalek L., Kalandra O., Du-fek F. Hutn. listy, 22, 53 (1967).
567. Brouwer Th. Chem. Weekblad, 60, 208 (1964).
568. Brown E. H., Cline J. E. Ana-, lyt. Chem., 17, 284 (1945).
569. Brown R. L., Brennen W. J. Chem. Phys., 47, 4697 (1967).
570. Bruner F., Di Corcia A. J. Chromatogr., 45, 304 (1969).
571. Buck R. P., Crowe T. J. Analyt. Chem., 35, 697 (1963).
572. Buckles M. F. Mikrochem. J., 9, 449 (1965).
573. Bilger P., Maierhofer J., Reis A. Z. analyt. Chem., 234, 176 (1968).
574. Buhl F., Gregorowicz Z. Zesz. nauk. Politechn. slaskiej, No 153, 55, 65 (1966).
575. Burford J. R. J. Chromatogr., 7, 760 (1969).
576. Burns E. A. Analysis of Ammonium Nitrate-Ш Assay. Progress Report No 20—365, jet Propulsion Laboratory, Pasadena, Calif, 1958; цит. no [868].
577. Burns E. A., Chang F. D. J. Phys. Chem., 63, 1314 (1959).
578. BurnsE. A., Lawler E. A. Analyt. Chem., 35, 802 (1963).
579. Burns E. A., Muraca R. F. Ibid., p. 1967.
580. Burrel F. M., Lucena F., Arri-bas S. J. Analyt. chim. acta, 11, 214 (1944).
276
581. Buscarous F., Alsina I. An. Real. Soc. esp. fis, у quim., B59, N 2, 101 (1963).
582. Busch M. Ber., 38, 861, 4055 (1905); 39, 1401 (1906).
583. Busvoid N. Chemikor-Ztg., 39, 28 (1914); 39, 214 (1915).
584. Caldwell B. J. Gas (USA), 39, 105 (1963).
585. Camm/in K. Beckman Rept., N 1, 25 (1970).
586. Campiglio A. Farmaco Educ. sci., 19, 1021 (1964); РЖХим, 1965, 20Г184.
587. Canib V.D., TurtU M. N., Bu-garski-Vojnovic M. B., Peristd N. U. Z. anal. Chem., 229, 93 (1967).
588. Carle G. С. J. Chromatogr. Sci., 8, 550 (1970).
589. Carrillo D., Urgell M., Iglesias J. An. quim. Real Soc. esp. ffs. у quim., 64, 841 (1968).
590. Carrillo D., Urgell M., Iglesias J. An. quim. Real Soc. esp. fis. у quim., 66, 461 (1970).
591. Cavard A., Andreani A. M., Stefani B. Rev. Group, avancem. methodes spectrogr., jan.— mars, 37 (1967).
592. Ceausescu D. Z. analyt. Chem., 208, 188 (1965).
593. Ceausescu D., Asteleanu M., Vlad L., Berger G. Rev. chim. (RSR), 19, 51 (1961).
594. Ceausescu D., Sarbu A. Rev. chim. (RSR), 23, No 10 (1972); Chim. anal., No 3, 187 (1972).
595. Ceausescu D., Sarbu A. Z. Chem., 10, 272 (1970).
596. Cerana L. A., Guler J. Rev. Fac. ingr. quim. Univ. nac. Litoral, 23-24, 189 (1964-1965).
597. Cerhardt A. Chem. Rundschau, 16, 502 (1963).
598. Chalmers R., Umar M. Analyt. chim. acta, 39, 521 (1967).
599. Chang Ta-Chang Lo. J. Chromatogr., 37, 14 (1968).
600. Chapman B., Gooke G. H., Whitehead R. Water Pollut. Control., 66, 280 (1967).
601. Chapman M. W., Broida H. P. NBS, Circ. No 575 (1956).
602. Chasan D. E., Norwitz G. Appl. Spectroscopy, 24, 283 (1970).
603. Chattergjee S. S., Banerjee В. K. Technology, 8, 285 (1971).
604. Chavtarova R. D. Докл. Бълг. АН, 26, 403 (1973).
605. Chem. analit. (Polska), 12, 915 (1967).
606. Chem. Process (Engl.), 9, No 3, 24 (1963).
607. Chem. Process (USA), 27, No 13, 34 (1964).
608. Cheng H. H., Bremner J. M., Edwards A. P. Sci., 146, No 3651, 1574 (1963).
609. Christian G. D., Jung P. D. J. Assoc. Offic. Analyt. Chemists, 49, 865 (1966).
610. Churacek J., Komarkova H., Komarek K. Chromatographia, 3, 465 (1970).
611. Clark J.D. Analytical Procedures for Rocket Propellants: IV. Hydrazine. Report N 9, U. S. Naval Air Rocet Test Station. New York, Lake Denmark, 1951; цит. no [868].
612. Clarke A. R., Cable M. Glass Technol., 8, 82 (1967).
613. Clear A. J., Roth M. In «Treatise on Analytical Chemistry», part 2, v. 5. Eds. I. M. Kolthoff, P. J. Elving. New York, Intersci. Publ., 1961, pp. 251, 284, 289.
614. Clerk J. T., Simon W. Mikro-chem. J., 7, 422 (1963).
615. Cachet L., Heitz G., Raguin J. Cent. Doc. Siderurg. Cirk. Inform. Techn., 28, 1071 (1971) C. A., 75, 29524h.
616. Coetzee С. I., Freiser H. Analyt. Chem., 40, 2071 (1968).
617. Cohen I. R., Purcell T. C., Alt-schuller A. P. Environ. Sci. a. Technol., 1, 247 (1967).
618. Collat J. W., Lingane J. J. J. Am. Chem. Soc., 76, 4214 (1954).
619. Colombo A., Vivian R., Rodart E. Analyt. chim. acta, 62, 472 (1972).
620. Conrad F. J. Taianta, 18, 952 (1971).
621. Conway E. J., ByrtieA. Biochem. J., 27, 419 (1933).
622. Conzemius R. J., Svec H. J. Analyt. chim. acta, 33, 145 (1965).
623. Capello M.A., Dorfman E. A. An. Soc. cient. argent., 181, 33 (1966).
624. Cornides I., Medzihradszky H., Bemat I. Haemotologia, 4, 21 (1970).
625. Cornu A., Bourguillot R., Stefani R. Energie nucl., 9, 386 (1967).
626. Cosgrove R. E., Mask C. A., Krull I.H. Anal. Lett., 3, 457 (1970).
277
627. Coulson D. M. J. Gas Chroma-togr., 4, 285 (1966).
628. Covington A. K., Thain J. M. Analyt. chim. acta, 55, 453 (1971).
629. Cremer E., Deutscher F. Aluminium (BRD), 46, 174 (1970).
630. Critchfield F. E., Johnson J. B. Analyt. Chem., 26, 1803 (1954).
631. Crutchfield C. A. Analyt. Chem., 41, 295 (1969).
632. Culmo R., Fyans R. Mikrochim. acta, 1968, 816.
633. Cundiff R. H., Marcunas P. C. Analyt. chim. acta, 21, 68 (1959).
634. Сиу E. J., Bray W. C. J. Am. Chem. Soc., 46, 1785 (1924).
635. Dagnall R. M., El-Ghamry M. T. West T. S. Taianta, 13, 1667 (1966).
636. Dagnall R. M., El-Chamry M. T., West T. S. Taianta, 15, 107 (1968).
637. Dagnall R. M., Thompson К. C., West T. S. Taianta, 14, 551 (1967).
638. Dambacher M., Guller A., Haas H. G. Clin. Chem., 14, 615 (1968).
639. Danchik R. S., Boltz D. F. Analyt. Chem.,“40, 2215 (1968).
640. Dan Manka P. Analyt. Chem., 36, 480 (1964).
641. Dard P., Guibourg J., Merli-vat J.-C., Bernard G. Advances Mass. Spectrom., 3, 797 (1966).
642. Davies I. E. W., Moody G. I., Thomas I. D. R. Analyst, 97, 87 (1972).
643. Davis D. G. Analyt. Chem., 35, 764 (1963).
644. Davis H. C., Allsopp H. /.Mem. sci. rev. metalurgie, 60, 209 (1963).
645. Dembeck H. Wasser, Luft u. Bet-rieb, 10, 822 (1966).
646. Dennis L. M., Isham H. Am. Soc., 29, 18 (1907); цит no [721], S. 63.
647. Dennis L. M., Nichols M. L. Gas Analysis. New York, Yan Nostrand, 1934, p. 279.
648. Derge K. Chem.-Ingr.-Techn., 37, 718, A945 (1965).
649. Derge K. Chemiker-Ztg., Chem. Apparat., 90, 283 (1966).
650. Dernbach C. J., Mehling J. P. Ind. Eng. Chem., Anal. Educ., 14, 58 (1942).
651. Desaty D., McGrath R., Vining L. C. An. Biochem., 29, 22 (1969).
§52. Deshmukh G. S., Murty S. V. S. S. Indian J. Chem., 1, 316 (1963).
653. Desjardins M. Advances Mass Spectrom., 4, 439 (1969).
654. Determination of Nitrate in Soils with the Nitrat Ion Activity Electrode. Orion Application Bulletin, No 6. Orion Research., Inc., 1968.
655. Devarda A. Chemiker-Ztg., 16, 1952 (1892).
656. Dewasner P., Tardy G. Rev. gen. therm., 1, N 11, 56 (1962); РЖХим, 1963, 13Г178.
657. Dey A. P. J. a. Proc. Inst. Chemists (India), 37, N 6, 249 (1965).
658. Diamond Ph. Am. Industr. Hyg. Assoc. J., 24, 399 (1963).
659. Dickinson G. W., Wheeler G. V. Appl. Spectroscopy, 18, 152 (1964).
660. Dietz R. N. Analyt. Chem., 40, 1576 (1968).
661. Dimitriades В. J. Air Pollut. Control. Assoc., 17, N 4, 238 (1967).
661a. DiGregorio I. S., Morris M. D. Analyt. Chem., 42, 94 (1970).
662. Docherty A. C. Lab. Pract., 18, 47 (1969).
663. Daring С. E., Geier R. Roske P. Acta chim. Acad. Sci. hung., 73, 399 (1972).
664. Doring С. E., Geyer R.t Roske P. Acta chim. Acad. Sci. hung., 75, 89 (1973).
665. Dragulesku C., Florea J. Studii $i. cercetari stiinte chim. acad. RPR. Baza Timisoara, 9, 205 (1962).
666. Driscoll I. H., Berger A. W., Becker I. H., Funkhouser I. T., Valentine I. R. J. Air. Pollut. Control Assoc., 22, 119 (1972).
667. Duff G. M. S., Wilson H. M. Chem. a. Ind., 1965, 1668.
668. Dukes E. K., Wallance R. M. Analyt. Chem., 33, 242 (1961).
669. Dulski T. R., McKaveney J. P. J. Metals, 19, 11, 15, 81, 84 (1967).
670. Du Preez L., Raal F. A. Diamond Res., No 6 (1965).
671. Dziegies J., Ignaezak M. Soc. sci. lodz., Acta chim., 16, 69 (1971); РЖХим, 1972, 5Г113.
672. Ehrenberger F., Weber. O. Gias-u. Instrum. Techn., 11, 502 (1967).
673. Ellis С. M., Vogel A. I. Analyst, 81, 693 (1956).
674. Emmot P., Wilson R. E. Talan-ta, 11, 1003, 1011 (1964).
675. Engelhardt H. Test., Instrum, a. Controls, 4, No 9, 29 (1967).
676. Engelmann Ch. Intern. J. Appl. Radiat. a. Isotopes, 18, 569 (1967).
278
677. Engelmann Ch. J. Radioanal. Chem., 6, 399 (1970); Bull, inform, sci. e. techn. Commissar energie atom, N 140, 65 (1969); Bull. Soc. chim. France, 1967, 2316; Rapp. CEA, N 2559 (1964).
678. Engelmann Ch. Radiochemical Methods of Analysis, v. 1. Vienne-ax, Intern. Atomic Energy Agency, 1965, p. 341.
679. Engelmann Ch., Calane G. Proc. Intern. Conf. Mod. Trends Activat. Analysis. College Station Texas, 1965, p. 333.
680. Engelmann Ch., Gosset J., Boissier M. Proc. Intern. Conf. Mod. Trends Activat. Analysis. NBS Spec. Publ., 1969, p. 819.
681. Engelmann Ch., Gosset J., Loeu-illet M. Menn. sci. Rev. metallur-gie, 63, 1081 (1966).
682. Em K. Wasser, Luft u. Betrieb, 8, 581, 585, 603 (1964).
683. Escoffier P. Rech, aerospatiale, No 106, 25 (1965).
684. Esther F. A., Taizo O. Bol. inst. oceanogr, Univ. Oriente, 5, No 1 — 2, 96 (1966/1967); РЖХим, 1968, 15Г178.
685. EulenhbferH. G. Z. analyt. Chem., 215, 31 (1966).
686. Evans Sh. J. Water Pollut. Centre Fed., No 4, 632, 758, 763, 768, 773 (1973).
687. Evens F. M., Fassel V. A. Analyt. Chem., 35, 1444 (1963).
688. Fadrus H., Malt) L. Z. analyt. Chem., 202, 164 (1964); Ceskosl. hyg., 13, 300 (1968).
689. Fadrus H., Maltj J. Z. analyt. Chem., 246, 239 (1969).
690. Fassel V. A., Evens F. M., Hill С. C. Analyt. Chem., 36, 2115 (1964).
691. Faulhaber E., LiebetrauL. Monats-ber. Dtsch. Akad. Wiss., Berlin, 7, 872 (1965).
692. Fay H., Mohr P., Cook .G. Analyt. Chem., 34, 1254 (1962).
693. Fay H., Mohr P., Cook G. Pat. USA No 3032654 (1962); РЖХим, 1965, 10Д16П.
694. Feigl F., Chan F. Mikrochim. acta, 1967, 339.
695. Feigl F., Souza de Campos I. I., Dalto S. L. Analyt. Chem., 36, 1657 (1964).
696. Fenstermacher J. E. J. Vac. Sci. Technol., 8, 380 (1971).
697. Fernandez A. E., Okuda T. Bui.
Inst, oceanogr. Univ. Oriente, 5, 96 (1966/1967); РЖХим, 1968, 15Г178.
698. Fest J. Ch., Schott B. Analyt. chim. acta, 63, 283 (1973).
699. Fielder R., Proksch G. Analyt. chim. acta, 60, 277 (1972).
700. Finkelson M. J. J. Assoc. Offic. Anal. Chem., 56, 119 (1973).
701. Fiorani M., Magno F. Analyt. chim. acta, 39, 285 (1967).
702. Fischer J. Z. analyt. Chem., 209, 176 (1965).
703. Fischer G. E., Becknell D. E. Analyt. Chem., 44, 863 (1972).
704. Fischer F. L., Ibert E. R., Beckman H. F. Analyt. Chem., 30, 1972 (1958).
705. Fishman M. J., SkougstadM. W., Scarbro G. F. J. Am. Water Works Assoc., 56, 633 (1964).
706. Fleet B., Ho A.Y. W. Ion-select. Electrod. Symp. Matrafiired, 1972, p. 17.
707. Fleet B., Stop H. von. Anal. Lett., 4, 425 (1971); Analyt. Chem., 43, 1575 (1971).
708. Fleps W. Gesundh. Ingr., 84, No 7, 209 (1963).
709. Fontaine R. Note CEA, No 1148, 44 (1969); РЖХим, 1970, 1Г67.
710. Foris A., Sweet Th. Analyt. Chem., 37, 701 (1965).
711. Fournet L. Ann. Chim. 7, 763 (1962).
712. Frank J., Trtik B., Placek K. J. Chromatogr., 36, 1 (1968).
713. Frant M. S., Ross J. W., Rie-seman J. H. Analyt. Chem., 44, 2227 (1972).
714. Frazer J. W. Mikrochim. acta, 1962, 993.
715. Frazer J. W. U. S. Atomic Energy Comm. Repts UCRL-5134; РЖХим, 1964, 9Г194.
716. Frazer J. M., Crawford R. Mikrochim. acta, 1963, ,561.
717. Frazer 'J. W., Jones G. D., Lim R., Waggoner M. C., Rogers L. B. Analyt. Chem., 41, 1485 (1969).
718. Frazier R. E. J. Am. Water Works Assoc., 55, 624 (1963).
719. Freddi R., Bombiciuseppe G., Fiorani M. Z. analyt. Chem., 222, 369 (1966).
720. Fresentus R., Jander G. Hand-buch der analytischen Chemie, Teil III, Bd. la. Berlin, Springer Verlag, 1940, S. 274—316; 356.
279
721. Presenilis R., Jander G. Hand-buch der analytischen Chemie, Bd. Va. Berlin — Gottingen — Heidelberg, Springer Verlag, 1957, S. 74.
722. Friedel R. A. Analyt. Chem., 28, 1806 (1956).
723. Friedel R. A., Schultz J.L., Sharkey A.C. Analyt. Chem., 31, 1128 (1959).
724. Friedel R.A., Sharkey A.E., Ir., Shultz J.L., Humbert C. R. Analyt. Chem., 25, 1314 (1953).
725. Friedrich K., Lassner E. J. Less-Common Metals, 13, 156 (1967).
726. Friedrich K., Lasner E., Pae-sold G. J. Less-Common Metals, 22, 429 (1970).
727. Frohberg M. G., Gerhardt A'., Kraus Th. Z. analyt. Chem., 227, 8 (1967).
728. Frutnovsky H. Ъ. analyt. Chem., 198, 331 (1963).
729. Fujinuma H., Shlmaba Y. Japan Analyst, 20, 1038 (1971).
730. Furman N. H. Standart Methods of Chemical Analysis, v. 1, 5th ed. Ed. Scotts. New York, Van Nostrand, 1958, p. 636.
731. Furst P., Johnson A. Acta Chem. Scand., 25, 930 (1971).
731a. Furuya K., Agahara I., Kama-da H. Analyst, 14, 330 (1965).
732. Gagliardi E., Pokorny G. Mikro-chim. acta, 1965, 699.
733. Galanos D. S., Kapoulas V. M. Analyt. chim. acta, 34, 360 (1966).
734. Galik V., Landa S. Sb. Vysoke Skoly chem.— technol. Praze. Technol. paliv., 8, 69 (1966).
735. Galwey A. K. Taianta, 9, 1043 (1962).
736. Garcia E. E. Analyt. Chem., 39, 1605 (1967).
737. Geetha R., Ramani A., Rajago-palan S. Curr. Sci., 41, 772 (1972).
737a. Gehring D.G., Dippl W. A.t Boucher R. S. Analyt. Chem., 42, 1686 (1970).
738. Gehrke Ch. W., Kaiser F. E„ Ussary J. P. J. Assoc. Offic. Analyt. Chemists, 51, 200 (1968).
739. Gelbwachs J. A., Birnbaum M., Tucker A. V., Fincher C. L. Optoelectron, 4, 155 (1972); РЖХим, 1973, 7И576.
740. Gerchman L. L., Rechnitz G.A. Z. analyt. Chem., 230, 265 (1967).
741. Gerhardt A., Kraus Th., Frah-
280
berg M. G. Z. analyt. Chem., 218, 192 (1966).
742. Germuth F. G. Ind. Eng. Chem., Anal. Educ., 1, 28 (1929).
743. Gertner A., Grdinic V. Acta phar-mac. jugosl., 15, 209 (1965).
744. Gibler G., Kempf Th. Z. analyt. Chem., 199, 23 (1964).
745. Gibson E. K., Moor С. B. Analyt. Chem., 42, 461 (1970).
746. Gierlinger W. Gas, Wasser, Warme, 17, No 2, 265 (1963).
747. Gilbault G. G., Kramer D. N. Analyt. Chem., 37, 1395 (1965).
748. Gilbert Th. R., Clay A. M. Analyt. Chem., 45, 1757 (1973).
749. Gillieson A. H. Science and Technology Tungsten, Tantalum, Molybdenium, Niobium and their Alloys. Oxford — London — Edinburg — New York — Paris — Frankfurt, Pergamon Press, 1964, p. 381—393.
750. Gimesi O., Rddy Gy., Erdey L. Acta chim. Acad. Sci. hung., 38, 303 (1963).
751. Ginderow D., Setton R. C. r., 257, 687 (1963).
752. Giry L., Chaigneau,Ricard L.—P. Analusis, 1, No 7, 475 (1972).
753. Glabisz U. Chem. analit.
(Polska), 9, 139 (1964).
754. Gnauck G. Abhandl. Dtsch.
Akad. Wiss. Berlin, KI. Chem., Geol. u. Biol., Nol, 133 (1962).
755. Gnauck G. Z. analyt. Chem., 189, 124 (1962).
756. Gnauck G., Krieg H. Pat. DDR, N 23207 (1962); РЖХим, 1963, 17Г164П.
757. Gnauck G., ScMn H. Abhandle Dtsch. Akad. Wiss. Berlin, KI. Chem., Geol. u. Biol., N 2, 319 (1966).
758. Gnauck G., ScMn H. Pat. DDR, N 44459 (1966); РЖХим, 1967, ЗГ24П.
759. Gnauck G., Schon H. Proc. Conf. Appl. Phys.-Chem. Methods Chem. Anal. (Budapest), No 2, 182 (1966).
760. Gokcen N.A., Tankins E. A. J. Metals, 14, 584 (1962).
761. Goldbaum L. R., Domanskl T. J., Schloegel E. L. J. Gas. Chroma-togr., 6, 394 (1968).
762. Goldbeck C. G., Turel S. P., Rodden C. J. Analyt. Chem., 40, 1393 (1968).
763. Goodfellow G. I., Webber H. M. Analyst, 97, 95 (1972).
764. Goodman B. L. Water a. sewage Works, 110, 27 (1963).
765. Goodspeed H. T., Holt B. D., Marsh J. H., Stoessel J. E. Analyt. Chem., 40, 682 (1968).
766. Goto H., Ban T. Ann. Rept. Fac. Pharmac. Sci., Nagoya City Univ., No 19, 59 (1971).
767. Goto H., Kakita Y., Atsuda I. Sci. Repts. Res. Inst. Tohoky Univ., A19, No 1, 50 (1967).
768. Gottlieb О. R., Magalhaes M. T. Analyt. Chem., 30, 995 (1958).
769. Goulden J. D. S., Manning D.J. J. Sci. Food a. Agric., 18, No 10, 466 (1967).
770. Goulden P.D., Alghan В. K., Brooksbank P. Analyt. Chem., 44, 1845 (1972).
771. GourisseD., Gautier A. J. Inorg, Nucl. Chem., 31, 839 (1969).
772. Goward G. W. Analyt. Chem;, 37, 117 (1965).
773. Goy al S. S., Tandon J. P.
Indian J. Chem., 5, 39 (1967).
773a. Graff S., Rittenberg D., Foster J. J. Biol. Chem., 133, 737, F 745 (1940).
774. Graham J. H. Mikrochem. J., 13, 327 (1968).
775. Grassholf K. Ber. Dtsch. Wiss. Komiss. Meeresforsch., 20, 155
(1969); Technicon Sympos., v. 1. Automat. Analyt. Chem. (New York City, 1966). New York, Medi-ad Inc., 1967, p. 573.
776. Grassholf K. Kieler Meeresforsch., 20, 5 (1964).
777. Greff C. Rev. fiz. ?i chim., A5, N 3, 148 (1968).
778. Gregoroivicz Z., Gbrka P. Chem. analit. (PRL), 16, 703 (1971).
779. Gregoroivicz Z., Gbrka P. Koks, smola, gaz, 18, 81 (1973).
780. Grefimann K. Mikrochim. acta, 1963, 782.
781. Grether C. Galvanotechn. u. Oberflachenschutz, 4, 281 (1963).
782. Griepink B., Terlouw J. K. Mikrochim. acta, 1968, 624.
783. Griess P. Chem. Ber., 12, 426 (1879).
784. Grine W. H., Chueh Ch. F. Water a. sewage Works, 110, 43, 254 (1963).
785. Gritzner G., Gutmann V., Schbber G. Mikrochim. acta, 1964, 193.
786. Gross D. J. Chromatogr., 10, 233 (1963).
787. Guilbault G. G., Kramer D. N. Analyt. Chem., 37, 918 (1965).
788. Guilbault G. G., Kramer D. N. Analyt. Chem., 38, 834 (1966).
789. Guilbault G. G., Smith R. K., Montalvo S. G. Analyt. Chem., 41, 600 (1969).
790. Guin V. P., Johnson R. A., Mull G. C. 6th World Petrol. Congr., Francfort/Main, 1963, Sec. 5 (Preprint), 5a, No 20.
791. Guiraud G., Berlier Y. Chim. anal. (Paris), 50, 379 (1968).
792. Gundiff R. H., Markunas P. C. Analyt. chim. acta, 20, 566 (1959).
793. Gunther F.A., Barkley J.H., Kolbesen M. J., Blinn, R. C., Staggs E. A. Analyt. Chem., 28, 1985 (1956).
794. Gupta H. K. L., Boltz D. F. Anal. Lett., 4, 277 (1971).
795. Haba F. R., Wilson C. L. Ta-lanta, 11, 21 (1964).
796. HachenbergH,,GutberletJ. Brenn-stoff-Chemie, 44, 235 (1963).
797. Hall T. C., Bacet F. E. J. Chem. Phys., 20, 1745 (1952).
798. Hamm R. E., Withrow C. D. Analyt. Chem., 27, 1913 (1955).
799. Hampton W. C. J. Gas Chromatogr., 3, 217 (1965).
800. Hanawalt R. B., Steckel J. E. Technicon Symps., v. 1. Automat. Analyt. Chem. (New York City, 1966). New York, Median Inc., 1967, p. 133.
801. Handy J. F. C. Chem. Process, 10, 54 (1964).
802. Handy J. F. C. Metallurgie (Belg.), 5, 179 (1964—1965).
803. Handy J. F. C. Wild-Barfield Heat — Treatm. J., 9, No 71, 1 (1964).
804. Hanin M., Jaudon E. Rev. metallurgie, 62, 37 (1965).
805. Hanin M., Villeneuve D. Chim. analyt., 48, 442 (1966).
806. Harley A. M., Curran D. J. Analyt. Chem., 35, 686 (1963).
807. Harmelin M. Chim. analyt., 48, 197 (1966).
808. Harrington W. L., Skoger-boe R. K., Morrison G.H. Analyt. Chem., 38, 821 (1966).
809. Harris L., Siegel В. M. Ind. Eng. Chem., 14, 258 (1942).
810. Hartinger L. Industrieabwasser, 1966, 52.
В. Ф. Волынец, M. П. Волынец
281
811. Hartley A. M., Asai B. I. Analyt. Chem., 35, 1207 (1963).
812. Hartley A. M., Asai H. I. Ibid., p. 1214. '
813. Hartley A. M., Bly В. M. Ibid., p. 2094.
814. Hartley A. M., Hehner A. Coll. Czech.Chem. Comm., 30,4250 (1965).
815. Hartmann H. J. Chromatogr., 7, 163 (1969).
816. Hartmann H., Bathke G. An-gew. Chem., 65, 107 (1953).
817. Harwood J. E., Kuhn A. L. Water Res., 4, 805 (1970).
817a. Hashmi M. H., AH E., Umar M. Analyt. Chem., 34, 988 (1962).
818. Hashmi M. H., Chughtai N. A. Mikrochim. acta, 1968, 1040.
819. Hashmi M. H. et al. Ibid., p. 1143, 1237.
820. Hauser Th. B., Shy С. M. Environ. sci. a. Technol., 6, 890 (1972).
821. Havas J., Huber M. ,Szabo I., Pungor E. Ing. Wiss. instrum., mai, 25 (1967).
822. Havlena E. J., Hutchinson K.
J. Gas Chromatogr., 6, 419
(1968).
823. Hawart H., Achtzehn H. K. Na-hrung, No 4, 359 (1972); РЖХим, 1973, 7P127.
824. Haymes J. G., Ollar A. Rept. Invest. Bur. Mines U. S. Dept. Inerior., No 6005 (1962).
825. Healy C., Parker A. Res. Group U. K. Atom. Energy Auth., NoAERE-R6449 (1970); РЖХим, 1971, 14Г161.
826. Hein W., Schneider H., Schumann H. Z. analyt. Chem., 237, 330 (1968).
827. Heistand B. N. Analyt. Chem., 42, 903 (1970).
828. Heitz G., Thomas F. Rev. metallurgic, 66, 641 (1969).
829. Helzel M. Am. Ceram. Soc. Bull., 48, 287 (1969).
830. Henriksen A. Analyst, 90, 83 (1965).
831. Henriksen A. Analyst, 95, 601 (1970).
832. Henrioul F. Rev. ferment, e. inds. aliment., 18, 51 (1963); РЖХим, 1965, 5P212.
833. Herecek M., Kozdk P. Mikrochim. acta, 1967, 480.
834. Hertens J., Massart P. L.
282
Bull. Soc. chim. belg., 82, No 3—4, 179 (1973).
835. Hetman J. S. Lab. Pract., 12, 920 (1963).
836. Hewitt P. J., Austin II. B. Water Pollut. Contr., No 4, 381 (1972).
837. Higam I. C., Sahasrabund-ne M., Davis T. W., McConnell, Bartlet J. C., Levi L. Perfum. a. Essent. Oil Rec., 53, No 9, 614 (1962); РЖХим, 1963, 10Д27.
838. Higson H. G., Bark L. S. Analyst, 89, 338 (1964).
839. Hinsvark O. N., Pat. USA, No 3304159 (1967); РЖХим, 1968, 11Г159П.
840. Hirokawa K., Goto H. Z. analyt. Chem., 234, 340 (1968).
841. Hirokawa K., Goto II. Z. analyt. Chem., 240, 311 (1968).
842. Hirsch B. F., Portock J. D. Analyt. chim. acta, 49, 473 (1970).
843. Hoffmann B. L., List G. B., Evans C. D. Nature, 211, 965 (1966).
844. Holt B. D., Goodspeed H. T. Analyt. Chem., 35, 1510 (1963).
845. Holty J. G., Potworowsky H. S.
Environ. Sci. a. Technol., 6, 835 (1972).
846. HortonA. D., Nucl. Sci. a. Engn., 13, 103 (1962).
847. Howell I. H., Boltz D. F. Analyt. Chem., 36, 1799 (1964).
848. Hozumi K. Analyt. Chem., 38, 641 (1966).
849. Hozumi K. Mikrochem. J., 10, 46 (1966).
850. Hozumi K., Umemoto K. Mikrochem. J., 12, 512 (1967).
851. Huhn W. Limnologica, 4, 427 (1966).
852. Humphrey В. E., Alvarez J. J. Mikrochem. J., 16, 652 (1971).
853. Hutchinson K., Boltz D. F. Analyt. Chem., 30, 54 (1958).
854. Hutton В. C., Salan S. A., Stephen W. I. J. Chem. Soc., A, 1966, 1573.
855. Hutton В. C., Stephen W. I. Analyst, 92, 501 (1967).
856. Hwang С. P., Forsberg C. B. Water a. sewage Works, 120, 71 (1973).
857. Hynek B. J., Nelen J. A. Analyt. Chem., 35, 1655 (1963).
858. Ikeda S., Hirata J. J. Chem. Soc. Japan, Chem. a. Ind. Chem., No 2, 425 (1973).
859. Ikeda S., Hirata J., Sataki H. J. Chem. Soc. Japan, Chem. a. Ind. Chem., No 10, 1980 (1970).
860. Ikeda S., Hirokawa K. Suzuki M. Z. analyt. Chem., 228, 180 (1967).
861. Ilosvay M. L. Bull. Soc. Chim., 2, 383 (1889).
862. Imai H. Technol. Repts Kensal Univ., No 5, 21 (1963).
863. Inform. Circ. Bur. Mines. U. S. Dept. Inerior, No 8397 (1968).
864. Inform, quim. analit., 18, 161 (1964).
865. Inokoshi Y., Ikeda M., Yamamoto T., Shiozaki Y. Pollut. Contr., 8, 7 (1973).
866. Inteinrich B., Draeger B. Pat. BRD, N 1127628 (1962).
867. In the Chemical Analysis Series. Ed. P. J. Elving, I. M. Kol-thoff. Determination of Caseous Elements in Metals, v. 40. Ed. La-ben M. Melnick, Lynn L. Lewis, Ben D. Holt. New York—London—Sydney—Toronto, Wiley—Interact Publ., 1974, p. 321—339.
868. In the Chemical Analysis Series. Ed. P. J. Elving, I. M. Kol-thoff. The Analytical Chemistry of Nitrogen and Its Compounds, v. 28, Ed. C. A. Streuli, Philip R. A. New York—London—Sydney — Toronto, Wiley—Intersci. Publ., 1970.
869. Ishikawa H., Baba H. Radioisotopes (Tokyo), 17, 195 (1968).
870. Israel Y., Vromen A. J. Elec-troanalyt. Chem., 11, 262 (1966).
871. Issa I. M., Issa В. M. Chem. anal., 45, 40 (1956).
872. Jaeger K. Linde Ber. Techn. Wiss., 1965, 38.
873. Jacob Z. L., Szpak S. Bull. Intern. Acad. Pol on. Sci., Classe Sci. Math. e. Nat., Ser. A, 91 (1950).
874. James A. T., Martin A. J. P. Biochem. J., 50, 679 (1952).
875. Janauer G. E., Carrano J. D., Johnston В. C. Mikrochim. acta, 1968, 61.
876. Jaudon E. Rev. metallurgie, 63, 1025 (1966).
877. Jaworowski B. J. Analyt. Chem., 38, 1605 (1966).
878. Jecko G. Beynaud B. Rev. metallurgie, 64, 681 (1967).
*'879. Jeffery P. G., Kipping P. J. Analyst, 88, 266 (1963).
880. Jelen. K., Lasa J., Ostrowski K.
Chem. analit. (Polska), 13, 1239 (1968).
881. Jenkins D., Medsker L. L. Analyt. Chem., 36, 610 (1964).
882. Jenkins S. H., Hey A. E., Cooper J. S. Intern. J. Air. a. Water Pollut., 10, 495 (1966).
883. Johnson С. M., Ulrich A. Analyt. Chem., 22, 1526 (1950).
884. Jiri Z. Chem. prumusl, 18, 104 (1968).
885. Johar G. S., Agarwala U. Sci. a. Culture, 36, 521 (1970).
886. Johar G. S., Tiwari G. D. Z. analyt. Chem., 265, 32 (1973).
887. Johnson M. G., Bobinson B. J. Analyt. Chem., 24, 366 (1952).
888. Johnes C. N. Analyt. Chem., 39, 1858 (1967).
889. Jones I. O. Mining a. Minerals Engn, 6, 25, 28 (1970).
890. Jones P., Nieless G. J. Chromatogr., 73, 19 (1972).
891. Jones B. N., Thorn G.D. Can. J. Res., 27B, 58 (1949).
892. Jovanovic, M. S., Sigulinsky F. D., Dragojevic M. Taianta, 13, 1275 (1966).
893. Jozefowicz E., Maslowska J. Zesz. nauk. Politechn. lodzk., No 74, 5, 15 (1965).
894. Junk G., Svec H. S. Geochim. cosmochim. acta, 14, 234 (1958).
895. JureSek M., Kalousovd J., Koz&k P. Mikrochim. acta, 1968, 1313.
896. Jurecek M., Kozak P. Mikrochim. acta, 1967, 480.
897. Jurion B. Chim. analyt., 50, 325 (1968).
898. Kaburaki Y., Mikami Y., Oka-bayashi Y., Saida Y. Japan Analyst, 18, 1100 (1969).
899. Kadic K. Chem. prftmusl., 13, 363 (1963).
900. Kahn L., Bresenski F. T. Environ. Sci. a. Technol., 1, 492 (1967).
901. Kainz G., Wachberger E. Mikrochem. J., 12, 584 (1967).
902. Kainz G., Wachberger E. Z. analyt. Chem., 222, 271 (1966).
903. Kajikarva M., Kawaguchi M., Amari T., Watanabe K. J. Japan Petrol. Inst., 15, 770 (1972).
904. Kakabadse G. J., Manohin B. Mikrochim. acta, 1965, 855.
905. Kala II. Pharmazie, 18, 29 (1963).
906. Kallmann S., Hobart E. W., Oberthin H. K., Brienza W. C.
11*
283
Analyt. Chem., 40, 332 (1968).
907. Kammori O., Hiyama Y., Hotto W. J. Japan Inst. Metals, 29, 126 (1965); Trans. Japan Inst. Metals, 8, 58 (1967).
908. Karmen A. J. Chromatogr. Sci., 7, 541 (1969).
909. Kashima J., Yamazaki T. Bull. Chem. Soc. Japan, 42, 542 (1969).
910. Kashima J., Yamazaki T. Chem. Soc. Japan, 39, 1453 (1966).
911. Kateman G., Willensen L. L. M., Wijenberg J. B. G., Storneb-rink P. J. Analyt. chim. acta, 31, 139 (1964).
912. Katz S. A., Rechnitz G. A. Z. analyt. Chem., 196, 248 (1963).
912a. Keeney D. R., Bremner J. M. Soil Sci. Soc. Am. Proc., 31, 317 (1967).
913. Keeney D. R., Byrnes В. H., Genson J. J. Analyst, 95, 383 (1970).
914. Keilin B., Otvos J. W. J. Am. Chem. Soc., 68, 2664 (1946).
915. Kekch A. Rev. chim. miner., 5, 1 (1968).
916. Kern W., Brauer G. Taianta, 11, 1177 (1964).
917. Kerridge J. F., Kaplan I. R., Petrowski C., Chang S. Geochim. cosmochim. acta, 39, 137 (1975).
918. Ketchum D. F. Analyt. Chem., 36,957 (1964).
919. Kielczewski W., Schneider M., Tomkowiak J. Chem. analit. (Polska), 11, 343 (1966).
920. Kienitz H., Kaiser R. Chem.-Ingr.-Techn., 36, 1187 (1964).
921. Kieruczenko A. Farmac. pol-ska, 22, 583 (1966).
922. Kieselbach K. Analyt. Chem., 16, 764 (1944).
923. Killhefter J. V., Jungermann E.
Analyt. Chem., 31, 581 (1959).
924. Kimoto Sh., Hert W. Mikro-
chim. acta, 1966, 108.
925. Kirshenbaum A. D., Grosse
A. V. Trans. Am. Soc. Metals, 45, 758 (1953).
926. Kirsten W. J., Hozumi K. Mik-rochim. acta, 1962, 777.
927. Kirsten W. J., Hozumi K., Nirk L. Z. analyt. Chem., 191, 161 (1962).
928. Kirtchik H. Analyt. Chem., 37, 1287 (1965).
929. Kiss B. A. Magyar kem. folyo-irat., 75, 302 (1969).
930. Kittner Z., Krdl J. Knizn. od-
284
bor a ved. spis. Vysok u£eni techn. Bme, 5, 263 (1966).
931. Kfeldahl J. G. С. T. Z. analyt. Chem., 22, 336 (1883).
932. Kleinova, J., Kadic K., Re-zag Z. Z.analyt. Chem., 199, 35 (1964).
933. Klingelhoefer W. C. Analyt Chem., 34, 1751 (1962).
934. Knecht E., Hibbert E. New Reduction Methods in Volumetric Analysis, ed. 2. London, Longmans Green, 1925.
935. Knie K. Osterr.-Abwasser-Rundschau, 11, No 4, 58 (1966).
936. Knorr M., Klatte O. J. Arch. Hyd. u. Bakteriol., 147, 94 (1963); РЖХим, 1974, 10И458.
937. Kobilarov N. Tehnika, 21, No 6 (1966); Hem. ind., 20, No 6, 136 (1966) (сербо-хорв.); РЖХим, 1966, 23Г171.
938. Koch О. G., Reinhard R. Z. analyt. Chem., 231, 420 (1967).
939. Koch W., Sauer K.-H. Z. analyt. Chem., 190, 81 (1962).
940. Kodura I., Lada Z. Chem. ana-lit. (PRL), No 4, 871 (1972).
941. Koh T., Iwasaki I. Bull. Chem. Soc. Japan, 40, 569 (1967).
942. Kolthoff I. M. J. Am. Chem.' Soc., 46, 2009 (1924).
943. Kolthoff I. M., Belcher R. Volumetric Analysis, v. 3. New York, Intersci. Publ., 1957, pp. Ill, 525, 582.
944. Kolthoff I. M., Harris W. E., Matsuyama G. J. Am. Chem. Soc., 66, 1782 (1944).
945. Kolthoff I. M., Naponen G. E. J. Am. Chem. Soc., 55, 1448
(1933).
946. Kolthoff I. M., Sandall E. B. Textbook of Quantitative Inorganic Analysis, 3nd ed. New York, Macmillan, 1946.
947. Kolthoff I. M„ Sandall E. B., Moskowitz B. J. Am. Chem. Soc. 55, 1454 (1933).
948. Kolthoff I. M., Stenger V. A. Ind. Eng. Chem., Anal. Educ., 1, 79 (1935).
949. Kolthoff I. M., Stenger V. A. Volymetric Analysis, v. 2. New York, Intersci. Publ., 1947, p. 125.
950. Kolthoff I. M., Stricks W., Morren Z. Analyst, 78, 405 (1953).
951. Komatsu M., Kakiyama H. Japan Analyst, 21, 315 (1972).
952. Komiya H. Ibid., p. 911.
953. Konishi F. Intern. J. Mass Spec-trom. a. Ion Phys., 9, 33 (1972).
954. Kowalska E., Sollorz J. Z. analyt. Chem., 210, 271 (1965).
955. Kowalska E., Sollorz J. Zesz. nauk Politechn. slaskiei, No 108, 53 (1964).
956. Kozak M., Кraj ci P. Chem. prhmusl., 13, 246 (1963).
957. Kraus Th. Schweiz. Arch, an-gew. Wiss. u. Techn., 28, 506 (1962); РЖХим, 1963, 17Д61.
958. Kraus Th., Winkler O. Rev. metallurgie, 61, 87 (1964).
959. Krausz I., Endrei A. H. Magyar kem. folyoirat., 67, 454 (1961).
960. Krawczyk D. F., McLane J., Vakghan R. D. РЖХим, 1968, 10И337.
961. Kreuzer L. B., Kenyon N. D., Patel С. K. N. Sci., 177, 447 (1972).
962. Krug H., Werner E. Abhandl. Dtsch. Akad. Wiss. Berlin. KI. Chem. Geol. u. Biol., No2, 323 (1966).
963. Kruse I. M., Mellon M. G. Analyt. Chem., 25, 1180 (1953).
964. Kubala J. Roczn. Chem., 38, 527 (1964).
965. Kubant J. Sb. praci UVP, 14, 62 (1969); РЖХим, 1969, 16Г116.
966. Kiihnel H. S. Fr., 83 164 (1931).
967. Kiihnert W., Bauer H. J.
Pharmaz. Zentralhalle, 102, 431 (1963); РЖХим, 1964, 10Г160.
968. Kumamoru T., Tao E., Okamoto N., Yamamoto Y. Bull. Chem. Soc. Japan, 38, 2204 (1965).
969. Kuroda D. Kagaku tokoguo (Osaka), 43, 361 (1969).
970. Laffolie H. de. Gias- u. Instrum. Techn., 10, 345 (1966).
971. Laffolie H. de., Lennartz G. DEW-Techn. Ber., 6, 80 (1966); РЖХим, 1967, 19Б73.
972. La Hue D., Axelrod H.D., Lodge J. P. Analyt. Chem., 43, 1113 (1971).
973. Lambert J. L., Manzo D. J. Analyt. Chem., 40, 1354 (1968).
974. Landova M., Machalkova D., Sommer L. Scripta Fac. ski. natur UJEP brun. chem., 2, 129 (1972/ /1973); РЖХим, 1973, 23Г140.
975. Lang H., Freedman R. W. Rept. Invest. Bur. Mines. U. S. Dep. Intern., No 7696 (1972).
976. Lange J. II. de. Chem. e. techn. rev., 21, 636 (1966).
977. Langmuir D., Jacobson R. L. Environ. Sci. Technol., 4, 834 (1970).
978. Lantelme F., Chemla M. Bull. Soc. Chim. France, 1968, 1314.
979. Larrat Ph. Chim. anal. (Paris), 51, 639 (1969).
980. Lasa J. Chem. analit. (Polska), 13, 1071 (1968).
981. Laserna G., Manalo J. Philippine J. Sci., 95, 281 (1968).
982. Lasztity A., Remport-Horvath Zs. Acta chim. Akad. sci. hung., 41, 161 (1964).
983. Latiman H. A., Woerner D. E. Analyt. Chem., 27, 215 (1955).
984. Lawson A., McAdie H. G. J. Chromatogr. Sci., 8, 731 (1970).
985. Lebout H. Ann. med. phys., 5, 196 (1962).
986. Lederer M. Aust. J. Sci., 11, 174 (1949); Цит. по I. M. Hais, K. Macek. Handbuch der Papier-chromatographie. Jena, VEB Gustav Fischer Verlag, 1958, S. 724.
987. Lederer M., Mancini P., Ossi-cini L. J. Chromatogr., 12, 89 (1963).
988. Lederer M., Ossicini L. J. Chromatogr., 15, 514 (1964).
989. Lee G. F. Water a. sewage Works, 116, 38 (1969).
990. Lee J., Sudworth G, B.,Gibson J. Analyst, 89, 103 (1964).
991. Lee R. E., Patterson A. K. At-mosph. Environ., 3, 249 (1969).
992. Legradi L. Acta chim. Acad, sci. hung., 47, 103 (1966); 57, 241, 247 (1968).
992a. Legradi L. Magyar kem. folyoirat., 70, 27, 333 (1964); 73, 525 (1967).
993. Leicknam J. P. Rev. Group, avancem. methodes spectrogr., 4, 319 (1968).
994. Leipzlger F. D., Quidoboni R. J. Appl. Spectroscopy, 21, 165 (1967).
995. Leithe W. Mikrochemie, 33, 149 (1947).
996. Leithe W. Z. analyt. Chem., 202, 102 (1964).
997. Leithe W. Z. analyt. Chem., 251, 185 (1970).
998. Lench A., Martin G. C. Trans. 8th Natl. Vacuum Sympos. a. 2nd Intern. Congr. Vacuum Sci. a. Technol. Washington, D. C., 1961, v.
2. Oxford — London—New York-Paris, Pergamon Press, 1962, p. 776.
285
999. Leonhardt J., Bothe H.-K. Ab-handl. Dtsch. Akad. Wiss. Berlin, KI. Chem. Geol. u. Biol., N 2, 307 (1966).
1000. Leschber R. Galvanotechnik, 58, 462 (1967).
1001. Lesser R. Trans. 8th Natl. Vacuum Sympos. a. 2nd Intern. Congr-Vacuum Sci. a. Technol. Wa.
shington, D. C., 1961. v. 2. Oxford —London — New York — Paris-Pergamon Press, 1962, p. 782, 787, 1002. Levitsky H., Horwitz G. Ana.
lyt. Chem., 34, 1167 (1962).
1003. Lewis D. C. Analyt. Chem., 33, 1127 (1961).
1004. Lingane J. J., Pecsok R. L. Analyt. Chem., 21, 622 (1949).
1005. Lord S. S., Rogers L. B. Analyt. Chem., 26, 284 (1954).
1006. Loubser H. N. Ryneveld W. P. van. Brit. J. Appl. Phys., 18, 1029 (1967).
1007. Luis P., Carducci C. N., Sd A Mikrochim. acta, 1969, 7.
1008. Luis P., Sa A., Mascaro A. Mikrochim. acta, 1968, 44.
1009. Luis P., Sd A. Mascaro A. Mikrochim. acta, 1969, 1.
1010. Lundeen С. V., Viscome A. S., Field T. Analyt. Chem., 45, 1288 (1973).
1011. Lustig E., Duy N., Diehl P., Kollerhals H. J. Chem. Phys., 48, 5001 (1968).
1012. Lilth E. Arch. Eissenhiitten-wesen, 35, 1151 (1964).
1013. Lynn S., Mason D. M., Sage В. H. Ind. Eng. Chem., 46, 1953 . (1954).
1014. Lysyj I. Pat. USA, N 3352644 (1967); РЖХим, 1968, 24Г161П.
1015. Lysyj I., Newton P. R., J. Chromatogr., 11, 173 (1963).
1016. Macejunas A. J. Am. Water Works Assoc., 59, 1190 (1967).
1017. Machova I., Doklddolovd J. Chem. Techn., 19, 767 (1967).
1018. Magee R. J., Shahine S. A., Wilson C. L. Mikrochim. acta, 1964, 1019.
1019. Magno F., Fiorani M. Ri-cerca sci., 38, 119 (1968).
1020. Mahenorappa M. K. Soil Sci., 108, 132 (1969).
1021. Majer J. R. Taianta, 17, 537 (1970).
1022. Majlat P. Acta phann. hung., 41, 278 (1971).
1023. Makino I. J. Ceram. Soc. Ja pan, 79, 312 (1971).
1024. Malamand F. Meth. phys. anal., 5, 227 (1969).
1025. Malissa H. Mikrochim. acta, 1965, 389.
1026. Malmstadt H. V., Vassalo D. A. Analyt. Chem., 31, 206 (1959).
1027. Maty J., Fadrus H. Ceskosl. hyg., 9, 367 (1964).
1928. Maly J., Fadrus H. Ceskosl. hyg., 11, 277 (1966).
1029. Man M., Cristescu L. Rev. chim. (RPR), 14, 602 (1963).
1030. Manahan S. E. Analyt. Chem., 42, 128 (1970).
1031. Mann L. T. Analyt. Chem., 35, 2179 (1963).
1032. Maras L., Szebeni Sz. Acta chim. Acad. sci. hung., 48, 11 (1966).
1033. Marchio J. L. J. Chromatogr., Sci., 9, 432 (1971).
1034. Marcu G. Sacelean V. Rev. roum. chim., 17, 705 (1972).
1035. Marcu G., Sacelean V. Studia Univ. Babes —Bolyai, ser. chem., 14, 19 (1969).
1036. Marianek H. Galvanotechnik, 63, 938 (1972).
1037. Marten J. F. Ind. chim. beige, 32, 1319 (1967).
1038. Martin A. E., Ross P. J. 9th Trans. Intern. Congr. Soil Sci., 3, 521 (1968).
1039. Martin A.P., Everaerts F.M. Analyt. chim. acta, 38, 233 (1967).
1040. Martin R. L., Flannery R. S. Pat. USA, No 3461042 (1969); РЖХим, 1970, 16Л343П.
1041. Mascini M. Analyt. Chem., 45, 614 (1973).
1042. Mason D. M., Taylor L. L., Vango S. P. Analyt. Chem., 27, 135 (1955).
1< 42a. Masson C. R., Pearce M. L. 18th Intern. Congr. Pure a. Appl. Chem., 6—12 Aug. 1961, Monreal, Canada, Rept. С. 1—1.
1043. Masson C. R., Pearce M. L. Trans. Metallurg. Soc. AIME., 224, . 1134 (1962).
1044. Matrka M., Vefisova E., Nav-ratil F. Chem. listy, 58, 1329 (1964).
1045. Matsunada K., Nishimura M. Japan Analyst, 20, 933 (1971).
1046. Mayer S. W., Kelly F. H., Morton M. E. Analyt. Chem., 27, 837 (1955).
286
1047. Mayolet F., Reimers P., Engelmann Ch. J. Radioanal. Chem., 12, 115 (1972).
1048. McConnayghey P. W. Pat. USA, No 3312528 (1967); РЖХим, 1968, 14Л65П.
1049. McGillivray R. Analyst, 87, 833 (1962).
1050. McGrath W. D., Magee R. J., Pickering W. F., Wilson C. L. Ta-lanta, 8, 892 (1961).
1051. McKinney G. L., Lau H. K. Y., Lott P. F. Mikrochem. J., 1972, 375.
1052. Meerkampvan E. J., Gatt F., Kolsek J. Chem. Rundschau, 19, 471 (1966).
1053. Mendelhall A. L., Fraser D. A. Am. Industr. Hyg. Assoc. J., 24, 555 (1964).
1054. Merz W. Z. analyt. Chem., 207, 424 (1965); 237, 272 (1968).
1055. Meyer K. Z. analyt. Chem. 140, 184 (1953).
1056. Meyer R.A., Parry E. P., Davis J. H. Analyt. Chem., 39, 1321 (1967).
1057. Midgley D., Torrance K. Analyst, 98, 217 (1973).
1958. Mihai Fr., Roch B., Moraru A. Bull, ^tiint si tehn. Inst, poli-techn. Timisoara, 12, 441 (1967); РЖХим, 1968, 18Г56.
1059. Miles M. J., Covington L. C. Metallurgie, 5, 367 (1964/1965).
1060. Milham P. J. Awad A. S., Pauli R. E., Bull J. E. Analyst, 95, 751, 758 (1970).
1061. Miller D. C., Winefordner J. D. Mikrochem. J., 8, 334 (1964).
1062. Miller E. H. Analyst, 37, 345 (1912).
1063. Milner G. W., Jones G., Cross-ley D. Res. Group U. K. Atomic Energy Author., No AERE-R 4713 (1964); РЖХим, 1965, 19Г138.
1064. Milner G. W., Jones G. Res. Group U. K. Atomic Energy Author., No AERE-R 5329 (1966).
1065. Milner G.L., Miller E. E. Analyt. Chem., 20, 481 (1948).
1066. Milner G. W. C., Rowe D. H., Fos‘er E., Phillips G.. Res. Group U. K. Atomic Energy Author., No AERE-R 4644 (1964);
РЖХим, 1965, 19Г137.
1067. Milner О. I., Zahner R. J. Analyt. Chem. 32, 294 (1960).
1068. Mirsch E. Fortschr. Wasser-hem., No 2, 120 (1965).
1069. Mitsui T. Microchem. J., 7, 277 (1963).
1070. Miyoshi K., Nakanishi S. New Nippon Elect. Techn. Rev., 7, 143 (1972); РЖХим, 1973, 5И213.
1071. Muzukami S., Miyahara K. Mikrochim. acta, 1962, 705.
1072. Mizutani Y. J. Earth Sci., Nagoya Univ., 10, 125 (1962).
1073. Moberg M. L., Knight W. P., Klndsvater H. M. Analyt. Chem., 28, 412 (1956).
1074. Moc&k J., Bustin D. I., Liako-v& M. Chem. zvesti, 26, 126 (1972).
1075. MUller G. Z. anal. Chem., 245, 155 (1969).
1076. Monar I. Mikrochim. acta, 1966, 934.
1077. Montenegro I., Gruz T., Lourdes M. Rev. port, guim, No 4, 217 (1971).
1078. Montgomery H. A. C., Quarm-ly C. Lab. Pract., 15, 538 (1966).
1079. Moore С. E., Meyer Th. S., Hudson J. W. Taianta, 13, 171 (1966).
1080. Moore F. B., Diehl II. Analyt. Chem., 34, 1638 (1962).
1081. Moore R. F., Harris D. L. Analyt. Chem., 36, 249 (1964).
1082. Morgantini M. J. Chromatogr., 9, 536 (1962).
1083. Morie G. P.,Ledjord С. T., Glover C. A. Analyt. chim. acta, 60, No 2, 397 (1972).
1084. Morris A. W., Riley J. P. Analyt. chim. acta, 29, 272 (1963).
1085. Morris G. F., Carson R. B., Shearer D. A., Jopkiewich W. T. J. Assoc. Offic. Analyt. Chemists, 51, 216 (1968).
1086. Morris G. M. Brit. Pat., No 896082 (1962); РЖХим, 1963, 15Г207П.
1087. Morris M. D., Lingane J. J. J. Electroanal. Chem., 8, 85 (1964).
1088. Morris M. S., Fleck S. A., Lichtenfels D. H. Analyt. Chem., 27, 1565 (1955).
1089. Mosen A. W., Kelley R. E., Mitchell II. P. Taianta, 13, 371 (1966).
1090. Mukherjee L. M., Suwala D., Schultz R. S. Analyt. chim. acta, 60, 247 (1972).
1091. Muller E. P., Freund W. Z. angew. Geol., 8, 304 (1962).
1092. Murakami T. Japan Analyst, 7, 766 (1958).
287
1093. Murakami T. Kogyo kagaku zasshi, 63, 1295 (i960).
1094. Myers R. J. K., Paul E.A. Can. J. Soil Sci., 48, 369 (1968).
1095. Nagasawa M. Bull. Univ. Osaka Prefect., B19, 12 (1964).
1096. Nakagawa Y., Обо F. Acta Med. Univ. Kagoshima, 12, 145 (1970).
1097. Nakamura T. J. Chem. Soc. Japan, Chem. a. Ind. Chem., N 8, 1429 (1972).
1098. Namiki M., Kakila Y., Goto H. Taianta, 11, 813 (1964).
1099. Narain C. J. Mines, Metals a. Fuels, 15, 236 (1967).
1100. Nardo L. U. de. C. r., 188, 563 (1929).
1101. Navome R. J. Am. Water Works Assoc., 56, 781 (1964).
1102. Navome R. J. Am. Waterworks Assoc., 59, 1193 (1967).
1103. Nazaki T., Yazawa, K. Bull. Chem. Soc. Japan, 37, 1891 (1964).
1104. Nergus O. Metallurgia, 18, 684 (1966).
1105. Nicksic S. W., Harkins J. Analyt. Chem., 34, 985 (1962).
1106. Nier A. O. Phys. Rev., 77, 789 (1950).
1107. Nightingale C. F., Walker J. M. Analyt. Chem., 34, 1435 (1962).
1108. Niki H., Warnick A., Lord R. SAE Preprints, s. a., No 710072.
1109. Nikolic K., Popovic R. Glasnik. hem. i tehnol. В i H., 15, 53(1967); РЖХим, 1968, 20Г85.
11 10., Nitric Acid, Fuming Technical Military Specification MIL-N-7254C, U. S. Government Printing Office, Washington, D. C., July 19, 1956.
1111. Nomura T., Takemura S., Nakamura 1'., Komatsu S. Japan Analyst, 22, 576 (1973).
1112. Norris T. A., Tlynn J. £. Analyt. Chem., 37, 152 (1965).
1113. Norton O. R., Mann С. K. Analyt. Chem., 26, 1180 (1954).
1114. Norwitz G. Analyt. Chem., 34 227 (1962).
1115. Novak J., Benes R. Пат. ЧССР, No 139138 (1970); РЖХим, 1972, 7Г115П.
1116. Nozaki T., Okuo T., Aku'su H., Fukurawa M. Bull. Chem. Soc. Japan, 39, 2685 (1966).
1117. Nozaki T., Yatsurugi Y., Akiya-ma N. J. Radioanaly.t., 4, 87 (1970).
1118. Nyman C. J., Johnson K. A, Analyt. Chem., 29, 483 (1957).
1119. Obermiller E. L., Charlier G. O. J. Gas Chromatogr., 7, 580 (1969).
1120. Obermiller E. L., Charlier G. O.
J. Gas Chromatogr., 6, 446 (1968).
1121. Obermiller E. L., Freedman R. W. J. Gas Chromatogr., 3, 242 (1965).
1122. O’Brieu J. E., Fiore J. Wastes Engn, 33, 238 (1962).
1123. Oda Sh., Ohashi Z., Furuya K., Kamada H. Taianta, 19, 779 (1972).
1124. Odler I. Analyt. Chem., 41, 1116 (1969).
1125. Oehme F. Chem. Rundschau, 21, 895 (1968); Metalloberflache, 19, 273 (1965).
1126. Oehme F. Gias- u. Instrum.-Techn., 10, 12, 17, 23, 28 (1966).
1127. Oehme F. Gias.- u. Instrum.-Techn., 11, 16 (1967).
1128. Oehme F., Wyden H., Hegner F. Galvanotechn. u. Oberflachenschutz, 6, 57 (1965).
1129. Okamoto N. J. Sci Hiroshima Univ., ser. A, Div. 2, 32 . 309(1968).
1130. Okuno I., Latham D. R., Haines W. E. Analyt. Chem., 37, 54 (1965).
1131. O’Laughlin J. W., Jlusn D. F. et al. Analyt. Chem., 40, 1931 (1968).
1132. Olson E. C. Analyt. Chem., 32, 1545 (1960).
1133. Ono K. Annual Rept. Sankyo Res. Labs., 19, 62 (1967).
1134. Ono K., Masuda H. Annual Rept. Sankyo Res. Labs., 17, 57, 72 (1965).
1135. Ordogh M., Atomtechn. tajekoz-tato, 11, 123 (1968); РЖХим,
1969, 2Г164.
1136. Orr С. I., Kordecki M. C. Appl. Spectroscopy, 15, 180 (1961).
1137. Orsos K. Szabvanyositas, 24, 304 (1972); РЖХим, 1973, 10И218.
1138. Orsos S. Egeszsegtudomang, 16, 388 (1972); РЖХим, 1973, 11И613.
1139. Orth R. C., Land H. B. J. Chromatogr., Sci., 9, 359 (1971).
1140. Oshry H. I. Proc. Intern. Conf, mod. Trends Activat. Analysis. College Station Texas, 1961, p. 2831; РЖХим, 1963, 9Д42.
1141. Ossicini L. J. Chromatogr., 9, 114 (1962).
1142. Overfield С. V., Winefordner J. D. J. Chromatogr., 30, 339 (1967).
1143. Paesold G., Muller K., Kieffer R. Z. analyt. Chem., 232, 31 (1967).
288
1144. Paltt S. R., Das M. N. Non-Agueous Titration. Calcutta, Indian Assoc. Cultivation Sci., 1954.
1145. Paluch J., Siekierzynska H. Pr.
i stud. Zakl. badan nauk. gomoS-Iqsk okrggu przemysl. PAN, N 6, 48 (1970); РЖХим, 1973 , 6И669.
1146. Pappalardo S. Chimica e Industrie, 46, 424 (1964).
1147. Pappenhagen J. M. Analyt. Chem., 30, 282 (1958).
1148. Pappenhagen J. M., Mellon M. (I. Analyt. Chem., 25, 341 (1953).
1149. Parekh K. J. M., Oza P. M. Salt Res. a. Ind., 5, No2, 44 (1968).
1150. Parker A., Healy C. Analyst, 95, 204 (1970).
1151. Parkinson R. T., Toft L. Analyst, 90, 220 (1965).
1152. Parkinson R. T., Wilson R. E. Taianta, 15, 931 (1968).
1153. Parsons M. L., Pennington S. N Walker J. M. Analyt. Chem., 35, 842 (1963).
1154. Paul A.D., Gibson J. A. J. Chem. Educ., 40, 417 (1963).
1155. Paul L., Carlson R. M. J. Agr. Food. Chem., 16, 766 (1968).
1156. Paul S. D., Krishnan M. S. Indian J. Chem., 3, 230 (1965).
1157. Pearce M. L., Trans. Metallurg. Soc. AIME, 227, 1393 (1963).
1158. Pearce M. L., Masson C. R. Iron a., Steel Inst., London, Spec. Rep., No68, 121 (1960); C. A. 55, 10196a.
1159. Penland T. L., Fischer G. Me-talloberflache, 26, 391 (1972).
1160. Penneman R. A., Andrieth L. F. Analyt. Chem., 20, 1058 (1948).
1161. Pennington S., Meloan C. Analyt. Chem., 39, 119 (1967).
1162. Perschke H., Keroe E., Proksch G., Muehl A. Analyt. chim. acta, 53, 459 (1971).
1163. Peterson С. O., Amrhein W. G. Analytical Instruments, v. 5, New York, 1968, p. 101.
1164. Pierson R. H., Fletcher A. H., Gantz E. S. Analyt. Chem., 28,1219 (1956).
1165. Pietrogrande A. Mikrochim. acta, 1964, 1106; Ann. chimica, 54, 843 (1964).
1166. Pietrogrande A., Dalia F. Farmace Educ. pract., 22, 723 (1967).
1167. Pippel G., Romer S. Chem. Techn., 15, 173 (1963).
1168. Piter C. W-, Wollish E. G. Analyt. Chem., 24, 519 (1952).
1169. Pittke E.-Ch. Messtechnik, 81, 77 (1973).
1170. Platte J. A. Pat. USA, N03195983 (1965); РЖХим, 1966, 17Г120П.
1171. Piecity V. Пат. ЧССР, No 139153 (1970); РЖХим, 1972, 8Г224П.
1172. Pohloudek-Fabini R., Gockeritz D., Schtifiler M. Mikrochim. acta, 1963, 668.
1173. Pokorny G., Likussar W. Analyt. chim. acta, 42, 253 (1968).
1174. Pollard F. H., McOmie J. F. W., Stewens H. M. Sch. Sci. Rev., 34, 21 (1952); Цит. по I. M. Hais, K. Macek. Handbuch der Papier-chromatogrphie. Jena, VEB Gustav Fischer Verlag, 1958, S. 724.
1175. Pollard F. H., Warrneder R. W., Nickless G. Some General Problems Paper Chromatographie. Praque, Chechosl. Acad. Sci., 1962, p. 175.
1176. Pommier C. Rev. chim. miner., 3, 401 (1966).
1177. Popa I., Stan T. Farmacia (RPR), 10, 745 (1962).
1178. Poppalardo S. Chimica e Industrie, 46, 424 (1964).
1179. Porkert H. Gias- u. Instrum.-Techn., 13, 557 (1969).
1180. Poschenrieder W. P., Warneck P. Analyt. Chem., 40, 385 (1968).
1181. Potrafke K.A., Kroll M., Blom L. Analyt. chim. acta, 31, 128 (1964).
1182. Potterson S. S., Shulls W. D. Anal. Lett., 1, 11 (1967).
1183. Prasad N. K., Asthana S. S. Chem. a. Ind., 1969, 262.
1184. Pregl F. Die quantitative or-ganische Mikroanalysc, 3 Aufl. Berlin, 1930.
1185. Prochazkova L. Analvt. Chem., 36, 865 (1964).
1186. Product. Group U. K. Atomic Energy Author., N Rept. 381 (S) (1962); N Rept. 412 (S) (1962); РЖХим, 1964, 9Г133, 9Г154.
1187. Proksch E., Bildstein H. Osterr. Chem. Ztg., 65, 41 (1964).
1188. Proksch G. Plant Soil, 31, 380 (1969).
1189. Propellant Hydrazin. Military Specification MIL-P-26536 B. U. S. Government Printing Office. Washington, D. C., March 3, 1964.
289
1190. Propellant Nitrogen Tetraoxide, Military Specification MIL-P-26539 B. U. S. Government Printing Office. Washington, D. C., October 15, 1965.
1191. Purer A. I., Seitz C.A. Analyt. Chem., 36, 1694 (1964).
1192. Qien A., Selmer — Olsem A. К. Analyst, 94, 888 (1969).
1193. Quon J. E., Lagvankar A. L. Intern. J. Air. a. Water Pollut., 9, 487 (1965).
1194. Qureshi M., Rawat J. P. Chemist-Analyst, 56, 89 (1967).
1195. Radford H. E., Evenson К. M. Phys. Rev., 168, 70 (1968).
1196. Rambeau G. Chim. anal. (Paris), 52, 774 (1970).
1197. Rappne S. S. Magyar allami-foldt invevigelencse, 1961, 377; РЖХим, 1963, 22Г93.
1198. Razlunas V., Loseke W. A., Grove E.L. Appl. Spectrosc., 20, 395 (1966).
1199. Reactor Mater., 9, No 4, 217 (1966—1967); РЖХим, 1967,19Л10.
1200. Reddy D.V., Rao B., Raju N. A. Z. analyt. Chem., 244, 123 (1969).
1201. Rees M., Nason A. Intern. J. Appl. Radiat. a. Isotopes, 16, 495 (1965).
1202. Reid D. J. Sci. Proc. Roy Dublin Soc., A3, No 6, 69 (1967); РЖХим, 1968 9Д26.
1203. Rei:h J. F., Bowman J. U. A. Pharm. Weekblad, 67, 475 (1930).
1204. Respi G., Pergola F, Chimica e Industrie, 45, 1398 (1963).
1205. Rexroad P. R., Krause G. F. J. Assoc. Offic. Anal. Chem., 53, 450 (1970).
1206. Rezdc Z., Kadic K. Z. analyt. Chem., 190, 305 (1962).
1207. Ricci E., Hahn R. L. Trans. Am. Nucl. Soc., 9, 104 (1966); Analyt. Chem., 37, 742 (1965); 39, 794 (1967).
1208. Ricci E., Hahn R. L., Strain J. E. Dyer F. F. Proc. Intern. Conf. mod. Trends Activat. Analysis. College Station Texas, 1965, No 200.
1209. RiceD. D., TrowellJ. M., Analyt. Chem., 39, 157 (1967).
1210. Richards F.A., Kleisch R.A. Recent Research Fields hydrosphere, atmosphere and nuclear geochemistry. Tokyo, Kenkyusba, 1964, p. 65.
290
1211. Ridden J. H. C., Barefoot R. R., Roy J. G. Analyt. Chem., 43, 1109 (1971).
1212. Rider B. F., Mellon M. G. Ind. Eng. Chem., Anal. Educ. 18, 96 (1949).
1213. Riedmann H., Eichner M. Pat. BRD, N 2108610 (1973); РЖХим, 1973,19Д67П.
1214. Riehm H. Z. analyt. Chem., 81, 439 (1930).
1215. Rison M. H., Barber W. H., Wilkniss P. E. Radiochim. acta, 7, 196 (1967).
1216. Riva M., Carisano A. Cron, chim., No 26, 3 (1969).
1217. Roberson С. E., Austin С. M. Analyt. Chem., 29, 854 (1957).
1218. Roberts A.G. Analyt. Chem., 21, 813 (1949).
1219. Robin J., Bazouin M., Blau C., Doumeng M. Rev. Group, avan-cem. methodes'spectrogr., \4, 1061 (1968).
1220. Robinson D. Proc. Soc. Analyt. Chem., 2, 142 (1965).
1221. Roboz J. Analyt. Chem., 38, 1629 (1966); 39, 175 (1967).
1222. Rodriguez A. R., Urgell M. M., Iglesias J. An. quim. Real Soc. esp. fis. ygium.,67,289 (1971); РЖХим, 1972, 24Б808.
1223. Roehrlng J. R., Simons J. C. Pat. USA, No 3065060 (1962).
1224. Roff J. A. Chem. a. Ind., 1963, 537.
1225. Romand J., Berneron R. Publ. Group, avancem. methodes spect-rogr., N 4, 327 (1963).’
1226. Romand J., Vodar B. Proc. Intern. Conf. Spectroscop., Bombay, Jan. 9—18, v. 2, sec. 1, p. 544, 1967; РЖХим, 1968, 16Г120.
1227. Rammers P. J., Visser J. Analyst, 94, 653 (1969).
1228. Rosenberg G. An. Fac. guim. у farmac. Univ. Concepcion, 11, 56 (1960); РЖХим, 1966, 1Г45.
1229. Roth E., Cornu A., Albert Ph. Z. analyt. Chem., 218, 24 (1966).
1230. Rothman H. Z. Erzbergbau u. Metallhiittenwesen, 20, No 10, 469 (1967).
1231. RottmannJ., Nickel H. Z. analyt. Chem., 247, 208 (1969).
1232. Rowe D. J. Gas J., 265, 49 (1951).
1233. Roy A. K., Pandey A. D. Indian J. Chem., 2, 204 (1964).
1234. NASA Spec. Publ. N SP-261, 221 (1971); C. A., 75, 9616 m.
1235. Rupe Ch. O. Pat. USA, N 3329486 (1967); РЖХим, 1968, 19Г14П.
1236. Rupe H., Becherer F. Helv., 6, 674 (1923).
1237. Sa A., 'Carducci C. N., Luis P. Mikrochim. acta, 1968, 53.
1238. Sabo J. G., Chemist-Analyst, 54, 110 (1965).
1239. Sadek F. S., Reilley C. N. Analyt. Chem., 31, 494 (1959).
1240. Sadowsky S. Roczn. Panstw. Zakl. hig., 20, 527 (1969); РЖХим, 1970, 13Г235.
1241. Saier E. I., Pozelsky A. Analyt. Chem., 26, 1079 (1954).
1242. Saito G., Sugimoto K., Hagi-no K. Japan Analyst, 20, 542 (1971).
1243. Safe I., Sipos B. Taianta, 19, 669 (1972).
1244. Sakata K., Matsuo S. Geochem. J., 6, 49 (1972).
1245. Sandhu S. S., Dhill Sh. S., Singh D. Indian J. Chem., 5, 434 (1967).
1246. Sansoni B., Stolz R. Angew. Chem., 75, 418 (1963).
1247. Sant B. R. Z. analyt. Chem., 145, 257 (1955).
1248. Santacesaria E., Guiffre L. Riv. Combust., 23, 438 (1969); C. A., 72, 45643q.
1249. Saran J., Khanna P. N., Ba-nerje S. Mikrochim. acta, 1972, 252.
1250. Sarudi I. Elelmiszerivzsg. kozl, 14, 47 (1968); РЖХим, 1968, 23Г125; Gesundh.-Ingr., 89, 86 (1968); РЖХим, 1968, 15Г173.
1251. Sato S., Tate K. J. Iron a. Steel Inst. Japan, 55, 366 (1969).
1252. Savelsberg W. Fachber. Oberf-lachen techn., 6, 63 (1968);
РЖХим, 1969, 4И544.
1253. Savic M. Glasnik hem. i teh-nol., В i H, 16, 51 (1968).
1254. Sawicki C. R., Scaringelli F. P. Microchem. J., 16, 657 (1971).
1255. Sawicki E., Johnson H., Stanley T. W. Analyt. Chem., 35, 1934 (1963).
1256. Sawicki E., Pfaff J., Stanley T. W. Rev. Univ, industr. san-tauder, 5, 337 (1963); РЖХим, 1964, 5П26.
1257. Sawicki E., Stanley T. W., Elbert W. C. Analyt. Chem., 34, 297 (1962).
1258. Sawluk T. Galvanotechnik, 64, 196 (1973).
1259. Scales F. M., Harrison A. P. Ind. Eng. Chem., 16, 571 (1924).
1260. Schaaf M., Rolle W. Isotopen-praxis, 8, 170 (1972).
1261. Schach H.-G. Limnologica, 4, 431 (1966).
1262. Scheurer P. G., Smith F. Analyt. Chem., 27, 1616 (1955).
1263. Schmidbauer H. J. Am. Chem. Soc., 85, 2336 (1963).
1264. Schneider W. Wiss. Z. Techn. Univ. Dresden, 12, 871 (1963).
1264a. Schoenheimer R., Ratner S., Rittenberg D. J. Biol. Chem., 130, 703 (1939).
1265. Schaler R. P., Simon W. Chi-mia, 24, 372 (1970).
1266. Schuller P. L., Уее1г E. J. Assoc. Offic. Analyt. Chemists, 50 1127 (1967).
1267. Schuy K. D. Z. Instrumenten-kunde, 75, 190 (1967).
1268. Schuy K. D., Franzen J., Hin-tenberger II. Z. Naturforsch., 19a, 153 (1964).
1269. Schwabe K., Berndt T. Pat. DDR, N 56655 (1967); РЖХим, 1969, 21И296П.
1270. Scibona G., Scuppa B. J. Phys. Chem., 68, 2003 (1964).
1271. Segui P. Chimica, 38, 297 (1962).
1272. Semel S., Helf S. J. Radioa-nal. Chem., 11, 91 (1972).
1273. Senegacnik M., Klafutar C., Palfk S., Smrekar L. J. Inorg. Nucl. Chem., 32, 1659 (1970).
1274. Sevcik J. Chromatographia, 5, 195 (1971).
1275. Severus-Laubenfeld H., Steiger J., Stahel R. Z. analyt. Chem., 21$, 241 (1966).
1276'. Shah R. A., Qadri A. A. Taianta, 10, 1083 (1963).
1277. Shaw E. C., Wiley P. Calif. Agr., 5, 11 (1969).
1278. Shearer G. B., Kohl D. H., Commoner B. Soil Sci., 118, 308 (1974).
1279. Shimizu R., Murata K., Shi-noda G. Technol. Repts. Osaka Univ., 16, 131 (1966).
1280. Shipley D. E. J. Inst. Fuel, 37, 510 (1964).
1281. Shiurba R. A., Jolly W. L. J. Am. Chem. Soc., 90, 5289 (1968).
1282. Shulek E., Vastagh G. Fr., 92, 352 (1933).
291
1283. Shulz W., Seehofer F. Beitr. Tabak forsch., 4, lol (1968).
1284. Siddiqui M. A., Bhatty M. K. Taianta, 15, 559 (1968).
1285. Sidgwick N. V. The Chemical Elements and their Compounds, v. 1. Oxford, 1951, p. 654.
1286. Simenauer A. Acta chim. Acad. sci. hung., 33, 295 (1962).
1287. Simon M. Trait, surface, 7, No 64, 9 (1966); РЖХим, 1967, 15И230.
1288. Simpson R. J. Metal Finish. J., 18, 265 (1972); РЖХим, 1973, 4Л273.
1289. Singh B., Rehmann A. J. Indian Chem. Soc., 17, 169 (1940).
1290. Singh B., Singh R. Analyt. Chim. acta, 11, 408 (1954).
1291. Singh D., Sherma Sh. Indian J. Chem., 8, 192 (1970).
1292. Singh E., Kashyap G. P. Z. analyt. Chem., 163, 338 (1958).
1293. Sisler H. H. Comprehensive Inorganic Chemistry, v. 5. Toronto—London—New York, D. van Nostrand Company, Inc., 1959, p. 11—100.
1294. Sloan W. J. Pat. USA, No 3656893 (1972); РЖХим, 1973, 2Л52П.
1295. Slobod R. L., Krogh M. E. J. Am. Chem. Soc., 72, 1175 (1950).
1296. Smith A. B. Pat. USA, No 3681027 (1972); РЖХим, 1973, 11Д116П.
1297. Smith A. I., Myers G., Cha-ner W. C. Mikrochim. acta, 1972, 217.
1298. Smith G. F., Wilcox C. S. Ind. Eng. Chem., Anal. Educ., 14, 49 (1942).
1299. Smith V. N., Fidiam J. F. Analyt. Chem., 36, 1739 (1964).
1300. Soczewinski E., Urban T., Wolski T. Chem. analit. (Polska), 12, 1231 (1967).
1301. Solbrig P., Saffert W., Schuberth H. Chem. Techn., 16, No 12, 745 (1964).
1302. Solomon P. J. Chromatogr., 30, 593 (1967).
1303. Somiya T., Hiran Sh., Ko-mada H., Ogahara I. Taianta, 11, 581 (1964).
1304. Sommerfeldt T. G., Milne R. A., Kozub G. C. Commun. Soil Sci., Plant. Anal., 2, 415 (1971).
1305. Sparks R. J., McCallum G. J.
292
N. Z. J. Sci., 12, 470 (1969); РЖХим, 1970, 9Г159.
1306. Spencer N. W., Reber C. A. Space Res., v. 3. Amsderdam, N.-Holland Publ. Co; New York, Wiley, 1963, p. 1151.
1307. Sperner F. Metallurgie (Belg.), 5, 355 (1964—1965).
1308. Sperner F., Koch K. Metall, 19, 742 (1965).
1309. Sprinson D. B., Rittenberg D. J. Biol. Chem., 180, 707 (1949).
1310. Stability Constants of Metallon Complexes, Special Publication, Nol7, Sect. I: Inorganic Ligands. London, The Chemical Society, 1964, p. 149.
1311. Staley H. G., Svec H. J. Analyt. chim. acta, 21, 289 (1959).
1312. Steel A. E. Nature, 173, 315 (1954).
1313. Stevens H. M. Analyst, 91, 743 (1966).
1314. Stewart B. A., Porter L. K., Beard W. E. Analyt. Chem., 35, 1331 (1963).
1315. Stock J. T., Bjork R. G. Mikrochem. J., 6, 219 (1962).
1316. Stock J. T., Bjork R. G. Taianta, 11, 315 (1964).
1317. Stockdale O. Analyst, 84, 667 (1959).
1318. Staffer R. Mikrochim. acta, 1972, 242.
1319. Stokstad E. L. R. Pat. USA, No 3174829 (1965); РЖХим, 1966, 10D88II.
1320. Stringat R., Floret A., Janin J. Rev. Group, avacem. methodes spectrogr., jan.-mars, 79 (1966).
1321. Strnad Z. Collect. Czech. Chem. Comm., 31, 3399 (1966).
1322. Strzondala V. Chem. pru-mysl., 14, 409 (1964).
1323. Stubblefield F. M. Ind. Eng. Chem., Anal. Educ., 16, 366 (1944).
1324. Sub B. W., HemkeH. Pharmaz. Zentralhalle, 106, 86 (1967).
1325. Sunghal J. P., Samiullah K., Gupta G. K. J. Indian Chem., Soc., 50, 23 (1973).
1326. Suttle E. T., Emerson D. E., Burfield D. W. Analyt. Chem., 38, 51 (1966).
1327. Svajgl O. Ropa a uhlie, 9, 220 (1967); РЖХим, 1968, ЗП220.
1328. Swift H. Mikrochem. J., 11, 193 (1966).
1329. Swinburn D. C., Melia P. J. Metallurgia, 81, 37 (1970).
1330. Szebelledy L., Somogyi Z. Z. analyt. Chem., 112, 400 (1938).
1331. Szekely E. Israel J. Chem., 4, 14 (1966).
1332. Szekely E. Taianta, 14, 941 (1967).
1333. Szekely E. Taianta, 15, 795 (1968).
1334. Szekielda K.-H. Chem. Labor, u. Betrieb., 13, 194 (1962).
1335. Szekielda K.-H. Ibid., p. 233.
1336. Tabata M., Funakashi Sh., Tanaka M. Analyt. chim. acta, 62, 289 (1972).
1337. Takeuchi T., Suzuki M., Ishii D. Mem. Fac. Engn. Nagoya Univ., 15, 1—51 (1963).
1338. Takitani Sh., Fukuoka N. et al. Japan Analyst, 15, 840 (1966).
1339. Tamura M. Genshiryoku ko-gyo, 16, 68 (1970); C. A., 74, 33246y.
1340. Tamura M. Radioisotopes, 18, 252 (1969).
1341. Tandon K. N., Merofra R. C. Analyt. chim. acta, 30, 407 (1964).
1342. Tant M. Kogaito taisaku, 8, 551 (1972); РЖХим, 1972, 24И678.
1343. Taylor B. L., Perrett B. S. Analyst, 92, 64 (1967); Res. Group U. K. Atomic Energy Author., No AERE-R 5247 (1967).
1344. Taylor R. C., Brown R. A., Young W. S., Hedington С. E. Analyt. Chem., 20, 396 (1948).
1345. Thielemann H. Mikrochim. acta, 1972, 28.
1346. Thompson J. E., Morrison G. R. Analyt. Chem., 23, 1153 (1951).
1347. Thiirauj W., Thiemann W. Brennstoff-Chemie, 44, 275 (1963).
1348. Tiwari P. N., Bergman R., Larsson B. Intern. J. Appl. Radiat. a. Isotopes, 22, 587 (1971).
1349. Tobia S. K., Gawargious Y. A., El-Shahat M. F. Taianta, 20, 513 (1973).
1350. Tokuoka M., Ruzicka J. Collect. Czech. Chem. Comm., 6, 339 (1934).
1351. Tokutomi M., Komiya S. Japan Analyst, 21, 81 (1972).
1352. T8lg G. Z. analyt. Chem., 205, 40 (1964).
1352a. Tolgyessy J., Braun T., Kyrs M. Isotope Dilution Analysis. Budapest, Akademiai Kiado, 1972, pp. 155—169.
1353. Tribalat S. C. r. 256, 5586 (1963).
1354. Trnjanln E. Remija u indust-riji, 12, 451 (1963); РЖХим, 1964, 7Г105.
1355. Trowell J. M. Analyt. Chem., 37, 1152 (1965).
1356. Truesdale V. W. Analyst, 96, 584 (1971).
1357. Tuchscheerer Th. Chemiker-Ztg., Chem. Apparat, 88, 111 (1964).
1358. Tustanowski S. J. Chromatogr., 31, 266 (1967).
1359. Uchikawa S. J. Sci. Hiroshima Univ., ADW 2, 33, 169 (1969).
1360. Uehara H., Arimitsu S. Analyt. Chem., 45, 1897 (1973).
1361. Uhlik L., Kuca Z. Huth, lis-ty, 23, 364 (1968).
1362. Ultee C. J. J. Chem. Phys., 43, 1080 (1965).
1363. Underwood A.L., Miller M. W., Howe L. H. Analyt. chim. acta, 29, 79 (1963).
1364. Urban W. C. Analyt. Chem., 43, 800 (1971).
1364a. Ussing H. H. Nature, 144, 977 (1939).
1365. Valente Soares M. I. Santos Pereira P. G., Antunes A. M. Rovta port. Quim., 13, 151 (1971).
1366. Van Huyssteen J. J. Water Res., 1, 237 (1967).
1367. Van Leuven H. С. E. Analyt. chim. acta, 49, 364 (1970).
1368. Уесега M. Mikrochim. acta, 1962, 896.
1369. Vecera M. Microchem. J., 10, 250 (1966).
1370. Vernotte Ph. Franc. pat., N 89056 (1967); РЖХим, 1968, 20Г41П.
1371. Vidal G., Galmard P., Lanus-se P. Analyt. Chem., 42, 98 (1970).
1372. Vidal G., Galmard P., Lanus-se P. Chim. analyt., 50, 369 (1968); Rech, aerospat., No 121, 33 (1967).
1373. Vidal G., Galmard P., Lanus-se P. Rech. a6rospat., No 130, 27 (1969).
1374. Vizard G. S., Wynne A. Analyst, 87, 810 (1962).
1375. Vokdc Z. Jenaer Rundschau, 12, 113 (1967).
1376. Vorlman G., Binder F. Z. analyt. Chem., 67, 269 (1925).
1377. Vrga V., Tomasevic M. Metallurgia, 9, No 3, 19 (1970).
1378. Wada E., Hattori A. Analyt. chim. acta, 56, 233 (1971).
293
1379. Wagner R. Vom Wasser, Bd. 36. Weinheim/Bergstr., 1969, S. 263.
1380. Walisch W. Pat. USA, No3241922 (1966); РЖХим, 1967, 10Д73П.
1381. Walters I. P. Analyt. Chem., 40, 1540 (1968).
1382. Walters С. I., Taylor A. McM. Biochim. Biophys. Acta, 82 , 423 (1964).
1383. WaVers G. K., Estle T. L. J. Appl. Phys., 32, 1854 (1961).
1384. Wankat C., Keyworth D. A., Brand V. A. Analyt. Chem., 35, 1090 (1963).
1385. Watts G. W., Chrisp I. D. Analyt. Chem., 24, 2006 (1952).
1386. Wayman D. H. Analyt. Chem., 28, 865 (1956).
1387. Webb M. S. W., Webb R. J. Analyt. chim. acta, 36, 403 (1966).
1388. Weinryb E., Hourler P. Chim. anal. (Paris), 48, 219 (1965).
1389. Weiss D. Analytical Chemistry and the Environment. Resume I. Intern., 9—12 Sept., 1974. Prague, The Czechoslovak Soc. For Sci. a. Technology, 1974, p. 21.
1390. Weiss H. G., Balls D. F. Analyt. chim. acta, 55, Nol (1971).
1391. Weiss R. F., Craig H. Deep-Sea Res., 20, 291 (1973).
1392. Weitkamp H., Korte F. Chem.-Ingr.-Techn., 35 , 429 (1963).
1393. Welcher F. J. Organic Analytical Reagents, v. 1. New York, Van Nostrand, 1947, p. 376.
1394. Wellings A. W. Trans. Faraday Soc., 23, 665 (1932).
1395. Wentworth W. E., Free-
man R. R. J. Chromatogr., 79, 322 (1973).
1396. Wernberg O., Lamm C. G., Nielsen T. J. Chromatogr. Sci., 9, 373 (1971).
1397. West Ph. W., Ramachand-ran T. P. Analyt. chim. acta, 35, 317 (1966).
1398. Westland A. D., Landford R. R. Analyt. Chem., 28, 1996 (1956).
1399. Wheeler Ph. P. Faith M. J. Microchem. J., 9, 309 (1965).
1400. Wiersma J. H. Anal. Lett., 3, 123 (1970).
1401. Wilkinson J., Hall D. J. Chromatogr., 10, 239 (1963).
1402. Willard H. H., Yung P. J. Am. Chem. Soc., 45, 3260 (1923); 50, 1379 (1928); 51, 139 (1929).
1403. Williams D. D., Miller R. R. Analyt. Chem., 34, 225 (1962).
1404. Williams E. F., Nash Th. P. J. Biol. Chem., 100, 737 (1933).
1405. Williams F. W., Findeis A. F. Analyt. Chem., 37, 857 (1965).
1406. Williams H. P., Wineford-ner J. D. J. Gas Chromatogr., 6, 11 (1968).
1407. Williams P. C., Analyst, 89, 276 (1964).
1408. Williams Ph. Lab. Pract., 22, 38 (1973).
1409. Williams W. G., Carritt D. E. Analyt. Chem., 44, 2119 (1972).
1410. Williamson R. C., Russee J. A. SAE Preprints, s. a., No 670945; РЖХим, 1968, 19И229.
1411. Willis R. R. Metallurgia, 79, No 473, 129 (1969).
1412. Wineburg J. P. Analyt. Chem., 40, 1744 (1968).
1413. Winge R. K., Fassel V. A. Analyt. Chem., 41, 1606 (1969).
1414. Wohlleben K., Schuster E. Radiochim. acta (BRD), 12, 75 (1969).
1415. Wolf B. Ind. Eng. Chem., Anal. Educ., 19, 334 (1947).
1416. Wolski T. Roczn. chem., 43, 209 (1969).
1417. Wood D. F., Wolfenden G. Analyt. chim. acta, 38, 385 (1967).
1418. Wood E. D., Armstrong F. A. J., Richards F. A. Marine Biol. Assoc. U. K., 47, 23 (1967);
РЖХим, 1967, 15П81.
1419. Woodbridge J. E. Pat. USA, No 3467499; РЖХим, 1970, 18Г94П.
1420. Wootis T. C., Cummings J. M. J. Assoc. Offic. Anal. Chem., 56, 373 (1973).
1421. Wright С. M., Orr A. A., Balling W. J. Analyt. Chem., 40, 29 (1968).
1422. Wright H. Explosivstoffe, 16, 176 (1968).
1423. Wronski M. Chem. analit.
(Polska), 9, 169 (1964).
1424. Wulfhekel H., Schaal G. Exptl Techn. Phys., 10, 231 (1962); РЖХим, 1963, 12Д55.
1425. Yamamoto Y., Okamoto W., Tao E. Analyt. chim. acta, 47, 127 (1969).
1426. Yamamoto Y., Uchikawa S., Akabori K. Bull. Chem. Soc. Japan, 37, 1718 (1964).
294
1427. Yashiki M., Deguchi M. Japan Anal., 20, 1192 (1971).
1428. Yatabe T. J. Environ. Pollut. Contr., 9, 233 (1973).
1429. Zaleiko N. S. Industr. Water Engng, 4, 32 (1967).
1430. Zdenek V. Chem. listy, 45, 293 (1951).
1431. Ziegler M., Glemser O. Z. analyt. Chem., 144, 187 (1955).
1432. Zielinski E. Chem. analit. (Polska), 8, 399 (1963).
1433. Zinggl H., Simmingskold B. Glastekn. tidskr., 20, 29 (1965); РЖХим, 1965, 24Г231.
1434. Zschiesche M., Maass I. Isoto-penpraxis, 9, 298 (1973).
1435. Zumbrunn J.-P. Surfaces, 12, No 7835, 41 (1973); РЖХим, 1973, 21Г179.
1436. Zuse M. Galvanotechnik, 57, 500 (1966).
1437. Zwarowska Z. Zesz. nauk, WSE. Krakowie, No 55, 81 (1972);
РЖХим, 1973, 17P95.
Дополнительная литература
1438. Аграпов X. И,, Некрашевич М. А., Парфенов В. Н., Рейтан Л. В. Зав. лаб., 34, 1445 (1968).
1439. Арнаутов И. В. Материалы 2-го Сибирского совещания по спектроскопии. Иркутск, изд. Инта геохимии СО АН СССР, 1967.
1440. Басаргин Н. Н., Чернова Е.А. Ж. аналит. химии, 23 102 (1968).
1441. Березин А. К., Витоженц Г. Ч., Сулин В. В., Шорников С. И. Radiochemical Methods of Analysis,’ v. 1. Vienna, Intern. Atomic Energy Agency, 1965;
РЖХим, 1966, 2Г107.
1442. Больберг H. Ш., Егорова Е. Д. Труды Главной геофиз. об-серв., вып. 293, 93 (1973).
1443. Горбачев В. М., Соболев Е. В. Изв. СО АН СССР, серия хим. наук, вып. 3, 161 (1970).
1444. Дмитриев М. Г., Соловьева Г. В., Сербина Л. П., Фомин'! С. А. Гигиена и санитария, № 9, 60 (1971).
1444а. Ильин В. Е., Соболев Е. В. Ж. прикл. спектроскопии, 10, 991 (1969).
1445. Кудеяров В. Н. Авт. свид. СССР, № 217685 (1968); Бюлл. изобр., № 16 (1968).
1446. Кудеяров В. И. Агрохимия. № 9, 127 (1970); Химия сельск, хоз., № 8(9), 708 (1970).
1447. Лаптева А. С., Новиков В. А., Бондаревская Е. А. Зав. лаб., 38, 32 (1972).
1448. Майборода Н. М. Почвоведение, № 2, 100 (1961).
1449. Мекеев Е. Е., Смирнова Г. Д. Изв. АН КазССР, серия хим., 19, № 5, 87 (1969).
1450. Могилевкина И. А. Почвове дение, № 8, 139 (1970).
1451. Новиков В. А., Бондаревская Е. А., Франгулян Л. А. Зав. лаб., 38, 925 (1972).
1452. Сб. «Исследование и разработка фотометрических методов для определения микроколичеств элементов в минеральном сырье». Алма-Ата, изд. КАЗИМС, 1967, стр. 164.
1453. Alm К. F., Andersson L. Н. FOA Repts, No 10, 6 (1972); РЖХим., 1973, 5П39.
1454. Allen J. D. J. Inst. Fuel, 46, No 384, 123 (1973).
1455. Butcher S., Ruff R. E. Analyt. Chem., 43, 1890 (1971).
1456. Cawse P. A. Analyst, 92, 311 (1967).
1457. Cleark A. L., Jennings A. C.
J. Agric. a. Food Chem., 13, 174 (1965).
1458. Comprchentive Analytical Chemistry. Eds. Wilson C. L., Wilson D. W. Amsterdam, Elsevier Publ. Comp., 1960, p. 467.
1459. Dimitriu A., Bugeag E. Dari seama sedint. Com. geol. RPR, 51, part 2, 217 [1963—1964 (1965)1; РЖХим, 1966, 22Г142.
1460. Dombrowski H. S. Analyt. Chem., 26, 526 (1954).
1461. Gael P. S. Proc. Chem. Symp., v. 2, 1969. Bombey, India, 1970, p. 85; РЖХим, 1971, 9Г136.
1462. Hargrove G. L., Shepard R. C., Farrar H. Analyt. Chem., 43, 439 (1971).
1463. Harman J. N., Neti R. M. USA Pat., No 3652227 (1972); РЖХим, 1973, 1И534П.
1464. Hassan S. S. M. Z. analyt. Chem., 255, 364 (1971).
1465. Keay J., Menage P. M. A. Analyst, 95, 379 (1970).
1466. Kneebone В. M., Freiser H. Analyt. Chem., 45, 449 (1973).
295
1467. Kiibler R., Padowetz W., Pavel J. Mikrochim. acta, 1972, 591.
1468. Laby R. H., Morton T. C. Nature (Engl.), 210, No 5033, 298 (1966).
1469. Macey D., Gilboy W. Nucl. Instrum. a. Methods, 92, 501 (1971).
1470. Malingerova N. Sb. Vysoke Skoly Zemed. Bme, A, Nol, 9 (1964); РЖХим, 1965, 12Г171.
1471. McClenny W., Hodgeson J. A., Bell J. P. Analyt. Chem., 45, 1514 (1973).
1472. Meyer R. A. J. Gas Chromatogr., 5, 486 (1967).
1473. Mori Masaki. J. Environ. Pol-lut. Contr., 9, 243 (1973).
1474. Premi P. R., Cornfield A. II. Analyst, 92, 196 (1967).
1475. Product. Group U. K. Atomic Energy Author. Rept. No225 (S), (1962); РЖХим, 1963, 5Г122.
1467. Rocco G. G., Garzon 0. G., Cali I. P. Intern. J. Appl. Radiat. Isotopes, 17, 433 (1966).
1477. Schluter E. C. Analyt. Chem., 31, 1576 (1959).
1478. Schbniger W. Advances in Analytical Chemistry and Instrumentation, v. 1. Ed. C. N. Reily. New York, Intersci. Publ., 1960, p. 196.
1479. Smals A. Lab. Pract., 16, 701 (1967).
1480. Treatice on Analytical Chemistry, part 2, v. 11. Eds. Kolthoff I. M., Elving P. J. New York, Intersci. Publ., 1965.
1481. Williams R. J. B. Chem. a. Ind., 1969, 1735.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбционный спектральный анализ 127 сл.
атомная абсорбция 127 сл.
молекулярная абсорбция 129 сл. атомная флуоресценция 129 ИК-спектроскопия 130 сл.
комбинационного рассеяния метод 130
УФ-спектроскопия 129 сл. Автоматические приборы для определения азота 117, 152, 182, 184—186, 190 сл., 205—208, 210, 221 сл., 225, 234 сл., 254, 256
Азокрасителей образование 84, 93 сл., 102
Азотистая кислота
отделение экстракцией 174 сл., 177 Азотистоводородная кислота определение
гравиметрическое 47 сл. титриметрическое
ацидиметрическое со щелочью 57
оксидиметрическое с нитратом 63 сл. перманганатом 63 церия(1У) солями 63 УФ-спектроскопическое 129 фотометрическое
по реакции с ионами Fe(III) 90 сл.
Азотная кислота определение титриметрическое 57 оксидиметрическое с железа(П) сульфатом 64 с электрохимической регистрацией КТТ кондуктометрически 82 отделение экстракцией 174 сл., 177
Азот молекулярный 7 сл., 147 сл., 153 сл.
атомная масса 7 взаимодействие с кислородом 16
металлами 16 сл.
изотопы 12 сл.
радиоактивные 12 стабильные 12
история открытия 7
круговорот в природе 9 сл. метастабильное состояние 16 нахождение в природе 7 сл. неорганические соединения с водородом 17 сл.
азотистоводородная кислота 24 амиды 21 аммиак 17 сл.
гидразин 22 сл. гидроксиламин 23 сл. имиды 21 нитриды 21 сл.
физико-химические свойства 19
кислородом 25
азотистая кислота 27 азотная кислота 27 сл. двуокись азота 25 сл. закись азота 25 окись азота 25 пятиокись азота 27 трехокись азота 25 физико-химические свойства 26
четырехокись азота 25 сл.
другими элементами 28 сл.
дициан 29
роданистоводородная кислота 31
физико-химические свойства 29
цианистый водород 30
циановая кислота 30 определение примесей 255 сл. положение в периодической системе элементов 7 промышленный метод получения 17 состояния окисления 11 спектральные аналитические линии 124
твердые растворы внедрения 16 сл.
297
физико-химические свойства 14 сл.
Азотометр 151 сл.
Активация
у-квантами 139
дейтронами 139
ионами 3Не 139
нейтронами 138 протонами 138 сл.
Аммиак 17 сл.
взаимодействие с водой 19 сл. кислородом 19 металлами 20 комплексы металлов с аммиаком
20
константы устойчивости 21 химико-аналитическая характери-
стика 18 сл.
физические свойства 17 сл.
Алмазы, определение азота 196 сл. Аммоний
обнаружение качественное едкими щелочами 32 сл. Несслера реактивом 33 определение
гравиметрическое 46
Несслера реактивом 47
нитробарбитуровой кислотой 46 сл.
платинохлористоводородной кислотой 46
тетрафенилбором 46 спектроскопическое
атомно-абсорбционное, косвенное 128
ИК-спектроскопическое 130 сл.
УФ-спектроскопическое 129 титриметрическое ацидиметрическое 54
Кьельдаля методом 54 сл. с сульфаминовой кислотой 55 сл.
формальдегидным методом 56 комплексометрическое с ионами меди 71 сл.
комплексонометрическое 72 оксидиметрическое с
гипобромитом 59 сл.
гипохлоритом 59
с электрохимической регистрацией КТТ 76 амперометрически 78 кондуктометрически 82 кулонометрически 80 фотометрическое
индотимоловый метод 88
индофенольный метод 86 сл.
Несслера метод 85 сл.
пиридин-пиразолоновый метод 89
электрохимическое кондуктометрическое 116 кулонометрическое 116 полярографическое 110 потенциометрическое (с ионоселективными электродами) 118
отделение и разделение отгонкой 178
хроматографией
бумажной 165 сл.
ионообменной 163 сл.
тонкослойной 169 сл.
экстракцией 174 сл., 177 Амперометрическое титрование 78 сл. Аргентометрия 74 сл., 77, 83, 223, 231 сл.
Атомно-флуоресцентный метод 129
Вакуум-плавление 238 сл., 250
Воды природные и производственные сточные, определение 208 сл.
азота органических веществ 233 аммония и аммиака 217 сл.
нитрат-ионов 227 сл.
нитрит-ионов 228 сл.
общего содержания азота 233 сл. роданид-ионов 232 сл.
цианид-ионов 229 сл.
Воздух, определение азота 205 сл. Волюмометрический метод 184 сл. Выделение азота
в виде аммиака 178 сл.
отгонкой миллиграммовых количеств 179 сл.
микрограммовых количеств 180 сл.
в виде молекулярного азота газохроматографическим мето-
дом 154, 157 сл., 182 сл.
методом Дюма 147 сл.
химическими методами 60 сл.,
62 сл., 70
Газовая хроматография 154, 157 сл., 182 сл.
Газо-жидкостная хроматография 116, 156, 178, 182 сл.
Газы и газовые смеси, определение азота 205 сл.
Гидразин
определение
гравиметрическое 47
косвенное с нитратом серебра 47
титриметрическое ацидиметрическое 56
Кьельдаля методом после превращения в аммиак 56
с фенилгидразином 57
298
с хлорной кислотой 56 оксидиметрическое с броматом 60 ванадатом 61 гипохлоритом 61 железа(Ш) солью 62 йодатом и иодом 60 монохлориодом 61 сл. нитритом 61 перманганатом 61 хлорамином Т 61 церия(Ш) сульфатом 61 с электрохимической регистра-
цией КТТ кулонометрически 80 сл. хронопотенциометрически
117
фотометрическое с
я-д иметил а минобенза ль дегидом 90 пикрилхлоридом 90 электрохимическое кулонометрическое 116 хронопотенциометрическое 117 Гидроксиламин определение титриметрическое оксидиметрическое с
броматом и гипобромитом 62
железа(Ш) солями 62 монохлориодом 63 титана(Ш) хлоридом 63 церия(1У) солями 62 сл. с электрохимической регистрацией КТТ амперометрически 78 кулонометрически 81 фотометрическое
по образованию окрашенного комплекса с V(V) и триэтаноламином 90 электрохимическое кулонометрическое 116 полярографическое 112 хронопотенциометрическое117 Горные породы, минералы, определение азота аммонийного 196 сл. нитратного 200 сл.
суммы аммонийного и нитратного 200
аммонийного, нитратного и нитритного 200
нитратного и нитритного 200 Грисса метод 84, 93 сл., 103, 253 Грисса—Илосвая метод 93
Двуокись азота
определение
спектроскопическое
ИК-спектроскопическое 131
У Ф-спектроскопическое 129 сл. титриметрическое
оксидиметрическое 64 фотометрическое 92 сл. электрохимическое
кулонометрическое 117
отделение
газохроматографическое 154 сл., 157 сл.
Деполяризации метод 83
Диазония соли образование 95 сл.
Дюма метод 147 сл., 184 сл., 194
Дюма—Прегля микрометод 148, 152, 194, 253
Закись азота
определение
гравиметрическое 53
ИК-спектроскопическое 131
отделение
газохроматографическое 154, 157 сл.
Изотопное разбавление стабильными изотопами 141 сл., 240
Изотопы азота 12 сл.
радиоактивные 12
стабильные 12
Индотимоловый метод 88 Индофенольный метод 86 сл. Ионных ассоциатов образование 84,
102
Ионоселективные электроды, определение 117 сл.
аммония 118
нитрат-ионов 118 сл.
нитрит-ионов 120 сл.
роданид-ионов 121 сл.
цианид-ионов 121
Карбиды, определение азота 242 Кинетический метод 90, 98 сл. Кондуктометрическое титрование
82 сл.
Константы устойчивости комплексов аммиачных 21 цианидных 30
Кулонометрическое титрование 80 сл.
Кьельдаля метод 54 сл., 178 сл., 181 сл.
Манометрический метод 154
Масс-спектальное определение азота 133 сл., 237
Материалы высокой чистоты, определение азота 242
Меркуриметрия 75
Металлы, сплавы, стали 234 сл.
методы выделения азота 235 сл.
299
анодное растворение 236
высокотемпературная экстракция 235 сл.
химические методы 236 сл.
методы определения азота 237 сл.
активационные 238 вакуум-плавление 238 сл. изотопное разбавление 240 сл. масс-спектральные 237 сл. спектральные 237 химические 237
Молекулярная абсорбция 129 сл.
ИК-спектроскопия 130 сл.
КР-спектроскопия 130 сл.
УФ-спектроскопия 129 сл.
Микрокристаллоскопические реакции 36 сл.
Несслера метод 84 сл.
Несслера реактива приготовление
86, 203, 226
Нитрат-ионы обнаружение качественное реакцией 33 сл., 39 сл.
восстановления до аммиака 34 нитрита 34
микрокристаллоскопической с нитроном 34 дифениламином 34 железа(П) сульфатом 33 марганца(П) хлоридом 34 медью и серной кислотой 33 определение
гравиметрическое 48 сл.
а-динафтодиметиламином 50 дициклогексилталлия(Ш) ионами 51
нитроном 48 сл.
га-толилизомочевиной 50 цинхонамином 51
спектроскопическое
атомно-абсорбционное, косвенное 129
ИК-спектроскопическое 130 сл.
КР-спектроскопическое 130 У Ф-спектроскопическое 130 титриметрическое ацидиметрическое 57 оксидиметрическое, восстановлением
ванадия(П) сульфатом 66 сл. железа(П) солями 64 сл. металлами 67 оксалатом 66
олова(П) солями 66 сульфаминовой кислотой 67 титана(Ш) солями 65 сл.
урана(1У) солями 67
хрома(П) солями 66 основанное на реакции оса-
ждения Pb(NOs)a 76 с электрохимической регистра-
цией КТТ амперометрически 80 кондуктометрически 82 потенциометрически 76 сл. фотометрическое с бруцином 99 сл. восстановлением до аммиака 102 нитрита 103 окиси азота 102
Грисса—Ромийна реактивом
га-диаминодифенилсульфоном 100 сл.
дифениламином 101 дифенил аминсульфокислотой 101
дифенилбензидином 101 комплексами меди(1) с 2,2'-дихинолилом и 2,3-диме-тил-1,10-фенантролином
кристаллическим фиолетовым 102
ксиленолами 100 метиленовым голубым 102 нильским голубым А 102 стрихнином 101 фенолдисульфокислотой 99 а-фурилдиоксимом 103 хромотроповой кислотой 101 электрохимическое
полярографическое 112 сл.
потенциометрическое (с ионоселективными электродами) 118 сл.
отделение и разделение хроматографией
бумажной 165 ионообменной 162 сл. тонкослойной 169 сл. экстракцией 177 электрофорезом 172 сл. Нитриды, определение азота 242 Нитрозосоединений образование 95 сл.
Нитрит-ионы
обнаружение качественное 33, 39 сл.
о-аминоанилидом бензолсульфокислоты 33
бензидином 33
иодидом калия 33 кобальта(П) нитратом 33 кислотами 3
300
перманганатом калия 33 сульфаниловой кислотой и а-нафтиламином 33
определение
гравиметрическое с амидосульфоновой кислотой, косвенное 52
броматом серебра 52 спектроскопическое
ИК-спектроскопическое 130
УФ-спектроскопическое 130 титриметр ическое
ацидиметрическое 58 сл.
оксидиметрическое с броматом калия 68 водорода перекисью 69 гидразином 70 гидроксиламином 70 гипохлоритом 67 сл. иодидом 68 перманганатом 68 сл. свинца(1У) тетраацетатом 69
титана(Ш) солями 70 феррицианидом 69 хлорамином Т 69 церия(1У) сульфатом 69
с электрохимической регистрацией КТТ амперометрически 78 сл. кондуктометрически 82 сл. кулонометрически 81 фотометрическое с
бриллиантовым зеленым 98 бруцином 96 сл.
2,3'-диаминонафталином 97 сл. К,К'-диметиланилином 95 сл. железа(П) сульфидом 98 кристаллическим фиолетовым 98
2,6-ксиленолом 96 риванолом 97
сульфаниловой кислотой 93 сл.
тиогликолевой кислотой 96 тиомочевиной 97
электрохимическое полярографическое ИЗ сл. отделение и разделение хроматографией
бумажной 165
ионообменной 162
тонкослойной 169 сл.
экстракцией 174 сл., 177 электрофорезом 172
Обнаружение качественное 32, 39 сл. аммония 32 сл. нитрат-ионов 33 сл. нитрит-ионов 33
роданид-ионов 34 сл.
цианид-ионов 35
Окись азота
определение
гравиметрическое 53
спектрофотометрическое 91 сл.
отделение
газохроматографическое 154, 157 сл.
Определение азота в алмазах 196 сл.
биологических материалах 254
взрывчатых веществах 210, 253
воздухе 205 сл.
водах природных и сточных 208 сл., 218 сл.
газах природных и промышленных 205 сл., 209 сл.
газовых смесях 205 сл., 209 сл.
горючих ископаемых 204 сл., 206 сл.
карбидах 242
кислотах 253
кормах 254
материалах высокой чистоты 242 металлах, сплавах, сталях 234 сл., 243 сл.
метеоритах 196 сл.
минералах 196 сл.
нефти и нефтепродуктах 204 сл., 206 сл.
нитридах 83, 242
окислах 242
органических соединениях 178 сл., 186
пищевых продуктах 254
полупроводниковых материалах 242
породах 196 сл.
почвах 196 сл.
растениях 254
рудах 196 сл.
серной кислоте 253
солях 253
стеклах 253
углях 204 сл., 206 сл.
удобрениях минеральных 82, 83, 120, 133, 191, 197, 253 сл. Органического вещества анализ 178 сл.
выделение аммиака 178 сл.
определение аммиака
титриметрическое 181 сл.
фотометрическое 181 сл.
определение молекулярного азота волюмометрическое 184 сл.
газохроматографическое 182 сл.
Пиридиниевых красителей образование 106
301
Пиридин-пиразолоновый метод 89
Полупроводниковые материалы, определение азота 242 сл.
Потенциометрическое титрование 76 сл.
Примесей определение
в азотной кислоте высокой чистоты 259 сл.
воды 259 сл.
фтористоводородной кислоты 260
в аммиаке жидком 257 воды 258 углекислоты 257 сл.
в газообразном азоте 255 водорода 256 воды 256 гелия 257 закиси азота 257 кислорода 255 окиси азота 257 окиси углерода 256
в четырехокиси азота 258 сл.
воды 258
двуокиси углерода 258 ионов металлов 259 окиси азота 259 органических веществ 258
Радиоаналитические методы 138, 193
Радиометрические методы 135 сл.
инструментальные 138 сл. косвенные 140
Радиохимические методы 139 сл.
Разложения методы металлов 235 сл. пород и минералов 201 сл. органических веществ 147, 178 сл.
Растворимость азота в воде 15 Реакции качественного обнаружения 32 сл., 39 сл.
Рентгенофазовый метод 133
Роданид-ионы
обнаружение качественное 34 сл., 39 сл.
железа(Ш) солями 34 кобальта(П) солями 34 меди(П) солями 34 натрия нитритом 35 пикриновой кислотой 35 реакцией образования берлинской лазури 35
ртути(П) нитратом 34 серебра нитратом 34 цинком металлическим 35 определение гравиметрическое, основанное на окислении SCN- до SO^_ и осаждении BaSOr 53 спектроскопическое
атомно-абсорбционное, косвенное 129
ИК-спектроскопическое 130 титриметрическое комплексонометрическое 72 оксидиметрическое 71 основанное на реакциях осаждения (аргентометрия) 74 сл.
с электрохимической регистрацией КТТ амперометрически 79 кондуктометрически 82 кулонометрически 81 потенциометрически 77 фотометрическое с бромфеноловым синим 109 ионами меди 109 кристаллическим фиолетовым
метиленовым голубым 108 сл. метилтимоловым синим 108 электрохимическое полярографическое 114 потенциометрическое (с ионо-селективными электродами) 121 сл.
отделение и разделение хроматографией бумажной 165 сл. ионообменной 162, 164 тонкослойной 169 сл.
экстракцией 176 сл. электрофорезом 172 сл.
Смеси азотсодержащих ионов и веществ, анализ аргона и азота 126 водорода, окиси углерода и азота 126
двуокиси и четырехокиси азота 93, 130
гидразина и аммиака 78 гидроксиламина и цианид-ионов 115
кислот азотной и азотистой 82 азотной и серной 82 азотной и хлорной 59 нитрат- и нитрит-ионов 58 сл., 67, 79, 81, 97, 100, 103, ИЗ сл.
роданид-ионов и тиомочевины 74 роданид- и цианид-ионов 79 селеноцианат- и цианид-ионов 79 цианид-, роданид-, цианат-ионов 79 сл.
Содержание азота в атмосфере 8 земной коре 8
302
Соединения азота, химические свойства азиды 24 азот молекулярный 15 сл. азотистая кислота 27 азотистоводородная кислота 24 азотная кислота 27 сл. амиды 21 сл. аммиак 18 сл. аммоний 20 гидразин 22 сл. гидроксиламин 23 сл. двуокись азота 25 сл. дициан 29 закись азота 25 имиды 21 окись азота 25 пятиокись азота 27 роданид-ионы 31 трехокись азота 25 цианид-ионы 30 четырехокись азота 25 сл.
Фазовый анализ 236 Физико-химические свойства азота молекулярного 14 сл. азотистоводородной кислоты 19 азотистой кислоты 26 азотной кислоты 26 аммиака 17 сл. гидразина 19 гидроксиламина 19 двуокиси азота 26 дициана 29 закиси азота 26 окиси азота 26 пятиокиси азота 26 роданистоводородной кислоты 29 трсхокиси азота 26 цианистого водорода 29 сл. циановой кислоты 29 сл. четырехокиси азота 26 Фиксация азота 10 Флуориметрические методы 107 сл.
Хемилюминесценция 161 Хроматография бумажная 165 сл. газовая 154, 157 сл., 182 сл. газо-жидкостная 116, 156, 178, 182 сл. ионообменная 162 сл. тонкослойная 169 сл.
Цериметрия 64 Цианид-ионы обнаружение по реакции капельной 45 образования цианистого водорода 35
определение спектроскопическое ИК-спектроскопическое 130
сл.
титриметрическое ацидиметрическое 59 комплексонометрическое 73 оксидиметрическое 71 основанное на реакциях осаждения (аргентометрия) 74 сл.
с электрохимической регистрацией КТТ амперометрически 79 деполяризации методом 83 кулонометрически 82 потенциометрически 77
фотометрическое
по образованию пиридиниевых красителей с аминами 106 по ослаблению светопоглоще-ния комплексными соединениями
ртути 104 серебра 105 тяжелых металлов 105 сл.
по реакции с
барбитуровой кислотой 107 органическими дисульфидами 106
флуориметричсское 107 электрохимическое
полярографическое 114 сл.
потенциометрическое (с ионо-селективными электродами) 121
отделение и разделение хроматографией бумажной 168 ионообменной 162, 164 тонкослойной 169 сл.
экстракцией 176 сл.
Четырехокись азота
определение
УФ-спектроскопическое 129 сл.
Экстракционно-спектрофотометрические методы определения аммония 86 сл., 88 нитрат-ионов 101, 102 нитрит-ионов 95, 97, 98, 101 роданид-иопов 108 сл.
цианид-ионов 104 сл., 231
Экстракция 174, 177
Электроаналитическпе методы амперометрия 78 сл. вольтамперометрия (полярография) 110
303
кондуктометрия 82 сл., 116
кулонометрия 80, 116
потенциометрия (ионоселективные электроды) 117 сл.
хронопотенциометрия 117
Электронно-зондовый микроанализ
132
Электронный парамагнитный резонанс 144 сл.
Электрофорез 172 сл.
Элементный органический анализ, определение азота 178, 186 сл.
Эмиссионный атомный анализ 124 сл.
вакуумная область спектра 124
дуговой разряд 126
искровой разряд 125 пламенная фотометрия 126 сл. тлеющий разряд 126
Ядерный магнитный резонанс 146
ОГЛАВЛЕНИЕ
От редколлегии................................................... 3
Предисловие...................................................... 5
Глава I
Общие сведения об азоте........................................ 7
Положение в периодической системе элементов. История открытия азота............................................................ 7
Нахождение в природе.........•................................... 7
Валентные состояния азота....................................... 11
Изотопы азота................................................... 12
Глава II
Химико-аналитическая характеристика азота и его соединений . . 14
Молекулярный азот............................................... 14
Неорганические соединения азота с водородом .................... 17
Неорганические соединения азота с кислородом.................... 25
Неорганические соединения азота с другими элементами............ 28
Глава III
Методы качественного обнаружения................................ 32
Аммоний (32). Нитрит-ионы (33). Ннтрат-ионы (33). Роданид-ионы (34). Цианид-ионы (35)
Глава IV
Гравиметрические методы......................................... 46
Неорганические соединения азота с водородом .................... 46
Аммоний (46). Гидразин (47). Азотистоводородная кислота (47)
Неорганические соединения азота с кислородом.................... 48
Нитрат-ионы (48). Нитрит-ионы (52). Окислы азота (53). Роданид-ионы (53)
Глава V
Титриметрические методы......................................... 54
Кислотно-основное титрование.................................... 54
Аммоний (54). Гидразин (56). Азотистоводородная кислота (57).
Нитрат-ионы и азотная кислота (57). Нитрит-ионы (58). Цианид-ионы (59)
Окислительно-восстановительное титрование....................... 59
305
Аммоний (59). Гидразин (60). Гидроксиламин (62). Азотистоводородная кислота (63). Двуокись азота. Азотная кислота (64).
Нитрат-ионы (64). Нитрит-ионы (67). Роданид-, цианид-ионы (71)
• Комплексометрическое титрование.............................. 71
Аммоний (71). Роданид-ионы (72). Цианид-ионы (73)
Методы титрования, основанные на реакциях осаждения............ 74
Роданид- и цианид-ионы (74). Нитрат-ионы (76)
Методы, основанные на электрохимической регистрации конечной точки титрования............................................... 76
Потенциометрическое титрование (76). Амперометрическое титрование (78). Кулонометрическое титрование (80). Кондуктометрическое титрование (82). Метод деполяризации (83)
Глава VI
Фотометрические методы......................................... 84
Аммоний (85). Гидразин (90). Гидроксиламин (90). Азотистоводородная кислота (90). Окислы азота (91). Нитрит-ионы (93).
Нитрат-ионы (99). Цианид-ионы (103). Роданид-ионы (108)
Глава VII
Электрохимические методы...................................... 110
Полярографический метод (вольтамперометрия).................... ПО
Аммоний (110). Гндроксиламин (112). Нитрат-ионы (112). Нитрит-ионы (ИЗ). Окислы азота (114). Роданид-ионы (114). Цианид ионы (114)
Кондуктометрический метод..................................... 116
Аммиак (116)
Кулонометрический метод....................................... 116
Аммиак (116). Гидразин (116). Гидроксиламин (116). Двуокись азота (117)
Хронопотенциометрический метод................................ 117
Ионоселективные электроды..................................... 117
Аммоний (118). Нитрат-ионы (118). Цианид-ионы (121). Роданид-ионы (121).
Глава VIII
Физические методы............................................. 123
Спектральные методы........................................... 123
Эмиссионный атомный спектральный анализ (124). Абсорбционный спектральный анализ (127)
Рентгеноспектральные методы................................... 132
Масс-спектральные методы...................................... 133
Изотопный анализ (133). Элементный анализ (135)
Активационный анализ.......................................... 135
Инструментальные методы (138). Радиохимические методы (139)
Изотопное разбавление стабильными изотопами.................. 141
Резонансные методы............................................ 144
Глава IX
Газометрические методы........................................ 147
Способы переведения различных соединений азота в измеряемую молекулярную форму.......................................... 147
Методы разложения образца при нагревании с окислителем (метод Дюма) (147)
306
Методы определения газообразных соединений азота............... 153
Манометрический и объемный методы (154). Газохроматографический метод (154)
Глава X
Методы разделения соединений азота в растворах................. 162
Хроматографические методы...................................... 162
Ионообменная хроматография (162). Хроматография на бумаге и в тонком слое сорбента (165)
Электрофоретические методы............................... 172
Экстракционные методы.................................... 177
Глава XI
Органический элементный анализ........................... 178
Выделение азота в виде аммиака........................... 178
Титриметрическое и фотометрическое определение аммиака .... 181
Газохроматографическое определение азота................. 182
Волюмометрическое определение азота...................... 184
Глава XII
Определение азота в природных и промышленных объектах .... 195
Минералы, руды, породы, почвы, алмазы, метеориты............... 196
Угли, горючие ископаемые, нефть и нефтепродукты................ 204
Воздух, природные и промышленные газы, газовые смеси........... 205
Природные и производственные сточные воды...................... 208
Аммоний и аммиак (217). Нитрат-ионы (227). Нитрит-ионы (228).
Цианид-ионы (229). Роданид-ионы (232). Азот органических веществ («органический азот») (233). Общее содержание азота («общий азот») (233)
Металлы, сплавы, стали......................................... 234
Методы выделения азота (235). Методы определения азота (237)
Прочие объекты................................................. 242
Глава XIII
Определение примесей в азоте и его соединениях................. 255
Газообразный азот.............................................. 255
Аммиак (жидкий)................................................ 257
Четырехокись азота (жидкая).................................... 258
Азотная кислота высокой чистоты................................ 259
Литература..................................................... 261
Предметный указатель........................................... 297
Аналитическая химия азотА. В. Ф. В о л ынец, М. П. В о л ынец. М., «Наука», 1977, стр. 307.
В монографии отражено современное состояние аналитической химии азота и его соединений, обобщен накопленный материал по идентификации, выделению и количественному определению различных химических форм азота при анализе природных и промышленных объектов. Наибольшее внимание уделяется физико-химическим экспрессным методам, особенно методам определения азота в металлах, природных водах, атмосфере и т. д. Приведены методы элементного анализа органических соединений на содержание азота. В связи со спецификой элемента в монографию введена глава по газометрическим методам определения азота. Рассматриваются вопросы изотопии, масс-спектрометрии азота в плане аналитической химии. Наиболее широко используемые методики определения даются полностью.
Монография рассчитана на широкий круг химиков-аналитиков, работников заводских и научно-исследовательских лабораторий, студентов и преподавателей вузов и техникумов.
Таблиц 29. Библ. 1481 назв.
Всеволод Федорович Волынец, Маргарита Павловна Волынец
Аналитическая химия азота
Серия: «Аналитическая химия элементов»
Утверждено к печати
Ордена Ленина институтом геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
Редактор издательства Р. А. Баранова Художественный редактор С. А. Литвак Технический редактор В. Д. Прилепская
Корректоры В. Г. Петрова, И. А. Талалай
Сдано в набор 13|Х-1976 г.
Подписано к печати 14'11-1977 г.
Формат бумаги 60х90‘|ц Бумага № 1
Усл. печ. л. 19,5 Уч.-изД. л. 21,3 Тираж 3300. Т-03339. Тип. зак. 1270
Цена 2 р. 04 к.
Издательство «Наука»
103717 ГСП, Москва, К-62, Подсосенский пер., 21
2-я типография издательства «Наука».
121099, Москва Г-99, Шубинский пер., 10
ИЗДАТЕЛЬСТВО «НАУКА»
Готовятся к выпуску следующие книги
СОВРЕМЕННЫЕ МЕТОДЫ АНАЛИЗА МИКРООБЪЕКТОВ И ТОНКИХ ПЛЕНОК. 22 л. (темплан выпуска литературы издательства «Наука» , 1976 г., кн. 2, Гй 127)
Сборник отражает последние достижения аналитической химии в области исследования микрообъектов, тонких слоев вещества и пленок. Рассматривается применение различных микрокомпонентов в современной технике. Излагаются возможности применения различных методов для исследования тонких слоев и пленок.
Книга рассчитана на химиков и геохимиков, занимающихся разработкой микрометодов, зондовых методов анализа пленочных структур, а также микрообразцов и включений в космохимических к геологических образцах.
Н. С. ФРУМИНА, Н. Н. ГОРЮНОВА, С. Н. ЕРЕМЕНКО. АНАЛИТИЧЕСКАЯ ХИМИЯ БАРИЯ, 15 л. (темплан выпуска литературы издательства «Наука»; 1976 г.; ки. 2, № 131)
В книге представлено современное состояние аналитической химии бария. Приведены физико-химические характеристики бария и его соединений. Рассмотрены различные способы определения и выделения бария из природных и промышленных объектов, а также определение примесей в самом барии.
Книга предназначена для работников химико-аналитических лабораторий научно-исследовательских институтов и предприятий, для преподавателей, аспирантов и студентов вузов.
Ю- А. ЗОЛОТОВ, Н. М. КУЗЬМИН. ЭКСТРАКЦИЯ МЕТАЛЛОВ АЦИЛПИРАЗОЛОНАМИ. 12 л. (темплаи выпуска литературы издательства «Наука»; 1977 г., кн. 2, JN1 105)
В монографии обобщены результаты исследования экстракции пятидесяти элементов при помощи ацилзамещенных пиразолона. Приведены сведения о самих реагентах и их свойствах, суммированы константы, характеризующие экстракцию металлов, подробно рассмотрена информация о поведении отдельных элементов. Представлены конкретные методы выделения и разделения металлов, в приложении даны наиболее интересные прописи.
Книга предназначена для химиков-аналитиков, радиохимиков и специалистов по экстракции.
Для получения книги почтой заказы просим направлять по адресу:
117464 МОСКВА, В-464, Мичуринский проспект, 12, магазин «Книга — почтой» Центральной конторы «Академкнига»;
197110 ЛЕНИНГРАД, П-110, Петрозаводская ул., 7, магазин «Книга — почтой» Северо-Западной конторы «Академкнига» или в ближайшие магазины «Академкнига».
АДРЕСА МАГАЗИНОВ «АКАДЕМКНИГА» :
480391 Алма-Ата, ул. Фурманова, 91/97
370005 Баку, ул. Джапаридзе, 13
320005 Днепропетровск, проспект Гагарина, 24
734001 Душанбе, проспект Ленина, 95
375009 Ереван, ул. Туманяна, 31
664033 Иркутск, 33, ул. Лермонтова, 289
252030 Киев, ул. Пирогова, 4
252142 Киев, проспект Вернадского, 79
277012 Кишинев, ул. Пушкина, 31
433900 Краматорск, ул. Марата, 1
443002 Куйбышев, проспект Лепина, 2
192104 Ленинград, Д 120, Литейный проспект, 57
199164 Ленинград, Универе, тет-ская наб., 5
199004 Ленинград, 9 линия, 16
103009 Москва, ул. Горького, 8
117312 Москва, ул. Вавилова, 55/7
630090 Новосибирск, Академгородок, Морской проспект, 22
630076 Новосибирск, 91, Красный проспект, 51
620151 Свердловск, ул. Мамина-Сибиряка,' 137
700029 Ташкент, ул. 50 лет Узбекистана, 11
700029 Ташкент, Л-29, ул. Ленина, 73
700100 Ташкент, ул. Шота Руставели, 43
634050 Томск, наб. реки Ушайки, 18
450075 Уфа, Коммунистическая ул., 49
450075 Уфа, проспект Октября,' 129
720001 Фрунзе, бульвар Дзержинского, 42
310003 Харьков, Уфимский пер., 4/6
ИСПРАВЛЕНИЯ И ОПЕЧАТКИ
Страница Строка Напечатано Должно быть
' 215 5 св. no2 NO,"
215 6 св. no2- no2
219 7 св. мг NO3~/.«e •msN 03~1мл
В. Ф. Волынец, М. П. Волынец Зак. 1270